/
























































































































































































































































































Текст
Ж I'M'JT
АКАДЕМИЯ НАУК СССР
М.В.ВОЛЬКЕНШТЕЙН
*
СТРОЕНИЕ
МОЛЕКУЛ
ИЗДАТЕЛЬСТВО АКАДЕМИИ НАУК СССР
МОСКВА • 1 9 47 • ЛЕНИНГРАД
АКАДЕМИЯ НАУК СССР
НАУЧНО-ПОПУЛЯРНАЯ СЕРИЯ-МОНОГРАФИИ
М.В. ВОЛЬКЕНШТЕЙН
СТРОЕНИЕ
МОЛЕКУЛ
ИЗДАТЕЛЬСТВО АКАДЕМИЙ НАУК СССР
МОСКВА <г 1947 <г ЛЕНИНГРАД
Иод общей редакцией Комиссии АН СССР
по изданию научпо-лоиуляриой литературы
Председатель комиссии Президент АН СССР
академик С. Л. ВАВИЛОВ
Зам. председателя члеп-корреспопдепт АН ('ССР
П. Ф. ЮДИН
ПРЕДИСЛОВИЕ
В основе современной физики и химии лежат
представления о строении вещества, полученные в результате
изучения атомов, молекул, жидкостей и кристаллов. Эта книга
посвящена строению молекул — главного объекта
химической науки, развитие которой привело ее к слиянию с
физикой именно в этой области. Основная задача книги —
охарактеризовать 'физические методы теоретического и
экспериментального исследования молекул и его результаты.
Теоретические построения, о которых будет итти речь, в настоящее
время не закончены; еще ведется большая работа по
физическому обоснованию химических воззрений, по определению
общего физического смысла разнообразных химических
явлений.
Тема книги настолько специальна, что популярное
изложение в полном смысле слова оказалось невозможным. Тем
не менее, предпринята попытка изложить материал с
наибольшей возможной простотой, избегая сложных математических
выкладок. Если, прочитав книгу, студент или научный
работник, физик или химик заинтересуется областью строения
молекул и обратится к более специальной литературе, задача
книги будет выполнена.
Яне стремился описать в этой книге все свойства молекул,
а ограничился лишь теми, которые с моей точки зрения
являются необходимыми для получения общих представлений.
Так, оставлены без внимания магнитные свойства, многие
вопросы, относящиеся к электронным спектрам и т. д.
Свойства жидкостей, высокополимерных веществ также выходят
за рамки настоящей работы. Вопросы строения атома
* з
в первой главе изложены конспективно, ибо им посвящен
целый ряд хороших книг. Библиография, приводимая в
конце книги, ограничена, в основном, избранными
монографиями и обзорами на русском языке, в которых
читатель найдет более детальные ссылки.
Сердечно благодарю О. В. Бланк-Волькенштейн, а также
Б. И. Степанова, П. П. Феофилова и М. А. Ельяшевича
прочитавших книгу в рукописи и сделавших ряд ценных
указаний.
Глава I
ВВЕДЕНИЕ
Развитие химической науки еще в начале прошлого века
привело к понятию о молекуле, как наименьшей частице —
носителе химических свойств вещества. В свою очередь,
в физике также оказалось необходимым применить
представление о молекулах; этого потребовало развитие учения
о теплоте — кинетическая теория газов. Однако, именно
химики научились первыми разбираться в строении молекул.
И если на первых порах и атом и молекула воспринимались
как рабочие гипотезы, то ныне уже не приходится
останавливаться на доказательствах их существования — они
общеизвестны. Молекула и атом существуют реально, мы имеем
право и должны говорить об их физических и химических
свойствах и строении.
В настоящее время физика и химия объединяются в
пограничной области знания, основная задача которой состоит
в изучении строения вещества. В строении молекул химик
находит объяснение особенностей в течении химических
реакций. Пользуясь общими сведениями о строении молекул,
химик-приходит к объяснению индивидуальных свойств
отдельных веществ, находит ответ на свой основной вопрос: что
получается в результате данного частного химического
процесса? С другой стороны, физик находит в строении атомов,
молекул, кристаллов объяснение общих свойств вещества:
механических, термических, электрических, оптических.
Физик решает, таким образом, задачу о том, как происходит
то или иное явление.
ФИЗИЧЕСКИЕ И ХИМИЧЕСКИЕ СВОЙСТВА ВЕЩЕСТВА
Строение молекул определяет физические и химические
свойства вещества. Обратно, исследование этих свойств есть
единственный метод познания строения молекул. Физический
и, особенно, химический эксперименты дают нам в большинстве
5
случаев сведения не об отдельных молекулах, а об их
совокупностях, содержащих огромное число молекул. Мы можем,
однако, перейти к свойствам отдельных молекул, пользуясь,
как это обычно делается в химии, представлением об
аддитивности, полагая, что отдельная молекула обладает теми
же свойствами, что и вся совокупность. Мы можем
оперировать средними свойствами молекул; так поступает,
например, кинетическая теория газов. Наконец, в ряде
физических явлений мы непосредственно встречаемся со
свойствами отдельных молекул.
Определив свойства молекулы, мы переходим к проблеме
ее строения. В основе химической и физической науки
лежит идея атомизма, непосредственно вытекающая из
результатов изучения окружающего нас мира. Молекула,
так же как и атом, построена из положительных ядер
и отрицательных электронов. Молекула есть сложное
образование. Поэтому при исследовании молекулы мы всегда,
прежде всего, выделяем элементы, из которых она строится:
ядра, электроны, атомы, группы атомов, химические связи.
Конечно, свойства молекулы не являются простой суммой
свойств составляющих элементов. Но в качестве первого
приближения мы и здесь можем руководствоваться идеей
аддитивности, т. е. определять свойство молекулы именно
ікак сумму свойств составляющих элементов, а уже затем
рассматривать отклонения от аддитивности. Такой метод
широко применяется в науке о природе, целесообразность
его очевидна.
Если строение молекулы считается известным, то это
означает, прежде всего, что известна ее геометрия.
Построенная из материальных частиц, молекула обладает
определенным объемом и формой. Но понятие „геометрия молекулы"
означает большее: оно включает в себя знание внутренней
структуры молекулы, расположения в ней тяжелых ядер
и электронов, длин химических связей.и значений углрв
между ними. Геометрия молекулы определяется теми
свойствами, которые условно можно назвать механическими. Это—
силы, действующие между атомами в молекуле, энергии
связей. К механическим свойствам молекул относятся и
частоты колебаний атомов в молекуле, угловые скорости
вращения молекулы как целого и отдельных ее частей-
Внутримолекулярные силы имеют электрическую
природу..Естественно перейти далее к электромагнитным свойствам молекул,
определяющим диэлектрическую постоянную и магнитную
проницаемость вещества. Здесь речь идет об относительном
расположении центров тяжестей положительных и
отрицательных зарядов в молекуле, о способности электронов
в молекуле смещаться под действием электрических и
магнитных сил. Этой же способностью, в силу электррмагнит-
6
ной природы света, обусловлены оптические свойства
молекул, проявляющиеся в преломлении, отражении, рассеянии,
поляризации, поглощении и испускании света веществом.
Из величины показателя преломления, из спектров
поглощения, испускания и рассеяния мы получаем особенно
денные и разнообразные сведения о строении молекул. И вообще,
в экспериментальных методах изучения физических свойств
и строения молекул мы, главным образом, имеем дело со
взаимодействием вещества со светом различных длин волн.
Изучение процессов рассеяния молекулами коротковолновых
рентгеновых лучей и спектров поглощения длинноволновых
инфракрасных лучей дает возможность определить
междуатомные расстояния—изучить геометрию молекулы.
Исследуя поглощение и рассеяние во всех областях спектра, мы
познаем природу внутримолекулярных сил. Определение
электромагнитных свойств неотделимо от исследования
преломляющей способности и ряда электро- и магнитооптических
явлений.
МЕТОДЫ ТЕОРИИ МОЛЕКУЛ
Современная физика показала, что к мельчайшим
частицам— электронам, атомам, вообще говоря, нельзя
применять те же классические представления, которыми мы с
полным правом пользуемся при изучении тел обычных размеров.
В своем движении, в процессах взаимодействия со светом
электроны подчиняются законам не классической, а
квантовой физики. Грубо говоря, классические законы применимы
в тем большей степени, чем больше масса и энергия
изучаемого тела.
Детали строения молекулы и те ее свойства, которые
непосредственно связаны с электронами в молекуле, не могут
быть поняты без помощи квантовой физики. Сюда относится
^природа химической связи, структура электронных спектров
и т. д. С другой стороны, классическая физика с успехом
применяется для объяснения ряда свойств молекул, причем
под объяснением мы разумеем не только качественное
толкование, но и количественный расчет. Классическая
физика объясняет те свойства молекул, которые связаны
с движением молекулы как целого или с движениями
весьма тяжелых по сравнению с электронами атомных ядер
внутри молекулы. Именно этими движениями определяется
структура инфракрасных спектров молекул и спектров
комбинационного рассеяния, диэлектрические свойства
молекул и т. д. Классическое описание имеет важное для
экспериментатора преимущество наглядности. В определенных
пределах классические методы применимы и для описания
явлений, по самой своей природе электронных, квантовых,
таких, как, например, дисперсия света, люминесценция,
оптическая активность. В-перечисленных случаях
представление о классическом колеблющемся электроне несомненно
помогает исследованию. Применимость такого представления
объясняется тем, что в этих явлениях речь идет о внешних
электронах молекулы, обладающих большими энергиями.
Более глубокая, количественная интерпретация этих явлений
требует, конечно, квантовой теории. Но применение
классических представлений в теории молекул определяется не
только их наглядностью и внешней привлекательностью.
Дело в том, что полное и строгое квантовомеханическое
описание возможно лишь в простейших случаях —
соответствующая математическая задача большей частью трудно
; разрешима. В то же время, задача физической теории состоит
, не в том, чтобы подсчитать a priori результаты. любого
1 опыта, но лишь в нахождении принципиальной сущности
явления и принципиального метода его количественного
исследования. Не имеет смысла, например, рассчитывать,
применяя сложные математические методы, величины,
которые могут быть легко определены из опыта, конечно при
условии, что теория вообще объясняет их сущность,
происхождение и порядок. Именно поэтому могут оказаться
полезными наглядные классические представления. С их
помощью мы получаем возможность особенно просто связать
одни явления с другими, найти, хотя бы качественно, их
общие причины. Впрочем, наглядность и простота — понятия
очень условные; можно не сомневаться, что в недалеком
будущем основные идеи квантовой механики станут и
привычными и наглядными для каждого физика и химика.
Качественное применение общих квантовомеханических
принципов, наряду со строгой математической теорией,
существенно содействует изучению строения и свойств
молекул.
( Кроме того, молекулы обладают и более простыми свой-
I ствами, исследование и правильный учет которых не зависят
от того, будем ли мы пользоваться классической или
квантовой механикой. Это — свойства симметрии. Молекулы, как
\ пространственные образования,имеют ту или иную симметрию.
'Зная симметрию молекулы, мы можем сразу решить многие
важные задачи строения, например установить, полярна или
неполярна молекула, способна она или не .способна вращать
плоскость поляризации, сколько линий и полос должны
содержать ее спектры поглощения, испускания,
комбинационного рассеяния. Все выводы, основанные на свойствах
симметрии, носят абсолютно строгий характер. Применение
свойств симметрии сильно упрощает теоретические расчеты,
понижает степени встречающихся при этом уравнений и т. д.
Именно с этих свойств необходимо начинать исследование
8
строения молекул. Знание симметрии позволяет
охарактеризовать молекулу в целом, но для детального изучения
особенностей строения его уже недостаточно. Здесь
приходится обратиться к другому важнейшему принципу
исследования — к идее аддитивности, уже упоминавшейся
выше.
Те физические явления, в которых находят свое
выражение свойства молекул, всегда происходят при изменении
энергии молекулы; т. е. энергии электронов, атомных ядер,
энергии молекулы в целом. Как мы указали, при этом особое
значение имеют процессы взаимодействия молекул со светом.
Во многих случаях энергия молекулы и ее составных
частей может быть представлена как энергия
периодических или приближенно периодических движений, роль
которых во всех физических явлениях исключительно
велика. Говоря на языке классической физики, мы имеем
здесь дело с колебаниями и вращениями электронов, ядер,
молекул и с взаимодействующими с ними колебаниями
световой волны. На протяжении всего дальнейшего изложения
нам придется постоянно иметь дело с колебательными
процессами.
Большая сложность задачи о строении молекулы
вынуждает пользоваться приближенными методами теоретического
расчета. Строгое решение квантовомеханической задачи,
а также задачи классической механики доступно лишь
в случае системы, состоящей из двух тел, двух материальных
точек. Уже самая простая молекула — ион молекулы
водорода содержит три таких точки: два протона и электрон.
Поэтому все расчеты основных свойств молекул—приближенные.
Но даже проведение приближенных расчетов в большинстве
случаев очень затруднительно. Здесь необходимо установить
целесообразное разделение сил между теорией и
экспериментом. Особого внимания в теории молекул заслуживают
именно полуэмпирические методы. Положим в основу
исследования некоторую систему теоретических представлений,
характеризующую явления при помощи физических величин,
находимых из опыта. Такая система позволяет расширить
объем исследуемого материала, так как, применяя величины,
полученные из некоторых опытов, мы можем предсказать»
результаты большего числа опытов. Ценность
полуэмпирической теории определяется ее способностью связать
разнообразные свойства молекул, найти их общее объяснение,
способностью, зная величины, относящиеся к одному виду
свойств, предсказывать результаты опытов в другой области
явлений.
Современная теория строения молекул широко применяет
изложенные методы и достигла на этом пути больших
и важных успехов.
ОСНОВНЫЕ ПОНЯТИЯ КВАНТОВОЙ МЕХАНИКИ
Прежде чем перейти к непосредственному рассмотрению
строения и физических свойств молекул, изложим вкратце
основные представления современного учения о свойствах
электронов, атомов и света.
Световые волны и кванты
Классическая физика показала, что свет представляет
собой переменное электромагнитное поле,
распространяющееся в пространстве со скоростью с = 3* 1010 см/сек в виде
поперечных к направлению распространения волн (рис. 1).
Электрическая сила Е периодически меняет свое значение
во времени и пространстве
X
Е = Еп cos 2^v
t )= EQ cos 2tt (kx — v/),
(i)
где t — время, x -~ координата в направлении
распространения, v — частота колебаний (число периодов в секунду),
k — волновое число, к — длина волны
" =т, (2)
* =
Е0 — амплитуда колебаний, наибольшее возможное значение
электрической силы. Волновые свойства света доказываются
рядом опытов и прежде всего явлениями интерференции
Рис. 1. Световая волна.
и диффракции. Электромагнитная природа света есть
следствие из всего учения об электричестве и магнетизме
(Максвелл, 1865).; она доказывается существованием и
свойствами радиоволн (Гертц, 1887), отличающихся от видимого
света лишь большими значениями X, и многими
экспериментами, из которых особенно существенны относящиеся
к давлению света (Лебедев, 1901).
Написав формулу (1), мы молча предположили, что
световая волна распространяется из минус бесконечности к плюс
бесконечности (рис. 2, а), и поэтому на всем своем
бесконечном протяжении может быть охарактеризована частотой v
Ю
и длиной волны X. В действительности, всякая световая
волна имеет начало и конец в пространстве и времени; она і
испускается атомом или молекулой в определенный момент ¦
и в определенной точке пространства. Испускание волны
длится не бесконечно, через некоторое время источник
гаснет. Поэтому реальная волна имеет в сущности форму
рис. 2, б.
Свойства световых волн, их длины X и частоты v могут
быть исследованы, -в частности, при помощи спектроскопа.
Спектральные приборы взаимодействуют со световыми
волнами таким образом, что во всех случаях выделяют
отдельные бесконечные синусоиды вида 2, а, характеризуемые
определенными X и v. Если на спектрограф попадает волна
2, а, то в спектре появится одна линия с длиной волны X.
лллККлллла-
Н s *і
Рис. 2,а — монохроматическая световая волна; б— ограниченный световой
импульс.
Если попадет реальный световой импульс 2, б конечной |
длины 5, то в спектре появится целый ряд линий с раз- j
ными X, образующих широкую полосу. Это объясняется тем, і
что импульс 2,6 может быть представлен целым набором '
синусоид с различными X, подобранных таким образом,
чтобы они гасили друг друга в тех областях пространства,
в которых волна отсутствует, и давали бы правильную
картину 2, б в области S. Таких синусоид потребуется тем
больше, интервал значений X будет тем шире, чем короче
импульс S.
Представим себе два опыта, производимые над
реальной световой волной. В первом из них мы хотим точно
определить время прохождения световой волны через дан^/
ную точку пространства. Для этой цели будем
регистрировать световую волну на фотографической пластинке;
падающей в своей вертикальной плоскости перпендикулярно
к горизонтально распространяющемуся лучу света. Если
волна бесконечна, то поперек всей пластинки пройдет темная
черта — след световой волны, и мы не сможем ничего ска-
П
загь о времени прохождения света. Чем короче будет
световой импульс, тем короче будет черта на фотопластинке,
тем точнее мы ' сможем определить время прохождения.
Очевидно, что точное определение времени возможно лишь
для бесконечно короткого светового импульса. Во втором
опыте мы хотим измерить частоту колебаний световой
волны. Для этого пропустим луч света через призму
спектрографа и вновь будем фотографировать проходящий свет,
на этот раз на неподвижной пластинке. Если волна
бесконечна и содержит колебание лишь с одной частотой v, на
пластинке будет наблюдаться одна точка — частота будет
точно определена. Но в этом случае мы ничего не можем
сказать о времени прохождения волны. Если волна
ограничена, то, как уже указывалось, она представляется
непрерывным набором синусоид — на пластинке спектрографа
точка расплывается в полоску. Точность определения частоты
упадет, но возрастет точность определения времени. Наконец,
в случае бесконечно короткого импульса неопределенность
частоты возрастает до бесконечности, но время
прохождения можно определить как угодно точно. Таким образом,
невозможно одновременное точное измерение частоты и
времени прохождения световой волны. Неточности связаны
соотношением
AvA* = l. (3)
Аналогичное соотношение неточностей имеет место для
длины волны и пространственного положения светового
импульса
А*Адг=1. (4)
Источник этих неточностей, свидетельствующих о строгой
применимости понятий частоты и волнового числа лишь
к бесконечному световому импульсу, лежит в самой
волновой природе света. Приведенные рассуждения принадлежат
академику Л. И. Мандельштаму.
Волновая теория света оказалась недостаточной для
описания ряда явлений взаимодействия света и вещества,
таких, как фотоэффект, излучение накаленного тела и т. д.
М. Планк (1900) показал, что лучистая энергия выделяется
накаленными телами в виде отдельных порций — квантов,
обладающих энергией
e = Av, (5)
где h = 6.628- 10т97 эрг. сек. — постоянная Планка. Это же
представление о корпускулярной природе света оказалось
необходимым для объяснения законов фотоэффекта,
фотохимических реакций (Эйнштейн, 1905, 1912).. В настоящее
время совершенно очевидно, что волновая и корпускулярная
теории света не исключают, а дополняют друг, друга, объяс-
12
няя разные стороны одних и тех же явлений. Для того
чтобы связать воедино обе теории, Эйнштейн предположил,
что энергия световых волн в единице объема,
пропорциональная, согласно электромагнитной теории света, величине
Я3 [ср. (1)], характеризует число световых квантов—фотонов,.-'
находящихся в единице объема. Иными словами ?2 есть'
мера вероятности нахождения фотона в точке х.
Волны материи
В 1927 г. американские физики Дэвиссон и Джермер,
изучая рассеяние электронов металлической поверхностью,
совершенно случайно сделали очень важное открытие
Аппарат, в котором исследовалась поверхность куска никеля,
испортился, и во время его исправления никель был
перегрет настолько, что закристаллизовался. Поверхность
кристаллического никеля рассеивала пучок электронов не
одинаково по всем направлениям. Электроны повели себя
подобно световым волнам, осуществляющим диффракцию.
Незадолго до этого Луи де-Бройль высказал
предположение, что движущийся электрон может рассматриваться
как некоторый волновой импульс. Указанные опыты
подтвердили эту возможность. Согласно теории де-Бройля,
движению частицы с массой т и скоростью v соответствует
длина волны
Медленному электрону, двигающемуся со скоростью 1 см/сек,
соответствует длина волны 7,3 см; быстрому электрону,
двигающемуся со скоростью, создаваемой ускоряющим
потенциалом в 100 вольт, соответствует А. = 1.2 А. Таким
образом, меняя скорости электронов, можно получить
электронные волны любой частоты. В настоящее время
электронные волны применяются в электронных микроскопах, где
особенно важно получать потоки быстрых электронов с малой
длиной волны.
Другая область применения электронных волн — это
изучение строения молекул, твердых тел и жидкостей при
помощи диффракции электронов (см, главу III).
Таким образом, оказалось, что не только свет, но и
материальные частицы — электроны — обладают- как
корпускулярными, так и волновыми свойствами. Здесь имеется далеко
идущая аналогия между светом и материей. В области
световых явлений мы сталкиваемся с волновыми свойствами
света при работе с предметами размеров, соизмеримых
с длиной волны (например, узкие щели диффракционной
решетки). Подобно этому волновые свойства электронов
13
проявляются при их взаимодействии с кристаллами, у которых
расстояния между атомами также соизмеримы с длиной
волны электронов. Так же как и свет, электроны могут
быть описаны при помощи некоторой волновой функции й
[аналогичной Е в формуле (1)], квадрат абсолютного
значения которой | <5* |2 характеризует вероятность найти электрон
в данной точке пространства.
Современная физика показывает, что реально измеряемой
величиной является именно вероятность найти материальную
частицу в данном состоянии, сданной энергией и импульсом,
в данной области пространства, в данный момент времени;
мы не имеем способа точно описать поведение частицы,
так, как это делается в классической механике
применительно к телам обычных размеров. Оказывается, что
невозможно одновременно определить со сколь угодно большой
точностью положение и импульс электрона, его энергию
и время существования в состоянии с этой энергией. Мы
встречаемся здесь с ограниченностью наших понятий
координаты и скорости, энергии и времени, подобной
ограниченности понятия частоты световой волны (см. выше), строго
применимого лишь в случае бесконечного светового импульса.
Эта ограниченность опять-таки определяется волновой
природой материи и находит'свое математическое выражение
в соотношениях неопределенности Гейзенберга, являющихся
следствием основных положений ,нашей науки. Эти
соотношения подобны классическим условиям (3), (4) и имеют вид
' bpHx^zh, (7)
ДеД*г&А, (8)
формально они могут быть получены из соотношений (3),
(4) путем умножения их на А и применения уравнения (5).
и доказываемого в квантовой механике соотношения р =й&.
Для иллюстрации этих соотношений можно предложить
следующее рассуждение.
Рассмотрим поведение электрона под некоторым
мысленным сверхмикроскопом. Мы тем лучше разглядим электрон,
тем точнее определим его положение, чем меньше длина
волны света, которым мы его осветили, ибо разрешающая
способность микроскопа возрастает с уменьшением длины
волны. Но с уменьшением длины волны растет частота*
а значит энергия и количество движения светового кванта.
Такой квант, соударяясь с электроном, меняет его импульс р>
который становится неопределенным. Очевидно, что эффект
столкновения тем больше, чем больше энергия кванта.
Расчет приводит к соотношению (7), Естественно, что эти
неточности слишком малы в силу малости величины А, чтобы
быть замеченными в мире предметов обычных размеров.
Мы видим, что в микромеханике обычные способы опре-
деления и измерения физических величин изменяют свой
смысл — сами эти величины получают иные^ определения.
В частности, мы не имеем права более говорить о траэк-
тории электрона внутри атома так же, как мы говорим
о траэкториях больших тел. Электрон в атоме
непосредственно проявляет свои волновые свойства.
Волновая механика
Таким образом, любая материальная частица или система
частиц может быть описана при помощи некоторой
волновой функции ф. Квадрат абсолютного значения этой функции
| ф |2 характеризует плотность частиц в данной области
пространства или вероятность найти частицу в этой области.
Шредингер (1927) разработал математическую теорию,
позволяющую найти эту функцию
вероятности для любых частиц,
находящихся в любых условиях —
под действием каких-либо
внешних или внутренних сил.
Функции ф определяются некоторым
уравнением — волновым
уравнением Шредингера, которое
одновременно со значениями ф как
функции координат частицы дает
энергию частицы е. Свободно
летящий электрон может иметь
любую скорость и любую
кинетическую энергию. Иначе обстоит
дело с частицей, находящейся
под действием каких-либо сил, обладающей потенциальной
энергией. Пусть вне некоторой-области пространства 0 —а
потенциальная энергия электрона возрастает до
бесконечности (рис. 3). Электрон не может вырваться из области
О —а, для этого он должен был бы обладать бесконечно
большой энергией. Это означает, что электронные волны
не проходят сквозь вертикальные стенки изображенного
на рисунке „потенциального ящика", но отражаются от этих
стенок, образуя систему стоячих волн, подобных
устанавливающимся при колебаниях струны с закрепленными
концами. Стоячие волны описываются уравнением
Рис. 3. Плотности
вероятностей ф2 для электрона в
потенциальном ящике.
X X
ф = A sin 2т. -j- -\- В cos 2тс у.
На границах области должны быть узлы волн при х
и х = а, ф = 0. Следовательно 5 = 0 и
(9)
= 0
4» = A sin 2іс -у.
(10)
15
С другой стороны,
sin 2* у = 0, (11)
или
¦-у-=діг, (Па)
где п — любое 'целое число. Электронные волны могут иметь
длину
•\ с\ 1а а. 2й /іл\
Х = 2а, а, у, у , \.. , -, (12)
где п — квантовое число. Но, согласно (6),
h hn /io\
V = HS = 2^' <13)
следовательно скорость, а значит и энергия электрона
mv* n-h
е~~2~ ~?та*
(14)
квантованы, т. е. могут принимать не любые, а лишь
определенные значения, отвечающие целым п.
На рис. 3 показаны первые уровни энергии е,
отвечающие л=1 и 2, а также вероятность ф2 [ср. (9), (12)] найти
электрон в той или иной точке ящика при этих энергиях.
При я=1 электрон вернее всего найти на расстоянии а/2
от начала координат, при /і=2— на расстояниях а/4 и За/4.
Этот пример мы заимствовали у Раиса.
Гармонический осциллятор
Рассмотрим еще один пример частицы, находящейся под
действием внешней силы. Пусть это будет сила упругости,
стремящаяся вернуть частицу в положение равновесия и по
величине пропорциональная
^штхтга _ смещению частицы х из этого
*етгтт*»т« -^ положения (рис. 4)
Рис. 4. Гармонический осциллятор. *= ' ^ ^
где k — коэффициент
упругости. Легко показать, что под действием такой силы
отклоненная частица совершает гармонические колебания. Сила
равна произведению из массы т на ускорение х. Имеем
тх — —~ kx. (16)
Но это уравнение имеет решение
x = x$cos2^t (17)
16
Действительно, скорость движения частицы, первая
производная х по времени t9 согласно (17), равна
-^~х = — 2wxQ sin 2-^v/, (18)
а ускорение — вторая производная
^ = х = — і*Ч*х0 cos 2Ы. (19)
Подставляя (17) и (19) в (16) и сокращая на jc0cos2^,
получим
4«V/» = Jfe. (20)
Значит уравнение (16) действительно удовлетворяется
гармоническим решением (17), при условии для частоты
колебаний
-кУк ¦ <2і>
Потенциальная энергия частицы, находящейся под действием
упругой силы /, как известно, равна
и=Щ-. (22)
В классической теории такая частица — гармонический
осциллятор—может иметь полную энергию любого численного
значения
Квантовая механика дает иной результат. Построим, так же
как и в случае „потенциального ящика", график
зависимости потенциальной энергии осциллятора от смещения х.
Согласно (22), он будет иметь форму параболы (рис. 5).
Такая парабола вновь образует „потенциальный ящик",
в котором устанавливаются стоячие волны. Энергия частицы
оказывается квантованной и, согласно решению уравнения
Шредингера, равной
e=(n + ±)fo, (23)
где v—частота осциллятора (21), п — колебательное
квантовое число. На рис. 5 показаны уровни энергии (23) и
вероят ности ^ для соответствующих состояний осциллятора.
Сравним эти вероятности с вероятностью найти классический
осциллятор — обычный маятник—на том или ином расстоянии
от положения равновесия. Так как маятник быстрее всего
2 Строение молекул. 17
проходит через положение' х = О, а дольше всего
задерживается в крайних точках, где его скорость меняет знак,
проходя через нуль, то кривая вероятности имеет вид,
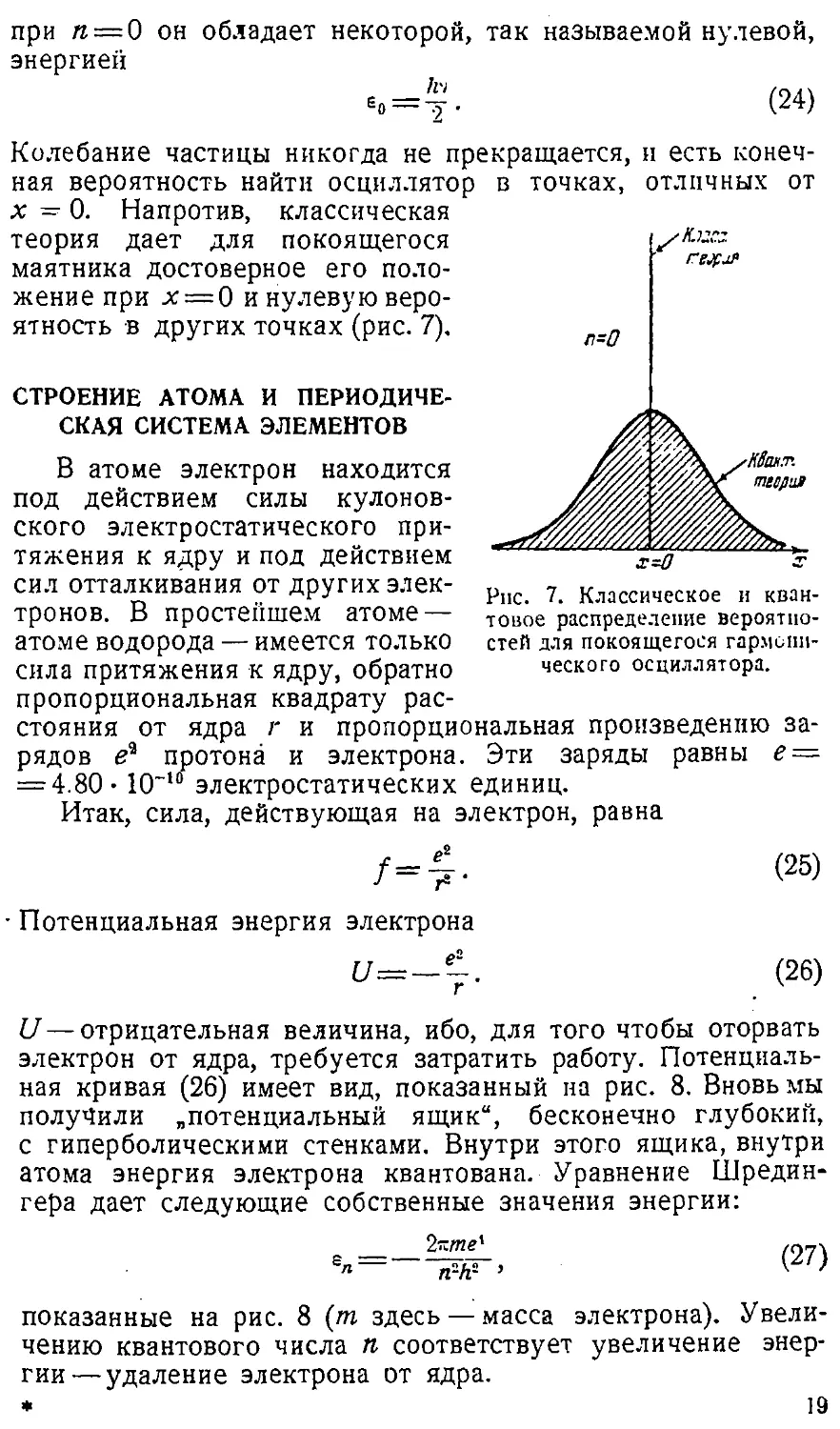
показанный на рис. 6 для случая п = 4. Классическая и кван-
Рис. 5. Плотности вероятностей &2 для гармонического
осциллятора.
товая кривые вероятности, как функции ху становятся тем
более сходными между собой, чем выше число п. Здесь
проявляется общий закон физики, согласно которому при
больших квантовых числах, при больших энергиях кван-
Ри:. 6. Классическое и квантовое распределение вероятностей для
колеблющегося гармонического осциллятора.
товые законы переходят в классические. Это так называемый
принцип соответствия Бора. Напротив, при малых значениях
п расхождение особенно явственное. Согласно (23),
квантовый осциллятор вообще никогда не останавливается, даже
18
в точках, отличных от
п*0
при л = 0 он обладает некоторой, так называемой нулевой,
энергией
*„ = !?. (24)
Колебание частицы никогда не прекращается, и есть
конечная вероятность найти осциллятор
х — 0. Напротив, классическая
теория дает для покоящегося
маятника достоверное его
положение при х = 0 и нулевую
вероятность в других точках (рис. 7),
СТРОЕНИЕ АТОМА И
ПЕРИОДИЧЕСКАЯ СИСТЕМА ЭЛЕМЕНТОВ
В атоме электрон находится
под действием силы кулонов-
ского электростатического
притяжения к ядру и под действием
сил отталкивания от других
электронов. В простейшем атоме —
атоме водорода — имеется только
сила притяжения к ядру, обратно
пропорциональная квадрату
расстояния от ядра г и пропорциональная произведению
зарядов е* протона и электрона. Эти заряды равны е =
= 4.80-10"10 электростатических единиц.
Итак, сила, действующая на электрон, равна
х*0
Рис. 7. Классическое и
квантовое распределение
вероятностей для покоящегося
гармонического осциллятора.
• Потенциальная энергия электрона
U=
?1
г
(25)
(26)
U — отрицательная величина, ибо, для того чтобы оторвать
электрон от ядра, требуется затратить работу.
Потенциальная кривая (26) имеет вид, показанный на рис. 8. Вновь мы
получили „потенциальный ящик", бесконечно глубокий,
с гиперболическими стенками. Внутри этого ящика, внутри
атома энергия электрона квантована. Уравнение Шредин-
гера дает следующие собственные значения энергии:
2тсте1
"Л
n-h-
(27)
показанные на рис. 8 {тп здесь — масса электрона).
Увеличению квантового числа п соответствует увеличение
энергии— удаление электрона от ядра.
* 19
Квантованная система может обладать только теми
значениями энергии — собственными значениями, которые
определяются условиями задачи. В случае „потенциального
ящика" это энергия (14), в случае осциллятора — (23) и
в случае атома водорода--(27). Атом может изменить свое
состояние только прерывным образом, перейти с одного
энергетического уровня на другой. Такой переход означает
прерывное возрастание или убывание энергии и может
сопровождаться поглощением или испусканием света. Частота
световой волны определится условием Бора [ср. (5)].
-п\
лп-і
2ътеА
л-г
/2,
пь яа = 1, 2, 3,
(28)
П=6
U
О
Рис. 8. Потенциальная кривая для атома водорода.
На рис. 9 представлена схема энергетических уровней
атома водорода и стрелками показаны квантовые переходы,
сопровождающиеся излучением или поглощением света.
Формула (28) таким образом описывает спектр атома.
Действительно, наблюдаемые на опыте частоты спектральных
/линий водорода очень точно передаются формулой (28).
Очевидно, что спектр атома должен быть линейчатым именно
[вследствие дискретности энергетических уровней.
Как видно из формулы (27) и рис. 8 и 9, уровни энергии
электрона в атоме сходятся к некоторому пределу. На
рис. 8 этот предел представлен осью абсцисс графика.
Предел соответствует энергии отрыва электрона. Очевидно, что
отрыв электрона — ионизация — произойдет, если мы
перейдем из самого глубокоа^-4ФО?ня #=1 на уровень л = оо.
т. е. затратим работу/'
( * = Ч^1 (29)
20
На рис. 9 приведены значения энергий уровней атома
водорода. Работа ионизации одного атома водорода равна
215 • 60 • 10"13 эргов. Значительно удобнее выражать энергию
в калориях на моль, относя ее не к атому или к молекуле,
а к грамм-атому или грамм-молекуле. Имеем
1 кг кал/моль = 4.185-10". 6.02- 10'23 =
= 25.04-1033 эрг/моль.
оо —
1
'.1-й
•
ос
Р
6
¦
1 ¦
¦ '
- і
9.28 & ч
О.ЗсгУ а
0?ч5# а
' 1.5ПГ 9
Серия
палет
' іЗёсУ *
Серая Боль мера
і
Тг 13.53 &
I
V
1
і
I
^
і
Серии
даймаио.
Рис. 9. Энергетические уровни и переходы между ними у атома
водорода.
Часто выражают энергию в электронвольтах eV, как это
и сделано на рис. 9. Электронвольт есть кинетическая
энергия, приобретаемая электроном при движении в
электрическом поле через разность потенциалов в 1 вольт. Имеем
leV = 23.07 кг кал/моль = 1.59 • 10"іа эрг.
Энергия ионизации водорода равна 13.53 eV, или 312
кг кал/моль.
Выше предельного уровня электрон оторван от ядра
и свободен в своем движении, а значит может испускать
21
и поглощать любые частоты. Таким образом, в спектре
испускания атома к серии сходящихся к пределу
спектральных линий примыкает непрерывный спектр — континуум
(рис. 10).
Наряду с работой ионизации — ионизационным
потенциалом— важной энергетической постоянной атома является
©о
е 5 y
1
а
I
300/ш? was т/.з
65Шк
Рис. 10. Серия Бальмера.
энергия перехода между первыми двумя уровнями, так
называемый резонансный потенциал возбуждения.
2%те* (I
[2*
_1_
Зкте*
(30)
Для атома водорода резонансный потенциал рав$н 10.16 eV.
Строение электронных оболочек атомов
Как же расположены электроны в атоме? Н. Бор (1913)
предполагал, что атом сходен с солнечной системой:
электроны двигаются по определенным круговым и
эллиптическим орбитам вокруг ядра, подобно планетам, движущимся
вокруг солнца. Несмотря на то, что теория Бора правильно
описывала большой экспериментальный материал и, в
частности, также приводила к формулам (27)— (30), мы
вынуждены в настоящее время отказаться от приписания
электрону в атоме определённой траектории движения. Волновые
свойства электрона и соотношения неопределенности
заставляют нас говорить лишь о вероятности найти электрон в той или
иной точке пространства, о плотности электронного облака.
Эта плотность оказывается наибольшей в тех местах, где
проходит боровская орбита. Поэтому мы будем пользоваться
понятием об орбитах, имея в виду его условный характер.
Пространственное распределение вероятности,
распределение плотности электронного облака описывается
собственной <!>-функцией задачи, "характеризующей состояние
системы. Оказывается, что вид этой функции зависит не
только от квантового числа п — главного квантового числа,
определяющего энергию (27), но и от квантового числа,
характеризующего момент количества движения электрона —
орбитального квантового числа /. Момент количества дви-
22
жения электрона, равный векторному произведению радиуса-
вектора на импульс [при движении по круговой орбите этот
вектор перпендикулярен к плоскости орбиты и численно
равен произведению из радиуса орбиты на импульс (рис. 11)],
может быть представлен в виде
p = VW+V-l-,
2-
(31)
где /=0, 1,2, ... Орбитальное квантовое число / не может
быть больше, чем главное квантовое число без единицы.
Оказывается, кроме того, что
электронная орбита не может занять
произвольное положение по отношению к
некоторому выделенному
направлению, например магнитного поля, а
располагается лишь под такими углами,
чтобы проекции момента количества
движения имели величину
?=тгхР
h
Рис. 11. Момент
количества движения электро-
(32) на, обращающегося по
орбите.
где т — магнитное квантовое число, которое может принимать
2/ —j— 1 различных значений
/и = —/, —/+1, ... , — 1, 0, 1, ... , /—1, / (33)
(рис. 12). Таким образом, квантован не только момент
количества движения ру но и его проекция на некоторую ось.
«E-V
геле
-2 -і О 1 2 3
Рис. 12. Пространственное квантование при / = 3.
Наконец, существует еще и четвертое квантовое число,
которое характеризует некоторый дополнительный момент
количества движения электрона. В наглядной модели
соответствующее движение может быть уподоблено вращению
электрона вокруг собственной оси (рис. 13). Такое вращение
23
называется спином. Проекция спинового момента на
выделенную ось равна
k (34)
%
причем спиновое квантовое число s может принимать только
значения: -(--к- и —-=-.
Рис. 13. Наглядное изображение
спина.
два
Таким образом, электрон
в атоме характеризуется
четырьмя квантовыми числами:
щ /, т и 5. В табл. 1
приведены возможные значения /, т
и s при различных п.
Собственные функции
электрона в атоме зависят, вообще
говоря, от всех четырех
квантовых чисел
Ф = Ф (я, К т, s). (35)
На рис. 14 изооражены формы электронного облака
водорода для некоторых значений щ I, т. На рисунке приведены
общепринятые условные обозначения состояний электронов:
значение главного
квантового числа П пишется спе- Таблица I
реди цифрой, а орбитальные
квантовые числа / = 0, 1, 2,
3 и т. д. обозначаются
соответственно буквами 5, р} d9
У и т. д. Мы видим, что
электронные облака,
соответствующие s-состояниям
(/=0), имеют сферическую
форму, в других случаях
они вытянуты различным
образом.
Энергия электрона в
атоме водорода зависит лишь
от главного квантового
числа п. Но в
многоэлектронных атомах на каждый
электрон действуют все
остальные электроны атома.
Энергия электрона оказывается зависящей не только от числа
я, но и от числа /. Она выражается-уже не формулой (27),
а формулой
• = 5«sr— (36>
о
о
1, 0, 1
-2,-1,0,1,2
"2
\_
2
J_
2
24
где Z — атомный номер, число положительных зарядов ядра,
равное числу электронов в атоме, а л* — эффективное
квантовое число, зависящее от п и /. Например, для п=3,
1=0 (35-состояние) я* = 1.63, для Ър п* = 2.12 и т. д.
1s т-0 2р т=1 2р т=0 3d я*1
Vf тО 2s m*Q 3s r=Q U m^D
Рис. 14. Плотность электронов в различных состояниях атома
водорода.
Принцип Паули
Строение электронной оболочки многоэлектронного атома.
подчиняется весьма важному и общему закону природы,
установленному Паули. Этот закон гласит, что в атоме не
может быть двух электронов, у которых все четыре
квантовых числа были бы одинаковы. Хоть одно из чисел /г, /,
т9 s должно отличаться. В данной системе не может быть
двух электронов с одинаковыми свойствами.
Если квантовые числа п, I, m двух электронов
одинаковы, то должны .отличаться их спины. Поэтому в атоме
могут быть лишь два электрона с одинаковыми п, I, т —
один с s= + -2"> другой с s = — у. Число возможных
различных значений /, /га "при данном п непосредственно
следует из табл. 1. Очевидно, что в атоме могут быть лишь
два электрона с /і=1, восемь с л = 2, восемнадцать сл = 3
и вообще 2д2 электронов с данным главным квантовым
числом га. Так как энергия электронов в основном зависит
от числа га, целесообразно классифицировать электроны по
его значениям. В атоме, содержащем большое число элек-
25.
тронов, два электрона образуют первую, так называемую
замкнутую, оболочку. Следующие 8 электронов находятся
уже во второй оболочке, они обладают значительно боль-
¦шей энергией. В третьей оболочке может содержаться
18 электронов и т. д.
Нериодическая система элементов
Именно этот принцип определяет строение периодической
системы элементов. Менделеев располагал элементы в ряд
по возрастающей массе. В настоящее время можно считать
установленным, что свойства элемента определяются не
массой, а строением электронной оболочки атома.
Важнейшим -признаком является порядковый номер в системе Z,
равный числу электронов атома или числу положительных
зарядов ядра. Рассмотрим, исходя из принципа Паулй,
какими электронами обладают атомы. Воспользуемся уже
указанными выше обозначениями квантовых чисел ли/. Число
электронов, имеющих одинаковые значения п и / (а значит
разные т и s), записывается в виде показателя степени.
Имеем:
«¦*/ 1. Н l-s
_ 2. Не Is2
3. Li ls*2s
4. Be 1$22$2
5. В ls*2s*2p
6. С ls*2se2p9
7. N ls22s22/73
A .. 8. О l$«2s*2/>*
* ~^ 9. F ls22s-2;?5
,_ 10. Ne ls22s22/>6
11.-Na ls*2s*2pe3s
12. Mg ls*2s22p63s2
13. Al ls*2s*2p*3s*3p
k ^ г 14. Si ls*2s22/763s*23/?2
15. P ls22s22p63s23p3
16. S ls22s22/?63s23/
17. CI 1$22522/?63$23/>3
18. Ar ls22s*2/?63s'23/?8
Физические и химические свойства атомов определяются
¦в первую очередь их внешними электронами, имеющими
наибольшие значения п и /. В самом деле, эти электроны
обладают наибольшей энергией и поэтому легче всего могут
быть возбуждены или даже отделены от атома. Легко видеть,
что строение внешних оболочек элементов, находящихся
в одной и той же группе периодической системы, весьма
сходно. Например, во внешней оболочке атомов водорода,
лития, натрия и других щелочных металлов содержится по
одному 5-электрону. В атомах щелочноземельных
металлов— по два 5-электрона, в атомах элементов третьей
группы по два 5 и по одному /^-электрону и т. д.' Атомы
благородных газов имеют замкнутую электронную оболочку.
Свойства электронной структуры атома, действительно,
повторяются периодически. В частности, такая
периодичность имеет место и для важнейшего свойства —энергии
электрона в атоме, определяемой потенциалом ионизации.
На рис. 15 показана кривая зависимости потенциала
ионизации первого электрона от порядкового номера элемента.
ИіММ!1
/ г 3 1 5 В 7 5 S1011121314151817181$2й312223гч%28Я282338Г ЗгЗЗЗіХЯЗ? г
Рис. 15. Потенциалы ионизации.
Дальнейшее заполнение оболочек, после 18-го элемента,
происходит не в такой последовательности, как в двух
первых периодах. Электроны в атоме располагаются, подчиняясь
принципу Паули, но так, чтобы их энергия была
наименьшей. В результате взаимодействия электронов в четвертом
периоде оказывается более выгодным начать заполнение
следующей оболочки до того, как будет заполнена
предыдущая. Этим и объясняется, что число элементов в периодах
не равно. 2л* = 2, 8, 18, 32, 50, а равно 2, 8, 8, 18, 18, 32.
Электронные структуры калия и кальция имеют вид
19. К ls*2s*2pe3sa3pe 4s
20. Са ls*2s22/?63s23/>$ 4s*
и лишь начиная со скандия, появляются 3<і-электроны
21. Sc 1^2s92^63523pG3i4^
и т. д. вплоть до 28. Ni с Зй?8-электронами. Именно
тождеством внешней оболочки при небольши^различиях
внутренней объясняется сходство элементов восьмой группы.
Еще большее сходство между редкоземельными элементами
объясняется тем, что все они имеют электроны
I&2s*2p93s*3p*3dl4s4p4d10 5s*5pe5i6s*
и разнятся лишь числом своих 4/-электронов, меняющимся
от 1 до 14.
Все эти сведения о свойствах электронов в атомах
получены, главным образом, из атомных спектров.
Глава II
ПРИРОДА ХИМИЧЕСКОЙ СВЯЗИ
МОЛЕКУЛЫ, АТОМЫ, ИОНЫ
Мы видели, что различия в свойствах атомов
определяются их электронным строением. В частности, и
химические свойства атомов — способность образовать те или
иные молекулы, вступив в соединение с другими атомами —
объясняются свойствами содержащихся в атоме электронов.
Очевидно, что в процессе химического взаимодействия
наибольшую роль должны играть легче всего возбудимые
внешние электроны атома — его валентные электроны*
Действительно, внутренние электроны, находящиеся на
внутренних заполненных оболочках, в образовании химической
связи практически не участвуют. Внутренние электроны
в молекуле ведут себя почти так же, как и в свободных
атомах. Наоборот, свойства валентных электронов в
молекуле заметно отличаются от свойств внешних электронов
в свободных атомах.
Существование молекул, как устойчивых систем, означает,
что атомы, соединенные химической связью, имеют меньшую
энергию, чем в свободном состоянии. Образование
устойчивой молекулы сопровождается выделением энергии
химической связи в виде теплоты или света. Здесь проявляется
общий закон природы, согласно которому любое тело
стремится к состоянию с наименьшей потенциальной энергией.
Наиболее естественно предположение, что силы,
действующие между атомами в молекуле, имеют электрическую
природу. Уже Берцелиус пришел к этой мысли. Сейчас,
зная, что все свбйства атомов коренятся в их электронном
строении, мы высказываем это положение с полной
уверенностью. Однако, химическая связь между атомами не
сводится к простой электростатике — притяжению
разноименных неподвижных зарядов. Это имеет место лишь в тех
29
случаях, когда в результате взаимодействия валентные
электроны в атомах не только меняют свои свойства, но
переходят от одного атома к другому. Атомы при этом
превращаются в заряженные частицы — и.оны.
Атом, присоединивший к~ себе электрон, приобретает
отрицательный заряд, делается отрицательным ионом. Атом,
отдавший один' или несколько своих электронов,
приобретает положительный заряд, образуя положительный ион..
Очевидно, что положительный и отрицательный ионы должны
притягиваться друг к другу с силой, описываемой законом'
Кулона — пропорциональной произведению зарядов ионов
и обратно пропорциональной квадрату расстояния между
ионами. Это — простейший тип химической связи, так
называемая ионная связь. Так как кулоновское притяжение не
искажает электронной структуры взаимодействующих ионов,
можно считать, что при ионной связи ионы в большой
степени сохраняют свою индивидуальность в молекуле. Число
ионных молекул велико, особенно в области неорганических
соединений.
Однако, основной тип химической связи — это не ионная,.
а гомеополярная или ковалентная химическая связь,
образованная не ионами, а незаряженными атомами. Здесь нельзя
приписать электроны отдельным атомам-партнерам, а
следует считать их принадлежащими молекуле или химической
связи в целом. Огромное большинство известных нам веществ
содержит связи такого типа, ибо таковы все органические
соединения. Гомеополярны связи в простейших
симметричных' молекулах Н2, 02, N2, С12, в которых мы не имеем
никаких оснований приписать одному атому знак -}-, а
другому—. Природа сил, образующих гомеополярные связи>
уже не может быть сведена к простой электростатике.
Всякая классификация явлений природы вносит в них
элементы условности и идеализации. Как мы увидим, реальные
химические связи — всегда и ионные и гомеополярные, и те
и другие силы определяют свойства связей. Логически
правильно говорить не о ионной или гомеополярной химической
связи, но о степени ее ионности и гомеополярности. Тем не
менее, как это всегда делается в физике, целесообразно
излагать вопрос, начиная с описания крайних, идеальных
случаев, применяя в качестве иллюстрации наиболее близкие
к этим крайним типам химические связи. Мы считаем ту или
иную химическую связь относящейся к типу ионной или
гомеополярной, исходя не из априорных представлений,,
а на основе сопоставления физических и химических свойств
реальных веществ. Ионные молекулы образуются из
атомов с заметно отличающимися электроотрицательностями
(см. ниже), потенциалами ионизации и сродством к
электрону. ;
30
Получив достаточную энергию, ионные молекулы
диссоциируют на свободные ионы. Кристаллические решетки,
ооразованные из ионов, обладают сравнительно высокими
точками плавления и в расплавленном состоянии эти вещества
проводят электрический ток. Гомеополярные молекулы
образуются из одинаковых атомов или из атомов, мало
разнящихся своими потенциалами ионизации и сродством
Зигргия
мвлекуль
Г0~ раднобеже
расстояние
/
\
Рссспсзчи*
Msoksy
jS/^cmomu
Рис. 16. Потенциальная кривая двухатомной молекулы
к электрону. При диссоциации они дают нейтральные атомы.
Гомеополярные соединения обычно не обладают
электропроводностью. Имеется еще целый ряд важных черт
различия, о которых мы будем говорить ниже.
Какова бы ни была химическая связь, она характеризуется
всегда некоторыми общими признаками.- Для того чтобы
два свободных атома или иона образовали двухатомную
молекулу, они должны притянуться друг к другу. Под
действием сил притяжения атомы сближаются. Но на малых
расстояниях между атомами в игру вступают силы
отталкивания. Дело в том, что атомы не являются геометрическими
3J
точками, они обладают электронными оболочками,
занимающими определенный объгм в пространстве. Электроны одного
атома не могут проникнуть сколько-нибудь глубоко в
электронную оболочку атома-партнера, хотя бы из-за
электростатического отталкивания электронов, на малых расстояниях
превышающего силы притяжения. Отталкивание может иметь
и другую — квантовомеханическую природу. Так или иначе,
благодаря одновременному действию двух сил
противоположного направления — сил притяжения и отталкивания —
на некотором расстоянии между'атомами (понимаемом как
расстояние между ядрами атомов) установится равновесие,
обе силы уравновесят друг друга и дадут в сумме нуль.
Очевидно, что на таком расстоянии потенциальная энергия
системы, состоящей из-двух атомов, будет наименьшей.
При дальнейшем уменьшении расстояния потенциальная
энергия будет быстро и безгранично возрастать. Напротив,
при увеличении расстояния потенциальная энергия должна
расти лишь до нуля, ибо свободные атомы или ионы,
бесконечно удаленные друг от друга, не взаимодействуют
и потенциальной энергией не обладают. Вид кривой
зависимости потенциальной энергии от расстояний между
взаимодействующими частицами, так называемой потенциальной
кривой, показан на рис. 16. Очевидно, что расстояние от
минимума кривой до оси абсцисс, к которой кр'ивая
асимптотически приближается в своей правой части, соответствует
работе, нужной для разрыва связи на составляющие ее атомы
и ионы и переносу их в бесконечность. Эта работа
называется энергией диссоциации или просто энергией связи.
Мы видим, таким образом, что химическая связь может
характеризоваться своим равновесным расстоянием, на
котором потенциальная кривая имеет минимум, и энергией.
ИОННАЯ СВЯЗЬ
Для определения размеров молекул, атомов и ионов, для
¦определения междуатомных расстояний существуют
различные методы, речь о которых будет итти ниже. Сейчас мы
прежде всего познакомимся с определением энергии ионных
молекул.
.С ионными связями приходится чаще всего встречаться
при рассмотрении свойств кристаллов солей щелочных и
щелочноземельных металлов. Так, каменная соль
кристаллизуется в кубической решетке, в узлах которой расположёны
попеременно ионы Na+ и СГ (рис. 17). В этой книге мы
будем главным образом говорить о свойствах свободных
молекул, существующих в разреженном газе, а не в
твердом теле или в жидкости. Однако, ионный кристалл весь.
может рассматриваться как одна большая молекула и свой-
ства свободной ионной двухатомной молекулы NaCl можно
лучше всего понять, именно разобравшись в силах
взаимодействия, существующих в кристалле.
Энергия ионной решетки
Энергия решетки, та работа, которую нужно затратить
на превращение кристаллического NaCl в свободные ионы
Na4" и СГ, может быть найдена на основе закона
сохранения энергии. Будем исходить из одного»грамм-атома
твердого натрия Na и
половины грамм-моле кулы
газообразного хлора
С12. Переведем натрий
и хлор в свободные
атомы. Для этого
нужно затратить,
во-первых, энергию
сублимации Na и перевести
его тем самым в
газообразное состояние.
Обозначим энергию
сублимации,
отнесенную к одному грамм-
атому Na буквой SNa.
Во-вторых, нужно
разорвать молекулы
хлора на атомы. Для этогс
•нужно затратить
работу диссоциации
молекул ХЛОра на атомы— Рис. 17. Расположение центров ионов Na+
Энергию СВЯЗИ атОМОВ и С1- в кристаллической решетке NaCl.
хлора» Отнесенная к
грамм-молекуле эта работа равна Va^cu- Превратим теперь
свободные атомы Na и С1 в ионы. Для отрыва электрона
от атома натрия нужно затратить работу, равную его
потенциалу ионизации /n3. При соединении оторванного от натрия
электрона с атомом хлора выделяется энергия так
называемого сродства к электрону ?сь ибо образование
отрицательного иона хлора энергетически выгодно. Наконец,
заставим свободные ионы Na+ и СГ соединиться и образовать
твердый ионный кристалл. Этот процесс также выгоден,
.цри его осуществлении выделяется энергия (рассчитанная
на грамм-молекулу) ?/иасі- Общий баланс энергии
разобранного процесса
— 5Na — VfrDci* — /n« + ?сі + ^NaCl.
3 Строение молекул. 33
Но мы могли бы и непосредственно соединить твердый
натрий с газообразным хлором, что также дало бы
кристаллический NaCl. При этом выделилась бы энергия — так
называемая теплота образования QNaci. Так как, согласно
закону сохранения энергии, разность энергий между
начальным и конечным состоянием какой-либо системы не
зависит от пути, по которому шел процесс перехода от одного
состояния к другому, получим соотношение
<?NaCl=— *$Na — %I%Dch — ^Na + Eq\ -f- ?Л*аС1, (1)
из которого, зная остальные величины, мы можем
определить энергию решетки ?Ліась непосредственное измерение
которой невозможно. Напротив, Q и S могут быть найдены
при помощи прямых калориметрических определений.
Величины D и /находятся из спектроскопических данных. Сродство
к электрону труднее всего найти, но и его удается
определить в отдельных случаях независимым путем (см. далее).
Представим описанную последовательность процессов в виде
схемы—так называемого цикла Бррна и Габера, впервые
предложивших этот метод определения энергии решетки:
[Na] крист -Ь V* (С13)газ Г— (Na)ra3-f-(Cl)ra3
— 7»Аі,
~Ь QNaCl — Л?а Ц~ Ес\
J J
[NaClWcx « — (Na%3 + (СГ)™
-f- ^NaCl
Можно применять и обратный путь — вычислять сродство
к электрону, определяя теоретически энергию решетки.
Исходя из представлений об электростатических силах
взаимодействия, Маделунг показал, что энергия решетки
имеет вид
U=-^ + ^r, (2)
где г—расстояние между ионами Na+ и СГ (постоянная
решетки), е — заряд иона, а, р, п — константы. Первый член
(2) характеризует притяжение, а для решеток типа NaCl
имеет значение 1.75. Второй член (2) —энергия
отталкивания. Она очень быстро растет с уменьшением расстояния г,
ибо п имеет для всех решеток значение от 6 до 10. Сродство
галоида к электрону -находилось из цикла согласно
уравнению (1). Из количественных данных, приведенных в табл, 2,
мы видим, что значения Е мало меняются в пределах
соединений одного и того же галоида. Этим доказывается
правильность примененных теоретических формул (табл. 2).
34
Таблица 2
LiF
NaF
KF
RbF
CsF
LiCI
NaCl
KCI
RbCI
CsCl
Q
145.6
136.0
134.5
133.2
131.7
97.6
98.3
104.4
105.1
106.3
-5
39.0
25.9
19.8
18.9
18.8
39.0
25.9
19.8
18.9
18.8
— /
123.8
118.0
99.7
95.9
89.4
123.8
118.0
99.7
95.9
89.4
-i°
31.8
31.8
31.8
31.8
31.8
28.9
28.9
28.9
28.9
28.9
и
245.1
216.4
193.2
183.4
175.9
201.1
184.0
168.3
162.1
153.2
E
95.1
95.3
92.6
96.4
95.8
88.2
97.1
84.5
86.7
90.2
LiBr
NaBr
KBr
RbBr
CsBr
LiJ
NaJ
KJ
RbJ
CsJ
Q
87.6
90.6
97.9
99.6
101.5
72.5
76.7
86.3
88.5
91.4
1
39.0
32.9
19.8
18.9
18.8
39.0
25.9
19.8
18.9
18.8
123.8
118.0
99.7
95.9
89.4
123.8
118.0
99.7
95.9
89.4
-4»
23.1
23.1
23.1
23.1
23.1
18.1
18.1
18.1
18.1
18.1
U
E
189.9 83.6
175.9 81.7
161.5 79.0
156.1; 81.4
149.6 83.2
176.2i77.2
164.4; 74.3
152.5'71.4
147.9 73.5
142.4 75.3
Силы, действующие в свободных ионных молекулах, носят
тот же характер; в этом случае также применима формула
типа (2).
Сродство к электрону
Сродство к электрону представляет собой взятый с
обратным знаком потенциал ионизации отрицательного иона.
В результате отрыва электрона от отрицательного иона
с единичным зарядом образуется свободный атом.
Непосредственное экспериментальное определение сродства к
электрону затруднительно и удалось пока в немногих случаях.
В частности, Сэттон и Майер исследовали скорость
испускания электронов и отрицательных ионов J" накаленной
вольфрамовой проволокой, находившейся в атмосфере паров
иода. Считая, что на поверхности проволоки имеет место
равновесие
J -)- электрон ~ J";
Сэттон и Майер нашли тепловой эффект этой реакции —
сродство иода к электрону, равный 72.4 кг кал/моль. Другие
авторы нашли для иода сродство к электрону равное 74.6,
для хлора 88 и для брома —80 кг кал/моль. Эти значения
близко сходятся' со средними величинами, найденными из
циклов Борна — Габера и теоретических расчетов.
Ионная валентность
Присоединяя электрон, атом галоида приобретает
электронную оболочку, аналогичную оболочке атома
благородного газа, соседнего с ним в периодической системе.
Например
F (l.s22sa2/?*) + электрон — F~ (l$*2s*2p*).
* ' ¦ 35.
Но это как раз оболочка атома неона, которая целиком
заполнена (стр. 26). Такая оболочка особенно устойчива,.
чем и объясняется большое сродство галоида к электрону:
от атома благородного газа особенно трудно оторвать
электрон. Очевидно, что образование ионной молекулы, типа
соединения щелочного металла с галоидом, энергетически
выгодно именно потому, что затрата энергии на ионизацию
щелочного металла с лихвой покрывается сродством галоида
к электрону и энергией, выделяемой при взаимном
притяжении разноименных ионов.
Пользуясь этими представлениями, Коссель (1917)
построил теорию электростатической ионной связи. Согласно
идее Косселя, при образовании ионной молекулы из
нейтральных атомов оба иона приобретают электронную
структуру атомов благородных газов. Так, в молекуле NaF и
ион Na+ и ион F~ имеют электроны
1522522^6,
т. е. воспроизводят структуру атома неона. Ионы К4* и С1"
имеют структуру аргона и т. д. Для'того чтобы приобрести
структуру благородного газа, металлы второй группы должны
отдать два электрона, образовав ионы типа Be"4", а
металлоиды шестой группы — приобрести два электрона,
образовав О™, S™, и т. д. Молекула СаО должна тем самым
содержать ион Са++, аналогичный аргону, и ион О™,
аналогичный неону. Коссель отождествил число валентных
штрихов в структурной химической формуле с числом
электронов, перекочевавших в данной связи от атома, получившего
положительный заряд к атому, получившему заряд
отрицательный.
Таким образом, положительная валентность элемента
равна числу электронов, которые нужно отдать,
отрицательная —числу электронов, которое необходимо приобрести
атому для образования оболочки благородного газа. Особая
роль благородных газов определяется замкнутостью,
устойчивостью их оболочек.
Теория Косселя удовлетворительно объяснила некоторые
свойства ограниченной группы ионных соединений, но
оказалась непригодной в большинстве других случаев. Об этом
речь пойдет далее. Сейчас можно указать, что соображения,
подобные изложенным, недостаточны уже потому, что
образование ионной молекулы не обязательно идет через
предварительную ионизацию атомов и, с другой стороны, при
разрыве ионной молекулы не обязательно образуются ионы.
В свою очередь ионы, соединяясь, могут дать молекулу,
в которой действуют не только простые электростатические
силы. Наконец, физически необоснованным является
предположение о существовании устойчивых ионов с несколь-
36
кими отрицательными зарядами. Отдельный атом не может
в свободном состоянии обладать более чем одним
избыточным электроном, ибо электроны отталкиваются друг от
друга. Таким образом, ионы О", S™ являются
фиктивными системами и представление об их
самостоятельном существований пригодно лишь в качестве рабочей
гипотезы.
