/



































































![§ 27. Изотопные сдвиги частот колебаний [5]](https://djvu.online/jpg1/b/8/1/b81hMmGuk04vy/068.webp)













































































































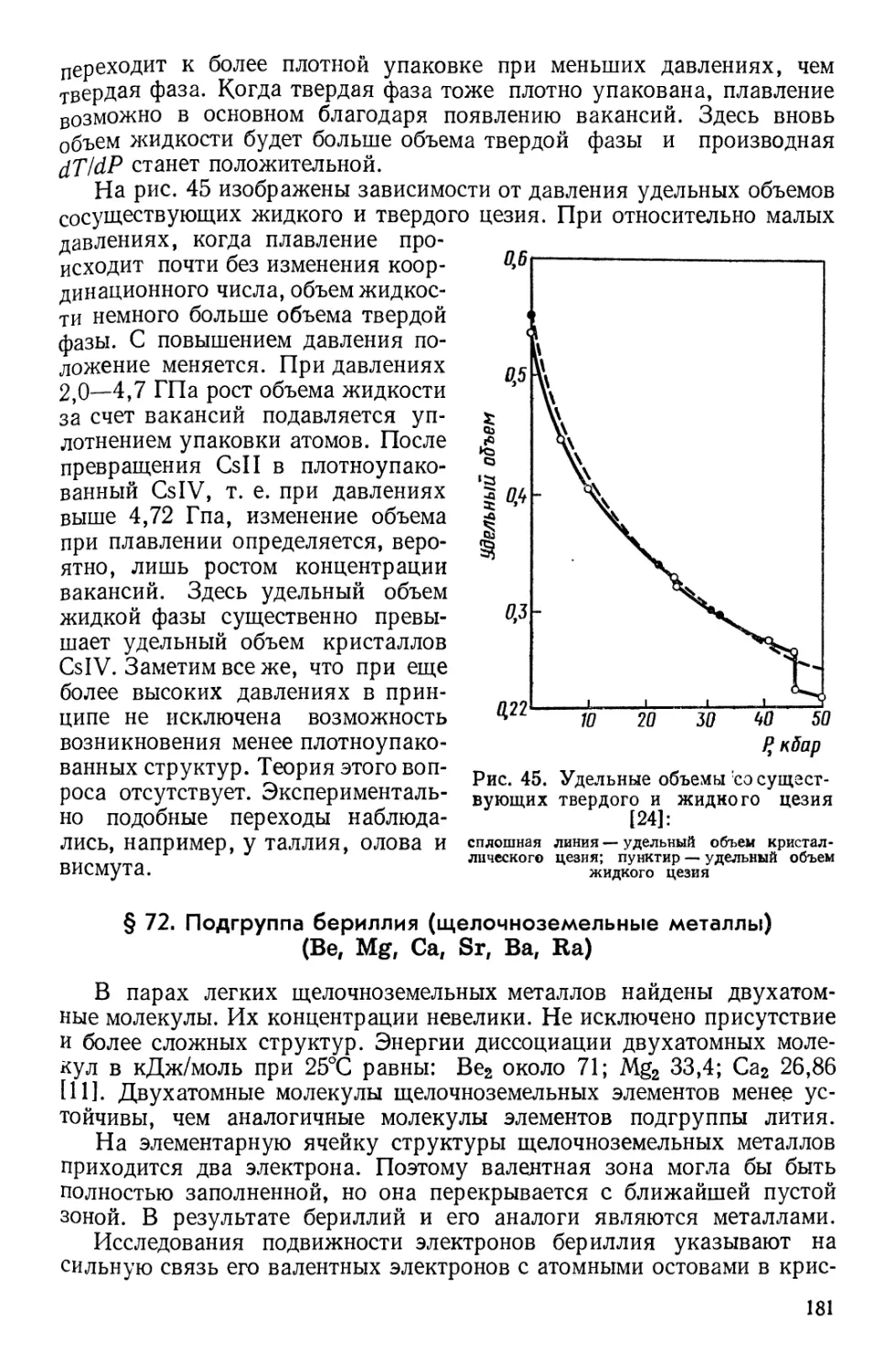













































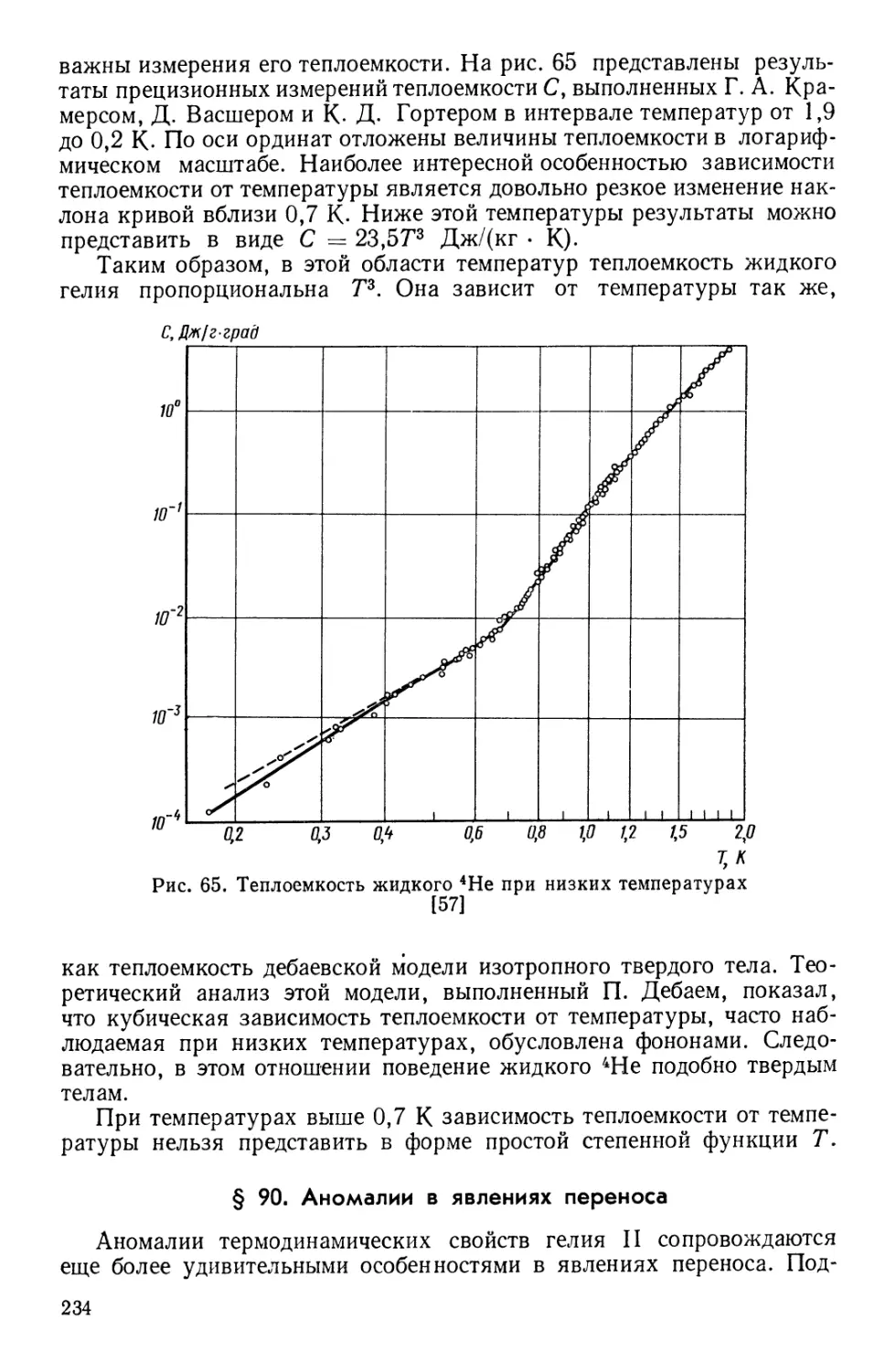








![§ 93. Причины сверхтекучести жидкого гелия [67]](https://djvu.online/jpg1/b/8/1/b81hMmGuk04vy/244.webp)



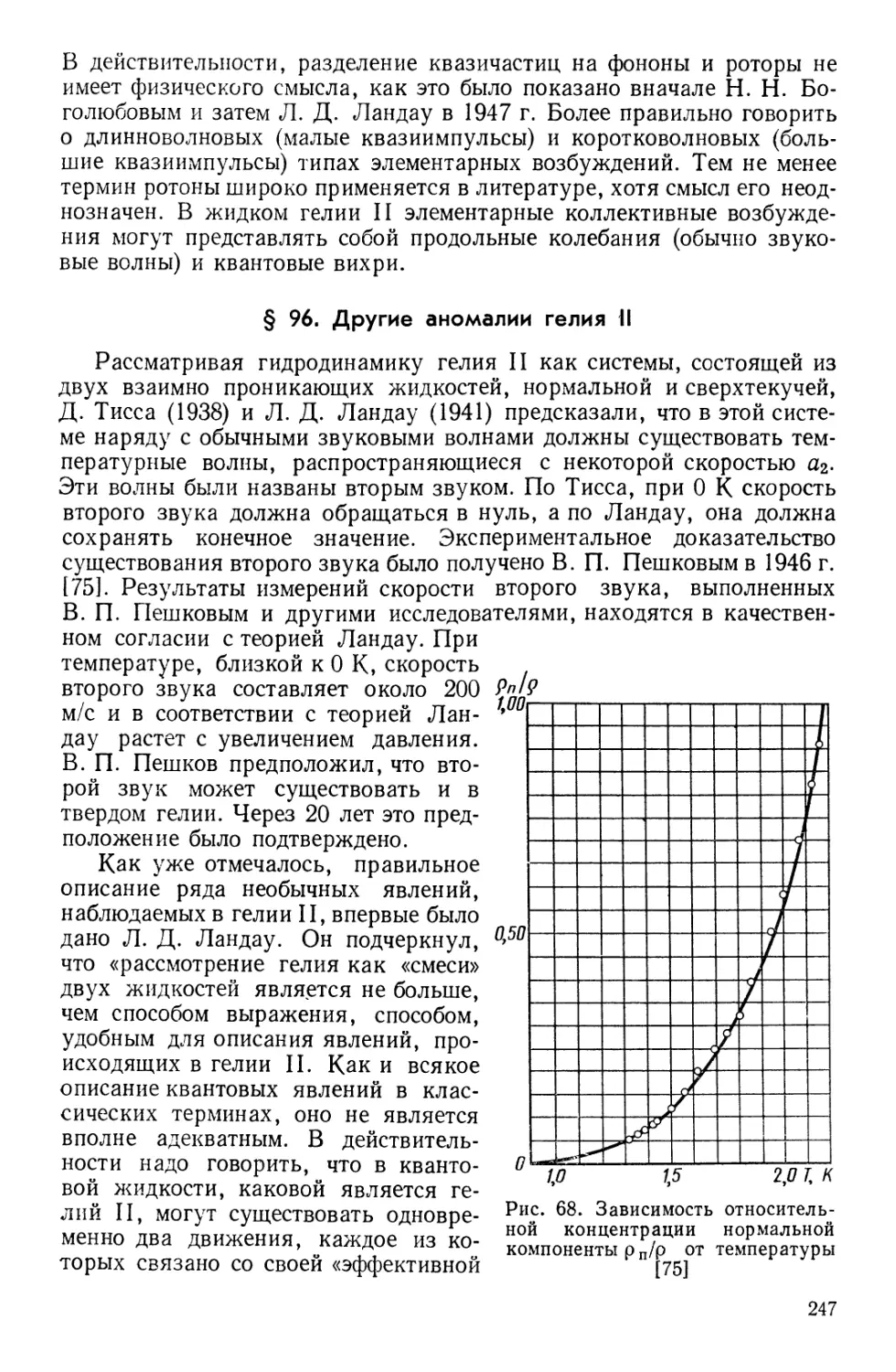
![§ 97. Сверхтекучесть и сверхпроводимость
§ 98. Изотоп 3Не [58]](https://djvu.online/jpg1/b/8/1/b81hMmGuk04vy/249.webp)


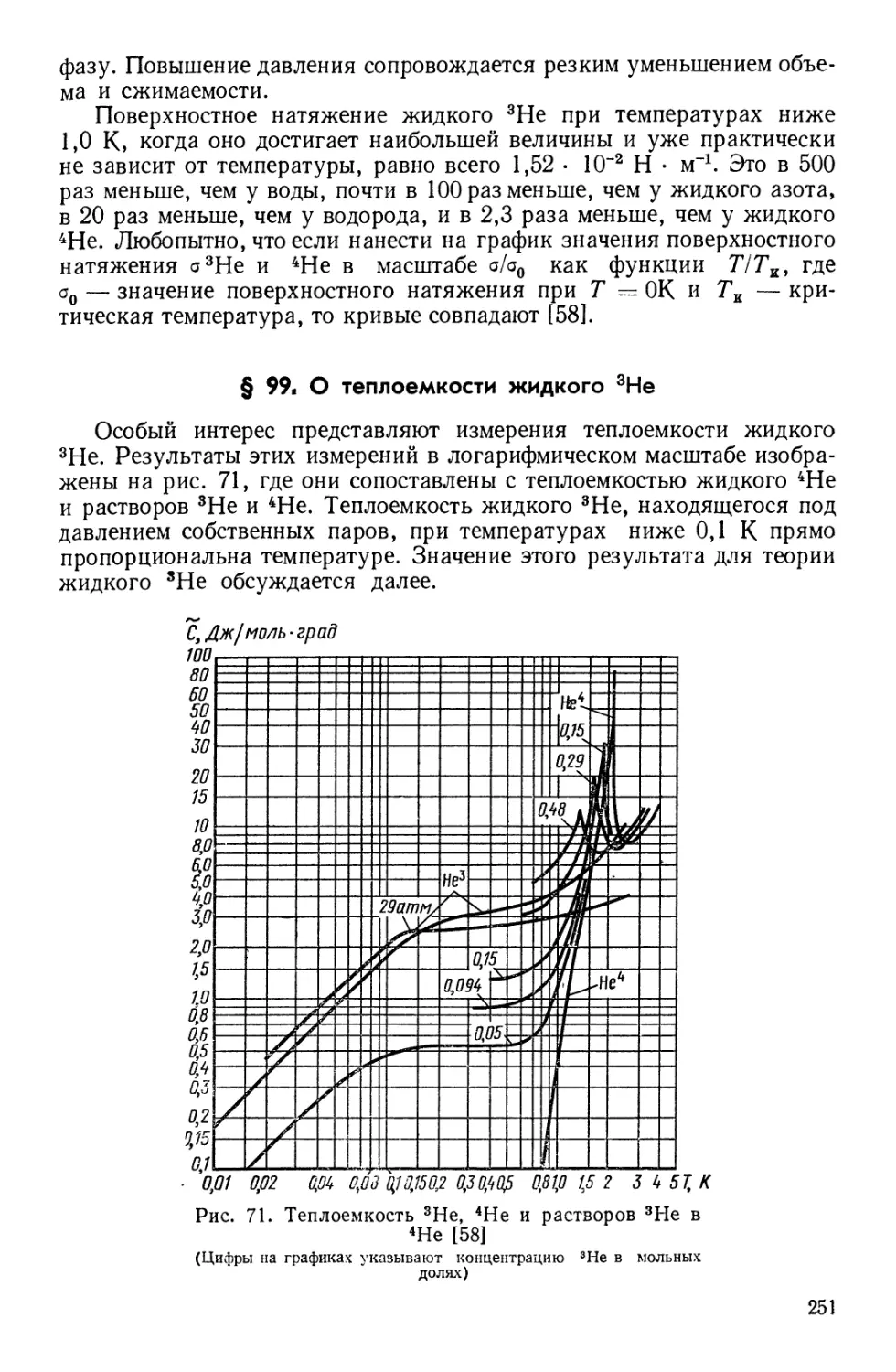



























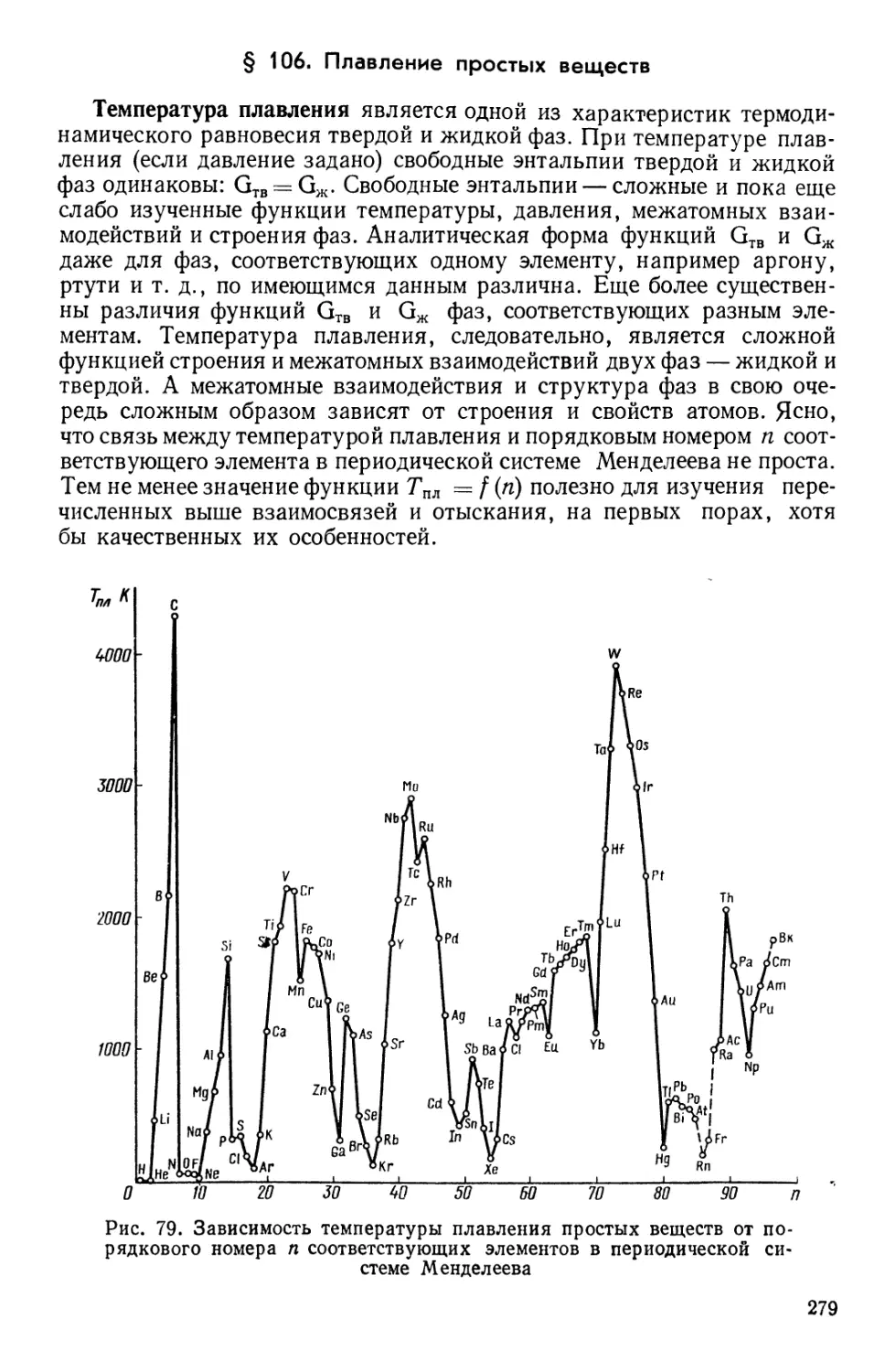




















