/




















































































































































































































































































































































































































































Текст
Acad. prof. RALUCA RIPAN
Lector ION CETEANU
CHIMIA METALELOR
Volumul II
EDITURA DIDACTICA SI PEDAGOGICA BUCURE§TI-1969
Р. Рипан,
И. Четяну
НЕОРГАНИЧЕСКАЯ ХИМИЯ
ТОМ 2
ХИМИЯ МЕТАЛЛОВ
Перевод с румынского
канд. хим. наук Д. Г. БАТЫРА и
канд. техн, наук X. Ш. ХАРИТОНА
Под редакцией
академика В. И. СПИЦЫНА
и
канд. хим. наук И. Д. КОЛЛИ
ИЗДАТЕЛЬСТВО «МИР»
МОСКВА 1972
УДК 546
2-5-2
88-72
Учебник по неорганической химии, предназначенный для
учащихся химических техникумов и студентов нехимических
вузов. Его можно использовать также как руководство к прак-
тическим занятиям, а обширный и удачно подобранный факти-
ческий материал делает книгу ценным справочным пособием,
краткой энциклопедией для младших научных сотрудников
и лаборантов химических лабораторий. По широте охвата
и разнообразию рассматриваемых вопросов книга полезна
не только химикам, но и специалистам других областей —
физикам, биологам, медикам, минерологам.
Том 1 вышел в свет в 1971 г.
Редакция литературы по химии
ВВЕДЕНИЕ
ХАРАКТЕРИСТИКА ПЕРЕХОДНЫХ МЕТАЛЛОВ
Этот том посвящен переходным металлам, у которых валент-
ный электрон находится на d-орбитали предпоследнего элект-
ронного уровня.
ЭЛЕКТРОННОЕ СТРОЕНИЕ
После элемента кальция с атомным номером 20, у которого
4в-орбиталь полностью заполнена, не происходит заполнения
электронами 4р-орбиталей внешнего электронного уровня, а начи-
нается комплектование Зй-орбиталей предпоследнего уровня.
Поскольку у элементов периодической системы имеются только
пять Зй-орбиталей, после кальция следует I серия из десяти
переходных металлов с атомными номерами от 21 до 30.
В табл. 1 представлено распределение электронов на 3d-
и 48-орбиталях переходных металлов I серии.
Таблица 1
Атомный номер 21 22 23 24 25 26 27 28 29 30
Элемент Sc Ti V Cr Мп Fe Со Ni Си Zn
Количество элек- тронов на 3d- орбитали 1 2 3 5 5 6 7 8 10 10
Количество элек- тронов на 4s- орбитали 2 2 2 1 2 2 2 2 1 2
После заполнения электронами Sd-орбиталей, начиная от эле-
мента галлия с атомным номером 31, происходит заполнение
внешнего электронного уровня, т. е. 4р-орбиталей.
Начиная с элемента иттрия с атомным номером 39, последо-
вательно заполняются электронами 4d-op6HTann и, таким образом,
образуется II серия из десяти переходных металлов. Распреде-
ление электронов на 4d- и 5з-орбиталях представлено в табл. 2.
После элемента бария с атомным номером 56 следует элемент
лантан с атомным номером 57, который имеет следующее распре-
6
ХАРАКТЕРИСТИКА ПЕРЕХОДНЫХ МЕТАЛЛОВ
Таблица 2
Атомный номер 39 40 41 42 43 44 45 46 47 48
Элемент Y Zr Nb Mo Тс Ru Rh Pd Ag Cd
Количество элек- тронов на 4d- орбитали 1 2 3 5 5 6 7 10 10 10
Количество элек- тронов на бх- орбитали 2 2 1 1 2 1 1 0 1 2
деление электронов по орбиталям: l№-2s22p6 •3№3p63d10-4s24p64d10•
• 5s25p®5d1 -6s2. Оно аналогично распределению у элементов скан-
дия и иттрия с одним электроном на d-орбитали и двумя электро-
нами на s-орбитали высшего электронного уровня.
В отличие от первых двух серий в III серии переходных
металлов заполнение электронами 5й-орбиталей, начатое у эле-
мента с атомным номером(57 (лантан), продолжается у элемента
с атомным номером 72 (гафний) и включает последующие элементы,
в том числе ртуть с атомным номером 80.
У семейства из 14 элементов от церия (атомный номер 58)
до лютеция (атомный номер 71), называемых редкоземельными
металлами *, или лантанидами, не происходит заполнения элект-
ронами 5й-орбиталей, а заполняются электронами 4/-орбитали.
В табл. 3 представлено распределение электронов на 5d-
и 6$-орбиталях переходных металлов III серии.
Таблица 3
Атомныймномер 57 72 73 74 75 76 77 78 79 80
Элемент La Hf Та W Re Os Ir Pt Au Hg
Количество элек- тронов на Ы- орбитали 1 2 3 4 5 6 7 9 10 10
Количество элек- тронов на 65- орбитали 2 2 2 2 2 2 2 1 1 2
♦ В советской литературе — редкоземельные элементы.— Прим, перев.
ХАРАКТЕРИСТИКА ПЕРЕХОДНЫХ МЕТАЛЛОВ
7
Из IV серии переходных металлов известен только актиний
с атомным номером 89*, электронная структура которого
Is2 -2s22p6 -З^З^Зс/10 •4s24p64d104/14 •5s25d65d10 •6s26p66di -7s2 анало-
гична скандию, иттрию и лантану.
У переходных металлов валентный электрон располагается
па предпоследнем электронном уровне, который находится в ста-
дии заполнения. Электронная структура переходных металлов
обеспечивает два наиболее устойчивых состояния: первое — когда
d-орбитали предпоследнего электронного уровня (3, 4, 5, 6) пол-
ностью заняты электронами, как в случае цинка, кадмия, ртути
(элементы с 18 электронами на предпоследнем уровне); второе —
когда d-орбитали предпоследнего электронного уровня полузапол-
нены (т. е. содержат по одному электрону на каждой d-орбитали),
как в случае марганца, технеция и рения.
При переходе одного электрона с внешней s-орбитали на d-ор-
биталь, непосредственно предшествующую ей, для металлов Сг,
Си, Nb, Mo, Ru, Rh, Ag, Pt, Au достигается более устойчивое
электронное состояние. Переход электронов с внешней s-орбитали
на d-орбиталь предпоследнего электронного уровня осуществ-
ляется без больших изменений энергии, поскольку энергии этих
двух подуровней различаются незначительно.
В химических реакциях электроны d-орбиталей участвуют
после того, как оказываются использованными s-электроны внеш-
ней орбитали.
В образовании химических соединений могут участвовать все
или часть электронов d-орбиталей предпоследнего электронного
уровня атомов переходных металлов; образуются соединения,
соответствующие различным валентным состояниям. Переменная
валентность переходных металлов, за исключением металлов
побочных подгрупп III и II групп, является их характерным
свойством.
Металлы побочных подгрупп IV, V, VI и VII групп образуют
соединения в высшем валентном состоянии (которое соответствует
номеру группы) и в более низких валентных состояниях. Металлы
побочной подгруппы IV группы могут быть двух-, трех- и четы-
рехвалентными, металлы побочной подгруппы V группы — одно-,
двух-, трех-, четырех- и пятивалентными, металлы побочной под-
группы VI группы — двух-, трех-, четырех-, пяти- и шестивалент-
ными, металлы побочной подгруппы VII группы — двух-, трех-,
четырех-, пяти-, шести- и семивалентными.
Из металлов VIII группы максимальную валентность прояв-
ляют только рутений в RuO4 и осмий в OsO4 или OsF8. Шести-
* Курчатовий с атомным номером 104 и следующей электронной струк-
турой: Is2 •2s22p6 •3s23p63c?104s24p64(P0 4/14-5s25p65d10-6s26p66d2.7s2. — Прим,
перев.
Переходные
Период
Уровень
Валентный электрон
4 М L К 44,956 2 Sc 9 Скандий 8 2 21 47,90 2 Ti 10 Титан 8 2 22 50,942 2 V 11 Ванадий 8 2 23 51,996 1 Ст 13 Хром 8 2 24
5 О N М L К 88,905 2 Y 9 Иттрий 18 8 2 39 91,22 2 Zr 10 Цирконий 18 8 2 40 92,906 1 Nb 12 Ниобий 18 8 2 41 95,94 1 Мо 13 Молибден 18 8 9 42
С Р О N М L К 138,91 2 La 9 18 Лантан 18 8 2 57 178,49 2 Hf 10 32 Гафний 18 8 2 72 180,948 2 Та 11 32 Тантал 18 8 2 73 183,85 W 32 Вольфрам 18 8 2 74
7 Q р О N м L К (227) 2 Ас 9 18 Актиний 32 18 8 2 89
Группа Ш-Б IV-Б V-Б VI-Б
Таблица 4
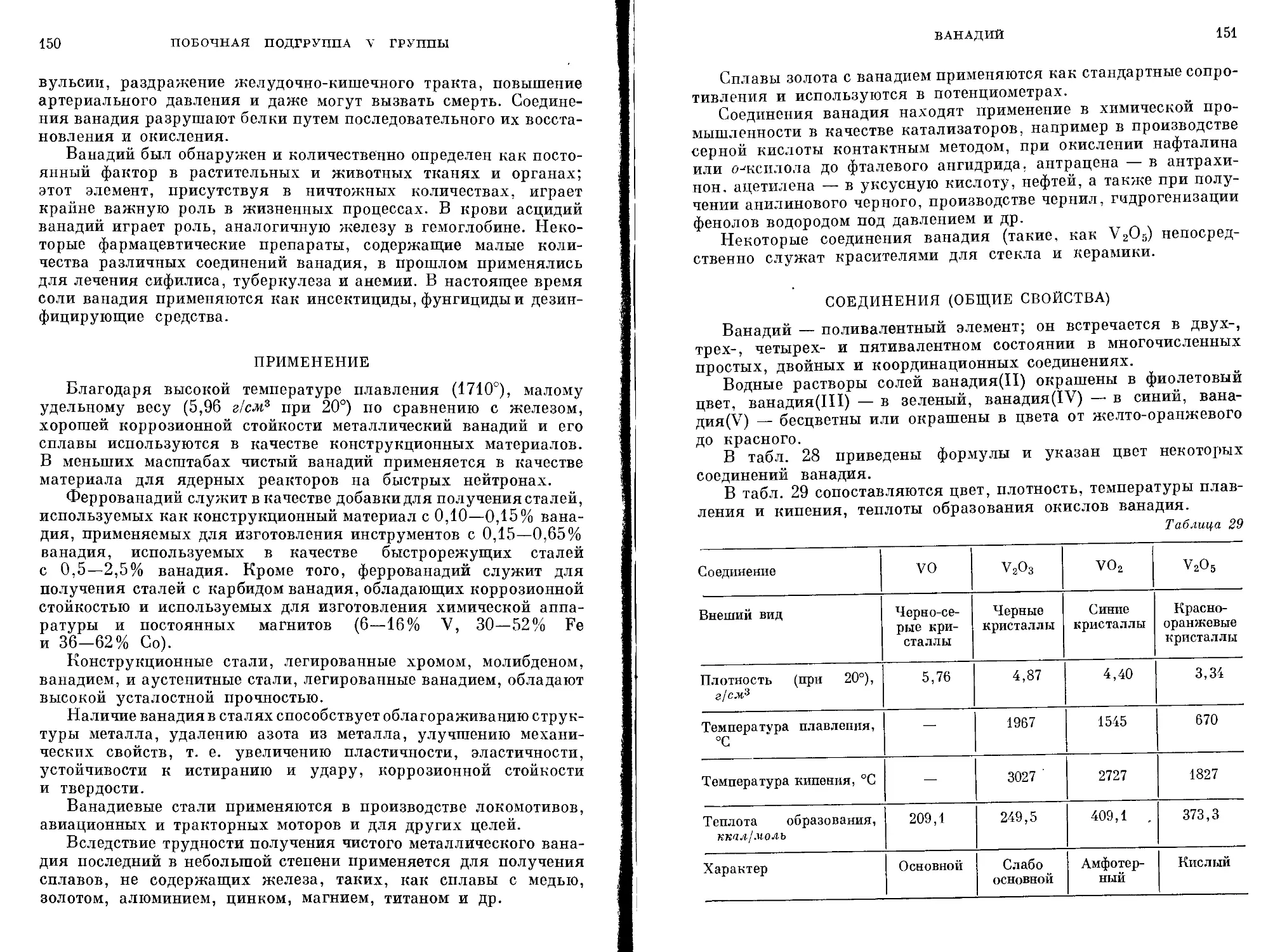
металлы
на d-орбитали
54,9381 2 Мп 13 Марганец 8 2 25 55,847 2 Fe 14 Железо 8 2 26 58,9332 2 Со 15 Кобальт 8 2 27 58,71 2 Ni 16 Никель 8 2 28 63,54 1 Си 18 Медь 8 2 29 65,37 2 Zn 18 Цинк 8 2 30
Тс 13 Технеций 18 8 2 43 101,07 1 Ru 15 Рутений 18 8 2 44 102,905 1 Rh 16 Родий 18 8 2 45 106,4 0 Pd 18 Палладий 18 8 2- 46 107,87 1 Ag 18 Серебро 18 8 2 47 112,40 2 Cd 18 Кадмии 18 8 2 4S
186,2 2 Re 13 32 Рений 18 8 2 75 290,2 2 Os 14 32 Осмий 18 8 2 76 192,2 2 1г 15 32 Иридий 18 8 2 77 195,09 1 Pt 17 32 Платина 18 8 2 78 196,967 1 Аи 18 32 Золото 18 8 2 79 200,59 2 Hg 18 32 Ртуть 18 8 2 80
VII-Б VIII 1-Б П-Б
10
ХАРАКТЕРИСТИКА ПЕРЕХОДНЫХ МЕТАЛЛОВ
валентными могут быть железо, рутений, родий, осмий, иридий,
платина. Четырех-, трех- и двухвалентными могут быть все
металлы этой группы. Одновалентными могут быть также все ме-
таллы этой группы, за исключением рутения и осмия.
Из всех переходных металлов только медь, серебро и золото
образуют соединения, в которых их валентность выше номера
группы.
Переходные металлы переменной валентности образуют ионные
соединения (с основными свойствами, восстановители) в низших —
одно-, двух- и трехвалентных состояниях. В высших —четырех-,
пяти-, шести- исемивалентных состояниях они образуют ковалент-
ные соединения (с кислотными свойствами, окислители), поскольку
катионы с большим зарядом и малым объемом сильно поляризуют
анион, обусловливая образование ковалентной связи.
Распределение электронов по уровням и атомным орбиталям
переходных металлов показано в табл. 4 и 5. Периодическое
повторение процесса заполнения электронами d-орбиталей пред-
последнего электронного уровня объясняет периодичность физи-
ческих и химических свойств переходных металлов.
КРИСТАЛЛИЧЕСКАЯ СТРУКТУРА
Переходные металлы Sc, La, Ac, Fe, Co, Rh, Ir, Ni, Pd, Pt,
Cu, Ag, Au кристаллизуются в кубической гранецентрированной
системе; металлы Ti, Zr, Hf, V, Nb, Ta, Cr, Mo, W, Fe при кристал-
лизации образуют кубическую объемноцентрированную решетку;
металлы Sc, Y, La, Ti, Zr, Hf, Cr, Mo, Tc, Re, Ru, Os, Co, Ni,
Zn, Cd имеют плотноупакованную гексагональную решетку;
металлы W, Мп образуют кубическую решетку особого типа,
а ртуть имеет ромбоэдрическую кристаллическую решетку (табл. 6).
Примерно 11 переходных металлов, а именно Sc, La, Ti, Zr,
Hf, Cr, Mo, W, Fe, Co, Ni, проявляют диморфизм, т. e. способ-
ность существовать в двух кристаллических формах; которые
имеют строго определенные температуры взаимного перехода и раз-
личную скрытую теплоту превращения. О типах кристаллических
решеток (гранецентрированной, объемноцентрированной и плот-
нейшей гексагональной) см. том I, стр. 9—17.
ВАЖНЕЙШИЕ ФИЗИЧЕСКИЕ, МАГНИТНЫЕ,
МЕХАНИЧЕСКИЕ И ЭЛЕКТРОХИМИЧЕСКИЕ СВОЙСТВА
ПЕРЕХОДНЫХ МЕТАЛЛОВ
Шкала атомных масс
В табл. 4 приведены значения относительных атомных масс
(атомных весов) переходных металлов по углеродной шкале изо-
топа ’2С — 12,00.
о '
Таблица 5
Переходные Металлы
Распределение электронов по уровням и орбиталям
K| L M | N 1 0 1 P | Q
ровсмь
1 1 2 3 1 4 1 5 1 6 1 7
Орбиталь s I s | p s | P | d | s | P I d I / I s P d | » | P | d | s | p
21 | Sc |2|2|6 2|6|1|2| 1 1 1 1 1 1 1 1
22 | Ti 2|2|6 2 1 6 1 2 1 2 1 1 1 1 1 1 1 1 1
23 | V 2|2|6 2 | 6 | 3 j 2 | 1 1 1 1 1 1 1 1
24 j Cr 2 | 2 | 6 2 | 6 j 5 | 1 1 1 1 1 Mill
25 | Мп 2 1 2 | 6 | 2 1 6 | 5 | 2 | Illi 1 1 1 1 1
26 | Fe 2 | 2 | 6 2 1 6 | 6 | 2 | 1 1 1 1 1 1 1 1
27 | Co 1 2 | 2 | 6 2 1 6 | 7 | 2 | 1 I- 1 1 1 1 1 1
28 j Ni 2 | 2 | 6 2 j 6 | 8 | 2 | 1 1 1 1 1 1 1 1
29 | Cu 1 2 | 2 | 6 2 | 6 110| 1 | 1 1 1 1 M 1 1
30 j Zn 1 2 | 2 | 6 2 | 6 110 | 2 | 1 1 1 1 1 1 1 1
39 | Y I 2 | 2 j 6 2 | 6 1101 2 | 6 [ 1 | — | 2 1 1 1 1 1
40 j Zr 1 2 | 2 | 6 2 | 6 1101 2 | 6 I 2 |-| 2 1 1 1 1 1
41 j Nb 1 2 | 2 | 6 2 | 6 110 j 2 j 6 I 4 |-| 1
42 | Mo 1 2 | 2 | 6 2 | 6 110| 2 | 6 | 5 |-| 1
43 | Tc 1 2 | 2 | 6 2 | 6 1101 2 | 6 | 5 | - | 2 1 1 1 1 1
44 | Ru 1 2 | 2 | 6 2 [ 6 1101 2 | 6|7|-|1
45 | Rh 1 2 | 2 | 6 2 j 6 f101 2 ( 6|8|-|1 1 1 1 1 1
46 | Pd 1 2 | 2 | 6 2 | 6 110 | 2 | 6 110 | - | 0 1 1 1 1 1
47 | Ag 2 | 2 | 6 2 j 6 110 | 2 | 6 |10| - 1 1 1 1 1 1 1
48 | Cd 2 | 2 | 6 2 | 6 | 10 | 2 | 6 110 | - | 2 1 1 1 1 1
57 j La 1 2 | 2 | 6 2 | 6 110 j 2 | 6 |Ю|-| 2 6 Ф1 | | |
72 | Hf 2 | 2 | 6 2 j 6 110 | 2 | 6 1101141 2 6 2 1 2 | | | |
73 | Ta 1 2 | 2 | 6 2 | 6 | 10 | 2 | 6 110114| 2 6 3|2| | | |
74 | W 1 2 | 2 | 6 2 | 6 110| 2 | 6 1101141 2 6 Ф1 | | |
75 j Re 1 2 | 2 | 6 2 | 6 |10j 2 | 6 |10|14| 2 6 5|2| | | |
76 | Os 2 | 2 | 6 2 | 6 [101 2 | 6 110114 | 2 6 6|2| | | |
77 | Ir 2 | 2 | 6 2 | 6 | 10 | 2 | 6 110 114 | 2 6 7 I 2 | | | |
78 | Pt 1 2 | 2 | 6 2 | 6 | 10 | 2 | 6 110114 j 2 6 9 1 1 1 1 1 1
79 | Au 1 2 | 2 | 6 2 j 6 j101 2 | 6 j10j14 j 2 6|10|l| I 1 |
80 | Hg 1 2 | 2 ] 6 | 2 | 6 1101 2 | 6 1101141 2 6 10|2| M 1
89 | Ac 1 2 | 2 | 6 2 | 6 110 | 2 | 6 110 114 | 2 6 10 | 2 | 6 | 1 | 2 |
12
ХАРАКТЕРИСТИКА ПЕРЕХОДНЫХ МЕТАЛЛОВ
Цвет, прозрачность и металлический блеск
В компактном состоянии (в виде блока, бруска, листа, про-
волоки, гранул и т. д.) металлы La, Ac, Ti, Zr, Hf, Nb, Ta, Mo,
W, Re, Ru, Os, Rh, Ir, Pd, Pt, Ag, Zn, Cd, Hg имеют сереб
ристо-белый цвет, металлы Sc, Y, V, Cr, Mn, Tc, Fe, Со, Ni —
серовато-белый, медь — медно-красный и золото — золотисто-жел-
тый (см. табл. 6). Будучи тонко измельченными, переходные метал-
лы обычно имеют черный или темно-серый цвет, за исключением
меди и золота, которые сохраняют окраску даже в пылевидном
состоянии.
Металлы непрозрачны, они не пропускают излучения в види-
мой области спектра (их валентные электроны полностью погло-
щают световую энергию в этой области спектра). Металлы обла-
дают характерным блеском благодаря отражению света от гладкой
и неокисленной поверхности.
Атомные радиусы
Приведенные в табл. 6 атомные радиусы (по Полингу) были
вычислены для координационного числа 12, чтобы их можно,
было сравнить между собой.
У переходных металлов значения атомных радиусов возра-
стают в пределах группы с увеличением атомного номера, умень-
шаются в периоде от побочной подгруппы III группы до VIII группы
и затем снова возрастают от VIII группы до побочной подгруп-
пы II группы.
На графиках зависимости атомных радиусов всех химических
элементов от атомных номеров видно, что переходные металлы,
обладающие малыми атомными радиусами, находятся на мини-
мумах кривой.
Ионные радиусы
В табл. 6 и 7 приведены значения ионных радиусов (по Аренсу)
(для электроположительных ионов переходных металлов в валент-
ном состоянии, соответствующем номеру группы, за исключением
Таблица 7
ЗНАЧЕНИЯ ИОННЫХ РАДИУСОВ (А)
Sc3+ Ti4+ V5+ Cr6+ Mn’+ Fe2+ Co2+ Ni2+ Cu+ Zn2+
0,81 0,68 0,59 0,52 0,46 0,74 0,72 0,69 0,96 0,74
уз+ Zr4+ Nb5+ Mo6+ Tc’+ Ru4+ Rh3+ Pd4+ Ag+ Cd2+
0,92 0,79 0,69 0,62 0,56 0,67 0,68 0,65 1,26 0,97
La3+ Hf4+ Ta3+ W6+ Re7+ Os4+ 1Г4+ Pt4+ Au+ Hg2+
1,14 0,78 0,68 0,62 0,77 0,65 0,68 0,65 1,37 1,10
Ас3+
1,18
ХАРАКТЕРИСТИКА ПЕРЕХОДНЫХ МЕТАЛЛОВ
13
элементов VIII группы). Ионные радиусы в группах возрастают
с увеличением атомного номера и уменьшаются в периодах с уве-
личением положительного заряда.
Радиусы катионов меньше соответствующих атомных радиусов,
они зависят от координационного числа кристаллической решетки
и от поляризации ионов.
Атомный объем. Плотность
Атомный объем, выраженный отношением атомной массы к плот-
ности элемента, только приблизительно отражает действительный
объем атомов, поскольку он включает и межатомное пространство
(в различных агрегатных состояниях элементов) при темпера-
туре измерения плотности.
В табл. 6 приведены значения атомных объемов переходных
металлов при температуре 20° (в см3/г-атом). Атомные объемы
переходных металлов возрастают в пределах группы с увеличе-
нием атомного номера. Наименьшие значения атомных объемов
у переходных металлов VIII группы.
Плотность, или удельный вес —это отношение атомной массы
к атомному объему, выраженное в граммах на 1 см3. По данным
табл. 6 можно проследить за изменением плотности переходных
металлов (измеренной при 20°) как в пределах группы, так и пери-
ода .
Температура плавления. Температура кипения.
Изменения температур плавления и кипения переходных метал-
лов в пределах группы и периода характеризуются данными
табл. 6.
Большинство переходных металлов обладают высокими темпе-
ратурами плавления и кипения, за исключением ртути — жидкой
в обычных условиях, а также кадмия, цинка, лантана и серебра,
температура плавления которых ниже 1000°. Самым тугоплавким
и высококипящим переходным металлом является вольфрам.
В процессе изучения переходных металлов были обнаружены
явления превращения порошков некоторых тугоплавких металлов
в компактную металлическую массу.
Потенциалы ионизации
В табл. 8 приведены значения первого, второго и т. д. потен-
циалов ионизации переходных металлов, выраженные в электрон-
вольтах. Можно легко проследить за изменением первого, второго
и т. д. потенциалов ионизации в подгруппах и периодах пере-
ходных металлов.
Оо
ч
Ьч
ЗНАЧЕНИЯ ПЕРВОГО, ВТОРОГО, ТРЕТЬЕГО И Т. Д. ПОТЕНЦИАЛОВ ИОНИЗАЦИИ (ев)
Ф СО ф 111 05 Ь- 05 44 СО 3 NJ О t— Ф^О^4^ ] J ) 1 СО со о? 44 СО о СО КО со III) о со сч 44 44 СО Ы) К Первый потенциал ионизации Второй » » Третий » » Четвертый » » Пятый » » Шестой » » Седьмой » »
СЧ Q5 со 1111 t- О СО СЧ со 3 и Г- СО СЧ ко^оо. |||| СМ СО ьп < 9,22 20,5 30,46 Au —
СОЮ ЮООО СО 4< ’НО 4< ф t— 00 1Л СО О СЧ I тчСОЮО^ч 1 СОСЧСЧГ— СО 05 I?- СО 05 со 05 СМ со КО 05 1 44 СО к? СО со 1 чз с. СО КО СО Ф К-О ко 4 со со 05 со со -4 ю I 4- СЧ *СГ КО г- 1 £
ЮЮФООО оо о сч сч Ф Г— г- СО СО СМ 00 I ^союссо 1 о о со о- ко со <^Ф Ф СО t-СЧ г- со н ю со КО 1 44 со со со 1 -Й cd сч Ф СО Г— Ф Г— СЧ | 44 СЧ СО КО с— 1 44
О 00 ** о о о Ф 44 еО ф ф О Г*СООЮ QsJ1 1 44 СО кО t— Ф 1 ф U-i со с© со сч СО С— М* КО 05 со г- со со со СЧ о | 44 СЧ СО СО 1 3 cd со ко ко Ф СО 1 44 СЧ ко со [ о
со со Ф S? СО Г— КО со СО Ф ’Г-» 1 44 СО кО СО Ф 1 й 00 с© сч сч Г- ко 05 СО 05 со 5 44 СЧ S? КО Г— I £ 00 со г- со со СО 44 кО I 4- сч СО ко со 1 ф cd ф КО СО СО ^ч^ня-нкОСОС-С- V- СО г- О ко 44 СЧ 44 СЧ КО Г- со о S
СО 05 КО Г- Kf 05 05 Vt* О СО со со Ф ф сч со t— 44 СО КО Г- 05 со о О ко со со 44 44 44 КО СО Г— t— г— СО Г* ф КО 44 сч 44 СЧ NT ко ь- со о СО Ф Ср со 05 Г- Ф СО f- t- К.О СО | 44 сч СО NT со 1
“<? о -г-< ьО О о с— 44 со со с— оо СО NT СОСО СОСЧ | 44 СМ -<г со со 1 > СО СЧ -<г ф о о со со ф г— ф со СО кО Г* 44 СО I 44 сч со КО О 1 л Z СО Г-СО со сч сч о Г— СО сч СО 1 1 I 44 сч со 1 1 1 н
00 КО СО М4 СО СО Г- СО 05 1 1 44 04 05 1 1 н СО СО со со 44 05 СО со СО СО СЧ СЧ 1 < 44 сч со со 1 1 N ф ф ф О CJ5 ф О 1 1 1
t- со 00 СО 1 1 1 о се ООсОО ко со ко СО СЧ О 44 | I I 44 сч со 1 1 ! 05 СО Ф С> КО СО 44 КО ^'ss 1 1 1 св Ь-З Ас (49)
ХАРАКТЕРИСТИКА ПЕРЕХОДНЫХ МЕТАЛЛОВ
15
В табл. 6 приведены общие характеристики групп переходных
металлов и первый потенциал ионизации (в ккал/моль) для каж-
дого металла.
Электропроводность и теплопроводность
В табл. 6 приведены данные о изменении проводимости пере-
ходных металлов в пределах групп и периодов.
Величина, обратная удельному сопротивлению (р), называется
удельной электропроводностью (у) и имеет размерность ом-1 -см'1.
В таблице в качестве общей характеристики переходных метал-
лов побочных подгрупп приводятся значения р-106 при 0°. Наибо-
лее высокой электро- и теплопроводностью обладают Ag, Си,
Au, Rh, Ir, Zn, Со, Ni. Для металлов Hg, Sc, La, Y, Ti, Zr, Hf.
V, Re, Мп и т. д. характерны высокое электросопротивление
и плохая теплопроводность.
Магнитные свойства
По величине удельной магнитной восприимчивости (/s-10"6
в электромагнитных единицах при 18°) переходные металлы
(см. табл. 6) делятся на ферромагнитные — Fe, Со, Ni, пара-
магнитные — Мп, Pd, V, Ст, Тс, W, Nb, Ti, Rh, Pt, Ir, Mo, Ta,
Ru, Re, Os, Sc, Y, La и диамагнитные — Cu, Ag, Au, Zn, Cd,
Hg, Zr.
Парамагнетизм переходных металлов выше, чем у металлов
главных подгрупп, поскольку в электронной конфигурации этих
элементов имеется большее число неспаренных электронов
с нескомпенсированным спином, парамагнетизм которых сумми-
руется с парамагнетизмом электронов, участвующих в образовании
металлических связей.
Количество электронов, которое участвует в образовании связей в метал-
ле и представляет валентность, изменяется от 1 до 6 и для переходных метал-
лов I серии имеет следующие значения:
Sc Ti V Cr Мп Fe Со Ni Си Zn
3 4 5 (Г 5 4
Из этих данных следует, что максимальное число металлических связей
образуют переходные металлы VI и VII побочных подгрупп и VIII группы,
для которых характерны малые атомные радиусы, большие плотность и твер-
дость, высокие температуры плавления и кипения, низкая летучесть, боль-
шое сопротивление разрыву и сжатию.
Парамагнитные металлы при определенной температуре, наз-
ванной Нэелем (1951 г.) температурой антиферромагнитного пере-
хода, обладают максимумом магнитной восприимчивости и ста-
новятся антиферромагнитными.
16
ХАРАКТЕРИСТИКА ПЕРЕХОДНЫХ МЕТАЛЛОВ
При нагревании ферромагнитных металлов до определенной
температуры, называемой точкой Кюри, ферромагнитные свойства
исчезают. Точка Кюри для железа равна 768°, для кобальта 1150°,
для никеля ~ 350°. В точке Кюри электроны с параллельно
ориентированными спинами в результате теплового движения
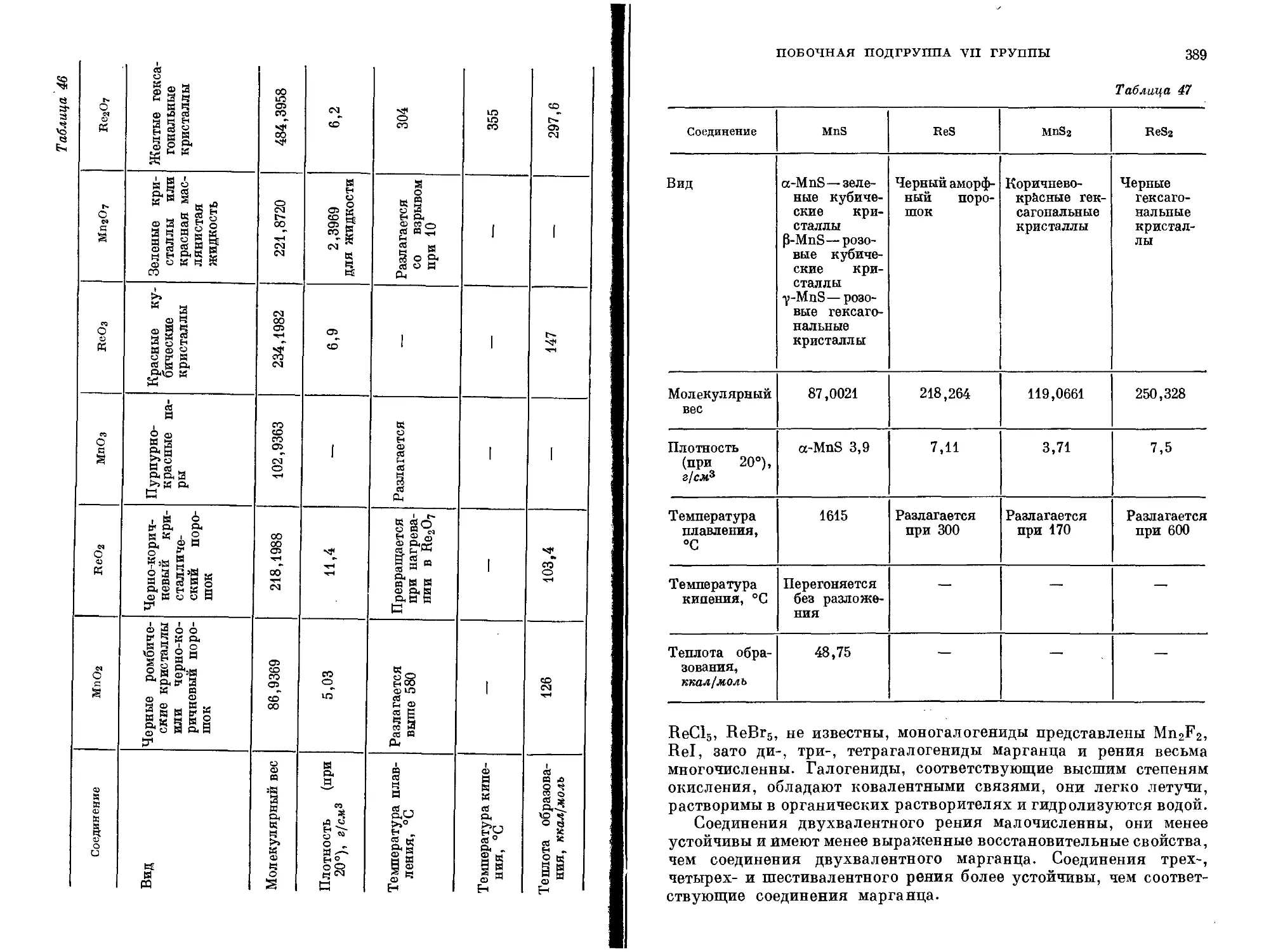
возвращаются в присущее им нормальное состояние неупорядо-
ченной ориентации, когда их магнитные моменты компенсируются
в большей степени, и металл становится парамагнитным. Поскольку
в точке Кюри проявляется непоследовательность и в изменении
других свойств, считают, что ферромагнетизм — свойство кристал-
лической структуры, а не атомов соответствующих металлов.
Механические свойства
По механическим свойствам (твердости, пластичности, ковко-
сти, тягучести, вязкости, эластичности, сопротивлению разрыву
и т. д.) переходные металлы и их сплавы можно классифицировать
самым различным образом.
Пользуясь табл. 6, можно судить об изменении твердости
(по шкале Мооса) переходных металлов в пределах группы
и периода.
В качестве примера мягких переходных металлов можно наз-
вать Hg, Cd, Au, Ag, Cu, Pt и др.; высокой твердостью обладают
Re, Os, Ru, Ir, V, Rh, Та. Хорошую ковкость обнаруживают
Cu, Ag, Au, Pt, Ni. Все переходные металлы и их сплавы имеют
особое значение в современной технике.
Электрохимические свойства
В табл. 9 представлен ряд электрохимических потенциалов
переходных металлов, установленный по стандартному потенциалу
окисления. Зная положение металла в ряду, можно точно харак-
теризовать отношение переходных металлов к воздуху, воде,
кислотам, щелочам, неметаллам.
Металлы, расположенные в начале ряда, химически более
активны, неустойчивы, легче окисляются (растворяются в раз-
бавленных кислотах), образуют соединения, которые с трудом
восстанавливаются, легко вытесняют металлы, стоящие в ряду
после них, из растворов их солей.
Металлы, расположенные после водорода, химически менее
активны, труднее окисляются (растворяются только в активных
окислительных средах при нагревании) и образуют химические
соединения, которые относительно легко восстанавливаются
до металла.
Металлы, проявляющие переменную валентность, в низшей
степени окисления образуют соединения восстановительного харак-
Таблица 9
РЯД ЭЛЕКТРОХИМИЧЕСКИХ ПОТЕНЦИАЛОВ
Система Нормальный потенциал, в (при 25°)
Ас + s Ac3+ + 3e_
La ? iLa3+ + 3e-
Y?--> = Y3+ + 3e-
Sc=₽ r- Sc3+-4-3e_
Hf? Hf4+ + 4e_
Ti <=s Ti3+-!-3e-
Zr?= t Zr4+ + 4e_
V?-> :V2++2e-
Mn = Mn2++ 2e_
Nb = =iNb3+4-3e-
V?± : уз+_|_3е-
Cr? ± Cr2+ + 2e~
Zn? Zn2++ 2e~
Cr? ?Cr3+ —3e~
Fe? *Fe2+4-2e-
Cd? ±Cd2+ + 2e-
Re? * Re3+ + 3e~
Co? =sCo2+ + 2e“
Ni?*Ni2+ + 2e-
Tc? =±= Tc2+ + 2e-
Mo = f2:M03+ + 3e_
H2? =±2H+ + 2e-
Fe? * Fe3+ + 3e_
W? * W3+ + 3e-
Cu = ^Cu2+ + 2e-
Co? ^=Co3+ + 3e-
Ru = r±Ru2+ + 2e-
Mil: ?± Mn3+ + 3e_
Cu? =ьСи+-|-е_
Rh = f±Rh2+ + 2e'
W? j: W3+ + 6e-
Rh = Rh3+ 3e~
Os? ^Os2+ + 2e-
Ag? =* Ag+ + e~
Pd? F±Pd2+ + 2e-
Hg = r*Hg2+ + 2e-
IT ?-! = Ir3+-|-3e_
Pt? sPt2+ + 2e-
Au = Au3+ + 3e"
Au- pt Au+ + e~
2-0101
—2,60 —2,52 —2,37 —2,08 И о 4 п о X S S к о 2 св П X
-1,70 к о л и св ч п
-1,63 * 3 X И Ф св X
—1,56 x ч св св g
—1,18 -1,18 5 ч П св 2 S X И S р- о и 3
—1,10 ф X 5
—0,87 -0,86 -0,761 d я вок S tr ф о S Рч воздухе X X о
—0,74 к й св X ' 2 св
-0,44 л о И X ф 5 п X
-0,402 -0,3 —0,277 О св СЦ емлени исляют § св X св Н О
—0,25 -0,24 £ £ © ч S & Я и о st стр X о о § й и
-0,20 0,000 о 2 л « X X п тах нагр НИИ растас Окис,
+0,036 и св го ©
+0,11 +0,346 ислот ь и В
+0,40 X О
+0,45 +0,47 +0,522 ХИЙКИВЕ сительно воздухе О X ф
+0,60 X X о св X ч
+0,68 X о ч о К X о
+0,70 м о X о с+ о © i
+0,70 +0,779 'ОЛЬК элект ИСЛЯ1 п X § св
+0,83 X X о X св
+0,854 +1,0 >яютс бывав ф к восст
+1,2 п >» 3 ч
+1,5 н о X
+1,7 св X < о
трудо:
18
ХАРАКТЕРИСТИКА ПЕРЕХОДНЫХ МЕТАЛЛОВ
тера с основными или амфотерными свойствами и химическими
связями главным образом ионного типа. Для соединений высших
степеней окисления характерны кислотный характер, окислитель-
ные свойства и ковалентные химические связи.
РАСПРОСТРАНЕННОСТЬ В ПРИРОДЕ
Химический состав твердой части земной коры может быть
выражен в весовых процентах, называемых показателями (пара-
метрами) Кларка, или кларками, и в атомных процентах.
Таблица 10
РАСПРОСТРАНЕННОСТЬ ПЕРЕХОДНЫХ МЕТАЛЛОВ
Атомный номер Металл Вес. % Атомный номер Металл Вес. %
26 Железо 4,70 48 Кадмий 5,0-10-4
22 Титан 0,65 78 Платина 5,0-10-з
25 Марганец 0,10 41 Ниобий 3,2-Ю-з
40 Цирконий 0,025 73 Тантал 2,4-Ю-з
30 Цинк 0,020 47 Серебро 1,0-10-5
29 Медь 0,01 44 Рутений 5,0-10-6
24 Хром 3,3-10-2 46 Палладий 5,0-10-6
23 Ванадий 2,0-10-2 76 Осмий 5,0-10-6
28 Никель 1,8-10-2 77 Иридий 5,0-10-6
74 Вольфрам 7,0-10-3 80 Ртуть 5,0-10-6
39 Иттрий 5,0-10-3 45 Родий 1,0-10-в
27 Кобальт 4,0-10-з 79 Золото 5,0-10-’
42 Молибден 1,0-10-з 75 Рений 1,0-10-7
57 Лантан 6,5-10-* 89 Актиний 2,3.10-15
21 Скандий 6,0-10-4 43 Технеций —
72 Гафний 4,0-10-4
Данные табл. 10 характеризуют распространенность переход-
ных металлов, выраженную в весовых процентах.
ПОБОЧНАЯ ПОДГРУППА III ГРУППЫ
|ПЕРИОДИЧЕСКОЙ<СИСТЕМЫ
^ПОДГРУППА СКАНДИЯ)
ХАРАКТЕРИСТИКА ПОДГРУППЫ
В побочной подгруппе III группы периодической системы
(соответственно в подгруппе скандия) находятся четыре переход-
ных металла: скандий (Sc), иттрий (Y), лантан (La), актиний (Ас),
14 лантанидов и 14 актинидов.
Лантаниды (редкоземельные элементы): Се, Pr, Nb, Pm, Sm,
Eu, Gd, Tb, Dy, Ho, Er, Tm, Yb, Lu и актиниды: Th, Pa, U,
Np, Pu, Am, Cm, Bk, Cf, Es, Fm, Md, No, Lw, обладая электрон-
ной структурой, отличной от электронной структуры четырех
переходных металлов, будут изучаться отдельно.
Раньше окислы элементов скандия, иттрия, лантана и ланта-
нидов называли редкими землями в отличие от обычных земель,
содержащих окись алюминия А12О3.
Электронная структура атомов скандия, иттрия, лантана
и актиния показана в табл. 11.
Таблица 11
Атомы этих элементов имеют по два электрона на «-орбитали
последнего электронного уровня. На s-, р- и d-орбиталях пред-
последнего уровня находятся 2, 6 и 1 электрон соответственно.
Третий внешний уровень атома иттрия, лантана и актиния содер-
жит 18 электронов, скандия — 8 электронов.
По сравнению с атомами щелочноземельных металлов атомы
металлов подгруппы скандия имеют избыточный электрон, кото-
рый располагается не на внешней р-орбитали по отношению
2*
Таблица 12
Элемент Скандий Sc Иттрий Y Лантан La Актиний Ас
Цвет Серовато-белый Серовато-белый ^Серовато-белый Серовато-белый
Кристаллическая структура a-Sc плотнейшая гексагональная; P-Sc кубическая гранецентриро- ванная Плотнейшая гек- сагональная a-La плотнейшая гексагональная; p-La кубическая гранецентриро- ванная Кубическая гра- нецентрирован- ная
Атомный номер 21 39 57 89
Атомный вес 44,956 88,905 138,91 227,035
Атомный радиус (по Полингу) 1,654 1,778 1,885 1,89
Радиус иона М3+, А (по Гольдшмидту, Полингу и Аренсу) 0,83; 0,81; 0,81 1,06; 0,93; 0,92 1,22; 1,15; 1,14 1,11; —; 1,18
Атомный объем (при 20°), смё/г-атом 14,74 19,886 22,45 —
Плотность (при 20°), г/см3 2,99 4,48 6,18 10,1
Температура плавления, °C 1539 1509 920 1050
1
Скрытая теплота плавления, кал/г 3,8 2,7 1,5 —
Температура кипения, °C 2727 2927 3469 3300
Удельная теплоемкость (при 20°), кал/г- град 0,148 0,071 0,047 —
Магнитная восприимчивость (при 18°) %s-10-6, эл.-магн. ед. Очень слабо пара- ми! нитпый Очень слабо пара- Mai нитный Очень слабо пара- магнитный Очень слабо пара- магнитный
Теплота перехода атомов в газообраз- ном состоянии, ккал/г-атом 93 103 88 —
Потенциал Me —> Ме+-|-е_ ионизации, Ме+ —> Me2 *-)-е- 'в Ме2+ —> Ме3+-|-е- Ме3+ —>- Ме4+~! е_ 6,7 12,8 24,8 73,6 6,50 12,30 20,46 61,50 5,59 11,38 19,10 52,50 (49)
Потенциал Me —> Me+ fe- ионизации, Ме+ —>- Ме2+-|- е“ ккал/г-атом Ме2+ —>- Ме3+-|-е” Ме3+ —>- Ме4+-|-е_ 154,5 295 571,8 1697 150 283,7 470 129 262,5 440,1 —
Нормальный потенциал для Ме/Ме3+ (при 25°), в -2,08 — 2,37 -2,52 — 2,60
Валентность III III III III
Массовые числа природных изотопов 45 89 139 227 и 228
Распространенность элементов в зем- ной коре, вес. % 6,0-10-4 5,0-10-3 6,5-10-4 2,3-10-45
22
ПОБОЧНАЯ ПОДГРУППА III ГРУППЫ
к валентным электронам, а на внутренней d-орбитали предпо-
следнего электронного уровня.
В химических реакциях атомы скандия, иттрия, лантана и акти-
ния отдают 3 электрона (2 электрона с внешней s-орбитали и 1 избы-
точный электрон с d-орбитали), образуя соединения, в которых
они проявляют степень окисления, равную трем. Электроположи-
тельные ионы элементов подгруппы скандия имеют электрон-
ную конфигурацию инертного газа и проявляют некоторое сход-
ство с ионами щелочноземельных металлов при заряде ядра на
единицу больше.
В табл. 12 приведены важнейшие константы скандия, иттрия,
лантана и актиния.
Элементы побочной подгруппы III группы периодической
системы — белые блестящие металлы, достаточно тугоплавкие,
очень слабо парамагнитные, с малым числом естественных изото-
пов. Кристаллические решетки иттрия и «-модификаций скандия
и лантана построены по типу плотнейшей гексагональной упа-
ковки, а Ас и fJ-формы Sc и La имеют кубическую гранецентри-
рованную решетку. Скандий и иттрий — легкие металлы, лантан
и актиний — тяжелые.
По физическим свойствам скандий и иттрий близки к тяжелым
редкоземельным элементам, а лантан и актиний — к легким ред-
коземельным элементам —металлам цериевой группы.
Скандий, иттрий и лантан мало распространены в природе
(на Скандинавском полуострове, в Бразилии, СССР, Австралии)
и встречаются в виде соединений в различных минералах вместе
с редкоземельными элементами и элементами побочной подгруппы
IV группы периодической системы.
В отличие от скандия, иттрия и лантана актиний радиоактивен
и встречается в минералах урана или тория наряду со свинцом,
висмутом, радием, протактинием, кальцием, барием и лантанидами
подгруппы церия. *
С химической точки зрения скандий, иттрий, лантан и актиний
(похожие на щелочноземельные металлы) являются активными
металлами: они окисляются во влажном воздухе при комнатной
температуре, превращаясь в соответствующие гидроокиси,
и растворяются в разбавленных кислотах с образованием солей
и выделением водорода. При нагревании скандий, иттрий и лантан
взаимодействуют с кислородом, серой, азотом, углеродом, крем-
нием, бором ит. д., образуя соединения типа Ме2О3, Me2S3, MeN,
МеС2, Ме4С3, MeSi2, МеВ6.
Электроположительные свойства металлов подгруппы скандия
усиливаются с увеличением атомного номера. В чистом виде
скандий, иттрий, лантан и актиний получить сравнительно трудно.
Обычно они образуются при электролизе расплавленных галоге-
нидов.
ПОБОЧНАЯ ПОДГРУППА Ш ГРУППЫ
23
Таблица 13
Соединение Sc»O3 y203 La2O3 Ас2О3
Вид Белый поро- шок или бес- цветные ку- бические кристаллы Белый поро- шок или бес- цветные ку- бические кристаллы Белый поро- шок или бес- цветные ку- бические ли- бо гексаго- нальные кристаллы Белый поро- шок или бес- цветные гек- сагональные кристаллы
Молекулярный вес 137,910 225,808 325,818 502,068
Плотность (при 20°), г/ыи3 Крист. 3,86 Крист. 4,84 Крист. 6,57 Крист. 9,19
Температура плав- ления, °C — 2417 2317 —
Температура кипе- ния, °C 4297 4197 —
Теплота образова- ния, ккал/моль 411 420 428,6 444
Растворимость в кислотах При нагре- вании Хорошая Хорошая Хорошая
Соединения металлов подгруппы скандия напоминают по свой-
ствам соединения щелочноземельных металлов, хотя их состав
аналогичен соответствующим соединениям алюминия.
В табл. 13—15 приведены наиболее важные константы окислов
и галогенидов элементов подгруппы скандия.
Окислы Sc2O3, У20з, La2O3 получают прокаливанием соответ-
ствующих гидроокисей, нитратов, карбонатов, оксалатов. Все
они бесцветны, тугоплавки и растворимы в разбавленных кислотах.
Гидроокиси Sc(OH)3, Y(OH)3, La(OH)3 получаютв виде студне-
образных белых осадков при обработке растворов солей скандия,
иттрия, лантана щелочами или аммиаком. Основные свойства
гидроокисей этих металлов усиливаются от Sc(OH)3 к La(OH)3.
Фториды, фосфаты, карбонаты и другие соединения металлов
подгруппы скандия плохо растворимы в воде, хлориды, сульфаты,
нитраты, ацетаты растворяются хороню.
Таблица 14
Соединение ScF3.i/2H2O ;YF3.1/2H2O ЬаЕз^/гНгО AcF3 ScCl3 YC13 LaCl3 АсС13
Вид Бесцветные гексаго- нальные кристаллы Бесцветные ромбиче- ские или кубические кристаллы Бесцветные гексаго- нальные кристаллы Бесцветные гексаго- нальные кристаллы Бесцветные ромбоэдри- ческие кристаллы Бесцветные ромбоэдри- ческие кристаллы Бесцветные гексаго- нальные кристаллы Бесцветные 1ексаго- нальные кристаллы
Молекулярный вес 110,958 154,907 204,912 284,030 151,315 195,264 245,269 333,394
Плотность (при 20°), г/сл3 — 4,01-5,06 5,93 7,88 2,39 2,67 3,84 4,81
Температура плавления, °C 1227 1387 1427 1327 960 721 852 927
Температура кипения, °C 1527 2227 2327 2277 1513 1507 1747 1757
Теплота обра- зования, ккал/молъ 367 397 421 420 220,8 234,8 263,6 260
Гидраты (число молекул Н2О) —- — — — 6 и 3 6 и 1 7
игил*». Ацидосоли (об- щая формула) MeT[ScF4] MeJfScFs] Me|[ScF6] MeI[YF4] Men[YF6] MeJlYFe] MeT[LaF4] MeJ[LaF6] Двойные соли 3ScCl3- •2AuCl3- 21II2O Двойные соли 2YC13- • 3PtCl2- • 4H2O,YClr • 2АиС13. • 16H2O MeJ[LaCle]- •nH2O Ме| [La2Clg]
Таблица 15
Соединение ScBr3 YBr3 LaBr3 АсВг3 Scl3 YI3 Lal3 Acl3
Вид Бесцветные кристаллы Бесцветные кристаллы Бесцветные гексаго- нальные кристаллы Бесцветные гексаго- нальные кристаллы Бесцветные кристаллы Бесцветные кристаллы Желтые ромбические кристаллы Бесцветные кристаллы
Молекулярный вес 284,683 328,632 378,637 466,762 425,669 469,618 519,623 607,748
Плотность, г/см^ 3,91 — 5,06 5,85 — — 5,26 —
Температура плявления,°C 960 904 783 827 945 1000 761 —
Температура кипения, °C — 1467 1577 1597 1307 1402 1407
Теплота обра- зования, ккал/моль 190,4 208 233 231 149 165 189 192
Гидраты (число молекул Н2О) 6 9 7 — 6 — — —
Ацидосоли (об- щая формула) — — Ме1[ЬаВг4] — — — Мет[Ьа14] Me|[LaI6] —
26
ПОБОЧНАЯ ПОДГРУППА III ГРУППЫ
Оксалаты скандия, иттрия и лантана осаждаются из кислых
растворов подобно оксалатам лантанидов или оксалату тория.
Галогениды, сульфаты, нитраты, карбонаты и оксалаты скан-
дия, иттрия и лантана со щелочными металлами образуют коор-
динационные соединения, которые могут быть представлены сле-
дующими формулами: Ме1[МешХ4], Ме|[МешХ5], Ме*[МешХв],
Ме1[Меш(8О4)3],Ме1[Ме1П(8О4)2],Ме1[Ме111(СОз)з],Ме1[МеП1(С2О4)2],
Ме*[МеП1(С2О4)3] (где Me1 = К+, Na+ и Меш = Sc3+, Y3+, La3+).
Склонность к образованию ковалентных соединений у скан-
дия и его гомологов выражена слабо. В качестве примеров кова-
лентных соединений можно назвать ацетилацетонаты и аддукты
эфира с триэтилскандием и триэтилиттрием.
СКАНДИЙ (ЭКАБОР) Sc
Z= 21; ат. вес= 44,96
Валентность III, заряд 3+
Массовое число природного изотопа 45
Электронная структура атома скандия: К -L •3s23p63<i1 -4s2.
Электронная структура атома и иона скандия для Зр-, 3d-
и 4,;-орбиталей:
Зре
U IU |п I
3d’
tl I
Sc
4s2
тг
Зр 4s
u I u IU I I I II |] C
Sc3+
ИСТОРИЧЕСКАЯ-СПРАВКА
В 1871 г. Д. И. Менделеев предсказал существование нового
химического элемента, названного им «экабором», с атомным
весом 44 и удельным весом примерно 3 г/см3. Менделеев предска-
зал также способность экабора к образованию двойных сульфатов,
не изоморфных квасцам, и малую растворимость трехокиси, кар-
бонатов и фосфатов зкабора.
В 1879 г. Нильсон выделил бесцветный окисел, металлический
элемент которого (открытый при помощи спектрального анализа)
был назван скандием (в честь Скандинавии). Физико-химические
свойства элемента, установленные Нильсоном (например, атомный
вес 44,96, удельный вес 3,1 г/см?, формула окиси Sc2O3 и др.),
были очень близки к свойствам экабора, предсказанным Менде-
леевым. Нильсон показал таким образом, что экабор и скандий
представляют собой один и тот же элемент.
РАСПРОСТРАНЕННОСТЬ В ПРИРОДЕ
В природе скандий встречается в виде соединений в поли-
металлических рудах редких земель, которые очень рассеяны.
Содержание скандия в земной коре примерно 6 НО-4 вес. %.
СКАНДИЙ
27
Важнейшие минералы скандия — тортвейтит, винчит и хло-
шинит.
Тортвейтит (Y,Sc)2Si2O7, содержащий 40% Sc (1—2% ZrO2
и НЮ2), является самым важным его минералом. Он встречается
в виде зеленых или красных моноклинных кристаллов с твер-
достью 6—7 по шкале Мооса. Залежи этого минерала находятся
в Норвегии, СССР, Мальгашской Республике; вместе с минера-
лами олова — в ЧССР, вольфрама — в ФРГ, циркония — в Бра-
зилии.
ПЕРЕРАБОТКА ТОРТВЕЙТИТА
В отличие от остальных силикатов редких земель тортвейтит
устойчив к обычным разрушающим реагентам (концентрирован-
ным кислотам НС1, HNO3, H2SO4, HF или расплавам Na2CO3,
Na2B4O7 и т. д.).
При нагревании смеси измельченных тортвейтита и угля в гра-
фитовом тигле в электрической печи при 1800° в течение 30—45 мин
образуется карбид скандия, загрязненный карбидами редкоземель-
ных элементов, карбидами циркония, железа и небольшим коли-
чеством карбида кремния. Под действием разбавленной (1 : 1)
соляной кислоты полученные карбиды, за исключением карбида
кремния, превращаются в хлориды. При обработке водных раство-
ров хлоридов избытком щавелевой кислоты выпадает Sc2(C2O4)3
и оксалаты редких земель:
2Sc2Si2O7 + 6С = Sc4C3 + 4SiO2 + ЗСО2
Sc4C3 + 12НС1 = 4ScCl3 + ЗС + 6Н2
2ScCl3 + ЗН2С2О4 = Sc2(C2O4)3 + 6НС1
Для отделения скандия от редкоземельных элементов суще-
ству ют различные способы, которые рассматриваются в разделах,
поев ященных редкоземельным элементам.
ПОЛУЧЕНИЕ МЕТАЛЛИЧЕСКОГО СКАНДИЯ
Для получения металлического скандия используют главным
образом его галогениды, которые могут быть получены из Sc2O3.
Последний в свою очередь образуется при прокаливании
Sc2(C2O4)3 "бЫгО при 400°.
Металлический скандий получают при металлотермическом вос-
становлении галогенидов скандия или при электролизе расплава
хлорида скандия в эвтектике КС1 — LiCl.
Металлотермическое восстановление галогенидов скандия
При магнийтермическом восстановлении смеси 62% ScCl3
и 38% КС1 (или смеси 50% ScF3 и 50% NaCl - КС1 - CaF2,
взятых в соотношении 10 : 10 : 1 по весу) металлическим маг-
28
ПОБОЧНАЯ ПОДГРУППА Ш ГРУППЫ
нием в небольшом избытке образуются магнийскандиевые сплавы
(содержащие примерно 14% скандия, когда восстанавливаются
хлориды, и 10% скандия при восстановлении фторидов). При
перегонке в вакууме при 1500° магний почти полностью удаляется
из таких сплавов.
При металлотермическом восстановлении фторида скандия ScFs,
с металлическим кальцием в танталовых тиглях образуется метал-
лический скандий, содержащий примерно 0,1% кальция:
2ScF3 + ЗСа — 2Sc + 3CaF2
-Электролиз расплава хлорида скандия
в эвтектике КС1 — LiCl
При электролизе расплава безводного хлорида скандия в эвтек-
тике КС1 — LiCl получают сплав Zn — Sc — К — Li. Электро-
лиз проводят при температуре 700—800° в графитовом тигле-
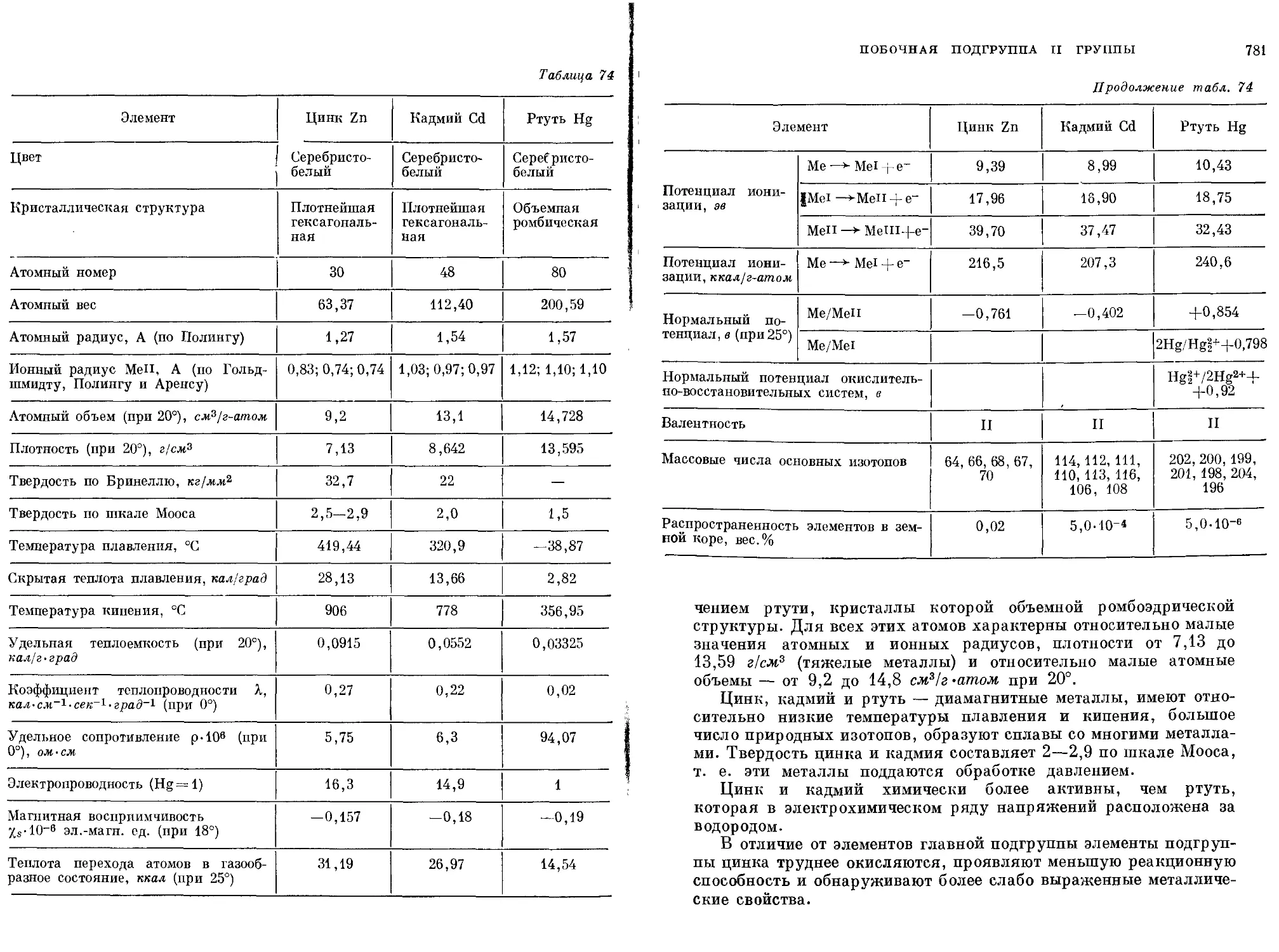
(рис. 1), который служит
Рис. 1. Ячейка для элект-
ролиза.
1 — графитовый тигель (анод);
2 — флюоритовый тигель; з —
катод из жидкого цинка или
кадмия; 4 — электролит; 5 —
вольфрамовый или молибдено-
вый стержень; 6 — защитная
обкладка из флюорита; 7 —
кварцевая трубка с боковыми
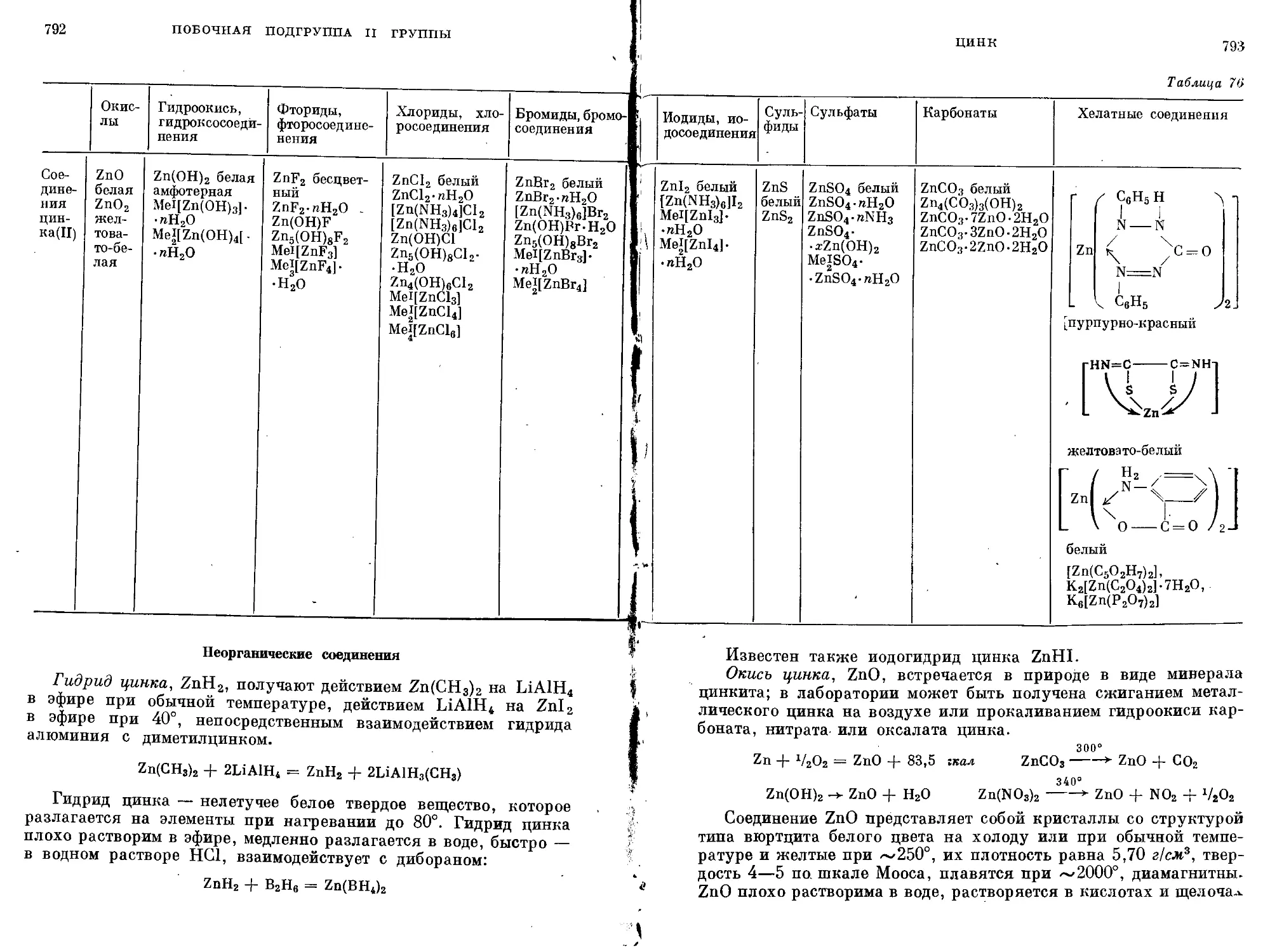
Отводами; 8 — термопара.
анодом; в качестве катода используют
расплавленный цинк. Сплав собирают
в тигель из флюорита (CaF2) (или оки-
си магния MgO), помещенный внутри
графитового тигля. Электрический ток
подводится к катоду при помощи воль-
фрамового (или молибденового) стерж-
ня, защищенного от действия выделя-
ющегося на аноде хлора обкладкой из
флюорита (или окиси магния).
При осуществлении электролиза
электролит предварительно расплавля-
ют, нагревая графитовый тигель, пос-
ле чего включают электрический ток,
доводя плотность тока на ка-
тоде до нескольких ампер на 1 см2.
Для измерения температуры графи-
тового тигля служит термопара с
милливольтметром. Вместо цинкового
катода используют также катод из
кадмия или из сплавов Mg — Zn и
Mg — Cd.
Для удаления хлора из анодного
пространства графитового тигля слу-
жит кварцевая трубка с двумя боковы-
ми отводами в выступающей части,
через эту трубку в анодное простран-
ство поступает СО2.
При нагревании сплава Zn — Sc —
К — Li в вакууме при 1250° отгоня-
СКАНДИЙ
29
ются цинк, калий, литий и остается металлический скандий
98 % -ной чистоты.
ФИЗИЧЕСКИЕ И ХИМИЧЕСКИЕ СВОЙСТВА
Скандий — серовато-белый металл, имеющий компактную гек-
сагональную кристаллическую решетку (a-Sc) или кубическую
гранецентрированную решетку (0-Sc). Металлический скандий
имеет удельный вес 2,99 г/см3 при 20° (легкий металл), плавится
при 1539°, кипит при 2727° (достаточно тугоплавкий), хрупок
{не поддается механической обработке), слабо парамагнитен.
По своим физическим свойствам металлический скандий про-
являет сходство с иттрием и редкоземельными элементами, такими,
как лютеций.
Нормальный потенциал скандия (в системе Sc/Sc3+) равен
—2,08 в (при 25°). Скандий — активный, легко окисляющийся
металл, разлагающий воду при нагревании, легко растворяю-
щийся при нормальной температуре в разбавленных кислотах
с образованием солей и выделением водорода:
Sc + ЗН2О = Sc(OH)3 + 3/2Н2 2Sc + 3H2SO4 = Sc2(SO4)3 + ЗН2
Sc + ЗНС1 = ScCl3 + 3/2H2 Sc + 3HNO = Sc(NO3)3 + 3/2H2
При нагревании металлический скандий взаимодействует с кис-
лородом, хлором, бромом, иодом, серой и азотом с образованием
соответственно Sc2O3, ScCl3, ScBr3, Scl3, Sc2S3, ScN.
Характеристика химической активности скандия приведена
ниже.
Комн. темп.
Нагревание
с НС1 —► ScCl3-6H2O
с H2S04 —► Sc2(SO4)3-6H2O
с HNO3 —> Sc(NO3)3-4H2O
Скандий ---
л с водой —► Sc (ОН)3 или Sc2O3
с кислородом —>- Sc2O3
с галогенами —► ScX3
(Х = С1~, F-, Вг-, Г)
с серой —* Sc2S3
X с азотом —+ ScN
СОЕДИНЕНИЯ (ОБЩИЕ СВОЙСТВА)
Известны многочисленные устойчивые простые или коорди-
национные соединения, в которых скандий постоянно электро-
положителен и трехвалентен.
30 FS
ПОБОЧНАЯ ПОДГРУППА III ГРУППЫ
В химических соединениях скандий присутствует либо в виде
катиона Sc3+, либо в составе комплексных анионов: IScF4]-,.
[ScFJ2-, [ScF6]3“,[Sc(SO4)2]_, [Sc(SO4)3]3-, [Sc(CO3)2J-, ISc(C2O4)2]",.
[Sc(C2O4)3]3- и т. д.
Соли скандия похожи на соли алюминия: они бесцветны,,
сильно гидролизуются в теплой воде, склонны к образованию ком-
плексных соединений, как-то: галогено-, псевдогалогено-, суль-
фате-, карбонате- или оксалатосоли.
С точки зрения растворимости и отношения к различным хими-
ческим реагентам соединения скандия напоминают соединения
щелочноземельных металлов.
В отличие от соответствующих соединений щелочноземельных
металлов окись скандия Sc2O3 является слабым основанием, хло-
рид ScCl3 более летуч, нитрат Sc(NO3)3 ‘4Н2О термически менее-
устойчив, сульфат Sc2(SO4)3’5Н2О растворим в воде.
Неорганические соединения
Окись скандия, SczO3, получают при нагревании металлат
в атмосфере кислорода, а также прокаливанием на воздухе гидро-
окиси, нитрата, карбоната или оксалата скандия:
2Sc + 3/2О2 = Sc2O3 -р411 ккал
200°
2Sc(OH)3----->- Sc2O3 + ЗН2О
Выше 200°
2Sc(NO3)3-4Н2О >- Sc2O3 +6NO2 + 3/2О2 + 8Н2О
1000°
Sc2(CO3)3.12Н2О Sc2O3 + ЗСО2 + 12Н2О
400°
Sc2(C2O4)3.5H2O----> Sc2O3 + ЗСО2 + ЗСО + 5Н2О
Окись скандия представляет собой рыхлый белый порошок
или бесцветные кубические кристаллы с плотностью 3,86 г!см\
Обе формы плохо растворимы в воде или в разбавленных кисло-
тах на холоду и поглощают двуокись углерода из воздуха. После
сильного прокаливания Sc2O3 плохо растворяется в концентри-
рованной НС1.
Гидроокись скандия, Sc(OH)3, получают обработкой растворов
солей скандия щелочами (или аммиаком) при pH 4,9—5,45 или
путем гидролитического осаждения из растворов солей скандия
тиосульфатом натрия:
Sc(NO3)3 + 3NaOH = Sc(OH)3 + 3NaNO3
2ScCl3 + 3Na2S2O3 + 3H2O = 2Sc(OH)3 + 6NaCl + 3SO2 +3S
На этой реакции основано количественное весовое определе-
ние скандия.
СКАНДИЙ
31
Гидроокись скандия получают в виде студнеобразного белого
осадка, трудно растворимого в воде, обнаруживающего слабо
основные свойства, в отличие от амфотерной А1(0Н)3.
Гидроокись скандия можно выделить в виде белого аморфного
порошка или в виде бесцветных кубических гранецентрированных
кристаллов с плотностью 2,6 г!сл?. Нагреванием при 250—255°
и давлении 0,1 мм рт. ст. получают ScOOH, а при более высокой
температуре — Sc2O3.
Разбавленные кислоты растворяют гидроокись скандия с обра-
зованием солей, а избыток гидроокиси или карбоната аммония
частично растворяет гидроокись скандия с образованием гексам-
минскандиевого катиона [Sc(NH3)el3+.
Гидроокись скандия с алюминоном C22H11O9(NH4)3 образует
темно-красный, хорошо растворимый в (NH4)2CO3 лак. Послед-
ний получают путем добавления концентрированного NH4OH
к смеси растворов нитрата скандия, алюминона и ацетата аммония.
Алюминон с ионом Sc3+ в слабо основной среде образует хелат
следующего строения:
Фторид скандия, ЗсГз-^НгО, получают обработкой растворов
солей скандия фтористоводородной кислотой или фторидом калия:
Sc(NO3)3 + 3HF = ScF3 + 3HNO3 Sc(NO3)3 + 3KF = ScF3 + 3KNO
Соль ScF3-x/2H2O представляет собой бесцветные гексагональ-
ные кристаллы с т. пл. 1227° и т. кип. 1527°, трудно растворимые
в воде. При обработке фторида скандия фторидами щелочных
металлов или аммония образуются растворимые фторосоли (ацидо-
соли) типов Me4ScF4], Me*[ScF5], MeJ[ScF6] (где Mer= К+, Na+).
При кипячении фтороскандиатов с гидроокисями щелочных
металлов осаждается гидроокись скандия:
(NH4)3[ScF6] + 3NaOH = Sc(OH)3 + 3NaF + 3NH4F
Безводный хлорид скандия, ScCl3, получают действием сухого
хлора на металлический скандий при нагревании, на сульфиды,
карбиды скандия или смесь Sc2O3 с углем. ScCl3 получают также
обработкой газообразным хлористым водородом сульфидов или
карбидов скандия либо нагреванием окиси скандия с хлоридом
32
ПОБОЧНАЯ ПОДГРУППА III ГРУППЫ
аммония:
Sc + 3/2С12 = ScCl3 + 220,8 ккал Sc2S3 + 6НС1 = 2ScCl3 + 3H2S
Sc2S3 + 3C12 = 2ScCl3 + 3S Sc2O3 + 6NH4C1 = 2ScCl3 + 6NH3 + 3H2O
Хлорид скандия представляет собой бесцветные ромбоэдриче-
ские кристаллы (плотность 2,3 г!смЛ, т. пл. 960°, т. кип. 1513°),
растворимые в спирте и гидролизующиеся легче, чем хлориды
редкоземельных металлов:
ScCl3 + ЗН2О ** Sc(OH)3 + ЗНС1
Гексагидрат хлорида скандия, ScCl3-6H2O, существующий
в виде бесцветных кристаллов, при нагревании превращается
в кристаллогидрат ScCl3-3H2O и в конечном итоге в оксихлорид
скандия ScOCl:
100° Выше 100°
ScCl3.6H2O ———> ScCl3-3H2O ----—Sc(OH)2C1+2HC1
— ЗН2О —Н2О
I
ScOCl+ H2O
Безводный хлорид скандия получают при дегидратации соеди-
нения ScCl3’6H2O в атмосфере сухого газообразного НС1.
Благодаря хорошей растворимости в воде, насыщенной газо-
образным НС1, или в растворах вода — спирт, вода — эфир,
насыщенных газообразным НС1, хлорид скандия можно легко
отделить от хлоридов алюминия, иттрия и лантана.
Хлорид скандия с хлоридами некоторых металлов образует
двойные соли, папример 3ScCl3 -2АиС13 ‘21Н2О — кристаллы жел-
того цвета.
Безводный бромид скандия, ScBr3, получают при нагревании
исходных компонентов либо хлорида или сульфида скандия в токе
бромистого водорода, а также при взаимодействии гидратирован-
ного бромида скандия с бромидом аммония в высоком вакууме
при 550—600°:
2Sc-|-3Br2=2ScBr3-{-380,8 калл
ScC 13 -ф 3HBr= ScBr3-P ЗНС1
Sc2S3 + 6HBr = 2ScBr3 4- 3H2S
Бромид скандия ScBr3 образуется в виде бесцветных кристал-
лов (плотность 3,91 г!см\ т. пл. 960°), гигроскопичных, раство-
римых в спирте, ацетоне и гидролизующихся в воде.
При растворении окиси скандия в бромистоводородной кислоте
образуется водный раствор бромида скандия, из которого на холоду
выделяются кристаллы ScBr3-6H2O.
Безводный иодид скандия, Scl3, получают при нагревании исход-
ных компонентов или при взаимодействии окиси скандия Sc2O3
с иодидом аммония при 350°. Scl3 можно получить также действием
СКАНДИЙ
33
иодистоводородной кислоты на нагретый примерно до температуры
плавления безводный хлорид скандия или нагреванием кристал-
логидрата ScI3-6H2O в атмосфере йодистого водорода до 350°,
а в присутствии иодида аммония — до 600—650°:
2Sc 3I2 = 2ScI3 -р 298 ккал ScCl3 -|- 3HI = Scl3 -|- ЗНС1
nh4i
Sc2O3 -|- 6NH4I = 2ScI3 4- 6NH3 -]- 3H2O ScI3*6H2O > Scl3
— 61120
Соединение Scl3 образуется в виде бесцветных кристаллов
с т. пл. 945°, растворимых в воде, пиридине, спирте, ацетоне и т. д.
Сульфид скандия, Sc2S3, получают действием паров серы
на металлический скандий, обработкой сероуглеродом окиси скан-
дия, восстановлением безводного сульфата скандия углем при на-
гревании и нагреванием безводного сульфата скандия в атмосфе-
ре сероводорода.
2Sc -р 38 = Sc2S3 Sc2(SO4)3 4~ 12С = Sc2S3 4- 12СО
2SczO3 4~ 3CS2 = 2Sc2S3 4- 3CO2 Sc2(SO4)s 4- 3H2S = Sc2S3 4~ 3H2SO4
Сульфид скандия — желтое твердое вещество; устойчив на воз-
духе, гидролизуется кипящей водой:
Sc2S3 4- 6Н0Н 2Sc(OH)3 4- 3H2S
Сульфит скандия, Sc2(SO3)3 -бНгО,— хлопьевидное белое веще-
ство, плохо растворимое в воде; его получают, пропуская газо-
образный SO2 через свежеприготовленный охлажденный во льду
раствор гидроокиси скандия:
2Sc(OH)3 4- 3SO2 = Sc2(SO3)3 4- ЗН2О
Сульфаты скандия. Безводный сульфат скандия,
Sc2(SO4)3, получают при нагревании окиси, гидроокиси или кар-
боната скандия с концентрированной H2SO4 или путем дегидра-
тации при 250° кристаллогидратов Sc2(SO4)3 -6Н2О, Sc2(SO4)3 -5Н2О:
Sc2O3 4~ 3H2SO4 = Sc2(SO4)3 4~ зн2о
2Sc(OH)3 4- 3H2SO4 = Sc2(SO4)3 4- 6H2O
Sc2(CO3)3 4- 3H2SO4 = Sc2(SO4)3 4- 3CO2 + 3H2O
250°
Sc2(SO4)3.6H2O--—~+ Sc2(SO4)3
— 0X120
Безводный сульфат скандия — белый гигроскопичный поро-
шок с плотностью 2,605 а/с№; при обработке 40%-ной H2SO4
образует соединение HlSc(SO4)2].
Гексагидрат сульфата скандия, Sc2(SO4)3-6Н2О, выделяют
из концентрированных (сиропообразной консистенции) водных
растворов сульфата скандия. При хранении на воздухе бесцветные
кристаллы гексагидрата Sc2(SO4)3'6Н2О превращаются в пента-
гидрат Sc2(SO4)3-5Н2О, а при 250° — в безводную соль Sc2(SO4)3.
3-0101
34
ПОБОЧНАЯ ПОДГРУППА III ГРУППЫ
При обработке сернокислых растворов сульфата скандия избыт-
ком сульфата натрия, калия или таллия образуются комплексные
сульфаты Na3[Sc(SO4)3l-5Н2О, K3[Sc(SO4)3], Tl3[Sc(SO4)31, которые
являются производными кислоты H3[Sc(SO4)3].
При низкой температуре, смешивая растворы сульфата скан-
дия с сульфатами щелочных металлов или сульфатом аммония,
можно выделить безводные трудно растворимые квасцы общей
формулы Me4Sc(SO4)2], где Me1 = Na+, К+, Rb+, Cs+, NH*.
Селенид скандия, Sc2Se3,— твердое фиолетовое вещество с плот-
ностью 4,52 г/см?.
Селенаты скандия. Безводный селенит скандия,
Sc2(SeO4)3, выделяется в виде тетрагональных кристаллов с плот-
ностью 3,27 г/см3 при упаривании (песчаная баня 100—110°) насы-
щенного водного раствора селената скандия, содержащего селе-
новую кислоту.
Упариванием смеси, полученной растворением окиси скандия
в разбавленной селеновой кислоте, получают моноклинные кри-
сталлы Sc2(SeO4)3 -5Н2О с плотностью 2,63 г/см3.
Октагидрат селената скандия, Sc2(SeO4)3-8Н2О, выпадает
на холоду при медленном упаривании раствора, полученного
растворением окиси скандия в селеновой кислоте. Добавле-
нием H2SeO4 к 10%-ному раствору Sc2(SeO4)3 (1 г кислоты
на 10 г соли) выделяют бесцветные устойчивые на воздухе кри-
сталлы в виде гексагональных призм Sc2(SeO4)3’10Н2О с плот-
ностью 2,21 г/см3.
' Диселенатоскандиееая кислота, H[Sc(SeO4)2] -Н2О, получается
добавлением соединения Sc2(SeO4)3-5Н2О в 60%-ный раствор
H2SeO4.
Известен гидратированный основной селенат скандия
Sc(OH)SeO4 -2Н2О — бесцветные кристаллы с плотностью 2,58 г!см\
Нитрид скандия, ScN, получают при нагревании исходных
компонентов или действием азота на гидрид скандия ScH3 при
800—900°, на карбид Sc4C3 при 1200° и на смесь окиси скандия
с углем при 1300° в присутствии катализатора (Fe2O3 или Na2CO3):
2Sc + N2 = 2ScN Sc4C3 + 2N2 = 4ScN + 3C
2ScH3 -p N2 ~ 2ScN -p 3H2 Sc2O3 -p 3C -J- N2 = 2ScN -p 3CO
Соединение включения ScN представляет собой устойчивые
и тугоплавкие (плавятся при 2900°) синие кубические кристаллы.
Вещество разлагается водой с образованием Sc(OH)3 и выделением
аммиака.
Нитрат скандия, Sc(NO3)3-4Н2О, выделяют, упаривая на водя-
ной бане раствор окиси или гидроокиси скандия в азотной кислоте:
Sc2O3 -р 6HNO3 = 2Sc(NO3)3 -р ЗН2О
Sc(OH)3 -р 3HNO3 — Sc(NO3)3 -Р ЗН2О
СКАНДИЙ
35
Бесцветные расплывающиеся на воздухе призмы Sc(NO3)3-4H2O
растворимы в воде. При нагревании превращается в основные
нитраты и в конечном итоге разлагается доЗс2О3:
2Sc(NO3)3 -4Н2О НаГУУ”е-> 2Sc(OH)(NO3)2.3H2O + 2NO2 + V2O2 + Н2О
над конц. H2SO4
100°
2Sc(NO3)3*4H2O Sc2O(NO3)4-0,5Н2О -J- 2NO2 -j- V2O2 *4" 7,5Н2О
Примерно 200° Выше' 2 0 0 °
2Sc2O(NO3)4-0,5H2O ----—------> Sc4O5(NO3)2----->
— Д2и
—6NO2—З/2О2
2Sc2O3-[- 2NO2+1/2Q2
Соединение Sc2O(NO3)4-0,5Н2О можно получить и нагреванием
при 90° раствора нитрата скандия со стехиометрически необхо-
димым количеством Sc2O3.
При взаимодействии нитрата скандия с нитратами щелочных
металлов или аммония образуются комплексные нитраты, например
KISc(NO3)4l.
Карбид скандия, Sc4C3, образуется в виде гексагональных
кристаллов восстановлением окиси скандия углем при нагрева-
нии.
Карбонат скандия, Sc2(CO3)3-12Н2О, получают обработкой
на холоду растворов солей скандия раствором бикарбоната щелоч-
ного металла, насыщенным газообразным СО2:
2Sc(NO3)3 + 6NaHCO3 = Sc2(CO3)3 + 6NaNO3 + 3CO2 + 3H2O
Карбонат скандия — белое твердое вещество, плохо раствори-
мое в воде, растворимое в минеральных кислотах и теплых раство-
рах карбонатов щелочных металлов. При нагревании карбонат
скандия ступенчато разлагается до Sc2O3:
350-550° 800-905°
Sc2(CO3)3.12H2O Sc2O(CO3)2
-1000°
>- Sc2O2CO3 ’ Sc2O3
Смесь карбоната и основных карбонатов скандия получают обра-
боткой растворов солей скандия карбонатами щелочных металлов
при нагревании. При упаривании этих растворов выделяют комп-
лексные карбонаты, например K[Sc(CO3)2] -Н2О.
Оксалат скандия, Sc2(02O4)3-5H2O, выделяется в виде кристал-
лического осадка при обработке слабо кислых растворов солей
скандия щавелевой кислотой:
2Sc(NO3)3 -р ЗН2С2О4 = Sc2(C2O4)3 -р 6HNO3
3»
36
ПОБОЧНАЯ ПОДГРУППА III ГРУППЫ
При нагревании оксалат скандия разлагается:
110° 280°
Sc2(C2O4)3-5H2O * Sc2(C2O4)3.H2O >
— 4x120 — И 2 О
400°
► Sc2(C2O4)3 > Sc2O3-|-3CO2+3CO
Известны также комплексные оксалаты общей формулы
Me4Sc(C2O4)2b(2-3)H2O, Mei[Sc(C2O4)3](5 - 6)Н2О, где Me1 =
= Na+, К+, NHJ и т. д.
Алкильные производные
Продукт присоединения эфира к триэтилскандию Sc(C2H5)3-
• О(С2Н5)2 получают действием магнийбромэтила на суспензию
безводного хлорида скандия в эфире в атмосфере азота:
2ScCl3 4“ 6C2H5MgBr -j- 2(С2Н5)2О =
= 2Sc(C2H5)3 -О(С2Н5)2 + 3MgBr2 + 3MgCl2
Соединение Sc(C2H6)3-O(C2H5)2 — желтая прозрачная жидкость
(плотность 1,071 г/см3) с неприятным запахом, дымящая на воз-
духе, смешивающаяся в любых соотношениях с бензолом, толуо-
лом, ксилолом, этилацетатом и т. д. Разлагается при соприкос-
новении с водой или спиртом, обрузуя Sc(C2H5)2(OH), и энер-
гично реагирует^ с бромной водой, давая бромид диэтилскандия
Sc(C2H6)2Br.
Хелатные соединения
А цетилацетонат скандия, Sc(CH3 — СО — СН = СО —
— СН3)3, получают обработкой растворов солей скандия спирто-
вым раствором ацетилацетона в присутствии аммиака:
/СН3
НО —(У
Sc(NO3)3 + 3 4CH + 3NH3 =
O = CZ
XCH3
+ 3NH4NO3
Ацетилацетонат скандия представляет собой бесцветные орто-
ромбические кристаллы, плавящиеся при 187,5°, возгоняющиеся
без разложения при 210—215°, растворимые в воде, спирте,
эфире, хлороформе, бензоле и четыреххлористом углероде.
Оксихинолят скандия, [Sc(C9H6NO)3-C9He-OH], получают обра-
боткой растворов солей скандия 5%-ным раствором 8-оксихино-
лина при pH, равном 8,5.
Оксихинолят скандия — желтое твердое вещество, плавя-
щееся при 197°, плохо растворимое в воде и очень хорошо раство-
ИТТРИЙ
37
римое в соляной кислоте, бензоле, толуоле, хлороформе, ацетоне,
метаноле, этаноле и т. д.
Помимо перечисленных, известны и другие соединения скандия, напри-
мер: гидрид ScH3, сульфит NH4[Sc(SO3)2]-3,5H2O, гидроксотиосульфат
Sc(OH)S2O3, тиоцианат Sc(SCN)3, гексатиоцианатоскандиат аммония
(NH4)3[Sc(SCN)6], ортоборат ScBO3, гексафторосиликат Sc2[SiF6]3, германат,
ортованадат ScVO4, формиат Sc(HCOO)3, формиаты Me|[Sc(HCOO)6], где
Mel — Li+, Na+, К+ и т. д., ацетаты Sc(CH3COO)3-2Н2О, Sc(OH)(CH3COO)2,
тартрат скандия — аммония NH4OOC — СНОН — СНОП — COOSc(OH)2.
ИТТРИЙ Y
Z = 39; ат. вес = 88,92
Валентность III, заряд 3+
Массовое число природного изотопа 89
Электронная [структура атома иттрия: К -L -М 'is24pe4dL «5s2.
Электронная структура атома и иона иттрия для 4р-, 4d-
и Ss-орбиталей:
ИСТОРИЧЕСКАЯ СПРАВКА
В 1835 г. Мозандер разделил иттриевые земли на собственно
иттриевую землю (которая обладает более основным характером
и дает бесцветные соли), тербиевые земли (менее основного харак-
тера, образующие розовые соли) и эрбиевые земли (дающие
желтые окислы и бесцветные соли).
В 1887 г. Крукс получил спектр катодной фосфоресценции
иттриевой земли. В 1900 г. Мутман и Баур описали фосфоресцент-
ный спектр чистого иттрия. Среди исследователей, изучавших
иттрий, упоминаются Лекок де Буабодран, Баур, Марк и др.
Металлический иттрий впервые был получен по способу Вёлера
путем восстановления хлорида иттрия YC13 металлическим калием
при нагревании.
РАСПРОСТРАНЕННОСТЬ В ПРИРОДЕ
В природе иттрий находится в малых количествах в полиметал-
лических минералах, содержащих соединения лантанидов, каль-
ция, скандия, титана, циркония, гафния, тория, ниобия, тантала,
урана, железа и т. д.
В земной коре содержится примерно 5,0 ЛО-3 вес. % иттрия.
Важнейшие минералы иттрия — ксенотим YPO4, самарскит
(Y, Er)J(Nb, Та)20т]3, гадолинит Be2Y2FeSi2Olo, таленит Y2Si2O7,
тортвейтит (Y, Sc)2Si2O7, иттриалит (Y, Th)2Si2O7, фергюсонит
38
ПОБОЧНАЯ ПОДГРУППА Ш ГРУППЫ
(Y, Ег, Са...) (Та, Nb)O4, иттрокальцит YF3-CaF2, евксенит, поли-
краз, монацит, хлопинит, иттротитанит, иттротанталит, лессин-
гит и г. д.
Описание некоторых из этих минералов можно найти в разделе,
досвященном нахождению в природе редкоземельных металлов.
ИЗВЛЕЧЕНИЕ ИТТРИЯ
Источником иттрия служат минералы ксенотим, самарскит,
тортвейтит, фергюсонит и др.
Извлечение иттрия из минералов весьма трудоемко, поскольку
этот элемент находится в рудах в малых количествах.
Руды, содержащие соединения иттрия, концентрируют и пере-
рабатывают различными химическими методами для получения
окиси иттрия или его галогенидов YX3, являющихся сырьем
для получения металлического иттрия.
Об обработке минералов, содержащих соединения иттрия, см.
раздел, посвященный редкоземельным металлам. Там же указаны
способы разделения катионов металлов, содержащихся в этих
ЙУДах.,
ПОЛУЧЕНИЕ МЕТАЛЛИЧЕСКОГО ИТТРИЯ
Металлический иттрий получают металлотермическим восста-
новлением окиси или галогенидов иттрия, а также электролизом
расплавленного хлорида иттрия.
Металлотермическое восстановление окиси
или галогенидов иттрия
При термическом восстановлении окиси Y2O3 или безводного хло-
рида иттрия YC13 металлическим магнием (взятым в 10%-ном
избытке по сравнению со стехиометрически необходимым для вос-
становления количеством) получают магнийиттриевый сплав,
содержащий 4—6% иттрия. Восстановление проводится в графи-
товом тигле, футерованном молибденом. Тигель нагревается
в электрической печи. Контроль температуры осуществляется
термопарой.
Магний удаляется из магнийиттриевого сплава перегонкой
в вакууме при 1300—1350°.
Восстановление фторида иттрия YF3 металлическим кальцием
(взятым в 10 %-ном избытке по сравнению со стехиометрически
необходимым для восстановления количеством) ведут в тантало-
вом тигле в атмосфере аргона примерно при 1550°. При этом
получают металлический иттрий с примесью 0,1% кальция и 0,1%
ИТТРИИ
39
тантала. При использовании смеси YF3 с СаС12 восстановление
проводят при 1300°.
Металлический иттрий можно получить (по способу Вёлера)
металлотермическим восстановлением безводного хлорида иттрия
YC13 (или YF3) с металлическим натрием, калием, рубидием
или цезием в вакууме (или в атмосфере сухого аргона) при 200—
350°, используя аппаратуру из йенского стекла или стекла супр е-
макс.
Электролиз хлорида иттрия
При электролизе расплава хлорида иттрия (или смеси YC13 —
NaCl) получают сплав из 24% иттрия, 56% магния и 20% кад-
мия.Электролиз проводят при 700—800° в графитовом тигле—аноде;
катодом служит жидкий сплав магния с25—30%кадмия. Электроли-
тическая ячейка подобна ячейке для электролитического получе-
ния скандия (рис. 1).
Для удаления магния и кадмия сплав Y — Mg — Cd нагре-
вают в вакууме до 900—1200°.
ФИЗИЧЕСКИЕ И ХИМИЧЕСКИЕ СВОЙСТВА
Физические и химические свойства иттрия похожи на свойства
скандия, лантана и тяжелых редкоземельных металлов, которым
он сопутствует в минералах.
Иттрий — метал серовато-белого цвета с плотно упакованной
гексагональной кристаллической решеткой и плотностью 4,48 г/сж3
при 20° (легкий металл). Иттрий плавится при 1509°, кипит при
2927° (тугоплавок, нелетуч, огнеупорен, как и эрбий или диспро-
зий), обнаруживает хорошую ковкость, очень слабо парамагнитен.
Металлический иттрий химически устойчив в сухом и неустой-
чив во влажном воздухе, так как покрывается пленкой Y2O3
или Y(OH)3.
Металлический иттрий окисляется значительно медленнее, чем
лантаниды группы церия.
Поскольку нормальный потенциал системы Y/Y3+ равен —2,37 в
при 25°, иттрий является активным металлом, разлагающим воду
при нагревании и легко растворяющимся при комнатной темпе-
ратуре в разбавленных кислотах с образованием солей и выде-
лением водорода:
Y -р ЗН3О — Y(OH)3 -р 3/2Н2 2Y -р 3H2SO4 = Y2(SOi)3 -р ЗН2
Y + ЗНС1 = YC13 + 3/2Н2 Y + 3HNO3 = Y(NO3)3 + з/2н2
При нормальной температуре иттрий взаимодействует с хлором,
образуя YCJ3, а при нагревании—с кислородом, иодом и серой,
образуя соответственно Y2O3, YI3, Y2S3.
40
ПОБОЧНАЯ ПОДГРУППА III ГРУППЫ
Химическая активность иттрия характеризуется следующей
схемой:
Комн. темп.
Нагревание
С хлором —>- YC13
с HCL —УС13-6Н2О
с H2SO4 -> Y2 (SO4)3-8H2O
с HNO3 —* Y(NO3)3-6H2O
Иттрий --
с водой —>- Y(OH)a или Y2O3
с кислородом —> YZO3
с иодом —► YI3
с серой —>- Y2S3
ПРИМЕНЕНИЕ
Металлический иттрий используется в качестве компонента
конструкционного материала для ядерных реакторов, поскольку
он обладает малым сечением захвата нейтронов.
СОЕДИНЕНИЯ (ОБЩИЕ СВОЙСТВА)
Известны многочисленные устойчивые простые и координацион-
ные соединения, в которых иттрий всегда электроположителен
и трехвалентен.
В химических соединениях иттрий может присутствовать в виде
катиона Y3+ или входить в состав комплексных анионов, напри-
мер, [YFJ-, [YF6]2-, [YF6]®-, [Y(SO4)313-, IY(CO3)2]" и т. д.
Соли иттрия бесцветны, гидролизуются, склонны к образо-
ванию двойных и комплексных солей.
Неорганические соединения
Окись иттрия, Y2O3, образуется при нагревании исходных
компонентов, а также прокаливанием на воздухе гидроокиси,
нитрата, карбоната или оксалата иттрия:
200°
2Y + 3/2О2----Y2O3 + 420 ккал
310-380°
2Y(OH)3-------> Y2O3 + ЗН2О
590°
2Y(NO3)3-6H2O-----> Y2O3 + 6NO2 + 3/2O2 + 12H2O
1100°
Y2(CO3)3.3H2O---> Y2O3 + 3CO2 + 3H2O
560°
Y2(C2O4)3-9H2O----> Y2O3 + 3CO2 + 3CO + 9H2O
ИТТРИЙ
41
Окись иттрия представляет собой белый порошок с плотно-
стью 5,046 г/см? или бесцветные кристаллы с плотностью 4,84 г/слЛ
Окись иттрия диамагнитна, плавится при 2417°, кипит при 4297°,
плохо растворяется в холодной воде и щелочах, растворяется
в кислотах, поглощает газообразный аммиак.
При 2000° Y2O3 восстанавливается водородом до металличе-
ского иттрия.
Раньше из смеси окислов иттрия и циркония (15 и 85% соот-
ветственно) изготовляли стержни ламп Нернста, которые при
сильном нагревании испускали интенсивный свет.
Гидроокись иттрия, Y(OH)3, получают взаимодействием солей
иттрия со щелочами при pH 6,83:
Y(NO3)3 + ЗКОН = Y(OH)3 + 3KNO3
Гидроокись иттрия можно выделить в виде студнеобразного
желтовато-белого осадка, аморфного порошка или желтовато-белых
гексагональных кристаллов. Гидроокись иттрия имеет более основ-
ной характер, чем А1(ОН)3. Она поглощает двуокись углерода
из воздуха, вытесняет аммиак из его солей, ббразует с перекисью
водорода перекись иттрия, при нагревании разлагается:
200-260° 310-380°
2Y(OH)3 ——----> 2YOOH--------> У2О3
' ' — 2Н2О -Н2О d
Фторид иттрия, YFgA^H^O, получают обработкой солей
иттрия фторидами щелочных металлов:
Y(NO3)3 + 3KF = YF3 + 3KNO3
Фторид иттрия можно выделить в виде студнеобразного белого
осадка или в виде бесцветных орторомбических (плотность
5,06 г/см3) или кубических (плотность 4,01 г/сж3) кристаллов; он
плавится при 1387°, кипит при 2227° и нагреванием с фторидом
аммония превращается в безводные соли.
Оксифторид иттрия, YOF, получают в виде бесцветных тетра-
гональных кристаллов с плотностью 5,18 г/см? взаимодействием
окиси иттрия с YF3’V2H2O в вакууме при 900° в течение несколь-
ких часов или гидролизом безводного фторида иттрия в токе
влажного воздуха при 800°.
Фторид иттрия образует следующие комплексные фториды
с фторидами щелочных металлов и кальция: NalYFJ, K3[YF8],
Rb3[YF6], Cs3[YFe], CalYFj.
Хлорид иттрия, YC13, получают действием хлора на суль-
фиды, карбиды или металлический иттрий, а также действием
газообразного НС1 на сульфиды или карбиды иттрия, нагрева-
нием Y2O3 с NH4C1, дегидратацией YC13’6H2O при нагревании
42
ПОБОЧНАЯ ПОДГРУППА III ГРУППЫ
в токе сухого газообразного НС1:
Y + 3/2С12 = YC13 + 234 ккал
YC2 + з/2С12 = YC13 + 2С
Y2S3 + 6НС1 = 2YC13 + 3H2S
YC2 + 3HC1 = YC13 + 2C + з/2н2
Y2O3+6NH4C1 = 2YC1s+6NH3+3H2O
hci
YC13-6H2O ——> YC13
—o112О
Хлорид иттрия представляет собой белые ромбоэдрические
кристаллы, растворимые в воде или спирте; плотность 2,67 г! см?,
т. пл. 721°, т. кип. 1507°.
При испарении водных растворов хлорида иттрия в условиях
нормальной температуры выпадают бесцветные гексагональные
призмы YC13"6H2O с плотностью 2,8 г! см?, растворимые в воде
и спирте и плохо растворимые в эфире. При нагревании YC13 -6Н2О
на водяной бане теряются 5 молекул воды, а при 160° в при-
сутствии газообразного НС1 образуется безводный YC13.
Хлорид иттрия с хлоридами некоторых металлов образует
двойные соли, например, 2YC13-3PtCl2 -4Н2О, YC13-2АиС13-16Н2О,
YCl3-3HgCl2-9H2O.
Оксихлорид иттрия, YOGI, образуется при нагревании безвод-
ного хлорида иттрия в атмосфере кислорода или хлорида иттрия
YC13>6H2O в токе воздуха или кислорода; YOGI плохо раство-
ряется в воде и растворяется в концентрированных кислотах.
Бромид иттрия, YBr3, получают нагреванием хлорида или
сульфида иттрия Y2S3 в токе НВг или нагреванием гидрата бро-
мида иттрия с NH4Br в вакууме при 550—600°:
YC13 + ЗНВг = YBr3 + ЗНС1 Y2S3 + 6HBr = 2YBr3 + 3H2S
Бромид иттрия представляет собой бесцветные, расплываю-
щиеся на воздухе кристаллы; т. пл. 904°, т. кип. 1467°; раство-
римы в воде и спирте, труднорастворимы в эфире. Известен
кристаллогидрат YBr3-9H2O.
’ Иодид иттрия, YI3, получают прямым взаимодействием исход-
ных компонентов при нагревании в атмосфере инертного газа
или нагреванием окиси Y2O3 с иодидом аммония или алюминия:
2Y + 3I2 = 2YI3 + 330 ккал
Y2O3 + 6MH4I = 2YI3 + 6NH3 + 3H2O
Y2O3 -|- 2AII3 = 2YI3 -|- A12O3
Иодид иттрия представляет собой бесцветные, расплывающиеся
на воздухе кристаллы с т. пл. 1000° и т. кип. 1307°; они раство-
ряются в воде и спирте и плохо растворяются в эфире.
Сульфид иттрия, Y2S3, получают при нагревании исходных
компонентов или восстановлением сульфата иттрия углем, а также
сильным нагреванием окиси иттрия Y2O3 в парах CS2 или, в атмо-
сфере H2S и сплавлением окиси иттрия с серой и карбонатом
ИТТРИЙ
43
натрия:
2Y + 3S = Y2S3 2Y2O3 + 3GS2 = 2Y2S3 + 3GO2
1450°
Y2(SO4)3 + 12C = Y2S3 + 12CO Y2O3 + 3H2S------>- Y3S3 + 3H2O
Сульфид иттрия представляет собой серовато-коричневые моно-
клинные кристаллы с плотностью 3,87 г/см?, плавящиеся при
1900—1950°, разлагающиеся водой и образующие двойные суль-
фиды с сульфидами щелочных металлов, например Y2S3*Na2S.
Оксисулъфид иттрия, Y2O2S, получают при действии серово-
дорода на сульфат иттрия; он образуется в виде серовато-белых
гексагональных кристаллов с плотностью 4,95 г!см?, с т. пл. 2120°.
Полисульфиды иттрия: дисульфид, YS2 (фиолетово-корич-
невый, плотность 4,25 г/см3), моносулъфид, YS (красный, плотность
4,51 г/см3, т. пл. 2040°), гептасулъфид, Y6S7 (моноклинные кри-
сталлы, плотность 4,10 г/см3, т. пл. 1630°).
Сульфат иттрия, Y2(SO4)3-8Н2О, выделяют из концентри-
рованных водных растворов сульфата иттрия, полученных обра-
боткой гидроокиси иттрия разбавленной H2SO4:
2Y(OH)3 + 3H2SO4 = Y2(SO4)3 + 6H2O
Соль Y2(SO4)3*8H2O выделяется в виде бесцветных моноклин-
ных кристаллов (плотность 2,558 г/см3), растворимых в воде
и разлагающихся при 700°.
Сульфат иттрия безводный, Y2(SO4)3, представляет собой
белый порошок (плотность 2,53 г/см3), растворимый в воде и раз-
лагающийся при нагревании.
Сульфат иттрия основной, 2Y203*S03*10H20, получают обра-
боткой сульфата иттрия разбавленным раствором аммиака.
Сульфат иттрия с сульфатами щелочных металлов образует
комплексные сульфаты общей формулы MeHY(SO4)3], Me*[Y2(SO4)6],
MeJ[Y2(SO4)7], где Me1 = Na+, K+, NH4. Известна кислота
H3[Y(SO4)3L
Нитрат иттрия, Y(NO3)3-6H2O, выделяется при комнатной
температуре из концентрированных водных растворов окиси или
гидроокиси иттрия в азотной кислоте:
Y2O3 + 6HNO3 = 2Y(NO3)3 + ЗН2О
Соль Y(NO3)3«6H2O образует бесцветные призмы, раствори-
мые в воде, спирте, азотной кислоте и разлагающиеся при нагре-
вании; при 100° соединение дегидратируется до Y(NO3)3«3H2O,
при 150—300° превращается в основной нитрат иттрия, а при 590°
разлагается на окись иттрия, двуокись азота и кислород.
Основной нитрат иттрия, Y8Oj4(NO3)4’17Н2О, выпадает при
кипячении водного раствора нитрата иттрия.
При концентрировании (25°) раствора, полученного действием
азотной кислоты на окись иттрия, выпадает основной нитрат
44
ПОБОЧНАЯ ПОДГРУППА III ГРУППЫ
YeO5(NO3)8-20Н2О в виде белых кристаллов с плотностью
2,431 г! см?.
Нитрат иттрия с хинолином образует аддукт Y(NO3)3 •
• 2C9H7N-10H2O.
Карбонат иттрия, Y2(CO3)3-ЗН2О, получают обработкой
растворов солей иттрия раствором бикарбоната щелочных метал-
лов, насыщенным СО2, или пропусканием газообразного СО2
через водную суспензию гидроокиси иттрия:
2Y(NO3)3 + 6NaHCO3 = Y2(CO3)3 + 6NaNO3 + 3CO2 -f- 3H2O
2Y(OH)3 + 3CO2 = Y2(CO3)3 + 3H2O
Карбонат иттрия представляет собой белый порошок, плохо
растворимый в воде, спирте, эфире и т. д. и растворимый в карбо-
натах щелочных металлов или в кислотах.
Кипячением растворов солей иттрия с карбонатами щелочных
металлов или аммония, гидролизом карбоната иттрия или дейст-
вием щелочей на карбонат иттрия получают основные карбонаты
переменного состава.
Карбонат иттрия с карбонатами аммония, натрия, таллия,
гуанидиния образует следующие соединения: NH4[Y(CO3)2]-Н2О,
Na[Y(CO3)2] >2Н2О, T1[Y(CO3)2-ЗН2О и т. д.
Ацетат иттрия, Y(CH3COO)3-4Н2О, выпадает при концен-
трировании растворов карбоната или окиси иттрия в уксусной
кислоте.
Ацетат иттрия представляет собой бесцветные триклинные кри-
сталлы, растворимые в воде и плохо растворимые в спирте и эфире.
Известны основной ацетат иттрия, Y(OH)(GH3COO)2, и ацетат
иттрия и гуанидиния, образующийся при смешивании концентри-
рованных растворов ацетата иттрия и ацетата гуанидиния.
Карбид иттрия, YC2, получают нагреванием в электрической
печи окиси иттрия с сахарным углем; он представляет собой серые
гексагональные кристаллы с плотностью 4,13 г!см?, довольно
твердые и хрупкие. Карбид иттрия взаимодействует с водородом,
кислородом, хлором, бромом, серой, селеном, сероводородом, амми-
аком, а также с хлористым водородом, бромистым водородом
и иодистым водородом.
Борид иттрия, YBe, получают электролизом расплава смеси
Y2O3-2B2O3 с MgO + MgF2 (1 : 15) при 990°; применяются воль-
фрамовые, молибденовые, ниобиевые, платиновые катоды и элект-
рический ток силой 20 а и напряжением 8,5 в.
Борид иттрия — окрашенное в темно-синий цвет веществ
(плотность 3,72 г/см?), реагирующее с расплавами щелочей, карбо-
натов, нитратов, гидросульфатов щелочных металлов, с концен-
трированной H2SO4 при нагревании и с концентрированной HNO3
на холоду.
ИТТРИЙ
45
Алкильные производные
Аддукт эфира и триэтилиттрия Y(C2H5)3-О(С2Н5)2 получают
действием магнийбромэтила на суспензию безводного хлорида
иттрия в эфире в атмосфере азота:
2YC13 + eQsHsMgBr 4- 2(С2Н5)2О =
= 2У(С2Н5)3-О(С2Н5)2 4- 3MgBr2 4" 3MgCl2
Соединение Y(C2H5)3-О(С2Н5)2 представляет собой бесцветную
жидкость (плотность 1,132 а/сл48 )с сильным сладковатым запахом;
окисляется на воздухе без воспламенения, смешивается в любых
пропорциях с бензолом, толуолом, ксилолом ит. д., разлагается
водой'иди спиртом с образованием Y(OH)(C2H5)2.
Хелатные соединения
А цетилацетонат иттрия,
получают обработкой водного раствора соли иттрия спиртовым
раствором ацетилацетона при значении pH 6,2, установленном
с помощью аммиака.
Ацетилацетонат иттрия, Y(C5H7O2)3,— белое твердое вещество
(плотность 1,403 г/см3), плавящееся при 133°, растворимое в воде,
эфире, хлороформе, бензоле, четыреххлористом углероде и серо-
углероде. Образует аддукты состава Y(C5H7O2)3 -NH3 (белые кри-
сталлы, т. пл. 129°), Y(G5H7O2)3 -C5H6N (белые иглы, т. пл. 121°),
Y(C5H7O2)3-CeH5NH2 (белые иглы, т. пл. 109°), Y(C5H7O2)3-СН3—
— CN (белое вещество, т. пл. 138°).
Гликолят иттрия, [Y(HO — СН2 — СОО)3], представляет
собой белое твердое вещество, образующееся при действии глико-
левой кислоты на окись или гидроокись иттрия.
Помимо перечисленных, известны и другие соединения иттрия, например:
гифид YH3, хлорит Y(C1O2)3, бромат Y(BrO3)3-9Н2О, иодат Y(IO3)3-3H2O,
перйодаты YIO5-8H2O, Y4I2O13-ИНгО, селениты Y2(SeO3)3-12Н2О,
Y2H2(SeO3)4-4Н2О, селенаты Y2(SeO4)3.8H2O, Y2(SeO4)3-9Н2О, Y2(SeO4)3.
•(NH4)2SeO4-6Н2О; фосфаты YPO4-2H2O, Y4(P2O7)3, Y(PO3)3, ортоарсенаты
YAsO4-2H2O, силикаты, иттросиликаты, хромит YCrO3, феррит YFeO3,
титанат Y2Ti2O7, ортованадат YVO4, ортониобат YNbO4, ортотанталат
YTaO4, молибдат Y2(MoO4)3 -4Н2О, ортоборат YBO3, комплексы Y2[Pt(CN)4]3 •
•21Н2О, Tl5Y[Cu(NO2)e]2, K5Y[Co(NO2)6]2, K5Y[Ni(NO2)6]2, NaY[Fe(CN)6] •
•5H2O, KY[Fe(CN)e]-8H2O, Y[Fe(CN)6] ^HjO, Y[Co(CN)e], лактат
Y(CH3 — CHOH — COO)3, оксалаты Y2(C2O4)3-9H2O, K3[Y(C2O4)3]-9H2O,
тартрат У2(С4Н4О6)3-5Н2О.
46
ПОБОЧНАЯ ПОДГРУППА III ГРУППЫ
ЛАНТАН La
Z == 57; ат. вес = 138,92
Валентность III, заряд 3+
Массовое число основного стабильного изото-
па 139
Электронная структура атома лантана: К -L -М •4s2ipe^d10 •
. 5з25рв5^-6s2.
Электронная структура атома и иона лантана (для 5р-, 5d-
и 6з-орбиталей):
5d‘ 6s2 5/>6 5d 6s
ПИШ ИI I I I I СЕЛ КФ ИЛ I I I I И □
La La31'
ИСТОРИЧЕСКАЯ СПРАВКА
В 1839 г. Мозандер выделил из цериевых земель желтую окись
церия и лантановую землю, разделенную им в 1841 г. на белую
окись лантана и красную землю, названную didim (двойник).
Лантан был выделен из «двойника» дробной кристаллизацией
двойных сульфатов или нитратов лантана с магнием или аммонием.
Название «лантан» происходит от греческого слова Lantanos,
что означает «скрытый». Действительно, он с трудом был обнару-
жен, так как образует бесцветные соли и обладает общими для
всех редкоземельных металлов реакциями.
Металлический лантан впервые был получен по методу Вёлера
восстановлением хлорида лантана LaCl3 металлическим калием
при нагревании. В 1875 г. Гиллебранд, Нертон и Мутман получили
металлический лантан электролизом расплавленного LaCl3.
РАСПРОСТРАНЕННОСТЬ В ПРИРОДЕ
Лантан встречается в природе в виде соединений (фосфата,
фторокарбоната, силиката и т. д.) в некоторых полиметаллических
минералах, которые кроме редкоземельных металлов, содержат
также кальций, иттрий, скандий, цирконий, гафний, торий, уран,
барий, железо и т. д.
Содержание лантана в земной коре составляет 6,5-IO-4 вес.%.
Важнейшими минералами лантана являются: монацит (La,
Се, Th, Са...)РО4, паризит Ca(La, Се, ...)2(CO3)3F2, корди-
лит Ba(La, Ce...)2(CO3)3F2, бастнезит (La, Ce)CO3F, синхизит
Ca(La, Се)(СО3)2Г2, ланханит (La, Се, Рг...)2(СО3)3-8Н2О, флюо-
церит (La, Ce)F3, ортит, лессингит, ринколит, роуландит и т. д.
Некоторые из этих минералов описаны в разделе, посвященном
распространенности в природе редкоземельных металлов.
ЛАНТАН
47
Процесс выделения лантана из соответствующих руд рассмат-
ривается при описании переработки руд редкоземельных металлов.
Там же указаны способы отделения лантана от сопутствующих
ему в природе элементов. (Методы отделения лантана от актиния
см. стр. 61.)
ПОЛУЧЕНИЕ МЕТАЛЛИЧЕСКОГО ЛАНТАНА
Металлический лантан получают металлотермическим восста-
новлением окиси или галогенидов лантана, термической диссоциа-
цией иодида лантана и электролизом расплавленных галогенидов
лантана.
Металлотермическое восстановление окиси
или галогенидов лантана
При магнийтермическом восстановлении окиси или хлорида
лантана (берется 10%-ный избыток магния по сравнению со сте-
хиометрически необходимым для восстановления количеством)
получают сплав магний — лантан:
La2O3 + 3Mg = 2La + 3MgO 2LaCl3 + 3Mg = 2La + 3MgCl2
Восстановление осуществляется в графитовом тигле, футеро-
ванном молибденом, в электрической печи.
Из магнийлантанового сплава магний удаляют перегонкой
в глубоком вакууме при 1300—1350°.
Металлотермическим восстановлением хлорида или бромида
лантана с металлическим кальцием (взятым в 10 %-ном избытке
по сравнению со стехиометрически необходимым для восстановле-
ния количеством) в танталовом тигле в атмосфере аргона или гелия
получают сплав кальций — лантан. Кальций из сплава удаляют
перегонкой в вакууме (нагревая сплав в тигле из окиси магния,
кальция или тантала).
Металлический лантан получают также металлотермическим
восстановлением хлорида лантана LaCl3 металлическим натрием,
калием, рубидием или цезием в вакууме (или в атмосфере аргона)
в аппаратуре из специального стекла при 250—350°.
Термическая диссоциация иодида лантана
Чистый металлический лантан получают термической диссо-
циацией иодида лантана Lal3 по способу Боера и Фаста.
Термическая диссоциация осуществляется в стеклянном сосуде
(в котором предварительно был создан вакуум), при соприкоснове-
нии паров Lal3 с вольфрамовой нитью, нагретой до 1300°.
48
ПОБОЧНАЯ ПОДГРУППА III ГРУППЫ
Иодид лантана Lal3, образующийся в результате прямого
взаимодействия исходных компонентов при 700°, превращается
в пар примерно при 800° и термически диссоциирует приблизи-
тельно при 1200°.
Электролиз расплава галогенидов лантана
Электролиз расплава хлорида (или фторида) лантана при 900°
проводят в графитовом тигле, являющемся анодом; катодом служит
молибденовый или вольфрамовый стержень, покрытый флюоритом
для защиты от хлора или фтора. На катоде выделяется металли-
ческий лантан 99,6—99,8 %-ной чистоты, который собирают в флюо-
ритовый тигель, помещенный внутри графитового тигля.
Последний обогревается снаружи, его температуру измеряют
при помощи термопары. Электролитическая ячейка для получения
лантана подобна ячейке для получения металлического скандия.
В качестве электролита можно использовать смесь LaCl3 — NaGl
(или КС1) — CaF2. В зависимости от катода, представляющего
собой расплавленный цинк или кадмий, получают сплавы Zn — La
или Cd — La, из которых цинк и кадмий удаляют перегонкой
в вакууме.
ФИЗИЧЕСКИЕ И ХИМИЧЕСКИЕ СВОЙСТВА
Лантан — блестящий серебристо-белый металл с плотно упако-
ванной кристаллической структурой типа плотнейшей гексаго-
нальной упаковки (модификация a-La, устойчивая между —195°
и 152°) или с кубической гранецентрированной кристаллической
решеткой (модификация P-La, устойчивая выше 352°). В интервале
между 152 и 352° могут существовать обе аллотропные модифи-
кации.
Лантан — тяжелый металл (плотность 6,18 г! см? при 20°),
очень ковкий, тягучий (в чистом состоянии), тверже олова, пла-
вится при 920°, кипит при 3469°, обладает очень слабыми пара-
магнитными свойствами.
С некоторыми металами, например с Ag, Си, TI, Са, Mg, Zn,
Cd, Hg, Al, Mn, Fe, Co, Ni, Pb, Sn и др., металлический лантан
при нагревании образует сплавы. С железом лантан образует
пирофорный сплав.
Нормальный потенциал системы La/La3+ равен —2,52 в (при
25°). Лантан является активным металлом, он разлагает воду при
комнатной температуре (быстрее при нагревании), растворяется
в разбавленных кислотах с образованием солей и выделением
водорода:
La + ЗН2О= La(OH)3 + з/2н2 La + ЗНС1 = LaCl3 + »/2Н2
ЛАНТАН
49
Металлический лантан хранят в бензоле, поскольку в сухом
воздухе он покрывается слоем La2O3, а во влажном воздухе
превращается в La(OH)3.
При комнатной температуре лантан взаимодействует с кисло-
родом и галогенами, образуя Ьа2О3 и LaX3 (где X = F“, С1",
Вг_, I-). При нагревании металлического лантана с водородом,
серой, азотом, фосфором, мышьяком, сурьмой и т. д. получают
соответственно LaH3, La2S3, LaN, LaP, LaAs, LaSb.
Приведенная ниже схема иллюстрирует химическую активность
лантана.
Комн. темп.
Нагревание
с водой —> La(OH)3
с НС1 -> LaCl3-7H2O
с H2SO4 —La2(SO4)3.9H2O
с HNO3 —La(NO3)3.6H2O
с кислородом —> La2O3
с галогенами —► галогениды
I лантана
Лантан —
' с водой —>- La2O3
с серой —>- La2S3
с азотом, фосфором, мышьяком,
сурьмой—>LaN, LaP, LaAs, LaSb
с водородом —>- LaH3
ПРИМЕНЕНИЕ
Металлический лантан используют при получении самария,
европия, иттербия и пирофорных сплавов.
Соли лантана применяют в стекольной промышленности при
изготовлении стекол для защитных очков (для работы с ультра-
фиолетовым излучением), поскольку такие стекла поглощают
ультрафиолетовые лучи.
Окись, гидроокись и галогениды лантана служат катализато-
рами.
Лантан входит в состав люминофоров в качестве активатора
флуоресцентных материалов.
СОЕДИНЕНИЯ (ОБЩИЕ СВОЙСТВА)
Известны многочисленные устойчивые простые, двойные или
координационные соединения, в которых лантан электроположи-
телен и трехвалентен. В химических соединениях лантан присут-
ствует в виде катиона La3+ или входит в состав комплексных
4—0101
50
ПОБОЧНАЯ ПОДГРУППА III ГРУППЫ
анионов [LaFJ , [LaFe]3 , [LaClgl3 , [LaBrJ , [LalJ , [Lale]3 ,
[La(SO4)2]-, [La(SO4)3]3-, [La(NO3)6]2-, [La(CO3)2J- и т. д.
Соли лантана бесцветны, гидролизуются, образуя гидроксосоли,
и проявляют склонность к образованию двойных и комплексных
солей. Основные соли лантана можно получить обработкой раство-
ров солей лантана малым количеством либо гидроокиси, либо
карбоната щелочного металла, либо NH4OH, (NH4)2S, (NH4)2CO3
и т. д. Окись и гидроокись лантана имеют более основной
характер, чем соответствующие соединения скандия и иттрия.
Неорганические соединения
Гидрид лантана, LaH3, с примесью ЬаН2 получают действием
чистого водорода на металлический лантан, нагретый до 210—290°:
2La ЗН2 = 2LaH3
Гидрид лантана (имеющий характер соединения включения)
образует хрупкие темно-синие кубические кристаллы с плотностью
5,14 г/см3, которые разлагаются при нагревании на составные
элементы, превращаются в La2O3nLaN при нагревании на воздухе,
а под действием воды (или в атмосфере влажного воздуха)
разлагаются с образованием La(OH)3 и выделением водо-
рода. По свойствам гидрид лантана напоминает гидриды щелочно-
земельных элементов.
Окись лантана, Ьа2О3, образуется при нагревании исходных
компонентов, при прокаливании на воздухе гидроокиси, нитрата,
карбоната, оксалата лантана или при сплавлении металлического
лантана с бурой:
2La -f- 3/2О2 = La2O3 -|- 428,6 ккал
310—380°
2La(OH)3-------> Ьа2О3+ЗН2О
200-250°
2La(NO3)3 • 6Н2О — La2O3 + 6N О2 + »/2О2
— bri2<J
900 — 1000°
La2(CO3)3-8H2O La2O3-p3CO2
—0H2U
800°
La2(C2O4)3. ЮН2О * Ьа2О3 + ЗСО2+ЗСО
— lUrl2U
Окись лантана диамагнитна, может быть получена в виде
белого аморфного порошка или в виде бесцветных кристаллов (куби-
ческая модификация устойчива до 400°, гексагональная — до 2317°)
с плотностью 6,57 г/см?, т. пл. 2317°, т. кип. 4197°. Вещество
обладает основным характером (приближается к окиси кальция),
поглощает двуокись углерода из воздуха, образуя карбонат; взаи-
модействует с водой, выделяя тепло; вытесняет аммиак из солей
аммония и в отличие от окислов редкоземельных элементов легко
ЛАНТАН
51
растворяется в разбавленных кислотах:
La2O3 + ЗСО2 = La2(CO3)3 La2O3 + 6NH4C1 = 2LaCl3 + 6NH3 + 3H2O
La2O3 + 3H2O = 2La(OH)3 La,O3 + 6HC1 = 2LaCl3 + 3H2O
Окись лантана хранят в условиях, исключающих доступ влаги
и СО2.
Гидроокись лантана, La(OH3)3, можно получить несколькими
способами:
La(NO3)3 + ЗКОН = La(OH)3 + 3KNO3 LaP + ЗН2О = La(OH)3+PHs
LaH3 + 3H2O = La(OH)3 + 3H2
4LaC2 + 12H2O = 4La(OH)3 + 3C2H2 + C2He
Гидроокись лантана — самое сильное основание в ряду гидро-
окисей редкоземельных металлов. Растворяется в разбавленных
минеральных кислотах или в водных растворах тартрата натрия
и калия, при обработке иодом дает подобно крахмалу синее окра-
шивание.
При нагревании гидроокись лантана дегидратируется, превра-
щаясь в конечном итоге в Ьа2О3:
200—260° 310—380°
2La(OH3) > 2LaOOH —— La2O3
—ZH2U —H2U
Если к соли лантана в растворе добавляют КОН и Н2О2,
образуется соединение Ьа(ОН)2-ООН -?гН2О.
Основной ацетат лантана получают обработкой при нагревании
раствора ацетата лантана малым количеством аммиака:
La(CH3COO)3 + МН4ОН = La(CH3COO)2(OH) + NH4CH3COO
Фторид лантана, LaF3, получают действием фтора на метал-
лический лантан или на карбид лантана при нагревании, а также
обезвоживанием кристаллогидрата ЬаЕэ^/гНгО с NH4F:
La + 3/2F2 = LaF3 -J- 421 ккал
LaF3-i/2H2O + NH4F = LaF3 + HF + NH3 + V2H2O
В слоистой гексагональной решетке кристаллов LaF3 (рис. 2)
каждый ион Las+ окружен пятью равноотстоящими (2,36 А)
ионами F-, образующими тригональную бипирамиду, и другими
шестью равноотстоящими (2,70 А) ионами F~, образующими три-
гональную призму.
Соединение LaF3 ^/гНгО получают обработкой концентрирован-
ных растворов хлорида лантана фтористоводородной кислотой
или фторидом калия, а также кипячением оксалата лантана
с раствором фторида щелочного металла. Этот гидрат фторида
выделяется в виде студнеобразного белого осадка или бесцветных
гексагональных кристаллов с плотностью 5,93 г!см\ т. пл. 1427°,
т. кип. 2327°; они плохо растворяются в разбавленных кислотах,
растворяются в концентрированных растворах фторидов щелочных
4*
52
ПОБОЧНАЯ ПОДГРУППА III ГРУППЫ
металлов с образованием октаэдрических кристаллов гексафтори-
дов (ацидосоли) Mei[LaFe],
Известны многочисленные примеры фторосолей: NalLaFJ,
KlLaFj, RbfLaFj, Na3[LaF6], K3[LaFe], RbJLaFJ, Gs3[LaFe]
и т. д.
Оксифторид лантана, LaOF, в виде бесцветных кубических
кристаллов (с решеткой типа фторида кальция) получают гидро-
лизом безводного фторида лантана в токе влажного воздуха при
Рис. 2. Структура LaF3.
800°, термическим разложением кристаллогидрата LaFg-1/^ Н2О
или действием окиси лантана на безводный фторид лантана в высо-
ком вакууме при 900°:
LaF3 + НОН LaOF + 2HF 2LaF3.V2H2O = LaOF + LaF3 + 2HF
LaF3 -p La2O3 3LaOF
Безводный хлорид лантана, LaCl3, получают действием газо-
образного хлора на металлический лантан, сульфид или карбид
лантана; действием сухого хлористого водорода на сульфид или
карбидлантана;действиемпаров монохлорида серына окись лантана;
действием хлористого аммония на окись лантана и дегидратацией
кристаллогидрата LaCl3 -ТНгО в токе сухого газообразного НС1:
2La -р ЗС12 = 2LaCl3 -р 263,6 ккал
La2S3 -р ЗС12 = 2LaCl3 -р 3S
LaC2 -p 3HC1 = LaCl3 -p 2C -p ®/2H2
LaCa -p »/2Cl2 = LaCl3 -p 2C
2La2O3 -p 6S2C12 = 4LaCl3 -p 3SO2 -p 9S
La2S3 -p 6HC1 = 2LaCl3 -p 3H2S
La2O3 + 6NH4C1 - 2LaCl3 + 6NH3 + 3H2O
ЛАНТАН
53
Хлорид лантана образует бесцветные гексагональные гигро-
скопичные кристаллы с плотностью 3,84 г/слг3, т. пл. 852°,
т. кип. 1747°, растворимые в воде, спирте, пиридине и плохо
растворимые в эфире и бензоле.
При охлаждении сиропообразного раствора, полученного
растворением Ьа2О3 в НС1, выделяются бесцветные, растворимые
в спирте триклинные кристаллы LaCl3 -7Н2О.
Хлорид лантана с хлоридами многих металлов образует двой-
ные соли, например, LaCl3 -PtCl4-13Н2О, LaCl3 -4HgGl2-12Н2О,
LaGl3-2BiCl3-8Н2О и комплексные хлориды Cs3[LaCle]-4Н2О.
Термическим разложением LaCl3 -7Н2О на воздухе получают
красные тетрагональные кристаллы оксихлорида лантана LaOCl,
устойчивые на воздухе (до 900°), плохо растворимые в воде или
разбавленных кислотах и легко растворимые в концентрирован-
ных кислотах.
LaCl3-7H2O = LaOCl + 2НС1 + 6Н2О
Бромид лантана, LaBr3, можно получить нагреванием хлорида
или сульфида лантана в токе газообразного НВг; нагреванием
окиси лантана в смеси паров S2C12 и НВг; нагреванием в вакууме
кристаллогидрата LaBr3-7H2O с NH4Br:
LaCl3 + ЗНВг = LaBr3 -f- ЗНС1 La2S3 + 6HBr = 2LaBr3 + 3H2S
La2O3 + 6HBr = 2LaBr3 + 3H2O
LaBr3.7H2O + NH4Br = LaBr3 -f- HBr + NH3 + 7H2O
Бромид лантана образует бесцветные гексагональные кри-
сталлы с плотностью 5,06 г! см?, т. пл. 783°, т. кип. 1577°, раство-
римые в воде, спирте, ацетоне, плохо растворимые в эфире. LaBr3
образует комплексные бромиды с бромидами щелочных металлов,
например MeHLaBrJ -nH2O.
При концентрировании на холоду (над H2SO4) раствора окиси
лантана Ьа2О3 в НВг выпадают бесцветные кристаллы LaBr3 •
7Н2О.
Нагреванием окиси лантана при 500—600° в парах брома
выделяют тетрагональные кристаллы оксибромида лантана LaOBr.
Иодид лантана, Lal3, получают при нагревании исходных ком-
понентов в инертной атмосфере; нагреванием окиси лантана
с NH4I при 350° или с АП3 при 450°; нагреванием окиси лантана
с алюминием и иодом в кварцевой трубке, а также действием
иодистоводородной кислоты на расплавленный безводный хлорид
лантана:
2La + 3I2 = 2LaI3 + 2 X 189,0 ккал
La2O3 -|- 6NH4I = 2LaT3 6NH3 -|- ЗН2О
La2O3 Ц- 2А1 -|- 3I2 = 2LaI3 -|- А12О3
La2O3 -|- 2АП3 = 2LaI3 -|- А12О3 LaCl3 -f- 3HI == Lal3 -J- 3HC1
54
ПОБОЧНАЯ ПОДГРУППА III ГРУППЫ
Иодид лантана образует желтые орторомбические кристаллы
с плотностью 5,26 г/сж3, т. пл. 761°, т. кип. 1402°, растворимые
в воде, спирте, ацетоне, пиридине и образующие с иодидами щелоч-
ных металлов комплексные иодиды общей формулы MeULalJ,
Mei[LaI6].
Антипирин с иодидом лантана образует соединение
[La(COC10H12N2)e]I3 в виде желтых, растворимых в воде кри-
сталлов.
Оксииодид лантана, LaOI, получают сплавлением в вакууме
иодида лантана с окисью лантана.
Сульфид лантана, La2S3, получают при нагревании исходных
компонентов, восстановлением La2(SO4)3 углем (при нагревании),
действием сероуглерода на окись лантана или нагреванием La2S3
с серой и карбонатом натрия:
2La 3S = La2S3 -4- 317,3 ккал
La2(SO4)3 + 12С = La?S3 + 12СО La2O3 + 3SC2 = La2S3 + 3CO + 3C
Сульфид лантана образует коричневые кубические кристаллы
с плотностью 5,01 г/см3 и т. пл. 2100—2150°.
Сульфид лантана, LaS, имеет вид золотистых кубических
кристаллов с плотностью 5,76 г/см3 и т. пл. 1970°.
Дисульфид лантана, LaS2, образует коричневые кубические
кристаллы с плотностью 4,775 г/см3.
Оксисулъфид лантана, La2O2S, представляет собой желтовато-
белые гексагональные кристаллы с плотностью 5,77 г/см3
и т. пл. 1940°. Для получения оксисульфида лантана сульфат
лантана нагревают при 750° в токе кислорода (для превращения
в La2O3-SO3), а затем La2O3*SO3 восстанавливают водородом
при 750° до La2O2S.
Сульфаты лантана. Безводный сульфат лантана,
La2(SO4)3, получают нагреванием окиси, гидроокиси, карбоната
или нитрата лантана с конц. H2SO4, а также дегидратацией
кристаллогидратов сульфата лантана.
Сульфат лантана представляет собой гигроскопичный белый
порошок с плотностью 3,60 г!см3, разлагающийся на воздухе при
1150°.
Известны кристаллогидраты La2(SO4)3-?гН2О, где п — 6, 8, 9
и 16.
Кристаллогидрат La2(SO4)3-9Н2О выделяют упариванием при
30—40° насыщенного водного раствора La2(SO4)3. Это бесцвет-
ные, слабо парамагнитные гексагональные кристаллы (плотность
2,82 г/см3), устойчивые при комнатной температуре.
Кристаллогидрат La2(SO4)3-8Н2О выделяется в виде бесцвет-
ных моноклинных кристаллов (устойчивых при комнатной темпе-
ратуре) при добавлении абсолютного спирта к раствору, получен-
ЛАНТАН
55
ному смешением стехиометрически необходимого количества сер-
ной кислоты с нейтральным раствором нитрата лантана.
Кристаллогидраты La2(SO4)3-6Н2О, La2(SO4)3-16Н2О неустой-
чивы при комнатной температуре.
Сульфат лантана образует комплексные сульфаты с многочис-
ленными сульфатами, например MeI[La(SO4)2], Mef[La(SO4)3], где
Me1 == К+, Т1+ и т. д. Известна кислота H3[La2(SO4)3].
Нитрид лантана, LaN, получают нагреванием металлического
лантана в атмосфере азота или в токе сухого NH3, нагреванием
окиси лантана с углем в атмосфере азота, действием азота на кар-
биды или гидриды лантана при нагревании:
900°
2La + N2--------->2LaN + 2x72,7 ккал
1200°
2LaC2 + N2-------> 2LaN + 4C
1200°
La2O3+ ЗС + N2-------> 2LaN + 3CO
800—900°
2LaH3 + N2-------> 2LaN + 3H2
Соединение внедрения LaN представляет собой белый аморфный
порошок, который при соприкосновении с водой разлагается
с образованием La(OH)3 и выделением NH3.
Нитраты лантана. Безводный нитрат лантана,
La(NO3)3, получают сплавлением окиси лантана с нитратом аммо-
ния:
La2O3 + 6NH4NO3 = 2La(NO3)3 + 6NH3 + 3H2O
Его нельзя получить дегидратацией кристаллогидратов нитрата
лантана, поскольку образуются основные соли.
Безводный нитрат лантана выделяется в виде белого порошка,
растворимого в воде, спирте, этилендиамине и т. д.; при нагре-
вании разлагается:
2La(NO3)3------>- 2LaONO34-4NO2+ О2
| 745°
La2O3-|- 2NO24- i/2O2
При концентрировании раствора окиси или гидроокиси лан-
тана в азотной кислоте выпадают бесцветные триклинные кри-
сталлы La(NO3)3-6Н2О — очень гигроскопичные, плавящиеся при
66,5°, разлагающиеся при температуре выше 126°, превращающие-
ся при хранении в эксикаторе над конц. H2SO4 в La(NO3)3 -Н2О.
Нитрат лантана образует двойные или комплексные нит-
раты с нитратами других металлов, например 2La(NO3)3 •
• 3MeIr(NO3)a.24H2O, где Me11 = Mg2+, Zn2+, Mn2+, Ni2+, Cu2+,
Fe2+, Cd2+ и t. д.; Mei[La(NO3)6]-nH2O, где Mei = NH+ Rb+,
Tl+ и т. д.; n — 1 или 4.
56
побочная подгруппа ш группы
Известны оксинитраты лантана: LaONO3, La8O5(NO3)8,
La2O3-LaONO3 и т. д.
Фосфид лантана, LaP, получают нагреванием при 400—500°
металлического лантана с избытком фосфора в вакууме.
Фосфид лантана образует очень неустойчивые черные кубиче-
ские кристаллы, которые под действием влаги воздуха разлагаются
с образованием La(OH)3 и выделением РН3.
Фосфаты лантана. Ортофосфат лантана,
LaPO4 -VsHsO, получают прокаливанием окиси лантана
с NH4NaHPO4 или смешиванием растворов сульфата лантана
и фосфата натрия:
La2O3 + 3NH4NaHPO4 2LaPO4 + Na3PO4 + 3NH3 + 3H2O
La2(SO4)3 + 2Na3PO4 2LaPO4 + 3Na2SO4
Соединение LaPO4-x/2H2O представляет собой белые гексаго-
нальные кристаллы с плотностью 4,28 г/см9 или белые моноклинные
кристаллы с плотностью 5,08 г/см9, плохо растворимые в воде.
Известен кислый ортофосфат лантана, Ьа2(НРО4)3, получен-
ный смешением растворов сульфата лантана с кислым фосфатом
натрия.
Дифосфаты лантана, Ьа4(Р2О7)3-8Н2О, ЬаНР2О7 •
• ЗН2О, Ьа2(Н2Р2О7)3, можно получить при взаимодействии
растворов сульфата лантана с различными количествами дифос-
фата натрия.
2La2(SO4)3 + 3Na4P2O7 + 8Н2О La4(P2O7)3 -8Н2О + 6Na2SO4
La2(SO4)3 + 2Na4P2O7 + 5Н2О 2LaHP2O7-3H2O + 3Na2SO4 + 2NaOH
La2(SO4)3 -p 3Na4P2O7 -p 6H2O —La2(H2P2O7)3 -p 3Na2SO4 -p 6NaOH
Триметафосфат лантана, LaP3O9, получают обработкой суль-
фата лантана гриметафосфатом натрия Na3P3O9:
La2(SO4)3 -р 2Na3P3O9 = 2LaP3O9 -р 3Na2SO4
Карбид лантана, LaC2, получают нагреванием смеси окиси
лантана с сахарным углем (или порошком графита) при 2000°
в электрической печи в атмосфере водорода:
La2O3 -Р 7С = 2LaCr + ЗСО
Карбид лантана образуется в виде желтых тетрагональных
кристаллов (плотность 5,02 г/см2), которые легко разлагаются
водой:
4LaC2 -р 12Н2О 4La(OH)3 + ЗС2Н2 -р С2Не
При нагревании карбид лантана взаимодействует с азотом,
хлором, бромом, серой, сероводородом, соляной, иодистоводород-
ной кислотами и аммиаком.
ЛАНТАН
57
Карбонат лантана, La2(CO3)3 -ЗНгО, образуется в виде белых
ромбических кристаллов, при обработке растворов соли лантана
раствором соды или поташа, насыщенными газообразным СО2, или
при пропускании двуокиси углерода в водную суспензию La(OH)3:
2La(NO3)3 + 6NaHCO3 = La2(CO3)3 + 6NaNO3 + 3CO2 + 3H2O
2La(OH)3 + 3CO2 = La2(CO3)3 + 3H2O
При кипячении растворов солей лантана с карбонатами щелоч-
ных металлов образуются основные карбонаты лантана переменного
состава, а в присутствии избытка карбоната щелочного метал-
ла образуются комплексные карбонаты: Na[La(CO3)2] -6Н2О,
K[La(CO3)2] -12Н2О, NH4[La(CO3)2J.
Карбонат лантана растворяется в разбавленных кислотах
с образованием соответствующих солей лантана и разлагается
при нагревании:
350—550° 800—905°
La2(CO3)3-8H2O------------> La2O(CO3)2----------->
—СО2—8Н2О 2 ' -СО2
- 1000°
* La2O2CO3 — ► La2O3
—GO2
Оксалат лантана, Ьа2(С2О4)3 ПОЩО, получают из растворимых
солей обработкой растворов нитрата (подкисленных 0,5 н. НС1)
избытком щавелевой кислоты:
2La(NO3)3 + ЗН2С2О4 = La2(C2O4)3 + 6HNO3
Оксалат лантана представляет собой белое вещество, плохо
растворимое в воде, которое при нагревании разлагается:
80° 320°
La2(C2O4)3-10H2O--—La2(C2O4)3-7H2O------—
—
400° 800°
—> La2(C2O4)3 ? La2O(CO3)2 —— > La2O3
—ZC(J2
При дегидратации оксалата лантана в интервале температур
80—320° можно получить его кристаллогидраты Ьа2(С2О4)3-ЗН2О,
Ьа2(С2О4)3-2Н2О, Ьа2(С2О4)3-Н2О.
Силицид лантана, LaSi2, выделяется на катоде при электро-
лизе расплава (1100°), состоящего из 30 г SiO2, 50 г СаСО3, 78 г
CaF2, 20 г СаС12 и 10 г Ьа2О3, в графитовом тигле (анод) с алюми-
ниевым катодом.
Силицид лантана образует гексагональные кристаллы серо-
стального цвета с плотностью 5,05 г/сл3, реагирующие с разбав-
ленными кислотами — HNO3, H2SO4, НС1.
•58
ПОБОЧНАЯ ПОДГРУППА III ГРУППЫ
Хелатные соединения
Ацетилацетонат лантана,
(,---------------------О н
z II I
СН3-С-С = С-С1
получают обработкой растворов солей лантана спиртовым раство-
ром ацетилацетона в присутствии небольшого количества аммиака
(pH = 7,8).
Ацетилацетонат лантана растворим в воде, спирте, эфире,
хлороформе, бензоле, четыреххлористом углероде. Он образует
аддукты с одной молекулой аммиака La(C5H7O2)3 -NH3, пиридина
La(C5H7O2)3-C5H5N и анилина Ьа(С5Н7О2)3-СвНбИНг-
Бензоилацетонат лантана, La(Gi0H9O2)3-ЗНгО, получают обра-
боткой растворов солей лантана спиртовым раствором бензоил-
ацетона с последующим добавлением спиртового раствора,
насыщенного аммиаком. Соль выделяется в виде желтых иголь-
чатых призм.
Купферонат лантана, [La{C8H5N(NO)O}3], получают в виде
желтых кристаллов с т. пл. 184° обработкой хлорида лантана
(рН = 3—4 устанавливают с помощью НС1) 0,2 М раствором
купферона (нитрозофенилгидроксиламин).
Купферонат лантана существует в двух таутомерных формах:
/ .О —N \
La(\ II )
\ XO = N-С8Н6/3
/ O = N \ -1
Lal \ I j
\ О — N — СвН5/3-
2
Кроме приведенных, известны и другие соединения лантана, например:
перхлорат La(C104)3, бромат La(BrO3)3-9H2O, иодат La(IO3)3, перйодаты
La(IO4)3-2Н2О, LaIO5-2H2O, сульфит La2(SO3)3-3H2O, тиосульфат
La2(S2O3)3, селенид La2Se3, арсенид LaAs, антимонид LaSb, борид LaBe,
бораты LaBO3, La2B4O9, германат La2GeO5, кобальтат LaCoO3, хромит
LaCrO3, манганит LaMnO3, феррит LaFeO3, метаванадат LaV3O9, хромат
La2(CrO4)3-8H2O, двойные хроматы La2(CrO4)3-K2CrO4-2H2O, молибдаты
La2(MoO4)3, La3MoO6, La2(MoO4)3 •Na2MoO4-2H2O, (NH4)3La[Mo7O24]-12H2O,
вольфраматы La2(WO4)3-3H2O, NaLa(WO4)2, Na5La(WO4)4, перренаты
La(ReO4)3-raH2O (« =2, 3), гексафтороплатинат La2[PtF6]3, гексацианофер-
рат(Ш) La[Fe(CN)e] -4,5H2O, гексацианоферрат(Н) Me+La[Fe(CN)6] -nH2O (где
Me+ =Na+, K+), гексацианокобальтат(Ш) La[Co(CN)6], цианамид La2(CN2)3,
ацетат La(CH3COO)3-l,5H2O, цитрат LaC6H5O7-7H2O.
АКТИНИЙ Ac
Z = 89; ат. вес = 227,035
Валентность III, заряд 3+
Массовые числа природных изотопов 227 и 228
Электронная структура атома актиния: К 'L -М -N •5s25pe5d10 •
* б^б/бй1-7s2.
АКТИНИЙ
59
Электронная структура атома
и 78-орбиталей:
и иона актиния для Qp-, Qd-
6р6 6dl 7s* 6р6 6d 7s
[КПЛЕ] И I I I I И 1ФФ+1 Г I I I I □
Ac Ac3 +
ИСТОРИЧЕСКАЯ СПРАВКА
В 1899 г. Дебьерн в остатках после переработки смоляной
обманки наряду с полонием и радием, открытыми в 1898 г. Пьером
и Марией Кюри, нашел новый радиоактивный элемент, названный
им actinium.
Независимо от него Гизель в 1902 г., исследуя фракцию, содер-
жащую редкоземельные элементы (выделенные из смоляной
обманки), нашел радиоактивный продукт, названный им emanium.
В 1904 г. Дебьерн и Гизель, сравнивая методы получения и свой-
ства вновь открытых ими радиоактивных элементов, пришли
к выводу, что это один и тот же элемент.
После установления закона радиоактивного распада, открытого
Содди и Фаянсом, актиний окончательно нашел свое место в перио-
дической системе элементов как аналог лантана.
Существование взаимосвязи между ураном и актинием (пред-
положенное в 1908 г. Болтвудом, который установил, что в мине-
ралах урана отношение Ac/U примерно постоянно) было оконча-
тельно установлено после открытия урана (Антонов, 1911 г.), прото-
актиния (Ган, Мейтнер и одновременно Содди, Крэнстон и Флекк,
1918 г.) и актино-урана 235U (Демпстер, 1935 г.).
Чистый актиний был впервые получен Хагеманном в 1947 г.
искусственным превращением радия под действием нейтронов.
Мезоторий 2 был открыт в 1908 г. Ганом расщеплением мезо-
тория 1, который является вторым природным изотопом актиния.
ИЗОТОПЫ
Все изотопы с атомным номером 89 и массовыми числами 221,
222, 223, 224, 225, 226, 227 (Ас), 228 (MsTh2), 229 и 230 выделены
и радиоактивны.
Важнейший изотоп 227Ас, названный актинием, и изотоп 228Ас
(или MsTh2) являются естественными продуктами радиоактивного
распада и принадлежат радиоактивным рядам актиния и тория
(см. т. I, стр. 135) и характеризуются массовыми числами (4и + 3)
и 4п.
Среди продуктов деления в ряду 238U нельзя выделить ни одного
элемента с атомным номером 89. Ряд нептуния (4и + 1), элементы
60
ПОБОЧНАЯ ПОДГРУППА III ГРУППЫ
которого существуют в природе только в ничтожных количествах,
содержит изотоп 225Ас.
В 1945 г. Петерсон, подвергая радий (1 мг) действию нейтронов
в ядерном реакторе, получил искусственным путем изотоп 227Ас:
я-
2||Ra(n, 7)227Ra->227 Ac
Искусственные изотопы актиния образуются в основном при
бомбардировке тория нейтронами, а-частицами, протонами или
ускоренными дейтронами.
РАСПРОСТРАНЕННОСТЬ В ПРИРОДЕ
Природный изотоп 227Ас присутствует во всех минералах урана
в очень малых количествах. Так, в 1 т смоляной руды содержатся
400 мг радия и 0,15 мг актиния.
Природный изотоп 228MsTh2 находится в минералах тория.
В земной коре актиний содержится в количестве 2,3 ПО-15 вес. %.
ВЫДЕЛЕНИЕ ИЗОТОПА 227Ас
Изотоп 227Ас выделяют из минералов урана и из выдержанных
длительное время отработанных препаратов протактиния.
Выделение актиния из минералов урана довольно затрудни-
тельно, поскольку его концентрация в этих минералах мала,
а по химическим свойствам он близок к редкоземельным элементам.
Химической переработкой минералов урана добиваются отде-
ления актиния и лантанидов от остальных сопутствующих эле-
ментов, а затем отделяют актиний от лантанидов.
Остатки от химической переработки смоляной обманки после
выделения урана содержат соединения свинца, висмута, меди,
железа, кальция, бария, тория, церия, актиния, лантана, празео-
дима, неодима, самария и др. Из содержащих актиний хлоридов
(остатков от переработки смоляной обманки) различными реак-
циями осаждения выделяют свинец, висмут, медь, железо, кальпий
барий, торий и, наконец, церий.
Отделение актиния и лантанидов от остальных сопутствующих
элементов осуществляют путем соосаждения с гидроокисью или
фторидом лантана (в отсутствие тория, циркония, радия) или
с йодатом циркония и сульфатом бария или свинца (когда актинию
сопутствуют малые количества лантанидов).
Актиний и лантан отделяют от празеодима, неодима и самария
дробной кристаллизацией двойных нитратов этих элементов с нит-
ратом аммония. Обогащение актинийсодержащего лантана воз-
можно дробным осаждением оксалатов в сильно азотнокислой
среде.
АКТИНИЙ
61
Отделение актиния от лантана осуществляют дробной кристал-
лизацией двойных нитратов этих элементов с магнием или марган-
цем; хроматографией на бумаге (ватман № 1) при 60° в сложном
растворителе (100 мл бутанола, 30 мл ацетилацетона, 5 мл уксус-
ной кислоты и 65 мл воды); электрохроматографией на бумаге
(ватман № 1), используя в качестве электролита 1 %-ный раствор
лимонной кислоты; разделением на катионитах.
Из отработанных препаратов протактиния можно выделить
актиний в чистом состоянии как с помощью органических раство-
рителей, так и с помощью ионообменных смол, поскольку в этом
случае актиний не загрязнен редкоземельными элементами.
ПОЛУЧЕНИЕ МЕТАЛЛИЧЕСКОГО АКТИНИЯ
Металлический актиний получают восстановлением в вакууме
фторида актиния парами лития при 1100—1275° или хлорида
актиния парами калия при 350°.
При электролизе спиртового раствора лантана и мезотория 2
в присутствии 15—25% ацетона используют, серебряный катод
при плотности тока 1—2 маЛсм?. В течение нескольких минут
на катоде осаждается лантан и мезоторий 2.
Искусственным путем актиний можно получить в ядерном
реакторе при облучении радия нейтронами.
ОЧИСТКА
Важнейшим изотопам актиния сопутствуют примеси других
изотопов, например 227RaAc, 22SAcX, 223АсК, 211АсВ в случае
изотопа 227Ас и 228MsTh!, 228RaTh, 224ThX, 22eRa в случае изотопа
228MsTh2.
Очистка изотопов актиния 227Ас, 228MsTh2 осуществляется
удалением названных примесей путем осаждения, которое про-
водят в присутствии веществ, играющих роль соосадителей; при-
меняют также экстракцию 0,25 М бензольным раствором теноил-
трифторацетона* при контролируемом pH и, наконец, ионный
обмен.
Изотоп 227RaAc отделяют осаждением тиосульфатом натрия
в слабокислом растворе или экстрагируют 0,25 М бензольным
раствором теноилтрифторацетона при pH 1.
* Тиофеикарбонилтрифторацетон, часто обозначаемый ТТА,
l-с СН2—С—CF3
62
ПОБОЧНАЯ ПОДГРУППА Ш ГРУППЫ
Экстракция актиния ТТА основывается на образовании в среде
неполярных органических растворителей (например, бензола) кри-
сталлического комплекса. Степень экстракции зависит от кислот-
ности растворов и от присутствия в водной среде неорганических
ионов, также образующих кристаллические комплексы с ТТА.
Изотоп 223АсХ выделяют соосаждением с хроматом бария в аце-
татной буферной смеси или он остается в водной фазе вместе
с 223АсК, когда 227Ас экстрагируется 0,25 М бензольным раство-
ром теноилтрифторацетона при pH 5,5—6.
Изотоп 228MsTh2 отделяют от 228MsTh1 элюированием на колонке
со смолой дауэкс 50 раствором цитрата аммония при pH 4,3 (или
лактатом аммония при pH 5—7), а также соосаждением с соедине-
ниями циркония, бария и т. д.
ФИЗИЧЕСКИЕ И ХИМИЧЕСКИЕ СВОЙСТВА
Актиний — серебристо-белый металл, имеющий кубическую
гранецентрированную решетку, т. пл. 1050°, т. кип. 3300°. В тем-
ноте заметно голубоватое излучение, обусловленное его радиоак-
тивностью.
Для наиболее интенсивного излучения актиния характерны
следующие длины волн: 2625,44; 2952,55; 3487,59; 3565,59; 3863,11;
4088,43; 4168,40; 4179,98; 4183,12; 4507,20; 4716,58.
Актиний проявляет сходство с лантаном, кальцием и металлами
цериевой группы; он радиоактивен и в результате радиоактивного
распада, в конечном итоге переходит в стабильный изотоп 2О’РЬ.
а-Активность чистого актиния очень слаба, но быстро растет
вследствие образования радиоактиния, актиния X и их производ-
ных. P-Активность, сильно пониженная у чистого актиния, также
растет по мере образования актиния X, причем спустя 2 часа между
ними устанавливается равновесие.
С химической точки зрения актиний является активным метал-
лом: он очень легко окисляется на воздухе и разлагает воду с обра-
зованием гидроокиси актиния и выделением водорода. Нормаль-
ный потенциал системы Ас/Ас3+ равен —2,6 в.
Актиний, так же как радий и плутоний, относится к радио-
активным элементам.
Идентификация и дозировка актиния осуществляются радио-
активными методами.
ПРИМЕНЕНИЕ
В лаборатории актиний служит источником получения ра-
диоактиния, актиния X и других продуктов радиоактивных пре-
вращений.
АКТИНИЙ
6»
СОЕДИНЕНИЯ (ОБЩИЕ СВОЙСТВА)
Известен ряд соединений, в которых актиний электроположи-
телен и трехвалентен.
Соединения актиния окрашены в белый цвет, за исключением
сульфида (черного цвета). В отличие от соответствующих соеди-
нений лантана соли актиния трудно гидролизуются, так как обла-
дают большей основностью.
Многие соединения актиния изоморфны соответствующим соеди-
нениям лантана.
К трудно растворимым в воде соединениям актиния относятся
гидроокись, фторид, фторосиликат, ортофосфат, карбонат, оксалат
и двойной сульфат калия и актиния.
Актиний количественно соосаждается с гидроокисью лантана
при pH 8,4—8,8 или с фторидом лантана в сильно кислой среде
(5 н. раствор HNO3), или карбонатом лантана и йодатом циркония
(в 0,2 н. растворе HNO3).
Неорганические соединения,
Окись актиния, Ас2О3, получают прокаливанием в кислороде
при 1100° оксалата актиния. Она представляет собой бесцветные
гексагональные кристаллы с плотностью 9,19 г/см3, основной
характер которых выражен более резко, чем у Ьа2О3.
Гидроокись актиния, Ас(ОН)3, получают в виде студнеобраз-
ного белого осадка при обработке растворов солянокислых или азот-
нокислых солей актиния аммиаком или щелочами.
Фторид актиния, AcF3, получают, обрабатывая плавиковой
кислотой гидроокись актиния (700°) или растворы солей актиния.
Ас(ОН)3 + 3HF = AcF3 + ЗН2О
Фторид актиния образует бесцветные гексагональные кри-
сталлы с плотностью 7,88 г/см?, т. пл. 1327° и т. кип. 2277°. При
нагревании до 800° на воздухе или до 900—1000° в атмосфере амми-
ака AcF3 превращается в оксифторид AcOF:
AcF3 + 2NH3 + Н2О = AcOF + 2NH4F
Оксифторид актиния образуется в виде белых кубических
кристаллов с плотностью 8,28 г/см3.
Хлорид актиния, АсС13, получают нагреванием при высокой
температуре гидроокиси или безводного оксалата актиния с СС14:
4Ас(ОН)3 + ЗСС14 = 4АсС13 + 6Н2О + ЗСО2
Хлорид актиния образуется в виде бесцветных гексагональных
кристаллов с плотностью 4,81 г/см?, т. пл. 927° и т. кип. 1757°.
При нагревании до 1000° в токе аммиака превращается в окси-
хлорид AcOCl.
64
ПОБОЧНАЯ ПОДГРУППА III ГРУППЫ
Оксихлорид актиния представляет собой белые тетрагональные
кристаллы с плотностью 7,23 г!см?.
Бромид актиния, АсВг3, получают при нагревании окиси или
оксалата актиния с бромидом алюминия при 750°:
Ас2О3 + 2А1Вг3 = 2АсВг3 -j- А12О3
Бромид актиния представляет собой бесцветные гексагональ-
ные кристаллы с плотностью 5,85 г!см?, т. пл. 827° и т. кип. 1597°.
Оксибромид актиния, АсОВг, выделен в виде белых тетраго-
нальных кристаллов с плотностью 7,89 г/см?.
Иодид актиния, Ас13, можно получить нагреванием окиси
актиния с иодидом алюминия при 700° или действием иодида
аммония на оксалат актиния при 500°:
Ас2О3 2АП3 = 2Ас13 А12О3
Ас2(С2О4)3 +;6NH4I = 2AcI3 + 6NH3 + ЗСО -f- ЗСО2 + ЗН,0
Иодид актиния — белое вещество, кипящее при 1407° и гидро-
лизующееся в парах аммиака при 700° с образованием оксиио-
дида AcOI.
Сульфид актиния, Ас2О3, получают нагреванием окиси или
оксалата актиния при 1400° в графитовом тигле в атмосфере смеси
H2S и CS2.
Сульфид актиния образует черные кубические кристаллы
с плотностью 6,75 г/см?.
Двойной сульфат калия и актиния выделяется в виде белого
кристаллического осадка при действии насыщенного раствора
K2SO4 на 0,003 н. раствор актиния в 0,1 н. растворе H2SO4.
Ортофосфат актиния, АсРО4 •1/2Н2О, образуется при высу-
шивании (70°) осадка, полученного обработкой растворов солей
актиния в 0,1 н. НС1 1н. раствором дигидрофосфата натрия:
АсС13 + NaH2PO4 = АсРО4 -f- NaCl -f- 2НС1
Ортофосфат актиния представляет собой белые гексагональные
кристаллы с плотностью 5,48 г/смД при нагревании до 700° пре-
вращается в безводную соль АсРО4.
Оксалат актиния получают в виде белого осадка обработкой
0,003 н. раствора актиния в 0,1н. НС1 1 н. раствором оксалата
аммония.
ПОБОЧНАЯ ПОДГРУППА IV ГРУППЫ
ПЕРИОДИЧЕСКОЙ СИСТЕМЫ
(ПОДГРУППА ТИТАНА)
ХАРАКТЕРИСТИКА ПОДГРУППЫ
В побочной подгруппе IV группы периодической системы нахо-
дятся титан (Ti), цирконий (Zr) и гафний (Hf) *.
В табл. 16 приведена электронная структура элементов титана,
циркония и гафния.
Таблица 16
Атомы этих элементов имеют на последнем электронном уровне
по 2 электрона и по 2.6.2 электрона на предпоследнем электронном
уровне. d-Орбитали предпоследнего уровня титана, циркония
и гафния заполнены неполностью и содержат по 2 электрона.
Вступая в химические реакции, атомы титана, циркония и гафния
отдают 2, 3 или 4 электрона, образуя соединения, в которых
они двух-, трех- или четырехвалентны.
Самым устойчивым соединениям элементов подгруппы титана
соответствует степень окисления, равная четырем, поскольку элек-
троположительные четырехвалентные ионы этих элементов имеют
на последнем электронном уровне устойчивую конфигурацию
из 8 электронов.
В табл. 17 приведены наиболее важные константы элементов
подгруппы титана.
В твердом состоянии титан, цирконий и гафний представляют
собой блестящие, серебристо-белые металлы. Титан обладает плот-
* К этой подгруппе относится и курчатовий (Ku), Z = 104 (см. при-
мечание на стр. 7).— Прим, перев.
5—0101
Таблица 17
Элемент Титан Ti Цирконий Zr Гафний Hf
Цвет Серебристо-белый Серебристо-белый Серебристо-белый
Кристаллическая структура Плотнейшая гексаго- нальная ниже 882°, кубическая гранецент- рированная выше 882° a-Zr — плотнейшая гек- сагональная; P-Zr — ку- бическая объемноцент- риро ванная Плотнейшая гексаго- нальная и кубическая объемноцептрирован- ная
Атомный номер 22 40 72
Атомный вес 47,90 91,22 178,49
Атомный радиус, А (по Полингу) 1,47 1,60 1,62
Ионный радиус Ме4+, А (по Гольд- шмидту, Полингу и Аренсу) 0,64; 0,68; 0,68 0,87; 0,80; 0,79 0,84; —; 0,78
Атомный объем (при 20°), см3/г-атом 10,7 14,6 13,43
Плотность (при 20°), г/см3 4,49 6,25 13,31
Твердость по Бринеллю, кг/мм2 160 107—200 78
Твердость по шкале Мооса 4 4,5 —
Температура плавления, °C 1677 1855 2222
1 I
Температура кипения, °C 3277 4474 5280
Удельная теплоемкость при 20°, кал/г • град 0,113 0,068 0,0341
Сопротивление р-106 при 0°, ом-см 43,5 41,0 30,0
Электропроводность (Hg = l) 2,1 2,3 3,1
Магнитная восприимчивость %-10-6 при 18°, эл.-магн. ед. 1,25 -0,45
Теплота перехода атомов в газообраз- ное состояние, ккал/г-атом 112 125
Потенциал ионизации, эв Me —> Ме+ + е~ Me1’ —>• Ме2++е~ Ме2+ —>- Ме3+4~е~ Ме3+ —>- Ме4+ ; е^ Ме4+ —>• Me5+-j-e~ 6,82 13,57 27,47 43,30 99,4 6,84 13,13 22,98 34,83 82,6 7,0 14,90 21,00 31,00
Потенциал ионизации, ккал[г-атом Me —>• Ме+4-е_ Ме+ —> Ме2+ + е_ Ме2+ —» Ме3+Ц е~ Ме3+ —>- Ме4+4~е_ 156,9 313,0 636,0 992,4 159,5 322,0 553,1 779,0 341
Нормальный потенциал (при 25°), в Ti/Ti3+—1,63 Zr/Zr4+—1,56 Hf/Hf4+—1,70
Нормальные потенциалы окислитель- но-восстановительных систем, в Ti2+/Ti3+—0,37 Ti3+/Ti4+— 0,20
Валентность II, III, IV (П), (III), IV (П), (III), IV
Массовые числа природных изотопов 48, 46, 47, 49, 50 9>, 92, 94, 91, 96 180, 178, 177, 179,176,174
Распространенность элементов в зем- ной коре, вес. % 0,65 0,025 4,0-10“4
68
ПОБОЧНАЯ ТПОДГРУППА IV ГРУППЫ
ностью 4,49 г/сл43 при 20° и относится к легким, а цирконий и гаф-
ний — к тяжелым металлам. Все они тугоплавки (температура
плавления от 1677 до 2222°, температура кипения от 3277 до 5280°).
В чистом состоянии обладают хорошими механическими свой-
ствами. При загрязнении кислородом, азотом, углеродом, бором,
водородом и т. д. теряют пластичность и становятся твердыми
и хрупкими.
Значения атомных радиусов и объемов циркония и гафния,
а также ионных радиусов Zr4+ и Ш4+ очень близки.
Для металлов подгруппы титана известно большое число при-
родных изотопов. Эти металлы образуют сплавы с железом, хро-
мом, марганцем, ванадием, алюминием, медью, углеродом, серой,
азотом, фосфором, бором и т. д. В порошкообразном состоянии
они способны поглощать большие количества водорода.
С химической точки зрения металлы подгруппы титана не
активны, устойчивы на воздухе или в воде при нормальных
условиях. При повышенных температурах становятся очень актив-
ными по отношению к кислороду, галогенам, сере, азоту, углероду,
бору и т. д.
Me + О2 = МеО2
Me + 2Х2 = МеХ4
Me + 2S = MeS2
Me + x/2N2 = MeN
Me + C = MeC
Me + 2B = MeB2
X = Cl, F, Br, I
Титан, цирконий и гафний растворяются в царской водке
или в смеси плавиковой и азотной кислот. Титан и цирконий
реагируют с расплавленным NaOH, а гафний взаимодействует
с расплавленным KHF2:
ЗМе + 12НС1 + 4HNO3 = ЗМеС14 + 4NO + 8Н2О
Me + 4NaOH = Na4MeO4 + 2Н2
Me + 4KHF2 = K2[MeFe] + 2KF + 2H2
При обработке титана, циркония и гафния концентрированной
плавиковой кислотой образуется TiF3, ZrF4 и HfF4.
В природе элементы подгруппы титана встречаются в виде
соединений (титан — в виде двуокиси, титанатов, ниобатов и т. д.,
а цирконий и гафний сопутствуют друг другу в виде двуокисей
и силикатов) в различных минералах, достаточно распространен-
ных в земной коре.
Обогащение титан-, цирконий- и гафнийсодержащих руд осуще-
ствляется гравитационными, электромагнитными, электростати-
ческими и флотационными методами.
Металлы подгруппы титана получают металлотермическим вос-
становлением тетрахлоридов или гексафторосолей в вакууме или
в атмосфере инертного газа (аргон, гелий), а также термической
диссоциацией тетраиодидов или электролизом расплавленных
ПОБОЧНАЯ ПОДГРУППА IV ГРУППЫ
69
в смеси с KG1 солей Me*[MeIVF0L Для очистки металлов применяют
метод Ван-Аркеля — де Бура, заключающийся в термической
диссоциации тетраиодида, полученного действием паров иода
на сырой металл. Превращение порошкообразного или губчатого
титана и гафния в плотный металл осуществляется прессованием,
а также спеканием или плавлением в вакууме (в атмосфере инерт-
ного газа) в индукционной или электродуговой печи.
Металлы подгруппы титана и их сплавы имеют огромное значе-
ние для современной техники, как это будет показано при рас-
смотрении этих элементов.
Двух- и трехвалентные соединения титана, циркония и гафния
с увеличением атомного номера становятся все менее устой-
чивыми. Они являются восстановителями и окрашены в черный,
черно-коричневый, фиолетовый и зеленый цвет.
Четырехвалентные соединения этих элементов, как правило,
устойчивы, бесцветны или окрашены в желтый цвет. Некоторые
соединения имеют фиолетовую или красную окраску.
В табл. 18—20 приведены наиболее важные константы двуоки-
сей и тетрагалогенидов этих элементов.
При сжигании металлов подгруппы титана в кислороде или
прокаливанием соединений общей формулы Н4МеО4, соответ-
ственно Ме(ОН)4, образуются устойчивые, тугоплавкие, плохо
растворимые в воде и разбавленных кислотах и щелочах двуокиси
МеО2. Последние взаимодействуют с конц. H2SO4 при нагревании
и с расплавленными щелочами.
Сплавлением двуокисей титана, циркония и гафния со щело-
чами получают соли общей формулы Me£MeIVO3 или MeJMeIVO4,
которые в большинстве трудно растворимы в воде, а растворимые,
переходя в раствор, полностью гидролизуются.
Двуокись титана обладает слабо кислыми свойствами, a ZrO2
и ЙГОг — слабо основными.
Соединения H4TiO4, Zr(OH)4, Hf(OH)4 (или в виде гидрати-
рованных двуокисей МеО2 -2Н2О) образуются при обработке
растворов соответствующих тетрагалогенидов щелочами и пред-
ставляют собой гелевидные белые осадки, плохо растворимые
в воде, обнаруживающие очень слабо выраженные кислые свойства,
вследствие чего они почти не реагируют со щелочами. Основной
характер соединений усиливается от титана к гафнию, одновре-
менно появляется способность растворяться в сильных кислотах.
Тетрагалогениды металлов подгруппы титана, за исключением
TiF4, ZrF4, растворяясь в воде, гидролизуются, образуют аддукты
с аммиаком, пиридином, анилином или другими аминами, а также
с РС13, РОС13, РС1а и др. Они образуют также комплексные соли
с галогенидами щелочных металлов. Координационные числа
у галогеносолей для титана 5, 6, 7, циркония — 5, 6, 7, 8, гаф-
ния — 6 и 7.
Таблица 18
Соединение TiO2 ZrO2 ню2
Вид Белый порошок или бесцветные кристаллы Белый порошок или бес- цветные кристаллы Белый порошок или бес- цветные кристаллы
тетраго- нальные (рутил) ромбиче- ские (брукит) тетраго- нальные (анатаз) моноклин- ные тетраго- нальные моноклин- ные тетраго- нальные
Молекулярный вес 79,899 123,219 210,489
Плотность (при 20°), г/сма 4,2-4,3 4,1-4,2 3,6-3,95 5,8 6,1 9,98 10,47
Температура плавления, °C 1855 2677 2790
Температура кипения, °C Разлагается выше 2927° 4300 —
Теплота образования, ккал/моль 225,5 261,5 266,1
Таблица 19
Соединение TiF4 ZrF4 HfF4 TiCl4 ZrCl4 HfCl4
Вид Расплываю- щийся белый по- рошок Бесцветные гек- сагональные кристаллы Бесцветные крис- таллы Бесцветная (чуть желто- ватая) жид- кость Блестящие белые кристаллы Белый поро- шок
Молекулярный вес 123,886 167,206 254,476 189,712 233,032 320,302
Плотность (при 20°), г/см" 2,79 4,43 7,13 1,76 2,80 —
Температура плавле- ния, °C 284 912 927 -21,4 437 (под давле- нием) 522 (под давле- нием)
Температура кипе- ния, °C 327 — — 136,5 Сублимируется при 331 Сублимируется при 317
Теплота образования, ккал/моль 393,6 456,8 435 194,9 234,5 237
Образует ацидосоли типа Me2[TiF0] Men[TiFe] Me|[TiF7] Me^ZrFsJ-HzO Me|[ZiFe]-nH2O Mel[ZrF7] Me|+[ZrF8]- (6 или 12)H2O Me2[HfF6] Me|[HfF7] Me2[TiCl0]-2Н2О Me|[ZrCl6] Me2[HfCl6]
Аддукты TiF4-4NH3 TiF4-2NH3 TiF4-C5H5N TiCl4-nNH3 (ra = 8, 6, 4) TiCl4-2C5H5N TiCl4-2CeH5NH2 TiCl4-PCl3 TiCl4-PCl5 TiCl4-2POCl3 TiCl4-SCl4 ZrCl4-raNH3 (га = 8, 4, 2) ZrCl4-4C5H5N ZrC]4-4CH3NH2 ZrCl4.4C2H5NH2 ZrCl4-4C3H7NH2 ZtC14.4C6H5NH2 3ZrCl4-2POCl3 2HfCl4-PCl5 3HfCl4-2POCl3
Таблица 20
Желтые кри- сталлы 686,108 1 477 (иол дав- лением) Сублимирует- ся при 427 175
’PZ Желтые окта- эдрические кристаллы 598,838 1 499 (под дав- лением) Сублимирует- ся при 431 160
’’III Красные ку- бические кристаллы 555,518 1 155 377 131
’Jain Бесцветные кристаллы 498,126 1 420 (под дав- лением Сублимирует- ся при 322 225
’-la-iz Белый порошок или бесцвет- ные гигро- скопичные кристаллы 410,856 1 450 (под давле- нием) Сублимируется при 357 181,6
m H Желтые окта- эдрические кристаллы 367,636 3,25 38,3 230 170
Соединение Внешний вид Молекулярный вес Плотность (при 20°), г/см3 Температура плавле- ния, °C Температура кипе- ния, °C Теплота образова- ния, ккал/моль
ПОБОЧНАЯ ПОДГРУППА IV ГРУППЫ
73
Действием теплой воды на тетрафториды TiF4, ZrF4, HfF4
получают кислоты H2[TiOF4], H2[ZrOF4], H2[HfOF4], из которых
при упаривании образуются кристаллогидраты TiF4-2H2O,
ZrF4-3H2O, HfF4-3H2O.
При гидролизе растворимых солей титана, циркония и гафния
образуются соли титанила TiO2+, цирконила ZrO2+, дицирконила
Zr2O2+ и гафнила НЮ2+, а при более глубоком гидролизе солей
титана можно получить H4TiO4 или H2TiO3:
MeIV + Н2О МеО2+ + 2Н+ MeIV = Ti4+, Zr4+, Hf4+
2MeIV + 3H2O Me2Of+ + 6H+ MeIV = Zr4+
MeIV + 4H2O H4MeO4 + 4H+ MeIV = Ti4+
Действием перекиси водорода на растворы солей элементов
подгруппы титана в присутствии щелочей получают пероксосоеди-
нения, являющиеся энергичными окислителями, устойчивость
которых растет в ряду титан, цирконий, гафний.
Бесцветные ионы Ti4+, Zr4+, Hf4+ образуют окрашенные хелат-
ные соединения с ацетилацетоном, ализарином, 8-оксихинолином,
купфероном и т. д.
Металлы подгруппы титана и их соединения проявляют сход-
ства в физических и химических свойствах как с элементами
побочных подгрупп III и V групп, так и с элементами главной
подгруппы IV группы периодической системы.
Элементы подгруппы титана имеют средние значения атомных
радиусов, атомных объемов и плотностей среди металлов побочных
подгрупп III, IV и V групп.
Металлы побочных подгрупп III и IV групп имеют серебристо-
белый цвет в отличие от светло-серого цвета металлов побочной
подгруппы V группы.
По химической активности титан, цирконий и гафний отли-
чаются от металлов подгруппы скандия и приближаются к метал-
лам подгруппы ванадия.
В то время как металлы побочной подгруппы III группы всегда
трехвалентны и большинство их соединений бесцветны или окра-
шены в белый цвет, металлы побочных подгрупп IV и V групп
проявляют переменную валентность и многие их соединения
окрашены.
В отличие от элементов главной подгруппы IV группы, которые
проявляют как металлические (олово, свинец), так и неметалличе-
ские (углерод, кремний, германий) свойства, элементы побочной
подгруппы IV группы обладают только металлическими свойствами.
Двуокиси металлов подгруппы титана похожи на окислы
металлов побочных подгрупп III и V групп, поскольку они устой-
чивы, тугоплавки и плохо растворимы в воде.
74
ПОБОЧНАЯ ПОДГРУППА IV ГРУППЫ
Продукты гидролиза соединений элементов подгруппы титана
отличаются от продуктов гидролиза соединений элементов побоч-
ной подгруппы III группы, имеющих амфотерный или основной
характер.
Элементы подгруппы титана подобны остальным переходным
металлам по своим физическим и химическим свойствам, обуслов-
ленным электронным строением (d -орбитали не заполнены)
и по склонности к образованию электроположительных ионов
и координационных соединений.
ТИТАН Ti
Z = 22; ат. вес = 47,90
Валентность II, III, IV, заряд 2+, 3+, 4+
Массовые числа природных изотопов 48, 46, 47,
49 и 50
Электронная структура атома титана К -L -М •3s23pe3d2 -4s2.
Электронная структура атома и ионов титана Ti2+, Ti3+, Ti4+
для 3d- и 45-орбиталей:
3d2 4s2 3d2 4s 3d* 4s 3d 4s
1TITI I I I и ITITI I I I □ Iti llll □ I l I I I I □
Ti Ti2+ Ti3+ Ti«+
ИСТОРИЧЕСКАЯ СПРАВКА
В 1789 г. Грегор открыл входящее в состав силикатных пород
неизвестное вещество, названное им меначином по названию
долины Меначан, в которой была найдена порода. В 1795 г. Клап-
рот пришел к выводу, что упомянутое вещество является окислом
металла, и назвал его «титановой землей» *. Затем почти столетие
соединения титана имели очень ограниченное применение. Однако
с быстрым развитием химии, металлургии и электротехники неко-
торые соединения титана, например двуокись титана (титановые
белила) ТЮг, стали широко применяться.
Титановые белила как пигмент обладают исключительно цен-
ными свойствами, благодаря которым они постепенно заменили
свинцовые и цинковые белила, малостойкие к атмосферным воз-
действиям и оказывающие вредное физиологическое действие
(свинцовые).
В 1825 г. Берцелиусу впервые удалось получить металличе-
ский титан восстановлением K2[TiFe] металлическим натрием.
В 1887 г. Нильсон и Петтерсон получили металлический титан
♦ Некоторые ошибки Клапрота исправил Ловиц, выделивший титан
из ильменита.— Прим. ред.
ТИТАН
75
05 %-ной чистоты восстановлением тетрахлорида титана метал-
лическим натрием в герметичном стальном сосуде.
В 1910 г. Гюнтер выделил металлический титан 99 %-ной
чистоты восстановлением тетрахлорида титана металлическим нат-
рием при 700° в стальной бомбе. Полученный металл обнаруживал
большую хрупкость и низкую механическую прочность, обуслов-
ленные примесями соединений с кислородом и азотом воздуха.
В 1925 г. Ван-Аркель и де Бур получили металлический титан
в результате термического разложения тетраиодида титана.
В 1938 г. Кроль разработал технологический процесс восста-
новления тетрахлорида титана металлическим магнием или каль-
цием, в вакууме или в атмосфере инертного газа, и предложил
необходимую аппаратуру.
Особо чистый металлический титан, обладающий превосход-
ными механическими свойствами, можно получить только при
использовании высоковакуумной техники.
С помощью вакуумной металлургии получают тугоплавкие
металлы высокой чистоты, поскольку при плавлении или переплав-
ке в вакууме осуществляется дегазация металлов. Вакуумная
металлургия возникла из потребности ядерной техники иметь
в промышленных количествах сверхчистые металлы (U, Be, Hf,
Th, Zr и др.) для строительства ядерных реакторов.
В наши дни мировое производство металлического титана
достигло десятков тысяч тонн в год.
Металлический титан высокой степени чистоты прочен, стоек
и играет важную роль как конструкционный материал в современ-
ной технике. Из термостойких и механически прочных сплавов
на основе карбида титана изготовляют различные детали реактив-
ных двигателей и ракет; корпуса сверхзвуковых самолетов делают
из сплавов или даже чистого титана, вместо алюминия, поскольку
последний при больших скоростях нагревается и теряет прочность.
РАСПРОСТРАНЕННОСТЬ В ПРИРОДЕ
В природе титан встречается только в виде соединений
(двуокиси, титанаты, силикаты, ниобаты и т. д.) в различных
распространенных в земной коре минералах. По распространен-
ности в земной коре (0,65 вес. %) титан занимает десятое место.
Важнейшими минералами титана являются следующие.
Ильменит, FeTiO3 (метатитанат железа или титанистый желез-
няк), впервые найденный в Ильменских горах на Южном Урале,
содержит 31,6% Ti и 36,8% Fe. Ему сопутствуют примеси в виде
изоморфных минералов MgTiO3 и MnTiO3. Ильменит встречается
в виде черных или серо-стальных ромбоэдрических кристаллов
с плотностью 4,72 г/см3 и твердостью 5—6 по шкале Мооса в вулка-
нических породах, часто вместе с магнетитом в титаномагнетитах
76
ПОБОЧНАЯ ПОДГРУППА IV ГРУППЫ
(которые являются растворами FeTiO3 в Fe3O4, загрязненном
МпО, СаО, А12О3, Сг2О3, Р2О5, V2O5 и другими окислами).
Залежи титаномагнетитов находятся в СССР, Канаде, США,
Южной Африке, Норвегии, Швеции, Финляндии, Румынии; тита-
номагнетитные наносные породы находятся в Индии, Австралии.
США и др.
Рутил, TiO2, содержит 60% Ti и имеет вид желтых (или корич-
невых, красных, черных — в зависимости от примесей) тетраго-
нальных кристаллов с плотностью 4,26 г/см3 и твердостью 6
по шкале Мооса. Рутил часто встречается вместе с кварцем, иль-
менитом, ильмениторутилом, гематитом, магнетитом, цирконом,
монацитом, пирохлором и др., реже — в залежах бокситов.
Залежи рутила найдены в СССР, США, Австралии, Бразилии,.
Индии и др. Рутил содержится в песках морских и океанских
побережий.
Рутил, брукит и анатаз служат для получения ферротитана;
они применяются также в керамической промышленности в каче-
стве коричневого пигмента, для получения титановых белил,
используемых как пигмент в масляных красках, в радиотехнике
в качестве детектора.
Брукит, TiO2, встречается в СССР, США, Австралии, Брази-
лии и др.; имеет вид желтых (красных, коричневых или черных)
ромбических кристаллов с плотностью 4,17 г/см? и твердостью
5—6 по шкале Мооса.
Анатаз, TiO2, находится в СССР, США, Бразилии, Швейцарии
и др.; имеет вид коричневых или черно-коричневых тетрагональных
кристаллов с плотностью 3,84 г/см3 и твердостью 5—6 по шкале
Мооса.
Переходы анатаза в брукит и рутил и рутила в анатаз могут
быть осуществлены термически:
860° |1040°
[Анатаз Брукит Рутил
(тетрагональ- (ромбиче- (тетрагональ-
ный) ский) ный)
Перовскит, CaTiO3 (метатитанат кальция), содержит 58,9%
TiO2; он встречается в СССР, Швейцарии и др.; имеет вид серовато-
черных или красновато-коричневых кубических кристаллов с плот-
ностьюЗ,97—4,04 г/см3и твердостью5,5—6 пошкалеМооса. Обычно
перовскит находится в залежах титаномагнетита и в хромовых
рудах.
Сфен (эмпирическая формула TiO2-СаО-SiO2) содержит 40,8%
TiO2; он встречается в СССР, Италии, Швейцарии и имеет вид
желтых (коричневых или серых) моноклинных кристаллов с плот-
ностью 3,29—3,56 г/см3 и твердостью 5—6 по шкале Мооса. Обычно
сфен встречается в малых количествах вместе с полевым шпатом.
ТИТАН
77
нефелином, цирконом, апатитом и другими минералами в гранитах,
сиенитах, андезитах и др.
К титансодержащим минералам относятся также пирофанит МпТ1О3,
лопарит (Na, Се, Са, Nb, Ti)O3, пирохлор (Na, Ca)2(Nb, Ti)2O6(F, ОН), микро-
лит (Na, Са, ...)2(Та, Ti, ...)2O6(F, ОН), эшинит (Се, Са, Th)(Ti, Nb)2O6.
В большинстве пород (глинах, песчаниках, базальтах, гранитах
и др.) земной коры титан находится в рассеянном состоянии.
Небольшие количества титана содержатся в некоторых минераль-
ных водах, в организме млекопитающих и в зеленых тканях
растений.
ОБОГАЩЕНИЕ И ПЕРЕРАБОТКА ТИТАНОВЫХ РУД
Титаномагнетитовые руды, содержащие 8—20% TiO2, 30—
40% FeO и Fe2O3, обогащают при помощи гравитационных, магнит-
ных или флотационных методов, в результате получают концент-
раты, содержащие 37—43% TiO2, 45—55% FeO и Fe2O3.
Если наносные породы ильменита, рутила, циркона, монацита
и др. обогащаются гравитационными или магнитными методами,
то образующиеся концентраты, содержащие до 98% TiO2, исполь-
зуются как техническая двуокись титана. Титан извлекают также
из остатков от переработки боксита.
Получение двуокиси титана
обработкой концентрата ильменита серной кислотой
В стальном реакторе, футерованном кислотоупорной плиткой,
смешивают сухой и тонко измельченный концентрат ильменита
с 92—96 %-ной H2SO4. Смесь перемешивается воздухом, а затем
в реактор добавляют небольшое количество воды, что приводит
к разогреванию смеси до температуры 170—190°, при которой
за 5—10 мин происходит бурное разложение ильменитового кон-
центрата. При дальнейшем добавлении в реактор холодной воды
растворимые сульфаты титана, железа, алюминия и других метал-
лов переходят в раствор и отделяются декантацией от остатка,
состоящего из кремнезема и неполностью разложившегося кон-
центрата ильменита.
Кристаллы FeSO4 -7Н2О выделяют из общего раствора суль-
фатов добавлением железной стружки и отделяют. Оставшийся
раствор разбавляют теплой водой (гидролиз при pH = 1,5) и отде-
ляют белый осадок метатитановой кислоты H2TiO3. Таким обра-
зом, кислота не загрязнена двухвалентным железом, поскольку
гидролиз FeSO4 осуществляется при pH, равном 6—7.
Дальнейшим прокаливанием при 500—800° дегидратированной
и высушенной метатитановой кислоты получают TiO2 99 %-ной
чистоты.
78
ПОБОЧНАЯ ПОДГРУППА IV ГРУППЫ
Этот процесс переработки концентрата ильменита не применяют
в широких масштабах, поскольку он сопровождается большими
потерями H2SO4 и повышенным расходом TiO2.
Схема переработки концентрата ильменита по этому методу
представлена ниже.
FeTiO3 + 2H2SO4 (конц.)TiOSO4-’r FeSO4 + 2H2O
Ильменит х —j
При выпаривании в вакууме из раствора,
выпадает FeSO4-7H2O
I
При кипячении раствора TiOSO4 с водой
Ч - J
Прокаливание I
ТЮ2 «---------H2TiO3 + H2SO4
Получение титановых шлаков из руд
и концентратов ильменита
Титаномагнетиты с флюсами (MgO или CaGO3-MgGO3) и коксом
перерабатывают в доменных печах примерно при 1600°. При этом
получают бедные титаном (до 45% TiO2) шлаки. Железо почти
полностью отделяется в виде чугуна, а в шлак переходят окислы
TiO2, А12О3, MnO, SiO2 и частично FeO в случае руд с большим;
содержанием SiO2.
Бедные титансодержащие шлаки в смеси с окислами железа
перерабатываются с коксом в доменных печах с целью получения
титансодержащих сталей.
Концентраты ильменита, содержащие 35—50% TiO2, перера-
батывают с флюсами и коксом в электрических печах при 1600—
1700° и получают шлаки, богатые титаном (70—80% TiO2). Темпе-
ратура плавления шлака растет с увеличением содержания TiO2,
SiO2, А12О3 и кокса.
Пирометаллургической переработкой ильменита с Na2GO3 или
NaOH и коксом получают шлак с низкой температурой плавле-
ния (1000—1100°) и губчатое железо. Этот процесс имеет ограни-
ченное применение из-за большого расхода Na2GO3 (или NaOH).
Переход двуокиси титана в шлак зависит от его содержания
в руде или концентрате, от состава и качества флюсов, от темпе-
ратуры плавления и от типа печи (доменная или электрическая).
ПОЛУЧЕНИЕ МЕТАЛЛИЧЕСКОГО ТИТАНА
Металлический титан можно получить несколькими способами:
восстановлением при нагревании тетрахлорида титана с щелочно-
земельными или щелочными металлами либо в присутствии водо-
рода в вакууме или в атмосфере инертного газа; восстановлением
K2[TiF6] металлическим натрием при нагревании в вакууме или
ТИТАН
79
атмосфере инертного газа; термической диссоциацией тетраиодида
титана; электролизом галогенидов TiCl2, TiGl3, TiCl4 в расплаве
галогенидов щелочных или щелочноземельных металлов; электро-
лизом расплавов окислов титана; алюмотермическим восстановле-
нием концентрата ильменита.
В чистом состоянии металлический титан получить сравни-
тельно трудно, поскольку он легко взаимодействует с восстанови-
телями: алюминием, углеродом, водородом или азотом, с которыми
образует соединения включения.
Для получения металлического титана применяют главным
образом тетрагалогениды титана и не используют окислы, гидро-
окиси, кислоты, нитриды и карбиды титана, поскольку для этих
соединений характерна высокая химическая устойчивость.
Металлотермическое восстановление тетрахлорида
титана (процесс Кроля)
Порошкообразный или губчатый металлический титан получают
магнийтермическим восстановлением тетрахлорида титана при
850° в молибденовом тигле, находящемся в реакторе (или реторте),
наполненном после предварительного создания глубокого ваку-
ума аргоном или гелием:
TiCl4 + 2Mg = Ti + 2MgCl2 + 122 ккал
Во избежание образования низших хлоридов титана магний
берут в 30 %-ном избытке по сравнению со стехиометрически необ-
ходимым по реакции количеством. Избыток магния удаляют плав-
лением и переплавкой сырого титана или промывкой сырого титана
разбавленными кислотами.
Для восстановления тетрахлорида титана вместо магния можно
использовать сплав Mg и Са (82 : 18), металлический кальций,
щелочные металлы в присутствии смеси галогенидов щелочных
металлов и гидрида натрия.
Натрийтермическим восстановлением тетрахлорида титана при
800—900° в вакууме (или атмосфере аргона, гелия) в реакторах
получают губчатый металл:
TiCl4 + 4Na = Ti + 4NaCl + 185,5 ккал
После отделения непрореагировавшего натрия спиртом и хло-
рида натрия водой металлический титан промывают спиртом
и сушат.
При металлотермическом восстановлении тетрахлорида титана
получается металлический титан 99,5 %-ной чистоты, который
в качестве примесей содержит железо, кислород, азот и др.
Восстановление тетрахлорида титана гидридом натрия в атмо-
сфере водорода при 300—350° осуществляется по уравнению
TiCl4 4- 2NaH = Ti + 2NaCl + 2HC1
«о
ПОБОЧНАЯ ПОДГРУППА IV ГРУППЫ
Восстановление гексафторотитаната(1У) калия K2[TiF6]
металлическим натрием (процесс Берцелиуса)
K2[TiFe]’+ 4Na = Ti + 2KF + 4NaF
Порошкообразный металлический титан получают восстанов-
лением фторосоли K2[TiF6] металлическим натрием в вакууме
или атмосфере аргона, гелия и др. при 800—900° в герметически
закрытом сосуде. Процесс ведут с избытком металлического
натрия.
После отделения непрореагировавшего натрия спиртом и фто-
ридов калия и натрия водой порошок титана промывают разбав-
ленной HG1, водой, спиртом и затем сушат.
Термическая диссоциация тетраиодида титана
(процесс Ван-Аркеля и де Бура)
Til4 Ti + 212
Тетраиодид титана получают прямым взаимодействием метал-
лического титана или титаноалюминиевых сплавов с элементарным
иодом при 200° в вакуумированном сосуде. Соединение Til4 суб-
Рис. 3. Сосуд для по-
лучения титана терми-
ческой диссоциацией
Til4.
лимируется при 377°, и его пары, сопри-
касаясь с титановой проволокой, нагретой
до 1100—1400°, разлагаются; металличе-
ский титан оседает на проволоку, а пары
иода конденсируются на холодной части
прибора. Таким путем получают прутки
металлического титана диаметром 5—30 мм.
На рис. 3 изображен сосуд из стекла
пирекс для получения металлического ти-
тана. Через отверстие А поступают по-
рошкообразный титан и иод, через от-
верстие Б откачивают воздух. Электроды
Ь\ и В2 — вольфрамовые, а проволока
Г — из титана. В ходе процесса сосуд
нагревают до 600° в электрической печи,
а проволока нагревается до 1100—1400°
электрическим током.
Этим путем получают металлический
титан высокой чистоты, поскольку боль-
шинство примесей, содержащихся в исходном металле, или не
реагирует с иодом, или не образует летучих при 377° иодидов.
Металлический титан, полученный этим способом, очень пласти-
чен, прочен и легко поддается механической обработке.
Методом термической диссоциации могут быть получены
в чистом состоянии и другие металлы: Zr, Hf, Ge, V, Nb, Та.
ТИТАН SI
Электролитическое получение металлического титана
Металлический титан в виде порошка или губчатой массы
получают электролизом галогенидов титана (TiCl2, TiGl3, TiCl4,
K2[TiF6], смесь TiCl2 c TiGl3 и т. д.) в расплаве галогенидов
щелочных или щелочноземельных металлов, а также электроли-
зом двуокиси титана в расплаве Na4P2O7 или Na2B4O7. Для этого
используют галогениды щелочных и щелочноземельных металлов,
образующих эвтектические смеси: NaCl — KG1, КС1 — Lid,
NaGl — КС1 — CaCl2, NaCl — KC1 — СаС12 и др. Когда метал-
лический титан получают электролизом тетрахлорида титана
TiCl4, пары последнего барбатируют через расплав электролита.
Электролитическое получение титана осуществляется в атмо-
сфере аргона при 500—950° в графитовом тигле, который служит
анодом; материалом катода служит молибден, сталь, никель, цир-
коний или даже титан, защищенный графитовой обкладкой в высту-
пающей над расплавом части.
Аргон, используемый при электролитическом получении титана,
должен быть чистым, т. е. не должен содержать кислорода, азота
и влаги. Для очистки его пропускают через кварцевые трубки
с нагретым примерно до 350° гидридом кальция СаН2, затем через
трубки с нагретой до 550° окисью меди(1) и через осушительные
колонки с перхлоратом магния Mg(C104)2 и Р4О10.
СаН2 -ф !/2О2 = СаО + Н2 СпО + Н2 = Си + Н2О
ЗСаН2 -j- N2 — Ca3N2 4- ЗН2 Р4Ою 4~ 2Н2О — H4P40i2
При плавлении титана, полученного электролитическим мето-
дом в вакууме (10-5—10“® мм pm. ст. при 950°) в виде губчатой
массы, образуется металлический титан 99,3—99,8%-ной чистоты;
он содержит в качестве примесей 0,03—0,12% углерода, 0,004—
0,017% азота и ничтожные количества Si, Fe, Mg, Мп, Al, Mo,
V, Gu, Zr, Ni, Pb, Ga, Gr, Co, Ta, W, Bi, Zn и др.
Получение металлического титана лабораторным методом
Металлический титан в небольших количествах можно полу-
чить восстановлением двуокиси титана металлическим кальцием
или гидридом кальция при нагревании без доступа воздуха:
TiO2 4- 2Са = Ti 4- 2СаО + 85,4 ккал TiO2 4- 2СаН2 = TiH2 4- 2СаО + Н2
Восстановление при помощи СаН2 осуществляется при 900—
1000° в аппаратуре из стали. Хотя гидрид титана почти полностью
диссоциирует на составные элементы при нагревании в вакууме,
все же металлический титан, полученный по этому методу, загряз-
нен гидридом.
<1—0101
82
ПОБОЧНАЯ ПОДГРУППА IV ГРУППЫ
В промышленности металлический титан не получают восста-
новлением двуокиси титана углем, поскольку параллельно обра-
зуется карбид титана TiC, который сильно изменяет физические
и химические свойства металла.
ПОЛУЧЕНИЕ ФЕРРОТИТАНА
Алюмотермическим восстановлением предварительно прока-
ленного при 1100—1200° (с целью удаления серы) концентрата
ильменита получают ферротитан, содержащий 18—45% титана,
5—8% алюминия, 3—5% кремния, 0,1% фосфора, 0,01% серы,
остальное — железо. Восстановление осуществляется в футеро-
ванном магнезитом тигле в электрической печи. Для связывания
окислов SiO2, А12О3 в виде шлаков добавляют СаО.
Ферротитан является одной из основных добавок при получе-
нии сталей специальных марок, поскольку добавка до 1 % титана
увеличивает коррозионную устойчивость и механическую проч-
ность стали при повышенных температурах.
Превращение порошкообразного или губчатого титана
в компактный металл
Получение брусков или стержней метал-
лического титана прессованием и спека-
нием порошкообразного или губчатого
титана (методом порошковой металлургии).
Смоченный чистым бензином крупнозернистый порошок метал-
лического титана, просеянный через сито, которое имеет от 12
до 60 отверстий на сантиметр длины, прессуют на гидравлических
прессах в обычных формах на холоду при больших давлениях —
7—8 т/см2, а при нагревании — при меньших давлениях. Тонко-
измельченный порошок титана имеет большую поверхность, более
высокую химическую активность и быстрее окисляется, покры-
ваясь пленкой окиси, поэтому его не используют. Цилиндрические
или кубические бруски, полученные прессованием титанового
порошка или губки, спекают при 950—1000° (по некоторым данным
даже при 1200°) в трубках из огнеупорных сплавов или фар-
фора, предварительно вакуумированных до давления 10-3—
—10~4 мм рт. ст., в течение 16 час. При этом удаляются магний,
водород, кислород и азот и остается металлический титан. Спекание
не проводят просто в атмосфере инертного газа (аргон или гелий),
поскольку в этом случае магний и водород не удаляются.
Когда прессование осуществляется при нагревании в графитовом
тигле, на поверхности металлического титана образуется твердый
тугоплавкий карбид. Можно провести спекание в вакууме при
1300—1500° под давлением 10 т/см2 за 2—3 час.
ТИТАН
83
Металлический титан, полученный прессованием и спеканием
крупных частиц, обладает высокой пластичностью и легко пере-
рабатывается на холоду в отличие от компактного металла, полу-
ченного прессованием и спеканием тонкого порошка титана,
который хрупок и с трудом поддается переработке на холоду.
Получение брусков или стержней метал-
лического титана сплавлением порошко-
образного или губчатого титана. Плавление
порошкообразного или губчатого титана осуществляют в вакууме
или атмосфере инертного газа (гелия или аргона) в графитовом
тигле или тигле из двуокиси тория, помещенного в индукционную
или электродуговую печь. Чаще применяют метод плавления
порошкообразного или губчатого титана в электродуговой печи
с медным (в форме тигля) и вольфрамовым электродами, которые
охлаждаются водой. Используются также и печи с электронным
сфокусированным пучком (с кольцеобразным катодом). Плавление
осуществляется в высоком вакууме. Электронный поток, излучае-
мый нагретой до 2000° вольфрамовой проволокой, фокусируют
на порошок (губку) расплавляемого металла и на поверхность
ванны. Расплавленный титан разливают в формы (прутки или
блоки) без доступа воздуха.
ФИЗИЧЕСКИЕ И ХИМИЧЕСКИЕ СВОЙСТВА
Титан — блестящий серебристо-белый металл: ниже 882° суще-
ствует модификация со структурой плотнейшей гексагональной
упаковки, выше — с кубической объемноцентрированной. Титан
относится к сравнительно легким (плотность 4,49 г/сж3 при 20°),
тугоплавким (т. пл. 1677°, т. кип. 3277°), парамагнитным металлам.
Металлический титан высокой чистоты обладает превосходными
механическими свойствами, особенно прочностью при низких
температурах — до —180°, а также при нагревании до 500°. Будучи
ковким и вязким, он легко поддается механической обработке,
при нагревании легко протягивается в тонкие нити или полосы.
Титан имеет преимущества перед алюминием, сплавы которого
быстрее теряют механическую прочность при нагревании. Значение
отношения между механической прочностью и плотностью метал-
лического титана высокой чистоты более значительно, чем те же
показатели для сталей или для легких сплавов алюминия. В связи
с этим титан находит все большее применение в самолетостроении.
Однако следует отметить, что пластичность, ковкость, тягучесть,
твердость, прочность на разрыв и другие механические свойства
титана резко ухудшаются в присутствии примесей кислорода,
азота, углерода, водорода и др.
Известны многочисленные сплавы, образуемые титаном с эле-
ментами Be, Al, Ga, In, TI, Ge, Sn, РЪ, C, As, Sb, Bi, Те, Cu,
6*
84
ПОБОЧНАЯ ПОДГРУППА IV ГРУППЫ
Ag, Au, Zn, Cd, Zr, Hf, V, Nb, Ta, Cr, Mo, W, U, Mn, Fe,
Co, Ni, Ru, Rh, Pd, Os, Ir, Pt и редкоземельными металлами.
Если классифицировать сплавы титана по свойствам, то разли-
чают сплавы, характеризующиеся низкой плотностью, твердо-
стью и высокой температурой плавления, повышенной механиче-
ской и химической стойкостью. Титан входит в состав углеродистых
и нержавеющих сталей и сверхтвердых сплавов.
К интерметаллическим соединениям титана относятся: TiBe4,
TiAl, TiAl3, TiAl4, Ti4Pb, TiSb, TiSb2, Ti4Bi, Ti3Cu, Ti2Cu3,
TiCu, TiCu3, Ti3Au, TiAu2, TiZn, TiZn2, TiCr2, Ti2Cr3, Ti2Mn,
TiMn2, TiFe, Ti2Fe, TiFe2, TiFe3, TiCo, Ti2Co, Ti2Ni, TiNi,
Ti3Pt и др. Среди важнейших тройных и четверных систем
титана можно назвать: Ti — Si — С, Ti — Си — С, Ti — Мп — С,
Ti - V - С, Ti - Al - Sn, Ti - Al - V, Ti - Al - Mn,
Ti - Cr - C - N, Ti - Cr - Al - C, Ti - Al - Nb - Ta.
Металлический титан в компактном состоянии химически устой-
чив на воздухе примерно при температурах до 610°, а в морской
воде — до 100°. В тонкоизмельченном состоянии при комнатной
температуре титан окисляется в кислороде (или на воздухе)
до TiO и TiO2- Титан энергично взаимодействует с кислородом
воздуха при 1200° и с чистым кислородом при 600°. С повышением
температуры увеличивается сродство титана к кислороду, азоту,
водороду, углероду; вследствие этого его механическая перера-
ботка осуществляется при температуре не выше 950°. Поглощая
кислород, азот или водород, металлический титан становится
хрупким.
Металлический титан разлагает воду при кипячении:
Ti + 4Н2О = H4TiO4 + 2Н2
Тонкий порошок титана реагирует с водой, галогенами, серой,
азотом, фосфором и углеродом следующим образом:
700—800°
Ti + 2H2O-----► TiO2 + 2H2
150°
Ti + 2F2------>- TiF4 + 393,3 ккал
300°
Ti + 2C12-----* TiCl4 194,9 ккал
350°
Ti + 2Br2----->-TiBr4+170 ккал
400°
Ti + 2I2----->-TiI4+131 ккал
800°
2Ti + N2------>2TiN + 160,6 ккал
800°
Ti + C-------► TiC +114 ккал
700° 1000°
Ti + 2S------* TiS2 Ti + ₽------> TiP
ТИТАН
85
В атмосфере сухого хлора титан горит, превращаясь в TiCl4,
а во влажном хлоре и в растворах гипохлоритов натрия, калия
или кальция он устойчив.
Металлический титан растворяется в царской водке, расплавах
щелочей и концентрированных кислотах HF, НС1, H2SO4 и т. д.
3Ti + 12НС1 + 4HNO3 = 3TiCl4 + 4N0 + 8Н2О
Ti + 4NaOH = Na4TiO4 + 2H2 Ti + 3HC1 = TiCl3 + 3/2H2
2Ti + 3H2SO4 = Ti2(SO4)3 + 3H2
Под действием концентрированной азотной кислоты металли-
ческий титан превращается в метатитановую кислоту H2TiO3 -Н2О:
3Ti + 4HNO3 + Н2О = ЗН2ТЮ3 + 4NO
Металлический титан очень медленно взаимодействует с разбав-
ленными кислотами НС1, HNO3 и растворами органических кис-
лот. Он устойчив к действию хромового ангидрида, формальде-
гида, лимонной, молочной, стеариновой кислот и др.
По отношению к большинству агрессивных сред металлический
титан проявляет коррозионную устойчивость.
Химическая активность титана иллюстрируется следующей
схемой:
Комн. темп.
Нагревание
с конц. HF —* TiF4
с конц. НС1 -> TiCl4
с конц. H2SO4 —* Ti2(SO4)3
с царской водкой —► TiCl4
Титан —
на воздухе или в кислороде —► TiO2
в воде —>- H4TiO4
с галогенами —> TiX4(X = F_, С1_, Вт-, I-)
с серой, азотом, углеродом —> TiS2, TiN, TiC
с распл. NaOH —> титанаты
ПРИМЕНЕНИЕ
Металлический титан входит в состав сплавов, не содержащих
железа, в количестве 0,1—0,6%, а также в состав углеродистых,
нержавеющих и высококачественных сталей.
В сплавах титан может быть легирующей добавкой (придающей
эластичность, механическую прочность, химическую стойкость,
плотность, твердость и другие свойства), или дегазантом, препят-
ствующим образованию пор в сплавах, поскольку он связывает
кислород, азот, водород и другие газы в трудно растворимые
соединения, удаляемые в шлак.
S6
ПОБОЧНАЯ ПОДГРУППА IV ГРУППЫ
Такие сплавы титана, как ферротитан, феррокарботитан, фер-
росиликотитан и др., очень важны для современной техники,
поскольку они обладают высокой механической прочностью, легки,
тугоплавки, тверды, устойчивы к коррозии.
Установки и приборы, изготовленные из титана или его спла-
вов, могут эксплуатироваться и при высоких и при очень низких
температурах, в морской воде и в сильно влажном воздухе. Бла-
годаря низкой плотности и высокой механической прочности даже
при температуре 500° титан и его сплавы являются очень ценным
материалом не только для авиации, но и для кораблестроения,
в производстве железнодорожных рельсов, вагонных осей и колес.
Вследствие большой химической стойкости металлический
титан (и его сплавы) идет на оборудование в производстве хлора
(в условиях контакта с влажным хлором), для изготовления ванн
для хромирования, никелирования, электрополировки, а также
как материал для оборудования в пищевой промышленности
и промышленности синтетических волокон.
Металлические детали, покрытые титаном методом титаниза-
ции (по подслою меди), приобретают большую поверхностную
прочность.
Сверхтвердые сплавы, полученные методом порошковой метал-
лургии из карбида титана, карбида вольфрама и металлического
кобальта, используются для изготовления резцов, сверл, калиб-
ров, фильер и т. д. Нитрид и борид титана могут быть также
использованы как твердые материалы с большой механической
прочностью на износ. Из борида титана изготовляются детали тур-
бин, турбореакторов и ракет.
Кристаллы титаната бария или бария и стронция с добавкой
0,003% лантана, редкоземельных элементов, висмута и тория
применяются как термисторы. Последние используются в простых
электронных системах регулирования температуры в качестве
прерывателей электрического тока, в автоматических коммутато-
рах и др.
СОЕДИНЕНИЯ (ОБЩИЕ СВОЙСТВА)
Известны двух-, трех- и четырехвалентные соединения титана.
Самые устойчивые (как простые, так и координационные) — соеди-
нения четырехвалентного титана. В современной учебной литера-
туре чаще применяется термин «степень окисления», что обозна-
чается римскими цифрами II, III, IV, например соедине-
ния Ti(IV).
Соединения титана в низшей степени окисления проявляют
основной характер, в высшей степени окисления — кислотный.
Хлориды титана в низшей степени окисления являются ионными
соединениями, а тетрахлорид титана — соединение ковалентное.
ТИТАН
87
Соли титана (цитраты, титанаты и др.), в малых количествах
не токсичны и являются стимуляторами дыхания, в больших
же количествах приводят к параличу органов дыхания. Салици-
латы титана применяются при лечении конъюнктивита и неко-
торых экзем.
В табл. 21 приведены формулы и указан цвет важнейших
соединений титана.
Соединения двухвалентного титана
Соединения двухвалентного титана достаточно редки и могут
быть получены путем энергичного восстановления соединений
трех- или четырехвалентного титана. Они неустойчивы, легко
окисляются на воздухе и превращаются в соединения более высо-
кой степени окисления.
Дигидрид титана, TiH2, получают восстановлением двуокиси
титана гидридом кальция при нагревании:
TiO2 + 2СаН2 = TiH2 + 2СаО +• Н2
Дигидрид титана — твердое серое вещество, разлагающееся
на составные элементы при нагревании до высоких температур
в вакууме. Применяется в качестве добавки к порошкам различных
металлов, которые переводят спеканием в компактное состояние.
Образующийся при разложении гидрида водород защищает поверх-
ность металла от окисления.
Моноокись титана, TiO, получают восстановлением при нагре-
вании в вакууме (или в атмосфере аргона) двуокиси титана с тита-
ном, цинком или углем; нагреванием карбида титана с окисью
цинка; электролитическим восстановлением смеси TiO2 (20%)
и СаС12 (80%) при 1000—1100° в атмосфере аргона при плотности
тока 200—400 а!дм2 и напряжении 4,5 е\
1550°
TiO2 + Ti---->- 2TiO 2TiO2 + Zn =ДЮ + ZnTiO3
2TiOj + Mg = TiO + MgTiO3 TiO2 + G = TiO + CO
TiC + 2ZnO = TiO + 2Zn -«CO
Моноокись титана образует кристаллы бронзового цвета с плот-
ностью 4,93 г/cat3, плавящиеся при 1750°, растворяющиеся в разб.
H2SO4 (или в разб. НС1) с образованием Ti2(SO4)3 и выделением
водорода:
2TiO + 3H2SOt = Ti2(SOt)3 + 2Н2О + Н2
Нагреванием моноокиси титана выше 800° получают двуокись
титана.
Гидроокись титана, Ti(OH)2, получается в виде черного осадка
при обработке галогенидов титана(П) щелочами.
Соединение Скис- лы Г идроокиси, кислоты, ти- танаты Фториды и фторосоеди- нения Хлориды и хло- росоединения Бромиды и бромо- соедине- ния
Соединения двухвалент- ного титана TiO цвета бронзы Ti(OH)2 черная TiF2 черный TiCl2 черно-коричне- вый TiBr2 черный
Соединения трехвалент- ного титана Ti2Og фиоле- товый Ti(OH)3 коричнево- фиолетовая TiF3 фиолетовый MeI[TiF5] Me|[TiFb] TiCl3 фиолетовый [Ti(H2O)e]Cl3 [Ti(H2O)5Cl]Cl2 [Ti(H2O)4Cl2]Cl [Ti(NH3)e]Cl3 Me2[TiCl5H2O] TiBr3 черно-си- ний fl
Соединения четырех- валентного титана тю2 бес- цвет- ный H4TiO4 белая студне- образная н2тю3 белая студне- образная H2TiO5 оранжево-жел- тая Me4TiO4 Me2TiO3 MenTiO3 Me2TiO3-3H2O Me*TiO8-6H2O TiF4 белый TiF4-2H2O или H2[TiOF4]-H2O H2[TiFe] MeI[TiFe] Men[TiFe] Me^[TiF7l TiCl4 бесцветная жидкость TiOCl2 H2[TiCle] H2[TiCl4Br2] H2[TiCl4I2] Me2[TiCle]-2H2O TiB4 желтый H2[TiBre] Me2[TiBr6]
Таблица 21
— Йодиды и иодосое- динения Суль- фиды Сульфаты и сульфа- тосоединения Хелатные соединения титана(1У) Соеди- нения вклю- чения
Til2 черный TiS черно- корич- невый pi/ о \ -| •Пи) L xx/xzAJ желтый Г ( 0=N \ " Til ~/N — СеН5 j L \ Уд- желтый г / он \ 1 / 1 \ I /°—€ I Til \ хz 1 1 =/ О 1 . ' \' Ь. фиолетово-красный Г / СН3\ / о = С/ \ Ti \ ^сн I L \ СНз/4. желтый TiH TiN медного цвета TiC черный
TiI3-6H2O фиолетовый ТЛ3 черно-фио- летовый TigSg черно- зеле- ный Ti2(SO4)3 зеленый Ti2(SO4)3*8H2O MeTTi(SO4)2-12H2O и двойные соли
Til4 коричнево- красный H2[Ti 16] TiS2 желтый Ti(SO4)2 бесцветный TiO(SO4)2-2H2O Me2[Ti(SO4)3] Mel(TiO(SO4)2] K2[TiO2(SO4)2]-3H2O пероксосоединение
90
ПОБОЧНАЯ ПОДГРУППА IV ГРУППЫ
Гидроокись титана — активный восстановитель; она легко окис-
ляется в присутствии воды с выделением водорода:
Ti(OH)2 + НОН = Ti(OH)3 + l/2H2 Ti(OH)2 + 2Н2О = H4TiO4 + Н2
Дифторид титана, TiF2,— черное твердое вещество с
т. пл. 1277° и т. кип. 2152°.
Дихлорид титана, TiCl2, получают восстановлением тетра-
хлорида титана амальгамой натрия на холоду или водородом под
давлением при 700°, а также диспропорционированием трихлорида
титана при 420° без доступа воздуха:
TiCl4 + Н2 = TiCl2 + 2НС1 2TiCl3 TiCl2 + TiCl4
Продукты реакции обладают разными температурами кипения
и могут быть разделены перегонкой.
Дихлорид титана можно выделить в виде черно-коричневого
порошка или в виде черных расплывающихся на воздухе кристал-
лов, которые способны к самопроизвольному воспламенению на
воздухе; плотность TiCl2 3,13 г/смъ, т. пл. 677°, т. кип. 1479°.
Он разлагается в вакууме при 600° на металлический титан и тет-
рахлорид титана, растворяется в спирте, воде, кислотах и др.;
плохо растворим в эфире, сероуглероде и др., является восстано-
вителем по отношению к HgCl2, разб. HNO3, TiCl4 (в солянокис-
лом растворе):
TiCl2 + 2HgCl2 == TiCl4 Hg2Cl2
TiCl2 + TiCl4 # 2TiCl3
Водный раствор дихлорида титана следует хранить в атмосфере
водорода или двуокиси углерода, поскольку вещество легко окис-
ляется, превращаясь в TiCl3 и TiCl4.
При нагревании раствора дихлорида титана в соляной кислоте
образуется трихлорид или тетрахлорид титана и выделяется
водород.
Дихлорид титана с жидким аммиаком образует серый порошок
состава [Ti(NH3)4]Cl2.
Дибромид титана, TiBr2, получают нагреванием трибромида
титана при 450° без доступа воздуха:
2TiBr3 TiBr2 + TiBr4
Дибромид титана представляет собой черный порошок с плот-
ностью 4,31 г/см3 и т. пл. 627°; при нагревании он разлагается
по уравнению:
2TiBr2 = Ti + TiBr4
Дииодид титана, Til2, получают нагреванием трииодида титана
без доступа воздуха или восстановлением тетраиодида титана
ТИТАН
91
металлическим серебром или ртутью при нагревании в токе водо-
рода:
2TiI3 =pt Til2 + Til4 Til4 + 2Ag = Til2 + 2AgI
Дииодид титана представляет собой черный порошок или очень
гигроскопичные пластинчатые кристаллы с плотностью 4,99 г/с№,
т. пл. 627° и т. кип. 1027°.
Моносульфид титана, TiS, получают нагреванием трисуль-
фида или дисульфида титана в токе водорода, а также пропуска-
нием паров TiCl4, смешанных с H2S, над раскаленной вольфрамо-
вой нитью’
Ti2S3 -|- Н2 = 2TiS -|- H2S TiS2 4- Н2 — TiS -|- H2S
TiCl4 + 2H2S = TiS + S + 4HC1
Моносульфид титана — черно-коричневое вещество, напоми-
нающее металл, устойчивое при комнатной температуре.
Соединения трехвалентного титана
Известно большое число соединений трехвалентного титана,
получающихся восстановлением соединений четырехвалентного
титана водородом при нагревании, цинком в кислой среде или вос-
становлением на катоде.
Водные растворы соединений трехвалентного титана окрашены
в фиолетовый цвет; они неустойчивы, обладают восстановительными
свойствами (хранение без доступа воздуха либо в атмосфере инерт-
ного газа или СО2), легко окисляются до четырехвалентного
титана. Благодаря восстановительным свойствам растворы солей
трехвалентного титана используются в титанометрии. Нормальный
потенциал системы Ti3+/Ti4+ равен —0,04 в. Ион Ti3+ — более
сильный восстановитель, чем ион Sn2+.
Известны как простые соединения трехвалентного титана
(например, Ti2O3, TiCl3, Ti2S3), так и координационные соедине-
ния — анионные и катионные, в которых трехвалентный титан
имеет координационные числа 5 и 6.
Окись титана (трехокись титана), Ti2O3, получают нагрева-
нием двуокиси титана с углем при 870° или действием смеси водо-
рода и тетрахлорида титана на двуокись титана при 1400° в ваку-
уме:
2TiO2 + С = Ti2O3 + СО 3TiO2 + TiCl4 + 2Н2 = 2Ti2O3 + 4НС1
Окись титана Ti2O3 представляет собой фиолетовые кристаллы
с решеткой, подобной корунду; они имеют плотность 4,601 г!см3,
т. пл. 2127°, т. кип. 3027°, плохо растворяются в воде и легко —
в H2SO4. Нагреванием Ti2O3 на воздухе при 1000° или кипячением
с азотной кислотой получают двуокись титана.
92
ПОБОЧНАЯ ПОДГРУППА IV ГРУППЫ
Гидроокись титана(Ш), Ti(OH)3, образуется в виде корич-
нево-фиолетового осадка при обработке соединений титана(1Щ
щелочами при pH = 4:
TiCl3 + 3NaOH = Ti(OH)3 + 3NaCl
Ti2(SO4)3 + 6K0H = 2Ti(OH)3 + 3K2SO4
Гидроокись титана обладает только основными свойствами,
не растворяется в щелочах и исключительно легко окисляется
кислородом воздуха, превращаясь в белый осадок H4TiO4:
2Ti(OH)3 + НОН + i/2O2 = 2Н4ТЮ4
Трифторид титана, TiF3, получают растворением металли-
ческого титана в фтористоводородной кислоте или восстановле-
нием тетрафторида титана кремнием или медью при нагревании.
Трифторид титана представляет собой фиолетовый порошок,
устойчивый на воздухе, плавящийся при 1227°, кипящий при 1727°,
растворяющийся в воде и образующий с фторидами щелочных
металлов комплексные фториды общей формулы Me*[TiF5I
и Me* [TiFe], например K2[TiF5], (NH4)3[TiF6].
Трихлорид титана, TiCl3, получают восстановлением тетрахло-
рида титана; водородом при 650°, металлическим серебром при
180—200°, металлической ртутью при 98°, диэтилцинком при 100°:
TiCl4 + i/2H2 = TiCl3 + НС1 TiCl4 + Ag = TiCl3 + AgCl
2TiCl4 + 2Hg = 2TiCl3 + Hg2Cl2
2TiCl4 + Zn(C2H5)2 = 2TiCl3 + ZnCl2 + C4HIO
Трихлорид титана представляет собой фиолетовые кристаллы,
которые сублимируются при 432°, диспропорционируют при 700°
до TiCl2 и TiCl4 и легко взаимодействуют с галогенами с образова-
нием соединений титана(1У); они растворимы в воде и спирте,
плохо растворимы в эфире, легко окисляются во влажном воздухе,
превращаясь в гидратированную двуокись титана:
2TiCl3 + 4Н2О = 2TiO2 + 6НС1 4- Н2
При восстановлении тетрахлорида титана металлической медью
в обычных условиях получают окрашенное в черный цвет соедине-
ние TiCl3 -CuCl.
Растворением металлического титана в конц. НС1 или восста-
новлением тетрахлорида титана в водном растворе активирован-
ным водородом (металлический Zn в растворе НС1 1 : 1) получа-
ют фиолетовый раствор, из которого выделяют кристаллогидрат
TiCl3-6H2O, существующий в виде гидратных изомеров:
[Ti(H2O)6]Cl3 — фиолетовый, [Ti(H2O)5Gl]Gl2 Н2О — зеленый,
[Ti(H2O)4Cl2]Cl -2Н2О — зеленый.
ТИТАН
93
Водные растворы трихлорида титана хранят в атмосфере водо-
рода или С02: их применяют как восстановители в объемном ана-
лизе.
Восстановлением в растворе трихлорида золота трихлоридом
титана получают гидратированную двуокись титана, которая адсор-
бирует коллоидное золото, окрашиваясь в фиолетовый цвет:
3TiCl3 + AuCl3 = 3TiCl4 + Au
3TiCl4 + 6H2O = 3TiO2 + 12HC1
Трихлорид титана восстанавливает также перманганат калия
до соединений двухвалентного марганца, соединения трехвалепт-
ного железа до двухвалентного, соединения двухвалентной меди
до одновалентной.
При 420° трихлорид титана диспропорционирует до TiCl4
и TiCl2 (см. реакцию получения дихлорида титана).
Трихлорид титана с хлоридами щелочных металлов образует
комплексные хлориды, например Cs2[TiCl5(H2O)J. Это соединение
в растворе имеет фиолетовый цвет, а в твердом состоянии окрашено
в зеленый.
Известен хлорид гексамминтитана(Ш) [Ti(NH3)e]Gl3, а также
аддукт TiCl3-2NH3, который получается из трихлорида титана
и жидкого аммиака.
Трибромид титана, TiBr3-6H2O, получают растворением
порошкообразного металлического титана в концентрированной
бромистоводородной кислоте или восстановлением тетрабромида
титана цинком в кислой среде (Zn + НС1):
2Ti + бНВг = 2TiBr3 + ЗН2
2TiBr4 + 2Н = 2TiBr3 + 2HBr
Трибромид титана выделяется в виде расплывающихся на воз-
духе фиолетово-красных кристаллов, которые плавятся при 115°,
растворяются в воде, спирте и ацетоне.
Безводную соль TiBr3 получают восстановлением TiBr4 водо-
родом при 400° или восстановлением TiBr4 металлическим сере-
бром, ртутью или титаном в бензольном растворе.
Соединение представляет собой черно-синие кристаллы, кото-
рые при 927° диспропорционируют на TiBr2 и TiBr4.
Трииодид титана, Til3 -6Н2О, выделяется в виде фиолетовых
кристаллов при упаривании в вакууме раствора, полученного
электролитическим восстановлением Til4 в иодистоводородной кис-
лоте (электроды из титана). При сильном нагревании в вакууме
трииодид титана диспропорционирует на Til2 и Til4 (см. реакцию
получения дииодида титана).
Сульфид титана (трисулъфид титана), Ti2S3, образуется при
осторожном нагревании дисульфида титана в токе водорода,
94
ПОБОЧНАЯ ПОДГРУППА IV ГРУППЫ
азота или двуокиси углерода, а также действием сероуглерода
на двуокись титана при 800°.
Сульфид титана представляет собой черно-зеленый порошок,
напоминающий металлический, плохо растворимый в воде, в NaOH
и разбавленных кислотах.
Сульфаты титан а(Ш). Безводный сульфат титана,
Ti2(SO4)3, получают обработкой фиолетовых кристаллов кислого
сульфата титана 3Ti2(SO4)3 -H2SO4 -25Н2О конц. H2SO4. Соль
представляет собой зеленый порошок, плохо растворимый в воде,
спирте, эфире и легко растворимый в разб. H2SO4 (образуются
фиолетовые растворы).
Кристаллы 3Ti2(SO4)3 -H2SO4 -25Н2О выделяются из фиолетового
раствора, получающегося при электролитическом восстановлении
сульфата титанила TiOSO4 в 50%-ной серной кислоте со свинцовым
катодом.
Соль Ti2(SO4)3-8Н2О получают упариванием в вакууме при 60°
раствора металлического титана в разбавленной H2SO4 или обра-
боткой трихлорида титана разб. H2SO4.
Известны титановые квасцы, например красные октаэдриче-
ские кристаллы RbTi(SO4)2-12Н2О, фиолетовые октаэдрические
кристаллы CsTi(SO4)2-12Н2О, а также двойные сульфаты
(NH4)2SO4 -3Ti2(SO4)3-18Н2О, Rb2SO4-3Ti2(SO)4)3-24Н2О, Na2SO4-
• Ti2(SO4)3-5Н2О.
Помимо перечисленных соединений титана(Ш), известны и другие,
например комплексы общей формулы Me|[TiF5], Me1j[TiCl6], Meij[TiC15(H2O)],
MeJ[Ti(SCN)6]-6H2O, Me+[Ti(C2O4)2]-2H2O, где Me+ = NHJ, К+, Rb+ и др.,
затем оксалат Т12(С2О4)з ЛОНгО, тартрат, цитрат, галлат, салицилат, сук-
цинат и др.
Соединения четырехвалентного титана
Наиболее многочисленные и устойчивые соединения (с простыми
и координационными связями) характерны для четырехвалент-
ного титана, который встречается в виде катионов Ti4+nTiO2 +
(радикал титанил) или анионов TiO2-, TiO4-, [TiFB]2-, [TiCl8]2-,
[Ti(SO4)3]2- и др.
Хлорид, бромид, иодид, сульфат, нитрат титана(1У) сильно
гидролизуются в водных растворах, превращаясь в соединения
титанила общей формулы TiOX2, где X = Cl-, Rr~, I-, NO^,
VaSOI" и т. д.
Соли титана(ТУ) бесцветны и относительно устойчивы только
в кислых растворах, поскольку в нейтральных растворах выпадает
ортотитановая кислота H4TiO4, а при кипячении — метатитановая
кислота H2TiO3.
Растворы солей титана(1У) в присутствии солей минеральных
кислот восстанавливаются металлическим цинком до солей тита-
на(П1), окрашенных в фиолетовый цвет.
ТИТАН
95
Неорганические соединения
Тетрагидрид титана, TiH4, получают электролизом 0,1 н.
раствора H2SO4 при 40° с титановыми электродами при силе тока
0,2 а и напряжении 240 в. Это газ без цвета и запаха, на воздухе
он горит:
ТШ4 + 2О2 = ТЮ2 + 2Н2О
Двуокись титана, TiO2, встречается в природе в виде минера-
лов рутила, анатаза (тетрагональные кристаллы) и брукита (ром-
бические кристаллы). В тетрагональной кристаллической решетке
рутила (рис. 4) и анатаза (рис. 5) каждый ион Ti4+ окружен шестью
ионами О2-, образующими октаэдр, каждый ион О2- окружен
Рис. 4. Структура рутила TiO2.
Рис. 5. Структура анатаза TiO2
тремя ионами Ti4+. Кристаллы рутила и анатаза различаются
соотношением осей симметрии, плотностью, цветом и некоторыми
другими свойствами.
Двуокись титана (титановые белила) получают сжиганием
металлического титана в избытке кислорода, прокаливанием орто-
или метатитановой кислоты, действием водяного пара на порош-
кообразный титан при 700—800°:
Ti О2 = TiO2 225,5 ккал H2TiO2 —> Ti02 Н2О
H4TiO4 -> TiO2 + 2Н2О Ti + 2Н2О = TiO2 + 2Н2
Двуокись титана представляет собой белый порошок, который
превращается в бесцветные кристаллы анатаза при охлаждении
после нагревания до 700° или в кристаллы рутила при охлаждении
после прокаливания выше 850°.
Двуокись титана с плотностью 3,6—3,95 г!ex’ (анатаз), 4,1 —
4,2 г/см® (брукит), 4,2—4,3 г/см® (рутил) плавится при 1855°,
96
ПОБОЧНАЯ ПОДГРУППА IV ГРУППЫ
разлагается выше 2927° (тугоплавка), плохо растворима в воде,
разбавленных кислотах или растворах щелочей; растворяется
в конц. H2SO4 при нагревании, в расплавах гидроокисей или кар-
бонатов щелочных металлов и в окислах металлов, проявляя кис-
лотные свойства. Способна к металлотермическому восстановле-
нию (с алюминием или кальцием) до металлического титана.
TiO2 + 2H2SO4 = Ti(SO4)2 + 2Н2О
TiO2 + 2NaOH = Na2TiO3 + H2O
TiO2 + 4NaOH = Na4TiO4 + 2H2O
TiO2 + Na2CO3 = Na2TiO3 + C02
TiO2 + 2Na2CO3 = Na4TiO4 + 2CO2
TiO2 + ZnO = ZnTiO3
TiO2 + NiO = NiTiO3
3TiO2 + 4A1 = 3Ti + 2A12O3
TiO2 + 2Ca = Ti + 2CaO
Титановые белила применяют в качестве пигмента для пласти-
ческих масс, масляных красок (обладающих большой кроющей
способностью и кислотостойкостью), при производстве молочного
стекла, тугоплавких стекол, фарфора, огнеупорного кирпича,
в производстве эмалей и глазурей.
В многочисленных реакциях органической химии двуокись
титана служит катализатором.
Кристаллы рутила, обладающие хорошим коэффициентом пре-
ломления, применяются в ювелирных изделиях.
Сосуды из листового железа покрываются глазурью из дву-
окиси титана и моноокиси железа. Из двуокиси титана, содержащей
незначительные количества моноокиси железа, изготовляют эмали
и материал для зубных протезов.
Природный продукт TiOa-FeO служит для футеровки пода
печей в металлургии.
Ортотитановая кислота (а-титановая кислота), TiO2-2H2O,
получается обработкой на холоду свежеприготовленных растворов
сульфата титанила или тетрахлорида титана (в солянокислой
среде) растворами щелочей (аммиака), содой или поташом, а также
сульфидом аммония. Гидролизом других солей четырехвалентного
титана на холоду также можно получить ортотитановую кислоту.
TiOSO4 + 2NaOH + Н2О = H4TiO4 + Na2SO4
TiCl4 + 4NaOH = H4TiO4 + 4NaCl
Ti(SO4)2 + 4NH4OH = H4TiO4 + 2(NH4)2SO4
Ti(SO4)2 + 2Na2CO3 + 2H2O = H4TiO4 -H 2Na2SO4 + 2CO2
TiCl4 + 2(NH4)2S + 4H2O = H4TiO4 + 4NH4C1 + 2H2S
TiCl4 + 4H2O H4TiO4 + 4HC1
Ортотитановая кислота образует студнеобразный белый осадок
(амфотерного характера: растворяется в минеральных кислотах),
который при нагревании или хранении ступенчато превращается
в метатитановую кислоту. Ортотитановая кислота слабее ортокрем-
ТИТАН
97
левой кислоты H4SiO4. Соли ортотитановой кислоты, называемые
ортотитанатами, имеют общую формулу Me4TiO4 или Me2+TiO4 где
Ме+ = Na+, К+, NH( и др., а Ме2+ = Mg2+, Zn2+, Mn2+, Со2+, Ni2+
и др.
Ортотитанаты получают сплавлением двуокиси титана со сте-
хиометрически необходимым количеством гидроокисей или карбо-
натов щелочных металлов либо различными окислами металлов.
В специальной литературе упоминаются соли производных
гетерополикислот H4[TiMo12O40], H4[TiWi204o], которые рассма-
триваются как производные кислоты H4TiO4.
Метатитановая кислота, H2TiO3 (^-титановая кислота TiO2-
•Н2О), выпадает в виде белого осадка (с микрокристаллической
структурой) при гидролизе тетрахлорида титана или сульфата
титанила при 80—100°, нагревании или хранении ортотитановой
кислоты, обработке солянокислого раствора тетрахлорида титана *
растворами щелочей, ацетата или тиосульфата натрия или суль-
фида аммония при нагревании:
TiCl4 + ЗН2О H2TiO3 + 4НС1
TiOSO4 + 2Н2О H2TiO3 + H2SO4 H4TiO4 = H2TiO3 + H2O
TiCl4 + 4KOH = H2TiO3 + 4KC1 + H2O
TiCl4 + 2Na2S2O3 + H2O = H2TiO3 + 4NaCl + 2SO2 + 2S
TiCl4 + 2(NH4)2S + 3H2O = H2TiO3 + 4NH4C1 + 2H2S
TiCl4 + 4NaCH3COO + 3H2O = H2TiO3 + 4NaCl + 4CH3COOH
Метатитановая кислота химически более инертна, чем ортоти-
тановая кислота, и растворяется
при нагревании. Соли метатитано-
вой кислоты, называемые метати-
танатами, можно представить сле-
дующими формулами: Me2TiO3,
Me2+TiO3, где Ме+ = Li+, Na+, К+,
NHJ и др., Ме2+ = Са2+, Ва2+,
Zn2+, Со2+, Ni2+ и др.
В кубической решетке перов-
скита CaTiO3 (рис. 6) ионы Ti4+
расположены в вершинах куба,
ионы О2- — в центре граней куба
и ионы Са2+ — в центре диаго-
налей куба.
Соединение BaTiO3 обладает очень высокой диэлектрической
постоянной и применяется для изготовления электрических кон-
* В солянокислом растворе Ti4+ существует в виде гексахлортитановой
кислоты H2TiCl6. Введение щелочных реагентов обусловливает фактически
сдвиг реакции гидролиза последней.— Прим. ред.
‘-0101
98
ПОБОЧНАЯ ПОДГРУППА IV ГРУППЫ
денсаторов большой емкости. Титанат стронция SrTiO3 служит
для изготовления батарей, используемых в автоматических метео-
рологических станциях.
Пероксотитановые соединения. При обра-
ботке нейтральных или кислых растворов сульфата титанила
или других солей титана(1У) перекисью водорода Н2О2 появляется
желтое (в случае разбавленных растворов) или интенсивное оран-
жевое (при использовании концентрированных растворов) окра-
шивание вследствие образования монопероксоортотитановой кис-
лоты H4TiO5, которая сама обладает желто-оранжевым цветом:
TiOSO4 + Н2О2 + 2Н2О = H4TiO6 + H2SO4
Колориметрическое определение титана основывается на этой
реакции.
В подкисленном H2SO4 растворе сульфат титанила с перекисью
водорода образует монопероксодисульфатотитановую кислоту
H2[TiO2(SO4)2], при нейтрализации которой (КОН) образуется,
желтый осадок K2[TiO2(SO4)2]-ЗН2О:
TiOSO* Н2О2 H2SO4 = H2[TiO2(SO4)2] Н20
H2[TiO2(SO4)2] + 2К0Н = K2[TiO2(SO4)2] + 2Н2О
Известен также аддукт ортотитаната калия с перекисью водо-
рода K4TiO4 -4Н2О2-2Н2О или K4TiO8-6H2O.
Пероксотитановые соединения используются как энергичные
окислители.
Тетрагалогениды титана. По своим физическим
свойствам тетрагалогениды титана похожи на тетрагалогениды
элементов подгруппы германия.
Цвет тетрагалогенидов титана и комплексных кислот общей
формулы H2[TiX8] (где X = F-, С1~, Вг~, I-), становится более
интенсивным с увеличением атомного номера галогена.
Тетрагалогениды титана характеризуются пониженными значе-
ниями температур плавления и кипения, отчетливо выраженной
склонностью к гидролизу* (за исключением тетрафторида), к ком-
плексообразованию с гидроокисями или галогенидами щелочных
металлов.
Тетрафторид титана, TiF4, получают взаимодействием титана
и фтора при нагревании или обработкой тетрахлорида титана
взятой в избытке безводной плавиковой кислотой при 100—120°:
Ti + 2F2 = TiFt + 393,6 ккал TiCl4 + 4HF = TiFt + 4HC1
Тетрафторид титана представляет собой расплывающийся
на воздухе белый порошок с плотностью 2,798 г/слг3 и т. пл. 284°;
♦ Эти соединения относятся к классу галоидоангидридов. Проявляют
свойства соединений с полярной связью; при гидролизе дают две кисло-
ты.— Прим. ред.
ТИТАН
99
он сублимируется при 327°, плохо растворяется в эфире, образует
аддукты с аммиаком и пиридином (TiF4-4NH3, TiF4-2NH3,
TiF4-C5H5N), реагирует с РОС13, SiHCl3 по уравнениям
3TiFt + 4РОС13 = 3TiCl4 + 4POF3
3TiFt + 4SiHCl3 = 3TiCl4 + 4SiHF3
При обработке теплой водой тетрафторида титана получается
кислота H2[TiOF4], из которой концентрированием на холоду
выделяют кристаллогидрат TiF4-2H2O.
Действием фтористоводородной кислоты на тетрафторид титана
получают бесцветную гексафторотитановую кислоту H2[TiFel,
для которой известны соли гексафторотитанаты
Me+[TiFe], Me2+[TiFe]-пН2О (где Ме+ = Na+, К+, Cs+, NH+, Ag+,
а Ме2+ = Са2+, Sr2+, Mg2+, Zn2+, Cd2+, Mn2+, Fe2+, Co2+, Ni2+ и др.).
Гексафторотитанаты можно получить обработкой TiF4 растворами
фторидов различных элементов или действием на H2[TiFe] раство-
рами солей различных элементов.
Известен также гептафторотитанат аммония (NH4)3[TiFjl.
Тетрахлорид титана, TiCl4, получают назреванием порошко-
образного металлического титана, карбида титана TiC или смеси
двуокиси титана с сахарным углем в токе хлора, а также пропуска-
нием паров СС14 над TiO2 при нагревании:
Ti + 2С12 = TiCl4 + 194,9 ккал TiO2 + СС14 = TiCl4 + СО2
TiO2 + 2С + 2С12 - TiCl4 + 2GO — 11 ккал TiC + 2С12 = TiCl4 + С
В промышленности технический TiCl4 получают хлорированием
в хлораторе богатых титановых шлаков или концентратов рутила
в присутствии углерода при 700°. Образующийся продукт загряз-
нен SiCl4, FeCl3, СоС13, VOC13 и др. Исчерпывающее хлорирование
титана из шлаков или концентратов рутила затруднено присут-
ствием окислов СаО, MgO, MnO, SiO2, которые хлорируются при
~ 1000°, превращаясь в СаС12, MgCl2, MnCI2, летучие только выше
1200°. Летучие хлориды удаляются из хлоратора по трубопроводу
в конденсаторы, а труднолетучие хлориды остаются в хлораторе
в расплавленном состоянии в виде эвтектических смесей.
Для очистки TiCI4 от FeCl3, SiCl4, VOC13 и др. используют
процессы, которые основываются на различиях физических и хими-
ческих свойств этих соединений.
Тетрахлорид титана — бесцветная (чуть желтоватая), сильно
преломляющая, с едким запахом жидкость, дымящая на воздухе,
гидролизующаяся при соприкосновении с водой; плотность
1,76 г!см?, т. затв. —24,1°, т. кип. 136,5°. С аммиаком, пириди-
ном, анилином, пиперидином, спиртами, эфирами, хлоридами РС13,
РС15, РОС13, SC14 и др. TiCl4 образует аддукты, а с гидроокисями
и хлоридами щелочных металлов — координационные соединения.
Несколько примеров аддуктов: TiCl4 -nNH3 (где п — 8, 6, 4),
7*
100
ПОБОЧНАЯ ПОДГРУППА IV ГРУППЫ
TiCl4-2С5Н5К, TiCl4.2CeH5NH2, TiCl4-С2Н6ОН, TiCl4-PCl3,
Т1С14-РС15, TiCl4-2РОС13, TiCl4-SCl4, 2TiCl4-SCl4; примеры гало-
генокомплексов: H2[TiCl8l, H2[TiCl4Br2], H2[TiCl4I2], Na2[TiCl8],
K2[TiCl8], (NH4)2[TiCl8]-2H2O.
Тетрахлорид титана можно восстановить при нагревании метал-
лическим магнием, кальцием, натрием, магниевокальциевым спла-
вом, гидридом натрия, водородом, серебром, сурьмой, алюминием,
ртутью, цинком медью и др.
При гидролизе тетрахлорида титана получают хлорид титанила
TiOCl2 (при небольшом количестве воды) и ортотитановую кислоту
H4TiO4 — при нормальной температуре — или метатитановую
кислоту H2TiO3 — при 80° (при большом количестве воды):
TiCl4 4- Н2О TiOCl2 + 2НС1
TiCl4 4- ЗН2О ч* H2TiO3 + 4НС1
TiCl4 + 4Н2О H4TiO4 + 4HG1
Известен кристаллогидрат TiCl4 -5Н2О, который выделяется
при выпаривании сильнокислого водного раствора тетрахлорида
титана в соляной кислоте. В вакууме соль TiCl4-5H2O теряет
3 молекулы воды.
Тетрабромид титана, TiBr4, получают нагреванием порошко-
образного металлического титана или карбида титана в парах
брома, действием бромистого водорода на тетрахлорид титана
при 130°, а также действием паров брома на смесь TiO2 с углем
(300°):
Ti + 2Вг2 = TiBr4 + 170 ккал TiC + 2Br2 = TiBr4 + С
TiCl4 + 4HBr = TiBr4 + 4НС1 TiO2 + 2С + 2Br2 = TiBr4 + 2СО
Тетрабромид титана представляет собой гигроскопичные, жел-
тые, октаэдрические кристаллы с плотностью 3,25 г/см3, гидроли-
зующиеся при растворении в воде; т. пл. 38,3°, т. кип. 230°.
С аммиаком дает аддукты, а с бромистоводородной кислотой —
окрашенную в красный цвет гексабромтитановую кислоту
Ha[TiBr8], образующую соли общей формулы Me+tTiBra], (где
Ме+ = Na+, К+, NHJ).
Тетраиодид титана, Til4, получают действием паров иода
на порошкообразный металлический титан при 400° или на карбид
титана, а также обработкой тетрахлорида титана сухим иодистым
.водородом:
Ti + 2I2 = Til4 4- 131 ккал TiC + 2I2 = Til4 + С
TiCl4 + 4HI = TiCl4 + 4НС1
Тетраиодид титана представляет собой металлоподобные крас-
ные кубические кристаллы с т. пл. 155° и т. кип. 377°; под дей-
ствием теплой воды происходит гидролиз, с иодистоводородной
кислотой Til4 образует гексаиодотитановую кислоту H2[TiI8l.
ТИТАН
101
Дисульфид титана, TiS2, получают прямым взаимодействием
исходных элементов при высокой температуре или пропусканием
смеси TiCl4 и H2S через фарфоровую трубку при 800 — 850°:
Ti + 2S = TiS2 TiCl4 + 2H2S = TiS2 + 4HC1
Дисульфид титана образует желтые кристаллы с металличе-
ским блеском; при обычных условиях они устойчивы на воздухе,
в воде и разбавленных кислотах (H2SO4, НС1 и др.), взаимодей-
ствуют с расплавленным едким кали или с двуокисью углерода при
нагревании; под действием HNO3 или конц. H2SO4 разлагаются
с выделением серы:
TiS2 + 6КОН = K2TiO3 + 2K2S + ЗН2О
TiS2 + 2СО2 = TiO2 + 2S + 2CO
При прокаливании дисульфида титана на воздухе образуется
двуокись титана и выделяется сернистый газ:
TiS2 + ЗО2 = TiO2 + 2SO2
Известны аддукты тетрахлорида и тетрабромида титана с серо-
водородом: 2TiCl4-2H2S, TiCl4-2H2S, TiBr4-H2S, TiBr4-2H2S.
Дисулъфат титана, Ti(SO4)2, образуется при действии SO2
в SO2C12 на TiCl4 и представляет собой бесцветное, сильно гигро-
скопичное вещество. В водном растворе образуется сульфат тита-
нила TiOSO4-2H2O.
Известны комплексные сульфаты, производные дисульфата
титана общей формулы MeJtTi(SO4)3], например K2[Ti(SO4)3J.
Сульфат титанила {оксисульфат титана), TiOSO4, получают
обработкой двуокиси титана концентрированной серной кислотой
или растворением в холодной воде охлажденного расплава двуоки-
си титана и пиросульфата калия:
TiO2 + H2SO4 TiOSO4 + Н2О TiO2 + K2S2O7 = TiOSO4 + K2SO4
Сульфат титанила представляет собой белый порошок, раствори-
мый в холодной воде и серной кислоте; в теплой воде происходит
его гидролиз:
TiOSO4 + 2Н2О H2TiO3 + H2SO4
С сульфатами щелочных металлов TiOSO4 образует комплекс-
ные соли общей формулы Me£[TiO(SO4)2] -пН2О, например,
K2[TiO(SO4)2l -10Н2О, Na2[TiO(SO4)2].
Хелатные соединения
Соли четырехвалентного титана взаимодействуют в растворе
при pH = 2,7 с 8-оксихинолином, образуя желтый осадок:
102
ПОБОЧНАЯ ПОДГРУППА IV ГРУППЫ
Ион Ti4+ с купфероном CeH5 — N(NO)ONH4 в холодных сла-
бокислых растворах образует желтый трудно растворимый хелат:
В нейтральной среде соли четырехвалентного титана взаимо-
действуют с ализарином (1,2-диоксиантрахиноном), образуя крас-
но-фиолетовый лак:
При обработке солей четырехвалентного титана в слабокислой
среде (pH = 2 — 3) 2,5%-ным раствором хромотроповой (1,8-диок-
синафталин-3,6-дисульфоновой) кислоты CioH4(OH)2(S03H)2 -2Н2О
образуется интенсивно окрашенное в коричнево-красный цвет
вещество.
Пирокатехин (1,2-диоксибензол) СвН4(ОН)2 с растворами солей
титана в слабокислой среде дает желтое окрашивание.
Обработкой солянокислых растворов соединений титана спир-
товым раствором морина (3,5,7, 2',4'-пентоксифлавона) получают
комплексное соединение оранжево-красного или красновато-корич-
невого цвета.
Известен также тетраацетилацетонат титана формулы
Ti
О = С
О—С
ZCH3
>сн
ХСН3
Помимо вышеприведенных соединений четырехвалентного титана изве-
стны и другие, например: титанаты Na2TiO3, Na4TiO4, K2TiO3-4H2O,
K2H2TiO4-H2O, CaTiO3, BaTiO3, Ba2TiO4, MgTiO3, Mg2TiO4, ZnTiO3,
Zn2TiO4, MnTiO3, FeTiO3, Fe4(TiO4)3, NiTiO3, CoTiO3, PbTiO3, селенид
TiSe2, селениты, селенаты, теллурид TiTe2, нитрид Ti3N4, амид Ti(NH2)4,
тиоцианаты Ti(SCN)4 -8HSCN, TiO(SCN)2-2H2O, комплексы общей формулы
Me+[TiO(SCN)4], Me$[TiO(C2O4)2], металлоорганические соединения
(C6H6)2TiCl2, (C5H6)2TiBr2, (C6H6)2Ti(CH3)2, (C6H6)2TiCl2AlR2 и др.
Соединения включения
Нитрид титана, TiN, образуется путем нагревания порошко-
образного металлического титана при 800° в атмосфере азота,
сильного нагревания тетрахлорида титана в токе аммиака, нагре-
ТИТАН
103
вания при 1205° смеси двуокиси титана с углем в атмосфере азота,
а также путем разложения нитрида Ti3N4 при нагревании:
2Ti + N2 = 2TiN + 2 X 80,3 ккал 2TiO2 4- 4С 4- N2 =2TiN + 4CO
3TiCl4 4- 16NH3 = 3TiN 4- 12NH4C1 4- V2N2 2Ti3N4 = 6TiN 4- N2
Нитрид титана представляет собой металлоподобный порошок
бронзового цвета; он обладает высокой твердостью (8—9 по шкале
Мооса), хорошо проводит электрический ток; плотность 5,1—
5,28 г/см3, т. пл. 2947°. Под действием водяных паров или при
•обработке щелочами TiN разлагается.
TiN 4- 2Н2О = TiO2 4- NH3 4- V2H2
TiN 4- 2KOH 4- H2O = K2TiO3 4- NH3 4- V2H2
В плавиковой кислоте в присутствии энергичных (окислителей
растворяется. Применяется в качестве абразивного материала при
шлифовке драгоценных камней.
Известен двойной нитрид 2Li3N-TiN, или Li8TiN3.
Карбид титана, TiC, всегда присутствует в титансодержащих
сталях. В лаборатории может быть получен нагреванием смеси
металлического титана с сахарным углем в электрической печи,
прокаливанием смеси двуокиси титана с углем в пламени электри-
ческой дуги (1620°), нагреванием угольной нити в парах TiCl4
и термической диссоциацией тетрахлорида титана в атмосфере
окиси углерода и водорода:
Ti 4- С = TiC 4- 114 ккал TiCl4 4- С = TiC 4- 2С12
TiO2 4- ЗС TiC4-2CO 4- 45,9 ккал
TiCl4 4- СО 4- ЗН2 = TiC 4- Н2О 4- 4НС1
Карбид титана образует металлоподобные черные кубические
кристаллы с плотностью 4,70—4,95 г/см3', он обладает высокой
твердостью (9 по шкале Мооса), тугоплавкостью (т. пл. 3137°),
устойчив к истиранию (будучи менее хрупким, чем карбид воль-
фрама). При нагревании карбид титана взаимодействует с кисло-
родом, галогенами и азотом. Нагреванием карбида титана при
1800—1900° в атмосфере водорода и азота получают нитрид титана
и образуется ацетилен:
2TiC 4- N2 4- Н2 = 2TiN 4- С2Н2
Карбид титана применяется в качестве раскислителя сталей,
при изготовлении угольных электродов для дуговых ламп и в
сочетании с карбидом вольфрама при получении сверхтвердых
сплавов.
104
ПОБОЧНАЯ ПОДГРУППА IV ГРУППЫ
ЦИРКОНИЙ Zr
Z= 40; ат. вес = 91,22
Валентность (II), (III), IV, заряд (2+), (3+), 4+
Массовые числа природных изотопов 90, 92, 94,
91, 96
Электронная структура атома циркония: К -L -М •4s24p64da -5s2.
Электронная структура атома и ионов Zra+, Zr3+, Zr4+ для 4d-
и 5я-орбиталей:
4d2 5s2 4<Z2 5s 4d! 5s 4d 5s
fhi m и пи rm □ hi i i i i □ i i i i i i □
Zr Zr2+ Zr3+ Zr>+
ИСТОРИЧЕСКАЯ СПРАВКА
В старых легендах Востока говорится о красоте золотисто-
желтых кристаллов минерала циркона и о красиво расцвеченных
его разновидностях — гиацинте, иацинте и иаргоне. Кристаллы
циркона ZrSiO4 (с греческого «zerc», что означает «драгоценный»,
или с персидского «(ar gun», означающее «цвет золота») применя-
лись как драгоценные камни. Среди античных редкостей упоми-
нается красный камень кристаллов эвдиалита (комплексный
силикат циркония, кальция и натрия) и матурские бриллианты,
попавшие в Европу с Цейлона. Из минерала циркона ZrSiO4,
привезенного с Цейлона в 1787 г., Клапрот впервые получил дву-
окись циркония.
В 1824 г. Берцелиус впервые получил загрязненный металли-
ческий цирконий восстановлением соединения K2[ZrF8] при нагре-
вании с металлическим калием или натрием.
Почти 100 лет металлический цирконий не удавалось получить
в чистом виде из-за его сродства к другим элементам. Загрязненный
металлический цирконий (будучи хрупким и обладая низкой
механической прочностью) находил весьма ограниченное примене-
ние. В большей степени он использовался как добавка при полу-
чении специальных сталей, а также огнеупорных материалов
(например, ZrO2).
Порошкообразный металлический цирконий начали получать
в 30-х годах. Металлический цирконий переводят в компактное
состояние из порошка методом порошковой металлургии.
Металлический цирконий ядерной чистоты, полученный совре-
менными методами вакуумной металлургии, относится к основным
материалам ядерной техники благодаря сочетанию химической
и термической стойкости с особыми механическими и ядерными
свойствами.
Соединения циркония в древности использовались как драго-
ценные камни, а в наши дни нашли применение для технических
ЦИРКОНИЙ
105
целей, как и сам металл, являющийся конструкционным материа-
лом для ядерных реакторов.
РАСПРОСТРАНЕННОСТЬ В ПРИРОДЕ
В природе цирконий встречается в виде соединений в различных
минералах, достаточно распространенных в земной коре. Цирко-
ний занимает 21-е место по распространенности элементов и его
содержание в земной коре (16-километровая зона литосферы)
составляет 0,025 вес. %. В земной коре циркония больше, чем
многих других металлов, таких, как медь, цинк, олово, никель
и свинец.
Соединениям циркония в природе сопутствуют главным обра-
зом соединения гафния.
К важнейшим минералам циркония относятся следующие.
Бадделеит (названный циркониевой землей), ZrO2, содержит
до 73,9% циркония; загрязнен HfO2, U3O8, ThO2, SiO2, TiO2, Fe2O3
и др.; минерал желто-коричневого цвета, плотность 4,8—6 г!см\
твердость 6—7 по шкале Мооса, обладает высокой химической
стойкостью. В природе бадделеит часто встречается вместе с иль-
менитом, рутилом, цирконом, пирохлором, магнетитом, апатитом,
турмалином и др. и образуется при выветривании циркона и эвдио-
лита. Самые большие залежи бадделеита находятся в Бразилии.
Циркон (гиацинт), ZrSiO4, содержит 49,5% Zr (загрязнен
HfO2, Fe2O3, СаО, иногда AI2O3, Y2O3, Се2О3, Р2О5, ThO2, Nb2O5,
Та2О5, U3O8), встречается в виде бесцветных или желтых (иногда
оранжевых, красных, редко зеленых в зависимости от примесей)
тетрагональных кристаллов с плотностью 4,56 г!см? и твердостью
7,5 по шкале Мооса. Разновидности циркония циртолит и мала-
кон обычно радиоактивны. В природе циркону сопутствуют черная
слюда, апатит, альбит, нефелин, флюорит и минералы редких
земель. Залежи циркона находятся в Калифорнии, Норвегии,
Бразилии, Австралии, Индии, США, Африке, Цейлоне и др. Цир-
кон — основной источник получения циркония, он часто служит
огнеупорным материалом при футеровке металлургических печей.
В числе других минералов циркония можно назвать эвдиалит (комплекс-
ный силикат циркония, кальция и натрия), эльпидит, катаплеит (комплекс-
ные силикаты циркония) и др. Цирконий встречается и в некоторых мине-
ралах редких земель — в монаците, ксенотиме, тортвейтите и др.
ОБОГАЩЕНИЕ И ПЕРЕРАБОТКА ЦИРКОНИЕВЫХ РУД
Для обогащения циркона применяется комплекс гравитацион-
ных, электромагнитных, электростатических и флотационных мето-
дов, а в случае бадделеита — гравитационные и электромагнитные.
Концентраты циркона можно разрушить спеканием с СаО или.
406
ПОБОЧНАЯ ПОДГРУППА IV ГРУППЫ
,СаСО3, сплавлением с NaOH или Na2CO3, спеканием с гексафторо-
силикатами, а также карбидизацией и хлорированием.
Переработка цирконовых концентратов спеканием с СаО или СаСОз
и обработка продуктов спекания серной кислотой
Спеканием тонкоизмельченного концентрата циркона с СаО
(взятого в 30 %-ном избытке по отношению к стехиометрически
необходимому количеству) или с СаСО3 при 1300° во вращающейся
цилиндрической печи получают CaZrO3 и CaSiO3:
ZrSiO4 + 2СаО = CaZrO3 CaSiO3
ZrSiO4 + 2СаСО3 = CaZrO3 + CaSiO3 + 2CO2
При действии на охлажденный продукт спекания конц.
H2SO4, взятой в 15%-ном (по весу) избытке, образуется раство-
римый в серной кислоте ZrOSO4, а также CaSO4 и H2SiO3, ча-
стично растворимые в воде:
CaZrO3 + 2H2SO4 = ZrOSO4 + CaSO4 + 2H2O
CaSiO3 + H2SO4 = CaSO4 + H2SiO3
Чтобы облегчить отделение метакремневой кислоты, добав-
ляют коагулянт.
Если к 2 об. ч. раствора ZrOSO4 прилить 1 об. ч. конц. H2SO4,
выпадают кристаллы H2[ZrO(SO4)2 -ЗН2О.
При действии на продукты спекания 15%-ным (по весу) избыт-
ком конц. НС1 при 80—90° протекают следующие реакции:
CaZrO3 + НС! = ZrOCl2 + СаС12 + 2Н2О
CaSiO3 -|- 2НС1 = СаС12 -|- H2SiO3
Путем добавления конц. НС1 в охлажденные растворы хлори-
дов при 20° получают кристаллы ZrOCl2 -8Н2О.
Последовательность переработки циркона при спекании с СаСО3
и обработки продукта спекания H2SO4 показана на следующей
«схеме:
ZrSiO4 2СаСОз = CaZrO2 CaSiO3 -j- 2СО2
Циркон 4 —1 '
-l-конц. H2SO4
ZrOSO4 CaSiO3 + H2SiO3
Растворим Частично растворим
| +конц. H2SO4
H2[ZrO(SO4)2]-3H2O
Трудно растворим
Последовательность переработки концентратов циркона мето-
ЦИРКОНИЙ
107
лом сплавления с NaOH изображена ниже.
ZrSiO4+4NaOH = Na2ZrO3+Na2SiO3 + 2H2O
< - ,--------------------- - >
При добавлении горячей воды
ZrO2*nH2O Na2SiO3
Трудно растворим Растворим
I +HC1
ZrOCl2 + SiO2
Растворим Трудно растворим
| +NHS
ZrOj^nHjO
Трудно растворим
Переработка концентратов циркона спеканием
с гексафторосиликатами
Спеканием измельченного концентрата циркона с K2[SiFe]
и КС1 при 650—700° в цилиндрической вращающейся печи полу-
чают K2[ZrFe] и K2[HfFe]:
ZrSiO* + K2[SiFe] = K2[ZrFe] + 2SiO2
HfSiO4 + K2[SiF6] = K2[HfF6] + 2SiO2
При охлаждении раствора, полученного обработкой продуктов
спекания 1%-ным раствором НС1 (на 1 вес. ч. продуктов спекания
7 вес. ч. 1%-ной НС1) при 85°, выпадают кристаллы K2[ZrFe]
и K2[HfF6],
Поскольку растворимость соединения K2[ZrFe] в 78%-ном
растворе HF при 20° равна 36,5 г/л, а соединения H2[HfFe] —
72 г/л, эти две соли разделяют методом дробной кристаллизации.
Прокаливанием при 900° ZrO2 -пНгО, образующегося при обра-
ботке раствора K2[ZrFe] аммиаком, получают ZrO2, который содер-
жит очень небольшие количества HfO2, Fe2O3 и TiO2.
Переработка концентратов циркона
путем карбидизации и хлорирования
Прокаливанием при 1900—2000° смеси концентрата циркона
с углем (20% по весу) получают карбид циркония ZrC, загрязнен-
ный SiO2:
ZrSiO* + ЗС = ZrC + SiO2 + 2СО
В действительности химический процесс протекает значительно
сложнее, поскольку в присутствии воздуха наряду с карбидом
циркония образуется и нитрид.
Хлорированием карбида и нитрида циркония при 350—450°
(в стальных резервуарах) получают тетрахлорид циркония, загряз-
ненный SiCl4 и Fe2Cle.
108
ПОБОЧНАЯ ПОДГРУППА IV ГРУППЫ
Для очистки ZrCl4 перегоняют при 450—600° после предвари-
тельного восстановления водородом Fe2Cl6 до FeCl2.
Поскольку реакции хлорирования карбида и нитрида циркония
изотермичны, нагревание необходимо только в начале процесса:
ZrC 2С12 = ZrCl4 -|- С -|- 202 ккал
ZrN 2С12 = ZrCl4 1/2N2 + 160 ккал
Переработка концентратов бадделеита
При кипячении концентратов бадделеита с НС1 двуокись цир-
кония остается в осадке, примеси же переходят в раствор. Затем
двуокись циркония сплавляют с NaHSO4 и холодный расплав
обрабатывают водой для получения раствора ZrOSO4, из которого
под действием аммиака выделяется белый осадок ZrO2-nH2O.
ОТДЕЛЕНИЕ ЦИРКОНИЯ ОТ ГАФНИЯ
В минералах цирконию сопутствует гафний. При переработке
концентратов руд гафний ведет себя так же, как и цирконий,
поскольку эти элементы очень близки по химическим свойствам,
обладают близкими значениями ионных радиусов (0,79 А у иона
Zr4+ и 0,78 А у иона Hf4+) и легко замещают друг друга в различ-
ных химических соединениях.
Чтобы получить «ядерно чистый» металлический цирконий,
соединения циркония, служащие исходным материалом, не должны
содержать соединений гафния.
Цирконий от гафния можно отделить дробной кристаллиза-
цией гексафторосолей K2[ZrF8], K2[HfF8] или (NH4)2[ZrF8],
(NH4)2[HfF8J, сульфатосолей калия K2[Zr(SO4)3], K2[Hf(SOt)3],
оксихлоридов ZrOCl2-8H2O, НЮС12-8Н2О, оксалатов, фосфатов
и др. Кроме того, разделение возможно путем термического
разложения смеси (NH4)2[ZrF8l и (NH4)2[HfFe], поскольку
вначале возгоняются NH4F, затем HfF4 и в конце ZrF4. Приме-
няются также дробная перегонка смеси 3ZrCl4 -ЗРОСЦ (кипит
при 360°) и 3HfCl4-2РОС13 (кипит при 355°), селективная экстрак-
ция органическими растворителями, такими, как трифторацетила-
цетон F3C — СОСН2 — СО — СН3, метилизобутилацетон, трибу-
тилфосфат и д£., адсорбция катионов Zr4+, Hf4+ на катионитах
и элюирование гафния соляной кислотой, а циркония — смесью
азотной и лимонной кислот, адсорбция комплексных анионов
циркония и гафния на ионообменных смолах с соляной кислотой
или смесью соляной и фтористоводородной кислот в качестве элю-
анта; электролиз расплава K2[ZrF8] с NaCl (этот метод сочетают
с процессами разделения, основанными на кристаллизации солей,
являющихся электролитами).
ЦИРКОНИЙ
109
ПОЛУЧЕНИЕ МЕТАЛЛИЧЕСКОГО ЦИРКОНИЯ
Металлический цирконий получают восстановлением тетрахло-
рида циркония металлическим магнием или натрием в атмосфере
гелия или аргона, восстановлением гексафторцирконата калия
металлическим натрием в вакууме (или в атмосфере водорода,
гелия, аргона и др.), термической диссоциацией тетраиодида цир-
кония, электролизом расплава K2[ZrF8] с КС1 в атмосфере аргона,
восстановлением двуокиси циркония металлическим натрием или
кальцием (соответственно смесью металлического натрия и каль-
ция) при нагревании в вакууме.
Магнийтермическое восстановление тетрахлорида циркония
(метод Кроля)
ZrCl4 2Mg = Zr 2MgCl2 + 52,5 ккал
Магнийтермическим восстановлением тетрахлорида циркония
в отсутствие ZrOCl2 при 650° в реакторе из сплава инконеля (72,7%
Ni, 15% Ст, 7% Fe, 1 % Nb и 2,5% Ti) в атмосфере гелия или аргона
получают губчатый металлический цирконий, загрязненный сле-
дами магния, железа, титана, алюминия, марганца, кремния, кис-
лорода, азота, углерода и хлора.
Из губчатого металлического циркония избыток магния и хло-
рида магния удаляют промывкой НС1 или разб. HNO3 и водой
либо перегонкой в вакууме примерно при 800°.
Плавлением губчатого циркония в вакуумной электродуговой
печи (соответственно в атмосфере инертного газа) или в печи с сфоку-
сированным пучком электронов в высоком вакууме получают ком-
пактный металлический цирконий.
Тетрахлорид циркония можно восстановить до металлического
циркония также металлическим натрием в атмосфере гелия или
аргона при нагревании.
Восстановление гексафторцирконата(ГУ) калия K2[ZrF8]
металлическим натрием (процесс Берцелиуса)
K2[ZrFe] + 4Na = Zr + 2KF + 4NaF
Восстановлением гексафтороцирконата(1У) калия металличе-
ским натрием при 800° в вакууме или атмосфере водорода, гелия,
аргона и др. в герметически закрытом сосуде получают порошок
металлического циркония, загрязненный главным образом
окислами.
Для очистки от KF и NaF порошкообразный металлический
цирконий промывают водой, разб. НС1, затем снова водой и сушат
при 50—60° в вакууме.
110
ПОБОЧНАЯ ПОДГРУППА IV ГРУППЫ
Металлический цирконий в компактном состоянии получают
плавлением в электродуговых печах (или в печах с сфокусирован-
ным пучком электронов) в вакууме или методами порошковой
металлургии (см. раздел, посвященный титану).
Термическая диссоциация тетраиодида циркония
(процесс Ван-Аркеля — де Бура)
Zrl4 5ft Zr + 2I2
Этот процесс применяется в промышленности при очистке
металлического циркония, полученного магнийтермическим вос-
становлением ZrCl4 или K2[ZrFe] металлическим натрием (как
описано выше).
При действии паров иода на порошкообразный или губчатый
металлический цирконий, нагретый выше 400°, образуется тетраио-
дид циркония Zrl4, который сублимируется при 431° и термически
диссоциирует при 1200°.
Для получения металлического циркония по этому методу при-
меняют аппаратуру, изображенную на рис. 3. В сосуд помещают
сырой цирконий и иод, создают вакуум и затем нагревают, при-
меняя наружный обогрев, примерно до 600°. Пары Zrl4, сопри-
касаясь с нагретой до 1400° циркониевой или вольфрамовой нитью,
разлагаются, и цирконий осаждается на нити, а пары иода вновь
вступают в реакцию с сырым цирконием.
Полученный по этому процессу металлический цирконий пласти-
чен и может подвергаться механической переработке (прокаты-
ваться, протягиваться, прессоваться и т. д.).
Электролиз расплава смеси K2[ZrFe] с КС1 или NaCl
Электролизом смеси 30 вес. % K2[ZrFe] и 70 вес.% КС1 при
750—800° в атмосфере аргона получают на катоде порошкообраз-
ный (или губчатый) металлический цирконий. На аноде выделяется
хлор, в электролите накапливается HF.
Электролиз расплава осуществляется в графитовом тигле, кото-
рый служит анодом. Катодом является металлический молибден.
Соединение K2[ZrFe] получают действием КС1 на раствор ZrO?
во фтористоводородной кислоте:
ZrO2 + 6HF + 2КС1 = K2[ZrFe] + 2НС1 + 2Н2О
Используемые далее соединения K2[ZrFe] и КС1 должны быть
чистыми и высушенными в вакууме примерно при 120°, поскольку
от степени их чистоты зависит чистота металлического циркония.
Для улавливания кислорода и азота аргон пропускают над
нагретым до 800—850° металлическим титаном или над нагретыми
до 400° брусками металлического урана, а для осушки аргон
ЦИРКОНИЙ
111
пропускают через колонки, содержащие P4Oi0. Металлический
цирконий, полученный этим способом, очищают от КС1, KF,
K2[ZrFe], промывая нагретой до 60° водой, 10%-ной НС1 и снова
водой.
Металлический цирконий можно также получить электролизом
расплава системы K2[ZrF8] — NaCl при 850° в атмосфере аргона.
Этот процесс в настоящее время используется в промышленности.
ФИЗИЧЕСКИЕ И ХИМИЧЕСКИЕ СВОЙСТВА
Цирконий — блестящий, серебристо-белый металл с плотней-
шей гексагональной (модификация a-Zr) и кубической объемно-
центрированной (модификация p-Zr) кристаллическими структу-
рами. Цирконий имеет плотность 6,25 г! см9 при 20° и твердость
4,5 по шкале Мооса; он тугоплавок (т. пл. 1855°, т. кип. 4474°),
диамагнитен, пластичен (ковкий, вязкий, тягучий); обладает высо-
кой механической прочностью. Значение отношения механической
прочности и плотности у металлического циркония меньше, чем
соответствующие характеристики у специальных сталей и сплавов
алюминия с магнием.
Для металлического циркония характерно большое удельное
электросопротивление, малая теплопроводность и низкий коэффи-
циент расширения.
Известны многочисленные сплавы металлического циркония
с Mg, Al, Sn, РЬ, Sb, Bi, Cu, Ag, Au, Cd, Ti, Hf, Th, V, Nb, Cr,
Mo, W, Mn, Fe, Ni, C, Si, P, N, В и др.
Амальгамы циркония неизвестны. К интерметаллическим соеди-
нениям циркония относятся: ZrCu3, ZrCu2, ZrMo3, ZrW2, Zr2Fe3,
ZrNi3, ZrNi4 и др.
Добавление небольших количеств Sn, Nb, Cr, Fe, Ni, Mo, Cu
к металлическому цирконию приводит к повышению его корро-
зионной устойчивости, а добавка алюминия или титана умень-
шает ее.
С химической точки зрения компактный металлический цирко-
ний обладает пониженной активностью (напоминает титан) и про-
являет высокую коррозионную устойчивость при температуре
ниже 200° (занимая второе место после тантала).
При нагревании порошкообразный металлический цирконий
взаимодействует с кислородом (600—800°) и галогенами (300—500°),
образуя соответствующие соединения четырехвалентного цирко-
ния. При 1100—1800° металлический цирконий вступает в реак-
цию с азотом или углеродом, образуя соответственно ZrN и ZrC.
Нагревая в вакууме металлический цирконий в парах серы, полу-
чают ZrS2.
При 1000° модификация P-Zr активно поглощает водород
с образованием гидрида ZrH2. По своей способности растворять
112
ПОБОЧНАЯ ПОДГРУППА IV ГРУППЫ
большие количества водорода при нагревании цирконий напоми-
нает палладий.
Металлический цирконий не взаимодействует с водой, хлорной
водой, разбавленными кислотами НС1, HNO3, H2SO4, концентри-
рованными (до 65%-ной концентрации) НС1, HNO3, разбавлен-
ными или концентрированными щелочами, растворами хлорида
натрия, хлорида кальция иди других хлоридов. Растворяется
только в расплавах гидроокисей щелочных металлов, царской
водке и концентрированной фтористоводородной кислоте:
Zr + 4NaOH = Na4ZrO4 + 2Н2
3Zr -J- 12НС1 + 4HNO3 = 3ZrCl4 + 4NO + 8H2O
Zr + 4HF = ZrF4 + 2H2
Химические свойства циркония иллюстрируются следующей
схемой:
Комн. темп.
Нагревание
с конц. HF —> ZrF4
с царской водкой —> ZrCl4
Цирконий —
в кислороде (или на воздухе) —> ZrO2
с галогенами —>• тетрагалогениды
с S, N2, С —> ZrS2, ZrN, ZrC
с распл. NaOH —* цирконаты
ПРИМЕНЕНИЕ
Ядерно чистый металлический цирконий используют в каче-
стве конструкционного материала для термоядерных реакторов,
поскольку он обладает низкой теплопроводностью, большой меха-
нической прочностью, высокой стойкостью к химической кор-
розии, очень низким сечением захвата тепловых нейтронов,
т. е. не задерживает нейтроны, высвобождающиеся в результате
цепной ядерной реакции при расщеплении топлива в реакторе.
С точки зрения потребности в металлах для ядерной техники цир-
коний превосходит алюминий, магний и бериллий в качестве кон-
струкционного материала для ядерных реакторов. Цирконий,
применяемый для этих целей, не должен содержать примесей
гафния, поскольку последний обладает большим эффективным
сечением захвата нейтронов.
Благодаря высокой коррозионной устойчивости металлический
цирконий применяется (вместо тантала и платины) для изготов-
ления аппаратов, резервуаров, конденсаторов, испарителей, тру-
бопроводов, насосов, кислотостойких клапанов и других изделий,
используемых в химической промышленности. В костно-черепной
хирургии пользуются скобками, шурупами и пластинками из цир-
кония. Циркониевая фольга толщиной 0,03 мм применяется в каче-
ЦИРКОНИЙ
ИЗ
стве радиационного фильтра в рентгеновских трубках с молибде-
новым антикатодом. Из циркония можно изготовлять свечи для
двигателей внутреннего сгорания, термоэлементы, пирометры и др.
Металлический цирконий используют в качестве адсорбента
в электронных трубках и высоковакуумной технике, поскольку
он обладает способностью адсорбировать большие количества раз-
личных газов. Смесь циркониевого порошка с двуокисью свинца
служит для покрытия нити накаливания в электронных лампах-
вспышках (блиц-лампы).
Из листков циркония изготовляют фильеры, используемые
в промышленности искусственных волокон. Порошок металли-
ческого циркония входит в состав патронного капсуля (он сокра-
щает время между ударом бойка и вылетом пули) и в состав люми-
несцентных пиротехнических смесей, применяемых при изготов-
лении осветительных ракет, трассирующих снарядов и др.
Металлический цирконий и ферроцирконий или ферросили-
коцирконий, добавленные в специальные стали, способствуют
увеличению пластичности, износоустойчивости и химической стой-
кости этих сплавов. В сталеплавильном деле цирконий служит
раскислителем и дегазантом, поскольку он проявляет большое
сродство к кислороду, азоту, сере, фосфору, углероду и др. Стали
с содержанием Zr 0,1% идут па изготовление броневых плит
и листов. Из нержавеющих сталей с цирконием изготовляют кор-
пуса газовых турбин.
В сплавы меди (латуни, бронзы) и никеля цирконий вводится
и как легирующая добавка, и как дегазапти антиоксидант. Добавка
циркония к этим сплавам обеспечивает увеличение твердости,
коррозионной стойкости, электро- и теплопроводности.
Сплавы медь — цирконий служат для изготовления электри-
ческих контактов, сегментов коммутаторов, коллекторных колец,
наконечников паяльников, поддерживающих крючков в электрон-
ных трубках и др.
Сплавы цирконий — алюминий — магний применяются в стро-
ительстве реактивных самолетов, космических ракет и в корабле-
строении.
Интерметаллическое соединение ZrPb используется в качестве
камней для зажигалок и для других пирофорных целей.
Соединения ZrO2, ZrSiO4,‘ ZrC, ZrB относятся к термостойким
материалам и широко применяются в современной технике.
СОЕДИНЕНИЯ (ОБЩИЕ СВОЙСТВА)
Известны многочисленные простые или координационные соеди-
нения циркония, в которых он выступает как четырехвалентный
элемент, и ограниченное число соединений (низкой устойчивости),
в которых он двух- И трехвалентен. Склонность к образованию коор-
8—0101
114
ПОБОЧНАЯ ПОДГРУППА IV ГРУППЫ
динационных соединений у циркония выражена сильнее, чем
у гафния, поскольку ионы циркония обладают большим поляри-
зующим действием.
В табл. 22 приведены формулы и указан цвет многих соедине-
ний циркония.
Соединения, двух- и трехвалентного циркония
Известно ограниченное число соединений (особенно галогени-
дов) двух- и трехвалентного циркония. Для них характерен черный
цвет, восстановительный характер и более низкая устойчивость
по сравнению с подобными соединениями титана.
Дифторид циркония, ZrF2,— твердое вещество черного цвета,
диспропорционирующее при нагревании (1402°) в ZrF4 и металли-
ческий цирконий.
Дихлорид циркония, ZrCl2, получается в результате диспро-
порционирования трихлорида циркония при 330° в вакууме или
при действии паров тетрахлорида циркония на нагретый до 1500°
металлический цирконий:
2ZrCl3 ZrCl2 + ZrCl4 Zr + ZrCl4 = 2ZrCl2
Дихлорид циркония представляет собой блестящее аморфное
вещество черного цвета с плотностью 3,6 г/см9', оно плохо раство-
ряется в спирте, эфире, бензоле, растворяется при нагревании
в концентрированных кислотах с выделением водорода. ZrCl2
не устойчив, разлагается при 727° на ZrCl4 и металлический цирко-
ний, разлагает воду с выделением водорода.
Дибромид циркония, ZrBr2, получают действием тетрабромида
циркония на металлический цирконий при 1500°, а также терми-
ческой диссоциацией трибромида циркония при 350°:
ZrBr4 Zr 2ZrBr2 2ZrBr3 =₽fc ZrBr2 + ZrBr4
Дибромид циркония — неустойчивое вещество черного цвета;,
он разлагает воду с выделением водорода и диспропорционирует
при нагревании (627°):
2ZrBr2 ZrBr4 Zr
Дииодид циркония, Zrl2,— неустойчивое вещество черного цвета,
разлагающееся при 427° на Zrl4 и металлический цирконий.
Трифторид циркония, ZrF3,— твердое вещество черного цвета,
разлагающееся при 1327° на ZrF4 и металлический цирконий.
Трихлорид циркония, ZrCl3, получают нагреванием тетрахло-
рида циркония с металлическим алюминием в запаянной трубке-
примерно при 350°:
3ZrCl4 -|- Al = 3ZrCl3 -|- А1С13
ЦИРКОНИЙ
115
Трихлорид циркония представляет собой черное кристалли-
ческое вещество с плотностью 3,0 г!смА, разлагающееся при 330°
в вакууме, разлагающее воду с выделением водорода и являющееся
восстановителем по отношению к HgCl2, РЬС12, BiCl3 и др.
Трибромид циркония, ZrBr3, получают восстановлением тетра-
бромида циркония водородом при 450°, металлическим цирконием
при 500° или металлическим алюминием примерно при 350°.
Трибромид циркония образуется в виде черного порошка,
он растворяется в воде с выделением водорода, распадается на
ZrBr4 и ZrBr2 при 350° и на ZrBr4 и металлический Zr при 827°,
восстанавливает хроматы до соединений трехвалентного хрома,
соли трехвалентного железа до двухвалентного, соединения двух-
валентной меди до одновалентной, соли четырехвалентного титана
до трехвалентного.
Трииодид циркония, Zrl3, представляет собой черный порошок,
который диспропорционирует при 927° на Zrl4 и металлический
цирконий.
Соединения четырехвалентного циркония
Известно много устойчивых соединений (простых или коор-
динационных) четырехвалентного циркония, в которых он входит
в состав катионов Zr4+, ZrO2+, Zr2O| + или анионов ZrO|_, ZrOt-,
[ZrFeJ2-, [ZrCl6l2-, [ZrBr6]2-, [Zr(SO4)3p-, [Zr(SO4)J4-, [Zr2O3(SO4)2]2-
и др.
При гидролизе солей четырехвалентного циркония (в зависи-
мости от концентрации ионов водорода в растворе) образуются
соли цирконила ZrO2+ или дицирконила Zr2O2+ и в конечном итоге
гидратированная двуокись циркония ZrO2 — соответственно
H4ZrO4 на холоду и H2ZrO3 при нагревании.
Большинство соединений четырехвалентного циркония — белые
порошки или бесцветные кристаллы.
Неорганические соединения
Двуокись циркония, ZrO2, встречается в природе как минерал
бадделеит, но может быть получена также нагреванием металли-
ческого циркония в кислороде или прокаливанием на воздухе
соединений H4ZrO4, H2ZrO3, Zr(NO3)4-5Н2О, ZrO(NO3)2-2H2O,
Zr(SO4)2, ZrN, ZrC и др.:
Zr + O2 = ZrO2 + 261,5 ккал H4ZrO4 ZrO2 + 2H2O
H2ZrO3 —> ZrO2 -|- H2O Zr(SO4)2 —> ZrO2 -|- 2SO3
Zr(NO3)4.5H2O —ZrO2 -f- 4NO2 -j- O2 -{- 5H2O
ZrO(NO3)2.2H2O ZrO2 + 2NO2 + V2O2 + 2H2O
Двуокись циркония существует в виде белого порошка, бес-
цветных моноклинных кристаллов с плотностью 5,8 г/см9, а также
8*
Соединение Окис- ли Гидро- окиси и кислоты Фториды и фторосоеди- нения Хлориды и хлоро- соединепия Бромиды и бромосоеди- нения
Соединения циркония (И) — — ZrF2 черный ZrCl2 черный ZrBr2 черный
Соединения циркония (III) — — ZrF3 черный ZrClo черный ZrBr3 ! черный I I
Соединения циркония (IV) ZrO2 бес- цвет- ная H4ZrO4 кислота или Zr(OH)4 студне- образная белая гидро- окись H2ZrO3 кислота или ZrO(OH)2 студне- образная белая гидро- окись H2ZrO5 белая кислота ZtF4 бесцветный ZrOF2-2H2O белый ZrF4-3H2O или H2[ZrOF4]-2H2O H2[ZrFe].3H2O Me^ZrFjbHzO Me2[ZrF8]-nH2O Me*[ZrF7] Ме^[ггР8]-(6или12)Н2О ZrCl4 белый ZrOCl2- •8Н2О белый Zr2O3Cl2 белый Me|[ZrCl6] ZrBr4 белый ZrOBr2-8H2O бесцветный (C5H5NH)2[ZrBre] в!
Таблица 22
Йодиды и иодосое- динения Суль- фиды Сульфаты и суль- фатосоединения Хелатные соединения циркония(1У) Сое- дине- ния вклю- чения
Zi 12 черный — — ’Zr/S О \ ' 1 । \ 1 zч/Ч 1 h । J L \ \/\z/4j желтый ZrHi,9 ZrN жел- тый
Zrl3 черный — — _ / O=N \ “ Zr(\ yN-CeHsj _ \ Q Л J желтый ZrB серый
Zrl4 желтый ZrOI2-8H2O желтый H2[ZrI6] ZrS2 серо- фио- лето- вый ZrOS жел- тый Zr(SO4)2-4H2O или H2[ZrO(SO4)2]-3H2O бесцветный Zr(SO4)2 белый H2[Zr(SO4)3].3H2O Me2[Zr(SO4)3] • nH2O MeJ[Zr(SO4)4] • пН2О MeJ[Zr4(OH)8(SO4)e]. 4Н2О K2[Zr2O3(SO4)2] K2[ZrO2(SO4)2].3H2O пероксосоединение Z Zr ! OH SO3Na 4 "I / 1 1 \ 1 /°~\ / 1 г I C / 1 1 =\ /= 0 1 ' C Z 4 красный R ? . \ °2 N|/TS0,H| kA/ I ' no2 ' желтый ZrC чер- ный
118
ПОБОЧНАЯ ПОДГРУППА IV ГРУППЫ
бесцветных тетрагональных кристаллов с плотностью 6,1 г!сл?.
Она тугоплавка и устойчива при повышенной температуре
(т. пл. 2677°, т. кип. 4300°), диамагнитна, плохо растворима
в воде, устойчива к действию различных химических реагентов,
обладает более основным характером, чем TiO2, и слабо выражен-
ными кислотными свойствами, разрушается под действием фтори-
стоводородной кислоты и сплавлением с гидроокисями или кар-
бонатами щелочных металлов:
ZrO2 + 4HF = ZrF4 + 2Н2О ZrO2 + 2NaOH = Na2ZrO3 + H2O
ZrO2 -|- Na2COg — Na2ZrO3 -{- CO2
Двуокись циркония с окислами и хлоридами щелочных метал-
лов образует метацирконаты общей формулы Me2+ZrO3.
При нагревании двуокись циркония восстанавливается каль-
цием, магнием, сплавами кальций — натрий, магний — натрий
и электролитически в расплаве с СаС12.
Двуокись циркония — превосходный огнеупорный материал
(обладает низкой теплопроводностью и малым коэффициентом рас-
ширения). Используется для изготовления тиглей, огнеупорных
и кислотостойких кирпичей, фарфора и стекла (из которых изго-
товляются электрические изоляторы повышенной термической
стойкости), высококачественных эмалей и глазурей и очень устой-
чивых красок. Стекла и эмали, содержащие ZrO2, устойчивы
по отношению к кислотам или щелочам, но не устойчивы к рез-
кому повышению температуры, поскольку при нагревании свыше
1000° моноклинная модификация переходит в тетрагональную.
Высококачественные огнеупорные материалы получают
из смеси ZrO2, ZrSiO4, MgO.
Из ZrO2 изготовляют нити накаливания, которые применяются
в проекционных лампах. Водная суспензия двуокиси циркония
может служить в рентгеноскопии как контрастное вещество.
а-Циркониевая (ортоциркониевая) кислота, H4ZrO4, или гид-
роокись циркония(1У), Zr(OH)4, получается обработкой растворов
солей четырехвалентного циркония щелочами, аммиаком, суль-
фидом аммония и др. (на холоду при pH = 2):
ZrCl4 + 4NaOH = H4ZrO4 + 4NaCl
ZrCl4 + 2(NH4)2S + 4H2O = H4ZrO4 + 4NH4C1 + 2H2S
а-Циркониевая кислота представляет собой гелевидный белый
осадок, легко образующий коллоидов растворы. Обладает амфо-
терным характером, растворяется в разбавленных кислотах и с али-
заринсульфонатом натрия
.СО.
C8HZ )CeH(OH)2SO3Na
ЦИРКОНИИ
119
дает красный осадок:
Колориметрическое определение циркония основывается
на взаимодействии четырехвалентного циркония с ализарином
.СО.
С6Н4< >С8Н2(ОН)2
хсси
при pH = 1—2, при котором образуется красный лак.
При сплавлении двуокиси циркония с избытком щелочи полу-
чаются ортоцирконаты щелочных металлов:
ZrO2 + 4MeIOH = MeJZrO4 + 2Н2О
^-Циркониевая (метациркониевая) кислота, H2ZrO3, или гид-
роокись цирконила, ZrO(OH)2, получается обработкой при нагре-
вании растворов солей четырехвалентного циркония с щелочами
или при гидролитическом осаждении (например, раствором
Na2S2O3 при нагревании), а также при продолжительном кипя-
чении ортоциркониевой кислоты.
p-Циркониевая кислота представляет собой студнеобразный
белый осадок, плохо растворимый в воде и разбавленных кислотах
и растворимый в спирте; по своим свойствам напоминает мета-
оловянную кислоту.
Сплавлением двуокиси циркония со стехиометрически необ-
ходимым количеством гидроокиси щелочного или щелочноземель-
ного металла получают метацирконаты:
ZrO2 + 2MeJOH = Me2ZrO3-|- Н2О ZrO2 + Меп(ОН)2 = MenZrO3 + Н2О
Метацирконаты CaZrO3 (т. пл. 2550°) и MgZrO3 применяются
в качестве огнеупорных материалов.
Пероксоциркониевые соединения. Обра-
боткой слабокислых растворов соединений четырехвалентного цир-
кония 30%-ной Н2Ог (в течение продолжительного времени) полу-
чают монопероксоциркониевую кислоту H4ZrO5 в виде объемистого
белого осадка.
Известен пероксодисулъфатоцирконатЦУ) калия
го.
| >Zr(SO4\2
О7
ЗН2О
К2
120
ПОБОЧНАЯ ПОДГРУППА IV ГРУППЫ
и тетрапероксоцирконатуУ) калия K4ZrO8 -6Н2О, который можно
рассматривать как аддукт K4ZrO4 -4Н2О2 ^ЩО.