Приведенные соображения существенны при оценке
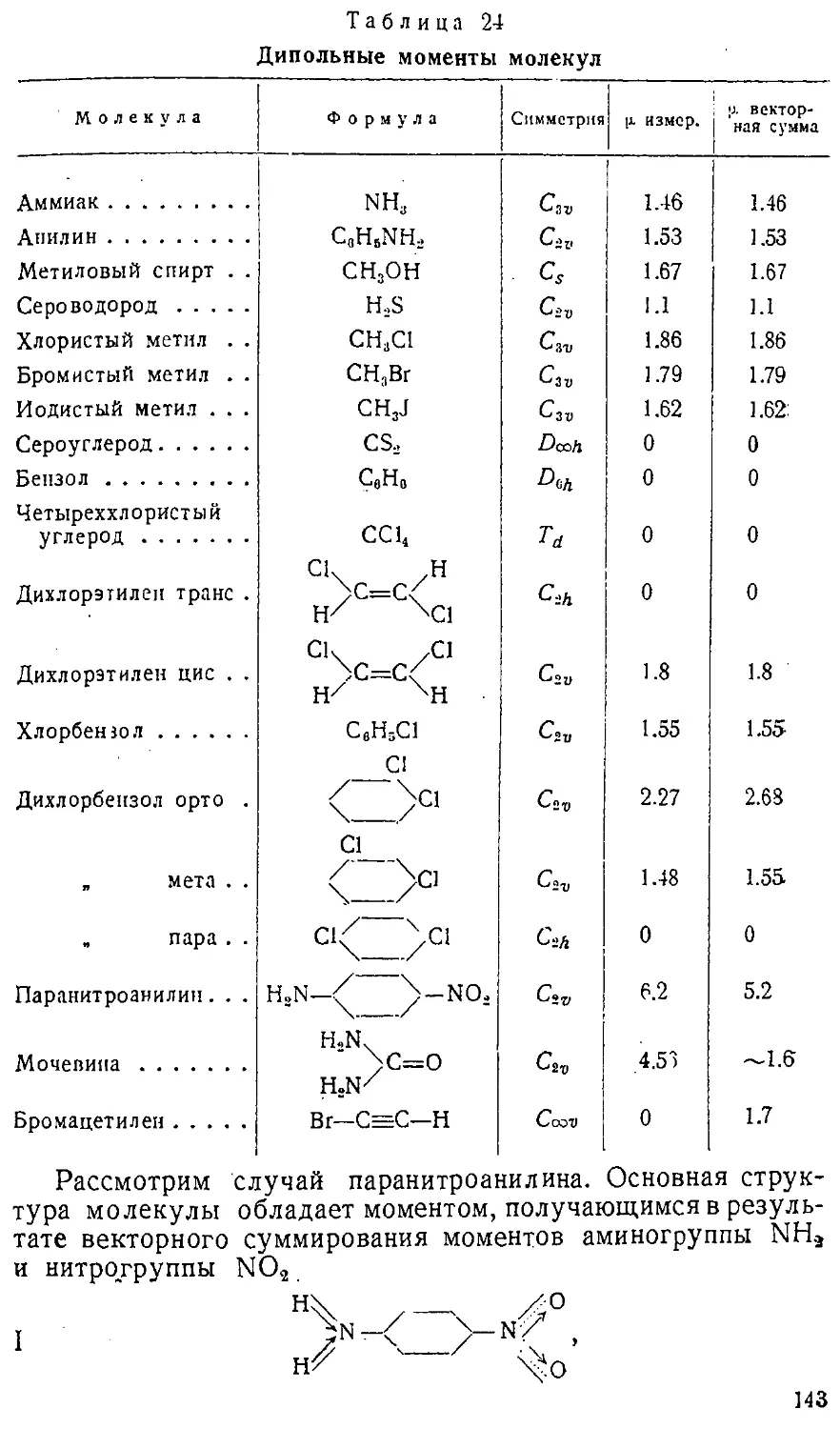
сравнительной электроотрицательности элементов. Вводя это
хорошо знакомое химикам понятие, мы можем, следуя
Мелликену, дать электроотрицательности количественную
оценку в форме суммы потенциала ионизации и сродства
к электрону /-f-?". Действительно, от того атома, у
которого эта величина больше, труднее отделить электрон,
например
/сі + Ес\ > /вг + ?вг, а значит /а — ?вг > /вг — Еа,
т. е. легче отделить электрон от Вг и передать его С1, чем
наоборот. В молекуле ВгСі хлор более электроотрицателен,
чем бром. Очевидно, что электроотрицательность в
периодической системе растет слева направо и снизу вверх.
Понятие электроотрицательности весьма важно для объяснения
свойств реальных молекул и связей.
Другие типы электростатических связей
Электростатическое притяжение может действовать не
только между разноименными зарядами. Заряженный ион
может притянуть молекулу электрически нейтральную, но
обладающую дипольным моментом (см. стр. 123), т. е. такую,
у которой центры тяжести положительного и
отрицательного заряда разведены на некоторое расстояние. Благодаря
этому обстоятельству может оказаться, например, что
положительный ион притягивает отрицательный конец молекулы,
ибо это притяжение не компенсируется отталкиванием
положительного конца, находящегося на большем расстоянии
от иона. Такая ион-дипольная связь имеет, повидимому,
место в ряде комплексных соединений с водой, аммиаком
и т. д.:вионах[Ре(Н90)6]+++, [№(ЫН3)^]++. Наконец,
электростатическая связь может возникнуть между ионом и без-
дипольной системой, в принципе даже с атомом
благородного газа, ибо ион способен поляризовать электронные
оболочки соседних частиц, возбуждая в них заряды
противоположного знака. Силы, вызванные поляризацией, имеют
значение во всех химических связях и являются
определяющими б- ряде случаев образования междумолекулярных
соединений и соединений растворенных ионов с
растворителем.
37
ГОМЕОПОЛЯРНАЯ СВЯЗЬ
Обратимся к рассмотрению молекул и связей другого
типа.
Известны многочисленные молекулы неорганических и
органических соединений, которые совершенно не удается
описать при помощи представлений об ионах. Таковы,
например, молекулы фтористого водорода HF, воды Н20,
аммиака NH3, метана СН4, бороводорода (ВН3)2, гидрида
лития LiH. Их часто изображали (и это попадало даже
в учебники) следующим образом:
H+F- (№%0— (Н+)3№- (Н+)4С4- (Н-)3В3+ Li+H"\
Все написанные ионы действительно имеют структуру
благородных газов — неона, гелия. Тем не менее, свойства этих
молекул не позволяют считать их ионными. В
действительности только молекула HF может с некоторым правом
рассматриваться как ионная.
Молекула водорода
Простейшей неионной молекулой является молекула
водорода Н2. Связь в такой молекуле называют гомеопо-
лярной — образованной атомами одинаковой полярности
в отличие от гетерополярной — ионной связи. Часто также
применяется термин ковалентная связь, в отличие от
электровалентной связи ионов. Молекула водорода характеризуется,
конечно, потенциальной кривой, подобной той, которая
изображена на рис. 16. Изучение физических свойств, главным
образом спектров Н2, показало, что равновесное расстояние
между двумя протонами равно 0.74 А, а энергия связи —
расстояние от минимума потенциальной кривой до ее
асимптоты 4.718 eV, или 108.84 кг кал/моль.
Теория Льюиса
Молекула водорода состоит из двух протонов и двух
электронов. Оба электрона находятся в атомах водорода
в состоянии Is и принимают равное участие в образовании
гомеополярной связи. В 1916 г. Г. Н. Льюис, сопоставляя
электронные структуры атомов и молекул, пришел к
важному выводу, что в образовании гомеополярной связи
обязательно участвуют два электрона. При этом они
располагаются таким образом, что около каждого из
взаимодействующих атомов находится два (водород) или восемь
электронов, образующих оболочку атома благородного газа —
гелия, неона и т. д. Отличие теории Льюиса от
электростатических представлений Косселя состоит в том, что при
38
образовании парной электронной связи атомы не
ионизируются, не приобретают локализованных зарядов
—электроны валентных связей обобществлены, принадлежат обоим-
взаимодействующим атомам или, точнее, связи ме^кду ними
Обозначая электроны точками, Льюис изображал формулы
молекул следующим образом:
.;'' Н ? Н \
v .
—ъ'ш
{ Н :
С1:':і Сі
\ •
*» г*
V 0 /
V П )
чж ^f
Около каждого атома водорода имеются два электрона, и т. д.
около атомов CI, N, С по восемь электронов, что мы
отметили пунктиром. Каждому штриху валентности обычной
химической структурной формулы соответствуют два
электрона. Отсюда делается вывод, что для двойной связи нужно
четыре, а для тройной — шесть электронов. Формулы эти-
ленаНу>С = с^н и ацетилена
Н-с=с-Н
записываются в теории Льюиса следующим образом:
\ н у.і А ;.\ н і
t :)« )
ч_и — —* *'
Условие о числе электронов, находящихся около каждого
атома, сохраняется попрежнему.
Рассмотрим, каким должно быть, согласно
представлениям Льюиса, строение молекулы хлористого аммония
(нашатыря) NH4C1. Так как около атомов азота и хлора
должно быть восемь электронов, азот не может дать пяти
одинаковых гомеополярных связей. Старая структурная формула
н а
\ /
химиков /N\ не совместима с представлениями Льюиса.
н J, н
Необходимо предположить, что электроны в NHiCl
располагаются так:
н;; и :1н
V..V''
н
і: и,:;
39
т. е. азот как бы отдает один электрон хлору и делается
однократно заряженным положительно. В действительности,
написанные здесь заряды носят скорее формальный
характер, в частности нельзя сказать точно, у какого из атомов
нехватает электрона — у азота или у одного из водородов.
Так или иначе, можно заключить, что четыре связи азота
с ¦ водородом гомеополярны, а пятая—связь с хлором —гетеро-
полярная, ионная. В самом деле, хорошо известно
индивидуальное существование иона аммония NH4+, ведущего себя
во многом сходно с ионами щелочных металлов. Возможны
также случаи, когда связь между одними и теми же
атомами осуществляется одновременно двумя способами — гомео-
полярным и гетерополярным. Так, например, связь между
N и О в молекуле
•• + ••"
HSC : N : О :
сн3
имеет именно такой характер. Это как бы двойная связь,
состоящая из одной гомеополярной и одной ионной. Такую
связь часто называют координационной или семиііолярной
и обозначают ее символом-*
сн3
сн3
В настоящее время взгляды Льюиса не утратили своего
значения. Его остроумное заключение относительно роли
двух спаренных электронов и сейчас лежит в основе
теории химической связи. Однако оно не всегда подтверждается.
Нам известны связи, образованные одним и, .может быть,
тремя электронами. Так, ион молекулы водорода Н+ также
может рассматриваться как молекула еще более простая,
чем Н2, самое простое из всех мыслимых соединений между
атомами. Эта молекула наблюдалась во многих случаях.
Очевидно, что в ней фигурирует лишь один электрон,
осуществляется одноэлектронная связь
Н-Н+.
Знак Н во всех формулах Льюиса означает протон; другие
символы химических элементов — соответствующие атомные
ядра (в первом периоде) или ядра совместно с внутренними
электронными оболочками.
Теория Льюиса, описав большое число фактов, не давала
возможности количественных расчетов свойств молекул на
40
основе представлений об их электронном строении. Однако
задачей физической теории является не только качественное
толкование явления, но, в еще большей мере, его
количественное объяснение. Количественную теорию химической
связи удалось построить лишь на основе квантовой,
волновой механики.
„РЕЗОНАНС" ЭЛЕКТРОНОВ
В простейших молекулах Н+ и Но имеются два протона
и, соответственно, один или два электрона. Несомненно, что
между всеми этими заряженными частицами действуют
электростатические кулоновские силы притяжения и
отталкивания. Этим силам в молекуле водорода Н, соответствует
общая потенциальная энергия (рис. 18)
ez
Г12 Г
eJ
At
В2
€-
А2
JL
'Г
е-
АВ
(3)<
Однако, имея в виду эти взаимодействия, нельзя свести
к'ним сущность химической связи в молекуле водорода. Здесь
"надо учитывать следующие
ЗлвЫрон
Злектво*
2
Пртсн
Рис. 18. К расчету связи в молекуле
водорода.
.принципиально важные
обстоятельства. Мы не имеем
права считать электроны в
молекуле неподвижными. В
то же время, в силу
волновых свойств электронов, в
силу соотношения
неопределенности, не имеет смысла
говорить об определенных
траекториях электронов в
молекулах. Более того, мы
не располагаем никакими практическими или мысленными
приемами для того, чтобы отличить первый электрон от
второго. Они неразличимы. Это также следует из общих
положений квантовой механики. Оторвав электрон от
молекулы водорода, превратив ее в ион Н+, мы, конечно, не
можем сказать, какой электрон нами оторван — тот,
который перед актом ионизации был у правого или у левого
ядра. Самая постановка вопроса в такой форме неправильна,
ибо нельзя определить, какой электрон у какого атома
находится. В данном случае и первый и второй электроны
1 Потенциальная энергия иона 1~ф состоящего из двух протонов.
и одного электрона, будет, соответственно, равна
Р- е1 е-
ГАВ
А1
Ві
41
молекулы водорода- произошли из ls-электронов свободных
водородных атомов. Оба электрона находятся в одном и
том же энергетическом состоянии. Но даже если бы
электроны отличались своей энергией, нельзя было бы их
отличить друг от друга, ибо перестановка электронов местами
не изменила бы свойств системы в целом. Эти
обстоятельства надо иметь в виду в дальнейшем.
*
Резонанс в классической и в квантовой механике
Постараемся понять сущность гомеополярной
химической связи, исходя из предложенной в свое время Гейзен-
бергом (1926) аналогии свойств двухэлектронной системы
со свойствами резонирующих маятников.
Представим себе два маятника,
обладающих одинаковыми длинами и массами,
v\\vj4\w\v^\NNVN\ одинаковыми периодами колебаний. Такие
маятники колеблются как гармонические
осцилляторы (ср. стр. 16). Заставим
маятники взаимодействовать. Для этого
соединим их упругой нитью или пружиной
(рис. 19). В результате взаимодействия,
маятники уже не будут колебаться неза-
<>иииО висимо друг от друга. Если привести в
. с колебательное движение один из
маятнике. . вязанные КОв,"оставив второй неподвижным, то через
маятники. некоторое время он раскачается, а
колебания первого затухнут, затем этот процесс
повторится в обратном направлении и т. д., пока энергия
колебаний не рассеется за счет трения, сопротивления воздуха и
других причин. В общем, оба маятника, рассматриваемые как
единая система, выполняют сложное колебательное движение.
Однако можем ли мы вообще говорить в этом случае
о колебаниях двух связанных маятников, как о колебании
-единой системы, с общей для обоих маятников частотой и
фазой? Оказывается, что можем. Сложное движение двух
маятников можно представить как сумму двух таких
колебательных движений, одно из которых имеет частоту
меньшую, а другое бблыпую первоначальной частоты маятника
в отсутствие связи. Одному колебанию соответствует
движение системы, при котором оба маятника смещаются
одновременно в одну сторону, другому — такое, при, котором
маятники расходятся (рис. 20). Колебания системы
связанных частиц, при которых все частицы колеблются с одной
и той же частотой и фазой, т. е. одновременно, проходят
через свои крайние положения, называются нормальными
колебаниями. Можно показать, что любое колебательное
42
движение сложной связанной системы слагается из
нормальных колебаний.
Иллюстрируем приведенный пример простым расчетом.
Пусть потенциальная энергия системы двух одинаковых
связанных маятников — двух гармонических осцилляторов —
имеет форму (ср. стр. 17).
іСХл лЛл
и=~ Н—~-—к'хгх.. (4)
U зависит не только от смещений первого и второго
осцилляторов порознь, но и от их произведения, содержит член
связи—k'xtx% с коэффициентом связи k\ Соответственно
У\\\\У\\\Ч\\\\\\\\\\У\Ч\\\\Ч\\\У\\\\\Ч\Ч\\\\\\\ЧЧ\\ЧЧ\\\\\Ч
Рис. 20. Колебания связанных маятников:
а — симметричное; б — антисимметричное.
силы, действующие на первой и второй осцилляторы,
равны
i 1 ¦- — ~і '-—=¦ — RiXj ' к> Хоу
/*2 ==::— ~77Г = Л Хі кХ.и
Будем искать частоты нормальных колебаний системы.
В этом случае обе силы должны привести осцилляторы
в колебания с одной и той же частотой co = 2*v
F^ — rfmxu /6ч
Fi = — <х?тхг.
Сравнивая (4) и (5), получаем систему двух уравнений
с двумя неизвестными
(k — со*/гс) хі — к'хъ = 0, rj\
—k'x^ik — tfmjx^O. v ;
Условие совместности и разрешимости этих уравнений
состоит в том, что отношение хк\хъ полученное из первого
43
уравнения, должно равняться этому отношению из второго
уравнения
Хі к' k — o>-m ,
-ЦТ — k — v-m ~ k' ' W
откуда получаем биквадратное уравнение для <о с решением
В отсутствие связи, т. е. при k' = 0, мы имели бы (ср. стр. 17)
е>*=(2™)» = А. (Ю)
Мы видим, что возможны две частоты нормальных
колебаний— одна со знаком + в (9), большая частоты
свободного осциллятора, другая со знаком — в (9), меньшая этой
частоты. Наложение связи k' привело к расщеплению
одинаковых частот ш на <о-(-Дсо и о — Дш. Меньшей частоте
соответствует симметричное движение осцилляторов х} = х%>
большей — антисимметричное хх =— х2і в чем легко
убедиться, подставив (8) в (7).1
Гейзенберг воспользовался аналогией с этими явлениями
для объяснения последовательности энергетических уров-^
ней в атоме гелия. Атом гелия содержит два электрона;
Оба они находятся вводном и том же состоянии. Систему,
в которой две или больше частиц имеют одинаковые
свойства, одинаковую энергию, в квантовой механике принято
называть вырожденной. В этом смысле вырождены два
наших маятника, пока на них не наложена связь. Однако,
как мы видели, введение связи уничтожает, снимает
вырождение— вместо двух совпадающих частот резонирующих
маятников возникают две новых, различных частоты
нормальных колебаний.
Можно условно говорить о резонансе электронов в атоме
гелия. Классическому равенству частот обращения двух .
электронов по орбите соответствует равенство — вырождение ^
их энергий. Однако благодаря электростатическому
взаимодействию — отталкиванию электронов — вырождение
снимается. Вместо совпадающих энергетических уровней невзаи-.
модействующих электронов получаются расщепленные —
1 В действительности формула (4) для приведенной модели имеет
несколько иной вид, и, соответственно, общий результат для связанных
маятников отличен от вычисляемого. Вместо (9) мы получаем частоту-
колебания рис. 20, а, равную частоте свободного маятника (10), а частота
рис. 20,0 повышена по сравнению с (10). В самом деле, очевидно, что
пружина в колебании рис. 20, а не сжимается и не растягивается — ее
отсутствие не сказалось бы на колебании. Мы пользуемся моделью
маятников исключительно с целью наглядной иллюстрации вычислений.
44
повышенные и пониженные уровни атома гелия как целого.
Более низкому уровню отвечает антисимметричное
движение электронов, антисимметричная ^-функция,
описывающая их состояние, более высокому уровню — симметричная
^-функция. Симметрия здесь понимается так, что в первом
случае ^-функция сохраняет знак при перестановке
электронов, во втором — меняет его. Исходя из принципа Паули,
мы приходим к следующему выводу. Если функция ф двух
электронов симметрична, не меняется при их перестановке,
то это значит, что квантовые числа обоих электронов
п, U т одинаковы. Следовательно, электроны должны
различаться своими спинами: при симметричной функции спины
равны -|- у и — у, направлены антипараллельно, напротив, при
«
антисимметричной функции спины направлены в одну
сторону.
Связь в молекуле водорода
Представления, примененные Гейзенбергом для
исследования свойств атома гелия, могут быть распространены
и на простейшую молекулу — молекулу водорода. Подобно
атому гелия, она содержит два электрона, но двигающихся
в поле уже не одного, а двух положительных ядер, двух-
протонов. Однако и здесь имеется вырождение электронов,
¦ снимаемое их взаимодействием. Энергетические уровни
молекулы водорода отличаются от уровней свободных атомов.
Взаимодействие электронов приводит к тому, что уровни
энергии ls-электронов свободных водородных атомов
оказываются как бы расщепленными. Один из уровней лежит
выше ls-уровня атома Н, другой ниже его. Таким образом,
соединение атомов водорода в молекулу энергетически
выгодно—электроны опускаются на более низкий
энергетический уровень. Разность между энергией свободных
атомов и энергией молекулы представляет собой энергию связи
в молекуле водорода. Как всегда, мы должны считать ее
отрицательной — со знаком минус.
Чему же соответствует повышенный уровень энергии
молекулы водорода? Так как в этом случае энергия более
высока, чем у свободных атомов, никакого соединения не
происходит, атомы водорода не образуют молекулы, а
отталкиваются.
Положение энергетических уровней молекулы Н2
естественно зависит от расстояния между атомами. Эта
зависимость для более низкого уровня, соответствующего
притяжению атомов, образованию реальной молекулы, имеет вид
обычной потенциальной кривой (рис. 16). Для более высо-
45
кого уровня кривая такого типа не может получиться. Так
как здесь происходит только отталкивание атомов, кривая
не имеет минимума, а непрерывно растет при уменьшении
междуатомного расстояния. Обе кривые для молекулы
водорода показаны на рис, 21. Расстояние между двумя кривыми
Ш при данном междуатомном расстоянии представляет собой
„расщепление, вызванное резонансом электронов".
Описанная картина получена впервые в 1927 г. Гейтле-
ром и Лондоном. Они применили к задаче о молекуле
водорода волновую механику и решили соответствующее урав-
fy *'Щ'ігсін
Рис. 21. Потенциальные кривые молекулы водорода.
нение Шредингера. Потенциальная энергия молекулы
записывалась в форме (2). Решение уравнения Шредингера для
системы двух частиц затруднительно, поэтому Гейтлеру
и Лондону пришлось воспользоваться приближенным
методом. Он состоял в следующем. За исходные состояния были
приняты собственные волновые функции свободных атомов
водорода, вид которых был описан выше (стр. 24), Каждая
из этих функций определяет вероятность найти электрон
в атоме водорода на том или ином расстоянии от ядра.
Обозначим функцию для первого электрона около ядра А
посредством <рд(1), для второго электрона около ядра В
посредством <?в(2). Будем далее рассматривать два атома
водорода как единую систему. Вероятность того, что одно-
46
временно первый электрон находится на данном расстоянии
г ах от ядра Л и второй — на расстоянии гт от ядра В
представится произведением функций ©д(1) ?в(2), ибо это есть
вероятность сложного события, равная произведению
вероятностей.1 Но, с тем же правом, в единой системе двух
атомов водорода мы можем говорить о вероятности
нахождения первого электрона на некотором определенном
расстоянии от ядра В и второго — на определенном расстоянии
от ядра Л. Значит волновая функция ?л(2) ?я(1) с
переставленными, обменявшимися местами электронами также будет
решением нашей задачи. Уравнение Шредингера обладает
тем свойством, что любая линейная комбинация его частных
решений (т. е. сумма этих решений, умноженных на любые
численные коэффициенты) вновь представляет решение
уравнения. Поэтому, согласно Гейтлеру и Лондону, мы можем
написать волновую функцию для молекулы водорода в виде
Ф = Сі?а(1) ?в(2) + едл(2) ?В(1); (И)
где с1 и сг — численные коэффициенты. Уже из
соображений симметрии очевидно, что абсолютные их значения не
должны отличаться: обмен электронов местами может
изменить в крайнем случае знак, но не величину функции.
Решение можно переписать в виде
*т=*М1) ?jb(2)±?a(2) W(l)) = <h±fc, (12)
где фі и фа — произведения атомных функций, зависящие,
конечно, от междуатомного расстояния. Мы получаем
симметричную или антисимметричную функции ф в зависимости
от того, какой знак примем в формуле (12). Очевидно, что
этот результат аналогичен найденному Гейзенбергом для
атома гелия. Мы вновь получили два решения, два
различных состояния. Появление двух различных состояний вместо
совпадающих и поэтому вырожденных состояний отдельных
атомов водорода вызвано взаимодействием ядер и
электронов. Волновым функциям (11) соответствуют значения
полной энергии, равные
Здесь члены С я А имеют знак минус. Си Л возникают
вследствие кулоновского электростатического взаимодействия
электронов и ядер, Л связано с возможностью перестановки,
обмена электронов, 5 на равновесном расстоянии равно
. х Вероятность того, что при бросании одной кости у нас выпадет
данное число, скажем 4, равна 1/в, Какова вероятность того, что при
одновременном бросании двух костей у нас на одной из них окажется 4,
а на другой, скажем, 5? Очевидно, что вероятность этого сложного
события равна произведению вероятностей, т. е. 1/9 х Vc — Vse-
0.347. Укажем на сходство этой формулы с (9). Верхние
знаки соответствуют симметричной функции (12) и дают
более низкий уровэнь энергии, нижние соответствуют
антисимметричной функции и дают более высокий уровень.
Все величины, входящие в (13), зависят от междуатомного
расстояния, равновесное значение которого может быть тем
самым найдено из условия минимума для энергии более
низкого уровня & = C-\-A/l-\-S. Гейтлер и Лондон нашли
величину энергии связи е = 3.14 eV и равновесное расстоя-
Рис. 22. Кривые равной электронной плотности атомов
водорода в случае их притяжения.
ние г0 = 0.86 А вместо опытных значений е = 4.70 eV
и г0 = 0.74 А. Учитывая изменение, вносимое в водородные
функции © благодаря влиянию соседнего атома, а также
рассчитав энергию взаимной поляризации атомов, Розен
нашел г = 4.02 eV и г0=0.74А в лучшем соответствии с
опытом.
Как же выглядят волновые функции (11), как
пространственно распределяются электроны в молекуле водорода?
Вероятность нахождения электрона в данной части объема
молекулы, плотность электронов, определяется величиной
<JA На основании (12) она равна
48
При взаимодействии двух атомов водорода электронная
плотность, в зависимости от состояния, увеличивается- или
уменьшается на 2^.2. Наибольшие значения этот член имеет
в пространстве между атомами водорода. Распределение
электронной плотности в случаях притяжения и
отталкивания показано на рис. 22 и 23 в виде своего рода
геодезических карт — проведены линии равной плотности, причем
большая цифра у линии обозначает более высокую
плотность, большую вероятность найти электрон. Мы видим,
что при образовании связи плотность электронов между
ядрами наибольшая; здесь осуществляется как-бы
наложение электронных облаков обоих атомов.
Рис. 23. Кривые равной электронной плотности атомов водорода
в случае их отталкивания.
Метод теории Гейтлера — Лондона приближенный, ибо
при описании молекулы применяются собственные функции
свободных атомов. В этом причина получающихся
расхождений с экспериментом. Переход к лучшим приближениям
достигается изменением волновых функций, к функциям
свободных атомов добавляются поправочные члены. Это —
естественный путь для решения любых физических
проблем, не поддающихся непосредственному исследованию
ввиду больших математических трудностей.
ВАЛЕНТНОСТЬ
Образованию связи соответствует симметричная
электронная функция, отталкиванию атомов — антисимметричная. Обе
эти функции мы получили без учета спина. Посмотрим,
как при этом должны быть направлены спиновые моменты
4 Строение молекул.
49
электронов. Совершенно так же, как в случае атома гелия
(стр. 45), мы получаем, что спины электронов при образовании
связи при симметричной собственной функции антисиммет*
ричны, антипараллельны. Наоборот, при антисимметричной
функции спины электронов должны быть направлены
одинаково. Параллельным спинам соответствует повышение
энергии — отталкивание. На рис' 21 указано направление
спинов. Таким образом, валентному штриху в молекуле
водорода отвечает пара электронов с антипараллельными
спинами. Имея в виду изложенное, мы можем попрежнему
пользоваться обозначением связи по Льюису.
Н:Н.
Антипараллельным спинам отвечают антипараллельные
магнитные моменты. Между ними осуществляется
притяжение. Часто делались попытки, основанные на непонимании
действительной природы химической связи, объяснить ее
притяжением спиновых магнитов. Между тем энергия
взаимодействия спинов оказывается ничтожно малой по сравнению
с энергией химической связи. Антипараллельность спинов
у электронов связи есть необходимое следствие принципа
Паули, но не причина связи. Тем не менее этим признаком можно
руководствоваться при классификации электронов, при
определении валентности атомов. Очевидно, что валентные связи
могут создаваться лишь теми электронами, спины которых
не установились попарно антипараллельно, не спарились
в самом атоме. Только свободные неспаренные электроны
атома участвуют во взаимодействии, приводящем к
расщеплению энергетических уровней и только при
антипараллельной установке спинов электронов взаимодействующих
атомов осуществляется химическая гомеополярная связь. Таким
образом, число свободных неспаренных электронов атома
должно равняться его валентности. Очевидно, что в
отличие от электростатических сил взаимодействия в ионных
соединениях, силы, определяющие гомеополярную связь,
обладают свойством насыщаемости. Электрон, уже
спаренный с другим электроном, не может участвовать в
образовании связи. Один ион, напротив, может притягивать любое
число ионов другого знака.
Изобразим, как это обычно делается, схематически
распределение электронов в атомах по энергетическим
уровням при помощи клеточек и стрелок. Клетки обозначают
•возможные состояния электронов (Is, 2s, 2р и т. д.),
а стрелки — самые электроны, причем направление стрелок
символизирует спины. В каждой клетке, согласно принципу
Паули, могут быть два электрона с антипараллельными
спинами. Получаем для элементов первых двух периодов табл. 3.
50
Таблица 3
Атом
Состояние электронов
Валентность
U 25
2р
Н
Не
Li
Be
В
С
N
О
F
Ne
t I
1
t
п
П U
' i !
h t
ti
n
n
t
ti
ti
I
t
t
ti
ti | ti | ti
t
t
t
n
1
0
I
0(2)
I (3)
2(4)
3
2
1
0
В полном соответствии с опытом и теорией Льюиса,
водород, литий и 'фтор одновалентны, азот трехвалентен,
кислород двухвалентен, ибо таково число свободных, не-
спаренных электронов в этих атомах. Атомы
благородных газов — гелия и неона — не образуют гомеополярных
(а также ионных) молекул, ибо у них все электроны
спарены и спин-валентность (смысл этого термина ясен из всего
изложения) равна нулю. Противоречие получается для
атомов Be, В, С, действительные валентности которых указаны
в скобках.
Валентные состояния атомов
Сущность этого противоречия заключается в том, что
мы молча предположили, что свободные атомы, вступая
в химическую связь, остаются в тех же состояниях. В
действительности для понимания' валентности недостаточно
распределить электроны в свободном атоме по возрастающей
энергии и подсчитать число неспаренных электронов.
Необходимо принять во внимание те изменения энергии,
которые возникают при образовании химической связи.
Развивая теорию Гейтлера и Лондона, Слэтер, Паулинг
и другие пришли к выводу, что валентное состояние атома,
* 51
: то состояние в котором он находится в молекуле, может и;
должно в ряде случаев отличаться от состояния свободного
j атома. Рассмотрим атом углерода. В свободном состоянии
он имеет два неспаренных электрона. Для того чтобы
разорвать пару 25-электронов, переведя один из них в свобод-,
ную 2/?-клетку, нужно, как это известно из
спектроскопических данных, затратить энергию, равную приблизительно
7eV = 161.5 кг кал/моль. Казалось бы, что такой переход-
исключен, очень невыгоден энергетически. Но мы не можем'
остановить свое рассмотрение на этом месте. Дело в том,
что при этом переходе углерод становится
четырехвалентным, он способен образовать уже не две, а четыре связи.1
Если считать, что, например, энергия связи С—Н равна
одной четверти энергии, нужной для диссоциации молекулы
метана на свободные атомы, то мы получим 374 :Ч =
= 93 кг кал/моль. Следовательно, образование двух
новых связей приводит к выигрышу энергии, равному
186 кг кал/моль, что превосходит затрату энергии на
возбуждение одного 25-электрона на 24.5 кг кал/моль. Таким
образом, переход углерода в четырехвалентное состояние
энергетически выгоден, и мы должны считать атом
углерода в этом состоянии имеющим электроны:
Is 2s 2р
tl t t
t
t
Аналогичные соображения применимы для Be, В и иных
случаев.
На первый взгляд может показаться, что четыре связи,
которые образуют атом углерода, различны, по своей
природе. В самом деле, три из них образуются 2р-электронами
и одна 2$тЭлектроном. Опыт показывает, что это не так:.
•в-молекуле метана СН4 все четыре связи С—Н одинаковы
по своим свойствам. В действительности, ввиду
относительной близости энергетических состояний 25- и 2р-электронов, ¦
они взаимодействуют, осуществляется резонанс, в
результате которого наиболее выгодное энергетическое состояние
соответствует не 2 s- или 2^-состояниям отдельно, а
некоторой их комбинации, подобно тому, что мы имели
в случае атома гелия и молекулы водорода. Все четыре
электрона атома С характеризуются четырьмя смешанными
собственными функциями, в которых участвуют и Us- и
2^-состояния. Эти четыре функции имеют, очевидно, форму
Ф = л +«, + b+ <V + Мар-+ *о-фір0, О5)
где К—Функция для 2$-электрона; <]*9р+> ^2р_, $9р0—фут-
ции для трех 2^-электронов с магнитными квінтовыми
52
числами -+.1, —1 и 0 (ср. стр. 24); а и Ъ — численные
коэффициенты, причем Ь+і Ь_ и Ь0 отличаются только
знаками. Всем четырем функциям (15) соответствует один и
тот же энергетический уровень, более низший, чем
функциям ф2, и фор, взятым порознь. Такое смешение,
гибридизация состояний, оказывается выгодным, оно дает
возможность образовать связи с большей энергией.
Мы можем представить себе и" другие, необычные
валентные состояния атомов. Углерод, присоединяя к себе
электрон, перейдет в трехвалентное состояние. При этом
выделяется за счет сродства к электрону энергия, равная
приблизительно 23 кг кал/моль. Таким образом, возможно
существование трехвалентного отрицательно заряженного
углерода
Is
25
2р
t t
t
Валентность 3
Напротив, в том случае, когда сродство атома-партнера
к электрону велико и может в некоторой степени
компенсировать энергию, нужную для отрыва 2р-электрона от
углерода (260 кг кал/моль), мыслим переход углерода
в трехвалентное состояние
Is
2s
2р
»+
\\ t t t
Валентность 3
В этом смысле мы можем представить себе двухвалентный
отрицательный и четырехвалентный положительный азот
№
N
+
Is
tl
2s
ft
ft
2p
t
t
tl
t
t
t
t
Валентность 2
Валентность 4
Напомним, что последнее состояние как раз и встретилось
нам в случае иона" аммония NH4+ (стр. 40).
Атом кислорода может существовать в одновалентном
отрицательном и в трехвалентном положительном
состояниях, атом фтора в нульва-лентном отрицательном и в
двухвалентном * положительном:
53
Is 2 s 2p Валентность
tl
tl
tl
tl
tl
tl
tl
tl
tl
t
tl
tl
tl
t
tl
t
t
t
tl
t
F" имеет структуру благородного газа — неона. В согласии
с рассказанным ранее, F" имеет ионную валентность, но не
способен образовать гомеопэляряые связи.
ТЕОРИЯ МОЛЕКУЛЯРНЫХ ОРБИТ
Изложенная теория гомеополярной химической связи
дает квантовохимическую интерпретацию концепции Льюиса.
Эта теория находится в соответствии с обычной валентной
схемой химии, с привычными представлениями о валентных
штрихах, при помощи которых изображается структурная
формула химического соединения. Можно развить и другой
метод рассмотрения химической связи, который, однако,
менее тесно связан с обычными представлениями химиков.
Такой метод — теория молекулярных орбит — разработан
Гундом, Герцбергом и Мелликеном.
Теория Гейтлера—Лондона исходит из электронных
состояний раздельных атомов и рассматривает, что происходит
с этими состояниями при соединении атомов в молекулу. Такое
рассмотрение приводит к появлению локализованных пар
электронов с антипараллельными спинами.
Поступим теперь иначе. Представим себе совокупность
двух протонов — ядерный остов молекулы водорода. Будем
помещать в поле этого остова последовательно один
электрон за другим и исследовать, какие при этом получатся
энергетические уровни. Такой метод совершенно подобен
методу изучения свойств электронов в атоме, с той
разницей, что теперь речь идет не об атоме, а о молекуле.
Принцип Паули остается попрежнему руководящим.
Очевидно, что один электрон, помещенный в поле двух
протонов А и В (при этом образуется ион молекулы водорода
Н2+), должен описываться волновой функцией
где вд(1) и <рв (1) имеют тот же смысл, что и выше (стр. 47).
Электрон характеризуется одновременно своими функциями
54
в поле ядра А и ядра В. Вводим второй электрон. Его
состояние описывается подобной же функцией
?а(2)+?*(2).
Полное состояние молекулы водорода должно выражаться
произведением функций для первого и второго электронов
(произвэдением вероятностей):
^ = (?дО) + Т1,(1К?л(2)+?в(2)) = ?д(1)?в(2) +
+ ?д (2)?в ОН ?л (!)?л (2) + ?в(1)?Л (2)- (16)
Собственная функция оказалась отличающейся от функции
Гейтлера—Лондона (12). Помимо первых двух членов,
совпадающих с теми, которые фигурировали в (12), появились
третий и четвертый. Каков их смысл? Очевидно, что член
?д(1)?д(2) определяет вероятность'того, что первый и
второй электроны находятся у ядра Л, член ?в(1)?в(2)—
вероятность нахождения и первого и второго электронов
у ядра 5. Если с первым:* двумя членами (15) можно
сопоставить формулу Льюиса,
НА:НВ,
то третий и четвертый члены характеризуют ионные
состояния соответственно
Нд • Нв и Нд '- Нд.
Теория молекулярных орбит не сводит, таким образом,
связь в молекуле водорода к чисто гомеопэлярной, но
указывает и на частично ионную природу этой связи.
Спаривание электронов с антипараллельными спинами не есть
в этой теории необходимый признак связи, что доказывается
уже самым фактом существования Н2+, непонятным с точки
зрения спин-валентности. В то же время, теория
молекулярных орбит не нарушает правильности соображений теории
локализованных пар. Она просто подходит к вопросу с
другой стороны, что приводит, как это явствует из сравнения
формул (12) и (16), к новым результатам. Каков же
критерий химической связи в этой теории, если критерий
спин-валентности здесь непригоден?
Связь в молекуле создается в том случае, когда
электроны опускаются на энергетический уровень, более
низкий, чем в свободных атомах. Как определить
энергетические уровни в молекуле, с чем можно их сравнить? Для
качественного решения вопроса Гунд, Герцберг, Мелликен
рассматривают электронную структуру того атома, который
55
получается при слиянии ядер молекулы воедино. Так, из
молекулы водорода получается атом гелия с двумя
15-электронами. Очевидно, что такой переход от
раздельных атомов водорода через молекулу водорода к атому
гелия сопряжен с понижением энергии электронов. Напротив,
слияние двух атомов гелия в атом бериллия неизбежно
должно привести к переходу двух 1 s-электронов в 2s-co-.
стояние, ибо этого требует принцип Паули. Электроны
повышают свою энергию, не создают связи. Следовательно,
два атома гелия соединиться в молекулу не могут. В
теории молекулярных орбит такие электроны называют
„разрыхляющими связь". Напротив, электроны, которые при
слиянии двух атомов в один понижают свою энергию,
называются связывающими. Электроны, энергетическое
состояние которых не меняется при переходе от разделенных
атомов к молекуле, в образовании связи участия не
принимают. Это—неактивные, несвязывающие электроны. Число
валентных штрихов естественно в этом случае отождествить
с числом пар связывающих электронов,- уменьшенным на
число пар разрыхляющих электронов.
Анализ электронных конфигураций молекул,
проводимый в теории молекулярных орбит, дает возможность
связать эти конфигурации с физическими и химическими
свойствами молекул. В частности, молекулы, имеющие
одинаковые электронные конфигурации при различных ядрах,
так называемые изоэлектронные молекулы, должны,
очевидно, обладать близкими свойствами. Пример таких
молекул— азот N2 и окись углерода СО. Число электронов в них
одинаково, и можно сказать, что распределение электронов
по энергетическим уровням также совпадает. Физические
'свойства этих соединений также очень близки друг к другу,.
как это видно из данных табл. 4.
Таблица 4
Физические свойства СО и N2
Температура плавления, абс
Температура кипения абс
Критическая температура, абс
Критическое давление, атм
Плотность жидкости
Вязкость -10е при 0° С
Частота колебаний молекулы, см-1 ....
Энергия диссоциации, eV
Равновесное междуатомное расстояние, А
со
66
83
133
33
0.793
163
2167
(10.0)
1.13
No
93
78
127
33
0.79&
166
2360
7.34
l.Ofr
56
Химические свойства СО и Na, в частности их большая
инертность, хорошо известны. Сопоставление свойств этих
молекул заставляет считать, что связь в молекуле СО,
также как и в N2, тройная, что в теории локализованных
пар может быть выражено формулой
-се;о+.
Теория молекулярных орбит, в которой непосредственно-
исследуется состояние молекулы как реальной системы,
имеет преимущество большей общности и физически более
обоснована, чем метод локализованных пар
Гейтлера—Лондона. Химическую связь в таких молекулах, как Ne, 02, SO,
и т. д., удалось впервые описать именно при помощи метода
молекулярных орбит. Однако этот метод — также
приближенный, и проведение его иногда сопряжено с большими
математическими трудностями. В то же время метод
Гейтлера— Л~індона, теория спин-валентности, имеет важное для
химиков преимущество наглядной интерпретации валентного
штриха, химической валентной схемы. Этим объясняется, что
большая часть приближенных квантовохимических расчетов
и оценок исходит из концепции локализованных пар,
дополненной представлениями об электронном резонансе.
РЕАЛЬНАЯ ХИМИЧЕСКАЯ СВЯЗЬ
Мы видели, что при описании молекулы водорода теория
молекулярных орбит, наряду с гомеополярным состоянием,
учитывает и ионные. Паулинг предположил, что реальная
связь в молекуле всегда представляе*собой наложение
гомеополярной и ионной связей, и предложил учитывать
это в расчетах по методу локализованных пар.
Внутренний электронный резонанс
Идея Паулинга является дальнейшим развитием
представлений о резонансе электронов, резонансе различных
энергетических состояний, представлений, впервые
введенных в науку Гейзенбергом и лежащих, как мы видели,
в основе теории Гейтлера и Лондона. Можно считать, что
если чисто ионному состоянию, характеризуемому
функцией фі и чисто гомеополярному в смысле
Гейтлера—Лондона состоянию §h отвечают близкие энергетические уровни
S; и ел, то в результате резонанса энергия системы
понижается и становится меньше и еі и еЛ. Резонанс
энергетически выгоден. В действительности, конечно, никакого
резонанса нет, электроны в молекуле находятся в некоторых,,
вполне определенных состояниях, которые можно в
принципе установить, не касаясь состояний свободных атомов и
57
ничего не говоря о чисто умозрительных состояниях ^ и фд
и соответствующих им энергиях е,- и еЛ. Однако, стремясь
сохранить наглядность описания и связь со структурными
формулами химии, мы разлагаем собственную функцию
реальной связи на собственные функции ионного и гомео-
полярного состояний. При этом мы принципиально не имеем
•способа наблюдать отдельно ионное и гомеополярное
состояния данной связи. Поэтому не имеет смысла говорить
о физическом явлении электронного резонанса, как это часто
делается. Электронный резонанс есть не явление, а способ
описания, сохраняющий ряд недостатков приближенного
метода Гейтлера и Лондона, но имеющий достоинство
наглядности— интерпретации квантовомеханической сущности
химической связи в терминах валентной схемы химии.
Отличие от классических воззрений химии на валентность здесь
сводится к тому, что вместо одной валентной схемы данной
связи, приходится пользоваться двумя или больше— гомеопо-
лярными и. ионными схемами.
Представление о резонансе здесь расширено по
сравнению со случаями, разобранными выше (Не, Н2). Резонанс
осуществляется не между совпадающими, вырожденными
состояниями, но между уровнями, отделенными друг от друга.
Легко • показать, что аналогия с классической задачей
о колебаниях связанной системы здесь попрежнему
сохраняется. В самом деле, нормальные колебания двух
связанных неодинаковых маятников, имевших в свободном
состоянии ча?тоты <oj и о>2, причем ^ij>^2, происходят с
частотами <оі>о>* и в>а<?юа'. Расстояние между частотами
увеличилось благодаря "взаимодействию. Аналогичным образом,
если гипотетическим ионному и гомеополярному состояниям
связи соответствовали энергии е,- и ?Л, где, скажем, е(->ед,
то реальная молекула имеет уровни ej и е3) расстояние
между которыми больше, чем между % и ед, ибо ?і>ег- и
€2<Сел (рис 24). Это „раздвижение" уровней тем больше,
чем ближе уровни ef и ел, оно наибольшее при совпадении
уровней ех- = еЛ и становится очень малым, если эти уровни
далеки друг от друга, если для превращения гомеополярной
связи в ионную, для переноса электрона нужна большая
затрата энергии.
Таким образом, согласно Паулингу, реальное состояние
связи, соответствующее наиболее низкому уровню еа,
возникает в результате резонанса ионного и гомеополярного
состояний— внутреннего резонанса в данной связи в отличие
от внешнего резонанса между состояниями различных связей
в многоатомной молекуле, о чем речь пойдет ниже. Реальное
состояние связи может быть записано при помощи функции
¦ = <*Фа + */Фі5 (17)
58
численные коэффициенты ct и ch характеризуют степень
ионности и гомеополярности связи. Теория молекулярных
орбит давала для молекулы водорода функцию (16),
которая может быть переписана в виде
+ = сн $н + сп фп + сй fe, (18)
где
ch = с и = с,-3 = 1.
Здесь фА описывает связь Н : Н, Аіп — связь Н~Н+ и
Фя — связь Н+Н~. Более строгий расчет показывает, что
коэффициенты ciU cw — доля ионных состояний —здесь
значительно меньше:
^іі = ^а = 0.175 при сд = 1.
Рис. 24. Схгма энергетических уровней реальной
связи.
Вообще теория молекулярных орбит всегда дает слишком
большие доли ионных состояний. Учитывая ионные
состояния, Вейнбаум улучшил расчет энергии молекулы Н2 и
получил e = 4.10eV вместо опытного 4.70 eV. Наконец, Джемс
и Кулидж, приняв во внимание все возможности
взаимодействий (поляризация и т. д.), нашли, в полном
соответствии с опытом, е = 4.697 eV. Этим была показана
возможность точного теоретического расчета энергии химической
связи, правда, в простейшем случае. Для более сложных
случаев, уже начиная с молекулы LiH с четырьмя
электронами, расчеты становятся весьма затруднительными, и
зачастую приходится ограничиваться ориентировочными
оценками нужных величин. Для такого рода оценок
концепция электронного резонанса, разработанная Паулингом,
очень полезна.
Рассмотрим, как это сделал Паулинг, природу связи
в молекулах галоидоводородов. Приближенные расчеты
позволяют построить потенциальные кривые гипотетических
„чистых" состояний молекул — ионного и гомеополярного.
59
Они показаны на рис. ;25 пунктиром. В результате резонанса
кривые раздвигаются и нижняя из них характеризует
реальную связь, а соответствующие верхние кривые
—возбужденное состояние свя-
імргияі 1 , , , зи. У молекулы HJ ионная
кривая лежит настолько'
выше гомеополярной, что
практически
взаимодействие отсутствует и
реальная связь является почти
нацело гомеополярной. У
НВг кривая для гомеопо-
лярного состояния выше
и еще более высока она
у НС1. Тем не менее и у
НВг и в НС1, ch больше
ch поскольку гомеополяр-
ная кривая лежит ниже
ионной. Однако у HF
соотношение обратное —
пунктирные кривые
пересекаются, и ионному
состоянию отвечает
меньшая энергия. Молекула
HF — преимущественно
ионная молекула. По
расчету Паулинга, процент
ионной связи для Н J, НВг,
НС1, HF равен
соответственно 5, 11, 17, 60. Мы
видели, что резонанс с
ионным состоянием
делает молекулу более
устойчивой, понижает
энергетический уровень
электронов, а значит
увеличивает энергию связи.
Это увеличение энергии
по сравнению с чисто
гомеополярной связью
можно назвать энергией
резонанса. Для
ориентировочной, эмпирической
оценки энергии резонанса
можно, по предложению Паулинга, воспользоваться
отклонением энергии связи от аддитивности. Предположим, что
энергия чисто гомеополярной связи атомов А и В есть
среднее арифметическое из энергий молекул А—А и В—В.
60
С
і>--
h
^
' -х
V ^
?—*'
ч— "г
і*?*5з*
h s
Л*
h
н+ г
Н F
н* сг
н сі
н^
н
н+
н
'
6г^-^
8г
1~ -**0*
J
о
3 ЧЯ
Расстояние
Рис. 25. Энергии ионных, гомеополярных
и реальных состояний молекул галоидо-
водородов (Паулинг).
Однако энергия реальной связи А—В может от этого
среднего отличаться, ибо атомы А и В имеют различную электро-
отрицатёльность— потенциалы ионизации и сродства к
электрону. Чем больше различие в электроотрицательности, тем,
очевидно, больше доля ионного состояния в реальной связи.
Итак величина
А = D (А-В) - V, {D (А-А) + D (В-В)},
(19)
где D— энергия диссоциации —может служить критерием
резонанса. В табл. 5 приведены значения А и D для
молекул галоидов и галоидоводородов.
Таблица
5
Энергия диссоциации
(в кг кал/моль) (Паулинг)
?>(А —А) и D(B —В)
н-н
103.4
?>(А — В)
Д
А'
1
Z)(A-B)
Д
А'
F —F 'сі— Сі Вг —Вг, J —J
63.5 57.8 46.1 36.2
Н — F
147.5
64.0
66.5
CI —F
86.4
25.7
25.8
Н —Сі Н — Вг! Н — J
102.7 87.31 71.4
22.1 12.5
25.4 18.3
1.6
10.2
Br —CI J —Br J —Сі
52.7 42.9
0.7 1.7
1.1
2.0
51.0
4.0
5.3
Наряду с отклонениями от среднего арифметического А,
в этой таблице приведены отклонения от среднего
геометрического А':
А' = D (А—В) — VD (A—A) DlB=B)~". (20)
Связь между А и разностью электроотрицательностей
атомов А и В легко выразить следующим образом:
Д(А-В) = (*А-*В)«,
(21)
где хк и хв — электроотрицательности атомов А и В,
выраженные в некоторых условных единицах. Сопоставляя
значения А для ряда молекул, Паулинг вычислил
электроотрицательности различных атомов. Необходимо
подчеркнуть крайнюю условность такого рода оценок.
Другими критериями внутреннего электронного
резонанса могут служить отклонения от аддитивности таких
свойств связей, как междуатомные расстояния, дипольные
моменты и т. д.
61
МНОГОАТОМНЫЕ МОЛЕКУЛЫ
Речь шла до сих пор о двухатомных молекулах. Строго
говоря, только в них и представлены отдельные химические
связи. В многоатомных молекулах осуществляется сложное
взаимодействие между электронными облаками всех атомов
и связей. Тем не менее, громадный опытный материал,
относящийся к свойствам именно многоатомных молекул,
удалось систематизировать при помощи валентной схемы,
оперирующей валентными штрихами, т. е. представлениями
об отдельных химических связях".
Основной вопрос теории строения молекул сводится
к тому, в какой мере физически обосновано выделение
взаимодействий в отдельных связях из сложной игры сил,
происходящей в многоатомной молекуле, в какой мере
справедлива валентная схема и каков физический смысл
валентного штриха? Уже то обстоятельство, что химические
свойства громадного числа молекул успешно описываются
при помощи этих представлений, свидетельствует о
законности валентной схемы. Совершенно очевидно, что
валентная схема, химические структурные формулы представляют
собой результат приложения к молекулам идеи аддитивности
свойств составляющих молекулу с грунтурных элементов.
Такими элементами здесь являются отдельные химические
связи, изображаемые валентными штрихами. Валентная схема
предполагает, что одни и те же связи в разных молекулах
сохраняют свои свойства, и, следовательно, свойства
молекулы в целом могут быть представлены как суммы свойств
^отдельных связей. Ниже мы увидим, что это во многих
случаях подтверждается физическими исследованиями:
отдельные связи сохраняют свои индивидуальные физические
качества в различных молекулах. Однако нам хорошо известны
также и такие случаи, когда имеются заметные отклонения
от аддитивности этих свойств, когда связи меняют свой
характер благодаря взаимодействию друг с другом. Это
было известно уже химикам и именно для объяснения
отклонений от аддитивности приходилось вводить
усложнения внутри самой структурной химии.
Примером таких усложнений может послужить гипотеза
частичных валентностей Тиле, разработанная для
объяснения аномальных свойств конъюгированных (чередующихся
с единичными) двойных связей и свойств связей в
ароматических соединениях, содержащих бензольное кольцо. Эти
связи обладают, коротко говоря, свойствами,
промежуточными между свойствами единичных и двойных связей —их,
по существу, нельзя изображать ни одним, ни двумя
валентными штрихами; а приходится считать, что этим связям
свойственны нецелочисленные валентности. В этом смысле
62
классическая валентная схема представляет собой лишь
первое приближение к действительности. Теоретически, как
мы видели, понятие валентности достаточно хорошо
обосновано. Теория спин-валентности подтверждает валентную
схему. Опираясь на представления, развитые Гейтлером и
Лондоном, на классическую валентную схему, Паулинг
создал метод истолкования неаддитивности свойств химических
связей в многоатомных молекулах — концепцию внешнего
электронного резонанса, о которой мы будем подробно
говорить в главе IV. Сейчас укажем только, что эта
концепция представляет собой дальнейшее развитие
структурной химии: для описания свойств молекул, не
укладывающихся в аддитивную схему, выписывается не одна, а
несколько структурных формул молекулы, подобно тому как
для отдельной связи выписываются ионная и гомеополярная
формулы. Эти идеи тесно связаны с методом
локализованных пар Гейтлера—Лондона. Напротив, последовательное
применение к многоатомным молекулам метода
молекулярных орбит означает по существу отказ от наглядных
химических формул. С точки зрения этой теории закономерно,
например, рассматривать группу СН3 не как совокупность
трех связей С—Н с двумя электронами в каждой из них
— С—Н
ч
а как одно целое — тройную связь углерода с тремя водо-
родами, образованную шестью электронами (Мелликен)
— Сгг Н3.
Метод молекулярных орбит также позволяет рассчитать
отклонения от аддитивности и, при последовательных
расчетах, приводит к тем же численным результатам, что и
метод локализованных пар. Однако, отсутствие прямой связи
с химической теорией делает метод молекулярных орбит
менее ценным, несмотря на его большую физическую
строгость.
Атомы в многоатомной молекуле имеют определенное
пространственное расположение. Уже химики, рисуя
валентные штрихи, приписывали им определенные направления
в пространстве. Современные физические методы
подтвердили правильность этих представлений классической
стереохимии (см. главу III). Теория химической связи дает
убедительную качественную интерпретацию направленности
валентностей, основанную на идеях Гейтлера и Лондона.
Направленные валентности
Атом с электронами в s-состоянии имеет распределение
электронной плотности со сферической симметрией, ибо у
^-электронов орбитальное квантовое число / = 0 и
соответственно' /я = 0; их орбиты не имеют выделенного
направления в пространстве и не имеют поэтому магнитного момента,
за исключением спинового. Два атома с s-электронами обра-
Рис. 26. Наложение
облаков s-электронов.
Рис. 27. Схема
облака р-элек-
трона.
зуют связь в том направлении, где плотность общего
электронного заряда делается наибольшей. Так обстоит дело в
молекуле .водорода (рис. 22). Мы можем упрощенно
изобразить получающуюся' картину в виде двух сферических
облаков отрицательных зарядов, перекрывающихся в некоторой
области (рис. 26).
Электроны, находящиеся в ^-состоянии, не образуют yjjce
облака со сферической симметрией (ср. рис. 14). Облако
Рис. 28. Направленные валентности.
/7-электрона имеет вид „восьмерки" (рис. 27), положение
которой в пространстве не фиксировано, пока не задано
некоторое выделенное направление. По отношению к
такому направлению „восьмерка" р-электрона располагается
либо вдоль него, либо вдоль двух других прямых, образующих
вместе с выделенным направлением систему прямоугольных
•осей (рис. 28) соответственно трем значениям магнитного
квантового числа т=0, -\-1, — 1, характеризующего
возможные проекции момента р-электрона с 1=1.
-64
Если развить далее представления, возникающие при
изучении молекулы водорода, то можно притти к
заключению, что химическая связь должна создаваться в том
направлении, в котором возможно наибольшее перекрывание
электронных облаков, наибольшее увеличение плотности:
электрического заряда. Для s-электронов все направления в!
этом смысле равнозначны: с какой бы стороны не
приблизился один атом водорода к другому, они с равным успехом
образуют связь. Для ^-электронов эта эквивалентность
исчезает— связь образуется по направлению восьмерок. Отсюда
получается, что угол между двумя связями, образованными
/7-электронами одного атома с s-электронами двух других,
должен быть прямым. Это—следствие перпендикулярности
восьмерок двух /7-электронов. Прямыми должны быть углы в
молекулах Н20, H2S, С120 и т. д: (рис. 29). Три/7-электрона
Рис. 29. Схема молекулы воды. Рис. 30. "Схема молекулы аммиака.
должны создавать также взаимно перпендикулярные связи,
например в молекулах NH3, РС13 и др. (рис. 30). Валентности,
образованные р-электронами—направленные валентности.
Изменение их направлений, отклонение значения угла между
связями от 90°, требует определенной затраты энергии, ибо
угол, равный 90°, является наиболее выгодным энергетически.
В действительности опытные значения углов между связями
заметно отличаются от теоретического. В табл. 6 приведены
экспериментальные значения углов {по Паулингу).
Расчет показывает, что единичные связи в насыщенных
соединениях четырехвалентного углерода должны быть
направлены под тетраэдрическими углами 109°28' (рис. ч 31).
Такое строение соединений типа СН4 было установлено еще
Вант-Гоффом и Лебелем на основе сопоставления их физико-
химических свойств. Эти выводы непосредственно
подтверждаются физическими опытами.
Несмотря на соответствие между валентной схемой химии
и теорией направленных валентностей, ни та ни другая не
5 " Строение молекул. 65
Таблица 6
Молекула
CUO
(СН",),0
NHS
Угол между
связями
Н—О—Н
С1—О—С1
Н—S—Н
С—О—С
Н—N—Н
Экспериментальное
значение величины
угла
105°
115°
92°2?'
111°
108°
Молекула
РС13
РВГз
PJ3
AsCl3
Угол между
связями
С1—Р—С1
Вг—Р—Вг
J—р—J
CI—As—С1
Экспериментальное
значение величины
угла
10\°
100°
98°
103°
Рис. 31.
Схема молекулы
метана.
дают нам достаточно общих и
надежно обоснованных физически
представлений о валентных силах.
Так, в результате исследования
колебательных спектров (см. главу
VIII) выяснилось, что если в
молекуле СН4 наибольшие сцлы
действуют вдоль валентных связей
С—Н, в согласии с валентной
схемой, то в молекулах СС14, CF4 это
уже не имеет места. Здесь не
меньшие силы действуют между
отдельными центрами, между
несвязанными валентными штрихами
атомами С1 — CI, F — F (Степанов).
Наличием сил, действующих по
направлениям, отличающимся от валентных, должно в частности
объясняться и отклонение значений реальных углов от
прямых (табл. 6). )
* Кратные связи
Единичные связи, образованные s и 5 (молекула HU, LiH)
или 5- и /7-электронами (молекулы в табл. 6, СН4 и т. д.),
обладают тем свойством, что наибольшая электронная
плотность находится на самой связи (рис. 26, 29, 30). Само
электронное облако отдельно взятой единичной связи
представляет собой тело вращения по отношению к направлению
связи, имеет осевую, цилиндрическую симметрию. Такие
единичные связи принято называть^ о (сигмд)-связями, я
образующие их электроны, оси <|>-функций~которых совпадают с
направлением связи, а-электронами, Мы уже видели, что в обычном
своем валентном состоянии в насыщенных соединениях
углерод образует четыре о-связи, расположенные под тетраэдри-
ческими углами друг к другу. Наряду с насыщенными
соединениями, в которых каждой связи соответствуют два
электрона, известны ненасыщенные соединения, в которых связь
между двумя атомами образована четырьмя или шестью
66 . '
электронами (ср. стр. 39). Атом может иметь только один
свободный (неспаренный) s-электрон и три неспаренных
/7-электрона. Поэтому между двумя атомами может образо- '
ваться лишь одна 55 или sp-связъ с осевой симметрией,
одна а-связь. Вторая и третья связи создаются уже только
за счет /^-электронов с т=-\-19
и /7і = — 1, т. е. образуются ме-.
жду „восьмерками",
расположенными перпендикулярно линии
связи АВ (рис. 32). В этом случае
наибольшая плотность
электронного облака уже не лежит на
самой связи. Такие связи
называются ъ (пи)-связями,. а
образующие их
электроны—^-электронами:. Углерод в соединениях
с кратными связями находится в
других валентных состояниях, и
направления валентностей здесь
иные, чем в насыщенных
соединениях. В молекуле с двойной связью — в
этилене—-первому валентному штриху С = С-связи соответствует о-связь.
Вторая связь между углеродами образована ^-электронами,
это гс-связь. Связи С—Н—а-связи. Они образуют между
собой и со связью С=С углы в 120°. Все пять связей
молекулы этилена лежат в одной плоскости, перпендикулярно
к которой расположены восьмерки ^-электронов (рис. 33).
В случае ацетилена мы встречаемся с третьим валентным
состоянием углерода. Первая связь в ацетилене — о-связь,
вторая и третья — те-связи, образованные ^-электронами с
Рис. 32. Схема --связи.
Рис. 33. Схема молекулы
этилена.
Рис. 3-4. Схема молекулы
ацетилена.
взаимно перпендикулярными восьмерками. Угол между С—Н
и С = С-связью равен 180°, молекула ацетилена линейна
(рис. 34). Расчет показывает, что к-связи менее прочны, чем
о-связи, но двойная связь имеет, конечно, большую энергию,
чем единичная, и тройная, большую, чем двойная. Все эти
выводы из теории химической связи полностью
подтверждаются опытом.
* 67
Глава III
ГЕОМЕТРИЯ МОЛЕКУЛ
РАЗМЕРЫ МОЛЕКУЛ
В кинетической теории газов молекулы обычно
рассматриваются как упругие твердые шарики конечных размеров.
Этим грубым предположением можно ограничиться при
объяснении многих свойств газов. В действительности, как
мы знаем, молекулы представляют собой сложные
пространственные структуры, в которых определенным образом
расположены атомные ядра и электроны. Спрашивается, какой
физический смысл можно придать размерам
идеализированных шарообразных частиц, находимым в газокинетических
исследованиях?
Взаимодействуют не только атомы внутри молекулы, но и
молекулы между собой. Именйо силы сцепления молекул,
силы междумолекулярного взаимодействия определяют
существование жидкостей, такие явления, как капиллярность,
адсорбция и т. д. Благодаря способности, большей или
меньшей, молекул к взаимному притяжению, между молекулами
создается потенциал, подо'бный по своей форме
внутримолекулярному (рис. 35). Имеется определенное
междумолекулярное расстояние, соответствующее минимуму
потенциальной энергии. На малых расстояниях появляются быстро
растущие силы отталкивания, в основном за счет
электростатического отталкивания электронных оболочек молекул.
Отличие междумолекулярного потенциала от
внутримолекулярного состоит в более пологом ходе кривой, меньшей
глубине минимума, ибо энергия междумолекулярного
взаимодействия много меньше энергии химической связи —
последняя имеет порядок величины 100 кг кал /моль,
первая— порядок 1 кг кал/ моль. Расстояние, на которое могут
сблизиться молекулы, их газокинетический диаметр,
определяется кривой рис. 35. Очевидно, что за счет своей
кинетической энергии молекулы могут сблизиться на расстояние
68
dry тем меньшее d0, чем выше температура среды, а значит
и скорости молекул. Поэтому диаметр, получаемый из
газокинетических измерений, должен, вообще говоря, зависеть
Рис. 35. Потенциальная кривая для междумолекулярного
взаимодействия.
от температуры. Таким образом, диаметры твердых,
шаровидных молекул в действительности представляют средние
расстояния, на которые могут сблизиться молекулы. Эти
диаметры в определенной мере характеризуют реальное
протяжение электронных оболочек молекул и должны
иметь тот же порядок величины.
Будем считать молекулы
шаровидными. Очевидно, что
центры одинаковых шаров могут
сблизиться на расстояние не
меньшее d=2r, а значит центры
соседних молекул не
могут.проникнуть в сферу радиуса 2г,
окружающую центр данной молекулы
(рис. 36). Объем такой сферы
v=^(2r)z = &v
О»
Рис. 36. Сфера действия мо-
П\ лекулы.
где v0 — объем отдельного шарика. Одна грамм-молекула
газа, содержащая iVA = 6.02 ¦ 1023 молекул, имеет
недоступный для взаимного проникновения молекул объем Nav =
= 8Navq. Эту величину нужно разделить на 2, ибо,
вычисляя vy мы считали первую молекулу шаровидной, а вторую —
69
точечной, говоря о невозможности проникновения центра
молекулы в объем v. Тем самым, в среднем, на каждую
молекулу приходится- объем v/2, В уравнение состояния
реального газа, уравнение ван-дер-Ваальса.