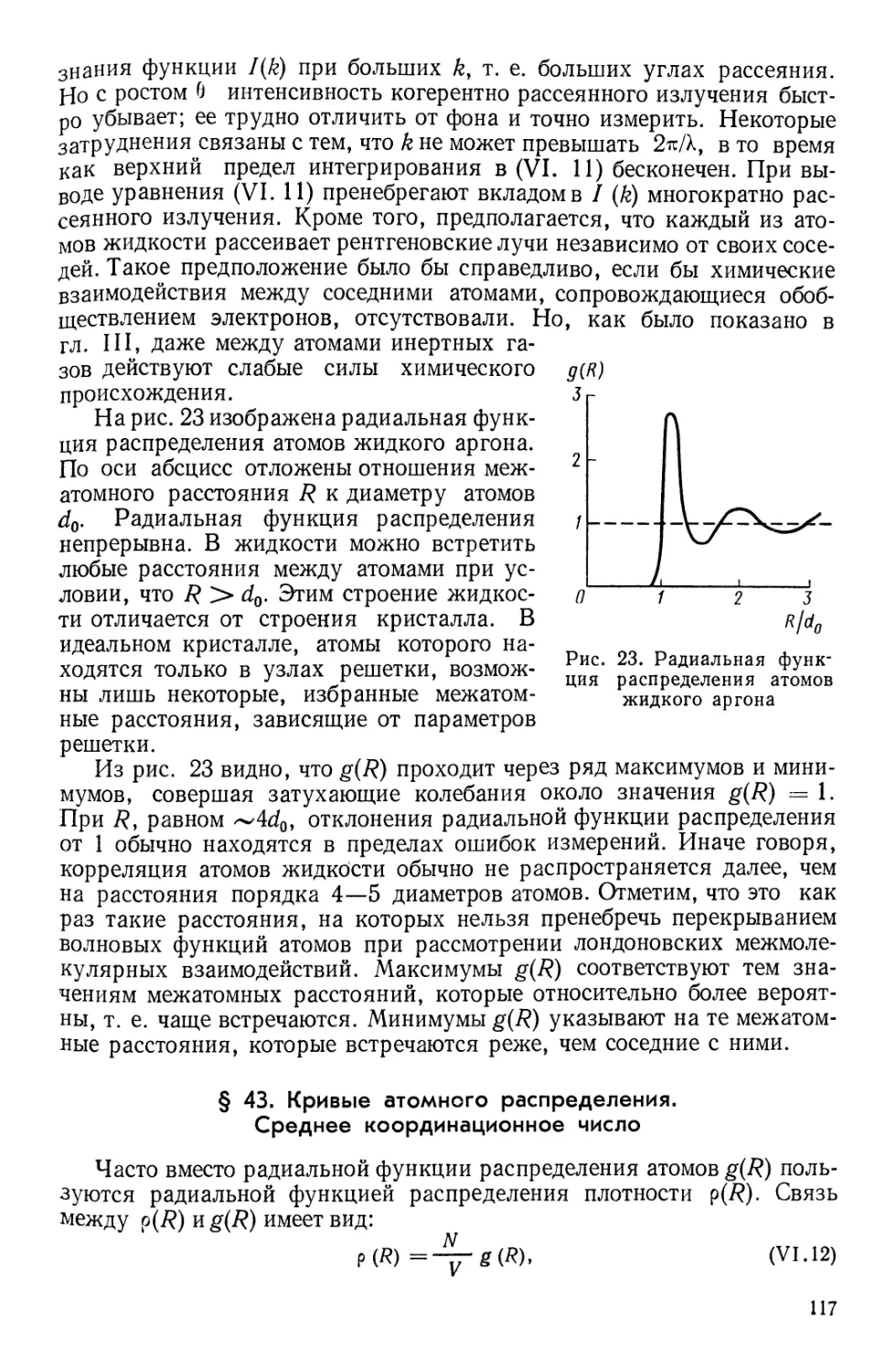
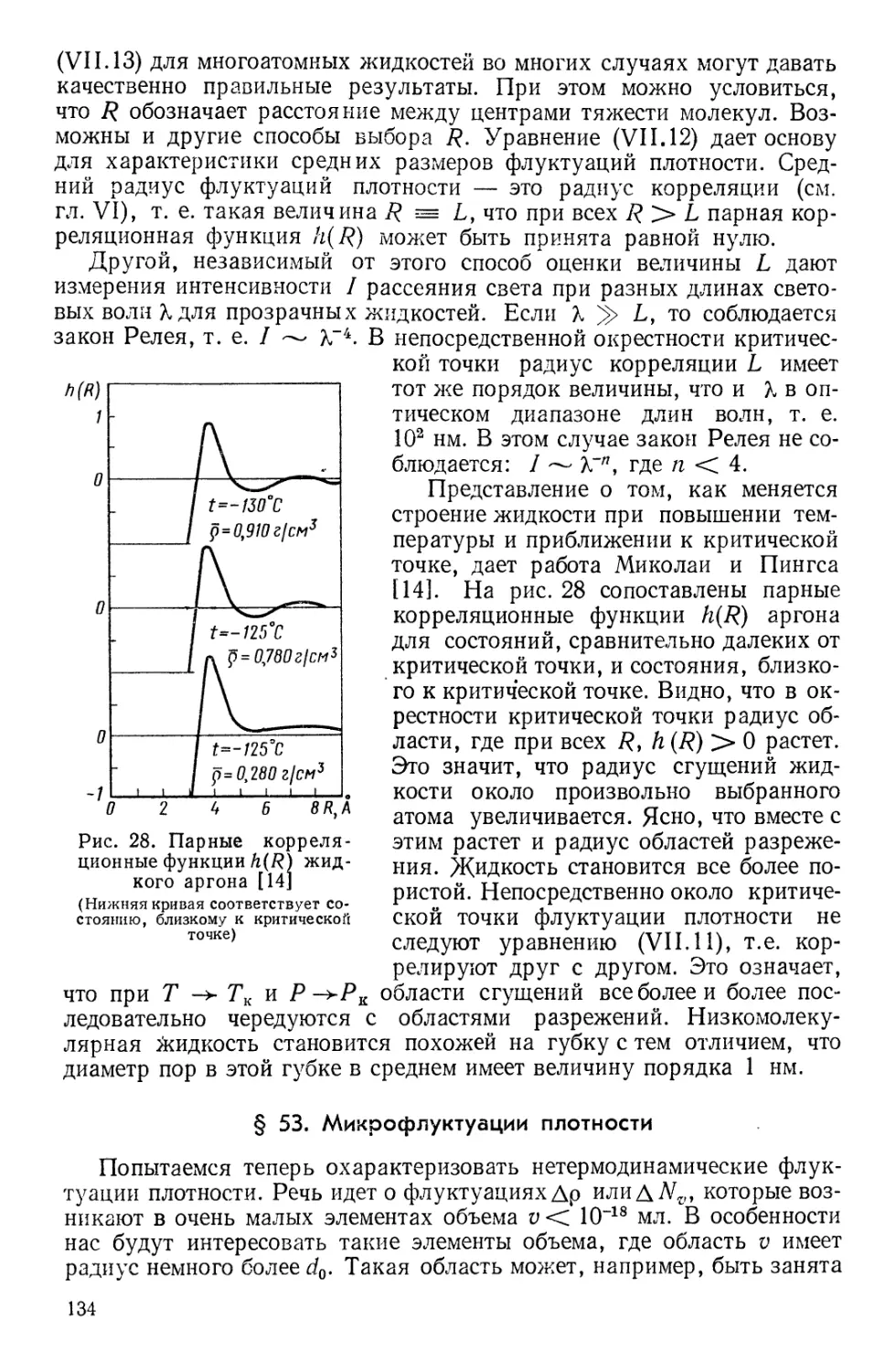
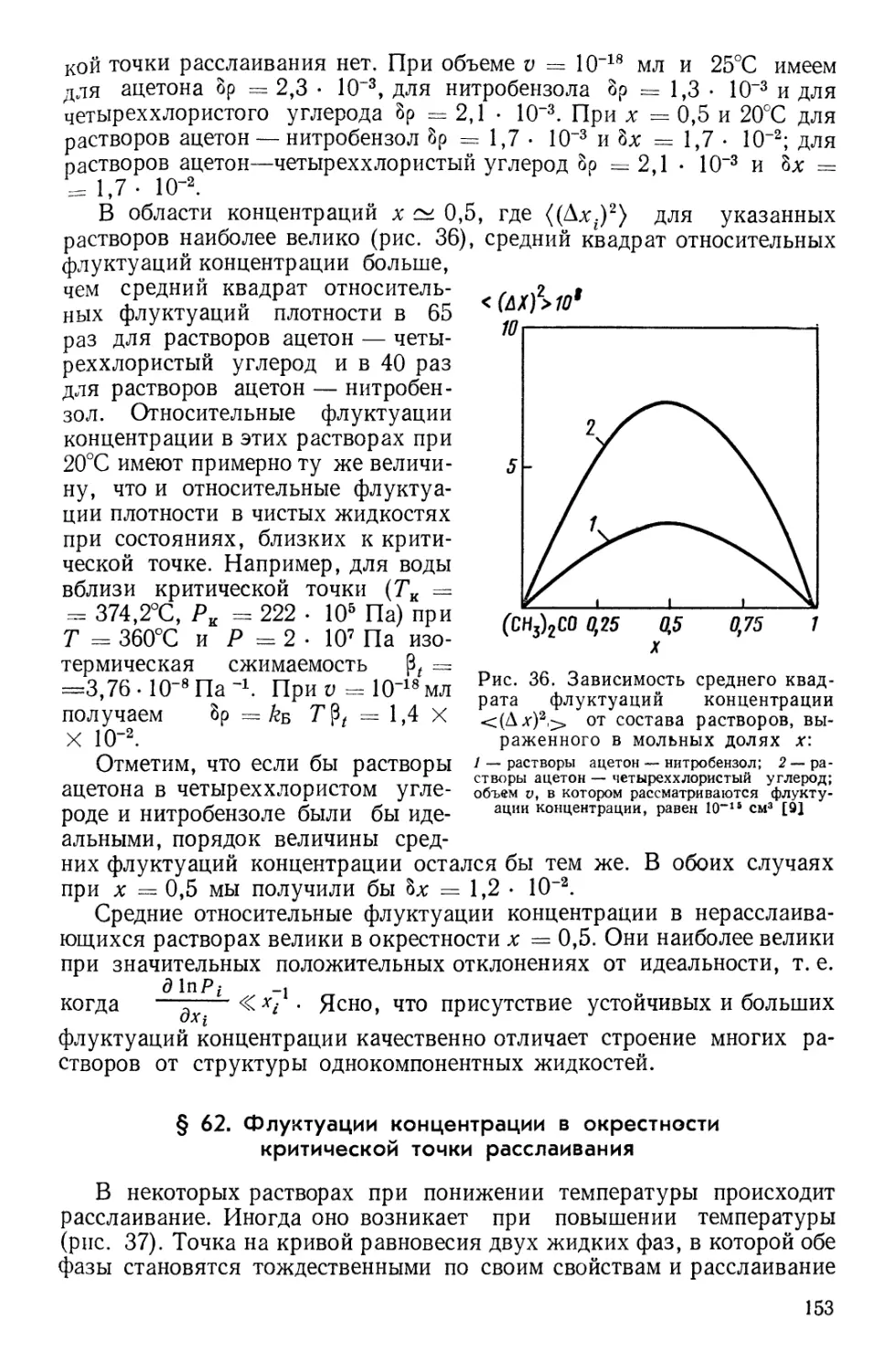
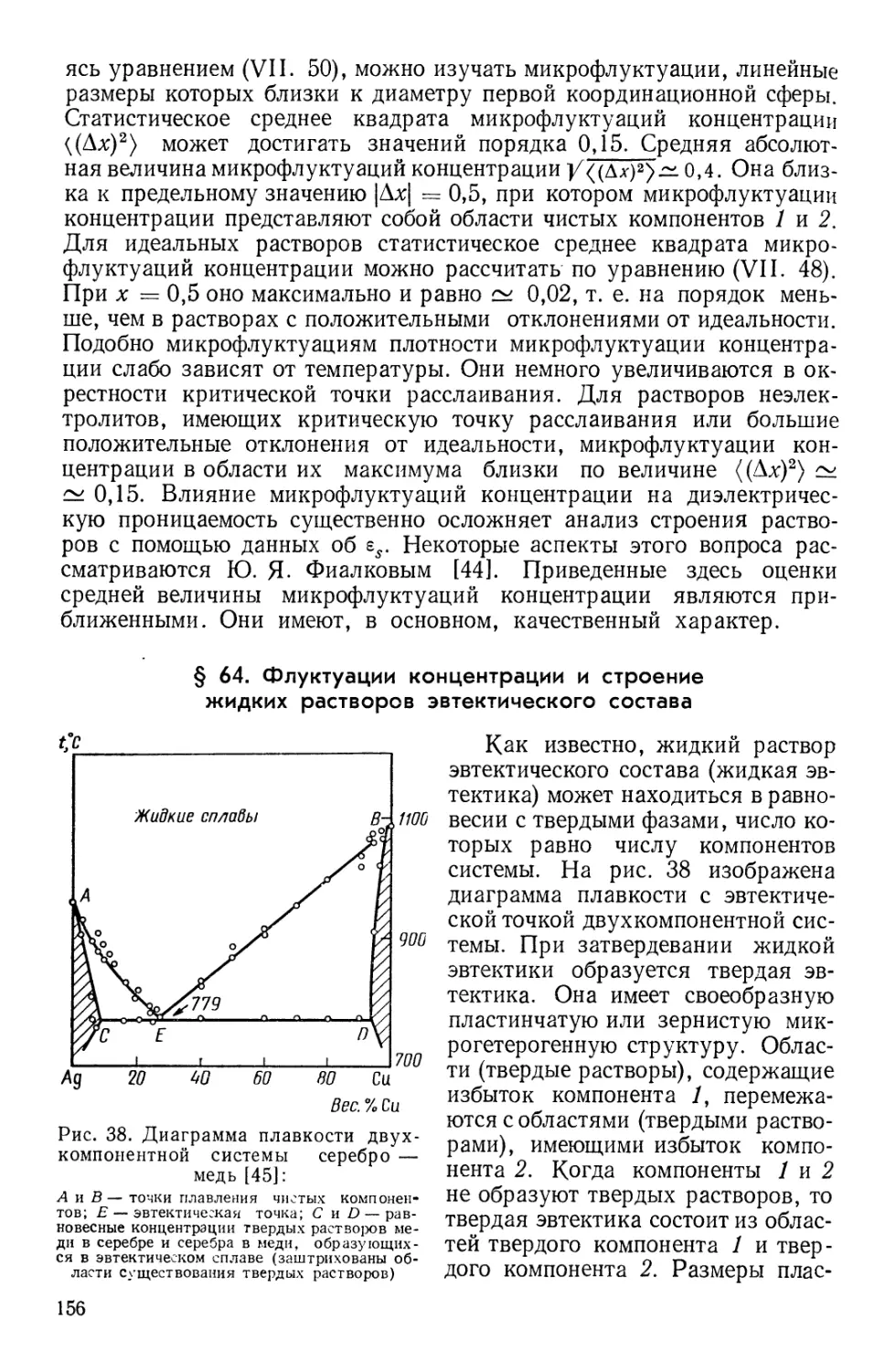
Текст
М. И. ШАХПАРОНОВ
ВВЕДЕНИЕ
В СОВРЕМЕННУЮ ТЕОРИЮ
РАСТВОРОВ
(МЕЖМОЛЕКУЛЯРНЫЕ
ВЗАИМОДЕЙСТВИЯ.
СТРОЕНИЕ.
ПРОСТЫЕ ЖИДКОСТИ)
Допущено
Министерством высшего и среднего
специального образования СССР
в качестве учебного пособия
для студентов
химических специальностей университетов
МОСКВА «ВЫСШАЯ ШКОЛА
1976
541
Ш91
УДК 541. 1(075)
Рецензенты: проф. Глазов В. М. (ин-т электронной
техники) и кафедра физической химии
Ростовского университета (зав. кафедрой
проф. Осипов О.А.).
Шахпаронов МИ.
Ш91 Введение в современную теорию растворов
(Межмолекулярные взаимодействия. Строение. Простые
жидкости). Учеб. пособие для вузов. М.,«Высш.