Тетрафторид циркония, ZrF4, получают действием при нагре-
вании фтора или фтористого водорода на порошкообразный метал-
лический цирконий, хлористого водорода на смесь металлического
циркония и фторида кальция, а также действием плавиковой кис-
лоты на ZrCl4 или нагреванием гексафтороцирконата аммония:
Zr + 2F2 = ZrF4 + 456,8 ккал
Zr + 4HF = ZrF4 + 2H2
Zr + 2CaF2 + 4HC1 = ZrF4 + 2CaCl2 + 2H2
(NH4)2[ZrF6] = ZrF4 + 2NH3 + 2HF
Тетрафторид циркония образует бесцветные гексагональные
кристаллы с плотностью 4,43 г/см3 и т. пл. 912°; он растворим
в плавиковой кислоте и фторидах щелочных металлов, менее
растворим в воде.
При концентрировании раствора, полученного обработкой гид-
ратированной двуокиси циркония ZrO2 -пН2О плавиковой кисло-
той, выделяются кристаллы ZrF4-3H2O или H2[ZrOFJ -2Н2О —
тетрафтороксоциркониевой кислоты.
Тетрафторид циркония образует фторосоли с многочислен-
ными фторидами. Общие формулы фторосолей: Me+[ZrF5] • Н2О,
Me2[ZrF8] -иНгО, Me3[ZrF7], Me2+[ZrF8] -6 или 12Н2О, где Ме+ = К+,
Rb+, Cs+, NH*, Т1+ и Ме2+ = Cu2+, Zn2+, Cd2+, Ni2+, Мп2+ и др.
Самый устойчивый анион фторосоединений циркония [ZrFe]2~;
он встречается как в кислоте H2[ZrF8] -ЗН2О, так и в ее солях.
K2ZrFe представляет собой бесцветные ромбические призмы с плот-
ностью 3,48 г/см3 и ограниченной растворимостью в воде.
Дифторид цирконила, ZrOF2-2H2O, при 200° теряет кристал-
лизационную воду.
Тетрахлорид циркония, ZrCl4, получают действием при нагре-
вании газообразного хлора на порошкообразный металлический
цирконий, карбид ZrC, нитрид ZrN или на смесь двуокиси цирко-
ния с углем, а также обработкой двуокиси циркония четырех-
хлористым углеродом при нагревании:
Zr + 2С12 = ZrCl4 + 234,7 ккал ZrO2 + 2С + 2С12 = ZrCl4 + 2СО
ZrC + 2С12 = ZrCl4 + С ZrO2 + СС14 = ZrCl4 + СО2
Тетрахлорид циркония имеет вид блестящих белых кристал-
лов с плотностью 2,80 г/см3', он сублимируется при 331°, раство-
ряется в спирте, эфире, концентрированной соляной кислоте,
хлоридах щелочных металлов и др., гидролизуется водой, образует
аддукты с аммиаком, многими органическими аминами, оксихло-
ридом фосфора, ацетоном и др.
ZrCl4 + 2КС1 = K2[ZrCle] ZrCl4 + 8NH3 = ZrCl4 8NH3
ZrCl4 + H2O ZrOCl2 + 2HC1 3ZrCl4 + 2POC13 = 3ZrCl4-2POC13
ЦИРКОНИЙ
121
Примерами аддуктов тетрахлорида циркония могут служить:
ZrCl4.nNH3 (где п = 8, 4, 2), ZrCl4-4CH3NH2, ZrCl4.4C2H5NH2,
ZrCl4 -3C3H7NH2, ZrCl4-4C8H5NH2, ZrCl4 -4С5Н5М, ZrCl4-2C10H7N,
3ZrCl4-2POC13, ZrCl4-CH3 - CO - CH3 и др.
Под действием безводной плавиковой кислоты ZrCl4 превра-
щается в ZrF4.
Известны гексахлороцирконаты натрия и калия.
Тетрахлорид циркония служит для получения металлического
циркония.
Дихлорид цирконила, ZrOCl2 -8Н2О, полученный гидролизом
тетрахлорида циркония, представляет собой выветривающиеся
белые тетраэдрические иглы; он растворим в холодной воде,
спирте, эфире, при 150° теряет 6 молекул Н2О, а при 210° превра-
щается в безводное соединение ZrOCl2.
Из смеси спиртового раствора ZrOCl2-8H2O с антраниловой
кислотой H2N — С8Н4 — СООН эфиром экстрагируется серовато-
розовый осадок состава H[ZrOCl2 — СОО]-5Н2О. При
обработке спиртового раствора ZrOCl2-8H2O эфирным раствором
бензидина H2N — С8Н4 — С6Н4 — NH2 отделяется бело-кремовый
осадок состава ZrOCl2 -2NH2 — С8Н4 — С8Н4 — NH2. При добав-
лении эфира к смеси спиртового раствора ZrOCl2 с диметилглиок-
симом HON = С(СН3) — С(СН3) = NOH выпадает желтый осадок,
в котором ZrOCl2 и диметилглиоксим находятся в соотношении
1 : 1. В спиртовом растворе ZrOCl2-8H2O взаимодействует с моче-
виной H2N — СО — NH2, образуя соединение ZrOCl2 •2CO(NH2)2,
которое осаждается при добавлении ацетона, эфира или хлоро-
форма.
Дихлорид бицирконила, Zr2O3Cl2, получают более глубоким
гидролизом тетрахлорида циркония или дихлорида цирконила:
2ZrCl4 + ЗН2О Zr2O3Cl2 + 6НС1 2ZrOCl2 + Н2О Zr2O3Cl2 + 2НС1
Дихлорид бицирконила представляет собой плохо раствори-
мое в воде белое твердое вещество.
Гидролизом тетрахлорида циркония можно получить и
Zr4(OH)8Cl8- 16Н2О:
4ZrCl4 + 24Н2О Zr4(OH)8Cl8-16H2O + 8НС1
Тетрабромид циркония, ZrBr4, получают обработкой при нагре-
вании порошкообразного металлического циркония, карбида цир-
кония или смеси двуокиси циркония с сахарным углем парами
брома:
Zr + 2Вг2 = ZrBr4 181,56 ккал ZrC + 2Br2 = ZrBr4 С
ZrO2 + 2С + 2Вг2 = ZrBr4 + 2СО
Тетрабромид циркония представляет собой белый (желтеющий
при нагревании) микрокристаллический порошок или бесцветные
кристаллы, сублимирующиеся при 357°, улетучивающиеся без
122
ПОБОЧНА!! ПОДГРУППА IV ГРУППЫ
разложения при 400°, растворимые в спирте или эфире, очень
гигроскопичные и гидролитически разлагающиеся на влажном
воздухе или в воде:
ZrBr4 + Н2О ZrOBr2 + 2НВг
Известна пиридиновая соль кислоты H2[ZrBr6] — очень
неустойчивое соединение с формулой (G5H5N)2[ZrBr6].
Среди аддуктов тетрабромида циркония известны ZrBr4 -SNHa,
ZrBr4-4G2H5NH2, ZrBr4 -4G6H5NH2, ZrBr4 .2G5H5N.
Дибромид цирконила, ZrOBr2 -8H2O, выделяется в виде расплы-
вающихся на воздухе бесцветных игл, растворимых в холодной
воде и в горячей конц. НВт.
Известен и дибромид дицирконила Zr2O3Br2.
Тетраиодид циркония, Zrl4, получают обработкой металличе-
ского циркония парами иода при 400—500°, действием паров иода
на нагретый карбид циркония, а также пропусканием йодистого
водорода над металлическим цирконием при 340° или над карбидом
циркония при 490°:
Zr + 2I2 = Zrl4 + 160 ккал Zr -f- 4HI = Zrl4 + 2H2
ZrC + 2I2 = Zrl4 + C ZrC + 4HI = Zrl4 -f- C + 2H2
Тетраиодид циркония образует желтые октаэдрические кри-
сталлы, сублимирующиеся при 431°, растворимые в эфире и бурно
разлагающиеся в воде или спирте:
Zrl4 + Н2О ZrOI2 + 2HI
Zrl4 + 2С2Н5ОН = ZrOI2 + 2C2H5I + Н2О
Известна также кислота H2[ZrI6] и аддукты Zrl4-8NH3, Zrl4-
•6 — 7NH3, ZrI4-4NH3, Zrl4-6G2H5NH2.
Тетраиодид циркония служит для получения чистого металли-
ческого циркония и приготовления нитрида циркония ZrN.
Дииодид цирконила, ZrOI2-8H2O, представляет собой гигроско-
пичные бесцветные иглы, превращающиеся в Zrl4 и ZrO2 при
нагревании, растворимые в воде, спирте и эфире.
Дисульфид циркония, ZrS2, образуется при нагревании исход-
ных элементов, а также взаимодействием двуокиси циркония
ZrO2 с парами CS2 или тетрахлорида циркония с сероводородом:
Zr + 2S = ZrS2 ZrO2 + CS2 = ZrS2 + CO2
ZrCl4 + 2H2S = ZrS2 + 4HC1
Дисульфид циркония образует серовато-фиолетовые гексаго-
нальные кристаллы с плотностью 3,87 г/см?, тугоплавкие и устой-
чивые на воздухе, в воде, HG1, разб. H2SO4. При прокаливании
дисульфида циркония на воздухе образуется ZrO2 и выделяется
SO2:
ZrS2 -}- ЗО2 = ZrO2 -}- 2SO2
ЦИРКОНИЙ
123
Сульфид цирконила (или оксисульфид циркония), ZrOS, пред-
ставляет собой желтый порошок с плотностью 4,87 г/см9, воспла-
меняющийся при нагревании.
Дисулъфат циркония, Zr(SO4)2 -4Н2О или H2[ZrO(SO4)2]-ЗН2О,
получают действием конц. H2SO4 на двуокись циркония или дихло-
рид цирконила:
ZrO2 + 2H2SO4 + 2Н2О = Zr(SO4)2-4Н2О = H2[ZrO(SO4)2]-3H2O
ZrOCl2 + 2H2SO4 + 3H2O = Zr(SO4)2-4H2O + 2HC1
Взаимодействие двуокиси циркония с трехокисыо серы при
400° приводит к образованию безводной соли Zr(SO4)2:
ZrO2 -{- 2S0g — Zr(SO4)2
Дисульфат циркония, рассматриваемый также как дисульфат-
оксоциркониевая кислота H2[ZrO(SO4)2] -ЗН2О, представляет собой
бесцветные ромбические кристаллы, трудно растворимые в спирте
и растворимые в воде, подкисленной H2SO4, поскольку при pH > 2
выпадают соли цирконила. При обработке дисульфата циркония
избытком H2SO4 получается трисульфатциркониевая кислота
H2[Zr(SO4)3] -ЗН2О, для которой известны соли аммония
(NH4)2[Zr(SO4)3] -ЗН2О и натрия Na2[Zr(SO4)3] -4Н2О.
Дисульфат циркония взаимодействует при нагревании с раство-
рами сульфатов щелочных металлов с образованием сульфатгидрок-
социрконатов общей формулы Me4[Zr4(OH)8(SO4)e] -4Н2О. При обра-
ботке дисульфата циркония растворами сульфатов щелочных
металлов в присутствии серной кислоты получают сульфатцир-
конаты общей формулы Me2[Zr(SO4)3], Me4[Zr(SO4)4], например
K4[Zr(SO4)4]-4Н2О, Na4[Zr(SO4)4]-тгН2О, где п = 3, 4, 11.
Известен дицирконилдисульфат калия K2[Zr2O3(SO4)2].
Тетранитрат циркония, Zr(NO3)4 -5Н2О, выделяется концент-
рированием в вакууме при 15° (в присутствии Р40ю и КОН)
раствора H4ZrO4 или H2ZrO3 в конц. HNO3.
Тетранитрат циркония представляет собой очень гигроскопич-
ные бесцветные кристаллы, растворяющиеся в воде с образованием
тетранитратоксоциркониевой кислоты H2[ZrO(NO3)4] -4Н2О, для
которой известна соль калия K2[ZrO(NO3)4] -4Н2О — бесцветные
кристаллы.
Концентрированием раствора кислот H4ZrO4, H2ZrO3 в разб.
HNO3 получают бесцветные кристаллы динитратацирконила
ZrO(NO3)2-2Н2О, растворимые в абсолютном спирте.
Если к спиртовому раствору динитрата цирконила добавить
эфир, выпадают бесцветные кристаллы динитрата дицирконила
Zr2O3(NO3)2-5H2O.
Ацетат циркония, Zr(CH3COO)4. При действии ледяной уксус-
ной кислоты на тетрахлорид циркония получают тетраацетат
циркония Zr(CH3COO)4, который гидролизуется в теплой воде,
124
ПОБОЧНАЯ ПОДГРУППА IV ГРУППЫ
образуя либо гидроксоацетат циркония Zr(GH3GOO)3(OH), либо
диацетат цирконила ZrO(GH3GOO)2:
ZrCl4 + 4СН3СООН = Zr(CH3COO)4 + 4НС1
Zr(CH3COO)4 + H2O Zr(CH3COO)3OH + СНзСООН
Zr(CH3COO)4 + Н2О ZrO(CH3COO)2 + 2СН3СООН
Ортосиликат циркония, ZrSiO4, встречается в природе в виде
различных разновидностей минерала циркона (см. раздел о рас-
пространенности циркония в природе).
Ортосиликат циркония представляет собой бесцветные кри-
сталлы с плотностью 4,56 г!см\ т. пл. 2550°, твердостью 7,5
по шкале Мооса, плохо растворимые в воде. В кристаллической
решетке циркона тетраэдры [SiOJ4- изолированы, т. е. ионы кис-
лорода, окружающие ион кремния, не принадлежат другим сосед-
ним тетраэдрам.
Известны многочисленные двойные силикаты, например
Na4SiO4 -2ZrSiO4, Na2SiO3-ZrSiO4.
Хелатные соединения
Ион Zr4+ в уксуснокислых (буферная смесь GH3GOOH
с NH4GH3GOO) или нейтральных растворах образует с 8-оксихи-
нолином флюоресцирующий желтый осадок
Флавоновая (1-окси-2,4-динитро-7-нафталинсульфоновая) кис-
лота в сильнокислой среде (7,5 н. HG1) образует с ионом Zr4+
желтый осадок
В сильнокислых растворах (4 н. Н2ЗО4или 6 н. HG1) купферон
G6H5N(NO)ONH4 с ионом Zr4+ образует хелатное соединение
ЦИРКОНИЙ
125
Известны также тетраацетилацетонат и тетрабензоилацетонат
циркония, соединение четырехвалентного циркония с ализарином
(лак), хелаты с эриохромцианином R, с пирокатехином фиолето-
вым, с ксиленолом оранжевым I, с ауромицином и др.
Тетраацетилацетонату и тетрабензоилацетонату циркония отве-
чают следующие формулы:
О = С
О —С
/СН3
Помимо приведенных известны и другие соединения четырехвалентного
циркония,например:метацирконаты Me|ZrO3, MenZrO3, где Мет= Li+, Na+,
К+, a Men = Ве2+, Mn2+, Са2+, Zn2+, хлорат ZrO(ClO3)2-6H2O, перхлорат
4Zr0(C104)2-HC104, иодат Zr(IO3)4, гексаиодатоцирконаты Me2(Zr(IO3)6],
где Me1 = Na+, К+, Rb+ и др., селенид ZrSe2, селенит Zr(SeO3)2, селенат
Zr(SeO4)2 -4Н2О, сульфит Zr(SO3)2-7Н2О, сульфатоцирконаты, амид Zr(NH2)4,
имид Zr(NH)2, гипофосфит Zr(H2PO2)4, ортофосфаты Zr3(PO4)4, Zr(HPO4)2,
Zr(H2PO4)4, ZrO(H2PO4)2, дифосфат ZrP2O7, арсенаты Zr3(AsO4)4, ZrOHAsO4,
силикоцирконаты, хроматы, молибдаты, вольфраматы, ацетаты Zr(CH3COO)4,
ZrO(CH3COO)2, формиат Zr(HCOO)4, бензоат Zr(C6H5COO)4, оксалаты
Zr(C2O4)2, ZrO(C2O4)-4Н2О, Zr(C2O4)2-2H4ZrO4, оксалатоцирконаты
Me, [Zr(C2O4)4]-nH2O, где Me1 = Na+, K+, Rb+, Cs+, NHJ, кислота
Н4ргО(С2О4)3]-7H2O, цитраты K[ZrO(C6H4O7)]-2,5H2O, K3C6H5O7-9,5H2O,
манДелаты Zr(CgH7O3)4, ZrO(C3H7O3)2, Zr(OH)(CgH7O3)3, Zr(OH)3(CgH7O3),
металлоорганические соединения (C5H5)2ZrCl2, (C5H5)2ZrBr2 и др.
Соединения включения
Известны гидрид ZrH2, нитрид ZrN, карбид ZrC и борид
ZrB циркония.
Нитрид циркония, ZrN, получают нагреванием в вакууме или
без доступа кислорода порошкообразного металлического цирко-
ния, карбида циркония, тетраиодида циркония или смеси дву-
окиси циркония с углем (сажей) в атмосфере азота при 1100—1200°:
Zr + V2N2 = ZrN + 82,2 ккал Zrl4 + l/2N2 = ZrN + 2I2
ZrC + V2N2 = ZrN + C ZrO2 + 2C + V2N2 = ZrN + 2CO
Нитрид циркония образуется в виде желтых очень хрупких
кристаллов с твердостью 8 по шкале Мооса и т. пл. 2985°; он устой-
чив при высокой температуре, обладает низкой химической актив-
ностью, легко хлорируется.
Карбид циркония, ZrC, получают нагреванием порошкообраз-
ного металлического циркония с углем в электродуговой печи
или восстановлением двуокиси циркония сажей при 1900—2100°
или при более низкой температуре в вакууме:
Zr -j- С (графит) = ZrC -f- 45 ккал
126
ПОБОЧНАЯ ПОДГРУППА IV ГРУППЫ
Карбид циркония образует блестящие серовато-черные метал-
лоподобные твердые (твердость 8—9 по шкале Мооса) кристаллы
с т. пл. 3532° и т. кип. 5100°. Он плохо растворим в воде, раство-
рим в HF, HNO3, H2SO4, царской водке и в расплавленных щело-
чах, устойчив к воде, легко взаимодействует при нагревании
с хлором, бромом, иодом, кислородом, азотом и др.; хороший
проводник электрического тока.
Карбид циркония служит шлифовальным материалом, замени-
телем алмаза для резки стекла, для изготовления огнеупорных
электродов и тиглей: он служит также исходным соединением для
получения галогенидов циркония.
Известен дикарбид циркония ZrC2.
Диборид циркония, ZrBr2, получают нагреванием порошкооб-
разных металлического циркония и бора без доступа воздуха.
Диборид циркония представляет собой гексагональные кри-
сталлы с плотностью 6,085 г! см? и т. пл. 2990°, хорошо проводя-
щие электричество. Известны бориды ZrB, Zr3B4.
ГАФНИЙ Hf
Z = 72; ат. вес = 178,49
Валентность (II), (III), IV; заряд (2—|-), (3+), 4+
Массовые числа природных изотопов 180, 178,
177, 179, 176 и 174
Электронная структура атома гафния: К-L -М -N-б^бр’б^-бз2.
Электронная структура атома гафния и ионов Hf2+, Hf3+, Hf4+
для 5d- и Gs-орбиталей:
StP 6s2
Itltl I I I EH
Hf
5d’ 6s 5d> 6s 5d 6s
цы i n □ in i та □ urn i □
Hf!+ Hf3+ Hf«+
ИСТОРИЧЕСКАЯ СПРАВКА
В прошлом веке считали, что цирконию в природе сопутствует
элемент, который называли «норием», а затем «жаргонием», «эвксе-
нием» и «океанием». Однако эти названия быстро исчезли, посколь-
ку существование предполагаемого элемента доказать не удалось.
Долгое время элемент с атомным номером 72 искали в минералах
редких земель.
В 1909 г. Урбан объявил об открытии в одном из минералов
редких земель элемента с атомным номером 72, который был
им назван «кельтием». Его считали трехвалентным.
ГАФНИЙ
127
На основании теории Нильса Бора о строении атомов было
показано, что ряд редкоземельных металлов должен заканчиваться
элементом с атомным номером 71 и что элемент с атомным номе-
ром 72 должен иметь свойства, отличные от редкоземельных метал-
лов. Согласно теории строения атомов, элемент с атомным номе-
ром 72 является гомологом циркония и его нужно искать среди
четырехвалентных элементов, соответственно в минералах цирко-
ния.
Хотя металлический цирконий, содержащий свыше 10% гаф-
ния, был получен Берцелиусом в 1824 г., все же металлический
гафний был выделен значительно позднее из-за очень близких
к цирконию физических и химических свойств.
В одной из лабораторий Копенгагена в 1923 г. Хевеши и Костер
на основании закона Мозли, подвергая рентгеноструктурному
анализу один из минералов циркония, открыли элемент с атом-
ным номером 72 и назвали его «гафнием» по старому латинскому
названию города Копенгагена (Hafniae).
Почти 40 лет после открытия гафния отделение циркония
от гафния было одной из наиболее важных проблем в технологии
редких элементов.
Если в металлургии некоторых металлов, электротехнике,
производстве керамики и других отраслях цирконий и гафний
или их соединения могут быть использованы без разделения,
то в ядерной технике разделение этих двух элементов абсолютно
необходимо, поскольку цирконий и гафний обладают сильно раз-
личающимися сечениями захвата нейтронов и их нельзя исполь-
зовать вместе.
РАСПРОСТРАНЕННОСТЬ В ПРИРОДЕ
Гафний более распространен, чем серебро и золото, и его
содержание в земной коре составляет 4,0-10-4 вес.%. В природе
гафний всегда встречается в виде соединений, особенно в минералах
циркония.
Содержание гафния в минералах циркония (бадделеит, циркон
и его разновидности — циртолит, малакон и др.) составляет
0,5—4 вес. % по отношению к содержанию циркония.
Тортвейтит, (Sc, Y)2Si2O7, содержит малые количества ZrO2
и НЮ2.
Присутствие гафния было установлено и в некоторых метеори-
тах.
ОБОГАЩЕНИЕ И ПЕРЕРАБОТКА ГАФНИЕВЫХ РУД
Руды циркония, содержащие гафний, обогащают комплексным
способом, включающим гравитационные, электромагнитные, элект-
ростатические и флотационные методы.
128
ПОБОЧНАЯ ПОДГРУППА IV ГРУППЫ
Концентраты силикатов циркония и гафния перерабатываются
спеканием с K2[SiFe] и KG1 при 650—700° (процесс описан в раз-
деле, посвященном переработке концентратов циркония), в резуль-
тате чего получают раствор, из которого дробной кристаллизацией
отделяют K2[ZrF6] от более растворимого K2[HfF6l.
При нагревании измельченных концентратов силикатов цирко-
ния и гафния с плавиковой кислотой и последующей обработке
KF или NH4F получают растворы, которые содержат K2[ZrF6],
K2[HfF6] или (NH4)2[ZrF6], (NH4)2[HfF6L Дробной кристаллиза-
цией сначала отделяют гексафтороцирконаты, поскольку они менее
растворимы, чем гексафторогафниаты.
Действием концентрированной H2SO4 на измельченные кон-
центраты силикатов циркония и гафния при нагревании с после-
дующей обработкой K2SO4 получают раствор, который содержит
K2[Zr(SO4)3] и K2[Hf(SO4)3]. Дробной кристаллизацией сначала
отделяют менее растворимый K2[Hf(SO4)3], затем K2[Zr(SO4)3].
Отделение гафния от циркония осуществляется методами опи-
санными при разделении циркония и гафния.
ПОЛУЧЕНИЕ МЕТАЛЛИЧЕСКОГО ГАФНИЯ
Производство губчатого металлического гафния по процессу
Кроля основано на металлотермическом восстановлении тетра-
хлорида гафния металлическим магнием или натрием в атмосфере
гелия или аргона при 850°:
HfCl4 + 2Mg = Hf + 2MgCl2 HfCl4 + 4Na = Hf + 4NaCl
Порошкообразный металлический гафний получают восстанов-
лением гексафторогафниата(1У) калия натрием при нагревании
в атмосфере гелия или аргона (процесс Берцелиуса):
K2[HfF6] + 4Na = Hf + 2KF + 4NaF
Бруски металлического гафния образуются в результате терми-
ческой диссоциации тетраиодида гафния в вакууме при 600°
(процесс Ван-Аркеля и де Бура). Тетраиодиды, образующиеся
в результате взаимодействия исходных элементов при 300°, субли-
мируются при 427° и подвергаются термической диссоциации
в интервале температур 1100—1400°.
Для получения гафния используют ту же аппаратуру, что и для
получения циркония соответствующим методом.
Металлический гафний в компактном виде получают плавле-
нием порошкообразного или губчатого металлического гафния
в электродуговых печах (или в печах с сфокусированным пучком
электронов) в вакууме, а также спеканием порошка методами
порошковой металлургии (см. раздел, посвященный титану).
ГАФНИЙ
129
ФИЗИЧЕСКИЕ И ХИМИЧЕСКИЕ СВОЙСТВА
Гафний — блестящий серебристо-белый металл с плотной гек-
сагональной или кубической объемно-центрированной структурой.
Он относится к тяжелым металлам: плотность 13,31 г/см3 при 20°.
Металл тугоплавкий, т. пл. 2222°, т. кип. 5280°. В чистом состоя-
нии металлический гафний очень пластичен, мягок (легко прока-
тывается и протягивается). Гафний хорошо проводит электриче-
ский ток, обладает высокой электронной эмиссией и в отличие
от циркония имеет большое эффективное сечение захвата тепловых
нейтронов, уступая в этом отношении только гадолинию. Эмис-
сионный спектр гафния богат спектральными линиями.
К элементам, образующим сплавы с гафнием, относятся железо,
медь, серебро, алюминий, цирконий, магний, молибден, вольфрам,
углерод, кремний, фосфор, азот и др.
С химической точки зрения металлический гафний проявляет
низкую активность и очень напоминает цирконий.
При нормальной температуре на воздухе поверхность метал-
лического гафния покрывается плотно прилегающей защитной
пленкой НЮ2.
При нагревании металлический гафний взаимодействует с кис-
лородом, галогенами, азотом и углеродом:
Hf + О2 = HfO2 + 266,1 ккал
Hf + 2F2 = HfF4 + 435 ккал
Hf + 2С12 = HfCl4 + 237 ккал
Hf +
Hf + 2Вг2 = HfBr4 4- 225 ккал
Hf + 212 = НП4 + 175 ккал
Hf + i/?N2 = HfN
С = HfC
Гафний растворяется в концентрированной фтористоводородной
кислоте и царской водке, а также в расплавленном KHF2:
Hf + 4HF = HfF4 + 2Н2
3Hf + 12НС1 + 4HNO3 = 3HfCl4 + 4NO + 8H2O
Hf + 4KHF2 = K2[HfF6] + 2KF + 2H2
Химическая активность
руется следующей схемой:
металлического гафния иллюстри-
Комн. темп.
Нагревание
( с HF ->• HfF4
t с царской водкой —> HfCl4
Гафний —
с кислородом —-* HfO2
с галогенами —>• тетрагалогениды
с N2, С -* HfN, HfC
с распл. KHF2 —-* K2[HfFe]
9—0101
130 ПОБОЧНАЯ ПОДГРУППА IV ГРУППЫ
Соединение Окис- ли Гидроокиси Фториды и фто- росоединения Хлорид и хлоро- соединения
Соединения двухва- лентного гафния — — — —
Соединения трехва- лентного гафния — —
Соединения четырех- валентного гафния ню2 Бес- цвет- ный Hf(OH)4 Белый НО. .О — ОН )Hf( HOZ ХОН Бесцветный HfF4 Бесцветный H2[HfOF4]-2H2O Me2[HfFe] Me|[HfF7] ШС14 Бесцветный HfOCl2-8M2O Бесцветный Me2[HfCla]
ПРИМЕНЕНИЕ
Стержни металлического гафния применяются в ядерной тех-
нике как материал, регулирующий мощность ядерного реактора
благодаря большой способности к захвату нейтронов.
Из металлического гафния изготовляют нити и катоды для
электронных трубок, поскольку он тугоплавок и обладает способ-
ностью к высокой электронной эмиссии.
Гафний — это металл современной техники; он применяется
в турбореактивных двигателях, ракетах, спутниках и др., а также
в высоковакуумной технике, обладая свойством сильно поглощать
газы (свойство, характерное и для титана, и циркония).
В производстве сталей, латуней и бронз металлический гафний
используется в качестве легирующей добавки, дегазанта и рас-
кислителя.
СОЕДИНЕНИЯ (ОБЩИЕ СВОЙСТВА)
Известно небольшое количество простых и координационных
неустойчивых соединений двух- и трехвалентного гафния и отно-
сительно большое число устойчивых соединений четырехвалент-
ного гафния.
В табл. 23 приведены формулы и указан цвет ряда соединений
гафния.
ГАФНИЙ
131
Таблица 23
1 1 Бромиды Йодиды Сульфогафниевые кислоты и сульфо- соединения Хелатные соединения гафния (IV) Соеди- нения вклю- чения
I HfBr2 । Черный — — ’Hf/к О \ - | / ч/Ч 1 HfH2 HfB2 Серые
ШВгз 'Черно-синий — — _ \UxJ/4. Желтый / ,СН3\ / /О = С( I Hfl < >СН I 1 о — сД I \ \сн3 /4 Желтый HfN Жел- тый
* HfBr4 Бесцветный (1ГОВг2-8Н2О ‘ Бесцветный i i Hfl4 Жел- тый H2[Hf(SO4)3l Белый H2[HfO(SO4)2].3H2O Me2[Hf(SO4)3] K2[HfO2(SO4)2]-3H2O Пероксосоединение HfC Корич- невый
Соединения двух- и трехвалентного гафния
Известно очень немного соединений (особенно галогенидов)
двух- и трехвалентного гафния; для них характерны восстанови-
тельные свойства и низкая устойчивость; окрашены они, как пра-
вило, в темные цвета.
Дибромид гафния, HfBr2, получают в результате диспропор-
ционирования трибромида гафния в вакууме при 300°:
2ШВг3 ШВг2 + ШВг4
Дибромид гафния — черное твердое вещество, самовоспламе-
няющееся на воздухе и разлагающееся при нагревании до 400°
в вакууме по уравнению
2HfBr2 = Hf 4- HfBr4
Трибромид гафния, ШВг3, получают восстановлением тетра-
бромида гафния при нагревании в атмосфере водорода или с метал-
лическим алюминием. Это твердое черно-синее вещество, которое
при нагревании в вакууме до 300° диспропорционирует на HfBr4
и HfBr2.
Соединения четырехвалентного гафния
Известны многочисленные устойчивые соединения (простые
и координационные) четырехвалентного гафния, в которых
9*
132
ПОБОЧНАЯ ПОДГРУППА IV ГРУППЫ
он может встречаться как в виде катиона Hf4+, HfO2+, так и в виде
аниона [HfF6]2~, [Hf(SO4)3]2_ и др.
Гидролизом солей четырехвалентного гафния получают соли
гафнила НЮХ2 (где X = С1~, Вт-, I-, 1/2SO2’ и др.). При более
глубоком гидролизе образуется гидратированная двуокись гафния
НЮ2-пН2О или Hf(OH)4.
Большинство соединений четырехвалентного гафния представ-
ляют собой бесцветные кристаллы или белые порошки.
Ион Hf4+ похож на ион Zr4+, поскольку оба иона обладают
аналогичным строением внешних электронных уровней и очень
близкими значениями атомных радиусов.
Координационные соединения гафния характеризуются более
низкой устойчивостью, чем аналогичные соединения циркония.
Неорганические соединения
Двуокись гафния, НЮ2, образуется при горении металлического
гафния в кислороде или при прокаливании гидроокиси, диокса-
лата, дисульфата гафния, дихлорида гафнила и других соедине-
ний гафния:
Hf 4- О2 = НГО2 + 266,1 ккал Hf(C2O4)2 = HfO2 + 2СО2 + 2СО
Hf(OH)4 = HfO2 + 2Н2О Hf(SO4)2 = HfO2 + 2SO3
HfOCl2 -8H2O = HfO2 + 2HC1 + 7H2O
Двуокись гафния может существовать в виде порошка, бесцвет-
ных моноклинных кристаллов с плотностью 9,98 г/см3 или бес-
цветных тетрагональных кристаллов с плотностью 10,47 г/см3',
последние имеют т. пл. 2790°, плохо растворимы в воде, диамаг-
нитны, обладают более основным характером, чем ZrO2, и обна-
руживают каталитические свойства.
Гидроокись гафния(1У), Ш(0Н)4, или гидратированная дву-
окись гафния, НЮ2-2Н2О, получается глубоким гидролизом солей
четырехвалентного гафния при нагревании; обработкой растворов
солей, четырехвалентного гафния растворами щелочей, тиосуль-
фата натрия, гидроокиси аммония и разложением пероксогафниа-
тов:
HfCl4 + 4Н2О Hf(OH)4 + 4НС1
Hf(SO4)2 + 4NaOH = Hf(OH)4 + 2Na2SO4
HfOCl2 + Na2S2O3 + 2H2O Hf(OH)4 + 2NaCl + S -f- SO2
H4HfOs -> Hf(OH)4 + V2O2
Гидроокись гафния получается в виде белого осадка, раство-
ряющегося при добавлении щелочей и перекиси водорода с обра-
зованием пероксогафниатов.
Пероксогафниевые соединения. Обработкой
при низкой температуре водного раствора дисульфата гафния
ГАФНИЙ
133
раствором щелочи или гидроокиси аммония с последующим
добавлением 30%-ного раствора Н2О2 получают пероксогидро-
окисъ гафния
НО. /О -ОН
>Hf<
нох хон
которая выделяется в виде бесцветных кристаллов при добавле-
нии спирта.
При нагревании ацетилацетоната гафния Hf(C5H7O2)4 с конц.
H2SO4 и K2SO4 в присутствии перекиси водорода получается
пероксодисулъфатогафниат()У) калия
ГО.
I /Hf (SO4)2 -ЗН2О
Тетрафторид гафния, HfF4, получается термическим разло-
жением соединения (NH4)2[HfF6] в токе азота при 300° в плати-
новой трубке:
(NH4)2[HfFe] = HfF4 + 2NH4F
Тетрафторид гафния образует бесцветные кристаллы с плот-
ностью 7,13 г/с.и3 и т. пл. 927°; соль растворима в воде и образует
координационные соединения с фторидами щелочных металлов
или аммония, например K2[HfF6], K3[HfF7], (NH4)2[HfF6],
(NH4)3[HfF7].
Из водного раствора тетрафторида гафния выпадают кри-
сталлы H2[HfOFJ -2Н2О, или HfF4-3H2O.
Г ексафторогафниат аммония, (NH4)2[HfF6], представляет
собой бесцветные псевдогексагональные призмы, которые, выпадают
при растворении гидроокиси гафния Hf(OH)4 в стехиометрически
необходимом количестве гидрофторида аммония или при обработке
тетрафторида гафния стехиометрически необходимым количеством
фторида аммония:
Hf(OH)4 + 4NH4HF2 = (NH4)2[HfFe] + 2NH4F +’4H2O
HfF4 + 2NH4F = (NH4)2[HfF6J
При обработке тетрафторида гафния избытком фторида аммония
получается (NH4)3[HfF7j, растворимость которого в воде ограни-
чена:
HfF4 + 3NH4F = (NH4)3[HfF7]
Тетрахлорид гафния, HfCl4, получают хлорированием при
нагревании металлического гафния, карбида гафния или смеси
двуокиси гафния с углем, а также действием паров СС14 на дву-
окись гафния примерно при 300°:
Hf + 2С12 = HfCl4 + 237 ккал HfO2 + 2С + 2С12 = HfCl4 -f- 2СО
HfC + 2С12 = HfCl4 + С HfO2 + СС14 = HfCl4 + СОа
134
ПОБОЧНАЯ ПОДГРУППА IV ГРУППЫ
Тетрахлорид гафния представляет собой белый порошок, кото-
рый сублимируется при 317° и образует аддукты с хлоридами и
оксихлоридами фосфора, например 2HfGl4-PGl5, ЗШС14 -2РОС13.
Металлотермическим восстановлением тетрахлорида гафния
с магнием или натрием получают металлический гафний.
Известны хлорогафниаты общей формулы MJlHfGlg].
Дихлорид гафнила, НЮС12-8Н2О, выпадает при концентри-
ровании раствора, полученного действием разб. НС1 на гидро-
окись гафния:
Hf(OH)4 + 2НС1 + 5Н2О = HfOCl2-8H2O
Дихлорид гафнила образует бесцветные тетраэдрические кри-
сталлы, растворимые в воде; их растворимость в соляной кислоте
меньше, чем ZrOCl2 -8Н2О.
Тетрабромид гафния, HfBr4, получают действием паров брома
на нагретую до 500° смесь двуокиси гафния с сахарным углем:
HfO2 + 2С + 2Вг2 = HfBr4 -р 2СО
Тетрабромид гафния представляет собой бесцветные кристаллы,
сублимирующиеся при 322°.
Дибромид гафнила, HfOBr2-2H2O, выпадает в виде бесцветных
кристаллов при концентрировании раствора, полученного дей-
ствием разб. НВт на гидроокись гафния:
Hf(OH)4 + 2НВг + 5Н2О = ШОВг2.8Н2О
Тетраиодид гафния, Hfl4, получают прямым взаимодействием
исходных элементов при 300°; он представляет собой желтые кри-
сталлы, сублимирующиеся при 427° и термически диссоциирую-
щие при 1400°:
300°
Hf + 2I2 ', ~ ~ > Hfl44-175 ккал
1100-1400°
Трисулъфатогафниевая кислота, H2[Hf(SO4)3], получается обра-
боткой тетрагалогенидов гафния конц. H2SO4 или их сплавлением
с KHSO4:
HfCl4 + 3H2SO4 = H,[Hf(SO4)3] 4- 4HC1
HfCl4 + 3KHSO4 = H2[Hf(SO4)3] 4- 3KC1 4- HC1
Трисулъфатогафниевая кислота представляет собой белое
растворимое в воде вещество. При концентрировании водного
раствора Н2[Ш(ЗО4)з1 выпадают кристаллы H2[Hf(SO4)3] -ЗН2О.
При обработке солей четырехвалентного гафния серной кисло-
той и сульфатами щелочных металлов образуются сулъфатогафниа-
ты общей формулы МДНДЗО^].
Гидрофосфат гафния, Hf(HPO4)2, получают обработкой сильно
солянокислых растворов солей гафния ортофосфорной кислотой:
ШС14 4- 2Н3РО4 = Ш(НРО4)2 4- 4НС1
ГАФНИЙ
135
Гидрофосфат гафния — белый осадок, растворимый в H2SO4
и HF.
Помимо описанных известны и другие соединения четырехвалентного
гафния, например диметафосфат гафнила HfO(PO3)2, дифосфат HfP2O7,
ортофосфат Hf3(PO4)4, дигидроортофосфат гафнила ШО(Н2РО4)2, ацетил-
ацетонат Hf(C5H7O2)4-10Н2О, тартрат и др.
Соединения включения
Нитрид гафния, HfN, получают нагреванием порошкообраз-
ного металлического гафния при 1100—1200° в атмосфере азота
или аммиака (в молибденовой лодочке) или путем термической
диссоциации тетраиодида гафния в атмосфере азота.
Нитрид гафния образует твердые металлоподобные желтые
кристаллы. Они тугоплавки (т. пл. 3310°) и хорошо проводят
электрический ток.
Карбид гафния, HfC, получают нагреванием порошкообраз-
ного металлического гафния или двуокиси гафния с углем (сажей)
в электрической печи либо пропусканием газообразной смеси
HfCl4, СН4 и Н2 над нагретой вольфрамовой проволокой:
Hf + С = HfC HfO2 + ЗС = HfC + 2СО
н2
HfCl4 + СН4 =-> HfC + 4НС1
Карбид представляет собой металлоподобные тяжелые корич-
невые кристаллы (плотность 14,19 г/сл«3); они тверды и тугоплавки
(т. пл. 3887°), обладают хорошей электропроводностью; могут
служить в качестве шлифовального материала или заменителя
алмаза при резке стекла.
Диборид гафния, HfB2, получают пропусканием газообразной
смеси HfCl4, ВВг3 (или ВС13) и Н2 над вольфрамовой проволокой,
нагретой примерно до 2300°:
HfCl4 + 2ВВг3 + 5Н2 HfB2 + 4НС1 + 6НВг
HfCl4 4- 2ВС13 + 5Н2 HfB2 + 10НС1
Диборид гафния образует твердые серые кубические кри-
сталлы с металлическим блеском; плавится при 3062°, хорошо
проводит электрический ток.
ПОБОЧНАЯ ПОДГРУППА V ГРУППЫ
ПЕРИОДИЧЕСКОЙ СИСТЕМЫ
(ПОДГРУППА ВАНАДИЯ)
ХАРАКТЕРИСТИКА ПОДГРУППЫ
В побочной подгруппе V группы периодической системы хими-
ческих элементов находятся переходные металлы ванадий (V),
ниобий (Nb) и тантал (Та).
В табл. 24 приведена электронная структура атомов ванадия,
ниобия и тантала.
Таблица 24
К L М О р
Элемент z Is 2s 2р 3s Зр 3d 4s 4р 4d 4У 5s 5р 5d 6s 6р 6d
V 23 2 2 6 2 6 3 2 3d34s2
Nb 41 2 2 6 2 6 10 2 6 4 — 1 4d45s!
Та 73 2 2 6 2 6 10 2 6 10 14 2 6 3 2 5d36s2
Атомы этих элементов имеют по 1—2 электрона на последнем
электронном уровне и 2.6.3 или 2.6.4 электрона на предпоследнем
электронном уровне. Орбитали 3d у ванадия, 4й у ниобия и 5d
у тантала заполнены электронами неполностью. В химических
реакциях атомы ванадия, ниобия и тантала могут участвовать
2, 3, 4 и 5 электронами, образуя соединения, в которых эти эле-
менты двух-, трех-, четырех- и пятивалентны. Помимо соединений,
в которых ниобий и тантал выступают как двух-, трех-, четырех -
и пятивалентные, известны окиси Nb2O и Та2О — единственные
соединения одновалентных ниобия и тантала. Наиболее устойчивы
соединения элементов подгруппы ванадия, в которых они пяти-
валентны, поскольку при отдаче 5 электронов структура атомов
этих элементов приобретает устойчивую конфигурацию из 8 элект-
ронов. У ванадия, ниобия и тантала склонность к проявлению
валентности ниже пяти уменьшается с увеличением атомного
номера.
В табл. 25 приведены наиболее важные константы элементов
подгруппы ванадия.
Таблица 25
Элемент Ванадий V Ниобий Nb Тантал Та
Цвет < в компактном виде Светло- серый Серебристо- белый Серебристо- белый
в порошке Темно-серый Серый Серый
Кристаллическая структура Кубическая объемно- центрирован- ная Кубическая объемно-цент- рированная Кубическая объемно-цент- рированная
Атомный номер 23 41 73
Атомный вес 50,942 92,906 180,948
Атомный радиус, А (по По- лингу) 1,36 1,47 1,49
Ионный радиус М5+, А (по Гольдшмидту, Полингу, Аренсу) 0,4; 0,59; 0,59 0,69; 0,70; 0,69 0,68; —; 0,68
Атомный объем (при 20°), см3/г-атом 8,35 10,83 10,9
Плотность (при 20°), г/см3 5,96 8,58 16,69
Твердость по Бринеллю, кг/мм3 264 210-250 200-250
Твердость по шкале Мооса 6 6 6
Температура плавления, °C 1710 2487 2997
Температура кипения, °C 3450 4930 5425
Удельная теплоемкость (при 20°), кал/г-град 0,120 0,065 0,036
Коэффициент линейной тепло- проводности % (при 0°), кал см~! • сек-1 град-1 — 0,125 0,130
Удельное сопротивление р-10в (При 0°), ОМ-СМ, 20,9 13,2 12,4
Электропроводность (Hg = 1) 4,5 7,2 7,6
138
ПОБОЧНАЯ ПОДГРУППА V ГРУППЫ
Продолжение таблицы 25
Магнитная восприимчивость Х-10~6 (при 18°), эл.-магн. ед. 4,5 пара- магнитный 2,34 пара- магнитный 0,84 пара- магнитный
Теплота перехода атомов в газообразное состояние, ккал/г-атол 106 184,5 185
Потенциал Me -+• Ме+ + е~ ионизации, Ме+—>- Ме2+---е- эв Ме2+ -> Ме3+ 4- е_ Ме3+ -> Ме4+ | е~ Ме4+ Ме5+ + е~ Ме5+ Ме6+4-е- 6,74 14,10 26,31 48,35 68,70 132,80 6,88 14,32 25,04 37,70 51,90 106,30 7,88 16,2 22,27 33,08
Потенциал ионизации Me -> Ме+ + е~, ккал/г-атол 156 156 138
Нормальный потенциал (при 25°), в V/V2+—1,18 V/V3+—0,87 Nb/Nb3+—1,10
Нормальный потенциал в оки- слительно-восстановитель- ных системах (при 25°), в у2+/уЗ+ —0,255 Nb6+/NM+4- +0,4 —
Валентность (II, III, IV), V (I, II, III, IV), V (I, II, III, IV), V
Массовые числа природных изотопов 51 93 181
Распространенность элементов в земной коре, вес. % 2,0-10-2 3,2-10-6 2,4-10-5
Элементы подгруппы ванадия — тяжелые парамагнитные
металлы (плотность 5, 6 —16,69 г/см3), светло-серого (ванадий)
и серебристо-белого (ниобий и тантал) цвета с кубической объем-
но-центрированной кристаллической структурой; т. пл. 1710—
2997°, т. кип. 3450—5425°, твердость 6 по шкале Мооса. Они
обладают ограниченным количеством изотопов. Для этих метал-
лов в чистом состоянии характерны превосходные механические
свойства, при загрязнении же кислородом, азотом, углеродом,
бором, водородом и др. они теряют пластичность и становятся твер-
дыми и хрупкими. Благодаря сходной электронной структуре
и близким значениям атомных и ионных Ме5+ радиусов и атомных
объемов ниобий и тантал проявляют большое сходство в химиче-
ском поведении и образуют многочисленные изоморфные соеди-
нения.
ПОБОЧНАЯ ПОДГРУППА V ГРУППЫ
139
Известны многие сплавы ванадия, ниобия и тантала с железом,
хромом, титаном, марганцем, молибденом, вольфрамом, цирко-
нием, алюминием, углеродом, бором, азотом и др.
Ванадий, ниобий и тантал проявляют пониженную химическую
активность (очень устойчивы к действию химических реагентов),
устойчивы на воздухе и в воде в обычных условиях и взаимодей-
ствуют при нагревании с галогенами, кислородом, серой, азотом,
углеродом и др. На ванадий действуют HF, HNO3, царская
водка, расплавленные щелочи, на ниобий и тантал — расплавлен-
ные щелочи и смеси HF с HNO3.
В природе ванадий, ниобий и тантал встречаются в виде соеди-
нений (ванадий в виде пентасульфида или ванадата свинца, калия,
уранила и др., а ниобий и тантал как ниобаты и танталаты) в раз-
личных минералах. Ниобий и тантал входят также в состав ком-
плексных минералов редкоземельных металлов. По распространен-
ности ниобий и тантал следуют за ванадием.
Обогащение ванадиевых, ниобиевых и танталовых руд осуще-
ствляется электромагнитными, гравитационными, флотационными
или химическими методами.
Металлы подгруппы ванадия получают путем металлотермиче-
ского восстановления соответствующих пентаокисей или галоге-
нидов. Кремне- или алюмотермическим восстановлением смеси
окислов этих металлов с окислами железа получают сплавы ферро-
ванадий, феррониобий, ферротантал.
Как будет показано в разделе о применении, мелаллы ванадий,
ниобий, тантал и сплавы на их основе — важнейшие материалы
современной техники.
Соединения ванадия, ниобия и тантала низких степеней окис-
ления (II, III, IV) получаются восстановлением соответствующих
соединений высших степеней окисления (V). Первые обладают
пониженной устойчивостью, легко окисляются (имеют восстанови-
тельный характер), окрашены в темные цвета (зеленый, синий,
черный).
Соединения пятивалентных ванадия, ниобия и тантала бесцвет-
ны либо окрашены в желтый, оранжевый, красный или коричне-
вый цвета.
В табл. 26 и 27 приведены наиболее важные константы соеди-
нений V2O5, Nb2O5, Та2О5, VF5, NbF5, TaF5. Пентаокиси V2O5,
Nb2O5, Ta2O5 получают прямым взаимодействием исходных эле-
ментов при нагревании или окислением окислов VO, V2O3, VO2,
NbO, Nb2O3, NbO2, TaO, TaO2 и др.; это соединения устойчивые,
тугоплавкие, плохо растворимые в воде, растворах разбавленных
кислот и щелочей. При сплавлении с щелочами превращаются
в ванадаты, ниобаты или танталаты. Пентаокись V2O5 является
ангидридом кислоты, Nb2O5 обладает слабокислым характером
Та2О5 проявляет амфотерные свойства.
140
ПОБОЧНАЯ ПОДГРУППА V ГРУППЫ
Таблица 26
Соединение v2o5 NbjOg Та2О5
Внешний вид Оранжевый порошок или красноватые ромбические кристаллы Белый порошок или: бесцветные ромбические кристаллы (a-Nb2O5) бес- цветные тетра- гональные кри- сталлы (g-Nb2O5) Белый порошок или: бесцветные ромбические кристаллы (а-Та2О5), бесц- ветные тетраго- нальные кри- сталлы (|3-Та2О5)
Молекулярный вес 181,881 265,809 441,893
Плотность (при 20°), г/см5 3,34 4,46 8,2—8,7
Температура плавле- ния, °C 670 1512 1877
Температура кипе- ния, °C 1827 — —
Теплота образования, ккал/моль 373,3 455,2 488,9
Пентагалогениды VF5, NbF5, NbBr5, Nbl5, TaF5, TaCl5, TaBr5,
Tal5 являются ковалентными легколетучими соединениями; они
плавятся при низких температурах, растворяются в органических
растворителях и гидролизуются в воде.
Известны многочисленные фторосоли общей формулы
MeI[MevFe], Me*[MevF7], Me*[MevF8] и оксофторосоли общей фор-
мулы Mei[MevOFJ, Mei[MevOF5], Mei[MevOFdJ.
Большой устойчивостью обладают простые ванадаты, ниобаты
и танталаты, изополи- и гетерополисоединения. В кислой среде
соединения пятивалентного ванадия могут восстанавливаться цин-
ком (или активным водородом) до соединений двухвалентного вана-
дия; соединения пятивалентного ниобия в этих условиях восста-
навливаются до трехвалентного ниобия, а соединения пятивалент-
ного тантала при этом не восстанавливаются.
При действии на соединения элементов подгруппы ванадия
перекисью водорода в присутствии щелочей получаются окрашен-
ные в желтый, красный или коричневый цвета пероксосоединения,
которые являются сильными окислителями.
Известны многочисленные хелатные соединения ванадия, нио-
бия и тантала с ацетилацетоном, бензоилацетоном, купфероном,
пирокатехином (в сильнощелочном растворе), щавелевой и винной
кислотами или их солями.
ВАНАДИЙ
141
Таблица 27
Соединение vf5 NbF5 TaF5
Внешний вид Белое Бесцветные Бесцветные
вещество кристаллы кристаллы
Молекулярный вес 145,934 187,898 275,940
Плотность (при 20°), г,'см3 2,18 3,29 4,74
Температура плавления, °C 102 75,5 96,8
Температура кипения, °C 111,2 236 229,4
Теплота образования, ккал/молъ 335 — —
Образует ацидосоли MeI[VF8] Me^NbFg] MeI[TaFe]
типа MeI[VOF4] Me£[NbF7] MeJ[TaF7]
Me*[VOF5] Me2[NbOF5].reH2O Me|[TaF8]
Me|[VO2F3] MeJ[NbOFe] MeI[TaOF4]
MeJ[VO2F4] Me4[NbOF7] Mei[TaOFe]
Среди металлоорганических соединений элементов подгруппы
ванадия наиболее важны производные циклопентадиена.
ВАНАДИЙ V
Z= 23; ат. вес = 50,942
Валентность (II, III, IV), V; заряд (2+, 3+, 4+), 5+
Массовое число природного изотопа 51
Электронная структура атома ванадия: К -L -3^3p63d3
Электронная структура атома ванадия и иона V5+ (для 3d- и
45-орбиталей):
4s2
3d3
t t t
V
3d
4s
vs+
142
ПОБОЧНАЯ ПОДГРУППА V ГРУППЫ
ИСТОРИЧЕСКАЯ СПРАВКА
В 1801 г. мексиканский минеролог Дель-Рио обнаружил в одном
свинецсодержащем минерале из Зимапана (Мексика) новое веще-
ство, металл которого он назвал «панхром» (вследствие различных
окрасок его окислов), а затем «эритроний» (из-за красной окраски,
приобретаемой его солями под действием кислот и при нагревании).
Новый элемент был открыт в 1830 г. Сефстрёмом (работавшим
у Берцелиуса) в минерале, найденном в Таберге (Швеция), и был
назван ванадием по имени Vanadis — богини севера. Дель-Рио
не уточнил, что за вещество он открыл, а Вёлер назвал свинцовый
минерал из Зимапана ванадинитом и показал, что эритроний
и ванадий — один и тот же элемент.
Ванадий рассматривали как аналог хрома до 1867 г., когда
английский химик Роско показал, что этот элемент обладает
свойствами, общими с элементами подгруппы фосфора. Впервые
металлический ванадий 98—99 %-ной чистоты получили Руфф
и Мартин путем металлотермического восстановления V2О3 в тигле
из MgO. Ванадий 99,3—99,8%-ной чистоты был получен в 1927 г.
Марденом и Риком путем восстановления пятиокиси ванадия
металлическим кальцием в присутствии хлорида кальция.
В 1934 г. Деринг получил порошкообразный чистый ванадий (по
процессу, описанному в 1870 г. Роско) нагреванием трихлорида
ванадия в токе водорода.
В компактном состоянии чистый ванадий получают по процессу
Ван-Аркела — де Бура.
РАСПРОСТРАНЕННОСТЬ В ПРИРОДЕ
В природе ванадий находится в виде соединений (сульфиды,
ванадаты, алюмосиликаты и др.), ассоциированных с некоторыми
из природных соединений железа, титана, алюминия, свинца,
урана, меди, цинка, вольфрама, циркония и др. в различных ком-
плексных минералах.
Содержание ванадия в земной коре составляет 2-10~2вес. %.
Наиболее важные минералы ванадия:
Патронит, V2S5, черного цвета, загрязнен SiO2, А12О3, серой,
сульфидами железа, никеля и др.; встречается в асфальтах Перу-
анских Анд.
Карнотит, K2(UO2)2(VO4)2-ЗН2О, пылевидная флуоресцирую-
щая масса желтого (или зеленовато-желтого) цвета; радиоактивен,
плотность 4,46 г/смК твердость 2—2,5 по шкале Мооса.
Ванадинит, Pb5(VO4)3Cl (изоморфен апатиту) или
3Pb3(VO4)2 -РЬС12, встречается в виде маленьких игольчатых
призм желтого (или коричнево-красного при наличии примесей)
цвета с плотностью 6,66—7,10 г/см3 и твердостью 3 по шкале
Мооса.
ВАНАДИЙ
14S
Роскоэлит (колорадская ванадиевая слюда), KV2[Al2Si3Oi0] (0Н)2,
черно-коричневого цвета, встречается в США.
Тюямунит, Ca(UO2)2(VO4)2-8Н2О, лимонного цвета, встре-
чается в песках Колорадо.
В числе других минералов ванадия можно назвать: ванадомаг-
нетиты, титанованадомагнетиты, ильменит с ванадием, деклуазит
(Zn, Cu)Pb(VO4)(OH) и его разновидности (купродеклуазит, рами-
рит, тритохлорит и др.), колсонит (Fe, V)3O4, ферванит
FeVO4-2H2O, россит CaV2O0-4H2O, сульванит Cu3(VS4], пухерит
BiVO4 и др.
Минералы, содержащие соединения ванадия, находятся в Перу,
Аргентине, Чили, Родезии, Южно-Африканском Союзе, Мозам-
бике, Мексике, США, Швеции, СССР, Франции, Англии, Испании,
Румынии, Австралии и в некоторых странах Азии.
В малых количествах соединения ванадия встречаются в неко-
торых растениях (сахарной свекле, табаке, виноградной лозе-
и др.), деревьях (бук, дуб), углях, нефтях, природных водах,
а также в молоке и некоторых тканях животных. В крови асцидий
(морских беспозвоночных) ванадий заменяет медь и фосфор
и играет ту же роль, что и железо в крови высших животных.
Соединения ванадия обнаружены в спектре Солнца, в метео-
ритах, в вулканической лаве, золе и породах, в базальтах раз-
личного происхождения.
ПЕРЕРАБОТКА РУД, СОДЕРЖАЩИХ ВАНАДИЙ
Переработка концентратов железных руд,
содержащих ванадий
♦
Примерно 50% всего производимого ванадия получается при
переработке ванадомагнетитов, титанованадомагнетитов и фосфо-
ристого гематита, содержащего ванадий. В процессе обогащения
ванадомагнетитов и титанованадомагнетитов электромагнитными
или гравитационными методами и фосфористого гематита — гра-
витационным методом ванадий переходит в железосодержащий
концентрат, из которого переработкой в высоких доменных печах
переводят в шлак примерно 80% ванадия. При окислительном
плавлении шлака (в конверторах или мартеновских печах) 50—
80% ванадия переходит в шлак в виде соединений типа шпинелей
(Fe2+, Mn2+)[(V3+, Cr3 + Fe3+)2O4]. Прокаливанием шлака с NaCl
при 800—900° во вращающихся печах получают триметаванадат
натрия:
6Fe[V2O4] + 12NaCl + 21/2О2 = 4Na3V3Oe + 3Fe2O3 + 6С12
Если содержание ванадия очень мало, разбавленный водный
раствор метаванадата натрия обрабатывают известковой водой
144
ПОБОЧНАЯ ПОДГРУППА V ГРУППЫ
для осаждения метаванадата кальция, который затем высушивается
примерно при 200° и служит для получения феррованадия.
Если содержание ванадия велико, водный раствор триметава-
надата натрия обрабатывают кислотами для осаждения V2O5.
Последний после высушивания и плавления (700—800°) исполь-
зуется для получения металлического ванадия или феррованадия.
Обработка водного раствора триметаванадата натрия известко-
вой водой или кислотами осуществляется в сосудах из дерева
хвойных пород или из кислотостойких материалов.
Если водный раствор триметаванадата натрия богат соедине-
ниями фосфора, последний выделяется в виде NH4MgPC>4.
Переработка карнотитных руд
Карнотитные руды K2(UO2)2(VO4)2-ЗН2О, содержащие 0,75—
1 вес. % ванадия в смеси с 6—9% Nad, подвергаются хлорирую-
щему прокаливанию при 850° в многоэтажных печах. После
охлаждения прокаленный материал проходит влажный размол
с 3%-ным раствором карбоната натрия и затем противоточную
промывку нагретым до 92—96° раствором карбоната натрия.
При подкислении раствора до pH = 6 выпадает желтый искус-
ственный карнотит, который после промывки водой и высуши-
вания сплавляют со смесью Na2CO3 и NaCl (с добавлением дре-
весных опилок). При обработке холодного расплава щелочами
получают ванадаты щелочных металлов, из которых сильным под-
кислением осаждают V2O5-nH2O. Последний превращают в тех-
нический V2O5 нагреванием.
Карнотитные руды, содержащие менее 0,7—1% ванадия, про-
каливают в смеси с NaCl при 550°, затем подвергают влажному
размолу с 3%-ным раствором карбоната натрия и подщелачивают
раствором, содержащим 7—9% Na2CO3 и 1% NaHCO3, при
107-121°.
Если растворы ванадатов щелочных металлов нейтрализовать
HNO3 и обработать сульфатом железа(П), то выпадает ванадат
железа (II).
Последовательность переработки карнотитных руд показана
на следующей схеме:
K2(UO2)2(VO4)2-3H2O+2NaCl-{-4Na2CO3--> 2Na3VO4+2Na2UO4+2KCl+CO2
Нагре-
вание /
/ Трудно растворимый
Нейтрализация HNO3/ +6HNO3 (рН=2,5)
и добавление FeSO47
Fe3(VO4)2 -<--------------/
2H3VO4 + 6NaNO3
I Нагревание
V2O54-3H2O
ВАНАДИЙ
145
Переработка концентратов ванадинитных руд
Концентраты ванадинитных руд (образовавшиеся при исполь-
зовании методов, основанных на разнице плотностей компонентов
руд) перерабатывают по следующей схеме:
Нагревание
2Pb5(VO4)3Cl-L36HCl, копц. •----------> 6VOCl3+10PbCl2+18H,O
Трудно
растворимый
2(NH4)3V3O9
| Нагревание
3V2O5+6NH3 + 3H,O
Экстракция ванадия органическими растворителями
Из водных растворов, полученных при измельчении ванадий-
содержащих концентратов железных руд, карнотитных руд или
ванадинита, ванадий можно извлечь, применяя (при pH ~ 3)
растворенные в хлороформе n-бензоилфенилгидроксиламин, 8-ок-
сихинолин, З-гидроксил-1,3-дифенилтриазин, п-диэтилдитиокар-
бамат натрия и др.
С вышеупомянутыми реагентами растворимые в воде соединения
пятивалентного ванадия образуют плохо растворимые в воде
соединения, которые, однако, хорошо растворяются в хлороформе
и благодаря этому экстрагируются в органическую фазу. VOG13
в 7,75 М растворе НС1 экстрагируется изопропиловым эфиром.
VF3 и VF5 в 20 М растворе HF экстрагируются этиловым эфиром.
Соединения четырехвалентного ванадия экстрагируются при
pH = 2,5 раствором ацетилацетона в СС14; из 0,5 М раствора
НС1 они экстрагируются 1,5—7 М раствором NH4SCN в этиловом
эфире.
Соединения трехвалентного ванадия при pH — 2 экстраги-
руются смесью ацетилацетона и хлороформа (1 : 1).
ПОЛУЧЕНИЕ МЕТАЛЛИЧЕСКОГО ВАНАДИЯ
Металлический ванадий получают в результате термической
диссоциации иодида ванадия(П) в вакууме, восстановлением окис-
лов VO, V2O3, VO2, V2O5 металлическим кальцием, сплавами
редкоземельных металлов, водородом под давлением при высокой
температуре или сахарным углем в атмосфере водорода в электри-
ческой печи, восстановлением хлоридов ванадия VC12, VC13,
10-0101
146
ПОБОЧНАЯ ПОДГРУППА V ГРУППЫ
VC14 водородом при длительном нагревании, а также катодным
восстановлением расплавов, содержащих V2O5.
Из-за трудности получения чистого металлического ванадия
широко применяется сплав железо — ванадий, названный ферро-
ванадием.
Термическая диссоциация иодида ванадия(П)
(процесс Ван-Аркеля — де Бура)
Металлический ванадий получают посредством термической
диссоциации иодида ванадия(П) VI2 при 900° в вакууме; для
этого используют аппаратуру, применяемую для получения метал-
лического титана по методу Ван-Аркеля — де Бура.
VI2 V + 12
Дииодид ванадия образуется при нагревании VI3 выше 280°
в вакууме. Это гексагональные пластинки фиолетово-розового
цвета с плотностью 5,44 г/с.н3, т. пл. 777° и т. кип. 927°.
Металлотермическое восстановление пятиокиси ванадия
Металлический ванадий можно получить металлотермическим
восстановлением пятиокиси ванадия металлическим кальцием при
900—950° в вакууме:
V2O5 + 5Са = 2V + 5СаО
Реакцию проводят в прочном металлическом сосуде, используя
СаС12 в качестве флюса, а также небольшое количество металли-
ческого натрия или калия (для освобождения от следов кислорода
или влаги).
Соединения кальция отделяют от порошкообразного металли-
ческого ванадия промыванием раствором (1 : 18) НС1.
Вместо пятиокиси ванадия могут быть использованы другие
его окислы — VO, V2O3 и VO2.
Металлический ванадий получают также восстановлением окис-
лов ванадия сплавами редкоземельных металлов в огнеупорном
тигле, футерованном окисью магния, или путем нагревания окис-
лов ванадия в атмосфере водорода под давлением при высокой
температуре.
При восстановлении окислов ванадия с сахарным углем в атмо-
сфере водорода (в электрической печи) получается металлический
ванадий, загрязненный карбидом ванадия; при алюмотермическом
восстановлении окислов ванадия металл получается также загряз-
ненным.
ВАНАДИЙ
147
Восстановление хлоридов ванадия VCI2, VCI3, VCI4
водородом, металлическим натрием или магнием
Восстановлением хлоридов ванадия VC12, VG13, VC14 водоро-
дом, натрием или магнием при нагревании в платиновой лодочке
получают порошкообразный металлический ванадий:
900° 900°
VC12 + H2-----> V4-2HC1 VCl3 + 3/2H2------> V + 3HC1
680-700°
VCI4 + 2H,---------► V-J-4HC1
в аргоне
Электролитическое получение металлического ванадия
Электролизом при 200° смеси А12Вг6 — КВг (взятых в молярном
соотношении 2 : 1) с V2O5 на катоде одновременно получают порош-
кообразные металлический ванадий и алюминий; в этом процессе
используют феррованадиевые аноды и медные катоды, плотность
тока 5—50 а/дм2.
Полученный порошкообразный металлический ванадий пре-
вращают в компактный металл методами порошковой металлургии
или переплавкой в вакууме (подобно превращению порошкообраз-
ного титана в компактный металл).
ПОЛУЧЕНИЕ ФЕРРОВАНАДИЯ
Феррованадий (сплав железо — ванадий) получают кремне-
или алюмотермическим восстановлением метаванадата кальция
Ga3(V30g)2 или смеси окислов ванадия с железом в присутствии
окиси кальция. Хотя для восстановления можно использовать
различные смеси окислов ванадия VO, V2O5, VO2, V2O3 и железа
F2O3, Fe3O4, все же чаще берут смесь V2O5 с Fe2O3.
В результате кремне- или алюмотермического восстановления
V2O5 в присутствии окиси кальция образуется загрязненный
ванадий; реакции протекают следующим образом:
2V2O5 + 5СаО + 5Si = 4V + 5CaSiO3 + Qt
(Qi = 244 ккал для 2V2O5 + 5Si)
3V2O5 + 5CaO + 10A1 = 6V + 5Ca(A102)2 + Q2
(Q2 = 619 ккал для 3V2O5 + 10A1)
Реакции восстановления смесей окислов V2O5 — Fe2O3 крем-
нием (ферросилицием) или алюминием, будучи сильно экзотермич-
ными, приводят к образованию феррованадия в компактном состоя-
нии.
При кремнетермическом восстановлении V2O5 без окиси каль-
ция или в присутствии небольшого количества СаО получается
V2O3, который реагирует с образовавшимся SiO2, давая силикаты
ванадия(Ш).
10*
148
ПОБОЧНАЯ ПОДГРУППА V ГРУППЫ
Практически восстановление V2O5 осуществляется ферросили-
цием (сплав железо — кремний, содержащий 75% Si) в электри-
ческой печи, футерованной СаО.
Шлак CaSiO3 или Са(А1О2)2, образующийся при получении
феррованадия, плавает на поверхности расплавленного сплава
и может быть легко удален.
ЭЛЕКТРОЛИТИЧЕСКАЯ ОЧИСТКА
Для электролитической очистки ванадия служит ванна из рас-
плава дихлорида ванадия с хлоридом натрия. Применяют желез-
ный катод и анод из загрязненного металлического ванадия. Очист-
ку ведут при плотности тока 108 а! дм2 и напряжении 0,35—1,0 в.
ФИЗИЧЕСКИЕ И ХИМИЧЕСКИЕ СВОЙСТВА
Ванадий в компактном состоянии — парамагнитный металл
светло-серого цвета (в порошкообразном состоянии темно-серый)
с кубической объемноцентрированной кристаллической решеткой
и плотностью 5,96 г/см3 при 20°. Он тугоплавок (т. пл. 1710°,
т. кип. 3450°), обладает твердостью 6 по шкале Мооса, очень
пластичен (легко вытягивается в нити) и прочен на разрыв. При
загрязнении углеродом и водородом (соответственно карбидом
и гидридом) металлический ванадий становится твердым и хрупким.
Коллоидный ванадий образуется в виде золя при дисперги-
ровании металлического ванадия под действием электрической
дуги в изобутиловом спирте.
Известны многочисленные сплавы (типа твердых растворов
или интерметаллических соединений), образуемых металлическим
ванадием с элементами Fe, Сг, Ni, Мп, Ti, Zr, Ge, Mo, W, Cu,
Al, Zn. Be, Mg, Ag, Hg, G, Si, N2, В, P и др. В качестве примера
интерметаллических соединений можно назвать: VFe, VMn, Ni3V,
V3A1. VAI, VA13, VBe2, V2Zr, V3Ge, VC, V2C, V4C3, V2Si, VSi2,
VN, V2N, V3N, VN2i VB2, VP, V2P, V3P, V3P2, VP2 и др.
При комнатной температуре металлический ванадий не окис-
ляется ни во влажном, ни в сухом воздухе. При нагревании
он взаимодействует с кислородом, галогенами, серой, азотом,
кремнием, углеродом и бором:
2V + S/2O2 = V2O5 V + 3/2I2 = VI3 V + С = VC
v + 6/2f2 = vf5
V + 2C12 = VC14
V A 3/2Br2 = VBr3
2V + 3S = V2S3
2V + N2 = 2VN
2V + V2N2 = V2N
2V + Si = V2Si
V + 2Si = VSi2
V + 2B = VB2
Обрабатывая водородом порошкообразный металлический вана-
дий, нагретый до 1300°, получают гидрид ванадия в виде черного
ВАНАДИЙ
149
порошка с плотностью 5,3 г/сж3 при 20°; последний устойчив,
не вступает в реакцию ни с водой, ни с соляной кислотой.
Металлический ванадий устойчив к действию воды, но реаги-
рует при комнатной температуре с концентрированными HF
и HNO3, с НС1О3, НС1О4, с царской водкой, а при нагревании —
с газообразным НС1, конц. H2SO4, разб. HNO3 и др.
V + 3HF = VF3 + 3/2Н2 2V + 8H2SO4 = 2(VOSO4-H2SO4) + 4SO2 6H2O
V + 2HC1 = VC12 + H2 3V + 12HC1 + 4HNO3 = 3VC14 + 4NO - - 8H2O
При анодном растворении металлического ванадия в 5 — 10%-
ном растворе щелочи образуются ванадаты щелочных металлов.
На воздухе расплавленные гидроокиси, карбонаты и нитраты
щелочных металлов медленно превращают ванадий в ванадаты
щелочных металлов.
При обработке металлического ванадия хлористым тионилом
SOC12, сульфурилом SO2C12 или фосгеном СОС12 образуется
тетрахлорид ванадия.
Устойчивость ванадия к действию многочисленных коррозион-
ных реагентов ниже, чем у металлического Ниобия или тантала.
Химическая активность ванадия характеризуется следующей
схемой:
Комн. темп.
Ванадий —
Нагревание
с конц. HF —> VF3
с конц. HNO3, НС1О3, НС1О4 —► соеди-
нение с VOJ
с царской водкой —VC14
с газообразным НС1 —>• VC12
с конц. H2SO4 —> VOSO4-H2SO4 или VOSO4
с кислородом —>• V2O5
с галогенами —► VF5, VC14, УВгз, VI3
с серой, азотом, углеродом, кремнием,
бором V2S3, VN, V2N, VC, V2Si, VSi2, VB2
с расплавленными щелочами, карбонатами,
нитратами щелочных металлов —► ванадаты
щелочных металлов
Металлический ванадий вытесняет платину, иридий, родий
изолото из растворов их солей и восстанавливает ионыН^2+, Сп2+,
Fe3+ до Hg2+, Gu+, Fe2+, превращаясь в соединения четырехва-
лентного ванадия:
PtCl4 + V + Н2О = VOC12 + Pt + 2НС1
4AuCl3 + 3V + 3H2O = 3VOC12 + 4Au + 6HC1
4HgCl2 + V + H2O = VOC12 + 2Hg2Cl2 + 2HC1
2Fe2Cl6 + V + H2O = VOC12 + 4FeCl2 + 2HC1
G физиологической точки зрения растворимые соединения вана-
дия в больших количествах токсичны для человека и животных.
Они вызывают раздражение кожи и слизистых оболочек, раздра-
жение центральной нервной системы, сонливость, паралич, кон-
150
ПОБОЧНАЯ ПОДГРУППА V ГРУППЫ
вульсии, раздражение желудочно-кишечного тракта, повышение
артериального давления и даже могут вызвать смерть. Соедине-
ния ванадия разрушают белки путем последовательного их восста-
новления и окисления.
Ванадий был обнаружен и количественно определен как посто-
янный фактор в растительных и животных тканях и органах;
этот элемент, присутствуя в ничтожных количествах, играет
крайне важную роль в жизненных процессах. В крови асцидий
ванадий играет роль, аналогичную железу в гемоглобине. Неко-
торые фармацевтические препараты, содержащие малые коли-
чества различных соединений ванадия, в прошлом применялись
для лечения сифилиса, туберкулеза и анемии. В настоящее время
соли ванадия применяются как инсектициды, фунгициды и дезин-
фицирующие средства.
ПРИМЕНЕНИЕ
Благодаря высокой температуре плавления (1710°), малому
удельному весу (5,96 г/см9 при 20°) по сравнению с железом,
хорошей коррозионной стойкости металлический ванадий и его
сплавы используются в качестве конструкционных материалов.
В меньших масштабах чистый ванадий применяется в качестве
материала для ядерных реакторов на быстрых нейтронах.
Феррованадий служит в качестве добавки для получения сталей,
используемых как конструкционный материал с 0,10—0,15% вана-
дия, применяемых для изготовления инструментов с 0,15—0,65%
ванадия, используемых в качестве быстрорежущих сталей
с 0,5—2,5% ванадия. Кроме того, феррованадий служит для
получения сталей с карбидом ванадия, обладающих коррозионной
стойкостью и используемых для изготовления химической аппа-
ратуры и постоянных магнитов (6—16% V, 30—52% Fe
и 36—62% Со).
Конструкционные стали, легированные хромом, молибденом,
ванадием, и аустенитные стали, легированные ванадием, обладают
высокой усталостной прочностью.
Наличие ванадия в сталях способствует облагораживанию струк-
туры металла, удалению азота из металла, улучшению механи-
ческих свойств, т. е. увеличению пластичности, эластичности,
устойчивости к истиранию и удару, коррозионной стойкости
и твердости.
Ванадиевые стали применяются в производстве локомотивов,
авиационных и тракторных моторов и для других целей.
Вследствие трудности получения чистого металлического вана-
дия последний в небольшой степени применяется для получения
сплавов, не содержащих железа, таких, как сплавы с медью,
золотом, алюминием, цинком, магнием, титаном и др.
ВАНАДИЙ
151
Сплавы золота с ванадием применяются как стандартные сопро-
тивления и используются в потенциометрах.
Соединения ванадия находят применение в химической про-
мышленности в качестве катализаторов, например в производстве
серной кислоты контактным методом, при окислении нафталина
или о-'ксилола до фталевого ангидрида, антрацена — в антрахи-
нон. ацетилена — в уксусную кислоту, нефтей, а также при полу-
чении анилинового черного, производстве чернил, гидрогенизации
фенолов водородом под давлением и др.
Некоторые соединения ванадия (такие, как V2O5) непосред-
ственно служат красителями для стекла и керамики.
СОЕДИНЕНИЯ (ОБЩИЕ СВОЙСТВА)
Ванадий — поливалентный элемент; он встречается в двух-,
трех-, четырех- и пятивалентном состоянии в многочисленных
простых, двойных и координационных соединениях.
Водные растворы солей ванадия(П) окрашены в фиолетовый
цвет, ванадия(Ш) — в зеленый, ванадия(1У) — в синий, вана-
дия^7) — бесцветны или окрашены в цвета от желто-оранжевого
до красного.
В табл. 28 приведены формулы и указан цвет некоторых
соединений ванадия.
В табл. 29 сопоставляются цвет, плотность, температуры плав-
ления и кипения, теплоты образования окислов ванадия.
Таблица 29
Соединение VO V2O3 vo2 v205
Внеший вид Черно-се- рые кри- сталлы Черные кристаллы Синие кристаллы Красно- оранжевые кристаллы
Плотность (при 20°), г/см3 5,76 4,87 4,40 3,34
Температура плавления, °C — 1967 1545 670
Температура кипения, °C — 3027 ‘ 2727 1827
Теплота образования, ккал/молъ 209,1 249,5 409,1 . 373,3
Характер Основной Слабо основной Амфотер- ный Кислый
152
ПОБОЧНАЯ ПОДГРУППА V ГРУППЫ
Одновременно с увеличением валентности у ванадия умень-
шается основность и усиливается кислотность и устойчивость
его соединений, как и способность к образованию координацион-
ных соединений. Переход от соединений пятивалентного ванадия
к соединениям низших степеней окисления рассматривается при
описании соединений пяти-, четырех- и трехвалентного ванадия.
Соединения двухвалентного ванадия
Известно немного соединений ванадия(П), которые обладают
отчетливо выраженными восстановительными свойствами; они
неустойчивы, на воздухе и в воде легко окисляются. Соединения
ванадия(П) в твердом состоянии черного, коричневого, жел-
того, зеленого или фиолетового цвета. Ион V2+, существующий
в водных растворах галогенидов VX2,— красно-фиолетового
цвета, синий в спиртовых растворах и желто-зеленый в эфире.
Окись и гидроокись ванадия(П) обладают основным характе-
ром и легко растворяются в кислотах с образованием солей.
В общем случае соединения ванадия(П) образуются при вос-
становлении соединений ванадия более высоких степеней окисле-
ния. Для восстановления иона V3+ до V2 + применяют Zn + HG1
или ион Сг2+ в кислой среде, поскольку восстановительный потен-
циал системы V2+/V3+ равен —0,255 в.
Электроположительный двухвалентный ванадий встречается
в виде катиона V2+, [V(H2O)8]2+ или аниона [V(CN)8]4~.
Соединения ванадия(П) во многом напоминают соединения
железа(П) с точки зрения устойчивости, восстановительного
характера кристаллографических систем.
Окись ванадия, VO или V2O2, получают восстановлением V2O5
или V2O3 тонко измельченным порошком металлического ванадия
при 1750°, восстановлением высших окислов ванадия V2O3, VO2,
V2O5 калием при нагревании и восстановлением хлористого
ванадила VOC13 водородом при 1700°.
V2O5 + 3V = 5VO 2V2O3 + 2К = 4VO + К2О2
V2O3 + V = 3VO VOC13 + 3/2Н2 = VO + ЗНС1
Окись ванадия образует черно-серые кубические кристаллы
с плотностью 5,76 г/см\ твердые, диспропорционирующие при
нагревании в вакууме на V2O3 и элементарный ванадий, плохо
растворимые в воде, обладающие основным характером и раство-
ряющиеся в разбавленных кислотах (НС1, HF, HNO3 и др.)
с образованием фиолетовых растворов соответствующих солей
ванадия(П).
Гидроокись ванадия, V(OH)2, образуется в виде коричневого
осадка при обработке щелочами водных растворов солей вана-
ВАНАДИЙ
153
дия(П). Гидроокись ванадия является очень слабым основанием,
неустойчивым и легко окисляющимся на воздухе.
Дифтпорид ванадия, VF2, представляет собой фиолетовые кри-
сталлы, плавящиеся при 1127° и кипящие при 2227°.
Дихлорид ванадия, VC12, получают восстановлением VC14 или
VC13 водородом при 750°, нагреванием VG13 в токе азота (900°),
обработкой нагретого до 350° ванадия (или феррованадия) газооб-
разным HG1, действием цинка и соляной кислоты на ванадиевый
ангидрид V2O5, нагреванием VSi2 в атмосфере хлора:
VC14 4- Н2 = VC12 + 2НС1 V + 2НС1 = VC12 + Н2
2уС13 = VC12 + VC14 VSi2 + 5C12 = VC12 + 2SiCl4
V2O5 4- 3Zn 4- 10HC1 = 2VC12 4- 3ZnCl2 4- 5H2O
При получении VC12 диспропорционированием VG13 (нагрева-
ние в атмосфере азота или СО2) дихлорид ванадия остается в твер-
дой фазе, а летучий VC14 отгоняется.
Дихлорид ванадия представляет собой расплывающиеся на воз-
духе зеленые гексагональные кристаллы с плотностью 3,23 г/см3,
т. пл. 1000°, т. кип. 1377°; они растворяются в воде с образованием
фиолетового раствора, из которого выделяется водород, при этом
раствор окрашивается в зеленый цвет вследствие перехода вана-
дия(П) в ванадий(Ш):
VC12 4- Н2О = VOC1 4- НС1 4- i/2H2
Спиртовый раствор дихлорида ванадия синего цвета, а эфир-
ный — желто-зеленого.
Дихлорид ванадия является восстановителем. Он окисляется
на воздухе, в водей атмосфере HF, осаждает металлы Sn, Cu, Ag
из водных растворов их солей и восстанавливает при 700°
двуокись углерода:
VC12 4- 3HF = VF3 4- 2НС1 4- V2H2
VC12 4- SnCl2 4- H2O = Sn + VOC12 4- 2HC1
VC12 4- CuSO4 4- H2O = Cu + VOC12 4- H2SO4
3VC12 4- 2CO2 = 2VOC1 4- VC14 4- 2CO
Дибромид ванадия, VBr2, представляет собой коричнево-
желтые гексагональные кристаллы с т. пл. 827° и т. кип. 1227°,
получаются восстановлением трибромида ванадия в токе водорода.
Электролитическим восстановлением бромистоводородного
и сернокислого раствора ванадиевого ангидрида получают дибро-
мид ванадия в сернокислом растворе.
Дииодид ванадия, VI2, получают нагреванием VI3 в вакууме
при температуре выше 280°. VI2 образует фиолетово-розовые гек-
сагональные пластинки с плотностью 5,44 г/см3, т. пл. 777°
и т. кип. 927°; плохо растворим в абсолютном спирте, бензоле,
четыреххлористом углероде, сероуглероде. Дииодид ванадия суб-
154
ПОБОЧНАЯ ПОДГРУППА V ГРУППЫ
лимируется при 800° в вакууме и превращается в V2O5 нагрева-
нием в кислороде при 500°.
Сульфид ванадия, VS или V2S2, получают сильным нагрева-
нием V2S3 в атмосфере водорода в течение нескольких дней или
в результате диссоциации V2S3 в атмосфере азота при 850—950°.
Сульфид ванадия представляет собой черно-коричневый порошок
с плотностью 4,20 г/смя; он неустойчив на воздухе (превращается
в V2O5 при прокаливании на воздухе). Устойчив к действию раз-
бавленных кислот НС1, H2SO4 и др., растворяется в (NH4)2S,
образуя пурпурно-красный раствор, или в конц. H2SO4 (при
нагревании) с образованием зеленовато-желтого раствора.
Известен двойной сульфид Cu3VS4 кубической структуры.
Сульфат ванадия(1Г), [V(H2O)8]SO4-H2O, выделяется при испа-
рении над Р4О10 без доступа воздуха раствора, полученного элек-
тролитическим восстановлением с применением ртутного катода
(или восстановлением амальгамой натрия, амальгамой цинка,
Zn + НС1, Zn 4- H2SO4 и др.) раствора V2O5 в H2SO4 или вод-
ного раствора V2(SO4)3-nH2O:
V2O5 + 3Zn 4- 5H2SO4 + 30H2O =
= 2[V(H2O)6]SO4-H2O + 3[Zn(H2O)6]SO4-H2O
V2(SO4)3 + Zn + 21H2O = 2[V(H2O)6]SO4.H2O + [Zn(H2O)6]SO4-H2O
Моногидрат сульфата гексаквованадия представляет собой
моноклинные фиолетово-красные кристаллы (изоморфные с
сульфатами ферромагниевого ряда [Men(H2O)8]SO4-Н2О, где
Me11 = Fe2+, Mg2+, Zn2+, Go2+ и др.), неустойчивые на воздухе
и образующие с сульфатами щелочных металлов или сульфатом
аммония двойные сульфаты Me2SO4-VSO4‘6H2O с ограниченной
растворимостью в воде.
(NH4)2SO4-VSO4‘6Н2О выделяется в виде аметистовых моно-
клинных кристаллов при испарении раствора, полученного элек-
тролитическим восстановлением смеси сернокислого раствора
V2O5 с (NH4)2SO4.
Гексацианованадат(1Г) калия, K4[V(GN)8]-ЗН2О, выделяется
при добавлении спирта к раствору, полученному обработкой
растворов солей ванадия(П) избытком KCN, или к раствору,
полученному восстановлением ацетата ванадия(Ш) амальгамой
калия. Это легко окисляющиеся на воздухе желтовато-коричневые
призмы (хранение без доступа воздуха). Водные растворы
K4[V(CN)8]-ЗН2О с ионами Fe2+ окрашиваются в коричневый
цвет, с ионами Cd2+ — в серый, с ионами Сп2+ — в белый, с ионами
Мп2+ — в желтый цвет.
Кроме описанных соединений ванадия(П) известны селенид VSe,
теллурид VTe и хелатные соединения с этилендиаминтетрауксусной кис-
лотой.
ВАНАДИЙ
155
Соединение трехвалентного ванадия
Известны многочисленные соединения трехвалентного вана-
дия, в которых электроположительный трехвалентный ванадий
встречается в виде катионов V3+, [V(H2O)8]3+, [V(H2O)6GI]2+,
[V(H2O)4C12] + , VO+ (радикал ванадил) или анионов [VFe]3~,
[VF5(H2O)p-, [V(SO4)21-, [V(CN)el3-, [V(SCN)6]3-, [V(C2O4)3]3- и др.
Водные растворы солей ванадия(Ш) окрашены в зеленый
цвет, обладают отчетливо выраженными восстановительными свой-
ствами (окисляются в присутствии воздуха и КМпО4 и восста-
навливают соединения серебра, золота, платины или ртути).
Соединения ванадия(Ш) хранят без доступа воздуха. В присут-
ствии кислот или щелочей они разлагают воду с выделением
водорода.
Соединения ванадия(Ш) получают восстановлением соедине-
ний ванадия(У) или (IV), а также окислением соединений вана-
дия^!). Растворы V2O5 или VO2 в H2SO4 или НС1 могут быть
восстановлены до соединений ванадия(Ш) металлическим маг-
нием, НВт, HI или электролитическим путем.
Все соли ванадия(Ш) легко гидролизуются.
В отличие от координационных соединений железа(Ш) коор-
динационные соединения ванадия(Ш) менее устойчивы.
Неорганические соединения
Окись ванадия(111), V2O3, получают восстановлением окиси
ванадия(У) окисью углерода, водородом, цианистым калием,
серой, оксалатом аммония и др. при нагревании:
V2O5 + 2СО = V2O3 + 2СО2 V2O5 + 2Н2 = V2O3 + 2Н2О
V2O5 + 2KCN = V2O3 + 2KCNO
Окись ванадия представляет собой парамагнитный блестящий
черный кристаллический порошок с плотностью 4,87 г/см3 и твер-
достью 5,5 по шкале Мооса. Она тугоплавка (т. пл. 1967°, т. кип.
3027°), плохо растворима в воде, обладает слабо основным харак-
тером и растворяется в кислотах (HGI, HNO3 и др.) с образова-
нием солей. На воздухе V2O3 медленно окисляется до VO2, а при
нагревании на воздухе превращается в V2O5.
При нагревании V2O3 взаимодействует с хлором:
3V2O3 + 6С12 = V2O5 + 4VOCL,
При 2500° V2O3 восстанавливается водородом под давлением
5 ат, а при температуре выше 1200° V2O3 восстанавливается углем.
Сплавлением V2O3 с окислами двухвалентных (MgO, ZnO,
CaO, SrO) или трехвалентных (А12О3, Fe2O3, Сг,О3) металлов без
156
ПОБОЧНАЯ ПОДГРУППА V ГРУППЫ
доступа кислорода получают двойные окислы, которые имеют
структуру шпинели или перовскита:
V2O3 + FeO = Fe[V2O4] V2O3 + A12O3 = 2A1VO3
Окись ванадия применяется для получения цветных стекол,
а также в качестве катализатора при разложении первичных
спиртов и дегидрогенизации циклопентана.
Гидроокись ванадия, V(OH)3, получают обработкой солей вана-
дия(Ш) щелочью или NH4OH без доступа воздуха. V(OH)3 полу-
чается в виде зеленого хлопьевидного осадка, легко окисляюще-
гося на воздухе, обладающего слабо основным характером п пре-
вращающегося в соли ванадия(Ш) при растворении в кислотах
без доступа воздуха.
Трифторид ванадия, VF3, получают нагреванием VF4 при
600° в токе азота, нагреванием VG12 с безводным фтористым водо-
родом при 700° и пропусканием тока сухого, нагретого до 600°
фтористого водорода (и азота) над VC13 или VBr3 в платиновой
аппаратуре.
2VF4 = VF3 + VF5 VC13 + 3HF = VF3 + 3HC1
VC12 + 3HF = VF3 + 2HC1 + i/2H2 VBr3 + 3HF = VF3 + 3HBr
Трифторид ванадия представляет собой зеленовато-желтый
порошок с плотностью 3,36 г!см3, т. пл. 1127°, т. кип. 1427°; он пло-
хо растворим в воде и органических растворителях (С2Н5ОН, CS2
и др.) и восстанавливается водородом при 500°:
VF3 + 3/2Н2 = V 4- 3HF
При упаривании без доступа воздуха раствора, полученного
электролитическим восстановлением V2O5 в водном растворе HF
либо растворением V(OH)3 или V2O3 в HF, образуются темно-
зеленые ромбические кристаллы VF3-3H2O или [VF3(H2O)3],
растворимые в теплой воде и плохо растворимые в абсолютном
спирте.
При 130° соединение [VF3(H2O)3] превращается в VF3 и V2O5.
Трифторид ванадия с фторидами многих металлов образует
зеленые комплексные соли типа MeI[VF4(H2O)2], Me*[VF5(H2O)l,
Me’ [VF6], где Me1 = NHJ, K+, Tl+; [Me11 (H2O)6][VF5(H2O)], где
Me11 = Zn2+, Cd2+, Ni2+, Fe2+.
Трихлорид ванадия, VG13, получают нагреванием тетрахлорида
ванадия при 150—160° в атмосфере азота или двуокиси углерода,
нагреванием порошкообразного металлического ванадия с избыт-
ком IG1, пропусканием газообразного HG1 над металлическим
ванадием (феррованадием), нагретого до 300—400°, перегонкой
при 150° в токе СО2 продукта, полученного пропусканием теплого
ВАНАДИЙ
157
хлора над V2S3, нагреванием смеси VOC13 с серой:
2VC14 2VC13 + Cl2 V + ЗНС1 = VC13 + 3/2Н2
V + 3IC1 = VC13 + 3/2I2 2VOC13 + S = 2VC13 + S02
Трпхлорид ванадия представляет собой расплывающиеся на
воздухе блестящие красновато-розовые пластинки с плотностью
3,00 г'см?-, они устойчивы в атмосфере газообразного СО2, раство-
рение их в воде сопровождается гидролизом. Соль растворима
в органических растворителях (спирте, эфире, ацетоне, бензоле,
CS2), обладает восстановительными свойствами по отношению
к многочисленным органическим веществам, восстанавливает при
700° СО2 до СО.
При нагревании в атмосфере азота или двуокиси углерода
трихлорид ванадия диссоциирует:
120-130° 150-160°
2VC1? . -----* УСП + УСЬ 2VC14 ,---------2УС13+С12
При более высокой температуре образующийся VC12 реагирует
с СО2 по уравнению, приведенному при описании дихлорида
ванадия.
При повышенной температуре VC13 восстанавливается водо-
родом до дихлорида ванадия, а затем до металлического ванадия.
С жидким или газообразным аммиаком VC13 реагирует по
уравнениям
VC13 + 4NH3 (газ) = VN + 3NH4C1
VC13 + 6NH3 (жидк.) = [V(NH3)6]C13
Хлорид гексамминванадия(111), [V(NH3)e]Cl3, представляет
собой твердое красно-коричневое вещество, плохо растворимое
в теплой воде, спирте и эфире, окисляющееся на влажном воздухе
и взаимодействующее с разбавленной НС1:
3[V(NH3)6]C13 -|- 12Н2О 3/2О2 = (NH4)3V30g 9NH4C1 6NH4OH
[V(N Н3)6]С13 + 6НС1 + 6Н2О = [V(H2O)6]C13 + 6NH4C1
Хлориды щелочных металлов образуют с VC13 комплексные
соли, например K[VC14], K2[VC15].
При упаривании в вакууме зеленого раствора, полученного
растворением трихлорида ванадия без доступа воздуха в воде,
подкисленной разб. НС1, солянокислых растворов V2O3 или
V2O5, восстановленных на платиновом катоде, выделяются зеленые
кристаллы хлорида гексаквованадия(Ш) [V(H2O)6]C13, образую-
щего три гидратных изомера, которые при прокаливании на воз-
духе превращаются в V2O5 и VOC13:
3[V(H2O)e]Cl3 + О2 = У2О5 + VOC1 + 8НС1 + 14Н2О
158
ПОБОЧНАЯ ПОДГРУППА V ГРУППЫ
Известен очень неустойчивый темно-фиолетовый аддукт
VCl3-2CeHe, растворяющийся в эфире с образованием синего
раствора.
Оксихлорид ванадия (монохлорид ванадила), VOC1, получают
гидролизом трихлорида ванадия в воде, нагреванием VG13 или
VC12 в атмосфере газообразного СО2 при 700°, нагреванием
VOG13 или VOC12 в атмосфере водорода, а также прокаливанием
кристаллогидрата [V(H2O)6]C13:
VC13 + Н2О VOC1 + 2НС1 VOC12 4- i/2H2 = VOC1 -- НС1
VOC13 + Н2 = VOC1 + 2НС1 [V(H2O)6]C13 = VOC1 + 2НС1 - 5Н2О
Оксихлорид ванадия представляет собой желто-коричневый
порошок с плотностью 3,6 г/см3 и т. кип. 127°; он плохо растворим
в воде и легко растворяется в разб. HNO3.
Трибромид ванадия, VBr3, получают прямым взаимодействием
элементов при нагревании или обработкой нитрида либо карбида
ванадия сухим бромом при нагревании:
V + 3/2Вг2 = VBr3 + 120 ккал VN + 3/2Br2 = VBr3 + i/2N2
VC + 3/2Вг2 = VBr3 + С
Трибромид ванадия образует расплывающиеся на воздухе
зеленовато-черные кристаллы, растворимые в спирте, эфире,
ацетоне, бензоле, сероуглероде, плохо растворимые в НВт и раз-
лагающиеся при нагревании. Водные растворы VBr3 подвергаются
гидролизу.
При упаривании в вакууме раствора, полученного взаимо-
действием V(OH)3 и НВг (без доступа воздуха) или электролити-
ческим восстановлением раствора V2O5 в конц. НВг, выпадают
зеленые кристаллы [V(H2O)8]Br3.
При упаривании в вакууме раствора безводного трибромида
ванадия в жидком аммиаке выпадает красно-коричневый бромид
гексамминванадия(Ш).
Известно устойчивое сине-зеленое соединение
[V{GO(NH2)2}6]Br3, образующееся при взаимодействии мочевины
с трибромидом ванадия.
Оксибромид ванадия, (монобромид ванадила), VOBr, получают
нагреванием VOBr2 в вакууме при 360°. Он представляет собой
фиолетовые октаэдрические кристаллы с плотностью 4,0 г/смя,
плохо растворимые в воде, ацетоне, хлороформе, толуоле, раз-
лагающиеся при 480° в вакууме по уравнению
3VOBr = VBr3 4- V2O3
Трииодид ванадия, VI3, получают прямым взаимодействием
элементов в вакууме при 150—280°. Это сильно гигроскопичный
темно-зеленый кристаллический порошок, который растворяется
в воде, гидролизуясь, растворяется в абсолютном спирте, не извле-
кается такими растворителями, как G6He, GG14, CS2, окисляется
ВАНАДИЙ
159
на воздухе при 130°, взаимодействует с хлором, превращаясь
в VC14 и 1С13, разлагается при 280° на VI2 и 12.
Известно устойчивое окрашенное в зеленый цвет соединение
[V{CO(NH2)2}8]I3, которое получают из безводного трииодида
ванадия и мочевины.
При упаривании в вакууме раствора, полученного электро-
литическим восстановлением раствора V2O5 в HI без доступа
воздуха, выпадает зеленый порошок VI3-6H2O (или [V(H2O)e]I3),
расплывающийся на воздухе, растворимый в спирте, воде и дру-
гих растворителях.
Сульфид ванадия, V2S3, получают нагреванием окиси V2O3
или VO2 в токе H2S либо нагреванием V2O5 при 700° в парах CS2.
Сульфид ванадия представляет собой черный порошок или
зеленовато-черные пластинки с плотностью 4,7 г!см9, плохо рас-
творимые в воде, щелочах, НС1, растворимые в HNO3 и сульфидах
щелочных металлов. При нагревании на воздухе, в водороде или
парах серы V2S3 реагирует по уравнениям
V2S3 + ii/2O2 = V2O5 + 3SO2
1200°
V2S3 + H2---> 2VS + H2S
400°
V2S3 + 2S---> V2S5
При растворении V2S3 в (NH4)2S образуется красный раствор,
а в K2S — фиолетовый.
Сульфаты ванадия (III). Дисулъфатованадиевая кис-
лота, H[V(SO4)2]-6H2O или V2(SO4)3-H2SO4-12H2O, выделяется
при концентрировании зеленого раствора, полученного восста-
новлением цинком раствора V2O5 в H2SO4. Нагреванием
H[V(SO4)2]-6Н2О при 180° в атмосфере газообразного СО2 полу-
чают безводную соль V2(SO4)3 в виде желтого порошка, трудно
растворимого в спирте и эфире и растворимого в воде с образо-
ванием зеленого раствора.
Безводный сульфат ванадия, V2(SO4)3, при нагревании в вакуу-
ме (примерно при 410°) превращается в сульфат ванадила по реак-
ции
V2(SO4)3 = 2VOSO4 + SO2
а выше 410° получается V2O5.
Известно несколько кристаллогидратов сульфата ванадия(Ш)
V2(SO4)3-nH2O (где п = 3, 4, 9, 10 или И), которые с сульфатами
щелочных металлов или аммония образуют соединения типа
квасцов MeIV(SO4)2-12Н2О.
Квасцы с ванадием в качестве трехвалентного элемента —
достаточно устойчивые фиолетовые октаэдрические кристаллы,
которые в водном растворе под действием кислорода воздуха
медленно окисляются. Несколько примеров квасцов с трехвалент-
160
ПОБОЧНАЯ ПОДГРУППА V ГРУППЫ
ным ванадием: KV(SO4)2-12Н2О (октаэдрические фиолетовые кри-
сталлы с плотностью 1,782 г/см3), RbV(SO4)2-12H2O (октаэдриче-
ские фиолетовые кристаллы с плотностью 1,915 г/см3),
CsV(SO4)2 -12Н2О (октаэдрические фиолетово-аметистовые кристал-
лы с плотностью 2,033 г/см3), T1V(SO4)2-12H2O (октаэдрические
красно-фиолетовые кристаллы), NH4V(SO4)2- 12Н2О (октаэдриче-
ские синие кристаллы).
Известны также соединения дисульфатованадиевой кислоты
с пиридином или гуанидином; зеленый H[V(SO4)21-(G5H5N)-3H2O,
фиолетовый H[V(SO4)2]-(CN3H5)2.
Гексацианованадат калия, K3[V(CN)e], осаждается при добав-
лении спирта к растворам, полученным при обработке растворов
солей ванадия(Ш) избытком KCN. Он представляет собой твердое
вещество, окрашенное в красный цвет (аналогичное K3[Fe(CN)6]),
растворимое в воде и плохо растворимое в спирте; при кипячении
со щелочами превращается в V(OH)3:
K3[V(CN)6] + ЗКОН = V(OH)3 + 6KCN
Хелатные соединения
При обработке трихлорида ванадия ацетилацетоном в при- '
сутствии карбоната натрия получают комплекс
который выделяется из раствора в виде темно-зеленых призм,
плавящихся при 185—190°, растворимых в воде, хлороформе,
бензоле и превращающихся на воздухе в
При действии бензоилацетона на трихлорид ванадия в при-
сутствии карбоната натрия получают комплексные хелаты
ВАНАДИЙ
161
При охлаждении фиолетового раствора, который получается
нагреванием в присутствии следов кислорода зеленого раствора,
полученного обработкой трихлорида ванадия с пирокатехином
в атмосфере азота, выпадают кристаллы (ХН4)з[У(С6Н4О2)31 ^ЩО.
Трибромид, трииодид или перхлорат ванадия(Ш) с мочевиной
образуют, как уже упоминалось, хелатные соединения общей
формулы [V{GO(NH2)2}e]X3—устойчивые, окрашенные в зеле-
новато-синий цвет.
Известны также хелатные соединения ванадия(Ш) с о-нитро-
зофенолом, и-нитрофенолом и нитрозо-Р-нафтолом.
Помимо описанных известны и другие соединения вападия(Ш), напри-
мер: гекса(тиоцпанато)вападаты Na3[V(SCN)6]-12Н2О, K3[V(SCN)c] -4Н2О,
(NTI4)3[V(SCN)6]-4Н2О, тетра(тиоцианато)производные [V(SCN)4(C5H5N)2] •
• C5H5N, [V(SCN)4(OH)2](C5H5NH)3, формиаты, ацетат V(CH3COO)3, окса-
латы Ме3 [V(C2O4)3]-ЗН2О, малонаты Мез[У(С3О4Н2)3] (где Mei = Na+, К+,
NHJ), тартраты, салицилаты и т. д.
Соединения четырехвалентного ванадия
Известны многочисленные соединения ванадия(1У), в которых
электроположительный четырехвалентный ванадий может вхо-
дить в состав анионов VO2-, V4O,~, [VOF5]3-, [VOF4(H2O)]2~,
[VO(SO4)2]2-, IV()(SCN)4]2', [VO(C2O4)2]2- или катионов VO2+
(радикал ванадил), V2O4+ (радикал диванадил) и реже V4+ или
tV(H2O)eB+.
Соли ванадия(1У) получаются восстановлением соединений
ванадия(У) (электролитическим или химическим путем — с водо-
родом, HNO2, H2SO3, H2S, НС1, HBr, HI), окислением кислородом
воздуха кислых растворов соединений ванадия низших валент-
ностей, растворением VO2 или V(OH)4 в кислотах.
В твердом состоянии соединения ванадия(1У) окрашены в ко-
ричневый, зеленый или синий цвета, а в водном растворе они
окрашены в синий цвет, обладают восстановительным характером
(например, относительно КМиО4 в кислой среде или КЮ3 в сильно
щелочной среде), устойчивы на воздухе и гидролизуются в воде:
10K2[VO(SO4)2J -I- 2КМпО4 + 10Н2О - K4[H2Vi0O28] + 2MnSO4+18KHSO4
6K2[VO(SO4)2] у KIO3 4- 24КОН = 3K4V2O7 + KI + 12K2SO4-L12H2O
В отличие от ванадия(П) и (III) у соединений ванадия(1У)
наблюдается более низкая основность и более резко выраженная
склонность к образованию координационных или двойных соеди-
нений.
Соединение V(OH)4 — настолько слабое основание, что заме-
щение четырех групп ОН- кислотными радикалами затруднено,
поэтому при обработке V(OH)4 кислотами образуются соли ради-
кала ванадила VO2+ или диванадила V2Oj+.
11—0101
162
ПОБОЧНАЯ ПОДГРУППА V ГРУППЫ
Из-за большой способности к комплексообразованию не изве-
стны соединения VBr4, VOSO3, VOC2O4, VOCO3, а только коор-
динационные соединения последних. Большинство комплексных
ионов ванадия (IV) содержит группу VO2+.
Неорганические соединения
Двуокись ванадия, VO2 или V2O4, получают сплавлением V2O5
с щавелевой кислотой, нагреванием без доступа воздуха экви-
молярной смеси V2O3 и V2O5, высушиванием в вакууме или в токе
двуокиси углерода соединения V(OH)4:
V2O5 + Н2С2О4.2Н2О = 2VO2 + 2СО2 + ЗН2О
V2O3 + V2O5 = 4VO2 V(OH)4 = VO2 + 2H2O
Двуокись ванадия образует темно-синие кристаллы (с кри-
сталлической решеткой, аналогичной рутилу) с плотностью
4,40 г/см\ плавящиеся при 1545° в атмосфере азота, кипящие
с разложением при 2727°, амфотерного характера, трудно рас-
творимые в воде и растворимые в кислотах или щелочах с образо-
ванием солей ванадила и ванадитов:
VO2 + 2НС1 = VOC12 4- Н2О 4VO2 + 2КОН = K2V4O9 + Н3О
При 530—600° водород восстанавливает VO2 по уравнению
2VO2 + Н2 V2O3 + Н2О
Двуокись ванадия окисляется при нагревании на воздухе
или под действием азотной кислоты.
Ортофосфорная кислота образует с VO2 следующие соединения
(синего цвета): VO2-2H3PO4, 2VO2'6Н3РО4-Н2О.
Известны гидраты: VO2-H2O (зеленый) в виде VO(OH)2 —
гидроокись 'ванадила или (H2VO3)2 — ванадиевая(1У) кислота
(димер); VO2-2H2O (синий) в виде V(OH)4 — гидроокись вана-
дия(1¥); 2VO2-H2O (черно-синий); 2VO2-7H2O.
Ванадиты Me^V4O9-wH2O, MexV2O8, MeIH3V2O8, в которых
Me1 = NH*, Na+, K+, x/2Ba2+, Ag+, x/2Pb2+, x/2Mn2+ и др. (рас-
сматриваются как соли кислот H2V4O9, H4V2O8), легко окисляют-
ся в ванадаты и превращаются в соли ванадила под действием
кислот:
Na2V4O9 + 10НС1 = 4VOC12 + 2NaCl + 5Н2О
Ванадиты щелочных металлов образуются при растворении
VO2 или V(OH)4 в щелочах или нагреванием растворов солей вана-
дила с избытком щелочей:
4V(OH)4 + 2КОН = K2V4O9 + 9Н2О
4VOC12 + lOKOH = K2V4O9 + 8KC1 + 5H2O
ВАНАДИЙ
163
Примеры ванадитов: (NH4)4V2O6 — зеленый, NH4H3V2O8 — ко-
ричневый, K2V4O9 — темно-зеленый, BaV4O9.5H2O — коричне-
вый, Na2V4O9*5H2O — зеленый.
Тетрафторид ванадия, VF4, получают действием плавиковой
кислоты на тетрахлорид ванадия при низкой температуре (от —28
до 0°) в платиновой цосуде:
VC14 + 4HF = VF4 + 4НС1
Тетрафторид ванадия представляет собой гигроскопичный
желто-коричневый порошок с плотностью 2,97 г/cjt3; он диспро-
порционирует в атмосфере азота при 325° на VF3 и VF3, раство-
ряется в ацетоне с образованием зеленого раствора, очень плохо
растворим в спирте и хлороформе. Растворение в воде сопровож-
дается гидролизом, цвет раствора — синий.
VF4 + Н2О = VOF2 4- 2HF
При обработке тетрафторида ванадия водными растворами фто-
ридов некоторых металлов получают комплексные оксифториды,
например K2[VOF4], (NH4)3[VOF5], или оксофтороаквосоли типа
Me*[VOF4(H2O)], [Меп(Н2О)6], [VOF4(H2O)],' где Me1 = Na+, К+,
NH+4, Т1+ и Me11 = Ni2+, Zn2+.
Дифторид ванадила [оксифторид ванадия(1У)], VOF2, полу-
чают нагреванием VOBr2 при 600—700° в токе фтористого водо-
рода.
Дифторид ванадила представляет собой желтый порошок с плот-
ностью 3,396 г/см?, растворимый в ацетоне и трудно растворимый
в других растворителях.
При упаривании растворов четырехвалентного ванадия, содер-
жащих HF (в присутствии конц. H2SO4), выделяются желтые
кристаллы в виде призм VOF2raH2O.
Тетрахлорид ванадия, VC14, получают нагреванием порошко-
образного металлического ванадия, феррованадия, нитрида VN,
карбида VC или силицида VSi2 в атмосфере сухого газообразного
хлора, при пропускании хлора и паров VOC13 над активирован-
ным углем, нагретым до красного каления, а также действием
хлористого тионила SOC12, сульфурила 8О2С12 или фосгена СОС12
на металлический ванадий:
V + 2С12 = VC14 + 141 ккал VOC13 + У2С12 + С = VC14 + СО
VN + 2С12 = VC14 4- V2N2 V 4- 2SO2C12 = VC14 + 2SO2
VC + 2C12 = VC14 + С V + 2COC12 = VC14 + 2CO
Тетрахлорид ванадия — самое устойчивое соединение четырех-
валентного ванадия; это маслянистая дымящая на воздухе красно-
коричневая жидкость с плотностью 1,82 г/ел3; затвердевает при
—26°, кипит при 148,5°, температура разложения 164°. Тетра-
хлорид растворяется в CS2, СС14, СНС13, С8Н8, С2Н5ОН,
11*
164
ПОБОЧНАЯ ПОДГРУППА V ГРУППЫ
С2Н5 — О — С2Н5 и воде. При растворении VC14 в спирте раствор
окрашивается в синий цвет, в этиловом эфире — в красный цвет,
а в воде образуется раствор VOC12, окрашенный в синий цвет
вследствие гидролиза:
VC14 + Н2О VOC12 + 2НС1
Тетрахлорид VC14 может служить хлорирующим агентом.
Дихлорид ванадила [оксихлорид ванадия(1¥)], VOC12, полу-
чают восстановлением VOC13 водородом или металлическим цин-
ком при нагревании, обработкой конц. НС1 пятиокиси ванадия,
действием цинка и разб. НС1 на ¥2О5, гидролизом тетрахлорида
ванадия:
2VOC13 + Н2 = 2VOCL, + 2НС1 V2O5 + 6НС1 = 2VOC12 + ЗН2О + С12
2VOC13 + Zn = 2VOCL, + ZnCl2 V2O5 + 6HC1 + Zn =2VOCl2-[-ZnCl2+3H2O
Дихлорид ванадила представляет собой расплывающиеся на
воздухе зеленые пластинки, разлагающиеся в воде и растворяю-
щиеся в разбавленных кислотах (HNO3, СН3СООН и др.) и спирте.
При действии воды на VOC13 или газообразного НС1 на Na3¥O4
при 440° получают темно-зеленую жидкость состава VOC12 •
• 2,5Н2О с плотностью 1,61 г/см?‘, водные растворы ее окрашены
в синий цвет:
Na3VO4 + 6НС1 = VOC12 + 3NaCl + V2C12 + 3H2O
Известны двойные соли дихлорида ванадила с двумя или
четырьмя молекулами гидрохлорида пиридина (C5H5NH)2[VOC14] •
• иН20 или гидрохлорида хинолина, как и VOCl2-PtCl4-10,5Н2О
или VO[PtCl6] -Ю,5Н2О.
Известны также зеленые кристаллы оксихлорида V2O3C12 •
4Н2О.
Тетрабромид ванадия, VBr4, не известен.
Двойной бромид ванадияДУ) и суръмыДН), VBr4-SbBr3-7Н2О,
выделяется при изотермическом обезвоживании над конц. H2SO4
раствора пятиокиси ванадия и трибромида сурьмы в бромистово-
дородной кислоте:
V2O5 + 2SbBr3 -J- ЮНВг 9Н2О = 2(VBr4 -SbBr3-7Н2О) + Br2
Соединение образует гигроскопичные черные призмы, которые
при растворении в НС1 окрашивают раствор в зеленый цвет.
Водный раствор соединения VBr4-SbBr3-7H2O неустойчив.
Дибромид ванадила {оксибромид ванадияДУ)], VOBr2, полу-
чают термическим разложением VOBr3 при 180° или действием
паров брома на смесь V2O5 с серой. Синтез проводится в нагретой
до 500—600° трубке для сожжения из тугоплавкого стекла.
Дибромид ванадила представляет собой расплывающийся на
воздухе желто-коричневый порошок, который растворяется в хо-
ВАНАДИЙ
165
лодной воде, образуя синий раствор, и разлагается при нагре-
вании:
360°
VOBr2 ---------->- VOBr + i/2Br2
вакуум
Нагревание
2УОВг2 + 3/2О2 воздух--------------->- ¥2О3 + 2Вг2
Тетраиодид ванадия, VI4, не известен.
Дииодид ванадила [оксииодид ванадияДУ)], VOI2 -4,51120, выде-
ляется в виде синих кристаллов при испарении в вакууме рас-
твора V2O5 в иодистоводородной кислоте.
Дисульфид ванадия, VS2, получают при добавлении H2SO4
к окрашенным в пурпурный цвет растворам солей ванадия(1¥)
в (NH4)2S.
Дисульфид ванадия представляет собой твердое коричневое
вещество, плохо растворяющееся в кислотах и растворяющееся
в щелочах или карбонатах щелочных металлов.
Известны тиованадаты общей формулы Me(S -2V0S пурпур-
ного цвета.
Сульфаты ванади я(1У). Сульфат ванадила, NOSO,,,
серовато-зеленого цвета получают нагреванием при 260° смеси
из 2VOSO4-H2SO4 и конц. H2SO4.
Сульфат ванадила существует в виде двух модификаций,
а именно: синяя, растворимая в холодной воде, и серовато-зеленая,
плохо растворимая в холодной воде. Нагреванием серовато-зеле-
ной формы при 130° в парах воды получают синюю модификацию.
Сульфат ванадила гидролизуется в воде. С сульфатами щелоч-
ных металлов образует комплексные соединения общей формулы
Mei[VO(SO4)2], Mei[V2O2(SO4)3], в которых четырехвалентный
ванадий входит в состав радикала ванадила VO2+ или диванадила
V2O‘+:
VOSO4 + H2O VO2 + H2SO4
VOSO4 + K2SO4 = K2[VO(SO4)2]
Темно-синий
2VOSO4 + K2SO4 = K2[V2O2(SO4)3]
Темно-синий
При нагревании до 610° сульфат ванадила диссоциирует:
2VOSO4 V2O5 + SO3 + SO2
Известны следующие гидраты сульфата ванадила: VOSO4 •
• 2Н2О, VOSO4-3H2O, VOSO4-5H2O, 2VOSO4-7H2O, 2VOSO4 •
• 13H2O.
Соединение 2VOSO4-H2SO4 (или кислоту H2[V2O2(SO4)3]) по-
лучают обработкой V2O5 серной кислотой:
V2O5 + 3H2SO4 = H2[V2O2(SO4)3] + 2Н2О + V2O2
166
ПОБОЧНАЯ ПОДГРУППА V ГРУППЫ
Известны сульфатные комплексы, содержащие как четырех-,
так и пятивалентный ванадий.
Хелатные соединения
Ацетилацетонат ванадила, [УО(С5Н7О2)г],
получают обработкой сульфата ванадила ацетилацетоном или
окислением на воздухе ацетилацетоната ванадия(Ш).
Ацетилацетонат ванадила представляет собой сине-зеленые
кристаллы, плохо растворимые в воде и легко растворимые в спир-
те, эфире, бензоле и хлороформе.
При обработке хлорида или гидроокиси ванадила бензоил-
ацетоном получают бензоилацетонат ванадила
плавящийся при 215° и легко образующий аддукты с пиридином,
пиперидином, пиколином, хинолином и анилином.
Аддукт пиридина
зеленого цвета (т. пл. 163°), пиколина — желтого цвета (т. пл. 146°),
хинолина — зеленовато-желтого (т. пл. 184°), анилина — серова-
то-желтого (т. пл. 212°).
Растворы солей ванадила с пирокатехином в щелочной среде
образуют красивые прозрачные коричневые кристаллы общей
формулы Ме*[УО(СвН4О2)21- С8Н4(ОН)2 (где Me1 == NH+, К+).
Помимо вышеприведенных известны и другие соединения V(IV), напри-
мер: VO(C1O4)2, VO1NO3)2, VOSeO3-2H2O, VO(CN)2, K2[VO(CN)4] • 5H2O,
K2[VO(SCN)4]-5H2O, (NH4)2[VO(SCN)4]-5H2O, (VO)3[Bi(SCN)6]2 • 7H2O,
Mel [VO(SO3)2].2HzO (где Mel = NHj, K+, Na+), VO(H2PO2)2-H2O,
(VO)2(AsO3)4-3H2O, (NH4)2[VO(C2O4)2].2H2O, (NH4)2[V2O2(C2O4)3]-5 —8H2O,
K2[V2O2(C2O4)3] -4H2O, (NH4)2[VO(OOC - CH2 - COO)2] -4H2O,
(NH4)2[VO(OOC - CH2 - COO)2]-3H2O.
ВАНАДИЙ
167
Соединения пятивалентного ванадия
Наиболее многочисленными и важными устойчивыми соеди-
нениями, природными и синтезированными, являются соединения
пятивалентного ванадия.
Соли ванадия(У) могут содержать радикалы [VO2]+, [VO]3+,
катион V5+ (в VF5) и различные анионы: [VO4]3~, [HVOJ2-,
IV2O7]4-, [V3O9]3-, [VO3]-, [V10O28]6~, [HV10O28]5-, [H2V10O28]*-,
(H3V10O28]3-, [VOFJ-, [VF6]-, IVO2(C2O4)213-, VS;, VS3;, [VOS3]3-,
[VO3S]3-, [V2OS8]4-, [V2O2S5]4~, [V2o5s2]4-. Среди соединений
ванадия^) следует упомянуть VFS, V2O5, VOX3 (где X == F~,
Cl-, Br-), ванадаты, изополиванадаты, гетерополиванадаты, перо-
ксованадаты.
В отличие от соединений двух-, трех- и четырехвалентного
ванадия соединения пятивалентного ванадия обладают ярковы-
раженными кислотными свойствами (V2O5 — ванадиевый ангид-
рид) и склонностью к комплексообразованию и конденсации.
Восстановлением соединений ванадия^) различными восста-
новителями (цинком в кислой среде, H2S, FeSO4, SnCl2, SO2,
СгС12 и др.) получают соединения четырех-, трех- и двухвалент-
ного ванадия, которые неустойчивы и под действием кислорода
воздуха снова превращаются в соединения пятивалентного вана-
дия:
Fe2+ Sn2+, Ti3+, SO2 Сг2+, Zn
VOJ------> VO2+------------► V3+-------► V2+
Бесцветный Синий Зеленый Фиолетовый
— 0,255e +0,3e +1,0в
Примеры окислительно-восстановительных реакций:
V2O5 + Zn + 6НС1 = 2VOC12 + ZnCl2 + 3H2O или
2VOC13 -f- Zn = 2VOC12 + ZnCl2 2VC13 + Zn = 2VC12 -f- ZnCl2
2VOCI3 + 2Zn + 4HC1 = 2VC13 + 2ZnCl2 + 2H2O
Неорганические соединения
Пятиокисъ ванадия (ванадиевый ангидрид), V2O5, получают
при сжигании порошкообразного металлического ванадия в кис-
лороде под давлением, прокаливанием на воздухе или в кислороде
окислов VO, V2O3, VO2, нитрида VN или триметаванадата аммо-
ния (NH4)3V3O9, а также при гидролизе трихлорида или трибро-
мида ванадила:
2V + 5/2О2 = V2O5 + 373,3 ккал 2VO2 + V2O2 = V2O5 + 28,2 ккал
2VО -|- 3/2О2 — V2O5 V2O3 -|- О2 = V2O5 87,80 ккал
2VN + 6/2О2 = V2O5 + N2
2(NH4)3V3O9 = 3V2O5 + 6NH3 + 3H2O
2VOC13 + 3H2O V2O5 + 6НС1
168
ПОБОЧНАЯ П0ДГРУПИ4 V ГРУППЫ
Подкислением водных растворов ванадатов щелочных металлов
до pH ~ 2 получают объемистый осадок (гелеобразный) V2O5 •
• пН2О с окраской от желтовато-красного до красно-коричне-
вого. Пятиокись в воде дает темно-красный коллоидный раствор,
растворяется в щелочах или кислотах с образованием солей.
Ванадиевый ангидрид представляет собой токсичный оран-
жевый порошок с т. пл. 670° и т. кип. 1827°; он плохо растворим
в абсолютном спирте, ограниченно растворим в воде.
При охлаждении расплавленного ванадиевого ангидрида выде-
ляются красноватые ромбические кристаллы V2O5 с плотностью
3,34 г/см3, обнаруживающие слабо парамагнитные свойства.
Пятиокись ванадия ведет себя как амфотерное вещество (преоб-
ладают кислые свойства): легко растворяется в щелочах и кон-
центрированных кислотах. При растворении V2O5 в избытке
холодной щелочи получаются орто- и пированадаты, а при нагре-
вании — триметаванадаты:
V2O5 + 6NaOH = 2Na3VO4 + ЗН2О
V2O5 + 4NaOH = Na4V2O7 + 2H2O
3V2O5 + 6NaOH = 2Na3V3O9 + 3H2O
Ортованадат-анион VO’“ в концентрированных растворах легко
превращается на холоду в пированадат-анион (или диванадат)
V2O74-, который при нагревании переходит в полимеризованный
метаванадат-анион iyQ~3)n. Переход мономерного ортованадата
натрия в полимер метаванадата натрия осуществляется следую-
щим образом:
nNaH2VO4-------->. -------> (NaVO3)n
-»/2н2о — n/2Na2H2V2O7 -»/2н2о
Если к растворам ортованадатов щелочных металлов добав-
ляют NH4C1, осаждается триметаванадат аммония:
3Na3VO4 + 9NH4C1 = (NH4)3V3O9 + 9NaCl + 3H2O + 6NH3
Растворением V2O5 в конц. НС1 при нагревании получают
VOC12, при этом выделяется хлор:
V2O5 + 6НС1 2VOC12 + ЗН2О + С12
Здесь V2O5 является окислителем.
Если V2O5 нагревать с сухим НС1 в присутствии Р4Ою, обра-
зуется VOC13:
V2O5 + 6НС1 2VOC13 + ЗН2О
Обработкой сульфатами щелочных металлов продуктов, обра-
зовавшихся при восстановлении раствора V2O5 в H2SO4 газообраз-
ным SO2, металлическим цинком или восстановлением на катоде,
получают комплексные сульфаты — квасцы или двойные суль-
ВАНАДИЙ
169
фаты ванадила, а также соединения, содержащие ванадий низших
степеней окисления.
V2O5 + 2H2SO4 + SO2 = 2VOSO4-H2SO4 + H2O
H2[V2O2(SO4)3]
| При 260°,
i над конц. H2SO4
voso4
+MeISO4
•'
Me2[VO(SO4)2]-raH2O
V2O5 + 5H2SO4+ Zn-|- 2H2O = 2H[V(SO4)2] -f- [Zn(H2O)8]SO4 • H2O
| 80°
i в CO2 (газообр.)
V2(SO4)3
| +Melso4
MeIV(SO4)2-12H2O
V2O5 + 5H2SO4 + 3Zn + 30H2O =
= 2[V(H2O)6]SO4-H2O + 3[Zn(H2O)6]SO4- H2O
| +2MeISO4
2(VSO4-Me2SO4-6H2O)
При’нагревании V2O5 взаимодействует с водородом, двуокисью
серы, окисью углерода, теллуром, хлором, хлористым тионилом
по уравнениям:
600°
+ ► V2O3-j-2H2O V2O5~|-Te V2O3-J-TeO2
1700° 650°
V2O5-h3H2------>- 2VO + 3H2O V2O5+3C12---------> 2VOCl3+3/2O2
75°
V2O5+SO2 2VO2 + SO3 V2o5 + 3SOC12-----► 2VOC13 + 3SO2
При сплавлении V2O5 co щелочами или различными окислами
металлов можно получить мета-, орто-, пиро- или поливанадаты.
Под действием перекиси водорода V2O5 в водном растворе
превращается в пероксованадиевые кислоты.
Пятиокись ванадия применяется при изготовлении стекла,
задерживающего ультрафиолетовые лучи, а также в качестве
катализатора при окислении двуокиси серы в трехокись серы
(получение H2SO4 контактным способом), при окислении спирта,.
170
ПОБОЧНАЯ ПОДГРУППА V ГРУППЫ
гидрогенизации олефинов, производстве анилинового черного
и др.
Ванадаты и поливанадаты. Из диаграммы состоя-
ния ванадатов в водных растворах (рис. 7), составленной в 1961 г.
Шиллером и Тило на основе результатов спектрофотометрических
исследований, вытекает, что в зависимости от значений концен-
траций и pH раствора могут существовать ионы [VOJ3-, [HVOJ2-,
IV2O7J2-, [V3O9]3-, [VO3]-, [V10O28Je-, [HV10O28J5-, [H2V10O28?-,
VO2+.
Рис. 7. Диаграмма состояния ванадатов в водном растворе.
В сильно щелочных растворах ванадатов при pH = 14—13,2
•существует ион VO3’; при pH 13,2 и 8,4 в разбавленных растворах
ванадатов существует анион HVO4~; в концентрированных раство-
рах присутствует пиро- или диванадат-анион V2O*“, образующий-
ся по уравнению:
2VO^- + 2Н+ У2(Д- + Н2О 2HVO1- ^V2O4< + Н2О
При уменьшении pH концентрированных растворов орто- или
пированадатов до значения 7,2 образуется триметаванадат-анион
(¥3О’_)и в разбавленных растворах — монометаванадат-ион VO':
3V2O*~ + 6Н+ 2V3O^~ + ЗН2О V3O3- =₽«= 3VO3
Смещение в ту или другую сторону равновесия между ионом
V3Og~ и VO3 зависит от концентрации ионов.
При pH ниже 7,2 в концентрированных растворах образуется
декаванадат [V10O28]6-, [HV10O28]s_, [H2V10O28]4-, а в разбавлен-
ных растворах — ионы VO3:
10V3O|- + 12Н+ 3[V10O28]e- + 6Н2О
Декаванадиевые ионы обнаруживают л'максимальную устой-
чивость при pH == 4,1.
ВАНАДИЙ
171
В сильно кислых растворах содержание ионов VO2 быстро
увеличивается до установления равновесия между декаванадат-
ионами и ионами ванадила, которое определяется концентрацией
и pH раствора:
[Vi0O28]«- + 16Н+ 10VO2+ + 8Н2О
На основании точек перегиба на кривых потенциометрического
титрования или разрывов на кривых кондуктометрического тит-
рования ванадатов при pH в интервале 3—6 Далберг и Дюре
предположили, что существуют гексаванадат-ионы [V80i7]4-,
IHV8O17]3~, [H2V8O17]2- ограниченной устойчивости. Шово пока-
зала, что гексаванадат-ионы неустойчивы и в сильно кислых рас-
творах быстро превращаются в ионы VO2:
IVeO17]4- + 10Н+ -> 6VO+ + 5Н2О [HVeOi7]3~ + 9Н+ -> 6VO£ + 5Н2О
[H2VeO17]«- 4- 8Н + =s=t 6VOj + 5H2O
Яндер установил существование неустойчивого пентаванадат-
иона [H4V50ie]3-, а Глемзер и Прайслер — неустойчивого доде-
каванадат-иона [V12O33]e~, сообщающего раствору коричнево-
красное окрашивание.
Пента-, гекса- и додекаванадат-ионы, обладая малой устой-
чивостью, быстро превращаются в устойчивые декаванадат-ионы.
Из водных растворов ванадатов в зависимости от pH и кон-
центрации раствора можно выделить орто-, пиро-, тримета-
и декаванадаты.
К ортованадатам относится Me|VO4, где Me1 = Na+, К+,
Li+, Ag+, Т1+, 1/2Са2+, V2Sr2+, х/2Ва2+, Х/2РЬ2+, x/2Hg2+, x/3Bi3 + и др.
Ортованадат натрия, Na3VO4, получают при сплавлении
V2O5 с Na2CO3, взятых в стехиометрических количествах; это
бесцветные гексагональные кристаллы с т. пл. 866°, плохо раство-
римые в спирте и растворимые в воде. При концентрировании
водных растворов, полученных обработкой соединений ванадия(У)
избытком NaOH или при растворении Na3VO4 в воде, выпадают
кристаллогидраты ортованадата натрия Na3VO4-rcH2O, где
п = 7, 9, 12, 13, 16.
Ортованадат натрия можно получить следующим образом:
V2O5 + 3Na2CO3 = 2Na3VO4 + ЗСО2
V2O5 4- 6NaOH = 2Na3VO4 4- 3H2O
VOC13 4- 6NaOH = Na3VO4 4- 3NaCl 4- 3H2O
Безводный ортованадат калия, K3VO4, получают сплавлением
V2O5 с К2СО3; он образует светло-желтые кристаллы, плохо
растворимые в спирте и растворимые в воде. Известны кристалло-
гидраты K3VO4-9H2O и K3VO4-12H2O.
Большинство ортованадатов трудно растворимы и окрашены,
например: Ca3(VO4)2 — желтый, Sr3(VO4)2 — желтый, Ва3(УО4)2 —
172
ПОБОЧНАЯ ПОДГРУППА V ГРУППЫ
желтый, Ag3VO4 — оранжевый, Т13УО4 — красный, Pb3(VO4)2 —
белый, Hg3(VO4)2 — оранжевый, BiVO4-Bi(OH)3 — цвета охры.
Ортованадаты свинца, кальция и кадмия с фторидом или хло-
ридом того же элемента образуют устойчивые соединения общей
формулы. 3Me*(VO4)2-MenX2 или Me|J(VO4)3X, где Me11 = Pb2+,
Са2+, Cda+, а X = F-, СП.
Пиро- или диванадаты Ме^УгОу (где Me1 = Na +
или К+) получают подкислением водных растворов ортованадатов
натрия или калия при значениях pH в интервале между 13,2
и 8, а также сильным прокаливанием кислых ортованадатов:
2Na3VO4 + H2SO4 = Na4V2O7 + Na2SO4 -f- H2O
2Na2HVO4 = Na4V2O7 + H2O
Соединение Na4V2O7-8H2O образует бесцветные гексагональ-
ные пластинки, плохо растворимые в спирте и растворимые в воде.
Триметаванадаты, MeiV3O9, обладают кристаллической
структурой, представляющей цепи неограниченной длины, в кото-
рых тетраэдры УО4 (с ванадием в центре тетраэдра) связаны меж-
ду собой через кислородный атом, общий для двух тетраэдров.
Триметаванадат аммония, (NH4)3V3O9, получают обработкой
солей ванадия(У) конц. NH4OH или действием NH4C1 на орто-
ванадаты щелочных металлов в водном растворе:
3VOC13 + 12NH4OH = (NH4)3V3O9 + 9NH4C1 + 6H2O
3Na3VO4 + 3NH4C1 + 3H2O = (NH4)3V3O9 + 3NaCl + 6NaOH
Это белое твердое вещество, плохо растворимое в воде, при
нагревании разлагается по уравнению, приведенному при полу-
чении У2О5.
Соли Na3V3O9 и К3У3О9 образуют бесцветные кристаллы,
плохо растворимые в спирте и растворимые в воде.
Триметаванадаты, Ag3V3O9 и Hg3V3O9, трудно растворимы
в воде и разбавленных минеральных кислотах.
Известна триметаванадиевая кислота Н3У3О9.
Декаванадаты. При обработке раствора Na3VO4, под-
кисленного 1 н. соляной кислотой до pH = 3,8, 0,1 М раствором
хлорида тетрафенилфосфина (СвН5)4РС1 получают желтое вещество
состава [(С8Н5)4Р]3[Н3У1()О28], трудно растворимое в воде и пере-
ходящее в органическую фазу при экстрагировании дихлорэтаном.
При обработке водного раствора Na3VO4, подкисленного
НС1 до pH = 2,5—3, гидрохлоридом хинолина получают
(C9H7NH)4[H2V10O28b (3—4)Н2О. Если к раствору Na3VO4, под-
кисленного НС1 до pH = 3,5—4,7, добавляют солянокислую соль
хинолиния, то образуется (C9H7NH)8[V10O28] •(!—9)Н2О.
Известны также соединения, которые образуются при обра-
ботке подкисленных НС1 растворов ортованадатов солянокислыми
ВАНАДИЙ
173
солями пиридиния или акридиния, например: (С5Н5УН)8[У10О28],
(C5H5NH)5lHVj0O28], (C5H5NH)4[H2V10O28],(C13H9NH)5[HV10O28l •
• (3—4)Н2О, (C13H9NH)4[H2V10O28] .(5-7)Н2О,
(C13H9NH)3[H3V10O28].
Известны также декаванадаты K8[V10O28], K5Na[V10O28] •
•10Н2О, K4Na[HV10O28H0H2O.
Гетерополисоединения. Известны многочислен-
ные гетерополисоединения, в которых ванадий присутствует как
центральный элемент — комплексообразователь или как кислый
радикал в форме координированной группы. Большинство гетеро-
полисоединений ванадия содержит в качестве координированных
групп различные кислотные радикалы, например У2О^~, W2O^_,
Мо2О*-, С2О£- и др.
К важнейшим гетерополисоединениям ванадия относятся: фос-
форнованадиевая кислота H7[PV12O36] или Н7(Р(У2О8)8], фосфоро-
ванадаты, например Na3H4[PV12O38] или Na3[PVi2O34(H2O)2],
фосфорнованадовольфрамовая кислота H7[P(V2O8)(W2O7)5]-^НгО
и ее калийная соль K8H[P(V2O8)(W2O7)5] фосфорнованадо-
вольфрамовые кислоты общей формулы H7[P(V2O8)ra(W2O7)m]-
•хН2О (где п = 2, 3, 4, а т = 4, 3, 2, поскольку п + т =6),
фосфорнованадовольфраматы аммония (NH4)8H[P(V2O8) 2(W2O7)4] •
•zH2O, (NH4)6H[P(V2O8)3(W2O7)3]-xH2O, (NH4)6H-
• [P(V2O8)4(W2O7)2]-a:H2O, фосфорнованадомолибденовая кислота
H7[P(V2O8)(Mo2O7)5] 'xHzO, фосфорнованадовольфрамомолибдено-
вые кислоты H7[P(V2O8)(W2O7)4(Mo2O7)|
H7[P(V2Oe)(W2O7)3(Mo2O7)2] -a;H2O, H7[P(V2O8)(W2O7)2(Mo2O7)3] •
•a;H2O, H7[P(V2O8)(W2O7)(Mo2O7)4] -xH2O и их соли, кремнева-
надовольфрамовая кислота H8[Si(V2O8)(W2O7)5] -а:Н2О и ее соли,
например K8[Si(V2O8)(W2O7)5]-a:H2O, Na8[Si(V2O6)(W2O7)5] -^Н2О,
кремневанадомолибденовая кислота H8[Si(V2O8)(Mo2O7)5]-а;Н2О,
германованадовольфрамовая кислота H8[Ge(V2Oe)(W2O7)5] -а:Н2О
и ее соли, например K5H3[Ge(V2O8)(W2O7)5] -a;H2O, германована-
домолибденовая кислота H8[Ge(V2O8)(Mo2O7)5]'а:Н2О и ее соли,
например K5H3[Ge(V2O8)(Mo2O7)5]-2:Н2О, мышьяковованадоволь-
фрамат аммония (NH4)eH[As(V2O8)2(W2O7)4] -а:Н2О, (NH4)8H-
• [As(V2O8)3(W2O7)3]-а:Н2О, желтые модификации ванадоволь-
фраматов MeJlV2W4O19] -a;H2O, например Na4[V2W40i9]-14Н2О,
K4[V2W4O19] -8Н2О, Ba2[V2W4O19|-13H2O, Ag4[V2W4O19]-2Н2О,
(CN3H6)8[V2W4O19]-а:Н2О, оранжевые формы типа Me?[V3W30i9]-
•;гН2О, например Na5[V3W30i9] •(17—19)Н2О, K5[V3W3O19J-
5Н<>0, (NH4)5[V3W3O19] -5Н2О, пурпурные соединения рядов
Mei[V3W7O31].^H2O и Met[V3W6O28]-а:Н2О, например (CN3H8)5-
• [V3W7O31] .6Н2О, (NH4)5[V3W8O28] • 18,5Н2О, ванадомолиб-
даты Me([V2Mo4Oi9]-хИгО, Me([V2Mo3Oi8]-а:Н2О, например
Na4[V2Mo4O19] -а;Н2О, Na4[V2Mo3O18] -а;Н2О, молибдооксалатована-
даты MeJ[V(C2O4)(MoO4)3]-а:Н2О, Me2H[VO2(C4O4)(MoO4)I -a:H2O,
174
ПОБОЧНАЯ ПОДГРУППА V ГРУППЫ
например К3[У(С2О4)(МоО4)з] • (3,5—4)Н2О, Na3[V(C2O4)(MoO4)3] •
•(8—12)Н2О, (NH4)3[V(C2O4)(MoO4)3].(4,5-5,5)Н2О,
Ва3[У(С204)(Мо04)3]2-17Н20, Na2H[VO2(C2O4)(MoO4)l -5Н2О.
Пероксосоединения. При обработке разбавленных
растворов ванадатов щелочных металлов перекисью водорода
в слабо кислой среде образуются желтые растворы, содержащие
моно- и дипероксованадат-анионы [VO3(O2)]3-, [VO2(O2)2]3-, или
фиолетово-синие растворы с тетрапероксоортованадат-анионом,
тетра- и пентапероксодиванадат-анионами [V(O2)4]3~, [V2O3(O2)4]4-,
[V2O2(O2)5]4-; в сильно кислой среде образуются коричнево-крас-
ные растворы, содержащие пероксосоли радикала
з+
Пероксоортованадаты или пероксодиванадаты в сильно кислой
среде превращаются в пероксосоли радикала
|VO2(O2)2]3- + 6H+^
- ,013+
VQ + Н2О2 + 2Н2О
Пероксосоединения ванадия плохо растворимы в спирте,
в воде подвергаются гидролизу с образованием перекиси водорода,
которая в свою очередь разлагается с выделением кислорода.
Известны монопероксометаванадаты MeI[VO2(O2)J, моноперо-
ксоортованадаты Me4[VO3(O2)J "иНгО, дипероксоортованадаты
МеЦУО2(О2)2]-wH20, тетрапероксоортованадаты МеЦУ(О2)4]-
•иН20, тетрапероксодиванадаты Mei[V2O3(O2)4], пентапероксо-
диванадаты Mei[V2O2(O2)5] (где Me1 = К+, Na+, NH*).
Пентафторид ванадия, VF5, получают разложением VF4 при
600° в платиновой трубке, через которую продувается азот, обра-
боткой фтором нагретого до 300° ванадия, действием трифторида
брома на металлический ванадий или трихлорид ванадия.
2VF4 = VF6 + VF3 V + s/2F2 = VF5
Пентафторид ванадия представляет собой белое твердое веще-
ство с плотностью 2,18 г/см3, т. пл. 102° и т. кип. 111,2°; он разъе-
дает стекло, на воздухе превращается в VOF3, реагирует с эфи-
ром и толуолом, растворяется в воде, спирте, хлороформе, аце-
тоне с образованием желтых растворов, плохо растворяется
в сероуглероде, образует с различными фторидами фторосоли
МеЧУРв) (где Me1 = К+, Na+, Ag+, V2Ba2+ и др.).
ВАНАДИЙ
175.
Гексафторованадат(У) калия, K[VFe], получают при действии
фторида калия на VF5 или действии BrF3 на смесь VC13 с KF:
VF5 + KF = K[VF6] 2VC13 + BrF3 + 9KF = 2K[VFe] + 6KC1 + KBr
Соль K[VFe] представляет собой белое твердое вещество,
дымящее на воздухе, растворяющееся в воде с гидролизом, при
нагревании разлагающееся на VF5 и KF, разлагающееся в 50 %-ной
H2SO4. Кристаллы имеют октаэдрическую структуру.
2VF5 + 5Н2О V2O5 + 10HF
Окситрифторид ванадия (трифторид ванадила), VOF3, получа-
ют нагреванием VF5 в платиновом сосуде в атмосфере кислорода,
обработкой при нагревании VOC13 концентрированной фтористо-
водородной кислотой или обработкой V2O5 фтором:
VF3 4- V2O2 = VOF3 VOC13 + 3HF = VOF3 + 3HCI
Окситрифторид ванадия представляет собой сильно гигроско-
пичное желтое твердое вещество с плотностью 2,46 г!см3, т. пл. 300°
и т. кип. 480°; он разрушающе действует на стекло, растворяется
в воде и органических растворителях, образует с фторидами
щелочных металлов комплексные соли MeI[VOF4], Mei[VOF5], где
Me1 = К+, Na+, NH4+ и др.
Тетрафторооксованадаты, MeI[VOF4], в которых
Me1 = К+, Na+, получают действием KF или NaF на VOF3 и обра-
боткой BrF3 метаванадатов калия или натрия:
VOF3 + MeJF = MeT[VOF4] Me*V3O9 + 4BrF3 = SMe^VOFJ + 3O2 + 2Br2
Оксифторид ванадия, VO2F, не выделен, хотя известны окси-
фторосоли общей формулы Me|[VO2F3], Me|[VO2FJ, например
K2[VO2F3], (NH4)2IVO2F3J, (NH4)3[VO2F4J.
Окситрихлорид ванадия (трихлорид ванадила), VOC13, полу-
чают нагреванием смеси V2O5 с активированным углем в атмосфере
хлора при 650°, нагреванием VC13 в кислороде, нагреванием V2O5.
с SOC12 при 75° или с НС1 при 60—80° (в присутствии Р40ю).
V,O5 + ЗС + ЗС12 = 2VOC13 + ЗСО V2O5 + 3SOC12 = 2VOC13 + 3SO2
VC13 +//2O2 = VOC13 V2O5 + 6HC1 "-7 2VOC13 + 3H2O
Ковалентное соединение VOC13 представляет собой желтую
жидкость с плотностью 1,76 г/см, т. затв. —79,5° и т. кип. 126,7°
(легко очищается перегонкой); оно растворяется в спирте, ацетоне,
уксусном ангидриде, восстанавливается водородом при нагрева-
нии, гидролизуется следами воды, взаимодействует с серой при
обычной температуре и со щелочными металлами при нагревании:
2VOC13 + ЗН2О V2O5 + 6НС1 VOC13 -f- Н2 = VOC1 + 2НС1
VOC13 + 1/2Н2 = VOC12 + HC1 2VOC13 + S = 2VC13 + SO2
176
ПОБОЧНАЯ ПОДГРУППА V ГРУППЫ
Окситрихлорид ванадия является растворителем для AsCl3,
РС13, TiCl4, SnCl4, РОС13.
Известен аддукт VOC13-(С^Нд^О, который получают из VOC13
с эфиром при 60—70°, и комплексное соединение трихлорида
ванадила с солянокислым пиридинием C5H5NH[VOC14J.
Окситрибромид ванадия (трибромид ванадила), VOBr3, полу-
чают при пропускании паров брома над сильно нагретым V2O3
или над смесью V2O5 с углем (также сильно нагретой):
V2O3 4- 6Br2 = 2VOBr3 -4 I/2O2 V2O5 + ЗС + ЗВг2 = 2VOBr3 + ЗСО
Окситрибромид ванадия представляет собой гигроскопичную
темно-красную жидкость с плотностью 2,93 г/см'Л, кипящую при
133° (при давлении 100 ммрт. ст.)-, он легко разлагается на VOBr2
и Вг2:
VOBr3 = VOBr2 4- i/2Br2
Разложение начинается при комнатной температуре и осуще-
ствляется полностью при 180°.
Известен оксибромид ванадия V2O3Br4.
Пентасулъфид ванадия, V2S5, встречается в природе в виде
минерала патронита. Его получают нагреванием V2S3 с серой
при 400° без доступа воздуха или обработкой водных растворов
тиованадатов разбавленными кислотами:
V2s3 + 2S = V2S5 2Na3VS4 + 3H2SO4 - V2S5 + 3Na2SO4 + 3H2S
Осаждение пентасульфида ванадия по этой реакции происхо-
дит не полностью, поскольку образующийся H2S частично вос-
станавливает пятивалентный ванадий. Пентасульфид ванадия
представляет собой черный порошок с плотностью 3 г/см3, легко
превращающийся в V2S3 при нагревании в инертной атмосфере
или в V2O5 при прокаливании на воздухе при высокой темпера-
туре; он плохо растворяется в воде и растворяется в кислотах
(JI2SO4, разб. HNO3 и др.), щелочах (NaOH, КОН), сульфидах
и полисульфидах щелочных металлов или аммония.
При растворении V2S5 в сульфидах или полисульфидах щелоч-
ных металлов или аммония образуются растворимые тиованадаты,
а при растворении V2S5 в щелочах образуются растворимые окси-
тиованадаты и тиованадаты.
Тиованадаты получают действием сульфида (полисульфида)
аммония или сульфидов щелочных металлов на пентасульфид
ванадия или на растворы ванадатов:
V2S5 + 3(NH4)2S = 2(NH4)3VS4
(NH4)3VO4 + 4(NH4)2S + 4H2O = (NH4)3VS4 + 8NH4OH
(NH4)3V3O9 + 12(NH4)2S + 9H2O = 3(NH4)3VS4 + 18NH4OH
ВАНАДИЙ
177
Известны орто-, мета- и гексаметатиованадаты VS3', VS3,
[H2V8Slg]4-, устойчивые на холоду, разлагающиеся при нагрева-
нии, растворяющиеся в воде с образованием пурпурных или крас-
но-коричневых растворов.
Соль (NH4)3VS4 образует черно-фиолетовые ромбические приз-
мы с плотностью 1,62 г/сл3; они растворяются в воде, окрашивая
раствор в фиолетовый цвет, плохо растворяются в спирте, под
действием H2S и (NH4)2S превращаются в темно-синие микрокри-
сталлы (NH4)VS3-2Н2О или в неустойчивые фиолетовые микро-
кристаллы (NH4)2[H2V8Si8] • 18Н2О:
(NH4)sVS4 -> NH4VS3 + (NH4)2S
6(NH4)3VS4 (NH4)4[H2VeS18l 4- 6(NH4)2S + 2NH3
При действии сильных кислот на (NH4)3VS4 в качестве конеч-
ных продуктов получают V2O5-ziH2O.
Окситиованадаты образуют анионы следующих типов: [VOS3]3-,
[VO3S]3“, [V2OSe]4-, [V2O2S5]4-, [V205S2]4-; они могут быть полу-
чены растворением V2S5 в щелочах или сплавлением соединений
ванадия(У) с К2СО3 и серой. Примеры окситиованадатов:
Na3[VOS3l -5Н2О— красно-коричневые кристаллы, Na3[VO3Sb
•10Н2О — оранжевые кристаллы, (NH4)4[V2OS8] — красные
кристаллы. K4[V2OS8]-ЗН2О —кристаллы, похожие на кри-
сталлы КМпО4.
Хелатные соединения
К хелатным соединениям пятивалентного ванадия относятся
VOCI3-(С2Н5)2О, НО — VO(C9H8NO)2 и производные переменного
состава, полученные путем обработки соединений пятивалентного
ванадия а-нитрозо-0-нафтолом и купфероном.
Помимо рассмотренных известны и другие соединения пятивалентного
ванадия, например оксалатованадаты MeJ [VO2(C2O4)2], в которых Mei ==
= Na+, К+ и др., хелатные соединения, получаемые путем обработки в кислой
среде соединений пятивалентного ванадия п-бензоилфенилгидроксиламином,
8-оксихинолином, З-окси-1,3-дифенилтриазином, п-диэтилдитиокарбаматом
натрия. Известны гексафторованадаты Mei [VFe], где Mei = Na+, Ag'*,
1/2Ba2+, оксппентафторованадаты Me2[VOF5], где Mei = К+, Na+, NHJ,
окситиованадаты и пероксованадаты, многочисленные гетерополисоединения.
Металлоорганические соединения
Известно ограниченное число металлоорганических соедине-
ний ванадия, как-то: V(CO)e, У(С5Н5)2, (C5H5)V(CO)4,
K2(C5H5)V(CO)3, V(C8H8)2, [1,3,5-(CH3)3C8H3]2V, (C5H5)2VBr,
(C5H5)2VBr2.
12 — 0 10 1
178
ПОБОЧНАЯ ПОДГРУППА V ГРУППЫ
Соединение V(CO)6, разлагающееся при 70°, получается по
реакции
Х2
2VCl3i-2Mg— Zn-;-12CO-----> 2V(CO)e + 2MgCl2+ZnCl2
пиридин
135°
20 атм
Соединение V(C5H5)2 представляет собой пурпурное вещество,
плавящееся при 168°. (C5H5)V(CO)4 — оранжевое вещество с т. пл.
138°. V(C6H6)2 — красновато-коричневое вещество, плавящееся
при 277°.
Соединения включения
Нитриды ванадия: VN и V2N.
Нитрид ванадия, VN, получают действием аммиака на VOC13
при нагревании, пропусканием газовой смеси, состоящей из азота
и паров галогенида ванадия, над нагретой до 1200—1400° нитью
из вольфрама, молибдена или рения, сильным нагреванием V2O3
или VC12 в атмосфере азота.
Нитрид ванадия — соединение цвета бронзы, тугоплавкое
(т. пл. 2320°), с плотностью 5,63 г/см3, практически нерастворимое
в воде, растворяется в конц. НС1 или Н2ЭО4и выделяет NH3 при
нагревании с расплавленной щелочью.
Карбиды ванадия, VC, V3C, V3C2, V4C3, получают
восстановлением окислов ванадия с углем, нагреванием V2O3
с углем при 1100° в атмосфере водорода, нагреванием смеси VC12,
Н2 и СО.
Карбид ванадия, VC, образует серые кубические кристаллы
с решеткой типа NaCl, плотностью 5,4 г/см3 и т. пл. 2810°; он
плохо растворим в воде, растворяется в конц. HNO3 и расплавлен-
ных KNO3, КС1О3. При нагревании VC взаимодействует с кис-
лородом, хлором, азотом. Карбид ванадия применяется для изго-
товления нитей ламп накаливания.
Силициды ванадия, VSi2, V2Si, можно получить
прямым взаимодействием элементов при нагревании или нагре-
ванием V2O5 с элементарным кремнием в электрической печи.
Дисилицид ванадия, VSi2, представляет собой металлоподобные
блестящие призмы с плотностью 4,42 г/см3, плохо растворимые
в воде, спирте, эфире, растворимые в плавиковой кислоте
и расплавленной щелочи, разрушаются хлором.
Салицид ванадия, V2Si, образует хрупкие очень твердые бле-
стящие черные призмы с плотностью 5,48 г/сл3; он взаимодей-
ствует при нагревании с хлором, бромом, иодом, плохо раство-
ряется на холоду в НС1, HNO3, H2SO4, растворяется в плави-
ковой кислоте и в смеси кислот HNO3 с H2SO4 или НС1 с H2SO4.
Борид ванадия, VB2, получают пропусканием газообразной
НИОБИЙ
17$
смеси галогенида ванадия и трибромида бора над нагретой до
1200—1400° проволокой из вольфрама, платины или рения, а так-
же электролизом пятиокиси ванадия, растворенной в борной
кислоте или расплавленных боратах, смешанных с фторидами
щелочных или щелочноземельных металлов.
Борид ванадия представляет собой серые кристаллы с плот-
ностью 5,28 г/см3 и твердостью 8 по шкале Мооса.
НИОБИЙ Nb
Z= 41; ат. вес = 92,906
Валентность (I, II, III, IV), V; заряд (1+, 2+,
3+, 4+), 5+
Массовое число природного изотопа 93
Электронная структура атома ниобия: Д'•£-М <4s24p64cZ4-5s1.
Электронная структура атома ниобия и иона Nba+ для 4d-
и 5»-орбиталей:
4d4 5s1
4<i ' 5s
Nb
Nbs+,
t
ИСТОРИЧЕСКАЯ СПРАВКА
Примерно в 1650 г. европейские завоеватели Колумбии при-
везли тяжелый черный минерал, пронизанный тонкими пластин-
ками блестящей, как золото, слюды. Поскольку этот минерал
не имел ничего общего с золотом, его поместили как неизвестный
минерал из Колумбии в Британский музей в Лондоне.
В 1801 г. английский химик Гатчет, проводя анализ этого
неизвестного минерала, открыл наряду с железом новый элемент,
который был им назван «колумбием», а минерал —«колумбитом».
Волластон и Берцелиус полагали, что колумбий, открытый
в 1801 г. Гатчетом, и тантал, открытый в 1802 г. Экебергом,
являются одним и тем же элементом, хотя заметили, что танталиты
и колумбиты различаются между собой по плотности (колумбиты
легче).
Название «ниобий» предложил Розе (по имени Ниобеи, дочери
Тантала, благодаря схожести этих двух элементов), который
впервые получил его в металлическом состоянии в 1846 г. путем
восстановления окиси Nb2O5.
В английской и американской литературе до сих пор часто
элемент называют колумбием в отличие от общепринятого —
ниобий.
12*
180
ПОБОЧНАЯ ПОДГРУППА V ГРУППЫ
РАСПРОСТРАНЕННОСТЬ В ПРИРОДЕ
В природе ниобий встречается только в виде соединений,
входящих в состав многочисленных комплексных минералов
вместе с танталом, железом, марганцем, оловом, вольфрамом,
рением, титаном, цирконием, торием, ураном, щелочными и щелоч-
ноземельными элементами.
Ниобий достаточно широко распространен в природе, осо-
бенно в шенитных и гранитных пегматитах; его содержание в зем-
ной коре составляет 3,2-Ю-5 вес.%. Часто ниобий встречается
в минералах редкоземельных элементов.
Наиболее важными минералами ниобия являются следующие.
Колумбит, (Fe, Mn)(NbO3)2, встречающийся в изоморфной сме-
си с танталитом (Fe, Мп)(ТаО3)2 под названием танталито-колум-
бита п представляющий собой пластинчатые или орторомбические
двупреломляющие кристаллы черного (или красновато-коричне-
вого при большом содержании марганца) цвета с плотностью
5,15 г/см3. Колумбит имеет твердость 6 по шкале Мооса, он устой-
чив к действию химических реагентов. Залежи колумбита нахо-
дятся в Норвегии, Финляндии, Франции, Австралии, Бразилии,
США и др.
Лопарит, (Na, Са, Ce)(Nb, Ti)O3, встречается в Африке,
США, Австралии, Норвегии и др. в виде серовато-черных кубиче-
ских кристаллов с плотностью 4,75—4,89 г/см3 и твердостью
5,6—6 по шкале Мооса.
Пирохлор, NaCaNb2O6F, имеет вид коричневых октаэдрических
кристаллов с плотностью 4,03—4,36 г/см3 и твердостью 5—
5,5 по шкале Мооса.
К другим минералам, содержащим ниобий, относятся: фергю-
сонит (Y, Ег, Се, U)(Nb, Та, Ti)O4, стибиоколумбит SbNbO4, эвксе-
нит (Y, Се, Са, Th . . .) (Nb, Та, Ti)2O8, ильменорутил
(Ti, Nb. Fe)O2, самарскит (Y, Er . . ,)[(Ta, Nb)2O7]3 и др., которые
встречаются в Африке, CIIIA, Австралии, Норвегии, Финляндии,
Канаде, Японии.
ПЕРЕРАБОТКА РУД, СОДЕРЖАЩИХ
НИОБИЙ И ТАНТАЛ
Ниобий и тантал выделяют из танталито- или титано-колумби-
товых концентратов, полученных с использованием электромаг-
нитных. гравитационных, флотационных и химических методов
обогащения руд, поскольку содержание ниобия и тантала в при-
родных рудах мало.
При переработке концентратов руд ниобия и тантала пресле-
дуется цель отделения второстепенных примесей (вольфрама,
олова, железа, марганца, свинца, кремния и др.), затем отделение
титана и, наконец, разделение ниобия и тантала.
НИОБИЙ
181
Металлургические процессы полной переработки руд ниобия —
тантала сложны как по характеру используемых реактивов, так
и по числу операций по разделению элементов, поскольку эти
концентраты могут содержать большое количество трудно отде-
ляемых примесей: Nb, Та, Ti, W, Sn, Fe, Mn, Pb.
При химической переработке концентратов руд ниобия —
тантала получают гидратированные пятиокиси, комплексные фто-
риды, оксифториды, галогениды ниобия и тантала.
Концентраты танталито-колумбитов разлагают сплавлением
со щелочами, карбонатами или гидросульфатами щелочных метал-
лов, с кислым фторидом калия либо проводят хлорирование при
600—700° в присутствии угля.
Переработка концентратов руд ниобия — тантала
Сплавлением тонко измельченных концентратов танталито-
колумбитов с щелочами или карбонатами щелочных металлов
получают ниобаты, танталаты, окиси железа, марганца и др.,
обладающие ограниченной растворимостью в воде, и силикаты,
алюминаты, станнаты, вольфраматы щелочных металлов, раство-
римые в воде:
Fe(NbO3)2 + 6NaOH = 2Na3NbO4 + FeO + 3H2O
Mn(NbO3)2 + 6NaOH = 2Na3NbO4 + MnO + 3H2O
После обработки охлажденного расплава водой и повторной
промывки осадка, отделенного фильтрованием, последний обра-
батывают горячей разб. НС1 (или разб. H2SO4) для растворения
окислов железа и марганца и для образования гидратированных
пятиокисей (Nb, Та)2О5-тгН2О; последние путем растворения
в плавиковой кислоте и обработки KHF2 превращаются в ком-
плексные соли Na2[NbOF5], K2[TaF7L
Процесс переработки танталито-колумбитовых концентратов
изображен на следующей схеме:
Сплавление
(Fe, Mn)(Nb, ТаО3)2 ---
Танта лито-колумбит
[ Na3NbO4
I Na3TaO4
| 4-H2O и HC1
(Nb, Ta)2O5.nH2O
+KF в HF
K2[NbOF5]
Более растворимый
K2[TaF7]
Менее растворимый
182
ПОБОЧНАЯ ПОДГРУППА V ГРУППЫ
При кипячении в воде охлажденного расплава, полученного
из концентратов танталито-колумбита с KHSO4 при повы-
шенной температуре, осаждаются гидратированные пятиокиси
(Nb, Та)2О5-иНоО, которые увлекают с собой соединения воль-
фрама, олова, железа, марганца, свинца, кремния и др. Для
очистки и разделения гидратированные пятиокиси ниобия и тан-
тала растворяют в оксалато-тартратном растворе, затем раствор
выпаривают досуха, остаток прокаливают до образования Nb2O5
и Та2О5, а затем последние превращают в комплексные фториды
калия обработкой KHF2.
При кипячении с разбавленной плавиковой кислотой охлаж-
денного расплава, полученного при высокой температуре из кон-
центратов танталито-колумбита и KHF2, выпадают CaF2, K2[SiFe]
и другие соединения, а в раствор переходят комплексные соли
ниобия (или тантала) с калием или другими катионами, которые
выделяются в виде кристаллов при охлаждении раствора.
Концентраты титано-ниобиевых руд обрабатывают 75%-ной
H2SO4 в стальных эмалированных резервуарах в течение 4 час
при 180°. Нейтрализацией и гидролизом раствора, содержащего
соединения ниобия, тантала, железа, титана, редкоземельных
металлов, отделяют гидратированные пятиокиси ниобия и тан-
тала, а затем гидроокись железа, сульфат титанила и т. д.
Обработкой концентратов руд ниобия и тантала или шлаков,
содержащих ниобий, при 650—900е хлором в присутствии угля
получают смесь хлоридов. Пятиокись ниобия или тантала и дву-
окись титана хлорируются в присутствии угля.
Nb2O5 + ЗС12 + ЗС = 2NbOCl3 + ЗСО Та2О5 + 5С12 + 5С=2ТаС15+5СО
TiO2 + 2CL, + 2С = TiCl4 + 2СО
Отделение ниобия от тантала
Ниобий можно отделить от тантала дробной кристаллизацией
комплексных фторидов, последовательным гидролизом некоторых
соединений, дробной перегонкой хлоридов с предварительным се-
лективным восстановлением, NbCL до хлоридов, у которых валент-
ность металла ниже пяти, селективной экстракцией органиче-
скими растворителями, адсорбцией на ионообменных смолах.
Ниобий селективно экстрагируется из солянокислых растворов
(тантал остается в солянокислом растворе) 5%-ным раствором
метилдиоктиламина [CH3(CH2)6CH2]2NCH3 в ксилоле, 8%-ным
раствором трибензиламина (C6H5CH2)3N в хлороформе или хло-
ристом метилене, 2%-ным водным раствором диэтилдитиокарба-
мата натрия при использовании в качестве растворителя СС14,
6%-ным водным раствором купферона при использовании в каче-
стве растворителя хлороформа. Часто холодный расплав соеди-
НИОБИЙ
183
нений ниобия и KHSO4 растворяют в растворе лимонной кислоты,
затем этот раствор разбавляют водой, нейтрализуют NH4OH
и обрабатывают 1%-ным раствором 8-оксихинолина в хлороформе
для экстрагирования ниобия в органическую фазу. Из 6—9 М
раствора HF и 6 М раствора H2SO4 ниобий экстрагируется
диизопропилкетоном или диизобутилкарбинолом.
Тантал экстрагируется из 1 М раствора HNO3 0,6 М раство-
ром ди-н-бутилфосфорной кислоты в н-бутиловом эфире; из 0,4 М
раствора HF и 3,7 М раствора НС1 или 3,9 М раствора HNO3 —
диизопропилкетоном; из 10 М раствора HF, 6 М раствора H2SO4
или 2,2 М раствора NH4F — метилизобутилкетоном.
Если осуществляется совместная экстракция ниобия и тан-
тала, соединения этих элементов, растворенные в смеси конц.
H2SO4 и HF и разб. NH4F, обрабатывают метилэтилкетоном.
После растворения сплавов или концентратов ниобий — танта-
ловых руд в смеси НС1 с HF и последующего окисления HNO3
раствор пропускают через колонку с сильно основной анионо-
обменной смолой для выделения комплексных (с HF) анионов.
Из смолы титан, ванадий, железо и олово выделяются элюирова-
нием раствором, содержащим NH4C1, НС1, HF, ниобий — рас-
твором NH4C1 и НС1, тантал — раствором NH4C1 и NH4F.
Ниобий можно также отделить от тантала хроматографи-
чески на активированной окиси алюминия.
ПОЛУЧЕНИЕ МЕТАЛЛИЧЕСКОГО НИОБИЯ
Металлический ниобий получают восстановлением соединений
Nb2O5, NbCl5, K2[NbF7], K2[NbOF5] или термической диссоциа-
цией Nbl5.
Пятиокисъ ниобия, Nb2O5, может быть восстановлена водо-
родом под давлением 7 ат при 1910°, металлическим кальцием
в присутствии небольшого количества щелочного металла и СаС12
в качестве флюса при 900—1000°, СаН2 или сплавом редкоземель-
ных металлов с использованием того же СаС12 в качестве флюса
при 950—1075°:
Nb,O5 + 5Н2 = 2Nb + 5Н2О Nb2O5 + 5Са = 2Nb + 5СаО
Восстановлением Nb2O5 углем при 1600° в вакууме получают
сплав ниобий — углерод, содержащий 2,5—3,4% углерода, а алю-
мотермическим восстановлением Nb2O5 получают сплав ниобий —
алюминий, из которого чистый ниобий выделяют повторным плав-
лением этого сплава в электрической дуге в вакууме.
Металлический ниобий получают также восстановлением NbCl5
или K2[NbF7] металлическим натрием или калием при нагре-
184
ПОБОЧНАЯ ПОДГРУППА V ГРУППЫ
вании без доступа воздуха (соответственно в вакууме и под слоем
флюса, например NaCl, КС1):
NbCl5 + 5Na = Nb + 5NaCl K2[NbF7] + 5Na = Nb + 2KF + 5NaF
Чистый металлический ниобий получают термической диссо-
циацией NbClj или Nbl5 при 1800—2000° в вакууме (по процессу
Ван-Аркеля и Бюргесса).
Кремне- или алюмотермическим восстановлением смеси Nb2O5
с окислами железа (или железным порошком) получают ферро-
ниобий.
Порошкообразный металлический ниобий можно получить
электролизом щавелевокислого раствора ниобиевой кислоты,
содержащего 3% НС1 (или H2SO4), с использованием угольного
или платинового катода или электролизом расплавов фторидов
ниобия с фторидами или хлоридами щелочных металлов с исполь-
зованием в качестве катода никелевого или железного тиглей,
а в качестве анода — графитового электрода.
Порошкообразный металлический ниобий превращают в ком-
пактный металл переплавкой в вакууме с помощью электрической
дуги или в печи со сфокусированным пучком электронов.
ФИЗИЧЕСКИЕ И ХИМИЧЕСКИЕ СВОЙСТВА
Ниобий в компактном состоянии представляет собой блестящий
серебристо-белый (или серый в порошкообразном виде) парамаг-
нитный металл с объемноцентрированной кубической кри-
сталлической решеткой, плотностью 8,58 г!смъ, т. пл. 2487°,
т. кип. 4930°.
По механическим свойствам металлический ниобий почти
идентичен танталу. Он обладает твердостью ~6 по шкале Мооса,
очень высокой пластичностью и как следствие очень легко про-
катывается и протягивается.
Известно большое количество сплавов, в состав которых вхо-
дит ниобий, например с Fe, Al, Та, Ti, Mo, W, Zr и щелочными
металлами. Амальгамы ниобия не известны.
С химической точки зрения ниобий можно отнести к элементам
с пониженной химической активностью или соответственно к ме-
таллам с очень большой устойчивостью к действию различных
химических реагентов.
При обычной температуре металлический ниобий в компактном
состоянии не разрушается на воздухе или при соприкосновении
с водой, однако тонкодисперсный порошок ниобия при повышен-
ной температуре энергично разлагает воду с выделением водорода.
При нагревании металлический ниобий взаимодействует с фто-
ром, хлором, бромом, кислородом, серой, селеном, азотом, водо-
НИОБИЙ
185
родом, углеродом и др.
300°
Nb-|-S/2F2 >- NbF5 2Nb -J- 5/2^2 —
180° Nb4-s/2Cl2 > NbCl5 Nb-|-1/2N2 = NbN
450-500° Nb-p/2Br2 > NbBr5 Nb-p/2H2 = NbH
Nb + С = NbC
Действием хлора на металлический ниобий при 350° получают
NbCl4 и при 450° - NbCl3.
На металлический ниобий не действует НС1, HNO3 и царская
водка, но он растворяется в HF, в смеси HF с HNO3 и в расплав-
ленных щелочах и карбонатах щелочных металлов.
Nb + 5HF = NbF5 + 5/2Н2 2Nb + 6NaOH + 5/2О2 = 2Na3NbO4 = ЗН2О
В отличие от тантала ниобий обладает более низкой корро-
зионной стойкостью; для него характерна также пониженная
термоэлектронная эмиссия.
С физиологической точки зрения соли ниобия относятся к слабо
токсичным.
ПРИМЕНЕНИЕ
Благодаря высокой температуре плавления, большой способ-
ности к эмиссии электронов, способности адсорбировать при
нагревании различные газы металлический ниобий применяется
в вакуумной технике, радиотехнике, радиолокационной и рентге-
новской аппаратуре. Поскольку металлический ниобий обладает
сверхпроводимостью, его используют в вычислительных машинах
(для криотронов).
Металлический ниобий применяется для получения жаропроч-
ных, тугоплавких, кислотостойких, магнитных, сверхтвердых,
термостойких и неокисляющихся сплавов.
Сплавы ниобия и тантала, будучи термостойкими сплавами
с превосходными техническими характеристиками, незаменимы
в областях техники высоких скоростей; это материал для сверх-
звуковых самолетов, ракет, межпланетных станций и др. Стали,
содержащие ниобий, используются в реактивных турбинах, цилин-
драх высокого давления и вращающихся деталях, подвергаю-
щихся различным сильным воздействиям. Поскольку металличе-
ский ниобий увеличивает прочность сварки, стали, содержащие
ниобий, служат для сварки металлов. При получении сталей
с ниобием используется феррониобий, а не металлический ниобий.
Благодаря своим свойствам металлический ниобий может
заменять платину; он служит для изготовления тиглей, капсул,
дистилляционных приборов в химических лабораториях.
186
ПОБОЧНАЯ ПОДГРУППА V ГРУППЫ
Карбид ниобия с карбидами вольфрама, молибдена, титана
и других металлов служит для получения самых термостойких
сверхтвердых сплавов.
Из перечисленных областей применения металлического ниобия
и его сплавов очевидно, что это один из наиболее ценных металлов
современной техники.
СОЕДИНЕНИЯ (ОБЩИЕ СВОЙСТВА)
Одно-, двух-, трех- и четырехвалентный ниобий встречается
лишь в ограниченном числе соединений, пятивалентный же нио-
бий — в виде многочисленных простых, двойных и координа-
ционных соединений.
С ростом валентности ослабляется основной характер соеди-
нений ниобия и увеличиваются устойчивость соединений и тен-
денция к образованию координационных соединений.
Соединения одно-, двух-, трех- и четырехвалентного ниобия
получают восстановлением соединений пятивалентного ниобия.
В табл. 30 приведены формулы и указаны цвета ряда соеди-
нений двух-, трех-, четырех- и пятивалентного ниобия.
Соединение Окислы Фториды и фторосоединения Хлориды и хлоросоеди- нения
Соединения ниобия (II) NbO черная — NbCh черный
Соединения ниобия (III) Nb2O3 сине-черная NbF3 синий NbCl3 черный
Соединения ниобия (IV) NbO2 сине-черная — NbCl4 коричневый
Соединения ниобия (V) Nb2O5 бесцветная Nb2O5-nH2O белая NbF5 бесцветный NbQF3 бесцветный MeI[NbF6] Me2[NbF7] Me*[NbOF5]-riH2O Me|[NbOF3] MeJ[NbOF7] NbClj желтый NbOCl3 бесцветный Me^NbCle] Me^NbOChl Me2[Nb()CL,]
НИОБИЙ
187
Соединения одновалентного ниобия
Закись ниобия, Nb2O, получают восстановлением Nb2O5 водо-
родом при 1700° в течение 44 час. Это черные гексагональные кри-
сталлы с плотностью 8,08 г!слС.
Соединения двухвалентного ниобия
Известно очень немного соединений двухвалентного ниобия,
а именно окись NbO, сульфид NbS и дихлорид ниобия NbCl2.
В отличие от соединений двухвалентного ванадия соединения
двухвалентного ниобия менее устойчивы.
Соединения двухвалентного ниобия окрашены в черный цвет;
они получаются восстановлением соединений пятивалентного
ниобия.
Окись ниобия, NbO или Nb2O2, получают пропусканием паров
NbOF3 или NbOCl3 над магниевой проволокой, восстановлением
соли K2[NbOF5] металлическим натрием или калием при нагре-
вании, восстановлением Nb2O5 водородом под давлением 150 ат
Таблица 30
Бромиды и бромосоеди- нения Йодиды и иодосое- динения Сульфиды Хелатные соединения ниобия (V) Соедине- ния включения
— — NbS черный MeJ[NbO(C2O4)3].nH2O MeI[NbO(C4H4O6)2]-nH2O Me’[NbO(CeH4O2)3]-nH2O Me2H[NbO(C6H4O2)3]- •2C6H4(OH)2-nH2O [Nb(OC2H5)2(C5H7O2)]Cl2 NbH серо-чер- ный NbN серый NbC серо-корич- невый NbB2 серый NbB
NbBr3 черный __ Nb2S3 черный
— — NbS2 черный
NbBr5 пурпурно- красный NbOBr3 желтый MeI[NbOBr4] Me?JNbOBr5] Nbl5 желтый (C5H5NH)6 [Nbln] Nb2S5 черный Nb2O2S3 коричнево- черный
188
ПОБОЧНАЯ ПОДГРУППА V ГРУППЫ
при 2500° и др.
2NbOF3 + 3Mg = 2NbO + 3MgF2
K2[NbOF5] + 3Na = NbO + 2KF + 3NaF
2NbOCl3 + 3Mg = 2NbO + 3MgCl2 Nb2O5 + 3H2 = 2NbO + 3H2O
Окись ниобия образует октаэдрические черные кристаллы
с плотностью 7,30 г/см? (или черный порошок с плотностью
6,3 г 1см3), растворяющиеся в HF, НС1 или в смеси HF с конц.
H2SO4 и окисляющиеся при нагревании на воздухе, в кислороде
или хлоре:
2NbO + 3/2О2 = Nb2O5 NbO + 3/2С12 = NbOCl,
Сульфид ниобия, NbS, получается в виде черного порошка
восстановлением Nb2S3 водородом при нагревании.
Соединения трехвалентного ниобия
Из ограниченного числа соединений трехвалентного ниобия
упомянем трехокисъ, трихлорид, трибромид и сульфид. В отли-
чие от ванадия соединения трехвалентного ниобия менее устой-
чивы и значительно менее многочисленны.
Эти соединения окрашены в черный цвет, обладают понижен-
ной устойчивостью; получаются при восстановлении соединений
пятивалентного ниобия.
Трехокисъ ниобия, Nb2O3, получают восстановлением Nb2O5
водородом при 1250° или металлическим магнием при легком
нагревании:
Nb2O5 + 2Н2 = Nb2O3 + 2Н2О Nb2O5 + 2Mg = Nb2O3 + 2MgO
Трехокисъ ниобия — черно-синий порошок, плавящийся при
1775°, трудно растворимый во всех кислотах (за исключением HF),
в царской водке и щелочах; превращается в Nb2O5 при нагревании
на воздухе, в NbC — при нагревании с углем при 1200° в атмо-
сфере водорода, в NbN — при нагревании с углем в атмосфере
азота.
Трифторид ниобия, NbF3, получают действием эквимолярной
смеси водорода и плавиковой кислоты на гидрид ниобия при 570°.
Трифторид ниобия представляет собой парамагнитные синие
кубические кристаллы с плотностью 4,02 г!смя, сублимирующиеся
в вакууме начиная с 570°, очень устойчивые на воздухе и превра-
щающиеся в Nb2O5 при прокаливании на воздухе или в кислороде.
Трихлорид ниобия, NbCl3, получают действием хлора на метал-
лический ниобий, нагретый до 450°, пропусканием смеси паров
NbCl5 и водорода через стеклянную трубку, нагретую до 400—
450°, восстановлением NbCl5 металлическим алюминием при нагре-
НИОБИЙ
189
вании, восстановлением NbCl4 металлическим ниобием при нагре-
вании или диспропорционированием NbCl4:
NbCl5 + Н2 = NbCl3 + 2НС1 3NbCl4 + Nb = 4NbCl3
3NbCl5 + 2A1 = 3NbCl3 + A12C16 2NbCl4 = NbCl3 + NbCl5
Трихлорид ниобия представляет собой слабо летучие черные
кристаллы; он почти не взаимодействует с водой, окисляется
азотной кислотой до Nb2O5, превращается в NbOCl3 при нагрева-
нии в кислороде, на воздухе или в СО2, разлагается на NbCl5
и металлический ниобий при нагревании до 1000°, медленно вос-
станавливает конц. H2SO4 до SO2 (300°).
Трибромид ниобия, NbBr3, получают восстановлением NbBr5
водородом (500°). Это черный порошок, который очищается субли-
мацией в вакууме примерно при 400° и разлагается при нагрева-
нии до 925°. Водой бромид гидролизуется.
5NbBr3 = 3NbBr5 + 2Nb
2NbBr3 + (n + 5)H2O = Nb2O5.reH2O + 6HBr + 2H2
Трииодид ниобия, Nbl3, представляет собой черное твердое
вещество.
Сульфид ниобия, Nb2S3, получают нагреванием при 1000°
в вакууме стехиометрической смеси металлического ниобия и серы;
это твердое вещество черного цвета.
Двойной сульфат ниобия с гидросульфитом аммония,
Nb2(SO4)3-(NH4)2SO4 -HaSOi -6Н2О, выделяется в виде красновато-
коричневых кристаллов при добавлении (NH4)2SO4 в красновато-
коричневый раствор, полученный электрохимическим восстанов-
лением (свинцовый катод) раствора, образовавшегося при обра-
ботке NbCl5 серной кислотой при нагревании.
Соединения четырехвалентного ниобия
Соединения четырехвалентного ниобия получают восстановле-
нием соединений пятивалентного ниобия; они окрашены в черно-
коричневый цвет, относительно неустойчивы.
Двуокись ниобия, NbO2 или Nb2O4, получают восстановлением
Nb2Os водородом при температуре около 1200°, восстановлением
порошкообразного Nb2O5 в смеси с парафином и углем при 1700°
и электролизом расплава K2[NbOF5].
Двуокись ниобия представляет собой черно-синий блестящий
порошок (кубическая структура кристаллов), плохо растворимый
в кислотах (НС1, HNO3 и др.) или щелочах; обладает восстанови-
тельным характером, окисляется до Nb2O5 на воздухе при повы-
шенной температуре, не взаимодействует с водой, восстанавли-
вается водородом при температуре выше 1300°, вступает в реакцию
190
ПОБОЧНАЯ ПОДГРУППА V ГРУППЫ
с хлором, нитритом, сульфитом и сульфатом натрия.
200-400°
10NbO2+5Cl2--------► 4Nb2O3-J-2NbCl5
250-350°
3NbO2 + 3NaNO2-------► Na3Nb3O9J-3NO
450-600°
12NbO2 + 3Na2SO3-------► 2Na3Nb3O9-|-3Nb2O5 + 3S
800-1100°
18NbO2 + 3Na2SO4-------->• 2Na3Nb3O94-6Nb2O5+3S
Тетрахлорид ниобия, NbCl4, получают действием хлора на на-
гретый до 450° металлический ниобий и металлотермическим
восстановлением NbCl5 металлическим ниобием, железом или
алюминием:
400°
Nb + 2Cl2=NbCl4 2NbCl3+Fe-------> 2NbCl4 + FeCl2
450-50 0°
4NbCl5 + Nb------> 5NbCl4
270°
3NbCl5 + Al ---> 3NbCl4+V2Al2Cle
Тетрахлорид ниобия образует коричневые иглы, устойчивые
при обычной температуре в сухом воздухе, разлагающиеся во
влажном воздухе или при нагревании, растворяющиеся в воде
без доступа воздуха или в атмосфере СО2. При нагревании проис-
ходит диспропорционирование NbCl4:
2NbCl4 NbCl3 + NbCl5 — 28,3 ккал
Концентрированный водный раствор NbCl4 окрашен в инди-
гово-синий цвет. При разбавлении раствор становится зеленым,
потом коричневым.
Дисульфид ниобия, NbS2, получается в виде черного порошка
при действии паров серы (сероуглерода как такового или в смеси
с H2S) на металлический ниобий или на сильно нагретую дву-
окись ниобия.
Соединения пятивалентного ниобия
Наиболее многочисленными и важными устойчивыми соеди-
нениями, встречающимися в природе или синтезированными
в лаборатории, являются соединения пятивалентного ниобия.
Соли ниобия(У) могут содержать радикал NbO3+, катион Nb5 +
и различные анионы, например: NbO3’, Nb2O?~, NbO3, NbnO3„,
[Nb5OieP~, [NbeO19]8-, [Nb(O2)4]3-, [Nb(O2)O3P-, [NbF8]-,[NbF7]2-,
[NbOF5]2-, [NbOFe]3", [NbOF7l4-, [NbCl6]-, [NbOClJ", [NbOCl5]2-,
[NbOBrJ-, [NbOBr5]2-.
В отличие от соединений одно-, двух-, трех- и четырехвалент-
ного ниобия вещества, в которых ниобий пятивалентен, более
НИОБИЙ
191
устойчивы, бесцветны или окрашены в оранжево-желтовато-
красный цвет и проявляют более резко выраженную склонность
к комплексообразованию.
Неорганические соединения
Пятиокисъ ниобия, Nb2O5, получают сжиганием металличе-
ского ниобия в кислороде, прокаливанием на воздухе или в кис-
лороде гидрида NbH, нитрида NbN, карбида NbC, сульфида
Nb2S5, низших окислов ниобия, оксифторониобатов аммония,
купфероната ниобия, а также дегидратацией ниобиевой кислоты
Nb2O5 -иНзО:
2Nb -f- 5/2О2 = Nb2O5 -|- 455,2 ккал
2NbN + 5/2О2 = Nb2O5 + N2
2NbH + ЗО2 = Nb2O5 + Н2О
Nb2Ss + 1о/202 = Nb2O5 -|- 5SO2
Пятиокись ниобия существует в виде двух диамагнитных бес-
цветных кристаллических модификаций (a-Nb2O5 — ромбическая
и p-Nb2O5 — тетрагональная) и в виде белого порошка (желтею-
щего при нагревании) с плотностью 4,46 г/см3, тугоплавкого
(т. пл. 1521°), амфотерного (в отличие от V2O5), плохо растворимо-
го в воде, кислотах (HF, НС1, H2SO4) и щелочах. При действии
расплавленных щелочей карбонатов и гидросульфатов щелочных
металлов пятиокись ниобия превращается в орто- или пиронио-
баты щелочных металлов.
При нагревании Nb2O5 с СС14, S2C12, РС15 или при действии
газообразного хлора на смесь Nb2O5 с углем получают NbCl5
и NbOCl3. Нагревание Nb2O5 с сероводородом или сероуглеродом
дает Nb2OS3.
При повышенной температуре Nb2O5 взаимодействует с газо-
образным HF или НС1 с образованием NbF5 или NbCl5 соответ-
ственно.
Пятиокись ниобия восстанавливается водородом при нагрева-
нии с образованием NbO2, Nb2O3, NbO или металлического
ниобия. Ее можно также восстановить при нагревании гидридом
кальция, углеродом, элементарным алюминием или кремнием.
Гидратированная пятиокисъ ниобия (ниобиевая кислота),
Nb2O5-wH2O, может содержать различное количество воды, она
образуется в виде белого коллоидного осадка при действии раз-
бавленной кислоты (H2SO4, НС1) на раствор ниобата щелочного
металла, при прокаливании окситрифторида ниобия с H2SO4
в присутствии соли аммония, при гидролизе пентагалогенидов
или окситригалогенидов ниобия в присутствии аммиака, при обра-
ботке водой охлажденного сплава Nb2O5 с KHSO4.
192
ПОБОЧНАЯ ПОДГРУППА V ГРУППЫ
Ниобиевая кислота Nb2O5*7zH2O представляет собой белое
твердое вещество, плохо растворимое в воде, NaHCO3, (NH4)2CO3,
жидком аммиаке, растворимое в HF, конц. H2SO4, щавелевой,
винной кислотах, кислом оксалате калия, в щелочах или
карбонатах щелочных металлов, в растворах, содержащих маннит,
пирокатехин, пирогаллол.
Присутствие перекиси водорода способствует растворению
Nb2O5-7zH2O в кислотах, щелочах, карбонате натрия, кислом
фосфате натрия и др.
При нагревании Nb2O5 -тгНгО постепенно теряет воду и пре-
вращается в безводный Nb2O5.
Ниобаты и полиниобаты. Известны метаниобаты
(MerNbO3)n -2:Н2О, где Me1 = Li+, Na+, К+, Ag+, Cu+, r/2Mgz+,
T/2Caz+, VzSr2*, T/2Ba2+. Hg|+, V^n2*, r/2Cd2+, ^РЬ^Л/гМп2*,
1/2Fe2+, 1/2Co2+, J/2Ni2+, 1/3A13+ и др., ортониобаты MeJNbO4
или MeIH2NbO4 •хН2О, где Me1 = Li+, Na+, K+, 1/2Mg2+, 1/3Y3+,
173Cr3+, 173Fe3+, 173Bi3+, 1/3Rh3+, пиро- или диниобаты MeINb2O7,
где Me1 = K+, 1/2Ca2+, I/2Mg2+, пентаниобаты Me*[Nb5O16] •zH2(),
гексаниобаты MeI[Nb6O13] •zHgO, где Me1 = Na+, K+, Rb + , Cs\
AaSn2*, x/2Be2+, V-jMg2*,' 1/2Ca2+, V2Srz+, ^Ba2*, V2Pbz+.
В качестве примеров пентаниобатов и гексаниобатов можно
назвать K7[Nb5O16] Л6Н2О, Na7H[NbeO19]-31Н2О, K8[NbeO19]
24Н2О, KeH2[Nb6O19]-10Н2О.
Мета-, орто- и пирониобаты получают сплавлением Nb2O5
с окислами, гидроокисями или карбонатами ряда металлов,
а пента- и гексаниобаты — конденсацией орто- или пирониобатов
в кислой среде.
Метаниобаты, как и метафосфаты, существуют в полимерной
форме.
Пероксониобаты. При обработке ортониобатов
Me*NbO4 перекисью водорода в щелочной среде (гидроокись или
карбонат щелочного металла) получают водные растворы желтого
цвета, из которых при добавлении спирта или спирто-эфирной
смеси выделяются пероксониобаты общей формулы Me|[Nb(O2)4]
(где Me1 = Na+, К+, Rb+, Cs+). При действии конц. H2SO4 на
пероксоортониобаты на холоду получают монопероксоортонио-
биевую кислоту H3[Nb(O2)O3 -иНгО в виде желтого порошка.
Известны также пероксониобаты общей формулы
МетМеп [Nb(C)2)4]-?гН2О (где Me1 = Na+, К+, Rb+, Cs+, a Me11 =
= Mg2+, Ca2+), метамонопероксониобиевая кислота
Nb—О —О
лимонно-желтого цвета.
НИОБИЙ
193
Пентафторид ниобия, NbF5, получают прямым взаимодей-
ствием элементов при 250—300° в платиновой трубке или нагре-
ванием пентахлорида ниобия с безводной плавиковой кислотой:
Nb + 5/2F2 = NbF5 NbCls + 5HF = NbF5 + 5HC1
Пентафторид ниобия образует двупреломляющие гигроско-
пичные бесцветные кристаллы с плотностью 3,29 г/см3, т. пл. 75,5°,
т. кип. 236° (очищают сублимацией в вакууме при 100°). Пента-
фторид ниобия растворяется в SiCl4, SbCl5, СС14, СвН5 — СН3,
С2Н5ОН, С2Н5 - О - С2Н5, СН3СООН, конц. H2SQ4, НС1, HNO3,
разлагается водой, взаимодействует при нагревании с бромом,
иодом, серой, разъедает при нагревании поверхность металлов
(Fe, Си, Sn, Zn, Pb, Ag).
При растворении NbF5 в избытке концентрированных раство-
ров гидроокисей или карбонатов щелочных металлов образуются
ниобаты щелочных металлов.
Известны комплексные соли общих формул MeI[NbFe],
Me|[NbF7], Me*[NbF8], например K2[NbF7], Rb[NbFe], Rb2[NbF,l,
Cs[NbF6], (NH4)2[NbF7l, Na3[NbF8], а также оксифторосоли общих
формул MeUNbOFJ, Mei[NbOF5]-nH2O, Mei[NbOFe], MeJ[NbOF7],
например, NHJNbOFJ, Na2[NbOF5] -2H2O, Na3[NbOF6],
K3[NbOF6], (NH4)3[NbOF6J, Rb2[NbOF5], Cs2[NbOF5],
(NH4)2[NbOF5], (NH4)3[NbOFe], Tl2[NbOF5], Zn[NbOF5] -6H2O,
Cu[NbOF5] -4H2O.
Гептафторониобат калия, K2[NbF7], получают сплавлением
NbF5 c KHF2 или концентрированием раствора K2[NbOF5] *Н2О
в плавиковой кислоте:
NbF5 -j- 2KHF2 = K2[NbF7] + 2HF K2[NbOF5] + 2HF = K2[NbF7]+H2O
Гептафторониобат калия представляет собой блестящие моно-
клинные бесцветные кристаллы с плотностью 3,21 г/см3, которые,
растворяясь в воде, превращаются в K2[NbOF5] -НгО.
Окситрифторид ниобия, NbOF3, получают нагреванием Nb2O5
в токе HF или сплавлением с избытком CaF2 в токе хлористого
водорода:
Nb2O5 + 6HF = 2NbOF3 + ЗН2О
Nb2O5 + 3CaF2 + 6НС1 = 2NbOF3 + 2СаС12 + 3H2O
Окситрифторид ниобия образует двупреломляющие бесцветные
кристаллы; с фторидами щелочных металлов дает комплексные
соли.
Пентахлорид ниобия, NbCl5 или Nb2Cl10, получают прямым
взаимодействием элементов при 180°, нагреванием (280—350°)
смеси Nb2O5 с углем в токе хлора, нагреванием (440°) Nb2O5
с СС14 или (200°) с S2C12 в закрытой трубке, нагреванием при 210°
Nb2O5 с РС15 в токе хлора, пропусканием паров NbOCl3 в смеси
13—0101 (
194
ПОБОЧНАЯ ПОДГРУППА V ГРУППЫ
с хлором над раскаленным углем:
Nb + 5/2С12 = NbCls Nb2O5 + 5С + 10С12 = 2NbCl5 + 5СОС12
Nb2O5 + 5СС14 = 2NbCl5 + 5COCL,
2Nb2O5 + 1OS2C12 = 4NbCl5 + 5SO2 + 15S
Nb2O5 + 5PC15 = 2NbCls + 5POC13
NbOCl3 + C + 2C12 «= NbCl5 + COC12
Пентахлорид ниобия представляет собой желтые игольчатые
кристаллы (с тригональной бипирамидальной структурой) с плот-
ностью 2,75 г/сл13, т. пл. 194°, т. кип. 241°; он растворяется вСС14,
S2C12, CS2, H2SO4, НС1, C2H5OH, C2H5 - О - С2Н5, СНС13,
СН3СООН, СН3 — СО — СН3, гидролизуется до NbOCl3 или
Nb2O5*7zH2O, взаимодействует при нагревании с Nb2O5, давая
NbOCl3, вступает в реакцию при нагревании с хлоридами щелоч-
ных металлов с образованием хлоросолей общей формулы
Me^NbClg], в расплавленном состоянии действует на платину.
Выше 250° NbCl5 взаимодействует с кислородом по уравнениям
250_330°
NbCl5 + ‘/2О2---->• NbOCl34-Cl2
Выше 5 0 0°
2NbCl5 + 5/2O2-------> Nb2O5 + 5Cl2
При действии пиридина, пиперидина, анилина, нафталина,
антрацена на спиртовый раствор NbCl5 образуются координа-
ционные соединения, например [МЬ(С5Н5М)в]С15,[КЬ(С5Н11 N)e]Cl5,
[Nb(CeH5NH2)e]Cl5 и др.
Известны соединения NbCl(OCeH5)4, КЬС12(ОС10Н7)3, обра-
зующиеся при обработке пентахлорида ниобия раствором фенола
или Р-нафтола в сероуглероде.
Обработкой газообразным аммиаком эфирного раствора NbCl5
получают Nb3N5 и NH4C1:
3NbCls + 20NH3 = Nb3N5 + 15NH4C1
В результате упаривания раствора, полученного путем экстрак-
ции теплой водой, слабо подкисленной НС1, продукта восстанов-
ления при нагревании NbCl5 (1 вес. ч.) 3 %-ной амальгамой натрия
(7 вес. ч.), выделяются зеленые кристаллы [NbeCl12JC12-7^0.
Хлорид хлорониобия, [NbeCl12]Cl2 *7Н2О, представляет собой
темно-зеленые, почти черные мелкие кристаллы, устойчивые
при температуре до 100°, плохо растворимые в холодной и раство-
римые в теплой воде, а также в разбавленных кислотах (НС1,
H2SO4) и в концентрированных щелочах.
При добавлении конц. НС1 к сильно щелочному раствору хло-
рида хлорониобия образуется коричневый осадок Nb6Cl14-9Н2О,
который превращается в зеленый хлорид [NbeCl12]C12 *7Н2О про-
стым разбавлением водой. Упариванием теплых водных растворов
НИОБИЙ
195
хлорида хлорониобия или бромида хлорониобия получают микро-
кристаллический черный осадок [NbeCl12](OH)2‘8Н2О, который
растворим в концентрированных щелочах и НС1.
При упаривании растворов, полученных обработкой гидро-
окиси хлорониобия избытком бромистоводородной кислоты, выде-
ляются растворимые в холодной воде кристаллы [NbeCl12]Br2.
• 7Н2О.
Окситрихлорид ниобия, NbOCl3, получают действием паров
NbCl5 на сильно нагретую Nb2O5, пропусканием газообразного
хлора над предварительно сильно нагретой в токе газообразного
СО2 смесью Nb2O5 с углем, действием паров СС14 или S2C12 (увле-
каемых током СО2 или С12) на нагретую Nb2O5, гидролизом NbCl5,
нагреванием при 200° Nb2O5 с SOC12, действием хлора на Nb2O5
при 800—850°:
3NbCl5 -J- Nb2O5 5NbOCI3 Nb2O5 + ЗС + 6С12 = 2NbOCl3 + ЗСОС13
2Nb2O5 + ЗСС14 = 4NbOCl3 -4- ЗСО2
2Nb2O5 + 6S2C12 = 4NbOCl3 + 3SO2 + 9S
NbCl5 + H2O NbOCl3 + 2HC1
Nb2O5 + 3SOC12 = 2NbOCl3 + 3SO2 2Nb2O5 + 6C12 4NbOCl3 + 3O2
Окситрихлорид ниобия образует шелковистые бесцветные
игольчатые кристаллы, которые очищают перегонкой при 332°
в атмосфере СО2 или водорода. Нагреванием при высокой темпе-
ратуре в атмосфере СО2 или Н2 NbOCl3 превращается в Nb2O5
и NbCl5; на воздухе он окисляется до Nb2O5, растворяется в H2SO4,
конц. НС1, спирте, эфире, в воде гидролизуется до Nb2O5 •тгН2О.,
С хлоридами щелочных металлов (с солями пиридиния или хино-
линия) образует оксихлоросоли типа Me+[NbOCl4], Me^INbOCljl.
Смесь хлора и NbOCl3, пропускаемая над нагретым до 700°
углем, полностью превращается в NbCl5.
Диоксихлорид ниобия, NbO2Cl, получают гидролизом NbOCl3
или действием газообразного НС1 на Nb2O5 при 700°. Это твердое
вещество коричневого цвета.
Комплексные соли ниобия, MeI[MbOCl4], MeJ[NbOCl5], окра-
шены в зеленовато-желтый цвет, гидролизуются в воде, полу-
чаются обработкой Nb2O5, NbOCl3 или NbCl5 конц. НС1, пири-
дином, хинолином или гидроокисями (хлоридами) щелочных
металлов. Примеры оксихлоросолей ниобия: С5Н5]\Н(КЬОС14],
2[C5H5NH)2[NbOCl5] -Н2О, C9H7NH[NbOCl4], (C9H7NH)2[NbOCl5J,
KlNbOClJ, K2[NbOCl5], Rb2[NbOCl5], Cs2[NbOCl5], (NH4)2[NbOCl5].
Пентабромид ниобия, NbBr5, получают прямым взаимодей-
ствием составляющих компонентов при 450—500°, действием
паров брома (увлекаемых током СО2) на нагретую смесь Nb2O5
с углем или нагреванием S2Br2 с Nb2O5.
13*
196
ПОБОЧНАЯ ПОДГРУППА V ГРУППЫ
Ковалентное соединение NbBr5 представляет собой гигроско-
пичные пурпурно-красные призмы, плавящиеся при 150°; пере-
ходят в газообразное состояние без разложения в токе СО2 или
N2 (270°), дымят на воздухе, превращаясь в NbOBr3 и затем
в Nb2O5'7zH2O, растворяются в абсолютном спирте, в безводном
С2Н5Вг, гидролизуются водой, при нагревании легко восстанав-
ливаются водородом до NbBr3.
Действием натриевого производного циклопентадиена на
NbBr5 в тетрагидрофуране можно получить трибромид ди(цикло-
пентадиен)ниобия Nb(C5H5)2Br3. Соединение окрашено в темно-
коричневый цвет, гидролизуется на воздухе, разлагается при
нагревании (примерно при 260е), растворяется в С2Н5ОН, СНС13,
плохо растворяется в петролейном эфире.
Окситрибромид ниобия. NbOBr3, получают гидролизом NbBr5
или пропусканием паров брома над предварительно нагретой
в атмосфере азота или СО2 смесью Nb2O5 с углем.
Окситрибромид ниобия — нелетучее желтое твердое вещество,
менее устойчивое, чем NbOCl3, разлагающееся на воздухе, раство-
римое в абсолютном спирте и безводном С2Н5Вг, гидролизующееся
во влажном воздухе или в воде, образующее с бромидами щелоч-
ных металлов или гидробромидом пиридина или хинолина окси-
бромосоли, например KlNbOBrJ, K2[NbOBr5], Rb2[NbOBr5],
Cs2[NbOBr5], C5H5NH[NbOBr4], C9H7NH[NbOBrJ.
Пентаиодид ниобия, Nbl5, представляет собой желтое твердое
вещество, разлагающееся при 327°; получается при нагревании
металлического ниобия в парах иода (давление 1 ат) без доступа
воздуха.
При добавлении солянокислого спиртового раствора KI к спир-
товой смеси NbClj и пиридина (при кипячении) получают корич-
невые иглы (С5Н5КН)6[МЬ1П].
Сульфаты ниобия (V). При нагревании (в сосуде,
снабженном обратным холодильником) 15 г SO3 с 3 a NbCl5, рас-
творенным в 250 мл SO2C12, в течение 40 час получают белый оса-
док Nb2O(SO4)4 по уравнению
2NbCl5 + 14SO3 = Nb2O(SO4)4 + 5S2O5C12
Известны также сульфаты Nb2O3(SO4)2, Nb2O4SO4.
Хелатные соединения
При добавлении ацетона или спирта к растворам, полученным
обработкой ниобатов щавелевой кислотой, выделяются бесцветные
кристаллы оксалатокомплексов общей формулы Me*[NbO(C2O4)3] •
• пН2О, которые гидролизуются в воде, превращаясь в Nb2O5 •
. пН2О. Примеры оксалатокомплексов: Na3[NbO(C2O4)3] -4Н2О,
НИОБИЙ
197
K3[NbO(C2O4)3] -2Н2О, Rb3[NbO(C2O4)3b2H2O, (NH4)3.
.[NbO(C2O4)3bl,5H2O.
Добавляя ацетон в растворы, полученные обработкой ниобие-
вой кислоты Nb2O5-nH2O тартратами щелочных металлов или
ниобатов щелочных металлов винной кислотой, выделяют тар-
тратокомплексы общей формулы MeI[NbO(C4H4O6)2] -иНЬО, где
Me1 = Na+, К+, Rb+, NHJ, ЧгСа2*, ЧгВа2*, ЧгРЬ2*.
Растворением свежеосажденной ниобиевой кислоты Nb2O5 •
• иН2О в горячем щелочном растворе пирокатехина в атмосфере
азота получают желтые или красные растворы, из которых можно
выделить кристаллы различных хелатов пирокатехина с ниобием,
например (NH4)3[NbO(C6H4O2)3J -9Н2О — красный,
(NH4)2H[NbO(C6H4O2)3]-0,5C6H4(OH)2.3H2O - желтый,
K2H[NbO(CeH4O2)3] -2СвН4(ОН)2.ЗН2О — оранжевый,
H(CN3He)2[NbO(CeH4O2)j]3 -6Н2О — красный и др.
Добавлением ацетил- или бензоил ацетона к NbCl5, растворен-
ному в спирте, или добавлением этилового эфира к раствору,
полученному кипячением избытка ацетил- или бензоилацетона
с солянокислым раствором K2H[NbO(C6H4O2)3] •2СвН4(ОН)2 -ЗЕЬО
или (CN3He)2H[NbO(C6H4O2)3l ‘6Н2О, получают ацетил- или бен-
зоилацетонаты ниобия общей формулы [Nb(OA)2B]Cl2, где А —
= СН3, С2Н5, а В — ацетилацетон или бензоилацетон.
Соединение [Nb(OC2H5)2(C5H7O2)]Cl2 образует желтые кри-
сталлы с т. пл. 74—75°, а соединение [Nb(OC2H5)2(C10H9O2)]Cl2 —
желтые кристаллы с т. пл. 110—112°.
Известны также хелатные соединения ниобия с пирогаллолом
или салицилатами.
Металлоорганические соединения
Чистые карбонилы ниобия неизвестны. Описаны только кар-
бонильные производные, содержащие анион [Nb(CO)e]“. Среди
немногих металлоорганических соединений ниобия упоминается
коричнево-красный, разлагающийся при 260° (C5H5)2NbBr3.
Соединения включения
Гидрид ниобия, NbH, получают нагреванием металлического
ниобия (в виде порошка) в атмосфере водорода, действием воды
на сплав ниобий — натрий, восстановлением NbOCl3 водородом.
Гидрид ниобия представляет собой серовато-черный порошок
с плотностью 6—6,6 г/сл3; он плохо растворяется в НС1, HNO3,
разб. H2SO4, растворяется в конц. H2SO4, устойчив на воздухе,
превращается в Nb2O5 при сильном нагревании в кислороде или
на воздухе, выделяет водород под действием HF или распл. KHSO4.
198
ПОБОЧНАЯ ПОДГРУППА V ГРУППЫ
Нитрид ниобия, NbN, получают взаимодействием азота с ме-
таллическим ниобием (1000°) или нагреванием в азоте при 1250°
смеси Nb20s с сажей.
Нитрид ниобия образует серые кристаллы с плотностью
8,4 г'с.и3, плавящиеся при 2300° в атмосфере водорода. Он хорошо
проводит электрический ток, окисляется до Nb2O5 в теплом воз-
духе. плохо растворим в HNO3, НС1, H2SO4, царской водке,
щелочами разрушается с выделением аммиака при нагревании.
Карбид ниобия, NbC, получают нагреванием при 1700° метал-
лического ниобия с углем, нагреванием при 1200° Nb2O5 с углем
в атмосфере водорода, нагреванием при 1450° смеси ниобата калия
с К2СО3 и углем, пропусканием газообразной смеси NbCl5 с СН4
или С2Н2 над вольфрамовой проволокой, нагретой до 2400— 3000°.
Карбид ниобия представляет собой кубические серовато-
коричневые кристаллы с решеткой типа NaCl, плотностью
8,20 г,см3, т. пл. 3497°, т. кип. 4300°. Кристаллы хорошо проводят
электрический ток, обладают твердостью 9—10 по шкале Мооса,
плохо растворимы в кислотах (H2SO4, НС1, HNO3), при сильном
нагревании на воздухе загораются и горят, превращаясь в Nb2O5.
Борид ниобия, NbB2. При действии разб. HNO3 на остаток,
получающийся при электролизе (~1000°) расплавленной смеси,
состоящей из Nb2O5 с боратом и NaF, CaF2 или MgF2 (сила тока
25 а), выделяются мелкие серые кристаллы NbB2 с плотностью
6,4 г.см3. Они разлагаются в расплавленных щелочах или кар-
бонатах щелочных металлов, не взаимодействуют с НС1, HNO3
и царской водкой, вступают в реакцию с H2SO4 и HF.
ТАНТАЛ Та
Z — 73; ат. вес = 180,948
Валентность (I), (II), (III), (IV), V; заряд (1+),
(2+), (3+), (4+), 5+
Массовое число природного изотопа 181
Электронная структура атома тантала: К-L -М -N ‘bs^bp^d3 •
• 6s3.
Электронная структура атома тантала и иона Та5+.для 5d-
и бя-орбиталей:
5d3 6s2
f I Т | Т | I [n
Ta
5d
6s
Tas+
ИСТОРИЧЕСКАЯ СПРАВКА
В 1802 г. шведский ученый Экеберг нашел в двух минералах
(один из Кимито в Финляндии, другой из Иттерби в Швеции)
новый окисел белого цвета, трудно растворимый в кислотах.
ТАНТАЛ
199
Экеберг назвал металл нового окисла «танталом» (в честь грече-
ского героя Тантала), а минералы, используемые для получения
окиси тантала,—«танталитами».
В чистом виде металлический тантал впервые был получен
в 1903 г.
РАСПРОСТРАНЕННОСТЬ В ПРИРОДЕ
В природе тантал встречается только в виде соединений в раз-
личных сложных минералах совместно с ниобием, титаном, цир-
конием, вольфрамом, германием, оловом, щелочными, щелочнозе-
мельными и редкоземельными металлами. Тантал достаточно
широко распространен в природе и его содержание в земной коре
составляет 2,4 •10~5 вес. %.
Наиболее важным минералом тантала является танталит.
Танталит, (Fe, Мп)(ТаО3)2, находится в изоморфной смеси
с колумбитом, называемой танта лито-колумбитом, и представляет
собой черные или коричнево-черные пластинчатые кристаллы
с плотностью 8,20 z/cmz и твердостью 6 по шкале Мооса. Залежи
танталита имеются в Финляндии, Швеции, Норвегии, Франции,
СССР, Бразилии, США, Индии, Японии, Австралии.
К другим минералам, содержащим тантал, относятся: торолит SnTa2O7,
моссит Fe(Nb, Та)2О6, тапиолит Fe(Ta, Nb)2O6, микролит Са2Та2О7, иттротан-
талит Y4(Ta2O7)3, стибиотанталит SbTaO4, висмутотанталит.
Переработка концентратов руд тантала и ниобия, как и про-
цессы отделения тантала от ниобия, рассматриваются при описа-
нии ниобия.
ПОЛУЧЕНИЕ МЕТАЛЛИЧЕСКОГО ТАНТАЛА
Металлический тантал получают путем восстановления соеди-
нений K2[TaF7], ТаС15, Та2О5 при нагревании без доступа воздуха
или термической диссоциацией ТаС15 в вакууме.
При восстановлении соединений K2[TaF7] или ТаС15 посред-
ством нагревания со щелочным металлом в инертной атмосфере
(или вакууме) при использовании галогенидов щелочных металлов
NaCl, КС1 и др. в качестве флюса получают порошкообразный
металлический тантал, загрязненный очень небольшим количе-
ством сплава тантал — щелочной металл:
K2[TaF7] + 5Na = Та + 5NaF + 2KF ТаС15 + 5Na = Та + 5NaCl
Галогениды щелочных металлов отделяют от порошкообраз-
ного металлического тантала промыванием водой, а сплав тантал —
щелочной металл превращают в гидрид ТаН0187, который остается
в металле как примесь.
В результате нагревания при 1000° в графитовом тигле смеси
K2[TaF7] с избытком алюминия получают интерметаллическое
200
ПОБОЧНАЯ ПОДГРУППА V ГРУППЫ
соединение А13Та, которое остается в избытке алюминия под
защитным слоем шлака фторидов алюминия и калия.
Губчатый металлический тантал получают по процессу Кроля
магнийтермическим восстановлением пентахлорида тантала (750°)
без доступа воздуха:
2ТаС15 + 5Mg = 2Та + 5MgCl2
Металлический тантал можно получить в результате терми-
ческой диссоциации ТаС15 (по процессу Ван-Аркеля — Бюргесса)
на нагретой до 1800—2000° вольфрамовой проволоке в вакууме
или при пропускании смеси паров пятихлористого тантала с водо-
родом над вольфрамовой проволокой, нагретой до 1200—1400°.
В последнем случае получают металлический тантал, содержащий
некоторое количество водорода, который можно удалить нагре-
ванием металла в вакууме.
Пятиокись тантала восстанавливают металлотермически
в инертной атмосфере с помощью натрия, калия, кальция и других
металлов; в качестве флюсов используют NaCl, КС1, СаС12ит. д.,
восстанавливают также углем в атмосфере водорода, LiH при
800—900° или СаН2 при 1025—1100° и сплавами редкоземельных
металлов при повышенной температуре.
Для получения металлического тантала не применяют алюмо-
термического восстановления Та2О5, поскольку в результате
этого процесса получаются сплавы тантал — алюминий.
Порошкообразный металлический тантал можно получить
электролизом оксалатных растворов Та2О5*пН2О, подкисленных
3%-ной НС1 или H2SO4; в качестве катода используют платину
или графит. Можно также проводить электролиз смеси K2[TaF7]
с Та2О5, КС1, KF при 750° в графитовом тигле, который служит
анодом; катод может быть из никеля, молибдена или графита;
плотность тока 90 а!дмг. Алюмо- или кремнетермическим восста-
новлением смеси окислов тантала с окислами железа в присут-
ствии окиси кальция получают сплав ферротантал.
Порошкообразный или губчатый металлический тантал можно
превратить в компактный металл при помощи процессов порош-
ковой металлургии: плавление в электродуговой печи (один элек-
трод вольфрамовый, другой из тантала) в высоком вакууме или
в печи со сфокусированным электронным пучком (используя коль-
цевой излучающий катод) также в высоком вакууме. Электронные
пучки, излучаемые нагретой до 2000° вольфрамовой проволокой,
фокусируются таким образом, чтобы облучать и материал, подле-
жащий расплавлению, и поверхность ванны. При плавлении под
действием электронного пучка можно добиться наилучшей дега-
зации тантал# и получить его монокристаллы.
Очистку металлического тантала осуществляют электролити-
чески или переплавкой в вакууме при высокой температуре.
ТАНТАЛ
201
ФИЗИЧЕСКИЕ И ХИМИЧЕСКИЕ СВОЙСТВА
Тантал — блестящий белый парамагнитный металл с кубиче-
ской объемноцентрированной кристаллической решеткой; плот-
ность 16,69 г!см\ т. пл. 2997°, т. кип. 5425°, твердость 6 по шкале
Мооса. Он ковок, вязок, тягуч, легко протягивается в очень
тонкие листы или нити. При загрязнении углеродом, кремнием,
бором, азотом, алюминием, оловом, титаном или следами водорода
и кислорода тантал становится твердым и хрупким.
В коллоидном состоянии тантал окрашен в черный цвет;
он образуется в электрической дуге между двумя танталовыми
электродами, погруженными в изобутиловый спирт.
Металлический тантал обладает высокой электропроводностью
(становится сверхпроводником при 4,2° К), высокой термоэлек-
тронной эмиссией и способностью абсорбировать газы.
Известны многочисленные сплавы тантала с Be, Mg, Al, Ga,
TI, Ge, Bi, Ti, Zr, V, Nb, Hf, Cr, Mo, W, U, Mn, Re, Fe, Co, Cu, Ag,
Zn, Cd, C, Si, В. Амальгамы тантала неизвестны.
С химической точки зрения тантал относится к металлам
с пониженной химической активностью и, следовательно, обла-
дает превосходной коррозионной стойкостью.
Металлический тантал в компактном состоянии не корродирует
на воздухе или при соприкосновении с водой на холоду и при
обычной температуре. Будучи нагретым до 1200°, порошкообраз-
ный тантал разлагает воду с выделением водорода.
При нагревании металлический тантал взаимодействует с фто-
ром, хлором, кислородом, бромом, серой, селеном, теллуром, азо-
том, углеродом, с различными органическими реагентами, напри-
мер, с бензолом, нафталином и др.:
Та + 5/2Е2 — TaFs 2Та 5/20г = ТВ2О5
Та + 5/2С12 = TaCls Та 2S = TaS2
Та + 5/2Вг2 = ТаВг6 Та + i/2N2 = TaN
Та 4- С = ТаС
Металлический тантал не взаимодействует с разбавленными
или концентрированными кислотами НС1, HNO3, H2SO4, царской
водкой и водными растворами щелочей и растворяется в плави-
ковой кислоте (в контакте с платиной), в смеси HF с HNO3 и в рас-
плавленных щелочах.
Действием газообразного НС1 на металлический тантал при
410° получают ТаС15, а при 600—750° — низшие хлориды. Метал-
лический тантал реагирует также с газообразным НВг (375°),
образуя ТаВг5, а выше 550° — ТаВг3. *
По своим физическим и химическим свойствам металлический
тантал обнаруживает большое сходство с платиной.
202
ПОБОЧНАЯ ПОДГРУППА V ГРУППЫ
ПРИМЕНЕНИЕ
Благодаря превосходной коррозионной стойкости, значитель-
ной механической прочности, высокой температуре плавления,
низкому давлению пара, малому коэффициенту термического
расширения, способности удерживать газы и другим ценным
свойствам металлический тантал находит самое разнообразное
применение.
В химической промышленности применяют изготовленные
из тантала дистилляционные аппараты, трубопроводы, змеевики
для охлаждения или нагревания, конденсоры, кипятильники,
абсорбционные емкости, резервуары, цистерны, электроды и т. д.
В химических лабораториях используются танталовые капсулы,
тигли, ячейки, мешалки, шпатели, сита, разновесы для аналити-
ческих весов.
В электропромышленности тантал используют как конструк-
ционный элемент в выпрямителях (с потенциалом до 12,5 в и очень
малым внешним сопротивлением) и в электролитических конден-
саторах (большой емкости и малого размера, работающих в широ-
ком интервале температур — от —60° до +120°).
Металлический тантал очень широко используют в электрон-
ной промышленности. В электронно-вычислительных машинах
применяют криотроны —мельчайшие детали из тантала и ниобия.
В области хирургии используют аппараты, инструменты, пла-
стинки и нити из тантала. Тонкие танталовые нити служат для
сшивания кровеносных сосудов и нервов, а более толстые нити
и пластины из тантала используют в костной хирургии.
В промышленности синтетических волокон тантал идет на изго-
товление прядильных фильер, а в пищевой промышленности
применяется прессовое оборудование из тантала для изготовления
синтетических мембран.
Металлический тантал успешно заменяет платину, золото,
серебро или их сплав в аппаратуре химической промышленности.
Он служит катализатором получения искусственных алмазов
из графита при давлении 50 000—130 000 ат и температурах
2000-2400°.
Металлический тантал служит для получения термостойких,
жаропрочных, сверхтвердых, нержавеющих сплавов, обладающих
большой химической стойкостью. Ферротантал является добавкой
в производстве нержавеющих и термостойких сталей с хромом
и никелем.
Области использования сплавов тантал — ниобий указывались
при описании ниобия.
Карбид тантала с карбидом вольфрама, молибдена, титана
и других металлов используют для получения сверхтвердых
термостойких сплавов.
ТАНТАЛ
203
СОЕДИНЕНИЯ (ОБЩИЕ СВОЙСТВА)
Известны многочисленные устойчивые соединения пятива-
лентного тантала; соединения одно-, двух-, трех- и четырехвалент-
ного тантала встречаются реже, и, как правило, они неустойчивы.
С увеличением валентности у тантала повышается устойчивость
соединений и склонность к образованию координационных соеди-
нений.
В табл. 31 приведены формулы и указан цвет многих соедине-
ний двух-, трех-, четырех- и пятивалентного тантала.
Соединение одно-, двух-, трех- и четырехвалентного тантала
обычно получают восстановлением соединений пятивалентного
тантала, за исключением дисульфида TaS2, который получают
прямым взаимодействием элементов при нагревании.
Соединения одновалентного тантала
Закись тантала, Та2О, представляет собой черные ортором-
бические кристаллы, получаемые восстановлением Ta2S5 углем
или металлическим танталом При температурах 1770° и выше
1300° соответственно.
Соединения двухвалентного тантала
Известно немного соединений двухвалентного тантала, обра-
зующихся при восстановлении соединений пятивалентного тан-
тала. Все они окрашены в темный цвет, преимущественно зеленый
или черный, и отличаются пониженной устойчивостью (окисляют-
ся в водных растворах).
Окись тантала, ТаО, получают в виде черного порошка,
состоящего из кубических кристаллов, при частичном восстанов-
лении Та2О5 углем в электрической печи при давлении 10—
20 мм рт. ст.
Дихлорид тантала, ТаС12, отгоняется при нагревании ТаС15
с металлическим алюминием и А12С16 в вакууме при 600° и давле-
нии 2—3 мм рт. ст.
Дихлорид тантала представляет собой темно-зеленое вещество
с плотностью 4,76 г! см?, плохо растворимое в холодной воде
и окисляющееся в теплой воде по уравнению
ЗТаС12 + ЗН2О = 2ТаС13 + Та(ОН)3 + 3/2Н2
Известны аддукты дихлорида тантала с НС1, НВг, H2SO4,
а также аддукты, содержащие одновременно галогеноводородные
кислоты и пиридин, например ЗТаС12-НС1 -4Н2О, ЗТаС12*НВг •
• 4Н2О, 3TaCl2-H2SO4-H2O, 3TaCl2-HCl-2C5H5N, ЗТаС12 •
- 3HC1-G5H5N, 3TaCl2-3HBr-C5H5N.
Таблица 31
Соедине- ния включе- ния C=S W . § «« К 3Z 2SUSO « о, cd аа £ а® а Н ф Н о «Но g § « ф U Д-ф К " gHo
Хелатные соединения пятивалентного тантала • —? °Лоо за ЬДЩ S ' нЁ
Суль- фиды 1 1 TaS2 Серый 1
Йодиды и другие иодсодер- жащие соединения 1 1 1 Та15 Коричне- вый
Бромиды и другие бромсодер- жащие соединения ТаВг2 Черный ТаВг3 Зеленый 1 ТаВг5 Желтый ТаОВг3
Хлориды и другие хлорсодержащие соединения ТаС12 Зеленый ТаС12-2Н2О Зеленый ЗТаС12-НХ.4Н2О ТаС13 Зеленый ЗТаС13-Н2О 3TaCl3-C5H5N Ta3OCl7.2CsH5N ТаС14 Зеленый “То То Z О „ Д’ д о д о д зи «Я Д cd Д cd Д Ю hi юЛ C-jU Lj U д § О Q е У и О с—1 О И О Ен Щ Щ Ю Q5 и и
Фториды и другие фторсодер- жащие соединения 1 TaF3 Серовато- белый 1 TaF5 Бес- цветный TaOF3TaO2F Бесцветный H[TaFeJ. 6Н2О Me[TaFe] Me’[TaF,] Meii[TaF,]
Окислы ТаО Черная Та2О3- •ЗН2О Зеленая ТаО2 1 Корич- невая Та2О3 Бесцвет- ная Ta2O5- • nH2O Белая
Соединение Соединения двухвалент- ного тан- тала Соединения трехвалент- ного тан- тала Соединения четырехва- лентного тантала Соединения пятивалент- ного тан- тала
ТАНТАЛ
205
Дибромид тантала, ТаВг2, представляет собой твердое
неустойчивое вещество черного цвета; реагирует с водой с выде-
лением водорода.
Соединения трехвалентного тантала
Соединения трехвалентного тантала получают в результате
восстановления соединений пятивалентного тантала; они окра-
шены преимущественно в зеленый цвет и окисляются легче, чем
соответствующие соединения ванадия.
Ион Та3+, относительно устойчивый в холодной воде, очень
легко окисляется в горячей.
Гидратированная окись тантала, Та2О3-ЗН2О или Та(ОН)3,
выделяется при обработке водного раствора трихлорида тантала
небольшим количеством NaOH или КОН.
Водная окись тантала Та(ОН)3 представляет собой зеленый
студнеобразный, очень легко окисляющийся осадок с амфотер-
ными свойствами.
Трифторид тантала, TaF3. Под действием смеси паров HF
с Н2 на нагретый до 310° порошкообразный металлический тантал
образуется смесь TaF5 с TaF3. Если эту смесь нагревать в вакууме
при 600°, то TaF5 сублимируется и остается трифторид тантала.
Он представляет собой серовато-белые кубические кристаллы,
плохо растворимые в воде и кислотах; при нагревании царская
водка и азотная кислота переводят его в Та2О5; при прокаливании
на воздухе или в кислороде он также переходит в Та2О5.
Трихлорид тантала, ТаС13, отгоняется при нагревании ТаС15
с металлическим алюминием и А12С16 при 350—400° в вакууме
(соответственно при давлении 2—3 мм рт. ст.):
ЗТаС15 + 2А1 = ЗТаС13 + А12С1в
Трихлорид тантала представляет собой темно-зеленое твердое
вещество с плотностью 3,84 г/см3, диспропорционирующее при
500° на ТаС12 и ТаС15, растворяющееся в воде и превращающееся
в Та2О3-ЗН2О под действием щелочей
ЗТаС13 = 2ТаС12 + ТаС15
Водный раствор трихлорида тантала неустойчив на воздухе,
поскольку ТаС13 превращается в Та2О5’5Н2О:
2ТаС13 + ЮН20 = Та2О5-5Н2О + 6НС1 + 2Н2
Если пропускать газообразный НС1 через водный раствор
трихлорида тантала, выпадают зеленые кристаллы Та3ОС17-ЗН2О.
Известны кристаллогидрат ЗТаС13-Н2О и аддукты трихлорида
тантала ЗТаС13 -C5H5N, Та3ОС17-2С2Н5ОН, Та3ОС17 -2CSH5N.
206
ПОБОЧНАЯ ПОДГРУППА V ГРУППЫ
Трибромид тантала, ТаВг3, получают восстановлением ТаВг5
водородом при 700° или обработкой НВг, нагретого до 550° метал-
лического тантала.
Зеленый порошок ТаВг3 плохо растворим в эфире, растворим
в воде и спиртах СН3ОН, С2Н5ОН; он обладает отчетливо выра-
женными восстановительными свойствами и мгновенно окисляется
во влажном воздухе до Та2О5.
При нагревании водного раствора трибромида тантала выпадает
ТаО2*2Н2О и выделяется водород.
Соединения четырехвалентного тантала
Соединения четырехвалентного тантала образуются при вос-
становлении соединений пятивалентного тантала или непосред-
ственно из элементов, как в случае TaS2. Они окрашены в корич-
невый, зеленый или серый цвет, относительно неустойчивы.
Двуокись тантала, ТаО2 или Та2О4, можно получить при
восстановлении Та2О5 (в смеси с парафином) углем при 1700°
или магнийтермическим путем.
Двуокись тантала — тугоплавкий порошок коричневого цвета,
при нагревании на воздухе или с KNO3 он превращается в Та2О5;
плохо растворяется в кислотах и царской водке, превращается
в танталаты путем сплавления со щелочами.
Тетрахлорид тантала, ТаС14, отгоняется вместе с А12С1в
при взаимодействии ТаС13 с ТаС15 или ТаС15 с металлическим
алюминием в вакууме (2—3 мм рт. ст.):
6ТаС15 + 2А1 = 6ТаС14 + А12С1в ТаС15 + ТаС13 = 2ТаС1,
Тетрахлорид тантала представляет собой твердое вещество
зеленого цвета с плотностью 3,57 г!см\ реагирующее с водой
и щелочами и обладающее четко выраженными восстановитель-
ными свойствами:
4ТаС14 + ЮН2О 2ТаС13 + Та2О5-5Н2О + 10НС1
ТаС14 + 7NaOH = Na3TaO4 + 4NaCl + ЗН2О + V2H2
Дисульфид тантала, TaS2, получают прямым взаимодействием
элементов при высокой температуре, действием паров CS2 или
смеси H2S с CS2 на пятиокись тантала при 900—1300° или сильным
нагреванием паров ТаС15 с H2S.
Дисульфид тантала образуется в виде металлоподобного твер-
дого вещества серого цвета. При сжигании на воздухе или кипя-
чении с HNO3, H2SO4, царской водкой дисульфид тантала пре-
вращается в Та2О5, а при кипячении с КОН — в танталат калия..
ТАНТАЛ
207
Соединения пятивалентного тантала
Наиболее многочисленные устойчивые соединения тантала
соответствуют электроположительному танталу(У), который встре-
чается как в виде катионов Та5+, ТаО3+, так и в виде анионов
ТаОГ, Та2О*~, ТапО^, [Та5О16]’-, [Та6О19]«-, [ТаСО^]3", [TaF6]“,
[TaF7]2-, [TaF8]3~, [TaOFJ-, [TaOF5]2-, [TaOF6]3-.
В отличие от соединений двух-, трех- и четырехвалентного
тантала соединения пятивалентного тантала устойчивы, бесцветны
или окрашены в желтый либо коричневый цвет, а также обладают
склонностью к комплексообразованию.
Соединения тантала(У) в отличие от аналогичных соединений
ванадия(У) и ниобия(У) не восстанавливаются в водных растворах
цинком в кислой среде.
Неорганические соединения
Пятиокисъ тантала, Та2О5, получают нагреванием металли-
ческого тантала в кислороде при 1600°, обезвоживанием при
нагревании гидрата Ta2Os-nH2O, нагреванием окислов ТаО,
Та2О3, ТаО2 в кислороде, прокаливанием TaS2, TaN, прокалива-
нием купфероната тантала или H[TaFe] -6Н2О:
2Та + 5/2О2 = Та2О5 + 488,9 ккал Та2О3 + О2 = Та2О5
Та2О5-пН2О = Та2О5 -|- нН2О 2ТаО2 -|- V2O2 — Ta2Og
2ТаО + 3/2О2 = Та2О5 2TaS2 + 1з/2О2 = Та2О5 + 4SO2
Пятиокись тантала существует в виде белого порошка, а также
двух кристаллических модификаций (а-Та2О5 — ромбическая
и Р-Та2О5 — тетрагональная), бесцветных, диамагнитных с плот-
ностью 8,2—8,7 г/см3 и т. пл. 1877°. Пятиокись тантала нераство-
рима в воде и кислотах, за исключением плавиковой кислоты.
При растворении Та2О5 в последней образуется H[TaF6] -6Н2О,
а при растворении Та2О5 в смеси H2SO4 с Н2О2 — пероксотанта-
ловая кислота.
Пятиокись тантала — амфотерное соединение (кислотные свой-
ства слабее, чем у Nb2O5), которое при сплавлении со щелочами
или карбонатами щелочных металлов превращается в танталаты.
Танталаты образуются также при сплавлении Та2О5 с окислами
различных металлов, например MgO, CaO, SrO, BaO, FeO, МпО.
В отличие от Nb2O5 пятиокись тантала не восстанавливается
при 1250° водородом, а восстановление Та2О5 углем или металли-
ческим магнием при нагревании приводит к образованию ТаО2.
Для полного восстановления Та2О5 при нагревании используют
гидрид кальция, силицид кальция или элементарный кремний.
Нагреванием Та2О5 с РС15 при 245°, с S2C12 при 330°, с СС14
при 300° получают ТаС15. Нагревание смеси Та2О5 с углем в парах
208
ПОБОЧНАЯ ПОДГРУППА V ГРУППЫ
брома приводит к образованию ТаВг5, а в атмосфере азота — TaN.
Под действием сероуглерода или смеси CS2 с H2S пятиокись тан-
тала превращается в TaS2 при температуре 900—1300°.
Пятиокись тантала используют для получения специальных
стекол, в производстве изоляционных материалов, для свечей
автомоторов и нитей накаливания, а также в качестве катализа-
тора окисления, этерификации и дегидратации.
Гидратированная пятиокись тантала {танталовая кислота),
Та2О5-иН2О, образуется под действием разбавленных кислот
НС1, H2SO4, HNO3 на водные растворы танталатов или фторо-
танталатов щелочных металлов, барботированием двуокиси угле-
рода через растворы танталатов щелочных металлов, обработкой
щелочами комплексных оксалатотанталатов:
2К3ТаО4 + 6НС1 = Та2О5-ЗН2О + 6КС1
2K3Ta3Oe -f- 6НС1 = 3(Та2О5 -Н2О) + 6КС1
К8[Та6О19] + 4H2SO4 = ЗТа2О5 -4Н2О + 4K2SO4
2K2[TaF7]+ 2H2SO4 + (га + 5)Н2О = Та2О5-гаН2О + 2K2SO4 + 14HF
Студнеобразный белый осадок Та2О5-иН2О с непостоянным
содержанием воды проявляет явно выраженную склонность
к образованию коллоидных растворов и растворяется в HF, НС1,
HNO3, H2[SiF6], горячей H2SO4, щавелевой кислоте и оксалатах
щелочных металлов, винной кислоте и тартратах щелочных
металлов, в щелочах и карбонатах щелочных металлов, в щелоч-
ных растворах пирокатехина.
Танталаты и политанталаты. Известны орто-
танталаты MeJTaO4, метатанталаты (Ме1ТаО3)п, пиро- или
дитанталаты МерТа2О7, пентатанталаты Mei[Ta5O16] -а:Н2О
и гексатанталаты Ме*[Та6О19] -а:Н2О, где Me1 = Na+, К+ и М2+ =
= Са2+, Sr2+, Mn2+, Со2+, Fe2+ и т. д.
Ортотанталаты и пиро- или дитанталаты образуются при
сильном нагревании Та2О5 со щелочами или карбонатами щелоч-
ных металлов, а также с окислами различных металлов, например
CaO, SrO, ВаО.
Та2О5 + 6КОН = 2К3ТаО4 + ЗН2О Та2О5 + ЗК2СО3 = 2К3ТаО4 + ЗСО2
Та2О6 2СаО = Са2Та2О7
Гексатанталаты, Na8[Ta6O19]-(24—25)Н2О, К8[Та6О19] -19H2O,
Rb8[Ta6O19] -14Н2О, Cs8[Ta6O19] -14Н2О, выделяются в виде бес-
цветных кристаллов (гексагональных или моноклинных), когда
расплавы, образующиеся при высокой температуре из Та2О5
с гидроокисями или карбонатами щелочных металлов, растворяют
в воде и концентрируют в вакууме.
6Ме3ТаО4 + 5Н2О = Ме*[ТавО19] + 10МетОН
ТАНТАЛ
209
При обработке водных растворов гексатанталатов щелочных
металлов растворами солей щелочноземельных металлов полу-
чают гексатанталаты щелочноземельных металлов, например
Ве4[Та6О19] -6Н2О. При прокаливании гексатанталатов получают-
ся полимеры метатанталатов:
Na3[TaeO19bxH2O = NaeTaeO18 + 2NaOH + (х — 1)Н2О
В качестве примеров метатанталатов можно назвать мономеры
Mn(TaO3)2, Fe(TaO3)2, Са(ТаО3)2, Со(ТаО3)2.
Пентатанталаты, Na7[Ta5O16] -(11 или 20)Н2О или
К7[Та5О16] >12Н2О, отделяют в виде бесцветных кристаллов при
концентрировании в вакууме над Р4О10 или при кипячении рас-
твора, полученного действием воды на расплав Та2О5 с большим
избытком NaOH или КОН. Известна также литиевая соль
Ы7[Та5О16] -20Н2О.
В качестве примеров пиро- или дитанталатов упоминаются
Са2Та2О7, Sr2Ta2O7, Ва2Та2О7.
Пероксотанталаты. При добавлении спирта в рас-
творы, полученные обработкой танталатов щелочных металлов
перекисью водорода в присутствии щелочей или карбонатов щелоч-
ных металлов, отделяются белые (или слегка желтоватые) кри-
сталлы тетрапероксоортотанталатов М+[Та(О2)4] -пН2О. Они рас-
творимы в воде, устойчивы при обычной температуре и разла-
гаются при нагревании или под действием конц. H2SO4. Примеры
тетрапероксотанталатов: К3[Та(О2)4] Д/гНгО, Na3[Ta(O2)4] •
• (1 или 14)Н2О, КСа[Та(О2)4]-4,5Н2О, NaCa[Ta(O2)4]-4,5Н2О,
KMg[Ta(O2)4]-7Н2О, NaMg[Ta(O2)4] -8Н2О, BbMg[Ta(O2)4]-9Н2О.
При действии разб. H2SO4 на тетрапероксоортотанталат калия
на холоду получают тетрапероксоортотанталовую кислоту
Н3[Та(О2)4] *пН2О — более устойчивую, чем тетрапероксоортонио-
биевая кислота.
Известна также и монопероксометатанталовая кислота
Г°Ч 1
та—О —ОН .
-0^ J
Пентафторид тантала, TaF5, получают в платиновых сосудах
прямым взаимодействием элементов при 250°, действием безводной
плавиковой кислоты на ТаС15 и термическим разложением
Ba3[TaF8]2 (1000°) в токе сухого воздуха:
Та + %F2 = TaFs TaCls + 5HF = TaF6 + 5HC1
Ba3[TaF8]2 = 2TaF3 -|- 3BaF2
Бесцветные двулучепреломляющие гигроскопичные призмы
TaF5 имеют плотность 4,74 г/см3, т. пл. 96,8° и т. кип. 229,4°, они
очищаются перегонкой в вакууме, гидролизуются водой, раство-
14-0101
210
ПОБОЧНАЯ ПОДГРУППА V ГРУППЫ
ряются в S2C12, CHGI3, СС14, BrF3, CS2, дымящей HNO3, конц.
НС1 или H2SO4 при нагревании, плохо растворяются в СН3СООН,
С2Н5ОН, С2Н5 — О — С2Н5. Пентафторид тантала взаимодействует
при нагревании с водородом, бромом, иодом, серой, цинком,
серебром, медью, железом, а также с хлоридами РС13, AsCl3,
SiCl4, SnCl4, TiCl4, SbCl5. При нагревании TaF5 c KHF2 или KF
получают K2[TaF7], а при обработке TaF5 щелочами образуется
бесцветный оксифторид тантала TaOF3:
TaF5 + NaOH = TaOF3 + NaF 4- HF
Гексафторотанталовая кислота, H[TaF6] -6H2O, выделяется
при концентрировании на водяной бане раствора Та205-пН2О
в 40%-ной плавиковой кислоте. Это бесцветные игольчатые кри-
сталлы с т. пл. 15°.
Комплексные фторотанталаты. Известны мно-
гочисленные фторо- и оксифторосоли общих формул MeI[TaF6],
MeJ[TaF7], МеЩТаР7], MeJ[TaF8], Me4TaOF4], MeJ[TaOF5]r
Mei[TaOFe], где Me1 = Li+, Na+, K+, Rb+, Cs+, NHJ, Tl+ и Me11 =
= Ba2+, Zn2+, Cu2+ и т. д.
Примеры фторотанталатов и оксифторотанталатов: Li[TaF6] •
• 2Н2О, Na[TaF6], Cs[TaF6], NHJTaFJ, Ag[TaF6], Ca[TaF6]2,
Ba[TaF6]2, Na2[TaF7]-H2O, K2[TaF7], Rb2[TaF7], Cs2[TaF7],
(NH4)2[TaF7], Tl2[TaF7], Zn[TaF7]-7H2O, Cu[TaF7b4H2O,
Na3[TaF8], (NH4)3[TaF8], Ba3[TaF8]2, K[TaOF4], K2[TaOF5],
K3[TaOFe], (NH4)3[TaOF6J.
Гептафторотанталат калия, K2[TaF7], получают при нагре-
вании TaF5 и KHF2 до 100° или продолжительным нагреванием
TaF5 с KF при 220-230°:
TaF5 + 2KHF2 = K2[TaF7] + 2HF TaF5 + 2KF = K2[TaF7]
Соль K2[TaF7] образует бесцветные моноклинные призмы
с плотностью 4,06 г!см3, устойчивые на воздухе, гидролизующиеся
в воде. Они могут быть перекристаллизованы из разб. HF, обла-
дают пониженной по сравнению с K2[NbF7] растворимостью в воде
и восстанавливаются при нагревании щелочными металлами.
2K2[TaF7] + (п + 5)Н2О Ta2O5-nH2O + 4KF -f- 10HF
K2[TaF7] + 5Na = Та + 5NaF + 2KF
Структура аниона [TaF7]2- изображена на рис. 8.
Оксигексафторотанталат калия K3[TaOF6], получают дей-
ствием фторидов щелочных металлов на пятиокись тантала в воде:
Та2О5 + 12KF + ЗН2О = 2K3[TaOF6] + 6КОН
Фтористоводородная кислота превращает оксифторотанталаты
во фторотанталаты:
K3[TaOFe] + 2HF = K2[TaF7] + KF + Н2О
ТАНТАЛ
211
Пентахлорид тантала, ТаС15 или Та2С110, получают непо-
средственно из элементов (при 600°), нагреванием нитрида, суль-
фида или карбида тантала в атмосфере хлора, действием газооб-
разного хлора на предварительно нагретую в токе СО2 смесь
Та2О5 с углем, нагреванием Та2О5 с СС14 при 300°, с РС15 при 245°
и с S2C12 при 330°:
Та + 5/2С12 = ТаС15 ТаС + »/2С12 = ТаС15 + С
Та + 5НС1 = ТаС15 + 5/2Н2 Та2О6 + 5С + 10С12 = 2ТаС15 + 5СОС12
TaN + 5/2С12 = ТаС15 + V2N2 Та2О5 + 5РС16 = 2ТаС15 + 5РОС13
TaS2 + 5/2С12 == ТаС15 + 2S 2Та2О6 + 1OS2C12 = 4ТаС15 + 5SO2 + 15S
Пентахлорид тантала выделяется в виде желтых гексагональ-
ных кристаллов с плотностью 3,68 г! см*, т. пл. 211,3° и т. кип.
241,6°, их очищают сублимацией при 200—220° в вакууме
Рис. 8. Структура анио-
на [TaF7]2~.
Рис. 9. Структура, пред-
ложенная для катиона
[ТавС112]2+ комплекса
[Та6С112]С12-7Н2О.
(1—2 мм рт. ст.). Эти кристаллы дымят на воздухе, бурно реа-
гируют с водой, превращаясь в Та2О5-иН2О и выделяя НС1,
растворяются в CS2, С2Н5ОН, СН3 — СО — СН3, СН3СООН,
насыщенных газообразным НС1. Пентахлорид тантала обладает
ограниченной растворимостью в СНС13, СС14, С2Н5Вг, плохо
растворим в С8Н6 и С2Н5 — О — С2Н5, в расплавленном состоя-
нии взаимодействуют с платиной.
Под действием сероводорода ТаС15 превращается в TaS2.
Натрий, магний, алюминий, цинк и олово восстанавливают ТаС15
при нагревании.
Известны аддукты ТаС15 ^CsHsN, TaCl5-raNH3, где п — 7, 10
или 12.
Если восстанавливать ТаС15 амальгамой натрия в атмосфере
азота, а затем продукт восстановления обработать разб. НС1
и испарять образовавшийся зеленый раствор на холоду, то выде-
ляются зеленые гексагональные кристаллы дихлорида хлоротан
14*
212
ПОБОЧНАЯ ПОДГРУППА V ГРУППЫ
тала, [Та6С112]С12*7Н2О. Они обладают ограниченной раствори-
мостью в холодной воде, растворяются в теплой воде, спирте или
в разбавленных растворах кислот НС1, H2SO4. Структура катиона
[Ta6Oi2]2+ показана на рис. 9.
Известны также гидроокись [Та6С112](ОН)2 -8Н2О и бромид
[Ta6Cli2]Br2 -7Н2О.
Оксихлориды тантала и оксихлоротан-
тал а т ы пиридина или хинолина. После сублима-
ции ТаС15 при 220° и давлении 2 мм рт. ст. остается нелетучий
диоксихлорид ТаО2С1.
Прямым окислением ТаС15 или его гидролизом можно получить
оксихлорид тантала.
Известны аддукты: 4ТаО2С1-7C5H5N, ТаО2С1 ^CsHjN, ТаОС13*
. 2(C5H5N -НС1) -2С2Н5ОН или (C5H5NH)2[TaOCl5] -2С2Н5ОН,
TaOCl3-2(C9H7N.HCl).2C2H5OH или (C9H7NH)2[TaOCl5l •
. 2С2Н5ОН.
Пентабромид тантала, ТаВг5, получают действием паров
брома на нагретый до 260—300° порошкообразный металлический
тантал или пропусканием паров брома над предварительно нагре-
той в токе СО2 смесью Та2О5 с углем.
Пентабромид тантала образует гигроскопичные пластинчатые
кристаллы желтого цвета; плотность 4,67 г!см?, т. пл. 280,2°,
т. кип. 348,8° (очищаются перегонкой в вакууме). Он дымит
на воздухе, гидролизуется водой, растворяется в СН3ОН, абсо-
лютном спирте и безводных С2Н5Вг, СС14, CS2.
Дибромид бромотантала, [Та6Вг12]Вг2-7Н2О, образуется при
испарении на холоду зеленого раствора, полученного обработкой
разб. НВг продукта восстановления ТаВг5 амальгамой натрия
в атмосфере азота. Соединение выделяется в виде зеленых кри-
сталлов.
Известны гидроокись [Та6Вг12](ОН)2-8Н2О и хлорид
[Та6Вг12]С12-7Н2О бромотантала.
Окситрибромид тантала, ТаОВг3, получают действием паров
НВг на Та2О5 или действием брома на смесь Та2О5 с углем. Это
твердое вещество желтого цвета.
Пентаиодид тантала, ТаТ5, получают при многократной пере-
гонке ТаВг5 с безводным HI. Пентаиодид тантала нельзя получить
действием паров иода на металлический тантал или на смесь
Та2О5 с углем.
Выделенный Та15 представляет собой темно-коричневые пла-
стинчатые кристаллы с т. пл. 365° и т. кип. 543°. Они очищаются
перегонкой в токе СО2, гидролизуются водой, плохо растворяются
во многих органических растворителях типа С6Н6, СС14, CS2
СНС13.
Сульфат тантала, Ta2(SO4)5, получают при нагревании в сосу-
де, снабженном обратным холодильником, ТаС15 с SO3, раство-
ТАНТАЛ
213
ренном в SO2C12, в течение 40 час:
2ТаС15 + 15SO3 = Ta2(SO4)5 + 5S2O5C12
Сульфат тантала выделяется в виде белого осадка, который
разлагается выше 100° и не восстанавливается цинком в серно-
кислом растворе.
Хелатные соединения
При добавлении ацетилацетона или бензоилацетона к спирто-
вому раствору ТаС15 образуются хелатные соединения общей
формулы [Та(ОА)2В]С12 (где А — СН3, С2Н5 и В = ацетилацетон
или бензоилацетон).
При взаимодействии Та2О5 -мН2О с горячими щелочными
растворами пирокатехина в атмосфере азота выделяются хелатные
соединения, например, К3Н[Та2О(С6Н4О2)6]-ЗС6Н4(ОН)2-ЮН20,
(NH4)3H[Ta2O(CeH4O2)6]-ЗС6Н4(ОН)2-7Н2О в виде серовато-зеле-
ных кристаллов.
При обработке танталатов щелочных металлов (или Та2Ов •
• пН2О) щавелевой кислотой (или оксалатами щелочных метал-
лов), винной кислотой (или тартратами щелочных металлов)
образуются хелатные соединения, например К3[ТаО(С2О4)3],
Ме1Н4[ТаО4(С4Н4О6)] -пН2О, которые осаждаются ацетоном или
этиловым спиртом.
Металлоорганические соединения
Нейтральные карбонилы тантала неизвестны. Описаны только
их производные, содержащие анион [Та(СО)6]~.
Из немногих металлоорганических соединений тантала упо-
минаются трибромид дициклопентадиентантала (С5Н5)2ТаВг3 —
твердое вещество цвета ржавчины, гидролизующееся на влажном
воздухе и разлагающееся при 280°.
Соединения включения
Гидрид тантала, ТаНО176, получают в виде хрупких серых
кристаллов при нагревании до 900° металлического тантала в водо-
роде или при пропускании смеси ТаС15 с водородом над раскален-
ной платиновой проволокой.
Нитриды тантала. Нитрид тантала, TaN, выде-
ляют нагреванием порошкообразного металлического тантала
в смеси азота с водородом (без доступа кислорода или паров
воды), сильным нагреванием Ta3N5B газообразном NH3, пропуска-
нием смеси паров ТаС1в с азотом и небольшим количеством
водорода над нагретой до 2500—2800° вольфрамовой проволокой.
Нитрид тантала представл яет собой темно-синие гексагональ-
ные кристаллы (в порошке черные), с плотностью 14,1 г!см3
214
ПОБОЧНАЯ ПОДГРУППА V ГРУППЫ
и т. пл. 3087°; соединение — хороший проводник электриче-
ского тока.
Нитрид тантала, Ta3N5, получают обработкой газообразным
аммиаком ТаС15 при высокой температуре. Он представляет собой
красный порошок, который загорается при сильном нагревании
на воздухе и взаимодействует с расплавленным КОН с выделе-
нием аммиака.
Известны и другие нитриды тантала: Ta2N, TaN2.
Карбиды тантала образуются при восстановлении
Та2О5 углем в электрической печи, нагревании TaN в угольном
тигле, нагревании порошкообразного металлического тантала
с углем до 1700—2100° в атмосфере водорода или до 1800—2550°
в парах бензола, нафталина и т. д. Желто-коричневые кубические
кристаллы ТаС плавятся при 3877°, обладают высокой твердостью,
значительной хрупкостью, хорошей электропроводностью.
Известны карбиды Та2С — серого цвета и Та6С5 — серебристо-
серого цвета.
Борид тантала, ТаВ2, образуется при обработке разб. HNO3
остатка, полученного в результате электролиза расплавленной
при 1000° смеси Та2О5 с боратом и NaF, CaF2, MgF2.
Борид тантала образует блестящие серые мелкие гексагональ-
ные кристаллы с плотностью 11,70 г!см3. Они тверды, хрупки,
обнаруживают хорошую электропроводность.
Известны также другие бориды тантала: ТаВ, Та2В, Та3В,
Та3В4.
Силициды тантала. Описано несколько силицидов
тантала: TaSi2, Ta5Si3, Ta2Si, Ta4,5Si.
ПОБОЧНАЯ ПОДГРУППА VI ГРУППЫ
ПЕРИОДИЧЕСКОЙ СИСТЕМЫ
(ПОДГРУППА ХРОМА)
ХАРАКТЕРИСТИКА ПОДГРУППЫ
В побочной подгруппе VI группы периодической системы
химических элементов (подгруппе хрома) находятся переходные
металлы хром (Сг), молибден (Мо) и вольфрам (W).
Таблица 32
В табл. 32 рассматривается электронная структура атомов
хрома, молибдена и вольфрама. Атомы хрома и молибдена имеют
по 1 электрону на последнем электронном уровне и по 2.6.5 элек-
тронов на предпоследнем электронном уровне. Атомы вольфрама
имеют 2 электрона на последнем электронном уровне и 2.6.4 элек-
тронов на предпоследнем электронном уровне. d-Орбитали пред-
последнего электронного уровня этих элементов укомплектованы
электронами неполностью. Участвуя в химических реакциях,
атомы хрома, молибдена и вольфрама образуют соединения,
в которых они двух-, трех-, четырех-, пяти- и шестивалентны.
Электроположительный характер этих элементов усиливается
в ряду хром, молибден, вольфрам, а устойчивость солей, в кото-
рых степень окисления указанных элементов меньше шести,
уменьшается с увеличением атомного веса.
В табл. 33 приведены наиболее важные константы элементов
этой подгруппы.
В компактном состоянии хром, молибден и вольфрам представ-
ляют собой парамагнитные блестящие серебристо-белые металлы
Таблица 33
Элемент Хром Сг Молибден Мо Вольфрам W
Цвет | в компактном состоянии Стальной Серебристо-белый Серебристо-белый
в порошке Серый Серый Серый
Кристаллическая структура а-Сг Кубическая объемно- центрированная ₽-Сг Г ексагональная компактная Кубическая объемно- центрированная Гексагональная компактная a-W Кубическая объемно- центрированная ₽-W Кубическая особого типа
Атомный номер 24 42 74
Атомный вес 51,996 95,94 183,85
Атомный радиус, А 1,30 1,39 1,41
Радиус иона Ме6+, А (по Гольдшмидту, Полингу, Аренсу) 0,35; 0,52; 0,52 Нет данных 0,62; 0,62 Нет данных 0,68; 0,62
Атомный объем (при 20°), см3/г-атом 7,28 9,41 9,50
Плотность (при 20°), г/см3 7,2 10,22 19,32
Твердость по Бринеллю, кг/мм2 90 120 70
Твердость по шкале Мооса 5 5,5 4,5
Температура плавления, °C 1855 2610 3380
Скрытая теплота плавления, кал/г 32 70 44
Температура кипения, °C 2642 4830 5930
Удельная теплоемкость при 20°, кал/г-град 0,1178 0,061 0,0321
Коэффициент теплопроводности X, кал-см^-сек^-град-1 (при 0°) — 0,346 0,40
Сопротивление р-ЮБ (при 0°), ом-см 3 5,70 4,91
Электропроводность (Hg = 1) 7,2 16,4 19,5
Магнитная восприимчивость х • 10—в, эл.-магн. ед. (при 18°) 3,7 0,93 0,252
Теплота перехода атомов в газообразное состояние, ккал (при 25°) 80,5 155,5 201,6
Потенциал иони- зации, эв Ме->Ме+-|-е~ Ме+->Ме2+-|-е~ Ме2+->Ме3+ + е_ Ме3+->Ме4+-|-е- Ме4+ -> МеБ+ + е- MeB+->Me6+-j-c~ МеБ+->Ме7+-|-е- 6,76 16,49 30,95 50,9 72,4 96,0 167,6 7,10 16,15 27,13 40,53 55,6 71,7 132,7 7,98 17,70 24,08 35,36 48 61
Потенциал иони- зации, ккал/г-атом Ме^-Ме++е- 155,4 162,7 183,0
Нормальный потенциал (при 25°), в Сг/Сг3+—0,74 Сг/Сг2+ —0,86 Мо/Мо3+—0,2 W/W3+4-0,ll W/W«++0,68
Нормальный потенциал окислительно- восстановительных систем, в Сг2+/Сг3+—0,41 Cr2O72/2Cr3+-f-1,33 Мо3+/Мо4+ —0,10 Мо3+/МоБ+—0,23
Валентность (I), II, III, (IV), (V), VI (II, III, IV, V), VI (II, III, IV, V), VI
Массовые числа природных изотопов 52, 53, 50, 54 98, 96, 95, 92, 97, 94, 100 184, 186, 182, 183, 180
Распространенность элементов в земной коре, вес.% 3,3-ю-2 1,0-10~3 7,0-Ю-з
218
ПОБОЧНАЯ ПОДГРУППА VI ГРУППЫ
с плотностью от 7,2 г/сл«3 до 19,32 г/сл«3 (тяжелые металлы), т. пл.
от 1855 до 3380° и т. кип. от 2642 до 5930° (тугоплавкие металлы).
Они обладают твердостью 4,5—5,5 по шкале Мооса и становятся
хрупкими, теряя пластичность, при загрязнении углеродом,
азотом, кремнием, фосфором, серой, бором. В чистом состоянии
эти элементы имеют превосходные механические свойства, позво-
ляющие любую обработку: они могут прокатываться, протяги-
ваться, штамповаться.
Известны многочисленные сплавы этих металлов с В, Al, С,
Si, Ti, Zr, N, P, As, V, Ta, S, U, Mn, Fe, Co, Ni, Pd, Pt, Cu, Zn.
С химической точки зрения металлы подгруппы хрома имеют
пониженную активность; они устойчивы на воздухе и в воде
в обычных условиях, взаимодействуют при нагревании с кисло-
родом, галогенами, серой, селеном, теллуром, азотом, фосфором,
углеродом, кремнием, бором и т. д.
По большинству физических и химических свойств молибден
и вольфрам похожи между собой и несколько отличаются от
хрома.
Металлический хром при обычной температуре растворяется
в разбавленных кислотах (HF, НС1, НВг, HI, H2SO4), а при
нагревании — в окислительно-щелочных расплавах. Под дей-
•Соединение Сг2С14 [Мо6С18]С14 [WeCl8]Cl4
Вид Белые кристаллы Желтый аморфный порошрк Серый аморфный порошок
Молекулярный вес 245,804 1001,076 1528,536
Плотность (при 20°), г[смЪ 2,930 3,714 5,436
Температура плавления, °C 815 Тугоплавкий Тугоплавкий
Температура кипения, °C 1302 » »
Теплота образования, ккал/моль 97,3 264 360
ПОБОЧНАЯ ПОДГРУППА VI ГРУППЫ
219
ствием конц. HNO3 или царской водки металлический хром
пассивируется. Металлические молибден и вольфрам растворяются
при нагревании в конц. HNO3, царской водке или окислительно-
щелочных расплавах. Под действием концентрированных кислот
(HNO3, H2SO4) при обычной температуре молибден и вольфрам
также пассивируются.
В природе хром, молибден и вольфрам находятся в виде соеди-
нений (хром — в хромитах, хроматах, бихроматах, силикатах;
молибден в виде дисульфида или молибдатов; вольфрам в виде
вольфраматов и дисульфида) в различных минералах. Хром более
распространен в природе, чем вольфрам и молибден.
Металлы подгруппы хрома получают восстановлением при
нагревании их окислов Cr2O3, а-МоО3, S-MoO2, WO3, W40n,
WO2, водородом, алюминием, кремнием, кальцием, магнием,
цинком, восстановлением на катоде некоторых расплавленных
солей хрома, молибдена или вольфрама, при термической диссо-
циации Сг12, Мо(СО)6, WC16, восстановлением при нагревании
СтС13, МоС15, WC18 водородом и т. д.
Порошкообразный металлический молибден или вольфрам
может быть переведен в компактное состояние спеканием в атмо-
сфере водорода, плавлением в вакуумных электродуговых печах
Таблица 34
СгВг2 [МовВг8]Вг4 [WeBr8]Br4 Сг12 [Мов18]4 [ WeT8]I4
Желтовато- белые кристаллы Оранжевый аморфный порошок Черно- синие кристаллы Коричнево- красные кристаллы Красновато- коричневый аморфный порошок Коричне- вый аморф- ный поро- шок
211,814 1534,548 2062,008 305,804 2098,492 2625,953
4,356 4,88 — 5,02 4,30 6,90
842 Туго- плавкий Разлагается при 400 793 —
1127 » — 827 — —
81 216 156 58 162 96
220
ПОБОЧНАЯ ПОДГРУППА VI ГРУППЫ
и плавлением в печах с сфокусированным электронным пучком
в атмосфере аргона или гелия.
Хром, молибден, вольфрам, а также сплавы на их основе —
очень ценные материалы современной техники. Стали, выплавлен-
ные с добавками хрома, молибдена или вольфрама, характери-
зуются высокой твердостью и стойкостью к действию различных
химических реагентов.
Большинство соединений этих металлов окрашено, они нелету-
чи и тугоплавки.
Наиболее устойчивы соединения трех- и шестивалентного
хрома, шести- и иногда четырехвалентного молибдена и шестива-
лентного вольфрама. Соединения двухвалентного хрома, двух-,
трех-, (четырех-) и пятивалентных молибдена и вольфрама неустой-
чивы, легко окисляются (являются восстановителями); получают
восстановлением соединений трехвалентного хрома и соответ-
ственно шестивалентных молибдена и вольфрама. В отличие
от молибдена и вольфрама для хрома известно координационное
соединение, отвечающее степени окисления I.
В табл. 34—38 приведены основные константы некоторых
соединений этих металлов.
Если в состоянии низших степеней окисления эти элементы
проявляют металлические свойства, то в состоянии высших сте-
пеней окисления соединения имеют кислотный характер и, как
правило, ковалентные связи (в случае простых соединений).
Трехокиси СгО3, МоО3, WO3 являются ангидридами кислот,
которые под действием расплавленных щелочей или карбонатов
щелочных металлов превращаются в хроматы, молибдаты, воль-
фраматы, а под действием галогеноводородов образуют окси-
галогениды общей формулы Ме6+ОХ4, Ме6+О2Х2 или соеди-
нения, содержащие анионы Ме6+ОХ“, Мев+О2Х", Ме6+О2Х^“,
Мев+О3Х|-.
В табл. 39 приведены формулы известных галогенидов хрома,
молибдена и вольфрама, сгруппированные по валентностям.
Гексагалогениды хрома не известны, единственным известным
гексагалогенидом молибдена является MoF6, гексагалогениды
вольфрама следующие: WFe, WC16, WBr6. Соединения MoF4,
WF6, WClg, WBr6 имеют ковалентные связи, они легко летучи
(обладают низкими температурами плавления и кипения),
растворимы в органических растворителях и гидролизуются
в воде.
По степеням окисления соединения двух-, трех-, четырех-
и шестивалентного молибдена проявляют большее сходство с соот-
ветствующими соединениями вольфрама, чем хрома. В трехвалент-
ном состоянии молибден отличается от вольфрама, поскольку для
последнего не известны тригалогенид, окись и сульфид. Известны
только координационные соединения.
Таблица 35
N СО £ Серые крис- таллы 247,978 7,75 1 1 84
MoS2 Черные кристал- лы 160,068 4,80 1185 (под давле- нием) 1 1
WC14 Серые кристал- лы 325,662 од 1 Разла - гае тс я при 477 121
M0CI4 Корич- невые кри- сталлы 237,752 1 317 322 79
41 и и Желтое га- зообразное или корич- невое твер- дое веще-1 ство 193,808 1 1 Диссоции- рует при -80 1
wf4 Красно- вато-ко- ричневые кристал- лы 259,843 1 1 ' Разла- гается при 800 1
CrF4 Корич- невый аморф- ный по- рошок 127,990 0 од 277 295 266
о £ Коричневые кристаллы 215,849 од 1500-1600 Разлагается при 1852 140,9
МоО2 Фиол етово- коричне- вые кри- сталлы 127,939 6,47 Сублими- руется в вакууме при 1100 1 1977 (с разл.) 132,5
о о 1 Черные кристаллы 83,994 1 1 1100 (разла- гается при 427) 142,5
Соединение Вид Молекулярный 1 вес Плотность (при 20°), г/см? Температура плав- ления, °C Температура ки- пения, °C Теплота образова- ния, ккал/моль
Таблица 36
Соединение СгО3 МоОз WO3 Н2СгО4 Н2МоО4 H2WO4
Вид Красные ром- бические кристаллы Белые ром- бические кристаллы Желтые ром- бические кристаллы Водный рас- твор: разбав- ленный— желтый, кон- центрирован- ный—красный Белые микро- кристаллы Оранжево-жел- тый аморф- ный порошок
Молекулярный вес 99,994 193,938 231,848 118,009 159,954 247,864
Плотность (при 20°), г /см3 2,80 4,69 7,16—7,22 — — 5,5
Температура плавле- ния, °C 197 795 1473 Разлагается при нагрева- нии Разлагается при 90 Разлагается при 150
Температура кипе- ния, °C Разлагается при 550 1257 1667 — — —
Теплота образования, ккал/молъ 137,7 180,3 201,5 — — —
Таблица 37
Соединение MoFe WFe MoOF4 wof4 МоОС14 WOC14
Вид Бесцветные кристаллы Бесцветный газ Бесцветные кристаллы Бесцветные кристаллы Зеленые кри- сталлы Красные кри- сталлы
Молекулярный вес 209,93 297,840 187,933 275,843 253,751 341,661
Плотность (при 20°), г/см3 Жидкость 2,55 Газ 12,3 г/л 3,00 — — —
Температура плавле- ния, °C 17,5 2,5 97 110 Сублимирует- ся при 100 209
Температура кипе- ния, °C 36 19,5 180 187,5 — 233
Гидролизуется в воде ДО MoO2F2, Н2МоО4 wof4, H2WO4 MoO2F2, Н2МоО4 H2WO4 МоО2С12, Н2МоО4 WO2C12, H2WO4
Таблица 38
eq к eq о £ Оранжево- красные кристаллы 375,658 1 Разлагается при на- гревании
eq га eq о о 2 Оранжево- красные кристаллы 287,747 1 Легко раз- лагается при обыч- ной тем-
eq к eq О о Коричнево- красные кристаллы 243,803 1 Легко раз- лагается при обыч- ной тем-
WO2C12 Желтые кристал- лы 286,755 1 265
МоО2С12 Желтовато- белые кристал- лы 198,844 3,31 Легко суб- лимирует- ся
eq -ч и eq о и Красная жидкость 154,900 1,912 -96,5
MoO2F2 Бесцветные кристал- лы 165,935 3,494 Сублими- руется при 270
eq eq о и § А о ® и 2 м 2 ® И S еб у О Сн О Он Д Л О О к д ф д 121,991 ф 6 ' с -ag „ Я И К с S н Ч н И о Сублими- руется при 29,6
Соединен не Вид Молекулярный вес Плотность (при 20°), г/см3 Температура плав- ления, °C
ПОБОЧНАЯ ПОДГРУППА VI ГРУППЫ
225
Таблица 39
CrF2 CrF3 CrF4 CrF5 —
— MoF3 — M0F5 MoF6
— — wf4 — WF6
Сг2С14 СгС13 СгС14 — —
[Мо6С18]С14 МоС13 МоС14 M0C15 —
[WeCl8]Cl4 __ WC14 WC15 WCle
СгВг2 СгВг3 — — —
[Мо6Вг8]Вг4 МоВг3 МоВг4 — —
[WeBr8]Br4 — — WBr5 WBre
Сг12 Сг13 — — —
[Мов18]14 — — —
[WeI8]I4 WI4 — —
Соединения трехвалентного хрома в отличие от соответствую-
щих соединений молибдена очень устойчивы и не обладают ни вос-
становительным, ни окислительным характером.
Из окислов СгО3, МоО3, WO3, кислот Н2СгО4, Н2МоО4 или
Н2МоО4-Н2О, H2WO4 и H2WO4-H2O можно получить изо- и гете-
рополисоединения. По сравнению с хромом степень конденсации
у соединений молибдена и вольфрама больше. .Многочисленные
координационные соединения содержат в составе аниона хром,
молибден и вольфрам в высшей степени окисления.
Согласно моделям Андерсона и Кеггина, изо- и гетерополи-
комплексы молибдена и вольфрама имеют в своей основе кольца
или цепи, составленные из октаэдров МоО6 или WO6, подобно
тому как в силикатах имеются кольца из SiO4 — тетраэдров.
15-0101
226
ПОБОЧНАЯ ПОДГРУППА VI ГРУППЫ
Известны металлоорганические и пероксосоединения элемен-
тов подгруппы хрома.
Помимо соединений, отвечающих определенным степеням окис-
ления, известно много нестехиометрических соединений.
В отличие от элементов главной подгруппы VI группы перио-
дической системы, имеющих (за исключением полония) неметал-
лический характер, элементы побочной подгруппы VI группы —
металлы.
По характерному электронному строению атомов с незапол-
ненной d-орбиталью, по физическим и химическим свойствам,
определяемым этой электронной структурой, по склонности к обра-
зованию электроположительных ионов и координационных соеди-
нений элементы подгруппы хрома относятся к переходным метал-
лам.
ХРОМ Сг
Z = 24; ат. вес = 51,996
Валентность (I), II, III (IV), (V), VI; заряд
(1-Ь)> 2-)-, 3-)- (4—|—), (5-|—), 6Ц-
Массовые числа природных изотопов 52, 53, 50,
54 и 56
Электронная структура атома хрома: К -L •3s23p63d5 -4/.
Электронная структура атома хрома и ионов Сг2+, Сг3+ для
3d- и 4«-орбиталей:
3d5 4s1 3<И 4s 3d3 4s
It It It It It I [3 |t|t|t|t| I □ Itltltl I I □
Cr Cr2+ Cr3+
ИСТОРИЧЕСКАЯ СПРАВКА
В 1765 г. русский геолог Паллас обнаружил на Урале оран-
жево-красный минерал, который он назвал крокоитом. После того
как было установлено, что крокоит содержит свинец, минерал
стали называть красным свинцом.
В 1797 г., разлагая крокоит минеральными кислотами, Воке-
лен впервые получил наряду с солью свинца кислоту красного
свинца, позднее названную хромовой кислотой; последняя при
сплавлении с бурой давала красивый зеленый перл.
Новый металл, полученный Вокеленом в виде серого порошка
путем сильного нагревания хромовой кислоты с углем в графито-
вом тигле, был назван хромом — от греческого слова chroma,
что означает цвет, поскольку соединения этого элемента много-
цветны.
Металлический хром впервые был получен Беккерелем в 1843 г.
электролитическим путем. В 1893 г. Муасан выделил металличе-
ХРОМ
227
ский хром, восстанавливая Сг2О3 углем в электрической печи,
а в 1898 г. Гольдшмидт получил хром из Сг2О3 алюмотермическим
способом.
Хотя производство феррохрома начало развиваться с 1907—
1908 гг., все же в промышленных масштабах сплавы хрома стали
получать только в 1915 г.
Первые предприятия по производству минеральных красите-
лей на основе хрома появились в 1816 г. в Англии и в 1818 г. во
Франции.
Начало использования электролитического хромирования или
хромирования при 1000° в атмосфере галогенидов хрома металлов
и сталей относится к 1925 г. Эти процессы имеют исключительно
большое значение, поскольку хромированные объекты нашли
широкое применение.
РАСПРОСТРАНЕННОСТЬ В ПРИРОДЕ
Хром относится к весьма распространенным элементам. Он
находится в виде соединений в различных минералах. Содержание
хрома в земной коре составляет 3,3-10-2 вес.%. Спектральным
методом доказано присутствие хрома на Солнце, звездах и в метео-
ритах. Часто хром сопутствует железу или замещает алюминий,
трехвалентное железо и другие элементы в различных минералах.
К наиболее важным минералам хрома относятся следующие.
Хромит, FeCr2O4, содержит 46,46% Сг. Он относится к классу
шпинелей и встречается в виде черно-коричневых октаэдрических
кристаллов с плотностью 4,5 г/см?, т. пл. 1545—1850° и твердо-
стью 5,5—7,5 по шкале Мооса. Залежи хромита находятся в США
(Пенсильвания, Калифорния, Северная Каролина), СССР (на
Урале), Канаде, Англии, Швеции, Турции, Греции, Югославии,
Румынии, Португалии, Египте, Родезии, Южной Африке,
Японии.
К разновидностям хромсодержащих шпинелей относятся маг-
нохромит (Mg, Fe)Cr2O4, алюмохромит Fe(Cr, А1)2О4, хромпико-
тит (Mg, Fe)(Cr, А1)2О4 и др.
Крокоит, РЬСгО4, уже упоминался в числе минералов свинца
и уваровит, Ca3Cr2(SiO4)3,— в числе минералов кальция.
Кочубеит является разновидностью клинохлора (который
представляет собой силикат алюминия, хрома, железа и магния);
встречается в виде зеленых призм с плотностью 2,61—2,78 г/см3
и твердостью 2—2,5 по шкале Мооса.
К другим минералам хрома относятся стихтит 2MgCO3 -5Mg(OH)2 •
2Сг(ОН)з, вокеленит (фосфат и хромат свинца и меди), волконскоит (гидра-
тированный силикат алюминия, хрома и железа), камерерит (силикат алю-
миния, хрома, железа и магния), лопецит К2Сг2О7, добрелит FeS-Cr2S3 и др.
В небольших количествах соединения хрома содержатся в во-
дах рек Сибири, Японского моря и в золе растений.
15*
228
ПОБОЧНАЯ ПОДГРУППА VI ГРУППЫ
ПЕРЕРАБОТКА ХРОМИТОВЫХ РУД
При сплавлении природного хромита FeCr2O4 с К2СО3 в отра-
жательной печи, куда подается избыток кислорода, получают
К2СгО4, Fe2O3 и СО2 по уравнению
2FeCr2O4 + 4К2СО3 + 7/2О2 = 4К2СгО4 + Fe2O3 + 4СО2
Охлажденный расплав перемешивают с горячей водой в авто-
клаве с целью растворения хромата калия. При обработке кон-
центрированного раствора К2СгО4 серной кислотой выпадают
кристаллы сульфата калия и в растворе остается бихромат калия,
который при концентрировании выделяется в виде оранжевых
кристаллов К2Сг2О7 -2Н2О:
2К2СгО4 H2SO4 = К2Сг2О7 K2SO4 -|- Н2О
Плохорастворимую в воде зеленую окись хрома Сг2О3 и рас-
творимый в воде K2SO4 получают восстановлением бихромата
калия серой при нагревании (в железных сосудах):
К2Сг2О7 -|- S = Сг2О3 -|- K2SO4
Благодаря различной растворимости в воде сульфат калия
легко отделяется от Сг2О3, который служит исходным продуктом
для получения металлического хрома.
Вместо серы для восстановления бихромата калия можно
использовать крахмал или углерод.
Схема переработки хромита приведена ниже.
Сплавление с 4К2СОз
2FeCr2O4 7/ n *" ^K2CrO4-|-Fe2O3-|-4CO2
Хромит Подача /2о2 (Плохо растворимый)
| +Н2°
К2СгО4
Конц, раствор
+H2SO4
К2Сг2О7 K2SO4
Раствор Кристаллы
Концентрирование
К2Сг2О7-2Н2О
| Сплавление с серой
Сг2О3 -)- K2SO4
(Плохо раство- (Растворимый)
римый)
ХРОМ
229
Хромит, смешанный с минералами железа (окислами или
карбонатом), служит для получения в доменных печах хромистых
сталей и феррохрома.
ПОЛУЧЕНИЕ МЕТАЛЛИЧЕСКОГО ХРОМА
Металлический хром можно получить восстановлением Сг2О3
при нагревании с алюминием, кремнием, кальцием, водородом,
катодным восстановлением растворов или расплавов солей хрома,
восстановлением при нагревании безводного СгС13 водородом,
щелочными или щелочноземельными металлами, цинком и маг-
нием, термической диссоциацией дииодида хрома.
Алюмотермическое восстановление окиси хрома
Сг2О3 + 2А1 = 2Сг + А12О3 + 120,30 кал
Для лабораторного получения металлического хрома в тигель
из огнеупорной глины или фарфора (высотой примерно 10 см)
помещают 10 г порошкообразного CaF2, затем сухую однородную
порошкообразную смесь из 35 г Сг2О3 и 25 г К2Сг2О7 (или меньшее
количество сухого СгО3) плюс 17 г тонко измельченного А1.
В середину тигля помещают зажигательную смесь из 15 г ВаО2
и 5 г порошкообразного алюминия и ленты металлического магния.
Подготовленный таким образом тигель помещают в вытяжной
шкаф (в песчанной бане или ящике с песком) и затем, надев защит-
ные очки с темными стеклами, зажигают магниевую ленту, закры-
вают тягу и наблюдают за реакцией на расстоянии. Металличе-
ский хром, полученный таким образом, остается на дне тигля
в виде шариков (корольков). Фторид кальцит (являясь флюсом)
снижает температуру плавления смеси и способствует образованию
металлического хрома в виде корольков. Бихромат калия является
источником кислорода и способствует повышению температуры
реакции.
Металлический хром, полученный промышленным алюмотер-
мическим способом, содержит 97,99% Сг. Основная примесь
в нем — железо.
Кремнетермическое восстановление окиси хрома
2Сг2О3 + 3Si + ЗСаО = 4Сг + 3CaSiO3
При сильном нагревании в электродуговой печи, футерованной
магнезитом, порошкообразной смеси Сг2О3, кремния, извести
и некоторого количества CaF2 получают металлический хром
и силикат кальция, который с фторидом кальция образует легкую
массу, отделяющуюся по охлаждении в виде шлака. Хотя избыток
элементарного кремния не употребляется, получающийст по этому
процессу металлический хром загрязнен силицидами хрома.
230
ПОБОЧНАЯ ПОДГРУППА VI ГРУППЫ
Металлотермическое восстановление окиси хрома
металлическим кальцием
При восстановлении смеси окиси хрома Сг2О3 с расплавлен-
ными СаС12 и ВаС12 металлическим кальцием в атмосфере аргона
получают ковкий металлический хром, который можно обраба-
тывать при нагревании.
Электролитическое получение металлического хрома
Электролитический хром получают электролизом водных рас-
творов его соединений или электролизом расплавов CrF3 с гало-
генидами щелочных металлов.
При электролизе раствора, содержащего 250 г/л СгО3 и 2,5—
3 г/л SOI’ в виДе H2SO4, применяют электрический ток при плот-
ности 27 а/дм2 и напряжении 5—6 в, свинцовый анод и катод
из алюминия, железа, меди или никеля. При этом на катоде
выделяется не пристающий к нему слой металлического хрома.
При использовании электролизера с пористой диафрагмой
можно применить в качестве питательного раствора водный рас-
твор NH4Cr(SO4)2-12H2O, который содержал бы 500 г/л Cr2(SO4)3
и 168 г/л (NH4)2SO4, в качестве катодного раствора смесь, содержа-
щую 64 г/л Cr2(SO4)3, 65 г/л CrSO4, 530 г/л (NH4)2SO4, а в качестве
анодного раствора — раствор, содержащий 290 г/л (NH4)2SO4,
290 г/л H2SO4, 8 г/л Cr2(SO4)3 и 39 г/л СгО^-. С этими растворами
процесс ведут при 50—60°, pH ~ 2,5, плотности тока 10,7 а/дм2,
используя алюминий или нержавеющую сталь (катод) и свинец
(анод).
Электролиз расплава CrF3 и смеси фторидов щелочных метал-
лов ведут при 800—950° в атмосфере азота или аргона. Графито-
вый тигель, в котором ведут процесс, служит одновременно
анодом; катодом является молибденовый брусок. В результате
на катоде образуется металлический хром в виде дендритов (при
напряжении выше 1 в) или одновременно хром и щелочные метал-
лы (при напряжении выше 4 в).
В качестве смесей фторидов щелочных металлов используют
2NaF -LiF (при 800°), 2NaF -3KF (при 800-950°) и 3,3NaF -l,7CaF2
(при 950°).
Другие процессы получения металлического хрома
Металлический хром можно также получить восстановлением
Сг2О3 чистым водородом при 1500°, термической диссоциацией
дииодида хрома (по процессу ван Аркеля — Кучмана — де Бура),
восстановлением безводного трихлорида хрома водородом при
1100—1200°, щелочными или щелочноземельными металлами
в атмосфере водорода, цинком, магнием и т. д., а также восста-
ХРОМ
231
новлением хроматов парами калия:
Сг2О3 + ЗН2 = 2Сг + ЗН2О СгС13 + 3/2Н2 = Сг + ЗНС1
Сг12 Сг + 12 СгС13 + 3Na = Сг + 3NaCl
2СгС13 + 3Zn = 2Сг + 3ZnCl2
Для получения металлического хрома непригоден процесс
восстановления Сг2О3 или СгО3 углем, так как в этом случае
неизбежно загрязнение его карбидами.
ПОЛУЧЕНИЕ ФЕРРОХРОМА
При алюмотермическом восстановлении смеси окислов Сг2О3
с Fe2O3 (или с FeO, Fe3O4) или при восстановлении хромита углем
в электродуговой печи получают феррохром, который является
сплавом железо — хром (60—72% хрома).
При алюмотермическом восстановлении окислов Сг2О3 с TiO2
или с МпОг, V2O5, МоО3 и т. д. получают сплавы хром — титан,
хром — марганец, хром — ванадий и хром — молибден.
ФИЗИЧЕСКИЕ И ХИМИЧЕСКИЕ СВОЙСТВА
В компактном состоянии металлический хром серого, как
сталь, цвета. Устойчивая модификация а-Сг имеет кубическую
объемно-центрированную кристаллическую решетку, а модифи-
кация Р-Сг — плотно упакованную гексагональную структуру
кристаллов. Последняя модификация постепенно переходит
в а-Сг.
При нагревании в вакууме амальгамы хрома до температуры
выше 300° получают тонко измельченный металлический хром,
который обладает пирофорными свойствами. Коллоидный раствор
хрома образуется в электрической дуге между двумя хромовыми
электродами, помещенными в изобутиловый спирт.
Плотность металлического хрома 7,2 г/сж3 при 20°, т. пл. 1855°,
т. кип. 2642°, твердость 5 по шкале Мооса (или « 90 кг!мм?
по Бринелю для электролитически полученного хрома). Хром
парамагнитен и в компактном состоянии ковок, тягуч, вязок,
поддается обработке давлением. При загрязнении (углеродом,
водородом и т. д.) хром становится твердым, хрупким, ломким;
обработка давлением при этом невозможна.
Известно очень много сплавов хрома с Fe, Ni, Со, Pd, Mn, Mo,
W, S, Se, Те, N, P, As, Sb, C, Si, Ge, Ti, Zr, Hf, B, Al, Be, Mg,
Zn, Cu, Hg. Примеры интерметаллических соединений хрома:
CrFe, Сг2Со2, СгСо, Cr3Pd2, СгМп3, CrS, Cr2S3, CrSe, Cr2Se3,
CrTe, Cr2Te3, Cr2N, CrN, Cr3P, Cr2P, CrP, CrP2, CrAs, CrSb,
CrSb2, Cr23C6, Cr7C3, Cr3C2, Cr3Si, Cr2Si, CrSi, CrSi2, Cr3Ge,
232
ПОБОЧНАЯ ПОДГРУППА VI ГРУППЫ
Cr3Ge2, CrGe, Cr2Ti, Cr2Zr, Сг2В, CrB, Сг3В4, СгВ2, Сг3В2,
СгА13, СтА17, Be2Cr, CrHg3, CrHg.
В обычных условиях металлический хром устойчив на воз-
духе, в воде и по отношению ко многим химическим реагентам.
При 1800—2000° металлический хром горит в кислороде (в то
время как порошок пирофорного хрома загорается при нагрева-
нии на воздухе выше 300°) и превращается в окись хрома Сг2О3.
При высокой температуре раскаленный хром реагирует с пара-
ми воды;
2Сг + ЗН2О Сг2О3 + ЗН2
Действием газообразного фтора на сильно нагретый метал-
лический хром получают CrF4 и CrF5. Галогеноводороды HF,
HCI, НВг, Ш при высокой температуре или их водные растворы
при обычной температуре, взаимодействуя с металлическим хро-
мом, дают соответствующие дигалогениды.
При нагревании до 600—800° металлический хром с хлором,
бромом и серой образует СгС13, СгВг3, Cr2S3 соответственно.
При действии паров иода на нагретый до 700—850° хром в вакууме
получают Сг12, а при высокой температуре в атмосфере азота
образуется Сг!3. Реакция металлического хрома с сероводородом
при 1200° дает CrS.
Азот, фосфор, углерод, кремний и бор реагируют с сильно
нагретым хромом, образуя CrN, Cr3P, Cr23C6, Сг7С3, Cr3Si, CrSi, CrB.
Разбавленная серная кислота действует на хром с выделением
водорода, а конц. H2SO4 действует на хром при нагревании
с выделением SO2.
Металлический хром пассивируется концентрированной азот-
ной кислотой, царской водкой, хлором, бромом и кислородом при
комнатной температуре; аналогичный процесс имеет место при
анодном окислении.
При высокой температуре хром реагирует с расплавленными
щелочами, нитратами и хлоратами щелочных металлов.
По отношению к химическим реагентам хром ведет себя то как
активный металл, подобно железу, цинку и др., то как пассивный
металл, соответственно с низкой химической активностью, подобно
золоту, платине и т. д. Для активного хрома нормальный потен-
циал системы Сг/Сг2+ равен —0,86 в, а для системы Сг/Сг3+ он
составляет —0,74 в (при 25° для раствора 1 г/л). Хром раство-
ряется при обычной температуре в разбавленных кислотах (HF,
НС1, НВг, HI, H2SO4) с выделением водорода и образованием
солей. На воздухе образуются соли хрома(Ш), а в отсутствие
воздуха (соответственно в атмосфере водорода) образуются соли
хрома(П).
Для пассивного хрома нормальный потенциал системы Сг/Сг2+
равен +1,19 в (при 25° для раствора 1 г/л); он не поддается дей-
ХРОМ
233
! ствию разбавленных кислот (HF, НС1, НВг, H2SO4) даже при
кипячении.
Пассивность хрома (и других металлов) объясняется образо-
ванием тонкой сплошной защитной пленки окиси на поверхности
металла. Явление пассивации можно приостановить с помощью
‘ । вибрации, ультразвука и просто очисткой поверхности металла,
i Химическая активность хрома иллюстрируется следующей
I схемой:
; с кислородом ИЛИ ВОДОЙ —► Сг2О3
с фтором —► CrF4 и CrF5
с хлором, бромом, серой —> СгС1з, СгВгз, Cr2S3
с иодом —* Сг13 и Сг12
Хром---------с HF, НС1, НВг, HI -> CrF2, СгС12, СгВг2, Сг12
Нагревание с Crg
с N2, Р, С, Si, В -> CrN, Сг3Р, Сг23С6, Сг7С3, Cr3Si, СгВ
с окислительно-щелочными расплавами —>• хроматы
щелочных металлов
! Металлический хром не токсичен, а соли хрома(Ш) и хроматы
I токсичны для человека и животных. Смертельная доза К2Сг2О7
I для человека равна 0,25—0,3 г.
ПРИМЕНЕНИЕ
Металлический хром используют для получения многих
сплавов. Наиболее важны феррохромы, хромистые чугуны и стали,
содержащие примерно 12% хрома; это нержавеющие, кислото-
упорные, термостойкие, жаропрочные, твердые материалы. Из
твердых хромистых сталей изготовляют инструменты для скорост-
ной резки металлов. Из нержавеющих химически стойких ста-
лей делают корпуса подводных лодок. Для нагревательных
элементов, используемых в электрических печах, и для термо-
элементов применяют сплавы, состоящие из 80 % Ni и 20% Сг
или 60% Ni, 24% Fe и 16% Сг.
Нагревательные элементы для электрических печей изготов-
ляют также из сплавов железо — хром — алюминий. Сплавы
медь — хром используют при изготовлении троллейбусных кабе-
лей и телекоммуникаций. Из сплавов платина — хром изготов-
ляют сетки, которые служат катализаторами при получении
азотной кислоты окислением аммиака.
Примерами других сплавов хрома могут служить хромель
(сплав Сг — Ni с Мп, Al, Si, Fe, используется для изготовления
термопар), нимоник (сплав Сг, Ni, Ti, Al применяется в произ-
водстве турбин), ваталит (сплав Сг — Со — Мо используется
в стоматологии и хирургии), стеллиты (сверхтвердые сплавы
на основе кобальта, содержащие Сг, W, С, Fe, Ni или Мо и слу-
жащие для изготовления резцов для скоростного резания метал-
234
ПОБОЧНАЯ ПОДГРУППА VI ГРУППЫ
лов). Многие сплавы на основе меди, титана, алюминия или воль-
фрама также содержат хром.
Тонкодисперсный порошок металлического хрома служит
катализатором реакции восстановления СОг водородом до СО
и Н2О, а также в реакции получения CS2 из паров серы и метана
и в других реакциях, особенно в области органической химии.
Благодаря устойчивости по отношению к агрессивным атмос-
ферным воздействиям и многочисленным химическим реагентам
металлический хром применяют в качестве защитного антикорро-
зионного покрытия металлов. Покрытие поверхности металла
хромом (хромирование) осуществляется электролитически (в вод-
ном растворе, содержащем 250—400 г/л СгО3, 2,5—4 г/л H2SO4,
при 27—60°; анод из свинца или сплава РЬ — Sb либо РЬ — Sn,
плотность тока 4—60 а/дм?) или цементацией (применяют поро-
шок хрома или феррохрома, смешанный с каолином, NH4C1, при
1000—1200° или порошок хрома, смешанный с окисью алюминия,
при 1300—1400° в атмосфере водорода). Хромовые покрытия,
используются и для получения отражательных поверхностей.
Широкое применение находят соединения хрома в живописи,
при дублении кож, в красильном деле, а также как фунгициды.
В живописи используются минеральные пигменты (характе-
ризующиеся устойчивостью и большой кроющей способностью)
следующих составов: РЬСгО4 — хромовый желтый, Сг2О3 — (или
РЬСгО4 с Fe4[Fe(CN)6]3) хромовый зеленый, РЬСгО4-РЬО — хро-
мовый оранжевый, ZnCrO4 с Fe4[Fe(CN)6]3 — цинковый зеленый,
3ZnCrO4 -К2Сг2О7 — цинковый и хромовый желтый.
При дублении кож используют основной сульфат хрома(Ш),
получаемый восстановлением водного раствора Na2Cr2O7 дву-
окисью серы (SO2) или сахарным углем в присутствии H2SO4.
Для окрашивания шерсти в качестве протравы применяют
хроматы и бихроматы натрия, калия и аммония, оксиацетат,
сульфоацетаты, нитроацетаты, нитросульфоацетаты, фториды,
оксихлориды, нитрат, формиат, лактат и тартрат хрома(Ш),
а также хромит натрия, двойной оксалат хрома(Ш) и калия.
В качестве сильных фунгицидов применяют хроматы и бихро-
маты щелочных металлов в сочетании с хлоридом цинка, сульфа-
том меди, арсенатом или фторидом натрия.
В многочисленных органических реакциях используют хрома-
ты, хромовую кислоту и Сг2О3 в качестве катализаторов.
СОЕДИНЕНИЯ (ОБЩИЕ СВОЙСТВА)
Известно очень много соединений двух-, трех- и шестивалент-
ного хрома, ограниченное число соединений четырех- и пятива-
лентного хрома и единственное координационное соединение, отве-
чающее одновалентному хрому.
ХРОМ
235
Трех- и шестивалентные соединения хрома наиболее устойчивы.
Соединения одно-, двух-, трех- и пятивалентного хрома обнару-
живают пониженную устойчивость.
На стр. 236 приведена схема окисления металлического хро-
ма до двух-, трех- и шестивалентного состояния и восстановле-
ния его до элементарного хрома.
Хром различной степени окисления, образует катионы Сг2+,
Сг34", [СгА6]3+, [СгХА5]2+, [СгХ2А4] + (где А — электронейтральная
молекула, X — кислотные одновалентные радикалы), СгО22+
и анионы: СгО2~, Сг2ОГ, [Сг(ОН)4]~, [Cr(OH)5J2", [Сг(ОН)6]3~,
[Сг(ОН)7]4-, [Сг(ОН)816-, [СгХ4А2]-, [СгХ5А]2-, [СгХ6]3", СгО2’,
Сг2ОГ, Сг3О2о', Сг4О2-.
Соединениям хрома (за исключением некоторых безводных
солей Сг2С14, СгВг2) присущ большой диапазон окраски — от
желтого, оранжевого, красного, коричневого, черного, зеленого,
синего до фиолетового.
Соединения хрома в низших степенях окисления обладают
основными свойствами и являются энергичными восстановителя-
ми. Соединения хрома высших степеней окисления имеют кислот-
ные свойства (СгО3, Н2СгО4, Н2Сг2О7, Н2Сг30ю, Н2Сг4О13)
и являются энергичными окислителями, например бихроматы
или пироксосоединения.
Много сходства можно найти между соединениями двухвалент-
ного хрома и двухвалентного железа или между соединениями
трехвалентного хрома и алюминия (или трехвалентного железа).
Число координационных соединений хрома особенно велико.
В табл. 40 приведены формулы и указан цвет наиболее важ-
ных соединений двух-, трех-, четырех-, пяти- и шестивалентного
хрома.
Помимо соединений хрома, отвечающих основным состояниям
валентности, известен ряд нестехиометрических соединений пере-
менного состава, например окислы с содержанием кислорода от
СгО117 до CrO)l9 и от СгО2>2 до СгО2,6.
Данные о металлоорганических соединениях и соединениях
включения хрома приведены в разделе, посвященном соединениям
шестивалентного хрома. Большинство соединений хрома токсичны
для человека и животных.
Неорганические соединения
Соединения одновалентного хрома
Перхлорат трис (а, а.'-дипиридил) хрома, [Cr(C5H4N —
C5H4N)31C1O4,— единственное известное соединение одновалент-
ного хрома. Его получают восстановлением перхлората трис(а, а -
дипиридил)хрома(П) [Cr(C5H4N — C5H4N)3](C1O4)2, растворен-
ettado покоим g
------------------>
i s
Nr- M
O:g
sited o ионьокэ1п g
еб О 2 n =1 с S б g 3 раство- 3+ лением восста-
S s J) я Э еб а я л о и a>s И Ф £ К Ч g S
о 4 Ф s -t 5- ** Я g gtsi § s Р» и s о s s & и и H д И Я Ф Ф 9 и к цинка) в кисл рах соединен! , Са, Н2, восс Са, Zn, Mg нений Сг3+
ч ф <л^ й
ф д
5 S и® §
’£ Ф S =г еб s a g ° а р- и ф 3 s о S к о ta3 со И ®
=Г Ф о о М Ф ЙВ.Р Г т м Ф и И д
tr 5ф § ф £ £ 2 Л <м 5 о ©п Сб SY Ри КО ф д Ч о „ И £ Я Ф Q Ф к 2 s Л § Я н ф Ф S К 2 й га
Ф Ен .2 а> о ffixo я
ХРОМ
237
ного в NH4GIO4, тонко измельченным магнием без доступа воз-
духа. Соединение окрашено в синий цвет, оно неустойчиво (при-
соединяет кислород воздуха), парамагнитно, растворяется в воде,
спирте, метаноле, ацетоне, пиридине.