а
V-
P + lk(V-b) = RT
(2)
входит поправка на объем Ь, характеризующая собственный
объем молекул, недоступный соседним молекулам. Это —
наименьший объем, который может занимать грамм-молекула
вещества. Согласно вышесказанному,
г. NV л »г , ж г 4т:г3
b = ~2~ = 4Mv0 = 4М • —
(3)
Постоянная b связана с объемом и давлением вещества в
его критическом состоянии, в том состоянии, в котором
никаким дальнейшим повышением давления нельзя обратить
газ в жидкость. Теория критического состояния дает
и— 3 ,
(4)
где Vk — критический объем грамм-молекулы вещества.
Измерив его, можно найти Ъу а значит г.
Другой способ определения г связан с измерением
внутреннего трения, вязкости газов. Газы являются тем более
вязкими, чем меньше длина свободного пробега молекул,
т. е. чем большее расстояние может пролететь молекула,
не сталкиваясь с другими. В свою очередь, это расстояние,
длина свободного пробега, обратно пропорциональна ъг*—
площади сечения сферы, окружающей молекулу,
недоступной остальным молекулам. Измеряя вязкость газов, можно
найти г. Некоторые результаты измерения rfT = 2r
приведены в табл. 7.
Таблица 7
Эффективный диаметр молекул (в А)
Молекула
Не'
Аг
Хе.
н2.
N*
dT из
внутреннего
трения
1.96
2.92
3.5
2.47
3.18
dr из
уравнения
состояний (4)
2.52
2.92
3.1
2.5
2.86
Молекула
о2
со
НС1
сн4
со2
dT из
внутреннего
трения
2.98
3.2-
3.0
3.34
3.32
dT из
уравнения
состоянии (4)
2.45
2.86
2.85
2.96
2.92
70
Размеры простых молекул, содержащих небольшое число
легких атомов, имеют порядок величины нескольких
ангстрем. Сложные молекулы, содержащие большое число
атомов, естественно имеют большие поперечники:' Так, для
ССіі d-т из внутреннего трения равно 22.0 А, для
нормальных углеводородов (гептана C7HiG, октана С8Н18 и нонана
С9Н.20) равно 26.7, 34.9 и 42.5 соответственно.
Сопоставляя газокинетические свойства ряда близких
молекул, отличающихся друг от друга определенным числом
атомов и групп атомов, удается найти не только
эффективный поперечник идеализированной шаровидной молекулы,
но также получить известную характеристику формы
молекулы и радиусы действия связанных атомов.
РАССЕЯНИЕ РЕНТГЕНОВЫХ ЛУЧЕЙ И ЭЛЕКТРОННЫХ ВОЛН
Действительное расположение атомов в молекулах
удалось найти, исследуя рассеяние рентгеновых лучей и
потоков электронов газами и твердыми телами.
Рентгеновы лучи отличаются от видимого света только
своей длиной волны, которая в несколько тысяч раз меньше
и имеет порядок величины ангстрема. Но таковы и
величины междуатомных расстояний в молекулах. Следовательно,
для рентгеновых лучей молекула
уже не является единым целым;
соизмеримость длин волн и
междуатомных расстояний делает
заметными диффракцию и
интерференцию рентгеновых лучей,
рассеянных отдельными атомами
молекулы. Пусть рентгенов луч
падает на двухатомную
молекулу АВ, свободно
существующую В газе. Каждый атом (а вер- Рис. 37. Рассеяние света (рент-
нее, каждый электрон) молекулы геновых лучей) молекулой.
явится источником рассеянных
рентгеновых волн (рис. 37), Будем фотографировать
рассеянный свет на пленке, помещенной вдоль дуги круга
с молекулой в его центре. Очевидно, что в разные точки
пленки придут лучи от А я В, проходящие разные пути.
Если в данную точку пленки С пришли лучи АС и ВС,
длина которых разнится- на целое число длин волн, то,
интерферируя, волны усилят друг друга и дадут на пленке
большое почернение. Напротив, если разность путей АС и
ВС равна половине волны или нечетному числу полуволн,
то волны погасят друг друга — почернения не будет. Таким
образом, на пленке должны возникнуть чередующиеся
светлые и темные пятна, причем из расстояния между ними —
71
если известна длина волны и радиус круга — можно
определить междуатомное расстояние АВ. Таков принцип иссле-.
дования строения молекул в газах. То обстоятельство, что*
молекулы находятся в непрерывном движении и хаотически
расположены в пространстве, не меняет дела, ибо
расстояния между рассеивающими атомами не меняются. Благодаря
этой беспорядочности интерференционная картина несколько-
сглаживается, но, как показывают расчет и опыт,
сохраняется в достаточной степени.
В основе структурного анализа молекул лежит
сохранение атомами в молекулах своей индивидуальности. И
рентгеновы лучи .и электронные волны рассеиваются '
электронами атомов. Рассеяние тем больше, следы на пленке тем:
сильнее, чем больше число рассеивающих электронов атома,
чем выше его атомный номер. В химической связи
участвуют и, тем самым, являются в молекуле обобществленными
немногочисленные внешние валентные электроны. Но эти
.электроны как раз очень слабо проявляются в рассеянии.
Поэтому рассеянный свет имеет своим источником
отдельный атом, его внутренние электроны и те междуатомные
расстояния, которые получаются этим методом, суть
расстояния между центрами тяжести электронных облаков атомов;
¦ положения этих центров тяжести мы можем принять
совпадающими с положениями атомных ядер.
Молекулы в.газе
На фотопленке, регистрирующей рассеяние рентгеновых
лучей или электронов, получается некоторое распределение
почернений, которое может быть представлено кривой
зависимости интенсивности рассеянного излучения /от угла
рассеяния ? (рис. 37) и длины волны X. Удобно пользоваться
зависимостью интенсивности от фактора
ибо это выражение фигурирует в теоретических формулах.
Кривая I(s) имеет ряд максимумов и минимумов (рис. 38),
положение и высота которых определяются, во-первых,
рассеивающей способностью атомов и, во-вторых, всеми
междуатомными расстояниями в молекуле. Рассеивающая
способность атома характеризуется так называемым атомным
фактором, тем. большим, чем болыііе число рассеивающих
электронов в атоме, чем выше атомный номер. Атомные факторы
можно вычислить теоретически. После этого оказывается
72
возможным определить все междуатомные расстояния из
экспериментальной кривой.
Обычно здесь пользуются так называемым методом проб
и ошибок: задаются некоторой формой молекулы и
междуатомными расстояниями и сравнивают вычисленную
кривую с полученной из опыта. Почти всегда имеется
возможность подобрать значения г таким образом, чтобы
добиться полного совпадения теоретической и
экспериментальной кривой. Конечно, принимаются во внимание сведения о
строении молекулы, полученные из других источников, а
частности проверяются химические представления. Так,
например, молекулу СС14 химики считают правильным
2D-
ID-
\ X 1 — ¦ 1 1 , ». -—у
О 0.5 s=s^ Я
Рис. с8. Кривая интенсивности рассеяния
рентгеновых лучей в парах четыреххлористого углерода.
тетраэдром (рис. 31). Тем самым устанавливается связь
между расстояниями гСісі и гссі> от которых зависит /;
очевидно гссі= —j-гсісь Вместо двух неизвестных, остается одна,
о
что сильно облегчает ее определение. Опыт дает rcici = 2.98 A
и гссі = 1-83 А. Точность определения расстояния 0.02—0.03 А-
Рассеяние электронов
Волновыми свойствами обладает не только свет, но и
электроны. Согласно уравнению де-Бройля [глава II ур-ие (6)],
длина электронной волны тем меньше, чем больше скорость
электрона. Электронам, получившим свою кинетическую
73
энергию в результате прохождения разности потенциалов
порядка десятков вольт, соответствуют длины волн в
несколько ангстрем, т. е. такие электронные волны должны
вести себя подобно рентгеновым. Действительно, они дают
сходную картину с тем отличием, что для получения электро-
нограммы нужна экспозиция порядка нескольких секунд, в
то время как для получения рентгенограммы требуются часы.
: С другой стороны, вследствие того что рассеяние электро-
, нов осуществляется главным образом атомными ядрами, а
¦ не электронами, получается несколько иная форма
зависимости атомного фактора от 0 и порядкового номера атома,
чем в случае рентгеновых лучей. Кривая I(s) для
электронов обладает не столь ярко выраженными максимумами.
Однако преимущества, доставляемые уменьшением
экспозиции, столь велики, что большая часть данных, относящихся
к свободным молекулам, получена электронографическим
.путем.
Рассеяние жидкостями и кристаллами
Интерференция лучей, рассеянных жидкостями, создается
не только отдельными атомами внутри молекулы, но и
молекулами, находящимися на разных расстояниях друг от
.друга. Таким образом, получаемая рентгенограмма характе-
¦ ризует не только внутримолекулярные, но и
междумолекулярные расстояния. В большинстве жидкостей эти
расстояния, меняются в широких пределах, но не являются вполне
произвольными — жидкости представляют собой не
совершенно беспорядочное скопление молекул, в них сохраняется
известная структура. Интерференционная картина более
размыта, чем в случае газа, максимумы менее отчетливы, но,
исследуя их расположение и интенсивности, удается все же
¦сделать ряд важных выводов о строении жидкости. Для
изучения внутреннего строения молекул пользоваться этим
методом неудобно.
Очень ценную информацию о строении молекул дает,
наряду с рассеянием газами, рассеяние кристаллическими
твердыми телами. Положение частиц в кристаллах строго
упорядочено и периодично. Существует несколько, типов
кристаллических решеток, соответственно тем частицам, из
которых они построены и природе сил связи между этими
частицами. Мы уже говорили о ионных кристаллах (стр. 32),
построенных из .отдельных ионов, связанных электростати-
, ческими силами. Наряду с ионными существуют атомные
решетки, в которых атомы соединены преимущественно го-
меополярными связями—; например алмаз или графит.
Особый тип связи встречается в решетках металлов. Наконец,
имеются молекулярные решетки, в узлах которых находятся
74
молекулы, связанные силами междумолекулярного
взаимодействия. Это —единственный тип решетки, в котором
отдельные молекулы сохраняют свою индивидуальность.
Молекулярные решетки образуются большинством органических
соединений — твердыми углеводородами и их производными
и т. д.
Рассмотрим, каково будет условие отражения
рентгеновских лучей от кристалла. \Пусть на кристаллическую
решетку падает пучок параллельных лучей АВ, AD (рис. 39).
Лучи, отраженные отдельными узлами решетки по
направлениям ВС, DC, дадут в результате интерференции
максимальную интенсивность, если разность путей ABC, ADC
равна целому числу длин волн и погасят друг друга, если
о о —о о о о о
Рис. 39. Брэгговское отражение.
эта разность будет равна нечетному числу полуволн. Из
рис. 39 видно, что разность путей равна
ADC — ABC = ED + DF=:2rsir\%
где г—расстояние между кристаллическими плоскостями,
а ? — угол скольжения между направлением падающего луча
и плоскостью. Условие наибольшего отражения, называемое
условием Брэгга, имеет, следовательно, вид
2rsin6 = ftX. (6)
Зная длину волны X и угол ? можно определить
расстояния г между частицами в кристалле. В настоящее время
хорошо разработаны методы получения интерференционных
картин как от правильных кристаллов (метод Лауэ), так и
от мелкокристаллических порошков (метод Дебая —Шерера).
На рис. 40 и 41 приведены характерные снимки. Сходные
результаты получаются при рассеянии электронов.
75
Исследуя расположение и интенсивности получаемых на
фотопленке пятен или колец, возможно, пользуясь
методами, в принципе не отличающимися от тех, которые
применяются для газов, установить расположение атомов в
кристаллической решетке. На рис. 42 и 43 приведены
структуры атомных решеток алмаза и графита. Алмаз
представляет собой однородную решетку атомов углерода,
расположенных под тетраэдрическими углами 109°23' друг ок
другу. Все расстояния С — С одинаковы и равны 1.54А.
Такой компактностью и однородностью решетки можйо
качественно объяснить исключительную твердость алмаза.
Напротив, в графите атомы углерода образуют плоские
шестиугольные сетки с расстояниями С — С 1.42 А. Расстоя-
Рис. 40. Лауэграмма. Рис. 41. Дебаеграмма.
ния между атомами С, лежащими в отдельных плоскостях,,
равны 3.40 А. Можно думать, что графит должен легко
раскалываться вдоль этих плоскостей.
Как показал еще в 1915 г. Брэтг, точное определение
интенсивностей в интерференционной картине от кристалла
дает возможность установить не только положение атомных
ядер, но и распределение электронной плотности. Этот
метод математического исследования кривых интерференции
известен под названием Фурье-анализа. На рис. 44 показаны
результаты, полученные для ионной решетки NaCl,
атомной— алмаза и молекулярной — гексаметилентетрамина
(уротропина). Приведены кривые равной электронной
плотности. Мы видим, что даже связь в NaCl не может
считаться чисто ионной: имеются электроны, общие для обоих
атомов и создающие электронное облако, обволакивающее
и Na+ и С1". Помимо того, что этот метод позволяет
непосредственно изучать химическую связь, он особенно важен
76
для нахождения положений атомов водорода. Дело в том,
что атом Н, имеющий один электрон, рассеивает очень
Рис. 42. Решетка алмаза, метан.
слабо, его атомный фактор очень мал. Поэтому обычные
методы для определения положения атомов водорода не-
Рис. 43. Решетка графита, бензол.
пригодны. На рис. 44,5 видно положение атомов водорода
по искажению кривых равной электронной плотности.
77
ій
Рис. 44. Кривые рзвной электронной плотности, полученные
методом Фурье-анализа.
а — NaCl, б — алмаз, в — уротропин.
78
Вращательные спектры
Очень важным методом определения междуатомных
расстояний является исследование вращательных
инфракрасных и рамановских спектров молекул (см. главу VII).
Положение линий в молекулярных спектрах зависит от
междуатомных расстояний. Значительная часть данных, приводимых
ниже, получена этим методом. Вращательные спектры
позволяют, в частности, надежно определять положение атомов
водорода в молекуле.
Сводка междуатомных расстояний и углов между связями
В табл. 8 приведены некоторые важнейшие значения
расстояний между атомами и углов между связями в
молекулах.
Точность большинства измерений при определении
расстояний равна 0.02 —0.05А. Углы найдены с меньшей
точностью — до ±2 — 5°.
ГЕОМЕТРИЯ МНОГОАТОМНЫХ МОЛЕКУЛ
Трехатомные молекулы
Молекулы, состоящие из трех атомов, могут быть
симметричными и несимметричными, связи в них могут
располагаться в одну линию или под углом. Согласно теории
направленных валентностей, молекулы, образованные
одиночными а-связями, должны быть прямоугольными (ср. стр.
64). Однако, отталкивание атомов, расположенных на концах
молекулы, значительно увеличивает углы. Наоборот, в
молекулах с тг-связями углы велики и приближаются к 180°.
Такие молекулы, как С0.2, CS2, являются линейными и
симметричными, NaO, HCN, HalCN — линейными,
несимметричными.
Четырехатомные молекулы
Молекулы типа АВ3 с тремя одинаковыми атомами В и,
тем самым, связями А — В, представляют собой либо
пирамиды с атомом А в вершине, либо треугольники с атомом
А в центре. Пирамидальные молекулы NH3, PC13 и другие.
Примеры треугольников: молекула ВС13, ионы СО?~, NO3.
Молекулы, содержащие четыре атома, могут иметь и
другую структуру. Ацетилен Н — С = С — Н— линейная моле-
79
Таблица 8
Tun молекул
Молекула
Строение
Двухатомные
Трехатомные
линейные
симметричные
Трехатомиые
линейные
несимметричные
Трехатомные
нелинейные
симметричные
Четырехатомная пирамида
Насыщенные
ррганические
соединения
Н*
N.
О.
С1.
Вг*
J2
СО
HF
НС1
НВг
HJ
СО.
HgCl,
HCN
N20
Н,0
SO*
СЦО
NHS
сн4
C2He
С8Н8
н—н
N=N
0=0
С1—С1
Вг—Ег
J—J
-с=о+
К—F
Н—C1
Н—Вг
H-J
о=с=о
Cl-Hg-Cl
H-C=N
N=N+—О"
н н
о
/Ч
о
сі/ \сі
N—Н
\
н
н\ Л
тетраэдр /^\
К NH
н\ /н
н-^с—с^-н
Н4С/ *\СН,
Связи и углы
н—н
N-^N
О—О
CI—С1
Вг—Вг
J—J
С—О
Н—F
Н—CI
Н—Вг
Н—J
С-О
Hg-ci
C-N
С-Н
N—N
N—О
О—Н
(НОН
S—О
<0S0
О—С1
<сюсі
N—Н
<HNH
С-Н
с-с
с—н
с-с
Междуатомные
расстояния
о
(в А) и углы
0.74
1.10
1.20
1.98
2.28
2.66
1.15
0.92
1.28
1.42
1.62
1.13
2.28
1.15
1.06
-1.10
-L22
0.97
105°
1.45
124°
1.68
115°
1.01
108°
1.09
1-55
1.09
1.54
Тип молекул
Насыщенные
органические
соединения
Ненасыщенные
органические
соединения
Ароматические
соединения
Молекула
СвН12
CH3F
CHSC1
CH5Br
CHSJ
COCl2
(NH8)jCO
•
C2H4
CSC14
C,H,
C8H4
CSH2
C8He
Cede
Строение
i
шестиугольник
H\
H->-C-Hal
CI
>C=0
QV
HfiN4
>C=0
H2NX
\p—r/
Ck /CI
>C=C<
ск \ci
h4 h H .и
>o=c—c=c<;
\c==c==c<
нх N{
H—С—С—Н
CH
нс/чч:н
не
CH
CH
CCI
скУ^ссі
etc
CCI
CCI
Связи и углы i
с—с
C~F
С—CI
С-Вг
C-J
С-0
С—CI
<С1СС1
С-0
C-N
с—с
C-H
с—с
C-Cl
(C1CCU
с—с
c=c
c-c
C-H
c-c
C-H
С—С
С—H
<coc
c-c
C—CI
Междуатомные
расстояния
о
(в А) и углы
1.53
1.42
1.77
2.06
2.28
1.28
1.68
117°
1.25
1.33
1.33
1.09
1.38
1.73
123.75°
152
1.43
1.31
1.09 ¦
1.22
1.06
1.39
1.08
120°
1.41
1.70
б Строение молекул.
81
кула. Фосген СОС1* содержит *-связь С=0 и поэтому,-
подобно этилену (ср. стр. 67), плоский
С1Ч .
>с=о.
ск
Угол С1СС1, как и следует из теории направленных
валентностей, близок к 120°.
Тетраэдрические молекулы и насыщенные органические
соединения
. Мы уже указали (стр. 65), что в соединениях типа СН4,
СС14 углы тетраэдрические. В молекулах хлористого
метилена СН2С12 и хлороформа СНС13 углы, С1СС1 несколько
увеличены вследствие отталкивания между атомами хлора
и, вероятно, притяжения между С1 и Н. Значения углов
112° ±2°. Приблизительно тетра-
эдрцческое расположение связей
сохраняется во всем ряду
насыщенных органических соедине- -
ний. Мы видим, что прототипом
насыщенных молекул является
атомная решетка алмаза с точным
тетраэдрическим расположением
* связей и расстоянием С—С 1.54А.
Это же расстояние С—С
сохраняется во всем ряду предельных
Рис. 45. Молекула циклопропана, углеводородов и их производных:
связь С—С в алмазе имеет ту же
самую природу. Отдельные связи С—С образует между
собой углы, также близкие к тетраэдрическим. .
В циклических насыщенных углеводородах имеются
отклонения углов от тетраэдрических. В плоском
треугольнике циклопропана (рис. 45) углы С—С1 равны не К)9°28',
а всего 60°. На такую деформацию угла в молекуле
затрачивается определенная энергия. То же, хотя и в меньшей
степени, имеет место в-молекулахциклобутана и циклопен-
тана. Большее содержание-энергии в таких молекулах, по,
сравнению с незамкнутыми в цикл, было замечено уже
химиками:и сформулировано, в виде эмпирических правил,
Бай^ррм в его теории напряжений. Для циклических
углеводородов, содержащих больше 5 атомов С„ напряжения
исчезают. Это объясняется тем, ,что циклогексан, например,
не* плоская молекула, углы между связями С—С близки к
.тетраэдрическим, а не равны. 120°. Возможны две
конфигурации циклргексана (рис. 46). Их обычно называют формой
ванны (а) ,и кресла (<5). Как показывают спектроскопические
82
и интерферометрические исследования, реальный циклогек-
сан, вероятно, существует в виде креслообразной формы.
Тетраэдрическое строение свойственно и другим
соединениям с четырехвалентными атомами. Укажем правильные
тетраэдры SiCl4l SnCl4, РЬС14, TiCl4 и т. д.
Четырехвалентный, положительно заряженный азот дает тетраэдрические
соединения типа ионов [NR4]+, получаемых замещением
атомов водорода в ионе -аммония NH4+ на те или иные
одновалентные атомы и группы. Аналогия между углеродом
а) 6) N^
Рис. 46. а —ванно- и б—креслообразные формы
молекулы С6Н12.
й четырехвалентным положительным азотом, вытекаюідая
из их электронного строения (ср. стр. 53), оказывается
полной.
Непредельные органические соединения
Мы квидели, что наличие ^-электронов определяет
плоскость, в которой располагаются атомы, ближайііше к
¦п-связи, и что отклонение от плоского их расположения
у соединений, с двойной и от линейного у соединений с
тройной связью энергетически невыгодно.
Большей прочности кратной связи соответствует
уменьшение ее длины. Расстояние между атомами углерода в
этане 1.55, в этилене 1.33 и в ацетилене 1.22А.
Циклические непредельные соединения строятся подобно
кристаллу графита. Расстояния С—С в этом случае равны
для бензола 1.39, для графита 1.42, для различных
производные бензола 1.41, т. е. в пределах ошибки
опыта»—одинаковы. Значения длин связей в ароматических соединениях
лежат между, длинами одиночной связи С —С и двойной
связи С = С. Молекула бензола плоская, шестиугольная,
углы между связями равны 120°.
Пространственная изомерия
Различное расположение в пространстве одних и тех же
атомов приводит к различию в физических и химических
¦свойствах молекул. Особенно наглядны примеры таких
молекул-с двойными связями, у которых все атомы лежат в
одной плоскости,. Здесь. возможны случаи так^ называемых
<цис- и трансйздм^ров^ Так, например, 1,2-дихлорэтилен
* # 83
может существовать в двух формах (рис. 47). Они
отличаются своими физическими и химическими свойствами. Элек-
тронографическое исследование дает расстояние между
атомами хлора в полном согласии с изображенными струк-
Рис. 47. а — транс- и б — цис-изомеры дихлорэтилена.
о
турами — в цисдихлорэтилене З.ЗОА, в трансдихлорэтилене
4.88Д.. Для превращения одного изомера в другой нужно,
очевидно, повернуть группу СНС1 вокруг к-связи С=С на
180?. Для этого требуется большая энергия порядка
десятков кг кал/моль — энергия
разрыва т>связи. Наоборот,
повороты вокруг а-связей
осуществляются легко.
ВНУТРЕННЕЕ ВРАЩЕНИЕ И
ПОВОРОТНЫЕ ИЗОМЕРЫ
В основе классической
стереохимии лежит так
называемый принцип свободного
вращения вокруг единичных
связей. Будучи образованы о-элек-
трбнами, единичные связи не
образуют выделенной
плоскости, обладают симметрией
вращения. Следовательно, в
отличие от дихлорэтилена, 1,2-дихлорэтан (рис. 48) не должен
давать, изомеров, ибо любой поворот СН.2С1 не меняет энергии
молекулы. Действительно, при обычных химических
исследованиях изомеры такого рода не встречаются. Однако более
подробное изучение физико-химических свойств вещества
показывает, что свободное вращение всегда в той или иной сте-
84
*іП
Рис. 48. Молекула 1, 2-дихлорэтана.
пени заторможено: повороты вокруг о-связей также требуют
определенной затраты энергии. Правда, затрата эта невелика,
и уже при комнатной температуре средний запас энергии у
молекулы достаточен для того, чтобы такие повороты
происходили очень часто. Торможение создается взаимодействием
остальных связей и атомов в молекуле. Рассмотрим,
например, молекулу этана Н3С—СН3. Атомы водорода,
связанные с различными углеродами, отталкиваются друг от друга,
ч что делает для этана невыгодной цисконфигурацию моле-
й кулы, в которой атомы Н расположены один над другим
(рис. 49,а). Как показывает исследование
термодинамических свойств этана, трансконфигурация молекулы (рис. 49,б)
устойчивее цисконфигурации на ~ 3 кг кал/моль. Очевидно,
что для свободного вращения одной группы СН3 по
отношению к другой вокруг С—С связи нужно, чтобы молекула
Рис. 49. а — цис- и б—транс-поворотные изомеры этана.
обладала энергией в 3 кг кал/моль. Запас энергии,
приходящейся в среднем на одну связь при комнатной
температуре, приблизительно в 10 раз меньше. Поэтому
внутреннее вращение заторможено. Расчет показывает, что повороты
на 120° в молекуле этана происходят примерно один раз
в КГ8 сек; зависимость потенциальной энергии молекулы
этана.от угла внутреннего вращения должна иметь вид,
изображенный на рис. 50. Эту кривую можно
приближенно выразить уравнением
I/=i^0(l-cos3?), (7)
где V0 = 3.3 кг кал/моль. Минимумам кривой соответствует
транс-, максимумам цис-конфигурация молекулы.
Торможение внутреннего вращения может привести при
несимметричных группах атомов к появлению так
называемых поворотных изомеров, даже в случае одиночных
о-связей. Так, для 1,2-дихлорэтана мыслимы следующие нерав-
85
позначные конфигурации (рис. 51). Потенциал в этом случае
более сложен и может изображаться кривой типа (50), но с
неодинаковыми максимумами и минимумами, соответственно
большему числу различных возможных конфигураций'
Рис. 50. Зависимость энергии от угла внутреннего вращения о
для этана.
(рис. 52). Более глубокому минимуму отвечает более
устойчивый, вероятно трансизомер, менее глубокому — менее
устойчивый, так называемый изогнутый изомер, отличающийся
от предыдущего поворотом одной группы СН2С1 на 120°.
а) ^ Л
Рис. 51. Поворотные изомеры I, 2-дихлорэтана:
а — транс, б— изогнутый.
Отличие поворотной изомерии от цис-трансизомерии
этиленовых соединений —в большой скорости превращения
одного изомера в другой в результате внутреннего
поворота в молекуле. Цис-трансизомеры производных этилена
легко выделить, так как необходимая энергия очень велика.
S6
Энергия, нужная для поворота вокруг с-связи, в десятки
раз меньше, а это означает ускорение процесса взаимного
перехода в несколько десятков тысяч раз. Скорость взаимного
превращения поворотных изомеров имеет порядок величины
10~8—10"10 сек.""1 Таким образом,не может быть химических ре-
Транс 60
Рис. 52. Зависимость энергии от угла внутреннего
вращения для 1,, 2-дихлорэтана.
акций, в результате которых удалось бы выделить
отдельные поворотные изомеры: они переходят друг в друга
слишком быстро. Но благодаря тому, что время
взаимодействия молекулы со светом является еще более коротким,
существование поворотных изомеров удается обнаружить
в) / г) Строя*
Рис. 53. Поворотные изомеры н-пентана.
прямыми физическими (оптическими) методами. Об э*том
еще будет речь впереди, в 'главе VIII. Здесь укажем, что
электронографическое исследование позволяет в' ряде
случаев установить присутствие и форму отдельных
поворотных изомеров. Вещества, способные давать такого рода
изомеры, представляют с^бой их равновесные смеси с тем
87
большим содержанием менее устойчивых изомеров, чем
выше температура. Электронография показала, например,
что 1,1,2,2-тетрахлорэтан
сі—с—с—сі
существует в основном в .виде трансизомера.
В молекулах углеводородов также наиболее
устойчивыми являются, очевидно, трансизомеры (рис. 53).
Конфигурации, подобные спирали, изображенной на этом же
рисунке, мало вероятны, и опыт не' подтверждает их
существования. Укажем, что ванно- и креслообразная формы
циклогексана также представляют собой поворотные
изомеры. Креслообразный изомер более устойчив.
СИММЕТРИЯ МОЛЕКУЛ
Молекулы обладают определенной симметрией. Мы уже
не раз пользовались соображениями этого рода. Рассмотрим
основные симметрические свойства молекул, их обозначения
и классификацию.
Будем говорить о молекуле как о системе точечных
атомов. Система точек может обладать тремя основными
типами элементов симметрии. Это, во-первых, плоскости
симметрии — плоскости зеркального отражения. Тачки могут
располагаться в пространстве таким образом, что между
ними можно провести одну или несколько плоскостей,
отражение в которых не меняет свойств системы. Например,
молекула Н20 имеет две плоскости симметрии (рис. 54),
плоскость самой молекулы и перпендикулярную ей
плоскость, проходящую по биссектрисе угла НОН. Отражение
атома Н во второй плоскости переводит его во второй
атом Н, не отличимый от первого, и тем самым не меняет
свойств молекулы. Плоскости симметрии обозначаются
буквой а (сигма). Второй вид элементов симметрии — это оси
вращения. Мы говорим, что система точек имеет ось
симметрии порядка п, если поворот системы вокруг такой оси
на угол 360°:п и кратный ему не меняет свойств системы.
Ось симметрии л-го порядка обозначается символом Сл. Так,
молекула НаО имеет ось симметрии второго порядка,
обозначаемую С2) проходящую по биссектрисе угла НОН в его
плоскости. Поворот связей ОН на 360°:2 = 180° вокруг
этой оси переводит связи друг в друга и свойств
молекулы не меняет.
Молекула аммиака NH3 имеет ось третьего порядка С3,
проходящую вдоль высоты правильной тригональной пира-
88
миды. Симметрическими операциями являются повороты
вокруг этой оси на 360°: 3 = 120°. Через .каждую из связей
NH и ось симметрии в молекуле NH3 проходит •
плоскость симметрии ov> где z> означает вертикальную плоскость,
плоскость, в которой лежит ось симметрии (рис. 55).
Молекула бензола имеет ось шестого порядка С6,
проходящую через центр молекулы перпендикулярно к ее
плоскости. Самосовмещение атомов дают повороты на
360° : 6 = 60° около оси. Сама плоскость бензола обозначается
од, где 'h — символ горизонтальной плоскости,
перпендикулярной оси. Оси линейных молекул, таких как С05, CICN,.
Z\
4
Рис. 54. Симметрия Civ.
Рис. 55. Ось
симметрии С3.
0==С==С=С=0, обозначаются символом С^. Это оси
бесконечного порядка, ибо поворот на любой угол вокруг
такой оси молекулы не меняет.
Третий вид элементов симметрии — это центр симметрии.
Центром симметрии системы точек называется точка,
отражение в которой любой из точек системы не меняет ее
свойств. Так, молекула трансдихлорэтилена имеет центр
симметрии, отмеченный на рис. 47 крестиком, а молекула
цисдихлорэтилена его не имеет. Напротив, цисдихлорэтилен.
имеет плоскость симметрии, проходящую перпендикулярно
плоскости молекулы через середину С=С-связи. Молекула
бензола имеет центр симметрии, NH3 и Н20 его не имеют.
Центр симметрии обозначается буквой I
Наконец, возможен еще один вид симметрии, более-
сложный, сочетающий вращение с отражением. Это — вра-
89'
щение около некоторых осей с последующим отражением
в плоскости. Так, молекула СС14 имеет зеркально-поворотные
оси, обозначаемые^ (рис.56). Поворачивая две С.—Сі-связи
на 360°: 4 = 90° вокруг вертикальной оси, проходящей
по общей биссектрисе двух тетраэдрических углов С1СС1
и затем отражая их от плоскости, проходящей через атом С
перпендикулярно этой оси, мы получим самосовмещение
связей и атомов.
Мы видим, что различные элементы симметрии,
сочетаясь, образуют новые элементы. Так, линия пересечения
двух взаимно перпендикулярных плоскостей симметрии
всегда является осью симметрии молекулы. Чем больше
число различных элементов симметрии, тем выше
симметрия молекулы. Комбинации, совокупности элементов сим-
Рис. 56. Ось симметрии S4.
Рис. 57. Симметрия Од
(молекула SFe).
метрии образуют так называемые точечные группы
симметрии, к которым всегда относятся те или иные молекулы.
Виды групп симметрии реальных молекул являются строго
определенными. В табл. 9 приведены некоторые примеры
симметричных молекул, расположенные по группам
возрастающей симметрии.
Симметрия молекул отличается от симметрии
кристаллов, ибо в молекулах мы встречаемся с замкнутыми
точечными группами, а не.с такой бесконечной симметрической
сеткой, і как кристаллическая решетка. Так, в молекулах
возможны оси пятого порядка Св, что невозможно в
кристаллах, ибо нельзя построить непрерывную сетку —
выложить паркет — из правильных пятиугольников.
Как мы увидим дальше, уже одно знание симметрии
без каких-либо представлений о природе сил химической
связи- и строении электронных оболочек позволяет
разобраться в ряде оптических и электрических явлений.
90
Таблица 9
Обозначе-
ние группы
Элементы симметрии
Примеры молекул и строение
Одна плоскость симметрии а
Центр симметрии і
«
Одна ось симметрии второго
порядка С2
Ось С2 и две плоскости а,,
проходящие-через ось
Ось Cs, горизонтальная
плоскость, перпендикулярная
оси ад, центр симметрии і
Три взаимноперпендикуляр-
ные оси С«, три плоскости
симметрии <т, центр
симметрии і
Ось С3, три эквивалентные
плоскости симметрии av
Ось С8, три эквивалентные оси
С2, три плоскости cv, одна
плоскость ал
Ось С8, три оси d, три
плоскости с и центр симметрии і
Ось Се, шесть осей С2,
распадающихся на два типа, шесть
плоскостей avt
распадающихся на два типа, плоскость
аЛ, центр і
Четыре оси Сг> три оси S4,
шесть плоскостей а
Четыре оси С8, три оси С4,
шесть плоскостей а, центр і
Хлорноватистая кислота,
НОС1— треугольник
Мезовинкая кислота (см. стр.
94)
Перекись водорода НООН
(строение показано на рис. 58)
Вода Н,0, фосген СОС1,
(строение Н20 см. на рис. 54)
Трансдихлорэтилен (рис. 47) —
плоская молекула
Этилен
Н
\с=о
(рис. 33) — плоская молекула
Аммиак NH, (рис. 56).
Хлороформ СНС13 — тригональные
пирамиды .
Плоский треугольник — трех-
хлористый бор ВС13. Ион
нитрата NOr, а также 1, 3,
5-трихлорбензол
С1
\ /и
С1
Цисконфигурация этана (рис.
49, а)
Трансэтан (рис. 49, б)
Бензол СвН„ (рис. 43) — плоский
шестиугольник
Тетраэдры: СН«, СС14 и т. д.
- (рис. 31)
Октаэдры: SF0, ион PtCIe2~
и т. д. (рис. 57)
91
АСИММЕТРИЧНЫЕ МОЛЕКУЛЫ
Молекулы, не имеющие центра и плоскости симметрии,
дают своеобразный вид пространственной изомерии.
Наиболее простая молекула, лишенная элементов симметрии о
и і, должна содержать четыре атома, ибо через три-атома
всегда можно провести плоскость, которая будет тем самым
и плоскостью симметрии молекулы. -Молекула перекиси
водорода, согласно Пенни и Сэзерленду, построена таким
образом, что две связи О—Н, разделенные связью О—О,
образуют между собой угол, близкий к прямому (рис. 58).
Молекулы, лишенные плоскостей и центра симметрии,
способны давать правый и левый изомеры, получаемые
отражением молекулы в некоторой зеркальной плоскости.
Очевидно, что совместить два изомера нельзя никаким
поворотом молекулы как целого: правый изомер Н202 может
«г
\
,-**'»
§?
-sc
J
Рис. 58. Оптические антиподы перекиси водорода.
превратиться в левый лишь в результате внутреннего
поворота одного из гидроксилов вокруг О—О-связи. Так как
это единичная о-связь, такие повороты происходят очень часто.
Особый интерес представляют молекулы с
асимметрическим центром, в частности с так называемым
асимметрическим атомом углерода. Заменим три из четырех атомов
водорода в метане на три отличающихся друг от друга
одновалентных атома или группы; получим, например,
молекулу C*HClBrJ (асимметрический центральный атом
принято отмечать звездочкой). Правый и левый изомеры такой
молекулы показаны на рис 59. Здесь никакое внешнее или
внутреннее вращение не может перевести один изомер в
другой. Изомеры относятся друг к другу, как правая и
левая рука.
В более сложных молекулах осуществляется асимметрия,
сходная с той, которая имеется у НаОч. Это асимметрия
алленового типа,'в основе которого лежат производные
ненасыщенного углеводорода аллена
>с=с=с/
в/ Nb
92
Плоскости, определяемые ^-электронами двух С=С связей
расположены под прямым углом друг к другу. Такие
молекулы не имеют плоскости и центра симметрии, но имеют
зеркально-поворотную ось 54. Сюда же относятся
асимметричные молекулы типа замещенных дифенила
/А А,
/ \__/ \
\ / \ /
\в.в/
в которых два бензольных кольца лежат во взаимнопер-
пендикулярных плоскостях. Они дают пространственные
изомеры, показанные на рис. 60.
В обычных химических реакциях правые и левые
изомеры, так называемые оптические антиподы, не отличаются
^/^"^@8г
Рис. 59. Оптические антиподы с асимметричным атомом углерода.
друг от друга. Однако их оптические свойства различны;
они вращают в противоположные стороны плоскость
поляризации проходящего света (см. главу VI). Заметные
различия в других физических свойствах и в химическом
поведении возникают при комбинировании в одной молекуле
нескольких асимметрических конфигураций, например
нескольких асимметрических атомов углерода. Рассмотрим
классический пример изомеров винной кислоты НС* (ОН)
(СООН) —(Н)С*(ОН)(СООН). Можно изобразить четыре
конфигурации ее молекул (рис. 61).
Формы I и II — типичные оптические антиподы. Напротив,
формы III и IV, также образованные двумя
асимметрическими группами, имеют центр симметрии и могут быть
превращены друг в друга простым поворотом. Формы I и
II химически не отличимы, но разнятся от формы III.
Правая и левая винная кислота [(I) и (II)] имеют температуру
•плавления 170°, мезовинная кислота (III) 140°. Мезовинная
кислота получается из правой конфигурации одной группы
93
НС*(ОН)СООН и левой конфигурации другой. Очевидно,
что никаким внутренним- поворотом ^около а-связи С—С
нельзя превратить один антипод в другой и антипод в ме-
зоформу. Способность оптических антиподов давать друг
с другом соединения, отличающиеся физико-химическими
свойствами, лежит в основе асимметрического синтеза —
метода выделения отдельных антиподов. Для получения
определенного изомера — оптически активной молекулы —
необходимо участие в реакции другой оптически активной
Я / ч В
-с=>о^=>
Рис. 60. Оптические антиподы при асимметрии алленового типа.
молекулы. Обозначая правый и левый изомеры одной
молекулы буквами d и /, правый и левый изомеры другой
молекулы буквами D и L, мы можем написать реакции
d,l + D-+dD-\-lD
В то время как молекулы d и I химически неотличимы,
молекулы dD и ID (а также dL и IV) разнятся друг от друга,
имея существенно отличное пространственное строение.
ОН Н<—/ \—тОН НО
ОН НО
/ а ж ш
Рис. 61. Пространственные изомеры винной кислоты.
Их изомерия уже не сводится к изомерии типа левый —
правый. dD и ID имеют обычно различные растворимости
и могут быть разделены кристаллизацией. Отделив затем Д
мы сможем получить чистые изомеры d и /, вместо
первоначальной их смеси d, I. В реакциях без участия чистых
изомеров получается всегда смесь антиподов, содержащая
равное их количество — рацемическая смесь. Спращивается,
откуда могут взяться первоначальные чистые изомеры D и
Ly с помощью которых разделяются рацемические смеси?
Оказывается; что чистые изомеры характерны для веществ
94
биологического происхождения, связанных тем или иным
¦способом с деятельностью и существованием живых
клеток.1 Вещества биологического происхождения могут
быть разделены на два класса. Первый класс—жизненна
необходимые вещества, участвующие в обмене (белки,
липоиды, углеводы), почти всегда встречаются в виде
определенных чистых изомеров, причем они всегда асимметричны.
Вещества второго класса — продукты переработки, экскреты и
запасные вещества (органические кислоты, алкалоиды, глюко-
зиды, терпены и т. д.)— могут встречаться в виде
рацемических смесей обоих изомеров. Их асимметрия не обязательна.
В круговороте жизни чистые антиподы приводят в ряде
случаев к более специфическим, ¦ более эффективным
биохимическим процессам, чем рацемические смеси. Можно
думать, что асимметрия протоплазмы живого вещества
сводится- к приобретенной им в процессе естественного отбора
способности избавляться от невыгодных для развития
антиподов. Эта асимметрия живого вещества приводит к
интереснейшим явлениям. Как установил еще Пастер, некоторые
виды бактерий способны питаться привой винной кислотой,
но не трогают левого изомера. Этим можно воспользоваться
для разделения антиподов. Существуют соединения, одни
антиподы которых ядовиты для некоторых живых существ,
а другие безвредны. Правая аспаргиновая кислота —
сладкая, левая — безвкусная. Асимметрия получающихся молекул
весьма характерна для биохимических процессов: на
различном содержании антиподов в продуктах
жизнедеятельности нормальной и раковой клеток мбжет основываться
метод диагностики рака. Укажем, что первый метод
разделения антиподов винной кислоты, которым пользовался
Пастер, наряду со способом, основанным на участии
бактерий, также носит биологический характер. Правая и левая
винные кислоты кристаллизуются в крупных кристаллах,
столь же отличающихся друг от друга, как правая и левая
рука. Пастер выделял антиподы, просто разбирая кристаллы
пинцетом под лупой, отбирая правые кристаллы от левых.
Таким образом, и в этом случае вмешательство живого
существа, человека, обладающего способностью асимметричного
выбора, оказалось необходимым для разделения антиподов.
Очевидно, что никаким способом нельзя выделить те
изомеры, которые превращаются друг в друга, рацемизу-
ются, слишком быстро. Это относится, в частности, к
переписи водорода, которая всегда представляет собой
рацемическую смесь, состоящую из равных количеств правого и
левого антиподов.
1 См. Гаузе. Асимметрия протоплазмы. Изд. АН СССР,
научно-популярная серия,
95
Глава IV
ХИМИЧЕСКИЕ СВЯЗИ В МНОГОАТОМНЫХ МОЛЕКУЛАХ
Многоатомная молекула представляет собой некоторое
обособленное целое, характеризуемое определенной
геометрической структурой, запасом энергии, рядом физических
и химических свойств. Однако все развитие науки о
строении вещества показывает, что целесообразно рассматривать
многоатомные молекулы, исходя из схемы аддитивности их
структурных элементов.. В качестве таких элементов могут
быть приняты либо атомы и ионы с их электронными
оболочками, либо химические связи, понимаемые как совокупности
валентных электронов, их образующих. Оказывается, что в
грубом приближении свойства молекул действительно могут
быть представлены, "как суммы свойств этих структурных
элементов. Прежде всего рассмотрим некоторые
относящиеся к этому вопросу экспериментальные данные.
МЕЖДУАТОМНЫЕ РАССТОЯНИЯ
Весьма характерны расстояния между атомами,
соединенными гомеополярными связями. Во всех насыщенных
органических соединениях длина С — С-связи практически одна
и та же. В алмазе, пропане, циклопентане и других
соединениях она равна 1.52 — 1.54А (табл. 8). Другие с-связи,
такие, как С — О, С — N, С— Hal, С — Н, N — Н также имеют
в больших группах соединений постоянные значения
междуатомных расстояний. То же относится к кратным связям:
атомы углерода и других элементов, соединенные тг-связями,
сохраняют постоянные расстояния во всем ряду
соответствующих молекул. В табл. 10 приведены некоторые значения
длин связей, причем каждое из этих значений получено в
результате исследования ряда соединений.
Таким образом, можно говорить о постоянных длинах
связей в многоатомных молекулах. Простое рассмотрение
96
Таблица 10
Длины связей
Связь
Молекулы
Расстояние, А
С—С
С—С
с=с
с=с
С—N
С—О
С—С1
С—Вг
с—н
о—н
Алмаз, насыщенные соединения . . .
Ароматические соединения, графит.
Этиленовые соединения
Ацетилен
Амины ....*.
Эфиры
Хлоропроизводные предельных
углеводородов
Бромопроизводные предельных
углеводородов
Насыщенные соединения
Спирты и вода
1.52—1.54
1.42
1.39 (бензол)
1.33
1.22
1.48
1.42—1.46
1.82—1.85
1.94—2.05
1.08
Ш
приводит к выводу, что во многих случаях постоянны не
только длины связей, соединяющих атомы между собой.
Оказывается, что во многих случаях длина связи атомов
А — В равна среднему арифметическому из длин
аналогичных связей атомов А—А и В — В. Считая условно, что
связь образуется между твердыми сферическими атомами,
мы скажем, что междуатомное расстояние равно сумме
радиусов атомов, образующих ковал ентную связь, — так
называемых козалентн^х радиусов. Последние тем меньше,
чем больше кратность связи; атомам, находящимся р разных
валентных состояниях, соответствуют оазные ковалентные
радиусы. В табл. 11, составленной Паулингом, в скобках
указаны значения этих радиусов, принятые в последнее
время Шомекером и Стивенсоном,
Таблица Д
о
Ковалентные радиусы атомов (в А)
В единичной связи
В двойной связи . ,
В тройной связи . .
В единичной связи
В двойной связи . ,
В тройной связи . ,
н
0.37 (0.37)
в
0.88
0.76
0.68
0.771
0.665
0.602
Si
1.17
1.07
1.00
N
0.74 (0.74)
0,60
0.547
1.10
1.00
0.93
о
0.66 (0.74)
0.55
0.50
1.04
0.94
0.87
-F
0.64(0.72)
0.54
Сі
0.99
0.89
Аналогичным абразсж, расстояния между ионами в
кристаллических решетках могут быть выражены в терминах
7 Строение молекул. *'
ионных радиусов. Очевидно, что в этих случаях уже не
имеет смысла говорить- о длинах связей — ионные силы
имеют не направленный, а центральный характер, и
одновалентный нон Na+ окружен- -в' кристалле шестью ионами'
хлора СГ, с ним взаимодействующими (рис. 17). Паулинг
предложил полуэмпирический метод определения размеров
шарообразных ионов, основанный; на том, что радиус иона
должен .быть тем меньше, чем больше положительный
заряд ядра, стягивающий. . внешнюю электронную оболочку
иона. Таким образом,- все • изоэлекіронные одновалентные
ионы имеют радиусвг
где Сп — постоянная, зависящая от " главного квантового
числа л, Z.—заряд ядра (атомный номер), 5 — поправка,
связанная с отталкивающим действием внутренних электронов.
Суммы этих-радиусов определяют м*еждуионные расстояния
в кристаллах с однозарядными ионами. Для нахождения
Зна**енййг, Паулинг восіюльзовйлся измеренными
расстояниями в' кристаллах NaF-, KCI, RbBr -и • CsJ (изоэлектронные
ионы ?- каждом' соединении со структурами, соответственно,
нео#а>*$ргона, криптона^ и-ксеыона). Для вычисления радиу-
с&? "ййогова'лентнык- ионов rv Паулинг получил
теоретические вьгр&жение,- позволяющее связать т,--с /*, и
валентностью 'v. Размеры ионов имеют .важнейшее значение для
кристаллохимии, ибо ими определяются 'структуры кристал1
лов. Мы -можем- представить себе кристалл получающймей
в результате плотной установки шарообразных ионов. В
зависимости от их относительных размеров поручится та или
иная форма пространственной решетки. Так, NflCl
кристаллизуется в виде кубической решетки, а элементарная ячейка
кристалла CsCI, также построенного из одновалентных
ионов щелочного металла и галоида,, имеет вид куба, в
вершинах которого расположены ионы СГ, а в центре—•
ион Cs"*\
ЭНЕРГИЯ МНОГОАТОМНОЙ МОЛЕКУЛЫ
.Перейдем теперь к энергетическим соотношениям..
Посмотрим, из чеію складывается запас энергии-многоатомной
молекулы и можно, ли распределить эту энергию по
отдельным, связям или атомам.
.Мы видели в главе II, как находится, энергия ионной
решетки при ломощи цикла Борна — Габера. В основе
определения энергии лежит первый закон термодинамики. Этим
..же" законом \ пользуются при вычислении "энергии много-
'атомйУх молекул, понижая под "&той величиной ""Знергйю,
"98 .
выделяемую при образовании молекулы из свободных атаг
мов. Теплоты образования, из атомов молекул.,
органических соединений, к которым главным образом и . относится
имеющийся экспериментальный материал, находятся из теп-
лот сгорания как самих этих соединений, так и тех атомов,
из которых они построены. Так, например, находя при.
помощи калориметрических измерений 'теплоту ' сгорания
какого-нибудь углеводорода и вычитая из нее: теплоты
сгорания углерода (р'еакция С + Ой->С03) и водорода,
(реакция Н2 + "2~ 0-2-^ Н.20), мы найдем энергию углеводорода.
Многочисленные измерения такого рода были проделаны
еще в прошлом веке Лугининым, Томсеном, Вертело, Свен-
тославским. В последнее время эти работы проводились
главным образом Россини. Необходимо подчеркнуть сильно
возросшую точность определения энергий, , доходящую
теперь до 0.2 кг кал/моль.
Вычислим Е— теплоту образования из. свободных
газообразных атомов молекулы предельного углеводорода
СдН.2п+2-
Очевидно, что из теплоты сгорания углеводорода
СлН.2/1 + 2 нужно вычесть, во-первых, теплоты сгорания ?
атомов С и п-\-\. .молекул Н2,. равных, соответственно
п 94.48 и (#+1) 68.35 кг кал/моль. Кроме того, нужно
вычесть энергий, необходимые для получения свободных
атомов углерода и водорода. Это теплота сублимации
(возгонки) алмаза s. и энергия молекулы водорода. Итак,
согласно Сыркину (Журн. -физ. хим., 17, 347, 1943), напишем
E=W — n 94.48 —<я -f1) 68.35 — ns —{п + 1) z9 (2)
где 2=,102.6 кг кал/моль, а 5 = 125 кг кал/моль. Таково
экспериментальное значение 'энергии молекулы СлН.2л + 2-
Можно ли: представить его • как- сумму энергии п — 1
связи С — С и 2 п + 2-связей С — И? Обозначив эти энергии
буквами х и у9 напишем
Е=^ — (it** 1) х — (2 п -f 2)з/. (3)
В алмазе для освобождения каждого атома С нужно"
разорвать по две связи,С — С, т.е. ns = 2nx. Подставив в (2)
это. значение .'и приравняв друг другу (2) и (3), получим
соотношение
' W +ffi 48 =94.48 -f 68.35 + х + г ^-2 у. .(4)
Если.свойства связей аддитивны и х и.у деііствительно по'*
.стоянные величины для всех связей G — С и С^гИ,кго (4)
* .до
должно быть также постоянной величиной. В табл. 12
приведены полученные из опытных данных значения величины,
стоящей в левой части уравнения (4);
Углеводород
л + 1
кг кал/моль.
Таблица 12
. СН4 C2He CjH8 СдНи С5Н18
. 153.6 155.8 156.3 156.5 156.5
QH14
156.6
т. е. энергии связей х и у действительно постоянны,
начиная с четвертого члена ряда. Определяя аналогичным
образом энергии других связей, мы найдем значения, которыми
можно пользоваться, для вычисления энергий соединений,
содержащих эти связи.
Таблица 13
Энергии связей
(Сыркин)
2 (С—С)
С—С
С=С
С=С
С—О
О—Н
С=0
С—С1
С-
с.
:—сі ^
:—Вг }
с—N \
c=n \
C=N
С—Д
Алмаз
Предельные углеводороды.
Этиленовые углеводороды
Ацетилен
Спирты, эфиры ,
Спирты
Кетоны
Галоидопроизводные углеводородов
Амины . .*
Цианиды .,..,.
Оредедьные углеводороды
125
62.77
101.16
128.15
75
ПО
156
76
57
43
53.5
84
149.2
85.6
-Применяя эти значения, во многих случаях удается
рассчитать энергии образования молекул в хорошем согласии
.с опытом. Очевидно, что принципиально возможно
приписать отдельным атомам, находящимся в определенных ва^
лентных состояниях, некоторые эффективные доли энергии
связей, подобно тому, как это делается при рассмотрении
междуатомных расстояний. Такой прием является, однако,
искусственным и формальным. Энергия молекулы есть,
прежде всего, энергия связей и физически осмысленно
говорить именно об энергиях связей, а не об энергиях атомов.
Аддитивную схему следует строить, исходя из химических
связей, а не из атомов как структурных элементов молекул.
100
Аддитивность энергий соблюдается далеко не всегда*
Особенно значительные отклонения имеются в соединениях,
содержащих я -связи, «- электроны (ср. стр. 67). Однако уже
непредельных соединениях, содержащих одни лишь
о-связи, схема аддитивности имеет ограниченное применение.
Теплоты изомеризации
В таблице мы привели энергии молекул насыщенных
неразветвленных углеводородов. Значения для первых
членов ряда несколько отклоняются от последующих. Только
начиная с бутана С4Н10 энергия, приходящаяся на одну
группу СН3 (добавление которой превращает каждый член
ряда в последующий), постоянна в пределах ошибки опыта.
Этого не получается, если обратиться к разветвленным
изомерам углеводородов. Известно, что, начиная с бутана С4Н,0>
все углеводороды обладают изомерами, отличающимися
расположением атомов и связей. Бутан имеет два изомера:
см,
пентан
•СН8Ч /СИ
н8с/ чсн/
нормальный бутан
три
і
Hs
няс/чл \сн
изобутан
н8(У \сн/ хсн,
н — пентан
н,сх
СНЧ /СН,
НіС/ чсн2/
иэопентан
(2 — нетилбутан)
H8Cv /СН3
тетраметнлиетан
гексан — 5 и т. д.
При помощи точных измерений, Россини установил, что
в разветвленных углеводородах аддитивность энергий
нарушается. Если бы энергии-Ьвязей во всех случаях сохраняли
свое значение, то энергии бутана и изобутана* например,* не
должны были бы отличаться, ибо обе молекулы имеют по
3 С-^-С- и по 10 С — Н- связей. В действительности, энергии
образования разветвленных изомеров больше, а именно в
реакциях изомеризации при 25°С должны выделяться теплоты
н — бутан-^нзобутан +1.64 кг кал/моль
н — пентан ^ йзоііентан +1.93 кг кал/моль
н -+- пентан —>-тетраметилметан + 4.67 кг кал/моль
Таким образом, строго говоря, аддитивность энергии
связей имеет место лишь в рядах однотипных соединений,
построенных по одному образцу.
101
Смысл аддитивности энергий
Пользуясь в указанных пределах аддитивной схекой, мы
должны., иметь в виду реальный ее смысл. Энергия молекулы
слагается из следующих частей: 1) энергии валентных
связей, 2) энергии взаимодействия атомов, не соединенных
валентными связями, 3) колебательной энергии и 4)
вращательной энергии молекулы как целого и ее составных
частей (внутреннее вращение). При этом потенциальная
энергия колебаний атомов в молекуле около их положений
равновесия .определяется первыми двумя частями, а
кинетическая—лишь относительным расположением атомов и
их массами. О колебаниях и вращениях мы будем говорить
подробно в Двух последних главах книги. Считая энергию
молекулы суммой энергий валентных связей, мы, по
существу, разлагаем по отдельным связям и энергии,
взаимодействия несвязанных атомов и колебательно-вращательную
энергию "молекулы. Аддитивность является, таким образом,
кажущейся, а значения энергии связей, приведенные в табл. 13,
эффективными. Эти эффективные значения слагаются из
действительных энергий связей .и долей .остальных частей
энергии молекулы/
Термохимические измерения, являющиеся - основным
источником сведений об эффективных энергиях- связей, не
дают возможности отличить действительную энергию связи
от остальных видов энергии молекулы. В настоящее время,
повидимому, -лишь ..шэлуэмпирическая теория колебаний
молекул может в конечно^ счете привести'к решению этой
задачи (см. -главу, VIII). Очевидно, что, толЫШргазобравпшсь
во всех видах энергии молекул, можно будет поставить
вопрос о причинах часто наблюдаемой кажущейся
аддитивности эффективных энергий связей. Легко видеть, что
квантовая химия также не да$т;искомого объяснения.
. Выделив в качестве отдельных структурных 'элементов
гомеополярные связи, мы должны применять к молекулам
соответствующую теорию, пользующуюся представлениями
об отдельных связях ^- метод Гейтлера — Лоидбна — Слэ-
тера—Паулинга:- Согласно этой теории, энергия- каждой
связи .получается из энергии-электростатического, кулонов-
ского .притяжения С* и'.энергии квантовомеханическо'го
резонанса — обмена электронов 4 (ср.. стр.* 47). Связь образуется
электронами с антипараллельными спинами, в этоі? случае
соответствующая энергия (отрицательная) ..входит щ знаком
плюс. Между несвязанными электронами разньіх,связей
осуществляется отталкивание; энергию которого мы -обозначим
буквой; /\. Кроме Toroj как мы видеди-, нужно.учесть энергии
валентных состояний,атомов» образовавших связи, ибо,, на
создание дополнительных возможностей связи,,- например,
102
pa перевод одного из 25-элекхронов углерода .в 2р-состояние,
нужно затратить определенную энергию. Обозначим энергии
валентных состояний атомов .посредством У\. Энергия
молекулы выразится: следующим образом.
E=C-2Vi + 2Au- ZFik.
i i і^Л (5)
Суммирование производится для Vt по всем атомам і
молекулы, для Ац — по всем связям і9 для Fik — no всем
электронам, • входящим в разные связи. Можно показать, что
Ffc = -TfAik. Очевидно, что в сумме УЛЦ уже фигурирует
разложение по связям, то же.относится К'?Рік.-Остальные
величины мы "также можем распределить "по
связям-вмолекуле. Таким образом, в формуле (5) уже содержится
предположение об аддитивности энергий. Именно пользуясь этим
предположением и находят, как мы видели, энергии
валентных состояний V?.
Следовательно, формула (5) может служить лишь для
выражения .факта аддитивности энергий в терминах метода
локализованных пар электронов,.который' по существу предг
лолагает такую .аддитивность.a priori. Объяснить же
.аддитивность энергий эта формула не может,
Таким образом, мы вынуждены пока ограничиться
признанием важного опытного факта кажущейся аддитивности
энергий. Разумно цоложить этот факт'в основу теории
молекулы в качестве дервого грубого приближения,-
Постоянство длин связей—междуатомндх расстояний носит.уже не
^кажущийся, а, вполне реальный характер.
Все сказанное относится к соединениям, ц которых
аддитивность— кажущаяся или реальная — действительно йм#ет
•место, главным образом к насыщенным соединениям.
Отклонения .от. аддитивности в соединениях, содержащих *?-связи,
значительно превосходит те, с которыми мы встречаемся
среди предельных углеводородов и их производных.
ОТКЛОНЕНИЯ ОТ АДДИТИВЙОСТИ-ДЛИН И" ЭНЕРГИЙ СВЯЗЕЙ
Рассмотрим подробно классический пример молекулы
-бензола.
Общепринятой в органической химии Н
является структурная формула Кекуле, н С Н
в которой единичные связи-С — С чере- \of ^С^
дуются с двойными. Однако,'прямое опре- }
деление1-' Структуры- из интерференции I I
рентгеновых и электронных.лучей пока- h/'C^C'/ \н
зывает, что все связи С—.Св бензоле .
имеют одинаковую длину 1.39 А, про- Н
ЮЗ
межуточную между длиной единичной связи С — С 1.5Ф А
и двойной связи С = С 1.33 А. Молекула бензола
представляет собой плоский правильный шестиугольник, имеет
ось симметрии С6, а не С3. С другой стороны, эксперимент
показывает, что формула Кекуле неправильна. В частности»
это доказывается тем, что имеется лишь одно
соединение типа
/\сі
С1
в то время как на основании формулы Кекуде можно было
ожидать двух изомеров с отличающимися свойствами:
С1
%/
С1
Ч/
ибо в первом случае между группами С — С1 имеется
двойная связь, во втором — единичная. Поэтому еще в прошлом
веке химикам пришлось пойти на нарушение классической
валентной схемы: Тиле предположил, что вторые связи
С — С в бензоле не локализованы, а распределены поровну
между всеми атомами. Энергия бензола не слагается
аддитивно из энергий трех единичных связей С — С, трех
связей С = С и шести связей С — Н. Имеется значительное
отклонение Ае, которое достигает 3°/0 от энергии молекулы
и равно 34.4 кг кал/моль. Эта величина Де найдена из-
сопоставления суммы энергий перечисленных связей с
энергией образования молекулы бензола. Близкое значение Де
получается также, если рассмотреть, наряду с теплотами*
сгорания, теплоты гидрогенизации бензола и циклогек-
сена QH^. Имеем
+ Н,
сн,
HjCf^CH,
н,с
сн*
+ 28.59 кг кал/моль
+ 3H,
сн,
Циклогексан
СН,
HjC/^CH,
н,с
сн,
сн,
+ 49.80 кг кал/моль
Цикл ore к сан
104
Связь С = С в диклогексене мало отличается от обычной-
двойной связи. .Поэтому можно считать, что на
гидрогенизацию трех связей С = С в бензоле Кекуле понадобится
3X28.59 = 85.77 кг кал/моль.
Разность между этим значением -и найденным дЛя бензола-
дает отклонение от аддитивности Ае=36 кг кал/моль.
Расхождение с первым значением можно объяснить тем,
что ход реакции гидрирования существенно иной, чем
процесса сгорания: во втором случае молекула разрушается,,
а в первом сохраняется шестичленное кольцо.
Величины As для молекул, содержащих несколько
ароматических колец, быстро возрастают с увеличением их.
числа и достигают весьма больших значений. Так, по Сыр-
кину, значения As для соединений такого типа имеют
следующие значения: бензол 34.4 кг кал/моль, нафталин 63.4,.
антрацен 86.2 и фенантрен 93.0.
Внешний электронный резонанс в бензоле
Паулинг применил концепцию электронного резонанса,,
с которой мы встретились при истолковании свойств
изолированных связей (стр. 57), к многоатомным молекулам и, в-
частности, к молекуле бензола* Очевидно, что две формулы:
Кекуле
I
II
соответствуют состояниям молекулы с одинаковой энергией^
Возможность написания двух таких формул определяется
тем обстоятельством, что тс-связи в молекуле бензола
создаются ^-электронами, чйи облака расположены пер-
пейдикулярно плоскости шестичленного кольца (рис. 62).
Следовательно, it-связи • в бензоле не локализованы, они
могут с равным успехом установиться соответственно
формуле (I) и формуле (И). Можно считать, что состояния
(1)'и (II) — вырожденные состояния с одной и той же энергией.
Мы имеем право рассматривать такую систему как
резонансную, подобно тому, кдк это делалось в случае молекулы
водорода. Благодаря подвижности ^-электронов имеется
возможность перехода от состояния (I) к состоянию (II) —
вырождение снимается, и уровень eft расщепляется.
Реальная молекула бензола находится на более низком уровне
энергии, возникающем в результате этого расщепления.
Поэтому энергия молекулы бензола равна не sft, а еъ
уменьшенному на некоторую величину — резонансную энергию А е.
Если энергия любой из структур Кекуле efe может быть
105
представлена суммой энергий связей, то резонансная
энергия как- раз и является отклонением от аддитивности]
Резонанс такого рода — внешний электронный резонанс,
поскольку в нем участвуют не только „внутренние" электроны
одной связи, но все • тт-электроны молекулы —
энергетически стабилизует молекулу бензола, увеличивает ее энергию
связи. В действительности молекула бензола не может быть
представлена -ни структурой (I),. ни структурой (II) порознь.
Валентная схема в .своей классической форме здесь более
непригодна, приходится описывать молекулу при помощи
нескольких, а не одной только структурных формул.
Квантовая химия дает нам воійожность связать каждую
структурную формулу с некоторой
^-функцией, описывающей состояние
системы. Эту функцию -можно
получить лишь приближенно.при
помощи метода локализованных пар
(более приспособленного для этой
цели) или метода молекулярных
орбит. Таким образоій, структурам
Рис. 62. Схема молекулы Кркуле (I) (II) будут СООТветство-
бензола. вать функции ^ь ^и и энергии
ej = еп — е^? а реальной молекуле
энергия еА — Ае и смешанная собственная функция
* = фі + фи,- ' (6)
т. е. действительное'состояние молекулы представляет
собой наложение отдельных структур, подобно тому, как
состояние реальной.связи представляет собой наложение
гомеополярной и ионной связей (стр. 58).