школа», 1976.
296 с. с ил.
Книга посвящена ряду вопросов теории жидких систем. Излагается
теория межмолекулярных взаимодействий, в том числе теория реактивного
взаимодействия полярных молекул. Рассмотрены методы описания
структуры жидкостей с помощью понятий об ассоциатах и комплексах, о
функциях распределения частиц и флуктуациях. Выполнен систематический обзор
структуры и свойств простых жидкостей, что дает основу для понимания
структуры и ряда свойств более сложных жидких систем: металлов,
полупроводников, диэлектриков, низкомолекулярных жидкостей, полимеров, сте-
клоподобных фаз. Приведено описание строения, свойств и теории простых
квантовых жидкостей.
20503 — 182
Ш 001(01)-76 80~76 541
© Издательство «Высшая школа», 1976 г.
СВЯЗЬ МЕЖДУ ИСПОЛЬЗУЕМЫМИ В КНИГЕ
ЕДИНИЦАМИ ИЗМЕРЕНИЙ В СИ И ДРУГИХ СИСТЕМАХ
Величина
Энергия
Длина
Объем
Давление
Сжимаемость
Удельная
электропроводность
Частота
Вязкость
Электрический момент
Единица
джоуль
метр
Гадетр)3; литр
паскаль=1 ньютон/метр"2
(паскаль)"1
Ом""1-метр"1 =
=сименс/метр
Герц
паскаль-секунда
Связь с другими применяемыми
единицами и пояснения
1 Дж = 0,238844 меж д.
кал = 107 эрг
1 кДж= 103 Дж; 1 МДж =
= 106 Дж
1 Нм = 10"9 м = 10А
1 см3 = 1 мл
1 Па= 10"5 бар=0,986923X
ХЮ-* атм=0,750062-10~2
мм.рт.ст. 1 МПа=106Па;
1 ГПа=109 Па
1 na-i= 10& бар"1
1 Om-i-m"1 = 1 См/м =
= Ю-2 Ом-!-СМ-1
3-1010 Гц = 1 см"1
1 Па-с = 10 пуаз
1 дебай = 10-18 см5/2 X
X г1/2 -сек-i - ID
ОСНОВНЫЕ ОБОЗНАЧЕНИЯ
В книге рассматривается большое число различных величин, поэтому иногда
возникает необходимость применять одно и то же буквенное обозначение для
разных величин. Эти случаи оговорены в тексте. Статистические средние величины
обозначены угловыми скобками <...>; векторные величины напечатаны жирным
шрифтом. Величины, отнесенные к одному молю вещества, обозначаются волной
над буквой, например V — молярный объем. Указатель не является
исчерпывающим. Узкоспециальные обозначения, употребляющиеся с соответствующими
пояснениями лишь в одном каком-либо месте текста, в указатель не входят.
А — интегральная интенсивность полосы спектра
а — скорость звука
В — второй вириальный коэффициент
С — теплоемкость
Су — теплоемкость при постоянном объеме
Ср — теплоемкость при постоянном давлении
с — концентрация в мол-л""1
D — энергия диссоциации молекулы
^о — диаметр изолированного атома или молекулы
d'o — минимальное межъядерное расстояние соседних атомов в твердой
фазе при нормальных условиях
Е — энергия межмолекулярного взаимодействия
£/? - — потенциальная энергия реактивного взаимодействия
8 — напряженность среднего макроскопического поля в диэлектрике
8о — напряженность внешнего поля
б — заряд электрона (ze — заряд иона)
F — свободная энергия
f — напряженность внутреннего поля
G — свободная энтальпия (термодинамический потенциал Гиббса)
g(R) — радиальная функция распределения атомов
Н — энтальпия
д //о — стандартная энтальпия образования
Н — оператор Гамильтона
Н' — оператор возмущения
На — матричный элемент возмущения t ._
h — постоянная Планка; h = 6,626-1 (Г34 Дж-с; h = ft/2rt = l,0545X
XlO"34 Дж-с.
h(R) __ парная корреляционная функция; h(R) = g(R) — 1
/ — ионизационный потенциал
k — волновой вектор
кв — постоянная Больцмана; fe= 1,3805-10"22 Дж-К"1
М — молекулярная масса
М — макроскопический электрический момент
N — число молекул
уул — число Авогадро; Na= 6,02322-1023 моль"1
п — показатель преломления
Р — давление
Рк — критическое давление
Р — электрический момент единицы объема, т. е. поляризация
р — импульс
R — газовая постоянная, Rr= 8314 Дж-К^-моль"1
R — межъядерное расстояние атомов или расстояние между
геометрическими центрами молекул
R — напряженность реактивного поля
г — радиус-вектор
5 — энтропия
S — СПИН
S(k) — структурный фактор
Т — температура, К
Гк — критическая температура; Гкр — температура в начале
конденсации Бозе—Эйнштейна
/ — время
U — внутренняя энергия
и — потенциальная энергия
V — объем системы
Ук — критический объем
v = _ — объем, приходящийся на одну молекулу
X, К, 2 — декартовы координаты
х — концентрация в молярных долях
z — координационное число
а — средняя поляризуемость молекулы
ар — объемный коэффициент расширения
Рг — изотермическая сжимаемость
Р,9 — адиабатическая сжимаемость
Y2 — анизотропия тензора поляризуемости молекул
8 — диэлектрическая проницаемость
8^ — статическая диэлектрическая проницаемость
6 — полярный угол в сферической системе координат
^ — длина волны
|л. — химический потенциал компоненты /
[а — дипольный момент молекул
v — частота в Гц
р — плотность
Р (R) — радиальная функция распределения плотности
о* — удельная электропроводность
9 — азимутальный угол в сферической системе координат
Ч1 — волновая функция
со = 2rtv — круговая частота
ПРЕДИСЛОВИЕ
В конце XIX столетия работы И. Г. Вант-Гоффа, С. Аррениуса,
Д.И. Менделеева и многих других исследователей привлекли к теории
растворов внимание широких слоев общества. Открытия термодинамики
растворов вызывали, примерно, такой же энтузиазм, какой
возникает сегодня, когда говорят о путешествиях в космос, удивительных
возможностях квантовой электроники, успехах молекулярной
биологии.
С тех пор в учебной литературе сложилась традиция ограничивать
теорию растворов законами Вант-Гоффа, Рауля, Генри, теорией
Аррениуса и другими вопросами, связанными с применением методов
термодинамики. Эта традиция поддерживалась тем, что работы по теории
растворов долгое время развивались преимущественно
термодинамическими методами. Но начиная с 50-х годов положение изменилось.
Постепенно ведущую роль стали играть спектроскопия, дифракционные
методы, рассеяние света, радиоспектроскопия, акустическая
спектроскопия. Резко расширились возможности изучения структуры жидких
систем. Стали доступны исследованию новые, ранее неизвестные
молекулярные процессы, в том числе даже такие, которые протекают в
жидкостях в течение 10~10—10~13 с. Не так давно об этом можно было
лишь мечтать.
Феноменологический подход в теории жидких веществ теперь
недостаточен. На передний план вышли проблемы молекулярной теории.
А это означает, в частности, стирание грани между растворами и так
называемыми чистыми жидкостями.
В итоге теория растворов быстрыми темпами двинулась вперед,
обогатилась огромным новым материалом и усложнилась.
Теперь она представляет собой целую систему областей знания, в
которой проблемы термодинамики, статистической физики и кинетики
переплетаются с вопросами квантовой механики, теории строения
молекул, молекулярной оптики, теории диэлектриков, полупроводников и
металлов, молекулярной акустики, квантовой электроники ь и многими
другими.
Поэтому перед каждым, кто рискнет написать учебное пособие по
теории растворов, возникнет необходимость «поднимать целину».
Потребуется излагать не только идеи—квинтэссенцию знания,
полагаясь на то, что дотошный читатель, если у него появятся вопросы,
сможет найти на них ответ в многочисленных толстых монографиях,
написанных на все вкусы. Придется прежде всего описывать факты,
приводить доказательства, на которые опирается теория. Рассмот-
рение всех разделов теории растворов в рамках одного, к тому же не
очень большого по объему учебного пособия вряд ли выполнимо.
В данном пособии мы были вынуждены ограничиться лишь
несколькими проблемами, без которых, на наш взгляд, обойтись невозможно.
В теории растворов имеются два направления. Перед одним из них
ставится задача охарактеризовать строение растворов, те силы, которые
действуют между частицами, по возможности глубже познать
молекулярный механизм множества процессов, протекающих в жидких
фазах. Перед другим — осуществить статистико-механический
расчет свойств жидких систем с помощью их моделей.
В этой книге речь идет только о первом из указанных двух
направлений.
Пособие состоит из трех частей.
Первая часть посвящена теории межмолекулярных сил. Теория
межмолекулярных взаимодействий в неэлектролитах в течение многих
лет выдвигала на первый план дипольные и дисперсионные силы.
Недооценивалась роль реактивного взаимодействия полярных молекул,
весьма существенная в жидких средах. При описании слабых сил
химического типа обычно ограничивались некоторыми, наиболее ярко
выраженными случаями образования водородной связи. Но
водородная связь — лишь одна из бесконечного множества форм слабых
химических взаимодействий, сопровождающихся перераспределением
электронной плотности. В последние десятилетия изучение этих
взаимодействий стало особенно интенсивным. Рассказать о них
необходимо потому, что их исследование имеет большое значение для химии
и ряда областей физики.
Во второй части книги рассмотрены основные понятия и
представления, с помощью которых можно охарактеризовать строение жидких
фаз. Здесь главную роль играют понятия об ассоциатах и комплексах,
о функциях распределения частиц и о флуктуациях. Эти понятия
дополняют друг друга. Каждому из упомянутых трех методов анализа
структуры жидкостей отведена отдельная глава.
Третья часть содержит систематизированное описание структуры
простых жидкостей, т. е. жидких водорода, гелия, лития и т. д.
Строение и свойства простых жидкостей в основном подобны строению и
свойствам растворов. Многие растворы, имеющие большое научное и
прикладное значение, состоят из простых жидкостей. Поэтому
исследование строения и свойств простых жидкостей — один из важнейших
путей изучения жидких растворов.
Предлагаемое в третьей части сжатое описание строения простых
жидкостей позволяет дать обзор особенностей, которые присущи
структуре не только простых, но в большинстве случаев и сложных жидких
систем: металлов, полупроводников, диэлектриков,
низкомолекулярных жидкостей, полимеров, стеклоподобных фаз. Большинство
химических процессов протекает в жидких средах, поэтому исследования
структуры жидкостей полезны для многих разделов химии. Отметим,
что XI глава книги посвящена простым квантовым жидкостям —
изотопам гелия. Этот очень интересный и важный раздел теории жидких
систем мало освещен в учебной литературе. Значение квантовых жид-
костей в науке и практике велико и быстро растет. Квантовые
жидкости, вероятно, — одна из широко распространенных форм
существования материи во вселенной. Можно думать, что квантовые жидкости
со временем займут надлежащее место не только в специальных, но и
в общих курсах строения вещества.
Автор признателен тем своим ученикам и сотрудникам, которые
принимали плодотворное участие в обсуждении проблем, излагаемых
в книге, и помогали при ее оформлении. Автор глубоко благодарен
М. Е. Мартыновой, М. И. Лупиной, Т. М. Усачевой, В. А. Сырбу и
М. М. Поплавской за их большую помощь в работе над рукописью, а
также проф. В. М. Глазову и проф. О. А. Осипову за полезные
критические замечания.
ВВЕДЕНИЕ
§ 1. Растворы
Растворами называют фазы, состоящие из двух или большего числа
независимых компонентов. Таковы, например, растворы солей или
сахара в воде.
Независимость компонентов означает, что концентрации их частиц
при заданных внешних условиях (например, температуре и давлении)
можно изменять независимо друг от друга.
Хорошо известно, что научные определения, как правило, отражают
лишь некоторые характерные особенности проблемы, поэтому они
ограничены и в какой-то мере условны. Это относится и к приведенному
определению растворов. В самом деле, при рассмотрении однокомпо-
нентных систем обычно отвлекаются от их изотопного состава. Если же
его учесть, то почти все встречающиеся в природе однокомпонентные
фазы —тоже растворы. Например, аргон является разбавленным
раствором изотопов 36Аг и 38Аг в 40Аг.
Для молекулярной теории растворов существенное значение имеет
то, что растворы состоят из неодинаковых частиц: атомов, ионов,
молекул. Частицы могут различаться составом, конформацией, состоянием
возбуждения. Поэтому для молекулярной теории растворов большой
интерес представляют не только двух- или многокомпонентные фазы,
но и однокомпонентные вещества, в том числе простые вещества. В
жидком аргоне, даже полностью очищенном от изотопных примесей,
как мы увидим далее, кроме атомов аргона имеются неустойчивые ассо-
циаты Аг2, Аг3, ..., Агп, где я может принимать большие значения. В
аргоне имеются возбужденные атомы, распределенные в среде,
состоящей из невозбужденных атомов аргона. Химически чистый я-гексан
состоит из различных конформеров. Молекулы гексана могут также
различаться изотопным составом и состоянием возбуждения. Около
13% молекул четыреххлористого углерода при 20°С находится в
возбужденных колебательных состояниях, поэтому химически чистый
СС14 тоже имеет общие черты с растворами. Кроме того, жидкий четы-
реххлористый углерод содержит не только мономерные молекулы СС14,
но и ассоциаты.
Молекулярная картина явлений, протекающих в
многокомпонентных фазах, во многом сохраняет особенности, присущие процессам,
идущим в однокомпонентных системах. Различие между однокомпо-
нентными системами и растворами в указанном узком смысле слова
существенно с позиций термодинамики. С точки зрения молекулярной
теории это различие не так уж принципиально.
Наука о растворах — одна из наиболее старых областей
естествознания. В древности вся химия и немалая часть физики были учением
о свойствах растворов. «Corpora non agunt nisi soluta» (тела не
действуют друг на друга, если они не растворены), — утверждали
алхимики. Надо сказать, что с известными ограничениями это" высказывание
не потеряло смысла и в наше время.
Теория растворов изучает свойства, строение, а также механизмы
тех процессов, которые протекают в растворах. Развитие науки о
растворах неразрывно связано с формированием неорганической и
органической химии, молекулярной физики, химической термодинамики,
химической кинетики, науки о полимерах и т.д. [1, 2]. Вместе с тем
менялось содержание теории растворов. Она продвигалась от изучения
сильных химических взаимодействий к слабым межмолекулярным
взаимодействиям химического и физического характера [1, 2].
Когда в жидких растворах идут химические процессы,
сопровождающиеся образованием прочных химических связей, то они играют
столь яркую роль, что многие другие сопутствующие им физические
и химические явления нередко не привлекают внимания. В растворах
со слабым химическим взаимодействием этого не происходит и задача
изучения других физико-химических явлений упрощается. За
последние десятилетия в теории растворов исследования систем со слабым
химическим взаимодействием стали занимать центральное место. Они
позволили не только лучше понять природу этих взаимодействий, но и
выявить их важную роль в обычных химических реакциях,
сопровождающихся образованием более устойчивых химических соединений.
Исследование систем со слабым химическим взаимодействием
позволило продвинуться вперед в понимании механизмов электрической
поляризации, вязкого течения, поглощения звука, переноса энергии
возбуждения, образования и исчезновения флуктуации и многих других.
Изучение этих явлений обогащает и углубляеть представления о
механизме обычных химических реакций.
Здесь и далее рассматриваются только жидкие растворы и, как
частный, но важный случай, так называемые чистые (однокомпонентные
жидкости). Всюду предполагается, что жидкие системы изотропны.
Как уже было сказано, речь идет о жидкостях, в которых есть
атомы, молекулы, ионы. Жидкости типа ядерной материи, быть может, со
временем тоже станут предметом теории растворов.
§ 2. Строение жидкостей
Жидкости — макроскопические тела, состоящие из атомных ядер
и электронов. Они имеют определенный объем, но текучи и способны
принимать форму тех сосудов, в которых находятся. Строение
жидкости означает способ распределения ядер и электронов в пространстве,
занятом жидкой фазой.
Строго говоря, все ядра и электроны любого образца жидкости
взаимосвязаны. Они — единое целое. Вопрос о том, каково строение жид-
кости, имеет следующий смысл. Во-первых, можно ли и с какой
степенью точности подразделить жидкую фазу на атомы, молекулы, ионы,
как структурные единицы? Что представляют собой эти частицы? Чем
отличается их состояние от состояния аналогичных атомов, молекул
или ионов, когда они почти изолированы друг от друга в разреженном
газе? Ответы на эти вопросы известны далеко не всегда.
Во-вторых, если жидкость состоит из атомов, молекул или ионов,
то как они распределены? Имеются ли в жидкой фазе упорядоченные
образования, состоящие из атомов, молекул или ионов? Что они собой
представляют и как связаны друг с другом? Например, жидкий аргон с
хорошей степенью точности можно считать состоящим из атомов аргона,
почти не отличающихся от его атомов в разреженном газе. Но в жидком
аргоне, как показывает опыт, каждый атом окружен в среднем
приблизительно восемью соседними атомами. Это напоминает расположение
атомов в объемно-центрированной кубической решетке. Вода состоит
из молекул Н2О, которые взаимосвязаны друг с другом так, что
образуют сложную пространственную сетчатую структуру, отчасти
напоминающую структуру льда.
Аргон и вода — жидкости, структурные элементы которых
выражены четко. Частицы этих жидкостей почти те же, что и в разреженных
газах. В металлах подобного сходства нет. Структурные элементы
металлов — это атомы и почти свободные коллективизированные
электроны, образующие с атомами единое целое. В парах металлов таких
структурных образований нет.
§ 3. Взаимодействия между молекулами
Классификация. Строение и свойства жидких систем зависят от
взаимодействий между молекулами. Жидкости занимают определенный
объем и сопротивляются сжатию потому, что между молекулами
действуют силы отталкивания. Образование жидкой фазы при конденсации
паров происходит благодаря силам притяжения молекул друг к другу.
Термин «молекула» здесь означает какую-либо частицу жидкости,
необязательно электрически нейтральную.
Силы можно подразделить на близкодействующие и дальнодействую-
щие. Близкодействующие силы возникают при соприкосновении
частиц; к ним относятся силы отталкивания и химические связи.