Соединения двухвалентного хрома
Большинство соединений двухвалентного хрома неустойчиво
и обладает ярко выраженным восстановительным характером
(нормальный потенциал системы Сг2+/Сг3+ = —0,41 в), очень
легко окисляется на воздухе, превращаясь в соединения хро-
ма(Ш).
Водные растворы солей хрома(П) получают без доступа воз-
духа растворением металлического хрома в разбавленных кисло-
тах (HF, НС1, НВг, HI, H2SO4) в атмосфере водорода или элек-
тролитическим восстановлением (либо восстановлением цинком
в кислых растворах) водных растворов солей хрома(Ш).
Катион Сг2+ бесцветен; его безводные соли белого, а водные
растворы солей синего цвета. В табл. 40 указана окраска соеди-
нений CrO, Cr(OH)2, CrF2, Сг2С14, CrBr2, Crl2, CrS, CrSO4-7H2O
и [Cr(C5H4N - C5H4N)3](C1O4)2.
Соли двухвалентного хрома парамагнитны, разлагают воду
с выделением водорода, восстанавливают соли олова, золота
и платины до соответствующих металлов, восстанавливают соли
двухвалентных ртути и меди до одновалентных соединений, вос-
станавливают индиго, метиленовый синий, ализарин с изменением
цвета, превращают ацетилен в этилен.
По отношению к определенным химическим реагентам (или
по изоморфизму сульфатов) соли хрома(П) напоминают соли
железа(II), от которых отличаются более ярковыраженным вос-
становительным характером.
Некоторые координационные соединения двухвалентного хро-
ма, например [Cr(C5H4N — C5H4N)3](C1O4)2, более устойчивы на
воздухе, чем простые соли, и труднее окисляются. В сильно
щелочных растворах и в отсутствие воздуха ион [Cr(C5H4N —
— C5H4N)3]2+ диспропорционирует по уравнению
2[Cr2+(C5H4N - C5H4N)3]2+
-> [Cr+(C6H4N - C6H4N)3]+ + [Cr3+(C6H4N - C6H4N)3]3+
Если к водному раствору катиона Сг2+ добавляют водный
раствор сульфо-2-нитро-4-фенилазо-1-окси-3-нафталин-3,6-дисуль-
фоновой кислоты, появляется синее окрашивание, а с о-арсенофе-
нилазо-1-окси-2-нафталин-3,6-дисульфоновой кислотой Сг2+ дает
розовое окрашивание.
Закись хрома, СгО, получают окислением амальгамы хрома
(CrHg3 или CrHg) кислородом воздуха или азотной кислотой.
238
ПОБОЧНАЯ ПОДГРУППА VI ГРУППЫ
После охлаждения расплава смеси NaF с 1% Сг2О3 в атмосфере
азота и водорода (в объемном соотношении 2:1) можно заметить
появление гексагональных пластинчатых кристаллов ярко-крас-
ного или коричнево-красного цвета — СгО, которые можно отде-
лить в отсутствие кислорода.
Закись хрома представляет собой пирофорный черный поро-
шок, который на воздухе при температуре выше 100° превращает-
ся в Сг2О3; она обладает основными свойствами, растворяется
в разб. НС1, образуя ди- и трихлориды хрома. Закись хрома
плохо растворима в разбавленных H2SO4 и HNO3 и восстанавли-
вается до металлического хрома водородом при 1000°.
Гидрат закиси хрома, Сг(ОН)2, получают в виде желтого
осадка, обрабатывая растворы солей хрома(П) щелочами (pH = 5)
в отсутствие кислорода.
При высушивании осадка Сг(ОН)2 над конц. H2SO4 в атмо-
сфере водорода получают коричневый порошок Сг(ОН)2, кото-
рый обладает основными свойствами, плохо растворим в воде
и в разбавленных кислотах, растворяется в концентрированных
кислотах, является восстановителем (восстанавливает бензойный
ангидрид до бензилового спирта), при прокаливании превращает-
ся в Сг2О3.
Дифторид хрома, CrF2, получают действием плавиковой кис-
лоты на нагретый до красного каления металлический хром или
на дихлорид хрома при обычной температуре.
Дифторид хрома образует зеленые нелетучие кристаллы с плот-
ностью 4,11 г! см°, т. пл. 1102° и т. кип. 2127°; он ограниченно рас-
творим в воде, плохо растворим в спирте, растворяется в горячей
НС1, восстанавливается водородом при повышенной темпера-
туре, во влажном воздухе превращается в Сг2О3, а в токе серо-
водорода — в CrS.
Дихлорид хрома, СгС12 или Сг2С14, получают нагреванием
до красного каления металлического хрома в токе газообразного
НС1, нагреванием безводного трихлорида хрома до 400—450°
в токе водорода, медленной и осторожной дегидратацией СгС12-
• пН2О (п — 4, 3 и 2), электролизом трихлорида хрома в соля-
нокислой среде
Сг + 2НС1 = СгС12 + Н2 СгС13 + V2H2 = CrCl2 + НС1
Димер дихлорида хрома, Сг2С14, представляет собой сильно
гигроскопичные блестящие белые кристаллы с плотностью
2,93 г!см3, т. пл. 815°, т. кип. 1302°. При растворении в воде димер
образует синий раствор при комнатной температуре и зеленый —
при нагревании.
Водный раствор дихлорида хрома получают восстановлением
без доступа воздуха раствора трихлорида хрома или солянокис-
лого раствора К2Сг2О7 амальгамой цинка.
ХРОМ
239
При быстром выпаривании водного раствора дихлорида хрома
в вакууме при температуре не выше 38° выпадают темно-синие
кристаллы СгС12-4Н2О, при медленном изотермическом выпари-
вании того же раствора (38—50°) выпадают темно-зеленые кри-
сталлы СгС12-4Н2О, при 60—70° — светло-синие кристаллы
СгС12-ЗН2О, при 84—113° — светло-зеленые кристаллы СгС12-
• 2Н2О, выше 115° — безводная белая соль Сг2С14.
Переход одного гидрата в другой * или в безводную соль
можно представить следующим образом:
38-50° 60-70° 84-113°
СгС12-4Н2О t СгС12.4Н2О ~" * СгС12-ЗН2О ~
Темно-синий Темно-зеленый -н2о Светло-синий -н2о
115°
СгС12-2Н2О t > СгС12
Светло-зеленый -2Н2о Белый
Дихлорид хрома поглощает кислород, превращаясь в Сг2ОС14,
и аммиак, превращаясь в зависимости от температуры в соеди-
нения [Cr(NH3)6]Cl2 темно-синего, CrCl2-5NH3 фиолетового,
CrCl2-3NH3 светло-фиолетового, CrCl2*2NH3 фиолетово-синего
и CrCl2’NH3 светло-зеленого цвета.
Водный раствор дихлорида хрома используется в газовом
анализе для количественного поглощения кислорода. В присут-
ствии кислорода дихлорид хрома в водных растворах взаимодей-
ствует с моноокисью ртути по уравнению
2СгС12 + 2HgO + ЗН2О + ЧгОг = 2Сг(ОН)3 + 3HgCl2
При высокой температуре пары дихлорида хрома восстанав-
ливаются металлическим железом. Реакция обратима.
СгС12 + Fe Cr + FeCl2
При медленном действии на дихлорид хрома конц. HCI на холо-
ду получают белые кристаллы Н[СгС13] -бДИгО.
Восстановительные свойства дихлорида хрома наглядно про-
являются по отношению к перманганату калия, иоду, индиго или
солям олова, золота и платины.
Известны аддукты дихлорида хрома: CrCl2-2N2H4 — фиолето-
вый, CrCl2’2C5H5N — зеленый, СгС12-О(С2Н5)2, [(NH4)3GrGl2 •
• О(С2Н5)2].
Дибромид хрома, СгВг2, получают действием бромистоводо-
родной кислоты на нагретый до красного каления металлический
хром, восстановлением трибромида хрома водородом при повы-
шенной температуре, нагреванием бромида гексамминхрома(Ш)
* Изменение окраски связано с различной степенью гидратации Сг2+.
Относительно явления гидратоизомерии см. стр. 248,— Прим. ред.
240
ПОБОЧНАЯ ПОДГРУППА VI ГРУППЫ
[Cr(NH3)6]Br3 до 150—350°в среде водорода и бромистого водорода:
Сг + 2НВг = СгВг2 + Н2 СгВг3 + V2H2 = СгВг2 + НВг
[Cr(NH3)e]Br3 + V2H2 = СгВг2 + NH4Br + 5NH3
Дибромид хрома представляет собой желтовато-белые гекса-
гональные кристаллы с плотностью 4,356 г/см3, т. пл. 842°,
т. кип. 1127°. Он растворим в воде и спирте, устойчив в сухом
и окисляется во влажном воздухе с переходом в зеленый окси-
бромид хрома(Ш).
Водный синий раствор дибромида хрома получают растворе-
нием безводного дибромида хрома в воде или действием водного
раствора НВг на металлический хром или ацетат хрома(П).
Известны следующие координационные соединения и аддукты
типа дибромида хрома: [Cr(NH3)6]Br2, CrBr2-5NH3, CrBr2-3NH3,
2СгВг2 -5NH3, 2CrBr2-3NH3, CrBr2.2N2H4.
Дииодид хрома, Crl2, получают действием паров иода на тон-
кодисперсный порошок металлического хрома, нагретый в квар-
цевом вакуумированном сосуде до 700—800°, растворением метал-
лического хрома в иодистоводородной кислоте, восстановлением
Сг13 водородом при нагревании:
Сг -j- 12 = Сг12 Сг 2HI =з Сг12 Н2 Сг13 V2H2 = Crl2 -|- HI
Дииодид хрома образуется в виде сильно гигроскопичных
коричнево-красных игольчатых кристаллов с плотностью
5,02 г/см3, т. пл. 793°, т. кип. 827°, хорошо растворимых в воде.
При нагревании в вакууме Сг12 перегоняется при температуре
выше 420°, термически диссоциирует при 500° с высвобождением
иода, а при 600° — с выделением металлического хрома. Реакцию
термической диссоциации дииодида хрома используют при хроми-
ровании железа и стали.
Для Сг12 известны координационные соединения [Cr(NH3)6]I2,
[Cr(N2H4)6]I2 и аддукты CrI2-reN2H4, где п = 4, 3 или 1.
Моносулъфид хрома, CrS, получают прямым взаимодействием
элементов при 700°, действием сероводорода на металлический
хром или трихлорид хрома при нагревании, действием водорода
на Cr2S3 при нагревании, обработкой двойного сульфида хрома
и алюминия Al2CrS4 азотной кислотой:
Сг + S = CrS Сг + H2S = CrS + Н2
2СгС!3 + 3H2S = 2CrS + S + 6НС1 Cr2S3 + H2 = 2CrS + H2S
Моносульфид хрома представляет собой парамагнитные черные
кристаллы в форме призм (или аморфный темно-серый порошок)
с плотностью 4,85 г/см3, т. пл. 1550°, которые разлагаются (перед
плавлением) при 1350°, легко окисляются в теплом воздухе и пре-
вращаются в СгС13 под действием хлора.
ХРОМ
241
Двойной сульфид алюминия и хромаДГ), Al2CrS4, выделяется
при сильном нагревании смеси A12S3 с CrS или при обработке
смеси хрома и алюминия сероводородом при высокой температуре.
Серые кубические кристаллы Al2CrS4 имеют плотность
3,09 г/см\ реагируют с кислотами с высвобождением CrS и выде-
лением H2S.
Сульфаты хром а(П). Водные растворы сульфата хро-
ма(П) получают в атмосфере СО2 путем восстановления водных
растворов сульфата хрома(Ш), а также квасцов хрома и калия,
подкисленных разб. H2SO4, металлическим цинком или амальга-
мой цинка, электролитическим восстановлением растворов суль-
фата хрома(Ш) (сильно подкисленных H2SO4) при использовании
свинцового или платинового катода, а также действием разб.
H2SO4 на металлический хром или ацетат хрома(П).
Водный раствор сульфата хрома(П) обладает восстановитель-
ными свойствами, быстро поглощает кислород из воздуха, окис-
ляется и восстанавливает соли висмута(Ш) или олова(1У).
Известны гепта-, пента- и моногидраты сульфата хрома(II).
Сульфат CrSO4 -7Н2О выделяется при концейтрировании водных
растворов сульфата хрома(П) при температуре ниже 40°; это
синие кристаллы, изоморфные FeSO4-7H2O. Они легко окисляют-
ся на воздухе, растворимы в воде и ограниченно растворимы
в спирте.
Сульфат CrSO4-H2O — белый порошок, который под действием
влаги превращается в CrSO4 -7Н2О. Пентагидрат сульфата
CrSO4-5H2O — синего цвета, по структуре аналогичен сульфату
меди CuSO4-5H2O.
Аммиачный раствор сульфата хрома(П) поглощает ацетилен.
Сульфат хрома(П) с сульфатами щелочных металлов или
некоторых двухвалентных металлов образует двойные сульфаты
общей формулы Me*Cr(SO4)2-бНгО, где Mer = K+, Rb+, Cs+, NH*
и MenCr(SO4)2 ’пН2О, где Me11 = Mg2+, Zn2+, Mn2+, Fe2+, a n может
иметь различные значения.
Координационные соединения
Известно небольшое число координационных соединений хро-
ма(П). Помимо уже упомянутых гексамминов [Cr(NH3)6]Cl2 сине-
го, [Cr(NH3)6]Br2 зеленого, [Cr(NH3)6]I2 зеленого цвета известны
перхлорат т(рис(а,а'-дипиридил)хрома(П) [Cr(C5H4N — C5H4N)3J-
•(С1О4)2 темно-фиолетового цвета, хромцианид калия
K4[Cr(CN)6] -ЗН2О синего цвета, комплексные аквосоли, карбо-
наты и оксалаты.
В отличие от соединений, в которых присутствует катион
Сг2+, координационные соединения с катионами [Cr(NH3)6]2+,
[Cr(N2H4)6]2+, [Сг(Н2О)6]2+ значительно более устойчивы.
16-0101
242
ПОБОЧНАЯ ПОДГРУППА VI ГРУППЫ
Кроме указанных известны и другие соединения двухвалентного хрома,
например: селенид CrSe, теллурид СгТе, нитрат Cr(NO3)2, ортофосфат
Сг3(РО4)2-пНгО, карбонат СгСО3, двойные карбонаты Li2Cr(CO3)2,
Na2Cr(CO3)2 •(! или 10)Н2О, K2Cr(CO3)2, (NH4)2Cr(CO3)2-Н2О, цианид Cr(CN)2,
гексацианоферрат(П) Cr2[Fe(CN)6], формиат Сг(НСОО)2-2Н2О, ацетат
Сг(СН3СОО)2, бензоат Сг(С6Н5СОО)2, оксалат Сг(СОО)2-Н2О, малонат
Сг(СОО — СН2 — СОО)-2Н2О, гликолят Сг(СН2ОН — СОО)2-Н2О, тартрат
Сг(СОО — СНОН — СНОН — СОО), салицилат СгС7Н4О2-ЗН2О.
Соединения трехвалентного хрома
Известно очень много устойчивых простых и координацион-
ных, моно- или полиядерных соединений хрома(Ш). В простых
и координационных моноядерных соединениях электроположи-
тельный трехвалентный хром может присутствовать в виде катио-
нов Сг3+, [СгА6]3+, [СгХА5]2+, [СгХ2А4]+ или в виде анионов Сг2О2",
[Сг(ОН)4]“, [Сг(ОН)6]3-, [Сг(ОН)7]4-, [Сг(ОН)8р-, [СгХ4А2]",
[CrXsAP-, [СгХ6]3~ (где А = Н2О, NH3, СН3 - NH2, С2Н5 - NH2,
С3Н7 - NH2, С4Н9 - NH2, C5H5N, x/2NH2 - СН2 - СН2 - NH2,
а X = F", Cl", Вт-, I", NO", NO", NCS", ОН", СН3СОО", X/2SO24",
1/2SeO'2, ^гСгО2'), а также в виде молекул типа неэлектролитов.
Водные растворы солей Сг3+, полученные растворением метал-
лического хрома в разбавленных кислотах или восстановлением
бихроматов различными восстановителями в кислой среде, окра-
шены в фиолетовый или зеленый цвет.
В водных растворах катион Сг3+ встречается только в виде
гидратированного иона [Сг(Н2О)6]3 + благодаря склонности трехва-
лентного хрома к образованию координационных соединений.
Фиолетовый цвет водных растворов хлорида, сульфата и нитрата
хрома(Ш) обусловлен катионом [Сг(Н2О)6]3+, который может
подвергаться частичному гидролизу * по уравнению
[Сг(Н2О)3]3+ + Н2О [Сг(Н2О)5ОНр+ + Н3О+
При нагревании твердых фиолетовых солей хрома(Ш) высво-
бождается вода и образуются соли, окрашенные в зеленый цвет:
[Сг(Н2О)6]С13 [Сг(Н2О)4С12]С1.2Н2О
Соединения хрома(Ш) очень разнообразны по цвету: желтые,
оранжевые, красные, коричневые, зеленые, синие, фиолетовые
и черные.
Соли хрома(Ш) парамагнитны, очень устойчивы в сухом
воздухе и проявляют более сильную тенденцию к гидролизу, чем
соли хрома(П). Благодаря этой тенденции при обработке солей
хрома(Ш) некоторыми химическими реагентами образуются
не соответствующие соединения, а гидроокись Сг(ОН)3.
* При комнатной температуре имеет место полимеризация гидроксил-
содержащих ионов за счет образования «оловых» мостиков.— Прим. ред.
ХРОМ
243
Соединения хрома(Ш) в кислой среде могут быть восстанов-
лены электрохимически либо цинком (амальгамой цинка) до соеди-
нений хрома(П), в щелочных же растворах могут быть окислены
Н2О2, РЬО2, хлорной или бромной водой, а также путем обра-
ботки расплавленными KNO3, КСЮ3, Na2O2 до соединений,
в которых хром электроположителен и шестивалентен.
По склонности к гидролизу и образованию двойных сульфатов
типа квасцов и различных изоморфных солей многие соединения
хрома(Ш) похожи на соответствующие соединения алюминия
и железа(Ш).
Известны металлоорганические соединения хрома(Ш), в кото-
рых органические радикалы присоединены к хрому тремя валент-
ными связями.
Соединения хрома(Ш) токсичны для человека и животных,
но благоприятствуют росту растений.
Окись хрома, Сг2О3, получают прямым взаимодействием эле-
ментов при повышенной температуре, нагреванием закиси хрома
на воздухе, прокаливанием хромата или бихромата аммония,
трехокиси хрома, гидроокиси или нитрата -хрома(П1), хромата
ртути(1), бихромата ртути, смеси сульфата хрома(Ш) с безводным
Na2CO3 либо смеси К2Сг2О7 с древесным углем или серой (или
их смесью), а также термическим разложением хлорида хромила
или хлорохромата кальция:
4Сг -{- ЗО2 = 2Сг2О3 545,4 ккал 4Hg2CrO4 — 2Сг20з 4- 8Hg -j- 5О2
2CrO + V2O2 = Сг2О3 4К2Сг2О7 = 2Сг2О3 + 4К2СгО4 + ЗО2
2(NH4)2CrO4 = Сг2О3 + 2NH3 + N2 + 5Н2О
Cr2(SO4)3 -f- 3Na2CO3 = Cr2O3 -|- 3Na2SO4 -f- 3CO2
(NH4)2Cr2O7 = Cr2O3 + N2 + 4H2O + 123 ккал
K2Cr2O7 -f- 2G = Cr2O3 -|- K2CO3 -f- CO
2CrO3 = Cr2O3 -f- 3/2O2 K2Cr2O7 S = Cr2O3 -f- K.2SO4
2Cr(OH)3 = Cr2O3 + 3H2O 2CrO2Cl2 = Cr2O3 + 2C12 + V2O2
2Cr(NO3)3 = Cr2O3 -f- 6NO2 3/2O2 Ca[CrO3Cl]2 = Cr2O3 -f- CaCl2 -|- 3/202
После выделения воды при прокаливании (NH4)2CrO4,
(NH4)2Cr2O7, Сг(ОН)3 происходит резкий подъем температуры,
в результате чего Сг2О3 сильно нагревается за счет выделяющейся
теплоты кристаллизации. Это явление наблюдается и при образо-
вании окислов TiO2, ZrO2, Sc2O3.
Окись хрома представляет собой зеленые гексагональные
микрокристаллы с плотностью 5,22 г!см3, т. пл. 2437° и т. кип.
3027°. Их твердость соизмерима с твердостью корунда, они амфо-
терны, Кристаллы Сг2О3 антиферромагнитны ниже 33° и пара-
магнитны выше 55°, растворяются в жидкой SO2 и плохо раство-
ряются в воде, разбавленных кислотах и растворах щелочей.
16*
244
ПОБОЧНАЯ ПОДГРУППА VI ГРУППЫ
При сплавлении Cr2O3 с NaOH, КОН, KHSO4, K2S2O7, Na2O2,
окислительно-щелочной смесью Na2CO3 с KNO3 или КОН с КС1О3
получаются соединения хрома(Ш) или (VI), растворимые в воде:
Сг2О3 + 2NaOH = 2NaCrO2 + Н2О Сг2О3 + 3Na2O2 = 2Na2CrO4 + Na2O
Cr2O3 + 6KHSO4 = Cr2(SO4)3 + 3K2SO4 + 3H2O
Cr2O3 -f- 2Na2CO3 + 3KNO3 = 2Na2CrO4 + 3KNO2 + 2CO2
Cr2O3 + 3K2S2O7 = Cr2(SO4)3 + 3K2SO4
Cr2O3 + 4K0H 4- KC1O3 = 2K2CrO4 + KC1 + 2H2O
При сплавлении окиси Cr2O3 с многочисленными окислами
двухвалентных металлов образуются окрашенные соединения
со структурой шпинелей Меп[Сг2О4].
При высокой температуре Сг2О3 восстанавливается алюминием,
кремнием, кальцием, магнием, натрием, калием, углеродом,
водородом, гексаном, циклогексаном и бензолом до металличе-
ского хрома.
Окись хрома(Ш) превращается в безводный трихлорид хрома
при сильном нагревании с СС14 или SC12 и при пропускании тока
хлора над нагретой до белого каления смесью Сг2О3 с углем:
2Сг2О3 — ЗСС14 = 4СгС13 + ЗСО2 Cr2O3 + ЗС + ЗС12 = 2СгС13 + ЗСО
При нагревании окиси хрома(Ш) в атмосфере H2S и CS2
образуется сульфид Cr2S3.
В щелочной среде Сг2О3 взаимодействует с КМпО4:
Сг2О3 + 2К0Н + 2КМпО4 = 2К2СгО4 + 2МпО2 + Н2О
Помимо кристаллической модификации окиси хрома(Ш) изве-
стна и аморфная модификация в виде порошка, растворяющегося
в кислотах и щелочах с образованием солей.
Окись Сг2О3 применяют в качестве зеленого пигмента в живо-
писи п для окрашивания фарфора и стекла. Кристаллический
порошок Сг2О3 используется в качестве абразива для полировки
металлов. Окись Сг2О3, образующая твердые растворы с Fe2O3,
служит для получения искусственных рубинов. В следовых коли-
чествах Сг2О3 окрашивает в красный цвет некоторые драгоценные
камни, как-то рубины и хризобериллы (александриты).
Окись хрома(Ш) служит катализатором процессов окисления
аммиака на воздухе, синтеза аммиака из элементов, получения
альдегидов окислением углеводородов и спиртов, образования
SO3 из SO2 и кислорода, разложения окиси углерода, формальде-
гида, метилового спирта, гидрогенизации и дегидрогенизации
многих углеводородов.
Гидраты окиси хром а (III), Сг2О3’мН2О (где п =
— 3, 2. 1). Тригидрат окиси хрома(ПI) [гидроокись хрома(Ш)],
Сг2О3-ЗН2О или Сг(ОН)з, получают в виде студнеобразного зеле-
ХРОМ
245
ного осадка при обработке растворов солей хрома(Ш) раствором
NaOH, КОН, NH4OH в интервале pH 4,8—8,5, при гидролити-
ческом осаждении солей хрома(Ш) карбонатами щелочных метал-
лов, сульфидом аммония или тиосульфатом натрия.
Cr2(SO4)3 + 6NaOH 2Cr(OH)3 + 3Na2SO4
Cr2(SO4)3 + 3Na2CO3 + 3H2O 2Cr(OH)3 + 3Na2SO4 + 3CO2
Cr2(SO4)3 + 3(NH4)2S + 6H2O 2Cr(OH)3 + 3(NH4)2SO4 + 3H2S
Cr2(SO4)3 + 3Na2S2O3 + 3H2O 2Cr(OH)3 + 3Na2SO4 + 3S + 3SO2
Гидроокись хрома обладает амфотерными свойствами, плохо
растворима в воде, легко переходит в коллоидное состояние
и легко растворяется в щелочах и кислотах с образованием солей
СгЗ+ + ЗОН- Сг(ОН)3 = НзСгОз НСгО2 + Н2О
Сго2+Н3о+
При растворении гидроокиси хрома в щелочах (pH ~ 11,8)
образуются гидроксохромиты типа MeI[Cr(OH)4], Ме*[Сг(ОН)5],
Ме*[Сг(ОН)6], Ме*[Сг(ОН)7], Ме^[Сг(ОН)8] по реакции
Сг(ОН)3 + КОН = К[Сг(ОН)4] Сг(ОН)3 + ЗКОН = К3[Сг(ОН)6]
Гидроксохромиты менее устойчивы, чем гидроксоалюминаты,
при кипячении их с водой выпадает гидроокись хрома.
Растворением гидроокиси хрома в жидком аммиаке получают
гидроокись гексамминохрома, а в присутствии какой-нибудь соли
аммония (например, NH4C1)—соответствующую соль гексаммин-
хрома(Ш):
Сг(ОН)3 + 6NH3 = [Cr(NH3)6](OH)3
Сг(ОН)3 + 3NH3 + 3NH4C1 = [Cr(NH3)6]Cl3 + ЗН2О
При растворении Сг(ОН)3 в кислотах образуются соли хро-
ма(Ш), например:
Сг(ОН)3 + ЗНС1 = СгС13 + ЗН2О 2Сг(ОН)3 + 3H2SO4 = Cr2(SO4)3 - 6Н2О
В результате нагревания Сг(ОН)3 превращается в Сг2О 3.
Гидроокись хрома в щелочном растворе под действием окисли-
телей (перекиси водорода, хлорной или бромной воды) превра-
щается в хромат. Сг(ОН)3 способен поглощать газы (водород,
азот и др.), красители и двухвалентные катионы, например Zn2+,
Cu2+, Ni2+, Cd2+.
Моногидрат окиси хрома, Сг2О3-Н2О или СгООН, представ-
ляет собой зеленые кубические кристаллы с плотностью 4,12 г/см3.
При нагревании до 430° превращаются в Сг2О3.
Дигидрат окиси хрома, Сг2О3-2Н2О, представляет собой зеле-
ные кристаллы с плотностью 2,9 г!см3, устойчивые на воздухе.
Они не реагируют с НС1, HNO3 и щелочами.
246
ПОБОЧНАЯ ПОДГРУППА VI ГРУППЫ
Хромиты и гидроксохромиты. При сплавлении
окиси хрома(Ш) с окислами, основаниями и карбонатами щелоч-
ных металлов или окислами многих других металлов образуются
хромиты. В зависимости от соотношения сплавляемых окислов
состав хромитов может сильно варьироваться. В соответствии
с общими формулами MeiCr2O4, МепСг2О4 можно назвать в каче-
стве примеров следующие хромиты: Li2Cr2O4 — коричневый,
Сн2Сг2О4 — синий, СпСг2О4 — черно-синий, ВеСг2О4 — зеленый,
MgCr2O4 — зеленый, СаСг2О4 — зеленый, ZnCr2O4 — зеленый,
CdCr2O4 — зеленый, РЬСг2О4 — зеленый, МпСг2О4 — зеленый,
FeCr2O4 — черный или коричнево-черный, СоСг2О4 — зеленый,
NiCr2O4 — серо-зеленый.
На основе природного хромита изготовляют огнеупорные
кирпичи, которые используются для внутренней обкладки метал-
лургических печей, требующих футеровки материалом, имеющим
основные свойства. В качестве огнеупорных химически стойких
материалов используют как природный хромит, так и хромит
в смеси с MgO или А12О3.
При обработке растворов солей хрома(Ш) избытком щелочей
(pH ~ 11,8) получают гидроксохромиты щелочных металлов
общих формул Ме1[Сг(ОН)4], Ме*[Сг(ОН)5], Ме|[Сг(ОН)6],
Ме1[Сг(ОЙ)7], Mei[Cr(OH)8], которые с растворами солей щелочно-
земельных металлов образуют соответствующие нерастворимые
гидроксохромиты.
В качестве примеров гидроксохромитов можно назвать
Na3lCr(OH)6], Na4[Cr(OH)7]-2—ЗН2О, Na5[Cr(OH)8]-4Н2О,
K3[Cr(OH)6] -Н2О, Sr3[Cr(OH)6]2, Ва31Сг(ОН)6]2.
Трифторид хрома, CaF3, получают действием плавиковой кис-
лоты на окись хрома(Ш), пропусканием фтористого водорода
над нагретым до 500—1100° трихлоридом хрома, а также сильным
нагреванием в атмосфере газообразного HCI смеси CaF2 и Сг2О3:
Сг2О3 + 6HF = 2CrF3 + ЗН2О СгС13 + 3HF = CrF3 + ЗНС1
Cr2O3 + 3CaF2 + 6НС1 = 2CrF3 + ЗСаС12 + ЗН2О
Соединение CrF3 образует парамагнитные зеленые ромбиче-
ские кристаллы с плотностью 3,78 г!см\ т. пл. 1100°, т. кип. 1427°,
растворимые в плавиковой кислоте и мало растворимые в воде.
Они превращаются в Сг2О3 при сильном нагревании на воздухе
и реагируют с парами воды при 400—600° по уравнению
2CrF3 + ЗН2О Сг2О3 + 6HF
Водные растворы трифторида хрома (используемые в произ-
водстве шелка, при переработке шерсти и фторировании галогено-
производных этана, пропана и т. д.) получают в результате вос-
становления глюкозой, спиртом, металлическим оловом или дре-
весным углем раствора СгО3 в плавиковой кислоте, нагретой
до 80°.
ХРОМ
247
Гидраты трифторида хрома — координационные соединения,
например: [Cr(H2O)e]F3 — фиолетовый, [Cr(H2O)6][CrFe] — зеле-
ный, [Cr(H2O)6][CrFe] -Н2О — зеленый, [Cr(H2O)6][CrFe] -2Н2О —
зеленый.
Известны фторо- и аквофторосоли хрома(Ш), например:
Me4CrF4] (где Me1 = К+, NHJ), Mei[CrF5(H2O)] (где Mei =
= Na+, К+, РЬ+, NH+), Me34CrF6] (где Mei = Na+, к+, NH|).
Оксифторид хрома, CrOF, представляет собой твердое зеленое
вещество с плотностью 4,20 г!см3, устойчивое при повышенной
температуре и разлагающееся при охлаждении; получается дей-
ствием фтористого водорода на окись Сг2О3 при 1100°.
Трихлорид хрома, СгС13, получают прямым взаимодействием
составляющих компонентов при 600°, действием хлора на нагре-
тую до 700—800° смесь Сг2О3 с углем (сажей) или на нагретый
до красного каления сульфид хрома(Ш), действием смеси хлора
и окиси углерода на окись хрома(Ш) при 700° или на пары хло-
рида хромила, а также действием СС14 на Сг2О3 при 700—800°:
Сг + 3/2С12 = СгС13 + 132 ккал Сг2О3 + ЗС12 -f- ЗСО = 2СгС13 + ЗСО2
Сг2О3 + ЗС + ЗС12 = 2СгС13 + ЗСО 2СгО2С12+ С12 + 4СО = 2СгС13 + 4СО2
Cr2S3 + ЗС12 = 2СгС13 + 3S 2Сг2О3 + ЗСС14 = 4СгС13 + ЗСО2
Гексагональные кристаллы безводного трихлорида хрома СгС13
имеют окраску цветов персикового дерева; они парамагнитны,
расплываются на воздухе, имеют плотность 2,87 г/см3, сублими-
руются при 1047°, плавятся при 1152°, трудно растворимы в воде,
спирте, эфире, ацетальдегиде, ацетоне, хлорокиси фосфора; вос-
станавливаются при высокой температуре до металлического
хрома кальцием, цинкоМ, магнием, водородом, железом, превра-
щаются в хлорид хромила при нагревании в атмосфере влажного
хлора, а при нагревании с H2S — в Cr2S3. Соединение СгС13
используется в качестве катализатора в многочисленных реакциях
органической химии.
Безводный трихлорид хрома плохо растворим в воде, но в при-
сутствии следовых количеств СгС12 растворяется очень быстро;
СгС12 вводят специально или он образуется при добавлении SnCl2,
FeSO4.
Гексагидрат трихлорида хрома известен в виде трех
гидратных изомеров: [Сг(Н2О)6]С13, [Сг(Н2О)5С1]С12 *Н2О,
[Сг(Н2О)4С12]С1-2Н2О; они различаются между собой цветом,
молекулярной электропроводностью, отношением к иону Ag+
и другими свойствами.
Хлорид гексаквохроМа(Ш') {фиолетовый хлорид Рекура),
[Сг(Н2О)6]С13, представляет собой серовато-синие моноклинные
или ромбоэдрические кристаллы с плотностью 2,76 г/см3, плохо
248
ПОБОЧНАЯ ПОДГРУППА VI ГРУППЫ
растворимые в спирте, эфире, ацетоне. При растворении в воде
образуется фиолетовый раствор.
Хлорид хлорпентаквохрома(Ш) (хлорид Бъеррума),
[Сг(Н2О)5С1]С12-Н2О,— гигроскопичный светло-зеленый крупно-
кристаллический порошок.
Хлорид дихлортетраквохрома(ПТ) (зеленый хлорид Рекура),
[Сг(Н2О)4С12]С1-2Н2О, представляет собой гигроскопичный темно-
зеленый порошок, растворимый в воде, сладкий на вкус.
В водном растворе между тремя изомерами устанавливается
равновесие, которое зависит от концентрации и температуры
раствора:
[Сг(Н2О)в]С13 [Сг(Н2О)5С1]С12 .Н2О [Сг(Н2О)4С12]С1.2Н2О
Фиолетовый Светло-зеленый Желто-зеленый
В разбавленном растворе на холоду устойчив фиолетовый
изомер, а при нагревании в концентрированном растворе в рав-
новесии находятся зеленые изомеры. По молекулярной электро-
проводности и по количеству хлора, который осаждается AgNO3
на холоду в сильно азотнокислой среде, можно легко определить
изомер.
При упаривании водного раствора трихлорида хрома выпадает
расплывающийся зеленый аморфный порошок, растворимый в во-
де. При нагревании выше 250° в атмосфере хлора или НС1 он пре-
вращается в безводную соль СгС13.
Хлориды щелочных металлов с трихлоридом хрома образуют
хлоросоли, например, соединения К3[СгС16] красновато-розового
цвета. Известны кислоты Н[СгС14] ’6,5Н2О, Н2[СгС15] -8,5Н2О,
Н3[СгС16] -10,5Н2О и оксихлориды CrOCl, СгОСЬЗН2О, Сг2ОС14,
Ст2ОС14.ЗН2О, Ст4О3С16, Сг10О3С124.24Н2О.
Трибромид хрома, СгВг3, получают действием паров брома
в присутствии азота на металлический хром или на смесь Ст2О3
с углем при высокой температуре.
Зеленые гексагональные кристаллы трибромида хрома имеют
плотность 4,25 г!см\ сублимируются при 927°, плавятся при
1127°, восстанавливаются до СгВг2 водородом при нагревании,
превращаются в Сг2О3 при нагревании на воздухе, разлагаются
щелочами и растворяются в воде только в присутствии солей
хрома(П).
Растворы трибромида хрома окрашены в зеленый цвет; их
можно получить растворением гидратированной окиси хрома(Ш)
в НВг при кипячении.
Известны изомеры [Сг(Н2О)6]Вг3 фиолетового
и [Сг(Н2О)4Вг2]Вг '2Н2О зеленого цветов.
Трииодид хрома, Сг13, получают действием паров иода на ме-
таллический хром, нагретый до красного каления, в атмосфере
азота или обработкой дииодида хрома парами иода под давлением
ХРОМ
249
1 ат при 500°:
Сг12 4“ V2I2 Crl3
Трииодид хрома образует фиолетово-черные кристаллы, устой-
чивые на воздухе при обычной температуре; при 200° реагируют
с кислородом с выделением иода, растворяются в воде в присут-
ствии солей хрома(П). Водные растворы трииодида хрома можно
получить действием иодистоводородной кислоты на Сг2О3, СгО3,
Ag2CrO4.
Сульфид хрома, Cr2S3, получают действием паров серы на
металлический хром, нагретый до температуры выше 700°, сплав-
лением Сг2О3 или СгС13 с серой или K2S, действием H2S на нагре-
тый до 440° Сг2О3, пропусканием H2S через сильно нагретый
СгС13, действием CS2 на металлический хром или соединения
Сг2О3, К2СгО4, К2Сг2О7 при высокой температуре:
2Cr + 3S = Cr2S3 2СгС13 + 3H2S = Cr2S3 + 6НС1
2Сг2О3 + 9S = 2Cr2S3 4- 3SO2 4Сг + 3CS2 = 2Cr2S3 + ЗС
Сг2О3 + 3H2S = Cr2S3 + 3H2O 2Cr2O3 + 3CS2 = 2Cr2S3 + 3CO2
Сульфид хрома выделяют в виде парамагнитных черных кри-
сталлов (или черно-коричневого порошка) с плотностью 3,60 г/с.м3;
Cr2S3 гидролизуется водой, превращается в Cr2O3, SO2 и основной
сульфат хрома (при нагревании на воздухе). Сульфид хрома плохо
реагирует с кислотами, но окисляется азотной кислотой, царской
водкой или расплавленными нитратами щелочных металлов.
В результате нагревания в токе водорода Cr2S3 восстанавливается
до CrS. Сульфид хрома служит катализатором гидрогенизации
ненасыщенных углеводородов и ароматических нитропроизводных.
С сульфидами щелочных металлов сульфид хрома образует
тиохромиты, например NaCrS2 — красного, KCrS2 — зеленовато-
серого, K2Cr4S7 — серовато-красного цветов. Известны также
тиохромиты: CuCr2S4, Ag2Cr2S4, FeCr2S4, SnCr2S4, CoCr2S4,
ZnCr2S4, PbCr2S4, NiCr2S4, CdCr2S4, MnCr2S4.
Безводный сульфат хрома, Cr2(SO4)3, получают дегидратацией
его кристаллогидратов на воздухе при 400° или в атмосфере СО2
при 280°, а также нагреванием окиси Сг2О3 с метил сульфатом
при 160—190°.
Соединение Cr2(SO4)3 образует парамагнитные фиолетово-крас-
ные микрокристаллы с плотностью 3,012 г!см\ плохо растворимые
в воде и кислотах, разлагающиеся при высокой температуре.
Водные растворы сульфата хрома(Ш) окрашены в фиолетовый
цвет на холоду и в зеленый — при нагревании, его получают
растворением гидратированной окиси хрома(Ш) в разб. H2SO4.
Известны кристаллогидраты Cr2(SO4)3’пН2О (п = 18, 17, 15,
14, 12, 9, 6, 3). Их применяют при дублении кож и в качестве
протравы при крашении в ситценабивном производстве.
250
ПОБОЧНАЯ ПОДГРУППА VI ГРУППЫ
Известны также гидросульфаты хрома(Ш), например:
Cr2(SO4)3-H2SO4, Cr2(SO4)3-H2SO4(16 или 24)Н2О, Cr2(SO4)3.
• 2H2SO4 ИЗНгО, 2Cr2(SO4)3,-H2SO4, 2Cr2(SO4)3 •7H2SO4, основные
сульфаты хрома(Ш), например Cr(OH)SO4, двойные сульфаты
общей формулы MeiCr(SO4)3, где Мет = Li+, Na+, К+, NH+,
квасцы общей формулы MerCr(SO4)2-12Н2О, где Me1 = Na+, К+.
Rb+, Cs+, Tl+, NH+.
Хромокалиевые квасцы, KCr(SO4)2 *12Н2О, выпадают при выпа-
ривании водного раствора, полученного либо смешением двух
растворов — сульфата калия и сульфата хрома(Ш) в стехиомет-
рически необходимых количествах, либо восстановлением с по-
мощью С2Н5ОН, H2SO3, Н2С2О4-2Н2О раствора К2Сг2О7, подкис-
ленного достаточно концентрированной H2SO4:
Cr2(SO4)3 -f- K2SO4 -}- 24Н2О = 2KCr(SO4)2-12Н2О
К2Сг2О7 4- ЗС2Н5ОН 4- 4H2SO4 4- 17Н2О =
= 2KCr(SO4)2-12H2O 4- ЗСН3 — СНО
К2Сг2О7 4" 3H2SO3 4~ H2so4 4~ 20Н2О = 2K.Cr(SO4)2 «12Н2О
К2Сг2О7 4- ЗН2С2О4 4- 4H2SO4 4- 17Н2О = 2KCr(SO4)2-12Н2О 4- 6СО2
Квасцы KCr(SO4)2 *12Н2О представляют собой фиолетовые
октаэдрические кристаллы с плотностью 1,83 г/см3-, при 78° пере-
ходят в зеленую модификацию (т. пл, 89°), обезвоживаются при
300°, растворимы в воде (нагревание не выше 350°), плохо рас-
творимы в спирте.
При нагревании до 50—70° фиолетовый водный раствор хромо-
калиевых квасцов становится зеленым.
Хромокалиевые квасцы применяются при дублении кож и в ка-
честве протравы в текстильном производстве.
Нитраты хром а(Ш). Фиолетово-синие водные рас-
творы нитрата хрома(Ш) получают растворением окиси Сг2О3
или гидроокиси Сг(ОН)3 с разб. HNO3, а также восстановлением
раствора хромовой кислоты каким-нибудь органическим веще-
ством в присутствии азотной кислоты.
Известны кристаллогидраты нитрата хрома(Ш): Cr(NO3)3.
• 12,5Н2О, Cr(NO3)3-9Н2О, Cr(NO3)3-7,5Н2О, Cr(NO3)3-4,5Н2О
и Cr(NO3)3-ЗН2О. Кристаллогидрат Cr(NO3)3-9Н2О представляет
собой пурпурные моноклинные кристаллы в форме призм ст. пл.
37°; они разлагаются при 125,5°, растворимы в воде, спирте, кис-
лотах. Кристаллогидрат Cr(NO3)3-7,5Н2О — коричневые моно-
клинные кристаллы в форме призм; т. пл. 100°, растворяются
в воде.
Известны основные нитраты хрома(Ш) Cr(OH)(NO3)2,
Cr(OH)2NO3, выделенные в виде зеленого аморфного порошка.
Ортофосфат хрома, СгРО4, получают прокаливанием его
кристаллогидрата при 946° в течение часа.
ХРОМ
251
Соединение СгРО4 представляет собой черный порошок с плот-
ностью 2,94 г!см? и т. пл. ~1800°; оно плохо растворимо в воде
и слабо взаимодействует с горячей H2SO4.
Известны кристаллогидраты СгРО4-мН2О, где п = 6, 4, 3 и 2.
Кристаллогидрат СгРО4-6Н2О представляет собой фиолетовые
триклинные кристаллы с плотностью 2,12 г: см?, растворимые
в воде и концентрированных минеральных кислотах.
Кристаллогидрат СгРО4-2Н2О образует зеленые кристаллы
с плотностью 2,42 г! см?, плохо растворимые в воде и растворимые
в концентрированных минеральных кислотах.
Координационные соединения
Известно очень много соединений трехвалентного хрома с коор-
динационным числом шесть, которые классифицируются по числу
координационных сфер на моно-, ди-, три-, тетра- или поли-
ядерные комплексы и по природе лигандов на аммины, аквоамми-
ны, ацидоаммины, аквосоли, ацидосоли, ацидоаквосоли, аци-
доамминосоли, гидроксосоли, аквогидроксосоли и т. д.
Обилие координационных соединений трехвалентного хрома
обусловлено склонностью иона Сг3* к образованию комплексов
с различными лигандами, способными входить во внутреннюю
координационную сферу. Существует множество изомеров этих
комплексов.
Моноядерные координационные
соединения
Они могут иметь следующие общие формулы: [CrA6]Y3,
ICtA5X]Y2, [CrA4X2]Y, [СгА3Х3], МеЧСгА2Х4], Mei[CrAX5],
Mei[CrX6], где А — электронейтральная молекула, X и Y —
кислотные радикалы, Me1 — различные катионы. В табл. 41
указаны основные типы моноядерных комплексов хрома(Ш).
1. Комплексы типа [CrA6]Y3, где А = NH3, Н2О, мочевина
NH2 — СО — NH2, антипирин COC10H12N2, этилендиамин
1/2NH2 — СН2 — СН2 — NH2, пропилендиамин X/2NH2 — СН2 —
— СН2 — СН2 — NH2, бензидин VaC^H^Na и Y = F-, С1~, Вг_,
I”, CN“, NCS~, NO~, СН3СОО“, V^OJ", V2SeO|~ включают гекс-
аммины [Cr(NH3)6]Y3 — желтые или оранжевые (называемые
также лутеосолями), где Y = Cl-, I-, NO3, 1/2SO'^, Х/3РО3-;
гексакарбамиды [Cr(NH2 — СО — NH2)6]Y3, например
(Cr(NH2 — СО — NH2)6]C13 -ЗН2О — зеленый; гексантипирины
[Cr(COC10H12N2)6]Y3; гексаквосоли [Cr(H2O)6]Y3 — фиолетовые,
например [Cr(H2O)6]F3, [Сг(Н2О)6]С13, [Сг(Н2О)6]13-ЗН2О,
ICr(H2O)6]ClSO4(3 или 4)Н2О, [Ct(H2O)6](NO3)3 *2Н2О,
{СКН2О)6](СН3СОО)3, [Cr(H2O)6]AsO4-НаО; аквопентаммины
252
ПОБОЧНАЯ ПОДГРУППА VI ГРУППЫ
[Cr(H2O)(NH3)5]Y3, например [Cr(H2O)(NH3)6]Cl3 — оранжевый;
диаквотетраммины [Сг(Н2О)2(ХН3)4]Х3, где Y = СГ, Вг-, NO",
VzSO*-, например [Cr(H2O)2(NH3)4]Y3 — красные; триаквотри-
1 [СгА6]У3 [Сг[(Н2О)А5]¥3 [Сг(Н2О)2А4]У3
2 [CrA5X]Y2 [Cr(H2O)A4X]Y2 [Сг(Н2О)2А3Х]У2
3 [СгА4Х2]У [Сг(Н2О)А3Х2]У [Сг(Н2О)2А2Х2]У
4 [СгАзХз] [Сг(Н2О)А2Х3] —
5 Ме1[СгА2Х4] — Ме1[Сг(Н2О)2Х4]
6 Ме2[СгАХ5] Ме2[Сг(Н2О)Х5]
7 MeJ[CrXe]
аммины [Gr(H2O)3(NH )31Y3, где Y = Cl". Вт", СЮ', напри-
мер [Сг(Н2О)з(МН3)з]Вгз — коричнево-красный; тетракводиамми-
ны [Cr(H2O)4(NH3)2]Y3 — красно-фиолетовые, где Y = СГ, Вг“,
1/2SO^_; трис(этилендиаммины) [GrEn3]Y3— желтые, например
[СгЕпз1С13-3,5Н2О, [СгЕпзПз’Н2О; диаквобис(этилендиаммины)
[Сг(Н2О)2Еп2]У3 — оранжево-желтые, например [Gr(H2O)2En2] •
• Вт3'2Н2О; трис(пропилендиаммины) [СгРп3]Хз, например
[СгРпзПз *Н2О — желтый; трибензидины [Cr(C12H12N2)3]Y3 —
светло-зеленые; тетракводипиридины [Cr(H2O)4Py2] Y3, например
[Cr(H2O)4Py2](NO3)3 — красный.
2. Комплексы типа [CrA6X]Y2, где А = NH3, Н2О, СНз — NH2,
С2Н5 — NH2, С4Н9 — NH2, изоамиламин С5НИ — NH2, пири-
дин C5H5N, V2NH2 - СН2 - СН2 — NH2 и X = F-, СГ, Вг",
Г, NO- NO", ОН-, V2SeO*-, V2SO«- и др.; Y = F-, СГ, Вт',
Г, NO", СЮ", NCS", V2SO4-, VzCzOI" и т. д. включают гидро-
ксопентаммины [Cr(NH3)5(OH)lY3 — розовые, где Y = СГ, Вг“,
Г, NO3, VzSO^', 1/2С2О^-; галогенопентаммины [Cr(NH3)5X]Y2,
где X = F-, СГ, Вт-, Г и Y = СГ, Вг", Г, NO", V^Of, напри-
мер [Cr(NH3)5F]F2 — красный, [Сг(ХН3)5Р]С12 — желтый,
[Gr(NH3)5Gl]Cl2 — пурпурный, [Сг(ННз)5Вг]Вг2 — фиолето-
вый, [Cr(NH3)5I]I2 — фиолетово-синий; нитропентаммины
ХРОМ
253
[Cr(NH3)s(NO2)]Y2, где Y = Cl-, Br", I", NO;, ^SO*-,
1/2C2O^-, например [Cr(NH3)5(NO2)]Cl2 — желтый; тиоцианато-
пентаммины [Cr(NH3)5(NCS)]Y2, где Y = Cl-, Br-, NO;, X/2C2O4-;
Таблица 41
[Cr(H2O)3A3]Y3 [Cr(H2O)4A2]Y3 — [Cr(H2O)6]Y3
[Cr(H2O)3A2X]Y2 — [Cr(H2O)5XjY2
— [Cr(H2O)4X2]Y
[Cr(H2O)3X3]
гидроксо- и галогеноаквотетраммины, например
[Cr(H2O)(NH3)4OH]Br2 - красный, [Cr(H2O)(NH3)4Cl]Cl2 -
красный, [Cr(H2O)(NH3)4Br]Br2 — карминово-красный,
[Cr(H2O)(NH3)4I]I2 — красный; гидроксо- и галогенидодиаквотри-
аммины, например [Cr(H2O)2(NH3)3(OH)]I2— фиолетово-красный,
[Cr(H2O)2(NH3)3Cl]Cl2 — красный, [Cr(H2O)2(NH3)3Br]Br2 — фио-
летовый; гидроксотриакводиаммины [Cr(H2O)3(NH3)2(OH)]Y2,
где Y = NCS-, X/2SO^-, хлорпентаметиламины [Сг(СН3 —
— NH2)5C1]Y2, хлорпентаэтиламины [Сг(С2Н5 — NH2)5C1]Y2,
хлорпентапропил амины [Сг(СН3 — СН2 — СН2 — NH2)5C1]Y2,
хлорцентабутиламины [Сг(С4Нэ — NH2)6C1]Y2. хлорпентаизо-
амиламины [Сг(С5Нп — NH2)5C1]Y2, где Y = Cl-, Br-, I-; гало-
гено- и сульфатопентаквосоли типа [Cr(H2O)5X]Y2, где X и Y
могут быть Cl-, Br-, T/2SO^-; гидроксо- и бромоакводиэтилендиа-
мины, например [Сг(Н2О)Еп2(ОН)]С12, [Сг(Н2О)Еп2Вг]Вг2.
3. Комплексы типа [CrA4X2lY (и их стереоизомеры), где А =
= NH3, Н2О, С3Н7 - NH2, С4Н9 — NH2, С5Нп - NH2, c5h5n,
V2NH, - СН2 - СН2 - NH2, 1/2C12H8N2, X = Cl-, Br-, NCS-,
ОН-, ’/2C2Of и Y = Cl-, Br-, I-, NCS-, NO;, X/2SO|- включают
дигалогено-, дитиоцианато- и оксалатотетраммины, напри-
мер [Cr(NH3)4Cl2]Br. [Cr(NH3)4Br2]Br, (Cr(NH3)4(NCS)2]Cl,
254
ПОБОЧНАЯ ПОДГРУППА VI ГРУППЫ
[Cr(NH3)4(C2O4)]NO3-НгО; дигалогеноаквотриаммины, например
[Cr(H2O)(NH3)3Cl2]Cl, [Cr(H2O)(NH3)3Br2]Br; дигидроксоаквотри-
аммины [Cr(H2O)(NH3)3(OH)2]Cl; дигидроксо- и дигалогенодиакво-
диаммины, например [Cr(H2O)2(NH3)2(OH)2]Br-Н20 — фиолето-
вый, [Cr(H2O)2(NH3)2Cl2]Cl — зеленый, [Cr(H2O)2(NH3)2Br2]Br —
зеленый; дигалогено-, дитиацианато- и оксалатодиэтилендиамины
типа [CrEn2X2]Y, где X = С1~, Вт-, I-, NCS-, и Y =
= С1~, Вт-, NO;, X/2SO2-; дигидроксодигалогено- и оксалатодиа-
квоэтилендиамины типа [Cr(H2O)2EnX2]Y, где X = ОН-, С1-,
Х/2С2О4- и Y = С1-, Вг-, I-, NCS-, NO3; дихлортетрамины
[Ст(С3Н7 - NH2)4C121Y, [Ст(С4Н9 - NH2)4C12]Y, [&№ -
— NH2)4C12]Y; дифтортетрапиридины [CrPy4F2]Y, где Y = NO;,
Cl-, Вт-, I-, NCS-; дигидроксо- и дигалогенодиакводипири-
дины, например [Ст(Н2О)2Ру2(ОН)2]С1, [Ст(Н2О)2Ру2С12]С1,
[Ст(Н2О)2Ру2Вт2]Вт-4Н2О — соединение зеленого цвета; дигидро-
ксо- и дигалогенотетраквосоли, например [Cr(H2O)4(OH)2]2SO4 —
зеленый, [Сг(Н2О)4С12]С1-2Н2О— зеленый, [Ст(Н2О)4Вт2]Вт •
• 2Н2О — зеленый.
4. Комплексы типа [СтА3Х3], где X = F-, С1-, Вт-, NCS-,
ОН-, X/2SO2- и А = NH3, Н2О, C2H5N, С2Н5 - ОН, X72NH2 -
— СН2 — СН2 — NH2 включают самые разнообразные неэлек-
тролиты, например тригалогенотриамминхром(Ш) [Cr(NH3)3Cl3] —
зеленый, [Cr(NH3)3Br3] — зеленый, [Cr(NH3)3Br2Cl] — зеленый,
[Cr(NH3)3Cl2Br] — зеленый; тритиоцианатотриамминхром(Ш)
[Cr(NH3)3(NCS)3] — розовый; галогеноксалатотриамминхром(Ш)
[Ct(NH3)3C1C2O4] — красный, [Cr(NH3)3BrC2O4] — темно-крас-
ный; тригидроксо- и тритиоцианатоакводиамминхром(Ш)
[Cr(H2O)(NH3)2(OH)3] -4Н2О—фиолетовый, [Cr(H2O)(NH3)2(NCS)3] •
•Н2О — пурпурный; тригалогенотрипиридинхром(Ш) [CrPy3F3] •
•Н2О — синий, [CrPy3F3] — фиолетовый, [СгРу3С13] — зеленый;
тригидроксо- и тригалогеноакводипиридинхром(Ш)
[Ст(Н2О)Ру2(ОН)3] -6Н2О — фиолетовый, [Cr(H2O)Py2F3] -Н2О —
фиолетовый, [Сг(Н2О)Ру2С13] — желтовато-зеленый; тригалогено-
и тритиоцианатотриаквохром(Ш) [Cr(H2O)3F3] -0,5Н2О — фио-
летовый, [Ст(Н2О)3С13] — фиолетовый, [Ст(Н2О)3Вт3] — красный,
[Cr(H2O)3(NCS)3] — красный; хелатные соединения хрома с аце-
тилацетоном [Сг(С5Н7О2)3]—фиолетово-красный, с 8-оксихиноли-
ном, гликоколем, аланином и пикратом [Cr(C6H2O7N3)3] >8Н2О —
желтовато-зеленый.
5. Комплексы типа Ме1[СгА2Х4] (и их стереоизомеры), где
А = NH3, Н2О, X72NH2 - СН2 - СН2 - NH2, C5H5N, С6Н5 —
- NH2, X = F-, Cl-, CN-, NCS-, OH-, X/2C2O2- и Mex = Na+,
К+, Cs+, NH;, x72Ca2+, x/2Sr2+ включают тетрациано- или тетра-
тиоцианатодиамминхроматы(Ш), например, Na[Cr(NH3)2(CN)4],
NH4[Cr(NH3)2(NSC)4] -Н2О соль Рейнеке; диоксалатодиамминхро-
маты(Ш) MeI[Cr(NH3)2(C2O4)2J; диоксалатодиаквохроматы(Ш)
ХРОМ
255
MeI[Cr(H2O)2(C2O4)2l; диоксалатоаквопиридинхроматы(Ш)
Мег[Сг(Н2О)Ру(С2О4)2]; хлор- или гидроксотритиоцианатодиам-
минхроматы(Н1)Ме1[Сг(ХН3)2С1(ХС8)з], MeI[Cr(NH3)2(OH)(NCS)3];
тетратиоцианато- или диоксалатоэтилендиаминхроматы(Ш)
Me^CrEn^NCS)^, МеЧСгЕ^СгО^г]; тетрахлор- или тетратиоциа-
натодипиридинхроматы МеЧСгРугСЦ, MeI[CrPy2(NCS)4]; дио-
ксалатодипиридин (дианилин-, дицинхонин-)хроматы(Ш)
МеЧСгРу2(С2О4)2], МеЧСг(С6Н5 - NH2)2(C2O4)2],
Мег[Сг(С19Н22М 2О) 2(С 20 4) 2]; дихлороксалатодиаквохроматы(Ш)
МеЧСг(Н2О)2(С2О4)С12].
6. Комплексы типа Mei[CrAX6], где А = NH3, Н2О, NH2 —
- СН2 - СН3, X = F-, Cl", Вт-, CN-, NCS-, СН3СОО', ОН",
Х/2С2О2- и Me1 = Li+, Na+, К+, Rb+, Cs+, NH4\ Ag+, V2Ca2+,
1/2Ba2+, V2Zn, включают пентагалогено-(пентатиоцианато-хрома-
ты(Ш) Mei[Cr(H2O)F5], Mei(Cr(H2O)Cl5], Me*[Cr(H2O)Br5],
Mei[Cr(H2O)(NCS)5]; диоксалатогидроксоаквохроматы(Ш)
Me*[ Сг(Н 2O)(C 2O 4) 2(OH) ]; диоксал атоацетатаквохроматы(111)
Mei[Cr(H2O)(C2O4)2(CH3 — COO)]; диоксалатохлораквохрома-
ты(Ш) Mei[Cr(H2O)Cl(C2O4)2L
7. Комплексы типа Me*[CrX6], где X = F“, Cl-, CN_, NCS ,
WJ-, V2C4O6H2-, ^/зРзО2- и M+ = Li+, Na+, K+, NHJ, Ag+,
x/2Ca2+, x/2Pb2+, 1/2Cd2+, 1/2Mn2+, 2/2Ni2+, 1/2Cu2+ включают гекса-
галогено-(или гексапсевдогалогено-)соли, например, K3[CrF6] —
зеленый, К3[СгС16] — фиолетовый, K3[Cr(CN)6] — желтый,
K3[Cr(NCS)6] -4Н2О — красный; триоксалато- (триселенато-, три-
хромато-, тритартрато-, трималонато-, трипирокатехинато-)соли,
например, К3[Сг(С2О4)3] — зеленый, (NH4)3[Cr(SeO4)3]-2,5Н2О—
зеленый, Сг[Сг(С2О4)3]-ЗН2О — коричневый, К3[Сг(С4О6Н4)3] •
• Х/2Н2О — фиолетовый, К3[Сг(С3Н2О4)3] — сине-зеленый,
К3[Сг(О — С6Н4 — О)3] — зеленый; диоксалатодихлор-(диацетато-,
дигидроксо-)соли типа Mei[CrCl2(C2O4)2],Me*[Cr(CH3COO)2(C2O4)2],
Me3i[Cr(OH)2(C2O4)2].
Ди-, три- и тетраядерные
координационные соединения
Известны многочисленные ди-, три- и тетраядерные координа-
ционные комплексы хрома(Ш), которые могут иметь в качестве
лигандов нейтральные молекулы NH3, NH2 — СН2 — СН2 — NH2
или кислотные радикалы SO2-, С2О^_, SeO2-, СН3СОО_, а в каче-
стве мостиковых групп — ОН — гидроксо, — О — оксо, — О — О —
пероксо, СН3СОО~.
В качестве примеров диядерных координационных соединений
хрома(Ш) можно назвать соли гидроксо-бггс(пентаммин)хрома(Ш)
(называемые родохромовыми солями) [(H3N)5Cr — ОН ->
-> Cr(NH3)5]X5, соли |л-оксо-бис(пентаммин)хрома(Ш) (называв-
256
ПОБОЧНАЯ ПОДГРУППА VI ГРУППЫ
мые эритрохромовыми солями) [(H3N)5Cr — О — Cr(NH3)5]X4-
• НХ, соли ди-ц-гидроксо-бис{диэтилендиамминхрома(Ш)} соли
три-ц-гидроксо-бис {триамминхрома(Ш)}
г Д
(H3N)3Cr^-O-Cr(NH3)3
L н
Х3
ди-[л-гидроксо-бис{диоксалатохроматы(Ш)}
MeJ
Н
z°\
(С2О4)2Сг^ Сг(С2О4)2
Х(/
н
сульфатохромовые кислоты
H2[Cr2(SO4)4], H4[Cr2(SO*)6], H6[Cr2(SO4)e]
селенатосульфатохромовые кислоты Н2[Ст2(ЗО4)(8еО4)з1,
H4[Cr2(SO4)2(SeO4)3] Н6[Ст2(ЗО4)3(8еО4)з]. В приведенных выше
общих формулах одновалентные кислотные радикалы обозначены
через X.
В качестве примеров триядерных комплексов хрома(Ш) можно
привести следующие формиатные, ацетатные, пропионатные, хлор-
ацетатные, бензоатные соединения: [Ст3(СН3—СОО)6(ОН)2]СН3СОО,
[Ст3(СН3 — СОО)6(ОН)(Н2О)](СН3СОО)2, [Сг3(СН3 - СОО)6(Н2О)2.
.(СН3СОО)31, [Стз(СН3 - СОО)з(ОН)3]Х3, [Сг3(СН3- СОО)6(ОН)2-
•Ру3]Х, [Ст3(СН3— СОО)6(Н2О)2Ру31Х3, [Ст3(С2Н5-СОО)6(ОН)2]Х,
[Сг3(С2Н5 - СОО)5(ОН)2(Н2О)2]Х2, [Сг3(С2Н5 - СОО)6(ОН)2]Х,
[Сг3(С1 - СН2 - СОО)6(ОН)2]Х.
При обработке растворов солей хрома(Ш) раствором ацетата
натрия или аммония образуется очень устойчивый как на холоду,
так и при нагревании растворимый комплекс:
ЗСгС13 + 9NaCH3COO + 2Н2О -
= [Сг3(СН3СОО)6(ОН)2]СН3СОО + 9NaCl + 2СН3СООН
При растворении гидроокиси Сг(ОН)3 в уксусной кислоте
образуются упомянутые выше комплексные ацетаты хрома(Ш).
ХРОМ
257
В качестве примеров тетраядерных комплексов хрома(Ш)
можно назвать соли гекса-ц-гидроксогексаэтилендиаминтетрахро-
ма(Ш) [Сг(ОН)6Еп6]Х6 и пента-р-гидроксогексамалонатотетрахро-
маты(Ш) Mei[Cr4(OH)5(C4H4O6)6].
Помимо перечисленных известны и другие соединения хрома(Ш), как-то:
хлорат Сг(С1О3)3, перхлорат Сг(С1О4)3, дихлорбромид СгС12Вг, бромат
Сг(ВгО3)3, дихлориодид СгС121, дибромиодид СгВг21, хромиты VCr2O4,
UCr2O5, бисульфит Cr(HSO3)3, дитионат Cr2(S2O6)3-18Н2О, селенид Cr2Se3,
селенат Cr2(SeO4)3, селенохромовые квасцы MeiCr(SeO4)2-12Н2О, теллурит
Сг2(ТеО3)2, теллурат Сг2(ТеО4)3-пН2О, азид Cr(N3)3, амид [Cr(NH2)3]n,
дихлоронитрат Cr(NO3)Cl2-пН2О, гидроксохлоронитрат Cr(OH)(NO3)Cl, суль-
фатонитраты Cr(NO3)SO4, Cr2(NO3)4SO4, гипофосфит Сг(Н2РО2)3-2Н2О,
фосфит СгРО3, двойные фосфиты NaCr(HPO3)2-14H2O, КСг(НРО3)2-
• 12Н2О, (NH4)Ct(HPO3)2-8Н2О, гидроортофосфат СгН3(РО4)2, двойные
фосфаты 2CrPO4(NH4)2HPO4 -ЗН2О, 2CrPO4-Na2HPO4-5Н2О, дифосфат
Сг4(Р2О7)3, двойные дифосфаты MeiCrP2O7-пН2О, где Mei = Na+, NHJ
и n = 5—8, метафосфат CrP3O9, тиофосфит Cr2P2S6, тиопирофосфат Cr2P2S7,
арсенид CrAs, арсенит CrAsO3, арсенаты Cr2Na3(AsO4)3, CrAsO4, Cr4(As2O7)3,
[Cr(H2O)6]AsO4-H2O, CrAs3O9, антимонид CrSb, антимонаты CrSbO4,
CrSb3O9-14H2O, ортованадат CrVO4, ортотанталат СгТаО4, гидроксокарбонат
[Сг2(ОН)5]2СО3-8H2O, тиокарбонат Cr2(CS3)3, цианид Cr(CN)3, тиоцианат
Cr(NCS)3, тетрафтороборат [Cr(H2O)6][BF4]3, безводный формиат Сг(НСОО)3,
гидратированный формиат Сг(НСОО)3-5Н2О, ацетат [Сг(Н2О)6](СН3СОО)3,
бензоат Сг(С6Н5СОО)3-пН2О, оксалат [Сг(Н2О)6]2(С2О4)3-пН2О (где
п = 0,14 или 13), лактат Сг(СН3 — СНОН — СОО)3, тартраты
Сг2(С4О6Н4)3-Н2О, Сг2(С4О6Н4)3, этилат Сг(ОС2Н5)3.
Соединения четырехвалентного хрома
Известно относительно немного соединений четырехвалентного
хрома. Они могут быть окрашены в желтый, коричневый, красный,
черный, зеленый и другие цвета, сравнительно неустойчивы,
проявляют главным образом окислительные свойства.
На основе исследования магнитной восприимчивости было
установлено, что хром в соединениях CrO2, K2[CrF6], Ва2СгО4,
Sr2CrO4, CrO4-3NH3 электроположителен и четырехвалентен.
Двуокись хрома, СтО2) получают нагреванием гидратированной
окиси хрома(Ш) в кислороде при 350—400°, прокаливанием
нитрата хрома(Ш) при 400° на воздухе и пиролизом хлористого
хромила СтО2С12 на стеклянных шариках при 360°.
Полученный сухим способом СгО2 представляет собой ферро-
магнитные (точка Кюри 116°) черные микрокристаллы со струк-
турой рутила, плохо растворимые в воде; они разлагаются при
427°, превращаются в Сг2О3 и СтО3 под действием воды при 100°,
растворяются в НС1 с выделением хлора, образуют хроматы
щелочных металлов при сплавлении со щелочами или карбо-
натами щелочных металлов, разлагаются при сильном нагревании
на Сг2О3 и кислород.
Известны следующие гидраты двуокиси хрома: 2СгО2’Н2О,
2СгО2-ЗН2О, СгО2-2Н2О, ЗСтО2-7Н2О. Полугидрат 2СтО2-Н2О
17—0101
258
ПОБОЧНАЯ ПОДГРУППА VI ГРУППЫ
образуется при прокаливании бихромата аммония (190°) на воз-
духе. Кристаллогидрат 2СгО2-ЗН2О получают действием хлора,
или иода на спиртовый раствор К2Сг2О7. Дигидрат СгО2-2Н2С>
можно приготовить взаимодействием К2Сг2О7 с KI в отсутствие;
кислот.
Гидратированная двуокись хрома с различным содержанием
воды получается в виде коричневого осадка обработкой водным
раствором хромового ангидрида СгО3 гидроокиси Сг(ОН)3, вос-
становлением СгО3 в растворе KI, а также восстановлением хро-
мата или бихромата с помощью Ag2O, С2Н5ОН, Na2S2O3 *5Н2О,
смеси HNO3 с Н2С2О4-2Н2О и других реагентов.
Тетрафторид хрома, CrF4, получают пропусканием фтора над,
металлическим хромом, над СгС13 или CrF3, помещенными в нагре-
тую до 350—500° трубку, изготовленную из окиси алюминия.
Расплывающееся на воздухе аморфное вещество CrF4 имеет
коричневый цвет, плотность 2,9 г!см\ сублимируется при 200°,
имеет т. пл. 277° и т. кип. 295°. Тетрафторид хрома восстанавли-
вается водородом при нагревании выше 230°, гидролизуется
водой, образуя соединения хрома(Ш) и (VI), действует на крем-
незем и взаимодействует при 100° с парафиновым маслом.
При нагревании CrF4 со смесью 2КС1 и СгС13 получается пара-
магнитное соединение K2[CrF6|.
Тетрахлорид хрома, СгС14, образуется в виде желтого газооб-
разного вещества при действии хлора на трихлорид хрома, нагре-
тый до 700° в кварцевой трубке.
При быстром охлаждении твердой углекислотой газообразный
тетрахлорид хрома превращается в твердое коричневое вещество,
которое начинает диссоциировать при —80°; в вакууме при обыч-
ной температуре диссоциация на СгС13 и С12 происходит быстрее:
СгС14 СгС13 + V2C12
Во влажной атмосфере СгС14 гидролизуется
ЗСгС14 + ЗН2О СгОз + 2СгС13 + 6НС1
Тетрахлорид хрома взаимодействует с водой с выделением кис-
лорода и окисляет растворы иодида калия с высвобождением иода.
Псевдохроматы хрома(1У) имеют общую формулу Ме*Сг4+О4
или MejrCrIVO4.
При нагревании в атмосфере водорода смеси ВаСгО4 с Сг20з;
и Ва(ОН)2, смеси Ва(ОН)2 с Ва3[Сг(ОН)6]2 или смеси BaCrOi.
с Ва(ОН)2 получают блестящий зеленый псевдохромат бария?
Ва2СгО4:
900-1000°
ВаСгО4-|-Сг2О3 + 5Ва(ОН)2------->• ЗВа^Сг1¥О4-|-5Н2О
1000° тт т,7
Ва3[Сг1И(ОН)6]2-|-Ва(ОН)2-> 2Ba£ICrIVO4+6Н2О + Н2
400-500°
ВаСгО4 + Ва(ОН)2+Н2-------> Ва2СгО4 + 2Н2О
ХРОМ
259
Аналогичным образом при 800° был приготовлен Sr^CrJVOi.
В подкисленной воде псевдохроматы диспропорционируют на
соединения трех- и шестивалентного хрома:
3CrIV01--|- 16Н+ ->• 2Crln4-CrVIO£- + 8H2O
Производное тетраокиси хрома с аммиаком CrO4>3NH3 обра-
зуется при действии (в течение нескольких часов) 30%-ного рас-
твора Н2О2 на 20%-ный раствор аммиака, насыщенный при 0°
(NH4)2Cr2O7 и газообразным NH3. Соединение CrO4-3NH3 пред-
ставляет собой сильно парамагнитные, двулучепреломляющие
красновато-коричневые кристаллы в виде ромбических призм
с плотностью 1,96 г!см3.
При действии сильных кислот и щелочей на CrO4-3NH3 выде-
ляется четверть общего содержания кислорода.
Аммиачное производное тетраокиси хрома имеет следующее
строение:
Г О. 1
H3N4|\0
H3N -> Сг/
При замещении аммиака этилендиамином, гексаметилентетра-
амином или цианидом калия получают соединения СтО4 -Ен -2Н2О,
CrO4 -C6H12N4, CtO4-3KCN.
Соединения пятивалентного хрома
Известно небольшое число соединений хрома(У), причем все
они сравнительно неустойчивы, ведут себя как окислители и окра-
шены в различные цвета: желтый, красный, пурпурный, коричне-
вый, зеленый.
Пятивалентность хрома была установлена как йодометрическим
методом, так и на основе исследования дифрактограмм его соеди-
нений и магнитной восприимчивости.
В металлоорганических соединениях хром, как правило, пяти-
валентен, например в производных с 3, 4 или 5 фенильными
группами.
Пентафторид хрома, CrF5, выделяют как побочный продукт
в .небольших количествах при получении тетрафторида хрома.
Он представляет собой красное твердое вещество, которое субли-
мируется при 100° и гидролитически разлагается водой на соеди-
нения трех- и шестивалентного хрома:
500°
4Cr-|-9F2--► 2CrF5-|-2CrF4
Окситетрафторохромат калия KlCrOFJ и окситетрафторо-
хромат серебра Ag[CrOF4],
17*
260
ПОБОЧНАЯ ПОДГРУППА VI ГРУППЫ
Соединение K[CrOF4] получают в виде пурпурного порошка
при обработке эквимолярной смеси СгО3 и КС1 с помощью BrF3
или при нагревании соединения K[CrOFJ-СгВгВз, полученного
взаимодействием BrF3 с K2Cr2O7.
Соединение Ag[CrOF4] получают по реакции между BrF3
и Ag2Cr2O7. Это пурпурный порошок, окисляющийся во влажном
воздухе.
Оксипентахлоро-и окситетрахлорохро-
маты, K2[CrOCI5], RbJCrOCLJ, Cs2[CrOCI5], (C5H6NH)[CrOCl4].
• Н2О, (C9H7NH)[CrOCl4] -Н2О,— твердые вещества красного
цвета.
Соединение К2[СгОС15] получают в виде рубиново-красного
осадка при обработке на холоду соляной кислотой смеси раство-
ров КС1 и концентрированного раствора СгО3, насыщенного
газообразным НС1.
Соединения типа Ме^[СгОС15] аналогичны соответствующим
соединениям молибдена Ме’[МоОС15], ванадия Me*[VOCl5], ниобия
Me^NbOClj] и тантала Ме*[ТаОС15].
Псевдохроматы хром a(V) типа Ме|СгО4 или
МеР(СгО4)2 (где Me1 — Li+, Na+ и Me11 = Ва2+, Sr2+).
Хромат Ва3(СгО4)2 получают в виде темно-зеленого мелко-
кристаллического порошка нагреванием до 800—1000° в течение
4 час в атмосфере азота соединения Ва5(СгО4)3(ОН) или смеси
ВаСгО4 с ВаСО3 либо Ва(ОН)2:
2Ва5(СгО4)3(ОН) = ЗВа3(СгО4)2 + ВаО + Н2О
2ВаСгО4 + ВаСОз = Ва3(СгО4)2 + СО2 + У2О2
Соединение Ва5(СгО4)3(ОН) получают нагреванием при 600°
в токе влажного азота смеси ВаСгО4 с Ва(ОН)2.
Хромат Sr3(CrO4)2 представляет собой темно-зеленый порошок
и получается по методикам получения Ва3(СгО4)2 (см. выше) при
использовании Sr5(CrO4)3(OH) или смеси SrCrO4 с SrCO3 либо
Sr(OH)2.
Хромат Na3CrO4 получают осторожным нагреванием (350—
500°) в токе азота смеси Na2CrO4 с NaN3:
Na2CrO4 + NaN3 = Na3CrO4 + 3/2N2
В подкисленной воде эти хроматы диспропорционируют на
соединения трех- и шестивалентного хрома:
3(CrvO43" + 8Н+ -> Сг111 + 2CrVIO^- + 4Н2О
Пер оксохроматы (красные, коричневые или желтые)
типа Me*CrO8 •nH2O, М1М1[СгО8 •пНзО, Мр(СгО8)2-пИзО более
устойчивы, чем синие хроматы хрома(У1). Они парамагнитны,
изоморфны пероксониобатам или пероксотанталатам и являются
ХРОМ
261
производными не выделенной в свободном состоянии кислоты
О О —ОН
^Сг—О—ОН
О7 ХО-ОН
Пероксохромат лития, Li3CrO8, выделяют в виде желтого
осадка при добавлении спирта к раствору, полученному действием
30%-ного раствора Н2О2 на смесь хромата и гидроокиси лития:
2Li2CrO4 + 2LiOH + 7Н2О2 = 2Li3CrO8 + 8Н2О
Пероксохромат натрия, Na3CrO8-14^0, получают путем обра-
ботки 30%-ным раствором Н2О2 смеси СтО3 и NaOH.
Соединение Na3CrOs-14Н2О представляет собой двупреломляю-
щие красновато-желтые кристаллы, образующие смешанные соли
с Na3TaO8-14Н2О, превращающиеся на воздухе в желтый порошок
Na3CrO8, который разлагается при 110° на Na2CrO4 и кислород.
Пероксохромат калия, К3СтО8, получают действием 30 %-ного
раствора Н2О2 на смесь СтО3 и КОН при 0°,или на растворы хро-
матов, содержащие большой избыток КОН. Соединение К3СтО8
образует двупреломляющие коричневые призматические кристал-
лы, устойчивые в сухом воздухе при обычной температуре, разла-
гающиеся со взрывом при 178°, а в водном или кислом растворах
разлагающиеся по уравнениям
2К3СгО8 + Н2О = 2К2СгО4 + 2КОН + 7/2О2
К3СгО8 + 6НС1 = СгС13 + ЗКС1 + ЗН2О + 6/2О2
Пероксохромат аммония, (NH4)3CrO8, получают действием
перекиси водорода на смесь СтО3 и аммиака при 0° или при обра-
ботке аммиаком синего эфирного раствора дипероксохромовой
кислоты Н2СтО6.
Соединение (NH4)3CrO8 представляет собой двупреломляющие
красные кристаллы, растворимые в воде, разлагающиеся со взры-
вом при 50° и образующие под действием аммиака коричневые
кристаллы CrO4-3NH3.
Известны также пероксохроматы NaMgCrO8-8Н2О красного,
KMgCrO8’7,5Н2О красного и Са3(СтО8)2-пНгО желтовато-белого
цвета.
Соединения шестивалентного хрома
Известны многочисленные соединения электроположительного
шестивалентного хрома, например: трехокисъ СгО3) перокись
СтО5, хроматы Ме*СгО4, МепСтО4, бихроматы Ме*Ст2О7, МепСг2О7,
трихроматы Ме*Ст3О10, тетрахроматы Ме*Ст4О13, пероксохроматы
MeiCr2O12, гелогенохроматы Мег[СгО3Х] (X = F-, С1~, Вт-, I”),
а также фтористый, хлористый и бромистый хромил СтО2Х2.
262
ПОБОЧНАЯ ПОДГРУППА VI ГРУППЫ
В высшей степени окисления хром проявляет в основном
неметаллические свойства и благодаря этому входит главным
образом в состав анионов СгО2-, Сг2О2-, [Сг3О10]2~, [Сг4О13]2“,
1СгОв]2~, [Сг2О12]2", [СгО3Х]“. Известно только несколько соеди-
нений, в состав которых хром входит в виде катиона хромила
СтО2+.
Соединения шестивалентного хрома получают окислением
металлического хрома или соединений двух- и трехвалентного
хрома. Они окрашены в желтый, оранжевый, красный или корич-
невый цвета и обладают окислительными свойствами.
Наиболее устойчивые соединения хрома(У1) — хроматы и би-
хроматы, а самые неустойчивые — пероксохроматы и галогениды
хромила.
Трехокисъ хрома (хромовый ангидрид), СгО3, получают дей-
ствием воды на хлористый хромил СгО2С12, обработкой сильно
концентрированных растворов хромата или бихромата калия
или натрия избытком конц. H2SO4, действием НС1 на хромат
серебра, обработкой хроматов РЬСгО4, ВаСгО4, SrCrO4 серной
кислотой, действием H2[SiFeJ на К2СгО4, обработкой ВаСгО4
избытком HNO3:
СгО2С12 + Н20 =₽* СгОз + 2НС1 Ag2CrO4 + 2НС1 = CrO3 + 2AgCl + Н2О
К2СгО4 -|- H2SO4 = СгОз -|- K2SO4 -|- Н2О
К2Сг2О7 -|- H2SO4 = 2СгО3 -|- K.2SO4 "Ь Н2О
РЬСгО4 + H2SO4 = СгОз + PbSO4 + Н2О
K2CrO4 + H2[SiF6] = СгОз + K2[SiF6] + Н2О
ВаСгО4 + 2HN03 = СгОз + Ba(NO3)2 + Н2О
Для очистки СгО3 перекристаллизовывают из водного рас-
твора и сушат при 70° на пористой пластинке.
Хромовый ангидрид представляет собой слабо парамагнитные
расплывающиеся на воздухе красные (с фиолетовым оттенком)
бипирамидальные орторомбические призмы с плотностью
2,80 гем? и т. пл. 197° (с легким разложением); при нагревании
выше температуры плавления превращается в красные пары,
хорошо растворим в воде, разлагается при 550° с выделением
2- додорода, обладает окислительными свойствами.
& При растворении СгО3 в воде образуются сильно кислые ток-
сичные растворы желтого, оранжевого или красного цвета, обла-
дающие окислительными свойствами. Очень разбавленные водные
растворы СгО3 желтого цвета; они содержат двухосновную хро-
мовую кислоту Н2СгО4, устойчивую в нейтральной среде и являю-
щуюся окислителем по отношению к SO2, H2S, SnCl2, FeSO4, HI
и др. Чем концентрированнее водный раствор СгО3, тем более
конденсированные кислоты (Н2Сг2О7, Н2Сг3О20, Н2Сг4О23,
ХРОМ
263
Н2Сг5О16) он содержит:
СгО3 + Н2О = Н2СгО4 ЗСгОз + Н2О = Н2Сг3О10
2СгО3 + Н2О = Н2Сг2О7 4СгО3 + Н2О = Н2Сг4О18
Трехокись хрома окисляет иод, серу, селен, фосфор, мышьяк,
углерод, окись углерода, спирты (С2Н5ОН, СН3ОН и др-), кис-
лоты (бромистоводородную, иодистоводородную, фосфорновати-
стую, фосфорноватую, муравьиную, щавелевую, малоновую,
янтарную, глутаровую, лимонную и др.) и водные растворы суль-
фата железа(П) и хлорида олова(П).
Из хромового ангидрида можно получить соли, содержащие
анионы типа (СгпО3п+1)2-. При п = 1 получаются хроматы,
п = 2 — бихроматы, п = 3 — трихроматы, п — 4 — тетрахрома-
-ты. При действии КОН или NaOH на СгО3 получают К2СгО4
или Na2CrO4, а при обработке СгО3 аммиаком образуется
<NH4)2Cr2O7 или (NH4)2CrO4.
Единственный известный гидрат трехокиси хрома 2СгО3 -7Н2О
устойчив в интервале температур от —ИЗ до —102°.
Хлористый водород и его концентрированные растворы обра-
зуют с СгО3 хлористый хромил СгО2С12.
Известны аддукты СгО3 с пиридином, пиколином и хинолином,
например СгО3-2С5Н5Х, СгО3 •2C5H4N(CH3), CrO3-2C9H7N и т. д.
Трехокись хрома катализирует автоокисление углеводородов
и ненасыщенных алифатических эфиров, восстанавливает чувстви-
тельность фотопластинок, десенсибилизированных метиленовой
синью, обладает прижигающим действием, что позволяет исполь-1(с
зовать ее в медицине. *
Хроматы (или монохроматы) типа Ме£СгО4 (где
.Ме1= Li+, Na+, К+, Rb+, NH+, Ag+, Т1+) или МепСгО4 (где
Me11 = Mg2+, Са2+, Sr2+, Ва2+, Pb2+, Hg2+) получают в щелочных
растворах окислением соединений хрома(Ш) с помощью С12,
Br2, I2, NaOCl, КОС1, Н2О2, PbO2, KMnO4, K3lFe(CN)e], сплавле-
нием соединений хрома(Ш) с окислительно-щелочными смесями,
обработкой водных растворов хромового ангидрида окислами,
основаниями или солями некоторых элементов, сплавлением
бихроматов с карбонатами щелочных металлов, нагреванием
трехокиси хрома с окислами или карбонатами некоторых элемен-
тов и обработкой растворов хроматов щелочных металлов раство-
рами солей серебра, бария и свинца, когда необходимо получить
плохо растворимые хроматы.
2К[Сг(ОН)4] + ЗС12 + 8КОН = 2K2CrO4 + 6КС1 + 8Н2О
2К[Сг(ОН)4) + ЗКС10 + 2КОН = 2К2СгО4 + ЗКС1 + 5Н2О
2К[Сг(ОН)4] + ЗН2О2 + 2КОН = 2К2СгО4 + 8Н2О
2К[Сг(ОН)4] + ЗРЬО2 + 8КОН = 2К2СтО4 + ЗК2[РЬ(ОН)4] + 2Н2О
2К[Сг(ОН)4] + 2КМпО4 = 2К2СгО4 + 2МпО2 + 4Н2О
264
ПОБОЧНАЯ ПОДГРУППА VI ГРУППЫ
К[Сг(ОН)4] + 3K3[Fe(CN)6] + 4К0Н == К2СгО4 + 3K4[Fe(CN)e] + 4Н2О
Cr2(SO4)3 + 5К2СО3 4- 3KNO3 = 2К2СгО4 + 3K2SO4 + 3KNO2 + 5СО2
Cr2(SO4)3 + ЮКОН -4 КС1О3 = 2К2СгО4 + 3K2SO4 + КС1 -j- 5Н2О
Сг2О3 + 2К2СО3 + 3KNO3 = 2К2СгО4 + 3KNO2 + 2СО2
Сг2О3 + 4КОН + КС1О3 = 2К2СгО4 + КС1 + 2Н2О
2FcCr2O4 + 4К2СО3 4- 7KNO3 = 4K2CrO4 + Fe2O3 + 7KNO2 + 4СОг
CrO3 + Li2O = Li2CrO4
CiO3 2MezOH = Me’CrO4 + H2O (где Me1 = Na+, K+, Rb+, Cs+, NHf)
CrO3 +Me£CO3 = Me*CrO4 + CO2 (где Me1 = Li+, Na+, K+, Rb+, Cs+)
CrO3 - BaCl2 + H2O + 2NaCH3COO = BaCrO4 Ц- 2NaCl -j- 2CH3COOH
CrO3 + 2AgNO3 + H2O — 2NaCH3COO = Ag2CrO4 + 2NaNO3 + 2CH3COOH
K2Cr2O, + K2CO3 = 2K2CrO4 + CO2 CrO3 + PbO = PbCrO4
2CrO3 + Cu2CO3(OH)2 = 2CuCrO4 + CO2 + H2O
K2CrO4 + 2AgNO3 = Ag2CrO4 + 2KNO3
K2CrO4 + BaCl2 = BaCrO4 + 2KC1
K2CrO4 + Pb(CH3COO)2 = PbCrO4 + 2KCH3COO
Хроматы представляют собой токсичные очень слабо пара-
магнитные желтые кристаллические вещества (изоморфные суль-
фатам, селенатам, молибдатам и вольфраматам), обладающие
окислительными свойствами, устойчивые в нейтральной и щелоч-
ной средах.
При подкислении водных растворов хроматов образуются
бихроматы, а в случае концентрированных растворов — три-,
тетра- или полихроматы.
Между хромат-(СтО^“) и бихромат-(Сг2О2~)анионами суще-
ствует равновесие, которое может быть сдвинуто в ту или иную
сторону с помощью кислот, оснований или растворов некоторых
солей, например, ВаС12*2Н2О, Bi(NO3)3-5Н2О, AgNO3 и др.
При обработке водных растворов некоторых хроматов или
бихроматов раствором хлорида бария, нитрата висмута или нит-
рата серебра образуется ВаСгО4 (но не бихромат, который более
растворим), (BiO)2Cr2O7 (но не хромат, который для этого катиона
более растворим), Ag2CrO4 и Ag2Cr2O7 (поскольку хромат и бихро-
мат серебра обладают близкими растворимостями в воде).
Хроматы щелочных металлов, аммония и магния растворимы
в воде в отличие от хроматов щелочноземельных и некоторых
тяжелых металлов, плохо растворимых в воде.
При сильном нагревании хроматы тяжелых металлов разла-
гаются с образованием Сг2О3.
Хроматы могут быть восстановлены галогеноводородными
кислотами — HCI, НВг, HI (с выделением хлора, брома или иода),
H2S (с выделением серы), SO2 (с образованием сульфата), солями
железа(П), спиртом и другими восстановителями.
ХРОМ
265
В уксуснокислом растворе хроматы окисляют бензидин
H2N—'У —NH2 до бензидиновой сини, а в слабо
х . „/NH-NHC8H5
сернокислом растворе — дифенилкарбазид (J = L ,
\ 1\ н IX ri
до соединения, окрашенного в интенсивный фиолетовый цвет.
Известны многочисленные двойные хроматы.
В очень разбавленных водных растворах бихроматов обра-
зуются гидрохроматы, содержащие анион НСгО~:
Сг2О?- + Н2О т 2HCrOj
Хромат лития, Li2CrO4, представляет собой желтые кристаллы
с плотностью 2,426 г/см3. При нагревании с Li2O или Li2CO3
до 700—800° в атмосфере азота они образуют хромат(У) Li3CrO4.
Известен дигидрат хромата лития Li2GrO4-2H2O в виде жел-
тых ромбических кристаллов и двойные хроматы, например,
Na3Li(CrO4)2 -6Н2О — желтые кристаллы и NH4LiCrO4 -2H2O —
желто-коричневые кристаллы.
Хромат натрия, Na2CrO4, представляет собой желтые ромби-
ческие бипирамидальные кристаллы с плотностью 2,723 г/см3'
и т. пл. 792°; они растворимы в воде, метаноле, плохо растворимы
в этаноле, превращаются в би-, три-, или полихроматы под дей-
ствием кислот и в хромат(У) Na3CrO4 при нагревании с Na3N
до 330—500° в атмосфере азота.
2Na2CrO4 + 2H2SO4 =s* Na2Cr2O7 + 2NaHSO4 + H2O
2Na2CrO4 + 2CO2 + H2O Na2Cr2O7 + 2NaHCO3
Известны кристаллогидраты Na2CrO/t-lOHoO — желтые моно-
клинные кристаллы, Na2CrO4-6H2O — темно-желтые триклинные
кристаллы, Na2CrO4-4H2O — желтые моноклинные кристаллы,,
а также гидроксохроматы Na2CrO4-2NaOH, Na2CrO4 -4NaOH.
При нагревании кристаллогидраты хромата(У) превращаются
в безводную соль:
20° 26°
Na2CrO4- 10Н2О •;...* Na2Ci'O4.6H2O ~ ~
-4Н2О — 2Н2О
62,8°
Na2CrO4-4H2O Na2CrO4
- 4H2O
Хорошо известен хромат калия, К2СгО4, который образует
очень слабо парамагнитные желтые ромбические бипирамидаль-
ные кристаллы с плотностью 2,732 г/см3, растворимые в воде,
в жидком SO2, в РОС13 и плохо растворимые в спирте. Хромат
калия обладает окислительными свойствами, превращается в би-,
три- или полихромат под действием кислот и при 666° переходит
в красную модификацию с т. пл. 971°.
266
ПОБОЧНАЯ ПОДГРУППА VI ГРУППЫ
Получены многочисленные двойные хроматы, например: КЫСгО4,
K3Na(CrO4)2, КРг(СгО4)2-яН2О, KNd(CrO4)2-яН2О, KSm(CrO4)2-ЗН2О,
KGd(CrO4)2-3,5H2O, КУ(СгО4)2-яН2О, K2(UO2)2(CrO4)3-6H2O,
K2UO2(CrO4)2-H2O, K4(UO2)3(CrO4)5-7Н2О, K6(UO2)4(CrO4)7.7H2O,
K2Fe2(CrO4)3.4H2O; K2Co(CrO4)2-2Н2О, K2Ni(CrO4)2 (2 или 6) Н2О,
К2Мп(СгО4)2-2Н2О.
Хромат, рубидия, Rb2CrO4 дает растворимые в воде желтые
орторомбические кристаллы с плотностью 3,525 г!см2.
Хромат, цезия, Cs2CrO4, представляет собой растворимые
в воде желтые орторомбические кристаллы с плотностью 4,35 г!см2.
Для рубидия и цезия известны двойные хроматы, например
Rb2Ni(CrO4)2 -6Н2О, Cs2Co(CrO4)2 -6Н2О, Cs2Ni(CrO4)2 -6Н2О и др.
Хромат аммония, (NH4)2CrO4,— желтые моноклинные кри-
сталлы с плотностью 1,866 г! см?, растворимые в воде и разлагаю-
щиеся при нагревании по реакции
2(NH4)2CrO4 = Cr2O3 + 2NH3 + N2 + 5Н2О
Известны двойные хроматы, например: LiNH4CrO4-
•2Н2О, NaNH4CrO4-2H2O, KNH4CrO4, K8(NH4)4(CrO4)5,
<NH4)2(UO2)2 (СгО4)з -6H2O, (NH4)2Fe2(CrO4)3 -4H2O,
(NH4)2Co(CrO4)2(2 или 6) H2O, (NH4)2Ni(CrO4)2-6H2O.
Хромат меди, CuCrO4, образует красные кристаллы в виде
ромбоэдрических призм, растворимые в кислотах, плохо раство-
римые в воде и разлагающиеся при нагревании.
Известны кристаллогидраты СпСгО4-2Н2О, СиСгО4*5Н2О, двой-
ные хроматы, например Na2Cu3(CrO4)4-nH2O, K2Cu(CrO4)2 "2Н2О,
а также аммиакаты CuCrO4"4NH3, CuCrO4 -4NH3 -Н2О.
Хромат серебра, Ag2CrO4, образуется в двух модификациях:
красной моноклинной с плотностью 5,625 г/см3 и зеленой с плот-
ностью 5,536 г! см?. Обе модификации растворяются в HNO3
и аммиаке и взаимодействуют с хлором при температуре выше
200°.
Ag2CrO4 -|- С12 = СгОз -|- 2AgCl -р */2О2
Известны двойные хроматы красного цвета Na2Ag4(CrO4)3,
K2Ag6(CrO4)4, (NH4)2Age(CrO4)4, (UO2)2Ag2(CrO4)3 и аммиакат
Ag2CrO4 -4NH3.
Хромат свинца, РЬСгО4, описан в числе соединений свинца(П).
Хромат бария, RaCrO4, представляет собой желтые кристал-
лы, плохо растворимые в воде и растворимые в минеральных
кислотах.
Помимо вышеприведенных, известны и другие хроматы, а именно:
BeCrO4, MgCrO4, СаСгО4, СаСгО4-яН2О (где я = 1/2, 1 или 2), SrCrO4,
BaCrO4, ZnCrO4, CdCrO4, Hg2CrO4, HgCrO4, Tl2CrO4, La2(CrO4)3, La2(CrO4)3-
• (1 или 8)H2O, Ce2(CrO4)3 (2 или 8)H2O, Pr2(CrO4)3-10H20, Nd2(CrO4)3-
• (8 или 9)H2O, Nd2(CrO4)3, Sm2(CrO4)3, Sm2(CrO4)3 (8 или 9) H2O, Gd2(CrO4)3-
• 8H2O, ТЬ2(СгО4)з-8Н2О, Dy2(CrO4)3-10H20, Er2(CrO4)3-яН2О.
УЬ2(СгО4)3-яН2О, У2(СгО4)3-яН2О, Sn(CrO4)2, Th(CrO4)2, Th(CrO4)2-яН2О
(где n = 1, 3 или 8), UO2CrO4, CoCrO4, NiCrO4, (UO2)2Pb(CrO4)3,
<UO2)2Hg2(CrO4)3.
ХРОМ
267
Изополисоединения
Бихроматы типа Ме|Сг2О7 или Me*Cr2O7 -rallgO (где Me1 =
= Li+, Na+, К+, Rb+, Cs+, NH+, Ag+, Ti+) и Me11 Cr2O7 или
МеПСг2О7-nH2O (где Me*1 = Cu2+, Mg2+, Ca2+, Sr2+, Ba2+, Zn2+,
Cd2+, Hg2+, Pb2+, Mn2+, Co2+, Ni2+) можно получить подкислением
растворов хроматов разб. H2SO4, обработкой водных растворов
СгО3 окислами, гидроокисями или карбонатами (например,
Na2O, Rb2O, NaOH, КОН, NH4OH, Li2CO3) при pH = 3,5, сме-
шением растворов хроматов с СтО3 (если необходимо получить
растворимые бихроматы) или обработкой водных растворов бихро-
матов растворами солей многих элементов (если необходимо полу-
чить бихроматы, плохо растворимые в воде).
Примеры реакций получения бихроматов:
2К2СгО4 + 2H2SO4 К2Сг2О7 + 2KHSO, + Н2О
2СгО3 -р Na2O — Na2Cr2O7 2СгО3 -j- 1л2СО3 = Li2Cr2O7 -|- СО2
2CrO3 + 2КОН = K2Cr2O7 + Н2О Na2CrO4 + СгО3 = Na2Cr2O,
Na2Cr2O7 + 2КС1 K2Cr2O7 + 2NaCl
K2Cr2O7 + 2NH4C1 (NH4)2Cr2O7 + 2КС1
K2Cr2O7 -j- 2AgNO3 = Ag2Cr2O7 -|- 2KNO3
Бихроматы устойчивы в кислой и неустойчивы в щелочной
средах, окрашены в оранжевый или красный цвет, как правило,
более растворимы в воде, чем соответствующие хроматы, являются
окислителями.
Превращение аниона Сг2О2- в СгО2- в щелочной среде обра-
тимо:
Сг2О?- + 2ОН- 2СгО£- + Н2О
Бодные растворы бихроматов токсичны, окрашены в оранже-
вый цвет, обладают кислой реакцией и являются окислителями
по отношению к НС1, НВг, HI, H2S, H2SO3, FeSO4, C2H5OH при
pH = 1 или 2.
K2Cr2O7 + 6НХ + 4H2SO4 = Cr2(SO4)3 + K2SO4 + 7H2O + 3X2
K2Cr2O7 + 14HX = 2CrX3 + 2KX + 7H2O + 3X2
K2Cr2O7 + 6KX 4-.7H2SO4 = Cr2(SO4)3 + 4K2SO4 + 7H2O + 3X2
K2Cr2O7 + 3H2S + 4H2SO4 = Cr2(SO4)3 + K2SO4 + 3S + 7H2O
K2Cr2O7 + 3H2SO3 + H2SO4 = Cr2(SO4)3 + K2SO4 + 4H2O
K2Cr2O7 4- 6FeSO4 + 7H2SO4 = Cr2(SO4)3 + 3Fe2(SO4)3 + K2SO4 + 7H2O
K2Cr2O7 + 3C2H5 — OH + 4H2SO4 =
= Cr2(SO4)3 + K2SO4 + ЗСН3 - CHO + 7H2O
В этих реакциях HX = НС1, НВг, HI; КХ = КВг, KI; X =
= V2C12, V2Br2, V2I2.
В результате перехода аниона Сг2О2 в катион Сг3+ по реакции
Сг2О2- + 14Н+ + бё -> 2Сг3+ + 7Н2О
268
ПОБОЧНАЯ ПОДГРУППА VI ГРУППЫ
оранжевый цвет раствора, свойственный бихроматам, переходит
в зеленый.
При подкислении растворов бихроматов до pH менее 6 или
при растворении СгО3 в воде получают двухромовую кислоту,
Н2Сг2О7, водный раствор которой окрашен в оранжевый цвет.
В очень разбавленных водных растворах бихромат-анион нахо-
дится в равновесии с анионом НСгО~.
Сг2О|- + Н2О 2HCrOj
При сильном подкислении концентрированных растворов
бихроматов анион Сг2О^_ превращается в анионы С3О,“, С4О1"
и раствор из оранжевого становится красным.
Дигидрат бихромата лития, Li2Cr2O7-2Н2О, выделяют из вод-
ных растворов в виде оранжево-красных кристаллов с плотностью
2,34 г!см3, которые плавятся при 187° с частичным разложением.
Бихромат лития превращается в Li2Cr2O7 при нагревании в тече-
ние нескольких дней при 110°; он плохо растворим в С2Н5 — О —
— С2Н5 и СС14, легко окисляет С2Н5ОН и превращается в ЫСг3О8,
если его нагревать с СгО3 при 350° в течение нескольких часов:
Li2Cr2O7 + 4CrO3 = 2LiCr3O8 -|- 3/2О2
Бихромат натрия, Na2Cr2O7, представляет собой слабо пара-
магнитные расплывающиеся на воздухе оранжево-желтые ромби-
ческие кристаллы с т. пл. 356,7°; соединение разлагается при
400° с образованием Na2CrO4, Сг2О3 и выделением кислорода,
является окислителем, растворяется в воде и спирте, превра-
щается в NaCr3O8 при нагревании с СгО3 при 350—400° в течение
нескольких часов.
Упаривая при температуре ниже 84,6° водные растворы бихро-
мата натрия, выделяют красные расплывающиеся на воздухе
кристаллы в форме моноклинных призм с плотностью 2,52 г!см?
дигидрата Na2Cr2O7-2Н2О. Они легко растворяются в воде,
плохо растворяются в спирте и выше 100° теряют две молекулы
воды, превращаясь в безводную соль Na2Cr2O7 до 100°.
Бихромат натрия получают в промышленном масштабе и ис-
пользуют при дублении кож и в электрических элементах.
Бихромат калия, К2Сг2О7, представляет собой оранжевые
триклинные (моноклинные выше 241,6°) кристаллы. Они токсич-
ны, горьки на вкус, имеют плотность 2,73 г! см? и т. пл. 398°,
разлагаются при 500° с образованием К2СгО4, Сг2О3 и выделением
кислорода. Бихромат калия растворяется в воде, жидком SO2,
РОС13, плохо растворим в жидком аммиаке и спирте, является
окислителем и превращается в хлористый хромил СгО2С12, будучи
нагрет с H2SO4 и каким-либо хлоридом. При нагревании К2Сг2О7
восстанавливается серой или углем по реакциям, приведенным
при описании получения окиси хрома(Ш).
ХРОМ
209
Реакции окисления НС1, НВг, HI, КВг, KI, H2S, H2SO3,
FeSO4 и С2Н5 — ОН с помощью К2Сг2О7 рассматривались при
описании бихроматов.
При подкислении концентрированных растворов К2Сг2О7 полу-
чают К2Ст3О10 и К2Сг4О13.
Бихромат калия применяют при дублении кож и в производ-
стве спичек.
Бихромат рубидия, ВЬ2Сг2О7, может существовать в виде
двух модификаций: оранжево-желтых моноклинных кристаллов
с плотностью 3,021 г/см3 и красных триклинных кристаллов
с плотностью 3,125 г/см3. Обе модификации растворимы в воде
и при нагревании до 300° с СгО3 в течение нескольких часов пре-
вращаются в ВЬСг3О8.
Бихромат цезия, Cs2Cr2O7, образует красные триклинные
кристаллы.
Бихромат аммония, (NH4)2Cr2O7, выделяется в виде оранжево-
красных моноклинных кристаллов с плотностью 2,15 г/см3, устой-
чивых на воздухе. Они растворяются в воде и спирте и разла-
гаются при нагревании на воздухе по реакции, приведенной при
описании получения Сг2О3.
Сульфат калия и перекись водорода реагируют с (NH4)2Cr2O7
по уравнению:
(NH4)2Cr2O7 ~|- K2SO4 -р 5Н2О2 = К2Сг2О12 (NH4)2SO4 ~|- 5Н2О
Помимо названных известны и другие бихроматы, например: Ag2Cr2O7 —
красный, Т12Сг2О7 — оранжевый, MgCr2O7 -5Н2О — оранжево-красный,
MgCr2O7-H2O — красный, СаСг2О7-иН2О (где п = 6, 5, 4) — оранжево-
красный, SrCr2O7-3H2O — коричнево-красный, ВаСг2О7 — коричневый,
ZnCr2O7-3H2O — оранжевый, CdCr2O7-H2O — оранжево-коричневый,
HgCr2O7 — красный, РЬСг2О7 — красный, МпСг2О7 — рубиново-красный,
€юСг2О7-Н2О — черный.
Трихроматы типа Ме*Сг3О10•nHgO или MeiCr3O10 (где
Me1 = Na+, К+, Rb+, Cs+, NH+) образуются при концентрирова-
нии растворов, полученных обработкой водных растворов бихро-
матов азотной кислотой (плотность 1,19—1.39 г/см3) или СгО3:
ЗМс2Сг2О7 + 2HNO3 = 2Ме1Сг3О10 4- 2Ме^О3 + Н2О
(Mei = Na+, К+, NHJ)
Ме2Сг2О7 + CrO3 = Me2Cr3O10 (Me1 = Na+, К+. Rb+, Cs+)
При обработке водных растворов трихроматов растворами
солей некоторых двухвалентных металлов получают трихроматы
липа МепСг3О10-пН2О (где Me11 = Sr2+, Ва2+, Cu2+, Zn2+, Cd2+,
Ni2+), плохо растворимы в воде.
Трихроматы окрашены в коричнево-красный или коричневый
цвет, слабо парамагнитны, разлагаются водой на соответствую-
щий бихромат и СгО3.
270
ПОБОЧНАЯ ПОДГРУППА VI ГРУППЫ
В качестве примеров трихроматов можно назвать Na2Cr3O10*
• Н2О, К2Сг3О10, ВЬ2Сг30ю, Cs2Cr3Oi0, (NH4)2Cr3Oi0, Т^СгзОю*
SrCr3O10, CuCr3O10, ZnCr3O10-3H2O, CdCr3O10-H2O, NiCr3O10.
Тетрахроматы Ме*Сг4О13 (где Me1 = Na+, K+, Rb+y
Cs+, NH4) выделяют из растворов, полученных обработкой кон-
центрированных водных растворов бихроматов СгО3 или HNOa
(плотность которой 1,39—1,41 г/см3):
Ме^Сг2О7 + 2СгО3 = Ме*Сг4О13
2Me*Cr2O7 + 2HNO3 = Ме^Сг4О13 + 2Ме^О3 + Н2О
Тетрахроматы образуют темно-красные кристаллы, разлагаю-
щиеся водой на бихромат и СгО3.
В качестве примеров тетрахроматов можно назвать Na2Cr4O13,
K2Cr4O13, Rb2Cr4O13, Cs2Cr4O13, (NH4)2Cr4O13, MgCr40i3,
CaCr4O13.
Пероксосоединения хром a(VI). При обработке
на холоду раствора хромата щелочного металла эфирным раство-
ром смеси 30%-ной Н2О2 и разб. H2SO4 получается неустойчивый
синий раствор, содержащий перекись хрома СгО5 или дипероксо-
хромовую кислоту Н2СгО6.
Перекись хрома, СтО5 или
°\ /°
является диамагнитным соединением, которое в водном растворе
взаимодействует со щелочами, окислами щелочных металлов,
с кислотами, с КМнО4 в кислой среде в присутствии следов молиб-
датов. Раствор СгО5 в эфире имеет синий цвет.
СгО5 + К2О = К2СгО4 4- О2 СгО5 4- 2КОН = К2СгО4 4- Н2О 4- О»
2СгО5 4~ 3H2SO4 ~ Cr2(SO4)3 4- ЗН2О 4- 7/2О2
4КМпО4 + 5СгО5 4- 6H2SO4 = 5Н2СгО4 4- 2K2SO4 4- 4MnSO4 4- Н2О + 1002.
При добавлении пиридина к эфирному раствору СгО5 при 0°
выпадают двупреломляющие синие игольчатые кристаллы СгО5-
C5H5N, которые обладают способностью взрываться, плохо,
растворимы в воде, растворимы в бензоле, нитробензоле и бромо-
форме.
Известны аддукты СгО5 с хинолином, пиперидином, анилином,
триметиламином и т. д.
При обработке эфирного раствора СгО5 или Н2СгО8 перекисью
водорода и спиртовым раствором КОН или аммиака, а также при
обработке К2Сг2О7 или (NH4)2Cr2O7 30%-ным раствором Н2О2
при 0° образуются пероксохроматы типа Ме^Сг2О12 *2Н2О (где-
ХРОМ
274
Me1 = К+, NH*, а хром шестивалентен):
0—0 0—0
Me1 — О -^Сг— 0 — 0 Ст— О — Me1 • 2Н2О
0^0 О-^0
Пероксохроматы (NH4)2Cr2O12— в виде фио-
летовых кристаллов и К2Сг2О12 -2^0 — в виде синих призмати-
ческих кристаллов взрываются при ударе, растворяются в воде
и спирте и разлагаются в водных растворах или под действием
щелочей и кислот с выделением кислорода и образованием бихро-
мата, хромата или соединения хрома(Ш):
В Н2О
Me2Cr20i2 Ме2Сг2О7 + 5/2О2
Ме2Сг2О12-|- 2Мет0Н = гМе^ОгО^ -|- Н20 + 5/202
Me*Cr2O12 + 4H2SO4 = Cr2(SO4)3 + Me*SO4+4Н2О + 4О2
При обработке фиолетового водного раствора пероксохромата
аммония (NH4)2Cr2O12-2Н2О аммиаком получают CrO4-3NH3,
а при обработке растворами солей бария или свинца — фиоле-
товые осадки.
Известен также пероксохромат таллия Tl2Cr20i2 синего цвета,,
который легко разлагается со взрывом.
Шестивалентное состояние хрома в вышеприведенных пероксо-
соединениях было установлено на основе магнито-химических
измерений.
Фтористый хромил [оксифторид хрома(у1)}, CrO2F2, полу-
чают действием конц. H2SO4 на смесь фторида кальция с хро-
матом свинца или с К2Сг2О7 при нагревании, обработкой безвод-
ной плавиковой кислотой СгО3 или К2Сг2О7 (в медных сосудах
при комнатной температуре), действием фтора на нагретый до 200°
хлористый хромил или пентафторида иода IF5 на трехокись хрома:
РЬСгО4 + CaF2 + 2H2SO4 = CrO2F2 + PbSO4 + CaSO4 + 2H2O
K2Cr2O, + 2CaF2 + 3H2SO4 = 2CrO2F2 + 2CaSO4 + K2SO4 + 3H2O
CrO3 + 2HF = CrO2F2 + H20 CrO2Cl2 + F2 = CrO2F2 + Cl2
K2Cr2O7 + 6HF = 2CrO2F2 + 2KF + 3H2O
Фтористый хромил — красновато-коричневое газообразное
вещество, которое при охлаждении превращается в фиолетово-
красные ромбоэдрические кристаллы с плотностью 4,2 г!см\
Соединение CrO2F2 легко летуче, очищается сублимацией (темпе-
ратура сублимации 29,6° при 760 мм рт. ст.), гидролизуется
водой и действует на стекло с образованием SiF4:
CrO2F2 + 2Н2О H2CrO4 + 2HF
.272
ПОБОЧНАЯ ПОДГРУППА VI ГРУППЫ
Металлический натрий и цинк восстанавливают красновато-
коричневые пары CrO2F2 с высвобождением хрома.
Известны фторохроматы типа MeIlCrO3F], Me4CrO->F(CrO4)],
MeI[CrOF(CrO4)2],
Фторохроматы, K[CrO3F] и NH4(CrO3F],— красные
соли, которые получают нагреванием плавиковой кислоты с хро-
матами или бихроматами калия или аммония.
Хлористый хромил, СгО2С12, получают нагреванием смеси
порошка хромата или бихромата щелочного металла с конц.
II2SO4 и каким-либо кристаллическим хлоридом (например, NaCl,
КС1), взаимодействием концентрированных растворов H2SO4
и СгО3 в НС1 или действием газообразного НС1 на СгО3, а также
действием РС15 на СгО3 и обработкой хлорохроматов кислотами:
К2Сг2О7 3H2SO4 -j- 4NaCl = 2СгО2С12 K2SO4 2Na2SO4 -|- ЗН2О
конц. H2SO4
CrO3-J-2HCl--------> СгО2С12 : Н2О
СгОз •{- РС15 = СгО2С12 + РОС13 К[СгО3С1] + 2НС1 = СгО2С12-|КС1Д-Н2О
Хлористый хромил представляет собой кроваво-красную диа-
магнитную жидкость с плотностью 1,9118 г!см\ затвердевающую
при —96,5° (образуются красные кристаллы в виде игл), кипящую
при 116,7° с выделением желтых паров. Он неустойчив на свету
и при нагревании, дымит во влажном воздухе вследствие гидро-
лиза водой, растворяется в С2Н5 — О — С2Н5, CS2, СС14 и СНС13,
является энергичным окислителем и хлорирующим агентом:
€1О2С12 + Н2О СгОз + 2НС1 СгО2С12 + 2Н2О Н2СгО4 + 2НС1
Хлористый хромил окисляет серу, фосфор, спирт, каучук,
вызывая их воспламенение.
При обработке хлористого хромила насыщенным раствором
КС1 образуются рубиново-красные кристаллы хлорохромата
калия, К[СтО3С1], называемого солью Пелиго:
СгО2С12 + КС1 + Н20 = K[CrO3Cl] -I- 2НС1
Щелочи превращают хлористый хромил в хроматы и хлориды
щелочных металлов:
СтО2С12 + 4КОН = К2СгО4 + 2КС1 + 2Н2О
При нагревании СгО2С12 до 180° он превращается в (СгО2)3С12,
а выше 300° — в Сг5О9. Если хлористый хромил нагреть до 360—
400° в атмосфере кислорода, он превращается в СгО3 и выде-
ляется хлор.
При действии трехокиси серы на хлористый хромил в растворе
CS2 образуется бесцветный сульфат хромила, CrO2SO4, который
разлагается при температуре 100—200°.
ХРОМ
273
В оксихлоридах: черном (СгО2)3С12, коричневом
(CrOohCle, черно-коричневом СгвО9С14 хром трех- и шестивалентен.
Известны также хлорохроматы Li[CrO3Cl], Na[CrO3Cl],
NH4[CrO3Cl], Са[СгО3С1]2-5Н2О, Sr[CrO3Cl]2-4Н2О.
Бромистый хромил, СгО2Вг2, получают действием избытка
сжиженного бромистого водорода на хлористый хромил в интер-
вале температур от —60 до —80°.
Соединение СгО2Вг2 представляет собой очень гигроскопичные
коричнево-красные игольчатые кристаллы, которые разлагаются
при комнатной температуре на воздухе (или нагревании в ва-
кууме).
Действуя конц. НВт на К2Ст2О7 при нагревании или хромовым
ангидридом на КВт, получают прозрачные красные кристаллы
бромохромата калия, К[СтО3Вт].
Иодохромат калия, К[СгО31], выделяется в виде неустойчивых
красных кристаллов по охлаждении горячего раствора К2Сг2О7
в конц. HI.
Кроме названных известны и другие соединения шестивалентного хрома,
например нитрат CrO2(NO3)2, изоцианат CrO^(OCN)2, изотиоцианат
CrO2(CNS)2 хромила, а также иодато-, сульфато-, ацетатохроматы типа
Mei[CrO3IO3], Me|[CrO3SO4], Mn*[CrO3SeO4], MeJ[CrO3(CH3COO)2] и др.
Металлоорганические соединения
Известны многочисленные ковалентные металлоорганические
соединения хрома, например гексакарбонил хрома, дибензол-
хром, дициклопентадиенилхром, тетрафенилхром, трифенилхром,
гидроокиси и соли пента-, тетра- и трифенилхрома, гексакарбо-
ниламины (изонитрилы) и др.
Гексакарбонил хрома, Ст(СО)8, получают пропусканием окиси
углерода под давлением 50 ат через смесь фенилмагнийброми-
да с безводным хлоридом хрома(Ш) в эфире при —70°, дейст-
вием кислот на производные (С8Н5)4Сг(СО)2, (С8Н5)3Сг(СО)3,
(С8Н5)2Сг(СО)4, обработкой эфирных растворов солей хрома(1П)
триэтилалюминием А1(С2Н5)3 и окисью углерода под давлением
при низкой температуре, действием окиси углерода под давле-
нием на производные дициклопентадиенилхрома, когда в ка-
честве промежуточных продуктов образуются [(С2Н5)Сг(СО)3]2,
1(С6Н5)2Сг][(С5Н5)Ст(СО)3].
Соединение Сг(СО)8 представляет собой диамагнитные бес-
цветные кристаллы в виде ромбических призм с плотностью
1,77 г/см3, растворимые в СС14, СНС13, СвН8, С2Н5 — О — С2Н5
и плохо растворимые в С2Н5 — ОН и СН3СООН. Гексакарбонил
хрома медленно разлагается на свету, быстро при 130—150°,
а со взрывом — при 210°, образуя металлический хром и окись
углерода; плавится при 149° в запаянной трубке, взаимодействует
1 S—0 10 1
274
ПОБОЧНАЯ ПОДГРУППА VI ГРУППЫ
с хлором, под действием HNO3 превращается в Cr(NO3)2 с выде-
лением СО и небольшого количества СО2:
Сг(СО)в 2C5H5N —► Cr(CO)4(C5H5N)2 2СО
2Cr(CO)6+5C5H5N -> Cr2(CO)7(C3H5N)24- 5СО
+c5h5n
2Cr(CO)3(C5H5N)j4-CO
Cr(CO)3(C5H5N)3 + 6HC1 = (C5H6N)3[CrCl6] + 3CO + s/2H2
При нагревании Cr(CO)3(C5H5N)3 в вакууме получают
Cr2(CO)3(C5H5N)3, при этом выделяется пиридин. Обработка
Сг(СО)6 металлическим натрием, растворенным в жидком аммиаке,
дает Na2[Cr(CO)5] и СО. Гидроокись калия, растворенная в мета-
ноле, взаимодействует с Сг(СО)8 при 100° (в трубке):
Сг(СО)6 + КОН + 5Н2О = К[Сг(СО)3(Н2О)2ОН] + ЗНСООН
Соединение К[Сг(СО)3(Н2О)2ОН] неустойчиво и гидролизуется
по уравнению
К[Ст(СО)3(Н2О)2ОН] + Н2О Сг(СО)3(Н2О)3 + КОН
В результате нагревания Ст(СО)8 с пиридином C5H5N при
200—210° в запаянной трубке получают коричнево-желтые приз-
матические кристаллы Cr(CO)4(C5H5N)2, а при 145—150° — кар-
минно-красные кристаллы Cr(CO)3(C5H5N)3, которые разлагаются
разб. НС1. Соединение Cr2(CO)7(C5H5N)5 красновато-желтого цвета
получают нагреванием Сг(СО)8 с C5H5N при 145° в течение 5 час.
Рубиново-красные игольчатые кристаллы Cr(CO)4(C12H8N2)
образуются при нагревании Сг(СО)8 с о-фенантролином C12H8N2
в запаянной трубке.
При действии Сг(СО)8 на производные циклопентадиена, такие,
какИаС5Н5 или КС5Н5, получают комплексы типа Мет1С5Н5Сг(СО)3].
При действии кислот на производные Ме1[С5Н5Ст(СО)3] обра-
зуется слабая кислота Н[С5Н5Сг(СО)3] — диамагнитные золотисто-
желтые кристаллы с т. пл. 57—58°; она растворяется в органиче-
ских растворителях, неустойчива, при нагревании разлагается
по уравнению
2Н[С5Н6Сг(СО)3] = [С5Н5Сг(СО)3]2 + Н2
Анион [С5Н5Сг(СО)3]- образует следующие соли:
[Со(С5Н5)2][С5Н5Сг(СО)3], [Сг(С5Н5)2] [С5Н5Сг(СО)3],
[Сг(С8Н8)2НС5Н5Сг(СО)3].
Известна кислота Н2[Сг(СО)5] и ее соли общей формулы
Ме*[Сг(СО)5].
Дибензолхром, Сг(С6Н8)2, получают действием безводного хло-
рида хрома(Ш) на бензол в присутствии безводного А12С1в, а так-
же восстановлением иона [Сг(С8Н8)]+, который образуется при
взаимодействии C8H5MgBr с СгС13 или СгО2С12.
ХРОМ
275
Соединение Сг(СвНв)2 представляет собой коричневые кри-
сталлы, которые плавятся при 285°, сублимируются при 150®
в вакууме, разлагаются при 300° на металлический хром и бензол,
плохо растворимы в воде и бензоле и легко окисляются во влаж-
ном воздухе до желтой гидроокиси [Сг(С8Н8)2]ОН.
Известны иодид и перхлорат дибензолхрома [Сг(С6Н8)2П,
[Сг(С8Н8)2]С1О4.
Дициклопентадиенилхром (или бнс-циклопентадиенилхром),
Сг(С5Н5)2, получают, пропуская пары циклопентадиена и гекса-
карбонила хрома, увлекаемые током азота, через стеклянную
трубку, нагретую до 280—350°, или действием безводного хлорида
хрома(Ш) на эфирный раствор бромида циклопентадиенилмагния
без доступа воздуха, а также нагреванием (80—120°) в вакууме
желтого комплекса [Cr(NH3)8](C5H5)3. Парамагнитные красные
игольчатые моноклинные кристаллы Сг(С5Н5)2 имеют плотность
1,42 г/см* и т. пл. 173°; они растворяются в органических раство-
рителях, неустойчивы и требуют хранения в темноте без доступа
воздуха.
Известно производное дициклопентадиенилхрома
[С5Н5Сг(СО)з]2, которое получают нагреванием Сг(С5Н5)2 с окисью
углерода под давлением или разложением Н[С5Н5Сг(СО)3] при
повышенной температуре. Сине-зеленую соль [Сг(С5Н5)2]С1 полу-
чают взаимодействием СгС13 с производным циклопентадиенил-
лития.
Если бромид пентафенилхрома обработать спиртовым раство-
ром КОН и удалить КВт эфиром, то, экстрагируя хлороформом
(0°) остаток, можно получить гидроокись пентафенилхрома,
Сг(СвН5)5ОН -4Н2О. Соединение Сг(СвН5)5ОН -4Н2О представляет
собой парамагнитные золотисто-желтые пластинчатые кристаллы,
которые превращаются в оранжево-красные кристаллы
Сг(СвН5)5ОН-2Н2О при высушивании над безводным СаС12 и в ко-
ричневые кристаллы Сг(С8Н5)5ОН при высушивании над Р4О10.
Гидроокись пентафенилхрома обладает слабо основными
свойствами. С кислотами она образует соли тетрафенилхрома
и иногда пентафенилхрома. Известны несколько солей пентафе-
нилхрома, например Сг(С8Н5)5Вг, Cr(CeH5)5Br-HgCl, 2Сг(С8Н5)5Вг -
• (С2Н5)2О, [Cr(C8H5)5]2SO4.4H2O, [Сг(С8Н5)5]2СО3-(1 или 6)Н2О,
[Cr(C8H5)5iHCO3 (1 или 3)Н2О.
Бромид пентафенилхрома, Сг(С8Н5)5Вг, представляет собой
оранжево-коричневый аморфный порошок, плохо растворимый
в воде и растворимый в спирте, ацетоне, пиридине и других орга-
нических растворителях.
Тетрафенилхром, Сг(С8Н5)4, получают электролизом раствора
иодида тетрафенилхрома Сг(С8Н5)41 в жидком аммиаке при —40
или —50°, причем платиновый катод и графитовый анод разделены
пористой перегородкой.
18*
276
ПОБОЧНАЯ ПОДГРУППА VI ГРУППЫ
Тетрафенилхром образует оранжевые кристаллы, плохо рас-
творимые в жидком аммиаке, растворимые в спирте и пиридине
и разлагающиеся при обычной температуре на металлический
хром и дифенил.
Гидроокись тетрафенилхрома, Сг(С8Н5)4ОН -ЗН2О, получают
дейст; г.ем Ag2O на Сг(С8Н5)41 в метиловом спирте или хлороформе
либо электролизом спиртового раствора (С8Н5)4Сг1.
Соединение Ст(С8Н5)4ОН -ЗН2О представляет собой оранжевые
кристаллы с т. пл. 104—105°, плохо растворимое в эфире и бен-
золе, хорошо растворимое в воде и спиртах. Оно превращается
в Сг(СвН5)4ОН-2Н2О высушиванием над безводным СаС12, имеет
более сильно выраженные основные свойства по сравнению с гид-
роокисью пентафенилхрома, но более слабые, чем у гидроокиси
трифенилхрома.
Соли тетрафенилхрома получают действием кислот на гидро-
окись тетрафенилхрома.
Примеры солей тетрафенилхрома: Сг(С8Н5)41,
Сг(С8Н5)41-(С2Н5)2О, Сг(С8Н5)4Вг, Сг(С8Н5)4С1О4,
[Сг(С8Н5)4]2Сг2О7.
Иодид тетрафенилхрома, Ст(С8Н5)41, представляет собой пара-
магнитные коричневые ромбические кристаллы с т. пл. 178°;
он растворяется в СНС13, С2Н5ОН, C5H5N и плохо растворим
в С2Н5О.
Трифенилхром, Сг(С8Н5)з, получают действием металлического
натрия на иодид трифенилхрома в жидком аммиаке.
Соединение Сг(С8Н5)3 представляет собой коричнево-желтый
тонко дисперсный порошок; неустойчиво, разлагается с выделе-
нием дифенила и превращается в гидроокись трифенилхрома под
действием воды или спирта.
Гидроокись трифенилхрома, Сг(С8Н5)3ОН, получают обработ-
кой спиртового раствора бромида трифенилхрома КОН.
Соединение Сг(С8Н5)3ОН — сильное основание, хорошо рас-
творимое в воде.
Соли трифенилхрома получают обработкой кислотами водных
растворов гидроокиси трифенилхрома. В качестве примера солей
трифенилхрома можно назвать Cr(C8H5)3Br, [Cr(C8H5)3]HgI3,
Сг(СвН5)3Ь(С2Н5)2О, [Сг(С8Н5)3] [Cr(NH3)2(SCN)4].2H2O.
На основании исследования магнитной восприимчивости было
установлено, что производные хрома с 3, 4 и 5 фенильными груп-
пами парамагнитны и хром в них пятивалентен.
Гексакарбиламины (изонитрилы) хрома,
Cr(CNR)e (где R = С8Н5 или их паразамещенные производные),
представляют собой красные твердые вещества, которые плавятся
без разложения, диамагнитны и медленно реагируют с раз-
бавленными кислотами. Примеры гексакарбиламинов хрома:
Cr(CN - С8Н5)8, Cr(CN - С8Н4 - С1)6 и Cr(CN - С8Н4 - СН3)8.
ХРОМ
277
Помимо указанных выше известны и другие металлоорганические
соединения, например: C6H6Cr(CO)3, NH2CeH5Cr(CO)3, НОСвН5Сг(СО)3,
HOOCCeH5Cr(CO)3, C6H4SCr(CO)3, Сг(СН3С6Н5)3, (С6Н5С6Н5)Сг(С6Н6),
Cr(C5H5)(CO)2(NO), Cr(C5H5)(NO)2(CH3), Cr(C5H5)(NO)2(C2H5),
[С6Н5СН2Сг(Н2О)5](С1О4)2, C4H4SCr(CO)3 и др.
Соединения включения
Гидрид хрома, СгН3, получают в виде черного осадка с плот-
ностью 6,77 г/смг при перемешивании в токе сухого водорода
суспензии СтС13 в эфирном растворе бромида фенилмагния.
Нитрид хрома, CrN, получают перегонкой амальгамы хрома
в атмосфере азота, действием азота или аммиака на металличе-
ский хром при высокой температуре, нагреванием бромида хрома
с аммиаком, действием нитрида лития, магния или аммиака в при-
сутствии NH4C1 на СгС13.
Черные кубические микрокристаллы CrN имеют плотность
5,8 г/см?, разлагаются на элементы при нагревании до 1500°
без доступа воздуха, превращаются в Сг2О3 с выделением азота
при нагревании на воздухе, в СгС13 (и азот) при нагревании в хло-
ре, подвергаются действию сухого НС1, конц. H2SO4 или цар-
ской водки.
Известен также нитрид хрома, Cr2N.
Фосфид хрома, СгР, получают обработкой парами фосфора
хромата натрия или калия, восстановлением фосфата хрома(Ш)
углем или действием фосфористого водорода на хлорид
хрома(Ш).
Соединение СгР представляет собой серый порошок с плот-
ностью 5,32 a/cjt3; тугоплавок, медленно окисляется на воздухе,
превращается в СгС13 и РС15 под действием хлора, а при сплавле-
нии с КОН дает К2СгО4 и выделяется водород.
Известен также фосфид хрома, Сг3Р.
Карбиды хрома, Сг213С8, Сг7С3, Сг3С2.
Карбид хрома, Сг213С8, получают карбидизацией хрома в элек-
трической печи или нагреванием металлического хфома в атмо-
сфере окиси углерода. Это красные кубические кристаллы с плот-
ностью 6,75 г!см?, которые разлагаются примерно при 1550°,
полностью сгорают в кислороде при 1200°, разлагают пары воды
при 750—800° и взаимодействуют с разб. H2SO4; карбид хрома
используют для получения нержавеющих сталей.
Карбид хрома, Сг7С3, получают действием метана на металли-
ческий хром при 600—800° или сплавлением металлического хрома
со сплавом хром — углерод при 1800° в тигле из MgO. Он пред-
ставляет собой серебристые ромбоэдрические кристаллы с плот-
ностью 6,92 г/см3, которые плавятся при 1665°, горят при 1150°
в кислороде, разлагают пары воды при 550°, плохо растворимы
в воде и взаимодействуют с горячей разб. H2SO4.
278
ПОБОЧНАЯ ПОДГРУППА VI ГРУППЫ
Карбид хрома, Сг3С2, получают нагреванием хрома или окиси
Сг2О3 с углем в электрической печи. При этом образуются очень
твердые блестящие ромбические кристаллы с плотностью 6,68 г!см?
и т. пл. 1890°; Сг3С2 плохо растворим в воде и кислотах, превра-
щается в СтС13 под действием хлора при нагревании, разлагает
воду при 500°; его применяют для получения различных сплавов.
Борид хрома, СгВ, получают действием бора или трихлорида
бора на металлический хром в присутствии водорода или элек-
тролизом сплава бората, фторида кальция и Сг2О3.
Соединение СгВ представляет собой слабо ферромагнитный
серебристо-белый тонко дисперсный кристаллический порошок
с плотностью 5 г! см? и твердостью 8 по шкале Мооса. СгВ является
хорошим проводником электрического тока, плавится при 1750°
и устойчив к действию кислот.
Известны такие бориды: Сг3В4, СгВ2, Сг4В, Сг2В, Сг5В3, Сг3В2
и др.
МОЛИБДЕН Мо
Z = 42; ат. вес — 95,94
Валентность (II), (III), (IV), (V), VI; заряд (2-)-),
(3+), (4+), (5+), 6+
Массовые числа природных изотопов 98, 96, 95,
92, 94, 100 и 97
Электронная структура атома молибдена: К •£ -М -4s2 •4//i •
•4ds «5s1.
Электронная структура атома молибдена и ионов Мо4+ и Мо8+
для 4d- и 5х-орбиталей:
4d5 5s1 4d2 5s 4d 5s
ЕЕШШ Ш ГтЩ I I I □ I I I i I I □
Mo Мо4+ Mo6+
ИСТОРИЧЕСКАЯ СПРАВКА
Значительно раньше, чем был получен металлический молиб-
ден, был известен природный сульфид молибдена (названный
молибденитом), из которого получали трехокись молибдена МоО3.
В древности молибденит, имеющий вид серых пластин, масля-
нистых на ощупь и оставляющих след на белых предметах, при-
меняли в качестве грифелей. Название минерала «молибденит»
происходит от греческого слова molibdos, что означает «свинец»,
подчеркивая тем самым сходство этого минерала со свинцом.
Молибденит в древности принимали за свинцовый блеск и графит,
поскольку и тот, и другой оставляли след на белых предметах.
В 1778 г. К. Шееле показал, что при обработке молибденита
(в отличие от графита) разб. HNO3 получается белый порошок
(terra molybdoeni) трехокиси молибдена МоО3.
МОЛИБДЕН
279
В 1785 г. Пеллтье показал, что молибденит является природным
сульфидом, при прокаливании которого образуется трехокись
молибдена.
Неочищенный металлический молибден впервые был получен
Хельмом в 1790 г. путем восстановления трехокиси молибдена
углем. Чистый металлический молибден (ковкий и тягучий)
впервые удалось получить в 1910 г.
Установлено, что в стали знаменитых средневековых японских
мечей содержится молибден.
С середины XIX в. молибден (соответственно ферромолибден)
используется как добавка к легированным сталям. В начале
XX в. были разработаны металлотермический метод превращения
порошкообразного молибдена в компактный металл и процесс
промышленного получения ферромолибдена.
Благодаря своим физическим и химическим свойствам метал-
лический молибден начиная с конца прошлого века используется
в очень многих отраслях современной техники.
Чистый молибден в компактном состоянии был получен срав-
нительно недавно; о нем можно сказать, что молибден является
новым металлом со старой историей.
РАСПРОСТРАНЕННОСТЬ В ПРИРОДЕ
В природе молибден находится в виде соединений (дисульфи-
дов, молибдатов, окислов и др.) в различных рассеянных мине-
ралах. Содержание молибдена в земной коре составляет
1,0-Ю-3 вес.%.
К наиболее важным минералам молибдена относятся следую-
щие.
Молибденит, MoS2, содержит 60% Мо. Это основной минерал
молибдена; он встречается в распыленном виде в различных кри-
сталлических породах в виде мягких (твердость 1,5 по шкале
Мооса) блестящих серых гексагональных пластинок, маслянистых
на ощупь, оставляющих черный след на бумаге или-белых пред-
метах, обладающих плотностью не меньше 4,7 г!см3. Молибденит
некоторых месторождений содержит до 0,2% рения.
Повеллит, СаМоО4, получается в результате взаимодействия
молибденовой кислоты (образующейся вследствие окисления
молибденита) с грунтовыми водами, содержащими растворимые
соли кальция. Он имеет вид желтых тетрагональных кристаллов
с плотностью 4,25—4,52 г/см3 и твердостью 3,5 по шкале Мооса.
Ферримолибдит (молибденовая охра), Fe2(MoO4)3-7,5Н2О, нахо-
дится в окисленных зонах залежей молибденовых и железных руд;
это серовато-желтые ромбические кристаллы с плотностью
4,5 г! см3 и твердостью 2 по шкале Мооса.
Вульфенит, РЬМоО4, описан среди минералов свинца.
280
ПОБОЧНАЯ ПОДГРУППА VI ГРУППЫ
К другим минералам молибдена относятся ильземанит МоО2-4МоО3,
белонозит MgMoO4, патераит (Са, Fe)MoO4, комплексные минералы свинец —
ванадий — молибден, свинец — вольфрам — молибден, свинец — мышьяк —
молибден и др.
В природе минералы молибдена вместе с минералами некото-
рых тяжелых металлов, таких, как медь, свинец, серебро, висмут,
золото, олово, железо, рений, образуют молибденовые руды
(в которых содержание Мо превышает 0,04%) или комплекс-
ные полиметаллические руды (содержащие примерно 0,01 % Мо).
Залежи молибденовых руд находятся в США, Чили, Мексике,
Канаде, Перу, Швеции, Норвегии, Финляндии, СССР, Франции,
Турции, Югославии, Австрии, ГДР, Италии, Испании, Порту-
галии, Японии, КНР, Индии, ДРВ, Марокко, Мальгашской Рес-
публике, Австралии. Руды Румынии содержат молибден наряду
со свинцом, серебром, золотом, висмутом, железом, рением и др.
В некоторых минералах молибдена из Норвегии и Южной Африки
содержатся в большом количестве рений и в небольшом — тех-
неций.
Металлический молибден был найден в некоторых метеоритах,
а соединения молибдена обнаружены в морской воде, в наземных
и морских растениях, в каменных углях, в нефтях и в животных
организмах.
ОБОГАЩЕНИЕ И ПЕРЕРАБОТКА МОЛИБДЕНОВЫХ РУД
Поскольку собственно молибденовые руды содержат 0,04 вес. %
молибдена, а полиметаллические комплексные руды — примерно
0,01% молибдена, необходимо эти минералы в размолотом виде
подвергать обогащению. Для обогащения полиметаллических
сульфидных руд до уровня молибденита применяют селективную
флотацию. Руды, содержащие вульфенит, повеллит, ферримолиб-
дит, обогащаются флотацией и подвергаются гидрометаллурги-
ческой переработке. Основную массу молибдена добывают из
концентратов молибдена и вульфенита.
Концентраты молибденовых руд перерабатываются пирометал-
лургическими или гидрометаллургическими способами. При пи-
рометаллургической переработке концентратов молибденита полу-
чают трехокись молибдена, молибдат натрия или ферромолибден.
При гидрометаллургической переработке богатых молибденом
концентратов можно получить трехокись молибдена, гептамолиб-
дат аммония (NH4)0[Mo7O24]-4^0, молибдат натрия Na2MoO4-2П.2О,
молибдат кальция СаМоО4, молибдат железа FeMoO4. Из бедных
концентратов обычно получают молибдат кальция СаМоО4.
Иногда концентраты молибденовых руд перед прокаливанием
обрабатывают кислотами для уменьшения содержания карбонатов
и фосфатов кальция и магния.
МОЛИБДЕН
281
При окислительном прокаливании концентратов молибденита
MoS2 (содержащих небольшие количества CuS, ZnS, FeS2 в каче-
стве сопутствующих сульфидов) при 550—600° в большом избытке
воздуха (многоярусные печи) получают в зависимости от условий
трехокись молибдена МоО3, двуокись молибдена Мо02. молибдаты
меди, цинка, железа и при этом выделяется сернистый газ:
MoS2 + ’/2О2 = МоО3 + 2SO2 + 266,2 ккал
При окислении сопутствующих сульфидов CuS, ZnS, FeS?
получают окислы металлов CuO, ZnO, Fe2O3 (и только частично
сульфаты), которые с трехокисью молибдена образуют молибдаты:
МоО3 + МепО = МепМоО4 (где Me11 = Cu2+, Zn2+, Fe2+)
ЗМоО3 -|- Ме2,пОз = Ме|п(МоО4)3 (где Me111 = Fe3+)
Двуокись молибдена МоО2 образуется в результате реакции,
которая протекает при прокаливанищбез доступа воздуха внутри
глыб концентратов молибденита:
MoS2 + 6МоОз = 7МоО2 + 2§Оа
Концентраты молибденита нагревают только вначале, а затем
подачу тепла прекращают, поскольку реакции окисления суль-
фидов экзотермичны.
При прокаливании молибденовых руд пустая порода (кварц,
силикаты, алюмосиликаты и т. п.) не претерпевает никаких
превращений.
Если прокаливание концентрата молибденита осуществляется
при 1000°, относительно летучую трехокись молибдена собирают
в специальных фильтрах и затем снова очищают сублимацией.
Спеканием при 650—750° бедных молибденом (5—20% Мо)
концентратов и остатков от выщелачивания, которые помимо
молибдена могут содержать заметные количества железа, меди
и других металлов, с окислительно-щелочной смесью (Na2CO3 -j-
+ избыток KNO3) получают молибдат и сульфат натрия, нитрит
калия и двуокись углерода: 7
MoS2 + 3Na2CO3 + 9KNO3 = Na2MoO4 + 2Na2SO4 + 9KNO2 + 3CO2
После охлаждения продукт спекания обрабатывают водой.
Раствор, полученный после отделения фильтрованием нераствори-
мого остатка, содержит почти весь молибден, кремний, фосфор
и мышьяк в виде соединений щелочных металлов. Из этого рас-
твора фосфор и мышьяк осаждают аммиачным раствором магния,
а кремний отделяют в виде H2SiO3, пропуская углекислый газ
или подкисляя раствор.
При добавлении конц. НС1 к раствору молибдата натрия, очи-
щенному и сконцентрированному до уд. веса 1,3 (pH = 4—3),
выпадает молибденовая кислота Н2МоО4-Н2О; последняя при
282
ПОБОЧНАЯ ПОДГРУППА VI ГРУППЫ
нагревании до 200° превращается в МоО3:
Na2MoO4 + 2НС1 + Н2О = Н2МоО4-Н2О + 2NaCl
I
МоО3 + 2Н2О
Иногда прокаленные концентраты молибденита промывают
водой для удаления растворимых в воде соединений, таких, как
сульфаты меди, цинка, железа, никеля и др.
Выщелачивая на холоду прокаленные концентраты молибде-
новых руд (для чего обычно применяют вращающиеся барабаны
с механическим перемешиванием) избытком 8—10%-ного водного
раствора аммиака в течение нескольких часов, отделяют гепта-
молибдат аммония (NH4)6[Mo7O24] (в растворе) от Fe(OH)3 (в виде
осадка):
7МоО3 + 6NH4OH = (NH4)e[Mo7O24] + ЗН2О
7CuMoO4 + 34NH4OH = (NH4)e[Mo7O24] + 7[Cu(NH3)4](OH)2 + 24H2O
7ZnMoO4 + 48NH4OH = (NH4)6[Mo7O24] + 7[Zn(NH3)6](OH)2 + 38H2O
7Fe2(MoO4)3 + 18NH4OH + 12H2O = 3(NH4)6[Mo7O24] + 14Fe(OH)3
После удаления осадка гидроокиси железа Fe(OH)3 фильтро-
ванием раствор, подкисленный НО до pH = 3,5—4, концентри-
руют при 60—70° до выделения кристаллов (NH4)6[Mo7O24l-4Н2О;
последние цри прокаливании в лодочке из нержавеющей стали
при температуре 450—500° разлагаются по уравнению
(NH4)6[Mo7O24].4H2O 7МоО3 + 6NH3 + 7Н2О
Концентрированием при 80—90° раствора парамолибдата аммо-
ния * после предварительного подкисления соляной кислотой
до pH =2—3 получают молибденовую кислоту Н2МоО4-2Н2О,
которую прокаливанием также превращают в МоО3.
При выщелачивании раствором карбоната натрия прокаленных
концентратов медномолибденовых руд (бедных молибденом) полу-
чают растворимый молибдат натрия Na2MoO4-H2O и трудно рас-
творимый основной карбонат меди Сн2СО3(ОН)2:
МоО3 -}- Na2CO3 ~ Na2MoO3 -|- СО2
2CuMoO4 + 2Na2CO3 + Н2О = 2Na2MoO4 -|- Cu2CO3(OH)2 + СО2
2CuO + Na2CO3 + 2Н2О = Cu2CO3(OH)2 + 2NaOH
Иногда применяют обработку раствора молибдата натрия
избытком 10—15%-ного раствора хлорида кальция при 80—90°.
В этом случае выпадает молибдат кальция СаМоО4, который при-
меняется для получения ферромолибдена.
Если слабо подкисленный раствор молибдата натрия обраба-
тывают хлоридом или сульфатом железа(П), то помимо FeMoO4
* Техническое название гептамолибдата аммония.— Прим. ред.
МОЛИБДЕН
283
выпадают фосфаты и арсенаты железа. При обработке слабо
щелочного раствора молибдата натрия сульфидом натрия полу-
чают растворимый тиомолибдат натрия Na2MoS4 и выпадают
сульфиды GuS, PbS. При нагревании раствора тиомолибдата
натрия, подкисленного разб. HG1 или H2SO4, осаждается три-
сульфид молибдена MoS3.
Вместо раствора карбоната натрия можно использовать едкий
натр, и из раствора молибдата натрия растворами солей железа(П)
можно осадить молибдат железа FeMoO4, который также при-
меняют для получения ферромолибдена.
Из комплексных медномолибденовых руд медь можно пере-
вести в раствор с помощью солей железа(Ш), например водным
раствором FeGl3 или Fe2(SO4)3. Из концентратов таких руд выде-
ляют и рений.
Поскольку из концентратов вульфенита РЬМоО4 добывают
как молибден, так и свинец, применяется метод переработки,
заключающийся в восстановительно-щелочном сплавлении кон-
центратов вульфенита со смесью соды (или карбоната магния)
<с углем (кокс) в электрической печи!
РЬМоО4 + Na2CO3 + С = Na2MoO4 + РЬ + СО2 + СО
РЬМоО4 -}- MgCOg -|- С = MgMoO4 РЬ -|- СО2 -|- со
При обработке молибдата натрия хлоридом кальция или
дихлоридом железа получают СаМоО4 или FeMoO4. Металличе-
ский свинец, полученный из концентратов вульфенита, содержит
серебро, золото и медь.
При сплавлении концентратов вульфенита (соответственно
концентратов молибденита с большим содержанием свинца, вис-
мута, меди и др.) с сульфидом натрия Na2S или смесью сульфата
натрия с углем получают тиомолибдат натрия и сульфиды свинца,
висмута, меди:
РЬМоО4 + 5Na2S = Na2MoS4 + PbS + 4Na2O
РЬМоО4 + 5Na2SO4 + 20C = Na2MoS4 + PbS + 4Na2O + 20CO
Благодаря различной растворимости в расплаве сульфида
натрия трудно растворимые сульфиды PbS, GuS, Bi2S3 легко отде-
ляются от растворимого тиомолибдата натрия Na2MoS4. В жидком
расплаве образуются два слоя: в верхнем находятся тиосоли
молибдена, мышьяка, олова, в нижнем — сульфиды свинца,
меди и висмута.
При обработке концентратов вульфенита теплым раствором
сульфида натрия получают Na2MoS4, PbS и NaOH:
PbMoO4 + 5Na2S + 4H2O -- Na2MoS4 + PbS + 8NaOH
284
ПОБОЧНАЯ ПОДГРУППА VI ГРУППЫ
Образовавшийся тиомолибдат в щелочной среде реагирует
с концентратом вульфенита:
Na2MoS4 + 4РЬМоО4 + 8NaOH = 5Na2MoO4 + 4PbS + 4Н2О
В заключение можно сказать, что при пирометаллургической
и гидрометаллургической переработке концентратов молибденовых
руд получают МоО3, МоО2, СаМоО4, FeMoO4, (NH4)e[Mo7O24l-
•4Н2О, Na2MoO4’2Н2О. Эти соединения служат для получения
металлического молибдена, ферромолибдена, молибденсодержа-
щих легированных сталей и различных соединений молибдена.
Как в анализе концентратов молибденовых руд и ферромо-
либдена, так и при выделении молибдена из продуктов, остав-
шихся от гидрометаллургической переработки молибденовых кон-
центратов, широко используются ионообменные смолы.
При использовании сульфонатного катионита в Н-форме для
анализа концентратов молибденовых руд и ферромолибдена из
кислых растворов ионообменник удерживает медь, железо, сви-
нец, ванадий (соответственно катион ванадила), а в растворе
остается молибдат-анион, который определяется 5%-ным раство-
ром 8-оксихинолина в уксусной кислоте. Адсорбированные катио-
ны элюируют последовательным промыванием смолы разбавлен-
ной (1 : 7) HNO3 — для свинца, разбавленной (1 : 9) H2SO4 —
для меди и ванадия, 4 н. HG1 — для железа.
Для извлечения молибдена из разбавленных растворов молиб-
датов применяют анионит, полученный конденсацией меламина
с формальдегидом. Из промывных вод от гидрометаллургической
переработки концентратов молибдена можно выделить до 200 кг
МоО3 на 1 № смолы.
Молибден может быть отделен от вольфрама при использова-
нии крупнопористого Н-катионита — пермутита ES. Последний
удерживает вольфрам и не удерживает молибден. Отделение
молибдена от железа, алюминия и кальция осуществляется на
анионообменной смоле в виде RNO3, которая удерживает молиб-
дат-анион и не удерживает растворимые нитраты железа, алю-
миния и кальция.
Для отделения молибдена от рения используют растворы
фосфорной кислоты и анионообменную смолу. Из 2М раствора
фосфорной кислоты смола извлекает 73% содержащегося молиб-
дена и только 4% рения. Вымывание молибдена осуществляется
кислотой, а рения — 10%-ным раствором NaOH.
ПОЛУЧЕНИЕ МЕТАЛЛИЧЕСКОГО МОЛИБДЕНА
Металлический молибден получают восстановлением окислов
молибдена водородом, кальцием, алюминием, магнием, цинком,
кремнием, углеродом, электролитическим восстановлением раз-
МОЛИБДЕН
285
личных соединений молибдена и термической диссоциацией гек-
сакарбонила молибдена.
Для получения металлического молибдена используют
следующие его соединения: а-МоО3, 6-МоО2, Н2МоО4-Н2О,
(NH4)6[Mo7O24l -4Н2О, К2[МоС161, MoS2.
Восстановление водородом окислов молибдена а-МоО3 и |}-Мо02
Восстановление трехокиси молибдена водородом при нагрева-
нии идет в две стадии:
600-700°
МоО3 + Н2 ----—* МоО24-Н2О (в течение 20—30 мин)
950-1 100°
МоО2Н-2Н2---------> Мо-|-2Н2О (в течение 15 — 20 мин)
Процесс осуществляется в металлических лодочках (из никеля
или железа), помещенных в цилиндрическую печь (соответственно
в кварцевой трубке), нагретую в противоположном конце до 1100°.
Температура, продолжительность восстановления и скорость
пропускания водорода в значительной степени влияют на раз-
меры гранул молибдена. Обычно работу ведут в сильном токе
водорода, чтобы выделяющиеся водяные пары не окисляли порош-
кообразный металлический молибден, нагретый до 800°. По это-
му процессу получают тонкий порошок металлического молиб-
дена, обладающий пирофорными свойствами. Водород должен
быть чистым и сухим; после восстановления окислов молибдена
порошкообразный металл охлаждают в токе водорода для пре-
дохранения от окисления.
Вместо трехокиси молибдена можно использовать гептамолиб-
дат аммония (NH4)e[Mo7O24] -4Н2О, который при 450—500° раз-
лагается на МоО3, NH3 и воду. После удаления воздухом аммиака
из кварцевой трубки (в трубчатой печи) трехокись молибдена
восстанавливают водородом при нагревании, как было описано
выше. Присутствие аммиака способствует не только восстановле-
нию трехокиси молибдена до двуокиси, но и образованию нитрида
молибдена в результате реакции между металлическим молибде-
ном и газообразным аммиаком при высокой температуре.
Поскольку в процессе прокаливания молибденовой кислоты
Н2МоО4-Н2О при 150° образуется трехокись молибдена и вода,
часто металлический молибден получают восстановлением молиб-
деновой кислоты водородом при нагревании.
Металлотермическое восстановление окислов молибдена
В бомбу из нержавеющей стали загружают смесь трехокиси
молибдена с равным количеством чистого металлического кальция,
сублимированного в вакууме, и небольшим количеством элемен-
286
[ПОБОЧНАЯ ПОДГРУППА VI ГРУППЫ
тарного иода, играющего роль катализатора. Применяется элек-
трообогрев; реакция идет в вакууме или в среде аргона по урав-
нению
МоО3 + ЗСа = Мо + ЗСаО
Получается порошкообразный металл.' После восстановления
избыток металлического кальция удаляют при обычной темпе-
ратуре последовательной обработкой абсолютным спиртом, водой
и разб. НС1.
При алюмо- или кремнетермическом восстановлении окислов
молибдена а-МоО3, 6-МоО2 в фарфоровом или шамотовом тигле
с добавками извести и флюорита получают загрязненный алю-
минием или кремнием металлический молибден:
МоОз -|- 2А1 = Мо 4- А12О3
2МоО3 4- 3Si = 2Мо 4- 3SiO2 + 263’ккал.
Алюмо- или кремнетермическое восстановление применяется
главным образом в процессе получения ферромолибдена.
Металлический молибден можно получить также восстановле-
нием при нагревании окислов молибдена цинком, магнием или
смесью металлов подгруппы церия:
МоО3 4- ЗМе = Мо + ЗМеО, где Me = Zn, Mg
МоОз + 2Ме = Мо + Ме2О3, где Me = металл подгруппы церия
При восстановлении окислов молибдена карбидами кальция,,
кремния, цинка, титана, бора получают карбидизированный
металлический молибден.
Восстановление окислов молибдена углеродом
Восстановлением при нагревании окислов молибдена либо
молибдатов углем получают металлический молибден, загряз^-
ненный карбидом молибдена МоС2:
МоО3 + ЗС = Мо + ЗСО МоО2 + 2С = Мо + 2СО
СаМоО4 4- ЗС = Мо + СаО 4- ЗСО
Карбид молибдена МоС2 получается по уравнениям
Мо 4- 2С = МоС2 Мо 4- 4СО = МоС2 4* 2СО2
Электролитическое получение металлического молибдена
Электролизом расплава, состоящего из 25% К3[МоС1в], 37,5%-
KG1 и 37,5% NaCl, при 900° в атмосфере аргона, используя вра-
щающийся катод и задавая плотность тока 3—10 а!дм2, получают
на катоде порошкообразный металлический молибден. Электро-
лизом расплава, состоящего из КС1, LiCl и примерно 4 мол. %
МОЛИБДЕН
287
К3[МоС16], при 600—900° в атмосфере аргона, задавая плотность
тока 3,2 а!дм2, получают на катоде металлический молибден
в виде компактного слоя.
Электролиз осуществляется без доступа воздуха и влаги.
В качестве катода применяют вольфрам или серебро, в качестве
анода — неочищенный молибден.
Порошкообразный металлический молибден можно получить
также электролизом раствора 20 г молибденовой кислоты
Н2МоО4-Н2О в 100 мл 28%-ного аммиака, куда добавляют 95—
100 мл ледяной уксусной кислоты (до pH == 6,0). Аноды плати-
новые или угольные, катоды стальные, медные или никелевые,
плотность тока 80—300 а!дм2, температура 30—50°.
При электролизе раствора МоО3 в конц. НСГ (или в конц.
H2SO4), используя ртутный катод, получают амальгаму, содер-
жащую 4,9% молибдена, из которой последний выделяют отгон-
кой ртути в вакууме при температуре 350—400°. Порошкообраз-
ный молибден, полученный после вакуумной отгонки ртути
из амальгамы молибдена, обладает пирофорными свойствами.
Термическая диссоциация гексакарбонила молибдена
Термической диссоциацией гексакарбонила молибдена при
600° и давлении 0,1 мм рт. ст. получают металлический молибден
высокой чистоты; при этом выделяется окись углерода:
Мо(СО)6 Мо + 6СО
При взаимодействии металлического молибдена с окисью угле-
рода при 200° и давлении 200—250 ат получаются бесцветные
ромбические кристаллы Мо(СО)в с плотностью 1,96 г!см2.
Другие процессы получения металлического молибдена
Металлический молибден можно также получить восстановле-
нием при нагревании без доступа воздуха хлоридов молибдена
МоС15, МоС13, (Мо8С18]С14 различными металлами, например Li,
Na, К, Са, Sr, Ва, а также восстановлением трифторида молиб-
дена MoF3 водородом при нагревании.
При термической диссоциации очень чистого молибденита
в вакууме при 1600—1700°, при прямом восстановлении чистого
молибденита водородом, магнием, кальцием, парами калия в атмо-
сфере инертного газа при нагревании или при сильном нагревании
молибденита с двуокисью молибдена, окисью кальция или смесью
окиси кальция с углем также получают порошкообразный метал-
288
ПОБОЧНАЯ ПОДГРУППА VI ГРУППЫ
лический молибден:
MoS2 = Мо + 2S
MoS2 -1- 2Mg = Мо + 2MgS
MoS2 -г 4К = Мо + 2K2S
3MoS2 + 4Cs
MoS2 + 2CaO + :
MoS2 + 2H2 = Mo + 2H2S
MoS2 + 2Ca = Mo + 2CaS
MoS2 + 2MoO2 = 3Mo + 2SO2
= 3Mo + 4CaS + 2SO2
= Mo + 2CaS + 2CO
ПОЛУЧЕНИЕ ФЕРРОМОЛИБДЕНА
Для получения ферромолибдена (сплав железа с молибденом,
содержащий 50—85% Мо) используют окисли молибдена, молиб-
дат кальция, молибдат железа и в меныней степени дисульфид
молибдена (чистый молибденит).
Ферромолибден получают в электрической печи восстановле-
нием алюминием, кремнием (ферросилицием) или углеродом при
нагревании смеси концентратов прокаленных молибденовых руд
или молибдата кальция с окислами железа в присутствии извести
СаО и флюорита GaF2. Окись кальция служит как для превраще-
ния трехокиси молибдена в СаМоО4 (чтобы исключить потери,
связанные с летучестью МоО3), так и для образования шлака
с SiO2 или А12О3.
Возможны различные реакции образования ферромолибдена:
2МоО3 + Fe2O3 + 6А1 + ЗСаО = 2Fe — Мо + ЗСа(А1О2)2
4МоО3 + 2Fe2O3 + 9Si + 9СаО = 4Fe — Мо + 9CaSiO3
2СаМоО4 + Fe2O3 + 6А1 + СаО = 2Fe — Мо + ЗСа(А1О2)2
4СаМоО4 -J- 2Fe2O3 + 9Si + 5СаО = 4Fe — Мо + 9CaSiO3
СаМоО4 + Fe + SiO2 + 3C = Fe — Mo + CaSiO3 + 3CO
2MoO3 + Fe2O3 + 9C = 2Fe — Mo + 9CO
В качестве катализатора при кремнетермическом восстановле-
нии применяют силицид кальция, который содержит 60% Si,
30% Са, остальное железо и алюминий.
Наибольшую часть молибдена, полученного из прокаленных
концентратов молибденовых руд, перерабатывают в ферромолиб-
ден, который применяется для получения молибденсодержащих
легированных сталей.
При алюмо- или кремнетермическом восстановлении смеси
окислов хрома с окислами молибдена в присутствии извести
и флюорита получают хромомолибденовый сплав, например
по уравнению
2МоО3 4- Cr2O3 + 6А1 + ЗСаО = 2Сг — Мо А ЗСа(А1О2)3
МОЛИБДЕН
289
ПРЕВРАЩЕНИЕ ПОРОШКООБРАЗНОГО МЕТАЛЛИЧЕСКОГО
МОЛИБДЕНА В КОМПАКТНЫЙ МЕТАЛЛ
Порошкообразный металлический молибден может быть превра-
щен в компактное состояние спеканием — процесс, аналогичный
принятому в металлокерамике,— в атмосфере водорода (или водоро-
да, насыщенного парами воды), сплавлением в электрической дуге
в вакууме и сплавлением в сфокусированном пучке электронов
в атмосфере аргона или гелия.
Молибденовые стержни или бруски, полученные прессованием
порошкообразного металлического молибдена на гидравлических
прессах в стальных матрицах, нагревают при 1100—1300° в атмос-
фере водорода, а затем спекают при 2200° в атмосфере водорода
или при 1600—1700° в атмосфере водорода, насыщенного парами
воды. Спекание осуществляют в установках для спекания или в
толстостенных молибденовых лодогках, используя промежуточный
слой из порошка А12О3 или ZrO2.
Бруски и блоки, полученные таким образом, применяются
в качестве электродов в печах для плавления стекла.
С помощью электрической дуги, образующейся между стерж-
нем из металлического молибдена (полученным спеканием прессо-
ванного порошка молибдена при 1000° в атмосфере водорода)
и медным электродом (в форме охлаждаемого водой тигля), полу-
чают молибден в компактном состоянии (плавление в вакууме).
Для электрической дуги применяют ток силой 7000 а и напря-
жением 54—60 в.
В случае молибдена и металлов или сплавов, имеющих высо-
кие температуры плавления и чувствительных к кислороду, азоту
и водороду, рекомендуется проводить плавление с помощью
сфокусированного пучка электронов в атмосфере аргона или
гелия.
ОЧИСТКА
К вредным примесям молибдена относятся сера, мышьяк,
фосфор, медь, кислород, азот, водород и углерод.
Очистка металлического молибдена осуществляется пироме-
таллургическим или электролитическим методом. При плавлении
молибдена в высоком вакууме выделяются летучие примеси.
Для удаления серы в молибден добавляют марганец, кремний,
кальций или силициды железа, кальция, алюминия. С целью
уменьшения содержания включенных в молибден азота, кисло-
рода, водорода и углерода к молибдену добавляют бор, торий,
иттрий, титан. Часто для раскисления порошкообразного молибде-
на при плавлении металла добавляют небольшие количества угля.
Упомянутые загрязнения вызывают уменьшение ковкости,
тягучести, вязкости, эластичности и механической прочности
19-0101
290
ПОБОЧНАЯ ПОДГРУППА VI ГРУППЫ
металлического молибдена, который становится более твердым
и хрупким.
ФИЗИЧЕСКИЕ И ХИМИЧЕСКИЕ СВОЙСТВА
В компактном состоянии (в форме проволоки, листов, лентг
пластинок, брусков, блоков и т. п.) молибден — металл серебри-
сто-белого цвета; он обладает объемно-центрированной кубической
или плотной гексагональной (неустойчивая модификация) кри-
сталлической решеткой. При нагревании амальгамы молибдена
выше 300° в вакууме получают тонкий порошок металлического
молибдена, который обладает пирофорными свойствами.
Молибден относится к тяжелым металлам (плотность 10,22 г/смя
при 20°); т. пл. 2610° (тугоплавок), т. кип. 4830°, твердость 5,5
по шкале Мооса (160 кг/см2 по Бринеллю). Он парамагнитен
и является хорошим проводником тепла и электричества. Значение
электропроводности молибдена составляет примерно 30% электро-
проводности серебра; это больше, чем у никеля, платины, железа,
ртути и других металлов.
В компактном состоянии чистый молибден обнаруживает отлич-
ные механические свойства: он пластичен, ковок, тягуч, вязок
и хорошо поддается прокатке, протяжке, штамповке и др.
Благодаря малому коэффициенту расширения и относительна
большой теплопроводности молибден обладает высокой механи-
ческой прочностью при изменении температуры. Механическая
прочность молибдена при повышенных температурах превосходит
прочность всех металлов.
Загрязнение углеродом, водородом, серой, азотом ухудшает
механические свойства молибдена: он становится твердым, хруп-
ким, ломким и не может легко перерабатываться в изделия.
Металлический молибден можно полировать, шлифовать и сва-
ривать серебром или медью.
Известно множество сплавов молибдена с Be, Al, Ge, Ti, Zr,
V, Nb, Ta, Cr, W, U, Mn, Fe, Co, Ni, Ru, Rh, Pd, Os, Ir, Pt, Cu,
Ag, В, C, Si, P, As, Те и др. Помимо большого числа твердых
растворов, которые молибден образует с рядом элементов, изве-
стны и многие интерметаллические соединения молибдена, напри-
мер: Ве2Мо, А1Мо3, А12Мо, А15Мо, FeMo, Fe3Mo2, Fe7Moe, СоМо,
Со2Мо3, NiMo, Ni3Mo, ZrMo, Mo3Ir, Mo3Os, Mo2B, MoB, MoB2,
Mo3B2, Mo2B5, Mo2C, MoC, MoSi2, Mo3Si, Mo3Si2, Mo3Ge, Mo3Ge2,
MoGe2, MoN, Mo2N, MoP, Mo3P, MoP2, MoAs2, MoAs, MoS2,
MoS3, MoTe2, MoTe3.
При получении сталей и других сплавов молибден добавляют
в виде брикетов МоО3, молибдата кальция СаМоО4, молибдата
железа FeMoO4, ферромолибдена или силикомолибдена. Брикеты
МоО3 получают прессованием смеси МоО3 и смолы на гидравличе-
ских прессах под давлением 7 кг/мм2.
МОЛИБДЕН
291
При обычных условиях металлический молибден устойчив
в сухом и влажном воздухе. Температура окисления в сухом
воздухе или кислороде определяется степенью измельчения метал-
ла. Порошкообразный металлический молибден устойчив на воз-
духе и в кислороде при комнатной температуре, но при 500®
очень быстро окисляется с образованием МоО3. В компактном
состоянии металлический молибден сохраняет блеск до 200°;
темнеет при 300° и медленно окисляется при 500°, превращаясь
в МоО3. При температуре выше 795° окисление происходит очень
быстро и в момент образования МоО3 сублимируется. Ниже 300°
окисная пленка, образующаяся на поверхности металлического
молибдена, состоит из МоО3, в интервале между 300—700° пленка
состоит из МоО3 и МоО2, при окислении молибдена выше 700°
образуется только летучий МоО3. При горении молибдена в кис-
лороде также образуется МоО3.
В обычных условиях вода медленно окисляет тонкий порошок
молибдена в молибденовую синь.
В результате нагревания молибдена (700—800°) в парах воды
сначала образуется МоО2, который при дальнейшем нагревании
превращается в МоО3 по уравнению
Мо + 2Н2О МоО2 + 2Н2 МоО2 + Н2О -> МоО3 + Н2
Металлический молибден взаимодействует с фтором (60°),
хлором (300°), бромом (600°) и иодом (выше 800°):
Мо + 3F2 = MoF6 Мо + 2Вг2 = МоВг4
Мо + 5/2С12 = МоС16 6Мо + 6I2 = [Мо618]14
Если фтор разбавлен азотом, реакция фторирования молибдена
идет примерно при 350°. При действии хлора на молибден (300°)
на воздухе (или в присутствии кислорода) либо во влажной атмо-
сфере получают оксихлориды молибдена МоОС12, МоОС13. Если
хлор, реагирующий при нагревании с молибденом, разбавлен
азотом или двуокисью углерода, пентахлорид молибдена вос-
станавливается избытком молибдена до трихлорида молибдена
МоС13, который диспропорционирует на МоС14 и [МовС18]С14:
ЗМоС15 + 2Мо = 5МоС13
12МоС13 6MoCi4 + [Mo6Cj8]C14
Тетрабромид молибдена МоВт4 обычно загрязнен МоВг3
и [МовВг8]Вг4. Нагреванием металлического молибдена с BF3
получают MoFe, а с СОС12 при 630° образуется [Мо8С18]С14.
В результате нагревания молибдена с серой, селеном или
теллуром в закрытой трубке могут образоваться сульфиды MoS2,
Mo2S3, Mo2S5, MoS3, селенид MoSe и теллурид МоТе2. При 1200°
взаимодействие сероводорода с молибденом дает MoS2. >
19*
292
ПОБОЧНАЯ ПОДГРУППА VI ГРУППЫ
При температуре выше 1500° молибден вступает в реакцию
с азотом. В этом случае могут образоваться нитриды Mo3N, Mo2N,
MoN, которые значительно легче получить действием аммиака
на металлический молибден, нагретый до 600—800°.
Путем взаимодействия молибдена с красным фосфором или
мышьяком при нагревании в закрытой трубке можно получить
соответственно фосфиды МоР, МоР2, Мо3Р или арсенид MoAs2.
При действии твердого углерода, окиси углерода, карбида
кремния и т. п. или углеводородов (смеси СН4 сН2в соотношении
1 : 1 по объему) на металлический молибден, нагретый выше 600°,
получают карбиды Мо2С, МоС, МоС2:
>боо°; >8оо°;
Mo-J-2G----► МоС2 Mo-f-C--------> МоС
При температуре выше 700° двуокись углерода окисляет молиб-
ден до МоО3, который восстанавливается окисью углерода по обра-
тимой реакцииЗ
Мо ЗСО2 МоО3 -|- ЗСО
Действием окиси углерода на металлический молибден, нагре-
тый до 200°, при давлении 200 ат получают бесцветные ромбиче-
ские кристаллы гексакарбонила молибдена Мо(СО)6.
Металлический молибден не реагирует с расплавленными
металлами Li, Na, К, Mg, Zn, Pb, Bi, Се даже при нагревании
до 800°, но взаимодействует с галлием (выше 300°) и оловом
(выше 1000°).
На холоду металлический молибден устойчив к разбавленным
кислотам (HG1, HF, HNO3) и водным растворам щелочей. Он рас-
творяется в HNO3 при концентрации ниже 10 н., в концентриро-
ванных горячих H2SO4, НС1, царской водке, смесях фтористоводо-
родной и азотной или серной и азотной кислот, в окислительно-
щелочных расплавах, образованных NaOH с KNO3, КС1О3 и др.,
или перекисях щелочных металлов Na2O2, К2О2. При растворе-
нии молибдена в окислительно-щелочных расплавах образуются
молибдаты.
При анодном растворении или обработке HNO3 (концентрация
выше 10 н.) металлический молибден пассивируется.
Молибден вытесняет медь и серебро из растворов их солей,
бурно реагирует с Na2O (600°), Na2O2 (240°), Na2SO4 (430°), Na2CO3
(950°), взаимодействует при температуре выше 1600° с тугоплав-
кими окислами А12О3, ZrO2, ВеО, MgO, TiO2, ThO2.
В интервале температур 400—1200° водород растворяется
в молибдене в небольших количествах.
Химические свойства молибдена иллюстрируются следующей
схемой:.
МОЛИБДЕН
293
Комн. темп.
Молибден
Нагревание
с водой при очень медленной реакции ->
-> молибденовая синь
с царской водкой-> Н2МоО4-Н2О
на воздухе или в кислороде -+• МоОз и МоО2
с F2, Cl2, Br2. I,^MoF8, M0CI5, M0CI3, M0CI4,
[Мо6С18]С14, МоВг4, МоВг3, [МовВг8]Вг4, [MoeIg]I4
с BF3->MoF6
с СОС12 -> [Мо6С18]С14
с S, Se, Те -► MoS2, Mo2S3, M02S5, M0S3, MoSe2,
МоТе2
с N2, Р, As, С ->Mo3N, Mo2N, MoN, МоР, MoP2,
М03Р, MoAs2, Мо2С, МоС, МоС2
с СО2 МоОз
с СО при давлении 200 ат —- Мо(СО)в
с распл. NaOH + KNO3->Na2MoO4
С физиологической точки зрения органические соединения
молибдена играют роль катализаторов во многих реакциях живых
клеток. Молибден в микроколичествах способствует фиксации
атмосферного азота в почве азотными бактериями, которые разви-
ваются только в присутствии соединений молибдена. В процессе
питания растений молибден играет очень важную роль. В присут-
ствии его солей растения могут противостоять действию многих
токсичных солей марганца, цинка, никеля, кобальта, меди и др.
Кормовые растения с большим содержанием молибдена токсичны
для животных. Внесение в почву ничтожно малых количеств
(порядка сотых долей грамма на гектар) молибдата аммония,
который считается молибденовым удобрением, вызывает быстрый
рост цитрусовых, льна, люцерны, цветной капусты.
Для людей продолжительное вдыхание белого дыма МоО3
опасно из-за его токсичности.
ПРИМЕНЕНИЕ
Примерно 90% всего количества молибдена применяется в чер-
ной металлургии в производстве легированных сталей, коррози-
онно- и термостойких сталей, инструментальных сталей и в ка-
честве добавок при литье изделий из сталей и чугуна. Из диа-
граммы термического равновесия сплавов железо — молибден
следует, что растворимость молибдена в железе меняется
с температурой, например при 1440° она составляет ~24%.
Добавка 0,1—2% молибдена в хромовые или хромоникелевые
стали способствует увеличению механической прочности при
высоких температурах, коррозионной стойкости, упругости,
твердости, термостойкости, способности к закаливанию и др.
Молибденсодержащие стали используются для изготовления
инструментов, в авто- и авиапромышленности, для изготовления
валов турбин, броневых плит, стволов огнестрельного оружия
294
ПОБОЧНАЯ ПОДГРУППА VI ГРУППЫ
и др. Стеллиты — инструментальные стали с содержанием молиб-
дена 5—15%. Стали с карбидом молибдена являются твердыми
сплавами и служат для изготовления резцов быстрорежущих
инструментов, а также вальцов. При сочетании карбида молиб-
дена с карбидом титана получают сверхтвердые сплавы.
Железо-никелевые сплавы с 2—3% молибдена (возможна
добавка меди) применяются при изготовлении трансформаторов,
кабельных экранов, сердечников для муфт в установках теле-
коммуникаций. Сплав из 45% Ni, 25% Со, 7% Мо, 23% Fe исполь-
зуется в технике телекоммуникаций.
Молибден используют для получения постоянных магнитов,
сплавов для часовых пружин, очень твердых цветных сплавов,
коррозионностойких сплавов. Из молибденсодержащих корро-
зионностойких сплавов изготовляют газовые турбины, трубы для
теплообменников, установки для получения вискозного шелка,
установки для очистки нефти, корпуса насосов для химической
промышленности, зеркала для корабельных сигнальных фонарей,
тигли, капсулы, диафрагмы и электроды, используемые при элек-
тролизе расплавов.
Благодаря большой механической прочности при температуре
выше 870° (но не в окислительной атмосфере!) молибден применяют
в ядерных реакторах.
Металлический молибден и его сплавы с железом, никелем,
марганцем используются при изготовлении электронных трубок.
Проволока из чистого молибдена или сплавов с 20% Мо, 20% Fe,
58% Ni, 2% Мп или 28% Мо, 5% Fe, 2% Мп (остальное никель)
служит для оснастки пультов управления флуоресцирующих
экранов, сеток накала электронных ламп, деталей рентгеновских
трубок и др. Нити, глазки и крючки, на которых крепятся волоски
и катоды накаливания, изготовляются из молибдена. Вольфра-
мовые спирали электрических лампочек накручиваются на сер-
дечник из молибденовой проволоки, который затем растворяется
при 60—70° в 50%-ной HNO3 с добавкой 5% HG1.
В электронной и электропромышленности применяют сплавы
из молибдена (20—90%) и вольфрама (80—10%).
Сплав из молибдена (30—60%) и серебра (70—40%) и неко-
торые сплавы из меди и молибдена используются при изготовле-
нии контактных выключателей. Сплавы из молибдена и тантала
применяют вместо платины.
Из молибдена изготовляют детали высокотемпературных печей,
работающих в инертной атмосфере или в вакууме.
Из молибдена делают электроды к печам для плавки стекла.
Молибденовые стержни служат электродами для сварки с ато-
марным водородом.
Благодаря малому коэффициенту термического расширения
молибден может применяться как материал для сварки металли-
МОЛИБДЕН
295
ческих деталей со стеклом. Термопары из чистого никеля и спла-
вы из никеля с 18% молибдена используются в обжиговых печах,
работающих в атмосфере водорода или в другой восстановитель-
ной среде. Для измерения температур выше 1900° используют
термопары из вольфрама и молибдена.
Молибден служит катализатором в процессах окисления, гид-
рогенизации, дегидрогенизации, изомеризации, полимеризации,
циклизации, конденсации, алкилировании, дегидратации и др.
Молибденовые красители, характеризующиеся высокой ста-
бильностью, интенсивным блеском и превосходной кроющей спо-
собностью, служат для изготовления различных красок, пласти-
ческих материалов, для окраски мехов, кож, волос и др. Из мине-
ральных пигментов очень большое значение имеет молибденовый
красный: 76% РЬСгО4 + 14% РЬМоО4 Д- 10% PbSO4. Для уве-
личения устойчивости органических красителей при хранении
рекомендовано применение диазониевых солей фосфомолибдено-
вой и фосфовольфрамомолибденовой кислот.
СОЕДИНЕНИЯ (ОБЩИЕ СВОЙСТВА)
Известно ограниченное число соединений двух-, трех-, четы-
рех- и пятивалентного молибдена и очень много соединений шести-
валентного молибдена. Наиболее устойчивы соединения шести-
и частично трехвалентного молибдена.
В табл. 42 приведены формулы и указан цвет соединений молиб-
дена, сгруппированных по валентностям. Помимо простых соеди-
нений, известно множество координационных соединений молиб-
дена в различных валентных состояниях.
Если в соединениях низших степеней окисления молибден про-
являет металлические свойства (основной и восстановительный
характер), то в соединениях шестивалентного молибдена в основ-
ном проявляются неметаллические свойства (кислотный характер
и ковалентные связи).
Несмотря на многообразие окраски соединений молибдена,
переход от одной валентности к другой можно проследить по изме-
нению цвета. При восстановлении шестивалентного молибдена
в солянокислом растворе получают ряд различно окрашенных
комплексов:
fMoe+O3Cl2]2- [Мо5+ОС16]2~ [Мо4+О2С14]4- [Мо3+С16р- [Мо2+С18С12]2+
Бесцветный Зеленый Коричневый Красный Желтый
Помимо соединений молибдена, отвечающих пяти состояниям
валентности, известны нестехиометрические окислы (с промежу-
точной валентностью между сг-МоО3 и 6-МоО2), названные молиб-
деновыми синями, металлоорганические производные и соедине-
ния включения.
Соединение Окислы Гидроокиси или кислоты Фториды, окси- фториды, фторо- соединения Хлориды, оксихло- риды, хлоросоеди- нения
Соединения молибде- на (II) МоО0,93-0,97 черная [МовС18]СЦжелтый Н2[Мо6С18С16] • •8Н2О Ме1[Мо6С18С16]- •пН2О желтый
Соединения молибде- на (III) MO2O3 черная Мо(ОН)3 черно- коричневая M0F3 розовый MoOF-4H2O желтый K[MoF4]-H2O фиолетовый M0CI3 кирпично- красный МоОСЬ4Н2О коричневый или зеленый К3[МоС16] корич- нево-красный К2[МоС15Н2О1 коричнево-крас- ный
Соединения молибде- на (IV) МоО2 фио- летово-ко- ричневая МоО2-2Н2О черно-ко- ричневая — — МоС14 коричневый
Соединения молибде- на (V) Мо2Ой фио- летовая МоО(ОН)3 красно- коричневая MoF5 оливко- во-зеленый MO2O3F4 крас- ный Mel[MoOF5)H2O зеленые M0CI5 черный МоОС13- 2(С2Н5)2О MoOC13-C5H5N К2[МоОС15] зеле- ный Ме1[МоОС14(Н2О)] Ме^МоОгСШгО);,] зеленые
Таблица 42'
Бромиды, оксибромиды, бромосоеди- нения Йодиды, оксииодиды, иодосоеди- нения Сульфиды, тио- соли, селениды, теллуриды Координационные соединения Соединения включения
[Мо8Вг8]Вг4 оранже- вый fMo6I8]I4 красновато- коричневый Mei[Mo6Cl8(OH)e] Mei[Mo6Cl8Br6] [МовС18(Н2О)2(ОН)4]. • 12Н2О [Мо6С18(Н2О)2С14]. • 6Н2О [Мо6С18(Н2О)2Вг4]. • (4—10)Н2О [МовС18(Н2О)214]. • 10Н2О [Mo6Cl8(SO4)2]-6H2O Mei[Mo6Br8(OH)e] MoN, Mo2N серые MoP2, MoP, Mo3P серо-черные- MoC, Mo2C серые MoSi2, Mo3Si2, Mo3Si серые- Mo2B, Mo3B2, MoB, MoB2 и Mo2B6 серые-
МоВг3 темно-зе- леный МоОВг* -4Н2О желтый — Mo2S3 серый Mo2Se3 серый Мо2Те3 серый K4[Mo(H2O)(CN)7]H2O [Mo(NH3)6]C13 K3[Mo(NCS)6]-4H2O [Mo2O(SO4)2]-5H2O [Mo2O(C2O4)2] • 6H2O
МоВг4 чер- ный — MoS2 черный K4[Mo(CN)8].2H2O H4[Mo(CN)8]-6H2O K4[Mo(OH)4(CN)4] K4[MoO2(CN)4] K3[Mo(OH)3(H2O)(CN)4] Mei[Mo(NCS)e]
Ме1[МоОВг5] Желтый Ме1[МоОВг4] красный — Mo2S5 корич- невый Mo2Se5 черный K3[Mo(CN)g] Ag3[Mo(CN)8] H3[Mo(CN)8]-3H2O Mei[MoO(NCS)5] Mei[MoO2(NCS)3]-nH2O MeI[MoO2(C2O4)(H2O)2]
298 ПОБОЧНАЯ ПОДГРУППА VI ГРУППЫ
Соединение Окислы Гидроокиси или кислоты Фториды, окси- фториды, фторо- соединения Хлориды, оксихло- риды, хлоросоеди- нения
Соединения молибде- на (VI) МоО3 белая Н2МоО4 белая Н2Мо2О7 белая Н2МоО4-Н2О лимонно-желтая или синевато-бе- лая Ме1МоО4 Ме1Мо2О7 МепМоО4 Пероксосоедине- ния Н2МоО5-3/2Н2О Н2МоО6, Н2МоО8 Me^IoOj Ме2[МоО8]-и.Н2О Ме^[МоО8] Ме1[Мо2Ои] • иН2О Ме1[Мо2О8] Mei[MoO3F4]H2O MoF6 белый MoOF4 белый MoO2F2 белый Mei[MeO3F2] Mel[MoO3F3] Me4MoO2F3]. •nH2O Mei[MoO2F4]- •nH2O Mei[MoOF5] Mei[MoOF6] МоОС14 зеленый МоО2С12 желто- ' вато-белый Н2[МоО3С12] бе- лый Ме1[МоО2С13]- •пН2О Ме1[МоО2С14]- • пН2О Ме1[М°3ОвС17]- • пН2О
Неорганические соединения
Соединения двухвалентного молибдена
Известно несколько соединений двухвалентного молибдена,
а именно окись молибдена МоО0193_0,97, гексамеры дигалогенидов
молибдена и ряд производных этих гексамеров. В соединениях
двухвалентный молибден встречается в виде катионов [МовГал8]4+,
анионов [МовГал8Х6]2- и соединений типа [Мо8Гал8(Н2О)2Х4],
где X = С1-, Br-, ОН-.
Гексамеры дигалогенидов молибдена окрашены в цвета от жел-
того до красновато-коричневого, тугоплавки и плохо растворимы
в воде. С помощью рентгеноструктурного анализа было установ-
лено, что гексамерам дигалогенидов молибдена следует приписы-
вать формулы [Мо8С18]С14, [Mo8Br8]Br4, [Мов18П4.
Окись молибдена, МоО0,93 — МоО0197. При действии паров
калия на кислородные соединения молибдена образуется ряд
продуктов, содержащих Kli5MoO3, K0i8MoO2, которые под дей-
ствием воды или спирта превращаются в растворимый молибдат,
водород и черный трудно растворимый остаток, обладающий силь-
ными восстановительными свойствами и отвечающий составу,
среднему между МоО0193 и МоО0,97.
МОЛИБДЕН
299
Продолжение табл. 42
Бромиды, оксиброми- ды, бромо- соединения Йодиды, ок- сииодиды, иодосоеди- нения Сульфиды, тио- соли, селениды, теллуриды Координационные соединения Соединения включения
МоО2Вг2 оранжево- красный Нз[МоОзВгз] желтый MoS3 коричне- вый MeiMoS4 красный 3MoS3-MenS оранжевый KM0S5 красный M0S4 корич- невый MeiMoO3S MeiMoO2S2 MeIH[Mo2O4S3] MoSe3 корич- невый Mei[Mo3O10l-nH2O Mei[Mo4O13]-nH2O Mei[Mo6O2i] • пН2О Mei[HMo6O2i] • пН2О Mei[H2MoeO2i]-nH2O MeI[H3Mo6O2i] • пН2О Ме|[МоМо6О24] • пН2О Mei[HMo7O24] • пН2О Mei[Mo8O26]-nH2O Mei[H2Moi2O40] • иН2О Mei[TeMo6O24] • иН2О Mel[PMo12O40]-nH2O MeI[AsMoi204o]-nH20 Mei[SiMo12O40] • tiH2O
Гексамер дихлорида молибдена, [МовС18]С14, получают дей-
ствием фосгена на нагретый до 630° металлический молибден,
пропусканием паров МоС13 (без доступа кислорода), увлекаемых
током газообразного СО2, через нагретую трубку, а также вос-
становлением пентахлорида молибдена МоС15.
6Мо + 6СОС12 = [Мо6С18]С14 + 6СО
12МоС13 = [Мо6С18]С14 4- 6МоС14
Отделение [МовС18]С14 от МоС14 основывается на разнице
в летучести этих двух соединений. Тетрахлорид молибдена МоС14
летуч, в то время как [МовС18]С14 нелетуч и остается в виде жел-
того аморфного порошка. Безводное соединение [Мо6С18]С14 имеет
плотность 3,714 г/см? при 25°, устойчиво на воздухе, тугоплавко,
плохо растворимо в воде, толуоле, уксусной кислоте и др., раство-
римо в спирте, эфире, спиртоэфирной смеси, концентрированных
кислотах (НО, НВг, HI) и щелочах. На рис. 10 показана струк-
тура катиона [Мо8С18]4+.
При обработке безводного соединения [Мо8С18]С14 избытком
щелочи получают растворимые соединения общей формулы
Ме^[МовС18(ОН)в], которые при обработке уксусной кислотой
или хлоридом аммония до pH = 9 превращаются
в [Мо6С18(Н2О)2(ОН)4]-12Н2О. Последние представляют собой
300
ПОБОЧНАЯ ПОДГРУППА VI ГРУППЫ
желтые ромбоэдрические кристаллы, растворимые при нагрева-
нии в различных кислотах:
[МовС181С14 + 6КОН = К2[МовС18(ОН)в] 4- 4KG1
К2[Мо6С18(ОН)в] + 2СН3СООН + 12Н2О =
= [Мо6С18(Н2О)2(ОН)4].12Н2О + 2КСНгСОО
При охлаждении раствора [Мо8С18(Н2О)2(ОН)4] «12Н2О в HG1
(растворение при нагревании) выпадают желтые кубические кри-
сталлы [Мо8С18(Н2О)2С14]-6Н2О или [Мо8С18]С14-8Н2О.
Рис. 10. Структура ка-
тиона [Мо6С18]4+.
Рис. 11. Структура иона
[Мо6С18Х6]2-.
С водным раствором нитрата серебра [Мо8С181С14 взаимодей-
ствует по уравнению
[Мо6С18]С14 + 4AgNO3 + 2Н2О = [MoeCl8(H2O)2(NO3)4] + 4AgCl
Из раствора [Мо8С18]С14 в НС1 (1 : 1) выпадают кристаллы
Н2[Мо8С18С18] >8Н2О или (Н3О)2[Мо8С18С18] -6Н2О. При обработке
соединения [Мо8С18]С14 хлоридами щелочных металлов получают
комплексные соли типа Ме*[Мо8С18С18] -6Н2О. При обработке
К2[Мо8С18(ОН)8] водным раствором HI образуется К2[Мо8С1818] •
•6Н2О.
Известны также соединения К2[Мо8С18С18] ‘4Н2О,
(NH4)2[Mo8Gi8Gl8l, К2[Мо8С18Вг8], [Мо8С18(Н2О)2Вг4]-4 или
10Н2О, [Мо8С18(Н2О)214Ы0Н2О, [Mo8Cl8(SO4)2].6H2O.
В ионе [Мо8С18]4+ все восемь атомов хлора расположены в углах
куба, а шесть атомов молибдена занимают несколько смещенные
внутрь позиции в центрах граней куба. Каждый атом молиб-
дена образует четыре почти копланарные связи с четырьмя ато-
мами хлора и обладает двумя неспаренными электронами — один
ориентированный к центру комплекса, другой — во внешнюю
сторону, что обусловливает октаэдрическую конфигурацию.
На рис. 11 показано строение иона [Мо8С18Х8]2-, где X может
быть галогеном или кислородом, молекулой воды или группой
МОЛИБДЕН
301
ОН". Шесть атомов хлора расположены в углах куба, а шесть
атомов молибдена — в углах большого октаэдра таким образом,
чтобы каждый X был связан с атомом молибдена.
Гексамер дибромида молибдена, [Мо8Вг8]Вг4, получают дей-
ствием разбавленных азотом паров брома на нагретый до 600—700°
металлический молибден (при этой температуре остальные бро-
миды молибдена сублимируют или диссоциируют), а также терми-
ческой диссоциацией трибромида молибдена:
2МоВг3 -> 1/6[Мо6Вг8]Вг4 МоВг4
Дибромид молибдена [МовВг8]Вг4 представляет собой аморфное
вещество оранжевого цвета с плотностью 4,88 г/см2 при 20°, туго-
плавкое, плохо растворимое в воде, кислотах, царской водке
и разбавленных щелочах.
При обработке соединения [Мо8Вг8]Вг4 избытком щелочи полу-
чают растворимые соединения типа Ме^[Мо8Вг8(ОН)8], где Me1 =
= Na+, К+.
Гексамер дииодида молибдена, [Мо618]14, получают на основе
обратимой реакции между металлическим йолибденом и парами
иода при нагревании или восстановлением безводного пентахло-
рида молибдена МоС15 газообразным Ш при 250°:
6Мо (тв.) + 612 (газ) [Mo6Ig]I4 (газ)
6МоС15 + 30HI = [MoeI8]I4 + 9I2 + 30НС1
Высвободившийся при этой реакции иод удаляют промыва-
нием CS2.
Дииодид молибдена [Мо818П4 представляет собой красновато-
коричневый аморфный порошок с плотностью 4,3 г!см? при 20°;
он плохо растворяется в воде или спирте, окисляется при тем-
пературе выше 250°, восстанавливается водородом при нагрева-
нии, реагирует с хлором, бромом и серой, разлагается под дей-
ствием перегретого пара или щелочей.
Соединения трехвалентного молибдена
Трехвалентное состояние не характерно для молибдена. Изве-
стно ограниченное число соединений молибдена, обладающих
восстановительным характером и устойчивостью, средней между
соединениями низшей и высшей степеней окисления. Наряду
с простыми соединениями Мо2О3, Мо(ОН)3, MoF3, МоС13, МоВг3,
Mo2S3 известны некоторые координационные соединения трехва-
лентного молибдена, характеризующиеся большей устойчивостью.
У соединений трехвалентного молибдена встречается большое
разнообразие расцветок — от желтой, розовой, красной, пур-
пурной, коричневой, зеленой, фиолетовой, серой до черной
302
ПОБОЧНАЯ ПОДГРУППА VI ГРУППЫ
Окись молибдена, Мо2О3, получают при дегидратации гидро-
окиси молибдена Мо(ОН)3, при восстановлении калием трехокиси
молибдена, растворенного в жидком аммиаке, и при осторожном
восстановлении МоО3 водородом при нагревании.
Окись молибдена представляет собой матово-черный порошок
с плотностью 7,07 г/см3 при 25°, плохо растворимый в воде. При
растворении в НС1 получается красный раствор, содержащий
катион Мо3+.
Гидроокись молибдена, Мо(ОН)3, получают при обработке
соединения молибдена(Ш) аммиаком или щелочами, восстанов-
лением молибдата аммония водородом в присутствии палладия,
катодным восстановлением слегка подкисленного раствора молиб-
дата аммония.
Гидроокись молибдена представляет собой черно-коричневое
аморфное вещество, плохо растворимое в воде и разбавленных
минеральных кислотах, разлагающее воду в щелочной среде
с выделением водорода.
Трифторид молибдена, MoF3, получают нагреванием трибро-
мида молибдена МоВг3 в токе сухого фтористого водорода при 600°:
МоВг3 + 3HF = MoF3 + ЗНВг
Трифторид молибдена представляет собой твердое вещество
розового цвета. Оно устойчиво при нормальных температуре
и давлении. При нагревании во влажном воздухе превращается
в МоО3 и HF. Восстанавливается водородом при нагревании,
образуя металлический молибден без стадии промежуточных
продуктов. По кристаллической структуре MoF3 изоморфен ReO3.
При электрическом восстановлении МоО3 в присутствии фто-
рида калия образуется соединение KlMoFJ -Н2О в виде фиолето-
вых кристаллов.
Оксифторид молибдена, MoOF -4Н2О, в виде светло-желтого
осадка получают при действии фторида аммония на MoOCl *4Н2О
в водном растворе без доступа воздуха.
Трихлорид молибдена, МоС13, получают восстановлением пен-
тахлорида молибдена водородом при 250°, пропусканием паров
МоС15 (увлекаемых инертным газом) над нагретым порошкообраз-
ным металлическим молибденом, диспропорционированием тетра-
хлорида молибдена при сильном нагревании, пропусканием газо-
образного НС1 над сильно нагретым МоВг3:
МоС16 + Н2 = МоС13 + 2НС1 2МоС14 = МоС13 + МоС16
ЗМоС15 + 2Мо = 5МоС13 МоВг3 + ЗНС1 = МоС13 + ЗНВг
Кирпично-красные игольчатые кристаллы трихлорида молиб-
дена МоС13 относительно устойчивы на воздухе, обладают плот-
ностью 3,578 г/см3 при 25°, плохо растворимы в воде, спирте
МОЛИБДЕН
303
эфире,[растворимы в серной кислоте, парамагнитны. При сильном
нагревании в атмосфере СО2 разлагаются по уравнению
2МоС з -* VelMoeClgJCU + МоС14
Нелетучий Летучий
Трихлорид молибдена хранят в герметичных сосудах, посколь-
ку на воздухе он медленно превращается в оксихлорид MoOCl.
При обработке МоС13 водными растворами щелочей получают
черно-коричневый осадок Мо(ОН)3, трудно растворимый в раз-
бавленных кислотах.
При электролитическом восстановлении МоО3 в соляной кисло-
те получают красный раствор соединения МоС13-ЗН2О, которое
восстанавливает соли серебра, а также двухвалентной меди и рту-
ти. При добавлении спирта или при упаривании раствора, полу-
ченного обработкой трихлорида молибдена раствором КС1, выпа-
дают коричнево-красные кристаллы К2[МоС15Н2О] или К3[МоС16].
Известны гексахлоромолибдаты типа Mei[MoCle], где Me1 = К+,
Rb+, Cs+, NH~; при гидролизе они превращаются в MoOCl -4Н2О:
Ме[[МоС16] + Н2О т Me*[MoCI6H2O} + Ме*С1
Ме[[МоС16Н2О] + Н2О =₽* Ме1[МоС14(Н2О)2] + МехС1
Ме1[МоС14(Н2О)2] + Н2О [МоС1з(Н2О)3] + МегС1
[МоС13(Н2О)3] + Н2О [МоС12(ОН)(Н2О)3] + НС1
[МоС12(ОН)(Н2О)3] + Н2О [МоОС1(Н2О)4] + НС1
При действии раствора KCN на комплекс К3[МоС1в] без доступа
кислорода в присутствии спирта осаждается черный комплекс
K4[MoIir(H2O)(CN)7] -Н2О, который растворяется в воде и окис-
ляется на воздухе в соединение желтого цвета K4[Mo(CN)8] «2Н2О,
в котором молибден четырехвалентен. При обработке плавиковой
кислотой твердого МоС13 последний превращается в MoF3.
Кислородные кислоты превращают трихлорид молибдена в мо-
либденовую кислоту.
Известно несколько соединений МоС13 с аммиаком и раз-
личными органическими аминами, например [Mo(NH3)e]Cl3,
[MoC12(NH3)JC1, [MoC13(NH3)3], [МоС13Амин3].
Оксихлорид молибдена, MoOCl-4Н2О или [МоОС1(Н2О)41,
существует в виде двух модификаций — коричневой и зеленой.
Обе они растворимы в воде, легко окисляются на воздухе и яв-
ляются восстановителями по отношению к соединениям меди(П),
ртути(П), железа(Ш) и серебра(Т).
Осадок коричневой модификации образуется при обработке
конц. НС1 ацетонового раствора соединений молибдена(Ш),
а зеленой модификации — при добавлении ацетона к солянокис-
лому раствору соединения молибдена(Ш). Обе модификации
получаются в отсутствие окислительных реагентов.
304
ПОБОЧНАЯ ПОДГРУППА VI ГРУППЫ
Трибромид молибдена, МоВг3, получают одновременно с МоВг4
при действии паров брома на нагретый до 350—400° металличе-
ский молибден, нагреванием смеси паров МоС15 и сухого НВг
при 425—525°, пропусканием паров брома, разбавленных азотом,
над нагретым до 450—500° металлическим молибденом с после-
дующим промыванием получившегося продукта холодным рас-
твором НВг.
Зеленые игольчатые кристаллы трибромида молибдена плохо
растворимы в воде и кислотах, диссоциируют при нагревании без
доступа воздуха на МоВг2 и бром, растворяются в пиридине
и NH4OH, превращаются в Мо(ОН)3 под действием щелочей.
Газообразный аммиак восстанавливает МоВг3 до металлического
молибдена. Из раствора трибромида молибдена в пиридине выпа-
дает соединение MoBr3 •3C5H5N.
Комплексный оксибромид молибдена, МоОВг -4Н2О или
[МоОВг(Н2О)4], осаждается при добавлении ацетона к раствору,
полученному при электролизе раствора молибденовой кислоты
в бромистоводородной кислоте.
Соединение МоОВг *4Н2О представляет собой светло-желтый
расплывающийся на воздухе порошок; он растворяется в воде,
окисляется на воздухе и обладает восстановительными свойствами.
Гекса[тиоцианато)молибдат калия, K3[Mo(NCS)e] ’4Н2О, полу-
чают действием KSCN на К3[МоС16] или электролитическим вос-
становлением солянокислых растворов МоО3 в присутствии тио-
цианатов щелочных металлов K3[Mo(NCS)e] образует жел-
тые псевдогексагональные кристаллы, которые очень легко окис-
ляются.
Известны также соединения Na3[Mo(NCS)e], (NH4)3[Mo(NCS)eJ.
Сульфид молибдена (трисулъфид молибдена), Mo2S3, получают
прямым взаимодействием элементов при 1100° или восстановле-
нием дисульфида молибдена водородом при нагревании:
1100
2Mo + 3S - * Mo2S3 2MoS2 + Н2 = Mo2S3 Ц-H2S
Выше 1200°
Сульфид молибдена образует иглы серо-стального цвета с плот-
ностью 5,91 г/см?, более твердые, чем молибденит, диссоциирую-
щие на молибден и серу при сильном нагревании, плохо раство-
римые в разбавленных кислотах и окисляющиесяТпод действием
конц. HNO3.
Iидратированный сульфид молибдена, Mo2S3-nH2O, выпадает
при пропускании H2S через подкисленные растворы солей молиб-
дена(П1).
Помимо перечисленных выше известны и другие соединения
молибдена(Ш), например Mo2Se3, Мо2Те3, Mo2O(SO4)2-5Н2О,
Мо2О(С2О4)2 -6Н2О, Мо4О3(С2О4) -12Н2О,Мо2(Сг2О7)3.
МОЛИБДЕН
305
Соединения четырехвалентного молибдена
Известно ограниченное число соединений четырехвалентного
молибдена, например б-МоО2, МоС14, MoBr4, MoS2, K4[Mo(CN)8L
• 2Н2О, K4[Mo(OH)4(CN)4], K4[MoO2(CN)4], Mei[Mo(NCS)e] и др.
Двуокись молибдена не проявляет ни кислотных, ни основных
свойств, кристаллизуется в решетке типа рутила. Тетрахлорид
и тетрабромид молибдена — легко летучие соединения, гидроли-
зующиеся при соприкосновении с водой. Дисульфид молибдена,
будучи самым устойчивым сульфидом молибдена, встречается
в природе в виде минерала молибденита. Наиболее устойчивый
комплекс молибдена(ТУ) — K4[Mo(CN)8] -2Н2О. Что касается цве-
та, то простые соединения четырехвалентного молибдена окра-
шены в коричневый или черный цвет, а координационные соеди-
нения — в желтый, красный, синий или фиолетовый.
б-Двуокись молибдена, МоО2, получают окислением металли-
ческого молибдена смесью паров воды и водорода при 700—800°,
легким нагреванием порошкообразного металлического молибдена
на воздухе, восстановлением трехокиси молибдена водородом
при 600—700°, восстановлением МоО3 при нагревании с аммиаком,
смесью СО СО2, MoS2, цинком, кадмием, магнием, металличе-
ским молибденом, а также восстановлением при нагревании молиб-
датов щелочных металлов металлическим цинком:
Мо + 2Н2О МоО2 + 2Н2 ЗМоО3 + 2NH3 = ЗМоО2 + ЗН2О + N2
Мо + О2 = МоО2 + 132,5 ккал!молъ МоО3 + СО = МоО2 + СО2
МоО3 + Н2 = МоО2 + Н2О 6МоО3 + MoS2 = 7МоО2 + 2SO2
2МоО3 4- Zn = МоО2 + ZnMoO4 Na2MoO4 4- Zn = МоО2 + Na2ZnO2
При восстановлении молибденовой кислоты кипячением с по-
рошкообразным металлическим молибденом получают раствор,
меняющий свою окраску с синей, зеленой, желтой до коричневой,
из которого при подкислении или подщелачивании выпадает
осадок МоО2 -2Н2О черно-коричневого цвета.
6-Двуокись молибдена образует серый аморфный порошок
или фиолетово-коричневые моноклинные кристаллы с плотностью
6,47 г!см3-, они устойчивы на воздухе, медленно сублимируются
в вакууме при 1100°, разлагаются при 1977° на МоО3 и металличе-
ский молибден, парамагнитны, обладают свойствами полупровод-
ников, плохо растворимы в воде, кислотах и щелочах.
ЗМоО2 = 2МоОз Мо
Водород восстанавливает нагретую до 950—1100° двуокись
молибдена до металлического молибдена. При высокой темпера-
туре пары воды и двуокись углерода окисляют МоО2 в МоО3.
Выше 300° хлор превращает двуокись молибдена в оксихлорид
молибдена (VI)'.
20-0101
306
ПОБОЧНАЯ ПОДГРУППА VI ГРУППЫ
Двуокись молибдена растворяется в растворе сульфата желе-
за(Ш) и в растворах кислот окислителей. При нагревании без
доступа воздуха или кислорода гидроокиси щелочноземельных
металлов превращают МоО2 в окрашенные молибдаты(1У), напри-
мер ВаМоО3 — красный, SrMoO3 — черный.
Азотная кислота и аммиачные растворы солей серебра окис-
ляют МоО2 до МоО3.
Тетрахлорид молибдена, МоС14, получают нагреванием при
250° в запаянной трубке двуокиси молибдена с раствором хлора
в СС14 или диспропорционированием сильно нагретого трихло-
рида молибдена в атмосфере газообразного СО2:
С12
МоО2 + СС14 —MOCI4 + CO2 2МоС13 VelMogClglCla + MoCU
Тетрахлорид молибдена представляет собой расплывающийся
очень тонкий коричневый порошок, который легко сублимируется
в желтые пары (т. пл. 317°, т. кип. 322°); он чувствителен к воз-
духу, свету, повышенной температуре и влажности, растворяется
в спирте и кислотах (HNO3, H2SO4), разлагается в воде, эфире
и при нагревании. Если нагревание тетрахлорида молибдена про-
водится в сухой инертной атмосфере, то образуются неустойчивые
галогениды МоС13, МоС15 и в конечном результате [МовС18]С14:
2МоС14 МоС13+ МоС15
''моС13+С12
—- M0CI4 + V4Mo6Clg]Cl4
При нагревании во влажном воздухе МоС14 превращается
в МоО2С12.
Тетрабромид молибдена, МоВг4, получают пропусканием паров
брома (увлекаемых током газообразного СО2) над нагретым до
525—625° металлическим молибденом:
Мо -|- 2Вг2 = МоВг4 + 60 кка._
Тетрабромид молибдена образует расплывающиеся на воздухе
блестящие черные иглы, которые плавятся при 337°, кипят при
347°, растворяются в воде и разлагаются щелочами. При сильном
нагревании тзтрабромид молибдена разлагается по уравнению
2МоВг4 -> 2МоВг3 + Вг2
Дисульфид молибдена, MoS2, встречается в природе в виде
минерала молибденита. Его получают нагреванием стехиометри-
ческой смеси молибдена и серы в стальной трубке, нагреванием
МоО3, МоО2 или (NH4)e[Mo7O24l с расплавленной смесью
К2СО3 и серы, нагреванием молибдата аммония с серой, обра-
боткой H2S нагретой трехокиси молибдена или кислой суспензии
МОЛИБДЕН
307
МоО2, а также разложением трисульфида молибдена или тиомо-
либдата аммония (~800°):
Mo + 2S (ромбич.) — MoS2 + 56,3 ккал МоО2 + H2S = MoS2 + 2Н2О
МоО3 + 3H2S = MoS2 + ЗН2О + S
2МоО3 + К2СО3 + 7S = 2MoS2 + К2О + СО2 + 3SO2
МоО2 + К2СО3 + 3S = MoS2 + К2О + СО2 + SO2
MoS3 — MoS2 + S (NH4)2MoS4 = MoS2 + H2S + S + 2NH3
Дисульфид молибдена представляет собой диамагнитные чер-
ные металлоподобные кристаллы с плотностью 4,80 г!см\ твер-
достью 1 —1,5 по шкале Мооса (оставляют черный след на бумаге),
сублимирующиеся в инертной атмосфере при 450°. Вещество пла-
вится под давлением при 1185°, при нагревании без доступа возду-
ха диссоциирует на Mo2S2 и серу (в вакууме диссоциация может
идти до металлического молибдена), реагирует с парами воды
или водородом при сильном нагревании, превращается в МоО3
при нагревании на воздухе при 550°:
MoS2 + 2Н2 Mo + 2H2S
550°
MoS2 + 7/2О2----> МоОз + 2SO2 + 264 ккал
Гексагональные кристаллы дисульфида молибдена имеют фор-
му пластинок и расслаиваются перпендикулярно главной оси
на очень тонкие лепестки. В кристал-
лической решетке MoS2 (рис. 12) каждый
атом молибдена расположен в центре
шести атомов серы, находящихся в уг-
лах треугольной призмы. Расстояние
Мо — S равно 2,41 А, расстояние Мо —
Мо в той же проекции 3,15 А, а между
соседними слоями — 6,26 А.
Концентрированные кислоты (HNOs,
H2SO4) при нагревании превращают ди-
сульфид молибдена в молибденовую кис-
лоту.
При сплавлении MoS2 с сульфидами
щелочных металлов или при растворении
MoS2 в полисульфиде аммония получают
растворимые тиосоли, например по урав-
О Мо
• 5
Рис. 12. Структура
MoS2.
нению
MoS2 + (NH4)2S2 = ( NH4)2MoS4
При комнатной или пониженной температуре молибденит
является полупроводником, который может быть использован
как детектор высокой частоты, выпрямитель или транзистор.
20*
308
ПОБОЧНАЯ ПОДГРУППА VI ГРУППЫ
Дисульфид молибдена используется в качестве сухой смазки,
в смесях с маслами или в жидких смазках, в качестве наполнителя
пластмасс, каучуков и др. Смазки с MoS2 на основе силикона
предназначены для эксплуатации при высоких температурах
(~400°).
Ацидосоединения молибден а(1У), содер-
жащие координированные CN~ -группы. Эти
соединения могут быть получены реакцией замещения из других
комплексов молибдена(1У), окислением комплексов молибдена(Ш)
или перегруппировкой соединений молибдена(У) в присутствии
цианидов щелочных металлов.
Октацианомолибдат калия, KJMo(CN)8] ^ЩО, получают
нагреванием на воздухе соли К3[МоС1в] с KCN, обработкой калие-
вого производного оксихлорида молибдена(У) К2[МоОС15] избыт-
ком KCN и КОН, обработкой KCN тетрацианотетрагидроксомо-
либдата(1У) калия:
К3[МоС16] + 8KCN + У2Н2О + V4O2 = K4[Mo(CN)8] 4- 6КС1 + КОН
2K2IMoOC15] + 8KCN + 4КОН = K4[Mo(CN)8] + К2МоО4 + ЮКС1+2Н2О
K4[Mo(CN)4(OH)4] + 4KCN = K4[Mo(CN)8] + 4КОН
Соединение K4[Mo(CN)8] "2Н2О4 образует диамагнитные желтые
ромбоэдрические кристаллы с плотностью 1,94 г!см3, которые
растворяются в воде и спирте и плохо
растворимы в эфире. Водные раство-
ры K4[Mo(CN)8] -2Н2О имеют щелоч-
ную реакцию, не разлагаются ще-
лочами или разбавленными кисло-
тами и при обработке водными
растворами солей серебра, таллия,
меди или кадмия образуют трудно
растворимые октацианомолибдаты.
При действии конц. НС1 на
K4[Mo(CN)8] -2Н2О образуются жел-
тые игольчатые кристаллы
H4[Mo(CN)8] .6Н2О:
K4[Mo(CN)8].2H2O + 4НС1 + 4Н2О = H4[Mo(CN)8]-6Н2О + 4КС1
Получающийся в этой реакции КС1 выпадает в осадок при
добавлении спирта.
Ион [Mo(CN)8]4- устойчив и имеет структуру, изображенную
на рис. 13.
Тетрацианотетрагидроксомолибдат калия, K4[Mo(OH)4(CN)4],
получают при обработке производного К2[МоОС15] строго опре-
деленным количеством KCN (на 1 моль комплекса 4 моля KCN)
и к последовательно изменяющему окраску (коричневый, зеленый,
МОЛИБДЕН
309
синий) раствору добавляют твердый КОН для осаждения ком-
плекса.
2К2[МоОС15] + 4KCN + 8КОН =
= K4[Mo(OH)4(CN)4] + К2МоО4 + 10КС1 + 2Н2О
Соединение K4[Mo(OH)4(CN)J, окрашенное в красно-фиолето-
вый цвет, менее устойчиво, чем K4[Mo(CN)8] ‘2Н2О. При нагре-
вании или при дегидратации в вакууме (над конц. H2SO4) соеди-
нения K4[Mo(OH)4(CN)4], вероятно, образуется соединение
K4[MoO2(CN)4] синего цвета, а при действии избытка цианида
калия на K4[Mo(OH)4(CN)4] получают K4[Mo(CN)8]’2Н2О.
Помимо описанных выше известны и другие соединения четырехвалент-
ного молибдена, например MoSe2, МоТе2, K3[Mo(OH)3(H2O)(CN)4].jiH2O,
Me2[Mo(NCS)e], МоОС2О4.nH2O, MejMoO4 и др.
Соединения пятивалентного молибдена
Для молибдена пятивалентное состояние не характерно,
и известно лишь немного соединений (особенно ковалентных)
пятивалентного молибдена. К простым соединениям относятся
Мо2О5, МоО(ОН)3, MoF5, МоС15 и Mo2S5.
Растворы соединений молибдена(У) получают из соединений
молибдена(У1) путем контролируемого электролитического вос-
становления или восстановлением иодистоводородной кислотой
в кислой среде.
Соединения молибдена(У) обладают коричневой, красной, фио-
летовой, зеленой или черной окраской.
Пятиокись молибдена, Мо2Оз, получается в виде темно-фио-
летового порошка при дегидратации гидроокиси молибдена
МоО(ОН)3 в токе газообразного СО2 (или азота), а также разло-
жением соединений Mo2O(SO4)2, Мо2О(С2О4)2 в токе азота:
2МоО(ОН)3 = Мо2О5 + ЗН2О
Mo2O(SO4)2 = Мо2О5 + 2SO2
Мо2О(С2О4)3 = Мо2О5 4СО
Гидроокись молибдена, МоО(ОН)3, получают дегидратацией
коричневого осадка Мо(ОН)5, полученного обработкой раствора
соединений молибдена(У) аммиаком или ацетатом натрия. Это
красновато-коричневое вещество, плохо растворимое в воде и рас-
творимое в кислотах, гидроокисях и карбонатах щелочных
металлов.
Пентафторид молибдена, MoF5, получают действием фтора
на гексакарбонил молибдена Мо(СО)в при —75°.
Пентафторид молибдена представляет собой оливково-зеленое
твердое вещество, плавящееся при 77° с превращением в желтую
жидкость с т. кип. 215,6°.
310
ПОБОЧНАЯ ПОДГРУППА VI ГРУППЫ
Оксифторид молибдена, Mo2O3F4. При пропускании HF над
нагретым до 300—400° МоО3 образуется красный сублимат
Mo2O3F4, который синеет на воздухе; он плохо растворим в воде,
а при растворении в плавиковой кислоте образует бесцветный
раствор.
Известны оксипентафторомолибдаты общей формулы
Me*[MoOF5] «Н2О зеленого цвета, где Me1 = К+, NH+, Tl+, x/2Zn2+,
V2Gu2+.
Пентахлорид молибдена, МоС15, получают действием сухого
хлора (в отсутствие кислорода и воды) на свежевосстановленный
порошкообразный металлический молибден, нагретый до 650—
700°, а также действием; раствор а хлора в СС14 на МоО3 при 240°
в запаянной стеклянной трубке:
Мо + 5/2С12 = МоС15 + 90,8 ккал
Во влажной атмосфере, насыщенной кислородом, образуется
не МоС15, а оксихлориды ‘МоОСЗз, МоО2С12.
Соединение МоС16 представляет собой расплывающиеся на воз-
духе черные кристаллы (похожие на кристаллы сублимирован-
ного иода) с плотностью 2,927 г/сж3;
они плавятся при 194°, превращаясь
У/п х. в черную жидкость, которая кипит
ПРИ 268° с выделением темно-крас-
О С1 ных паР0В-
Пентахлорид молибдена очищают
перегонкой в атмосфере хлора или
Рис. 14.5Структура1МоС15. двуокиси углерода.
В кристалле МоС15 пять атомов
хлора (рис. 14) расположены в вер-
шинах треугольной бипирамиды и находятся на равном рас-
стоянии от центрального атома молибдена; расстояние Мо — С1
равно 2,27 А.
Пентахлорид молибдена парамагнитен, растворяется в без-
водном эфире (образуя красновато-коричневый раствор), спиртах,
кетонах, альдегидах, аминах, углеводородах; он гидролизуется
при соприкосновении с водой или во влажном воздухе, превра-
щаясь в оксихлорид молибдена МоОС13, а при нагревании в сухом
воздухе превращается в МоО2С12.
МоС16 + Н2О МоОС13 + 2НС1
Для предотвращения гидролиза МоС15 хранят в герметичных
склянках или в запаянных стеклянных ампулах.
При хранении на воздухе кристаллы МоС15 разрушаются, ста-
новятся серыми, затем расплываются в жидкость, выделяют НС1
и цвет последовательно меняется через желтый, зеленый до синего.
МОЛИБДЕН
311
При нагревании водород восстанавливает пары МоС15 по урав-
нениям
250°
* МоС1з+2НС1
900°
МоС15 + 5/гН2 *" МоЦ-5НС1
Пропусканием нагретой смеси паров МоС15 с водородом над
стальными деталями осуществляется цементация стали молиб-
деном.
При сильном нагревании МоС13 в атмосфере инертного газа
образуются МоС13 и хлор:
МоС15 -> МоС13 + С12
Пентахлорид молибдена может быть восстановлен при 350°
металлическим молибденом по уравнению
12МоСУ+ 18Мо> 5[МовС13]С14
Азотная и концентрированная серная кислоты окисляют МоС15
до молибденовой кислоты:
ЗМоС15 + HNO3 + 10Н20 = ЗН2МоО4 + 15НС1 + NO
2МоС15 +(H2SO4 + 6Н2О;= 2Н2МоО4 + 10НС1 + SO2
Известны аддукты: МоС15-РОС13, МоС15«РС15, M0CI5 >9NH3,
M0CI5-2(С2Н5)2О и др.
Пентахлорид молибдена является катализатором в реакциях
Фриделя — Крафтса и в реакциях галогенирования.
Оксипентахлоромолибдат калия, К2[МоОС15]. Если добавить
КС1 и пропускать ток газообразного НС1 через солянокислый
раствор молибденовой кислоты Н2МоО4«Н2О, к которому была
добавлена HI, образуется зеленый осадок К2[МоОС15]; последний
растворяется в воде с гидролизом:
К2[МоОС15] + Н2О ^К[МоОС14(Н2О)]+ КС1
К2[МоОС15] + ЗН2О МоО(ОН)3 + 2КС1 + ЗНС1
Соединение К2[МоОС15] можно также получить электролизом
солянокислых растворов молибдата калия, используя в качестве
катода платиновую чернь.
Известны также оксихлороаквомолибдаты общей формулы
МеЧМоОС14(Н2О)], МеЧМоОгСЦНгО)^, где Me1 = К+, Rb+, Cs+,
NH+, CSH6NH+, C9H7NH+ и др. Это соли зеленого цвета, гидро-
лизуются во влажном воздухе или при соприкосновении с водой.
Аддукты оксихлорида молибдена (V).
МоОС13 «2(С2Н5)2О получают действием влажного этилового эфира
на пентахлорид молибдена. Он образует зеленые игольчатые кри-
сталлы. MoOCl3«C5H5N также представляет собой зеленые кри-
сталлы.
312 ПОБОЧНАЯ ПОДГРУППА VI ГРУППЫ
Производные оксибромида молибдена (V).
Известны производные оксибромида молибдена(У) общей формулы
Mei[MoOBr5] желтого цвета и Ме1[МоОВг4] красного цвета, где
Me1 = NH+, Li+, К+, Rb+, Cs+, ^Са2*, V2Mg2+, C5H5NH+,
C9H7NH+.
Пентасулъфид молибдена, Mo2S5'3H2O. При пропускании H2S
через раствор, полученный восстановлением молибдата аммония
цинком в сернокислой среде, получают коричневый осадок
Mo2S5-3H2O, который растворим в сильных кислотах или раство-
рах щелочей и полностью дегидратируется до Mo2S5 при нагре-
вании в атмосфере СО2.
Октацианомолибдаты (V). При анодном окислении
или окислении перманганатом калия в кислой среде соединения
K4[Mo(GN)8] получают октацианомолибдат(У) калия K3[Mo(CN)8]—
соль красного цвета. Известен также комплекс Ag3[Mo(GN)8],
который под действием соляной кислоты превращается
в H3[Mo(GN)8j -ЗН2О. Последний неустойчив и разлагается на све-
ту (как и соль калия).
Комплексы молибден a(V), содержащие
координированную тиоцианатную группу.
При обработке производных Mei[MoOGls] различными тиоциана-
тами можно получить соединения общей формулы Me*[MoO(NCS)5l
зеленого цвета или Me*[MoO2(NGS)3] «пН2О красного цвета. Были
получены кислоты H2[MoO(NCS)5], H2[MoO2(NCS)3] «2Н2О,
H4[Mo2O3(NGS)8] и плохо растворимые соли этих кислот с различ-
ными органическими основаниями.
Помимо приведенных выше известны и другие соединения молибде-
na(V), например Mo2Se5, Mei[MoO2(C2O4)(H2O)2], Н[МоО2(С2О4)(Н2О)2],
(NH4)2[MoOC15], MejMoO*.
Соединения шестивалентного молибдена
Известно очень много устойчивых соединений электроположи-
тельного шестивалентного молибдена. Большинство из них
являются координационными соединениями.
К соединениям молибдена высшей степени окисления отно-
сятся трехокись (или молибденовый ангидрид) МоО3, гексафторид
MoF6 (единственный галогенид), трисульфид MoS3, триселенид
MoSe3, оксигалогениды МоОХ4 (где X — фтор или хлор), МоО2Х2
(где X — фтор, хлор, бром), оксигалогеносоли МеЧМоО2Х3] -иН2О,
Ме*[МоО3Х2], Ме*[МоО2Х4]’иНгО (где X — фтор или хлор),
Me4MoOF5], Me|[MoOFe], тиосоли Me*MoS4, окситиосоли, молиб-
деновые кислоты Н2МоО4, Н2МоО4-Н2О, Н2Мо2О7 Н2[Мо4О13],
оксигалогеномолибденовые кислоты Н2[МоО3С12], Н3[МоО3Вг3],
молибдаты, изополисоединения, гетерополисоединения и пероксо-
соединения.
МОЛИБДЕН
313
Соединениям шестивалентного молибдена присуща широкая
гамма расцветок — от белого, желтого, оранжевого, красного,
коричневого до зеленого цветов.
У соединений шестивалентного молибдена (в отличие от соот-
ветствующих соединений низших степеней окисления) более
отчетливо выражены кислотные свойства и ковалентный характер
связей, в большей степени проявляется склонность к комплексо-
образованию.
Соединения шестивалентного молибдена могут быть восста-
новлены в кислой среде металлическим цинком, дихлоридом
олова, сернистым газом; его растворы последовательно окраши-
ваются в синий, зеленый, коричневый, красный и желтый цвета.
Частичным восстановлением МоО3 или Н2МоО4 были получены
молибденовые сини, которые являются нестехиометрическими
соединениями.
а-Трехокись молибдена (молибденовый ангидрид), МоО3, полу-
чают прокаливанием молибдена на воздухе при 570°, окислением
на воздухе примерно при 600° порошкообразного металлического
молибдена или окислов MoO0i90_0i97, Мо2О3,МоО2, Мо2О5, про-
каливанием на воздухе (или в токе кислорода) при 450—500°
гептамолибдата аммония (NH4)e[Mo7O24J ’4H2O или в ячейке
из нержавеющей стали при 150° молибденовой кислоты, а также
окислением двуокиси молибдена азотной кислотой или нитратом,
серебра.
Мо + 3/2О2 = МоО3 + 180,3 ккал Н2МоО4 МоО3 + Н2О
MoS2 + 7/2О2 = МоО3 + 2SO2 -|- 266,2 ккал
ЗМоО2 + 2HNO3 = ЗМоОз + 2NO + Н2О
(NH4)e[Mo7O24] 7МоО3 + 6NH3 + ЗН2О
Прокаливание гептамолибдата аммония осуществляется на воз-
духе с целью дополнительного окисления продуктов восстановле-
ния МоО3 аммиаком *. Очищенный сублимацией а-МоО3 пред-
ставляет собой слабо парамагнитные двулучепреломляющие белые
и блестящие, как снег (желтеющие при нагревании), орторомбиче-
ские кристаллы слоистой структуры с плотностью 4,69 г!см3
при 21°; т. пл. 795° (сублимируются при 740°), т. кип. 1257°, раз-
лагаются при нагревании до ~1700°. Если трехокись молибдена
содержит в коллоидном состоянии окислы молибдена низших
степеней окисления, то она окрашена в зеленовато- (или синева-
то-) белый цвет.
Очистка молибденового ангидрида осуществляется сублима-
цией в фарфоровых или кварцевых трубках (открытых с обеих
сторон) при 800—850° в течение 2—3 час.
* В процессе разложения аммонийных соединений Mo(VI) происходит
очень незначительное восстановление аммиаком шестивалентного молибдена
и в препаратах наблюдается образование следов синей.— Прим. ред.
314
ПОБОЧНАЯ ПОДГРУППА VI ГРУППЫ
Водород, окись углерода, аммиак, метан и другие углеводо-
роды при нагревании восстанавливают трехокись молибдена
до двуокиси по уравнениям
600-700°
МоО3 + Н2------► МоО2ГН2О
3MoO3+2NHs;--------> 3Mo024-3H20 + N2
400°
МоОз+СО--------» MoO2-J-CO2
700°
4MoO3 + CH4-------► 4MoO2-(-CO2 + 2H2O’
Восстановление водородом возможно до металлического молиб-
дена, а восстановление окисью^углерода или метаном — до метал-
лического молибдена, загрязненного карбидами молибдена.
Натрий, калий, магний, кальций, алюминий, металлы под-
группы церия, кремний, углерод и др. при нагревании восстанав-
ливают трехокись молибдена до металлического молибдена. Вос-
становление МоО3 щелочными металлами или магнием происходит
с большим выделением энергии и сопровождается взрывом. При
описании получения металлического молибдена рассматривались
процессы, основанные на восстановлении МоО3 кальцием, алюми-
нием, металлами цериевого ряда и углеродом. При электро-
литическом восстановлении МоО3 в НС1 получают растворы
МоС13.
Действием газообразного хлора, хлоридов MerCl (Me1 = Na+,
К+ и др.), СС14, РС15, FeCl3 на трехокись молибдена при нагрева-
нии получают оксихлорид молибдена МоО2С12. При нагревании
трехокиси молибдена в токе H2S образуется MoS2.
При нагревании трехокиси молибдена при 150—300° в токе
сухого НС1 получается соединение МоО3’2НС1 или Н2[МоО3С121,
которое сублимируется в виде белых игл, растворимых в воде;
гидролизом последних получают молибденовую кислоту Н2МоО4«
•Н2О.
Трехокись молибдена растворима в воде (1,5 г/л при 100°).
При ее растворении в избытке растворов щелочей или карбонатов
щелочных металлов получают молибдаты общей формулы
MeiMoO4 «nH2O. Из раствора МоО3 в NH4OH при разбавлении
выделяется (NH4)e[Mo7O24l *4Н2О.
При сплавлении смеси МоО3 с щелочами, карбонатами и бора-
тами щелочных металлов можно получить в зависимости от взя-
тых соотношений молибдаты или изополимолибдаты:
МоО3 + 2NaOH = Na2MoO4 + Н2О
2МоО3 + 2NaOH = Na2Mo2O7 + Н2О
ЗМоОз + 2NaOH = Na2[Mo3O10] -|- Н2О
4МоО3 + 2NaOH = Na2[Mo4O13] + Н2О
6МоОз1+ 6NaOH = Nae[MoeO2i] + ЗН2О
МОЛИБДЕН
315
7МоО3 + 6NaOH = Nae[Mo7O24] + ЗН2О
8МоО3 + 4NaOH = Na4[Mo8O2e] + 2Н2О
Трехокись молибдена взаимодействует со многими минераль-
ными и органическими кислотами. Она используется при полу-
чении металлического молибдена и его многочисленных соедине-
ний, а также ферромолибдена.
Молибденовые кислоты (гидраты трехокиси
молибдена). Известны четыре молибденовые кислоты или гидраты
молибденового ангидрида, а именно: Н2МоО4 или МоО3>Н2О,
Н2Мо2О7 * или 2МоО3-Н2О, Н2МоО4>Н2О или МоО3«2Н2О
и Н2[Мо4О131 или 4МоО3 ’H2O. Последняя существует в тримерной
форме и называется метамолибденовой.
Молибденовая кислота, Н2МоО4, представляет собой мелкие
белые кристаллы (кажущиеся гексагональными), выделяется кон-
центрированием при 40—70° водных растворов кислоты Н2МоО4 •
• Н2О.
Молибденовая кислота, Н2Мо2О7, выпадает в виде белых кри-
сталлов при нагревании (90—95°) в течение нескольких дней
1 вес. ч. Н2МоО4 с 10 вес. ч. смеси НС1 (1 об. ч.) с водой (4 об. ч.)
и несколькими каплями HNO3.
Молибденовая кислота, Н2МоО4*Н2О, получается при обра-
ботке теплых (60—70°) концентрированных растворов молибдатов
(Na2MoO4’2Н2О, К2МоО4) или гептамолибдата аммония разбав-
ленными растворами кислот (НС1, HNO3, H2SO4) до pH = 3—2
или при гидролизе соединения Н2[МоО3С12]:
Na2MoO4 + 2НС1 + Н2О = Н2МоО4 -Н2О + 2NaCl
(NH4)e[Mo7O24l + 6HNO3 + 11Н2О = 7Н2МоО4-Н2О -f- 6NH4NO3
Н2[МоО3С12] + 2Н2О Н2МоО4.Н2О + 2НС1
Кислота Н2МоО4-Н2О существует в виде двух модификаций —
одна лимонно-желтая, плохо растворимая в воде, другая —
голубовато-белая, растворимая в воде. Обе модификации раство-
ряются в водных растворах щелочей или концентрированных кис-
лотах. В зависимости от взятых соотношений молибденовой кис-
лоты и щелочи или кислоты можно получить молибдаты щелочных
металлов, изополисоединения и гетерополисоединения.
При хранении Н2МоО4-Н2О в эксикаторе над конц. H2SO4
образуется Н2МоО4, который во влажном воздухе вновь гидра-
тируется.
Водный раствор растворимой в воде модификации Н2МоО4-Н2О
при 40° полностью прозрачен, а через несколько дней начинает
опалесцировать, что связано с выпадением Н2МоО4.
Молибденовая кислота, Н2[Мо4О431,— сильная кислота, она
образуется в растворе при подкислении водных растворов молиб-
* Вопрос о существовании димолибденовой кислоты является предметом
научной дискуссии.— Прим. ред.
316
ПОБОЧНАЯ ПОДГРУППА VI ГРУППЫ
датой (до pH = 2) минеральной кислотой (например, НС1). Сле-
дует отметить, что в более кислых растворах образуется не
Н2[Мо4О13], а МоО2С12.
Предполагается также существование додекамолибденовой (или
метамолибденовой) кислоты Нв[Н2Мо12О401.
Молибдаты, изополимолибдаты и гетеро-
полимолибдаты. При растворении трехокиси молибдена
МоО3 в растворах щелочей или карбонатов щелочных металлов
образуются простые молибдаты щелочных металлов типа Ме*МоО4,
где Me1 = Na+, К+:
МоО3 + 2NaOH = Na2MoO4 + Н2О
МоО3 + 2Na2CO3 + Н2О = Na2MoO4 + 2NaHCO3
При добавлении отдельными порциями минеральных кислот
к растворам простых молибдатов, полученных растворением
трехокиси молибдена в растворах молибдатов щелочных металлов,,
можно получить за счет'конденсации различные изополисоедине-
ния молибдена, состав которых зависит от условий реакции —
концентрации, температуры, pH, времени.
Наиболее устойчивыми анионами изополисоединений молиб-
дена являются [МовО21]6-, [HMoeO21]s-, [Н2МовО21]4-, [Н3МовО21]3",
[Мо7О24]6 _ или [МоМовО24]в-, [НМо7О24]5~, [Мо4О13]2-, [Мо8О2в]4-,
[Н2Мо8О26]2-, [Н2Мо12О40)в- *.
В щелочных растворах с pH > 6,5 устойчивы простые молиб-
даты, содержащие анион МоО2-; при pH == 6,5—5,5 простые-
молибдаты находятся в равновесии с гексамолибдатами (анионы
которых упоминались выше); при pH = 5,5—4 в равновесии
находятся гексамолибдаты и гептамолибдаты; при pH = 4—1,2
в равновесии находятся гексамолибдаты с октамолибдатами, геп-
тамолибдаты с тетрамолибдатами и октамолибдаты с тетрамолиб-
датами; при pH = 1,25 устойчивы додекамолибдаты (соответ-
ственно додекамолибденовая кислота) и при pH ~ 1 возможно-
образование соединений с радикалом МоО2+, а при pH ~ 0,9'
выпадает трехокись молибдена.
Конденсация в кислой среде аниона МоО2- в гекса-, гепта-,
тетра- или молибдат-анионы может осуществиться по следующим
уравнениям:
6МоО2- + 6Н+ [MoeO21]e- + ЗН2О
7МоОГ + 8Н + [Мо7О24]в- + 4Н2О
4МоО|- + 6Н+ [Мо4О13]2- + ЗН2О
* В отечественной научной литературе (в случае Мо и W) полиионы,
выделяемые из водных растворов, называются аквополиионами. Поли-
соединения, полученные сплавлением МоО3 с окислами металлов, называются
изополисоединениями.— Прим. ред.
МОЛИБДЕН
317
3[Мо7О24]6- 4[Мо4О13]2- + 5МоО2-
Разбавл. раствор
'8МоО|- + 12Н+ [Мо8О2в]4- + 6Н2О 2[Мо4О13]2- [Мо8 026]4-
Конц. раствор
[Мо6О21]в- + МоО2- + 2Н+ [Мо7О24]в- + Н2О
[НМо6О21]6- + МоО*- + Н+ =₽* [Мо7О24]в- + Н2О
[НМо6О21]5- + 2МоО1~ + 5Н+ [Мо8О2в]<- + ЗН2О
При снижении pH и увеличении концентрации раствора гекса-
молибденовой или тетрамолибденовой (соответственно октамолиб-
деновой) кислоты можно получить додекамолибденовую кислоту.
Помимо названий, данных изополисоединениям по количеству
атомов молибдена в соответствующей молекуле, применяют такие
названия, как парамолибдат для гекса- и гептамолибдатов и мета-
молибдаты для додекамолибдатов. Классификация изополисоеди-
нений (изополикислот, изополисолей) молибдена осуществляется
по количеству атомов молибдена в анионе соответствующего
соединения.
При подкислении смеси растворов двух или нескольких про-
стых солей, например Na2MoO4 Na2SiO4 или Na2MoO4 Ц-
-)-Na2HPO4, или при смешивании кислот этих солей (взятых
в определенных соотношениях) можно получить гетерополисоеди-
нения ряда 12; 11; 9; 8г/2; 6; 5; 3; 21/2. Наиболее многочисленные
и устойчивые соединения принадлежат ряду 12 и 6.
По номенклатурной системе гетерополисоединений сначала
называют центральный анион (комплексообразователь), например
кремне-, мышьяко-, теллуро-, иодо- и др., затем число и природу
координационных групп (например, H4[SiMo12O40l "reH2O — крем-
не-12-молибденовая кислота) либо название гетерополисоединений
начинают с греческого числительного, которое показывает соот-
ношение между координационными группами и центральным
атомом комплекса, затем следует название координированных групп
и название простого аниона или соответствующей центральному
атому кислоты соответствующей степени окисления (например,
H4[SiMo12O40] -пН2О — 12-молибдокремневая кислота).
Ниже приводятся несколько реакций получения 12-гетеро-
полимолибдатов :
12Na2MoO4 + Na2SiO3 + 22HNO3 = Na4[SiMo12O40] + 22NaNO3 + 11H2O
12Na2MoO4 + H3PO4 + 21HNO3 = Na3[PMo12O40] + 21NaNO3 + 12H2O
12Na2MoO4 + H3AsO4’+[21HNO3 = Na3[AsMo12O40] + 21NaNO3 + 12H2O
Все 12-гетерополимолибдаты образуются в виде желтых
осадков.
Известно много 6- и 12-гетерополисоединений, которые содер-
жат комплексные анионы с молибденом и кислородом, и значи-
тельно меньше содержащих теллур, фосфор, мышьяк, кремний,
318
ПОБОЧНАЯ ПОДГРУППА VI ГРУППЫ
германий, церий и других кислотообразователей. Несколь-
ко примеров 6- и 12-гетерополисоединений с молибденом:
Кв[ТеМовО24] -7Н2О, (NH4)6[TeMo8O24l-7Н2О, Н3[РМо12О401 •
• пН2О, H4[SiMo12O40] -пНгО, Ме|г [РМо12О40]2-58Н2О (где Me11 =
= Mg2+, Са2+, Sr2+, Ва2+, Zn2+, Mn2+, Со2+, Ni2+), Меп[РМо12О40]2.
• 48Н2О (где Me11 = Са2+, Sr2+, Ва2+, Cd2+, Mn2+, Ni2+, Со2+);
Me«[SiMo12O40] -31Н2О (где Me11 = Cu2+, Mg2+, Zn2+, Mn2+, Ni2+,
Co2+), Meir[SiMoj2O40b24H2O (где Me11 = Ca2+, Sr2+, Ba2+),
H4[GeMo12O40] ’nH2O, Me* [GeMo12O42], H2Mej[CeMo12O42] (где
MeI=NHt).
6-Г етерополисоединения (кислоты или их соли)
имеют анионы кольцевой структуры, состоящей из шести окта-
эдров МоОв (координационное число молибдена шесть), располо-
женных таким образом, чтобы каждый октаэдр имел два общих
с соседними ребра. В кольце
шести октаэдров МоОв каждый
атом молибдена окружен дву-
мя атомами кислорода, не свя-
занными с другими атомами
молибдена, и четырьмя атома-
ми кислорода, которые явля-
ются общими с двумя соседни-
ми октаэдрами. Кольца шести
октаэдров МоО8 содержат шесть
атомов молибдена с 12 атомами
кислорода, которыми связыва-
ются октаэдры между собой, и
12 атомами кислорода, не свя-
О О
Рис. 15. Структура иона занными с другими атомами
[ТеМо6О241в-. молибдена, т. е. всего на шесть
атомов молибдена приходятся
24 атома кислорода и таким образом достигается конфигура-
ция Мо8О*2". В кольце, образованном из шести октаэдров
МоОв, внутреннее пространство имеет точную форму и величину
одного октаэдра МоОв, в центре которого помещается атом
комплексообразователя (например, Те6+) с координационным
числом шесть.
Существует несколько вариантов расположения шести октаэд-
ров в кольцо таким образом, чтобы в центре образовался седьмой
октаэдр. Андерсон на основе экспериментальных данных доказал,
что анион [ТеМо8О24]6- соли К8[ТеМо8О24] -7Н2О имеет структуру,
изображенную на рис. 15. Эванс доказал, что этот же анион соли
(NH)e[TeMoeO24] -6Н2О имеет ту же самую структуру.
Когда в модели Андерсона центральным атомом является
Мов+, получают структуру аниона [МоМо8О24]6-, в которой центры
всех шести октаэдров кольца не находятся в одной плоскости.
МОЛИБДЕН
319
Центры октаэдров 1, 2, 3 и 4 образуют прямоугольник, на котором
расположены остальные три октаэдра таким образом, чтобы окта-
эдр 5 имел грани, общие с 1, 3 и 6, октаэдр 7 был связан с окта-
эдрами 2, 4, 6 общими гранями и октаэдр 6 имел бы общие грани
со всеми шестью соседями. 12-Гетерополисоединения имеют струк-
туру тетраэдра, окруженного 12 октаэдрами МоОв, расположен-
ными четырьмя группами по три октаэдра. Центральный атом
тетраэдра окружен четырьмя атомами кислорода, которые рас-
положены в вершинах тетраэдра и каждый из них является общим
для группы, состоящей из трех октаэдров МоОв. Все три октаэдра
Рис. 16. Структура аниона [РМо12О40]
в
МоОв каждой группы расположены таким образом, чтобы иметь-
один общий кислород (который совпадает с кислородом тетраэдра)
(рис. 16) и чтобы каждый октаэдр имел по два кислорода, общих
с каждым из соседних октаэдров.
В анионе [РМо^СЦо!3- соли Каз[РМо12О40] по модели Кеггина,
предсказанной Полингом, фосфор, расположенный в центре тетра-
эдра, окружен 12 октаэдрами, сгруппированными в четыре группы,,
содержащие по три октаэдра. Атомы молибдена расположены
симметрично вокруг атома фосфора, находящегося в центре куба,
у которого атомы молибдена занимают середину сторон (рис. 16, а).
Каждый атом молибдена (например, 2) окружен четырьмя ближай-
шими соседями (1, 3, 4, 5). Каждый из четырех кислородов тетра-
эдра (рис. 16, а) находится на равном расстоянии от трех атомов
молибдена (1, 2, 3). Группа из трех атомов молибдена располо-
жена таким образом (рис. 16, б), чтобы каждый октаэдр имел одно
ребро, общее с каждым соседом, и каждый кислород тетраэдра
РО3_ был общим с тремя октаэдрами МоО6. Если иметь в виду
атомы кислорода, общие трем октаэдрам, двум октаэдрам, а также-
не связанные с другими, то можно установить, что общее число-
кислородных атомов комплексного аниона равно 40:
320
ПОБОЧНАЯ ПОДГРУППА VI ГРУППЫ
Атомы кислорода, общие трем октаэдрам 12 X х/з = 4
Атомы кислорода, общие двум октаэдрам 12 X 4 X г/2 = 24
Атомы кислорода, не связанные с другими = 12
Всего 40
Октаэдры МоОв являются несколько искаженными и располо-
жены симметрично вокруг центрального атома.
Из рис. 16, в видно, что атом кислорода — общий тетраэдру
и трем октаэдрам, атомы кислорода а3 принадлежат двум окта-
эдрам, не относящимся к этой группе из трех октаэдров, атомы
кислорода а2 являются общими двум октаэдрам той же группы,
а кислород й4 изолирован.
Для установления структуры и типа ионной диссоциации гете-
рополисоединений молибдена (и других элементов) применяют
различные физико-химические методы, как-то: кондуктометриче-
ский, потенциометрический, полярографический, хроматографи-
ческий, рентгенографический метод меченых атомов.
Изополикислоты молибдена кристаллизуются с различным
числом молекул воды, растворимы в воде, спирте, эфире, ацетоне,
образуют трудно растворимые соли с различными органическими
основаниями, алкалоидами и др., при обработке щелочами пре-
вращаются в простые молибдаты.
Гетерополикислоты молибдена кристаллизуются с различным
числом молекул воды, обладают большим молекулярным весом,
растворимы в воде и различных органических растворителях,
осаждают белки, образуют плохо растворимые соединения с рас-
творами солей серебра, свинца, бария, стронция, кальция, цезия
или с различными органическими аминами, алкалоидами и др.
Характерное свойство гетерополикислот молибдена (а также
вольфрама и ванадия) — диссоциация нескольких ионов водорода
с почти одинаковой константой диссоциации, например:
Н3[РМо12О40] + ЗН2О [РМо12О40]з- + ЗН3О+
H4[SiMo12O40] + 4Н2О [SiMoi2O40]4- + 4Н3О+
12-Гетерополимолибдаты обладают окислительно-восстанови-
тельным потенциалом, примерно равным потенциалу хромовой
кислоты; они окисляют соли железа(П), сульфиты, гидрохинон.
Восстановленные формы, окрашенные в синий цвет, сохраняют
то же соотношение между центральным атомом и координирован-
ными группами и могут быть вновь окислены до первоначального
состояния перманганатом калия, перекисью водорода, бромной
водой и другими окислителями.
Ниже приводятся несколько примеров комплексных ионов
из различных рядов гетерополимолибдатов, содержащих фосфор
в качестве центрального атома: [РМо12О40]3-, [РМоиОзд]7-,
1Р2Мо18Ов21в~, [Р2Мо17Ов1]10-, [Р2Мо5О231в-, [НР2Мо5О23]5-.
МОЛИБДЕН
321
12-Гетерополисоединения молибдена могут превращаться в не-
насыщенные гетерополисоединения* 11; 9; 8V2; 2х/2 в результате
деструкции при увеличении pH и, наоборот, при подкислении
из гетерополисоединений 21/2; 5; 8L/2; 9; 11 можно получить 12-ге-
терополисоединения или соединения с катионом МоО**.
Молибдат натрия, Na2MoO4 *2H2O, выделяется при концен-
трировании раствора, полученного обработкой при нагревании
трехокиси молибдена избытком раствора NaOH или Na2CO3 или
обработкой водой продукта сплавления концентрата молибденита
MoS2 с Na2CO3 либо вульфенита РЬМоО4 с Na2CO3 и коксом, а так-
же продукта прокаливания вульфенита с NaNO3 при 700°:
МоО3 -i- 2NaOH = Na2MoO4 + Н2О; МоО3 + Na2CO3 = Na2MoO4 + СО2
MoS2 + 3Na2CO3 + 9/2О2 = Na2MoO4 + 2Na2SO4 + 3CO2
PbMoO4 + Na2CO3 + C = Na2MoO4 + Pb + CO + CO2
PbMoO4 + 2NaNO3 = Na2MoO4 + PbO + 2NO2 + i/2O2
Соединение Na2MoO4‘2H2O образует бесцветные ромбические
пластинки, которые плавятся при 687° и легко растворяются в во-
де. Водный раствор молибдата натрия применяют для определения
в кислой среде анионов РО9_, AsO9-, SiO|_ в виде гетерополи-
соединений.
Известен также кристаллогидрат Na2MoO4*10H2O.
При дегидратации кристаллогидратов Na2MoO4 -пН2О, где
п = 2 или 10, получают триболюминесцентные октаэдрические
кристаллы Na2MoO4 с плотностью 3,28 г/см3 при 18°.
Молибдат калия, К2МоО4, получают при сплавлении МоО3
с избытком К2СО3. Он представляет собой расплывающийся
на воздухе белый порошок с плотностью 2,91 г/см3 при 18°, т. пл.
926°, плохо растворимый в спирте и растворимый в воде. Известен
кристаллогидрат К2МоО4-5Н2О.
При обработке гидроксиламином смеси растворов К2МоО4
и KCN получают K4[Mo(CN)8], который выделяют из водного
раствора в виде желтых кристаллов K4[Mo(CN)8] -2Н2О:
К2МоО4 + 8KCN + 2NH2 — ОН = K4[Mo(CN)8] + 6КОН + N2
Если берут небольшое количество KCN, получают соединение
K4[MoO2(GN)4] фиолетового цвета. При обработке октацианомолиб-
дата(1У) калия конц. НС1 образуются желтые иглы H4[Mo(CN)8] •
. 6Н2О.
При обработке молибдатов щелочных металлов (в растворе,
содержащем в качестве буфера ацетат щелочного металла) 5%-ным
раствором 8-оксихинолина в ледяной уксусной кислоте получают
желтый осадок оксината молибдена MoO2(C9H7NO).
* Например, так называемые соединения И ряда.— Прим. ред.
21—0101
322
ПОБОЧНАЯ ПОДГРУППА VI ГРУППЫ
Молибдаты щелочных металлов образуют трудно растворимые
соединения с цинхонином Ci9H22ON2, о-дианизидином (3,3'-диме-
токсибензидином) H2N-(СНзО^вНз •СвНз(СНзО)-NH2, акридином
C13H9N, ацетилацетоном, основными красителями — производны-
ми трифенилметана.
Молибдат аммония, (NH4)2MoO4, выпадает при добавлении
спирта к сильно аммиачным растворам трехокиси молибдена.
Он образует белые кристаллы, которые на свету синеют вслед-
ствие восстановления шестивалентного молибдена.
Молибдат кальция, СаМоО4, получают нагреванием трехокиси
молибдена с окисью или карбонатом кальция:
МоОз + СаО = СаМоО4 МоО3 + СаСО3 = СаМоО4 + СО2
Молибдат кальция представляет собой белый порошок с огра-
ниченной растворимостью в воде, разлагающийся под действием
кислот или щелочей; применяется для полуке-
ды ния ферромолибдена.
Гидраты СаМоО4-Н2О или СаМо04-2Н20
получают обработкой молибдатов щелочных
металлов хлоридом или гидроокисью кальция:
jig Na2MoO4 4- СаС12 = СаМоО4 + 2NaCl
жЦ Na2MoO4 + Са(ОН)2 = СаМоО4 + 2NaOH
Молибдаты PbMoO4, Hg2MoO4,
CuMoO4, FeMoO4, Fe2(MoO4)3 трудно раство-
римы в воде и разлагаются при нагревании и
Рис. 17. Структура П0Д Действием кислот или щелочей. Они обра-
аниона [Мо2О7]2-. зуются при сливании растворов молибдатов
щелочных металлов или молибдата аммония с
растворами солей свинца, ртути(1), меди(П), железа(П), же-
леза (HI).
Известны и другие молибдаты, например ВеМоО4*2Н2О,
MgMoO4-nH2O (п =2, 5, 7), ВаМоО4-ЗН2О, Т12МоО4, Ag2MoO4,
Th(MoO4)2, а также молибдаты пиридина, этиламина, анти-
пирина и др.
Димолибдат натрия, Na2Mo2O7-6Н2О, выделяется в виде
бесцветных кристаллов при упаривании (30°) водного раствора
NaOH (2 моля) и Н2МоО4-Н2О (1,4—1,5 моля).
Анион [Мо2О7]2- имеет сложную конечную структуру, состоя-
щую из тетраэдров и октаэдров, имеющих определенные общие
атомы кислорода (рис. 17).
Димолибдат аммония, (NH4)2Mo2O7-Н2О, получают обработ-
кой разбавленным раствором аммиака соединения (NH4)2[MoO2F4k
2(NH4)2[MoO2F4] + 6NH4OH = (NH4)2Mo2O,-Н2О + 8NH4F + 2H2O
МОЛИБДЕН
323
Соединение (NH4)2Mo2O7 -Н2О представляет собой бесцветные
моноклинные кристаллы, которые при 105° превращаются в геп-
тамолибдат аммония (NH4)6[Mo7O24]-4H2O. Из водных растворов
димолибдата аммония также выпадают кристаллы гептамолибдата
аммония.
Известны димолибдаты Li2Mo2O7, К2Мо2О7, Rb2Mo2O7.
Тримолибдат натрия, Na2[Mo3O10] -7Н2О, выпадает в виде
бесцветных игл из раствора, полученного подкислением НС1
до кислой реакции (по метилоранжу) раствора нейтрального
молибдата натрия, или из раствора, содержащего 3 моля МоО3
и 2 моля NaOH.
Тримолибдат калия, К2[Мо3О10]-ЗН2О, выпадает в виде бес-
цветных моноклинных кристаллов, плохо растворимых в холод-
ной воде. Его получают упариванием раствора, содержащего
3 моля МоО3 и 2 моля КОН.
Известны и другие тримолибдаты, например (NH4)2[Mo3O10] •
• 2Н2О, Mg[Mo3O10]-10H2O, Са[Мо3О10]-6Н2О, Ва[Мо3О10]-ЗН2О.
Тетрамолибдат натрия, Na2[Mo4O13]-пН2О, где п = 7 или 6.
При добавлении спирта к раствору, содержащему 4 моля
МоО3 на 2 моля NaOH, выпадают бесцветные призматические
иглы Na2[Mo4O13] -7Н2О, которые легко теряют одну молекулу
воды.
Известны также тетрамолибдаты: К2[Мо4О13] -ЗН2О,
(NH4)2 [Мо4О13] -пН2О (где 3 > п > 2), Са[Мо4О13] -9Н2О,
Ва[Мо4О13] ’пН2О (где п =5 или 9), Т12[Мо4О13]-Н2О.
Гептамолибдат (парамолибдат) аммония, (NH4)elMo7O24l •
• 4Н2О или (NH4)e[MoMo6O24]-4Н2О, выпадает при упаривании
раствора димолибдата аммония или раствора молибденового
ангидрида в избытке аммиака.
Соединение (NH4)e[MoMoeO24] -4Н2О представляет собой бес-
цветные моноклинные призмы, растворимые в воде и разлагаю-
щиеся при нагревании по уравнению
110° 200°
4(NH4)6[Mo7O24].4H2O — - 4(NH4)6[Mo7O24]
280-500°
2SM«O,
Его используют для качественного и количественного определения
фосфорной кислоты.
Гетерополисоединения молибдена) VI), которые содержат
в качестве координированных групп радикалы органических кислот,
образующие две координационные связи
(хелатные соединения)
При комплексирующем действии концентрированных раство-
ров кислот щавелевой Н2С2О4-2Н2О, гликолевой (оксиуксусной)
НО — СН2 — СООН, молочной СН3 — СНОН — СООН, мин-
21*
324
ПОБОЧНАЯ ПОДГРУППА VI ГРУППЫ
дальной (оксифенилуксусной) J С6Н6СНОН -СООН, галловой
(НО)3С6С2— СООН-Н2О, салициловой НО-СООН, винной
НООС - СНОН - СНОН - СООН, яблочной НООС - СНОН —
— СН2 — СООН, лимонной С6Н8О7 и др. на водные растворы
простых или конденсированных молибдатов получают гетеро-
полисоединения молибдена, которые содержат анионы, образую-
щиеся при замещении одного атома кислорода в радикалах МоО2-,
Мо2О2- или двух атомов кислорода в радикале МоО2- радикалами
упомянутых кислот, которые присоединяются по двум координа-
ционным связям.
Большинство этих соединений содержит кристаллизационную
воду и катионы Na+, К+, NH+, Ag+, Ва2+.
В твердом состоянии были получены три типа оксалатомолиб-
датов, а именно:
а) Ме*[МоО3(С2О4)], где Me* = Li+, Ag+
Mej[MoO3(C2O4)]-nH2O, где Me* = Na+, К+, NHj, г/2Ва2+
Ме|Н[МоО3(С2Н4)] • гаН2О, где Me* = Li+, K+, NH+
анионы которых имеют строение
-H-о /О-СО-
XMoZ |
-Об) 7 II хо-со
о
б) Ме*1Мо2О6(С2О4)] (где Me1 = Li+, Na+, К+), напри-
мер (C9H8N)2 [МоО2 (С2О4)21 — оксалатомолибдат хинолина,
в) (C10H10N)2[MoO2(C2O4)2] — оксалатомолибдат хинальдина,
(C2iH23N2)2[MoO2(C2O4)2] — оксалатомолибдат стрихнина.
Реакциями нейтрализации и осаждения было доказано суще-
ствование в водном растворе оксалатомолибдатов, в анионе кото-
рых соотношение Мо : С2О4- равно 1:1, 1:2, 1 : 4 и 1 : 6.
Наибольшей устойчивостью обладают оксалатомолибдаты,
в анионе которых соотношение Мо : С2О2- равно 1 : 1 или 1 : 2.
В водном растворе между оксалатомолибдатами общих фор-
мул Ме*[МоО3(С2О4)] и Ме*[МоО2(С2О4)2] существует равновесие:
-Н20
Ме*[МоОз(С2О4)] + Н2С2О4 Ме’[МоО2(С2О4)2]
+Н2О
Известны гликолятомолибдаты Ме^[МоО2(ОСН2СОО)2], лакта-
томолибдаты Ме*[МоО2(С3Н4О3)2], манделатомолибдаты
Me* [ МоО з (С8Н6О 3) ], гал латомо либдаты Me* [МоО 2 {С6Н 2 (ОН) 2ОСО 2} 2 ],
салицилатомолибдаты Ме*Н[МоО3(С6Н4ОСОО)],
Ме*[МоО2(С6Н4ОСОО)2], тартратомолибдаты Ме*[МоО3(С4О6Н4)],
Ме*[МоО2(С4О6Н4)2] -nH2O, Ме*[Мо2О6(С4О6Н4)], малатомолибда-
ты Ме*[МоО2(С4Н4О5)2]-nH2O, Ме*[Мо2Ое(С4Н4О5)1, цитратомо-
МОЛИБДЕН
325
либдаты Ме*[МоО2(С6Н6О7)2], где Me1 = Na+, К+, NH* и реже
^гВа2*.
При обработке молибдатов полифенолами, особенно пирокате-
хином С6Н4(ОН)2, образуются хелатные соединения красного
цвета, которые могут иметь следующее строение:
'-оч У,. ,0 _/Ч"| оч -о 1 /О _/ч
Me* -О' >Мо/ 11 О О и -М или Me* 1 L L\/ О' )Мо( 1 -о X) :и
Пероксосоединения
Известны пероксомолибденовые кислоты Н2МоО5-3/2Н2О или
Н2[МоО3(О2)] ^/гНгО, Н2МоО6 или Н21МоО2(О2)2], Н2МоО8 или
Н2[Мо(О2)4] и пероксомолибдаты типа Ме*МоО5, Ме*[МоО2(О2)2] •
• пН2О, Ме*Н[МоО2(О2)2], Ме*[Мо(О2)4], Ме*[МоО(О2)3] -пН2О,
Ме*[Мо2О(О2)5] -нН2О.
Монопероксомолибденовая кислота, Н2МоО5-3/2Н2О, в виде
желтого трудно растворимого в эфире порошка образуется при
концентрировании раствора, полученного обработкой пероксомо-
либдата бария ВаМоО5 серной кислотой или раствора молибде-
новой кислоты Н2МоО4.Н2О перекисью водорода:
ВаМоО5 ф H2SO4 = H2MoOs ф BaSO4
Н2МоО4 ф Н2О2 “ H2MoOs ф Н2О ф 8,08 ккал
Перекись водорода превращает желтую монопероксомолибде-
новую кислоту в оранжевую дипероксомолибденовую кислоту
Н2[МоО2(О2)2]:
H2MoOs ф Н2О2 — Н2[МоО2(О2)2] ф Н2О ф 4,30 ккал
Тетрапероксомолибденовую кислоту, Н2[Мо(О2)4], получают
действием избытка перекиси водорода на молибденовые кислоты
Н2МоО4, Н2Мо2О7, Н2МоО4’Н2О. Соединение Н2[Мо(О2)4] окра-
шено в красный цвет, оно очень хорошо растворимо в воде, в сухом
виде взрывает.
При добавлении перекиси водорода к бесцветным растворам
молибдатов образуются пероксомолибдаты, которые в кислой
среде устойчивы (оранжево-желтого цвета), а в щелочной —
неустойчивы (темно-красного цвета).
При упаривании в вакууме водного раствора молибдата аммо-
ния с конц. NH4OH и Н2О2 получают неустойчивый дипероксомо-
либдат аммония, (NH4)2[MoO2(O2)2], красного цвета:
(NH4)2MoO4 ф 2Н2О2 = (NH4)2[MoO2(O2)2] ф 2Н2О
Обработкой при —18° сильно аммиачного раствора молибдата
аммония пергидролем (30%-ной перекисью водорода) и КС1 полу-
326
ПОБОЧНАЯ ПОДГРУППА VI ГРУППЫ
чают красные кристаллы тетрапероксомолибдата калия,
К2[Мо(О2)4], который при нагревании или ударе детонирует,
а при комнатной температуре разлагается.
Тетрапероксомолибдат натрия, Na2MoO8 -яНгО (сп = 2, 3, 5),
можно получить по методу, аналогичному получению соли калия,
или действием 30%-ной Н2О2 на насыщенный раствор Na2MoO4
при 0°. Соль Na2[Mo(O2)4bnH2O представляет собой вещество
красного цвета, которое взрывается при нагревании или ударе,
а при комнатной температуре превращается; в желтую соль
Na2[MoO2(O2)2].
Обработкой перекисью водорода гептамолибдата аммония
в кислой среде получают раствор пероксопроизводного
(NH4)2 [МоО(О2)3] -2Н2О, из которого упариванием при 45° выде-
ляют (NH4)2MoO5.
Монопероксомолибдат бария, ВаМоО5, получают обработкой
соединения (NH4)2MoO5 хлоридом бария.
Фторопероксомолибдаты. Известны фтороперо-
ксомолибдаты типа Mei[MoO(O2)F4]-Н2О (где Me1 = К+, Rb+,
Cs+), представляющие собой желтые устойчивые соли, теряю-
щие при 150° кислород и превращающиеся в оксифторосоли
Me*[MoO2F4] -Н2О.
При добавлении спирта к раствору, полученному действием
перекиси водорода на фторомолибдат калия, выпадает фторопер-
оксомолибдат K2[MoO2(O2)F2] -H2O.
Оксалатопероксомолибдаты. Известны окса-
латопероксомолибдаты типа Н2[МоО2(О2)2] •Ме*С2О4-nH2O, где
Me1 = К+, NHt, 1/2Ва2+, МеИМоО(О2)2(С2О4)1, где Mel = К+,
NHJ.
Гексафторид молибдена, MoFe, получают прямым воздействием
фтора на металлический молибден, помещенный в платиновую
трубку и нагретый до 60°, действием разбавленного азотом фтора
на нагретый до 350° металлический молибден, действием трифто-
рида брома BrF3 на металлический молибден или [МовВг8]Вг4
при 350°, действием сухого фтористого водорода на [Мо6Вг8]Вг4
при 800—860°, действием фтора на гексакарбонил молибдена
при 50—60°:
Мо -[- 3F2 = MoFe [Мо8Вг8]Вг4 -ф 12BrF3 = 6MoFe -ф 12Вг2
Мо -ф 2BrF3 = MoFe -ф Вг2 Мо(СО)в -ф 3F2 = MoF8 -ф 6C0'j
[Мо8Вг8]Вг4 -ф 36HF ='46MoF8 -ф 12НВгфф 12Н2
На холоду MoFe представляет собой бесцветные кристаллы,
которые, плавясь при 17,5°, превращаются в летучую жидкость
с плотностью 2,55 г!смъ и т. кип. 36°; ее очищают перегонкой
в вакууме при 40°.
В гексафториде молибдена шесть атомов фтора расположены
в вершинах октаэдра, в центре которого находится атом молибдена.
МОЛИБДЕН
327
Гексафторид молибдена растворяется в аммиаке и щелочах
и гидролизуется в воде по уравнениям
MoFe + Н2О MoOF4 + 2HF MoFe + 2Н2О MoO2F2 + 4HF
MoFe + 5H2O & H2MoO4-H2O + 6HF
Фториды щелочных металлов образуют с MoF6 фторомолибдаты
общей формулы Me*[MoF8], где Me1 = К+, Rb+, Cs+.
Окситетрафторид молибдена, MoOF4, получают обработкой
сухим фтористым водородом окситетрахлорида молибдена МоОС14
(в платиновом приборе) сначала при охлаждении, а затем при
постепенном нагревании:
МоОС14 + 6HF = MoOF + 4НС1 + 2HF
При постепенном повышении температуры сначала выделяется
НС1, затем избыток HF и в конце перегоняется MoOF4.
Можно также получить MoOF4 действием фтора на покрытый
окислами молибден или обработкой металлического молибдена
смесью азотной и плавиковой кислот:
Мо + 4HF + 2HNO3 = MoOF4 + 2NO -f- ЗН2О
Окситетрафторид молибдена представляет собой расплываю-
щиеся на воздухе бесцветные кристаллы с плотностью 3,00 г!см\
т. пл. 97° и т. кип. 180°; он растворяется с разложением в воде,
спирте, эфире и хлороформе.
MoOF4 + 4Н2О Н2МоО4-Н2О + 4HF
При упаривании бесцветного водного раствора MoOF4 выпа-
дают гидраты трехокиси молибдена.
Диоксидифторид молибдена, MoO2F2, получают действием
сухого фтористого водорода на МоО2С12 либо нагреванием МоО3
с PbF2 или Na3[AlFe],
Соединение MoO2F2 получается в виде расплывающихся бес-
цветных кристаллов с плотностью 3,494 г/см2, сублимирующихся
при 270°, гидролизующихся в воде и растворяющихся в AsCl3,
SiCl4, SO2C12.
Оксифторомолибдаты. При растворении молибда-
тов в плавиковой кислоте или при пропускании фтора в фтористо-
водородный раствор трехокиси молибдена получают оксифторо-
молибдаты. Последние растворимы в воде, хорошо кристалли-
зуются, в большинстве бесцветны, устойчивы на воздухе при
обычной температуре, при нагревании во влажном воздухе теряют
фтор в виде фтористоводородной кислоты.
Ниже приведены общие формулы оксифторомолибдатов:
Me*[MoO3F2], Me*[MoO3F3], Me4MoO2F3] -nH2O, Me*[MoO2F4] •
• nH2O, Men [MoO2FJ-6Н2О, Me4MoOFB] и Me*[MoOFe], где
Mei = NHJ, К+, Rb+, Tl+, x/2Zn2+, V2Ca2+, ^Со2*, r/2Ni2+. Изве-
328
ПОБОЧНАЯ ПОДГРУППА VI ГРУППЫ
стны в основном соли аммония и калия, отвечающие этим общим
формулам.
Окситетрахлорид молибдена, МоОС14, представляет собой рас-
плывающиеся зеленые кристаллы, сублимирующиеся ниже 100°;
образуются при действии паров РС15 на МоО2С12.
Диоксидихлорид молибдена, МоО2С12, получают пропусканием
хлора над нагретым до 500—700° МоО2, нагреванием МоС15 на
воздухе, действием смеси хлора и кислорода на металлический
молибден, нагретый без доступа влаги.
Соединение МоО2С12 образует желтовато-белые кристаллы
с плотностью 3,31 г!см? при 17°, легко сублимирующиеся, раство-
римые в воде, спирте, эфире, ацетоне и превращающиеся под дей-
ствием безводной фтористоводородной кислоты в MoO2F2.
При действии трехокиси серы на МоО2С12, растворенный
в SO2C12, получают бесцветное твердое вещество MoO(SO4)2,
которое гидролизуется в воде.
Оксихлоромолибденовая кислота, Н2[МоО3С12], с эмпирической
формулой МоО3 -2НС1 образуется при пропускании безводного
газообразного НС1 над нагретой до 150—250° трехокисью молиб-
дена или при действии НС1 на какой-либо молибдат, нагретый
до 440°.
Кислота Н2[МоО3С12] образует расплывающиеся белые иголь-
чатые кристаллы, которые диссоциируют при 160° и растворяются
в воде, эфире и ацетоне.
При концентрировании водного раствора Н2[МоО3С12] выпа-
дает Н2МоО4-Н2О. При упаривании в вакууме эфирного раствора
Н2[МоО3С12] выпадают белые кристаллы Н2[МоО3С12] «2(С2Н5)2О,
которые плавятся при 75° и разлагаются при 90°.
Оксихлоромолибдаты. Известно несколько бесцвет-
ных оксихлоромолибдатов щелочных металлов, которые гидро-
лизуются в воде; их состав отвечает общим формулам
МеЧМоО2С13] -nH2O, Mei[MoO2Cl4bnH2O, МеЧМо3О6С17]-пН2О.
Диоксидибромид молибдена, МоО2Вг2, получается в виде оран-
жево-красных кристаллов при действии паров брома на двуокись
молибдена или при действии на трехокись молибдена смеси В2О3
с КВг при нагревании
МоО3 -J- В2О3 + 2КВг = МоО2Вг2 + 2КВО2
Оксибромомолибденовая кислота, Н3[МоО3Вг3], с эмпириче-
ской формулой МоО3 -ЗНВг представляет собой желтые игольча-
тые кристаллы, растворимые в воде и образующиеся при действии
газообразного НВг на МоО3.
Трисулъфид молибдена, MoS3, образуется при действии серо-
водорода на кислые (солянокислые, сернокислые, муравьино-
МОЛИБДЕН
329
кислые) растворы молибдатов при нагревании *, при разложении
тиомолибдата кислотой или при обработке кислотой (на водяной
бане) двойного сульфида молибдена(VI) и пиперазина:
К2МоО4 + 2НС1 + 3H2S = MoS3 + 2КС1 + 4Н2О
(NH4)2MoS4 + H2so4 = M0S3 + (NH4)2SO4 + H2S
Трисульфид молибдена представляет собой черно-коричневый
порошок; он плохо растворим в воде, растворяется в щелочах,
сульфидах и полисульфидах щелочных металлов с образованием
тио-, тиоокси- и политиомолибдатов, разлагается при нагревании
по уравнению
MoS3 MoS2 + VsSg
При нагревании водород восстанавливает трехокись молибдена
до металлического молибдена:
MoS3 + ЗН2 = Мо -J- 3H2S
Тиомолибдаты, политиомолибдаты и окси-
тиомолибдаты. Тиомолибдаты щелочных металлов
общей формулы MeiMoS4 получают при растворении MoS3 в суль-
фидах или полисульфидах щелочных металлов и при сплавлении
МоО3 со смесью карбоната щелочного металла, серы и угля.
Тиомолибдаты Me|MoS4, где Me1 = К+, NH4, Gs+, Na+, окра-
шены в красный цвет, плохо растворяются в спирте и эфире, легко
растворяются в воде и разлагаются при подкислении до pH = 6
с выделением MoS3.
Известен также тиомолибдат K6Mo2S9 в виде оранжево-красных
кристаллов, медленно разлагающихся до K2MoS4:
K6Mo2S9 = 2K2MoS4 + K2S
Тиомолибдаты щелочноземельных металлов в отличие от тио-
молибдатов щелочных металлов образуются только сухим путем;
они окрашены в оранжевый цвет, их состав отвечает эмпирической
формуле 3MoS3 -MenS.
Политиомолибдат калия, K2MoS5, образует красные призмы,
которые разлагаются в сухом воздухе, плохо растворяются в хо-
лодной и растворяются в теплой воде (30—40°). При обработке
водного раствора K2MoS5 уксусной кислотой выпадает кислота
H2MoS5, а при обработке соляной кислотой выпадает тетрасуль-
фид молибдена MoS4. Кислота H2MoS5 получается в виде корич-
нево-красного осадка, который в токе H2S при 140° разлагается,
образуя MoS4, и растворяется в щелочах. Сульфид MoS4 пред-
* Для получения MoS3 удобнее сначала насыщать раствор молибдата
сероводородом, а затем подкислять его, в противном случае выпадает молиб-
деновая синь.— Прим. ред.
330
ПОБОЧНАЯ ПОДГРУППА VI ГРУППЫ
ставляет собой коричневый осадок, который медленно окисляется
на воздухе и при растворении в сульфидах щелочных металлов
образует политиомолибдаты красного цвета.
При добавлении спирта к золотисто-желтым растворам, полу-
ченным действием сероводорода или сульфидов щелочных метал-
лов на растворы молибдатов, выпадают кристаллические осадки
окситиомолибдатов общих формул MeJMoSO3, Me*MoS2O2,
MeIH[Mo2S3O4], где Me1 = Na+, K+, NH+.
Металлоорганические соединения
Известно ограниченное число металлоорганических соединений
молибдена.
Гексакарбонил молибдена, Мо(СО)6, получают при обработке
МоС15 окисью углерода в присутствии бромида фенилмагния,
qq действием окиси углерода под дав-
Ф лением 200—500 ат на металли-
ческий молибден в присутствии
железа или меди, действием окиси
углерода при низком давлении на
00 9Мо К3[МоС16] в присутствии железа
или меди.
Соединение Мо(СО)в представ-
ляет собой двупреломляющие диа-
С0 магнитные бесцветные ромбиче-
Рис.^18. Структура'}Мо(СО)в. ские кристаллы с плотностью
1,96 г/см3, сублимирующиеся без
плавления при 40° (при 156°
давление пара составляет 1 ат). При Температуре 600° и давлении
0,1 мм рт. ст. разлагаются на металлический молибден и СО,
реагирует со спиртовым раствором КОН и окислителями (хлор,
бром и др.):
Мо(СО)е 2С12 и МоС14 -j- 6СО
Методом дифракции электронов^было установлено, что в струк-
туре гексакарбонила молибдена (рис. 18) атом молибдена распо-
ложен в центре правильного октаэдра, в вершинах которого
находятся группы СО.
При действии гексакарбонила молибдена на пиридин, о-фенан-
тролин, этилендиамин образуются соединения Мо(СО)3-SCsHsN
желтого, Mo(CO)4"C12H8N2 красного и Mo2(CO)e(H2N — С — С —
Н2 Н2
— NH2) желтого цвета.
Со спиртовым раствором КОН гексакарбонил молибдена обра-
зует соединение К2Мо(СО)5, которое под действием кислот пре-
вращается в летучее и неустойчивое соединение Н2Мо(СО)5.
МОЛИБДЕ Н
331
При действии растворенного в жидком аммиаке металлического
натрия на Мо(СО)6 получают Na2[Mo(CO)5],
В качестве металлоорганических производных молибдена
упоминаются также (С5Н5)Мо(СО)5Мо(С5Н5), С5Н5Мо(СО)3СН3,
(С5Н5)2МоХ2, (С5Н5)2Мо2(СО)в, (С5Н5)2МоХ3 (где X = кислотный
радикал), (С6Н6)2Мо, CeH6Mo(CO)3, Mo(CO)2(C5H5N)SC6H5.
Окислы молибдена, промежуточные меж-
fl у а-МоО3 и 6-МоО2. При нагревании различных смесей а-МоО3
с 6-МоО2 или с металлическим молибденом в вакууме ниже 580°
получают смесь невосстановленного а-МоО3, 6-МоО2 и у-Мо4Оц.
Если нагревание осуществляется при 600—650°, дополнительно
образуется окись р-Мо8О23, при 650—700° — окись Р'-Мо9О26;
выше 750° окись 6-МоО2 устойчива, а выше 1100° неустойчива.
Общую формулу окислов Мо4Оц, Мо8О23, Мо9О26 можно выразить
как МопОзп^, где п — 4, 8 или 9.
Окись у-Мо^и нестехиометрического состава (между МоО2>75
и МоО2>70) образует фиолетово-красные орторомбические пла-
стинки с плотностью 4,18 г/см\
Окись р-Мо8О23 представляет собой темно-синие моноклинные
кристаллы с металлическим блеском и плотностью 4,32 г!см\
Окись Р'-Мо9О26 имеет вид металлоподобных фиолетовых или
темно-синих моноклинных кристаллов с плотностью 4,26 г! смй.
Молибденовые сини. При восстановлении МоО3
или МоО3-2Н2О в водной суспензии НС1 + Zn, Мо, Al, Fe, SO2,
HI, N2H4, SnCl2, C6H12O6 получают молибденовую синь
Mo4Oi0(OH)2, не реагирующую со щелочами. При более глубоком
восстановлении получают соединение Мо5О7(ОН)8 или МоО2|20 •
• 4Н2О бордово-красного цвета, которое на воздухе окисляется
до молибденовой сини состава Мо2О4(ОН)2 — устойчивой на воз-
духе и по отношению к щелочам и превращающуюся в смесь
МоО3 с МоО2 при дегидратации. То же соединение Мо2О4(ОН)2
синего цвета можно получить и восстановлением МоО3 водородом
в момент его выделения.
При восстановлении дигидрата МоО3-2Н2О (110°) порошкооб-
разным металлическим молибденом в течение нескольких недель
получают молибденовую синь Mo8Oi5(OH)i6, которая также не реа-
гирует со щелочами.
Дигидрат трехокиси молибдена МоО3 -2Н2О при продолжи-
тельном действии дихлорида олова в солянокислом растворе пре-
вращается в молибденовую синь MoO2j60 -nH2O, которая в отли-
чие от остальных молибденовых синей взаимодействует со ще-
лочами.
При восстановлении кислых растворов соединений молибде-
на^!) соединениями молибдена(У), металлическим молибденом
или при электролитическом восстановлении образуются аморфные
молибденовые сини, которые под действием щелочей превращаются
332
ПОБОЧНАЯ ПОДГРУППА VI ГРУППЫ
в растворимый молибдат и МоО(ОН)3 — коричневый осадок,
а при нагревании — в окислы у-Мо4Оц и б-МоО2.
При контролируемом восстановлении молибдатов в 0,06 н. кис-
лоте получают растворимую молибденовую синь, которая содер-
жит анион [Мо4+Мо2+О1812~. В кислой среде (pH — 3) при частич-
ном восстановлении молибдена(У1) образуется коричнево-красный
анион [НМоз+Мо|+О18]2~, который при добавлении NaOH в при-
сутствии Мо5+ превращается в вышеупомянутый синий анион.
Если восстановление молибдатов осуществляется при pH ~ 4,
образуется анион [НМо^Мо^О^!- коричнево-желтого цвета;
он неустойчив и при понижении кислотности разлагается.
Молибденовые сини являются диамагнитными полупроводни-
ковыми соединениями, которые при полной дегидратации (320°)
превращаются в окислы молибдена состава между МоО2>6(>
и МоО2193, а в слегка подкисленной воде (pH ~ 4) образуют кол-
лоидные растворы, используемые для крашения перьев, волос,
мехов и шелка.
Соединения включения
Нитриды молибдена, MoN, Mo2N. При действии
газообразного аммиака на тонко измельченный металлический
молибден при 800° образуются соединения включения — нитриды
молибдена Mo2N, MoN. Эти твердые соединения являются провод-
никами электрического тока.
В кристаллической решетке нитрида MoN (рис. 19) атомы азота
находятся в центре треугольных призм, образованных шестью
атомами молибдена, и каждый атом молибдена окружен шестью
атомами азота.
В кубической гранецентрированной решетке нитрида Mo2N
атомы азота расположены в октаэдрических пустотах.
Фосфиды молибдена, МоР2, МоР, Мо3Р.
Фосфид молибдена, МоР2, получают нагреванием при 550°
в закрытой трубке смеси металлического молибдена с красным
р
фосфором, взятых в соотношении = 2,2. Он представляет
собой черный порошок, плохо растворимый в НС1 или аммиачном
растворе Н2О2, взаимодействующий с HNO3, царской водкой,
конц. H2SO4 при нагревании и термически диссоциирующий:
МоР2 = МоР + г/|Р4 + 10 ккал
Фосфид молибдена, МоР, получают электролизом расплавлен-
ной смеси МоО3 с метафосфорной кислотой или смеси с гексаме-
тафосфатом натрия и NaCl, а также нагреванием смеси МоО3
с метафосфорной кислотой в графитовом тигле. Он представляет
собой черные игольчатые кристаллы с плотностью 4,8 г!см9\ при
МОЛИБДЕН
333
нагревании на воздухе разлагается, не взаимодействует с НС1
и NaOH.
Фосфид молибдена, Мо3Р, получают электролизом расплава
гексаметафосфата с трехокисью молибдена и хлоридом натрия
в графитовом тигле. Это серый порошок с плотностью 6,5 г!см3,
который не горит при нагревании на воздухе и не взаимодей-
ствует с НС1 и NaOH.
Карбиды молибдена, МоС, Мо2С.
Карбид молибдена, МоС, получают нагреванием составляющих
элементов при 1200—1600° в атмосфере водорода или плавлением
молибдена в присутствии кокса.
Карбид МоС представляет собой серое вещество с металличе-
ским блеском, плотно упакованной гексагональной решеткой,
твердостью 7—8по шкале Мооса, плотностью
8,4 г! см3. Он тугоплавок (т. пл. 2700°),
хорошо проводит электрический ток (ста-
новится сверхпроводником при 9,26 °К),
инертен с химической точки зрения.
Карбид молибдена, Мо2С, получают взаи- •
модействием металлического молибдена с са-
харным углем при 1400—1500° в атмосфере
водорода в графитовой трубчатой печи,
карбидизацией трехокиси молибдена при
900—950° в восстановительной атмосфере,
содержащей НС1, а также карбидизацией
нитридов молибдена при температуре 1200°.
Карбид Мо2С представляет собой серое
Рис. 19. Структура
MoN.
Карбид Мо2С представляет собой серое вещество с плотно
упакованной гексагональной решеткой, плотностью 8,9 г1см3,
твердостью 7 по шкале Мооса и т. пл. 2310°. При 2,70° К он ста-
новится сверхпроводником, устойчив к действию НС1 и щелочей,
образует твердые растворы с карбидами титана.
Силициды молибдена, MoSi2, Mo3Si2, Mo3Si.
Дисилицид молибдена, MoSi2, получают нагреванием металли-
ческого молибдена при 1100—1800° в атмосфере SiCl4 и водорода,
прямым взаимодействием составляющих компонентов при 1400—
2600°, кремнетермическим восстановлением окислов молибдена
и алюмотермическим восстановлением смеси кремнезема и МоО2.
Силицид MoSi2 образует серые тетраэдрические кристаллы
с плотностью 6,24 г!см3, устойчивые по отношению к расплавлен-
ным металлам — Na, Bi, Sn, Ga и др. Дисилицид молибдена
можно перерабатывать спеканием или литьем; его применяют
при изготовлении управляемых снарядов, электронагревательных
элементов, электродов зажигания и т. п.
Известны также силициды молибдена Mo3Si2, Mo3Si.
Бориды молибдена, Мо2В, Мо3В2, а- и р-МоВ,
МоВ2 и Мо2В5, получают нагреванием в графитовом тигле
334
ПОБОЧНАЯ ПОДГРУППА VI ГРУППЫ
составляющих элементов при 1500—1600° в атмосфере водорода
или при нагревании металлического молибдена с В4С и В2О3 при
2000° в атмосфере водорода.
Борид молибдена, Мо2В, представляет собой серые тетраэдри-
ческие кристаллы с плотностью 9,31 г!см3 и твердостью 8—9 по
шкале Мооса. В этом бориде каждый атом бора находится в пусто-
тах между восемью атомами молибдена, которые расположены
в виде тетраэдрических пластинок.
Борид молибдена, Мо3В2, образует тетраэдрические кристаллы
с плотностью 7 г/см3, устойчивые при температуре выше 1850°.
Модификация а-МоВ имеет вид серых кубических кристаллов
с плотностью 8,77 г!см3, устойчивых до 2000°.
Модификация р-МоВ представляет собой серые орторомбиче-
ские кристаллы с плотностью 10,1—10,7 г!см3 и т. пл. 2180°;
устойчива при более высокой температуре.
Борид молибдена, Мо2В5, устойчивый при 1600°, представляет
собой ромбоэдрические кристаллы с плотностью 7,48 г/см3..
Борид молибдена, МоВ2, является метастабильным соедине-
нием гексагональной структуры; плотность 7,78 г!см3, т. пл. 2100°;
образуется по уравнению
7Мо + ЗВ4С + В2О3 - 7МоВ2 + ЗСО
ВОЛЬФРАМ W
Z = 74; ат. вес = 183,85
Валентность (II), (III), (IV), (V), VI; заряд (2Ц-),
(3+), (4+)- (5+), 6+
Массовые числа природных изотопов 184, 186,
182, 183 и 180
Электронная структура атома вольфрама: К-L-M >N •
• 5s25p65d4 -6s2.
Электронная структура атома и ионов четырех- и шестивалент-
ного вольфрама для 5d- и 6«-орбиталей:
ИСТОРИЧЕСКАЯ СПРАВКА
Красиво окрашенные вольфрамовые руды применялись в Китае
как пигменты для знаменитых фарфоров значительно раньше,
чем стал известен их химический состав и был получен металли-
ческий вольфрам.
Соли вольфрама издавна использовались в качестве огнестой-
кой пропитки тканей.
ВОЛЬФРАМ
335
Средневековые мастера плавки металлов встречались с боль-
шими трудностями при добывании олова из касситерита, посколь-
ку последнему часто сопутствовали вольфрамовые руды — черные
или серо-желтые комья, которые называли «тунгстеном», что
означает «тяжелый камень». При восстановлении касситерита
углем «тяжелые камни» образовывали пену, адсорбировавшую
часть олова. Металлург XVI в. Агрикола назвал этого «врага»
расплавленного олова «волчьей пеной»— wolfram (от слов Wolf —
волк и Rahm — пена).
В 1781 г. К. Шееле впервые получил из «тяжелых камней»,
называемых также «проклятыми камнями», вольфрамовую кисло-
ту. В 1783 г. испанцы — братья Жозе и Фаусто Д’Эльухар —
впервые выделили металлический вольфрам и определили неко-
торые его свойства. В чистом виде металлический вольфрам был
получен Вёлером в 1850 г. В 1896 г. под руководством профессора
Липина на Путиловском заводе в Петербурге было начато произ-
водство вольфрамовых сталей.
В Англии и Франции вольфрам и в настоящее время назы-
вают тунгстеном.
Вольфрам используют в электротехнике начиная с 1910 г.,
т. е. с того времени, когда был разработан металлокерамический
процесс превращения порошкообразного металла в компактный
металл. В современной технике твердые и сверхтвердые сплавы
вольфрама имеют особо важное значение.
Если в прошлом вольфрамовые руды рассматривались как
досадное загрязнение, то в настоящее время они служат источ-
ником одного из самых важных металлов современной техники.
РАСПРОСТРАНЕННОСТЬ В ПРИРОДЕ
В природе вольфрам не встречается в свободном состоянии,
а только в виде соединений (главным образом вольфраматов)
в различных минералах. Содержание вольфрама в земной коре
равно 7ПО-3 вес.%.
Основные минералы вольфрама.
Вольфрамит, (Mn, Fe)WO4, встречается в виде хрупких корич-
нево-черных моноклинных кристаллов с плотностью 6,7—7,5 г/см3
и твердостью 4,5—5,5 по шкале Мооса. В разновидности воль-
фрамита, названной ферберитом, преобладает железо, а содер-
жание марганца не превышает 20%. В разновидности гюбнерит
преобладает марганец, содержание железа ниже 20%. Залежи
вольфрамита (ферберита, гюбнерита) имеются в СССР, США,
Мексике, Бразилии, Аргентине, Китае, Таиланде, Бирме, КНДР,
Австралии, Испании, Португалии.
Шеелит, CaWO4, встречается в виде тетрагональных призма-
тических кристаллов, обычно в виде включений, реже в компакт-
336
ПОБОЧНАЯ ПОДГРУППА VI ГРУППЫ
ной массе серого цвета. В зависимости от примесей кристаллы
могут быть бесцветными или окрашены в желтый, зеленовато-
желтый, коричневый и красный цвета. Плотность кристал-
лов 5,4—6,1 г!см?, твердость 4,2—4,5 по шкале Мооса. Под
действием ультрафиолетовых лучей шеелит флюоресцирует
ярким синим цветом. Залежи шеелита находятся в США, Тасма-
нии и др.
Ферритунгстит, Fe2+(WO4)(OH)4-4Н2О,—продукт окисления
ферберита; имеет вид трубчатых гексагональных кристаллов или
чешуйчатой массы желтого или коричнево-желтого цвета.
В числе других минералов вольфрама можно назвать тунгстенит WS2,
штольцит PbWO4, распит PbWO4, чиллагит Pb(W, Мо)О4.
ОБОГАЩЕНИЕ И ПЕРЕРАБОТКА ВОЛЬФРАМОВЫХ РУД
Из-за низкого содержания вольфрама его природные руды
не могут служить непосредственным металлургическим сырьем.
Обогащение вольфрамовых руд осуществляется гравитационными,
электростатическими, электромагнитными, флотационными или
химическими методами.
Концентраты вольфрамовых минералов промывают 5—
10%-ным раствором НС1 для удаления кальция и фосфора. Чтобы
освободиться от серы, мышьяка и сурьмы, присутствующих в виде
сульфидов, арсенидов и антимонидов, концентраты шеелита про-
каливают. Серу, мышьяк и сурьму необходимо удалять из кон-
центратов вольфрамовых руд, поскольку эти примеси вредны
как для металлического вольфрама, так и для его сплавов. Про-
каливание концентратов осуществляется таким образом, чтобы
сера, мышьяк и сурьма не окислялись до сульфата, арсената
и антимоната, поскольку в этом случае после окислительного
прокаливания концентраты необходимо промывать водой.
Концентраты богатых вольфрамом руд могут непосредственно
перерабатываться в ферровольфрам. Концентраты бедных воль-
фрамом руд перерабатывают гидрометаллургическим способом
в вольфрамовую кислоту H2WO4, вольфрамовый ангидрид WO3
или вольфрамат кальция CaWO4 (синтетический шеелит), которые
используются для получения металлического вольфрама или
ферровольфрама.
Соединения вольфрама, из которых получают металлический
вольфрам, служащий материалом для нитей накаливания в
электрических лампочках и электронных трубках, должны
быть очень чистыми; общее содержание примесей (сера,
мышьяк, сурьма, фосфор, кальций и др.) не должно превы-
шать 0,06%.
ВОЛЬФРАМ
337
Переработка концентратов шеелитных руд
Концентраты шеелитных руд перерабатывают спеканием с кар-
бонатом натрия и кварцевым песком, а также обработкой при
нагревании концентрированным раствором карбоната натрия
в автоклаве или конц. НС1 в керамических сосудах либо сталь-
ных резервуарах.
Концентраты шеелитных руд размалывают в фарфоровых
мельницах с фарфоровыми или кварцевыми шарами.
При спекании порошка концентрата шеелита с карбонатом
натрия (взятого в избытке) и кварцевым песком при 800—900°
в течение 2—3 час в пламенных или вращающихся трубчатых
печах вольфрамат кальция превращается в вольфрамат натрия:
CaWO4 + Na2CO3 + SiO2 = Na2WO4 + CaSiO3 + CO2
Примеси в шеелите, такие, как сульфиды железа, мышьяка,
и молибдена, фосфат кальция, силикаты алюминия, взаимодей-
ствуют с карбонатом натрия при температуре спекания:
2FeS2 + 5Na2CO3 4- 15/2О2 = 2NaFeO» + 4Na2SO4 + 5СО2
As2S3 + 6Na2CO3 4- 7O2 = 2Na3AsO4 + 3Na2SO4 + 6CO2
MoS2 + 3Na2CO3 4- 9/2O2 = Na2MoO4 + 2Na2SO4 4- 3CO2
Ca3(PO4)2 4- 3Na2CO3 = 2Na3PO4 4- ЗСаО 4- 3CO2
При многократном промывании водой охлажденного продукта
спекания в раствор переходят вольфраматы, сульфаты, фосфаты,
арсенаты и силикаты натрия и остаются основные карбонаты тяже-
лых металлов, а также гидроокиси алюминия и железа, образо-
вавшиеся в результате гидролиза алюминатов и ферритов щелоч-
ных металлов, силикат и гидроокись кальция. Раствор вольфрама-
та натрия, свободный от соединений кремния, фосфора, мышья-
ка, молибдена, обрабатывают при нагревании 10%-ным раствором
хлорида кальция с целью осаждения вольфрамата кальция CaWO4.