Строгая квантовомеханическая теория шестиэлектронной
задачи бензола (бензол .имеет 6 ^-электронов, которыми
и определяются его неаддитивные свойства), показывает,
что для полного описания свойств молекулы нужно
написать не. две структуры, а пять. Можно доказать, jrro к
структурам Кекуле (I) и (II) следует добавить еще и
структуры Дьюара:
/
Ж Ш I
В структурах (III).— (V) имеются химические связи между
несоседними атомами. Это энергетически менее выгодно:
структурам. Дьюара должна отвечать энергия е/>, больщая,
?06
V
чем энергия структур. Кекуле в,г Тем не менее, теория
показывает, что 'следует учитывать все пять структур и только
пять, пока мы ведем расчет в данном приближении."
Благодаря тому, что структуры Дьюара менее выгодны, их доля
в действительной молекуле бензола меньше, чем у
структур Кекуле, но участие их еще более понижает
энергетический уровень молекулы, увеличивает резонансную
энергию As. Волновая функция бензола должна быть записана
в виде
'[й; = ^(Фі + Фіі) + ^(6щ+^у + ^), (7)
где <ьш, ^iv, $v— собственные функции, соответствующие
структурам Дьюара. Таким образом, в резонансе- участвуют
структуры с неодинаковой энергией, подобно тому, как это
имеет место в отдельной связи (ср. стр. 59, рис. 24, 25).
Численные коэффициенты ск и cD дают нам доли участия
структур Кекуле и Дьюара. Согласно расчетам Паулинга,.
ск — 0.625 и Со — 0.270. Это значит, что „процент"
структур Кекуле равен
2ск =0.781 (8)
и „процент* структур Дьюара
ъЪСЬ 2 =0.219. (9)
Рассмотрим энергетические соотношения, определяемые
этими Структурами бензола, в'томвйде, в котором они
фигурируют в расчетах, выполненных по методу локализованных
рар. Энергию обменного, резонансного взаимодействия,
энергию спаривания двух ^-электронов, в однод ^-связи
атомов углерода мы обозначим буквой А (ср. стр. 102). Это
величина отрицательная., Два u-электрона, находящиеся у .
соседних* атомов, образуя связь,, понижают своим
взаимодействием 'энергию- системы на—Л, Наоборот,, если я-элек-.
троны атомов углерода не спарены, то каждая пара их,
отталкиваясь^.повышает энергию системы .на величину,
равную, согласно расчету, — -g-A В каждой из структур
Кекуле .(I) и "(II) шесть спаренных и шесть неспаренных
^-электронов в. С — С-связях. Следовательно, отдельная
структура Кекуле имеет по- отношению к свободным
атомам С-энергию
¦ЗА — 3/2Л'=1.5А.
В каждой из структур- Дьюара четыре спаренных, и восемь
неспаренных ^-электронов. Отдельная структура имеет
энергию
2Л — 4/2А = 0,
т. е. энергия структуры Дьюара выше, чем энергия
структуры Кекуле на 1.5А Смесь структур, представляющая,
согласно (7), реальную молекулу, имеет уровни —
повышенный по сравнению со структурой Кекуле, с энергией О,
и пониженный —с энергией 2.6Л (рис. 63). Расстояние
между уровнем, соответствующим отдельной структуре Кекуле,
и пониженным уровнем реальной молекулы, очевидно,
представляет собой отклонение энергии бензола от аддитив-
%so
ША
м=\ия
Рис. 63. Схема энергетических уровней бензола (Склар).
ности — резонансную энергию Де. Согласно расчету*
Де = 2.6Л —1.5А = 1.1 Л. Но, согл.аснО-.і).сішвной._ идей-
концепции электронного^ резонанса, резонансная энергия Ае
может быть приравнена" к" отклонению энергии молекулы
от аддитивности. Следовательно, значение As может быть най-
денб'изопыта. Если принять для As значение 36 кгкал/моль,
получим Л = — 32.7 кг кал/моль. Паулинг и Веланд дают
Л = — З4.в, а Склар Л =— 44.4 кг кал/моль. Не
приходится удивляться ?аким большим расхождениям' между
разными авторами. Расчеты разонансной энергии носят
весьма грубый и приближенный характер и получаемые
результаты определяются применяемыми методами. Здесь
существенно иное. Как мы увидим, одно и то же
значение определенной полуэмпирическим путем энергии
обмена А может быть применено для характеристики свойств
целого ряда однотипных соединений. Именно этим
доказывается, что излагаемые представления имеют некоторый фи-
108
зический смысл, хотя и лишены пока строго
количественного обоснования.
Мы видим, что молекула бензола описывается не одвой,
а пятью валентными схемами структурной химии. Это,
конечно, не значит, что реальный бензол представляет собой
смесь изомерных молекул (I) — (V), существующих раздельно.
Такое понимание совершенно неправильно и никак не
следует из концепции электронного резонанса. С другой
стороны, Паулинг справедливо критикует понятие мезомерии,
которым в ряде случаев химики заменяют понятие
электронного резонанса. Считая формулы (I) — (V) мезомерами
бензола, предполагают, что молекула бецзола непрерывно
превращается из одной формы в другую. Иными словами,
•если бы мы сфотографировали бензол на кинопленке с
числом кадров порядка 1030 сек."1,1 то на одном кадре мы
увидели бы молекулу бензола в состоянии (I), на другом — в
состоянии (III) и т. д. При этом относительное число
кадров, на которых окажется снимок молекулы в состоянии,
описываемом формулами (I) и (II), даст нам число ск — долю
структур Кекуле, вес этого состояния. Такая точка зрения
также неверна. В действительности этот мысленный опыт
дал бы на всех кадрах одно и то же смешанное состояние
молекулы бензола, характеризуемой функцией ф (7).
Состояния (I) — (V) („мезомеры") порознь не существуют, и
действительное состояние молекулы отличается и от структуры
Кекуле и от структуры Дыоара, на что указывает и рис. 63.
Следовательно, не имеет смысла говорить, что молекула
резонирует, колеблется между разными мезомерными
состояниями.
Сходные соотношения наблюдаются в ряде других
молекул, близких к бензолу. Так, большое сходство между
бензолом и „ неорганическим бензолом*4 B3N3He объясняется
тем, что, наряду с гомеополярной структурой (I), в нем
представлена и внутриионизованнад (И)
Н
/-ві /В-ч
нЛ>\„ н V н
к к
I II
Структура (II) содержит четырехвалентные отрицательный
€ор и положительный азот, изоэлектронные с углеродом
1 Формальный расчет дает для молекулы водорода частоту обмена
алектронамн порядка 10" сек.-1.
109
в его'четырехвалентном состоянии. Очевидно, что, наряду
со структурой Кекуле (II), может быть написана и вторая
структура Кекуле и- три структуры Дьюара. Бензол и
BaN3H6 — изоэлектронные молекулы. ¦
В молекулах с несколькими ароматическими кольцами
возможностей резонанса больше. Так, Хля описания свойств
нафталина
Н Н
I I;
н\с^\с/\с/н
н
ее
н н
необходимо учитывать 42 структуры соответственно числу
различных возможностей образования ^-связей. Из этих
структур, важнейшие
f\/%
S\S\
/Х/\
\^\/
\/\S Х/\/
Энергия резонанса, грубо говоря, тем больше, молекула
тем устойчивее, чем больше число резонирующих структур
(ср. численные данные, приведенные на стр. 105).
Электронный резонанс и длины связей
Отклонения от постоянных „аддитивных" значений длин
связей могут быть с успехом истолкованы при помощи
этих же самых представлений.
Увеличение кратности связи, увеличение числа валент-
ных^штрихов всегда сопровождается упрочнением связи и
•уменьшением ее длины (ср. табл. 10). При прочих равных
условиях, более прочным связям соответствуют .меньшие
междуатомные расстояния. Внутренний электронный
резонанс ионной и гомеополярной структуры в пределах одной
связи упрочняет связь (стр. 60) и, следовательно, приводит
к уменьшению ее длины по сравнению с суммой
аддитивных атомных радиусов. Мы видели, что резонанс с ионным
состоянием тем сильнее, чем больше разнятся
электроотрицательности атомов, образующих связь (ср. стр. 61). Пау-
линг оценивал электроотрицательности, исходя из длин
реальных связей. Шомекер и Стивенсон дают следующую
эмпирическую формулу для длины связи
/*ав = гА -f- гв — 0.016 і-яса — *в|,
(Ю)
ПО
где га и гв — атомные радиусы в ангстремах, хл и'^-
электроотрицателъности в электронвольтах.
Мы видели, что внешний электронный резонанс,
подобный рассмотренному выше на примере бензола, означает
изменение кратности и энергии связей. В бензоле, благодаря
наложению нескольких структур, все связи С-г-С
одинаковы. Длина их естественно должна лежать между длиной
единичной (1.54А) и двойной (1.33А) связи. Действительно,
длина связи в бензоле равна 1.39А. Очевидно, что
недостаточность классической валентной схемы, выражаемая при
помощи представлений об электронном резонансе, означает,
что реальные связи могут быть не целого порядка, не
единичными, двойными и тройными, а промежуточными.
Условно можно говорить о полуторной связи в бензоле.
Неаддитивность сводится здесь и в других подобных случаях к
тому, что единичные'связи делаются частично двойными и
длины их уменьшаются и, наоборот, длины двойных и
тройных связей увеличиваются, так как понижается их порядок.
При этом, однако, заметные изменения длины наблюдаются
именно для единичных, а не кратных связей. Имеется ряд
'попыток найти зависимость между длиной связи и ее
дробным порядком. Попытки эти носят главным образом
спекулятивный характер, в них почти всегда не учитывается то
существенное обстоятельство, что точность определения
длины связи есть 0.02 — 0.03 А и поэтому, например, не
имеет смысла считать, что в бензоле (г=1.39) длина связи
и ее порядок иные, чем в графите-или в других
ароматических соединениях (г = 1.42).
Рассмотрим ряд примеров. Начнем с некоторых
соединений углерода и азота. Следует-иметь я виду, что," наряду
с резонансом типа бензольного, принципиально возможен
внешний резонанс с --внутриионизованными структурами,
в которых атомы заряжены, находятся в необычных
валентных состояниях (ср. стр. 53). Несмотря на то, что
энергии этих структур .повышены,-их участие может- ,быть
выгодным, ибо увеличение числа структур приводит к общему
вьщг?ышу энергии. Мы уже видели, что внутриионизован-
"ные структуры существенны для описания- свойств'
молекулы 'B^N3H6. . . .
Молекула ¦' углекислого газа С02 построена линейно и
симметрично. Расстояния С — О равны -і.ібАі В то же время
суммы ковалентных'' -радиусов для G — 0 = 1.43А, для
С = 0 1.22А и для С^О I.IOA. Связь в С02 занимает
положение промежуточное между двойной и тройной связями.
Здесь имеется резонанс структур
о^с=о, о=с—о, о-— с=о, о=с—6,5—с=о.
I 'II III IV V
111
Структурами (II) и (III) изображен внутренний резонанс
с ионным состоянием в пределах одной связи, структуры
(IV) и (V) списывают внешний электронный резонанс связей.
Валентные состояния С и О написаны в соответствии с
изложенным ранее.
В молекуле СО длина связи 1.13А. Резонансные структуры
+ - +
С —О, С = 0, С = 0
i и .ш
с расстояниями (I)— 1.25А> (II) — 1.13А и (III) — і.іоА.
Молекула С02 очень похожа на линейную, но
несимметричную молекулу N20, которая имеет две структуры '
N = N = 0, N = N — 0.
i и •
В линейной, симметричной молекуле закиси углерода С302
расстояния С —С 1.29А и С —О 1.20 А, в то время как
двойные связи С = С и С = 0 имеют длины соответственно
1.33 и 1.22А. Небольшое уменьшение длин связей
определяется резонансом структур
о=с=с=с=о, о=с —с=с—о, о—с=с—с=о
I II III
о=с=с—с=о, о—с—с=с=о
IV V
и структур, отвечающих внутреннему резон.ансу типа
о—с=с=с=о.
Электронный резонанс в углеводородах и их производных
Сыркин объясняет изменения энергии при переходе от
неразветвленных к разветвленным углеводородам
изменением возможностей резонанса. В группе СН3 (метильной)
в принципе возможны три резонансных структуры с
двухвалентным углеродом, в которых связи устанавливаются
.непосредственно между атомами водорода:
н Н нч " н .
I I хн хч
/|Хн /С/Н /С\н \н
н н/ н
I На lli 11с
112
В группе СН.2 возможна лишь одна такая структура
Н н
I с\н
/|\н /\
II
МТЯ V UTTf* ТТП ГЧ-\\7ГГТТ C4-J
Н.2С С Н2 - Н /
Так как в разветвленных углеводородах число групп СНД
относительно выше (ср. стр. 101), в них имеется больше
резонансных структур, и их молекулы должны обладать
большей энергией — находиться на более низком
энергетическом уровне.
Значительно большие отклонения от аддитивности
наблюдаются в углеводородах с ^-связями — двойными и тройными.
Эти отклонения более заметны в энергиях молекул, чем
в междуатомных расстояниях, что связано, в частности,
и с большей относительной точностью измерения энергий.
Резонансная энергия бутадиена
н2с = сн—сн=сн,
равна 4.03 кг кал/моль. Она возникает за счет структур
СН = СН
\ н+с-с=сн —сн=сн3.
-l-гц и /
II Л/ III, IV, V, VI
В галоидозамещенных этиленовых углеводородов связи
С — Hal отличаются от аналогичных связей в предельных
соединениях, благодаря ионным структурам
С1+ Н
н н
Длина связи С — С1 здесь 1.69А, в то время как в С2Н5С1
она равна 1. 8іА. В еще большей степени внутриионизован-
ные резонансные структуры такого рода представлены
среди производных ацетилена
Сі+=:С = С — Н.
Электронный резонанс и химические свойства молекул
Излагаемые представления расширяют валентную схему
химии, выражают ее современное развитие. Они оказались
пригодными для истолкования и ряда химических свойств
"молекул, неотделимых, конечно, от их физических свойств.
Так, в химическое поведение бензола — его устойчивость,
S Строение молекул. 113
отсутствие изомеров одихлорбензола (стр. 104)—
объясняется изложенным. Здесь мы приведем еще один очень
яркий пример, принадлежащий Я- К. Сыркину.
Последовательное гидрирование молекулы фенантрена
і_!1/—V1 ю
У \ / \9
2\ /ь 6\ /9
3 4 7 8
происходит таким образом, чтобы сохранить наибольшую
резонансную энергию. Фенантрен имеет Ае=93.02 кг кал/моль.
Присоединение первых двух атомов водорода идет
в среднее кольцо, в положения 12, 13, ибо при этом
крайние кольца сохраняют структуру бензола и остается As =
= 2-34.4 = 68.8. Если бы водороды присоединились в
крайнем кольце, сохранилась бы структура нафталина с
меньшей энергией As = 63.4. Следующие два водорода могут
направиться, конечно, только в крайнее кольцо. Однако
оказывается, что при этом первые два атома также
перекочевывают в это же кольцо, гидрируются положения 7, 8., 9, 10.
При этом остается энергия нафталина Де=63.4. Пятый
и шестой атомы Н вновь направляются в положения 12, 13
с сохранением структуры бензола и энергии As = 34.4, но
при дальнейшем гидрировании переходят во второе крайнее
кольцо; среднее сохраняет структуру бензола. Таким
образом, гидрирование идет через стадии с сохранением
наибольшей резонансной энергии — неаддитивной энергии. Пункти-'
ром отмечены связи, участвующие в резонансе.
НН Н'ННН НННН'
Можно было бы привести множество других примеров.
Так, Сыркин и Дяткина объяснили на основе этих
представлений химические свойства нафталина и антрацена. Ряд
правил органической химии получает здесь убедительное
истолкование. В то же время, было бы неверным требовать
от концепции электронного резонанса исчерпывающего
объяснения во всех случаях. С одной стороны, концепция эта
носит качественный, приближенный, химический,, а не
физический характер. С другой стороны, направление и ход
114
химических реакций зависит от множества внутренних
и внешних факторов. Небольшие изменения в значениях
относительных энергий конечных и промежуточных
состояний реагирующих молекул могут повести к громадным
изменениям скорости реакций, к изменению ее направления.
Задача концепции электронного резонанса состоит не в
истолковании любых свойств молекул, а в наглядном объяснении
отклонений от аддитивности, перед которыми становилась
в тупик классическая валентная схема. Ниже мы рассмотрим
различные физические свойства молекул, для описания
которых концепция электронного резонанса часто оказывается
очень полезной.
ОКРАСКА ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
Современная теория цветности исходит из тех же
представлений об электронном резонансе, которые применяются
для описания неаддитивности.
Известно, что многие органические соединения как
природного, так и синтетического происхождения очень ярко
и разнообразно окрашены. Это означает, что они поглощают
свет в видимой области. До недавнего времени не удавалось
объяснить этого обстоятельства. В результате рассмотрения
обширного опытного материала были найдены лишь
некоторые закономерные признаки, которыми обладают молекулы,
поглощающие свет в видимой области. Важнейший признак,
подмеченный еще в 18.88 г. Виттом, это— присутствие в
молекулах окрашенных соединений, так называемых 2ФРЖ)г
форных гр_упп. Таковы группы атомов и связи: бензольное
кольцо, — С = С —,Чс=0, —N==N —, —N02 и др. Мы
видим, что хромофорные, группы содержат т>связи. Однако
одного наличия хромофоров недостаточно для появления
окраски. Хромофоры, согласно Витту, проявляют свое
действие в. присутствии групп второго рода—ауксохромов.
Таковы группы: —NH2,— ОН,— ОСН3идр. С другое стороны,
в настоящее время известно, что как раз в тех молекулах,
которые содержат одновременно хромофоры и ауксохромы,
особенно велики -отклонения от аддитивности.
Поглощение коротковолновых лучей света, видимых
и ультрафиолетовых, происходит в молекулах, так же как
и в свободных атомах, при переходе электронов с
некоторого низкого, невозбужденного уровня на более высокий,
возбужденный уровень. Частоты, соответствующие этим
переходам, подчиняются, конечно, условию Бора (ср. стр. 20),
имеющему универсальное значение
*
115
В атоме переходу между электронными уровнями еі и efe
соответствуют отдельные линии, атомный спектр линейчатый.
В молекулах, наряду с электронными переходами, возможны
квантованные изменения колебательной и вращательной
энергии молекулы, о которых мы будем подробно говорить
в последних главах. В молекуле рядом с каждым'уровнем
^расположены колебательно-вращательные уровни и, вместо
отдельных линий, получаются широкие полосы. Говоря
о частоте, определяемой формулой (И), мы имеем в виду
центр полосы поглощения. Разности энергий электронных
уровней &і — еЛ у неокрашенных огранических веществ
(например^ насыщенных углеводородов) соответствуют длинам
волн порядка 1000—2000 А. Полосы поглощения
расположены в далекой ультрафиолетовой области спектра, в так
называемой вакуумной, шумановской области. У соединений,
содержащих хромофоры и ауксохромы, полосы поглощения
резко сдвинуты в сторону длинных волн, к видимой области
спектра. Спрашивается, как можно объяснить этот сдвиг —
уменьшение разностей е? —efe?
В 1937 г. Склар вычислил положение полос поглощения
ряда углеводородов, содержащих хромофор — С = С — и
ароматические кольца, исходя из неаддитивностей энергии этих
соединений. Обратимся вновь к рис. 63, на котором дано
расположение энергетических электронных уровней
молекулы бензола. Два самых низких уровня реальной молекулы
е и е' расположены на расстоянии 2.6 А. Следующий
возбужденный уровень е" лежит значительно выше. Между
уровнями е, е' возможен электронный переход с
поглощением света. Очевидно, что этому переходу соответствует
наименьшая частота света, наибольшая длина волны. Именно
этот переход определяет положение самой длинноволновой
полосы — окраску бензола. Об окраске мы здесь говорим
условно, так как бензол бесцветен, но его полоса
поглощения уже сдвинута из шумановской в ближайшую
ультрафиолетовую область. Для вычисления длины волны этой полосы
цужно знать А. Очевидно, что длина волны равна
Но, как мы видели, значение А может быть найдено из
совершенно независимых данных по теплотам сгорания и
гидрогенизации. Выше мы оценили А из резонансной энергии
Де== —1.1 Л и нашли Л = — 32.7 кг кал/моль. Склар
находит из теплот гидрогенизации бензола и циклогексадиена
большую величину А, а именно 44.4 кг кал/моль.
Расхождение объясняется тем, что Склар исходил из другого способа
оценки энергии двойной связи С = С, учитывая ее
изменения под действием соседних атомов. Здесь сказывается не-
Ш
которая произвольность в отнесении всей неаддитивности
энергий за счет электронного резонанса.
Величина А — — 44.4 кг кал/моль является разумным
эффективным значением. Энергия А фигурирует не только
в случае бензола, но и в случаях других молекул,
содержащих нзлокализованные ^-электроны (в случаях
бутадиена и ряда циклических соединений). Пользуясь этим
значением А, Склар получил значения длин волн для ближайших
к видимой области спектра полос поглощения (табл. 14).
Т
а бл ица
14
(Склар)
Молекула
Бутадиен
Структура
/ \
\ /
^>=СН3
/\
/
\
\_/
С=С-
-с=
С
>*,
вычислено
2470
3645
6914
1900
о
А
измерено
' 2590
3550
7000
¦
1 . 2100
Азулен — единственный известный углеводород, имеющий
синюю окраску. Совпадение с опытом хорошее, и это
подтверждает правильность оценки А. Укажем, чтов расчет
с А = 32.7 кг кал/моль дает для бензола ^ = 3 500 А и для
других соединений — также повышенные значения.
Ферстер продолжил работы Склара, воспользовавшись
упрощенным методом расчета. Во всех случаях при расчете
учитывались только самые важные структуры. Так, для
бензола отбрасывались структуры Дьюара и сохранялись
структуры Кекуле. Полученные результаты весьма
убедительны. Они приведены в табл. 15. Для оценки А Ферстер
применил остроумный полуэмпирический метод. Теория
дает возможность выразить в' — ев Л-величинах. Согласно
(11) и (12),
А
(13)
откуда
ig
ъЧ-W;
(14)
117
ig
he
величина' постоянная. Очевидно, что зависимость
е' — s
рассчитанных lg—-^ от измеренных lgX представится графи-
чгски прямой линией, проходящей под углом в 45° к осям
координат.
Таблица 15
(Ферстер)
Бензол . .
Нафталин.
Антрацен.
Нафтацен
Пентацен .
Фенантрен
Пирен . . .
2.40
1.97
і.ео
1.31
1.08
L94
1.70
2450
2950
3650
4500
5450
3000
3450
2590
2750
3700
4600
5800
2950
3300
Как видно из рис. 64, полученные данные хорошо ложатся
на такую прямую. Ферстер находит А = — 49 кг кал/моль,
v. де
3d
22-
іО
' хо Бензол
Нафталин О
G\PeHat/njpett
о
Пирен
Янтрсце*
Нофтацвн
Опетще*
±
J i_l
X.
IgA
Ш8 то то sooo вооо /5
Рис. 64. Зависимость lg Де/Л от lgX (Ферстер).
в хорошем соответствии со значением, вычисленным Скла-
ром.
Расчеты, основанные на этих идеях, были проведены для
.118
многих других молекул типичных красителей. Удалось
разобраться в характере влияния тех или иных заместителей —
ауксохромов на положение полосы поглощения красителя.
Роль ауксохрома сводится к увеличению возможностей
электронного резонанса и к повышению веса внутриионизован-
ных структур. Например, в трифенилметановых красителях,
содержащих в качестве хромофоров бензольные кольца,
резонанс осуществляется между этими кольцами и ауксо-
хромами. Фиолетовая краска кристаллвиолет имеет
следующие важнейшие структуры иона:
S\ ?\ S\
н3с сн3 Щ Щ
1 структура 9 структур 3 структуры
Группа N(CH3)2 — ауксохром именно потому, что создает
дополнительные возможности резонанса.
Недавно (1942) Склар вычислил не только положение
полос поглощения ряда органических соединений, но и
интенсивности этих полос, причем его результаты находятся
в хорошем согласии с опытом.
ЭЛЕКТРОННЫЙ РЕЗОНАНС. И НЕАДДИТИВНОСТЬ
Специфически неаддитивные свойства молекул,
содержащих тг-связи, определяются невозможностью локализации
^-электронов, тс-электроны обладают большой подвижностью; :
говоря на квантовомеханическом языке, они размазаны ¦
между различными связями в молекуле. Современная химия \
описывает-свойства молекул с нелокализованными электро-
нами при помощи концепции электронного резонанса. При
этом вся неаддитивная энергия приравнивается энергии
•Ае электронного резонанса. Применяя такой прием,следует
иметь в виду его условность.
Выше мы говорили о смысле аддитивности энергий
(стр. 102). Аддитивные значения энергий — кажущиеся.
Поэтому кажущейся является и резонансная энергия. Целый
ряд факторов здесь не учитывается, и в первую очередь
119
колебательная энергия молекул. С другой стороны, в
обычных оценках As пренебрегают тем, что на изменение длины
связей в реальной молекуле, по сравнению с длинами связей
в составляющих структурах, также требуется определенная
энергия. Это было отмечено Леннард-Джонсом. Поэтому,
согласно его расчетам, резонансная энергия бензола равна
не 34.4, а всего лишь 30.2 кг кал/моль.
Смысл и ценность идеи электронного резонанса состоят
в дальнейшем развитии наглядной химической валентности
схемы. Было бы неправильным требовать от этой концепции
физической строгости, точности и однозначности расчетов.
Квантовомеханическая теория сложных молекул в настоящее
время невозможна и, вероятно, не нужна. Концепция
электронного резонанса позволяет связать разнообразные факты,
относящиеся к свойствам молекул на полузмпирической
основе. Формальные квантовомеханические расчеты не имеют
здесь большой ценности. По существу говоря, весьма
затруднительно приписать той или иной структурной формуле
волновую функцию <{/. Нахождение такой функции в
многоэлектронной проблеме невозможно. В действительности не
существует ни отдельных структур молекулы (например,
структур Кекуле, Дьюара), ни их ф-функций. Однако,
„резонансная" теория современной химии несомненно более
совершенна, чем какая-либо из ранее существовавших теорий
валентности. ,И если квантовая механика и не дает
возможности строгого обоснования теории и точных расчетов, то
во всяком случае она объясняет химику принципиальную
возможность существования „смешанных" валентных формул
и выигрыша энергии вследствие такого смешения.
Весьма плодотворной нам представляется разработка
физической теории „неаддитивных* молекул на основе
аналогии с металлом и, в частности, со сверхпроводником
(Лондон). Физические свойства металлов — их
электропроводность и теплопроводность, излучение,
поглощение и отражение света и т. д. — объясняются относительной
свободой, нелокализоваиностью части валентных электронов.
С химической точки зрения кусок металла представляет
собой одну громадную молекулу. В результате „резонанса
электронов" металла их энергетические уровни размываются в
широкую полосу. Для перехода валентного электрона с
низкого энергетического уровня на более высокий, на котором
электрон „свободен" и может двигаться, перенося ток, нужна
небольшая энергия. В „неаддитивных" молекулах ті-электроны
также могут рассматриваться как свободные. В результате
энергетические уровни сближаются, вещество поглощает свет
.в видимой области и зачастую настолько интенсивно, что
обладает здесь металлическим блеском (метилвиолет).
Аналогом тока является перенос ^-электронов с одного конца
120
молекулы на другой, возникновение большого дипольного-
момента и сильного электронного поглощения. Молекулы
с конъюгированными двойными связями-г-своего рода про*
водники, и это находит свое выражение в их магнитных.
свойствах. Напротив, насыщенные, обладающие аддитивными
свойствами соединения ведут себя подобно диэлектрикам,
в которых для появления электропроводности нужна
значительная затрата энергии. Физическая теория органических
соединений, основанная на этих соображениях, сможет,
очевидно, дать общую характеристику этих молекул, но в
отдельных случаях не будет в состоянии предсказать их
индивидуальные свойства. Физическая теория всегда является
более строгой и общей, но тем самым менее конкретной^
чем химическая. Этим определяется право на существование
таких теорий, как концепция электронного резонанса.. Но
для подлинного понимания общей сути явления пригодна
лишь физическая теория. Создание такой теории
многоатомных молекул есть'дело ближайшего будущего.
Глава V
ЭЛЕКТРИЧЕСКИЕ СВОЙСТВА МОЛЕКУЛ
Электрические, магнитные и оптические свойства молекул
определяются поведением электронов и положительных ядер
молекулы в электромагнитном поле — статическом поле,
создаваемом между заряженными пластинами конденсатора
или полюсами магнита, и переменном поле световых и
радиоволн. Электромагнитные свойства молекул неразрывно
связаны с их строением и дают поэтому возможность его
исследовать.
ДИЭЛЕКТРИЧЕСКАЯ ПОСТОЯННАЯ
Вещество, помещенное между пластинами конденсатора,
увеличивает его емкость. Вместо емкости С0, которую имел бы
конденсатор в вакууме, создается емкость
С=аС0, (1)
где е — диэлектрическая постоянная, характеризующая
вещество. Для газов она не многим превосходит единицу, для
жидкостей меняется в широких пределах — от 1.05 для
жидкого гелия и 1.56 для жидкого воздуха, до 79.5 для воды
и г^ 95 для синильной кислоты (при 25° С). Увеличение
-емкости объясняется поляризацией диэлектрика. Под
действием электрической силы в молекулах диэлектрика
происходит смещение отрицательных и положительных
зарядов (рис. 65), диэлектрик поляризуется. Благодаря этому
усиливается электрическое поле между пластинами
конденсатора. Если в отсутствие вещества это поле, имело силу
Е, то, согласно теории электричества, диэлектрик
увеличивает ее до величины
D = *E. (2)
Так как е больше единицы, D больше Е. Можно показать, .что
D = sE = E~\- 4*Р, (3)
122
где Я—некоторая характеристика диэлектрика, называемая
поляризацией.
Рассмотрим молекулярный механизм поляризации. Для
того чтобы разобраться в том, что происходит с отдельной
молекулой между пластинами конденсатора, необходимо
оценить, какая сила действует на нее. Помимо общей силы
(2), на молекулу действует поле соседних молекул, оценить
которое в общем .виде достаточно трудно; оно зависит от
относительного расположения и, в сущности, от формы
молекул. Можно доказать, что для хаотически
расположенных молекул, удаленных друг от друга на расстояния,
значительно превышающие размеры мо-
лекул, поле, действующее на
отдельную молекулу в газе, характеризуется
силой
F=?-f-$A
(4)
Поляризация Р 1 см3 диэлектрика,
содержащего N молекул, складывается
из дипольных моментов всех молекул
(дипольный момент равен
произведение величины смещенного заряда на
расстояние между центрами тяжести Рис- 65* Поляризация мо-
положительного и отрицательного за- лекулы.
рядов). Обозначив индуцированный в молекуле дипольный
момент буквой р, получим
P = Np.
(5)
С другой стороны, индуцированные моменты молекул р
создаются действующим полем F и должны быть
пропорциональны его силе
p = aF,
(6)
где а — характерная для молекулы физическая постоянная —
поляризуемость молекулы. Из уравнений (2)—(6) получаем
4іс
«(і — т Щ
1+Цма,
откуда
е — 1 4тс ..
7+2 = ТШ>
здесь N—число молекул в 1 см3. Переходя к
грамм-молекуле, напишем
*=лъ4,
м
і2а
где Ma— число Авогадро, М — молекулярный вес, с?
—плотность. Получаем
= МАа = Р. (I)
е + 2 d 3
Это выражение молярной поляризации Р—формула Клау-
зиуса—Мосотти. Величина Р имеет важное эмпирическое
значение, так как она дает возможность единообразно
охарактеризовать поляризуемость и дипольные моменты молекул
(см. ниже).
Опыт показывает, что существуют два типа молекул,
различающихся своим поведением в электростатическом
поле. Молекулы первого типа имеют в соответствии с
формулой (7) диэлектрическую постоянную, не зависящую от
температуры. В самом деле, весь ход вывода этой формулы
таков, что ни а, ни Р не должны зависеть от теплого
движения молекул. К первому типу относятся молекулы
одноатомные, симметричные двухатомные молекулы 02, N2, моле-,
кулы СОа. CS2, большинство углеводородов. Напротив, для
молекул второго типа наблюдается резкая зависимость е от
температуры, не объясняемая формулой (7). У этих веществ
6 падает с ростом температуры. В то же время
диэлектрические постоянные веществ второго типа обычно
значительно превосходят е для веществ первого типа. Ко второму
типу относится большинство известных соединений, в
частности Н20, HCN, СН3На1, СН2На12, СННа13. Разница между
молекулами первого и второго типа этими двумя
признаками, однако, не ограничивается. Ниже мы сопоставим
свойства этих типов молекул и в таблице и рассмотрим причины
различия между ними.
ПОЛЯРИЗУЕМОСТЬ МОЛЕКУЛ
Поляризуемость а непосредственно характеризует
свойства молекулы и непосредственно связана с размерами ее
электронного облака. Это следует уже из соображений
размерности. Согласно (6) a = plF. Но р есть произведение из
заряда на расстояние, a F, согласно закону Кулона, имеет
размерность заряда, деленного на квадрат расстояния. Итак,
[а]=іш=іш[Р]=[Р]} (8)
т. е. поляризуемость должна измеряться в кубических
сантиметрах. Это естественно, так как поляризуемость определяет
смещаемость электронного сблака под действием
электрической силы, т. е. объем, который может занять это облако.
Очевидно, что и численное значение поляризуемости должно
быть того же порядка, что и размеры электронного облака,
124
размеры молекулы. Действительно, легко показать, что
проводящий шар в электрическом поле приобретает дипольный
момент p = r*F, где г—радиус шара. Иными словами,
поляризуемость проводящего шара равна кубу его радиуса.
Считая молекулы такими проводящими шариками, мы
придем к выводу, что а должно иметь порядок величины
кубического ангстрема (Ю-24 см3). Благодаря этой простой связи
между объемом молекулы и поляризуемостью можно думать,
что молярная поляризацияР должна быть близка к
величине объема всех молекул в моле, т. е. к объемной
поправке ван-дер-Ваальса Ь, деленной на 4 (ср. стр. 70)
b = 4NA^r*^4NA^a = 4P. (9)
Соотношение (9) приблизительно соблюдается для молекул
первого типа, но у молекул второго типа Р значительно
превосходит 6/4.
Следует различать поведение молекул в
электростатическом поле, характеризуемое диэлектрической
постоянной е, и поведение в электромагнитном поле световой волны,
определяемое показателем преломления п. В области длин
волн, достаточно удаленной от полос поглощения молекулы,
должно соблюдаться соотношение, вытекающее из
электромагнитной теории света Максвелла, согласно которому для
немагнитных веществ
Л = /а. (10)
Из (7) и (10) получаем
л2 — 1 М 4тс Л. п ¦ л л ч
l?+T4'=-5'NAa = R- (11)
Это формула Лорецтц—Лоренца, полученная в оптике
независимо от вывода формулы (7). Здесь а — оптическая
поляризуемость, R носит название молекулярной, рефракции.
Оказывается, что для веществ первого типа приблизительно
соблюдается соотношение (10) и молекулярная рефракция
мало отличается от молекулярной поляризации. Напротив,
у веществ второго типа Р значительно больше R. *
В табл. 16 сопоставлены все признаки, которыми
разнятся молекулы названных двух типов.
1 Строго говоря, Р может равняться R лишь для бесконечно
длинных волн: е = п2оэ. Однако, практически, для веществ первого типа
соблюдается условие е = л2 в области длин волн, удаленной от полос
еоглощения. "
125
Таб
Первый тип
P~bj\
P=z COnst
П* = г
P^R
лица 16
Второй тип
Р>Ы±
Р = Р(Т)
Л2<е
Р>Я
Данные табл. 17, относящиеся к конкретным химическим
веществам, подтверждают справедливость сделанных выше
выводов.
Таблица 17
Вещество
Я
Ь/4
1-Й ТИП
НИ. ,
сё*..
свй14
С0Н0 .
2.0
21,7
29.8
27.5
2.0
21.7
29.7
25.9
5.0
19.5
44.0
37.0
2-Й тип
Н.О
NrV .
ССН5С1
17.6
63.0
86.0
3.7
5.6
31.1
7.7
9.5
36.1
П*
1.0003
2.62
1.87
2.29
81
22
11
1.00
2.54
1.89
2.23
1.78
1.75
2.31
ПОЛЯРНЫЕ МОЛЕКУЛЫ
Вещества второго типа — это полярные молекулы. Они
построены таким образом, что центры тяжести
положительных и отрицательных зарядов в отдельной молекуле не
совпадают, а находятся на некотором расстоянии / даже при
отсутствии внешнего поля (рис. 66). Так, например, в
молекуле НС1 атомы Н и С1 обладают различными электро-
отрицательностями. Поэтому, в НС1 представлено в
значительной мере ионное состояние Н+СГ, и центр тяжести
электронного облака смещен по направлению к хлоруг
Следовательно, молекула НС1 обладает постоянным диполь-
ным моментом, для нее характерным. То же самое относится
ко всем молекулам второго типа. Очевидно, что молекулы,
имеющие центр симметрии, дипольным моментом не
обладают, ибо симметрия относится не только к атомам, но и
к электронам молекулы и центр симметрии является центром
тяжести и для положительных и для отрицательных зарядов.
Так же неполярны плоские молекулы с осью симметрии»
перпендикулярной плоскости.
1 Газ.
2 Жидкость при комнатной температуре.
126
Вещества, состоящие из молекул, имеющих постоянные-
дипольные моменты, поляризуются полем не только
вследствие индукции, т. е. появления наведенного момента»
определяемого поляризуемостью, но и вследствие ориентации
молекул в поле. Молекулы стремятся установиться в поле
таким образом, чтобы их дипольные моменты были
параллельны силе поля. При этом молекулы
имеют наименьшую потенциальную
энергию. Однако, параллельной установке
диполей препятствует вращательное тепловое
движение молекул. Благодаря ему,
диэлектрическая постоянная полярных веществ
должна падать при увеличении
температуры. При данной температуре
поляризация среды Р0, создаваемая ориентацией
диполей, определяется средним значением
проекции дипольного момента молекулы
на направление поля, т. е. косинусом угла между
средним направлением диполя и направлением поля (рис. .67),
согласно формуле
Рис. 66. Схема
диполя НС1.
P0 = Afy.cos*>,
(12)
где N—попрежнему число молекул в 1 см3, ц
—постоянный дипольный момент молекулы, я> — угол между направлен
-^-г
Рис. 67. Ориентация диполей.
нием диполя и слой поля F. Черта сверху означает
усреднение по всем возможным направлениям при данной
температуре. Очевидно, что средний угол 0 тем ближе к 0° и
cos 0 тем ближе к единице, чем больше ^ и Fz чем
меньше 7V Согласно расчетам, зависимость cos 9 от температуры
изображается следующим графиком (рис. 68, кривая-а). По
127
оси абсцисс отложено -у, по оси ординат cos ft (или Р0). При
низких температурах, т. е. при больших у, кривая
асимптотически приближается к прямой линии, отвечающей
cost>=l. Это — выражение того факта, что при прекращении
теплового движения происходит насыщение поляризации:
все диполи устанавливаются вдоль поля, и дальше
поляризация повыситься не может. Наоборот, при высоких Г
малых
1
т J кривая практически сливается с прямой, идущей
лод углом из начала координат. Это имеет место при
комнатной и более высоких температурах. Здесь соблюдается
-соотношение
3kT >
COS
(13)
где k — постоянная
Больцмана,
характеризующая тепловую
энергию молекул, равная
1.33-10"16 эрг/град. На
основании (12) и (13)
ориентационная
поляризация равна
Рис. 68. Зависимость ориентации диполей P0 = N-~jt F. (14)
от температуры.
По аналогии с (6) мы
можем принять |і.2/3&Гза своего рода поляризуемость — ори-
¦ентационную „поляризуемость" молекулы. Общая
поляризация полярных веществ представляет собой'""сумму.
индукционной поляризации Р, "о которой мы говорили выше и
ориёнтацйонной.поляризации Р0. Поэтому для полярных
веществ следует в формуле Клаузиуса—Мосотти (7) написать не
-а, а сумму а и ориёнтацйонной поляризуемости \?/3kT. Для
полярных веществ мы получим, вместо (7), выражение
е—1
d
4т:
NA[a +
3kT
= Р.
(15)
В то же время поведение полярных молекул в поле
световой волны не отличается от поведения неполярных — веществ
первого типа. Электрическое поле световой велны—быстро
переменное поле. Направление его силы F меняет знак
с частотой (в видимом свете) порядка 4— 8-1014 сек."1
Напротив, частота вращения молекул, вызванного тепловым
движением, имеет порядок 1011—1019 сек."1-За время одного
светового колебания молекула не успевает повернуться,
диполь не успевает ориентироваться в поле световой волны.
128
Поэтому выражение для молекулярной рефракции остается
прежним, оно содержит только обычную индукционную
оптическую поляризуемость
Л? — 1 р.
/z- + 2 d
4-
NAa = %.
(16)
Сопоставление формул (7) и (11), с одной стороны, и (15) и
(16), с другой, полностью объясняет причины всех различий
между веществами первого и второго типов, перечисленных
в табл. 16.
Изложенная теория ориентационной поляризации была
предложена Дебаем. Квантовая механика, учитывающая
квантованность энергии вращения молекул, не вносит в
формулы (14) и (15) существенных изменений.
Легко видеть, что приведенные соотношения (15) и (16)
дают способ определения дипольных
моментов молекул. Можно,
во-первых, воспользоваться соотношением
*-*-?*&¦
О?)
а
CflCl,
¦са
Зная е, п> М, d и Т можем найти \і.
Во-вторых, исследование
зависимости диэлектрической постоянной, а
значит и молекулярной
поляризации Р от температуры дает нам,
согласно (1#), возможность
определить р.. На рис. 69 приведены
графики зависимости Я от у для
типичных веществ первого и второго типов. Для СС14 Я от Г
не зависит, и прямая идет параллельно оси абсцисс. Для
СН3С1 прямая идет наклонно, под углом у к оси абсцисс.
Тангенс этого угла равен
Рис. 69. Зависимость
поляризации от температуры.
а — бездипольная молекула,
б— дипольная молекула.
4тс
tgT = x^fe
(18)
Измерив Я при разных температурах и найдя наклон графика,
мы определим р. Прямая пересекает ось ординат в точке
p — ^-j\/Aa, Найдя эту ^точку, определим а. Определение
дипольных моментов молекул производится либо в газах,
либо, чаще всего, в разбавленных растворах в неполярных
растворителях. В чистых жидкостях взаимодействие между
молекулами настолько сильное, что заметным образом
сказывается на величине диэлектрической постоянной. Молекулы,
связанные друг с другом силами взаимодействия, не могут
ориентироваться в поле независимым образом.
9 Строение молекул. *?*
ДИСПЕРСИЯ ПОЛЯРИЗАЦИИ
Как мы только что видели, молекулярная поляризация
полярных молекул отличается от молекулярной рефракции
из-за того, что диполи не успевают ориентироваться в поле
световой волны. Тем самым мы встретились с двумя
крайними случаями зависимости поляризации от частоты
колебаний поля. Совершенно естественно возникает вопрос о
поведении диполей при небольших частотах колебаний
электрического поля, соизмеримых с частотами вращения
диполей. Таковы частоты ультракоротких радиоволн.
Оказывается, как и следовало ожидать, что в той области, в
которой частоты колебаний поля делаются больше частот
вращения дипольных молекул, ориентационная поляризация
резко уменьшается (рис. 71). Эти явления особенно важны
для изучения свойств жидкостей, ибо частоты вращения
диполей непосредственно связаны с вязкостью: чем больше
вязкость жидкости, тем медленнее вращаются диполи, тем
быстрее падает ориентационная поляризация с возрастанием
частоты. Однако не только ориентационная, но и
индукционная оптическая поляризация зависит от частоты колебаний
электромагнитного поля.
Электронная поляризация
Известно, что показатель преломления веществ в
сильной мере зависит от длины волны — частоты падающего
света. Этим объясняется спектральное разложение света.
Иными словами и рефракция R и поляризуемость а зависят
от частоты.
Сущность явления может быть понята при помощи чисто
классических представлений. Будем считать внешние
электроны молекулы, определяющие ее поляризуемость,
осцилляторами, способными колебаться около положения
равновесия с собственными частотами колебаний vf. Это как раз те
частоты, которые может поглотить или испустить молекула.
Вычислим поляризуемость отдельного электрона,
колеблющегося как гармонический осциллятор (стр. 16).
В отсутствии поля электрон находится под действием
внутренней упругой силы — kx, и его движение описывается
уравнением (16) (глава I)
mx = — kx. (19)
В электрическом поле F на электрон действует, кроме того,,
сила eF, пропорциональная напряженности поля F и заряду.
электрона е. Движение электрона описывается уравнением
mx =—kx-\-eF. (20)
130
Уравнение (19) имеет решение (ср. стр. 16)
x = x0cos2itv0?, (21)
где v0 — собственная частота колебаний электрона, равная
v
2г. У м?
(22)
Будем искать решение (20) в случае поля световой волны,
периодически меняющегося с частотой v
F=Fqcos2tM. (23)
Легко видеть, что уравнение (20) при условии (23)
удовлетворится решением, подобным (21).
x = x0cos2nvt, (24)
т. е. под действием внешнего поля, колеблющегося с
частотой v, электрон сам колеблется с этой частотой. Подставив
(24) в (20), получим
X = T=Z?Z?F- (25)
Умножим обе части (25) на е. Получаем значение диполь-
ного момента, индуцированного внешним полем
P = ex = T=WF=aF- (2б>
Тем самым, мы нашли выражение поляризуемости.
Подставив значение k из (22), имеем
(27)
е- \
п —
— Аъ*т V-
В постоянном поле v = 0 и
е°-
~° 4iramV
[
-V2 *
е*
" к '
(28)
т. е. статическая поляризуемость обратно пропорциональна
коэффициенту упругости электронного осциллятора.
Молекула обладает целым набором электронов,
способных колебаться с различными частотами v,-. Каждый из
электронов вносит свою долю в общую электронную
поляризуемость молекулы. Поэтому дисперсионная формула (27)
в случае многоэлектронной системы может быть переписана
в виде суммы по всем электронным частотам
і
* 131
Здесь ft— так называемая сила осциллятора —характеризует
степень участия электрона в данном .колебании, а значит и
интенсивность соответствующей полосы поглощения. Сумма
всех // равна числу колеблющихся электронов в молекуле.
Очевидно, что, когда v становится близкой к одной из
v;—к частоте полосы поглощения молекулы, действие
световой волны особенно значительно, происходит резонанс,
электроны усиленно раскачиваются, и поляризуемость, а значит и
показатель преломления, сильно возрастает. При v = vz.
показатель преломления резко меняет свое значение (рис. 70).
Квантовая механика приводит к подобной же зависимости
от частоты внешнего поля, с той разницей, что, вместо
частот колебаний v?, появляются частоты квантовых
переходов с нулевого на і-тый уровень v/0, а в числителях —
коэффициенты/^, характеризующие вероятности таких пёре-
Рис. 70. Дисперсия показателя преломления и полоса
поглощения.
ходов, т. е. интенсивности соответствующих полос
поглощения
Такова, следовательно, зависимость электронной поляризации
от частоты падающего света—дисперсия электронной
поляризации. У большинства прозрачных веществ частоты
электронных переходов v/0 попадают в далекую ультрафиолетовую
область спектра. Поэтому аномальные явления — аномальная
дисперсия, возникающая при v = v/0, в видимом свете не
наблюдается.
Атомная поляризация
Не только электроны, но и положительные ядра смещаются
под действием поля. Смещение происходит, конечно, в
противоположную сторону и на значительно меньшее расстоя-
ние. Атомная поляризация является небольшой долей общей
индукционной поляризации Р. В быстропеременном поле
видимого света тяжелые ядра не успевают сместиться за
время колебания световой волны. Поэтому, так же как и
ориентационная, атомная поляризация не входит в
молекулярную рефракцию.
Следовательно, атомная поляризация Ра может быть
найдена, если известная общая молекулярная поляризация Р,
ориентационная поляризация Ро~-^-^а^и молекулярная
рефракция /?, равная электронной поляризации Ре. Имеем
P=Pe+Pa + Po = R +
4тс
3 ^ЛМТ
+р«.'
(31
Зная п, s и у., найдем Ра. В табл. 18 приведены некоторые
значения Ре и Ра (в см3) для неполярных молекул, у которых
я0=о.
Т а б л и ц г
Вещество
Бензол
Гексан
Четыреххлористый углерод
Сероуглерод
L 18
Ре
25.05
29.33
25.78
19.38
ра
0.55
0 26
1.2
2.3
Для атомной поляризации также наблюдаются
дисперсионные явления. Здесь, однако, существенны не быстрые
колебания электронов, а медленные колебания ядер около их
положений равновесия, частоты которых попадают в далекую
инфракрасную область спектра. Атомная поляризация
выражается формулой, аналогичной (29), с тем отличием, что
здесь V,-—инфракрасные частоты ядерных колебаний. В целом
мы можем написать
Р = Ре + Ра + Ро=У,-1^~+У-^Ч+Ро. (32)
'Еі~
'Аі
электронные частоты ядерные частоты
Все численные множители введены в коэффициенты
Общий ход -дисперсии Р показан на рис. 71 (стр. 134).
МОЛЕКУЛЯРНАЯ РЕФРАКЦИЯ И СТРОЕНИЕ МОЛЕКУЛ
Молекулярная рефракция характеризует поляризуемость
всех электронов молекулы и тем самым представляет собой
важную молекулярную постоянную, подобно энергии
молекулы и другим ее свойствам. На прежних основаниях можно
133
подойти к рассмотрению молекулярной рефракции с точки
зрения аддитивности. Действительно оказалось, что
рефракцию молекулы можно в первом приближении представить
как сумму рефракций составляющих элементов — атомов,
ионов и связей.
10* 107 10s 10s 101С 10" 101? 1Q1S 13й 10f5 Wf*
У ИВ ^5-1010
Инфра-крт. SIO^-wo**
сёк
Видимые У-8-10'?
УльттшислЕт.8-1014' 10іе
Ряс. 71. Зависимость показателя преломления п от частоты
электрического поля.
Ионные рефракции
В ионных молекулах выделение в качестве структурных
элементов отдельных ионов является, как мы уже видели,
физически оправданным приемом. Приблизительное
постоянство поляризуемостей отдельных положительных ионов
можно установить, сравнивая рефракции солей, содержащих
данный ион с рефракциями соответствующих кислот. Дело
в том, что, грубо говоря, можно считать положительный
ион водорода имеющим поляризуемость и рефракцию,
равными нулю, ибо Н+ есть протон, лишенный электронного
облака, способного смещаться в электромагнитном поле.
Вазастьерна определил разности рефракций в следующих
случаях:
134
NaCl — HC1 =* 0.74, KC1 — HC1 = 2.85,
NaN03 — HN03 = 0.76, KN03 — HN03 = 2.80,
i- Na2S04 — j H2S04 = 0.71, \ K.2S04 — ~ H,S04 = 2.89.
Средняя R для Na+=0.74 см3, средняя R для K+=2.85 см3.
Мы видим, что здесь действительно имеется постоянство
рефракций ионов. Однако, если протон сам и не поляризуется,-
то, притягивая электроны отрицательного иона, он влияет
на поляризуемость последнего, уменьшает ее.
Заранее очевидно, что поляризуемость иона должна расти
с увеличением объема электронного облака, с увеличением
атомного номера элемента. С другой стороны,
поляризуемость должна быть тем большей, чем больше электронов
содержит ион, и тем меньше, чем больше у иона
положительных зарядов. Как мы только что сказали, влияние
соседнего иона противоположного знака уменьшает
поляризуемость. Естественно, что смещаемость электронной
обол-очки должна быть тем меньше, чем больше она
деформирована соседним ионом. Поэтому в различных соединениях
рефракции, ионов понижены по сравнению с наибольшей
рефракцией свободного иона.
Борн и Гейзенберг предложили простой метод
вычисления поляризуемостей ионов из спектральных данных. Мы
видели, что энергетический уровень многоэлектронного
атома и иона определяется не только главным квантовым
числом я, но и поправками к нему, определяемыми
орбитальным квантовым числом / (стр. 22). Борн и Гейзенберг
связали эти поправки с поляризуемостью иона и, таким
образом, нашли значение ряда поляризуемостей. Применяя
результаты, полученные при изучении рефракции солей,
удалось построить полную таблицу поляризуемостей всех
ионов, подобных благородным газам. Проверкой служит
вычисление теплот сублимации решеток галогенидов
щелочных металлов, давшее хорошее совпадение с опытом. Эти
вычисления возможны здесь именно потому, что связи
в ионных решетках сводятся к взаимной . поляризации
и электростатическому притяжению. Одновременно
получилось хорошее совпадение вычисленных и измеренных
инфракрасных частот колебаний ионов решеток. Исходя из этих
работ, Фаянс и Иосс условно определили рефракции ряда
свободных ионов. Непосредственно свободные ионы не
наблюдаются, ибо даже в разбавленных растворах ионы
окружены молекулами растворителя, которые с ними
взаимодействуют и меняют поляризуемость*
Аналогичные расчеты проводил и Паулинг. Приводимые
в литературе значения рефракции ионов имеют, конечно,
условное значение. Так, физически нелепо говорить о мно-
135
гозарядных отрицательных ионах типа О2-, N3~ и т. д.:
несколько свободных электронов не могут удерживаться на
одном атоме. Кроме того, за малыми исключениями, во всех
соединениях существенны гомеополярные силы и,
следовательно, нельзя трактовать рефракции большинства молекул
как ионные. Влияние гомеополярных сил, а также соседних
атомов и ионов сильно уменьшает „ионные" рефракции. Так,
рефракция F", равная для „свободного иона" 2.65 см3
(Паулинг) уменьшается в MgF2 до 2.28, а в SF6 (заведомо
гомеополярные связи) до 1.94 см3. Практически такие
расчеты мотут оказаться очень нужными, ибо рефракция
и дисперсия — основные свойства таких материалов, как
оптическое стекло (которое содержит ионные молекулы
Si02, окислов металлов и др.). До настоящего времени не
существует метода теоретического расчета рефракции
стекол.
Гомеополярные молекулы
Таким образом, частые попытки вычисления рефракций
соединений неионного характера, исходя из рефракций ионов,1
следует признать недостаточно обоснованными. Не имеет
смысла говорить о рефракции иона О9" в молекулах
С02, S03, а по существу и Si02, иона F" в SF6 и тем более
иона С4+ или С4~ в органических молекулах. Мы уже видели,
что при характеристике свойств, связанных с внешними
электронами в гомеополярных соединениях, более разумно
выделять в качестве структурных единиц не отдельные
атомы или ионы, а электронные облака междуатомных сея-
зей. Соответствующая аддитивная схема для поляризуемо-
стей связей широко применяется в органической химии.
Обычно выделяют некоторые эффективные значения для
атомов и поправки на кратные связи — так называемые
инкременты. Из этих величин можно вычислить рефракции
связей. Некоторые данные, относящиеся к .D-линии Na,
приведены в табл. 19.
Таблица 19
(Эйзенлор, Фаянс и Кнорр)
*d
Атомы Н
F.
С1
Вг
J .
С .
о.
1.100
0.997
5.967
8.865
13.900
2.418
1.525
Инкременты Hjj
Связи С — Н
С —С
С —О
С=С .
СееС .
С=0.
Rd
1.733
2.398
1.705
1.209
1.971
4.15
6.025
3.42
1 Ср. ван-Аркедь и де-Бур, .Химическая связь".
136
Символы |~~ и 1= означают инкременты, т. е. увеличе-
ния рефракции, вызываемые соответственно присутствием
двойной и тройной связи.
Расчет по аддитивной схеме применяется для вычисления
молярных рефракций, что может служить критерием
правильности химической, структурной формулы.
Соответствующие примеры приведены в табл. 20.
Таблица 20
Молекула
Этиловый
спирт.
Углекислота. .
Закись
углерода .
Ацетоук-
сусный
эфир .
Бензол .
Структурная формула
я
С2Нь-ОН
о=с=о
0 = С=С=:С=0
с
с4о
• 0 .О
^ ^и
сн8—с—сна—с—ос,н5
/°-н/°
с н3—с=сн—с—ос?н5
<z>
Расчет RD
С—С + 5 (С—Н) +
-+С—0—Н
2(С=0)
2(С=С) + 2(С=0)
С~С + С-С + С=0
_j_C-0-C
4 (С—0+С-0-С+
+ Ю(С-Н) + 2(С=0)
3(С—Q + C-0—С +
+ 9(С-Н)+С—С +
+ с=о+с~о-н
3(С-0) + 3(С=С) +
+ 6 (С—Н)
*D
вычислено
12.96
6.84
15.49 ч
13.57/
31.57]
1
' 1
32/)2)
25.93
найдено*
і2.7а
6.71
16/0
32.00
25.76
В случае закиси углерода значение Rd подтверждает
правильность линейной формулы и непригодность формулы
с замкнутым кольцом, поедлагавшейся некоторыми
исследователями. Значение рефракций двух изомеров — таутоме-
ров ацетоуксусного эфира —показывает, что реальное веще-*
ство представляет собой их смесь.
Как и следовало ожидать, во многих соединениях,
содержащих тс-связи, имеется отклонение рефракции от
аддитивности—так называемая экзальтация. Однако отклонения
эти большей частью невелики и не столь чувствительны
к явлениям, интерпретируемым в теории электронного
резонанса, как другие свойства молекул. Примерами могут
служить значения R для бензола, С09 и других соединений.
137
^Следует подчеркнуть, что если показатель преломления
измеряется с очень большой точностью, то это не относится
к значению плотности dy также фигурирующему в
выражении для рефракции (16).
Молекулярная дисперсия — разность рефракций при
определенных длинах волн —также характерна для молекулы,
и ее значение часто применяется в качестве структурного
признака наряду с /?z>
Поляризуемость
Из значений рефракции мы можем вычислить величину
электронной поляризуемости по формуле Лорентц—Лоренца
(16). Очевидно, что значение поляризуемости, найденное из
рефракции, будет отличаться от значения, определенного
из диэлектрической постоянной по формуле Клаузиуса—
.Мосотти, благодаря дисперсионной зависимости от частоты
поля. Следует поэтому различать оптическую и статическую
поляризуемость молекулы. Оптическая поляризуемость
переходит в статическую при частотах колебаний
электромагнитного поля v, стремящихся к нулю, при бесконечно
больших длинах волн. Если известна кривая зависимости
показателя преломления от длины волны света, то можно
•оценить его значение Ясо при бесконечно длинных волнах
и вычислить статическую поляризуемость, не пользуясь
.диэлектрической постоянной.
Поляризуемость атома или иона растет с уменьшением
¦его электроотрицательности (стр. 37). Иными словами,
поляризуемость тем меньше, чем больше потенциал
ионизации и сродство к электрону. Вывод естественный, ибо,
чем труднее оторвать электроны, тем, очевидно, труднее
и деформировать электронную оболочку, поляризовать ее.
Пользуясь данными табл. 21, можно сравнить
электроотрицательности (суммы потенциала ионизации и сродства
к электрону) атомов галоидов и водорода, рефракции
и поляризуемости соответствующих отрицательных ионов.
Таблица 21
Атом
F
С1
Вг
J
Н
I+E, eV
25.3
19.6
18.1
16.5
14.25
Ион
F"
сі-
Вг-
J-
н-
R, смз
2.65
9.30
12.14
18.07
25.65
о
0.96
3.5/
4.99
7.57
С другой стороны, поляризуемость растет с увеличением
размеров электронной оболочки атома, иона или молекулы.
J 38
Квантовая механика дает приближенную формулу для
поляризуемости
где N— число внешних электронов, /— потенциал ионизации,
выраженный в электронвольтах. Эта формула относится
к статической поляризации и является аналогом формулы (28),
но с учетом того обстоятельства, что электроны в
действительности не колеблются как гармонические осцилляторы,,
но имеют, так сказать, предел упругости — могут быть
оторваны от атома или молекулы при ионизации.
ДИПОЛЬНЫЕ МОМЕНТЫ
Обратимся теперь к рассмотрению второй физической
величины, которая наряду с поляризуемостью характеризует
электрическое поведение молекулы. Это — дипольный
момент.
Дипольный момент есть свойство всей молекулы. Он
создается распределением всех электронов и положительных
зарядов. Тем не менее, оказывается возможным истолковать
измеренные значения дипольных моментов многоатомных
молекул при помощи свойств отдельных связей в молекуле,
при помощи схемы аддитивности. Заранее очевидно, что
целесообразно разложение дипольного момента молекулы
именно по связям, а не по атомам, ибо отдельно взятые
атомы дипольных моментов не имеют; относительное
смещение центров тяжести положительных и отрицательных
зарядов возникает именно благодаря взаимодействию атомов.
Дипольные моменты двухатомных молекул
Легко видеть, что порядок величины дипольного момента
есть 10"18 электростатических единиц. В самом деле, заряд
электрона равен 4.3-10"10, а расстояние между атомами
исчисляется в ангстремах (10~8 см). Поэтому величины
дипольных моментов всегда выражаются в 10~18, в так
называемых единицах Дебая (Z)).
Дипольный момент двухатомной молекулы тем больше,
чем больше отличаются электроотрицательности
составляющих ее атомов. Естественно, что особенно большими должны
быть моменты ионных молекул, так как в них электроны
перемещены от положительного иона к отрицательному.
Значения дипольных моментов нескольких двухатомных
молекул приведены в табл. 22.
В ряду галоидоводородов \і растет с увеличением доли
ионного состояния (ср. стр. 60). Можно думать, что именно
Т а 6 л и ц а 22
Молекула
Р-Ф)
er(D)
|і :ег
СО.
НС1
НВг
ш .
NaJ
ксі
KJ .
0.11
1.04
0.79
0.38
4.9
6.8
6.8
5.42
6.14
б.ні
7.7 S
14.90
15.05
16.70
0.02
0.18
0.12
0.05
о.за
0.42
0.41
ионное состояние связи вообще ответственно за наличие
у молекулы дипольного момента.
Дяткина и независимо Уолл, предположили, что чисто
гомеополярная связь дипольного момента не имеет. Это
подтверждается и теоретическими расчетами — по
необходимости приближенными. С другой стороны, если пренебречь
поляризацией, дипольный момент чисто ионной связи
должен быть просто равен произведению из заряда каждого
из ионов на междуатомное расстояние ег. Таким образом,
если, например, двухатомная молекула НС1 имеет структуры
внутреннего резонанса — гомеополярную и ионную —
н-сі ' н+сг
h i
и описывается функцией состояния
Ъ = сн\+Ъ? " (34)
то расчет дает для ее дипольного момента
_ сдУд + chCjS (\ih + \ц) + сгрі
где s—численный фактор, а у-А и-^ — дипольные моменты
чистых гомеополярной и ионной структуры. Но, как мы сказали,
^л=0, ъ = ег9 (36)
откуда окончательно
_ _ chcts + с?
ц=
ch*+2ctchs + c?
en
Ф7)
С увеличением с$. возрастает и при наибольшем значении
сі = 1у ^л —О, у- = ег. Можно, следовательно, сказать, что
отношение р:ег9 всегда меньшее единицы, является мерой
ионности молекулы. Мы привели величины этих отношений
в таблице. Действительные доли ионных состояний выше
приведенных, так как дипольные моменты уменьшаются
благодаря взаимной поляризации ионов.
140
До сих пор мы ничего не говорили о направлении
дипольного момента. Между тем его направление
существенно. Дипольный момент есть величина векторная. Обычно
его считают направленным от положительного заряда
к отрицательному. Так, в НС1 момент имеет направление
н_сГ.
В симметричных двухатомных молекулах представлены с
равным весом структуры с противоположными зарядами:
Н —Н Н+ Н" Н~ Н+
tiA = 0 ъ~ег=2Ж
Позтому большие моменты ионных структур компенсируют
друг друга. Малый момент молекулы СО объясняется также
взаимной компенсацией структур
с=о с+ —о" с~=о+
Дипольные моменты многоатомных молекул
Из сказанного ясно, что при построении аддитивной
схемы расчета дипольных моментов молекул необходимо
учитывать направления
моментов отдельных связей.
Дипольные моменты суммируются по
правилам векторного
сложения, по закону, аналогичному
закону параллелограма сил
(рис. 72). Если две связи с
моментами щ и Н расположены
ПОД УГЛОМ », ТО, СЧИТая, ЧТО рис> ^ Векторіюе сложение Ди-
является разумным допуще- польных моментов.
нием, эти моменты
направленными вдоль связей, определим суммарный момент по формуле
^ = ^ -f Н* + 2№Cos». (38)
Зная углы 0, можно вычислить моменты отдельных связей.
Приведем примеры таких расчетов. Молекула воды имеет
угол fr=105° (табл. 6) и р. = 1.84 D. Вычислим момент
ОН-связи ^ = ^2
^ = ^(2+2008105°)= 1.482|іД ^ = 1.5Ш.
Линейные трехатомные симметричные молекулы (например,
С02), обладая центром симметрии, имеют. момент, равный
нулю. Это непосредственно следует из формулы (38), дающей
141
О при &= 180°— два противоположно направленных
момента компенсируют друг друга. То же самое, конечно,
относится к любым молекулам с центром симметрии.