Химическая связь образуется в результате обобществления электронов
при сближении молекул. Энергия химических связей может достигать
величин порядка 400 кДж-моль"1 — это сильные химические связи.
Химическая связь может быть и очень слабой с энергией порядка
1 кДж- моль"1. Близкодействующее отталкивание, как мы увидим,
в основном вызвано ростом кинетической энергии электронов при
сближении молекул.
Дальнодействующие силы подразделяются на электростатические
взаимодействия между ионами, металлическую связь и силы Ван-дер-
Ваальса, названные так в честь голландского физика Я. Д. Ван-дер-
Ваальса, известного своими исследованиями газов и жидкостей.
10
Металлическая связь представляет собой взаимодействие ионов с
электронным газом в конденсированной фазе—жидкости или
кристалле. В отличие от химической связи число частиц, непосредственно
взаимодействующих друг с другом, здесь очень велико. В принципе
каждый из электронов связан со всеми ионами, имеющимися в жидкости.
Такая связь наблюдается в жидких металлах и отчасти в
полупроводниках.
Энергия взаимодействия. Пусть две молекулы cud находятся
на расстоянии R друг от друга. Примем, что молекулы пребывают в
стационарных состояниях, т. е. их свойства не зависятот
времени.Предположим далее, что положения атомных ядер в молекулах
фиксированы. Молекулы не изменяют своей конфигурации и не вращаются.
Атомные ядра в молекулах не колеблются. В этом приближении
полная энергия изолированной молекулы зависит только от состояния ее
электронов. Колебания ядер и другие их перемещения обычно не
вносят существенных изменений в энергию молекул, поэтому в данном
случае ими можно пренебречь. Такой способ приближенного
описания молекул был предложен М. Борном и Д. Р. Оппенгеймом в 1927 г.
и назван ими адиабатическим приближением. Адиабатическое
приближение справедливо, когда движение электронов не испытывает резких
изменений при малых перемещениях ядер.
Пусть <|>с и tyd обозначают волновые функции молекул cud, когда
они изолированы друг от друга, т. е. когда R ->- <х>. В соответствии с
общими требованиями квантовой механики волновые функции ^си <Ъа
есть однозначные и непрерывные функции координат электронов,
имеющихся в молекулах cud соответственно. Квадрат модуля каждой
волновой функции должен быть интегрируемым. Каждая из волновых
функций антисимметрична, т. е. меняет знак при перестановке
(взаимном обмене пространственными и спиновыми координатами) любых
двух электронов в молекуле. Для операторов Гамильтона каждой из
Л Л
молекул введем обозначения Н с и Нd\ энергии молекул обозначим через
Ес и Ed. Поскольку молекулы находятся в стационарных состояниях,
уравнения Шредингера для них имеют вид:
(B.I)
Оператор Гамильтона системы, состоящей из двух изолированных мо-
Л Л
лекул end, представляет собой сумму операторов Н с и Hd:
АЛЛ
Н^^Нс + На, (В.2)
где индекс оо означает, что молекулы с и d бесконечно удалены друг от
друга. Энергия системы равна сумме энергий изолированных молекул:
Е^^Ес + Еа. (В.З)
Это соотношение удовлетворяется, если волновая функция системы
II
представляет собой произведение волновых функций изолированных
молекул
«К» = 4^. (В.4)
что легко показать с помощью уравнения Шредингера:
л /л AV . . л . . л ( (
: Е<» ^оо • (В.5)
Допустим теперь, что расстояние /? между молекулами с и d пе
настолько велико, чтобы можно было пренебречь взаимодействием между
л
электронными облаками этих молекул. Тогда оператор Гамильтона Я
л
не равен #«>:
л л л
H = Hm + H't (B.6)
л
где оператор Нг учитывает изменение (возмущение) состояния еистемы
в результате взаимодействия между молекулами.
До тех пор, пока электронные облака молекул с и d не
перекрываются, волновая функция системы в принципе может быть представлена с
помощью (В.4) как произведение волновых функций изолированных
молекул. Если же расстояние R таково, что электронные облака
молекул end перекрываются (точнее сказать, если'их перекрыванием
нельзя пренебречь), то волновая функция системы <]> отличается от <]>«> хотя
бы уже потому, что должна быть антисимметричной по отношению к
перестановкам пространственных и спиновых координат электронов
молекул как с, так и d, поскольку электронные облака этих молекул
теперь образуют единое целое.
Полная энергия системы Е в принципе может быть найдена с
помощью уравнения Шредингера:
л
Яф = £<|/, (В.7)
откуда следует, что
р
р л | л
Е = ] ф*Я^о = <ф I H
(В.8)
где интегрирование производится по координатам всех электронов*»
Условимся, что разность
AE = E-EW (B.9)
представляет собой'энергию взаимодействия молекул end. Так как
л
оператор возмущения Н' и волновая функция системы ф зависят от
расстояния R между молекулами, то энергия взаимодействия тоже явля-
* В уравнении (В.8) и далее использована символика, предложенная
Дираком. (См. П. А. М. Дирак. Принципы квантовой механики. М., Физматгиз, I960.
12
ется некоторой функцией /?. Энергия системы может быть определена с
точностью до некоторой постоянной, выбор которой обычно диктуется
соображениями удобства. Мы можем выбрать эту постоянную так,
чтобы Е°о была равна нулю. Тогда Д £ = Е и знак разности Д можно
опустить. В дальнейшем символ Е будет обозначать энергию
взаимодействия. Поскольку энергия Е определена с помощью уравнения (В.8),
она (с точностью до произвольной постоянной) есть полная энергия
системы, т. е. сумма квантовомеханической средней* кинетической
энергии электронов [Гэ] и квантовомеханической средней
потенциальной энергии электронов и ядер [иэ,я 1
Я = [Гэ]+[м8.яЬ (вл°)
С другой стороны, энергия Е есть функция R и может рассматриваться
как потенциальная энергия взаимодействия между молекулами end.
Как известно, сила, с которой каждая из молекул действует на
другую, связана с E(R) соотношением:
В адиабатическом приближении [3]
dE(R)
dR
= <ф
ди
э.я
Ф > . (В.12)
где ф — волновая функция системы, состоящей из молекул с и d (ф
есть функция координат всех электронов; ф зависит также и от
координат ядер, как от параметров, характеризующих состояние системы);
&э,я — полная потенциальная энергия всей системы, т. е. сумма
попарных кулоновских электростатических энергий взаимодействия между
всеми возможными парами частиц, иэ,я зависит от координат всех ядер
и электронов, имеющихся в молекулах end; абсолютная величина
производной ^э'я равна электростатической силе, которая действо-
dR
вала бы между молекулами end, если бы не только положения ядер,
но и координаты электронов были бы фиксированы. Интегрирование в
(В.12) производится по координатам всех электронов, при этом
происходит квантовомеханическое усреднение электростатической силы,
действующей между молекулами с и d\ R, как и прежде, расстояние
между центрами молекул.
Когда R настолько велико, что перекрыванием электронных
облаков молекул end можно пренебречь, взаимодействие между моле-
* Термин «квантовомеханическое среднее» означает здесь пространственное
усреднение по волновой функции основного состояния, например
13
пулами называется дальнодействующим. При перекрывании
электронных облаков возникает близкодействующее отталкивание и, кроме того,
может образоваться химическая связь.
Литература
1. П. И. В а л ь д е н. Теории растворов в их исторической
последовательности. Петроград, 1921.
2. Ю. И. Соловьев. История учения о растворах. М., Изд-во АН СССР,
1959.
3. А. С. Давыдов. Квантовая механика. М., «Физматгиз», 1973.
ЧАСТЬ ПЕРВАЯ
МЕЖМОЛЕКУЛЯРНЫЕ ВЗАИМОДЕЙСТВИЯ
ГЛАВА 1
ДАЛЬНОДЕЙСТВУЮЩИЕ МОЛЕКУЛЯРНЫЕ СИЛЫ
§ 4. Оператор возмущения #'
Предположим, что R велико и перекрыванием электронных облаков
молекул cud можно пренебречь. В этом случае, как уже говорилось в
§ 3, волновая функция ф системы, состоящей из молекул end, может
быть представлена как произведение волновых функций отдельных
изолированных молекул.
Примем, что обе молекулы электрически нейтральны и находятся в
своих основных квантовых состояниях, тогда
Гамильтониан системы:
л л
(1.2)
л
Следовательно, оператор возмущения Н' равен изменению
потенциальной энергии системы приуменьшении расстояния от бесконечно
большого до R:
Н' = Аи = и — ит , (1.3)
где //со — потенциальная энергия двух изолированных молекул с и
d. Для вычисления энергии системы надо найти оператор возмущения
Н' = ди.
Потенциальная энергия системы и зависит от положений зарядов в
молекулах с и d и расстояния между молекулами; и вычисляется с
помощью закона Кулона методами классической электростатики. Будем
считать, что обе молекулы находятся в вакууме. Осложнения,
возникающие, когда молекулы с и d окружены другими частицами,
рассмотрим позже.
Взаимодействие двух атомов водорода. Рассмотрим самый простой
случай. Пусть каждая молекула состоит из электрона и протона, т. е.
15
является атомом водорода. Допустим, что положение зарядов
фиксировано, например, так, как показано на рис. 1. Тогда, пользуясь
обозначениями, смысл которых ясен из этого рисунка, получим:
1 ^ ll 1N (1.4)
,1 1
и = е*[ — + — —
1 R г
г12
•е*
Так как
— е2( + ) = */«..
ТО
1
/"21
(1.5)
Преобразуем это выражение к виду, удобному для расчетов.
Построим систему декартовых координат X, У, Z с началом в точке, где
расположен заряд +ес>
ориентировав ее так, чтобы ось Z была
направлена по линии, соединяющей заряды
-\-ес и +ed. В точке, где
помещается заряд -\-ed, расположим начало
другой, вспомогательной системы
координат X', Y', Z'. Пусть оси Z
и Z' совпадают, а оси X и Х'\ Y и
У параллельны. Обозначим
координаты зарядов в системе
координат XYZ: + г,(0,0,0); + ed (0,0,
^)» ес \Ас> YC,ZC)\ ed (Xd, Yd9
Zd).
Так как Х'*=Х; Y'=Y и
Zr = Z—R, то расстояния г, г12 и
г21 между зарядами можно представить с помощью следующих
соотношений:
у У
Рис. 1. К расчету энергии взаим о
действия ^двух атомов водорода
r= [(xd-
(1.6)
Следовательно,
+ и —
R
Пусть расстояние R между + ес и + ^ много больше, чем ги и г22.
У Z' Р 1 1 \
у р у с ^
Тогда R > Хс, У с, ...? Z'd. Разложим г"
и r\x в ряд по степе-
16
ням малых параметров а = 2^£-,... , * по формуле
R R
(1+af1 + na + a+... .
Подставляя результат в уравнение (1.5), получим:
H'=-JjT (XcXd + YcY'd - 2Z* zd) + ° (*~4). (l-7>
Таким образом, потенциальная энергия взаимодействия двух молекул
ди = Н' при расстояниях i?, значительно превышающих размеры
молекул, может быть представлена в виде ряда по степеням R'1. В
правой части уравнения (1.7) приведен лишь первый отличающийся от
нуля член этого ряда.
Взаимодействие двух молекул. Теперь усложним задачу.
Предположим, что в молекуле с имеется та атомных ядер с зарядами е с1 ,..., е ста
и mv электронов. В молекуле d содержится па атомных ядер с зарядами
edi> •••> edna и ^v электронов.
Допустим, что условие электронейтральности каждой из молекул
сохраняется. Тогда
2 ew = ec = -mve; 2 eda = ed=-n*e> (1.8)
a=l a=l
где е — заряд электрона.
Примем, что положения всех зарядов в молекулах end
фиксированы. Расстояния между атомными ядрами в каждой из молекул
соответствуют экспериментальным данным о строении этих молекул.
Электроны расположены неподалеку от атомных ядер.Величины
eda rda
(1.9)
представляют собой радиусы-векторы, определяющие
местонахождение центров тяжести положительных зарядов в некоторых двух
произвольно выбранных декартовых системах координат, которые мы будем
обозначать индексами «с» и «rf». В уравнениях (l.9)rcanrda —
радиусы-векторы атомных ядер, имеющих заряды еса и eda
соответственно.
Поместим начала обеих координат в точки, где находятся центры
тяжести положительных зарядов молекул с и d. Тогда rta и /va будут
равны нулю. Ориентируем оси координат вновь так, как на рис. 1:
оси Zc и Zd пусть совпадают; оси Xс, Хд иУс, Yd параллельны.
Координаты центров тяжести электронов будут определяться
следующими соотношениями:
*с= 2 eXcJm,e = Xc/nn; (1.I0)
17
Zd =
где
Формально мы можем рассматривать молекулу с как систему,
состоящую из двух зарядов. Заряд +гюе находится в центре тяжести
положительных зарядов; заряд — mv e—■ в центре тяжести отрицательных
зарядов. Точно так же и молекула d формально может
рассматриваться как система, состоящая из зарядов -\-п*> ей — п^ еу располагающихся
в центрах тяжести положительных и отрицательных зарядов этой
молекулы. Если расстояние R между центрами тяжести положительных
зарядов молекул cud велико, то это упрощение не повлияет на
результаты наших расчетов. Таким образом, мы, по существу, снова
возвращаемся к задаче, рассмотренной в начале этого параграфа. Проводя
вычисления так же, как и при выводе формулы (1.7), увидим, что
множители rav и /2V сокращаются. Первый отличный от нуля член
разложения Аи в ряд по степеням R'1 имеет вид:
Уравнение (1.11) по форме совпадает с (1.7). Отличие состоит в том, что
Xс, ..., Zd представляют собой суммы координат электронов, если
начала декартовых систем cud находятся в центрах тяжести
положительных зарядов молекул с и d и системы координат ориентированы
так, как на рис. 1. Если бы обе молекулы были ионами, то первый член
ряда в разложении Да по степеням R~l был бы пропорционален R~l.
Если бы одна из молекул была бы электронейтральна, а другая нет,
то ряд начинался бы с члена, пропорционального R~2.
§ 5. Метод возмущений
Молекулы cud могут находиться в основном квантовом состоянии
или возбужденных квантовых состояниях:
О, 1, 2, ..., /г, ..., т, для молекулы с
О, 1, 2, ...,/, ...,п, для молекулы d
Соответственно и система, состоящая из взаимодействующих друг с
другом молекул cud, может находиться в основном или каком-либо
возбужденном состоянии, когда одна или обе молекулы возбуждены.
Обозначим электронные состояния системы индексами
0,1, ...,/,...,/>.••
Л
Если оператор возмущения Н' представляет собой лишь малую по-
правку к оператору Н^ невозмущенной системы, то энергия взаимодей-
18
ствия молекул системы, находящейся в /-том квантовом состоянии,
может быть представлена в виде
Ег = Е'£+Е]9 (1.12)
где Ei и Et — вклады в энергию взаимодействия, вычисляемые с
помощью метода возмущений в первом или соответственно втором
приближении этого метода. Когда собственные значения оператора
л
Гамильтона #<» не вырождены, то [1]
.1,(0)
(1.13)
^i ~~ LJ £(0) __
S\ij\
£(0) Ы0) . (1Л4>
Л
где ф(9> — собственная функция оператора //«> невозмущенной
системы, находящейся в /-том квантовом состоянии; ЕР и £;0) —
энергия невозмущенной системы, находящейся в квантовых состояниях i
и /; индекс (0) здесь и далее указывает, что речь идет о
невозмущенном состоянии системы; Ни- — матричный элемент возмущения:
Я'|ф)0)>. (1.15)
Уравнения (1.13) и (1.14), как уже было сказано, справедливы для
невырожденных невозмущенных систем, но они формально применимы и в
случае вырожденных систем, если при вычислении матричных
элементов возмущения в качестве волновых функций выбрать некоторые
линейные комбинации функций ф^. Отметим также, что метод
возмущений применим при условии, что
I //•• I <^\ Е^ Е^ I (I 16)
т. е. матричные элементы возмущения должны быть малы по сравнению
с соответствующими разностями невозмущенных уровней энергии
системы.
§ 6. Энергия взаимодействия
в первом приближении метода возмущений
Взаимодействие двух молекулярных диполей. Оператор дипольного
момента молекулы в принятых нами обозначениях имеет вид:
где X = ^ xv ; Y = 2 rv ; z = 2 z* ; ix, iy и iz — единичные век-
V V V
торы, направленные вдоль осей X, Y и Z.
19
В энергетическом представлении, т. е. когда волновые функции
молекулы являются собственными функциями энергии системы,
значения оператора дипольного момента образуют матрицу М, элементы
которой имеют вид:
где h — постоянная Планка.
Если i Ф /, то амплитуда матричного элемента дипольного момента
носит название дипольного момента перехода ц .;-:
Пусть молекулы cud находятся в основных квантовых состояниях.
Если их дипольные моменты не равны нулю, то
I** = < feo 12 ercv | Фсо > Ф 0; (1.20)
ltd =«М 2 erd* | <Ы > ф 0.
Энергия взаимодействия молекул £од, обусловленная присутствием в
них постоянных дипольных моментов, может быть найдена с помощью
первого приближения метода возмущений:
£од = Ео = < № |
л
Подставляя значение Я' = Д и из уравнения (1.11) и вводя согласно
(1.17) и (1.20) следующие обозначения компонент постоянных
дипольных моментов молекул cud, находящихся в основных квантовых
состояниях:
v-cox = < feo I еХс | Фсо > ;
(1.22)
PdOZ = < Нч/о I eZrf | <Ы >.
.получим
Переходя к сферическим координатам, имеем
РсОХ = ^ sin ^ cos Чс\ V-dOX
^сОУ = V-c sin ^c sin ?c5 V'dOY = ^rf sin ^ sin ?rf'»
H-cOZ = ^ cos ^» ^0Z = ^ COS &rf.
Подставляя значения ц сОх , ..., [i doz в (1.23), получим
£од == ^°^° [sin Ьс sin ^ cos (yc - yd) - 2 cos bc cos ^]. (1.24)
Полярные углы 0- c и ft d и азимутальные углы ср с и ср d
определяют направления дипольных моментов молекул ji с0 и ц rf0.
Дипольные моменты молекул могут быть определены экспериментально.
:20
Зная величины дипольных моментов, с помощью формулы (1.24) можно
вычислить энергию дипольного взаимодействия молекул, если заданы
расстояния R между диполями и углы, определяющие ориентацию
диполей в пространстве.