Последний под действием конц. НС1 при нагревании в присут-
ствии небольшого количества HNO3 превращается в вольфрамо-
вую кислоту H2WO4, из которой путем прокаливания получают
трехокись вольфрама WO3:
CaWO4 4- 2НС1 = H2WO4 4- СаС12
I
WO3 4- H2O
Для удаления мышьяка и фосфора раствор вольфрамата
натрия обрабатывают на холоду аммиаком и хлоридом магния
с целью осаждения фосфатов и арсенатов магния и аммония.
Метасиликат натрия Na2SiO3, содержащийся в растворе воль-
фрамата натрия, гидролизуется при pH = 8—9, а образовав-
шуюся кремневую кислоту H2SiO3 отделяют фильтрованием.
При переработке концентратов шеелита с концентрированным
раствором карбоната натрия в автоклавах с механической мешал-
22 — 0101
538
ПОБОЧНАЯ ПОДГРУППА VI ГРУППЫ
кой при 200—225° и давлении 14—15 ат вольфрамат кальция
превращается в вольфрамат натрия:
CaWO4 + Na2CO3 = Na2WO4 + СаСО3
Процесс ведут с 3—4-кратным избытком карбоната натрия
по сравнению со стехиометрически необходимым количеством.
В этом случае вольфрамат натрия загрязнен значительно меньше,
поскольку сульфиды и силикаты в этих условиях меньше под-
вергаются действию карбоната натрия.
Охлаждением теплого концентрированного раствора воль-
фрамата натрия осаждают кристаллы Na2WO4-2H2O. При обра-
ботке нагретого до 80° раствора вольфрамата натрия конц. НС1
выпадает H2WO4.
При обработке концентрата шеелита избытком конц. НС1
(в керамических или стальных резервуарах) при 80—110° и нор-
мальном (или слегка повышенном) давлении и механическом
перемешивании (возможно перемешивание и сжатым воздухом)
вольфрамат кальция превращается в вольфрамовую кислоту:
CaWO4 + 2НС1 = H2WO4 + СаС12
Некоторые соединения, содержащиеся в концентрате шеелита,
такие, как СаСО3, Са3(РО4)2, FeS, Fe2O3, взаимодействуют с соля-
ной кислотой и переходят в раствор:
СаСО3 + 2НС1 = СаС12 + СО2 + Н2О FeS + 2НС1 = FeCl2 + H2S
Са3(РО4)2 + 6НС1 = ЗСаС12 + 2Н3РО4 Fe2O3 + 6НС1 = Fe2Cle + ЗН2О
Кварц, касситерит, большинство алюмосиликатов и некоторые
сульфиды не вступают в реакцию с соляной кислотой и, оставаясь
нерастворимыми, загрязняют вольфрамовую кислоту.
После промывания горячей водой загрязненную вольфрамовую
кислоту обрабатывают аммиаком, и при pH = 6,6 выделяются
кристаллы паравольфрамата аммония:
6H2WO4 + 5NH4OH = (NH4)6[HW8O21b4H2O + 4Н2О
При прокаливании паравольфрамата аммония образуется WO3
и выделяются NH3 и Н2О:
250-300°
(NH4)5[HWeO21]-4H2O------> 6WO3 + 5NH3+7H2O
При нагревании раствора паравольфрамата аммония с соляной
кислотой переосаждается чистая вольфрамовая кислота H2WO4:
(NH4)s[HW6O21] + 5НС1 + ЗН2О = 6H2WO4 + 5NH4C1
При нагревании раствора паравольфрамата аммония с едким
кали выделяется аммиак и в растворе образуется вольфрамат
калия:
(NH4)s[HWeO21J + 12КОН = 6K2WO4 + 5NH3 + 9Н2О
ВОЛЬФРАМ
339
Переработка концентратов вольфрамитных руд
Концентраты вольфрамитных руд (Fe, Mn)WO4 перерабаты-
вают путем спекания с карбонатом и нитратом натрия либо обра-
боткой 35—40%-ным раствором NaOH или КОН.
Для измельчения концентратов вольфрамита применяют сталь-
ные шаровые мельницы.
При спекании концентратов вольфрамита с карбонатом натрия
(взятым в 10—15 %-ном избытке по сравнению со стехиометрически
необходимым количеством) и NaNO3 при температуре 980—1000°
во вращающейся трубчатой железной печи образуются воль-
фрамат натрия, нитрит натрия и окислы Fe2O3, Мп3О4, СО2:
2FeWO4 + 2Na2CO3 + NaNO3 = 2Na2WO4 + NaNO2 + Fe2O3 + 2CO2
3MnWO4 + 3Na2CO3 + NaNO3 = 3Na2WO4 + NaNO2 + Mn3O4 + 3CO2
В отсутствие нитрата натрия спекание осуществляется в посто-
янном токе воздуха. Карбонат натрия при температуре спекания
взаимодействует с примесями концентратов вольфрамита, как
это было описано при переработке концентратов шеелитных руд.
При многократной обработке холодного' продукта спекания
водой образуется раствор вольфрамата натрия, загрязненный
растворимыми соединениями кремния, фосфора, мышьяка, молиб-
дена и др. При описании способа переработки концентратов
шеелита было указано, что вольфрамат натрия превращается
в вольфрамат кальция CaWO4, затем в вольфрамовую кислоту,
в паравольфрамат аммония (NH4)5[HW6O2il-4Н2О и, наконец,
в WO3. Иногда из паравольфрамата аммония получают воль-
фрамат калия K2WO4, вольфрамовую кислоту H2WO4 и в конеч-
ном итоге тот же WO3.
Ниже схематически показана переработка концентратов воль-
фрамитных руд по этому способу.
(Fe, Mn)W04 спекается с Na2CO3-|-NaNO3
Концентрат
вольфрамита
Охлажденный пек: Na2WO4, NaNO2, Fe2O3, Мп3О4-|-загрязнения, которые-
извлекаются при многократной обработке водой
Раствор вольфрамата натрия очищают от соедпненпй кремния, фосфора,
мышьяка, молибдена
ч___ >
Раствор вольфрамата натрия -> Выделение кристаллов Na,VVO4-2H;O>
+10%-ный СаС12 при нагревший
CaWO4
।
। -f-конц. НС1 при наг ре ваии
2 2*
340
ПОБОЧНАЯ ПОДГРУППА VI ГРУППЫ
Прокаливание
H2WO4--------------> WO3 + H2O
I 4-NH4OH (до pH— 6,6)
400°
(NH4)5[HW6O21].4H2O —> 6WO3+5NH3+7H2O
I +KOH
K2WO4
I Ч-конц. HCl при нагревании
Прокаливание
H2WO4--------------► wo3
(чистый)
При нагревании порошкообразных концентратов вольфрамит-
ной руды с избытком 35—40%-ного раствора NaOH или КОН
в стальных резервуарах при температуре кипения и непрерывном
перемешивании в течение 4—8 час образуются вольфрамат натрия
и гидроокись железа и марганца:
(Fe, Mn)W0* + 2NaOH Na2WO4 + (Fe, Mn)(OH)2
При разбавлении раствора водой до удвоения объема кри-
сталлизации Na2WO4-2H2O не происходит, и осаждаются Fe(OH)2
и Мп(ОН)2. Под действием кислорода воздуха происходит окис-
ление гидроокисей Fe(OH)2 и Мп(ОН)2:
2Fe(OH)2 + V2O2 + Н2О = 2Fe(OH)3
Мп(ОН)2 + ^гОг = Н2МпО3 (или МпМпО3)
Этот метод основан на том, что в процессе переработки воль-
фрамитных концентратов образуются растворимый вольфрамат
натрия, а также соединения железа и марганца ограниченной
растворимости. И в этом случае вольфрамат натрия может быть
превращен в различные соединения вольфрама.
Для выделения вольфрама из разбавленных растворов воль-
фраматов, полученных при гидрометаллургической переработке
концентратов вольфрамовых руд, могут быть использованы раз-
личные анионообменные смолы. Вольфрам можно отделить от мо-
либдена с помощью Н-катионита — крупнопористого пермутита
ES, который удерживает вольфрам и пропускает молибден.
При хлорировании концентратов вольфрамовых руд образуют-
ся летучие хлориды и оксихлориды WC16, WOC14, WO2C12, кото-
рые гидролизуются водой, давая H2WO4 и НС1. Температура
реакций зависит от природы минерала и хлорирующего агента
(Cl2, HCl, СС14, SC12 и т. д.).
ВОЛЬФРАМ
341
ПОЛУЧЕНИЕ МЕТАЛЛИЧЕСКОГО ВОЛЬФРАМА
Металлический вольфрам получают восстановлением при
нагревании трехокиси вольфрама водородом, углеродом или
окисью углерода, металлотермическим восстановлением окислов
вольфрама металлическим алюминием, цинком, кальцием или
магнием, термической диссоциацией гексахлорида вольфрама,
восстановлением при нагревании гексахлорида вольфрама водо-
родом, нагреванием трисульфида вольфрама с окисью кальция,
наконец, катодным восстановлением некоторых соединений воль-
фрама.
Восстановление трехокиси вольфрама водородом,
углеродом или окисью углерода
При восстановлении трехокиси вольфрама WO3 водородом
при 860—920° образуется порошкообразный металлический воль-
фрам:
WO3 ЗН2 —> W ЗН2О
Восстановление ведут в электрических или газовых трубчатых
печах, в которые помещают металлические лодочки с WO3. При
этом подают чистый водород и принимаются меры для макси-
мально быстрого удаления образующихся паров воды, чтобы
не допустить образования рыхлых гранул вместо порошкообраз-
ного вольфрама. С этой целью сначала нагревают печь до 480°,
затем до 760° и, наконец, до 920°.
Грануляцию порошкообразного металлического вольфрама
регулируют скоростью пропускания водорода, толщиной слоя
в лодочке, продолжительностью и температурой восстановления.
При действии водорода на трехокись вольфрама в зависимости
от температуры получают W40n, WO2 или металлический воль-
фрам:
550°
4WO3 + H2 ~Z± W4On + H2O
W4OH + 3H2 4wO2 + 3H2O WO2 + 2H2 W + 2H2O
' Вместо трехокиси вольфрама можно взять вольфрамовую
кислоту H2WO4 с добавкой 0,8—1,2% двуокиси тория ThO2 или
нитрата тория Th(NO3)4, которые препятствуют росту кристаллов
вольфрама в процессе кристаллизации.
Восстановление трехокиси вольфрама сажей осуществляется
в графитовых тиглях и протекает постадийно в зависимости
от температуры:
650-850°
4WO3 + C----------► W4OU + CO
900-1050°
W40u + 3C--------->- 4WO2 + 3CO
1200°
4WO2 + 8C---------► 4W-J-8CO
1200°
WO3 + 3C----------> W + 3CO
342
ПОБОЧНАЯ ПОДГРУППА VI ГРУППЫ
Металлический вольфрам, образующийся при восстановлении
WO3 сажей, представляет собой серый порошок, загрязненный
1—2% углерода.
Металлический вольфрам может быть получен и восстановле-
нием вольфраматов CaWO4, Na2WO4 углем (сажей) при нагрева-
нии. Был предложен способ получения металлического вольфра-
ма нагреванием смеси Na2WO4 и NH4C1 с углеродом:
1000-1150°
Na2WO4+2NH4Cl + 3C--------->- W+2NaCl + 2NH3-|-H2O + 3CO
Расплавленный хлорид натрия, оставаясь на поверхности,
предотвращает окисление металлического вольфрама.
Восстановление трехокиси вольфрама окисью углерода осу-
ществляется в электрических трубчатых печах и протекает
по этапам:
4WO3 + СО W4On + СО2
W4OH + ЗСО 4WO2 + ЗСО2
4WO2 + 8СО -+ 4W + 8СО2
WO3 + ЗСО -> W + ЗСО2 — при более высокой температуре
В результате восстановления WO3 окисью углерода при нагре-
вании также получается серый порошок металлического воль-
фрама.
Металлотермическое восстановление окислов
вольфрама
При металлотермическом восстановлении окислов вольфрама
WO3, W40n, WO2 алюминием, цинком, кальцием или магнием
получают загрязненный порошок металлического вольфрама.
Реакции, лежащие в основе этих восстановительных процессов,
протекают быстро и сопровождаются большим выделением энер-
гии. Избыток цинка и магния удаляется перегонкой или промы-
ванием разб. НС1, а избыток кальция — теплой водой. Окислы
вольфрама, не восстановленные до металлического вольфрама,
удаляются щелочами.
Металлотермическое восстановление окислов вольфрама при-
меняется главным образом для получения сплавов с вольфрамом.
Термическая диссоциация гексахлорида вольфрама
(процесс Ван-Аркеля — Лимпта)
В сосуд из стекла пирекс, снабженный двумя вольфрамовыми
крючками, соединенными тонкой вольфрамовой нитью, вводят
WC16, затем в сосуде создают вакуум и нагревают. Вольфрамовые
крючки снаружи включают в электрическую цепь, чтобы тонкая
вольфрамовая нить нагрелась до 1600°.
ВОЛЬФРАМ
343
При термической диссоциации гексахлорида вольфрама обра-
зующийся металлический вольфрам оседает на нагретой докрасна
нити:
1600°
WCle —^2 W + 3C12
Металлический вольфрам можно получить и при пропускании
смеси водорода с парами WC16 над нагретой до 1000° проволокой
из металлического вольфрама.
Получение металлического вольфрама путем нагревания
трисульфида вольфрама с окисью кальция
При нагревании трисульфида вольфрама с окисью кальция
в электрической печи получают порошкообразный металлический
вольфрам:
WS3 + 2СаО = W + 2CaS + SO2
Порошкообразный металлический вольфрам отделяют от суль-
фида кальция, последовательно промывая продукт реакции водой,
разб. НС1 и, наконец, горячей водой.
Электролитическое получение металлического
вольфрама
Металлический вольфрам получают электролизом расплавлен-
ных вольфраматов в слабо щелочной или нейтральной среде,
поскольку в кислой среде образуются вольфрамовые бронзы.
При электролизе расплавленных вольфраматов в слабо щелочной
или нейтральной среде получают кристаллический металличе-
ский вольфрам 99,5—99,9%-ной чистоты.
Для создания слабо щелочной среды в электролитическую
ванну загружают дегидратированную буру Na2B4O7, смесь пиро-
и метафосфатов натрия, небольшие количества перекиси натрия
Na2O2 или несколько большие количества вольфрамата лития
Li2WO4. Для увеличения электропроводности расплава добавляют
хлорид натрия.
Электролиз смеси из 9 ч. чистого WO3, 19 ч. Na4P2O7-10H2O,
3 ч. NanPnOsn и 9 ч. NaCl осуществляется в графитовом тигле при
900° и плотности тока на катоде 35 а!см2.
Электролиз смеси 1 ч. технического WO3 (или концентратов
вольфрамитных руд), 19 ч. Na4P2O7-10Н2О и 3,1 ч. Na„P„O3n
или смеси 1 ч. концентратов вольфрамитных руд и 1 ч. Na2B4O7
осуществляют в графитовом тигле, который служит одновременно
анодом, при температуре 1100—1300° и плотности тока на катоде
50 а!см2. Анодом может служить также тигель или несколько гра-
фитовых стержней, расположенных вокруг стального катода.
344
ПОБОЧНАЯ ПОДГРУППА VI ГРУППЫ
Примеси (Fe, Мп, Sn и пр.), источником которых являются кон-
центраты вольфрамитных руд, оседают на дно электролизера
в виде шлама.
После окончания электролиза расплав из электролизера сли-
вают в форму и порошок металлического вольфрама отделяют
от затвердевшего электролита, последовательно промывая смесь
горячей водой, разб. НС1 и затем опять водой.
В металлическом вольфраме, полученном электролитическим
методом, в качестве примесей содержатся соединения кремния,
молибдена, щелочных металлов и кислород.
ПОЛУЧЕНИЕ ФЕРРОВОЛЬФРАМА
Для получения ферровольфрама, представляющего собой сплав
железо — вольфрам (75—85% вольфрама), служат окислы воль-
фрама WO3, WO2, синтетический шеелит CaWO4 или концентраты
вольфрамовых руд, которые содержат 55—65% WO3, 3—5% SiO2,
0,03-0,08% Р, 0,03-0,08% S, 0,04-0,2% As, 0,10-0,22% Си,
2-4,5% Мо.
Восстановление смеси вольфрамата кальция и окиси железа
Fe2O3 углем (1700—1750°) проводят в электродуговых печах
и получают ферровольфрам, загрязненный карбидом вольфрама
W2C:
CaWO4 + Fe2O3 + 6С = Fe2W + СаО + 6СО
Ферровольфрам может быть получен восстановлением при
нагревании смеси окислов вольфрама и железа и фторида каль-
ция металлическим алюминием или кремнием (а также ферроси-
лицием). Практически восстановление смеси окиси или окислов
вольфрама (из которых состоит концентрат вольфрамитных руд)
с окисью или окислами железа и фторидом кальция осуществляет-
ся смесью порошкообразного алюминия и ферросилиция (25 : 75)
в специальном тигле.
Зажигательной смесью служит порошок алюминия и перекись
бария.
При восстановлении одним алюминием происходят потери
вольфрама, так как образуется ферровольфрам, загрязненный
марганцем и кремнием, и реакция протекает очень быстро. С ука-
занной восстановительной смесью реакция восстановления идет
медленнее и уменьшается переход примесей в ферровольфрам.
В лабораторной практике для восстановления берут смесь
из 100 г концентрата вольфрамовых руд (в которых содержится
55—65% WO3), 6 г Fe3O4 и 6 г CaF2, куда добавляют 15 г порош-
кообразного алюминия и 9 г ферросилиция. Полученный ферро-
вольфрам содержит в качестве примесей 0,4—0,8% Si, 0,1— 0,2%
Al, 0,05-0,1% С и 0,3-0,5% Мп.
ВОЛЬФРАМ
345»
Ферровольфрам получают также электролизом расплавленной
смеси безводной буры, фторида натрия, окислов вольфрама и же-
леза.
Превращение порошкообразного металлического
вольфрама в компактный металл
Порошкообразный металлический вольфрам может быть пре-
вращен в компактный металл путем спекания (металлокерамиче-
ский процесс), плавления в электрической дуге или в сфокусиро-
ванном пучке электронов.
Порошковая металлургия (металлокерамический метод) позво-
ляет получить вольфрам в виде слитков — кубиков или брусков
(основание 450 X16 мм). При этом порошкообразный металличе-
ский вольфрам прессуют в стальных матрицах на гидравлическом
прессе под давлением 1,5—4«103 кг!см2. Спрессованные слитки
спекают в два этапа — при 1300—1400° и затем при 3000—3200°
в атмосфере водорода или инертного газа. Нагревание осуще-
ствляется постепенно и спекание длится примерно 30 мин.
После спекания хрупкие бруски вольфрама подвергаются ков-
ке на круглых кузнечных молотах при температурах 1400—1500е
и 1800°. При этом из брусков диаметром 16 мм получают прутки
толщиной 5 мм, диаметр которых при повторной ковке на кузнеч-
ных молотах может быть доведен до 1 мм.
Для получения монокристаллов металлического вольфрама
по методу Пинша порошок металлического вольфрама (частицы
0,5 мк) смешивают с органическим связующим и пропускают под
давлением через алмазную фильеру. Полученную нить нагревают
до 2200°, пропуская через индукционную печь, находящуюся
в атмосфере водорода, что способствует росту крупных частиц.
Медленное протягивание нити позволяет получить монокристалл
длиной несколько метров за счет укрупнения маленьких частиц.
При плавлении порошкообразного металлического вольфрама
в электрической дуге получают крупнокристаллический металл,
который растрескивается при переработке.
При плавлении порошка вольфрама с помощью сфокусирован-
ного электронного пучка получают металлический вольфрам высо-
кой чистоты с хорошими физико-химическими свойствами.
ФИЗИЧЕСКИЕ И ХИМИЧЕСКИЕ СВОЙСТВА
В компактном состоянии вольфрам — блестящий металл сере-
бристо-белого цвета, с объемно-центрированной кубической кри-
сталлической решеткой для модификации a-W и кубической
решеткой специального типа (рис. 20) для модификации |3-W.
Модификация |3-W пирофорна и получается при нагревании моди-
фикации a-W до 600—700°. Порошок металлического вольфрама
346
ПОБОЧНАЯ ПОДГРУППА VI ГРУППЫ
имеет серый цвет, а коллоидный раствор — черно-коричневый
цвет в проходящем свете и черный — в отраженном. Коллоидный
вольфрам образуется в электрической дуге между двумя вольфра-
мовыми проволоками, опущенными в изобутиловый спирт или
при чередующейся обработке тонкого порошка металлического
вольфрама 15%-ной НС1 и 15%-ным NaOH. Гидрозоль вольфрама
малоустойчив и всегда заряжен отрицательно.
Вольфрам — тяжелый металл (плотность 19,32 гкм3 при 20°);
твердость его 4,5 по шкале Мооса, он тугоплавок (т. пл. 3380°,
т. кип. 5930°), обладает малой упругостью пара, большой элек-
тронной эмиссией и способностью к
светоизлучению в раскаленном состоя-
нии. Теплоемкость вольфрама 0,0321
кал/г-град при 20°, он парамагнитен.
Электро- и теплопроводность вольфрама
меньше, чем у меди, и значительно
больше, чем у железа (стали) или ни-
келя.
Металлический вольфрам в ком-
пактном состоянии обладает прекрас-
ными механическими свойствами, но
Рис. 20. Структура 0-W. труднее поддается переработке под дав-
лением, чем, например, молибден. Ме-
таллический вольфрам, полученный в компактном состоянии метал-
локерамическим методом, ковок, может прокатываться в листы тол-
щиной 0,003 мм и протягиваться в нити диаметром меньше
0,010 мм. Вольфрамовая нить диаметром 0,007 мм, используемая
для изготовления спиралей в электрических лампочках, может
быть изготовлена из проволоки диаметром 0,010 мм путем
обработки ее различными расплавленными солями, например
K3lFe(CN)6], (NH4)2S2O8 или смесью NaNO3 и KNO3.
Известны многочисленные сплавы вольфрама с Be, В, А1,
С, Si, РЬ, Ti, Zr, Hf, N, P, As, Sb, Bi, V, Ta, S, Se, Cr, Mo, U,
Re, Fe, Co, Ni, Pt, Zn, Hg и другими металлами.
К интерметаллическим соединениям вольфрама относятся:
Be9W, W2B, WB, A14W, W2C, WC, W3Si„ WSi2, W2Zr, W2N,
WP, WP2, WAs2, WS2, WS3, WSe2, WSe3, CrW, W2Re3, Fe2W,
Fe7W6, Co3W, Co7W6, Ni4W, PtW.
С химической точки зрения металлический вольфрам устойчив
в обычных условиях на воздухе, а при нагревании до 900° на воз-
духе (или в кислороде) превращается в WO3. Тонкодисперсный
порошок металлического вольфрама пирофорен.
При комнатной температуре металлический вольфрам не реа-
гирует с водой, но при температуре выше 600° пары воды дей-
ствуют на металл:
W + 2Н2О WO2 + 2Н2
вольфрам 347
Металлический вольфрам взаимодействует с фтором при обыч-
ной температуре, с хлором при 250—300°, с бромом при 800°
и с иодом при 950°:
w + 3F2 = WFe + 422 ккал W + 6/2Вг2 = WBrs + 60 ккал
W + ЗС12 = WCle + 163,1 ккал 6W + 6I2 = [W6I8]I4 + 96 ккал
Взаимодействие галогенов с металлическим вольфрамом на
воздухе (кислород, влага) приводит к образованию не галогени-
дов, а оксигалогенидов, например WOC14.
Нагреванием металлического вольфрама с серой при темпера-
туре ~880° получают дисульфид вольфрама WS2, который под
действием паров серы превращается в WS3.
При нагревании металлический вольфрам взаимодействует
с селеном, теллуром и азотом:
750°
W+2Te--------► WTe2
в вакууме
2000-2500°
*W + N2 --------> wn2
При нагревании порошка металлического вольфрама в атмосфе-
ре газообразного аммиака образуется нитрид W2N (плотность
6,3 г/см3), который разлагается водой или при нагревании.
Взаимодействуя с парами фосфора при 700—900°, металличе-
ский вольфрам превращается в WP2, а при 900—1000° — в WP.
При 800—1500° порошкообразный металлический вольфрам
реагирует с сажей, тонкодисперсным графитом, окисью углерода
или углеводородами (СН4, С2Н2, C6He, C10Hi2) с образованием кар-
бидов W2C, WC. При загрязнении углеродом вольфрам становится
твердым, но хрупким и не поддается механической обработке.
Силициды вольфрама WSi2, W3Si2 и его бориды W2B, WB,
W2B5 получают нагреванием металлического вольфрама с крем-
нием или бором при 1000—2000°.
Порошкообразный металлический вольфрам растворяется при
80—100° в конц. HNO3, царской водке и смеси концентрированных
HNO3 и HF.
2W + 4HNO3 + 10HF = WF6 + WOF4 + 4NO + 7Н2О
Окислительно-щелочные расплавы (КОН + KNO3, КОН +
+ КС1О3 и Na2CO3 + NaNO3) растворяют металлический воль-
фрам с образованием вольфраматов:
W + 2КОН + 3KNO3 = K2WO4 + 3KNO2 + Н2О
Горячие концентрированные минеральные кислоты (НС1,
HNO3, H2SO4) действуют на вольфрам очень медленно. Растворы
348
ПОБОЧНАЯ ПОДГРУППА VI ГРУППЫ
хлорида меди(П) или хлорида железа(Ш) вызывают коррозию»
вольфрама.
Будучи нагретым до 570—650°, металлический вольфрам
устойчив к действию ртути, натрия, калия, галлия, магния и дру-
гих металлов.
Вольфрам является восстановителем по отношению к окислам
РЬО, СнО, А12О3, ZrO2, ThO2, которые он восстанавливает при
нагревании, а также по отношению к ионам Ag+, Au3+ в аммиач-
ных растворах и но отношению к ионам Hg2+, Pt4+, Pb4+, Pd4+
в солянокислых растворах, которые восстанавливаются им до
Hg22+, Pt2+, Pd2+.
Химическая активность вольфрама иллюстрируется следую-
щей схемой:
Комн. темп.
с фтором -»- WFe
192
Вольфрам
Нагревание
с кислородом или на воздухеWO3
с парами bohh-»-WO2
с Cl2, Br2, I2-»-WCle, WBr5, [WeI8]I4
с S, Se, Te^WS,, WS3, WSe2, WTe2
c N2, P, С, углеводородами-»-WN2, WP, \VP2,
W2C, wc
c Si, B^WSi2, W3Si2, W2B, WB, W2B5
c HNO3 -J-HF^ WF6 -i-WOF4
с расплавленной смесью KOH-J-KNO3->-K2WO4
, с газообразным NH3 ->}W2N
С физиологической точки зрения вольфрам очень мало токси-
чен и не вызывает отравлений.
ПРИМЕНЕНИЕ
Значительная часть мирового производства вольфрама (в виде
ферровольфрама) используется для получения высококачествен-
ных сталей. Вольфрамсодержащие стали характеризуются боль-
шой прочностью на истирание, устойчивостью к высоким темпе-
ратурам и химическим реагентам, хорошей пластичностью и упру-
гостью, большой твердостью (до относительно высоких температур)
и способностью к самозакаливанию, благодаря которой сталь
сохраняет свои превосходные механические свойства до 500—
600°.
Вольфрамсодержащие стали разделяются на быстрорежущие
(16—20% W), инструментальные (12—19% W), немагнитные,
броневые, оружейные и конструкционные (0,15—2% W).
В состав вольфрамсодержащих сталей входят главным образом
элементы Fe, W, Сг, V, Мо, Ni, Со, С, Si.
Сплав 90% W, 6% Ni и 4% Си, названный «тяжелым сплавом»,
имеет плотность 16,5—18,5 г! см*. перерабатывается легче, чем
металлический вольфрам, мало проницаем для различного рода
ВОЛЬФРАМ
349
излучений (служит для изготовления экранов против у-лучей).
Сплав вольфрам — уран (также относящийся к «тяжелым спла-
вам») обладает высокой плотностью и очень малой проницаемо-
стью для у-лучей.
В современной технике очень важны сверхтвердые сплавы,
в которых вольфрам присутствует в виде карбида. Сверхтвердые
сплавы делятся на металлокерамические и литые.
Металлокерамические сплавы содержат 88—90% W и обра-
зованы спеканием порошков моно- или поликарбидов. Для полу-
чения металлокерамических сплавов монокарбидного типа тонкий
порошок карбида вольфрама с диаметром частиц 2—3 мк смеши-
вают с 3—15% порошкообразного цементирующего металла
(кобальт или никель) и с раствором каучука в качестве связую-
щего. Смесь прессуют и спекают при 1500°. Эти сплавы («побе-
дит» в СССР, «видна» в ФРГ, «карболой» в США и др.) применяют
для обработки металлов резанием, для изготовления фильер
(используемых при вытягивании проволоки), штампов, инстру-
ментов и буров.
Металлические сплавы поликарбидного типа содержат 5—30%
карбида титана, 4—10% металлического кобальта, остальное —
карбид вольфрама. Эти сплавы термостойки, тверды и более
хрупки, чем сплавы монокарбидного типа; они служат для изго-
товления инструментов для резания легированных сталей.
Полученные литьем сплавы различных карбидов (содержащие
W. Со, Сг) наносят на рабочие поверхности деталей, подвергаю-
щихся большому износу (режущие поверхности бура, плуга
и т. д.).
При впрессовывании алмазной крошки в массу, содержащую
кристаллы карбида вольфрама и карбида кобальта, получают
материалы, используемые как «алмазы» для резки стекла, шлифо-
вальные и полировальные инструменты.
Металлический вольфрам применяют в виде пластинок и про-
водов в электро- и радиотехнике, в виде спиралей в электронных
трубках и электрических лампочках, а также в виде трубок,
стержней, пластинок и лент нагревательных элементов в элек-
тропечах и в электровакуумных приборах. Вольфрам служит так-
же для изготовления электроконтактов и электродов, используе-
мых при сварке с атомарным водородом. Вольфрамовые сверх-
чистые электроды служат для сварки в авто- и авиапромышлен-
ности.
Металлический вольфрам используют для изготовления анодов
к рентгеновским трубкам.
Измерение температур в интервале 1700—2500° осуществляется
термопарами W — Мо (1700°), W — С (графит) (1800—1900°),
W — Nb (2000°), W — Re (2500°) в вакууме, водороде, азоте,
аргоне или в других инертных атмосферах.
Соедине- ние Оки- слы Гидроокиси или кислоты И их соли Фториды, оксифториды, фторосоеди- нения Хлориды, оксихлориды, хлоросоеди- нения Бромиды, оксиброми- ды, бромо- соединения
Соединения ВОЛЬ- фрама(П) — — — [W6C18]C14 серый [WeBr8]Br4 черно- синий
Соединения воль- фрама(Ш) — —_
Соединения воль- фрамату) wo2 ко- рич- невая — WF4 красно- коричне- вый wof2 WC14 серый —
Соединения воль- фрама^) WC15 зеле- ный WB г5 черно- коричне- вый
Соединения воль- фрама^!) wo3 жел- тая H2WO4 оранжево- желтая H2WO4-H2O белая H2W2O7 белая MeiWO4-nH2O MejW2O7 MelW3O10 MeiW4O13 MeiW5OJe H5[BW12O40].nH2O H4[SiW12O40]-raH2O H4[GeW)2O40] • nH2O H,[PW12O40]-nH2O H2[WO3(O2)] MeI[W(O2)4]-raH2O Mei[W2O3(O2)4].«H2O WFe бес- цветный WOF4 бес- цветный Mei[WO2F3] Mei[WO2F4] Mei[WO3F3] WCld фио- летово- синий WOC14 красный WO2C12 желтый WBrb черно' синий WOBr4 черно- коричне- вый WO2Br2 оранжево- красный
Таблица 43
Йодиды, оксииодиды, иодосоеди- нения Сульфиды, тиосоли, селениды, туллуриды Координационные соединения Соединения включения
[Wglgllj коричне- вый — H2[W6Cl8Cle]-8H2O Mei[W6Cl8Cle]-nH2O H2[WeBr8Cl6].2H2O Mei[W6Br8Cle]-nH2O C5HeN)2[WeBr8Cle] WN2 черно- коричне- вый W2N корич- невый WP2 чер- ный WP серый WAs2 чер- ный W2C серый WC серый WSi2 серо- синий W2B серый WB серый W2B5 серый
H3[W2C19] зеленый или желтый Mei[W2Cl9] зеленовато-желтый Mei[W2Clio] красный Mei[WF4]-nH2O (C5H5N)3[W2CleJ (C6H5NH2)[W2Cle] K3[W2Br9]
WI4 чер- ный WS2 черный WSe2 серый WTe2 серый Mei[W(CN)8]-nH2O H4[W(CN)8]-6H2O K4[W(OH)3(CN)5].6H2O K2[W(OH)C15] K2[W(NCS)e]
Mei[WOCl5]-nH2O Mei[WOCl4]-nH2O Mei[WOBr5] Mei[WOBr4] Mei[W(CN)8]-nH2O H3[W(CN)8].6H2O
WS3 коричневый MeiWS4 MeiWS3O-H2O MeiWS2O2-H2O MeiWSO3-H2O жел- тые WSe3 черный Mei[HWeO21]-nH2O MliH3WeO21].nH2O Mei[H2W]204o]-nH20 Mei0[W.2O41] MeilW-Aj] Mei[BW12O40]-nH2O Mei[SiW1204o]-nH20 Mei[GeW1204o]-raH20 Mei[P W t204o] nH2O Mei[AsW)2O40]-nH2O Mei[CoHWi2O40]-nH2O MeiHfCoin W12O40] • nH2O и гетеросоединения воль- фрама с органическими кис- лотами
352
ПОБОЧНАЯ ПОДГРУППА VI ГРУППЫ
Из листового вольфрама изготовляют лодочки, в которых
производят испарение веществ в высоком вакууме.
Благодаря высокой электропроводности и малому коэффи-
циенту термического расширения металлический вольфрам при-
меняют для сварки с твердыми стеклами.
Использование металлического вольфрама как конструкцион-
ного материала в ядерных реакторах ограничено из-за относи-
тельно большого сечения захвата нейтронов.
Вольфрамовые бронзы служат для покрытия некоторых метал-
лических деталей.
Соединения вольфрама применяются в качестве химических
реактивов, катализаторов, красителей для масляных красок или
керамики, огнезащитной пропитки древесины и тканей, а также
для получения металлического вольфрама или вольфрамсодер-
жащих сталей.
СОЕДИНЕНИЯ (ОБЩИЕ СВОЙСТВА)
Известно небольшое число соединений двух-, трех-, четырех-
и пятивалентного вольфрама и очень много соединений шестива-
лентного вольфрама, причем последние наиболее устойчивы.
В табл. 43 приведены формулы и указан цвет некоторых соеди-
нений вольфрама, сгруппированных по валентностям.
Вольфрам в соединениях низших степеней окисления обнару-
живает металлические свойства (образует основания с восстанови-
тельными свойствами), а в шестивалентном состоянии проявляет
главным образом неметаллический характер (образует кислоты
с ковалентными связями). Среди соединений вольфрама(П) гало-
гениды существуют в виде гексамеров, а вольфрам(Ш) встре-
чается только в координационных соединениях.
Помимо соединений вольфрама, отвечающих названным состоя-
ниям валентности, известны нестехиометрические окислы, воль-
фрамовые бронзы, металлоорганические соединения и соединения
включения.
Неорганические соединения
Соединения двухвалентного вольфрама
Известно немного соединений вольфрама(П). Они относитель-
но неустойчивы на воздухе, быстро окисляясь. Выделены
гексамеры дихлорида, дибромида и дииодида вольфрама
типа [W6X8]X4 или W6X12, галогеновольфрамовые кислоты
H2[W6X8X6] "nH2O, а также их соли.
Гексамер дихлорида вольфрама, [W6C18]C14, получают вос-
становлением гексахлорида вольфрама водородом или алюминием
ВОЛЬФРАМ
353
при нагревании, амальгамой натрия при обычной температуре
и термической диссоциацией тетрахлорида вольфрама в токе
сухого СО2:
12WCle + 18Н2 = [ W6C18]C14 + 6WC14 + 36НС1
I8WCI4 [WeCl8]Ch + 12WCl5
Серое аморфное соединение [W6C181C14 с плотностью 5,436 г/см?
неустойчиво на воздухе, гигроскопично, тугоплавко, нелетуче,
разлагается при нагревании выше 627°; является восстанови-
телем и реагирует с водой с образованием WO2, HCl и выделе-
нием водорода:
627°
5[WeCl8]Cl4---12WC15 + 18W
[W6C1S]C14 + 12Н2О = 6WO2 + 12НС1 + 6Н2
Известен также кристаллогидрат гексамера [W6C18]C14 -2Н2О
и его соли [WeCl8](NO3)4, [W6C18](SO4)2.
Хлороволъфрамовая кислота, H2[W6Cl8Cle] -8Н2О или
(H3O)2[W6C18C161 -6Н2О, образуется, если продукт восстановления
WC16 металлическим алюминием или амальгамой натрия (в атмо-
сфере азота и в присутствии кремнезема в качестве ускорителя
реакции) обработать соляной кислотой, насыщенной хлористым
водородом.
Кислота H2[WeCl8Cle] -8Н2О образует желтые игольчатые кри-
сталлы, которые гидролизуются водой и растворяются в щелочах
с выделением водорода.
При потере шести молекул воды и двух молекул соляной кис-
лоты хлоровольфрамовая кислота превращается в кристалло-
гидрат [W6C18]C14 -2Н2О.
Известны соли типа M*[W6C18C16] -нНгО.
Гексамер дибромида вольфрама, [W6Br8]Br4, получается вос-
становлением пентабромида вольфрама WBr5 водородом при
400—450° в присутствии ZnCl2:
6WBrs + 9Н2 = [WeBr8]Br4 + 18HBr
Соединение [W6Br8]Br4 представляет собой черно-синие иголь-
чатые кристаллы, разлагается на элементы при сильном нагре-
вании, взаимодействует с водой на холоду и реагирует с амидом
калия KNH2.
[WeBr8]Br4 + 12Н2О = 6WO2 + 12НВг + 6Н2
При охлаждении раствора, полученного обработкой хлоро-
вольфрамовой кислоты H2[W6C18C16] "8Н2О горячей конц. НВг,
выпадают коричнево-желтые кристаллы хлоробромовольфрамо-
вой кислоты H2[W6Br8Clg] -2Н2О, которая легко гидролизуется
водой. При нейтрализации хлоробромовольфрамовой кислоты
23 -0101
354
ПОБОЧНАЯ ПОДГРУППА VI ГРУППЫ
щелочами или органическими основаниями образуются соли
типа Mei[W6Br8Cl6].nH2O, (BH)2[W6Br8Cl6l, где Mei = Na+? K+,
а В = C5H5N.
Гексамер • дииодида вольфрама, [W6I8]I4, получают пропуска-
нием паров иода над нагретым до 950° порошкообразным метал-
лическим вольфрамом, термической диссоциацией тетраиодида
вольфрама и нагреванием гексахлорида вольфрама с иодистово-
дородной кислотой при 400—450°:
6W + 6I2 = [WelsHi + 96 ккал
6WI4 ** [WelgJI* + 612
6WCle + 36HI = [ W6IS]I4 + 36НС1 + 12I2
Соединение W6I8]I4 имеет вид коричневого аморфного порошка)
(плотность 6,9 г/см3); оно плохо растворимо в холодной воде,
спирте, сероуглероде и растворяется в щелочах, разлагается теплой
водой, окисляется при нагревании на воздухе с образованием WO®
и выделением иода, восстанавливается водородом при темпера-
туре выше 500° до металлического вольфрама. Из [WeI8]I4 иод
вытесняется хлором при 250° и бромом при 350°.
Соединения трехвалентного вольфрама
Трехвалентное состояние не характерно для вольфрама. Из-
вестно немного координационных соединений вольфрама(Ш) типа
Mei[W2Cl9l, R3[W2C16] (где Mei = Н+, Na+, К+, Rb+, Cs+, NH+
[Ag(NH3)2l+, 1/3[Co(NH3)6]3+ и R = C5H5N или C6H5NH2).
При электролитическом восстановлении водных растворов
вольфраматов щелочных металлов в конц. НС1 и при восстанов-
лении солянокислых растворов вольфраматов амальгамой цинка,
свинца или кадмия образуются растворимые координационные-
соединения вольфрама(Ш), которые содержат анионы [W2C19]3-
зеленовато-желтого, [W2C11O]4~ красного и [W2Cl10(H2O)2]4- крас-
ного цвета.
Известную только в растворе кислоту H3[W2C19] получают
при действии иодистоводородной кислоты на соль таллия
T13[W2C19], Концентрированный раствор кислоты H3[W2C19] окра-
шен в зеленый цвет и проявляет сильно кислотные свойства;,
разбавленный раствор имеет желтую окраску.
Соли типа Mei[W2Cl9] (где Mei —щелочной металл) кристалли-
зуются в гексагональной системе (изоморфны друг другу), окра-
шены в зеленовато-желтый цвет, диамагнитны, медленно окисля-
ются на воздухе, растворимы в воде, если катионами являются Na+,
К+, Rb+, Cs+, а также в случае комплексных катионов.
Диамагнитное желтое соединение K3[W2C19] образуется путем
восстановления K2WO4 в растворе НС1; оно превращается в W5Og,
ВОЛЬФРАМ
355
при нагревании в водном растворе КОН или в K4[W(CN)8] под
действием цианида калия. При нагревании K3[W2C19] взаимодей-
ствует с водой с образованием соединения вольфрама высшей
степени окисления и выделением
водорода.
Анион I W2Cl9]3- (рис. 21) состоит
из двух октаэдров WC16, имеющих
одну общую грань. Расстояние
W—С1 равно 2,46 А, расстояние
W—W 2,45 А.
Комплексные соединения типа
Mei[W2Cl9] рассматриваются как
производные гипотетического хло- Рис. 21. Структура аниона
рида W2Cle. [W2C19]3-.
Известно также соединение
K3[W2Br9l.
При действии плавиковой кислоты на комплексы типа
Me*[W2Cl9], где Me1 = Li+, К+, NH+, получают координационные
соединения вольфрама(Ш) типа Me[WF4]
Ковалентные соединения (C5H5N)3[W2C16J, (C6H5NH2)3[W2Clel
плохо растворимы в воде и частично растворимы в неполярных
растворителях (С6Н6, С2Н5 — О — С2Н5); они образуются при
действии пиридина или анилина на K3[W2C191:
K3[W2C]9] + 3C5HsN = (CsH5N)3[W2C16] + 3KC1
K3[W2C19] + 3C6H5NH2 = (C6HsNH2)3[W2C16] + 3KC1
Соединения четырехвалентного вольфрама
Известно довольно ограниченное число устойчивых соедине-
ний вольфрама(1У), которые получают как восстановлением соеди-
нений вольфрама(У1) [иногда вольфрама(V)], так и окислением
металлического вольфрама или соединений двух- или трехва-
лентного вольфрама.
Простые соединения вольфрама(1У) WO2, WF4, WC14, WI4,
WS2, WSe2, WTe2 имеют темный цвет — от коричневого до черного.
Наиболее устойчивыми координационными соединениями воль-
фрама(1У) являются ацидосоли, в состав которых входит анион
[W(CN)8P~.
Двуокись вольфрама, WO2, получают восстановлением трех-
окиси вольфрама металлическим вольфрамом при 1000° в токе
азота или водородом при 600°.
Можно восстанавливать трехокись вольфрама смесью водо-
рода с парами воды (в соотношении 0,8 : 0,45 по объему) при
800—900°, а также углеродом при 900—1000° или цинком в кис-
лом растворе. Двуокись вольфрама образуется также при дей-
23*
356
ПОБОЧНАЯ ПОДГРУППА VI ГРУППЫ
ствии паров воды на нагретый выше 600° порошкообразный метал-
лический вольфрам.
Мелкие тетрагональные коричневые кристаллы WO2 имеют
плотность 12,11 г!см\ т. пл. 1500—1600° и т. кип. 1730°. Они раз-
лагаются при 1852° и в значительной степени сублимируются при
1000°. Соединение WO2 устойчиво на воздухе, окисляется до WO3
нагреванием до 500° на воздухе (в кислороде) или при 300° в атмо-
сфере NO2, окисляется до W40n (синего цвета) при нагревании
до 500° в атмосфере N2O и NO, трудно растворимо в воде, но
растворяется при нагревании в концентрированных минеральных
кислотах и щелочах.
WO2 + i/2O2 = WOS 4WO2 + 3N2O = W4On + 3N2
WO2 4- NO2 = WOS 4- NO WO2 4- 2HC1 = WO2C12 4- H2
WO2 4- 2KOH= K2WO4 4- H2
В результате нагревания WO2 в парах СС14 при 250° полу-
чают WC14; при прямом хлорировании VVO2 образуется WO2C12,
а при высокой температуре — WC16. При 800—860° водород вос-
станавливает WO2 до металлического вольфрама. Действием серы
на двуокись вольфрама при нагревании получают WS2.
Тетрафторид вольфрама, WF4, получают в результате вос-
становления WF6 бензолом при 110° в закрытом никелевом сосуде
в течение 3—9 дней.
Соединение WF4 представляет собой гигроскопичное краснова-
то-коричневое твердое вещество; гидролизуется в щелочных рас-
творах при нагревании, окисляется в кислой среде с образованием
WO3 и разлагается на элементы при температуре 800° в вакууме.
При действии плавиковой кислоты на WO2 при 600° в течение
3 час получают оксифторид WOF2:
WO2 4- 2HF = WOF2 4- Н2О
Оксифторид устойчив к действию концентрированных кислот и го-
рячих щелочей.
Тетрахлорид вольфрама, WC14, получают обработкой четырех-
хлористым углеродом нагретого до 250° WO2; он также образу-
ется в смеси с [W6C18]C14 при восстановлении гексахлорида воль-
фрама WC18 водородом при высокой температуре:
WO2 4- СС14 = WC14 4- СО2
12WC16 4- 18Н2 = 6WC14 4- [WeCls]Cl4 4- 36НС1
Серые кристаллы WC14 с плотностью 4,624 г/см? при 25° гигро-
скопичны, тугоплавки, нелетучи, гидролизуются водой. При на-
гревании до 475° без доступа воздуха WC14 разлагается на WC15
и [WeCl8]Cl4 и восстанавливается до WC12 или до металлического
вольфрама нагреванием в атмосфере водорода.
18WC14 12WC15 4- [W6C18]C14
ВОЛЬФРАМ
357
Тетраиодид вольфрама, WI4, получают при нагревании иоди-
стоводородной кислоты с гексахлоридом или тетрахлоридом воль-
фрама в запаянной трубке:
WC16 4- 6HI = WI4 + 6НС1 + I2; WC14 4- 4HI = WI4 4- 4HC1
Соединение WI4 образует тугоплавкие черные кристаллы с плот-
ностью 5,2 г/см\ медленно выветривающиеся на воздухе, плохо
растворимые в холодной воде, эфире и хлороформе, растворяю-
щиеся в абсолютном спирте на холоду и в расплавленных щелочах,
карбонатах щелочных металлов или кислого сульфата калия.
В теплой воде тетраиодид вольфрама гидролитически разлагается,
а при сильном нагревании диссоциирует на элементы.
Газообразный хлор вытесняет иод из WI4 при 18°, а пары бро-
ма — при 100°. Водород восстанавливает WI4 до металлического
вольфрама только при сильном нагревании.
Дисульфид вольфрама, WS2, получают прямым взаимодейст-
вием элементов при800—900° в кварцевойтрубке, запаянной в атмо-
сфере азота, термическим разложением трисульфида вольфрама
в атмосфере азота, действием паров серы, сероводорода или серо-
углерода на трехокись вольфрама, нагреванием сначала до 609°,
затем до 1400° смеси WO3, S и К2СО3 в атмосфере H2S, действием
сероводорода на WC16 сначала при 375°, а затем при 500°, электро-
лизом расплавленной смеси WO3, Na2B4O7, CaF2 и Na2SO4:
W J- 2S WS2 2WO3 4- 7S = 2WS2 4- 3SO2
WS3 WS2 4- S WO3 + 3H2S = WS2 4- S 4- 3H2O
wo, 4- 3S 4- K2CO3 = WS2 4- K2SO4 4^ co2
WClg 4- 3H2S = WS2 4- 6HC1 4- S
Дисульфид вольфрама образует парамагнитные темно-серые
(с синеватым отливом) кристаллы с гексагональной структурой
решетки; плотность 7,75 г/смя. Соединение диссоциирует на эле-
менты при нагревании до 1200° в вакууме, плохо растворимо в воде
и спирте, растворимо в смеси азотной и плавиковой кислот или
в окислительно-щелочных расплавах (КОН 4- KNO3, К2СО3 4-
4-KNO3, КОН +КС1О3).
При 800° водород восстанавливает WS2:
WS2 4- 2Н2 W + 2H2S
Диселенид вольфрама, WSe2, получают взаимодействием эле-
ментов при 950° в атмосфере азота или термической диссоциацией
WSe3. Образуются серые кристаллы с решеткой типа MoS2 и плот-
ностью 9,22 г 1см?.
Дителлурид вольфрама, WTe2, получают взаимодействием эле-
ментов в вакууме при температуре выше 750°; это диамагнитные
серые кристаллы с гексагональной структурой решетки и плот-
ностью 9,44 г/см?. Соединение устойчиво к воде, аммиаку и НС1,
358
ПОБОЧНАЯ ПОДГРУППА VI ГРУППЫ
разлагается H2SO4 и HNO3 при нагревании, окисляется кисло-
родом при 650—700°.
Аци д осое динени я вольфрам a(IV), содержа-
щие координированные CN~-r руппы. Соединения
этой группы получают окислением комплексов W(III) или
восстановлением комплексов W(V) в присутствии цианида щелоч-
ного металла и реакцией замещения других комплексов W(IV).
Соли типа MeJ[W(CN)8]-nH2O, где Me1 = Na*, Rb+, x/2Mg2+,
^гСа2*, 1/2Sr2+, 1/2Pb2+, образуют красивые кристаллы устойчивые
на воздухе. Соединения получают обработкой октациановольфрама-
та серебра Ag4[W(CN)8l концентрированным раствором соответ-
ствующего хлорида-
Соли Me|[W(CN)8l -иН2О (где Mi = Na+, К+, Rb*. Cs+, NH+
1/2Mg2+, 1/2Ca2+, ^Sr2*, 1/2Ba2+) растворимы в воде. В тех случаях,
когда Me1 = Tl+, 1/2Cd2+, растворимость солей в воде ограничена.
Если же Me1 = Ag+, 1/2Zn2+, ^Мп2*, соли в воде нерастворимы.
Октациановолъфрамат калия, K4[W(CN)8] -2Н2О, осаждается
из раствора, полученного действием концентрированного раст-
вора KCN на оксихлоровольфрамат(У) типа Me*[WOCl5] «nH^O,
MeHWOClJ -nH2O.
Желтый кристаллический порошок K4[W(CN)8]-2Н2О диа-
магнитен, устойчив на воздухе, имеет плотность 1,989 г/см\ раст-
ворим в воде и ограниченно растворим в спирте. Вещество де-
гидратируется при 110°. Концентрированные водные растворы
этого соединения, окрашенные в красный цвет, становятся жел-
тыми при разбавлении.
В водном растворе довольно устойчивый анион [W(CN)8]4-
может быть окислен до [W(CN)8]5- бромной или хлорной водой,
а также теплой HNO3 или КМнО4 в кислой либо щелочной среде.
Анион [W(CN)8]4- под действием концентрированных минераль-
ных кислот (НС1, H2SO4) при нагревании разрушается.
Пентацианотригидроксоволъфрамат калия, K4[W(OH)3(CN)5] •
•6Н2О, представляет собой коричнево-желтые кристаллы, устой-
чивые в сухом воздухе и растворяющиеся в воде с образованием
красновато-фиолетовых растворов.
При обработке соединения Ag4[W(CN)8] избытком разб. НС1
образуется раствор желтого цвета, из которого при охлаж-
дении и насыщении газообразным НС1 выпадает кислота
H4[W(CN)8] -6Н2О.
HJW(CN)8]-6Н2О, образующаяся в виде желтого кристалли-
ческого порошка, является относительно сильной кислотой. Она
устойчива в сухом воздухе, растворима в воде и абсолютном спирте,
плохо растворима в эфире и бензоле, дегидратируется при 60°
и разлагается при нагревании до более высокой температуры.
Известны и другие координационные соединения вольфрама(1У),
например K2[W(OH)C1 51 — зеленый, K2(W(NCS)6] и др.
ВОЛЬФРАМ
359
Соединения пятивалентного вольфрама
Известно немного соединений вольфрама(У), которые более
устойчивы, чем аналогичные соединения молибдена(У).
Наиболее важными координационными соединениями воль-
фрама^) являются оксигалогеновольфраматы и октациановоль-
фраматы.
Пентахлорид вольфрама, WC15, получают термическим разло-
жением WC16 или контролируемым восстановлением WCle во-
дородом.
Пентахлорид вольфрама представляет собой гигроскопичные
темно-зеленые игольчатые кристаллы с плотностью 3,87 г/сж3,
т. пл. 248° и т. кип. 286°. Это соединение плохо растворимо в CS2
и гидролитически разлагается холодной водой:
WC15 + Н2О WOC13 + 2НС1
При сжигании WC15 в кислороде (или на воздухе) образуется
окситетрахлорид вольфрама WOC14.
При перегонке WC15 в токе водорода образуются WC14 и
IWeCl8]Cl4.
Оксихлоровольфрамат ы(У). При электролизе ра-
створов вольфраматов в конц. НС1 (с применением ртутного катода)
получают неустойчивые синие растворы, которые окисляются на
воздухе; они содержат анион [WOC15]2-. При обработке этих раст-
воров солями щелочных металлов или органическими основаниями
(пиридином C5H5N, анилином C6H5NH2, хинолином C9H7N) обра-
зуются оксихлоровольфраматы типа Me^[WOCl5] (где Me1 = NH^,
Rb+, Cs+), Mei[WOCl5] ./iH2O (где MeI= K+), MeHWOClJ или
Mei[WOCl4]-H2O (где Mei = K+), BH[WOC14] или BH[WOC14] •
•H2O (где В = C5H5N, C6H5NH2, C9H7N).
В качестве примеров можно назвать K2[WOC15] зеленого,
KfWOCl4] -Н2О светло-синего и KIWOC1J коричнево-желтого цвета.
Оксихлоровольфраматы устойчивы в сухом воздухе, гидроли-
зуются и окисляются во влажном воздухе и при соприкосновении
с водой, окисляются также под действием Cl2, KMnO4, Н2О2, HNO3
и т. д.
Кислые растворы оксихлоровольфраматов устойчивы на воз-
духе, а в атмосфере газообразного СОг выдерживают нагревание
до температуры кипения.
Пентабромид вольфрама, WBr5, получают бромированием по-
рошкообразного металлического вольфрама парами брома (раз-
бавленными СО2) при 800° без доступа кислорода и влаги, а также
восстановлением WC16 сухим НВг при 250—300° в стеклянной
трубке без доступа воздуха.
Соединение WBr5 образует гигроскопичные черно-коричневые
игольчатые кристаллы с т. пл. 295°, т. кип. 392° (кипение сопро-
360
ПОБОЧНАЯ ПОДГРУППА VI ГРУППЫ
вождается частичным разложением), растворимые в абсолютном
спирте, СС14, СНС13, СНВг3, С6Н6, С2Н5 - О - С2Н5 и щелочах,
гидролизующиеся во влажном воздухе или холодной водой.
При сильном нагревании WBr5 частично теряет бром и пре-
вращается в [W6Br8]Br4. При нагревании WBr5 в кислороде обра-
зуется WO2Br2.
При 350° водород восстанавливает WBr5 до [W6Br8]Br4, а при
более высокой температуре — до металлического вольфрама.
При температуре 400° WBr5 восстанавливается иодистоводо-
родной кислотой до металлического вольфрама.
При действии конц. HNO3 на WBr5 образуется WO3:
6WBr5 + 12HNO3 = 6WOS + 12NO + 15Вг2 + 6H2O
Оксибромовольфрамат ы(У). Известны оксибромо-
вольфраматы следующих типов: Me*[WOBr5], Me^WOBrJ,
MeHWOBrJ -Н2О, где Mei = Rb+, Cs+, NH+, C5H6N+.
Эти соли окрашены в коричневый цвет, устойчивы в сухом
углекислом газе, гидролизуются водой, растворяются в конц. НВг
с образованием раствора зеленого цвета.
Ацидосоединения вольфрам a(V), содержа-
щие координированные CN'-г руппы.
Октациановолъфрамат\У), K3[W(CN)8] *10,5Н2О, получают
следующим образом.
Если раствор K4[W(CN)8]-2Н2О, подкисленный H2SO4, окис-
лять перманганатом калия, образуется красный осадок, который
можно растворить в КС1 и при упаривании выделить из него кри-
сталлы K3[W(CN)8bKCb5H2O.
Из соединения K3[W(CN)8] •КСЬбНгО можно осадить ионы С1~
с помощью Ag+ (из AgNO3), тогда при упаривании выпадут жел-
тые кристаллы K3[W(CN)8] -10,5 Н2О.
Соединение K3[W(CN)8]-10,5 Н2О представляет собой желтые
кристаллы, относительно устойчивые во влажном воздухе в отсут-
ствие света, растворимые в воде и в растворе КС1. Под действием
света или HI, H2S, SO2 это соединение восстанавливается до окта-
циановольфрамата(1 V):
2[W(CN)s]s- + 21- 2[W(CN)S]‘- + 12
2[W(CN)S]3- + S3- 2[W(CN)S]‘- + S
2[W(CN)S]3" + S4+ 2[W(CN)8]<- + Se+
Соли типа Me*[W(CN)8] -пН2О (где Ме+ = Na+, Rb+, Cs+,
1/2Sr2+, 1/2Ba2+) образуются при действии концентрированных
растворов соответствующих хлоридов на Ag3[W(CN)8J; они раст-
воримы в воде.
Соль Cu3[W(CN)8] зеленого, Ag3[W(CN)8] красного цвета, обе
трудно растворимы в воде.
ВОЛЬФРАМ
361
Для выделения желтых кристаллов кислоты H3[W(CN)8] *6Н2О
раствор октациан овольфрамата(1У), окисленного КМпО4, обра-
батывают разб. НС1 и охлаждают, предварительно насытив газо-
образным НС1.
Соединения шестивалентного вольфрама
Известно очень много устойчивых соединений вольфрама) VI).
Большинство из них являются координационными соединениями.
В табл. 43 приведены формулы и указан цвет наиболее важных
соединений (или типов соединений) вольфрама(У1). В отличие
от молибдена, для которого известен лишь галогенид MoF6, воль-
фрам образует несколько гексагалогенидов — WF6, WC16, WBr6.
Соединения вольфр'ама(У1) окрашены в самые разные цвета:
белый, желтый, оранжевый, красный, коричневый, фиолетовый,
синий и черный.
У соединений вольфрама(У1) (в отличие от соединений низших
степеней окисления) сильнее выражены кислотные свойства, более
четко проявляется ковалентный характер связей и наблюдается
большая тенденция к комплексообразованию.
При частичном восстановлении WO3, H2WO4 или вольфрама-
тов Me^WO4 получают нестехиометрические соединения, такие,
как вольфрамовые сини и вольфрамовые бронзы.
Трехокись вольфрама (вольфрамовый ангидрид), WO3, полу-
чают нагреванием порошкообразного металлического вольфрама
при 900° на воздухе или в токе кислорода, а также прока-
ливанием вольфрамовой кислоты H2WO4 (полученной об-
работкой НС1 водного раствора Na2WO4) или вольфраматов.
Hg2WO4, (NH4)5[HW6O21b4H2O:
800°
W + 8/2O2 = WO3-|-201,5 ккал Hg2WO4--> WO3-|-2Hg-|-i/2O2
500° 250-300°
H2WO4 —WO3 (NH4)5[HW6O21] —6WO3 + 5NH3
—-Нзи —0X12^
При прокаливании паравольфрамата аммония (NH4)5[HW6H21J -
•4Н2О наряду с WO3 в небольших количествах образуются и зеле-
новато-синие окислы вольфрама низших степеней окисления,
поскольку аммиак восстанавливает трехокись вольфрама. Чтобы
избежать этого, продукт реакции увлажняют разб. HNO3 и затем
прокаливают при 700—800°.
Вольфрамовый ангидрид представляет собой парамагнитные
желтые (оранжевые при нагревании) ромбические мелкие кристал-
лы с плотностью 7,16—7,22 г/см\ которые плавятся при 1473°,
превращаясь в зеленую жидкость, кипящую при 1667°. Они субли-
мируются, начиная от 1357°, плохо растворимы в воде и кислотах,
растворимы в расплавах и растворах щелочей с образованием
вольфраматов или поливольфраматов.
362
ПОБОЧНАЯ ПОДГРУППА \1 ГРУППЫ
При восстановлении WO3 водородом получают в зависимости
•от температуры вольфрамовую синь \У4ОИ, WO2 или металличе-
ский вольфрам.
При восстановлении WO3 цинком в кислой среде при легком
нагревании образуется соединение W4O10(OH)2, окрашенное в
фиолетовый цвет. При 240° оно превращается в W4O11«1/3H2O
или W12O32(OH)2, а при 500° — в W4OH.
Окись углерода при 800° восстанавливает WO3 до WO2, а при
более высокой температуре — до металлического вольфрама.
Метан взаимодействует с WO3 при 670°, давая W4OH, при
825° — WO2, при 950° — металлический вольфрам.
При нагревании WO3 с газообразным НС1 или С12 образуется
летучий оксихлорид вольфрама WO2C12:
WO3 + 2НС1 = WO2C12 + Н2О WO3 + Cl2 = WO2C12 + i/2O2
Под действием четыреххлористого углерода WO3 превраща-
ется при 200° в WOC14, а при 280° — в WC16:
WO3 + СС14 = WOC14 + СО2 4WO3 + 6СС14 = 4WCle + 6СО2
При нагревании WO3 с WC16 получают WOC14:
2WO3 + 4WCle = 6WOC14
Нагревание (500°) WO3 с фосфором дает WP2 и Р4О10:
10WO3 + 8Р4 = 10WP2 + ЗР4О10
При взаимодействии паров серы с WO3 образуются WS2 и SO2:
2WO3 + 7S = 2WS2 + 3SO2
Нагреванием трехокиси вольфрама с различными окислами
металлов (Ме*О, МепО, Ме*пО3, MelvO2), взятых в различных соот-
ношениях, можно получить вольфраматы или поливольфраматы:
WO3+Ме2О = Ме2 WO4 2WO3 + Ме2О = Me^W2O7
WO3-|~MeIIO = MeIIWO4 3WO3 + Me2O = MeJW3O10
В химической, стекольной и керамической промышленности
трехокись вольфрама используют для получения металлического
вольфрама и его сплавов.
Гидраты трехокиси вольфрама (вольфра-
мовые кислот ы). Гидраты трехокиси вольфрама, рассмат-
риваемые как вольфрамовые кислоты WO3-H2O или H2WO4,
WO3-2H2O или H2WO4-H2O, WO3-V2H2O или 2WO3-H2O =
= H2W2O7, образуются при действии сильной кислоты на раствор
вольфрамата щелочного металла. Они различаются цветом, устой-
чивостью при нагревании, степенью гидратации и конденсации.
Моногидрат трехокиси вольфрама, WO3-H2O или H2WO4,
получают обработкой паравольфрамата аммония (NH4)5[HW6O21] •
ВОЛЬФРАМ
363
'4Н2О горячей конц. НС1 или путем медленного смешения 1 М ра-
створа Na2WO4-2H2O с горячей смесью 3 объемов раствора (1 : 2)
НС1 и 1 объема раствора (1 : 2) HNO3:
2(NH4)5[HWeO21]-4H2O + 10НС1 = 12H2WO4 + 1ONH4C1-|- 2H2O
В результате образуется H2WO4 в виде оранжево-желтого
осадка с плотностью 5,5 г/см3. Он диамагнитен, плохо растворим
в кислотах, растворим в растворах щелочей и NH4OH, устойчив
до 150°, но выше этой температуры теряет гидратную воду:
H2W04 + 2NaOH = Na2WO4 + 2Н2О
6H2WO4 + 5NH4OH = (NH4)5[HW6O21]-4H2O + 4H2O
Дигидрат трехокиси вольфрама, WO3-2H2O или H2WO4-H2O,
получают, добавляя в разбавленные растворы вольфраматов нат-
рия или калия небольшими порциями минеральные кислоты при
20°. Это белый осадок с плотностью 4,613 г{см?, устойчивый в кис-
лой среде; при промывании водой легко образует коллоидный ра-
створ, а при высушивании в эксикаторе над Р4О10 (20°) —
WO3-H2O.
При медленном добавлении (с перемешиванием) раствора (1 : 2)
H2SO4 в 1 М раствор Na2WO4-2H2O (20°) образуется вольфрамо-
вый гель (аморфный гидрат) светло-желтого цвета. После декан-
тации гель фильтруют под пониженным давлением и перемеши-
вают с водой. Затем его ультрацентрифугируют до тех пор, пока
в промывных водах не исчезнут ионы SO2“. После высушивания
в вакууме над Р4О10 (содержание воды 18—30%) гель приобретает
стеклянный блеск.
Полугидрат трехокиси вольфрама (ди- или пироволъфрамовую
кислоту), WO3>1/2H2O, получают высушиванием при 100° моно-
или дигидрата трехокиси вольфрама.
Соединение ^Оз^/гНгО представляет собой белый порошок,
плохо растворимый в воде и растворимый в растворах щелочей
или NH4OH. Он устойчив при нагревании и дегидратируется толь-
ко при температурах выше 250°.
Вольфраматы, изополивольфраматы и г е -
тер опо л ив ол ь ф рама ты. При растворении трехокиси
вольфрама в щелочах или растворах карбонатов щелочных метал-
лов образуются простые вольфраматы щелочных металлов типа
Me*WO4 (где Mel = Na+, К+ и т. д.).
WO3 + 2NaOH = Na2WO4 + Н2О
WO3 + 2Na2CO3 + Н2О = Na2WO4 + 2NaHCO3
При постепенном подкислении водных растворов вольфрама-
тов или растворов WO3 в растворах вольфраматов щелочных ме-
таллов в зависимости от концентрации, pH, температуры, вре-
мени и др., вследствие конденсации могут образовываться раз-
364
ПОБОЧНАЯ ПОДГРУППА VI ГРУППЫ
личные изополисоединения вольфрама.
Наиболее устойчивыми анионами изополисоединений воль-
фрама являются [HWeO2)]5-, [H3W6O21]3- и [H2W12O40]6-.
Помимо названий, данных изополисоединениям по числу атомов
вольфрама в соответствующей молекуле, употребляются названия:
паравольфраматы А (для гексавольфраматов, содержащих анион
[HWjO21]6"), паравольфраматы Б (для соединений с анионом
[W^OiJ10-), метавольфраматы (для додекавольфраматов с анио-
ном [H2AVf2О4q]3~ или [H2(W301o)4]6_) и Т-метавольфраматы (для
соединений с анионом [H3W6O21]3-).
В щелочных растворах (pH > 8) устойчивы простые вольфра-
маты, содержащие анион WO3-; при pH — 6,6 устойчивы гекса-
вольфраматы (паравольфраматы типа А): при pH = 6,3—6,1 пара-
вольфраматы типа А находятся в равновесии с паравольфраматами
типа Б (имеющими анион [W^O^]10-); при pH = 4 устойчивы
додекавольфраматы (метавольфраматы) с анионом [H2W12O40]6_;
при pH = 3,3 образуются Т-метавольфраматы [H3W6O21I3-; при
pH < 1 возможно образование соединения с радикалом WO|+,
при этом выделяется WO3 или его гидраты.
На следующей схеме показаны превращения аниона WO*-
в различные вольфрамовые изополианионы или WO3 в зависимости
от pH водных растворов:
рН^8
wo4_--------->- w3ot;
Вольфрамат
рН=6,6
рН=6,6 рН=6,2-6,1
----------” [HWeO21]3- — [W12041]io~
рН=3,3
Нагревание
Паравольфрамат А
рН=4
рН<3
[H3We021]3- —------>- [H2W12O40]e-
рН>3
ф-Метавольфрамат Метавольфрамат
Паравольфрамат Б
Кристаллические
соли
рН<1
WO3
При подкислении водных растворов вольфрамата натрия мине-
ральной кислотой (HNO3 или НС1) до pH = 6,6 получают гекса-
вольфрамовую кислоту *:
6Na2WO4 + 12HNO3 = H5[HW6O21] + 12NaNO3 + 3H2O
* Существование гексавольфрамовой кислоты является предметом науч-
ной дискуссии.— Прим. ред.
ВОЛЬФРАМ
365
В зависимости от pH раствора между анионами WO4_
и [HW8O21]5- устанавливается равновесие:
6WO^- + 7Н+ [HWeO21]s- + ЗН2О
При нагревании или осторожном подкислении до pH = 3 све-
жеприготовленных растворов соединений, содержащих анион
{HW8O21I5-, получают соединения с ф-метавольфрамат-анионами.
Из концентрированных, подкисленных до pH = 6,3—6,1 рас-
творов паравольфраматов А с анионом [HWeO2i)s‘ выпадают кри-
сталлы паравольфраматов Б с анионом [W12O41]10-, паравольфра-
мат Cyme — Na10W12O41-28Н2О. В водном растворе анион
lW12O4il10- медленно деполимеризуется в анион [HW6O21]5-:
[W12Otlp<>- + Н2О 2[HWeO21]6-
При подкислении свежеприготовленных растворов гексаволь-
фраматов IHW8O21]5- до pH = 4 образуются метавольфраматы,
содержащие анион [H2W1204o]6“, а при разбавлении растворов
последний медленно превращается (вследствие гидролиза) в ф-мета-
вольфраматы:
2[HWeO21]6- + 4Н+ [H2W12O40]’- + 2Н2О
[H2W12O40]’- + 2Н2О 2[H3W6O21]3-
При pH < 3 гидролиз не происходит.
Точно не известно, сохраняется ли пространственное располо-
жение атомов в комплексе, если кристаллический параволь-
фрамат Б (например) переходит в водный раствор.
Путем прокаливания смеси трехокиси вольфрама с безводным
карбонатом натрия или с окислами щелочных металлов при тем-
пературе 600—800° можно получить различные изополивольфра-
маты *, состав которых зависит от соотношения реагентов:
2WO3 Na2CO3 = Na2W2O7 4- CO2 4WO3 4- Na2CO3 = Na2W4O43 -j- CO2
3WOS + Na2CO3 = Na2W3O10 + CO2 5WO3 + Na2CO8 = Na2W6O16 + CO2
При подкислении растворов смеси двух или нескольких про-
стых солей, например Na2WO4 4-Na2HPO4 или Na2WO4 4-
ф- Na2SiO3, или при смешении кислот этих солей (взятых в сте-
хиометрических соотношениях) можно получить гетерополисоеди-
нения ряда 12, 11, 101/2, 9, 8г/2, 6 и 3. Наиболее многочисленные
и устойчивые гетерополисоединения принадлежат ряду 12 (т е.
содержат 12 атомов W).
Примеры реакций образования 12-гетерополивольфраматов:
12Na2WO4 4- Na2SiO3 4- 22HNO3 = Na4[SiW12O40] -f- 22NaNO3 + 11H2O
12Na2WO4 4- H3PO4 4- 21HNO3 = Na3[PW12O40] 4- 21NaNO3 4- 12H2O
* Полученный сплавлением сухих компонентов по этой реакции тетра-
вольфрамат совершенно не идентичен выделяющемуся из растворов мета-
вольфрамату, являющемуся формальным тримером тетравольфрамата.—
Прим. ред.
366
ПОБОЧНАЯ ПОДГРУППА VI ГРУППЫ
В номенклатуре гетерополисоединений вольфрама применяют
ту же индексацию, что и в номенклатуре гетерополисоединений
молибдена.
Известны многочисленные 12-гетерополисоединения, например-
кислоты боро-, кремне-, германо-, фосфо-12-вольфрамовые:
H5[BW12O40bnH2O (п = 24 или 30,5), HJSiW12O40l-пН2О (п =
= 5, 14, 24 или 30), HJGeW12O40] -nH2O (п — 6, 17, 24 или 30),
H3[PW12O40] -nH2O (п = 5, 14, 21, 24, 29 или 30) и их соли:
Me|[BW12O40]-пН2О и Me*[SiW1204ol-иН2О, где MeT=Li+, Na+,
К+, NH+, 1/2Mg2+, 1/2Са2+, 1/2Ва2+, V-jCu2*, V2Cd2+; MeniH[SiW12O40l
•28Н2О, где Меш= Fe3+, Сг3+ или Al3+; Me*[GeW12O40l -пН2О,
где Mei = NH+, Na+, Rb+, Cs+, Ag+, VaBa2*; MeUPW12O40b
•nH2O, где Mei = Na+, K+, NH+, TH, 1/-Alg2+, x/2Ca2+, !/2Sr2+,
г/2Ва2+, 1/2Zn2+, 1/2Cd2+, 1/2Co2+, 1/2Ni2+. Получены также арсено-
12-вольфраматы Me*[AsW12O40] -пН2О, где Me1 = К+, NH+,
Т1+, и кобальто-12-вольфраматы Me*H[Co3+W12O40l -пН2О, и
Me*H[Co3+W12O40] -пН2О, где Mei = Na+, К+.
По модели Кеггина 12-гетерополисоединения обладают струк-
турой тетраэдра, окруженного 12 октаэдрами, которые распределе-
ны в четыре группы по 3 октаэдра. В соответствии с этой моделью-
формулы 12-гетерополисоединений вольфрама записываются сле-
дующим образом: Mei[B(W3O10)4] -nH2O, Me*[Si(W3O10)4] -nH2O,
MeilGe(W3O10)4bnH2O, MIJP(W3O10)4bnH2O, Mei[As(W3O10)4b
•nH2O.
Для установления структуры и типа ионной диссоциации
гетерополисоединений вольфрама применялись кондуктометриче-
ский, потенциометрический, полярографический, хроматографи-
ческий, рентгенографический методы, а также метод изотопного
обмена и др.
Гетерополикислоты вольфрама растворимы в воде, спирте,
эфире, ацетоне, образуют трудно растворимые соли с алкалоидами
и органическими основаниями, превращаются в простые вольфра-
маты при обработке щелочами.
Гетерополисоединения вольфрама кристаллизуются с большим
числом молекул воды и имеют высокие молекулярные веса, осо-
бенно в концентрированных растворах, где может происходить
полимеризация этих сложных ионов.
Гетерополикислоты вольфрама диссоциируют ступенчато с вы-
делением нескольких ионов водорода, причем константы диссо-
циации каждой ступени равны между собой.
Примерами комплексных ионов различных рядов гетерополи-
вольфраматов с фосфором в качестве центрального атома могут
служить: [PW12O40l3-, [PW1fO39]7-, [P2W21O71]6", [P2W18O62]6-,
[p2w17oeli10-, [PW3O1313-.
При деструкции, связанной с повышением pH, 12-гетерополи-
соединения вольфрама могут превращаться в 11-, КР/г-, 9-, 81/2-,_
ВОЛЬФРАМ
367
6- и 3-гетерополисоединения *, и наоборот, с увеличением кислот-
ности 3-, 6-, 84 2-, 9-, Ю1/^ и И-гетерополисоединения превраща-
ются в 12-гетерополисоединения.
Вольфрамат натрия существует как в виде безводной соли
Na2WO4, так и в виде двух кристаллогидратов — Na2W04-2H20‘
и Na2WO4 -10Н2О.
Бесцветные кубические кристаллы безводной соли Na2WO4
имеют плотность 3,685 г/см3 и т. пл. 697°. Соль растворима в воде,
при 1100° восстанавливается водородом до металлического воль-
фрама, реагирует при 350° с газообразным HCI, а при нагревании
в атмосфере хлора превращается в оксихлориды вольфрама:
Na2WO4 + ЗН2 = W + 2NaOH + 2Н2О
Na2WO4 + 4НС1 = WO2C12 + 2NaCl + 2Н2О
Кристаллогидрат Na2WO4-2H2O выделяется при концентри-
ровании (20°) растйора WO3 в NaOH. Его диамагнитные ортором-
бические кристаллы с плотностью 3,5 г/см3 устойчивы на воздухе,
превращаются в безводную соль при 100°; кристаллогидрат плохо-
растворим в С2Н5ОН, C8H5NO2, GeH5NH2, C5H5N и других орга-
нических растворителях.
При концентрировании водных растворов вольфрамата натрия
при температурах ниже 6° получают бесцветные кристаллы
Na2WO4-10H2O.
Вольфрамат калия, K2WO4, получают путем сплавления
эквимолярной смеси WO3 и К2СО3. Это соединение представляет-
собой расплывающиеся на воздухе бесцветные кристаллы (три-
морфные) с плотностью 2,83 г/см3, очень хорошо растворимые
в воде и поглощающие газообразный СО2.
При концентрировании водного раствора вольфрамата калия
(температура выше 10°) выделяются бесцветные плохо растворимые-
в спирте призмы кристаллогидрата K2WO4-2H2O с плотностью
3,11 г/см3. При обработке смеси растворов вольфрамата калия
и цианида калия гидроксиламином NH2 — ОН образуется
K4[W(CN)8J.
Нагреванием водного раствора вольфрамата калия получают
паравольфрамат калия K5[HWeO21] -5Н2О:
12K2WO4 + 18Н2О = 2K5[HWeO21].5H2O + 14КОН
Вольфрамат рубидия, Rb2WO4, и вольфрамат цезия, Cs2WO4,
получают, обрабатывая вольфрамат серебра Ag2WO4 хлоридом
рубидия RbCl или хлоридом цезия CsCl. Эти соединения образуют
гигроскопичные бесцветные кристаллы, устойчивые к нагреванию
(плавятся примерно при 959° и разлагаются при 1200°). Они рас-
творимы в воде и легко связывают ее с образованием неустой-
чивых кристаллогидратов Rb2WO4 ^/гНгО, Cs2WO4*2H2O.
* По числу атомов вольфрама в комплексе отличают, например, ряд 9s
от ряда 6.— Прим. ред.
368
ПОБОЧНАЯ ПОДГРУППА VI ГРУППЫ
Вольфрамат кальция, CaWO4, существует в природе в виде
минерала шеелита. В лаборатории может быть получен сплавле-
нием Na2WO4 с избытком СаС12 или путем обработки солянокис-
лого раствора хлорида кальция СаС12 вольфраматом натрия.
При этом CaWO4 образуется в виде белого аморфного осадка или
белых тетраэдрических кристаллов с плотностью 6,062 г/см3,
плохо растворимых в воде. Вольфрамат кальция легко восста-
навливается алюминием при нагревании:
CaWO4 + 2А1 = W + Са[А12О4]
Вольфраматы, стронция, бария, свинца, цинка, SrWO4, BaWO4,
PbWO4, ZnW04, образуются в виде белых осадков в результате
обработки водных растворов вольфраматов щелочных металлов
растворами соответствующих солей. К трудно растворимым в воде
вольфраматам относятся Hg2WO4, Ag2WO4, CuWO4, FeWO4,
MnW04, Cr2(WO4)3 и др.
Диволъфрамат натрия, Na2W2O7, получают сплавлением трех-
окиси вольфрама со стехиометрически необходимым количеством
Na2O или Na2CO3. Он образует белые кристаллы. Анион W2O,_
обладает сложной цепочечной структурой, состоящей из тетра-
эдров и октаэдров, связанных между собой атомами кислорода.
Параволъфрамат натрия, Na6[HWeO21]-13,5^0, выделяется
при хранении раствора, полученного в результате добавления НС1
к горячему водному раствору Na2WO4-2H2O до достижения
pH = 6,5.
Соединение Na6[HWeO2i] -13,5Н2О представляет собой бес-
цветные триклинные кристаллы с плотностью 3,987 г!см3, рас-
творимые в воде и перекиси водорода, плохо растворимые в С2Н5ОН,
C6H6NO2, C6H6NH2, C6H5N, устойчивые при 20°, дегидратирую-
щиеся при нагревании и восстанавливающиеся до металлического
вольфрама нагреванием до 900° в атмосфере водорода.
Параволъфрамат калия, K5[HW6O2J '5Н2О, выпадает при
охлаждении теплого водного раствора КОН, К2СО3 (или нор-
мального вольфрамата), насыщенного WO3 или H2WO4-H2O, а
также при обработке раствора паравольфрамата натрия хло-
ридом калия.
Соединение KB[HW6O21]-5Н2О образует триклинные кристал-
лы, устойчивые на воздухе, мало растворимые в воде при 20° и де-
гидратирующиеся при нагревании.
Параволъфрамат аммония, (NH4)6[HW6O21]-4Н2О, выпадает
из растворов WO3, H2WO4, H2WO4>H2O, H2W2O7 в аммиаке
при pH = 6,6. Соединение (NH4)6[HWeO21] -4Н2О представляет
собой бесцветные триклинные кристаллы, растворимые в воде.
Под действием горячей конц. НС1 превращается в H2WO4.
ty-Метаволъфрамат калия, K3[HsWeO2J >9Н2О, осаждается при
добавлении раствора КС1 к подкисленному до pH = 3,3 раствору
ВОЛЬФРАМ
369
вольфрамата натрия. Соединение K3[H3W6O21] -ЭНгО образует
растворимые в воде бесцветные кристаллы. При обработке вод-
ного раствора ф-метавольфрамата калия солями бария выпадает
белый осадок ф-метавольфрамата бария Ba3[H3W6O2J2*21H2O.
В отличие от солей калия и бария ф-метавольфрамат натрия
очень хорошо растворим в воде.
Метаволъфрамовая (или додекаволъфрамовая) кислота,
H6[H3W12O40| "«НгО, образуется при обработке метавольфрамата
бария серной кислотой или метавольфрамата свинца сероводоро-
дом:
Ваз[Н2Д\з2О40] - 3H2SO4 — H6[H2Wi204ol + 3BaSO4
Pb3[H2w)2o40] 3H2s - H6[H2w12o40] 4- 3Pbs
Метавольфрамовая кислота образует легко растворимые в воде
бесцветные кристаллы.
Метавольфраматы щелочных металлов,
MeJ[H2W12O40] -геН2О, выкристаллизовываются из сиропообразных
растворов, полученных кипячением растворов простых вольфра-
матов с вольфрамовой кислотой, и в отличие от простых вольфра-
матов или паравольфраматов обладают более высокой раствори-
мостью в воде.
Структура аниона [H2W12O40]6~ сходна со структурой аниона
[РМо12О40]3-, но центральная тетраэдрическая пустота в нем не за-
нята никакими атомами. В качестве примеров метавольфраматов
можно назвать K6[H2W12O40l •18H20,Ba3[H2W1204o],Pb3[H2W120 40].
Фосфорно-12-волъфрамовая кислота, H3[PW12O40] -nH2O (где
п = 5, 14, 21, 24, 29, 30), выкристаллизовывается из растворов,
полученных обработкой теплого водного раствора, содержащего
смесь фосфата и вольфрамата (или паравольфрамата), сильной
кислотой, действием серной кислоты на фосфоровольфрамат бария
или царской водки на аммонийную соль фосфорновольфрамовой
кислоты.
При контролируемой нейтрализации фосфорно-12-вольфрамо-
вой кислоты (которая очень хорошо растворима в воде) получают
12-гетерополисоли типа Me|[PW12O40] -иЩО, где п = 4 для
Me1 = К+, NH+, Т1+ и п = 24 для Mei = 1/2(Mg2+, Са2+, Sr2+,
Ва2+, Zn2+, Cd2+, Mn2+, Co2+, Ni2+).
Калиевые, аммонийные, рубидиевые и цезиевые соли фосфор-
но-12-вольфрамовой кислоты мало растворимы в воде. Многие
органические основания (алкалоиды, аминокислоты и др.) обра-
зуют с фосфорно-12-вольфрамовой кислотой плохо растворимые
соли. Под действием щелочей фосфорно-12-вольфраматы претер-
певают деградацию и превращаются в гетерополисоединения рядов
11, 10V2, 9, 8V2, 3.
Кобальто-6-вольфраматы и кобальто-12-
вольфраматы. При нагревании солей Со2+ с почти нейтраль-
24-0101
370
ПОБОЧНАЯ ПОДГРУППА VI ГРУППЫ
ными растворами, содержащими ионы вольфрама, образуются
соединения изумрудно-зеленого цвета, содержащие анион
[Co|+W12O42]8_. В окислительной среде в состав этих соединений
входит анион [Co2+Co3+W12O42]7“*.
Если соли Со2 + обрабатывают при нагревании кислыми воль-
фрамовыми растворами, образуются соединения изумрудно-зе-
леного цвета, содержащие анион H[Co2+W12O40]5~. При проведе-
нии реакции в окислительной среде образуются желтые соеди-
нения с анионом H[Co3+W12O40] 4“.
При увеличении кислотности среды кобальто-6-вольфраматы
превращаются в кобальто-12-вольфраматы, а введение щелочей
вызывает обратный переход.
Взаимосвязь между рядами кобальто-6-вольфраматов и ко-
бальто-12-вольфраматов иллюстрируется следующей схемой:
Н+ Окислители
Со2++ 12WOj- + Н+ ~---H[Co2+Wi2O40]5- ч H[Co3+Wi204o]4--
ОН~ Восстановители
Зеленый Желтый
-(-5Н+
- Со2+
-2Н2О
+он-
4-Со2+
-ЗН2О
4-12Н+ Окислители
2Со2++ 12WO|~ ;-----* [Co|+W12O42]8- , [Co2+Co3+W12O42]7-
— 6Н2О Восстановители
Соответствующие гетерополикислоты, присутствующие в рас-
творе кобальто-6-вольфраматов и кобальто-12-вольфраматов в виде
примесей, выделяют ионообменным методом.
Периодатовольфраматы. При действии H2WO4
на перйодаты в присутствии необходимого количества карбонатов
образуются периодато-1-вольфраматы и периодато-6-вольфраматы
типа MeJ[I(WO4)6], где Me1 = Na+. К+, 1/2Sr2+, 1/2Ва2+.
При описании гетерополисоединений ванадия приводились
примеры смешанных гетерополисоединений, которые имеют коор-
динационные группы с ванадием, молибденом и вольфрамом.
Пер оксосоединения. Если вольфрамовую кислоту
H2WO4 или трехокись вольфрама растворять при кипячении
в перекиси водорода, то образуется монопероксовольфрамовая
кислота, H2WO6 или H2[WO3(O2)], желтого цвета:
H2WO4 + Н2О2 = H2WO5 + Н2О
При обработке нейтральных или слабо щелочных растворов
вольфраматов щелочных металлов избытком концентрированной
* Некоторые исследователи считают эти соединения кобальто-11-воль-
фраматами.— Прим. ред.
ВОЛЬФРАМ
371
перекиси водорода получают устойчивые тетрапероксовольфра-
маты типа Me*[W(O2)J *wH2O, окрашенные в желтый цвет.
Из слабо кислых растворов вольфраматов с избытком перекиси
водорода или слабо щелочных растворов вольфраматов щелочных
металлов с малым количеством перекиси водорода выпадают бес-
цветные тетрапероксодивольфраматы Me*[W2O3(O2)J -пН2О.
Образование тетрапероксовольфраматов, тетрапероксодиволь-
фраматов или пероксополивольфраматов зависит от pH растворов
вольфраматов и от количества и концентрации добавленной пере-
киси водорода:
2W0£- -г 2Н++ 4Н2О2 [W2O3(O2)4]2- + 5Н2О
[W2O3(O2)4]2-+ 20Н- + 4Н2О, 2[W(O2)4]2- + 5Н2О
2[W(О2)412- + 2Н+ + ЗН2О [W2O3(O2)4?- + 4Н2О2
[HWeO21?- + 12Н2О2 3[W2O3(O2)J2- + Н+ + 12Н2О
Гетерополикомплексы вольфрам a(VI)\ со-
держащие в качестве координированных
групп радикалы органических кислот,
которые занимают два координационных
места. При комплексующем действии концентрированных
растворов щавелевой, гликолевой, миндальной, галловой, сали-
циловой, винной, яблочной, лимонной и других кислот на вод-
ные растворы простых или конденсированных вольфраматов
образуются координационные соединения вольфрама(У1), которые
содержат анионы, образующиеся замещением одного или двух
кислородов в WO2~ радикалами названных кислот, присоединя-
ющихся по двум координационным связям. Примерами координа-
ционных соединений вольфрама с радикалами органических кислот
в качестве координированных групп могут быть оксалатоволь-
фраматы Ме*[АУОз(С2О4)] •nH2O, Me^[WO2(C2O4)2], гликолято-
вольфраматы Mei[WO2(OCH2COO)2], манделатовольфраматы
Mei[WO3(C8H6O3)2], галлатовольфраматы Me^[WO2{C9H2(OH)2.
• ОСОО}2], салицилатовольфраматы МеПП\УО3(СвП4ОСОО)],
Mei[WO2(C6H4OCOO)2], тартратовольфраматы Me*[WO3(C4H4Oe)j,
MeJ[WO3(C4O6H4)2], малатовольфраматы Me|[WO2(C4H4O5)2] -nH2O,
цитратовольфраматы Mej[WO2(C6H6O7)2] •Ме1С6Н7О7-пН2О, где
Mei = Na+, К+, NH+ реже ^Ва2*.
При обработке вольфраматов пирокатехином образуются хе-
латные соединения типа
Ме2
24*
372
ПОБОЧНАЯ ПОДГРУППА VI ГРУППЫ
Известны также маннитовольфраматы типа Mei[W2O7(CeH14O6)x].
Реакцией нейтрализации и осаждения было установлено суще-
ствование в водном растворе оксалатовольфраматов, в анионе кото-
рых соотношение W : С2О®_ составляет 1:6, 1:4; 1:2; 1:1.
Гексафторид вольфрама, WF0, получают при обычной темпе-
ратуре действием фтора на порошкообразный металлический воль-
фрам, помещенный в платиновую трубку, обработкой плавиковой
кислотой (трифторидом мышьяка или пентафторидом сурьмы)
гексахлорида вольфрама WC16 (в платиновом сосуде, сильно охлаж-
даемом снаружи):
W + 3F2 = WF6 4- 422 ккал WC16 + 2AsF3 = WF6 + 2AsCl3
WC16 + 3H2F2 = WF6 + 6HC1 WC16 + 3SbF5 = WF6 + 3SbCl2F3
При нормальных условиях WF6 представляет собой бесцветный
газ. дымящий на воздухе, гидролитически разлагающийся водой,
растворимый в щелочах и фторидах щелочных металлов и во мно-
гих органических растворителях. При температуре 19,5° он превра-
щается в жидкость, а при 2,5° и давлении 375 мм рт. ст. затвер-
девает. Растворение в органических растворителях WFe сопро-
вождается окрашиванием растворов: в красный цвет — в бензоле,
коричнево-фиолетовый — в диоксане и т. п. Из бензольного раст-
вора гексафторида вольфрама при —78° выпадает белое твердое
вещество WF6-C6H6. При растворении WFe в щелочах или фтори-
дах щелочных металлов образуются двойные соединения.
Гексафторид вольфрама — химически активное вещество, оно
разрушающе действует на стекло и все металлы, за исключением
платины и золота.
На основании спектров комбинационного рассеяния и ИК-
спектров было установлено, что WF6 имеет октаэдрическую струк-
туру кристаллической решетки.
Окситетрафторид вольфрама, WOF4, получают действием пла-
виковой кислоты на WOC14. Для этого охлажденную до —20°
смесь указанных веществ в платиновом сосуде медленно нагре-
вают до -|-20°. Взаимодействие PbF2 с WO3 при температуре ~700°
также приводит к образованию WOF4.
При действии смеси HF с HNO3 на порошкообразный металли-
ческий вольфрам получают смесь WF6 с WOF4:
2W + 10HF + 4HNO3 = WFe -|- W0F4 + 4NO + 7Н2О
Окситетрафторид вольфрама образует гигроскопичные бес-
цветные кристаллы с т. пл. 110° и т. кип. 187°. Соединение гидро-
лизуется теплой водой с выделением HF, плохо растворимо в СС14
и CS2, растворяется в СНС13 или абсолютном спирте.
На холоду WOF4 связывает аммиак с образованием оранжевого
аддукта WOFWgNHs, который растворим в воде, плохо раство-
ВОЛЬФРАМ
373
рим в жидком аммиаке, а при нагревании разлагается на NH4F.
HF и W4OH.
Фторокси вольфраматы, как правило, образуются
при действии плавиковой кислоты на вольфраматы и в зависимости
от состава разделяются на следующие три группы: MeI[WO2F.!|.
где Mei = Na+, К+. NH4+. Tl+. ^Zn2*, ^Cd2*, 1/2Mg2+. 1 '2Мн2+;
Me24WO2F4]. где Mei = K+, NH+, Tl+; Me’[WO3F3], где Mei -
= NH+
Большинство фтороксивольфраматов представляет собой бес-
цветные кристаллы, растворимые в воде и разъедающие стекло.
Фтороксиволъфрамат калия, K[WO2F3] -Н2О. выпадает из ра-
створа K2[WO2F4] -Н20 в плавиковой кислоте или образуется при
обработке паравольфрамата калия K5[HW6O21] -5Н2О избытком
плавиковой кислоты. Он представляет собой бесцветные ортором-
бические кристаллы, которые при нагревании превращаются в
K2[WO2F41 -н2о.
Фтороксиволъфрамат аммония, NH4[WO2F3]-Н2О, выпадает
в виде бесцветных кристаллов в форме призм из раствора,
полученного действием избытка плавиковой кислоты па
(NH4)5[HW6O21].4H2O,
Фтороксиволъфрамат таллия, T1[WO2F3], образуется в виде
белых плохо растворимых в воде пластинчатых кристаллов при
упаривании смеси T1F, избытка WO3 и плавиковой кислоты.
Фтороксиволъфрамат калия. K2[WO2F4] -Н20, получают обра-
боткой КОН раствора WO3 в плавиковой кислоте или при насы-
щении раствора вольфрамата калия плавиковой кислотой. Он
представляет собой бесцветные моноклинные кристаллы, плохо
растворимые в холодной воде, растворимые в горячей, дегидра-
тирующиеся при 100° и превращающиеся в WO3 в результате
нагревания до 600°.
Фтороксиволъфрамат аммония, (NH4)2[WO2F4|,получают дей-
ствием плавиковой кислоты на вольфрамат аммония. Это соеди-
нение представляет собой бесцветные орторомбические кристаллы,
растворимые в воде и устойчивые при температурах до 100°.
Фтороксивольфраматы (NH4)3[WO3F3], Na3[WO3F3], K3[WO3F3J,
Rb3[WO3F3], Cs3[WO3F3] представляют собой бесцветные куби-
ческие кристаллы, растворяющиеся в воде (с разложением).
Гексахлорид вольфрама. WC16, получают путем пропускания
сухого хлора над нагретым до 250—300° порошком металлического
вольфрама (не содержащего окислов), а также действием паров СС14
на нагретую до 300° (без доступа воздуха, кислорода и паров воды)
трехокись вольфрама:
W + ЗС12 = WCle + 163,1 ккал WO2 + ЗСС14 = WC16 + ЗСОС12
Взаимодействие РС15 с WO3 или WS2 дает загрязненный гекса-
хлорид вольфрама.
374
ПОБОЧНАЯ ПОДГРУППА VI ГРУППЫ
Соединение WC16 образует фиолетово-синие кубические кри-
сталлы с плотностью 3,52 г/см3 при 25°. т. пл. 284°, т. кип. 346.7°.
Они растворимы в С2Н6ОН, СНС13, СС14, CS2, С6Нс, С2Н6 — О —
— С2Н5, СН3 — СО — СН3 и т. д. Кристаллы WC16 гидролитически
разлагаются под действием влажного воздуха или воды с обра-
зованием оксихлоридов, а при нагревании в атмосфере водорода
или в среде галогеноводородов превращаются в галогениды воль-
фрама низших степеней окисления:
WC16 + Н20 WOC14 + 2НС1 WC16 + 2Н2О WO2C12 + 4НС1
При действии НВг на WC1O (275°) образуется WBr5, при реак-
ции HI с WCle (400°) — [WeI8]14, а при действии водорода на WC16
(высокая температура) — смесь WC14, [W6C181C14 и WC16.
При растворении WC16 в жидком аммиаке образуется соедине-
ние, состав которого точно не известен.
Металлы, которые проявляют большое сродство к хлору (на-
пример, Na, К, Са, Mg, Al), при нагревании восстанавливают WC16
до хлорида вольфрама низшей степени окисления или даже до ме-
талла. При восстановлении гексахлорида вольфрама с помощью
N2S4 в хлороформе получают неустойчивые соединения WC14 -N2S4.
При обработке спиртового раствора WC16 фенолом получают
красный осадок гексафенолята вольфрама:
WC16 + 6С6Н5ОН = W(OCeH5)6 + 6НС1
Методом дифракции электронов установлено, что WC16 имеет
октаэдрическую структуру и что расстояние W — С1 равно 2,26 А.
Гексахлорид вольфрама хранят в герметически закрытых сосу-
дах. Его применяют в качестве катализатора разложения хлор-
сульфоновой кислоты HSO3C1 и в других органических реакциях
2HSO3C1 -> SO2C12 + H2SO4
Окситетрахлорид вольфрама, WOC14, получают действием
хлора на металлический вольфрам при нагревании в присутствии
кислорода (воздуха) или паров воды, гидролизом WC16 в теплой
воде, окислением WC16 кислородом (воздухом), действием паров
WClg на трехокись вольфрама WO3, пропусканием смеси паров
SC12 и хлора над нагретым до 150—300° WO3, нагреванием WO3
(200°) с парами СС14 и хлора в запаянной трубке, действием смеси
хлора и азота (2С12 + 3N2) на нагретую до 540° WO3, пропускани-
ем газообразного хлора над нагретой до 700—800° смесью метал-
лического вольфрама с WO2, действием хлористого тионила SOC12
на WO2 при 150—300°, взаимодействием паров WC16 с WO2C12,
реакцией между дифтордихлорметаном и WO2 при 527°, сильным
ВОЛЬФРАМ
375
нагреванием WO2CI2:
2W + 4d2 + О2 = 2WOC14
W + WO2 + 4C12 = 2WOC14
WCIe + l/2O2 =» WOC14 + Cl2
W + 3C12 + H2O = WOC14 + 2HC1
2WC16 + WO3 = 3WOC14
WO2C12 + WC16 = 2WOC14
2WO2C12 = WOC14 + WO3
WCle + H2O WOC14 + 2HC1
WO3 + SC12 + Cl2 = WOC14 + so2
WO3 + 2CC14 = WOC14 + COC12
3WO2 + 7SOC12 = 3WOC14 + S2C12 + 5SO2
2WO2 + 2CC12F2 = WOC14 + WOF4 + 2CO
Окситетрахлорид вольфрама представляет собой блестящие
красные игольчатые кристаллы с т. пл. 209° и т. кип. 233°.Соеди-
нение очищают сублимацией. Оно растворимо в CS2 и S2C12. WOC14
гидролизуется теплой водой, превращается в WO2C12 и затем в WO3
под действием кислорода, образует нитриды вольфрама при взаимо-
действии с газообразным NH3, восстанавливается до металлическо-
го вольфрама при нагревании в атмосфере водорода, превращается
в WC18 при нагревании с углеродом. При высокой температуре
WOCI4 взаимодействует с SiO2 по уравнению
2W0C1* 4- SiO2 = 2WO2C12 + SiCl4
Диоксидихлорид вольфрама, WO2C12, получают, пропуская
хлор над нагретой примерно до 700° WO3, действуя НС1 на WO3
при 400°, пропуская смесь хлора и азота над нагретым до 540°
WO2, обрабатывая СОС12 трехокись вольфрама при 350°, действуя
СаС12 (или хлоридами MgCl2, СоС12, NiCl2) на нагретую до 800—
900° WO3 в атмосфере газообразного СО2:
2WO3 4~ 2С12 2WO2C12 -]- О2 WO2 4- С12 = WO2C12
WO3 4~ 2НС1 — WO2C12 -|- Н2О WO3 -|- СОС12 = WO2C12 4- СО2
2WO3 + СаС12 = WO2C12 + CaWO4
Соединение WO2C12 образует желтые кристаллы, которые пла-
вятся при 265°, сублимируются до начала плавления, плохо раст-
воряются в спирте. Диоксидихлорид вольфрама гидролизуется
теплой водой, растворяется в щелочах с образованием вольфрама-
тов, разлагается на WOC14 и WO3 при сильном нагревании, обра-
зует двойные соли с хлоридами щелочных металлов и восстанав-
ливается до металла при нагревании в атмосфере водорода. На-
гревание WO2C12 с WC18 дает WOC14. На воздухе WO2C12 более
устойчив, чем WOC14.
Гексабромид вольфрама, WBre, получают путем пропускания
сухих паров брома, разбавленных азотом, над нагретым примерно
до 600° порошком металлического вольфрама.
376
ПОБОЧНАЯ ПОДГРУППА VI ГРУППЫ
Соединение WBr0 образует черно-синие игольчатые кристаллы
с плотностью 6,9 г!см3, которые дымят на воздухе, плавятся при
309°, разлагаются при нагревании выше 400° или под действием
воды, растворяются в обычных органических растворителях с обра-
зованием фиолетово-красных растворов.
Известны также смешанные галогениды вольфрама(¥1) WC16-
• WBr6 или WBr3Cl3, WCle-3WBr0 или 5, которые пред-
ставляют собой кристаллические соединения, неустойчивые к дей-
ствию кислорода воздуха и воды.
Окситетрабромид вольфрама, WOBr4. получают действием
паров брома на смесь WO3 (иди WO2) с углем; это гигроскопичные
черно-коричневые кристаллы, которые разлагаются на воздухе;
т. пл. 277°, т. кип. 327°.
Диоксидибромид вольфрама, WO2Br2, получают пропусканием
смеси паров брома с воздухом над нагретым до 700—800° метал-
лическим вольфрамом; образующиеся красные (или оранжевые)
кристаллы при нагревании разлагаются по уравнению
2WO2Br2 = WOBr4 + WO3
Трисулъфид вольфрама, WS3, получают в виде коричневого
осадка при подкислении H2SO4 или НС1 тиовольфраматов щелоч-
ных металлов или аммония Me*WS4 до pH < 5:
Na2WS4 + H2SO4 = WS3 + Na2SO4 д- H2S
(NH4)2WS4 + 2HC1 = WS3 + 2NH4C1 + H2S
Соединение WS3 плохо растворимо в воде, растворяется в суль-
фиде аммония, (NH4)2S и в щелочах, разлагается на WS2 и серу
при нагревании до 170°, восстанавливается до металла при нагре-
вании в атмосфере водорода или с окисью кальция:
WS3 + (NH4)2S = (NH4)2WS4 WS3 + 3H2 = W + 3H2S
WSs = WS2 + S WS3 + 2CaO = W -}- 2CaS + SO2
4WS3 4- 8NaOH = 3Na2WS4 4- Na2WO4 + 4H20
Известны двойные сульфиды вольфрама и пиперазина:
C4H12N2S-WS3 •C4Hi0N2 — желтые игольчатые кристаллы и
C4Hi2N2S-2WS3>C4H10N2 — желтые пластинчатые кристаллы.
Тиовольфраматы и окситиовольфраматы типа MeiWS4,
MeiWS3O-H2O, MeiWS2O2-H2O, MeiWSO3-H2O, где Me1 = Na+,
K+, NH4, получают обработкой водных растворов вольфраматов
щелочных металлов (при определенных значениях pH) с помощью
H2S, (NH4)2S, Meis.
Пропускание сероводорода через растворы вольфрамата натрия
при pH = 7 дает окситиовольфраматы с анионом WS^2- в том
случае, когда отношение H2S/Na2WO4 равно 2, и окситиовольфра-
маты с анионом WS3O2-, когда это отношение равно 3. Тиовольфра-
ВОЛЬФРАМ
377
маты с анионом WS|~ образуются при действии H2S на слабо кислые
(pH > 6) растворы паравольфраматов IHW6O2J5~.
Примеры реакций получения тио- и окситиовольфраматов:
Na2WO4 + 4H2S = Na2WS4 4- 4Н2О
Na2WO4 4- 4(NH4)2S 4- 8HC1 = Na2WS4 8NH4C1 4H2O
Na2WO4 + 3Na2S 4- 6HC1 = Na2WS3O 4- 6NaCl 4- 3H2O
Тиовольфраматы и окситиовольфраматы растворимы в воде;
их водные растворы окрашены в красный, оранжевый или желтый
цвет.
В качестве примеров тиовольфраматов и окситиовольфраматов
можно назвать Na2WS4, K2WS4, (NH4)2WS4, K2WS3O-H2O,
K2WS2O2-H2O, K2WSO3-H2O.
Триселенид вольфрама, WSe3, получают в виде черного осадка
при действии разбавленных кислот (H2SO4, HCl) на растворы селе-
новольфраматов Me*WSe4, которые образуются при насыщении
растворов вольфраматов щелочных металлов H2Se.
Соединение WSe3 — черное аморфное вещество, которое раз-
лагается при 220° на WSe2 и элементарный селен.
Металлоорганические соединения
Известно небольшое число металлоорганических соединений
вольфрама.
Гексакарбонил вольфрама, W(CO)e, получают действием фенил-
магнийбромида, насыщенного окисью углерода, на гексахлорид
вольфрама, действием окиси углерода на металлический воль-
фрам в присутствии железа или меди при давлении 200 атм, а так-
же действием окиси углерода на K3[W2C19] в присутствии железа
или меди при низком давлении.
Гексакарбонил вольфрама образует бесцветные ромбические
кристаллы, которые сублимируются при температурах 50° и выше,
разлагаются при 100°, выделяя металлический вольфрам и W4On,
не взаимодействуют с водой и разбавленными кислотами, разла-
гаются дымящей HNO3 и плохо растворимы в С2Н5 — ОН, С2Н6 —
— О — С2Н6 и СсНв.
Методом дифракции электронов было установлено, что все
шесть групп СО расположены в вершинах октаэдра, в центре кото-
рого находится атом вольфрама.
При обработке гексакарбонила вольфрама спиртовыми раст-
ворами щелочей получают соли типа Me*W(CO)5, которые под
действием кислот превращаются в неустойчивый летучий гид-
рид H2W(CO)5.
К металлоорганическим производным W относятся также
(C5H5)2W2(CO)6, C6HeW(CO)3, (C6H6)2WX3 (где X — кислотный
радикал), W(CO)4(C6H6N)2, W(CO)3(C6H5N)3, W(CO)4(C12H8N2).
378
ПОБОЧНАЯ ПОДГРУППА VI ГРУППЫ
Вольфрамовые бронзы
Вольфрамовые бронзы, MexWO3, где Me1 = Na+, Li+, К+,
Rb+, Cs+ и 1 > x > 0, являются нестехиометрическими соедине-
ниями (аналогами твердых растворов интерметаллических фаз),
устойчивыми на воздухе и интенсивно окрашенными. Вольфрамо-
вые бронзы химически инертны, обладают высокой тепло- и элект-
ропроводностью. Они получаются при частичном восстановлении
поливольфраматов щелочных металлов водородом, металлическими
вольфрамом или натрием при нагревании, электролитическим вос-
становлением расплавленных вольфраматов щелочных металлов
с WO3 и частичным восстановлением расплавленных вольфраматов
щелочных металлов и WO3 металлическим натрием в инертной
атмосфере.
Различно окрашенные вольфрамо-натриевые бронзы полу-
чают частичным восстановлением поливольфраматов натрия водо-
родом при 620—630°
Ч~Н2 4-Нз
Na2W5O16 —5Na014WO3 Na2W3O10 —3Na0,66WO3
— l±2v —XlgO
Фиолетово-синий Красный
+H2 +H2
Na2W4O13—4Na0,5WO3 Na2W2O7 —-> 2NaWO3
— H2V —X12O
Фиолетово-красный Золотисто-желтый
Как показано выше, нагревая пентавольфрамат натрия
Na2W5Ole в атмосфере водорода, можно последовательно полу-
чить вольфрамо-натриевые бронзы, окрашенные в фиолетово-
синий, фиолетово-красный, красный или золотисто-желтый цвета.
При этом выделяется металлический вольфрам.
+н2 +зн2 +зн2
Na2W5Ole —->• 5Na0,4WO3 - -> 4Na015WO3 —-
— Н2О —ЗН2О—W —ЗН2О—w
+ЗН2
-+ 3Na0,66WO3 „ -> 2NaWO3
— D X12<J— W
Вольфрамо-натриевые бронзы можно получить в результате
восстановления расплавленной смеси Na2WO3 с WO3 порошко-
образным металлическим вольфрамом:
3zNa2WO4 + (6 — iz)WOs + zWSNa^WOs
Цвет вольфрамо-натриевых бронз зависит от значений х в фор-
муле NaxWO3. При 0,3 > х > 0,1 бронзы имеют синий цвет,
0,40 > х > 0,32 — фиолетово-синий, 0,5 > х > 0,46 — фиоле-
тово-красный, 0,66 > х > 0,64 — красный, 1 > х > 0,93 — золо-
тисто-желтый.
По данным кристаллографических исследований было уста-
новлено, что вольфрамо-натриевые бронзы NaxWO3, в которых
ВОЛЬФРАМ
379
0,93 > х > 0,26, кристаллизуются в кубической системе, а при
х < 0,26 — в тетраэдрической системе.
Известны также в ольфрамо-литиевые LixW03, вольфрамо-ка-
лиевые KXWO3 (х = 0,27), вольфрамо-рубидиевые RbxWO3 (х =
= 0,27) и вольфрамо-цезиевые Cs^-WOs (х = 0,32) бронзы.
В вольфрамовых бронзах Me^W03 атомы щелочных металлов
находятся в ионном состоянии (Na+, Li+, К+, Rb+, Cs+) и валент-
ность вольфрама должна была бы равняться 6 (при х = 0) и 5
(при х = 1). В действительности, в вольфрамовых бронзах не су-
ществует атомов пяти- и шестивалентного вольфрама. Они экви-
валентны, поскольку один электрон атома вольфрама статистиче-
ски делокализован в кристаллической решетке подобно электрону
в металлах. Благодаря этому вольфрамовые бронзы обладают свой-
ствами, характерными для металлов.
Вольфрамовые бронзы используются для получения высокока-
чественных типографских красок.
Окислы вольфрама, средние между WO3
и WO2- При нагревании смесей WO3 с WO2 или WO3 с метал-
лическим вольфрамом, взятых в различных Соотношениях, в ва-
кууме или в инертной атмосфере, при нагревании WO3 (800°)
со смесью 1 об. ч. водорода и 2 об. ч. водяных паров при давлении
1 ат, при восстановлении WO3 водородом (550°) или СО (при тем-
пературах ниже 800°) получают синие окислы вольфрама общей
формулы W„O3n_| (где ге= 4, 10, 11, 12 и т. д.), т. е. W4OH, W10O29
или W20O58, W^O^, W12O36, которые имеют недостаток кисло-
рода по сравнению с WO3.
Окись y-W4Ofl с нестехиометрическим составом в интерва-
ле WO2176 — WO2>85 представляет собой фиолетово-синие моно-
клинные кристаллы, которые сублимируются при 900° и реагируют
с водой и газообразным НС1 при повышенной температуре.
3W4OU+ 22НС1 = W + 11WO2C12 + 11Н2О
Окись p-W20O68 с нестехиометрическим составом в интервале
WO2195 — WO2,90 образует темно-синие кристаллы.
Для a-фазы известна синяя окись вольфрама, нестехиометри-
ческий состав которой находится в пределах между WO3 и WO2i96.
Босстановлением при легком нагревании WO3 или H2W04
в водной суспензии с помощью НС1 + Zn, Al, Fe, HI, SnCl2 и др.
или электролитическим восстановлением WO3 в кислом растворе
при 60—70° (используя платиновый катод и никелевый анод),
получают фиолетовый раствор W4Oi0(OH)2, который при 240°
превращается в W40n «ПзНгО или W12O32(OH)2, устойчивые
до 500°:
240°
12WO3 + 6H+ 3W4O10(OH)2----> Wi2O32(OH)24-2H2O
5 00°
3W4On + H2O
380
ПОБОЧНАЯ ПОДГРУППА VI ГРУППЫ
Восстанавливая WO3 водородом, можно получить вольфрамо-
водородные бронзы: H0j5WO3 — в виде кубических фиолетовых
кристаллов, H0>33WO3 — в виде синих тетраэдрических кристал-
лов и H0jlWO3 — в виде ромбических кристаллов, которые раз-
лагаются под действием растворов щелочей или воды.
Соединения включения
Нитриды вольфрама. WNa, W2N и W2N3.
Когда нить металлического вольфрама нагревают при 2500°
в атмосфере азота, образуются черно-коричневые кристаллы WN2,
кристаллизующиеся в кубической гранецентрированной системе.
Они имеют плотность 5 г/см3, гидролизуются водой с выделением
NH3, устойчивы в вакууме до температуры 400° и окисляются
по реакции
2WN2 + ЗО2 = 2WO3 + 2N2
При действии газообразного аммиака на нагретый до 700—
800° порошок металлического вольфрама или на нагретый до
825—875° WO3 образуется коричневый нитрид W2N, который суще-
ствует в виде двух кристаллических модификаций.
Нитрид вольфрама, W2N3 (теоретически получаемый дейст-
вием газообразного NH3 на WOC14 или WC16 на холоду), вряд
ли существует.
Фосфиды вольфрама, WP2, WP, W2P и W3P4.
Дифосфид вольфрама, WP2, получают прямым взаимодействи-
ем порошкообразного металлического вольфрама с красным фос-
фором при 700—950° в кварцевой трубке, действием газообраз-
ного РН3 HaWGlenpH 250—450°, восстановлением WO3 фосфором
при 550°. Дифосфид выделяют в виде черных кристаллов с плот-
ностью 9,17 г/см3; они плохо растворимы в воде, спирте и эфире.
Фосфид вольфрама, WP, получают в результате термической
диссоциации WP2 или при восстановлении WP2 фосфидом меди
Сн2Р3 при 1200°. Образуется серый кристаллический порошок
с плотностью 12,3 г/см3, плохо растворимый в воде, НС1 и щелочах
и растворимый в смеси конц. HNO3 и HF.
Существование фосфидов W2P, W3P4 сомнительно.
Диарсенид вольфрама, WAs2, получают прямым взаимодей-
ствием элементов при 620° в запаянной стеклянной трубке или
взаимодействием сухого AsH3 с WC16 при 350° без доступа воздуха.
Соединение включения WAs2 представляет собой черные кри-
сталлы с плотностью 6,9 г!смъ. Соединение устойчиво на воздухе,
окисляется до WO3 и As2O3 при нагревании на воздухе (или кис-
лороде), плохо растворимо в воде, HF, НС1, растворяется в цар-
ской водке с образованием H2WO4h образует вольфраматы и арсе-
наты при сплавлении со щелочами.
ВОЛЬФРАМ
381
В результате действия газообразного AsH3 на WC16 при 70°
в запаянной трубке образуется твердое соединение W2AsCI9;
оно гигроскопично, неустойчиво на воздухе и плохо растворимо
в обычных органических растворителях.
Карбиды вольфрама, W2C, WC, получают несколь-
кими способами: путем карбидизации порошкообразного метал-
лического вольфрама окисью углерода, смесью СН4 и Н2 при 840°
или углеродом при 1400° (в графитовой трубчатой печи) в токе
водорода, карбидизацией нити металлического вольфрама при 1800°
парами нафталина, при 1500° окисью углерода или при 1500—
1900° углеродом в атмосфере водорода или углеводородов (обра-
зующихся при действии водорода на графит, из которого изготов-
лена трубка) при нагревании, нагреванием выше 1500° WO3 с из-
бытком угля, нагреванием галогенидов вольфрама с углеводоро-
дами при 1500°, пиролизом W(CO)e и нагреванием смесиW(C0)e
с водородом при 400—800° и пониженном давлении (10 мм рт. ст.).
2W + 2СО = W2C + С02 W + 2СО = WG + СО2
2W + СН4 W2C А 2Н2 W + СН4 WC А 2Н2
2W + С = W2C W + С = WC
WO3 + 4С = WC А ЗСО
W(C0)6 = WC + СО2 + 4СО
2WC + 2Н? W2C + СН4
Карбид вольфрама, W2C, представляет собой серые кубические
кристаллы (a-W2C) или серые гексагональные кристаллы (|3-W2C)
с плотностью 17,34 г!см3. Они тугоплавки (т. пл. 2880°), имеют
твердость 9 по шкале Мооса, обладают низкой электропровод-
ностью. Соединение W2C окисляется до WO3 при нагревании
(550°) в кислороде, превращается в WC14 и графит при нагревании
(250°) в атмосфере хлора, реагирует с фтором (18°), растворяется
в смеси HNO3 — HF (20°), восстанавливает при нагревании дву-
окись титана с образованием карбида титана TiC и металлического
вольфрама-
Карбид вольфрама, WC, представляет собой серовато-синий
порошок или серо-синие кристаллы с плотностью 15,77 г!см3.
Они плохо проводят электрический ток (становятся сверхпровод-
никами при 2,5° К), парамагнитны, имеют твердость 9 по шкале
Мооса, плавятся при 2600° с выделением графита, устойчивы к дей-
ствию кислот и смеси HNO3 — HF. Карбид вольфрама взаимодей-
ствует с фтором при 18°, с хлором при 450°, окисляется до WO3
при нагревании на воздухе, взаимодействует при нагревании
с многочисленными окислами металлов (F2O3, MnO2, MnO. TiO2)
с образованием двойных или тройных карбидов, образует очень
однородные твердые растворы с TiC и ТаС.
382
ПОБОЧНАЯ ПОДГРУППА VI ГРУППЫ
Карбид WC является главной составной частью жаропрочных
твердых сплавов. Из сверхтвердых сплавов, содержащих WC —
TiC — Со, делают наконечники для сверл.
Специальные стали содержат двойные карбиды железа и воль-
фрама; их примерный состав: FeWC, Fe2W2C, Fe3W3C, Fe3W2C,
Fe6W6C.
Двойной карбид вольфрама, FeWC, представляет собой черно-
серые мелкие кубические кристаллы с плотностью 17,5 г/слг1;
они растворимы в смеси HNO3 — HF при 18° и окисляются до WO3
и Fe2O3 при нагревании до 880° в атмосфере кислорода.
Силициды вольфрама, WSi2 и W3Si2.
Дисилицид вольфрама, WSi2, получают нагреванием в атмо-
сфере аргона порошкообразного металлического вольфрама с эле-
ментарным кремнием при 1000—1200° и повышенном давлении,
разложением SiCl4 (в присутствии водорода) на раскаленных воль-
фрамовых нитях, алюмотермическим восстановлением смеси WO3
и SiO2.
Соединение WSi2 образует блестящие серо-синие гексаэдриче-
ские призмы с плотностью 9,75 г!см3 и т. пл. 2050°. Они устойчивы
на воздухе до 900°, окисляются чистым кислородом, превращаются
в WC16 и SiCl4 под действием хлора при 450°, не реагируют с мине-
ральными кислотами (НС1, HNO3, H2SO4), растворяются в смеси
HNO3 с HF и расплавленных щелочах.
Силицид вольфрама. W3Si2, слабо изучен.
Бориды вольфрама, W2B, Wfi, W2B5, получают пря-
мым взаимодействием элементов при 1200—2000° в вакууме или
в атмосфере инертного газа, алюмотермическим восстановлением
смеси WO3 с В2О3, электролизом при 960° расплава, состоящего
из WO3, Na2B4O7 и NaF.
Борид вольфрама, W2B, представляет собой серые тетраэдри-
ческие кристаллы с плотностью 16,72 г!см3 и т. пл. 2770°. Они
превращаются в WB при нагревании до 1900° с углем и восстанав-
ливаются до металлического вольфрама при сильном нагревании
с металлическими титаном, танталом или цирконием.
Борид вольфрама, WB, существует как в виде серого порошка,
так и в виде блестящих серых тетраэдрических (6-WB) или орто-
ромбических ф-WB) кристаллов, которые имеют плотность
~16 г!смъ, т. пл. 2860° и твердость 9 по шкале Мооса. Борид реа-
гирует с HNO3 и H2SO4 при нагревании, растворяется в царской
водке, превращается в W2N нагреванием до 1100° в атмосфере
газообразного аммиака.
Борид вольфрама, W2B5, представляет собой серые гексаго-
нальные кристаллы с плотностью 13,1 г! см?.
ПОБОЧНАЯ ПОДГРУППА VII ГРУППЫ
ПЕРИОДИЧЕСКОЙ СИСТЕМЫ
(ПОДГРУППА МАРГАНЦА)
ХАРАКТЕРИСТИКА ПОДГРУППЫ
В побочной подгруппе VII группы периодической системы хи-
мических элементов находятся переходные металлы марганец (Мп),
технеций (Тс) и рений (Re).
В табл. 44 показана электронная структура атомов марганца,
технеция и рения.
ронном уровне по 2 электрона и на предпоследнем электронном
уровне 2.6.5 электронов. d-Орбитали предпоследнего электронно-
го уровня неполностью укомплектованы электронами. Участвуя
в химических реакциях, атомы марганца, технеция и рения обра-
зуют соединения, в которых они одно-, двух-, трех-, четырех-,
пяти-, шести- и семивалентны.
В табл. 45 приведены наиболее важные константы элементов
подгруппы марганца.
В компактном состоянии марганец имеет светло-серый, тех-
неций — серебристо-серый и рений — серебристо-белый цвета.
Кристаллические решетки металлических технеция и рения по-
строены по типу плотнейшей гексагональной упаковки.
Таблица 45
Элемент Марганец Мп Технеций Тс Рений Не
Цвет Светло-серый Серебристо-серый Серебристо-белый
Кристаллическая структура а-, |3- и 6- Мп—ку- бическая, у-Мп— тетраго- нальная гране- центрированная Гексагональная плотнейшая Гексагональная плотнейшая
Атомный номер 25 43 75
Атомный вес 54,9381 98,918 186,22
Атомный радиус, А 1,28 1,36 1,38
Радиус иона Ме7+, А (по Гольд- шмидту, Полин- гу и Аренсу) 0,46; 0,46 -; 0,56 0,77; -
Атомный объем (при 20°), см3/г-атом а-Мп 7,39 Р- и у-Мп 7,6 8,60 8.85
Плотность (при 20°), г/см3 а-Мп 7,44 Р-Мп 7,29 у-Мп 7,21 11,49 20,99
Твердость по шка- ле Мооса 5—6 — 7,4
Температура плав- ления, °C 1245 2127 3170
Температура ки- пения, °C 2150 — 5630
Удельная теплоем- кость (при 20°), кал[г-град а-Мп 0,114 Р-Мп 0,155 у-Мп 0,120 — 0,033
Сопротивление р-106 (при 0°), ом- см а-Мп 150-260 Р-Мп 90 у-Мп 40 — 31
Электропровод- ность (Hg= 1) 5,5 — 3
Продолжение табл. 45
Магнитная вос- приимчивость %’Ю-6 (при 18°), эл.-маги. ед. 9,66 2,5 0,33
Теплота перехода атомов в газо- образное состоя- ние (при 25°), ккал 68,34 -— 189
Электроотрица- тельность (по Полингу) 1,5 1,9 1,9
Потенциал ионизации, ав| Ме->-Ме+-|-е- Ме+-*Ме2+-|-е- Ме2+->Ме3++е_ Ме3+->Ме4++е- Me4+->Me6+-J-e- Ме6+-*Ме6+-|-е_ 7,43 15,46 33,69 53 76 101 7,28 15,26 29 43 59 76 7,87 16,6 26 38 51 65
Потенциал ионизации, ккал/г-атом\ Me Ме+4-е~ 171,2 — ~ 184
Нормальные по- тенциалы (при 25°), в Мп/Мп2+—1,18 Мп/МпЗ++0,47 Тс/Тс2+ 0,24 Re/Re3+—0,3
Нормальный по- тенциал окисли- тельно-восстано- вительных си- стем, в Мп2+/Мп3++1,51 Мп+4Н2О5± MnOf+8H-^- +7е-4-0,49 Тс+4Н2О TcOj+8H++ +7е--|-0,472 Re+/Re2+0,02 и Re2+/Re3+— 0,23 Re+4H2O ReOj-|-8H+-|- +7e~+0,36
25—0101
386
ПОБОЧНАЯ ПОДГРУППА VII ГРУППЫ
Продолжение табл. 45
MnO2+2H2O MnOj+4H++ +Зе--|-1,695 Мп+2Н2О 5±МпО2+4Н++ +4е_4-0,30 ТсО2+2Н2О ТсОу+4Н++ +3е-+0,771 Тс+2Н2О ТсО2+4Н++ +4е“+0,28 ReO2+2H2O ReOj+4H++ +Зе~+0,51 Re+2H2O ReO2+4H++ +4е~+0,26
Валентность (I), II, III, IV, (V), VI, VII (I, II, III), IV, (V, VI), VII (I, II, III), IV, (V, VI), VII
Массовые числа природных изо- топов 55 99, 97, 96, 95, 93, 94, 92, 98, 101, 100, 107, 102 187, 185
Распространен- ность элементов в земной коре, вес. % 0,10 1,0-10-7
Модификации а-, 0- и 6-марганца имеют кубическую струк-
туру, а у-марганец — гранецентрированную тетрагональную
структуру.
Марганец, технеций и рений относятся к тяжелым металлам
(их плотность от 7,21 до 20,99 г/см3), они парамагнитны, туго-
плавки (т. ил. в пределах от 1245 до 3170°, т. кип. — от 2150 до
5630°), обладают твердостью 6—7,4 ио шкале Мооса, становятся
твердыми и хрупкими, теряя пластичность при загрязнении бором,
кремнием и фосфором.
Технеций менее изучен, чем марганец и рений; он имеет более
ограниченное применение; для него известно большее число при-
родных изотопов.
Известны многочисленные сплавы марганца и рения с Al. Sn,
Ti, Сг, Мо, W, U, Fe, Ni, Pd, Pt, Ag, Cu, Zn, Hg и другими метал-
лами.
В зависимости от степени чистоты и измельчения металлы
подгруппы марганца по-разному ведут себя в отношении хими-
ческих реагентов. С точки зрения химической активности реак-
ционная способность уменьшается в ряду марганец, технеций,
рений.
При комнатной температуре в порошкообразном состоянии эти
элементы окисляются во влажном воздухе, превращаясь в окислы,
а при нагревании (как в порошкообразном, так и в компактном
состояниях) эти металлы взаимодействуют с кислородом, фтором,
хлором, серой, селеном, фосфором, кремнием и бором.
ПОБОЧНАЯ ПОДГРУППА VII ГРУППЫ
387
Марганец растворяется в разбавленных кислотах НС1, HNO3,
H2SO4, технеций — в HNO3, царской водке и смеси концентри-
рованных кислот HNO3 и H2SO4, а рений растворяется на холоду
в разб. HNO3 или при нагревании в конц. H2SO4, НСЮ4, хлорной
и бромной воде.
Шелочи не реагируют с марганцем и технецием и медленно
действуют на рений.!
В природе марганец очень редко встречается в свободном со-
стоянии, но часто в виде соединений (окислы, карбонаты, фосфаты,
силикаты, сульфаты, сульфиды, вольфраматы, бораты и т. п.)
в различных минералах. Рений находится в природе и в свободном
состоянии, и в виде соединений в некоторых минералах молибдена,
ниобия, тантала, железа, меди, иттрия и платины.
Металлы подгруппы марганца можно получить путем восста-
новления водородом (при нагревании) следующих соединений:
MnF2, МпС12, МпВг2, Mnl2, NH4TcO4, As(C6H5)4TcO4, Тс2О7,
ТсО2, Tc2S7, MeiReO4 (где Mei = К+, NH+), ReO2, Re2O7, ReS2,
Re2S7, ReOCl4, ReO3Cl, ReOF4, и ReO2F2; разработаны также спосо-
бы термического восстановления соединений MnF2, МпС12, МпВг2,
Мп12 с помощью Na или Mg, термического восстановления окис-
лов МпО2, Мп3О4, Мп2О3, МпО углеродом или алюминием, а также
термического разложения соединений Re3Cl9, ReO2, Mei[ReI6]
(где Mei = К+, NH+) и катодного восстановления некоторых
соединений марганца (MnSO4*7Н2О), технеция (As(C6H5)4TcO4),
рения (MeiReO4) в кислых растворах.
Марганец, рений и их сплавы широко используются в совре-
менной технике.
Наиболее устойчивы соединения двух-, четырех- и семивалент-
ного марганца, четырех- и семивалентных технеция и рения. Сое-
динения одно- и пятивалентного марганца, одно-, двух-, трех-,
пяти- и шестивалентных технеция и рения менее устойчивы.
В табл. 46 и 47 приведены некоторые константы соединений
этих элементов.
Если в состоянии низших степеней окисления проявляются
металлические свойства этих элементов, то в состоянии высших
степеней окисления обнаруживается неметаллический кислотный
характер и способность к образованию ковалентных связей.
Высшие окислы Mn2O7, Тс2О7, Re2O7 являются ангидридами,
дающими при растворении в воде соответствующие кислоты НМпО4
НТсО4, HReO4, а при взаимодействии с основаниями щелочных
металлов — соответствующие соли MeiMnO4, MeiTcO4, MeiReO4,
MeiReO5. (
В табл. 48 приведены формулы галогенидов металлов подгруп-
пы марганца (сгруппированные по валентностям). Из всех воз-
можных гептагалогенидов выделен только ReF7, из гексагалоге-
нидов TcF6, ReF6, ТсС16, ReCl6, пентагалогениды, кроме ReF5,
25*
Таблица 46
О <я & Желтые гекса- гональные кристаллы 484,3958 6,2 304 355 297,6
МП2О7 1 1 Зеленые кри- сталлы или | красная мас- лянистая жидкость 221,8720 2,3969 для жидкости Разлагается со взрывом при 10 1 1
ReO3 Красные ку- бические кристаллы 1 234,1982 6*9 1 1 147
МпОз j Пурпурно- красные па- ры 102,9363 1 Разлагается 1 1
! ReO2 Черно-корич- невый кри-1 сталличе- ский поро- шок 218,1988 11,4 1 Превращается при нагрева- нии в Не2С>7 1 103,4
М11О2 Черные ромбиче- ские кристаллы или черно-ко- ричневый поро- шок 86,9369 5,03 Разлагается выше 580 1 126
Соединение j Вид Молекулярный вес Плотность (при 20°), г/см3 Температура плав- ления, °C Температура кипе- ния, °C Теплота образова- ния, ккал/моль
ПОБОЧНАЯ ПОДГРУППА VII ГРУППЫ
389
Таблица 47
Соединение MnS ReS MnSa ReSg
Вид a-MnS—зеле- ные кубиче- ские кри- сталлы p-MnS—розо- вые кубиче- ские кри- сталлы у-MnS — розо- вые гексаго- нальные кристаллы Черный аморф- ный поро- шок Коричнево- красные гек- сагональные кристаллы Черные гексаго- нальные кристал- лы
Молекулярный вес 87,0021 218,264 119,0661 250,328
Плотность (при 20°), г/сл3 a-MnS 3,9 7,11 3,71 7,5
Температура плавления, °C 1615 Разлагается при 300 Разлагается при 170 Разлагается при 600
Температура кипения, °C Перегоняется без разложе- ния — — —
Теплота обра- зования, ккал /моль 48,75 — — —
ReCl5, ReBr5, не известны, моногалогениды представлены Mn2F2,
Rel, зато ди-, три-, тетрагалогениды марганца и рения весьма
многочисленны. Галогениды, соответствующие высшим степеням
окисления, обладают ковалентными связями, они легко летучи,
растворимы в органических растворителях и гидролизуются водой.
Соединения двухвалентного рения малочисленны, они менее
устойчивы и имеют менее выраженные восстановительные свойства,
чем соединения двухвалентного марганца. Соединения трех-,
четырех- и шестивалентного рения более устойчивы, чем соответ-
ствующие соединения марганца.
390
ПОБОЧНАЯ ПОДГРУППА VII ГРУППЫ
Таблица 48
Mn2F2 MnF2 M11F3 MnF4 — .— —
— — — TcF6 ——
— — —. ReF4 ReF5 ReFe ReF?
MnCl2 MnClg МпСЦ .
— — — TcCl4 TcClg .—
— ReCl2-4H2O RejClg Re3Cli2 ReCl5 ReClfj —
— MnBr2 — — .— .— .—
——— — _— __
— — RegBrg ReBr4 ReBr5 — —
— MnJ2 —. — — — .—
— — __ —
ReJ BeJ2 RegJg ReJ4 •— — —
Помимо простых соединений марганца и рения существуют ме-
таллоорганические соединения и соединения включения.
В отличие от элементов главной подгруппы VII группы перио-
дической системы, которые обнаруживают отчетливо выраженные
неметаллические свойства, элементы подгруппы марганца проявля-
ют металлический характер.
По электронному строению и физико-химическим свойствам,
определяемым этим строением, по склонности к образованию элект-
роположительных ионов и координационных соединений металлы
подгруппы марганца относятся к переходным металлам.
МАРГАНЕЦ Мп
Z = 25; ат. вес = 54,9381
Валентность (I), II, III, IV (V), VI, VII
Массовое число природного изотопа 55
Электронная структура атома
Электронная структура атома
Мп3+ для 3d- и 4з-орбиталей:
марганца: K-L ’3&3p*3d5 *4s2.
марганца и катионов Мп2+,
3ds
t t t t t
Мп
4s’ _________за» 4s ______________за* 4s
til t t t t t Г“| Ititltiti I П
Mn’+ Mn’.-
МАРГАНЕЦ
391
ИСТОРИЧЕСКАЯ СПРАВКА
Пиролюзит МпО2 был известен еще в древности. Так, например,
у Плиния есть упоминание об использовании пиролюзита при
изготовлении стекла и эмалей. Долгое время пиролюзит при-
нимали за магнетит Fe3O4, а затем его рассматривали как минерал,
в состав которого входят железо, цинк, олово и кобальт.
В 1774 г. Шееле установил состав природной двуокиси мар-
ганца и в том же году Ган впервые получил из нее металлический
марганец. Новый металл, полученный восстановлением при на-
гревании черной окиси МпО2 (пиролюзита) углем в 1785 г., Гитон
де Морво назвал марганцем в отличие от Бергмана, который назы-
вал его магнием, потому что в прошлом минералы марганца назы-
вались magnezia nigra.
НАХОЖДЕНИЕ В ПРИРОДЕ
В природе марганец редко встречается в свободном состоянии,
но очень распространен в виде соединений (окислы, карбонаты,
фосфаты, силикаты, сульфаты, сульфиды, вольфраматы, бораты)
в различных минералах. В малых количествах некоторые соеди-
нения марганца находятся в организме человека (в печени, сердце,
железах и т. п.), в организмах животных, насекомых, бактерий
и в растениях.
Содержание марганца в земной коре составляет 0,10 вес.%.
К наиболее важным минералам марганца относятся следующие.
Гаусманит, Мп3О4 или Mn2+Mng+O4, содержит 72% Мп и обра-
зует блестящие черные тетрагональные кристаллы с плотностью
4,7—4,9 z!cms и твердостью 5 по шкале Мооса. В природе гаусма-
ниту сопутствуют браунит, магнетит и гематит.
Браунит, Мп2О3, существует в виде черных тетрагональных
кристаллов с плотностью 4,75—4,82 г/см3 и твердостью 6 по шкале
Мооса. Окислением этого минерала получают псиломелан.
Пиролюзит, МпО2, содержит 63,2% Мп, встречается в виде
черных тетрагональных кристаллов со структурой типа рутила:
плотность 4,75 г/см3, твердость 2,5 по шкале Мооса. В качестве
разновидностей двуокиси марганца, встречающихся в природе,
можно назвать рамсделит (черные орторомбические кристаллы),
псиломелан (коричнево-черные моноклинные кристаллы) и крип-
томелан (черные моноклинные кристаллы).
В пиролюзите в виде примесей содержатся Fe, Al, Ba, Sr, Са,
Cu, Ni, Co, Zn, Bi, In, K, Li и другие металлы.
Пиролюзит и его природные разновидности находятся в окис-
ленных зонах залежей марганцевых руд.
Манганит, Мп2О3 -Н2О или МпООН, встречается в виде темно-
серых (почти черных) моноклинных призматических кристаллов
с плотностью 4,2—4,33 г!см? и твердостью 3—4 по шкале Мооса.
392
ПОБОЧНАЯ ПОДГРУППА VII ГРУППЫ
Родохрозит (или марганцевый шпат), МиСО3, содержит 47,8%
Мп, часто загрязнен Fe, Mg, Са, Zn, Со; встречается в виде розовых
тригональных кристаллов с плотностью 3,6—3,7 г!см3 и твердо-
стью 3,7—4,5 по шкале Мооса.
Литиофилит (трифилит), Li(Mn2+, Fe3+)PO4, встречается
редко (обычно в сопровождении кварца, берилла и минералов
лития); представляет собой желтовато-розовые (или коричнево-
красные) ромбические кристаллы с плотностью 3,5 г!см? и твер-
достью 4,5—5 по шкале Мооса.
Триплит, (Mn2+, Fe2+)2PO4F, встречается в виде коричневых
или розовых моноклинных кристаллов с плотностью 3,44—
3,87 г!см3 и твердостью 4—5 по шкале Мооса; ему сопутствуют
кварц, берилл, родохрозит, аппатит, флюорит и др.
Родонит, (Mn2+, Са2+, Fe2+)SiO3, встречается в виде розовых
(или серовато-красных) триклинных кристаллов с плотностью
3,40—3,75 г!см3 и твердостью 5—5,5 по шкале Мооса; ему сопут-
ствуют родохрозит и сульфиды MnS, ZnS, PbS.
Спессартин, Mn3Al2[SiO4]3, относится к группе гранатов
и встречается в виде красных (оранжево-желтых или коричневых
в зависимости от загрязнений) кубических кристаллов с плот-
ностью 4,18 г!см3 и твердостью 7—7,5 по шкале Мооса.
К другим минералам марганца относятся: пирохроит Мп(ОН)2, манга-
нозит МпО, вернадит МпО2-пН2О, креднерит CuMn2O4, хюбнерит MnWO4,
якобсит MnFeOi, биксбиит (Mn, Fe)2O3, алабандин MnS, гауерит MnS2, ман-
ганокальцит (Мп, Са)СО3, малладрит MnSO4-7H2O, смикит MnSO4-H2O,
пурпурит (Mn, Fe)PO4, натрофилит NaMnPO4, стюартит Мп3(РО4)2-4Н2О,
реддингит (Мп, Fe)3(PO4)2-3H2O, тенроит Mn2SiO4, бустамит
(Мп, Ca)(SiO3)2, пиросмалит (Мп, Fe)g(OH, Cl)10,SieO15, суссексит МпНВО3.
Кроме того, минералы железа и некоторые минералы цинка
содержат различные количества марганца.
Залежи марганцевых руд находятся в СССР, Югославии,
Болгарии, Португалии, Австрии, Италии, Индии, Китае, Индо-
незии, Турции, ДРВ, ОАР, Гане, Марокко, Кубе, США, Арген-
тине, Бразилии, Румынии, на Филиппинах, в Африке (ЮАР,
Ангола, Берег Слоновой Кости и др.).
Большинство минералов марганца служат для получения
ферромарганца и марганцевых чугунов и сталей.
ПОЛУЧЕНИЕ МЕТАЛЛИЧЕСКОГО МАРГАНЦА
Металлический марганец получают в процессе термического
восстановления безводных галогенидов марганца(П) натрием,
магнием или водородом, а также электролизом водных растворов
сульфата марганца(П) с сульфатом аммония. Получение чистого
металлического марганца достаточно сложно, так как со многими
восстановителями (Al, С, Mg и т. д.) марганец образует сплавы.
МАРГАНЕЦ
393
Восстановление окислов марганца углем
или алюминием
В результате восстановления окислов марганца МпО2, Мп3О4
Мп2О3, МпО углем при высокой температуре в тигле из MgO
или СаО получают металлический марганец, загрязненный угле-
родом. Для получения некарбидизированного марганца берут из-
быток окиси марганца. Обычно восстановление окислов марган-
ца углем осуществляется в тиглях из MgO в электрических печах
в присутствии сульфата натрия. Этот процесс служит главным
образом для получения ферромарганца путем восстановления
смеси окислов железа и марганца.
Для получения металлического марганца методом алюмотер-
мического восстановления берут Мп3О4, получаемый прокалива-
нием при 1000—1100° окислов МпО2, Мп2О3:
580-620° 950-1100°
6МпО2----^77“* ЗМп2О3 ---77——2Мп3О4
-3/2O2 — 1/2O2
ЗМп3О4 + 8А1 = 9Мп + 4А12О3 + 503,76 ккал (~1979°)
Чтобы уменьшить потери марганца в виде шлака, в реакцион-
ную смесь добавляют СаО, который уменьшает вязкость шлака
и облегчает расслоение реакционной массы. В шлак переходит не
более 20% марганца в виде алюмината марганца(П) МпА12О4.
Полученный алюмотермическим способом металлический мар-
ганец содержит 94—96% Мп, 6—4% примесей Fe, Si, Al. При
работе с избытком алюминия получают сплав марганец — алю-
миний.
Алюмотермический способ служит также для получения спла-
вов железо — марганец, хром — марганец, никель — марганец,
ванадий — марганец и др.
Восстановление безводных галогенидов марганца(П) натрием,
магнием или водородом
В результате восстановления (при нагревании без доступа
воздуха) безводных галогенидов марганца(П) (MnF2, МпС12,
MnBr2, Мп12) натрием, магнием или водородом получают кри-
сталлы металлического марганца, а также соответствующий гало-
генид или галогеноводород:
MnX2 + 2Na = Mn + 2NaX (X = F", С1~, Вг-, I-)
МпХ2 + Mg = Мп + MgX2
МпХ2 + Н2 = Мп + 2НХ
394
ПОБОЧНАЯ ПОДГРУППА VII ГРУППЫ
Электролитическое получение
Электролиз водного раствора, содержащего 70 г MnSO4-6H2O
и 150—210 г (NH4)2SO4 на каждый литр и нейтрализованного ам-
миаком или известковой водой до pH = 6,5, ведут в электролизере
с диафрагмой из алунда (асбеста или пористой керамической пла-
стинки), анодами из свинца и катодами из нержавеющей стали
(18% Сг, 12% Ni, 2% Мо) при плотности тока на катоде 2а!дм2.
На катоде образуется порошкообразный металлический у-мар-
ганец. Для электролиза можно использовать также водный ра-
створ, который содержит 95—120 г MnSO4 >6Н2О и 125 г (NH4)2SO4
на каждый литр и нейтрализован аммиаком до pH = 7—7,4;
стенки электролизера покрыты свинцом, аноды изготовлены из
сплава РЬ — Ag, катоды — из меди или латуни, плотность тока
на катоде 4 а!дм2. В результате получают металлический марга-
нец 99,7—99,9%-ной чистоты с примесями свинца, меди, серы и т. д,
ОЧИСТКА
Для получения пластичного (ковкого и вязкого) металлического
марганца сырой металл подвергают двойной перегонке в вакууме,
затем переплавляют в атмосфере аргона и закаливают при высокой
температуре. Перегонка в вакууме осуществляется в высокочастот-
ной печи при 1—2 мм рт. ст.
ФИЗИЧЕСКИЕ И ХИМИЧЕСКИЕ СВОЙСТВА
Цвет металлического марганца в компактном состоянии светло-
серый, а в порошкообразном — серовато-черный.
Металлический марганец известен в четырех аллотропных моди-
фикациях, которые отличаются между собой типом кристалличе-
ской решетки, плотностью, механическими свойствами и термиче-
ской устойчивостью. Модификация а-Мп в виде кубических кри-
сталлов с плотностью 7,44 г!см? сохраняет твердость и хрупкость
до 700° и устойчива при температурах ниже 730°. Модификация
Р-Mn также в виде кубических кристаллов обладает плотностью
7,29 г!см? и механическими свойствами, промежуточными между
а- и у-модификациями. Модификация у-Mn в виде гранецентриро-
ванных тетрагональных кристаллов с плотностью 7,21 г!смъ пла-
стична до 1150°. Модификация 6-Мп, менее изученная, существует
в пределах температур от 1150° до точки плавления.
По данным многочисленных работ установлены следующие
средние значения температур, при которых осуществляются обра-
тимые превращения вышеперечисленных модификаций:
742° 1101’ 1160° 1245°
а-Mn Р-Mn ~ у-Mn ~ 6-Мп-----------------► Мп
700° 1079° 1143°
(Расплавленный)
МАРГАНЕЦ
395
Металлический марганец имеет т. пл. 1245°, т. кип. 2150°, твер-
дость 5—6 по шкале Мооса (при загрязнении становится более
твердым), проявляет триболюминесцентные свойства, характе-
ризуется необычно высоким электрическим сопротивлением (при-
мерно в 2 раза больше, чем у висмута), парамагнитен.
Нестабильные изотопы марганца с массовыми числами 50, 51,
52, 54 и 56 получают в результате бомбардировки соседних эле-
ментов Fe, Cr, V, Со нейтронами, протонами, а-частицами и фо-
тонами.
Известны многочисленные сплавы марганца с элементами Fe,
Ni, Cr. W, V, U, Pd, Pt, Ag, Cu, Au, Hg, Zn, Mg, Al, B, Sn, Ge,
Ti, Zr, Th, Bi, Sb, As, C, Si, N, P, Se, Те. Наиболее важны спла-
вы Fe — Мп, Cr — Мп, Мп — Си, Мп — Ni, Си — Мп — Ni,
Си — Zn — Мп. Ферромарганец представляет собой сплав железа
с марганцем и содержит 60—90% Мп. Состав марганцовистой стали
примерно следующий: 83—87% Fe, 12—15% Мп, 1—2% С, а содер-
жание Мп в зеркальном чугуне составляет 12—20%.
Химические свойства марганца в значительной степени зави-
сят от его чистоты, поскольку даже малые количества примесей
существенно меняют его реакционную способность.
При обычной температуре в атмосфере сухого воздуха метал-
лический марганец окисляется только с поверхности. Во влажном
воздухе этот процесс идет в объеме.
При высокой температуре марганец горит на воздухе или в кис-
лороде, образуя окислы, состав которых зависит от температуры.
Мп 1/2О2 = МпО -|- 96,5 ккал 2Мп 3/2О2 = Мп2О3 -)- 233 ккал
Мп + О2 = МпО2 + 125,5 ккал ЗМп + 2О2 = Мп3О4 + 345 ккал
Чистый металлический марганец не разлагает воду при ком-
натной температуре и очень медленно взаимодействует с парами
воды, причем образуется Мп(ОН)2 и выделяется водород. Если
металлический марганец загрязнен углеродом, азотом и т. д.,
он медленно реагирует с холодной водой и быстрее при нагревании:
Мп + 2Н2О = Мп(ОН)2 + Н2 Мп3С + 6Н2О = ЗМп(ОН)2 + СН4 + Н2
MnxNy + 2хН2О = хМп(ОН)2 + V/2N2 + хН2
При обработке фтором нагретого порошка металлического мар-
ганца образуются MnF2 и MnF3. В результате взаимодействия при
нагревании газообразного хлора, паров брома и галогеноводородов
НС1, НВг с металлическим марганцем получают МпС12 или соот-
ветственно МпВг2. При взаимодействии рассчитанного количества
брома с марганцем, нагретым на водяной бане, в присутствии абсо-
лютного эфира образуется оранжево-желтая масса, которая при
хранении в эксикаторе над конц. H2SO4 превращается в прозрач-
ные блестящие желтые иглы МпВг2-(С2Н5)2О.
396
ПОБОЧНАЯ ПОДГРУППА VII ГРУППЫ
Прямым взаимодействием при нагревании марганца с серой
в зависимости от их соотношения получают MnS или MnS2.
При нагревании марганца в атмосфере азота (температура
выше 1200°) образуется Mn5N2, а при действии газообразного NH3
на марганец (при нагревании) — Mn3N2.
Кремний, сурьма, висмут и цинк образуют при нагревании
с марганцем сплавы типа твердых растворов и интерметаллических
соединений. При растворении углерода в расплавленном марганце
образуется карбид Мп3С.
При нагревании порошка марганца с фосфором в закрытой
вакуумированной трубке образуется МпР2, который разлагается
при 400°, превращаясь в энергично окисляющийся на воздухе МиР.
При нагревании пирофорный марганец разлагает окись и дву-
окись углерода:
Мп + СО = МпО + С + 33,3 ккал 2Мп + СО2 = 2МпО + С + 46,3 ккал
При действии смеси СО2 и Н2 на порошкообразный металличе-
ский марганец образуются МпО и Мп3С:
4Мп 4- СО2 + Н2 = МпО + Мп3С + Н2О
Если марганец обрабатывать при нагревании разбавленными
кислотами (НС1, HNO3, H2SO4), то получаются соответствующие
соли марганца(П) и выделяется водород. Концентрированная
серная кислота, медленно на холоду и быстро при нагревании,
растворяет марганец:
Мп + 2H2SO4 == MnSO4 + SO2 + Н2О
Концентрированная азотная кислота восстанавливается марган-
цем до NO и NO2.
Концентрированные растворы щелочей NaOH, КОН не вза-
имодействуют с марганцем. При нагревании порошкообразного
металлического марганца с расплавленным КС1О3 образуется
КМпО4, а с расплавленным KNO3 — К2МпО4.
Химическая реакционная способность марганца показана на
следующей схеме:
' с Н2О Мп(ОН)2
с О2 —> МпО, МпО2, Мп2О3, Мп3О4
с F2 —> MnF2, MnF3
с С12 или НС1 —* МпС12
с Вг2 или НВг —> МпВг2
Марганец--------► с s> N2> р> с “MnS, MnS2, Mn5N2, МпР2, МпР, Мп3С
Нагревание с NHs Mn3N2
с СО или СО2 —> МпО
с СО2-|-Н2 —> МпО4-МпзС
сразбавл.НС1,НКО3 ->МпС12-6Н2О, Mn(NO3)2-6H2O
с конц. H2SO4 —► MnS04
с распл. КС1О3 —> КМпО4
МАРГАНЕЦ
397
С физиологической точки зрения марганец является важным
элементом для жизнедеятельности растений.
ПРИМЕНЕНИЕ
В металлургии сплавы марганца с железом (ферромарганец)
или с алюминием и кремнием используются в качестве раскислите-
лей и десульфурантов чугунов и сталей, а также как специальные
легирующие добавки. Окислы и сульфиды марганца образуют
легкий шлак, который полностью отделяется от стали. В сталях
марганец способствует росту зерен, уменьшению теплопровод-
ности и увеличению коэффициента линейного расширения.
Стали, легированные 1,0—2,0% Мп и менее 0,5% С, применя-
ются для изготовления рельсов, валов моторов, зубчатых колес
и т. п. Из легированных сталей аустенитного типа, содержащих
10-15 % Мп и 0,9—1,4% С, изготавливают детали, обладающие
большим сопротивлением удару и истиранию (всевозможные щеч-
ки, тормозные колодки, шары к мельницам). Аустенитные стали
с марганцем не магнитны.
В сплавах цветных металлов марганец используют в качестве
раскислителя или одного из компонентов. Он способствует увели-
чению их твердости и коррозионной устойчивости. Сплавы Си —
— Мп применяются для изготовления турбин, из сплавов Ni —Мп
изготавливают электроды свечей зажигания. Известны латуни
с марганцем и алюминий-марганцевые бронзы.
В качестве примера сплава с большой пластичностью можно
привести композицию состава: 72% Мп, 18% Си и 10% Ni, а в ка-
честве очень твердого сплава — 80% Мп и 20% Си.
Марганец используется в промышленном масштабе для защит-
ного покрытия металлов; такое покрытие лучше цинкового, так
как марганец имеет более отрицательный, чем цинк, электродный
потенциал. По микротвердости марганцевое покрытие находится
между хромовым и никелевым покрытиями. Покрытия из мар-
ганца (или сплава марганец — никель) обладают большой кор-
розионной устойчивостью.
СОЕДИНЕНИЯ (ОБЩИЕ СВОЙСТВА)
Известно относительно мало соединений одно-, трех-, четы-
рех-, пяти-, шести- и семивалентного марганца и очень много
соединений двухвалентного марганца.
В табл. 49 приведены формулы и указан цвет ряда соединений
марганца, сгруппированных по валентностям.
Для одновалентного марганца известны только координацион-
ные соединения. Помимо простых соединений известно много коор-
398
ПОБОЧНАЯ ПОДГРУППА VII ГРУППЫ
динационных соединений двух-, трех- и четырехвалентного мар-
ганца.
Соединения марганца(П) и в меньшей степени марганца(Ш)
проявляют металлические свойства (образуют основания и явля-
ются восстановителями). Соединения марганца(У1) и (VII) про-
являют неметаллический характер (образуют кислоты и соеди-
нения с ковалентными связями).
Переход марганца из одного валентного состояния в другое
сопровождается изменением цвета его соединений (в порядке воз-
растания валентности): бесцветный, розовый, красный, корич-
невый, черный, зеленый, фиолетовый. За эту гамму цветов, при-
сущих соединениям марганца, его называют «химическим хаме-
леоном».
Соединения семи-, шести-, четырех- и трехвалентного марганца
являются окислителями, и их окислительная способность зависит
от pH раствора. Ниже указаны окислительно-восстановительные
потенциалы некоторых реакций, где принимают участие ионы
МпО~, МпО*-, MnOg" (соответственно МпО2,) Мп3+, в кислой,
нейтральной или щелочной среде при 25°.
M11O4 ф 8Н+ ф 5е~ Мп2+ ф 4Н2О ф!,52 в
MnOj + 4Н+ + Зе- МпО2 ф 2Н2О ф!,695 в
MnOj ф 2Н2О ф Зе- МпО2 ф 4ОН“ ф0,57 в
MnOj ф е‘ч± MnOJ- ф0,564 в
МпО2~ ф 4Н+ ф 2е“ ч* МпО2 ф 2Н2О ф!,26 в
МпО|“ ф 2Н2О ф 2е~ ш МпО2 ф 4ОН- ф0,71 в
МпО2 ф 4Н+ ф е- Мп3+ ф 2Н2О ф0,95 в
МпО2 ф 4Н+ ф 2е- Мп2+ ф 2Н2О ф!,23 в
Мп3+ ф е~ Мп2+ ф!,51 в
Помимо соединений марганца, отвечающих семи состояниям
валентности, известны нестехиометрические окислы, металлоорга-
нические соединения и соединения включения.
Неорганические соединения
Соединения одновалентного марганца
Для марганца не характерно электроположительное однова-
лентное состояние. Известно небольшое число соединений марган-
ца(1), например гекса- и трицианокомплексы Me|(Mn+(CN)6],
Me*[Mn+(CN)3], где Mei = К+, Na+, гексаизонитрильные комплек-
сы [Mn+(CNR)6]X, где X = 1“, 1“, С1О~, и димер монофторида
Мп 2?2*
МАРГАНЕЦ
399
При действии порошкообразного алюминия, сплава Деварда,
амальгамы натрия или амальгамы алюминия на фиолетовый щелоч-
ной раствор Na4[Mn2+(CN)e]-8Н2О или K4[Mn2+(CN)6] в атмо-
сфере водорода при температуре ниже 22° получают коричнево-
желтый раствор, из которого при добавлении раствора КС1, со-
держащего КОН и KCN (или раствора цианида щелочного металла
с ацетатом щелочного металла), выделяют бесцветные кристаллы
K5[Mn+(CN)6l:
3Na4[Mn2+(CN)e] + Al + 4NaOH + 2H2O =
= 3Na5[Mn+(CN)e] + Na[Al(OH)4(H2O)2]
3Na6[Mn+(CN)e] + 15K+ = 3Ks[Mn+(CN)e] + 15Na+
Соединение Ks[Mn+(CN)6] представляет собой бесцветные тет-
раэдрические мелкие кристаллы, плохо растворимые в воде.
На воздухе это соединение медленно, а в воде быстро окисляется.
Кристаллы окрашены в синий цвет, растворимы в спирте или
ацетоне, диамагнитны. Комплекс восстанавливает аммиачные
растворы серебра и двухвалентные катионы Pb2+, Cd2+ до металла.
Соединение Na5[Mn+(CN)e] образует бесцветные кристаллы,
растворимые в воде и легко окисляющиеся на воздухе и в воде;
растворы его в спирте или ацетоне окрашены в синий цвет.
Была получена двойная соль K2(Mn+(CN)3] -K4[Mn2+(CN)e],
почти не растворимая в воде.
Известны гексаизонитрильные комплексы марганца(1)
[Mn+(CNR)e]C104, [Mn+(CNR)e]I, [Mn+(CNR)e]I5; это устойчивые
и диамагнитные соли желтого (полииодид — коричневого) цвета.
Димер монофторида марганца, Mn2F2, получают термической
диссоциацией тетрафторида марганца MnF4 (полученного при
действии плавиковой кислоты на МпО2) при 450—500°:
2MnF4 = Mn2F2 -J- 3F2
Соединения двухвалентного марганца
Известны многочисленные соединения, в которых электропо-
ложительный двухвалентный марганец находится в виде катио-
на Мп2+ или анионов [Мп(ОН)4]2~, (Мп(ОН)6]4-, [MnF3]_, [MnF4]2-,
[MnX3]_, [MnXJ2-, [MnXe]4-, где X = Cl- или Вг_, (Mn(PO4)2]4~,
[Mn(SCN)6]4~, [Mn(CN)e]4~, [Mn(CN)3]-, [Mn(C2O4)3l4-, [Mn(C2O4)el10-,
[MnN(CH2 —-CH2OH)3(OH)4]2~, [MnN(CH2 —COO)3(OH)2]3-. Суще-
ствование большого числа соединений марганца(П) свидетель-
ствует о том, что двухвалентное состояние марганца можно рас-
сматривать как одно из самых устойчивых состояний.
Катион Мп2+ (электронная структура Is2 •2s22pe •3s23p63d5),
обладая наполовину заполненной 3</-орбиталью (3d5), проявляет
большую устойчивость; соединения сильно парамагнитны, окра-
400
ПОБОЧНАЯ ПОДГРУППА VII ГРУППЫ
шены в розовый цвет, имеют отчетливо выраженный основной
характер, окисляются только энергичными окислителями, ката-
лизируют окисление двуокиси серы, дихлорида олова, щавеле-
вой и винной кислот, льняного масла и т. д.
Ниже приведены примеры окислительно-восстановительных
реакций, по которым осуществляется окисление катиона Мп2 +
в кислой и щелочной средах:
2MnSO4 + 5РЬО2 + 6HNO3 = 2HMnO4 + 3Pb(NO3)2 + 2PbSO4 + 2Н2О
2Mn(NO3)2 + 5KBiO3 + 16HNO3 = 2HMnO4 + 5Bi(NO3)3 + 5KNO3 + 7H2O
2MnSO4 + 5Br2 + 16KOH = 2KMiiO4 + lOKBr + 2K2SO4 + 8H2O
Для окисления катиона Mn2+ в кислой среде можно исполь-
зовать (NH4)2S2O8 при каталитическом действии иона Ag+, а в ще-
лочной среде окислителями могут быть Н2О2, Cl2, NaOBr и др.
2Mn(NO3)2 + 5(NH4)2S2O8 + 8Н2О =
= 2НМпО4 + 5(NH4)2SO4 + 5H2SO4 + 4HNO3
Окисление катиона Мп2+ до аниона Мп()“ сопровождается
изменением окраски от светло-розовой (бесцветной) до фиолетовой.
При прокаливании соединений марганца(П) с окислительно
щелочной смесью (К2СО3 KNO3, КОН + KNO3, К2СО3 +
+ КС1О3, КОН + КС1О3) получают манганат щелочного металла
зеленого цвета:
MnSO4 + 2К2СО3 + 2KNO3 = K2MnO4 + 2KNO2 + K2SO4 + 2СО2
В щелочной среде катион Мп2+ неустойчив и очень легко окис-
ляется кислородом воздуха или другими окислителями.
МпС12 + 2КОН + V2O2 = Н2МпО3 + 2КС1
МпС12 + 4КОН + С12 = Н2МпО3 + 4КС1 + Н2О
Mn(OH)2 -|- V2O2 = Н'2МпО3
В слабо кислой, нейтральной или щелочной среде соли мар-
ганца(П) окисляются анионом перманганата до соединений мар-
ганца(ГУ):
3MnSO4 + 2КМпО4 + 7Н2О = бН-ДОпОз + K2SO4 + 2H2SO4
Соединения марганца(П) могут быть получены восстановле-
нием соединений марганца более высоких степеней окисления
как в кислых растворах, так и в твердом виде, i
Примеры окислительно-восстановительных реакций, по кото-
рым при восстановлении аниона МпО^ в кислой среде получают
соединения марганца(П), приведены при описании свойств пер-
манганата калия.
Восстановление аниона МпО' до катиона Мп2+ сопровожда-
ется изменением окраски от фиолетовой до светло-розовой или
бесцветной.
МАРГАНЕЦ
401
Среди наиболее важных соединений марганца(П) следует
назвать галогениды, сульфиды, сульфаты, нитраты, ортофосфаты,
карбонаты, силикаты, ацетаты.
Некоторые соединения марганца(П), такие, как гидроокись,
карбонат, оксалаты, фосфаты, проявляют сходство с соответствую-
щими соединениями магния. Соединения марганца(П) напоминают
соединения железа(П) по устойчивости в кислой среде, легкости
окисления в щелочной среде и по способности к образованию
координационных соединений, содержащих координированные
CN--группы. В табл. 49 приведены формулы и указан цвет неко-
торых соединений марганца(П).
Благодаря сильно основному характеру двухвалентный марга-
нец образует координационные соединения, большинство из кото-
рых довольно неустойчивы. Лигандами для марганца(П) явля-
ются аммиак, этилендиамин, тиоцианаты, цианиды, этилендиамин-
тетрауксусная кислота, фенантролин, формальдоксим, салицил-
альдегид, дитизон, а также кислоты щавелевая, малоновая,
винная, лимонная и другие.
Окись марганца, МпО, встречается в приррде в виде минерала
манганозита в форме зеленых мелких кристаллов с плотностью
5,18 г/см3 и твердостью 5—6 по шкале Мооса.
Соединение МпО получают прокаливанием основания или окса-
лата марганца(П) при 300° в токе азота или водорода, термиче-
ским разложением Mn(NO3)2 при 300° или МпСО3 ниже 330°
в стеклянной трубке, восстановлением окислов МпО2, Мп2О3,
Мп3О4 водородом, окисью углерода или углеродом при 700—900°,
нагреванием металлического марганца в атмосфере окиси или
двуокиси углерода.
Окись марганца МпО представляет собой парамагнитные зеле-
ные кубические кристаллы с решеткой типа NaCl (расстояние
Мп — О 2,21 А) и плотностью 5,091 г/см3, кристаллы плавятся
при 1780°, превращаясь в черную жидкость. МпО плохо раство-
рима в воде, обладает основными свойствами и под действием
кислот превращается в соединение марганца(П).
При нагревании на воздухе МпО может переходить в МпО2,
Мп2О3, Мп3О4:
200° —300° 580-620“ 850-1100°
6МпО + ЗО2------> 6МпО2 --—-> ЗМп2О3----—-> 2Мп3О4
-3/гО2 -1/гО2
Если МпО обрабатывают хлором в среде четыреххлористого
углерода, то образуются Мп2О3 и МпС12. При нагревании МпО
с SiCl4 получают MnCl2, SiO2 и С12. При 100° сульфид аммония
превращает МпО в MnS розового цвета. При охлаждении расплав-
ленной смеси МпО с NiO или СоО (в определенных интервалах
концентрации) выделяются смешанные кристаллы соответствую-
щих окислов.
26-0101
402
ПОБОЧНАЯ ПОДГРУППА VII ГРУППЫ
При действии Р0С13 на МпО образуется МпО-ЗРОС13 или
МпО-2РОС13, а в присутствии ацетона или этилацетата можно
выделить аддукты МпО-Р2О3С14 ^СгНеО и МпО-Р2О3С14 •
• 2СН3СООС2Н5.
Окись марганца является катализатором реакций дегидрогени-
зации пиперидина (600°), синтеза метана из СО и Н2 и превра-
щения уксусной кислоты в ацетон.
Гидроокись марганца(11), Мп(ОН)2, встречается в природе
в виде минерала пирохроита. В лаборатории его получают обра-
боткой растворов солей марганца щелочами при pH = 8,5 в атмо-
сфере водорода. В присутствии солей аммония осадок Мп(0Н)2
не выпадает, поскольку при этом концентрация ионов ОН- слиш-
ком мала и образуются аммиачные комплексы марганца(П).
Соединение Мп(ОН)2 представляет собой студнеобразный белый
осадок, плохо растворимый в воде и обладающий слабо основ-
ными свойствами. Гидрат окиси окисляется на воздухе с образо-
ванием соединений MnOOH, МпМпО3 или Мп(НМпО3)2, Н2МпО3,
МпО2, Мп3О4. Гидроокись марганца легко окисляется перекисью
водорода, хлором, бромом, гипохлоритами щелочных металлов
и другими окислителями.
Мп(ОН)2 + Н2О2 = Н2МпО3 + Н2О
Mn(OH)2 + 2КОН + С12 = H2MnO3 -J- 2КС1 + Н2О
Мп(ОН)2 + Н2МнО3 == МпМпО, -]- 2Н2О
Мп(ОН)2 + 2Н2МнО3 =. Мп(НМпО3)2 + 2Н2О
При растворении Мп(ОН)2 в щелочах без доступа воздуха
получают гидроксосоединения типа Ме|[Мп(ОН)4] (где Me1 =
—Na+, К+). Эти соединения при обработке растворами солей бария
или стронция превращаются в Меп[Мп(ОН)4] или Ме^1[Мп(ОН)6]
(где Me11 = Ba2+, Sr2+). При пропускании тока кислорода через
нагретый до 30 — 50° щелочной раствор Mn(OH)2 с NaOH обра-
зуются Na3[Mn(OH)6] >2,5Н2О и Na4[Mn(OH)7] -5Н2О. Гидроокись
Мп(ОН)2 в присутствии избытка щелочи при кипячении окис-
ляется кислородом воздуха до МпО2. На воздухе вместо Мп(ОН)2
образуется окисел у-Мп2О3, который превращается в а-MnOOH
и Мп5О9-мН2О.
Из раствора хлорида аммония Мп(ОН)2 вытесняет аммиак.
При растворении Мп(ОН)2 в растворе хлорида кальция или ди-
хлорида марганца образуются гидроксокомплексы.
Под действием Na2S гидроокись марганца(П) медленно пре-
вращается в MnS, с (NH4)2S эта реакция идет быстро. При нагре-
вании Мп(ОН)2 с серой и водой образуется MnS2O3. В отсутствие
кислорода воздуха Мп(ОН)2 дает хелатные соединения марган-
ца^!) с ацетилацетоном, бензоилацетоном и ацетоуксусным
эфиром.
МАРГАНЕЦ
403
Дифторид марганца, MnF2, получают нагреванием (NH4)2[MnF4J
до 300° в атмосфере СО2, действием фтористого водорода на метал-
лический марганец, нагретый до красного каления, и нагрева-
нием Mn[SiF6] -6Н2О до 1000° в токе фтористого водорода:
(NH4)2[MnF4] MnF2 + 2NH4F
Mn[SiF6].6H2O -» MnF2 + SiF4 + 6H2O
При обезвоживании (в эксикаторе над конц. H2SO4 при ком-
натной температуре) кислого раствора металлического марганца
или МпСО3 в небольшом избытке плавиковой кислоты выделяются
кристаллы MnF2-4H2O, которые мало устойчивы и почти пол-
ностью дегидратируются при 50°. При обезвоживании раствора
Мп(0Н)2 в избытке плавиковой кислоты выпадают ромбоэдри-
ческие кристаллы MnF2-5HF'6Н2О.
Безводное соединение MnF2 представляет собой розовые тетра-
гональные кристаллы с решеткой типа рутила. Они имеют плот-
ность 3,92 г!смъ, т. пл. 929,5° и т. кип. 2027°. Соединение мало
растворимо в воде, причем растворенная часть гидролизуется
с образованием оксифторида, плохо растворимо в спирте, эфире,
жидком аммиаке, растворяется в сильных кислотах при нагре-
вании, восстанавливается водородом до металлического марганца
и на холоду под действием фтора превращается в MnF3.
При температуре около 1000° дифторид марганца под действием
кислорода образует Мп3О4, с серой — MnS, а с кремнием или
бором — соответствующие силициды или бориды марганца.
При обработке водного раствора дифторида марганца фтори-
дом аммония NH4F образуется мало растворимое в воде соедине-
ние (NH4)2[MnF4] розового цвета.
Известны также фторосоли Na2IMnF4], K2[MnF4), K[MnF3],
Na[MnF3], NH4[MnF3]. Анионы [MnF3]_, [MnFJ2- малоустойчивы.
В качестве катиона внешней сферы марганец(П) входит в состав
ряда гекс а-и тетрафторосолей. Гексафторосиликат
марганца, Mn[SiF6]-6Н2О, представляет собой розовые ромбо-
эдрические кристаллы с плотностью 1,9037 г/см3, очень хорошо
растворимые в воде. Упоминаются и другие гекса- и тетрафторо-
соли: MnlZrFj-5Н2О, Mn[TiF6] -6Н2О, MnlSnFj-6Н2О, Mn[BF4]2.
• 6NH3, Mn[BF4]2.(NH4)2[BeF4].6H2O, Mn[BF4]2.6(COC10H12N2).
Дихлорид марганца, MnCl2, получают обработкой сухим хло-
ром металлического марганца, газообразным НС1 — МпО или
МпСО3. Можно применить дегидратацию кристаллогидратов
МпС12-пН2О (где п= 6, 4, 2) в токе НС1. Используется также
действие газообразного хлора на нагретые до 500—900° природ-
ные силикаты (Мп, Са, Fe)SiO3 или MnSiO3.
Безводное соединение МпС12 представляет собой парамагнит-
ные розовые гексагональные расплывающиеся на воздухе кри-
сталлы с решеткой типа CdCl2; при высушивании (160°) кристаллы
26*
•404
ПОБОЧНАЯ ПОДГРУППА VII ГРУППЫ
белеют; плотность 2,977 г/см\ т. пл. 650°, т. кип. 1290°. Дихлорид
марганца растворим в воде и спирте, плохо растворим в эфире
и аммиаке. При нагревании восстанавливается водородом до метал-
лического марганца, под действием фтора при комнатной темпе-
ратуре превращается в MnF3, обладает каталитическими свой-
ствами.
При высокой температуре кислород или пары воды переводят
дихлорид марганца в Мп 3О4 с промежуточным образованием Мп2О3:
700° 1100°
ЗМпС12+3/2О2------>- Мп2О3 + 2С12 ЗМпС12 + 2О2----->- Мп3О4 + ЗС12
920 — 1080°
2МпС12 + ЗН2О---------->- Мп2О3+4НС1 + Н,
1160—1200°
ЗМпС12 + 4Н2О----------> Мп3О4 + 6НС1 + Н2
Реакция сероводорода с МпС12 обратима:
МпС12 + H2S MnS + 2НС1
При взаимодействии МпС12 с NOC1 (—10°) образуется соеди-
нение MnCl3NO, которое бурно реагирует с водой и разбавлен-
ными кислотами; оно неустойчиво — разлагается на МпС12, С12
и NO.
Дихлорид марганца с хлоридами многих элементов образует
двойные соли, например 2MnCl2-NaCl, МпС12-6К[Н4С1-2Н2О,
MnCl2 -2Т1С13 -6Н2О, MnCl2 -2IC13 -8Н2О, MnCl2.2CdCl2.6CUH12ON2.
Известны аммиакаты MnCl2-raNH3 (где п = 6, 2, 1), аддукты
со спиртами МпС12 "иСНзОН (где п = 1, 2, 3, 4), МпС12-гаС2Н5ОН
(где п= 2 или 3). МпС12-пСДРуОИ (где 1 или 5), МпС12.
• 2С4Н9ОН, МпС12 .«CjHhOH (и = 2 или 3).
Известны кристаллогидраты дихлорида марганца МпС12-гаН2О
(где п — 6, 4, 2, 1).
Гексагидрат дихлорида марганца, МпС12'6Н2О, выделяется
из насыщенного водного раствора дихлорида марганца при —21°;
нагревание МпС12-6Н2О вызывает ступенчатую дегидратацию
до безводной соли:
а-МпС12-4Н2О
Устойчивый
МпС12-6Н2О
— 21120
>• Р-МпС12-4Н2О
Метастабильный
57,8°
- 2Н2О
— 2Н2О
150° 190°
МпС12-2Н2О -----—МпС12-Н2О------------► МпС12
— Н2О
МАРГАНЕЦ
405
Водные растворы дихлорида марганца окрашены в розовый
цвет; они образуются при растворении металлического марганца,
а также карбоната, окислов или гидроокисей марганца в соляной
кислоте (1 : 1). При обработке соляной кислотой окислов мар-
ганца высших степеней окисления в растворе образуется дихлорид
марганца и выделяется хлор. Металлические цинк, алюминий
и магний вытесняют марганец из водных растворов дихлорида
марганца в виде мелких кристалликов.
Дихлорид марганца восстанавливает хлорное золото:
2AuCl3 + ЗМпС12 = 2Аи + ЗМпС14
МпС14 + 4Н2О = Мп(0Н)4 + 4НС1
4
МпО2 + 2Н2О
При контролируемом подщелачивании раствора дихлорида мар-
ганца можно получать различные основные соли, например
Мн(0Н)С1, Мн2(ОН)3С1, Мп3(ОН)6С1. При действии ионов С1_
на дихлорид марганца образуются ацидосоединения, которые
могут содержать малоустойчивые анионы [МнС13]~, [МпС14]2-,
[МпС16]4“. Примеры комплексных солей: ЬНМпС13]*2 или 5Н2О,
К[МпС13].2Н2О, Cs[MnCl3].2H2O, Li2[MnCl4].2 или 4Н2О,
CsJMnClJ-2Н2О, (NH4)2[MnCl4].2H2O, Hg[MnCl4].4H2O,
Li4[MnCle].10H2O, Na4[MnCl6], K4[MnCl6], Mg2[MnCl6] .12H2O,
Са2[МпС16Ы2Н2О и CdJMnClJ.
При электролизе дихлорида марганца в избытке соляной
кислоты образуются МнС13 или МнС14.
Дибромид марганца, МнВг2, получают действием паров брома
на порошкообразный металлический марганец или при нагрева-
нии соединения МнВг2 •(С2Н6)2О.
При действии стехиометрически необходимого количества бро-
ма в безводном эфире на тонкий порошок металлического мар-
ганца (нагревание на водяной бане) без доступа следов воды
образуется полужидкая желтая масса, которая в эксикаторе
над конц. H2SO4 превращается в желтые прозрачные кристаллы
МиВг2 .(С2Н6)2О.
Соединение МпВг2 представляет собой парамагнитные расплы-
вающиеся на воздухе розовые пластинчатые гексагональные кри-
сталлы с решеткой типа Cell2, плотностью 4,385 г!смъ и т. пл. 698°;
МнВг2 хорошо растворим в воде и плохо — в жидком аммиаке.
Известны кристаллогидраты МпВг2-иН2О (где и = 6, 4, 2, 1).
При охлаждении насыщенного раствора дибромида марганца до
температуры ниже 13° выпадают кристаллы МнВг2-6Н2О, которые
при нагревании ступенчато дегидратируются:
13° 80°
MnBr2-6H2O _2Н (а- или 0-) МпВг2-4Н2О —~~Q> МпВг2-2Н2О
406
ПОБОЧНАЯ ПОДГРУППА VII ГРУППЫ
Водные растворы дибромида марганца получают при действии
бромной воды на порошкообразный металлический марганец или
обработкой бромистоводородной кислотой карбоната марганца(П).
Известны аммиакаты MnBr2-nNH3 (где п = 6, 2, 1) и аддукты
MnBr2.ROH (где ВОН = СН3ОН, С2Н5ОН, С3Н7ОН, С4Н9ОН),
двойные бромиды, например 2МпВг2-2MgBr2 >12Н2О, и неустойчи-
вые ацидосоли, содержащие анионы [МпВг3]_, [MnBrJ2-. В каче-
стве примеров бромосолей можно указать на CalMnBrJ-4Н2О,
HglMnBrJ -4Н2О, Sn4+[MnBr6] -6Н2О.
Дииодид марганца, Мп12, получают дегидратацией его кри-
сталлогидратов MnI2-nH2O (где п = 8, 6, 4, 2, 1) на холоду в ва-
кууме, нагреванием избытка Сп212 с тонким порошком металли-
ческого марганца при 400—500° в атмосфере азота или обработкой
тонкого порошкообразного металлического марганца иодом в без-
водном эфире. Соединение Мп12 представляет собой ферромаг-
нитные быстро расплывающиеся на воздухе розовые (темнеющие
при хранении) пластинчатые гексагональные кристаллы с решет-
кой типа Cdl2; плотность 5,01 г/см3, т. пл. 638°, т. кип. 827°.
Дииодид марганца легко растворим в воде, ограниченно раство-
рим в Asl3; обладает хорошими каталитическими свойствами.
Известны кристаллогидраты Мп12-пН2О (где п = 8, 6, 4, 2, 1),
аммиакаты MnI2'«NH3 (где п = 6, 2), двойные соли, например
Mnl2>2HgI2-6Н2О, MnI2-4As2O3-12H2O, и основной иодид Мп!2 •
• МпО-6Н2О.
Моносулъфид марганца, MnS, существует в виде трех различ-
ных кристаллических модификаций, которые различаются как
способом получения, так и свойствами.
Устойчивая модификация а-MnS иногда встречается в природе
в виде минерала алабандина (или марганцевой обманки). Может
быть получена путем обработки твердой соли марганца(П) (хло-
рида, фосфата, оксалата и нитрата или сульфата) сульфидом
аммония, восстановлением при нагревании MnSO4 серой (или угле-
родом), действием H2S на МпО, МпСО3 или MnSO4, сплавлением
Мп3О4 с KSCN, обработкой растворов солей марганца(П) суль-
фидом щелочного металла при 187°, взаимодействием аммиачного
раствора соли марганца с (NH4)2S, а также нагреванием в инерт-
ной атмосфере модификации у-MnS выше 200°. Для очистки a-MnS
кипятят со свежеприготовленным разбавленным раствором (NH4) 2S,
затем промывают водным раствором H2S, спиртом, эфиром и сушат
при 120° в вакууме.
Модификация a-MnS представляет собой зеленые кубические
гранецентрированные кристаллы с кристаллической решеткой
типа NaCl (межионное расстояние 2,60 А), плотностью 3,9 г/см3,
т. пл. 1615° и твердостью 3,5 — 4 по шкале Мооса. В вакууме
а-MnS возгоняется без разложения, а под действием паров воды
и разбавленных кислот разлагается с выделением H2S.
МАРГАНЕЦ
407
Мета стабильную модификацию р-MnS получают путем обра-
ботки разбавленных растворов солей марганца(П) сульфидами
щелочных металлов или сероводородом в ацетатных буферных
растворах при pH = 5,1. Для очистки (З-MnS промывают водным
раствором H2S, затем спиртом, эфиром и сушат в вакууме при 80°.
Модификация [З-MnS представляет собой розовые гранецентри-
рованные кубические кристаллы с кристаллической решеткой
типа ZnS. На воздухе под действием света кристаллы быстро
темнеют, разлагаются слабыми кислотами, превращаются в a-MnS
при нагревании с СО2 (320°), с NH3 (250°) и H2S (220°), а также
с небольшим количеством воды (305°); при нагревании на воздухе
выше 747° 0-MnS превращается в Мп3О4.
Метастабильную модификацию у-MnS получают при барботи-
ровании H2S через раствор 20 г МпС12-4Н2О в 500 мл воды,
не содержащей растворенного кислорода. Очистка та же, что
и для 0-MnS.
Модификация у-MnS образует розовые гексагональные кри-
сталлы с решеткой типа вюртцита ZnS; в результате нагревания
в инертной атмосфере выше 200° у-модификация превращается
в a-MnS.
Моносульфид марганца образует с сульфидами щелочных метал-
лов двойные соли, например 2MnS •Na2S, 5MnS-2Na2S, 3MnS-
•Na2S и 2MnS-K2S.
Сульфат марганца, MnSO4*H2O, можно выделить при сильном
нагревании смеси МпО2 с углеродом в конц. H2SO4. Восстановле-
ние МпО2 (мокрым путем) сульфатом железа(П) и взаимодействие
анилина с двуокисью марганца в сернокислой среде также приво-
дит к получению сульфата марганца.
2МпО2 + 2H2SO4 + С = 2MnSO4 -Н2О + СО2
3MnO2 + 6FeSO4 + 9Н2О = 3MnSO4-H2O + Fe2(SO4)3 + 4Fe(OH)s
4MnO2 + 2C6H5NH2 + 5H2SO4 -> 4MnSO4-H2O +
+ 2O=CeH4=O + (NH4)2SO4
Моногидрат представляет собой бесцветные моноклинные кри-
сталлы, растворимые в воде, плохо растворимые в спирте, превра-
щающиеся при температурах выше 152° в безводную соль MnSO4.
Соединение MnSO4 образуется при дегидратации кристалло-
гидратов или при нагревании MnO2 с FeSO4:
2MnO2 + 2FeSO4 = 2MnSO4 + Fe2O3 + ’/2О2
Сульфат марганца представляет собой бесцветный, горький
на вкус порошок с плотностью 3,19 г!см9 и т. пл. 700°, он раство-
рим в воде, плохо растворим в спирте, разлагается при нагревании.
1020-1050°
•ЗМпЭОд Mn3O4 -|- 2SO3+SO2
408
ПОБОЧНАЯ ПОДГРУППА VII ГРУППЫ
Известны кристаллогидраты MnSO4-nH2O (где п = 7, 5, 4, 2),
аммиакаты MnSO4-nNH3 (где п = 6, 5, 2, 1 и 0,5), двойные суль-
фаты типа MeiMn(SO4)2-пН2О (где Mei = Na+, К+, Rb+, Cs+,
реже Т1+ и п— 12, 4, 6, 2), кислые сульфаты MnSO4-H2SO4,
MnSO4-3H2SO4 и основной сульфат 2MnSO4 .Мп(ОН)2.
При нагревании кристаллогидраты и аммиакаты сульфата мар-
ганца^!) ступенчато теряют воду (или аммиак) и превращаются
в безводную соль MnSO4:
9° 27° 43°
MnSO4.7H2O --—MnSO4-5H2O —MnSO4-4H2O ------------—
——JH.2O —xiX12C>
60-100° 152-280°
—► MnSO4-2H2O -------> MnSO4-H2O ---тут* MnSO4
— H2O — H2O
53° 70,5° 201°
MnSO4-6NH3 —MnSO4-5NH3 -------—MnSO4-2NH3 —
—NH3 —0NH3 — NHg
220° 276°
-> MnSO4.NH3 MnSO4.0,5NH3 —- MnSO4
Известны также псевдоквасцы MnSO4-A12(SO4)3-(24 или
22) H2O и двойные сульфаты 3MnSO4 -Cr2(SO4)3, 2MnSO4 -Fe2(SO4)3.
Нитрат марганца, Mn(NO3)2, получают при нагревании (165—
200°) смеси МпО2 с NH4NO3 или при дегидратации моногидрата
Mn(NO3)2’H2O в присутствии Р4О10 при комнатной температуре.
Соединение Mn(NO3)2 представляет собой розовато-белые мел-
кие кристаллы, растворимые в воде или жидком NH3 и разлагаю-
щиеся при нагревании выше 195°:
Mn(NO3)2 МпО2 2NO2
Водные растворы нитрата марганца образуются при действии
разб. HNO3 на Мп(ОН)2 или МпСО3 или при действии конц.
HNO3 на МпО2 в присутствии щавелевой кислоты.
Известны кристаллогидраты Mn(NO3)2(где п = 6, 4, 3,
2, 1), аммиакат Mn(NO3)2-9NH3, Mn(NO3)2-2N2H4, двойные нит-
раты типа Mn(NO3)2-Meiv(NO3)4-8Н2О, 3Mn(NO3)2-2Meiii(NO3)3.
• 24Н2О (где MeIV = Се4+, Th4+ и Меш = Bi3+, La3+, Рг3+,
Nd3+, Sm3+), основной нитрат Mn(OH)NO3>H2O.
Кристаллогидрат, Mn(NO3)2-6Н2О, представляет собой быстро
расплывающиеся на воздухе бесцветные моноклинные игольчатые
кристаллы с плотностью 1,82 г! см? и т. пл. 28,5°; он растворим
в воде и спирте и выпадает из раствора, содержащего64г Mn(NO3)2
в 100 г воды при температуре от —36 до 24,7°.
Ортофосфат марганца, Мп3(РО4)2-7Н2О, получают обработ-
кой растворов солей марганца(П) избытком гидроортофосфата
натрия.
Соединение Мп3(РО4)2 >7Н2О имеет вид белого аморфного
порошка; он плохо растворим в воде, хорошо растворим в раство-
МАРГАНЕЦ
409
pax хлорида, сульфата, нитрата, сукцината аммония и в разбав-
ленных кислотах, разлагается щелочами.
При нагревании Мп3(РО4)2 -7H2O ступенчато дегидратируется:
100° 250°
Мп3(РО4)2.7Н2О , ’ Мп3(РО4)2-ЗН2О ’
— 4x120 —ZX12U
До красного каления
-* Мп3(РО4)2-Н2О -------—------->- Мп3(РО4)2
— Х12О
Тригидрат, Мн3(РО4)2-ЗН2О, встречается в природе в виде
минерала реддингита и представляет собой розовые и светложел-
тые орторомбические мелкие кристаллы с плотностью 3,102 г!см9.
Его можно получить кипячением МпСО3 с Н3РО4 или разложе-
нием МпНРО4 теплой водой.
Гидроортофосфат марганца, МпНРО4, получают дегидрата-
цией его кристаллогидратов МпНРО4-ЗН2О, МпНРО4-0,5Н2О
при 200° или действием горячего спирта на дигидроортофосфат
марганца Н4[Мп(РО4)2] -ЗН2О:
100° 200°
МпНРО4-ЗН2О ——.МпНР04-0,5Н20 —МпНРО4
— 2,5x120 —0,5x120
Кристаллогидрат МпНРО4-ЗН2О выпадает из раствора, полу-
ченного обработкой сульфата марганца(П) гидрофосфатом
натрия.
Соединение МпНРО4 растворяется в растворах хлорида, суль-
фата, нитрата аммония, переходит в ортофосфат марганца при
длительном воздействии воды и превращается в Мн2Р2О7 при
нагревании.
2МпНРО4 -> Мп2Р2О7 + Н2О
Дигидроортофосфат марганца, Н4[Мп(РО4)2] -ЗН2О, выде-
ляется из раствора ортофосфата или гидроортофосфата марганца(П)
в ортофосфорной кислоте.
Соединение Н4[Мп(РО4)2] 'ЗН2О представляет собой бесцвет-
ные призматические кристаллы, которые разлагаются во влаж-
ном воздухе и теряют одну молекулу воды при нагревании до 100°.
Известны соли K3HlMn(PO4)2]-5Н2О, Na2H2[Mn(PO4)2] -4Н2О.
Карбонат марганца, МпСО3, встречается в природе в виде
минерала родохрозита (марганцевый пшат). В лаборатории МпС03
можно получить путем нагревания в вакууме раствора карбоната
или бикарбоната натрия с дихлоридом марганца при 160°, а также
по реакциям взаимодействия дихлорида марганца с СаСО3 при
140—170° или сульфата марганца(П) с мочевиной при 180°.
Безводная соль МпСО3 представляет собой розовые ромбо-
эдрические кристаллы (изоморфны с СаСО3, FeCO3 и ZnCO3)
с плотностью 3,125 г/см3 и твердостью 3,5—4,5 по шкале Мооса;
они плохо растворимы в воде. При нагревании на воздухе МпСО3
410
ПОБОЧНАЯ ПОДГРУППА VII ГРУППЫ
разлагается, а при нагревании с сульфидами щелочных металлов
или с (NH4)2S превращается в MnS:
70-330'
[МпСО3--------- МпО + СО2
330°
ЗМпСОз — > Мп3О4 -f- СО -|- 2СО2
Когда растворы солей марганца обрабатывают при обычной
температуре карбонатами или бикарбонатами щелочных металлов
при pH = 7,6, выпадает белый осадок МпСО3-2Н2О, который
темнеет на воздухе и при 70—100° превращается вМпСОз-КгНгО.
Известны смешанные карбонаты типа Ме|1Мп(СО3)4,
МепМп(СО3)2, МеПМп3(СО3)4, где Me” = Со2+, Zn2+, Mg2+. Их
получают обработкой смеси хлоридов двухвалентных металлов
карбонатами щелочных металлов при 100—120°. Известен также
двойной карбонат К2СО3-МпСО3-4Н2О фиолетового цвета.
Ортосиликат марганца, Mn2SiO4, встречающийся в природе
в виде минерала тефроита, представляет собой серовато-розовые
орторомбические кристаллы с плотностью 4,043 г/см3, твердостью
5,5—6 по шкале Мооса и т. пл. 1313°. Его можно получить про-
стым смешением растворов дихлорида или сульфата марганца(П)
и ортосиликата натрия.
Метасиликат марганца, MnSiO3, встречающийся в природе
в виде минерала родонита, представляет собой розовые триклин-
ные игольчатые кристаллы с плотностью 3,715 г/см3 (или 3,48 г/см3
для стеклообразной модификации) и т. пл. 1273°.
Дитиоцианат марганца, Mn(SCN)2 -4Н2О, выделяется при
упаривании (ниже 15°) раствора МпСО3 в HSCN или при обра-
ботке тиоцианата бария сульфатом марганца(П).
Соединение Mn(SCN)2-4Н2О образует зеленые кристаллы, кото-
рые при дегидратации превращаются в зеленовато-желтые орто-
ромбические пластинчатые кристаллы Mn(SCN)2 «ЗН2О, затем
в желтые гексагональные пластинчатые кристаллы Mn(SCN)2 •
• 2Н2О, а при 160° — в желтую безводную соль Mn(SCN)2.
При сильном нагревании Mn(SCN)2 разлагается:
3Mn(SCN)2 Mn3C + (CN)2 + 3CS2 + 2N2
При действии избытка ионов SCN- на тиоцианат марганца(II)
образуются ацидосоли, содержащие анион [Mn(SCN)e]4-, напри-
мер Na4[Mn(SCN)e]-12H2O, K4[Mn(SCN)6] .ЗН2О, Cs4[Mn(SCN)6],
Cs2Ag2[Mn(SCN)e].2H2O, Hg2[Mn(SCN)6]-3 или 4H2O.
Ацетат марганца, Мп(СН3СОО)2 •‘ШгО, образуется при насы-
щении водного раствора уксусной кислоты карбонатом марганца.
Соединение образует розовые моноклинные кристаллы с плотно-
стью 1,59г/сл13, легко растворимые в воде (с гидролизом) и плохо
растворимые в спирте.
МАРГАНЕЦ
411
При обработке Mn(NO3)2 ледяной уксусной кислотой обра-
зуется безводная соль Мп(СН3СОО)2— очень легко окисляющиеся
белые кристаллы с плотностью 1,75 г!см3, плохо растворимые
в спирте.
Ацетат марганца(II) является катализатором реакций превра-
щения ацетальдегида в уксусную кислоту в присутствии кисло-
рода и окисления хинолина перекисью водорода.
Ацидосоединения марганц а(П), содержа-
щие координированные CN“-группы
При обработке растворов солей марганца(П) цианидом калия
выпадает цианид марганца, Mn(CN)2, который не удается выде-
лить в чистом виде.
При растворении Mn(CN)2 в избытке цианида щелочного метал-
ла образуются окрашенные координационные соединения опре-
деленного состава, содержащие анионы [Mn(CN)e]4-, [Mn(CN)3]“.
Ацидокомплексы, которые являются производными кислоты
H4[Mn(GN)e], получают в присутствии большого избытка цианида,
а производные гипотетической кислоты H[Mn(CN)3]— в при-
сутствии небольшого избытка цианида или гидролизом комплек-
сов типа MeJ[Mn(CN)6].
Гексацианомарганцовая (марганцовоцианистоводородная) кисло-
та, H4[Mn(CN)e], образуется при действии H2S на суспензию
Pb2[Mn(GN)6] в воде. При быстром упаривании в вакууме водного
раствора HJMn(CN)6] выпадают бесцветные кристаллы, неустой-
чивые и плохо растворимые в эфире. Разбавленная кислота
H4[Mn(GN)6] на воздухе при обычной температуре легко разла-
гается.
Соли гексацианомарганцовой(1Г) кислоты. При охлаждении
раствора, полученного обработкой МпО, МпСО 3 или Мп(СН3СОО)2-
•4Н2О большим избытком KCN на водяной бане при 70—80°,
в атмосфере азота выделяются фиолетово-синие кристаллы
K4[Mn(CN)e]-ЗН2О, которые, будучи неустойчивыми, окисляются
на воздухе до K3[Mn(CN)6] и затем до Мп2О3. При старении вод-
ный раствор ацидосоли K4[Mn(CN)6] -ЗН2О мутнеет и из него
выделяется зеленый осадок K[Mn(CN)3].
Реакцией двойного обмена из KJMn(CN)6] -ЗН2О были полу-
чены трудно растворимые, окрашенные в различные цвета ацидо-
соединения: Zn2[Mn(GN)e] — фиолетовое, Cd2[Mn(CN)8] — фиолето-
вое, Co2[Mn(CN)s] — красновато-коричневое, Cu2[Mn(CN)6] — жел-
тое, Pb2[Mn(CN)6]—желтое, Ca2[Mn(CN)6]—синее, Ba2[Mn(CN)6]—
синее, Al4[Mn(GN)6]3 — синее.
Известны также соли типа Me*Men[Mn(CN)6], где Me1 = К+,
Na+, NH’ и Ме1Г = Ва2+ или Си2+.
7 4
412
ПОБОЧНАЯ ПОДГРУППА VII ГРУППЫ
Соединения типа Mei[Mn(CN)6] отличаются от соединений типа
Mej!Fe(CN)6] меныпей устойчивостью и большей склонностью
к гидролизу.
Соли гипотетической трицианомарганцовой(1 Г) кислоты,
H[Mn(CN)3] или H2[Mn2(CN)6L При гидролизе ацидосоли
K4[Mn(CN)6l -ЗНгО в растворе с малой концентрацией CN~ или
при обработке раствора дихлорида марганца небольшим избыт-
ком KCN образуются соли K[Mn(GN)3] или димер K2[Mn2(GN)6]:
K4[Mn(CN)6] -> K[Mn(CN)3] 4- 3KCN
Зеленая ацидосоль, K[Mn(CN)3], плохо растворима в воде
и спирте, неустойчива во влажном воздухе, разлагается щелочами
2K[Mn(CN)3] 4- КОН 4- Н2О = K3[Mn(CN)6] + Мп(ОН)г 4- V2H2
Известны зеленые ацидосоли Na[Mn(CN)3], NH4[Mn(CN)3]..
А цетилацетонат марганца,
(/СНз\ '
/°-% \
СН I
К\0 = С/ I
ХСН3/2J,
получают в виде желтого осадка (растворимого в бензоле) при
обработке МпО или МнСО3 ацетилацетоном в атмосфере азота.
Известны также аналогичные соединения с бензоилацетоном,
салициловым альдегидом и т. д.
Кроме приведенных выше известны и другие соединения марганца (II):
хлорат Мп(СЮ3)2 или Мп(С1О3)2-6Н2О, перхлорат Мп(СЮ4)2 или Мп(С1О4)2 •
• пН2О, перренат Ми(ВеО4)2-иН2О, перхлораты [Mn(NH3)e](C104)2,
[Mn(NH3)5H20](C104)2, [Mn(COC10H12N2)6](ClO4)2, бромат Мп(ВгО3)2, иодат
Мп(Ю3)2, сульфит MnSO3-?iH2O, MnSO3-(NH4)2SO3, MnSO3-Na2SO3-H2O,
MnSO3-K2SO3, тиосульфат MnS2O3-5H2O, дитионат MnS2O6-?гН2О, кис-
лый тетратионат MnS4O6-H2S4O6, аддукт персульфата с гексаметилентетра-
мином MnS2O8-2C6H12N4-8Н2О, селенид MnSe, оксиселенид MnSe-MnO,
теллурид МпТе, селенит MnSeO3-2H2O, пироселенит MnSe2O5-?iH2O,
селенат MnSeO4-?гН2О, двойные селениты MnSe04-Me2Se04.nH2O
(где Mel = К+, Rb+, Cs+, NH£), теллурит МпТеО3-геН2О, фосфит МпНРО3-
• Н2О, пирофосфаты Мп2Р2О7, МпН2Р2О7-4Н2О, метафосфаты Мп3(Р3О9)2т
Мп2Р4О12, Мп3Р6О18, двойные ортофосфаты NH4MnPO4-Н2О, КМпРО4,
NaMnPO4, Na4Mn(PO4)2, ЫМпРО4, двойные пирофосфаты К2МнР2О7,
K2MnP2O7-8Н2О, Na2MnP2O7, Na4Mn2(P2O7)2-9Н2О, двойные метафосфаты
NaMn(PO3)3, Na2Mn3(PO3)8, тиофосфат Mn3(PS4)2, арсениты Mn3(AsO3)2 или
Mn3(AsO3)2 -5Н2О, Mn3H6(AsO3)4.2Н2О, Mn3(AsO3)2-ЗН2О, арсенаты
Mn3(AsO4)2-H2O, MnHAsO4, H4[Mn(AsO4)2], пироарсенат Mn2As2O7, двойные
арсенаты NH4MnAsO4-6Н2О, KMnAsO4, пиротиоарсенаты Mn2As2S7, метаанти-
монит Mn(SbO2)2, метаантимонат Mn(SbO3)2-нН2О, тиоантимонит Mn3(SbS3)2,
тиоантимонат Mn3(SbS4)2, силикаты (Мп, Fe)SiO3, (Мп, Ca)2SiO4, Mn3Al2(SiO4)3,
Mn2CaBe3(SiO4)3, Mn5(SiO4)2(OH)2, Mn3Ca3(Si3O9)2, MnAl2(Si2O6)(OH)4,
Mn2Na2(AlSiO4)6S2SO4, метатитанат MnTiO3, ортотитанат Mn2TiO4, фор-
миат Mn(HCOO)2-2H2O, оксалат MnC2O4-?iH2O, оксалатокомплексы
Me2[Mn(C2O4)2] -2H2O (где Mei = K+, NHJ), (NH4)4[Mn(C2O4)3]-4H2O,
(NH4)10[Mn(C2O4)6]-8H2O, (NH4)14[Mn(C2O4)8]-8H2O, бораты Mn(BO2)2,
МАРГАНЕЦ
413
МпВ4О7-иН2О, Мп3(ВО3)2, МпН4(ВО3)2-Н20, анионные комплексы, полу-
ченные обработкой соединений марганца(П) различными третичными
аминами (триэтаноламином, диэтанолмоноацетамином, этанолдиаце-
тамином, нитрилтриуксусной кислотой), и содержащие анионы
[MnN(CH2 — СН2ОН)з(ОН)4]2-, [MnN(CH2 - СН2ОН)2(СН, — СОО)(ОН)2]-.
(MnN(CH2 — СН2ОН)(СН2 — СОО)2(ОН)2]2~, [MnN(CH2 — СОО)3(ОН)2]3-.
Соединения трех валентно го марганца
Известны многочисленные соединения, в которых электропо-
ложительный трехвалентный марганец находится в виде катиона
Мп3 + или анионов [Мп2О4]2-, [МпО3]3", [Ми021-, [Мп(ОН)6]3-,
!Мп(ОН)7]4-, [MnFJ". [MnFs]2~, [MnF6(H2O)]2_, [MnF6l3-,
[M11CIs]2-, [MnCl5(H2O)l2-, [MnClJ3-, [Mn(SO4)2]-, [Mn(HPO4)2]",
IMn(HPO4)2(H2O)2J-, I Mn(CN)J3 , [Mn(C2O4)3]3-, [Mn(C2O4)2(H2O)2]-,
!Mn(OOC — CH2 — COO)3]3-, [Mn(OOC — CH2 — COO)2(H2O)2]",
IMn(O - C6H4 — COO)2(H2O)2]-, [MnN(CH2 — CH2OH)3(OH)4)-.
Катион Mn3+ неустойчив, обладает окислительными свойствами
и склонностью к образованию координационных соединений.
Соединения, содержащие катион Ми3+, легко гидролизуются
и диспропорционируют. При окислении бензидина H2N •С6Н4-
• C6H4-NH2 и 4,4'-диамино-3,3'-диметилдифенил-о-толуидина
соединениями марганца (III) образуются продукты синего цвета.
Под действием воды соединения марганца(III) гидролизуются до
МиООН, который диспропорционирует:
2Мп3+4-4Н2О 2МпООН + 6Н+ 1
S —>• Мп2++2Н2О4-2Н+
MnO2 Mn(OH)2 J
Соединения марганца(Ш) получают окислением соединений
марганца(П) хлором, перманганатом калия, анодным окислением,
а также контролируемым восстановлением соединений марганца
высших степеней окисления (например, МпО2, КМпО4) в сильно
кислой среде.
К важнейшим соединениям марганца(Ш) следует отнести без-
водную окись Мп2О3 или гидратированную окись МпООН, манга-
ниты Меп[Мп2О4], галогениды MnF3, МпС13, сульфат, квасцы,
дифосфатоманганит, ацетат, гексациано-, оксалато-, малонатоман-
ганиты, ацетилацетонат. Соединения марганца(Ш) похожи на
соединения железа(Ш) склонностью к гидролизу, окислитель-
ными свойствами и тенденцией к образованию координационных
соединений.
В табл. 49 приведены формулы и указаны цвета некоторых
соединений марганца(Ш).
Окись марганца(11Г), Мп2О3, встречается в природе в виде
минерала браунита и может быть получена в виде двух модифи-
каций: а-Мп2О3 и у-Мп2О3.
414
ПОБОЧНАЯ ПОДГРУППА VII ГРУППЫ
Модификация а-Мп2О3 образуется при окислении МпО, вос-
становлении МпО2 или Мп3О4, прокаливании MnQO3, Mn(NO3)i2,
Мп(СН3СОО)2, МнС2О4, МпС12-пН2О на воздухе (600—80$°)
или МпС12, МпВг2, Мп12 в кислороде, нагревании у-Мп2О3-моди-
фикации.
Модификация у-Мп2О3 образуется при нагревании (около 500°)
у-МпО2 в вакууме в течение 78 час, нагревании (250°) в вакууме
в течение 72 час осадка МпООН, полученного в результате добав-
ления аммиака к сульфату марганца(П) в присутствии Н2О2-
Соединение Мп203 представляет собой парамагнитное корич-
нево-черное вещество с плотностью 4,57—4,60 г!смъ. Оно превра-
щается в Мн3О4 нагреванием (950—1100°) на воздухе (нагрева-
нием в высоком вакууме при 300—600° или в атмосфере водорода
при 230°); обладает основными или слабо кислотными свойствами,
растворяется в плавиковой кислоте, диспропорционирует в раз-
бавленных кислотах (H2SO4, HNO3), неустойчиво в сильно щелоч-
ной среде.
Мп2О3 + H2SO4 МпО2 + MnSO* + Н2О
Мп2О3 + 2HNO3 MnO2 + Mn(NO3)2 + Н2О
Под действием двуокиси серы Мп2О3 превращается в MnS,
MnSO4 и MnS2O6, а под действием H2S—в MnS.
При нагревании Мп2О3 может быть восстановлена водородом,
окисью углерода, алюминием или углеродом до металлического
марганца.
Взаимодействием Мп2О3 с многочисленными окислами метал-
лов при высокой температуре получают манганиты типа шпинели
Меп1Мп2+О4] или перовскиты Меш[Мп3+О3]. Примеры манганитов
обоих типов: Zn[Mn2O4], Co[Mn2O4], Cd[Mn2O4], Ni[Mn2O4],
Mn[Mn2O4], La[MnO3], Pr[MnO3l, Nd[MnO3], DylMnOJ.
Некоторые манганиты обладают полупроводниковыми свой-
ствами.
Гидрат окиси марганца(11Г), Мп2О3-Н2О или МпООН, встре-
чается в природе в виде минерала манганита и может быть получен
действием газообразного хлора или КМпО4 на суспензию МпСО3
в воде:
ЗМпСОз + С12 + Н2О — 2МпООН + МпС12 + ЗСО2
8МпСО3 + 2КМпО* + 6Н2О «= ЮМпООН + 2КОН + 8СО2
Соединение МпООН представляет собой парамагнитные серые
моноклинные кристаллы с плотностью 4,335 г/см3, твердостью 4
по шкале Мооса, дегидратирующиеся до Мп2О3 при нагреванид
(365—400°). Под действием разбавленных кислот (HNO3, H2SO4,
НС1, НВт) МпООН диспропорционирует:
2MnOOH + 2HNO3 = MnO2 + Mn(NO3)2 + 2Н2О
МАРГАНЕЦ
415
Под действием, щавелевой, малоновой, винной, салицило-
вой и других кислот МпООН дает устойчивые соединения мар-
ганца(Ш).
В водном растворе двуокиси серы МпООН образует дитионат
и сульфат марганца(П).
Известны манганиты типа Ме'МпОа (где Mei =|Li+, Na+)
и гидроксоманганиты, например Na3[Mn(OH)6]-2,5Н2О,
Na4[Mn(OH)7]-5H2O, Ba2[Mn(OH)7], Sr2[Mn(OH)7].
Трифторид марганца, MnF3, получают действием фтора на ме-
таллический марганец или дигалогениды MnF2, МпС12, МпВг2,
Мп12.
Трифторид марганца выделяется в виде красных моноклинных
кристаллов с плотностью 3,54 г!смг и т. пл. 1077°; он растворяется
в воде без разложения, если в результате растворения образуются
концентрированные растворы на холоду, и гидролитически разла-
гается в разбавленных растворах с образованием HF, MnF2
и МпООН. При нагревании MnF3 образуется MnF2 и выделяется
фтор.
Трифторид марганца взаимодействует при нагревании с водо-
родом, серой, фосфором, мышьяком, кремнием, углеродом, бором,
трихлоридом фосфора и четыреххлористым углеродом с образова-
нием MnF2 и различных фторсодержащих соединений соответ-
ствующих элементов.
Из растворов, полученных действием плавиковой кислоты
на Мп2О3 или восстановлением КМпО4 в присутствии избытка
NH4F солями (NH4)2SO4 -FeSO4-6Н2О или KjFe(CN)6] при pH =
= 3—4, Hg2(NO3)2 при pH ~ 2, NaNO2 при pH — 6, выделяются
рубиново-красные призматические кристаллы MnF3*2H2O.
Известны фторсоединения типов MeT[MnF4], MeI[MnF4] "пИгО,
MeifMnFJ, Mei[MnF5(H2O)], Mei[MnF5(H2O)]-пН2О и Mei[MnFe].
Трихлорид марганца, МпС13, получают в виде коричневого
кристаллического осадка при добавлении СС14 к зеленому раство-
ру, полученному насыщением суспензии МпО2 в эфире газообраз-
ным НС1 при —70° или действием конц. НС1 на Мп(СН3СОО)3
при —100°.
Соединение МпС13 растворяется в органических растворите-
лях, разлагается при —40° на МпС12 и С12, очень неустойчиво
в присутствии следов воды.
В отличие от МпС13 ацидосоли типов МеИМпС15] (где Me1 = К+,
Rb+, NH+, Tl+), Mei[MnCl5(H2O)] (где Me1 = К+, NHJ) и
Mei[MnCl6] (где Mer=Cs+) значительно более устойчивы. В качестве
примеров хлоросолей можно привести К2[МпС15] — коричневые
кристаллы, (NH4)2[MnCl5(H2O)J — коричнево-красные кристаллы
и Сз3[МпС1б] — коричневые кристаллы.
Известны аддукты гидрохлоридов пиридина и хинолина и хло-
рида марганца(Ш) 2(C5H5N -НС1) -MnCl3, 2(C9H7N-НС1)-МпС13.
416
ПОБОЧНАЯ ПОДГРУППА VII ГРУППЫ
Аддукт трибромида марганца с этиловым эфиром МпВг3 •
• 3(С2Н5)2О представляет собой аморфное твердое вещество
оранжево-желтого цвета, растворимое в воде.
Трибромид и трииодид марганца не выделены.
Сульфат марганца, Mn2(SO4)3, получают нагреванием МпО2
с конц. H2SO4 при 138°:
4МпО2 + 6H2SO4 = 2Mn2(SO4)3 + 6Н2О + О2
Соединение Mn2(SO4)3 представляет собой гигроскопичные
темно-зеленые тетраэдрические кристаллы с плотностью 3,24 г!см?',
сульфат марганца очень устойчив в отсутствие влаги, мало раство-
рим в конц. H2SO4, обладает окислительными свойствами, разла-
гается при 300° по уравнению
Mn2(SO4)g —> 2MnSO4 -|- SO3 -j- V2O2
При разбавлении зеленого раствора Mn2(SO4)3 в конц. H2SO4
в растворе образуется гидросульфат марганца(Ш) коричнево-
красного цвета, а затем выпадает гидроокись МпООН.
Гидросулъфат марганца, Mn2(SO4)3•H2SO4-пН2О (где п — 4,
6, 8) или HlMn(SO4)2] • ~ Н2О, выделяется в виде коричнево-крас-
ных кристаллов; при температуре выше 150° превращается в ней-
тральную соль Mn2(SO4)3:
2H[Mn(SO4)2J. -^-Н2О -> Mn2(SO4)3 + H2SO4 + nH2O
Марганец входит в состав квасцов CsMn(SO4)2-12Н2О,
RbMn(SO4)2-12Н2О, NH4Mh(SO4)2-12H2O, KMn(SO4)2-12Н2О,
TlMn(SO4)2- 12Н2О; они окрашены в красный цвет и легко дегид-
ратируются, превращаясь в безводные комплексные соли типа
Me4Mn(SO4)2l.
Известны также двойные сульфаты: синий Mn2(SO4)3 ’A12(SO4)3,
зеленый Mn2(SO4)3-Fe2(SO4)3 и зеленовато-желтый Mn2(SO4)3 •
• Cr2(SO4)3.
Ортофосфат марганца, МпРО4-Н2О. При растворении
Мп(СН3СОО)3 в 92%-ном растворе Н3РО4 образуется кислота
Н[Мп(НРО4)2]-ЗН2О или Н3О[Мп(НРО4)2(Н2О)2], которая выде-
ляется в виде фиолетовых кристаллов, неустойчивых во влажном
воздухе. Известны ди(гидрофосфато)манганиты щелочных металлов
и аммония типа Mei[Mn(HPO4)2] •ЗНгО (где Mei = Li+, Na+, К+,
NH+) — фиолетово-красные кристаллы и гуанидиновая соль
HN = C(NH2)2H[Mn(HPO4)2] желтого цвета. Нейтральный орто-
фосфат марганца(Ш) МпРО4*Н2О получают разбавлением раство-
ров Н[Мп(НРО4)2) -ЗН2О, растворением МпО2 в сиропообразной
Н3РО4, окислением раствора MnSO4 перманганатом калия в при-
сутствии кислот Н3РО4 и СН3СООН при 100° или нагреванием
МАРГАНЕЦ
417
(200—240°) КМпО4 со смесью концентрированных Н3РО4 и H2SO4
(в соотношении 1 : 1 или 5 : 1 по объему).
Соединение МпРО4-Н2О представляет собой серовато-зеленый
порошок, плохо растворимый в воде и растворимый в Н3РО4
при температурах выше 110°, а также в НС1 с выделением хлора.
Ацетат марганца, Мп(СН3СОО)3, образуется при нагревании
уксусного ангидрида с нитратом марганца(П):
2Mn(NO3)2-6Н2О + 15(СН3СО)2О =
= 2Мп(СН3СОО)3 + 4NO2 + 24СН3СООН + V2O2
Соединение Мп(СН3СОО)3 представляет собой очень гигроско-
пичные коричневые кристаллы (хранятся в герметически закры-
тых склянках); оно окисляет а-гликоли^спирты, а-кетоны, y-ацети-
леновые гликоли и применяется для получения различных соеди-
нений марганца(Ш).
Известны кристаллогидраты Мп(СН3СОО)3-пН2О (где п = 2
или 4).
Дигидрат Мп(СН3СОО)3-2Н2О получают действием КМпО4
или хлора на раствор Мп(СН3СОО)2 -4Н2О в уксусной кислоте:
4Мп(СН3СОО)2.4Н2О + КМпО4 + 8СН3СООН =
= 5Мп(СН3СОО)3-2Н2О + КСН3СОО + 10Н2О
Соединение Мп(СН3СОО)3-2Н2О образует коричневые кри-
сталлы, устойчивые в сухом воздухе, гидролизующиеся при сопри-
косновении с водой. Это соединение окрашивает в коричневый
цвет конц. HNO3, в фиолетовый — конц. H2SO4 и превращается
в MnS при взаимодействии с сульфидом аммония. При раство-
рении Мп(СН3СОО)3-2Н2О в ледяной уксусной кислоте получают
коричневый раствор, содержащий соединение [Мп4(СН3СОО)6].
• (СН3СОО)6.
Гексацианоманганит ы, Me*[Mn(CN)e] (где Mei =
= Li+, Na+, К+, Rb+, Cs+, NH+, x/2Ca2+), представляют собой
кристаллические соединения темно-красного цвета, устойчивые
в сухом воздухе, гидролизующиеся в разбавленных растворах
с образованием Мп2О3>Н2О, изоморфные ферроцианатам(Ш),
по сравнению^ которыми они менее устойчивы.
Гексацианоманганит калия, K3[Mn(CN)6], получают действием
Н2О2 на МпСО3, растворенный в KCN, обработкой цианистым
калием Мп(СН3СОО)2 в водном растворе в присутствии воздуха
при легком нагревании в течение нескольких дней, действием
дициана (CN)2 и KCN на металлический марганец в присутствии
воздуха:
2MnCO3 + 12KCN + Н2О2 = 2K3[Mn(CN)e] + 2К2СО3 + 2КОН
2Мп(СН3СОО)2 + 12KCN + Н2О + V2O2 =
= 2K3[Mn(CN)e] + 4КСН3СОО + 2КОН
27—0101
418
ПОБОЧНАЯ ПОДГРУППА VII ГРУППЫ
Орторомбические коричнево-красные кристаллы K3[Mn(CN)6]
устойчивы на воздухе и в концентрированных растворах циани-
дов, гидролизуются при комнатной температуре в сильно разбав-
ленных растворах цианидов:
2K3[Mn(CN)e] + 10Н2О Mn2O3.H2O + 6КОН + 12HCN
При восстановлении} K3[Mn(CN)e] металлическим калием
в жидком аммиаке образуется желтый плохо растворимый про-
дукт, содержащий K5[Mn(CN)6], K6[Mn(CN)e]-2NH3. При катод-
ном восстановлении K3[Mn(CN)6] сначала получают K4[Mn(CN)e],
затем K[Mn(CN)3] cjKCN и, наконец, Мп(ОН)2.
Известны также кристаллогидраты Na3[Mn(CN)6] • пН2О (где
п = 5, 4, 3 или 2).
Т р и о к с а л а т о ма н г а н и т ы, Mei[Mn(C2O4)3], пур-
пурно-красного цвета и диоксалатодиаквоманга-
н и т ы, Mei[Mn(C2O4)2(H2O)2], желтого цвета.
При растворении МпООН в концентрированном растворе
Н2С2О4-2Н2О, охлажденном льдом, получается фиолетово-крас-
ный раствор, который неустойчив на свету и, вероятно, содержит
кислоту Н3[Мп(С2О4)3].
Триоксалатоманганит калия, К3[Мп(С2О4)3] >ЗН2О, выделяется
при добавлении спирта к холодному раствору, приготовленному
в темноте путем восстановления КМпО4 рассчитанным количест-
вом щавелевой кислоты в присутствии К2СО3:
КМпО4 + 5Н2С2О4.2Н2О + К2СО3 = К3[Мп(С2О4)3].ЗН2О + 12Н2О + 5СО2
Соединение К3[Мп(С2О4)3]-ЗН2О представляет собой темные
фиолетово-красные моноклинные кристаллы с плотностью
2,149 г!см9, устойчивые в темноте, растворимые в воде, плохо
растворимые в различных органических растворителях. При нагре-
вании это вещество разлагается:
2К3[Мп(С2О4)3]-ЗН2О —2К3[Мп(С2О4)3] — 2МпС2О4 + ЗК2С2О4 + 2СО2
Были получены триоксалатоманганиты натрия, аммония, тал-
лия и триоксалатоманганиты с комплексными катионами, напри-
мер [Co(NH3)e][Mn(C2O4)3]-nH2O.
При разбавлении концентрированных растворов триоксалато-
манганитов пурпурно-красного цвета получают диоксалатодиакво-
манганиты желтого цвета:
Ме3[Мп(С2О4)3] + 2Н2О Ме1[Мп(С2О4)2(Н2О)2] +|Ме2С2О4
Путем взаимодействия МпООН с щавелевой кислотой Н2С2О4-
• 2Н2О на холоду в темноте была получена кислота
Н[Мп(С2О4)2(Н2О)2] оливково-зеленого цвета.
шранс-Изомер соли К[Мп(С2О4)2(Н2О)2] зеленого цвета выде-
ляется при добавлении спирта к красному раствору, получен-
МАРГАНЕЦ
419
ному действием стехиометрически необходимого количества
Н2С2О4>2Н2О на КМпО4. Известен также г^ыс-изомер соли
К[Мп(С2О4)2(Н2О)2] желтого цвета.
При растворении диоксалатодиаквоманганитов в большом коли-
честве воды анион [Ми(С2О4)2(Н2О)2]_ гидролитически разла-
гается с образованием МпООН:
Ме®[Мп(С2О4)2(Н2О)2] + Н2О 5* МпООН + 2Н2С2О4 + МегОН
Трималонатоманганиты, Mei[Mn(OOC — СН2 —
— СОО)3],— вещества красного цвета, а дималонатоди-
а к в ом а н г а н и т ы, Mei[Mn(OOC — СН2 — СОО)2(Н2О)2],—
зеленого.
Дималонатодиаквоманганиты Mei[Mn(OOC — СН2 — СОО)2-
• (Н2О)2] выделяются в виде зеленых кристаллов из рас-
творов МпООН в малонате щелочного металла или при восстанов-
лении КМпО4 стехиометрически необходимым количеством мало-
ната щелочного металла.
При обработке дималонатодиаквоманганитов щелочных метал-
лов избытком малонатов щелочных металлов образуются красные
трималонатоманганиты:
Мет[Мп(ООС — СН2 — СОО)2(Н2О)2] + Ме£С2О4СН2 г*
Ме}[Мп(ООС — СН2 — СОО)3] + 2Н2О
Известна кислота Н3О[Мп(ООС — СН2 — СОО)2(Н2О)2] в виде
зеленого, устойчивого на воздухе и разлагающегося в воде тонко
дисперсного порошка.
Ацетилацетпонат марганца(11Г),
(/СН3\ 1
/О=С\ \
Z ,сн I .
\O-cZ I
чсн3/з.
получают растворением водной суспензии МпООН или
Мп(СН3СОО)3 в ацетилацетоне, а также обработкой сульфата
марганца(П) с КМпО4 при pH = 5—6, ацетилацетоном и затем
аммиаком:
8MnSO4 + 2КМпО4 + 30С6Н8О2 + 14NH3 -+
Ю[Мп(С6Н7О2)3] + K2SO4 + 7(NH4)2SO4 + 8Н2О
Соединение [Мп(С5Н7О2)3] (перекристаллизованное из эфира)
представляет собой блестящие черные кристаллы с т. пл. 172°;
они плохо растворимы в воде и хорошо растворяются в эфире,
бензоле, хлороформе, этилацетате.
Кроме описанных выше известны и другие соединения марганца(Ш),
например: иодат Мп(Ю3)3, селенит Mn2(SeO3)3 -5Н2О, пирофосфаты
Мп4(Р2О7)3»пН2О (где п = 8, 14), НМпР2О7, NaMnP2O7-5Н2О,
27*
420
ПОБОЧНАЯ ПОДГРУППА VII ГРУППЫ
KMnP2O7-3 — 5Н2О, RbMnP2O7, NH4MnP2O7, NH4MnP2O7-3H2O,
AgMnP2O7-3H2O, Ba(MnP2O7)2-5H2O, Na3Mn(H2P2O7)3, метафосфат MnP3O9,
арсенат MnAsO4-H2O, триарсенатомарганцевая кислота H6Mn(AsO4)3,
формиат [Mns(HCOO)6](HCOO)3-2H2O, дисалицилатодиаквоманганиты
Mei[Mn(O — С6Н4 — СОО)2(Н2О)2]-21^0, анионные комплексы, получен-
ные путем обработки соединений марганца(Ш) в щелочной среде различ-
ными третичными аминами (такими, как триэтаноламин, диацетатэтанола-
мин и др.) и содержащие анионы [MnN(CH2 — СН2ОН)3(ОН)4]“ — зеленый
и [MnN(CH2 — СН2ОН)(СН2СОО)2(ОН)4]3- — желтый, или третичными диа-
минами типа комплексона!П( двунатриевая соль этилендиаминтетрауксус-
ной кислоты), с которым образуется анион рубиново-красного цвета:
Соединения четырехвалентного марганца
Известно относительно немного соединений, в которых четырех-
валентный марганец встречается в виде катиона Мн4+ или в виде
анионов МпО2-, Мн2О2-, Мн3О2~, Мп5О41_, Мп7О25“, [MnF5]_,
[MnF6]2~, [МпС16]2-, [Mn(CN)6J2-, [Mn(CN)8]4-, [Mn(IO3)e]2-,
[Mn(PO4)2]2', [Mn(C2O4)2(OH)2]2~ и [Mn(C3H5O3)2]2-.
Среди соединений марганца(1У) наиболее устойчивы двуокись
МнО2 и дисульфид MnS2, которые встречаются в природе в виде
минералов.
Катион Мн4+ неустойчив в воде. При гидролизе солей, содер-
жащих этот катион, образуется гидратированная двуокись мар-
ганца. Благодаря амфотерному характеру двуокиси марганца
легко могут быть получены соединения марганца (IV) действием
кислот (HCl, H2SO4) или щелочей (гидроокисей щелочноземель-
ных металлов, различных окислов металлов) на двуокись мар-
ганца.
Известны тетрагалогениды MnF4 и МнС14, характеризующиеся
малой устойчивостью. В отличие от этих тетрагалогенидов ацидо-
соли типов МеЧМпХ5], Mei[MnX8] (где X = F-, Cl-, CN-) значи-
тельно более устойчивы. В табл. 49 приведены формулы и ука-
зана окраска некоторых соединений марганца(1У).
Двуокись марганца, МпО2, встречается в природе в форме
четырех кристаллических разновидностей, а именно: пиролюзита,
рамзделита, псиломелана и криптомелана. Искусственным путем
МАРГАНЕЦ
421
были получены пять разновидностей двуокиси марганца, а именно:
а-МпО2 (соответствующая криптомелану), р-МпО2 (напоминающая
пиролюзит), у-МпО2 (соответствующая рамзделиту), б-МпО2
и е-МпО2, не имеющие природных аналогов.
Двуокись марганца получают прокаливанием (195—300°) на воз-
духе Mn(NO3)2, окислением солей марганца(П) в щелочной среде
с помощью С12, Вт2, НОС1, Ме+ОС1, анодным окислением разбав-
ленных растворов нитрата или ацетата марганца(П), гидролизом
солей марганца(ГУ), разложением НМпО4, действием конц. HNO3
на раствор НМпО4, восстановлением КМпО4 в щелочной среде
различными восстановителями:
Mn(NO3)2 = MnO2 + 2NO2 ЗМпО + КС1О3 = 3MnO2 + КС1
2МпООН + ‘/2О2 = МпО2 + Н2О ЗМп2О3 + КС1О3 = 6МпО2 + КС1
2НМпО4 = 2МпО2 + Н2О + О3 МпС14 + 2Н2О = MnO2 + 4НС1
ЗМпСО3 + КС1О3 = 3MnO2 + КС1 + ЗСО2 МпО + = МпО2
Мп(ОН)2 + С12 + 2КОН = МпО2 4-J2KC1 + 2Н2О
Мп(ОН)2 + НОС1 + КОН = МпО2 + КС1 + 2Н2О
При получении двуокиси марганца в растворе образуются
гидраты с различным содержанием воды.
Двуокись марганца представляет собой твердое парамагнитное
черное вещество (в виде порошка или гранул) амфотерного харак-
тера, обладающее как окислительными, так и восстановитель-
ными свойствами. Она проявляет свойства как основного окисла
(образуя соли — производные гипотетического основания^Мп(ОН)4,
которые очень неустойчивы, легко восстанавливаются и гидроли-
зуются в растворе), так и кислотного окисла (образуя соли —
производные гипотетических кислот Н2МппО2п+1, которые очень
легко гидролизуются в растворе).
При действии кислот или щелочей на двуокись марганца полу-
чают неустойчивые соли марганца(1У):
MnO2 + 4НС1 = МпС14 + 2Н2О МпО2 + 2NaOH = Na2MnO3 + Н2О
Двуокись марганца окисляет Н2, H2S, С, СО, SO2, HF, HCl,
хлориды в присутствии сильных кислот, бромиды и иодиды в при-
сутствии слабых кислот, NH3, NH4NO3; может быть восстанов-
лена нагреванием с конц. H2SO4 или МнС12:
Выше 170° Выше 220°
2а-МпО24-Н2 ----- п > Мп2О3 2Р-МпО2-|-Н2 -------Мп2°з
450 — 500°
2MnO2+4HF---------> 2MnF2 + O24-2H2O
90°
МпО2 + 4НС1----► МпС12+С12 + 2Н2О
2МпО2 + 3H2S + ЗО2 = MnSO* + MnS2O3 + ЗН2О
MnO2 + 2NaCl + 3H2SO4 = MnSO4 + 2NaHSO4 + 2H2O + Cl2
422
ПОБОЧНАЯ ПОДГРУППА VII ГРУППЫ
MdO2 + 2КВг + 4СЩСООН - Мп(СН3СОО)2 + ЖСН3СОО + 2Н2О + Вг2
МпО2 4- 2KI 4- ЗСО2 4- Н2О = МпСО3 + КНСО3 + 12
6МпО2 4- 2NH3 = ЗМп2О3 4- N2 4- ЗН2О
МпО2 4- 4NH4NOa = Mn(NO3)2 4- 8Н2О 4- 3N2
4MnO2 4- 6H2SO4 = 2Mn2(SO4)3 4- 6Н2О 4- О2
МпО2 4- 2MgCl2 = МпС12 4- 2MgO 4- С12
Двуокись марганца обладает восстановительными свойствами
но отношению к окислительно-щелочному расплаву KNO3
4- К2СО3, а также по отношению к хлору или кислороду в при-
сутствии щелочи:
МпО2 4” KNO3 4~ К2СО3 d К2МпО4 -|- I4NO2 4~ СО2
2МпО2 4- ЗС12 4- 8КОН = 2КМпО4 4- 6К.С1 4- 4Н2О
МпО2 4- V2O2 + 2КОН - К2МпО4 4- Н2О
В вакууме (10 мм рт. ст.) МпО2 превращается в Мп2О3 (600°),
Мп3О4 (1000°) и МпО (1300°).
При прокаливании на воздухе МпО2 образует Мп2О3
(580-620°) и Мп3О4 (950-1100°).
В порошкообразном состоянии МпО2 служит катализатором
в реакциях разложения]КС1О3, KMnO4, Н2О2, NaOCl, окисления
аммиака до HNO3, превращения уксусной кислоты в ацетон.
Двуокись марганца, введенная в состав стекла, уничтожает
зеленую окраску, обусловливаемую силикатом железа(П), и при-
дает стеклу розовый цвет (или черный цвет, будучи добавлена
в большем количестве). В керамической промышленности двуокись
марганца используется ^для окрашивания эмалей и глазурей
в коричневый, пурпурный и черный цвега.
Тонко дисперсный порошок двуокиси марганца или коллоид-
ная двуокись марганца обладает адсорбирующими свойствами:
поглощает хлор, двуокись серы, а также соли бария, радия,
алюминия, серебра и калия.
Коллоидную модификацию двуокиси марганца можно полу-
чить продолжительным кипячением Мп3О4 с HNO3, действием
HOG1 на Мп(ОН)2, тиосульфата натрия на КМпО4, восстановле-
нием 1,6%-ного раствора КМиО4 3%-ной перекисью водорода,
восстановлением КМиО4 ортомышьяковистой кислотой H3AsOs
в щелочном или сернокислом растворе, восстановлением КМпО4
сульфитом натрия в присутствии желатины.
Двуокись марганца используется в качестве деполяризатора
в элементах Лекланше.
Манганит ы, Me*MnnO2ri0. Как уже говорилось, гидра-
тированная двуокись марганца проявляет слабо основные свойства
и взаимодействует с сильными основаниями, образуя манганиты,
т. е. соли гипотетических кислот общей формулы H2MnnO2n+i
(где п = 1, 2, 3, 5, 7).
МАРГАНЕЦ
423
Манганиты получают также термическим разложением перман-
ганатов и манганатов, окислением солей марганца(П) в окисли-
тельно-щелочных расплавах и сильным нагреванием МпО2 с раз-
личными окислами металлов, взятыми в строго определенных
количествах:
До 600° Выше 500°
2КМпО4-------- К2Мп2О5 + 3/2о2 К2МпО4--------► К2МпО3+1/3О2
МпС12 + 4NaOH + NaNO3 = Na2MnO3 + NaNO2 + 2NaCl + 2H2O
2МпС12Й+ 6NaOH + 2NaNO3 = Na2Mn2O5 + 2NaNO2^+ 4NaCl + 3H2OJ,
Известны манганиты типов Me*MnO3, Me*Mn2O5, MeJMn3O7,
MejMnaOn, MeJMn70i5, Me*MnnO2n+i.
Гипотетические кислоты, соответствующие манганитам, боль-
шей или меньшей степени конденсации образуются при дегидра-
тации моногидрата двуокиси марганца МпО2-Н2О, рассматривае-
мого как гипотетическая кислота Н2МпО3:
-(п-1)Н2б
, МпО24-Н2О^НгМпОз!' лН2МвО3---------* Н2МппО2п+1
Выделены следующиеЫанганиты: К2МпО3, К2Мп2О5, К2Мп5Ои,
Na2MnO3, Na2Mn2O5, Na2Mn50n "иНгО, (NH4)2MnO3, NH4HMnO3,
СаМпО3, CaMn2O5, СаМп3О7,|СаМп5О11, СаМп7О15-иН2О.
Манганиты мало изучены, не имеют строго определенного
состава, особенно при получении в растворах, очень легко гидро-
лизуются и обладают плохой растворимостью-
При действии Н2О2 на К2МпО4 в щелочном растворе полу-
чают пер оксоманганит К2Н2[МпО(О2)3]-иН2О, из которого были
получены К3Н[МпО(О2)3] "иН2О и Na2H2[MnO(O2)3]-иН2О.
Тетрафторид марганца, MnF4, в твердом состоянии не выде-
лен; он образуется в водном растворе при?растворении МпО2
в концентрированной плавиковой кислоте или при^действии
последней на эфирный раствор МпС14.
Водный раствор MnF4 обладает окислительными свойствами,
обесцвечивает индиго и образует с анилином окрашенные соеди-
нения.
Пента - и гексафторосоли Mei[MnF5], Me|lMnFeJ.
При"действии BrF3 на КМпО4 была получена соль K[MnF3] розо-
вого цвета. При этом способе избыток BrF3 удаляется упарива-
нием в вакууме при 20°.
При обработке эквимолярных смесей КМпО4 и KG1 фторидом
брома, КМпО4 —’перекисью водорода в присутствии ионов F-
и К2МпО4 —^40%-ной плавиковой кислотой образуется K2[MnF6].
Соединение K2[MnFe] представляет собой слабо двупреломляю-
щие золотисто-желтые гексагональные пластинчатые кристаллы,
которые становятся красными при нагревании, гидролизуются
424
ПОБОЧНАЯ ПОДГРУППА VII ГРУППЫ
(медленно в холодной, быстро в теплой воде/ с образованием
МпО2 и разлагаются на Мп3О4 и KF при сильном нагревании.
При обработке МпО2 раствором RbF в HF получают Rb2[MnFel
в виде желтых гексагональных пластинчатых кристаллов.
Известны также гексафторосоли Na2[MnFe], Mg[MnFe],
Sr[MnF6], Ba[MnFe] золотисто-желтого цвета, переходящего в крас-
ный при нагревании.
Тетрахлорид марганца, МпС14, получается в виде красновато-
коричневого кристаллического осадка при добавлении CS2 к фио-
летовому раствору, охлажденному до —80°. Последний получают
растворением МпО2 в ацетилхлориде при 0° в течение 15 час
или обработкой суспензии МпО 2 в эфире газообразным HG1 при
—70°:
МпО2 + 4СН3СОС1 =ТМпС]4 + 2(СН3СО)2О
Безводный МпС14 в виде черного осадка, загрязненного неболь-
шим количеством МпС12, получают также добавлением смеси
СНС13 с СС14 к зеленому раствору, образовавшемуся при барбо-
тировании газообразного HG1 через суспензию МпО2 в эфире
при —70°.
Кристаллы МпС14 неустойчивы, очень легко плавятся; превра-
щаясь в зеленые капли, разлагаются при —10° с выделением
хлора и образованием розовато-белого остатка МпС12.
ГексахлороманганитДУ) калия, К2[МпС1в], получают при низ-
кой температуре обработкой солянокислого раствора КС1 солью
Са(МпО4)2 или пропусканием газообразного HG1 в уксуснокислый
раствор КМпО4.
Ca(MnO4)2 + 4КС1 + 16HGL =g2K2[MnCle] + СаС12 + 8Н2О 4- ЗС12
Соединение К2[МпС18] представляет собой неустойчивые темно-
красные кристаллы, которые сохраняются в эксикаторе непро-
должительное время и быстро разлагаются на МпС12, К.С1 и хлор.
Были также получены Rb2[MnGle] и (NH4)2[MnGl6L
Дисульфид марганца, MnS2, встречается в природе в виде
минерала гауерита; это коричнево-красные или черно-коричневые
гексаэдрические кристаллы с плотностью 3,71 г/см2 и твердостью 4
по шкале Мооса.
Дисульфид марганца получают нагреванием (160—170°) в за-
паянной трубке смеси MnSO4 с полисульфидом щелочного металла
в водном растворе. Он представляет собой парамагнитный крас-
ный аморфный порошок, устойчивый в сухом воздухе, слабо'
разлагающийся во влажном воздухе, легко разлагающийся на MnS
и серу при 170° и превращающийся в MnCl2, H2S и серу под дей-
ствием соляной кислоты.
Сульфат марганца, Mn(SO4)2, выпадает при охлаждении чер-
ного раствора, полученного обработкой при 50—60° смеси MnSOt-
МАРГАНЕЦ
425
• 4Н2О и порошка КМпО4 55%-ной H2SO4, или при окислении
(50—60°) сернокислого раствора MnSO4 двуокисью свинца РЬО2:
3MnSO4 + 2KMnOt + 8H2SO4 = 5Mn(SO4)2 +SK2SO4 + 8Н2О
MnSO4 4- PbO2 + 2H2SO4 = Mn(SO4)2 + PbSO4 + 2H2O
При растворении на холоду черных кристаллов Mn(SO4)2
в 40—70%-ной H2SO4 образуются коричневые растворы, которые
при разбавлении гидролизуются с образованием Mn2(SO4)3 и выде-
лением кислорода:
2Mn(SO4)2 + Н2О -> Mn2(SO4)3 + H2SO4 + l/2O2
Известны двойные сульфаты Mn(SO4)2 -Mn(HSO4)2 -8Н2О,
6Mn(SO4)2 -4MnSO4 -5K2SO4.
Помимо приведенных известны и другие соединения марганца(1У),
например: диселенид MnSe2, дителлурид МпТе2, селенит Mn(SeO3)2,
пироселенит MnSe2O5, координационные соединения К2[Мп(103)3],
Ме4[Мн(ТеО4)4] (где Mei = Na+, К+ и др.), H4[Mn(HAsO4)4],
(NH4)2H2[MnO(AsO4)2], (NH4)2H2[MnO(PO4)2] или (NH4)2Mn(PO4)2-Н2О,
K2[Mn(CN)6]-2KCN или K4[Mn(CN)8], K2[Mn(C2H4)2(OH)2]-2H2O,
Ме5Мп(СзН5О2)2].
Соединения пятивалентного марганца
Известно очень немного соединений, в которых электрополо-
жительный марганец входит в состав анионов [MnF6]-, МпО®-.
На основании магнитных измерений было установлено, что в анио-
не МпО|“ марганец пятивалентен. Соединения пятивалентного'
марганца легко разлагаются водой.
Гексафтороманганаты, Me4Mn5+Fe] (где Me1 = К+,
Na+), получают действием плавиковой кислоты на манганаты
или двуокись марганца в присутствии фторидов|щелочных метал-
лов, восстановлением КМпО4 30%-ным раствором Н2О2 в кон-
центрированной плавиковой кислоте, содержащей KF, действием
фтора или трифторида брома на дихлорид марганца.
Гексафторогипоманганат калия, K[MnFe], представляет собой
желтые кристаллы, легко гидролизующиеся даже холодной водой.
Безводные или гидратированные (гипоманганаты) MeJMnOt
могут быть получены восстановлением сильно щелочных раство-
ров КМпО4 с помощью Na2SO3-7H2O или KI, разложением манга-
ната натрия в избытке 10 н. раствора NaOH или нагреванием
выше 175°, обработкой конц. NaOH охлажденного продукта,
полученного добавлением МпО2 и Na2O2 в расплавленный NaNO2
(500°), нагреванием МпО2 с избытком К2СО3 на воздухе при тем-
426
ПОБОЧНАЯ ПОДГРУППАМИ ГРУППЫ
пературах выше 700°, сплавлением соединений марганца (II)
с'окислительно-щелочными смесями:
2КМпО4 + 2Na2SO3 + 6NaOH + 18Н2О =
= 2Na3MnO4-10H2O + 2Na2SO4 + 2KOH
4Na2MnO4 + 4NaOH = 4Na3MnO4 + 2H3O + O2
2MnO2 + 2Na2O2 + NaNO2+ 2NaOH + 19H2O - 2Na3MnO4«10H2O4-NaNO3
2MnO2 -|- 2K2CO3 -J- l/2O2 = 2K3MnO4 -f- 3CO2
Известны гипоманганаты Na3MnO4-ЮНгОТтемно-синего цвета,
Na3MnO4-7H2O темно-синего, Na3MnO4 сине-зеленого, Na3MnO4-
• NaOH зеленого, K3MnO4 темно-синего, Li3MnO4 сине-зеленого,
Ca3(MnO4)2 сине-зеленого, Sr3(MnO4)2 сине-зеленого, Ba3(MnO4)3-
• H2O зеленого цвета.
Гипоманганаты представляют собой синие, сине-зеленые или
зеленые кристаллы, устойчивые в сухом воздухе (в отсутствие
газообразного СО2) и в сильно щелочных растворах. Они обра-
зуют смешанные соли с фосфатами, арсенатами, ванадатами
и разлагаются в разбавленных растворах или под действием
кислот по уравнениям
2МпО^- + 2Н2О MnO|- + MnO2 + 4ОН
2MnO’ + 4H+ МпОГ + MnO2 + 2H2O
Известна также основная соль Ba5(MnO4)3(OH).
Соединения шестивалентного марганца
Известно относительно немного соединений шестивалентного
марганца, например трехокись МпО3, марганцевая кислота
и безводные (Ме*МпО4) или гидратированные (Ме*МпО4-пН2О)
манганаты (Me1 = Li+, Na+, К+, 1/2Ca2+, VaSr24", VaBa24", 1/2Cda+).
Трехокись марганца (марганцовистый ангидрид), МпО3, полу-
чают в виде пурпурно-красных (фиолетовых) паров при нагре-
вании зеленого раствора, образующегося при растворении КМпО4
в конц. H2SO4.
При конденсировании пурпурно-красных паров МпО3 полу-
чают темно-красную массу с характерным запахом, вызывающим
кашель; при 50° она разлагается на МпО2 и кислород.
Марганцовистый ангидрид растворяется в воде с образованием
НМпО4 и МпО2, разлагается в абсолютном эфире с образованием
МпО2 и кислорода, растворяется” в щелочах с образованием манга-
натов зеленого цвета, а при’обработке HG1 в избытке эфира превра-
щается в зеленый МпС13 или синий МпС14.
ЗМпО3 -f- Н2О 2НМпО4 -|- МпО2 МпО3 —> МпО2 ^/2О2
МАРГАНЕЦ
427
Марганцовистая кислота, Н2МпО4, существует только в вод-
ном растворе. Это слабая кислота, соли ее (манганаты) легко
разлагаются гидролитически или при нагревании.
Манганаты, MeiMnO4, образуются при сплавлении солей
марганца(П) или МпО2 с окислительно-щелочной смесью (КОН 4-
4- KNO3, КОН 4- КСЮ3, Na2GO3 + KNO3, Na2GO3 4- KGIO3),
окислении кислородом воздуха смеси МпО2 с распл. КОН, нагре-
вании перманганатов с концентрированными щелочами, электро-
лизе 40—50%-ных растворов NaOH или КОН (анод из метал-
лического марганца, температура 50—65°, плотность тока 2—
5 а/дм2).
MnSO4]4- 2Na2CO3 4- 2KNO3 = Na2MnO4 4- 2KNO2 4- Na2SO4 4- 2CO2
MnO2 -f- 2KOH 4- KNO3 =. K2MnO4 -f- KNO3 4- H2O
3MnO2 4- 6KOH 4- KClOa = 3K2MnO4 4-jKCl 4- 3H2O[]
MnO2 4- 2KOH + V2O2 - K2MnO4 + H2O
2KMnO4 4- 2K.OH = 2K2MnO4 4- V2O2 4- H2O
Манганаты щелочных металлов Me*MnO4 устойчивы на холоду
в щелочной среде, окрашены в зеленый (или черно-зеленый)
цвет, разлагаются под действием разбавленных кислот или при
нагревании выше 500°:
ЗК.2МпО4 4 2Н2О 2КМпО4 4- MnO2’4- 4К.ОН
’ЗК2МпО4Г4- 4СН3СООН = 2КМпО4 4- Н2МпО3 +- 4КСН3СООИ4-’|Н2О’
ЗК2МпО4 = 2К3МпО4 + МпО2 4- Ог
В сильно щелочных растворах (10 н. NaOH или КОН манга-
наты превращаются в гипоманганаты:
4МпО1- 4- 4ОН- 4МпО|_ 4- 2Н2О 4- О»
Манганаты являются окислителями по отношению к сере, серо-
водороду, двуокиси серы, солям железа(П) или различным орга-
ническим веществам и восстановителями по отношению к хлору
2К2МпО4 4- 5S 4- 2Н2О == 2МпООН 4- 2K2S2O3 4- H2S
2K.2MnO4 4- С12 => 2КМпО4 4- 2KG1
Окислительно-восстановительные системы, в которых ион
МпО|~ является окислителем или восстановителем, указаны ниже:
МпО|- 4- 4Н+ 4- 2е~ МпО2 4- 2Н2О
МпО|~ —•
МпО|- 4- 2Н2О 4- 2е~ МпО2 4- 4ОН-
Под действием двуокиси углерода или азотной кислоты манга-
наты диспропорционируют следующим образом:
6К2МпО4 4- 4СО2 = 4КМпО4 4- 4К2СО3 4- Мп2О3 4- V2O2
ЗК2МпО4 4- 4HNO3 = 2КМпО4 4- МпО, 4- 4KNO3 4- 2Н2О
428
ПОБОЧНАЯ ПОДГРУППА VII ГРУППЫ
Манганат калия, К2МпО4, представляет собой темно-зеленые
кристаллы, растворимые в разбавленных щелочах и изоморфные
с кристаллами Na2SO4-10H2O, Na2MnO4-ЮН2О. К2МпО4 диспро-
порционирует в воде.
Манганат натрия, Na2MnO4, представляет собой черные кри-
сталлы с фиолетовым отблеском, он растворим в щелочах и дис-
пропорционирует в воде. Известны кристаллогидраты Na2MnO4-
• иН2О (где п = 10, 6, 4).
Известны следующие манганаты: Li2MnO4, (NH4)2MnO4,
Rb2MnO4, Cs2MnO4, CaMnO4, SrMnO4, BaMnO4, CdMnO4-H2O,
PbMnO4.2H2O.
Соединения семивалентного марганца
Известно относительно небольшое число соединений семива-
лентного марганца, который имеет электронную структуру инерт-
ного газа аргона (25— 7—18). В качестве примеров соединений
марганца(УП) можно привести окись марганца(УП) Мп2О7
(марганцовый ангидрид), марганцовую кислоту НМпО4 и ее соли
МетМпО4, Меп(МпО4)2.
Катион Мп7+ не известен. Известен только анион МпО", кото-
рый в водных растворах имеет фиолетовый цвет, является окисли-
телем и более устойчив, чем анионы МпО|" и МпО|“. Фиолетовый
анион МпО" превращается при подщелачивании в зеленый анион
МпО“", который в кислой среде опять переходит в анион МпО".
За эту способность ион МпО" называют «химическим хамелеоном».
2MnOj + 2ОН- -> 2МпО^- + Н2О + !/2О2
2МпО=- + 4Н + -> MnOj + МпО2 + 2Н2О
Окись марганца(уН) (марганцовый ангидрид), Мп2О7, полу-
чают действием конц. H2SO4 на КМпО4 при —-20° или обработкой
раствора КМпО4 фторсульфоновой кислотой при легком нагре-
вании.
Конц. H2SO4
KMnO4 + H2SO4=HMnO4 + KHSO4 2НМпО4----------► Мп2О7
— Н2О
Окись Мп2О7 представляет собой темно-зеленые кристаллы,
устойчивые при —5° и растворяющиеся без разложения в конц.
GH3GOOH.
При плавлении кристаллов Мп2О7 образуется темно-красная
в проходящем свете и зеленая в отраженном свете жидкость
с плотностью 2,396 г!см2, которая относительно устойчива в сухом
воздухе ниже 0°, разлагается со взрывом при 10° на МпО2 и О3
и реагирует с многочисленными неорганическими восстановите-
лями и различными органическими соединениями.
МАРГАНЕЦ
429
В присутствии H2SO4 окись марганца(УП) окисляет на холоду
различные разновидности углерода.
Марганцовая кислота, НМпО4, известна только в водном
растворе и образуется при растворении Мп2О7 в большом коли-
честве холодной воды, при обработке Ва(МпО4)2 разб. H2SO4,
при действии H2[SiF6] на КМпО4, при окислении соединений
марганца(П) в сильно азотнокислой среде РЬО2 или K2S20g,
(NH4)2S2O8 в присутствии иона Ag+.
2КМпО4 + H2[SiFe] = 2HMnO4 + K2[SiFe]
2MnSO4 + 5РЬО2 + 6HNO3 =а[2НМпО4 + 3Pb(NO3)2 + 2PbSOt + 2Н2О
2Mn(NO3)2 -|- 5(NH4)2S2O8 -|- 8Н2Ол=
= 2НМпО4 + 5(NH4)2SO4 + 5H2SO4 + 4HNO3
Разбавленные растворы НМпО4 относительно устойчивы и могут
быть’доведены до 20%-ной концентрации упариванием в вакууме.
Концентрированные растворы НМпО4 не устойчивы и разлагаются
с образованием МпО2 и выделением озона:
2НМпО4 -> 2МпО2 + 03Н- Н2О
Марганцовая кислота является сильной кислотой, она разла-
гается под действием концентрированных кислот (H2SO4, Н3РО4,
СН3СООН) или при каталитическом действии платинового порош-
ка. Соли марганцовой кислоты, названные перманганатами, имеют
общие формулы МетМпО4 или МетМпО4"гаН2О (где MeI = Li+,
Na+, К+, Rb+, Cs+, NH+, Ag+, ^Mg2*, ^Ca^, 1/2Sr2+, ^Ba2*).
Перманганаты — устойчивые соединения, они изоморфны пер-
хлоратам, при нагревании разлагаются легче последних и в вод-
ных растворах являются’сильными окислителями.
Кристаллогидраты LiMnO4-3H2O, NaMnO4 -ЗН2О, Mg(MnO4)2.
• 6Н2О, Са(МпО4)2 «5Н2О, Sr(MnO4)2 *ЗН2О очень хорошо раство-
римы в воде в отличие от безводных перманганатов NH4MnO4,
KMnO4, RbMnO4, CsMnO4, AgMnO4, Ba(MnO4)2.
Перманганаты используются в качестве окислителей в водных
растворах, при дистилляции воды, в качестве дезинфицирующего
средства, при отбеливании шерсти, хлопка или текстильных воло-
кон, для очистки газов и т. п. При смешивании перманганатов
с серой, фосфором или различными органическими веществами
образуются взрывчатые смеси.
Наряду с нормальными перманганатами известны и основные
перманганаты, например Hg(MnO4)2 *3HgO.
Перманганат калия, КМпО4, получают длительным кипяче-
нием К2МпО4 в воде, действием разбавленных кислот (H2SO4,
Н2СО3), хлора или озона (в присутствии катализатора AgNO3)
на К2МпО4, электролитическим окислением растворов манга-
натов (60°, катод железный или никелевый, анод из сплава 77%
430
ПОБОЧНАЯ ПОДГРУППА VII ГРУППЫ
Мп, 14% Fe, 7% С), а также окислением солей марганца(Н)
бромной водой в щелочной среде:
ЗК2МпО4 +[2Н2О -= 2КМпО4 + МпО2 + 4КОН
ЗК2МпО4 + 2H2SO4 -= 2КМпО4 + МпО2 + 2K2SO4 + 2Н2О
2К2МпО4 + С12 = 2КМпО4 + 2КС1
2К2МпО4 + О3 + Н2О = 2КМпО4 + 2КОН + О2
2Мп(ОН',г + 5Вг2 + 12КОН = 2КМпО4 + 10КВг + 8Н2О
Перманганат калия КМпО4 представляет собой пурпурные
орторомбические призматические кристаллы, устойчивые на воз-
духе, изоморфные с КС1О4, BaSO4, ВаСгО4, ВЬМпО4, KBF4,
имеющие плотность 2,703 г/см3; он растворим в воде, жидком
аммиаке, пиридине и метаноле и разлагается при нагревании
выше 200°. КМпО4 обладает окислительными свойствами:
200°
2КМпО4------► К2МпО4-|-МпО2+О2
До красного каления
2КМпО4------------------► К2МпО3-|-МпО2-|-3/2О2"
В кислой среде КМпО4 окисляет НС1, H2S, Н3Р, SO2, FeSOt»
Н2С2О4-2Н2О, НСООН, HNO2, H3AsO3, Н2О2 и т. д.:
MnOj + 8Н3О+ + 5е- Мп2+ + 12Н2О
2КМпО4 + 10НС1 + 3H2SO4 = 2MnSO4 + K2SO4 + 5С12 + 8Н2О-
2KMnO4 + 5H2S + 3H2SO4 = 2MnSO4 + K2SO4 + 5S + 8H2&
8KMnOt + 5H3P + 12H2SO4 = 8MnSO4 + 4K2SO4 + 5H3PO4 -f- 12H2O
2KMnO4 + 5H2SO3 + 3H2SO4 = 2MnSOt + K2SO4 + 5H2SO4 + 3H2O
2KMnO4 + 10FeSO4 + 8H2SO4 = 2MnSO4 + K2SO4 + 5Fe2(SO4)3 + 8H2O
2KMnO4 + 10KI + 8H2SO4 = 2MnSO4 + 6K2SO4 + 5I2 + 8H2O
2KMnO4 + 5H2C2O4 + 3H2SO4 = 2MnSO4 + K2SO4 + 10CO2 + 8H2O
2KMnOt + 5H2O2 + 3H2SO4 = 2MnSO4 + K2SO4 + 50, + 8H2O
В нейтральной среде KMnO4 окисляет иодиды до йодатов
и различные органические соединения (спирты, оксалаты, формиа-
ты и др.) до двуокиси углерода:
MnOj + 4Н3О+ + Зе - МпО2 + 6Н2О
В щелочной среде КМпО4 окисляет SO2, KI, КЮ3, восста-
навливаясь до К2МпО4 или МпО2.
Активный водород (выделяющийся при действии разб. H2SO4
на Zn) восстанавливает КМпО4:
2КМпО4 + 8H2SOs +’ 5Zn = 2MnSO4 + K2SO4 + 5ZnSO4 + 8H2O
Перманганат калия образует смешанные кристаллы с NH4MnO4
ВЬМпО4, КС1О4, BaSO4.
МАРГАНЕЦ
431
Перманганат серебра, AgMnO4, образуется при нагревании
растворов КМпО4 и AgNO3 до 80° с последующим охлаждением.
Соединение AgMnO4 представляет собой блестящие черные
моноклинные игольчатые кристаллы. Оно обладает окислитель-
ными свойствами и разлагается при длительном хранении или
нагревании.
Перманганат натрия, NaMnO4-3H2O, получают обработкой
AgMnO4 хлоридом натрия; это расплывающиеся на воздухе пур-
пурные кристаллы с плотностью 2,47 г/см3-, соединение очень
хорошо растворимо в воде и разлагается при 170° с выделением
кислорода.
Перманганат лития, LiMnO4-3H2O, получают обработкой
AgMnO4 хлоридом лития или LiC104; он представляет собой пур-
пурные тетраэдрические кристаллы с плотностью 2,06 г/см3.
LiMnO4-3H2O разлагается при 190° с выделением кислорода.
Известны также перманганаты: NH4MnO4, RbMnO4, CsMnO4,
Mg(MnO4)2-6H2O, Ca(MnO4)2-5H2O, Sr(MnO4)2-3H2O, Ba(MnO4)2,
Pb(MnO4)2, Cu(MnO4)2 -8H2O, Zn(MnO4)2 -6H2O, Cd(MnO4)2 -6H2O,
[Ag(NH3)2]MnO4, [Ag(C5H5N)2]MnO4, [Cr(NH3)6](MnO4)3,
[Co(NH3)6](MnO4)3, [Cu(NH3)4](MnO4)2, [Cu(C5H5N)4](MnO4)2,
[Cd(C5H5N)4](MnO4)2, [Al(COC10H12N2)6](MnO4)3,
[Cr(COC10H12N2)6](MnO4)3 и др.
Окислы марганца нестехиометрическо-
го состава и солеобразная окись Мп3О4.
Известны многочисленные нестехиометрические окислы марганца,
например MnOli20, MnO1>33, MnO1>60, MnO1>75, MnOlj80, MnOlj83,
MnOli90, MnO4j93, MnO1>95, характеризующиеся сильно отличными
свойствами.
Солеобразная окись марганца встречается в природе в виде
минерала гаусманита; она получается нагреванием окислов МпО2,
Мп2О3 (950—1100°), восстановлением МпО2, Мп2О3 водородом
(200—230°), а также нагреванием при высокой температуре на воз-
духе МпО, МпСО3, МпС2О4’иН2О, MnSO4:
[580-620° '950-1 100°
6МпО2 —ЗМп2О3 -----------—-—> 2Мп3О4
—°/зО2 — 1/2O2
/200° 230°
ЗМпО2-|-2Н2 > Мп3О44“2Н2О ЗМп2О3 -|- * 2Мп3О4-|-Н2О
ЗМпО + !/2О2 =/Мп3О. ЗМпСО3’+ V2O2 = Мп3О4 + ЗСО2
ЗМпС2О4-пН2О -|- 2О2 = Мп3О4 -|- 6СО2 -|- иН2О
В зависимости от способа получения Мп3О4 выделяют в виде
кубических или тетраэдрических кристаллов желтого, красно-
коричневого или черного цвета.
Парамагнитное соединение Мп3О4 может быть восстановлено'
при нагревании водородом, окисью углерода, серой, сероводоро-
432
ПОБОЧНАЯ ПОДГРУППА VII ГРУППЫ
дом; оно превращается в безводный МпС12 при нагревании с избыт-
ком NH4G1 без доступа воздуха и превращается в соли марган-
ца^!) и (IV) под действием кислот, поскольку само вещество
рассматривается как соединения марганца(И) и (IV) Мп2+[Мп4+О4].
Металлоорганические соединения
Известно большое число металлоорганических соединений мар-
ганца, например, димер пента карбонила марганца, натриевые
и калиевые производные пентакарбонила марганца, иодид или
бромид пентакарбонила марганца, алкильные или ариль-
ные производные пентакарбонила марганца, производные типов
Mn(CO)4PR3, Mn(CO)4AsR3, Mn(CO)4SbR3.
Димер пентакарбонила марганца, Мп2(СО)10, получают в авто-
клавах при обычной температуре действием окиси углерода (под
давлением 20 ат) на смесь Мп12 с порошком металлического
магния в безводном этиловом эфире.
Ковалентное соединение Мп2(СО)10 представляет собой диамаг-
нитные прозрачные золотисто-желтые моноклинные кристаллы,
которые плавятся при 155° в закрытой трубке, разлагаются при
нагревании выше 250° под давлением 20 ат (или при 110° в отсут-
ствие окиси углерода) и растворимы в обычных органических
растворителях. Мп2(СО)10 очищают сублимацией при обычной
температуре и давлении меньше 0,5 мм рт. ст.
Со щелочами Мп2(СО)10 образует соли общей формулы
Ме1Ми(СО)5, которые содержат парамагнитный анион [Мп(СО)5]":
13Мп2(СО)10 + 40ОН- 2Mna+ + 24[Mn(CO)J- + lOCOg- + 20Н2О
Пентакарбонил натрия-марганца, NaMn(CO)5, получают дей-
ствием раствора металлического натрия в жидком аммиаке на
Ми2(СО)10, а также обработкой раствора Мп2(СО)10 в тетрагидро-
фуране избытком 1%-ной амальгамы натрия или металлическим
натрием в инертной атмосфере.
При действии амальгамы натрия на бензольный раствор
Мп2(СО)10 образуется белый осадок, состоящий из NaMn(CO)5
и Мп(СО)5Н.
Действием иода на Ми2(СО)10 при 130—140° в закрытой трубке
получают прозрачные рубиново-красные кристаллы Мп(СО)51,
которые плавятся на воздухе с разложением при 115°, трудно-
растворимы в воде, растворимы в обычных органических раство-
рителях. Это соединение менее устойчиво, чем Мп2(СО)10. Известно
также производное Мп(СО)5Вг.
При действии замещенных фосфинов PR3, арсинов AsR3 или
стибинов SbR3 на Ми2(СО)10 получают парамагнитные соединения
типов Mn(CO)4PR3, Mn(CO)4AsR3, Mn(CO)4SbR3.
МАРГАНЕЦ
433
Пентакар бонилметилмарганец, СН3Мп(СО)5, получают обра-
боткой раствора NaMn(GO)5 в тетрагидрофуране сульфатом или
иодистым метилом или пиролизом пентакарбонилацетилмарганца
СН3 - СО — Мн(СО)5.
Пентакарбонилметилмарганец представляет собой бесцветные
летучие кристаллы, устойчивые на воздухе.
Были также выделены производные С6Н5Мп(СО)5,
С6Н5 - СН2Мп(СО)5, С6Н5 - СОМп(СО)5.
Соединение СН3 — СОМп(СО)5 получают обработкой раствора
NaMn(CO)5 в тетрагидрофуране хлористым ацетилом СН3СОС1;
это белые летучие кристаллы.
Известны также производные (С5Н5)2Мп, С5Н5Мн(СО)3,
(CH3C5H4)Mn(CeHe),(CH3COC5H4)Mn(CO)3,[Ni(C12H8N2)]3[Mn(CO)5l.
Соединения включения
Нитриды марганца, Mn4N, Mn2N, Mn3N, Mn5N3,
можно получить действием азота или аммиака на металлический
марганец при 600—850°.
По диаграмме термического равновесия системы марганец —
азот, предложенной У. Цвикером (U. Zwicker, Z. Metallkunde,
42, 277, 1951), были установлены промежуточные фазы, соответ-
ствующие нитридам Mn4N, Mh2N, Mn3N2.
Нитрид марганца, Mn4N, серого цвета, имеет кубическую
структуру, содержит 6% азота; он ферромагнитен, вязок при тем-
пературе выше 700°, хрупок при низких температурах; Mn4N
может быть получен нагреванием металлического Мп при 690°
и давлении 100 мм рт. ст. в атмосфере азота в течение 12—24 час.
Нитрид марганца, Mn2N, серого цвета, обладает сложной
структурой и содержит 9—12% азота.
Нитрид марганца, Mn3N2, серого цвета, имеет тетраэдриче-
скую структуру, содержит 13,5—15% азота, обладает плотностью
6,21 г/см3, диамагнитен; образуется в результате сжигания мар-
ганца в чистом азоте (1210—1220°).
Нитрид марганца, Mn5N2, серого цвета, с плотностью 6,6 г/см3-
получают нагреванием пирофорного марганца в атмосфере азота
в калориметрической бомбе под давлением 10 ат при 850° в тече-
ние 2 мин.
Фосфиды марганца, Мп3Р, Мп2Р, МпР, выявленные
на диаграмме термического равновесия системы марганец — фос-
фор (/. Berak, Т. Неитапп, Z- Metallkunde, 41, 19, 1950), пред-
ставляют собой металлоподобные серые тетраэдрические, гексаго-
нальные или орторомбические кристаллы.
Арсениды марганца, Mn2As, Mn3As2, MnAs, полу-
чают прямым взаимодействием элементов при 800—1400°; это
блестящие серые кристаллы.
28—101
434
ПОБОЧНАЯ ПОДГРУППА VII ГРУППЫ
Антимониды марганца, Mn2Sb, Mn3Sb2, MnSb,
образуются в виде серебристо-серых кристаллов прямым взаимо-
действием элементов при нагревании, а также при нагревании
амальгамы марганца с металлической сурьмой в атмосфере водо-
рода.
Карбиды марганца, Мп3С, Мп23С6, Мп7С2, Мп7С3,
Мп6С2.
Карбид марганца, Мп3С, получают нагреванием смеси Мп3О4
с углеродом при 1500° в электрической печи или нагреванием
металлического марганца с углем при 1600° в вакууме (20 мм
рт. ст.).
Карбид Мп3С представляет собой кристаллы в виде орторомби-
ческих призм (изоморфные цементиту Fe3G) с плотностью 6,89 г!см'л
при 17° и малой твердостью. Мп3С разлагается водой с образо-
ванием Мп(ОН)2 и выделением СН4 и Н2, взаимодействует с кис-
лотами, галогенами и аммиаком.
Карбиды Мп23С6, Мп7С3 изоморфны соответствующим соеди-
нениям хрома.
Были выделены двойные карбиды 4Mn3C«Fe3C, 2Mn3G-Fe3C.
Силициды марганца, Mn3Si, Mn5Si3, MnSi, MnSi2,
представляют собой темно-серые тугоплавкие твердые кристаллы;
реагируют с галогенами, плавиковой кислотой и концентрирован-
ными щелочами.
Бориды марганца, Мп4В, Мп2В, МпВ, Мп3В4, обра-
зуются в виде коричневых тетраэдрических или орторомбических
кристаллов, плотность которых находится в пределах от 6,12
до 7,20 г/см2. Бориды марганца взаимодействуют с фтором, хло-
ром, бромом, разбавленными кислотами и аммиаком. Их полу-
чают прямым взаимодействием элементов при нагревании или
боротермическим восстановлением окислов марганца.
ТЕХНЕЦИЙ (ЭКАМАРГАНЕЦ) Тс
Z — 43; ат. вес = 98,918
Валентность (I), (II), (III), IV, (V), (VI), VII
Электронная структура атома технеция: А- Л-Л7 •4s24p64cZ3-5s2.
Электронная структура атома технеция и катиона Тс4+ для 4rf-
и 5х-орбиталей:
43s
5s2
n
43’
Tc
Tc4+
5s
t t t t t
t t t
Известно примерно 19 изотопов технеция, как это показано
в табл. 50.
ТЕХНЕЦИЙ
435
Таблица 50
Атомный номер Атомный вес Период полураспада Вид радиоактивного излучения
92 43,5+1 мин
92 4,3 мин №
93 2,75+0,5 час №
94 52,5 мин №
95* 60 сутки р+Ке_у
95 20+0,5 час Ку
96* 51,5 мин е~ и продукты распада
96 4,20+0,04 сутки Ку
97* 90+2 сутки е~ и продукты распада
43 97 >104 лет к
98 42+2 мин р-Ае-у
99* 6,09+0,03 час Р~уе- и продукты распада
99 2,12-105 лет р-
100 1,33+0,2 мин р-
100 15,8±0,2 сек р-
101 16,5+0,5 мин P~Y
102 <1 мин Р-
105 Короткоживущий Р-
107 <1,5 мин Р-
ИСТОРИЧЕСКАЯ СПРАВКА
На основе спектральных данных, полученных при исследова-
нии концентратов минералов ниобия и платины, в 1925 г. Ноддак,
Такке и Берг объявили об открытии элемента с атомным номе-
ром 43, названного ими «мазурием». Однако эти результаты
позднее не подтвердились.
В 1937 г. Перье и Сегре получили элемент 43 искусственным
путем при облучении молибдена *|Мо в течение нескольких меся-
цев дейтронами с энергией 5 Мэв в циклотроне. Название «техне-
ций» было дано элементу 43 по греческому слову tehnetos, что
означает «искусственный».
ПОЛУЧЕНИЕ МЕТАЛЛИЧЕСКОГО ТЕХНЕЦИЯ
Технеций можно получить либо при распаде урана 2’|U, либо
при бомбардировке молибдена “®Мо подходящими частицами (дей-
тронами, протонами, нейтронами и а-частицами), а также при
облучении рутения 44Вн нейтронами, у-частицами и т. д. либо
28*
436
ПОБОЧНАЯ ПОДГРУППА VII ГРУППЫ
восстановлением соединений, таких, как NH4TcO4, ТсО2, Тс2О7,
Tc2S7, As(C6H5)4TcO4.
Наиболее устойчивые изотопы технеция 9’Тс, ||Тс обладают
большим периодом полураспада и могут быть получены по сле-
дующим реакциям:
J|Ru(n, ?)$JRu tq—---► «Тс*
** 2,8 суток
6- в-
ЦМо(п, у)$»Мо —ч-^- «Тс* 2>2И0, ®’Ru (устойчивый)
В этих реакциях при захвате одного нейтрона и испускании
одного фотона (и, у) получают радиоактивный атом Тс*, масса
которого больше на одну единицу массы атома облученного эле-
мента.
При ядерных реакциях, протекающих под действием космиче-
ских лучей на некоторые природные элементы, непрерывно обра-
зуется технеций в следовых количествах. Серебристо-белый поро-
шок металлического технеция получают восстановлением при
нагревании NH4TcO4, Тс2О7, ТсО2 и Tc2S7 водородом в плати-
новой (или серебряной) лодочке, помещенной в трубку из туго-
плавкого стекла:
600°
NH4TcO4 + ’/2H2---> Tc + NH3+ 4Н2О
600°
Тс2О74-7Н2------ 2Тс-|-7Н2О
600° 1100°
ТсО24-2Н2 ——► Тс4-2Н2О Tc2S7 + 7H2---► 2Tc-|-7H2S
Если вместо NH4TcO4 берут As(CeH5)4TcO4, восстановление
проводят не в платиновой, а в медной лодочке.
При электролизе суспензии, состоящей из 3—4 г As(GeH5)4G104
и 0,2 г Тс в виде As(GeH5)4TcO4 в 1 л разб. H2SO4 (электролиз
ведут с платиновыми электродами при плотности тока 10 а/дм*
и напряжении 2—3 в), выделяется порошкообразный металли-
ческий технеций, загрязненный собственными окислами низших
степеней окисления- Очистка осуществляется нагреванием при
~б00° в атмосфере водорода.
Металлический технеций можно также получить вытеснением
из кислых растворов его соединений амальгамой цинка.
ФИЗИЧЕСКИЕ И ХИМИЧЕСКИЕ СВОЙСТВА
Технеций представляет собой парамагнитный металл серебри-
сто-серого цвета с плотной гексагональной упаковкой кристалли-
ческой решетки, плотностью 11,49 г/см3 (тяжелый металл) и т. пл.
2127° (тугоплавкий металл); технеций имеет много изотопов.
С химической точки зрения металлический технеций устойчив
при обычной температуре в сухом воздухе, но во влажном воздухе
ТЕХНЕЦИЙ 437
покрывается слоем окислов (соответствующих низшим степеням
окисления). При нагревании (500°) на воздухе или в кислороде
превращается в летучую окись Тс2О7.
В отличие от рения технеций обладает меньшим сродством
к хлору, и ТсС14, получаемый прямым взаимодействием элементов
при нагревании, мало устойчив.
Металлический технеций растворяется в HNO3, царской
водке, смеси концентрированных кислот HNO3 Д- H2SO4 и в отли-
чие от рения не подвергается действию Н2О2 или НС1.
СОЕДИНЕНИЯ (ОБЩИЕ СВОЙСТВА)
Известен ряд соединений одно-, двух-, трех-, четырех-, пяти-,
шести- и семивалентного технеция. Самыми устойчивыми являются
соединения семи- и четырехвалентного технеция. Соединения
технеция(УП) менее устойчивы при нагревании и легче гидроли-
зуются, чем соответствующие соединения рения(УП). Как в семи-
валентном, так и в четырехвалентном состояниях соединения
технеция обнаруживают окислительные свойства.
Окислительно-восстановительные потенциалы для нескольких
систем в кислой среде при 25° приведены ниже:
ТсОу Д- 8Н+ Д- le~ Тс + 4Н2О Д-0,472 в
TcOj + 4Н+ + Зе- ТсО2 + 2Н2О Д-0,771 в
ТсО2 + 4Н+ -: 2е-Тс2++ 2Н2О Д-0,60 в
Соединения технеция и рения могут быть легко разделены
благодаря различной растворимости их сульфидов Tc2S7 и Re2S7
в 9—10 н. НС1, а также вследствие различной устойчивости их
хлоридов и хлоросолей.
Соединения четырехвалентного технеция
Из соединений технеция(1У) известны ТсО2, ТсС14, К2[ТсС16]
и К2[Тс16]. Соединения технеция(1У) в присутствии избытка
энергичного окислителя, например K2S2O8, (NH4)2S2O8, Ce(SO4)2,
окисляются до пертехнетатов.
Двуокись технеция, ТсО2, получают термическим разложением
NH4TcO4, термическим восстановлением NH4TcO4, восстановле-
нием активным водородом (вытесняемым Zn из 3—4 н. раствора
НС1) при нагревании или электролитическим восстановлением
NH4TcO4 при строго определенном потенциале (электроды из Pt):
NH4TcO4 ТсО2 Д- 2Н2О Д- l/2N2
Соединение ТсО2 выделяют в виде черного порошка; оно очи-
щается перегонкой при 900—1100° в отличие от МпО2 и ReO2,
которые при этой температуре разлагаются по уравнениям
ЗМпО2 Мп3О4 Д- О2 7ReO2 2Re2O7 Д- ЗБе
438
ПОБОЧНАЯ ПОДГРУППА VII ГРУППЫ
Кислород и воздух окисляют ТсО2 ДО Тс2О7.
При действии газообразного хлора на ТсО2 (300°) образуется
черно-синий продукт, который очищают перегонкой при 80°.
При 900° получают коричневый продукт, который очищают пере-
гонкой при ~500°. Гидролиз этих продуктов дает черный осадок
ТсО2 -2Н2О.
Тетрахлорид технеция, ТсС14, образуется в виде красных
кристаллов при действии СС14 на Тс2О7 (400°) в закрытой трубке.
При сильном нагревании ТсС14 разлагается на элементы.
Гексахлоротехнетат калия, К2[ТсС16], получают восстановле-
нием NH4TcO4 иодидом калия в сильно солянокислой среде
при нагревании.
Соединение К2[ТсС1в] представляет собой желтые кубические
кристаллы. Термически оно менее устойчиво, чем Ka[ReCl6l,
и легко гидролизуется, превращаясь в ТсО2-2Н2О:
1000°
К2[ТсС16]--->• Тс + 2КС1 + 2С12
В отличие от Ка[ТсС1в] соединение К2[ВеС16] перегоняется
без разложения.
Соединения семивалентного технеция
Из соединений технеция(УП) известны Тс2О7, НТсО4, МетТсО4
(где Me1 = NH+, К+, As(C6H5)*, Р(С6Н5)+), а также TcO3F,
ТсО3С1 и Tc2S7.
Анион ТсО“ бесцветен, относительно устойчив, обладает окис-
лительным характером и сильно ингибирующим действием по отно-
шению к коррозии.
Металлы Zn, Fe, Ni, Sn, Pb, Cu, Hg могут восстанавливать
(в определенных условиях) соединения технеция(УП) до метал-
лического технеция. В солянокислой среде дихлорид олова вос-
станавливает соединения технеция(УП) до соответствующих соеди-
нений технеция(П). Галогеноводороды HG1, НВг, HI также
обладают восстановительным действием по отношению к соеди-
нениям технеция(УП).
Окись технеция (пертехнециевый ангидрид), Тс2О7, получают
действием чистого сухого кислорода на металлический технеций
при 500°.
Соединение Тс2О7 представляет собой парамагнитные гигро-
скопичные желтые кристаллы с т. пл. 119,5° и т. кип. 310,6°.
Тс2О7 легко растворяется в воде с образованием технециевой
кислоты НТсО4. Благодаря хорошей летучести Тс2О7 можно
очищать перегонкой при 550°. При нагревании Тс2О7 может вос-
станавливаться водородом до металлического технеция и метал-
ТЕХНЕЦИЙ
439
лическим технецием до ТсО3. Взаимодействие аммиака с Тс2О7
в присутствии воды дает NH4TcO4:
Тс2О7 + 2NH3 + Н2О = 2NH4TcO4
Технециевая кислота, НТсО4, образуется при действии воды
на окись технеция(УП).
Обезвоживание розовых водных растворов технециевой кис-
лоты над конц. H3SO4 приводит к осаждению красных гигроско-
пичных кристаллов НТсО4.
Технециевая кислота является сильной кислотой, ее можно
экстрагировать из водных растворов многими органическими
растворителями.
Соли технециевой кислоты, называемые пертехнетатами, имеют
общую формулу МетТсО4 (где Me1 = NH*, К+, As(C6H5)+ или
Р(С6Н5)Д; они обладают сильными окислительными свойствами.
Для экстракции пертехнетатов используют главным образом
хлороформ и метилэтилкетон.
Триоксифторид технеция, TcO3F, получают действием фтора
на ТсО2 при 150°. Он представляет собой желтое вещество с т. пл.
18,3°.
Триоксихлорид технеция, ТсО3С1, экстрагируют хлороформом
в виде бесцветного вещества из раствора, полученного при добав-
лении 12 М НС1 к раствору КТсО4 в 18 М H2SO4.
Сульфид технеция, Tc2S7, осаждается в результате пропуска-
ния H2S через солянокислые (2—4 н. НС1) растворы пертехне-
татов .
Соединение Tc2S7 представляет собой темно-коричневый поро-
шок, трудно растворимый в воде. Сульфид технеция восстанав-
ливается водородом при 1000° до металлического технеция, пол-
ностью улетучивается в токе хлора при 100°, окисляется под
действием Н2О2 в щелочной среде до пертехнетатов и в отличие
от Re2S7 растворяется в 9—10 н. НС1.
К другим известным соединениям технеция относятся ТсО3 (пурпур-
ный), TcF6 (золотисто-желтый), ТсС16 (зеленый) и Тс2(СО)10.
Отделение технеци я(УП) от молибден а(У1)
и р е н и я(УП) с помощью ионообменных смол.
В слабо щелочной среде Тс7+ и Мо6+ удерживаются амберли-
том IRA 400. Для того чтобы разделить их, молибден элюируют
10%-ным раствором NaOH или 1 М раствором К2СаО4, а техне-
ций — 0,5 М раствором тиоцианата или 0,25 М раствором пер-
хлората.
Смола дауэкс-2 в сульфатной форме удерживает Тс7+ и Re7+.
Технеций элюируют раствором сульфата и тиоцианата аммония,
доведенного с помощью NaOH до pH = 8,5. Re7+ элюируют
440
ПОБОЧНАЯ ПОДГРУППА VII ГРУППЫ
0.1 М растворами (NH4)2SO4 и NH4SGN, а 1 М растворы тех же
веществ вымывают Тс7+. Для элюирования Тс7 + можно приме-
нять 0,25 М раствор, содержащий ион С1О~.
Отделение Тс7+ о т Мо6+ и U6+ экстракцией.
Органические пертехнетаты очень хорошо экстрагируются
хлороформом независимо от pH раствора. Если осаждение прово-
дится в щелочной среде, то отделение Тс7+ от Мо6+ или U6+ экстрак-
цией происходит количественно.
Было предложено осуществлять экстракцию пертехнетатов
в щелочной среде метилэтилкетоном, а НТсО4 — изобутилкето-
ном или амиловыми спиртами.
РЕНИЙ Re
Z = 75; ат. вес = 186,2
Валентность (I), (II), (III), IV, (V), (VI), VII
Массовые числа основных природных изотопов 187 и 185
Электронная структура атома рения.’ К-L -M-N-5s25p65d5 -6s2.
Электронная структура атома
и бх-орбиталей:
рения и катиона Re4+ для 5d-
5ds
6s2
N
________5<Р
t t t
6s
t т т т т
Re
Re4+
ИСТОРИЧЕСКАЯ СПРАВКА
В 1870 г. Д. И. Менделеев предсказал существование двух
гомологов марганца — экамарганца и двимарганца, которые тоже
располагаются в побочной подгруппе VII группы периодической
системы. Ноддак и Такке попытались установить свойства дви-
марганца по аналогии со свойствами соседних элементов. Они
брали руды платины, молибдена, рутения, вольфрама, осмия
и других близко расположенных к элементу 75 металлов, в кото-
рых предполагали существование двимарганца, обогащали их
самыми подходящими способами и затем пытались идентифици-
ровать там новый элемент с атомным номером 75.
В 1925 г. Ноддак, Такке и Берг по данным спектрального
анализа концентратов платиновых руд объявили об открытии
элемента с атомным номером 75, который они назвали «рением»
в честь реки Рейн.
В том же, 1925 г. Лоринг и Дрюке, Гейровский и Долейшек
заметили присутствие элемента 75 в соединениях марганца.
В 1927 г. в Германии впервые получили 120 мг металличе-
ского рения из колумбита и гадолинита, которые содержат
10-6—10-7 вес. % рения.
РЕНИЙ
441
В 1928 г. супруги Ноддак получили металлический рений
из норвежского молибденита, который содержал 2—4 -10“® вес. %
рения.
РАСПРОСТРАНЕННОСТЬ В ПРИРОДЕ
В природе рений встречается в свободном состоянии в метео-
ритном железе, найденном в Южной Африке, Мексике, СССР
и других странах, и в виде соединений в минералах других эле-
ментов. Соединения рения в природе встречаются довольно редко,
обычно они рассеяны в минералах ряда элементов. Содержание
рения в земной коре равно 1,0-10"’ вес. %.
К минералам, содержащим 10~3—10-4 вес. % рения, относятся
молибденит MoS2 (из Норвегии, Японии, США, СССР, Румынии),
колумбит (Fe, Mn)Nb2O6 и танталит (Fe, Mn)Ta2Oe (из Норвегии,
Франции, Гренландии), халькопирит CuFeS2 (из СССР, Чили,
Конго, Родезии), борнит Cu5FeS4, халькозин Cu2S, пирит FeS2,
гадолинит Y2FeBe2[Si2Oi0J, тортвейтит (Sc, Y)2Si2O7 и минералы
платины.
ИЗВЛЕЧЕНИЕ РЕНИЯ ИЗ МОЛИБДЕНИТА
Поскольку сульфидные полиметаллические руды, содержащие
молибден, рений, медь, цинк и железо, характеризуются малым
содержанием молибдена и очень малым содержанием рения, их
необходимо обогащать. Сульфидные полиметаллические руды
молибденита MoS2, содержащие сульфид рения, обогащают селек-
тивной флотацией. Для извлечения рения концентраты молибде-
нита перерабатывают либо пирометаллургическим, либо гидро-
металлургическим методом.
Концентраты молибденита MoS2, содержащие сульфиды рения,
меди, цинка, железа и других металлов, подвергают окислитель-
ному прокаливанию при температуре 600—650° в большом избытке
воздуха. При этом получают летучие (SO2, Re2O7, МоО3) и неле-
тучие (CuO, ZnO, Fe2O3) окислы, которые под действием МоО3
частично превращаются в молибдаты. Прокаливание проводят
в печах таким образом, чтобы в камерах обеспыливания, кото-
рыми снабжены эти печи, Re2O7 максимально отделялся от S02
и МоО3 на основе разницы в летучести.
Полагая, что рений в молибдените находится в виде ReS2,
можно представить себе реакции, которые проходят при прока-
ливании MoS2 и ReS2 следующим образом:
M0S2 -р V2O2 = MoOg -р 2SO2 -р 266,2 ккал
2ReS2 -р 16/2О2 = Re2O7 -р 4SO2 -р 206,14 ккал
Поскольку температуры кипения равны для SO2 —10,1°,
для Re2O7 355°, а температура сублимации МоО3 740°, необхо-
442
ПОБОЧНАЯ ПОДГРУППА VII ГРУППЫ
димо осуществлять ступенчатый подъем температуры прокалива-
ния концентратов молибденита, чтобы сначала удалилась SO2.
затем Re2O7 с остатком SO2 и наименьшим количеством МоО3
и в конце — МоО3.
Необходимо, чтобы концентрат в процессе прокаливания воз-
можно полнее соприкасался с воздухом и температура была
около 650° для полного выделения Re2O7.
Для отделения рения от молибдена можно использовать раз-
ницу в температурах сублимации окислов Re2O7, МоО3, можно
осадить молибден в виде фосфоромолибдата или молибдата бария
или осадить рений в виде KReO4, NH4ReO4, наконец, можно
экстрагировать HReO4 или органические метаперренаты
As (С6Н5)4 ReO4, Р (С6Н6)4 ReO4 органическими растворителями
{амиловыми спиртами, трибутилизооктиламинфосфатом).
Как уже упоминалось, для разделения этих элементов можно
использовать ионообменные смолы.
При растворении Re2O7 (на холоду или при нагревании под
давлением) образуется HReO4, которая экстрагируется органи-
ческими растворителями, удерживается на ионообменных смолах,
превращается в KReO4 под действием КС1 или КОН и в летучий
Re2O7 при сильном нагревании.
В гидрометаллургическом процессе концентраты молибденита
измельчаются на холоду, а затем на водяной бане с конц. HNO3.
Измельченный высушенный продукт вместе с конц. H2SO4
подвергается дистилляции при 310° в токе воздуха или 200°
в токе НС1.
Дистиллят, состоящий из Re2O7 и МоО3, растворяют в воде
и раствор насыщают на холоду газообразным NH3 и затем газо-
образным H2S с целью осаждения сульфида рения и молибдена.
Смесь сульфидов растворяют в конц. HNO3 на водяной бане,
раствор выпаривают досуха, сухой продукт вновь обрабатывают
водой и к отфильтрованному раствору добавляют КС1 или КОН
для осаждения KReO4.
По другому варианту продукт, полученный при измельчении
концентрата молибденита конц. HNO3, после высушивания на во-
дяной бане и в сушильном шкафу при 120° нагревается до 600—
650° в трубке из тугоплавкого стекла для получения Re2O7;
последняя получается загрязненной небольшим количеством МоО3.
По третьему варианту раствор, полученный при измельчении
концентратов молибденита конц. HNO3, обрабатывается орто-
фосфатом натрия или аммония для неполного осаждения фосфо-
ромолибдата, который удерживает небольшое количество рения.
При обработке азотнокислого раствора (полученного после филь-
трования фосфоромолибдата) аммиаком (для нейтрализации),
а затем с помощью (NH4)2S и H2SO4 осаждаются сульфиды рения
и молибдена. Смесь сульфидов рения и молибдена растворяется
РЕНИЙ
443
в HNO3, после чего отделение рения от молибдена осуществляется
способами, описанными выше.
ПОЛУЧЕНИЕ МЕТАЛЛИЧЕСКОГО РЕНИЯ
Металлический рений получают восстановлением метаперре-
натов щелочных металлов KReO4, NH4ReO4, окислов ReO2,
Re2O7, сульфидов ReS2, Re2S7 или оксигалогенидов рения ReOCl4,
ReO3Cl, ReOF4, ReO2F2 водородом при нагревании, а также
термическим разложением соединений Re3Clg, ReO2, (NH4)2[ReIe],
K2[ReIe] или разложением на нагретом катоде димера Re2(CO)10
и электролизом кислых растворов метаперренатов.
Восстановление метаперренатов
щелочных металлов водородом при нагревании
Когда метаперренат калия KReO4 в серебряной лодочке поме-
щают в трубку для сжигания и нагревают в токе водорода при 250°
(2 час) и затем при 400—500° (еще 2 час), образуются ReO2, КОН
и Н2О:
KReO4 + з/2Н2 = ReO2 + КОН + Н2О
Гидроокись калия удаляется после промывания водой 6 н. НС1
и затем спиртом.
Нагревая в токе водорода при 1000° двуокись рения, поме-
щенную в лодочке из SiO2, А12О3 или ZrO2 в трубку для сжига-
ния, получают серовато-черный порошок металлического рения:
ReO2 + 2Н2 = Re + 2Н2О
Если ReO2 содержит КОН, то порошкообразный металличе-
ский рений имеет черный цвет и расплывается на воздухе, посколь-
ку он загрязнен окислами рения низших степеней окисления
и метаперрениевой кислотой HReO4.
Для получения более чистого металлического рения рекомен-
дуется восстановление метаперрената аммония водородом при
нагревании:
-300°
2NH4ReO4 + 3H2---> 2ReO2 + 2NH3 + 4H2O
1000°
ReO2 + 2H2---->- Re + 2H2O
При температуре около 1000° соединения Re2O7, ReS2, Re2S7,
ReOCl4, ReO3Cl восстанавливаются водородом:
Re2O7 + 7H2 = 2Re + 7H2O
ReOCl4 + 3H2 = Re + 4HC1 + H2O
Re2S7 + 7H2 = 2Re + 7H2S
ReO3Cl + 7/2H2 = Re + HC1 + 3H2O
ReS2 -|- 2H3 = Re -|- 2H2S
444
ПОБОЧНАЯ ПОДГРУППА VII ГРУППЫ
Термическая диссоциация
тримера трихлорида рения Re3Cl9
При нагревании вольфрамовой нити (диаметр 50 мк) до 18000,
(или платиновой нити того же диаметра до 1200—1400°) в парах
Re3Cl9 она равномерно покрывается слоем металлического рения.
Нагревание проводят в ампуле из стекла пирекс, которая имеет
отверстие для подачи Re3Cl9 и металлического рения и отверстие
для откачивания. В ампулу впаяны два никелевых контакта,
соединенных внутри ампулы вольфрамовой или платиновой
нитью, которые подсоединяются к электрической цепи. При обо-
гревании ампулы снаружи высвободившийся при термической
диссоциации Re3Cl9 хлор вновь вступает в реакцию с металли-
ческим рением, образуя Re3Cl9, который в свою очередь подвер-
гается термическому разложению.