Молекулы, не имеющие центра, но обладающие высокой
степенью симметрии,— плоские, с осью симметрии, проходящей
перпендикулярно плоскости [треугольник (ВС13)], тетраэдри-
ческие молекулы—также бездипольны. Следовательно,
моменты отдельных связей компенсируют друг друга в
молекулах, принадлежащих к группам симметрии Сь Coh> V9, Dzh>
&ш Tdi Od. Напротив, дипольны молекулы групп CSi Съ
C2V, Czv- Измеряя дипольные моменты полярных молекул
и вычисляя значения ^ отдельных связей по формулам
векторного сложения, удалось определить \і для ряда связей.
Полученные таким образом значения р приведены в табл. 23.
Таблица 23
Дипольные моменты связей
Связи
Молекулы
V-(D)
О—Н
N—Н
с-н
с—с
с—о
с=о
СА-С1
Вода, спирты
Аммиак
Предельные соединения
Эфир (С2Н5)20
Кетоны ....
Хлорбензол. .
1.8
1.5
0.4
0.0
1.2
2.7
1.55
Связи, которые не участвуют во внешнем электронном
резонансе, сохраняют значения своих моментов во всех
молекулах. Так, во всем ряду алифатических кетонов С = 0
имеет момент 2.7D. В свою очередь, при помощи значений
моментов связей можно вычислить моменты любых молекул,
содержащих эти связи. Сравнение измеренных дипольных
моментов некоторых молекул с рассчитанными по правилу
векторной суммы приведено в табл. 24.
Отклонения от аддитивности и электронный резонанс
Во многих случаях измеренный момент отличается от
векторной суммы моментов связей. Эти различия опять-таки
могут быть с успехом истолкованы при помощи концепции
электронного резонанса, исходя из представлений о резонансе
с внутриионизованными структурами. Очевидно, что
значение таких структур для дипольного момента особенна
велико, ибо в чистом виде внутриионизованны^ структуры
должны были бы обладать очень большими моментами.
Вопросу о влиянии электронного резонанса на дипольный
момент посвящен ряд работ Сыркина, Васильева и др.
142
Таблица 24
Дипольные моменты молекул
Молекула
Формула
Симметрия
; ;j. вектор-
іі. нзмер. ная СуМна
Аммиак
Анилин
Метиловый спирт .
Сероводород . . . .
Хлористый метил .
Бромистый метил .
Йодистый метил . .
Сероуглерод
Бензол
Четыреххлористый
углерод
Дихлорэтилен транс
Дихлорэтилен цис .
Хлорбензол
Дихлорбеизол орто
мета
пара
Паранитроатжлин.
Мочепииа
Бромацетилен . . .
NH,
CGH5NH,
СН3ОН
H,S
СНаС1
СН3Вг
CH3J
cs,
свн0
СС14
С1 Н
y/ \сі
січ /Сі
\с=с<
Y/ ХН
СЙН5С1
CI
/ хсі
С1
\__/с1
С1<^ ^С1
H2N-/ ^>-NO:
H„N4
>c=o
HgNx
Br-C=C-H
Lav
L-2v
cs
L>*V
^.4-U
^3U
Dcoh
Aft
с
th
Co у
C*v
Liv
CooV
1.46
1.53
1.67
1.1
1.86
1.79
1.62
0
0
0
0
1.8
1.55
2.27
1.48
0
6.2
4.53
0
1.46
1.53
1.67
1.1
1.86
1.79
1.62:
0
0
0
0
1.8
1.55
2.68
1.55
0
5.2
~1.6
1.7
Рассмотрим случай паранитроанилина. Основная
структура молекулы обладает моментом, получающимся в
результате векторного суммирования моментов аминогруппы NH*
и нитрогруппы N02.
I
НЧ
^N-<
>N'
¦¦'/і
О
^о
из
причем отдельно взятая нитрогруппа имеет структуры
и момент 3.8 D. Момент аминогруппы 1.5 D. Они
направлены в одну сторону и в сумме дают 5.3 D. Опытное
значение выше на 0.9 D. Это связано с участием структуры
II 4N = < > = NA ,
которая должна обладать значительно большим моментом,
чем (I). Аналогичным образом, значительное увеличение
дипольного момента мочевины по сравнению с векторной
суммой определяется резонансом ітруктур
с-о с-о +/С-0
и
Векторная сумма моментов С = 0-связи (2JD) и двух
NH3 групп дает~1.6?> Измеренное значение заметно выше.
Увеличение у объясняется участием внутриионизованных
структур, в чистом виде имеющих моменты порядка 15D.
Ничтожно малый момент бромацетилена получается
в результате компенсации моментов отдельных структур
Вг—С==С—Н Вг=С = С-Н.
(h (И)
Момент хлорацетилена должен быть выше вследствие
большей электроотрицательности С1. Доля структуры (II)
поэтому уменьшается в ряду J, Вг, С1.
• Этих немногих примеров достаточно, чтобы убедиться
в большой чувствительности дипольного момента к не
аддитивным свойствам молекул. Результат, как мы "видели,
¦естественный. Отклонение значения дипольного момента
от векторной суммы можно считать своего рода
„индикатором электронного резонанса".
144
Внутреннее вращение и дипольный момент
Внутреннее вращение частей молекулы вокруг
единичных связей, (стр. 84) должно сильно влиять на измеряемое
значение дипольного момента молекулы, ибо при
относительных поворотах меняется расположение моментов
связей и их векторная сумма. Мы измеряем, конечно,
некоторое среднее значение, соответствующее всем возможным
поворотам — за время измерения молекула много раз
успевает повернуться. Средний результирующий момент
молекулы дихлорэтана (рис. 73) должен, согласно расчету, при
свободном вращении равняться
H- = l/2[xsin*>,
где jj- — момент отдельной С—С1-связи, а & — угол между
С — С и С — Cl-связями. Это дает ? =
— 2AD. В действительности, при обыч- ,'~~~~ ~"y^N,
ных температурах свободного вращения Ч^ у*^'
нет, оно заторможено. Дихлорэтан "Х;:-"/^Г
представляет собой смесь поворотных
изомеров — более устойчивого
трансизомера и менее устойчивого
изогнутого. Транс-изомер имеет центр
симметрии, и его момент равен нулю. При
повышении температуры увеличивается
содержание менее устойчивого изомера,
так как, приобретая тепловую энергию ^-__
вращательного движения, молекула ока- ^*й~/ ~~~^
зывается способной повернуться в менее (v / ;
устойчивое положение, перейти из более ^--^ *'
глубокого минимума потенциальной кри- рис^ 73. Диполи в
молевой в менее глубокий (рис. 52). Так как куле і, 2-дихлорэтана.
дипольный момент менее устойчивого
-изогнутого изомера значителен, измеряемый средний
момент оказывается зависящим от температуры, растет с ее
повышением, не достигая, впрочем, теоретического
значения р. = 2,4?)'для свободного вращения, ибо при
доступных опыту температурах оно невозможно. Возрастание
дипольного. момента дихлорэтана с температурой есть однозначное
доказательство того, что бездипольный транс-изомер более
устойчив.
Влиянием внутренних поворотов объясняются
своеобразные дипольные свойства асимметрических молекул. Мы уже
говорили выше о молекулах, содержащих два
асимметрических атома углерода и возможных их формах: правом
и левом ч изомерах и мезоформе винной кислоты (стр. 94).
Антиподы построены асимметрично, их моменты всегда
10 Строение молекул. 145
отличны от нуля. Мезоформа, напротив, имеет центр
симметрии и поэтому ее момент должен быть равен нулю.
Опыт дает и для антиподов и для мезоформы отличные от
нуля значения моментов. Мезоформы имеют моменты,
отличные от нуля опять-таки вследствие внутренних
поворотов, уничтожающих центр симметрии. Однако моменты
мезоформ всегда ниже, чем у антиподов, ибо последние ни
при каком внутреннем повороте не могут перейти в безди^
польную конфигурацию с центром симметрии.
Поляризуемость и дипольный момент
Изложенное показывает, что поляризуемость и
дипольный момент—важнейшие характеристики молекулы.
Значения рь и а определяют электрические и оптические свойства
молекул:-как мы покажем в последующих главах,
поляризуемость и дипольный момент представляют собой
основные величины не только в преломлении, но и в рассеянии
света, в колебательных спектрах, во всех явлениях
молекулярной оптики. Это объясняется, конечно, тем, что
поляризуемость и дипольный момент наиболее непосредственно
выражают свойства электронной оболочки молекулы в ее
нормальном состоянии. При возбуждении электронных
переходов существенными становятся уже и другие величины,
характеризующие молекулу: потенциал ионизации, энергии
электронных уровней, вероятности переходов между ними.
Однако, как мы уже указывали, поляризуемости тесно
связаны с этими величинами; тем самым на поведении
невозбужденной молекулы сказываются свойства ее возбужденных
электронных уровней. И то и другое есть выражение
строения электронной оболочки. Квантовая механика строго
и последовательно учитывает эти обстоятельства.
Величины \і и а являются, очевидно, важными
структурно-аналитическими признаками. На основании
измеренных значений и их сопоставления для ряда молекул
возможны детальные выводы'о строении молекулы. — как о ее
симметрии в целом, так и о присутствии тех или иных
характерных групп и связей и их расположении.
Применение молекулярной рефракции, основанное на простом
измерении показателя преломления и плотности, прочно вошло
в обиход химиков. Измерение дипольных моментов более
затруднительно, но можно думать, что в недалеком
будущем и значения \і будут широко применяться в структурном
анализе вещества.
Междумолекулярное взаимодействие
Дипольный момент и поляризуемость определяют также
и физическое взаимодействие молекул — те силы, которые
146
при уменьшений расстояний между молекулами создают их
сцепление, превращают газы в жидкости, заставляют
молекулы газа прилипать к поверхностям твердых тел в
явлениях адсорбции и т. д. (ср. стр. 69). Мы рассматривали
выше объемную поправку в уравнении ван-дер-Ваальса,
описывающем зависимость объема реального газа от давления
и температуры. Благодаря силам междумолекулярного
взаимодействия, в газе создается дополнительное внутреннее
давление, характеризуемое в уравнении ван-дер-Ваальса величиной
^2. Поэтому эти силы называют ван-дер-ваальсовыми силами.
Представим себе две дипольные молекулы. Очевидно,
что электростатическое притяжение заставит их установиться
таким образом, чтобы положительный конец одного диполя
расположился как можно ближе к отрицательному концу
другого диполя и как можно дальше от его
положительного конца. Молекулы стремятся встать либо антипарал-
лельио, либо „в хвост" друг другу (рис. 74). Этой уста-
+
Рис. 74. Ориентационное взаимодействие молекул.
новке препятствует тепловое движение. Ориентационное
взаимодействие такого рода тем больше, чем больше
дипольные моменты, чем меньше расстояние г между
диполями и чем ниже температура. Расчет дает для
выигрыша энергии, получаемого в результате притяжения
диполей, величину
и»=-Т1$і-> (39)
где щ и н-2 — моменты первого и второго диполя. Очевидно,
что энергия очень быстро падает с увеличением расстояния.
Наряду с ориентационным, возможно и индукционное
взаимодействие. Каждый диполь индуцирует в соседней
молекуле диполь того же направления с тем большим
моментом, чем больше поляризуемость молекулы (рис. 75).
Расчет дает
¦Ut = - *?. (40)
Зависимость энергии от расстояния здесь та же самая;
а, как и прежде —поляризуемость молекулы.
Однако, хорошо известно, что ван-дер-ваальсовы силы
действуют и между бездипольными молекулами. Если бы,
* іЯ
это было не так, то никаким повышением давления и
понижением температуры нельзя было бы обратить в жидкости
благородные газы, а также, например, воздух, состоящий
из смеси бездипольных симметричных молекул N.,, О.,, С02.
Это явление удалось объяснить Лондону при помощи кван-
товомеханических соображений. Пусть два атома
благородного газа находятся на расстоянии г. Дипольных моментов
они не имеют. Будем рассматривать их электроны в грубом
приближении как гармонические осцилляторы. Тогда, даже
в невозбужденном состоянии они имеют нулевую энергию
колебаний (стр. 17)
Н" (41)
и колеблются с частотой v. Когда электроны во время
„нулевых" колебаний отклоняются от положения равновесия,
в атомах создаются мгновенные диполи, -которые
притягиваются — колебания
электронов двух атомов оказываются
связанными. Но как мы
видели (стр. 43), связь между
колебаниями приводит к расще-
Рнс. 75. Индукционное взаимодей- плению частот, а значит к воз-
ствие молекул. можности перехода на более
низкий энергетический
уровень, к выигрышу энергии. Этот выигрыш, согласно
Лондону, составляет
+
Ua = — -t
asAv
2ru
(42)
Энергия взаимодействия бездипольных молекул — так
называемого дисперсионного взаимодействия — также обратно
пропорциональна шестой степени расстояния и определяется
поляризуемостью молекул. Табл. 25, в которой сопоставлены
все три эффекта для нескольких молекул, мы приводим по
Лондону.
Таблица 25
Газ
MD)
а A3
U0r*
Uirt
Udr*,
эрг см9
CO.
ш .
НВг
НС1
NHS
НоО
0.12
0.38
0.78
1.03
1.5
1.84
1.99
5.4
3.58
2.63
2.21
1.48
0.0034
0.35
6.2
18.6
84
190
0.057
1.68
4.05
5.4
10
10
67.5
388
176
105
93
74.7
Ван-дер-ваальсовы силы, в свою очередь, заметно влияют
на измеряемые значения у-, а и других физических посто-
148
янных молекул. Взаимодействующие молекулы поляризуются
и ориентируются не независимо друг от друга. Эти вопросы
относятся, однако, не к свойствам отдельных молекул,
а к свойствам жидкостей, растворов, твердых тел и т.^д.,
которых мы здесь почти не касаемся.
МАГНИТНЫЕ СВОЙСТВА МОЛЕКУЛ
Поведение молекул в магнитном поле характеризуется
величинами, аналогичными ^ и а — парамагнитными
моментами атомов и молекул и диамагнитными восприимчиво-
стями. Магнитные моменты — очень тонкие показатели
природы связи в молекуле, ибо уже для отдельных, атомов они
часто не равны нулю и непосредственно выражают свойства
электронов. Диамагнитные восприимчивости характеризуют
смещаемости электронных оболочек в магнитном поле и
подобно поляризуемостям укладываются приблизительно
в аддитивную схему (Паскаль). Не ставя своей задачей
исчерпывающее описание всех свойств молекул, мы
ограничимся здесь этими указаниями и отошлем читателя
к специальной литературе.г
1 См. В. К л емм. Магнетохимия, Госхимиздат, 1939; Е. Раб инович.
Парамагнетизм и химическая связь, „Успехи химии", т. 14, стр. 669 (1934 г.).
Глава VI
АНИЗОТРОПИЯ МОЛЕКУЛ
Изучая поляризацию молекул в электрическом поле и
преломление световых волн, мы знакомимся с общими
свойствами электронного облака молекулы, характеризуемыми
величинами у и а. Однако существуют физические явления,
которые позволяют разобраться в строении электронного
облака более подробно.
Очевидно, что электронные облака большинства молекул—
за исключением обладающих высокой симметрией тетраэдра
Td или октаэдра Od — имеют неодинаковую протяженность,
неодинаковые свойства по разным направлениям в молекуле.
Так, в двухатомной молекуле с единичной связью
электронное облако вытянуто вдоль связи и имеет меньшую
протяженность в поперечном направлении. Молекулы
анизотропны, они имеют разные свойства по разным
направлениям.
АНИЗОТРОПИЯ ПОЛЯРИЗУЕМОСТИ
До сих пор мы говорили о величине а, не учитывая
того обстоятельства, что вследствие разной протяженности
электронных облаков, сама поляризуемость молекулы должна
быть различной по разным направлениям. Так как в явлении
обычной рефракции—преломлении света— молекулы никак
не ориентированы, а расположены совершенно беспорядочно,
измеряемое значение молекулярной рефракции, а значит
и величина а характеризует среднюю поляризуемость
молекулы по всем возможным направлениям. В действительности
следует определять порознь значения а для разных
направлений. Можно всегда выделить три главных направления
для поляризуемости молекул и рассматривать электронную
оболочку не как шар, а как эллипсоид (рис. 76).
Поляризуемости по трем осям эллипсоида обозначим посредством
150
аи аъ аг. Так, например, для линейной молекулы (НС1, СО»,
HCN) обозначим через аг поляризуемость вдоль линий
связи, а2 и а3 будут поляризуемости по двум
перпендикулярным направлениям. В этом случае выбор перпендикуляра
совершенно произволен и поэтому а3 = а3. Для высокосим-
метри'чных молекул, например для октаэдра SF6,
поляризуемости по всем трем главным направлениям одинаковы,
аі = аі==^аг) и эллипсоид поляризуемости обращается в шар.
Но, вообще говоря, все три значения аи аъ az обычно не
равны друг другу. Как мы уже сказали, величина а,
определяемая из рефракции, есть среднее значение
п fli + ga + ля
а— -
(1)
Анизотропию поляризуемости, различие между аи а,, аг
можно характеризовать при
помощи величины
^ = 1{(а1-а2)2 + (а2_а3)2 +
+ («.-*і)'}; (2)
Ь называют просто анизотропией.
Очевидно, что для
высокосимметричных молекул Ь = 0} а при
а2 = а3 (например, линейные мо- Рис* 76- Элл™д П0ЛЯРизУе"
лекулы)
Ь = аг — аіл (3)
Какие же опыты могут нам дать возможность определить
порознь аи аъ а?7
Явление Керра
Выше мы рассматривали раздельно поведение молекул
в электростатическом поле между обкладками конденсатора
и в электромагнитном поле световой волны. Если
подвергнуть молекулы одновременному воздействию
электростатического поля и лучей света, распространяющихся
перпендикулярно .полю, то обнаружится своеобразное явление.
Поместим вещество (газ, жидкость) между пластинами
конденсатора и будем освещать его вдоль пластин
поляризованным светом, такими световыми волнами, у которых
имеется определенное направление колебаний в
пространстве, например направление, составляющее 45° с плоскостью
конденсатора. Для этого нужно пропустить свет через
поляризатор, например через призму Николя. После выхода
луча из конденсатора поставим вторую призму Николя —
151
анализатор, повернутую под прямым углом к первой (рис.77).
Поляризованный свет через вторую призму не пройдет, луч
будет погашен. Если теперь включить конденсатор, наложить
на вещество электрическое поле, часть света пройдет через
скрещенные николи. Чем это объяснить?
К
^-(F^-a-lsH---:
і
fP\ В)
П
В) 8)
-0?Е
г)
ь-
1
I
* а)
Рис. 77. Схема для наблюдения явления Керра:
а — источник света; б—монохроматор; в — диафрагмы; г — зрители'
ная труба, П — поляризатор, А — анализатор, А"—кювета с
конденсатором.
Благодаря анизотропии поляризуемости, даже те молекулы
которые не имеют дипольного момента, ориентируются
в поле. Они стремятся установиться таким образом, чтобы
их поляризация была наибольшей, т. е. чтобы ось
максимальной поляризуемости расположилась вдоль поля (рис. 78).
Вещество в электрическом
поле в целом приобретает
анизотропные свойства, так как
молекулы располагаются в
определенном порядке. Но это
означает, что и диэлектрическая
постоянная и показатель
преломления должны быть
различны вдоль поля и
перпендикулярно к нему.
Следовательно, должны отличаться и пока-
пп л затели преломления для тех
Рис. 78. Ориентация анизотропных лучей) волны которых колеб-
лются вдоль поля и поперек
поля. Опыт и теория показывают, что разница в
показателях преломления двух взаимно перпендикулярных лучей
пропорциональна квадрату напряженности поля Е
& 4k^
¦*-?
п,
п,
я
КЕ\
(4)
152
где п — показатель преломления в отсутствие поля,
непостоянная величина для данного вещества — константа
Керра. Это явление двойного лучепреломления газов и
жидкостей в электрическом поле было открыто Керром
в 1875 г. Благодаря неодинаковой скорости распространения
двух перпендикулярно поляризованных лучей в
анизотропной среде при прохождении луча, поле которого
первоначально направлено под углом 45° к Е, поляризация луча
меняется, и мы видим свет, прошедший через скрещенные
николи. Так как явление практически безинерционно и
двойное лучепреломление устанавливается почти мгновенно
вслед за включением конденсатора, то, накладывая на
конденсатор переменное электрическое поле можно в
описанной установке гасить и включать свет в такт с колебаниями
электрического поля. Такая модуляция света, основанная на
Рис. 79. Схема анизотропной молекулы.
явлении Керра, широко применяется в современных
аппаратах для телевидения и звукового кино, в частности
в советской системе Тагефон.
Константа Керра определяется, следовательно,
анизотропией поляризуемости. Если молекулы обладают, кроме
того, и дипольньш моментом, то для их ориентировки будут
.существенны также значения трех составляющих общего
вектора дипольного момента по трем главным направлениям
в молекуле: ри р9, \lz (рис. 79). Суммарный дипольный.
момент молекулы равен
i^W+i^+h9.
(5>
Ориентации молекул препятствует, как мы видели, тепловое
движение. Постоянная Керра тем ниже, чем выше
температура. Теория дает следующее значение К в газах, как
функцию температуры и молекулярных постоянных
155
+ (а, — а,)^,, — а1о) + ^2 [fai2 — р.,») («і — а4) +
+ GV - !*з4)(я« - *э) + GV —«О К-«і)] }, (6)
где N—число молекул в 1 см3, а,, аъ а3 — оптические, а10,
лі0, а30 — статические поляризуемости.
Для бездипольных молекул получаем
К=-^т^(а1 — аі)(аі0 — а20)^-(аі — а3)(а20 — а30)^
+ (й3— ai)(fl30 — аіс)і- (7)
Для дипольных линейных молекул, а2 = а3 иу.2 = р.3 = 0
K=^{^f{ax-a%) (aio-a^ + jtfsv? {<*!-**)} (8)
К имеет порядок величины 10~16—10"13 электростатических
-единиц. Сходные явления наблюдаются при ориентации
•молекул в магнитном поле и даже при механической
ориентации молекул в потоке жидкости (явление Максвелла).
Деполяризация рассеянного света
Второе явление, в котором сказывается анизотропия
поляризуемости — это деполяризация . рассеянного света.
Выше (стр. 71) мы говорили о рассеянии молекулами
рентгеновых лучей. Длины волн видимых лучей в тысячи раз
превосходят размеры молекул, поэтому интерференционная
картина в рассеянии видимого света не наблюдается.
Единственным результатом интерференции должно быть
отсутствие рассеянного света в направлении, перпендикулярном
направлению падающего луча — когерентные волны,
рассеиваемые отдельными молекулами должны гасить друг
друга. Однако, благодаря непрерывно возникающим в
результате теплового движения неоднородностям плотности газа,—
отклонениям в каждое данное мгновение числа молекул
в единице объема от среднего числа N—не все рассеянные
волны гасят друг друга, и остается свечение даже в
направлении, перпендикулярном падающему лучу. Именно
рассеянием солнечного света на таких неоднородностях
плотности воздуха в атмосфере объясняется голубое сияние
небосвода. Если бы рассеяния света не было, то даже днем
мы видели бы черное небо со звездами и необычайно ярким
солнцем. Голубая окраска небосвода объясняется тем,
что интенсивность рассеяния зависит от длины волны:
154
более короткие волны рассеиваются сильнее, чем более
длинные.
Разберемся подробнее в процессе взаимодействия
молекулы и световой волны при рассеянии. Электрическое поле
световой волны индуцирует в молекуле электрический
момент, тем больший, чем больше поляризуемость.
Индуцированный в молекуле диполь колеблется стой же частотой,
что и световая волна, — молекула испускает свет этой частоты
и с направлением колебаний волны, определяемым
направлением колебаний диполя. Осветим молекулу естественным,
неполяризованным светом и будем наблюдать рассеянный
свет под прямым углом (рис. 80). В естественном свете
направление колебаний световой волны произвольное, но
всегда, конечно, перпендикулярное к лучу. Если бы моле-
А
.Г
Наблюди-
, мель
Рис. 80. Рассеяние света изотропной молекулой.
кула поляризовалась изотропно, то индуцированный диполь
М в каждое мгновение совпадал бы по направлению с
электрическим полем волны Е. Под прямым углом мы увидели
бы поляризованную светом волну с направлением колебаний
по оси г, перпендикулярной лучу и линии наблюдения.
В силу поперечности световых волн в этом случае не будет
составляющей поля рассеянной световой волны по линии луча
х, и не будет восприниматься составляющая по направлению
наблюдения^. Однако если молекула поляризуется
анизотропно, то направление индуцированного момента М может не
совпасть с /Г (рис. 81); у рассеянной волны появится
составляющая колебаний вдоль направления падающего луча х, и под
прямым углом мы увидим свет частично деполяризованный.
Степенью деполяризации р обычно называют отношение
составляющих интенсивности по осям х и z
Р =
*х
(9)
155
Очевидно, что степень деполяризации будет тем выше, чем
больше анизотропия молекулы. Расчет дает
б*2
Р — 45а2 + lb"
(10)
Для совершенно изотропных молекул ? = 0 и р = 0 свет
полностью поляризован.
Для совершенно анизотропных молекул (нереальный
случай) а.2 = а3 = 0 И следовательно
Ь* = ах\ а9 = і-дД ? = ±. (II)
Во всех остальных случаях р больше 0, но меньше у.
Сопоставляя выражения для р (10), /С(7) и для моле-
НаБлюёв*
тель
Рис. 81. Рассеяние света анизотропной молекулой.
кулярной рефракции, мы находим из трех уравнений три
неизвестных аи а.2, аъ и полностью определяем эллипсоид
поляризуемости молекулы. При этом следует применить
соотношение между статическими и оптическими поляри-
зуемостями
Дао
[зо
а..
Ясо — 1
п — 1 '
где Поо — показатель преломления для бесконечно длинных
волн. В случае неполярных веществ приближенно
Про—1
Л —1
П2 — 1»
г&е е — диэлектрическая постоянная.
156
Анизотропия поляризуемости и строение молекул
В результате ряда измерений постоянной Керра,
деполяризации рассеянного света и молекулярной рефракции в газах
удалось вычислить эллипсоиды поляризуемости многих
молекул. В табл. 26 приведены полученные таким образом
значения а.
Т а б л и ц a 26
Эллипсоиды поляризуемости молекул
(Стюарт)
Вещество
Формула
Постоянная!
Керра
#¦1013
<«л ' а,-102-і!вл-і0=* яз-1024
lUUp t „,,з ; "СМ3 гыЗ
см-
см-
ВоДОрОД ; Н»
Хлор CL
Углекислота С02
Цианистый водород. HCN
Сероводород HoS
Аммиак NH3
Ацетон і(СЬ3)2СО
Четыреххлористый 1
углероа | СС1*
Бензол ¦ С0Н8
Нитробензол ..... I C0H5NOo
Dcok
Dcoh
Dooh
С оси
С «-у
c„
^Зи
Td
<0.045
2.3
1.42
93.0
1.59
3.48
32.1
<0 2
5.56
146
1.7
4.3
8.0
1.0
1.0
1.7
0.5
4.2
5.6
1.04;
6.60 |
4.10!
3.92
4.21
2.42
7.10
10.5
12.81
13.25
0.68
3.62
1.93
1.92
3.21
2.18
4.82
0.6S
3.62
1.93
1.92
3.93
2.18
7.08
10.5 I 10.50
16.35; 12.81
7.751 17.76
Мы видим, что у всех линейных молекул поперечные
поляризуемости а2 и а3 равны друг другу. Наибольшая
поляризуемость направлена вдоль линии связи, именно в этом
направлении сосредоточена наибольшая плотность электронного
облака. Эллипсоид поляризуемости представляет собой
эллипсоид вращения. То же имеет место при наличии осей
симметрии, начиная с третьего порядка CZv у аммиака,
бензола. В последнем случае ав — поляризуемость в
направлении, перпендикулярном плоскости молекулы. Молекула СС14
имеет высокую симметрию Td и изотропную поляризуемость:
эллипсоид вырождается в шар. Наибольшие отклонения К
и р от нуля, вероятно, объясняются искажениями симметрии
молекулы, вызванными колебаниями атомов. Особенно
велика постоянная Керра для нитробензола, что объясняется
не только большой'анизотропией поляризуемости, но и
значительным дипольным моментом молекулы. В этом случае
расположение главных осей эллипсоида поляризуемости
следующее
157
и а^ перпендикулярно плоскости молекулы. Если в диполь-
ной молекуле наибольшая составляющая дипольного
момента р-! имеет направление, отличное от наибольшей
поляризуемости аь как, например, в хлороформе СНС13
Н
С1 ^ CU
¦>а,,
то постоянная Керра, согласно (6), может иметь
отрицательное значение (/Сснсі3 = — 7*5 - Ю"1В).
Определяя эллипсоид поляризуемости молекулы, мы
непосредственно познаем структуру и свойства электронного
облака.
Явления Керра и деполяризации рассеянного света имеют
большое структурно-аналитическое значение.
Междумолекулярное взаимодействие очень сильно влияет
на эти явления. Значения /Сир, полученные для жидкостей,
резко отличаются от значений для газов. Здесь сказывается
то обстоятельство, что
молекулы в жидкости
ориентируются не независимо друг
от друга, а обычно целыми
группами. Бездипольные
молекулы с анизотропной
поляризуемостью, например
углеводороды, стремятся
установиться параллельно
Другу, вдоль направления
этом заметно меняется
Рис. S2. Междумолекулярное
взаимодействие в случае анизотропных
молекул.
(рис. 82) или в хвост друг
наибольшей поляризуемости. При
общий характер расположения молекул—у соседних
молекул возникает „ближний порядок", хотя в целом жидкость
изотропна и однородна. Теория приводит к существенным
изменениям формул (6) и (10). Как показывает расчет, среднее
значение поляризуемости — рефракция — при этом меняется
много меньше. Этим объясняется относительно малая
чувствительность молекулярной рефракции к
междумолекулярному взаимодействию. ¦
Эллипсоиды поляризуемости связей
Недавно были предприняты попытки (Оттербайн, Заксе,
Денби, Шен-Ниен-Ванг) применить идею аддитивности и к
анизотропным поляризуемостям молекул. В предлагаемой схеме
каждой химической связи приписывается собственный
эллипсоид поляризуемости. Обозначим его главные значения
158
буквами aj (вдоль связи), а2 и а3 (перпендикулярно связи).
Для отдельно взятой единичной связи будем считать ^ = а^
(что можно принять лишь в качестве грубого приближения,
ибо взаимодействие с соседними связями нарушит это
условие). Тогда, например, поляризуемости трехатомной
симметричной нелинейной молекулы
H.2S выразятся через аи а2, и угол
между связями ft следующим
образом (рис. 83):
ах = 2с4 cos3 8/а + 2*« sin°" ^«>
а.2 = 2а, sin2 t>/2 + 2з2 cos30/s, (12)
Сопоставляя значения аи aif д3»
полученные из постоянных Кер-
ра, с значениями рефракции и
Р, удалось вычислить а? для
ряда связей. Обратное- вычисле- Рис> 53. Эллипсоиды пояяризге-
ние /Сир дало хорошее согласие мости отдельных связей. *
с опытом. В табл. 27 приведены
значения поляризуемостей некоторых связей.
Среднее значение а = у («і 4-а>> ~Ь аз) Дает рефракцию
отдельной связи.
Экспериментальные данные по АГи р недостаточно богаты и.
надежны, чтобы полностью проверить здесь аддитивную схему.
Расчеты проведены пока-что для немногих связей и
показывают применимость этих
представлений в качестве грубого
приближения. Можно быть
уверенным, что встретятся и
значительные отклонения от аддитивности,
в особенности в случаях с
резонансными связями. Но есть также
основания полагать, что
отклонения средних значений
поляризуемостей отдельных связей а о г
аддитивности должны бытьмнога
меньше, чем их анизотропии.
Таблица 27
Поляризуемости связей
(Денби)
Связь
С?Г1024,
смз
(а2=а3М024,
СМ3
с-н
с—а
С-Вг
0.79
3.67
5.04
0.58
2.08
2.88
АНИЗОТРОПИЯ ЭЛЕКТРОННЫХ ПЕРЕХОДОВ
Анизотропия поляризуемости молекулы означает и
анизотропию поглощающих и излучающих электронных
переходов.
В самом деле, как мы видели (стр. 132),
поляризуемость определяется частотами и вероятностями электронных
переходов, согласно дисперсионной формуле
159-
i l0
Очевидно, что для трех составляющих поляризуемости аи
¦аъ а3 должны быть написаны такие же формулы, но с
различными fi0 или v-0. Это значит, что по разным
направлениям молекула в принципе способна испускать и поглощать
¦свет разных частот и интенсивностей, что имеются
преимущественные пространственные направления электронных
переходов.
Анизотропное поглощение света *
Косвенным доказательством этого положения служит
приблизительное постоянство частот электронных переходов
определенных групп атомов и связей в молекулах. Так, при
введении в насыщенный углеводород атома галоида,
в спектре поглощения наряду с далекими ультрафиолетовыми
полосами, характерными для углеводородного остатка,
появляются более близкие к видимой области полосы,
свойственные связи С — Hal. Положение и вид этих полос
практически не зависят от того, в какой углеводород введен
атом галоида, т. е. от длины углеродной цепочки. Полосы
электронных переходов С — Hal характеристичны для этоей
связи. С — С1 имеет полосы с длинами волн 1600—1540А,
С —Вг 2850—1900А, и С —J 3600—2100А. Следовательно,
соответствующие электронные переходы происходят в
определенных связях, имеющих определенное пространственное
положение в молекуле. Отсюда можно заключить, что и сами
электронные переходы также должны носить направленный
характер.
Для непосредственной проверки этого положения нужно
исследовать спектр поглощения среды, состоящей из
молекул, ориентированных каким-нибудь способом, пропуская
свет в различных направлениях. Конечно, при
беспорядочном расположении молекул наблюдаемое поглощение будет
по всем направлениям одинаково. Можно указать разные
способы ориентировки молекул: ориентация в электрическом
и магнитном поле, ориентация в потоке жидкости,
ориентация при адсорбции молекул на кристаллическом или
волокнистом веществе анизотропного строения. Наиболее
полная ориентация молекул достигается при кристаллизации;
в молекулярной решетке -все молекулы имеют строго
заданное направление, в то время как в газах и жидкостях
внешним ориентирующим факторам препятствует тепловое
движение. Однако, результаты, полученные при
исследовании анизотропного поглощения в кристалле, не могут
быть истолкованы вполне однозначно, ибо всегда возникает
160
вопрос, не является ли, например, наблюдаемое различие
в спектрах поглощения вдоль и поперек кристаллической
оси свойством кристалла в целом, возникающим при
взаимодействии молекул в решетке, а не свойством отдельных
молекул.
Кун с сотрудниками исследовали анизотропию
поглощения— дихроизм в случае молекулы паранитрозодиметил-
анилина
\N / \_N = 0.
НзСУ \ /
Молекулы, находившиеся в растворе, ориентировались
электрическим полем. Действительно, удалось установить
небольшие различия в интенсивностях и положениях полос
поглощения при прохождении света вдоль оси молекулы
и перпендикулярно к ней. Однако, доступные эксперименту
электрические поля не в состоянии создать сколько-нибудь
полную ориентировку молекул при комнатной температуре.
Поэтому наблюдаемый дихроизм был очень мал и лишь
в 3 — 4 раза превышал вычисленную ошибку опыта. Шайбе
с сотрудниками установил очень большие различия в
поглощении кристаллического гексаметилбензола
сн.
няс
по направлениям параллельному и перпендикулярному
к бензольному кольцу (рис. 84). Если отвлечься от
указанной выше неоднозначности, то из этих результатов можно
заключить, что поглощающие электронные переходы
происходят' в плоскости кольца, -говоря языком классической
физики, в этой плоскости расположены электронные
осцилляторы. К такому же выводу приходит Склар на основании
теоретического рассмотрения вопроса о спектрах
поглощения бензола и его производных. Об основных положениях
этой теории мы говорили раньше (стр. 116).
Наиболее тонким методом определения анизотропных
свойств электронных переходов является исследование поля-
11 Строение молекул. 161
ризационных спектров люминесценции, начатое впервые
и продолжаемое в настоящее время академиком С. И.
Вавиловым с сотрудниками.
то ворс то збоо зооо
2500
тс*
5Q«W°c#*
Л)
Рис. 84. Анизотропное поглощение света. Кривые
поглощения твердого гексаметилбензола:
а — электрическая сила световой волны параллельна
плоскости кольца; б — перпендикулярна к ней.
Черточками отмечено положение максимумов (Шайбе).
Люминесценция молекул
В 1904 г. Р. Вуд обнаружил, что пары Na, освещенные
в определенном направлении желтым светом D-линии Na
с длиной волны X 5896 А, в свою очередь излучают свет
с этой же длиной волны. Аналогичное явление^ наблюдается
для паров ртути; при освещении линией 2537 А они излу-
162
чают ту же длину волны. Это явление было названо
резонансной флюоресценцией. Сущность егопроста-.поддействием
кванта Av электроны переходят на первый резонансный
уровень, а затем, возвращаясь в невозбужденное состояние,
излучают этот же квант (рис. 85). Наблюдаются однако
и другие более сложные процессы. Так, излучение желтой
линии D Na можно возбудить, освещая Na линией 3303А:
при освещении квантами с большей энергией излучаются
кванты с меньшей энергией. Поглощая квант, электрон
переходит на более высокий уровень, лежащий выше
резонансного. При столкновениях с другими атомами
возбужденный атом отдает часть своей энергии: она превращается
в кинетическую энергию, в теплоту, и атом переходит на
более низкий, резонансный уровень.
Переход с резонансного уровня на невозбужденный
сопровождается излучением соответствующего кванта (рис, 86).
/1
'
/
to
t
Рис. 85. Резонансная Рис. 86. Стоксова флюо-
флюоресценция. ресцснция.
Такое испускание более длинноволнового света при
освещении коротковолновым называется стоксовой флюоресценцией.
С этими явлениями приходится часто встречаться;
достаточно напомнить синее свечение керосина при его
освещении ультрафиолетовой частью дневного света, яркозе-
леное свечение красителя флюоресцеина. Термин
люминесценция имеет более общий характер: он включает как
флюоресценцию, так и фосфоресценцию —длительное
"излучение после прекращения освещения.
Мы рассказали об атомной люминесценции. Случаи
керосина, флюоресцеина относятся уже к молекулярной
люминесценции. Характер явления здесь принципиально тот же
самый, различие состоит в том, что благодаря присутствию
ряда очень близких энергетических уровней, связанных
с колебаниями и вращениями, молекула излучает
совокупность спектральных линий, сливающихся в широкие полосы
(рис. 87). Обычно и здесь происходит испускание меньших
частот, чем частота света, падающего на вещество (закон
* . 163
Хй
WO 3000 SW 3800
» 5000 5W 5800
Веизм
Нафталан
Дафеиал
¦Л
^Ч
Трцфзнил-
•мшп-лх
Стильбзм
Дцфзнил-
Оу/паОием
йифвнил-
геШтрие*
¦ГЦл.
«*<\
Рис. 87. Полосы флюоресценции некоторых ароматических
соединений (по Константиновой-Шлезингер).
Стокса), причем вид излучаемой полосы — распределение
интенсивностей по длинам волн — воспроизводит форму
Рис. 88. а — полоса поглощения и б— полоса флюоресценции.
полосы поглощения (рис. 88). В некоторых случаях
возможно испускание и более коротких волн, чем падающая;
такая люминесценция называется
антистоксовой, и происходит она благодаря тому,
что молекула предварительно
находившаяся в возбужденном состоянии и
поглотившая квант, испускает свет с большей
частотой, ибо ее внутренняя энергия
выделяется в акте излучения (рис. 89).
Поляризация люминесценции
„ Люминесценция молекул, возбужден-
Рис. 8Э. Антистоксопа ных светом — фотолюминесценция или
флюоресценция, флюоресценция — определяется, таким
164
образом, двумя раздельными (последовательными)
процессами; поглощением и испусканием света. Время, проходящее
между этими процессами, время существования
возбужденного состояния, имеет порядок величины равно 10~9 сек. Мы
'видели, что эти процессы носят направленный характер. При
освещении вещества, например молекул в растворе,
поляризованным светом определенной длины волны молекула сможет
поглотить световую энергию и возбудиться, если направление
колебаний световой волны будет не перпендикулярным к
направлению „поглощающего осциллятора" в молекуле.
Поглощенная энергия тем больше, чем меньше угол между
направлениями колебаний световой волны и поглощающего
осциллятора. В свою очередь, испускаемое излучение оказывается
частично поляризованным, частично потому, что молекулы
ориентированы беспорядочно и находятся в состоянии
непрерывного теплового движения. Кроме того, возможность
передачи энергии возбуждения соседним молекулам также
оказывает деполяризующее влияние. Удобно наблюдать
поляризованную флюоресценцию при ориентации молекул каким-либо
способом, например при адсорбции их в анизотропных
пленках целлофана. Исследование поляризации
флюоресценции показывает, что направление излучающих
переходов, направление колебаний излучающих
электронных осцилляторов далеко не всегда совпадает с
направлением поглощающих переходов. Это доказывается открытой
в 1929 г. СИ. Вавиловым зависимостью степени
поляризации флюоресценции от длины волны возбуждающего
света. Мы видели, что существует дихроизм поглощения —
разные длины волн неодинаково поглощаются молекулой
в разных направлениях. Если предположить (Ф. Перрен),
что излучающий осциллятор повернут на определенный угол
по отношению к поглощающему, то разным длинам волн
возбуждающего света будут соответствовать разные углы,
ибо для разных длин волн поглощающие осцилляторы
расположены различно.
Анизотропные свойства молекул характеризуются
степенью поляризации р
р=Ы> (14)
где іі — интенсивность флюоресценции с тем же
направлением колебаний, что и падающего света, а /.2 — с
перпендикулярным направлением колебаний. Согласно расчету, р
зависит от угла о между направлениями поглощающих и
излучающих' переходов по, формуле Левшина—Перрена
•3cos2csx — l
Р= coso)+3-- (15)
3 65
Очевидно, что при совпадении направлений
поглощающего и излучающего осциллятора <р = 0 и /? = у. Если эти
направления расположены под прямым углом, то р =— --
В остальных случаях р больше — 7з> но меньше 7-2* Так
как, в силу только что сказанного, 9 зависят от длины
волны возбуждающего света, р также должно от нее
зависеть. Кривые зависимости р от X, так называемые
поляризационные спектры, многочисленных органических соединений,
главным образом красителей, были изучены П. П. Феофи-
ловым в лаборатории С. И. Вавилова. Молекулы изучались
как в растворах, так и ориентированные в целлофановых
пленках. В последнем случае многие красители
обнаруживают сильный дихроизм поглощения. Примем за меру
дихроизма D соотношение
я=?5&. . (16)
где Кі и /Со — коэффициенты поглощения (логарифм
отношения интенсивности прошедшего света к интенсивности
падающего света) вдоль и поперек оси ориентации
целлофана. D, конечно, есть функция длины волны. Естественно,
что спектральные зависимости дихроизма и поляризации
флюоресценции оказываются подобными, как это показано
на рис. 90, для случая красителя бензофлавина
+
СГ
Исследование поляризации флюоресценции более простых
молекул сразу позволяет установить направления
осцилляторов поглощения и излучения. Повидимому, в плоских
молекулах, содержащих резонирующие двойные связи, все
поглощающие и излучающие переходы происходят в
плоскости молекулы. Это доказывается и расчетами Склара.
Именно в плоскости молекулы тг-электроны имеют
наибольшую подвижность. П. Феофилов показал расчетом, что при
плоскостной ориентации осцилляторов для молекул,
обладающих симметрией Dnh (плоскость симметрии, центр
симметрии, ось Сп, проходящая через центр перпендикулярно
плоскости), при п} равном или большем трех, степень поля-
166
ризации р всегда должна равняться V?- Напротив, при
симметрии с осью С.2 общие соображения показывают, что
угол © между направлениями излучающего и поглощающего
осциллятора равен либо 0, либо "/.,, а значит /? = у или
-0-. Молекула анилина
.н
_/
/
N
\
Н
имеет симметрию С3„. Тем не менее, дяя нее р оказалось
2000
зооо
5000Д
Рис. 90. Поляризационной спектр и дихроизм
бензофлавина (Феофилов).
близким к 7?» а не к 7-2, что может объясняться резонансом
структур
//
*/
н
•н
Ч / \
н
н
_/—~\=Й/
н
•н
Симметрия для самого движения электрона, ответственного
за отрицательные заряды углеродов в кольце есть Сзл.
Очевидно, что аминная группа вообще должна мало участвовать
в процессе флюоресценции и вызывать лишь небольшие
отклонения р от 111.
ОПТИЧЕСКАЯ АКТИВНОСТЬ
Специфические явления, связанные с анизотропией,
возникают при взаимодействии со светом молекул,
построенных антисимметрично. Мы уже говорили (стр. 93), что ве-
167
щества, состоящие из молекул, не имеющих центра или
плоскости симметрии, являются оптически активными — они
способны вращать плоскость поляризации проходящего света.
Угол поворота тем больше, чем больше молекул активного
вещества расположено на пути луча, т. е. чем длиннее путь
света в веществе и чем выше концентрация. Оптическую
активность обычно выражают величиной так называемой
молекулярной вращающей способности
где М — молекулярный вес, d— плотность, ? — угол
вращения в градусах на дециметр пути луча в активной среде.
Значением <? пользуются в сахарном производстве для
определения концентрации сахара, молекулы которого
оптически активны, в его растворах. Для этой цели применяются
специальные поляризационные приборы — сахариметры.
Френель показал, что можно свести оптическую
активность к циркулярному двойному преломлению, вызываемому
неодинаковыми скоростями распространения лучей,
поляризованных по кругу в правую и левую сторону. Поляризация
по кругу — циркулярная поляризация — имеет следующий
смысл. Пусть вдоль некоторого направления z
распространяется световая волна, составляющие колебаний которой
по осям х я у выражаются так:
Ех — А cos 2izvtt
?ч'=:рА sin2mtf. (18)
Возводя в квадрат и складывая, получим
ЕХ* + Е/ = А*, (19)
т. е. уравнение окружности. Вектор Е описывает
окружность: при знаке — правую, по часовой стрелке, при знаке
-[- левую, против часовой стрелки (рис. 91). Наложение
двух волн, циркулярно поляризованных вправо и влево,
дает линейно поляризованную волну. Действительно,
результирующая движения двух векторов, обращающихся по
окружности, в разные стороны, совершает колебания вдоль
диаметра этой окружности, соединяющего точки, в которых
встречаются векторы Е> т. е. волна линейно поляризована.
Очевидно, что при более быстром вращении одного из
векторов направление колебаний результирующей волны
будет поворачиваться вслед за ним. Следовательно, при более
быстром распространении одной из циркулярно
поляризованных волн в среде плоскость поляризации будет
поворачиваться. Таким образом, в среде, в которой показатели
168
преломления правой и левой волн различны, направление
колебаний повернется на угол
Т = * (Щ — nr) j-, (20)
тем больший, чем больше разность показателей
преломления п1 и пп длина пути в среде z и чем меньше длина
волны X. Относя у к единице длины пути (обычно дециметр),
получаем
* = i = f (*'-«'>• (21)
Борн показал, что в среде, состоящей из асимметричных
молекул, должно иметься циркулярное двойное
лучепреломление при условии, что размеры молекул не исчезающе
малы по сравнению с длиной волны. Рассмотрим оптическое
г
(3
а) 5)
Рис. 91. а — правая и 6 — левая круговая
поляризация света.
поведение простейшей модели асимметричной молекулы,
предложенной Куном. Эта модель состоит из двух связанных
и находящихся на расстоянии г электронных осцилляторов,
направленных под прямым углом друг к другу (рис. 92).
Колебания двух связанных электронных осцилляторов можно
рассматривать как колебания связанных маятников (стр. 42).
Модель имеет два нормальных колебания —симметричное 5,
в котором оба электрона движутся в положительных
направлениях координатных осей, и антисимметричное as, в
котором первый электрон движется в положительном, а второй
в отрицательном направлении. Эти два колебания относятся
друг к другу, как правый и левый антиподы молекулы
перекиси водорода (рис. 93). Будем воздействовать на модель,
находящуюся в состоянии одного из этих колебаний, скажем
5, правым и левым лучом, распространяющимся вдоль оси
модели (рис. 94). Очевидно, что модель будет различно
реагировать на правую и левую волну. Направление колебаний-
в распространяющемся луче движется по спирали. В случае
правой волны это правовинтовая спираль, в случае левой —
169-
левовинтовая. Электромагнитная волна действует на
электроны, смещает их в направлении своих колебаний. Правая
волна сместит и первый и второй электроны по
положительным направлениям, совпадающим с направлением
приложенных к ним сил. Левая волна может
сместить в направлении своих
колебаний лишь первый электрон, а
направление движения второго
электрона противоположно
приложенной к' нему силе. Таким образом,
работа, производимая над
электронами модели силой поля световой
волны различна для правой и левой
волны и, следовательно, правая и
левая волны должны
распространяться через среду, состоящую из
асимметрических моделей с
неодинаковой скоростью. Для того чтобы
оптическая активность модели
имела место, необходимо, чтобы осцил-
.ляторы находились на некотором конечном расстоянии друг
от друга. Очевидно, что модель, находящаяся в состоянии
.второго нормального колебания, должна вращать плоскость
лоляризации в противоположном направлении. Если бы оба
Рис. 92. Модель оптически
активной молекулы (Кун).
+х
Рис. 93. Симметричное и антисимметричное колебания модели.
колебания происходили с одинаковыми частотами и
амплитудами, то они компенсировали бы взаимно оптическую
активность, и среда не вращала бы плоскость поляризации. Однако,
даже при одинаковых электронных осцилляторах, их
взаимодействие снимает вырождение, и частоты, как мы знаем, рас-
170
щепляются. Поэтому они вносят в величину вращающей
способности разные доли. Действительно, как показывает
теория и опыт, вращающая способность <р зависит от длины
волны. Зависимость эта выражается дисперсионной
формулой, подобной той, которая получается для поляризуемости
^ v? — -г
(22)
с тем отличием, что в формуле для поляризуемости все
числители положительны, а в (22) LL имеют разные знаки
и в сумме дают нуль
2*і=о.
(23)
Рис. 94. Влияние света на модель Куна.
Разобранная модель имеет всего две частоты:
симметричного и антисимметричного колебания. Сумма (22) сводится
к двум членам
9=L
V*<
¦V-
V-
as
ъ* У
(24)
Ь и v'as — собственные частоты колебаний модели. Она
способна поглощать свет с этими частотами. Так как s- и as-
колебания различно реагируют на правый и левый свет, он
должен и поглощаться 'по-разному. Оптически активные
молекулы должны неодинаково поглощать правый и левый
циркулярно поляризованный свет. Это явление —
циркулярный дихроизм— действительно было обнаружено Коттоном
в 1896 г. Циркулярный дихроизм можно охарактеризовать
величиной отношения разности коэффициентов поглощения
171
правого и левого света (Кг и К?) к коэффициенту
поглощения естественного света К
і=^Кі ¦ " (25)
Кун показывает, что g тем больше, чем больше расстояние
между осцилляторами г. Числитель L в формуле (24)
пропорционален произведению силы осциллятора (стр. 132) на
фактор циркулярного дихроизма g и обратно пропорционален
собственной частоте колебаний
I = cf-& = — С ?&&¦, (26)
^s 'as
С — постоянный множитель.
При помощи очень упрощенной модели Куна удается
качественно объяснить дисперсию оптической активности
молекул и зависимость ее от свойств полос поглощения.
Как мы указывали, электронные спектры поглощения
органических молекул содержат интенсивные полосы в далеком
ультрафиолете. Одновременно, в большинстве случаев
наблюдаются очень слабые полосы поглощения в ближней
ультрафиолетовой или даже в видимой области. Можно
думать, что у молекул, содержащих хромофорные группы
с я-электронами, происхождение этих полос связано с
внешним электронным резонансом (ср. стр. 116). Допустим, что
оптически активная молекула действительно описывается
моделью Куна. Тогда одному из колебаний соответствует
далекая интенсивная полоса, с большой силой осциллятора fy
второму — слабая близкая полоса с малым /. Согласно (26),
фактор циркулярного дихроизма жданной полосы тем
больше, чем меньше ее интенсивность. Поэтому именно слабые
близкие полосы особенно существенны для оптической
активности. Рассмотрим с этой точки зрения оптическую
активность метилового эфира а-азидопропионавой кислоты
(Кун и Браун)
Н
Н,С *С СООСН,
N3
Вещество имеет слабую ближнюю полосу поглощения при
А. 2800 А с /=3.9 • 10 "4. В этой области молекулярная
вращающая способность 9 = 200°. Полоса связана с
хромофорной азидогруппой N3 и, вероятно, определяется резонансом
— N = N = N, — N — N =N.
172
Исследование хода циркулярного дихроизма g в этой полосе
(в центре полосы # = — 0.0065) дает возможность вычислить с,
а значит и [М\ по формулам (22), (26). Таким образом,
находится зависимость той доли оптической вращающей
способности, которая определяется полосой группы N3 от
длины волны.
На рис. 95 приведена кривая 1 — кривая поглощения,
дающая логарифм коэффициента поглощения А* как функцию
длины волны, кривая 2 — наблюдаемой молекулярной
вращающей способности, кривая 3 — вычисленной вращающей
способности группы N3. Легко видеть, что основной ход
кривой [М] определяется этой группой.
Особенно велика оптическая активность именно тех
соединений, которые содержат хромофоры —
электронно-резонансные группы. Сравним молекулы
н н
і
СН3 С* ОН Н,С С* ОН
CqHs С0Пц
Первая из них содержит хромофор—бензольное кольцо, во
втором это кольцо гидрировано, т. е. лишено своих
хромофорных свойств. [М] для этих двух молекул равны сответ-
ственно 52° и —7°.
Оптическая активность и циркулярный дихроизм не суть,
конечно, свойства отдельных групп N3 или С6НЙ, но
молекулы в целом, ибо только молекула имеет нужную
асимметрию. Хромофоры становятся активными под действием
соседних атомов и групп в молекуле. Это, так называемое,
вицинальное действие тем сильнее, чем ближе к
хромофорной группе расположен асимметричный остаток.
Аддитивность в этих явлениях имеет место в том смысле, что
зачастую сходные соединения претерпевают изменения вращения
в том же направлении и на ту же величину, если в
молекулы вводятся одни и те же замещающие атомы или группы.
Приведем пример:
СООН CONH2
н—с*—он н—с*—он
QHg
—240°
свн5
-137°
173
соон
CONH*
н—с*—он
свни
—40°
н—с*—он .
сена
-Ь7б°"
Замена группы ОН на NR2 вызывает в обоих случаях
изменение [М] примерно на 100°, замена С6Н3 на CfiHn —
16
20
2?
28
31
«'•? т См ш
Рис. 95. Оптическая активность метилового эфира
ct-азидопропионовой кислоты (Кун и Браун).
на 200°. Для того, чтобы оптическая активность была велика,
необходимо фиксированное асимметричное строение
молекулы. Поэтому внутреннее вращение, которое может
уменьшить асимметрию, всегда уменьшает оптическую
активность. С этим, конечно, также связана большая активность
соединений с двойными связями, вокруг которых
внутреннее вращение невозможно.
Мы рассказали о классической модели оптически
активной молекулы. Имеется ряд квантовомеханических исследо-
174
ваний, среди которых следует упомянуть работы Кирквудаг
Алтара и Эйринга, дающих истолкование оптической
активности, в принципе не отрицающее правильность теории
Куна —Борна. Повидимому, можно свести оптическую
активность к взаимодействию асимметрично расположенных
анизотропно поляризующихся атомов и групп со светом. В
принципе можно выразить [М] через анизотропные
поляризуемости частей молекулы, но достаточно простое и универсальное
выражение здесь пока получить не удалось. Полной теории
этого замечательного явления еще нет; мы не располагаем
даже полуэмпирическими методами предварительного
расчета оптической активности новых соединений. Изложенные
классические представления объясняют ряд весьма важных
обстоятельств: зависимость оптической активности от
асимметрии и размеров молекул, связь с циркулярным дихроизмом
и роль хромофорных групп. Однако, будущая теория без
сомнения должна быть квантовой, ибо явление —по существу
электронное и не может быть сведено к классическим
модельным представлениям. Оптическая активность представляет
особый интерес еще и потому, что, вероятно, изо всех
молекулярно-оптических явлений она наиболее чувствительна
к междумолекулярному взаимодействию.
Расскажем в заключение об одной интересной
возможности возникновения оптически активных веществ — чистых
антиподов—-в природе. Мы видели, что правый и левый
свет не одинаково поглощается асимметричными
молекулами. Если они способны в результате фотохимической
реакции разлагаться под действием света, то, освещая,
например, рацемическую смесь антиподов светом,
поляризованным вправо, мы вызовем разложение одного из
антиподов и сохраним неизменным второй. Такой
„асимметрический синтез" был осуществлен в одном случае Куном
и Кнопфом. Так как свет, рассеянный земной атмосферой,
согласно данным опыта, частично циркулярно поляризован
вправо, можно думать, что чистые антиподы,
встречающиеся в живой природе, впервые возникли именно в
результате такой фотохимической реакции. Более вероятно, однако,
их возникновение просто вследствие случайных нарушений
равновесия между правыми и левыми антиподами в
рацемических смесях. Что касается самой асимметрии, то в
сложных молекулах она, конечно, встречается очень часто.
Глава VII
СПЕКТРЫ МОЛЕКУЛ
Мы не раз уже имели дело со спектрами молекул,
говоря об окраске органических соединений, о явлениях
люминесценции и оптической активности/ Способность молекулы
поглощать и излучать свет тех или иных длин волн
определяет по существу все ее оптические свойства; с этой
способностью связана основная оптическая константа молекулы —
ее поляризуемость. Как мы уже указывали, энергетические
переходы в молекуле, в отличие от атома, могут
происходить не только при изменении состояния электронной
оболочки, но и при колебаниях атомных ядер в молекуле и при
ее вращении как целого. Конечно, и колебания и вращения
сопровождаются некоторыми изменениями в положениях
и энергиях электронов, и поэтому, строго говоря,
неотделимы от них. Тем не менее, приближенно можно считать
колебательные и вращательные переходы независимыми от
электронных, так как движение тяжелых ядер происходит
в тысячи раз медленнее, чем движение электронов. За время
изменения электронного состояния ядра не успевают
переместиться, поэтому можно рассматривать колебания и
вращения независимо. Разделение на три вида переходов
соответствует различию в энергии, требуемой для их
осуществления. Легче всего возбудить вращение молекул; для
этого достаточно энергии теплового движения; все молекулы
газа в обычных температурных условиях вращаются. Энергии
вращательных переходов равны по порядку величин 0.03 —
0.3 кг кал/моль, чему соответствуют излучаемые и
поглощаемые частоты порядка 10—100 см-1 или длины волн 1000—
100 ц, лежащие в очень далекой инфракрасной области,
перекрывающейся с областью коротких радиоволн.
Колебательным переходам отвечают энергии порядка 0.3—12 кг кал/моль,
частоты 100—4000 см-1, длины волн 100—2.5 у. в
более близкой инфракрасной области. Для возбуждения
176
же электронов нужны энергии порядка десятков и сотен
кг кал/моль; соответствующие частоты исчисляются
десятками тысяч см-1 и лежат в видимой и
ультрафиолетовой областях спектра. Таким образом, при постепенном
возбуждении молекулы она будет испускать сначала
линейчатый
колебательно-вращательный спектр •/¦*
(ибо одновременно с ко- | •
лебаниями всегда проис- ' "— W
ходят и вращения—около *
каждого колебательного | ===z~
уровня имеется совокуп- " у*г
ность вращательных) в * —
инфракрасной области и \ "
лишь затем электронный ' ' —v-f
4 ¦ .
3
спектр в видимой и
ультрафиолетовой областях,
состоящий из систем
полос, так как около
каждого электронного уровня *
имеется набор колеба- з
тельных и вращательных ?
уровней. На рис. 96 при- 1
ведена схема уровней
молекулы, а на рис. 112
фотография электронного
спектра.
У*цеш
т '
г
і
*
з
2
*V
V4
КОЛЕБАТЕЛЬНЫЕ И
ВРАЩАТЕЛЬНЫЕ
ПЕРЕХОДЫ
Начнем с колебатель- " ' " *Н*
ных переходов. Силы ,, „ <,- п
F „ м ллл Рис. 9о. Схема энергетических уровней
химической связи, соеди- молекулы,
няющей атомы, стремятся
при отклонении атомов из положения равновесия вернуть их
в это положение. Под действием этих сил атомы колеблются
около положения равновесия. Смещения атомов вызываются
внешним воздействием — оптическим, тепловым. Наиболее
проста картина,колебательного движения двухатомной
молекулы, так как в ней возможно лишь одно колебание вдоль
линии связи атомов.
Колебания двухатомных молекул
Кривая зависимости потенциальной энергии двухатомной
молекулы от междуатомного расстояния уже приводилась
нами ранее (рис, 16), В части кривой вблизи ее минимума,
12 Строение молекул. 177
соответствующей малым смещениям из положения
равновесия, можно заменить кривую потенциальной энергии
параболой (рис. 97)
и=
Иными словами, малые колебания
двухатомной молекулы могут
рассматриваться как колебания
гармонического осциллятора,
потенциальная энергия которого всегда
квадратично зависит от смещения (стр. 16).
Можно представить себе молекулу
состоящей из двух тяжелых частиц,
соединенных пружиной. При
увеличении расстояния между атомами, на каждый из них
действует возвращающая упругая сила, пропорциональная
коэффициенту упругости k и смещению атома из положения
равновесия. Если массы двух атомов т1 и тъ то силы,
действующие на них, равны произведениям их масс на ускорения.
Имеем (рис- 98)
Рис. 97. Замена части
потенциальной кривой параболой.
тхх1 — к (г—г0) = — k (хх — х2)}
ЩХъ
k(r — r0) = k{xl — x,l)y
или
*і —х*. = — k ( ~ + J-) (хх — х2).
Заменяя х,—х% — х,
получаем уравнение движения щ
гармонического осциллятора
х = х
т
(3')
с частотой (стр. 17):
г -*'
'О
(2)
(3)
•-1
!779
—U
Рис. 98. Колебание двухатомной
молекулы.
= ±і/а
2к у т '
(4)
где т — приведенная масса молекулы
т =
1
1 | J__ mi-\~mi
(5)
тпі
178
Мы видели, что, согласно квантовой механике, такой
осциллятор может иметь полную энергию
- е„= (tf+-g")Av' (6)
где v = 0, 1, 2, 3, .. . — колебательное квантовое число,
которому соответствуют разные колебательные
энергетические уровни. Разность энергий двух соседних уровней всегда
равна h и свет частоты v может, следовательно, поглощаться
и испускаться молекулой при этих переходах.
Ангармоничность
При больших амплитудах колебаний уже нельзя считать
двухатомную молекулу гармоническим осциллятором. В самом
деле, возвращающая сила в гармоническом осцилляторе
растет безгранично с увеличением амплитуды; гармонический
осциллятор поэтому никогда не может разорваться. Напротив,
в реальной молекуле сравнительно легко достигается „предел
упругости". Когда энергия 'колебаний достигнет значения
энергии диссоциации D, молекула разорвется на
составляющие атомы лли ионы. Поэтому действительные колебания
молекулы ангармоничны. Для больших амплитуд выражение
потенциальной энергии (1) уже не пригодно! Потенциальная
энергия зависит от расстояния между атомами более
сложным образом. Удобно пользоваться для приближенного
выражения реальной потенциальной кривой формулой Морзе
U=D{\ — е-Ж'-'о))2, (7)
где D — энергия диссоциации, а — постоянная. При больших
г—г0-потенциальная энергия стремится к значению D9 при
малых формула (7) переходит в (1). Минимум кривой имеет
место при г = г0. Решение уравнения Шредингера дает в этом
случае значения колебательной энергии в виде
Колебательные энергетические уровни реальной молекулы
располагаются не на равных расстояниях Av, а сгущаются па
мере возрастания энергии, пока не сольются при ?V=D
(рис..99). Чем больше энергия диссоциации D, тем круче
поднимается кривая, и для больших D формула (8)
переходит в формулу (6) колебаний гармонического осциллятора.
Очевидно, что более крутой параболе соответствует
больший коэффициент упругости ку большая частота колебаний.
Зная выражения потенциальной энергии (7), мы можем
* 1пЬ
связать величину коэффициента к с энергией диссоциации —
энергией связи В и с постоянной а. Именно
к = 2Da\ (9)
т. е. коэффициент упругости прямо пропорционален энергии
связи. В главе II мы видели, что и энергия связи и
междуатомное расстояние в молекуле г0 определяются строением
и свойствами электронной оболочки и поэтому не независимы
друг от друга. Следует ожидать, что к зависит не только
от D, но и от г0. Действительно, удалось найти эмпирические
формулы, характеризующие
эту зависимость. Приведем
формулу Беджера
и 1.85 - 10е , /1ЛЧ
к=г—rf-тзДин/см, (10)
\ го aij)
где dy — постоянная для
данных значений / и /-рядов
периодической системы, в
которых расположены
атомы. Дляо второго ряда d(J =
= 0.680А. Это значение в
равной степени пригодно
Рис. 99. Колебательные энергетиче- ДЛЯ молекул Na, 02, N0,
¦ские уровни двухатомной молекулы. СО. При і = 2, / = 3 (CS,
PN, SiO), du = 0.900 и т. д.