Если, например, дипольные моменты молекул ц с и \i d равнонаправ-
лены вдоль оси Z, т. е. &с=0 , В^=0или &с=180°, ftd = 180° и <?c= cpd,
то £Од отрицательно и имеет минимальное значение. Тогда молекулы
притягиваются друг к другу и энергия их притяжения — -Бд.тахмакси-
мальна. Если дипольные моменты молекул противонаправлены вдоль
ochZ, т. еЛс = 0, $d =180° или Ьс =180°, bd = Ои срс = cpd, то
они отталкиваются друг от друга с энергией Еп.тах. Когда & с = 90°,
Ьа = 90° и срс—cpd — 90°, то энергия дипольного взаимодействия
молекул end равна нулю, и т. д.
Уравнение (1.24) можно записать и в следующей форме:
£од = R'3 Ко № - 3 (|»с /?) (rirf Л) / Л«]. (1.25)
Если воспользоваться сферической системой координат, общей для обеих моле*
кул, то уравнение (1.24) преобразуется к такому виду [2]:
У [cos Ъ'е cos b'd + sin ^ sin *'d cos ( cp; - ^ ) -
— 3 (cos ^ cos ^ + sin §'c sin ^^ cos (cp^ — cp^) . (cos bd cos ^^ +
+ sin bd sin ^ cos ( ?j* — <pi )] . (1.26)
Здесь ^с, фс; &d, Ф^; ^, ф/? — углы, определяющие ориентацию векторов
l*roi ^o и /? в этой, общей для обеих молекул системе координат.
Усреднение по ориентациям. Допустим,что молекулы с и Смогут
принимать по отношению друг к другу любые ориентации. Если
молекулы полярны, т. е. имеют постоянные дипольные моменты, то
энергия взаимодействия молекул зависит от их взаимной ориентации и
поэтому различные ориентации в общем имеют разную вероятность. В тех
случаях, когда ЕОл определяется уравнением (1.24), представляет
интерес ее значение, усредненное по любым ориентациям, при некотором
постоянном значении R между точечными дипольными моментами:
-ОД
= f f Еоле kbT d*c d<*d / [\ e kbT d<»cdcs>di (1.27)
где dco = sin&d&dxp; T — температура, К; ks — постоянная Больцма-
на. Интегрирование в (1.27) производится по ft от 0 до тс и по <р от 0 до
2тг. Здесь и^далее, если не будет специально оговорено, скобки <...>
обозначают операцию статистического усреднения.
Примем, что £од <^С&б Т (как будет показано ниже, это
допущение хорошо соответствует действительности), тогда е ~Е°^ kb T можно
разложить в ряд
Еол
— k T "•"•••
21
Ограничиваясь первыми двумя членами ряда и учитывая, что
< cos 9- > = < cos 9 > = < sin 9- > = < sin cp > = 0;
1 2 1
<cos2ft>= — ; <sin2 9>=—-; <Cos2<p>= , (1.29)
о 3 2
получим
2 2
<£Од > — — • (1.30)
3/гБ Г #6
Пример. Дипольный момент молекул пиридина С5Н5 N равен 2,2 дебая
(D). Пусть расстояние R между двумя небольшими молекулами сравнительно
велико — R = 2 нм. Расчеты по формуле (1.24)_дают £Од>тах = 0,12- КГ21 Дж =
= 0,03 кв Т при Т =300 К- Среднее расстояние R между центрами соседних
молекул жидкого пиридина может быть оценено по формуле:
гдер — плотность; М — молярная масса; Na — число Авогадро. При 300 К
плотность жидкого пиридина р = 980 кг/м3 и R = 0,63 нм.
Сопоставим R с вандерваальсовыми размерами молекулы пиридина. Эта
молекула напоминает плоский шестиугольник, ее «толщина» в среднем составляет
около 0,4 нм. Поэтому R имеет тот же порядок величины, что и размеры молекул.
Уравнение (1.24) применимо, если R на порядок превышает размеры молекул.
Расчет по формуле (1.24) взаимодействия между соседними молекулами, т. е.
при R— 0,6 нм для молекул пиридина, не имеет смысла.
Но если такой расчет все-таки произвести, то получится, что £од,тах=
= 0,95 кв Т и разность энергий дипольного взаимодействия при
параллельном и антипараллельном расположении диполей двух соседних
молекул пиридина в жидкой фазе при 300 К составляет около 2 къ Т>
что по порядку величины близко к энергии слабого химического
взаимодействия молекул. Такого рода совпадение (при отсутствии до
недавних пор надежных экспериментальных методов исследования
слабых химических взаимодействий) является одной из причин того, что
влияние дипольного взаимодействия между молекулами в жидкой фазе
сильно переоценивалось. Другая причина, как будет показано в гл.
II, состоит в том, что при описании межмолекулярных взаимодействий
обычно не учитывалось влияние реактивного поля, создаваемого
полярными молекулами.
При R > 0,4 нм энергия дипольного взаимодействия £од <
<&б Т. Средняя энергия дипольного взаимодействия молекул во много
раз меньше £од. Для молекул пиридина при R =2 нм <£од>= 6,7х
Х10"25 Дж, т. е. 1,6-10"4 кБТ при Т - 300 К.
Отметим, что вопрос о том, при каких расстояниях между
молекулами разложение в ряд (1.7) справедливо, до сих пор не изучен. Ясно,
что такое разложение справедливо лишь, когда расстояние между
молекулами много больше суммы радиусов молекул R > d0 и теряет
силу при R порядка d0. Более точные критерии расстояний, при
которых возможно разложение в ряд (1.7), отсутствуют.
Другие члены разложения энергии вандерваальсового взаимодействия в ряд па
степеням/?"1. Формулы (1.23)—(1.26) выражают лишь ту часть степенного ряда,
22
описывающего зависимость £Од от R, которая связана с дипольным членом
(1.7) при разложении потенциальной энергии кулоновского взаимодействия Аи
в ряд по степеням R'1. Другие члены степенной функции Ео' = f(R) могут быть
получены тем же путем с помощью первого приближения метода возмущений.
Приведем результаты расчетов без вывода [2]. Пусть ес' и е'd — заряды частиц
с и d, Qd и Qc — квантовомеханические средние квадрупольных моментов* этих
частиц. Энергия кулоновского взаимодействия двух заряженных частиц (ионов)
Вклад в энергию, обусловленный взаимодействием свободного заряда е ' частицы
с с дипольным моментом \i d частицы d
Ео =__!i^L
1 ' • ' #2
Энергия взаимодействия заряда ес' с квадрупольным моментом Qd
Здесь предполагается, что квадрупольный момент цилиндрически симметричен.
Углы 9- иф характеризуют ориентацию оси симметрии квадрупольного момента.
Взаимодействие дипольного момента^ и квадрупольного момента Q, вносит
следующий вклад в £0':
--.cp^]. (1.36)
Квадруполь-квадрупольное взаимодействие имеет энергию:
E0(Q, Q) = ^Г1 {1 -5cos* Ъс-5 созП^ с ^
+ 2 [sin Ьс sin bd cos (cpc — yd) — 4 cos bc cos ^]2} . (1.37)
Статистическое усреднение по всем углам для фиксированных значений R дает:
ес)
* Пусть имеется система зарядов е. (i = 1, 2, 3, ...). Квадрупольный
момент этой системы — симметричный тензор, компоненты которого Qa3 определяют
уравнениями:
где г,—радиус-вектор, определяющий положение г-того заряда г., Х.а , Х$
(а, р = 1, 2, 3) — декартовы компоненты радиуса-вектора г.; да$ —г символ
Кронекера. 6*$ равно единице при а = р и равно"нулю при a =j= P-
Предполагается, что начало координат находится внутри рассматриваемой системы
зарядов.
23
iL42>
§ 7. Лондоновские (или дисперсионные)
взаимодействия
При больших расстояниях между молекулами, когда
обобществление электронов еще не происходит, движение электронов в молекулах
все-таки до известной степени взаимосвязано. Электроны молекулы с
как бы отталкиваются от электронов молекулы d или «избегают» их.
Эта корреляция движений электронов является еще одной причиной
возникновения межмолекулярных взаимодействий. Теория таких
взаимодействий впервые была развита Ф. Лондоном в 1930 г.
Рассмотренный им вид вандерваальсовых сил Ф.Лондон назвал дисперсионными
силами. В настоящее время распространен термин «лондоновские» силы.
Для отыскания энергии лондоновского взаимодействия между
молекулами применяется второе приближение метода возмущений, т. е.
уравнение (1.14):
Числитель в членах суммы (1.14) всегда положителен, а знаменатель
может быть как положительным, так и отрицательным, в зависимости
от знака разности Е\0)—£(0). Когда обе молекулы находятся в
основных квантовых состояниях, разность £]0)—£"|°> отрицательна при
любых значениях /, отличающихся от нуля. Поэтому энергия Е"о
отрицательна, т. е. молекулы, находящиеся в основном квантовом
состоянии, притягиваются друг к другу под влиянием лондоновских сил.
Если же одна или обе молекулы находятся в возбужденных состояниях,
то одни члены суммы (1.14) будут отрицательными, а другие
положительными. Для молекул, находящихся на достаточно высоких уровнях
возбуждения, большая часть суммы (1.14) будет состоять из
положительных слагаемых, так что в итоге энергия Е". будет положительной,
т. е. лондоновское взаимодействие будет сопровождаться отталкиванием
молекул.
При комнатной температуре подавляющее большинство молекул
обычно находится в основном квантовом состоянии, поэтому
наибольший интерес представляет расчет величины Е"о:
\Ног\2
где
24
= Есо + Ed*', E}V = Eck + Edl. (1.44)
Матричный элемент возмущения
I *Л + ^ ^ - 2ZC Zd I ^ 'Ы. 5(1.45)
Как уже говорилось, такое выражение для матричного элемента
возмущения применимо, если предполагается отсутствие перекрывания
между волновыми функциями молекул с и d. Так как здесь
фигурируют волновые функции не только основных, но и всех возбужденных
состояний молекул, то приближения, на которых основывается вывод
уравнения (1.45), применимы лишь при значительно больших
расстояниях, чем это обычно предполагается.
Рассмотрим, например, два атома водорода, имеющих одинаковые
спины, расположенные на расстоянии R = 0,3 нм. Их боровские
радиусы равны 5,29-10~2 нм. При таком расстоянии между атомами
перекрывание орбиталей основных состояний пренебрежимо мало. Но
величина Я, при которой нельзя пренебречь перекрыванием волновых
функций, быстро растет с ростом главного квантового числа п. Если атомы
водорода находятся не в основном Is, а в одном из ближайших
возбужденных состояний 2р, то перекрыванием волновых функций можно
пренебречь лишь при R = 0,9 нм.
Амплитуды компонент дипольного момента перехода между
квантовыми состояниями «0» и «Ь молекулы с и дипольного момента
перехода между квантовыми состояниями «0» и «/» молекулы d имеют вид:
(1.46)
eZd I tydl) = <<htt I eZd I 1W •
В (1.46) учтено, что в силу эрмитовых свойств матрицы М, \ъсОкх =
"м|1лй*= vbuz- Подставляя (1.46) в ■ (1.45), получим:
Hoi = Н*о = ^"3 (V-cokx Pdoix + Pcoky Pdoiy ~ fycokz Vdoiz)- (I -47)
Переходя от декартовых координат к сферическим (см. § 6), будем
иметь:
/t ^оч
(1.48)
[s\nbcok si
V-doi
где ф (ft, <p) = [sin $cOk sin ^o/ cos (<?cOk — cprf0/) — 2 cos ftcOjfe cos ftdod.
Углы $cOk, <?cOkf Ьт и <?dOl определяют ориентацию дипольных
моментов перехода (i^ n\idOl . Подставляя (1.48) и (1.44) в (1.43), придем
к выражению:
фЬ
25
В уравнении (1.49) ориентация дипольных моментов перехода
учитывается множителем Ф|/(&,<р). Если частицы имеют сферическую форму,
то все ориентации дипольных моментов перехода можно считать
равновероятными. Усредняя Ф^ОК <р) по всем возможным значениям углов
О- и <р, находим:
2
(1.00)
^Г
I асос асо^ о>
где rf(oc = sin $cOk dbcOk dycOk; dud = sin bdol dbd{n dyd(H .
Таким образом, энергия лондоновского взаимодействия молекул cud
в основном квантовом состоянии, усредненная по всем возможным ори-
ентациям дипольных моментов перехода, имеет вид:
*
aJi Jmd EcQ + Ed0 — Eck — Edt
k I
k -h / > 1
Уравнение (1.51) можно написать подробнее, выделив в нем члены,
содержащие (л^до u\i2m, т.е. постоянные дипольные моменты
невозбужденных молекул с и d. ixc00 ^ jj.^ и^аоо = iid определяются
уравнениями (1.20). Тогда
jimd c^ ch
k>\
}- (1.52)
}
EcQ + Ed0 — £сЛ — Edt I
Если одна или обе молекулы в основном квантовом состоянии неполяр-
ны, то один или оба члена в фигурных скобках уравнения (1.52),
зависящие от \х с и ii d, обращаются в нуль. Вклад этих членов в Е^
незначителен и мы рассмотрим его позже. Основной вклад в
вносит третий член в фигурных скобках этого уравнения. Он не зависит
от того, есть или нет у молекул постоянные дипольные моменты. Эта
часть < Е"о >> обусловлена лондоновским взаимодействием молекул,
поэтому будем обозначать ее символом Еол:
3 Zi
со + dQ Eck — Edi
k>\ l\
(1.53)
Эта формула неудобна для расчетов, потому что величины yLcQk и
рт , как правило, неизвестны. Чтобы преобразовать уравнение (1.53)
к виду, пригодному для вычислений, Ф. Лондон воспользовался
результатами квантовой теории показателя преломления света,
описывающей дисперсию показателя преломления, т. е. зависимость его от
частоты колебаний волн света. Отсюда и возник термин «дисперсионные»
26
силы. Если поместить молекулу в слабое внешнее электрическое поле
8, то молекула поляризуется. В молекуле индуцируется электрический
момент, пропорциональный 8:
A|i = agf (I.54)
где a — поляризуемость молекулы; a — скаляр, когда молекула
поляризуется одинаково, независимо от направления внешнего поля.
Такая молекула обладает изотропной поляризуемостью. Примерами
могут служить атомы инертных газов. Многоядерные молекулы, как
правило, поляризуются неизотропно. Поляризуемость молекулы в
этом случае представляет собой тензор второго ранга. Тогда под а
подразумевают так называемую среднюю поляризуемость, т. е. одну треть
следа этого тензора,
1
Зная среднюю поляризуемость молекул и плотность жидкости или газа,
можно с помощью уравнения Лоренца — Лоренца вычислить
показатель преломления. Поэтому теория дисперсии показателя преломления
в основном сводится к отысканию зависимости а от частоты световых
колебаний.
Когда поле 8 постоянно и изотропная молекула находится в
основном квантовом состоянии, ее поляризуемость
2
3 жшА E0-Et
1 + 0
где |i0/ — амплитуда дипольного момента перехода между
квантовыми уровнями 0 и /. Наибольший вклад в сумму (1.56) вносят те члены,
для которых разность энергий Ео— Et мало отличается от энергии
ионизации молекулы /. Они соответствуют таким состояниям, когда
электрон наиболее слабо связан с молекулой и поэтому легче меняет
состояние движения при наложении внешнего поля. Строгого
теоретического доказательства этого утверждения в литературе нет, но
экспериментальные данные для многих молекул, по-видимому, ему не
противоречат. Следовательно, можно приближенно принять, что
1Ф0
где суммирование производится по тем состояниям молекулы, которые
близки к ионизированному. Точно так же и в уравнении (1.53)
основную роль играют состояния молекул с и d, при которых Eck—Ecq~I с и
Edl—Ed0 ~ I d. Поэтому для изотропных молекул уравнение (1.53)
можно преобразовать при помощи соотношения (1.57) к виду
Это и есть формула Лондона. Величины ас, ady /c и Idопределяются
экспериментально.
27
Уравнение (1.58) применяется для полуколичественной оценки
энергии лондоновских взаимодействий молекул, форма которых не
очень сильно отличается от сферической. Для несимметричных молекул
теория лондоновских взаимодействий пока что слабо развита.
Энергия лондоновского взаимодействия по порядку величины
близка к энергии дипольного взаимодействия, когда полярные молекулы
имеют большие дипольные моменты ц > 2D и расстояние R
между молекулами не очень велико. С ростом R энергия лондоновского
взаимодействия уменьшается так же, как и средняя энергия
дипольного взаимодействия <£1ОД>, но гораздо быстрее, чем неусредненная по
ориентациям молекул энергия £Од. Формула Лондона в принципе
непригодна для расчета взаимодействий между соседними молекулами в
жидкой фазе. Значения R, при которых применима формула Лондона,
по меньшей мере на порядок должны превышать сумму радиусов
молекул.
Примеры. Рассмотрим снова в качестве примера пиридин.
Ионизационный потенциал молекул пиридина / — 9,76 э. в. [3] (225 ккал/моль). Средняя
поляризуемость а=9,31- КГ24 см3. Пусть R — 2 нм. Учитывая сказанное, при
такой величине R перекрыванием волновых функций возбужденных молекул
вряд ли можно пренебречь. Расчет по формуле Лондона дает Еол = — 1,4-10""24 Дж,
т. е. ЕОл = — 4-10~4 кв Т при Т = 300 К. Если же принять R = 0,63 нм,
то Еол = — 0,4 кв Т. Но для таких значений R, по порядку сравнимых с
размерами невозбужденных молекул, формула Лондона заведомо неприменима.
Взаимодействие при очень больших расстояниях между молекулами.
При очень больших расстояниях R, порядка 103 нм и более,
удовлетворяющих неравенству
Я»-у-, (1.59)
где с — скорость света; / — ионизационный потенциал, необходимо
учитывать, что скорость распространения электромагнитных волн
конечна [4]. Поэтому нельзя предполагать, что корреляции между
движениями электронов в молекулах мгновенны*. Возникают эффекты
«запаздывания» взаимодействий. Эти эффекты ослабляют лондоновс-
кие притяжения между молекулами. Уравнение Лондона (1.58)
заменяется следующим [4]:
23hc ac аа
Е—^-^г- (L60>
§ 8. Поляризационное взаимодействие .