При термическом разложении соединений ReO2, (NH4)3[ReIe]r
K3[ReIe] также получают металлический рений:
700°
7ReO2----->• 3Re-|'2Re2O7
700°
(NH4)2[ReIe] Re+2NH4I + 2I3
Разложение димера пентакарбонила рения
Металлический рений получают разложением на нагретом
катоде димера пентакарбонила рения Re3(CO)10. При этом Re3(CO)1(>
предварительно пропускают в потоке газа-носителя (Не или СО3)
через ионизационную камеру, содержащую радий или уран.
Электролитический способ получения
металлического рения
При электролизе (25—45°) раствора, содержащего 3,3 г/л H3S04
(d = 1,84 г/см2) и 11 г/л KReO4, с катодом из Мо, W, Pt, Rh,
Сг, Ni при плотности электрического тока 10—14 а/д.ч? на катоде
осаждается блестящий слой металлического рения. Если в элек-
тролитическую ванну, которая содержит соли никеля, железа
и кобальта, ввести метаперренаты, то происходит электролити-
ческое осаждение сплавов Re — Ni, Re — Fe, Re — Co.
Для удаления примесей окислов рения электролитически
полученный металлический рений нагревают при 1000° в токе
водорода.
ФИЗИЧЕСКИЕ И ХИМИЧЕСКИЕ СВОЙСТВА
R компактном состоянии металлический рений имеет сереб-
ристо-белый цвет (в порошкообразном состоянии — черный, в кол-
лоидном — коричневый), для него характерны плотнейшая гекса-
РЕНИЙ
445
тональная решетка, плотность 20,99 г/см2 (тяжелый металл),
т. пл. 3170° (тугоплавкий металл), т. кип. 5630°. Он парамагни-
тен, обладает высокой электронной эмиссией, является электри-
ческим полупроводником. При 1630° электронная эмиссия рения
выше, чем у вольфрама.
Известны природные изотопы 187Re, 185Re и искусственные
изотопы 186Re, 188Re, 188Re, 184Re.
Металлический рений обладает превосходными механическими
свойствами, он более ковок и тягуч, чем вольфрам. Обработка
рения прессованием сопряжена с трудностями, так как рений
допускает лишь холодное прессование, становясь хрупким при
нагревании.
Порошок рения превращают в компактный металл теми же
методами, которые были описаны для вольфрама и титана.
Известны многочисленные сплавы рения с Re, Al, Sn, Cu,
Ag, Zn, Hg, Ti, Nb, Ta, Cr, Mo, W, U, Fe, Co, Ni, Rh, Pd, Os,
Ir, Pt. Сплавы рения получают сплавлением компонентов в вакуу-
ме, спеканием порошка металлического рения с порошком другого
тугоплавкого металла в инертной (аргон) или в восстановитель-
ной атмосфере, нагреванием NH4ReO4 с порошком легирующего
металла в атмосфере водорода, электролизом раствора, содержа-
щего соли рения и другого металла. R качестве примеров интер-
металлических соединений можно указать Re2Re, Fe3Re2, Fe2Re3,
TisRe24, W2Re3, URe2, U3Re.
Спектр рения очень богат характеристическими линиями,
в частности в видимой области наблюдается синяя линия 4889,15 А,
зеленые линии 5275,57 А, 5270,89 А и красные линии 7912,94 А,
7640,93 А.
Металлический рений обладает относительной химической
инертностью, и его поведение по отношению к различным хи-
мическим реагентам зависит от степени его чистоты и из-
мельчения.
R компактном состоянии при обычной температуре металли-
ческий рений не взаимодействует с кислородом воздуха. Тонко
измельченный металлический рений окисляется при обычной
температуре во влажном кислороде с образованием рениевой
кислоты. При нагревании порошка металлического рения выше
150° (или компактного металла до 350°) на воздухе или в токе
кислорода образуется летучая окись Re2O7. При неполном окисле-
нии рения на воздухе или в кислороде образуется ReO2. Метал-
лический рений значительно более устойчив по отношению к кис-
лороду, чем вольфрам или молибден. Рениевая проволока подвер-
гается действию смеси воздух + азот, содержащей 10% кисло-
рода, только при температурах выше 1600°. При легировании
устойчивость рения по отношению к кислороду значительно
понижается.
446
ПОБОЧНАЯ ПОДГРУППА VII ГРУППЫ
При нагревании металлический рений взаимодействует с фто-
ром, хлором, бромом, серой и селеном:
125° 500 — 650°
Re + 3F2-------► ReF6 3Re-|-9/2Br2-----------► Re3Brg
400 — 600° Нагревание
Не + 5/гС12 ReCis i/sRegClg + C^
Нагревание
Re + 2S (или Se)-----------> ReS2 (или ReSe2)
Нагревание хлора или брома с металлическим рением в при-
сутствии кислорода воздуха приводит к образованию наряду
с галогенидами и оксигалогенидов ReOCl4, ReO3Cl, ReO3Br,
ReO2Br2.
Ниже 1000° азот и аммиак не взаимодействуют с рением,
выше 1900° азот, насыщенный парами воды или содержащий
15% СО2, действует на проволоку из металлического рения.
При действии фосфора (выше 750°), мышьяка (выше 600е),
кремния (1500—1600°) и германия (повышенная температура)
на металлический рений образуются Re2P, ReP, ReP2, ReP3,
Re3As7, ReSi2, ReSi, Re3Si, ReGe2 и др.
В отличие от молибдена или вольфрама рений при высокой
температуре не образует карбидов с углеродом, окисью углерода,
метаном (или с различными углеводородами). Нагревание рение-
вой проволоки до белого каления в атмосфере углеводородов
делает ее ломкой, при этом электросопротивление проволоки
не меняется.
В нормальных условиях рений устойчив к действию воды.
Металлический рений не взаимодействует с галогеноводорода-
ми HF, НС1, НВг, растворяется на холоду в HNO3 (разбавленной
или концентрированной), при нагревании — в концентрирован-
ных H2SO4, НС1О4, а также в пергидроле, хлорной или бромной
воде.
3Re + 7HNO3 = 3HReO4 + 7NO + 2Н2О
2Re + 7H2SO4 = 2HReO4 + 7SO2 + 6H2O
8Re + 7HC1O4 + 4H2O = 8HReO4 + 7HC1
2Re + 7H2O2 = 2HReO4 + 6H2O
2Re + 7C12 + 8H2O = 2HReO4 + 14HC1
Водные растворы щелочей на воздухе медленно действуют
на металлический рений в компактном состоянии.
РЕНИЙ
447
Химическая активность металлического рения иллюстрируется
следующей схемой:
Комн. темп.
Рений
Нагревание
( во влажном воздухе —>- HReO4
t с HNO3 HReO4
(в воздухе или кислороде —>- Re2O7, ReO2
I с F2, Cl2, Вг2, —ReF6, ReCl5, ReCl6, Re3Clg, Re3Br9
I c S, Se, P, As, Ge —>- Re2P, ReP, ReP2, ReP3, Re3As7,
ReSi2, ReSi, Re3Si, ReGe2
конц. H2SO4, конц. HC1O4 —>- HReO4
с пергидролем, хлорной или бромной водой —>- HReO4
„ в окислительно-щелочном расплаве —>• метаперренаты
При высокой температуре в неокислительной атмосфере метал-
лический рений очень устойчив к действию углерода и некоторых
расплавленных металлов.
С физиологической точки зрения рений и его соединения не
токсичны для человека и животных, но токсичны для растений.
ПРИМЕНЕНИЕ
Благодаря пониженной летучести, хорошей электропроводно-
сти, превосходной термоэлектронной эмиссии металлический рений
используют для изготовления различных электродов в электрон-
ных рентгеновских трубках и радиолампах.
Металлический рений применяют в качестве добавки при
получении многих сплавов: W, Мо, Та, Ni, Rh, Os, Ir, Pt. Добав-
ка рения к платиновым металлам способствует изменению твер-
дости, ковкости, тягучести и механической прочности. Сплавы
рения с W, Мо, Та, Ir, Pt, Os, Ni тугоплавки, нелетучи, механи-
чески прочны, трудно окисляются, устойчивы к действию различ-
ных химических реагентов.
Как металлический рений, так и сплавы Re — Pt, Re — Pd,
Re — Rh, Re — Ir применяют для изготовления термопар. Из
сплавов платиновых металлов с Re делают ювелирные изделия.
Из металлического рения и его сплавов изготавливают кон-
такты мощных электрических переключателей.
Сплавы тройной системы Ir — Rh — Re допускают прокаты-
вание в тонкие листы и протягивание в проволоку очень малого
диаметра. Сплавы рения с W, Мо, Сг, Та, Со обладают значитель-
ной твердостью и кислотостойкостью. Благодаря устойчивости
рения к кислотам было предложено покрывать им стенки резер-
вуаров и цистерн для хранения и транспортировки кислот.
Осаждением металлического рения получают зеркала с боль-
шой отражательной способностью.
Б виде порошка (или в коллоидном состоянии) металлический
рений может быть использован как катализатор гидрирования
(для соединений с этиленовыми связями), дегидрирования (для
448
ПОБОЧНАЯ ПОДГРУППА VII ГРУППЫ
спиртов), хлорирования (при превращении метана в хлороформ),
в окислительно-восстановительных реакциях (при окислении
NH3 в NHO3, SO2 в H2SO4, нитритов в нитраты, пропана в акро-
леин и др.), при изотопном обмене (получение тяжелого аммиака
обменом с дейтерием).
СОЕДИНЕНИЯ (ОБЩИЕ СВОЙСТВА)
Известно относительно небольшое число соединений одно-,
двух-, трех-, пяти- и шестивалентного рения, и все они малоустой-
чивы. Наиболее устойчивы соединения четырех- и семивалентного
рения.
В табл. 51 приведены формулы и указан цвет некоторых соеди-
нений рения, сгруппированных по валентностям.
Соединения одно- и двухвалентного рения можно получить
только без доступа воздуха, поскольку они устойчивы в отсут-
ствие воздуха.
Соединения трех-, четырех-, пяти- и шестивалентного рения
диспропорционируют при нагревании или под действием воды:
9Re3+ Re7 + + 2Re<*+ + 6Re2+ 3Re5+ =₽* Re7+ + 2Re«+
5Re«+ 2Re7+ + 3Re2+ 3Re’+ 2Re7+ + Re<+
В отличие от марганца соединения семивалентного рения более
устойчивы, чем четырехвалентного. Окислительные свойства сое-
динений рения(УП) выражены слабее, чем у соединений марган-
ца (VII): анион ВеО~ бесцветен, а МпО“ фиолетовый. Под дейст-
вием щелочей метаперренаты MerReO4 превращаются в мезопер-
ренаты Ме*ВеО5, но не в соединения рения(У1), как в случае
превращений аналогичных соединений марганца.
В щелочной или очень слабо кислой среде воздух (в большей
или меньшей степени) окисляет все соединения рения низших
степеней окисления до соединений рения(УП). Чем меньше pH
растворов, в которых находятся соединения рения(УП), тем
ниже степень окисления соединений, получаемых в результате
восстановления этих соединений.
Растворы соединений одно-, двух- и трехвалентного рения
устойчивы только в отсутствие воздуха.
Если соединения рения в низших степенях окисления прояв-
ляют основной и восстановительный характер, то соединения
шести- и семивалентного рения имеют кислотные неметалличе-
ские свойства и ковалентные связи.
Соединения рения могут быть самых разнообразных цветов,
причем соединения в низших степенях окисления преимущественно
окрашены в черный цвет.
Помимо соединений рения указанных степеней окисления
известны металлоорганические производные и соединения вклю-
чения.
РЕНИЙ
449
Соединения одновалентного рения
Известно небольшое число соединений электроположительного
одновалентного рения. Соединения эти малоустойчивы и полу-
чаются (соответственно хранятся) без доступа воздуха.
Соединения рения(1) получают восстановлением KReO4
в слабо сернокислой (или солянокислой) среде амальгамой цинка,
восстановлением сильно кислых растворов соединений рения(Ш)
и (IV) цинком или амальгамой натрия и термическим разложением
Rel4 при 110° в токе азота.
В качестве примеров соединений рения(1) можно привести
Re2O-2H2O, Rel, Me* [Re(CN)e]-rH2O (где Me1 = Na+, K+),
DipyH[ReDipy2(CN)2l, H3[RePhn(CN)4] (где Dipy = а, а'-дипи-
ридил и Phn — о-фенантролин) и галогенокарбонилы Re(CO)5X,
[Re(CO)4X]2 (где X = СР, Br", I"), Re(CO)3PyCl, Re(CO)3PhnX
(где X = СР и Ру = C5H5N).
Под действием энергичных окислителей соединения рения(1),
окисляются до перренатов. С галогеноводородами НС1, НВг
рений(1) образует комплексы [ReXn]n-1.
Дигидрат окиси рения, Re2O-2H2O, образуется в виде черного
осадка при восстановлении HReO4 в 0,2 н. НС1 порошкообраз-
ным цинком в атмосфере СО2-
Моноиодид рения, Rel, представляет собой черные кубические
кристаллы, которые при нагревании в вакууме разлагаются на
элементы.
Соединения двухвалентного рения
Известно небольшое число соединений электроположитель-
ного двухвалентного рения. Эти соединения обладают малой устой-
чивостью и образуются в отсутствие воздуха как в смеси с соеди-
нениями рения(1), так и при диспропорционировании соединений
рения(Ш) и (IV) по уравнениям
9Re3+ 6Re2 + + 2Re4+ + Re’+
5Re4+ 3Re2+ + 2Re7+
Соединения двухвалентного рения менее устойчивы и более
малочисленны по сравнению с аналогичным состоянием марганца.
В качестве примеров соединений рения(П) можно при-
вести ReO фН2О, Rel2, ReS, ReCla'^HaO, H2[ReCl4]-2Н2О,
H2[Re(OH)3Cl], ReX2«(AsR3)3, ReX2-(AsR3)4, ReX2-(PR3)3.
Моногидрат окиси рения, ReO-H2O, образуется в отсутствие
воздуха в результате восстановления HReO4 металлическим кад-
мием в разбавленном солянокислом растворе. Соединение ReO -Н2О
представляет собой черный порошок; оно плохо растворимо
в воде, легко растворяется в HNO3 и бромной воде, не взаимо-
действует с НС1 или щелочами, окисляется на воздухе.
29-0101
450
ПОБОЧНАЯ ПОДГРУППА VII ГРУППЫ
Дииодид рения, Rel2, получают термическим разложением
Rel4. Он представляет собой парамагнитный серовато-черный
Порошок.
Моносулъфид рения, ReS, получают действием смеси водорода
и сероводорода на нагретый до 90° ReO3:
ReO3 + 2Н2 + H2S = ReS + ЗН2О
Соединение ReS представляет собой черный аморфный порошок
с плотностью 7,11 г/см3. Оно превращается в ReS2 при нагрева-
нии до 300—500° с серой, диспропорционирует на ReS2 и метал-
лический рений при нагревании выше 680° в вакууме или токи
азота, окисляется кислородом воздуха в присутствии воды по-
уравнению
2ReS + 9/2О2 + 2Н2О -» HReO4 + HReO3S + H2SO4
Соединения трехвалентного рения
Известно относительно мало соединений электроположитель-
ного трехвалентного рения: Re2O3*raH2O, Re3Cl9, H[Re3Cl10],
Me4Re3Cl10], Ме1[Не3С1и], Mei[Re3Cl12], Mei[Re2Cl8], Re3Rr9,
H[Re3Rr10], Me4Re3Br10], Me4Re4Br13], Re3I9, Re (CH3)3,
Re(C2H5)3 и др.
При обработке тримеров галогенидов рения(П1) галогенида-
ми щелочных металлов в присутствии соответствующих галогено-
водородов образуются ацидосоли типа MeI[Re3X10], MeJ[Re3XH],
Mei[Re3X12] (где Me1 = К+, Rb+, Cs+ и Х = С1", Вг").
Большинство соединений рения(Ш) интенсивно окрашена
и склонно к комплексообразованию.
Соединения рения(Ш) окисляются до соединений рения(1У}
и (VII) преимущественно в щелочной среде.
Соединения рения(Ш) более устойчивы, чем соответствующие
соединения марганца, и проявляют меньшую способность к дис-
пропорционированию.
Растворы соединений рения(Ш), как и растворы соединений
железа(Ш), окрашиваются в интенсивно синий цвет в присут-
ствии K4[Fe(CN)6] и в интенсивно красный цвет в присутствии
KSCN.
Окись рения(11Т), Re2O3-raH2O, получают в отсутствие воз-
духа путем обработки Re3Cl9 щелочами.
Re2O3*raH2O представляет собой черное твердое вещество, не
обладающее кислотными свойствами, мало растворимое в воде,
легко окисляющееся до ReO2*raH2O в воде и диспропорциониру-
ющее в щелочной среде при нагревании по уравнению
9Re3+ 6Re2+ +[2Re4+ + Re7*
Тример трихлорида рения, Re3Cl9, получают в отсутствии
воды и кислорода действием хлористого сульфурила SO2C12 на
РЕНИЙ
451
порошкообразный металлический рений и термическим разложе-
нием ReCl5 или Ag2[ReCle] в атмосфере азота:
6Re + 9SO2C12 = 2Re3Cl8 + 9SO2
3ReCl5 Resell + 3C12
3Ag2[ReCl6] Re3Cl9 6AgCl -|- 3/2Cl2
Соединение Re3Cl9 представляет собой парамагнитные блестя-
щие красные гексагональные кристаллы, устойчивые в сухом
воздухе, растворяющиеся в воде, ацетоне, ледяной уксусной кис-
лоте, спирте, эфире, диоксане; т. пл. 727°, т. кип. 827°. Re3Cl9
очищают перегонкой при пониженном давлении, поскольку при
атмосферном давлении тример разлагается на металлический
рений и хлор. Из водных или спиртовых растворов Re3Cl9 выде-
ляют гидраты и сольваты трихлорида рения. Иодный раствор трехъ-
ядерного комплекса Re3Cl9 не дает осадка с AgNO3, поскольку
в этом соединении хлор связан неионогенно.
При гидролизе Re3Cl9 водой (холодной или теплой) образуется
Re2O3-nH2O, а при растворении Re3Gl9 в НС1 — H[Re3Cl10],
который можно экстрагировать эфиром из очень сильно соляно-
кислой среды.
При обычной температуре Re3Cl9 очень плохо растворим
в плавиковой кислоте, а при 350° Re3Cl9 взаимодействует с HF
по уравнению
Re3Cl9 + 9HF = 3Re + 9НС1 + 9/2F2
Водород восстанавливает (250—300°) Re3Gl9 до металличе-
ского рения. Нагревание Re3Cl9 на воздухе или в кислороде
(400°) приводит к образованию оксихлоридов ReOCl4, ReO3Cl.
В нейтральных или щелочных водных растворах в присут-
ствии воздуха Re3Cl9 окисляется до соединений рения(1У).
Обработка Re3 С19 растворами, содержащими анион CN-, дает
соединения с комплексным анионом [Re3+(GN)4]_, легко диспро-
порционирующие по уравнению
2[Re3+(CN)4]~ -> [Re2+(CN)4 •Re4+(CN)4]2-
Трихлорид рения Re3Cl9 применяют для покрытия металлов
рением путем нагревания при 450—500° в атмосфере азота.
Ацидосоли общей формулы MeI[Re3Cl10], Ме^ВеэСХц],
Me|[Re3Cl12] (где Me1 = К+, Rb+, Cs+) представляют собой крас-
ные ромбоэдрические мало растворимые в воде кристаллы. Они
образуются в результате добавления KG1, RbGl, CsGl к соляно-
кислому раствору Re3Cl9 (который является кислотой H[Re3Cl10]),
восстановлением метаперренатов в 8—9 н. растворе НС1 дихлори-
дом хрома или катодным восстановлением солянокислого раство-
ра K2[ReCl6J.
29*
452
ПОБОЧНАЯ ПОДГРУППА VII ГРУППЫ
При нагревании до 550—600° ацидосоли Me|[Re3Cl12] диспро-
порционируют по уравнению
6Mej[Re3Cl12] 9MeJ[ReCl6] + 2Re3Cl9 + 3Re
Пиридиновая соль, C5H5N •H[Re3Cl10], имеет темно-красный
цвет и легко растворяется в воде.
При восстановлении метаперренатов ReO~ в НС1 водородом
или НвР2О4 образуются соединения, содержащие анион [Re2Cl8]2-,
т. е. соединения типа Me£[Re2Cl8]. Монокарбоновые органические
кислоты взаимодействуют с соединениями, содержащими анион
[Re2Gl8]2-, по уравнению
[Re2Cl8]2- + 4RCOOH - Re2(RCOO)4Cl2 + 4НС1 + 2С1-
Тример трибромида рения, Re3Br9, получают в отсутствие
воды и кислорода нагреванием до 450° металлического рения
в смеси паров брома с азотом или разложением Ag2[ReBr6] в ваку-
уме при умеренной температуре:
3Ag2[ReBre] —>- Re3Br9 -f- 6AgBr 4- 3/2Вг2
Соединение Re3Br9 представляет собой коричнево-красные
кристаллы с т. пл. 627° и т. кип. 727°. Это соединение очищают
многократной сублимацией в вакууме при 500°, оно устойчиво
на воздухе, гидролизуется водой, растворяется в ацетоне, спирте
и эфире с образованием устойчивых красных растворов.
При растворении окиси Re2O3-nH2O в НВг образуется кисло-
та H[Re3Br10]. Были получены соли этой кислоты: K[Re3Br10],
C5H5N-H[Re3Br10], МеЧВе4Вг13].
Восстановление метаперренатов ReO* в НВг водородом или
Н6Р2О4 приводит к соединениям, содержащим анион [Re2Br8]2".
Тример трииодида рения, Re3I9, образуется при нагревании
Rel4 до 350° в закрытой трубке. Он представляет собой фиолетово-
черные кристаллы, плохо растворимые в воде, спирте, ацетоне,
эфире и разбавленных кислотах.
При нагревании в вакууме Re3I9 ступенчато разлагается
с выделением иода.
Помимо описанных известны и другие соединения рения(Ш), например
K3[Re(CN)6], K3[Re(OH)3(CN)3], алкоголяты Re(OR)3, металлоорганические
производные Re(CH3)3, Re(C2H5)3, продукты взаимодействия Re3Cl9
с (С6Н5)3Р, (С6Н3)3Аз, с бис-(диметилдиарсином) (CH3)4As2, производные
(H-C5H5)2ReH, (ft-C5H5)2ReH+.
Соединения четырехвалентного рения
Известны многочисленные устойчивые соединения электро-
положительного четырехвалентного рения. Соединения семива-
лентного рения наиболее устойчивы, однако соединения рения(1У)
РЕНИЙ
453
немногим уступают им и они гораздо более устойчивы, чем соот-
ветствующие соединения марганца.
В табл. 51 приведены формулы и указан цвет некоторых соеди-
нений рения(ГУ).
В отличие от соединений марганца(ТУ) соединения рения(1У)
легче превращаются в соединения рения(УП). Окисление соеди-
нений рения(ТУ) в соединения рения(УП) проводят в слабо кислой
(НС1) или щелочной средах кислородом воздуха, С12, Н2О2,
К2Сг2О7, КМпО4, солями Fe3+ и Се4+.
При нагревании ВеО2 на воздухе образуется Ве2О7 (в отличие
от соответствующего соединения МпО2, которое отдает кислород
и превращается в МпО), а при нагревании в вакууме ВеО2 дис-
пропорционирует на Ве2О7 и металлический рений.
Нагревая соединения рения(ТУ) со щелочами, получают
ВеО2-гаН2О.
Соединения рения(1У) получают окислением металлического
рения кислородом при нагревании или восстановлением метапер-
ренатов сульфатом хрома(П) в H2SO4 (концентрация меньше 6 н.).
В качестве восстановителей можно применять KI, SnCl2, TiCl3
и СгС12 в 3—4 н. растворе НС1.
Рений(ГУ) входит в состав ряда анионов, например ReO|~,
[ReXeF-, [Re(OH)X5l2- или [Re(OH)n Xe_J2-, [Re2OX10l4- (где
X = галоген), [ReO2(CN)4]4-.
Двуокись рения, ReO2, получают нагреванием металлического
рения в небольшом количестве кислорода, восстановлением
Re2O7 или ReO3 водородом или металлическим рением при нагре-
вании в тугоплавкой стеклянной трубке и восстановительным
пиролизом NH4ReO4 с металлическим рением:
300°
Re-|-О2= ReO2-|-103,4 ккал Re2O7-|-3H2 t * 2ReO24-3H2O
-600°
4NH4ReO4 + 3Re = 7ReO2-|-4NH3 + 2H2O 2Re2O7-|-3Re 7ReO2
Соединение ReO2 -2H2O получают гидролизом при нагрева-
нии ацидосолей типа Mex[ReCl6], добавлением небольшого коли-
чества щелочей к ацидосолям Mex[ReCl6], из соединений рения(У)
при растворении их в воде, химическим или электрохимическим
восстановлением метарениевой кислоты или метаперренатов
в слабо кислых растворах:
Me2[ReCl6] + 4Н2О ReO2.2H2O + 2МехС1 + 4НС1
Me2[ReCle] + 4NaOH = ReO2-2H2O 2MezCl + 4NaCl
2MezReO4 + 3SnCl2 + 8HC1 -= 2ReO2.2H2O + 3SnCl4 + 2MezCl
2MezReO4 + 3Zn + 8HC1 = 2ReO2-2H2O + 3ZnCl2 + 2MezCl
454
ПОБОЧНАЯ ПОДГРУППА VII ГРУППЫ
Двуокись рения ReO2, полученная сухим путем или дегидра-
тацией ReO2-2H2O в вакууме при 650°, представляет собой слабо
парамагнитный нелетучий коричнево-черный кристаллический
порошок с плотностью 11,4 з/см3; она устойчива в отсутствие
кислорода и диспропорционирует на Re2O7 и металлический
рений при нагревании без доступа воздуха или в вакууме. В при-
сутствии кислорода воздуха нагревание ReO2 ведет к превраще-
нию в Re2O7 — соединение желтого цвета.
Под действием окислителей, таких, как HNO3, Н2О2, хлорная
или бромная вода, двуокись рения превращается в метарениевую
кислоту HReO4. При обработке двуокиси рения без доступа
воздуха конц. НС1 и растворами хлоридов щелочных металлов
(образуются ацидосоли общей формулы Me*[Re4+Cle] (где Mei =
= К+, Rb+, Cs+).
При нагревании двуокиси рения до 100—150° в смеси фтора
с азотом образуются ReOF5 и ReO2F3.
Нагревание ReO2 в атмосфере хлора, брома, иода или газо-
образного НС1 дает оксигалогениды рения(УП).
При алюмотермическом восстановлении или восстановлении
водородом (500°) двуокись рения превращается в металлический
рений.
У двуокиси рения кислотные свойства преобладают над основ-
ными.
Ре ни ты, MeiReO3 (где Mez = Na+, К+), образуются в отсут-
ствие воздуха при сплавлении ReO2 с гидроокисями или окис-
лами щелочных металлов:
ReO2 + 2МетОН Me^ReO3 + Н2О
Рениты представляют собой диамагнитные черно-коричевые
мелкие кристаллы, плохо растворимые в воде. Рениты превраща-
ются в ReO2*nH2O под действием разбавленных кислот в присут-
ствии воздуха и окисляются до гипоренатов MexReO3, метаперре-
натов Me4ReO4 или мезоперренатов MeiReO5 под действием возду-
ха, Н2О2 или HNO3.
Тетрафторид рения, ReF4, получают восстановлением ReF6
водородом (200°), металлическим рением (500°) или SO2 (400°)
в платиновой трубке.
Соединение ReF4 представляет собой синее твердое вещество
с плотностью 5,38 г/смэ, т. пл. 124,5° и т. кип. 795°. ReF4 очищается
перегонкой при 500° в приборе из платины в токе SO2, растворяет-
ся в воде с разложением и разъедает стекло при нагревании до
'—'80°.
При растворении ReF4 в 40 %-ном растворе HF образуется
темно-зеленый раствор, из которого при добавлении KF выпадают
зеленые тригональные кристаллы K2[ReFe], Гексафтороренат калия
обладает той же кристаллической структурой, что и K2[PtCl6],
РЕНИЙ
455
и также может быть получен восстановлением КИеО4 иодисто-
лодородной кислотой (или KI) в присутствии конц. HF, нагрева-
нием K2[ReRre] с фторидом одновалентного металла МеЧГ или
K2[ReIe] с KHF2.
2KReO4 4- 6KI + 16HF = 2K2[ReF6] + 4KF + 3I2 + 8H2O
Известны также соединения Rb2[ReFe], Cs2[ReF6].
Тример тетрахлорида рения, НезС112, получают в виде черного
вещества обработкой ReO2-2H2O хлористым тионилом.
Гексахлорорениевая кислота, H2[ReCle], является сильной
кислотой, которая образуется в водном растворе при присоедине-
нии 2 молей HG1 к 1 молю ReCl4, растворении ReO2 в НС1 или
восстановлении метаперренатов иодистоводородной кислотой HI
в конц. НС1 без доступа воздуха.
Гексахлорорениты, Mei[ReCle] (где Me1 = К+, Rb+,
€s+, NH*, Tl+, Ag+, Hg2+, (As(CeH5)4]+), являются солями кислоты
H2[ReCl6J. Соединения K2[ReCle], Rb2[ReCle], Cs2[ReClel,
(NH4)2[ReCle] окрашены в зеленый цвет, Tl2[ReCle] — в желтый
и Ag2[ReCle] — в оранжевый. При нагревании с растворами
щелочей гексахлорорениты диспропорционируют в соединения
рения(П) и (VII):
5Re<+ 3Re2+ + 2Re’+
Гексахлороренит калия, K2[ReCl6], получают из растворов
в результате восстановления KReO4 иодидом калия или НвР2О4,
СгС12, N2H4’HC1 в присутствии конц. НС1 или концентрированного
раствора КС1, добавлением КС1 к раствору ReO2 в конц. НС1
в отсутствие воздуха, обработкой конц. НС1 продукта сплавления
КС1 с металлическим рением в токе хлора, нагреванием Re3Cl9
или ReCl5 с КС1:
2KReO4 + 6KI + 16НС1 = 2K2[ReCl6] + 4К.С1 + 3I2 + 8Н2О
8KReO4 + ЗН6Р2О4 + 26КС1 + 22НС1 = 8K2[ReCl6] + 6К3РО4 + 20Н2О
ReO2 + 4НС1 + 2КС1 = K2[ReCl6] + 2Н2О
HCl
Re + 2KC1 + 2C12 K2[ReCl6]
4Re3Clg + 18K.C1 9K2[ReCl6] + 3Re
ReCl5 + 2K.C1 K2[ReCle] + V2C12
Соединение K2[ReCle] представляет собой желтовато-зеленые
октаэдрические кристаллы с плотностью 3,34 г!смэ. Это соединение
гидролизуется в нейтральных растворах до ReO2. -«Н2О, дис-
пропорционирует в щелочных растворах, перегоняется при 1100°
без разложения, восстанавливается нагреванием с металлическим
калием до металлического рения, а при обработке избытком KCN
456
ПОБОЧНАЯ ПОДГРУППА VII ГРУППЫ
и Н2О2 превращается в K3[Res+O2(CN)4], который представляет
собой оранжевые кристаллы, содержащие рений(У).
При растворении K2[ReCle] в воде раствор приобретает интен-
сивно зеленый цвет благодаря образованию продукта гидролиза
K2[Re(OH)Gl5] и только спустя некоторое время или при нагре-
вании выпадает ReO2-nH2O.
Взаимодействие водного раствора KCN с K2[ReCl6] дает серое
вещество K4[ReO2(CN)4].
Известна также соль K4[Re2OCl10] в виде коричневых мелких
кристаллов, изоморфных аналогичному соединению рутения,
которые растворяются в воде, гидролизуясь.
Общие формулы оксохлороренитов и гидроксохлороренитов —
MeJ[Re2OCl10], Mei[Re(OH)Cl5].
Т етрабромид рения, ReBr4, получают прямым взаимодействи-
ем элементов при нагревании, термическим разложением кислоты
H2[ReBre], диспропорционированием ReBr5 под действием воды
и упариванием досуха раствора HReO4 с избытком НВг.
H2[ReBre] ReBr4 + 2НВг
3ReBr5 + ЗН2О -> 2ReBr4 + ReO3Br + бНВг
Существование соединения ReBr4 не доказано, однако в неко-
торых работах сообщается, что оно окрашено в темно-красный
цвет, под действием кислорода при 100—120° превращается
в ReO3Br, образует аддукты с пиридином или о-фенантролином.
Гексаброморениты, Me|[ReBr6l (где Me1 = К+, Rb + ,
Cs+), представляют собой темно-красные или кирпично-красные
кристаллы.
Гексаброморенит калия, K2[ReBre], получают нагреванием
металлического рения с КВг в парах брома, добавлением КВг
к раствору, полученному путем растворения без доступа воздуха
ReO2 в НВг, восстановлением KReO4 иодидом калия или Н6Р2О4
в присутствии конц. НВг и концентрированного раствора КВг.
Соединение K2[ReBre] представляет собой темно-красные куби-
ческие кристаллы с плотностью 4,34 г/см3-, оно менее устойчиво,
чем К2[ВеС1в].
Известны оксиброморениты типа Me2[Re(OH)Br5].
Тетраиодид рения, Rel4, получают в результате упаривания
досуха раствора, полученного восстановлением HReO4 иодисто-
водородной кислотой:
HReOi + 9HI = H2[ReI6] + з/212 + 4Н2О
H2[ReI6] -> Rel4 + 2HI
Соединение Rel4 представляет собой гигроскопичное неустой-
чивое черное аморфное вещество, которое гидролизуется водой,
превращаясь в ReO2*2H2O, термически разлагается на Re3I9,
РЕНИЙ
457
Rel2, Rel и иод при хранении в вакууме при комнатной темпера-
туре, растворяется в ацетоне и эфире, образуя коричневый раствор.
Гексаиодоренит калия, K2[ReI6], получают восстановлением
KReO4 иодистоводородной кислотой в присутствии KI без досту'
па воздуха.
Соединение K2[ReIe] представляет собой черно-коричневые
кристаллы. При растворении в воде оно гидролизуется, образует
фиолетовый раствор при растворении в ацетоне, реагирует с 20%-
ной H2SO4 по уравнению
K2[ReI6] + H2SO4 = H[ReIe] + HI + K2SO4
Согласно уравнению этой реакции K2[ReIe] можно рассматри-
вать как продукт взаимодействия K[ReI5] и KI.
Соединение K2[ReIe] разлагается при нагревании (300°) в азоте
и превращается в K2[ReFe] при нагревании (250°) в плавиковой
кислоте.
K2[ReI8] -* Re + 2KI + 2I2
Добавление KCN к раствору K2[ReIe] в метаноле приводит
к образованию K3[Re5+(CN)8].
Дисульфид рения, ReS2, получают сильным нагреванием
порошкообразного металлического рения с избытком серы в атмо-
сфере H2S, нагреванием при 1000° (18 час) смеси стехиометриче-
ски необходимых количеств тонко измельченного рения и серы
в закрытой кварцевой трубке, термическим разложением Re2S7
при температуре выше 250° в токе азота, нагреванием Re2S7
с серой в вакууме.
Re + 2S = ReS2 + 42 ккал Re2S7 = 2ReS2 + 3S
Дисульфид рения получают и в растворе путем пропускания
H2S через 0,1—2 н. солянокислый (0,1—10 н. сернокислый) раст-
вор K2[ReCl6].
Соединение ReS2 сходно с MoS2, WS2 и представляет собой
черные гексагональные кристаллы с решеткой типа Cdl2, плот-
ностью 7,5 г!см3 (25°), низкой твердостью (кристаллы мягкие).
Это соединение устойчиво на воздухе при обычной температуре,
легко летуче. При температуре выше 600° в избытке воздуха оно
превращается в Re2O7 и SO2, в растворах NaOH, Na2S, НС1, H2SO4
растворяется с трудом. ReS2 является катализатором реакции
дегидрирования спиртов.
Под действием азотной кислоты дисульфид рения превращает-
ся в НЙеО4и серу или серную кислоту. При 1000° ReS2 восстанав-
ливается водородом до металлического рения.
458
ПОБОЧНАЯ ПОДГРУППА VII ГРУППЫ
Хелатные соединения
Кипячение ReO2 или K2[ReCle] с щавелевой, винной, лимонной,
галловой кислотами или пирокатехином дает различные устой-
чивые хелатные соединения рения: H2[Re(OH)4C2O4],
Н
I \
С—СООН
I /
ОН /2-1
К4
/ н
/ I
ReO(OH)2 I -ООС —С —
\ I
\ -о
к2н2
Н2
(-ООС С —СООН\ -
-О/ХС—СООН/2_
H2[Re(C6H2.O2. ОН-СОО)21 -6Н2О, H[Re(OH)3C6H4O2(H2O)].
L (Н2О)2]
Эти соединения обладают диамагнитными свойствами и разла-
гаются щелочами, причем осаждается ReO2-nH2O.
Помимо приведенных известны и другие соединения рения(1У), напри-
мер диселенид ReSe2, гексатиоцианаторенаты(1У) Me2[Re(NCS)6],
Соединения пятивалентного рения
Известно небольшое число соединений рения(У). Эти соеди-
нения устойчивы в кислой среде (~8 н.), легко диспропорциони-
руют в воде на соединения рения(1У) и (VII):
3Re6+ 2Re4+ + Re7+
Большая скорость реакции диспропорционирования опреде-
ляет малую устойчивость соединений рения(У). Вследствие склон-
ности к диспропорционированию большинство соединений ре-
ния(У) легко разлагается в нейтральных щелочных или слабо
кислых растворах.
Примеры соединений рения(У): Re2O6, ReF5, ReCl5, ReBr5,
соли, содержащие анионы ReO~, [ReOClJ-, [ReOGl5]2-, [ReFe]_,
[Re(CN)8]3~, [ReO2(CN)4]3-, [ReO2(NCS)4]3-, соли, содержащие
катионы [ReO2Py4]+,[ReO(OH)Py2]2+, [ReO2En2] +,[ReO(OH)En2]2+,
IRe(OH)2En2]3+, соединения типов ReOX3(PR3)2, N = ReX2(PR3)2,
ReO(OEt)X2(PR3)2 (где Py = C5H5N, En = этилендиамин, X =
= галоген).
Соединения рения(У) образуются путем восстановления соеди-
нений рения(УП) в сильно кислой среде. Для восстановления Re7 +
РЕНИЙ
459
в конц. H2SO4 применяют Сг2+ или амальгаму висмута, а в разб.
НС1 — SnCl2 в присутствии оксалат- или тартрат-анионов.
На воздухе соединения рения(У) легко окисляются до соеди-
нений рения(УП). Для окисления соединений рения(У) исполь-
зуют KMnO4, KIO3, HgCl2 и цериевые соли.
Рений(У) проявляет склонность к образованию окрашенных
координационных соединений. Многие из комплексов рения(У)
содержат кислород.
Окись рения(У), Re2O5, получают электролитическим восста-
новлением метаперренатов (или 0,24 н. раствора HReO4) в конц.
H2SO4 или термическим разложением As(CeH5)4ReO4.
Соединение Re2O5 представляет собой синее вещество, плохо
растворимое в воде, ралагающееся примерно при 200°.
Гипоренаты, MeIReO3 (где Me1 = Na+, К+, УгВа24"),
мало устойчивы, их получают сплавлением смеси стехиометриче-
ски необходимых количеств ReO2 и MeTReO4 с избытком щелочи
без доступа воздуха.
Гипоренат натрия, NaReO3, получают в результате продол-
жительного нагревания смеси ReO2, NaReO4 и NaOH при 700°
без доступа воздуха.
Соединение NaReO3 выделяют в виде желтых кристаллов, оно
диспропорционирует на Na2ReO3 и NaReO4 в отсутствие щелочей,
окисляется на воздухе до NaReO4, а в присутствии воды или
кислот превращается в ReO2 и NaReO4. В твердом состоянии
NaReO3 хранится в водном или спиртовом растворе NaOH без
доступа воздуха.
Пентафторид рения, ReF5, представляет собой желтовато-
зеленое вещество с т. пл. 48° и т. кип. 221,3°.
Известен окситрифторид рения(У), ReOF3,— нелетучее соеди-
нение черного цвета.
Пентахлорид рения, ReCl5, получают действием СС14 на
нагретый до 400° Re2O7 в закрытой трубке из тугоплавкого стекла,
а также в результате взаимодействия избытка хлора с нагретым
до 120—200° порошкообразным металлическим рением и после-
дующей перегонкой в вакууме образовавшегося продукта. При
хлорировании рения образуются ReOCl4 (т. кип. 223°), ReCl5
(т. кип. 327°) и Re3Cl9 (т. кип. 827°).
Соединение ReCl5 представляет собой черное твердое вещество
с т. пл. 275° и т. кип. 327° (пары красновато-коричневого цвета);
его очищают перегонкой в атмосфере хлора или в вакууме. При
перегонке в атмосфере азота ReCl5 превращается в Re3Cl9 и хлор,
при нагревании на воздухе окисляется до ReOCl4 и ReO3Cl, про-
являет склонность к диспропорционированию на соединения
рения(ТУ) и (VII), гидролитически разлагается водой на
ReO2-2H2O, HReO4 и хлор, растворяется в конц. НС1, образуя
зеленый раствор, содержащий HReO4, H2[ReCl6] и хлор, вое-
460
ПОБОЧНАЯ ПОДГРУППА VII ГРУППЫ
станавливается при нагревании под действием КС1, превращаясь
в K2[ReCle] с высвобождением хлора.
Оксопентахлорорениевая кислота, H2[ReOCl5], мало устойчи-
ва и не была выделена в свободном состоянии; она образуется
в водном растворе при восстановлении HReO4 в конц. НС1.
Оксопентахлороренат калия, K2[ReOCl5] -Н2О, получают обра-
боткой HReO4, растворенной в конц. НС1, иодидом калия или
добавлением КС1 к зеленому раствору, полученному электро-
литическим восстановлением KReO4 в конц. HG1.
Соединение K2[ReOCl5] -Н2О представляет собой зеленовато-
желтые кристаллы, устойчивые в твердом состоянии, гидролити-
чески разлагающиеся на ReO2 -2Н2О и метаперренаты под действием
воды, легко окисляющиеся на воздухе до KReO4, плохо раствори-
мые в конц. НС1.
Оксопентахлороренат аммония, (NH4)2[ReOCl5], получают дей-
ствием NH4I на HReO4, растворенную в конц. НС1; это зеленовато-
желтые кристаллы, растворимые в конц. НС1.
Пентабромид рения, ReRr5, получают действием паров брома
на нагретый до 650° металлический рений.
Соединение ReRrj представляет собой зеленовато-белое тведое
вещество, которое очищают перегонкой в токе брома; оно разла-
гается на Re3Rr9 и бром при нагревании в вакууме или токе азота
и гидролитически разлагается водой на соединения рения(1У)
и (VII).
Диоксотетрацианоренаты, Me|[ReO2(CN)4l (где
Me1 = К+, Na+, Т1+).
Диоксотетрацианоренат калия, K3[ReO2(CN)4], получают об-
работкой K2[Re4+Cl6] избытком KCN и Н2О2, действием спирто-
вого раствора KCN на K2[Re5+OCl5], окислением K4[Re4+O2(CN)4J
в среде KCN или восстановлением KReO4 в присутствии KCN.
Соединение K3[ReO2(CN)4] представляет собой оранжевые
кристаллы, которые легко растворимы в воде, устойчивы в щело-
чах и неустойчивы в кислотах.
Диоксотетрационоренат натрия, Na3[ReO2(CN)4) *2Н2О, по-
лучают в результате восстановления NaReO4 гидразином в при-
сутствии NaCN. Это также оранжевые кристаллы.
Диоксотетрационоренат таллия, Tl3[ReO2(CN)4], неустойчив
в воде при обычной температуре и образуется в результате обра-
ботки K3[ReO2(CN)4] растворами солей таллия(1).
Помимо приведенных, известны и другие соединения рения(У), напри-
мер: K3[Re(CN)8bH2O, K3IReO2(NCS)4], ReOX3-(PR3)2, ReOX3 .(AsR3)2,
ReOX3 -(SbR3)2 (где X = Cl", Br_, NCS"), нитридокомплексы N =
== ReX2(PR3)e, Mel[ReOCl4], Mei[ReF6] (где Mel = Na+, K+ и т. д.),
[ReO2(C5H5N)4]C1.2H2O, [ReO2En2]Cl-2H2O, [ReO(OH)En2]Cl2,
[Re(OH)2En2]Cl3 (где En = этилендиамин), [ReO(OH)(C5H5N)2]Cl2,
[Re2O3(C5H5N)4]Cl4, [ReO(OC2H5)(C5H5N)2]Cl2, ReO(OC2H5)X2(PR3)2, окса-
лато- и тартратокомплексы.
РЕНИЙ
461
Соединения шестивалентного рения
Известно небольшое число соединений рения(У1), мало устой-
чивых и, следовательно, химически активных.
Примерами соединений рения(У1) могут служить: ReO3, рена-
ты Me*ReO4 (где Me1 = К+, Na+, 1/2Ва2+), гексагалогениды ReFe,
ReCle, оксигалогениды ReOF4, ReO2F2, ReOCl4, ReOBr4, ReO2Br2,
октацианоренаты Me2[Re(CN)8] (где Me1 = K+, Na+).
Для соединений рения(У1) характерна склонность к диспро-
порционированию на соединения рения(1У) и (VII):
3Re’*^ 2Re’++ Re4+
Диспропорционирование происходит в результате нагревания
соединений Re6+ под действием воды и в некоторых случаях под
действием щелочей.
В отличие от соединений марганца(У1) соединения Ree+ мало
устойчивы в водном растворе. Их получают восстановлением
соединений рения(УП) или окислением соединений рения(Ш),
(IV) или (V).
Трехокись рения, ReO3, получают окислением без доступа
воздуха порошкообразного металлического рения окисью ре-
ния(УП) Re2O7 при 300° в кварцевой трубке, нагреванием (145°)
смеси Re2O7 с ReO2, восстановлением Re2O7 углеродом, окисью
углерода, серой или двуокисью серы при нагревании и неполным
окислением на воздухе или в кислороде тонко измельченного
металлического рения или окислов рения низших степеней окис-
ления:
Re -|- 3Re2O7 7ReO3 Re2O7 -|- СО —>- 2ReO3 4- СО2
ReO2 + Re2O7 3ReO3 2Re2O7 + S 4ReO3 + SO2
Re2O7 + C 5ft 2ReO8 + CO Re2O, + SO2 2ReO3 + SO3
Соединение ReO3 представляет собой очень слабо парамагнит-
ные блестящие красные кубические кристаллы с плотностью
6,9 г/см3; они устойчивы на воздухе при температурах ниже 110°,
окисляются до Re2O7 на воздухе при повышенной температуре.
Это соединение диспропорционирует на ReO2 и Re2O7 при темпе-
ратурах выше 300° в вакууме (в отсутствие воздуха) или на рени-
ты MeJReO3 и метаперренаты MeTReO4 при кипячении с концен-
трированными щелочами. Оно не реагирует с водой, разб. НС1
или разбавленными растворами щелочей и превращается в ренаты
Me*ReO4 при сплавлении со щелочами без доступа воздуха:
3Re6+O3 + 4NaOH = 2NaRe7+O4 + Na2Re4+O3 + 2НгО
Re’+O3 + 2NaOH = Na2Re6+O4 + H2O
Трехокись рения восстанавливается при нагревании водоро-
дом или SnCl2, KI в кислой среде и окисляется до HReO4 азот-
ной кислотой.
ReO3 + 2HNO3 •= HReO4 + 2NO2 + H2O
462
ПОБОЧНАЯ ПОДГРУППА VII ГРУППЫ
ВеО3 имеет кубическую центрированную кристаллическую
решетку (рис. 22), в которой каждый атом рения октаэдрически
окружен атомами кислорода. Такой же тип решетки у WO3 и СгО3.
Рениеватая кислота, H2ReO4, была получена только в водном
Рис. 22. Структура ReO3.
растворе восстановлением
раствора HReO4 актив-
ным водородом или раст-
ворением в азотной кис-
лоте металлического ре-
ния, его сульфидов или
окислов низших степеней
окисления.
Родный раствор H2ReO4
имеет красновато-желтый
цвет, он неустойчив из-за
склонности Re6+ к дис-
проиорционированию.
Ренаты, Me^ReO4
(где Me1 = К+, Na+, 1/2Ra2+), соответствуют манганатам. Они
окрашены в зеленый цвет, в большей степени являются окис-
лителями, чем восстановителями, были получены в растворах,
но не выделены в свободном виде из-за неустойчивости и склон-
ности к диспропорционированию в воде и спиртах.
3Me^ReO4 + 2Н2О 2MeTReO4 + ReO2 + 4МеЮН
При сплавлении (500°) метаперренатов щелочных металлов
с ReO2 и NaOH (или КОН) образуются гипоренаты MeTRe5+O3,
которые при охлаждении окисляются в неустойчивые ренаты
Me^ReO4 зеленого цвета.
Ренат бария, BaReO4, имеет зеленый цвет, устойчив в при-
сутствии щелочей и выделяется в виде остатка, когда холодный
продукт сплавления Ba(ReO4)2 с ReO2 nNaOH промывают спиртом.
500°
Ba(ReO4)2 + ReO2 + 4NaOH-----> BaReO4 + 2Na2ReO4 + 2H2O
Спирт удаляет избыток гидроокиси натрия, после чего под.
действием воды BaReO4 разлагается по уравнению
3BaReO4 + Н2О = Ba(ReO4)2 + BaReO3 + Ва(ОН)2
Если поддерживать избыток NaOH, то при добавлении малых
количеств воды гидролиз и диспропорционирование BaReO4 не
происходят.
Гексафторид рения, ReFe, получают действием фтора (в отсут-
ствие кислорода и хлора) на порошкообразный металлический
рений, нагретый до 125° в трубке, сделанной из флюорита. Для
РЕНИЙ
463
конденсирования паров ReFe трубка охлаждается снаружи жид-
ким воздухом.
Re + 3F2 = ReF6 + 27,5 ккал
Светло-желтые кристаллы ReFe плавятся при 18,7°, превра-
щаясь в светло-желтую жидкость с плотностью 3,62 г 1см3
и т. кип. 35,6°.
Соединение ReFe очень реакционноспособно, действует на
кремнезем (соответственно на стекло) при ^30°, реагирует с водой
и жирами:
3ReF6 + 3SiO2 = ReF4 + 2ReO3F + 3SiF*
| 3ReF6 + 10H2O = 2HReO4 + ReO2 + 18HF
Кислород c ReFe образует Re7+O2F3 и Re7+OF5.
Гексафторид рения восстанавливается до ReF4 водородом
(200°), окисью углерода (300°), двуокисью серы (380—400°) или
металлическим рением (500°). При еще более высокой температуре
ReF6 может быть восстановлен до металлического рения.
Жидкий ReFe с фторидами щелочных металлов образует фторо-
соли, содержащие анион [ReF8]2-.
При гидролизе K2[ReF8] получают синее соединение K[ReOF5l;
оно растворимо в эфирах и кетонах и гидролитически разлагается
водой.
Оксифториды, Ree+OF4 и Re6+O2F2. При действии
смеси фтора с кислородом на порошкообразный металлический
рений, нагретый до 125—300° в трубке из флюорита, образуется
смесь ReFe с ReOF4 и FeO2F2.
Компоненты смеси ReFe, ReOF4, ReO2F2 разделяют фракцион-
ной перегонкой в вакууме.
Окситетрафторид рения, ReOF4, представляет собой синий
порошок с т. пл. 39,7° и т. кип. 62,7°.
Диоксидифторид рения, ReO2F2, получают в виде белого
порошка, который плавится при 156° без разложения.
Гексахлорид рения, ReCle, получают пропусканием хлора
(очищенного от кислорода) над нагретым до 600° в атмосфере азота
металлическим рением.
Соединение ReCle представляет собой коричнево-зеленые кри-
сталлы, которые плавятся при ^22°, переходя в темно-коричневую
жидкость, превращающуюся при легком нагревании в зеленые
пары. Очищают ReCle перегонкой в токе хлора. Для конденсации
используют U-образную стеклянную трубку, охлаждаемую снару-
жи водой и льдом.
R результате гидролитического разложения ReCle образуются
HReO4, ReO2 и НС1:
3ReCI6 + 10Н2О = 2HReO4 + ReO2 + 18НС1
•464
ПОБОЧНАЯ ПОДГРУППА VII ГРУППЫ
Действием кислорода на ReCle при нагревании получают
Re2O3Cl6.
Окситетрахлорид рения, ReOCl4, получают нагреванием (150°)
«меси ReCl5 и Re3Cl9 в атмосфере кислорода:
3ReCl5 + Re3Cl9 + ЗО2 = 6ReOCl4
Твердое коричнево-зеленое соединение ReOCl4 имеет т. пл. 28°,
т. кип. 223°, растворяется в конц. НС1, образуя коричневый рас-
твор кислоты H2[Ree+OCi6], которая в водной среде диспропорцио-
нирует:
3ReOCl4 + 6НС1 = 3H2[ReOCl6]
3H2[ReOCl6] + 5Н2О = H2[ReCl6] + 2HReO4 + 12HC1
Была выделена калиевая соль этой кислоты K2[ReOGle], кото-
рая неустойчива и легко гидролизуется.
При действии газообразного или жидкого аммиака на ReOGl4
при 300° образуются хлорид оксидиамидорения(У1) ReO(NH2)9Cl2
и NH4C1:
ReOClt + 4NH3 = ReO(NH2)2Cl2 + 2NH4C1
Окситетрабромид рения, ReOBr4, получают действием паров
брома на ReO2. Он представляет собой синие кристаллы, которые
разлагаются при нагревании выше 80° или под действием воды:
6ReOBr4 = 2Re3Br9 -]- 3Br2 -]- ЗО2
3ReOBr4 + 7Н2О = 2HReO4 + ReO2 + 12НВг
Диоксидибромид рения, ReO2Br2, получают действием паров
брома на Re2O7. Он представляет собой неустойчивое соединение,
которое разлагается при 60—70° до начала плавления.
Помимо приведенных соединений известны октацианоренаты типа
Me2[Re(CN)8].
Соединения семивалентного рения
Семивалентное состояние более характерно для рения, чем
для марганца. Известны многочисленные соединения рения(УП).
В качестве примеров соединений рения(УП) можно назвать:
гидрид K2[ReH9], окись рения(УП) Re2O7, метаперренаты с анио-
ном ReO“, мезоперренаты с анионом ReO|~, тиоперренаты с анио-
нами ReO3S_, ReS~, гептафторид ReF7, оксигалогениды ReOF5,
ReO2F3, ReO3F, ReO3Cl, ReO3Br, гептасульфид Re2S7, гептаселе-
нид Re2Se7. Из этих соединений Re2O7 и перренаты (мета- и мезо-)
наиболее устойчивы.
При гидролизе оксигалогенидов рения(УП) в воде образуют-
ся метарениевая кислота и соответствующий галогеноводород.
При описании различных состояний валентности было показа-
но, что диспропорционирование соединений рения средних степе-
РЕНИЙ
465
ней окисления приводит в большинстве случаев к образованию
соединений рения(УП) и (IV).
Соединения семивалентного рения более устойчивы, чем
четырехвалентного, в противоположность аналогичным соедине-
ниям марганца. Кроме того, ReO~ не фиолетовый, как МпО^,
а бесцветный анион. В присутствии щелочей метаперренаты пре-
вращаются в мезоперренаты MeJReO5, а не в соединения рения(У1),
как можно было бы ожидать по
аналогии с соединениями марганца.
Комплексный гидрид рения и ка-
лия, K2[ReH9]. При восстановлении
KReO4 металлическим калием в эти-
лендиамине с последующей экстрак-
цией изопропиловым спиртом (для
удаления гидроокиси калия) было
получено диамагнитное белое твер-
дое вещество, соответствующее фор
муле K2[ReH9],
На основании спектров ЯМР,
данных рентгеноструктурного ана-
лиза и дифракции нейтронов было
доказано наличие связи Re — Н.
Анион [ReH9]2- имеет структуру, изображенную на рис. 23.
Расстояние Re — Н равно 1,68 А.
При восстановлении метаперренатов MeIReO4 в солянокислом
или сернокислом растворе амальгамой цинка или натрия при
температуре ~5° в атмосфере газообразной СО2 были получены
бесцветные растворы с восстановительными свойствами, которые
содержали анион [ReH9]2-. Наличие связи Re — Ну этих соеди-
нений в растворе было доказано при помощи спектрофотометри-
ческих измерений в инфракрасной области.
Раньше ошибочно считали (по аналогии с галогенами, кото-
рые образуют галогениды), что рений образует рениды, т. е.
вместо K2[Re7+H9] писали KRe, K[Re(H2O)n], соответственно
K[Re(H2O)4], K[HRe(OH)(H2O)3], K[H2Re(OH)3(H2O)L
Безводная окись рения(уН) (ангидрид метарениевой кислоты},
Re2O7, получается нагреванием порошкообразного металличе-
ского рения в кислороде или сухом воздухе выше 150° или осто-
рожным выпариванием метарениевой кислоты досуха:
2Re -|- 7/2О2 = Re2O7 + 296,7 ккал
Соединение Re2O7 представляет собой слабо диамагнитные
желтые (или бесцветные при —80°) гексагональные кристаллы
с плотностью примерно 6,2 г!см3, т. пл. 304° и т. кип. 355°; оно
сублимируется, начиная от 220°, гигроскопично и растворяется
в воде, спирте, эфире, хлороформе, ацетоне и уксусной кислоте.
30-0101
466
ПОБОЧНАЯ ПОДГРУППА VII ГРУППЫ
При растворении Re2O7 в воде образуется метарениевая
кислота:
Re2O7 И- Н2О ч* 2HReO4
При действии диоксана на Re2O7 образуется гигроскопичный
серый порошок, который плавится при 90—100°.
Действием сухого H2S на Re2O7 при 80° получают черный
Re2S7.
При нагревании Re2O7 может быть восстановлен водородом до
ReO2 или до металлического рения, а металлическим рением — до
ReO3 или ReO2; SO2 и СО восстанавливают Пе2О7'до ReO3.
300° 500°
Re2O7 + 3H2 ->- 2ReO2+3H2O Re2O7 + 7H2---► 2Re+7H2O
Перекись рения, ReO4 или Re2O8, получают нагреванием мелко
измельченного металлического рения в кислороде при 150° или
нагреванием Re2O7 в кислороде.
Соединение ReO4 представляет собой красновато-желтый кри-
сталлический порошок с плотностью ^8,4 г/см3, который под
действием ультрафиолетового облучения или при нагревании пре-
вращается в Re2O7 с выделением кислорода; ReO4 восстанавли-
вается при нагревании водородом или SO2 до металлического
рения, растворяется в воде, образуя раствор, из которого выде-
ляется кислород и остается HReO4, окисляет H2S, титановые
и ванадиевые кислоты.
Метарениевая кислота (часто называемая рениевой кислотой),
HReO4, образуется при растворении Re2O7 в воде, нагревании
водной суспензии порошкообразного металлического рения
с хлорной водой или перекисью водорода (избыток окислителя
разрушают кипячением), растворении порошка металлического
рения в 30%-ной HNO3 на холоду или в конц. H2SO4 при нагре-
вании (уравнения реакций приведены на стр. 446), пропускании
водного раствора метаперренатов щелочных металлов через катио-
нообменную смолу типа R — Н.
Re2O7 + Н2О 2HReO4
2Re + 7С12 + 8Н2О = 2HReO4 + 14НС1
2Re + 7Н2О2 = 2HReO* + 6Н2О
Для разрушения избытка Н2О2 водный раствор HReO4 кипя-
тят с платиновой чернью.
Водный раствор HReO4 — сильная кислота. Она бесцветна,
может быть экстрагирована амиловым спиртом, не выделяется
в свободном состоянии, растворима в органических основаниях,
растворяет металлы, обладающие восстановительными свойства-
ми, и образует соли, именуемые метаперренатами.
РЕНИЙ
467
Анион ReOl может быть экстрагирован из кислой среды три-н-
бутилфосфатом РО(ОС4Н9)3, а из]щелочной среды — циклически-
ми аминами.
Метаперренаты (соли метарениевой кислоты) образу-
ются при сплавлении порошка металлического рения с щелочами
в токе кислорода, действием HReO4 на окислы, гидроокиси
или карбонаты многих металлов или на различные органические
основания. Получение трудно растворимых метаперренатов осно-
вано на обработке растворов, содержащих анион ReO4, раствора-
ми солей многих катионов. Для промышленного получения
NH4ReO4 метаперренат калия пропускают через анионообмен-
ную смолу, а затем элюируют анион водным раствором аммиака.
R зависимости от природы катиона могут существовать сле-
дующие метаперренаты: MeTReO4 (где Me1 = Na+, К+, Rb+, Cs+,
NH+, Ag+, Tl+, Cu+ и x/2 Hg2+), МеП<еО4-2Н2О (где Me1 = Li+),
Men(ReO4)2 (где Me11 = Re2+, Mg2+, Ca2+, Sr2+, Ra2+, Zn2+, Cd2+,
Hg2+, Cu2+, Pb2+, Mn2+, Fe2+, Co2+ и Ni2+), Men(ReO4)2-2H2O (где
Men= Ca2+, Sr2+, Hg2+, Pb2+, Mn2+, Ni2+), Me11 (ReO4)2-4H2O (где
MeII= Mg2+, Ba2+, Zn2+, Cu2+, Co2+, Ni2+, Fe2+), Me11* (ReOJs-(2
или 4)H2O (где Me111 = Fe®+), [Meni(NH3)6](ReO4)3 (где Mein=
= Co3+, Cr®+), [Me11 (NH3)J(ReO4)2 (где Me11 = Zn2+, Cd2+),
[Me1 (NH3)2]ReO4 (где Me1 = Ag+).
Известны также метаперренаты, в состав катионов которых
входят различные органические основания, например метаперрена-
ты нитрона [C20HieN4H]ReO4, 2,2'-дипиридила [C10H8N2H]ReO4,
2,2',2"-трипиридила [C15H11N3H]ReO4, бруцина, стрихнина,
пиридина, тетрафенилфосфония P(C6H5)4ReO4. тетрафенилар-
сония As(C6H5)4ReO4.
Известны основные метаперренаты, например Hg2(ReO4)2 ‘HggO
Как правило, метаперренаты имеют более низкую раствори-
мость и большую термическую устойчивость, чем соответствующие
перхлораты и перманганаты. Если проследить за поведением при
нагревании KReO4, КМпО4, КС1О4 и КЮ4, можно установить,
что KReO4 плавится при 518° и кипит при 1370°, в то время как
КМпО4 разлагается при 200°, КС1О4 — при 400° и КЮ4 — при
600°. Примерами соединений рения с низкой растворимостью
являются метаперренаты калия, рубидия, цезия, таллия, серебра,
нитрона и стрихнина.
Метаперренаты щелочных металлов и аммония бесцветны,
плавятся без разложения, за исключением NH4ReO4, кристал-
лизуются без воды, за исключением LiReO4-2H2O, восстанавли-
ваются при 1000° водородом до металлического рения, восстанав-
ливаются до Me^[Re4+Gl6], Mei[Re3+Cig], Ме*[Ре2+С14] при нагре-
вании под давлением в автоклавах с водородом и конц. НС1, пре-
вращаются в оксигалогениды Re7+ под действием галогенов.
30*
468
ПОБОЧНАЯ ПОДГРУППА VII ГРУППЫ
В табл. 52 приведены значения плотностей и температур плав-
ления метаперренатов щелочных металлов.
Таблица 52
Метаперренаты Плотность, 2/CAt3 Температура плавления, °C
LiReO4-2H2O 3,69 87,5
NaReO4 5,24 414
KReO4 4,38 518
RbReO4 4,73 598
CsReO4 4,76 616
При обработке метаперренатов в кислой среде смесью KSCN
с SnCl2 образуется оранжево-желтое соединение ReO(SCN)4,
которое может быть экстрагировано эфиром, бутилацетатом или
циклогексаноном.
2KReO4 + 8KSCN + SnCl2 + 12НС1 = 2ReO(SCN)4 + SnCl4+10KCl+6H2O
Анион ReO“ обладает меньшей окислительной способностью,
чем анион МпО^, более устойчив и труднее восстанавливается.
В общем случае, чем в более кислой среде осуществляется
восстановление метаперренатов (или HReO4), тем больше валент-
ность рения в восстановленном соединении.
В щелочной среде анион ReOj может быть восстановлен элек-
тролитически до металлического рения или до соединений ре-
ния(1) с помощью металлов Na, К, Cs и др. Цинк и кадмий вос-
станавливают анион ReO” в 0,2 н. солянокислом растворе до
ReO2; как побочные продукты реакции образуются ReO и Re2O.
В 3—4 н. солянокислом растворе анион ReO^ восстанавливают
с помощью Sn2+, Cr2+, Ti®+, V2+ и других металлов до ReCl4.
В сильно солянокислой среде ReO~ восстанавливается SnCl2
только до ReCi5. Дихлорид хрома может восстановить ReO4
в солянокислой среде до Re3Cl9. При описании получения метал-
лического рения были приведены реакции восстановления KReO4
и NH4ReO4 водородом при нагревании.
Мезоперренаты, Ме^(ВеО5)2 (где Me11 = Ва2+, Sr2+,
2Na+, 2К+), являются солями гипотетической кислоты H3ReO5,
которая не была выделена в свободном состоянии.
При нагревании металлического рения или ReO2 с расплав-
ленной щелочью в присутствии воздуха, перекиси водорода,
KNO3 или КС1О3 образуется мезоперренат красного цвета
MeJReOs (где Me1 = К+, Na+).
При сплавлении Ba(ReO4)2 с NaOH или при смешивании насы-
щенных растворов Ва(ОН)2 (или концентрированного раствора
РЕНИЙ
469
NaOH) с Ba(ReO4)2 при 100° в отсутствие двуокиси углерода обра-
зуется Ba3(ReO5)2. Мезоперренат бария Ba3(ReO5)2 представляет
собой зеленовато-желтые кристаллы, устойчивые на воздухе
и разлагающиеся под действием воды и двуокиси углерода:
Ba3(ReO5)2 + 2Н2О т Ba(ReO4)2 + 2Ва(ОН)2
Ba3(ReO3)2 + 2СО2 2ВаСО3 + Ba(ReO4)2
Гептафторид рения, ReF7, получают пропусканием фтора под
давлением ~250 мм рт. ст. над порошком металлического рения,
нагретого до 300—400°.
ReF7 представляет собой светло-желтое твердое вещество
с т. пл. 48,3°.
Оксипентафторид рения, ReOF5, получают при обработке
ReO2 или KReO4 смесью фтора с азотом при 150°.
ReOF5 представляет собой кристаллы кремового цвета
с т. пл. 34,5°, т. кип. 55°; они дымят во влажном воздухе, гидро-
лизуются в воде с образованием HReO4 и HF и действуют на
влажное стекло.
Диокситрифторид рения, ReO2F3, образуется наряду с ReOF5
по способу, описанному выше, и отделяется от последнего фрак-
ционной перегонкой. Соединение ReO2F3 представляет собой
желтый порошок, дымящий во влажном воздухе, с т. пл. 95°,
т. кип. 200°; он гидролизуется водой с образованием HReO4
и HF и действует на влажное стекло.
При взаимодействии трифторида брома с KReO4 получают гиг-
роскопичные желтые кристаллы диоксотетрафтороперрената
калия K[ReO2F4], которые гидролизуются водой.
Были получены соли общей формулы Mer[ReO2F4] (где Me1 =
= Rb+, Cs+, Ag+, ^гСа2*, V2 Sr2+).
Триоксифторид рения (фторид перренила), ReO3F, получают
действием IF5 на KReO4 или плавиковой кислоты на ReO3Cl. Он
представляет собой желтый порошок с т. пл. 147° и т. кип. 164°.
Триоксихлорид рения (хлорид перренила}, ReO3Cl, получают
окислением воздухом (или кислородом) или окисью рения Re2O7
смеси ReCl5 с Re3Cl9, действием СС14 на Re2O7 и прямым хлориро-
ванием ReO3 при 160—190°.
ReCig -|- Re3Cle -|- 6О2 = 4ReO3Cl -j- 5С12
Re2O7 + СС14 = 2ReO3Cl + СОС12 2ReO3 + Cl2 = 2ReO3Cl
ReO3Cl представляет собой бесцветную двупреломляющую
жидкость, которая кипит при 131°, затвердевает при 4,5°, стано-
вится пурпурной под действием солнечного света, превращается
в HReO4 и НС1 при гидролитическом разложении во влажном
воздухе.
ReO3Cl + Н2О = HReO* + НС1
470
ПОБОЧНАЯ ПОДГРУППА VII ГРУППЫ
Триоксибромид рения {бромид перренила), ReO3Br, получают
действием кислорода на ReBr4 при 100—120° или перегонкой над
Re2O7 продукта, полученного нагреванием порошкообразного
металлического рения в смеси кислорода и паров брома.
ReO3Br представляет собой белое твердое вещество с т. пл.
39,5° и т. кип. 163°, которое гидролизуется водой, превращаясь
в HReO4 и НВг.
Сульфид рения, Re2S7, получают прямым взаимодействием
элементов при нагревании или нагреванием газообразного H2S
с Re2O7.
Re2S7-nH2O образуется при насыщении сероводородом 2—6 н.
растворов НС1, содержащих перренат-ион ВеО4, при добавлении
Na2S2O3-5H2O к горячему раствору 2—7 н. H2SO4 или 1—4 н.
НС1, содержащих перренат-ионы ReO4, и при подкислении раст-
воров тиоперренатов MexReO3S:
гМеЪеО* + 7H2S -Р2НС1 2 Re2S7-nH2O + 2МегС1
2MezReO4 + 7Na2S2O3-5H2O + H2SO4
=₽* Re2S7-nH2O + 6Na2SO4 + 2MeJSO4
7MeIReO3S + 2HC1 Re2S7-«H2O + SMe^eOi + 2MeiCl
Соединение Re2S7 представляет собой черно-коричневый поро-
шок (или тетраэдрические кристаллы) с плотностью 4,87 г/см®;
разлагается на ReS2 и серу при нагревании (600°) в вакууме, вос-
станавливается при нагревании в токе водорода до металлического
рения, превращается в Re2O7 и SO2 при сжигании на воздухе,
окисляется до HReO4 азотной кислотой, бромной водой или
Н2О2, превращается в тиоперренаты при растворении в растворах
сульфидов щелочных металлов и обладает каталитическими
свойствами.
Тиоперренаты MeTReO3S (где Me1 = К+, Na+, Rb+,
Cs+, NHJ, Tl+, V2Pb2+).
При длительном пропускании H2S через нейтральный кон-
центрированный холодный раствор метаперрената калия образуется
зеленовато-желтый раствор, который содержит тиоперренат
калия, KReO3S:
KReO* + H2S =₽t KReO3S -j- H2O
При обработке раствора KReO3S нитратом таллия выпадает
желтая соль TlReO3S, которая мало растворима в воде и спирте.
Перекись водорода, бромная вода и HNO3 окисляют анион
ReO3S_ до перренат-аниона ReO4.
Подные растворы тиоперренатов при подкислении разлагают-
ся с образованием Re2S7.
РЕНИЙ
471
Тиорениевая кислота, HReO3S, не выделена в свободном
состоянии. Она получена только в водном растворе и является
сильной кислотой.
При действии H2S на перренаты в щелочной среде можно
получить анионы, содержащие больше серы, например ReS^“.
Селенид рения, Re2Se7, получают действием H2Se на раствор
KReO4 в присутствии КОН. Он представляет собой черный поро-
шок, который при нагревании восстанавливается водородом до
металлического рения и разлагается на ReSe2 и элементный селен
при 330°.
Помимо приведенных известны и другие соединения рения(УП), напри-
мер, ReO2(SO3F)3, ReO3(SO3F), ReO3Cl2-SO2C12, NH4[ReO2Cl4]SO2Cl2.
Карбонилы рения. Димер пентакарбонила рения,
Re2(CO)10, получают действием окиси углерода на Re2O7 при
250° в автоклаве под давлением 200 ат:
Re2O7 + 17СО = Re2(CO)i0 + 7СО2
Вместо Re2O7 можно использовать ReO2, ВеО3иметаперренаты.
Сульфид рения Re2S7 превращается в Re2(CO)10 под действием
окиси углерода в присутствии водорода или влаги.
Ковалентное соединение Re2(CO)10 представляет собой бес-
цветные мелкие кристаллы, которые плавятся при 177° в закры-
той трубке, разлагаются при нагревании выше этой температуры
(а именно при 400°), сублимируются при нагревании выше 140°
и растворяются в гексане или диоксане. Это соединение превра-
щается в галогенид карбонила Re(CO)5X под действием галогенов,
разлагается концентрированными кислотами (HNO3, H2SO4) при
нагревании, не взаимодействует с разбавленными растворами
кислот или щелочей и взаимодействует с аминами, превращаясь
в аминопроизводные, например Re(CO)3Py2, Re(CO)3Phen (где
Ру = C5H5N, и Phen — фенантролин).
В соединении Re2(CO)10 атомы рения расположены в центрах
двух октаэдров, соединенных общей вершиной, в остальных вер-
шинах которых расположены группы СО. Октаэдрические груп-
пы молекулы Ве2(СО)10 повернуты на 45°. В соединении Re2(CO)10
расстояние Re — Re равно 3,02 А, а ковалентная связь Re — СО
равна 2,56 А.
Галогенокарбонилы рения, Re(CO)5X, пред-
ставляют собой бесцветные (или светло-желтые) кристаллы, не
имеющие запаха, устойчивые на воздухе, сублимирующиеся без
разложения в атмосфере СО, трудно растворимые в воде и раст-
воряющиеся в бензоле и лигроине.
1
472 ПОБОЧНАЯ ПОДГРУППА VII ГРУППЫ
Галогенокарбонилы могут быть получены по следующим
реакциям:
При 200°
2K2[ReI6] + 10СО ----* 2Re(CO)5I + 4KI + 31,
Порошок Си '
При 200° и 10 атм
2K2[ReBre] + 10CO Порошок Cu 2Re(CO)5Br+4KBr + 3Br2
KReO4 + СС14 + 8СО = Re(CO)5Cl + СОС12 + ЗСО2 + КС1
ReCl5 + 4Cu + 9СО = Re(CO)5Cl + 4CuCbCO
Re + CuCl2 + 6CO = Re(CO)5Cl + CuCbCO
С избытком цианидов щелочных металлов галогенокарбонил
Re(CO)5X образует очень хорошо растворимые соли общей фор-
мулы MeI[Re(CO)4(CN)2L При нагревании галогенокарбонилов
Re(CO)5X в инертном растворителе образуются галогенокарбони-
лы типа [Re(GO)4X]2:
2Re(CO)5X [Re(CO)4X]2 + 2СО
Известны также галогенотетракарбонильные производные
Re(CO)4X2.
Аминозамещенные карбонильные про-
изводные. При действии жидкого аммиака или различных
органических аминов на растворенные в бензоле или лигроине
галогенокарбонилы рения образуются аминозамещенные карбо-
нильные производные:
Re(CO)6Cl + NH3 -► Re(CO)4(NH3)Cl + HCONH2
Re(CO)5Cl + 2NH3 -> Re(CO)3(NH3)2Cl + 2HCONH2
С анилином было получено соединение Re(CO)3(CeH5NH2)2Cl
в виде кристаллов, которые сублимируются при 200°.
С ароматическими фосфинами и изонитрилами были полу-
чены производные Re(CO)3[P(CeH5)3]2Cl, Re(CO)4(RCN)2Cl.
Известны также карбонильные производные С5Н5Ре(СО)3,
C5H5Re(CO)2H.
Соединения включения
При действии аммиака (300—350°) на NH4ReO4 или на Re3Clg
образуются серые кубические кристаллы нитрида рения, состав
которых колеблется между Re2N и Re3N. Поскольку нитрид рения
разлагается при 280°, получить его прямым синтезом невозможно.
Эта особенность приближает рений к платиновым металлам.
Фосфиды Re2P, ReP, ReP2 и ReP3 получают прямым
взаимодействием фосфора с металлическим рением при темпера-
туре выше 750°. Самым устойчивым фосфидом является ReP, кото-
рый представляет собой синие кристаллы.
Известны также бориды Re3B, Re7B3 и ReB3, с и л и ц и -
д ы ReSi2, ReSi, Re3Si, арсенид Re3As7 и германид ReGe2-
ПОБОЧНАЯ ПОДГРУППА VIII ГРУППЫ
ПЕРИОДИЧЕСКОЙ СИСТЕМЫ
В побочной подгруппе VIII группы периодической системы
химических элементов находятся переходные металлы железо
(Fe), кобальт (Со), никель (Ni), рутений (Ви), родий (Rh), пал-
ладий (Pd), осмий (Os), иридий (1г) и платина (Pt), которые рас-
положены в трех триадах.
Эти переходные металлы могут быть сгруппированы по общим
химическим свойствам в подгруппу железа (Fe, Со, Ni) и в под-
группу платины (Ru, Rh, Pd, Os, Ir, Pt).
ХАРАКТЕРИСТИКА ПОДГРУППЫ ЖЕЛЕЗА
В подгруппе (триаде) железа находятся переходные металлы
железо (Fe), кобальт (Со) и никель (Ni).
В табл. 53 показано электронное строение атомов железа,
кобальта и никеля.
Атомы элементов железа, кобальта, никеля имеют на послед-
нем электронном уровне по два электрона, которые они легко
отдают в химических реакциях с образованием соединений, где
эти элементы электроположительны двухвалентны. Для того
чтобы образовать соединения, в которых эти элементы обладают
Таблица 54
Элемент Железо Fe Кобальт Со Никель Ni
Цвет в компактном со- стоянии Серебристо-серый Серебристо-серый Серебристо-серый
в порошке Серый Серый Серый
Кристаллическая структура сс-Р-6-Fe кубическая объемноцентрированная y-Fe кубическая гранецентрированная а-Со плотнейшая гексагональная (ниже 492°) Р-Со кубическая гранецентрированная (выше 482°) oc-Ni кубическая гранецентрированная p-Ni плотнейшая гексагональная
Атомный номер 26 27 28
Атомный вес 55,847 58,9332 58,71
Атомный радиус, А (по Полингу) 1,26 1,257 1,25
Радиус иона, А (по Гольдшмидту, Полингу, Аренсу) Fe2+0,82; 0,80; 0,74 Fe3+ 0,67; -; 0,64 Со2+ 0,82; 0,72; 0,72 Со3+ 0,64; —; 0,63 Ni2+ 0,78; 0,69; 0,69
Атомный объем (при 20°), см3/г-атом 7,10 6,82 6,59
Плотность (при 20° , г/см3 7,876 8,83 8,907
Прочность по Бринеллю, кг/мм3 60-77 132 85
j 1 ИТП иг’
Твердость по шкале Мооса 4-5 5,5 5
Температура плавления, °C 1536 1492 1455
Скрытая теплота плавления, кал/град 65,65 63,6 72,1
Температура Кипения, °C 3250 3185 3075
Удельная теплоемкость (при 20°), кал/г-град 0,1077 0,1001 0,1050
Коэффициент линейной теплопровод- ности X, кал-см-1-сек-1-град-1 (при 0°) 0,21 — 0,142
Сопротивление р-10® (при 0°), ом-см 8,8 5,60 6,14
Электропроводность (Hg = 1) 9,6 16,6 15,3
Магнитная восприимчивость /•10е эл.-магн. ед. (при 18°) Ферромагнитные металлы
Теплота перехода атомов в газооб- разное состояние, ккал (при 25°) 96,68 105 101,61
Продолжение табл. 54
7,63 18,15 35,16 56,00 79,10 112,90 175,8 -0,25 1 1 (I), II, (III), (IV) 58, 60, 62, 61, 64
7,86 । МО ф 05 sr со со О см со мо О см см со 8 § МО 8 f й о* 1 О* + Со2+/Со3+ +1,843 > ИН, мн мн кн" СЛ МО
| 7,90 СО S м* со о* со о о МО* МО о <о оэ о кР* е> МО со ХР ф* 1 с© со О* 1 Fe2+/Fe3+ +0,771 (I), II, III, (IV), (VI) со мО й МО с© МО
Ф + % 2 t о 2 ф + еэ Ф S t + ф S ф + + п ф § t £ ф + + м* ф § t + со ф § ! Ф + & о S t i ф 2 1 ф + + се Ф S о ф S ф + t © S + еэ ф S ф S + со ф S ф S Нормальные потенциалы окислитель- но-восстановительных систем Ме2+/Ме3+, в й о и В ф Я сб m Массовые числа природных изотопов
Потенциал иони- зации, эв Потенциал иони- зации, ккал)моль Нормальный по- тенциал, в (при 25°)
<?1
Распространенность элементов н зем- 4,7 4,0-10~3 1,8-10~'
ной коре, вес. %
ПОБОЧНАЯ ПОДГРУППА VIII ГРУППЫ
477
валентностью выше двух, их атомы отдают электроны неполностью
укомплектованной Зй-орбитали.
В табл. 54 приведены наиболее важные константы элементов
подгруппы железа.
В компактном состоянии железо, кобальт и никель представ-
ляют собой ферромагнитные серебристо-серые металлы с плотно-
стью от 7,87 до 8,90 г/см* (тяжелые металлы), с т. пл. от 1455 до
1536° и т. кип. от 3075 до 3250° (тугоплавкие металлы). Они имеют
твердость 4—5,5 по шкале Мооса и теряют пластичность при
загрязнении углеродом, кремнием, фосфором, серой и бором.
Металлы подгруппы железа обладают превосходными механиче-
скими свойствами, большой механической прочностью и могут
прокатываться, протягиваться и штамповаться.
По значению атомных весов в триаде железа существует ано-
малия Со — Ni, которую можно объяснить как тем фактом, что
свойства элементов меняются периодически в зависимости от
зарядов их ядер, так и изменением свойств металлов побочной
подгруппы VIII группы по вертикали, поскольку кобальт сходен
с родием и иридием, а никель — с палладием и платиной.
Для железа известны четыре аллотропные модификации: а-
(3-, у- и б-Fe, а для кобальта и никеля — по две аллотропные
модификации: а- и |3-Со, а- и (3-Ni.
Помимо сходства, существующего между металлами подгруп-
пы железа, существует некоторое сходство между железом и мар-
ганцем, а также никелем и медью.
Известны многочисленные сплавы железа с металлами — Со,
Ni, Mn, Cr, Мо, W, V, Nb, Zr, Ti, Си и с неметаллами — углеро-
дом, кремнием, азотом, фосфором, бором, серой.
Для элементов триады железа характерна склонность к свя-
зыванию окиси углерода, закиси азота или других нейтральных
молекул.
С химической точки зрения кобальт и никель менее реакцион-
носпособны, чем железо, и проявляют стойкость к коррозии
(при обычной температуре) на воздухе, в воде и в различных
растворах.
Разбавленные кислоты (HCl, HNO3, H2SO4) растворяют железо
и кобальт при обычной температуре, а никель — при нагревании.
Металлы подгруппы железа пассивируются конц. HNO3 при
обычной температуре и реагируют при нагревании с кислородом,
парами воды, галогенами, серой, фосфором, мышьяком, кремни-
ем, углеродом и бором.
В природе железо, кобальт и никель редко встречаются в сво-
бодном состоянии и очень распространены в виде соединений
(окислы, сульфиды, арсениды, тиоарсениды, сульфаты, арсенаты,
силикаты и карбонаты) в различных минералах. По распростра-
ненности в природе за железом следует никель, а затем кобальт.
Таблица 55
NiSO4 Желтые ортором- бические кристал- лы 154,7716 3,64 Разлагаются при нагревании 1 216,0
C0SO4 ф । ф • 3£3§ й « и “ 3 О О Л О Е й К Ч S н F ф св & 154,9948 3,666 1 216,7
FeSO4 Белые ортором- бические кристал- лы 151,9086 3,14 1 221,30
ОТ 2 СО. И 2 CD 5 о> Св о g ф о И g f? Ч 3 Й Q, S е. « к 90,774 4,60 797 1 20,4
ОТ О О ф 1 ф ' З°з§ я £ и Е- з O.Q ДОС Og Ч S Я tr< Ф св О< к к 90,9972 5,45 В атмо- сфере азота 1100 1 22,77
FeS £ х а ₽; и О S о се S и 3 О В О,® о Я g R О Л £ о, Ми« к 87,911 4 ,84 1193 Разла- гается 22,80
NiO ф 1 о и Ч g « 74,794 6,827 1957 1 57,30
СоО Серо- зеленые кубиче- ские кристал- лы 74,9326 6,45 1935 Разла- гается при 2860 57,20
FeO Черные кубиче- ские кристал- лы 71,8464 1 - 1360 1 00 CD
Соединение Вид Молекулярный вес Плотность (при 20 °), г/см3 Температура плавле- ния, °C Температура кипения, °C Теплота образования, ккал/моль
железо 479
Металлические железо, кобальт и никель получают восста-
новлением окислов Fe2O3, Fe3O4, FeO, Со2О3, Со3О4, СоО, NiO,
Ni2O3, NiO2, Ni3O4 водородом, окисью углерода, углеродом,
алюминием, кремнием, бором или другими восстановителями при
нагревании. В промышленности металлические железо, кобальт
и никель не получают в чистом виде, а только в виде сплавов.
В чистом виде металлические железо, кобальт и никель полу-
чают термическим разложением карбонилов Fe(CO)5, Со2(СО)8,
Со4(СО)12, Ni(CO)4 или электролитическим путем.
Для очистки металлов подгруппы железа применяют плав-
ление в высоком вакууме, зонную плавку и электролитическое
рафинирование.
Железо, кобальт, никель и их сплавы (чугуны, стали и др.) —
очень важные конструкционные материалы современной техники.
Наиболее устойчивыми являются соединения железа(Ш),
кобальта(П) и никеля(П). Железо менее склонно к образованию
координационных соединений, чем кобальт и никель. Однако
координационные соединения железа(Н) и (III), кобальта(Ш)
и никеля(Н) достаточно устойчивы, а простые соединения желе-
за(Н), кобальта(1) и никеля(1) гораздо менее устойчивы и имеют
восстановительный характер.
В состоянии высших степеней окисления соединения металлов
подгруппы железа проявляют окислительные свойства.
В табл. 55 и 56 приведены основные константы галогенидов
двухвалентных железа, кобальта и никеля.
По состояниям валентности соединения двухвалентных железа,
кобальта и никеля проявляют большое сходство.
Такие соединения, как гемоглобин и витамин В12, в состав
которых входят двухвалентные железо и кобальт, обеспечивают
нормальную жизнедеятельность человека и животных.
Помимо соединений, отвечающих различным состояниям окис-
ления, известны многочисленные нестехиометрические соединения.
Известно довольно большое число металлоорганических про-
изводных металлов подгруппы железа.
По всем приведенным выше характеристикам элементы под-
группы железа относятся к переходным металлам.
ЖЕЛЕЗО Fe
Z = 26; ат. вес = 55,847
Валентность (I), II, III, (IV) и (VI)
Массовые числа основных природных изотопов 56, 54, 57,
58
Электронная структура атома железа: K-L •3s23p63cZ6 -4s2.
Электронная структура атома железа и катионов Fe2+, Fe®+
480
ПОБОЧНАЯ ПОДГРУППА VIII ГРУППЫ
Соединение FeF2 CoF2 NiF2 FeCl2 СоС12
Вид Белые тетраго- нальные кристаллы Розовые тетраго- нальные кристаллы Зеленые тетраго- нальные кристаллы Бесцветные ромбо- эдрические кристаллы Синие гексаго- нальные кристаллы
Молекулярный вес 93,8438 96,9300 96,7068 126,753 129,8392 '
Плотность (при 20°), г/см3 3,95-4,09 4,43 — 2,988 3,36
Температура плав- ления, °C 1102 1202 1027 672 727 i
Температура кипе- ния, °C 1827 1727 1627 1026 1049 Г
Теплота образова- ния, ккал/моль 154 158 158 82,05 74,8
Гидраты (число мо- лекул Н2О) 4, 8 2, 3, 4 2, 3, 4 1, 2, 4, 6 1, 1,5, 2, 4, 6
для 3d- и 4з-орбиталей:
34* 4s2 346 4s 3d5 4s
Itllt Itltltl и Itlltltltlt I □ [t It It It It I □
Fe Fe2+ Fe3+
ИСТОРИЧЕСКАЯ СПРАВКА
Железо было известно еще до нашей эры. Древние египтяне
и индейцы Северной Америки пользовались предметами, сделан-
ными из метеоритного железа. В те времена изделия из железа
были наиболее распространенными после изделий из бронзы.
ЖЕЛЕЗО
481
Таблица 56
1 1 NiCl2 FeBr2 СоВг2 NiBr2 Fel2 а-Со12 Nil2
Золотисто- желтые ромбоэдри- ческие кристаллы Желтые гексаго- нальные кристаллы Зеленые гексаго- нальные кристаллы Коричневые ромбоэдри- ческие кристаллы Желтые гексаго- нальные кристаллы Черные гексаго- нальные кристал- лы Серые ромбо- эдриче- ские кристал- лы
129,616 215,665 218,7512 218,528 309,6558 312,7420 312,5188
3,53 4,636 4,91 4,6 5,315 5,584 5,80
987 684 678 963 587 ' 520 —
— 927 927 — 827 570 747
73 60 58 59 40 36 38
1, 2, 4, 6, 7 2, 4, 6, 9 1, 2, 4, 6 2, 3, 6, 9 2, 4, 6, 9 2, 4, 6, 9 6
Впервые металлическое железо было получено из минералов
(время точно не установлено) народами, населявшими Кавказ
и Туркестан. Позже железо распространилось в Вавилон, Египет,
Грецию и затем Рим. Железо, полученное примитивным способом
(который состоял в нагревании железной руды с древесным углем
в ямах или печах из камня или глины), было загрязнено шлаком
и очищалось продолжительной ковкой.
На древнем Востоке железо выплавляли в печах, оснащенных
мехами для вдувания воздуха. В XV в. удалось поднять темпера-
туру плавки за счет усиления тока воздуха и получить чугун,
который ломался под ударами молотка. Позднее нагреванием
31-0101
482
ПОБОЧНАЯ ПОДГРУППА VIII ГРУППЫ
чугуна с древесным углем в горне в токе воздуха было получено
ковкое железо.
Качественным скачком в металлургии железа оказалась заме-
на древесного угля коксом, являющимся одновременно горючим
материалом, восстановителем и источником углерода. В XIX в.
были разработаны технологические процессы получения стали
и построены электропечи для производства легированных (спе-
циальных) сталей.
РАСПРОСТРАНЕННОСТЬ В ПРИРОДЕ
В природе железо редко встречается в свободном состоянии.
Обычно оно входит в виде соединений (окислы, карбонаты, суль-
фиды, сульфаты, арсениды, тиоарсениды, фосфаты, силикаты и др.)
в состав различных минералов. Содержание железа в земной коре
составляет 4,7 вес.%, а в космических телах железа еще больше.
В свободном состоянии железо встречается либо в метеоритах
(представляющих собой сплавы железа с 5—20% никеля), либо
в теллургических образованиях, в которых железо (a-Fe, феррит)
почти чистое и содержит максимум 2% Ni, 0,3% Со, 0,4% Си
и 0,1% Pt. Природное железо представляет собой серые объемно-
центрированные кубические кристаллы с плотностью 7—7,8 г!см3,
твердостью 4—5 по шкале Мооса и ярко выраженными магнит-
ными свойствами.
Наиболее типичными минералами железа являются сле-
дующие:
Магнетит, Fe3O4, содержит до 72% железа и представляет
собой черные кубические кристаллы со слабым металлическим
блеском, плотностью 4,9—5,2 г!см3, твердостью 5,6—6 по шкале
Мооса и магнитными свойствами.
Гематит, a-Fe2O3 (от греческого слова hematikos, что озна-
чает «кровавый»), содержит до 65% железа и представляет собой
красно-черные ромбоэдрические кристаллы с плотностью
5—5,3 г/сл13, твердостью 5,5—6 по шкале Мооса. a-Fe2O3 легко
восстанавливается при нагревании под действием Н2, С, СО, А1,
Si и др.
Лимонит (гетит), Fe2O3-H2O или HFeO2, содержит до 60% же-
леза и представляет собой кристаллы, гранулы, оолиты или
конкреции черно-коричневого цвета. Плотность лимонита 3,3—
4 г!см3, твердость 1—4 по шкале Мооса. Плотность гетита 4—
4,4 г!см3, твердость 4,5—5,5 по шкале Мооса. В отличие от других
железосодержащих руд лимонит и гетит легче всех восстанавли-
ваются до металлического железа.
Сидерит, FeCO3, содержит примерно 35% железа, обладает
желтовато-белым (с серым или коричневым оттенком в случае
ЖЕЛЕЗО
483
загрязнения) цветом,
4,5 по шкале Мооса.
Пирит, FeS2 или
плотностью 3,9 г!см? и твердостью 3,5—
S
Fe^ |, содержит 46,6% железа и встре-
XS
чается в виде кубических кристаллов, желтых, как латунь, с
металлическим блеском (желтовато-коричневым оттенком), плот-
ностью 4,9—5,2 г/см3, твердостью 6—6,5 по шкале Мооса. Он
содержит в небольшом количестве Со, Ni, As, Sb и иногда Си,
Ag, Аи.
Марказит, FeS2, также содержит 46,6% железа, но встречает-
ся в виде желтых, как латунь, бипирамидальных ромбических
кристаллов с плотностью 4,6—4,9 г!смъ и твердостью 5—6 по шка-
ле Мооса. При температуре 450° он превращается в пирит.
Леллингит, FeAs2, содержит 27,2% железа и встречается
в виде серебристо-белых бипирамидальных ромбических кристал-
лов с плотностью 7,0 — 7,40 г/сл3 и твердостью 5 — 5,5 по шкале
Мооса.
Миспикель, FeAsS, содержит 34,3% железа и встречается в виде
белых моноклинных призм с плотностью 5,6—6,2 г!см3 и твер-
достью 5,5—6 по шкале Мооса.
Мелантерит, FeSO4-7H2O, реже встречается в природе
и представляет собой зеленые (или серые из-за примесей) хмоно-
клинные кристаллы, обладающие стеклянным блеском, хрупкие,
с плотностью 1,8—1,9 г!см3.
Вивианит, Fe3(PO4)2 -8Н2О, встречается в виде сине-серых
или зелено-серых моноклинных кристаллов с плотностью 2,95 г/сл3
и твердостью 1,5—2 по шкале Мооса.
Помимо описанных, известны и другие минералы, например: ильменит
FeTiO3 (см. минералы титана), магномагнетит (Fe, Mg)[Fe2O4], фиброферрит
FeSO4(OH)-4,5Н2О, ярозит KFe3(SO4)2(OH)6, кокимбит Fe2(SO4)3-9Н2О,
рёмерит Fe2+Fe|+(SO4)4-14Н2О, графтонит (Fe, Мп)3(РО4)2, скородит
Fe3+AsO4-2Н2О, штренгит FePO4-2H2O, фаялит Fe2SiO4, альмандит
Fe3Al2[SiO4]3, андрадит Ca3Fe2[SiO4]3, гиперстен (Fe, Mg)2[Si2O6], геденбергит
(Са, Fe)[Si2O6], эгирин (Na, Fe) [Si2Oe], шамозит Fe^+Al[AlSi3O1()](OH)6 -
• пН2О, нонтронит (Fe3+, Al)2[Si4O1()](OH)2-nH2O.
Помимо этих минералов, многие алюмосиликаты, какими явля-
ются глины, загрязнены соединениями железа.
Залежи полезных ископаемых, богатые минералами железа,
находятся в СССР, Швеции, Норвегии, Финляндии, ГДР, ФРГ,
Австрии, Швейцарии, Чехословакии, Франции, Греции, Италии,
Югославии, Люксембурге, Англии, Польше, Румынии, Испании,
Венгрии, Афганистане, Китае, на Кипре, в КНДР, Индии, Япо-
нии, МНР, Таиланде, Турции, ДРВ, Гвинее, Марокко, Сьерра-
Леоне, Тунисе, Канаде, Кубе, Мексике, США, Аргентине, Брази-
лии, Чили, Колумбии, Перу, Венесуэле, Австралии.
31*
484
ПОБОЧНАЯ ПОДГРУППА VIII ГРУППЫ
Содержание железа в минералах колеблется от 25 до 70%.
К бедным рудам относятся руды, содержащие менее 45% железа,
в промышленных целях они не используются. Обогащают руды
главным образом магнитным способом. Руды, содержащие гематит
или лимонит, превращают в магнетит Fe3O4 термической обработ-
кой в восстановительной атмосфере, а затем их пропускают через
магнитное поле для отделения от пустой породы.
Железо входит в состав гемоглобина, который является ком-
понентом эритроцитов крови.
Существуют организмы, у которых вместо железа в молекуле
гемоглобина содержится медь.
ПОЛУЧЕНИЕ МЕТАЛЛИЧЕСКОГО ЖЕЛЕЗА
Чистое мало окисляемое металлическое железо может быть
получено восстановлением окиси железа(Ш) Fe2O3 (которую полу-
чают прокаливанием FeC2O4 •а/4Н2О на воздухе) водородом при
нагревании:
Fe2O3 + ЗН2 = 2Fe + ЗН2О — 25 ккал
Восстанавливая Fe2O3 водородом при 278—340°, получают
пирофорное железо, при 550—650° — железо, которое только
частично окисляется с поверхности, и выше 700° — спекшуюся мас-
су губчатого металлического железа.
Чистое железо получают также термическим разложением
пентакарбонила железа Fe(CO)5 без доступа воздуха при темпера-
туре выше 140° и электролизом при 30° водного раствора FeCl2-
'4Н2О с добавкой NH4C1 (в 1 л раствора содержится 30 г FeCl2-
-4Н2О и 100 г NH4C1). При этом используют анод из электро-
литического железа и катод из нержавеющей стали; плотность тока
на катоде 10 а/дм2.
Загрязненное металлическое железо получают алюмо- или
кремнетермическим восстановлением окислов железа Fe2O3HFe3O4:
Fe2O3 + 2А1 = 2Fe + А12О3 + 198,12 ккал (-2920°)
3Fe3O4 + 8А1 = 9Fe + 4А12О3 + 773,80 ккал (-2677°)
Для получения феррохрома, ферровольфрама, ферромолибде-
на, (феррониобия, ферротитана, ферроциркония или других фер-
росплавов применяют алюмо- или кремнетермическое восстанов-
ление смеси Fe2O3 с окислами соответствующих металлов, напри-
мер Cr2O3, WO3, Мо03, Nb2O5, TiO2 и ZrO2. При кремнетермиче-
ском восстановлении в качестве восстановителя используют эле-
ментарный кремний или ферросилиций.
Сверхчистое железо, содержащее 10-6% примесей, получают
методом зонной плавки.
ЖЕЛЕЗО
485
ПОЛУЧЕНИЕ ЧУГУНА ИЛИ СЫРОГО ЖЕЛЕЗА
Для получения чугуна или сырого железа (которое является
сплавом железо — углерод с сопутствующими элементами: серой,
фосфором, кремнием, марганцем) используют гематит, магнетит,
лимонит, сидерит и не применяют минералы, содержащие серу
(пирит FeS2 или марказит FeS2), мышьяк (леллингит FeAs2 или
миспикель FeAsS) и фосфор (вивианит Fe3(PO4)2-8Н2О). Мнералы
с содержанием серы больше 0,3—0,4 вес. % не пригодны для до-
менных процессов. Известно, что пирит, который является самым
распространенным минералом железа в природе, используют для
производства серной кислоты.
Для получения сырого железа можно применять F2O3, обра-
зовавшийся при прокаливании на воздухе пирита, мелантерита
или сидерита:
2FeS2 4" И/2О2 ~ Fe2O3 4SO2 2FeSO4 — ^/2^2 ~ Ге20з 4~ 2SO3
2FeCO3 +1 /2О2 = Fe2O3+2СО2
Сырое железо, помимо углерода и сопутствующих элементов
(S, Р, As и Si), которые имеются в рудах, содержит ряд элементов,
специально добавляемых в процессе производства.
Промышленное получение железа включает следующие про-
цессы: восстановление железных руд до металла (производство
чугуна), окисление для удаления вредных примесей и добавление
компонентов, необходимых для получения ковкого железа и стали.
Процесс получения чугуна восстановлением железных руд
осуществляется в доменных печах.
Процесс превращения чугуна (сырого железа) в ковкое железо
и сталь осуществляется путем аффинажа (т. е. окисления и час-
тичного удаления примесей—S, Р, As, Si и др.) по процессу
Пудделя, Бессемера, Томаса или Мартена.
На рис. 24 изображена доменная печь в разрезе, а также пыле-
уловительная камера и установка для предварительного нагрева
воздуха (каупер). Доменная печь имеет форму двух усеченных
конусов, соединенных основаниями, высота ее 65 м, диаметр 5—
11 м. Рабочий объем печи в самой широкой части 200—2000 м3,
производительность ~2000 т чугуна в сутки. Верхний усеченный
конус, называемый шахтой, сделан из огнеупорных кирпичей
и имеет в верхней части, называемой колошником, автоматическое
загрузочное устройство и приспособление для удаления доменных
газов. Снаружи печь имеет стальную оболочку или стянута обру-
чами. Печь поддерживается металлоконструкциями, так как
сама по себе обладает большим весом.
Нижний усеченный конус (заплечики) сделан из огнеупорного
силикатного кирпича, он уже и короче верхнего. В его нижней
части находится горн, который имеет цилиндрическую форму.
486
ПОБОЧНАЯ ПОДГРУППА VlH ГРУППЫ
В нижней части горна (металлосборнике) накапливаются чугун
и шлак, которые удаляются через отверстия с желобами (через
верхнее отверстие выпускают шлак, а через нижнее — чугун).
В верхней части горна находятся отверстия для вдувания воздуха
(фурмы). Воздух вводят под давлением 0,5—1,5 ат и температуре
800—900°. Горн сделан из огнеупорного кирпича и снабжен спе-
циальным охлаждающим устройством. Снаружи он опирается на
толстую стальную плиту (20—30 жж). Опора домны должна быть
Рис. 24. Схема домны с воздушным обогревателем.
А — зона предварительного обогрева; Б — зона восстановления I; В — зона прокалива-
ния флюсов и восстановления II; Г — зона карбидизации железа; Д — зона плавления
чугуна и образования шлака; Е — зона горения кокса; 1 — железная руда и флюсы;
2 — стальной кожух; з — стенка из огнеупорного кирпича; 4 — отверстие для вдувания
воздуха; 5 — шлак; в — металлургический кокс; 7 — отверстие для удаления газов;
s — пылеуловительная камера; 9 — тигель; 10 — чугун; 11 — обогреватель воздуха;
12 — кожух из листовой стали; 13 — огнеупорный кирпич.
очень прочной, чтобы выдержать ее огромную тяжесть. Внутрен-
ний объем домны между верхним загрузочным уровнем и фурмами
составляет ее полезный объем.
Отношение между полезным объемом домны (в кубических
метрах) и суточной производительностью (в тоннах) есть коэффи-
циент использования полезного объема домны.
Через загрузочное отверстие в домну последовательно подают
слой кокса и слой железной руды (куски рамером 10—15 см или
ЖЕЛЕЗО
487
брикеты), смешанной с флюсом. Первый и последний слои загру-
женной домны должны состоять из кокса.
В процессе работы загружаемый материал постепенно опу-
скается в домну и по пути нагревается газами, поднимающимися
снизу. Эти газы образуются при горении кокса в нижней части
заплечиков в среде нагретого в воздухонагревателе до 800—
900° воздуха, который поступает в домну под давлением 0,5 —
1,5 ат. В процессе прохождения газов через домну они охлажда-
ются от 1800° (температура газов в нижней части заплечиков) до
200—400° (температура газов при выходе из домны).
Выходящие из домны через колошник газы являются горючими
газами, они имеют температуру 200—400°, теплотворную способ-
ность примерно 800 ккал/м2 и содержат примерно 25% СО, осталь-
ное СО2, Н2 и N2. Водород образуется в результате разложения
паров воды из воздуха, вводимого в домну. Азот — тоже из воз-
духа. Выходящие из домны газы проходят через пылеуловитель-
ную камеру и сжигаются в воздухонагревательном аппарате.
Каждая домна имеет 3—4 обогревателя воздуха (каупера), кото-
рые действуют последовательно: когда один отдает тепло, осталь-
ные нагреваются. Каупер имеет высоту 25—35 м, диаметр 6—8 м,
сделан из огнеупорного кирпича и покрыт снаружи толстой сталь-
ной оболочкой. Сначала нагревают каупер в течение 2 час (сжига-
нием очищенных доменных газов), затем через него пропускают
воздух, который нагнетается компрессором или другими устрой-
ствами. Нагретый до 800—900° воздух поступает через фурмы
в домну. Воздух и доменные газы движутся через каупер противо-
током. В доменной печи происходят следующие процессы: термиче-
ское разложение некоторых соединений (СаСО3, MgCO3, FeCO3
и др.), восстановление окислов железа Fe2O3, Fe3O4, FeO до твер-
дого металлического железа, введение углерода в металлическое
железо (или образование эвтектического чугуна), образование
шлака и горение кокса. В соответствии с этими превращениями
объем домны делится на различные зоны, как это показано на
рис. 24.
В верхних зонах домны при нагревании до 500° загруженное
сырье полностью теряет влагу и химически связанную воду.
При температуре выше 400° FeCO3 термически разлагается на FeO
и СО2, а выше 850° СаСО3 (введенный в качестве флюса) термиче-
ски разлагается на СаО и СО2.
Восстановление окислов железа начинается в верхних зонах
домны и заканчивается в заплечиках. Окислы железа восстанав-
ливаются окисью углерода по уравнениям
Выше 400°
3Fe2O3 + CO------► 2Fe3O4 + CO2-rl0,7 ккал
488
ПОБОЧНАЯ ПОДГРУППА VIII ГРУППЫ
Выше 500°
ГезО4 + СО •— —>- 3FeO4-CO2 — 3,6 ккал
Выше 650°
FeO-j-CO —------> Fe-]-CO2 + 3,8 ккал
Fe2O3 + 3CO -----2Fe + 3CO2 + 8,8 ккал
В результате этих реакций образуется пористое твердое желе-
зо и СО2.
Во второй восстановительной зоне при температурах выше
900—950° окислы железа Fe3O4 и FeO эндотермически восстанав-
ливаются коксом до металлического железа и СО:
Fe3O4 + С -*• 3FeO + СО — 43,3 ккал
FeO -|- С -» Fe + СО — 34,8 ккал
По этим реакциям СО2 не образуется, поскольку выше 900°
двуокись углерода в присутствии углерода полностью переходит
в окись углерода:
Выше 900°
C02-j-C 1 * 2СО — 41 ккал
Ниже 900°
Для того чтобы восстановление окислов железа осуществля-
лось с наименьшей затратой горючего, необходимо, чтобы перво-
начально прошел процесс восстановления окислов Fe2O3, Fe3O4
окисью углерода до FeO, а затем процесс образования метал-
лического железа в результате восстановления половины FeO
окисью углерода и половины — металлургическим коксом.
В средней зоне домны пористое твердое железо поглощает
углерод и науглероживается. При науглероживании железа
в области температур 1000—1200° получают эвтектический чугун,
который плавится при 1145° и является сплавом железо — угле-
род, содержащим 4,3% углерода. Когда железу в минералах
сопутствует марганец, науглероживание приводит к образованию
чугуна с большим содержанием углерода, а когда железу сопут-
ствуют кремний или фосфор, науглероживание осуществляется
меньшим количеством углерода. Содержащийся в чугуне углерод
может быть в виде цементита Fe3C (карбид железа) или графита.
Карбидизация металлического железа до цементита осуществляет-
ся по уравнениям
3Fe -|- С = Fe3C 3Fe -f- 2СО — Fe3C -f- CO2
При науглероживании пористого железа образуется жидкий
эвтектический чугун, стекающий в металлосборник горна; его
периодически выпускают через выпускное отверстие в нижней
части горна.
ЖЕЛЕЗО
489
После восстановления окислов железа и стекания жидкого
чугуна на дно горна флюсы взаимодействуют с пустой породой,
сопутствующей окислам железа, и коксовой золой, образуя жид-
кий шлак (менее подвижный, чем чугун), который скапливается
в горне поверх чугуна, откуда периодически выпускается через
отверстие для выпуска шлака.
Если порода имеет кислый характер (т. е. содержит SiO2
и А12О3), то для образования шлака железная руда перемеши-
вается с основными флюсами — СаСО3 (известковый камень) или
CaCO3-MgCO3 (доломит), а если порода имеет основной характер
(т. е. содержит СаСО3 и MgCO3) — с кислыми флюсами (песок
или гранит).
Нагревая до 1000—1300° смесь кислых окислов (SiO2, А12О3)
с основными окислами (СаО, MgO, образовавшимися при терми-
ческом разложении известкового камня или доломита), получают
расплавленный шлак, который стекает в горн и плавает на поверх-
ности расплавленного чугуна. Шлак состоит из силикатов и алю-
минатов или из алюмосиликатов кальция и магния и использует-
ся для получения цемента, бетона, кирпичей и панелей. Эти
материалы применяют в строительном и дорожном деле. Темпера-
тура плавления и вязкость зависят от соотношения окислов
(SiO2, А12О3 и СаО) в шлаке. Кислый или основной характер
шлака зависит от отношения СаО + MgO/SiO2 + А12О3.
В нижней части заплечиков на уровне фурм находится зона
горения кокса, где образуется СО2 и выделяется большое коли-
чество тепла. Двуокись углерода, проходя через слои кокса при
температуре 900°, восстанавливается до окиси углерода.
При обогащении вдуваемого в домну воздуха кислородом
увеличивается производительность печи и улучшается качество
шлака и состав доменных газов.
Чугун представляет собой сплав железа с углеродом (углеро-
да более 1,7%) и сопутствующими элементами — серой, фосфором,
мышьяком, кремнием и марганцем. Чугун отличается хрупко-
стью, его нельзя перерабатывать такими методами, как прокатка
и ковка.
Чугуны классифицируют по характеру применения на серые,
белые и специальные, а по содержанию кремния и марганца —
на сырые обычные (с содержанием кремния и марганца менее 5%)
и сырые легированные (в которых содержание одного или обоих
элементов выше 5%). Из общего количества доменного чугуна 15—
20% составляет серый чугун, небольшое количество — специаль-
ный чугун, остальное — белый чугун. Получение чугуна опре-
деленного типа зависит от состава и качества исходных материалов,
расхода горючего, температуры зон в домне, количества и тем-
пературы вводимого воздуха и состава шлака.
490
ПОБОЧНАЯ ПОДГРУППА VIII ГРУППЫ
Серые чугуны (литые) образуются при большом расходе горю-
чего. Они содержат 1,7—4,3% углерода в виде графита, 1,25—
4% кремния, 0,5—1,3% марганца, менее 0,6% серы и 0,2—
0,6% фосфора. Большое количество кремния уменьшает раство-
римость углерода в железе, увеличивает подвижность расплавлен-
ного чугуна, уменьшает усадку чугуна при охлаждении (следо-
вательно, улучшает его литьевые свойства) и способствует полу-
чению мягкого чугуна, который легче поддается переработке.
Кремний благоприятствует выделению углерода в виде графита.
В изломе эти чугуны обладают серым цветом, так как содержат
углерод в виде графита. Чем меньше размер частиц графита и чем
равномернее их распределение в чугуне, тем лучше механические
свойства последнего.
Серые чугуны применяются для отливки различных деталей,
поскольку в расплавленном состоянии они очень подвижны, хоро-
шо льются и обладают малой усадкой при охлаждении.
Белые чугуны образуются при малом расходе горючего. Они
содержат 1,7—4,3% углерода в виде цементита Fe3C, более 4%
марганца, 0,9—1,6% кремния и 0,08% серы.
Марганец способствует образованию цементита, препятствует
образованию графита, способствует удалению серы в виде суль-
фида MnS и помогает раскислению железа. В изломе эти чугуны
имеют белый цвет благодаря цементиту (Fe3C), обладающему
белым цветом и блеском.
Белые чугуны — твердые и хрупкие, они трудно поддаются
механической обработке и служат для изготовления некоторых
деталей или в качестве исходного сырья для получения сталей.
Специальные, или легированные чугуны являются ферроспла-
вами с большим содержанием кремния, марганца и др. Они при-
меняются как раскислители при получении сталей. Примерами
специальных чугунов служат ферросилиций с 12—14% кремния,
ферромарганец с 60—80% марганца, зеркальный чугун с 12%
кремния и 20% марганца. Сера, фосфор и мышьяк—вредные для
чугунов примеси. Сера благоприятствует образованию цементита,
уменьшает подвижность чугуна и способствует увеличению вяз-
кости и ломкости при высокой температуре. Фосфор уменьшает
усадку чугуна, увеличивает тведрдость и ломкость и понижает
температуру плавления чугуна. Мышьяк вызывает также увели-
чение твердости и хрупкости чугуна.
СТАЛЬ
Сталь является сплавом железо — углерод, содержащим крем-
ний, марганец, фосфор и серу в следующих максимально возмож-
ных количествах: 0,1—1,7% С, 0,3% Si, 0,6% Мп, 0,05% Р
и 0,05% S.
ЖЕЛЕЗО
491
Сталь получают аффинажем белых чугунов. Под аффинажем
чугунов понимают частичное окисление и частичное же удаление
углерода, кремния, марганца, серы и иногда фосфора. Продукты
окисления СО, СО2, SO2 и др. выделяются в виде газов, a SiO2
и МпО2 накапливаются в шлаке. Сталь отличается от чистого
железа своими механическими и физико-химическими свойствами,
такими, как твердость, эластичность, сопротивление разрыву,
химической стойкостью и т. и.
Стали можно классифицировать по способам получения, по
химическому составу, по свойствам и областям применения.
Для получения сталей из белого чугуна применяют процессы
окисления Пудделя, Бессемера, Томаса или Мартена. Эти про-
цессы позволяют получать сталь в жидком состоянии.
Процесс Пудделя имеет только историческое значение и давно
уже не применяется, поскольку позволяет получать сталь в смеси
со шлаком, из которой последний удаляется ковкой. Чугун поме-
щают в печь, футерованную окислами железа, в которой пламя
отдельно сжигаемого угля соприкасается с поверхностью чугуна.
Окисление углерода и остальных примесей чугуна осуществляет-
ся пламенем печи и кислородом из окислов железа, составляющих
обкладку печи.
Процесс Бессемера, называемый кислым процессом (по кисло-
му характеру внутренней обкладки конверторов), состоит в про-
дувании холодного воздуха под давлением 2—2,5 ат через жид-
кий чугун, который находится в специальном сосуде (конверторе).
При получении сталей в конверторах нет необходимости в допол-
нительном обогреве, поскольку в результате окисления примесей
S, Р и особенно Si кислородом воздуха выделяется много тепла
и температура расплавленного металла поднимается от 1250
до 1750°.
Конвертор (рис. 25) представляет собой большой грушевид-
ный металлический сосуд (емкостью 20—60 т), изготовленный из
стальных листов толщиной -~30 мм. Он может вращаться вокруг
горизонтальной оси. Днище конвертора снабжено отверстиями,
через которые вдувается воздух, а корпус его футерован изнутри
силикатным кирпичом.
Для аффинажа по этому процессу применяют чугун, который
содержит 0,9—1,6% Si, меньше 0,06% S и меньше 0,06% Р,
т. е. чугун с малым содержанием серы и фосфора. При окислении
кремния и марганца и частично железа и углерода получается
кислый шлак, который содержит силикаты железа и марганца,
и выделяются СО и СО2. Для снижения температуры в конвертор
часто добавляют небольшое количество железного лома, а для
охлаждения расплавленного металлического материала в кон-
вертор подают водяной пар.
492
ПОБОЧНАЯ ПОДГРУППА VIII ГРУППЫ
За процессом аффинажа можно следить по виду пламени над
конвертором или по шуму в нем, по дыму, выходящему из кон-
вертора, и по пробам, которые отбирают из расплавленного метал-
ла. Процесс аффинажа длится 10—15 мин и заканчивается, когда
пламя над конвертором становится коричневым (что указывает
на начавшееся окисление марганца), а в самом конверторе слы-
шен глухой шум.
Рис. 25. Конвертор.
По окончании аффинажа в конвертор вводят марганец, крем-
ний или алюминий (соответственно ферромарганец, ферросилиций
или металлический алюминий), чтобы восстановить образовав-
шуюся в процессе окись железа. Марганец служит как для вос-
становления окиси железа, так и для удаления серы в виде MnS.
После полного сгорания углерода и окисления кремния и мар-
ганца в конверторе остается мягкое железо.
Для получения стали вдувание воздуха в конвертор прекраща-
ют до полного сгорания углерода или к мягкому железу в кон-
верторе добавляют углеродистый чугун, освобожденный от вред-
ных примесей.
Преимущество бессемеровского процесса состоит в том, что он
не требует дополнительного нагревания, и сталь, полученная по
этому способу, хорошо сваривается, прокатывается и протяги-
вается на холоду.
К недостаткам бессемеровского способа следует отнести потери
как за счет частичного окисления железа и перехода его в шлак,
так и за счет частичного выбрызгивания стали; кроме того, сера
и фосфор удаляются неполностью, вследствие чего получаемая
сталь уступает по качеству мартеновской и электростали.
ЖЕЛЕЗО
493
Бессемеровская сталь служит для изготовления проволоки,
гвоздей, труб, сельскохозяйственных орудий и машин.
Процесс Томаса называют основным процессом (так как вещест-
ва, из которых состоит внутренняя обкладка конверторов, имеют
основной характер); он используется для получения стали аффи-
нажем фосфористых чугунов, которые содержат 1,8—2,5% фосфора
и 0,15% серы. Аффинаж фосфористого чугуна осуществляют в кон-
верторе Томаса, который в отличие от конвертора Бессемера футе-
рован доломитным кирпичом основного характера. В процессе
аффинажа чугуна в конвертор добавляют негашеную известь СаО.
При аффинаже фосфористого чугуна выделяется большое количе-
ство тепла (благодаря окислению фосфора окисью железа, раство-
ренной в расплавленном металле) и образуется основной томас-
шлак состава Са3(РО4)2'СаО:
Р4 4~ 50г = Р40ю 4“ 716,8 ккал Р404о 4“ 6FeO = 2Fe3(PO4)2
Fe3(PO4)2 + 4СаО = Са3(РО4)2-СаО + 3FeO 4- 108,34 ккал
Томасова мука, представляющая собой размолотый шлак, слу-
жит в качестве химического удобрения в сельском хозяйстве; она
образуется по уравнению
5СаСО3 + 1/2P4Oi0 4- SiO2 = Са3(РО4)2-СаО-CaSiO3 + 5СО2
Управление процессом аффинажа фосфористого чугуна осу-
ществляется по тем же критериям, что и управление процессом
Бессемера.
В отличие от бессемеровской томасова сталь содержит больше
окиси железа и шлака.
К недостаткам томасовского процесса относится высокая
стоимость доломита, используемого при футеровке конвертора.
Томасова сталь применяется для тех же целей, что и бессемеров-
ская.
Процесс Мартена состоит в окислении примесей (таких, как
Si, Mn, С, S и Р) доменных чугунов кислородом воздуха (который
пропускают над расплавленным металлом) и кислородом, выделя-
ющимся из окислов железа; последние добавляют при аффинаже
чугуна в виде металлолома или гематита. Этот процесс получения
стали осуществляется в пламенных печах, в которых при сжига-
нии горючего газа в кислороде (воздухе) достигаются высокие
температуры (1700—1750°). Воздух и горючий газ предваритель-
но нагреваются в так называемых регенераторах, сделанных из
огнеупорного кирпича, соответственно до 1100—1200° и 1200—
1350°. Мартеновская печь имеет четыре таких регенератора. Два
из них предназначены для нагрева воздуха и два других — для
нагрева горючего газа. Через каждый регенератор поочередно
пропускают газы, отводимые из мартеновской печи, и затем,
в обратном направлении,— воздух или горючий газ. Когда два
494
ПОБОЧНАЯ ПОДГРУППА VIII ГРУППЫ
регенератора нагреваются, другие два отдают накопленное тепло.
При получении мартеновской стали применяют чугуны различного
качества (главным образом те, которые не пригодны для конвер-
торной переработки), а также железный и стальной лом. По этому
процессу можно получить стали высокого качества и желаемого
состава, поскольку процесс аффинажа длится несколько часов
и хорошо контролируется и управляется.
Существуют два типа мартеновских печей: неподвижные и ка-
чающиеся. В зависимости от характера примесей в чугуне исполь-
зуют мартеновские печи с кислой или основной футеровкой.
В мартеновских печах с кислой футеровкой перерабатывают чугу-
ны с малым содержанием фосфора и серы, а в мартеновских печах
с основной футеровкой — чугуны с большим содержанием фос-
фора и серы, которые дешевле и доступнее.
Углерод, кремний и марганец удаляются из чугуна с помощью
окиси железа(П) под слоем шлака, который предохраняет рас-
плавленный металл от окисления:
FeO + С = Fe + СО — 34,8 ккал FeO + Мп = Fe + МпО+ 32,29 ккал
2FeO + Si = 2Fe + SiO2 + 78,99 ккал
При аффинаже чугуна в печь добавляют СаО, который в слу
чае основного процесса способствует удалению фосфора и серы,
а в случае кислого процесса превращает SiO2 в CaSiO3. При кислом
процессе получающиеся FeO и МпО взаимодействуют с SiO2
футеровкой печи, образуя FeSiO3 и MnSiO3, которые выделяются
в виде шлака.
При завершении процесса аффинажа чугуна для раскисления
стали в печь добавляют ферросилиций или ферромарганец. В про-
изводстве стали мартеновский процесс используется шире, чем
бессемеровский и томасовский, поскольку он позволяет получать
более вязкую и ударопрочную сталь. Производство стали коли-
чественно превосходит производство чугуна, так как мартенов-
ским способом перерабатывают большие количества железного
и стального лома.
Для получения легированных (специальных) сталей, обладаю-
щих превосходными свойствами, применяют электродуговые или
индукционные печи, в которых нет прямого контакта стали с пла-
менем. Для получения легированных сталей используют мягкое
железо, а также бессемеровские или мартеновские стали, к кото-
рым для раскисления добавляют углерод и остальные компоненты
в виде железа или чугуна, ферросилиция, ферромарганца, ферро-
хрома и ферроалюминия.
Легированные стали содержат железо, углерод, сопутствуюг
щие (Si, Мп, Р, S) и элементы, которые специально добавляют
ЖЕЛЕЗО
495
например: Сг, Ni, Мп, V, Nb, Zr, Мо, W, Ti, Al, Cu, В, Si, N2, H2
и др. Поскольку специально добавляемые элементы реагируют
с железом, углеродом и остальными компонентами стали, образу-
ются легированные стали с различными механическими и хими-
ческими свойствами. Многообразие свойств сталей, полученных
легированием, обусловлено количеством легирующего элемента
и отношением легирующей добавки к основным элементам сплава
железо — углерод. Элементы Со, Ni, Мп, Сг, Мо, W, V, Nb, Zr,
Ti, Al, Be, добавляемые при получении легированных сталей,
могут образовывать с железом твердые растворы или интерметал-
лические соединения. Элементы В, N2, О2, Н2 образуют с железом
твердые растворы включения. Когда железо с легирующими до-
бавками дает интерметаллические соединения, меняются главным
образом механические свойства сталей, в случае же образования
твердых растворов меняются главным образом химические свой-
ства сталей. Элементы Мп, Сг, Мо, W, V, Zr, Nb, Ti образуют
с углеродом карбиды (простые, комплексные или легированные
цементиты), устойчивые в стали. Элементы Ni, Со, Al, Cu, Si, N2
образуют с железом не карбиды, а только твердые растворы. На
свойства стали оказывают существенное влияние добавки различ-
ных элементов. Например, кремний приводит к увеличению эла-
стичности и прочности, марганец способствует увеличению вяз-
кости и понижает пластичность, вольфрам способствует увели-
чению твердости, хром — увеличению прочности.
Для придания стальным деталям поверхностной твердости
и большей стойкости по отношению к действию вызывающих
коррозию химических реагентов или к истиранию часто приме-
няют термохимическую обработку (цементацию и азотирование).
Вязкая и эластичная сталь с низким содержанием углерода цемен-
тируется путем насыщения поверхности углеродом при нагрева-
нии (900°) в присутствии угля, цианидов или окиси углерода. Для
образования нитридов железа в поверхностном слое сталь нагре-
вают продолжительное время в атмосфере аммиака при 600—
700° или в амосфере азота при высоком давлении. Известны нитри-
ды феррита, аустенита и мартенсита.
ФИЗИЧЕСКИЕ И ХИМИЧЕСКИЕ СВОЙСТВА
Чистое железо в компактном состоянии представляет собой
серебристо-серый металл с синеватым отливом, имеющий плот-
ность 7,867 г/см3, твердость 4—5 по шкале Мооса, т. пл. 1536°
и т. кип. 3250°. Железо существует в виде четырех аллотропных
модификаций, а именно: a-Fe, устойчивое до 768°, [J-Fe, устойчивое
в интервале 768—906°, y-Fe, устойчивое в интервале 906—1401°,
496
ПОБОЧНАЯ ПОДГРУППА VIII ГРУППЫ
и б-Fe, устойчивое в интервале 1401—1536°.
a-Fe Модификации а, р, 6 имеют кубическую
j 1768° объемноцентрированную кристаллическую струк-
P-Fe туру, а модификация у — гранецентрированную
'll906° кубическую структуру решетки. Модификации а
y-Fe и б — ферромагнитны, а Р и у — диамагнитны. Ферро-
|| 1401° магнетизм исчезает, когда железо нагрето до точки
б-Fe Кюри 768°.
1 1 1536° В тонко измельченном состоянии металлическое
Fe железо обладает пирофорными свойствами и обра-
(Расплав) зуется в результате перегонки амальгамы железа
или восстановления Fe2O3 водородом примерно
при 270°.
Горячее пирофорное железо самопроизвольно загорается на
воздухе, поскольку, будучи тонкоизмельченным и имея водород
включения, энергично взаимодействует с кислородом воздуха.
Известно очень большое число сплавов (чугуны, стали и др.),
образуемых железом с различными металлами (Со, Ni, Ми, Сг, Мо,
W, V, Nb, Zr, Sb, Ti, Sn, Pb, Al, Be, Mg, Zn и Си), а также с неме-
таллами — углеродом, кремнием, азотом, фосфором, серой, водо-
родом.
Было замечено, что железо в легированных сталях образует
твердые растворы, эвтектические твердые сплавы и интерметал-
лические соединения с многочисленными элементами. Примеры
интерметаллических соединений: Fe3Mo2, Fe5Nb3, FeCr, FeZn7,
Fe5Zn21, Fe2Sn, FeSn, FeSn2, Fe2W, Fe3W2, Fe3Zr2, Fe3Ti, FeAl3,
Fe2N, Fe4N, Fe3P, Fe3C, Fe2C, Fe3Si2, FeSi, FeSi2 и Fe2Si.
Основным легирующим элементом железа является углерод.
Введенный в относительно небольших количествах углерод суще-
ственно изменяет характер и свойства железа. Уже говорилось
о том, что сплавы железа с другими элементами, содержащие
0—1,7% углерода, относятся к сталям, а содержащие 1,7—6,7%
углерода,— к чугунам. Углерод может находиться в железе
в виде графита (элементный углерод) или в форме цементита.
На рис. 26 приведена диаграмма термического равновесия
неустойчивой легированной системы Fe — Fe3C (сплошные ли-
нии) и устойчивой легированной системы Fe—С (пунктирные
линии). На этой диаграмме чистое железо расположено в левой
части, а цементит — в правой части. Цементит Fe3C содержит
6,67% С, обладает сложной кристаллической структурой (появ-
ляется в виде независимого структурного составляющего в форме
первичного, вторичного, третичного цементита), большой твер-
достью, плотностью 7,86 г!см3, магнитными свойствами при
комнатной температуре, которые теряет при 215°. Отклонения на
этой диаграмме появляются из-за существования различных
ЖЕЛЕЗО
497
аллотропных модификаций железа и различной растворимости
углерода в этих аллотропных модификациях.
В сплавах железо — углерод существуют твердый раствор
6 (б-феррит), твердый раствор у (аустенит) и твердый раствор а
(феррит). Растворимость углерода в у-твердом растворе, кото-
рый обладает гранецентрированной кубической структурой, боль-
ше, чем в а- и б-твердых растворах с объемноцентрированной
кубической структурой.
& 1536
§ 1500
Ц. 1300-
Твердый раствор S
Жидкий раствор?твердый раствор S
Жидкий раствор
О
1153
е|
§ I
1145°
Твердый раствор?
цементит(втор.)н
ледебурит
(твердый
раствор t) /
Цементит в
(первичный)? g
ледебурит §
Жидким раствор
аустенит
идти раствор?
'цементит (первич)
'Г
а~ 12DD- дуСтенит\1й_
^1100-
s
М fiOOO-
\121°Цементши(вторичный)\ Цемешпт(первичный)'
\?перлит?ледеоуригп ; ледебурит
.... , V 4.3
*=».& Феррит? дернит? Содержание углерода,°L
перлит цементит ‘ ??>/•>
6,67
Рпс. 26. Диаграмма термического равновесия Fe — С и Fe — Ре3С.
В сплавах железо — углерод появляются гомогенные кри-
сталлы a-Fe, y-Fe, б-Fe, Fe3C и гетерогенные составляющие, такие,
как перлит, ледебурит I и II. Перлит представляет собой эвтек-
тоидную смесь 86,5% феррита и 13,5% цементита с т. пл. 721°.
Ледебурит I является эвтектической смесью 48% аустенита
и 52% цементита с т. пл. 1145°. Ледебурит II является эвтектиче-
ской смесью 48% перлита и 52% цементита с т. пл. 721°.
Выше линии ликвидуса ABCD сплавы железо — углерод жид-
кие; ниже линии солидуса AHJECF все сплавы железо — углерод
полностью кристаллизованы. Между кривыми ликвидуса и соли-
дуса сплавы пастообразны и представляют собой гетерогенную
смесь расплава и твердых кристаллов б-Fe, y-Fe, Fe3C различного
состава. Из расплавов с 1,7—6,67% С вдоль линии ВС выделяется
твердый раствор у и вдоль линий CD — первичные кристаллы
Fe3C. При закаливании аустенит превращается в мартенсит.
В зависимости от положения сплавов железо — углерод (на
диаграмме термического равновесия) относительно эвтектической
.32-0101
498
ПОБОЧНАЯ ПОДГРУППА VIII ГРУППЫ
точки С и эвтектоидной точки S различаются доэвтектоидные
стали, эвтектоидная сталь, заэвтектоидные стали, доэвтектические
чугуны, эвтектические чугуны и заэвтектические чугуны.
Системы железо — углерод с содержанием углерода более
6,67% не рассматриваются, поскольку в технике используются
только сплавы, содержащие примерно до 5% углерода. Углерод
способствует увеличению прочности, твердости, сопротивления
и уменьшению пластичности.
Механические свойства железа зависят от степени его чистоты.
В чистом состоянии железо достаточно мягко, ковко, тягуче, вязко,
оно хорошо проводит тепло и электричество. При загрязнении
железа различными неметаллами или металлами механические
свойства его в значительной степени меняются.
В компактном состоянии чистое железо устойчиво в сухом
воздухе и ржавеет во влажном воздухе, превращаясь в Fe2O3 •
• пН2О (где п имеет значение, близкое к единице) — ржавчину;
последняя образует пористую, рыхлую пленку, которая не пре-
дохраняет железо от действия кислорода.
2Fe + S/2O2 + nH2O = Fe2O3.nH2O
Когда загрязненное железо соприкасается с влагой и дву-
окисью углерода из атмосферы, на поверхности металла обра-
зуются гальванические пары, в которых железо, являясь отрица-
тельным элементом, разрушается. Из-за электрохимической
коррозии поверхность загрязненного железа во влажном воздухе
за короткое время покрывается ржавчиной. Ионы Fea+ с аниона-
ми ОН- (из воды) или СО3“ (образовавшимися при растворении
СО2 в воде) образуют Fe(OH)2 или FeCO3, которые в водной среде
и в присутствии кислорода превращаются в Fe(OH)3 (или
в Fe2O3-3H2O).
Для предохранения железа от действия коррозионных агентов,
его покрывают слоем масляной краски, эмали или другого метал-
ла: Zn (цинкование), Sn (лужение), Сг (хромирование), Ni (нике-
лирование), Cd (кадмирование), РЬ (свинцевание) — либо проводят
поверхностное окисление расплавленным NaNO3 или KNO3.
Пары воды разлагаются нагретым докрасна железом выше
700° по обратимому уравнению
3Fe + 4Н2О ч* Fe3Oi -f- 4Н2 35,7 ккал
При комнатной температуре железо растворяет примерно
0,005% водорода. При этом образуется гидрид включения FeH,
который способствует увеличению твердости железа:
2Fe -р Н2 ч± 2FeH — 2 X 7,2 ккал
Один грамм железа растворяет при 1530° 0,272 см3водорода,
при 1550°—0,279 см3 водорода и при 1650°—0,310 см3 водорода.
ЖЕЛЕЗО
499
Насыщенное водородом железо при нагревании на. воздухе до
900° теряет значительную часть этого газа.
При обычной температуре сухой кислород не взаимодействует
с железом. При нагревании полированной железной пластинки
в кислороде выше 150° наблюдается потемнение ее поверхности,
а при нагревании до белого каления образуется магнетит:
3Fe + 2О2 = Fe3O4 + 266,5 ккал
При 1900° в присутствии кислорода железо полностью пре-
вращается в окислы.
При ^900° растворимость кислорода в a-Fe равна 0,18%,
а в fi-Fe и y-Fe она больше. Железо с небольшим содержанием
кислорода образует твердые и хрупкие сплавы.
При нагревании железо взаимодействует с газообразным хло-
ром, превращаясь в Fe2Cl6, но не вступает в реакцию с жидким
хлором. При действии паров брома или иода на порошкообразное
кристаллическое железо получают Fe3Br8 (или 2FeBr3 -FeB^)
и Fe3I8 (или 2FeI3-FeI2):
2Fe + ЗС12 = Fe2Cle + 191,4 ккал
3Fe + 4Br2 = Fe3Br8 3Fe + 4I2 = Fe3I3
Нагреванием порошкообразного металлического железа в па-
рах брома при 190° получают Fe2Br6:
2Fe + ЗВг2 = Fe2Bre + 152 ккал
Тонко измельченное железо взаимодействует при нагревании
с серой, образуя сульфиды FeS, FeS2:
Fe + S (ромбич.) = FeS + 22,8 ккал Fe + 2S (ромбич.) = FeS2 + 38,8 ккал
Сера плохо растворима в железе, но сульфид железа FeS (из
сплавов системы железо — сера) образует с железом эвтектику,
которая содержит 30% серы, плавится при 985°, обладает ломко-
стью при красном калении и ухудшает качество железа (чугуна
или стали).
При нагревании железного порошка в токе аммиака образуют-
ся нитриды Fe2N, Fe4N:
[400-500° 430°
2Fe+NH3---------► Fe2N + 3/2H2 4Fe-f-NH3-----> Fe4N4-3/2H2
600°
2Fe2N-------
Вакуум
Fe4N + i/2N2
Фосфор, мышьяк и кремний образуют с железом при нагрева-
нии интерметаллические соединения, например Fe3P, Fe2P,
FeP, Fe3As2, Fe2As, Fe3As4, Fe3Si2, FeSi, FeSi2, Fe2Si.
Поскольку нормальный потенциал системы Fe/Fe2+ равен
—0,44 в, железо относится к легко окисляемым металлам. Под
действием разбавленных неорганических кислот (НС1, H2SO4 и др.)
32*
500
ПОБОЧНАЯ ПОДГРУППА VIII ГРУППЫ
оно превращается в соответствующие соли железа(П) с выделе-
нием водорода:
Fe + 2НС1 = FeCl2 + Н2 Fe + H2SO4 = FeSO4 + H2
Разбавленная азотная кислота, взятая в избытке, взаимодей-
ствует с железом по уравнению
4Fe + IOHNO3 = 4Fe(NO3)2 + NH4NO3 + ЗН2О
Концентрированные кислоты, такие, как HNO3 и H2SO4, вза-
имодействуют с железом при нагревании по уравнениям
Fe + 4HNO3 = Fe(NO3)3 + NO + 2Н2О
2Fe + 6H2SO4 = Fe2(S04)3 + 3SO2 + 6H2O
Нормальный потенциал системы Fe/Fe3+ равен —0,036 в.
Под действием концентрированных кислот HNO3, H2SO4,
Н2СгО4 на холоду железо становится пассивным благодаря обра-
зованию плотной защитной пленки на поверхности металла, меня-
ющей значение электрохимического потенциала.
Железо подвергается действию концентрированных растворов
щелочей. Разбавленные растворы щелочей действуют на железо
только в присутствии двуокиси углерода.
Железо вытесняет металлы Bi, Sb, Pb, Sn, Cu, Ag, Hg, и Au
из растворов их солей.
Химическая активность железа иллюстрируется следующей
схемой:
Железо
Комн. темп.
Нагревание
с кислородом во влажном воздухе -^Fe2O3-nH2O
с парами брома или иода Fe3Br8 и Fe3I8
с разбавленными кислотами HCl, H2SO4->
—>-FeCl2-nH2O, FeSO4-nH2O
с парами воды Fe3O4
с кислородом-»-Fe3O4
с газообразным хлором Fe2Cl6
с парами бромаFe2Brfi
с парами серы—^FeS, FeS2
с фосфором, мышьяком, кремниемFe3P, Fe2P,
FeP, Fe3As2, Fe2As, Fe3As4, Fe3Si2, FeSi, FeSi2, Fe2Si
с аммиаком-> Fe2N, Fe4N
с конц. кислотами HNO3, H2SO4 -^Fe(NO3)3-nH2O,
Fe2(SO4)3-nH2O
С физиологической точки зрения железо имеет особое значение
для организма человека и животных, поскольку является катали-
затором процесса дыхания. Как уже упоминалось, железо входит
в состав молекулы гемоглобина. Молекула гемоглобина состоит
из интерциклического соединения гема, которое содержит двух-
валентное железо, и белка глобина. В легких человека гемоглобин
присоединяет кислород, превращаясь в оксигемоглобин, который
разносится кровью, снабжая кислородом все клетки организма.
ЖЕЛЕЗО
501
ПРИМЕНЕНИЕ
Железо и его сплавы широко применяются во всех отраслях
промышленности благодаря сохранению хороших механических
и физико-химических свойств при высоких температурах (до 900°).
Сплавы железа (стали) бывают различных типов: магнитные,
немагнитные, кислотостойкие, твердые, нержавеющие, жаропроч-
ные (устойчивые при высоких температурах) и др. Не перечисляя
всех областей применения сталей, достаточно сказать, что они
используются в производстве электровозов, вагонов, железно-
дорожных рельсов, автомашин, тракторов, экскаваторов, буровых
установок, железнодорожных мостов и подъемных кранов.
Помимо сплавов, в которых преобладает железо (чугуны
и стали) известны такие сплавы, как латуни и бронзы, в которых
содержится 4—6,5% железа. В этих сплавах железо служит
добавкой для модификации прочности, пластичности, твердости,
ковкости, антифрикционных свойств, скорости старения и т. д.
Для современной техники железо и его сплавы имеют огром-
ное значение. Мировой расход железа превышает 100 млн. тонн
в год.
СОЕДИНЕНИЯ (ОБЩИЕ СВОЙСТВА)
Известно небольшое число соединений одно-, четырех- и шести-
валентного железа и очень много соединений двух- и трехвалент-
ного железа.
В табл. 57 приведены формулы и указан цвет некоторых соеди-
нений железа, сгруппированных по состояниям валентности.
Для одновалентного железа известны только карбонильные
и нитрозильные производные.
Самыми устойчивыми являются соединения трехвалентного
железа. Склонность к образованию координационных соединений
у железа выражена слабее, чем у кобальта, платины, хрома и дру-
гих металлов VIII группы, и проявляется как в двух-, так и в трех-
валентном состояниях. Известны главным образом ацидокомплек-
сы (галогено-, циано-, тиоцианато-, сульфато-, нитрато-, фосфато-,
формиато-, оксалато-, тартрато- и цитратокомплексы), а также ряд
хелатных соединений.
Двухвалентное железо проявляет в соединениях металлический
характер (оно образует основания и имеет восстановительные
свойства); в соединениях высших степеней окисления железо
проявляет неметаллический характер (образует ковалентные соеди-
нения и имеет окислительные свойства). Нормальный потенциал
окислительно-восстановительной системы Fe2+/Fe3+ равен 0,771 в.
По многим свойствам катион Fe2+ похож на катионы Мп2+,
Zn2+,Coa+, Ni2+, катион Fe3+ — на А13+, анион FeO2- — на МпО2-,
сто:-.
502
ПОБОЧНАЯ ПОДГРУППА VIII ГРУППЫ
Помимо соединений железа, отвечающих пяти состояниям
валентности, известны металлоорганические соединения и соедине-
ния включения.
Соединения двухвалентного железа
Известны многочисленные соединения, в которых электрополо-
жительное двухвалентное железо находится в виде катионов Fe2+,
IFe(H2O)6]2+, [Fe(NH3)6]2+, [Fe(C10H8N2)3l2+, [Ре^^Н.^з)^2-,
[Fe(C12H8N2)3]2+ или анионов [Fe(OH)6]4-, [FeF3]“, [FeFj2-,
[Fe(H2O)3Cl3]-, [Fe(H2O)2Cl4]2-, [FeCle?-, [FeBr3]-, [Fe(CN)e]4-,
lFe(SCN)6]4-, [Fe(CN)5X]n~ и lFe(NO3)6]4-
Катион Fe2+ (с электронной структурой Is2•3s23p63de)
бесцветен, парамагнитен, неустойчив в щелочной среде и очень
легко окисляется кислородом воздуха. Он склонен образовывать
устойчивые координационные соединения с о-фенантролином,
а,а'-дипиридилом и цианидами щелочных металлов.
Катион [Fe(H2O)6]2+ имеет зеленый цвет, находится в гидрати-
рованных солях железа(П) и их водных растворах.
Соли железа(П) получают растворением металлического желе-
за, окиси, гидроокиси, карбоната или сульфида железа(П)
в различных кислотах без доступа воздуха.
Соединения железа(П) обладают восстановительным характе-
ром и малой устойчивостью из-за склонности к переходу в соедине-
ния железа(Ш). Соединения железа(П) проявляют восстанови-
тельные свойства в щелочной среде, так как в щелочах образует-
ся Fe(OH)2, который легко окисляется до Fe(OH)3. В кислой среде
соединения железа (II) могут быть окислены до соединений желе-
за(Ш) только энергичными окислителями (КМпО4, К2Сг2О7,
HNO3, Н2О2 и С12), как это показано дальше. Окисление соедине-
ний железа(П) в водных растворах до соединений железа(Ш)
сопровождается изменением зеленой окраски на желтую или
коричневую.
Многие безводные соли железа(П) присоединяют аммиак с обра-
зованием амминокомплексов, которые мало устойчивы и разла-
гаются водой.
Соединения железа(П) похожи на аналогичные соединения
марганца(П), кобальта(П), никеля(П) и цинка с точки зрения
устойчивости в кислой среде, легкости окисления в щелочной
среде и склонности к образованию координационных соединений,
содержащих, например, координированную CN-rpynny.
Окись железа, FeO, получают окислением металлического желе-
за, термическим разложением FeC2O4*2H2O без доступа воздуха,
восстановлением окиси железа(Ш) Fe2O3 окисью углерода при
500° или водородом при 700— 800°, прокаливанием (650—700°
в отсутствие воздуха) смеси стехиометрически необходимых коли-
ЖЕЛЕЗО
503
честв Fe2O3 и порошка металлического железа, нагреванием
FeCO3 при 490-581°:
Fe + V2O2 — FeO 63,8 ккал Fe2O3 СО = 2FeO СО2
Fe2O3 + Н2 = 2FeO + Н2О Fe2O3 + Fe = 3FeO
FeC2O4.2H2O = FeO -p CO CO2 2H2O FeCO3 == FeO -J- CO2
Соединение FeO представляет собой диамагнитный черный
неустойчивый кристаллический порошок (структура типа NaCl).
Оно превращается в Fe2O3 при нагревании до 200—250° на воздухе,
диспропорционирует на Fe3O4 и металлическое железо при 570°,
плавится примерно при 1360°, трудно растворимо в воде и щело-
чах, легко растворяется в кислотах с образованием солей желе-
за(П), восстанавливается до металлического железа под действи-
ем Н2 или СО при нагревании, разлагает при нагревании воду
с образованием Fe2O3 и выделением Н2.
2FeO -|- Н2О Fe2O3 -f- Н2
Окись железа(П) образует с многочисленными окислами
металлов соединения типа шпинелей Fe2+[Me|+O4] или перов-
скита Fe2+Me4+O3.
Гидроокись железа, Fe(OH)2, образуется в виде хлопьевидного
желтовато-белого осадка при обработке растворов солей желе-
за(П) щелочами без доступа воздуха, начиная с pH = 7,7:
Fe2+ + 2ОН- - Fe(OH)2
В присутствии воздуха образуется коричнево-зеленый осадок,
который содержит Fe(OH)2*Fe(OH)3. Последний превращается
ж красновато-коричневый осадок Fe(OH)3:
2Fe2+ + ЗОН- + i/2O2 + Н2О = Fe(OH)2-Fe(OH)3
2Fe2+ + 4ОН- + Х/2О2 + Н2О = 2Fe(OH)3
В присутствии перекиси водорода, хлора или гипохлоритов
окисление Fe(OH)2 в Fe(OH)3 происходит мгновенно.
В кристаллическом состоянии Fe(OH)2 имеет такую же сло-
истую структуру, как и CdCl2.
Поскольку Fe(OH)2 проявляет основные свойства, соли желе-
за(П) гидролизуются незначительно.
Соединение Fe(OH)2 плохо растворимо в щелочах и легко
растворяется в минеральных кислотах с образованием солей.
При кипячении тонкого железного порошка с 50%-ным рас-
твором NaOH без доступа воздуха получают раствор, из которого
выпадают голубовато-зеленые кристаллы NaJFe(OH)e]. При обра-
ботке Na4[Fe(OH)e] растворами солей бария выпадает гексагидро-
ксоферрат(П) бария Ba2[Fe(OH)6] в виде бесцветных кристаллов.
Дифторид железа, FeF2, получают действием фтора на метал-
лическое железо, действием газообразного HF на металлическое
51)4
ПОБОЧНАЯ ПОДГРУППА VIII ГРУППЫ
железо или FeCl2, восстановлением Fe2Fe металлическим железом, 1
NH3 или водородом, нагреванием кристаллогидратов FeF2-4H2O, |
FeF2-8H2O в токе газообразного HF: |
5 0 0 ° ?
Fe + F2 = FeF2~rl54 ккал Fe + 2HF-----► FeF2—Н2 »
На холоду t
Fe2Fe + H2=2FeF2T-2HF FeCl2 + 2HF--------> FeF24-2HCl |
Соединение FeF2 представляет собой белые (слегка желтова-
тые) тетрагональные кристаллы с решеткой типа рутила, плотно-
стью 3,95—4,09 г/см3, плавящиеся при 1102°, легко растворимые
в воде, восстанавливающиеся до металлического железа под дей-
ствием водорода при высокой температуре, превращающиеся
в FeCl2 под действием газообразного НС1 и в FeS под действием
H2S. Известны кристаллогидраты FeF2-4H2O в виде ромбоэдри-
ческих белых кристаллов с плотностью 2,095 г!см3 HFeF2-8H2O
в виде зеленых призм.
Известны ацидосоли K[FeF3], K2[FeF4], NH4[FeF3]-2Н2О, сме- \
шаяный фторокомплекс [Fe2+(H2O)6][Fe3+F5(H2O)] и аквопентаммин
[Fe(H2O)(NH3)5]F2.
Дихлорид железа, FeCl2, получают действием газообразного.
НС1 на стружку металлического железа (500°), на цементит (600°)
или на Fe2O3 (1000°), обработкой железа водным раствором НС1,
восстановлением безводного Fe2Cle избытком водорода при 550—
630° или порошкообразным металлическим железом при нагре- 1
вании, действием порошкообразного железа на раствор Fe2Cl6,
разложением 2FeCl2-NO при нагревании или FeCl2-4CO под
действием света, действием смеси паров SiCl4 с газообразным НС1
на тонкий порошок железа, действием хлора на пирит FeS2 при
250—1000°, нагреванием кристаллогидратов FeCl2-«H2O (где п =
— 6, 4, 2) в токе газообразного НС1:
Fe+2HC1 = FeCl2 + Н2
Fe3C + 6НС1 = 3FeC]2 + ЗН2 + С
2FeCl2.NO -> 2FeCl2 + 2NO
FeS2 + Cl2 = FeCl2 J- 2S
Соединение FeCl2 представляет собой парамагнитные бесцвет-
ные ромбоэдрические кристаллы с кристаллической решеткой типа
CdCl2, плотностью 2,988 г!см3, т. пл. 672° и т. кип. 1026°, которые
сублимируются без разложения при нагревании в токе азота.
Это соединение превращается в Fe2Cl6 и Fe2O3 при нагревании
в сухом воздухе или кислороде, растворяется в метаноле, ацетоне,
ацетонитриле и плохо растворяется в воде:
6FeCl2 + V2O2 2Fe2Cl6 + Fe2O3
Известны кристаллогидраты FeCl2>nH2O (где п = 6, 4, 2, 1),
ацидоаквосоли MeIIFe(H2O)3Cl3] (где Me1 = Li+, К+, Rb+, Cs+),
Fe2Cl6 + H2 = 2FeCl2 + 2HC1
Fe2Cl6 + Fe = 3FeCl2
FeCl2 -4CO -> FeCl2 + 4CO
ЖЕЛЕЗО
505
Mei[Fe(H2O)2Cl4] (где Me1 = Rb+, Cs+), ацидосоли K3Na[FeCl6],
[Cd(H2O)6]2[FeCl6] и координационные соединения с сс,сс'-дипири-
дилом [Fe(C10H8N2)3]Cl2 ‘7H2O, с сс,сс',сс"-трипиридилом
ZF,e(C15H11JV3)3]Cl2, с о-фенантролином [Fe(C12H8N2)3]Cl2.
Гексагидрат, FeCl2-6H2O, выделяется в отсутствие воздуха
(в атмосфере СО2) в виде зеленых кристаллов из насыщенного
водного раствора дихлорида железа в интервалах температур от
—36,5 до 12,3°.
По данным рентгеноструктурного анализа было установлено,
что FeCl2-6H2O содержит транс-октаэдры [Fe(H2O)4Cl2], соединен-
ные между собой, и не содержит гексаквоионов.
Тетрагидрат, FeCl2-4H2O, выделяется в отсутствие воздуха
(в атмосфере СО2) из насыщенного раствора дихлорида железа
при температуре выше 12,3° (соответственно при 30—40°); пред-
ставляет собой зеленовато-синие моноклинные кристаллы с плот-
ностью 1,937 г/см3 и твердостью 2 по шкале Мооса.
Дигидрат, FeCI2-2H2O, получают дегидратациейFeCl2‘4Н2О
при 70—90°. Он представляет собой гигроскопичные бесцветные
моноклинные кристаллы с плотностью 2,358 г/см3. Моно-
гидрат, FeCl2*H2O, был получен термическим путем при темпера-
туре выше 120°.
При высокой температуре FeCl2 реагирует с водородом:
950— 1 1 00°
FeCl2-!-H2 ~ Fe-H2HC1
При действии хлора на FeCl2 образуется Fe2Cl6.
Дихлорид железа реагирует с аммиаком и WO3 или МоО3_
в токе СО2:
Номн. температура
FeCl2 + 6NH3-------------> [Fe(NH3)e]Cl2
Нагревание
6FeCl2 + 7NH3-------> 3Fe2N4-12HC14-2N2+»/2H2
FeCl2 + 2WO3 (или 2MoO3) = FeWO4 + WO2C12 (или FeMoO4 + MoO2Cl2>
Известны координационные соединения, например
Fe(C5H5N)4]Cl2, [Fe(H2N — CH2—CH2—NH2)3]C12, и аддукты
FeCl2-2C6H5-NH2, FeCle-6CH3NH2, FeCl2-4СНзОН, FeCl2.
• 2CeH12N4-9H2O, 10FeCl2-NO, 12FeCl2-NO, имеющие малук>
устойчивость.
Дихлорид железа применяется как лечебное средство при
анемии, также в качестве тонизирующего средства и ускорителя
образования эритроцитов.
В реакциях Фриделя — Крафтса хлорид железа(П) применяют
в качестве катализатора.
Дибромид железа, FeBr2, получают действием брома, НВг или
NH4Br на тонкие железные пластинки, нагреванием Fe3Br8 при
506
ПОБОЧНАЯ ПОДГРУППА VIII ГРУППЫ
400—500°, нагреванием NaBr с железным порошком без доступа
воздуха, дегидратацией кристаллогидратов FeBr2-nH2O в кварце-
вой трубке в токе газообразного НВг:
Fe 4- Вг2 = FeBr2 4* 60 ккал Fe 4- 2NaBr = FeBr2 4* 2Na
Fe3Br8 -> 3FeBr2 4- Br2
Соединение FeBr2 представляет собой желтые гексагональные
кристаллы с кристаллической структурой типа Cdl2, плотностью
4,636 г! см3, т. пл. 684°, т. кип. 927°, растворимые в воде, спирте,
эфире, уксусной кислоте, бензоле, ацетонитриле, хлористом эти-
лене, пиридине ит. п., трудно растворимые в феноле на холоду,
неустойчивые на воздухе и превращающиеся в Fe2O8 и Вг2 при
нагревании до 300° на воздухе.
При нагревании FeBr2 восстанавливается водородом до метал-
лического железа.
Известны кристаллогидраты FeBr2-nH2O зеленого цвета (где
п ~ 9, 6, 4, 2), ацидосоли, например NH4[FeBr3] -6Н2О, и много-
численные аддукты, например FeBr2*nNH8 (где п = 6, 2, 1),
FeBr2.6CH3-NH2, FeBr2.2C5H5N-2Н2О, FeBr2-2C5H5N, FeBr2-
• 6C6H5 — NH — NH2, FeBr2-C2H4-2H2O, 2FeBr2-CNBr.
Соединения бромида железа с шестью молекулами аммиака или
пиридина, с тремя молекулами а,а'-дипиридила или о-фенантро-
лина могут быть представлены следующими формулами:
[Fe(NH8)6]Br2, [Fe(C5H5N)6]Br2, [Fe(C10H8N2)3]Br2-6Н2О,
!Fe(C12H8N2)3]Br2.7H2O.
Водные растворы бромида железа образуются при растворении
железа в водном растворе бромистоводородной кислоты.
Дииодид железа, Fel2, получают взаимодействием порошкообраз-
ного железа с Nal при нагревании, термической диссоциацией
Fe(CO)4I2, действием иода на различные сульфиды железа и пря-
мым взаимодействием элементов при нагревании.
Соединение Fel2 представляет собой красновато-коричневую
(часто черную) массу или парамагнитные желтые гексагональные
кристаллы с плотностью 5,315 г! см3, т. пл. 587°, т. кип. 827°. Оно
превращается в Fe2O3 и 12 при нагревании на воздухе, устойчиво
в присутствии восстановителей и растворимо в воде.
Известны кристаллогидраты FeI2-nH2O (где п = 9, 6, 4, 2)
и аддукты FeI2-6CH3 — NH2, FeI2-6CeH5 — NH — NH2, Fel2-
•2C5H5N.
Соединения иодида железа c 6NH3, 6C5H5N (пиридином),
3C1oH8N2 (а,а'-дипиридилом) и 3C12H8N2 (о-фенантролином)
могут быть выражены формулами [Fe(NH3)6]I2, [Fe(C5H5N)e]I2,
{Fe(Ct0H8N2) 3] 12 -5H2O, [Fe(Ct2H8N2)3]12 -7H2O.
Водный раствор иодида железа образуется при обработке вод-
ной суспензии железа иодом или водным раствором HI.
Иодид железа применяется для лечения анемии.
ЖЕЛЕЗО
507
Цианид железа, Fe(CN)2, образуется в виде коричнево-желто-
го осадка при обработке растворов солей железа(П) растворами
цианидов щелочных металлов по уравнению
Fe2+ + 2CN- = Fe(CN)2
При действии избытком KCN на Fe(CN)2 образуется гекса-
цианоферрат(П) калия (ферроцианид калия):
Fe(CN)2 + 4KCN = K4[Fe(CN)e]
Гексацианоферрат ы(П) (ферроцианиды),
Mei[Fe(CN6)l -nH20, Me”[Fe(CN)e] -nH20, MeJ11 [Fe(CN)6]3-nH20,
MelV[Fe(CN)6] -«H2O (и их безводные производные) (где Me1 =
= Li+, Na+, К+, Rb+, Cs+, NH+, Tl+, Ag+, Cu+, Hg2+; Men= Be2+,
Mg2+, Ca2+, Sr2+, Ba2+, Cu2+, Zn2+, Cd2+, Hg2+, Pb2+, V2+, Mn2+,
Fe2+, Ni2+, MoO3+, UO2+; Me111 = Al3+, Ga3+, In3+, Sb3+, Bi3+,
V3+, Fe3+) представляют собой красиво кристаллизующиеся коор-
динационные соединения, окрашенные в желтый, белый, красно-
вато-коричневый, синий, серый и другие цвета и обладающие диа-
магнитными свойствами.
Гексацианоферраты(П) щелочных и щелочноземельных (за
исключением бария) металлов растворимы в воде, устойчивы в обыч-
ных условиях, разлагаются при высокой температуре или под
действием серной кислоты и легко окисляются в кислой или ней-
тральной среде.
При обработке растворов гексацианоферратов(П) щелочных
металлов растворами солей тяжелых металлов, таких, как CuSO4*
• 5Н2О, NiSO4-7H2O, Co(NO3)2-6H2O, AgNO3 и Pb(NO3)2, обра-
зуются полностью или частично замещенные катионами тяжелых
металлов окрашенные и трудно растворимые соединения.
Примеры растворимых в воде гексацианоферратов(П):
Li4[Fe(CN)e].9H2O, Na4IFe(CN)e] -10H2O, K4[Fe(CN)e]-ЗН2О,
Rb4[Fe(CN)6] -2H2O, Cs4[Fe(CN)6] -3 и 6H2O, (NH4)4[Fe(CN)e] -3H2O,
Li2K2[Fe(CN)e]-3H2O, Li2Cs2[Fe(CN)6], LiCs3[Fe(CN)6],
Na3K[Fe(CN)e]-9 или 12H2O, NaK3[Fe(CN)6]-4H2O, Mg2[Fe(CN)6].
• 12H2O, Ca2[Fe(CN)6] -12Н2О, Sr2[Fe(CN)6] -15H2O, Na2Mg[Fe(CN)B],
K2Mg[Fe(CN)6], (NH4)2Mg[Fe(CN)6J и др.
Примеры мало растворимых в воде гексацианоферратов(П):
AgJFe(CN)B] -Н2О, Cu4[Fe(CN)6), Cu2[Fe(CN)6] -nH2O, Ba2[Fe(CN)6] •
• 6Н2О, Zn2[Fe(CN)6] -nH2O, Cd2[Fe(CN)6l -7 и 12Н2О, Hg4[Fe(CN)6],
Hg2[Fe(CN)e], Ga4[Fe(CN)6]3, Mn2[Fe(CN)e], Pb2[Fe(CN)B],
Pb2[Fe(CN)6].(3 или 5) H2O, Li2Cu2[Fe(CN)6], Li2Cu[Fe(CN)e)],
Na2Cu[Fe(CN)6]-2H2O, K2Cu(Fe(CN)6l -H2O, (NH4)2Cu[Fe(CN)e] •
• H2O, MgCu2[Fe(CN)B], MgCu[Fe(CN)B], K2Ba[Fe(CN)6l -3H2O,
MeiZn3[Fe(CN)6)2 (где Me1 = Na+, K+, Cs+, NH+),Na2Cd[Fe(CN)e] •
•H2O, K2Cd[Fe(CN)6], (NH4)2Cd[Fe(CN)e], K2Hg[Fe(CN)6J,
K2Hg3[Fe(CN)6]2, MeJFe2+[Fe(CN)e] (где Mei = Na+, K+)
508
ПОБОЧНАЯ ПОДГРУППА VIII ГРУППЫ
KIn5[Fe(CN)6]4, Sb4[Fe(CN)6]3 -25Н2О, Bi J Fe(CN)6]5, KBi[Fe(CN)6| -4
и 7H2O, V2[Fe(CN)6], V4[Fe(CN)6]3, Th[Fe(CN)6]-4H2O,
(MoO2)2[Fe(CN)6] -2H2O, (UO2)2[Fe(CN)6l, [Cr(NH3)6]4[Fe(CN)6]3,
[CrEn3]4[Fe(CN)6]3, [Cr(NH3)5H2Ol4lFe(CN)6]3.0,5H2O, [Cr(NH3)3.
• Cl]2[Fe(CN)6] -4H2O.
При обработке концентрированных растворов гексацианофер-
ратов(П) концентрированным раствором хлорида железа(Ш) обра-
зуется синий осадок, отвечающий формуле Fe^II[FeII(CN)6]3 и наз-
ванный берлинской или прусской лазурью:
3K4[Fen(CN)e] + 2Fe2nCl6 = Fe“I[FeII(CN)6]3 + 12КС1
Берлинская лазурь обладает малой растворимостью в воде
и кислотах и разлагается при кипячении с концентрированными
растворами щелочей с выделением Fe(OH)3:
Fe4[Fe(CN)6]3 + 12КОН = 3K4[Fe(CN)6] + 4Fe(OH)3
Анион [Fe(CN)6]4- в солянокислой (12 н.) или сернокислой
(10 н.) средах окисляется анионом [Ce(SO4)3]2- по уравнению
[Fe(CN)6F- + [Ce(SO4)3F- = [Fe(CN)6p- + + 3S0J-
Анион [Fe(CN)6]4~ можно окислять иодом при pH = 8 в при-
сутствии большого избытка NaHCO3 или КМпО4.
Гексацианоферрат калия, KjFe(CN)6]-ЗН2О, выпадает из
раствора Fe(CN)2 в KCN, при нейтрализации кислоты H4[Fe(CN)6l
едким кали, выделяется при электролизе KCN (железные электро-
ды и переменный ток) или разложении берлинской лазури под
действием КОН, К2СО3 и Са(ОН)2, при действии KCN на FeS,
восстановлении K3[Fe(CN)6] в щелочной среде перекисью водорода,
действии К2СО3 и Са(ОН)2 на Na2Fe[Fe(CN)6]:
H4[Fe(CN)6] + 4КОН = K4[Fe(CN)6] + 4Н2О
Fe4[Fe(CN)6]3 + 12КОН = 3K4[Fe(CN)6] + 4Fe(OH)3
le4[Fe(CN)6]3 -p 6Na3CO3 -J- 6Ca(OH)2 — 3Na4[Fe(CN)3] 4Fe(OH)3-j-6CaCO3
2K3[Fe(CN)6] + 2KOH + H2O2 = 2K4[Fe(CN)6] + 2H2O + O2
Na2Fe[Fe(CN)6] + 2K2CO3 + 2Ca(OH)2 =
= K4[Fe(CN)6] + 2CaCO3 + Fe(OH)2 + 2NaOH
Соединение K4[Fe(CN)6] -3H2O представляет собой диамагнит-
ные желтые моноклинные кристаллы, нетоксичные, соленые
и горькие на вкус; плотность 1,94 г/см3 при 25°, растворимы в этил-
амине, ацетоне. Оно дегидратируется при 87—90°, меняя окраску
на белую, разлагается при 100° на Fe(CN)2 и KCN, устойчиво по
отношению к кислороду воздуха и щелочам, реагирует с хлором,
ЖЕЛЕЗО
509
металлическим натрием, NH4C1 и разб. H2SO4.
2K4[Fe(CN)6] + Cl2 = 2K3[Fe(CN)6] + 2KCI
2K4[Fe(CN)6] + 4Na = 2Fe + 8KCN + 4NaCN
K4[Fe(CN)e] + 4NH4C1 = Fe(CN)2 + 4NH4CN + 4KC1
2K4[Fe(CN)6] + 3H2SO4 = K2Fe(Fe(CN)e] + 3K2SO4 + 6HCN
При обработке K4[Fe(CN)6] диметилсульфатом (CH3)2SO4 обра-
зуется [(CH3)6Fe(CN)6]SO4:
K4[Fe(CN)6] + 3(CH3)2SO4 = [(CH3)6Fe(CN)6]SO4 + 2K2SO4
K4lFc(CN)6]-3H2O применяют в процессе изготовления фото-
графической бумаги, как химический реактив при определении
железа, цинка, меди, урана, метиленовой сини и в производстве
минеральных красителей.
Железистосинеродистая кислота, H4[Fe(CN)6], образуется при
обработке Ba2[Fe(CN)6] -6Н2О серной кислотой, действии H2S на
Pb2[Fe(CN)6] -(3 или 5) Н2О, обработке концентрированного рас-
твора K4[Fe(CN)6] конц. НС1:
Ba2[Fe(CN)6] + 2H2SO4 = H4[Fe(CN)6] + 2BaSO4
Pb2[Fe(CN)6] + 2H2S = H4[Fe(CN)6] + 2PbS
K4[Fe(CN)6] + 4HC1 = H4[Fe(CN)6] + 4KC1
H4[Fe(CN)6] является сильной кислотой с восстановительными
свойствами и представляет собой белые (или светло-желтые) куби-
ческие кристаллы с плотностью 1,536 г!см\ нерастворимые в воде
и хорошо растворимые в конц. H2SO4 (без разложения), устойчи-
вые в сухом состоянии и разлагающиеся в присутствии влаги,
а также при температуре 100° по уравнению
3H4[Fe(CN)6] -> Fe2[Fe(CN)e] + 12HCN
Кислород, озон, хлор, бром и иод окисляют H4IFe(CN)6] до
H3[Fe(CN)6]. С окисью углерода H4[Fe(CN)6] реагирует по урав-
нению
H4[Fe(CN)6] + СО + 2Н2О = H3[Fe(CN)5(CO)] + НСООН + NH3
Известны аддукты: H4[Fe(CN)6]-10СН3 — ОН, H4[Fe(CN)6]-
• 4С3Н5ОН, H4[Fe(CN)6] -2(С2Н5)2О, H4[Fe(CN)6] -СН3 - СО -
— сн3.
Пруссидные соединения, Me^[Fe2+(CN)5X],
являются координационными соединениями железа(II); окра-
шены в желтый или оранжево-желтый цвет и содержат пять коор-
динированных цианогрупп и одну группу из нижеприведенных:
NO. СО, NH3. Н2О, N2H4, NO2, NOS, SCN, SO3, AsO2 и др.
Примеры пруссидных соединений: K3[Fe(CN)5NO],
Li3[Fe(CN)5CO]-4Н2О, Na3[Fe(CN)5CO]-7Н2О, K3[Fe(CN)5CO] •
. 3,5H2O, Sr3[Fe(CN)5CO]-4H2O, Cu3lFe(CN)5C012-nH2O,
510
ПОБОЧНАЯ ПОДГРУППА VIII ГРУППЫ
Zn3[Fe(CN)5CO]2-7H2O, Cd3[Fe(CN)5CO]2-7Н2О, Mn3[Fe(CN)5CO], •
•18Н2О, (UO2)3[Fe(CN)5CO]2-5Н2О, Na3[Fe(CN)5NH3] -пНД
Zn3[Fe(CN)5NH3]2 -12Н2О, Cd3[Fe(CN)5NH3]2 -10Н2О,
Cu3[Fe(CN)5NH3]2, Pb3[Fe(CN)5NH3]2.5H2O, Na3[Fe(CN)5H2O] •
•7H2O, K3[Fe(CN)5H2O], Na3[Fe(CN)5N2H4l -H2O, Na4[Fe(CN)5NOJ •
•rcH2O, Na2K2[Fe(CN)5NO2l-H2O, K4[Fe(CN)5NO2],
Ag4[Fe(CN)5NO2]-2H2O, Na4[Fe(CN)5NOSl, K4[Fe(CN)5NOS],
Na4[Fe(CN)5(SCN)], Na5[Fe(CN)5SO3]-9H2O, K5[Fe(CN)5SO3l-3H2O,
Fe5[Fe(CN)5SO3l3, Na4[Fe(CN)5AsO2] -10H2O.
Тиоцианат железа, Fe(SCN)2-3H2O, выпадает из раствора, при-
готовленного без доступа воздуха при обработке Ba(SCN)2 суль-
фатом железа(П) FeSO4, нейтрализации HSCN гидроокисью желе-
за(П) Fe(OH)2, восстановлении Fe(SCN)3 железом в виде чешуек
в спирте или солями катионов Hg2+, Sn2+, сульфитами и арсенитами.
Соединение Fe(SCN)2 -ЗН2О представляет собой зеленые клино-
ромбические призмы, которые становятся красноватыми на возду-
хе: оно растворимо в воде, спирте, эфире, аммиаке и образует коор-
динационные соединения типа Me*[Fe(SCN)6] -?гН2О с тиоцианатами
некоторых металлов.
Примеры гексатиоцианатоферратов(П): Na4[Fe(SCN)6]-12Н2ОГ
K4[Fe(SCN)6]-4H2O, (NH4)4[Fe(SCN)6], Hg2[Fe(SCN)6]-4Н2О.
Известны аддукты: Fe(SCN)2-3SC(NH2)2, Fe(SCN)2^CsHsN,
Fe(SCN)2-2C6H5 — NH2, Fe(SCN)2-4C9H7N, Fe(SCN)2 .3C1oH8N2,
Fe(SCN)2-3C12H8N2, Fe(SCN)2-6C6H5 - NH - NH2.
Сульфид железа, FeS, встречается в природе в виде минерала
пирротина, представляющего собой желтые гексагональные кри-
сталлы с магнитными свойствами, плотностью 4,58 — 4,70 г! см3,
твердостью 4 по шкале Мооса; иногда сопутствует природным
сульфидам меди, никеля, кобальта и др.
Сульфид железа(П) получают в результате нагревания желез-
ного порошка с серой, пропусканием смеси H2S и Н2 над нагре-
тым до 750—1000° Fe2O3, действием сульфида аммония (или рас-
твора H2S, забуференного ацетатом натрия или ацетатом аммония)
на растворы солей железа(П) при pH > 4,5:
Fe + S = FeS + 22,8 ккал Fe2O3 + 2H2S + Н2 = 2FeS + ЗН2О
FeSO4 + (NH4)2S = FeS + (NH4)2SO4
FeSO4 + H2S + 2NaCH3COO FeS + Na2SO4 + 2CH3COOH
Соединение FeS представляет собой темно-коричневые гекса-
гональные кристаллы, которые обладают кристаллической струк-
турой типа арсенида никеля, плавятся при 1193°, трудно раство-
римы в воде, растворяются в разбавленных кислотах с образова-
нием солей железа(П) и выделением H2S.
Влажный осадок FeS черного цвета окисляется на воздухе,
превращаясь в Fe(OH)3:
2FeS + ЗН2О + 3/2О2 = 2Fe(OH)3 + 2S
ЖЕЛЕЗО
511
При нагревании сульфид железа(II) взаимодействует с парами
воды:
3FeS + 4Н2О = Fe3O4 + 3H2S + Н2
S
Дисульфид железа, Fe(f |, встречается в природе в виде мине-
\S
рала пирита или марказита. Пирит обладает блестящим желтым
(как латунь) цветом, кристаллической структурой типа NaCl
и является самым распространенным минералом железа в природе.
Дисульфид железа получается нагреванием порошкообразного
железа с серой, пропусканием H2S через суспензию гидроокиси
железа(Ш) и действием Fe2Cl6 на P2S5:
a-Fe + 2S (ромбич.) = FeS2 + 38,8 ккал
2Fe(OH)3 + 3H2S = FeS2 + FeS + 6H2O
3Fe2Cle + 2P2S5 = 3FeS2 + 3FeCl2 + 4PSC13
Прокаливанием пирита при 400—500° получают Fe2O3 и SO2,
которую применяют в промышленномпроизводстве серной кислоты.
2FeS2 + n/2O2 = Fe2O3 + 4SO2
В присутствии влаги марказит окисляется по уравнению:
FeS2 + ’/2О2 + Н2О = FeSO4 + H2SO4
Сульфат железа, FeSO4, получают прокаливанием пирита
(250—290°), нагреванием PbSO4, с металлическим железом (540°),
нагреванием пирита FeS2 и халькопирита FeCuS2 с окисью меди
СпО при температуре выше 300° на воздухе, дегидратацией FeSO4-
•7Н2О в вакууме (280°), в токе СО2 (300°) или Н2 (350°), дегидрата-
цией FeSO4*7H2O в вакууме (540—550°) или в водороде (320°),
нагреванием Fe3O4 с SO2 на воздухе, восстановлением Fe2(SO4)3
двуокисью серы SO2 при нагревании, обработкой FeSO4-7H2O
конц. H2SO4 при комнатной температуре:
FeS2 + ЗО2 = FeSO4 + SO2 FeS2 + CuO + 3/2O2 = FeSO4 + CuS
PbSO4 + Fe = FeSO4 + Pb Fe2(SO4)3 + SO2 = 2FeSO4 + 2SO3
Fe3O4 + 2SO2 + O2 = FeSO4 + Fe2O3 + SO3
Безводная соль FeSO4 токсична. Она представляет собой
парамагнитный, очень гигроскопичный орторомбический кристал-
лический порошок белого цвета с плотностью 3,14 г! см3. FeSO4
превращается в Fe2(SO4)3 в атмосфере кислорода под действием
ультразвука и в Fe2O3 и SO3 при нагревании на воздухе:
2FeSO4 + !/2О2 = Fe2O3 + 2SO3
Известны кристаллогидраты FeSO4’?zH2O (где п = 7, 4, 1).
Гептагидрат, FeSO4*7H2O, называемый железным купоросом,.
512
ПОБОЧНАЯ ПОДГРУППА VIII ГРУППЫ
известен очень давно и встречается в природе в виде минерала
мелантерита.
Кристаллогидрат FeSO4-7Н2О или [Fe(H2O)6]SO4-Н2О выпадает
в интервале температур от —1,82 до —56,7° из раствора, получен-
ного (без доступа воздуха) в результате растворения железа (в виде
порошка, проволоки, опилок, пластинок) в H2SO4 (1 : 4 или 1 : 5)
при нагревании, растворения окиси, гидроокиси или карбоната
железа (II) в разб. H2SO4, пропускания СО2 через суспензию
гипса CaSO4-2H2O и окиси железа(П) FeO, окисления пирита
или марказита во влажном воздухе при нагревании:
Fe Ч- H2SO4 + 7Н2О = FeSO4-7H2O + Н2
FeO + H2SO4 + 6Н2О = FeSO4-7H2O
CaSO4-2H2O 4- FeO - CO2 4- 5H2O = FeSO4-7H2O 4- CaCO3
FeSO4-7H2O представляет собой парамагнитные зеленовато-
синие моноклинные кристаллы с плотностью 1,898 г!см? при 18°,
т. пл. ~64°, т. кип. 118,3°; кристаллогидрат растворяется
в СН3ОН, С2Н5ОН и глицерине, плохо растворяется в жидком
NH3, уксусной кислоте, метилацетате и полностью дегидратирует-
ся при 280° в вакууме.
При нагревании FeSO4-7H2O претерпевает следующие пре-
вращения:
60-80° 110-160°
FeSO4-7H2O--------► FeSO4-4H2O ----
4 2 -зн2о 1 2 -зн2о
540 — 550° Выше 580°
FeSO4-H2O FeSO4----------►
1/2Fe2O3 4- V2SO2 + VsSOs
Соединение FeSO4-7H2O хранится в склянках из коричневого
стекла с герметической пробкой для предотвращения окисления
воздухом. Водный раствор сульфата железа устойчив в присут-
ствии серной кислоты (на воздухе окисляется до FeSO4OH) и слу-
жит восстановителем в очень многих реакциях:
10FeSO4 4- 2КМпО4 4- 8H2SO4 = 5Fe2(SO4)2 4- 2MnSO4 4- K2SO4 4- 8H2O
6FeSO4 + K2Cr2O7 + 7H2SO4 = 3Fe2(SO4)3 4- Cr2(SO4)3 4- K2SO4 4- 7H2O
4FeSO4 + H2[PtCle] 4- 14KOH = 4Fe(OH)3 + Pt 4- 4K2SO4 4- 6KC1 4- 2H2O
41eSO4 -J- H2SeO3 -J- 2H2SO4 — 2Fe2(SO4)3 4“ So "b 3H2O
3FeSO4 4- 3AgNO3 = Fe2(SO4)3 4“ Fe(NO3)3 4“ 3Ag
6FeSO4 4- 3Hg(NO3)2 = 2Fe2(SO4)3 4- 2Fe(NO3)3 4- 3Hg
2FeSO4 4- 2Ce(SO4)2 = Fe2(SO4)3 + Ce2(SO4)3
При добавлении концентрированного раствора сульфата железа
к слабо кислому раствору нитрита образуется коричневый аддукт
(FeSO4)x(NO)y:
2FeSO4 4- 2NaNO2 4- 2H2SO4 = 2NO 4- Fe2(SO4)3 + Na2SO4 4- 2H2O
a:FeSO4 4- jNO = (FeSO^NO^
ЖЕЛЕЗО
513
Известны и другие аддукты: FeSO4'l,5CH3OH, FeSO4*
•2С6Н5 — NH — NH2-Н2О, FeSO4 • 2C6H5NH2, FeSO4-4C5H5N.
Известны также координационные соединения, образуемые
сульфатом железа с а,а'-дипиридилом, а,а',а"-трипиридилом
и о-фенантролином [Fe(C10H8N2)3]SO4, [Fe(C15H11N3)3]SO4,
[Fe(C12H8N2)3]SO4.
Сульфат железа служит для получения черных чернил (кото-
рые после высыхания с трудом растворяются водой), для приго-
товления многочисленных синих, зеленых, красных, фиолетовых,
черных и других пигментов, в качестве химического реагента при
получении элементов Pt, Au, Ag, Hg, Se, Те и др., а также в объем-
ном анализе.
Двойной сульфат железа(П) и аммония (соль Мора), (NH4)2SO4-
•FeSO4‘6H2O, выделяется из раствора, полученного растворени-
ем 139 г FeSO4-7H2O и 66 г (NH4)2SO4 в минимальном количестве
воды, подкисленной несколькими каплями H2SO4.
Соединение (NH4)2SO4-FeSO4-6H2O представляет собой пара-
магнитные сине-зеленые моноклинные кристаллы, растворимые
в воде, устойчивые на воздухе и окисляющиеся труднее, чем
сульфат железа(П).
Соль Мора применяют в объемном анализе для приготовления
стандартных растворов железа(П) и для калибровки веществ при
магнитных измерениях.
Нитрат железа, Fe(NO3)2-6Н2О, выпадает в виде кристал-
лов в интервале температур между —12 и 60,5° в атмосфере инерт-
ного газа (например, СО2) из раствора, полученного либо раство-
рением металлического железа (в виде порошка, стружки или
проволоки) в разб. HNO3 (d = 1,034 г/см3), либо обработкой рас-
твора сульфата железа(П) раствором нитрата бария.
4Fe + IOHNO3 + 21Н2О = 4Fe(NO3)2-6H2O + NH4NO3
При концентрировании этих же растворов выпадает кристалло-
гидрат Fe(NO3)2-9Н2О (в интервале температур —28 и —12°).
Под действием HNO3 (d = 1,073 г/см3) металлическое железо
образует смесь нитратов железа(П) и железа(Ш).
Известны гексанитроферраты(П) общей формулы
Me*Men[Fe(NO2)6], например K2Ca[Fe(NO2)6l, K2Sr[Fe(NO2)6],
K2Ba[Fe(NO2)6l, MeiCd[Fe(NO2)6] (где Me1 = K+, NH+, Tl+),
K2Pb[Fe(NO2)6], Me*Hg[Fe(NO2)6] (где Me1 = K+, NH+).
Ортофосфат железа, Fe3(PO4)2-8Н2О, встречается в природе
в виде минерала вивианита и образуется путем концентрирования
раствора, состоящего из 12 ч. (NH4)2SO4 •FeSO4*6H2O, 10 ч.
Na2HPO4-12Н2О, 2 ч. NaCH3COO-ЗНаО и 150 ч. дистиллирован-
ной воды:
3(NH4)2SO4-FeSOi + 2Na2HPO4 + 2NaCH3COO =
= Fe3(PO4)2 + 3(NH4)2SO4 + 3Na2SO4 + 2CH3COOH
33-0101
514
ПОБОЧНАЯ ПОДГРУППА VIII ГРУППЫ
Соединение Fe3(PO4)2-8Н2О представляет собой голубовато-
белые моноклинные кристаллы с плотностью 2,58 г/см3, трудно
растворимые в воде и уксусной кислоте, растворимые в минераль-
ных кислотах. Вещество хранят в герметически закрытых склян-
ках, поскольку оно легко окисляется на воздухе. При восстанов-
лении ортофосфата железа(П) водородом при нагревании обра-
зуется Fe3P2.
Известны кристаллогидраты Fe3(PO4)2 «6Н2О, Fe3(PO4)2 *Н2О,
гидроортофосфаты FeHPO4'2Н2О, Fe(H2PO4)2 *2Н2О и гидрокси-
ортофосфаты, например 2Fe3(PO4)2 •Fe(OH)2-8H2O.
При действии ортофосфорной кислоты на ортофосфат желе-
за^!) получают дигидроортофосфат железа (II) Fe(H2PO4)2-2Н2О.
Карбонат железа, FeCO3, встречается в природе в виде мине-
рала сидерита или железного пшата. FeCO3 может быть приготов-
лен обработкой растворов солей железа(П) растворами карбоната
(или бикарбоната) натрия без доступа воздуха при 80° или нагре-
ванием до 150° смеси FeSO4 с Na2CO3 в предварительно вакууми-
рованной закрытой трубке:
FeSO4 + Na2CO3 = FeCO3 + Na2SO4
FeSO4 + 2NaHCO3 = FeCO3 + Na2SO4 + CO2 + H2O
Соединение FeC03 представляет собой парамагнитный блестя-
щий белый порошок с плотностью 3,82—3,90 г!см3, трудно раство-
римый в воде. Оно окисляется во влажном воздухе (или в водной
суспензии в присутствии воздуха), разлагается на FeO и СО2
нагреванием при 490—581°, восстанавливается до металлического
железа под действием водорода при нагревании, превращается
в Fe3O4 под действием КОН при высокой температуре в отсут-
ствие воздуха, растворяется в минеральных кислотах (H2SO4,
HNO3, НС1) и в растворах бикарбоната натрия.
4FeCO3 + 6Н2О + О2 = 4Fe(OH)3 + 4СО2
FeCO3 "F 2NaHCO3 Fe(HCO3)2 -J- Na2CO3
Карбонат железа растворяется в воде, содержащей двуокись
углерода, благодаря образованию растворимого бикарбоната желе-
за Fe(HCO3)2, который на воздухе превращается в Fe(OH)3:
FeCO3 -J- СО2 4“ Н2О 4=^ Fe(HCO3)2
2Fe(HCO3)2 + Н2О + VaOa = 2Fe(OH)3 + 4CO2
Учитывая это свойство, бутылки с железосодержащими (бикар-
бонат железа) минеральными водами заполняются до отказа,
чтобы исключить присутствие воздуха и, следовательно, возмож-
ность образования гидроокиси железа(Ш) Fe(OH)3.
Известна ацидоаквосоль Na3[Fe(H2O)(HCO3)5].
ЖЕЛЕЗО
515
Оксалат железа, FeC2O4-2H2O, образуется в виде желтовато-
белого осадка при обработке растворов солей железа(П) раство-
ром оксалата щелочного металла.
Известны комплексные оксалаты K2[Fe(C2O4)2l ‘Н2О,
(NH4)2[Fe(C2O4)2l -ЗН2О, (NH4)2Sr[Fe(C2O4)3] -6Н2О.
Хелатные соединения
При обработке слабо кислых растворов солей железа(П)
0,5 %-ными водными растворами о-фенантролина образуются окра-
шенные в интенсивно-красный цвет комплексы, которые содержат
катион:
При действии 2%-ного раствора а,а'-дипиридила в 3 н. НС1
на растворы солей железа(П) (pH ~ 4,5) образуются раствори-
мые комплексы красного цвета, которые содержат катион:
При обработке растворов солей железа(П) в аммиачной среде
(и в присутствии тартратов для связывания катиона Fe8+ при
возможном его появлении) 1 %-ным спиртовым раствором а-ди-
метилглиоксима образуется растворимый красный комплекс:
Н 3С- с---------С— СН з
II II
/О-ЪГ /N=O^
/
XO=n/ xn-oz
ll и
н3с-с-------с-сн3
н
33*
516
ПОБОЧНАЯ ПОДГРУППА VIII ГРУППЫ
При действии ацетилацетона на соединения железа(П) в при-
сутствии пиридина образуется соединение:
Помимо перечисленных, известны и другие соединения Fe(II): двойные
хлориды 2RbCl-FeCl2-2Н2О, RbCl-FeCl2-2Н2О, FeCl2-HgCl2-4Н2О,
FeCl2-4IE>iCl3-12H2O, хлорат Fe(C103)2, известный только в растворе, перхло-
рат Fe(C104)2-6Н2О, двойные и тройные бромиды FeBr2-HgBr2, FeBr2-
• Al2Br6, RbBr-FeBr2-2FeBr3-3H2O, бромат Fe(BrO3)2, двойные иодиды
2FeI2-РЫ2-ЗН2О, Fel2-2HgI2-6Н2О, перйодаты Fe5(IO6)2, FeH3IO6, сульфиты
FeSO3-3H2O, FeSO3 .T12SO3, двойные сульфаты Rb2SO4-FeSO4-6H2O,
MgSO4 -7H2O -FeSO4 -7H2O, ZnSO4 -7H2O -FeSO4 -7H2O, FeSO4 -7H2O -MnSO4 •
• 7H2O, FeSO4-Al2(SO4)3.24H2O, FeS04-Fe2(S04)3-10H20, 4FeSO4-
• Fe2(SO4)3-12H2O, тиосульфаты FeS2O3-5H2O, 3Na2S2O3-FeS2O3-8H2O,
дитионаты FeS2O6-7H2O, 3(NH4)2S2O6-FeS2Oe-6H2O, тетратионат FeS4O6,
пиросульфат FeS2O7, селенит FeSeO3, селенаты FeSeO4-5 или 7H2O,
K2SeO4-FeSeO4-6H2O, (NH4)2SeO4-FeSeO4-6H2O, гипофосфит Fe(H2PO2)2-
• 6H2O, гипофосфат Fe2P2O6-4,5H2O, тиогипофосфат Fe2P2S6, метафосфат
FeP2O6-4H2O, ортофосфат NH4FePO4-H2O, тиоортофосфат Fe3(PS4)2, пиро-
фосфат (NH4)2FeP2O7-2H2O, тиопирофосфат Fe2P2S7, арсенит Fe(AsO2)2,
ортоарсенаты Fe3(AsO4)2-8H2O, 5Fe3(AsO4)2-Pb3(AsO4)2, пироарсенат
Na2FeAs2O7, антимонит Fe(SbO2)2, метаванадат FeV2O6, фторосоль
Fe[VF6]-7H2O, метаниобат FeNb2O6, метатанталат FeTa2O6, тетрафтороборат
Fe[BFJ2-6H2O, тиокарбонат FeCS3, селеноцианат Fe(SeCN)2, силикаты
Fe2SiO4, (Mg, Fe)SiO3, титанаты Fe2TiO4, FeTiO3, галогеносоли
[Fe(H2O)6][SiF6], [Fe(H2O)6][TiF6], [Fe(H2O)6][SnCl6], [Fe(H2O)6][SnBr0],
молибдат FeMoO4, тиомолибдат FeMoS4, вольфрамат FeWO4, перренат
Fe(ReO4)2-4H2O, формиат Fe(HCOO)2-2H2O, ацетаты Fe(CH3COO)2-4H2O,
(UO2)2Fe(CH3COO)6.7H2O, Na(Li)(UO2)3Fe(CH3COO)9-9H2O, оксалат
FeC2O4-2H2O, тартраты FeC4H4O6-5H2O, Na2[Fe(C4H4O6)2]-2H2O,
K2[Fe(C4H4O6)2], цитраты FeC6H6O7-H2O, NH4FeC6H5O7.
Соединения трехвалентного железа
Известны многочисленные соединения, в которых электро-
положительное трехвалентное железо находится в виде катионов
Fe3+,[Fe(NH3)6]3+, [Fe(H2N — СО — NH2)613+ или анионов FeO;,
Fe2O*-,FeS- [Fe(OH)e]3-, [FeFj-, [FeF6]3-, [Fe(H2O)F5]2-, [FeClJ-,
[FeClJ3-, [Fe(H2O)Cl5]2-, [Fe2Cl9]3-, [FeBrJ-, [FeBr6]3-,
[Fe(H2O)Br3Cl2P-, [Fe(H2O)Br2Cl3]2-, [Fe(CN)6]3-, [Fe(CN)5X]n-,
[Fe(SCN)e]3-, [Fe(SO4)3]3- и [Fe(SO4)2]-.
Катион Fe3+ (с электронной структурой Is2 -2s22pe -3s23p63d5)
имеет 5 электронов на Зй-орбитали, устойчив, бесцветен, пара-
магнитен, является слабым окислителем и образует с анио-
ном SCN- интенсивно окрашенное кроваво-красное соединение.
ЖЕЛЕЗО
517
Чем выше концентрация ионов водорода в растворе, тем сильнее
выражены окислительные свойства соединений железа(Ш).
Соли железа(Ш) могут быть получены окислением порошко-
образного металлического железа галогенами, окислением солей
железа(П), растворением окиси или гидроокиси железа(Ш)
в кислотах.
Для галогенидов железа(Ш) характерна склонность к обра-
зованию димеров Fe2F6, Fe2Cl6, Fe2Br6, смешанных галогенидов
Fe3Cl8-ЮНгО, Fe3Br8-16Н2О, Fe3I8 и галогенокомплексов.
Соли железа(Ш) устойчивы на воздухе, гидролизуются водой,
и их водные растворы имеют кислую реакцию:
Fe2Cl6 + 6Н2О 2Fe(OH)3 + 6НС1
При гидролизе солей железа(Ш) могут образоваться основ-
ные соли, которые выделяются в коллоидном состоянии и окраше-
ны в коричнево-желтый цвет.
При восстановлении соединений железа(Ш) H2S, HI, N2H4,
NH2OH, SO2, водородом в момент выделения (Zn + 2НС1) обра-
зуются соединения железа (II).
Окись железа, Fe2O3, является самым устойчивым природным
кислородсодержащим соединением железа, которое встречается
в форме минералов гематита или красного железняка.
Известны три модификации окиси железа(Ш), а именно:
a-Fe2O3, y-Fe2O3 и 6-Fe2O3. Модификация a-Fe2O3 (соответ-
ствующая гематиту) парамагнитна и образуется окислением желе-
за на воздухе при температуре выше 200°, а также нагреванием
6-Fe2O3 в течение 3 час при 110° или сжиганием пирофорного железа
на воздухе при комнатной температуре и давлении меньше
760 мм рт. ст. Модификация y-Fe2O3 ферромагнитна и образуется
окислением железа на воздухе при температуре ниже 200°, а так-
же окислением Fe3O4 или нагреванием a-Fe2O3 при 300°. Модифи-
кация 6-Fe2O3 ферромагнитна и образуется окислением растворов
солей железа(П) в щелочах.
Модификации окиси железа(Ш) могут также быть получены
прокаливанием гидроокиси Fe(OH)3 при 700°, нитрата Fe(NO3)3-
•6Н2О при 600—800°, карбоната FeCO3 при 500° на воздухе,
сульфата FeSO4 или пирита FeS2 на воздухе:
2Fe 3/2О2 = Fe2O3 -|- 196,8 ккал
2Fe(OH)3 = Fe2O3 + ЗН2О 2FeCO3 + V2O2 = Fe2O3 + 2CO2
2FeSO4 = Fe2O3 + SO2 + SO3 2FeS2 + H/2O2 = Fe2O3 + 4SO2
2Fe(NO3)3-6H2O = Fe2O3 + 6NO2 + 3/2O2 + 12H2O
Нагревание порошкообразного железа или трихлорида железа
в водном паре также дает Fe2O3.
Соединение a-Fe2O3 представляет собой красный порошок
с плотностью 5,24 г! см3, который плавится примерно при 1550°,
трудно растворим в воде и может быть восстановлен до Fe3O4,
518
ПОБОЧНАЯ ПОДГРУППА VIII ГРУППЫ
FeO или металлического железа водородом, углеродом, окисью
углерода, металлическим алюминием или элементным кремнием.
При восстановлении а-, у-, 6-Fe2O3 водородом (280—340°) обра-
зуется пирофорное металлическое железо, а при алюмо- или крем-
нетермическом восстановлении модификаций а-, у-, 6-Fe2O3 обра-
зуется загрязненное металлическое железо.
Растворимость а-, у-, 6-Fe2O- модификаций в кислотах зави-
сит от температуры и продолжительности прокаливания окиси
перед растворением. Если окись железа была слегка прокалена,
она растворяется в кислотах.
Окись железа под названием железный сурик, охра, мумия,
применяется как пигмент приготовления для красок.
Гидроокись железа, Fe(OH)3 или Fe2O3*3H2O, образуется
в виде красновато-коричневого осадка при обработке растворов
солей железа(Ш) гидроокисями или карбонатами щелочных
металлов (или аммиака) при нагревании:
Fe2Cl6 + 6NaOH = 2Fe(OH)3 + 6NaCl
Fe2Cl6 + 3Na2CO3 + 3H2O = 2Fe(OH)3 + 6NaCl + 3CO2
Соединение Fe(OH)3 начинает осаждаться при pH = 2,2
и остается в коллоидном состоянии до pH ~ 7.
Гидроокись железа трудно растворима в воде, легко образует
коллоидные растворы, является слабым основанием почти амфо-
терного характера, поскольку растворяется в разбавленных кис-
лотах и теплых концентрированных растворах сильных основа-
ний (но не аммиака) с образованием солей.
2Fe(OH)3 + 6НС1 Fe2Cl6 + 6Н2О Fe(OH)3 + ЗКОН = K3[Fe(OH)e]
Fe(OH)3 применяют для очистки газов от H2S, а также в слу-
чае отравлений соединениями мышьяка, поскольку он образует
с ними трудно растворимые соединения.
При частичной дегидратации Fe(OH)3 образуется железистая
кислота, HFeO2, которая встречается в природе в виде двух
кристаллических модификаций: a-HFeO2 (гетит) и y-HFeO2 (лепи-
докрокит). Гидрогетит или лимонит HFeO2-nH2O имеет плотность
3,3—4 г/см3 и окраску, меняющуюся от желтой до темно-коричне-
вой или черной.
Ферриты (соли железистой кислоты), MeIFeO2 или
Men[Fe2O4] (где Me1 = Li+, Na+, К+, Ag+ и Me11 = Mg2+, Zn2+),
получают сплавлением Fe2O3 с окислами, гидроокисями или
карбонатами соответствующих металлов:
Fe2O3 2NaOH = 2NaFeO2 Н2О Fe2O3 Na2CO3 «= 2NaFeO2 CO2
Гидроксоферриты, Me*[Fe(OH)6] или Me“[Fe(OH)6]2
(где Me1 =Na+, K+ и Me11 = Sr2+, Ba2+), получают действием
избытка NaOH, KOH, Sr(OH)2, Ba(OH)2 на соли железа(Ш)
ЖЕЛЕЗО
519
при нагревании:
2Fe(C104)3 + 6Sr(OH)2 = Sr3[Fe(OH)6]2 + 3Sr(C104)2
Соединения Na3[Fe(OH)6] желтого, K3[Fe(OH)6] красного,
Sr3[Fe(OH)6]2 и Ba3[Fe(OH)6]2 белого цвета. Последние два —
кристаллические порошки.
Окись железа(11, III), Fe3O4, со структурой шпинели
Fe2+[Fe2+O4] встречается в природе в виде минерала магнетита.
Соединение Fe3O4 можно получить прокаливанием других
окислов железа или восстановлением Fe2O3 (400—500°) водородом,
насыщенным парами воды, или окисью углерода (800°).
Fe3O4 представляет собой ферромагнитные ломкие (твердость
5,6—6,5 по шкале Мооса) черные кубические кристаллы с метал-
лическим блеском; они трудно растворимы в воде и кислотах,
имеют т. пл. 1538° и разлагаются при 1787°. Окись железа(П, III)
устойчива в сухом воздухе и служит для изготовления электродов,
поскольку является хорошим проводником электрического тока
и устойчива к действию химических реагентов.
Димер трифторида железа, Fe2F6 или
получают взаимодействием фтора с порошкообразным металличе-
ским железом, FeCl2 или Fe2Cle, а также нагреванием Fe2O3 в ат-
мосфере HF и нагреванием Fe2Cl6 с NH4F:
2Fe -J- 3F2 = Fe2F6 -J- 466,50 ккал 2FeCl2 -J- 3F2 = Fe2Fg -J- 2C12
Fe2Cl6 + 6NH4F = Fe2Fe + 6NH4C1
Соединение Fe2F6 представляет собой зеленые триклинные
кристаллы с кристаллической решеткой типа ReO3, плотностью
3,87 г!см9, т. пл. 1027°, т. кип. 1327°, трудно растворимые в спирте
и эфире. Это соединение гидролизуется водой, восстанавливается
до FeF2, а затем до металлического железа при нагревании в токе
водорода и превращается в Fe2O3 с высвобождением фтора при
нагревании на воздухе.
Водный раствор Fe2F6 получают действием плавиковой кисло-
ты на гидроокись железа(Ш).
Известны кристаллогидраты FeF3-nH2O (где п = 9; 6; 4,5; 3)
в виде красноватых (FeF3‘3H2O), розовых (FeF3-4,5Н2О) и почти
бесцветных (FeF3-9H2O) кристаллов.
Известны ацидосоли общей формулы MeJ[FeF4] (где Me1 = К+,
NHJ), Mei[FeF6] (где Me1 = Li+, Na+, К+, Rb+, Cs+, NH^),
Mei![FeF6l2 (где Me11 = Ba2+), Mep[FeF6]2-6H2O (где Me11 = Cu2+),
Mei[Fe(H2O)F5] (где Mei = K+, Rb+, NH+), Mei[Fe(H2O)F5]-raH2O
520
ПОБОЧНАЯ ПОДГРУППА VIII ГРУППЫ
(где Me1 = Tl+, Ag+), Me11 [Fe(H2O)F5] (где Me11 = Sr2+, Ba2+)
и [Men(H2O)6][Fe(H2O)F5] (где Me“=Zn2+, Cd2+).
Димер трихлорида железа, Fe2Cl6 или Fe[FeCl6], получают дей-
ствием сухого хлора на металлическое железо (в виде порошка,
проволоки, стружки и др.) при 400—500°, на смесь Fe2O3 с углем
(400—450°), на расплав FeCl2 (500°), на пирит (900°), действием
S2C12, SOC12, СС14, СОС12, NH4C1, MgCl2 или РОС13 на Fe2O3 при
нагревании, действием SOC12, SO2C12 на металлическое железо
при нагревании, нагреванием пирита (марказита) FeS2 или миспи-
келя FeAsS с хлористым тионилом при 150° в закрытой трубке:
2Fe + ЗС12 = Fe2Cl6 + 192,08 ккал
Fe2O3 + ЗС12 + ЗС = Fe2Cl6 + ЗСО
2FeCl2 + С12 = Fe2Cl6
2FeS2 -|- 5С12 — Fe2Cle -j- 2S2C12
Fe2O3 + 3SOC12 = Fe2Cl6 + 3SO2
Fe2O3 + 3COC12 = Fe2Cl6 + 3CO2
Fe2O3 2POC13 = Fe2Cl6 -j- 1/2Р40ю
2Fe2O3 + 3CC14 = Fe2Cl6 + 3CO2
Соединение Fe2Cl6 представляет собой парамагнитные гигро
скопичные гексагональные темно-красные с зеленоватым оттен-
ком кристаллы с плотностью 2,898 г!см3 при 25°, т. пл. 306°,
т. кип. 316°. Оно гидролизуется водой, растворимо в жидком SO2,
жидком хлоре, AsCl3, PBr3, CS2, превращается в Fe2O3 с выделе-
нием хлора при нагревании на воздухе. В результате гидролиза
Fe2Cl6 образуется коллоидный раствор Fe(OH)3:
F.j2C16 + 6Н2О X 2Fe(OH)3 + 6НС1
При сильном нагревании Fe2Cl6 разлагается:
Fe2Cle 2FeCl2 + С12
При действии газообразного NH3 на Fe2Cl6 образуется гексам-
мин [Fe(NH3)6]Cl3, который разлагается водой.
По значению плотности паров хлорида железа(Ш) при 400—
446° ему соответствует формула Fe2Cl6, а при 750° в избытке хло-
ра — формула мономера FeCl3.
Методом дифракции электронов установлено, что расстояние
С1 zci С1
Fe — Cl в ,Fe. Fe равно 2,17 А.
C1Z ^С1/ 4 Cl
Fe2Cl6 экстрагируется из 7 н. НС1 амилацетатом или из <; 8 н.
раствора изопропиловым эфиром. Для экстракции Fe2Cl6 мож-
но использовать также амиловый спирт, бутилацетат и смесь
эфира с бутанолом.
Хлорид железа(Ш) является окислителем по отношению к KI,
SnCl2, H2S, NH4C1, Na2S2O3, N2H4, NH2OH, водороду, фосфору,
теллуру, магнию, кальцию, алюминию, железу и др.
Fe2Cl6 + 2KI = 2FeCl2.+ I2 + 2КС1
Fe2Cl6 + H2S = 2FeCl2 + 2HC1 + S
ЖЕЛЕЗО
521
Fe2Cl6 + SnCJ2 = 2FeCl2 + SnCl4
3Fe2Cl6 + 2NH4C1 = 6FeCl2 + 8HC1 + N2
Fe2Cl6 4- 2Na2S2O3 = 2FeCl2 + Na2S4Oe + 2NaCl
Окись CuO и сульфид Cu2S (которые часто встречаются в при-
роде) взаимодействуют с Fe2Cl6 по уравнениям
Fe2Cl6 + ЗСиО = Fe2O3 + ЗСиС12 2Fe2Cl6 + Cu2S = 4FeCl2 + 2СиС12 + S
Известны кристаллогидраты FeCl3-nH2O (где п = 12, 7, 5, 4),
ацидосоли общей формулы Me^[FeCl6] (где Me1 = Cs+, NH4, Tl+),
MeHFeClJ ->гН2О (где Me1 = Cs+, NH4+), Me* [Fe(H2O)Cl5] (где
Me1 = Cs+, NH;), Men[Fe(H2O)Cl5] (где Men=Be2+, Mg2+),
Ме}[Ре2С191 (где Me1 == Rb+, Cs+) и многочисленные аддукты, на-
пример, FeCl3'2С2Н5ОН, FeCl3 •(СгНУаО, 2FeCl3-ЗСнН^НгО
(с антипирином), FeCl3-4C5H5N (с пиридином), FeGl3-SC^H^Na •
• 2Н2О (с бензидином), FeCl3 -CgEUNHa (с анилином), FeCl3 •
•С6Н5 — СО — С6Н5 (с бензофеном), ВеС13-С6Н5 — СОС1 (с хлори-
стым бензоилом).
С увеличением содержания кристаллизационной воды цвет
кристаллогидратов хлорида железа(Ш) меняется от красного до
лимонно-желтого и температура плавления понижается.
Известна кислота H[FeCl4] -2Н2О, которая представляет со-
бой расплывающиеся на воздухе желтовато-коричневые пластин-
чатые кристаллы. Получено также соединение [Fe(H2N —СО —
NH2)6]C13.
Соль Fe2Cl6 обладает кровеостанавливающим действием и ис-
пользуется как катализатор (подобно А12С16) в реакциях Фриде-
ля — Крафтса. Она применяется также в качестве химического
реактива, а также при лечении анемии, для коагуляции коллоидов
в сточных водах и при получении некоторых органических кра-
сителей.
Смешанный хлорид железа Д1) и железаДП}, Fe3Cl8 ИОНгО
или 2FeCl3-FeCl2 -10Н2О, представляет собой желтые гексагональ-
ные кристаллы с т. пл. 45°.
Димер трибромида железа, Fe2Br6, получают прямым взаимо-
действием элементов при 170—200° в закрытой трубке или дей-
ствием брома на FeBr2:
2Fe + ЗВг2 = Fe2Br6 152 ккал 2FeBr2 + Вг2 Fe2Bre
Бромид железа представляет собой сильно расплывающиеся на
воздухе темно-красные гексагональные кристаллы с т. пл. 227°
и т. кип. 627° (их очищают сублимацией в токе паров брома).
Fe2Br6 используют как бромирующий агент, легко высвобождаю-
щий бром. Соединение Fe2Br6 растворяется в спирте, эфире и уксус-
ной кислоте, имеет окислительные свойства.
522
ПОБОЧНАЯ ПОДГРУППА VIII ГРУППЫ
Известен аддукт с антипирином Fe2Cl6-ЗСиИ^К^О и гексагид-
рат FeBr3-6H2O (т. пл. 27°), растворимый в спирте и эфире.
Известны ацидосоли общей формулы MeHFeBrJ, Me*[FeBr8]
(где Me1 = Rb+, Cs+). Также известны ацидоаквосоли общей
формулы Me*[Fe(H2O)Br3Cl2], Me*[Fe(H2O)Br2Cl3] (где Me1 =
= Rb+, Cs+).
При действии газообразного NH3 на Fe2Br6 образуется
[Fe(NH3)6]Br3, разлагающийся водой.
Смешанный бромид железа(11) и железа{111), Fe3Br8-16H2O или
2FeBr3-FeBr2-16Н2О, получают прямым бромированием железа
в виде чешуек в присутствии паров воды или термической дис-
социацией соединения Fe3Br8(CN)2 -SCNBr, которое получают по
уравнению
3FeBr2 + 5CNBr = Fe3Br8(CN)2-3CNBr
Безводная соль Fe3Br8 представляет собой коричневые моно-
клинные кристаллы, которые плавятся при 107—109° с разло-
жением.
Смешанный иодид железа(Н') и железа(Ш), Fe3I8 (или 2FeI3-
Fel2), получают обработкой порошка металлического железа
иодом; представляет собой черно-коричневое твердое вещество,
которое с К2СО3 образует KI по уравнению
Fe3Ig IK.2CO3 = 8KI + Fe3O4 4СО2
Гексацианоферрат ы(Ш) (ферроцианид ы),
MeJ[Fe(CN)6]2-иЩО (или безводные), Me|I[Fe(CN)6]2-иНаО (или
безводные), MeIMeII[Fe(CN)6] -иНгО, MeIn[Fe(CN)6] (где Me1 =
= Li+, Na+, К+, Rb+, NH4+, Tl+, Ag+, Hg2+, Me1* = Be2+, Mg2+,
Ca2+, Sr2+, Ba2+, Zn2+, Cd2+, Hg2+, Cu2+, Pb2+, Mn2+ и Me111 =
= Bi3+, Fe3+).
Известно также несколько гексацианоферратов(Ш) общей
формулы Me1Mep[Fe(CN)6]3-иНаО (или безводные) (где Me1 = К+,
Rb+ и Me11 — Zn2+, Cd2+), Me|MeiI[Fe(CN)6]5 (где Me1 = Cs+ и Me11 =
= Zn2+) и аддукты гексаметилентетрамина и гексацианоферра-
тов(Ш) общей формулы Men[Fe(CN)e]2 -(3-4)C6H12N4 • (18-20)Н2О
(где Меп=Са2+, Ва2+) и Me'Men[Fe(CN)6l-(3-4)С6[112А4-(4
или 5)Н2О (где Me1 = Li+, Na+, К+, NHJ и Me11 = Ва2+, Sr2+).
В общем случае гексацианоферраты(Ш) имеют красивую окраску
и меньшую растворимость, чем гексацианоферраты(П).
Гексацианоферраты(Ш) щелочных или щелочноземельных ме-
таллов легко растворимы в воде, а ферроцианиды остальных эле-
ментов мало растворимы в воде и имеют характерные цвета.
Гексацианоферрат калия, K3[Fe(CN)6], выделяют из концен-
трированных растворов, полученных в результате нейтрализации
H3[Fe(CN)6] гидроокисью калия, окислением K4[Fe(CN)6] или
K3NH4[Fe(CN)g] хлором, взаимодействием K4[Fe(CN)6] с избытком
ЖЕЛЕЗО
523
KFe[Fe(CN)8l, обработкой растворов солей железа(Ш) избытком
раствора KCN:
H3[Fe(CN)6] + ЗКОН = K3[Fe(CN)6] + ЗН2О
2K4[Fe(CN)6] + С12 = 2Ks[Fe(CN)6] + 2КС1
2K3NH4[Fe(CN)6] + PbO2 = 2K3[Fe(CN)6] + PbO + 2NH3 + H2O
K4[Fe(CN)6] + KFe[Fe(CN)6] = K3[Fe(CN)6] + K2Fe[Fe(CN)6]
Fe2Cl6 + 12KCN = 2K3[Fe(CN)6] + 6KC1
При обработке растворов солей железа(Ш) небольшим коли-
чеством KCN в результате гидролиза выпадает Fe(OH)3, который
в избытке KCN превращается в K3[Fe(CN)8]:
Fe2Cl6 + 6KCN + 6Н2О = 2Fe(OH)3 + 6КС1 + 6HCN
Fe(OH)3 + 6KCN = K3[Fe(CN)6] + ЗКОН
Соединение K3[Fe(CN)6] представляет собой токсичные пара-
магнитные оранжево-красные (или желтые при —190°) моноклин-
ные призмы с плотностью 1,85 г!см^ при 25°, твердостью 1—2 по
шкале Мооса; оно растворяется в воде и мало растворимо в жид-
ком SO2, гидразине и спирте. Это соединение окисляет соли
церия(Ш), аммиак, иодиды, щавелевую кислоту, гипофосфиты,
фосфиты, сульфиты, селеноводород, теллуроводород, арсин, сти-
бин и элементы As, Sb, Bi, Pb, Cu, Ag, Cd и Hg:
[Fe(CN)6]3- + Ces+ = [F (CN)ep- + Ce4+
12[Fe(CN)6]3- + 16NH3 = 12[Fe(CN)6]4- + 12NHJ + 2N2
pB=U-l
2[Fe(CN)6]3- + 2I- ---2[Fe(CN)6]<- + I2
pH=8
В щелочной среде K3[Fe(CN)8] восстанавливается до K4[Fe(CN)8]:
2K3[Fe(CN)6] + 2KOH = 2K4[Fe(CN)6] + H2O + V2O2
На основе этой реакции можно получить непрерывный ток
кислорода, обрабатывая твердый гексацианоферрат(Ш) калия
15—20%-ным раствором КОН или NaOH.
При обработке растворов солей железа(П) раствором
K3[Fe(CN)6] получают осадок, окрашенный в интенсивно-синий
цвет (соответствует формуле Fe3+[Fe3+(CN)8]2), называемый турн-
буллевой синью.
4Fe2+SO4 + 3K3[Fe3+(CN)6] = Fe2+[Fe3+(CN)e]2 + Fe3+K[Fe2+(CN)6] + 4K2SO4
Железосинеродистая кислота, H3[Fe(CN)6], получается дей-
ствием H2S или разб. H2SO4 на Pb3[Fe(CN)8]2-4Н2О, действием
конц. НС1, H2[SiF8] или винной кислоты НООС — СНОН —
СНОН — СООН на K3[Fe(CN)6], количественным окислением
H4[Fe(CN)8] кислородом, озоном, хлором, конц. HNO3, СаОС12,
524
ПОБОЧНАЯ ПОДГРУППА VIII ГРУППЫ
ВаО2, Н2О2, МетОС1, Me*S2O8, солями церия(1У) или анодным
окислением:
Pb3[Fe(CN)e]2 + 3H2S = 2H3[Fe(CN)6] + 3PbS
Pb3[Fe(CN)6]2 + 3H2SO4 = 2H3[Fe(CN)6] + 3PbSO4
K3[Fe(CN)6] + 3HC1 = H3[Fe(CN)6] + 3KC1
2K3[Fe(CN)6l + 3H2[SiF6] = 2H3[Fe(CN)6] + 3K2[SiF6]
2H4[Fe(CN)6] + V2O2 = 2H3[Fe(CN)6] + H2O
2H4[Fe(CN)6] + H2O2 = 2H3[Fe(CN)6] + 2H2O
Железосинеродистая кислота представляет собой парамагнит-
ные коричнево-желтые блестящие игольчатые кристаллы, которые
очень хорошо растворимы в воде и спирте, трудно растворимы
в эфире. Это соединение превращается в берлинскую лазурь под
действием света или в зеленую массу при нагревании.
H3[Fe(CN)6] сильно диссоциирует в воде (будучи сильной кисло-
той) и легко разлагается с выделением HCN, поскольку она менее
устойчива, чем H4[Fe(CN)6],
Известны следующие производные кислоты H3[Fe(CN)6]:
СН3 •H3[Fe(CN)5NO2]-Н2О, С2Н5 .H3[Fe(CN)5NO2] -2Н2О, С3Н7-
•H3[Fe(CN)5NO2b2H2O.
Пруссидные соединения, Me*[Fe3+(CN)5X], яв-
ляются координационными соединениями железа(Ш); они окра-
шены в фиолетово-красный или рубиново-красный цвет и содер-
жат пять координированных цианогрупп, в то время как шестое
координационное место занято другой группой, например NO,
NH3, Н2О, NO2, SO3 и др. Примеры пруссидных соединений:
Na2lFe(CN)5NOb2H2O, Ag2[Fe(CN)5NO], (NH4)2[Fe(CN)5NO],
K2[Fe(CN)5NObH2O, Ca[Fe(CN)5NO], Ba[Fe(CN)5NOb3 или 6H2O,
Zn[Fe(CN)5NOb8H2O, Cd[Fe(CN)5NO], Fe[Fe(CN)5NObnH2O,
Na2[Fe(CN)5NH3)].H2O, Na2[Fe(CN)5H2O], K2[Fe(CN)5H2O],
Na3[Fe(CN)]5NO2b2H2O, (CH3)3[Fe(CN)5NO2l, NaK2[Fe(CN)5NO2],
K3[Fe(CN)5NO2l.
Нитропруссид калия, K2[Fe(CN)5NO], получают действием
30%-ной HNO3 на гексацианоферрат(Ш) или (II) или действием
NaNO2 на растворы K4[Fe(CN)6],
K4[Fe(CN)6] + KNO2 + Н2О = K2[Fe(CN)5NO] + KCN + 2KOH
Соединение K2[Fe(CN)5NO] представляет собой гигроскопич-
ные токсичные красные кристаллы, устойчивые на воздухе; окис-
ляется в щелочном растворе под действием КМнО4, не окисляется
в нейтральной или кислой среде и реагирует (как и нитропруссид
натрия) с анионами S2~, HS~, окрашиваясь в фиолетово-красный
цвет.
K2[Fe(CN)5NO] + K2S = K4[Fe(CN)5NOS]
ЖЕЛЕЗО
525
Нитропруссиды щелочных и щелочноземельных металлов
растворяются в воде.
Тиоцианат железа, Fe(SCN)3-ЗН2О, выпадает при упаривании
в вакууме (над H2SO4) раствора, полученного нейтрализацией
HSCN свежеосажденной гидроокисью железа(Ш), или при обра-
ботке Ba(SCN)2 раствором сульфата железа(Ш).
Раствор тиоцианата железа получают обработкой растворов
солей железа(Ш) раствором тиоцианата в отсутствие комплекси-
рующих с железом компонентов, таких, как фториды, метафос-
фаты, пирофосфаты, фенолы, лимонная, винная, молочная и ща-
велевая кислоты.
Соединение Fe(SCN)3-ЗН2О представляет собой парамагнит-
ные красные с зеленым оттенком кубические кристаллы, которые
обладают неприятным металлическим вкусом, очень гигроскопич-
ны, легко растворяются в воде, эфире, спиртах (этиловом, амило-
вом, бензиловом) и жидком SO2 и трудно растворимы в CS2, С6Н6,
СНС13, СНВтз, СС14, С2Н5Вт и С6Н5СН3.
Для экстрагирования тиоцианата железа(Ш) из водного рас-
твора используют смесь амилового спирта с эфиром или смесь спир-
та, ацетона или этилацетата.
По значению молекулярного веса, определенного эбуллиоско-
пическим методом, для тиоцианата железа(Ш) была установлена
формула Fe[Fe(SCN)6] -6Н2О.
Известны аддукты Fe(SCN)3-(3 или 4)C5H5N, Fe(SCN)3-
• 3C9H7N, Fe(SCN)3.C6H12N4 (уротропин), Fe(SCN)3 •3C11H12ON2
(антипирин) и др.
Известны ацидосоли Li3[Fe(SCN)6] -nH2O, Na3[Fe(SCN)6] -12Н2О,
K3[Fe(SCN)eb4H2O, Cs3[Fe(SCN)6]-2Н2О, (NH4)3[Fe(SCN)6]-4H2O.
Сульфид железа, Fe2S3, получают действием сульфида аммония
на растворы солей железа(Ш) в щелочной среде, действием H2S
на водную суспензию Fe(OH)3 без доступа воздуха, нагреванием
Fe2O3 не выше 100° в атмосфере H2S.
Fe2Cle + 3(NH4)2S = Fe2S3 + 6NH4C1
Fe2S3 не образуется при пропускании H2S через кислые рас-
творы солей железа(Ш), поскольку в этих условиях образует-
ся FeS:
В кислой среде
Fe2Cl6 + 3H2S----------► 2FeS + S + 6HCl
Fe2S3 представляет собой ферромагнитное серовато-черное
аморфное вещество, которое при нагревании превращается в маг-
нитный пирит, при высушивании над Р4Ою становится пиро-
форным, во влажном состоянии разлагается на воздухе и раство-
ряется в разб. НС1:
Fe2S3 + ЗН2О + S/2O2 = 2Fe(OH)3 + 3S
Fe2Ss + 4HC1 = 2FeCl2 + 2H2S + S
526
ПОБОЧНАЯ ПОДГРУППА VIII ГРУППЫ
Известны двойные сульфиды общей формулы Me*S-Fe2S3 или
MerFeS2 (где Me1 = Na+, К+).
Сульфат железа, Fe2(SO4)3, получают прокаливанием (окисле-
нием или сульфированием) FeSO4, прокаливанием пирита (или
марказита), имеющего формулу FeS2, с NaCl на воздухе,
дегидратацией Fe2(SO4)3-9Н2О при температуре выше 175°, тер-
мическим разложением (NH4)2SO4-FeSO4 -6Н2О, окислением
FeSO4 азотной кислотой при 120—150°, нагреванием диметил-
сульфата с Fe2O3.
6FeSO4 -> Fe2(SO4)3 + 2Fe2O3 + 3SO2
6FeSO4 + 3/2O2 2Fe2(SO4)3 + Fe2O3
2FeSO4 + 2SO3 -> Fe2(SO4)3 + SO2
2FeS2 + 2NaCl + 8O2 -> Fe2(SO4)3 + Na2SO4 + Cl2
Соединение Fe2(SO4)3 представляет собой парамагнитный очень
гигроскопичный желтовато-белый кристаллический порошок с плот-
ностью 3,097 г!слС при 18°, который растворяется в воде или разб.
H2SO4 и трудно растворим в спирте. Это соединение разлагается
на Fe2O3 и SO3 при 600—773° и взаимодействует при нагревании
с различными окислами металлов (CuO, Сп2О, МепО, где Меп=
щелочноземельные металлы), с безводным СаС12, серой и др.
Fe2(SO4)3 + ЗСиО = Fe2O3 -|- 3CuSO4
Fe2(SO4)3 + ЗМепО = Fe2O3 + 3MenSO4
Fe2(SO4)3 + ЗСаС12 = Fe2Cl6 + 3CaSO4
Безводная соль Fe2(SO4)3, будучи диморфным соединением,
может существовать в виде белых орторомбических или желтых
гексагональных кристаллов.
При растворении Fe2(SO4)3 в воде образуются растворы,
которые меняют окраску от желтой до красновато-коричневой
вследствие гидролиза и разрушения под действием света. Водные
растворы сульфата железа(Ш) можно получить растворением
Fe2O3 или Fe(OH)3 в конц. H2SO4 при нагревании или окислением
сульфата железа(П) азотной кислотой.
Насыщенный водородом палладий, платиновая чернь, металлы
Mg, Al, Sn, Hg и фосфорноватистая кислота Н6Р2О4 действуют
как восстановители по отношению к сульфату железа(Ш) в вод-
ном растворе.
Fe2(SO4)3 взаимодействует в водном растворе с сульфидами,
иодидами и окислами меди, причем медь переходит в раствори-
мый сульфат меди:
Cu2S + 2Fe2(SO4)3 = 4FeSO4 + 2CuSO4 4- S
CuS + Fe2(SO4)3 = 2FeSO4 + CuSO4 + S
2(CuI + V2I2) + 2Fe2(SO4)3 = 4FeSO4 + 2CuSO4 + 2I2
3Cu2O -f- 4Fe2(SO4)3 = 6FeSO4 -{- Fe2O3 -f- 6CuSO4
ЖЕЛЕЗО
527
Известны кристаллогидраты Fe2(SO4)3-иЩв (где п =12, 10,
9, 7, 6, 4, 3, 1), аммиакат Fe2(SO4)3-12NH3, квасцы МеФе(8О4)2*
• 12Н2О (где Me1 = Na+, К+, Rb+, Cs+, NH+, Т1+), основные суль-
фаты 2Fe2O3-SO3, Fe2O3-SO3, Fe2O3*2SO3, Fe2O3 -2SO3-15H2O
и комплексные сульфаты общей формулы Me*[Fe(SO4)3] -ЗН^О,
Men[Fe(SO4)2]2-иН2О (где Mei = Na+, К+ и Me" = Ва2+).
В природе встречается Fe2(SO4)3 "ЭНгО в виде минерала коким-
бита. В зависимости от температуры Fe2(SO4)3-9Н2О дегидрати-
руется следующим образом:
Fe2(SO4)3.9H2O -Fe2(SO4)3-4H2O —
— ЭЛ-2О — оП2И
175°
-* Fe2(SO4)3-H2O -—Fe2(SO4)3
— rl2U
Сульфат железа применяют при декопировании нержавеющих
аустенитных сталей или сплавов меди, как химический реактив
при гидрометаллургической переработке медных руд, при коа-
гуляции и осаждении загрязнений воды, как окислитель в различ-
ных реакциях и катализатор в многочисленных синтезах.
Нитрат железа, Fe(NO3)3-9Н2О, выпадает при растворении
металлического железа в HNO3 (d — 1,29 г/см3) и последующем
упаривании полученного раствора (40—45° при нормальном дав-
лении или в вакууме при обычной температуре). Нитрат железа
представляет собой фиолетовые (аметистовые) орторомбические
призмы, растворимые в воде и эфире.
Кристаллы Fe(NO3)3-9Н2О хранятся в герметических склян-
ках, поскольку поглощают влагу из воздуха.
Гексагидрат, Fe(NO3)3-6Н2О, представляет собой парамагнит-
л ные расплывающиеся кубические кристаллы, известные как драго-
( ценный камень аметист, с плотностью 1,68 г/см3, т. пл. 47,2°
и т. кип. 125,1°. Гексагидрат дегидратируется при нагревании
выше 100°, растворим в воде, спирте и ацетоне.
Существование кристаллогидратов Fe(NO3)3-ЗН2О, Fe(NO3)3-
•Н2О не было подтверждено.
Известны основные нитраты различного состава, например,
Fe(NO3)2OH (бесцветный), FeNO3(OH)2 (бесцветный), Fe2O(NO3)4
(желтый), а также гидронитрат 2Fe(NO3)3-2HNO3-17Н2О и двой-
ные нитраты, например CsNO3 -Fe(NO3)3 -7Н2О.
Ортофосфат железа, FePO4-(2 или 2,5)Н2О, образуется в виде
желтого осадка при обработке растворов солей железа(Ш) гидро-
фосфатом натрия в присутствии ацетата натрия:
Fe2Cl6 + 2Na2HPO4 + 2NaCH3COO = 2FePO4 + 6NaCl + 2CH3COOH
Соединение FePO4-(2 или 2,5)H2O растворяется в минераль-
ных кислотах и трудно растворимо в уксусной кислоте.
528
ПОБОЧНАЯ ПОДГРУППА VIII ГРУППЫ
Известны комплексные ортофосфаты NaH2[Fe(PO4)2] -ЗН2О,
NaH5[Fe(PO4)3]-Н2О, гидроортофосфаты Fe(H2PO4)3, FeH3(PO4)2-
•2,5Н2О, основные ортофосфаты 2FePO4-Fe(OH)3-2,5Н2О и двой-
ные ортофосфаты 2FePO4-KgPO^
Ацетаты железа. При обработке растворов солей
железа(Ш) при обычной температуре раствором ацетата щелоч-
ного металла (или ацетата аммония) образуется растворимый
покрашенный в коричнево-красный цвет полиядерный комплекс —
ацетат ди-ц-гидроксогекса(ацетато)трижелеза(Ш):
3Fe2Cl6 + 18NaCH3COO + 4Н2О =
= 2[Fe3(CH3COO)6(OH)2]CH3COO + 4СН3СООН + 18NaCl
При разбавлении и кипячении раствора полиядерного ацетата
железа(Ш) образуется основной ацетат железа в виде красновато-
коричневого осадка:
[Fe3(CH3COO)6(OH)2]CH3COO + Н2О 3Fe(OH)2CH3COO + 4СН3СООН
В присутствии большого количества ионов Сг3+ эта реакция
затруднена, а в присутствии малых количеств ионов Сг3+ послед-
ние соосаждаются с железом.
Известны полиядерные комплексные ацетаты
[Fe3(CH3COO)6OH(H2O)](CH3COO)2, [Fe3(CH3COO)6(H2O)2] (СН3СОО)3
и были получены полиядерные комплексы, содержащие катионы
[Fe2Cr(CH3COO)6(OH) 2]+, [FeCr2(СН3СОО)6(ОН) 2]+.
Известны также следующие полиядерные комплексы:
Mg[Fe3(CH3COO)8(OH)3] -10Н2О, Zn[Fe3(CH3COO)8(OH)3] -5Н2О,
Cd[Fe3(CH3COO)8(OH)3] -7Н2О, Mn[Fe3(CH3COO)8(OH)3] -5Н2О.
Хелатные соединения
При действии ацетилацетона на свежеприготовленный Fe(OH)3
или водный раствор Fe2Cl6, содержащий ацетат натрия, образует-
ся ацетилацетонат железа(Ш).
Соединение [Fe(C5H7O2)3] представляет собой парамагнитные
двупреломляющие гранатово-красные призмы с плотностью
1,33 г!см3, которые плавятся при 184°, мало растворимы в воде,
растворимы в органических растворителях (С2Н5ОН, С6Н6,
СНС13, СН3СОСН3), устойчивы в кислотах и разлагаются при
кипячении со щелочами.
ЖЕЛЕЗО
529
При обработке растворов солей железа (III) 0,5%-ным водным
раствором о-фенантролина образуется хелатное соединение, окра-
шенное в синий цвет и содержащее катион
При обработке растворов солей железа(Ш) 6%-ным раствором
купферона 1 (нитрозофенилгидроксиламин) получают коричнево-
красное хелатное соединение, которое трудно растворимо в воде
и кислотах, растворяется в эфире, разлагается под действием
щелочей и отвечает формуле
.•ТТОЙ
Феррон (7-иодо-8-оксихинолин-5-сульфоновая кислота) при
pH ~ 2,6 образует с растворами солей железа(Ш) зеленый хелат,
который растворим в воде и отвечает формуле
При действии салициловой кислоты (НО — С6Н4 — СООН) на
растворы солей железа(Ш) образуются хелатные соединения,
окрашенные в зависимости от значения pH раствора в различные
цвета. При pH ~ 1 образуется фиолетовый хелат, который содер-
жит катион
Те—ООС
О
34-0101
530
ПОБОЧНАЯ ПОДГРУППА VIII ГРУППЫ
при pH ~ 4 образуется оранжево-красный хелат
при pH 9 образуется желтый хелат, который содержит анион
При обработке растворов солей железа(Ш) сульфосалицило-
вой кислотой (2-окси-5-сульфобензойной кислотой) HO3S —
С6Н3(ОН)СООН при pH ~ 2 образуется пурпурно-красный, при
pH ~ 5 — коричневый и при pH ~ 10 — желтый хелаты. Фор-
мулы этих хелатов, вероятно, аналогичны хелатам железа с сали-
циловой кислотой.
Растворы солей железа(Ш) образуют с оксином (8-оксихиноли-
ном) зеленовато-черный хелат, который трудно растворим в воде
и отвечает формуле [Fe(C9H6ON)3].
Хелатные соединения были получены также обработкой рас-
творов солей железа(Ш) пирокатехином, бензоилацетоном, метил-
бензоилацетатом, этилбензоилацетатом, диметилглиоксимом,
дифенилкарбазидом, дитизоном, гуанидином и этилендиаминтетра-
уксусной кислотой.
Помимо приведенных выше известны и другие соединения железа(Ш),
например: двойные хлориды 3CsCl-Ре2С16, Fe2Cl6-2РС15, Fe2Cl6-ЗРОС13,
хлорбромид FeBrCl2, хлориодид FeICl2, полииодид Fel5, хлорат Fe(C103)3
(известен только в растворе), перхлораты Fe(C104)3-(9 или 10)Н2О,
Ме1[Ре(С1О4)4]-гаНгО (где Mei = Na+, К+), иодат Fe(IO3)3, перйодаты
LiKFeIO6, FeH2IO6-4H2O, Fe(IO4)3, кислота H[Fe(H2O)(SO4)2]-3H2O и ее
соль (C2H5)[Fe(H2O)(SO4)2]-Н2О, хлорсульфат FeSO4Cl-6Н2О, двойные
сульфаты общей формулы Me2SO4•Fe2(SO4)3-nH2O, Mell SO4-Fe2(SO4)3.
• 24H2O (где Mei = K+, Cs+ и Mell = Mg2+, Zn2+), безводные двойные
сульфаты (NH4)2SO4-Fe2(SO4)3, 3(NH4)2SO4 •Fe2(SO4)3, сульфитосульфаты
Na3Fe(SO3)2SO4-6H2O, KFeSO3SO4, K3Fe(SO3)2SO4, K4Fe2(SO3)4SO4 -5H2O,
селенит Fe2(SeO3)3-nH2O (где n = 10, 9, 7, 4, 3, 1), сульфатоселенат
K2SO4-Fe2(SeO4)3 •24H2O, двойные селенаты MelSeO4-Fe2(SeO4)3-24H2O,
теллурит Fe2(TeO3)3-4H2O, азид Fe(N3)3, гипофосфиты [Fe3(H2PO2)6]•
• (H2PO2)3, [Fe3(H2PO2)6OH](H2PO2)3NO3-24H2O, Mel [Fe(H2O)(H2PO2)5]
(где Mei = Na+, K+), гипофосфат Fe4(P2O6)3-20H2O, пирофосфаты Fe4(P2O7)3,
KFeP2O7, Fe2H6(P2O7)3-7,5H2O, Na6Fe2(P2O7)3-9H2O, Na8Fe4(P2O7)5-14H2O,
Cu3Fe2(P2O7)3-12H2O, метафосфаты Fe2P60i8, Na3FeP6O18, Ag2FeP5O15, арсе-
нит FeAsO3, арсенаты FeAsO4, FeAsO4-raH2O (где n = 4, 2, 1), FeH3(AsO4)2-
• 2,5H2O, K3Fe2(AsO4)3, Ba3H6[Fe(AsO4)3]2, 6FeAsO4.2Fe(OH)3-12H2O,
KFeAs2O7, FeAsO4-NH4H2AsO4, метаантимонат FeSb3O9-6,5H2O, ортоантимо-
нат FeSbO4 -3,5H2O, галогеносоли Fe[SbCl8] -8H2O, Fe[SbBr6]3 -14H2O, тиокар-
бонат Fe2(CS3)3, титанаты Fe4(TiO4)3, Fe2Ti3O9, силикаты KFeSi2O6, KFeSi3O8,
хроматы MeiFe(CrO4)2 (где Mei = K+, NHJ), молибдат Fe2(MoO4)3-42H2O,
фосфо-12-молибдат Fe[PMo12O40]-58H2O, силико-12-молибдат Fe4[SiMo12O40]3-
ЖЕЛЕЗО
531
• 93Н2О, силико-12-вольфрамат Fe4[SiW12O40]3-93Н2О, дитиокарбамат
Fe(CS2NR2)3, ксантогенат Fe(CS2OC2H5)3, этилат Fe(OC2H5)3, тиоэтилат
Fe(SC2H5)3, формиаты Na3[Fe(HCOO)6], [Fe3(HCOO)6(OH)2]HCOO-4Н2О,
оксалаты Fe2(C2O4)3-5Н2О, Ме3[Ре(С2О4)3] (где Mei = Li+), Me|[Fe(C2O4)3]-
• пН2О (где Mei = Na+, К+, Rb+, NH4, T1+), MeJ [Fe(C2O4)3]2-nH2O (где
Mei = Na+, NH4), Mei[Fe(H2O)2(C2O4)2]-nH2O (где Mel = Na+,1 K+),
Мер [Fe(C2O4)3]2-nH2O (где Men = Ra2+), Fe2(C2O4)3-2Fe(OH)3-4H2O, тар-
траты Fe(C4H5O6)3 -5H2O, Na[FeC4H2O6]-5H2O, K[FeC4H2O6]-0,5H2O,
Me|[Fe(C4H4O6)3]-0,5H2O (где Mei = Na+, K+, NHJ), MeJ [Fe(C4H3O6)3]
(где Mei = Na+, NHJ), цитраты (NH4)3H3[Fe2(C6H4O7)3],
[Fe3(C6H5O7)2(OH)2(H2O)]3 -CeH5O7 -18H2O.
Соединения четырехвалентного железа
Известно очень небольшое число соединений, в которых элек-
троположительное четырехвалентное железо находится в составе
аниона [Fe4+O4]4~.
В результате окисления при 800—900° смеси гексагидроксо-
ферратов и гидроокислов стронция и бария образуются твердые
ферраты Sr2FeO4 и Ba2FeO4 коричневого цвета:
Me3I[Fe3+(OH)6]2 + Меп(ОН)2 + V2O2 = 2Me^Fe4+O4 + 7Н2О
Соединения шестивалентного железа
Железо в шестивалентном состоянии входит в состав ферратов,
которые содержат анион [FeO4]2-.
Ферраты общей формулы MeiFeO4 (где Me1 =Na+, К+, 1/2Ва2+)
получают окислением соединений железа(Ш) в щелочной среде.
Феррат калия, K2FeO4, пурпурно-красного цвета, изоморфен
К2СгО4, К2МоО4 и K2SO4; образуется при действии хлора на сус-
пензию Fe(OH)3 в концентрированном растворе КОН, при сплав-
лении Fe2O3 с окислительно-щелочной смесью и при анодном
окислении металлического железа в концентрированном раство-
ре КОН:
2Fe(OH)3 -f- ЗС12 -|- ЮКОН = 2K2FeO4 6КС1 8Н2О
Fe2O3 -f- 3KNO3 -|- 2К2СО3 = 2K2FeO4 -|- 3KNO2 2СО2
Fe2O3 + 3KNO3 + 4КОН = 2K2FeO4 + 3KNO2 + 2H2O
Fe2O3 + KC1O3 + 4KOH = 2K2FeO4 + KC1 + 2H2O
Феррат бария, BaFeO4*H2O, образуется в виде фиолетового
осадка при обработке K2FeO4 концентрированным раствором хло-
рида бария. Соединение BaFeO4-H2O хранят без доступа влаги,
поскольку оно ступенчато разлагается во влажном воздухе. При
нагревании выше 120° BaFeO4-H2O разлагается по уравнению
2BaFeO4-H2O = Fe20s -f- 2ВаО -J- 2Н2О -J- 3/2О2
34*
532
ПОБОЧНАЯ ПОДГРУППА VIII ГРУППЫ
Как правило, ферраты имеют окраску от пурпурно-красной до
фиолетовой, проявляют сильные окислительные свойства в кислой
среде (окисляют аммиак на холоду до элементного азота) и способ-
ны разлагаться при нагревании с выделением кислорода:
2[FeO4]2- + 10Н+ = 2Fe3+ + :t/2O2 + 5Н2О
В водной среде ферраты щелочных металлов неустойчивы
и разлагаются по уравнению
4Na2FeO4 + ЮН2О = 4Fe(OH)3 + 8NaOH + ЗО2
Кислота H2FeO4 и ее ангидрид FeO3 не известны, поскольку
при подкислении растворов ферратов шестивалентное железо вос-
станавливается до трехвалентного состояния и выделяется кис-
лород.
Металлоорганические соединения
Карбонильные соединения железа. Изве-
стны моно- и полиядерные карбонильные соединения железа.
В качестве примеров моноядерных карбонильных соединений
железа можно привести пентакарбонил железа Fe(CO)5, тетракарбо-
нилгидрид железа(П) H2Fe(CO)4, тетракарбонилферраты(П)
общей формулы Me*[Fe(CO)4], Men[Fe(CO)J, MeI[HFe(CO)4],
Men[HFe(CO)4]2, галогениды карбонилжелеза Fe(CO)5X2,
Fe(CO)4X2, Fe(CO)2X2, Fe(CO)2X. В качестве примеров полиядер-
ных карбонильных соединений железа можно назвать эннеакарбо-
нил дижелеза Fe2(CO)9, додекакарбонил трижелеза Fe3(CO)12
с многочисленными их производными и галогениды карбонил-
железа общей формулы Fe3(CO)gX6.
Пентакарбонил железа, Fe(CO)5, получают действием окиси
углерода на порошкообразное металлическое железо при давле-
нии 200 ат и температуре 150—200°, действием окиси углерода на
Fel2 (или на FeS) в присутствии металлической меди при давле-
нии 200 ат и температуре 200°, а также разложением Fe(CO)4I2
или Fe2(CO)9:
Fe 4- 5СО Fe(CO)5
5Fe(CO)4I2 — 4Fe(CO)6 + Fel2 + 4I2
Fel2 + 5CO + 2Cu = Fe(CO)5 -f- 2CuI
3Fe2(CO)g —► 3Fe(CO)6 -|- Fe3(CO)i2
Соединение Fe(CO)5 представляет собой маслянистую светло-
желтую диамагнитную жидкость с плотностью 1,4664 г!см3, которая
затвердевает при —20° (в виде почти бесцветных моноклинных игл),
кипит при 103°, обладает антидетонационными свойствами. Оно
разлагается на металлическое железо и СО при нагревании до
200—250° без доступа воздуха, растворяется в спирте, эфире, аце-
ЖЕЛЕЗО
533
тоне, бензоле, уксусной кислоте, разлагается водой, превра-
щается в Fe2(CO)9 под действием ультрафиолетовых лучей при
обычной температуре.
2Fe(CO)6 Fe2(CO)9 + СО
При сжигании паров Fe(CO)5 на воздухе образуется тонкий
порошок чистого Fe2O3. В эфирном растворе Fe(CO)5 энергично
взаимодействует с конц. H2SO4 при обычной температуре:
Fe(CO)5 +jH2SO4 == FeSO4 + 5СО + Н2
Обработка галогенами (при низкой температуре) растворен-
ного в неводных растворителях Fe(CO)5 дает неустойчивые соеди-
нения общей формулы Fe(CO)5X2, которые самопроизвольно выде-
ляют СО, превращаясь в устойчивые галогениды тетракарбонила
железа Fe(CO)4X2.
В обычных условиях Fe(CO)5 образует аддукты с аммиаком,
пиридином и этилендиамином Fe(CO)5-NH3, Fe(CO)5 -CsHsN,
Fe(CO)5 -Еп. В присутствии пиридина при 60° и под действием
аммиака Fe(CO)5 превращается в Fe3(CO)8(NH3)6.
Пентакарбонил железа взаимодействует со многими галогени-
дами металлов по уравнениям
Fe(CO)5 + 2СС14 = FeCl2 + С2С16 4- 5СО
Fe(CO)5 + SO2C12 = FeCl2 + SO2 + 5CO
Fe(CO)5 + SnCl4 = Fe(CO)4Cl2 + SnCl2 + CO
Fe(CO)5 + SbCl5 = Fe(CO)4Cl2 + SbCl3 + CO
Между Fe(CO)4Cl2 и SnCl2 или SbCl3 образуются аддукты
Fe(CO)4Cl2 .SnCl2, Fe(CO)4Cl2 -SbCl3.
По отношению к солям катионов Ag+, Hg9+, Cu2+, Fe3+ или
к некоторым органическим красителям Fe(CO)5 является вос-
становителем.
При действии HgCl2 на растворенный в ацетоне Fe(CO)5 обра-
зуется Hg[Fe(CO) 4] -HgCl2:
Fe(CO)5 + 2HgCl2 + Н2О = Hg[Fe(CO)4]-HgCl2-i- 2HC1 + CO2
При действии растворенных в жидком аммиаке щелочных
металлов на Fe(CO)5 образуются тетракарбонилферраты (II) общей
формулы Me*Fe(CO)4:
Fe(CO)6 + 2Na = Na2Fe(CO)4 + CO
Обработкой Fe(CO)4 трифенилфосфином (C6H5)3P получают
(C6H5)3PFe(CO)4 или [(C6H5)3P]2Fe(CO)3.
Сильные основания, какими являются гидроокислы щелочных
и щелочноземельных металлов, превращают Fe(CO)5 в H2Fe(CO)4
по уравнению
Fe(CO)6 + Ва(ОН)2 = H2Fe(CO)4 + ВаСО3
534
ПОБОЧНАЯ ПОДГРУППА VIII ГРУППЫ
и
и
С
С
II
о
Рис. 27. Структура Fe(CO)5.
для получения Fe2O3 — пигмента,
Перекись водорода и КМпО4 окисляют Fe(CO)5 в ацетоновом
растворе до коллоидного Fe2O3, а хлорная или бромная вода
превращают Fe(CO)5 в галогениды железа(П).
Пентакарбонил железа образует л-комплексы с циклопентадие-
ном С5Н6 при 300°, с диметилацетиленом СН3 -С = С -СНз, с бута-
диеном СН2 = СН -СП = СН2
ит. д., например ферро-
цен Fe(C5H5)2, Fe2(CO)3(C5H5)2,
Fe(CO)3C4H6.
По данным рентгенострук-
турного анализа и дифракции
электронов было установлено,
что Fe(CO)5 обладает бипира-
мидальной тригональной струк-
турой (рис. 27).
Fe(CO)5 применяют для
приготовления гранулирован-
ного металлического железа,
которое используется в ка-
честве катализатора, а также
применяемого для приготовле-
ния красок.
Эннеакарбонил железа, Fe2(CO)9, получают при облучении
ультрафиолетовым светом Fe(CO)5, растворенного в эфире или
уксусной кислоте, при обычной температуре или разложением
Fe(CO)4Br2 под действием света:
2Fe(CO)6 Fe2(CO)9 + СО
6Fe(CO)4Br2 2Fe2(CO)9 + Fe2Br6 + 3Br2 + 6СО
Fe2(CO)9 представляет собой диамагнитные блестящие золоти-
стые триклинные кристаллы с плотностью 2,085 г/см? при 18°,
которые растворяются в спирте, ацетоне, толуоле, пиридине, труд-
но растворимы в воде, эфире, бензоле. Это соединение очень чув-
ствительно к действию кислорода воздуха при обычной температуре
и разлагается при 90—100° по уравнению
3Fe2(CO)9 3Fe(CO)6 -|- Feg(CO)i2
Эннеакарбонил железа взаимодействует с меркаптанами,
с растворами щелочей, с ацетиленовыми производными и др.
Fe2(CO)9 -|- 2HSC2H5 — Fe2(CO)e(SC2H5)2 4- ЗСО -|- Н2
Fe2(CO)9 + 4ОН- = [Fe2(CO)8]2“ + СОВ" + 2Н2О
При обычной температуре и в темноте Fe2(CO)9 связывает СО,
превращаясь в Fe(CO)5. При действии окиси азота на Fe2(CO)9
(70—85°) образуется FeNO "3Fe(CO)5 и выделяется СО.
ЖЕЛЕЗО
535
Рис. 28. Структу-
ра Fe2(CO)9.
Действие о-фенантролина на раствор Fe2(CO)9 в ацетоне
при 80° приводит к образованию [FePhen3][Fe(CO)8], а на
раствор Fe2(CO)B в пиридине при повышенной температуре —
[FePhen3][Fe4(CO)13].
По данным рентгеноструктурного анализа было установлено,
что в молекуле Fe2(CO)9 два атома железа связаны между собой
через три СО-группы и к каждому атому железа присоединены
еще три СО-группы (рис. 28). Вероятно существование связи
и непосредственно между обоими атомами
железа, расстояние между которыми состав-
ляет всего 2,46 А.
Додекакарбонил железа, Fe3(CO)12, полу-
чают разложением растворенного в толуоле
Fe2(CO)9 при 70° в атмосфере СО2, разложением
Fe(CO)5 в присутствии NaOH или аммиака,
окислением тетракарбонилдигидрид железа(П)
H2Fe(CO)4 перекисью водорода Н2О2 или МиО2:
3Fe2(CO)9 = Fe3(CO)12 -|- 3Fe(CO)3
3H2Fe(CO)4 -f- 3H2O2 = Fe3(CO)i2 6H2O
3H2Fe(CO)4 -f- 3MnO2 -f- 3H2SO4 = Fe3(CO),2 -f-
+3MnSO4 + 6H2O
Fe3(CO)i2 представляет собой диамагнит-
ные темно-зеленые моноклинные кристаллы с
плотностью 1,996 г'см? при 18°, которые растворяются в спирте,
эфире, пиридине, ацетоне, толуоле, этилацетате и др. Это соеди-
нение разлагается на металлическое железо и СО при нагревании
выше 140° и превращается в Fe2(CO)7Br4 под действием брома.
Додекакарбонил железа реагирует с пиридином, метанолом,
щелочными металлами, растворенными в жидком NH3, ацетиле-
новыми производными, тиоэфирами.
2Fe3(CO)12 -|- 3C5H5N — Fe3(CO)9(C5H5N)3 -f- 3Fe(CO)3
2Fe3(CO)12 + ЗСН3ОН = Fe3(CO)9(CH3OH)3 + 3Fe(CO)5
Fe3(CO))2 -|- 6Na = Na6Fe3(CO)j2
При действии конц. H2SO4 на Fe3(CO)12 при нагревании обра-
зуется FeSO4 и выделяются СО и Н2.
Кристаллическая решетка Fe3(CO)i2 состоит из тригональных
бипирамид, соединенных по три. Атомы железа находятся в цен-
тре каждой бипирамиды, а группы СО — в вершинах трех триго-
нальных бипирамид.
Тетракарбонилдигидрид желеааДГ), H2Fe(CO)4, получают
действиемFe(CO)5 на концентрированные растворы щелочей (NaOH,
КОН или Ва(ОН)2) в вакууме, подкислением раствора соединения
[FeEn3HFe(CO)4] или действием окиси углерода на
536
ПОБОЧНАЯ ПОДГРУППА VIII ГРУППЫ
[Ni(NH3)6l[HFe(CO)4l2:
Fe(CO)5 + Ва(ОН)2 = H2Fe(CO)4 + ВаСО3
[FeEn3][Fe(CO)4] + 2НС1 = H2Fe(CO)4 + FeCl2 + ЗЕп
3[Ni(NH3)6][HFe(CO)4]2 + 12CO =
= 3H2Fe(CO)4 4" Fe3(CO)i2 4- 3Ni(CO)4 4" 18NH3
Соединение H2Fe(CO)4 — неприятно пахнущая светло-желтая
жидкость, устойчивая ниже —10°, затвердевающая при —70°.
Выше —10° вещество разлагается с выделением водорода, пента-
карбонила железа и полиядерных карбонилов железа. Под дей-
ствием кислорода жидкость самопроизвольно загорается.
Соединение H2Fe(CO)4 растворяется в воде, пиридине и жидком
аммиаке, обладает восстановительными свойствами и превращает-
ся в Fe3(CO)i2 под действием мягких окислителей (смесь МпО2
с H2SO4) или в Fe(OH)3, СО2 и СО под действием энергичных окис-
лителей (Н2О2).
3H2Fe(CO)4 ЗМпО2 4- 3H2SO4 = Fe3(CO)j2 4- 3MnSO4 4- 6Н2О
2H2Fe(CO)4 4- 5Н2О2 = 2Fe(OH)3 4- 8СО 4- 4Н2О
При действии пиридина или о-фенантролина на H2Fe(CO)4
образуются соли [C5H5NH]2[Fe(CO)4], [PhenH2]Fe(CO)4l.
При полном или частичном замещении водородов H2Fe(CO)4
различными одно- или двухвалентными катионами Na+, К+, Сн+,
Ag+, Mn2+, Са2+, Ва2+, Zn2+, Cd2+, Hg2+, [Men(NH3)e]2+ (где Меп =
= Mn2+, Fe2+, Co2+, Ni2+) образуются тетракарбонилферра-
ты (II) общей формулы Me*Fe(CO)4, MenFe(CO)4, MeHHFe^OJJ,
Me11 [HFe(CO)4]2:
H2Fe(CO)4 4- 2KNH2 = K2Fe(CO)4 + 2NH3
H2Fe(CO)4 4- Bal2 4~ 2NH3 = BaFe(CO)4 4- 2NHJ
2H2Fe(CO)4 4- [Men(NHs)6]SO4 = [Men(NH3)6][HFe(CO)4]2 4- H2SO4
H2Fe(CO)4 4- HgCl2 = HgFe(CO)4 4- 2HC1
H2Fe(CO)4 4- 2HgCl2 = HgFe(CO)4-HgCl2 -|- 2HC1
H2Fe(CO)4 4- CdCl2 = CdFe(CO)4 4- 2HC1
HgFe(CO)4 — очень устойчивое твердое вещество желтого цве-
та; оно трудно растворимо в воде и многочисленных органических
растворителях, кипит при 101°, разлагается на Hg, Fe, СО при
нагревании до 150°, образует аддукты с HgCl2 или Hgl2, взаимо-
действует с иодом при комнатной температуре.
HgFe(CO)4 4- 2I2 = Fe(CO)4I2 4- Hgl2
Известны многочисленные аддукты тетракарбонилферратов(П),
например CdFe(CO)4-2NH3, ZnFe(CO)4-3NH3, Cu2Fe(CO)4"2NH3,
CdFe(CO) 4 -2C5H5N, Ag2Fe(CO) 4 .C12H8N2.
ЖЕЛЕЗО
537
При замещении водородов H2Fe(CO)4 хлором, бромом или иодом
образуются галогениды тетракарбонила железа общей формулы
Fe(CO)4X2 (где X = С1-, Вт-, 1~).
H2Fe(CO)4 ” Fe(CO)4I2 -J- Н2, или
H2Fe(CO)4 2I2 — Fe(CO)4I2 -f- 2HI
HgFe(CO)4 + 2I2 = Fe(CO)4I2 + Hgl2
При взаимодействии H2SO3, H2SeO3, H2TeO3 c H2Fe(CO)4
выделяются соединения общей формулы Fe3(CO)2X9 (где X =
= S2-, Se2-, Те2-); они относительно устойчивы и диамагнитны.
При действии ацетилена на H2Fe(CO)4 в щелочных растворах
образуется Fe2C10OgH4.
Галогениды карбонилжелеза. Хорошо извест-
на группа дигалогенидов тетракарбонилжелеза Fe(CO)4X2; гало-
гениды карбонилжелеза типов Fe(CO)2X2, Fe(GO)2X, Fe(CO)5X2,
Fe3(CO)9X6 известны в меньшей степени.
Дииодид тетракарбонилжелеза, Fe(CO)4I2, получают действием
на холоду иода на Fe(CO)5, H2Fe(CO)4 или HgFe(CO)4 и действием
СО на Fel2 в эфирном растворе при давлении 110 ат:
Fe(CO)5 + I2 = Fe(CO)4I2 + СО H2Fe(CO)4 + 2I2 = Fe(CO)4I2 -j- 2HI
Fel2 + 4СО Fe(CO)4I2 + 38,94 ккал
HgFe(CO)4 + 2I2 = Fe(CO)4I2 + Hgl2
Fe(CO)4I2 представляет собой красно-коричневый порошок,
который сублимируется в вакууме и растворяется в неполярных
органических растворителях. Это соединение разлагается на Fel2
и СО водой или пиридином в избытке при обычной температуре
и реагирует с SOC12, хлором, амальгамами щелочных метал-
лов и т. д.
Fe(CO)4I2 + 2SOC12 = Fe(CO)2Cl2 + I2 + 2CO + SO2 + SC12
Fe(CO)4I2 + 4C12 = Fe(CO)4Cl2 + 2IC13
Fe(CO)4I2 + 4Na = Na2Fe(CO)4 + 2NaI
При нагревании Fe(CO)4I2 в атмосфере водорода образуется
Fe(CO)2I2, а в токе СО2 — Fe2I6 и Fe(CO)2I2.
Действием пиридина или о-фенантролина на Fe(CO)4I2 получа-
ют продукты замещения Fe(CO)2(C5H5N)2I2, Fe(CO)2(Ci2HgN2)2I2.
Хлорид и бромид тетракарбонилжелеза, Fe(CO)4Cl2
и Fe(CO)4Br2, окрашены в желтый и красновато-коричневый цвет;
они малоустойчивы, диамагнитны и образуются на холоду по
уравнениям
FeCl2 + 4СО Fe(CO)4Cl2 -f- 17,86 ккал
FeBr2 -|- 4CO Fe(CO)4Br2 -|- 28,30 ккал
538
ПОБОЧНАЯ ПОДГРУППА VIII ГРУППЫ
Известны также смешанные галогениды Fe(CO)4ICl, Fe(CO)4IBr.
Дииодид дикарбонилжелеза, Fe(CO)2I2,— красное твердое веще-
ство, которое в спиртовом растворе взаимодействует с пиридином
и о-фенантролином, образуя соединения Fe(CO)2(C12H8N2)I2 темно-
красного и Fe(CO)2C5H5NI2 зеленого цвета.
Нитрозильные и тионитрозильныесое-
динения железа.
Тетранитрозилжелезо, Fe(NO)4, получают действием окиси
азота на Fe(CO)5 под давлением при температуре 45°.
Fe(NO)4 представляет собой черные неустойчивые кристаллы,
которые разлагаются на Fe(NO) и Fe(NO)2 выше 0° и превращаются
в (FeSO4)x(NO)z/ под действием разб. H2SO4.
Тетратионитрозилжелезо, Fe(NS)4, получают действием
S4N4 на Fe(CO)5 в спиртовом или бензольном растворе при 75°,
действием порошка железа на (S2N2)3 в растворе тетрагидрофура-
на, а также действием безводных галогенидов железа на S4N4
в спиртовом растворе.
Fe(CO)6 + N4S4 = Fe(NS)4 + 5СО
Соединение Fe(NS)4 выделяется в виде черного порошка, кото-
рый трудно растворим в бензоле, разлагается выше 100е, горит со
взрывом на воздухе и гидролизуется до Fe2O3 •пН2О.
Галогениды нитрозилжелеза. Известны хло-
риды, бромиды и иодиды нитрозилжелеза, например Fe(NO)2Cl,
Fe(NO)2Cl3, Fe(NO)3Cl, FeNOCl, Fe(NO)2Br, Fe(NO)2I и др.
Галогениды нитрозилжелеза являются восстановителями, они
малоустойчивы и могут быть получены по уравнениям
2Fe(CO)2(NO)2 + I2 = 2Fe(NO)2I + 4СО
2FeI2 + 4NO = 2Fe(NO)2I + I2 2FeBr2 + 4NO = 2Fe(NO)2Br + Br2
FeCl2 + Fe + 6NO = 2Fe(NO)3Cl
Динитрозилдикарбонилжелезо, Fe(CO)2(NO)2, получают дей-
ствием сухой окиси азота на Fe2(CO)9 или Fe3(CO)12 при 85° или
обработкой какой-либо кислотой смеси стехиометрически необхо-
димых количеств Na2Fe(CO)4 и NaNO3:
Fe3(CO)12 -j- 2NO = Fe(CO)2(NO)2 + 2Fe(CO)5
Na2Fe(CO)4 + 2NaNO3 -f- 4HC1 = Fe(CO)2(NO)2 + 4NaCl + 2CO2 + 2H2O
Fe(CO)2(NO)2 образует темно-красные кристаллы с плотностью
1,56 г1см3, т. пл. 18,4° и т. кип. 110°; они легко окисляются на
воздухе и растворяются в органических растворителях. Под дей-
ствием иода Fe(CO)2(NO)2 превращается в Fe(NO)2I, а при обработ-
ке о-фенантролином или толуолом выделяет окись углерода.
Известны и другие нитрозильные производные Fe(NO)2(SC2H5),
Fe(NO)2(SC6H5), а также соединения общей формулы
MeI[Fe(NO)2S]-иН2О, MeI[Fe(NO)2S2O3]-nH2O, Me4Fe4(NO)7S3] •
ЖЕЛЕЗО
539
•(1 или 3)Н2О, MeI[Fe4(NO)7S3] (где Mei = Na+, К+, Rb+, Cs+,
nh;, ti+).
л-К о индексы железа. л-Комплексы содержат моле-
кулу ненасыщенного органического соединения, связанную с ато-
мом металла за счет взаимодействия атомных орбиталей металла
с обобщенными л-орбиталями ненасыщенной органической молеку-
лы. Обычно молекула ненасыщенного органического соединения
циклична, например ферроцен (см. ниже), но могут также образо-
ваться и комплексы с ациклическими ненасыщенными органиче-
скими соединениями типа бутадиена СН2 = CH-СН = СН2.
НС-СН
Циклопентадиен || || образует с натрием, магнием и мно-
нс сн
ХСН2
гимн из переходных металлов циклопентадиенильные соединения
Na(C5H5), Mg(C5H5)2, Мн(С5Н5)2, обладающие структурой сандви-
ча, в которой атом металла оказывается связанным с кольцом
в целом и не связан с каждым атомом углерода в отдельности.
В жидком или газообразном состоянии кольца могут свободно
вращаться вокруг общих осей, переходя с одной стороны на дру-
гую вокруг атома металла. В твердом состоянии свобода вращения
колец ограничена и они закреплены в положении, в котором они
движутся одновременно, как в ферроцене.
Дициклопентадиенилы переходных металлов могут быть полу-
чены действием разбавленного азотом циклопентадиена на нагре-
тый до 300° металл, циклопентадиена на карбонилы металлов при
~300°, циклопентадиенилмагнийбромида на хлориды металлов
или на ацетилацетонаты соответствующих металлов, циклопента-
диенила натрия на безводные хлориды металлов в растворе тетра-
гидрофурана и др.
Дициклопентадиенилжелезо (ферроцен), n-Fe(C5H5)2, получа-
ют по реакциям
Растворитель
FeCl2 + 2C5H5MgBr Fe(C5H5)2-pMgBr2-|-MgC12
FeCl2 + 2C5H6 + 2(C2H5)2NH -> Fe(C5H5)2 + 2(C2H5)2NH2Cl
В автоклаве
2Fe(CO)5-|-2C5Hg ► 2C5H5Fe(CO)2-|-6CO -рН2
Нагревай ие
6C5H5Fe(CO)2--------> 3Fe(C5H5)2+Fe3(CO)12
n-Fe(C5H5)2 представляет собой диамагнитные оранжевые кри-
сталлы с т. пл. 173° и т. кип. 249°; они трудно растворимы в воде,
химически стойки. При окислении n-Fe(C5H5)2 нитрат-анионом
образуется катион [Fe(C5H5)2]+ синего цвета.
540
ПОБОЧНАЯ ПОДГРУППА VIII ГРУППЫ
Рис. 29. Структура
ферроцена.
Методами рентгеноструктурного анализа и дифракции электро-
нов была подтверждена Сандвичева структура ферроцена (рис. 29)
и установлено, что расстояние С—С равно 1,4 А, а расстояние
Fe - С = 2,04 А.
Известны также следующие циклоиентадиенильные л-соеди-
нения железа: Ре(С5Н5)2С1, (С5Н5)2Ре2(СО)4, (C5H5)2Fe(CO)2,
(C5H5)Fe(CO)2Cl, (C5H5)Fe(CO)2CN.
Известны следующие олефиновые соединения железа:
C7H8Fe(CO)3 (желтая жидкость), C6H8Fe(CO)3 (бесцветные кристал-
лы), C8H8Fe(CO)3 (красные кристаллы).
Соединения включения
Нитриды железа, Fe2N и Fe4N.
Нитрид железа, Fe2N, образуется при
прокаливании (600—700°) порошка метал-
лического железа в токе аммиака или при
нагревании до 420—425° Fe2O3 [полученного
прокаливанием гидроокиси железа(Ш)] в
токе газообразного аммиака в течение
нескольких дней. Соединение Fe2N представ-
ляет собой черный порошок с плотностью
6,35 г!см2, который проявляет магнитные свойства, обладает
очень большой твердостью, окисляется на воздухе при 200°,
разлагается на элементы при 600° и превращается в Fe4N при
нагревании до 440—550° в вакууме.
Fe2N растворяется в разб. НС1, разлагается под действием
хлора или брома, взаимодействует с водородом при нагревании;
его хранят в запаянных стеклянных ампулах.
2Fe2N + ЗН2 4Fe + 2NH3
Нитрид железа, Fe4N, образуется при нагревании (440—550°)
Fe2N в вакууме или нагревании (430°) железного порошка в токе
аммиака в течение примерно 24 час.
Поскольку нитриды железа обладают высокой твердостью,
стальные детали нитруются аммиаком примерно при 600°.
Фосфиды железа, Fe3P, Fe2P, FeP, являются тверды-
ми и хрупкими интерметаллическими соединениями, которые часто
встречаются в чугунах в виде вредных примесей.
Фосфид железа, Fe2P, образуется при прокаливании фосфата
железа с сажей; фосфид железа, FeP, можно получить по уравнению
2FeS + 2РН3 = 2FeP + 2H2S + Н2
Фосфиды железа устойчивы по отношению к кислотам и цар-
ской водке.
КОБАЛЬТ
541
Арсениды железа, Fe3As2, Fe2As, Fe3As4, можно найти
в некоторых чугунах в виде вредных примесей. Они ухудшают
качества чугуна.
Арсенид железа, Fe3As2, представляет собой черное твердое
вещество, которое образуется при нагревании (700°) порошко-
образной смеси железа и мышьяка в течение 8 час.
Карбиды железа, Fe3C (цементит), Fe2C, FeC, FeC2,
FeC3, Fe3C4.
Цементит, Fe3C,— наиболее распространенный карбид желе-
за; он встречается в белых чугунах, поскольку является компонен-
том сплавов железо — углерод. Данные о получении и свойствах
цементита были приведены при описании чугунов и диаграммы
термического равновесия системы железо — углерод.
КОБАЛЬТ Со
Z — 27; ат. вес = 58,9332
Валентность (I), II, III, (IV)
Массовое число природного изотопа 59
Искусственный радиоактивный изотоп с массовым числом 60
имеет период полураспада, равный 3,5 года, и испускает у-л учи
и |3-частицы.
Электронная структура атома кобальта: K-L-3s23ps -3d’-4s2.
Электронная структура атома кобальта и катионов Со2+, Со3 +
для 3d- и 4з-орбиталей:
3d’
1 1 1
Со
4s2
3d’
11 11 1 1 1
Со’+
6s
3d6
11 1 1 1 1
Со’+
ИСТОРИЧЕСКАЯ СПРАВКА
Название «кобальт» происходит от немецкого слова Kobold,
что означает «карлик, охраняющий клады» (горный дух или
нечистая сила), или от греческого слова kobalo, что означает
«талантливый имитатор». Впервые термин kobelt (что эквивалентно
слову Kobold) упоминается в труде Агриколы «О горном деле
и металлургии».
Археологи нашли ожерелье из стекла, окрашенного кобальто-
вой синью, которое было изготовлено за 2500 лет до н.э. На стату-
этках, появившихся в царствование V династии в Египте (или
2680—2530 лет до н. э.), были обнаружены красивые кобальтовые
краски. За 1480 лет до н. э. ассирийцы отправляли в Египет стекло,
окрашенное в синий цвет, под названием «вавилонский камень».
В древних захоронениях, которые датируются XIII—XII вв. до
н. э., были найдены статуэтки, окрашенные кобальтовыми краси-
542
ПОБОЧНАЯ ПОДГРУППА VIII ГРУППЫ
телями. В развалинах Трои и Помпеи также были найдены синие
кобальтовые стекла. Знаменитые кобальтовые стекла были известны
в Александрии, Византии, Греции и Риме со времен Адриана.
Красители, содержащие кобальт, применялись в Китае за
907—618 лет до н. э.
В средние века кобальтами называли руды, служившие для
получения металлического серебра, свойства которого отлича-
лись от свойств обычного серебра. Считалось даже, что различия
в свойствах чистого серебра и сплавов серебро — кобальт (кото-
рые получали из серебряных руд, содержащих соединения кобаль-
та) обусловлены вмешательством сверхъестественной силы.
Позднее кобальтом стали называть элемент, соединения кото-
рого использовались при получении синих эмалей и при окраске
в синий цвет расплавленного стекла.
В 1520 г. Шнеберг разгадал секрет кобальтовых синей (кото-
рый был ранее утерян) и установил, что они являются сложны-
ми арсенидами серебра, кобальта, никеля и висмута.
В 1540 г. Христиан Шеррер использовал двойной силикат
кобальта и калия для получения синих эмалей, применяемых
в керамике.
Впервые металлический кобальт (загрязненный) был получен
в 1735 г. шведским химиком Брандтом. В 1792 г. Копф опублико-
вал труд, посвященный металлическому кобальту и его соеди-
нениям.
Серьезное изучение кобальта началось с работ Тенара, Пруста,
Берцелиуса, Митчерлиха, Бернарди и др.
РАСПРОСТРАНЕННОСТЬ В ПРИРОДЕ
В природе кобальт редко встречается в виде самородков, однако
соединения его очень распространены (арсениды, сульфиды, тио-
арсениды, сульфаты, арсенаты, карбонаты, окислы и др.) в раз-
личных минералах. Природный кобальт, как земного, так и метео-
ритного происхождения, находится в виде сплавов с Fe, Ni, Си,
Ag, Pt, Bi, Sb, Mn, Zn. Железные метеориты содержат 0,01 —
2% Co и 90% Fe (остальное никель).
Поскольку кобальт необходим для жизнедеятельности людей,
животных и растений, он находится в небольших количествах
в виде соединений в организме человека, животных и в различ-
ных растениях. Человеку и животным необходим витамин В12
(C63H88O14N14PCo), в состав которого входит кобальт.
Спектральным анализом было установлено присутствие кобаль-
та в атмосфере Солнца и различных звезд.
В небольших количествах соединения кобальта встречаются
в некоторых пахотных землях, минеральных водах и морской воде.
КОБАЛЬТ
543
В земной коре содержится 4,0 -10-3 вес. % кобальта. К наиболее
важным минералам кобальта относятся следующие.
Кобальтин, CoAsS, содержит 35,4% Со; он представляет
собой блестящие, как сталь, белые или серые кубические кристал-
лы с плотностью 6—6,5 г!см3 и твердостью 5,5 по шкале Мооса. Это
важный источник добычи кобальта. Обычно кобальтин сопут-
ствует халькопириту, пириту, цинковой обманке, молибдениту,
магнетиту, кальциту, апатиту, кварцу, скуттерудиту, смальтину.
Скуттерудит, CoAs3, встречается в виде серых кубических
кристаллов с плотностью 6,5 г!см3 и твердостью 6 по шкале Мооса.
В природе этот минерал сопутствует пиротину, миспикелю и глау-
кодоту (Со, Fe)AsS; последний является разновидностью миспи-
келя, но с большим содержанием кобальта.
Смальтин, CoAs3_2 (названный синей кобальтовой краской),
встречается в виде блестящих серых кубических кристаллов с пло-
тностью 6,5 г!см3 и твердостью 6 по шкале Мооса.
Сафлорит, CoAs2 или (Со, Fe)As2, сопутствует смальтину
и встречается в виде серых орторомбических кристаллов с плот-
ностью 6,5 г/см3 и твердостью 5 по шкале Мооса.
Карролит, CuCo2S4, имеет вид серых, как сталь (или серебри-
сто-белых), октаэдров с плотностью 4,85 г!см3 и твердостью 5,5
по шкале Мооса.
Линеит, Co3S4, образует серые октаэдрические кристаллы
с плотностью 4,8 г!см3 и твердостью 5 по шкале Мооса.
Эритрин, Co3(AsO4)2 -8Н2О, содержит 37,5% СоО; образуется
при выветривании скуттерудита или смальтина и встречается в ви-
де малиново-розовых моноклинных призматических кристаллов
с плотностью 2,95 г!см3 и твердостью 1,5—-2,5 по шкале Мооса.
К другим минералам кобальта относятся: данаит (Со, Fe)AsS, герсдор-
фит (Со, Ni, Fe)AsS, раммельсбергит (Со, Ni)As2, маухерит (Со, Ni)3As2,
феррокобальтит FeAsS-nCoAsS, висмутосмальтит Co(As, В1)2, баденпт
(Со, Ni, Fe)(As, Bi)3, джейпурит CoS, каттиерит CoS2, никелокаттиерит
(Co, Ni)S2, бравоит (Co, Ni, Fe)S2, виоларит (Co, Ni, Fe)3S4, полицинпт
(Co, Ni, Fe)4S6, аллоклазит (Co, Fe)(As, Bi)S, вильямит CoS2-NiS2-CoSb2 •
• NiSb2, биберит CoSO4-7H2O, форбезит (Co, Ni)HAsO4-4H2O, аннабергит
(Co, Ni)3(AsO4)2-8H2O, кабрерит (Co, Ni, Mg)3(AsO4)2 •ЩО, розелит
Ca2(Co, Mg)(AsO4)2-2H2O, корнетит (Co, Cu)3(PO4)2-3Cu(OH)2, сфероко-
бальтин CoCO3, кобальтсмитсонит (Co, Zn, Mg)CO3, трансвалит Co2O3-reH2O,
стениерит (Co, Al, Fe)2O3-H2O, гейбахит 3(Co, Ni, Fe)2O3-4H2O, миндигпт
2Co2O3-CuO-3H2O, шульценит 2CoO •Co2O3-CuO-4H2O, триёит 2Co2O3-
• CuO-6H2O, винклерит 2Co2O3-Ni2O3-2H2O, асболан m(Co, Ni)O-MnO2-
• nH2O.
В рудах соединения кобальта находятся вместе с соединениями
никеля, железа, меди, свинца, марганца, висмута и серебра.
Залежи кобальтовых руд известны в Норвегии, Швеции, Фин-
ляндии, Великобритании, СССР, Франции, ГДР, ФРГ, Италии,
Югославии, Румынии, Польше, Чехословакии, Венгрии, Бель-
544
ПОБОЧНАЯ ПОДГРУППА VIII ГРУППЫ
гии, Китае, Японии, Австралии, Республике Конго, Марокко,
Нигерии, ЮАР, Родезии, Уганде, Канаде, на Кубе, в США, Арген-
тине, Бразилии, Чили.
ИЗВЛЕЧЕНИЕ КОБАЛЬТА ИЗ РУД
Извлечение кобальта из руд представляет определенные труд-
ности, поскольку содержание кобальта в рудах очень мало,
а отделение окиси кобальта от загрязняющих ее окислов других
металлов прокаливанием концентратов кобальтовых руд затруд-
нено.
Кобальт может быть извлечен из концентратов собственно
кобальтовых руд или из остатков после прокаливания пиритов,
а также в ходе рафинирования никеля, цинка или переработки
свинцовых руд.
Арсениды, сульфиды, тиоарсениды кобальта обогащают гидро-
механическим способом или селективной флотацией, а окись или
арсенаты кобальта, проявляющие магнитные свойства, обогащают
магнитным методом.
При прокаливании концентратов арсенидов, сульфидов или
тиоарсенидов кобальта, загрязненных теми же соединениями нике-
ля, меди, свинца, висмута, железа, мышьяка и др., образуются
летучие окислы SO2, As2O3 и окислы соответствующих металлов.
При обработке смеси окислов соляной кислотой удаляется свинец
в виде РЬС12, а затем под действием H2S в солянокислой среде
осаждаются CuS и Bi2S3. Далее, при пропускании через раствор
хлора и добавлении Са(ОН)2 осаждаются CaHAsO4 и Fe(OH)3.
Если в раствор после удаления мышьяка и железа добавляют
СаОС12, то сначала отделяются Co2O3*nH2O, а затем Ni2O3-nH2O.
При обработке Со2О3 -nH2O серной кислотой в присутствии
сульфата железа(П) образуется сульфат кобальта(П):
СО2О3 4~ 2FeSO4 4~ 3H2SO4 = 2COSO4 4~ Fe2(SO4)3 4- ЗН2О
Когда кобальт извлекают из руд, содержащих медь, железо,
никель, висмут и платиновые металлы, то сначала разрабаты-
вается схема отделения различных металлов и извлечения
кобальта в зависимости от состава руды.
Кобальт может быть отделен путем электролиза очищенных
растворов сульфатов, полученных обработкой H2SO4 концентра-
тов прокаленных кобальтовых руд.
Для отделения кобальта от железа или никеля можно исполь-
зовать ионообменные смолы.
Из солянокислых растворов при pH = 0,8—3,0 Н-катиониты
удерживают как железо, так и кобальт. При pH = 1,5—1,8 осу-
ществляется элюирование железа 0,05 М раствором щавелевой
КОБАЛЬТ
545
кислоты, а после удаления следов щавелевой кислоты кобальт
элюируют 2 н. НС1.
В солянокислых растворах образуются анионы [СоС14]2-, кото-
рые могут количественно удерживаться на сильно основных анион-
ных смолах, в то время как катионы никеля Ni2+ или [Ni(H2O)6]2 +
переходят в элюент.
ПОЛУЧЕНИЕ МЕТАЛЛИЧЕСКОГО КОБАЛЬТА
Металлический кобальт получают восстановлением водородом
(при нагревании) соединений Со2О3, Со3О4, СоО, СоС2О4,
Со(НСОО)2, СоС12, CoBr2, [Co(NH3)5C1]C12, [Co(NH3)5C11CO3, вос-
становлением (при нагревании) окислов кобальта углеродом,
окисью углерода или метаном, алюмо- или кремнетермическим
восстановлением окислов кобальта, термическим разложением
карбонилов Со2(СО)8, Со4(СО)12 и элекролизом водных растворов
солей CoSO4-7H2O или (NH4)2SO4-CoSO4-6H2O.
Восстановление водородом окислов, галогенидов,
оксалата и формиата кобальта
Окислы, галогениды, оксалат и формиат кобальта восстанав-
ливаются водородом при нагревании по уравнениям:
ЗСо2О3+ Н2 — 2Со3О4 + Н2О
~200°
Со2О3 + Н2-----> 2СоО + Н2О
-200°
Со3О4-(-Н2-----► ЗСоО+Н2О
250-1100°
Со2О3+ЗН2-------------> 2Со + ЗН2О
250-1100°
Со3О4 + 4Н2-----------> ЗСо + 4Н20
250-1 100°
СоО-}-Н2-----------► Co- Н, >
~ооо°
СоС12 + Н2-----> Co-j-2HCl
-350°
СоВг2 + Н2-----> Со + 2НВг
-750°
СоС2О4 + 2Н2-------► Со+2С0-|-2Н20
250-700°
Со(НСОО)2 + Н2--------> Со + 2СО + 2Н2О
[Co(NH3)5C1]C12 + з/2н2 Со + 3NH4C1 + 2NH3
При восстановлении окислов Со2О3, Со3О4, СоО водородом
при температуре 250—380° образуется порошкообразный метал-
лический кобальт, обладающий пирофорными свойствами, чего
35-0101
546
ПОБОЧНАЯ ПОДГРУППА VIII ГРУППЫ
не происходит в случае восстановления при температуре выше
700°. Если восстановление соединений кобальта водородом осу-
ществляется при температуре ниже 492°, образуется модификация
а-Со с плотной гексагональной кристаллической решеткой, а при
температуре выше 492°— модификация р-Со с кубической гране-
центрированной кристаллической решеткой.. Восстановлением со-
единений кобальта водородом при нагревании можно получить
металлический кобальт 99,86 %-ной чистоты.
Восстановление окислов кобальта углеродом
(коксом или древесным углем), окисью углерода
или метаном при нагревании
Восстановлением окислов Со2О3, Со3О4, СоО углеродом (кок-
сом, антрацитом, древесным углем) или окисью углерода при
нагревании в электрических печах получают металлический
кобальт, загрязненный углеродом или карбидами кобальта.
Реакции восстановления окиси Со3О4 углеродом или окисью
углерода обратимы:
900-1100°
Со3О4+4С t ЗСо + 4СО
300-900°
Со304-|-4СО t ~ > ЗСо + 4СО2
При сильном нагревании концентратов кобальтовых и медных
руд, смешанных со шлаком (полученным при рафинировании
меди), содержащим 15% Со, и с 10% кокса в электрических печах
(5000—5500 а, 120 в), образуется два сплава: один более тяжелый,
красного цвета, содержит 89% Си, 4,5% Со, 4% Fe, другой более
легкий, белого цвета, имеет состав: 42% Со, 39% Fe, ~15% Си,
1,6—2% Si; образующийся шлак содержит 15% Со.
Действием метана на Со2О3 при разных температурах получа-
ют окись кобальта, металлический кобальт или карбиды кобальта:
450°
ЗСо2О3 + СН4----► 6СоО + СО + 2Н2О
800°
Со2О3 + СН4-----► 2Со + СО + 2Н2О
Алюмо-, кремне- или боротермическое восстановление
окислов кобальта
Алюмо-, кремне- или боротермическим восстановлением окис-
лов кобальта получают сплавы кобальта с соответствующим эле-
ментом. Алюмотермическое восстановление окислов Со3О4, СоО
осуществляется в тигле из окиси магния или корунда по урав-
КОБАЛЬТ
547
нениям
ЗСо3О4 + 8А1 = 9Со + 4А12О3 (при 3303°)
ЗСоО + 2А1 = ЗСо + А12О3 (при 2495°)
При восстановлении окислов кобальта Со3О4,СоО ферросили-
цием образуется сплав железо — кобальт — кремний.
Получение металлического кобальта термическим разложением
карбонилов Со2(СО)8, Co4(CO)i2
При термическом (60°) разложении карбонилов Со2(СО)8,
Co4(CO)i2 образуется черный мелкодисперсный порошок метал-
лического кобальта и выделяется окись углерода:
51° 60°
2Со2(СО)8 — -> Со4(СО)12 -— -* 4Со
— — 16(Л)
Методы получения карбонилов Со2(СО)8, Со4(СО)12 приведены
при описании карбонильных соединений кобальта.
Электролитическое получение металлического кобальта
Металлический кобальт можно получить электролизом вод-
ного раствора, содержащего 190—480 г/л CoSO4*7H2O, при темпе-
ратуре 50—60°; аноды из чистого свинца, катоды из нержавеющей
стали, плотность тока 2,3—3,0 а/дм2. Можно проводить также
электролиз водного, слегка подкисленного раствора (NH4)2SO4’
•CoSO4’6H2O при температуре 20° и ограниченной плотности тока.
Металлы, химически более активные, чем кобальт, например
магний или цинк, вытесняют кобальт из кислых растворов его
солей.
ОЧИСТКА
Сырой кобальт очищают плавлением в высоком вакууме, мето-
дом зонной плавки или электролитическим рафинированием.
ФИЗИЧЕСКИЕ И ХИМИЧЕСКИЕ СВОЙСТВА
В компактном состоянии кобальт представляет собой серебри-
сто-серый металл (похожий на железо) с плотностью 8,83 г/см3,
т. пл. 1492° и т. кип. 3185°. Кобальт обладает твердостью 5,5 по
шкале Мооса (он тверже железа), более хрупок, чем сталь, обла-
дает ферромагнитными свойствами (которые исчезают при темпе-
ратуре выше 1150° и образуется парамагнитная модификация),
тягуч и плохо поддается ковке.
35*
548
ПОБОЧНАЯ ПОДГРУППА VIII ГРУППЫ
Металлический кобальт известен в двух кристаллических
модификациях: а-Со — с плотной гексагональной структурой
и р-Со — с кубической гранецентрированной кристаллической
решеткой.
Пирофорный кобальт (получается восстановлением окислов
кобальта водородом при 250—380°) представляет собой черный
порошок, который окисляется на воздухе при обычной темпера-
туре, разогреваясь при этом до белого каления. Коллоидный
кобальт (образуется при добавлении воды к пиридиновым раство-
рам солей кобальта, восстановленных пирогаллолом) окрашен
в золотисто-коричневый цвет.
Известны многочисленные сплавы кобальта с металлами — Fe,
Ni, Мп, Cr, Мо, W, V, Nb, Та, Ti, Zr, Th, Sn, Pb, Al, Be, Zn, Hg,
Си и с неметаллами — C, Si, N, P, S.
При обычной температуре металлический кобальт в компакт-
ном состоянии устойчив к действию сухого и влажного воздуха,
воды, сильных щелочей и разбавленных растворов органических
кислот.
В сухом или влажном воздухе металлический кобальт в ком-
пактном состоянии окисляется только при температуре выше 300°,
образуя окислы Со2О3, Со3О4, СоО. При действии паров воды на
нагретый до красного каления металлический кобальт образуется
СоО. Кобальт-магниевый сплав энергично разлагает воду на
холоду.
Порошкообразный металлический кобальт взаимодействует при
нагревании с галогенами, серой, фосфором, мышьяком, сурьмой,
углеродом, кремнием, бором, но не реагирует с азотом:
2Со + 3F2 = 2CoF3 + 2 X 190 ккал
Со + F2 = CoF2 +158 ккал
Со + С12 = СоС12 + 74,8 ккал
Со + Вг2 = СоВг2 + 58 ккал
Со + 12 = Со12 + 36 ккал
Со + S = CoS + 20,5 ккал
При нагревании (380°) порошка металлического кобальта
(обладающего пирофорными свойствами) в струе аммиака в тече-
ние 3—4 час образуется черный нитрид Co3N, а при 276° — Co2N;
оба эти нитрида легко растворяются в разбавленных кислотах.
Если порошкообразный металлический кобальт нагревают
в атмосфере аммиака при 470°, образуется черный нитрид кобаль-
та Co3N2.
При нагревании кобальта в парах фосфора (или при действии
фосфористого водорода на дигидрид кобальта) образуются фосфи-
ды Со2Р, Со3Р2, Со2Р3, СоР, СоР3. Взаимодействие при нагрева-
нии углерода с кобальтом аналогично его реакции с железом,
однако при охлаждении сплавов кобальт — углерод выделяется
не карбид Со3С, а графит.
КОБАЛЬТ
549
Газообразный хлористый водород взаимодействует с кобальтом
по уравнению
Со + 2HCJ СоС12 + Н2
Сероводород при 100° и SO2 при сильном нагревании (до крас-
ного каления) превращают металлический кобальт в CoS.
При действии окислов азота N2O (230°), NO (150°), NO2 (ком-
натная температура) на кобальт образуется окись СоО.
При нагревании РС13, AsCl3, SbCl3 с металлическим кобальтом
образуются фосфид, арсенид и антимонид кобальта. В условиях
температуры 220° и давления 250 ат окись углерода реагирует
с тонкодисперсным порошком кобальта, образуя карбонил
Со2(СО)8.
Металлический кобальт медленно растворяется в разбавлен-
ных кислотах НС1, H2SO4 и быстро — в разб. HNO3, поскольку
нормальный потенциал системы Со/Со2+ равен —0,277 в:
Со + 2НС1 + гаН2О = СоС1?.пН2О + Н2
Со + H2SO4 + гаН2О = CoSO4.reH2O + Н2
8Со + 20HNO3 + (п — 10)Н2О = 8Со(КОз)2.пЙ2О + 2NO + N2
Под действием дымящей HNO3 на холоду кобальт пассивирует-
ся. Плавиковая кислота и царская водка реагируют с кобальтом
на холоду. При комнатной температуре ледяная уксусная, щаве-
левая, лимонная и винная кислоты очень медленно растворяют
металлический кобальт. Расплавленное едкое кали (550°) также
растворяет металлический кобальт.
Кобальт вытесняет медь, серебро, ртуть, палладий, платину
и золото из растворов их солей.
Химическая реакционная способность кобальта наглядно
иллюстрируется следующей схемой:
Комн. темп.
Кобальт
Нагревание
с разб. НС1-> СоС12-пН2О
с разб. HNO3->-Co(NO3)2-reH2O
на воздухеСо2О3, Со3О4, СоО
с водяными парами-> СоО
с галогенами —CoF3, CoF2, СоС12, СоВг2, Со12
с серой -> CoS
с NH3->Co3N, Co2N, Co3N2
с фосфором или РН3-> Со2Р, Со3Р2, Со2Р3, СоР, СоР3
с H2S —> CoS
ч с СО -> Со2(СО)8
С физиологической точки зрения в небольших дозах кобальт
не токсичен для человека, животных или растений; он относится
к олигоэлементам, необходимым для жизнедеятельности человека
и животных. Однако в относительно больших количествах соли
кобальта все же токсичны.
50
ПОБОЧНАЯ ПОДГРУППА VIII ГРУППЫ
Радиоактивный изотоп '7С0 получают бомбардировкой изотопа
2®Со тепловыми нейтронами. Он используется в качестве источни-
ка у-лучей (в меньшей сепени р-частиц) в области радиохимиче-
ских исследований, в медицине и в различных промышленных
процессах.
ПРИМЕНЕНИЕ
Металлический кобальт применяют для электролитического
покрытия различных металлических деталей, поскольку он очень
устойчив к действию кислорода воздуха и воды.
Кобальт используют в больших количествах при получении
твердых сталей типа стеллитов (содержат кобальт и хром в соот-
ношении 3:1, устойчивы к истиранию, к действию химических
реагентов, обладают высокой температурой плавления), быстро-
режущих сталей (для резцов, сверл и др.), сверхтвердых металло-
керамических сплавов, образованных из карбидов W, Ti, Мо, Та,
Ni, V, сцементированных кобальтом, сплавов с постоянными
магнитными свойствами, кислотостойких, огнеупорных (до тем-
пературы 900°) сплавов и др. Сплавы кобальта находят очень
широкое применение. Из них изготовляют катоды, электрические
сопротивления, зубные протезы; они применяются в автомобиле-
строении, турбореактивной, турбокомпрессорной и ракетной тех-
нике и т. д.
Еще с древних времен известно применение окислов СоО
и СО3О4 при изготовлении синих эмалей и для окраски в синий
цвет расплавленного стекла. Способность окислов СоО, Со3О4
образовывать твердые растворы (красиво окрашенные в синий,
зеленый, розовой и другие цвета) с окислами различных металлов
обусловила промышленное применение окислов кобальта в кера-
мической и стекольной промышленности.