С увеличением і9 ] величина dtj возрастает. Эмпирическая
формула Беджера хорошо передает полученные из опыта
соотношения между к и г0.
Частоты колебаний и силовые коэффициенты к
Из сказанного ясно, что, чем прочнее химическая связь
в молекуле, тем больше D и тем меньше г0, а значит тем
большее.Силовые коэффициенты к непосредственно
характеризуют прочность химических связей. Величины к могут
быть найдены, согласно (4), из значений колебательных
частот, получаемых из спектров молекул (см. ниже).
Колебательные частоты зависят не только от к, но и от масс
колеблющихся атомов. Частоты тем больше, чем меньше
эти массы; быстрее всего колеблются двухатомные молекулы,
содержащие водород. Оценим порядок величины силовых
коэффициентов k. Колебательные частоты лежат, как мы
указывали, в области сотен и тысяч см~Л Масса атома
водорода равна 1.65 • 10 ~24 г, для других атомов ее нужно
множить на атомный вес. Переходя от волновых чисел,
выражаемых в см-1, к частотам в сек-1 (ддя чего нужно
180
і
умножить их на скорость света с = 3-1010 см/сек), получим
частоты порядка 3- 1013 — 1014сек~1 . Согласно (4), порядок
величины к есть 105 дин/см. В табл. 28 приведены значения
v, т и к для некоторых двухатомных молекул.
Таблица 28
(Кольрауш)
Молекула
Наблюд.
частота, см
-1
н2 .
D* .
HF.
НС1
НВг
HJ.
С12.
о, .
N. .
СО
N0
4161
2993
3935
2886
2558
2233
556
1555
2329
2155
1877
т, атомн.
един.
0.5
1.0
0.95
0.97
0.99
0.994
17.75
8.0
7.0
6.85
7.48
k. 10—\
дин/см
^эксп.
кг кал/моль!
5.07
5.24
8.65
4.74
3.78
¦2.89
3.21
11.3
22.2
18.6
15.4
103.23
105.03
147.9
102.2
86.7
70.4
57.2
117.2
170.2
210.8
122.0
П, А
0.74
0.74
0.92
1.27
1.41
1.60
¦1.99
1.21
1.10
1.13
1.15
Приведенные массы всех молекул, содержащих водород,
близки к единице за исключением /я = 0.5 в случае
молекулы водорода. Поэтому частоты всех гидридов
относительно высоки.
Сопоставление значений, указанных в таблице,
иллюстрирует изложение закономерности. Мы видим, что к для^
кратных связей (Ns, СО, 02, NO) выше, чем для единичных.
Изотопы
Замена атома в молекуле его изотопом почти не влияет
на строение электронной оболочки, природу связи, и, тем
самым, на коэффициент к, а сказывается только на массе.
Наиболее' яркий случай — это изотоп водорода с массой 2 —
дейтерий. Замена водорода на дейтерий увеличивает
приведенную массу почти в_два раза и, следовательно, уменьшает
частоту колебаний в j/Траз. Различие в свойствах соединений
дейтерия и водорода определяется именно разницей в
колебательных частотах. Действительно, нулевые энергии этих
соединений, рассматриваемых как гармонические осциля-
торы, равные, согласно стр.-19,
2 »
(И)
181
также разнятся в у* 2 раз. Наблюдаемая энергия
диссоциации Н2 и D2, например, равна
D —D--
(12)
ибо подъем на уровень диссоциации происходит не с самой
низкой точки потенциальной кривой, а с уровня г» = 0,
обладающего энергией е0 (рис. 100). Поэтому наблюдаемая D
дейтерия выше, чем D водорода.
Рис. 100. Энергия диссоциации водорода и дейтерия.
Колебания многоатомных молекул
В многоатомных молекулах число связей и тем самым
возможностей различных колебаний уже не равно единице.
Молекула способна выполнять целый ряд колебаний с
различными частотами. Если в спектре.двухатомной молекулы
фигурирует одна частота, то в спектре сложных молекул
таких частот несколько, вообще говоря,.. тем больше, чем
больше атомов в молекуле. Численные значения этих частот,
равно как и интенсивности соответствующих спектральных,
линий, полностью определяются строением молекулы —ее
геометрией, природой связей между атомами, их
электрическими свойствами. Колебательные спектры многоатомных
молекул дают исключительно ценную информацию об их
строении и свойствах. Этим вопросам будет посвящена
заключительная глава книги.
Вращение молекул
Перейдем теперь к вращению молекул. Отвлечемся
вначале от колебательного движения; будем считать атомы
находящимися на постоянных расстояниях г0 друг от друга.
Тогда двухатомная молекула представится в виде твердого,
тела, напоминающего по своей форме гимнастическую
№
гирю, гантель (рис. 101). Такая молекула способна вращаться
вокруг осей, проходящих через ее центр тяжести, —это так
называемый .ротатор. Ротатор не имеет потенциальной,
а только кинетическую энергию. Для двух атомов с массами
тх и Ш} эта энергия
К=~
m,v
m»v\
(13)
Но каждая точка ротатора описывает окружность с
радиусом, равным ее расстоянию от оси вращения.
Следовательно, скорости движения атомов vx и v* равны
произведениям этих расстояний на
угловую скорость — круговую
частоту (о
V
r(2^vrot — fx
О).
V.2 = r227cvrot = r3a>. V ;
Подставляя в (13), получим
/о,» С15)
Рис. 101. Вращение двухатомной
молекулы.
где
<o = 2itvrot — круговая частота.
Выражение, взятое в скобки, есть момент инерции
/ жесткой молекулы относительно данной оси. Так как г,
и г% — расстояния до центра тяжести, то должны
соблюдаться условия
тігх = /?г,г.,, (16)
и
гх + Ъ = Гь. (17)
Следовательно,
fYl\TYla О __ О /"1 Q\
/ т ЛЛ-т г1 — і s г2 = тг2
где т — приведенная масса. Очевидно, что лишь для осей
перпендикулярных линии связи в двухатомной, да и вообще
в линейной молекуле момент инерции, а значит, и энергия
отличны от нуля. Для оси, совпадающей с линией связи
/ = 0, ибо эта ось проходит через все атомы и тем самым
равны нулю расстояния гх и г2. Линейная молекула имеет
всего два вращения, третье не сопровождается изменениями
энергии и никак не может проявиться. Любая мнргоатом-
ная нелинейная молекула имеет, напротив, три вращения.
183
Согласно классическим представлениям, молекула может
вращаться с любой частотой со, • тем большей, чем выше
температура среды. Квантовая механика показывает, что это
не так: вращающаяся двухатомная молекула может
находиться лишь в строго определенных энергетических
состояниях
/г2
= ¦537 УС/+1Х 7 = 0,1, 2, ...
8*2/
(19)
у—вращательное квантовое число. Выражение (19) нам уже
знакомо. Мы видели (стр. 23), что момент количества
движения электрона в атоме есть
(20)
P = ^Vi(i+D.
•?=2
Момент количества движения ротатора
равен
р = mir1vl + лі2г3і>2 = /<о. (21)
Если мы заменим в (19) -g^ Yj(j +1) на /со,
то снова получим классическое выражение
энергии (15).
Частоты вращений подчиняются условию
Л
Рис. 102.
Вращательные
энергетические уровни
двухатомной
молекулы.
(і) П , —;
Vf0t — "2^ = 4^7 VJ'(J+ !)¦
(22)
Частоты ротатора квантованы. Напротив,
у гармонического осциллятора квантуется
не частота, а. энергия.
Энергетические уровни ротатора
расположены, согласно (19), не на одинаковых расстояниях друг
от друга, а на расстояниях, возрастающих с ростом/(рис-102).
На основании условия частот Бора, мы заключаем, что
частоты, излучаемые и поглощаемые ротатором, не совпадают
с его собственной частотой вращения, но равны
iLirL=m.{/V+V-M+1)}-
(23)
На рис. 102 приведена схема уровней ротатора.
Фактически вращение происходит не независимо отколе*
баний, и нельзя считать двухатомную молекулу твердым
телом. Расстояние г между атомами меняется в процессе
колебания и так как потенциальная кривая молекула идет
круче при r<V0, чем при г^>г0, большую часть времени
молекула проводит лри г^>г0 — ангармоничные колебания
увеличивают расстояние между атомами. С другой стороны,,
вращение само по себе также увеличивает это расстояние,.
184
ибо на вращающиеся атомы действует центробежная сила.
Таким образом, колебания взаимодействуют с вращениями
и происходят одновременно с ними: каждому
колебательному состоянию соответствует ряд вращательных.
Вращательный спектр многоатомной молекулы весьма усложнен
благодаря тому, что в общем случае она ведет себя как
асимметрический волчок, имеет три различных момента
инерции /ь Л, /3 по отношению к трем перпендикулярным
осям и, соответственно, три набора вращательных линий,
которые могут перекрываться.
ИНФРАКРАСНЫЕ СПЕКТРЫ
Колебательно-вращательные полосы наблюдаются
в.инфракрасной области спектра. Инфракрасные лучи не
действуют на фотопластинку и поглощаются стеклом и
кварцем. Поэтому спектральные приборы для изучения
инфракрасного спектра строятся с оптикой, изготовленной из таких
материалов, как флюорит-и каменная холь. Для еще более
далеких областей спектра применяются приборы, в которых
призмы заменены отражательными дифракционными
решетками, а линзы — вогнутыми зеркалами. Регистрация инфра- *
красных лучей основана на их тепловом действии.
Инфракрасные лучи создают электрический ток в термоэлементе»
меняют сопротивление в болометре. Тело, подвергнутое
прерывистому освещению инфракрасными лучами тех длин
волн, которые оно может поглотить, расширяется и
сжимается в такт с прерыванием света и поэтому звучит. Это
явление Тиндаля—Рентгена лежит в основе
оптико-акустического газового анализатора и спектрофона М. Вейнгерова
и применяется в различных методах изучения
инфракрасных спектров (А. Н.Теренин). Инфракрасные спектры молекул
обычно наблюдаются как спектры поглощения, и в
результате опыта получается кривая, дающая зависимость
коэффициента поглощения (или пропускания) вещества от длины
волны (рис. 103).
Каким же образом происходит поглощение и
испускание инфракрасных волн молекулой? Мы рассмотрели
энергетические уровни колебаний и вращений двухатомной
молекулы, спрашивается, какие переходы возможны между
этими уровнями, и будут ли эти переходы всегда
сопровождаться испусканием или поглощением света?
Начнем с колебаний. Квантовая механика показывает,
что излучающие и поглощающие переходы возможны лишь
между двумя соседними, колебательными уровнями
гармонического осциллятора —в других случаях вероятность
перехода равна нулю. Колебательное квантовое число осцил-
185
лятора при поглощении может возрасти только на единицу,
а при испускании света на единицу уменьшиться.
Разрешенным переходам
v
v
v+1'
v — V
соответствует частота [ср. (6)]
V h ~ h 2- У т '
(24),
равная классической частоте собственных колебаний
осциллятора. Значит ли это, что молекула — гармонический
осциллятор,— колеблющаяся с частотой v всегда способна испу-
Рис. 103. Кривые инфракрасного поглощения — бутана и изо-
бутана.
стить или поглотить свет соответствующей длины волны?
Оказывается, что это не так.
Электромагнитная теория света говорит нам, что лишь
заряженные тела при своих колебаниях около положения
равновесия способны испускать и поглощать
электромагнитные волны. Амплитуды этих волн тем больше, а
излучаемые и поглощаемые энергии тем значительнее, чем больше
заряд колеблющейся частицы и чем больше амплитуда ее
колебаний, т. е. чем больше изменение дипольного момента
осциллятора во время колебания, характеризуемое
величиной
!*'=№)„ ' (25)
drj r~rQ.
Это значит, что колебание молекулы, не сопровождающееся
изменением ее дипольного момента, никак не проявляется
186
в инфракрасном спектре: раз момент не меняется, молекула
не испускает и не поглощает света. Наоборот, чем сильнее
меняется момент, чем больше величина |х', тем сильнее
испускание и поглощение, тем интенсивнее инфракрасная
полоса. Для симметричных двухатомных молекул (Н.2, D.2,
N.2, 0.2 и т. д.) р-' = 0; они не дают, следовательно,
инфракрасных колебательных полос, и их колебания могут быть
возбуждены термическим или электрическим воздействием,
¦но не в результате непосредственного поглощения света.
Наиболее интенсивны колебательные инфракрасные
.полосы у тех молекул, чьи дипольные моменты меняются
с расстоянием особенно сильно (у ионных молекул). Мы
видели (стр. 140), что есть основания считать чисто гомео-
полярные молекулы имеющими нулевой дипольный момент.
Очевидно, что интенсивность колебательной инфракрасной
полосы тем больше, чем выше ионносЛ молекулы, и может
служить в известной степени критерием степени ионности.
Величина |і-' — производная дипольного момента по
расстоянию между атомами, взятая при r=rQ, имеет смысл и
размерность электрического заряда и называется
эффективным зарядом молекулы. Интенсивность инфракрасной
колебательной полосы пропорциональна квадрату произведения
у*' на амплитуду колебаний /**, ,-H-'2/V2 и> определяя
интенсивность, можно найти значения у.'.
T а б л и ц а 29
(Бартоломе)
Молекула
V-'
га, А
И-Ао
НС1
НВг
HJ .
0.086
0.075
0.033
1.04
0.79
0.38
1.28
1.42
1.62
0.184
0.115
0.049
В табл. 29 сопоставлены значения р, выраженные в
зарядах электрона с у, г0 и их частным, у-' близко к у: г0
я быстро растет с увеличением степени ионности. Если бы
молекула была чисто ионной, то очевидно
Р = ег (26)
Р=е,. (27)
т. е. эффективный заряд равнялся бы просто заряду
электрона. Кривая зависимости ^ от г представилась бы прямой
линией (рис. 104). С другой стороны, для чисто гомеопо-
лярной молекулы и ^ и р* на всех расстояниях равны нулю.
Однако, эти чистые случаи в природе отсутствуют. Реальная
молекула, реальная химическая связь, представляет собой,
как мы знаем, наложение ионной и гомеополярной связей
187
(стр. 57). Поэтому реальная кривая ^(г) идет не вдоль
прямой ъ = ег или оси абсцисс \^ = 0, но приближается к этим
двум крайним случаям в зависимости от того, к какому из
двух чистых типов связь ближе. Если связь приближается
к ионной, в том смысле, что на больших расстояниях
молекула диссоциирует на два свободных иона, представляющих
собой, конечно, систему с р = ег, то кривая у-(У),
отклоняясь каким-то образом от прямой р. = ег в области малых
г, сливается с ней на больших расстояниях. Напротив, если
молекула диссоциирует на свободные незаряженные атомы
и поэтому может считаться гомеополярной, то кривая
сливается на больших расстояниях с осью абсцисс.
Определенные из интенсивностей
значения ^' дают наклоны кривой при
г=г0, но нам не известен даже
знак р' (ибо интенсивность
пропорциональна у.'2), и поэтому о
виде реальной кривой |х(г) мы
можем получить лишь самое
общее представление. ¦
При вращении молекулы ди-
польный момент ее не меняется,
если отвлечься от малых
изменений, вызванных центробежным
растяжением. Однако, проекция
"% момента вращающейся молекулы
М1, ._, на любые направления, фиксиро-
нис. 104. Зависимость диполь- ^ ' ^ F
ного момента от междуатом- ванные в пространстве, меняется
ного расстояния. периодически с частотой vrotj.n,
как показывает классическая
теория, этого достаточно для поглощения и испускания света
стой же частотой vrot и интенсивностью, пропорциональной
квадрату дипольного момента (Л Бездипольные,
симметричные молекулы не излучают и не поглощают при вращении.
Рассмотрим, как должна выглядеть
колебательно-вращательная полоса. Пусть двухатомная молекула с дипольным
моментом у вращается в плоскости чертежа вокруг оси г
(рис. 105). Проекции момента на оси х, у я z равны
tJLjc==H- cos o = t*. cos 27rvrot t>
\iy = ]i sin<? = fx sin 2*vrot?, (28)
Если молекула одновременно колеблется с частотой v, то
момент ее меняется с этой же частотой
jjL.= ix'rvsin2^v/, (29)
где rv — амплитуда колебания. Подставив (29) в (23),
получим
188
V-x = V-'rv sin 2^ cos 2^vrot t=~ jx'rfl [sin 2ъ (v -f vrot) t+
+ sin2*(v —vrot)*], (30)
ty = Iх 4 sin 2~v/ sin 2тг;го1 tf = -g- V-'rv [cos 2т: (v — vrot) t—
— COs2i:(v-|-Vrot)*].
Благодаря одновременному вращению дипольный момент
колеблется не с частотой v, а частью с частотой v + vrot,
¦частью с частотой v — vrot, ибо в выражении проекции момента
фигурируют периодические функции с этими частотами.
Вращение создает, вместо одной колебательной
спектральной линии с частотой v, колебательно-вращательную полосу
-с двумя ветвями, простирающимися от v в обе стороны
/
/
t
/
1
і
А
\
\ .
\ /
\ /
V
л \
\
\
\
\
IV.
%
Рис. 105. Вращение дипольной Рис. 106. Схема вращательной струк-
молекулы. туры колебательной полосы.
{рис, 108). Квантовая механика меняет этот результат лишь
в том отношении, что полосы оказываются не непрерывными,
а распадающимися на ряд линий, ибо частоты вращения не
произвольны, а, как мы видели, квантованы. Теория и опыт
показывают, что вращательные линии подчиняются условию,
подобному высказанному для осциллятора: вращательные
излучающие и поглощающие переходы возможны лишь
между соседними вращательными уровнями и поэтому
сопровождаются изменением вращательного квантового числа на
единицу. Расстояние между двумя соседними
вращательными линиями, соответствующими переходам
У-у + 1, y + W + 2,
можно вычислить по формуле (23)
vy.y + t=-83/(2y+2),-
^4-і./ + а=8Э/(2У + 4) (31)
189
J
^.+ і.У + а —Ул/ + і= 4^7- (32>
Измерив это расстояние, мы, следовательно, найдем момент
инерции молекулы и, значит, междуатомное расстояние.
Многие междуатомные расстояния были измерены таким
способом с большой точностью. Это особенно важно для
водород-содержащих молекул, не поддающихся рентгено-
и электронографическому исследованию (ср. стр. 79).
Ангармоничность реальных колебаний молекул приводит
к существенному изменению правил о допустимых
переходах между колебательными уровнями — правил отбора. Для
ангармонического осциллятора излучающими и
поглощающими оказываются любые колебательные переходы с
изменением колебательного квантового числа на несколько
единиц. Поэтому в спектре реальной молекулы наблюдается не
только частота v, связанная с переходом между соседними
уровнями, но также и частоты, соответствующие переходам
и т. д. Если бы речь шла о гармоническом осцилляторе, то
этим переходам, согласно (6), соответствовали бы частоты
обертонов 2v, 3v и т. д., кратные основной частоте.
Благодаря ангармоничности [ср. (8)], частоты обертонов несколько
отличаются от кратных v, но обычно незначительно.
Интенсивности обертонов всегда много ниже, чем основных частот,
тем не менее в инфракрасной спектроскопии молекул они
имеют большое значение, ибо, чем выше порядок обертона,
тем выше его частота, и полоса поглощения ближе к
видимой области спектра. Ближняя инфракрасная область более
доступна эксперименту- Большая часть исследований
инфракрасных спектров относится ко вторым, третьим и даже
четвертым обертонам.
КОМБИНАЦИОННОЕ РАССЕЯНИЕ СВЕТА (ЯВЛЕНИЕ РАМАНА)
Не только инфракрасные спектры поглощения дают нам
способ изучения колебаний и вращения молекул.
Колебательно-вращательные переходы ярко проявляются в
спектрах рассеянного света в видимой области. Это создает
богатые экспериментальные возможности, которые
действительно легли в настоящее время в основу исследования
колебаний нескольких тысяч молекул.
В предыдущей главе мы уже говорили о молекулярном
рассеянии света, о поляризации рассеянного света и о том,
что его интенсивность тем больше, чем меньше длина волны
падающего света. До 1928 г. физики ограничивались
исследованием этих общих свойств рассеяния — интенсивности
190
и поляризации. В начале 1928 г. .советские физики проф.
Г. С* Ландсберг и академик Л. И. Мандельштам впервые
исследовали спектр света, рассеянного кристаллическим
кварцем. К этому эксперименту их привела теория
рассеяния света в кристаллах, на протяжении ряда лет
разрабатывавшаяся Л. И. Мандельштамом. Эксперимент был
поставлен следующим образом. Кубик, выточенный из
кристаллического кварца, освещался светом ртутной лампы, имеющей
спектр, состоящий из нескольких раздельных интенсивных
линий. Свет, рассеянный под прямым углом, конденсировался
на щели светосильного спектрографа. После преодоления
трудностей, связанных с очень малой интенсивностью
рассеянного света, была получена спектрограмма, на которой,
помимо линий ртутного спектра, обнаружился ряд более
слабых линий, в спектре ртутной лампы не содержавшихся.
•В первом своем сообщении (10/V 1928) авторы дали
правильное и полное объяснение найденному явлению. Явление
изменения спектрального состава рассеянного света было
обнаружено в органических жидкостях индусом Раманом,
опубликовавшим свое открытие (16/11 1928) на три
месяца раньше московских ученых. Однако, несмотря на то,,
что независимость проведения этих работ —
общепризнанный факт, а также несмотря на ошибочное
истолкование явления, данное в двух первых сообщениях Рамана,
и более случайный характер его работы, явление получило
название явления Рамана. Как мы увидим, физическая
сущность явления позволяет назвать его комбинационным
рассеянием света. Но из-за краткости терминов — эффект Рамана,
рамановские спектры, раман-линии — они укоренились в
литературе. Здесь мы также часто будем ими пользоваться,
имея в виду факты, относящиеся к истории открытия.
Дальнейшее исследование показало, что
дополнительные слабые линии в спектре рассеянного света имеют
различные положения для разных молекул и, тем самым,
характерны не для- источника света, а для рассеивающего
вещества. Как же можно объяснить их появление?
Квантовая теория
Простейшая теория явления — квантовая. Пусть квант
видимого света Av0 попадает в молекулу. При этом может
произойти следующее. Молекула возбудится квантом Ь0
и немедленно излучит его вновь без изменения энергии.
В спектре рассеянного света появится та же самая частота
v0, которая фигурировала в спектре падающего света. Здесь
можно говорить об упругом рассеянии кванта — это обычное
классическое релеевское рассеяние, с которым мы уже
встречались раньше (стр. 154). Далее может иметь место не-
191
упругое рассеяние. Квант Av0 отаает часть своей энергии
молекуле и будет излучен молекулой с уменьшенной
частотой. При этом энергия, воспринятая молекулой, истратится
на ее колебание и вращение, которые строго квантованы.
Уменьшение энергии рассеянного кванта будет точно
соответствовать энергии, необходимой для того или иного
колебательного или вращательного перехода.
Обозначив, например, частоту падающего кванта v0,
рассеянного V=v0 — Avo> и собственную частоту колебаний
молекулы v, получим
Av0 + e,e0 = Av0, + e,.1, (33)
откуда, согласно (6),
Av0 = v0 — v'0 = ~(s1 —s0) = v. (34)
^
Мы видим, что разница между частотой падающего и
рассеянного света точно равна частоте колебаний молекулы,
которая обычно наблюдается в инфракрасной области. Часть
энергии падающего света затратилась на возбуждение
колебания молекулы.
Возможен, наконец, и третий процесс—„сверхупругое"
соударение кванта с молекулой. Если молекула"
предварительно уже находилась в колеблющемся состоянии с
энергией ?и = і, то квант света может увеличить свою энергию,
отобрав "от молекулы энергию колебаний. В этом случае
частота рассеянного кванта больше первоначальной, но
попрежнему на ту же самую величину v
Av0 -f *„ я! = Av0' + e„ _ о, (35)
Av0 = v0— v0' = -^ (s0 — «i) = — v. (36)
Таким образом, в спектре рассеянного света должны
наблюдаться, наряду с частотой v0, две частоты, смещенные
в разные стороны на одну и ту же величину v; стоксова
раман-линия с уменьшенной частотой и антистоксова —
с увеличенной. На рис. 107 приведены вид спектра и схема
соответствующих спектральных переходов. Расстояние между
линиями дает собственную частоту колебаний молекулы v.
Это было ясно показано Мандельштамом и Ландсбергом
уже в их первой работе. Естественно, что для чисто
вращательных и колебательно-вращательных переходов
получается в принципе то же самое. Однако, практическое
наблюдение вращательной структуры рамановского спектра
затруднительно. Большая часть опытов проводится над жидкостями
и твердыми телами, в которых вращение затруднено
192
и обычно мало проявляется. Поэтому мы будем говорить
преимущественно о колебательном Раман-эффекте.
Интенсивности антистоксовых линий при обычной
температуре много ниже, чем для стоксовых; это показано
схематически на рис. 107. Объяснение состоит в том, что число
молекул, в которых колебания уже возбуждены,
относительно мало, а ведь именно в результате „сверхупругих
столкновений" с такими молекулами излучается антисток-
сова линия с частотой
Классическая теория
Мы уже видели, что рассеяние света молекулой
определяется ее поляризуемостью, смещаемостью электронной
оболочки. Под действием электрического поля световой
волны оболочка смещается, в ней создается электрический
момент, колеблющийся с той же частотой v0, с которой
меняется внешнее поле. Такая колеблющаяся оболочка сама
излучает свет с частотой v0. Однако, электронная оболочка
молекулы сама по себе не находится в покое — колебания
молекулы периодически ее деформируют, и поляризуемость
молекулы периодически меняется при изменении
междуатомного расстояния в процессе колебания. Такая
относительно медленно колеблющаяся оболочка молекулы
модулирует рассеиваемый молекулой свет, подобно тому, как
колебания мембраны микрофона модулируют радио-волны.
Математически это можно выразить следующим образом.
Индуцированный в молекуле момент равен
М = аЕ= аЕ0 cos 2ъ\і, (37)
где v0 — частота колебания световой волны. Но, как мы
сказали, сама поляризуемость есть функция междуатомного
расстояния
а = а(г) = а0-\-агг±... , (38)
которое периодически меняется при колебаниях с частотой v
r=rvcos 2nvt , (39)
где rv амплитуда колебания.' Подставляя (38) и (39) в (37),
получим
М — (а0 -f a'r^cos 2-rcv^-j- . ..)?0 cos 2wQt=
= a0?0cos 2*v0* + ~2 a'r* Eocos 2* (vo + v)t +
+ ^-a'r,?0 cos 2* (ve — v)* +... (40)
13 Строение молекул. *^
В спектре модулированного света, наряду с неизменной
„несущей" частотой v0, появляются стоксова частота v0 — у
и антистоксова v0-J-v. Результат тот же, что и в квантовой
теории. Подробное исследование показывает полное
соответствие двух теорий; в частности формулы квантовой
механики также приводят к связи интенсивности
римановских линий с поляризуемостью молекулы. Выражение (40)
показывает, что интенсивность (квадрат амплитуды)
пропорциональна (a'rvy. Выражение (40) непригодно для
объяснения наблюдаемых различий в интенсивности стоксовых
и антистоксовых линий: согласно (40) их интенсивности
должны быть одинаковы. Здесь сказывается недостаточность
классической теории. Тем не менее, как это строго доказал
Плачек, классическими представлениями при истолковании
Раман-эффекта можно пользоваться в широких пределах.
Объяснение этого обстоятельства в том, что речь здесь
St.
Н/ \ь% Ы\ \Н
a-$t
•М —I И=Г
v-v v v5 v yv
У-J 1 VbQ
J5-4^v г)
6) 8J
Рис. 107. Схема энергетических переходов при комбинационном рассеянии.
а — рэлеевское рассеяние; б— стоксов случай; в — антистоксов случай;
г—схема спектра.
идёт о процессах сравнительно медленных, q колебаниях
тяжелых ядер, лишь косвенным образом воздействующих
на состояние электронной оболочки.
Главное и очень существенное отличие комбинационного
рассеяния от инфракрасного поглощения состоит в том,
что первое определяется поляризуемостью молекулы а и ее
изменениями при колебаниях и вращениях, второе — диполь-
ным моментом молекулы ^ к его изменениями.
Поляризуемость и интенсивности рамановских линий
Величина а', определяющая интенсивность рамановских
линий, имеет смысл
da\ (41)
a=\-KFjr=r,
производной поляризуемости по междуатомному расстоянию»
взятой в точке г==г0 — равновесного расстояния, а' тем
самым характеризует изменчивость поляризуемости под
194
влиянием колебаний молекулы. В случае инфракрасных
спектров мы имели дело не с а', а с ц/ — производной ди-
польного момента. Мы видели, что у симметричных без-
дипольных молекул дішольный момент при колебаниях не
меняется, у-' = 0, инфракрасного спектра эти молекулы не
имеют. Но поляризуемость а этих же самых симметричных
молекул меняется сильно: а' не равно нулю. Очевидно,
что а' тем больше, чем в большей степени деформируется
электронная оболочка при колебаниях. У чисто ионных
молекул, если пренебречь .взаимной поляризацией ионов,
а' должно равняться нулю. Внешняя электронная оболочка,
содержащая оптические электроны в таких молекулах,
целиком стянута к отрицательному иону, при колебаниях
следует за движениями этого иона и не деформируется.
Напротив, электронная оболочка гомеополярной связи
обволакивает оба атома и поэтому меняется при колебаниях
особенно сильно.
Таким образом, свойства рамановских линий прямо
противоположны свойствам инфракрасных полос. Раманов-
ские линии наиболее интенсивны для гомеополярных,
симметричных, бездипольных молекул, интенсивности
инфракрасных полос равны в этом случае нулю.
Наоборот,-интенсивности рамановских линий ионных молекул очень малы,
у инфракрасных полос они. наибольшие. Ход интенсивностей
рамановских линий при изменении степени ионности
молекулы обратен ходу интенсивностей инфракрасных полос.
Многоатомные молекулы
В спектре комбинационного рассеяния двухатомной
молекулы рядом с каждой линией спектра возбуждающего
света (обычно — ртутной лампы) имеется один стоксов
и один антистоксов спутник. Обертоны обычно настолько-
слабы, что они не наблюдаются. Смысл термина
„комбинационное рассеяние" теперь ясен: в этом явлении обычный
процесс рассеяния комбинируется с внутренними
движениями молекул. Многоатомная молекула имеет целый ряд
колебательных частот, поэтому рамановский спектр может
содержать и большое число линий. Обычно получают ра-
мановские спектры жидкостей. Схема экспериментальной
установки дана на рис. 10S. Исследуемое вещество
помещается в сосуды с плоским дном и загнутым в виде рога,
зачерненным другим концом, для того чтобы исключить
паразитный отраженный свет. Свет лампы падает на
сосудик сбоку, а рассеянный свет конденсируется линзой на
щели светосильного спектрографа. Несмотря на очень малук>
общую интенсивность рассеянного света, хорошие раманов-
ские спектры в настоящее время получают за несколько
* 195
минут, так как применяются очень мощные ртутные лампы
и светосильные спектрографы. На рис. 109 приведен снимок
5
і ! і і ' 8
гт.1 і I і еэ лг
Рис. 108. Схема установки для исследования комбинационного
рассеяния:
а — сосуд; б— лампа; в — светофильтр; г — конденсорндя линза;
д —спектрограф.
рамановского спектра. Буква р — означает рамановские
линии, буква Hg — линии первоначального ртутного спектра.
4—гп щ г
г им шипит і птшшпі I
Рис. 109. Рамановский спектр (углеводород).
ЭЛЕКТРОННЫЕ СПЕКТРЫ
До сих пор мы говорили о колебаниях и вращениях
молекул, электроны которых находятся в своем основном,
а невозбужденном состоянии. При обычных температурах
это всегда имеет место, — для возбуждения электронных
переходов нужны энергии, значительно превосходящие
энергию теплового движения. Электронные переходы могут
быть возбуждены при поглощении молекулой световых
квантов с частотами, лежащими в видимой и
ультрафиолетовой областях, при соударении молекулы с электронами
в электрическом разряде, при соударении с другими
молекулами, обладающими высокой кинетической энергией,
соответствующей высоким температурам. При переходе на
более высокий электронный уровень оболочка молекулы
меняет свои свойства и строение: в случае двухатомной
молекулы это означает изменение вида потенциальной
кривой. При этом возникают различные возможности: в
возбуждённом состоянии может иметь место увеличение или
(чаще) уменьшение энергии диссоциации Д увеличение или
уменьшение равновесного расстояния г0, наконец, возбуж-
196
=-^зю->
денное состояние может вообще оказаться неустойчивым,
обладать потенциальной кривой, не имеющей минимума!
Каждой потенциальной кривой соответствует своя
колебательная частота v, следовательно, при переходе в
возбуждённое состояние эта частота меняется. Меняются и
вращательные уровни, так как меняются г0 и /. Каждой
потенциальной кривой, каждому электронному уровню отвечает
своя совокупность колебательных и вращательных уровней,,
как это показано на рис. 96.
Основной вопрос, на который нужно ответить для того»
чтобы разобраться в молекулярном электронном спектре»
состоит в следующем. Пусть молекула, находящаяся в
основном электронном состоянии и некотором определённом
колебательном состоянии, характеризуемом числом v,
получает энергию электронного возбуждения ее в виде
светового кванта /zv0 или каким-либо другим способом. Допустим,
что форма потенциальных -кривых — строение электронной
оболочки основного и возбуждённого состояний — нам
известны. Спрашивается, в какое именно колебательное
состояние перейдет молекула при поглощении кванта Av0, уце-
.леет она при этом или диссоциирует? Иными словами, на
какую точку потенциальной кривой возбуждённого
состояния мы попадём, исходя из заданной потенциальной кривой
основного состояния и колебательного уровня v?
Принцип Франка —Кондона
Франк и Кондон сформулировали и обосновали принцип,
согласно которому электронный переход, изменение
состояния электронной оболочки,происходит так быстро по
сравнению с колебаниями ядер, что при электронном переходе
не успевают измениться ни скорость движения ядер, ни их
положения. Этим дается' ответ на вопрос, поставленный
выше. Рассмотрим случай, приведённый на рис. 110. Пусть
молекула находится в основном невозбуждённом
электронном состоянии на колебательном уровне v = l. Это значит,
что молекула колеблется с частотой v и обладает колеба-
тельной энергией—«-(6). При колебании молекула больше
всего времени проводит в точках наибольшего
отклонения— в точках пересечения потенциальной кривой с
уровнем Е1 = —^~> ибо в этих точках скорость движения
наименьшая, в них колебательное движение меняет свое
направление на обратное. Таким образом, вероятнее всего
найти молекулу в этих точках. При возбуждении
электронной оболочки квантом Ь0 молекула перейдет в
электронное состояние,характеризуемое верхней кривой на рис.110.
197
Для того чтобы найти, каково будет колебательное
движение молекулы в этом верхнем состоянии, необходимо,
согласно принципу Франка — Кондона, предположить, что
скорость и положения ядер не изменились, т. е. провести из
точек наиболее вероятного состояния молекулы на нижней
кривой вертикали до их пересечения с верхней кривой. Те
состояния ядер, которые соответствуют этим точкам
пересечения, и осуществятся в. верхнем возбужденном состоянии.
Смысл принципа Франка — Кондона и свойства
потенциальной кривой можно пояснить при помощи наглядной
механической модели (Герцберг). Представим себе жестяную
полосу, изогнутую по форме потенциальной кривой и
маленькое цилиндрическое тело, катающееся по этой
изогнутой полосе без трения (рис.
111). Если несколько поднять
цилиндрик над минимумом, то,
предоставленный самому себе,
он будет кататься взад и
вперед, вып?лнять
колебательное движение. Если' поднять
цилиндрик влево до точки,
Рис. ПО. Принцип Франка —Кон- Рис. 111. Наглядная модель по-
дона. тенциальной кривой (Герцберг).
лежащей выше горизонтальной асимптоты—уровня
диссоциации, то он скатится вниз, вкатится на горизонталь
и уйдет в бесконечность с кинетической энергией, равной
превышению потенциальной энергии его высшей точки над
энергией горизонтали. Этим иллюстрируется процесс
диссоциации. Теперь изобразим электронный переход. В этой
модели ему соответствует мгновенное изменение формы изгиба
жестяной полосы. Если при этом изменении минимум
остался на месте, то цилиндрик, покоившийся в минимуме, не
перейдет в колебательное состояние. Если минимум
сместится вправо, то цилиндрик скатится вправо и начнет
колебаться. Если, наконец, полоса изогнется таким образом,
что вправо от покосившегося цилиндрика возникнет спуск
на более низкую горизонталь, то цилиндрик укатится
в бесконечность — молекула диссоциирует.
198
Спектры поглощения галоидов содержат в видимой
области отдельные полосы, которые сходятся к
коротковолновому пределу, за которым начинается непрерывный спектр—
континуум. На рис. 112 приведена спектрограмма
паров иода, стрелкой указана граница полосатого спектра
и континуума. Объяснение этой картины заключается в том,
что возбужденному электронному состоянию молекулы J,
отвечает потенциальная кривая с менее глубоким
минимумом и увеличенным равновесным расстоянием (рис. 113).
Молекула J.2 в возбужденном состоянии менее устойчива.
Наиболее вероятно найти невозбужденную молекулу в
минимуме потенциальной кривой при v = 0. Вертикаль,
проведенная из этой точки, пересекает верхнюю кривую на
уровне более высоком, чем уровень диссоциации молекулы.
Это значит, что молекула диссоциирует на атомы, которые
разлетятся с кинетической энергией, равной превышению
над уровнем диссоциации: К' — Е' — D'. Но кинетическая
, 25 30 35 40 45 j
іііііііішшцн
Рис. 112. Электронный спектр иода.
энергия свободных атомов не квантована — они могут
поглотить и излучить любую энергию. Следовательно, во всей
области переходов, приводящих к точкам, лежащим выше
уровня диссоциации, должен получаться непрерывный
спектр. Граница его отвечает точному переходу на уровень
диссоциации. Переходы на дискретные колебательные
уровни менее вероятны, поэтому полосатый спектр молекул
в этом случае менее интенсивен, чем континуум. Очевидно,
что, зная место схождения полос, границу континуума,
можно определить энергию диссоциации молекулы в ее
возбужденном состоянии.
Молекулярный водород под действием электрического
разряда испускает непрерывный спектр в ультрафиолетовой
области. Поэтому водородными лампами часто пользуются
как источниками света при изучении спектров поглощения.
Объяснение этого явления следующее. Мы видели, что
невозбужденные электроны двух атомов водорода образуют
связь, когда их спины антипараллельны, чему соответствует
основная потенциальная кривая молекулы Н.2.
Отталкиванию атомов с параллельными спинами соответствует кривая
отталкивания, не имеющая минимума (цилиндрик немед-
199
ленно скатывается в бесконечность по такой кривой).
Возбужденному электронному состоянию молекулы Н2
соответствует кривая с минимумом, расположенная выше кривой
отталкивания. Излучая свет, возбужденная молекула может
перейти на кривую отталкивания, диссоциировать и тем
самым дать непрерывный спектр. Согласно принципу Франка —
Кондона, границы непрерывного спектра определятся
переходами, показанными на рис. 114; другие переходы менее
вероятны.
Рис. 113. Потенциальные кри- Рис.114. Возникновение непрерывно-
вые для J2 (Франк). го спектра молекулярного водорода.
Мы видим, что принцип Франка — Кондона является
своего рода ключом к сложной картине полосатого спектра
двухатомной молекулы.
Диссоциация молекул
Электронные спектры дают важнейший непосредственный
способ определения энергии диссоциации молекул.
Рассмотрим, упомянутый уже случай J2 (Франк).
Граница дискретного спектра и континуума лежит при Х =
4995 = А. Мы исследуем пары иода при низкой
температуре. Поэтому следует положить, что • поглощающие
молекулы J.2 находятся в самом низком, колебательном
состоянии v = 0. Поглотив квант с энергией 2.4 eV
(55.37 кг кал/моль), соответствующий Хд, молекула пе-
200
решла из основного электронного состояния с^ = 0в
возбужденное электронное состояние и диссоциировала. Про*
дукты распада —атомы иода — сами, следовательно,
находятся в возбужденном состоянии по сравнению с атомами,
которые получились бы в результате диссоциации молекулы
JLj в невозбужденном состоянии. Часть энергии кванта faD
затратилась на возбуждение молекулы, часть на
диссоциацию в возбужденном состоянии
faD = Ee + D\ (42)
где Ег — энергия электронного перехода, равная расстоянию
по вертикали между уровнями г> = 0 нижней и верхней
потенциальной кривой, D'— энергия диссоциации в
возбужденном состоянии. Но к тому же результату мы придем,.
сначала разорвав молекулу, а затем возбудив один из
получившихся атомов J
faD = D + E„ (43)
где D—энергия диссоциации ^
в основном состоянии, Еа—
энергия ' возбуждения атома
(рис. 115). Энергетические
уровни, а значит величины Еа Рис. 115. Схема определения
для атома известны из атом- энергии диссоциации.
ных спектров и теоретических
расчетов. В частности, атом иода обладает уровнем с
энергией 0.94 eV. Подставив это значение Еа, находим D
D = hvD — Яа= 2.4—0.94 = 1.06 eV = 34.2 кг кал/моль.
Химический опыт дает близкую величину: 34.5 кг кал/моль.
В других случаях получается столь же хорошее
совпадение. Энергии диссоциации многих двухатомных молекул
были определены спектроскопическим путем. Очевидно,
что на основании зависимости, имеющейся между частотами
колебаний и энергиями диссоциации [формула (9)], молено
найти D из частот нескольких .полос даже в тех случаях,
когда не удается' непосредственно наблюдать место слияния
полос в континуум.
Диссоциация молекулы в результате поглощения света
есть, очевидно, типичная фотохимическая реакция. Преды*
дущий случай'мы можем описать химическим уравнением
Ja + Avz>=J* + J,
где звездочкой обозначен возбужденный атом иода. В том
случае, когда возбужденный атом способен испускать свет
(в -приведенном примере J* света не испускает), процесс
201
диссоциации наблюдается непосредственно по испусканию
соответствующей линии атомного спектра, как это было
показано акад. А. Н. Терениным ! на примере реакции
NaJ + faD -: Na* + J,
Na:i: —Na + Av.
Каждый квант взаимодействует с одной молекулой. Число
свободных атомов, возникших при реакции и приходящихся
на один квант (квантовый выход реакции), равен двум. Мы
встретились здесь с частным случаем основного закона
фотохимии Эйнштейна, гласящего, что число молекул или
атомов, непосредственно взаимодействующих с квантом света,
всегда равно единице. Первичный фотохимический процесс
состоит в поглощении света молекулой; молекулы, не
поглощающие света не возбуждаются, не диссоциируют, свет
в этих случаях не вызывает никаких фотохимических
действий. Отсюда ясно, сколь важное значение для фотохимии
имеют электронные спектры поглощения молекул.
3889 3872 3862 3855 3850
Рис. 116. Электронный спектр — полосы циана CN и С±
в спектре угольной дуги.
Свободные радикалы
Разрушенные действием света или электрического
разряда, или нагревания, или, наконец, просто в результате
химической реакции сложные молекулы могут, диссоциируя,
дать недолговечные образования, осколки молекул с
ненасыщенными валентностями — свободные радикалы. Так, при
крекинге углеводородов возникают радикалы СН, СН2, СН3,
С, которые неустойчивы и быстро соединяются друг с
другом. Химия знает лишь косвенные пути доказательства
существования таких свободных радикалов и практически
не имеет возможности установления их структуры. Впервые
такие свободные радикалы удалось изучить при помощи
молекулярных спектров. Так как свободные радикалы не
успевают исчезнуть за время акта поглощения или
испускания света, спектры их легко наблюдаются. В настоящее время
хорошо изучены свойства многих радикалов, известны их
энергии и междуатомные расстояния. Укажем случаи двух-
1 А. Н. Те ренин. Фотохимия паров солей. ОНТИ, 1934.
•202
атомных радикалов СН, С2, CN, NH, ОН. Свободный гидро-
ксил встречается почти всегда в пламенах и разрядах
в среде, содержащей пары воды (Кондратьев). На рис. 116
приведены- характерные полосы испускания радикалов CN
и С>, получающихся при горении угольной дуги в воздухе.
Интересно отметить, что спектры испускания комет также
показывают, что в этих газовых образованиях содержатся
свободные радикалы: CN, С и ионизированные молекулы
СО+, N«+. В ряде случаев удалось также обнаружить
свободные радикалы в спектрах звезд. Напротив, исследование
спектра солнечного света, отраженного планетами,
свидетельствует о том, что в атмосферах планет содержатся
молекулы. Так, спектр Венеры указывает на присутствие
в ее атмосфере углекислого газа, в атмосфере больших
планет — Юпитера, Сатурна,
Урана и Нептуна — имеется
метан, а в атмосфере Юпитера
еще и аммиак. Атмосфера
Марса, согласно спектральным
исследованиям, содержит
ничтожное количество кислорода,
менее одной тысячной от его
содержания в земной атмосфере.
Рис. 117. Потенциальные кривые
п случае предиссоциации.
Предиссоциация
Специфические
обстоятельства возникают в тех случаях,
когда молекула может без
излучения изменить свое
электронное состояние и диссоциировать. Это возможно, если
'различным электронным состояниям соответствуют
пересекающиеся потенциальные кривые. На рис. 117 изображен
такой случай. Кривая устойчивого состояния А пересекается
кривой отталкивания В. Следовательно, имеется конечная
вероятность для молекулы перейти в точке пересечения С
с кривой А на кривую В и диссоциировать. В наглядной
модели Герцберга (ср. рис. 111) это означало бы наличие
в изогнутой полосе прорези, сквозь которую катающийся
цилиндрик может провалиться на вторую полосу, лишенную
минимума. Явление перехода с устойчивой кривой на
кривую отталкивания называется предиссоциацией.
Предиссоциация означает, следовательно, уменьшение времени
существования молекулы в данном устойчивом состоянии,
уменьшение времени жизни, времени излучения. Соответственно
должна возрасти неопределенность в значении энергии,
а значит, и частоты излучающего 'электронного перехода
{ср. главу 1, (8)]. В результате электронные полосы в спектрах
203
размываются, исчезает их колебательно-вращательная
структура. Это явление особенно часто встречается среди
многоатомных молекул.
Многоатомные молекулы
Подробный анализ электронных спектров многоатомных:
молекул весьма труден. Потенциальная энергия
многоатомной молекулы в данном электронном состоянии зависит уже
не от одного только расстояния г, но от всех
междуатомных расстояний. Здесь следует говорить не о потенциальных:
кривых, а о совершенно ненаглядных потенциальных
поверхностях. Тем не менее, в ряде случаев удалось
разобраться во всех деталях спектров молекул. Особенно
помогает применение свойств симметрии молекулы, значительно
упрощающих спектр. В настоящее время хорошо изучены
электронные спектры бензола и его производных, спектры
ряда более простых трехатомных молекул и т. д.
Электронные спектры сложных молекул наблюдаются,
главным образом, как спектры поглощения в видимой и
ультрафиолетовой области и спектры испускания —
люминесценции. Не занимаясь подробностями строения спектров —
колебательными и вращательными линиями в полосах —
уже из одного общего вида и положения полос можно
сделать важные выводы о строении молекул. Эксперимент
дает нам спектральную кривую поглощения или испускания
света молекулой. Мы уже говор-или (стр. 115) о том, что
наиболее длинноволновые максимумы кривой поглощения
принадлежат хромофорным группам и могут быть объяснены
при помощи концепции электронного резонанса. Мы видели
также, что часто есть возможность связать определенную
спектральную полосу с определенной группой или
химической связью в молекуле и установить
направление.электронного перехода в молекуле (стр. 160). Если в спектре
наблюдаются такие характеристические полосы, относящиеся
к определенным группам, то на основании изменения
положения и вида этих полос при введении в молекулу других
атомов можно судить о том, что происходит с данной
группой. На рис. 118 показаны кривые поглощения ацетона
СН3СОСН3 и его производных. Максимум поглощения при
X .2754 А связан с хромофорной группой >СО, состояние
которой очевидно меняется при введении в ацетон
замещающих атомов и групп, в частности вследствие резонанса,
невозможного в ацетоне:
сі сі+ сі-
^> с = о ^) с - о~ / с = 0+-
н3с н3с н8с
I II Ш
204
Весьма интересны и характерны также изменения
полосатого спектра, создаваемые внешними влияниями
(температура, давление) и междумолекулярным взаимодействием,
например влиянием растворителя. Последнее может быть
¦очень значительным и проявляться заметным образом
в изменении окраски. Так, растворы иода в четыреххлори-
стом ^углероде, бензоле имеют интенсивную пурпурную
окраску, а в воде и спиртах —бурую.
Положение и вид полос поглощения широко применяются
ъ качестве эмпирического признака при анализе строения
молекул: на основании присутствия или отсутствия тех или
иных.характерных полос делаются выводы о присутствии
в молекуле определенных структурных элементов. С еще
7J
то зооо то ~*~
т_- 1 1 1 1
сн.соон
СН3С0СНгС1
ClCHjCOCHjCl/ Х^4
сн3сосн3
і I i i і
Z5DD0 35000 W00 55000
v $см'1 >-
Рис. 118. Кривые электронного поглощения ацетона
и его производных.
большим успехом пользуются спектрами поглощения
и люминесценции для идентификации сложных веществ и
обнаружения их малых количеств. Молекулярный
спектральный анализ нашел богатые и разнообразные применения
в биохимии для определения таких сложных веществ, как
алкалоиды, гормоны и витамины. Определяя интенсивности
полос, можно проводить и количественный анализ. В
настоящее время существуют, например, промышленные приборы
.для количественного спектрального анализа витамина А
по его полосам поглощения 2800 и 3260 А. Однако, в целом,
молекулярный спектральный анализ по электронным
спектрам еще очень мало разработан. Его теоретическое обосно-.
ванне находится в начальной стадии. В то же время
молекулярный спектральный анализ —область, которая обещает
лать очень многое как теоретической химии и биохимии,
так и их приложениям.
lg/r
30
10
205
Глава VIII
КОЛЕБАНИЯ МНОГОАТОМНЫХ МОЛЕКУЛ
Заключительную главу нашей книги мы посвятим
подробному описанию колебаний многоатомных молекул и
явлений, с ними связанных.
Строение молекулы мы рассматривали как строение
ее электронной оболочки, определяющее и геометрическое
расположение ядер. Все физические свойства молекул —
электрические, магнитные, оптические — являются
выражением строения. Но колебания молекул, колебательные
спектры занимают здесь особое положение. Колебание-
молекулы есть внутримолекулярный процесс, происходящий
без большого' изменения свойств электронной оболочкь
и в то же время всецело этими свойствами определяемый.
Поэтому, изучая инфракрасные и рамановские спектры,
мы можем получить несравненную по ценности информацию
о строении молекулы. Мы уже видели, что частоты
колебаний характеризуют силы связей между атомами, а
интенсивности колебательных спектральных линий — природу
этих связей, степень их ионности. В свою очередь, ряд
физических и физико-химических свойств вещества
определяется теми колебательными процессами, которые
происходят в молекуле. Любой вид энергии, приобретаемый
молекулой, превращается в той или иной степени в энергию
колебательного движения.- Поэтому термодинамические,
тепловые свойства вещества неотделимы от колебаний:
Ниже мы увидим, что теплоемкости не могут быть
вычислены, если частоты колебаний не известны.
Колебательные движения очень важны во всех кинетических
явлениях, в процессах распада сложных молекул, в
процессах передачи энергии при соударениях между
молекулами.
Область колебаний и колебательных спектров
заслуживает нашего специального внимания еще и потому, что в ней
206
нашли свое наиболее ясное выражение основные
методологические идеи теории строения молекул,
охарактеризованные в главе I. Теория колебаний и колебательных
спектров— прежде всего полуэмпирическая теория. Исходя
из общей картины явления, полученной из квантово-меха-
нической теории строения молекул, мы находим общие
закономерности, связывающие частоты и интенсивности
колебательных спектров со структурой молекулы, но
конкретные величины, через которые эти закономерности
выражаются, определяются из опыта. Теория колебаний —
приближенная теория. В ней последовательно и критически
применяются приближенные методы, выработанные на основе
всех имеющихся сведений о строении молекул и в качестве
первого, наглядного и физически обоснованного
приближения принимается аддитивная валентная схема. Благодаря
этому, мы получаем такое истолкование колебательных
процессов и свойств колебательных спектров, которое
непосредственно смыкается с другими данными физического
и химического исследования. В теории колебаний особенно
широко применяются свойства симметрии молекул. Наконец,
существенная особенность колебательных процессов
выражается в том, что они поддаются исследованию
наглядными методами классической физики, что объясняется
сравнительной медленностью этих процессов, большими массами
ядер и тем, что частоты гармонических колебаний не
квантуются.
Естественно, что интересны главным образом
колебания многоатомных молекул. Многоатомные молекулы
представляют собой основной объект исследования химика,
биохимика, физика, занимающегося вопросами строения
вещества.
Однако не только теория, но и практика требуют изучения
колебаний многоатомных молекул. Колебательные спектры,
инфракрасные и рамановские, в настоящее время широко
применяются для молекулярного спектрального анализа,
для определения структуры молекул, их идентификации,
количественного и качественного анализа смесей. Эти
спектры значительно легче расшифровать, чем спектры
электронные. С другой стороны, знание колебательных
спектров необходимо для расчетов термических свойств
молекул, их свободных энергий и констант равновесий для
химических процессов.
В этой главе мы будем излагать теорию колебательных
спектров в том виде, в котором она сложилась в работах
М. А. Ельяшевича, Б. И. Степанова и М. В. Волькенштейуа.
Методы исследования молекулярных колебаний,
применяемые в этих работах, видимо, наиболее просты
и универсальны.
20?
СВЯЗАННЫЕ КОЛЕБАНИЯ И НОРМАЛЬНЫЕ КОЛЕБАНИЯ
Сколько и какие колебания может иметь многоатомная
молекула? В ./V-атомной молекуле каждый из /V атомов
может передвигаться в трех направлениях в трехмерном
пространстве. Всего, таким образом, Af точек имеют 3N
различных независимых движений, 3N степеней свободы.
Но N атомов объединены в молекулу. Сама молекула, как
целое, имеет три степени свободы поступательного
движения и три вращения (в частном случае линейных молекул^
как мы видели, — два вращения). Поэтому для независимых
перемещений атомов в молекуле по отношению, друг
к другу остается 3 7V—б (в линейных молекулах 3/V—5)
степеней свободы. Всего, следовательно, iV-атомная молекула
имеет 3iV—6(3jV—5) независимых колебаний. В
двухатомной молекуле АГ=2; 3/V — 5 = 1, имеется одно колебание.
Но уже в трехатомной молекуле таких колебаний 3-3—6 = 3,
¦а в линейной трехатомной 3-3—5 = 4. Отделив движения
молекулы как целого, мы выделили колебания,
происходящие в неподвижной, невращающейся молекуле. Сложную
молекулу можно представить себе как совокупность ряда
осцилляторов — маятников, движения которых связаны
между собой. Энергия, попадающая на один из
осцилляторов, скажем на отдельную связь в молекуле,
перераспределяется через некоторое время по всем степеням свободы,
все атомы и связи вовлекаются в колебание.
Мы уже рассматривали связанные колебания в частном
случае двух степеней свободы (стр. 42). Мы видели, что
наличие связи между колебаниями приводит к .расщеплению
частот, что система как целое способна выполнять так
называемые нормальные колебания, в которых все атомы
движутся с одинаковой -частотой и фазой в тех или иных
направлениях. Любое колебательное движение молекулы
представится как наложение ее нормальных колебаний.
Число нормальных колебаний, конечно, равно числу
взаимодействующих осцилляторов, числу степеней свободы.
В общем случае jV-атомной молекулы число колебательных
¦степеней свободы и число нормальных колебаний равно
3N— 6. Это означает, что, например, в спектре трехатомной
нелинейной молекулы Н20 должны быть представлены три
частоты, три колебания. Может оказаться, что некоторые
из этих колебаний не проявляются в инфракрасных или
рамановских спектрах, так как они не сопровождаются
изменением дипольного момента или поляризуемости.
Возможны также случаи, в которых частоты отдельных
нормальных колебаний совпадают друг с другом и поэтому разным
нормальным колебаниям соответствует одна спектральная
линия. Такого рода явления поддаются полному объяснению
208
пои помощи одних лишь свойств симметрии молекулы, без
каких-либо представлений о реальных внутримолекулярных
силах.
СИММЕТРИЯ КОЛЕБАНИЙ
Мы видели, что нормальные колебания системы с двумя
степенями свободы распадаются на симметричное и
антисимметричное. Во всех тех случаях, когда молекула имеет
какие-нибудь элементы симметрии — центр, плоскости,
оси — все нормальные колебания должны быть симметричны
или несимметрлчны как по отношению к симметрии
м.олекулы в целом, так и по отношению к отдельным
ее элементам, ибо они происходят около положений
равновесия, обладающих данной симметрией. Колебание может
быть симметричным к некоторой плоскости симметрии; это
значит, что во время колебания одинаковые атомы одинаково
Рис. 119. Колебания молекулы hLO.
смещаются в разные стороны по отношению к этой плоскости
и эти смещения переходят друг в друга при отражении.
Например, показанные .на рис. 119 колебания v(s) и 8(s)
молекулы типа Н20 (симметрия Си) симметричны по
отношению к вертикальной плоскости симметрии,
перпендикулярной плоскости чертежа — отражение смещений атомов
з этой плоскости не меняет картины. Напротив, колебание
v(as) антисимметрично по отношению к этой плоскости —
отражение меняет направления смещений атомов на
обратные. В то же время это колебание симметрично по
отношению ко второй плоскости симметрии, плоскости рисунка,
ибо оно само происходит в этой плоскости. Таким образом,
в зависимости от того,, сохраняются или меняются на
обратные знаки смещений из положений равновесия.у
колеблющихся атомов при выполнении симметрических операций
отражений и поворотов, мы говорим о симметричных или
антисимметричных колебаниях. Возможны, однако, и такие
случаи, в которых при -симметрической операции
нормальное колебание не переходит ни само в себя, как в случаях
v (s), Ь (s)9 ни в колебание с обратными знаками, как
14 Строение молекул 209
в случае v(us), но вообще переходит в новую формул
движения. Рассмотрим поперечное колебание линейной
молекулы СО-2 (рис. 120). При повороте вокруг оси молекулы
ОСО на 90° мы получаем новое колебание, происходящее
в другой плоскости—перпендикулярной плоскости чертежа.
Это новое нормальное колебание имеет, однако, прежнюю
частоту, значит два разных нормальных колебания имеют
здесь одинаковые частоты. Такие колебания называются
вырожденными.
Разделение нормальных колебаний по их свойствам
симметрии может быть проведено заранее, до всякого
расчета частот и смещений и очень облегчает такие расчеты.
Зная число атомов в молекуле и ее симметрию, можно
установить, сколько будет симметричных колебаний, сколько
антисимметричных, сколько вырожденных (Брестер, Вигнер,
Плачек).
Правила отбора
Применяя свойства симметрии, возможно указать также,
сколько линий мы будем наблюдать в инфракрасном или
v/s/ vfasj SfasJ
Рис. 120. Колебания молекулы С02.
рамановском колебательном спектре. В спектре проявляются
лишь те колебания, которые сопровождаются изменением
электрического дипсльного момента (инфракрасные спектры)
или поляризуемости (рамановские спектры). Очевидно, что
если молекула имеет центр симметрии и бездипольна,
то при полносимметрическом колебании дипольный момент
все время останется равным нулю (рис. 120); такое
колебание будет неактивным в инфракрасном спектре. Напротив,
в рамановском спектре такое колебание будет наблюдаться,
ибо поляризуемость электронной оболочки сильно меняется
при пульсирующем полносимметрическом колебании —
эллипсоид поляризуемости сжимается и растягивается при
колебании молекулы. Антисимметричное по отношению
к центру симметрии колебание молекулы сопровождается,
появлением дипольного момента (рис. 120, v(as)) и поэтому
в инфракрасном спектре наблюдаемо. Однако в рамановском
спектре оно не активно. Можно показать, что действительно
величина а' равна здесь нулю. И вообще, исходя из свойств
симметрии эллипсоида а и вектора щ можно доказать, что
симметричные колебания молекул с центром симметрии
210
разрешены в рамановском и запрещены в' инфракрасном
спектре, а антисимметричные — наоборот. Правила отбора
в других случаях также получаются при помощи
соображений симметрии (Плачек).
Поляризация рамановских линий
Совершенно такие же рассуждения, как примененные
нами при описании свойств обычного молекулярного
рассеяния (стр. 154), приводят к тому, что рамановские линии
в зависимости от свойств молекулы и ее колебаний должны
быть полностью или частично поляризованы. Это объясняется
анизотропией поляризуемости а, а значит и ее
производной а'. Действительно, мы можем говорить о различных
.величинах а' в различных направлениях, о значениях
а'и а'2, а'3. Очевидно, что степени деполяризации
рамановских линий не должны совпадать с р обычного рэлеевского
рассеяния. Так, рэлеевское рассеяние тетраздрической
молекулы СС14 должно иметь р = 0. Та же величина получится,
конечно, при комбинационном рассеянии молекулой,
находящейся в состоянии полносимметрического колебания.
Но антисимметрическое колебание нарушает симметрию
системы и поэтому степень деполяризации возрастает. Для
рамановских линий имеет место соотношение,, подобное
прежнему:
Р ~45а'2+7Ь'2' \ }
где а' и V — среднее значение и анизотропия эллипсоида
л/ именно
Д'=4(а'і + я« + я'з), (2)
Ь' = % [<*'* ~ а'»)* + (а'* - а'^ + (а'з - а'^] V'2- (3)
В зависимости от симметрии колебания величины а'и V могут
быть равны или не равны нулю. Если, например, колебание
антисимметрично по отношению к какому-нибудь элементу
симметрии, можно показать, что а' = 0, но" Ь' отлично
от нуля. Следовательно, р' = у-. То же имеет место для
всех вырожденных колебаний. Напротив, если колебание
симметрично, а' не равно нулю и р' меньше 6/7. В частном
случае, когда молекула высокосимметрична, изотропна,
ее эллипсоид поляризуемости обращается в шар.
Следовательно, Ь = 0, при полносимметрическом колебании равно
нулю и Ь\ откуда получаем р' = 0 для полносимметричных
• 211
колебаний молекул с изотропной .поляризуемостью. Эти
правила, относящиеся к поляризации рамановских линий,
сформулированы Плачеком.
Некоторые примеры
Разберем несколько примеров. Линейная молекула ССХ2
имеет 3*3 — 5 = 4 нормальных колебания. Одно из них
полносимметричное, одно антисимметрично, одно двукратно
вырождено и также несимметрично (рис. 120). В
инфракрасном спектре должны наблюдаться две полосы
антисимметричного и вырожденного колебания, в рамановском одна
полоса полносимметричного с р' большим нуля и меньшим
V, (0,86).
Молекула S02 изогнутая, симметричная. Она имеет
3-3 — 6 = 3 колебания: два симметричных, одно
антисимметричное- Все колебания разрешены и в инфракрасном
и в рамановском спектрах, ибо молекула не имеет центра
симметрии'. Две рамановские линии имеют р' меньше 0.86,
одна р' = 0.86.
Трансдихлорэтилен имеет в отличие от цисдихлорэтилена
центр симметрии (стр. 84). Поэтому инфракрасный и рама-
новский спектры первого беднее линиями, часть из них
запрещена. Большая часть рамановских линий трансизомера
поляризована, имеет р' меньше 0.86, ибо она принадлежит
колебаниям, симметричным относительно центра. -
Если четырехатомная молекула типа XY3 построена как
пирамида, то она имеет четыре рамановских линии (две из них
двукратно вырождены). Две линии имеют р' меньше 0.86, две
р1 = 0.86. Таковы случаи NH3, РС13. Если же молекула —
плоский треугольник, то колебание, антисимметричное
относительно плоскости в рамановском спектре запрещено и
наблюдаются только три линии (ВС13, ионы С032", N03~).
Напротив, в инфракрасном спектре запрещено
полносимметричное колебание. Таким образом, один лишь подсчет числа
наблюдаемых линий дает в этом случае возможность
установить, структуру молекулы.
Тетраэдрическая молекула типа XY4 имеет 3-5 — 6 = 9
нормальных колебаний, но всего четыре различные частоты,
ибо высокая симметрия приводит к вырождению. Имеется
одно полносимметрическое колебание ср' = 0,
невырожденное, одно вырожденное двукратно и два вырожденные
трехкратно. Все эти колебания имеют р' =0.86. В рамановском
спектре разрешены все четыре частоты, в инфракрасном —
лишь две.
Двукратное вырождение частот появляется при наличии
оси симметрии порядка не ниже третьего, трехкратное —
в группе тетраэдра и октаэдра.
212
Мы видим, что одних соображений симметрии достаточно
для того, чтобы сделать весьма далеко идущие выводы
о колебательных спектрах и строении молекул.