Вернемся к уравнению (1.52) и рассмотрим вклад в <£о> первых
двух членов, находящихся в фигурных скобках. Вклад этих членов
называют энергией поляризационного взаимодействия молекул:
(1.61)
k>\ J
* Предположение о мгновенных корреляциях хотя и не оговаривается, но
содержится в теории Ф. Лондона.
28
Пользуясь уравнением (1.56), получим:
( £ £ ) . (1.62)
Уравнение (1.62) имеет простой физический смысл. Молекула с,
имеющая дипольный момент ^с,поляризует молекулу d. Энергия поляризации
пропорциональна квадрату дипольного момента молекулы с и
средней поляризуемости молекулы d. Точно так же молекула dy
имеющая дипольный момент [a di поляризует молекулу с. Это изменяет
энергию поляризации на величину —\i%acR~e.
Энергия поляризационного взаимодействия между молекулами
примерно на порядок меньше энергии лондоновского и дипольного
взаимодействия. Например, для двух молекул пиридина при R = 2 нм,
Еоп~ 116- 10~б къ Т при 300 К. Тем не менее, поляризационное
взаимодействие между молекулами оказывает существенное влияние на
свойства полярных жидкостей. Полярная молекула поляризует всю
окружающую ее массу молекул и создает (индуцирует) в этом окружении
некоторый дипольный момент Ац, величина которого зависит от
поляризуемости и диэлектрической проницаемости среды. Поляризация
окружающей среды создает поле («реактивное» поле) в том элементе
объема, где находится полярная молекула. В результате происходит
дополнительная поляризация полярной молекулы. Реакция
окружающей среды на присутствие в ней полярной молекулы приводит к
появлению реактивного поля, действующего на молекулу. В итоге
возникает существенный дополнительный вклад в энергию взаимодействия
полярных молекул со средой. Нетрудно понять, что этот вклад
пропорционален числу молекул в единице объема. Он значителен в жидкой
фазе и мал в разреженных парах. Влияние этого фактора будет
рассмотрено в гл. II.
§ 9. Об аддитивности вандерваальсовых сил
Если энергия взаимодействия частиц может быть представлена в
виде суммы энергий взаимодействия всех возможных пар частиц
Я- ЦЯу, (1.63)
то силы называются аддитивными. В теории газов, жидкостей и твердых
тел часто допускают, что межмолекулярные силы аддитивны. Для
химических взаимодействий это допущение неверно. Электростатические
взаимодействия между постоянными диполями или в более общем случае
между мультиполями аддитивны. Энергия поляризации практически
аддитивна. Энергия лондоновского взаимодействия, рассчитанная с
помощью второго приближения метода возмущений, тоже аддитивна. Но если
воспользоваться третьим приближением метода возмущений, то
появляются отклонения от аддитивности д£ол. При рассмотрении трех
молекул эти отклонения могут составлять несколько процентов от ЕОл.
ОтношениеАЕОя/ЕОл ~ a/R3; знакд£Ол зависит от взаимного
расположения молекул [4].
29
§ 10. Дипольное, лондоновское и поляризационное
взаимодействия молекул в жидкой фазе
До сих пор мы считали, что молекулы cud находятся в вакууме
или очень разреженном газе. Формулы, выведенные в § 1—9,
справедливы только при этом условии. Несмотря на это, в ходе статистико-
механических расчетов свойств жидких систем часто допускают, что
потенциальная энергия взаимодействия E(R)m между молекулами cud
в чистой жидкости или растворе такая же, как в вакууме:
В действительности, когда рядом с молекулами cud находятся другие
молекулы, условие (1.64) не выполняется.
В приведенных выше расчетах всюду предполагалось, что
расстояние между центрами молекул с и d много больше размеров самих
молекул, так что перекрыванием электронных облаков можно пренебречь.
Для жидких фаз это означает, что молекулы cud разделены слоем
жидкости, толщина которого составляет несколько молекулярных
диаметров. В жидкой фазе энергия дипольного, лондоновского и
поляризационного взаимодействия между молекулами end для заданного
значения R будет меньше, чем в вакууме, при том же расстоянии.
Изменения потенциальной энергии при переходе от вакуума к
жидкой фазе зависят от вида жидкости. Точные его теоретические расчеты
пока еще не выполнены. Приближенные оценки показывают, что,
например, энергия лондоновского взаимодействия между молекулами
end, погруженными в жидкий гелий, меньше, чем в вакууме, примерно
на 2%. В среде жидкого аргона уменьшение составляет около 15%,
в среде жидкого метана — почти на 20%, в четыреххлористом
углероде,— почти на 33% (О. Синаноглу [4]).
Так как расчеты потенциальной энергии дипольного, лондоновского
и поляризационного взаимодействий имеют смысл только для тех рас-
Таблица 1
Слагаемые энергии вандерваальсова взаимодействия
двух одинаковых молекул (по Лондону)*
Молекулы
СО
HI
HBr
НС1
NH3
H2O
C5H5N
0
0
0
1
1
1
2
D
,12
,38
,79
,08
,48
,84
,22
a-1024, см3
1,99
5,5
3,58
3,1
2,4
1,48
9,3
/, э. в.
14,01
10,38
11,62
12,74
10,15
12,60
9,76
X10*1,
Дж-м«
(при 20°С)
0,0034
0,34
6,4
22,4
79
188
400
66
377
179
147
70
33
1013
м« '
,6
,1
,1
Дж-мв
0,057
1,60
4,50
7,23
10,5
10,0
91,6
* Величиьы дипольного момента а, средней поляризуемости а и ионизационного
потенциала I исправлены в соответствии с данными, приведенными в справочниках [3J и [6].
30
стояний, которые намного превышают размеры молекул, значения этих
потенциальных энергий малы. Как мы сейчас увидим, их вклад в
потенциальную энергию жидкости почти всегда ничтожен. Поэтому учет
влияния среды здесь не существенен. Пользуясь уравнениями
потенциальной энергии вандерваальсова взаимодействия молекул в вакууме,
мы получаем представление о верхнем пределе величин потенциальной
энергии этих взаимодействий в жидких фазах.
Оценим вклады различных типов вандерваальсовых взаимодействий
в среднюю статистическую потенциальную энергию одного моля
жидкой фазы Е. (Здесь и далее волна над символом Е или каким-либо другим
буквенным обозначением указывает, что речь идет о величине,
отнесенной к одному молю вещества.) Для чистых жидкостей расчет может быть
выполнен по формуле
+ £Оп; (1.65)
SVRl
2л/Л _ 2~N2A „ 2nN2A
В уравнении (1.65) Na обозначает число Авогадро; V — мольный
объем жидкости; RB— расстояние от центра произвольно выбранной
молекулы, начиная с которого применимы уравнения (1.30) для <с.Еол>,
(1.58) для £ол и (1.62) для ЕОп. Как уже говорилось, RB должно
примерно на порядок превышать размеры молекул. Формулу (1.65) мы
даем без вывода. Ее вывод будет приведен в § 48, так как для вывода
(1.65) необходимо воспользоваться понятием о функции распределения
молекул.
В (1.65) фигурирует дипольное взаимодействие <£од>,
усредненное по всем возможным ориентациям молекул, так как при расчете Е
выполняется статистическое усреднение по координатам всех молекул.
В табл. 1 приведены вклады <£од>, £Ол и ЕОп в энергию
вандерваальсова взаимодействия между двумя одинаковыми молекулами.
Зная указанные величины, можно с помощью формулы (1.65) оценить
вклады дипольного, лондоновского и поляризационного
взаимодействий в Е жидкостей, перечисленных в табл. 1. Так, для одного моля
воды при RB = 1 нм (диаметр молекулы воды d0 ~ 0,28 нм) получаем
<СЕОд> = 797Дж/моль, ЕОл = 140 Дж/моль, Еоп = 42 Дж/моль;
Е = 979 Дж/моль. Изменение энергии при испарении одного моля
воды составляет около 42 000 Дж/моль . Следовательно, потенциальная
энергия вандерваальсова взаимодействия молекул представляет собой
лишь небольшую часть потенциальной энергии воды.
Для пиридина при RB = 2 нм имеем <£од>= 47 Дж/моль, £оя =
= 119 Дж/моль; ЕОп =11,0 Дж/моль, Е = 177 Дж/моль, а теплота
испарения пиридина составляет около 33 000 Дж/моль. Средняя
энергия теплового движения одной молекулы жидкости по порядку
величины близка к къ Т. Для одного моля она в Na раз больше, т. е. RT.
31
При комнатной температуре, т. е. около 300 К, средняя энергия
теплового движения одного моля жидкости по порядку величины составляет
около 2500 Дж. Эта энергия в несколько раз превышает общую энергию
дипольного, лондоновского и поляризационного взаимодействий.
Ясно, что ни дипольное, ни лондоновское, ни тем более
поляризационное взаимодействия не могут быть причиной появления каких-
либо устойчивых молекулярных ассоциатов или комплексов в жидкой
фазе. Даже постановка вопроса о возникновении ассоциатов за счет
дипольных или дисперсионных взаимодействий не имеет смысла,
потому что представление о таких взаимодействиях в принципе приемлемо
только при больших расстояниях между молекулами. Совершенно
очевидно, что энергия вандерваальсова взаимодействия играет лишь
небольшую роль в свойствах жидкостей. Эта роль нередко
преувеличивается, так как расчеты производятся для слишком малых значений RBy
равных размерам молекул, что необоснованно. Заметим, что Еоп не
следует отождествлять с энергией реактивной поляризации молекул.
Как уже говорилось, дипольные, лондоновские и поляризационные
взаимодействия имеют общую, электромагнитную природу. В этом
смысле различия между ними условны. Но они имеют и объективное
содержание, так как дипольные взаимодействия непосредственно
обусловлены постоянными дипольными моментами; лондоновские —
корреляцией движений электронов; поляризационные — влиянием
постоянных электрических моментов молекул на движение электронов в
окружающей среде.
При уменьшении расстояния R между центрами молекул резко
усиливается перераспределение электронной плотности. При малых R
его ролью пренебречь нельзя. Взаимодействующие молекулы образуют
единую систему. Взаимодействие усложняется. Его уже нельзя
подразделить на три независимые составляющие, как при больших R.
Формулы и понятия о точечных моментах, пригодные для больших R,
теряют смысл. На смену дипольным, лондоновским и дисперсионным силам
приходит более сложное взаимодействие, которое принято называть
химическим.
§11. Резонансное взаимодействие между молекулами
Если какая-либо полоса в эмиссионном спектре (спектре
испускания света) молекулы с накладывается на одну из полос в спектре
поглощения молекулы d (рис. 2), то между этими молекулами возникает
индуктивный резонанс. Электромагнитное поле молекулы с индуцирует
в молекуле d вынужденные колебания той же частоты.
Взаимодействие между молекулами напоминает взаимодействие между двумя
одинаковыми маятниками, когда они соединены пружиной. Заставим
колебаться один из маятников, а второй пусть будет неподвижным.
Постепенно второй маятник начнет колебаться, колебания первого будут
затухать. Затем первый маятник остановится, а второй сильно
раскачается. Произойдет перекачка энергии колебаний от первого маятника
ко второму. Потом этот процесс повторится в обратном направлении и
32
так будет продолжаться до тех пор, пока энергия колебаний не перейдет
в тепло за счет трения.
Впервые на существование резонансных явлений при
взаимодействии между молекулами обратили внимание Эйзенштейн и Лондон.
Индуктивный резонанс при возбуждении электронных состояний
подробно исследован Перреном,
Вавиловым Фёрстером, Гала- Подт g e Полт в е
ниным [7,8]. Возможность ин- испускания Г\ Г\ поглощения
дуктивного резонанса при
возбуждении колебательных
состояний молекул впервые
была рассмотрена автором [9].
Если молекулы end
одного и того же сорта, причем
одна из них более возбужде- —
на, а другая менее, то между —^
ними всегда возможен индук- Рис 2 наложение полос в спектрах испус -
тивныи резонанс. При этом кания и поглощения света при индуктив-
нужно лишь, чтобы они сбли- ном резонансе
зились на расстояние
порядка десятка молекулярных
диаметров (иногда^ 10 нм). Чем полнее накладываются полосы в
спектрах испускания и поглощения, тем сильнее индуктивный резонанс.
Рассмотрим случай острого резонансного взаимодействия между
одинаковыми молекулами end.
Пусть, например, молекула с находится в основном, а молекула d
в возбужденном состоянии, которое мы обозначим индексом k. Поглотив
квант света fr(dki молекула с может перейти в то состояние, которое
имеет молекула d. И наоборот, испустив квант ^со^, молекула d может
перейти в основное состояние, которое имеет молекула с:
c + h<*k^ d. (I.66)
Здесь % = hl2n\ со = 2лv.
В возбужденных квантовых состояниях расстояния между
молекулами, при которых нельзя пренебречь перекрыванием их волновых
функций, как уже говорилось, резко возрастают. Но когда между
молекулами может возникнуть состояние острого резонанса, интервал
значений /?, при которых надо учитывать наложение волновых
функций, увеличивается еще больше. Здесь даже слабые возмущения
становятся существенными. Поэтому волновая функция %к системы,
состоящей из невозбужденной молекулы с и возбужденной молекулы d,
должна быть антисимметризована при расстояниях, значительно
больших, чем в случае лондоновских взаимодействий. Она имеет вид:
Фок = ^о^/г, (1.67)
Где А — оператор антисимметризации*.
* Простейший пример: антисимметризация произведения электронных
волновых функций двух атомов водорода (без учета спиновых состояний электронов)
АЬС (1) ^ (2) = У-Г №' (1) ^ (2) -+' (2) ** (1)]*
2—503 33
Если бы молекула с получила квант ДсОд,, а молекула d его
потеряла, волновая функция системы имела бы вид
Собственные значения оператора энергии системы для волновых
функций ФОк и фк0 одинаковы и, следовательно, состояния %к и фк0
вырождены. Так как молекулы с и-d представляют собой единую
систему, то излучения и поглощения фотонов в действительности не
происходит. Имеет место резонансный обмен энергий возбуждения без актов
излучения и поглощения света. Поскольку состояния ф0Л и tyk0
вырождены, волновая функция системы должна быть линейной комбинацией
функций f^0K и <]>к0 . Для четных состояний (g) волновая функция не
меняет знак при изменении знака координат всех электронов:
Для нечетных состояний (и) волновая функция меняет знак при
изменении знака координат всех электронов:
^(«)=Фок-Фкв. (1-70)
Пользуясь первым приближением метода возмущений, получим:
£'(«) = СП*) I Я'I **(*)>; (I.71)
Функции ЧГ (g) nW (u)y как обычно, нормированы так, что
<*' (в) I ъ (*)> =1 и (W (и) | w (и)) = 1.
Л
Подставляя в (1.71) и (1.72) значение оператора возмущения //' в
рассмотренном ранее дипольном приближении, найдем, что кроме диполь-
ного взаимодействия между молекулами в Е' входят выражения,
зависящие от компонент момента перехода \л$к (индексы cwd здесь мы
опускаем, так как молекулы одинаковы):
£р (g) - - £р (и) = - R* 1Ых)2 + Ыу)2 - 2 (Nftz)"] . (1.73)
где Е' — энергия резонансного взаимодействия.
Уравнение (1.73) аналогично уравнению (1.47). Из него следует, что
сильное резонансное взаимодействие возможно только при отличном
от нуля дипольном моменте перехода, связанном с излучением или
поглощением фотона fi(Dk. А это как раз и имеет место, если
соответствующий переход наблюдается в спектрах испускания или поглощения
света. Когда полосы в спектрах испускания и поглощения молекул
cwd перекрываются лишь частично, резонансное взаимодействие
ослабевает. Если полосы не перекрываются, резонансный обмен энергий
может наблюдаться при непосредственных контактах («столкновениях»)
молекул.
Пример. Если оба атома водорода находятся в основном состоянии, то
резонансное взаимодействие между ними отсутствует. Индуктивный резонанс воз-
34 ^
никает, когда один атом находится в Is состоянии, а другой в 2р состоянии. Для
переходов атома водорода из Is в различные 2р состояния отличны от нуля
следующие матричные элементы дипольного момента: (\хх) ,; (\iy)is, ;
х ls'2px 2py
Расчет показывает [2], что
| (^)ls, 2px f = | (^y)is, 2ру |2 = | ^z) is, 2pz |2 = 0,555 е2 q\ ,
где До == 5,29* 10"2 нм (боровский радиус атома водорода). Следовательно,
для'переходов Is ч^ 2рх и Is -&. 2ру резонансная энергия равна:
Е'р= ±0,Б55е2а20/ R*,
а для перехода Is ^ 2pz
В одной шестой всех возможных случаев взаимодействия между парой атомов
водорода, один из которых находится в Is состоянии, а другой в 2р
состоянии, энергия резонансного взаимодействия равна 1,110 e2a20/R3; в одной шестой
случаев она равна —1,110 e2a3JRz\ в одной третьей 0,555 е2а20//?3, и еще в одной
третьей —0,555 e2a2JRz.
При R — 1,0 нм величина (e2a20/R3 = 6,45-10~22 Дж, и для переходов Is ч£
^> 2р энергия резонансного взаимодействия двух атомов водорода не превышает
7,0-10~22 Дж. Эту величину можно сопоставить с энергией лондоновского
взаимодействия между двумя невозбужденными атомами водорода. Потенциал ионизации
/н = 13,59 э.В. = 2,18-10~18 Дж. Поляризуемость ан = 0,66-10"24 см3.
E0Jl=-JL^JjL==7,\ . 10-25 Дж
4 R*
при R = 1,0 нм.
Таким образом, энергия резонансного взаимодействия двух атомов водорода
при расстоянии между их ядрами, равном 1 нм, в тысячу раз больше энергии
лондоновского взаимодействия. Так как £'р пропорционально R~3, а Еол
пропорционально R~6, то с ростом R отношение Е'р/Еол возрастает пропорционально Rs.
Резонансная энергия равна къ Т при 300 К и расстоянии между
атомами, изменяющемся в зависимости от сорта атомов, от 0,6 до 2 нм [2].