Тонкий порошок металлического кобальта применяют в каче-
стве катализатора в реакциях гидро- и дегидрогенизации. В каче-
стве катализаторов используются следующие соединения кобаль-
та: СоО, Со3О4, Со(ОН)2, Co2O3*nH2O, CoF2, СоМоО4, Со2(СО)8
и др. Кобальтовые катализаторы нашли промышленное примене-
ние в качестве сиккативов (для олиф) и в процессах переработки
нефтей.
СОЕДИНЕНИЯ (ОБЩИЕ СВОЙСТВА)
Известно очень много соединений двух- и трехвалентного
кобальта и довольно ограниченное число соединений одно-
и четырехвалентного кобальта. Наиболее устойчивыми среди
простых соединений являются соединения двухвалентного кобаль-
та, а среди координационных — соединения трехвалентного
кобальта. В табл. 58 приведены формулы и указан цвет некоторых
соединений кобальта разных степеней окисления.
КОБАЛЬТ
551
Помимо названных соединений кобальта разных степеней
окисления известны соединения включения, металлоорганические
соединения и соединения нестехиометрического состава, к кото-
рым относятся окислы: Со3О5, Со3О5-пН2О, Со4О7-7Н2О, Со4О5,
Со4О5-пН2О, Со6О7, Со8О9, Со8О9-пН2О, Со16017, Со5О7, Со5О8-
•wH2O, Со7О10, Со7Оц, Со9О14«пН2О, Со12О19, Со13О49 •zzH2O и др.
При описании гетерополисоединений вольфрама были названы
кобальт-6-вольфраматы и кобальт-12-вольфраматы, содержащие
анионы [Co2+WI2O42l8-, [Co2+Co3+W12O42]7~, [Co2+WI2O40]6-,
[Co3+W12O40]s_.
Соединения одновалентного кобальта
Соединения кобальта(1), число которых ограничено, довольно
неустойчивы, обнаруживают восстановительные свойства. В каче-
стве примеров соединений одновалентного кобальта можно на-
звать Co2Se, K3[Co(CN)4], MeJ[Co(CO)(CN)3l.
Соединения двухвалентного кобальта
Большинство простых солей кобальта(П) образуется при
обработке окиси СоО или гидроокиси Со(ОН)2 различными кисло-
тами. Соли двухвалентного кобальта, полученные при использо-
вании сильных кислот, в большинстве растворимы, их разбавлен-
ные растворы окрашены в розовый цвет и имеют кислую реакцию
благодаря гидролизу. Разбавленные растворы солей двух-
валентного кобальта содержат катион 1Со(Н2О)8]2+.
Координационные соединения кобальта(П) довольно неустой-
чивы и легко окисляются до соединений кобальта(Ш). Координа-
ционные соединения кобальта(П) нельзя отнести к типичным
комплексам; они образуются в отсутствие окислителей (кислорода
воздуха). Исключением являются некоторые трудно растворимые
комплексы и ряд соединений, у которых лигандами служат моле-
кулы с восстановительными свойствами, например гидразин.
Известны координационные соединения кобальта (II) с ами-
нами, акво-, ацидо-, гидроксо-, ацидоаммино-, ацидоакво-, ацидо-
аквоаммино-, аквоамминокомплексы и др.
Многочисленные безводные соли кобальта(II), такие, как
галогениды, тиоцианат, сульфат, могут присоединять шесть моле-
кул аммиака, образуя неустойчивый комплексный катион
[Co(NH3)6]2+, который разлагается водой по уравнению
[Co(NH3)6]C12 + 6Н2О Со(ОН)2 + 4NH4OH + 2NH4C1
Поскольку равновесие реакции смещено влево, аммиак в при-
сутствии солей аммония не осаждает Со(ОН)2.
552
ПОБОЧНАЯ ПОДГРУППА VIII ГРУППЫ
В присутствии солей аммония [Co(NH3)6]C12 окисляется на
воздухе до пурпуреосоли [Co(NH3)5C11C12.
Из класса ацидосоединений известны трех-, четырех- и шести-
координированные соединения, содержащие анионы [СоХ3]-,
[СоХ412-, [СоХ6]4-.
Координационные соединения кобальта(П) приведены вместе
с простыми соединениями.
Окись кобальта, СоО, получают действием кислорода или паров
воды на металлический кобальт при температуре выше 940° (чтобы
исключить образование Со3О4), окислением металлического кобаль-
та окислами азота N2O, NO или NO2, разложением Со2О3 или Со3О4
при температуре выше 940° в токе N2 или СО2, прокаливанием
соединений Со(ОН)2, CoSO4, Co(NO3)2, СоСО3, СоС2О4,
Со(НСОО)2 без доступа воздуха (в вакууме или в токе азота),
поскольку на воздухе прокаливание нужно вести при тем-
пературе выше 940°, чтобы исключить образование Со3О4, про-
каливанием CoS на воздухе и восстановлением при нагревании
Со2О3 водородом, метаном или аммиаком.
Со + 1/2О2 ► СоО+ 57,20 ккал Со(ОН)2 200° СоО + Н2О
Со + Н2О > СоО + Н2 COSO4 550° СоО -[* SO24~ ^/2^2
230° Co+N2O > CoO + N2 Co(NO3)2 105° CoO+2NO2+V2O2
150° Co+NO > CoO + V2N2 СоСО3 477° СоО + СО2
Комн. 2Co-|-NO2 ► темп. 2СоО ^/2^2 Со(НСОО)2 175° СоО+2СО + Н2О
940° Оо2Оз 2СоО -I-I/2O2 СоС2О4 300-400° СоО + СО + СО2
940° Со3О4 ► ЗСоО CoS + 3/2O2 750° CoO + SO2
Соединение СоО представляет собой серовато-зеленый кристал-
лический порошок со структурой типа NaCl с плотностью 6,45 г!см?
и т. пл. (без разл.) 1935°. Окись кобальта устойчива до 2860°
(выше этой температуры разлагается на элементы), ферромагнит-
на выше 19° (антиферромагнитна ниже этой точки Кюри), плохо
растворима в воде и других растворителях, превращается в Со3О4
при нагревании в кислороде выше 100° и восстанавливается до
металлического кобальта водородом, углеродом, окисью углеро-
да, кремнием, бором, алюминием, железом, спиртом или аммиа-
ком (при нагревании).
Окись кобальта является основным окислом; СоО вытесняет
аммиак из теплых растворов солей аммония и образует соли
кобальта(П) при растворении на холоду в сильных минеральных
кислотах или при нагревании в большинстве органических кислот.
КОБАЛЬТ
553
При сплавлении СоО с избытком КОН или NaOH образуются
кобальтиты Ме*СоО2 ярко-синего цвета, а при растворении СоО
в концентрированных теплых растворах щелочей (КОН, NaOH)
образуются ярко-синие растворы гидроксокобальтатов(П)
Ме*[Со(ОН)4], которые очень легко гидролизуются и окисляются.
В результате действия газообразного хлора, S2C12 или СС14
на СоО при нагревании образуется СоС12. При повышенной тем-
пературе сера и сероводород превращают СоО в CoS. Обработка
СоО смесью SO2 и О2 при 600° дает CoSO4.
При сильном нагревании (800—1500°) СоО с окислами различ-
ных металлов — А12О3, Cr2O3, Fe2O3, SiO2, TiO2, SnO2, MnO2,
PbO2, ZnO, MgO, MnO, NiO—образуются соединения строго опре-
деленного состава или твердые растворы, интенсивно окрашен-
ные в синий, зеленый или розовый цвет. К красящим пигментам
относятся тенарова синь (в основе которой лежит Со[А12О4] или
твердый раствор Со[А12О4] с Со3О4 или СоО), лазурно-синий станнат
(смесь Co2SnO4 с SnO2 и SiO2 в определенных соотношениях),
зелень Ринмана CoZnO2, краситель, который получают прокалива-
нием смеси СоО, Сг2О3, А12О3, и кобальтовый розовый (твердый
раствор СоО с MgO).
Алюминат кобальта, Со[А12О4], в виде синих октаэдрических
кристаллов образуется при прокаливании (1200—1300°) смеси
СоО с А12О3 или CoSO4 с NH4A1(SO4)2 ПЗНгО в течение 30 мин.
Цинкат кобальта (зелень Ринмана, или турецкая зелень),
CoZnO2, также относится к красящим веществам. Он образуется
при прокаливании (1000—1100°) смеси СоО или СоСО3 с ZnO.
Окись кобальта применяют для изготовления отрицательных
электродов аккумуляторов, для получения окрашенных стекол,
фарфора или эмалей и для многих других целей. В частности,
в качестве катализатора СоО применяется во многих реакциях
[как, например, окисления (окисление NH3, HCN), дегидрогени-
зации (получение бутадиена из бутана, альдегидов из спиртов)
и в реакциях разложения (как в случае Н2О2, N2O и др.)].
Гидроокись кобальта, Со(ОН)2, существует в виде двух моди-
фикаций, а именно а-Со(ОН)2 и 0-Со(ОН)2.
Метастабильная модификация а-Со(ОН)2 образуется в виде
синего осадка при добавлении растворов щелочей к солям кобаль-
та^!) (около 0°).
Устойчивая модификация |3-Со(ОН)2 образуется в виде розового
осадка при добавлении растворов солей кобальта(П) к растворам
щелочей, при нагревании a-модификации и при кипячении рас-
твора CoSO4-«H2O со смесью К! с К1О3. Переход а-Со(ОН)2
в р-Со(ОН)2 не происходит в присутствии лактозы, мальтозы,
сахарозы, рафинозы и желатины.
Соединение 0-Со(ОН)2 розового цвета, имеет ромбоэдрическую
кристаллическую решетку (типа брукита) с плотностью 3,597 г!смэ.
554
ПОБОЧНАЯ ПОДГРУППА VIII ГРУППЫ
Обе модификации слабо растворимы в воде, растворимы в теп-
лых концентрированных растворах щелочей, минеральных кисло-
тах и большинстве органических кислот, превращаются в СоО
при нагревании в вакууме или в токе азота и образуют Со3О4 •
•иН2О при нагревании на воздухе. Из охлажденных растворов,
полученных растворением Со(ОН)2 в теплых концентрированных
щелочах, выделяются красные устойчивые на воздухе кристал-
лы Со(ОН)2.
При растворении а-Со(ОН)2 и р-Со(ОН)2 в аммиаке в присут-
ствии солей аммония образуются желтые гексаммины кобальта(П);
они довольно неустойчивы и при хранении на воздухе или в при-
сутствии окислителя превращаются в устойчивые вишнево-крас-
ные пурпуреосоли кобальта(Ш).
Со(ОН)2 + 4NH3 + 2NH4C1 [Co(NH3)e]Cl2 + 2Н2О
4[Co(NH3)e]Cl2 + 4NH4C1 + 02 = 4[Co(NH3)5C1]C12 + 2Н2О + 8NH3
2[Co(NH3)6]G12 + 2NH4C1 + H2O2 = 2[Co(NH3)5C1]C12 + 2H2O + 4NH3
В коллоидном состоянии Co(OH)2 окрашена в коричневый
цвет. Хлорная или бромная вода, гипохлориты щелочных метал-
лов, перекись водорода и K3[Fe(CN)6l окисляют Со(ОН)2в щелоч-
ной среде до Со2О3-пН2О — соединение черно-коричневого цвета:
2Со(ОН)2 + 2K3[Fe(CN)el + 2КОН = Со2О3-ЗН2О 4- 2K4[Fe(CN)6]
Гидроокись кобальта катализирует окисление сульфита натрия
кислородом воздуха.
Гидроксокобальтиты, Mei[Co(OH)4] (где Me1 =
= Na+, К+), образуются при насыщении Со(ОН)2 концентрирован-
ным теплым раствором щелочи; это фиолетово-красные кристаллы,
которые гидролизуются водой и легко окисляются кислородом
воздуха. Действием на Na2[Co(OH)4] концентрированного раствора
Sr(OH)2 или Ва(ОН 2 были получены Sr2[Co(OH)6] и Ва2[Со(ОН)6]
в виде красно-фиолетовых призматических кристаллов.
Дифторид кобальта, CoF2, получают обработкой газообраз-
ным HF СоС12 (при комнатной температуре) или СоО (500°), тер-
мическим разложением (NH4)2[CoF4] в токе СО2 и дегидратацией
CoF2-4H2O при нагревании в токе HF или в присутствии водо-
отнимающих средств (конц. H2SO4 или Р4Ою).
Соединение CoF2 представляет собой токсичные парамагнит-
ные (антиферромагнитные при низкой температуре) розовые тетра-
гональные призмы с плотностью 4,43 г! см9-, растворяется в воде,
плохо растворимо в спирте, эфире, бензоле, пиридине, устойчиво
в воде или аммиаке при комнатной температуре, но превращается
в СоО с выделением HF под действием горячей воды, взаимо-
действует с водородом при нагревании по уравнению
CoF2 -I- Н2 =₽t Со + 2HF
КОБАЛЬТ
555
Натрий, магний, алюминий при нагревании с CoF2 воспла-
меняются.
Дифторид кобальта катализирует фторирование различных
органических соединений.
Известны кристаллогидраты CoF2*nH2O (где п = 2, 3, 4),
комплексные аммины [Co(NH3)6]F2, [Co(NH3)5H2O]F2, фторосоли
Na[CoF3I -Н2О, K[CoF3].H2O, KJCoFJ, (NH4)2[CoFJ, Co[SiF6] •
•6H2O, Co[TiF6] .6H2O, Co[GeFg] *6H2O, Co[SnF6] -6H2O,
Со[А1(Н2О)Г5], основная соль СоГ2"СоО"Н2О и кислая соль
CoF2.5HF.6H2O.
Кристаллогидраты CoF2*hH2O выделяются в виде красновато-
розовых кристаллов при упаривании растворов металлического
кобальта, окиси, гидроокиси или карбоната кобальта(П) в плави-
ковой кислоте.
Дихлорид кобальта, СоС12, получают действием хлора на
порошок или стружку металлического кобальта при 550—600°,
термическим разложением соединения [Со(ХН3)5С1]С12 и дегидра-
тацией кристаллогидратов СоС^-гаНгО (где п =(6, 4, 2, 1) в атмо-
сфере НС1 при 150-175°.
6[Co(NH3)5CI]C12 -> 6СоС12 + 6NH4C1 + 22NH3 + N2
Соединение СоС12 образует парамагнитные сильно гигроскопич-
ные голубые гексагональные кристаллы со структурой типа Cdl2;
т. пл. 727°, т. кип. 1049° (очищается сублимацией в токе хлора
или СО2), растворимо в воде, спирте, ацетоне и практически не
растворимо в пиридине и метилацетате.
Цвет водных растворов дихлорида кобальта определяется
концентрацией и температурой растворов. Холодные разбавлен-
ные водные растворы дихлорида кобальта окрашены в красновато-
розовый цвет, а при нагревании становятся пурпурно-красными,
фиолетовыми и затем синими. Водный 50%-ный раствор СоС12
при 60° имеет аметистовый цвет, при 77° он становится пурпурным
и при 100—134° — синим.
Известны кристаллогидраты СоС12 -тгНгО (п = 6,4) — розовый
и СоОг’/гНгО (п — 2, 1, 5, 1) — синий.
Гексагидрат, СоС12'6Н2О (или аквосоль [Со(Н2О)6]С12), выде-
ляется в виде красных кристаллов при испарении (при комнатной
температуре) раствора окиси, гидроокиси или карбоната кобаль-
та(П) в разб. HCl (1 : 1). Если раствор испаряют при 30—35°,
выделяется кристаллогидрат [Со(Н2О)4С12] -2H2O фиолетово-сине-
го цвета. При обработке концентрированных растворов дихлорида
кобальта конц. НС1 или концентрированными растворами хлори-
дов щелочных металлов образуются синие растворы, содержащие
анионы [СоС13]", [CoClJ2-; при разбавлении или под действием
HgCl2 благодаря обратному переходу к катиону [Со(Н2О)6]2+
556
ПОБОЧНАЯ ПОДГРУППА VIII ГРУППЫ
растворы становятся розовыми:
[Co(H2O)e]2+ + 4С1- -> [СоС1412- + 6Н2О
Розовый Синий
[СоС14]2- + 2HgCl2 + 6Н2О -> [Co(H2O)e]2+ + 2[HgCl4]2-
В общем случае координационные соединения кобальта(П)
с координационным числом 4 имеют синий цвет, а с координаци-
онным числом 6 — розовый.
Водный раствор дихлорида кобальта применяется в метеоро-
логии для изготовления индикаторной бумаги, которая служит
для определения степени атмосферной влажности, поскольку
в сухом воздухе эта бумага синего цвета, а во влажном — розового.
При нагревании водород и металлический магний (в атмосфере
СО2) восстанавливают СоС12:
СоС12 Н2 Со -р 2НС1 СоС)2 -р Mg = Со -р MgCl2
Производное NaC10H8 восстанавливает СоС12 в тетрагидро-
фуране по уравнению
ЗСоС12 + 6NaC10H8 = ЗСо + 6С10Н8 + 6NaCl
Дихлорид кобальта реагирует с хлором при обычной темпера-
туре и кислородом при нагревании:
2СоС12 + С12 2СоС13 ЗСоС12 + 2О2 Со3О4 + ЗС12
В присутствии аммиака и солей аммония (этилендиамина
и солянокислого этилендиамина) дихлорид кобальта окисляется
кислородом воздуха:
4СоС12 + 20NH3 + 4NH4C1 + О2 = 4[Co(NH3)6]Cl3 + 2Н2О
4СоС12 + 8Еп -р 4Еп-НС1 + О2 = 4[СоЕп3]С13 + 2Н2О
Дихлорид кобальта реагирует с цианидом натрия при нагре-
вании по уравнению
345-550°
СоС12 + 2NaCN--------> CoCN2 + С + NaCl
Известны синие хлоросоли типа МеЧСоС13], Mei[CoCl4] -2Н2О,
Ме|[СоС16]-10H2O (где Me1 = Li+, К+, Rb+, Cs+), комплексные
аммины [Co(NH3)6]C12, [Co(NH3)2C12], [СоРу2С12] и двойные хлори-
ды 2СоС12 -7ЫСЫ8Н2О, 2СоС12 -3LiCl-6Н2О, CoCl2-SnCl4-6H2O
или ColSnClg] -6Н2О.
Хлорид гексамминкобалъта(11), [Co(NH3)6]C12, можно полу-
чить действием NH4OH на Со(ОН)2 в присутствии NH4C1 и без
доступа воздуха, поскольку в присутствии кислорода образуется
пурпуреосоль [Co(NH3)5C1]C12:
Со(ОН)2 + 4NH4OH + 2NH4C1 = [Co(NH3)e]Cl2 + 6Н2О
4[Co(NH3)e]Cl2 + 4NH4C1 + O2 = 4[Co(NH3)5C1]C12 + 2H2O + 8NH3
КОБАЛЬТ
557
Соединение [Go(NH3]6]G12 в твердом состоянии красного цвета,
а водный раствор его окрашен в розовый цвет.
Дибромид кобальта, СоВг2, получают действием паров брома
на нагретый металлический кобальт или на измельченный поро-
шок кобальта под слоем эфира, действием НВг на СоО или СоС12
(500°), дегидратацией СоВг2-6Н2О в течение нескольких часов
(140—160°) и разложением СоВг2(С2Н5)2О при 60°.
Соединение СоВг2 представляет собой парамагнитные зеленые
блестящие гексагональные кристаллы с решеткой типа Cdl2,
с плотностью 4,91 г/см? при 25° т. пл. 678° (в токе азота) и
т. кип. 927°. Дибромид кобальта очень гигроскопичен, растворяется
в воде, спиртах, ацетоне, ацетальдегиде, бензальдегиде, метил-
ацетате, ацетонитриле и плохо растворим в хлороформе, уксусном
ангидриде, этилацетате, ацетофеноне. Разбавленный водный рас-
твор дибромида кобальта красного цвета, а концентрированный рас-
твор при низкой температуре синего цвета. При 575° СоВг2 вос-
станавливается водородом по уравнению
СоВг2 + Н2 =Ft Со + 2НВг .
Известны кристаллогидраты СоВг2-пН2О (п = 6, 4, 1, 2), бромо-
соли типа Ме2[Со(Н2О)2Вг4], MeJ[CoBr6]-пН2О, гексабромостанпаты
[Co(H2O)6][SnBr6] -4Н2О, [Co(CsHsN)6][SnBr6], [Co(C6H12N4)6][SnBr6] -4Н2О,
различные аддукты, например СоВг2-пСН3ОН (п = 6, 4, 3, 2), СоВг2 •
• пС2Н5ОН (п = 3, 2, 1), CoBr2.nNH3 (п = 6, 4, 2, 1), CoBr2.2N2H4,
СоВг2-6CH3NH2, CoBr2.nC6H6N (п = 4, 2), СоВг2.2C9H7N, СоВг2.
• 3C12HSN2-8Н2О, основные бромиды СоВг2-9Со(ОН)2, СоВг2-ЗСо(ОН)2.
Дииодид кобальта, Со12, получают нагреванием металличе-
ского кобальта в парах иода или в токе HI при 400—450°, дей-
ствием водного раствора иода на тонко измельченный кобальт,
дегидратацией кристаллогидрата Со12-6Н2О при 130° и нагрева-
нием СоО с А1216 при 230—300°.
Дииодид кобальта существует в виде двух модификаций —
сг-Со12 и р-Со12.
Модификация а-Со!г представляет собой парамагнитные чер-
ные гексагональные кристаллы с решеткой типа Cdl2, с плотно-
стью 5,584 г/см3, т. пл. 520° (в вакууме), т. кип. 570° (с разложе-
нием и выделением иода), очень гигроскопичные. Модификация
р-Со12 образует желтые игольчатые кристаллы с плотностью
5,45 г/см3', они менее устойчивые, чем а-модификация.
Обе модификации растворимы в воде, спирте, эфире, ацетоне,
метилацетате, диметилсульфате, ацетонитриле, пиридине, хиноли-
не, хлориде мышьяка(Ш), хлорокиси фосфора, хлористом тиони-
ле, разлагаются при ~600° с выделением иода, взаимодействуют
на холоду с водородом по уравнению
Col2 + Н2 Со + 2HI
558
ПОБОЧНАЯ ПОДГРУППА VIII ГРУППЫ
Концентрированные водные растворы а-Со12 красного цвета
при температурах ниже 20°, оливково-зеленого — в интервале
между 20 и 40° и зеленого — выше 40°.
Известны кристаллогидраты Со12-пН2О (п = 9, 6, 4, 2), иодосоли типа
Me2[CoI4] (Mei = Na+, Cs+, V2Cd2+, V-jHg*-1-, ^РЬ*4-), MeJlCoIe], аддукты
CoI2-nNH3 (n = 6, 2), CoI2-nC5H5N (n = 6, 4, 2), CoI2-2N2H4, CoI2-nCH3OH
In = 6, 5), основная соль CoI2-3CoO, полииодид кобальта Col4.
Дииодид кобальта применяется в качестве катализатора
в многочисленных синтезах, например при получении спиртов,
уксусной кислоты и др.
Дицианид кобальта, Co(CN)2, получают нагреванием СоС12
с безводным NaCN в присутствии порошка железа (как катализа-
тора), нагреванием при 250° в токе азота осадка, полученного обра-
боткой раствора нитрата кобальта(П) стехиометрически необходи-
мым количеством KCN.
Соединение Co(CN)2 — гигроскопичный сине-фиолетовый
порошок с плотностью 1,872 г!см3.
Кристаллогидрат Co(CN)2'311получают в виде красновато-
коричневого осадка при обработке раствора соли кобальта(П)
(в избытке) раствором HCN или KCN.
Соединение Co(CN)2-3H2O парамагнитно, приобретает фиоле-
товую окраску при 100°, бурно разлагается при 250° на воздухе,
плохо растворимо в воде, растворяется в аммиаке и растворах
цианидов щелочных металлов.
ГексацианокобалътатД I) калия, K4[Co(CN)6], выделяется при
упаривании раствора дицианида кобальта в растворе KCN при
обычной температуре или в результате обработки растворов солей
кобальта(П) избытком KCN.
Соединение K4[Co(CN)6] представляет собой фиолетовые кри-
сталлы, которые необходимо хранить в твердом состоянии без
доступа кислорода, очень хорошо растворимые в воде. Водный
раствор K4[Co(CN)6] легко окисляется при нагревании под дей-
ствием кислорода, хлора или брома до соединения K3[Co(CN)6I
желтого цвета:
2K4[Co(CN)e] + Н2О + 1/2О2 = 2K3[Co(CN)e] + 2КОН
2K4[Co(CN)e] + Cl2 = 2K3[Go(CN)6] + 2КС1
В отсутствие воздуха K4[Co(CN)e] разлагает воду с образованием
K3[Co(CN)6] и выделением водорода:
2K4[Co(CN)e] + 2Н2О = 2K3[Co(GN)e] + 2КОН + Н2
Известна кислота H4[Co(CN)6] — бесцветная, растворимая
в воде и трудно растворимая в спирте, эфире и хлороформе.
Известны гексацианокобальтаты(П) типа Me*[Co(CN)6],
Ме^[Со(СМ)61, Me™[Co(CN)6]3 (где Me1 = Na+, К+, Ag+; Me11 =
КОБАЛЬТ
559
=Ca2+,Sr2+, Ва2+, Zn2+,Cd2+, Hg2+, Pb2+, Mn2+, Co2+, Ni2+; Meni=
= Al3+, Fe3+), Me*Men[Co(CN)6] (где Me1 = K+; № = Zn2+,
Co2+, Ni2+). Гексацианокобальтаты(П) тяжелых металлов плохо
растворимы в воде, имеют разнообразную окраску. В отличие от
устойчивых гексацианокобальтатов(Ш) они отличаются более низ-
кой устойчивостью.
Дитиоцианат кобальта, Co(SCN)2 -4Н2О, выпадает из раствора,
полученного при обработке сульфата кобальта(П) раствором
тиоцианата бария.
Соединение Co(SCN)2-4Н2О представляет собой фиолетово-
красные ромбические кристаллы, которые гигроскопичны и рас-
творимы в воде или спирте.
В присутствии тиоцианата калия KSCN пиридин образует
с ионом Со2+ кристаллический осадок розового цвета:
Со*+ + 2SCN- + 4C5H5N = [Co(C6H5N)4](SCN)2
Известны тетра-, пента- и гексатиоцианатокобальтаты(П) типа
Me*[Co(SCN)4], Me|[Co(SCN)5], MeJ[Co(SCN)6],
Соединение (NH4)2[Co(SCN)4] -4Н2О предста'вляет собой синие
игольчатые кристаллы и может быть получено по уравнению
СоС12 + 4NH4SCN = (NH4)2[Co(SCN)4] + 2NH4C1
Реакция протекает легко в присутствии смеси изоамилового
спирта и эфира или ацетона.
Соединения K2[Co(SCN)4] -4Н2О и Na2[Co(SCN)4] -8Н2О пред-
ставляют собой синие растворимые в воде кристаллы.
Соединение Hg[Co(SCN)J синего цвета, плохо растворимо
в воде.
Сульфид кобальта, CoS, встречается в природе в виде минерала
джейпурита и может быть получен сухим путем нагреванием (475—
700° без доступа воздуха) порошкообразного металлического
кобальта с серой (взятых в необходимых соотношениях), нагрева-
нием (700°) порошка кобальта в токе H2S, нагреванием серы с СоО,
восстановлением (при высокой температуре) CoSO4 окисью угле-
рода или водородом, углем, серой в токе водорода или восстанов-
лением силикатов Co2SiO4, CoSiOs серой (600—800°).
Со + S = CoS + 22,77 ккал CoS04 + 4С = CoS + 4СО
2СоО + 3S = 2CoS + SO2 2CoSiO3 -|- 3S = 2CoS + 2SiO2 + SO2
В растворах CoS получают действием (NH4)2S или Na2S на
соли кобальта(П) в щелочной среде, обработкой сероводоро-
дом растворов солей кобальта(П) при pH = 4,54, действием вос-
становителей (водород, SO2, СО, СН2О, СН3ОН, С2Н5ОН, НСООН,
Н2С2О4-2Н2О) на теплые растворы сульфата кобальта(П).
Сульфид кобальта представляет собой черные (или серо-сталь-
ные) гексагональные кристаллы с решеткой типа NiAs, с плотно-
560
ПОБОЧНАЯ ПОДГРУППА VIII ГРУППЫ
стью 5,45 г/с№ и т. пл. 1100° (в азоте); он плохо растворим в воде,
разбавленных минеральных кислотах и уксусной кислоте, вос-
станавливается при высокой температуре до металлического
кобальта водородом, углеродом, алюминием.
При нагревании CoS на воздухе (или в кислороде) образуются
СоО, Со3О4 и SO2:
684° 950°
3CoS + 5O2---► Co3O4+3SO2 CoS + 3/2O2--► Co0 + S02
Свежеприготовленный сульфид кобальта мгновенно раство
ряется в хлорной воде и царской водке:
2CoS + ЗС12 2СоС13 + 2S
2CoS + 6НС1 + 3HNO3 = 2СоС13 + 2NO + 2S + 4Н2О
Соединение CoS играет роль катализатора при окислении H2S
на воздухе или в парах воды, при гидрогенизации фенолов, полу-
чении тиофенолов и др.
Известны двойные сульфиды CoSJn2S3, Na2S-CoS.
S
Дисульфид кобальта, CoS2 или Со(^ | , получают длительным
XS
нагреванием с серой (175—225°) порошкообразного металличе-
ского кобальта или сульфида кобальта в течение 30—40 час.
После удаления избытка серы сероуглеродом продукт CoS2 высу-
шивают при 150°.
Дисульфид кобальта представляет собой парамагнитные куби-
ческие кристаллы (аналогичные пириту) с плотностью 4,27 г/см3;
взаимодействует с азотной кислотой и царской водкой.
Сульфат кобальта, CoSO4, получают продуванием смеси возду-
ха и SO2 над нагретым до 550—600° порошком СоО или дегидра-
тацией (~ 250°) кристаллогидратов CoSO4 "«НгО (п = 7, 6, 5, 4,
2, 1).
При прокаливании CoS (300—600°) образуется CoSO4, загряз-
ненный СоО. Сульфат кобальта CoSO4 представляет собой пара-
магнитные гексагональные кристаллы с плотностью 3,666 г/см3;
розовые кристаллы становятся фиолетовыми выше 500°, разлагают-
ся при нагревании на воздухе при 690—720°, превращаясь в СоО
и Со3О4, восстанавливаются до CoS при нагревании с серой, угле-
родом, смесью Н2 и H2S, металлическим кобальтом и др., гигро-
скопичны и растворимы в воде. При нагревании с сульфидом
кобальта(П) CoSO4 восстанавливается, причем реакция обратима:
3CoSO4 + CoS ч* 4СоО + 4SO2
Сульфат кобальта взаимодействует с СаО при 533°, с SrO при
431°, с ВаО при 328° и с MgO при 592°.
КОБАЛЬТ
561
Известны кристаллогидраты CoS04-nH20 (п = 7, 6, 5, 4, 2, 1), двойные
сульфаты типа Me2SO4-CoSO4-6H2O (где Me1 = К+, Rb+, Cs+, NH^).,
Me2SO4-CoSO4-(2 или 3) C5H6N .«H2O (где Mei = K+, NHJ), CoSO4 -MeIV(SO4)2
(где MeIV = Ti4+, Zr4+), различные аддукты, например CoSO4-(2 или 3)С6Н5М,
CoSO4-3N2H4, CoSO4-NH2OH-2H2O и основные соли CoSO4-4CoO-4H2O,
CoSO4 -5CoO-10H2O, CoSO4-3Co(OH)2.4H2O (синяя), 2CoSO4-3Co(OH)2 .
• 5H2O (фиолетовая)
Гептагидрат, CoSO4-7H2O или [Co(H2O)6]SO4 "НгО, выпадает
при упаривании (ниже 40,7°) растворов окиси, гидроокиси или
карбоната кобальта(П) в разб. H2SO4. Соединение CoSO4-7H2O
представляет собой парамагнитные красные моноклинные кристал-
лы, изоморфные сульфатам MenSO4 -7Н2О (где Me11 = Mg2+, Fe2+,
Ni2+, Mn2+), с плотностью 1,889 г/см3 и т. пл. 96—98°; они устой-
чивы на воздухе, легко растворяются в воде и плохо — в спирте.
В природе CoSO4-7H2O встречаются в виде минерала биберита.
При нагревании, а также под действием конц. H2SO4 или абсолют-
ного спирта CoSO4-7H2O дегидратируется с образованием низших
кристаллогидратов.
CoSO4 -6Н2О представляет собой моноклинные кристаллы
с плотностью 2,019 г/сл3; пентагидрат CoSO4>5H2O — красные
триклинные кристаллы с плотностью 2,134 г/сл3; тетрагидрат
CoSO4-4H2O — красновато-розовые кристаллы с плотностью
2,368 г/см3; дигидрат CoSO4 -2Н2О — красновато-розовые кри-
сталлы с плотностью 2,668 г/см3‘, моногидрат CoSO4 • Н2О — розо-
вые кристаллы с плотностью 3,125 г/см3.
Нитрат кобальта, Co(NO3)2 -6Н2О, выпадает в осадок при
упаривании (ниже 50—55°) растворов, полученных растворени-
ем металлического кобальта, окиси или карбоната кобальта(П)
в разб. HNO3.
Соединение Co(NO3)2-6Н2О представляет собой красные моно-
клинные призматические кристаллы с плотностью 1,80—1,83 г/см3
и т. пл. 38°. Оно гигроскопично, растворимо в воде, спирте, ацето-
не, метилацетате, диоксане, тетрагидрофуране, ацетофеноне,
ацетонитриле, дибутилфосфате, диметилформамиде и плохо раст-
воримо в конц. HNO3. При нагревании Co(NO3)2-6Н2О до ~55,5°
происходит потеря трех молекул воды, а при 100° оно разлагается
до окиси кобальта и выделяются пары окислов азота.
Известны также кристаллогидраты Co(NO3)2-пН2О (п = 9, 5, 4, 3, 2),
безводная соль Co(NO3)2, различные аддукты Co(NO3)2-nNH3 (п = 9, 6, 2),
Co(NO3)2-nCsHsN (п = 6, 4, 3, 2), двойные нитраты 3Co(NO3)2-2Bi(NO3)3
. 24Н2О, Co(NO3)2-MeIV (NO3)4-8H2O (где MeIV = Ce*+, Th*+), Co(NO3)2 •
• 6Cu(NO3)2 -42H2O, основные соли Co(NO3)2-3Co(OH)2-3H2O, Co(NO3)2 •
• 5Co(OH)2, Co(NO3)2-6Co(OH)2.
Ортофосфат кобальта, Co3(PO4)2-8H2O, образуется в виде
розового осадка с плотностью 2,769 г/см3 в результате обработки
растворов солей кобальта(П) раствором Na2HPO4 при комнатной
36-0101
562
ПОБОЧНАЯ ПОДГРУППА VIII ГРУППЫ
температуре. Если осаждение проводят из горячих растворов,
образуется кристаллогидрат Со3(РО4)2-4Н2О фиолетового цвета,
который после 4—5 дней хранения под маточным раствором пре-
вращается в Со3(РО4)2-8Н2О.
При нагревании окта- и тетрагидратов ортофосфата кобаль-
та образуются кристаллогидраты с меныпим содержанием кристал-
лизационной воды или безводная соль Со3(Р04)2 фиолетового
цвета:
140° 600°
Со3(РО4)2.8Н2О -— Со3(РО4)2.2Н2О Со3(РО4)2
— OX12V *“лх12М
Восстановлением ортофосфата кобальта водородом (550—1000°)
были получены фосфиды СоР, Со2Р.
Известны двойные ортофосфаты NaCoPO4, КСоРО4,
Na4Co(PO4)2, К6Со3(РО4)4, CsCoPO4-6Н2О, NH4CoPO4-6Н2О,
Co(U02)2(P04)2 и гетерополифосфаты Со3(РМо12О40]2-58Н2О,
Co3[PW4204o]2 -48Н2О.
Карбонат кобальта, СоСО3-6Н2О, кристаллизуется в виде
фиолетово-красных кристаллов из растворов солей кобальта(П),
насыщенных двуокисью углерода, карбонатами щелочных метал-
лов. Если растворы солей кобальта(П) недостаточно насыщены
углекислым газом, образуются основные соли [СоСО3]х[Со(ОН)21а,
состав которых зависит от концентрации и температуры исходных
растворов.
Известны также кристаллогидрат СоС03-0,5Н20, безводная соль СоСО3,
двойные карбонаты типа Ме2 СО3-СоСОз‘геН2О (где Me1 = Na+, К+,
NHp, основные карбонаты СоСО3-reCoO-mH20 (п = 1, 2, 3), например
2СоСО3-ЗСоО-4Н2О, которые могут быть выражены общей формулой
[СоСО3]х[Со(ОН)2]у.
Безводная соль СоСО3 образуется при нагревании (140°) СоС03-6Н20;
это парамагнитные двупреломляющие розовые ромбоэдрические мелкие кри-
сталлы, изоморфные MgCO3 или МпС03, с плотностью 4,2 г/см3', они плохо
растворимы в воде, растворяются в H2F2, НС1, H2SO4, HNO3 при нагревании,
разлагаются в интервале температур 427—477° в две стадии:
2СоСО3 —СоС03.СоО —2СоО
— L(j2
Хелатные соединения
Действием рубеановодородной кислоты на растворы солей Со2+
в присутствии ацетата натрия может быть выделен коричнево-
желтый осадок:
H2N-C-C-NH2 HN=C-C=NH + Со2+—>
II Н II
S S HS SH
rHN=C-C=NHl
При растворении Со(ОН)2 в ацетилацетоне или при действии
ацетилацетона на смесь растворов карбоната натрия и нитрата ко-
бальта(П) образуется ацетилацетонат кобальта(П) [Со(С5Н7О2)2].
КОБАЛЬТ
563
Соединение [Со(С6Н702)21 представляет собой блестящие крас-
ные игольчатые кристаллы, которые сублимируются без плавле-
ния, растворимы в воде, спирте, ацетоне, уксусной кислоте,
хлороформе (очищаются перекристаллизацией из спиртовых или
хлороформных растворов) и плохо растворимы в эфире. Известен
дигидрат [Со(С5Н7О2)2] -2H2O и аддукты [Со(С5Н7О2)21 ’ZNHg,
[Со(С6Н7О2)2] .2C5H6N, [Со(С6Н7О2)2] •2C6H5NH2.
Обработкой Na3[Co(NO2)e] избытком ацетилацетоната натрия
можно осадить Na[Co(C5H7O2)3] — розовые кристаллы, плохо
растворимые в воде и спирте. Известно также соединение
К[Со(С5Н7О2)3].
Помимо рассмотренных известны и другие соединения кобальта(П),
например: хлорат Со(С1О3)2-ггН20 (и = 6, 4, 2), аддукты Со(С1О3)2.2N2H*,
Со(С1О3)22C6H)2N4-10Н20, перхлорат Со(С1О4)2-nH2O (п = 9, 7, 6, 5, 2),
аддукты Со(С1О4)2-nNH3 (п = 6, 5, 4, 3, 2), Со(С1О4)2-nN2H4, Со(СЮ4)2.
• 6C5H5N-4Н2О, Со(С1О4)2 ^CsHsN, бромат Со(ВгО3)2-6Н2О, безводный
иодат Со(1О3)2 или кристаллогидрат Со(1О3)2-пН2О (п = 4, 2), аддукт
CoS2O4 ^CsHsN, тиосульфат CoS2O3-6H2O, дитионат CoS2Oe-8H2O, тритионат
CoS3O6-6H2O, тетратионат CoS4O6 ^CsHsN, сульфит CoSO3, пероксосульфат
CoS2O8•2C6HJ2N4 -8H2O, селенид CoSe, селенит CoSeO3, диселенит CoSe2O6,
селенат CoSeO4, двойные селенаты Me2SeO4-CoSeO4-6H2O (где Me1 = K+,
Rb+, Cs+, NHJ), теллурит CoTeO3-H2O, теллурат CoTe04, азид Co(N3)2,
двойные азиды Co(N3)2-KN3, Co(N3)2-NH4N3, амид Co(NH2)2, нитрит
Co(NO2)2, аддукты нитрита 2Co(NO2)2-3N2H4, Co(NO2)2-2С6Н12М4-10H2O,
Co(NO2)2-reCjHsN (n = 6, 3, 2), ацидосоли типа Me2MeII[Co(NO2)6] (где Me1 —
= K+, Rb+, Cs+ и Me11 = Ca2+, Sr2+, Ba2+, Pb2+), гипофосфит Co(H2PO2)2-
• 6H2O, фосфит CoHPO3-2H2O, гипофосфат Co2P2O6-8H2O, ортофосфаты
CoHPO4-nH2O, Co(H2PO4)2-nH2O, пирофосфат Co2P2O7, метафосфатыCo2P4O)2,
Co3P6O18-9H2O, Na2Co6(PO3)14, арсенит Co3(AsO3)2.4H2O, ортоарсенаты
Co3(AsO4)2-8H2O, Co3(AsO4)2-CoO, Co3(AsO4)2.CoO-H2O, Co3(AsO4)2-3NH3-
• 5H2O, Me2[Co2(AsO4)2] (где Me1 = Na+, K+), MeI[CoAsO4]-геНгО (где
Me1 = Cs+, NHJ), CoHAsO4-2H2O> Co2As2O7, антимонит CoSb2O4, тиоанти-
монит CoSb2S4, метаантимонат CoSb2O6, ванадаты CoV2O6-6/2H2O, Co2V2O7,
фторо- и оксифторованадаты Co[VF5]-7H2O. Co[VF4O]-7H2O, ниобаты
CoNb2O6, Co4Nb2O9, танталаты CoTa2O6, Ва3СоТа2О9, тиокарбонат CoCS3,
цианат Co(CNO)2, тетратиоцианатосоли Me2[Co(SCN)4] или Men[Co(SCN)4]
(где Me1 = К+, NHJ и Me11 = Hg2+), Me2[Co(SCN)4]-пН20 (где Me1 =
Na+, К+, Cs+,Ag+, NH4, x/2Ba2+), селеноцианат Co(SeCN)2, силикаты Co2SiO4,
CoSiO3-2H2O, Co3Si2O7 .H2O, K.2CoSi3O8, CaCoSiO4, CaCoSi2O8, CoZnSiO4,
гетерополисоли Co2[SiMo)2O40] ЛвЩО, Co2[SiW)2O40]-18H2O, германат
Co2GeO4, титанаты Co2TiO4, CoTiO3, тиотитанаты Co2TiS4, CoTiS3, борогидрид
Co(BH4)2, алюмогидрид Со(А1Н4)2, бораты Co3(BO3)2, Co(BO2)2, CoB6O)0,
CoB8O)3, CoB407-10H20, Co3B4O9-4H2O, фторобораты Co[BF4]2-6H2O,
Co[BF4]2-6NH3, формиат Co(HCOO)2 или Co(HCOO)2-2H2O, Co(HCOO)2.
• 2C6H5N, ацетаты Co(CH3COO)2, Co(CH3COO)2-4H2O, MeI[Co(CH3COO)3],
Me3ICo(CH3COO)4], Co(CH3COO)2-2N2H4, Co(CH3COO)2 .nC6H6N (n = 2,5),
оксалаты CoC2O4, CoC2O4-(2 или 4)H2O, CoC2O4-reNH3 (n = 6, 4, 2),
CoC2O4.2N2H4, CoC2O4«2C6H6N, MelC2O4-CoC2O4-nH20 (где Me1 = Na+,
K+, NHJ), малонаты CoC3H2O4-2H2O, Me2[Co(C3H2O4)2]-nH2O (где Me1 =
= K+, Cs+, NH|), сукцинат K2Co(C4H4O4)2, лактат Co(C3H5O3)2-3H2O, малат.
36*
564
ПОБОЧНАЯ ПОДГРУППА VIII ГРУППЫ
Со(С4Н5О5)2 (2 или 3) Н2О, тартраты Со(С4Н4О6)-2,5Н2О, К2Со(С4Н4О6)2-
• 4Н2О, цитраты Со3(СвН5О7)2-14Н2О, MeI[Co(CeH6O7)2]-nH2O (где Me1 =
= К+, NHJ), пальмитат Со(С)6Н3)О2)2, стеарат Со(С)8Н35О2)2, олеат
Со(С)8Н33О2)2, бензоат Со(С7Н6О2)2 .4Н2О.
Соединения трехвалентного кобальта
Известно ограниченное число простых соединений трехвалент-
ного кобальта. Они относительно неустойчивы, обнаруживают
окислительные свойства и легко гидролизуются с образованием
солей кобальта(П) и выделением кислорода.
Хотя квасцы кобальта(III) относятся к устойчивым соединениям
кобальта, все же они менее устойчивы, чем алюминиевые, желез-
ные(Ш) и хромовые(Ш) квасцы.
Известно очень много устойчивых координационных соедине-
ний кобальта(Ш), которые проявляют некоторое сходство с коор-
динационными соединениями хрома(Ш).
Безводная окись ко б альта (III), Со2О3, получается дегидрата-
цией Со2О3*иН2О (250°) или прокаливанием Co(NO3)2 при 180°.
Окись Со2О3 представляет собой черные гексагональные мел-
кие кристаллы с плотностью 5,34 г/см3, она превращается в Со3О4
при 265°, в СоО при 940° с выделением кислорода, окисляет соля-
ную кислоту до элементарного хлора, при нагревании взаимо-
действует с SiCl4 и восстанавливается водородом или метаном,
с различными окислами двухвалентных металлов образует соеди-
нения типа: Меп[Со’+О4].
2Со2О3 + 3SiCl4 = 4СоС12 + 3SiO2 + 2С12
Восстановление Со2О3 водородом или метаном рассматривалось
при описании получения металлического кобальта.
Кобальтат ы(Ш), Mg[Co2O4], Zn[Co2O4], Mn[Co2O4],
Fe[Co2O4], Со[Со2О41 или Co3O4, Ni[Co2O4], Cu[Co2O4], обладают
структурой шпинелей и получаются в виде черных порошков.
Кобалътат(Ш) кобалъта(1Г), Со2+[Со|+О4] или Со3О4, полу-
чают нагреванием порошкообразного металлического кобальта
при 300 —400° на воздухе, сжиганием на воздухе пирофорного
порошка кобальта, нагреванием СоО или Со(ОН)2 на воздухе
(или в кислороде) при температуре выше 100°, прокаливанием
Со2О3-нН2О при 300°, СоСО3 при 450°, СоС2О4 при 600°, Co(NO3)2
при 600—650°.
Соединение Со3О4 образует парамагнитные черные октаэдри-
ческие кристаллы со структурой шпинели и с плотностью
6,1—6,2 г/см3; при нагревании (940°) превращается в СоО
с высвобождением кислорода; восстанавливается до металличе-
ского кобальта при нагревании с Н2, С, СО, Na, К, А1, взаимо-
действует с C1F3, BrF3, H2S, S2C12 при нагревании, растворяется
в НС1 (с выделением хлора), в H2SO4 и HNO3 (с выделением кисло-
КОБАЛЬТ
565
рода) и в расплавленных щелочах.
СО3О4 ЗСоО 4“ F2O2 СО3О4 8НС1 = ЗСоС1з 4Н20 CI2
Со3О4 + 3H2SO4 = 3CoSO4 + ЗН2О + 1/202
Известны гидраты Со3О4>иН2О (п = 7, 6, 3).
Соединение Со3О4 применяется для изготовления стекла,
сильно поглощающего ультрафиолетовые лучи, а также в качестве
катализатора реакций: термического разложения КС1О3 и КМпО4,
окисления NH3, а также синтеза HCN из СН4, СО2, NH3, N2
и воздуха.
Гидраты окиси кобалъта{11Г), Со2О3-(3—1)Н2О, получают
окислением СоО или Со3О4 во влажном воздухе, окислением
в щелочной среде водной суспензии Со(ОН)2 под действием кисло-
рода воздуха, хлорной или бромной воды, Н2О2, K2S20s, КОВг,
NaOBr, действием растворов щелочей на фториды, хлориды, суль-
фаты или селенаты кобальта(Ш), продолжительным кипячением
водных растворов комплексов кобальта(Ш) или нагреванием
комплексов кобальта(Ш) со щелочами, гидролизом солей кобаль-
та(Ш) (хлорида, сульфата, нитрата), полученных окислением
соответствующих солей кобальта(П).
Соединение Со2О3-(3—1)Н2О представляет собой твердое чер-
ное вещество, плохо растворимое в воде или аммиаке, раствори-
мое в НС1 с выделением хлора и в большом избытке щелочей
с образованием гидроксокобальтатов(Ш) Ме*[Со(ОН)6]. Под
действием кислот на Со2О3-(3—1)Н2О образуются соединения
кобальта(П).
Гидраты окиси кобальта(Ш) обладают окислительными свой-
ствами (восстанавливаются до металлического кобальта водоро-
дом, углеродом, окисью углерода при нагревании) и служат
катализаторами разложения Н2О, NaOCl, Са(ОС1)2 и обесцвечи-
вания индигокармина.
Соединение Со2О3-Н2О или СоО(ОН) представляет собой чер-
ные ромбоэдрические кристаллы с плотностью 4,72 г!см3.
Трифторид кобальта, CoF3, получают действием фтора на
металлический кобальт или на безводные CoF2 или СоС12 при
300—400° в герметичном никелевом сосуде, а также действием
C1F3 на металлический кобальт при нагревании в автоклавах
из специальной стали.
Трифторид кобальта представляет собой парамагнитные корич-
невые триклинные кристаллы с плотностью 3,88 г/см3, т. пл. 1027°
и т. кип. 1327°; разлагаются при нагревании до 250° или под дей-
ствием воды, плохо растворимы в спирте, эфире, бензоле.
При нагревании CoF3 до 300° в токе СО2 или до 600—700°
в атмосфере фтора образуются CoF2 и фтор.
При действии кислорода воздуха на нагретый до 250—300° CoF3
образуется океифторид кобальта, а при 450—500°— окись Со3О4
566
ПОБОЧНАЯ ПОДГРУППА VIII ГРУППЫ
и выделяется фтор. При нагревании CoF3 с серой идет бурная
реакция:
4CoF3 + S = 4CoF2 + SF4
В атмосфере водорода CoF3 восстанавливается по уравнению
2CoF3 + Н2 2CoF2 + 2HF
При нагревании CoF3 восстанавливается до CoF2, а в присут-
ствии металлов Na, Mg, Zn, Fe, Al, Cu — до металлического
кобальта.
Известны фторосоли K3[CoF6], Na3[CoFe], [CoF3(NH3)3],
Кристаллогидрат, CoF3-3,5H2O или [Co(H2O)8HCoF6] -H2O,
образуется на аноде при электролизе насыщенного раствора дифто-
рида кобальта в 40%-ном растворе HF (платиновые электроды)
или при действии фтора на раствор дифторида кобальта в водном
растворе HF.
Соединение CoF3-3,5H2O представляет собой зеленый порошок
с кристаллической структурой типа ReO3 и с плотностью 2,314 г/см3,
оно плохо растворимо в воде, спирте, эфире и бензоле, является
окислителем по отношению к KI, Н2С2О4 -2Н2О, Me+NO2, NH2OH,
N2H4, превращается в CoF2 и Со2О3-ЗН2О под действием влаж-
ного воздуха.
Трифторид кобальта применяется в качестве фторирующего
агента и катализатора в реакциях фторирования многих органи-
ческих соединений.
Трихлорид кобальта, Со2С18 или Со[СоС16], получают пропу-
сканием хлора через спиртовый раствор дихлорида кобальта,
охлажденный до —60°, действием сухого НС1 на Со2О3 при —5°
в темноте и под слоем абсолютного эфира, действием ацетилхлори-
да на Со2О3 при низкой температуре.
Трихлорид кобальта представляет собой темно-зеленый (жел-
тый при —60°) порошок; Со2С16 довольно неустойчив, окисляет
KI до элементарного иода и разлагается под действием света, теп-
ла, спирта или воды:
СО2С1() 2C0CI2 Н- CI2
ЗСо2С16 + 15Н2О 2Со[Со(Н2О)3С13]2 + 6НС1 + 3/2О2
Во влажном воздухе Со2С18 превращается в кислоту
Н[Со(Н2О)3С13] синего цвета.
Известна неустойчивая хлоросоль Cs[CoCl4] зеленого цвета.
Цианид кобальта, Co(CN)3, получают в виде твердого синего
вещества нагреванием кислоты H3[Co(CN)6] до 120°. При концен-
трировании водного раствора цианида кобальта(Ш) выделяется
дигидрат Co(CN)3-2H2O красного цвета.
Сульфид кобальта, Co2S3, образуется совместно с CoS и CoS2
при нагревании металлического кобальта с серой (350—400°)
или Со2О3 с H2S (600—650°), а также при нагревании солей
кобальта(П) (СоСО3, СоС12 и др.) с серой и К2СО3 или Na2CO3
КОБАЛЬТ
567
(1200—1300°). В растворе Co2S3 (загрязненный CoS2) получают при
действии (NH4)2S, K2S или Na2S на гексаммины кобальта(Ш).
Соединение Co2S3 представляет собой диамагнитные серые,
как сталь, блестящие кристаллы с плотностью 4,8 г/см3; они плохо
растворимы в воде, с трудом взаимодействуют с концентрирован-
ными кислотами, при нагревании превращаются в CoS и CoS2.
Тиокобалътат(11Г) кобалыпа(1Г), Co2+[Co2+S4] или Co3S4,
встречается в природе в виде минерала линнеита и может быть
получен сильным нагреванием смеси (Co2S3 с S и К2СО3, а также
прокаливанием CoS в токе H2S в интервале 400—500°.
Соединение Co3S4 представляет собой черно-серый аморфный
порошок или парамагнитные кубические кристаллы с плотностью
4,86 г!смя, разлагающиеся при 680° по уравнению
Co3S4 2CoS + CoS2
Сульфат кобалыпа(11 Г), Co2(SO4)3-18Н2О, выделяется на охла-
жденной! до 0° аноде в ходе электролиза насыщенного раствора
сульфата кобальта(П) в 40%-ной H2SO4; другой метод получения
состоит в окислении фтором или озоном насыщенного раствора
CoSO4*7H2O в 8 н. H2SO4 (ниже 0°). Электролиз осуществляют
в платиновой ячейке, которая служит анодом, катод также пла-
тиновый.
Соединение Co2(SO4)3-18Н2О представляет собой зеленовато-
синие игольчатые кристаллы, относительно неустойчивые (не
изменяются в течение нескольких дней в отсутствие воды на
холоду), разлагающиеся водой с выделением кислорода, раство-
ряющиеся без разложения в разб. H2SO4 на холоду и проявляю-
щие окислительное действие по отношению к HCl, KI, НСООН,
альдегидам и спиртам.
Co2(SO4)3 Н20 = 2CoS04 -)- H2SO4 V2O2
При взаимодействии сульфатов щелочных металлов или суль-
фата аммония с сульфатом кобальта(Ш) образуются квасцы
MeICo3+(SO4)2-12Н2О (где Me1 = К+, Rb + , Cs+, NH*); это синие
октаэдрические кристаллы, плохо растворимые в воде, устойчивые
в сухом воздухе при комнатной температуре.
Карбонат кобальта, Со[Со(СО3)3], выделяется в виде зеленого
осадка из раствора, полученного обработкой Н2О2 или NaOCI
раствора карбоната щелочного металла с солью кобальта(Ш).
Координационные соединения трехвалентного кобальта
Известно очень много комплексных соединений кобальта(Ш)
с координационным числом шесть, которые по числу координа-
ционных сфер классифицируются на моно-, би, три-, тетра- или
полиядерные, а по природе координированных групп — на аммины,
аквоаммины, ацидоаммины, аквосоли, ацидосоли, ацидоаквосоли,
ацидоамминосоли, гидроксосоли, аквогидроксосоли.
568
ПОБОЧНАЯ ПОДГРУППА VIII ГРУППЫ
Благодаря сильно выраженной склонности кобальта(Ш) к об-
разованию координационных соединений, разнообразию лигандов,
входящих во внутреннюю координационную сферу, и суще-
ствованию изоморфных форм имеется очень большое число коор-
динационных соединений кобальта(Ш). Большинство их полу-
чают окислением простых или комплексных соединений кобаль-
та(П) кислородом воздуха, Н2О2 или КМпО4 в щелочной или
нейтральной среде.
Моноядерные координационные
соединения
Моноядерные координационные соединения кобальта(Ш) могут иметь
следующие общие формулы: {CoA6]Y3, [CoA6X]Y2, ICoA4X2]Y, [СоА3Х3],
МеЧСоАзХз], Ме|[СоА3Х3], Ме^СоАгХ*], Ме^[СоАХ6], MeJ[CoXe], MeJ[CoYXs],
Me|[CoY2X4], в которых А — электронейтральные молекулы, У и X —
кислотные радикалы, Me1 — различные одновалентные катионы.
1. Комплексы типа [CoA6]Y3 включают следующие соединения. Гекс-
аммины [Co(NH3)6]Y3 (желтые или коричневые, называемые также лутеосоля-
ми), где Y = F-, С1-, Вт-, I", ОН", SCN-, С1О2, СЮ3, СЮр, Ю3,
NOj, NOj, MnOj, ReOj, HCOj, CH3COO-, VzSO3", V2SO?", J/2S2Oi-, ^O3",
V2S2O2-, V2SeO|-, V2CO|", 1Г2С202-, i/2CrO|", V2Cr2O?", V2MoOr, V2WO|-,
1/2HPO|", 1/зРО|”, 1/3[Fe(CN)6]3", 1/4[Fe(CN)6]!1-. Гексагидроксиламины
[Co(NH2OH)6]Y3 (желтые), где Y = Cl~, Br", N03, OH", V2S0?", VzCzO3".
Аквопентаммины [Co(NH3)5(H2O)]Y3 (красновато-розовые), где Y = F", Cl",
Вг-, I-, C1O2, BrO3, ClOj, NOj, i/2SOl-, V2C2OJ-, VsPO^", V4P2O*",
1/3[Fe(CN)6]3-. Аквоаммины, содержащие катионы
(NH3)4 13+
NH3"13+
Co C2H5NH2
H2o
Co En2
H2O_
Диаквотетраммины [Co(NH3)4(H2O)2]Y3 (пурпурно-красные, иначе розео-
тетраммины), где Y = Cl", Вг", I", ClOj, ОН", NOj, VzSOI", V2C2O|-,
CH3COO". [BF4]-, VsITlClg]3-, 1/4P2Oj". Диакводипиридиндиаммины
[Co(NH3)2Py2(H2O)2]Y3, где Y = Cl", Br", HSOj, NOj, V2S2O2- Диакводи-
этилендиамины [CoEn2(H2O)2JY3, где Y = C1-, Bi", ClOj, NO3, NCS",
VzSOI". Триаквотриаммины [Co(NH3)3(H2O)3]Y3 (с соответствующими сте-
реоизомерами), где Y = Cl", NOjjVaSO^", ClOj, VjPtCle], триаквоамминоэти-
лендиамины (CoEn(NH3)(H2O)3]Y3, где Y = Cl", Br", NOj. Триэтиленди-
амины [CoEn3]Y3 (желтые), где Y = F", Cl", Br~, I", OH", NO3, SCN",
1/2SO|~, !/2С0^~. Трипропилендиамины [CoPr3]Y3, где Pr = H2N — CH2 —
— CH — CH3 и Y = Cl", Вг-, I". Трибутилен-2,3-диамины [CoBu3]Y3,
NH2
где Bu = H3C — CH — CH — CH3 и Y = Br-, I", SCN". Три-тгарамс-цикло-
I I
nh2 nh2
пентан-1,2-диамины [Co(C6Hi2N2)3]Y3, где Y = Cl", I", ClOj. Три-о-фени-
лендиамины [Co(O — C6H8N2)3]Y3, где Y = C1-, I", NO3, OH", 1/3[Fe(CN)6]3".
Три-транс-циклогександиамины{Со{С6Н)0(НН2)2}3]У3, где Y=C1", Br", ClOj,
NO3. Тритриметилендиамины, три-1-стильбендиамины, соли тридипиридилко-
КОБАЛЬТ
569'
бальта(Ш) [Co(C)oH8N2)3]Y3, где Y = С1-, ClOj, r/2SO|-. Соли тригуанидинко-
бальта(Ш), соли бис-триаминпропанкобальта(Ш) [Co(H2N — СН2 —
—CH СН2 — NH2)2]Y3, где Y = С1~, Вг-, I”, SCN-. Соли бис-ди-
I
nh2
где Y =
С1
(SCN)2’
ЗН2О, [Со(КНз)в]^Оз,
•6Н2О, [Co(NН3)в](С1О2)з • Н2О,
[Co(NH3)6] gQ®, [Со(ГШз)в](ВеО4)3.
[Со(МНз)б]2(8О4)3 • 5 или 4Н2О,
[Co(NH3)eI2g%\
’------------------------- ------),
[Co(NH3)8][Fe(CN)8],
атилентриаминкобальта(Ш) [Co(H2N — CH2 — CH2 — NH — CH2 — CH2 —
NH2)2]Y3. Амминсодержащие катионы [Eo^^s^^2^2J , j ,
Нъ,]”. №У,1Г- KT‘
[CoEn2(C6H)2N2)]3+, [CoEh2(C12H8N2)]3+, [CoEn2(C12H)2N2)]3+.
Как правило, гексаммины получить очено трудно. Они образуются при
действии аммиака под давлением или в присутствии катализаторов, таких,
как активированный уголь.
Примеры гексамминов: [Со(ННз)6]Р3-2Н2О; [Co(NH3)e]Y3,
= С1-, Вг-, I-, SCN-, ClOj, IO3, NO2, NOj, MnOj, HCOO-; [Co(NH3)6^
[CO(NH3)6]g1()^, [Co(NH3)6]g2, [Co(NH3)e]^3-
[Co(NH3)e]gOe • H20, [СоГННз)6](ОН)з
[Со(КНз)в](С103)з-Н20, [Со(ННз)6]^°з)2’
• 2H2O, [Со(КНз)6](СНзСОО)з • 3H2O,
[Co(NH3)e]|Os [Co(NH3)6]f°4, [Со(ННз)б1§Й4,
[Co(NH3)6]2(SeO4)3-5H2O, [Co(NH3)6]2(C03)3-7H2O, [Со(ННз)в]2(С2О4)3.4Н2О,
[Co(NH3)6]2(HPO4)3 • 4H2O, [Co(NH3)6]P04 H20, [Co(NH3)e][Fe(CN)e],
[Co(NH3)6]4[Fe(CN)6]3.
Примеры аквопентамминов: [co^q3^5Jy3, где Y = F-, Cl-, Br-, I-,
NO3; [Со^2оз)б]с13-nNH3 (где n = 6, 3, 2, 1); [со^з)5]Вг3-nNH3 (где-
n = 10, 3); [Co(NJJ3)5]l3.10NH3, [Со^з)б](СЮ3)з -H2O, ['
•H2°, [co^q3^5](C1O4)3-H2O, [со^з)з]2(8О4)з-ЗН2О,
Y = Br-, I-, ClOj, ClOj, NOj; [со^дз)б]РО4-2H2O, [со(^з)5]4(Р2О7)3 •
[COH2O3)5]Ire(CN)el'0’5H2°- [Co^JJ3)5]2(C2O4)3.4H2O.
диакводиэтилендиаминов: [СоЕн2(Н2О)2]С1з • 2H2O,.
триэтилендиаминов: [СоЕп3]С13-ЗН2О, [СоЕп3]Вг3.
[СоЕпз]У3, где Y = ОН-, NOj, SCN-, !/2СОз2-;
[СоЕпз]^4-5Н2О, И-соЕпз]С^4?в -5Н2О,
трибутил-2,3-диаминов: (СоВпз]Вг3 -2Н2О,
LoH2O
> n(NH3)5]SO4
’ Loh2o Jy 1
(ВгОз)з •
где
• 12Н2О,
Примеры
[СоЕп2(Н2О)2]Вг3-2Н2О;
• ЗН2О, [СоЕпзПз-Н2О,
[СоЕп3]2(8О4)з-4Н2О,
Г1
[Z-CoEn3]z _С4н4Об и
[СоВпз]13-2Н2О, [СоВп3](8СК)з-6Н2О. Координационные соединения типа
[СоЕн3]Уз проявляют оптическую изомерию.
2. Комплексы типа [CoA6XY2] охватывают следующие соединения. Гидро-
ксопентаммины [Co(NH3)6OH]Y2, где Y = С1—, Вг-, I-, ОН-, NOj, r/2SO^-,
1/282О|-,1/2820|-.Гидроксоакводипиридиндиаммины[Со(КНз)2Ру2(Н20)0Н]'У2
где Y = Cl-, Вг-, NOj, NCS-, 1/2S2Og-. Ацидопентаммины [Co(NH3)5X]Y2,
где X = F-, Cl-, Вг-, I-, С1О2, СЮ3, С1О4, BrOj, IOj, NO2, ONO-, NO3,
NCS-, HCOj, HCOO-, CH3COO-, SO|-, ЙОГ, S2O!-, S2O2-, SeOr, SeO2-.
CO§-, HPO^-, C2O^-, CrO2-, MoO£- и Y = F-, Cl-, Вг-, I-, NO3, ClOj,
570
ПОБОЧНАЯ ПОДГРУППА VIII ГРУППЫ
ClOj, ClOj, IOj, ReOj, 1/2SOI-, V2SO2-, i/2S20f-, i/2SeO«-, V2CO§-,
i/2Cr2O?-, i/2MoOl', V2WOJ-, 1/3[Co(NO2)6]3-, i/4P2O|-, i/2[PtCl6]2-, CH3COO-,
V2C2OJ-. Нитрозопентаммины [Co(NH3)5NO]Y2, где Y = Br~, I-, ClOj,
IO", NOj, 1/2SO|", i/2CrO?", Ацидоаммины ICoEn2NH3X]Y2, где
X = F-, C1-, Вт-, OH-, NO3, NO2, NCS- и Y = F-, Cl", Br-, ClOj, NOj,
NO2, NCS-, V^OI-, d — CjoH14BrS04 с соответствующими стереоизомерами,
нейтральными комплексами типа [Co(NH3)5X], где X = РО|~, [Fe(CN)6]3",
С6Н6О|-. Комплексы, производные пентамминов, содержащие анионы
{Со(КНз)6Р207]-, [Co(NH3)5C6H(C02)5]2-, [Со(ХНз)6С6(С02)6]3-. Ацидоаквотет-
раммины [Co(NH3)4(H20)X]Y2 (с соответствующими изомерами), где X =
= С1-, Вт-, ОН", NO3, NO2, CN-, NCS- и Y = С1-, Вг~, NOj, VzSO^,
V2S2O;J-, V2SeO|". Ацидоакводиэтилендиамины [CoEn2(H20)X]Y2 (с соответ-
ствующими изомерами), где X == Cl-, Вт-, 0H-, NO3, NCS" и Y = C1-, Вг-,
I", NOi, V2S0|", V2S2O|", !/2HP0|-, VzCsOf". Ацидоаквотетраммины
[Co(NH3)4(H2O)xrY (с соответствующими стереоизомерами), где X = SOI-,
SOJ-. S20f-, SeO|", и Y = V2SO§-, OH", CN~, NCS", Cl", Вт", I", ClOj,
HSOf, NO3, V2SeO|-, V2S2O|", V^Og-, V2CrO|-. Ацидодиаквотриаммины
tCo(NH3)3(H2O)2Cl]Y2, где Y = Cl", Br-, NO7, V2SO^-, VzSeO2-. Ацидодиак-
воэтилендиаминаммины, например [CoEn(NH3)(H2O)2Cl]C2O4. Ацидодиакво-
триаммины, например [Co(NH3)3(H20)2S04]2S04-Н20. Ацидотриакводиам-
мины, например [Со(ХН3)2(Н2О)зС1]8О4-Н20.
Примерыгидроксопентамминов: [Co(NH3)50H]Cl2 -Н2О, [Co(NH3)5OH]Br2-
• Н2О, [Co(NH3)5OH]I2, [Co(NH3)5OH](OH)2.1,5H2O, [Со(ХН3)50Н]8О4 •
• 2Н2О, [Со(ХН3)6ОН]82О6-2Н20; ацидопентамминов: [Co(NH3)5F]F2-2Н2О,
[Co(NH3)5F]C12.2H2O, [Co(NH3)6F]Bt2 -Н2О, [Co(NH3)5F](NO3)2 -Н20,
!Со(МНз)6С1]С12, [Со(ХНз)5С1]Вг2, [Co(NH3)5C1]I2, [Co(NH3)5C1](NO3)2,
[Co(NH3)5C1]SO4-2H2O, [Co(NH3)5C1]CO3 -4,5H2O, [Co(NH3)5C1]2P2O7-3 или
4H2O, [Со(ХНз)5С1]С2О4, [Co(NH3)5Br]Cl2, [Co(NH3)5Br]Br2, [Со(ХНз)6Вг]12,
[Co(NH3)5Bt](C1O4)2 -H2O, [Co(NH3)5Br](NO3)2, [Co(NH3)6I]C12, [Со(МНз)61]Вг2,
[Co(NH3)6I]I2, [Co(NH3)5I](C1O3)2, [Co(NH3)6I](C104)2, [Co(NH3)6I](NO3)2,
[Co(NH3)5I]SO4, [Со(ННз)61](СНзСОО)2, [Co(NH3)5NO3]Y2, где Y = Br-, I-,
СЮ3, CIO7, NOj, VzSeO?-; нитрозопентамминов: [Co(NH3)5NO]Br2-1,5H2O,
ICo(NH3)6NO]I2 -2H2O, [Co(NH3)5NO](NO3)2 -1,5H2O, [Co(NH3)5NO]SO4 •
H2o.
3. В комплексы типа [CoA4X2]Y с соответствующими стереоизомерами
(празео- и виолеосоли, кроцео- и флавосоли) включены следующие соедине-
ния. Ацидогидроксотетраммины [Co(NH3)4(OH)X]Y, где X — F", С1~ и Y =
= С1-, N03, NO2, V2S2O|-. Ацидонитротетраммины [Co(NH3)4N02X1Y,
где X = С1-, Br-, NCS", НСО2, C6H4Nj и Y = C1-, Br~, I", NCS", NO3.
Диацидодипиридиндиаммины [Co(NH3)2Py2X2]Y, где X = C1", NO2, ONO-
и Y = C1-, Br-, NOjb NCS", V2S20|". Диацидоэтилендиаминдиам-
мины [Co(NH3)2EnX2]Y (с соответствующими стереоизомерами), где
X = C1-, Вг- и Y = С1-, Br-, I-, HSOj, NO3, NCS-, V2S20§-
Диацидотетрапиридины [CoPy4X2]Y, где X = Cl- и Y = C1-, Br-, NO3, HSO4,
HSeO4, MnOj, VzSzO;?-, V2CrO|". Диацидотетрапиколины, например
[Co(C6H7N)4Cl2]CL Диацидодиэтилендиамины [CoEn2X2]Y (с соответствую-
щими стереоизомерами), где X = F-, Cl-, Br-, I-, OH-, NO2, ONO", CN_,
NCS-, HCOO-, CH3COO" и Y = F-, Cl", Br", I-, ClOj, NO3, NCS" ClOj,
V2S2O2-, i/2sO2-, V2S2Or, V2SeO|", V2HPO42-, VzCrO2-, V2MoO2- динитро-
пропилендиаминэтилендиамины, содержащие катионы [CoPnEn(NO2)2]+.
Дихлор-(или динитро-)триметилендиаминэтилендиамины, содержащие катио-
ны [СоТпЕпС12]+, [CoTnEn(NO2)2]+, где Tn = H2N — (СН2)3 — NH2- Диаци-
додипропилендиамины, содержащие катионы [СоРп2С12]+, [CoPn2(NO2)2]+,
[CoPn2(NCS)2]+. Диацидотетраммины [Co(NH3)4X2]Y, (с соответствующими
стереоизомерами), где Х2 = СО|~, С20|", СгО?-, МоО^~, [BeF4]+,
СзН2О£- и Y = F", С1-, Br-, I-, С103, СЮ4, ОН-, NO5, IO3, ReOl, HSOj,
VaSO2-, x/2S2O2-, VzSeOj-, V2CO2-, HCOj, ^CrO2-, VzCrzO2-,
КОБАЛЬТ
571
1/2МоО?*, 1/2WO^_. Диацидодиэтилендиамины, содержащие катионы
[CoEn2SO3]+, [CoEn2S04]+, [CoEn2S2O3]+, [CoEn2CO3]+, [CoEn2C2OJ+. Диаци-
додипропилендиамины, содержащие катионы [CoPn2S03]+, [CoPn2CO3]+,
1СоРп2С204]+. Комплексы, содержащие анионы [Co(NH3)4(SO3)2]_,
ICo(NH3)4(CrO4)2]-, [CoEn(NH3)2(SO3)2]~, [Co(En)2(S2O3)2]~, [CoPn2(SO3)2]~
Диацидоаквотриаммины, содержащие катионы [Co(NH3)3(H2O)C12]+,
ICo(NH3)3(H2O)(NO2)2]+, [Co(NH3)3^H2O)BrCl]+, [CoEd(NH3)(H2O)C12]+,
(Co(En)(NH3)(H2O)Br2]+, [CoEn(NH3)(H2O)BrCl]+, [Co(NH3)3(H2O)SO3]+,
(Co(NH3)3(H2O)C2O4]+, [Co(NH3)3(H2O)CrO4]+.
Примеры диацидодипиридиндиамминов: [CoPy2(NH3)2Cl2]Cl,
[CoPy2(NH3)2Cl2]NO3, [CoPy2(NH3)2(NO2)2]Br, [CoPy2(NH3)2(NO2)2]NO3,
ICoPy2(NH3)2(ONO)2]Br.H2O, [CoPy2(NH3)2(ONO)2]I -H20,
fCoPy2(NH3)2(ONO)2J2S2O6 -4H2O, [CoPy2(NH3)2(ONO)2]NO3 -H2O,
tCoPy2(NH3)2Cl(NO2)]NO3 -H20, диацидоэтилендиаминдиамминов:
lCoEn(NH3)2Cl2]C1.0,5H2O, [CoEn(NH3)2Cl2]HSO4-H20, [CoEn(NH3)2Br2]Br;
диацидо диэтилендиаминов: [CoEn2Cl2]Cl-H20, [СоЕп2С12]Вг-Н2О,
[CoEn2Cl2]2SO4-2H2O, [CoEn2Cl2]2S2O6.H2O, [CoEn2BrCl]Cl, [CoEn2BrCl]Br,
[CoEn2BrCI]2SO4, [CoEn2BrCl]NO3, [CoEn2(OH)Cl]Br, [CoEn2(0H)Cl]NO3,
fCoEn2(NO2)Cl]Cl, [CoEn2(NO2)Cl]Br, [CoEn2(NO2)Cl]NO3, [CoEn2(NCS)Cl]Cl,
ICoEn2(NCS)Cl]Br, [CoEn2(NCS)Cl]I, [CoEn2(NCS)Cl]ClO4, [CoEn2(NO2)Br]NO2,
[CoEn2(N02)Br]Br, [CoEn2(NO2)Br]I, [CoEn2(NO2)Br]NO3, [CoEn2(NCS)Br]Br,
[CoEn2(NCS)Br]NO3, [CoEn2(NCS)Br]2SO4, [CoEn2(N03)(N02)]N03,
[CoEn2(NO2)(NCS)]Cl -2H2O, [CoEn2(NO2)(NCS)]Br, [CoEn2(N02)(NCS)]I,
iCoEn2(0NO)(NCS)]NO2-H2O, [CoEn2(ONO)(NCS)]NCS; диацидотетрамминов:
[Co(NH3)4C03]F.3H20, [Co(NH3)4CO3]C1, [Co(NH3)4C03]N03.0,5H20,
(Co(NH3)4C03]2S04-3H20, [Co(NH3)4C2O4]C1, [Co(NH3)4C204]Br,
[Co(NH3)4C2O4]C1O4, [Co(NH3)4C2O4]IO3 -H2O, [Co(NH3)4C204]2S04 -2H2O,
[Co(NH3)4C204]N03-H20, [Co(NH3)4CrO4]NO3-0,5H2O, [Co(NH3)4Cr04]2Cr04-
• 2,5H2O, [Co(NH3)4CrO4]2Cr2O7-2H2O, [Co(NH3)4Mo04]N03-0,5H20,
[Co(NH3)4MoO4]2MoO4 -3H2O, [Co(NH3)4BeF4]Cl .5H2O, [Co(NH3)4(C3H2O4)]C1 •
•H2O.
4. Комплексы типа [CoA3X3] охватывают весьма разнообразные ком-
плексы типа неэлектролитов, например триацидотриамминкобальт(Ш)
lCo(NH3)3F3], [Co(NH3)3Cl3], [Co(NH3)3(IO3)3], [Co(NH3)3(NO2)3],
[Co(NH3)3(NO2)2C1], [Co(NH3)3(NO3)3], [CoEn(NH3)(NO2)3], [CoPn(NH3)(NO2)3],
[Co(NH3)3(C2O4)C1] -0,5H2O, [Co(NH3)3(C2O4)(NO2)], [Co(NH3)3(C2O4)(NCS)],
[Co(NH3)3BO3]; триацидоакводиаммины, например [Co(NH3)2(H2O)(IO3)3];
триацидобепзотриазолдиаммины, например [Co(NH3)2(C6H5N3)(NO2)3]; хелат-
ные соединения с аланином [Со{СН3 — CH(NH2) — СОО}3], с пиколиновой
кислотой [Co(NC6H4COO)3], с оксином [Co(C9H6ON)3].
При действии а-нитрозо-Р-нафтола на ион Со3+ в солянокислой среде
образуется объемистый пурпурно-красный осадок [Co(C6H6O2N)3], плохо
растворимый в кислотах и слабых щелочах и растворимый в спирте, уксус-
ном эфире, пиридине и хлороформе.
Со3* + 3
Обрабатывая растворы солей кобальта(Ш) ацетилацетоном, можно
выделить ацетилацетонат [Со(С6Н7О2)3], который представляет собой зеленые
572
ПОБОЧНАЯ ПОДГРУППА VIII ГРУППЫ
кристаллы, плавящиеся при 241° и растворимые в органических раство-
рителях.
5. Комплексы типа Ме1[СоА3Х2], Ме3[СоА3Х3], где А = NH3 и X =
= С2О|_, SOj~, включают соединения, содержащие анионы
[Co(NH3)3(C2O4)2]-, [Co(NH3)3(SO3)3]3-
6. Комплексы типа Ме1[СоА2Х4] (с соответствующими стереоизомерами)
известны для следующих классов соединений. Тетрацидодиамминсоедине-
ния, где А = NH3, V2En, X = Cl~, NO2, V2SO2-, V2C20i-, !/2C3H2O2-,
Mei = Na+, K+, NHJ, Rb+, Cs+, Ag+, T1+, Hg|+, !/2Mg2+, ^Ba2*, V2Cd2+,
V2Zn2+, 1/2Pb2+, Cu+, ^Mi2*, 1/3[Co(NH3)6]3+. Тетрацидодиаквосоли
Mei[Co(H2O)2(CN)4], где Mel = Li+, Na+, K+, NHj, Ag+, */2Ca2+, !/2Sr2+,
x/2Ba2+. Этилендиаминтетрацетатокобальтаты Mei[Co(OOC — CH2)2N —
— CH2 — CH2 — N(CH2COO)2], где Mei = Li+, Rb+, Cs+, V2Mg2+, ^Ca2*,
V2Ba2+, V2Co2+, 1/3[Co(NH3)8]3+.
7. Комплексы типа Me2[CoAX5] охватывают следующие соединения.
Пентацианоаквокобальтаты Me2[Co(H2O)(CN)5]-геНгО, где Mel = Li+, Na+,
К+, NHJ', Ag+, !/2Ca2+, x/2Sr2+, V2Ba2+, V2Cd2+. Тетранитронитрозилакво-
кобальтаты Me2i[Co(H2O)(NO)(NO2)4], где Mel = Na+, K+. Диоксалатогид-
роксоаквокобальтаты Me|[Co(H2O)(OH)(C2O4)2], где Mei = Na+, K+.
8. Комплексы типа Ме|[СоХ6] включают следующие соединения. Без-
водные или гидратированные галогено- (или псевдогалогено)соли, где X =
= F-, С1-, CN- и Mel = К+, NHJ, Ag+, Т1+, Cu+, V2Ba2+, V2Sr2+, V2Zn2+,
VzCd2*, V^n2*, V2Ni2+, 1/3[Co(NH3)5H2OP+, 1/3[Co(NH3)e]3+, i/3[CoPn3]3+,
1/3[CoPnEn2]3+. Пентацианогалогенокобальтаты, содержащие анионы
[Co(CN)5Br]3~, [Co(CN)5I]3-. Пентацианогидроксокобальтаты, содержащие
анион [Co(CN)5(OH)]3~. Безводные или гидратированные гексанитрокобаль-
таты Me2[Co(NO2)6], где Mel = Li+, Na+, К+, Rb+, Cs+, NH|, Tl+, Ag+,
V2Sr2+, V2Ba2+, V2Cd2+, V2Pb2+, !/3[Со(КН3)6]3+, 1/2[Co(NH3)6(NO2)]2+,
1/2[Co(NH3)5(C1O3)]2+, [Co(NH3)4(NO2)2]+. Трисульфито-, трикарбонато-,
триоксалато-, трималопато-, дитиосульфатодинитрозил-, тринистеинатоко-
бальтаты, содержащие анионы [Co(SO3)3]3-, [Со(СО3)3]3-, [Со(С2О4)3]3-,
[Co(C3H2O4)3]3-,[Co(NO2)2(S2O3)2]3-,
/ ,h2nx
Со I * ^СН — CH2S
\ \ ОСЯХ
Примеры гексагалогено-(или гексапсевдогалогено-)солей: K3[CoF6],
Co[CoCle], Na3[Co(CN)e]-(2 или 4) Н20, (NH4)3[Co(CN)6]-Н2О, Tl3[Co(CN)6] •
• 6Н2О или безводных солей [Co(NH3)6][Co(CN)6], [CoPn3][Co(CN)6],
[CoPnEn2][Co(CN)6], [Co(NH3)5H2O][Co(CN)6] -Н2О; гексанитрокобальтатов:
Li3[Co(N02)e] -8Н2О, Na3[Co(NO2)e], K3[Co(NO2)e] -4Н2О, [Co(NH3)e][Co(NO2)e],
[Co(NH3)6(NO2)]3[Co(NO2)6]2.2H2O, [Co(NH3)4(NO2)]3[Co(NO2)6],
[Co(NH3)5(C1O3)]3[Co(NO2)6]2; трисульфитокобальтатов: Li3[Co(SO3)3]-4H2O,
Na3[Co(SO3)3].4H2O, K3[Co(SO3)3]-6H2O, (NH4)3[Co(SO3)3-H2O,
Ca3[Co(SO3)3]2*nH2O, Ba3[Co(SO3)3]2-nH2O, Bi2[Co(SO3)3]2*27H2O.
Гексацианокобальтат(П1) калия, K3[Co(CN)6], выпадает в виде светло-
желтых кристаллов при упаривании раствора, полученного действием избыт-
ка KCN на раствор соли кобальта(П) в присутствии кислорода воздуха
или при нагревании. При обработке K3[Co(CN)e] минеральными кислотами
(H2SO4 или HNO3) образуется гексацианокобальтовая(Ш) кислота
H3[Co(CN)e] -5Н2О в виде бесцветных кристаллов, растворимых в воде и спир-
те и устойчивых в водном растворе (ниже 50°).
Гексанитрокобальтат(111) натрия, Na3[Co(NO2)e], получают барбати-
рованием воздуха через раствор, полученный смешением концентрирован-'
КОБАЛЬТ
573
ных растворов нитрата кобальта(П), NaNO2 и 50%-ной СН3СООН:
Co(NO3)2 + 2NaNO2 = Co(NO2)2 + 2NaNO3
2NaNO2 + 2CH3COOH = 2NaCH3COO + 2HNO2
Co(NO2)2 + 2HNO2 = Co(NO2)3 + NO + H20
Co(NO2)3 + 3NaNO2 = Na3[Co(NO2)e].
Соединение Na3[Co(NO2)6] представляет собой желтый микрокристалли-
ческий порошок; оно растворяется в воде и служит для количественного
определения ионов К+, Rb+, Cs+, поскольку соединения K3[Co(NO2)6],
Rb3[Co(NO2)e], Cs3[Co(NO2)6] имеют желтый цвет и плохо растворяются
в воде, спирте или эфире.
9. Комплексы типа MeJ[CoYX5] охватывают пентацианотиосульфато-
(или сульфито-)кобальтаты (III), содержащие анионы [Co(S2O3)(CN)6]4-,
(Co(SO3)(CN)6]4-.
10. Комплексы типа Me3[CoY2X4] включают тетрацианодисульфитоко-
бальтаты(Ш), содержащие анион [Co(SO3)2(CN)4]5~.
По приведенной классификации моноядерных координацион-
ных соединений кобальта(Ш) были представлены соединения ряда
алгминов, аквосолей, ацидосолей и соединения, являющиеся
переходными между этими рядами, т. е. ацидоаквосоли, ацидо-
амминосоли, аквоамминосоли, ацидоаквоамминосоли и др. По
природе координированных групп были приведены соединения,
у которых комплексный ион — катион или анион, и координаци-
онные соединения типа неэлектролитов.
Полиядерные координационные
соединения
Известны многочисленные полиядерные соединения кобальта,
в которых центральные атомы координационных сфер связаны
между собой лигандами, служащими связывающими мостиками.
Полиядерные комплексы могут быть получены методом конденса-
ции моноядерных координационных соединений или окислением
аммиачных растворов солей кобальта(П).
Примеры полиядерных соединений кобальта типа гексамминов, пен-
тамминов, тетрамминов, триамминов: [Co3En6{(H2N — C2H4)3N}2]Y9-геНгО,
где Y = СП, I-, NOj, С104, i/2S0|-, [(NH3)5CoCO3Co(NH3)5](SO4)2.
. 4Н2О, [(N02)2(NH3)3CoS04Co(NHs)3(N02)2]-2H20,
[(NO2)2(NH3)3CoSeO4Co(NH3)3(NO2)2]-2H2O,[(NH3)3CoX3Co(NH3)3], где X -
двухвалентный бидентатный лиганд*.
Примеры двухядерных комплексных ионов кобальта:
[(NH3)5CoNH2Co(NH3)5]5+, [(NH3)6Co3+ - О - О - Co4+(NH3)6]5+,
[(NH3)6Co - О - О - Co(NH3)5]4+, [(H2O)(NH3)4Co -N Co(NH3)4C1]4+,
н2
* Бидентатным лигандом называют заместитель во внутренней сфере
комплекса, занимающий два координационных места.— Прим. ред.
574
ПОБОЧНАЯ ПОДГРУППА VIII ГРУППЫ
[(H2O)(NH3)4Co - N Co(NH3)4Br]4+, [(H2O)(NH3)4Co — N ->
H2 H2
Co(NH3)4(NO3)?+, [C1(NH3)4Co - N -> Co(NH3)4(NO3)]3+,
H2
[Br(NH3)4Co - N Co(NH3)4(NO3)]3+, [C1(NH3)4Co - N
H2 H2
Co(NH3)4(NCS)]3+, [Br(NH3)4Co - N -> Co(NH3)3(NCS)]3+
H2
(NH3)4Co^ /Co(NH3)4
N
H2
H
0
(NH3)4C<X Xo(NH3)4
N
H2
4-b
En2Co^ CoEn2
NO2
Ея3Со CoEn2
О
H
КОБАЛЬТ
575
En2Co
r(NH3)3
L ci2
Г(ЯНз)з
L Вг2
(NH3)3
н2о
Н2
N
СоЕп2
SO4
Н2
Со—N ->
Н2
Со —N
Со'
з+
(ЯНз)з Н2 (NH3)3-]2+
Со—N -*• СоН2О
С12 С1
Н2
N
Со
(NH3)3 ,
Со
Н20
о
н
н
о
о
и
(NH3)3']+
Со
Cl2 J
(NH3)3”]+
Со
Вг2 _
4+
(NH3)3
Н20
4+
(NH3)3
Со
н20
H 1 0 3+
(NH3)3 Co H2O 4 / \ (NH3)3 Co 4 Z NOs 0 H J J
(NH3)3 . Co NO3 H 0 Z \ (NH3)3 Co \ Z NO3 0 H 2+
(NH3)3Co—ОН ->• Co(NH3)3
СН3СОО
н
о
з+
(NH3)3Co <-OH-Co(NH3)3
no2
r(NH3)3 Н2 (NH3)31+
Со—N -*• СоС1
_ Cl2 NO3 J
Г (NH3)3 з+ 4+ (NH3)31+
I Со—О —О-Co CI
ОН
С12
(NH3)3
Со:
(NH3)3
н20
Н2
N
\ (NH3)3
СНзСОО
Н2О
4+
Со'
Н2
N
о
н
Со
(NH3)3
N03
з+
СНзСОО 1 3+
(NH3)3 H20 Co^ 0 H (NH3)3 Co 7 CH3COO r
- H2 N 3+
(NH3)3Co— ОН -> Co(NH3)3
ОН
н
о
3+
(NH3)3Co ОН—Co(NH3)3
о
н
ОН
(NH3)3Co OH —Co(NH3)3
CH3COO
з+
576
ПОБОЧНАЯ ПОДГРУППА VHI ГРУППЫ
ОН
(NH3)3Co- NO2 -> Co(NH3)3
N02
ОН
34-
34-Z \ 44-
(C3H7NH2)3Co-OH -> Co(C3H7NH2)3
O—f)/
34-
OH -]44-
(CjO4)2Co /Со(С2О4)2 и др.
ОН
Примеры триядерных комплексных ионов кобальта:
ОН Н2О ОН
/ \// \
(NH3)4Co3+ Со2+ Co3+(NH3)4
\ /\\ Z
ОН Н2О он
ОН Н20 он
z \zz \
Еп2Со Со СоЕп2
\ А\ Z
ОН Н2О он
44-
ОН
ОН
44-
44-
Со
Н2О
Со—ОН —► Co(NH3)3
ОН Н20 ОН
ОН
ОН
34-
(NH3)3Co— ОН —► Со— ОН —Co(NH3)3
ОН
ОН
Примеры
кобальта:
катионных
тетраядерных координационных
соединений
,ОН^Со(КНз)4
Co<OH-Co(NH^
k 'Co(NH3)4
OHZ
6+
КОБАЛЬТ
577
Соединения четырехвалентного кобальта
Известно ограниченное число соединений четырехвалентного
кобальта, которые, как правило, довольно неустойчивы.
К соединениям кобальта(ТУ) относятся двуокись СоО2-Н2О,
диселенид CoSe2, гексафторокобальтат(ТУ) цезия Cs2[CoF6] и неко-
торые полиядерные соединения, например
(NH3)4Co4+'
Co3+(NH3)4
С14
Двуокись кобальта, СоО2-Н2О, в виде черного осадка получа-
ют анодным окислением металлического кобальта в процессе
электролиза щелочных растворов или при действии иода на холод-
ный раствор сульфата кобальта(Ш) в присутствии избытка КОН.
Соединение СоО2’Н2О относительно неустойчиво, относится
к энергичным окислителям и обладает четко выраженным основ-
ным характером. При взаимодействии двуокиси кобальта с окисла-
ми некоторых металлов (например, с Na2O, К2О, Cs2O, MgO,
SrO, ВаО) образуются кобальтаты(1У), такие, как Na2CoO3,
К2СоО3, Cs2CoO3, MgCoO3, Sr2CoO4, Ва2СоО4, ВаСо2О5.
Металлоорганические соединения
Карбонильные соединения кобальта. Из-
вестны моно- и полиядерные карбонильные соединения кобальта.
В качестве примера моноядерных карбонильных соединений ко-
бальта можно назвать тетракарбонилкобальтат водорода НСо(СО)4,
тетракарбонилкобальтаты типа Мет[Со(СО)4], Меп[Со(СО)4]2,
МеП1[Со(СО)4]3, дииодид карбонила кобальта Со(СО)12 и др.
В качестве примера полиядерных карбонильных соединений
кобальта можно назвать октакарбонил Со2(СО)8 и додекакарбонил
кобальта Со4(СО)12.
Октакарбонил кобальта, Со2(СО)8, получают действием окиси
углерода на порошкообразный металлический кобальт при
220° и 250 ат, действием окиси углерода на CoS, Со12 или СоВг2
при 200° и 200 ат в присутствии металлической меди, обработкой
СоСО3 смесью водорода и окиси углерода при 120—200° и 230—
300 ат, разложением НСо(СО)4 кислотами или нагреванием:
2CoS + 8СО + 4Cu = Со2(СО)8 + 2Cu2S
2CoI2 + 8СО + 4Cu = Co2(CO)8 + 4CuI
2CoCO3 + 10CO = Co2(CO)8 + 4CO2
37—0101
578
ПОБОЧНАЯ ПОДГРУППА VIII ГРУППЫ
Соединение Со2(СО)8 представляет собой диамагнитные оранже-
во-красные прозрачные кристаллы с плотностью 1,73 г!см5 при
18° и т. пл. 51°. Карбонил разлагается на Со4(СО)12 и СО при
51—60° (или на металлический кобальт и окись углерода при
температуре выше 60°), плохо растворим в воде, растворяется
в С2Н5ОН, С2Н6 — О — С2Н6, CS2, гидролизуется водой, взаимо-
действует со щелочами, галогенами, ацетиленом (или его произ-
водными), изонитрилами и др.
ЗСо2(СО)8 + 4Н2О = 4НСо(СО)4 + 2Со(ОН)2 + 8СО
6Со2(СО)8 + 8NaOH = 8НСо(СО)4 + 4Na2CO3 + Со4(СО)12
Со2(СО)8 + 2Вг2 = 2СоВг2 8СО
Со2(СО)8 + НС = СН С2Н2Со2(СО)6 + 2СО
Действием водорода на Со2(СО)8 при 165° и 120 ат получают
НСо(СО)4, который взаимодействует с HgCl2, аммиачным раство-
ром NiCl2 с о-фенантролином.
При действии пиридина, метанола, этанола, фенилтио-
ла или ацетона на Со2(СО)8 образуются соответственно
[Co(C5H6N)6] [Со(СО)4]2, Со2(СО)5-1,5С2Н5ОН, Co(CO)3SC6H5.
Додекакарбонил кобальта, Co4(CO)i2, образуется путем нагре-
вания (51—60°) Со2(СО)8 или окисления НСо(СО)4 при температу-
ре выше —26°.
2Со2(СО)3 Со4(СО))2 + 4СО
Соединение Со4(СО)12 представляет собой черные кристаллы,
которые при температуре выше 60° разлагаются на металлический
кобальт и окись углерода, растворяются в обычных растворите-
лях и взаимодействуют с натрием, растворенным в жидком
аммиаке.
Со4(СО)12 + 3Na = 3NaCo(CO)4 + Со
Тетракарбонилкобальтовая, кислота, НСо(СО)4, получается
нагреванием металлического кобальта, CoS или Со12 со смесью
СО и Н2 при 50 ат (в случае CoS или Со12 в присутствии меди),
действием гидроокислов на Со2(СО)8, обработкой координационных
соединений кобальта(П) окисью углерода.
2Со + 8СО + Н2 = 2НСо(СО)4
2CoS + 8СО + Н2 + 4Cu = 2НСо(СО)4 + 2Cu2S
ЗСо2(СО)8 -J- 2Ва(ОН)2 = 4НСо(СО)4 -J- 2ВаСО8 2СоСО8
18К2[СоХ4] + 24СО = ЗСо2(СО)8 + 12Кг[СоХв]
1 + 2Н2О
2HCo(CO)4 + Со(ОН)2 + 8СО
Соединение НСо(СО)4 представляет собой токсичную, непри-
ятно пахнущую желтую жидкость, которая затвердевает при
—26,2°, кипит при 10°, легко разлагается на Со2(СО)8 и Н2, облада-
КОБАЛЬТ
579
ет отчетливо выраженными восстановительными свойствами и пре-
вращается в Со4(СО)12 под действием кислорода воздуха или МпО2,
взаимодействует в водных растворах с аммиаком или с аммиачными
растворами солей никеля или кобальта. В последнем случае обра-
зуются [Co(NH3)6] [Со(СО)4]2, [Ni(NH3)6][Co(CO)4]2.
Известны соединения типа МетСо(СО)4, Меп[Со(СО)4]2;
МеПт[Со(СО)4]3 (где Ме*= Т1+, МеП= Zn2+, Cd2+, Hg2+, Sn2+,
Pb2+ и Ме1п = Ga3+, In3+, Tl3+). Эти соединения плохо раствори-
мы в воде, растворяются в органических растворителях и разла-
гаются при 70°.
Дииодид карбонила кобальта, Со(СО)12, получают действием
окиси углерода на Со12 при обычной температуре и давлении
100 ат.
Соединение Со(СО)12 представляет собой темно-коричневые
кристаллы, летучие при комнатной температуре и разлагающие-
ся с выделением окиси углерода.
Нитрозильные производные кобальта.
Известны следующие нитрозильные производные кобальта:
Co2(NO)4C12, Co(NO)C1, Co(CO)3NO и др.
Циклопентадиен ильные производные
кобальта. Циклопентадиенилдикарбонил кобальта,
(С5Н5)Со(СО)2, представляет собой диамагнитную темно-красную
жидкость с т. кип. 75° (22 мм рт. ст.); в вакууме выделяет окись
углерода.
Известны соли бис(циклопентадиенил)кобальта типа
[Со(С5Н5)21У, где Y = F", CN", NO', СЮ;, ^СО2-.
Соединения включения
Гидриды кобальта, СоН2 и СоН.
Дигидрид кобальта, СоН2, получают перемешиванием раство-
ра фенилмагнийбромида с безводным СоС12 в атмосфере сухого
водорода:
СоС12 + 2C6H6MgBr + 2Н2 = СоН2 + 2C6He + MgBr2 + MgCl2
Соединение СоН2 представляет собой темно-серые кристаллы
с плотностью 0,533 г/см3; оно устойчиво под слоем эфира ~ ниже 5°;
при обычной температуре окисляется медленно в сухом и быстро
во влажном воздухе, разлагается на элементы под действием
воды или спирта, превращается в соли кобальта(П) (с выделением
водорода) под действием разбавленных кислот.
Гидрид кобальта, СоН, получают термическим разложением
СоН2 при 44—45,8°; это серый порошок, устойчивый при нагре-
вании до температуры 150°.
Нитриды кобальта, Co4N, Co3N, Co2N, Co3N2 и CoN.
Нитрид кобальта, Co4N, представляет собой черно-серые
кубические кристаллы, которые образуются при азотировании
37*
580
ПОБОЧНАЯ ПОДГРУППА VIII ГРУППЫ
гексагональной модификации металлического Со при 350—450°.
Нитрид кобальта, Co3N, образуется в виде серых гексагональ-
ных кристаллов при действии газообразного NH3 на пирофорный
металлический кобальт при 276°.
Нитрид кобальта, Co2N, получают действием газообразного
NH3 на пирофорный металлический кобальт при 380—390°; это
черный порошок с орторомбической структурой кристалличе-
ской решетки; при 276° он превращается в Co3N, а при высокой
температуре разлагается на элементы. Соединение Co2N гидро-
лизуется водой и растворяется в кислотах с образованием солей
кобальта(II) и аммония.
Нитрид кобальта, Co3N2, образуется в виде черного порошка
при нагревании (2000°) Co(CN)2 с СоО в атмосфере азота или при
выделении аммиака из Co(NH2)2.
Нитрид кобальта, CoN, получают нагреванием (40—50°)
Co(NH2)3; он представляет собой пирофорный черный аморфный
порошок, который растворяется в щелочах с выделением аммиака
и в разб. H2SO4 с образованием CoSO4, (NH4)2SO4 и выделением
водорода.
Фосфиды кобальта, Со2Р, Со3Р2, Со4Р3, Со2Р3, СоР,
СоР2, СоР2>37 и СоР3.
Фосфид кобальта, Со2Р, получают нагреванием металлического
кобальта в парах РС13, РВг3, Р13, плавлением в электрической дуге
пластинок металлического кобальта с фосфидом меди. Соединение
Со2Р образует серые игольчатые кристаллы с плотностью 6,4 г/смъ,
т. пл. 1386° и твердостью 6 по шкале Мооса; Со2Р растворим
в кислотах.
Фосфид кобальта, Со3Р2, может быть выделен в виде черного
порошка по реакции СоС12 с фосфином РН3 или при восстановле-
нии ортофосфата кобальта водородом. Соединение плохо раство-
римо в НС1.
Фосфид кобальта, Со4Р3, образуется в виде черного аморфного
порошка при действии паров фосфора на металлический кобальт
при 500°.
Фосфиды кобальта, СоР и Со2Р3, имеют вид черных кристаллов.
Арсениды кобальта, Co3As, Co5As2, Co2As, Co3As2.
CoAs, Co2As3, CoAs2, CoAs3, представляют собой серые блестящие
кристаллические порошки.
Антимониды кобальта, CoSb, CoSb2, CoSb3, также
имеют вид серых кристаллических порошков.
Карбиды кобальта, Со2С и Со3С.
Карбид кобальта, Со2С, получают действием окиси углерода
на металлический кобальт при 208—230° или обработкой метаном
металлического кобальта при 200—440°. Соединение Со2С пред-
ставляет собой парамагнитные серые орторомбические кристаллы
с плотностью 7,76 г! см3, которые разлагаются в атмосфере водо-
НИКЕЛЬ
581
рода при 198—275°, азота — выше 297—369° и двуокиси углеро-
да — около 400°.
Силициды кобальта, Co2Si, Co3Si2, CoSi, CoSi2.
Силицид кобальта, Co2Si, получают алюмотермическим вос-
становлением смеси SiO2 с СоО или пропусканием паров SiCl4
над нагретым до 1200—1300° металлическим кобальтом. Он пред-
ставляет собой серые кристаллы с плотностью 7,1 г/см3 и т. пл.
1327°; он взаимодействует с царской водкой, а при сплавлении
с щелочами превращается в кобальтит и образуется силикат щелоч-
ного металла.
Силицид кобальта, CoSi, выделяют нагреванием (1500°) метал-
лического кобальта с силицидом меди; это парамагнитные серые
кристаллы с плотностью 6,3 г/см5 и т. пл. 1393°.
Силицид кобальта, CoSi2, образуется в результате нагревания
в электрической печи смеси металлического кобальта, силицида
меди и элементарного кремния. Соединение CoSi2 — это синевато-
черные кубические кристаллы с плотностью 5,3 г/см3 и т. пл. 1277°.
Бориды кобальта, Со3В, Со2В, СоВ.
Борид кобальта, Со3В, представляет собой ферромагнитные
серые орторомбические кристаллы.
Борид кобальта, Со2В,— продукт взаимодействия (1100—
1200°) металлического кобальта с 5% бора в атмосфере водорода.
Он представляет собой серые игольчатые кристаллы с плотностью
7,9 г/см3, растворимые в азотной кислоте и царской водке, окисля-
ющиеся на воздухе.
Борид кобальта, СоВ, получают нагреванием в электриче-
ской печи металлического кобальта с элементарным бором; это
серые орторомбические кристаллы с плотностью 7,25 г/сл3, устой-
чивые на воздухе, растворимые в азотной кислоте, царской водке
и расплавленных щелочах, взаимодействующие при нагревании
с галогенами, парами серы и парами воды.
НИКЕЛЬ Ni
Z =[28; ат. вес = 58,71
Валентность (I), II, (III), (IV)
Массовые числа природных изотопов 58, 60,
62, 61, 64
Массовые числа искусственных изотопов 56, 57,
59, 63, 65, 66
Электронная структура атома никеля: К -L -3.s23p63(/8 •4s2.
Электронная структура атома никеля и катиона Ni2+ для
3d- и 4я-орбиталей:
4s2 3d’ 4s
3d8
u u u t t
Ni
tl u H t t
Ni2 +
582
ПОБОЧНАЯ ПОДГРУППА VIII ГРУППЫ
ИСТОРИЧЕСКАЯ СПРАВКА
В древности в Китае применяли сплав, названный «пакфон-
дом» (packfond), следующего состава: 13—18% Ni, 50—66% Си,
остальное цинк или олово. Этот сплав получали из медно-никеле-
вых руд, добывавшихся на юге Китая, и он служил для изготов-
ления предметов искусства, монет, а позднее — для изготовления
огнестрельного оружия.
Металлический никель в загрязненном состоянии впервые был
получен Кронштадтом в 1751 г. и в чистом состоянии — Бергма-
ном в 1775 г. Название «никель» было дано по минералу купфер-
никель, что означает «чертова медь».
РАСПРОСТРАНЕННОСТЬ В ПРИРОДЕ
В природе никель находится в самородном состоянии или
в виде соединений (сульфидов, арсенидов, тиоарсенидов, антимо-
нидов, арсенатов, силикатов, сульфатов, основных карбонатов)
или же входит в состав различных минералов. Содержание его
в земной коре 1,8 НО-2 вес.%.
Никель встречается иногда в металлических метеоритах, содер-
жащих твердые растворы (сплавы) железо-никель с 2—7% Ni
(камасит) или 30—75% Ni (таенит). Метеориты, кроме металличе-
ского никеля, содержат его соединения, например (Fe, Ni)8Sg—
пентландит, (Fe, Ni)3C — когенит и (Fe, Ni)3P — шрейберзит.
При описании распространенности железа в природе упоминалось,
что металлическое железо метеоритного происхождения содер-
жит никель.
В сернистых минералах никеля часто присутствуют такие эле-
менты, как медь, железо, кобальт, платина, платиновые металлы,
золото, селен и теллур.
Наиболее важные минералы никеля следующие.
Миллерит, NiS,— минерал с наибольшим содержанием никеля;
встречается в виде желтых тригональных кристаллов с плотно-
стью 5,2—5,6 г!смг и твердостью 3—4 по шкале Мооса.
Пентландит, (Fe, Ni)9S8, встречается в виде желтых кубиче-
ских кристаллов с плотностью 4,5—5 г/cjn3 и твердостью 3 —
4 по шкале Мооса.
Никколит, NiAs, сопутствует сульфидам или арсенидам нике-
ля и самородному серебру; этот арсенид образует медно-красные
гексагональные кристаллы с плотностью 7,6—7,8 г1см% и твердо-
стью 5 по шкале Мооса.
Хлоантит, NiAs3_2, имеет вид белых кубических кристаллов
с металлическим блеском, плотностью 6,4—6,8 гем? и твердостью
3,5—6 по шкале Мооса.
НИКЕЛЬ
583
Герсдорфит, NiAsS, находится в виде серебристо-белых, почти
серых кубических кристаллов с плотностью 5,6—6,2 г/смг и твер-
достью 5,5 по шкале Мооса.
Аннабергит, Ni3(AsO4)2-SHaO, встречается в окисленных
зонах природных арсенидов никеля и представляет собой зеленые
моноклинные кристаллы, обладающие стеклянным блеском,
с плотностью 3 г!см9 и твердостью 2,5—3 по шкале Мооса.
Гарниерит, Ni4Si4O10(OH)4 "4Н2О, встречается в виде сине-
зеленых гелей.
Ревдинскит, (Ni, Mg)6Si4O10(OH)8, образуется в зонах вывет-
ривания силикатов магния с низким содержанием никеля и пред-
ставляет собой сине-зеленые моноклинные кристаллы с плотно-
стью 2,5—3,2 г!смг и твердостью 2—2,5 по шкале Мооса.
К другим минералам никеля относятся: полидимит Ni3S4, бравоит (Ni, Fe)S2,
виоларит Ni2FeS4, моренозит NiSO4-7H2O, ретгерсит NiSO4-6H2O, динерит
Ni3As, маухерит Ni3As2, раммельсбергит NiAs2, брейтгауптит NiSb, ульма-
нит NiSbS, форбезит (Ni, Co)HAsO4-4Н2О, меланит NiTe2, бунзенит NiO,
треворит NiFe2O4, заратит Ni3(OH)4CO3 ^ЩО, пимелит (Ni, Mg)3Si4O10(OH)2.
Залежи минералов никеля находятся в Финляндии, Норвегии,
Великобритании, СССР, ФРГ, Франции, Испании, Италии, Чехо-
словакии, Румынии, Греции, Южно-Африканской Республике,
США, Канаде, на Кубе, в Бразилии, Индонезии.
В малых количествах никель был обнаружен в спектре Солнца,
нефтях, морской воде, в организмах животных, многочисленных
земных и морских растениях и в некоторых насекомых.
ИЗВЛЕЧЕНИЕ НИКЕЛЯ ИЗ РУД
Никель извлекают из сернистых, мышьяковистых или гидро-
силикатных руд с помощью различных пиро- и гидрометаллурги-
ческих процессов.
Во многих случаях в процессе пирометаллургической перера-
ботки никелевых руд (или рудных концентратов) преследуют цель
получить сырой пек (состоящий из сульфидов никеля, меди и желе-
за), затем конверторный (бессемеровский) пек и, наконец, сырой
никель, содержащий 95—98% металла. Гидрометаллургическим
методом перерабатываются концентраты сульфидов никеля, гидро-
силикатные никелевые руды, сульфидные или арсенидные пеки
никеля, меди и железа. Для отделения никеля от меди, кобальта
и железа из растворов их солей применяют электролитический
метод или используют ионообменные смолы.
Извлечение никеля из сернистых медных и никелевых руд
В случае сернистых руд с низким (менее 1,5% Ni) содержани-
ем никеля используют концентраты этих руд, получаемые маг-
584
ПОБОЧНАЯ ПОДГРУППА VIII ГРУППЫ
нитным обогащением с последующей селективной флотацией.
При неполном прокаливании концентратов сульфидных мине-
ралов никеля и меди (особенно пирротина) в механических печах
осуществляется удаление примерно 1/8—1/5 части общего коли-
чества серы. В прокаленном продукте необходимо оставить доста-
точно серы, чтобы на следующем этапе можно было получить
массу, которая содержала бы весь никель и всю медь (соответ-
ственно и часть железа) в виде сульфидов Ni3S2 — Cu2S — FeS.
За прокаливанием следует плавление пека с добавкой SiO2
(в качестве флюса и шлакообразователя) в шахтных (типа ватер-
жакетных) или отражательных печах. Из концентратов прокален-
ных никелевых и медных руд железо переходит (хотя и не полно-
стью) в шлак, который содержит очень незначительные количества
никеля и меди.
После удаления части серы из концентратов (содержащих 7—
10% Ni, 4—5% Си и 25% S) образуется сырой пек с 10—30% Ni,
45-65% Си, 25-28% S и шлак с 0,3-0,4% Ni + Си, 35% SiO2,
35% FeO, ~10% А12О3, <10% СаО + MgO и 1,5% Si. Сырой
никелево-медный пек (Ni3S2 — Cu2S — FeS) перерабатывается
в конверторах для увеличения содержания никеля-меди (40—
58% Ni, 24—41% Си, 16—22% S и не более 0,1—0,5% Fe). Кон-
вертор имеет форму горизонтального цилиндрического барабана,
футерованного материалом основного характера. При бессемеро-
вании сырого пека сульфид железа FeS превращается в FeO, а суль-
фиды никеля — меди не изменяются. Для перевода окиси желе-
за(П) в шлак в конвертор загружают SiO2. Образующийся шлак
содержит 3% Ni и 1—1,5% Си.
Пек с большим содержанием никеля и меди выливают в чугун-
ные формы и после охлаждения перерабатывают различными спосо-
бами для отделения никеля от меди.
Разделение сульфидов никеля и меди
с помощью сульфида натрия по процессу Орфорда
Конверторный никелево-медный пек после охлаждения, измель-
чения (в шаровых мельницах) и смешения с сульфидом натрия
(или с Na2SO4, NaHSO4 и коксом) плавится в шахтной (типа
ватержакетной) печи. При охлаждении расплава после перенесе-
ния в чугунные отстойники образуются два легко разделяющихся
слоя. В верхнем слое находится двойной сульфид меди — натрия
Cu2S-Na2S (с небольшим количеством Ni3S2), в нижнем — двойной
сульфид никеля —натрия Ni3S2>Na2S (с небольшим количеством
Cu2S и FeS).
Перерабатывая верхний слой в конверторах, получают сырую
медь, а сульфид никеля Ni3S2 окисляется до NiO, который с SiO2
НИКЕЛЬ
585
превращается в шлак:
NigSg -j- V2O2 3NiO 4- 2S0g 2NiO -f- SiO2 “ Ni2SiO4
Из сырой меди электролитическим рафинированием получают
чистую медь, а в анодном шламе остаются золото и серебро.
Нижний слой, измельченный и промытый горячей водой (для
отделения солей натрия), прокаливают в печах при 1200° с целью
превращения в NiO (загрязненный 0,1 % Си, 0,2% Fe, 0,1% Si,
0,015% S и следами драгоценных металлов). При восстановлении
окиси никеля(П) углем в электрических печах образуется сырой
металлический никель, который так же, как и медь, подвергают
рафинированию. В анодном шламе остаются платина и платино-
вые металлы.
Отделение никеля от меди из конверторного пека
по процессу Монда
Конверторный никелевомедный пек, измельченный и промы-
тый горячей водой (с целью удаления солей натрия), превращается
в окислы прокаливанием при 800°. Если над сплавом, полученным
восстановлением окислов никеля и меди водяным газом (56% Н2
и 25% СО) при 350—400°, пропускать окись углерода, нагретую
до 50—60°, при атмосферном давлении, образуется летучий тетра-
карбонил никеля Ni(CO)4. При 180—200° происходит термическая
диссоциация Ni(CO)4 на металлический никель и окись углерода.
Последняя снова вводится в процесс. Металлический никель,
полученный по процессу Монда, содержит 99,8% Ni, очень не-
большие количества железа и углерода, следы серы и кремния;
медь и кобальт отсутствуют. Процесс Монда применим при давле-
нии 200 am, когда образующийся в жидком состоянии Ni(CO)4
отделяется от Fe(CO)3 дробной перегонкой.
Отделение никеля от меди из конверторного пека
электролитическим путем
Прокаливанием при 800° никелево-медного конверторного пека
получают окислы никеля и меди. При обработке серной кислотой
(60 г/л) окислов никеля и меди (75°) образуется раствор сульфата
меди, а окислы никеля и других металлов остаются в осадке.
Раствор сульфата меди служит для электролитического получения
металлической меди.
Сплавлением NiO с углем (коксом) и известью в электрических
печах получают сырой никель (содержащий ~65% металла),
который после отливки в виде стержней используется в качестве
анодов. При электролизе разб. H2SO4 с использованием анодов из
586
ПОБОЧНАЯ ПОДГРУППА VIII ГРУППЫ
сырого никеля в раствор переходят никель, медь, железо, а в анод-
ном шламе остаются серебро, золото и платина.
Для осаждения на катоде металлического никеля электролит
не должен содержать ионов меди [Си(Н2О)4]2+, поэтому медь осаж-
дают из раствора порошкообразным металлическим никелем.
Для удаления железа из раствора к нему добавляют гидро-
окись никеля и образующийся в присутствии воздуха Fe(OH).
отделяют фильтрованием:
FeSO4 + Ni(OH)2 = Fe(OH)2 + NiSO4
2Fe(OH)2 + H2O + V2O2 = 2Fe(OH)3
Извлечение никеля из природных силикатов
(из гарниерита и ревдинскита)
Измельченную руду смешивают с избытком гипса (или с пири-
том FeS2 в отсутствие сульфида меди), затем высушивают и гра-
нулируют или брикетируют. Размол осуществляется в молотко-
вых мельницах, гранулирование — в обычных аппаратах, а бри-
кетирование — на цилиндрических прессах.
При плавлении брикетов (агломерата) в смеси с коксом и флю-
сами (известь, магнезит, флюорит) в шахтных печах образуется
никелевый пек Ni3S2 'FeS (с небольшим количеством растворен-
ного ферроникеля) и шлак, состоящий из силиката кальция
и железа. При 1100—1200° часть гипса разлагается:
2CaSO4-2H2O -* 2СаО + 2SO2 + 4Н2О + О»
Смесь сульфата кальция и кокса превращает силикаты тяжелых
металлов в сульфиды, а известь и магнезит образуют шлаки из
окислов SiO2 и А12О3.
Сульфид кальция и моносульфид железа образуются при нагре-
вании по уравнениям
CaSO4 + 4СО = CaS + 4СО2 FeS2 FeS + S
Действием CaS и FeS на гидросиликаты никеля получают нике-
левый пек Ni3S2-FeS. Промышленные пеки (плавящиеся при
~900 ) содержат 25—40% Ni, 40—50% Fe, 15—25% S, а шлаки
состоят из 38-44% SiO2, 20-25% СаО, 15-20% FeO, 7% MgO
и ~0,2—0,3% Ni. Расплавленный никелевый пек периодически
удаляют и загружают в конверторы, где он перерабатывается
в присутствии кварца до получения чистого сульфида никеля
Ni3S2 (с небольшим количеством металлического никеля), а же-
лезо переходит полностью в шлак, который содержит до 2% Ni.
В конверторах не получают металлический никель, поскольку
реакция образования металлического никеля из NiO и Ni3S2 осу-
ществляется при температуре выше 1500°. Конверторный пек
НИКЕЛЬ
587
выливают в виде слитков и после охлаждения и измельчения
прокаливают при 1200° (во вращающихся печах) для превращения
в окись никеля(П) (получается загрязненной).
Нагреванием (1200°) окиси никеля(П), смешанной с древесным
углем, в электрических печах получают сырой расплавленный
никель, который отливают в виде слитков или гранулируют.
Восстановлением окиси никеля(П) в специальных печах полу-
чают никелевый порошок, а в ретортах — круги металлического
никеля.
ПОЛУЧЕНИЕ МЕТАЛЛИЧЕСКОГО НИКЕЛЯ
Металлический никель можно получить восстановлением при
нагревании окислов никеля NiO, Ni2O3, Ni3O4(Ni2O, Ni4O) водо-
родом, окисью углерода, углеродом, алюминием, кремнием, бо-
ром или другими восстановителями. В результате восстановления
окиси никеля водородом при 270—280° образуется порошкообраз-
ный пирофорный никель, а при 350—400° — порошкообразный,
но довольно устойчивый металлический никель. Для ускорения
восстановления окиси никеля(П) водородом процесс ведут при
600-700°.
Окись углерода восстанавливает NiO, начиная от температу-
ры 250—300°. Процесс протекает быстро и полностью заканчи-
вается к 700—900°.
При прокаливании (1250°) брикетов, образованных из NiO
и пасты, состоящей из пшеничной муки и древесного угля, обра-
зуется порошок металлического никеля, а при сильном прокали-
вании в электрических печах смеси NiO с древесным углем и из-
вестью образуется расплавленный металлический никель. Вос-
становление окиси никеля(П) твердым углем начинается примерно
при 600° и полностью завершается при 1000°.
Алюмо- и кремнетермическое восстановление окиси никеля(П)
описывается уравнениями
3NiO 2А1 = 3Ni + А12О3 + 218,10 ккал
2NiO + Si = 2Ni SiO2 + 91,44 ккал
Алюмо- или кремнетермическим восстановлением, а также
восстановлением углем (при нагревании) смеси окиси никеля
с окислами железа получают сплавы железо — никель (ферро-
никель), которые обычно содержат и элемент-восстановитель.
Сильно карбидизированные сплавы железо-никель используют
непосредственно для получения сталей.
Металлический никель можно получить восстановлением без-
водного хлорида никеля NiCl2 водородом при ~600°.
Металлический никель получают также электролитическим
путем. Осадки электролитического никеля содержат значительное
588
ПОБОЧНАЯ ПОДГРУППА VIII ГРУППЫ
количество водорода (поскольку никель осаждается в условиях
высокой катодной поляризации) и образованы из мелких кри-
сталлов, твердость которых превосходит твердость плавленого
или отпущенного металла. Электролитическим методом можно
получить порошок, чешуйки или хрупкую массу никеля. Для полу-
чения порошка электролитического никеля используют электро-
литы, содержащие простые или двойные соли (Ni2SO4-7Н2О,
(NH4)2Ni(SO4)2 -6Н2О, NiC12-raH2O), и аммиачные электролиты,
содержащие соединения никеля.
Порошкообразный металлический никель получается легче,
чем порошкообразное железо, поскольку электролит более устой-
чив, а порошок никеля обладает меньшей склонностью к окисле-
нию. Для электролитического получения металлического никеля
можно использовать различные растворы, содержащие соли нике-
ля. Электролиз осуществляется в условиях определенных pH,
температуры и катодной плотности тока, как это видно из сле-
дующих четырех примеров:
а) 70—95 г/л NiSO4-7H2O, 40 г/л NH4C1, 8-50 г/л NaCl, pH = 6,5j— 7,2,
температура 20—30°, плотность тока 10—50 а/дм2;
б) 50—95 г/л NiSO4-7H2O, 20—80 г/л (NH4)2SO4, pH = 4,5, температура
30—40°, плотность тока 10—30 а!дм2;
в) 40 г/л NiSO4-7H2O, 50 г/л (NH4)2SO4, 150—300 мл!л 25%-ного раствора
NH4OH, температура 25—60°, плотность тока 10—15 а!дмг',
г) 180 г/л NiSO4-7H2O, 25 г/л NH4C1, 30 г/л Н3ВО3, pH = 5,6 — 5,9, тем-
пература 45—60°, плотность тока 2,5—5 а!дмг.
Анодный шлам, образующийся при электролитическом полу-
чении или рафинировании никеля, служит, как уже отмечалось,
для извлечения химически неактивных металлов, например пла-
тины или платиновых металлов.
ОЧИСТКА
Примесями в металлическом никеле являются кобальт, медь,
железо, цинк, углерод, кремний, мышьяк, сера, кислород и водо-
род. К вредным примесям относятся сера (вызывает хрупкость
при нагревании), углерод (понижает тягучесть) и кислород (ухуд-
шает механические свойства).
Сырой металлический никель очищают электролитическим пу-
тем, переплавкой или по процессу Монда путем превращения
в Ni(CO)4, который термически разлагается при 200°. Электро-
литическое рафинирование сырого никеля осуществляется в элект-
ролизерах с диафрагмой в очень слабо кислых растворах (pH ~
~ 5) сульфата или хлорида никеля при ~90° с анодами из сырого
никеля при плотности тока 20—40 а!дм2.
НИКЕЛЬ
589
С целью частичного окисления серы, углерода, кремния, цин-
ка, железа и др. сырой никель переплавляют в электрической печи
с футеровкой основного характера в присутствии воздуха.
ФИЗИЧЕСКИЕ И ХИМИЧЕСКИЕ СВОЙСТВА
В компактном состоянии никель — металл блестящего сереб-
ристо-серого цвета (после полировки появляется красивый ме-
таллический блеск); может существовать в двух кристаллических
модификациях: p-Ni — структура с плотной гексагональной упа-
ковкой и a-Ni — с кубической гранецентрированной решеткой.
Никель в коллоидном состоянии получают восстановлением
водородом коллоидного раствора Ni(OH)2 при каталитическом
действии коллоидного палладия, восстановлением различных
соединений никеля водородом, гидразином, фенилгидразином,
дихлоридом олова и др. в присутствии спирта, бензола, диоксана,
эфира или ацетона, а также при образовании электрической дуги
между двумя никелевыми электродами в воде в присутствии вос-
становителя или защитного коллоида, обладающего восстанови-
тельными свойствами.
Никель — тяжелый металл, его плотность 8,907 г/см3 при 20°,
твердость 5 по шкале Мооса, он ковок, тягуч и может перераба-
тываться при нагревании под давлением, тугоплавок (т. пл. 1455°,
т. кип. 3075°), имеет относительно низкую тепло- и электропровод-
ность.
Плавление металлического никеля осуществляется под за-
щитным слоем флюса (битое стекло с добавкой извести или флю-
орита), поскольку многие газы (пары воды, двуокись и окись угле-
рода, водород, углеводороды, двуокись серы) оказывают вредное
действие на расплавленный металл.
Пирофорный порошок металлического никеля может быть полу-
чен одним из приведенных ниже способов: электролитическим
путем, нагреванием амальгамы никеля, восстановлением раство-
ренных в жидком аммиаке дихлорида, дибромида или дииодида
никеля металлическим натрием, калием или кальцием, а также
восстановлением окислов никеля водородом при 270—280°.
Кристаллическая модификация a-Ni, в отличие от модифика-
ции p-Ni, ферромагнитна, для нее точка Кюри близка к 350°.
Известно множество сплавов, которые никель образует с же-
лезом, кобальтом, медью, марганцем, цинком, хромом, молибде-
ном, вольфрамом, бериллием, углеродом, кремнием, фосфором,
серой и др.
С химической точки зрения металлический никель не активен,
он не корродирует в воде, на воздухе и в различных растворах.
При обычной температуре воздух и вода не действуют на ме-
таллический никель в компактном состоянии. На воздухе (в кисло-
590
ПОБОЧНАЯ ПОДГРУППА VIII ГРУППЫ
роде) металлический никель легко превращается в NiO (зеленого
цвета) при ~500°, если он находится в компактном состоянии,
или при 150—200° — в виде порошка.
Помимо окиси NiO известны также окислы Ni2O, Ni3O, Ni4O,
/°
Ni( |-nH2O, Ni3O4-2H2O, Ni2O3-H2O.
XO
При 600—1000° никель реагирует с водяным паром:
Ni + Н2О NiO + Н2
Металлический никель в твердом состоянии (компактном, губ-
чатом или порошкообразном) или в расплаве поглощает водород
лучше, чем железо, кобальт или медь.
При нагревании металлический никель взаимодействует с гало-
генами, серой, селеном, теллуром, фосфором, мышьяком, сурьмой,
углеродом, кремнием и бором, образуя различные соединения:
NiF2, NiCl2, NiBr2, Nil2, NiS, Ni3S2, Ni6S5, Ni7S6, NiS2, Ni2S,
Ni3Se4, Ni2Se3, NiSe2, Ni2Te3, NiTe2, NiP2, NiP3, Ni5As2, NiSb,
Ni3C, Ni2Si, NiB и др.
В условиях обычной температуры и влажного воздуха хлор
или бром с металлическим никелем дают соответствующие дига-
логениды.
При нагревании в атмосфере сероводорода поверхность метал-
лического никеля покрывается пористой пленкой NiS, которая
не прилегает плотно к металлу и не оказывает защитного действия.
В результате взаимодействия двуокиси серы с никелем обра-
зуются NiO и сульфид никеля, который растворяется в расплав-
ленном металле, образуя сплавы, хрупкие при нагревании. Для
удаления серы в сплавы никеля добавляют металлические мар-
ганец, магний или литий. Сульфид никеля NiS образует с метал-
лическим никелем легкоплавкую при 645° эвтектику, в то время
как сульфиды марганца, магния или лития плавятся при высокой
температуре и кристаллизуются в виде изолированных включений.
Двуокись азота при 200° окисляет металлический никель до NiO,
сама восстанавливаясь до NO.
При пропускании газообразного аммиака над тонкодисперс-
ным порошком металлического никеля, нагретого до 500°, обра-
зуется черный кристаллический порошок Ni3N.
При 900° никель взаимодействует с СО2:
Ni + СО2 NiO + СО
Пропусканием окиси углерода над нагретым до 50—60° метал-
лическим никелем получают летучее соединение Ni(CO)4.
В результате действия окиси углерода, метана, ацетилена,
бензола, гексана и др. на никель при высокой температуре обра-
НИКЕЛЬ
591
зуется карбид никеля Ni3C и выделяется водород (в случае при-
менения углеводородов), который растворяется в расплавленном
металле, образуя сплавы.
При нагревании металлический никель восстанавливает много-
численные окислы или гидроокиси металлов, сульфиды, тиоци-
анаты и нитраты щелочных металлов.
Никель взаимодействует с расплавленными щелочами (темпера-
тура выше 600°). При нагревании (550—600°) металлического нике-
ля с NaOH в вакууме образуются NiO, металлический натрий
и выделяется водород.
Галогеноводороды в газообразном состоянии взаимодействуют
при нагревании с никелем по общему уравнению
Ni + 2НХ ч* NiX2 + Н2
Разбавленные кислоты HCl, H2SO4, HNO3 медленно растворяют
металлический никель. Чем более разбавлен раствор кислоты,
тем более высокая температура требуется для растворения никеля.
В конц. HNO3 (d = 1,42) металлический никель при 15° пас-
сивируется, а при 72° — энергично растворяется. В царской водке
никель растворяется, образуя хлорид никеля(П).
Никель медленно растворяется в кислотах Н2СО3 и Н3РО4,
а с уксусной, щавелевой, винной и лимонной кислотами взаимо-
действует только после длительного контакта.
Растворы NaCl, СаОС12, (NH4)Fe(SO4)2-12Н2О вызывают кор-
розию (растворяют) металлического никеля при комнатной тем-
пературе.
При действии растворов персульфатов щелочных металлов на
порошок металлического никеля образуются двойные сульфаты
никеля и щелочных металлов.
Химические свойства никеля наглядно иллюстрируются сле-
дующей схемой:
Комн. темп.
Никель
Нагревание
во влажном воздухе с С12 или Вг2 -> дигалогениды
на воздухе (в кислороде) или с водой-*NiO
с галогенами ->- NiF2, NiCI2, NiBr2, Nil2
с серой -»• NiS, Ni2S3, Ni6S5, NiS-,, Ni2S
с C, Si, B->Ni3C, Ni2Si, NiB
c H2S—►NiS
с P, As, Sb—► NiP2, NiP3, Ni5As2, NiSb
c SO2—►NiS и NiO
c NO2 или CO2-*NiO
c CO-*Ni(CO)4 или Ni3C
c CH4, C2H2, CgHg, CeH12-> Ni3C
c NaOH в вакууме—►NiO
с галогеноводородами HX->NiX2
592
ПОБОЧНАЯ ПОДГРУППА VIII ГРУППЫ
С физиологической точки зрения металлический никель не
токсичен для человека, животных и растений.
ПРИМЕНЕНИЕ
Никель относится к самым широко используемым металлам,
но его применение ограничено, поскольку это один из наиболее
дорогих технических металлов.
Из никеля изготовляют коррозионноустойчивые изделия, ап-
параты для физико-химических измерений, детали машин и др.
Примерно 10% общего количества никеля используется для
никелирования, т. е. для покрытия никелем железа, стали, меди,
латуни и других металлов или сплавов. Никелирование осуществ-
ляется как гальваническим способом, так и плакированием.
Наибольшее количество никеля идет на получение сплавов,
имеющих исключительно важное значение в технике.
В качестве примеров сплавов никеля можно назвать никель-
содержащие стали, бронзы, латуни, монетные сплавы, сплавы для
электрических сопротивлений (константан — 40% Ni, 60% Си;
никелин —31%Ni, 56% Си, 13% Zn, манганин — 4% Ni, 12% Мн,
84% Си), сплавы, используемые для изготовления деталей, устой-
чивых к коррозии и высоким температурам, сплавы для зубных
протезов (тикониум — 68,2% Со + Ni, 30% Cr и 1,8% Be), спе-
циальные сплавы, такие, как монель-металл (65—70% Ni, 25—
30% Си, остальное Fe + Мп + Si + С + S + Р), аргентанили алпа-
ка (13—36%Ni, 46—66% Си, 19—31% Zn), платинит (40—46% Ni,
остальное железо), инвар (35—37% Ni, остальное железо),
кислотоупорный сплав (58—61% Ni, 12—19,5% Fe, 15% Cr,
2% Мп, Мо, W, Со, Be).
В технике особое значение имеют сплавы Ni — Си, Ni — Fe,
Ni — Cr (нихром) и Fe — Ni —Cr (ферронихром). Сплавы нике-
ля с хромом, медью и железом не окисляются и проявляют высо-
кую стойкость к коррозионному действию многочисленных хими-
ческих веществ.
Еще в древности были известны и применялись для изготовле-
ния монет сплавы никель — медь.
Тонкодисперсный порошок металлического никеля приме-
няется для щелочных аккумуляторов, в химической промышленно-
сти в качестве катализатора, как пигмент в антикоррозионных
красках, при изготовлении постоянных магнитов и получении
инвара. Тонкодисперсный никель, многочисленные сплавы нике-
ля и некоторые соединения никеля служат катализаторами в реак-
циях гидрогенизации, полимеризации, циклизации, изомеризации
и в различных реакциях обмена.
Некоторые соли никеля применяются в керамической промыш-
ленности в качестве пигментов.
НИКЕЛЬ
593
СОЕДИНЕНИЯ (ОБЩИЕ СВОЙСТВА)
Известно ограниченное число соединений одно-, трех- и четы-
рехвалентного никеля и очень много соединений двухвалентного
никеля.
В табл. 59 приведены формулы и указан цвет некоторых соеди-
нении никеля, сгруппированных по валентности. Наиболее устой-
чивы соединения двухвалентного никеля.
Большинство соединений одно-, трех-и четырехвалентного нике-
ля являются координационными, за исключением окислов, никела-
тов и сульфидов. Соединения никеля(1) неустойчивы и обладают
восстановительными свойствами, в то время как соединения ни-
келя(Ш) и (IV) проявляют окислительный характер.
Помимо соединений, соответствующих указанным четырем со-
стояниям валентности, известны металлоорганические соедине-
ния и соединения включения.
Соединения одновалентного никеля
Известно ограниченное число соединений одновалентного нике-
ля, при этом большинство из них неустойчивы, легко выветрива-
ются на воздухе; соединения окрашены в желтый, красный, зеле-
ный и синий цвета, получаются восстановлением соединений
никеля(П). Примеры соединений никеля(1): окись Ni2O — оран-
жево-желтая, гидроокись NiOH — синяя, цианид NiCN — оран-
жевый, сульфид Ni2S — желтый, селенид Ni2Se — желтый,
комплексы K2[NiCl3] — красный, Na2[Ni(CN)3] — красный,
K2[Ni(CN)3] — красный, K2[Ni(NO)(CN)3] — красный, [Ni(NO)-
• SC2H5] — красный, [Ni(NO)SC6H5] — красный, K2[Ni(CO)-
• (CN)3] — желтый, K3[Ni(NO)(S2O3)2] *2Н2О — сине-зеленый.
Гидроокись никеля, NiOH, получают обработкой кристалли-
zSO3Ni
ческого фиолетового соединения Н — Nf -нНгО водными
XSO3H
растворами щелочей.
Гидроокись никеля образуется в виде темно-синего осадка,
который легко выветривается на воздухе, растворяется в рас-
творе KCN с образованием K2[Ni(CN)3] и превращается в Ni2S
под действием Na2S.
SO3Ni
Н — Nx "нНгО получают путем обработки
XSO3H
Соединения
дигалогенидов никеля эквимолярной смесью дитионата натрия
Na2S2O4-2H2O с нитритом натрия NaNO2.
Трицианоникелат(1) калия, K2[Ni(CN)3], получают восстанов-
лением K2[Ni(CN)4] дихлоридом олова, гипофосфитом натрия
38-0101
594
ПОБОЧНАЯ ПОДГРУППА VIII ГРУППЫ
NaH2PO2, амальгамой калия или металлическим цинком на холоду
без доступа воздуха в кислой или щелочной среде.
Соединение K2[Ni(CN)3] представляет собой диамагнитную жид-
кость красного цвета; оно плохо растворимо в спирте, легко окис-
ляется кислородом воздуха и даже водой, не содержащей рас-
творенного воздуха:
4K2[Ni(CN)3] + 2Н2О = 3K2[Ni(CN)4] + 2К0Н + Ni + Н2
В водном растворе K2lNi(CN)3] поглощает окись углерода
и превращается в комплекс K2[Ni(CO)(CN)3] желтого цвета. При
подкислении красного раствора K2[Ni(CN)3] выпадает оранже-
вый осадок NiCN. При восстановлении K2[Ni(CN)4] водородом
в сильно щелочной среде образуется комплекс K3[Ni(CN)4].
Соединения двухвалентного никеля
Известно очень много соединений, в которых электроположи-
тельный двухвалентный никель находится в виде катиона Ni2+,
[Ni(H2O)4]2+, [Ni(H2O)6]2+, [Ni(NH3)2]2+, [Ni(NH3)3]2+, [Ni(NH3)4]2+
[Ni(NH3)6]2+, [Ni(NH3)5(H2O)]2+, [Ni(NH3)4(H2O)2]2+, lNi(NH3)3.
• (H2O)3]2+, [NiEn]2+, [NiEn2]2+, [NiEn3]2+, [Ni(H2O)2En2]2+,
[Ni(H2O)4En]2+, [NiPy2l2+, [NiPy3]2+, [NiPy4]2+, [NiPye]2 + ,
[Ni(H2O)2Py4]2+, [Ni(H2O)Py5]2+, [Ni(N2H4)2]2+, [Ni(N2H4)3]2+,
[Ni(NH2OH)6]2+, [Ni(CeH12N4)6]2+, [Ni(C6H5NH2)6]2+,
[Ni(C6H5NHNH2)el2+, [Ni(C12HgN2)3]2+ или в виде аниона [NiX3] ,
[NiXJ2- (где X = F", C1-, Вг").
Катионы [Ni(H2O)4]2+, [Ni(H2O)6]2+ зеленого цвета входят
в кристаллические решетки различных кристаллогидратов солей
никеля. В водных растворах солей никеля встречается катион
[Ni(H2O)6]2+.
Безводные соли никеля окрашены в желтый [NiCl2, NiSO4,
Ni(CN)2], коричневый (NiBr2), серый (Nil2) или зеленый (NiF2)
цвет.
Соли двухвалентного никеля могут быть получены растворе-
нием металлического никеля в разбавленных минеральных кис-
лотах при нагревании или в царской водке при обычной темпера-
туре, действием галогенов, серы, селена, теллура на металличе-
ский никель, растворением окиси, или карбоната никеля(П)
в различных кислотах.
Большинство солей никеля(П) с сильными кислотами раство-
римы в воде, и их водные растворы имеют кислую реакцию. В ка-
честве примеров солей никеля(П), ограниченно растворимых
в воде, можно назвать сульфид, карбонат, фосфат, основные соли.
Последние образуются при добавлении небольшого количества
щелочи к растворам солей никеля; окрашены в зеленый цвет.
НИКЕЛЬ
595
Большинство безводных солей никеля(Н) присоединяют воду,
образуя аквосоли, или аммиак, образуя комплексные аммиакаты,
которые разлагаются водой по уравнению
[Ni(NH3)6]Cl2 + 6Н2О =,* Ni(OH)2 + 4NH4OH + 2NH4C1
Большинство комплексных соединений никеля(П) имеют коор-
динационные числа шесть или четыре.
Известны координационные соединения никеля типа аквосо-
лей, амминосолей, аквоамминосолей, ацидосолей, амминоацидо-
солей, а также хелатных соединений.
Соли никеля не токсичны для человека и животных, являются
антисептиками.
Окись никеля, NiO, встречается в природе в виде минерала
бунзенита; это непрозрачные темно-зеленые октаэдрические кри-
сталлы с плотностью 6,7 г/см3. Они устойчивы к действию кислот
и расплавленных щелочей.
Соединение NiO можно получить, прокаливая при 1000—1100°
(в токе азота или в вакууме) нитрат Ni(NO3)2, карбонат NiCO3,
сульфаты NiSO4, [Ni(NH3)6]SO4 или окислы Ni2O3, Ni3O4, NiO2,
восстанавливая при нагревании окислы Ni2O3, Ni3O4, NiO2 во-
дородом, аммиаком или окисью углерода, дегидратируя Ni(OH)2
при 230—240° или окисляя порошкообразный металлический
никель кислородом при 500° или двуокисью азота при 200°.
Окись никеля представляет собой парамагнитные серо-зеленые
кубические кристаллы с решеткой типа NaCl, плотностью
6,827 г/см3 и т. пл. 1957°; она плохо растворима в воде, обладает
основными свойствами, растворяется в кислотах с образованием
солей никеля(П).
Соединение NiO вытесняет аммиак из солей аммония при на-
гревании, взаимодействует при нагревании с кислотными окисла-
ми, такими, как МоО3, WO3, оказывает каталитическое действие,
способствуя разложению КМпО4 при 100—105°, окислению NHa
при 300—360° и др.
При нагревании окись никеля может быть восстановлена до ме-
таллического никеля водородом, окисью углерода, неметаллами
С, Si, В, метаном СН4, металлами Al, Mg, Zn, Be, Cu, Pb, Fe, Co,
как было описано в разд. «Получение металлического никеля».
Окись никеля применяется в качестве катализатора и зелено-
го пигмента в керамической промышленности.
О
| -nH2O, получают при действии Н2О2
О
на спиртовый раствор NiCl2 в присутствии спиртового раствора
КОН.
Соединение NiO2-nH2O представляет собой серовато-зеленый
аморфный порошок, плохо растворимый в воде; проявляет окис-
38*
Перекись никеля, Ni^
596
ПОБОЧНАЯ ПОДГРУППА VIII ГРУППЫ
пительные свойства, неустойчиво под действием разб. H2SO4;
выделяет Н2О2.
Водная окись, Ni3O4-2H2O или Ni3O2(OH)4, образуется при
промывании водой холодного продукта, полученного нагрева-
нием до красного каления порошкообразного металлического
никеля с перекисью щелочного металла, при термическом разло-
жении Ni2O3-H2O и при анодном окислении металлического ни-
келя (40—60°).
Соединение Ni3O4-2H2O или Ni3O2(OH)4 представляет собой
черные гексагональные кристаллы с плотностью 3,33 г/см?- проя-
вляет окислительные свойства, при температуре выше 40° прев-
ращается в NiO1>ie.
Гидроокись никеля Ni(OH)2, получается в виде объемистого
зеленого осадка при обработке растворов солей никеля(П) ще-
лочью (или небольшим количеством NH4OH в отсутствие солей
аммония), начиная от pH = 6,8 в 0,01 М растворе, а также при
кипячении раствора сульфата никеля со смесью иодида и йодата ка-
лия:
3NiSO4 + 5KI + КЮ3 + ЗН2О = 3Ni(OH)2 + 3K2SO4 + 3I2
При легком нагревании или при хранении осадка в контакте
с раствором выпадают зеленые кристаллы Ni(OH)2, имеющие плот-
ность 4,1 г!см3.
Соединение Ni(OH)2 в виде зеленых ромбоэдрических кристал-
лов (а также в коллоидном состоянии) плохо растворимо в воде
и концентрированных растворах NaOH, КОН, при нагревании
до 230—240° превращается в NiO, обладает основными свойствами,
растворяется в кислотах с образованием солей никеля(П) или
в NH4OH (как в присутствии солей аммония, так и без них) с об-
разованием синих растворов, содержащих катион [Ni(NH3)6]2+:
Ni(OH)2 + 6NH4OH = [Ni(NH3)6](OH)2 + 6H2O
Ni(OH)2 + 2NH4C1 + 4NH4OH [Ni(NH3)6]Cl2 + 6H2O
Аммиак в присутствии солей аммония не осаждает Ni(OH)2,
поскольку равновесие приведенной реакции смещается в сторону
образования комплексных аммиакатов [Ni(NH3)6]Cl2.
В отличие от Fe(OH)2 и Со(ОН)2 гидроокись никеля устойчива
на воздухе и окисляется только энергичными окислителями
(например, хлорной или бромной водой в щелочной среде) с об-
разованием Ni2O3-H2O:
2Ni(OH)2 + Cl2 + 2NaOH = Ni2O3-H2O + 2NaCl + 2H2O
Дифторид никеля, NiF2, получают термическим разложением
соединения (NH4)2[NiF4], дегидратацией кристаллогидратов NiF2-
•пН2О (где п = 4, 3, 2) в токе фтористого водорода в медной трубке
при 700—800° действием HF или фтора на NiCl2 при 150°, нагре-
ванием C1F3 с NiO, обработкой фтором при нагревании металли-
НИКЕЛЬ
597
ческого никеля, окиси NiO или сульфида NiS:
Ni + F2 = NiF2 + 158 ккал (NH4)2[NiF4] = NiF2 + 2HF + 2NH3
Соединение NiF2 представляет собой зеленые тетраэдрические
призмы с кристаллической решеткой типа рутила, т. пл. 1027°
и т. кип. 1627°; дифторид никеля очищают сублимацией в атмо-
сфере HF, он плохо растворим в воде, спирте, эфире, превраща-
ется в NiO при нагревании на воздухе или в NiS при нагревании
с серой, образует фторосоли с KF или NH4F, восстанавливается
водородом при нагревании по уравнению
NiF2 + Н2 Ni + 2HF
Известны кристаллогидраты NiF2-nH2O (где ге = 4, 3, 2)
зеленого цвета, фтороникелаты(П) K[NiF3], K2[NiF4l,
K[Ni(H2O)F3], (NH4)2[NiF4], комплексные фториды [Ni(NH3)5 •
• (H2O)]F2, [Ni(H2O)2Py4]F2, кислые фториды NiF2-5HF-6H2O
и различные фторосоли, в которых никель является катионом,
например Ni[BeF4]-6Н2О, Me*Ni[BeF4]2-6Н2О, Ni[BF4]2’6H2O,
Ni[SiF6]-6Н2О, Ni[SnF6]-6Н2О.
Дихлорид никеля, NiCl2, получают действием сухого хлора
на нагретый до 600—800° порошкообразный металлический никель,
длительным действием хлора на металлический никель в атмо-
сфере влажного воздуха при обычной температуре, нагреванием
соединения [Ni(NH3)6]Cl2 до 450°, нагреванием кристаллогидра-
тов NiCl2*nH2O (где п = 7, 6, 4, 2, 1) при 500—600° в токе газо-
образного НС1:
Ni + С12 = NiCl2 + 73 ккал [Ni(NH3)e]Cl2 -+ NiCl2 + 6NH3
Соединение NiCl2 представляет собой блестящие золотисто-
желтые ромбоэдрические кристаллы с плотностью 3,53 г/см3
и т. кип. 987°, очищается сублимацией в токе НС1, растворяется
в воде, спирте, эфире, превращается в NiO при прокаливании
на воздухе или в кислороде, восстанавливается водородом при
нагревании:
2NiCl2 + О2 2NiO + 2С12 NiCl2 + Н2 Ni + 2НС1
В водном растворе дихлорид никеля слабо гидролизуется:
NiCl2 + НОН Ni(OH)Cl + HCl
Известны кристаллогидраты NiCl2-nH2O (п = 7, 6, 4, 2, 1)
зеленого цвета, которые для координационных чисел 4 и 6 могут
быть записаны в виде [Ni(H2O)4]Cl2, [Ni(H2O)6]Cl2, комплексные
хлориды [Ni(NH3)2]Cl2, [NiEn]Cl2-nH2O, [Ni{C6H4(NH2)2}2]Cl2•
•nH2O, [Ni{C10H8(NH2)2}2]Cl2, [Ni(C4H8O2)2]Cl2, [Ni(N2H4)2]Cl2,
[Ni(NH2CONHNH2)2]Cl2, [Ni(C6H6CSNHNH2)2]Cl2,[Ni(C3H6NH2)4] •
• Cl2, [NiEn,]Cl2, [Ni(NH3)6]Cl2, [Ni(a,a' -C6H4N - C5H4N)3]G12-
598
ПОБОЧНАЯ ПОДГРУППА VIII ГРУППЫ
•nH2O, [NiPy4]Cl2, хлоросоли Li[NiCl3] -6Н2О, K[NiCl3]-5Н2О,
Rb2[NiGl4], Cs[NiCl3], Cs2[NiGl4], NH4[NiCl3] -nH2O (n = 2 или 6),
Ni[SnCl6], [Ni(H2O)6][BiCl5], [Ni(NH3)6][SbCl6]2, основные соли
NiCl2.mNi(OH)2(7n = 1, 2, 3), NiCl2(3 или 4) Ni(OH)2-nH2O,
NiCl2 (6 или 7) Ni(OH)2-nH2O. Хлорид гексамминникеля(П)
INi(NH3)6]Cl2 разлагается водой по уравнению
[Ni(NH3)6]Cl2 + 6Н2О Ni(0H)2 + 4NH4OH + 2NH4C1
При концентрировании водного раствора дихлорида никеля
(28,8°) выпадают зеленые кристаллы [Ni(H2O)4]Cl2, а при 64° —
зеленые кристаллы NiCl2*2H2O. Водный раствор дихлорида нике-
ля получают растворением металлического никеля в царской
водке или окиси либо карбоната никеля(П) в соляной кислоте.
Дибромид никеля, NiBr2, получают нагреванием тонкодисперс-
ного порошка металлического никеля в парах брома, нагрева-
нием соединения NiBr2-(C2H6)2O, пропусканием бромистого водо-
рода над нагретым докрасна NiO:
Ni + Вг2 = NiBr2 + 59 ккал NiO + 2НВг = NiBr2 + Н2О
Соединение NiBr2-(C^Hj^O желтого цвета получается дей-
ствием сухого брома на измельченный металлический никель под
слоем эфира.
Дибромид никеля представляет собой гигроскопичные блестя-
щие коричневые ромбоэдрические кристаллы со структурой типа
CdCl2, плотностью 4,6 г/см\ т. пл. 963°; растворим в воде и мета-
ноле, очищается сублимацией в токе НВг, превращается в NiO
при сильном нагревании на воздухе или в водяных парах, восста-
навливается водородом при нагревании:
NiBr2 + Н2 Ni + 2НВ1
Растворенные в жидком аммиаке щелочные металлы быстро
восстанавливают NiBr2 до тонкодисперсного металлического нике-
ля.
Известны кристаллогидраты NiBr24iH2O (где п = 9, 6, 3, 2),
комплексные бромиды [Ni(NH3)2]Br2, [NiEn]Br2,
[Ni{(CH2)6N4}2]Br2-10H2O, lNi{C6H4(NH2)2}2]Br2.nH2O,
[Ni{C10H6(NH2)2}2]Br2, [Ni(C4H8O2)2]Br2, [Ni(N2H4)2]Br2,
[Ni(NH3)6]Br, [NiEn3]Br2.reH2O, [NiPn3lBr2, [Ni(a,a'-C5H5N -
— C6H5N)3]Br2‘nH2O, бромосоли MeJ[NiBr3], Me*[NiBr4],
(C2H5)2[NiBr4l-2H2O, основные бромиды NiBr2 3Ni(OH)2, NiBr2-
•mNi(OH)2 •nH2O (m = 2, 5, 7).
Дииодид никеля, Nil2, получают нагреванием тонкодисперс-
ного порошкообразного металлического никеля в парах иода
и дегидратацией кристаллогидрата NiI2*6H2O в токе HI:
Ni -j- I2 = Nil2 -j- 38 ккал
НИКЕЛЬ
599
Соединение Nil2 представляет собой серые ромбоэдрические
кристаллы со структурой типа CdCI2, плотностью 5,8 г/см3 и т. кип.
747°; очищается сублимацией в токе HI, растворяется в воде
и спиртах, восстанавливается водородом при нагревании:
Nil2 + Н2 Ni + 2HI
Водные растворы иодида никеля при старении окрашиваются
в коричнево-красный цвет вследствие образования полииодида ни-
келя.
Известен гексагидрат [Ni(H2O)6]I2 в виде зеленовато-синих
призм, комплексные иодиды [Ni(C4H8O2)2]I2, [Ni(NH3) 4]12,
INi(N2H4)2H2, [Ni(NH3)6]I2, [NiEn3]I2-nH2O, [NiPn3]I2, комп-
лексные полииодиды [NiEn3]I4, [Ni(NH3)e]I9, аддукты Nil2-
•Hgl2 -4NH3 -4H2O, Nil2-2HgI2-4NH3 -2H2O.
Цианид никеля, Ni(CN)2 или Ni[Ni(CN)4], получают нагрева-
нием гидратов Ni(CN)2-hH2O (n — 7, 4, 3, 5/2, 2) или аммиака-
тов [Ni(NH3)4](CN)2-2H2O, Ni(CN)2-(2 или 3) NH3, Ni(CN)2-
•ИН3Д/2Н2О при 180—200° в атмосфере азота.
Соединение Ni(CN)2 — коричнево-желтый'порошок с диамаг-
нитными свойствами, плохо растворимый в воде.
При обработке растворов солей никеля(П) стехиометрическими
количествами растворов цианидов щелочных металлов образу-
ется светло-зеленый осадок гидратированного цианида никеля
Ni(CN)2'?zH2O, интересного тем, что он является парамагнитным
веществом.
При растворении зеленого осадка Ni(CN)2 *пН2О в избытке
KCN образуется золотисто-желтый раствор K2[Ni(CN)4] ’нНгО,
из которого при упаривании в зависимости от температуры,
могут быть выделены оранжевые орторомбические призмы
K2[Ni(CN)4I-Н2О или оранжевые моноклинные кристаллы
K2[Ni(CN)4l -ЗН2О.
Путем упаривания раствора Ni(CN)2 ’тгН2О в NaCN получают
желтые кристаллы Na2[Ni(CN)4]-ЗН2О.
Соединения K2[Ni(CN)4]-Н2О, K2lNi(CN)4] 'ЗН2О,
Na2[Ni(CN)4] ’3H2O растворимы в воде, легко дегидратируются
и превращаются под действием сильных кислот в Ni(CN)2*nH2O.
При обработке растворов тетрацианоникелатов(П)
Me*[Ni(CN)4] ‘пНгО хлорной или бромной водой в щелочной среде
образуется гидратированная окись никеля(Ш) в виде черного
осадка:
2Me|[Ni(CN)4] + 9С12 + 6MerOH = Ni2Os-3H2O + lOMe^l + 8C1CN
Следует отметить, что растворы гексацианокобальтатов(П)
более устойчивы и не образуют гидратированной окиси кобаль-
та(Ш).
600
ПОБОЧНАЯ ПОДГРУППА VIII ГРУППЫ
При обработке раствора тетрацианоникелата(П)
Me*[Ni(CN)4] • nil 2С) большим избытком цианида KCN или NaCN
образуются оранжево-красные растворы гексацианоникелатов(П)
Me*[Ni(CN)6] .пН2О.
При действии металлического калия на раствор K2[Ni(CN)4l
в жидком аммиаке образуется соединение K4[Ni(CN)4] в виде
оранжево-желтого аморфного порошка; это очень сильный восста-
новитель, легко окисляющийся на воздухе и выделяющий водо-
род из воды:
2K4[Ni(CN4)] + 2Н2О 2K2[Ni(CN)3] + 2KCN + 2КОН + Н2
Соединение K2[Ni(CN)3] образуется и при действии цинка
в кислой среде или амальгамы натрия (калия) в атмосфере водо-
рода на раствор K2[Ni(CN)4] •пНгО; выделяется из водного
раствора при добавлении спирта. Обработка соединения
K2[Ni(CN)3] окисью азота или окисью углерода дает неустой-
чивые оранжево-красные соединения K2[Ni(NO)(CN)3],
K2[Ni(CO)(CN)3].
Известны тетрацианоникелаты(П) Men[Ni(CN)4] -nH2O, а также
соединение Cr2[Ni(CN)4]3.
Тиоцианат никеля, Ni(SCN)2 "иЩО (и = 3/2 или 1/2), выде-
ляется в виде зеленых кристаллов из раствора, полученного обра-
боткой раствора сульфата никеля(П) тиоцианатом бария или кар-
боната никеля роданистоводородной кислотой.
Безводная соль Ni(SCN)2 окрашена в темно-коричневый цвет.
Тиоцианат никеля образует с тиоцианатами щелочных метал-
лов координационные соединения типа Me*[Ni(SCN)J *пН2О,
MeJ[Ni(SCN)6]-иНгО (где Ме+ = Na+, К+, NH4), например
Na2[Ni(SCN)4] -8Н2О — зеленое, K4[Ni(SCN)6] «4Н2О — синее,
(NH4)4[Ni(SCN)6] «4Н2О — синее.
Известны также соединения Cs2Ag2[Ni(SCN)e]’2Н2О,
Cs2Cu2[Ni(SCN)6] -2Н2О.
В присутствии тиоцианата щелочного металла или аммония
пиридин образует с ионом Ni2+ синий осадок [Ni(C5H5N)4](SCN)2,
плохо растворимый в спирте.
Ni2+ + 4CSHSN + 2SCN- = [Ni(C5H6N)4](SCN)2
Сульфид никеля, y-NiS, встречается в природе в виде минерала
миллерита и представляет собой блестящие бронзово-желтые
тригональные кристаллы с плотностью 5,2—5,6 г/см3, твердо-
стью 3—4 по шкале Мооса и т. пл. 797°. В природе сульфиду ни-
келя часто сопутствуют сульфиды других металлов в различных
минералах, таких, как пирротин, пентландит и др.
Были получены три аллотропные формы моносульфида никеля,
а именно а-, р- и y-NiS.
НИКЕЛЬ
601
Модификацию a-NiS (всегда сопровождающуюся модифика-
циями p-NiS и y-NiS) получают при pH>3,6 добавлением Na2S
или (NH4)2S к нейтральному раствору соли никеля(П); это чер-
ный аморфный порошок, растворимый в разбавленных (2 н.)
кислотах (НС1, H2SO4).
Модификация |3-NiS (всегда сопровождающаяся y-NiS) полу-
чается прямым взаимодействием элементов при нагревании без
доступа воздуха, нагреванием раствора хлорида никеля(П) с K2S
при 160—180° в запаянной трубке, действием H2S на нагретый
докрасна никель, обработкой H2S растворов солей никеля(П)
(в уксуснокислой среде). Модификация p-NiS, полученная из вод-
ных растворов, представляет собой черные гексагональные кри-
сталлы, которые растворяются в 2 н. НС1 при нагревании и вы-
ветриваются на воздухе при обычной температуре. Модифика-
ция p-NiS, полученная по сухому способу, представляет собой
бронзово-желтые гексагональные кристаллы с плотностью
4,60 г/см\ они проявляют сходство с миллеритом y-NiS, плохо
растворимы в НС1 или H2SO4, растворяются в HNO3, царской
водке, смеси КС1О3 с НС1:
3NiS + 6НС1 4- 2HNO3 = 3NiCl2 + 3S + 2ND + 4H2O
3NiS + КСЮз + 6HC1 = 3NiCl2 + 3S + KC1 + 3H2O
Модификация y-NiS — единственная, которая может быть
выделена в чистом состоянии (идентична природному миллериту);
имеет вид черных ромбоэдрических кристаллов. При обработке
смеси а-, Р~ и y-NiS 2 и. НС1 на холоду растворяется a-NiS, при
нагревании — p-NiS, остается нерастворимой модификация y-NiS.
В аналитической практике (при получении из растворов) вы-
падает смесь трех модификаций: а-, Р~ и y-NiS.
Среди таких модификаций сульфида никеля y-NiS обладает
самой низкой растворимостью в воде, она плохо растворяется
в 2 н. НС1 и растворяется только в присутствии окислителей.
Все модификации сульфида никеля растворяются в хлорной
воде, в твердом виде взаимодействуют с фтором, плавятся при
797° в атмосфере азота, превращаются в NiO при прокаливании
на воздухе (с промежуточным образованием NiSO4), восстанав-
ливаются при нагревании с водородом или углеродом до металла,
превращаются при нагревании в Ni3S4 и NiS2, взаимодействуют
в щелочной среде с окисью углерода:
NiS + 5СО + 4NaOH = Ni(CO)4 + Na2C03 -j- Na2S -f- 2H2O
Сульфид никеля имеет важное значение как катализатор.
Известны основной сульфид 2NiS -NiO, кислый сульфид Ni(HS)2
и двойные сульфиды, например 3NiS >K2S — серые кристаллы,
4NiS -BaS — красные кристаллы.
602
ПОБОЧНАЯ ПОДГРУППА VIII ГРУППЫ
Дисульфид никеля, NiS2 или , в природе не встре-
чается; может быть получен сплавлением смеси карбоната нике-
ля NiCO3, серы и карбоната калия К2СО3 или нагреванием сме-
си NiS с серой при 170° в течение 30—40 час.
Дисульфид никеля представляет собой серое вещество с кри-
сталлической структурой, аналогичной пириту, с плотностью
4,31 г!см\
В природе встречается только двойной сульфид (Ni, Fe)S2,
называемый бравоитом.
Сульфат никеля, NiSO4, получают прокаливанием кристалло-
гидратов NiSO4-7zH2O (п = 7, 6, 5, 4, 3, 2, 1) при 280°, упарива-
нием раствора сульфата никеля(П) в конц. H2SO4, действием ме-
тилсульфата при 160—190° на NiO или на какую-либо неорганиче-
скую (или органическую) соль никеля(П).
Сульфат никеля представляет собой лимонно-желтые ортором-
бические кристаллы с плотностью 3,64 г/см3; он растворим в воде,
разлагается при высокой температуре, восстанавливается при
нагревании водородом, алюминием, магнием.
Известны кристаллогидраты NiSO4-7zH2O (п = 7, 6, 5, 4, 3,
2, 1). Кристаллогидрат NiSO4-7H2O или [Ni(H2O)e]SO4 «Н2О
встречается в природе в виде минерала моренозита. Может быть
получен упариванием при обычной температуре (до 31°) ней-
трального раствора окиси, гидроокиси или карбоната никеля(П)
в разб. H2SO4. Кристаллогидрат [Ni(H2O)e]SO4 -Н2О представляет
•собой зеленые орторомбические кристаллы с плотностью 1,931 г/см3
Модификация a-NiSO4-6H2O или a-[Ni(H2O)e]SO4 встречается
в природе в виде минерала ретгерсита. Упариванием нейтраль-
ного водного раствора сульфата никеля(П) при 31,5—53,5° полу-
чают синие тетрагональные кристаллы с плотностью 2,036 г/см3.
Упариванием водного раствора сульфата никеля(П) при 53,3 — 70°
получают зеленые моноклинные кристаллы (изоморфные MgSO4<
•6Н2О).
Известны двойные сульфаты Ме$эО4-NiSO4 *6Н2О (где
Me1 = К+, Rb+, Gs+, NH+, Т1+), которые изоморфны соответст-
вующим соединениям железа, кобальта, магния, цинка, NiSO4*
•Ti(SO4)2, основные сульфаты NiSO4 -3Ni(OH)2 ’иНгО, 3NiSO4-
•4Ni(OH)2, аддукты NiSO4-3N2H4, NiSO4-N2H4.3H2O,
комплексные сульфаты [NiEn]SO4-иНгО, [Ni(CeH7N)2]SO4,
lNi{C8H4(NH2)2}2]SO4 -nH2O, [Ni{C10H6(NH2)2}2SO4]SO4,
[Ni(NH3)4]SO4 -2H2O, [Ni(NH2CONHNH2)2]SO4,
[Ni(G6H5GSNHNH2)2]SO4, [Ni(G3H6N2H4)2]SO4, [Ni(NH3)6]SO4,
(NiEn3]SO4 "иНгО.
Нитрат никеля, Ni(NO3)2, получают нагреванием до плавления
кристаллогидрата Ni(NO3)2 -6Н2О или обработкой дымящей HNO3
НИКЕЛЬ
603
водного раствора нитрата никеля(П), сконцентрированного до
сиропообразной консистенции. Нитрат никеля представляет собой
желто-зеленый порошок, растворимый в жидком аммиаке с обра-
зованием фиолетово-розового раствора и превращающийся при
прокаливании до 550° в кислороде в остаток, состав которого варьи-
рует между NiO и NiO2.
Известны кристаллогидраты Ni(NO3)2 ’пН20 (и = 9, 6, 4 и 2),
которые выделяются при упаривании (между —30 и 120°) раство-
ра, полученного действием на металлический никель, окись, гидро-
окись или карбонат никеля(П) разбавленной азотной кислоты.
Соединение Ni(NO3)2-6Н2О или [Ni(H2O)e](NO3)2 осаждается
при упаривании водного раствора нитрата никеля(П) при ком-
натной температуре; это расплывающиеся на воздухе зеленые
кристаллы с плотностью 2,02 г/см\ т. пл. 56,7° и т. кип. 136,7°.
Из водного раствора нитрата никеля при температуре ниже —3°
выпадают кристаллы Ni(NO3)2-9Н2О, при температуре выше 54°—
кристаллы Ni(NO3)2-4Н2О, при температуре выше 85,4° —
дигидрат Ni(NO3)2-2Н2О.
Известно тпранс-производное [Ni(H2O)2(NH3)4](NO3)2.
Нитрат никеля(П) применяют в керамической промышленности
Известны комплексные нитраты [NiEn](NO3)2 -пН2О,
lNi{CeH4(NH2)2}2](NO3)2 -nH2O, [Ni(H2N - СеН4 - СеН4 - NH2)2](NO3)2,
lNi(C9H8N2)2](NO3)2 -nH2O, [Ni(NH3)4](NO3)2, [Ni(CeH5CSNHNH2)2](NO3)2,
(Ni(NH3)e](NO3)2, [NiEn3](NO3)2, [Ni(H2O)2, En2](NO3)2,[Ni(C5H6N)e](NO3)2,
нитратосоли типа Me2[Ni(NO3)4], двойные нитраты 3Ni(NO3)2-Mein (NO3)3 •
• 24H2O, 3Ni(NO3)2-2Ce(NO3)3, Ni(NO3)2 .Ce(NO3)4-8H2O, основные нитра-
ты Ni(0H)NOa-3H2O, Ni(NO3)2-3Ni(OH)2, Ni(NO3)2-4Ni(OH)2.7H2O,
Ni(NO3)2.3Ni0-2,5H2O, Ni(NO3)2-2NiO, Ni(NO3)2-4NiO-6H2O.
Карбонат никеля, NiCO3, получают нагреванием NiCl2 с CaCO3
при 150° в запаянной трубке или нагреванием при 140° NiCl2
с раствором NaHCO3, насыщенным СО2.
Карбонат никеля образует зеленовато-белые ромбоэдрические
мелкие кристаллы, плохо растворимые на холоду в HCl HNO3
и разлагающиеся при сильном нагревании на NiO и СО2.
Известны кристаллогидраты NiCO3 -6Н2О, NiCO3 -ЗН2О. Гекса-
гидрат NiCO3-6H2O образуется в виде зеленого кристаллического
осадка при обработке растворов солей никеля(П) раствором
NaHCO3, насыщенным СО2, или обработкой суспензии Ni(OH)2
в воде двуокисью углерода при 40° под давлением в течение не-
скольких дней. Тригидрат NiCO3*3H2O образуется в виде зеле-
ного осадка при добавлении большого избытка раствора КНСО3
к раствору соли никеля(П).
Карбонат никеля применяется в керамической промышленности
в качестве пигмента.
При получении карбоната никеля нельзя применять карбонат
щелочного металла, поскольку образуется основной карбонат ни-
604
ПОБОЧНАЯ ПОДГРУППА VIII ГРУППЫ
келя переменного состава, например 2NiCO3 -3Ni(OH)2 -4Н2О,
2NiCO3-5Ni(OH)2, 4NiCO3-Ni(OH)2, 3NiCO3-Ni(OH)2.
Карбонат никеля с карбонатами щелочных металлов или ам-
мония образует комплексные карбонаты типа Me^[Ni(CO3)2] •
• пН2О, например Na2[Ni(CO3)2] *10Н2О, K2[Ni(CO3)2] -4Н2О, в виде
кристаллических осадков зеленого цвета.
Известны аммиакаты NiCO3-5NH3 -4Н2О, NiCO3-3NH3.
Ацетат никеля, Ni(CH3COO)2 -4Н2О, выпадает из раствора,
полученного обработкой гидроокиси или карбоната никеля уксус-
ной кислотой, в виде зеленых кристаллов с плотностью 1,80 г/см3;
они плохо растворимы в спирте, растворяются в воде на холоду
и гидролизуются водой при нагревании.
Известна безводная соль Ni(CH3COO)2 и аддукт Ni(CH3COO)2-
•2C5H5N.
Помимо приведенных известны и другие соединения двухвалентного
никеля, например хлорит Ni(C102)2, безводный и гидратированные хлораты
Ni(C103)2, [Ni(H20)4](C103)2, [Ni(H20)6](C10s)2, комплексные хлораты
[Ni(NH3)e](C103)2, [Ni(NH3)6H20](C103)2, [Ni(C5H6N)4](C103)2, безводный
и гидратированные перхлораты Ni(C104)2, Ni(C104)2-nH2O (п = 9, 6, 4, 2),
комплексные перхлораты [Ni(NH3)6](C104)2, [Ni(C5H5N)6](C104)2-4Н2О,
бромат Ni(BrO3)2-6Н2О, безводный и гидратированные йодаты Ni(IO3)2,
Ni(IO3)2-пН2О (п = 6, 4, 2), перйодаты Ni2I2O9, Ni5(IO6)2, сульфит
NiSO3, аддукты сульфита NiSO3-3NH3- ЗН2О, NiSO3-3N2H4,
NiSO3-2N2H4-2Н2О, основной сульфит 2NiSO3-Ni(OH)2-6Н2О, комплексные
сульфиты Me2{Ni(SO3)2], дитионат NiS2O6, политионаты NiS3Oe-6H2O,
NiS4O6, NiS50e, тиосульфаты NiS2O3, NiCu(S2O3)2-4Н2О, аддукты пероксо-
сульфата NiS2O8 ^CsHjN, NiS2O8-6NH3, селенид NiSe, селениты NiSeO3,
NiSeO3-nH2O (n = 2, 1, 1/2), NiSeO3-SeO2-3H2O, NiSeO3 -3SeO2 .H2O,
3NiSeO3-NiO-H2O, селенаты NiSeO4, NiSeO4-rtH2O (n = 6, 4, 1),
Me2SeO4-NiSeO4-6H2O (где Mei = K+, Rb+, Cs+, NHp, 2NiSeO4-3Ni(OH)2-
• 5H2O, NiSeO4-6NH3, теллурид NiTe, теллурит NiTeO3-4H2O, теллурат
NiTeO4, амид Ni(NH2)2, азиды Ni(N3)2-H2O, Ni(OH)N3, KN3-Ni(N3)2, нитри-
ты Ni(NO2)2, Ni(NO2)2-NiO, Ni(NO2)2-4NH3, Ni(NO2)2 .reNH3 (n = 6, 4),
(Ni(NO2)2 -2N2H4, Ni(NO22-reC6H5N (n = 6, 4, 2), MeI[Ni(NO2)6] (где Mei =
= Li+, Na+, K+, T1+, V2Mg«+, V-iCa2*, i/2Sr2+, i/2Ba2+, J/2Cd2+, ^Hg2*, !/2Pb2+,
Ме2МеП [Ni(NO2)el (где Mei = K+, Rb+, Gs+, NHJ, T1+ и Mell = Mg2+,
Ca2+, Sr2+, Ba2+, Zn2+ или Hg2+), гипофосфит Ni(H2PO2)2-6H2O, фосфиты
NiHPO3.H2O, K2Ni3(HPO3)4-32H2O, (NH4)2Ni3(HPO3)4-18H2O, ортофос-
фаты Ni3(PO4)2-8H2O, NH4NiPO4 (1 или 6) H2O, NaNiPO4-7H2O, LiNiPO4,
2K3PO4.Ni3(PO4)2, пирофосфаты Ni2P2O7, Ni2P2O7-6H2O, Na2NiP2O7-6H2O,
метафосфаты Ni2P40i2, Ni2P4O12-12H2O, Ni3P60i8, трифосфаты NiH3P3O10 •
• 3H2O, Na3NiP3Oio-12H20, NaNi2P3O10, тиофосфит Ni3(PS3)2, тиофосфаты
Ni3(PS4)2, Ni2P2S7, NinPnS3jl, арсенит Ni3(AsO3)2-nH2O, арсенаты Ni3(AsO4)2 •
•8H2O, Ni3(AsO4)2, Ni3(AsO4)2mNH3-nH2O, K2Ni2(AsO4)2, Na2Ni2(AsO4)2,
Na2NiAs2O7, антимонит Ni[Sb2O4], антимонат NiSb2O6-пИгО, тиоантимонаты
[NiEn3]2SbS4Cl-5H2O, [NiEn3]2SbS4NO3, [NiEn3]2SbS4Br-5H2O, [NiEn3]2-
• SbS4I-5H2O, lNiEn3]2SbS4SCN-2H2O, аддукт тиокарбоната с аммиаком
NiCS3-3NH3, цианамид NiCN2, цианат Ni(CNO2-(4 или б^ЩК, фульми-
нат Na2[Ni(CNO)4], тиоцианаты [NiEn2][Ag(SCN)4], [NiEn2][Hg(SCN)4], селе-
ноцианаты Ni(SeCN)2-6NH3, [NiEn2][Ag(SeCN)2]2, [NiEn3][Hg(SeCN)4], си-
ликаты Ni_2SiO4, NiSiO3, CaNiSi2O6, Ni3Si2O6(OH)4, (Ni, Mg)3Si4O10(OH)2,
(Ni, Mg)3Si2O6(OH)4, германат Ni2GeO4, станнат NiSn03-5H20, бораты
НИКЕЛЬ
605
Ni(BO3)2, NiB4O7-6H2O, 4NiB4O7-2NiO .Nil2, алюминат Ni[Al2O4], галлат
Ni[Ga2O4], индат Ni[In2O4], тиоиндат NiIn2S4, ниобат NiNb2O6, танталат
Ni2Ta2O6, хромит NiCr2O4, хроматы NiCrO4, K2Ni(CrO4)2-nHO2 (n = 2
или 6), дихромат NiCr2O7-3/2H2O, молибдат NiMoO4, вольфрамат NiWO4,
гетерополисоединения Ni3[PMo12O40]2-58H2O, Ni2[SiMo12O40]-31H2O,
Ni3[PW1204o]2 -48H2O, Ni2[SiW1204oI-18H20, манганат NiMnO4-6NH3, без-
водный и гидратированные перренаты Ni(BeO4)2, Ni(BeO4)2-nH2O (га = 5,
4, 2), аддукты перрената Ni(BeO4)2-(4 или 6)NH3, феррит Ni[Fe2O4], феррат
NiFeO4, многочисленные комплексные цианиды Ni2[Fe(CN)6], K2Ni[Fe(CN)6],
Na2Ni3[Fe(CN)6]2, NiCu[Fe(CN)6], KNi[Fe(CN)6], NiPt(CN)4, Ni[Zn(CN)4] и др.,
оксалаты NiC2O4-2H2O, 2NiC2O4.NH3-6H2O, NiC2O4-2NH3-5H2O,
NiC2O4-2N2H4, K2[Ni(C2O4)2]-6H2O, формиат Ni(HCOO)2-2H2O, малонат,
сукцинат, глутарат, адипинат, тартрат, цитрат, лактат, малат, гликолят и т. и.
Координационные соединения
Известно большое} число моноядерных и довольно мало ди-
и полиядерных координационных соединений никеля(П).
Хотя между никелем и кобальтом наблюдается большое
сходство, все же координационные соединения первого менее
устойчивы, чем соответствующие координационные соединения
кобальта.
Классификация моноядерных
координационных соединений
Моноядерные координационные соединения никеля(П) могут
быть классифицированы по числу координированных групп как
ди-, три-, тетра-, гексакоординационные комплексы или по при-
роде координированных групп как аммины, аквосоли, ацидосоли
и комплексы типа неэлектролитов.
1. К комплексам с координационным числом 2 относятся диам-
мины [Ni(NH3)2]X2 (где X = Cl", Вг", N;), (C7H4NS2)-, VjBeFj2-,
этилендиамины [NiEnlX2-иЩО (где X = С1-, Вг-, NO;, 1/2SO2-),
дипиридины [NiPy2]X2 (где X = N;, (CH3NNO2)-, (C7H4NS2)“,
VjFefCNJsNOl2-. дипиколины [Ni(C6H7N)2]X2 (где X = ^SO2-),
дианилины [Ni(C6H5NH2)2]X2 (где X = VgSeO2-, (C7H4NS2)-, дифе-
нилгидразины [Ni(C8H5 — NH — NH2)2]X2, дигексаметилентетра-
мины [Ni{(GH2)0N4}2]X2-lOH2O (где X = Cl- или Br-), ди-п-апи-
зидины [Ni{n-C8H4(OCH3)NH2}2]X2, дифенилендиамины
[Ni{C8H4(NH2)2}2]X2-nH2O (где X = Cl-, Br-, NO;, V2SO2-),
динафтилендиамины [Ni{C10H8(NH2)2}2]X2 (где X = Cl-, Br-,
^SO2-), дибензидины [Ni(H2N — C8H4 — C8H4 — NH2)2]X2 (где
X = NO;, NCS-, 1/2[CdCl4]2-, VJSiFeJ2-), диамино-8-хинолины
[Ni(C9H8N2)2]X2-иНгО (где X = Cl-, NO;), дидиоксаны
[Ni(C4H8O2)2]X2 (где X = Cl", Br-, I-).
2. Трикоординированные комплексы включают триаммины
.[Ni(NH3)3]X2 (где X = SCN-), трипиридины [NiPy3]X2 (где X =
606
ПОБОЧНАЯ ПОДГРУППА VIII ГРУППЫ
= SCN-); дицианомоноаммины [Ni(NH3)(CN)2] -0,5 Н2О, дициано-
m - NH^CW (гд. R = СН„ сл, С,Н„
С4Н9), [Ni ( NH) (CN)2] (где R = R' = CH3), ацидосоли
Me4NiCl3], Me4NiBr3], Me1 [Ni(CN)3] (где Me1 = Rb+, Cs+, NH*).
3. К комплексам с координационным числом 4 относятся те-
траммины [Ni(NH3)4]X2-nH2O (где X = I-, NO;, CN", VzSOI-,
VztBeFJ2-), дианилиндиаммины [Ni(NH3)2(CeH5NH2)2]X2, дитолу-
идиндиамины [Ni(NH3)2(CH3 — CeH4 — NH2)2]X2, дигидразины
[Ni(N2H4)2]X2 (где X = Ci", Br-, SCN-, CIO;, ^SO^, VAOJ-),
диаквогидразины [Ni(H2O)2(N2H4)]X2-H2O, дисемикарбазиды
[Ni(NH2CONHNH2)2]X2 (где X = Cl-, VzSOJ-), дитиобензогидра-
зиды [Ni(C6H5CSNHNH2)2]X2 (где X = Cl-, NO;, VzSOJ-), тетра-
квосоли [Ni(H2O)4]X2 (где X = Cl", CN", CH3COO-, PO;, VzSO2-),
дигликоляты [Ni(CH2OH — CH2OH)2]X2, этаноламины
[Ni(C2H4OHNH2)2]X2, тетраэтиламины [Ni(C2H5NH2)4]X2, тетра-
аллиламины [Ni(C3H5NH2)4]X2, диэтилендиамины [NiEn2]X2 -?zH2O
(где X = C1-, CIO;, SCN", SeCN", ^[H^SON),]2-, 1/2[Hg(SCN)2 •
• (CN)2]2“, 1/2S20j_), ди-а-ацетамидопиридины [Ni(C3HeN2H4)2]X2
(где X = SCN-, CIO;, VzSO2-), тетрапиридины [NiPy4]X2, ди-
акводипиридины [Ni(H2O)2Py2]X2, тетрапиколины [Ni(C5H4N —
— CH3)JX2, ди-а-ацетамидопиридины [Ni(NC5H4 — NH — ОС —
— CH3)2]X2, тетранилины [Ni(C6H5NH2)4]X2, диакводибензилами-
ны [Ni(H2O)2(C6H5 — CH2 — NH2)2]X2, тетра-о-фенилендиамины
[Ni{CeH4(NH2)2}4]X2, диэтанолдитолуидины [Ni(C2H5OH)2(CH3 —
— C6H4 — NH2)2]X2, тетрафенилгидразины [Ni(CeH5 — NH —
— NH2)4]X2, диакв одибензидины [Ni(H2O)2(NH2 — C6H4 —
— HN2)2]X2, тетрахинолины [Ni(C9H7N)4]X2, дифенан-
тролины, диакводиантипирины, тетрамминантипирины, бмс-ди-
аминбутаны, Р, Р',Р"-триаминтриэтиламины, Р,Р',Р"-триаммин-
пропиламины, ди-р-аминтриэтиламины, диамингуанидины, тетра-
этил ентиокарбамиды, цианотриаммины [Ni(NH3)3CN]X, сульфа-
тотриаллиламины [Ni(C3H5NH2)3SO4] •2C3HsNH2, дииододигидра-
зин [Ni(N2H4)2I2], дицианодиаммин [Ni(NH3)2(CN)2], дицианоакво-
бензиламин [Ni(H2O)(NH2 — СН2 — C6H5)(CN)2], дицианодиметил-
(этил, пропил, бутил)амины, трицианомоноаммины MeI[Ni(NH3)-
• (CN)31, тетрацидосоли Me£[NiF4] (где Me1 = К+, NH^)r
Me|[NiCl4], (где Me1 = VzCd2*, VzNi2*), Mei[NiBr4]„
Me*[Ni(NO3)4], Me|[Ni(CN)4]-пНгО (где Me* = Na+, K+, ^Ca2^
V2Sr2+, 1/2Ba2+, V-sZn2*, 1/2Cu2+, V-sCo2*, VzNi2*), Mei[Ni(SCN)4],
Me^[Ni(CNO)4]-raH2O, диацидосоли Me£[Ni(SO4)2], Me£[Ni(S03)2[,
Mei[Ni(SeO4)2]-nH2O, Mei[Ni(CO3)2bnH2O, MeJ[Ni(C2O4)3],
Mei[Ni(BeF4)2] -nH2O, Mei[Ni(P4O12)2L
4. Комплексы с координационным числом 6 включают гексам-
мины [Ni(NH3)6]X2 (где X = Cl", Br-, I-, I;, NO;, N;, H2PO;,.
НИКЕЛЬ
607
[SbCl6]МпО;, ReO4-, V2SO42-, ^CrO^, V2S2O62-, VAO2-,
1/2SeO|-, VzWOJ", VzMnO2-, VjBeFJ2-, аквопентаммины
[Ni(NH3)5(H2O)]X2 (где X = F", V2MnO2-, V^O2-, VzCO2-),
диаквотетраммины [Ni(NH3)4(H2O)2]X2, триаквотриаммины
[Ni(NH3)3(H2O)3]X2, тригидразины [Ni(N2H4)3]X2, гексафенил-
гидразины [Ni(C6H5 — NH — NH2)e]X6, трисемикарбазиды
[Ni(NH2CONHNH2)3]X2, трифенилсемикарбазиды [Ni(CeH5).
• NHCONHNH2)3]X2, тритиосемикарбазиды
[Ni(NH2CSNHNH2)3]X2, гексагидроксиламины [Ni(NH2OH)e]X2,
гексаквосоли [Ni(H2O)e]X2 (где X = Cl-, ClOj, BrO^, IO"r
V2SO2-, V2SO|-, V2S2O|-, V^O2-, VzSeO*-, ^CO2-, VJBiC^]2-,
1/2[SiFe]2-, VzISrBrg]2-), триэтаноламины [Ni(0H — C2H4 —
— NH2)3]X2, тригликоляты Mex[Ni(CO2 — CH2 — NH2)3], три-
этилендиамины [NiEn3]X2-иНгО (где X = Cl-, Br-, I-, NO7 ClO^r
NJ, OH-, [BFJ-, [Agl2]-, V2SO2-, ^CrO2-, V2MoSr, WS2-,
VzlPtClJ2-), диакводиэтилендиамины [Ni(H2O)2En2]X2 (где X =
= NO^, СЮ7, P-C10H7COO-, CeHsCOO-), тетраквоэтилендиами-
ны [Ni(H2O)4En]X2, тримоноэтилэтилендиамин [Ni(C2H5NH —
— CH2 — CH2 — NH2)3](C1O4)2, трипропилендиамины [NiPn3]X2t
тригликоли [Ni(CH20H — CH2OH)3]X2 (где X = Cl-, Br-, I-,
CN-, SCN-), триглицероли [Ni(CH2OH - CHOH - CH2OH)3]X2,
гексаметилентетрамины [Ni(C6Ht2N4)]X2,гексапиридины [NiPy6]X?,
аквопентапиридины [Ni(H2O)Py5]X2, диакв отетрапиридины
[Ni(H2O)2Py4]X2, три-сс, а!-дипиридилы [Ni(a,a'-C5H4N —
- C5H4N)3]X3.«H2O (где X = Cl-, Br-, NO- SCN-, V2SO2-),
трифенантролины [Ni(C12HgN2)3], Х2>иН2О, гексафениламины
[Ni(C6H5NH2)e]X2, гексафенилгидразины [Ni(CeH5 — NH —
— NH2)6]X2, тетракводитолуидины [Ni(H2O)4(CH3 — C6H4 —
— NH2)2]X2, гексантипирины [Ni(COCI0H12N2)e]X2, триаквотри-
антипирины [Ni(H2O)3(COC10H12N2)3]X2, дипиридинтетратиоци-
анаты
Г {(CH2)gN4}2-|
Fe
L РУ4 J
(SCN)4
Ni
₽y2
комплексы типа неэлектролитов [NiEn2(SCN)2], [NiPy4Cl2],
[Ni(C12H8N2)2X2], диаквокомплексы Me|[Ni(H2O)2(C2O4)2],
Mei[Ni(H2O)2(C6H4O2)2], Mei[Ni(H2O)2(C12H8O2N2)]-3H2O, ацидо-
соли MeJ[NiCl6l, Mei[Ni(NO2)6], MeJ[Ni(NO3)e], Mei[Ni(CN)6h
MeJ[Ni(SCN)6], MeJ[Ni(HCOO)6], Mei[Ni(SO4)3], Mei[Ni(C6H4O2)3],
Mei[Ni(C6H5N - CH2 - NH2)2(C2O4)2L
Хелатные соединения типа неэлектролитов
Катион Ni2+ (соответственно [Ni(H2O)e]2+) образует интенсивно
окрашенные хелатные соединения типа неэлектролитов с много-
численными органическими соединениями, содержащими оксим-
608
ПОБОЧНАЯ ПОДГРУППА VIII ГРУППЫ
ную группу = N — ОН, например с диметилглиоксимом HON =
= С(СН3)-С(СНз) = NOH, дифенилглиоксимом HON = С(С6Н5) •
•С(С6Н5) = NOH, диацетилмоноксимом CH,-СО-CfNOH)-СНз,
о-циклогександиоксимом
Н2С — СН2 — С = NOH
I I
Н2С - СН2 - С = NOH
Диметилглиоксим с катионом?И2+(соответственно с [Ni(H2O)6]2+)
образует в аммиачной среде хелатное соединение, окрашенное
в красный цвет. В случае малых концентраций раствор имеет
интенсивно красный цвет, а из концентрированных растворов
выпадает красный осадок:
Н3С-C=N —ОН
NI2+ + 2 | +2NH3 =
Н3С — C = N —ОН
+ 2NH+
II
С —сн3
Рубеановодородная кислота
HN = C—SH
I
HN = C—SH
(в кислой таутомерной форме) с катионом Ni2+ образует синий
хелат, плохо растворимый в воде и в минеральных кислотах:
HN=C-SH
NP+ + |
HN=C-SH
При действии оксина на растворы солей никеля(П) при pH =
= 4,3—14,6 образуется зеленый осадок оксината никеля
[Ni(C9HeON)2] -ЗНгО, который дегидратируется при 123° и безвод-
ная соль которого устойчива при 219—340°. Если раствор какой-
либо соли никеля в 1 н. НС1 обработать Z-цистином (бпс-р-ами-
но-р-карбокси-этилдисульфидом), а затем добавить аммиак, то
выпадает хелатное соединение
’S— НС2— CH—C = oq
I I
H2N о
h2n о
I I
LS-CH2—НС —c = o
H2O
НИКЕЛЬ
609
Известны многочисленные хелатные соединения никеля(П)
с ацетилацетоном, этилендиамином, а- и р-нафтойными кислотами,
с n-метоксибензойной, амино-3-нафтойной, нитрилтриуксусной
и этилендиаминтетрауксусной кислотами, с изонитрозобензоила-
цетоном, изонитрозоацетофеноном и др.
Ди-итриядерные комплексы
Известно немного ди- и триядерных комплексов никеля(П),
например [Ni2tren3]X4 (где tren = три-0,0',Р"-триамминотриэтила-
мин), [Ni2(H2N - С2Н4 — NH — С2Н4 — NH — С2Н4 -
— NH2)3]X4 -пИгО, дигликолевые соли [Ni2{H2C(OH) — СОО}2]Х2
фосфаты Me*[Ni3(HPO4)4] -иНгО.
Соединения трехвалентного никеля
Соединения трехвалентного никеля довольно малочисленны.
Они неустойчивы, проявляют окислительные свойства, образу-
ются при энергичном окислении некоторых соединений никеля(П).
В качестве примеров соединений никеля(Ш) можно назвать гидра-
тированную окись Ni2O3 -НгО.никелаты LiNiOE, NaNiO2, Ba2Ni2O5,
аддукт NiCl3 •2C6H4[As(CH3)2]2, сульфид Ni2S3, ацетаты
Ni(CH3COO)3, [Ni3(CH3COO)6](CH3COO)3, [Ni3(CH3COO)6](OH)3,
NaNi(UO2)3(CH3COO)9-8H2O, а также координационные соеди-
нения Mei[Ni(CH2 = NO)e], [Ni{P(C2H5)3}2Br3],
Окись никеля, Ni2O3-H2O или NiOOH, существует в виде трех
модификаций: а-, [}- и y-Ni2O3 -H2O.
В результате окисления соли никеля(П) в щелочной среде
пероксосульфатом, гипохлоритом или гипобромитом щелочного
металла, при электролитическом окислении Ni(OH)2 и нагрева-
нии Ni(NO3)2-бНгО или NiCO3-6H2O при 250—300° на воздухе
образуется модификация [J-Ni2O3-H2O — черные гексагональные
кристаллы с плотностью 4,15 г!см3, которые устойчивы на воздухе
и растворяются в НС1 с выделением хлора или в кислородных кис-
лотах с выделением кислорода:
К12Оз*Н2О -j- 6НС1 -|- 8Н2О = 2NiCl2*6H2O -р С12
Окислением щелочного раствора K2[Ni(CN)4] пероксосульфа-
том щелочного металла при 70° или гипобромитом щелочного ме-
талла при обычной температуре получают |3-Ni2O3-Н2О, который
довольно быстро превращается в a-Ni2O3-H2O — модификацию
черного цвета с плотностью 3,20 г/см3.
Гидролиз никелата NaNiO2 приводит к образованию модифи-
кации y-Ni2O3-H2O — черные игольчатые гексагональные кри-
сталлы с плотностью 3,85 г/см3.
Окись никеля неустойчива, обладает окислительными свойст-
вами, при легком нагревании или при обычной температуре в вод-
ной среде превращается в Ni3O4-2H2O.
39—0101
610
ПОБОЧНАЯ ПОДГРУППА VIII ГРУППЫ
На принципе анодного окисления Ni(OH)2 до Ni2O3«H2O осно-
вывается действие аккумуляторов Эдисона, в которых использу-
ется щелочной электролит КОН; они дают электрический ток
напряжением ~1,8 в по реакции
Разрядка
Ni2O3-H2O + Fe + 2H2O , ---2Ni(OH)2-|-Fe(OH)2
Зарядка
Такой аккумулятор имеет анод из Ni2O3 -Н2О и катод из порош-
кообразного железа. Электролитом служит раствор КОН, обычно
содержащий LiOH — для увеличения емкости аккумулятора.
При разрядке аккумулятора анод превращается в Ni(OH)2, а катод
покрывается слоем Fe(OH)2- В настоящее время аккумуляторы
заполняются щелочным электролитом, при этом вместо железа
в качестве катода используется металлический кадмий.
Аккумуляторы с щелочным электролитом довольно легки,
устойчивы к ударам, легко переносят перегрузку и могут хра-
ниться продолжительное время в разряженном состоянии.
Соединения четырехвалентного никеля
Четырехвалентное состояние не характерно для никеля. Извест-
но очень немного соединений четырехвалентного никеля. Все они
имеют окислительный характер и неустойчивы. В качестве при-
меров соединений никеля(1У) можно привести никелаты BaNi2O5,
K2Ni2+[Ni 4+О3]2, Na2Ni2+[Ni4+O3]2, ортопериодаты Me1 NiIO8 (где
Me1 = Na + , K+), а также координационные соединения
Металлоорганические соединения
К металлоорганическим производным никеля относятся карбо-
нильные соединения, галогениды нитрозилникеля и циклопента-
диенильные л-соединения. Примеры металоорганических произ-
водных никеля: Ni(CO)4, [H2Ni(CO)3]2, Ni(CO)2[P(C6H5)3]2,
Ni(CO)2[P(GF3)2]2, Ni(CO)3[As(C6H5)3], Ni(CO)3[Sb(C6H5)3l,
[Ni(NO)Cl]4, INi(NO)I]4, Ni(C5H5)2, [Ni(C5H5)2]Cl, C5H5NiNO,
(C5H5NiCO)2.
НИКЕЛЬ
611
Тетракарбонил никеля, Ni(CO)4, получают пропусканием оки-
си углерода над порошкообразным (или губчатым) металлическим
никелем, нагретым до 50—60°, при атмосферном давлении, дей-
ствием окиси углерода под давлением 50—100 ат на концентри-
рованный раствор гексаммина никеля, нагретый до 80°, обработкой
соединения K2[Ni(CO)(CN)3] кислотами, действием окиси угле-
рода и фенилмагнийбромида на хлорид никеля(П):
Ni + 4СО Ni(CO)4 -|- 52 ккал
[Ni(NH3)elCl2 + 5СО + 2Н2О = Ni(CO)4 + (NH4)2CO3+2NH4C1 + 2NH3
4K2[Ni(CO)(GN)3] + 2HC1 = Ni(CO)4 + 3K2[Ni(CN)4] + 2KC1 + H2
NiCl2 + 2CeH6MgBr + 4CO = Ni(CO)4 + MgBr2 + MgCl2 + CeH5 - C6H5
Соединение Ni(CO)4 представляет собой диамагнитную бес-
цветную жидкость, очень летучую и исключительно токсичную;
имеет плотность 1,356 г/см3 при 0°, т. кип. 43°, затвердевает при
—23°, полностью разлагается на металлический никель и окись
углерода при нагревании до 180—200° или под действием ультра-
фиолетовых лучей. Тетракарбонил никеля плохо растворим в воде,
растворяется в эфире, хлороформе, бензоле,-толуоле, не взаимо-
действует с разбавленными кислотами и щелочами.
При действии хлора, брома или иода на Ni(CO)4 образуются
дигалогениды никеля.
Кислород или воздух окисляют Ni(CO)4 до NiO и СО2:
2Ni(CO)4 + 5О2 = 2NiO + 8СО2
Реакция сопровождается воспламенением.
Под действием окиси азота NO тетракарбонил никеля в газооб-
разном состоянии или в растворе хлороформа превращается
в соединение Ni(NO)OH, а в спиртовом растворе — в Ni(NO)OH-
•С2Н5ОН.
Концентрированная серная кислота бурно (со взрывом) реа-
гирует с Ni(CO)4:
Ni(CO)4 + H2SO4 = NiSO4 + 4CO + H2
Азотная кислота и царская водка превращают Ni(CO)4 в соли
никеля(П).
При действии о-фенантролина C12H8N2 на спиртовый раствор
Ni(CO)4 образуется соединение Ni(CO)2 -C12H8N2 рубиново-крас-
ного цвета. Сера, растворенная в органических растворителях,
с Ni(CO)4 дает NiS.
Взаимодействие тетракарбонила никеля с P(CF3)3 при ком-
натной температуре приводит к образованию Ni((CO)3P(CF3)3
и Ni(CO)2[P(CF3)3]2.
При действии метилизонитрила CH3NC или фенилизонитрила
C6H5NC на эфирный раствор Ni(CO)4 образуются желтые кристал-
лы Ni(CO)(CH3NC)3 и Ni(C6H6NC)4.
39*
612
ПОБОЧНАЯ ПОДГРУППА VIII ГРУППЫ
При действии PF3, РС13 или РВг3 на Ni(CO)4 образуются соот-
ветственно Ni(PF3)4, (NiPCl3)4 или Ni(PBr3)4.
Тетракарбонил никеля служит для никелирования поверх-
ностей стекла и для приготовления коллоидных растворов нике-
ля путем нагревания Ni(CO)4 в толуоле.
Соединения включения
Гидрид никеля, NiH2, получают действием водорода на соеди-
нение Ni(C6H5)2 или на коричневый коллоидный раствор, полу-
ченный при растворении безводного NiCl2 в эфирном растворе
фенилмагнийбромида:
Ni(CeHs)2 + 2Н2 = NiH2 + 2CeHe
NiCl2 + 2CeH5MgBr + 2Н2 = NiH2 + 2СеНе + MgBr2 + MgCl2
Соединение NiH2 представляет собой черный кристаллический
порошок, устойчивый в эфирном растворе, разлагающийся в спирте
или воде с высвобождением водорода. Он служит катализатором
реакций гидрирования.
Нитриды никеля, Ni3N, Ni4N, Ni3N2, получают на-
греванием тонко измельченного металлического никеля в токе
аммиака при 500° или разложением амида Ni(NH2)2. Это пара-
магнитные черные гексагональные мелкие кристаллы, которые
устойчивы на воздухе, в воде или щелочах при обычной темпе-
ратуре, разлагаются при температуре выше 585° или в воде при
нагревании и взаимодействуют с концентрированными кислотами
при нагревании.
Нитрид никеля, Ni4N, получают обработкой аммиака тонко-
дисперсного металлического никеля, нагретого до 170°, он пред-
ставляет собой ферромагнитные кубические кристаллы, которые
превращаются в Ni3N при нагревании до 190° в вакууме.
Нитрид никеля, Ni3N2, получают при нагревании Ni(NH2)2
(120°) или нагреванием смеси Ni(CN)2 с NiO в атмосфере азота
при температуре электрической дуги. Соединение Ni3N2 пред-
ставляет собой черный кристаллический порошок, который выде-
ляет аммиак при нагревании с расплавленными щелочами, раст-
воряется в кислотах, превращается в NiCI2 при нагревании в атмо-
сфере хлора и в NiO — при нагревании с кислородом.
Известны также двойные нитриды, Ni2Ta4N5,
Ni0,88Ta4N5, Nio,3Ti8,7N и Nio,2Mo0t8 NOi9.
Фосфиды никеля, Ni3P, Ni5P2, Ni7P3, Ni3P2, Ni8P5,
Ni2P3, NiP2, NiP3, NiP, NiP4.
Фосфид никеля, Ni3P, образуется на катоде при электролизе
расплавленной смеси фосфата щелочного металла с NiO; это серые
тетраэдрические кристаллы с плотностью 7,89 г!см3. В некоторых
НИКЕЛЬ
613
метеоритах содержатся твердые растворы (Fe, Ni, Со)3Р в виде
игольчатых или трубчатых кристаллов.
Фосфид никеля, Ni5P2, можно получить сплавлением смеси
гидрофосфата кальция с NiO и с порошком угля; это блестящие
серые кристаллы с плотностью 7,283 г!см3.
Фосфид никеля, Ni2P, получают при сильном нагревании ме-
таллического никеля с РС13, РВг3 или Р13 или восстановлением
ортофосфата никеля водородом при 450° или метафосфата никеля
(600°). Соединение Ni2P представляет собой серые тетраэдриче-
ские кристаллы в плотностью 7,2 г/см3, плохо растворимые в конц.
HNO3 при нагревании и легко растворимые в разб. HNO3.
Фосфид никеля, Ni2P3, образуется при нагревании NiCl2 с па-
рами фосфора; он представляет собой блестящие серые кристаллы,
которые не реагируют с НС1, HNO3 и царской водкой.
Фосфид никеля, NiP2, получают нагреванием сплава Sn — Ni
с фосфором при 700° в запаянной трубке. Это серые тетрагональные
кристаллы с плотностью 4,62 г/см3, растворимые в HNO3 и не вза-
имодействующие с NaOH.
Фосфид никеля, NiP3, можно получить при нагревании сплава
Sn — Ni с большим количеством фосфора; он представляет собой
блестящие серые кристаллы с плотностью 4,19 г/см3, плохо раст-
воримые в конц. HG1 и растворимые в разб. HNO3.
Арсениды никеля, Ni5As2, Ni2As, Ni3As, NiAs, NiAs2,
NiAs3, получают нагреванием порошкообразного металлического
никеля с мышьяком при 925° в атмосфере водорода; они пред-
ставляют собой гексагональные кристаллы, образующие двойные
арсениды с Fe2As.
Арсенид никеля, Ni3As2, получают прямым взаимодействием
элементов (взятых в стехиометрически необходимых количествах)
при 925°, нагреванием до 500—850° арсенида NiAs и восстановле-
нием при нагревании NiO цианидом калия в присутствии эле-
ментарного мышьяка. Соединение Ni3As2 представляет собой
серовато-черный порошок с плотностью 7,86 г/см3, плавящийся
примерно около 1000°.
Арсенид никеля, NiAs, встречается в природе в виде минерала
никелина и представляет собой блестящие медно-красные гекса-
гональные кристаллы с плотностью 7,6—7,8 г/см3. Этот арсенид
можно получить действием паров AsCl3 на металлический никель
при 500—800° или пропусканием смеси паров As2O3 с водородом
над сильно нагретым NiCl2. Он имеет кристаллическую струк-
туру, изображенную на рис. 30. Каждый атом никеля октаэдри-
чески окружен шестью атомами мышьяка и двумя атомами никеля,
близко расположенными по вертикали.
Арсениды NiAs2 и NiAs3 также встречаются в природе в виде
минералов.
614
ПОБОЧНАЯ ПОДГРУППА VIII ГРУППЫ
рала ореитгауптита; его можно
Рис. 30. Структура (соединения NiAs.
Антимониды никеля, NiSb, Ni5Sb2, Ni3Sb, Ni4Sb,
Ni13Sb4, Ni2Sb, Ni7Sb3, Ni9Sb4, NiSb2, Ni5SbH, Ni2Sb5, Ni4Sb5,
Ni2Sb3, MgNiSb, NiCoSb.
Антимонид никеля, NiSb, встречается в природе в виде мине-
>лучить пропусканием паров
сурьмы над нагретым нике-
лем или действием паров
SbCl3 на нагретый до 600—
800° порошкообразный ни-
кель. Он представляет собой
фиолетовые гексагональные
кристаллы с плотностью 8,69
г!см9 и т. пл. 1150°; NiSb
взаимодействует при нагрева-
нии с кислородом, хлором,
HNO3 и на холоду с конц.
HNO3.
Карбиды никедя.
Ni3C, (Ni, Fe)3C, MgNiC,
Ni3Mo3C, Ni2Mo4C, Ni3W3C, (V, Ni)3Nb3C, (V, Ni)3Ta3C.
Карбид никеля, Ni3C, известен в двух формах — одну полу-
чают при растворении углерода в расплавленном никеле (при
высокой температуре), другая образуется при действии СО, СН4,
С2Н2, СвН6, С6Н14 на металлический никель при умеренной тем-
пературе.
Карбид никеля, Ni3C, представляет собой серый порошок с плот-
ностью 7,97 г!см?, разлагающийся при нагревании в токе водорода,
азота или окиси углерода.
К соединениям включения относятся также силициды Ni3Si,
Ni5Si2, Ni2Si, Ni3Si2, NiSi, NiSi2, германиды Ni3Ge, NiGe, стан-
ниды Ni4Sn, Ni3Sn, Ni3Sn2, бориды Ni2B, Ni3B2, NiB, Ni2B3,
NiMo2B2.
МЕТАЛЛЫ ПОДГРУППЫ ПЛАТИНЫ
ХАРАКТЕРИСТИКА ПОДГРУППЫ
В подгруппу платины входят переходные металлы рутений (Ru),
родий (Rh), палладий (Pd), осмий (Os), иридий (1г) и платина (Pt).
В табл. 60 схематически показана электронная структура ато-
мов рутения, родия, палладия, осмия, иридия и платины.
Таблица 60
К L M 0 p
Эле- мент z 18 2s 2p 3s 3p 3d 4s 4p 4d 4/ 5s 5p 5d 6s 6p 6d
Ru 44 2 2 6 2 6 10 2 6 7 — 1 4dW
Rh 45 2 2 6 2 6 10 2 6 8 — 1 4<№5si
Pd 46 2 2 6 2 6 10 2 6 10 — 0 4d105s°
Os 76 2 2 6 2 6 10 2 6 10 14 2 6 6 2
Ir 77 2 2 6 2 6 10 2 6 10 14 2 6 7 2 5d'6s2
Pt 78 2 2 6 2 6 10 2 6 10 14 2 6 9 1 5d96si
Сумма электронов незаполненных d- и s-орбиталей следую-
щего электронного слоя равна восьми у Ru и Os, девяти у Rh
и Ir и десяти у Pd и Pt,
По числу электронов на 4d- и Ss-орбиталях у Ru, Rh, Pd и 5d-
и бя-орбиталях у Os, Ir, Pt и по аналогии химических свойств пла-
тиновые металлы можно разделить на три группы: Ru — Os,
Rh - Ir и Pd - Pt.
В табл. 61 приведены наиболее важные константы металлов
подгруппы платины.
Таблица 61 2
Элемент Рутений Ru Родий Rh Палладий Pd Осмий Os Иридий 1г 05 Платина Pt
Цвет в компактном состоя- нии Серебри- сто-белый Серебри- сто-белый Серебри- сто-белый Серебри- сто-белый Серебри- сто-белый Серебри- сто-белый
в порошке Серый Серовато- черный Серовато- черный Черный Черный Черный
Кристаллическая структура Гексаго- нальная плотней- шая Кубическая гранецент- рированная Кубическая гранецент- рированная Гексаго- нальная плотнейшая Кубическая гранецент- рированная Кубическая гранецент- рированная
Атомный номер 44 45 46 76 77 78
Атомный вес 101,07 102,905 106,4 190,2 192,2 195,09
Атомный радиус, А (по Полингу) 1,34 1,34 1,37 1,35 1,36 1,39
Ионный радиус, А (по Гольдшмидту, Полингу и Аренсу) Ru4+ 0,65; 0,63; 0,67 Rh3+0,68; 0,69; 0,68 Pd2+—; -; 0,80 Pd4+--;— ; 0,65 Os4+ 0,67; 0,65 1г4+ 0,66; 0,64; 0,68 Pt2+ —; -; 0,80 РИ+—; -; 0,65
Атомный объем (при 20°), см3/г-атом 8,27 8,29 8,86 8,43 8,54 9,10
Плотность (при 20°), г /см3 12,30 12,42 11,97 22,70 22,65 21,45
Твердость по Бринеллю, кг/мм3 220 139 49 — 172 64
Твердость по шкале Мооса 6,4 6 4,8 7 6,25 4,3
Температура плавления, °C 2450 1966 1552 2727 2454 1769
Температура кипения, °C 3727 3729 3127 4230 4130 3827 .
Удельная теплоемкость (при 20°), 0,059 0,058 0,056 0,031 nnrfl17 0,031 '\ 0,032
Коэффициент теплопроводности %, кал-см~1-секХ~-град-1 (при 0°) — 0,21 0,17 — 0,14 0,17
Удельное сопротивление р*106 (при 0°), ом* см 7,64 4,70 10,88 9,5 6,10 10,96
Электропроводность (Hg = — 1) 12,3 20,0 8,6 9,9 15,4 8,5
Магнитная восприимчивость Ха-10~в, эл. магн. ед. (при 18°) 0,50 1,11 5,4 0,05 1,04 1,10
Теплота перехода атомов в газообраз- юе состояние, ккал (при 25°) 160 138 93 174 165 121,6
1отенциал иони- зации, эв Ме —► Ме+ + е- Ме+ —* Ме2+ + е~ Ме2+—► Ме3++е_ Ме3+ —► Ме<++ е~ Ме4+ —► Ме5+ + е_ Ме5+ —► Ме6+ 4- е_ 7,36 16,76 28,46 46,52 62,9 80,6 7,46 18,07 31,05 45,63 66,7 85,2 8,33 19,42 32,92 48,77 65,6 89,9 8,7 15 25 40 54 68 -9,2 16 27 39 57 72 9,0 18,56 28,55 41,13 54,8 75,3
Потенциал иониза- ции, ккал/г-атом Ме —► Ме+ + е- 173 177 187 -200 -212 -205
Нормальный по- Ме/Ме2+ тенциал, в (при 20°) Ме/Ме3+ +0,45 +0,6 +0,7 +0,83 +0,7 +1,0 +1_,2
Нормальный потенциал окислительно- восстановительных систем, в — Rh4+/Rh3+ = = 1,5 Rh4+/Rh3+== = 1,43 — — — —
Залептность (П), (III), IV, (V), VI,(VII), VIII (I),(II), III, (IV), (VI) (I), II, (III), IV (II), (III), IV, (V), VI, (VIII) (I), (II), III, IV, (V), (VI) (I), II, (III), IV, (VI)
Массовые числа природных изотопов 102,101,104, 100,99, 96, 98 103 106, 108,105, НО, 104,102 192, 190, 189, 188,187,186, 184 193, 191 195, 194, 196, 192
Распространенность элементов в земной коре, вес. % 5,0-10-е 1,0-10-в 5,0-10-e | 5,0-10-е j 1,0-10-8 5,0-10-5
618
ПОБОЧНАЯ ПОДГРУППА VIII ГРУППЫ
В компактном состоянии рутений и осмий представляют собой
блестящие серебристо-белые металлы с плотнейшей гексагональ-
ной кристаллической решеткой. Они относятся к тяжелым метал-
лам, тверды, хрупки и тугоплавки. При действии кислорода (воз-
духа или паров воды) на металлические рутений и осмий обра-
зуются летучие четырехокиси RuO4, OsO4.
Под действием окислительно-щелочных расплавов металличе-
ские рутений и осмий превращаются в рутенаты МезВиО4 и осма-
ты Me*OsO4.
Некоторые соединения рутения и осмия, отвечающие степеням
окисления шесть и семь, проявляют свойства, напоминающие
свойства соответствующих соединений марганца.
Известны многочисленные галогенидные и псевдогалогенидные
координационные соединения, соответствующие различным сте-
пеням окисления рутения и осмия.
Металлические родий и иридий в компактном состоянии также
являются серебристо-белыми металлами с гранецентрированной
кубической структурой. Они относятся к тяжелым, относительно
твердым, хрупким и тугоплавким металлам, устойчивы к дей-
ствию кислот и царской водки. Характерной особенностью этих
элементов является образование окислов, соответствующих степени
окисления, равной трем, а не восьми, как у рутения и осмия.
Многочисленные координационные соединения трехвалентных
родия и иридия проявляют большое сходство с комплексами трех-
валентных хрома и кобальта.
Палладий и платина в компактном состоянии являются сере-
бристо-белыми металлами с гранецентрированной кубической
структурой кристаллической решетки. Они относятся к тяжелым
тугоплавким металлам. Они мягкие, ковкие и легко прокатываются.
Из платиновых металлов осмий имеет самую высокую, а пал-
ладий наиболее низкую температуры плавления.
Для металлического палладия характерно свойство поглощать
большое количество водорода.
В общем случае платиновые металлы обнаруживают большое
сходство в свойствах. Все они относятся к тяжелым тугоплав-
ким металлам, обладают низкой химической реакционной способ-
ностью, проявляют высокую устойчивость к действию многих
химических реагентов, характеризуются переменной валентно-
стью и способностью к образованию многочисленных координа-
ционных соединений, соединения их, как правило, окрашены.
Способность к образованию координационных соединений вы-
ражена очень сильно, поскольку они слабо электроположительны
и имеют неполностью заполненные d-орбитали. Платиновые ме-
таллы являются типичными комплексообразователями, образуя
координационные соединения, отвечающие различным степеням
окисления и координационным числам.
са
Я
Й
й
сб
в
"§
3
Я
О
в
=в
о
Й
са
Я
« Я
Я
Я
2
S
Ф >с
S’ § ©н
и^;
s
О S’ Я
*?\о ®
со
я
сб
2
йЙ
G
я
Я
Я
3
й*
Я
ч
ч
сб
2
Сб
a
’Я
я
й
я 2.
сб
ч
X
о _Д 2 Ч[Ч
.eS
W ф
§ рач
ф о Я
§«
S «п
CS
й
5 о ©,
2 й о
ч
«б
я
S
a
s
«
S
S
=s
д ”
Сб
В
3
a
2- «
-Р S
J? s
Я
Ч
§
S
а
s
к
сб Д Сб
Ч £< Я
О-
Я
Я
й
К
К
St P?N 5Й Ю
о S5.
X я S3
ч
2
§
S
3
д2
« g
.7 S
й ч
О. сб
«: и
сб
о
К Й
2 О нч
Ч со К
S'S.u S
3 л^я
О W О' в
^SSS
* Рч Рч
:Я
й =s
S
н
S
й
о
Й
й
9
2
И
3
a
ё
в
Й
СР
яо Е
И
X
н хб
я °
Я
в «о
О СП
Я
Еч
Сб
Й
я
о so
14 зИ
2 « 2
*>£•*
Я со ф
« W
Я 2 S
л S
Я Й
SH о
>Я
с
2
я
3
ф
Я
я a
Я
СР я
й оСР
W 5Й
R §
£3
Я Я
>Я И-’Й
S 80
о в Д
мН иЧ
Я сб .
О Сч’Н ео
S'gBQ.
сб
§ °
оЩ
к
-О о
s S ^"хо
сл ф Л
g-ffi 3 S
sS&§
И-В н н
й СС О сб
ш й й
л А
* S «
K=su
z s з g
2 я
о Q
Ч о
Ч й2
й
§ S §
§ §к
>я
3
я
3
S н:
- о
=S л
3
ьзЯ
Сб s
S- ®
Сб в
£< са
§ д
К =Н и
3 И
р-1
Й Р й
м 2 S
Йрр Я
"“сз й
tc S- S
« g
Й и о
Я
•—14F—( ZJ
«и g
к
2
Й
Ft
о
Й
Я
я
Я
й
я
§
5S
я
й
0
ч
a
Я
Я
я
ф
5
О
я
сб
§Я
с2
3
сб :й
Д Я
VO g
?5
0^8
дг: a
й
В
Я
ч
ч
сб
a
й
сб
:S
И
Я
2
2
о
И
Я й
и р_
Я
2 3
S-
5 S
®JS
сб В
«
PC
ca й «:
620
ПОБОЧНАЯ ПОДГРУППА VIII ГРУППЫ
Платиновые металлы взаимодействуют с фтором, хлором, кис-
лородом, серой и фосфором только при высоких температурах.
Они образуют сплавы с очень многими металлами.
Характеризуя образование окислов и сульфидов платиновых
металлов, следует отметить, что их сродство к кислороду умень-
шается, а к сере увеличивается в ряду Ru, Rh, Pd и Os. Ir. Pt.
Эти элементы проявляют сходство с металлами побочных
подгрупп I и II групп периодической системы, так как они обра-
зуют неустойчивые окислы и устойчивые сульфиды.
Платиновые металлы обнаруживают важные каталитические
свойства и являются катализаторами гидрирования и окисления.
По внешнему виду (цвету), высоким температурам плавления
и кипения, склонности к образованию координационных соедине-
ний платиновые металлы сильно отличаются от металлов подгруп-
пы железа.
В природе платиновые металлы встречаются в самородном
состоянии и в виде соединений в различных минералах. К эле-
ментам, сопровождающим платиновые металлы в природе, отно-
сятся Fe, Со, Ni, Си, Ag, Au, Zn, Sn, Pd, Мп, Мо, Re, S. As. Sb,
Те.
Для извлечения платиновых металлов применяются различ-
ные методы, зависящие от исходного сырья.
Платиновые металлы извлекают из самородной платины, из
различных минералов этих элементов и из анодного шлама после
очистки золота, серебра, меди, никеля и др.
Обработка платиновых руд осуществляется либо царской вод-
кой (см. схему на стр. 619), либо сплавлением с окислительно-
щелочными смесями.
Разделение платиновых металлов сопряжено с некоторыми
трудностями, обусловленными сходством свойств этих элементов.
РУТЕНИЙ Ни
Z = 44; ат. вес = 101,97
Валентность (II), (III), IV, (V), VI, (VII), VIII
Массовые числа природных изотопов 102, 101,
104, 100, 99, 96 и 98
Электронная структура атома рутения: А? •A/-4s24p64d7-os1.
Электронная структура атома рутения и катионов Ru2+, Ru3+,
Ru4+ для 4d- и 5$-орбиталей:
5s’ 4d6 5s 4d5 5s 4d« 5s
0 Ш310 0 0ШЕШ □ i n
Ru Ru2+ Ru3+ Ru4 +
РУТЕНИЙ
621
ИСТОРИЧЕСКАЯ СПРАВКА
Рутений был открыт в 1844 г. русским химиком К. Клаусом,
выделившим его из остатков от переработки платиновых руд.
Название происходит от слова Ruthenia, что по латыни означает
Россия.
РАСПРОСТРАНЕННОСТЬ В ПРИРОДЕ
В природе рутений встречается как в самородном виде вместе
с осмием, иридием и родием, так и в виде сульфида, называемого
лауритом, BuS2, который содержит сульфиды и других платиновых
металлов. Среди платиновых элементов рутений наименее рас-
пространен в природе — его содержание в земной коре равно
.5-10 е вес.%.
ИЗВЛЕЧЕНИЕ И ПОЛУЧЕНИЕ МЕТАЛЛИЧЕСКОГО
РУТЕНИЯ
Рутений извлекают из растворов, которые образуются при
обработке платиновых руд (или концентратов платиновых руд)
царской водкой. Извлечение рутения из руд, в которых он нахо-
дится вместе с платиной, иридием и осмием, основано на лету-
чести четырехокиси рутения RuO4. Последняя образуется при
прокаливании хлоридов платиновых металлов, а эти в свою оче-
редь — при сплавлении трудно растворимых остатков после
переработки платиновых руд с NaOH и NaCl в токе газообраз-
ного хлора. Для отделения RuO4 от OsO4 смесь обоих окислов
сплавляют с щелочами и затем обрабатывают царской водкой
с целью получения одного OsO4, который отделяется перегонкой.
Металлический рутений может быть получен восстановле-
нием водородом при нагревании соединений RuO4, Ru(OH)3,
(NH4)3[RuC161.
ФИЗИЧЕСКИЕ И ХИМИЧЕСКИЕ СВОЙСТВА
В слитках рутений представляет собой блестящий серебристо-
белый металл с плотнейшей гексагональной кристаллической
структурой. В порошкообразном состоянии металл серого цвета.
Металлический рутений имеет плотность 12,30 г!см3 (тяжелый
металл), т. пл. 2450°, т. кип. 3727° С (тугоплавкий металл). Его
твердость равна 6,4 по шкале Мооса (твердый и хрупкий металл,
с трудом поддающийся переработке давлением). Известны четы-
ре аллотропные модификации металлического рутения, а именно:
a-Ru — устойчив от 0 до 1035°, 0-Ru — от 1035 до 1190°, y-Ru —
от 1190 до 1500° и 6-Ru — от 1500 до 2450°.
622
ПОБОЧНАЯ ПОДГРУППА VIII ГРУППЫ
При растворении сплавов Ru — Zn в НС1 образуется тонко-
дисперсный металлический рутений, называемый взрывчатым руте-
нием. Взрывчатость порошков платиновых металлов обусловлена
тем, что гремучая смесь (образующаяся при насыщении плати-
новых металлов водородом и кислородом) взрывается под ката-
литическим действием платиновых металлов. Порошкообразный
металлический рутений способен поглощать водород, причем
количество поглощенного водорода зависит от степей дисперсности
и температуры.
Известны сплавы рутения с Pt, Pd, Ir и Os.
С химической точки зрения металлический рутений достаточ-
но инертен, он не взаимодействует с многочисленными химическими
реагентами.
Металлический рутений, нагретый до ~600° в кислороде (или
на воздухе), покрывается с поверхности синевато-черной пленкой
RuO2, а при сжигании в кислороде (на воздухе) при ~1000° пре-
вращается в летучий RuO4.
При нагревании металлический рутений взаимодействует с фто-
ром, хлором, серой, фосфором и мышьяком, образуя RuF5. RuC12,
RuCl3, RuS2, RuP2, RuP, Ru2P, RuAs2.
При действии окиси углерода на металлический рутений
при 200° и давлении 200 ат образуется Ru(CO)5.
Поскольку нормальный потенциал системы Ru/Ru2+ равен
+0,45 в, металлический рутений устойчив к действию кислот и даже
царской водки в отсутствие кислорода, но образует рутенаты
MejRuO4 под действием расплавленных окислительно-щелоч-
ных смесей (КОН + KNO3, КОН + КСЮ3, К2СО3 + КС1О3,
КОН + KMnO4, NaOH + Na2O2).
ПРИМЕНЕНИЕ
Поскольку рутений — редкий металл, к тому же твердый,
хрупкий и с трудом поддается переработке давлением, его при-
меняют только в виде сплавов, в состав которых он входит в малых
количествах. Сплавы Ru — Pt и Ru — Pt — Pd используются
для изготовления украшений, кончиков «вечных» перьев, элек-
трических контактов и игл звукорегистрирующих аппаратов.
Для изготовления электрических контактов применяются также
сплавы Ru — Ir, Ru — Os.
СОЕДИНЕНИЯ (ОБЩИЕ СВОЙСТВА)
Получены соединения двух-, трех-, четырех-, пяти-, шести-,
семи- и восьмивалентного рутения. Больше всего известно соеди-
нений трехвалентного рутения, а наиболее устойчивы соединения
четырех-, шести- и семивалентного рутения.
РУТЕНИЙ
623
В табл. 62 приведены некоторые соединения рутения, сгруп-
пированные по валентности.
Таблица 62
Соединения двух- валентного руте- ния Ru(OH)2, RuC12, RuBr2, RuS2, RuSe2, RuTe^. K4[Ru(CN)e]-3H2O, KFe[Ru(CN)6]-3H2O, [Ru(C10H8N2)3]X2-6H2O, [Ru(NH3)5SO2]C12, [Ru(NH3)4SO2C1]C1 (где X=Cb, Вг~, I")
Соединения трех- валентного руте- ния Ru(OH)3, RuF3, RuCl3, RuBr3, Rul3, Ru2S3, Mei[RuX4]- • nH20, Mel[RuX5], Mei[Ru(H2O)X5], Mei[Ru(NO)X5], Mei[RuX6] (где Х = С1“, Вт", I-, 1/2С2О1~ иМе!=К+, Na+, NHt), (RuAm6]X3, [RuAm5X]X2, [RuAm4X2]X, [RuAm3X3], [RuAm4(H2O)2]X3 (где Am = NH3, г/2Ен и X = C1-, Br-, I-), [Ru(C5O2H7)3], [Ru(NH3)4(OH)C1]C1
Соединения четы- рехвалентного рутения RuO2, RuO2-2H2O илиВи(ОН)4,ВнС14, RuC14-5H2O, RuS2, Ru(SO4)2, Me^RuCl6], Me^RuCl5(OH)], Me^[Ru(C2O4)3], MeifClgRu—0 —RuClj) (где MeI = K+ или Na+)
Соединения пяти- валентного ру- тения RuF5 и Cs[RuFe]
Соединения шести- валентного ру- тения MeiRuO4-nH2O или MeiRuO4, Mei[RuO2Cl4] (где Mel = = K+, Rb+), RuF6
Соединения семи- валентного ру- тения МетВиО4 (где MeJ = K+ или Na+)
Соединения вось- мивалентного рутения RuO4
Кроме простых известны многочисленные координационные
соединения рутения, которые, как правило, устойчивы, а также
его металлоорганические производные.
Соединения двухвалентного рутения
Известно ограниченное число соединений двухвалентного руте-
ния, причем большинство их неустойчиво и окрашено в темные
цвета. Примеры соединений рутения(П); Ru(OH)2, RuC12, RuBr2,
624
ПОБОЧНАЯ ПОДГРУППА VIII ГРУППЫ
RuS2, RuSe2, RuTe2, K4[Ru(CN)e| -3H2O, KFe[Ru(CN6)l -3H2O,
[Ru(C10H8N2)3|X2-6H2O (где X = Cl-, Br-, I-). [Ru(NH3)6SO2]Cl2,
[Ru(NH3)4SO2C1|C1. Комплексы [Ru(C10H8N2)2|C12,[Ru(C]0H8N2)3]C12
получают действием а,а'-дипиридила на RuGl3. Они устой-
чивы к нагреванию и действию кислот и щелочей.
Гидроокись рутения, Ru(OH)2, образуется в виде коричнево-
го осадка при обработке щелочью синего раствора RuC12. Сое-
динение Ru(OH)2 мало устойчиво и легко окисляется, превращаясь
в Ru(OH)3 черного цвета.
Хлорид рутения, RuC12, получают действием хлора на порош-
кообразный металлический рутений, нагретый до 250° в трубке
из тугоплавкого материала. Он представляет собой коричневый
порошок, плохо растворимый в холодной воде, кислотах и щело-
чах и растворимый в спирте.
Гексацианорутенат(1Г) калия, KjRu(CN)6|-ЗН2О, образует
бесцветные кристаллы, плохо растворимые в воде.
Соединения трехвалентного рутения
Известно небольшое число простых и координационных соеди-
нений электроположительного трехвалентного рутения. В отли-
чие от координационных простые соединения рутения(Ш) мало
устойчивы.
Примеры соединений рутения(Ш): Ru(OH)3, RuF3, RuCl3,
RuBr3, Rul3, Ru2S3, ацидосоли типа Mex[RuX4] -nH2O, Me^[RuX5],
Me|[Ru(H2O)X5], Mei[Ru(NO)X5], Mei[RuXel (где Me1 - K+, Na+,
NH* и X = Cl-, Br-, I-, V2C2O24-), аммины, ацидоаммины, акво-
аммины (с их изомерами) типа [RuAme]X3, [RuAm5X]X2,
[RuAm4X2]X, [RuAm3X3], [RuAm4(H2O)2|X3 (где Am = NH3
или 1/2En и X = Cl-, Br-, I-), ацетилацетонат [Ru(C5H-O2)3l.
При нагревании гексамминов и пентамминов рутения(Ш) с раство-
рами щелочей получают рутениевый красный [Ru(NH3)4(OH)C1]C1,
который применяется в качестве редокс-индикатора *. Хлоросоли
рутения(Ш) окрашены в оранжевый или красный цвет, аце-
тилацетонат — в кроваво-красный.
Гидроокись рутения, Ru(OH)3, образуется в виде черного осад-
ка при обработке растворов солей рутения(Ш) щелочами или
при окислении гидроокиси рутения(П).
Хлорид рутения, RuCl3, получают действием хлора на нагре-
тую до 125° Ru(OH)3 или на нагретый до 450° порошкообразный
металлический рутений, а также при упаривании раствора RuO4
в НС1 в токе газообразного НС1:
Ru -|- 3/2С12 = RuCl3 -|- 62,84 ккал
* Индикатор окислительно-восстановительных реакций. —Прим. ред.
РУТЕНИЙ
625
Ковалентное соединение RuCl3 представляет собой блестящий
коричнево-черный кристаллический порошок с плотностью
3,11 г!см3\ оно плохо растворимо в воде и кислотах, гидролизуется
теплой водой, разлагается на элементы при нагревании до 627°,
восстанавливается при нагревании водородом или металлическим
цинком в кислой среде. При действии хлоридов щелочных метал-
лов на трихлорид рутения образуются хлоро- и хлороаквосоли
типа MeJ[RuCl5] и Me*[Ru(H2O)Cl5].
Бромид рутения, RuBr3, получают нагреванием смеси исход-
ных элементов или упариванием раствора, полученного обработ-
кой Ru(OH)3 бромистым водородом НВг.
Ru + 8/2Вг2 RuBrs + 35 ккал
Ковалентное соединение RuBr3 представляет собой черный по-
рошок, который разлагается на элементы при нагревании до 600°.
Иодид рутения, Rul3, образуется прямым взаимодействием
элементов при нагревании или обработкой RuCl3 иодидом калия
или Ru(OH)3 иодистым водородом.
Ковалентное соединение Rul3 представляет собой черный кри-
сталлический порошок, разлагающийся на элементы при нагре-
вании до 127°.
Соединения четырехвалентного рутения
Известно небольшое число устойчивых соединений четырехва-
лентного рутения. В качестве примеров можно привести RuO2,
RuO2-2H2O или Ru(OH)4, RuC14, RuC14-5H2O, RuS2, Ru(SO4)2,
ацидосоли типа Me*[RuCl6], Me|[RuCl5(OH)], Me^[Ru(C2O4)3l (где
Me1 = K+ или Na+) и полиядерные комплексы MeJ[Cl5Ru — О —
- RuC15].
Окись рутения, RuO2, получают нагреванием порошкообраз-
ного металлического рутения в кислороде при 600° или прока-
ливанием сульфида рутения(ГУ) в токе кислорода:
Ru + О2 = RuO2 + 52,6 ккал RuS2 + ЗО2 = RuO2 4- 2SO2
Окись рутения(1У) представляет собой синие тетраэдрические
кристаллы со структурой кристаллической решетки типа рутила;
(плотность 6,97 г!см\ т. пл. 955°, очень устойчива, разлагается
только при нагревании до 1127°, плохо растворима в воде и кис-
лотах, восстанавливается до металлического рутения водородом
или окисью углерода при нагревании.
Известно соединение RuO2*2H2O или Ru(OH)4 черного цвета,
плохо растворимое в воде и спирте, растворимое в кислотах, раз-
лагающееся при нагревании.
40—0191
626
ПОБОЧНАЯ ПОДГРУППА VIII ГРУППЫ
Хлорид рутения, RuC14, образуется при действии НС1 на четы-
рехокись рутения:
RuO4 + 6HCI = H2[RuO2C14] + Cl2 + 2Н2О
H2[RuO2C14] + 2НС1 = RuC14 + Cl2 + 2Н2О
RuO4 + 8НС1 = RuC14 + 2С12 + 4Н2О
Ковалентное соединение RuC14 коричневого цвета, разлагает-
ся при нагревании на элементы, превращается в Ru(OH)Cl3
под действием воды или в RuO2 при прокаливании в кислороде,
восстанавливается до металла при нагревании в токе водорода.
При действии хлоридов щелочных металлов на RuC14 обра-
зуются гексахлорорутенаты(1У) Me*[RuCl6] и декахлоро-р-оксо-
дирутенаты(1У) Me*[Cl5Ru — О — RuC15],
Пентагидрат RuC14 -5Н2О представляет собой гигроскопич-
ные коричневые ромбоэдрические кристаллы, легко растворимые
в воде и спирте.
Сульфид рутения, RuS2, встречается в природе в виде мине-
рала лаурита и может быть получен при нагревании смеси порош-
кообразного металлического рения с серой в кварцевой трубке
без доступа воздуха или при действии сероводорода на растворы
солей рутения(1У):
Ru -J- 2S (ромбич.) = RuS2 + 47,0 ккал
Соединение RuS2 представляет собой серовато-черные кри-
сталлы с плотностью 6,99 г/см2', оно плохо растворимо в воде,
кислотах, царской водке, растворяется в расплавленных щелочах
и разлагается выше 1185° на элементы.
Соединения пятивалентного рутения
Пятивалентное состояние не характерно для рутения, и как
следствие известно очень немного соединений рутения(У), напри-
мер RuF5 и Cs[RuF6].
Фторид рутения, RuF5, получают действием фтора на нагре-
тый до 300° в платиновой трубке порошок металлического рутения.
Ковалентное соединение RuF5 представляет собой прозрач-
ные темно-зеленые кристаллы с плотностью 2,963 г/см3, т. пл. 101°
и т. кип. 272°; RuF5 разъедает стекло, разлагается водой и вос-
станавливается иодом по уравнению
3RuF5 + I2 = 3RuFs + 2IF3
Соединения шестивалентного рутения
Соединения шестивалентного рутения, например рутенаты
типа Me*RuO4-nH2O или Me*RuO4,—наиболее устойчивые сое-
динения рутения.
РУТЕНИЙ
627
Известны также такие соединения рутения(У1), как хлорору-
тенаты(У1) Mei[RuO2Cl4] (где Me1 = Rb+ или К+)и гексафторид
рутения RuF6.
Рутенат калия, K2RuO4-H2O, осаждается при концентриро-
вании оранжевого водного раствора, полученного растворением
в воде холодного расплава, образовавшегося при действии
расплавленной окислительно-щелочной смеси (КОН + KNO3,
КОН КС1О3, К2СО3 + KNO3) на порошок металлического
рутения или окись рутения(1У):
Ru + 2К0Н + 3KNO3 = K2RuO4 + 3KNO2 + Н2О
Ru + 2К0Н + КСЮ3 = K2RuO4 + KC1 + H2O
RuO2 + 2K0H + KNO3 = K2RuO4 + KNO2 + H2O
Соединение K2RuO4>H2O представляет собой темно-зеленые
тетраэдрические кристаллы; они растворимы в воде, дегидрати-
руются при нагревании до 200°, разлагаются в вакууме выше 400°.
Это соединение восстанавливается водородом до RuO2 или метал-
лического рутения и превращается в KRuO4 под действием раз-
бавленных кислот.
Соединения семивалентного рутения
В качестве примеров соединений семивалентного рутения можно
назвать перрутенаты MexRuO4 (где Ме1 = К+ или Na+), которые
проявляют некоторое сходство с перманганатами и перренатами.
Перрутенат калия, KRuO4, выделяется при упаривании на хо-
лоду окрашенного в зеленый цвет раствора, полученного окисле-
нием концентрированного раствора K2RuO4*H2O газообразным
хлором или жидким бромом.
K2RuO4 + V2C12 = KRuO4 + КС1
При длительном пропускании газообразного хлора через рас-
твор рутената калия образуется четырехокись рутения RuO*
в виде золотистых игл.
Соединение KRuO4 представляет собой черные тетрагональные
кристаллы, выше 200° оно разлагается с выделением кислорода
(аналогично перманганатам), под действием щелочей превращается
в рутенаты:
2KRuO4 + 2КОН = 2K2RuO* + Н2О + У2О2
Соединения восьмивалентного рутения
Четырехокись рутения, RuO4, получают прокаливанием ме-
таллического рутения в кислороде при температуре выше 1000°,
действием гипохлоритов щелочных металлов на порошкообраз-
ный металлический рутений при комнатной температуре, обра-1-
40*
628
ПОБОЧНАЯ ПОДГРУППА VIII ГРУППЫ
боткой хлором растворов рутенатов щелочных металлов или раст-
воров солей|рутения (соответствующих различным степеням окис-
ления), обработанных избытком едкого кали:
Ru + 2О2 = RuO4 + 52 ккал Ru + 4КОС1 = RuO4 + 2КС1
K2RuO4 Cl2 => RuO4 2КС1
RuC14 + 2C12 -J- 8K0H = RuO4 + 8KC1 + 4H2O
Соединение RuO4 представляет собой золотисто-желтые (или
оранжевые) ромбоэдрические кристаллы с плотностью 3,29 г/см.
и т. пл. 25,5° (27° в случае оранжевой модификации); разлага-
ется со взрывом на RuO2 и кислород при нагревании ниже тем-
пературы кипения (т. кип. 108°) и превращается в рутенаты под
действием щелочей.
RuO4 + 2КОН = K2RuO4 + V2O2 + Н2О
Пары четырехокиси рутения имеют сильный запах озона, они
токсичны. Ковалентное соединение RuO4 — сильный окислитель,
попадая на кожу, вызывает почернение, со спиртом взаимодей-
ствует со взрывом, окисляет конц. НС1 и растворяется в воде
и четыреххлористом углероде.
RuO4 + 8НС1 = RuC14 + 2С12 + 4Н2О
Металлоорганические соединения
Известны карбонильные соединения рутения Ru(CO)5, Ru2(CO)9
Ru3(CO)12, галогениды карбонилрутения Ru(CO)2Br2, Ru(CO)2I2,
Ru(CO)Br и галогениды дициклопентадиенилрутения [(C5H5)2Ru]X
(где X = Cl", Br", I-).
Пентакарбонил рутения, Ru(CO)5, получают действием окиси
углерода на порошкообразный металлический рутений при тем-
пературе 200° и давлении 200 ат или на трииодид рутения в при-
сутствии порошка металлического серебра (170°, давление 450 ат).
Соединение Ru(CO)5 представляет собой летучую бесцветную
жидкость, которая затвердевает при температуре —22°, превра-
щаясь в бесцветные кристаллы, растворимые в спирте, хлороформе
и бензоле. Ru(CO)5 превращается в Ru2(CO)9 под действием ультра-
фиолетовых лучей и в Ru2(CO)9 и Ru3(CO)]2 при нагревании до 50°.
Эннеакарбонил рутения, Ru2(CO)9, образуется при нагрева-
нии (50°) раствора Ru(CO)5 в бензоле или при облучении Ru(CO)5
ультрафиолетовыми лучами.
Соединение Ru2(CO)9 представляет собой оранжевые гекса-
гональные кристаллы, устойчивые на воздухе и на свету, раст-
воримые в воде и бензоле; при нагревании до 150° разлагаются,
под действием иода образуют Ru(CO)2I2, с окисью азота дают
Ru(NO)5.
ОСМИЙ
629
Додекакарбонил рутения, Ru3(CO)12, представляет собой зеле-
ные кристаллы.
Дииодид дикарбонилру тения, [Ru(CO)2I2]n, получают дей-
ствием углерода на трииодид рутения при нормальном давлении
или обработкой иодом карбонила Ru2(CO)9.
Ruls -f- 2С0 = Ru(CO)2I2 -|- 4/2I2
Диамагнитное соединение [Ru(CO)2I2]n коричневого цвета,
очень устойчиво, обладает низким давлением пара, взаимодей-
ствует с пиридином и цианидом калия, образуя Ru(CO)2Py2I2
и K2[Ru(CO)2(CN)2l21 соответственно.
Бромид карбонилрутения, Ru(CO)Br, может быть получен
действием окиси углерода на RuBr3 при температуре 183° и дав-
лении 350 ат. Это бесцветные кристаллы, которые медленно раз-
лагаются водой, восстанавливают аммиачный раствор нитрата
серебра и разлагаются выше 200°.
2Ru(CO)Br = Ru(CO)2Br2 + Ru
ОСМИЙ Os
Z = 76, ат. вес = 190,2
Валентность (II), (III), IV, (V), VI, (VIII)
Массовые числа природных изотопов 192, 190,
189, 188, 187, 186, 184
Электронная структура атома осмия: К -L-M-N-Б8-БряБ<Б •№.
Электронная структура атома осмия и катиона Os4+ для 5d- и 6s-
орбиталей:
5d«
н 1 1 1 1
6s2
0
Os
SdS
1 1 1 1
6s
□
Os«+ .
ИСТОРИЧЕСКАЯ СПРАВКА
Осмий был открыт в 1803 г. Теннантом в нерастворимой фрак-
ции, полученной при действии царской водки на сырую платину.
Название «осмий» происходит от греческого слова osme, что озна-
чает «пахучий», поскольку пары четырехокисй осмия ОзО4 обла-
дают очень неприятным запахом.
РАСПРОСТРАНЕННОСТЬ В ПРИРОДЕ
В природе осмий находится в смеси с иридием в минерале не-
вьянскит (осмиридий), который содержит 21,0—49,3% осмия
и представляет собой белые или светло-серые гексагональные пла-
стинчатые кристаллы с плотностью 17—22 г!см3 и твердостью
630
ПОБОЧНАЯ ПОДГРУППА VIII ГРУППЫ
6—7 по шкале Мооса. Вместе с невьянскитом часто встречается
минерал сысертскит, содержащий осмий, иридий (иногда руте-
ний) и представляющий собой светло-серые гексагональные кри-
сталлы с плотностью 17,8—22,5 г! см? и твердостью 6 по шкале
Мооса. Осмий часто встречается в минералах платины, иногда
в остатках от переработки золотоносных руд. Содержание осмия
в земной коре равно 5-10-е вес.%. Залежи, содержащие осмий,
встречаются в СССР, США, Тасмании, Австралии и Южной Африке.
ИЗВЛЕЧЕНИЕ И ПОЛУЧЕНИЕ МЕТАЛЛИЧЕСКОГО
ОСМИЯ
Осадок, полученный после обработки царской водкой сырой
платины, невьянскита или сысертскита, нагревают с царской вод-
кой в специальном дистилляционном аппарате или прокаливают
в трубке из тугоплавкого стекла в токе NO2 при 300° с целью
получения летучей четырехокиси осмия OsO4. Отгоняющаяся OsO4
улавливается раствором NaOH (или смесью растворов NaOH
с Na2S) в виде осмата натрия Na2OsO4 -пН2О (или смеси Na2OsO4 •
•пН2О с OsS4). При обработке раствора осмата натрия тиосуль-
фатом натрия и хлоридом аммония выпадает осадок хлорида тет-
рамминодиоксоосмия(У1) [OsO2(NH3)4]Cl2.
При прокаливании комплекса [OsO2(NH3)4]C12 в атмосфере
водорода образуется губчатый металлический осмий, который
для очистки промывают соляной и плавиковой кислотами, а затем
сильно нагревают в токе водорода.
Металлический осмий получают также восстановлением окис-
лов OsO, Os2O3, OsO2, OsO4 водородом при нагревании или гек-
сахлороосмата (IV) калия K2[OsCl6] цинком в кислой среде:
K2[OsCl6] + 2Zn = Os + 2ZnCl2 + 2KC1
ФИЗИЧЕСКИЕ И ХИМИЧЕСКИЕ СВОЙСТВА
В слитках осмий представляет собой блестящий серебристо-
белый металл с плотнейшей гексагональной решеткой. В порошко-
образном состоянии металл черного цвета.
Металлический осмий имеет плотность 22,70 г!см? (самый тяже-
лый среди платиновых металлов), т. пл. 2727°, т. кип. 4230° (на-
иболее тугоплавкий среди платиновых металлов) и твердость 7
по шкале Мооса (твердый и хрупкий металл, с трудом поддающий-
ся переработке давлением).
Известны сплавы осмия с иридием, платиной, палладием и ру-
тением.
С химической точки зрения металлический осмий во многом
сходен с металлическим рутением; это инертный металл, обладаю-
осмий
631
щий высокой стойкостью к действию многих химических реа-
гентов.
При обычной температуре порошкообразный металлический
осмий медленно окисляется на воздухе до OsO4. Порошок метал-
лического осмия, нагретый до 212—500° на воздухе, быстро прев-
ращается в летучую четырехокись осмия. На стенках сосудов,
в которых хранится металлический осмий, оседает черный поро-
шок OsO2, который образуется в результате восстановления OsO4
под действием следов органических соединений.
Осмиевая чернь (порошок) поглощает водород.
При нагревании металлический осмий взаимодействует с гало-
генами, серой, фосфором и мышьяком, образуя OsF8, OsFe, OsF5,
OsF4, OsC14, OsC13, OsS2, OsP2 и др.
Расплавленные окислительно-щелочные смеси (КОН 4-KNO3,
КОН + КС1О3, КОН + К2О2, К2СО3 + KNO3) растворяют ме-
таллический осмий с образованием осматов Me|OsO4 (где Me1 = К +
или Na+). При подкислении водных растворов осматов получа-
ются четырехокись и двуокись осмия.
Нормальный потенциал системы Os/Os2+ равен -[-0,85 в, поэто-
му металлический осмий не взаимодействует с кислотами и даже
царской водкой в отсутствие кислорода.
ПРИМЕНЕНИЕ
Благодаря большой твердости и высокой температуре плав-
ления и кипения металлический осмий используется в качестве
легирующего элемента для платины, палладия, иридия и иногда
рутения.
Сплавы Os — Ir применяются при изготовлении перьев для
авторучек, иголок для циркулей, электрических контактов и др.
Сплавы Os — Pt, как и порошок металлического осмия, служат
катализаторами при синтезе аммиака, окислении углеводородов,
гидрировании ацетона и в других реакциях.
Раньше из металлического осмия изготовлялись нити нака-
ливания для электрических ламп.
СОЕДИНЕНИЯ (ОБЩИЕ СВОЙСТВА)
Известны двух-, трех-, четырех-, пяти-, шести- и восьмивалент-
ные соединения осмия. Наиболее многочисленны и устойчивы сое-
динения четырех- и шестивалентного осмия.
В табл. 63 приведены формулы некоторых соединений осмия,
сгруппированных по валентности.
Наряду с простыми известны многочисленные комплексные
соединения осмия, а также его металлоорганические производные.
632
ПОБОЧНАЯ ПОДГРУППА VIII ГРУППЫ
Таблица 63
Соединения ос- мия(П) OsO, OsCl2, OsBr2, Osl2, OsS2, OsSe2, OsTe2, OsSO,, K4[Os(CN)e].3H2O
Соединения ос- мия(Ш) Os2O3, (OsCl3)n, OsCl3-3H2O, Os(NO3)3, K3[OsCl6], K2[Os[NO2)5], K2[Os(NO)C15]
Соединения ос- мия(1 V) OsO2-2H2O, OsO2, OsF4, 0sC14, OsBr4, Osl4 K2[OsXe] (где X = F~, Cl~, Br-) OsS2
Соединения ос- мия(У) OsF5, Na[OsFe]
Соединения ос- мия(У1) OsF6, MeiOsO4, MelOs04-raH20, MelfOsOaCU], Mei[OsO3Cl2], Mei[OsNCl4], Mei[OsNCl5], Mei[OsO2(C2O4)2], Mei[OsO2(NO2)4] (где Mei = K+ и Na+), [OsO2(NH3)4]C12
Соединения ос- мия(УШ) OsO4, 0sFg, 0sS4
Соединения двухвалентного осмия
Известно ограниченное число соединений двухвалентного ос-
мия, которые, как правило, неустойчивы и окрашены в темный
цвет. Примеры соединений осмия(П): OsO, OsCl2, OsBr2, Osl2,
OsS2, OsSe2, OsTe2, OsSO3, K4IOs(CN)6] -3H2O.
Окись осмия, OsO, получают нагреванием смеси порошкооб-
разного осмия, сульфита осмия(П) OsSO3 и карбоната натрия
в токе двуокиси углерода.
Окись OsO представляет собой серовато-черный порошок,
плохо растворимый в воде и кислотах.
Хлорид осмия, OsCl2, получают нагреванием хлорида осмия
OsCl3 при 500° и пониженном давлении:
20sCl3 “ 20sCl3 -|- С12
Соединение OsCl2 представляет собой расплывающийся на воз-
духе темно-коричневый порошок, плохо растворимый в воде
и щелочах, растворимый в спирте, эфире и азотной кислоте, обра-
зующий Os(CO)3Cl2 при нагревании с окисью углерода выше 220°.
Иодид осмия, Osl2, представляет собой зеленое твердое веще-
ство, которое образуется при обработке кислых растворов солей
ОСМИЙ
633
осмия(1¥) иодидом калия:
K2[OsCl6] 4- 4KI = Osl2 + 6КС1 + I2
Гексацианоосмат(1 Г) калия, K4[Os(CN)6] -ЗН2О, осаждается
в виде желтых моноклинных кристаллов при обработке цианистым
калием щелочного раствора четырехокиси осмия.
Соединения трехвалентного осмия
Трехвалентное состояние не характерно для осмия. Известно
небольшое число соединений осмия(Ш), большинство которых
окрашено в темные цвета. В качестве примеров соединений ос-
мия(Ш) можно назвать Os2O3, (OsCl3)n, OsCl3-3H2O, Os(NO3)3,
K3[OsCle], K2[Os(NO2)5], K2[Os(NO)C15] и др.
Окись осмия, Os2O3, получают восстановлением OsO4 порошко-
образным металлическим осмием при нагревании или нагрева-
нием солей осмия(Ш) с карбонатом натрия в токе двуокиси угле-
рода.
Окись Os2O3 представляет собой темно-коричневый порошок
(или медно-красные чешуйки), плохо растворимый в воде.
Полимер трихлорида осмия, (OsCl3)n, получается при быстром
охлаждении паров, выделяющихся при нагревании металлического
осмия в хлоре при 1050°.
Полимер (OsCl3)n представляет собой гигроскопичные корич-
невые кубические кристаллы, которые легко растворяются в воде
и спирте. Он разлагается на OsCl2 и С12 выше 500°, превращается
в OsO4 при продолжительном кипячении с HNO3.
Известен тригидрат ОзС13-ЗН2О — зеленые кристаллы, раст-
воримые в воде и спирте и разлагающиеся при сильном нагревании.
Соединения четырехвалентного осмия
Известно много устойчивых соединений четырехвалентного
осмия, например OsO2*2H2O, OsO2, OsF4, OsCl4, OsBr4, Osl4,
K2[OsX6] (где X = F-, Cl", Br“), OsS2.
Гидратированную окись осмия(/У), OsO2*2H2O, получают на-
греванием солей ocmhh(IV) с едким натром или карбонатом натрия
в токе двуокиси углерода или гидролизом гексахлороосматов
K2[OsCl6], (NH4)2[OsCle] при нагревании:
K2fOsCle] + 4NaOH = OsO2.2H2O + 4NaCl + 2K.C1
(NH4)2[OsCle) + 4H2O - OsO2-2H2O + 2NH4C1 + 4HC1
Соединение OsO2-2H2O представляет собой темно-синий поро-
шок; оно плохо растворимо в воде и кислотах, дегидратируется,
превращаясь в OsO4 и металлический осмий при нагревании
634
ПОБОЧНАЯ ПОДГРУППА VIII ГРУППЫ
выше 460°, восстанавливается до металла водородом при нагре-
вании.
Безводная окись осмия(1У), ОзО2, получается нагреванием тон-
кодисперсного металлического осмия в парах ОзО4 и представляет
собой коричнево-красные гексагональные кристаллы с плотностью
7,91 г/см3, плохо растворимые в воде и кислотах, разлагающиеся
выше 500°.
Фторид осмия, OsF4, получают пропусканием фтора над нагре-
тым до 280° металлическим осмием. При этом OsF4, как правило,
загрязнен OsF5, OsF6 и OsF8.
Фторид осмия представляет собой коричневый порошок, кото-
рый разлагается водой или при сильном нагревании.
Хлорид осмия, OsCl4, получают обработкой четырехокиси осмия
конц. НС1 или охлаждением коричнево-желтых паров, образую-
щихся при пропускании хлора над нагретым до 650—700° метал-
лическим осмием.
Хлорид осмия представляет собой коричнево-красные иголь-
чатые кристаллы, которые водой медленно переводятся в OsO2-
•2Н2О, разлагаются при нагревании до 323° и образуют с хлори-
дами щелочных металлов гексахлороосматы Ме*[ОзС16] (где Ме1 =
= К+, Na+, NH*).
Гексахлороосматы, Ме*[ОзС16], восстанавливаются цинком
в кислой среде до металлического осмия.
Бромид осмия, OsBr4 и иодид осмия, Оз14, получают нагре-
ванием на водяной бане четырехокиси осмия с концентрирован-
ным водным раствором НВг или HI.
Известны гексабромоосматы типа Me|[OsBr6] (где Ме1 = К+
или Na+).
Сульфид осмия, OsS2, получают прямым взаимодействием
элементов при нагревании, пропусканием сероводорода через
нейтральный раствор осмиевой кислоты или через раствор гекса-
хлороосмата(1У) калия, а также действием сероводорода на окси-
сульфид осмия при высокой температуре.
H2OsO4 + 3H2S = OsS2 + S + 4H2O
K2[OsCle] + 2H2S = OsS2 + 4HC1 + 2KC1
Сульфид осмия представляет собой черные кубические кри-
сталлы; он плохо растворим в воде и спирте, разлагается при силь-
ном нагревании, при нагревании на воздухе превращается в ОзО4,
в атмосфере водорода восстанавливается до металла.
Соединения пятивалентного осмия
Пятивалентное состояние не характерно для осмия. Известно
очень немного соединений пятивалентного осмия, например OsF5,
Na[OsF6],
осмий
635
Соединения шестивалентного осмия
Известны многочисленные соединения шестивалентного осмия.
Примеры соединений осмия (VI): OsFe (единственный гексагалоге-
нид), осматы Me*OsO4 или Me|OsO4-пН2О, хлорооксоосматы
Me*[OsO2Cl4], Me*[OsO3Cl2], хлоронитридоосматы Me*[OsNCl4],
Mei[OsNCl5], оксалатооксоосматы Me*[OsO2(C2O4)2], нитрооксо-
осматы K2[OsO2(NO2)4] (где Me1 = К + и редко Na+), хлорид тетрам-
минодиоксоосмия(У1) [OsO2(NH3)4]C12.
Наиболее устойчивы из соединений осмия(У1) осматы.
Гексафторид осмия, OsFe, получают вместе с OsF4 и OsF8 при
нагревании металлического осмия в среде фтора.
Фторид осмия представляет собой желтые кристаллы с т. пл.
34,5° и т. кип. 47,5°; OsF6 действует на стекло, гидролизуется
водой, превращаясь в OsO4, OsO2 и выделяя HF, взаимодейству-
ет со многими металлами и неметаллами. Пары фторида осмия
бесцветны и токсичны.
Осмат калия, K2OsO4>2H2O, получают восстановлением нитри-
том калия или спиртом четырехокиси осмия в растворе едкого кали:
OsO4 + 2К0Н + KNO2 + Н20 = K2OsO4.2H2O + KNO3
Соединение K2OsO4«2H2O представляет собой фиолетовые
октаэдрические кристаллы, которые выше 200° теряют кристалли-
зационную воду в инертном газе, превращаются в OsO4 при нагре-
вании на воздухе и разлагаются кислотами:
2K2OsO4 + 2H2SO4 = OsO4+ OsO2 + 2K2SO4 + 2H2O
K2OsO4 + 4HX ** K2[OsO2X4] + 2H2O
Соединения восъмивалентного осмия
Известно небольшое число соединений восьмивалентного осмия,
обладающих сильными окислительными свойствами.
Четырехокись осмия, OsO4, получают окислением тонкоизмель-
ченного порошка металлического осмия кислородом при комнат-
ной температуре, на воздухе или в парах воды при температуре
выше 212°, действием азотной кислоты на окислы осмия низших
степеней окисления.
Соединение OsO4 представляет собой диамагнитные светло-
желтые моноклинные кристаллы с плотностью 4,906 г!см?', т. пл. 42°
и т. кип. 131° (легко летуче, очищается перегонкой); обладает
резким запахом, растворимо в воде, спирте и эфире. Пары четырех-
окиси осмия очень токсичны и действуют на слизистые оболочки
глаз и органов дыхания.
Водный раствор четырехокиси осмия не обладает кислотными
свойствами; он служит для окрашивания биологических препара-
тов, изучаемых под микроскопом при идентификации жиров, кото-
636
ПОБОЧНАЯ ПОДГРУППА VIII ГРУППЫ
рые восстанавливают OsO4 до OsO2>2H2O — соединения синего
цвета.
Четырехокись осмия является энергичным окислителем (по
отношению к конц. НС1 и различным органическим веществам)
и может быть восстановлена до OsO2 или металлического осмия:
OsO4 + 8НС1 = OsCl4 + 2С12 + 4Н2О
При действии OsO4 на гидроокись калия образуется
K2[OsO4(OH)2].
Четырехокись осмия превращается в хлоро-, нитро- или окса-
латооксоосматы по уравнениям:
OsO4 + 2КС1 + 4НС1 = K2[OsO2Cl4] + Cl2 + 2Н2О
OsO4 + 2KNO2.+ 2NO = K2[OsO2(NO2)4]
OsO4 + 2К0Н + ЗН2С2О4 == K2[OsO2(C2O4)2] + 2СО2 + 4Н2О
Во многих реакциях четырехокись осмия служит катализа-
тором.
Октафторид осмия, OsF8, образуется при быстром охлаждении
паров, полученных пропусканием фтора над нагретым до 250°
металлическим осмием.
Соединение OsF8 представляет собой желтые кристаллы
с т. пл. 34,4° и т. кип. 47,3°; OsF8 разлагается при нагревании
выше 225°, обладает окислительными свойствами, при попадании
на кожу вызывает ожоги, взаимодействуют с водой по уравнению
OsF8 + 4Н2О OsO4 + 8HF
Тетрасулъфид осмия, OsS4, осаждается в виде темно-коричне-
вого осадка сероводородом из подкисленных растворов ОзО4,
а также может быть получен действием (NH4)2S или Na2S на ще-
лочные растворы четырехокиси осмия.
Металлоорганические соединения
Известны карбонилы осмия Os(CO)5, Os2(CO)9, Os3(CO)12,
галогениды карбонила осмия Os(CO)4X2, Os(CO)3X2 (где X = Cl-,
Br", I") и галогениды дициклопентадиенила (C5H5)20sX (где X =
= Cl", Вт-, I-).
Пентакарбонил осмия, Os(CO)5, получают действием окиси
углерода на Osl3 в присутствии металлической меди (темпера-
тура 120°, давление 200 ат) или действием окиси углерода на че-
тырехокись осмия при 100° и 50 ат.
Osl3 + 5СО -J- 3Cu = Os(CO)5 + 3CuI OsO4 + 9СО = Os(CO)6 + 4CO2
Пентакарбонил осмия представляет собой бесцветную, лету-
чую при комнатной температуре жидкость, которая затвердевает
при —15°.
РОДИЙ
637
Эннеакарбонил осмия, Os2(CO)8, получают обработкой Osl3
окисью углерода в присутствии меди; это желтые кристаллы,
которые плавятся при 224° и очищаются сублимацией.
Дихлорид трикарбонилосмия, Os(CO)3Cl2, получают действием
окиси углерода на OsCl3 при температуре 120° и давлении 200 ат.
Соединение Os(CO)3Cl2 представляет собой бесцветные кри-
сталлы, которые плавятся примерно при 270° и разлагаются выше
280°; дихлорид плохо растворим в воде и большинстве кислот, при
нагревании в кислороде превращается в OsO4.
РОДИЙ Rh
Z = 45; ат. вес — 102,905
Валентность (I), (II), III, (IV), (VI)
Массовое число единственного природного изо-
топа равно 103
Электронная структура
Электронная структура
и 55-орбиталей:
атома родия: K'L’M •4s2«4p6.4d8’5s1.
атома родия и катиона Rh3+ для 4d-
4d8
н 1 1
5s’ 4d6 5s ,
Rh Rh3+
ИСТОРИЧЕСКАЯ СПРАВКА
Родий был открыт (1803 г.) Волластоном, который, исследуя
самородную платину, выделил красный комплексный хлорид
и, восстанавливая последний водородом, получил новый металл;
его название происходит от греческого слова rhodon, что означает
«розовый», поскольку большинство солей родия окрашены в розо-
вый цвет.
РАСПРОСТРАНЕННОСТЬ В ПРИРОДЕ
В природе родий встречается в качестве примеси в самород-
ной платине (~1,3% Rh), в невьянските (~0,5—7,7%), в пла-
тиниридии, в лаурите RuS2, в самородном золоте некоторых место-
рождений, а также в ряде полиметаллических руд. Содержание
родия в земной коре равно 1 -10-6 вес.%. Залежи родия находятся
в Мексике, Бразилии, Южной Америке, СССР.
ИЗВЛЕЧЕНИЕ И ПОЛУЧЕНИЕ
МЕТАЛЛИЧЕСКОГО РОДИЯ
Родий может быть извлечен из самородной платины, руд или
концентратов платиновых руд посредством различных химических
процессов или с помощью ионообменных смол.
638
ПОБОЧНАЯ ПОДГРУППА VIII ГРУППЫ
а) При действии царской водки на самородную платину, пла-
тиновые руды или их концентраты в раствор переходят родий,
платина, палладий, железо и медь в виде простых хлоридов
и хлоросоединений. Для удаления элементов неплатиновой груп-
пы к раствору добавляют железный или цинковый порошок. Оса-
док родия, платины, палладия и меди после промывания горячей
водой, сушки и прокаливания обрабатывают разб. H2SO4 при
нагревании, чтобы перевести в раствор железо и медь в виде
FeSO4‘nH2O, CuSO4-nH2O. Добавляя НС1 и NH4C1 к раствору,
полученному растворением родия, платины и палладия в цар-
ской водке, переводят в осадок платину в виде (NH4)2[PtCl6],
а в раствор переходят анионы [RhCl6]3- и [PdCl4]2-. При обра-
ботке избытком разб. NH4OH в осадок выпадает [Rh(NH3)5Cl]Cl2,
а в растворе остается [Pd(NH3)4]Cl2.
б) Раствор, полученный действием царской водки на самород-
ную платину, платиновые руды или их концентраты, выпарива-
ют досуха, затем твердый остаток растворяют в воде и обрабаты-
вают смесью карбоната и нитрита натрия для осаждения катио-
нов Fe2+, Cu2+, РЬ2+ (в виде карбонатов) и с целью перевода в ра-
створ анионов [Rh(NO2)6]3-, [Pt(NO2)6]2-, [Pd(NO2)4]2“. После
обработки раствора газообразным хлором и NH4C1 осаждается
(NH4)3[Rh(NO2)6], а в растворе остаются платина и палладий
в виде комплексных анионов.
в) Смолой дауэкс-50 в Na-форме катионы неплатиновых ме-
таллов извлекаются из водных растворов, содержащих также
и комплексные анионы [RhCl6]3~, [PtCle]2-, [PtCl4]2-. С помощью
ионообменных смол можно извлечь и ионы амминохлоридов пла-
тиновых металлов.
Металлический родий получают в виде прошка восстановле-
нием его солей водородом или щавелевой кислотой в кислой среде
либо формальдегидом или гидразингидратом в щелочной среде.
Действуя водородом на [Rh(NH3)5Cl]Cl2, при нагревании полу-
чают губчатый металлический родий, который охлаждают в токе
СО2, чтобы предохранить от окисления.
ФИЗИЧЕСКИЕ И ХИМИЧЕСКИЕ СВОЙСТВА
В компактном состоянии родий представляет собой блестящий
серебристо-белый металл с гранецентрированной кубической кри-
сталлической решеткой. Порошкообразный родий серовато-чер-
ного цвета, вследствие чего его часто называют родиевой чернью-
Тонкодисперсный металлический родий обладает пирофорными
свойствами. Известен также коллоидный родий, который полу-
чают восстановлением гидразином трихлорида родия.
Металлический родий имеет плотность 12,42 г!см3 (тяжелый
металл), т. пл. 1966°, т. кип. 3729° (тугоплавкий металл), твер-
РОДИЙ
639
дость 6 по шкале Мооса, поддается переработке, его можно вытя-
гивать в проволоку и прокатывать при нагревании.
Расплавленный родий растворяет до 7% углерода, который
выделяется в виде микроскопических кристаллов графита при
охлаждении.
Известны сплавы родия с платиной, палладием, иридием и др.
Измельченный металлический родий поглощает водород.
С химической точки зрения металлический родий — неактив-
ный элемент. При обычной температуре на воздухе металлический
родий очень устойчив, а при нагревании примерно до 1000° или
под действием окислительно-щелочных расплавов (КОН 4- KNO3,
К2СО3 4- KNO3, К2СО3 4- КС1О3) родий образует окислы Rh2O3,
RhO2.
При нагревании металлический родий взаимодействует с фто-
ром, хлором, бромом, серой, фосфором, давая соответствующие
соединения: RhF3, RhF4, RhCl3, RhBr3, Rh2S3, Rh2S5, Rh2S4,
RhaS8, RhP3, RhP3, RhP2, Rh5P4, Rh2P.
Сплавлением металлического родия c NaCl в атмосфере хлора
можно получить Na3[RhCI6], а при сильном нагревании металли-
ческого родия с K2S2O7 или KHSO4 получается Rh2(SO4)3.
Металлический родий в виде порошка растворяется в царской
водке, в H2SO4 и НС1 в присутствии кислорода воздуха.
ПРИМЕНЕНИЕ
Металлический родий применяют для покрытия поверхновтей
рефлекторов, поскольку такая поверхность обладает большой отра-
жательной способностью. В радиотехнике применяют контакты,
изготовленные из металлического родия или сплавов родий —
платина. Металлический родий применяют в ювелирной промыш-
ленности, так как электролитически осажденный родий дает бле-
стящие покрытия.
Родиевая чернь широко применяется в качестве катализатора
(при гидрировании или дегидрировании многих соединений) или
черного пигмента для нанесения рисунков на фарфор.
Сплавы родий — платина применяются в качестве катализато-
ров, в промышленности синтетических волокон, в ювелирном деле,
для изготовления термопар и кончиков перьев в авторучках.
Из родий-иридиевых сплавов изготовляются термоэлементы.
СОЕДИНЕНИЯ (ОБЩИЕ СВОЙСТВА)
Известны многочисленные устойчивые соединения родия(Ш);
небольшое число соединений родия(П) и (IV) и несколько соеди-
нений родия(1) и (VI) мало устойчивы.
640
ПОБОЧНАЯ ПОДГРУППА VIII ГРУППЫ
Таблица 64
Соединения двух-
валентного ро-
дия
RhO, RhF2, RhCI2, RhBr2, Rhl2, RhS, [Rh(C5H5N)e]Cl2,
[Rh{As(C2H6)3}4]Cl2
Соединения трех- валентного ро- дия Rh2O3, Rh2O3.3H2O или Rh(OH)3, RhF3, RhCl3, RhCl3.4H2O, RhBr3, Rhl3, Rh(SCN)3, Rh(NO3)3, Rh(NO3)3.2H2O, Rh2(SO3)3-6H2O, Rh2S3, Rh2(SO4)3. •aH20, MelRh(SO4)2-12H2O, Rh(CH3COO)3-2i/2H20, Rh2(C2O4)3, Mei[RhX6] (i де X = C1“, NOj, CN“, ^SOJ-, 1/2SO§-, КгСгОГ), Mei[RhCl5], [RhAm6]X3, [Rh(Am)5(H2O)]X3, [RhAm5X]X2, [RhAm4X2]X (где Am = NH3 или 1/2En и Х = С1~, Br-, I~), [Rh(NH3)3Br3]
Соединения четы- рехвалентного родия RhO2, RhO2-nH2O, RhF4
В табл. 64 приведены формулы некоторых соединений двух-,
трех- и четерыхвалентного родия. Кроме простых и координа-
ционных соединений родия, сгруппированных по валентности,
указаны и его металлоорганические соединения.
Соединения двухвалентного родия
Известно немного соединений двухвалентного родия, напри-
мер RhO, RhF2, RhCl2, RhBr2, Rhl2, RhS, [Rh(C5H5N)6]Cl2,
[Rh{As(C2H5)3}4]Cl, хелатное соединение с диметилглиоксимом.
Окись родия, RhO, представляет собой черно-коричневое веще-
ство, плохо растворимое в воде и кислотах.
Хлорид родия, RhCl2, получают нагреванием хлорида родия(Ш)
при 950°; это прошок, который может быть окрашен в различные
цвета — от темно-коричневого до фиолетово-красного.
Сульфид родия, RhS, получают нагреванием металлического
родия до красного каления в парах серы; он представляет собой
темно-серые кристаллы, плохо растворимые в воде, кислотах
и царской водке.
Соединения трехвалентного родия
Известно большое число устойчивых соединений трехвалент-
ного родия. В качестве примеров можно назвать Rh2O3, Rh2O3*
•ЗН2О или Rh(0H)3, RhF3, RhCI3, RhCl3.4H2O, RhBr3, Rhl3,
Rh(SCN)3, Rh(NO3)3, Rh(NO3)3-2H2O, Rh2(SO3)3-6H2O, Rh2S3,
Rh2(SO4)3.ziH2O (где n = 4, 12, 15), MezRh(SO4)2-12H2O (где
РОДИЙ
641
Me1 = К+, Rb+, NH4+), Rh(CH3COO)3-21/2H2O, Rh2(C2O4)3, аци-
досоли типа Me*[RhX6] (где Me1 = K+, Na+, NH4 и X == Cl-,
NO-, CN-, WJ’, Mei[RhX5] (где X == СГ),
гексаммины, аквопентаммины, ацидопентаммины, диацидотетрам-
мины и триацидотриаммины типа [RhAm6]X3, [RhAm5(H2O)]X3,
[RhAm5X]X, [RhAm4X2|X (где Am = NH3, или 1/2En, a X = Cl-,
Br-, I-) и [Rh(NH3)3Br3], Многие координационные соединения
родия(Ш) обнаруживают сходство с аналогичными соединениями
хрома(Ш) и кобальта(Ш).
Окись родия, Rh2O3, получают нагреванием порошкообраз-
ного металлического родия, нитрата родия Rh(NO3)3 или хло-
рида родия RhCl3 на воздухе при 800°.
Окись родия представляет собой зеленые кристаллы со струк-
турой корунда; она плохо растворима в воде, кислотах и царской
водке, восстанавливается до металлического родия водородом
при нагревании, разлагается на элементы при нагревании до 1200°,
образуя промежуточные неустойчивые окислы RhO, Rh2O.
При обработке растворов солей родия(Ш) [Rh2(SO4)3-nH2O,
RhCl3, Na3[RhCI6] щелочами или карбонатами щелочных металлов
получается гидратированная окись родия, Rh2O3-3H2O, или
гидроокись родия, Rh(OH)3, в виде желтого студнеобразного осад-
ка; последний растворим в кислотах или избытке щелочи и прев-
ращается в Rh2O3 при обезвоживании:
Rh2(SO4)3 + 6NaOH = 2Rh(OH)3 + 3Na2SO4
Фторид родия, RhF3, можно получить действием фтора на на-
гретый до 500—600° металлический родий:
Rh + 3/2F2 = RhF3 + 143,6 ккал
Фторид родия представляет собой красные ромбические кри-
сталлы с плотностью 5,28 г/см\ т. пл. 1127°, т. кип- 1227°, плохо
растворимые в воде, кислотах и спирте.
Хлорид родия, RhCl3, получают действием газообразного хлора
на порошкообразный металлический родий, нагретый до 250—
300°, или дегидратацией RhCl3-4H2O выше 180°.
2Rh -j- ЗС12 2RhCl3 + 80,2 ккал
Хлорид родия представляет собой расплывающийся на воз-
духе красновато-коричневый порошок; он плохо растворим в воде
и кислотах, сублимируется при 800° и выше 948° разлагается на
элементы.
При упаривании солянокислого раствора Rh(OH)3 выпадают
темно-красные кристаллы RhCl3-4H2O; соединение растворимо
в воде и превращается в RhCl3 путем нагревания выше 180°.
Действие металлического цинка на водный раствор хлорида
родия приводит к образованию металлического родия.
41-0101
642
ПОБОЧНАЯ ПОДГРУППА VIII ГРУППЫ
При обработке раствора хлорида родия гидроокисью аммония
получается хлорид хлоропентаммина родия(Ш) [Rh(NH3)5Cl]Cl2
— желтый кристаллический осадок, растворимый в соляной
кислоте:
RhCl3 + 5NH4OH = [Rh(NH3)3Cl]Cl2 4- 5Н2О
Бромид родия, RhBr3, получают прямым взаимодействием
элементов (~250°):
2Rh + ЗВг2 2RhBr3 -|- 100 ккал
Бромид родия образуется в виде коричневого порошка, который
разлагается на элементы при нагревании до 527°. Комплексные
аммины [Rh(NH3)8]Br3, [Rh(NH3)5(H2O)]Br3, [Rh(NH3)5Br]Br2,
[Rh(NH3)3Br3] являются производными RhBr3.
Иодид родия, Rhl3, получают действием иодида калия на раст-
воры солей родия(1П) при кипячении:
Rh2(SO4)3 + 6KI = 2RhI3 + 3K2SO4
Иодид родия представляет собой черное вещество, плохо
растворимое в воде, разлагающееся при 327°; служит для при-
готовления некоторых комплексных амминов родия(Ш).
Сульфид родия, Rh2S3, получают действием H2S на RhCl3
(360°); это серовато-черный порошок, устойчивый до 500°; выше
этой температуры на воздухе или в кислороде он воспламеняется
и горит с образованием металлического родия.
При пропускании сероводорода через нагретый водный раст-
вор RhCl3-4H2O (~100°) выпадает Rh2S3-3H2S — соединение
серого цвета.
Известен также гидросулъфид родия, Rh(HS)3, черного цвета,
разлагающийся при нагревании.
Гексанитрородат{Ш)калия, K3[Rh(NO2)eh образуется в виде
светло-желтого, растворимого в НС1 осадка при обработке суль-
фата родия(Ш) нитритом калия:
Rh2(SO4)3 + 12KNO2 = 2K3[Rh(NO2)6] + 3K2SO4
Гексахлорородат^И) натрия, Na3[RhCl8], в виде красных три-
клинных кристаллов получают нагреванием металлического родия
и хлорида натрия в атмосфере газообразного хлора:
2Rh + 6NaCl + ЗС12 = 2Na3[RhCl6]
Соединения четырехвалентного родия
Четырехвалентное состояние не характерно для родия. Из не-
многих соединений можно назвать RhO2, RhO2-nH2O и RhF4.
Окись родия, RhO2, получают сплавлением металлического
родия с гидроокисью и нитратом калия (800—900°); это твердое
черное вещество.
ИРИДИЙ
643
Гидратированная окись родия, RhO2*nH2O, получается элект-
ролитическим окислением Rh(OH)3 в избытке щелочи или при
окислении растворов солей родия(Ш) хлором в щелочной среде.
Соединение RhO2-nH2O представляет собой оливково-зеленое
твердое вещество, плохо растворимое в воде, растворимое в кис-
лотах и щелочах, превращающееся в Bh2O3 при нагревании в атмо-
сфере инертного газа.
Фторид родия, BhF4, получают нагреванием металлического
родия до 50° в атмосфере фтора или пропусканием фтора над фто-
ридом или хлоридом родия(Ш) (нагретыми до 500°).
Фторид родия представляет собой коричнево-красное вещество,
которое очищается сублимацией при 550°.
Металлоорганические соединения
Известны карбонилы родия Bh2(CO)8, Bh4(CO)12, ВЬ4(СО)ц,
галогениды карбонилродия Bh2(CO)4X2 (где X = Cl-, Br-, I-),
галогениды дициклопентадиенилродия [(С5Н5)2ВЫХ (где X = С1~,
Вг-), а также соединение HBh(CO)4.
Октакарбонил родия, Bh2(CO)8, получают действием окиси
углерода на металлический родий при высоком давлении или про-
пусканием окиси углерода под давлением над иодидом родия(Ш)г
нагретым до 70—75°, в присутствии металлической меди.
Соединение Bh2(CO)8 представляет собой оранжевые кристаллы,,
которые плавятся с разложением (76°).
Додекакарбонил родия, Rh4(CO)12, получают действием окиси
углерода при низком давлении в присутствии металлической меди
на RhCl3, нагретый до ~100°. Это красные кристаллы, разлагаю-
щиеся при нагревании до 150°.
Тетракарбонилродиевая кислота, HBh(CO)4, получается дей-
ствием окиси углерода и водорода на металлический родий при
низком давлении.
Соединение HBh(CO)4 представляет собой желтую летучую
жидкость, которая затвердевает при —10° и образует белый осадок
с раствором HgCl2.
ИРИДИЙ 1г
1— 77; ат. вес = 192,2
Валентность (I), (II), III, IV, (V), (VI)
Массовые числа природных изотопов 193 и 191
Электронная структура атома иридия: К -L-М-N>5s25pe5cF •
• 6s2.
41*
644
ПОБОЧНАЯ ПОДГРУППА VIII ГРУППЫ
Электронная структура атома иридия и катиона 1г3+ для 5d-
и ба-орбиталей:
54’
11 t 1 1
546
1 1 1 1
1г3+
6s
ИСТОРИЧЕСКАЯ СПРАВКА
В 1803 г. Теннант отделил иридий от осмия из черного порош-
ка, который остается после растворения сырой платины в царской
водке. Название «иридий» происходит от греческого слова iridis,
что означает «радуга», поскольку растворы солей иридия много-
цветны.
РАСПРОСТРАНЕННОСТЬ В ПРИРОДЕ
В природе иридий встречается в самородном состоянии и вместе
с осмием, платиной, рутением и родием в минералах невьянските
и сысертските, упомянутых при описании распространенности
в природе осмия. В невьянските содержится 46,8—77,2 вес. %
иридия.
Иридий был найден в метеоритном железе, его присутствие
установлено в фотосфере Солнца.
Содержание иридия в земной коре 1-Ю-8 вес.%. Залежи ири-
дия находятся в СССР, США, Канаде и Тасмании.
ИЗВЛЕЧЕНИЕ И ПОЛУЧЕНИЕ
МЕТАЛЛИЧЕСКОГО ИРИДИЯ
Иридий извлекают из различных видов осмиридия из нераст-
воримой в царской водке фракции самородной платины, из анод-
ного шлама и из карбонильных остатков, образующихся в про-
цессе рафинирования’никеля.
Разновидности осмиридия или трудно растворимые в царской
водке фракции от обработки самородной платины сплавляются
с ВаО2. После охлаждения расплав обрабатывают водой, раство-
ряют в царской водке и многократным упариванием с соляной
кислотой осаждают двуокись кремния. Затем отфильтровывают
BaSO4 (образующийся при осаждении иона Ва2+ серной кисло-
той) и хлоридом аммония осаждают из раствора ацидосоль
(NH4)2[IrCl8J — соединение красновато-желтого цвета.
При термическом разложении (или при нагревании в атмо-
сфере водорода) соединения (NH4)2[IrCl8] образуется губчатый
металлический иридий.
ИРИДИЙ
645
Для отделения иридия от рутения сплавы иридий — рутений
сплавляют с окислительно-щелочной смесью КОН + KNO3
и после охлаждения обрабатывают водой для перевода в раствор
рутения в виде K2RuO4-nH2O и для осаждения иридия в виде
1т(ОН)3. Затем рутенат калия K2RuO4>H2O превращают
в (NH4)2[RuC16], а 1г(ОН)3 — в (NH4)2[FrCl8].
Для удаления неплатиновых металлов из водных растворов
гексахлоросолей платиновых металлов применяют смолу дауэкс-
50 в Na-форме.
ФИЗИЧЕСКИЕ И ХИМИЧЕСКИЕ СВОЙСТВА
В компактном состоянии иридий представляет собой блестя-
щий серебристо-белый металл с кубической гранецентрированной
структурой. В порошкообразном состоянии он черного цвета.
Металлический иридий имеет плотность^ 22,65 г!см3 (тяжелый
металл), т. пл. 2454°, т. кип. 4130° (тугоплавкий), твердость 6,25
по шкале Мооса. Иридий хрупок и с трудом перерабатывается дав-
лением (прокатывается и протягивается при 1200—1300°). Электро-
проводность иридия примерно в 16 раз больше, чем у ртути.
При действии соляной кислоты на сплавы 1г — Zn образуется
тонкодисперсный порошок иридия, который может взрываться
и теряет эту способность после продолжительного нагревания
при 100—200°.
В отличие от металлического иридия в компактном состоянии
порошкообразный иридий (соответственно иридиевая чернь) погло-
щает водород.
Расплавленный металлический иридий растворяет углерод,
который при охлаждении выделяется в виде кристалликов гра-
фита, сильно увеличивая хрупкость металла.
Известны сплавы иридия с платиной, осмием, рутением, ро-
дием, медью и цинком.
С химической точки зрения иридий — инертный металл; он
очень устойчив к действию многих химических реагентов.
На воздухе при обычной температуре металлический иридий
не изменяется; при нагревании до 700° превращается в 1гО2.
При повышенной температуре порошкообразный металличе-
ский иридий взаимодействует с фтором, хлором, бромом, иодом,
серой, фосфором, образуя соответствующие соединения: IrF8,
IrCl4, IrCl3, IrCl2, IrCl, IrRr3, Irl3, Ir2S3, IrS2, Ir3S8 — коричне-
вый, Ir2P — серо-синий, IrP2 — серовато-черный.
Металлический иридий не взаимодействует с кислотами и цар-
ской водкой, но растворяется в окислительно-щелочных расплавах
(КОН + KNO3, NaOH + Na2O2) и в расплавленном K2S2O7.
При сильном нагревании металлического иридия с КС1 в атмо-
сфере хлора образуется К2[1тС18].
646
ПОБОЧНАЯ ПОДГРУППА VIII ГРУППЫ
ПРИМЕНЕНИЕ
Из металлического иридия изготовляют катоды (которые не
амальгамируются), контакты для магнето, провода для свечей
двигателей внутреннего сгорания, лабораторную посуду и инст-
рументы.
Порошкообразный иридий применяют в качестве катализа-
тора в реакциях превращения кислорода в озон, хлорной воды
в хлорноватистую кислоту и в других реакциях.
Из иридий-осмиевых сплавов делают кончики перьев в авто-
ручках, опорные оси для стрелок морских компасов, а также эта-
лоны для точного измерения длины.
Сплавы иридий — платина применяются для изготовления тер-
моэлементов, эталонов для измерения длины кислотостойкой хими-
ческой аппаратуры и в ювелирном деле.
СОЕДИНЕНИЯ (ОБЩИЕ СВОЙСТВА)
Известны многочисленные устойчивые соединения трех- и че-
тырехвалентного иридия и ограниченное число соединений одно-,
двух-, пяти- и шестивалентного иридия с низкой устойчивостью.
Таблица 65
Соединения двух- валентного ири- дия 1гС12, IrBr2, Irl2, IrS, K4[Ir(CN)8]
Соединения трех- валентного ири- дия 1г2О3, 1г2О3-ЗН2О или 1г(ОН)з, 1гС13, 1гВг3, 1гВг3-4Н2О, Irl3, Ir(CN)3, Ir(SCN)3, Ir2S3 Ir2(SO4)3, Ir2(SO4)3-nH2O, MeiIr(SO4)2-12H2O (где Mei = K+, Rb+, Cs+, NHJ, T1+), K3[IrXe]-nH2O (где X=C1-, Br-, NOi, CN", VaSOf-, i/2SO|-), Na3[IrCle].12H2O, (NH4)3[IrCle], K3[Ir(C2O4)3], H3[Ir(CN)e], [IrAme]X3, [IrAm5X]X2, [IrAm4X2]X, [IrAm5(H2O)]X3 (где Am = NH3, 1/2En, Ру, и X=C1“, Br-, I-), [Ir(NH3)e][IrCl8], [Ir(NH3)5(H2O)][IrCle]
Соединения четы- рехвалентного иридия IrO2, 1гО2-2Н2О или Ir(OH)4, IrF4, IrCl4, Irl4, IrS2 Mei[IrXe] (где Mel = Na+, K+, NHJ и X=F~, Cl", Br-), [Ir(NH3)4Cl2]Cl2
Соединения шести- валентно го ири- дия IrO3, IrFe, IrOF4, IrS3
В табл. 65 приведены формулы некоторых соединений двух-,
трех-, четырех- и шестивалентного иридия.
ИРИДИЙ
647
Помимо простых и координационных соединений иридия,
сгруппированных по валентности, указаны и его металлооргани-
ческие соединения.
Соединения одновалентного иридия
Одновалентное состояние не характерно для иридия. Известно
немного соединений иридия(1), например IrCl и галогениды карбо-
нилиридия типа 1г(СО)3Х (где X = Cl~, Br-, И).
Хлорид иридия, IrCl, получают действием газообразного хлора
на металлический иридий, нагретый до температуры выше 773°,
а также термическим разложением 1гС13 или 1гС12 при темпера-
туре выше 773°.
Хлорид иридия представляет собой медно-красные кристаллы,
которые разлагаются на компоненты при нагревании выше 798°.
Соединения двухвалентного иридия
Известно ограниченное число соединений двухвалентного ири-
дия. В качестве примеров можно назвать 1гС12, 1гВг2, Irl2, IrS,
K4lIr(CN)8], [IrD2H2NH4X], где D2H2 означает две молекулы ди-
метилглиоксима без двух атомов водорода.
Хлорид иридия, IrCI2, получают нагреванием губчатого ме-
таллического иридия или хлорида иридия 1гС13 в токе хлора
при 763°.
Соединение 1гС12 представляет собой блестящие темно-зеленые
кристаллы, плохо растворимые в кислотах и щелочах и разлагаю-
щиеся при нагревании до 773° на IrCl и хлор, а выше 798° —
на составные элементы.
Сульфид иридия, IrS, получают нагреванием металлического
иридия в парах серы; это блестящее темно-синее твердое веще-
ство, которое плохо растворимо в воде и кислотах, растворяется
в K2S и взаимодействует с влажной окисью углерода в присутст-
вии меди:
2IrS 4- 9СО + 4Cu + Н2О = 2HIr(CO)4 + 2Cu2S + СО2
Соединения трехвалентного иридия
Известно большое число устойчивых соединений трехвалент-
ного иридия. Примеры соединений иридия(Ш): Ir2O3, 1г2О3-
•ЗН2Оили1г(ОН)3, IrCl3, IrBr3, IrBr3-4H2O, Irl3, Ir(CN)3, Ir(SCN)3,
Ir2S3, Ir2(SO4)3, Ir2(SO4)3-nH2O, Me4r(SO4)2-12H2O (где Me1 =
= K+, Bb+, Cs+, NH+, Tl+), K3[IrX8]-nH2O (где X = Cl~, Br-,
NO", CN-, V'.SO*-, 1/2SO42-), Na3IIrCle]-12H2O, (NH4)3[IrCl8],
K3[Ir(C2O4)3], H3[Ir(CN)e], [IrAme]X3, [IrAm5X]X2, [IrAm4X2]X,
[IrAm5(H2O)]X3 (где Am = NH3, 1/2En, Ру и X = Cl-, Br-, I'),
648
ПОБОЧНАЯ ПОДГРУППА VIII ГРУППЫ
а также диядерные комплексы [Ir(NH3)8][IrCl8], [Ir(NH3)5(H2O)b
•11гС16].
Координационные соединения трехвалентного иридия отно-
сятся к наиболее устойчивым соединениям иридия.
Окись иридия, 1г2О3, получают при легком прокаливании суль-
фида иридия(Ш) или при нагревании К3[1гС16] с Na2CO3:
Ir2Ss -|- в/2О2 = Ir2O3 -|- 3SO2
2К3[1гС1в] + 3Na2CO3 = 1г2О3 + 6КС1 + 6NaCl + ЗСО2
Соединение 1г2О3 представляет собой твердое темно-синее
вещество, плохо растворимое в воде и спирте, растворимое в H2SO4,
разлагающееся при нагревании выше 400°:
Выше 400° - ИВО’
21г2О3--------> 31гО2-|-1г 1г2О3 —-+ 21г-р/2О2
При действии щелочей на раствор Na3[IrCl6] в атмосфере дву-
окиси углерода получают 1г2О3-ЗН2О или 1г(ОН)3 в виде олив-
ково-зеленого осадка; последний реагирует со щелочами, превра-
щается в 1гО3*2Н2О под действием азотной кислоты или кисло-
рода, при нагревании разлагается на 1гО2-2Н2О и металлический
иридий.
Хлорид иридия, 1гС13, образуется при действии хлора на порош-
кообразный (или губчатый) металлический иридий, нагретый
до 600°:
1г з/2С12 = 1гС13 -|- 60,45 ккал
Соединение 1гС13 оливково-зеленого цвета, имеет плотность
5,30 г/см3', оно летуче, плохо растворимо в воде, кислотах и щело-
чах, разлагается на 1гС12 и хлор при 765°, на IrCl и хлор при 773°,
на элементы при температуре выше 798°.
Бромид иридия, 1гВг3-4Н2О, осаждается при упаривании
раствора 1гО2'2Н2О в бромистоводородной кислоте. Бромид
1гВг3-4Н2О образует оливково-зеленые кристаллы, растворимые
в воде, плохо растворимые в спирте, дегидратирующиеся при на-
гревании до 105—120°. Безводная соль 1гВг3 при сильном нагре-
вании разлагается на элементы.
Иодид иридия, Irl 3, представляет собой твердое зеленое веще-
ство, слабо растворимое в холодной воде и спирте, растворимое
в теплой воде и разлагающееся при нагревании до 427°.