МЕХАНИКА КОЛЕБАНИЙ
Мы познакомились с тем, что можно сказать о спектре
молекулы, зная ее симметрию и, обратно, с тем, что можно
сказать о симметрии, зная спектр. Однако к свойствам
симметрии отнюдь не сводится та информация о строении
молекулы, которую можно получить из колебательных
спектров. Общие свойства симметрии не зависят от природы
электронного облака молекулы, от природы химической
связи. Из колебательных спектров можно сделать выводы
о силах, действующих между атомами в молекуле. Для этого
необходима такая теория, которая может истолковать
механические и электрооптические свойства колебательных
спектров — численные значения частот и интенсивностей
спектральных линий.
Начнем-с частот колебаний. Задача состоит в том, чтобы
из их значений найти вид колебаний и численные
коэффициенты сил k, определяющие внутримолекулярные
взаимодействия. Считая, в первом приближении, колебания
гармоническими (это приближение допустимо для малых
амплитуд), можно применить к задаче методы классической
механики связанных колебаний.
Очень важен разумный выбор колебательных координат,
решение вопроса о том, что именно колеблется в молекуле
в первую очередь, как направлены возвращающие силы. После
всего изложенного в этой книге, должно быть ясным, что
имеются известные основания считать силы направленными
прежде всего по валентным связям в молекуле. Это по
крайней мере так для большинства гомеополярных молекул.
Таким образом, естественно, исходя из валентной схемы
химии, считать основными структурными элементами
молекулы валентные связи и разделить упругие силы на валентные —
действующие вдоль валентных штрихов, растягивающие или
сжимающие валентные связи, и деформационные —
действующие перпендикулярно валентным штрихам, изменяющие
валентные углы. При расчетах колебаний применяются
естественные координаты q— изменения валентных
расстояний и у—изменения валентных углов. Конечно, выделение
таких сил есть приближенный метод, ибо все
изменения связей и углов не независимы друг,от друга, но в тех
случаях, когда сила, действующая при колебании вдоль
самой связи больше, чем сила взаимодействия с другими
связями, этот метод законен. Ниже мы увидим, что такая
валентно-силовая схема применима не во всех случаях:
213
будучи хорошим приближением для СН4, она теряет смысл
для СС14; иными словами, применимость этих представлений
определяется индивидуальными особенностями молекулы.
Тем не менее, свойства большого числа молекул удается
истолковать на основе валентно-силовой схемы, понимаемой
в том смысле, что в качестве естественных координат
выбираются изменения длин связей и углов между ними.
Расчет колебаний молекулы
Простейший случай многоатомной молекулы —
симметричная, нелинейная трехатомная молекула XY2*V(PHC- 121).
В покоящемся состоянии она имеет длины связей XY — г
и валентный угол Ь. Естественных координат три: изменение
длины первой связи ди длины второй связи д^ и валентного
угла 7- Если бы все эти изменения происходили независимо
друг от друга, то потенциальная энергия колебаний
молекулы представилась бы суммой энергии трех гармонических
осцилляторов ди ft и у
1/в = **-+**Ц_А?. (4)
Силовые,- динамические коэффициенты k для одинаковых
связей одинаковы. Для угла &т, конечно, имеет другое
значение.
Такая запись полностью соответствует классической,
аддитивной валентной схеме химии. Здесь предполагается,
что валентные связи и углы не взаимодействуют друг
с другом, а колеблются независимо друг от друга. Однако
такое приближение недостаточно; необходимо учитывать
все взаимодействия между связями и углами при
колебаниях. Полное выражение потенциальной энергии имеет вид
гj kg? i kg* -i &тт2 !
+ h qxq% +'dq^( -j- dq^. (5)
Число динамических коэффициентов, характеризующих
молекулу не два, а четыре. Но для того чтобы найти
частоты и формы колебательных движений молекулы,
недостаточно знать динамические коэффициенты k. Частоты
определяются не только упругими силами, но и инертными
массами колеблющихся атомов. Частота колебания
отдельной связи между двумя атомами, так же как частота коле-
214
бания двухатомной молекулы, выражается коэффициентом
к связи и ее приведенной массой
*=***=VI > ^
где
т т^ пи* V*
Мы назовем величину 1/т кинематическим коэффициентом.
Динамический коэффициент k определяет упругую силу,
действующую на колеблющиеся частицы; кинематический
коэффициент характеризует их инерцию,то ускорение,
которое может возникнуть под действием упругой силы. Зная
динамические и кинематические коэффициенты, можно написать
уравнение движения
колеблющейся системы. Для
двухатомной молекулы это
уравнение имеет вид (ср. стр. 178)
тх = — kxy (8)
откуда
а^=|*, (9)
и мы получаем соотношение (6).
Можем ли мы и в случае
многоатомной молекулы
считать, что каждая связь характеризуется своим независимым
кинематическим коэффициентом (7) и колеблется независимо
от других связей, если отвлечься от динамических
взаимодействий? Конечно, нет. Свлзи в молекуле исходят из общих
атомов и взаимодействуют через посредство этих атомов
друг с другом. Смещение одного из атомов неизбежно
изменяет длины всех его связей — наряду с динамическим
взаимодействием имеет место кинематическое. Поясним это
рис, 119. Из атома М исходят две связи под углом ft.
Растянем одну из этих связей и предоставим ей колебаться,
считая, что она" динамически не взаимодействует со второй
связью и с углом, т. е. h = d=0 в уравнении (5).
Изменение длины этой связи выразится как сумма смещений ее
крайних атомов с массами т и Ж, спроэктированных на
направление связи
Я\ = ЯтіЛ-Ями (10)
qmi и дм% — смещения атомов т и М9 отсчитываемые вдоль
215
Рис. 121. Молекула XY2.
связи <7і. Составим уравнение движения для каждого из
атомов т и М. Для крайнего атома
inqml= — kqu (И)
ибо на него при колебании действует возвращающая сила —
kq1 только одной связи. Напротив, атом М стремится
вернуться в положение равновесия как под действием силы
связи qu так и под действием силы второй связи, которая
также растягивается при изменении положения М (рис. 121).
Так как мы ищем уравнение для колебания связи qu нужно
спроектировать силу, действующую на М со стороны второй
связи, на первую связь, т. е. умножить ее на cos &. Имеем
Mqm = — kxqx — koq,2 COS &. (12)
Разделим уравнения (11) и (12) соответственно на т и М
и сложим эти уравнения. Получим
-gcos&.ft. (13)
Но, согласно (10),
Имеем окончательно
9і = -Ьі(і + м)яі — %со*ъ-д*, (14)
т. е. колебание связи q} кинематически связано с колебанием
связи q.2. Уравнение (14) содержит коэффициент
кинематического, инерционного взаимодействия
ij cos 0. (15)
Взаимодействие тем сильнее, чем легче атом М и чем
больше угол Ь отличается от прямого:
В действительности нужно учитывать кинематические
взаимодействия не только соседних связей, но и связей
с соседними углами и т. д. Кинематические коэффициенты
зависят от масс атомов и их расположения — длин связей и углов
между ними. М. А. Ельяшевич составил таблицы
кинематических коэффициентов для всех возможных случаев
инерционного взаимодействия. С помощью этих коэффициентов
можно, зная вид потенциальной энергии (5), составить
уравнения движения колеблющейся молекулы. Выпишем эти
уравнения для симметричной молекулы XY2, ограничившись
216
для простоты кинематическими взаимодействиями, т. е.
считая в (5) k = d = 0
Icosa-^ + K^+i^-
— -ш sin*>• kqi—j^-sm 0 • Л?4-f J2
Л4/- " Mr ~~~ '' - ' (
+ I^r(l-cos{})lAY-^lT = 0.
>?,-^sina-Vr=o,(i6)
/ЛГ- j
Это три уравнения с тремя неизвестными, дающие
уравнение третьей степени относительно оА Однако применение
свойств симметрии может упростить задачу. Симметричное
колебание рис. 119 характеризуется комбинацией смещений
атомов #і-}~<72' сохраняющей знак при отражении от
плоскости, равносильном замене qx на #2. Антисимметричное
колебание, наоборот, характеризуется разностью смещений
Яі — Яі- Обозначив
Яз = Яі + Яъ
Яа* = Чі — Яг,
мы сможем переписать (12) в виде уравнений
(17)
{[^ + м)к-У°'Ь-к
^<q
as
о,
{(і+
м
(18>
Первое из уравнений (18) получено вычитанием второго
уравнения (16) из первого, второе уравнение (18) —
сложением двух первых уравнений (16), третье уравнение
осталось неизменным. Мы видим, что антисимметричное
колебание qas отделилось от симметричных qs и у. Первое
уравнение (18) дает для частоты антисимметричного колебания
значение
CD'
* й-яfl-«*»>}'
(19)
а два других уравнения (18) дают частоты симметричных
колебаний. В более сложных случаях приходится, конечно,
встречаться с уравнениями высоких степеней относительно оД.
Однако возможность составления уравнений непосредственно
217
в численном виде на основе далеко идущей схемы
аддитивности и применение свойств симметрии дают возможность
разобраться и в колебаниях весьма сложных многоатомных
молекул.
Зная частоты и коэффициенты, можно, очевидно, найти
из уравнений типа (14) отношения между qu q* и -у и тем
самым определить форму колебания, соответствующую
каждой частоте — относительные величины смещений
колеблющихся атомов. В нашем частном случае
антисимметричное колебание происходит так, что одна из связей
удлиняется, а другая сокращается при постоянном значении угла.
Действительно при антисимметричном колебании
qs = 0, откуда q1= — q,. (20)
Напротив, при симметричных колебаниях растяжения связей
сопровождаются изменением угла. Имеем
Яа* = 0, ?1 = ?* (21)
qs = 2qi = 2q2 = 2q, (22)
и на основании второго (или третьего) уравнения (18) мы
получим.
q_ Mr~_ *
""(і+іі'+л-"-»1' (23)
В зависимости от значения со, мы получим это отношение
большим или меньшим единицы. Если со относительно
велико и q много больше уг, можно говорить о
преимущественно валентном колебании, в котором меняются главным
образом длины связей. Обозначим такое валентное
симметричное колебание v($) (рис. 119). Антисимметричное
колебание— чисто валентное, v(as). Если со относительно мало
и уг больше qy можно считать колебание деформационным,
таким, в котором меняется главным образом валентный
угол. Обозначим деформационное симметричное
колебание Ъ{$). За немногими исключениями, колебания всегда
носят смешанный характер, в них участвуют все связи и углы
молекулы. Разделение колебаний на валентные и
деформационные имеет, как мы только что показали, условный
смысл, но все же оно физически обосновано и сильно
облегчает анализ колебательных спектров, .
218
Нормальные колебания и нормальные координаты
Данному нормальному колебанию соответствует
определенная комбинация естественных координат, определяющая
относительную величину и характер смещений.'Мы уже
вводили координаты (17) и видели, что антисимметричному
колебанию молекулы XY2 соответствует совокупность
смещений
Яі = — Яъ 7 = 0,
а симметричным — смещения
Ч\ — Чъ Ct = q1 = q* = q,
где С характеризует отношение q:^r [ср. (23)]. Объединяя
эти смещения в одно выражение, получим координаты,
полностью описывающие нормальные колебания молекулы
v(as): Qas=Cas(q1—q^)J
v(s): Qsi = Csl(qi + q^)-\-CsiClt9 (24)
8 (s): Qs* = Cja (qx + qu) -f- CsX^y
где Casy Сst9 Csi — численные коэффициенты. Координаты
Q выражаются через естественные координаты q и у. Если
произвести замену (24), то выражение потенциальной энергии
5) перейдет в
2Л2 2/->2 2л2
— 2 * 2 I 2 ' ^ '
т. е. в нем исчезнут члены взаимодействия. Одновременно
в сумму квадратных членов перейдет и выражение
кинетической энергии. Коэффициенты С находятся при помощи
выражения (25). Координаты Q называются нормальными
координатами. Мы видели, что задача определения частот
и формы колебаний сводится именно к нахождению
нормальных координат.
Некоторые примеры
Приведем примеры нормальных колебаний
многоатомных молекул. В табл. 30 даны типы колебаний, их частоты
и степени деполяризации р' в рамановских спектрах. На
рис. 122 и 123 изображены формы колебаний
пирамидальной молекулы XY3. и линейной ацетилена.
219
¦о
о*-
vfs/ v(as) bis)
Рис. 122. Колебания молекулы NH3.
&
ьш-
о*
?
\ts)
к-^О
О^і
н-4
%ш
vfas)
о> <•—•*—*о
?
V*'
і~Л
ЬгШ
Рис. 123. Колебания молекулы ацетилена C..R..
?
Молекула
SO,
Ср. рис. 119
РСі3
Ср. рис. 122
- СС14
Ср. рис. 31
"
СпНо
Ср. рис. 123
Та
Симметрия и
число
колебаний
Civ
3
Czv
6
т$
Dcoh
7
блица ,30
Тип нормального
колебания и
степень вырождения
v(s)
S(5)
n (as)
v(s)
0(5)
м (as)^2
о (as)—2
N(S)
о (as)-2
v (as)—3
о (as)—3
Vl (5)
v, (s)
ч (as)
0! (as)—2
o, (as)—2
Частота,
1146
525
1340
511
258
484
190
459
217
760 и 790
313
1975
3370
3277
600
729
P'
0.18
0.50
0.86
0.16
0.28
0.86
0.86
0.05
0.86
0.86
0.86
<0.2
P
—
Динамические коэффициенты
Наибольший интерес для проблемы строения молекул
представляют динамические коэффициенты k, определяемые
междуатомными силами. Частоты колебаний выражаются
через эти коэффициенты и кинематические коэффициенты
(ср. стр. 215). Однако число нормальных колебаний и,
следовательно, могущих быть наблюдаемыми частот у
отдельной многоатомной молекулы всегда меньше, чем число
динамических коэффициентов. Так, симметричная молекула
XY2 имеет три частоты, а уравнение (5) содержит четыре
различных коэффициента. Если бы молекула была
несимметричной XYZ, то она имела бы попрежнему три
частоты, но уже шесть разных коэффициентов. Вообще Л/-атом-
ная нелинейная молекула имеет TV—1 связей и значит
N—1 естественных координат q и 2N—5 независимых уг-
.лов между связями — координат у.
Если молекула описывается п — ЪЫ—6 естественными
координатами, то в ней возможны п1 попарных
взаимодействий каждой координаты с каждой. Однако при этом мы
два раза сосчитали все взаимодействия разных координат
типа кц&ч и один раз коэффициенты отдельных
координат. Возможное число различных коэффициентов
взаимодействия мы получим, вычитая из л2 п коэффициентов
отдельных координат и разделив полученную разность на 2:
/г2 — п п (п — 1)
2 ~~ 2 "
Общее число различных коэффициентов получим,
прибавив п коэффициентов отдельных координат
JL^lll +л==МгИіі)> (26)
тогда как наибольшее возможное число различных частот
есть
л = ЗЛЛ-6.
Благодаря симметрии молекулы, некоторые из
коэффициентов равны друг другу. Число неизвестных уменьшается.
Тем не менее, невозможно по спектру молекулы
определить все ее динамические постоянные; Поэтому часто
пренебрегают некоторыми из этих постоянных; мы поступили
таким образом при выводе (16). Это, в сущности,
незаконный прием. Для того, чтобы вычисленные динамические
коэффициенты имели реальный физический смысл,
необходимо иметь возможность оценки всех этих коэффициентов.
Здесь на помощь приходит идея аддитивности.
221
На основании всех имеющихся в нашем распоряжения
данных о строении и свойствах молекул, мы можем в
первом приближении предположить, что свойства отдельных
связей сохраняются в различных молекулах. Применительно
к разбираемому вопросу, это означает, что одни и те же
связи можно охарактеризовать постоянными значениями
динамических коэффициентов k> что в одинаковых группах
атомов все постоянные k сохраняют свое значение. Так,
можно предположить, что д^я вычисления частот всего
ряда молекул СС14, СНС13, СН.2С1^, СН3С1, СН4 достаточен
ограниченный набор постоянных, характеризующих связи
С —С1 и С —Н, углы
и взаимодействия между ними. Но целая, совокупность
молекул дает уже значительно большее число наблюдаемых
частот, превышающее число искомых динамических
постоянных. Рассматривая спектры всей совокупности молекул,
мы находим приближенные значения величин &,
сохраняющие свой физический смысл для всех молекул,
содержащих данные связи. Уточняя приближенные значения,
полученные таким образом, гмы находим отклонения от
постоянства коэффициентов k в отдельных молекулах. Такой
полуэмпирический метод определения k был разработан и
применен во многих случаях Ельяшевичем и Степановым.
Молекулы углеводородов и их производных
Задача нахождения динамических коэффициентов
облегчается в тех случаях, когда известны спектры молекул,
содержащих изотопы. Замена атома его изотопом меняет
только массу и кинематический коэффициент.
Динамические коэффициенты —междуатомные силы для молекул
с изотопными атомами те же самце. Так, во всем ряду
молекул СН4, CH3D, CHXD>, CHD3; CD4 фигурируют одни и
те же значения k. Число различных постоянных здесь равно
пяти. Это — коэффициенты ¦ связи С — Н (или С — D) ka9
С С С ' ч
угла ц/ \н (или ц/ \;о,или о/ \d) ?т, коэффициенты
взаимодействий двух соседних связей kq Qtt> двух смежных
углов &т т, связи с прилежащим углом kqV Эти постоянные
независимы друг от друга.
Таким образом, достаточно пяти" частот, чтобы найти
все k. Между тем пять молекул метанов имеют 29
различных частот. Применив для расчета постоянных 5 частот, мы
предскажем 24 остальных. В табл. 31 сведены полученные
значения k для дейтерометанов, а также вычисленные и
измеренные частоты. Совпадение оказалось очень хорошим.
222
Все эти молекулы имеют 3-5 — 6 = 9 нормальных
колебаний, но с увеличением симметрии появляется вырождение,
уменьшающее число различных частот. Аналогичным
образом определяются постоянные для этана С2Нб; наряду с
перечисленными тут еще добавляются коэффициенты связи
С — Си угла С — С — Н, а также соответствующих
взаимодействий. Таким образом удалось вычислить спектры
нормальных парафинов вплоть до пентана, изобутана и тетра-
метилметана и сравнить их с экспериментальными
данными (табл. 31).
Таблица 31
Колебательные спектры метанов
(Степанов)
Постоянные,
105 дин/см
?,7 = 5.31
k-[=QA6
kqiq2 = 0.03
6^ = 0.22
?TlT2 = -0.02
,
Молекула
сн4
CH3D
CH2Do
CHD3
CD,
Тип колебания и степень
вырождения
s(s, С-Н)
о (as, Н-С—Н)—2
v (as, С-Н)—3
bias, Н-С—Н)-3
v(s, С—Н)
n(s, С—D).
о (s, Н-С—Н)
n (as, С—Н)—2
о (as, Н-С—Н)—2
Ь (as, Н—С—D)-2
v (s, С—Н)
v(s. C-D)
о (s, Н-С-Н)
b(s, D—С—D)
b(as, H-C-D)
v(as, С—Н)
b(ast Н-С—D)
v (as, С—D)
о (as, Н—С—D)
v(s, С—Н)
ч (s, С—D)
Ь($, D-C-D)
s(as, С—D)— 2
5 (as, Н—С—D)—2
о (as, D—С—D)—2
v(s, C-D)
о (as, D-C-D)—2
v (as, C—D)—3
о (as, D—C—D)-3
\
'' ВЫЧ» CM
2914
1536
3022
'1304
2962
2196
1309
3021
1486
3143
2973
2164
1434
1032
1336
3020
10Э1
2255
1260
2990
2147
1010
2255
1300
1035
2085
1098
2255
1000
!
VH3ST CM
2914
—
30 >2
1304
2950
2200
1307
3031
1474
1156
2974
2139
1450
1033
1333
3020
1091
2255
1286
3000
2141
988
2260
1299 •
—
2085
2258
988
Оказалось возможным также разобраться в спектрах
любых других парафинов и предсказать спектры
углеводородов, которые еще" не исследовались (изононаны). Это
очень важно, в частности, и для практических применений
223
спектрального анализа смесей углеводородов — бензинов
(см. ниже, стр. 259).
Б. Степанов рассчитал также все коэффициенты k и
частоты для всех бромо- хлоро- и фторозамещенных метана
и для некоторых производных этана. В табл. 32 приведены
полученные им динамические коэффициенты связей С — Вг,
С —С1, С —F, С —Н в молекулах СНЪ ,,CHaHal, CH,HaIa,
СННаіз, СНаі4, где Hal = F, CI, Br.
Таблица 32
Молекулы
к
C-C1
lC-F
'с-н'10""5-
дин/см
сн, ..
СН3На1
СНоНаі,
СННаі,"
СНа14 .
3.0
3.05
ЗЛ
3,2
3.7
3.8
3.9
3.25
-
6.2
6.8
6.9
7.8
5.31
5.43
5.43
5.43
Сила связи растет в ряду С — Вг, С — С1, С — F, причем
«связь С — F стоит особняком: она значительно больше
отличается от С — С1 и С —Вг, чем они разнятся между собой.
Сила связи С — Н одинакова во всех молекулах,
содержащих галоид. Связь С — Hal укрепляется при увеличении
числа атомов галоида в молекуле. Значения частот
валентных симметричных колебаний v(s) для связей С — Ни
С — Hal приведены в табл. 33.
Молекула
Таблица 33
v<*)C-Hab CM_1
Вг
C1
СОс-н- см
-1
Вг
С1
сн4 ..
СНаНа1
СН«На1.
СННа13
СНаЦ .
610
576
539
265
-
732
700
668
459
1049
1078
937
904
2914
2973
2988
3021
2Л4
2967
2985
3019
—
2914
2965
2964
3062
Частоты СН во всех случаях много больше остальных
частот ^что объясняется малой массой Н и большим k) и
мало меняются при замене одного галоида другим.
ХАРАКТЕРИСТИЧЕСКИЕ ЧАСТОТЫ И КОЛЕБАНИЯ
Частота С — Н в приведенных примерах является
характеристичной. Это значит, что независимо от того, какие
атомы и связи входят в состав молекулы, помимо С — Н-
224
связи, существует такое колебание — характеристическое
колебание, в котором участвует главным образом С — Н-
связь, а остальные углы и связи изменяются мало. Такое
колебание отдельной связи — характеристическое
колебание — относительно мало взаимодействует с изменениями
других естественных координат молекулы; энергия
характеристического колебания в основном сосредоточена в
данной связи.1
Взаимодействие колебаний может быть динамическим и
кинематическим. Динамическое взаимодействие относительно
невелико в молекулах с аддитивными свойствами для таких
электронных * оболочек, которые локализованы в данной
связи. Кинематическое взаимодействие зависит от масс
атомов и их конфигурации. Бартоломе и Теллер впервые
рассмотрели условия, . необходимые для того, чтобы данное
нормальное колебание было характеристичным для
определенной связи в молекуле. Представим себе нормальный
насыщенный углеводород — цепочку С — С - связей. Будем
замещать группу СН3 на конце цепочки различными
атомами и группами, вводить в молекулу наряду с С—С и
С — Н-связями связи С — X. Спрашивается, в каких
случаях возникнет характеристическая частота С — Х-связи,
т. е. такая частота, которая не зависит от длины
углеродной цепочки? Бартоломе и Теллер, проанализировав
колебания таких систем, отвечают на этот вопрос следующим
образом. Частоты валентных колебаний самой углеродной
цепочки всегда попадают в определенный интервал
значений, ибо все связи имеют одинаковые коэффициенты k.
Этот интервал простирается от~800 до 1200 см~Ч
Колебание, а значит и частота С — X, будет характеристичной,
если собственная частота отдельно взятой С — Х-связи не
попадает в интервал С — С - частот. Механика колебаний
действительно показывает, что взаимодействия между
связанными колебаниями тем меньше, чем больше разнятся
между собой их частоты; если, напротив, частоты
колебаний отдельных степеней свободы близки, они
взаимодействуют особенно сильно вследствие явления механического
резонанса. С этим мы уже не раз встречались. Частоты
С—.Х-связей, взятые из соединений СН3 — X, в которых
С — С-связей, нет, приведены в табл. 34.
Частоты С — С1, С — Вг, С — J, С —SH,
С—Н-связей характеристичны, частоты С — О, С — N, С — F,
близкие к С — С-частотам—нехарактеристичны. Действительно,
1 Строго говоря, возможны и такие случаи, когда колебание не
характеристично; в нем участвуют и другие связи, но частоты сохраняют
характеристические значения. В этих случаях заметно меняются
интенсивности.
15 Строение молекул.
225
Таблица Si
Частоты С—X,
см~1
С1 712 . Н 2914
Вг 594 ОН 1032
J 522 NH 1037
SH 704 F. . ; 1049
опыт показывает, что во всем ряду СлН.2я + 1Х частоты
С — X с Х = С1, Br, J, SH сохраняют свое значение
независимо от п, начиная с /г = 3. По наличию в колебательном
спектре соответствующей частоты можно с большой*
точностью заключить о присутствии в молекуле данной С — X-
связи. Напротив, обнаружить связи С — N, С— О, С — F по
их колебательным частотам при наличии С — С - связей
невозможно. Благодаря взаимодействию, в любом валентном
колебании всегда участвуют иС — С-и С — О- связи и не имеет
никакого смысла приписать ту или иную частоту только
колебанию С — О-связи или С — С-связи. Частоты С — Сі
в молекулах СлН2л + 1С1 приведены в табл. 35. О причинах
удвоения С — С1-частот мы скажем дальше (стр. 234).
Таблица 35
(Кольрауш)
Молекула
сн3сі....
С2Н5С1 . . .
С3Н7С1 . . .
С4НвС1 . . .
Частоты С-
_
656
650
651
-С1,см—1
712
—
726
722
Молекула
С5НИС1 . .
свн18сі • ¦
С7Н15С1 . .
[
Частоты С—Сі, см-*
656
651
651 ¦
722
724
725
Вопрос о характеристичности групповых частот в общей
форме, конечно, не имеет смысла. Следует говорить об
относительной, а не об абсолютной характеристичности.
С —Cl-колебания характеристичны в молекулах, содержащих
наряду с С — Сі только С — С и С — Н - связи, но не
характеристичны в молекулах, содержащих связи с частотами,
близкими к С — Сі: С — Вг, С — S.. Однако, когда речь идет о
характеристических связях, имеются в виду практически
наиболее важные случаи —связи С —X в замещенных
углеводородах. Наряду с перечисленными, характеристичны
колебания связей с высокими частотами. Это либо связи,
содержащие водород (малая масса), либо кратные связи (большие k).
В табл. 36 приведены характеристические частоты
колебаний некоторых связей в многоатомных молекулах.
225
Частоты С—X,
Таблица 36
Связи
Соединения
Частоты, см
—\
Связи
Соединения
Частоты, см~1
с-с
с=с
с=с
с—о
с=о
С—N
C=N
C=N
Этан ....
Этилен . .
Ацетилен •
Метиловый
спирт . . .
Ацетон . .
Метиламин
Различные
HCN ....
992
1621
1973
1032
1710
1037
1560—1660
2094
C-S
N=0
С-Н
C-D
S—Н
N-H
О—Н
Меркаптаны
X-ONO.
х-сн3 .
CHS.D .
X-SH . .
X-NH. .
Х-ОН. .
. 650
1640 '
2700—3000
2200
2575
3310-3370
340
Очевидно, что характеристичные колебания можно с
известным правом рассматривать как колебания отдельных
связей. Здесь непосредственно выражается аддитивность
свойств молекул. Поэтому, при грубых расчетах законно
трактовать характеристические колебания связей так же,
как колебания двухатомных молекул, отвлекаясь от
остальных атомов в молекуле. Иными словами, можно грубо
оценить коэффициенты k для этих связей по формуле
k = u>*
ТПлТПч
^1 + т2
Расчеты по этой формуле сразу. показывают, что
коэффициенты k до некоторой степени характеризуют силу
химической связи. Действительно, k для одиночной, двойной и
тройной связей относятся примерно, как 1:2:3. Это очень
важное обстоятельство для применений колебательных
спектров к изучению структуры молекул.
Не только колебания отдельных связей, но и колебания
целых групп в молекуле могут обладать свойствами
характеристичности. Так, в спектрах всех производных бензола
сохраняются некоторые частоты, типичные для бензольного
кольца.
Характеристичность колебаний связей и групп имеет
основное значение для молекулярного спектрального анализа.
ЧАСТОТЫ КОЛЕБАНИИ И ЭЛЕКТРОННЫЙ РЕЗОНАНС
Частоты- характеристичны в отсутствие механического
резонанса. Если кинематические взаимодействия в молекуле
незначительны, приведенные массы и коэффициенты k
различных связей заметно отличаются друг от друга. Однако
в отсутствие механического резонанса колебаний
характеристические частоты могут меняться благодаря отклонениям
от аддитивности в свойствах молекул. Рассмотрим эти
явления с точки зрения концепции электронного резонанса.
Введение в молекулу таких связей, которые способны вступать в
* 227
электронный резонанс с исследуемой связью, меняет
динамический коэффициент этой последней, так же как меняет длину
связи, дипольный момент и т. д. Несомненно, что для того
чтобы на основании изменений характеристических частот
делать выводы об электронном резонансе, необходимо сначала
убедиться в характеристичности этих частот, в том, что их
изменения при введении тех или иных атомов или групп
объясняются именно электронным, а не механическим резонансом.
Такое исследование требует, строго говоря, полного анализа
картины колебаний и далеко не всегда доступно. Тем не менее,
уже на основании эмпирической характеристичности можно
сделать интересные выводы о строении электронной
оболочки молекулы.
Карбонильная связь
Рассмотрим частоты карбонильной связи С = 0 в
различных соединениях. Подробные расчеты для молекул
не были сделаны, но опыт показывает (табл. 37), что в
отсутствие электронного резонанса частота С = 0
действительно характеристична.
Таблица 37
Частоты vc_0 в кетонах
Молекула
н8с-со.сн8
н8с.со-с2н5
Н8С-СО-С8Н7
Н5Со" СО*СоН5
НаОСО-СД,
(H,C)sCHCHs-CO.CH2CH(CH8)s
Н8С-СО-СвН1а
¦ Среднее
Частота С=0,
см *
1708
. 1712
1710
1711
1709
1706
1710
1708
Колебания в значениях частоты не превышают
нескольких обратных сантиметров. Однако во многих замещенных
кетонах частота С=0 меняется от 1650 до 1810 см"1, как
это видно из табл. 38.
Как показали Волькенштейн и Сыркин, эти факты
действительно можно качественно объяснить при помощи кон-
228
Таблица 38
Частоты vc=:0 в молекулах ХСОХ', см"1
он
NH.
сбн5
с«н
2П5
н
ОСоНз
С!
ОН .
NH2 .
СвН3
С2Н5.
Н . .
ОС2Н5
С1 . .
1647
1651
1657
1655
1652
1664
1671
1692
1731
1647
1652
1653
1682
1696
1715
1768
1651
1654
1682
1657
1671
1696
1692
1715
1711
1722
1733
1792
1722 , 1733
1768 : 1715
1715 ' 1743
, 1772
1731
1768
1792
1772
1810
цепции электронного резонанса. Мы знаем, что увеличение
кратности связи должно вести к увеличению kt а значит,
при неизменных массах, и к возрастанию частоты.
Напротив, при понижении кратности k и v уменьшаются. Жирная
черта в таблице отделяет частоты, пониженные по
сравнению с карбонильной частотой в (С2Н8)аСО от повышенных.
Введение групп ОН, NH.2, С6Н8 понижает частоту, введение
групп ОС2Н8, С1 ее увеличивает. Легко видеть, что первые
группы создают возможность резонансных структур с
пониженной кратностью СО-связи, а вторые — с
повышенной. За изменения частот ответственны следующие структуры:
Группа ОН >С —0-
н3с/
Группа NH2
н,с/
о-
Эта- возможность еще более вероятна в молекуле мочевины,
где таких структур две (ср. стр. 144).
Группа ССН5 >С — 0~— три структуры
Н3(У
Напротив, в случаях
Группа OC«Hs
НКС, - 0-
15^2
н3с/
с = о+
сі-
Атом С1 /С ^Н 0+ .
229
Интересно сопоставить С = 0-частоты в соединениях
Н,СЧ С1СН„Ч' С1аСНч . С13СЧ
\с = о • >с = о >с = о >с=о.
н3с/ н3с/ н3с/ н3с/
1708 1725 1743 1762
Увеличение числа атомов С1 заметно увеличивает частоту,
что объясняется резонансом типа
нгс- сі—сн2- ' сьсн- сі3с-
/С = 0+ X = 0+ /С = 0+- /С = 0+
н3сх н3с/ н3с/ н3с/
I II Ш IV
Структура (I) мало вероятна. Структура (II) уже более
вероятна, ибо возможность резонанса
/Н /Н
сі - с-< сі- с<
стабилизует ее. Еще более вероятна структура (III), ибо в
ней две возможности стабилизации
/CI /CI CI-
сі — с-< сі- с( сі — сч
\н хн \н
и относительно наиболее вероятна структура (IV), которая
стабилизуется трижды:
/СГ Сі" Сі /Сі
сі — с-< сі — с\ сі —с^ сі- с< .
ЧС1 Сі Сі" ХС1
Ход частот тем самым хорошо объясняется.
Двойная связь в группе X или X' естественно вызывает
понижение СО-частоты (v = 1685 см"1)
(Н3С)2 С = СНЧ (H3CjoC+ - СНчЧ
>С = О >С — О- .
н8с/ . н3с/
Здесь мы имеем дело с обычной конъюгацией двойных
связей.
Таким образом, значение частоты карбонильной связи
может служить своего рода „индикатором на электронный
резонанс". Еще.раз повторяем, что предварительно нужно
убедиться в характеристичности частоты. Увеличение
частоты СО в альдегидах (v = 1768 см"1)
н\
>с = о
W
220
объясняется не электронным резонансом, а механическими
изменениями, вносимыми особенно легкими атомами водорода.
Рассмотрим еще некоторые молекулы, содержащие СО-
связи.
В окиси углерода СО-частота и k сильно повышены
по сравнению с карбонильной связью. Мы уже приводили
структуры, которыми это объясняется (стр. 112).
В спектре С0.2 нет частоты, близкой к 1700 см"1.
Частоты 1336 и 2350 получаются вследствие расщепления
частот двух связанных степеней свободы С = 0. Здесь
частота СО теряет свою характеристичность, так как две связи
СО очень сильно взаимодействуют. То же имеет место в
случае иона СОз2" [v(5) = 1072, v(as)=1429, 8(os) = 710], где,
наряду с механическим" взаимодействием трех одинаковых
связей, существен резонанс
о о-
II J
3 структуры
Напротив, в диметилкарбонате •
*
н5с - оч
>с = о
Н3С - (К
частота vG = 0 =1750 наблюдается.
Еще более примечательны случаи органических
карбоксильных кислот, аминокислот и их солей. Муравьиная,
уксусная, хлоруксусная и т. д. кислоты с общей формулой
ноч
>с = о
х/ .
дают характерные частоты в области 1700 см"1. Напротив,
их ионы таких частот не имеют, что объясняется резонансом
>с = о >с —о- >с+ —о-,
у/ Xх , у/
фактически уничтожающим двойную связь в СО-группе.
Особенно своеобразно поведение аминокислот (Эдсалл).
Нейтральный глицин H2N • СН2 • СООН не имеет карбонильной
частоты. Напротив, гидрохлорид глицина имеет частоту 1743,
которой соответствует интенсивная линия. Это можно объяс-
231
нить тем, что нейтральная аминокислота существует в виде
биполярного иона
H,N+ • СНл • С/
(2 структуры),
в котором обе С = 0-связи не двойные, а „полуторные".
Добавляя НС1, мы уничтожаем биполярный ион
H3N+ * СН2. COO" + НС1 -> H3N+CH2COOH + СГ.
Ион
H3N+ - СНа - С
/
о-н
^о
содержит двойную С = О -связь и обладает характерной
частотой.
Другие случаи
Мы подробно рассказали о частотах СО-групп в
различных молекулах, так как они представляют особенно
яркий пример влияния электронного резонанса. Изменения
частот.вследствие этого влияния наблюдаются также и во
многих других случаях, в частности при наличии
гс-связей. Так, в производных этилена (табл. 39) зачастую
наблюдаются пониженные значения характеристической частоты
С = С-связи. Если при замене водорода на метил или
дейтерий изменение объясняется механическими причинами, то
в других случаях главную роль играет, очевидно,
электронный резонанс.
Молекула
Н2С=СН»
н2с=снЬ
Н2С=СНСН#
Н2С=СНОС2Н5
Н2С=СНС0Н5
Таблица 39
vc=C см"1
1621
1609
1647
1635
1631
Молекула
H.C=CH.CN
Н2С=СНС1
Н2С=СНВг
Н2С=СШ
vc-c см~1
1608
1608
1598
1581
Понижение связано со структурами типа
Н2С— "СН = С1+-
Частоты этиленовых соединений подробно исследованы в
работе Прилежаевой, Сыркина и Волькенштейна. Они
установили, что органические радикалы можно расположить в
232
следующий ряд по степени их растущего влияния
(понижающего) на частоту двойной связи в монозамещенных:
К«Иф<СООН, СООС9НВ, ОС,Н8<С6Н5<СОН<
<COCl<CN<Br<C = CH.
Упомянем в связи с этим, что частота двойной связи сильно
понижена в комплексах, содержащих ион серебра. Так,
чистый циклогексен имеет vc«c =1653 см"1, в комплексе
1584, цис-2-бутен соответственно 1660 и 1598 и т. д.
Это, очевидно, объясняется резонансом
\с-с/
Ag+
XAg
\
>с+
/
Ag
С<
Большой материал по частотам тройных С = N- и С = С-
связей также поддается убедительному истолкованию
при помощи концепции электронного резонанса.
Частоты циановых соединений весьма разнообразны и
характерны, как это видно из табл. 40. В галоидцианах внутри-
ионизованная структура становится вероятнее при
уменьшении электроотрицательности в ряду CI, Br, J.
Таблица 40
Соединения
Формула
*C-N' CM
-1
Алифатические нитрилы
Ароматические нитрилы
Цианамид
Горчичные масла
Изонитрилы
Тиоцианиды
Ион циана
Ион родана
Цианистая ртуть
Цианистое серебро
Хлорциан
Бромциаи ,
Иодциан ,
R-CN
Ar-CN
HoN-CN
R-CNS'
¦R-NC
R-SCN
CN~
SCN-
Hg(CN)2
AgCN
C1CN
BrCN
JCN
R-C—N
CflH5+=C=N-
H,N4-=C=N-
R-N-—C==S+
R—N=C, R~N-^C-
R-S+=C=N~
C=N-, C-==N
S-—C=N, S=C=N-
/CN ,CN
"Кем Hg+/C=N-
/CN
Ag-C—N Ag+ C=N"
Ag+ c-=N
Hal—C=N
Hal+=C=N-
2250
2227
2233
2100-2180
2165
2152
2080
2060
2192
2178
2201
2187
2158
233
Другие свойства молекул полностью подтверждают
изложенное. Уменьшение характеристической частоты,
уменьшение коэффициента к, а значит кратности связи, всегда
сопровождается увеличением междуатомного расстояния и
отклонениями дипольного момента от аддитивности. Однако
значения колебательных частот во многих случаях более
чувствительны к электронному резонансу, чем, например,
междуатомные расстояния.
Исследование химической связи — ее неаддитивных,
„электронно-резонансных" свойств—методом колебательных
спектров обещает очень многое. Конечно, рассмотрение
численных значений частот есть лишь начальная стадия этой
работы, ибо смысл физических констант имеют не частоты,
а динамические коэффициенты связей и взаимодействий.
Для их нахождения необходимы последовательные расчеты
колебательных спектров, подобные тем, о которых мы
говорили выше.
КОЛЕБАТЕЛЬНЫЙ СПЕКТР И ИЗОМЕРИЯ МОЛЕКУЛ
Колебательный спектр в целом и характеристические
колебания в частности определяются структурой молекулы.
Поэтому почти все виды изомерии находят свое выражение
в колебательных спектрах. Нет разницы, собственно говоря,
только между спектрами оптических антиподов,
содержащих, как мы видели, одни и те же, одинаковым образом
по отношению друг к другу расположенные связи.
Изомерия положения, конечно, решительным образом сказывается
на спектрах. Это весьма существенно для молекулярного
спектрального анализа (см. стр. 258). Не менее важны и
интересны случаи таутомерии. Так, кето- иэнольная форма ацето-
уксусного эфира (см. стр. 137) дают различные частоты,
соответственно карбонильной (С = О) и этиленовой (С = С) связи.
Измеряя относительную интенсивность этих линий в раманов-
ских спектрах смесей таутомеров, Кольрауш смог
проследить за кинетикой таутомерного превращения.
Поворотная изомерия
Особенного внимания заслуживают рамановские и
инфракрасные спектры поворотных изомеров, ибо сама эта
изомерия была впервые установлена Кольраушем именно
спектральными методами (ср. стр. 86).
Поворотные изомеры отличаются друг от друга только
расположением атомов. Поэтому динамические
коэффициенты колебаний в них следует принять одинаковыми-
Напротив, кинематические коэффициенты могут заметно
разниться и обусловить различия в частотах тех колебаний,
234
которые этими кинематическими коэффициентами
определяются. Так, характеристические частоты С—Hal в галои-
допроизводных парафинов имеют различные значения для
разных поворотных изомеров. Более низкая частота С —С1,
приведенная в табл. 35, принадлежит трансдзомеру, более
высокая —изогнутому (рис. 124). Так как хлористый метил
и этил не могут иметь поворотных изомеров, в их
спектрах представлена лишь одна частота С —С1. Число
примеров такого рода очень велико. Эмпирические выводы о
поворотной изомерии полностью подтверждаются расчетом.
Ельяшевич и Степанов вычислили частоты колебаний транс-
и изогнутого изомера н- бутана в хорошем согласии с
опытом. Шесть частот н-бутана заметно отличаются у его
поворотных изомеров.
Hal
R
Транс Изогнутый.
Рис. 124. Поворотные изомеры н-галоидопарафшюв.
Важно отметить, что колебательные спектры дают в
настоящее время наиболее эффективный способ определения
константы равновесия смеси поворотных изомеров. По
относительным интенсивностям соответствующих
спектральных линий и их изменениям с температурой мы можем
судить об относительном содержании поворотных изомеров
в смеси и его изменении с температурой. Так, Мицусима
с сотрудниками установил, что при охлаждений н- бутана
часть линий в его спектре ослабляется и исчезает,
„вымораживается". Это — линии менее устойчивого, в данном случае
изогнутого, изомера.
В молекулах с заторможенным внутренним вращением
имеются крутильные колебания, сопровождаемые
изменением одной лишь естественной координаты — угла поворота
тех или иных групп в молекуле вокруг единичной связи.
При высоких температурах крутильные колебания могут
перейти в свободное вращательное движение. Крутильные
колебания непосредственно в колебательных спектрах часто
не наблюдаются, так как они почти не меняют ни дипольного
235
момента, ни поляризуемости. Частоты крутильных
колебаний малы, порядка сотен-десятков см"1. Они находятся
косвенным путем из.термических свойств молекул (см. ниже,
стр. 248).
• КОЛЕБАТЕЛЬНЫЙ СПЕКТР И МЕЖДУМОЛЕКУЛЯРНОЕ
ВЗАИМОДЕЙСТВИЕ
Значения частот и вид (ширина, контур) колебательных
спектральных линий и полос могут изменяться под
действием сил междумолекулярного взаимодействия. При
переходе от газа к жидкости частоты, как правило, понижаются,
а ширина полос возрастает. В табл. 41 даны частоты
некоторых рамановских линий в газе и жидкости и
относительное понижение частоты (в %)¦
Таблица 41
Вещество
V, СМ
—1
газ
жидкость
Дч
:-. %
V-(D)
Н* . .
'Nl . .
о:..
РС13 .
AsCl3
H.,S .
HCl .
НВг .
HJ. .
4162
2331
1555
523
422
2615
2886
2558
' 2233
4149
2326
1550
511
405
2576
2770
2466
2165
0.3
0.2
0.3
2.3
4.0
1.5
4.0
3.6
3.1
о
о
о
1.1
2.1
0.93
1.04
0.79
0.38
Теория этого явления рассматривалась Бухгеймом, Кирк-
вудом и Волькенштейном. Последний показал, что заметное
понижение частот может возникнуть лишь при действии
ориентационных, но не индукционных или дисперсионных
междумолекулярных сил (ср. стр. 147). Причиной
понижения частоты .служит то обстоятельство, что колебания ядер
во взаимодействующих молекулах не могут рассматриваться
как независимые, но как связанные ван-дер-ваальсовыми
силами, что приводит к расщеплению и, в частности, к
понижению частот.
Под действием междумолекулярных сил молекула
может изменить свою форму, свою симметрию. Так,
молекула С S2 в газе линейная, очевидно, изгибается в жидкости,
ибо в рамановском спектре жидкого сероуглерода
наблюдаются частоту колебаний, запрещенных правилами отбора
для линейных молекул с центром симметрии (ср. стр. 212).
Изменение симметрии может привести к снятию
вырождения—увеличению числа различных частот. Этим, очевидно,
объясняется тот факт, что вместо одной линии в рама-
236
новском спектре газообразного аммиака в спектрах его
растворов и в спектре жидкого аммиака наблюдаются три
линии (табл. 42).
Таблица 42
Частоты аммиака NH3, см
-і
Газ —
Жидкость і 3210
Водный раствор
Комплексное соединение
Ag(NH3)2Cl
3225
3228
3336 ! —
3300 I 3380
3310 I 3395
3308
3396
Влияние растворителя на значение частоты колебаний
может быть значительным, особенно в дипольных
растворителях. Дело в том, что междумолекулярное
взаимодействие может изменять даже природу связи в молекуле —
степень ее ионности. Это находит' свое выражение, конечно,
не только в колебательных спектрах, но и в значениях
дипольных моментов и в t других физических свойствах
молекул. Так, в газе НС1 имеет ц.=1.03Д в растворах
в СС14 у-=1.32.0 и в бензоле у. =1.261). Частоты связи
C = N цианистой ртути следующие: в метиловом спирте 2204,
в воде 2194, в пиридине 2180 и в аммиаке 2164.
Водородная связь
Особенно велико взаимодействие между молекулами,
содержащими, с одной стороны, гидроксильный или амин-
ный водород, а с другой — кислород, азот, фтор, хлор.
В этих случаях силы взаимодействия по порядку величины
приближаются к силам химической связи, и молекулы
находятся в ассоциированном, тесно связанном друг с
другом состоянии. Так, молекулы муравьиной, уксусной
и других кислот существуют в виде димеров — попарно
соединенных молекул. На разрыв димера нужно затратить около
13—14 кг кал/моль — величину, значительно превосходящую
энергию обычных ван-дер-ваальсовых сил. Изобразим димер
муравьиной кислоты схематически следующим образом:
н-с Ы с-и
No —н —о
Пунктир обозначает междумолекулярную связь через
гидроксильные водороды. Расстояние О —Н---0 имеет
определенную величину, характерную для водородной связи.
237.
Вещества, ассоциированные благодаря водородным
связям, ведут себя аномально во всех отношениях.
Соответствующие жидкости имеют особенно большие
температурные интервалы между точками плавления и точками
кипения, большие теплоты испарения, большие диэлектрические
постоянные. Все эти свойства объясняются сильными
междумолекулярными связями, возможностью образования
длинных цепочек из ассоциированных молекул, разорванных
и замкнутых. Например, синильная кислота, отдельные
молекулы которой имеют дипольный момент 3D, в жидком
состоянии обладает диэлектрической постоянной 118,
что соответствует, если исключить электронную
поляризации», дипольному моменту около 9 D. Это объясняется
полимеризацией молекул синильной кислоты, образующих
цепочки
....Н — C = N...H — C = N...H — C = N...
со средней длиной в три молекулы. Вода и лед, как
показали Берналь и
Фаулер, представляют
собой как бы одну
молекулу, ибо все
молекулы Н20 в них
связаны (рис. 125). В этом
смысле справедливо
выражение Лэнгмюра,
писавшего, что весь
океан есть одна
молекула, а ловля рыбы в
нем — процесс
диссоциации.
Водородная связь
имеет энергию порядка
ч—7 кг кал/моль. Она,
очевидно, не может
быть сведена к ван-дер-ваальсовым силам, ибо носит вполне
специфический, полухимический характер. Наряду с
водородом такого рода связи способен создавать лишь дейтерий.
Природа водородной связи в настоящее время еще не ясна.
Можно утверждать лишь, что эти междумолекулярные связи
стабилизуются электронным резонансом, подобно
внутримолекулярным связям: В приведенных примерах существенны
возможности следующих структур:
Рис. 125. Структура воды (Берналь и
Фаулер).
4-
'О
н_с/о-н.. о\
'чо7
..H-V
238
С —Н И
н-с<°
о
Н 0\
Й <*>
с — н,
X0 H 0X
— H
со значительным электростатическим притяжением, которое
укрепляет связь, так же как и, сама возможность
нескольких резонансных структур. Для водородной связи, очевидно,
существенно также, что водород имеет малый атомный
объем и малую массу.
Водородная связь и колебательный спектр
Вероятно наиболее ярки и типичны проявления
водородной связи в колебательных спектрах. Образование
водородной связи всегда сопровождается очень значительным
понижением колебательной частоты смежной химической связи
и большим расширением полосы. В рамановских спектрах
воды, спиртов и т. д. наблюдаются широкие, до
нескольких' сот см—1 шириною, полосы ОН-связей, в то время
как для паров этих веществ получаются вполне резкие
линии ОН-связи с шириною лишь в несколько см-1.
Положение и ширина полосы в сильной степени зависят
от температуры, от растворителя. Г. С. Ландсберг и его
сотрудники— Ухолин, Малышев, Яковлев —изучили раманов-
ские спектры воды и спиртов в весьма широком интервале
температур, давлений и концентраций в растворах, начиная
от температуры жидкого- воздуха вплоть до температуры
выше критической и давления в несколько сот атмосфер.
Некоторые результаты этих исследований приведены
в табл. 43.
Таблица 43
Гидроксильная полоса СН3ОН
(Ландсберг, Ухолин, Яковлев)
Г, °К
Положение
максимума,
см~1
Смещение,
¦1
см
Ширина,
см *
Г, °К
Положение
максимума,
см"*
Смещение,
см~*
Ширина,
см~^
83
203
293
323
3280
3300
3402
3427
390
370
268
243
270
325
350
373
413
463
533
3473
3507
3535
3670
197
163
135
—
350
Линия
На рис. 126 представлена 'фотография рамановского
спектра СН3ОН при ~500°. Стрелками показаны полоса и
линия связи. При высоких температурах они наблюдаются
одновременно.
Объяснение этих явлений недавно было дано
Б. И. Степановым. Не входя в исследование природы водо-
239
родной связи, он рассмотрел вопрос о колебаниях двух
связей О — Н---0, соединяемых водородом. Одна из этих
связей — гидроксильная — имеет большую энергию Don =
100 кг кал/моль, другая — водородная связь — много слабее,
Z)o4,,h^6 кг кал/моль. Свободной ОН-связи соответствует
линия 3670 см-1, группе 0---Н соответствует полоса. При
колебаниях О—Н-связи энергия этих колебаний может
перейти в водородную связь, ибо обе связи соединены
легким атомом водорода, являющимся причиной особенно
большого коэффициента кинематического взаимодействия;
динамическое их взаимодействие также значительно. Этой
энергии колебаний достаточно для того, чтобы связь 0---Н
разорвалась. Таким образом, осуществляется своеобразный
процесс предиссоциации (см. стр. 203), как всегда приводящий
к расширению, размытию спектральных линий. Все
количественные закономерности, установленные в работах
-J
Рахимовская
полоса
он.
Рйлюмоккая
линия
ОН
Романовская
лини*
СН.
Рис. 12). Рамановский спектр метилового спирта при высокой температуре
(Малышев и Ландсберг).
по рамановским спектрам спиртов, получают в свете этих
представлений удовлетворительное истолкование.
Возможность изолированного рассмотрения связей О — Н---0
определяется тем, что, как показывают опыт и расчеты,
колебание О —Н в спиртах и других гидроксильных
соединениях характеристично.
Водородная связь может осуществиться не только между
различными молекулами, но и внутри одной молекулы.
Такая внутримолекулярная водородная связь имеет место,
например, в гваяколе
/\/°\
н
\
сн,
Если группы ОН и ОСН3 находятся не в орто-, а в мета- или
пара-положении, водородная связь не образуется, так как
расстояние между О и Н становится слишком большим.
В спектре гваякола частота ОН несколько понижена, и по-
240
лоса расширяется (Батуев), но значительно меньше, чем
в случае междумолекулярной связи. Это понятно с точки
зрения теории Степанова — связь 0---Н здесь не может
диссоциировать.
Интересны спектры оксониевых соединений. Молекула
диметилового эфира вступает в прочное
междумолекулярное соединение с молекулами НС1, НВг и т. д. Ранее это
объяснялось возможностью существования
четырехвалентного кислорода
ttsC4 iv /Н
Н3(У ХС1.
Сейчас мы знаем, ' что такое написание не более
осмысленно, чем изображение пятивалентного азота в хлористом
аммонии (ср. стр. 39).- Как показали Гантмахер, Волькенштейн
и Сыркин на основании изучения рамановских спектров
(СН3)2О.НС1 в широком интервале температур, между
кислородом эфира и водородом НС1 создается водородная
связь, стабилизуемая резонансом
н,с>°-н-сі н3с>°-Н а
Частоты междумолекулярных колебаний
В тех случаях, когда потенциальная энергия
междумолекулярного взаимодействия обладает достаточно резко
выраженным минимумом (ср, рис. 35), возможны колебания
молекул по отношению друг к другу около этого минимума.
Так как потенциальная кривая в этом случае очень
пологая и энергия диссоциации весьма мала,
междумолекулярные частоты невелики, имеют порядок величины
вращательных частот — десятков и сотен см"1. Вода имеет частоты
.60, 176, 500, 700 „см-1, система диметилэфир-хлористый
водород 493 см"1 (деформационное колебание группы
СГ . .. Н — С1). Здесь особенно существенны исследования
Е. Ф. Гросса и его сотрудников.
Е. Ф. Гросс изучал спектры рассеяния жидкостей и
кристаллов (молекулярные решетки) целого ряда веществ. Во
многих жидкостях несмещенная рэлеевская линия рассеяния
имеет интенсивный фон — так называемые „крылья* линии
(рис. 127). Индийские ученые, сотрудники Рамана, считали,
что эти „крылья" вызываются заторможенным вращением
молекул в жидкости. Гросс и Вукс показали, что в
действительности „крылья" представляют спектр низкочастотных
.междумолекулярных колебаний. При замораживании жидко-
16 Строение молекул 241
сти в спектре полученного кристалла на месте „крыльев"
удалось наблюдать ряд линий, характерных для данного
вещества (рис. 128). В кристаллах междумолекулярные колеба-
«8 н0 Hfl Hg
Рис. 127. Крылья рэлеевской линии, рассеянной
жидким нафталином (Гросс).
ния имеют вполне определённые частоты, в жидкости эти
частоты размываются, так как нарушается упорядоченность
решетки. Нафталин имеет частоты 45, 73, 109, 124 см"1,
1
11
і
0
I J
\
і
р
і
Р Н
1
9 р
Р Р
¦,
Р Нд
РР F
4
н
3
р
РР Р р
Рис. 128. Низкочастотный рамановский спектр
кристаллического нафталина (Гросс).
бензол 62, 104 см"1, парадибромбензол 20, 33, 93 см"1
Изменение кристаллической модификации заметно
сказывается на низкочастотном спектре.
АНГАРМОНИЧНОСТЬ КОЛЕБАНИЙ
Колебания двухатомных молекул в действительности
ангармоничны, и это приводит к появлению обертонов в их
спектрах. То же самое относится, конечно, и к колебаниям
многоатомных молекул. Здесь, наряду с обертонами одного
колебания, возможны еще и комбинационные частоты —
суммы и разности частот различных нормальных колебаний.
242
Четырехбромистоэ олово SnBr4 имеет основные частоты
Vj = 64, va = 88, v3 = 220, v4 = 279 см"1. Наряду с ними,
в римановском спектре удалось наблюдать (Ландсберг
и Малышев) следующие обертоны и комбинации: 2vt = 132,
2v, = 176, v4 — v=193, v2+v3 = 305, v2-f v4 = 370, 2v, = 438,
v3 + v4 = 479 см *. Гораздо легче наблюдаются
комбинационные частоты в инфракрасных спектрах. Поглощая
комбинационную частоту, например, v2 -f-v3, молекула переходит на
колебательный уровень, соответствующий сумме энергий &v.2-|-Av3.
Можно показать, что такой процесс возможен лишь
вследствие ангармоничности взаимодействующих друг с другом
колебаний.
Ферми-резонанс
Особый интерес представляют те случаи, в которых
обертон или комбинация каких-либо частот случайно совпадает
по численному значению с частотой другого нормального
колебания. Это — так называемый Ферми-резонанс. В
сложной молекуле такие случаи всегда могут встретиться.
Наиболее хорошо изучен резонанс в С02 (Ферми) и СС14
(Гориути). Линейная молекула С02 имеет основные частоты
Vj = 1330, v2 = 667.5 и v3=2350 см-1. Первая принадлежит
ч^(5)-колебанию, вторая S(as), третья v(7zs). В рамановском
спектре должна наблюдаться лишь частота v(s)
полносимметричного колебания. В действительности наблюдаются
две линии 1286 и 1388 см'1 (и еще две более слабых 1265
и 1408 см"1). Согласно Ферми, это объясняется совпадением
уровней vt я^ 2v2. Здесь имеет место резонанс, вырождение
частот— случайное, в отличие от необходимого, определяемого
симметрией молекулы (стр. 210). Так как колебания с частотами
Vj и 2v2 ангармоничны и взаимодействуют, вырождение
снимается, и частота V!^2v2 расщепляется на две (рис. 129).
Аналогичное явление наблюдается в спектре СС14 (см. табл. 30),
где частота 775 см"1 расщепляется на две: 760 и 790
вследствие резонанса с комбинацией 459 -f- 313. Ферми-резонанс
имеет место во всех молекулах, содержащих СН2 и СН3-груп-
пы, ибо удвоенная частота деформационного колебания угла
НСН 1450-2 близка к частоте валентного колебания ~
2900 см"1. Здесь всегда возникает расщепление.
Л. И. Мандельштам пояснил сущность Фермигрезонанса
в молекуле С0.2 весьма изящной механической аналогией.
Представим себе маятник с растяжимой нитью — пружиной-
или резинкой (рис. 130). Его груз может совершать два
движения, имеет две колебательные степени свободы —
обычное качание и колебание вдоль нити подвеса.
Колебания эти воздействуют друг на друга, колебание нити
подвеса периодически меняет период качаний. Колебание
* 243
в одной степени свободы меняет параметр другого
колебания. Анализ этой механической картины показывает, что
наибольшее взаимодействие, приводящее к расщеплению
частот — параметрический резонанс,—происходит здесь при
v. = 1336
2\= '338
vz=!3#3
>л=іж
Vp = 668 с.**
-t
v/s)
БЫ
Расщеплете
Рис. 129. Схема колебательных уровней молекулы С02 (Ферми-
резонанс).
соотношении частот колебания и качания Vj = 2v2. В молекуле
С02 имеет место подобное же соотношение, как мы.только что
видели; в ней частота поперечного деформационного
колебания („качания") вдвое меньше
частоты продольного, валентного
колебания.
ЧАСТОТЫ КОЛЕБАНИЙ И
ТЕРМИЧЕСКИЕ СВОЙСТВА МОЛЕКУЛ
Таким образом мы
рассмотрели, чем определяются частоты
колебаний и как они проявляются
в спектрах. Однако колебания
молекул сильно сказываются не
только на оптических, но и на
термических и кинетических
свойствах вещества. Значительная
часть энергии, которой обладает молекула, есть энергия ее
колебаний, В процессах соударений между молекулами, при
химических реакциях и т. д. играет существенную роль
обмен колебательной энергии между молекулами,
накопление энергии в какой-либо из связей молекулы и
последующий разрыв связи." Эти весьма сложные явления еще
недостаточно, изучены в настоящее время. Напротив, хорошо
разработана теория термодинамических свойств многоатом-
244
Рис. 130. Параметрический
резонанс.
ных молекул. Зная частоты колебаний и строение
молекулы, можно вычислить теплоемкость молекул и другие
их термодинамические характеристики (энтропии,
внутренние энергии и т. д.). Очевидно, что колебательный спектр
является основным источником сведений о колебательных
частотах, необходимых для такого рода расчетов.
Внутренняя энергия и теплоемкость молекул
Молекула может запасти при данной температуре тем
больше энергии, чем больше внутренних степеней свободы
у молекулы и чем меньше кванты, нужные для перевода
молекулы из невозбужденного на возбужденные уровни.
Кванты, необходимые для возбуждения электронных
переходов в молекуле, всегда слишком велики, чтобы эти переходы'
могли осуществиться при обычных температурах. Молекула
находится в основном электронном состоянии и в этих
условиях может иметь колебательную внутреннюю энергию.
Согласно основному положению классической статистики,
мы с тем большей вероятностью найдем молекулу на
данном колебательном уровне, чем меньше энергия, нужная
для его возбуждения.
Первый возбужденный колебательный уровень
двухатомной молекулы расположен на расстоянии Ъ над
нулевым уровнем (см. стр. 179). Если в невозбужд'ённом состоянии
находится М0 молекул, то число молекул, обладающих
колебательной энергией, на величину hv более высокой, равно
Nt = N0e~^ ' . ¦ (27)
Но каждый гармонический осциллятор может иметь энергию,
на Av, 2Ь9 3Av, . . . более высокую, чем энергия нулевого
уровня, равная Av/2. Общее число молекул, находящихся во
всех этих состояниях, равно
х (28)
М=М0 \е кТ+е кТ+е кТ + . . ..
Относительная энергия молекулы- на л-ом уровне равна
en = nkv. (29)
Среднюю энергию молекулы мы получим, умножив энергии
молекул во всех состояниях на их число в этих состояниях
и разделив на общее число молекул
со
2 Nnnh»
е = —
N
п=1
со hi
N0 2 nhsc кТ
п = 1 _
со nhs
N0 2 е кТ
to
(30)
245
Для многоатомной молекулы, имеющей 3/V—6 степеней
свободы и 3N—б колебательных частот, выражение (30)
надо просуммировать по всем 3N—6 частотам
3N — 5 fcti /Oi\
. екТ-1
Внутренняя колебательная энергия одного моля газа будет
в Na (число Авогадро) раз больше.
Теплоемкость газа (ту энергию, которую нужно затратить
на нагрев его на один градус при постоянном объеме) мы
получим, продифференцировав выражение энергии по
температуре
C, = d^- = NAk46KML • (32)
(e^-l)9
На рис; 131 показана зависимость теплоемкости от
температуры. При высоких температурах колебательная
теплоемкость равна (ЗЛ/ — 6)Л/д&==
= (3N — 6)/?, (R = Nak —
постоянная Клапейрона), а при
абсолютном нуле обращается
в нуль.
Колебательные степени
свободы как бы вымораживаются
у при понижении температуры,
Рис. 131. Зависимость теплоем- и это происходит тем легче,
кости от температуры. чем выше частоты V,-
нормальных колебаний. Другие виды
энергии молекулы — энергия поступательного движения,
вращения, электронных переходов (последние при обычных
температурах несущественны) — также поддаются учету.
Мы видим, что, зная частоты колебаний, можно вычислить
внутренние энергии и теплоемкость молекул, а также
связанные с ними основные термодинамические функции
вещества— энтропии и свободные энергии. Эти функции
существенны для определения физико-химических свойств
молекул, в частности констант равновесия при химических
реакциях— отношения количеств веществ, получающихся при
реакция, к количествам веществ, вступающих в реакцию при
данной температуре. Приведем несколько примеров. В табл.
44 сопоставлены вычисленные на основании
спектроскопических данных и найденные экспериментально теплоемкости
нескольких. веществ.
24&
T,
Вещество
i б л и ц а 44
! с
v"» ВЫЧ.
СО. (газ)
SO." (газ)
PCf3117° С
AsCl3 217
SiCl4 97
ТіС14 167
SnCl4 157 - .
0.159
0.117
0.118
0.095
0.123
0.108
0.084
^*і) набл.
0 154
0.117
0 119
0.098
0.117
0.125
0 087
Такое же хорошее совпадение получается и в других
случаях, в частности для молекул галоидоэтанов, некоторые
из которых имеют практическое значение в качестве тепло-
уносителей именно благодаря своей большой теплоемкости,
наряду с низкой температурой кипения. Спектры таких
веществ — фреонов (например, CF2C1 — CF2C1) — подробно
изучались Глоклером. Расхождения между опытными и
вычисленными значениями теплоемкости, не превышающие 1—
3°/0, следует приписать упрощениям, применяемым в расчете,
пренебрегающем взаимодействием колебаний и вращений и
ангармоничностью колебаний.
В качестве примера определения постоянной равновесия
приведем реакцию каталитической гидрогенизации этилена
СоН4 -|- Н2 ч^ С2Н6.
Опыт дает константу равновесия при 450°, равную 5.16 -10"4
расчет — 5.19- 10 "4. Зная константу равновесия, можно
вычислить теплоту, выделяемую или поглощаемую при
реакции.