При больших расстояниях между атомами или молекулами сумма
резонансных энергий, умноженная на априорные вероятности
резонансного взаимодействия, как и в случае с двумя атомами водорода,
равна нулю. По этой причине, а также потому, что возбужденные
электронные состояния молекул обычно маловероятны, энергия
резонансного взаимодействия не вносит существенного вклада в энергию вандер-
ваальсова взаимодействия. Но учет резонансного взаимодействия
между молекулами важен при рассмотрении процессов переноса
энергии.
§ 12. Влияние изотопных замещений
При изотопных замещениях дипольные моменты молекул
меняются незначительно. Средние поляризуемости тяжелой изотопной
разновидности молекул несколько меньше, чем легкой. Средняя
поляризуемость C6D6 на 0,54% меньше, чем средняя поляризуемость С6Н6,
7cdci3 меньше, чем асна, , на 0,17% и т. д. Потенциал
ионизации тяжелых изотопных разновидностей молекул обычно боль-
2* 35
ше, чем легких. Потенциал ионизации /nh3 равен 10, 15 э. в., a /nd3
11,52 э. в.; /сн4 = 12,99 э. в., a/cd4 = 13,25 э. в. и т. д. Уменьшение
поляризуемости снижает энергию лондоновского взаимодействия, а
рост ионизационного потенциала увеличивает ее [5]. В итоге влияние
изотопного замещения на лондоновскую энергию взаимодействия двух
молекул незначительно.
Еще меньше изотопное замещение сказывается на энергии
поляризационного и дипольного взаимодействия двух молекул. Вандервааль-
сово взаимодействие является дальнодействующим, поэтому энергия
вандерваальсова взаимодействия зависит от молярного объема. Для
жидкостей, состоящих из многоатомных молекул, изменения
молярного объема при изотопном замещении обычно не превышают 0,2%.
В одних случаях замещение легкого изотопа на тяжелый ведет к росту
V, в других — к уменьшению V. В итоге влиянием изотопного
замещения на вандерваальсово взаимодействие можно пренебречь.
Для этого есть все основания. Мы видели, что вклад всех видов
вандерваальсова взаимодействия в потенциальную энергию одного моля
жидкости весьма мал и по порядку величины близок к 0,5 кДж.
Изотопное замещение может изменять общую величину энергии
дипольного, лондоновского и поляризационного взаимодействия на
величину порядка 10"2 Дж/моль. Если изменение потенциальной
энергии жидкости при изотопном замещении существенно превышает эту
величину, следует полагать, что оно вызвано не изменением
лондоновского, дипольного или поляризационного взаимодействий, а другими
причинами.
ГЛАВА II
РЕАКТИВНОЕ ПОЛЕ И ЕГО ВЛИЯНИЕ
НА МЕЖМОЛЕКУЛЯРНЫЕ ВЗАИМОДЕЙСТВИЯ
§ 13. Реактивное взаимодействие
Расчеты, приведенные в предыдущей главе, показывают, что
энергия дипольного взаимодействия между полярными молекулами мала.
Дипольные взаимодействия между молекулами не могут иметь
существенного влияния на свойства жидких систем. Экспериментальные
данные о свойствах жидкостей, содержащих полярные молекулы, на
первый взгляд, противоречат этому заключению. В табл. 2 сопоставлены
свойства трех пар жидкостей: m/кшс-дихлорэтилена с ^wc-дихлорэти-
леном; траяс-дибромэтилена с ^г/с-дибромэтиленом; бензола с
пиридином. В каждой из этих трех пар одна жидкость состоит из полярных
молекул, а другая из неполярных. Первые две пары жидкостей состоят
из молекул-изомеров.Молекулы бензола и пиридина имеют почти
одинаковую молекулярную массу и одинаковое число электронов; они близки
по форме и межядерным расстояниям. Из табл. 2 следует, что при
прочих сходных условиях полярность молекул сочетается с более вы-
36
сокой плотностью и температурой кипения жидкости, более низкой
температурой плавления и, следовательно, большим температурным
интервалом устойчивости жидкой фазы.
Подобное поведение наблюдается и во многих других случаях.
Значит, дипольный момент молекул существенно влияет на энергию
межмолекулярного взаимодействия. Иначе свойства жидкости «не
замечали бы»: имеют или не имеют молекулы дипольный момент. Природа
влияния дипольного момента молекул следующая.
Если молекула полярна, то под влиянием поля, создаваемого ее
дипольным моментом, молекулы жидкости поляризуются. Во всех
молекулах, окружающих данную полярную молекулу, происходят
смещения электронов и атомных ядер, возникают небольшие
электрические моменты.
Поляризация окружающей среды есть ее реакция на присутствие
в ней данной полярной молекулы. Эта реакция, т.е. поляризация,
приводит к тому, что в элементе объема, занимаемом данной полярной
молекулой, возникает реактивное поле R, равнонаправленное с
дипольным моментом данной молекулы. Реактивное поле поляризует ту
полярную молекулу, благодаря которой оно возникло. Если дипольный
момент молекулы ji велик, то реактивное поле будет больше. Оно будет
относительно сильно поляризовать эту полярную молекулу. Тогда
энергия реактивного взаимодействия Е% будет значительна.
Под влиянием реактивного поля жидкость сжимается. Энергия
реактивного взаимодействия увеличивает потенциальную энергию
жидкой фазы. Жидкость становится термодинамически более устойчивой.
Это одна из причин роста температуры кипения и понижения
температуры плавления полярных жидкостей по сравнению с
соответствующими неполярными жидкостями, приведенными в табл. 2.
Для расчета Е% необходимо воспользоваться понятиями и методами
теории диэлектриков, такими, как среднее макроскопическое поле 8,
внутреннее поле/, поле Лоренца, реактивное поле 8r и др. Прежде
чем перейти к описанию вывода той формулы, по которой можно
вычислять £д , мы рассмотрим связанные с этим выводом вопросы теории
диэлектриков.
§ 14. Среднее макроскопическое поле
и внутреннее поле
Пусть жидкость представляет собой диэлектрик, т. е.
электропроводность жидкости настолько мала, что ею можно пренебречь.
Рассмотрим образец жидкости, объем которого равен V. Жидкость состоит из
электронов и ядер и каждая из этих частиц есть внутренний источник
электромагнитного поля. Кроме того, могут быть внешние источники
поля, находящиеся вне образца жидкости. Можно, например,
электрически зарядить конденсатор и между его пластинами поместить жидкий
Диэлектрик. Электрические заряды на поверхности пластин
конденсатора будут представлять собой внешние источники поля. Нас
интересует электрическая составляющая поля внутренних и внешних
источников.
37
Таблица 2
Влияние реактивного поля на свойства жидкостей*
Жидкость
Траяс-дихлорэтилен
Дш>дихлорэтилен
Транс-ддбромэтлен
Дис-дибромэтилен
Бензол
Пиридин
0
1
0
1
0
2
Q
,89
,35
,20
и
V при 25
77,
76,
83,
82,
89,
81,
28
12
0
0
40
31
о
-50
—80
—6,5
-53
5,533
—41,8
о
с
47,67
60,36
108
112,5
80,10
115,58
о
Г-ч
97,7
140,4
114,5
165,5
74,567
157,38
о
UO
1
I8
2,35
2,35
2,47
2,47
2,28
2,28
о
ю
о,
• 'Л
2,35
9,31
2,47
7,08
2,28
12,51
QS
0
—4
0
—1
0
-5
л
•5
| кДж/мо
,П
,83
,44
•См. [11].
Электрическое поле в образце жидкого диэлектрика слагается из
полей, созданных внутренними и внешними источниками. В теории
диэлектриков часто пользуются понятиями «среднее макроскопическое
поле» и «внутреннее поле». Определим эти понятия.
Выберем какой-либо физически малый не обязательно
макроскопический элемент объема dV внутри (т. е. не около поверхности)
диэлектрика. В этом элементе объема имеется некоторое электрическое поле.
Оно зависит от положений внутренних и внешних (по отношению ко
всей жидкости) источников поля. Сохраним положения внешних
источников поля неизменными. Внутренние источники будут менять свои
положения благодаря тепловому движению, поэтому поле в элементе
объема будет изменяться. Усредним это поле по всем возможным
положениям внутренних источников в макроскопическом образце
диэлектрика. Мы получим среднее макроскопическое поле S, действующее
в данном элементе объема жидкости dV. Это поле можно измерить. При
отсутствии внешних источников поля, среднее макроскопическое поле
равно нулю, и жидкость будет неполяризована.
Определяя среднее макроскопическое поле 8, мы учитывали вклады
всех источников поля, как тех, которые находятся внутри элемента
объема dV, так и тех, которые расположены вне этого элемента объема.
Изменим постановку задачи. Будем учитывать влияние только тех
источников поля, которые находятся вне элемента объема dV.
Электрическое.поле, создаваемое всеми источниками поля, за
исключением тех у которые находятся в самом элементе объема dV,
называется внутренним полем f в этом элементе объема dV. Мгновенные
значения внутреннего поля отличны от нуля и могут достигать 108В/м и
более. Если внешнего поля (внешних источников) нет, то в неполяри-
зованном диэлектрике среднее внутреннее поле/ = 0, так как жидкость
изотропна и электрически нейтральна. Если же внешние источники
поля имеются, то среднее внутреннее поле отлично от нуля. Измерить его
нельзя, но вычислить можно.
38
§ 15. Поле Лоренца
Рассмотрим макроскопический шар объемом F, состоящий из
непрерывного изотропного жидкого диэлектрика. Шар находится в
вакууме, на него действует постоянное однородное внешнее электрическое
поле ё0. Простой электростатический расчет (см., например, [10]) ведет
к следующему выражению для среднего макроскопического поля 8
внутри шара:
«=—ГТ~*<ь (НЛ)
где £у — статическая диэлектрическая проницаемость шара.
Напомним, что диэлектрическая проницаемость жидкости е зависит от
частоты v. Статическая диэлектрическая проницаемость — это значение s
в статическом поле, т. е. при v = 0.
Будем считать, что поле 8 слабое по сравнению с молекулярными
полями, и его влиянием на диэлектрическую проницаемость,
внутреннюю энергию, плотность жидкости можно пренебречь. Однако как бы
ни было мало поле 80t под его влиянием молекулы жидкости в шаре
будут поляризоваться. В каждой из них возникает некоторый малый
индуцированный дипольный момент. Векторная сумма всех этих
индуцированных дипольных моментов дает макроскопический
электрический момент всего шара M$v Так как жидкость изотропна, то
направление М$v будет совпадать с 80> поэтому
(скаляр ату есть макроскопическая поляризуемость шара).
Макроскопический электрический момент M$v зависит от диэлектрической
проницаемости г и 8:
(IL3)
Это весьма общее соотношение электростатики [10]. Оно справедливо
для любого изотропного диэлектрика, причем не только в тех случаях,
когда поле 8 постоянно, но и когда оно переменно. Поэтому в
уравнении (II.3) фигурирует не es, a &.
Комбинируя соотношения (II. 1), (II.2) и (II.3), получаем
макроскопическое уравнение Клаузиуса—Мосотти:
£s 1 „ 4
t+2V"T^y (IL4)
Эта формула вполне точна, если жидкость изотропна и сферическая
область жидкости достаточно велика, чтобы ее можно было считать
макроскопической. Но для молекулярной теории формула (II.4) не
представляет интереса, если связь между макроскопической
поляризуемостью шарообразного объема жидкости ату и поляризуемостью
отдельных молекул неизвестна. В простейшем случае для неполярных
39
однокомпонентных жидкостей применимо приближенное соотношение
*mV~N*. (II.5)
где N — число молекул жидкости в объеме V; а — средняя поляризуе-
мостьодной молекулы [см. уравнение (1.55)]. Подставляя (II.5) в (II.4),
получаем молекулярное уравнение Клаузиуса—Мосотти:
j£zJLye.L „м,. (IL6)
Это уравнение в отличие от (II.4) приближенное, потому что при его
выводе приходится пользоваться неточным соотношением (II.5).
Отметим, что вывод уравнения (II.6) не опирается на какие-либо
утверждения о характере среднего внутреннего поля жидкости.
Уравнение Лоренца—Лоренца получается из (II.6) заменой г3 на
/г2оо, где Яоо — предельное значение показателя преломлении п в
оптическом диапазоне волн, полученное экстраполяцией п в область длин
волн, стремящихся к бесконечности. Уравнение Лоренца—Лоренца
следует, таким образом, из приближения (11.5) и не связано с какими-
либо допущениями о среднем внутреннем поле в жидкой фазе.
Не останавливаясь более подробно на характеристике уравнения
(11,6), перейдем к вычислению среднего внутреннего поля в жидкости.
Рассмотрим теперь внутри достаточно большого образца того же
жидкого диэлектрика макроскопическую сферическую область
объемом v. Примем, что поверхность этой области проведена так, что ни
одна из молекул ею не пересекается. Поэтому образец диэлектрика
внутри сферы электрически нейтрален. Если сфера велика, то
связанными с этим слабыми отклонениями от строго сферической поверхности
можно пренебречь.
Нас интересует внутреннее поле /, т. е. среднее поле, действующее
на любой, сколь угодно малый элемент dv объема v этой сферы (мы
будем называть ее сферой v):
f=-i-j/rfo. (II.7)
V
Источники внутреннего поля / подразделяются на две группы:
источники, находящиеся внутри сферы v, и источники, расположенные
вне ее, которые соответственно дают вклады ftnfeB напряженность
внутреннего поля /:
f = fi+fe- (И.8)
Усредняя (И.8) по объему v, имеем
7=fi +fe- (H-9)
Так как объем сферы v макроскопический, то средние по объему и
средние статистические величины совпадают, т. е. / = </>; это же
относится к ft n~fe.
Вычислим сначала средний вклад от источников поля, находящихся
вне сферы v, т. е. fe. Для этого нужно отнять от среднего
макроскопического поля 8 «собственное поле» 8si обусловленное источниками поля,
40
находящимися в сфере v:
/* = * — «*. (ii.io)
Сфера v, как и весь макроскопический объем V жидкости, однородно
поляризована. Пусть Mv обозначает средний электрический момент
сферы v. Векторы Mv и 8S связаны простым соотношением:
Вектор Ps представляет собой средний макроскопический
электрический момент, приходящийся на единицу объема жидкости. Ps можно
назвать собственной поляризацией диэлектрика.
Доказательство уравнения (11.11) несложное (см. [10]). Но оно
требует изложения таких вопросов теории диэлектриков, которые выходят
за рамки этой книги. Поэтому мы приводим уравнение (11.11) без
вывода.
Так как согласно (П.З)
4я
то из (П.11) и (11.10) следует, что
(ПЛ2)
Вернемся к сфере v, заполненной жидким диэлектриком. На каждый
элемент объема dv этой сферы действует среднее внутреннее поле /,
под влиянием которого жидкость в элементе объема поляризуется.
Поле/одно и то же для всех одинаковых элементов объема dv, иначе
жидкость была бы неизотропной.
Здесь следует сказать,что при усреднении поля/, действующего на сферу v
жидкого диэлектрика, не предполагается, что источники, находящиеся внутри
сферы v или какого-либо ее элемента объема ^фиксированы относительно
лабораторной системы координат. Любая молекула или ее часть в сфере v может с
равной вероятностью занимать любое положение относительно лабораторной системы
координат. Это диктуется условием изотропности жидкого диэлектрика. Для
кристаллов такое условие не выполняется, поэтому результаты расчетов/ зависят
от их строения.
Суммарный средний макроскопический электрический момент Mv
сферы v пропорционален /
Mv = amv7, (Н.13)
где amv — макроскопическая поляризуемость сферы v. Согласно (Н.З)
Mv= *s~~ vS. (П.14)
4 я
Так как макроскопические поляризуемости единицы объема для
всего образца диэлектрика, имеющего объем ]/, и диэлектрика в
макроскопической сфере v одинаковы, то из уравнения (11.4) получаем
~r==~r-~~^r'ZT2 * (ПЛ5)
41
Из уравнений (11.13) и (11.14) следует, что
Подставляя значение amz/v из уравнения (11.15), получаем:
7 = !ly^ (ПЛ6)
Сопоставление (11.16) и (11.12) показывает, что для изотропного
диэлектрика / =fe. Отсюда вытекает физически очевидное следствие:
Таким образом, средний вклад в среднее внутреннее поле от источников
поля, находящихся внутри сферы v, для любого изотропного
диэлектрика равен нулю.
Среднее внутреннее поле/, подчиняющееся уравнению (11.16),
называют полем Лоренца. Для слабых полей 8, и изотропных
диэлектриков поле Лоренца точно соответствует действительности. В ходе
расчета поля Лоренца была принята для объема v сферическая форма.
Ясно, что результат расчета не должен зависеть от того, какая форма
выбрана для объема v, мысленно выделенного в жидком диэлектрике.
Сферическая форма выбрана только потому, что в этом случае
расчеты принимают самый простой вид. Можно показать, что поле
Лоренца применимо и к некоторым кристаллам, имеющим кубическую
решетку.
Иногда считают, что применение формулы Лоренца для среднего
внутреннего поля жидкостей не вполне оправдано, потому что в
жидкостях есть корреляция между ориентациями молекул, находящихся
неподалеку друг от друга. Такая корреляция действительно
существует. Об этом будет подробно сказано далее при описании структуры
жидкостей. Но надо иметь в виду, что при расчетах среднего внутреннего
поля / усреднение положений источников поля, находящихся вне
элемента объема dvy производится относительно лабораторной системы
координат, никак не связанной ни с положениями источников поля вне
dv, ни с положениями источников поля внутри dv. Иными словами,
элемент объема dv есть однородная часть однородной макроскопической
системы. Если в элементе объема dv есть полярная молекула, то ее
корреляция с окружением, конечно, будет приводить к отклонениям / от
/. С течением времени эта полярная молекула вследствие изотропии
жидкости будет с равной вероятностью принимать любые ориентации
относительно лабораторной системы координат. Поэтому при
усреднении/отклонения/от/будут взаимно уничтожаться и в итоге влияние
корреляции исчезнет.
Среднее внутреннее поле / в изотропных жидкостях есть поле
Лоренца. Оно отличается от среднего макроскопического поля 8 только
тем, что при расчете / не учитывается вклад источников поля,
находящихся внутри изотропного элемента объема dv.
42
§ 16. Реактивное поле и поле полости
Поле Лоренца / в общем случае слагается из поля полости G и
реактивного поля R:
Обе составляющие поля Лоренца есть средние величины. Знак"усредне-
ния обычно опускают и мы последуем этому правилу. Нужно только
помнить, что G и R — не мгновенные локальные величины полей,
а статистические средние макроскопические характеристики.