Сульфид иридия, Ir2S3, получают пропусканием сероводорода
через подкисленный соляной кислотой раствор хлорида ири-
дия(Ш), нагретый до 100°, барботированием сероводорода через
растворы солей иридия(1У) и нагреванием порошкообразного ме-
таллического иридия с серой при температуре не выше 1050°
в вакуумированной кварцевой трубке:
21гС13 + 3H2S = Ir2Ss + 6НС1
2H2[IrCl6] + 4H2S = Ir2Ss + S + 12HC1
ИРИДИЙ
649
Соединение Ir2S3 — твердое коричневое вещество, которое
разлагается на элементы при нагревании выше 1050°, плохо раст-
воримо в воде и растворяется в HNO3 и растворе K2S.
Соединения четырехвалентного иридия
Известны многочисленные устойчивые соединения иридия(1У);
в качестве примера можно назвать 1гО2, 1гО2-2Н2О или 1г(ОН)4,
IrF4, 1гС14, 1гВг4, Irl4, IrS2, Ме*[1гХ8] (где Me1 = Na+, К+, NH*
и X = F-, С1-, Br-), [Ir(NH3)4Cl2]Cl2.
Окись иридия, 1гО2, получают нагреванием порошкообразного
металлического иридия на воздухе или в кислороде при темпе-
ратуре ~700°, нагреванием дигидрата 1гО2 -2Н2О при 300° в азоте
и прокаливанием К2[1гС18] с Na2CO3.
1г -|- О2 = 1гО2 -|- 50,2 ккал
К2[1гС16] 4- 2Na2CO3 = 1гО2 + 2КС1 + 4NaCl + 2СОг
Окись иридия представляет собой чердые тетрагональные
кристаллы с кристаллической решеткой типа рутила и плотно-
стью 3,15 г/см3; она плохо растворима в воде, спирте и кислотах,
восстанавливается до металла водородом при нагревании и тер-
мически диссоциирует на элементы при нагревании до 1100°.
При обработке раствора 1гС14 или Na2[IrCl6] горячими щело-
чами образуется 1гО2-2Н2О или 1т(ОН)4 в виде синего осадка,
который растворяется в НС1 или НВг с образованием Н2[1гС16)
или Н2[1гВг8].
Фторид иридия, IrF4, получают нагреванием IrF6 с порошком
металлического иридия при 150°. Это желтая маслянистая жид-
кость, которая разлагается на воздухе и гидролизуется водой:
IrF4 + 4НОН = 1г(ОН)4 + 4HF
Хлорид иридия, 1гС14 (с примесью 1гС13), получают нагрева-
нием (600—700°) металлического иридия с хлором под давлением.
1гС14 представляет собой гигроскопичное коричневое твердое
вещество, растворимое в холодной воде и разлагающееся теплой.
При нагревании порошкообразного металлического иридия
с NaCl или КС1 в атмосфере хлора образуется Na2[IrCl8] или
К2[1гС161:
1г + 2МетС1 + 2С12 = Ме|[1гС1в]
Гексахлороиридат{1У) аммония, (NH4)2[IrCl6], получают обра-
боткой раствора Na2[IrCl8] или К2[1гС16] хлоридом аммония.
Соединение (NH4)2[IrCl6] образует темно-красные октаэдриче-
ские кристаллы, плохо растворимо в холодной воде, растворяется
в горячей, восстанавливается до металлического иридия при
650
ПОБОЧНАЯ ПОДГРУППА VIII ГРУППЫ
нагревании с углем или в токе водорода, разлагается при прока-
ливании по уравнению
(NH4)2[IrClel Ir + 2NH4C1 + 2С12
Бромид иридия, 1гВг4, получают путем растворения при низкой
температуре окиси иридия(1У) в бромистоводородной кислоте.
Он представляет собой расплывающееся на воздухе синее вещество,
которое растворяется в спирте, растворяется в воде с разложением
и диссоциирует при нагревании на элементы.
Иодид иридия, Irl 4, может быть получен кипячением раствора
(NH4)2[IrCl8l с иодидом калия:
(NH4)2[IrCl6] + 4KI = Irl4 + 2NH4C1 + 4КС1
Черное твердое соединение Irl4 плохо растворимо в воде и раз-
лагается выше 100°.
Сульфид иридия, IrS2, получают нагреванием порошкообраз-
ного металлического иридия с серой (или с полисульфидами ще-
лочных металлов) в кварцевой трубке без доступа воздуха (в ва-
кууме) или пропусканием сероводорода через растворы солей
иридия(1У).
Сульфид IrS2 — коричневое твердое вещество, плохо раство-
римое в воде; под действием окиси углерода при нагревании вос-
станавливается сначала до Ir2S3, а затем до металлического иридия.
Соединения шестивалентного иридия
Соединений шестивалентного иридия известно довольно мало,
и все они, как правило, мало устойчивы. В качестве примеров
соединений иридия(У1) упоминаются IrO3, IrF6, IrOF4, IrS3.
Фторид иридия, IrF6, получают нагреванием порошкообраз-
ного металлического иридия в атмосфере фтора в трубке из флю-
орита (CaF2) (250°).
Соединение IrF6 представляет собой желтые тетрагональные
кристаллы с плотностью 6,0 г/см5, т. пл. 44,4° и т. кип. 53°; под
действием металлического иридия при нагревании превращается
в IrF4, восстанавливается водородом до металлического иридия,
разъедает влажное стекло (образуется IrOF8), реагирует с водой
по уравнению
IrF6 + 5Н2О = 1гО2-2Н2О + 6HF + Ч2О2
Сульфид иридия, IrS3, получают нагреванием (600°) 1гС1в
с избытком серы в вакууме; это серый, плохо растворимый в кис-
лотах порошок.
ПАЛЛАДИЙ
651
Металлоорганические соединения
Известны карбонильные соединения иридия 1г2(СО)8, 1г4(СО)12,
галогениды карбонилиридия 1г(СО)3Х, 1г(СО)2Х2 (где X — С1",
Br-, I-), галогениды дициклопентадиенилиридия (С5Н6)21гХ (где
X = С1_, Вг_), а также соединение Н1г(СО)4.
Октакарбонил иридия, 1г2(СО)8, получают действием окиси
углерода под давлением на порошок металлического иридия,
нагретый до 160—180°, или действием окиси углерода под давле-
нием 200 ат на соединения К2[1гВг6], К2[1г1в].
Соединение 1г2(СО)8 представляет собой зеленые кристаллы,
которые сублимируются при 160° и растворяются в хлороформе,
оно превращается в 1г4(СО)12 при нагревании со щелочами или кис-
лотами.
Додекакарбонил иридия, 1г4(СО)12, получается действием оки-
си углерода на 1г13 при пониженном давлении; образует оранжево-
желтые кристаллы, которые разлагаются при 210°.
Галогениды карбонилиридия, 1г(СО)3Х,
1г(СО)2Х2. Нагреванием галогенидов иридия- 1гХ3 до 150° с оки-
сью углерода при давлении 1 ат получают оба типа галогенидов
карбонилиридия —1г(СО)3Х, 1г(СО)2Х2 (где X = Cl~, Вт-, I-).
Галогениды типа 1г(СО)3Х окрашены в коричневый цвет, субли-
мируются при ~-115°, разлагаются водой и более устойчивы на
воздухе, чем галогениды 1г(СО)2Х2, которые легко теряют окись
углерода, сублимируются (~150°) и окрашены в светло-желтый
цвет.
ПАЛЛАДИЙ Pd
Z = 46; ат. вес = 106,4
Валентность (I), II, (III), IV
Массовые числа природных изотопов 106, 108,
105, 110, 104, 102
Электронная структура атома палладия: К -L -М •4s24p84d10 -5s0.
Электронная структура атома палладия и катиона Pd2+ для
4d- и 58-орбиталей:
4d»°
h|u|hIu|w
Pd
4d> 5s
U|n|U| t | ?] £2
Pd2+
ИСТОРИЧЕСКАЯ СПРАВКА
Палладий был открыт в 1803 г. Виллостоном и назван в честь
маленькой планеты (астероида) Pallas, открытой в 1802 г. другом
Волластона Олберсом.
652
ПОБОЧНАЯ ПОДГРУППА VIII ГРУППЫ
НАХОЖДЕНИЕ В ПРИРОДЕ
В природе палладий встречается как в самородном'состоянии
вместе с платиной (в палладийсодержащей платине присутствует
0,1—7% Pd и 80—88% Pt), золотом (в палладийсодержащем золоте,
названном порпецитом, имеется до 5—11% Pd и 4% Ag), ртутью
(палладийсодержащая ртуть потарит), так и в виде соединений,
например стибиопалладините Pd3Sb, станнопалладините Pd3Sn2,
палладите PdO, браггите (Pd, Pt, Ni)S и др.
Содержание палладия в земной коре 5-10-в вес. %. Залежи,
содержащие палладий, находятся в СССР, Бразилии, Колумбии
и Сьерра-Неваде.
ИЗВЛЕЧЕНИЕ И ПОЛУЧЕНИЕ
МЕТАЛЛИЧЕСКОГО ПАЛЛАДИЯ
При обработке царской водкой сырой платины или платиновых
руд (соответственно концентратов платиновых руд) палладий,
платина и родий переходят в раствор в виде анионов [PdClJ2-,
[PtCl8]2', [RhClgl3-. После отделения платины и родия палладий
выделяется в виде желтого осадка [Pd(NH3)2Cl2],
Металлический палладий (в порошкообразном или губчатом
состоянии) получают термическим разложением соединений PdO,
PdF2, PdCl2, PdBr2, Pdl2, Pd(NO3)2, PdO2, H2[PdCl6], H2[PdBre],
действием CO, CH4, C2H4 или H2 в кислой среде (Zn + HC1) на вод-
ный раствор хлорида палладия(П), обработкой водой соедине-
ния [PdCOCl2]2, восстановлением водородом соединений PdF3,
[Pd(NH3)2Cl2], [Pd(NH3)4]Cl2, Mei[Pd(CN)J (где Mei = Na+, K+),
действием цинка, железа, магния на водные растворы солей пал-
ладия.
Металлический палладий получают также восстановлением
формальдегидом различных солей палладия в щелочной среде.
ФИЗИЧЕСКИЕ И ХИМИЧЕСКИЕ СВОЙСТВА
В компактном состоянии палладий представляет собой бле-
стящий серебристо-белый металл с гранецентрированной кубиче-
ской кристаллической решеткой. В порошкообразном состоянии
он имеет серовато-черный цвет. Металлический палладий обла-
дает плотностью 11,97 г1см3 (тяжелый металл), т. пл. 1552°, т.
кип. 3127° (тугоплавкий металл, хотя в платиновой группе у него
самые низкие температуры плавления и кипения); твердость рав-
на 4,8 по шкале Мооса (металл ковок, тягуч, способен перерабаты-
ваться давлением). В ряду металлов, поддающихся катодному рас-
пылению, палладий стоит впереди Au, Ag, Pb, Sn, Pt, Си, Cd,
Ni, Ir, Fe.
ПАЛЛАДИЙ
653
Металлический палладий (в виде тонкой пластинки, порошка,
губки или в коллоидном состоянии) обладает большой абсорбцион-
ной способностью по отношению к различным газам — водороду,
окиси углерода, кислороду и др.
В отличие от остальных платиновых металлов абсорбция водо-
рода палладием сопровождается образованием твердого раствора
или образованием гидридов включения Pd2H, PdH0j6. Наибольшей
абсорбционной способностью по отношению к водороду обладает
коллоидный палладий, затем порошкообразный и, наконец, губча-
тый палладий. При комнатной температуре один объем металличе-
ского палладия поглощает 2800 объемов водорода, и поглощающая
способность возрастает при добавлении к металлическому палла-
дию серебра, золота или бора. Палладий может отдавать водород
при нагревании в вакууме до 300°.
Благодаря поглощенному атомарному водороду палладий про-
являет восстановительные свойства по отношению к HgCl2, Fe2Gl6,
K3lFe(CN)6], КСЮз, KNO3 и др.
Известны многочисленные сплавы палладия с платиной, ири-
дием, золотом, серебром, медью, никелем, кобальтом и ртутью.
С химической точки зрения палладий — инертный металл,
он устойчив к действию многих химических реагентов.
При обычной температуре на воздухе или в кислороде метал-
лический палладий не изменяется, а при нагревании до красного
каления превращается в окись палладия(П) PdO черного цвета.
Фтор, хлор, иод, сера и селен взаимодействуют с металличе-
ским палладием при нагревании, образуя PdF2, PdF3, PdCl2,
Pdl2, PdS, PdSe. При действии азота, фосфора и мышьяка на ме-
таллический палладий при высокой температуре образуются раз-
личные соединения включения. Расплавленный металлический
палладий растворяет углерод, который при охлаждении выделя-
ется в виде микроскопических частиц графита.
При высокой температуре металлический палладий взаимо-
действует с расплавленными Na2O2 и K2S2O7:
Pd -|- Na2O2 = PdO -|- Na2O
3Pd + 6K2S2O7 = 3PdSO4 + 6K2SO4 + 3SO2
Поскольку нормальный потенциал системы Pd/Pd2+ равен
-j-0,987, металлический палладий растворяется в конц. HNO3,
горячей конц. H2SO4 и в царской водке.
ПРИМЕНЕНИЕ
Металлический палладий служит для защитных и декоратив-
ных покрытий деталей из меди и серебра. Процесс палладирования
осуществляется электролитическим путем.
654
ПОБОЧНАЯ ПОДГРУППА VIII ГРУППЫ
В химической промышленности металлический палладий в виде
пластинок, листов, в губчатом, порошкообразном и коллоидном
состояниях применяется как катализатор многочисленных хими-
ческих реакций. В качестве примеров таких реакций можно на-
звать синтез воды из элементов, окисление NH3 до окислов азота,
окисление углеводородов кислородом, гидрирование жиров. Ката-
лизаторами могут служить различные соли палладия и паллади-
рованный асбест, который получают прокаливанием асбеста,
пропитанного раствором хлорида палладия(П) и небольшим коли-
чеством спирта. В присутствии палладированного асбеста окисля-
ются водород (20°), спирт (150°), бензин (250°), окись углерода
(290°), метан (404°).
К веществам, оказывающим вредное действие на палладий
при использовании его в качестве катализатора, относятся 12г
H2S, HgCl2, HCN, AsH3.
Из металлического палладия и сплавов Pd — Rh, Pd — Au,
Pd — Au — Pt, Pd — Au — Ag, Pd — Ag — Co, Pd — Pt — Ir
изготовляют электрические контакты, которые используются в раз-
личных областях электротехники. Из сплавов Pd — Au — Pt
делают термоэлементы. Сплавы Pd — Au, Pd — Au — Pt, Pd —
— Pt — Ir применяются для изготовления хирургических инст-
рументов. В зубопротезной технике используют сплавы палла-
дия с золотом или серебром, медью, иридием.
В ювелирном деле применяются сплавы палладия с платиной
или золотом, которые иногда содержат иридий, медь и серебро.
СОЕДИНЕНИЯ (ОБЩИЕ СВОЙСТВА)
Известны многочисленные устойчивые соединения двухва-
лентного палладия и ограниченное число соединений одно-, трех-
и четырехвалентного палладия, для которых характерна низка»
устойчивость.
В табл. 66 приведены формулы некоторых соединений двух-,
трех- и четырехвалентного палладия. Помимо простых указыва-
ется довольно много координационных соединений палладия.
Соединения одновалентного палладия
Одновалентное состояние не характерно для палладия. В ка-
честве примера можно назвать соединение Pd2S.
Сульфид палладия, Pd2S, получают нагреванием [Pd(NH3)2]Cl2
с 25 вес. % серы под слоем буры при 1065°. Он представляет собой
зеленовато-серое аморфное вещество с плотностью 9,3 г/сл43; плохо
растворимо в воде, кислотах и царской водке.
ПАЛЛАДИЙ
655
Таблица 66
Соединения двух- валентного пал- ладия PdO, PdO-H2O или Pd(OH)2, PdF2, (PdCl2)„, PdCl2- • 2H2O, H2[PdCl4], Na2[PdCl4]-3H2O, K2[PdCl4], K2[Pd(NO2)2Cl2], [Pd(NH3)4]Cl2, [Pd(NH3)2Cl2], [PdRCl2]2 (где R = третичные фосфины, третичные арсины, окись углерода), PdBr2, H2[PdBr4], Mei[PdBr4], [Pd(NH3)4]Br2, [Pd(NH3)2Br2), [PdR2Br2], [PdRBr2]2 (где R = третичные фосфины, третичные арсины), Pdl2, H2[PdI4], Mei[PdI4], [Pd(NH3)4]I2, [Pd(NH3)2I2], [PdRI2] (где R = третичные фосфины, третичные арсины), Pd(CN)2, H2[Pd(CN)4], K2[Pd(CN)4], Ba[Pd(CN)4]-4H2O, PdS, PdSO4-2H2O, Pd(NO3)2, [Pd(NH3)4](NO3)2, [Pd(NH3)2(NO3)2], ацетилацетонат, диметилглиоксимат и др.
Соединения трех- валентного пал- ладия Pd2O3.«H2O, PdF3, H2[PdCl6], Rb2[PdCl6], Cs2[PdCl5]
Соединения четы- рехвалентного палладия PdO2-2H2O, PdS2, PdSe2, ,Mei[PdCle], K2[PdBr„], [Pd(NH3)JX4 (где X = C1", Br-), [Pd(C10H8N2)2Cl2]Cl2
Соединения двухвалентного палладия
Известны многочисленные устойчивые соединения, в которых
двухвалентный палладий находится в виде катионов Pd2+,
[Pd(NH3)4]2- или аниона [PdX4]2~ (где X = Cl-, Br-, I-, CN-,
SCN-, NO-, VAO2-).
Помимо простых соединений палладия(П) упоминаются много-
численные координационные соединения класса тетрамминов,
Тетрацидосолей, хелатных соединений, соединений типов
[Pd(NH3)2X2] (где X = Cl", Br', I~, NO;, NO;, ОН-, X/2SO2-,
VaSO2-, ЧгСО^-) и [PdBX2]2, (где X = Cl“, I", NO;, SCN", SC2H5
и В = третичные фосфины, третичные арсины, окись углерода,
этилен и др.).
Окись палладия, PdO, получают сильным нагреванием метал-
лического палладия в кислороде, прокаливанием Pd(NO3)2, на-
греванием (600°) PdCl2 с NaNO3, сильным нагреванием амаль-
гамы палладия, действием расплавленной Na2O2 на металлический
палладий и нагреванием (200°) PdO2-2H2O.
Pd + V2O2 PdO + 22,9 ккал Pd(NO3)2 = PdO + 2NO2 + V2O2
Соединение PdO — наиболее устойчивый окисел палладия;
PdO представляет собой черный порошок с плотностью 8,31 г!см\
плохо растворимый в воде и кислотах. Оно разлагается на эле-
656
ПОБОЧНАЯ ПОДГРУППА VIII ГРУППЫ
менты при 877° и обладает окислительными свойствами (окисляет
водород в воду и окись углерода до двуокиси).
Гидроокись палладия, Pd(OH)2 или PdO«H2O, образуется в ре-
зультате гидролиза Pd(NO3)2 горячей водой или при кипячении
растворов солей палладия(П) со щелочами.
Гидроокись палладия представляет собой коричнево-красный
порошок; она плохо растворяется в воде, растворяется в кислотах,
может быть обезвожена нагреванием до PdO и обладает слабо
выраженными окислительными свойствами.
Гидроокись тетрамминопалладия(1 Г), [Pd(NH3)4](OH)2, обра-
зует бесцветные кристаллы.
Известно соединение [Pd(NH3)2(OH)2] желтого цвета; оно
получается при обработке [Pd(NH3)2SO4] гидратом окиси бария.
Фторид палладия, PdF2, получают прямым взаимодействием
элементов при высокой температуре или нагреванием PdF3 со
стехиометрически необходимым количеством порошкообразного
металлического палладия:
Pd -J- F2 = PdF2 + 101,9 ккал 2PdF3 + Pd = 3PdF2
Фторид палладия представляет собой коричневые кристаллы
со структурой кристаллической решетки типа рутила, плотностью
5,8 г!см\ т. пл. 727° и т. кип. 1227°; он плохо растворим в воде
и растворяется в плавиковой кислоте с образованием тетрафторо-
палладиевой кислоты H2[PdF4].
Полимер хлорида палладия,
Cl CL С1 /С1 f
)87° / Pd 3.18А
ХС1/ Х С1 4з,4А- С1 !
получают нагреванием порошкообразного или губчатого палла-
дия в токе хлора при высокой температуре:
Pd + С12 = PdCl2 + 43,49 ккал
Соединение (PdCl2)rt представляет собой расплывающиеся на
воздухе красные кристаллы, которые плавятся при ~940°,
сублимируются при 738° и хорошо растворяются в воде.
Хлорид палладия, PdCl2, безводный или растворенный в спирте,
поглощает сухую окись углерода, образуя аддукт [PdCOCl2]2
коричневого цвета, который разлагается водой с выделением ме-
таллического палладия.
При действии сероводорода на хлорид палладия(П) образуется
сульфид палладия(П). Водород, окись углерода, метан, этан вос-
станавливают хлорид палладия(П) до металла.
PdCl2 + СО + Н2О = Pd + 2НС1 + СО2
При упаривании водного раствора хлорида палладия(П) вы-
падают в осадок расплывающиеся на воздухе коричневые призма-
тические кристаллы PdCl2 -2Н2О.
ПАЛЛАДИЙ
657
При растворении металлического палладия в соляной кислоте
в присутствии хлора или при действии соляной кислоты на поли-
мер хлорида палладия (II) образуется H2[PdCl4].
Обработка водных растворов хлорида палладия(П) избытком
аммиака дает хлорид тетрамминопалладия(П):
PdCl2 + 4NH4OH = [Pd(NH3)4]Cl2 + 4Н2О
Известны стереоизомеры комплекса типа неэлектролита
[Pd(NH3)2Cl2], которые образуются при обработке K2[PdCl4]
аммиаком или [Pd(NH3)]4Cl2 соляной кислотой; они окрашены
в коричнево-желтый цвет и плохо растворимы в воде. При заме-
щении молекул аммиака в соединении [Pd(NH3)2Cl2] молекулами
аминов, диаминов, гидразинов, фосфинов PR3 (где R = алкильная
группа), арсинов AsR3 или тиоэфиров SR2, селеноэфиров SeR2
и т. д. образуются многочисленные координационные соединения
аналогичного типа.
Известны нейтральные координационные соединения типа
[PdRClJ2 (где R = третичные фосфины, третичные арсины, этилен
или окись углерода). Для дихлоро(триметиларсин)палладия(П)
приведена формула
r(H3C)3As Cl Cl
)pd( )pd/
L Clz XC1Z xAs(CH3)3J
Известны тетрахлоропалладаты(П) Na2[PdCl4] -3H2O красно-
вато-коричневого цвета и K2[PdCl4l коричнево-красного цвета;
они образуются при действии хлора на металлический палладий
в присутствии хлоридов NaCl, КС1 или при обработке солянокис-
лого раствора хлорида палладия(П) NaCl или КС1.
Известны также дихлородинитропалладаты(П), например
K2[Pd(NO2)2Cl2], которые получаются при действии KNO2 на
соединение KJPdClJ.
Бромид палладия, PdBr2, получают действием бромной воды
на металлический палладий или действием КВг на PdCl2.
Бромид палладия представляет собой коричнево-красное ве-
щество с т. пл. 627°; оно разлагается на элементы при нагревании
до высокой температуры, плохо растворимо в воде и растворяется
в водном растворе НВг или бромидов щелочных металлов с обра-
зованием тетрабромопалладиевой кислоты HJPdBrJ или тетра-
бромопалладатов(П) Me|[PdBr4],
Известен бромид тетрамминопалладия(П) [Pd(NH3)4]Br2 и сте-
реоизомеры комплексов [Pd(NH3)2Br2], [PdR2Br2] (где R = амин,
диамин, гидразин, фосфин, арсин). Получены также нейтральные
42-0101
658
ПОБОЧНАЯ ПОДГРУППА VIII ГРУППЫ
комплексы типа [PdRBr2]2 (где R = третичные фосфины, тре-
тичные арсины, этилен).
Иодид палладия, Pdl2, получают нагреванием порошка метал-
лического палладия в парах иода, действием раствора иода на ме-
таллический палладий или обработкой иодистым калием водного
раствора хлорида палладия(П).
Иодид палладия представляет собой темно-красный порошок,
который разлагается на элементы при нагревании до 360°, плохо
растворим в воде и спирте и растворяется в HI или в растворах
иодидов щелочных металлов с образованием тетраиодопалладие-
вой кислоты H2[PdI4] или тетраиодопалладатов(П) Me*[PdI4]
(где Me1 = К + , Na+).
Известны иодид тетрамминоцалладия(П) [Pd(NH3)4]I2, сте-
реоизомеры соединения [Pd(NH3)212], а также соединения с ана-
логичной формулой, в которых молекулы аммиака замещены моле-
кулами аминов, диаминов, гидразинов, фосфинов, арсинов и др.
Упоминается также нейтральное координационное соединение
типа [PdRI2]2 (где R = третичные фосфины или третичные арсины).
Цианид палладия, Pd(CN)2, представляет собой желтовато-
белое вещество, слабо растворимое в воде и спирте и растворяю-
щееся в HCN hhhKCN с образованием H2[Pd(CN)4 или K2[Pd(CN)4],
Известен тетрацианопалладат(П) бария Ba[Pd(CN)4] -4Н2О.
Сульфид палладия, PdS, получают прямым взаимодействием
элементов при нагревании, термическим разложением сульфида
палладия PdS2 или пропусканием сероводорода через водный
раствор солей палладия(П), например PdCl2 или [Pd(NH3)4]Cl2,
нагретый до 70—80°.
Сульфид PdS2 представляет собой темно-коричневое твердое
металлоподобное вещество с т. кип. 950°; он плохо растворим
в воде, соляной кислоте, сульфиде аммония и растворяется в азот-
ной кислоте и царской водке.
Сульфат палладия, PdSO4-2H2O, осаждается при упаривании
на холоду раствора Pd(OH)2 в серной кислоте или металлического
палладия в горячей конц. H2SO4.
Соединение PdSO4>2H2O образует расплывающиеся корич-
нево-красные кристаллы.
Нитрат палладия, Pd(NO3)2, выпадает в осадок при упари-
вании на холоду раствора металлического палладия в азотной
кислоте.
Нитрат палладия представляет собой расплывающиеся на воз-
духе желто-коричневые призматические кристаллы; при терми-
ческом разложении Pd(NO3)2 образуется PdO, с аммиаком он дает
соединение [Pd(NH3)4](NO3)2. Горячей водой гидролизуется
Pd(NO3)2, образуя Pd(OH)2.
Известны стереоизомеры комплексного соединения
Pd(NH3)2(NO3)2].
ПАЛЛАДИЙ
659
Хелатные соединения
При действии ацетилацетона СН3-СОСН2>СО-СН3, диметил-
глиоксима HON = С(СН3)-С(СН3) = NOH, дифенилглиоксима
HON = С(С6Н5) •QCgHg) = NOH, тиосемикарбазида H2N-NH>
•CS-NH2 на растворы солей палладия(П) образуются окрашен-
ные, плохо растворимые хелатные соединения
СН3
СН
СН3
Желтые кристаллы
‘ Н3С —С------С- СН3 Л
II II
О —N N = O
н< )Pd/ )н
XO = NZ XN-OZ
II II
Н3С —С-----С —сн3
Желтые кристаллы
Соединения трехвалентного палладия
Известно небольшое число соединений трехвалентного пал-
ладия, причем все они плохо растворимы. В качестве примеров
можно назвать Pd2O3-nH2O, PdF3, H2[PdCl3], Rb2[PdGl5],
Cs2[PdCl5].
Гидратированная окись палладия, Pd2O3-nH2O, получается
окислением водного раствора Pd(NO3)2 озоном (8°) или электро-
литическим путем.
Соединение Pd2O3-raH2O представляет собой темно-коричневый
порошок, который взрывается при нагревании, плохо растворим
в HNO3 и H2SO4 и растворяется в НС1 с выделением хлора.
Фторид палладия, PdF3, получается действием фтора на ме-
таллический палладий или на PdF2 (200°), а также обработкой
фтористым бромом BrF3 иодида палладия Pdl2 при нагревании:
Pd + 3/2F2 PdF3 + 99,6 ккал PdF2 + X/2F2 = PdF3
Фторид палладия представляет собой гигроскопичные пара-
магнитные черные ромбические кристаллы с плотностью 5,06 г/см3',
при нагревании выше 227° он разлагается, растворим в плавиковой
кислоте, обладает окислительными свойствами, восстанавлива-
ется до металла водородом при нагревании, окисляет соляную кис-
лоту до элементарного хлора.
Соединения четырехвалентного палладия
Хотя соединения четырехвалентного палладия более устой-
чивы, чем соединения палладия(Ш), все же они обладают окисли-
тельными свойствами и проявляют склонность к переходу в соеди-
нения палладия(П). В качестве примеров соединения четырех-
валентного палладия можно назвать PdO2-2H2O, PdS2, PdSe2,
42*
660
ПОБОЧНАЯ ПОДГРУППА VIII ГРУППЫ
Mei[PdGl6] (где Me1 = К+, Rb+, Cs+), K2[PdBre],
[Pd(C10H8N2)2Cl2]C12, [Pd(NH3)8]X4 (где X = Cl“, Br-).
Гидратированная окись палладия, PdO2'2H2O, получается
обработкой гексахлоропалладата(1У) калия K2[PdCl8] щелочью
или действием озона на растворы солей паладия(П) (в присут-
ствии избытка щелочей или карбонатов щелочных металлов).
K2[PdCl6] + 4КОН = PdO2.2H2O + 6КС1
K2[PdCl4] + 2КОН + О3 + Н2О = PdO2-2H2O + 4КС1 + О2
Соединение PdO2 "2Н2О образуется в виде темно-красного осад-
ка; оно растворимо в разбавленных кислотах и концентрированных
растворах щелочей, обладает окислительными свойствами, вос-
станавливается до металла водородом при нагревании, разлага-
ется выше 200°.
PdO2-2H2O PdO + 1/2О2 + 2Н2О
Сульфид палладия, PdS2, получают сплавлением PdCl2 с серой
(450—500°) или обработкой спиртом, а затем разб. НС1 холодного
продукта, полученного при нагревании докрасна в инертном газе
смеси 1 ч. (NH4)2[PdCl6], 12 ч. серы и 12 ч. NaOH.
Сульфид палладия(1У) представляет собой темно-коричневые
кристаллы, которые превращаются в сульфид палладия(П) нагре-
ванием до температуры выше 600° и образуют двойные сульфиды
с сульфидами щелочных металлов.
Гексахлоропалладат(1У) калия, K2[PdCl8], получают, пропу-
ская ток хлора через раствор K2IPdCl4] или при обработке
K2[PdCl4] царской водкой.
K2[PdCl4] + ЗНС1 + HNO3 = K2[PdCl6] + !/2С12 + NO + 2Н2О
Соединение K2[PdCl8] образует красные октаэдрические кри-
сталлы; оно растворяется в воде, а под действием горячей воды
высвобождает хлор, превращаясь в K2[PdCl4].
Гексахлоропалладат ы(1У), Rb2[PdCl6], Cs2[ PdCl6],
представляют собой красные кристаллы, плохо растворимые в воде.
Хлорид дихлор-бис-(дипиридил)палладия(1У),
lPd(C10H8N2)2Cl2lCl2, в виде оранжевого осадка получается при
действии хлора на суспензию [Pd(Ci0H8N2)2Cl2] в хлороформе.
ПЛАТИНА Pt
Z = 78; ат. вес = 195,09
Валентность (I), II, (III), IV, (VI)
Массовые числа природных изотопов 195, 194,
196, 198, 192
Электронная структура атома платины: K-L-M-N-5s25p6"
• SdMs1.
ПЛАТИНА
661
Электронная структура атома платины, катиона Pt2+ и анио-
нов [PtX4]2-, [PtX8]2- для 5d-, 6s- и бр-орбиталей:
5d’ 6s«
luiuinitii t| Ш
Pt
5d
ИНЕЕ
6s 6p
[Ptx4p-
5d«
|tl|U|tl|t | t |
Pt*+
6s
5d 6s 6p
ЕЕЕЕИ □ EEE
[Ptx6p-
ИСТОРИЧЕСКАЯ СПРАВКА
Платина была открыта Антонием Улио в 1735 г. в золотонос-
ных песках р. Пинто (Колумбия) и описана как элемент Уэстоном
в 1750 г.
Название «платина» происходит от испанского слова platina —
уменьшительное от plata, что означает серебро. Длительное время
платину рассматривали как фальсифицированное серебро, посколь-
ку ее не могли расплавить и растворить как серебро.
В чистом виде платина стала известна с 1845 г., после того
как были открыты другие платиновые металлы. Вначале платина
имела ограниченное применение. В первой половине XIX в. в Рос-
сии из нее чеканили монеты. Позднее из платины стали отливать
слитки и делать оправу для драгоценных камней. Промышленное
использование платины начинается одновременно с изучением
ее свойств.
РАСПРОСТРАНЕННОСТЬ В ПРИРОДЕ
В природе платина встречается в наносных залежах, в виде
сплавов с различными металлами и иногда в виде соединений с мы-
шьяком, серой и др. в различных минералах.
В качестве примеров сплавов платины можно назвать ферро-
платину, купферроплатину, никель-, палладий-, иридий- и родий-
содержащую платину и др. Содержание платины в этих сплавах
составляет 75—88 вес.%. Обычно сплавы платины с палладием
содержат иридий и родий и не содержат рутения и осмия.
Платиновыми минералами являются поликсен (самородная
платина), невьянскит (сплав осмиридия, содержащий0,1—55% пла-
тины), сперрилит PtAs2 (содержит 56,6 % | Pt), куперит PtS, брэг-
гит (Pt, Pd, Ni)S и др.
Поликсен содержит до 80—88% платины и представляет собой
серебристо-белые или светло-серые кубические кристаллы с плот-
ностью 15—19 г! см? и твердостью 4—4,5 по шкале Мооса.
Содержание платины в земной коре 5-10-5 вес.%. Залежи
платины находятся в Колумбии, Бразилии, Канаде, СССР, Южной
Африке, Калифорнии, Новой Зеландии.
662
ПОБОЧНАЯ ПОДГРУППА VIII ГРУППЫ
ИЗВЛЕЧЕНИЕ И ПОЛУЧЕНИЕ МЕТАЛЛИЧЕСКОЙ ПЛАТИНЫ
Платину извлекают гидрометаллургическим путем из сырой
платины, из платиновых руд (или их концентратов) и из анодного
шлама, образующегося при электролитическом рафинировании
никеля или меди.
При обработке хлоридом аммония раствора, полученного дей-
ствием царской водки на сырую платину или на концентраты пла-
тиновых руд, образуется желтый осадок (NH4)2[PtCl6].
Черный порошок металлической платины получается при тер-
мическом разложении соединений PtO, PtO2, PtX2, PtX4, H2[PtCl6]
(где X = F~, СГ, Br-, Г), (NH4)2[PtCl6], [Pt(NH3)6]Cl4, PtS,
Pt2S3, PtCl3, восстановлением слабо подкисленных растворов
солей платины металлическим цинком, алюминием, кадмием,
железом, а также восстановлением растворов солей платины в ще-
лочной среде при нагревании их с FeSO4-7H2O:
H2[PtCl6] -> Pt + 2НС1 + 2С12 (NH4)2[PtCle] -> Pt + 2NH4C1 + 2C12
H2[PtCl6] + 4FeSO4 + 14K0H = Pt + 4Fe(OH)3 + 4K2SO4+6KC1+2H;O
H2[PtCl6] + 3Zn = Pt + 3ZnCl2 + H2
Чтобы отделить порошкообразную платину от гидроокиси желе-
за, последнюю растворяют в разбавленной соляной кислоте.
Для восстановления соединений платины в щелочной среде
до порошкообразного металла можно также использовать фор-
мальдегид и гидразин.
При действии окиси углерода на растворы H2[PtCl6] образу-
ется коллоидная платина красного цвета, которая при хранении
превращается в черную порошкообразную платину.
Действием хлорида олова(П) на H2[PtCle] можно также полу-
чить коллоидную платину (красного цвета), которая с гексагидро-
ксооловянной(1У) кислотой H2[Sn(OH)e] образует адсорбционное
соединение кроваво-красного цвета, названное пурпуреоплатиной.
Кислота H2[Sn(OH)6] образуется при гидролизе хлорида олова(1У).
H2[PtCl6] + 2SnCl2 + 12Н2О = Pt -J- 2H2[Sn(OH)6] + 10НС1
Прокаливанием сульфида платины(П) в токе водорода полу-
чают губчатую металлическую платину.
Для очистки сырую платину растворяют в царской водке и хло-
ридом аммония осаждают соединение (NH4)2[PtCl6], которое при
термическом разложении дает металлическую платину 99 %-ной
чистоты (1% — платиновые металлы).
ФИЗИЧЕСКИЕ И ХИМИЧЕСКИЕ СВОЙСТВА
В компактном состоянии платина представляет собой блестя-
щий серебристо-белый металл с гранецентрированной кубической
ПЛАТИНА
663
кристаллической решеткой. В порошкообразном состоянии метал-
лическая платина черного цвета, в коллоидном — красного. При
нагревании порошка платины (платиновой черни) до 700—800°
образуется губчатая платина, которая при 1200—1300° превраща-
ется в компактную металлическую платину.
Если растворять сплав Zn — Pt в соляной кислоте, то обра-
зуется взрывающаяся (гремучая) платина.
Металлическая платина имеет плотность 21,45 г! см? (тяжелый
металл), т. пл. 1769°, т. кип. 3827° (тугоплавкий металл) и твер-
дость 4,3 по шкале Мооса (самый мягкий среди платиновых метал-
лов). Платина легко перерабатывается давлением. Электропровод-
ность ее в 9,7 раза больше, чем у ртути.
Металлическая платина хорошо сваривается со стеклом, обла-
дая тем же коэффициентом расширения.
Известны многочисленные сплавы платины с оловом, свинцом,
медью, серебром, золотом, иридием, палладием, железом, кобаль-
том, никелем, цинком и др.
Водород легко диффундирует через нагретую платину. В отли-
чие от палладия в платине водород растворяется в очень малых
количествах. Платина поглощает атомарный водород и отдает его
значительно труднее, чем палладий. Метан, кислород, азот, аргон,
гелий, хлор, окись углерода, аммиак и другие газы не диффунди-
руют через платину, благодаря чему они могут быть очищены
от водорода.
С химической точки зрения платина обладает низкой реак-
ционной способностью, она очень устойчива к действию много-
численных химических реагентов.
Из всех платиновых металлов у платины наименьшее сродство
к кислороду, она устойчива на воздухе при обычной температуре.
При нагревании (350—450°) платины в кислороде образуется PtO2
или РЮ.
При нагревании металлической платины в атмосфере фтора,
хлора или паров иода образуется PtF4, PtF2, PtCl4, PtCl2, Ptl4.
Металлическая платина взаимодействует при нагревании с се-
рой, фосфором, мышьяком, сурьмой, углеродом (или органиче-
скими соединениями, которые выделяют углерод), кремнием,
бором, образуя PtS, Pt2P, Pt5P4, PtP2, арсениды, антимониды,
карбиды, силициды и бориды — хрупкие и ломкие соединения.
Вследствие этого запрещается пользоваться сосудами и инстру-
ментами из платины при прокаливании сульфидов, сульфитов,
тиосульфатов и смесей, содержащих фосфор, мышьяк, сурьму,
углерод, кремний, бор в элементарном состоянии или в виде сое-
динений. Расплавленная платина растворяет углерод, который
при охлаждении выделяется в виде микрочастиц графита.
Газообразная смесь хлора и окиси углерода превращает пла-
тину в [PtCOCl2].
664
ПОБОЧНАЯ ПОДГРУППА VIII ГРУППЫ
Поскольку нормальный потенциал системы Pt/Pt2+ при 25°
равен +1,2 в, платина растворяется в царской водке и в НС1 в при-
сутствии кислорода:
3Pt + 18НС1 + 4HN03 = 3H2[PtCle] + 4NO + 8Н2О
Pt + 6НС1 + О2 H2[PtCl6] + 2Н2О
Цианиды щелочных металлов действуют на платину в присут-
ствии паров воды выше 500—600°:
Pt + 4KCN + 2Н2О = K2[Pt(CN)4] + 2КОН + Н2
Поскольку платина реагирует с окислительно-щелочными
расплавами (КОН + KNO3, К2СО3 + KNO3, КОН + КС1О3,
NaOH + Na2O2), не рекомендуется плавление перекисей металлов
или окислительно-щелочных смесей в платиновых сосудах.
Сосуды (чашки, тигли и др.) из платины чистят механическим,
а затем химическим способом — кипячением в НС1 или HNO3
(но не с их смесью), либо расплавляя в них одно из соединений
Na2CO3, KHSO4, K2S2O7. После механической и химической
очистки сосуды из платины очень тщательно промывают горячей
дистиллированной водой, а затем прокаливают в окислительном
пламени.
ПРИМЕНЕНИЕ
Благодаря исключительной химической стойкости, высокой
температуре плавления, превосходным механическим и особым
каталитическим свойствам платина применяется в самых разнооб-
разных отраслях.
В химической промышленности распространены чашки, тигли,
ячейки, шпатели, сита, фильтры, катоды, перегонные аппараты
и другая аппаратура, изготовленная из платины, а металлическая
платина (порошкообразная или губчатая, а также в виде сит,
пластинок или в коллоидном состоянии) применяется в качестве
катализатора при получении азотной кислоты окислением аммиака,
серной кислоты контактным методом, при дегидрировании спирта
и в других реакциях. Каталитическая способность платины умень-
шается в присутствии металлов Al, Со, Bi и полностью исчезает
в присутствии Си, Zn, Ag, Sn, Fe. К веществам, отравляющим пла-
тиновые катализаторы, относятся окись углерода, цианистоводо-
родная кислота, иод, фосфор, мышьяк, сера и др.
В электротехнической промышленности платиновые сплавы
идут на изготовление электрических контактов, плавких предо-
хранителей для различных приборов, катодов, антикатодов для
рентегеновских трубок и катушек для электрических печей.
В современной пирометрии платина служит для изготовления
электрических термометров сопротивления и термоэлементов для
измерения температур в интервале между 1200 и 1600 °.
ПЛАТИНА
665-
Сплавы платина — иридий и платина — золото применяются'
в зубоврачебной технике и для изготовления шприцов.
Из сплава платины с 10% иридия был изготовлен международ-
ный эталонный метр, который находится на хранении в Междуна-
родном бюро мер и весов.
Платина и ее сплавы служат для изготовления украшений,
и оправы для драгоценных камней в ювелирных изделиях.
СОЕДИНЕНИЯ (ОБЩИЕ СВОЙСТВА)
Известно очень много устойчивых соединений двух- и четырех-
валентной платины и ограниченное число соединений одно-, трех-
и шестивалентной платины.
Благодаря склонности к комплексообразованию платина обра-
зует очень много координационных соединений.
В табл. 67 приведены формулы многих соединений платины,
сгрупированных по валентности.
Таблица 6Т
Соединения двух- валентной пла- тины PtO, PtF2, PtCl2, PtBr2, Ptl2, Pt(CN)2, PtS, H2[PtCl4], H2[Pt(CN)J, Mel[PtX4], Mei[PtX4]-raH2O (где Mei = = K+, NHJ, Na+, x/2Ca2+, 1/2Ва2+ и X = C1-, CN~, NO2), Mel[Pt(NH3)Cl3] (где Mel=K+, NHJ), [PtAm4]Cl2, [Pt(NH3)2Am2]Cl2 (где Am = NH3, NH2OH, N2H4), [Pt(NH3)3Cl]Cl, [PtAm2Cl2], [PtAmCl?]2 (где- Am = C2H4, C5H5N), H2[Pt(CN)4]-5H2O, Mei[Pt(CN)4]- • nH2O, MeiI[Pt(CN)4]-raH2O, MeII[Pt(CN)4], [Pt(C5H7O2)2], [Pt(NH3)4][PtCl4], [Pt(NH3)4[Pt(NH3)Cl3]2
Соединения трех- валентной пла- тины Pt2O3.raH2O, PtF3, PtCl3, PtBr3, Ptl3, Pt(CN)3, K2[PtCl5], K[PtCl4], [Pt{N(C2H5)3}4Cl]Cl2, [Pt{N(C2H6)3}2Cl3]
Соединения четы- рехвалентной платины PtO2-raH2O, PtF4, PtCl4, PtBr4, Ptl4, PtS2, Pt(SO4)2- • 4H?O, PtP2O7, H2[PtXe]-H2O (где X = C1“, Br~, I", OH-),Mei[PtXe] или Mei[PtX6]-raH2O (где Mei = K+, Na+, Rb+, Cs+, Li+, NHJ, Ag+ и X = C1", Br", I", CN-, SCN", OH", VaC-jOZ-), Mei[PtCl5OH], Mei[PtCl4(OH)2], Mei[PtCl3(OH)3], Mei[PtCl2(OH)4], Mei[PtCl(OH)5], Mei[PtO2X2], Mei[PtNH3Cl5], [PtAme]X4, [PtAm5X]X3, [PtAm4X2]X2, [PtAm3X3]X, [PtAm2X4]
Соединения шести- валентной пла- тины 3PtO3-H2O, PtF6
<366
ПОБОЧНАЯ ПОДГРУППА VIII ГРУППЫ
Для соединений платины характерна большая гамма расцве-
ток желтого, оранжевого, красного, коричневого, зеленого и чер-
ного. Разбавленные растворы солей двух- и четырехвалентной
платины окрашены в светло-желтый цвет, а концентрированные —
от красновато-желтого до коричнево-желтого.
В таблице приведены также некоторые алкильные производ-
ные платины.
Соединения двухвалентной платины
Известны многочисленные устойчивые соединения двухва-
лентной платины. Кроме простых получено множество коорди-
национных соединений платины(П) класса ацидосолей, амминов,
ацидоамминов и хелатов.
Окись платины, PtO, получают нагреванием губчатой пла-
тины при 450° в токе кислорода:
Pt -|- V2O2 = PtO 17 ккал
Окись PtO представляет собой серовато-черный кристалличе-
•ский порошок с плотностью 14,9 г/см3; она плохо растворима в воде,
растворяется в кислотах с образованием солей платины(П), раз-
лагается на элементы при нагревании выше 507°, восстанавливается
.до металла водородом при нагревании.
Гидроокись платины, Pt(OH)2, получают обработкой едким
кали раствора K2[PtCl4] в атмосфере газообразной СО2.
Соединение Pt(OH)2 представляет собой черный порошок,
который слабо растворим в воде, растворяется в концентрирован-
ных кислотах (HCl, HNO3, H2SO4), восстанавливается до металла
перекисью водорода и окисляется озоном или КМнО4 до окиси пла-
тины(1У) PtO2-2H2O.
Pt(OH)2 + 4НС1 = H2[PtCl4] + 2Н2О
Фторид платины, PtF2, получают нагреванием тонкой пла-
тиновой проволоки до 500—600° в токе газообразного фтора в труб-
ке из флюорита.
Фторид платины представляет собой зеленовато-желтое твер-
дое вещество, плохо растворимое в воде и разлагающееся на эле-
менты при нагревании до высокой температуры.
Хлорид платины, PtCl2, получают нагреванием губчатой пла-
тины в токе хлора (500°), нагреванием кислоты H2[PtCl4] при 240°
или прокаливанием хлорида платины(1У) до 350°.
Pt -|- С12 — PtCl2 + 29 ккал
Хлорид платины представляет собой оливково-зеленое твер-
дое вещество с плотностью 5,87 г/см3; он плохо растворим в воде,
выше 581° разлагается на элементы, растворяется в разб. НС1
ПЛАТИНА
667
с образованием H2[PtCl4), присоединяет окись углерода, давая
карбонильные соединения Pt(CO)Cl2, Pt(CO)2Cl2.
Известны тетрахлороплатинаты(П) Me2[PtCl4]-гаН2О, (где Mei = К+,
NH£, Na+, 1/2Са2+, 1/2Ва2+), хлороамминплатпнаты(П) MeI[Pt(NH3)Cl3]
(Me1 = К+, NHJ), комплексные хлориды типа [PtAm4]Cl2 (где Ат = NH3,
NH2OH, N2H4, C5H5N, CH3 - C6H4 - NH2, C2H5 - NH2, H2N - CS -
— NH2), [Pt(NH3)2Am2]Cl2 (где Am = N2H4, C5H5N), хлорид
[Pt(NH3)3Cl]Cl, а также координационные соединения типа неэлектролитов
общей формулы [PtAm2Cl2] (где Am = NH3, С2Н4, R3P, R3As, R2S, C6H5NC,
C4H9NC, a R = алкильная группа), [PtAmCl2]2 (где Am = C2H4, C5H5N).
Бромид платины, PtBr2, получают нагреванием соединений
PtBr4, H2[PtBr6] выше 180°:
PtBr4 3=* PtBr2 + Br2
он разлагается на эле-
Рис. 31. Структура соединения PtS.
Бромид платины представляет собой коричневое твердое веще-
ство с плотностью 6,65 г/см3; в
менты, плохо растворим в воде
и растворяется в НВг, КВт и
бромной воде.
Иодид платины, PtI2, полу-
чают нагреванием PtCl2 с раст-
вором иодида калия.
Черное твердое соединение
Ptl2 имеет плотность 6,4 г!см3,
разлагается на элементы наг-
реванием до 327° плохо раст-
воримо в воде и растворяется
в Ш.
Цианид платины, Pt(CN)2,
получают в виде желто-корич-
невого студнеобразного осадка обработкой раствора K2[Pt(CN)4]
• ЗН2О тетрахлороплатинатом(П) калия K2[PtCl4]:
K2[Pt(CN)4] + K2[PtCl4] = 2Pt(CN)2 + 4KC1
Известна кислота H2[Pt(CN)4] -5Н2О и несколько тетра-
цианоплатинатов(П): K2[Pt(CN)4]-ЗНгО, Ba[Pt(CN)4]-4Н2О,
Cu[Pt(CN)4], Hg[Pt(CN)4].
Сульфид платины, PtS, получают действием расплавленной
серы на порошкообразную платину под слоем буры, сплавлением
PtCl2 с серой и Na2CO3, нагреванием смеси платины с пиритом
(1200—1400°), пропусканием газообразного сероводорода через
раствор тетрахлороплатинатов или платино(П)-тетрахлористово-
дородной кислоты.
Сульфид платины представляет собой темно-зеленый кристал-
лический порошок с плотностью 8,847 г/см3-, он плохо растворим
в воде, устойчив к действию кислот и царской водки, разлагается
668
ПОБОЧНАЯ ПОДГРУППА VIII ГРУППЫ
на элементы при нагревании до высокой температуры, восстанав-
ливается до металла водородом при нагревании.
На рис. 31 изображена структура кристаллической решетки
сульфида платины(П), в которой каждый атом платины имеет
четыре копланарные связи и каждый атом серы тетраэдрически
координирован.
Координационные соединения
Известны многочисленные моноядерные и ограниченное число
полиядерных координационных соединений платины(П). Моно-
ядерные комплексы платины(П) относятся к классам ацидосоедине-
ний, амминов, ацидоамминов и хелатов.
1. Координационные соединения типа H2[PtXJ, H2[PtX4]-
•nH2O, Me*[PtX4], Me*[PtXJ-nH2O, Men[PtX4]-иН2О включают
ацидосоединения платины(П). Примеры: H2[PtCl4], H2[Pt(CN)4]«
•5Н2О, K2[PtCl4], (NH4)2[PtCl4], K2[Pt(CN)4] -3H2O, NaK[Pt(CN)4l •
•3H2O, Ca[Pt(CN)4] -5H2O, Ba[Pt(CN)4] -4H2O, K2[Pt(NO2)4] -2H2O,
K[PtC2H4Cl3].
Тетрахлороплатинатп^П') калия, K2[PtCl4], выпадает в осадок
при концентрировании раствора, полученного кипячением раст-
вора K2[PtCle] с К2С2О4-Н2О или N2H4:
K2[PtCle] + К2С2О4 = K2[PtCl4] + 2КС1 + 2СО2
2K2[PtCle] + N2H4 = 2K2[PtCl4] + 4HC1 + N2
Эти реакции восстановления рекомендуется проводить в ней-
тральной среде, поскольку в щелочной среде они протекают до
образования металлической платины.
Соединение K2[PtCl4] образует красные кристаллы, которые
под действием KNO2 превращаются в бесцветные K2[Pt(NO2)4] •
•2Н2О, а после обработки KCN переходят в желтое кристалличе-
ское вещество K2[Pt(CN)J -ЗН2О. Пропуская этилен Н2С = СН2
над K2[PtCl4], получают K[PtC2H4Cl3], который при обработке
аммиаком превращается в [Pt(NH3)(C2H4)Cl2].
Тетрахлороплатинат(П) аммония, (NH4)2[PtClJ, выпадает
в виде красных кристаллов при концентрировании раствора, полу-
ченного восстановлением (NH4)2[PtCle] оксалатом аммония
(NH4)2C2O4 в присутствии небольшого количества К2СО3.
(NH4)2[PtCle] + (NH4)2C2O4 = (NH4)2[PtCl4] + 2NH4C1 + 2CO2
Для восстановления (NH4)2[PtCle] до (NH4)2[PtCl4] можно
также использовать в качестве восстановителя гидразин, серни-
стую кислоту и хлорид меди(1), поскольку окислительно-восста-
новительный потенциал уравнения
[PtCle]2- + 2е- [PtCl4]2- + 2С1-
равен-]-0,72 в.
ПЛАТИНА
669
Платино(И)-тетрацианистоводородная кислота, H2[Pt(CN)4]-
•ЗН2О, выделяется в виде красных кристаллов из раствора, полу-
ченного при обработке Ba[Pt(CN)4] *4Н2О серной кислотой или
при пропускании сероводорода через растворы солей Cu[Pt(CN)4],
Hg[Pt(CN)4J.
Тетрацианоплатинат(11) калия, K2[Pt(CN)4] -ЗН2О, выпадает
в виде желтых кристаллов из раствора, полученного действием
K.CN на раствор K2[PtCl4] или обработкой теплой водой остывше-
го продукта, образующегося при прокаливании губчатой метал-
лической платины с K4[Fe(CN)6] -ЗН2О.
Тетрацианоплатинат(П) бария, Ba[Pt(CN)]4]-4Н2О, пред-
ставляет собой зеленовато-желтые кристаллы, которые светятся
под действием рентгеновских и канальных лучей, а также в ре-
зультате радиоактивного облучения; на этом свойстве основано
их применение для флюоресцирующих экранов.
Тетранитроплатинат(11) калия, K2[Pt(NO2)J-2Н2О, выде-
ляется в виде бесцветных кристаллов при упаривании растворов,
полученных кипячением 45—50%-ного раствора KNO2 с K2[PtCl0]
или K2[PtCl4l.
K2[PtCle] + 6KNO2 = K2[Pt(NO2)4] + 2NO2 + 6KC1
K2[PtCl4] + 4KNO2 = K2[Pt(NO2)4] + 4KC1
2. Координационные соединения типа [PtAm4]X2 (где Am =
= NH3, NH2OH, N2H4, C5H5N, CH3 - C6H4 - NH2, C2H5 - NH2,
H2N - CS - NH2, H2N - CH2 - CH2 - NH2 и X = C1-, NO",
NOj, OH-, включают тетраммины платины(П).
Хлорид тетрамминплатины(11), [Pt(NH3)4]Cl2 (/ основание
Рейве), представляет собой бесцветные кристаллы. Его полу-
чают действием аммиака на ^мс-дихлородиамминплатину(П)
[Pt(NH3)2Cl2] (соль Пейроне) или обработкой (NH4)2[PtCl4] хло-
ридом аммония:
[Pt(NH3)2Cl2] + 2NH3 = [Pt(NH3)4]Cl2
(NH4)2[PtCl4] + 2NH4C1 = [Pt(NHs)4]Cl2 + 4HC1
Хлорид тетрагидроксиламинплатины(И), [Pt(NH2OH)4]Cl2,
•образует бесцветные кристаллы. Его получают обработкой
K2[PtCl4] избытком солянокислого гидроксиламина NH2OH-HC1
или обработкой основания Александера [Pt(NH2OH)J(OH)2 раз-
бавленной соляной кислотой:
K2[PtCl4] 4- 4NH2OH-HC1 = [Pt(NH2OH)4]Cl2 + 2КС1 + 4НС1
[Pt(NH2OH)4](OH)2 + 2HC1 = [Pt(NH2OH)4]Cl2 + 2H2O
3. Координационные соединения типа [Pt(NH3)2Am2]Cl2 (где
Am = N2H4, C5H5N) включают хлориды диамминдигидразин-
<(или дипиридин-)платины(П) и их стереоизомеры.
670
ПОБОЧНАЯ ПОДГРУППА VIII ГРУППЫ
4. Координационные соединения типа MeI[Pt(NH3)Cl3] (где
Me1 = К+, NH4) охватывают ацидоамминплатинаты(П), напри-
мер оранжевые соли Косса K[Pt(NH3)Cl3], NH4[Pt(NH3)Cl3],
5. Координационные соединения типа [PtAm3X]X (где X = С1-),
например соль Клеве [Pt(NH3)3Cl], включают галогениды монога-
логенотриамминплатины(П).
6. Координационные соединения типа [PtAm2X2] (где Ат =
= NH3, С2Н4, C6H5NC, C4H9NC, R3P, R3As, R2S, a R = алкильная
группа; X = Cl-, Вг-, I-, SCN-, NOj, 1/гС2О4-) и [PtAmCl2]2 (где
Am = C2H4, C5H5N), как и ацетилацетонат платины(П), явля-
ются комплексными соединениями неэлектролитного типа.
цис-Д ихлородиамминплатина(11) (солъПейронё), [Pt(NH3)2Cl2],
образуется в виде кристаллов интенсивно-желтого цвета
в результате обработки насыщенного раствора (NH4)2[PtClJ
18%-ным аммиаком или при обработке K2[PtCl4] 20%-ным раство-
ром NH4CH3COO:
(NH4)2[PtCl4] + 2NH3 = [Pt(NH3)2Cl2] + 2NH4C1
K2[PtCl4] 4- 2NH4CH3COO = [Pt(NH3)2Cl2] + 2CH3COOH + 2KC1
транс-Дихлор одиамминплатина{1 Г) {II основание Рейве),
[Pt(NH3)2Cl2],— соединение светло-желтого цвета, растворимость
которого меньше растворимости ^мс-изомера, образуется в ре-
зультате обработки [Pt(NH3)4]Cl2 концентрированной соляной кис-
лотой при нагревании:
[Pt(NH3)4]Cl2 + 2НС1 = [Pt(NH3)2Cl2] + 2NH4C1
Дихлородигидразинплатина{11), [Pt(N2H4)2Cl2],— белое веще-
ство, устойчивое на воздухе; можно получить действием безвод-
ного гидразина на суспензию (NH4)2[PtCle] в метаноле:
2(NH4)2[PtCle] + 7N2H4 = 2[Pt(N2H4)2Cl2] + 4NH4C1 + 2N2H4-2HC1 + N2
Динитродигидразинплатина{11), [Pt(N2H4)2(NO2)2], получа-
ется при обработке K2[Pt(NO2)4 гидразингидратом; представляет
собой бесцветные кристаллы, плохо растворимые в холодной воде
и разлагающиеся со взрывом при нагревании.
Димер,
- С1 С1 С1 -
\pt^ /pt\
_Н4С2 Cl с2н4..
, представляет
собой оранжевые кристаллы, которые растворяются в хлоро-
форме и эфире, разлагаются при 125 —130° и превращаются
в [Pt(C5H5N)2Cl2] под действием избытка пиридина.
Ацетилацетонат платины(П),
/СН3\ "1
/О=С< 1
\о-с^СН I
'СНз'2-»
ПЛАТИНА
67i
в виде желтых кристаллов получают обработкой соединения
K2[PtCl4] ацетилацетоном в присутствии щелочей.
Гликоколятп платины в yuc-форме представляет собой бесцветные
игольчатые кристаллы, а в транс-форме — бесцветные пластинки.
0 = С —0ч О —С = 0"]
I >pt\ I
H2C-Nz xn—сн2
Н2 н2 J
^ис-Форма
Н2
Г0 = С — О N —СН2 -]
I >pt\ I
Н2С —Nz х0-С = 0
L ' н2 J
транс-Форма.
7. Диядерные координационные соединения.
Тетрахлороплатинат{11) тетрамминплатины(Н) {соль Маг-
нуса), [Pt(NH3)4][PtClJ, получают обработкой раствора
[Pt(NH3)4]Cl2 10%-ным раствором K2[PtCl4]:
[Pt(NH3)4]Cl2 + K2[PtCl4] = [Pt(NH3)4][PtCl4] + 2KC1
Соединение [Pt(NH3)4][PtCl4] образует темно-зеленые кри-
сталлы, плохо растворимые в воде, спирте и,соляной кислоте.
Трихлороамминплатинат(1Г) тетрамминплатины(17),
[Pt(NH3)4][Pt(NH3)Cl3]2, выделяется в виде оранжево-желтого-
твердого вещества.
Соединения трехвалентной платины
Известно небольшое число соединений трехвалентной платины,
причем все они относительно неустойчивы. В качестве примеров
соединений платины(1П) можно назвать Pt2O3-nH2O, PtF3, PtCl3,
PtBr3, Ptl3, Pt(CN)3, K2[PtCl5], K[PtCl4], [Pt{N(C2H5)3}4Cl]Cl2,
[Pt{N(C2H5)3}2Cl3] и др.
Гидратированная окись платины, Pt2O3-nH2O, образуется
в виде темно-коричневого осадка при обработке раствора PtCl3.
раствором карбоната натрия.
Хлорид платины, PtCl3, получают нагреванием (390°) PtCl4
в токе газообразного хлора.
Хлорид платины представляет собой темно-зеленое твердое
вещество с плотностью 5,256 а/сж3; при нагревании выше 435°
он разлагается на элементы, растворяется в воде и взаимодействует
с соляной кислотой при нагревании по уравнению
2PtCl3 + 4НС1 = H2[PtCl4] + H2[PtCle]
Бромид платины, PtBr3, представляет собой темно-зеленое
твердое вещество, плохо растворимое в воде; при нагревании
до 450° разлагается на элементы.
Иодид платины, Ptl3,— черное твердое вещество, по внешнему
виду напоминающее графит; разлагается при нагревании до 270°,
плохо растворяется в воде.
<672
ПОБОЧНАЯ ПОДГРУППА VIII ГРУППЫ
Сульфид платины, Pt2S3, представляет собой серый порошок
с плотностью 5,52 г/см3, плохо растворимый в воде, кислотах и цар-
ской водке, разлагающийся на элементы выше 225°.
Соединения четырехвалентной платины
Известны многочисленные устойчивые соединения четырехва-
лентной платины.
В качестве примеров соединений платины(1У) можно назвать
PtO2-nH2O, PtF4, PtCl4, PtBr4, Ptl4, PtS2, Pt(SO4)2-4H2O, PtP2O7
и многочисленные ацидосоединения, ацидодиоксосоединения, ам-
мины и ацидоаммины.
Гидратированная окись платины, PtO2-3H2O, получается
кипячением водного раствора PtCl4 с едким натром.
Соединение PtO2*3H2O представляет собой желтый порошок,
плохо растворимый в воде; обладает амфотерными свойствами,
^растворяется в соляной кислоте с образованием H2[PtCle]-6Н2О
или в щелочах с образованием гексагидроксоплатинатов(1У)
Me|[Pt(OH)6] (где Me1 — Na+, К+) и ступенчато дегидратируется,
превращаясь при 400° в PtO:
конц. H2SO4 100° 400°
PtO2-3H2O ---—PtO2.2H2O —PtO2-H2O —Pto-p/2o2
Желтая — H2O Коричневая —н2о Н2о Черная
Подкисление H2[Pt.(OH)6] уксусной кислотой вызывает обра-
зование коричневого осадка PtO2 *2Н2О.
При действии кислорода на нагретую до ~350° металлическую
платину получается черный порошок PtO2, который плохо раст-
ворим в воде и кислотах и разлагается на элементы при нагрева-
нии до 450°.
Фторид платины, PtF4, получают пропусканием газообразного
фтора над нагретой докрасна металлической платиной. Он пред-
-ставляет собой красновато-желтые кристаллы, которые гидроли-
зуются водой, плавятся при 627° и разлагаются на элементы
выше 700 — 800°.
Хлорид платины, PtCl4, образуется путем хлорирования
металлической платины или нагревания (360°) кислоты H2[PtCle]
в атмосфере хлора.
Хлорид платины представляет собой гигроскопичные красно-
вато-коричневые кристаллы с плотностью 2,43 г/сж3; он легко
растворим в воде, разлагается на элементы при высокой темпе-
ратуре и на PtCl2 и хлор — при нагревании до 370°.
Нагревание PtCl4 в атмосфере водорода приводит к образованию
металлической платины, а действие сероводорода на PtCl4 при
температуре ниже 100°— к образованию PtS2. Из водных раство-
ров PtCl4 выпадают кристаллы H2[Pt(OH)2ClJ.
ПЛАТИНА
673
Известны кристаллогидраты PtCl4-nH2O (п — 1, 4, 5, 7).
Бромид платины, PtBr4, образуется при растворении платины
в бромистоводородной кислоте, содержащей элементарный бром,
или при нагревании H2[PtBr6] до 180°.
Бромид платины представляет собой темно-коричневый поро-
шок с плотностью 5,69 г/см3; он растворим в воде, спирте, эфире,
бромистоводородной кислоте и разлагается на элементы при силь-
ном нагревании.
Растворение PtBr4 в воде дает кислоту H2[Pt(OH)2Br4J.
Иодид платины, Ptl4, получают прямым взаимодействием
элементов в запаянной трубке или путем обработки концентри-
рованного раствора H2[PtCle] горячим раствором_К1:
H2[PtCl6] + 4KI = Ptl4 + 4КС1 + 2НС1
Иодид платины (IV) представляет собой темно-коричневый
порошок с плотностью 6,064 г/см3; он разлагается на элементы
при нагревании до 370° и растворяется в HI с образованием
H2[PtI6l.
Сульфид платины, PtS2, получают прокаливанием соединения
(NH4)2[PtCl6] с серой без доступа воздуха или пропусканием серо-
водорода через водный раствор PtCl4, H2[PtCl6] или гексахлоро-
платинатов(1У)Ме2[Р1С1в1, Me^[PtGl6l -пН20.
Сульфид платины представляет собой черно-коричневый поро-
шок с плотностью 7,22 г/см3; он плохо растворим в воде и сульфи-
дах щелочных металлов, растворяется в конц. HNO3, царской
водке и полисульфидах щелочных металлов, разлагается на PtS
и серу при нагревании.
Сульфат платины, Pt(SO4)2-4H2O, образует желтые пластин-
чатые кристаллы, растворимые в воде, спирте, эфире и кислотах.
Пирофосфат платины, PtP2O7, представляет собой желтовато-
зеленое твердое вещество с плотностью 4,85 г/см3, разлагающееся
выше 600°.
Координационные соединения
Известны многочисленные моноядерные координационные сое-
динения платины(1У), относящиеся к ацидосоединениям ацидо-
диоксосоединениям, амминам и ацидоамминам.
Число координационных соединений платины(1У) очень велико,
поскольку у многих комплексов встречаются изомерные формы.
В качестве примеров комплексов класса ацидосоединений упо-
минаются гексацидокислоты H2[PtX6l-иН2О (где X = С1-, Вг-,
I-, ОН-) и гексацидосоли Me*[PtX6] или Me*[PtX6]-иН2О (где
Me1 = К+, Na+, Rb+, Cs+, Li+, NH*, Ag+ и X = Cl-, Br-, I-,
CN-, SCN-, ОН-, ЧгСгО^").
43-101
674
ПОБОЧНАЯ ПОДГРУППА VIII ГРУППЫ
Платинохлористоводородная кислота, H2[PtCl6] -бНгО, осаж-
дается при упаривании раствора, полученного растворением губ-
чатой платины (или платиновой черни) в царской водке, раство-
рением PtCl4 или PtO2-nH2O в соляной кислоте, обработкой губ-
чатой платины соляной кислотой, содержащей хлор, и электроли-
зом раствора НС1 с использованием платинового анода.
Соединение Н2[PtCle] -6Н2О представляет собой расплываю-
щиеся на воздухе коричнево-красные кристаллы, легко раствори-
мые в воде, спирте и эфире.
Хлорид олова(П), сульфат железа(П), окись углерода, метал-
лический цинк и др. восстанавливают платинохлористоводород-
ную кислоту до металлической платины.
Гексахлороплатинат(ГУ) натрия, Ха2[Р1С18] -бНгО, выпадает
в осадок при упаривании раствора, полученного нейтрализацией
15—20%-ного раствора H2[PtCl8]-6Н2О насыщенным раствором
карбоната натрия.
Соединение Na2[PtCl6]-6Н2О образует оранжево-красные кри-
сталлы, легко растворимые в воде.
Гексахлороплатинат(1У) калия, K2[PtCl8], получают обра-
боткой 15—20%-ного раствора H2[PtCl8]-6Н2О 1—12%-ным рас-
твором КС1, подкисленным НС1.
Соединение K2[PtCl6] образуется в виде желтых кристаллов,
плохо растворимых в холодной воде, спирте и эфире. В кристалли-
ческой решетке K2[PtCl8l шесть атомов хлора сгруппированы во-
круг атома платины, с которым они связаны октаэдрически.
Охлаждением раствора, полученного кипячением K2[PtCl8J
с одним из растворов КВг, KI, KSCN, выделяют кристаллы
K2[PtBr6], K2[PtIe], K2[Pt(SCN)8] соответственно красного, чер-
ного и красного цвета.
Гексахлороплатинат{1У) аммония, (NH4)2[PtCl8], получают
обработкой 15—20%-ного раствора H2[PtCl8]-6Н2О 12—15%-ным
раствором NH4C1.
Соединение (NH4)2[PtCl8] образуется в виде желтых кристаллов,
которые растворяются в холодной воде, спирте и эфире и разла-
гаются при прокаливании по уравнению
(NH4)2[PtCle] Pt + 2NH4C1 + 2С12
Гексагидроксоплатинат(1У) водорода, H2[Pt(OH)8], получают
действием перекиси водорода на основание Александера
[Pt(NH2OH)4](OH)2:
[Pt(NH2OH)4](OH)2 +'k3H2O2 = H2[Pt(OH)e] + 2N2 + 6H2O
Соединение H2[Pt(OH)e] имеет вид желтых игольчатых кри-
сталлов: оно плохо растворяется в воде, теряет молекулу воды
при 100° и три молекулы воды при 120°.
ПЛАТИНА
675
Гексагидроксоплатинат ы(1У), Me*[Pt(OH)6], по-
лучают обработкой гексахлороплатинатов щелочами или раство-
рением PtO2-3H2O в щелочах:
K2[PtCle] + 6К0Н = K2[Pt(OH)e] + 6КС1
Известны хлорогидроксоплатинаты типов Me^[PtCl5OH]r
MeJ[PtCl4(OH)2],MeJ[PtCl3(OH)3],MeJ[PtCl2(OH)4],Me|[PtCl(OH)5],
диацидодиоксоплатинаты Me^[PtO2Cl2] и ацидоамминплатинаты
Me4PtNH3Cl5] (где Me1 = К+, Na+, NH+, и X = СГ, Вг").
Имеются сообщения о координационных соединениях класса
амминов [PtAm6]X4 и ацидоамминов [PtAm5X]X3, [PtAm4X2]X2,
[PtAm3X3]X, [PtAm2X4], например [Pt(NH3)6]Cl4, [Pt(NH3)5Cl]Cl3,
[Pt(NH3)4Cl2]Cl2, [PtEnNH3NO2Cl2]X, [PtEnNH3ClBrNO2]X,
[Pt(NH3)2Cl4].
Соединения шестивалентной платины
Известно ограниченное число соединений шестивалентной пла-
тины, причем все они устойчивы только в определенных условиях.
Окись платины, ЗРЮ3-Н2О, образуется йа аноде при элект-
ролизе холодного раствора PtO2*nH2O в 2 н. растворе КОН.
Соединение 3PtO3«H2O представляет собой коричнево-красный
порошок, растворимый в НС1, HNO3, H2SO4; оно относительно
неустойчиво и разлагается на окись платины(1У) с выделением кис-
лорода.
Фторид платины, PtF6, представляет собой темно-красные
кристаллы с т. пл. 56,7°.
Алкильные производные платины
Иодид триметилплатины, (CH3)3PtI, получают обработкой
PtCl4 метилмагнийиодидом:
PtCli + 3(CH3)MgI = (СНз)зРН + 2MgCl2 + Mgl2
Соединение (CH3)3PtI образует оранжевые кристаллы; оно ра-
створяется в воде и бензоле, разлагается при 250°, не взаимодей-
ствует на холоду с минеральными кислотами, превращается:
в (CH3)3PtOH под действием окиси Ag2O, будучи растворенным
в бензоле, поглощает аммиак, образуя (CH3)3Pt(NH3)2I, реаги-
рует с ацетилацетонатом таллия(1), давая ацетилацетонат три-
метилплатины.
Тетраметилплатина, (CH3)4Pt, образуется при обработке1
(CH3)3PtI метилнатрием; это светло-желтое твердое вещество,
которое растворяется в горячем бензоле и разлагается при высо-
кой температуре.
Гексаметилплатина, (CH3)3Pt — Pt(CH3)3, представляет собой
светло-желтое твердое вещество, которое растворяется в бензоле
и превращается в (CH3)3PtI под действием иода.
43»
ПОБОЧНАЯ ПОДГРУППА I ГРУППЫ
ПЕРИОДИЧЕСКОЙ СИСТЕМЫ
(ПОДГРУППА МЕДИ)
ХАРАКТЕРИСТИКА ПОДГРУППЫ
В побочной подгруппе I группы периодической системы (в под-
группе меди) находятся переходные металлы медь (Си), серебро
(Ag) и золото (Ап).
В табл. 68 показана электронная структура атомов меди,
серебра и золота.
Атомы этих элементов имеют по одному электрону на послед-
нем электронном уровне и по 18 электронов на предпоследнем
электронном уровне в отличие от щелочных металлов, которые
на предпоследнем электронном уровне имеют по 8 электронов
(за исключением лития, у которого 2 электрона).
В химических реакциях атомы металлов подгруппы меди могут
терять как валентные электроны, так и электроны предпослед-
него электронного уровня. При этом образуются соединения,
в которых медь может иметь валентность I, II или III, серебро —
I, реже II или III, золото — I или III и очень редко II. В отличие
от ионов щелочных металлов (которые всегда электроположитель-
ны, одновалентны и имеют устойчивую конфигурацию инертного
газа), ионы металлов подгруппы меди могут проявлять перемен-
ную валентность и не обнаруживают устойчивой конфигурации.
ПОБОЧНАЯ ПОДГРУППА I ГРУППЫ
677
В табл. 69 приведены наиболее важные константы металлов
побочной подгруппы I группы периодической системы.
Металлы подгруппы меди окрашены: медь в медно-красный,
серебро в серебристо-белый, золото в золотисто-желтый цвет; они
обладают гранецентрированной кубической решеткой, имеют
небольшие значения ионных и атомных радиусов, являются тяже-
лыми металлами; это металлы с высокой плотностью (от 8,96 до
19,3 г/см3 при 20°) и малым значением атомных объемов (от 7,1
до 10,23 см31г-атпом при 20°).
Медь, серебро и золото — вязкие, ковкие и тягучие металлы
(легко протягиваются, прокатываются и штампуются); имеют
твердость ~2,5 по шкале Мооса, плавятся при температуре около
1000е, характеризуются низкой летучестью, кипят в области тем-
ператур 2177—2707?. Элементы подгруппы меди имеют высокую
электро- и теплопроводность, они диамагнитны, легко образуют
сплавы как между собой, так и с другими химическими элемента-
ми, для них характерно небольшое число природных изотопов.
С химической точки зрения металлы подгруппы меди устой-
чивы, относительно инертны (в ряду напряж'ений они расположе-
ны после водорода). Их химическая активность уменьшается от
меди к золоту, а потенциалы ионизации находятся между 7,72
и 9,23 эв (потенциалы ионизации щелочных металлов значитель-
но меньше).
С кислородом непосредственно взаимодействует только медь,
с серой — медь и серебро, с галогенами — все три металла (медь
при комнатной температуре, золото при нагревании). Медь
и серебро растворяются в разб. HNO3 или конц. H2SO4 при нагре-
вании, а золото — в царской водке, в хлорной воде и в безводной
H2SeO4 при нагревании.
Устойчивые соединения металлов подгруппы меди соответ-
ствуют двухвалентной меди, одновалентному серебру и трех-
и одно-валентному золоту. Многочисленные соединения металлов
подгруппы меди легко восстанавливаются до металла.
Большинство солей, содержащих ионы Сп+ и Ag+, плохо раст-
воримо в воде и неустойчиво во влажном состоянии. Некоторые
соединения меди(1) легко окисляются, а ряд соединений сереб-
ра(1) легко восстанавливается.
Растворимые соединения меди, серебра и золота токсичны.
Галогениды и псевдогалогениды меди, серебра и золота, соответ-
ствующие степени окисления I, растворяются в разбавленном
аммиаке и концентрированных растворах галогеноводородов или
их солей с образованием координационных соединений.
По физико-химическим свойствам металлы подгруппы меди
и щелочные металлы (которые обладают большим атомным объе-
мом, малой плотностью и являются мягкими, легко летучими
и высоко активными металлами) и их соединения существенно раз-
Таблица 69
Элемент Медь Си Серебро Ag Золото Au
Цвет Медно-красный Серебристо-белый Золотисто-желтый
Кристаллическая структура Гранецентрирован- ная кубическая Гранецентрирован- ная кубическая Гранецентрирован- ная кубическая
Атомный номер 29 47 79
Атомный вес 65,542 107,870 196,967
Атомный радиус, А (для координационного числа (12) 1,28 1,44 1,44
Ионные радиусы, А (по Гольдшмидту, Полингу и Аренсу) Си+ 1,0; 0,96; 0,96 Си2+ —; —; 0,72 Ag+1,13; 1,16; 1,26 Аи+ -; 1,37; 1,37 Аи3+-; о,85
Атомный объем (при 20°), см3/г-атом 7,1 10,27 10,23
Плотность (при 20°), г/см3 8,96 10,50 19,3
Твердость по Бринеллю, кг/лслс2 37,4-42,0 25 18,5
Твердость по шкале Мооса 2,5-3 2,5-2,7 2,5
Температура плавления, °C 1083 960,5 1063,4
Скрытая теплота плавления, кал/град 42 21,07 15,8
Температура кипения, °C 2582 2177 2707
1
Удельная теплоемкость (при 20 °C), кал)г-град 0,092 0,056 0,031
Коэффициент теплопроводности Л, кал-слг1 -сек-1- • град~х (при 0°) 0,94 1,0 0,75
Сопротивление р-108 (при 0°), ом-см 1,692 1,47 2,44
Электропроводность (Hg=l) 55,6 63,9 38,5
Магнитная восприимчивость Xs-10-e, эл.-магн. ед. (при 18°) —0,086 -0,20 -0,15
Теплота перехода атомов в газообразное состоя- ние, ккал (при 25°) 81,52 69,12 82,29
Потенциал ионизации, эв Me —* Ме+ + е~ Ме+—>- Ме2+-|-е~ Ме2+—>• Ме3+ + е~ 7,72 20,29 36,83 7,57 21,48 34,82 9,22 20,5 30,46
Потенциал ионизации, ккал/г-атом Me —»- Ме+ 178,0 174,7 212,7
Нормальные потенциалы (при 25°), в Cu/Cu+ -|-0,522 Cu/Cu2+ + 0,346 Ag/Ag+ 4 0,799 Au/Au+ 41,7 Au/Au3+ |-1,5
Нормальные потенциалы окислительно-восстано- вительных систем, в Cu+/Cu2+ 4 0,153 Au+/Au3+-| 1,29
Валентность I, П, (П1) I, (И), (Ш) I, Hl
Массовые числа природных изотопов 63,65 107, 109 197
Распространенность элементов в земной коре, вес. % 0,01 1,0-10-5 5,0-10-’
680
ПОБОЧНАЯ ПОДГРУППА I ГРУППЫ
личаются. Поскольку в изменении свойств элементов подгруппы
меди наблюдается вторичная периодичность (что не имеет места
в случае щелочных металлов), серебро нельзя рассматривать как
элемент, промежуточный между медью и золотом. В электрохимиче-
ском ряду металлы подгруппы меди расположены среди элементов
с пониженной активностью, а щелочные металлы — среди наибо-
лее активных.
В отличие от щелочных металлов элементы подгруппы меди
значительно труднее теряют электрон последнего электронного
уровня (и, следовательно, труднее окисляются), образуют ионы
различной валентности, причем последние окрашены (даже если
молекула содержит бесцветный анион). Все эти элементы способны
образовывать координационные соединения, большинство которых
легко растворимо в воде.
Элементы подгруппы меди высших степеней окисления обра-
зуют окрашенные соединения, поскольку при образовании этих
катионов появляются незаполненные й-орбитали.
Соединения металлов подгруппы меди и щелочных металлов
различаются устойчивостью, растворимостью, окраской, магнит-
ными свойствами и химическим характером. Поскольку медь,
серебро и золото — слабо электроположительные элементы, они
образуют соединения кислотного характера в отличие от щелочных
металлов.
Окислы меди, серебра и золота плохо растворимы в воде, обла-
дают слабо выраженными основными свойствами, с водой не вза-
имодействуют, окрашены, очень легко восстанавливаются, в то
время как окислы щелочных металлов белого цвета, хорошо раст-
воримы в воде (с образованием сильных оснований), с трудом вос-
станавливаются. Все галогениды щелочных металлов легко рас-
творимы в воде, в то время как некоторые галогениды металлов
подгруппы меди в воде практически нерастворимы. Сульфиды меди,
серебра и золота окрашены и плохо растворимы в воде, в то время
как сульфиды щелочных металлов частично гидролизуются водой
с образованием щелочей и выделением сероводорода.
В отличие от щелочных металлов элементы подгруппы меди
встречаются в природе в самородном состоянии; медь и серебро'
в виде сульфидов, вместе с сульфидами других металлов (Pb, Zn
и др.), а золото в виде теллурида.
По отношению к металлам VIII группы и побочной подгруппы
II группы (между которыми они расположены в периодической
системе элементов) металлы подгруппы меди обладают средними
значениями в ряду физико-химических свойств (атомный радиус,
атомный объем, плотность, твердость, температура плавления,
температура кипения, химическая активность и др.), и наблюдает-
ся сходство по горизонтали, т. е. Ni — Си — Zn, Pd — Ag — Cd,
Pt — Au — Hg.
МЕДЬ
681
МЕДЬ Си
Z = 29; ат. вес = 63,542
Валентность I, II, (III)
Заряд 1+, 2+, 3+
Массовые числа природных изотопов 63 и 65
Электронная структура атома меди: К -L -М -4s1.
Электронная структура атома меди и катионов Си+ и Си2
для 3d- и 4$-орбиталей:
3dio 4S1 3d 10 4s 3<Z9 4s
|п|Ж1Ш1 0 ItllnlWWl □ |N|U|H|n| 11 □
Cu Cu+ Cu2+
ИСТОРИЧЕСКАЯ СПРАВКА
Медь была известна еще в древние времена: о ней упоминается
и в «Илиаде» и старинных персидских рукописях. В Египте, Асси-
рии, Финикии и на Американском материке были найдены изде-
лия из меди, «возраст» которых превышает 6000 лет. Самые древ-
ние предметы были изготовлены из почти чистой меди, а несколько'
позднее появляются изделия из бронзы (сплав медь — олово) —
наступает бронзовый век, из которого до наших дней дошли мно-
гие изделия из этого металла.
Название «медь» было дано в честь острова Кипр, где в древно-
сти и было развито производство медных изделий. Богатые залежи
меди привлекали внимание к этому острову, и Кипр последова-
тельно завоевывался египтянами, ассирийцами, финикиянами,
греками, персами и римлянами.
В Азии были найдены остатки медных копей.
РАСПРОСТРАНЕННОСТЬ В ПРИРОДЕ
В природе медь находится в самородном состоянии, а также
в виде соединений (главным образом сульфидов) в различных
полиметаллических минералах.
Самородная медь образовывалась в результате различных гео-
логических процессов, путем восстановления таких соединений,
как сульфиды, куприт и малахит. В качестве примесей самородная
медь содержит окись Си2О, иногда железо, свинец, серебро, реже
ртуть или золото. Для металлургической промышленности само-
родная медь не представляет интереса, поскольку она редко встре-
чается в природе.
Залежи самородной меди имеются в СССР, США, Румынии.
К наиболее важным минералам меди относятся следующие.
Халькозин-, Cu2S, содержащий 79,8% меди; встречается в виде
компактных масс, состоящих из серовато-черных бипирамидаль-
682
ПОБОЧНАЯ ПОДГРУППА I ГРУППЫ
ных призматических кристаллов с плотностью 5,7 г!см5 и твердо-
стью 2—3 по шкале Мооса.
Халькопирит, CuFeS2, содержащий 34,57% меди, встречается
в виде желтых (как латунь) тетраэдрических кристаллов с плотно-
стью 4,2 г!см? и твердостью 3—4 по шкале Мооса.
Борнит, 3Cu2S -FeS2 -FeS, содержащий 63,3% меди, встречает-
ся в виде хрупких компактных масс медно-красного цвета. Его
плотность равна 5 г/см?, твердость 3 по шкале Мооса.
Ковелин, CuS, содержащий 66,5% меди, встречается в виде
мелких пластинчатых кристаллов, окрашенных в индиго-синий
цвет, с плотностью 4,6 г!см? и твердостью 1,5—2 по шкале Мооса.
Куприт, Сп2О, содержащий 88,8% меди, встречается в виде
серовато-красных кубических кристаллов с плотностью 5,85—
6,16 г/см? и твердостью 3,5—5 по шкале Мооса.
Мелаконит (или тенорит), СпО, черного цвета, часто встре-
чается вместе с купритом в железных и марганцевых рудах.
Малахит, Си2(СО3)(ОН)2 или СиСО3-Си(ОН)2, содержащий
57,4% меди, встречается в виде зеленых сталактитных масс с плот-
ностью 3,9—4,1 г/см? и твердостью 3,5—4 по шкале Мооса.
Азурит, Сп3(СО3)2(ОН)2 или 2СпСО3 >Си(ОН)2, содержащий
55,3% меди, встречается в виде темно-синих игольчатых призма-
тических кристаллов с плотностью 3,7—3,9 г! см? и твердостью
3,5—4 по шкале Мооса.
Хризоколл, CuSiO3-2H2O, встречается в виде типичного геля
светло-синего цвета с плотностью 2—2,3 г/см? и твердостью 2—4
по шкале Мооса.
Помимо этих минералов известны также кубанит CuFe2S3, теннантит
Cu3AsS3 или 3Cu2S-As2S3, тетраэдрит Cu3SbS3 или 3Cu2S-Sb2S3, энаргит
Cu3AsS4 или 3Cu2S-As2S5, халькостибит CuSbS2, атакамит СпС12 •ЗСп(ОН)2,
катангит 5CuSiO3-Сп(ОН)2, халкантит CuSO4-5H2O, оливенит, туркоаза,
халькосидерит, тиролит, халькофиллит.
В природе большинство соединений меди встречается вместе
с соединениями других металлов, образуя полиметаллические
руды, которые кроме ценной части содержат пустую породу кис-
лого (редко щелочного) характера. В среднем содержание меди
в рудах составляет 1—2 вес.%.
Важные залежи медной руды находятся в СССР, США, Югосла-
вии, Испании, Португалии, ГДР, Конго и Румынии.
Медь относительно широко распространена в природе, ее содер-
жание в земной коре равно 0,01 вес.%.
Методом спектрального анализа было доказано наличие меди
в спектрах Солнца и многих звезд. В метеоритах содержание меди
велико — примерно в 16 раз больше, чем в земной коре.
Соединения меди в следовых количествах были обнаружены
в большинстве растений и в крови некоторых моллюсков, кара-
МЕДЬ
683
катиц, устриц, ракообразных и др. В частности, гемоцианин —
это белок, в состав которого входит медь и который является
синим дыхательным пигментом крови упомянутых существ.
Загрузка
Удаление
nopodoi
Рис. 32. Флотационная ячейка с
перемешиванием со сжатым возду-
хом.
ПЕРЕРАБОТКА МЕДНЫХ РУД
Почти вся мировая добыча металлической меди в настоящее
время основывается на переработке сульфидных, окисных и кар-
бонатных медных руд. Поскольку содержание меди в рудах мало
(в среднем примерно 1—2%), руды измельчают (в щековых дро-
билках, пневматическими молотками и др.), размалывают (в мель-
ницах различного типа), обогащают с помощью гравитационных
(основанных на разностях плотностей руд и пустой породы)
и флотационных методов. Изме-
льченную и фракционирован-
ную по размерам частиц мед-
ную руду промывают водой,
при этом пустая порода в
значительной части удаляется.
Обогащение руд методом
флотации осуществляется на
флотационных установках и ос-
новывается на разнице свойств
поверхности частиц сульфидов
и пустой породы.
Во флотационную ячейку
(рис. 32), где порошкообразная
руда (природный сульфид меди)
перемешивается с водой, содер-
жащей небольшое количество скипидара, креозота и других слабо
полярных веществ, названных пенообразователями, подают воз-
дух, при этом частицы сульфида меди всплывают вместе с пузырь-
ками воздуха и пенообразователем; образующаяся пена выливается
через края флотационной ячейки, а частицы породы (силикаты,
алюмосиликаты и др.) оседают на дно ячейки.
Пенообразователи представляют собой маслянистые вещества,
интенсивно способствующие увлечению и селективному накапли-
ванию в пене на поверхности воды частиц сульфидной руды.
Для увеличения адгезионной способности флотируемых материа-
лов к пленке пенообразователя добавляют коллекторы (ксанто-
генаты, пальмитат натрия, диэтилдитиофосфат натрия, щавелевую
кислоту, керосин), которые способны превратить гидрофильный
(нефлотабильный) в гидрофобный (флотабильный) материал.
Поскольку коллекторы оказывают селективное действие, они
специфичны для определенных веществ.
684
ПОБОЧНАЯ ПОДГРУППА I ГРУППЫ
Вещества, обладающие свойствами, противоположными кол-
лекторам (т. е. вещества, превращающие флотабильные материа-
лы в нефлотабильные), называются депрессантами. С помощью-
депрессантов (это известковое молоко для пирита, сода для суль-
фидов металлов, силикат натрия для глины, цианид натрия для
обманки, бикарбонат натрия для галенита) осуществляется диффе-
ренциальная флотация.
Вещества, которые сводят на нет действие депрессантов, назы-
ваются активаторами. Наиболее известными активаторами являют-
ся серная кислота (для пирита), сульфат меди (для обманки),
сульфид натрия, ксантогенаты, ароматические производные тио-
мочевины (для окисленных сульфидов).
Дифференциальной флотацией комплексных руд, осуществляе-
мой при помощи депрессантов и активаторов, отделяют ценную
часть от пустой породы и выделяют минералы из полиметалличе-
ских руд. Дифференциальная (селективная) флотация основана
на свойстве частиц различных сульфидов и пустой породы по-раз-
ному смачиваться водой и маслами определенных плотностей.
В дифференциальной флотации важную роль играет pH и темпе-
ратура смеси, находящейся в флотационной ячейке, поскольку
при определенных значениях pH и температуры может флотиро-
ваться только один компонент.
Флотация применяется для обогащения и разделения сульфи-
дов, арсенидов, антимонидов цветных металлов (меди, свинца,
цинка, молибдена и др.), а также для обогащения кислородсодер-
жащих минералов меди, угольной пыли, графита, каолина.
Для извлечения меди концентраты руд перерабатываются пиро-
или гидрометаллургическими методами. Выбор метода переработ-
ки зависит от характера руды и от содержания в ней меди.
Пирометаллургический метод наиболее старый; он использует-
ся при переработке руд (сульфидов, окислов или карбонатов}
с большим содержанием меди (концентратов медных руд). При
плавлении концентратов полиметаллических сульфидных руд
(таких, как смеси Cu2S, CuFeS2, FeS2, As2S3, Sb2S3, ZnS) с флюса-
ми (например, SiO2) в шахтных печах медь с определенной частью
железа образует медный пек Cu2S-FeS, остальное железо, цинк
и другие металлы переходят в шлак в виде силикатов, а мышьяк,
сурьма, фосфор и частично сера превращаются в летучие-
окислы.
Сульфид меди, Cu2S, имеет т. пл. 1228°; он устойчив при высо-
кой температуре, в расплавленном состоянии растворяет сульфи-
ды щелочных и щелочноземельных металлов, а также сульфиды
свинца, цинка, железа, никеля, кобальта. При затвердевании
растворов или эвтектик, полученных растворением вышеупомя-
нутых сульфидов в сульфиде меди(1), образуются пеки с плотно-
стью 4—5 г!смР.
МЕДЬ
685
Расплавленный сульфид меди(1) смешивается в любых пропор-
циях с сульфидом железа(П) FeS (т. пл. 1168°). При затвердевании
эта система ведет себя как состоящая из двух твердых растворов,
которые образуют эвтектику с содержанием меди 28%.
При прокаливании сульфидов меди (1200°) образуется окись
меди(П), которая при нагревании с сульфидом железа(П), песком
(SiO2) и углем взаимодействует по уравнению
2CuO + FeS + SiO2 + С -> Cu2S + FeSiO3 + СО
В случае закиси меди имеет место следующая реакция:
Си2О FeS 4- SiO2 Cu2S 4- FeSiOg
При растворении сульфида железа(П) в сульфиде меди(1)
образуется медный пек Cu2S -FeS (называемый также медным кам-
нем), который содержит 20—45% меди и отделяется от шлака
вследствие более высокой плотности.
При переработке медных пеков с двуокисью кремния в каче-
стве флюса в конверторах (вертикальных или горизонтальных)
типа бессемеровских (футерованных магневитовым ! кирпичом)
образуется газообразный SO2 и силикатный шлак FeSiO3, который
плавает на поверхности «черной меди» (сырая медь с содержанием
96—98% Си). При переработке пека в конвертор через расплавлен-
ный пек пропускают сильную струю воздуха.
Сырая медь и шлак образуются в конверторе в результате сле-
дующих реакций, которые протекают в две стадии:
( FeS + 3/2О2 = FeO + SO2 f 2Cu2S + 3O2 = 2Cu2O + 2SO2
I ( II i Cu2S 4" 2Cu2O 6Cu -j- SO2
(FeO + SiO2 = FeSiO3 [ Cu2S + O2 = 2Cu + SO2
Когда в конверторе окисляется сульфид железа(П), пламя
окрашивается в желто-зеленый цвет; начало окисления сульфида
меди вызывает изменение цвета пламени — оно становится синим;
исчезновение синего пламени — сигнал для прекращения подачи
воздуха во избежание окисления сырой меди, которая остается
в расплавленном состоянии благодаря теплоте, выделяющейся
при горении серы.
Сырую медь можно получить и при нагревании пека (сильно
прокаленного) с коксом и двуокисью кремния:
4Cu2S-FeS + 15О2 = 8CuO + 2Fe2O3 + 8SO2
8CuO + 2Fe2O3 —5C = 8Cu 4- 4FeO 4- 5CO2
FeO 4- SiO2 = FeSiOa
При сплавлении руд, содержащих окислы меди, с двуокисью
кремния, сульфидом железа(П) и другими сернистыми минера-
лами также образуется медный пек, который в дальнейшем пере-
рабатывается в конверторах по процессу, описанному выше.
686
ПОБОЧНАЯ ПОДГРУППА I ГРУППЫ
Из минералов, содержащих окислы металлов, можно извлечь
медь путем нагревания этих минералов примерно до 600° (в шахт-
ных печах) с SiO2 и углем (коксом), поскольку при 600° углерод
или окись углерода восстанавливают окислы меди до металла,
а двуокись кремния с окисью железа(П) и окислами других метал-
лов образуют шлак, который собирается на поверхности сырой
меди.
Гидрометаллургический метод применяют при переработке
бедных медных руд (главным образом окисленных) и содержащих
медь отходов металлургической переработки других металлов.
В процессе гидрометаллургической переработки с помощью неко-
торых химических реагентов (H2SO4, NH4OH, NaCN, Fe2(SO4)3) пло-
хо растворимые соединения меди (и самородная медь) переводят
в легко растворимые соединения, а затем различными способами
их извлекают из раствора.
Если руды и отходы содержат медь в виде легко растворимых
при обычной температуре соединений, медь извлекают простым
выщелачиванием руд и отходов этими растворителями. При выбо-
ре реагентов, при помощи которых медь переводится в раствор,
учитывается характер как медных руд или отходов, так и пустой
породы.
При обработке порошкообразных медных руд, содержащих
малахит CuCO3-Си(ОН)2, азурит 2CuCO3-Си(ОН)2, куприт Сп2О,
мелаконит СиО, хризоколл CuSiO3-2H2O, разб. H2SO4 (в больших
емкостях со свинцовым покрытием) образуется водный раствор
сульфата меди.
Если руды содержат комплексные сульфиды меди, сначала
осуществляют сульфатирующее или хлорирующее прокаливание
этих руд, а затем прокаленный продукт обрабатывают разб. H2SO4,
в которую добавлено небольшое количество сульфата железа(Ш).
Сульфатирующее прокаливание комплексных сульфидов меди
осуществляется при температуре 450—480° в токе воздуха:
Cu2S + S/2O2 = CuSO4 + CuO CuS + 2O2 = CuSO4
При хлорирующем прокаливании смесь комплексных сульфи-
дов меди с избытком хлорида шелочного (или щелочноземельного)
металла нагревают до красного каления, подавая избыточное
количество воздуха. При хлорирующем прокаливании соедине-
ния меди, серебра, свинца и др. переходят в хлориды, соединения
железа — в окислы.
Самородная медь и сульфидные медные руды медленно раство-
ряются в серной кислоте, содержащей ионы Fe3+.
Си + Fe2(SO4)3 = CuSO4 + 2FeSO4
Cu2S + 5Fe2(SO4)3 + 4H2O = 2CuSO4 + 10FeSO4 + 4H2SO4
CuS + 4Fe2(SO4)3 + 4H2O = CuSO4 + 8FeSO4 + 4H2SO4
МЕДЬ
687
Эти реакции активируются кислородом воздуха, который вновь
окисляет сульфат железа (II).
При обработке медьсодержащих руд и отходов водным раство-
ром аммиака медь переходит в раствор в виде комплексного катио-
на [Cu(NH3)4]2+. Растворению соединений меди в аммиаке благо-
приятствует присутствие соли аммония (карбоната или цианида).
В аммиачных растворах с большим или меньшим содержанием кар-
боната наряду с комплексным ионом меди находятся и комплекс-
ные ионы цинка, кадмия, серебра, никеля и кобальта.
Медные руды растворяют в водных растворах цианидов щелоч-
ных металлов в присутствии окислителя (кислорода или гипохло-
ритов):
2Cu + 4KCN + Н2О + l/2O2 2K[CufCN)2] + 2К0Н
Для цианирования медных руд применяют растворы цианида
(NaCN, KCN), более концентрированные, чем растворы, приме-
няемые для растворения золота и серебра.
Из кислых водных растворов солей меди (таких, как растворы
сульфата меди) медь можно извлечь восстановлением SO2 под дав-
лением при 150°, катодным восстановлением (с применением труд-
норастворимых анодов из графита, магнетита, свинца, силицида
железа, сплава медь — сурьма) и адсорбцией на сульфированных:
катионообменных смолах (или сульфированном угле).
CuSO4 + Fe Cu + FeSO4
CuSO4 -|- SO2 -|- 2H2O Cu 4- 2H2SO4
При осаждении элементарной меди присутствие ионов Fe3+ спо-
собствует растворению:
Cu -J- 2Fe3+ -» Си2+ + 2Fe2+
Для извлечения меди из растворов, содержащих и другие-
катионы, применяют катионообменные смолы дуолит С-20 в Н-фор-
ме (регенерирование осуществляют 5 %-ной НС1, при этом металлы
переходят в элюент в виде хлоридов) и амберлит ХЕ-77 в Са-фор-
ме (регенерируют 9,4%-ным раствором Са12 при 25—27°).
При сжигании сульфированного угля, насыщенного солями
меди(П), образуется элементарная медь высокой чистоты.
Из аммиачных растворов, в которых медь находится в виде
комплексного катиона [Cu(NH3)4]2+, ее можно извлечь фенолфор-
мальдегидными катионнообменными смолами типа R — СН2 —
— SO3H или путем катодного восстановления.
Из растворов комплексных цианидов медь извлекают цинком
или алюминием, катодным восстановлением или адсорбцией
на активированном угле либо сильно основных анионообменных
смолах типа RC1, которые удерживают комплексный анион меди
и регенерируются смесью НС1 с бутанолом или ацетоном.
688
ПОБОЧНАЯ ПОДГРУППА I ГРУППЫ
АФФИНАЖ И РАФИНИРОВАНИЕ
Аффинаж — металлургический процесс, целью которого
является получение чистого металла из сырого путем удаления
примесей. Методы аффинажа у различных металлов различны,
поскольку они могут основываться на окислении или восстановле-
нии примесей, на ликвации (примеси с более высокой температурой
плавления остаются нерасплавленными), на сегрегации (примеси
с более низкой температурой плавления выделяются селективным
отверждением), на адсорбции (примеси адсорбируются без уча-
стия химической реакции).
Для получения чистого металла после процесса аффинажа про-
водят электролитическое или пирометаллургическое рафиниро-
вание, например перегонку в вакууме или атмосфере инертных
газов.
Рафинирование меди
Сырая медь (называемая «черной медью»), которая образуется
при металлургическом получении, содержит 93—98,5% Си
и загрязнена кислородом, железом, мышьяком, сурьмой, свинцом,
цинком, никелем, кобальтом, оловом, висмутом, серой и, возможно,
серебром, золотом, платиной. Свинец, сера, селен, теллур, висмут
и кислород — примеси, вредные для меди, а мышьяк, фосфор,
никель, железо, марганец и кремний улучшают механические
свойства меди.
Пирометаллургическое рафинирование меди
Если сырую медь расплавляют в отражательной печи, в кото-
рую вдувают сжатый воздух, происходит окисление элементов S,
Fe, Ni, Zn, Со, Sn, Pb, As, Sb (частично металлической меди)
и связывание двуокисью кремния (из кислой обкладки печи)
с превращением в шлак (силикаты).
Несколько реакций, протекающих в отражательной печи, при-
ведено ниже:
2Cu2O + Cu2S 6Cu + SO2
2Fe + 2Cu + SO2 Cu2S + 2FeO
2Ni 4- 2Cu + SO2 Cu2S 4- 2NiO
2Zn + 2Cu + SO2 Cu2S + 2ZnO
FeO + SiO2 -> FeSiOs
NiO 4- SiO2 NiSiOs
При нагревании расплава двуокись серы полностью элимини-
руется, окислы As2O3 и Sb2O3 частично улетучиваются, а большая
часть сурьмы остается в меди.
После удаления шлака для раскисления частично окисленной
меди в расплав бросают сырые березовые чурки, которые при тем-
пературе печи образуют пары воды, водород и окись углерода.
МЕДЬ
689
Все эти газы перемешивают расплав, способствуют удалению дву-
окиси серы, восстанавливают окись меди(1) до металла.
Медь, рафинированная пирометаллургическим методом, содер-
жит 1 % Си2О, висмут, олово и иногда серебро, золото, платину
и платиновые металлы. Из такой меди отливают аноды, которые
в дальнейшем служат для получения электролитической меди.
Электролитическое рафинирование меди
Электролизерами для электролитической очистки меди являют-
ся бетонные чаны, стенки которых обложены свинцовыми пла-
стинами. В них наливают электролит — раствор сульфата меди
с серной кислотой и добавкой сульфата натрия. В отсутствие сер-
ной кислоты при электролизе сульфата меди в большей или мень-
шей степени образуется Сп2О.
В электролизер определенным образом помещают аноды из
пирометаллургически рафинированной меди и катоды из чистой
меди. При пропускании электрического тока (определенного
напряжения) на катоде осаждается чистая меДь, а аноды, состоя-
щие из сырой меди (с примесями цинка, железа, олова, никеля,
висмута), растворяются в результате процессов окисления. Неме-
таллические примеси и металлы, менее активные, чем медь (сереб-
ро, золото, платина, платиновые металлы), находящиеся в анодах,
выпадают в виде шлама на дно электролизера. Из анодного шлама
извлекают серебро, золото, платину и платиновые металлы.
При электролизе водного раствора сульфата меди на катоде
оседает чистая медь, а на аноде выделяется кислород:
CuSO4 -> Cu2+ + SOJ- Н2О =pt Н+ + он-
На катоде: Сп2+ -|- 2е~ Си
На аноде: 2ОН- — 2е~ 2ОН Х/2О2 + Н2О
Выделяющийся на аноде кислород вместе с серной кислотой
электролита растворяет медь, наиболее активные сопутствующие
металлы и окись меди(1) анода:
Cu -f- H2SO4 -]- 1/2О2 — CuSO4 -]- Н2О
Си2О + 2H2SO4 + V2O2 = 2CuSO4 + 2Н2О
ФИЗИЧЕСКИЕ И ХИМИЧЕСКИЕ СВОЙСТВА
В компактном состоянии металлическая медь медно-красного
цвета с ярким металлическим блеском в хорошо отполированном
состоянии (отражательная способность соизмерима с отражатель-
ной способностью алюминия). Очень тонкие листы меди просве-
чивают. Цвет меди в коллоидном состоянии может быть от зеленого
до фиолетового.
44-0101
690
ПОБОЧНАЯ ПОДГРУППА I ГРУППЫ
Медь относится к тяжелым металлам (плотность равна 8,96 z!cms
при 20°) с твердостью 2,5—3 по шкале Мооса; имеет гранецентри-
рованную кубическую кристаллическую решетку. Количество
природных изотопов у нее невелико. Медь пластична, исключи-
тельно легко куется (можно получить фольгу толщиной 2,6 мк),
способна к вытягиванию.
Механические свойства меди ухудшаются при загрязнении
висмутом, свинцом, серой, теллуром, кислородом, поскольку
образуются хрупкие и легко летучие сплавы.
Металлическая медь плавится при 1083°, кипит при 2582°,
диамагнитна, является очень хорошим тепло- и электропроводни-
ком. Наиболее вредное влияние на электропроводность меди ока-
зывает фосфор, который часто намеренно добавляют при металлур-
гическом получении меди в качестве раскислителя.
Известно большое количество сплавов, которые медь образует
с элементами Zn, Sn, Al, Ni, Pb, Mn, Be, Fe, Mg, Hg, Ag, Au и Si.
Сплавы меди с цинком, называемые латунями, могут быть
классифицированы по составу на красные латуни (содержащие
менее 20% цинка и обладающие хорошей тягучестью), желтые
латуни (содержащие 20—50% цинка), белые латуни (хрупкие
с содержанием цинка 50—80%) и специальные латуни (которые
помимо меди и цинка содержат свинец, никель, марганец, железо
олово и алюминий).
Сплавы меди с оловом (или алюминием), называемые бронза-
ми, классифицируются по составу на оловянные, алюминиевые,
фосфористые и специальные бронзы; последние кроме меди и оло-
ва могут содержать алюминий, свинец, никель, марганец, железо,
кремний, бериллий и др.
К наиболее широко применяемым сплавам меди следует отне-
сти: латуни (80—20% Си, остальное цинк), бронзу (90% Си и 10%
Sn), томпак (90% Си и 10% Zn), аргентан (65% Си, 20% Zn, 15%
Ni), «новое серебро» (50% Си, 25% Zn, 25% Ni), константан (60%
Си, 40% Ni), никелин (56% Си, 31% Ni, 13% Zn), манганин
(85% Си, 12% Мп, 3% Ni), мельхиор (80?6 Си, 20% Ni), сплав
Деварда (50% Си, 45% А1, 5% Zn).
С химической точки зрения медь относительно инертна, хотя
она непосредственно взаимодействует с кислородом, серой, гало-
генами и другими элементами, реагирует с HNO3, конц. H2SO4,
H2S, NaCl, NaCN, с сульфатами и нитратами щелочных металлов,
аммиаком, солями аммония и др.
Порошок металлической меди, полученный при 100° восстанов-
лением окиси меди окисью углерода, обладает пирофорными
свойствами.
Во влажном воздухе (не содержащем СО2, H2S, SO2) металли-
ческая медь покрывается плотной защитной пленкой, состоящей
из смеси металлической меди и закиси меди и образующейся
МЕДЬ
691
по уравнениям
2Сп 4- О2 + 2Н2О -> 2Cu(OH)2 Cu(OH)2 -J- Cu -> Cu2O + H2O
Если во влажном воздухе присутствуют газообразные СО2,
H2S, SO2, то медь покрывается защитной пленкой, состоящей
из основного карбоната и основного сульфата меди.
При нагревании до 100—130° на воздухе медь покрывается пур-
пурной пленкой окиси меди(1). Около 200° образуется смесь Сп2О
и СпО. Очень сильно нагретая на воздухе или в кислороде, медь
горит ярко-зеленым планенем и превращается в СпО.
В присутствии двуокиси углерода водные растворы хлоридов
щелочных металлов действуют на медь с образованием основного
карбоната (идентичного малахиту), а в отсутствие СО2 образуется
оксихлорид меди (идентичный природному атакамиту). Металли-
ческая медь взаимодействует с фтором (500°), хлором, бромом
(300°), а на холоду — с водными растворами иода.
Нагретые концентрированные галогеноводородные кислоты
реагируют с порошкообразной металлической медью на воздухе
или в кислороде:
2Cu + 4НС1 + О2 = 2СиС12 + 2Н2О
Сера, селен и теллур легко взаимодействуют с медью при нагре-
вании или при растирании в ступке.
Медь сульфидируется в результате соприкосновения с H2S
(газообразным или в виде водного раствора) или с сульфидами
металлов на воздухе или в кислороде.
Медь взаимодействует с сероводородом в присутствии сероугле-
рода с образованием сульфида меди(1) и метана:
8Cu + 2H2S + CS2 -> 4Cu2S + СН4
Хлорид селена(1У) SeCl4 реагирует с металлической медью
по уравнению
2Cu + SeCl4 -> 2CuC12 + Se
Металлическая медь с азотом непосредственно не взаимодей-
ствует, но при действии аммиака на нагретую до красного кале-
ния медь образуется нитрид Cu3N. Действием на медь азотистово-
дородной кислоты или азида натрия на сульфат меди получают
азид меди CuN3 — коричнево-красный осадок, неустойчивый, спо-
собный разлагаться со взрывом.
При обычной температуре металлическая медь восстанавли-
вает двуокись азота, а около 200° — окись азота:
2Cu + NO2 -» Cu2O + NO 4Cu + 2NO 2Cu2O + N2
Нагревание без доступа воздуха металлической меди с фосфо-
ром, мышьяком, сурьмой, углеродом и кремнием приводит к обра-
зованию фосфидов Сп3Р, Сп3Р2, арсенидов CusAs2, Gu3As, Gu5As2,
карбида и силицидов.
44*
692
ПОБОЧНАЯ ПОДГРУППА I ГРУППЫ
Металлическая медь растворяется в HNO3, конц. H2SO4 при
нагревании, а также в водных растворах цианидов щелочных
металлов, в аммиаке или растворах (расплавах) солей аммония
и в растворах солей железа(Ш):
2Cu + 8HNO3 = 3Cu(NO3)2 + 2NO + 4Н2О
Си + 2H2SO4 = CuSO4 + SO2 + 2Н2О
2Си + 4KCN + Н2О + ‘/2О2 = 2K[Cu(CN)?] + 2К0Н
2Си + 8KCN + Н2О + = 2K3[Cu(CN)4] + 2КОН
Си + 4NH4OH + V2O2 = [Cu(NH3)4](OH)2 + ЗН2О
2
Си + (NH4)2CO3 + i/2O2
Си
СО3 -
(NH3)2.
СО3 -
(NH3)2.
+ н2о
+ 2Н2О = СиСО3 • Си(ОН)2 + (NH4)2CO3 + 2NH3
Си + 2NH4NO3 = Cu(NO3)2 + 2NH3 + H2
Си + Fe2(SO4)3 = CuSO4 + 2FeSO4
Ортофосфорная кислота медленно растворяет нагретый поро-
шок меди. Органические кислоты (как в водных, так и в спирто-
вых, альдегидных, кетонных и других растворах) взаимодействуют
с металлической медью в присутствии кислорода воздуха. С уксус-
ной кислотой медь легко образует основной ацетат меди
Си(СН3СОО)2-Сп(ОН)2, называемый ярь-медянкой:
2Си + 2СН3СООН + О2 = Cu(CH3COO)2.Cu(OH)2
Поскольку медь — металл с невысокой химической активно-
стью (нормальный потенциал системы Cu/Cu2+ равен +0,346 в),
она может быть вытеснена из растворов своих солей более актив-
ными металлами или путем катодного восстановления.
Химические свойства меди наглядно иллюстрируются следую-
щей схемой:
Комн. темп.
Медь
Нагревание
во влажном воздухе -> Си2О
во влажном воздухе в присутствии СО2, H2S,
SO2 -»• основные карбонат и сульфат меди
с NH4OH -> [Си (NH3)4](OH)2
с KCN +кислород + вода -* K[Cu(CN),] или
K3[Cu(CN)4]
с HNO3 -> Cu(NO3)2.nH2O
на воздухе -»• Си2О и CuO
с F2, С12, Вг2, 12 —> CuF2, CuC12, CuBr2, Cul2
c S, Se, Те -> CuS, CuSe, CuTe
c H2S -> CuS
с P, As, Sb, C, Si Cu3P, Cu3P2, Cu3As2,
СизАз, Cu5As2, Cu2Sb, Cu3Sb, карбид и силициды
с конц. НС1 на воздухе -»• СиС12
с конц. H2SO4 CuSO4-5H2O
с NH3 -> Cu3N
с NO2 или NO -»• Cu2O
МЕДЬ
693
Хотя большинство соединений меди очень токсично, некото-
рые из них применяются для лечения проказы, анемии, диабета
и др. Разбавленные растворы сульфата меди служат рвотным сред-
ством. Летальная доза для человека 2—3 г медной соли.
Медь играет особо важную биологическую роль, являясь,
вероятно, катализатором окислительных процессов в клетках.
Соединения меди (например, сульфат меди) служат для приго-
товления противоспоровых препаратов, которые используются
как средства защиты растений (например, виноградной лозы)
от грибковых заболеваний.
К наиболее известным из числа применяемых для опрыскива-
ния растений препаратов относятся смеси сульфата меди с изве-
стью или сульфата меди с карбонатом натрия (также основной
карбонат меди).
ПРИМЕНЕНИЕ
Медь, ее сплавы и соединения благодаря исключительно цен-
ным физико-химическим свойствам в настоящее время применяют-
ся во всех отраслях современной техники.
В электротехнической промышленности электролитическая
медь (обладающая высокой электропроводностью) служит для
изготовления электрических проводов. Порошкообразная медь
идет на изготовление коллекторных щеток электромоторов и дина-
момашин.
В теплообменных системах благодаря высокой теплопроводно-
сти металлическая медь применяется для производства котлов,
радиаторов, змеевиков и др.
В металлургической промышленности медь служит для полу-
чения различных сплавов (латуни, бронзы и др.). Из латуни изго-
товляют детали машин, автоклавы, водопроводные краны, краны
на газовых линиях, дверные ручки, петли и др. Из бронзы отли-
вают медали, статуи, различные декоративные сосуды. Из арген-
тана делают посуду и музыкальные инструменты.
Часто металлическую медь применяют в качестве легируюшей
добавки к сплавам алюминия или железа.
В химической промышленности медь служит катализатором
процесса разложения метана и аналогичных углеводородов. Сплав
Деварда разлагает воду и восстанавливает в кислой среде NO
и NO2 до NH3. Благодаря своей коррозионной устойчивости медь
применяется для защитных покрытий других металлов.
В производстве минеральных красителей применяется малахи-
товая и швейнфуртская зелень.
Соединения меди служат красителями в стекольной промыш-
ленности.
В сельском хозяйстве некоторые соединения меди применяются
как фунгициды, в частности для виноградников.
694
ПОБОЧНАЯ ПОДГРУППА I ГРУППЫ
Медь необходима для нормального развития растений; ее отсут-
ствие вызывает болезнь, называемую хлорозом, и задерживает
рост растений.
Медь широко используется при лечении анемии у животных.
СОЕДИНЕНИЯ (ОБЩИЕ СВОЙСТВА)
Известны многочисленные устойчивые соединения двухвалент-
ной меди, ограниченное число соединений одновалентной меди
и всего несколько соединений трехвалентной меди.
Наряду с простыми получены многочисленные координацион-
ные соединения, несколько металлоорганических производных
и соединения включения.
В табл. 70 приведены формулы и указан цвет некоторых соеди-
нений меди различных степеней окисления.
Благодаря ярко выраженной склонности к образованию коор-
динационных соединений для меди известны многочисленные аци-
до- и аквосоли, комплексные аммины, хелатные соединения и др.
Соединения одновалентной меди
Большинство соединений одновалентной меди белые (CuCl,
CuBr, Cui, CuCN, CuSCN, СпСН3СОО), некоторые же красного
(Cu2O, CuF, СпгСг-НгО, Cu2[SiF6]), желтого (СпОН), синего (Cu2S)
и черного (Cu2Se, Cu2Te) цвета. Как правило, соединения меди(1)
устойчивы при высокой температуре, плохо растворимы в воде,
во влажном состоянии легко окисляются кислородом воздуха
(обладают отчетливо выраженными восстановительными свой-
ствами), проявляют склонность к образованию координационных
соединений.
Из водных растворов солей меди(1) выпадают кристаллы солей
меди(П), поскольку в растворе равновесие смещается вправо.
2Сп+ Сп2+ -|- Си
Благодаря этому смещению в водном растворе соли меди(1)
образуются всегда в присутствии восстановителей.
При растворении плохо растворимых соединений (Cu2O, CuCl,
CuBr, Cui, CuCN) в избытке аммиака, солей аммония, галогено-
водородов, цианидов щелочных металлов, органических аминов
(пиридина, пиперидина, хинолина) образуются растворимые
координационные соединения, большинство которых бесцветно.
В координационных соединениях ион меди(1) может иметь
координационные числа 2, 3 и 4.
МЕДЬ
695
Неорганические соединения
Закись меди, Си2О, встречается в природе в виде минерала
куприта и представляет собой красные октаэдрические кристаллы.
В лаборатории Сп2О получают обработкой растворов солей меди (II)
растворами щелочей или карбонатов щелочных металлов в присут-
ствии восстановителей (глюкозы, гидроксиламина). Вначале обра-
зуется желтый осадок СпОН, который при кипячении переходит
в красный осадок Си2О:
CuSO4 + 2NaOH = Cu(OH)2 + Na2SO4
2Cu(OH)2 -j- C6H12O6 = Cu2O + C6H12O7 + 2H2O
Глюконовая
кислота
Соединение Cu2O представляет собой диамагнитные кубические
кристаллы, цвет которых меняется от коричневого до карминово-
красного, плавятся при 1229° (без
в воде, растворяются в аммиаке
или галогеноводородах с обра-
зованием координационных сое-
динений:
Cu2O + 4NH3 + Н2О =
= 2[Cu(NH3)2]OH
Cu2O + 4НХ = 2H[CuX2] + Н2О
(где X = СР, Br-, I-)
Cu2O + 8НС1 (конц.) = 2Н3[СиС14]+
+ н20
разложения), плохо растворимы
Рис. 33. Структура закиси Сн2О.
Кристалл закиси меди(1) образован как бы путем взаимопро-
никновения двух кубов. В кубической решетке (рис. 33) ионы кис-
лорода расположены тетраэдрически, а ионы меди(1) находятся
в каждой тетраэдрической группе между ионами кислорода.
При нагревании закись меди легко восстанавливается до метал-
ла водородом, окисью углерода или магнием, цинком, алюминием,
щелочными металлами.
Cu2O4-H2 — 2Cu + H2O Cu2O-j-Mg = 2Cu-j-MgO
200°
Cu2O-|-CO----> 2Cu4-CO2 3Cu2O+2A1 = 6Cu + A12O3
Если восстановление осуществляется магнием, цинком или
алюминием, образуется не чистая медь, а ее сплавы.
При 1025° Сп2О превращается в СпО, а выше 1050° — терми-
чески диссоциирует на элементы:
2Сн2О О2 —> 4CuO 2Cu2O —> 4Cu -j- О2
696
ПОБОЧНАЯ ПОДГРУППА I ГРУППЫ
Закись меди взаимодействует с хлоридом меди(П), хлоридом
ртути(П) и иодидом мышьяка(Ш):
3Cu2O + 4CuC12 + ЗН2О 6CuCl + CuCl2.3Cu(OH)2
3Cu2O 3HgCl2 3H2O —> 2CuC12 CuCl2.3Cu(OH)2 3Hg
3Cu2O + 2AsI3 6CuI + As2O3
Прокаливанием Cu2O с серой в атмосфере водорода (либо дей-
ствием полисульфидов или тиосульфатов щелочных металлов
на Си2О) получают сульфид меди(1) Cu2S синевато-черного цвета.
Из закиси меди и металлической меди изготовляют выпрями-
тели (купроксид) переменного тока.
Закись меди Сп2О применяется в качестве пигмента в стеколь-
ной и керамической промышленности.
Гидрат закиси меди, СиОН, получают обработкой солей
меди(1) (или смеси соли меди(П) с восстановителем) щелочью
при низкой температуре и pH = 3:
CuCl + КОН = СиОН + КС1
При кипячении СиОН легко превращается в красную закись
меди(1):
2CuOH -> Си2О + Нр*
Желтое соединение СиОН имеет плотность 3,368 г/см3, оно
неустойчиво, легко окисляется, плохо растворимо в воде и лег-
ко растворяется в аммиаке с образованием бесцветных ионов
[Cu(NH3)n]+, которые быстро окрашиваются в синий цвет бла-
годаря окислению иона меди(1).
Фторид меди (полуфтористая медь), CuF, получается дей-
ствием газообразного фтористого водорода на сильно нагретый
хлорид меди(1) или нагреванием (1100—1200°) фторида меди(П):
CuCl + HF = CuF + НС1 2CuF2 = 2CuF + F2
Соединение CuF образует рубиново-красные кристаллы с т. пл.
908°; они устойчивы в сухом воздухе, плохо растворимы в воде,
спирте, безводном фтористом водороде, разлагаются во влажном
воздухе или водой по уравнению
2CuF -> CuF2 + Cu
Хлорид меди {полухлористая медь), CuCl, получается раство-
рением Си2О в НС1, нагреванием СиС12, восстановлением СиС12
двуокисью серы [хлоридом олова(П), солями гидразина или гидро-
ксиламина, цинком, алюминием, серебром], действием хлора
на суспензию меди в органической жидкости, которая не взаимо-
действует с галогенами, обработкой металлической меди соляной
кислотой в присутствии кислорода воздуха, азотной кислоты или
МЕДЬ
697
хлората калия.
Cu2O + 2НС1 = 2CuCl + Н2О 2СиС1г 2СиС1 + С12
2CuGl2 + SO2 + 2Н2О = 2CuCl + H2SO4 + 2НС1
2CuCl2 + SnCl2 = 2CuCl + SnCl4 2Cu + 2HC1 + V2O2 = 2CuCl + H2O
При сгорании тонкодисперсной меди (или тонкой медной про-
волоки), нагретой в атмосфере хлора, образуется смесь хлоридов
меди(1) и (II).
Хлорид одновалентной меди представляет собой бесцветное
твердое вещество с кристаллической структурой типа цинковой
обманки ZnS, плотностью 3,7 г!см\ т. пл. 434°, т. кип. 1367°; он
устойчив в сухой атмосфере, плохо растворим в воде, эфире, аце-
тоне, окисляется и разлагается на свету и во влажной атмосфере.
Осадок полухлористой меди растворяется в аммиаке, соляной
кислоте, хлоридах щелочных металлов или NH4C1, тиосульфатах,
пиридине, пиперидине, хинолине за счет процесса комплексооб-
разования.
CuCl -J- НС1 = H[CuCl2]—перламутрово-серые иглы
CuCl-j-2HCl = H2[CuCl3]
CuC 1 + MezCl = Ме:[СиС12]
CuCl + 2MeICl = Me|[CuCl3]
2CuCl + Me1 Cl = MeI[Cu2Cl3]
2CuCl + 2MerCl = Me£[Cu2Cl4]
3CuCl+2MeICl = Me2[Cu3Cl5]
бесцветные кристаллы
(MeI=K+, Rb+, Cs+, NH+)
одновалентной меди содержат
Аммиачные растворы хлорида
ионы [Cu(NH3)n]+Cl-.
При действии газообразного NH3 на CuCl на холоду или при
обработке солянокислого раствора CuCl небольшим количеством
аммиака [при действии на порошок меди горячего раствора NH4C1
или аммиачного раствора хлорида меди(П)] образуются бесцвет-
ные кристаллы [CuNH3]Cl. Если хлорид меди(1) нагревают
в атмосфере газообразного NH3, образуются черные кристаллы
[CuNH3].[CuC12] или 2CuC1-NH3.
Солянокислые или аммиачные растворы хлорида одновалент-
ной меди легко поглощают окись углерода, образуя димер
ci zco 1
'Cu Cu^
r ОС
OH2J
красный осадок, который разлагается водой, в вакууме или при
нагревании.
Солянокислые растворы хлорида меди(1) поглощают газо-
образный фосфин с образованием [СиРН3]С1.
-698 ПОБОЧНАЯ ПОДГРУППА I ГРУППЫ
Хлорид меди в аммиачном растворе легко поглощает ацетилен
и углеводороды типа R — С == С — Н с образованием красных
осадков, которые становятся взрывчатыми при высушивании и раз-
лагаются под действием кислот.
R _ С == С — Н + CuCl R — С С — Си 4- НС1
Поскольку солянокислые или аммиачные растворы хлорида
меди(1) очень легко окисляются, их хранят в присутствии метал-
лической меди в герметически закрытых сосудах.
Хлорид меди(1) применяется в газовом анализе для поглоще-
ния окиси углерода, фосфористого водорода и ацетилена.
Бромид меди (полубромистая медь), СпВг, образуется при
действии брома на тонкий порошок металлической меди (взятой
в избытке), при сильном нагревании металлической меди в токе
газообразного НВг, путем термического разложения СпВг2,
нагревания сульфата двухвалентной меди и бромида щелочного
металла в присутствии уксусной кислоты и медных опилок или
действия SO2 на раствор сульфата меди и бромида щелочного
металла, а также взаимодействием бромистоводородной кислоты
с Сп2О:
2Cu + Br2 = 2CuBr 4- 49,7 ккал 2Cu + 2HBr = 2CuBr + Н2
2СиВг2 = 2СиВг + Вг2
CuSO4 + 2KBr + Си = 2CuBr + K2SO4
Си2О 4- 2НВг (газ.) = 2СиВг 4- Н2О 4- 60,6 ккал
Си2О 4~ 2НВт (раствор) = 2СиВг 4- Н2О -)- 20,76 ккал
Соединение СпВг представляет собой светло-желтые тетраэдри-
ческие микрокристаллы с плотностью 4,72 г/см3, т. пл. 483°
и т. кип. 1345°; они синеют на свету, плохо растворимы в воде
и растворяются в НС1, НВг, аммиаке, солях аммония, пиридине,
концентрированных растворах хлоридов, бромидов или тиосуль-
фатов щелочных металлов с образованием координационных
соединений.
Под действием азотной или серной кислоты бромид меди(1)
разлагается, а при нагревании на воздухе конечным продуктом
разложения является CuO.
По многим химическим свойствам бромид меди(1) аналоги-
чен хлориду меди(1).
Бромид меди(1) с бромидами щелочных металлов или аммо-
ния образует бромосоли типа Ме^СиВгг], Me* [СиВг3].
При охлаждении слабо кислого раствора бромида меди(1) с бро-
мидом аммония выпадают бесцветные ромбоэдрические кристаллы
2NH4[CuBr2]-Н2О. Охлаждение бромистоводородного раствора
бромида меди(1) с бромидом аммония в присутствии металличе-
ской меди в атмосфере СО2 приводит к осаждению бесцветных кри-
сталлов (NH4)2ICuBr3].
МЕДЬ
699
В результате кипячения гидроокиси тетрамминмеди(П) с по-
рошкообразной медью и спиртовым раствором брома получается
соединение [Cu(NH3)2]Br.
Иодид меди, Cui, встречается в природе в виде минерала мар-
шита; в лаборатории он может быть получен обработкой металли-
ческой меди иодом (в парах или растворе), действием иодистово-
дородной кислоты на металлическую медь, закись меди или соли
меди(1), обработкой растворов солей меди(П) раствором иодида
щелочного металла (при этом в качестве промежуточного продукта
образуется неустойчивое соединение Cul2).
2Cu -j- I2 = 2CuI 4- 32,8 ккал 2Си 4- 2HI = 2CuI 4- Н2
2СиО 4- 2HI = 2CuI + Н2О 4- 1!2G2 CuSO4 4- 2KI = Cul2 4- K2SO4
2CuI2 -> 2CuI -|- I2
Свежеприготовленное соединение Cui представляет собой мел-
кие бесцветные или белые тетраэдрические кристаллы с плотно-
стью 5,65 г/см3, т. пл. 588° и т. кип. 1293°; при нагревании они
становятся коричнево-красными, а затем черными, плохо раство-
римы в воде, сероуглероде, в растворах хлоридов, бромидов,
сульфатов и нитратов щелочных металлов, растворяются в циани-
дах, растворах аммиака и в жидком аммиаке.
Шелочи и карбонаты щелочных металлов превращают иодид
меди(1) в гидроокись СиОН, а кислоты его разлагают.
Суспензия Cui в воде может быть восстановлена до металличе-
ской меди кипячением с железом, цинком или оловом.
Иодид меди(1) образует с различными иодидами иодосоли, на-
пример 2NH4[CuI2]-Н2О, Hg[CuI3] и др.
По охлаждении раствора, полученного добавлением спиртово-
го раствора иода к раствору [Cu(NH3)4](OH)2 при кипячении,
выпадают зеленые кристаллы [Cu(NH3)4][CuI2].
В результате действия спиртового раствора иода на
[Cu(NH3)4](OH)2 при кипячении в присутствии порошкообразной
металлической меди образуется соединение [Cu(NH3)2]I.
Известно также соединение [Cu{(C2H5)3As}]I.
Сульфид меди, Cu2S, встречается в природе в виде минерала
халькозина; в лаборатории может быть получен прямым взаимо-
действием элементов при сильном нагревании, растиранием смеси
серного цвета с восстановленной медью, прокаливанием окислов,
оксисульфидов, сульфидов и некоторых солей меди(П) с серой
в атмосфере водорода (или инертного газа), действием сероводоро-
да (на металлическую медь, взаимодействием многих сульфидов
металлов (или паров сероуглерода) с металлической медью, вос-
становлением сульфида меди(П) спиртом, арсенитом натрия,
700
ПОБОЧНАЯ ПОДГРУППА I ГРУППЫ
водородом и восстановлением сульфата меди серой.
2Cu + S = Cu2S + 24,17 ккал
400-500°
2CuS + Н2------------------------+ Cu2S + H2S
4Gu + CS2 = 2Cu2S + G
2CuSO4 + 3S = Cu2S + 4SO2
2GuO + 2S = Cu2S + SO2
2Cu + H2S = Cu2S + H2
2CuCl + H2S = Cu2S + 2HC1
Сульфид меди(1) представляет собой синие (или синевато-чер-
ные) октаэдрические кристаллы с плотностью 5,97 г/см3 и т. пл.
1100°; он плохо растворим в воде и растворяется в кипящей разб.
HNO3. При нагревании Cu2S в атмосфере водорода до 800° обра-
зуется металлическая медь и сероводород:
Cu2S + Н2 = 2Cu + H2S
Нагревание сульфида меди(1) на воздухе (или в кислороде)
приводит к образованию окиси и сульфата меди(П):
Cu2S 6/2О2 “ Си О -|- CuSO4
Концентрированная серная кислота и нитрат серебра взаимо-
действуют с сульфидом меди(1) по уравнениям:
Cu2S + 2H2SO4 = GuS + CuSO4 + SO2 + 2H2O
Cu2S -|- 4AgNO3 = 2Gu(NO3)2 Ag2S -|- 2Ag
Сульфид меди(1) взаимодействует с окислами меди(1) и (II)
и окисью свинца(П):
Cu2S + 2Cu2O = 6Cu + SO2 Cu2S + 2GuO = 4Cu + SO2
Cu2S + 6CuO = 4Cu2O + SO2 Cu2S + ЗРЬО = Cu2O + 3Pb + SO2
Соединение Cu2S образует с различными сульфидами
координационные, двойные или тройные сульфиды, например
NalCuS], KICuS], 4Cu2S-K2S, Cu2S-CuS-Na2S, 3Cu2S-2CuS-K2S,
4Cu2S*A12S3, 3Cu2S*As2S5, K2Cu3Si0, Rb2Cu3S10.
Сульфит меди, Cu2SO3’H2O, образуется в результате пропу-
скания S02 через раствор ацетата меди(П), к которому добавлена
уксусная кислота:
2Gu(CH3GOO)2 + 2SO2 + ЗН2О = Cu2SO3 + 4CH3GOOH + H2SO4
Перламутрово-белое соединение Cu2SO3-H2O неустойчиво, под
действием горячей воды превращается в основную сольСп2ЗО3-
•9СпО фиолетового цвета, а затем в Сп2О.
Сульфит меди(1) образует с сульфитами щелочных и некоторых
трехвалентных металлов двойные соли типов Me4CuSO3] (где
Me1 = Na+, К+, NH+) и Me3+[Cu(SO3)2]-8Н2О (где Ме3+ = La3+,
Се3+, Nd3+, Рг3+).
Сульфат меди, Cu2SO4, получают нагреванием сульфата ме-
ди(П) с металлической медью или диметил-, либо диэтилсульфата
МЕДЬ
701
с Cu2O при 160° в отсутствие следов воды:
CuSO4 -|- Cu = CU2SO4 (СНз)2ЗО4 4- Си2О —> CU2SO4 4- СНз — О — СН3
Соединение Cu2SO4 представляет собой серый порошок, который
устойчив в сухом воздухе на холоду и разлагается под действием
влаги. При нагревании сульфата меди до 100° в сухом воздухе
образуется основная соль CuSO4’CuO.
| Сульфат меди растворяется в конц. НС1, уксусной кислоте
и аммиаке.
Известны также сульфаты [Cu(NH3)2]2SO4-2Н2О, [CuCO]2SO4-
•Н2О.
Селенид меди, Cu2Se, образуется путем восстановления селе-
нида меди(П) CuSe водородом или селената меди(П) CuSeO4 углем
при нагревании, а также действием закиси меди(1) на двуокись
селена.
2CuSe + Н2 = Cu2Se 4- H2Se
2CuSeO4 + 6G = Cu2Se + SeO2 + 6GO
4Cu2O 4- 7SeO2 = Cu2Se 4- 6CuS§03
Соединение Cu2Se представляет собой черный или темно-зеле-
ный порошок; он способен разлагать растворимые соли золота
и серебра.
Теллурид меди, Си2Те, получают действием паров теллура
на сильно нагретую медь или действием теллурида алюминия
на солянокислые растворы хлорида меди(1), а также действием
закиси меди на бромид теллура.
Соединение Сп2Те существует либо в виде мелких октаэдриче-
ских кристаллов серого цвета, либо в виде черного порошка.
Нитрат меди, CuNO3, получают в виде белого порошка, всег-
да загрязненного Cu(NO3)2, в результате' диссоциации нитрата
диамминмеди(Т) при потере аммиака. Нитрат диамминмеди(1)
получают восстановлением аммиачных растворов нитрата меди(П)
металлической медью:
[Си(ГШз)4](КОз)2 + Си 2[Gu(NH3)2]NO3
[Си(КНз)2]МОз -> CuNO3 4- 2NH3
Цианид меди, CuCN, получают действием водных растворов
цианистоводородной кислоты на закись меди, цианистоводородной
кислоты или цианидом калия на хлорид меди(1), обработкой рас-
твора хлорида меди(П) цианистоводородной кислотой в присут-
ствии газообразного SO2 или действием цианида щелочного метал-
ла на сульфат меди(П) в присутствии сульфита щелочного металла:
Си2О 4- 2HCN = 2GuCN 4- Н2О
CuCl 4- HCN = CuCN 4- HCl CuCl -|- KCN = CuCN 4- КС 1
2CuC12 4- 2HCN 4- SO2 4- H2O = 2CuCN 4- H2SO4 4- 4HC1
2CuSO4 4- 2KCN 4- Na2SO3 4- H2O = 2CuCN 4- 2KHSO4 4- Na2SO4
702
ПОБОЧНАЯ ПОДГРУППА I ГРУППЫ
Цианид меди(1) представляет собой белые клиноромбические
кристаллы с т. пл. 473°; при сильном нагревании он разлагается,
плохо растворим в воде и растворяется в НС1, H2SO4, аммиаке,
солях аммония, цианидах щелочных металлов.
При растворении хлорида меди(1) в растворах, содержа-
щих избыток цианидов щелочных металлов, образуются комп-
лексные цианиды типов MeI[Cu(CN)2], Me*[Cu(CN)3], Me*[Cu(CN)4],
MeI[Cu3(CN)4], Mei[Cu2(CN)5l, Mei[Cu3 (CN)5], Me4Cu2(CN)3] (где
Me1 = Na+, K+, Rb+, Cs+, NH4+, ^Ca2*, VjSr2*, ^Ba2*, ^Mg2^
1/2zn2n.
Тиоцианат меди, CuSCN, образуется в виде белого осадка
при обработке растворов солей меди(11) тиоцианатом калия в при-
сутствии таких восстановителей, как сульфат железа(П), двуокись
серы или сульфиты щелочных металлов:
2CuSO4 + 2KSCN + SO2 + 2Н2О = 2CuSCN + 2KHSO4 + H2SO4
Растворением тиоцианата меди в растворах тиоцианатов щелоч-
ных металлов или аммония получают комплексные тиоцианаты
одновалентной меди, например Cs[Cu(SCN)2] и K8[Cu(SCN)7].
Ацетат меди, СнСН3СОО, образуется в результате нагрева-
ния ацетата меди(П) при 270°; это белые игольчатые кристаллы,
которые разлагаются в воде и выветриваются на воздухе.
Ацетиленид меди, Сн2С2-Н2О, получают пропусканием ацети-
лена через аммиачный раствор соли меди(1).
Соединение Сн2С2-Н2О образуется в виде коричневого осадка,
который при высушивании (80—100°) над безводным СаС12 дегид-
ратируется до Сн2С2; ацетиленид меди плохо растворяется в раз-
личных органических растворителях, разлагается со взрывом
при нагревании (120°), при ударе или под действием сильного
света, а также в случае соприкосновения с окислителями, напри-
мер галогенами.
При растворении Сн2С2*Н2О в растворе KCN или в разб. НС1
выделяется ацетилен.
Гексафторосиликат меди, Cu2[SiF8], получают действием крем-
нефтористоводородной кислоты на металлическую медь:
H2[SiF6] + 2Cu = Cu2[SiF6] + Н2
Соединение Cu2[SiF8] представляет собой твердое вещество
красного цвета, которое разлагается при высокой температуре
по уравнению
Cu2[SiF6] -> 2CuF + SiF4
Координационные соединения
Известны многочисленные координационные соединения ме-
ди(1). Одновалентная медь входит в состав комплексных катионов,
анионов и комплексов типа неэлектролитов.
МЕДЬ
703
Примеры комплексных катионов и анионов меди(1):
[Cu(NH3)J+, IGuG12]-, [CuC13]2-, [CuCl4]3-, [CuBr2]-, [CuBr3]2-,
[Cul2]-, [CuS]-, [Cu4S3]2-," [Cu(SO3)2]3-, [Cu(CN)2]-, [Cu(CN)3]2-,
[Cu(CN)413-, [Cu2(CN)3]-, [Cu3(CN)5]2-, [Cu(SCN)2]-, [Cu(SCN)7]6-.
Растворы солей меди(1) (в которые бросают кристалл Na2SO3
для удержания меди в одновалентном состоянии) окрашиваются
в оранжево-красный цвет при добавлении а,а'-дипиридила, в фио-
летовый цвет при добавлении о-фенантролина, а с п-диметиламино-
бензилиденродамином они образуют красный осадок, отвечающий
формуле
Соединения двухвалентной меди
Известны многочисленные устойчивые соединения двухвалент-
ной меди, которые отличаются от соединений меди(1) и (III) цве-
том, устойчивостью, растворимостью в воде и других растворите-
лях.
Соединения меди(Н) синего цвета в гидратированном состоянии
благодаря гидратированному иону [Сп(Н2О)4]2+ и белого цвета
в безводном (дегидратированном) состоянии; они имеют характер-
ный спектр поглощения, обладают неприятным «металлическим»
вкусом, токсичны.
Большинство солей меди(П) растворимо в воде. Пониженная
растворимость наблюдается у CuS, Cu2[Fe(CN)8], CuC2O4<H2O'
и основных солей, например Cu(NO3)2 •3Cu(OH)2, Cu3(PO4)2-
Cu(OH)2 -2H2O, Cu3(AsO4)2 •Cu(OH)2 -6H2O, Cu3(AsO4)2-
•2Cu(OH)2-иН2О, CuCO3 •Cu(OH)2, 2CuCO3-Cu(OH)2,
Cu(CH3COO)2•Cu(OH)2 -5H2O, Cu(CH3COO)2-шСп(ОН)2-raH2O.
Катион Cu2+ обладает отчетливо выраженной способностью,
к образованию координационных соединений в солянокислой или
водно-аммиачной среде. Образование координационных соедине-
ний меди(П) в большинстве случаев сопровождается изменением
цвета и растворимости.
Одно и то же химическое соединение в зависимости от степени
гидратации и температуры может иметь различную окраску. Так,,
хлорид меди(Н) в твердом безводном состоянии зеленого цвета,
поскольку является аутокомплексом Cu[CuCl4], с небольшим коли-
чеством воды он коричневато-зеленого цвета, в то время как с боль-
шим количеством воды — синий за счет гидратации Си2+
704
ПОБОЧНАЯ ПОДГРУППА I ГРУППЫ
(Сп(Н2О)4]С12:
Cu[CuCl4] + 8Н2О 2[Cu(H2O)4]C12
При добавлении воды равновесие реакции смещается слева
направо, а при добавлении соляной кислоты (или хлорида калия)
равновесие сдвигается в обратную сторону.
При обработке растворов солей меди(Н) очень небольшими
количествами щелочей (или несколькими каплями аммиака) обра-
зуются зеленые осадки основных солей. В случае избытка щелочи
выпадает студнеобразная зеленовато-синяя Сп(ОН)2, которая при
легком нагревании превращается в окись меди(Н).
При действии винной или лимонной кислоты на растворы солей
меди(П) образуются растворимые хелатные соединения, которые
окрашены в интенсивно-синий цвет и не осаждаются щелочами.
В результате обработки растворов солей (или трудно раствори-
мых соединений) меди(Н) избытком аммиака образуются лазурно-
синие растворимые соединения, цвет которых обусловлен комп-
лексным катионом [Cu(NH3)4]2+.
Многие из плохо растворимых соединений меди(П) раство-
ряются в азотной кислоте (при нагревании) или в избытке раство-
ров цианидов щелочных металлов с образованием комплексных
цианидов. Среди органических реагентов на медь(П) упоминаются
щелочные ксантогенаты, динитрорезорцин (в присутствии серной
кислоты и ацетата натрия), пирогаллол (в присутствии сульфита
натрия), пиридин (в присутствии тиоцианата калия), бензидин
(в присутствии иодида или цианида), ацетон (в присутствии диз-
тилоксалата) и т. д.
В координационных соединениях ион меди(П) имеет координа-
ционное число 4 и редко 6.
Неорганические соединения
Окись меди, CuO, встречается в природе в окисленных зонах
залежей медных руд и называется черной медью, мелаконитом
или теноритом. Окись меди получают нагреванием металлической
меди выше 80°, нагреванием (50°) водной суспензии Сп(ОН)2, про-
каливанием нитрата или основного карбоната меди(П).
Си г/2О2 = СиО + 37,6 ккал Си(ОН)2 = СиО -|- Н2О
Cu(NO3)2 = СиО 2NO2 -|- V2O2 СиСО3-Си(ОН)2 = 2СиО -|- Н2О -|- СО2
Окись меди представляет собой парамагнитный черный поро-
шок (или черные кубические кристаллы) с плотностью 5,83—
6,45 г!см3, т. пл. 1026° и твердостью 3—4 по шкале Мооса; она
плохо растворима в воде и растворяется в концентрированных
кислотах, при нагревании или в иодиде аммония.
МЕДЬ
705
При нагревании CuO с Cr2O3, Fe2O3, UO3 образуются хромиты,
ферриты и уранаты меди.
Окись меди растворяется в стекле, эмалях и т. д., придавая им
зеленовато-синюю окраску. Стекла, содержащие коллоидную медь,
окрашены в пурпурно-красный цвет.
Окись меди может быть восстановлена водородом, окисью угле-
рода, различными углеводородами, углеродом, кремнием, бромом,
бериллием, магнием, кальцием, натрием, калием, карбидом каль-
ция, карбидом алюминия.
СиО + Н2 — Си -|- Н2О + 30 ккал
2СиО -|- С — 2Си СО2 + 20,6 ккал
СиО -|- СО = Си -|- СО2 ЗСиО 4- 2Fe = ЗСи 4~ Fe2O3
СиО 4- Be = Си -|- ВеО ЗСиО 4- 2А1 = ЗСи 4- 2А12О3
5СиО 4- СаС2 = 5Си 4- СаО 4- 2СО2
12СиО 4- С3А14 = 12Си 4- 2А12О3 -}- ЗСО2
Горячие растворы хлорида олова(П), солей железа(П) и арсе-
нитов взаимодействуют с окисью меди:
2СиО 4- SnCl2 = 2CuCl 4- SnO2
ЗСиО 4- 2FeCl2 = 2СиС1 4- СиС12 -|- Fe2O3
2СиО 4- Na3AsO3 = Си2О 4- Na3AsO4
Под действием полисульфидов щелочных металлов или аммо-
ния окись меди превращается в сульфид CuS.
При нагревании окись меди взаимодействует также с треххло-
ристым фосфором, аммиаком и цианидами щелочных металлов:
17СиО 4- 5РС13 = 2Си3(РО4)2 4- CuCl2 4- lOCuCl 4- РОС13
ЗСиО 4- 2NH3 = ЗСи 4- N2 4- ЗН2О
Она каталитически восстанавливает перекись водорода, гипо-
хлориты и гипобромиты щелочных металлов.
Окись меди применяется в производстве стекла и эмалей в ка-
честве зеленого пигмента; в микроанализе она используется для
определения углерода, водорода и азота в органических соеди-
нениях.
Гидроокись меди, Сп(ОН)2, образуется в виде студнеобразного
синего осадка при обработке на холоду растворов солей меди(П)
растворами щелочей при pH = 5—5,5:
CuSO4 4- 2NaOH = Cu(OH)2 4- Na2SO4
Реакцию проводят на холоду, поскольку при нагревании обра-
зуется окись меди по уравнению
Cu(OH)2 = СиО 4- Н2О
Трудно растворимое в воде соединение Сп(ОН)2 имеет довольно
слабые основные свойства; оно растворяется в кислотах с образо-
45-0101
706
ПОБОЧНАЯ ПОДГРУППА I ГРУППЫ
ванием солей или в растворах щелочей, карбонатов, дианидов или
тиосульфатов щелочных металлов, не обладающих восстанови-
тельными свойствами сахаров, крахмала, глицерина, белков
с образованием комплексных соединений.
Cu(OH)2 + 2HNO-, = Cu(NO3)2 + 2Н2О
Cu(OH)2 + 2NaOH = Na2[Cu(OH)4]
2Cu(OH)2 + 6KCN = 2K[Cu(CN)2] + 4KOH + (CN)2
Cu(OH)2 + 4NH4OH = [Cu(NH3)4](OH)2 + 4H2O
При растворении Cu(OH)2 в аммиаке образуется реактив Швей-
цера [Cu(NH3)4](OH)2, который служит для растворения целлю-
лозы, гидроцеллюлозы и нитроцеллюлозы.
Устойчивый порошок Сн(ОН)2 используется в качестве пиг-
мента под названием «бремовой сини».
Фторид меди, CuF2, получают нагреванием топко измельчен-
ной меди в атмосфере фтора. При обработке спиртом раствора
окиси, гидроокиси или основного карбоната меди(П) в плавиковой
кислоте образуется синий, плохо растворимый дигидрат фторида
меди CuF2-2H2O. Упариванием над конц. H2SO4 раствора основ-
ного карбоната меди в избытке концентрированной плавиковой
кислоты выделяют синие кристаллы CuF2-5HF-6H2O.
Соединение CuF2 представляет собой белые мелкие кристаллы
с т. пл. 927°, фторид меди растворим в аммиаке, плавиковой, соля-
ной и азотной кислотах, в пиридине и этилацетате, восстанавли-
вается до металлической меди водородом при нагревании, разла-
гается перегретыми парами воды по уравнению
CuF2 + Н2О =₽* CuO + 2HF
При обычной температуре гидратированный фторид меди мед-
ленно разлагается с выделением фтористого водорода и образова-
нием основного фторида меди(Н):
CuF2-2H2O CuOHF + HF + Н2О
Фторид меди с фторидами щелочных или других металлов,
а также аммония образует фторосоли типов Me4CuF3l и Me*[CuF4]
(где Me1 = К+, Rb+, NH4).
Соединение (NH4)2[CuF4]-2Н2О выделяется в виде нераствори-
мых в воде синих кристаллов в результате упаривания концен-
трированного раствора NH4C1, насыщенного свежеприготовлен-
ным Сн(ОН)2.
Основные фториды (оксифториды) мед и(П).
Соединение CuF2-CuO черного цвета образуется при действии фто-
МЕДЬ
707
ра на CuO или на нагретый докрасна сульфат CuSO4.
Соединение CuF2-Cu(OH)2 в виде бледно-зеленого порошка
образуется путем разложения CuF2-2H2O (при нагревании),
в результате обработки раствора плавиковой кислоты избытком
окиси или основного карбоната меди(П), а также при действии
KF на раствор сульфата или хлорида меди(Н).
Димер безводного хлорида меди, Сн2С14 или Сн[СиС14], получают
действием избытка хлора на тонко измельченную медь или хлорид
меди(1), дегидратацией гидрата хлорида меди(П) (нагреванием
или в вакууме), действием газообразного НС1 на безводный суль-
фат меди, обработкой гидратированного хлорида меди конц.
H2SO4.
2Cu 4- 2С13 = Cu2Cl4 4~ 106,8 ккал
2CuCl -f- С12 = Си2С14 4~ 31,8 ккал
2CuSO4 4- 4НС1 = Cu2Cl4 4- 2H2SO4
Нагревание *
2CuCl2-nH2O ----—Cu2Cl4 + 2nH2O
КОНЦ. Н2ЬС>4
Соединение Сн2С14 представляет собой моноклинные кристаллы
темно-коричневого цвета с плотностью 3,054 г!см3, т. пл. 537°
и т. кип. 954—1032° (в атмосфере хлора); при нагревании на воз-
духе превращается в оксихлорид и окись меди.
Нагревание Си2С14 выше 344° в атмосфере инертного газа при-
водит к образованию хлорида меди(1) и хлора:
Cu2Cl4 2СиС1 4- С12
Хлорид меди растворяется в воде, жидком аммиаке, пиридине,
пиперидине, хинолине, ацетоне, эфире, этилацетате, метаноле,
этаноле и др. с образованием координационных соединений.
При нагревании водород и окись железа(П) восстанавливают
Сн2С14 до металлической меди:
Си2С14 4- 2Н2 2Си 4- 4НС1
Си2С14 4- 6FeO -> 2Cu 4- 2FeCl2 4- 2Fe2O3
Известно несколько кристаллогидратов СнС12-гаН2О (где п =
= 1, 2, 3, 4). Моногидрат хлорида меди СнС12-Н2О представляет
собой желтовато-синие кристаллы.
Дигидрат хлорида меди, СнС12-2Н2О или
Г С1 п
I
Н2О -> Си он2 ,
С1
образует зеленые орторомбические кристаллы плотностью
2,47 г/см3 и т. пл. 110°; при обычной температуре в присутствии
45*
708
ПОБОЧНАЯ ПОДГРУППА I ГРУППЫ
конц. H2SO4 или Р4О10 происходит дегидратация, а выдерживание
Сн2С14 во влажном воздухе или кристаллизация хлорида меди(П)
из горячих спиртовых растворов вновь приводит к образованию
дигидрата.
Три- и тетрагидраты хлорида меди, СнС12 -ЗН2О и СнС12 >4Н2О,
выпадают в виде синих кристаллов при концентрировании водных
растворов хлорида меди(П) (ниже 0°).
Цвет водных растворов хлорида меди(Н) зависит от температу-
ры и концентрации. Водные растворы средних концентраций окра-
шены в зеленый цвет на холоду и желтый или даже коричневый
при нагревании. Концентрированные растворы, будучи нагреты,
обладают желтым цветом, а при разбавлении теплой водой ста-
новятся зелеными, зеленовато-синими и даже синими. В водных
растворах хлорида меди(П) средней концентрации присутству-
ют зеленые ионы [СнС14]2-, а в разбавленных — синие ионы
[Сн(Н2О)4]2+.
Cu[CuC14] + 8Н2О 2[Си(Н2О)4]С12
Если к синему разбавленному раствору хлорида меди(П) доба-
вить хлорид калия, появляется зеленая окраска.
Хлорид меди образует с соляной кислотой соединения СиС12-
• НСЬ2Н2О, СнС12-2НС1 -5Н2О, СнС12-ЗНС1-иН2О.
Красные кристаллы CuCl2 -НС1 -2Н2О выделяются при действии
газообразного НС1 на гидратированный хлорид меди(11) или при
охлаждении до 0° концентрированного раствора хлорида меди(П),
насыщенного газообразным НС1.
Игольчатые кристаллы гранатового цвета СнС12-2НС1 >5Н2О
выпадают при охлаждении до —10° раствора хлорида меди(П),
насыщенного газообразным НС1.
Соединение СнС12-ЗНС1-гаН2О представляет собой красные
кристаллы.
Хлорид меди образует с хлоридами щелочных металлов, аммо-
ния, ртути(П), свинца(Н) хлоросоли типов МеЧСиСЦ], МеЧСиС13] •
• 2Н2О, Ме« [СиС14]-2Н2О (где Me1 = Li+, К+, Cs+, NH4+, x/2Hg2+,
x/2 Pb2+).
Соединение Li[CuCl3] «2Н2О осаждают путем упаривания кон-
центрированного раствора смеси хлоридов меди(П) и лития; это
призматические кристаллы гранатового цвета с т. пл. 130°.
Красные игольчатые кристаллы соединения К[СнС13] выпа-
дают в осадок из солянокислых растворов смеси хлоридов меди(П)
и калия.
Соединение NHJCuClJ ’2Н2О осаждается в виде синих кри-
сталлов при добавлении NH4C1 к соляной кислоте, насыщенной
основным карбонатом меди(П).
Упаривание раствора, полученного действием аммиака на теп-
лый насыщенный раствор хлорида меди(П), приводит к осажде-
МЕДЬ
709
нию темно-синих октаэдрических кристаллов [Cu(NH3)41C12-2Н2О.
Фосфор, ртуть, серебро, хлорид олова(П) и глюкоза являются
восстановителями по отношению к хлориду меди(П).
Растворы хлорида меди в присутствии металлической меди
интенсивно поглощают окись углерода.
Хлорид меди служит для приготовления некоторых красите-
лей, а также применяется в качестве катализатора и в пиротех-
нике.
Известно несколько основных хлоридов меди, например СпС12.2
или ЗСпО, СпС12-СпО-НгО, СпС12 -4СпО -6Н2О, СпС12-6СпО*
• 9Н2О, СиС12-8СпО-12Н2О.
Бромид меди (бромная медь), СпВг2, образуется в водном рас-
творе при обработке металлической меди избытком жидкого брома
в присутствии воды или при нейтрализации бромистоводородной
кислоты закисью, гидроокисью или основным карбонатом меди(П):
Си + Вг2 (жидкий) = СиВг2 -|-41,3 ккал
2НВг + Cu(OH)2 = СиВг2 + 2Н2О 2НВг + СиО = СиВг2 + Н2О
4НВг + СиСО3-Си(ОН)2 = 2СиВг2 + ЗН2О + СО2
Концентрированием водного раствора бромной меди (20°)
выделяют его тетрагидрат СиВг2>4Н2О, а упариванием того же
раствора в вакууме над конц. H2SO4 — кристаллы СиВг2.
Черные клиноромбические кристаллы цепной структуры
плавятся при 327°, растворяются в воде или спирте^с образова-
нием коричневого раствора, в ацетоне с образованием зеленого
раствора и в пиридине с образованием синего раствора.
Дигидрат бромида меди, СпВг2-2Н2О, представляет собой бле-
стящие зеленые клиноромбические иглы, выветривающиеся в су-
хом воздухе и расплывающиеся во влажном воздухе.
Концентрированные водные растворы бромной меди окрашены
в красно-коричневый цвет, разбавленные же растворы на холо-
ду — в синий и при нагревании — в зеленый цвет.
Пропусканием газообразного НВг через концентрированный
раствор бромной меди можно осадить черные клиноромбические
кристаллы СпВг2, которые растворяются в малом количестве воды,
а из пурпурных растворов на холоду выпадают черные кристаллы
Н[СпВг3] '2Н2О. Известен также декагидрат этой кислоты
Н[СпВг3Ы0Н2О.
Бромосоли типа МеЧСпВг3] (где Me1 = Li+, К+, Cs+) окрашены
в пурпурно-красный цвет.
710
ПОБОЧНАЯ ПОДГРУППА I ГРУППЫ
В результате действия аммиака на бромную медь можно полу-
чить соединения
Вг2 л
Си
L (NH3)d ,
Г Вг 1
Си
L (NH3)3J
[Cu(NH3)4]Br2,
nh4
Вг3 ~|
Си
NH3J
(NH4)2[CuBr4]-H2O
По химическим свойствам бромид меди похож на хлорид меди.
При добавлении окиси или гидроокиси меди(П) к холодным
растворам бромной меди(Н) получается основная соль СпВг2 •
• ЗСиО-ЗН2О.
Иодид меди, Сп12 (иодная медь), и полииодиды меди.
До настоящего времени иодид меди(П) не выделен, поскольку
при обработке растворов солей меди(П) растворами иодидов
щелочных металлов образуется иодид меди(1) и выделяется иод:
2CuSO4 + 4KI = 2CuI 4- 2K2SO4 + I2
При энергичном перемешивании раствора сульфата меди
(средней концентрации) с твердым иодидом калия выпадает (через
определенное время) плотное зеленое вещество, частично раство-
римое в воде. Его формула Сп14. Нагревание Сн14 до 80° с иодом
вызывает образование Сп18, который окрашивается в синий цвет
(характерный для иона Сн2+) при перемешивании с иодидом одно-
валентной меди:
Cul6 + 4CuI 5CuI2
Обработка аммиаком дииодида или полииодидов меди дает
соединения [Cu(NH3)4]I2, [Cu(NH3)4]I4, [Cu(NH3)4]Ie.
Известна также соль К2(Сп14].
Взаимодействие раствора иодида калия с гидроокисью меди(Н)
приводит к образованию различных основных солей (зеленого
цвета): CuI2.Cu(OH)2, Cul2-3Cu(OH)2-9Н2О.
Хлорат меди, Сн(СЮ3)2’6Н2О, осаждается при упаривании
в вакууме раствора Си(ОН)2 в хлорноватой кислоте или при дей-
ствии Ва(С1О3)2 на раствор сульфата меди(П):
Си(ОН)2 + 2НС1О3 = Си(СЮ3)2 + 2Н2О
CuSO4 + Ва(СЮ3)2 = Си(СЮ3)2 + BaSO4
Соединение Сп(С1О3)2-6Н2О представляет собой расплываю-
щиеся на воздухе синие октаэдрические кристаллы, которые пла-
вятся при 65° и растворяются в воде и спирте.
Основной хлорат меди, Си(С1О3)2-ЗСпО-ЗН2О, получают раз-
ложением кристаллогидрата Сн(С1О3)2-6Н2О при 100° или дей-
ствием Сп(ОН)2 на водный раствор хлората меди(Н).
МЕДЬ
711
Соединение Cu(CIO3)2-ЗСиО-ЗН2О представляет собой зеленые
кристаллы (или синий порошок), плохо растворимые в воде и рас-
творимые в разбавленных минеральных кислотах.
Бромат меди, Сн(ВгО3)2-6Н2О, выпадает в осадок из раство-
ра, полученного нейтрализацией основного карбоната меди(П)
водным раствором бромноватой кислоты.
Соединение Сп(ВгО3)2-6Н2О представляет собой сине-зеленые
кристаллы, которые при нагревании до 200° превращаются в зеле-
ный порошок Си(ВгО3)2, а при нагревании до более высокой тем-
пературы — в СпО и основной бромид меди(П). Известен основной
бромат меди(П), Сп(ВгО3)2-5СпО-ЮНгО,— зеленый порошок,
плохо растворимый в воде.
Иодат меди, Сп(Ю3)2-Н2О, получают добавлением соли
меди(П) к концентрированному раствору йодноватой кислоты или
обработкой азотной кислотой (добавляемой по каплям) смеси
иодида калия с нитратом меди(П).
Соединение Сн(Ю3)2-Н2О представляет собой синие триклин-
ные кристаллы, которые плохо растворяются в воде, растворимы
в аммиаке, превращаются в зеленую безводную соль Cu(IO3)2
при нагревании до 240° и в СпО при нагревании до более высокой
температуры.
Обработка окиси меди(П) йодноватой кислотой дает основной
иодат меди(Н) СпОНЮ3 зеленого цвета.
Перйодаты мед и(Н). Ортопериодат меди, Си2НЮ8-
•нН2О (где n = 1, 2, 3), в виде зеленовато-желтого кристалли-
ческого порошка образуется при высушивании (45°) осадка, полу-
ченного действием ортопериодной кислоты на суспензию основного
карбоната меди(Н) в воде или ортопериодата натрия на избыток
сульфата меди(П):
CuCO3-Cu(OH)2 + H5IO6 = Cu2HIO6 + ЗН2О + СО2
2CuSO4 + Na2H3IOe = Cu2HIOe + 2NaHSO4
Ортопериодат меди, Cu5(IO6)2-нНгО (где п = 3, 5, 7), пред-
ставляет собой зеленый порошок.
Дипериодат меди, Сп212О9-6Н2О, выпадает в виде зеленых
кристаллов в результате упаривания раствора гидроокиси меди(П)
в водном растворе ортоиодной кислоты:
2Си(ОН)2 + 2Н5Юв = Cu2I2O9 + 7Н2О
Метапериодат меди, Cu(IO4)2, получают в виде синего осадка
при кипячении раствора ацетата меди(П) с ортоиодной кислотой:
Cu(CH3COO)2 + 2H6IO6 = Cu(IO4)2 + 2СН3СООН + 4Н2О
Сульфид меди, CuS, встречается в природе в виде минерала
ковеллина; в лаборатории может быть получен действием серо-
водорода (или сульфидов щелочных металлов) на растворы солей
712
ПОБОЧНАЯ ПОДГРУППА I ГРУППЫ
меди(П) в кислой среде, нагреванием металлической меди в парах
серы, нагреванием сульфида Cu2S с серным цветом, действием
тиоцианата аммония на сульфат меди(П) при 810°.
CuSO4 + H2S = CuS + H2SO4 Си + S CuS
Cu2S S = 2CuS
CuSO4 + 2NH4SCN = CuS + 2SO2 + 2NH3 + 2HCN
Обработка аммиачных растворов солей меди(П) сероводородом
вызывает образование коллоидного сульфида меди(П) коричнево-
черного цвета, который флюоресцирует зеленым цветом и коагули-
рует под действием электролитов.
Сульфид меди представляет собой черный порошок с плотно-
стью 3,9 г/сл3; он плохо растворим в воде, щелочах, аммиаке и рас-
творяется в растворах цианидов щелочных металлов, при нагре-
вании в конц. H2SO4 или HNO3.
CuS + 2H2SO4 = CuSO4 4- SO2 + S + 2H2O
6CuS + 22HNO3 = 6Cu(NO3)2 + 3H2SO4 + 3S + 10NO + 8H2O
Взаимодействие сульфида меди с раствором AgNO3 дает нитрат
меди(П) и сульфид серебра:
CuS + 2AgNO3 = Cu(NO3)2 + Ag2S
Во влажном воздухе под действием озона сульфид меди пре-
вращается в сульфат меди(П).
Нагревание сульфида меди(Н) до температуры красного кале-
ния без доступа воздуха приводит к образованию Cu2S, а нагрева-
ние до более высокой температуры вызывает разложение CuS
на элементы.
Водород восстанавливает сульфид меди (600°) до металличе-
ской меди.
Известны полисульфиды меди: Cu2S3, Cu2S5, Cu2S8,
Cu2S7 — все аморфные вещества коричневого цвета. Получен
также основной сульфид 5CuS •CuO.
Сульфит меди, CuSO3,— очень неустойчивое соединение, полу-
чается обработкой основного карбоната или гидроокиси меди(П)
водой, насыщенной газообразным SO2.
Известны основные сульфиты: 2CuSO3 •Си(ОН)2 •1/2Н2О жел-
того и CuSO3-ЗСиО-7Н20 зеленовато-желтого цвета.
Безводный сульфат меди, CuSO4, получают дегидратацией его
гидратов при 250° или действием конц. H2SO4, обработкой конц.
H2SO4 металлической меди при нагревании, действием диметил-
или диэтилсульфата на окись меди(П).
Конц. H2SO4
CuSO4-5H2O —2^5"* CuSO4
Си + 2H2SO4 = CuSO4 + SO2 + 2H2O
CuO + (CH3)2SO4 = CuSO4 + CH3 — О — CH3
МЕДЬ
713
Сульфат меди представляет собой белые орторомбические приз-
матические кристаллы с плотностью 3,6 г/сл3; под действием
влаги воздуха они превращаются в CuSO4 -5Н2О, растворяются
в воде, глицерине, серной кислоте и различных аминах, плохо
растворимы в жидком аммиаке.
При нагревании CuSO4 образуется основная соль, а затем чер-
ная окись меди(П).
Водород, окись углерода и углерод при нагревании восстанав-
ливают сульфат меди.
Безводный сульфат меди, обладая свойством жадно поглощать
воду и будучи плохо растворим в спирте, служит для его абсолю-
тирования.
Известно несколько кристаллогидратов сульфата меди CuSO4-
• гаН2О (где п = 1, 3, 5, 6 и 7).
Моногидрат, CuSO4-H2O, образуется дегидратацией CuSO4-
• 5Н2О при 100° или в вакууме над конц. H2SO4 (или Р4О10)
и представляет собой зеленовато-белый порошок с плотностью
3,2 г/см3, жадно поглощающий воду и разлагающийся при нагре-
вании выше 200°.
Тригидрат, CuSO4-3H2O, в виде синего порошка с плотностью
2,66 г/см3 образуется при хранении CuSO4-5H2O на воздухе при
обычной температуре или в результате кристаллизации сульфата
меди(П) из раствора, содержащего 50—55% H2SO4.
Пентагидрат, CuSO4-5H2O или [Cu(H2O)4]SO4-Н2О, назы-
ваемый медным купоросом,— наиболее устойчивый кристалло-
гидрат сульфата меди; он представляет собой триклинные кристал-
лы в форме параллелепипедов, синего цвета, с плотностью
2,27 г/см3; при хранении на воздухе пентагидрат превращается
в растворимое в воде соединение CuSO4-3H2O.
При нагревании пентагидрат сульфата меди дегидратируется
следующим образом:
100° 150° 250°
CuSO4.5H2O —CuSO4-3H2O ——> CuSO4.H2O —CuSO4
— ZH2O — 2H2O — H20
Бесцветный
Водные растворы сульфата меди окрашены в синий цвет, обла-
дают кислой реакцией благодаря гидролизу и могут быть полу-
чены растворением меди, окиси, гидроокиси или основного карбо-
ната меди(П) в серной кислоте.
CuSO4 + 2Н2О Cu(OH)2 + H2SO4
Из концентрированных растворов сульфата меди(П) выпа-
дают синие кристаллы CuSO4-5H2O.
Сульфат меди образует с сульфатами щелочных металлов или
аммония изоморфные двойные сульфаты типа MeISO4 -CuSO4 -6Н2О
(где Mei = NHJ, К+, Rb+, Cs+).
714
ПОБОЧНАЯ ПОДГРУППА I ГРУППЫ
Известны смешанные сульфаты, образованные из сульфата
меди(П) с сульфатом марганца, цинка, кадмия, кобальта, никеля,
магния или железа(П).
Действием избытка газообразного NH3 на гидратированный
сульфат меди(П) получают соединение [Cu(NH3)4]SO4 -Н2О.
При обработке водного раствора сульфата меди сильно раз-
бавленным раствором NH4OH осаждается основной сульфат
Cu2(OH)2SO4 зеленого цвета:
2CuSO4 + 2NH4OH = Cu2(OH)2SO4 + (NH4)2SO4
Сульфат меди образует с гидроксиламином комплекс
[CuNH2OH]SO4, который разлагается в воде и устойчив на воз-
духе, в эфире и абсолютном спирте.
При действии окиси углерода на раствор сульфата меди, в ко-
тором суспендирована тонко измельченная медь, образуется соеди-
нение [CuCO]2SO4-Н2О.
Сульфат меди служит для получения многих соединений меди.
Кроме того, он применяется в элементах Даниэля, в гальвано-
пластике, при электролитическом рафинировании меди, в каче-
стве антисептика, для обработки семян и получения химических
средств защиты растений от грибковых заболеваний.
Известны основные сульфаты меди, например SO3-4CuO •гаН2О,
SO3-8CuO-12Н2О, SO3 ПОСнО-гаН2О (где п = 2; 2,5; 3; 3,5; 4;
5; 16).
Селенид меди, CuSe, образуется при барботировании селеново-
дорода H2Se через раствор сульфата меди(Н), действии паров
селена на металлическую медь или нагревании хлорида меди(П)
в токе селеноводорода.
CuSO4 + H2Se = CuSe + H2SO4 Cu + Se = CuSe
CuCl2 + H2Se = CuSe + 2HC1
Селенид меди представляет собой черные игольчатые кристал-
лы, плохо растворимые в воде и разлагающиеся при нагревании
по уравнению
2CuSe Cu2Se -ф Se
Селенит меди, CuSeO4-5H2O, осаждается в результате упари-
вания (40—50°) раствора окиси или гидроокиси меди(Н) в селе-
новой кислоте.
Соединение CuSeO4-5H2O представляет собой очень устойчи-
вые синие кристаллы с плотностью 2,6 г/см3‘, они растворимы
в воде и разлагаются при нагревании:
110° 250°
CuSeO4-5H2O —CuSeO4-H2O —
— 4Н2О —М2О
—► CuSeO4 — CuSeO34-i'2O2 —SeO2 + CuO
МЕДЬ
715
Дигидрат селената меди CuSeO4-2H2O представляет собой
бесцветные микрокристаллы, а моногидрат CuSeO4-H2O — белый
порошок.
Известны двойные селенаты типа Me|SeO4 -CuSeO4 -6Н2О (где
Me1 = К+, Na+, NH+) и основной селенат SeO3-4CuO-гаН2О (где
п = 2, 3, 4).
Нитрат меди, Cu(NO3)2 -лгН2О (где п = 3, 6, 7), выпадает
из концентрированных растворов металлической меди в концен-
трированной азотной кислоте или растворов окиси, гидроокиси,
основного карбоната меди(П) в разб. HNO3.
3Cu + 8HNO3 = 3Cu(NO3)2 -i- 2NO + 4Н2О
СиО + 2HNO3 = Cu(NO3)2 + Н2О Си(ОН)2 + 2HNO3 = Cu(NO3)2 + 2Н2О
Зелено-белая безводная соль, Cu(NO3)2, выпадает в осадок при
охлаждении раствора тригидрата Cu(NO3)2-ЗН2О в дымящей
HNO3 или при перемешивании (0°) ацетилнитрата с окисью
меди(П).
Тригидрат, Cu(NO3)2 -ЗН2О, осаждается из концентрирован-
ных растворов нитрата меди(П) при температуре ниже 25° и пред-
ставляет собой синие призматические кристаллы с плотностью
2,047 г!см3 и т. пл. 114,5°; они неприятны на вкус, оказывают
разъедающее действие, легко растворяются в воде, спирте, разб.
HNO3.
Гексагидрат, Cu(NO3)2-6Н2О, осаждается в результате кон-
центрирования водных растворов нитрата меди(П) в интервале
температур между —20 и 24,5°; это синие, расплывающиеся на воз-
духе кристаллы, которые плавятся при 26,4°, очень хорошо раст-
воримы в воде, спирте и разб. HNO3.
Cu(NO3)2-ЗН2О ЗН2О (тверд.) -> Cu(NO3)2-6Н2О 3,9 ккал
Синий кристаллогидрат, Cu(NO3)2 -9Н2О, очень хорошо рас-
творим в воде и спирте и выпадает из водных растворов нитрата
меди(П) при температуре —24°.
Сильное нагревание вызывает термическое разложение нитрата
меди.
Cu(NO3)2 —> CuO 2NO2 -J- 1/2O2
Нитрат меди является энергичным окислителем, например он
окисляет фосфор в фосфорноватую кислоту. Известны двойные
нитраты типа Me*nCu3(NO3)12-24Н2О (где Меш = Се3+, Рг3+,
Nd3+, Sm3+).
Нитрат тетраммина меди, [Cu(NH3)4](NO3)2, образуется в ви-
де синих орторомбических кристаллов при пропускании аммиака
в теплый насыщенный раствор нитрата меди.
Основной нитрат меди, Cu(NO3)2-ЗСп(ОН)2, получают кипя-
чением разбавленных растворов нитрата меди(П), обработкой
716
ПОБОЧНАЯ ПОДГРУППА I ГРУППЫ
гидратированной коричневой окиси СпО-гаН2О, гидроокиси или
основного карбоната меди(П) раствором нитрата меди(П), добав-
лением небольшого количества щелочи (или NH4OH) к раствору
нитрата меди(П), нагреванием (~186°) гидратированного нитрата
меди(П):
Cu(NO3)2-3H2O + ЗСиО = Cu(NO3)2-3Cu(OH)2 + 12,1 ккал
4Cu(NO3)2 + 6NaOH = Cu(NO3)2.3Cu(OH)2 + 6NaNO3
Соединение Cu(NO3)2-ЗСи(ОН)2 представляет собой зеленый
кристаллический порошок, плохо растворимый в воде и раство-
римый в разбавленных кислотах.
Фосфаты мед и(П). Ортодигидрофосфат меди,
Сп(Н2РО4)2, выпадает из очень кислых водных растворов Р4О10,
обработанных СпО; это гигроскопичные и легко гидролизующиеся
зеленые кристаллы.
Ортогидрофосфат меди, СпНРО4 -Н2О, осаждается из слабо
кислых водных растворов Р4О10, обработанных СпО, и представ-
ляют собой зеленые кристаллы с плотностью 3,4 г! см? и твердостью
3—4 по шкале Мооса. Если кислотность водного раствора
Р4О10, обработанного СпО, невысока, образуется основная соль
Сп3(РО4)2 -Сп(ОН)2.
При обработке фосфорной кислотой гидроокиси меди(П) обра-
зуется метастабильное соединение 2Cu3(PO4)2 -2СиНРО4 -7Н2О.
В природе встречается соединение Cu3(PO4)2 -Си(ОН)2 -2Н2О —
зеленые клиноромбические кристаллы с плотностью 4 г/см3.
Известны также многочисленные двойные фосфаты, например
KCuPO4-Cu3(PO4)2, Na2HPO4-Cu3(PO4)2, NaH2PO4 -3Cu3(PO4)2.
Пирофосфат (дифосфат) меди, Сп2Р2О7 -5Н2О, образует зеле-
ные кристаллы; он плохо растворим в воде, растворяется в аммиа-
ке (с образованием комплексов), разбавленных минеральных кис-
лотах, пирофосфатах щелочных металлов (с образованием двойных
солей) и разлагается щелочами, сероводородом или сульфидами
щелочных металлов.
Известен также основной пирофосфат меди Сп2Р2О7 -2Сп(ОН)2 •
• ЗН2О.
При нагревании окиси меди с фосфорной кислотой (плотность
1,68—1,75 г!смА) или нагревании до 316° продукта, полученного
упариванием досуха разбавленного раствора фосфорной кислоты
и нитрата меди(П), образуется метафосфат меди Сп(РО3)2 в виде
синего порошка, плохо растворимого в щелочах и большинстве
кислот.
Обработка спиртом смеси растворов сульфата меди и метафос-
фата натрия вызывает осаждение синего кристаллического порош-
ка Сп(РО3)2 •4Н2О.
При смешении концентрированных растворов хлорида меди(П)
и гексаметафосфата натрия образуется гексаметафосфат меди
МЕДЬ
717
Cu3(P3O9)2 в виде синего осадка, который превращается в стекло-
образную массу, плохо растворимую в воде.
Арсениты и арсенаты мед и(П). Известны много-
численные арсениты и арсенаты меди(П), например зелень Шееле
HCuAsO3, HCuAsO3-H2O, Cu2As2O5, Cu(AsO3)2-2Н2О, Cu(AsO2)2-
• 3CuO, Cu(H2AsO4)2-2H3AsO4, Cu(H2AsO4)2 -2H3AsO4-2H2O,
Cu(HAsO4) -H2O, Cu3(AsO4)2 -4H2O, Cu3(AsO4)2-Cu(OH)2,
Cu3(AsO4)2 -Cu(OH)2 -6H2O, Cu3(AsO4)2 -2Cu(OH)2, Cu3(AsO4)2 •
• 2Cu(OH)2 -7H2O, Cu3(AsO4)2 -5Cu(OH)2 -7H2O.
Карбонаты мед и(П). При действии карбонатов щелоч-
ных металлов на растворы солей меди(П) образуются основные
карбонаты меди(П), состав которых зависит от условий осаждения.
Малахит, СиСО3 -Сп(ОН)2 или О = С /2 2й 22 ,
\ О—xjll—\_/1±
встречается в природе в виде минерала; в лаборатории может
быть получен путем обработки растворов сульфата меди(П) сте-
хиометрически необходимым количеством карбоната натрия, кипя-
чением окиси меди(П) с раствором (NH4)2CO3, нагреванием СаСО3
до 200—250° с хлоридом меди(П) или с раствором сульфата
меди(П), электролизом растворов карбонатов щелочных металлов
с анодом из металлической меди.
Соединение СпСО3-Сп(ОН)2 представляет собой светло-зеле-
ный порошок, плохо растворимый в воде, жидком аммиаке, пири-
дине и хорошо растворимый в водных растворах газообразного
СО2, в растворах цианидов щелочных металлов, солей аммония
и в разбавленных кислотах. Известен также гидратированный
малахит СпСО3 -00(011)2 •1/2Н2О.
О—Си—ОН
Азурит, 2СиСО3-Си(ОН)2 или 0 =
/Си
Q
О = с/
х0 —Си—ОН,
образуется при обработке малахита газообразным СО2 (под дав-
лением 50 ат) или при нагревании карбоната кальция в растворе
нитрата меди(П); он представляет собой двупреломляющие кли-
норомбические кристаллы синего цвета с плотностью 3,5—
3,9 г/см* и твердостью 4 по шкале Мооса.
Известны двойные карбонаты типа Ме*[Си(СО3)2] (где Ме1 =
= Na+, К+), представляющие собой синие клиноромбические кри-
сталлы. При нагревании или под действием воды зти двойные кар-
бонаты разлагаются:
Me2[Cu(CO3)2] СиО + Ме2СО3-|-СО2 (при нагревании)
718
ПОБОЧНАЯ ПОДГРУППА I ГРУППЫ
Me2[Cu(CO3)2] [Cu(CO3)2]2- + 2MeI'|
> (в воде)
Cu2+ + 2CO|~ J
Цианид меди, Cu(CN)2, образуется в виде желто-коричневого
осадка в результате обработки сульфата меди(П) стехиометриче-
ски необходимым количеством цианида калия или действия циани-
стоводородной кислоты на ацетат меди(П):
CuSO4 + 2KCN = Cu(CN)2 -|- K2SO4
Cu(CH3COO)2 + 2HCN = Cu(CN)2 + 2CH3COOH
Цианид меди неустойчив, легко превращается в Cu[Cu(CN)2].
• Н2О и затем в цианид меди(1) с выделением дициана:
2Cu(CN)2 2CuCN + (CN)2
Тиоцианат меди, Cu(SCN)2, получают в виде черного кристал-
лического осадка путем обработки растворов солей меди(П) сте-
хиометрически необходимым количеством тиоцианата калия:
CuSO4 + 2KSCN = Cu(SCN)2 + K2SO4
Соединение Cu(SCN)2 мало устойчиво при соприкосновении
с водой (быстрее в присутствии сульфита или двуокиси серы
в водном растворе), превращается в тиоцианат меди(1):
2Cu(SCN)2 -> 2CuSCN -j- (SCN)2
2Cu(SCN)2 + H2SO3 + H2O = 2CuSCN + H2SO4 + 2HSCN
После быстрого высушивания тиоцианат меди(П) может сохра-
няться при обычной температуре длительное время.
Если к раствору соли меди(П) добавляют пиридин до появле-
ния синего окрашивания, а затем несколько капель раствора
тиоцианата щелочного металла, образуется горохово-зеленый оса-
док, обусловленный образованием комплекса [CuPy2(SCN)2]:
CuSO4 + 2Ру + 2KSCN = [CuPy2(SCN)2] + K2SO4
Соединение [CuPy2(SCN)2] растворяется в хлороформе и кисло-
тах, плохо растворимо в воде, применяется для количественного
определения меди.
Ацетат меди, Сп(СН3СОО)2-Н2О, выпадает в осадок в резуль-
тате концентрирования (30°) растворов окиси, основного карбоната
или основного ацетата меди(Н) в уксусной кислоте или при обра-
ботке сульфата меди(П) ацетатом кальция, бария или свинца.
СиО + 2СН3СООН = Си(СН3СОО)2 + Н2О
СиСО3-Си(ОН)2 + 4СН3СООН = 2Си(СН3СОО)2 + ЗН2О + СО2
CuSO4 + Ва(СН3СОО)2 = Си(СН3СОО)2 + BaSO4
МЕДЬ
719
Соединение Сн(СН3СОО)2 -Н2О представляет собой клинором-
бические кристаллы темно-зеленого (с синим оттенком) цвета,
легко растворимые в воде.
Кроме моногидрата ацетата меди(П) известен и пентагидрат
Сп(СН3СОО)2 -5Н2О — синие кристаллы, которые теряют кристал-
лизационную воду при 140° и разлагаются выше 250°.
Кипячение водных растворов ацетата меди(П) вызывает осаж-
дение синих кристаллов (растворимых в большом избытке аммиа-
ка) — основных ацетатов меди(П), например Сн(СН3СОО)2 •
• Сн(ОН)2 -5Н2О, 2Сн(СН3СОО)2 -Сп(ОН), -5Н2О, Сн(СН3СОО)2 •
• 2Сп(ОН)2, Сп(СН3СОО)2-ЗСн(ОН)2-2Н2Ь.
Основные ацетаты меди(П) применяются для борьбы с болез-
нями растений и в качестве пигмента для масляных и водно-эмуль-
сионных красок. Ацетат меди(П) с ацетатом аммония или ацета-
тами других элементов образует двойные ацетаты 2Сп(СН3СОО)2 •
• NH4CH3COO Н2О, Сп(СН3СОО)2-Са(СН3СОО)2-6Н2О.
При взаимодействии ацетата меди(П) с арсенитом меди(П)
образуется швейнфуртская зелень состава Сп(СН3СОО)2 •
• 3Cu(AsO2)2 или [Cu{Cu(AsO2)2}3](CH3COO)2,' которая, несмотря
на очень красивый оттенок, не используется в живописи, посколь-
ку под восстановительным действием плесени пигмент выделяет
токсичный AsH3.
Оксалат меди, СпС2О4-Н2О, в виде зеленовато-синего осадка
получают путем обработки растворов солей меди(П) стехиометри-
чески необходимым количеством щавелевой кислоты.
Взаимодействие соединения СпС2О4-Н2О с раствором оксалата
щелочного металла (или аммония) приводит к получению оксала-
тосоли типа Mei[Cu(C2O4)2] -2Н2О.
Ацетиленид меди, СпС2 •1/2Н2О, можно получить пропуска-
нием ацетилена через аммиачный раствор соли меди(П’.
Соединение СнС2 1/2Н2О представляет собой красно-коричне-
вый осадок, который при высушивании разлагается со взрывом,
а в свежеприготовленном виде легко разлагается разбавленными
кислотами с выделением ацетилена.
Гексафторосиликат меди, Cu[SiFe] -4Н2О, выделяется в виде
синих клиноромбических кристаллов при концентрировании (50°)
раствора, полученного действием гексафторокремниевой кислоты
на гидроокись меди(П):
Си(ОН)2 + H2[SiF6] = Cu[SiFe] + 2Н2О
Если концентрирование осуществляется при обычной темпера-
туре, выделяются синие гексагональные призматические кристал-
лы состава Gu[SiFe] -6Н2О, очень хорошо растворимые в воде.
При нагревании кристаллогидратов гексафторосиликата меди(П)
до 100° образуется безводная соль, которая при 120° разлагается
с выделением SiF4.
720
ПОБОЧНАЯ ПОДГРУППА I ГРУППЫ
Метаборат меди, Сп(ВО2)2, получают прокаливанием (900°)
соли, полученной упариванием досуха раствора, содержащего две
молекулы борной кислоты на молекулу нитрата меди(П).
Соединение Сн(ВО2)2 представляет собой двупреломляющие
синие кристаллы, очень твердые, плохо растворимые в воде и кис-
лотах, разлагающиеся при нагревании до 1150° с образованием
метабората меди(1) СнВО2.
Координационные соединения
Ион двухвалентной меди образует большое число устойчивых
координационных соединений с многочисленными химическими
реагентами, как органическими, так и неорганическими. Двухва-
лентная медь может входить и в состав комплексных катионов или
анионов, и в состав комплексов типа неэлектролитов.
Примеры комплексных анионов и катионов меди(П):
[Cu(OH)4]2~, [Cu(OH)e]4~, [CuCl3]-, [CuClJ2-, [CuBr3]-, [СиВгД",
[Cu(NH3)Br3]~, [Cu(CO3)2]2-, [Cu(H2O)4]2+, [Cu(NH3)4]2+,
[Cu(NH3)3Br]+, [Cu(CH3 — OH)2]2+, [Cu(C2H5 — OH)2]2+.
При обработке растворов солей меди(П) винной, лимонной или
аминоуксусной кислотой либо другими органическими оксипроиз-
водными образуются хелатные соединения. В присутствии винной
кислоты едкий натр не осаждает медь, раствор окрашивается
в интенсивно-синий цвет и образуется комплекс
ОН НО
II I I II
NaO —С —С —О —Си—О —С —С —ONa
I Z \ |
NaO —С —С —О О — С —С —ONa
II I I I I II
О Н Na Na Н О
Нагревание гидроокиси меди(П) с водным раствором амино-
уксусной кислоты приводит к получению синих кристаллов гли-
коколята меди:
Cu(OH)2+2H2N —СН2 —СООН
О = С —О О —С = О_.
zGu\
Н2С —N N — СН2
L Н2 Н2 J
+2Н2О
Ксантогенаты щелочных металлов образуют с растворами солей
меди(П) коричневый осадок ксантогената меди, который восста-
МЕДЬ
721
навливается до желтого ксантогената меди(1):
/ОС2Н5
CuSO4 + 2C = S
XSNa
/ОС2н5
c=s
Xs\
Cu +
/Sz
c=s
xoc2H5
/OC2H5
s=c
xsx
Cu ->
/S'7
s=c
xoc2H5
/OC2H5
C = S
xs
Cu-f-Na2SO4
S
cCs
xoc2H5
/ОС2Н5
s=c
/SCu xs
2C = S + |
XOC2H5 /S
S = C
XOC2H5
Дисульфид
ксантогената
Рубеановодородная кислота образует с подкисленными уксус-
ной кислотой растворами солей меди(П) зеленовато-черный оса-
док рубеаната меди(П):
NH = C — S NH = C — S NH = C—
ЛСи\ I ZCu\ | ZCu\ |
' Xs C = NH Xs C = NH Xs C = -
Обработка растворов солей меди(П) раствором нитрозофенил-
гидроксиламина (купферона) дает серовато-белый осадок вещества,
строение которого можно представить следующим образом:
гС8Н5 —N = O. .0 — N п
II >< II
L N —0х xO = N-C6H5J
или
гС8Н5—N —О 0 = N
I zCu\ ।
N = OZ x0 — N — C8H5J
Известен также диоксалатокупрат натрия Na2[Gu(C2O4)2] -2Н2О.
Соединения трехвалентной меди
По сравнению с соединениями меди(1) и (II) соединения трех-
валентной меди весьма немногочисленны.
Известно несколько координационных соединений меди(Ш),
очень устойчивых и строго определенного состава, которые обра-
46-0101
722
ПОБОЧНАЯ ПОДГРУППА I ГРУППЫ
зуются путем анодного окисления
ления солей меди(П) в щелочной
теллурат.
соединений меди(П) или окис-
среде, содержащей иодат или 1
Неорганические соединения
Окись меди, Си2О3, образуется окислением Сн(ОН)2 (—20®)
в 35%-ном растворе КОН с помощью K2S2Og, Na2S2, С12, Вг2,
КОС1, КОВг. Сначала раствор окрашивается в фиолетово-красный
(или коричнево-черный) цвет, а затем выпадают красные кристал-
лы Си2О3, которые растворимы в серной или соляной кислоте
и разлагаются при нагревании (начиная с 75°) по уравнению
Cu2O3 = 2СиО ^/2О2
Купрат(Ш) калия, КСиО2, синего цвета образуется при нагре-
вании (400—500°) окиси меди(П) с К2О4.
Гексафторокупрат(Ш.) калия, K3[CuF6], синего цвета, полу-
чается нагреванием до 300° смеси порошка металлической меди
и фторида калия или нагреванием в атмосфере фтора смеси метал-
лического калия и порошка металлической меди.
Диортопериодато-соединения мед и(П1).
Соединение К7[Си(Ю6)2] -7Н2О является производным кислоты
Н7[Си(Ю6)2] и может быть получено окислением надсернокислым
калием K2S2O8 (100°) 15%-ного щелочного раствора перйодата
калия и сульфата меди(П).
Соединения Na5K2[Cu(IO6)2] -(12 или 15) Н2О, Na7[Cu(IOe)2] •
• (12 или 16) Н2О, K3H4[Cu(IO6)2] -4Н2О представляют собой твер-
дые вещества коричневого цвета, а соединение Na4KH2[Cu(IO6)2] •
• 13,5Н2О окрашено в желтый цвет. Водные растворы перйодатов
меди(Ш) имеют интенсивную окраску и являются сильными окис-
лителями.
Дигексателлуратокупрат(111) натрия, Na9[Cu(TeO6)2] -20Н2О,
выпадает в процессе медленной кристаллизации из прозрачного
раствора, полученного кипячением суспензии ТеО2 в 28%-ном
растворе КОН, к которому добавлен 12,5%-ный раствор сульфата
меди и K2S2Og.
Металлоорганические соединения
Известно ограниченное число металлоорганических соединений
меди. В качестве примера можно назвать этил- и фенилмедь, для
которых характерна относительная неустойчивость.
Этилмедъ, СиС2Н5, получают обработкой эфирного раствора
иодида меди(1) бромидом этилмагния при —18°; это зеленое, легко
разлагающееся вещество.
МЕДЬ
723
Фенилмедъ, CuC6H5, образуется при обработке эфирного раство-
ра иодида меди(1) фенилмагнийиодидом в атмосфере водорода или
азота.
Фенилмедь представляет собой серовато-зеленое твердое веще-
ство, которое при 80° разлагается на медь и дифенил, плохо рас-
творимо в бензоле, медленно гидролизуется водой с образованием
бензола и окиси меди (I).
Соединения включения
Гидрид меди, СиН, получают восстановлением сульфата меди
фосфорноватистой кислотой Н6Р2О4 при нагревании.
Гидрид меди представляет собой коричневые кристаллы с ку-
бической гранецентрированной решеткой; он медленно окисляется
на воздухе (на холоду) и выделяет водород при легком нагревании.
4CuH + ЗО2 = 4CuO + 2Н2О 2CuH = 2Си + Н2
Нитрид меди, Cu3N, образуется путем пропускания аммиака
над нагретой докрасна медью или над CuF2, Cu2O, СпО, нагреты-
ми до 260—280°.
Нитрид меди, Cu3N, можно получить, действуя аммиаком
на Сп(ОН)2 при нагревании:
12Cu(OH)2 + 6NH3 = 2Cu3N + 3Cu2O + 2N2 + 21H2O
Натрид меди представляет собой темно-зеленый порошок
с плотностью 5,5 г/см3-, он разлагается при нагревании до 300°
и бурно реагирует с кислотами, образуя соли меди(П) и металли-
ческую медь.
Фосфиды меди, Сп3Р, Сп2Р, Сп3Р2.
Фосфид меди, Сп3Р, можно получить взаимодействием металли-
ческой меди с красным фосфором или нагреванием фосфида Сп2Р
до 1000° в токе водорода. Он представляет собой игольчатые кри-
сталлы медного цвета с т. пл. 1030° и плотностью 7,09 г/см3.
Фосфид меди, Сп2Р, получают восстановлением ортофосфата
меди углем в электрической печи; это коричневый кристалли-
ческий порошок с плотностью 6,4 г!см3.
Фосфид меди, Сп3Р2, в виде коричневого порошка образуется
при сильном нагревании металлической меди в парах фосфора,
двуокиси углерода и азота.
Силицид меди, Cu2Si, получают путем сильного нагревания
смеси порошкообразной металлической меди с элементарным крем-
нием или двуокисью кремния; это серое, очень твердое вещество
с плотностью 6,9 г!см3.
46*
724
ПОБОЧНАЯ ПОДГРУППА I ГРУППЫ
СЕРЕБРО Ag
Z — 47; ат. вес = 107,870
Валентность I, (II), (III); заряд 1+, (2+), (3+)
Массовые числа природных изотопов 107 и 109
Электронная структура атома меди: К -L -М -iszip6itP0-5s1.
Электронная структура атома меди и катиона Ag+ для 4d-
и 5х-орбиталей:
4<Г° 5s< 4d10 5s
н 11 11 11 11
1! 11 11 11 11
Ag Ag+
ИСТОРИЧЕСКАЯ СПРАВКА
Серебро является одним из тех металлов, которые привлекли
внимание человека еще в древние времена.
История серебра тесно связана с алхимией, поскольку уже
в те времена был разработан метод купелирования серебра.
За 2500 лет до н. э. в Древнем Египте носили украшения
и чеканили монеты из серебра, считая, что оно дороже золота.
В X в. было показано, что между серебром и медью существует
аналогия, и медь рассматривалась как серебро, окрашенное
в красный цвет. В 1250 г. Винсент Бове высказал предположение,
что серебро образуется из ртути при действии серы.
В средние века кобалдом называли руды, которые служили
для получения металла со свойствами, отличными от уже извест-
ного серебра. Позднее было показано, что из этих минералов добы-
вается сплав серебро — кобальт, и различие в свойствах опреде-
лялось присутствием кобальта.
В XVI в. Парацельс получил хлорид серебра из элементов,
а Бойль определил его состав. Шееле изучал действие света на хло-
рид серебра, а открытие фотографии привлекло внимание и к дру-
гим галогенидам серебра.
В 1663 г. Глазер предложил нитрат серебра в качестве прижи-
гающего средства. С конца XIX в. комплексные цианиды серебра
используются в гальванопластике.
РАСПРОСТРАНЕННОСТЬ В ПРИРОДЕ
Серебро является редким металлом, его содержание в земной
коре равно 1-10-5 вес.%. В природе серебро встречается как само-
родное, так и в виде соединений — сульфидов, селенатов, теллу-
ратов или галогенидов в различных минералах.
Серебро встречается также в метеоритах и содержится в мор-
ской воде.
Серебро в виде самородков встречается в природе реже, чем
самородная медь или золото, и часто это бывают сплавы с золотом
СЕРЕБРО
725
(кюстелит), медью (медьсодержащее серебро), сурьмой (сурьмусо-
держащее серебро), ртутью и платиной. Образование самород-
ного серебра связано с действием воды или водорода на сульфид
серебра (соответственно на аргентит).
Металлическое серебро представляет собой гранецентрирован-
ные кубические кристаллы серебристо-белого цвета, часто покры-
тые черным налетом. Залежи самородного серебра находятся
в СССР, Норвегии, Канаде, Чили, ФРГ и других странах.
Наиболее важными минералами серебра являются следующие.
Акантит, Ag2S,— серые ромбические кристаллы, устойчивые
при температуре ниже 179°. Обе модификации природного сульфи-
да серебра содержат 87,1% Ag, имеют плотность 7,2—7,4 г/см3
и твердость 2—2,5 по шкале Мооса.
Аргентит, Ag2S,— серые кубические кристаллы, устойчивые
при температуре выше 179°. Аргентит — основной источник сереб-
ра. В природе он сопутствует самородному серебру, кераргиту
(AgCl), церусситу (РЬСО3), арсенидам и антимонидам серебра; его
залежи часто находятся рядом с сульфидами свинца, цинка и меди.
Такие руды находятся в Норвегии, Мексике,' Перу, СССР, Чили,
Румынии. Галенит PbS, добываемый в Румынии, Франции, стра-
нах Скандинавского полуострова, содержит серебро.
Прустит, Ag3AsS3 или 3Ag2S-As2S3, содержит 65,4% серебра;
он имеет вид красных гексагональных призматических кристаллов
с плотностью 5,57—5,64 г/см3 и твердостью 2—2,5 по шкале Мооса.
Залежи прустита имеются в СССР, Мексике, Чили, Перу, Боливии.
Пираргирит, Ag3SbS3 или 3Ag2S -Sb2S3, содержит 68,4%
серебра, сопутствует пруститу в виде темно-красных кристаллов.
Стефанит, Ag5SbS4 или 5Ag2S -Sb2S3, содержит 68,5% серебра
и представляет собой серовато-черные призматические кристаллы
с плотностью 6,2—6,3 г/см3 и твердостью 2,5 по шкале Мооса.
Залежи находятся в Мексике.
Полибазит, 8(Ag, Cu)2S -Sb2S3, содержит 62,1—74,9% Ag;
он представляет собой серовато-черные призматические или пла-
стинчатые кристаллы с плотностью 6,27—6,33 г/см3 и твердостью
2—3 по шкале Мооса. Залежи имеются в ЧССР, Венгрии, Мексике.
Кераргирит, AgCl, содержит 75,3% серебра и имеет вид плот-
ных бесцветных (или желтоватых, серовато-фиолетовых, даже
черных в случае длительного воздействия прямого света) пластов
(редко в виде кубических кристаллов). Плотность кераргирита
5,55 г/см3, твердость 1,5—2 по шкале Мооса. Залежи встречаются
в СССР, Чили, Боливии, Мексике, Перу, Австралии.
К другим минералам серебра относятся бромаргирит AgBr, иодаргирит
Agl, дискразит Ag3Sb, штромейерит Cu2S.Ag2S, ялпаит 3Ag2S-Cu2S, наума-
нит Ag2Se, гессит Ag2Te, петцит (Ag, Au)2Te. миаргирит AgSbS2, смитит
AgAsS2, аргиродит 4Ag2S -GeS2, канфильдит 4Ag2S-SnS2, эмболит Ag(Cl, Br),
штернбергит Ag2S.2Fe2S3.
726
ПОБОЧНАЯ ПОДГРУППА I ГРУППЫ
При окислении аргентита (акантита) Ag2S образуется сульфат
серебра Ag2SO4, который, будучи частично растворим, вымывается
водой. Когда на пути вод, содержащих сульфат серебра, встре-
чается сульфат железа(П), выделяется свободное серебро, а если
встречаются хлориды (т. е. ионы С1_), то образуется кераргирит:
Ag2SO4 -f- 2FeSO4 = 2Ag -|- Fe2(SO4)j
Ag2SO4 -|- 2NaCl = 2AgCl -|- Na2SO4
Если воды, содержащие сульфид серебра, встречают сульфиды
других элементов, то образуются скопления двойных сульфидов
подобно встречающимся смесям серебро — мышьяк, серебро —
сурьма, серебро — медь, серебро — свинец, серебро — германий.
ПЕРЕРАБОТКА СЕРЕБРЯНЫХ РУД
И ПОЛУЧЕНИЕ МЕТАЛЛИЧЕСКОГО СЕРЕБРА
Примерно 80% от общего мирового количества добываемого
серебра получается как побочный продукт переработки комплекс-
ных сульфидов тяжелых цветных металлов, содержащих сульфид
серебра (аргентит) Ag2S. При пирометаллургической переработке
полиметаллических сульфидов свинца, меди, цинка, серебра
последнее извлекается вместе с основным металлом в виде серебро-
содержащих свинца, меди или цинка.
Для обогащения серебросодержащего свинца серебром приме-
няют процесс Паркеса или Паттинсона.
По процессу Паркеса серебросодержащий свинец плавится
вместе с металлическим цинком. При охлаждении тройного сплава
свинец — серебро — цинк ниже 400° отделяется нижний слой,
состоящий из жидкого свинца, который содержит небольшое коли-
чество цинка и серебра, и верхний твердый слой, состоящий из сме-
шанных кристаллов цинк — серебро с небольшим количеством
свинца. Образование смешанных кристаллов цинк — серебро осно-
вывается на более высокой растворимости серебра в цинке, чем
в свинце, и на разделении при охлаждении серебросодержащего
цинка и свинца на два слоя. При отгонке цинка (точка кипения
которого 907е) из сплава свинец — цинк — серебро остается сви-
нец, который содержит 8—12% серебра и служит для получения
сырого серебра путем купелирования. Из тройного сплава свинец—
цинк — серебро цинк может быть удален в виде Na2ZnO2 сплавле-
нием с Na2CO3.
По процессу Паттинсона расплавленный серебросодержащий
свинец медленно охлаждается. Свинец, который кристаллизуется
первым, отделяется до тех пор, пока расплав не достигнет состава
эвтектики с содержанием 2,25% серебра. Эвтектика затвердевает
при 304° и служит затем для получения сырого серебра методом
купелирования.
СЕРЕБРО
727
При купелировании свинец, содержащий 2,25—12% серебра,
плавится в купелях в печи, куда подают воздух или кислород
на поверхность расплавленного металла. Окись свинца (свинцо-
вый глет) РЬО вместе с окислами мышьяка, сурьмы, цинка и меди,
образовавшимися при полном окислении серебросодержащего
свинца (с большим содержанием серебра), удаляют с поверхности
сырого серебра, который содержит примерно 95% Ag.
Отделение серебра от серебросодержащего свинца возможно
также электролитическим путем, применяя аноды из серебросодер-
жащего свинца, а в качестве электролита — гексафторокремне-
вую кислоту H2[SiFe] с гексафторосиликатом свинца Pb[SiF6].
При электролизе свинец осаждается на катоде, а серебро вме-
сте с золотом, платиной и платиновыми металлами переходят
в анодный шлам. Аналогично при электролитическом рафиниро-
вании серебросодержащей меди, которую используют в качестве
анодов (применяя при этом разбавленную серную кислоту как
электролит), на катоде электролитически осаждают медь, а сереб-
ро, золото и платиновые металлы также переводят в анодный
шлам.
Извлечение серебра, золота и платиновых металлов из анод-
ного шлама легко осуществляется химическим путем. В отличие
от золота и платиновых металлов серебро легко растворяется
в азотной кислоте.
Из нитрата серебра AgNO3 металлическое серебро можно оса-
дить сульфатом железа(П), металлическим цинком, формальдеги-
дом в аммиачной среде или нитратом марганца(П) в щелочной
среде:
3AgNO3 + 3FeSO4 = 3Ag + Fe(NO3)3 + Fe2(SO4)3
2AgNO3 -|- Zn = 2Ag -f- Zn(NO3)2
2[Ag(NH3)2]OH + HCHO = 2Ag -f- 3NH3 + HCOONH4 + H2O
2AgNO3 + Mn(NO3)2 + 4NaOH = 2Ag + MnO2 + 4NaNO3 + 2H2O
Примерно 20% мирового количества серебра получают перера-
боткой собственно серебряных руд и рекуперацией серебра из
монет, изделий или серебряного лома.
Измельченную, размолотую и обогащенную (в случае низкого
содержания серебра) серебряную руду перерабатывают методами
цианирования, амальгамирования, хлорирования и др.
В случае переработки методом цианирования тонко измельчен-
ную руду (природное серебро, аргентит или кераргирит) смеши-
вают с 0,4%-ным раствором NaCN и перемешивают струей возду-
ха. В водном растворе цианида натрия в присутствии кислорода
воздуха серебро и аргентит растворяются медленнее, чем керар-
гирит:
2Ag + 4NaCN + Н2О + V2O2 2Na[Ag(CN)2] + 2NaOH
Ag2S + 5NaCN + H2O + V2O2 -* 2Na[Ag(CN)2] + 2NaOH + NaSCN
AgCl + 2NaCN Na[Ag(CN)2J + NaCl
728
ПОБОЧНАЯ ПОДГРУППА I ГРУППЫ
Сульфид серебра Ag2S растворяется в тетрацианоцинкате(П)
натрия по реакции
Ag2S + Na2[Zn(CN)4] 2Na[Ag(CN)2] + ZnS
Количество взятого для переработки серебряных руд цианида
натрия больше теоретически необходимого, поскольку серебря-
ные руды часто содержат соединения меди, железа и цинка, кото-
рые также реагируют с цианидом Натрия.
Цианирование осуществляется в деревянных чанах диаметром
10-12 м.
Из растворов комплексных цианидов серебра серебро может
быть осаждено в виде металла тонко измельченным металлическим
цинком или алюминием. Осаждение металлического серебра
из растворов комплексных цианидов серебра металлическим цин-
ком или алюминием осуществляется по уравнениям
2Na[Ag(CN)2] + Zn = 2Ag + Na2[Zn(CN)4]
3Na[Ag(CN)2] + Al + 4NaOH + 2H2O = 3Ag + Na[Al(OH)4(H2O)2]+6NaCN
Сырое серебро плавится, отливается в виде брусков и затем
рафинируется электролитическим или химическим методом.
Можно также извлечь комплексный анион [Ag(CN)2]_ с помо-
щью анионообменных смол. Применяют анионообменные сульфои-
рованные смолы R2SO4 (предварительно обработанные 5%-ным
водным раствором серной кислоты). Реакцию ионного обмена
в процессе извлечения анионов [Ag(CN)2]~ с помощью анионооб-
менных смол (предпочтительно в виде пористых анионитов) можно
представить следующим образом:
R2SO4 + 2[Ag(CN)2]- -> 2R[Ag(CN)2] + SO*-
Чтобы реакция обмена протекала с большой скоростью, соз-
дают кислую среду (pH = 3,5).
Комплексные цианиды вымывают из анионообменной смолы
селективным элюентом, например 2 н. раствором цианида калия
или натрия.
Процесс амальгамирования применяют к рудам, содержащим
самородное серебро, аргентит или кераргирит, он основывается
на образовании амальгамы серебра.
Для амальгамирования тонко измельченные серебряные руды
обрабатывают небольшим количеством воды и ртутью (1 вес. ч.
ртути на 6 вес. ч. серебра).
Сульфид серебра Ag2S под действием хлорида меди(1) [который
образуется при восстановлении хлорида меди(П) ртутью] превра-
щается в хлорид серебра:
Ag2S -J- 2CuGl = 2AgCl + Cu2S 2GuG12 + 2Hg = 2GuGl + Hg2Cl2
Последний под действием ртути и хлорида меди(1) восстанавли-
вается до металлического серебра, которое образует амальгаму
СЕРЕБРО
72»
со ртутью:
2AgCl + 2Hg = 2Ag + Hg2Cl2 AgCl + CuCl = Ag + CuCl2
Амальгаму серебра фильтруют под давлением. При отгонке
ртути остается сырое серебро, которое очищают химическим или
электрохимическим способом.
При прокаливании смеси сульфида серебра и хлорида натрия
(500—600е) в окислительной атмосфере образуется хлорид серебра г
Ag2S -|- 2NaCl -|~ 2О2 = 2AgCl -f- Na2SO4
Для извлечения серебра из AgCl или из Na[AgCl2] применяют
амальгамирование, осаждение металлического серебра медью
и осаждение сульфида серебра из соединения Na2[Ag2(S2O3)2]i
AgCl + NaCl = Na[AgCl2]
Na[AgCl2] Cu — Ag -f- Na[CuCl2]
2AgCl 2Na2S2O3 = Na2[Ag2{S2O3)2] -j~ 2NaCl
Na2[Ag2(S2O3)2] -p Na2S = Ag2S -f- 2Na2S2O3
Сульфид серебра Ag2S затем перерабатывают с целью получе-
ния элементарного серебра.
РАФИНИРОВАНИЕ СЕРЕБРА
Сырое серебро можно рафинировать химическим или электро-
литическим путем.
В химическом процессе сырое металлическое серебро раство-
ряют в азотной кислоте, очищенный перекристаллизацией нитрат
серебра обрабатывают аммиаком, превращая его в гидроокись
диамминосеребра; последнюю восстанавливают сульфитом аммо-
ния (берут точно рассчитанное количество) при 70° до чистого
металла. Серебро плавят над негашеной известью в токе водорода
и затем в вакууме:
3Ag + 4HNO3 = 3AgNO3 + NO + 2H2O
AgNO3 + 3NH4OH = [Ag(NH3)2]OH + NH4NO3 + 2H2O
2[Ag(NH3)2]OH + (NH4)2SO3 + 3H2O = 2Ag + (NH4)2SO4 + 4NH4OH
При электролитическом рафинировании применяют аноды из
сырого серебра, а в качестве электролита берут раствор нитрата
серебра. По мере пропускания постоянного тока через электролит
чистое серебро электролитически осаждается на катодах, а метал-
лы, более активные, чем серебро, переходят (из анодов) в раствор
в виде ионов. При этом золото, платина и платиновые металлы
остаются в анодном шламе.
В случае необходимости получить порошкообразное серебро
(в отличие от компактной массы) в качестве электролита приме-
няют 4%-ный раствор AgNO3 с 1% HNO3 и 2—3%-ный раствор
'730
ПОБОЧНАЯ ПОДГРУППА I ГРУППЫ
гексафторокремневой кислоты. Можно использовать раствор,
содержащий 5—6 г!л Ag2SO4, сульфаты щелочных металлов или
раствор комплексных цианидов серебра (при высокой плотности
тока и низкой концентрации).
Электролитический метод эффективен при извлечении серебра
из монет и серебросодержащих сплавов.
ФИЗИЧЕСКИЕ И ХИМИЧЕСКИЕ СВОЙСТВА
Серебро проявляет большее сходство с палладием (за которым
он следует в периодической системе), чем с рубидием (с которым он
находится рядом в I группе периодической системы и в том же
пятом периоде).
Расположение серебра в побочной подгруппе I группы перио-
дической системы определяется электронной структурой атома,
которая аналогична электронной структуре атома рубидия. Боль-
шое различие в химических свойствах серебра и рубидия опреде-
ляется разной степенью заполненности электронами 4с/-орбитали.
Атом серебра отличается от атома палладия наличием одного
электрона на 5</-орбитали.
По большинству физических и химических свойств серебро
приближается к меди и золоту. В подгруппе меди серебро (средний
элемент) обладает наиболее низкими температурами плавления
и кипения и максимальным значением коэффициента расширения,
максимальной тепло- и электропроводностью.
Физико-химические свойства серебра в значительной степени
зависят от его чистоты.
Металлическое серебро в компактном полированном виде (бру-
ски, трубки, проволока, пластинки, листы) представляет собой
белый блестящий металл, обладающий большой отражательной
способностью по отношению к инфракрасным и видимым лучам
и более слабой — к ультрафиолетовым лучам. Серебро в виде
тонких листочков (они кажутся синими или фиолетовыми в про-
ходящем свете) обладает электрическими и оптическими свойства-
ми, отличными от свойств металлического серебра в слитках.
Коллоидные растворы серебра окрашены в розовый (до корич-
невого) цвет и могут быть получены восстановлением суспензии
Ag2O водородом при 50° [или другими восстановителями, например
сахаром, окисью углерода, цитратом железа(П), цитратом аммо-
ния, хлоридом олова(П), пирогаллолом, фенолом, фосфором
в эфире, фосфорноватистой кислотой, формальдегидом, гидрази-
ном, фенилгидразином и др.], а также путем создания электриче-
ской дуги в воде между двумя серебряными электродами. Для
стабилизации коллоидных растворов серебра применяют белки,
желатину, гуммиарабик, агар-агар и другие органические веще-
ства, играющие роль защитных коллоидов.
СЕРЕБРО
731
Белковое коллоидное серебро (протаргол и колларгол) приме-
няется как фармацевтический препарат.
В нейтральных или слабо щелочных растворах гидрозоль
серебра ведет себя как отрицательный коллоид, а в слабо кислых
растворах — как положительный.
Коллоидное серебро является энергичным восстановителем
(по отношению к Fe2Ci6, HgCl2, КМпО4, разб. HNO3), обладает
хорошей адсорбционной способностью (по отношению к кислороду,
азоту, водороду, метану, этану и др.), является катализатором
и сильным бактерицидом (до появления антибиотиков применялся
для обработки слизистых оболочек) и служит для лечения некото-
рых трудно излечиваемых кожных болезней.
Вода, хранящаяся в серебряных сосудах, стерилизуется и не
портится длительное время благодаря наличию иона Ag+,
образующегося в результате контакта воды со стенками посуды.
Металлическое серебро обладает кубической гранецентрирован-
ной решеткой с плотностью 10,50 г/см? при 20°, т. пл. 960,5°,
т. кип. 2177° (пары желтовато-синие); оно диамагнитно, является
очень хорошим проводником тепла и электричества (удельное
сопротивление при 20° равно 1,59 мком/см). В числе физико-
механических свойств следует отметить пластичность, относитель-
ную мягкость (твердость 2,5—3 по шкале Мооса), ковкость и тягу-
честь (легко протягивается и прокатывается), малую прочность.
Серебро образует сплавы типа твердых растворов с золотом
и палладием и интерметаллические соединения с элементами Li,
Be, Mg, Са, Sr, Ba, Zn, Cd, Hg, Al, Ga, In, TI, Pr, Sn, Zr, Th, P,
As, Sb, S, Se, а также сплавы типа эвтектик с элементами Bi,
Cu, Ge, Ni, Pb, Si, Na, TI.
При легировании устраняются основные недостатки серебра,
такие, как мягкость, низкая механическая прочность и высокая
реакционная способность по отношению к сере и сульфидам.
Некоторые газы, например водород, кислород, окись и дву-
окись углерода, растворяются в серебре, причем растворимость
их пропорциональна квадратному корню от давления. Раствори-
мость кислорода в серебре максимальна при 400—450° (когда
1 объем серебра поглощает до 5 объемов кислорода). Рекомен-
дуется избегать охлаждения серебра, насыщенного кислородом,
поскольку выделение этого газа из охлаждаемого серебра может
сопровождаться взрывом.
При поглощении кислорода или водорода серебро становится
хрупким.
Азот и аргон с трудом растворяются в серебре при температуре
выше —78°.
С химической точки зрения серебро достаточно инертно, оно
не проявляет способности к ионизации и легко вытесняется из
соединений более активными металлами или водородом.
732
ПОБОЧНАЯ ПОДГРУППА I ГРУППЫ
Под действием влаги и света галогены легко взаимодействуют
с металлическим серебром образуя соответствующие галогениды.
Соляная и бромистоводородная кислоты в концентрированных
растворах медленно реагируют с серебром:
2Ag + 4НС1 = 2H[AgCl2] + Н2 2Ag + 4НВг = 2H[AgBr2] + Н2
Кислород взаимодействует с нагретым до 168° металлическим
серебром при разных давлениях с образованием Ag2O. Озон при
225° в присутствии влаги (или перекиси водорода) действует
на металлическое серебро, образуя высшие окислы серебра.
Сера, реагируя с нагретым до 179° металлическим серебром,
образует черный сульфид серебра Ag2S. Сероводород в присутствии
кислорода воздуха и воды взаимодействует с металлическим сереб-
ром при комнатной температуре по уравнению
2Ag + H2S + !/2О2 = Ag2S + Н2О
Металлическое серебро растворяется в H2SO4 (60° Be) при
нагревании, в разб. HNO3 на холоду и в растворах цианидов
щелочных металлов в присутствии воздуха (кислорода или другого
окислителя):
2Ag -|- 2H2SO4 = Ag2SO4 -|- SO2 -|- 2Н2О
3Ag + 4HNO3 = 3AgNO3 + NO + 2H2O
2Ag + 4NaCN + H2O + V2O2 = 2Na[Ag(CN)2] + 2NaOH
Селен, теллур, фосфор, мышьяк и углерод реагируют с металли-
ческим серебром при нагревании с образованием Ag2Se, Ag2Te,
Ag3P, Ag3As, Ag4C. Азот непосредственно не взаимодействует
с серебром.
Органические кислоты и расплавленные щелочи или соли щелоч-
ных металлов не реагируют с металлическим серебром. Хлорид
натрия в концентрированных растворах и в присутствии кислорода
воздуха медленно взаимодействует с серебром с образованием
хлорида серебра.
В солянокислом растворе серебро восстанавливает некоторые
соли металлов, такие, как CuCl2, HgCl2, Fel2, VOC12.
Химическая реакционная способность серебра наглядно иллю-
стрируется следующей схемой:
с галогенами в присутствии Света и влаги
-> AgF, AgCl, AgBr, Agl
c HCl, НВг на воздухе H[AgCl2], H[AgBr2J
с разб. HNO3 -> AgNO3
с MeiCN в присутствии кислорода ->
-> Mel[Ag(CN)2]
с кислородом -> Ag2O
с озоном во влажной атмосфере ->
-»• AgO, Ag2O3
с S, Se, Те, Р, As, С-> Ag2S, Ag2Se, Ag2Te,
Ag3P, Ag3As, Ag4C
c H2S во влажном воздухе Ag2S
с конц. H2SO4 -► Ag2SO4
Комн. темп.
Серебро
Нагревание
СЕРЕБРО
733
ПРИМЕНЕНИЕ
Значительная часть мирового производства серебра идет
на изготовление монет.
В химической промышленности применяются аппараты
из серебра (для получения ледяной уксусной кислоты, фенола
и др.), лабораторная посуда (тигли или лодочки, в которых пла-
вятся чистые щелочи или соли щелочных металлов, оказывающие
разъедающее действие на большинство других металлов), лабора-
торные инструменты (шпатели, щипцы, сита и др.).
В пищевой промышленности применяются серебряные аппара-
ты, в которых приготовляют фруктовые соки и другие напитки.
Металлическое серебро служит для изготовления зеркал путем
термического испарения, для получения солей, а также для изго-
товления предметов домашнего обихода, украшений, безделушек.
Бруски (или электролитический порошок) серебра служат
положительными электродами в аккумуляторах, в которых отри-
цательными электродами являются пластинки из окиси цинка,
а электролит — едкое кали.
В медицине известен ряд фармацевтических препаратов, содер-
жащих коллоидное серебро.
Серебро применяется в качестве катализатора при синтезе
воды (на холоду), разложении и восстановлении закиси азота
N2O, разложении окиси серебра, растворенной в аммиаке, в реак-
циях обмена водород — дейтерий, детонации смеси воздух — аце-
тилен, при сжигании окиси углерода, окислении спиртов в альде-
гиды и кислоты и др.
Сплавы серебра широко применяются для изготовления монет,
зубных пломб, мостов и протезов, столовой посуды, в холодильной
и химической промышленности.
СОЕДИНЕНИЯ (ОБЩИЕ СВОЙСТВА)
Известны соединения, в которых серебро одно-, двух- и трех-
валентно. В отличие от устойчивых соединений одновалентного
серебра соединения двух- и трехвалентного серебра немногочис-
ленны и мало устойчивы.
В табл. 71 приведены формулы и указан цвет некоторых соеди-
нений серебра, сгруппированных по степеням окисления. При-
водятся также и металлоорганические соединения.
Соединения одновалентного серебра
Известны многочисленные устойчивые соединения (простые
и координационные) одновалентного серебра. Ион одновалентного
серебра Ag+ с радиусом 1,55 А диамагнитен, бесцветен, гидрати-
рован, легко поляризуется, является окислителем (легко восста-
734
ПОБОЧНАЯ ПОДГРУППА I ГРУППЫ
навливается различными восстановителями до металлического
серебра) и играет роль катализатора в реакции окисления иона
марганца(II) анионом S2O3_.
Большинство соединений серебра(I) плохо растворимо в воде.
Нитрат, перхлорат, хлорат, фторид растворяются в воде, а ацетат
и сульфат серебра растворимы частично. Соли серебра(1) белые
или слегка желтоватые (когда анион соли бесцветен). Вследствие
деформируемости электронных оболочек иона серебра(I) некото-
рые его соединения с бесцветными анионами окрашены.
Многие из соединений серебра(I) окрашиваются в серый цвет
под действием солнечного света, что обусловлено процессом восста-
новления до металлического серебра.
У солей серебра(I) мало выражена склонность к гидролизу.
При нагревании солей серебра со смесью карбоната натрия
и угля образуется металлическое серебро:
2AgNO3 + Na2CO3 + 4С = 2Ag + 2NaNO2 + 5СО
Известны многочисленные координационные соединения сереб-
ра^), в которых координационное число серебра равно 2, 3 и 4.
Неорганические соединения
Окись серебра, Ag2O, получают при обработке растворов AgN03
щелочами или растворами гидроокисей щелочноземельных метал-
лов:
2AgNO3 + 2К0Н = Ag2O + 2KNO3 + Н2О
Окись серебра представляет собой диамагнитный кристалличе-
ский порошок (кубические кристаллы) коричнево-черного цвета
с плотностью 7,1—7,4 г'смй, который медленно чернеет на свету,
высвобождая кислород, и разлагается на элементы при нагревании
до 200°:
Ag2O -► 2Ag -j- 1/2О2
Водород, окись углерода, перекись водорода и многие металлы
восстанавливают окись серебра в водной суспензии до металли-
ческого серебра:
40° 10°
Ag2O Н2 —► 2Ag -(- Н2О Ag2O -f- СО —> 2Ag СО2
Ag2O Н2О2 = 2Ag -|- Н2О -|- О2
При окислении Ag2O озоном образуется окись серебра(П) AgO.
Окись серебра(I) растворяется в плавиковой и азотной кислотах,
в солях аммония, в растворах цианидов щелочных металлов,
в аммиаке и т. д.
Ag2O -|~ 2HF = 2AgF -|- Н2О Ag2O -f- 2HNO3 = 2AgNO3 -f- H2O
Ag2O + 2(NH4)2CO3 = [Ag(NH3)2]2CO3 + 2H2O + CO2
СЕРЕБРО
735-
Ag2O + 4KCN + Н2О = K[Ag(CN)2] + 2К0Н
Ag2O + 4NH4OH = 2[Ag(NH3)2]OH + 3H2O или
Ag2O + 4NH3 + H20 = 2[Ag(NH3)2]OH
При хранении гидроокись диамминсеребра [Ag (NH3)2]OH
(которая является растворимым основанием с окислительными
свойствами) превращается в способный взрываться имид серебра:
2[Ag(NH3)2]OH -+ Ag2NH + 3NH3 + 2Н2О
Растворы хлоридов щелочных металлов превращают окись-
серебра(1) в хлорид серебра(I), а при действии избытка Hgl2
на Ag2O образуется AgJHglJ.
Окись серебра — энергичный окислитель по отношению
к соединениям хрома(Ш), альдегидам и галогенопроизводным
углеводородов:
5Ag2O -|~ Сг2О3 = 2Ag2CrO4 -|- 6Ag
3Ag2O + 2Сг(ОН)3 + 4NaOH = 2Na2CrO4 + 6Ag + 5H2O
Окисление галогенопроизводных углеводородов! приводит
к образованию спиртов, а окисление альдегидов — соответствую-
щих кислот.
Растворы сульфидов щелочных металлов и водные суспензии
сульфидов тяжелых металлов превращают окись Ag2O в суль-
фид Ag2S.
Суспензии окиси серебра применяются в медицине как анти-
септическое средство. Смесь, состоящая из окиси серебра с легко
восстанавливающимися окислами (например, меди или марганца),
является хорошим катализатором окисления окиси углерода кис-
лородом воздуха при обычной температуре. Смесь состава 5% Ag2O,
15%Со2О3, 30% СиО и 50% МпО2, названная «гопкалитом», служит
для зарядки противогазов в качестве защитного слоя против окиси
углерода.
Гидроокись серебра, AgOH, образуется в виде неустойчивого
белого осадка в результате обработки AgNO3 спиртовым раствором
калиевой щелочи при pH = 8,5—9 и температуре — 45°.
Соединение AgOH обладает амфотерными свойствами, легко
поглощает двуокись углерода из воздуха и при нагревании с Na2S
образует аргентаты эмпирических формул Ag2O -3Na2O и Ag2O •
• 2Na2O.
Основные свойства гидроокиси серебра усиливаются в присут-
ствии аммиака вследствие образования гидроокиси диаммин-
серебра [Ag (NH3)2]OH.
Фторид серебра, AgF, получают прямым взаимодействием
элементов при нагревании, действием плавиковой кислоты на окись.
736
ПОБОЧНАЯ ПОДГРУППА I ГРУППЫ
или карбонат серебра(1), термическим разложением (200°) Ag[BF4],
причем наряду с AgF образуется BF3:
2Ag + F2 = 2AgF + 97,4 ккал Ag2CO3 + 2HF = 2AgF + H2O + CO2
Ag2O + 2HF = 2AgF + H2O Ag[BF4] = AgF + BF3
Выделение кристаллов AgF из водного раствора осуществляется
путем концентрирования в вакууме в темноте.
Соединение AgF представляет собой расплывающиеся на воз-
духе бесцветные гранецентрированные кубические кристаллы
с плотностью 5,85 г/cw? и т. пл. 435°; фторид серебра плохо
растворим в спирте, легко растворим в воде (в отличие от остальных
галогенидов серебра) и в аммиаке; его нельзя хранить в стеклянной
посуде, поскольку он разрушает стекло.
Под действием паров воды и водорода при нагревании фторид
серебра восстанавливается до металлического серебра:
2AgF + Н2О = 2Ag + 2HF + V2O2 2AgF + Н2 = 2Ag + 2HF
Ультрафиолетовые лучи вызывают превращение фторида
серебра в полуфторид Ag2F. Водный раствор фторида серебра
служит для дезинфекции питьевой воды.
Известны кристаллогидраты AgF -пН20 (где п = 1, 2, 4) и фто-
рокислоты H[AgF2], H2[AgF3], H3[AgF4],
Моногидрат AgF-H2O осаждается в виде светло-желтых куби-
ческих кристаллов при упаривании в вакууме раствора безводного
AgF в воде.
Дигидрат AgF -2Н2О, представляющий собой твердые бесцвет-
ные призматические кристаллы с т. пл. 42°, выпадает из концент-
рированных растворов AgF.
Из раствора, полученного растворением Ag2O в 20%-ной пла-
виковой кислоте, выпадают кристаллы AgF -4Н2О. При охлажде-
нии раствора AgF в плавиковой кислоте осаждаются бесцветные
кристаллы H3[AgF4], которые при 0° в токе воздуха превращаются
в белые кристаллы H[AgF2].
Хлорид серебра, AgCl, встречается в природе в виде минерала
кераргирита и может быть получен обработкой металлического
серебра хлорной водой, взаимодействием элементов при высокой
температуре, действием газообразного НС1 на серебро (выше 1150°),
обработкой соляной кислотой серебра в присутствии воздуха (кис-
лорода или другого окислителя), действием растворимых хлори-
дов на серебро, обработкой растворов солей серебра соляной кис-
лотой или раствором какого-либо хлорида.
Ag + 1/2С12 = AgCl -|- 30,3 ккал AgNO3 + НС1 = AgCl + HNO3
Ag + НС1 = AgCl + 1/2H2 AgNO3 + NaCl = AgCl + NaNO3
2Ag + 2HC1 + i/2O2 = 2AgCl + H2O
СЕРЕБРО
737
При работе с разбавленными водными растворами образуются
коллоидные растворы хлорида серебра.
Соединение AgCl представляет собой диамагнитные белые куби-
ческие гранецентрированные кристаллы с т. пл. 455°
и т. кип. 1554°.
Хлорид серебра растворяется в растворах хлоридов (NaCl,
КС1, NH4C1, СаС12, МпС12), цианидов, тиосульфатов, нитратов
щелочных металлов и аммиаке с образованием растворимых и бес-
цветных координационных соединений:
AgCl + КС1 = K[AgCl2] AgCl + 2Na2S2O3 = Na3[Ag(S2O3)2] + NaCl
AgCl + 2KCN = K[Ag(CN)2] + KC1 AgCl + 2NH3 = [Ag(NH3)2]Cl
Под действием света хлорид серебра восстанавливается (окра-
шиваясь в фиолетовый, а затем в черный цвет) с высвобождением
серебра и хлора:
AgCl -*• Ag VgCIa
На этой реакции основывается применение хлорида серебра
в фотопленках.
Водород медленно взаимодействует с AgCl при нагревании,
а бром и иод реагируют с AgCl при нагревании в присутствии воды
по уравнениям)
AgCl + !/2Н2 = Ag + НС1
5AgCl + ЗВг2 + ЗН2О = 5AgBr + НВгО3 + 5НС1
5AgCl + 3I2 + ЗН2О = 5AgI + HIOs + 5НС1
Кипячение хлорида серебра с серной кислотой дает Ag2SO4:
2AgCl -I- H2SO4 = Ag2SO4 + 2НС1Ц
Известны следующие бесцветные аддукты хлорида серебра
с аммиаком: AgCl-NH3, 2AgCl-3NH3, AgCl-3NH3.
При сплавлении AgCl с Na2CO3 образуется металлическое
серебро:
4AgCl + 2Na2CO3 = 4Ag + 4NaCl + 2CO2 + O2
Хлорид серебра восстанавливается в результате нагревания
с металлическими свинцом, магнием, цинком, ртутью, медью или
амальгамой цинка, а в аммиачных растворах восстанавливается
на холоду щелочными металлами:
230°
2AgCl + Pb-----► 2Ag + РЬС12
Из AgCl делают линзы для приборов, работающих в области
инфракрасного излучения, и радарные экраны. Большие коли-
чества AgCl идут на изготовление фотопленки. Часто AgCl приме-
няют для фиксирования космических лучей.
47—0101
738
ПОБОЧНАЯ ПОДГРУППА I ГРУППЫ
Поскольку AgCl обладает бактерицидным действием, на его
основе готовят препараты, применяемые для обработки слизистых
оболочек глаза. «Серебряная вода», которая образуется при обра-
ботке дистиллированной воды AgCl, служит для стерилизации
и консервирования некоторых пищевых продуктов.
Бромид серебра, AgBr, встречается в природе в виде минерала
бромаргирита; в лаборатории может быть получен (в темноте)
обработкой раствора AgNO3 раствором НВг (или бромида щелоч-
ного металла) либо непосредственным взаимодействием брома
с металлическим серебром. Получение AgBr осуществляется в тем-
ноте, чтобы исключить фотовосстановление:
AgNO3 + КВг = AgBr + KNO3 Ag + l/2Br2 = AgBr + 27,4 ккал
Соединение AgBr может существовать либо в коллоидной форме,
либо в виде диамагнитных желтых кубических гранецентрирован-
ных кристаллов с плотностью 6,47 г!смй, т. пл. 434° и т. кип. 1537°;
бромид серебра плохо растворим в воде и растворяется в аммиаке,
тиосульфатах щелочных металлов и в конц. H2SO4 при нагревании.
AgBr + 2NH4OH = [Ag(NH3)2]Br + 2Н2О
2AgBr + H2SO4 = Ag2SO4 + 2HBr
AgBr -|- 2Na2S2O3 = Na3[Ag(S2O3)2] -f- NaBr
Бромид серебра более чувствителен к свету, чем хлорид серебра,
и под действием света разлагается на элементы:
AgBr -> Ag + i/2Br2
Бромистое серебро восстанавливается цинком в кислой среде
или металлами (такими, как свинец или медь) при нагревании,
а также сплавлением с безводным карбонатом натрия:
2AgBr + Na2CO3 = 2Ag + 2NaBr + CO2 + V2O2
На холоду AgBr поглощает аммиак, причем могут образовы-
ваться различные аддукты: AgBr-NH3, 2AgBr-3NH3, AgBr-3NH3.
Бромид серебра применяется для изготовления фотопленок
и в качестве катализатора при получении монокарбоновых жирных
кислот или олефинов с помощью реактива Гриньяра.
Применение галогенидов серебра
в фотографии
Бромид (хлорид или иодид) серебра(1), диспергированный до коллоид-
ного состояния в желатине, наносится в темноте на тонкую пленку, стеклян-
ные пластинки и бумагу, которые также хранятся в темноте.
Под действием кванта света hv бромид (галогенид) серебра(1) на свето-
чувствительной пленке разлагается на элементы:
Вг- — /iv -> 1/2Br2 + е~ Ag+ + е- Ag
СЕРЕБРО
739
Элементный бром (галоген) химически связывается с желатиной, а кол-
лоидное серебро образует очень мелкие зерна. Для того чтобы невидимое
изображение сфотографированного объекта стало видимым на фотографи-
ческой пленке или пластинке, их подвергают проявлению. В процессе про-
явления галогенид серебра (частично восстановленный) восстанавливается
химическим путем с помощью органических восстановителей до металличе-
ского серебра. Восстановление галогенида серебра проявлением осуществ-
ляется быстрее в соседстве с первоначально существующими зернами коллоид-
ного серебра. После того как при проявлении видимое изображение стало
достаточно ясным, проводят процесс закрепления (фиксирования), при кото-
ром с фоточувствительного слоя пленки или пластинки извлекаются неразло-
жившиеся галогениды серебра.
В качестве фиксатора применяют водный раствор тиосульфата натрия,
который легко растворяет оставшиеся галогениды серебра.
Видимое, устойчивое на свету изображение, полученное проявлением
и закреплением (негатив), является обратным изображением реального
объекта. Для получения реального изображения негатив проектируется
(в течение короткого времени) с помощью копировального аппарата или
увеличителя на фотобумагу. Проявлением и закреплением фотобумаги,
на которой было спроектировано обратное изображение сфотографирован-
ного объекта, получают действительное изображение (позитив).
Путем введения некоторых специальных добавок в состав светочув-
ствительного слоя можно увеличить чувствительность пленки к свету, селек-
тивную восприимчивость к различным областям спектра. Можно так же при-
готовить фоточувствительные составы получения цветных изображений.
Иодид серебра, Agl, встречается в природе в виде минерала
иодаргирита; в лаборатории может быть получен (в темноте) обра-
боткой раствора AgNO3 раствором HI или иодида щелочного
металла, путем непосредственного взаимодействия паров иода
с металлическим серебром, хлоридом или бромидом серебра при
нагревании, действием HI на металлическое серебро на холоду.
AgNO3 -(- HI = Agl -|- HNO3 Ag -|- 1/2^2 = Agl -|- 29,8 ккал
AgNO3 + KI = Agl + KNO3 Ag 4- HI = Agl + V2H2
Иодид серебра может существовать либо в виде прозрачных
двулучепреломляющих лимонно-желтых гексагональных призмати-
ческих кристаллов, либо в виде двулучепреломляющих красных
октаэдров. Известна также коллоидная форма Agl.
Иодид серебра диамагнитен, плавится с разложением при 555°,
плохо растворим в воде и растворяется в концентрированных
растворах HI, иодидах щелочных металлов и в Hgl2 с образо-
ванием координационных соединений:
Agl + HI = Н[ Agl2] Agl + Mell = Mei[AgI2]
2AgI + Hgl2 = Ag2[HgI4] Agl + 2MeTI = Me*[AgI3]
(где Me1 =~ K+, Rb+, Cs+, NH4+).
Под действием света или при нагревании Agl разлагается
на элементы.
Известны аддукты 2AgI-NH3, Agl -NH3.
47*
740
ПОБОЧНАЯ ПОДГРУППА I ГРУППЫ
В аммиачных растворах Agl может быть восстановлен до метал-
лического серебра щелочными или щелочноземельными металлами.
Иодид серебра служит для получения светочувствительных
пленок.
Хлорат серебра, AgC103, получают растворением Ag2O или
Ag2CO3 в НС1О3, пропусканием хлора через водную суспензию
Ag2O или Ag2CO3, обработкой раствора AgNO3 хлоратом калия
при 85°:
Ag2O + 2НС1О3 = 2AgC103 + Н2О
Ag2CO3 + 2НС1О3 = 2AgC103 + СО2 + Н2О
Ag2O + 6С12 + 5Н2О = 2AgC103 + 10НС1
AgNO3 -j- Kd О3 = AgC103 -|- KNO3
Хлорат серебра представляет собой белые кристаллы с плот-
ностью 4,43 г/см3 и т. пл. 230° (разлагается при 270° на AgCl
и кислород); он растворим в воде и аммиаке и обладает окислитель-
ными свойствами. Смесь хлората серебра и серы взрывается при
ударе, трении или нагревании.
Перхлорат серебра, AgC104, получают действием избытка
НСЮ4 на раствор AgNO3 при нагревании, обработкой раствора
Ag2SO4 перхлоратом бария Ва(С1О4)2, обработкой Ag2O 60%-ным
раствором НС1О4.
2AgNO3 + 2НС1О4 = 2AgC104 + 2NO2 + Ч2О2 + Н2О
Ag2SO4 -|- Ва(СЮ4)2 = 2AgC104 -|- BaSO4
Перхлорат серебра представляет собой быстро расплывающийся
на воздухе белый порошок с плотностью 2,8 г/см3, он очень хорошо
растворим в воде, уксусной кислоте, эфире, нитробензоле, гликоле,
аммиаке и др.
Известен кристаллогидрат AgC104-H20 и аддукты перхлората
серебра с бензолом, анилином и пиридином.
При концентрировании раствора, полученного действием
NaC104 на аммиачный раствор нитрата серебра или растворением
AgC104 в аммиаке, выпадают кристаллы [Ag(NH3)2]C104-Н2О.
Перхлорат серебра применяется в качестве катализатора в орга-
нической химии.
Бромат серебра, AgBrO3, получают обработкой растворов
солей серебра броматом калия КВгО3 или действием жидкого
брома на водную суспензию Ag2O:
AgNO3 + КВгО3 = AgBrO3 + KNO3
Ag2O + 6Br2 +J5H2O = 2AgBrO3 + ЮНВг
^Бромат серебра представляет собой двупреломляющие бесцвет-
ные кристаллы с плотностью 0,9987 г!см3-, он растворим в спиртах
и плохо растворяется в воде. При нагревании AgBrO3 разлагается
СЕРЕБРО
741
по уравнению
AgBrO3 -> AgBr + 3/2О2
Иодат серебра, AgIO3, получают обработкой растворов солей
серебра раствором йодата щелочного металла, действием ШО3
на металлическое серебро или действием иода на водную суспен-
зию Ag2O.
AgNO3 + KIOS = AgIO3 + KNO3
Ag2O + 6I2 + 5H2O = 2AgIO3 + 10HI
Иодат серебра представляет собой бесцветные орторомбические
кристаллы с плотностью 5,52 г/см3 и т. пл. 200° (при более сильном
нагревании он разлагается на Agl и О2); растворяется в воде
и глицерине.
Перйодаты серебра. Известны следующие перйодаты
серебра: AgIO4 — оранжевый, Ag2H3IO6 — лимонно-желтый,
Ag3IO5 и Ag5IO6 — черные.
Сульфид серебра, Ag2S, встречается в природе в виде минерала
аргентита или акантита; в лаборатории может быть получен очень
легко, когда металлическое серебро или его соединения реагируют
(в присутствии влажного воздуха) с серой, сероводородом или
различными сульфидами. Металлическое серебро может взаимо-
действовать с серой в твердом виде (при нагревании до белого
каления) и в растворах (в горячей воде или на холоду при исполь-
зовании раствора серы в сероуглероде).
Примеры реакции получения сульфида серебра:
2Ag + S = Ag2S 3Ag2O + 4S + H2O = 3Ag2S + H2SO4
2AgNO3 + H2S = Ag2S + 2HNO3 4Ag + 2H2S + O2 = 2Ag2S + 2H2O
Сульфид серебра при температуре ниже 179° представляет
собой орторомбические (или моноклинные) черные (или темно-
серые) призматические кристаллы, которые соответствуют природ-
ному акантиту, а при температуре выше 179° — это черные (или
темно-серые) кубические кристаллы, которые соответствуют при-
родному аргентиту. Орторомбическая модификация имеет плот-
ность 7,326 г!см\ а кубическая — 7,317 г!см\
Сульфид серебра диамагнитен, плавится при 842°, плохо раство-
рим в воде, аммиаке, тиосульфатах щелочных металлов и раство-
ряется в азотной кислоте и растворах цианидов щелочных металлов.
3Ag2S + 8HNO3 = 6AgNO3 + 2NO + 3S + 4H2O
Ag2S + 4NaCN = 2Na[Ag(CN)2] + Na2S
При нагревании в вакууме выше 350° Ag2S разлагается на эле-
менты.
Соединение Ag2S восстанавливается до металлического серебра
нагреванием выше 260° в атмосфере водорода, сплавлением с КОН,
742
ПОБОЧНАЯ ПОДГРУППА I ГРУППЫ
нагреванием с Ag2SO4 до 300° или с металлическим свинцом
(выше 400°):
Ag2S + Н2 = 2Ag + H2S Ag2S + Ag2SO4 = 4Ag -|- 2SO2
Ag2S "Ь 2К.ОН = 2Ag -|- K2S -|- H2O -|- V2O2 Ag2S -|- Pb — 2Ag -|- PbS
Галогены энергично взаимодействуют c Ag2S при высокой тем-
пературе с образованием AgF, AgCl, AgBr, Agl.
Концентрированная соляная кислота реагирует с Ag2S по урав-
нению
Ag2S + 2НС1 = 2AgCl + H2S
При температуре выше 1000° кислород окисляет Ag2S до Ag2SO4.
Сульфид серебра с сульфидами многих элементов образует
двойные сульфиды, например 4Ag2S -K2S -2Н2О — красный,
3Ag2S -Na2S-2Н2О— красный, 4Ag2S-5A12S3 — желтый, 3Ag2S.
• Sb2S3 (или Ag3SbS3), 5Ag2S-Sb2S3 (или Ag5SbS4), 3Ag2S-As2S3,
Ag2S3-Bi2S3.
Соединение Ag2S применяется в качестве катализатора во мно-
гих химических реакциях.
Сульфат серебра, Ag2SO4, осаждается при охлаждении кон-
центрированных растворов, полученных обработкой порошкообраз-
ного металлического серебра, Ag2O или Ag2CO3 концентрированной
серной кислотой, или при нагревании и упаривании до полного
выделения HNO3 раствора AgNO3 в избытке H2SO4.
2Ag -|- 2H2SO4 = Ag2SO4 -|- SO2 -|- 2Н2О
Ag2CO3 -|- H2SO4 = Ag2SO4 -|- СО2 -|- Н2О
Ag2O "Ь H2SO4 = Ag2SO4 -|- Н2О
2AgNO3 + H2SO4 = Ag2SO4 + 2HNO3
Сульфат серебра представляет собой диамагнитные белые ор-
торомбические мелкие кристаллы, которые плавятся при 652°
на воздухе (разлагаются выше 1085°) и частично растворяются
в воде
Выше 1085°
Ag2SO4 ► 2Ag + SO2-|-O2
При нагревании водород восстанавливает сульфат серебра:
В ы ттт с 12 5 о
Ag2SO4-|-H2 2Ag-—H2SO4
350°
Ag2SO4+5H2-----> 2Ag + H2S+4H2O
Хлор взаимодействует с сильно нагретым Ag2SO4 по уравнению
Ag2SO4 + С12 = 2AgCl + SO2 + О2
При растворении сульфата серебра в аммиаке получают ком-
плексные аммины [Ag(NH3)2]2SO4 и [Ag2(NH3)3]SO4.
СЕРЕБРО
743
Известны сульфате- и дисульфатоаргентаты(1), содержащие
анионы [AgSOJ-, [Ag(SO4)2]3-.
Из растворов, полученных взаимодействием Ag2SO4 с H2SO4
и содержащих немного воды, в зависимости от температуры выпа-
дают желтые призмы a-AgHSO4, устойчивые ниже 66°, или
желтые иглы 0-AgHSO4, устойчивые выше 66°. Помимо а-
и 0-AgHSO4 известны и другие гидросульфаты серебра, например,
Ag2SO4-2H2SO4, Ag2SO4-3H2SO4.
Сульфат серебра в серной кислоте может быть восстановлен
до металлического серебра сульфатом железа(П), медью, цинком,
железом и другими металлами.
Ag2SO4 + 2Cu = 2Ag + SO2 + 2CuO Ag2SO4 + 2FeSO4 = 2Ag + Fe2(SO4)3
Тиосульфат серебра, Ag2S2O3, получают обработкой раствора
AgCH3COO или AgF раствором Na2S2O3-5Н2О, взятым в коли-
честве, меньшем стехиометрически необходимого.
Тиосульфат серебра представляет собой неустойчивый белый
порошок, плохо растворимый в воде и растворимый в аммиаке
и в растворах тиосульфатов щелочных металлов с образованием
координационных соединений
Ag2S2O3 -|- 3Na2S2O3 = 2Na3[Ag(S2O3)2]
Соединение Na3[Ag(S2O3)2] образуется также при растворении
AgCl в концентрированном растворе Na2S2O3-5H2O и разлагается
при кипячении в разбавленных растворах, при подкислении или
старении:
2Na3[Ag(S2O3)2] = Ag2S Na2SO4 -|- 2Na2S2O3 S -|- SO2
2Na3[Ag(S2O3)2] + 4HNO3 = Ag2S + Na2SO4 + 4NaNO3 + 3S+3SO2 + 2H2O
Кипячение водной суспензии Ag2S2O3 приводит к образованию
Ag2S и H2SO4.
Нитрид серебра, Ag3N, получают разложением аммиачных
растворов фторидов серебра или электролизом нитрида аммония
в жидком аммиаке, используя серебряные электроды.
Нитрид серебра представляет собой коричневые кубические
кристаллы, он плохо растворим в воде, растворяется в аммиаке,
разлагается со взрывом при нагревании, взаимодействует с HNO3
и растворами цианидов щелочных металлов
Ag3N + 4HNO3 = 3AgNO3 + NH4NO3
Ag3N + 6KCN + 3H2O = 3K[Ag(CN)2] + NH3 + ЗКОН
Амид серебра, AgNH2, образуется в результате действия KNH2
на AgNO3 (реакция идет в жидком аммиаке) или упаривания
аммиачного раствора Ag2O в присутствии конц. H2SO4.
KNH2 + 'AgNO3 = AgNH2 + KNO3 [Ag(NH3)2]OH = AgNH2 + NH4OH
ПОБОЧНАЯ ПОДГРУППА I ГРУППЫ
744
Амид серебра представляет собой неустойчивое белое вещество,
чувствительное к действию света; он разлагается со взрывом при
высушивании, растворяется в жидком аммиаке.
Имид серебра и калия, KAgNH, разлагается без взрыва при
температуре выше 100°.
Из гидроокиси диамминсеребра(1) можно получить также
взрывчатый имид серебра, Ag2NH:
2[Ag(NH3)2]OH = Ag2NH + 3NH3 + 2H2O
Нитрит серебра, AgNO2, получают обработкой AgNO3 нитри-
том щелочного металла (взятого в количестве, меньшем стехиоме-
трически необходимого) или обработкой Ag2SO4 нитритом бария.
Нитрит серебра представляет собой желтые игольчатые кри-
сталлы с плотностью 4,493 г/сл43; он растворим в теплой (60°)
воде, в аммиаке и растворах нитритов щелочных металлов.
При упаривании раствора нитрита серебра в аммиаке выпадают
желтые кристаллы нитрита амминсеребра [AgNH3]NO2; послед-
ний имеет т. пл. 70° и растворим в воде. При добавлении эфира
к водному раствору соединения [AgNH3]NO2 выпадают расплываю-
щиеся на воздухе белые кристаллы [Ag(NH3)2]NO2.
Нитриты щелочных металлов растворяют нитрит серебра
с образованием ацидосоединений типа MeI[Ag(NO2)2] (где Ме* =
= Na+, К+). Нитрит серебра служит для получения алифатических
нитропроизводных.
Нитрат серебра, AgNO3, осаждают путем концентрирования
растворов металлического серебра, Ag2S, Ag2Onnn Ag2CO3 в HNO3.
3Ag + 4HNO3 = 3AgNO3 + NO + 2H2O
Ag2O + 2HNO3 = 2AgNO3 + H2O
Ag2S -|- 2HNO3 = 2AgNO3 -J- H2S
Ag2CO3 -J- 2HNO3 = 2AgNO3 -|- CO2 -|- H2O
Упариванием нейтральных водных растворов нитрата серебра
при температуре ниже 157° получают бесцветные орторомбические
кристаллы AgNO3, а при температуре выше 157° — бесцветные
ромбоэдрические кристаллы AgNO3.
Соединение AgNO3 диамагнитно, обладает плотностью
4,352 г/см3, плавится при 208,6°, растворяется в воде, метаноле,
этаноле, ацетоне, пиридине, разлагается при нагревании примерно
до 300° по уравнению
AgNO3 = Ag -|- NO2 -f- V2O2
Нитрат серебра в водном растворе может быть восстановлен
водородом (80°) или бором на холоду, а в аммиачном растворе —
различными щелочными металлами, альдегидами, спиртами или
СЕРЕБРО
745>
сахарами, обладающими восстановительными свойствами:
AgNO3 + »/2Н2 = Ag + NH3 + ЗН2О
2AgNO3 + 3NH4OH + НСНО = 2Ag + 2NH4NO3 + HCOONH4 + 2H2O
2[Ag(NH3)2]OH + HCHO = 2Ag + 3NH3 + HCOONH4 + H2O
Реакция восстановления нитрата или гидроокиси диаммин-
серебра формальдегидом лежит в основе серебрения стекла при
изготовлении зеркал.
Ртуть восстанавливает нитрат серебра с образованием кристал-
лических амальгам (дерево Дианы) состава Ag3Hg4, Ag3Hg2,
Ag3Hg. Свинец, цинк и другие металлы, более активные, чем
серебро, вытесняют его из растворов солей. Фосфористый водород
РН3 восстанавливает соединение AgNO3 до металлического серебра
или фосфида серебра Ag3P и фосфорноватистой кислоты НвР2О4.
При нагревании фтора или хлора с AgNO3 образуется AgF
или AgCl.
Обработка водных растворов AgNO3 растворами галогеново-
дородов (за исключением плавиковой кислоты) приводит к осаж-
дению галогенидов AgCl, AgBr, Agl. Нитрат- серебра взаимодей-
ствует также со многими аминами (с моно-, ди- и триэтиламинами,
этилендиамином, анилином, пиридином и др.).
Смесь AgNO3 с серой при ударе взрывается, а смесь AgNO3
с углем при растирании в ступке загорается без детонации. Азот-
нокислое серебро связывает аммиак, образуя белую соль
[Ag(NH3)3JNO3, которая разлагается при нагревании.
Нитрат серебра реагирует с нитратами щелочных или других
металлов, образуя двойные соединения. Изучены системы с раз-
личными нитратами, например MexNO3AgNO3 (где Me1 = Li+,
Na+, К+, Rb+, NHi); установлено существование соединений
2MeINO3-AgNO3 и ЗМеИМО3-AgNO3 (где Mei = Li+, К+, Rb+,
NH+ Т1+, x/2Ra2+, Х/2РЬ2+).
Нитрат серебра применяется для получения самых различных
соединений серебра, в производстве зеркал, для получения свето-
чувствительных эмульсий, красителей для хлопчатобумажных
тканей, краски для волос; 0,95—1,05%-ные растворы нитрата
серебра применяются в медицине, а также в качестве аналитиче-
ского реактива и для электролитического осаждения серебра.
Карандаши твердого нитрата серебра (ляпис) состоят из 1 ч.
AgNO3 и 2 ч. KNO3 и служат для прижигания.
Нитрат серебра применяется в качестве катализатора при полу-
чении перекиси водорода,восстановлении К282О8солями хрома(Ш),
марганца(Н) или церия(Ш), в реакциях полимеризации.
Фосфид серебра, Ag3P, серого цвета, может быть получен взаи-
модействием элементов при сильном нагревании или сплавлением
металлического серебра с метафосфорной кислотой НпРпО3п
и углем.
746
ПОБОЧНАЯ ПОДГРУППА I ГРУППЫ
Ортофосфат серебра, Ag3PO4 в виде желтого осадка образуется
при обработке раствора AgNO3 подкисленным раствором Na3PO4>
• 12Н2О или Na2HPO4-12 Н2О
3AgNO3 + 2Na2HPO4 Ag3PO4 + 3NaNO3 + NaH2PO4
Ортофосфат серебра представляет собой желтые кубические
кристаллы с плотностью 6,37 г!см3 и т. пл. 849°; он чувствителен
к свету, плохо растворим в воде и растворяется в аммиаке, мине-
ральных кислотах.
Гидрофосфат серебра, Ag2HPO4, выпадает в осадок при концент-
рировании растворов Ag3PO4 в Н3РО4, содержащей 50% Р4О10.
Желтые кристаллы Ag2HPO4 чувствительны к свету и превра-
щаются в Ag3PO4 под действием воды:
3Ag2HPO4 = 2Ag3PO4 + Н3РО4
Дигидрофосфат серебра, AgH2PO4, выпадает при концентри-
ровании растворов Ag3PO4 в 85%-ной Н3РО4 или растворов
Ag2HPO4 в 80%-ной Н3РО4.
Соединение AgH2PO4 представляет собой двупреломляющие
бесцветные кристаллы, чувствительные к свету; под действием
воды, спирта, эфира или при нагревании оно разлагается
2AgH2PO4 Ag2HPO4 + Н3РО4 3AgH2PO4 -> Ag3PO4 + ЗН3РО4
Метафосфаты серебра. Известны ди-, три-, тетра-,
гекса- и декаметафосфаты серебра Ag2P2O6-H2O, Ag3P3O9-H2O,
Ag4P4O12, Ag6P6O18, Ag10P10O30 -H2O; все они твердые белые веще-
ства.
Арсенид серебра, Ag3As, черного цвета, образуется при дей-
ствии мышьяковистого водорода AsH3 на раствор AgNO3.
Ортоарсенат серебра, Ag3AsO4, получают обработкой раство-
ров AgNO3 раствором H3AsO4 или Na2HAsO4-7Н2О.
Это фиолетово-красные кубические кристаллы с плотностью
45,65 г/см3, они плохо растворимы в воде, растворяются в аммиаке,
карбонате аммония, уксусной кислоте, при нагревании до 800°
разлагаются по уравнению
2Ag3AsO4 —> 6Ag -j- As2O5 -|- 3/2O2
Антимонид серебра, Ag3Sb, получают взаимодействием элемен-
тов при нагревании или пропусканием сурьмянистого водорода
SbH3 через концентрированный холодный раствор AgNO3.
Антимонид серебра представляет собой белые орторомбические
кристаллы, плохо растворимые в НС1 и H2SO4.
Хромат серебра, Ag2CrO4, в виде коричнево-красного осадка
-образуется при обработке растворов солей серебра растворами
хроматов щелочных металлов; он легко растворим в аммиаке
СЕРЕБРО
747
и минеральных кислотах.
Ag2CrO4 -|- 4NHa = [Ag(NH3)2]2CrO4
2Ag2CrO4 + 4HNO3 = 4AgNO3 + Н2Сг2О7 + Н20
Бихромат серебра, Ag2Cr2O7, в виде коричнево-красного осадка
образуется при обработке растворов солей серебра растворами
бихроматов щелочных металлов; при кипячении он превра-
щается в хромат серебра:
2Ag2Cr2O7 + НОН 5* 2Ag2CrO4 -|- Н2Сг2О7
Ацетиленид серебра, Ag2C2, получают действием ацетилена
на порошкообразное серебро или на аммиачный раствор нитрата
серебра, обработкой ацетиленида натрия или ртути теплым раство-
ром нитрата серебра, взаимодействием карбида кальция с раство-
ром нитрата серебра в метаноле:
2Ag + С2Н2 = Ag2G2 -|- Н2 2AgNO3 -|- С2Н2 = Ag2G2 -|- 2HNO3
Ацетиленид серебра образуется в виде коричневого аморфного
порошка; он плохо растворим в воде, легцо взрывается при
ударе, под действием яркого света или при нагревании выше
140°, разлагается под действием НС1 или KCN:
Ag2C2 + 2НС1 = 2AgCl + С2Н2
Ag2C2 + 4KCN + 2Н2О = 2K[Ag(CN)2] + 2КОН + С2Н2
Карбонат серебра, Ag2CO3, получают обработкой раствора
AgNO3 (взятого в избытке) раствором NaHCO3 или Na2CO3-10H2O.
Карбонат серебра представляет собой желтовато-белые ромбо-
эдрические кристаллы с плотностью 6,077 г/см3-, он плохо раство-
рим в воде, растворяется в аммиаке или растворах цианидов и тио-
сульфитов щелочных металлов, разлагается при кипячении с боль-
шим количеством воды или нагревании на воздухе.
Ag2CO3 —>- Ag2O -|- СО2 Ag2CO3 —> 2Ag -|- СО2 -|- 1/2О2
Упаривание (в темноте) раствора Ag2CO3 в NH4OH приводит
к осаждению бесцветных двулучепреломляющих орторомбических
кристаллов [Ag(NH3)2]2CO3, которые растворимы в воде, но
плохо растворяются в спирте.
Ag2CO3 -|- 4NH4OH = [Ag(NH3)2]2CO3 -|- 4НОН
Карбонат серебра с карбонатами щелочных металлов образует
двойные карбонаты, например K2CO3-Ag2CO3 — бесцветные кри-
сталлы с плотностью 3,769 г!см3.
Цианид серебра, AgCN, получают обработкой растворов солей
серебра цианидом щелочного металла, взятым в стехиометрически
необходимом количестве:
AgNO3 + KCN = AgCN + KNO3
748
ПОБОЧНАЯ ПОДГРУППА I ГРУППЫ
Цианид серебра представляет собой бесцветные ромбоэдриче-
ские кристаллы с плотностью 3,95 г/см3 и т. пл. 320—350°; он плохо
растворим в воде, растворяется в аммиаке или растворах солей
аммония, цианидов и тиосульфатов щелочных металлов с образо-
ванием координационных соединений.
AgCN + 2NH4OH = [Ag(NH3)2]CN + 2Н2О
AgCN + KCN = K[Ag(CN)2]
Уксусная кислота и сероводород взаимодействуют с дициано-
аргентатами Me1 [Ag(CN)2] по уравнениям
K[Ag(CN)2] + HNO3 = AgCN + KNO3 + HCN
2K[Ag(CN)2] + 2H2S = Ag2S + K2S + 4HCN
При обработке K[Ag(CN)2] нитратом серебра образуется дициа-
ноаргентат серебра Ag[Ag(CN)2], представляющий собой димерную
форму моноцианида серебра.
Известны цианоаргентаты типов Me*[Ag(CN)3] и MeJ[Ag(CN)4].
Тиоцианат серебра, AgSCN, получают обработкой раствора
AgNO3 раствором тиоцианата щелочного металла, взятым в стехио-
метрически необходимом количестве.
Тиоцианат серебра представляет собой белое твердое вещество,
он плохо растворим в воде, растворяется в избытке тиоцианатов
щелочных металлов с образованием тиоцианатоаргентатов
MeI[Ag(SCN)2], Me£[Ag(SCN)3], MeJlAg(SCN) J и восстанавливается
в аммиачных растворах металлами (Na, К, Са) или гидроксил-
амином.
Ацетат серебра, AgCH3COO, осаждают путем концентрирова-
ния раствора Ag2CO3 в уксусной кислоте; это бесцветные кри-
сталлы, растворимые в воде и спирте.
Оксалат серебра, Ag2C2O4, получают обработкой раствора
AgNO3 раствором Н2С2О4-2Н2О или оксалатов щелочных ме-
таллов.
2AgNO3 -|- Na2C2O4 = Ag2G2O4 -|- 2NaNO3
Оксалат серебра представляет собой белые моноклинные кри-
сталлы с плотностью 5,029 г/см3', он плохо растворим в воде, чув-
ствителен к свету, разлагается при нагревании до 100°. При
140° Ag2C2O4 разлагается со взрывом.
Координационные соединения
Большинство простых соединений одновалентного серебра
с неорганическими и органическими реагентами образуют коор-
динационные соединения. Благодаря образованию координацион-
ных соединений многие плохо растворимые в воде соединения
СЕРЕБРО
749
серебра превращаются в легко растворимые. Серебро может иметь
координационные числа 2, 3, 4 и 6.
Известны многочисленные координационные соединения,
у которых вокруг центрального иона серебра координированы
нейтральные молекулы аммиака или аминов (моно- и диметил-
амин, моно- и диэтиламин, пиридин, этилендиамин, анилин, толуи-
дин, тиомочевина и др.) и различные кислотные радикалы, напри-
мер, С1-, Вг-, I-, CN-, SCN-, NOj, NOj, Х/282ОГ и др.
При действии аммиака или различных органических аминов
на окись, гидроокись, нитрат, сульфат, карбонат серебра обра-
зуются соединения с комплексным катионом, например,
[Ag(NH3)2]+, [AgEn]+, [AgEn2]+, [AgPyP, [AgPy2]+.
Устойчивость комплексных катионов серебра ниже устойчи-
вости соответствующих катионов меди(П).
Hpnj растворении галогенидов серебра (AgCl, AgBr, Agl)
в растворах галогенидов, псевдогалогенидов или тиосульфатов
щелочных металлов образуются растворимые в воде координа-
ционные соединения, содержащие комплексные анионы, напри-
мер, [AgCl2]-, [AgCl3]2-, [AgClJ3-, [AgBr3]2-, [Agl2]-, [Agl3]2~,
{AglJ3-, [Ag2I4]2-, [Ag2Is]3-, [Ag2I6]4", [Ag2I7]5-, [Ag(CN)2]-,
{Ag(CN)3]2-, [Ag(CN)J3-, [Ag(SCN)2]", [Ag(SCN)3]2-, Ag(SCH)4]3-,
[Ag(S2O3)l-, [Ag(S2O3)2]3-, (Ag(S2O3)3l5-, [Ag2(S2O3)6]10-,
(Ag3(S2O3)4l5 , [Ag2(S2O3)3]4 .
Дитизон образует с солями серебра в кислой среде желтый
дитизонат, растворимый в СС14:
N-NH-C6H5
Ag--S---С
N = N —С6Н5^
В щелочной среде образуется фиолетовый дитизонат, плохо
растворимый в СС14.
n-Диметиламинобензилиденродамин образует с концентриро-
ванными растворами солей серебра фиолетовый осадок:
С разбавленными растворами солей серебра п-диметиламино-
бензилиденродамин не образует осадка, а только окрашивает
раствор в интенсивно фиолетовый цвет.
750
ПОБОЧНАЯ ПОДГРУППА I ГРУППЫ
Соединения двухвалентного серебра
Известно немного соединений двухвалентного серебра. Для
них характерна низкая устойчивость и способность разлагаться
водой с выделением кислорода.
Неорганические соединения
Окись серебра, AgO, получают действием озона на металличе-
ское серебро или на Ag2O, AgNO3 или Ag2SO4, обработкой раствора
AgNO3 раствором K2S2O8, обработкой щелочной суспензии Ag2O
перманганатом калия, анодным окислением металлического
серебра с использованием в качестве электролита разбавленного
раствора H2SO4 или NaOH.
Ag2O Н- Оз —► 2AgO -|- О2
2AgNO3 + K2S2O8 +4КОН = 2AgO + 2K2SO4 + 2KNO3 + 2H2O
Ag2O + 2KMnO4 + 2KOH = 2AgO + 2K2MnO4 + H2O
Обработка Кг82О8 соединений серебра в слабо кислой среде
и в присутствии пиридина приводит к образованию оранжевого
кристаллического осадка [AgPy4]S2O8.
Окись серебра представляет собой диамагнитный серовато-
черный кристаллический порошок с плотностью 7,48 г/см3', она
растворима в H2SO4, НС1О4 и конц. HNO3, устойчива при обыч-
ной температуре, разлагается на элементы при нагревании до 100°,
является энергичным окислителем по отношению к SO2, NH3,
Me+NO2, обладает свойствами полупроводника.
Фторид серебра, AgF2, получают действием газообразного
фтора на металлическое серебро при 250—300° или на галогениды
серебра(1) при 200—300°.
Ag + F2 = AgF2 + 84,5 ккал
Фторид серебра представляет собой парамагнитный коричнево-
черный порошок с т. пл. 690°; он разлагается под действием воды
или влажного воздуха и обладает окислительным действием
по отношению к иодидам, спирту, солям хрома(Ш) и марганца(П).
6AgF2 + ЗН2О = 6AgF + 6HF + О3
Сульфид серебра, AgS, образуется в виде коричневого осадка
при обработке раствора AgNO3 в бензонитриле раствором серы
в сероуглероде.
Нитрат серебра, Ag(NO3)2, получают окислением AgNO3 озо-
ном. Это бесцветные кристаллы, разлагающиеся водой:
4Ag(NO3)2 + 2Н2О 4AgNO3 + 4HNO3 + О2
СЕРЕБРО
751
При анодном окислении раствора AgNO3 в пиридине мож-
но получить оранжево-красные призматические кристаллы
[AgPy4](NO3)2, которые разлагаются под действием воды или
аммиака.
Координационные соединения
Известен ряд координационных соединений двухвалентного-
серебра типов [Ag(C5H5N)4]X2 и [AgAm2]X2 (где Ат = фенант-
ролин Ci2H8N2, дипиридил Ci0H8N2 и X = NOj, СЮ", СЮ",.
hso;,
Соединения трехвалентного серебра
Известно небольшое число соединений трехвалентного-
серебра, например Ag2O3, K8H[Ag(IO6)2] *ЮН2О, K7[Ag(IO6)2],
Na7H2[Ag(TeO6)2]-14Н2О и др.
Окись серебра, Ag2O3, образуется в смеси'с окисью серебра(П)’
при анодном окислении серебра или при действии фтора (или перо-
ксосульфата) на соль серебра(1).
Черная кристаллическая смесь Ag2O3-AgO неустойчива, обла-
дает окислительными свойствами и при легком нагревании превра-
щается в AgO.
Диортопериодатоаргентат ы(Ш), Ме*Н[ Ag(IO6)2] -
•пН2О, являются диамагнитными солями оранжевого цвета
с кристаллами красивой формы; их рассматривают как производ-
ные гипотетической кислоты H7[Ag(IO6)2J.
При окислении смеси водных растворов AgNO3, KsIO6 и КОН
надсернокислым калием K2S2O8 образуется коричневый раствор,
из которого при концентрировании путем медленного испарения
выпадают оранжевые кристаллы K6H[Ag(IO6)2] -10Н2О, а при быст-
ром упариваниии— K7[Ag(IO6)2]-КОН-8Н2О. Обработка соеди-
нения K6H[Ag(IO6)2] карбонатом натрия приводит к осаждению
оранжево-желтых кристаллов Na5KH[Ag(IO6)7]-16Н2О.
Диортотеллуратоаргентаты, MegH3[Ag(TeO6)2] -
• пН2О, Me+HJAg^eOshl'«Н2О представляют собой красиво кри-
сталлизующиеся желтые диамагнитные соли — производные гипо-
тетической кислоты H9[Ag(TeO6)2].
Окисление водного раствора смеси Ag2SO4, Na2CO3 и ТеО2
пероксосульфатом калия K2S2O8 приводит к образованию корич-
невого раствора, из которого при концентрировании путем изо-
термического испарения осаждаются желтые кристаллы
Na6H3[Ag(TeO6)2l’18Н2О. При использовании больших количеств
карбоната натрия выпадают кристаллы Na7H2[Ag(TeO6)2] •14Н2О.
Нитрат диэтиленгуанидинсеребра, [Ag(elig)2](NO3)3, представ-
752
ПОБОЧНАЯ ПОДГРУППА I ГРУППЫ
ляет собой диамагнитные красные призматические кристаллы,
устойчивые при обычной температуре. Известны соли, в которых
кислотный радикал NO~ заменен радикалом С1О7, ОН- или
х/2 sof.
Металлоорганические соединения
Известно ограниченное число металлоорганических производ-
ных серебра. В качестве примеров упоминаются AgC2H5, AgC6H5,
устойчивость которых ниже, чем у соответствующих соединений
меди.
Фенилсеребро, AgC6H5, получают действием финилмагнийодида
на суспензию AgCl в эфире. Оно мало устойчиво и быстро раз-
лагается.
ЗОЛОТО Ан
Z — 79; ат. вес = 196,967
Валентность I, (II), (III); заряд 1+, 2+, 3+
Массовое число основного природного изотопа 197
Электронная структура атома золота: К ~L-М-N-О-6s1.
Структура атома золота и катиона Ап3+ для 5d- и бх-орбиталей:
5d10
tl tl tl tl u
6s1
t
Au
Au*+
5d>
ИСТОРИЧЕСКАЯ СПРАВКА
Золото — один из самых древних металлов, известных человеку.
Древние греческие легенды и египетские папирусы повествуют
•о войне за золото в Средиземном море. В произведениях Геродота,
Страбона, Плиния приводятся легенды о золоте. Золото считалось
символом богатства и долгой счастливой жизни. Алхимики обозна-
чили золото знаком Солнца.
Название «золото» имеет индоиранское происхождение.
В Египте иероглиф, означающий золото, имел изображение
платка, мешка или лотка (что отображало способ извлечения золота
из наносных залежей).
В старых руководствах встречаются описания золотых копей
и примитивные методы добычи золота.
После открытия Америки в XV в. испанские завоеватели при-
везли в Европу в качестве трофеев огромные количества золота.
Богатые золотые залежи были найдены в Бразилии (1719 г.),
на Урале (1745 г.), в Калифорнии (1848 г.), в Южной Африке
и в Трансваале (1886 г.) и на Аляске (1900 г.).
золото
753
РАСПРОСТРАНЕННОСТЬ В ПРИРОДЕ
В природе золото встречается большей частью в самородном
состоянии и в виде теллуридов золота или двойных теллуридов
золота и серебра.
Содержание золота в земной коре равно 5-Ю-7 вес.%. В при-
роде золоту часто сопутствуют кварц SiO2 (иногда дацит, родохро-
зит МпСО3, гематит Fe2O3), а также сульфиды и арсениды (пирит,
халькопирит, галенит, обманка, стибнит, миспикель и др.). Руды
многих цветных металлов (меди, олова, никеля, цинка, свинца,
серебра, кобальта, хрома, ртути, платины и палладия) содержат
золото в качестве примесей. Самородное золото встречается в двух
формах — ископаемое золото в золотоносных жилах, и намывное
золото в наносных скоплениях золотоносного песка.
Обычно самородное золото содержит 5—15% серебра, меди
и реже железа, ртути, платиновых металлов, висмута и др. Среди
разновидностей самородного золота [встречаются купроаурид
содержит до 20 вес. % меди), порпецит (содержит 5—11 % палладия
и 4% серебра), бисмутоаурит (содержит до 14% висмута).
Ископаемое золото образует кубические гранецентрированные
кристаллы (вкрапление в кварц или руду) золотисто-желтого
цвета с плотностью 15,6—18,3 г!смг и твердостью 2,5—3 по шкале
Мооса. Золото обладает высокой электро- и теплопроводностью.
Сплавы золото — серебро, которые содержат 15—30% серебра
и небольшие количества меди, называют электрум; они встречаются
в природе в виде светло-желтых кубических кристаллов с плот-
ностью 12—15 г!смг и твердостью 2—3 по шкале Мооса.
К наиболее важным минералам золота относятся следующие.
Калаверит, АиТе2, содержит 39,2—42,8% золота, встречается
в виде серебристо-белых (со светло-желтым оттенком) моноклин-
ных кристаллов, которые часто сопровождают самородное золото
в кварцевых жилах пирита.
Креннерит, (Au, Ag)Te2, содержит 30,7—43,9% золота и пред-
ставляет собой непрозрачные серебристо-белые (иногда желтые,
как латунь) ромбические кристаллы с металлическим блеском.
Сильванит, (Au, Ag)Te4, является двойным теллуридом золота
и серебра, содержащим 25,4—29,9% золота; встречается в виде
непрозрачных серебристо-серых (иногда желтоватых) моноклин-
ных кристаллов с интенсивным металлическим блеском.
Петцит, (Au, Ag)2Te, содержит 19—25,2% золота, 50% сереб-
ра; встречается в виде серых кубических кристаллов с краснова-
тым оттенком.
Нагиагит, Au2Pb10Te6Sb2S15, содержит 5,8—12,8% золота;
встречается в виде серых ромбических кристаллов.
Гессит, Ag2Te,— теллурид серебра, но часто содержит значи-
тельные количества золота.
48-0101
754
ПОБОЧНАЯ ПОДГРУППА I ГРУППЫ
Золотоносные залежи находятся в Южно-Африканской Рес-
публике, Конго, Южной Родезии, Гане, Канаде, СССР, США,
Австралии, Румынии, Франции.
Золото было обнаружено в спектре Солнца; оно найдено также
во многих метеоритах, в каменных углях, морской воде. В морской
воде золото встречается либо в виде очень мелких вкраплений
на органических или минеральных частицах, либо в виде ионов
[AuClJ-
ПЕРЕРАБОТКА ЗОЛОТОНОСНЫХ РУД
Процесс переработки золотоносных руд включает измельчение,
обогащение и извлечение золота из руд и концентратов. Золото-
носные руды измельчают в щековых дробилках, молотковых
и шаровых мельницах, а также с помощью пневматических
молотов.
Поскольку золотоносные руды содержат обычно 3—15 г золота
на тонну, их перерабатывают только после обогащения гравита-
ционными или флотационными методами.
Обогащать выгодно только руды, содержащие не менее 3,63 г
золота на тонну. Измельченную руду сначала классифицируют
по размерам гранул и затем промывают водой.
Процессы гравитационного обогащения золотоносных руд
основываются на разности плотностей самородного золота
(15,6—18,3 г/Ам3) и пустой породы (2,6—6 г/сл43). Когда измель-
ченные руды, содержащие золото, промывают водой, порода почти
полностью вымывается. Обогащение золотоносных руд проводят
в деревянном желобе, выложенном камнем (который местами пре-
рывается горизонтальными бассейнами для сбора золотого песка),
а также на транспортерных лентах или на столах (длиной 1,5—6 м,
шириной 1—2 м), наклоненных под углом 50° и покрытых мягким
сукном (это может быть также рубчатый бархат, палаточная ткань,
мешковина, пористая жатая хлопчатобумажная ткань, рубчатый
пористый каучук и др.). Для обогащения золотоносных руд (оло-
вянных, вольфрамовых, железных) существует метод сортировки.
Этот метод заключается в разделении руд на минеральные состав-
ляющие по разности их удельных весов. Разделение осуществляют
с помощью струи воды переменного напора (в восходящем и нисхо-
дящем направлениях). В сортировочной машине минеральные
частицы то взмучиваются водой, то оседают на сите, расслаиваясь
в соответствии с их плотностями (самые плотные образуют нижние
слои).
При этом методе необходимо предварительное разделение, тем
более тщательное, чем ближе значения удельных весов минералов,
подвергающихся разделению.
Метод флотации применяют в промышленном масштабе для
извлечения сильно рассеянного ископаемого золота из сульфид-
золото
755
ных руд (таких, как сульфиды меди и свинца) или для удаления
минералов (таких, как сульфиды мышьяка, сурьмы, минерала
серебра, меди и др.), которые увеличивают расход цианида и за-
трудняют процесс извлечения золота цианидным выщелачиванием.
В качестве пенообразователей при флотации используют сосно-
вую смолку или крезолы, а коллекторами служат ксантогенаты.
Присутствие серебра и меди в золотоносных минералах снижает
эффективность флотации. Концентраты, полученные методом фло-
тации, всегда содержат серебро и медь.
ИЗВЛЕЧЕНИЕ ЗОЛОТА
ИЗ КОНЦЕНТРАТОВ ЗОЛОТОНОСНЫХ РУД
Из концентратов золотоносных руд золото извлекают амаль-
гамированием, цианированием, а также с помощью хлорной воды,
царской водки или тиомочевины. Существует и пирометаллургиче-
ский способ.
Извлечение золота амальгамированием
С помощью ртути ископаемое золото и серебро извлекают в виде
амальгам из золотоносных руд или золотосодержащих остатков,
которые находятся в водной суспензии. Амальгамирование золота
осуществляют смешиванием концентратов руд или золотосодержа-
щих остатков в водной суспензии со ртутью на слегка наклоненных
амальгамированных медных листах. При амальгамировании
извлекают только крупные (>0,3 мм) частицы с чистой поверхно-
стью, поскольку только в этом случае ртуть амальгамирует золото
или серебро. Очень мелкие частицы золота или частицы, покрытые
инертным слоем (силикатами или сульфидами), на амальгамиру-
ются. При амальгамировании золото не извлекается полностью,
а теллуриды золота не амальгамируются совсем.
Амальгама золота представляет собой вязкую пасту, содер-
жащую 20—50% золота. Ртуть с золотом может образовать интер-
металлические соединения AuHg2, Au2Hg, Au3Hg и твердый рас-
твор, содержащий 16,7% ртути.
При нагревании амальгамы золота и серебра выше 357° (точка
кипения ртути) ртуть отгоняется и накапливается в конденсато-
рах, а золото и серебро полностью остаются в дистилляционном
аппарате. Сырое золото, полученное после отгонки ртути, пла-
вится с флюсами (Na2CO3, Na2B4O7 и KNO3) в графитовых тиглях
и отливается в виде слитков весом 6—8 кг.
Металлы, более активные, чем золото, которые остаются после
отгонки ртути, окисляются при окислительно-щелочном сплав-
лении и превращаются в шлак.
До 1936 г. этот процесс был широко распространен. В настоя-
щее время его применение очень редко.
48*
756
ПОБОЧНАЯ ПОДГРУППА I ГРУППЫ
Извлечение золота цианированием
Важнейшим в настоящее время способом извлечения золота
является цианидное выщелачивание, которое применяется как
при переработке очень бедных руд, так и при переработке концент-
ратов золотоносных руд, полученных промыванием или флотацией.
Бедные руды, концентраты золотоносных руд или остатки
от амальгамирования растворяют в воде, содержащей 0,03—0,25%
цианидов щелочных (или щелочноземельных) металлов при 80°
и перемешивают, пропуская через раствор кислород (или воздух)
под давлением. При этом образуются комплексные цианиды
золота(1).
Обычно для экстракции применяют растворы цианидов нат-
рия NaCN, калия KCN или кальция Ca(CN)2.
При использовании цианида натрия процесс растворения золо-
та описывается следующим уравнением:
4Au + 8NaCN + 2Н2О + О2 = 4Na[Au(CN)2] + 4NaOH
Водные растворы цианидов помимо золота растворяют и другие
металлы, такие, как серебро, медь, никель, кобальт, цинк, алюми-
ний и железо.
Обычно работают с 0,25%-ными водными растворами цианида,
которые подщелачивают известью, поскольку в таких растворах
золото обладает максимальной растворимостью, а растворимость
сопутствующих металлов мала. Нейтральные или кислые растворы
цианидов не применяют. В отличие от самородного золота теллу-
риды золота не растворяются в растворах цианидов.
Цианирование проводят в чанах и растворы комплексных циа-
нидов золота(1) отделяются от плохо растворимых веществ декан-
тацией и фильтрованием.
Для извлечения золота из растворов комплексных цианидов
можно применять металлический цинк, амальгаму цинка, метал-
лический алюминий, элементарный кремний, некоторые силициды,
хлорид меди(1), активированный уголь и ионообменные смолы.
В случае использования металлического цинка (в виде порошка
или мелких стружек) золото извлекают из раствора комплексных
цианидов при температуре 40° и перемешивании.
2Na[Au(CN)2] + Zn = 2Au + Na2[Zn(CN)4]
Для извлечения часто применяют цинковые стружки, покрытые
свинцом [для чего металлический цинк в виде стружки предвари-
тельно помещают в раствор Pb(NO3)2 или РЬ(СН3СОО)2].
Золото, осажденное цинковой стружкой, отделяют промыва-
нием водой, а затем очищенная цинковая стружка вновь вводится
в реакцию.
Металлическое золото, выделенное с помощью цинка из водных
растворов комплексных цианидов золота, очищают обработкой
золото
757
кислотами (НС1 или H2SO4) для удаления цинка, а затем сплав-
ляют с бурой или содой и отливают в слитки.
Технологический процесс извлечения золота из золотоносных
руд в виде комплексного цианида натрия Na[Au(CN)2] связан
с определенными трудностями. Эффективность этого процесса
зависит от ряда факторов, таких, как концентрация, температура,
основность раствора цианида, степень измельчения руды и т. д.
В случае тонкого измельчения образуется большое количество
шлама, который трудно фильтруется.
Металлы, образующие растворимые комплексы с цианидами
щелочных металлов, затрудняют процесс осаждения золота.
Из суспензии золотоносной руды в воде золото можно удержать
на активированном угле, а затем извлечь горячим раствором NaCN.
Промытый и высушенный активированный уголь может быть исполь-
зован заново. Более выгодно, однако, применять ионообменные
смолы.
Из растворов комплексного цианида Na[Au(CN)2] золото можно
извлечь с помощью сильно основных анионообменных смол с боль-
шой обменной емкостью.
Анионообменная смола амберлит IRA — 400 удерживает комп-
лексные цианиды всех тяжелых металлов, включая золото, медь,
серебро, железо, кобальт, никель и цинк. Для селективного извле-
чения применяют смесь органических растворителей с минераль-
ными кислотами (например, ацетон и азотная кислота).
При использовании анионообменной смолы в хлоридной форме
скорость извлечения комплексного аниона золота [Au(CN)2]_ мак-
симальна при pH = 3,5.
RC1 + [Au(CN)2]“ -* R[Au(CN)2] + Cl-
Для качественного определения золота в элюенте послед-
ний обрабатывают 10%-ным раствором бензидина в уксусной
кислоте.
Почти селективное извлечение золота из сильно основной ионо-
обменной смолы осуществляется смесью ацетона с 5% HNO3
и 5% воды, поскольку в этом случае медь и железо экстрагиру-
ются плохо.
Для элюирования следов золота применяют смесь равных объе-
мов 4 н. НС1 и 4 н. HNO3.
Цианид меди(1), цианиды железа(П) и (III) могут быть извле-
чены раствором NaCN. Цианиды серебра и кобальта элюируют
2 н. раствором KSCN, а цианид цинка — 0,1 н. НС1.
Для извлечения золота из морской воды используют сульфи-
рованную полистирольную смолу, которая обладает большим срод-
ством к тяжелым металлам и малым сродством к щелочным и ще-
лочноземельным металлам.
758
ПОБОЧНАЯ ПОДГРУППА I ГРУППЫ
Извлечение золота хлорированием
Длительное время золото извлекалось из бедных золото-
носных руд и остатков от амальгамирования хлорной водой. В
настоящее время этот процесс почти не применяется, за исклю-
чением тех случаев, когда золото сильно рассеяно в каменистых
породах.
Хлорирование золотоносных руд осуществляется в цилиндрах
с внутренней свинцовой обкладкой. При хлорировании водной
суспензии золотоносных руд золото превращается в растворимый
Ан2С16. При этом не применяют соляную кислоту, чтобы исклю-
чить возможность образования сероводорода, который осаждает
золото в виде сульфида. В этом случае золото не извлекается из
раствора.
Когда золотоносные руды содержат сульфиды, теллуриды,
арсениды и антимониды, их предварительно прокаливают, чтобы
избежать избыточного расхода хлора. В присутствии хлорида
натрия прокаливание осуществляется при температуре не выше
300°, чтобы исключить улетучивание хлорида золота(Ш).
Технологические схемы промышленной переработки золото-
носных руд довольно сложны и разнообразны, поскольку в пре-
делах каждой схемы комбинируется несколько процессов кон-
центрирования и извлечения золота в зависимости от химического
и минералогического состава руды.
В большинстве случаев комбинируют гравитационные и флота-
ционные методы обогащения с методами экстракции золота амаль-
гамированием и цианированием. При правильном выборе процесса
переработки золотоносных руд можно достигнуть количественно-
го извлечения золота.
При гравитационном обогащении последовательно применяют
сортировочную машину и сортировочные столы (наклонные
поверхности, покрытые мягким сукном).
Флотация используется при обогащении золотоносных руд,
остатков от амальгамирования или цианирования руд либо золо-
тоносных концентратов. При флотации осадков от амальгамирова-
ния отделяются золото и сульфиды металлов неиспользуемых
отходов или минералы, затрудняющие цианирование. При флотации
остатков от цианирования в пену переходит золото, которое трудно
цианируется. Затем пена перерабатывается пирометаллургическим
способом.
Комплексные концентраты металлов Ап — Си, Аи — Ag, Аи —
— РЬ, полученные при флотации, перерабатывают пирометаллур-
гическими способами.
В зависимости от элементов, сопутствующих самородному золо-
ту, можно легко выбрать технологическую схему переработки,
обеспечивающую наиболее полное извлечение золота.
золото
759
ОЧИСТКА
Среди примесей самородного золота есть благородные (сере-
бро, платина, платиновые металлы) и неблагородные (медь, никель
и др.) металлы. Очень важно не только извлечь из самородного
золота чистое золото, но и выделить сопутствующие ему благо-
родные металлы. Золото отделяют от серебра на основе их раз-
личной растворимости в азотной кислоте (в которой серебро раст-
воримо, а золото почти не растворяется). В Древнем Египте был
известен способ выделения золота из сплава золото — серебро
(3 вес. ч. Ag на 1 вес. ч. Ан) методом квартации. При обработке
этого сплава азотной кислотой (плотность 1,2) серебро и небла-
городные металлы превращаются в нитраты, а золото (с платино-
выми металлами) остается в виде трудно растворимого черно-
коричневого порошка. Из раствора нитратов серебро выделяют
обработкой медным порошком. По этому процессу золото отделя-
ется (хотя и не полностью) от основной массы примесей. При обра-
ботке самородного золота конц. H2SO4 при нагревании золото
и платиновые металлы остаются в виде губчатой массы, а серебро
и обычные металлы превращаются в сульфаты, которые растворя-
ются при последовательной обработке разб. H2SO4 и теплой водой.
При обработке ископаемого золота царской водкой в раствор
переходят золото, медь, платина и платиновые металлы, а серебро
отделяется в виде осадка AgCl. Из фильтрата металлическое золото
осаждают восстановителем [хлоридом или сульфатом железа(П),
щавелевой кислотой, муравьиной кислотой, двуокисью серы].
Этот процесс, заключающийся в растворении сырого золота
в царской водке и в отделении элементарного золота с помощью вос-
становителей, применяют для переработки сплавов золота с со-
держанием серебра менее 5%.