Очень большой интзрес представляют молекулы с
заторможенным вращением, так же как и обладающие
поворотными изомерами. И те и другие имеют крутильные
колебания, которые, как правило, не наблюдаются в спектрах.
Однако на эти колебания также затрачивается
определенная энергия; они делают свой взнос в величину теплоемкости,
энтропии и т. д. Сравнивая данные расчета, в котором
применены значения колебательных частот и данные
калориметрических измерений, мы можем найти частоты
крутильных колебаний и потенциальную энергию внутреннего
вращения молекулы (ср. стр. 85), Приведем, не вдаваясь в
подробности, пример такого определения потенциала
внутреннего вращения. Энтропия, т. е. запас внутренней энергии
вещества при данной температуре, деленный на эту
температуру, находится путем измерений теплоемкости и расчета.
247
Для метилмеркаптана CH3SH, при 279.12° К и одной
атмосфере, имеем
Энтропия поступательного и вращательного
движения 57.545 кал/моль град
Колебательная энтропия 0.592
Всего 58.137 нь 0.10 ; '
Опытные данные 60.16 ±0.10 „
Разность, связанная с внутренним вращением 2.02
При свободном вращении 2.50 я
При торможении вращения, соответствующем
потенциальной энергии в 1460 кал/моль . . 2.02
Для расчета вращательной доли теплоемкости, энтропии
и т. д. необходимо знать моменты инерции молекулы, т. е.
расположение масс и расстояния между ними. Для
сложных молекул эти данные получаются главным образом
электронографическим путем (стр. 73). Таким образом,
современная статистическая теория термических свойств
молекул, а значит и их химических свойств, широко
применяет результаты исследования строения молекул
разнообразными физическими методами.
Все потенциалы торможения внутреннего вращения для
ряда молекул, начиная с этана, определялись таким же
способом*, как и в приведенном примере. Подробная рецептура
расчетов для этих случаев была разработана Питцером.
В тех случаях, когда вещество представляет собой смесь
поворотных изомеров, необходимо, исходя из
колебательных частот каждого из изомеров, вычислить их
термодинамические функции, а из отношения интенсивностей
колебательных спектральных линий определить константу
равновесия смеси. Лишь зная эти величины, возможно найти
величины потенциала внутреннего вращения — частоты
крутильных колебаний, которые у разных изомеров разные.
Применяя потенциал вида, изображенного на рис. 52,
и данные по колебательным спектрам, Волькенштейн
рассчитал энтропию н-бутана, представляющего смесь транс-
и изогнутого изомеров (на рис. 124 следует заменить R
и Hal на СН3).
Колебательная энергия
Каждая молекула, содержащая более одного атома
и поэтому имеющая колебательные степени свободы,
обладает нулевой энергией колебаний, лишиться которой она
не может и при абсолютном нуле. При температурах,
отличных от нуля, колебательная энергия повышается.
Колебательная энергия играет важную роль в свойствах молекул.
Мы уже указывали, что все различие между свойствами
соединений водорода и дейтерия сводится к различию нуле-
248
вых энергий. Нулевые энергии могут быть весьма
значительны, они тем больше, чем больше атомов в молекуле,
чем больше частот имеет молекула. В самом деле, нулевая
энергия многоатомной молекулы равна сумме нулевых
энергий всех ее нормальных колебаний
3N — 6 l4J 3N-6
?0= Б ^ = 1.423 ? (v?см"0кал/моль. (33)
Для вычисления е0> а также полной энергии колебаний
нужно, следовательно, знать частоты всех нормальных
колебаний молекулы. Колебательная энергия, несомненно,
важнейший фактор при решении вопросов об аддитивности
энергий связей и об отклонениях от нее — электронном
резонансе. Разрывая молекулу на отдельные связи, мы
уничтожаем целый, ряд возможностей внутримолекулярных
колебаний, в частности все деформационные колебания.
При этом колебательная энергия молекулы, конечно, резко
уменьшается, в сложных молекулах — на десятки кал/моль.
Приписывая отдельной связи некоторую энергию и пользуясь
этой величиной как аддитивной постоянной, мы приписываем
связи некоторую долю общей колебательной энергии
молекулы. Сравнивая сумму энергий отдельных связей,
найденных из различных молекул, с энергией изучаемой молекулы
= и считая отклонения от аддитивности вызванными
электронным резонансом, мы всегда молча предполагаем, что либо
колебательные энергии, "доли которых распределены по
связям, аддитивны, либо их неаддитивность также
определяется электронным резонансом. Последнее означает, что
различие в колебательных энергиях суммы связей и
молекулы сводится к различию в динамических коэффициентах,
определяющих соответствующие частоты. Мы видели, что
для некоторых характеристических частот это, вероятно,
действительно имеет место — частоты, а значит и
колебательные энергии изменяются вследствие электронного
резонанса. Однако возможны и такие отклонения от
аддитивности колебательных энергий, которые определяются
кинематическими коэффициентами — изменениями конфигурации
молекулы. Выше (стр. 101) мы привели разности энергии
изомеров предельных углеводородов. На основании расчетов частот
колебаний, сделанных Ельяшевичем и Степановым, можно
вычислить нулевые энергии этих молекул и их разность
(табл. 45, в кг кал/моль).
Динамические коэффициенты в этих случаях одинаковы,
и различие сводится к изменению кинематических
коэффициентов. Следовательно, неаддитивность здесь не может
быть полностью объяснена электронным резонансом. Для
того чтобы получить истинную энергию электронного резо-
249
Таблица 45
Молекула
Нулевая
энергия
Разность
нулевых энергий
Н-бутан (транс)
Изобутан
Н-пентан
Тетраметилметан
нанса, необходимо во всех случаях исключать ту долю
неаддитивности, которая приходится на „кинематическую"
разность нулевых энергий, а также, разумеется, и энергий
возбужденных колебательных состояний, поскольку при
обычных температурах заметно возбуждены колебания
с малыми частотами. Для этого нужно знать все
динамические и кинематические коэффициенты колебаний
сравниваемых молекул и все их частоты. До настоящего времени
соответствующие расчеты не проводились.
ИНТЕНСИВНОСТИ И ПОЛЯРИЗАЦИИ
В КОЛЕБАТЕЛЬНЫХ СПЕКТРАХ
Из колебательных спектров мы можем определить не
только частоты колебаний, но также и интенсивности
спектральных линий. Эти величины в случае двухатомной
молекулы непосредственно характеризуют природу химической
связи — степень ее ионности. Спрашивается, каким образом
интерпретировать данные по интенсивностям (а также
поляризации) в спектрах многоатомных молекул, как связать эти
данные со свойствами' связей, имеющихся в молекуле. Для
этого необходимо прежде всего установить зависимость
интенсивности колебательных спектральных линий от формы
колебания, а уже затем найти значения эффективных
зарядов ji' и изменений поляризуемости^ расстоянием а' (ср. стр.194)
для отдельных связей в многоатомной молекуле.
Соответствующая теория -интенсивностей и поляризаций —
электрооптических свойств колебаний — была предложена в
работах М. Волькенштейна и М. Ельяшевича.
Интенсивность инфракрасной колебательной полосы
зависит от величины изменения электрического дипольного
момента, интенсивность рамановской линии—от величины;
изменения поляризуемости при данном нормальном колеба-|
нии. В каждом нормальном колебании участвуют, вообще
говоря, все атомы и связи молекулы, изменяются все
междуатомные расстояния. Необходимо выделить из общего
изменения р. и а при нормальном колебании те изменения,
которые происходят в определенной связи. Для нахождения ^ я а'
мы можем итти'тем же путем, который мы применяли при
250
7Я.85
78.12
95.89
95.24
0.73
0.75
определении динамических постоянных связей. В
механической задаче мы сначала исходили из валентной
аддитивной схемы, а затем уже дополняли ее членами
взаимодействия в выражении для потенциальной энергии (5). В
электрооптической задаче можно поступить так же — исходить из
аддитивности дипольных моментов и поляризуемостей,
приписав определенные значения р. и а отдельным связям.
Это уже было сделано при вычислении дипольного момента
молекулы (стр. 141) и эллипсоида ее поляризуемости (стр. 159).
Здесь мы пойдем дальше, предположив, что аддитивными
свойствами обладают не только дипольные моменты и
поляризуемости у- и а, но и их изменения при изменениях длины
связи: эффективные заряды связей р/ и изменения
поляризуемости связей а. Решая механическую задачу о
колебаниях, мы находим нормальные координаты колебаний,
которые выражаются через естественные координаты—
изменения расстояний и углов [ср. (24)]. Общее изменение
дипольного момента или поляризуемости при данном нормальном
колебании многоатомной молекулы представляет собой
производную соответствующей величины по нормальной
координате этого колебания:
"'"?' (34)
* dpi v J
Vi—dQ *
Такие соотношения мы должны написать для каждой из
компонент а и щ ибо это величины анизотропные. Зная
форму молекулы и решение механической задачи, мы выразим
величины (34) через изменения поляризуемости и дипольного
момента при изменениях уже не нормальных, а имеющих
непосредственный физический смысл естественных
координат. Это попрежнему — изменения длин связей и валентных
углов. Каждая связь характеризуется при этом, в общем
случае восемью постоянными—р, у/, аи а2, а3, а'и а'2, а;3..В
двухатомных молекулах эллипсоид поляризуемости есть
эллипсоид вращения (ср. стр. 157) и а2 = а3, а также а'2 = а'3.
Число различных постоянных уменьшается до шести. В
первом приближении можно считать, что это условие сохраняется
и для единичных о-связей в многоатомных молекулах.
Легко видеть, что интенсивности для чисто
деформационных колебаний зависят лишь от самих величин у и а,
но не от их производных у/ и а'. В самом деле, при
изменении угла изменяется лишь относительное расположение.
эллипсоидов поляризуемости и векторов дипольных моментов
связей, но не сами эти величины, ибо длины связей остаются
неизменными. Для чисто валентных колебаний интенсивности
зависят, напротив, только от р/ и а\ но не от р. и а. В вы-
251
ражения интенсивностей для смешанных колебаний, при
которых меняются и углы и расстояния, войдут все
перечисленные постоянные связей. Интенсивность инфракрасной
колебательной полосы есть, в общем случае, функция \і и р'
всех связей в молекуле
Л* = -/0іь---.1ія; V-'u*--* V-'n)- (35)
Для рамановской линии имеем
Jp=: J (а1ЬаШ ,а13; *2Ь af-9 а23>'-'і anU ап*> аяЗ»
a'lU а'і2) а'із! а'зЬ а'22і а23»*--? а'яЬ а'тШ а'лз)- (36)
Все а и а' нумеруются дважды — номером связи 1, 2,... , ?
и номером 1, 2, 3, соответственно трем главным
направлениям эллипсоида поляризуемости каждой связи. Значения ^
и а можно взять из независимого, не спектроскопического
источника. Мы уже видели, что дипольные моменты
отдельных связей находятся из сопоставления дипольных моментов
различных молекул, содержащих эти связи. Поляризуемости
связей вычисляются из значений рефракций, постоянных
Керра и степеней деполяризации р рэлеевского рассеяния
(стр. 159). Значения^' и а можнэ найти, конечно, только из
интенсивностей колебательных спектральных линий и
связанных с ними физических величин.
Применяя идею аддитивности, мы предположили, что
одинаковые связи в различных молекулах характеризуются
не только одними и теми же у и <*, но также и \і' и а'.
Сравнивая между собой интенсивности (а в случае
комбинационного рассеяния также и степени деполяризации р')
колебательных спектральных линий, мы можем определить
^ и а' определенных связей и с помощью этих величин
вычислять интенсивность (и поляризации) в спектрах других
молекул, содержащих прежние связи. Таким образом, и здесь
применяется полуэмпирический метод вычисления
электрооптических постоянных связей.
Теория поляризаций и интенсивностей в рамановских
спектрах
Степень деполяризации рамановской линии равн а
(ср. стр. 156)
, _ 6 Ь'*
а интенсивность пропорциональна сумме интенсивностей
обеих перпендикулярно поляризованных компонент,
числителя и знаменателя (1)
Ур~45а'2 + 13&'2, (37)
252
где а' и Ь' — среднее значение и анизотропия эллипсоида а' —
зависят от а и а' всех связей в молекуле. Можно показать, что,
независимо от конкретных значений ее и а для чисто
деформационных колебаний, среднее значение эллипсоида а'=0.
Действительно, при изменениях угла, эллипсоиды связей
только поворачиваются. Если бы эллипсоиды были шарами,
то их поворот не изменил бы поляризуемости всей
молекулы (рис. 132). Значит, интенсивность для деформационного
колебания может определяться только отклонением
эллипсоидов поляризуемости связей от шарообразности, их
анизотропией
р = {Н(в| - а*у+(в* ~аз)9+(*3 ~~ *іГ} V2 * (38)
Но среднее значение а' для всей молекулы зависит только
от средних значений а отдельных связей, а анизотропия Ь' —
Рис. 132. Сферическая часть эллип- Рис. 133. Эллипсоиды поляризуе-
¦соидов поляризуемости при дефор- мости при валентном колебании,
мационном колебании.
только от величин (3. Следовательно, для чисто
деформационных колебаний а' равно нулю. Мы получаем для чисто
деформационных колебаний, даже если они
полносимметричны (ср. стр. 211),
р'= 77 = 0.36.
Валентные колебания, напротив, сопровождаются
значительными изменениями среднего значения а, ибо эллипсоиды
поляризуемости. при этих колебаниях сильно
деформируются— растягиваются и сжимаются (рис. 133). Поэтому
для симметричных валентных колебаний а' не равно нулю,
и р'<0.86. Отсюда следует, что, чем больше доля участия
углов в колебании, чем колебание „деформационнее", тем
больше р\ Опыт подтверждает это положение. Мы видели
(стр. 212), что симметричная трехатомная молекула имеет два
симметричных колебания, одно из которых в большей
степени валентное, второе — деформационное. Как следует из
табл. 46, р' для второго колебания всегда больше, чем для
первого.
Таким образом, р' может служить мерой
„деформационное™ колебания". Эти соображения, полученные на основе
253
Таблица 46
(Волькенштейн и. Ельяшевич)
Соединение
Трехатомная
молекула или группа
Частота,
Колебание
CHXL . . . . :
СНоВг,
CHJ.
(С.Н5)-0
SO*
Насыщенные органические
соединения
С1
С1
с\
хВг
О
хн
283
700
174
577
121
483
43S
840
525
1145
1450
2850
4s)
v(s)
N(S)
4s)
v(s)
*(s)
V(S)
b(s)
0.43
0.09
0.35
0.11
0.42
0 19
0.48
0.29
0.50
0.18
0.86
0.10
схемы аддитивности, существенно дополняют правила Пла-
чека (стр. 211), выведенные из свойств симметрии.
Как зависит р'*от симметрии молекулы в случае
валентных полносимметрических колебаний? v(s)— колебания групп
СН, СНо, СН3, СН4 — характеристичны (стр. 224). Плачек
показал, что v(s) для метана имеет р' = 0. В остальных случаях
р'>0, но меньше 0.86. Между тем симметрия падает от
СН4 к СН. Значит ли это, что р' одновременно должно
расти? Оказывается, что да. Если для отдельной СН-связи
мы имеем анизотропию р' и среднее значение а' изменения
поляризуемости с расстоянием, то для п одинаковых связей,
образующих друг с другом одинаковые углы 0, получается
формула
п— 1
ер (і
?п
2п
3 sin2 &)
45а,4 + 7Р'9(1 —-^3 sin2»)
(39)
2л
Приведем пример связи SH. В метилмеркаптане H3CSH
линия 2573 см-1 характеристического колебания S — Н-связи
имеет р/ =0.30. Считая для единичной связи S — Н а2' = а3',
имеем
?і
_ 6?*- _
6(а'і — а2Т
45 а'* + 7 §'* 5 (в/ + 2а.')2•+ 7 (а/ — oa')s
= 0.30, (40)
254
откуда as' :а1' = 0.7. Вычислим теперь ps' для v(s)
сероводорода H.2S, содержащего две связи S — Н под углом 92°20':
Рі =
6р'*(1 ^sin292°20')
= 0.11
45а"2 +7р (1— :f sin2 92°20')
(41)
опыт дает р/ = 0.15. Совпадение достаточно хорошее.
Все эти соображения и расчеты делались без применения
строгой картины колебаний, без знания нормальных
координат и величин а'.. Однако, цель исследования поляризаций
и интенсивностей состоит именно в том, чтобы, с одной
стороны, находить а/, а с другой — вычислять сами
интенсивности и р\ Такие расчеты пока произведены лишь для
хлоро- и бромометанов, для дейтерометанов, для С0.2 и CS.2
(Волькенштейн).
Применяя схему аддитивности, можно сказать, что во
всем ряду молекул СС14, СНС1Я, СН9С13, СН3С1, СН4, СН3Вг,
СН2 Вго, СНВгз, СВг4 фигурируют лишь три связи —
единичные связи С — Н, С — С1, С — Вг. Примем а.2 = а3 и а.2' ==сс:/.
Значения поляризуемостей а^ и а2 = а3 мы имеем из
независимого источника. Остается определить шесть
постоянных, по две для каждой связи а/ и аЛ Они могут
быть найдены из сравнения степеней деполяризации.
Определив электрооптические постоянные, можно рассчитать
интенсивности и остальные р' всех рамановских линий
перечисленных молекул. Данные, приведенные в табл, 47,
показывают, что рассчитанные и найденные экспериментально
значения р' очень близки.
Таблица 47
Рамановский спектр СН2С12
(Волькенштейн и Ельяшгвич)
Колебания
v(s, С—С1) . . .
v (s. С—Н)
3 (s, С1—С—С1) ,
5 (s, Н-С—Н) .
v(as, С— Н) . . .
b(asy Н-С-С1)
n (as, С—CI). . ,
В (as, H—C—Cl)
о (as, Н—С—CI)
Частоты,
СИ"1
700
2986
283
1418
3046
896
736
1266
1149
Интенсивность
относительная
вычисл.
нзмер. 'вычисл.
измер,
(1.00)
0.25
0.63
0.08
0.10
0.04
0.23
0.01
0.06
(1.00)
0.31
0.63
0.09
—
—
0.20
—
0.05
(0.09)
0.11
0.77
0.75
0.86
0.86
0.86
0.86
0.86
(0.09)
0.10
0.43
0.88
0.86
0.79
0.89
255
Повышенное значение р' для b(s) 283 см"1 получилось
вследствие пренебрежения взаимодействиями поляризуемо-
стей отдельных связей. Надо сказать, что количество и
качество экспериментальных данных по поляризациям и интен-
сивностям еще крайне неудовлетворительно. Это, в частности,
мешает расширению и уточнению теории, которая
принципиально пользуется полуэмпирическим методом. Поэтому
на первых порах поневоле приходится ограничиваться
приближением чистой аддитивности — валентной схемой.
Значения постоянных а/ и а3' приведены в табл. 48.
Таблица 48
(Волькеиштейн и Ельяшевич)
Связь
аа, А»
0.79
3.67
5.04
а3, A3
0.58
2.08
2.88
«1 о
0.73
2.04
2.52
- *А2
г0
0.53
1.15
1.44
аі', А2
1.28
2.82
3.23
сс2',А2
с-н
с—а
С—Вг
О.Зі
0.68
0.83
а растет с ростом поляризуемости. Поляризуемость С — Н-
связи здесь наименьшая, ибо у водорода наименьший объем
электронного облака. Но а' для С — Н относительно велико;
отношение а*: а для С — Н больше, чем для С — С1 и С — Вг.
Это, очевидно, определяется более гомеополярным
характером С — Н-связи.
Инфракрасные спектры
Экспериментальные данные по интенсивностям в
инфракрасных спектрах многоатомных молекул пока что
отсутствуют. Тем не менее удалось определить эффективный
заряд С — Н-связи и вычислить интенсивности всех
инфракрасных полос метана и дейтерометанов, применяя данные
по атомной поляризации и дисперсии метана в инфракрасной
области. Числитель в дисперсионной формуле для атомной
поляризации (стр. 132) содержит силы осцилляторов,
пропорциональные интенсивностям соответствующих
инфракрасных полос, выражающиеся через у. и ^'. Интерпретируя
экспериментальные значения сил осцилляторов в метане при
помощи излагаемой теории, получаем для С — Н-связи
р/ = 0.3.10-10 CGSE = 0.062 е,
т. е. С — Н-связь по своей ионности занимает промежуточное
место между НВг и HJ (табл. 29). Отношение ц/г0 для
С —Н равно 0.077 е.
Для дальнейших расчетов в этом направлении нужны
новые экспериментальные данные.
256
Интенсивности характеристических колебаний
и электронный резонанс
Если колебание характеристично и, тем самым, в нем
участвует главным образом одна определенная связь, то
интенсивность соответствующей спектральной линии
определяется в основном {і/ или а' данной связи. Сопоставляя
интенсивность характеристических колебаний, мы можем
сделать выводы о влиянии электронного резонанса на
интенсивности, на jj/ и «'. Оказывается, что интенсивности' в коле-
бательных.спектрах исключительно чувствительны к
электронному резонансу (табл. 49).
Таблица 49
Интенсивности рамановских линий
Молекула v, см-1 ; J
Группа С=0 (Волькенштейн)
СН8СОС1 1 1798 0.8
СН8СОСН8 1 1708 (1.0)
СНяСОСвНб і 1678 | 10.0
С6Н6СОСвН5 \ 1653 ! 18.5
Группа C—N (Шорыгин)
CH8CN 2249 (1.0)
HSC=CHCN 2229 4.2
CeH6CN 2227 10.0
Резонансные сі руктуры соединений этого типа были
рассмотрены ранее. Мы видим, что ход частот и
интенсивности в общем противоположен, причем интенсивности
меняются от соединения к соединению во много раз
сильнее, чем частоты. Это качественно можно объяснить тем,
что в структурах, дающих повышенную частоту
/
поляризуемость электронного облака С = О-связи
уменьшена благодаря дополнительному притяжению электрона.
Напротив, поляризуемость структуры
Ч
С—О
/
с дополнительным отрицательным зарядом должна быть
большей. Вероятно, играет дакже заметную роль изменение
17 Строение молекул 257
весов внутриионизованных структур при колебаниях С = 0
и CsN-связей (Шорыгин).
Весьма существенно для молекулярного спектрального
анализа то обстоятельство, что некоторые линии, не
характеристичные по частотам, имеют характеристичные
интенсивности. Это особенно проявляется" в инфракрасных спектрах
(Гопштейн). В цепочке атомов, содержащей наряду сС-С-
связями С —О или С — N-связь, невозможно определить ее
присутствие по частотам (стр. 225). Но введение С — О-связи,
имеющей в отличие от С — С-связей заметный дипольный
момент }і и эффективный заряд іі', сразу резко повышает
интенсивности всех линий, относящихся к колебаниям, в
которых С — О-связь участвует. Таким образом, спектр спирта
отличается по интенсивностям от спектра соответствующего
углеводорода, хотя разница в частотах цепочки незначительна.
Изучение интенсивностей и поляризаций в
колебательных спектрах очень обогатит науку о строении молекулы.
В настоящее время мы располагаем громадным
экспериментальным материалом по частотам, но несоизмеримо малым
по интенсивностям. Не будет преувеличением сказать, что
в области колебательных спектров мы вступаем сейчас в
„эпоху интенсивностей". Этой области обеспечено мощное
развитие.
МОЛЕКУЛЯРНЫЙ СПЕКТРАЛЬНЫЙ АНАЛИЗ
Наряду со многими другими физическими и химическими
методами исследования колебательные спектры —
инфракрасные и рамановские, нашли разнообразные и важные
применения в^анализе вещества. Колебательные спектры помогают:
1) определять строение молекул, устанавливать, из каких
атомов состоят молекулы, как эти атомы расположены и
какие между ними существуют связи;
2) идентифицировать вещества;
3) определять качественный и количественный состав
смесей различных веществ.
Структурный анализ
При решении вопроса о структуре молекулы
колебательные спектры имеют преимущества по сравнению с другими
физическими методами, В самом деле, такие постоянные
молекулы, как дипольный момент, рефракция
(поляризуемость) и т. д., могут дать лишь косвенную информацию о
деталях строения, ибо относятся к молекулам в целом.
Напротив, колебательные спектры непосредственно
отображают структуру молекулы. Электронные спектры также
могут быть с успехом применены к структурно-аналити-
258
ческим задачам, но в общем они труднее поддаются
расшифровке и интерпретации. Совершенно очевидна важная
роль характеристических колебаний при исследовании
структуры. По частотам, интенсивностям и степеням
деполяризации соответствующих спектральных линий можно
непосредственно судить о присутствии в молекуле определенных
групп и связей. Типичные изменения частот и особенно
интенсивностей говорят о взаимодействии электронных
облаков соседних групп и связей в молекуле, об
электронном резонансе. Ряд примеров, показывающих, как
отражается строение молекулы на спектральных линиях
характеристических колебаний, мы привели выше. На основании
подробного исследования колебаний ряда молекул, содержащих
типичные группы и связи, оказалось возможным даже
предсказывать колебательные спектры других молекул,
содержащих эти группы, еще не исследованных
экспериментально. Это, в частности было сделано для высших парафинов —
нонанов — Б. Степановым, установившим характерные
признаки в спектрах нормальных и изопарафинов.
Качественный и количественный анализ
В тех случаях, когда требуется проанализировать
неизвестное вещество или смесь неизвестных веществ, также
можно применять колебательные спектры.
Наиболее существенны эти возможности при анализе
органических веществ, где эмиссионный спектральный
анализ на отдельные элементы не имеет широкого применения, а
работа обычными химическими методами сопряжена с очень
большими трудностями. Колебательные спектры в
частности дают возможность анализировать смеси сходных
веществ— углеводородов, что особенно важно для
промышленности моторного топлива. Процессы облагораживания
естественных бензинов и получения искусственного
горючего высокого качества необходимо направлять таким образом,
чтобы в горючем содержалось наибольшее количество
разветвленных углеводородов, сгорающих в моторе без
преждевременной детонации. В настоящее время хорошо
разработана методика качественного анализа бензинов при
помощи рамановских спектров; сравнивая интенсивности линий,
удается количественно анализировать сложные
многокомпонентные смеси. Инфракрасные спектры в далекой области
основных частот не менее полезны. Работы в этом
направлении ведутся главным образом в США и в Советском
Союзе— в Оптическом институте, в Физическом институте
Академии Haytf и в Физико-химическом институте им. Карпова.
*
259
ЛИТЕРАТУРА
Глава 1
1. Блэквуд и др. Очерки по физике атома. Пер. с англ. Е. Л. Файн-
берга, изд. 2-е, ОНТИ, 1940.
2. Э- В. Шпольский. Атомная физика. ОНТИ, 1944.
3. С. Э. Фриш. Атомные спектры. ГТТИ, 1933.
4. Н. White. Introduction to atomic spectra. 1934.
5. Я. К. Сыркин. Периодическая система. Усп. химии,.3, 358, 1934.
6. К. Дарроу. Введение в волновую механику Шредингера. Усп. ф :3.
наук 9, 437,' 1929.
7. В. Кондратьев. Структура атомов и молекул. АН, 1946.
8. М. Борн. Современная физика. ОНТИ, 1935.
Глава II
См. 5,7.
9. О. Rice. Electronic Structure and Chemical Binding.
10. L. Pauling. The Nature of the Chemical Bond.
11. Я. К. Сыркин, M. Д я т к и h а. Химическая связь и строение молекул,
Госхимиздат, 1946.
12. А. Жуховицкий, Я. Сыркин, М. Дяткина. Современное
состояние теории валентности. Усп. химии, 9, 930, 1940.
13. Они же. Общая теория многоэлектронной проблемы в квантовой
химии. Усп. химии, 9, 1038, 1940.
14. Они же. Валентные состояния и аддитивная схема органической химии.
Усп. химии, 9, 1143, 1940.
15. Р. Кроииг. Оптические основы теории валентности. ОНТИ, 1937.
16. Г. Гельман. Квантовая химия. ОНТИ, 1937.
17. Пенни, ван-Флек, Шерман.Квантовая теория валентности. ОНТИ,
1938.
Глава III
См. 9, 10, 11, 15.
18. Г. Стюарт. Структура молекул. ГНТИУ, 1937.
19. Гольдшмидт. Стереохимия. Химиздат, 1940.
20. П. Дебай. Строение вещества ОНТИУ, 1936.
21. П. Дебай. Стереохимия и физика. Усп. химии,3, 522, 1934.
22. Г. Гиллеман. Молекулярная асимметрия. Усп. химии,77, 161, 1938.
23. М. В о л ь кенштейн. Внутреннее вращение и ротационная
изомерия. Усп. химии, 13, 1944.
24. В. Гюккель. Теоретические основы органической химии. ОНТИ,
т. I, 1933; т. II, 1934. - - ,
25. Л. Б р о к у э й. Диффракция электронов газовыми молекулами. Усп.
физич. наук, 17, 175, 280, 1937, ..
260
Глава IV
См. 9, 10, 11, 13, 14, 15, 16, 17, 24.
2\ Я. Сыркин. Энергии связей в органических соединениях. ЖФХ, 17,
347, 1913.
27. Я Сыркин, М. Дяткин а, А. Жуховицкий. Резонанс в
органической химии. Усп. химии, 10, 121, 1941.
28. Л. П а у л и н г. Значение резонанса для природы химической связи
и структуры молекул. Усп. химии, 7, 1312, 1938.
29. J. Lennard-Jones, С. Couison. Trans. Farad. Soc, 35, 811, 1939.
30. E. Lloyd, W. Penney. Trans. Farad. Soc, 35, 835, 1939.
31. Т. Фэрстер. Окраска и строение органичес.-шх соединений. Усп.
химии, 9, 72, 1940.
32. М. В о л ьк еншт е й н. Окраска органических соединений. Природа,
№ 5, 1943.
Глава V
См. И, 16, 18, 20, 23, 24, 26.
33. Ч. С майе. Диэлектрическая постоянная и структура молекул. ОНТИ,
1937.
34. П. Дебай, Г. 3 а к к. Теория электрических свойств молекул. ОНТИ,
1936.
35. Р. Б е к к е р. Электронная теория.
36. Van Vleck. Theory of electric and magnetic susceptibilities.
37. Le Fevre. Dipole moments. 1938.
38. F. Wall. J. Am. Chem. Soc, 62, 800, 1940.
39. M. Дяткина. ЖФХ, 14, 1589, 1940; 15, 597, 1941.
40. M. Бори. Оптика. ОНТИУ, 1937.
41. Лондон. Общая теория молекулярных сил. Усп. физ^.иаук, 18, 421,
1937.
42. Р. К р е м а н, М. П е с т е м е р. Зависимость между физическими
свойствами и химическим строением. ГОНТИ, 1939.
Глава VI
'См. 8, 24, 35, 36, 40, 41, 42 и особенно 18.
43. К. Denbigh. Trans. Farad. Soc, 36, 936, 1940.
44. П. Ф e о ф и л о в. ЖЭТФ, 12, 328, 1942.
45. Kuhn и др. Анизотропия поглощения света. Zs.Phys. Chem. (В.), 45,
121, 1939.
46. G. S с h е і b е и др. Зависимость поглощения света от направления.
Zs. Elektroch., 49, 372, 1943.
47. М. Во л ькенште йн. Современная теория оптической активности.
Усп. химии, 9, 1089, 1252, 1940.
48. Е. Кондон. Теория оптической вращающей способности. Усп. физ.
наук, 19, 380, 1938.
49. Г. Ландсберг. Оптика. ОНТИ, 1940,
Глава VII
См. 7, 18, 24, 40, 42, 49 и особенно 15.
50. К. Шеф ер, Ф. Мат ос си. Инфракрасные спектры. ОНТИ, 1935.
51. Г. Ландсберг. Комбинационное рассеяние света и его значение для
химических проблем. Усп. химии, 1, 464, 1932.
52. Sutherland. Infrared and Raman-Spektra. 1935.
53. G. H e r 2 b e r g. Molekuispektren. 1939.
54. E. Рабинович. Полосатые спектры. Усп. физ. наук, 11, 554, 1931;
13, 253, 1932; 13, 854, 1932.
261
55. Э. Шпольский. Диффузные спектры и химические проблемы. Усп-
физ. наук, 1933.
56. Е. Teller. Molekulspektren. Hand-Jahrb. Chem. Phys., 9, Т. 2.
57. К. Б онге ф фер; Г а рте к. Фотохимия. ОНТИ, 1935. ^
58. Е. В art hoi о те. Характер связи в галоидоводородах на основании
измерения абсорбции, Zs. Phys. Chem. (В.), 23, 131, 1938.
Глава VIII
См. 15, 18, 23, 42, 50, 51, 52, 53 и особенно 56.
59. М. Е л ь я ш е в и ч. Механика колебаний молекул. Усп. физ. наук, 1946.
60. М. В о л ь ке н ш тей н. Электрооптика колебаний молекул. Усл. физ.
наук, 1946.
61. М. В о ль к е нш т е йн.» Раман-эффект и междумолекулярное
взаимодействие. Усп. физ. наук, 18, 153, 1937.
62. М. Волькенштейн, Я. К. Сыркин. Раман-эффект пироновых
соединений и электронный резонанс. ЖФХ, 13, 948, 1939.
63. Е. Прилежаева, Я. Сыркин, М. Волькенштейн. Раман-
эффект этиленовых соединений и электронный резонанс. ЖФХ, 14,
1396, 1940.
64. М. Волькенштейн, М. Ельяшевич, Б. Степанов. Теория
колебательных спектров многоатомных молекул, I. Задачи теории.
ЖЭТФ, 15, 35, 1945.
65. Г. Ландсберг, С. Ухолин. ДАН СССР, 16, 391, 403, 1937.
/66. В. Малышев. Междумолекулярные силы и комбинационное
рассеяние света. Изв. АН СССР, сер. физ., 5, 13,1941.
67. Е. Гросс. Изв. АН СССР, сер. физ., 5,№ 1, 1941.
68. М. В о л ькеншт е йн. Раман-эффект углеводородов и анализ
жидкого топлива. Усп. химии, 8, 970, 1939.
69. П. Ш о р ы г и н. Молекулярный спектральный анализ. Усп. химии,
№ 3, 1944.
70. М. Волькенштейн. Молекулярный спектральный анализ. Природа,
Ш 2, 1945.
71. М. Волькенштейн, М. Ельяшевич, Б. Степанов.
Колебания молекул, монография (готовится к печати).
72. Hibben. Raman-Effekt and its Chemical Applications. 1939.
73. K. W. F. Kohlrausch. Der Smekal-Raman-Effekt, Erg. B. 1938.
74. G. Herzbe*g Jnfrared and Raman Spectra of Polyatomic Molecules.
1945.
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Аддитивность б, 9, 61—63, 99—
104, 119—121, 133—137, 141—
144, 3 73, 207, 221, 227, 249—256.
Азидо-группа 172.
Азот 26, 30, 51, 53, 56, 57, 70, 80.
Активность оптическая 93, 167—
175.
Аллеи 81, 94.
Алмаз 74, 76—78,' 97, 99.
Аммиак 38, Ь5, 66, 80, 88, 91, 143,
148, 203, 220, 237.
Аммоний 39, 40, 53.
Аммоний хлористый 39, 241.
Ангармоничность 179, 190, 242.
Анизотропия 151, 211, 253.
Анизотропия молекул 150—175.
Анизотропия поляризуемости 150—
159, 211, 251—253.
Анизотропия электронных
переходов 160—162.
Анилин 143, 167.
-Антиподы оптические 92—95, 145,
170—175, 234.
Антисимметричные колебания
44, 170, 209—212.
Антисимметричные функции 47.
Антистоксова флюоресценция
164.
Антистоксовы Раман-лииии 193.
Аргон 26, 36, 70, 98. "
Ароматические соединенияч62, 8Ц "
161, 164, 227.
Асимметрический атом углерода
92, 93, 145, 172, 173.
Асимметрический синтез 95, 175.
Асимметричные молекулы 92—95,
169.
Атома строение 19—28.
Атомная поляризация 132.
Атомная рефракция 136, 256.
Атомные спектры 21, 22.
Атомный радиус 97, 110.
Атомный фактор 72, 77.
Ауксохром 115.
Ацетилен 39, 67, 79, 81, 83, 113,
220.
Ацетон 204, 228.
Ацетоуксусный эфир 137, 234.
Бензол 77, 80, 83, 89, 103—109,
116—118, 133, 157, 161, 227.
Бензол неорганический 109—111.
Бензофлавин 166, 167.
Биполярный ион 232.
Благородные газы 27, 36, 39, 51,
98.
Борна — Габера цикл 33, 34, 98.
Бромацетилен 144.
Бромистый водород 60, 80, 140,
148, 181, 187.
Брэгга условие 75.
Бутадиен 81, 113, 117.
Бутан 101, 186, 248, 250.
Валентности направленные 64—66.
Валентность 49—54.
Валентные колебания 213—218,
253.
Валентные состояния 51—54.
Ваи-дер-Ваальса 'поправка на
объем 70, 125, 147.
Ван-дер-Ваальса уравнение 70.
Векторная сумма 141—145, 251.
Вероятность перехода 132.
Винная кислота 94, 145.
Вода 38, 65, 66, 80, 88, 126, 141,
148, 209, 238, 241.
Водород 19, 38—41, 51, 54, 55, 59,
80, 99, 126, 136, 157, 181, 199,
200, 236.
Водородная связь 237—241.
Волновая механика 15*—22.
Волновая функция 15, 47, 48, 58,
59, 66, 106, 107.
Волновое уравнение 15, 17, 19,
46, 47, 179.
Волны световые 10—13, 131, 154—
156, 170, 193.
263
Волны электронные 13, 73.
Вращательные спектры 79, 184.
Вращение внутреннее 84—88, 145,
174, 248.
Вращение молекул 127, 129, 130,
176, 182—185, 208, 247, 248. .
Вращение плоскости поляризации
158—175.
Вырождение 45, 58, 210, 223, 236,
243.
Газы 68, 72, 126, 148, 157, 236.
Галоидоводороды 60, 80, 140, 148,
181, 187.
Галоидометаны 224, 255.
Галоиды 61, 80, 138, 181, 199.
Гармонический осциллятор 16—
19, 42, 130, 148, 170, 178, 214.
Гваякол 240.
Гексаметилбензол 161.
Гелий 26, 28, 44, 51, 5% 70.
Гибридизация 52.
Глицин 231.
Гомеополяриая связь 30, 45—67,
136, 187, 195, 213, 256.
Графит 74, 76, 77, 111.
Двойная связь 39, 66, 83, 97, 100,
112, 136, 227, 232.
Двойное лучепреломление 153,
168.
Двухатомные молекулы 60, 80,
139, 178—190.
Дебаеграмма 76.
Дейтерий 181, 222.
Дейтерометаны 223.
Деформационные колебания 209,
218, 220, 249, 253.
Диамагнетизм 149.
Диаметр молекулы 68—71.
Динамические коэффициенты 221,
234
Диполь 123, 147, 155.
Дипольный момент 123—129,139—
148, 153, 154.
Дисперсионное взаимодействие
148, 236.
Дисперсия 130—133.
Дисперсия оптической активности
172.
Диссоциация 60, 179, 196, 200—
202.
Дифенил 94.
Диффракция электронов 13, 73.
Дихлорбензол 104.
Дихлорэтан 84—88, 145.
Дихлорэтилен 84, 143, 212.
Дихроизм 161, 165, 166. . .
Дихроизм циркулярный 171.
Диэлектрик 121—133.
264
Диэлектрическая постоянная 6
122—129, 156.
Дьюара структуры 106, 117, 120.
Жидкости 74, 129, 154, 158, 192
236—242.
Закись азота 80, 112.
Закись углерода 89, 112.
Заряд иона 30, 98, 135, 136.
Заряд эффективный 186—188,251.
Изобутан 101, 250.
Изомеризация 101, 249.
Изомерия поворотная 84—88, 234,
248.
Изомерия пространственная 83,
234
Изотопы 181, 223.
Индукционная поляризация 123.
130.
Индукционное взаимодействие
147, 148, 238.
Инкремент 136.
Интенсивности в спектрах 186,
194, 250—258.
Интерференция 71, 154.
Инфракрасные спектры 79, 185—
190, 195, 205, 210, 212, 250, 256,
258
Ион 29, 97, 98,134—136,138,188,195.
Ион биполярный 232.
Ион-дипольная связь 37.
Ионизации потенциал 22, 27, 33,
37.
Ион молекулы водорода "9, 40,
41, 55.
Ионная молекула 30, 59, 98, 136,
187, 195.
Ионная рефракция 134.
Ионная связь 30—37, 50, 55, 60,
98, 134, 140, 188, 195.
Ионное состояние 59, 60, 140.
Ионности степень 60,98, 140, 187,
195, 237.
Ионный радиус 98.
Испускание света 11.
Испускания спектры 20.
Карбонильные соединения 142,
228—232, 257.
Квантовая механика 15—28, 42—
49, 120, 129, 132, 148, 179, 184,
191.
Квантовое число вращательное
184.
Квантовое число главное 20, 22,
26, 45, 135.
Квантовое число колебательное
179, 186.
Квантовое число магнитное 23, 45.
Квантовое число орбитальное 23,
26, 45, 135.
Квантовое число спиновое 23, 45.
Кварц 185, 191.
Кекуле структуры 105—110, 117,
120.
Керра постоянная 152, 157—159,
252.
Керра явление 151—154.
Кетоны 142, 228.
Кинетическая теория газов 5, 6,
68.
Кинетич еская энергия 68, 163,
196 199 219.
Кислород'26, 51, 54,57, .70,80,
181.
Клаузиуса — Мосотти уравнение
124.
Колебания асимметричные 43, 169,
209, 212.
Колебания валентные 213, 253.
деформационные 213,
253.
Колебания крутильные 235, 248.
маятников 43, 169,208.
молекул 177—182,
206—259.
Колебания нормальные 43, 169,
208, 219, 251.
Колебания нулевые 19, 148, 181.
связанные 43, 169, 208.
„ симметричные 43, 169,
209, 212.
Колебания характеристические
224—234, 257—259.
Колебательно-вращательные
спектры 177—190.
Колебательные спектры 178—182,
185—188, 206—259.
Комбинационное рассеяние света-
190—196, 210—212, 236, 239—
244, 252—259.
Комбинационный тон 242.
Кометы 203.
Комплексные соединения 37, 237.
Константа равновесия 235, 247,
248.
Конъюгированные связи 62, 104,
113, 117, 120, 230.
Координаты естественные 214, 251.
Координаты нормальные 219, 251.
„ симметрии 217.
Координационная связь 40.
Коэффициенты динамические 180,
214, 221—223.
Коэффициенты кинематические
215, 216.
Красители 115—120.
Кратные связи 66, 67, 97, 101,
105—121, 181, 227—234.
Кристалл 32, 74, 161, 242.
Кристаллическая решетка атомная
74—78.
Кристаллическая решетка ионная
32, 74—78.
Кристаллическая решетка
молекулярная 74—78, 242.
Лауэграмма 76.
Левшина-Перрена формула 165.
Линейные молекулы 80, 151, 208,
210, 212, 219—221.
Локализованных пар метод 45—
54, 57, 58, 63.
Лорентц — Лоренца формула 125.
Люминесценция 162, 167, 205.
Люминесценции поляризация
164—167.
Магнитные свойства 6, 121, 149.
Максвелла явление 154.
Масса приведенная 178—181, 183,
215.
Междумолекулярное
взаимодействие 146—148, 175, 236—242.
Мезовшшая кислота 91, 93—95,
145.
Мезомерия 109.
Мезоформа 94, 146.
Меркаптан 248, 254.
Металл 120.
Метан 38, 52, 65, 70, 77, 80, 91,
203, 222—224.
Метил 112, 202, 254.
Метил бромистый 81,143,224, 226.
„ йодистый 81, 143, 224, 226.
„ хлористый 81, 143, 224,
226, 235.
Метиловый спирт 143, 239, 240.
Метиловый эфир сс-азидопропио-
новой кислоты 172.
Модуляция 153, 193.
Молекулы асимметричные 92—95,
169.
Молекулы двухатомные 60, 80,
140, 178—190.
Молекулы линейные 80, 151, 208,
210, 212, 219—221.
Молекулы многоатомные 79, 96—
119, 141, 183, 185, 195, 204—259.
Молекулы нелинейные 79, 209,
212, 214—219.
Молекулы трехатомные 79, 209,
212, 214—219.
Молекулы четырехатомные 79,
212, 220.
Молекулярная дисперсия 130.
„ поляризация 124—
129.
Молекулярная решетка 74—78,242.
265
Молекулярная рефракция 125—
138, 150, 156, 158.
Молекулярный спектральный
анализ 204, 253, 259.
Молекулярных орбит теория 54—
57, 63.
Момент количества движения
электрона 23, 184.
Морзе формула 179.
Мочевина 144, 229.
Направленные валентности 64—66.
Насыщенные соединения 80—88,
97—103, 120, 222, 225, 234.
Нафталин 105, 114, 118, 242.
Неон 26, 36, 51, 70, 98.
Неопределенностей соотношение
14, 203.
Неорганический бензол 109.
Нормальные колебания 43, 169,
208, 219, 251.
Нормальные координаты 219, 251.
Нулевая энергия 19, 148, 181,
248—250.
Обертоны 179, 190, 243.
Обмен электронов 47.
Оболочка электронная 22, 26, 48,
49, 98.
Окись углерода 56, 57, 80, 112,
141, 148, 181, 231.
Оксониевые соединения 241.
Оптическая активность 93—95,
167—175.
Ориентационная поляризация
126—129.
Ориентационное взаимодействие
147, 148, 236.
Ориентация молекул 126—129, 154,
160—161.
Осциллятор 16—19, 42, 130, 148,
161, 165, 166, 169, 170, 178, 214.
Осциллятора сила 132,160,172, 256.
Отбора правила 185, 189, 210, 212.
Парамагнетизм 149.
Параметрический резонанс 243,
244.
Параиитроанилин 143.
Паранитрозодиметиланилин 161.
Паули принцип 25, 27, 45, 49, 54, 56.
Перекись водорода 91, 92, 169.
Периодическая система элементов
26.
--связь 67, 84, 96, 101, 105—121,
181, 227—234.
тс-электроны 67, 93, 101, 105—121.
Планеты 203.
Поворотная изомерия 84—88,145.
234.
266
Поглощение света 20, 115—119,
160—175, 185—190, 196—205.
Полуторная связь 111, 232.
Поляризация атомная 132.
индукционная 123,130.
„ люминесценции 164—
167.
Поляризация молекулярная 124—
129.
Поляризация ориентационная
126—129.
Поляризация рамановских линий
194—195, 211, 250-256.
Поляризация рэлеевского
рассеяния 154—157, 211, 252.
Поляризация света 6, 151—152,
154—156.
Поляризуемости связей 159, 251—
256.
Поляризуемость 124—139, 146—
159, 194—195, 210, 211, 250—256.
Порядковый номер 26, 72, 98.
Потенциал ионизации 22, 27, 33,
37, 146.
Потенциал резонансный 22.
Потенциальная кривая 32, 45, 69,
178, 180, 182, 197—204.
Потенциальная энергия 17, 19, 32,
85, 127, 178—182, 198, 214, 219,
245, 249.
Потенциальный ящик 15, 17.
Предиссоциация 203, 240.
Правила отбора 185, 189, 210, 212.
Приведенная масса 178—181, 183,
215.
Принцип Паули 25, 27, 45, 49, 54,
56.
Производная поляризуемости 194,
211, 250—256.
р-электрон 24—28, 50—54, 65.
Радикалы свободные 202.
Радиусы атомные 97, 110.
ионные 98.
Размеры молекул 68, 79—82.
Рамана явление 190—196, 210—
212, 236, 239—244, 252—259.
Рассеяние света 154,157,190—196,
210, 212, 236,239—244,252-259.
Рассеяние электронов 73.
Расстояния междуатомные 76, 79—
84, 110-112, 119.
Рацемическая смесь 94, 175.
Редкоземельные элементы 28.
Резонансная флюоресценция 163.
Резонанс механический 42, 225.
„ параметрический 243,244.
Резонанс электронный внешний
1G5—120, 137, 142—144, 172, 204,
227—234, 238, 241, 249, 257.
Резонанс электронный внутренний
41—49, 57—61, 112, 140.
Рентгеновы лучи 71—78.
Рефракция атомная 136, 138.
ионная 134, 135, 138.
молекулярная 125—138,
150, 156, 158.
Ротатор 183.
Рэлеевское рассеяние 154—156,
211.
Связь водородная 237—241.
„ гомеополярная 30, 45—67,
136, 187, 195, 213, 256.
Свяіъ двойная 39, 66—87, 97, 100,
112, 136, 227, 232.
Связь ионная 30—37, 50, 55, 60,
98, 134, 140, 195.
Связь коиъюгированная 62, 104,
113, 117, 120, 230.
Связь координационная 40.
„ кратная 66, 67, 97, 101, 105—
121, 181, 227—234.
Связь полуторная 111, 232.
„ семиполярная 40.
„ тройная 39, 63—67, 97, 227,
229, 233.
Свободное вращение 84, 145, 248.
с-связь 66, 67, 84, 159, 251.
а-электрои 66, 67. *
Сила осциллятора 132,160, 172,256.
Симметрия 8, 45, 47, 88—9], 142—
146, 204, 207, 209—212, 217, 220,
254.
Симметричные колебания 43, 169,
209—212, 217, 220, 254.
Синильная кислота 80, 151, 157,
227, 238.
Собственные значения 19, 48, 59,
179, 184.
Соответствия принцип 18.
Спектры атомов- 21, 28.
„ видимые 115—119.
. вращательные 79, 176,
177, 182—185, 188—190, 192.
Спектры инфра-красные 79, 185—
190,195,206, 210, 212,250,256,258.
Спектры испускания 20.
колебательные 178—181,
185—188, 206—259.
Спектры молекул 115—119, 132,
176—260.
Спектры поглощения 115—119,
132, 160—162, 172—190, 196—
206, 210, 212, 250, 256, 258.
Спектры рассеяния 79, 190—196,
210—212, 236, 239—244, 252—259.
Спектры ультрафиолетовые 115—
119, 132, 160—162, 172-175,
196—205.
Спектры электронные 115—119,
132, 160—162,172—175, 196-205.
Спин 24, 45, 49—54.
Спин-валентность 51—54.
Среднее значение поляризуемости
151, 211, 252.
Сродство к электрону 33, 37.
Стекло 136, 185.
Степень свободы 208, 246.
деполяризации 155—157,
165, 211.
Степень деполяризации Рама:і-
линии-211, 212, 251—256.
Стокса закон 163.
Стоксова флюоресценция 163.
Стоксовы раман-линии 191—194.
Структуры внутриионизованные
109, 111—113, 142—144, 172, 204,
229-234, 257.
Структуры резонансные 105—121,
142—144, 172, 204, 229—234,257.
s-электрон 24—28, 50—54, 64—67.
Тагефон 153.
Таутомерия 137, 234.
Теплоемкость 245—248.
Теплота изомеризации 101.
образования 99—101.
сгорания 99, 100.
„ сублимации 99.
Термодинамические свойства 98—
101, 236, 244—250.
Тетраэдрические молекулы 82, 142,
157, 212, 220.
Тиндаля—Рентгена явление 185.
Трехатомные молекулы 79, 80,
209, 212, 214—219.
Тройная связь 39, 63—67, 97, 227,
229, 233.
Углеводороды 71, 79—88, 97, 99—
101, 112, 117, 124, 164, 222—224,
248—250, 259.
Углекислый газ 71, 80, 89, 111,
137, 157, 220.
Углеяод 26, 51—53, 99, 136.
Углерода окись 71, 80, 112,140, 181.
Угол валентный 66, 80—83,141, 215.
Уровни энергии 15—24, 45—48,
57—61, 108, 116, 162—164, 179—
184, 189—194, 197—203, 244.
Уротропин 70, 78.
Фактор атомный 72, 77.
„ циркулярного дихроизма
172.
Фенантрен 114, 118.
Ферми-резонанс 243.
Флюоресценция 162—167.
Фосфоресценция 163.
267
Фоток 13.
Фотохимии основной закон 12,
202.
Фотоэффект 12.
Франка — Кондона принцип 197—
200.
Фреон 247.
Фтор 26, 35, 51, 54, 136, 138, 224,
226, 237.
Фтористый водород 60, 80, 181.
Функция волновая 15, 47, 48, 58,
59, 66, 106, 107.
Фурье-анализ 76.
Характеристичность 204, 224—234,
240, 257-259.
Хлор 26, 81, 136, 138, 224—226,
237.
Хлористый водород 60, 71, 80, 126,
134, 140, 148, 181, 187, 236, 237,
241.
Хлористый натрий 33, 76, 78.
Хлороформ 91, 158, 224, 255.
Хромофор 115, 172, 173, 204.
Цветность 115—119.
Циан 203.
Цианиды 100, 233, 237.
Цикл Борна —Габера 33, 34, 98.
Циркулярное двойное
лучепреломление 168, 169.
Циркулярный дихроизм 171, 172. *
Частоты вращения 183, 184.
колебаний 17, 44, 133,
148, 171, 178, 190, 192, 197, 215,
217, 223—250, 259.
Четырехатомные молекулы 79,
212, 220.
Четыреххлористый углерод 66, 71,
73, 90, 91, 129, 157, 224.
Шредингера уравнение 15, 17, 19,
46, 47, 179.
Экзальтация 137.
Электрон 7, 13—15, 19—28, 35—42,
45—67, 93, 101, 105—121, 130,
139, 148, 159, 169, 195—205.
Электрон-вольт 21.
Электронный резонанс 57—61,
105—120, 137, 140—144, 172, 204,
227—234, 238, 241, 249, 257.
Электроны валентные 29, 39.
„ внешние 29, 39.
внутренние 29.
„ разрыхляющие 56.
связывающие 56.
Электроотрицательность 37, 61,
111, 126, 138.
Эллипсоид поляризуемости 151,
157—159, 211, 252, 253.
Энергия внутренняя 245, 247.
вращательная 176, 183,
184.
Энергия диссоциации 56, 61, 179,
182, 196, 198-202.
Энергия кинетическая 68, 163,
196, 199, 219.
Энергия колебательная 17, 43, 179,
186, 192, 206, 214, 219.
Энергия кулоновская 19, 34.
нулевая 19, 148, 181, 248—
ж250.
Энергия обменная 47.
„ потенциальная 17, 19, 3.\
85, 127, 178—182, 198, 214, 219,
245, 249.
Энергия свободная 248.
связи 6, 34, 47, 56—61,
98—110, 120.
Энергия электронная 19, 20, 24,
58, 98—110, 120, 197.
Энтропия 247, 248.
Этан 80, 83, 85, 91, 100.
Этилен 39, 67, 81, 91, 227.
Этиленовые соединения 81, 83,91,
232.
Эффективный заряд 186—188,251.
ИМЕННОЙ УКАЗАТЕЛЬ
Алтар 175.
Байер 82.
Бальмер 22.
Бартоломе 187, 225.'
Батуев 241.
Беджер 180.
Берналь 238.
Бертело 99.
Берцелиус 29.
Бор 18, 20, 22, 184.
Борн 34, 98, 135, 169, 175.
Браун 172, 174.
Брестер 210.
Брэгг 75, 76.
Бухгейм 236.
Вавилов 162, 165t 166.
Вазастьерна 134.
Ван-Аркель 136.
Ван-дер-Ваальс 70, 125.
Вант-Гофф 65.
Васильев 142.
Вейибаум 59.
Вейигеров 185. ¦
Бе ланд 108.
Вигиер 210.
Витт 115.
Волькенштейн 207, 228, 232, 236,
241, 248, 250, 254—256.
Вуд 162.
Букс 241.
Габер 34, 98.
Гаптмахер 241.
Га узе 95.
Гейзенберг 14, 42, 44, 45, 57,
135.
Гейтлер 46—51, 54—58, 63, 102.
Гертц 10.
Герцберг 54, 55, 198, 203, 204.
Глоклер 247.
Гопштейн 258.
Гориути 243.
Гросс 241, 242.
Гунд 54, 55.
Дебай 75, 76, 129, 139.
Де-Бройль 13, 73.
Де-Бур 136.
Денби 158, 159.
Джемс 59.
Джермер 33.
Дьюар 105—110, 117.
Дэвиссон 13.
Дяткина 114, 140.
Ельяшевич 207, 216, 222, 235, 249,
250, 254—256.
Заксе 158.
Иосс 135.
Кекуле 103—110, 117.
Керр 151—153, 157—159.
Кирквуд 175, 236.
Клаузиус 124, 128, 138.
Клемм 149.
Кнопф 175.
Кнорр 136.
Кольрауш 181, 226, 234.
Кондон 197, 198, 200.
Кондратьев 203.
Константинова-Шлезингер 164.
Коссель 36, 39.
Коттон 171.
Кулидж 59.
Кун 161, 169—172, 174, 175.
Ландсберг 191, 192, 239, 240, 213.
Лауэ 75, 76.
Лебедев 10.
Лебель 65.
Левшин 165.
Леннард-Джонс 120.
Лондон 46—49, 51, 54, 55, 57, 58,
63, 102, 120, 148.
269
Лорентц 125, 138.
Лоренц 125, 138.
Лугинин 99.
Льюис 38-40, 50, 54, 55.
Лэнгмюр 238.
Маделунг 34.
Майер 35.
Максвелл 10, 124, 154.
Малышев 239, 240, 243.
Мандельштам 12, 191, 192, 243.
Мелликеи 37, 54, 55, 63.
Менделеев 26.
Мицусима 235.
Морзе 179.
Мосотти 124, 128, 138.
Оттербайн 158.
Паскаль 149.
Пастер 95.
Паули 25, 45, 50, 54, 56.
Паулинг 51, 57—61, 65, 97,
102, 108, 135.
Пенни 92.
Перрен 165.
Питцер 248.
Планк 12.
Плачек 194, 210-212.
Прилежаева 232. *
Рабинович 149.
Райе 16.
Раман 191, 241.
Рентген 71, 185.
Розен 48.
Россини 99.
Свентославский 99.
Склар 108, 116-119, 161.
Слэтер 51, 102.
Степанов 66, 207, 222—224, 235
239, 241, 249.
Стивенсон 97, ПО.
Стоке 164.
Стюарт 157.
Сыркии 99, 100, 105, 114, 14*>
228, 232, 241.
Сэзерленд 92.
Сэттон 35.
Теллер 225.
Теренин 185, 202.
Тиле 62.
Тиндаль 185.
Томсен 99.
Уолл 140.
Ухолин 239.
Фаулер 238.
Фаянс 135, 136.
Феофилов 166, 167.
98, Ферми 243.
Ферстер 117, 118.
Франк 197, 198, 200.
Френель 168.
Шайбе 161, 162.
Шен-Ниен-Ванг 158.
Шерер 75.
Шомекер 97, ПО.
Шорыгин 257, 258.
Шредингер 15, 46, 47, 179.
Эдсалл 231.
Эйзенлор 136.
Эйнштейн 12, 202.
Эйринг 175.
Яковлев 239.
ОГЛАВЛЕНИЕ
Стр.
Предисловие 3
Глава L Введение 5
¦Физические и химические свойства вещества 5
Методы теории молекул 7
Основные понятия квантовой механики 10
Световые волны и кванты 10
Волны материи 13
Волновая механика ¦ 15
Гармонический осциллятор 16
Строение атома и периодическая система элементов 19
Строение электронных оболочек атомов 22
Принцип Паули 25
Периодическая система элементов 26
Глава П. Природа химической связи 29
Молекулы, атомы, ионы 29
Ионная связь 32
Энергия ионной решетки 33
Сродство к электрону. . . і 35
Ионная валентность 35
Другие типы электростатических связей 37
Гомеополярная связь 38
Молекула водорода '38
Теория Льюиса 38
„Резонанс" электронов 41
Резонанс в классической и в квантовой механике 42
Связь в молекуле водорода 45
Валентность " 49
Валентные состояния атомов 51
Теория молекулярных орбит 54
Реальная химическая связь 57
Внутренний электронный резонанс ' 57
Многоатомные молекулы 62
Направленные валентности 64
Кратные связи 66
Глава III. Геометрия молекул 68
Размеры молекул 68
Рассеяние рентгеновых лучей и электронных волн 71
Молекулы в газе 72
Рассеяние электронов 73
* Рассеяние жидкостями и кристаллами 74
Вращательные спектры 79
Сводка междуатомных расстояний и углов между связями... 79
Геометрия многоатомных молекул 79
Трехатомные молекулы 79
Четырехатомные молекулы 79
Тетраэдрические молекулы и насыщенные органические
соединения 82
Непредельные органические соединения 83
Пространственная изомерия * . . . 83
Внутреннее вращение и поворотные изомеры 84
Симметрия молекул 88
Асимметричные молекулы 92
Глава IV. Химические связи в многоатомных молекулах 96
Междуатомные расстояния 96
Энергия многоатомной молекулы 98
Теплоты изомеризации 101
Смысл аддитивности энергии 102
Отклонения от аддитивности длин и энергий связей 103
Внешний электронный резонанс в бензоле " 105
Электронный резонанс и длины связей 110
Электронный резонанс в углеводородах и их производных. . . 112
Электронный резонанс и химические свойства молекул 113
Окраска органических соединений 115
Электронный резонанс и неаддитивность 119
Глава V. Электрические свойства молекул 122
Диэлектрическая постоянная 122
Поляризуемость молекул 124
Полярные молекулы 129
Дисперсия поляризации 130
Электронная поляризация 130
Атомная поляризация 132
Молекулярная рефракция и строение молекул ; 133
Ионные рефракции 134
4 Гомеополярные молекулы . . і . , 136
Поляризуемость . . > 138
Дипольные моменты 139
272
Дипольные моменты двухатомных молекул 139
Дипольные моменты многоатомных молекул 141
Отклонения от аддитивности и электронный резонанс 142
Внутреннее вращение и дипольный момент 145
Поляризуемость и дипольный момент 146
Междумолекулярное взаимодействие 146
Магнитные свойства молекул 149
Глава VI. Анизотропия молекул 150
Анизотропия поляризуемости 150
Явление Керра 151
Деполяризация рассеянного света 154
Анизотропия поляризуемости и строение молекул 157
Эллипсоиды поляризуемости связей. 158
Анизотропия электронных переходов 159
Анизотропное поглощение света 160
Люминесценция молекул 162
Поляризация люминесценции 164
Оптическая активность 167
Глава VII. Спектры молекул 176
Колебательные и вращательные переходы 177
Колебания двухатомных молекул 178
Ангармоничность 179
Частоты колебаний и силовые коэффициенты 180
Изотопы 181
Колебания многоатомных молекул 182
Вращение молекул 182
Инфракрасные спектры " 185
Комбинационное рассеяние света (явление Рамана) 190
Квантовая теория 191
Классическая теория 193
Поляризуемость и интенсивности рамановских линий 194
Многоатомные молекулы 195
Электронные спектры 196
Принцип Франка-Кондона 197
Диссоциация молекул 200
Свободные радикалы 202
Предиссоциация 203
Многоатомные молекулы 204
Глава VIII. Колебания многоатомных молекул 206
Связанные колебания и нормальные колебания 208
Симметрия колебаний 209
910
Правила отбора ^іи
Поляризация рамановских линий 211
Некоторые примеры 212
Механика колебаний
Ve"18 Строение молекул. __
Расчет колебаний молекулы 214
Нормальные колебания и нормальные координаты 219
Некоторые -примеры 219
Динамические коэффициенты 221
Молекулы углеводородов и их производных 222
Характеристические частоты и колебания 224
Частоты колебаний и электронный резонанс 227
Карбонильная связь • 228
Другие случаи 232
Колебательный спектр и изомерия молекул 234
Поворотная изомерия ... * 234
Колебательный спектр и междумолекулярное взаимодействие . . . 236
Водородная связь 237
Водородная связь и колебательный спектр 239
Частоты междумолекулярных колебаний 241
Ангармоничность колебаний 242
Ферми-резонанс 2іЗ
Частоты колебаний и термические свойства молекул 244
Внутренняя энергия и теплоемкость молекул 245
Колебательная энергия 248
Интенсивности и поляризации в колебательных спектрах 250
Теория поляризаций и интенсивностей в рамановских спектрах 252
Инфракрасные спектры 256
Интенсивности характеристических колебаний и электронный
резонанс 257
Молекулярный спектральный анализ 258
Структурный анализ 258
Качественный и количественный анализ 259
Литература 260
Предметный указатель 263
Именной указатель 269
М. В. ВОЛЬКЕНШТЕЙН. СТРОЕНИЕ МОЛЕКУЛ.
*
Печатается по постановлению
Ргдакционно-издательского совета
Академии Наук СССР
*
РИСО АН СССР № 2544, подписано к печати
9/1—1947 г. М-00552, типаж 5000, печ. л. 17V*.
уч.-изд. л. 19. Зак. *& 476
*
2-я типография , Печатный Двор" им.
А. М. Горького треста .Полиграфкннга"
ОГИЗа при Совете Министров СССР.
Ленинград, Гатчинская, 26.
ЗАМЕЧЕННЫЕ ОПЕЧАТКИ
Страница
41
58
70
129
Строка
14 сверху
20 снизу
23 сверху
13 сіызу
Напечатано
»! > «j И <*!<. <o's
п г*
СН.С1
Должно быть
е*
я и*
сна.
С-rpotn» молекул. Заж. 476