Электролитическим способом (рафинированием) можно полу-
чить золото 99,99 %-ной чистоты. При этом применяют анод из не-
очищенного золота, катод из чистого золота (в виде тонкой пла-
стинки), а в качестве электролита — раствор хлорида золота(Ш)
Ан2С16 в разб. НС1 (с содержанием золота 7—9%). При электро-
лизе происходят следующие процессы.
На катоде
H[AuC14] + ЗН+ = Au + 4НС1
Au3+ -р Зе- -> Au
H[AuC12] + H+ = Au + 2HC1
Au+ -|- e- Au
никель, платина, палладий),
На аноде
Au + HCl + ЗС1- = H[AuC14]
Au — 3e_ Au3+
Au + HCl + Cl- = H[AuC12]
Au — e~ = Au+
Большинство металлов (медь,
содержащихся в сыром золоте, растворяются на аноде и лишь
немногие металлы (осмий, иридий и родий) остаются в виде анод-
ного шлама.
760
ПОБОЧНАЯ ПОДГРУППА I ГРУППЫ
Электролиз проводят при температуре 70°, анодной плотности
тока 1500—2000 а/м2 и напряжении 1,2 в.
В процессе электролиза на дно электролизера (которое выло-
жено фарфором или термостойким стеклом) оседает наболыпое
количество элементарного золота, поскольку на аноде одновременно
образуются ионы золота(1) и золота(Ш):
3Au+ —2Au Au3+
Если сырое золото содержит больше 5% серебра (или содержит
свинец, селен и теллур), то в качестве электролита применяют
раствор нитрата серебра AgNO3. В этом случае серебро электро-
литически осаждается на катоде, а золото переходит в анодный
шлам. Для рекуперации золота анодные шламы обрабатывают
серной кислотой, плавят и отливают в виде слитков, последние
подвергают электролитическому рафинированию.
Для получения спектрально чистого золота рафинированное
золото обрабатывают царской водкой. Из раствора отделяют пла-
тину (действием хлорида натрия и спирта), а затем выделяют (с по-
мощью таких восстановителей, как газообразная SO2, щавелевая
кислота и др.) металлическое золото в виде порошка, который пла-
вят и отливают в формы в атмосфере чистого водорода.
ФИЗИЧЕСКИЕ И ХИМИЧЕСКИЕ СВОЙСТВА
Золото — металл золотисто-желтого (иногда коричневого) цве-
та. Его цвет зависит от толщины слоя и степени измельчения.
Золото имеет кубическую гранецентрированную структуру кри-
сталлической решетки. Окраска коллоидных растворов золота
зависит от размера частиц и может быть пурпурно-красной, фио-
летово-синей или коричнево-черной.
В компактном состоянии золото блестит (вследствие большой
отражательной способности), имеет плотность 19,3 г!см3 (тяжелый
металл), твердость 2,5 по шкале Мооса (мягкий металл), т. пл.
1063,4°, т. кип. 2707°. Оно диамагнитно и очень пластично.
Золото хорошо куется (из него можно получить листочки тол-
щиной 1/12500 мм) и способно вытягиваться в очень тонкие нити:
из 1 г золота можно получить нить длиной 2 км. Такие примеси,
как медь, свинец, мышьяк, очень сильно ухудшают ковкость
золота.
При температуре жидкого воздуха (—186°) золото становится
хрупким.
Теплопроводность золота в 2 раза меньше, чем серебра, а элект-
ропроводность — в 38,5 больше, чем ртути.
Эмиссионный спектр золота очень широк, он имеет линии в обла-
стях от ультрафиолетовой до инфракрасной.
золото
761
Известны сплавы золота с Ag, Cu, Sn, Hg, РЬ, Zn и другими
металлами.
При 450° 1 объем золота поглощает до 40 объемов кислорода.
При охлаждении кислород выделяется со взрывом.
С химической точки зрения золото мало активно, потенциал
системы Au/Au+ равен 1,7 в, системы Au/Au3+--h 1,50 ей системы
Au+/Au3+----hl,29 в.
В химических реакциях золото образует одно- и трехвалентные
соединения. Золото — типичнейший благородный металл. Оно не
растворяется в обычных реактивах.
На воздухе, в воде, галогеноводородах, азотной и серной
кислотах при низкой или комнатной температуре золото ус-
тойчиво.
Металлическое золото взаимодействует с фтором, хлором (газо-
образным или хлорной водой) и иодом при нагревании, образуя
соответствующие галогениды. На холоду хлор взаимодействует
с золотом только в присутствии влаги. Металлическое золото при
обычной температуре легко растворяется в жидком броме, в бром-
ной воде или в эфирных растворах брома с образованием трибро-
мида золота.
При концентрировании раствора золота в царской водке при
избытке НС1 выпадает золотохлористоводородная кислота:
3Au + 12НС1 + 3HNO3 = 3H[AuClJ + 3NO + 6Н2О
Галогеноводороды (HF, НС1, НВг, HI) взаимодействуют с зо-
лотом в присутствии окислителей, таких, как нитраты, гипохлори-
ты, хлораты, перманганаты, перекиси.
Кислород, сера, азот и бор непосредственно не взаимодей-
ствуют с золотом. При действии смеси кислорода и озона на метал-
лическое золото образуется окись золота(Ш) Аи2О3.
При нагревании золото вступает в реакцию с теллуром, фосфо-
ром, мышьяком и сурьмой, образуя теллурид АпТе2, фосфиды
Ан3Р4 и Ап4Р6, арсениды и антимониды.
В присутствии окислителей (нитратов, перманганатов, хро-
мовой кислоты, йодатов, перйодатов, двуокиси марганца, двуокиси
свинца) золото подвергается действию концентрированной серной
кислоты при температуре выше 300° или ортофосфорной кислоты
при температуре выше 250°.
Золото растворяется в смеси конц. H2SO4 с гидросульфатами
или сульфатами щелочных металлов, в 98%-ном растворе H2SeO4
при температуре выше 130°, в очень чистой кипящей конц. HNO3,
в расплавах, состоящих из оснований и нитратов щелочных метал-
лов, в перекиси натрия Na2O2 (или бария ВаО2) при нагревании,
в растворах цианидов щелочных металлов в присутствии кислорода
762 ПОБОЧНАЯ ПОДГРУППА I ТРУП ПЫ I
4
(или других окислителей). s
2Au + 3H2SO4 + 3Na2SO4 = Au2(SO4)3 + 3Na2SO3 + 3H2O |
2Au + 6H2SeO4 = Au2(SeO4)3 + 3H2SeO3 + 3H2O 5
Au + 4HN03 = Au(NO3)3 + NO + 2H2O |
2Au + 2NaOH + 3NaNO3 = 2Na[AuO2] + 3NaNO2 + H20 |
2Au + 3Na2O2 = 2Na[AuO2] + 2Na2O I
2Au + 3BaO2 = Ba[AuO2]2 + 2BaO |
2Au + 4KCN + V2O2 + H20 = 2K[Au(CN)2] + 2K0H |
При анодном растворении золота в растворе КОН образуется |
аурат калия К[АиО2] и анодный осадок Au2O3. |
Золото не взаимодействует с кислотами НСЮ, Н103, H2S2O8, ?
HN3, СН3СООН и др. *
Реакционная способность золота иллюстрируется следующей ’
схемой:
Комн. темп.
Золото-----
Нагревание
( с С12 в присутствии влаги -> Аи2С18
с жидким бромом или бромной водой -> Аи2Вг6
с царской водкой -> Н[АиС14]
с H2SO4 + Na2SO4 -> Au2(SO4)3
V с раствором KCN + О2 K[Au(CN)2] '
Г с F2, С12, Вг2, I2 -> AuF3, Au2C16, Au2Bre, Au2Ie |
I с O2 в смеси с О3 -> Аи2О3
1 с Те, Р, As, Sb AuTe2, Au3P4, Au4Pe, apce- У
•[ ниды, антимониды ;
i с конц. HNO3 Au(NO3)3
I c 98%-ной H2SeO4 Au2(SeO4)3
(. с расплавом NaOH-|-NaNO3 -> Na[AuO2]
Металлическое золото, его сплавы и соединения не токсичны.
Некоторые соединения золота используются в качестве медика-
ментов.
ПРИМЕНЕНИЕ
Золото является главным валютным металлом большинства
стран.
Металлическое золото и его сплавы применяют для изготов-
ления лабораторных приборов, деталей аппаратов, используемых
в физико-химических исследованиях, а также для покрытия раз-
личных предметов, изготовленных из стекла, фарфора или метал-
лов. В ювелирной промышленности применяют сплавы золота,
содержащие 37,5; 58,3; 75,0; 91,6% золота (содержание меди и се-
ребра колеблется в пределах 0—62,5; 0—41,7; 0—25; 0—8,4%),
а также интерметаллические соединения АнА12 пурпурно-фиоле-
тового цвета, AuZn2 синего, Au2Na желтого, Ан4К оливково-зе-
леного, Ан2К фиолетового цвета. Ювелирные изделия из чистого
золота не делают, поскольку металл очень мягок и слишком дорог.
золото
763
Золото благодаря его химической инертности находит приме-
нение в микроэлектронике. Сплав золота с 6% платины и 24%
серебра служит для изготовления электрических контактов в теле-
фонной аппаратуре. Электрические контакты изготовляют также
из сплава золота с 20% серебра, 5% никеля и 3% циркония.
Сплавами золото — серебро — медь, золото — медь — кадмий
— цинк или золото — серебро — медь — платина пользуются
в стоматологии. В прошлом из сплавов золота делали
монеты.
Коллоидное золото применяется в медицине как антисептик.
Золото играет роль катализатора в следующих процессах:
синтез воды из элементов, окисление окиси углерода кислородом,
разложение перекиси водорода и закиси азота, термическое раз-
ложение металоорганических соединений, восстановление двуоки-
си хлора СЮ2 аммиаком в щелочных растворах, фторирование угле-
водородов, гидрирование ацетилена и нитробензола и др.
СОЕДИНЕНИЯ (ОБЩИЕ СВОЙСТВА)
Известны многочисленные соединения золота(1) и (III) и не-
большое число соединений золота(П). Последние обладают малой
устойчивостью. Одно- и трехвалентное золото склонно к образо-
ванию устойчивых координационных соединений.
Растворы солей золота устойчивы только в отсутствие восста-
новителей, поскольку в их присутствии легко выделяется метал-
лическое золото или образуются различно окрашенные коллоидные
растворы золота.
Многие соединения золота мало устойчивы и разлагаются при
низких температурах.
Золото легко вытесняется из растворов его солей более актив-
ными металлами, такими, как цинк, магний, алюминий, никель,
например:
Аи2С16 + 3Zn — 2Au -|- 3ZnCl2
В табл. 72 приведены формулы и указан цвет ряда соединений
золота разных степеней окисления.
Соединения одновалентного золота
Известно небольшое число простых соединений одновалентного
золота, которые, как правило, трудно получить в чистом состоянии,
поскольку они образуются в смеси с соединениями золота(Ш)
или с металлическим золотом. Они мало устойчивы и быстро раз-
лагаются водой:
3Au+ Au3+ + 2Au
К оординационные соединения зол ота (I), например Me1! Au (GN) 2],
устойчивы в водных растворах.
764
ПОБОЧНАЯ ПОДГРУППА I ГРУППЫ
Гидрид золота, АиН, образуется в небольшом количестве при
нагревании металлического золота (1400°) в атмосфере водорода
или при действии атомарного водорода на золотую фольгу.
Гидрид золота представляет собой белый порошок; АиН пре-
вращается в АиОН под действием NaOH ив Au2S под действием H2S.
Окись золота, Аи2О, получают кипячением AuCl, Au2C18 или
К[АиВг2] с КОН или к карбонатом щелочного металла, а также
восстановлением Au2C18 с Hg2(NO3)2 «2Н2О в разбавленных рас-
творах:
2AuCl + 2К.ОН = Au2O ф- 2КС1 ф- Н2О
2K[AuBr2] ф- 2К.ОН = Аи2О ф- 4КВг ф- Н2О
Au2Cle ф- 6КОН = Au2O + 6КС1 + ЗН2О + 02
Au2C16 + 2Hg2(NO3)2-2H2O = Au2O ф- 2HgCl2 ф- 2Hg(NO3)2 ф- 2HC1 ф- 3H2O
Окись золота представляет собой синий гидрозоль или неустой-
чивый фиолетовый порошок. Она легко диспропорционирует при
225° по уравнению
3Au2O = 4Au ф- Au2O3
Поэтому окись золота Аи20 можно рассматривать как смесь,
состоящую из золота и Аи203.
При действии концентрированного раствора NH4OH на Аи2О
образуется черный осадок Au3N -NH3, который мало устойчив
в горячей воде и разлагается со взрывом при нагревании.
Известно также соединение 3Au2O-4NH3 черного цвета, не-
устойчивое в горячей воде и взрывающееся при ударе.
Хлорид золота, AuCl, получают действием газообразного хлора
на нагретое до 254—282° металлическое золото термическим раз-
ложением Аи2С18 (175°) или нагреванием HlAuClJ до 160—200°
в вакууме.
Au ф- !/2С12 <=s AuCl ф- 8 ккал Аи2С18 2 AuCl ф- 2С12
H[AuC14] AuCl + HCl ф- Cl2
Соединение AuCl представляет собой желтый аморфный поро-
шок с плотностью 7,4 г/см3. Он плохо растворим в воде, диспропор-
ционирует на Аи2С18 и металлическое золото под действием теплой
воды, спирта, эфира, ацетона или света, разлагается на элементы
при нагревании до 287°.
6AuCl = Au2C16 ф- 4Au 2AuCl 2Au ф- Cl2
В порошкообразном состоянии хлорид золота представляет
собой димер Аи2С12.
Хлорид золота взаимодействует с разб. НС1 и КВг по уравне-
ниям
3AuCl ф- HCl = H[AuC14] ф- 2Au ф- 4,98 ккал
12АиС1 ф- 4КВг = ЗК[АиС14] ф- KJAuBrJ ф- 8Аи
золото
765
Дихлороаурат ы(1), МеЦАиС12] (где Me1 = Na+, К+,
NH4), образуются при действии хлоридов щелочных металлов
на хлорид золота(1), путем восстановления хлорида золота(Ш)
в присутствии хлоридов щелочных металлов и нагреванием
МеЧАиС14] до плавления. Дихлороаураты — соединения желтого
цвета, они мало устойчивы и легко разлагаются водой:
ЗМе^АиОг] Me^AuClJ + 2Au + 2МетС1
Известны аммиакаты AuCl-nNH3 (где п ~ 12, 6, 4, 2, 1).
При действии жидкого аммиака на AuCl образуется белый поро-
шок AuCl*12NH3, который при 20° превращается в AuCl-3NH3.
При пропускании окиси углерода через суспензию Аи2С18
в тетрахлорэтилене при 120° либо AuCl в СС14 (иногда в С8Н8)
или над нагретым до 90° AuCl образуется аддукт AuCl -СО.
Соединение АиСЬСО представляет собой бесцветные кристал-
лы, которые растворимы в бензоле или эфире и самопроизвольно
разлагаются в присутствии воды, воздуха, хлора, окиси углерода
и улетучиваются при температуре выше 75° с частичным разло-
жением на золото, хлор и фосген.
Известно соединение [AuS = C(NH2)2]C1, которое образуется
из окиси золота(1) и тиомочевины.
Бромид золота, АиВг, получают нагреванием Аи2Вг8 при 115°
или разложением H[AuBr4] -3H2O в вакууме при 10°.
Аи2Вг6 -> 2АиВг -|- 2Вг2 Н[АиВг4] -> АиВг + НВг + Вг2
Бромид золота часто содержит примесь металлического золота
и Аи2Вг8, поскольку он легко диспропорционирует по уравнению
6АиВг = Аи2Вгв 4Аи
Соединение АиВг получают в виде желтого твердого вещества
с плотностью 7,9 г!см3. Оно разлагается на элементы при нагре-
вании до 212° и диспропорционирует на Аи2Вг8 и элементное золо-
то под действием теплой воды, спирта, эфира или ацетона.
Бромистоводородная кислота взаимодействует с АиВг по урав-
нению
ЗАиВг + НВг = Н[АиВг4] + 2Аи + 3,65 ккал
Свежеприготовленные водные растворы АиВг с растворами
бромидов щелочных металлов образуют дибромоаураты(1)
МеЧАиВг2] (где Me1 = К+, Na+), которые превращаются в тетра-
бромоаураты(Ш) МеЦАиВг4] и металлическое золото.
Дибромоаураты(1) МеЧАиВг2] могут также быть получены вос-
становлением тетрабромоауратов Мет[АиВг4].
Известны аммиакаты AuBr*nNH3 (где п = 6, 4, 3, 2).
Иодид золота, Aul, получают прямым взаимодействием эле-
ментов при 100°, действием водных растворов иода на металличе-
766
ПОБОЧНАЯ ПОДГРУППА I ГРУППЫ
ское золото, восстановлением Аи2С18 или Н[АцВг4] раствором KI,
действием Ш на Аи2О3.
Au + l/2I2 Aul + 7,2 ккал Au2Cle + 6KI = 2AuI + 6КС1 + 2I2
2H[AuBr4] + 6KI = 2AuI + 2HBr + 6KBr + 2I2
Au2O3 + 6HI = 2AuI + 3H2O + 2I2
Иодид золота — твердое вещество лимонно-желтого цвета,
образующее кристаллы с плотностью 8,25 г/см\ разлагающееся
на элементы при нагревании до 177° или под действием воды.
Двуокись серы и окись углерода восстанавливают Aul до метал-
лического золота.
Иодид золота реагирует с иодидами щелочных металлов (или
с HI) по уравнениям
3AuI -|- Me1! = MeT[AuI4] + 2Au
3AuI + HI = H[AuI4] + 2Au
Известны аммиакаты Aul -nNHs (где n = 8, 6, 4, 3, 2, 1)
и координационные соединения, которые Aul образует с третич-
ными фосфинами и арсинами, когда последние добавляют к водно-
спиртовым растворам HlAuClJ, содержащим KI.
Сульфид золота, Au2S, получают в результате барботирова-
ния H2S через концентрированный раствор K[Au(CN)2] или
К[АиВг2], а также путем нагревания золотосодержащих пиритов:
2K[Au(CN)2] + H2S = Au2S + 2KCN + 2HCN
Коричнево-черное твердое вещество Au2S плохо растворимо
в воде и разбавленных кислотах и растворяется в царской водке,
цианидах, сульфидах и полисульфидах щелочных металлов.
В последних двух случаях образуются тиосоли:
Au2S + Na2S = 2Na[AuS] Au2S + 3Na2S = 2Na3[AuS2]
Дитиосулъфатоаурат(Д) натрия, Na3[Au(S2O3)2]-2Н2О, полу-
чают обработкой Au2C18, тетрахлороауратов(Ш) Ме!([АиС14] или
дихлороауратов(1) Ме1[АиС12] раствором Na2S2O3.5H2O.
Au2C16 + 8Na2S2O3 = 2Na3[Au(S2O3)2] + 2Na2S4O6 + 6NaCl
Cs[AuCI4] + 4Na2S2O3 = Na3[Au(S2O3)2] + Na2S4O6 + 3NaCl + CsCl
Na[AuCl2] + 2Na2S2O3 = Na3[Au(S2O3)2] + 2NaCl
Соединение Na3[Au(S2O3)2] "2H2O окрашено в красно-корич-
невый цвет, дегидратирует при 150°, разлагается при нагревании
до высокой температуры на воздухе и превращается в Аи^
под действием галогеноводородов, серной и щавелевой кислот.
2Na3[Au(S2O3)2] + 5О2 = 2Au + 3Na2SO4 + 5SO2
золото
767
Обработка ВаС12 -2H2O соединения Na3[Au(S2O3)2] -2Н2О в при-
сутствии спирта или ацетона вызывает осаждение Ba3[Au(S2O3)2]2 •
• 6Н2О.
Известна также соль калия K3[Au(S2O3)2].
Фосфид золота, Ан3Р, получают действием фосфина на AuCl
или Аи2С18:
3AuC1 + РН3 = Au3P + ЗНС1
6Au2C16 + 7РН3 + 12Н2О = 4Au3P + ЗН3РО4 + 36НС1
Фосфид золота представляет собой черный порошок; он разла-
гается водой и щелочными растворами и образует РН3 под дей-
ствием НС1. При восстановлении Аи2С18 фосфидом золота обра-
зуется красный гидрозоль золота:
ЗАи3Р + V2Au2C16 + 6Н2О = 10Аи + ЗН3РО2 + ЗНС1
Цианид золота, AuCN, выделяется в результате обработки
Ан2С18 стехиометрически необходимым количеством KCN, выпа-
риванием досуха раствора, содержащего AuOQH и HCN в стехио-
метрически необходимом соотношении, обработкой растворов
Mer[Au(CN)2] и Men[Au(CN)2]2-nH2O соляной или азотной кисло-
той, нагреванием К [Au(CN)2] с НС1 при 50° и термическим раз-
ложением Au(CN)3.
!/2Аи2С16 + 3KCN = AuCN + ЗКС1 + (CN)2
Au(OH)3 + 3HCN = AuCN + 3H2O + (CN)2
K[Au(CN)2] + HC1 = AuCN + KC1 + HCN Au(CN)3 -> AuCN + (CN)2
Цианид золота представляет собой желтые гексагональные
мелкие кристаллы с плотностью 7,122 г!см3-, он плохо растворим
в воде и растворяется в растворах цианидов щелочных металлов
(или в растворах K4[Fe(CN)8-3H2O) на воздухе с образованием
дицианоауратов:
AuCN + KCN = K[Au(CN)2]
8AuCN + 2K4[Fe(CN)6].3H2O + V2O2 =
= 8K[Au(CN)2] + 2Fe(OH)3 + 4HCN + H2O
При сильном нагревании AuCN разлагается на золото и ди-
циан:
2AuCN -> 2Au + (CN)2
ДицианоауратыМе1[Аи(СН)2], Me11 [Au(CN)2]2 -иНгО (где Me1 =
== К+, Na+ и Me11 = Са2+, Sr2+, Ва2+, Zn2+, Ni2+) получают дей-
ствием цианидов щелочных металлов на металлическое золото
в присутствии кислорода, обработкой AuCl или AuCN цианидом
калия, действием K4[Fe(CN)8] •ЗНгО на AuCN в присутствии кис-
лорода и действием HCN на смесь, состоящую из AuCN и карбо-
768
ПОБОЧНАЯ ПОДГРУППА I ГРУППЫ
ната щелочного металла.
4Au + 8KCN + 2Н2О + О2 = 4K[Au(CN)2] + 4КОН
Соединение K[Au(CN)2] представляет собой бесцветные окта-
эдрические кристаллы с плотностью 3,45 г!см9-, разлагается выше
200°, плохо растворим в эфире, растворяется в воде.
Примеры дицианоауратов: Na[Au(CN)2], Ca[Au(CN)2]2 -ЗИгО,
Sr[Au(CN)2]2 -ЗН2О, Ba[Au(CN)2]2.2H2O, Ni[Au(CN)2].
Золото(1) дитиоцианатоводородная кислота, H[Au(SCN)2], име-
ет вид густой маслянистой жидкости с красноватым оттенком; она
образуется обработкой Н[АиС12] тиоцианатом калия в присут-
ствии сульфита натрия.
ДитиоцианатоауратД) калия, K[Au(SCN)2], получают путем
нейтрализации кислоты H[Au(SCN)2] избытком КНСО3 или дейст-
вием KSCN на AuCN; это бесцветные кристаллы, которые пла-
вятся при 100° и разлагаются при сильном нагревании на золото
и роданид калия.
Хелатные соединения
Соли золота(1) под действием п-диметиламинобензилиденрода-
мина окрашиваются в фиолетовые цвета вследствие образования
хелатного соединения
|-Аи----N —С = О -1
S = С С = СН N(CH3)2
Соединения двухвалентного золота
Экспериментально доказано, что соединения золота(П) состоят
из смеси соединений золота(1) и (III). Раньше они рассматрива-
лись как индивидуальные соединения.
Окись золота, АиО или Au[AuO2], получают нагреванием
(155—165°) Ап2О3. Она представляет собой гигроскопичный корич-
невый порошок, разлагающийся при нагревании.
Димер хлорида золота, Ан2С14, красного цвета, является про-
дуктом насыщения порошкообразного металлического золота газо-
образным хлором.
Бромид золота, АиВг2, в виде красного твердого вещества
получают действием раствора брома в сероуглероде на порошок
металлического золота.
Сульфид золота, AuS, представляет собой твердое черное веще-
ство, неустойчивое при нагревании; получается действием серо-
водорода на растворы солей золота(Ш) или добавлением Аи2С1в
к раствору Mei[Au(S2O3)2]’2Н2О.
золото
769
Сульфат, золота, AuSO4 или Au[Au(SO4)2], в виде оранжевого
порошка образуется при действии смеси H2SO4 и Н1О3 на тонкий
порошок золота, нагретый до 300°.
Соединения трехвалентного золота
Известны многочисленные устойчивые соединения трехвалент-
ного золота, большинство которых образуются легко; в водных
растворах существуют в виде четырехкоординационных комплекс-
ных ионов.
При действии восстановителей на растворы соединений золо-
та(1П) образуются соединения золота(1) или металлическое золото.
Склонность к гидролизу солей золота(1П) в водных растворах
часто приводит к осаждению АнООН.
Помимо простых соединений золота(Ш) выделены многочис-
ленные координационные ацидо-, гидроксо- и аминосоединения,
а также хелатные соединения.
Окись золота, Аи2О3, получают нагреванием (140—150°)
АнООН в вакууме или действием смеси кислорода с озоном на ка-
тодно диспергированное золото.
Коричневое твердое соединение Аи2О3 мало устойчиво, разла-
гается на свету или при нагревании до 155°, растворяется в КОН
с образованием. К[Ан(ОН)4]-Н2О.
Au2O3 —2AuO + i/2O2
Гидроокись золота, АиООН, получают обработкой растворов
солей золота(Ш) стехиометрически необходимым количеством
щелочи, действием азотной кислоты на аурат магния Mg[AuO2]2,
гидролизом некоторых соединений золота(Ш), например
Au2(SO4)3, Au(NO3)3, Na[AuF4].
Au2C16 + 6KOH = 2AuOOH + 6KC1 + 2H2O
Mg[AuO2]2 + 2HNO3 = 2AuOOH + Mg(NO3)2
Au2(SO4)3 + 4H2O 2AuOOH + 3H2SO4
Au(NO3)3 -j- 2H2O AuOOH + 3HNO3
Гидроокись золота представляет собой красно-коричневый
порошок; при нагревании до 140—150° в вакууме она превращает-
ся в Аи2О3, обладает слабо амфотерными свойствами (проявляет
слабо кислотный характер НАиО2), растворяется в кислотах и ще-
лочах, плохо растворима в воде.
AuOOH + 4НС1 Н[АиС14] + 2Н2О
АиООН + 2H2SO4 H[Au(SO4)2] + 2Н2О
АиООН + 4НВг Н[АиВг4] + 2Н2О
АиООН + КОН + Н2О = К[Аи(ОН)4]
49-0101
770
ПОБОЧНАЯ ПОДГРУППА I ГРУППЫ
Известны основные аураты и аураты типов MeI[Au(OH)4] •
• пН2О, МеЧАиОД-иНгО, Меп[АиО2]2-гаНгО (где Мет= К+, Na+
и Men = Mg2+> Са2+, Sr2+j Ва2+).
Тетрагидроксоаурат(1АЛ) калия, К[Ан(ОН)4]-НгО, выделяется
в виде желтых кристаллов при концентрировании в вакууме рас-
твора АиООН в КОН.
Аурат(Ш) натрия, Na[AuO2] 'Н2О, получают кипячением
АиООН с NaOH или обработкой Ва[АиО2]2 раствором Na2SO4-
. ЮН20 при нагревании.
Соединение Na[AuO2]-H2O представляет собой зеленые кри-
сталлы, которые растворяются в воде и плохо растворимы в спир-
те. Двуокись серы и щавелевая кислота восстанавливают при
нагревании в водных растворах аураты и основные аураты щелоч-
ных металлов до металлического золота.
Аурат магния, Mg[AuO2]2, получают обработкой раствора
Ап2С18 или H[AuC14) гидроокисью магния; образуются хорошо
растворимые в воде зеленые кристаллы.
Фторид золота, AuF3, получают термическим разложением
(120°) соединения AuF3>BrF3. Он представляет собой оранжевый
порошок, который разлагается водой или нагреванием выше 500°.
С фторидами щелочных металлов AuF3 образует тетрафтороаураты,
которые легко гидролизуются.
AuF3 + 2Н2О АиООН + 3HF Na[AuF4] + 2Н2О АиООН + NaF-|-3HF
2AuF3 -> 2Аи -j- 3F2 AuF3 -)- NaF == Na[AuF4]
Соединение AuF3-BrF3 окрашено в лимонно-желтый цвет; его
получают действием BrF3 на порошок металлического золота.
Димер хлорида золота(11Г),
ГС1 С1 С1-,
>< >< I
LC1Z XC1Z XC1J ’
получают обработкой металлического порошкообразного (или че-
шуек) золота газообразным хлором при 245 —260°, жидким хло-
ром при 80—100°, хлорной водой или царской водкой, а также
нагреванием Н[АнС14].4Н2О до 120°.
2Au -|- ЗС12 = Аи2С16 56 ккал
2Au + 6НС1 + 2HNO3 = Au2C16 + 2NO + 4Н2О
Димер представляет собой сильно двулучепреломляющие, гигро-
скопичные рубиново-красные блестящие призматические кристал-
лы с плотностью 4,67 г! см?-, он растворяется в воде, спирте и эфире
и разлагается при нагревании выше 175°.
175° 288°
Au2Cle----► 2AuC1 + 2C12' Au2C16----> 2Au + 3C12
золото
771
Разбавленные растворы Ап2С18 оранжевого цвета вследствие
гидролиза имеют более кислую реакцию, чем концентрированные
растворы Ап2С18, окрашенные в коричнево-красный цвет.
1/2Au2Cle + Н2О H2[AuOC13] [AuOCl3]2- + 2Н +
VaAuaCls + Н2О H[AuOHCl3] [AuOHCl3]- + Н +
Соединение Н[АпОНС13] может быть экстрагировано эфиром.
При действии соляной кислоты или хлоридов щелочных метал-
лов на Аи2С18 образуется Н[АиС14] -нНгО. При подщелачивании
водных растворов Ан2С18 избытком NaOH или КОН образуются
тетрагидроксоаураты MeI[Au(OH)4] -пН2О.
Из водных растворов Аи2С16 под действием металлов (Zn, Mg,
Fe, Al) легко выпадает элементарное золото.
При 55° окись углерода взаимодействует с Ап2С18:
Au2C16 + 4СО = 2AuCOCl + 2СОС12
При обычной температуре иод восстанавливает Ап2С18:
Au2Cle + 4I2 = 2AuI + 6IC1'
Между фосфином РН3 и Ап2С18 протекают следующие реакции:
2Au2Cl6 + РН3 + 4Н2О = 4AuCl + Н3РО4 + 8НС1
ЗАпС1 А РН3 = Au3₽ А ЗНС1
Au2C16 + 6Au3P + 12Н2О = 20Au + 6Н3РО2 + 6НС1
Au2Cle + ЗН3РО2 + ЗН2О = 2Аи + ЗН3РО3 + 6НС1
Аи2С16 + ЗН3РО3 + ЗН2О = 2Аи + ЗН3РО4 + 6НС1
Перекись водорода восстанавливает Ац2С18 до металлического
золота тем легче, чем больше pH раствора.
Au2C16 + ЗН2О2 + 6К0Н = 2Au + 6КС1 + 6Н2О + ЗО2
Соли железа(П) восстанавливают Ап2С18 в кислых или щелоч-
ных растворах до металлического золота:
Au2C16 + 6FeCl2 = 2Au + 3Fe2Cle
В нейтральных или кислых растворах Аи2С18 восстанавливает-
ся сернистой кислотой по уравнению
Au2C16 + 3H2SO3 + ЗН2О = 2Au + 3H2SO4 + 6НС1
При восстановлении Au2C18 [или других соединений золота(III)]
хлоридом олова(П) в концентрированных сильно кислых раство-
рах образуется коричнево-черный осадок тонко дисперсного
золота, а в слабо кислых разбавленных растворах — пурпурно-розо-
вый осадок коллоидного золота. Продукт адсорбции гексагидро-
ксооловянной кислотой коллоидного золота называется «кассие-
вым пурпуром».
Au2CJe + 3SnCl2 = 2Au + 3SnCJ4 SnCl4 + 6H2O H2[Sn(OH)6] + 4HC1
49*
772
ПОБОЧНАЯ ПОДГРУППА I ГРУППЫ
Хлорид сурьмы(Ш) количественно осаждает Аи2С18:
Au2C16 + 3SbCl3 = 2Au + 3SbCl5
Окислы PbO, Pb3O4 также восстанавливают нейтральные или
щелочные растворы Ан2С18 с выделением металлического золота.
В качестве восстановителей для Аи2С18 применяют и различные
органические соединения, например бензидин, индиго, сахара.
Димер хлорида золота служит для получения многих соедине-
ний золота, гидрозолей и пурпура.
Известен аддукт АиС13 •Р(С2Н5)3.
Золотохлористоводородная, кислота, Н[АиС14]-геНгО, обра-
зуется при растворении металлического золота в царской водке
с избытком НС1 или при обработке Ан2С18 соляной кислотой:
Au + 4НС1 + HNO3 = H[AuC14] + NO + 2Н2О
Au2C16 -J- 2НС1 = 2H[AuC14]
При концентрировании водных растворов золотохлористоводо-
родной кислоты выпадают кристаллогидраты Н[АиС14]-ЗН2О,
Н[АиС14] -4Н2О в виде гигроскопичных, горьких на вкус кристал-
лов желтого цвета.
Конечным продуктом гидролиза золотохлористоводородной
кислоты является соединение Н[Ан(ОН)4]. Реакция протекает
по следующим стадиям:
H[AuCl4] + Н2О +±. Н[АиС130Н] + НС1
H[AuCl3OH] + Н2О H[AuC12(0H)2] + НС1
H[AuC12(OH)2] + Н2О H[AuCl(OH)3] + НС1
H[AuCl(OH)3] + Н2О H[Au(OH)J + НС1
Соединение Н[Аи(ОН)4] извлекается из подкисленных НС1
водных растворов этиловым эфиром или алифатическими эфирами.
При действии аммиака на Н[Аи(ОН)4] образуются бесцветные кри-
сталлы [Au(NH3)4]Cl3.
Тетр а хлоро а у р а т ы, МеЧАиСД] или MeI[AuCl4]
образуются путем нейтрализации Н[АнС14]-иНгО или добавлении
хлоридов щелочных металлов (в эквивалентных количествах)
к водным растворам Аи2С18 или HfAuClJ-иНгО.
H[AuC14] + MeIOH = Me^AuClJ + Н2О
Аи2С16 + 2Ме+С1 = гМеЧАиСЦ] Н[АиС14] + МетС1 = Me^AuClJ + НС1
Me1 = Li+, Na+, K+, Rb+, Cs+, NHJ
Тетрахлороаурат(11Г} лития, LilAuClJ *2H2O, образует оран-
жевые кристаллы; он очень гигроскопичен, дегидратирует при
нагревании до 90° и разлагается при 105°:
90° 105°
Li[AuCl4].2H2O -Li[AuCl4]----------> Au + LiCl + 3/2Cl2
золото
773
ТетрахлороауратДП) натрия, Na[AuCl4]-2Н2О, называемый
также золотой солью, выделяют в виде желто-оранжевых ромбо-
эдрических кристаллов, устойчивых при нагревании до 100°.
Водные растворы тетрахлороаурата натрия обладают слабо кислой
реакцией и могут быть восстановлены до элементного золота
с помощью РН3, AsH3, SbH3, SO2, гидрохинона. Золотая соль
служит для гальванического золочения, росписи на фарфоре или
стекле, для получения цветных фотографий.
ТетрахлороауратДП) калия, К[АнС14]-2Н2О, представляет
собой диамагнитные выветривающиеся желтые ромбоэдрические
кристаллы с плотностью 3,3—3,9 г/см3 и т. пл. 357°; при нагрева-
нии разлагается по уравнению
K[AuC14].2H2O -> Au + КС1 + 3/2С12 + 2Н2О
Тетр ахлороаураты, Rb[AuCl4l, Cs[AuC14] и NHJAuClJ •
• ЗН2О, представляют собой желтые моноклинные кристаллы.
Тетрахлороаурат серебра, Ag[AuCl4], получают обработкой
раствора золотохлористоводородной кислоты разбавленным рас-
твором AgNO3. Соль выделяется в виде оранжево-красных призма-
тических кристаллов.
Димер бромида золота, Ан2Вг8, получают действием жидкого
брома, паров брома или бромной воды на металлическое золото
при обычной температуре, обработкой Ан2С18 избытком НВг
при 70°, а также при медленном нагревании Н[АиВг4] -иНгО:
2Au + ЗВг2 = Au2Br6 Au2Cle + 6HBr = Аи2Вг6 + 6HCJ
При работе избегают высоких температур, чтобы предотвра-
тить разложение Аи2Вг8 на АиВг и Вг2:
150°
Аи2Вгв ~ 2АиВг + 2Вг2
Темно-коричневые блестящие пластинки Ап2Вг8 растворяются
в воде, эфире, броме, бромоформе, метилацетате и трихлориде
сурьмы, плохо растворимы в сероуглероде и четыреххлористом
углероде. Водные растворы Аи2Вг8 мало устойчивы и при нагре-
вании разлагаются с выделением золота. Соединение Ан2Вг6
образует с бромидами некоторых металлов тетрабромоаураты
MeHAuBrJ, MeUAuBrJ «иН2О; последние окрашены в коричнево-
красные цвета и в воде растворяются хуже, чем тетрахлороаураты.
Известны аддукты AuBr3.(C8H5CH2)2S, AuBr3-Py, а также
соединения Аи2Вг4(С2Н5)2, Ап2Вг2(С2Н5)4.
Золотобромистоводородная кислота, Н[АнВг4] -иНгО (где п =
= 4, 5 или 6), выпадает при концентрировании растворов, полу-
ченных обработкой Ан2Вг8 или АнООН бромистоводородной кис-
лотой.
Аи2Вг6 + 2НВг = 2H[AuBr4] АиООН + 4НВг = Н[АиВг4] + 2Н2О
ПОБОЧНАЯ ПОДГРУППА I ГРУППЫ
Соединение H[AuBr4]-nH2O образует красные кристаллы
ст. пл. 27°, которые очищаются перекристаллизацией из эфира
и гидролизуются горячей водой.
Н[АпВг4] + НОН ** H[AuOHBr3] А НВг
Тетрабромоаурат калия, К[АпВг4], представляет собой крас-
ные кристаллы, легко растворимые в воде.
Димер иодида золота, Au2I8, образуется при обработке Аи2С18
стехиометрически необходимым количеством раствора KI.
Au2G13 A 6KI = Au2I8 A 6KI
Димер выделяют в виде серовато-зеленого порошка; он мало
растворим в воде, растворяется в избытке KI с образованием
K[AuI4] и K[AuI2] и превращается при хранении на воздухе
в Aul с выделением элементного иода:
Au2I3 A 2K.I = K[AuI4] А К[Ап12] А I2
Au2Ie 2AuI А 212
ТетраиодоауратДП) калия, K[AuI4], получают обработкой
Ап2С18 раствором KI, взятом в большом избытке; это черные
блестящие кристаллы, которые при нагревании до 60° разлагают-
ся по уравнению
K[AuI4] Au A KI А 3/212
Иодат золота, Au(IO3)3, образуется при действии К1О3
на Аи2С18 и представляет собой желтые кристаллы, хорошо рас-
творимые в воде.
Сульфид золота, Au2S3, получают пропусканием сероводорода
через кислые растворы соединений золота(Ш), например:
Au2C16 A 3H2S = Au2S3 А 6НС1
2H[AuG14] A 3H2S = Au2S3 А 8HG1
2Li[AuCl4] A 3H2S = Au2S3 A 2LiCl A 6HC1
Сульфид золота — черный (похожий на графит) аморфный
порошок с плотностью 8,754 г/см3', при нагревании до 200° разла-
гается на элементы; плохо растворим в спирте и растворяется
в цианидах и сульфидах щелочных металлов, а также в царской
водке.
Au2S3 A 8KCN = 2K[Au(CN)4] A 3K2S Au2S3 A Na2S = 2NaAuS2
Au2S3 A 8HC1 A12HNO3 = 2H[AuC14] A 2NO A 3S A 4H2O
Сульфат золота, Аи2(ЗО4)з, образуется при растворении
АиООН в стехиометрически необходимом количестве серной кис-
лоты; это желтое твердое вещество, которое легко гидролизуется
золото
775
и превращается в дисульфатозолотую кислоту H[Au(SO4)2] серной
кислотой, взятой в избытке.
ДисулъфатоауратДП) калия, K[Au(SO4)2l,— соединение жел-
того цвета, легко гидролизуется.
Гидросульфит аурила, HAuOSO4, выделяется в виде желтых
кристаллов.
Двойной сульфат, K2SO4*Au2(SO4)3, образуется в виде желтых
кристаллов.
K[Au(SO4)2] + 2Н2О AuOOH + KHSO4 + H2SO4
Нитрат золота, Au(NO3)3-nH2O, выпадает в виде желтых
кристаллов при добавлении ацетона к концентрированному рас-
* твору, полученному при нагревании тонкого порошка металли-
' ческого золота в конц. HNO3.
Под действием влажного воздуха или света Au(NO3)3*nH2O
. разлагается:
Au(NO3)3 + Н2О =₽£ AuONO3 + 2HNO3
i Au(NO3)3 + 2H2O AuOOH + 3HNO3
При концентрировании на холоду в присутствии дегидратанта
из раствора АиООН в HNO3 (d = 1,49) при 20° выпадают желтые
кристаллы тетранитратозолотой кислоты H[Au(NO3)4l -ЗН2О
с плотностью 2,84 г/смг, которые выветриваются в сухом воздухе,
расплываются во влажном воздухе.
Концентрирование в вакууме раствора АиООН в HNO3 (d =
= 1,40) дает осадок нитрата аурила AuONO3 — соединение корич-
невого цвета.
Тетранитратоаурат(11Г) калия, K[Au(NO3)4], получают обра-
боткой H[Au(NO3)J -ЗН2О (растворенного в конц. HNO3) нитра-
том калия. Это гигроскопичные блестящие желтые кристаллы,
которые разлагаются водой в результате нагревания при 105°.
Нитрат тетрамминзолотаД11), [Au(NH3)4](NO3)3, полу-
(! чают на холоду обработкой аммиаком смеси Н[АиС14] -иНгО
( и NH4NO3 в растворе. При этом образуются бесцветные кристаллы,
j HfAuClJ + 3NH4NO3 + 5NH3 = [Au(NH3)4](NO3)3 + 4NH4C1
I Тетраацетатоаураты, Men[Au(CH3COO)4]2 «2Н2О (где
f Me11 = Mg2+, Ca2+, Sr2+, Pb2+), полученные растворением ауратов
l двухвалентных металлов в горячей уксусной кислоте; представ-
' ляют собой бесцветные моноклинные кристаллы, легко раство-
: римые в воде.
Тетрацианозолотая кислота, H[Au(CN)4] -ЗН2О, получается
действием синильной кислоты на золотохлористоводородную кис-
лоту; зто бесцветные кристаллы, легко растворимые в воде, спир-
те и эфире.
i
776
ПОБОЧНАЯ ПОДГРУППА I ГРУППЫ
Известны тетрацианоаураты(Ш) и дицианогалогеноау-
раты(Ш) типов MeI[Au(CN)4]’пНгО, MeI[Au(CN)2X2]-nH2O,
MenlAu(CN)2X2]2-пН2О (где Mei = К+, Na+ и Me1* = Са2+, Ва2+,
Zn2+, Со2+, Cd2+).
Тетрацианоаурат(111) натрия, Na[Au(CN)4] -2Н2О, в виде бес-
цветных кристаллов применяют для гальванического золочения
и окрашивания в золотистый цвет фарфора и стекла.
Тетрацианоаурат(11Г) калия, K[Au(CN)4]выпадает
при концентрировании раствора, полученного смешиванием
Аи2С18 с концентрированным раствором KCN.
Au2C16 + 8KCN = 2K[Au(CN)4] + 6КС1
Соединение K[Au(CN)4]-^H^O представляет собой бесцвет-
ные пластинчатые кристаллы; при 200° они теряют кристаллиза-
ционную воду, а при более сильном нагревании разлагаются
по уравнению
K[Au(CN)4] -> K[Au(CN)2] + (CN)2
Тетрацианоаурат(111) аммония, NH4[Au(CN)4] -Н2О, выде-
ляется при концентрировании раствора АиООН в NH4CN; это
легко растворимые в воде бесцветные пластинчатые кристаллы.
Тетрацианоаурат^Ш) кобалътаЦI), Co[Au(CN)4]2 >9Н2О,
осаждают путем концентрирования раствора K[Au(CN)4]-3/2Н2О
в избытке Co(NO3)2-6Н2О; соединение представляет собой оранже-
вые кристаллы, которые при 150° теряют кристаллизационную
воду, а при 210° разлагаются:
2K[Au(CN)4] + Co(NO3)2 = Co[Au(CN)4]2 + 2KNO3
Тетратиоцианатозолотая кислота, H[Au(SCN)4]-2Н2О, пред-
ставляет собой красные кристаллы, устойчивые в сухом воз-
духе и гидролизующиеся под действием влажного воздуха или
воды.
Тетратиоцианатоаурат(11Г) калия, K[Au(SCN)4], осаждается
в виде оранжево-красных кристаллов в результате концентриро-
вания раствора Аи2С1в, обработанного KSCN и нейтрализован-
ного КНСО3
Тетратиоцианатоаурат(11Г) натрия, Na[Au(SCN)4], образует-
ся в виде красного осадка при обработке раствора Н[АиС14]-Н2О
или Н[АиС14] -нН2О тиоцианатом натрия.
H[AuC14] + 5NaSCN = Na[Au(SCN)4] + 4NaCl + HSCN
Na[AuCl4] + 4NaSCN = Na[Au(SCN)4] + 4NaCl
зол ото
777
Хелатные соединения
Известно небольшое число хелатных соединений золота(Ш),
например
Н5С2\ О-С = О zC2H5
/AU\ I /AU\
Н6С2 ХО = С— 0х хс2н6
Н6С2 /0\ /С2Н5
XAU)f ^Au
Н6С2/ \oZ ^С2Н5_|2
Металлоорганические соединения
Неустойчивые арильные производные золота(1) получают
в эфирных растворах при действии арилмагнийбромида на кар-
бонилхлорид золота AuCICO. Алкильные производные золота(1)
не выделены.
Известны многочисленные алкильные производные золота(Ш),
которые имеют исключительно важное значение. По числу алкиль-
ных групп они могут быть классифицированы на триалкильные,
диалкильные и моноалкильные производные:
где R = СН3, С2Н6, алкильный радикал, X = пиридин или толу-
идин, Y = Cl", Вг-, CN-, ^СгО*-. к
Амины и эфираты триалкильных производных золота трудно
выделить вследствие их неустойчивости. В качестве примеров
триалкильных производных золота можно упомянуть Аи(СН3)3*
• С2Н5 — О — С2Н5, Аи(С2Н5)з-С2Н5 — О С2Н6, Ап(СН3)з"
•c5h5n.
Диалкильные производные золота(Ш) — жидкие или твердые
бесцветные вещества с низкой температурой плавления; они плохо
растворимы в воде и легко растворяются во многих органических
растворителях. Получаются действием реактивов Гриньяра
на Аи2С18, Ан2Вг8 или Н[Аи(0Н)С13] в среде эфира или пиридина.
778
ПОБОЧНАЯ ПОДГРУППА I ГРУППЫ
Примеры диалкильных производных золота(Ш):
Н5С2 О-С = О С2Н5
| Ч/
_Н5С2/ Ч = С —(Ч Ч2н5
Н2
Н5С2 N —СН2
ЧЧ I
Н5С2/ Ч-сн2
Н2
Вг
Пример моноалкильного производного золота(Ш) — этилди-
бромид золота, (G2H6)AuBr2; соединение красного цвета, разла-
гается при нагревании до 80°, взаимодействует с этилендиамином
(Ен) по уравнению
2С2Н5АиВг2 + ЗЕп — [AuEn2]Brs -f- [Au(C2Hs)2En]Br,
ПОБОЧНАЯ ПОДГРУППА П ГРУППЫ
ПЕРИОДИЧЕСКОЙ СИСТЕМЫ
(ПОДГРУППА ЦИНКА)
В побочной подгруппе II группы периодической системы нахо-
дятся переходные металлы цинк (Zn), кадмий (Cd) и ртуть (Hg).
В табл. 73 показано электронное строение атомов цинка, кад-
мия и ртути.
Атомы этих элементов имеют по 2 электрона на последнем
и по 18 электронов на предпоследнем электронных уровнях в отли-
чие от атомов щелочноземельных металлов, которые содержат
по 8 электронов на предпоследнем уровне, за исключением берил-
лия, имеющего 2 электрона.
В химических реакциях атомы металлов подгруппы цинка
теряют два валентных электрона, образуя соединения, в которых
эти элементы двухвалентны. В отличие от ионов щелочноземель-
ных металлов ионы металлов подгруппы цинка не обладают устой-
чивой конфигурацией инертного газа и в отличие от ионов осталь-
ных переходных металлов ионы металлов подгруппы цинка имеют
полностью укомплектованные 3d- (для Zn2+), 4d- (для Cd2+) и 5d-
(для Hg2+) орбитали.
В табл. 74 приведены наиболее важные константы элементов
побочной подгруппы II группы.
Все металлы подгруппы цинка имеют серебристо-белый цвет
и плотнейшую гексагональную упаковку кристаллов, за исклю_
Таблица 74
Элемент Цинк Zn Кадмий Cd Ртуть Hg
Цвет Серебристо- белый Серебристо- белый Cepef ристо- белый
Кристаллическая структура Плотнейшая гексагональ- ная Плотнейшая гексагональ- ная Объемная ромбическая
Атомный номер 30 48 80
Атомный вес 63,37 112,40 200,59
Атомный радиус, А (по Полингу) 1,27 1,54 1,57
Ионный радиус Meli, А (по Гольд- шмидту, Полингу и Аренсу) 0,83; 0,74; 0,74 1,03; 0,97; 0,97 1,12; 1,10; 1,10
Атомный объем (при 20°), см2/г-атом 9,2 13,1 14,728
Плотность (при 20°), г/см2 7,13 8,642 13,595
Твердость по Бринеллю, кг/мм2 32,7 22 —
Твердость по шкале Мооса 2,5—2,9 2,0 1,5
Температура плавления, °C 419,44 320,9 —38,87
Скрытая теплота плавления, кал/град 28,13 13,66 2,82
Температура кипения, °C 906 778 356,95
Удельная теплоемкость (при 20°), кал/г-град 0,0915 0,0552 0,03325
Коэффициент теплопроводности X, кал-см-1 сегг^ град~^ (при 0°) 0,27 0,22 0,02
Удельное сопротивление р-108 (при 0°), ом-см 5,75 6,3 94,07
Электропроводность (Hg=l) 16,3 14,9 1
Магнитная восприимчивость Xs-10-8 эл.-магн. ед. (при 18°) -0,157 -0,18 —0,19
Теплота перехода атомов в газооб- разное состояние, ккал (при 25°) 31,19 26,97 14,54
ПОБОЧНАЯ ПОДГРУППА II ГРУППЫ
781
Продолжение табл. 74
Элемент Цинк Zn Кадмий Cd Ртуть Hg
Потенциал иони- зации, эв Me —> Mei -ре- 9,39 8,99 10,43
|Mei —*МеП + е~ 17,96 16,90 18,75
МеИ —>- Melii_|-e_ 39,70 37,47 32,43
Потенциал иони- зации, ккал/г-атом Me —>• Mei e- 216,5 207,3 240,6
Нормальный по- тенциал, в (при 25°) Me/Meii —0,761 —0,402 +0,854
Me/Mei 2Hg/Hgi++0,798
Нормальный потенциал окислитель- но-восстановительных систем, в Hgi+/2Hg2++ +0,92
Валентность II II II
Массовые числа основных изотопов 64, 66, 68 , 67, 70 114, 112, 111, 110, 113, 116, 106, 108 202, 200,199, 201, 198, 204, 196
Распространенность элементов в зем- ной коре, вес. % 0,02 5,0-10-4 5,0-10-6
пением ртути, кристаллы которой объемной ромбоэдрической
структуры. Для всех этих атомов характерны относительно малые
значения атомных и ионных радиусов, плотности от 7,13 до
13,59 г!смй (тяжелые металлы) и относительно малые атомные
объемы — от 9,2 до 14,8 смЛ!г -атом при 20°.
Цинк, кадмий и ртуть — диамагнитные металлы, имеют отно-
сительно низкие температуры плавления и кипения, большое
число природных изотопов, образуют сплавы со многими металла-
ми. Твердость цинка и кадмия составляет 2—2,9 по шкале Мооса,
т. е. эти металлы поддаются обработке давлением.
Цинк и кадмий химически более активны, чем ртуть,
которая в электрохимическом ряду напряжений расположена за
водородом.
В отличие от элементов главной подгруппы элементы подгруп-
пы цинка труднее окисляются, проявляют меньшую реакционную
способность и обнаруживают более слабо выраженные металличе-
ские свойства.
782
ПОБОЧНАЯ ПОДГРУППА II ГРУППЫ
Соединение ZnF2 CdF2 HgF2 ZnCl2 CdCl2
Вид Бесцвет- ные моно- клинные кристал- лы Белые кубиче- ские кристал- лы Бесцвет- ные окта- эдриче- ские кри- сталлы Белые рас- плываю- щиеся ку- бические кристаллы Бесцвет- ные гек- сагональ- ные кри- сталлы
Молекулярный вес 103,38 150,41 238,61 136,294 182,324
Плотность (при 20°), г/ел3 4,84 6,64 8,95 2,91 4,047
Температура плав- ления, °C 875 1110 645 270 568
Температура ки- пения, °C 1758 650 732 964
Теплота образова- ния, ккал/моль 193,0 127,7 53,43 99,28 93,24
Гидраты (число молекул Н2О) 4 — 2 4,3, 5/2, 3/2 4, в/2,1
Сродство к кислороду у металлов подгруппы цинка больше,
чем у металлов побочной подгруппы I группы, и значительно мень-
ше, чем у металлов главной подгруппы II группы.
Окислы и основания щелочноземельных металлов проявляют
основные свойства, а окислы и гидроокиси металлов подгруппы
цинка обладают амфотерными или очень слабо основными свой-
ствами.
В природе цинк встречается в виде сульфидов, карбонатов,
силикатов или окислов, кадмий — в виде сульфидов, карбонатов,
или окислов, а ртуть — в виде сульфидов, селенидов, теллуридов,
окислов и очень редко — в самородном состоянии. Цинк добывает-
ся из обманок (сфалеритов), вюртцита или смитсонита, кадмий —
из кадмийсодержащих обманок или гринокита, ртуть — из кино-
вари.
ПОБОЧНАЯ ПОДГРУППА II ГРУППЫ
783
Таблица 75
HgCl2 ZnBr2 CdBr2 HgBr2 Znl2 Cdl2 Hgi2
Бесцвет- ные ром- биче- ские иглы Бесцветные гигроско- пичные ромбиче- ские кри- сталлы Бесцвет- ные кри- сталлы Бесцвет- ные ром- бические кристал- лы Бесцвет- ные гек- сагональ- ные кри- сталлы Бесцвет- ные гек- сагональ- ные кри- сталлы Ниже 127° кри- сталлы от красного до желто- го цвета
271,524 225,212 275,242 360,20 319,20 366,23 454,43
5,45 4,22 5,196 6,064 4,67 5,56 6,276
277 394 585 238,5 446 388 259
304 670 863 319 624 713 354
55,00 78,234 75,2 40,50 50,76 48,8 25,2 (красная модифика- ция) 24,55 (желтая модифика- ция)
2 3,2 4 — 4,2 — —
Очистка металлических цинка, кадмия и ртути осуществляется
главным образом перегонкой в вакууме или в атмосфере инерт-
ного газа.
Что касается соединений металлов подгруппы цинка, то с уве-
личением атомного номера проявляется все возрастающая склон-
ность к образованию аутокомплексов, гидролизу и появлению
окраски.
В отличие от цинка и кадмия ртуть образует диртутные соеди-
нения, которые содержат катион (Hg — Hg)2+. Существуют также
соединения ртути, подобные соединениям цинка и кадмия.
Известно очень много основных солей этих элементов, посколь-
ку их соединения легко гидролизуются.
В табл. 75 приведены некоторые константы галогенидов цинка,
кадмия и ртути.
784
ПОБОЧНАЯ ПОДГРУППА II ГРУППЫ
Соединения металлов подгруппы цинка отличаются от соеди-
нений щелочноземельных металлов устойчивостью, способностью
растворяться в различных растворителях, окраской и химическим
характером соединений.
Известно довольно много металлоорганических соединений
этих элементов.
В соответствии с приведенными выше характеристиками эле-
менты подгруппы цинка являются переходными металлами.
ЦИНК Zn
Z = 30; ат. вес = 65,37
Валентность II; заряд 2Д-
Массовые числа основных природных изотопов
64, 66, 68, 67, 70
Электронная структура атома цинка: K’L-M-4s2.
Электронная структура атома цинка и катиона Zn2* для 3d-
и 48-орбиталей:
3di0 4s2 3d10 4s
luiumitiiui |w] |и|Щ|Щ| □
Zn Zn*+
ИСТОРИЧЕСКАЯ СПРАВКА
С древних времен известны латуни, являющиеся сплавами
Си — Zn. Металлический цинк был впервые получен в 1746 г.
в Англии нагреванием каламина с древесным углем.
РАСПРОСТРАНЕННОСТЬ В ПРИРОДЕ
В природе цинк находится только в виде соединений в различ-
ных минералах и не встречается в свободном состоянии. В земной
коре содержится 0,02 вес-% цинка.
К наиболее важным минералам цинка относятся следующие.
Цинковая обманка (сфалерит) имеет вид кубических желтых,
коричневых или черных кристаллов; плотность 3,9—4,2 г/см3,
твердость 3—4 по шкале Мооса. В качестве примесей кристаллы
содержат Cd, In, Ga, Mn, Hg, Ge, Fe, Cu, Sn, Pb. Из цинковой
обманки одновременно с цинком добывают кадмий, индий, галлий
и германий.
В кристаллической решетке цинковой обманки (рис. 34), кото-
рая относится к кубической системе, атомы цинка чередуются
с атомами серы, а именно: каждый атом цинка тетраэдрически
окружен четырьмя атомами серы, и наоборот.
цинк
785
Расстояние между атомами Zn — Sb решетке обманки равно
2,35 А. В решетке цинковой обманки атомы серы образуют плот-
ную кубическую упаковку, в которой различают три идентичных
слоя, кристаллографически различно расположенных в порядке
1, 2, 3, 1, 2, 3. Атомы цинка расположены в тетраэдрических
пустотах решетки.
Вюртцит, ZnS, представляет собой коричнево-черные гекса-
гональные кристаллы с плотностью 3,98 г! см3 и твердостью 3,5—
4 по шкале Мооса; обычно содержит цинка больше, чем цинковая
обманка.
Рис. 34. Структура цинковой Рис. 35. Структура вюртцита,
обманки.
В решетке вюртцита (рис. 35), которая относится к гексаго-
нальной системе, каждый атом цинка тетраэдрически окружен
четырьмя атомами серы и каждый атом серы — четырьмя атомами
цинка. Характер расположения слоев атомов в вюртците отличает-
ся от расположения в цинковой обманке.
Смитсонит {цинковый шпат), ZnCO3, встречается в виде белых
(или зеленых, коричневых, серых в зависимости от примесей)
тригональных кристаллов с плотностью 4,3—4,5 г! см3 и твердо-
стью 5 по шкале Мооса. Цинковый шпат образуется из сульфата
цинка (последний получается в результате выветривания цинко-
вой обманки), взаимодействующего с известковым камнем.
Каламин, Zn2SiO4-Н2О-ZnGO3 или Zn4[Si2O7](OH)2 *Н2О •
• ZnCO3, представляет собой смесь карбоната и силиката цинка;
образует белые (или желтые, коричневые, зеленые, синие в зави-
симости от примесей) ромбические кристаллы с плотностью 3,4—
3,5 г!см3 и твердостью 4,5—5 по шкале Мооса.
Виллемит, Zn2SiO4, залегает в виде бесцветных или желто-
коричневых ромбоэдрических кристаллов с плотностью 3,89—
4,18 г! см? и твердостью 5—5,5 по шкале Мооса.
Цинкит, ZnO,— гексагональные кристаллы желтого, оран-
жевого или красного цвета с решеткой типа вюртцита и твердо-
стью 4—4,5 по шкале Мооса; обычно им сопутствуют кристаллы
виллемита и франклинита (Zn, Mn)[Fe2OJ.
50—0101
786
ПОБОЧНАЯ ПОДГРУППА II ГРУППЫ
Ганит, Zn[Al2O4], имеет вид темно-зеленых кубических кри-
сталлов с плотностью 4—4,6 г!см3 и твердостью 7,5—8 по шкале
Мооса.
Помимо приведенных известны и другие минералы цинка, например:
монгеймит (Zn, Fe)CO3, гидроцинкит ZnCO3-2Zn(OH)2, трустит (Zn, Mn)SiO4,
гетеролит Zn[Mn2O4], франклинит (Zn, Mn)[Fe2O4], халькофанит
(Mn, Zn)Mn2O5-2Н2О, госларит ZnSO4-7Н2О, цинкхалькантит (Zn, Cu)SO4-
• 5Н2О, адамин Zn2(AsO4)(OH), тарбуттит Zn2(PO4)(OH), деклуазит
(Zn, Cu)Pb(VO4)OH, леграндит Zn3(AsO4)2-3H2O, гопеит Zn3(PO4)2-4Н2О.
Природные соединения цинка входят в состав полиметалличе-
ских руд, которые помимо цинка содержат соединения элементов
Pb, Gu, Fe, Sn, Cd, In, Ga, As, Al, Ge и Hg.
Залежи цинковых руд имеются в СССР, Финляндии, Норвегии,
Австрии, ГДР, ФРГ, Бельгии, Чехословакии, Италии, Греции,
Югославии, Польше, Англии, Испании, Швеции, Бирме, Китае,
Таиланде, Турции, ДРВ, Конго, Марокко, Юго-Западной Африке,
Тунисе, Родезии, Канаде, Мексике, США, Боливии, Австралии,
Румынии.
ПЕРЕРАБОТКА ЦИНКОВЫХ РУД
И ПОЛУЧЕНИЕ МЕТАЛЛИЧЕСКОГО ЦИНКА
Цинк добывают из концентратов цинковой обманки, смитсо-
нита и каламина.
Сульфидные полиметаллические руды, которые содержат пирит
FeS2, галенит PbS, халькопирит CuFeS2 и в меньшем количестве
цинковую обманку ZnS, после измельчения и размалывания под-
вергаются обогащению цинковой обманкой методом селективной
флотации. Если цинковая руда содержит в качестве примеси маг-
нетит, то для его удаления применяют магнитный метод.
При прокаливании (700°) концентратов сульфида цинка в спе-
циальных печах образуется ZnO, который служит для получения
металлического цинка и сернистого газа, используемого в боль-
ших количествах для производства H2SO4.
2ZnS + ЗО2 = 2ZnO + 2SO2 + 221 ккал
Для превращения ZnS в ZnO измельченные концентраты цин-
ковой обманки часто предварительно нагревают в специальных
печах горячим воздухом, который подают встречным потоком.
Окись цинка также получают прокаливанием смитсонита
при 300°.
Металлический цинк образуется путем термического восста-
новления ZnO углеродом, окисью углерода, водородом, метаном,
карбидом кальция, ферросилицием, а также сильным нагрева-
нием ZnS с железом или углеродом в присутствии СаО в качестве
цинк
787
флюса и с карбидом кальция; получают металлический цинк
и электролизом водного раствора сульфата цинка.
1100-1400°
ZnO + C--------->• Zn-|-CO — 57 ккал
Выше 1000°
ZnO + H2 Zn + H2O
1000-1100°
ZnO-CO , Zn-|-CO2+47,80 ккал
Выше 900°
2ZnO + CH4---------> 2Zn + 2Н2О + С
ZnO + СаС2 = Zn + СаО + 2С 2ZnO + FeSi = 2Zn + Fe + SiO2
ZnS + CaC2 = Zn + CaS + 2C ZnS + Fe 2Zn + FeS
2ZnS + 2CaO + 7C = 2Zn + 2CaC2 + 2CO + CS2
Чтобы избежать окисления металлического цинка, температура
кипения которого равна 906°, восстановление ZnO углем, окисью
углерода и водородом проводят в шамотных ретортах и конден-
сация паров цинка осуществляется как можно быстрее в сосудах
из глины или листового железа без доступа воздуха. В печи, обо-
греваемой метаном, можно нагревать одновременно несколько
реторт. Если температура конденсационных сосудов ниже 420°,
оседающий порошок металлического цинка загрязнен ZnO
(10 вес.%) и металлическим кадмием. При температуре конденса-
ционных сосудов выше 420° металлический цинк образуется в рас-
плавленном состоянии.
Металлургический процесс получения металлического цинка,
применяемый в промышленном масштабе, заключается в восста-
новлении ZnO углеродом при нагревании. В результате этого про-
цесса ZnO восстанавливается не полностью, не полностью конден-
сируются пары цинка, теряется некоторое количество цинка,
идущего на образование Zn[Al2O4] с шамотом реторты, и получают
загрязненный цинк.
Восстановление ZnS углем с добавкой флюса осуществляют
в электрических печах.
1Цинк 99,99%-ной чистоты получают электролизом 1 М раство-
ра ZnSO4-7H2O, подкисленного H2SO4 (0,5 М раствор H2SO4)[
при 35°, при этом используют катоды из чистого алюминия,
а аноды — из свинца; плотность электрического тока 2—4 а'дм*
(на катоде). Электролиз осуществляют в деревянных или цемент-
ных ваннах. Потенциал разложения сульфата цинка в обычных
условиях 2,35 в.
Для электролиза применяют как можно более чистый раствор
сульфата цинка. Сульфат цинка получают растворением окиси
цинка в разб. H2SO4 (ZnO в свою очередь получают прокалива-
нием концентратов цинковой обманки или смитсонита) и затем
50*
788
ПОБОЧНАЯ ПОДГРУППА II ГРУППЫ
подвергают очистке- При добавлении порошкообразного цинка
к загрязненному раствору сульфата цинка осаждаются металлы
меньшей, чем у цинка, химической активности. Для отделения
цинка и кадмия от других металлов чаще всего используют ионо-
обменные смолы. Отделение цинка и кадмия от лития, натрия,
калия, рубидия, цезия, меди, магния, кальция, стронция, бария,
алюминия, титана, железа и никеля основано на образовании
в солянокислых растворах хлоридов цинка и кадмия, которые
можно количественно удержать на сильно основных анионообмен-
ных смолах типа RC1. Для элюирования цинка применяют 2 н.
раствор NaOH, содержащий 2% NaCl, а для элюирования кадмия
— 1 н. раствор HNO3.
ОЧИСТКА СЫРОГО ЦИНКА
Рафинирование сырого цинка (с примесями Pb, Fe, Cd, As, Ga,
Ge, Cu и Мп) осуществляют переплавкой в пламенной печи, фрак-
ционной перегонкой в вакууме или в атмосфере азота, а также
электролитическим путем. Очистка цинка методом переплавки
основывается на свойстве расплавленного цинка не смешиваться
с железом, свинцом, кадмием и другими металлами. Это свойство
позволяет легко отделять цинк от упомянутых металлов простой
декантацией, поскольку он обладает меньшим удельным весом.
Перед перегонкой удаляют летучие примеси, такие, как мышьяк,
сурьма и большая часть свинца и кадмия, которые превращаются
в окислы, сульфиды и хлориды, собирающиеся над поверхностью
металлического цинка, после чего их отгоняют.
Металлический цинк отливают в форме брусков, прутков, пла-
стинок, листков, проволоки; получают его в виде порошка (цин-
ковая чернь, цинковое зеркало) или в коллоидном состоянии.
ФИЗИЧЕСКИЕ И ХИМИЧЕСКИЕ СВОЙСТВА
Цинк — блестящий серебристо-белый металл с плотностью
7,13 г!см3 при 20° (тяжелый металл), т. пл. 419,44°, т. кип. 906°,
твердость 2,5—2,9 по шкале Мооса. Имеет плотную гексагональ-
ную структуру кристаллов. Цинк становится пластичным (ковким,
тягучим, вязким) при 100—150° и хрупким — выше 200° (при этой
температуре он может быть превращен в порошок); он диамагни-
тен и имеет пять природных изотопов. Загрязнение цинка свин-
цом или мышьяком вызывает появление хрупкости.
Известны многочисленные сплавы цинка с Al, Cu, Sn, Mg, Са,
Ni, Pb, Cd, Fe, Mn, Sb, Bi, Ag, Hg, Au, Pd, Pt, W, TI, Hg, Si,
La, Ce, Li, Na, Rb и Cs.
С химической точки зрения цинк менее активен, чем щелочные
и щелочноземельные металлы, и более активен, чем олово, свинец,
медь, ртуть, серебро и золото.
цинк
789
При хранении в сухом воздухе цинк длительное время остается
блестящим, однако во влажном воздухе поверхность цинка покры-
вается тонкой защитной пленкой окисла и основного карбоната,
которая в дальнейшем предохраняет металл от коррозионного дей-
ствия атмосферных реагентов. Благодаря этому свойству цинком
покрывают железные листы или пластины (методами электролити-
ческого или горячего цинкования) для предохранения их от раз-
рушения.
При сильном нагревании на воздухе цинк воспламеняется
и горит ярким сине-зеленым пламенем, образуя
Zn + 1/2О2 = ZnO + 83,5 ккал
Цинк окисляется кислородом воздуха при нагревании выше
225°, а в токе сухого кислорода — выше 150°.
Металлический цинк не разлагает воду при комнатной темпе-
ратуре или кипячении, поскольку в присутствии воды поверх-
ность цинка покрывается Zn(OH)2, которая препятствует даль-
нейшему разрушению металла. Цинк, нагретый почти до темпе-
ратуры плавления, энергично разлагает пары воды с образова-
нием ZnO и выделением водорода:
Zn -|- Н2О ZnO Н2
При комнатной температуре во влажном воздухе галогены
образуют с цинком соответствующие галогениды. В сухом состоя-
нии галогены взаимодействуют с металлическим цинком при
высокой температуре:
Zn -j- F2 = ZnF2 4- 172,7 ккал Zn 4- Вг2 = ZnBr2 4~ 78,4 ккал
Zn -}- С12 = ZnCl2 + 99,55 ккал Zn I2 = Znl2 -f- 49,8 ккал
Нагревание порошкообразного металлического цинка с порош-
ком серы приводит к образованию ZnS. Реакция экзотермична:
Zn 4- S = ZnS -}- 44 ккал
Под действием сероводорода поверхность металлического цинка
покрывается плотной защитной пленкой ZnS:
Zn -j- H2s ZnS 4~ н2
Нагревание металлического цинка в атмосфере газообразного
аммиака дает нитрид Zn3N2. Двуокись серы во влажной атмосфере
и хлористый тионил взаимодействуют с цинком при нагревании
по уравнениям
3Zn 4- SO2 = ZnS 4- 2ZnO Zn 4- SOC12 = ZnCl2 4- SO
Цинк непосредственно не вступает в реакции с азотом, углеро-
дом, водородом и другими элементами. Обработка парами фосфора
нагретого до 440—780° металлического цинка в атмосфере азота
или водорода приводит к образованию Zn3P2 и ZnP2.
Поскольку нормальный потенциал системы Zn/Zn2+ равен
—0,761 в, металлический цинк легко растворяется в кислотах
790
ПОБОЧНАЯ ПОДГРУППА II ГРУППЫ
и щелочах. При растворении цинка в разбавленных кислотах
(HCl, H2SO4 и др.) образуются соответствующие соли цинка
и выделяется водород:
Zn + 2НС1 = ZnCl2 + Н2 Zn + H2SO4 = ZnSO4 + H2
Чистый металлический цинк медленно взаимодействует с раз-
бавленными кислотами, поскольку образующийся атомарный
водород покрывает поверхность цинка и выделение водорода
уменьшается. При образовании гальванического элемента Zn — Си
в HCl (1 : 1) выделение водорода происходит очень интенсивно,
и цинк легко растворяется, превращаясь в хлорид. Для образо-
вания гальванического элемента Zn — Си в раствор (1:1) НС1
добавляют несколько капель сульфата меди. Медь, стоящая в элек-
трохимическом ряду напряжений правее цинка, осаждается им,
покрывает поверхность цинка и ускоряет таким образом раство-
рение Zn в НС1 за счет образования гальванических элементов.
По отношению к конц. H2SO4 или HNO3 цинк является вос-
становителем:
Zn + 2H2SO4 = ZnSO4 -ф SO2 + 2H2O
4Zn + IOHNO3 = 4Zn(NO3)2 + NH4NO3 + 3H2O
При действии щелочей на металлический цинк образуются
гидроксоцинкаты и водород:
Zn + 2NaOH + 2Н2О = Na2[Zn(OH)4] + Н2
Металлический цинк вытесняет менее активные металлы
(Cu, Ag, Hg, Аи, Pt и др.) из растворов их солей и восстанавливает
хромовую, марганцовую и молибденовую кислоты, соли железа(Ш)
и олова(1У). С помощью цинка можно осуществить цинкотермиче-
ское разложение многих окислов, например CdO, РЬО, СиО и NiO-
Восстанавливая сероуглерод, цинковый порошок раскаляется.
Химические свойства Zn иллюстрируются следующей схемой:
во влажном воздухе -»• покрывается окисью
J. и основным карбонатом
с галогенами в присутствии влаги соответ-
ствующие галогениды
| в НС1 (1:1) или H2SO4 (1:1) -> ZnCl2-nH2O,
I ZnSO4-nH2O
l вытесняет Cu, Ag, Hg, Au и Pt из растворов их солей
цинк i на воздухе (в кислороде) горит ZnO
в парах воды при 420° -> ZnO
с галогенами -> ZnF2, ZnCl2, ZnBr,, Znl2
c S или H2S -> ZnS
Нагревание c SO2 -»• ZnS ZnO
-------------*- J с селеном, теллуром, фосфором -> ZnSe, ZnTe,
' Zn3P2, ZnP2
c NH3 -» Zn3N2
c NaOH (или KOH) -► Na2[Zn(OH)4]
c CuO, PbO, NiO, CdO -> ZnO + соответству-
, тощие металлы
цинк
791
С точки зрения физиологии цинк — необходимый элемент
как для организма человека и животных, так и для растений.
Цинк входит в состав фермента, имеющегося в красных кровяных
тельцах, с помощью которого происходит перенос двуокиси угле-
рода в крови. Для человека и животных чистый цинк не токсичен.
Токсичным он становится будучи загрязнен мышьяком, сурьмой,
свинцом, кадмием и др. Вдыхание ZnCl2 в больших дозах смер-
тельно.
ПРИМЕНЕНИЕ
Металлический цинк применяется для покрытия железных
(стальных) листов и проволоки с целью предохранения их от кор-
розии, для извлечения серебра из серебросодержащего свинца
по процессу Паркеса, для получения водорода в результате раз-
ложения HCl (1 : 1), для вытеснения металлов (с более низкой
химической активностью) из растворов их солей (например, вытес-
нение Ag или Аи из растворов K[Ag(GN)2], K[Au(CN)2] или РЬ
из растворов Pb(NO3)2, РЬ(СН3СОО)2), для изготовления галь-
ванических элементов, в качестве восстановителя во многих
химических реакциях, для получения многочисленных сплавов
с медью (латуни), алюминием, магнием, свинцом, оловом и дру-
гими металлами.
СОЕДИНЕНИЯ (ОБЩИЕ СВОЙСТВА)
Известно много устойчивых соединений электроположитель-
ного двухвалентного цинка. Наряду с простыми соединениями
цинка(П) выделили многочисленные координационные соедине-
ния, в которых цинк входит в состав катиона или аниона.
В соединениях цинк может присутствовать в виде катионов
Zn2+, fZn(H2O)6]2+, [Zn(NH3)J2+, [Zn(NH3)6]2+ и в виде анионов
[ZnF3]-, [ZnF4]2~, [ZnCl3]-, [ZnClJ2-, [ZnBr3l", [ZnBrJ2-,
[Znl3/-, [ZnlJ2-, [Zn(CN4)]2-, [Zn(CN)6l4-, fZn(OH)3]-, [Zn(OH)4]2-.
В табл. 76 приведены формулы и указан цвет ряда соединений
цинка(П).
Из простых соединений цинка легко растворяются в воде
хлорид, бромид, иодид, сульфат, нитрат и ацетат; плохо раство-
ряются фторид, сульфид, карбонат, ортофосфат и основные соли.
Большинство соединений цинка бесцветно.
Водные растворы растворимых солей цинка имеют кислую
реакцию вследствие гидролиза.
Наряду с неорганическими известны хелатные и металлоорга-
нические соединения цинка.
792
ПОБОЧНАЯ ПОДГРУППА II ГРУППЫ
ЦИНК
793
Окис- ли Гидроокись, гидроксосоедй- нения Фториды, фторосоедине- нения Хлориды, хло- росоединения Бромиды, бромо- соединения I
Сое- дине- ния цин- ка(П) ZnO белая ZnO2 жел- това- то-бе- лая Zn(OH)2 белая амфотерная Mei[Zn(OH)3l- пН2О Mei[Zn(OH)4[. •пН2О ZnF2 бесцвет- ный ZnF2-nH2O . Zn(OH)F Zn5(OH)8F2 Mei[ZnF3] Mei[ZnF4]- •H2O ZnCl2 белый ZnCl2-nH2O [Zn(NH3)4]Cl2 [Zn(NH3)e]Cl2 Zn(OH)Cl Zn5(OH)8Cl2. •H2O Zn^OH^Cl, Mel[ZnCl3] Mei[ZnCl4] Mei[ZnCle] ZnBr2 белый ZnBr2-nH2O [Zn(NH3)e]Br2 Zn(0H)Br-H20 Zn5(OH)8Br2 Mel[ZnBr3]- •nH2O Mei[ZnBr4]
Неорганические соединения 1'
Гидрид цинка, ZnH2, получают действием Zn(CH3)2 на LiAlH4
в эфире при обычной температуре, действием LiAlH4 на Znl2
в эфире при 40°, непосредственным взаимодействием гидрида
алюминия с диметилцинком.
Zn(CH3)2 4- 2LiAlH4 = ZnH2 -f- 2LiAlH3(CH3)
Гидрид цинка — нелетучее белое твердое вещество, которое
разлагается на элементы при нагревании до 80°. Гидрид цинка
плохо растворим в эфире, медленно разлагается в воде, быстро —
в водном растворе HG1, взаимодействует с дибораном:
ZnH2 4- B2He = Zn(BH4)2
Таблица 76
Йодиды, ио- досоединения Суль- фиды Сульфаты Карбонаты Хелатные соединения
Znl2 белый [Zn(NH3)e]I2 Mei[ZnI3]- • nH2O Mei[ZnI4]- •nH2O ZnS белый ZnS2 ZnSO4 белый ZnSO4-nH2O ZnSO4-nNH3 ZnSO4- zZn(OH)2 MelSO4- • ZnSO4-nH2O ZnCO3 белый Zn4(CO3)3(OH)2 ZnCO3.7ZnO-2H2O ZnCO3-3ZnO-2H2O ZnCO3.2ZnO-2H2O г / CeH5 Н > -г 1 1 N N Zn /С = О N=N 1 Ч Свн5 У2. [пурпурно-красный rHN=C C = NHt 1 1 1 1 \s s J L желтовато-белый Г / H2 1 Znl z ? j L \ O C = O 72- белый [Zn(C5O2H7)2], K2[Zn(C2O4)2] • 7H2O, K-e[Zn(P 307)2]
Известен также иодогидрид цинка ZnHI.
Окись цинка, ZnO, встречается в природе в виде минерала
цинкита; в лаборатории может быть получена сжиганием метал-
лического цинка на воздухе или прокаливанием гидроокиси кар-
боната, нитрата или оксалата цинка.
300°
Zn 4- V2O2 = ZnO 4- 83,5 гкал ZnCO3-------► ZnO 4- СО2
340°
Zn(OH)2 -4- ZnO 4- Н2О Zn(NO3)2------> ZnO 4- NO2 4- */2O2
Соединение ZnO представляет собой кристаллы co структурой
типа вюртцита белого цвета на холоду или при обычной темпе-
ратуре и желтые при ~250°, их плотность равна 5,70 г!см\ твер-
дость 4—5 по. шкале Мооса, плавятся при ~2000°, диамагнитны.
ZnO плохо растворима в воде, растворяется в кислотах и щелоча^
ПОБОЧНАЯ ПОДГРУППА II ГРУППЫ
794
с образованием солей, обладает полупроводниковыми, люминес-
центными и фотохимическими свойствами.
ZnO + 2НС1 = ZnCl2 + Н2О ZnO + NaOH + Н2О = Na[Zn(OH)3]
При нагревании (~1000°) ZnO восстанавливается до металли-
ческого цинка углеродом, окисью углерода, водородом, метаном,
карбидом кальция и ферросилицием.
ZnO -|- С = Zn + СО — 57 ккал ZnO + СО Zn -|- СО2 47,80 ккал
ZnO + Н2 Zn + Н2О
Окись цинка растворяется при нагревании в SiO2 или В2О3,
образуя стекловидную массу.
2ZnO SiO2 = Zn2SiO4 ZnO -|- В2О3 = Zn(BO2)2
Нагревание ZnO с некоторыми окислами металлов приводит
к образованию различных цинкатов, например зелени Ринманна
CoZnO2.
Окись цинка (называемая иначе цинковыми белилами) служит
для приготовления масляных красок, для получения некоторых
препаратов, используемых в медицине и косметике, в резиновой
и керамической промышленности, а также в качестве катализа-
тора при синтезе метанола.
О
Перекись цинка, Zn^ |, получают в виде желтовато-белого
порошка действием Н2О2 на эфирный раствор диэтилцинка или
амида цинка. Обработка перекисью водорода Zn(OH)2 дает гидрат
перекиси цинка неопределенного состава.
Гидроокись цинка, Zn(OH)2, образуется в виде белого объеми-
стого осадка, если растворы солей цинка обработать щелочами^
В кристаллическом состоянии Zn(OH)2 существует в виде
пяти (ос-, р~, у-, б-, Е-) модификаций, из которых при комнатной
температуре только e-Zn(OH)2 устойчива в воде. Модификация
oc-Zn(OH)2 имеет гексагональную структуру, p~Zn(OH)2 —
ромбоэдрическую или гексагональную, y-Zn(OH)2 — призма-
тическую, 6-Zn(OH)2 — ромбоэдрическую, e-Zn(OH)2 — бипирами-
дальную структуру.
Гидроокись цинка мало растворима и не разлагается водой
при температуре ~39°; она имеет амфотерный характер и раство-
ряется в кислотах и щелочах с образованием солей:
^Zn(OH)2 + 2H2O^[Zn(OH)3]-4-H3O<-
Zn2+ + 2ОН- Zn(OH)2^
^Zn(OH)2 + 4H2O 4=t [Zn(OH)4]2- + 2H3O+
Zn(OH)2 + 2HC1 = ZnCl2 4- 2H2O Zn(OH)2 + 2NaOH = Na2[Zn(OH)4]
цинк
795
При действии конц. NaOH на Zn(OH)2 могут образоваться
гидроксоцинкаты Na[Zn(OH)3], Na[Zn(OH)3]-ЗНаО, Na2[Zn(OH)4],
Na2[Zn(OH)4] -2Н2О, которые осаждаются спиртом.
Растворение Zn(OH)2 в избытке аммиака дает гидроокись
гексамминцинка IZn(NH3)e](OH)2.
Фторид цинка, ZnFz, получают нагреванием ZnF2-4H2O при
700—800° в токе фтористого водорода или действием газообраз-
ного фтора на металлический цинк, ZnBr2 или ZnS.
Ковалентное соединение ZnF2 имеет вид бесцветных моноклин-
ных кристаллов с решеткой типа рутила, плотностью 4,84 г/см3,
т. пл. 875° и т. кип. 1500°, фторид цинка плохо растворим в воде
и спирте.
ZnF2 + Н2О т ZnO + 2HF
При действии плавиковой кислоты на ZnO, Zn(OH)2
и Zn4(CO3)3(OH)2 в присутствии воды или при обработке растворов
солей цинка концентрированным раствором KF в платиновой
посуде получают бесцветные кристаллы ZnF2-4H2O с плотностью
2,53 г!см3\ соединение плохо растворимо в воде.
Известны фтороцинкаты NH4[ZnF3], Na[ZnF3], K[ZnF3],
(NH4)2[ZnF4l-Н2О и основные фториды, например Zn(OH)F,
Zn5(OH)8F2.
Хлорид цинка, ZnCl2, получают нагреванием гранулирован-
ного металлического цинка при 420° в токе хлора, действием газо-
образного хлора на нагретые до 700° ZnO, ZnS, нагреванием (450°)
ZnO со смесью С12 и S2C12, нагреванием цинка с HgCl2, нагрева-
нием (600—610°) кристаллогидратов ZnCl2-raH2O (где п = 4, 3,
2V2, IV2, 1) в токе газообразного НС1.
Zn + С12 = ZnCl2 99,55 ккал
Хлорид цинка очищают сублимацией при 600—700° в токе хлора.
Ковалентное соединение ZnCl2 представляет собой расплавляю-
щиеся на воздухе белые кубические кристаллы с решеткой типа
CdCl2, плотностью 2,91 г/см3, т. пл. 270° (расплав желтого цвета)
и т. кип. 732°. Хлорид цинка легко растворяется в воде, эфире
и спирте; благодаря гидролизу его водный раствор имеет кислую
реакцию.
Известны кристаллогидраты ZnCl2-raH2O (где п = 4, 3, 21/2,
П/г, 1) и хлоросоли типов MeI[ZnCl3], Me|[ZnCl4], Me*[ZnCle],
например (NH4)2[ZnCl4l, Cs2[ZnCl4], Na2[ZnCl4]-3H2O, K4[ZnCl8],
основные хлориды Zn(OH)Cl, Zn5(OH)8Cl2-H2O, Zn4(OH)eCl2,
аммиакаты ZnCl2-raNH3 (где n = 1, 2, 10), комплексные аммиа-
каты [Zn(NH3)4]Cl2-H2O, [Zn(NH3)e]Cl2,
'(C2H5)H2N Cl"
^Zn/
(C2H5)HzN/f XC1
796
ПОБОЧНАЯ ПОДГРУППА II ГРУППЫ
Тетрагидрат хлорида цинка, ZnCl2>4H2O, его вернеров-
ская формула [Zn(H2O)41Cl2.
Кристаллогидраты хлорида цинка осаждают концентрирова-
нием (при различных температурах) растворов металлического
цинка, окиси, гидроокиси или карбоната цинка в разбавленной
соляной кислоте.
Хлорид цинка применяют в медицине, им пропитывают древе-
сину для предотвращения гниения, используют при изготовлении
пергамента и для очистки металлов перед пайкой.
Бромид цинка, ZnBr2, получают действием паров брома
на нагретый докрасна цинк; это гигроскопичные бесцветные
ромбические кристаллы с плотностью 4,22 г/см\ т. пл. 394°,
т. кип. 670°, легко растворимые в воде, эфире и спирте. /Известны
кристаллогидраты ZnBr2-3H2O, ZnBr2-2H2O, комплексные бро-
миды [Zn(NH3)6]Br2, основные бромиды
'(CH3)2S. .Вг"
*Zn
-(CH^S^ ^Вг
Zn2(OH)2Br2 '2Н2О, Zn5(OH)8Br2, аммиакаты ZnBr2-raHN3 (где
71 = 2, 10), многочисленные аддукты, например, ZnBr2-2N2H4,
ZnBr2^CsHsN, ZnBr2•2G7H9N, ZnBr2-C8H12N4, ZnBr2-CH3OHT
ZnBr2«C5H5OH и бромосоли NHjZnBrJ •(! или 2) H2Or
(NH4)2[ZnBrJ, Na[ZnBr3]-H2O, Na[ZnBr3] -5H2O, K[ZnBr3]-2H2O.
Иодид цинка, Znl2, получают действием паров иода на рас-
плавленный цинк при 600° или обработкой однородной смеси
порошка цинка с иодом несколькими каплями воды. Иодид цинка
выделяется в виде бесцветных гексагональных кристаллов
с решеткой типа Cdl 2, плотностью 4,67 г!см3, т. пл. 446°
и т. кип. 624°. Он растворим в воде, эфире, спирте и кислотах.
Известны кристаллогидраты ZnI2-4H2O, ZnI2«2H2O, комплекс-
ный иодид [Zn(NH3)e]I2, аммиакаты ZnI2-raNH3 (где п = 2, 4, 5),
аддукты ZnI2-2C6H14N4-8H2O, ZnI2-2N2H4 и иодосоли
Na[ZnI3b(2—3)Н2О, K2[ZnI4]-2Н2О, (NH4)2[ZnI4l.
Цианид цинка, Zn(CN)2, образуется путем обработки раство-
ров солей цинка стехиометрически необходимым количеством
KCN или NaCN; соединение представляет собой бесцветные ром-
бические кристаллы, плохо растворимые в воде. При растворении
Zn(CN)2 в избытке KCN или NaCN образуются растворимые
комплексные цианиды типов Me*[Zn(CN)4], Me*Zn[Zn(CN)6] -nH2O.
Zn(CN)2 + 2KCN - K2[Zn(CN)4]
Соединение K2[Zn(CN)4] получают действием металлического
цинка на K2[Cu(CN)2], K2[Cu(CN)3l, K[Ag(CN)2], K[Au(CN)2].
Оно мало устойчиво и разлагается кислотами или сульфидами
цинк
797
аммония.
K2[Zn(CN)4] + 4НС1 = ZnCl2 + 2К.С1 4- 4HCN
K»[Zn(CN)4] 4- (NH4)2S = ZnS 4- 2K.CN 4- 2NH4CN
Известно соединение K2Zn[Zn(CN)e] "бИгО.
Сульфид цинка, ZnS, встречается в природе в виде минерала
цинковой обманки (сфалерита) или вюртцита; в лаборатории может
быть получен нагреванием смеси порошкообразного цинка, ZnO
или ZnCO3 с серой, пропусканием смеси азота с CS2 над нагретым
докрасна цинком, действием H2S на пары хлорида цинка, обработ-
кой растворов солей цинка сульфидом аммония (NH4)2S при
pH = 2-3.
Zn 4- S = ZnS 4- 44 ккал 2Zn 4- CS2 2ZnS 4- C
Сульфид цинка представляет собой белое аморфное вещество,
которое при 1020° в токе сероводорода превращается в вюртцит,
прокаливанием при 430—550° на воздухе превращается в ZnO
и ZnSO4, при 600° сульфид с сульфатом цинка дают ZnO и SO2,
а при 940° ZnSO4 превращается в ZnO и SO3. Сульфид цинка
обладает люминесцентными свойствами, он плохо растворим
в воде, щелочах и уксусной кислоте и хорошо растворяется
в минеральных кислотах с выделением H2S.
ZnS 4- 2НС1 ZnCl2 4- H2S
Под давлением 150 ат и при температуре 1850° вюртцит пла-
вится, а при атмосферном давлении и температуре 1180° он субли-
мируется.
Известен оксисульфид ZnS -ZnO.
Сульфид цинка служит для изготовления экранов, чувстви-
тельных к действию рентгеновских лучей и радиоактивного излу-
чения-
Смесь ZnS с BaSO4, называемая «литопоном», применяется
в качестве пигмента для приготовления масляных красок.
При действии Na2S2O3-5H2O на растворы солей цинка при 100°
осаждается ZnS2, который при ~120° разлагается на ZnS и серу:
zs
Zn2+4-2S2O|- = Znz | 4-S2O|-
XS
Дитионат в водной среде разлагается по уравнению
S2O|-4-ЗН2О = SO2-4-SO2-4-2Н3О+
Сульфат цинка, ZnSO4’7H2O или [Zn(H2O)6]SO4-Н2О, осаж-
дают путем концентрирования (при 38,12°) растворов металли-
ческого цинка, окиси, гидроокиси или карбоната цинка в разб.
H2SO4.
798
ПОБОЧНАЯ ПОДГРУППА II ГРУППЫ
Соединение ZnSO4 -7Н2О представляет собой бесцветные орто-
ромбические кристаллы, изоморфные с кристаллами FeSO4-7H2O,
MgSO4-7H2O, CoSO4.7H2O, MhSO4-7H2O.
Сульфат цинка имеет плотность 1,97 г/см3, он легко раство-
ряется в воде, плохо растворим в спирте, полностью дегидрати-
руется при 280°, превращается в ZnO при нагревании выше 770°
(точнее при 940°).
Известны также кристаллогидраты ZnSO4-raH2O (где п = 6, 4,
2, 1), безводная соль ZnSO4 и двойные сульфаты типа Me^SO4-
•ZnSO4-6H2O (где Me1 = Na+, К+, NH4). Безводная соль ZnS04
может быть получена сульфатирующим прокаливанием цинковой
обманки.J
Упоминаются многочисленные основные сульфаты ZnSO4-
•mZnO-иНгО (где тип имеют различные значения), аммиакаты
ZnSO4-raNH3 (где п = 5, 4, 3, 2) и двойные соли, например
Na2SO4-ZnSO4-4Н2О, 3Na2SO4 -ZnSO4, K2SO4-ZnSO4-6H2Or
(NH4)2SO4-ZnSO4-6H2O.
Сульфат цинка служит для получения большинства соединений
цинка, из него готовят электролиты для цинкования; ZnS04
применяется также для пропитки древесины, предотвращающей
гниение, и для приготовления фармацевтических препаратов.
Нитрид цинка, Zn3N2, образуется в результате нагревания
порошкообразного металлического цинка до 600° в токе аммиака
или нагревания амида цинка Zn(NH2)2 до 330°; это серые кубиче-
ские кристаллы с плотностью 6,22 г/см3; они разлагаются водой
с выделением аммиака и растворяются в разбавленных кислотах
(НС1, H2SO4):
3Zn + N2 = Zn3N2 4- 5,3 ккал Zn3N2 + 6Н2О = 3Zn(OH)2 -f- 2NH3
Известен также нитрид LiZnN.
Амид цинка, Zn(NH2)2, получают нагреванием (150°) диэтил-
цинка в эфирном растворе с аммиаком; это белый порошок, кото-
рый разлагается водой и кислотами, превращается в Zn3N2 при
нагревании до 330° и в ZnO2 при обработке перекисью водорода
в эфирном растворе.
Нитрат цинка, Zn(NO3)2-6Н2О, выделяется упариванием
при обычной температуре раствора, окиси, гидроокиси или кар-
боната цинка в разб. HNO3.
Соединение Zn(NO3)2-6H2O получают в виде бесцветных
тетрагональных кристаллов с плотностью 2,130 г/см3 и т. пл. 36,4°;
при нагревании до 105° они полностью дегидратируются, легко
растворяются в воде и спирте.
Известны также кристаллогидраты Zn(NO3)2 •иН2О (где
п = 9, 4, 2, 1), основные соли Zn(OH)NO3, Zn5(OH)8(NO3)2 и много-
численные двойные соли mMeINO3-ZnSO4, аммиакаты Zn(NO3)2-
цинк
799
•raNH3 и аддукты Zn(NO3)2 •3C2H4(NH2)2, Zn(NO3)2-2C6H5NH2,
Zn(NO3)2 -2C5H5N -2H2O, Zh(NO3)2 -SCeHsNHNH;,.
Безводный нитрат цинка, Zn(NO3)2, получают действием
жидкой тетраокиси азота N2O4 на металлический цинк:
Zn + 4N2O4 = Zn(NO3)2-2N2O4 + 2NO
Zn(NO3)2 *2N2O4 = Zn(NO3)2 4~ 2N2O4
Ортофосфат цинка, Zn3(PO4)2, получают, обрабатывая раство-
ры солей цинка в нейтральной среде раствором Na2HPO4 Л2Н2О.
3ZnCl2 + 4Na2HPO4 = Zn3(PO4)2 + 2NaH2PO4 + 6NaCl
Соединение Zn3(PO4)2 представляет собой бесцветные ромбиче-
ские кристаллы с плотностью 4,0 г!см? и т. пл. 900°; они плохо
растворимы в воде и спирте.
Тетрагидрат ортофосфата цинка, Zn3(PO4)2-44^0. пред-
ставляет собой диамагнитные синие кристаллы с плотностью
3,11 г/сл3; они плохо растворимы в воде,, легко растворяются
в разбавленных кислотах с образованием Zn(H2PO4)2, а также
в аммиаке и карбонате аммония.
При обработке растворов солей цинка раствором Na2HPO4*
•12Н2О в слабо аммиачной среде в присутствии солей аммония
выпадает ортофосфат цинка и аммония:
ZnCl2 + Na2HPO4 4- NH4C1 NH4ZnPO4 + 2NaCl + HC1
Карбид цинка, ZnC2, получают путем пропускания ацетилена
через раствор диэтилцинка в лигроине. Он представляет собой
белый порошок, который разлагается водой или кислотами с выде-
лением ацетилена.
ZnC2 + 2Н2О = Zn(OH)2 + С2Н2
Карбонат цинка, ZnCO3, встречается в природе в виде мине-
рала смитсонита. Получают его обработкой растворов солей
цинка раствором КНСО3, насыщенным СО2, при 3° или действием
двуокиси углерода на водную суспензию Zn(OH)2. Приготовлен-
ный ZnCO3 промывают водой методом декантации и сушат при 110°.
Бесцветные тригональные кристаллы ZnCO3 имеют плотность
4,44 г/смъ, они плохо растворимы в воде, растворяются в кислотах
и щелочах, разлагаются при 300°, превращаясь в ZnO.
Обработка растворов солей цинка растворами карбонатов
щелочных металлов дает основные карбонаты цинка, состав кото-
рых меняется в зависимости от условий эксперимента. В качестве
примера можно привести следующее уравнение:
4ZnSO4 4- 4Na2CO3 4- Н2О = Zn4(CO3)3(OH)2 4- 4Na2SO4 4- СО2
800
ПОБОЧНАЯ ПОДГРУППА II ГРУППЫ
В состав основных карбонатов цинка входят карбонат цинка
и гидроокись или окись цинка и вода, например ZnCO3•3Zn(OH)2,
ZnCO3-7ZnO .2Н2О, ZnGO3-3ZnO-2H2O, ZnCO3-2ZnO-2H2O,
ZnCO3 -ZnO -H2O.
Хелатные соединения
Дитизонат цинка пурпурно-красного цвета образуется при
/NH- NH<- СвН5
действии раствора дитизона SCr в четыреххло-
4N = N- СвН5
ристом углероде или хлороформе на соединения цинка в ней-
тральной или слабо кислой среде.
Н СвН5 СвН5Н
II II
/N — N ,N —N
s=cz xznZ X=S
4'4N = N/X ''N = N//
CeH5
Дитизонат цинка
Подобные хелатные соединения получают и в случае использо-
вания солей кадмия, ртути, меди, серебра.
При действии рубеановодородной кислоты на растворы солей
цинка, забуферованные ацетатом натрия, образуется желтовато-
белый осадок рубеаната цинка:
При обработке растворов солей цинка антраниловой (о-амино-
бензойной) кислотой образуется белый осадок антранилата цинка:
7n( ___Z |
- \хо — c=o./2J
бис-Ацетилацетонатцинк, lZn(C5O2H7)2], представляет собой
бесцветное вещество с т. пл. 138°; оно легко растворяется в воде
и бензоле.
цинк
801
В качестве примеров хелатных соединений можно назвать
также диоксалатоцинкат K2lZn(C2O4)2 -7Н2О и дипирофосфато-
цинкат Ke[Zn(P2O7)2].
Металлоорганические соединения
Известны следующие цинкоорганические соединения: Zn(CH3)2,
Zn(C2H3)2, Zn(C3H7)2, Zn(C4Hg)2, Zn(C5H11)2, Zn(C6H5)2, CeHgZnl,
Na[Zn(C2H5)3] и др.
Диметилцинк, Zn(CH3)2, образуется обработкой металличе-
ского цинка иодистым метилом при обычной температуре; это
бесцветная жидкость с неприятным запахом, которая имеет плот-
ность 1,245 г!см\ затвердевает при —29,2°, самопроизвольно
загорается на воздухе и разлагается водой по уравнению
Zn(CH3)2 + 2Н2О -> Zn(OH)2 + 2СН4
Диэтилцинк, Zn(C2H5)2, получают без доступа воздуха
(в атмосфере СО2) по уравнениям:
Zn + Hg(C2H5)2 = Zn(C2H5)2 +'Hg
ZnCl2 + 2(C2H5)MgI = Zn(C2H5)2 4- Mgl2 + MgCl2
2(C2H5)ZnI — Zn(C2H5)2 Znl2
Диэтилцинк представляет собой бесцветную жидкость, которая
затвердевает при —30°, кипит при 118°, устойчива, но реакционно-
способна, разлагается при температуре выше 200°. Соединение
Zn(C2H5)2 превращается в Zn(C2H5)2O2 при окислении на воздухе
и в ZnO2 под действием эфирного раствора Н2О2. Диэтилцинк
взаимодействует с водой, спиртом, аммиаком и кислородсодержа-
щими органическими соединениями.
Zn(C2H5)2 + 2Н2О = Zn(OH)2 + гаде
Дифенилцинк, Zn(CeH5)2, образуется при действии дифенил-
ртути’на металлический цинк; это белые кристаллы с т. пл. 107°
и т. кип. 283°.
Иодид этилцинка, C2H5ZnI, получают нагреванием металличе-
v ского цинка с иодистым этилом при 72,3° в атмосфере СО2. Он
представляет собой белые кристаллы, которые разлагаются на воз-
духе или в воде, растворяются в смеси этилацетата с толуолом
и взаимодействуют с органическими галогенидами, эфирами, кето-
нами и соединениями, содержащими двойные связи.
Н3С С2Н5 ЩС
''c = O-f-Zn--►'c^O + ZnlCl
а 1 H5cf
51—010’
802
ПОБОЧНАЯ ПОДГРУППА II ГРУППЫ
Триэтилцинкат(11) натрия, Na[Zn(C2H5)3], получают раство-
рением натрия в диэтилцинке. Он представляет собой бесцветное
твердое вещество ст. пл. 27°.
Известен триэтилцинкат(П) лития Li[Zn(C2H5)3L
Помимо приведенных известны и другие соединения цинка, например:
хлорит Zn(ClO2)2, хлорат Zn(ClO3)2-4H2O, перхлорат Zn(C104)2, бромат
Zn(BrO3)2-6H2O, иодат Zn(IO3)2-2Н2О, сульфит ZnSO3, дитионит ZnS2O4,
политионаты ZnS2Oe, ZnS3Oe, ZnS4Oe, ZnS5Oe, пероксосульфат ZnS2O8,
тиосульфат ZnS2O3, селенид ZnSe, селенит ZnSeO3 или ZnSeO3-2H2O, селенат
ZnSeO4, или ZnSeO4-raH2O, теллурид ZnTe, теллурат ZnTeO4, азид Zn(N3)2,
нитрит Zn(NO2)2-3H2O, фосфиды Zn3P2, Zn3P4, ZnP2, ZnP4, фосфин ZnHP,
гипофосфит Zn(H2PO2)2, фосфит ZnHPO3, ортофосфаты Zn(H2PO4)2-2H2O,
ZnHPO4-3H2O, NaZnPO4, или NaZnPO4-H2O, KZnPO4, метафосфаты
Zn2P4O12, ZnPeO18, дифосфат Zn2P2O7, арсениды Zn3As2, ZnAs2, арсенит
Zn3(AsO3)2, ортоарсенат Zn3(AsO4)2-8H2O, диарсенат Zn2As2O7, антимониды
ZnSb, Zn3Sb2, Zn4Sb3, антимонит ZnSb2O4, антимонат ZnSb2Oe, тиоантимонат
Zn3(SbS4)2, роданид Zn(SCN)2, гексацианоферрат(П) Zn2[Fe(CN)e], цианамид
ZnCN2, силикат Zn2SiO4, гексафторосиликат Zn[SiFe]-6H2O, германат
Zn2GeO4, станнат ZnSnO3-4H2O, хромат ZnCrO4, бихромат ZnCr2O7-3H2O,
молибдат ZnMoO4, вольфрамат ZnW04, танталат ZnTa2Oe, ванадат ZnV2Oe-
• 2H2O, титанат Zn2TiO4, перренат Zn(ReO4)2-4H2O, борат ZnB2O4 или
Zn(BO2)2, тетрафтороборат Zn[BF4]2, алюминат Zn[AI2O4], галлат
Zn[Ga2O4], индат Zn[In2O4], формиат Zn(HCOO)2, ацетаты Zn(CH3COO)2-
•2H2O, Zn4O(CH3COO)e, оксалаты ZnC2O4.2H2O, ZnC2O4.2C2H5NH2,
ZnC2O4-mNH3-raH2O, тартрат ZnC4H4Oe.
КАДМИЙ Cd
Z = 48; ат. вес — 112,40
Валентность II; заряд 2+
1 Массовые числа основных природных изото-
пов 114, 112, 111, 110, 113, 116, 106, 108
Электронная структура атома кадмия: К -L -М •4s24pe 4d10 -5s2.
Электронная структура атома кадмия и катиона »Cd2+ для
id- и 5а-орбиталей:
да1’
Ss*
1Г
4<f10
11 н 11 11 11
Cd2+
н 11 11
са
ИСТОРИЧЕСКАЯ СПРАВКА
Кадмий был открыт в 1817 г. Штромейером, который устано-
вил, что при действии H2S на некоторые образцы ZnO, растворен-
ные в НС1, осаждается желтый сульфид какого-то нового металла.
Название «кадмий» происходит от греческого слова kadmes —
название минерала, позднее переименованного в «каламин».
КАДМИЙ
803
РАСПРОСТРАНЕННОСТЬ В ПРИРОДЕ
В природе кадмий не встречается в свободном состоянии,
а только в виде соединений (сульфида, окиси, карбоната) в раз-
личных минералах, которые сопутствуют почти всем минералам
цинка и свинца. Содержание кадмия в земной коре равно 5,0»
• 10-4 вес.%.
К наиболее важным минералам кадмия относятся следующие.
Пршибрамит, ZnS,— разновидность цинковой обманки (сфа-
лерита); содержит примерно 5% Cd и встречается в виде корич-
нево-желтых кубических кристаллов с твердостью 3—4 по шкале
Мооса.
Эритроцинкит, ZnS, является разновидностью вюртцита,
содержащей кадмий; встречается в виде черно-коричневых гекса-
гональных кристаллов с твердостью 3,5—4 по шкале Мооса.
Гринокит, CdS, редкий минерал, он изоморфен вюртциту,
сопутствует цинковой обманке и вюртциту; представляет собой
оранжево-желтые гексагональные призматические кристаллы
с плотностью 4,9 г/сл3 и твердостью 3—3,5 по шкале Мооса.
В числе других минералов кадмия упоминаются монтепонит
CdO и отавит CdCO3.
Залежи минералов кадмия найдены на территории СССР, США,
Австралии, Бельгии, Чехословакии, Норвегии, Швейцарии, ФРГ„
ГДР и Боливии.
ИЗВЛЕЧЕНИЕ КАДМИЯ ИЗ РУД
ПОЛУЧЕНИЕ МЕТАЛЛИЧЕСКОГО КАДМИЯ
При прокаливании концентратов цинковой обманки (сфале-
рита) или вюртцита, которые содержат CdS, либо гринокита,
смитсонита, содержащих CdCO3, наряду с ZnO получается и CdO.
Последний, будучи очень летучим, оседает на наиболее удаленных
зонах конденсационных установок. При нагревании концентратов
цинковых руд, содержащих соединения кадмия (или смеси ZnO
и CdO), с NaCl и углем (коксом) при 964° отгоняется CdCl2.
Сырой металлический кадмий можно получить сухим путем,
т. е. восстановлением (при нагревании) CdO углеродом, окисью
углерода, водородом или металлическим магнием, а также вос-
становлением CdCO3 углем, CdCl2 или CdBr2 — водородом или
металлическим цинком, CdS или CdSO4 — водородом.
Сырой металлический кадмий получается также в результате
вытеснения из растворов его солей металлическим цинком. Чистый
металлический кадмий получают электролизом его солей.
Восстановление CdO (загрязненного ZnO) углем осущест-
вляется при 850—900° в шамотной реторте. При этой температуре
уголь восстанавливает лишь небольшое количество ZnO; образо-
51*
804
ПОБОЧНАЯ ПОДГРУППА II ГРУППЫ
вавшийся в очень небольшом количестве металлический цинк
перегоняется с трудом, поскольку процесс ведут при температуре
ниже его точки кипения.
При растворении CdO (загрязненного ZnO) в разб. H2SO4
образуется раствор сульфата кадмия, из которого металлический
кадмий выделяют порошкообразным цинком или электролитиче-
ским путем.
Для получения металлического кадмия катодным восстановле-
нием используют водный раствор CdSO4-nH2O (или раствор
CdCl2-2,5H2O с NaCl); катоды алюминиевые, аноды свинцовые,
ток плотностью 5—90 а! дм'.
ОЧИСТКА
Сырой металлический кадмий очищают физическим, химиче-
ским или электролитическим путем.
Физический метод очистки состоит в перегонке сырого кадмия
в присутствии угля в вакууме или в атмосфере водорода.
Для очистки химическим путем сырой кадмий растворяют
в разб. H2SO4 и через образовавшийся раствор барбатируют H2S,
чтобы осадить CdS; последний получается загрязненным неболь-
шими количествами CuS и ZnS. Растворенную в НС1 смесь суль-
фидов CdS, CuS, ZnS обрабатывают избытком (NH4)2CO3 для
осаждения CdCO3. При этом в растворе остаются соединения
меди и цинка. После промывания и высушивания CdCO3 превра-
щают в CdO прокаливанием. Окись кадмия восстанавливают
углем при 850—900° в шамотной реторте.
Для электролитической очистки сырой кадмий растворяют
в разб. H2SO4 и подвергают электролизу, используя свинцовые
аноды и алюминиевые катоды.
ФИЗИЧЕСКИЕ И ХИМИЧЕСКИЕ СВОЙСТВА
Кадмий — блестящий серебристо-белый металл (напоминаю-
щий олово) с плотной гексагональной структурой кристалличе-
ской решетки и плотностью 8,642 г/см3 (тяжелый металл),
с т. пл. 320,9° и т. кип. 778°; пары кадмия желтого цвета. Кадмий
токсичен, сублимируется в вакууме при 164°, диамагнитен, имеет
твердость 2 по шкале Мооса, мягок, очень ковок и тягуч в чистом
состоянии.
Сгибание кадмиевых стержней сопровождается особым звуком,
который напоминает так называемый «крик олова».
Металлический кадмий входит в состав многих сплавов наряду
с медью, оловом, серебром, висмутом и цинком. Диаграмма состоя-
ния системы Cd — Bi приведена в 1-м томе (стр. 513).
КАДМИЙ
805
С химической точки зрения металлический кадмий во многом
напоминает цинк; последний вытесняет кадмий из растворов его
солей.
При обычной температуре на воздухе поверхность металличе-
ского кадмия быстро становится матовой, покрываясь плотной
тонкой защитной пленкой окиси.
Металлический кадмий не взаимодействует с водой при обычной
температуре или температуре кипения, поскольку в воде поверх-
ность кадмия также покрывается тонкой защитной пленкой
Cd(OH)2. При высокой температуре кадмий энергично разлагает
пары воды с образованием CdO и водорода:
Cd + 2Н2О Cd(OH)2 + Н2
Металлический кадмий взаимодействует с галогенами, кисло-
родом, серой, селеном, теллуром, фосфором и мышьяком при
нагревании, образуя соответствующие соединения. Сильное нагре-
вание на воздухе или в кислороде сопровождается воспламенением
кадмия, который горит красновато-желтым пламенем, превра-
щаясь в окись CdO коричневого цвета.
Восстановительные свойства кадмия слабее, чем у цинка;
кадмий взаимодействует только с водородом и азотом.
В результате нагревания кадмия в токе SO2 получается смесь
CdSO4 и CdS.
Нормальный потенциал системы Cd/Cda+ равен —0,402 в при
25°, т. е. металлический кадмий в электрохимическом ряду распо-
ложен между железом и никелем; он медленно растворяется в раз-
бавленных кислотах (НС1, H2SO4) при нагревании с образованием
солей и выделением водорода.
В горячей разб. HNO3 кадмий растворяется:
3Cd + 8HNO3 = 3Cd(NO3)2 + 2NO + 4Н2О
При 350° кадмий вытесняет медь и железо из Cu2S и FeS.
С физиологической точки зрения металлический кадмий и его
растворимые соли токсичны.
При вдыхании паров или порошков металлического кадмия
органические кислоты организма образуют с кадмием очень ток-
сичные растворимые соли. Нельзя допускать контакта металли-
ческого кадмия с пищевыми продуктами. Отравление кадмием
сопровождается нарушением общего состояния организма, ане-
мией, различными симптомами со стороны дыхательной, нервной
и сосудистой систем, а также повреждением костей.
Учитывая токсичность кадмия и его соединений, необходимо
строго соблюдать правила техники безопасности при работе
с кадмием.
806
ПОБОЧНАЯ ПОДГРУППА II ГРУППЫ
ПРИМЕНЕНИЕ
Металлический кадмий служит для приготовления многих
сплавов, используется при пайке или сварке серебра и его сплавов,
для изготовления фотоячеек, чувствительных к ультрафиолетовым
лучам, для замещения железа в щелочных аккумуляторах типа
эдисоновских, для изготовления подшипников в автомобильной
промышленности и для покрытия (кадмирования) железа, стали
и сплавов на основе меди или алюминия.
Пары кадмия могут быть использованы для обогрева лабора-
торных бань, если необходимо поддерживать постоянную тем-
пературу.
Амальгаму кадмия применяют при изготовлении нормальных
элементов Вестона. Из сплавов Си — Cd (содержание Cd
0,9—1,2%) изготовляют проводники для систем, применяемых
в трамваях и троллейбусах. Сплавы Sn — Cd служат для сварки
электрических проводов. Сплавы Ag — Cd применяются в юве-
лирном деле.
Металлический кадмий входит также в состав легкоплавких
Сое- ди- не- ние Окись, гидро- окись. гидро- ксосое дипения Фтори- ды, фторосо- едине- ния Хлориды, хлоросоеди- нения Бромиды, бромосоедине- ния
Сое- дине- ния кад- мия (II) CdO коричне- во-желтая или коричнево- красная, ос- новного харак- тера Cd(OH)2 белая [Cd(NH3)4(OH)2 Mei[Cd(OH)4] MeH[Cd(OH)e] CdF2 белый Cd(OH)F CdF2- nCd(OH)2 MeilCdFs] CdCl2 бесцветный Cd[CdCl4] CdCl2-nH2O CdCl2-«NH3 CdCl2-B, 2B или 6B (где В — органическое основание) CdCl2- raCd(OH)2 Mei[CdCl3] Mei CdCl3]-nH2O Mei[CdCl4].nH2O Mel[CdCJ6]-raH2O rR3P. ,С1т лса\ LR3P'/ XC1J - Cl Cl Cl - \cd^ /Gd\ _R3P/ 4 Cl XPR3. [Cd(NH3)6]Cl2 CdBr2 бесцветный Cd[CdBr4] CdBr2-nH2O CdBr2-ziNH3 Cd(OH)Br CdBr2.Cd(OH)2-rcH2O Mei[CdBr3] Mel[CdBr3j.nH2O Mei[CdBr4] Meii[CdBr4] • raH2O Mei[CdBr6] rR3P /Br 1 лса\ ЬВэр/ 'Вг . - Br Br Br " \cd^ /Gd\ _R3PZ XBrZ ^PR3. ]
КАДМИЙ
807
и типографских сплавов; из легкоплавких сплавов делают, напри-
мер, предохранители для автоклавов.
Кадмий применяют также в урановых реакторах.
СОЕДИНЕНИЯ (ОБЩИЕ СВОЙСТВА)
Известны многочисленные устойчивые соединения электро-
положительного двухвалентного кадмия.
Наряду с простыми и двойными соединениями известны многие
координационные соединения кадмия, поскольку кадмий прояв-
ляет склонность к комплексообразованию. В комплексных соеди
нениях кадмия координационные числа могут быть 3, 4 и 6.
Известны координационные соединения кадмия типа ацидосолей,
гидроксосоединений амминов и хелатных соединений.
Хлорид, бромид, иодид и сульфат кадмия в водных раство-
рах образуют аутокомплексы Cd[CdCl4], Cd[CdBr4], Cd[CdI3]2)
Cd[CdI4l, Cd[Cd2(SO4)3l.
В табл. 77 приведены формулы и указан цвет некоторых соеди-
нений кадмия.
Таблица 77
Йодиды, иодосое- динения Сульфи- ды, тио- соли Сульфаты и кар- бонаты Хелатные соединения
Cdl2 бесцветный Cd[CdI3]2 Cd[CdI4] Cd(OH)I-H2O CdI2-mCd(OH)2- •raH2O CdI2-raCdO CdI2-raNH3 Mel[CdI3] Mei[CdI3]raH2O MeI[CdI4] Mei[CdI4]-nH2O Meli[CdI4]-nH2O -(НзС)23 Г ZCd\ _(H3C)2SZ XI. rR3P I-i >d) Lr3pz z iJ CdS жел- тый Cd2SO4S оранже- во-жел- тый Cd2SCl2 оранже- во-жел- тый 1 CdSO4 бесцветный CdSO4-nH2O CdSO4. • mCd(OH)2- raH2O CdSO4-z?NH3 MeiSOrCdSO4- •nH2O MeiSO4- • 2MeiiSO4. • CdSO4-nH2O Cd[Cd2(SO4)3] CdCO3 бесцветный raCdCO3-mCd(OH)2 г N-NH —CeH5, Cd ( — S—j - ' N = N —CeH5 4- красный Г /' zN€ S\ 1 cd z ( L желтый [Cd(CsH7O2)2], [CdEn3] X2 [Cd(C10H8N2)3] X2 [Cd(Ci2H3N2)3] X2 Mei[Cd(C2O4)2]-2H2O
808
ПОБОЧНАЯ ПОДГРУППА II ГРУППЫ
Большинство соединений кадмия бесцветно или белого цвета,
за исключением CdO (коричневый), CdS (желтый), Cd2SO4S
(оранжево-желтый), Cd2SCl2 (оранжево-желтый) и многочислен-
ных окрашенных хелатных соединений.
Соли кадмия и сильных кислот растворимы, имеют металли-
ческий вкус и очень токсичны, они не склонны к гидролизу.
Известно большое число основных солей кадмия переменного
состава, они плохо растворимы в воде. Помимо простых, двойных
и координационных соединений выделены металлоорганические
производные кадмия.
Многие из соединений кадмия проявляют сходство с соответ-
ствующими соединениями цинка.
Неорганические соединения
Гидрид кадмия, (CdH2)n, получают при температуре —70°
действием раствора Cdl2 или Cd(CH3)2 в тетрагидрофуране на эфир-
ный раствор LiAlH4:
Cdl2 + 2LiAlH4 =» CdH2 + 2A1H3 + 2LiI
Соединение (CdH2)n представляет собой белое твердое очень
неустойчивое вещество, которое разлагается на элементы
при —20°, медленно разлагается водой и взаимодействует с ВН3,
превращаясь в Cd(BH4)2.
Окись кадмия, CdO, встречается в природе в виде минерала
монтепонита. Она может быть получена путем сжигания металли-
ческого кадмия на воздухе или прокаливания Cd(OH)a, CdCO3,
Cd(NO3)2:
Cd + 1/2O2 = CdO + 57,0 ккал
Окись кадмия представляет собой коричнево-желтый аморф-
ный порошок с плотностью 6,95 з/слс3; она может быть выделена
также в виде коричнево-красных кубических кристаллов с кри-
сталлической решеткой типа NaCl и плотностью 8,18 г!смъ.
Окись кадмия тугоплавка (т. пл. 1426°), плохо растворима
в воде, имеет основной характер, растворяется в кислотах с обра-
зованием солей, превращается в CdCl2 под действием хлора при
сильном нагревании, образует CdS при нагревании с серой до 300°,
восстанавливается до металлического кадмия углеродом, магнием,
водородом и окисью углерода при нагревании. Может использо-
ваться в качестве катализатора.
CdO + 2НС1 = CdCl2 + Н2О CdO + 2HNO3 = Cd(NO3)2 + H2O
CdO + H2 Cd + H2O CdO + CO Cd + CO2
Известна смесь CdO -3CdO2 или Cd4O7.
КАДМИЙ
809
Гидроокись кадмия, Cd(OH)2, образуется в виде студнеобраз-
ного белого осадка, если растворы солей кадмия обработать
избытком щелочи (NaOH или КОН) при pH ;> 7,8 или подверг-
нуть электролизу раствор хлорида кадмия (анод кадмиевый,
катод платиновый).
Гидроокись кадмия может существовать либо в виде белого
геля, либо в виде белых гексагональных пластинчатых кристаллов,
которые имеют слоистую структуру, напоминающую структуру
Cdl2; плотность 4,79 г; см3. При прокаливании (300°) Cd(OH)2
образуется CdO.
Гидроокись кадмия является веществом амфотерного харак-
тера; кислотные свойства у нее выражены значительно слабее,
чем у Zn(OH)2; CdO плохо растворима в воде, при обычной тем-
пературе растворяется в кислотах, аммиаке и солях аммония,
а при нагревании — в концентрированных растворах гидроокис-
лов щелочных или щелочноземельных металлов.
Cd(OH)2 + 2НС1 = CdCl2 + 2Н2О Cd(OH)2 + 2NaOH = Na2[Cd(OH)4]
Cd(OH)2 + 4NH4OH = [Cd(NHs)4](OH)2 + 4H2O
Cd(OH)2 + 2Ba(OH)2 = Ba2[Cd(OH)e]
Cd(OH)2 -J- 4NH4C1 = [Cd(NH3)4]Cl2 + 2HC1 -|- 2H2O
При разбавлении водой или при кипячении соединения
[Cd(NH3)4](OH)2 вновь осаждается Cd(OH)2.
Гидроокись тетрамминкадмия, [Cd(NH3)4](OH)2, взаимо-
действует с H2S или с цианидами щелочных металлов по уравне-
ниям:
[Cd(NH3)4](OH)2 + H2S = CdS + 4NH3 + 2H2O
[Cd(NH3)4](OH)2 + 4KCN = K2[Cd(CN)4] + 2KOH + 4NH3
Известны гидроксосоединения^2Кл1(ОН)4], Na3[Cd(OH)5H2O]-
•H2O, Sr2[Cd(OH)e], Ba2[Cd(OH)eJ.
Известны также аддукты гидроокиси кадмия с перекисью
кадмия, например Cd(OH)2 -4CdO2, Cd(OH)2 -5CdO2 -2H2O,
3Cd(OH)2 -5CdO3 -2H2O.
Фторид кадмия, CdF2, получают упариванием раствора метал-
лического кадмия в растворе HF или обработкой при нагревании
металлического кадмия, CdO или CdCl2 в газообразном HF.
Cd F2 = CdF2 + 127,7 ккал
Фторид кадмия представляет собой белые кубические кри-
сталлы с плотностью 6,64 г!см3, т. пл. 1110° и т. кип. 1758°; он
плохо растворим в воде, растворяется в разб. HF и превращается
в CdO под действием паров воды при 600—800°.
810
ПОБОЧНАЯ ПОДГРУППА II ГРУППЫ
Известны фторосоли NH4[CdF3], 3Cd[BeF4] -8Н2О, Cd[BeF4] •
• (NH4)2[BeF4]-6Н2О и основные фториды Cd(OH)F, CdF2 •
• nCd(OH)2 (где п = 2, 4, 6, 9).
Хлорид кадмия, CdCl2, получают действием хлора на нагретые
до 586° гранулы металлического кадмия, нагреванием CdO в токе
газообразного НС], нагреванием кристаллогидратов CdCl2 -пН2О
(где п = 5, 4, 21/2, 2, 1) при 120° в атмосфере газообразного НС1.
Cd + С12 = CdCl2 + 93,24 ккал CdO + 2НС1 = CdCl2 + Н2О
Хлорид кадмия представляет собой бесцветные гексагональ-
ные кристаллы со слоистой ионной структурой; плотность
4,047 г!см3, т. пл. 568°, т. кип. 964°; CdCl2 очищают сублимацией
в атмосфере азота, соединение очень гигроскопично и легко
растворяется в воде и спирте. В водном растворе хлорид кадмия
образует аутокомплекс Cd[CdCl4].
Водород, свинец и цинк восстанавливают при нагревании
хлорид кадмия до металлического кадмия.
CdCl2 + Н2 Cd + 2НС1
CdCl2 + Pb 5* Cd + PbCl2 CdCl2 + Zn =₽*= Cd + ZnCl2
Известны кристаллогидраты CdCl2 -nH2O (где n = 5, 4, 21/2,
2, 1), основные хлориды CdCl2 -nCd(OH)2 (где n = 1, 2 и 4), хло-
росоли типа MeI[CdCl3] (где Me1 = К+, Rb+, Cs+), MeI[CdCl3] •
•nH2O (где Me1 = NH+), Me*'[CdCl4] (где Me1 = Na+, K+, Rb+,
Cs+), Me*[CdCl4] -nH2O (где Me1 = Na+), Me^tCdC^l-nH2O (где
Me11 = Mga+, Caa+, Sra+, Baa+), аммиакаты CdCl2-nNH3 (где
n — 10, 6, 4, 2), аддукты CdCl2 -B, CdCl2 -2B или CdCl2 -6B (где
В — молекула пиридина, а-пиколина, анилина, толуидина, фенил-
гидразина или пропилендиамина).
Известны координационные соединения:
R3As
R3As
где PR3 — третичный фосфин, AsR3 — третичный арсин.
Диаммиакатп. хлорида кадмия, CdCl2 -2NH3, имеет цепочечную
структуру, в которой атомы кадмия расположены в центрах
октаэдров:
NH3 NH3 NH3
С]/ t ^С]/I ^Cl^t ^Cl
NH3 NH3 NH3
КАДМИЙ
811
Хлорид гексамминкадмия, [Cd(NH3)e]Cl2, представляет собой
белый кристаллический порошок, плохо растворимый в воде;
при хранении на воздухе он превращается в CdCl2 -2NH3, а при
нагревании теряет весь аммиак.
Бромид кадмия, CdBr2, получают обработкой парами брома
нагретого докрасна металлического кадмия, а также нагреванием
CdBr2 -2Н2О до 200° и нагреванием CdCO3 с газообразным НВг.
Cd + Вг2 = CdBr2 + 75,2 ккал
Бромид кадмия представляет собой бесцветные кристаллы
со слоистой ионной структурой типа Cdl2; плотность 5,196 г/см3,
т. пл. 585°, т. кип. 863°. Бромид кадмия очищают сублимацией
в токе азота; он растворим в воде, спирте, эфире и ацетоне, вос-
станавливается при нагревании водородом или металлическим
цинком, оказывает каталитическое действие. В водном растворе
бромид кадмия образует аутокомплекс CdfCdBrJ.
Известен кристаллогидрат CdBr2 -4Н2О, бромиодид CdlBr
и основные бромиды кадмия Cd(OH)Br-nH2O, CdBr2 -Cd(OH)2-
• пН2О (где п = 1, 2, 6), бромосоли Мет[СйВг3](где Me1 = Na+,
К+, Rb+), Me4CdBr3]-пН2О (где Me1 = Na+, К+, NH+),
MeHCdBrJ (где Me1 = К+, NH+), Me11 [СйВг4]-пН2О(где Me11
= Ваа+), Me*[CdBe] (где Me® = К+, NH+), аммиакаты CdBr2 •
• nNH3 (где п = 6, 4, 3, 2) и аддукты бромида кадмия с пиридином,
а-пиколином, анилином, толуидином, ксилидином, фенилгидра-
зином, бромистоводородной кислотой.
Бромид кадмия образует с третичными фосфинами или арси-
нами координационные соединения, например:
Иодид кадмия, Cdl2, получают действием иода на металличе-
ский кадмий при нагревании или в присутствии воды, высушивани-
ем в атмосфере HI продукта, полученного выпариванием досуха
раствора металлического кадмия или CdCO3 в растворе HI, и упа-
риванием спиртового раствора Cdl2, полученного в результате
обработки спиртом упаренного досуха продукта взаимодействия
CdSO4-nH2O с KI.
Cd -|- I2 = Cdl2 -|- 48,8 ккал
Соединение Cdl2 представляет собой блестящие бесцветные
пластинчатые кристаллы с плотностью 5,56 г!смг, т. пл. 388°
и т. кип. 713°; Cdl2 растворяется в воде, спирте, эфире, почти
нерастворим в аммиаке.
В слоистой решетке Cdl2 (рис. 36) каждый ион Cda+ окружен
шестью ионами I- (которые расположены в плоскости с плотней-
812
ПОБОЧНАЯ ПОДГРУППА II ГРУППЫ
шей гексагональной упаковкой) и каждый ион I-—тремя ионами
Cd2+. Ионы кадмия расположены в параллельных плоскостях,
каждая из которых заключена между двумя плоскостями ионов
I-. Каждый слой решетки состоит из трех плоскостей ионов,
а именно: центральная плоскость ионов Cda+ и две наружные
плоскости ионов I Каждый слой представляет собой компактную
электронейтральную решетку. Связь между слоями осуществляет-
ся за счет вандерваальсовых
сил. Кристалл раскалывает-
ся вдоль плоскостей ионов
иода.
В водном растворе иодид
кадмия образует аутокомп-
лексы:
3CdI2 Cd[CdI3]2
2CdI2 Cd[CdI4]
При действии соляной
или бромистоводородной кис-
лоты на эфирные растворы
Cdl2 образуются соответст-
венно CdCl2 и CdBr2.
Известны основные иодиды
Cd(OH)I-H2O,CdI2-mCd(OH)2.
•nH2O, Cdl2 -nCdO, иодосоли Me1 [Cdl3] (где Me1 = Cs+, NH*),
Me4CdI3] -nH2O (где Me1 = Na+, K+, NHJ), MeJ[CdI4] (где
Me1 = K+, Cs+), Me^CdlJ-nH2O (где Me1 = Na+, K+),
Me11 [CdI4]-nH2O (где Me11 = Sr2+, Baa+), аммиакаты CdI2-nNH3
(где n = 6, 4, 2), аддукты иодида кадмия с пиридином, а-пиколи-
ном, анилином, толуидином, ксилидином и иодистоводородной
кислотой.
Растворение иодида кадмия в диметилсульфиде дает координа-
ционное соединение
'(H3C)2S г
\dz
(НзС)^^ ^1
При обработке растворов солей кадмия KI и N2H4 выпадает
в осадок соединение Cdl2-2N2H4.
Иодид кадмия образует с третичными фосфинами и арсинами
координационные соединения, аналогичные описанным для хло-
рида кадмия.
Цианид кадмия, Cd(CN)2, получают в виде белого осадка обра-
боткой растворов солей кадмия стехиометрически необходимым
количеством цианида щелочного металла:
CdSO4 + 2K.CN = Cd(CN)2 + K2SO4
КАДМИЙ
813
Цианид кадмия представляет собой белый порошок или бес-
цветные октаэдрические кристаллы с плотностью 2,23 г!см3',
он плохо растворим в воде и растворяется в метиламине или
в избытке цианидов щелочных металлов с образованием циано-
солей.
Cd(CN)2 + 2K.CN = K2[Cd(CN)4]
Известны также тетрацианокадматы Sr[Cd(CN)4] -ЗН2О,
Ba[Cd(CN)J-Н2О.
Тетрацианокадматы разлагаются сероводородом с образова-
нием сульфидов кадмия, например:
K2[Cd(CN)4] + H2S = CdS + 2KCN + 2HCN
Тиоцианат кадмия, Cd(SCN)2, получают обработкой Cd(OH)2
или CdCO3 тиоциановой кислотой HSCN или действием раствора
сульфата кадмия на раствор тиоцианата бария.
CdCO3 + 2HSCN = Cd(SCN)2 + СО2 + Н2О
CdSO4 + Ba(SCN)2 = Cd(SCN)2 -|- BaSO4
Соединение Cd(SCN)2 представляет собой блестящие бесцвет-
ные мелкие кристаллы, плохо растворимые в воде, растворимые
в растворах тиоцианатов щелочных металлов или аммония с обра-
зованием тетратиоцианатокадматов(П) Me*[Cd(SCN)4] -2Н2О (где
Me1 = К+, Rb+, NH+).
При действии тиоцианатов щелочных металлов или аммония
на растворы солей кадмия в присутствии пиридина осаждается
соединение [Cd(C5H5N)2](SCN)2 белого цвета, растворимое
в избытке пиридина.
CdSO4 + 2KSCN + 2C5H6N = [Cd(C5H5N)2](SCN)2 -|- K2SO4
Сульфид кадмия, CdS, встречается в природе в виде редкого
минерала гринокита. Он может быть получен в виде желтых гек-
сагональных кристаллов нагреванием металлического кадмия
с порошком серы либо пропусканием сероводорода над металли-
ческим кадмием или над CdO, CdCl2 при нагревании.
Cd + S = CdS + 34,36 ккал CdCl2 -|- H2S = CdS + 2НС1
Cd + H2S =₽£ CdS + H2 CdO -|- H2O =₽*= CdS + H2O
Обработка сероводородом растворов солей кадмия при
pH « 0,4 вызывает осаждение аморфного сульфида кадмия интен-
сивно-желтого цвета. Если через 0,6 н. раствор сульфата кадмия
барбатировать сероводород, образуется оранжево-желтый осадок,
состоящий из CdS и Cd2SO4S:
zCd—О /О
2CdSO4^H2S -> SZ SZ + H2SO4
XCd-OZ V)
814
ПОБОЧНАЯ ПОДГРУППА II ГРУППЫ
Если сероводород пропускают через 0,6 н. раствор хлорида
кадмия, образуется оранжево-желтый осадок CdS и Cd2SCl2:
zCd—Cl
2CdCl2 + H2S = Sz + 2HC1
^Cd-Cl
При барбатировании сероводорода через щелочные (аммиач-
ные) разбавленные растворы солей кадмия получают коллоидный
сульфид CdS золотисто-желтого цвета, флуоресцирующий и трудно
фильтруемый.
Сульфид кадмия плавится при 1750° в атмосфере водорода,
превращается в сульфат кадмия под действием влажного воздуха
или света и в CdO при прокаливании на воздухе; восстанавли-
вается до металлического кадмия парами воды при высокой
температуре, плохо растворим в аммиаке и в растворах солей
аммония, растворяется при нагревании в конц. НС1 и в разб. HNO3
и H2SO4, а также в растворах цианидов щелочных металлов:
CdS + Н2 Cd + H2S CdS + Н2О CdO + H2S
CdS + 4HC1 = H2[CdCl4] + H2S
3CdS + 8HNO3 = 3Cd(NO3)2 + 2NO + 3S + 4H2O
CdS + 4KCN = K2[Cd(CN)4] + K2S
Сульфид кадмия применяется в качестве желтого пигмента
при изготовлении масляных красок.
Известны двойные сульфиды 3CdS -Na2S, 3CdS -K2S.
Сульфат, кадмия, CdSO4, образуется путем дегидратации кри-
сталлогидратов CdSO4 -гаН2О (где п — 7, 6, 4, 6/2, 8/3) при нагре-
вании, нагреванием CdS в токе SO2 или металлического кадмия
в расплаве K2S2O8 или (NH4)2S2O8, нагреванием до 100° 2CdSO4-
• (NH4)2SO4 с конц. H2SO4, нагреванием (CH3)2SO4 с CdO, CdCO3,
Cd(NO3)2 или CdCl2.
Соединение CdSOt образует бесцветные орторомбические
призматические кристаллы с плотностью 4,72 г!см3', они люминес-
цируют при облучении катодными лучами, растворимы в воде,
плохо растворимы в СН3ОН, С2Н5ОН или С2Н5СН3СОО, пре-
вращаются в CdSO4 -CdO и затем в CdO при сильном нагревании.
При нагревании водород восстанавливает CdSO4 до CdS и затем
до металлического кадмия.
Известны кристаллогидраты CdSO4-raH2O (где га = 7, 6, 4,
5/2, 8/3, 1), основные сульфаты CdSO4-mCd(OH)2-гаН2О (где
т = 1, 2, 3, 7/2), аммиакаты CdSO4-raNH3 (где га = 6, 3, 2),
кристаллогидраты сульфата тетрамминкадмия [Cd(NH3)4]SO4 •
• гаН2О (где га = 4, 5/2, 2), двойные сульфаты Me*SO4-CdSO4 •
• гаН2О (где Me1 = Na+, К+, Rb+, Cs+, NHJ), Mez2SO4-2Men SO4-
•CdSO4-raH2O (где Me1 = Na+, K+ и Me11 = Ca2+), CdSO4-MgSO4-
КАДМИЙ
815
•пН2О и аддукты сульфата кадмия с пиридином, фенилгидрази-
ном, анилином, толуидином и ксилидином.
В водных растворах сульфат кадмия образует аутокомплексы:
3CdSO4 Cd[Cd2(SO4)3]
Упаривание водного раствора сульфата кадмия при обычной
температуре сопровождается выпадением клиноромбических приз-
матических кристаллов 3CdSO4 -8Н2О с плотностью 2,94—
3,09 г!см3.
При нагревании до 104° кристаллогидрата 3CdSO4 -ЭНзО
образуются мелкие бесцветные кристаллы CdSO4-H2O с плот-
ностью 3,786 г/сл3; при нагревании докрасна они разлагаются
с образованием CdO.
Сульфат кадмия, 3CdSO4 -8Н2О, применяют для получения
многих соединений кадмия, при изготовлении нормального эле-
мента Вестона, а также для получения фармацевтических пре-
паратов.
Нитрид кадмия, Cd3N2, получают нагреванием металлического
кадмия в атмосфере аммиака или амида кадмия Cd(NH2)2 в вакууме
при 180°. Это черный аморфный порошок с плотностью 6,85 г/с№;
во влажном воздухе он становится оранжевым и самопроизвольно
разлагается при соприкосновении с водой:
3Cd (NH2)2 Cd3N2 -I- 4NH3 Cd3N2 + 6H2O 3Cd(OH)2 + 2NH3
Амид кадмия, Cd(NH2)2, образуется при действии амида калия
KNH2 на родамид кадмия Cd(SCN)2 в жидком аммиаке и пред-
ставляет собой желтовато-белое твердое вещество с плотностью
3,09 г/см3, разлагающееся при нагревании или под действием воды.
Cd(SCN)2 -]- 2KNH2 = Cd(NH2)2 + 2K.SCN
Нитрат кадмия, Cd(NO3)2 -4Н2О, выпадает при концентриро-
вании (ниже 59,5°) раствора металлического кадмия, окиси, гидро-
окиси или карбоната кадмия в разбавленной азотной кислоте.
Соединение Cd(NO3)2-4Н2О представляет собой гигроскопич-
ные бесцветные игольчатые кристаллы с плотностью 2,45 г/см3,
т. пл. 59,5° и т. кип. 132°; оно легко растворяется в воде, спирте,
жидком аммиаке и этилацетате и плохо растворимо в пиридине.
При нагревании Cd(NO3)2-4Н2О ступенчато теряет кристалли-
зационную воду и превращается в безводную соль Cd(NO3)2, кото-
рая плавится при 360° и разлагается при высокой температуре.
Cd(NO3)2 — CdO + 2NO2 + !/2О2
816
ПОБОЧНАЯ ПОДГРУППА II ГРУППЫ
Известны также кристаллогидраты Cd(NO3)2 -тгН2О (где
п = 9 или 2), аммиакаты Cd(NO3)2 -nNH3 (где п = 6, 4) и основ-
ные нитраты Cd(NO3)2-7nCd(OH)2-тгН2О (где т = 1 или 3,
а п = 2 или 5), Cd(NO3)2 -7СйО и др.
Карбонат кадмия, CdCO3, встречается в природе в виде ред-
кого минерала отавита и обычно сопутствует смитсониту ZnCO3;
имеет вид белого порошка или белых кристаллов с плотностью
4,96 г!см\ плохо растворим в воде и органических растворителях,
растворяется в минеральных кислотах. Карбонат кадмия восста-
навливается до металлического кадмия углеродом при нагревании.
При обработке растворов солей кадмия карбонатами щелоч-
ных металлов образуются основные карбонаты кадмия, состав
которых определяется концентрацией реагирующих растворов
и температурой, при которой протекает реакция
(т ziJCdSOj -|- (m -|- ra)Na2CO3 + mH2O = reCdCO3-mCdfOH^ +
+ (m + zi)Na2SC>4 -]- ttiCC)2
Если 77i=l и п = 3, уравнение принимает следующий вид:
4CdSO4 -]- 4Na2CO3 -]- Н2О = Cd4(CO3)3(OH)2 -}- 4Na2SO4 -|- COj
Хелатные соединения
Соединения кадмия с дитизоном (pH ~ 12,2) образуют дитизо-
нат кадмия; последний устойчив в щелочной среде и окрашен
в красный цвет:
,NH — NH — CeH5
Cd2+ + 2S = C/ -*
XN = N- CeH5
//----7N-NH-C6H5\ -]
Cd— — S—(Г +2H+
\ XN = N —C6H5 / 2 J
При обработке солей кадмия в слабо кислой или щелоч-
ной среде о-хинолином образуется осадок оксината кадмия
Cd(C9HeON)2 -2Н2О лимонно-желтого цвета:
В числе органических реактивов, используемых для получения
хелатных соединений кадмия, можно назвать ацетилаце-
тон СН3 -СОСН2 -СО -СН3, этилендиамин H2N -СН2 -СЩ -NH2,
а,а'-дипиридил CioH8N2, дифенилкарбазон C8H5N = N«CO-NH-
КАДМИЙ
817
•NH-CgHj, о-фенантролин C12H8N2, ацетат бруцина, оксалаты
и тартраты щелочных металлов. Примеры некоторых хелатных
соединений кадмия с упомянутыми органическими реагентами:
ICd(C5H7O2)2], [CdEn3]X2, [Cd(C10H7N2)3]X2, [Cd(C12H8N2)3]X2,
MeI[Cd(C2O4)2]-2H2O (где Me1 = Na+, K+, NHJ).
Металлоорганические соединения
Известны алкильные Cd(CH3)2, Cd(C2H5)2, Cd(C3H7)2, Cd(C4H9)2,
Cd(ii30-C4H9)2 и арильное Cd(C6H5)2 производные кадмия.
Алкильные производные кадмия получают действием порошка
CdCl2 или CdBr2 на реактив Гриньяра в эфирном растворе или
порошка металлического кадмия на иодистый этил без доступа
воздуха.
CdBr2 + 2RMgBr -> CdR2 + 2MgBr2 (где В = СН3, С2Н6, С3Н7, С4Н9)
При электролизе раствора NaC2H5 в Zn(C2H5)2 на кадмиевом
катоде выделяется Cd(C2H5)2.
Алкильные производные кадмия — при комнатной темпера-
туре маслянистые жидкости, которые разлагаются на свету, выде-
ляя черный порошок металлического кадмия; на воздухе они
медленно окисляются, самопроизвольно возгораются при нагре-
вании на воздухе, превращаясь в CdO, взаимодействуют с много-
численными органическими соединениями, имеющими различные
функциональные группы.
CdR2 + 2R'COC1 -> CdCl2 + 2R — СО — R'
Дифенилкадмий, Cd(CeH5)2, получают при нагревании CdCl2
с фениллитием ЫС8Н5 или при нагревании опилок металлического
кадмия с дифенилртутью без доступа воздуха.
CdCl2 + 2LiC6H5 -н- Cd(CeH5)2 + 2LiCl Cd + Hg(C6H5)2 Cd(C6H5)2 + Hg
Соединение Cd(C6H5)2 представляет собой бесцветные кристал-
лы с т. пл. 174°; оно взаимодействует с HgCl2 и SbCl3 по уравне-
ниям:
Cd(CeH6)2 -}- HgCl2 = CdCI2 -f- Hg(C6H5)2
3Cd(CeH5)2 + 2SbCl3 = 3CdCl2 + 2Sb(C6H5)3
Помимо приведенных известны и другие соединения кадмия, например:
хлорит Cd(C102)2-2H20, хлорат Cd(C103)2-2Н2О, перхлорат Cd(C104)2, бро-
мат Cd(BrO3)2, аммиакат бромата Cd(BrO3)2 -4NH3. иодат Cd(IO3)2, аммиа-
кат йодата Cd(IO3)2-4Н2О, перйодаты Cd(IO4)2, CdHIO5, Cd3(IO5)2-6Н2О,
перманганат Cd(MnO4)2-7Н2О, дитионат CdS2O6-6H2O, тетратионат CdS4Oe,
тиосульфаты CdS2O3-2H2O, Na4[Cd(S2O3)3], сульфит CdSO3 или CdSO3-2H2O,
2CdSO3-3H2O, амидосульфонат Cd(NH2SO3)2-2Н2О, селенид CdSe, иодоселе-
нид CdI2-3CdSe, селениты CdSeO3, 3CdSeO3-H2SeO3, 2CdSeO3-H2SeO3i
CdSeO3-NH3, селенаты CdSeO4-2H2O, Me2SeO4-CdSeO4-6H2O, теллурид
52-0101
818
ПОБОЧНАЯ ПОДГРУППА II ГРУППЫ
CdTe, тиотеллурид TeS -3CdS, теллурит CdTeO3 или 3CdTeO3.2H2O, теллурат
CdTeOj, хроматы CdCrO4, K2CrO4•3CdCrO4-CdO, CdCrO4-6NH3, хромит-
Cd[Cr2O4], феррит Cd[Fe2O4], молибдат CdMoO4, вольфрамат CdWO4, боро-
12-вольфрамат Cd5[BW12O40]2-51H2O, азид Cd(N3)2, нитриты Cd(NO2)2,
Cd(NO2)2-H2O, Mei[Cd(NO2)3], Cd(NO2)2,-CdO, фосфиды Cd2P, Cd3P, CdP2,
гипофосфит Cd(H2PO2)2, Cd2H2P2Oe-3H2O, ортофосфаты Cd5H2(PO4)4-4H2O,.
CdH4(PO4)2-2H2O, апатиты [Cd1o(P04)6](OH)2,[Cd1o(P04)e]F2,[Cd5Ca5(P04)6]F2,
пирофосфат Cd2P2O7 или Cd2P2O7-21^0, метафосфат CdP2O6-3H2O, тиофос-
фат Cd3(PS4)2, арсенид Cd3As2, арсениты Cd3As2Oe, Cd2As2O4, арсе-
наты 2Cd3(AsO4)2-3H2O, Cd3(AsO4)2-2CdHAsO4.4H2O, CdHAsO4-H2Or
CdH4(AsO4)2-2H2O, CdAs20e, CdAs2O7, тиоарсенат Cd3(AsS4)2, антимонид
CdSb, антимонат CdSb2Oe,nH2O, тиоантимонат Cd3(SbS4)2, тиокарбонат
CdCS3, гексацианоферраты(П) Cd2[Fe(CN)e], K2Cd[Fe(CN)6], K4Cd4[Fe(CN)6]3,
гексацианоферраты(Ш) Cd3[Fe(CN)6]2.reH2O, KCd10[Fe(CN)6]7, тетратиоциа-
натомеркурат Cd[Hg(SCN)4], цианамид Cd(CN2)2, силикаты Cd2SiO4,
CdSiO3, гексафторосиликат Cd[SiFe]-6H2O, бораты Cd3(BO3)2, Cd(BO2)2,
ванадаты K4Cd4[V10O28)2.27H2O, формиаты Cd(HCOO)2, Cd(HCOO)2-2H2O,
ацетаты Cd(CH3COO)2 или Cd(CH3COO)2-иН20 (где я=1, 2, 3, 15),
Cd(CH3COO)2-яСН3СООН (где п = 0,5, 1, 1,5), оксалаты CdC2O4, CdC2O4 -
• яН20 (где п = 2, 3), CdC2O4-6(NH4)2C2O4.9H2O, CdC2O4.8(NH4)2C2O4 •
• 11Н2О, какодилат Cd{(CH3)2AsO}2.
РТУТЬ Hg
Z = 80; ат. вес = 200,59
Валентность II
Массовые числа основных изотопов 202, 200,
199, 201, 198, 204, 196
Электронная структура атома ртути: К -L-М-N-О-6s2.
Электронная структура атома ртути и катиона Hg2+ для bd-
ти 6«-орбиталей:
5d«
П
П Н U п
Hg
6s2
5d'a
Н Н
6s
ИСТОРИЧЕСКАЯ СПРАВКА
Ртуть является одним из первых известных человеку металлов.
Она отличается от других металлов агрегатным состоянием при
обычной температуре (жидкость), особым блеском и высокой плот-
ностью.
В египетских захоронениях, относящихся к XVI или XV вв.
до н. э., были найдены драгоценности из сплавов золота, серебра
и ртути. С конца VI в. до н. э. ртуть применяют для извлечения
золота или серебра из руд методом амальгамирования, а также-
для золочения серебра или меди.
В Древнем Риме применялась киноварь для получения пиг-
ментов и косметических препаратов.
РТУТЬ
819
Алхимики рассматривали ртуть как «spirit metalic» (т. е. носи-
тель металлических свойств); ее считали обязательным компонен-
том всех металлов. Алхимики открыли методы получения красной
окиси HgO, сулемы HgCl2 и основного сульфата ртути. Они же
предложили название «ртуть» в честь планеты Меркурий
(VI в. н. э.). Знак Hg для ртути был введен Берцелиусом.
РАСПРОСТРАНЕННОСТЬ В ПРИРОДЕ
В природе ртуть редко встречается в самородном виде, но
гораздо чаще в виде соединений (сульфида, селенида, теллурида,
окиси, хлорида и оксихлорида) в различных минералах. Содержа-
ние ртути в земной коре равно 5,0НО-6 вес. %.
В свободном состоянии ртуть встречается в верхних слоях
залежей киновари или иногда она сопутствует золоту, серебру
и палладию в виде амальгам; образуется при восстановлении
природных соединений ртути под действием восстановительных
газов или битуминозных пород.
К наиболее важным минералам ртути относятся следующие.
Киноварь, HgS, которая встречается в виде хрупких блестя-
щих красных тригональных кристаллов с плотностью 8,09—
8,20 г!см3 и твердостью 2—2,5 по шкале Мооса. Киновари часто
сопутствуют сульфиды железа, мышьяка, сурьмы, свинца или
цинка, селенид и теллурид ртути, сульфат кальция или бария,
кремнезем и известняк. Залежи киновари встречаются на глубине
не более 300—400 м.
Киноварь — основной источник получения ртути.
Метакиноварь, HgS, как правило, сопутствует киновари в при-
роде и представляет собой черные кубические кристаллы, которые
в отличие от кристаллов киновари хорошо проводят электриче-
ский ток.
К другим минералам ртути относятся тиманнит HgSe, колорадоит HgTe,
ливингстонит HgSb4S7, каломель Hg2Cl2, монтроидит HgO, эглестонит
Hg4OCl2, терлингуаит Hg2OCl.
Залежи минералов ртути найдены на территории Испании,
Италии, Югославии, СССР, Франции, Австрии, Чехословакии,
США, Канады, Мексики, Аргентины, Перу, Чили, Китая, Японии,
Австралии, Филиппинских островов.
В очень малых количествах соединения ртути встречаются
в некоторых термальных водах штатов Калифорния и Невада
(США), а также в морской воде.
Спектральным путем было установлено присутствие ртути
в газообразных и жидких продуктах извержения некоторых вул-
канов.
Обнаружена ртуть в некоторых метеоритах.
52®
820
ПОБОЧНАЯ ПОДГРУППА II ГРУППЫ
ПОЛУЧЕНИЕ
Ртуть извлекают из минералов или из концентратов минералов
(киноварь или ливингстонит). Для обогащения руды киновари
применяют ручную сортировку или селективную флотацию.
При прокаливании (600—700°) концентратов киновари в струе
воздуха образуются ртуть (пары) и двуокись серы:
HgS + О» Hg + SO2 + 60 ккал
Пары ртути конденсируются в трубках из нержавеющей стали
или монель-металла, и жидкая ртуть стекает в коллекторы. Прока-
ливание концентратов ртути осуществляют в шахтных, цилиндри-
ческих вертикальных или наклонных вращающихся печах.
Между печами для прокаливания и конденсационными труб-
ками помещают пылеуловительные камеры, температура которых
должна быть выше температуры конденсации ртути.
При температуре прокаливания киновари (350—400°) осуще-
ствляется сульфатирование ртути по уравнениям:
2HgS + ЗО2 = Hg2SO4 + SO2 HgS + 2О2 = HgSO4
Сульфаты Hg2SO4, HgSO4 разлагаются только при высокой
температуре.
В случае богатых руд, а также при получении небольших
количеств ртути, когда нет необходимости в больших печах,
ртуть получают нагреванием киновари (или концентратов кино-
вари) с известью или железными опилками при 600—700° в чугун-
ных ретортах.
4HgS + 4СаО = 4Hg -j- CaSO4 3CaS 4- 30 ккал
HgS Fe = Hg -|- FeS 4- 12 ккал
Если киноварь содержит Fe2O3, то ртуть может быть получена
и по уравнению
7HgS 4- 2Fe2O3 = 7Hg 4- 4FeS 4- 3SO2
Обычно применяют батарею из 12 реторт, которые помещают
в слегка наклоненном виде, в нагретые до 600—700° печи.
В случае очень богатых киноварью руд или руд, содержащих
много сульфидов (пириты, марказиты и стибниты), над поверх-
ностью содержимого реторт пропускают слабый ток воздуха,
чтобы избежать возможности конденсации киновари в более
холодных частях установки.
Гидрометаллургический процесс переработки киновари (или
концентратов киновари) не имеет промышленного применения.
Киноварь растворяют в щелочном растворе сульфида натрия
и затем отделяют ртуть с помощью алюминиевого порошка или
электролитическим путем.
HgS 4- Na2S -► Na2[HgS2]
3Na2[HgS2] 4- 8NaOH 4- 2A1 4- 4H2O - 3Hg 4- 2Na{Al(OH)4(H2O)2J4-6Na2S
РТУТЬ
821
Для электролиза можно использовать растворы киновари
в азотной кислоте.
В лаборатории металлическую ртуть можно получить восста-
новлением диртутных или ртутных соединений хлоридом олова(П)
в солянокислой среде, сульфатом железа(П), двуокисью серы,
цинком, алюминием, магнием и другими металлами, химически
более активными, чем ртуть.
ОЧИСТКА
Сырая ртуть содержит в качестве примесей Pd, Zn, Cd, Sn,
Bi, Cu, Ag, Au, Mg, Al, Fe, Co, Ni, Cr, Mo, W, Мп и TI.
Очистку осуществляют физическими способами — фильтро-
ванием или перегонкой, а также химическим или электрохими-
ческим путем.
Ртуть фильтруют через фильтровальную бумагу, наколотую
иголкой, замшу, сукно или через крупнозернистые тигли для
фильтрования.
Перегонка при пониженном давлении с введением небольшого
количества воздуха через капилляр в сырую ртуть позволяет
получить чистую ртуть, поскольку при этом сочетается эффект
окисления воздухом с эффектом перегонки, что обеспечивает
максимальное удаление примесей. Для перегонки ртути при
пониженном давлении в автоматических аппаратах используют
ртуть, предварительно очищенную фильтрованием или химиче-
ским путем.
Химическая очистка ртути заключается в барбатировании
воздуха через загрязненную ртуть (12—16 час), налитую в сосуд
с 10%-ным раствором HNO3, и окислении примесей сернокислым
раствором К2Сг2О7 либо щелочным (или сернокислым) раствором
КМпО4. После обработки указанными реактивами ртуть промы-
вают дистиллированной водой и перегоняют. Спектрально чистую
ртуть получают электролизом растворов солей ртути, используя
анод из загрязненной ртути и катод из платины при контролируе-
мом напряжении.
ФИЗИЧЕСКИЕ И ХИМИЧЕСКИЕ СВОЙСТВА
Ртуть представляет собой блестящий серебристо-белый металл
с ромбоэдрической решеткой; плотность ее 13,595 г/см3 (тяжелый
металл), температура затвердевания —38,87° (ртуть — единствен-
ный жидкий металл при обычной температуре), т. кип. 356,95°,
твердость 1,5 по шкале Мооса (в твердом состоянии).
Электропроводность ртути достаточно мала. В качестве едини-
цы электросопротивления (1 ом) было принято сопротивление
ртутного столба длиной 1,063 м и сечением 1 м,м3 при 0°.
822
ПОБОЧНАЯ ПОДГРУППА II ГРУППЫ
Существуют методики приготовления коллоидных растворов
ртути в жирах, дисперсной ртути (с диаметром частиц 80—300 ммк)
и порошка металлической ртути.
Под действием электрических разрядов пары ртути начинают
светиться, причем в излучении значительная доля ультрафиоле-
товых лучей. Благодаря этому свойству кварцевые лампы, запол-
ненные парами ртути, служат источником ультрафиолетового
излучения.
Ртуть образует сплавы со многими металлами; сплавы ртути
получили название амальгам. К металлам, образующим амаль-
гамы, относятся Na, К, Ag, Au, Zn, Cd, Sn, Pb, Си. В зависимости
от содержания ртути амальгамы бывают жидкие, твердые и пасто-
образные. Амальгамы получают простым введением металла (кусоч-
ков или порошка) в ртуть или растиранием металла со ртутью
в фарфоровой ступке. Амальгамы являются сплавами типа
твердых растворов или интерметаллических соединений.
Известны следующие интерметаллические соединения ртути
с натрием: NaHg4, NaHg2, Na7Hg8, NaHg, Na3Hg2, Na3Hg.
При электролизе водных растворов солей аммония с ртутным
катодом или при перемешивании амальгамы натрия с раствором
соли аммония образуется мало устойчивая амальгама аммония.
С химической точки зрения ртуть — наименее активный
металл из подгруппы цинка.
Под действием электрических разрядов на смесь Не, Ne, Аг,
Кг или Хе с парами ртути образуются соединения HgHe, HgNe,
HgAr, HgKr, HgXe, в которых ртуть связана с указанными газами
вандерваальсовыми силами.
При обычной температуре сухой воздух и кислород не дей-
ствуют на ртуть, но в присутствии следов воды поверхность ртути
становится матовой и покрывается серым слоем окиси. При нагре-
вании ртути на воздухе до 350° образуется красная окись HgO,
которая при 500° термически разлагается на элементы:
350°
Hg-|-1/2O2 ( HgO -|- 21,5 ккал
500°
Металлическая ртуть взаимодействует с галогенами при обыч-
ной температуре, образуя сначала галогениды Hg2X2, а затем
галогениды HgX2.
Озон легко окисляет ртуть до Hg2O — соединение черного
цвета.
При растирании ртути с порошкообразной серой, селеном или
теллуром в фарфоровом тигле образуются соединения HgS, HgSe,
HgTe черного цвета. Ртуть взаимодействует с серой при высокой
температуре, образуя красный сульфид ртути HgS.
Азот, фосфор, углерод, кремний и бор непосредственно
не взаимодействуют со ртутью.
РТУТЬ
823
Хлористый водород в присутствии влаги медленно реагирует
со ртутью при обычной температуре, образуя Hg2Cl2.
При обычной температуре ртуть вступает в реакцию с газо-
образными НВг или HI, а ртуть при нагревании — с хлористым
иодом, хлористым сульфурилом, хлористым тионилом и четырех-
хлористым углеродом:
2Hg + 2ICI = HgCl2 + Hgl2
2Hg + SO2C12 = Hg2Cl2 + SO2 3Hg+4SOC12 = 3HgCl2 + 2SO2 + S2C12
Hg + SO2C12 = HgCl2 + SO2 4Hg + CC14 = 2Hg2Cl2 + C
Ртуть растворяется в конц. H2SO4, в разб. или конц. HNO3
и в царской водке при нагревании:
Hg + 2H2SO4 = HgSO4 + SO2 + 2H2O
3Hg + 8HNO3 = 3Hg(NO3)2 + 2NO + 4H2O
3Hg + 6HCi + 2HNO3 = 3HgCl2 + 2NO + 4H2O
6Hg + 8HNO3 = 3Hg2(NO3)2 + 2NO + 4H2O
Ртуть легко восстанавливает хлорид и перхлорат железа(III)
в растворах:
Fe2Cl6 -f- 2Hg = Hg2Cl2 2FeCl2
2Fe(C104)3 + 2Hg = Hg2(C104)2 + 2Fe(C104)2
G физиологической точки зрения ртуть и ее растворимые
соединения токсичны. Помня о большой токсичности паров ртути,
образующихся даже при комнатной температуре, необходимо
соблюдать все меры предосторожности при работе с этим металлом.
ПРИМЕНЕНИЕ
Широкое применение ртути в самых различных областях
основано на таких свойствах ртути, как малая упругость пара
при обычной температуре и большая упругость пара при высокой
температуре, большой удельный вес, малая удельная теплоем-
кость, низкая тепло- и электропроводность.
Ртуть применяют во многих физических и физико-химических
приборах, например, в термометрах, барометрах, манометрах,
денситометрах, ареометрах, капиллярных электрометрах, гиро-
скопах для авиации, в элементах Вестона и др. Ртуть исполь-
зуется также в установках для собирания газов, в поляро-
графических установках и для изготовления специальных ламп.
В электрических устройствах и приборах применяются ртутные
переключатели, прерыватели, предохранители и выпрямители
электрического тока.
В многочисленных типах ячеек для электролиза (например,
ячейки для получения гидроокисей щелочных металлов) ртуть
•служит катодом.
824
ПОБОЧНАЯ ПОДГРУППА II ГРУППЫ
Ртуть используют в металлургии для извлечения золота и
серебра из руд методом амальгамирования.
Ртуть применяют также для получения амальгам натрия,
цинка, висмута, алюминия, свинца, олова, кадмия и других,
металлов, имеющих разнообразное промышленное применение.
В области органического синтеза ртуть часто служит катали-
затором.
В плутониевых реакторах жидкая ртуть используется в каче-
стве хладоагента.
СОЕДИНЕНИЯ (ОБЩИЕ СВОЙСТВА)
Известно большое число соединений, в которых ртуть может
быть в виде катиона (Hg — Hg)2+ или Hg2+, а также катиона Hg2+.
Соединения, содержащие катион (Hg — Hg)2+, называют
диртутными, а содержащие катион Hg2+ — ртутными. Между
обоими катионами и металлической ртутью существует равновесие:
(Hg - Hg)2+ ч* Hg2+ + Hg,
которое при обычной температуре и в водных растворах смещено
влево, а при нагревании или под действием света и химических
реактивов, образующих с катионом Hg2+ трудно растворимые
и слабо диссоциированные соединения, смещено вправо.
В табл. 78 приведены формулы и указан цвет некоторых ди-
ртутных и ртутных соединений.
Помимо диртутных и ртутных известно несколько металло-
органических соединений ртути.
Диртутные соединения (соединения одновалентной ртути)
Диртутные соединения содержат катион (Hg — Hg)2+ или
Hg|+, в которых атомы ртути соединены между собой ковалент-
ными связями.
Наличие катиона (Hg — Hg)2+ было доказано различными
физико-химическими методами во многих диртутных соединениях
в твердом, газообразном и растворенном (в воде) состояниях.
Диртутные соединения могут быть получены из металлической
ртути или ее соединений.
Hg2+ + Hg = (Hg - Hg)2+
В водном растворе катион (Hg — Hg)2+ устойчив при pH < 2,5
и в присутствии металлической ртути.
Диртутные соединения имеют склонность к полимеризации
и комплексообразованию, а также к образованию соединений,
плохо растворимых в воде, и окрашенных соединений.
РТУТЬ
825-
Примеры реакций, в которых диртутные соединения разла-
гаются под действием тепла, света или химических реактивов:
Hg2I2 Hgl2 + Hg Hg2O HgO + Hg
Hg2(CN)2 Hg(CN)2 + Hg Hg2S HgS + Hg
Hg2Cl2 + 2NH4OH HgNH2Cl + Hg + NH4C1 + 2H2O
Цинк, алюминий, магний, железо, медь и другие металлы,
химически более активные, чем ртуть, вытесняют ртуть из ди-
ртутных соединений:
(Hg - Hg)2+ + Zn = 2Hg + Zn2+ (Hg — Hg)2+ + Cu = 2Hg + Cu2+
Под действием таких восстановителей, как дихлорид олова
в солянокислом растворе, сульфат железа(П) и двуокись серы,
соединения диртути восстанавливаются до элементной ртути.
Из растворимых в воде солей диртути можно назвать нитрат,
хлорат и перхлорат, а в качестве трудно растворимых — хлорид,,
бромид, иодид, сульфат, карбонат и все основные соли.
Окись диртути, Hg2O, образуется (в загрязненном состоя-
нии) при действии щелочей на диртутные соли или сухого воздуха
(кислорода) на амальгаму калия при обычной температуре.
Hg2(NO3)2 + 2NaOH = Hg2O + 2NaNO3 + H2O
Окись диртути представляет собой оливково-зеленый порошок
с плотностью 9,8 г/см3. Соединение неустойчиво и легко разла-
гается под действием света или тепла на HgO и металлическую
ртуть.
Фторид диртути, (Hg2F2)n, получают обработкой ртути фто-
ром на холоду, действием 40%-ного раствора HF на Hg2CO3
или обработкой Hg2Cl2 фторидом серебра.
Полимер фторида диртути представляет собой желтые тетраго-
нальные мелкие кристаллы с плотностью 9,93 г/см3; они сублими-
руются при 383° и нормальном давлении, плавятся при 570°,
растворяются в воде. Растворение фторида диртути в воде сопро-
вождается гидролитическим разложением, которое ускоряется
на свету:
Hg2F2 + НОН HgO + Hg + 2HF
Газообразный аммиак или аммиак в водном растворе взаимо-
действует с (Hg2F2)n по уравнению
Hg2F2 + 2NH3 ** HgNH2F + Hg + NH4F
Хлорид диртути, (Hg2Cl2)n, встречается в природе в виде
минерала каломели. Он может быть получен действием хлора
на избыток ртути, растиранием смеси из 3 ч. ртути и 4 ч. HgCl^
с небольшим количеством воды (или смеси HgSO4, NaCl, Hg),
обработкой раствора Hg2(NO3)2 разб. HG1 или хлоридом щелоч-
826
ПОБОЧНАЯ ПОДГРУППА II ГРУППЫ
ного металла, восстановлением хлорида ртути солянокислым
раствором хлорида олова(П) или водным раствором двуокиси серы.
2Hg + С12 = HgCl2 + 63,32 ккал HgCl2 + Hg Hg2Cl2
HgSO4 + 2NaCl + Hg = Hg2Cl2 + Na2SO4
Hg2(NO3)2 + 2NaCl = Hg2Cl2 + 2NaNO3
2HgCl2 + H2[SnCl4] = Hg2Cl2 + H2[SnCl6]
2HgCl2 + SO2 + 2H2O = Hg2Cl2 + H2SO4 + 2HC1
Из растертой смеси HgSO4 + Hg -|- 2NaGl (или HgCl2 +
+ Hg + H2O) каломель (Hg2Cl2)n выделяют сублимацией при
500°. Полимер хлорида диртути, полученный в растворе, промы-
вают дистиллированной водой, фильтруют с помощью водоструй-
ного насоса и сушат при 105° в темноте.
Соединение (Hg2Cl2)n представляет собой белые игольчатые
микрокристаллы, которые быстро желтеют при нагревании или
становятся серыми, разлагаясь на Hg и HgCl2. Плотность кри-
сталлов 7,15 г!см5, твердость 1—2 по шкале Мооса, они сублими-
руются при 383° и 760 мм рт. ст., плавятся при 543° (темпера-
тура определена экстраполированием), плохо растворяются
в воде.
Определением молекулярного веса методами радиокристалло-
графии, магнетохимии и криоскопии было доказано, что формула
каломели соответствует Hg2Cl2. По данным рентгеноструктурного
анализа установлено, что тетрагональная решетка каломели
состоит из рядов линейных молекул G1 — Hg — Hg — Cl.
Соединение (Hg2Cl2)n растворяется при нагревании
в конц. HNO3, царской водке, хлорной и бромной воде и в кон-
центрированных растворах хлоридов, бромидов или иодидов
щелочных металлов.
3Hg2Cl2 -j- 8HNO3 = 3HgCl2 4- 3Hg(NO3)2 -р 2NO 4- 4Н2О
3Hg2Cl2 4- 6НС1 4- 2HNO3 = 6HgCl2 4- 2NO 4- 4H2O
Hg2Cl2 4- Cl2 = 2HgC]2 Hg2Cl2 -j- Br2 = HgCl2 4* HgBr2
Hg2Cl2 4- 2KC1 = K2[HgCl4] -p Hg
Hg2Cl2 4- 4NaBr = Na2[HgBr4] 4- Hg 4- 2NaCl
При нагревании сера реагирует с каломелью, давая HgCl2
и HgS.
Двуокись серы в слабо кислой среде восстанавливает хлорид
диртути:
Hg2Cl2 4- SO2 4- 2Н2О 2Hg 4- H2SO4 4- 2НС1
Щелочи взаимодействуют с хлоридом диртути по уравнению
Hg2C]2 4- 2MerOH = Hg 4- HgO 4- гМе^! Н2О
РТУТЬ
827
При действии водного раствора аммиака на хлорид диртути
образуется черный плохо растворимый продукт, состоящий из хло-
С1
рида амидортути Hg^ белого цвета и тонко измельченной
ХМ12
металлической ртути черного цвета:
Hg2Cl2 + 2NH4OH HgNH2Cl + Hg + NH4C1 + 2H2O
Эта реакция в прошлом служила для определения хлорида
диртути, названного «каломелью» по греческому названию calos —
melos, что означает «черный красивый».
Бромид диртути, Hg2Br2, образуется в результате обработки
разбавленных растворов Hg2(NO3)2 растворами бромидов щелоч-
ных или щелочноземельных металлов, действия бромной воды
на избыток ртути (на холоду), действия брома на слабо азотно-
кислый раствор Hg2(NO3)2, нагревания HgBr2 со стехиометри-
чески необходимым количеством ртути:
2Hg + Вг2 = Hg2Br2 + 49,42 ккал Hg2(NO3)2 + 2КВг = Hg2Br2 + 2KNO3
Соединение Hg2Br2 представляет собой бесцветные тетраго-
нальные кристаллы (плотность 7,71 г/см3), которые желтеют при
нагревании. Бромид диртути плохо растворим в воде, спирте,
эфире, ацетоне, метил- и этилацетате; растворяется при нагре-
вании в концентрированных кислотах (HNO3, H2SO4) с образо-
ванием солей ртути.
Реакция между аммиаком и Hg2Br2 идет по уравнению
Hg2Br2 + 2NH4OH HgNH2Br + Hg + NH4Br + 2H2O
Известен двойной бромид Hg2Br2-А^Бгв-
Иодид диртути, Hg2I2, получают в результате взаимодействия
растворов иодида щелочного металла и Hg2(NO3)2, при действии
ртути на Hgl2 или иодид, смоченный спиртом, путем смешивания
(Hg2Cl2)n с иодом в присутствии небольшого количества воды,
действием амальгамы свинца на иод, растворенный в органиче-
ском растворителе, обработкой металлической ртути метилиоди-
дом или этилиодидом:
2Hg + I2 = Hg?I2 + 28,91 ккал Hg2(NO»)2 + 2KI = Hg2I2 + 2KNO3
Иодид диртути выделяется в виде диамагнитных сильно дву-
преломляющих тетрагональных кристаллов, окрашенных в цвет
от желтого до зеленого; плотность их 7,75 г!см3. Под действием
ультрафиолетовых лучей кристаллы флуоресцируют оранжевым
цветом, они плохо растворимы в воде, спирте, эфире, хлороформе
и сероуглероде, растворяются при нагревании в концентриро-
ванных кислотах (HNO3, H2SO4) с образованием солей ртути,
разлагаются при нагревании или под действием света по
828
ПОБОЧНАЯ ПОДГРУППА II ГРУППЫ
уравнению
Hg2I2 Hgl2 + Hg
Соединение Hg2I2 взаимодействует с нитратом ртути, аммиаком
и иодидом калия, взятых в избытке:
Hg2I2 + Hg(NO3)2 Hg2(NO3)2 + Hgl2
Hg2I2 + 2NH4OH HgNH2I + Hg + NH4I + 2H2O
Hg2I2 + 2KI = K2[HgI4] + Hg
Сульфид диртути, Hg2S, получают обработкой сероводоро-
дом Hg2(CH3COO)2 или смесью H2S + СО2 хлорида диртути
(Hg2Cl2)n при 0°; Hg2S представляет собой коричневый порошок,
плохо растворимый в воде, разлагающийся выше 0° на HgS и метал-
лическую ртуть.
Сульфат диртути, Hg2SO4, получают действием конц. H2SO4
на избыток металлической ртути при нагревании, обработкой
растворов Hg2(NO3)2 разб. H2SO4 или раствором сульфата щелоч-
ного металла, электролизом 5%-ного раствора H2SO4 с ртутным
анодом, действием диметилсульфата (CH3)2SO4 на соединения
диртути:
2Hg + 2H2SO4 = Hg2SO4 + SO2 + 2H2O
Hg2(NO3)2 + H2SO4 = Hg2SO4 + 2HNO3
Сульфат диртути представляет собой диамагнитный желтовато-
белый кристаллический порошок, кристаллы которого относятся
к моноклинной системе, имеют плотность 7,56 г!см3, плохо раство-
римы в воде и разлагаются при нагревании на HgSO4 и ртуть.
Известны кристаллогидрат Hg2SO4-2H2O, кислая соль
Hg2SO4.H2SO4, основная соль Hg2SO4-Hg2O-Н2О и двойная
соль (NH4)2SO4-Hg2SO4-H2O.
Нитрат диртути, Hg2(NO3)2-2Н2О, выпадает из раствора,
полученного действием HNO3 (плотность 1,39) на избыток ртути
при комнатной температуре или действием металлической ртути
на раствор Hg(NO3)2 или AgNO3.
6Hg + 8HNO3 = 3Hg2(NO3)2 + 2NO + 4H2O
Hg(NO3)2 + Hg — Hg2(NO3)2 2AgNO3 + 4Hg = Hg2(NO3)2 + 2AgHg
Соединение Hg2(NO3)2-2H2O образует диамагнитные бесцвет-
ные орторомбические кристаллы с плотностью 4,785 г/см3, которые
плавятся при 70° с разложением, растворимы в холодной воде,
жидком NH3 и сероуглероде. Нитрат диртути растворяется в воде
с образованием основных солей вследствие гидролиза:
Hg2(NO3)2 + НОН Hg,(OH)NO3 + HNO3
Чтобы избежать образования основных солей, нитрат диртути
растворяют в воде, подкисленной HNO3.
РТУТЬ
829
При действии аммиака на нитрат диртути получается нитрат
окиси амидодиртути Hg2ONH2NO3:
/Hg\
2Hg2(NO3)2+4NH4OH^=O/ xNH2NO3 + 2Hg + 3NH4NO3 + 3H2O
XHg/
Соединение
/Hg
О7 'NH2NO3
\Hg/
является производным ангидрооснования Миллона, которое
имеет формулу
/Hg\
о7 4nh2oh
xHgz
Нитрит натрия ваимодействует с нитратом диртути:
Hg2(NO3)2 + 2NaNO2 = Hg(NO2)2 + Hg + 2NaNO3
Известны двойные нитраты Me11 (NO3)2-Hg2(NO3)2 (где
Me11 = Sr2+, Ba2+, РЬ2+) и основные соли 3Hg2(NO3)2-2Hg2O
(2 или 3) Н2О, Hg2(NO3)2-Hg2O-H2O, Hg2(NO3)2-Hg2O-Н2О.
Основные нитраты ртути применяются для приготовления
косметических препаратов.
Карбонат диртути, Hg2CO3, образуется при действии основ-
ных карбонатов щелочных металлов на растворы нитрата диртути
на холоду; это белый порошок с плотностью 5,07 г!смъ, плохо
растворимый в воде, разлагающийся теплой водой, чувствитель-
ный к действию света.
Нагревание Hg2CO3 до 130° вызывает его разложение:
Hg2CO3 HgO + Hg + СО2
Помимо приведенных известны и другие соединения диртути, например:
хлорит Hg2(C102)2, хлорат Hg2(C103)2, перхлорат Hg2(C104)2, бромат
Hg2(BrO3)2, иодат Hg2(IO3)2, перйодаты Hg2(IO4)2, Hg2(IO4)2-2Hg2O,
(Hg2)sI2O12, селенид Hg2Se, тиосульфат 3Hg2S2O3-5CuS2O3, дитионат Hg2S2Oc,
сульфит,;£^28О3, селениты znHg2SeO3-Hg2O-геН2О, 3Hg2SeO3-SeO2, селенаты
Hg2SeO4, 5Hg2SeO4-Hg2O, теллурат Hg2(HTeO4)2-6H2O, азид Hg2(N3)2,
нитрид (Hg2)3N2, нитрит Hg2(NO2)2, фосфид (Hg2)3P2, ортофосфат
(Hg2)3(PO4)2, метафосфат Hg2(PO3)2, пирофосфат (Hg2)2P2O7-Н2О, арсенид
(Hg2)3As2, ортоарсенит (Hg2)3(AsO3)2, метаарсенит Hg2(AsO2)2, ортоарсе-
наты (Hg2)3(AsO4)2, Hg2AgAsO4, метаарсенат Hg2(AsO3)2, ортоантимонат
Hg2HSbO4-7H2O, ортотио антимон ат (Hg2)3(SbS4)2, карбид Hg2C2-H2O,
цианид Hg2(CN)2, аммиакат цианида Hg2(CN)2-4NH3, цианат Hg2(CNO)2,
тиоцианат Hg2(CNS)2, селеноцианат Hg2(SeCN)2.
Ртутные соединения (соединения двухвалентной ртути)
Известно большое число соединений двухвалентной ртути Hg2+,
называемых ртутными соединениями, в отличие от соединений
одновалентной ртути — диртутных. Ртутные соединения полу-
830
ПОБОЧНАЯ ПОДГРУППА II ГРУППЫ
чают окислением ртути, разложением соединений диртути или
обработкой галогенами диртутных соединений.
У ртутных соединений ярко выражена способность к ком-
плексообразованию и гидролизу. Хлорид и цианид ртути из-за
склонности к образованию аутокомплексов очень слабо ионизи-
рованы в воде, хотя и растворяются в ней. Легкость гидролиза
обусловливает большое число основных соединений ртути; они
плохо растворимы в воде, окрашены в различные цвета — от жел-
того до коричневого.
Известны координационные соединения ртути, относящиеся
к классам ацидосолей, амминов и хелатных соединений. Коорди-
национное число ртути может быть 2, 3, 4 и 6.
При действии восстановителей на соединения ртути образуются
соединения диртути, а затем элементарная ртуть.
Гидрид ртути, HgH2, получают по уравнению
Hgl2 + 2LiAlH4 = HgH2 + (А1Н3)г +[2LiI
Как Hgl2, так и LiAlH4 растворяют в смеси этилового эфира,
тетрагидрофурана и бензина, затем растворы, охлажденные
жидким воздухом, смешиваются. Температура постепенно повы-
шается до —125°, при этом образуется белый объемистый осадок
HgH2, который легко разлагается на элементы при —90°.
Окись ртути, HgO, встречается в природе в виде довольна
редкого минерала монтроидита. Ее получают окислением ртути
при 350° на воздухе (или в кислороде) в присутствии катализа-
торов — платины или окиси NiO, термическим разложением
Hg(NO3)2, электролизом 6%-ного раствора NaOH (с ртутным
анодом и железным катодом), обработкой щелочами растворов
солей ртути, гидролизом нитрата ртути большим избытком горя-
чей воды.
350°
Hg+V2O2 HgO + 21,68 ккал Hg(NO3)2=HgO + 2NO2+1/2O2
500°
HgCl2 + 2KOH = HgO + 2KC1 + H2O
Hg(NO3)2 + H2O HgO + 2HNO3
При обработке растворов солей ртути небольшими количе-
ствами щелочей образуются основные соли ртути, мало раствори-
мые в воде и окрашенные в коричнево-красный цвет:
2HgCl2 + 2КОН = Hg2Cl2O + 2КС1 + Н2О
3HgCl2 + 4КОН = Hg3Cl2O2 + 4КС1 + 2Н2О
Кипячение растворов солей диртути с карбонатом натрия
приводит к образованию окиси ртути:
Hg2(NO3)2 + Na2CO3 = HgO + Hg + 2NaNO3 + CO2
РТУТЬ
831
Окись ртути представляет собой диамагнитные желтые (при
диаметре частиц 2 мк) или красные (при диаметре частиц 10—20 мк)
орторомбические кристаллы с плотностью 11,08 г!см\ плохо
растворимые в воде. Окись ртути имеет основной характер (раство-
ряется в галогеноводородах и в азотной кислоте и образует основ-
ные соли со многими солями ртути), обладает окислительными
свойствами, разлагается на элементы при нагревании до 500°.
Водород восстанавливает желтую окись ртути при 50—127°
и красную окись ртути при 90—220°.
HgO + Н2 = Hg + Н2О + 46,75 ккал
Восстановление HgO водородом катализируется СпО или
А12О3.
Нагревание HgO с фтором дает HgF2, при этом выделяется
кислород.
При действии сухого хлора на HgO на холоду образуются
окись хлора С12О и оксихлорид ртути, а при сильном нагревании
образуется HgCl2 и выделяется кислород:
/Hg-Cl
2HgO + 2Cl2=O/ +С12О HgO + Cl2 = HgCl2 + i/2O2
Hg—Cl
Обработка хлором водной суспензии HgO приводит к образо-
ванию НОС1 и Hg2Cl2O:
,Hg—Cl
2HgO + 2C12+ H2O = O< +2HOC1
XHg-Cl
Окись ртути, взаимодействуя с жидким бромом, парами брома
или раствором брома в четыреххлористом углероде, превращается
в оксибромид ртути Hg2Br2O. При нагревании HgO с иодом
до 100° образуются Hgl2 и Hg(IO3)2.
Монохлорид серы S2C12 и хлористый тионил SOC12 взаимодей-
ствуют с окисью ртути по уравнениям:
2HgO + 2S2C12 = 2HgCl2 + SO2 -f- 3S (комн, темп.)
HgO + 5SOC12 = HgCl2 + 3SO2C12 + S2C12 (при 160°)
Окись ртути окисляет натрий или калий в расплаве, магний
или алюминий при нагревании, а также элементарный фосфор,
фосфорноватистую кислоту Н8Р2О7 и окись мышьяка(Ш) As2O3.
При перемешивании HgO с NH4OH образуется желтый мелко
кристаллический порошок, называемый основанием Миллона:
НО —Hg,
2HgO + NH4OH= )NH2OH
НО —Hgz
«32
ПОБОЧНАЯ ПОДГРУППА II ГРУППЫ
Соединение плохо растворимо в воде. Нейтрализуя это основание
с помощью HI или HNO3, получают следующие соли:
НО —Hg^
НО—HgZ
NH2I
НО —Hg.
>nh2no
НО —Hgz
3
Если основание Миллона дегидратируют в атмосфере аммиака
твердым КОН или нагревают до 125°, образуются гидроокись
пксиамидортути (ангидрооснование) и имид оксидиртути:
НО —Hg -н2о /Hg. -н2о zHg.
)NH2OH------>- О< )NH2OH--------> О< >NH
НО — Hgz ^Hgz XHg'
Имид оксидиртути представляет собой коричневый порошок,
который разлагается со взрывом при ударе или при нагревании
до 130°, не обладает основными свойствами и не образует соли
с кислотами.
Окись ртути применяется в медицине, в сельском хозяйстве
в качестве фунгицида, как катализатор.
Гидроокись ртути, Hg(OH)2, осаждается при обработке
растворов солей ртути щелочами (pH = 5); соединение обладает
амфотерным характером, неустойчиво и легко превращается
в HgO.
Диспергированная до коллоидного состояния Hg(OH)2 может
•служить бактерицидным средством.
Перекись ртути, HgO2, образуется при действии 30% -ного
раствора Н2О2 на HgO при 0° или на смесь HgCl2 с КОН в спир-
товом растворе.
Соединение HgO2 представляет собой красный малоустойчивый
порошок; он легко разлагается при ударе или при нагревании.
Фторид ртути, HgF2, получают обработкой металлической
ртути или HgCi2 газообразным фтором, термическим разложе-
нием Hg2F2 при 450° и давлении 10 мм рт. ст., действием газо-
образного хлора на нагретый до 270° Hg2F2, обработкой ртути
с помощью CIF3, взаимодействием газообразного HF с HgS при
нагревании.
Hg + F2 = HgF2 + 53,429 ккал Hg2F2 HgF2 + Hg
Hg2F2 + Cl2 = HgF2 + HgCl2 HgS + 2HF ч* HgF2 + H2S
Соединение HgF2 представляет собой бесцветные октаэдриче-
ские кристаллы с плотностью 8,95 г!см5, т. пл. 615° и т. кип. 650°.
Оно устойчиво в сухом воздухе даже на свету, но легко превра-
щается в основной фторид ртути Hg(OH)F под действием влаги.
При упаривании на холоду раствора HgO в избытке плавиковой
кислоты выпадают желтые кристаллы HgF2"2H2O.
Известны также фтор хлорид ртути HgClF, фторбромид ртути
HgBrF и фторид амидортути HgNH2F.
РТУТЬ
833
Хлорид ртути (сулема), HgCl2, получается прямым взаимо-
действием элементов при нагревании в цилиндрах из эмалирован-
ного чугуна, нагреванием смеси HgSO4 с NaCl при 300°, упарива-
нием раствора ртути в царской водке, растворением HgO в экви-
валентном количестве HG1, а также обработкой HgO хлором при
нагревании, действием газообразного HG1 на HgSO4 при 120°
и, наконец, обработкой металлической ртути или HgS с помощью
S2C12: *
Hg + Cl2 HgCl2;+ 55 ккал HgSO4 + 2NaCl = HgCl2 + Na2SO4
HgO + 2HC1 — HgCl2 + H2O HgSO4 + 2HC1 = HgCl2 + H2SO4
Hg + S2C12 = HgCl2 + 2S HgS + S2C12 = HgCl2 + 3S
3Hg + 6HC1 + 2HNO3 = 3HgCl2 + 2NO + 4H2O
Сулема образует бесцветные орторомбические мелкие кри-
сталлы с плотностью 5,45 г/см3, соединение является диэлектри-
ком, т. пл. 277° и т. кип. 304°, легко очищается сублимацией,
очень устойчиво на воздухе, токсично, растворяется в воде, спир-
тах, эфире, ацетоне, бензоле, толуоле, диоксане, хлороформе,
пиридине и сероуглероде.
Водные растворы хлорида ртути имеют слабо кислый харак-
тер вследствие гидролиза:
HgCl2 + НОН Hg(OH)Cl + НС1
Для объяснения некоторых аномальных свойств растворов
хлорида ртути (например, понижение точки замерзания и др.)
было высказано предположение, что в растворах молекулы хло-
рида ртути ассоциированы (HgCl2)n и, следовательно, склонны
к образованию аутокомплексов:
72HgCl2 ч* Hg[HgCl4] 3HgCl2 Hg[HgCl3]s
Хлорид ртути в воде находится в очень слабо ионизированном
состоянии.
При сильном нагревании хлорид ртути разлагается на эле-
менты!
HgCl2 ч* Hg + С12
Обработка водных растворов хлорида ртути H2S, H2Se или
РН3 вызывает осаждение ряда соединений: HgCl2*2HgS (белое),
HgS (черное), HgCl2«2HgSe (белое), HgSe (черное), P2(Hg2Cl2)3
(желтое).
Щелочи взаимодействуют с хлоридом ртути:
HgCl2 + 2МегОН ч* HgO +[2МетС1 + Н2О
На холоду реакция хлорида ртути с NaHSO3 идет по урав-
нению
HgCl2 + 2NaHSO3 ч* Na2[Hg(SO3)2] + 2НС1
53-0101
834
ПОБОЧНАЯ ПОДГРУППА II ГРУППЫ
При действии хлоридов щелочных металлов на HgCl2 обра-
зуются хлоромеркураты типов Me^HgClg] и Me^lHgClJ, например
K[HgGl3], K[HgCl3].H2O, K2[HgCl4bH2O, Na[HgCl3b2H2O,
Cs[HgCl3],~ NH4[HgCl3],
При действии аммиака на HgCl2 можно получить либо неплав-
кий белый осадок HgNH2Gl, либо плавкий белый осадок HgCl2’
• 2NH3:
+ 2Н2О
HgCl2 + 2NH4OH =
,C1
HgCl2 + 2NH4OH = Hg< + NH^Cl + 2H2O
xnh2
~H3N Cl"
X
H3NZ XC1
Соединение [Hg(NH3)2]Gl2 получают в избытке NH4C1.
Сильное нагревание хлорида амидортути вызывает разло-
жение:
6HgNH2Cl -> 3Hg2Cl2 + 4NH3 + N2
Солянокислый раствор дихлорида олова восстанавливает
хлорид ртути до хлорида диртути и затем до элементной ртути.
2HgCl2 + H2[SnCl4] = Hg2Cl2 + H2[SnCl6]
Hg2Cl2 + H2[SnCl4] = 2Hg + H2[SnCl6]
Металлы, более активные, чем ртуть, восстанавливают хлорид
ртути до хлорида диртути и затем до металлической ртути.
Гидразин и двуокись серы в водном растворе также восстана-
вливают HgCl2 до Hg2Cl2, а затем до металлической ртути.
Для восстановления HgCl2 до Hg2Cl2 можно использовать
формиат натрия, щавелевую кислоту, оксалаты щелочных метал-
лов, аскорбиновую кислоту и фосфористую кислоту.
2HgCl2 + NaHCOO = Hg2Cl2 + NaCl + HCl + CO2
2HgCl2 + H2C2O4 = Hg2Cl2 4- 2HC1 + 2CO2
2HgCl2 -j- (NH4)2C2O4 = Hg2Cl2 4- 2NH4C1 + 2CO2
2HgCl2 CgO3H3 = Hg2Cl2 4- C6O6He 2HC1
Известны смешанные галогеномеркураты, содержащие анионы
[HgX4P-, [HgX3Y]2-, [HgX2Y2]2-, [HgXY3]2-, [HgY4]2- (где
X и Y = Cl , Br , I , GN ); известны также оксихлориды
Hg3Cl2O2, Hg3Cl4O, Hg4Cl2O4, Hg4Cl2O3 и комплексные хлориды
[Hg(NH2OH)2]Cl2, [Hg(C2H5NH2)2]Gl2, [HgEn2]Cl2, [HgEn3]Cl2,
fHg(CeH5NH2)2Cl2 [Hg(C5H5N)2]Gl2,
X X Xx PR3
\lg^ \lg^ _Hg^
Й3Р^ ^X^ ^x^ ^x
(где X = C1~, Br-, I-).
РТУТЬ
835
Бромид ртути, HgBr2, образуется при действии избытка
брома на слегка нагретую под слоем воды металлическую ртуть,
обработкой металлической ртути в нагретой стеклянной трубке
парами брома, увлекаемыми током азота, действием бромидов
щелочных металлов на растворы Hg(NO3)2 или Hg(C104)2, рас-
творением HgO в эквивалентном количестве НВг.
Hg + Вг2 HgBr2 + 40,50 ккал HgO + 2НВг = HgBr2 + Н2О
Hg(C104)2 + 2КВг = HgBr2 + 2КС1О4
Соединение HgBr2 представляет собой диамагнитные блестящие
бесцветные орторомбические игольчатые кристаллы с плот-
ностью 6,064 г/см3, т. пл. 238,5° и т. кип. 319°. Бромид ртути
разлагается на элементы при высокой температуре и растворяется
в воде, спиртах, эфире, ацетоне, бензоле, диоксане, пиридине,
сероуглероде и этилендиамине.
Гидролиз бромида ртути дает основной бромид ртути Hg(OH)Br.
Обработкой HgBr2 аммиаком получают бромид амидортути
.Вг
Hg( :
XNH,
.Br
HgBr2 + 2NH4OH = Hg< + NH4Br + 2H2O
xNH2
Известны бромомеркураты Me4HgBr3] (где Me1 = Na+, K+,
NHJ), Mer[HgBr3]-H2O (где Me1 = Li+, Na+, K+, Cs+, NHf),
Me11 [HgBrJ (где Me11 = Pb2+), смешанные галогеномеркураты,
содержащие анионы [HgBrCl3]2-, [HgBr2Cl2]2-, [HgBr3Cl]2-,
[HgIBr3]2-, [HgI2Br2]2-, [HgI3Br]2~, и комплексные бромиды
[Hg(NH3)2]Br2, R3 (P или As)2(HgBr2)n (где n = 1, 2, 3, 4).
Иодид ртути, Hgl2, получают в результате растирания ртути
с иодом в присутствии неболыпого’количества воды или спирта,
при действии иода на HgO или Hg(NO3)2 в воде, по реакции KI
с HgCl2, взятых в эквивалентных количествах в виде 0,02 М
растворов, при действии СН31 или С2Н51 на соли ртути, нагрева-
нием до 230° смеси HgO и А1218.
Hg + Б ** Hgl2
+ 25,20 ккал (красная модификация)
+ 24,55 ккал (желтая модификация)
HgCl2 + 2KI = Hgl2 + 2К.С1
А12О6 + 3HgO = 2HgI2 + А120д
Ковалентное соединение Hgl2 представляет собой блестящие
красные кристаллы с тетрагональной решеткой и плотностью
6,276 г/см3, устойчивые при температурах ниже 127°, или желтые
53*
836
ПОБОЧНАЯ ПОДГРУППА II ГРУППЫ
орторомбические кристаллы с плотностью 6,225 г/см3, устойчивые
в интервале от 127° до температуры плавления. Обе модификации
иодида ртути находятся в равновесии:
Hgl2 (красный) Hgl2 (желтый) — 0,61 ккал
Иодид ртути диамагнитен, имеет т. пл. 259°, т. кип. 354°,
разлагается при высокой температуре на элементы, плохо раство-
рим в воде, растворяется в растворах иодидов щелочных металлов,
спирте, эфире, ацетоне, бензоле, диоксане, пиридине, хлороформе,
бромоформе, четыреххлористом углероде и этилендиамине. Если
Hgl2 растворяют в избытке KI, то получают почти бесцветный
раствор, из которого при хранении в эксикаторе над конц. H2SO4
выпадают светло-желтые кристаллы K2[HgI4].
Щелочной раствор соединения K2[HgI4] называется реактивом
Несслера; он служит для качественного определения аммиака
или солей аммония, например:
/Hg.
2K,[HgI4)+4KOH + NH4Cl =О< )NH2-I + 7KI + KC1+3H2O
\Hg/
Иодид оксиамидодиртути, Hg2ONH2I, образуется в виде
коричневого осадка.
Известно большое число иодомеркуратов, соответствующих
следующим формулам: MeI[HgI3] (где MeI = K+, CeH5NH+),
MeI[HgI3(H2O)] (где Me1 = К+, Rb+, Cs+, NH4), Me^ [Hgl3]2-
•nH2O (где Me2+ = Mg2+ или [Ni(NH3)e]2+), Meni[HgI3]3-??H2O
(где Меш=А13+), Me*[HgI4] (где Me1 = К+, Rb+, Cs+, Cu+),
Mei[HgI4bnH2O (где Me1 = Li+, Na+, K+, Ag+), Me11 lllglj
(где Me11 = Zn2+, Cd2+, [NiEn3]2+), Men[HgI4]-тгНгО (где Меп=
= Mg2+, Ca2+, Sr2+, Ba2+).
Соединение Ag2[HgI4] плохо растворимо в воде и существует
в виде двух модификаций: а именно: a-Ag2[HgI4l — золотисто-
желтые тетрагональные кристаллы с плотностью 6,02 г!см3,
устойчивые ниже 50,7°, и fJ-Ag2[HgI4] — красные кубические
кристаллы, устойчивые в интервале от 50,7° до температуры
разложения (158°).
Известны оксииодиды Hg3I2O2, Hg3I4O, Hg4I2O3, иодид амидо-
^|тути HgNH2I, двойная соль HgI2-2HgS и комплексные иодиды
Hg(NH3)2]I2, [Hg(N2H4)2]I2, [Hg(C8H5NH2)2]I2, [Hg(C5H5N)2]I2,
[Hg(C9H7N)2H2, R3(P или As)2(HgI2)n (где n = 1, 2, 3 и 4).
Цианид ртути, Hg(CN)2, получают растворением HgO в рас-
творе HCN, обработкой раствора HgSO4 (слабо подкисленного
H2SO4) 10%-ным раствором KCN, обработкой берлинской лазури
Fe4[Fe(CN)e]3 окисью ртути или хлоридом ртути и горячей водой.
Fe4[Fe(CN)6]3 + 9HgO = 9Hg(CN)2 + 2Fe2O3 + 3FeO
Белые тетрагональные диамагнитные кристаллы Hg(CN)2 очень
токсичны, имеют плотность 4,0 г!см\ разлагаются на Hg и (GN)2
РТУТЬ
837
при 320°, растворяются в спиртах, анилине, пиридине, хинолине,
жидком NH3 и аммиачных растворах, в воде растворяются хуже,
чем хлориды ртути.
Цианид ртути взаимодействует с концентрированными раство-
рами галогеноводородов по уравнению
Hg(CN)2 + 2НХ HgX2 + 2HCN
Металлическое олово разлагает цианид ртути в присутствии
NaCl или нитратов щелочных металлов:
(NaCl)
Hg(CN)2 + Sn + 2Н2О---Hg + Sn(OH)2 + 2HCN
Сероводород c Hg(CN)2 образует HgS.
Известны цианомеркураты типов Me1 [Hg(GN)3] (где Me1 = Na+,
К+, 1/2Са2+, x/2Sr2+, х/2Ва2+), Mex[Hg(CN)4] (где Мех = Na+, К+,
Tl+, x/2Sr2+, х/2Ва2+, x/2Zn2+, Х/2РЬ2+ и др.) и смешанные ацидомер-
кураты типов Me1 [Hg(CN)2X] (где Mex=Na+, К+, Cs+, NH4
и X = Cl", Br-, I-, SCN"), Mex[Hg(CN)2X2], Men[Hg(GN)2X]2,
(где Me11 = Mg2+, Zn2+, Cd2+, Mn2+, Co2+, Ni2+ и X = NOj,
SON-), Meni[Hg(CN)2X]3 (где Me111 = Ce3+, La3+, Sm3+, Y3+
и X = SCN”).
Получены также аммиакаты Hg(GN)2-2NH3 (где п — 1 или 2)
и оксицианид ртути Hg(CN)2-HgO.
Тиоцианат ртути, Hg(SCN)2, получают обработкой водного
раствора Hg(NO3)2 или HgCl2 pacTBopoM^KSCN, взятым в эквива-
лентном количестве, или действием раствора HSCN на окись
ртути.
Соединение Hg(SCN)2 представляет собой белые игольчатые
кристаллы, которые легко разлагаются при нагревании или под
действием света, плохо растворимы в воде и растворяются в ацетоне
и концентрированных растворах тиоцианатов щелочных металлов.
Если тиоцианат ртути смешать с небольшим количеством раство-
ра крахмала, а затем полученную пасту высушить в виде палочек,
то при нагревании приготовленный таким образом роданид горит
синим пламенем, образуя объемистый остаток, по внешнему виду
напоминающий змею («змея фараона»).
Известен основной тиоцианат ртути Hg(SCN)2 -2HgO и ряд
тиоцианатомеркуратов, содержащих анионы [Hg(SCN)3]~,
[Hg(SCN)4]2-, [Hg(SCN)5l3-, Hg(SCN)2Cl2]2-, [Hg(SCN)2Br2]2-.
Сульфид ртути, HgS, встречается в природе в виде минерала
киновари (красные тригональные кристаллы) или в виде минерала
метакиновари (черные кубические кристаллы).
Красный сульфид ртути HgS (киноварь) можно получить суб-
лимацией смеси ртути (или HgO, 2HgSO4"HgO и др.) с серой при
600° в токе азота, обработкой ртути водными растворами сульфи-
дов или полисульфидов щелочных металлов при 50—60°; он также
выделяется при хранении черного сульфида ртути HgS в теплых
838
ПОБОЧНАЯ' ПОДГРУППА II ГРУППЫ
концентрированных растворах полисульфидов щелочных металлов
и при действии раствора тиосульфата натрия на хлорид ртути.
Черный аморфный сульфид ртути HgS выпадает в осадок при
барботировании сероводорода через водные растворы солей ртути,
в результате действия раствора сульфида щелочного металла
на соль ртути, при взаимодействии разбавленного раствора
Na2S2O3-5H2O с раствором HgCl2 -j- NaCl -}- H2SO4, при расти-
рании смеси ртути с серным цветом.
Под действием H2S из раствора HgCl2 сначала выделяется
белый осадок хлоросоли ртути Hg2SCl2 или Hg3S2Cl2, который
затем становится желтым, коричневым и, наконец, превращается
в черный сульфид HgS:
3HgCl2 + 2H2S = Hg3S2Cl + 4HC1 Hg3S2Cl2 + H2S = 3HgS + 2HC1
Красная модификация HgS представляет собой тригональные
кристаллы с плотностью 8,1 г!см3 и твердостью 2—2,5 по шкале
Мооса; эта модификация очень устойчива.
Черная модификация HgS обладает кристаллической структу-
рой, аналогичной цинковой обманке; плотность ее 7,67—7,69 г/см2,
твердость 3,3 по шкале Мооса. При нагревании до 344° черная
модификация превращается в красную, она плохо растворяется
в воде, разбавленных кислотах, сульфиде аммония и щелочах при
обычной температуре, лучше — в сульфидах щелочных металлов,
при нагревании же растворяется в конц. HNO3 и царской водке:
9HgS + 8HNO3 = 3Hg3S2(NO3)2 + 3S + 2NO + 4H2O
3Hg3S2(NO3)2 + 16HNO3 = 9Hg(NO3)2 + 6S + 4NO + 8H2O
3HgS + 6HC1 + 2HNO3 = 3HgCl2 + 3S + 2NO + 4H2O
HgS + Me2S = MeI[HgS2]
Тиосоли ртути в водных растворах гидролизуются:
NajHgSil + Н2О т HgS + NaHS 4- NaOH
При нагревании сульфид ртути вступает в реакции с водородом
и кислородом:
HgS + Н2 Hg + H2S
HgS 4" 2О2 “ HgSO4 или 2HgS -|- ЗО2 = Hg2SO4 SO2
Он также взаимодействует с водным раствором иода в KI
и с S2C12:
HgS + I2 + 2KI = K2[HgI4] + S HgS + S2C12 = HgCl2 + 3S
В присутствии воды некоторые металлы, например Zn, Си —
при обычной температуре или Sn, Pb, Ag, Bi, Fe — при нагрева-
нии, вытесняют ртуть из сульфида ртути и соединяются с серой.
Известны многочисленные двойные сульфиды, например
K2S-HgS (5 или 7) Н2О — серебристые иглы, K2S-HgS-H2O —
РТУТЬ
839
золотисто-желтые пластинки, K2S -5HgS -5Н2О — черные или крас-
ные кристаллы, K2S -2HgS — зеленые пластинки, а также HgS •
•MoS2, HgS -WS2, HgS -MoS3, HgS 'WS3, 3HgS -Sb2S5 и др.
Сульфат ртути, HgSO4, выделяют упариванием при высокой
температуре раствора, полученного растворением при нагрева-
нии ртути или HgO в конц. H2SO4:
Hg -J- 2H2SO4 = HgSO4 + SO2 + 2H2O
Можно также получить HgSO4 обработкой (CH3)2SO4 соедине-
ний ртути (HgO, HgCl2, Hg(NO3)2, HgCO3-2HgO).
Сульфат ртути представляет собой диамагнитные белые орто-
ромбические мелкие кристаллы, которые чернеют на свету, жел-
теют и затем становятся красными при нагревании; плотность
HgSO4 6,47 г/см3, он разлагается при нагревании на воздухе
до ~550° (т. пл. 850°) и гидролизуется в воде, превращаясь в трудно
растворимый желтый основной сульфат HgSO4-2HgO:
3HgSO4 + 2Н2О HgSO4.2HgO + 2H2SO4
Хлор или бром с HgSO4 в присутствии воды образуют хлорид
или бромид ртути.
Известны также основные соли 2HgSO4-HgO, HgSO4-HgO-
•Hg2O, двойные сульфаты Me*SO4 -3HgSO4-2Н2О (где Me1 = К +
или NH*), а также аддукты HgSO4-2NH3-ЩО, 2HgSO4-HgO*
•HgS -4Н2О, 3HgSO4 -HgO -2HgS -4H2O.
Сульфат ртути применяют в качестве катализатора.
Нитрат ртути, Hg(NO3)2 -пН2О, образуется при растворении
ртути илг HgO в ~ 26 %-ной HNO3 при 30°:
3Hg + 8HNO3 = 3Hg(NOa)2 4- 2NO + 4Н2О
Из водного раствора нитрата ртути при различных температу-
рах и в присутствии определенных дегидратантов можно выделить
кристаллогидраты Hg(NO3)2 -тгН2О (где п = х/2, 1, 2 и 8).
Соединение Hg(NO3)2представляет собой прозрачные
бесцветные кристаллы с плотностью 4,3 г/см3 и т. пл. 145°.
Соединение Hg(NO3)2 -8Н2О — диамагнитные бесцветные ром-
бические кристаллы с т. пл. 16°.
Нитрат ртути растворяется в разб. HNO3 или ацетоне, мало
растворим в конц. HNO3, гидролизуется водой, превращаясь
в трудно растворимые основные соли Hg(OH)NO3, Hg3O2(NO3)2-
• Н2О или Hg3O2(NO3)2 желтого цвета.
Обработка насыщенного раствора нитрата ртути аммиаком
в присутствии NH4NO3 приводит к образованию комплексного
нитрата [Hg(NH3)2](NO3)2, [Hg(NH3)4](NO3)2. При действии аммиа-
ка на разбавленные растворы нитрата ртути (в отсутствие солей
840
ПОБОЧНАЯ ПОДГРУППА II ГРУППЫ
аммония) выпадает осадок белого нитрата оксиамидодиртути:
zHgx
2Hg(NO3)2+4NH4OH = )NH,NO3 + 3NH4NO3 + 3H,O
Пропускание фосфина через раствор нитрата ртути сопровож-
дается осаждением соединения Hg3P2 -3Hg(NO3)2 -HgO белого
цвета, которое разлагается со взрывом при нагревании или при
Ударе.
Известны также основные нитраты Hg(NO3)2-МепО-izHgO
(где Me11 = Zn2+, Gd2+, Cu2+, Ni2+, Go2+) и аддукты Hg(NO3)2 •
•N2H4, Hg(NO3)2 -2HgS, Hg(NO3)2HgBr2, Hg(NO3)2-Hgl2.
Из нитрата ртути получают большинство соединений ртути.
Основные карбонаты ртути. При обработке раствора нитрата
ртути кислыми карбонатами щелочных металлов или раствором
хлорида ртути с большим избытком карбоната щелочного металла
образуется коричнево-красный осадок основных карбонатов ртути.
44Hg(NO3)2 + 8NaHCO3 = Hg4O3CO3 + 8NaNO3 + 4H2O + 7CO2
4HgCl2 + 4Na2CO3 = Hg4O3CO3 + 8NaCl + 3CO2
Известен также основной карбонат HgGO3 -2HgO или Hg3O2CO3.
Хелатные соединения
Обработка нейтральных или слабо кислых растворов солей
диртути или ртути 1%-ным спиртовым раствором дифенилкарба-
зида в присутствии твердого карбоната натрия приводит к обра-
зованию растворимых хелатных соединений фиолетово-синего
цвета!
.NH-NH —СвН5
Hg2+ + O = C/ +U2 ->
XNH —NH —С6Н5
При взаимодействии нейтральных или слабо азотнокислых
растворов солей ртути /г-диметиламинобензилиденродамином обра-
зуется хелатноеj соединение красного цвета:
HS
/ ж-----с=о
| I
1-S=C4 ,с=сн
\
Реакция ацетилацетоната натрия с хлоридом ртути в водном
растворе дает ацетилацетонат ртути [Hg(C5O2H7)2] белого цвета;
РТУТЬ
841
соединение плохо растворимо в органических растворителях
и легко растворяется в воде.
В качестве примеров хелатных соединений ртути упоминаются
диоксалатомеркураты Mei[Hg(C2O4)2], галогениды трисэтиленди-
аминртути [HgEn3]X2 (где X = Cl-, Br-, I-) и соединения, полу-
ченные обработкой растворов солей ртути дитизоном С8Н5-
•N = N -GS -NH -NH -С8Н5, аллоксаном
.СО —МИ.
О = С( )СО-4Н2О,
ХСО —NHZ
Р-аминонафталидтиогликолевой кислотой, антраниловой кисло-
той H2N -С8Н4-СООН и др.
Металлоорганические соединения
Известны многочисленные алкильные и арильные производ-
ные ртути, галогениды алкил- и арилртути, гидроокислы алкил-
ртути, циклопентадиенильные соединения, галогениды цикло-
пента диенилртути и ацетиленовые производные.
Примеры алкильных и арильных производных ртути:
Hg(CH3)2, Hg(C2H5)2, Hg(C3H7)2, Hg(C4H9)2, Hg(C5Hu)2,
Hg(C7H15)2, Hg(C8H17)2, Hg(C8H5)2, Hg(C8H5 — CH2)2. Известны
галогениды алкил- и арилртути CH3HgX, C2H5HgX (где X = Cl-,
Br-, I-), C4H9HgX (где X = Cl-, Br-), С5НиН&Вг .C6H5HgCl,
гидроокиси алкилртути CH3HgOH, C2H5HgOH, циклопентадие-
нильные соединения Hg(C5H5)2, C5H5HgX (где X = Cl-, Br-,
I-, CN-) и ацетиленовые производные R — Hg — С = C —
- Hg - R (где R = CH3, C2H5, C3H7, C4H9, C5HU, C8H13, C7H15,
C9H19).
Диметилртутъ, Hg(CH3)2, представляет собой бесцветную
жидкость с т. кип. 92° (пары токсичны); ее можно получить по
реакциям:
HgCl2 + 3CH3MgBr =. Hg(CH3)2 + MgBr2 + MgCl2
HgBr2 + 2CH3MgBr = Hg(CH3)2 + 2MgBr2
2HgBr2 + Sn(CH3)4 = 2Hg(CH3)2 + SnBr4
3HgCl2 + 2A14C3 + 18HC1 = 3Hg(CH3)2 + 4A12C16
Диэтилртутъ, Hg(C2H5)2, представляет собой бесцветную
жидкость с т. кип. 159° (пары токсичны); соединение раство-
ряется в бензоле или хлороформе, может быть получено по урав-
нениям:
HgCl2 -|- 2C2HsMgBr = Hg(C2H6)2 -|- MgBr2 -f- MgCl2
2C2H5HgCl + 2Na = Hg(C2H6)2 + 2NaCl + Hg
Na2Hg + 2C2HsI = Hg(C2H6)2 + 2NaI
Na2Hg (C2H5)2SO4 = Hg(C2H6)2 -f- Na2SO4
842
ПОБОЧНАЯ ПОДГРУППА II ГРУППЫ
Нагревание Hg(C2H5)2 вызывает ее разложение на Hg и С4Ню-
Под действием водорода при 200° и давлении 50 атм Hg(C2Hs)2
разлагается, образуя Hg и С2Нв.
Соединение Hg(C2Hs)2 взаимодействует с галогенами, соляной
кислотой и различными хлоридами, например РС13, СН3СОС1,
SiCl4, AsCl3, SbCl3 и др.
Hg(C2Hs)2 + 2С12 = HgCl2 + 2С2ЩС1
Hg(C2H5)2 + 2НС1 = HgCl2 + 2С2Н6
3Hg(C2H6)2 + 2РС13 = 3HgCl2 + 2Р(С2Н5)3
Металлы Li, Na, Be, Zn, Cd, Al, Sb, Bi вступают в реакцию
c Hg(C2H5)2, при этом выделяется ртуть и образуются этильные
производные соответствующих металлов.
Соединение Hg(C2H5)2 не взаимодействует с водой, спиртами
или эфирами.
Дифенилртутъ, Hg(CeH5)a, выделяют в виде белого твердого
вещества с т. пл. 125°, т. кип. 204°.
Хлорид метилртути, CH3HgCl, представляет собой белое
твердое вещество с т. пл. 167°; получают его по реакциям:
Hg + СН3С1 = CHaHgCl Hg(CH3)2 + Cl2 = CH3HgCl + СН3С1
Хлорид этилртути, C2HsHgCl, желтовато-белое твердое
вещество с т. пл. 192°, образуется по уравнению
Hg(C2H5)2 + HCl = C2H6HgCl + CjHe
При действии ацетилена на хлорид этилртути в щелочном
растворе получается С2Н5 — Hg — С = С — Hg — С2Н5.
Помимо приведенных известны и другие соединения ртути, например:
хлорит Hg(C102)2, хлорат Hg(C103)2, гидроксихлорат Hg(OH)ClO3, пер-
хлораты Hg(C104)2, [Hg(NH3)4](C104)2, Hg(C104)2-2HgO, Hg302[C104]2,
гипобромит Hg(BrO)2, бромат Hg(BrO3)2, гидроксибромат Hg(OH)BrO3,
иодат Hg(IO3)2, перйодаты Hg(IO4)2, Hg(IO4)2,-4HgO, Hg5(IO6)2, селенид
HgSe, теллурид HgTe, тиосульфаты HgS2O3, Na2[Hg(S2O3)2], дитионат
HgS2O6, сульфиты HgSO3, HgSO3-2HgO-H2O, MelSO3-HgSO3-nH2O,
Me2[Hg(SO3)2] (где Mei = Na+, K+, NH+), селениты HgSeO3, Mei[Hg(SeO3)2]t
персульфат [Hg(NH3)4]S2O8, селенаты HgSeO4-H2O, HgSeO4, (NH4)2SeO4 •
• HgSeO4-2H2O, теллураты HgTeO4-2H2O, HgTeO4-2HgO, нитрид Hg3N2,
нитриты Hg(NO2)2, Me2[Hg(NO2)4] и Me2[Hg(NO2)4]-nHgO, Mei[Hg(NO2)3]
и MeI[Hg(NO2)3bH2O, (где Me1 = Na+, K+, Rb+, Cs+, T1+), Pb[Hg(NO2)4],
фосфид Hg3P2, двойная соль Hg2(NO3)2-Hg2(H2PO4)2-2H2O, ортофосфаты
Hg3(PO4)2, Na2HPO4-Hg3(PO4)2-ЗНгО, дифосфат Hg2P2O7, метафосфаты
Hg3PeO18, Hg2P4O12, Hg6P12O36, тиоортофосфат Hg3(PS4)2, арсенид Hg3As2,
ортоарсенит Hg3(AsO3)2, ортоарсенат Hg3(AsO4)2, метаарсенат Hg(AsO3)2,
антимонид Hg3Sb2, метаантимонат Hg(SbO3)2-nH2O, ортотиоантимонат
Hg3(SbS4)2, карбид HgC2-H2O, ацетиленид Hg2C2-H2O, цианат Hg(NCO)2,
фульминат Hg(CNO)2, тиоцианат, селеноцианат Hg(SeCN)2, селеноцианато-
меркураты Mei[Hg(SeCN)4], Mell [Hg(SeCN)4]-nH2O (где Mell = Cu2+,
Zn2+, Ni2+, Co2+), Mei[Hg(SeCN)3]. гексацианоферраты(П) Hg2[Fe(CN)el,
K2Hg[Fe(CN)6], гексацианоферраты(Ш) Hg3[Fe(CN)6]2, MeiHg[Fe(CN)6], тио-
алкильные производные Hg(SC2Hs)2, C2H5 — S — Hg — Cl.
СПИСОК ЛИТЕРАТУРЫ
1. Atanasiu I. A., Facsko G., Electrochimie, Bucuresti, Editura tehnica,
1958.
2. Бектуров А. Б., Илясова А. К., Труды хим. наук АН Казах. ССР, 10
(1964).
3. Beral E.,Zapan М., Chimie anorganica, Bucuresti, Editura tehnica, 1968.
4. Бетехтин А. Г., Минералогия, Гоегеолиздат, M., 1950.
5. Блок Н. И., Качественный химический анализ, ГХИ, М.— Л., 1952.
6. Cascenco G. A., Bazele metalurgiei fizice, Bucuresti, Editura tehnica,
1952.
7. Cdlu^aru A., Depunerea electrolitica a metalelor in forma dispersa, Editura
Academiei Republicii Socialiste Romania, 1962.
8. Chariot G., L’analyse qualitative et les reactions en solutions, Ed. IV,
Paris, Ed. Masson, 1957.
9. Chauveau Fr., Bull. Soc. chim. France, fasc. I, 1960, p. 834.
10. Chauveau Fr., Souchay, Pierre, Bull. Soc. chim. France, fasc. Ill, 1963,
p. 561-563.
11. Ciogolea Gh., Morait Gh., Baloescu C., Analiza medicamentelor (Bazele
teoretice si practice), Bucuresti, Editura medicala, 1961.
12. Коттон Ф., Уилкинсон Дж., Современная неорганическая химия,
«Мир», М., 1969.
13. Currant Р. J., Durrant В., Introduction to Advanced Inorganic Chemistry,
London and Beccles Longmans, 1962.
14. Ephraim F., Inorg. Chem., London, 1954, p. 527— 534.
15. Evans R. C., Chimie et la structure cristalline, Paris, 1954.
16. Firman M. A., Tehnologia substanfelor minerale utile, Bucuresti, Editura
tehnica, 1953.
17. Gallais F., Chimie minerale theoretique et experimentale, Paris, Masson,
1957.
18. Gimblett F. G. R., Inorganic polymer chemistry, 1963.
19. Гринберг А. А., Введение в химию комплексных соединений, Госхим-
издат, М.— Л., 1951.
20. Guliaev А. Р., Metalurgia fizica, Bucuresti, Editura tehnica, 1953.
21. Heslop R. B., Robinson P. L., Inorganic Chemistry, Elsevier Publishing
Company, Amsterdam, London, New York, Princeton, 1960.
22. Holleman A. F., Wiberg E., Lehrbuch der anorganischen Chemie, Berlin,
1956.
23. Jander G., Wendt H., Lehrbuch der analytischen Chemie und preparativen
anorganischen Chemie, 1954.
24. Ключников H. Г., Руководство по неорганическому синтезу, Госхим-
издат, М.— Л., 1953.
25. Киреев В. А., Курс физической химии, Госхимиздат, М.— Л., 1951.
26. Кисляков И. П., Металлургия редких металлов.
27. Кокорин А. И., Ж. анал. хим., 27, 549 (1957).
28. Larsen Е. М., Transitional Elements, Inc. New York, Amsterdam, 1965.
29. Michel A., Benard J., Chimie Minerale, Paris, VI-ё. Masson et Cie, Edi-
teurs Libraires et 1’Academie de Medecine, 120, Boulevard Saint-Germain,
1965.
844
СПИСОК ЛИТЕРАТУРЫ
30. Naray-Szabo Istvan, Kristaly-kemia, Budapest, Akademiai Kiado, 1965.
31. Naray-Szabo Istvan, Szervetlen kemia, I, II, III, Budapest Akademiai
Kiado, 1958.
32. Неницеску К., Общая химия, «Мир», М., 1968.
33. Неницеску К., Органическая химия, т. 1—2, Издатинлит, М., 1962,
1963.
34. Некрасов Б. В., Курс общей химии, Госхимиздат, М.— Л., 1952.
35. Nesmeianov A. N., «Analele romano-sovietice» (seria chimie), 2 (33),
aprilie-iunie, 1960, anul XV, seria a Ш-a, p. 3—50.
36. Несмеянов A. H., Баранов В. И., Заборенко К. Б., Руденко Н. П., При-
селков И. А., Практикум по радиохимии.
37. Pannetier G., Chimie generale (Atomistique et liaisons chimiques), Masson
et Cle Editeurs, 1966.
38. Pariselle H., Cours de chimie — Metaux, Paris, Masson et Cie Editeurs,
1956.
39. Pascal P., Nouveau traite de chimie minerale, Paris, Editeurs Masson
et Ci0, tom V, VII fasc. 1 et 2, IX, XII, XIV, XVI, XVII, XVIII, XIX,
XX.
40. Полинг Л., Общая химия, «Мир», М., 1964.
41. Phillips С. S. G., Williams R. I. P., Inorganic Chemistry, vol. 2, Oxford
At. the Clarendon Press, 1966.
42. Нозин M. E., Технология минеральных удобрений и солей.
43. Реми Г., Курс неорганической химии, Издатинлит, М., 1960.
44. Pribil R., Complexonii in chimie analitica, Bucuresti, Editura tehnica,
1961.
45. Ripan R., Popper E., Liteanu C., Chimia analitica calitativa, semimicro-
analiza, Bucuresti, Editura de stat didactica si pedagogica, 1961.
46. Ruttewit K., Massing G., Z. Metall Kunde, Zen’tralblatt, II, 865 (1940).
47. Сауков А. А., Геохимия, Госгеолиздат, M., 1951.
48. Schiller К., Thilo E., Z. Anorg. Allg. Chemie, 310, 274 (1961).
49. Schumann H., Metalurgie fizica, Bucuresti, Editura tehnica, 1962.
50. Schreiter W., Metale rare, Bucuresti, Editura tehnica, 1966.
51. Slavinski M. P., Proprieta(ile fizico-chimice ale elementelor, Bucuresti,
Editura tehnica, 1955.
52. Sidgwich N. V., The chemical elements and their compounds, 1950.
53. Spacu P., Curs de chimie anorganica, Bucuresti, 1950 (litografiat).
54. Solacolu S., Chimia fizica a silica(ilor tehnici, Bucuresti, Editura tehnica,
1957.
55. Souchay M. P., Polianions et polycations, 1963.
56. Спицин В. И., Кодочигов Р. Н., Голутвина М. М., Кузина А. Ф., Соко-
лова 3. А., Методы определения радиоактивности.
57. lonescu, Tudor D., Schimbatori de ioni, Bucuresti, Editura tehnica, 1961.
58. Trzebiatowski W., Lehrbuch der anorganischen Chemie, Berlin, Veb.
Deutscher Verlag der Wissenschaften, 1963.
59. Wells A. F., Structural Inorganic Chemistry, Oxford at the clarendon
Press, 1962 (есть русский перевод: Уэллс А. Ф., Строение неорганиче-
ских веществ, Издатинлит, М., 1948).
60. Winnacker Kiichler, Tehnologia chimica anorganica, Bucuresti Editura
tehnica, 1962.
61. Manualul inginerului chimist, Vol. I, si II, Bucuresti, Editura tehnica,
1951______1952
62. Handbook of Chemistry and Physics, Fort-First Edition, 1959—1960.
63. Tabele de calcul in chimia analitica, Bucuresti, Editura tehnica, 1961.
64. Manualul chimistului, Vol. I si II, Ed. AGIR, 1948.
65. Landolt-Bornstein, Physik Chemie, Astronomie, Geophysik, Tehnik, Ber-
lin, Gottingen, Heidelberg, Springer-Verlag, 1950—1959.
66. Comptes Rendus, 88—645, 1879.
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Азурит 682, 686, 717
Акантит 725
Активаторы 684
Актиний 58 и сл.
бромид 25, 64
гидроокись 63
изотопы 59, 60, 61, 62
— выделение 60, 61
иодид 25, 64
окись 23, 63
оксалат 64
оксибромид 64
оксифторид 63
оксихлорид 64
ортофосфат 64
очистка 61, 62
получение 61
применение 62
распространенность в природе 60
сульфат двойной 64
сульфид 64
физические свойства 20—22, 62
фторид 24, 63
химические свойства 22—26, 62
хлорид 24, 63
электронная структура 19, 58
Анатаз 76
структура 95
Аннабергит 583
Аргентит 725, 726
А урат
магния 770
натрия 770
Бадделеит 105, 115, 127
переработка 108
Берцелиуса процесс получения
гафния 128
титана 80
циркония 109, 110
Бессемера процесс 491, 492
Бихромат
аммония 269
калия 268, 269
лития 268
натрия 268
рубидия 269
цезия 269
Бицирконила дихлорид 121
Борнит 441, 682
Браунит 391
Брукит 76
Ванадий 141 и сл.
бориды 178, 179
карбиды 178
минералы 142, 143
нитриды 178
получение 145—147
— восстановлением хлоридов
147
— металлотермическим восста-
новлением 146
— термической диссоциацией
иодида 146
— электролитическое 147
применение 150, 151
руды ванадинитные 145
— железные, переработка 143,
144
— карнотитные, переработка
144
силициды 178
соединения включения 178, 179
физические свойства 137, 138,
148
химические свойства 139, 140,
148-150
экстракция из растворов 145
электролитическая очистка 148
электронная структура 136, 141
Ванадий(П)
гидроокись 152, 153
дибромид 153
дииодид 153, 154
— термическая диссоциация 146
846
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
дифторид 153
дихлорид 147, 153
окись 151, 152
селенид 154
сульфат 154
сульфид 154
теллурид 154
Ванадий (III)
ацетилацетонат 160
бензоилацетонат 160
гидроокись 156
окись 151, 155, 156
оксибромид 158
оксихлорид 158
сульфаты 159, 160
сульфид 159
трибромид 158
трииодид 158, 159
трифторид 156
трихлорид 156, 157
— восстановление 147
хелатные соединения 160, 161
Ванадий(1У)
бромид двойной [с Sb(III)] 164
двуокись 162, 163
дисульфид 165
сульфаты 165, 166
тетрафторид 163
тетрахлорид 163, 164
хелатные соединения 166
хлорид, восстановление 147
Ванадий(У)
ванадаты 170
гетерополисоединения 173, 174
декаванадаты 172, 173
диванадаты 172
металлоорганические соединения
177, 178
окситиованадаты 176, 177
пентасульфид 176
пентафторид 141, 174
пероксосоединения 174
пированадаты 172
поливанадаты 170—172
пятиокись 140, 147, 148, 151,
167-169
— металлотермическое восста-
новление 146
тетрафтороксованадаты 175
тиованадаты 176, 177
триметаванадаты 172
хелатные соединения 177
Ванадила
ацетилацетонат 166
бензоилацетонат 166
дибромид 164, 165
дииодид 165
дифторид 163
дихлорид 164
сульфат 165
трибромид 176
трифторид 175
трихлорид 175, 176
Ванадинит 142
Ван-Аркела — де Бура процесс по-
лучения
ванадия 146
гафния 128
титана 80
циркония 110
Вивианит 483
Виллемит 785
Винчит 27
Вольфрам 334 и сл.
бориды 382
гексакарбонил 377
диарсенид 380, 381
карбиды 381, 382
металлоорганические соедине-
ния 377
минералы 335, 336
нитриды 380
получение 341—344
— восстановлением трехокиси
341, 342
— металлотермическим восста-
новлением окислов 342
— нагреванием трисульфида
с СаО 343
— термической диссоциацией
гексахлорида 342, 343
— электролитическое 343, 344
превращение порошка в ком-
пактный металл 345
применение 348, 349, 352
руды, обогащение и переработка
336—340
силициды 382
соединения включения 380—382
физические свойства 216—218,
345, 346
фосфиды 380
химические свойства 218—226,
346—348
электронная структура 215, 334
Вольфрам(П)
дибромид 350, 353
дииодид 351, 354
дихлорид 350, 352, 353
Вольфрам(Ш)
координационные соединения
351, 354, 355
Вольфрам(1У)
ацидосоединения, содержащие
координированные CN-группы
358
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
847
двуокись 221, 350, 355, 356
диселенид 357
дисульфид 221, 357
дителлурид 357
сульфиды 221, 351
тетраидодид 351, 357
тетрафторид 221, 356
тетрахлорид 221, 350, 356
Вольфрам(У)
ацидосоединения, содержащие
координированные CN-rpyn-
пы 360
• пентабромид 350, 359, 360
пентахлорид 350, 359
Вольфрам(У1)
вольфраматы 363—369
гексабромид 350, 375, 376
гексафторид 223, 350, 372
гексахлорид 350, 373, 374
— восстановление 342, 343
гетерополивольфраматы 363—369
гетерополикомплексы 371, 372
гидроокиси 350, 362, 363
диоксидибромид 350, 376
диоксидихлорид 350, 375
изополивольфраматы 363—365
кобальто-6-вольфраматы 369, 370
кобальто-12-вольфраматы 369,
370
метавольфраматы 369
окситетрабромид 350, 376
окситетрафторид 350, 372
окситетрахлорид 350, 374, 375
окситиовольфраматы 376, 377
периодатовольфраматы 370
пероксосоединения 370, 371
тиовольфраматы 376, 377
трехокись 222, 350, 361, 362
— восстановление 341, 342
триселенид 377
трисульфид 343, 351, 376
фторооксивольфраматы 373
Вольфрамат
бария 368
калия 367
кальция 369
рубидия 367
свинца 368
стронция 368
цезия 367
цинка 368
Вольфрамит 335
переработка концентратов 339,
340
Вольфрамовые бронзы 378, 379
Вольфрамовые кислоты 362, 363
Вольфрамовый ангидрид см. Воль-
фрам(У1), трехокись
Вульфенит 279, 283, 284
Вюртцит 785
Гадолинит 37
Ганит 786
Гарниерит 583, 586
Гаусманит 391
Гафнила
дибромид 134
дихлорид 134
Гафний 126 и сл.
карбид 135
минералы 127
нитрид 135
получение 128
применение 130
руды, переработка 127, 128
соединения включения 135
физические свойства 66, 67, 68,
129
химические свойства 68, 69, 73,
129
электронная структура 65, 126
Гафний(П)
дибромид 131
Гафний(Ш)
трибромид 131
Гафний(1У)
гидроокись 130, 132
гидрофосфат 134, 135
двуокись 70, 130, 132
диборид 135
сульфогафниаты 134
тетрабромид 72, 131, 134
тетраиодид 72, 131, 134
тетрафторид 71, 130, 133
тетрахлорид 71, 130, 133, 134
Гексаброморенит калия 456
Гексаиодорепит калия 457
Гексаметилплатина 675
Гексамминванадия(Ш) хлорид 157
Гексамминтитана(Ш) хлорид 93
Гексанитрородат калия 642
Гексатантал аты 208, 209
Гексафторованадат калия 175
Гексафторогафниат аммония 133
Гексафторогафниат калия, восста-
новление 128
Гексафторогипоманганат калия 425
Гексафторокобальтат цезия 577
Гексафторокупрат(Ш) калия 722
Гексафторотанталовая кислота 210
Гексафторотитанат калия, восста-
новление 80
Гексафторотитановая кислота 99
Гексафтороцирконат(ГУ) калия,
восстановление 109
848
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Гексахлороиридат аммония 649, 650
Гексахлороманганит(1У) калия 424
Гексахлоропалладат калия 660
Гексахлороплатинат(1У)
аммония 674
калия 674
натрия 674
Гексахлороренат калия 455, 456
Гексахлорорениевая кислота 455
Гексахлорородат натрия 642
Гексахлоротехнетат калия 438
Гексацианованадат калия 154, 160
Гексацианокобальтат калия 558, 559
Гексацианомарганцовая(П) кисло-
та 411
соли 411
Гексацианоосмат калия 633
Гексацианорутенат калия 624
Гексацианоферрат калия 508, 509,
522, 523
Гематит 482
Гептамолибдат аммония 323
Гептафторониобат калия 193
Гептафторотанталат калия 210
Гептафторотитанат аммония 99
Герсдорфит 583
Гессит 753
Гипоренат натрия 459
Гринокит 803
Гюбнерит 335
Двухромовая кислота 268
Депрессанты 684
Дибензолхром 274, 275
Дивольфрамат натрия 368
Дигексателлуратокупрат(Ш) нат-
рия 722
Дикарбонилжелеза дииодид 538
Диметилртутъ 841
Диметилцинк 801
Димолибдат
аммония 322, 323
натрия 322
Динитродигидразинплатина(П) 670
Динитрозилдикарбонилжелезо 538
Диоксотетрацианоренат
калия 460
натрия 460
таллия 460
Дипероксомолибдат аммония 325
Дироданоаурат(1) калия 768
Диселенатоскандиевая кислота 34
Дисульфатоаурат(Ш) калия 775
Дисульфатованадиевая кислота
159, 160
Дитиосульфатоаурат(1) натрия 766,
767
Дифенилкадмий 817
Дифенилртуть 842
Дифенилцинк 801
Дихлор-бнс-(дипиридил)палладия
хлорид 660
транс- Дихлоро диамминплатина(П)
670
ци.с-Дихлородиамминплатина(П) 670
Дихлородигидразинплатина(П) 670
Дициклопентадиенилжелезо 539, 540
Дициклопентадиенилхром 275
Дицирконилдисульфат калия 123
Диэтиленгуанидинсеребра нитрат
751, 752
Диэтилртутъ 841, 842
Диэтилскандия бромид 36
Диэтил цинк 801
Железистая кислота 518
Железистосинеродистая кислота 509
Железо 479 и сл.
арсениды 541
додекакарбонил 535
карбиды 541
л-комплексы 539
металлоорганические соедине-
ния 532—540
минералы 482—484
модификации 495, 496
нитриды 540
нитрозильные соединения 538
пентакарбонил 532—534
получение 484
применение 501
соединения включения 540, 541
сырое, получение 485—490
тетракарбонилгалогениды 537,
538
тионитрозильные соединения 538
физические свойства 474—477,
495—498
фосфиды 540
химические свойства 477—479,
498—500
электронная структура 473, 479
эннеакарбонил 534, 535
Железо(П)
бромид 481, 505, 506
гидроокись 503
дисульфид 511
иодид 481, 506
карбонат 514
нитрат 513
окись 478, 502, 503
оксалат 515
ортофосфат 513, 514
пруссидные соединения 509, 510
предметный указатель
849
сульфат 478, 511—513
— двойной 513
сульфид 478, 510, 511
тетракарбонилдигидрид 535, 536
тетракарбонилферраты 533, 536
— галогениды 537
тиоцианат 510
ферроцианиды 507—509
фторид 480, 503, 504
хелатные соединения 515, 516
хлорид 480, 504, 505
— гидраты 505
цианид 507
Железо (II), (III)
бромид смешанный 522
иодид смешанный 522
окись 519
хлорид смешанный 521
Железо (III)
ацетаты 528
ацетилацетонат 528
гидроксоферриты 518, 519
гидроокись 518
нитрат 527
окись 517. 518
— модификации 517, 518
ортофосфаты 527, 528
пруссидные соединения 524, 525
сульфат 526, 527
сульфид 525, 526
тиоцианат 525
трибромид 521, 522
трифторид 519
трихлорид 520, 521
ферриты 518
ферроцианиды 522—524
хелатные соединения 528— 530
Железо(1У)
ферраты 531
Железо (VI)
ферраты 531, 532
Железный купорос 511, 512
Железосинеродистая кислота 523, 524
Земли
иттриевые 37
тербиевые 37
эрбиевые 37
Золото 752 и сл.
извлечение из концентратов руд
— — — амальгамированием 755
— — — хлорированием 758
— — — цианированием 756,757
минералы 753
очистка 759, 760
применение 762, 763
руды, переработка^754, 755
1/« 64-0101
физические свойства 677— 679,760
химические свойства 677, 680.
761, 762
электронная структура 676, 752
Золото(1)
бромид 765
гидрид 764
дироданоаурат 768
дитиосульфатоуарат 766, 767
дихлороаураты 765
дицианоаураты 767, 768
золотодиродановодородная кис-
лота 768
иодид 765, 766
окись 764
сульфид 766
фосфид 767
хелатные соединения 768
хлорид 764
цианид 756, 767
Золото (II)
бромид 768
окись 768
сульфат^ 769
сульфид 768
хлорид 768
Золото(Ш)
аураты 770
бромид 773
гидроокись 769
гидросульфат аурила 775
дицианогалогенаураты (III) 776
золотобромистоводородная кис-
лота 773, 774
золотохлористоводородная кис-
лота 772
иодат 774
иодид 774
металлоорганические соедине-
ния 777, 778
нитрат 775
окись 769
сульфат 774, 775
— двойной 775
сульфид 774
тетраацетатоаураты 775
тетрабромоаураты 774
тетраиодоаураты 774
тетратиоцианатоаураты 776
тетрахлороаураты 772, 773
тетрацианоаураты 776
фторид 770
хелатные соединения 777
хлорид 770—772
Ильменит 75, 76
переработка 77, 78
Иридий 643 и сл.
850
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
додекакарбонил 651
извлечение 644
октакарбонил 651
получение 644, 645
применение 646
распространенность в природе
644
физические свойства 616—618
645
химические свойства 618, 620.
645
электронная структура 618, 643,
644
Иридий(1)
хлорид 647
Иридий(П)
сульфид 646, 647
хлорид 646, 647
Иридий(Ш)
бромид 646, 648
иодид 646, 648
окись 646, 648
сульфид 648, 649
хлорид 646, 648
Иридий(1У)
бромид 646, 650
гексахлороиридат 649, 650
иодид 646, 650
окись 646, 649
сульфид 646, 650
фторид 646, 649
хлорид 646, 649
Иридий(У1)
сульфид 646, 650
фторид 646, 650
Иттриалит 37
Иттрий 37 и сл.
алкильные производные 45
ацетат 44
— основной 44
ацетилацетонат 45
бромид 24, 42, 44
гидроокись 41
гликолят 45 .
извлечение из минералов 38
иодид 25, 42
карбид 44
карбонат 44
нитрат 43
т— основной 43, 44
окись 23, 40, 41
— металлотермическое восста-
новление 38
оксисульфид ,43
оксифторид 41
оксихлорид 42 ,
полисульфйдц 43
получение 37—39 ; ,
получение металлотермическим
восстановлением 38, 39
— электролизом хлорида 39
применение 40
распространенность в природе
37, 38
сульфат безводный 43
— основной 43
сульфид 42, 43
физические свойства 20—22, 39
фторид 24, 38, 41
хелатные соединения 45
химические свойства 22—26, 40
хлорид 24, 39, 41, 42
— электролиз 39
электронная структура 19, 37
Кадмий 802 и сл.
бромид 783, 806, 811
гидрид 808
гидроокись 809
— тетрамминкадмия 809
иодид 783, 807, 811, 812
карбонат 816
металлоорганические соедине-
ния 817
минералы 803
нитрат 815, 816
нитрид 815
оксинат 816
очистка 803, 804
получение 803
применение 806, 807
руды, переработка 803
сульфат 807, 814, 815
сульфид 807, 813, 814
тиоцианат 813
физические свойства 780, 781,
804
фторид 782, 806, 809
хелатные соединения 816, 817
химические свойства 782—784,
805
хлорид 782. 806, 810
— гексамминкадмия 811 , . .
— диаммиакат 810
цианид 812, 813
электронное строение 779, 802
Калаверит 753 . ,
Каламин 785
Карнотит 142 -
Карролит 543
«Кассиевый пурпур» 771 - .
Кераргирит 725, 726
Киноварь 819, 820, 821
Кобальт 541 и сл.
антимониды 580
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
851
арсениды 580
бориды 581
гидриды 579
дииодид карбонила 579
додекакарбонил 578
извлечение из руд 544, 545
карбиды 580, 581
карбонильные соединения 577—
579
металлоорганические соедине-
ния 577—579
минералы 543
модификации 548
нитриды 579, 580
нитрозильные соединения 579
октакарбонил 577, 578
очистка 547
получение 545—547
применение 550
силициды 581
соединения включения 579—581
тетракарбонилкобальтовая кис-
лота 578, 579
физические свойства 474—477,
547, 548
фосфиды 580
химические свойства 477—479,
548, 549
циклопентадиенильные соеди-
нения 579
электронная структура 473, 541
Кобальт(1)
селенид 551
Кобальт(П)
алюминат 553
ацетилацетонат 562, 563
гидроксокобальтиты 554
гидроокись 553, 554
дибромид 481, 557
дииодид 481, 557, 558
дисульфид 560
дифторид 480, 554, 555
дихлорид 480, 555
дицианид 558
карбонат 562
кобальтат 564
нитрат 561
окись 478, 552, 553
ортофосфат 561, 562
ортофосфаты двойные 562
сульфат 478, 560, 561
сульфиды 478, 559, 560
тиокобальтат 567
тиоцианат 559
хелатные соединения 562, 563
хлорид гексамминкобальта 556,
557
цинкат 553
Кобальт(Ш)
гидроокись 565
карбонат 567
кобальтаты 564
координационные соединения
567-576
— — моноядерные 568—573
— — полиядерные 573—576
окись 564, 565
сульфат 567
сульфид 566, 567
тиокобальтат 567
трифторид 565, 566
трихлорид 566
цианид 566
Кобальт(1У)
гексафторокобальтат 577
двуокись 577
диселенид 577
Кобальтин 543
Ковелин 682
Колумбит 180, 441
Кочубеит 227
Креннерит 753
Криптомелан 391
Крокоит 226, 227
Кроля процесс получения
гафния 128
титана 79
циркония 109
Ксенотин 37, 38
Купрат калия 722
Куприт 682, 686
Лантан 46 и сл.
ацетилацетонат 58
бензоилацетонат 58
бромид 25, 53
гидрид 50
гидроокись 51
дисульфид 54
дифосфаты 56
иодид 53, 54
— термическая диссоциация 47,
48
карбид 56
карбонат 57
купферонат 58
минералы 46
нитраты 55
нитрид 55
окись 23, 50, 51
— металлотермическое восста-
новление 47
оксалат 57
оксибромид 53
оксииодид 54
54*
852
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
оксинитраты 56
оксисульфид 54
оксифторид 52
ортофосфат 56
получение 46—48
— металлотермическим восста-
новлением 47
— термической диссоциацией
иодида 47, 48
— электролизом 48
применение 49
распространенность в природе 46
силицид 57
сульфаты 54,55
сульфиды 54
триметафосфат 56
физические свойства 20—22, 48
фосфаты 56
фосфид 56
фторид 24, 51, 52
хелатные соединения 58
химические свойства 22—26, 49
хлорид 24, 52, 53
электролиз 48
электронная структура 19, 46
Леллингит 483
Лимонит 482
Линеит 543
Литиофилит 392
Лопарит 180
Магнетит 482, 519
Магнуса соль 671
Малакон 105, 127
Малахит 682, 686, 717
Манганит 391
Марганец 390 и сл.
антимониды 434
арсениды 433
бориды 434
карбиды 434
нитриды 433
металлоорганические соедине-
ния 432, 433
минералы 391, 392
модификации 395
очистка 394
пентакарбонилы 432
получение 392—394
— восстановлением галогени-
дов Мп(П) 393
— — окислов 393
— электролитическое 394
применение 397
силициды 434
соединения включения 433, 434
физические свойства 384—386,
394, 395
390, 399
411
фосфиды 433
химические свойства 386—390,
395, 396
электронная структура 383,
390
Марганец(1)
монофторид
Марганец(П)
ацетат 410,
ацетилацетонат 412
ацидосоединения, содержащие
координированные CN-rpyn-
пы 411, 412
гексафторосиликат 403
гексафторосоли 403
гидроокись 402
гидроортофосфат 409
дибромид 390, 405, 406
дигидроортофосфат 409
дииодид 390, 406
дитиоцианат 410
дифторид 390, 403
дихлорид 390, 403—405
зеленая ацидосоль 412
карбонат 409, 410
метасиликат 410
моносульфид 389, 406, 407
нитрат 408
окись 401, 402
ортосиликат 410
ортофосфат 408, 409
сульфат 407, 408
тетрафторосоли 403
цианид 411
Марганец(Ш)
ацетат 417
ацетилацетонат 419
гексацианоманганиты 417, 418
гидроокись 414, 415
гидросульфат 416
диоксалатодиаквоманганиты
418, 419
окись 413, 414
ортофосфат 416, 417
сульфат 416
трималонатоманганиты 419
триоксалатоманганиты 418
трифторид ?90, 415
трихлорид 390, 415
Марганец(1У)
гексафторосоли 423, 424
двуокись 388, 391, 420—422
дисульфид 389, 420, 424
манганиты 422, 423
пентафторосоли 423
сульфат 424, 425
тетрафтрид 390, 423
тетрахлорид 390, 424
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
853
Марганец(У)
гексафтороманганаты 425
гипоманганаты 425, 426
Марганец(У1)
манганаты 427, 428
трехокись 388, 426
Марганец(УП)
окись 388, 428, 429
— солеобразующая 431, 432
перманганаты 429—431
Марганцевый пшат 392
Марганцовая кислота 429
Марганцовистая кислота 427
Марганцовый ангидрид см. Марга-
нец(УП), окись
Марказит 483
Мартена процесс 493, 494
Медь 681 и сл.
аффинаж 688, 689
гидрид 723
металлоорганические соедине-
ния 722, 723
минералы 681, 682
нитриды 723
применение 693, 694
рафинирование пирометаллур-
гическое 688, 689
— электролитическое 689
руды, переработка 683—687
силицид 723
соединения включения 723
физические свойства 677—679,
689, 690
фосфиды 723
химические свойства 677, 680,
690—693
«черная» 688
электронная структура 676, 681
Медь (I)
ацетат 702
ацетиленид 702
бромид 698, 699
гексафторосиликат 702
гидроокись 696
закись 695, 696
иодид 699
координационные соединения
702, 703
нитрат 701
селенид 701
сульфат 700, 701
сульфид 699, 700
— переработка 684—686
сульфит 700
теллурид 701
тиоцианат 702
фторид 696
хлорид 696—698
цианид 701, 702
Медь(П)
арсенаты 717
арсениты 717
ацетат 717, 719
ацетиленид 719
бромат 711 (
— основной 711
бромид 709, 710
гексафторосиликат 719
гидроокись 705, 706
гликолят 720
диоксалатокупрат 721
дипериодат 711
иодат 711
— основной 711
иодид 710
карбонаты 717, 718
координационные соединения
720, 721
ксантогенат 720, 721
метаборат 720
метапериодат 711
нитрат 715
— гидраты 715
— основной 715, 716
— тетраммина 715
окись 704, 705
оксалат 719
оксифториды 706, 707
ортопериодат 711
полисульфиды 712
рубеанат 721
селенат 714, 715
селенид 714
сульфат 712—714
— гидраты 713
сульфид 711, 712
сульфит 712
тиоцианат 718
фосфаты 716, 717
фторид 706
хлорат 710
— основной 710, 711
хлорид 707—709
— гидраты 707, 708
цианид 718
Медь(Ш)
диортопериодаты 722
купраты 722
окись 722
Мезоторий 2 59
Мелаконит 682, 686
Мелантерит 483
Меначин 74
Метавольфрамат калия 368,
369
Метавольфрамовая кислота 369
854
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Метакиноварь 819
Метамонопероксониобиевая кислота
193
Метарениевая кислота 466
Метатитановая кислота 97
Метацирконаты 119
Метилртути хлорид 842
Миллерит 582
Миллона основание 831
Миспикель 483
Молибдат
аммония 322
калия 321, 322
кальция 322
натрия 321
свинца 322
Молибден 278 и сл.
бориды 333, 334
гексакарбонил 330
— термическая диссоциация 287
карбиды 333
металлоорганические соедине-
ния 330—332
минералы 279, 280
— обогащение и переработка
280—284
нитриды 332
очистка 332
получение 284—288
— восстановлением металлотер-
мическим 285, 286
— окислов водородом 285, 286
— термической диссоциацией
гексакарбонила 287
— электролитическое 286, 287
применение 293—295
силициды 333
соединения включения 332—
334
физические свойства 216—218,
290
фосфиды 332, 333
химические свойства 218—226,
290—293
электронная структура 215, 278
Молибден(П)
дибромид 297, 301
дииодид 297, 301
дихлорид 296, 299, 300
окись 296, 298
Молибден(Ш)
гидроокись 297, 302
окись 297, 302
оксибромид 304
оксифторид 302
оксихлорид 303
сульфиды 297, 304
трибромид 297, 304
трифторид 296, 302
трихлорид 296, 302, 303
Молибден(ГУ)
ацидосоединения, содержащие
координированные CN-rpyn-
пы 308
двуокись 221, 296, 305, 306
дисульфид 221, 297, 306—308
тетрабромид 297, 306
тетрахлорид 221, 296, 306
Молибден(У)
бромид 297
гидроокись 296, 309
комплексы, содержащие коорди-
нированную CNS-rpynny 312
оксибромид 312
оксифторид 310
оксихлорид 311
октацианомолибдаты 312
пентасульфид 297, 312
пентафторид 296, 309
пентахлорид 296, 310, 311
пятиокись 296, 309
Молибден (VI)
гексабромид 299
. гексафторид 223, 298, 326, 327
гексахлорид 298
гетерополимолибдаты 316—323
гидроокиси 298, 315, 316
диоксидибромид 328
диоксидифторид 327
диоксидихлорид 328
изополимолибдаты 316, 317,
320
молибдаты 316, 317
окситетрафторид 327
окситетрахлорид 328 >
пероксосоединения 325, 330
сульфиды 299, 329, 330 ’ г.
трехокись 222, 278, 279, - 288,
289, 298, 313-315
— восстановление 285, 286
трисульфид 299, 328 329
Молибденит 279—283, 441, 442:
Молибденовая охра, см. Ферримо-
либдит
Молибденовые кислоты 315, 316
Молибденовые сини 331, 332
Молибденовый ангидрид см. Молиб-
ден (VI), трехокись
Монацит 46
Монда процесс извлечения никеля
585
Монопероксометатанталовая кисло-
та 209
Монопероксомолибдат бария 326
Монопероксомолибденовая кислота
325
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
855
Монопероксоортотитановая кисло-
та 98
Монопероксоциркониевая кислота
119
Мора соль 513
Нагиа гит 7 53
Невьянскит 629, 630, 637, 644,
661
Никель 581 и сл.
антимониды 614
арсениды 613
гидрид 612
извлечение из руд 583—587
карбиды 614
координационные соединения
. 605—607
металлоорганические соедине-
ния 610—614
минералы 582, 583
модификации 589
нитриды 612
отделение от меди 584—586
очистка 588, 589
получение 587, 588
применение 592
тетракарбонил 610—612
физические свойства 474—477,
' ' '589 589
, фосфиды 612, 613
химические свойства 477—479,
589-591
электронная структура 473, 581,
582
Никедь(1)
гидроокись 593
окись 593
селенид 593
сульфид 593
трицианоникелат 593, 594
цианид 593
Никель (II)
ацетат 604
гидроокись 596
дибромид 481, 598 1
дииодид 481, 598, 599
дисульфид 602
дифторид 480, 596, 597
дихлорид 481, 597, 598
карбонат 603, 604
нитрат 602, 603
окись 478, 595
— водная 596
перекись 595, 596
сульфид 478, 600, 601
тетрацианоникелаты 599, 600
тиоцианат 600
хелатные соединения 607—609
цианид 599
Никель(Ш)
окись 609, 610
Никель(1У)
никелаты '610
ортопериодаты 610
Никколит 582, 583
Ниобиевая кислота см. Ниобий(У),
пятиокись гидратированная
Ниобий 179 и сл.
борид 198
гидрид 197
карбид 198
минералы 180
нитрид 198
отделение от тантала 182, 183
получение 183, 184
применение 185, 186
руды, переработка 180—182
соединения включения 197, 198
физические свойства 137, 138,
184
химические свойства 139, 140,
184, 185
электронная структура 136, 179
Ниобий(1)
закись 187
Ниобий(П)
окись 186, 187, 188
сульфид 187, 188
хлорид 186
Ниобий(Ш)
сульфат двойной 189
сульфид 187, 189
трехокись 187, 188
трибромид 187, 189
трииодид 189
трифторид 186, 188
трихлорид 186, 188, 189
Ниобий(1У)
двуокись 186, 189, 190
дисульфид 187, 190
тетрахлорид 186, 190
Ниобий(У)
ацетилацетонат 197
бензоилацетонат 197
диоксихлорид 195
металлоорганические соедине-
ния 197
ниобаты 192
окситрибромид 196
окситрифторид 193
окситрихлорид 195
пентабромид 187, 195, 196
пентаиодид 187, 196
пентафторид 141, 186 193
пентахлорид 186, 193, 194
856
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
пероксониобаты 192
полиниобаты 192
пятиокись 140, 183, 186, 191
— гидратированная 191, 192
сульфаты 187, 196
сульфид 187
хелатные соединения 196, 197
Нитрозилжелеза галогениды 538
Нитропруссид калия 524
Оксалатомолибдаты 324
Оксалатопероксомолибдаты 326
Оксопентахлороренат
аммония 460
калия 460
Оксопентахлорорениевая кислота 460
Оксибромовольфраматы 360
Оксибромомолибденовая кислота 328
Оксигексафторотанталат калия 210
Оксидиртути имид 832
Оксипентахлоромолибдат калия 311
Окситиомолибдаты 330
Оксифторомолибдаты 327
Оксихлоровольфраматы 359
Оксихлоромолибдаты 328
Оксихлоромолибденовая кислота 328
Октациановольфрамат калия 358, 360
Октацианомолибдат калия 308
Ортованадат
калия 171
натрия 171
Ортотитановая кислота 96, 97
Орфорда процесс извлечения никеля
584, 585
Осмий 629 и сл.
дихлорид трикарбонилосмия 637
извлечение из руд 630
карбонил 637
пентакарбонил 636
получение 630
применение 631
распространенность в природе
629, 630
физические свойства 616, 617,
618, 630
химические свойства 618, 620,
630, 631
электронная структура 618, 629
Осмий(П)
гексацианоосматы 633
иодид 632, 633
окись 632
хлорид 632
Осмий(Ш)
окись 632, 633
трихлорид 632, 633
Осмий(1У)
бромид 632, 634
гексахлороосматы 632, 634
гидроокись 632—634
иодид 632, 634
окись 632, 634
сульфид 632, 634
фторид 632, 634
хлорид 632, 634
Осмий (V)
фторид 632, 634
Осмий(У1)
гексафторид 632, 635
осмат 632, 635
Осмий(УШ)
октафторид 632, 636
тетрасульфид 632, 636
четырехокись 632, 635, 636
Палладий 651 и сл.
извлечение из руд 652
получение 652
применение 653, 654
распространенность в природе
652
физические свойства 616—618,
652, 653
химические свойства 618, 620,
653
электронная структура 618, 651
Палладий(1)
сульфид 654
Палладий(П)
бромид 655, 657
— тетрамминпалладия 657
гидроокись 655, 656
— тетрамминпалладия 656
иодид 655, 658
— тетрамминпалладия 658
нитрат 655, 658
окись 655, 656
сульфат 655, 658
сульфид 655, 658
фторид 655, 656
хелатные соединения 659
хлорид 655—657
цианид 655, 658
Палладий(Щ)
гидроокись 655, 659
фторид 655, 659
Палладий(ТУ)
гексахлоропалладаты 660
гидроокись 655, 660
сульфид 655, 660
Паравольфрамат
аммония 368
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
857
калия 368
натрия 368
Паркеса процесс получения серебра
726
Патронит 142
Пейроне соль 670
Паттисона процесс получения сереб-
ра 726, 727
Пентакарбонилметилмарганец 433
Пентафенилхром 275
Пентацианотригидроксовольфрамат
калия 358
Пентландит 582
Переходные металлы
атомные массы 8—10
атомный объем 13
кристаллическая структура 10
магнитные свойства 15, 16
металлический блеск 12
механические свойства 16
плотность 13
потенциалы ионизации 13, 14
прозрачность 12
радиусы атомные 12
— ионные 12, 13
распространенность в природе 18
температура кипения 13
— плавления 13
теплопроводность 15
цвет 12
электронное строение 5—10
электропроводность 15
электрохимические свойства
16—18
Перманганат
калия 429, 430
лития 413
натрия 431
серебра 431
Перовскит 76
структура 97
Пероксогафниевые соединения 132,
133
Пероксодисульфатогафниат(1¥) ка-
лия 133
Пероксодисульфатоцирконат (IV)
калия 119
Пероксотитановые соединения 98
Пероксохромат
аммония 261
калия 261
лития 261
натрия 261
Пероксоциркониевые соединения
119, 120
Перрутенат калия 627
Пертехнециевый ангидрид см. Тех-
неций(УП), окись
55-0101
Петцит 753
Пираргирит 725
Пирит 441, 483, 511
Пиролюзит 391
Пирохло_р 180
Платина 660 и сл.
алкильные производные 675
извлечение из руд 662
очистка 662
получение 662
применение 664, 665
распространенность в приро-
де 661
физические свойства 616—618
662, 663
химические свойства 618, 620,
663, 664
электронная структура 618.
660, 661
Платина(П)
бромид 665, 667
гидроокись 665, 666
гликоколят 671
иодид 665, 667
координационные соединения
668—671
окись 665, 666
сульфид 665, 667, 668
фторид 665, 667
хлорид 665—667
цианид 665, 667
Платина(Ш)
бромид 665, 671
гидроокись 665, 671
иодид 665, 671
сульфид 665, 672
хлорид 665, 671
Платина(1У)
бромид 665, 673
гексагидроксоплатинаты 675
гидроокись 665, 672
иодид 665, 673
координационные соединения
673—675
пирофосфат 665, 673
сульфат 665, 673
сульфид 665, 673
фторид 665, 672
хлорид 665, 672
Платина(У1)
окись 665, 675
фторид 665, 675
Платиновые металлы, разделение 619
Платино (11)тетрацианистоводородиая
кислота 669
Платино хлористоводородная кисло-
та 674
Повеллит 279
858
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Полибазит 725
Поликсен 661
Политиомолибдат калия 329
Прустит 725
Пршибромит 803
Псиломелан 391
Пудделя процесс 491
Пурпуреоплатина 662
Рамсделит 391
Ревдинскит 583, 586
Ренат бария 462
Рениеватая кислота 462
Рений 440 и сл.
арсенид 472
бориды 472
галогенокарбонилы 471, 472
германид 472
извлечение из молибденита 441
карбонилы 471
минералы 441
нитрид 472
получение 443, 444
— восстановлением метапер-
ренатов 443
— разложением Re2(CO)i0 444
— термической диссоциацией
Не3С19 444
— электролитическое 444
применение 447, 448
силициды 472
соединения включения 472
физические свойства 384—386,
444, 445
фосфиды 472
химические свойства 386—390,
445—447
электронная структура 383, 440
Рений (I)
гидроокись 449
моноиодид 390, 449
Рений(П)
гидроокись 449
дииодид 390, 450
моносульфид 389, 450
Рений (III)
окись 388, 450
трибромид 390, 452
трииодид 452
трихлорид 390, 450—452
— диссоциация 444
Рений(1У)
гексаброморениты 456
двуокись 388, 453, 454
дисульфид 389, 457
рениты 454
тетрабромид 350, 456
тетраиодид 350, 456, 457
тетрафторид 390, 454, 455
хелатные соединения 458
Рений(У)
гипоренаты 459
диоксотетрацианоренаты 460
окись 459
окситрифторид 459
пентабромид 390, 460
пентафторид 390, 459
пентахлорид 390, 459
Рений(У1)
гексафторид 390, 462, 463
гексахлорид 390, 463, 464
диоксидибромид 464
диоксидифторид 463
окситетрабромид 464
окситетрафторид 463
окситетрахлорид 464
оксифториды 463
ренаты 462
трехокись 461, 462
Рений(УП)
гептафторид 390, 469
гидрид комплексный 465
диокситрифторид 469
мезоперренаты 468, 469
метаперренаты 467, 468
окись 388, 465, 466
перекись 466
селенид 471
сульфид 388, 470
тиоперренаты 470
триоксибромид 470
триоксифторид 469
триоксихлорид 469
Рейзе основание I 669
Рейзе основание II 670
Ринмана зелень 553
Родий 637 и сл.
додекакарбонил 643
извлечение из руд 637, 638
октакарбонил 643
получение 638
применение 669
распространенность в природе
637
физические свойства 616—618,
638, 639
химические свойства 618, 620,
639
электронная структура 618 637
Родий(П)
окись 640
сульфид 640
хлорид 640
Родий(Ш)
бромцд 640, 642
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
859
гексанитрородат 642
гексахлорородат 642
гидроокись 640, 641
гидросульфид 642
иодид 640, 642
окись 640, 641
сульфид 640, 642
фторид 640, 641
хлорид 640—642
Родий(1 V)
гидроокись 640, 643
окись 640, 642
фторид 640, 643
Родонит 192
Родохрозит см. Марганцевый пшат
Роскоэлит 143
Ртуть 818 и сл.
металлоорганические соедине-
ния 841, 842
минералы 819
очистка 821
получение 820, 821
применение 823, 824
физические свойства 780, 781,
821, 822
хелатные соединения 840, 841
химические свойства 782—784,
822, 823
электронное строение 779, 818
Ртуть(1)
иодид 827, 828
карбонат 829
нитрат 828, 829
окись 825
сульфат 828
сульфид 828
фторид 825
хлорид 825—827
Ртуть(П)
бромид 783, 835
гидрид 830
гидроокись 832
иодид 783, 835, 836
— оксиамидодиртути 836
карбонаты основные 840
нитрат 839, 840
— оксиамидодиртути 840
окись 830—832
перекись 832
сульфат 839
сульфид 837—839
тиоцианат 837
фторид 782, 832
хлорид 783, 833, 834
хлоромеркураты 834
цианид 836, 837
цианомеркураты 837
Рутенат калия 627
Рутений 620 и сл.
бромид карбонилрутения 629
дииодид дикарбонилрутения
додекакарбонил 629
извлечение из руд 621
карбонил 628
металлоорганические соедине-
ния 628, 629
пентакарбонил 628
получение 621
применение 622
распространенность в природе
621
физические свойства 616—618,
621, 622
химические свойства 618, 620,
622
электронная структура 618, 620
Рутений(П)
гексацианорутенат 623, 624
гидроокись 623, 624
хлорид 623, 624
Рутений(Ш).
бромид 623, 625
гидроокись 623, 624
иодид 623, 625
хлорид 623—625
Рутений(1У)
окись 623, 625
сульфид 623, 626
хлорид 623, 626
Рутений(У)
фторид 623, 626
Рутений(У1)
рутенаты 623, 626, 627
Рутений(УП)
перрутенаты 623, 627
Рутений(УШ)
четырехокись 623, 627, 628
Рутил 76
структура 95
Самарскит 37, 38, 180
Сафлорит 543
Серебро 723 и сл.
металлоорганические соедине
ния 752
минералы 725, 726
применение 733
рафинирование 729, 730
руды, переработка 726—729
физические свойства 677—679,
730, 731
химические свойства 677, 680,
731, 732
электронная структура 676, 724
55*
860
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Серебро(1)
амид 743, 744
антимонид 746
арсенид 746
ацетат 748
ацетиленид 747
бихромат 747
бромат 740, 741
бромид 738
галогениды, .применение в фо-
тографии 738, 739
гидроокись 735
гидрофосфат 746
дигидрофосфат 746
дитизонат 749
имид 744
— калия и Ag 744
иодат 741
иодид 739, 740
карбонат 747
координационные соединения
748, 749
метафосфаты 746
нитрат 744, 745
нитрид 743
нитрит 744
окись 734, 735
оксалат 748
ортоарсенат 746
ортофосфат 746
перйодаты 741
перхлорат 740
сульфат 742, 743
сульфид 741, 742
тиосульфат 743
тиоцианат 748
фосфид 754
фторид 735, 736
хлорат 740
хлорид 736—738
хромат 746, 747
цианид 747, 748
Серебро(П)
координационные соединения
751
нитрат 750, 751
окись 750
сульфид 7 50
фторид 750
Серебро(Ш)
диортопериодатоаргентаты 751
диортотел л уратоаргентаты 751
окись 751
Сидерит 482, 483
Сильванит 7 53
Скандий 26 и сл.
алкильные производные 36, 37
ацетилацетонат 36
бромид 25, 32
гидроокись 30, 31
— взаимодействие с алюми-
ноном 31
иодид 25, 32, 33
карбид 35
карбонат 35
нитрат 34, 35
нитрид 34
окись 23, 30
оксалаты 35, 36
оксихинолят 36, 37
получение 27—29
— металлотермическим восста-
новлением галогенидов 27,
28
— электролизом расплава хло-
рида 28, 29
распространенность в природе
26, 27
селенат, октагидрат 34
— безводный 34
селенид 34
сульфат безводный 33
— гексагидрат 33, 34
сульфид 33
сульфит 33
физические свойства 20—22, 26,
29
фторид 24, 31
— металлотермическое восста-
новление 28
химические свойства 22—26, 29
хлорид безводный 24, 31, 32
— гексагидрат 32
— электролиз 28, 29
электронная структура 19, 26
Скуттерудит 543
Смальтит 543
Смитсонит см. Цинковый пшат
Спессартин 392
Сталь 490—495
Стефанит 725
Сфалерит см. Цинковая обманка
Сфен 76
Таленит 37
Тантал 198 и сл.
борид 214
гидрид 213
карбиды 214
минералы 199
нитриды 214
отделение от ниобия 182, 183
получение 199, 200
применение 202
руды, переработка 180—182
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
861
силицид 214
соединения включения 204, 213,
214
физические свойства 137,138, 201
химические свойства 139, 140
201
электронная структура 136, 198
Тантал(1)
закись 203
Тантал(П)
дибромид 204, 205
дихлорид 203, 204
окись 203
Тантал(Ш)
окись 204, 205
трибромид 204, 206
трифторид 204, 205
трихлорид 205
Тантал(1У)
двуокись 204, 206
дисульфид 206
тетрахлорид 204, 206
Тантал(У)
ацетилацетонат 213
бензоилацетонат 213
дибромид бромотантала 212
дихлорид хлоротантала 211, 212
металлоорганические соедине-
ния 213
окситрибромид 212
оксифторид 210
оксихлориды 212
оксихлоротанталаты 212
пентабромид 204, 212
пентаиодид 204, 212
пентатантал аты 209
пентафторид 141, 204, 209, 210
пентахлорид 204, 211
пероксотанталаты 209
пятиокись 140, 200, 204, 207,
208
— гидратированная 208
сульфат 212, 213
фторотанталаты 210
хелатные соединения 213
Танталаты 208
Танталит 199, 441
Танталовая кислота см. Тантал(V),
пятиокись гидратированная
Температура антиферромагнитного
перехода 15
Тетрабромоаурат калия 774
Тетрагидроксиламинплатины(11)
хлорид 669
Тетрагидроксоаурат(Ш) калия 770
Тетраиодаурат(Ш) калия 774
Тетракарбонилжелеза
бромид 537
дииодид 537
хлорид 537
Тетракарбонилкобальтовая кислота
578, 579
Тетракарбонилродиевая кислота 643
Тетраметилплатина 675
Тетрамминзолота(Ш) нитрат 775
Тетрамминплатины(П) хлорид 669
Тетрамолибдат натрия 323
Тетранитратоаурат(Ш) калия 775
Тетранитрозилжелезо 538
Тетранитроплатинат(П) калия 669
Тетрапероксомолибдат
калия 326
натрия 326
Тетрапероксомолибденовая кислота
325
Тетрапероксоортотанталовая кис-
лота 209
Тетрароданоаурат(111)
калия 776
натрия 776
Тетрароданозолотая кислота 776
Тетратионитрозилжелезо 538
Тетрафенилхром 275, 276
Тетрахлороаурат(Ш)
калия 773
лития 772
натрия 773
серебра 773
Тетрахлороплатинат
аммония 668
калия 668
тетрамминплатины(П) 671
Тетрацианоаурат(Ш)
аммония 776
калия 776
кобальта(П) 776
натрия 776
Тетрацианозолотая кислота 775
Тетрацианоплатинат(П)
бария 669
калия 669
Тетрацианотетрагидроксомолибдат
калия 308, 309
Технециевая кислота 439
Технеций 434 и сл.
изотопы 435
получение 435, 436
физические свойства 384—386,436
химические свойства 386—390,
436 , 437
электронная структура 383, 434
Технеций(1У)
двуокись 437, 438
тетрахлорид 390, 438
Технеций(УП)
окись 438, 439
862
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
отделение от Mo(VI) и Re(VII)
439, 440
— от U(VI) и Mo(VI) 440
сульфид 439
триоксифторид 439
триоксихлорид 439
фторид 390, 439
Технеций(УШ)
хлорид 390, 439
Тиомолибдаты 329
Тиорениевая кислота 471
Титан 74 и сл.
карбид 103
минералы 75—77
— переработка 77
нитрид 102, 103
получение 74, 75, 78—82
— восстановлением гексафторо-
титаната 80
— лабораторный метод 81
— металлотермическим восста-
новлением тетрахлорида 79
— термической диссоциацией
тетраиодида 80
— электролитическое 81
превращение порошка в ком-
пактный металл 82, 83
применение 85, 86
распространенность в природе 75
соединения включения 102, 103
сплавы 83, 84, 86
физические свойства 66, 67, 68,
83
химические свойства 68, 69, 73,
84, 85
шлаки, получение 78
электронная структура 65, 74
Титан(П)
гидроокись 87, 88
дибромид 88, 90
дигидрид 87
дииодид 89, 90, 91
дифторид 88, 90
дихлорид 88, 90
моноокись 87, 88
моносульфид 89, 91
Титан(Ш)
гидроокись 88, 92
окись 88, 91
сульфаты 89, 94
сульфид 89, 93, 94
трибромид 88, 93
трииодид 89, 93
трифторид 88, 92
трихлорид 88,’_92, 93
Титан(ТУ)
ацетилацетонат 102
двуокись 70, 88, 93, 95, 96
двуокись, восстановление 81
— получение 77, 78
дисульфат 89, 101
дисульфид 89, 101
купферонат 102
оксисульфат 101
тетрабромид 72, 88, 100
тетрагидрид 95
тетраиодид 72, 75, 89, 100
— термическая диссоциация 80
тетрафторид 71, 88, 98, 99
тетрахлорид 71, 75, 88, 99,
100
— металлотермическое восста-
новление 79
хелатные соединения 101, 102
Титанизация 86
«Титановая земля» 74
Титановые белила см.Титан, двуокись
Титановые квасцы 94
Томаса процесс 493
Тортвейтит 27, 37, 38, 127, 441
переработка 27
Триметаванадат аммония 168
Триметаванадиевая кислота 172, 173
Триметилплатины иодид 675
Тримолибдат
калия 323
натрия 323
Триоксалатоманганит калия 418
Триплит 392
Трисульфатогафниевая кислота 134
Трифенилхром 276
Трихлороамминплатинат(П) тетра-
амминплатины(П) 671
Трицианомарганцовая(П) кислота
412
Трицианоникелат калия 593, 594
Триэтилцинкат натрия 802
Тюямунит 143
Ферберит 335
Фергюсонит 37, 38, 180
Феррат
бария 531
калия 531
Ферримолибдит 279
Ферритунгстит 336
Феррованадий, получение 147, 148
Ферровольфрам, получение 344
Ферромолибден 279, 284
получение 288, 289
Ферротитан, получение 82
Феррохром, получение 231
Ферроцен см. Дициклопентадиенил-
железо
Флотация дифференциальная 684
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
863
Фосфорно-12-вольфрамовая кислота
369
Фтороксивольфрама г
аммония 373
калия 373
таллия 373
Фторопероксомолибдаты 326
Фторотанталаты комплексные 210
Халькозин 681
Халькопирит 551, 682
Хлоантит 583
Хлопинпт 27
Хлоровольфрамовая кислота 353
Хлорониобия хлорид 194, 195
Хризоколл 682
Хром 226 и сл.
бориды 278
гексакарбонил 273, 274
гидрид 277
карбиды 277, 278
металлоорганические соедине-
ния 273—277
минералы 227
нитриды 277
получение 229—231
— алюмотермическим восстано-
влением Сг2О3 229
— восстановлением хроматов 231
— кремнетермическим восста-
новлением Сг2О3 229
— металлотермическим восста-
новлением Сг2О3 230
— электролитическое 230
применение 233, 234
соединения включения 277, 278
физические свойства 216—218,
231
фосфиды 277
химические свойства 218—226,
232, 233, 235, 236
электронная структура 215, 226
Хром(1)
перхлорат трис(а, а'-дипири-
дил)хрома 235, 237
Хром(Н)
дибромид 219, 239, 240
дииодид 219, 240
дифторид 225, 238
дихлорид 218, 238, 239
закись 237, 238
координационные соединения 241
моносульфид 240
сульфаты 241
сульфид двойной 241
Хром(Ш)
восстановление водородом 230
гидроксохромиты 245, 246
гидроокись 244, 245
зеленый хлорид Рекура 248
квасцы хромокалиевые 250
координационные соединения
251—257
нитраты 250
окись 243, 244
— алюмотермическое восстано-
вление 229
— кремнетермическое восста-
новление 229
— металлотермическое восста-
новление 230
оксифторид 247
ортофосфат 250, 251
сульфаты 249, 250
сульфид 249
трибромид 248
трииодид 248, 249
трифторид 225, 246, 247
трихлорид 247, 248
фиолетовый хлорид Рекура 247,
248
хлорид Бьеррума 248
хромиты 227, 246
Хром(1У)
двуокись 221, 257, 258
псевдохромат бария 258
тетрафторид 221, 225, 258
тетрахлорид 221, 258
Хром(У)
оксипентахлорохроматы 260
окситетрафторохромат калия
259, 260
— серебра 259, 260
окситетрахлорохроматы 260
пентафторид 225, 259
пероксохроматы 260, 261
псевдохроматы 260
Хром(У1)
бихроматы 264, 265, 267—269
бромистый хромил 273
бромохромат калия 273
гидрохроматы 265
иодохромат 273
перекись 270, 271
пероксосоединения 270, 271
тетрахроматы 270
трехокись 222, 262, 263
трихроматы 269, 270
фтористый хромил 271, 272
фторохроматы 272
хлористый хромил 272
хроматы 263—266
Хромат
аммония 266
бария 266
864
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
калия 265
лития 265
меди 266
рубидия 266
свинца 266
серебра 266
цезия 266
Хромит, переработка 228, 229
Хромовый ангидрид см. Хром(У1),
трехокись
Цементит 541
Циклопентадиенилдикарб нил ко-
бальта 57 9
Цинк 784 и сл.
амид 798
duc-ацетилацетонат 800
бромид 783, 792, 796
гидрид 792
гидроксоцинкаты 792, 795
гидроокись 792, 794
дитизонат 800
иодид 783, 793, 796
карбид 799
карбонат 793, 799, 800
металлоорганические соедине-
ния 801, 802
минералы 784—786
нитрат 798, 799
нитрид 798
окись 792—794
ортофосфат 799
— тетрагидрат 799
очистка 788
перекись 794
получение 787, 788
применение 791
рубеанат 800
руды, переработка 786 787
сульфат 793, 797, 798
сульфид 793, 797
фторид 782, 792 795
физические свойства 780, 781,
788, 789
хелатные соединения 800
хлорид 782, 792, 795
— тетрагидрат 796
химические свойства 782—784,
789, 790
цианид 796
электронное строение 779, 784
Цинкит 785
Цинковая обманка 784, 785
Цинковый шпат 785
Циркон 105, 127
Циркониевая земля см. Бадделеит
а-Циркониевая кислота 118, 119
P-Циркониевая кислота 119
Цирконий 103 и сл.
диборид 126
дикарбид 126
карбид 125, 126
минералы 105
нитрид 125
отделение от гафния 108
получение 104, 109—111
— восстановлением гексафторо-
цирконатаЦУ) 109
— магнийтермическим восста-
новлением тетрахлорида 109
— термической диссоциацией
тетраиодида 110
— электролизом НО, 111
применение 112, 113
распространенность в природе
105
руды, переработка 105—108
соединения включения 125, 126
физические свойства 66, 67, 68.
111
химические свойства 68, 69, 73,
111, 112
электронная структура 65, 104
Цирконий(П)
дибромид 114, 116
дииодид 114, 117
дифторид 114, 116
дихлорид 114, 116
Цирконий(Ш)
трибромид 115, 116
трииодид 115, 117
трифторид 114, 116
трихлорид 114, 115, 116
ЦирконийЦУ)
ацетат 123, 124
ацетилацетонат 125
бензоилацетонат 125
гидроокись см. а-Циркониевая
кислота
двуокись 70, 115, 116, 118
дисульфат 117, 123
дисульфид 117, 122
купферонат 124
оксихинолят 124
ортосиликат 124
тетрабромид 72, 116, 121, 122
тетраиодид 72, 117, 122
— термическая диссоциация
110
тетранитрат 123
тетрафторид 71, 116, 120
тетрахлорид 71, 116, 120, 121
— магнийтермическое восстано-
вление 109
хелатные соединения 123, 125
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
865
Цирконила
гидроокись см. f-Циркониевая
кислота
дибромид 122
дииодид 122
дифторид 120
сульфид 123
тетрахлорид 121
Циртолит 105, 127
Чугуны
классификация 489, 490
получение 485—490
Шеелит 335, 336
переработка концентратов 337
Эдисона аккумулятор 610
Экабор см. Скандий
Экамарганец см. Технеций
Эритрин 543
Эритроцинкит 803
Этилдибромид золота 778
Этилртути хлорид 842
Этилцинка иодид 801
СОДЕРЖАНИЕ
Введение......................................................
Характеристика переходных металлов............................
Электронное строение.................................
Кристаллическая структура............................
Важнейшие физические, магнитные, механические н элек-
трохимические свойства переходных металлов............
Распространенность в природе ........................
Побочная подгруппа III группы периодической системы (подгруппа
скандия) .....................................................
Скандий (экабор)..........................................
Историческая справка ................................
Распространенность в природе.........................
Переработка тортвейтита .............................
Получение металлического скандия . ..................
Физические и химические свойства.....................
Соединения (общие свойства)..........................
Иттрий ...................................................
Историческая справка ................................
Распространенность в природе.........................
Извлечение иттрия....................................
Получение металлического иттрия......................
Физические и химические свойства.....................
Применение ..........................................
Соединения (общие свойства)..........................
Лантан ...................................................
Историческая справка ................................
Распространенность в природе.........................
Получение металлического лантана.....................
Физические и химические свойства.....................
Применение ..........................................
Соединения (общие свойства)..........................
Актиний ..................................................
Историческая справка ...............................
Изотопы ............................................
Распространенность в природе ........................
Выделение изотопа 227Ас.............................
Получение металлического актиния....................
Очистка.............................................
Физические и химические свойства....................
Применение .........................................
Соединения (общие свойства).........................
5
5
5
10
10
18
19
26
26
26
27
27
29
29
37
37
37
38
38
39
40
40
46
46
46
47
48
49
49
58
59
59
60
60
61
61
62
62
63
СОДЕРЖАНИЕ 867
Побочная подгруппа IV группы периодической системы (подгруппа
титана) ................................................... 65
Титан ....................................................... 74
Историческая справка..................................... 74
Распространенность в природе............................. 75
Обогащение и переработка титановых руд................... 77
Получение металлического титана.......................... 78
Получение ферротитана.................................... 82
Физические и химические свойства......................... 83
Применение .............................................. 85
Соединения (общие свойства).............................. 86
Цирконий..................................................... 104
Историческая справка .................................... 104
Распространенность в природе............................. 105
Обогащение и переработка циркониевых руд................. 105
Отделение циркония от гафния............................. 108
Получение металлического циркония........................ 109
Физические и химические свойства......................... 111
Применение .............................................. 112
Соединения (общие свойства).............................. ИЗ
Гафний ...................................................... 126
Историческая справка .................................... 126
Распространенность в природе............................. 127
Обогащение и переработка гафниевых руд................... 127
Получение металлического гафния ......................... 128
Физические и химические свойства......................... 129
Применение .............................................. 130
Соединения (общие свойства).............................. 130
Побочная подгруппа V группы периодической системы (подгруппа
ванадия) ................................................. 136
Ванадий ..................................................... 141
Историческая справка .................................... 142
Распространенность в природе............................. 142
Переработка руд, содержащих ванадий...................... 143
Получение металлического ванадия......................... 145
Получение феррованадия................................... 147
Электролитическая очистка................................ 148
Физические и химические свойства......................... 148
Применение .............................................. 150
Соединения (общие свойства).............................. 151
Ниобий ...................................................... 179
Историческая справка .................................... 179
Распространенность в природе............................. 180
Переработка руд, содержащих ниобий и тантал.............. 180
Получение металлического ниобия.......................... 183
Физические и химические свойства......................... 184
Применение............................................... 185
Соединения (общие свойства).............................. 186
Тантал....................................................... 198
Историческая справка .................................... 198
Распространенность в природе............................. 199
Получение металлического тантала......................... 199
Физические и химические свойства......................... 201
Применение .............................................. 202
Соединения (общие свойства).............................. 203
868
СОДЕРЖАНИЕ
Побочная подгруппа VI группы периодической системы (подгруппа
хрома)........................................................ 215
Хром ..................................................... 226
Историческая справка ................................. 226
Распространенность в природе ......................... 227
Переработка хромитовых руд............................ 228
Получение металлического хрома ....................... 229
Получение феррохрома.................................. 231
Физические и химические свойства...................... 231
Применение ........................................... 233
Соединения (общие свойства)........................... 234
Молибден ................................................. 278
Историческая справка ................................. 278
Распространенность в природе ......................... 279
Обогащение и переработка молибденовых руд............. 280
Получение металлического молибдена ................... 284
Получение ферромолибдена ............................. 288
Превращение порошкообразного металлического молиб-
дена в компактный металл.............................. 289
Очистка............................................... 289
Физические и химические свойства...................... 290
Применение ........................................... 293
Соединения (общие свойства)........................... 295
Вольфрам.................................................. 334
Историческая справка ................................. 334
Распространенность в природе ......................... 335
Обогащение и переработка вольфрамовых руд............. 336
Получение металлического вольфрама.................... 341
Получение ферровольфрама ............................. 344
Физические и химические свойства ..................... 345
Применение ........................................... 348
Соединения (общие свойства)........................... 352
Побочная подгруппа VII группы периодической системы (подгруппа
марганца)..................................................... 383
М арганец ................................................ 390
Историческая справка ................................. 391
Нахождение в природе.................................. 391
Получение металлического марганца..................... 392
Очистка............................................... 394
Физические и химические свойства...................... 394
Применение ........................................... 397
Соединения (общие свойства)........................... 397
Технеций (экамарганец).................................... 434
Историческая справка ................................. 435
Получение металлического технеция .................... 435
Физические и химические свойства...................... 436
Соединения (общие свойства)........................... 437
Рений .................................................... 440
Историческая справка ................................. 440
Распространенность в природе.......................... 441
Извлечение рения из молибденита....................... 441
Получение металлического рения........................ 443
Физические и химические свойства...................... 444
Применение ........................................... 447
Соединения (общие свойства)........................... 448
СОДЕРЖАНИЕ 869
Побочная подгруппа VIII^ группы периодической системы.......... 443
Железо .................................................... ЬТЭ
Историческая справка .................................. 480
Распространенность в природе .......................... 482
Получение металлического железа ....................... 484
Получение чугуна или сырого железа..................... 485
Сталь......................~........................... 490
Физические и химические свойства....................... 495
Применение ............................................ 501
Соединения (общие свойства)............................ 501
Кобальт ................................................... 541
Историческая справка .................................. 541
Распространенность в природе .......................... 542
Извлечение кобальта из руд............................. 544
Получение металлического кобальта ..................... 545
Очистка....................;........................... 547
Физические и химические свойства....................... 547
Применение ............................................ 550
Соединения (общие свойства) ........................... 550
Никель .................................................... 581
Историческая справка .................................. 582
Распространенность в природе........................... 582
Извлечение никеля из руд............................... 583
Получение металлического никеля ....................... 587
Очистка................................................ 588
Физические и химические свойства....................... 589
Применение ............................................ 592
Соединения (общие свойства)............................ 593
Металлы подгруппы платины ..................................... 615
Рутений ................................................... 620
Историческая справка .................................. 621
Распространенность в природе .......................... 621
Извлечение и получение металлического рутения .... 621
Физические и химические свойства....................... 621
Применение ............................................ 622
Соединения (общие свойства)............................ 622
Осмий ..................................................... 629
Историческая справка .................................. 629
Распространенность в природе .......................... 629
Извлечение и получение металлического осмия............ 630
Физические и химические свойства....................... 630
Применение ............................................ 631
Соединения (общие свойства) ........................... 631
Родий ..................................................... 637
Историческая справка .................................. 637
Распространенность в природе .......................... 637
Извлечение и получение металлического родия............ 637
Физические и химические свойства....................... 638
Применение ............................................ 639
Соединения (общие свойства)............................ 639
Иридий .................................................... 643
Историческая справка................................... 644
Распространенность в природе .......................... 644
Извлечение и получение металлического иридия........... 644
Физические и химические свойства....................... 645
870
СОДЕРЖАНИЕ
Применение ...................................... 646
Соединения (общие свойства)............................. 646
Палладий ................................................... 651
Историческая справка .................................. 651
Нахождение в природе ................................... 652
Извлечение и получение металлического палладия .... 652
Физические и химические свойства........................ 652
Применение ............................................. 653
Соединения (общие свойства) ............................ 654
Платина .................................................... 660
Историческая справка ................................. 661
Распространенность в природе ......................... 661
Извлечение и получение металлической платины............ 662
Физические и химические свойства........................ 662
Применение ............................................. 664
Соединения (общие свойства)............................. 665
Побочная подгруппа I группы периодической системы (подгруппа меди) 676
Медь........................................................ 681
Историческая справка ................................... 681
Распространенность в природе ......................... 681
Переработка медных руд ............................... 683
Аффинаж и рафинирование .............................. 688
Физические и химические свойства ..................... 689
Применение ............................................. 693
Соединения (общие свойства)........................... 694
Серебро .................................................... 724
Историческая справка ................................. 724
Распространенность в природе ......................... 724
Переработка серебряных руд и получение металлического
серебра . . . х....................................... 726
Рафинирование серебра.................................. 729
Физические и химические свойства........................ 730
Применение ............................................. 733
Соединения (общие свойства)............................. 733
Золото...................................................... 752
Историческая справка ................................... 752
Распространенность в природе ........................... 753
Переработка золотоносных руд............................ 754
Извлечение золота из концентратов золотоносных руд . . . 755
Очистка................................................ 759
Физические и химические свойства....................... 760
Применение ............................................ 762
Соединения (общие свойства) ............................ 763
Побочная подгруппа II группы периодической системы (подгруппа
цинка) ................................................. 779
Цинк ....................................................... 784
Историческая справка................................... 784
Распространенность в природе .......................... 784
Переработка цинковых руд и получение металлического
цинка................................................. 786
Очистка сырого цинка................................... 788
Физические и химические свойства....................... 788
Применение ........................................... 791
Соединения (общие свойства)............................ 791
Кадмий ................................................... 802
СОДЕРЖАНИЕ 871
Историческая справка.................................. 802
Распространенность в природе.......................... 803
Извлечение кадмия из руд. Получение металлического
кадмия ............................................... 803
Очистка............................................... 804
Физические и химические свойства...................... 804
Применение............................................ 806
Соединения (общие свойства)........................... 807
Ртуть ..................................................... 818
Историческая справка ................................. 818
Распространенность в природе.......................... 819
Получение ............................................ 820
Очистка............................................... 821
Физические и химические свойства...................... 821
Применение............................................ 823
Соединения (общие свойства)........................... 824
Список литературы ......................................... 843
Предметный указатель....................................... 845
РРипан, И.Четяяу
НЕОРГАНИЧЕСКАЯ
ХИМИЯ