Чтобы объяснить физический смысл полей О и /?, вернемся к
описанному в § 16 однородно поляризованному сферическому образцу
жидкого диэлектрика. Его объем равен v и средний дипольный момент
Мv = Ps v. Образец находится в вакууме. Если внешнее поле
отсутствует, то среднее макроскопическое поле в объеме v есть «собственное»
поле Ss, связанное с Ps уравнением (11.11).
Допустим теперь, что этот сферический образец находится внутри
жидкого электрически нейтрального диэлектрика. Внешнее поле, как
уже было сказано, отсутствует. Поляризованная сфера v будет
воздействовать на молекулы окружающей ее жидкости; они будут
поляризоваться. Это приведет к изменению среднего макроскопического
поля внутри сферы и изменению ее поляризации. Обозначим новые
значения среднего макроскопического поля, электрического момента и
поляризации в сфере v символами 8, М и Р соответственно. Реактивное
поле, по определению, есть разность между 8 и Ss. Электростатический
расчет [10] показывает, что
<"">
где es—статическая диэлектрическая проницаемость жидкости,
окружающей сферу v. Реактивное поле определяется поляризацией Р
диэлектрика в сфере v и диэлектрической проницаемостью жидкости,
окружающей сферу v.
Допустим теперь, что жидкости вокруг сферы v нет, а есть внешнее
поле, которое, не меняя электрического момента MVJ создает в сфере
v среднее макроскопическое поле, равное 8, т. е. такое же поле, как в
уравнении (11.19). Тогда разность 8 и 8S носит название поля полости
G, причем [10]
В общем случае поле внутри однородно поляризованной сферы tr,
обусловленное частицами окружающей жидкости и источниками
внешнего поля, есть
Если диэлектрик в сфере v — жидкость и ее диэлектрическая прони-
43
цаемостьтоже равна ss1 то Р удовлетворяет равенству 4пР = (es — 1)8
[см. ур. (II.3)]. Тогда правая часть равенства (11.21) совпадает с
формулой (11.16), т. е. сумма G и R дает поле Лоренца.
§ 17. Отличие дипольного момента полярных молекул
в жидкой среде от дипольного момента в вакууме
Пусть молекула в основном квантовом состоянии имеет дипольный
момент. Если она изолирована, т. е. находится в вакууме, то ее
дипольный момент равен jiv. Поместим эту молекулу в центр сферы,
заполненной изотропным диэлектриком (жидкостью), статическая
диэлектрическая проницаемость которого равна е52. Тогда поле,
создаваемое дипольным моментом, изменится. Оно будет равно полю
дипольного момента ji, отличающегося от щ множителем [10]:
_1_ О
(11.22)
Допустим теперь, что первая сфера окружена сферой с
диэлектрической проницаемостью ел, как это показано на рис. 3. Поле диполя в
среде, имеющей диэлектрическую проницаемость esl, будет такое, как
если бы внутренняя сфера на рис. 3 была бы заполнена средой с
диэлектрической проницаемостью e5l, а в центре сферы находился
точечный дипольный момент, равный ре [10]:
Зз е (з 4- 2)1
Потенциал Ф этого поля в среде с диэлектрической проницаемостью
ел равен [10]
jvcos_e_t
•si**
(11.24)
здесь г — радиус-вектор, проведенный от центра сферы в точку, где
определяется Ф; 6 — угол между г и осью Z, вдоль которой направлен
дипольный момент; г — модуль г; ре называют внешним моментом
молекулы в среде с
диэлектрической проницаемостью esl. В
вакууме, т. е. при e5l = 1, внешний и
внутренний моменты равны, ре = pv.
Нов диэлектрике, где esl > 1, на
полярную молекулу влияет
реактивное поле, под действием
которого молекула поляризуется,
поэтому ее внутренний момент ji|
отличается от вакуумного момента
щ,, {л. = jiv + а/?, где а —
средняя поляризуемость молекул.
Чтобы определить ц,.,
воспользуемся рассуждением,
принадлежащим Фрелиху [10]. Поле, созда-
Рис. 3. К расчету величины диполь
ного момента в жидкой фазе
44
ваемое вне сферы, не зависит от того, создано ли оно точечным
диполем или равномерно поляризованной сферой, если она имеет
тот же момент. Поэтому молекула, представленная в виде сферы
с диэлектрической проницаемостью е25, в центре которой помещен
диполь р., должна иметь тот же момент, что и сфера с
диэлектрической проницаемостью esU которая находится в среде с той же
диэлектрической проницаемостью esl и имеет точечный диполь ре в ее
центре, так как в соответствии с определением \ле в обоих случаях поле
вне внутренней сферы одинаково. Следовательно,
сфера
Из соображений симметрии следует, что fx. имеет то же направление,
что и ре> так что интеграл в (11.25) и вектор ре равнонаправлены,
а именно вдоль оси Z. Следовательно, вектор поляризации Р в
уравнении (11.25) тоже направлен вдоль оси Z, т. е. Р = Pz. Поляризация
Pz = Mgv/v есть электрический момент единицы объема диэлектрика,
имеющего диэлектрическую проницаемость esl. В изотропной среде Pz
и напряженность среднего макроскопического поля ez равнонаправле-
ны. 8zyl потенциал поля ф связаны соотношением 8z = — . От-
dZ
сюда, пользуясь уравнением (П.З), имеем
Pz = 8z = — "^ »—- (11.26)
и, согласно рис. 3,
cos в __ __z_ __a_ м
г2 "" г3 = ~ dZ [ г
Так как
С д2 / 1 \ рд2/1\ рЭ2/1\
i — Ыо = \ — Ыо = \ — dv
J ох2 \ г J J ду2 \ г / J ^2 \ г j
И
Г1 / 1 \ Р д ( \ \ Г Г \
) = — 4я,
—)d*>= J\"4-(—)dQ = -'[ [ — r*sin
r j J dr \ r ) J J r2
2 0 0
J
0 0
то, подставляя значение Ф в (11.25) из уравнения (11.24), находим
nsl
Здесь используется теорема Остроградского—Гаусса, с помощью
которой интеграл по объему v преобразуется в интеграл по замкнутой
поверхности Q, в которой заключен объем v. Таким образом, уравнение
45
(11.25) преобразуется к виду
J^L) ^L±V (Ц.28)
3s5l
Уравнение (11.28) показывает, что »ju не зависит от радиуса сферы,Ъкру-
жающей точечный диполь. Следовательно, дипольный момент,
содержащийся между какими-либо двумя сферическими поверхностями внутри
однородного изотропного диэлектрика, равен нулю. Это справедливо
даже в том случае, если две сферы не концентричны ([10] стр. 213—214).
§ 18. Энергия реактивного взаимодействия
Примем, что полярная молекула жидкости представляет собой
сферу с диэлектрической проницаемостью е^2 = 1, погруженную в среду
с диэлектрической проницаемостью esl = es. В центре молекулы
расположен точечный диполь р,. Радиус сферической области,
занимаемой молекулой, равен а. Реактивное поле /?, создаваемое молекулой в
том элементе объема, который она занимает, согласно (11.19) имеет вид
так как объем, занимаемый одной молекулой, равен 4/3 па?. Но, за
редкими исключениями, молекулы не имеют сферической формы. Кроме
того, они не обладают свойствами макроскопических тел. Поэтому
может показаться, что уравнение (11.29) основано на неприемлемых,
слишком грубых упрощениях. Такое мнение было бы ошибочно.
Вспомним, что интересующее нас поле R не есть локальное
свойство данной конкретной молекулы. R — это средняя характеристика
полей, создаваемых полярными молекулами в изотропном
диэлектрике, отнесенная к одной полярной молекуле, на которую в среднем
приходится объем v = VIN а =(4/3) naz. Такую сферу v следует считать
очень малым объемом, сохраняющим свойства макроскопического
объекта. Ее можно представлять себе как область вакуума, в центре
которой находится точечный диполь ji. Она может рассматриваться и как
непрерывная среда с диэлектрической проницаемостью esV имеющая
момент, равный ц., и результат будет тем же.
Основное упрощение, принимаемое при выводе уравнения (11.29),
заключается не в том, что полярная молекула принимается
сферической, а в том, что среда, непосредственно
окружающая молекулу, считается бесструктурным континуумом. Это
приближение здесь нет оснований считать очень грубым, так как доля,
вносимая соседними молекулами в реактивное' поле, невелика.
При отсутствии внешнего поля среднее реактивное поле полярных
молекул в изотропной среде равно нулю. Это объясняется тем, что все
диполи могут с равной вероятностью иметь любые ориентации в
лабораторной системе координат, т. е. такой системе, положение которой
никак не связано с положениями молекул диэлектрика. Но
потенциальная энергия реактивного поля Е%отлична от нуля, так как
реактивное поле и дипольный момент молекулы равнонаправлены. Потенци-
46
альная энергия Е% состоит из потенциальной энергии—fi. R сферы v
с моментом [А. в поле R и изменения потенциальной энергии жидкости
вследствие ее поляризации под влиянием поля, создаваемого |а.. При
этом учитывается и энергия, затрачиваемая на поляризацию той
молекулы, в которой расположен диполь pi..
Потенциальная энергия ER— скаляр; она является функцией
R* [10]:
Er = - ft Л + №* = - 1h К + Р'Л1. (11.30)
где (3 — некоторая постоянная и |3' = а + |3. Чтобы найти (3,
воспользуемся следующим методом. Реактивное поле должно быть таким,
чтобы потенциальная энергия £дбыла минимальна, т. е.
dER _ dER _ dE^ _ Q
dRx dRy dR2
Поэтому
Подставляя значение R из (11.29), получим:
Уравнение (11.32) неудобно для расчетов, потому что содержит
внутренний дипольный момент {лг-, который надо заменить на
вакуумный момент jiv, определяемый экспериментально. Чтобы перейти от
|i2 к {iw> следует воспользоваться уравнением
<"■*>
Это уравнение следует из (11.23) и (11.28). Обычно принимается, что
г8г обусловлена электронной и атомной поляризуемостью молекул,
экстраполированной к бесконечно большой длине волны. Тогда для
диэлектрика из теории Максвелла следует, что
s,2 = 8oo^n^, (11.34)
где /г», — показатель преломления жидкости, экстраполированный в
область статических полей, т. е. бесконечно больших длин волн % -+-
->■ оо.Нопри экстраполяции показателя преломления не учитывается
влияние атомной поляризуемости. Если прямых экспериментальных
данных о влиянии атомной поляризуемости нет, то приближенно
можно принять, что
е« -LI nD ' (IL35)
где по — показатель преломления жидкости для D-линии натрия.
47
Отсюда и из (11.32) и (11.33), полагая &sl = &si получаем:
* 2*s + *co 3 a»
Уравнению (11.36) можно придать иной вид, если воспользоваться
формулой Лоренца—Лоренца
(п.»)
имеющей тот же смысл, что и уравнение (II.6) для изотропных сред,
состоящих из неполярных частиц. Тогда, учитывая, что
v а
^А 3
получим
Энергия реактивного взаимодействия полярных молекул с
окружающей средой, как показывает формула (11.38), в основном
определяется величиной дипольного момента ц^и статической диэлектрической
проницаемостью &s. В не очень плотных парах es мало отличается от
единицы, поэтому ER мала, хотя и сравнима по величине с энергией
дипольного и лондоновского межмолекулярных взаимодействий. В
жидкостях, если ss ^> 1, энергия реактивного взаимодействия
полярных молекул с окружением может быть довольно большой.
Например, статическая диэлектрическая проницаемость чистого жидкого
N, N-диметилформамида (CH3)2NCHO при 20° С е, = 38 [11].
Молярный объем V = 77\4 мл, дипольный момент ^ = 3,8D, показатель
преломления по = 1,429. Подставляя эти значения в уравнение (11.38),
получим Е# = —19,4 кДж/моль. Благодаря реактивному
взаимодействию потенциальная энергия молекул диметилформамида в жидкой
фазе на 19,4 кДж/моль ниже, чем в паре. Это одна из причин того, что
температура кипения N,N-диметилформамида равна 1539 С, т. е. на
117° превышает температуру кипения пентана. Хотя пентан имеет
почти ту же молярную массу, дипольный момент его молекул равен 0,083D,
т. е. в 46 раз меньше, чем дипольный момент молекул
N,N-диметилформамида. Следовательно, реактивное взаимодействие в пентане очень
слабое. Потенциальная энергия жидкого пентана мала, его летучесть
значительно выше, чем летучесть N,N-диметилформамида.
Когда es > 1, энергия реактивного взаимодействия полярных
молекул, согласно (11.36), в основном определяется отношением \л1 к
объему v, приходящемуся на одну молекулу:
а2 а2
ER ~1±-~ -LfL. (II 39)
а и
48
При растворении полярных жидкостей в неполярном растворителе
диэлектрическая проницаемость жидкости es уменьшается и ER
падает. Так, например, бесконечно разбавленный раствор М,Ы-диметилфор-
мамида в я-гексане имеет es = 1,87. В этом случае энергия
реактивного взаимодействия молекул Ы,1М-диметилформамида Е% = —3,45
кДж/моль.
В табл. 2, о которой говорилось в начале этой главы, приведены
значения Е% жидкостей, вычисленные по уравнению (И.38). Из таблицы
видно, что энергия реактивного поля велика и может существенно
влиять на свойства жидких систем.
В заключение подчеркнем, что реактивное поле полярных молекул
само по себе не может быть причиной возникновения каких-либо
упорядоченных структур в жидкой фазе. Упорядоченные структуры, т. е.
ассоциаты и комплексы, могут возникать лишь в результате
взаимодействий химического типа, рассматриваемых в следующей главе.
Пользуясь уравнением (11.38), можно показать, что под влиянием
реактивного взаимодействия энергия образования ассоциатов и
комплексов обычно уменьшается; их устойчивость снижается.
ГЛАВА III
БЛИЗКОДЕЙСТВУЮЩИЕ СИЛЫ
§ 19. Близкодействующее отталкивание
Близкодействующее отталкивание всегда возникает при
соприкосновении молекул. Если бы не было близкодействующего отталкивания,
то молекулы не имели бы объема. Они были бы проницаемы подобно
вакууму, и тела не могли бы существовать. Столкновений, обмена
кинетической и потенциальной энергий между молекулами не было бы.
Состояние термодинамического равновесия было бы недостижимо.
Эмпирические соотношения, описывающие зависимость энергии
отталкивания Е от расстояния R между молекулами, получают,
исследуя свойства газов, жидкостей и твердых тел и процессы столкновений
между молекулами.
При любом расчете E(R) необходимо учесть, что отталкивание
противодействует притяжению молекул. Если зависимость энергии
притяжения от R хорошо известна, то расчеты энергии отталкивания
существенно облегчаются. Лучше других изучены силы притяжения,
действующие между атомами инертных элементов и между ионами с
полностью заполненными электронными оболочками, например такими,
как Na+, K+, Cs+, Cl~, Вг~ и т. д. Но, как мы увидим, даже для этих
частиц сведения об энергии притяжения не очень точны. Этого
достаточно, чтобы эмпирические выражения для энергии отталкивания тоже
были неточными.
Простейшее эмпирическое соотношение для потенциальной энергии
близкодействующего отталкивания имеет следующий вид:
E(R) = AR-", (II 1.1)
49
где А и п — эмпирические постоянные. Отметим, что значение п =
= 12, принимаемое в некоторых расчетах, не имеет теоретического
обоснования; оно выбрано с целью упрощения вычислений.
Лучше описывает экспериментальные данные экспоненциальная
функция, предложенная Борном и Мейером:
E(R) = Ce-Rl? , (111.2)
где С и р — эмпирические постоянные. Значения р обычно лежат в
интервале (1,5—3,5)* 10~2 нм. Для электрически нейтральных молекул
р можно принять равным примерно 2-10~2 нм. Для отрицательно
заряженных ионов получаются несколько большие величины р, чем в
случае нейтральных молекул.
При очень малых расстояниях R порядка 1 • 10~2 нм пригоденэкрани-
рованный кулоновский потенциал
(Ш.З)
где е — заряд электрона; гхе и ztf — заряды атомных ядер,
находящихся на расстоянии R друг от друга; а — постоянная.
Близкодействующее отталкивание между атомами (или молекулами)
вызвано, в основном, ростом кинетической энергии электронов при
сближении атомов. Это можно доказать с помощью теоремы о вириале.
§ 20. Теорема о вириале и ее применение
для анализа близкодействующего отталкивания
Вириал и его свойства. Рассмотрим систему, состоящую из одной или
нескольких частиц (например, электронов или ядер, или электронов и
ядер), расположенных в конечной области пространства. Пусть Ft —
вектор силы, действующей на /-тую частицу. Положение частицы
относительно произвольно выбранного начала координат определено
радиусом-вектором г..
Вириалом сил (Клаузиус), или вириалом, называют выражение
^=^iriFi. (III.4)
По теореме о вириале удвоенное среднее значение кинетической
энергии системы <7>равно отрицательному среднему значению вириала
системы:
.5)
Скобки <...> означают усреднение.
Для систем, подчиняющихся законам классической механики,
усреднение производят по времени, для квантовых систем — по
прост*
ранству*.
* Пусть Ft — равнодействующая всех сил, приложенных к t-той частице
системы, имеющей массу /я.. В момент времени t частица имеет скорость v^
Дифференцируя по времени выражение m.r.v., получим
50
Пусть система является консервативной и ее потенциальная
энергия и зависит только от координат частиц. Тогда сила, действующая на
/-тую частицу, /^ = ———. Если и — однородная функция степени
п от координат частиц, то по теореме Эйлера об однородных функциях*
W = — пи.
Применение теоремы о вириале. Рассмотрим теперь систему,
состоящую из двух атомов: с — атом гелия, d — атом водорода. С
позиций классической электростатики эта
система может быть представлена
схемой, изображенной на рис. 4. Начало
координат расположим в точке, где
помещается ядро атома гелия. Радиусы-
векторы гъ г2, г3 и г4 =3 R
определяют одно из возможных положений
протона и электронов по отношению к
ядру гелия. Между частицами действуют
кулоновские силы Ftj=
Их по-
Рис. 4. К расчету вириала
системы, состоящей из атомов
водорода и гелия
тенциал utj ~ гт:\ гдегг-;- — расстояние
между частицами i и /.
Потенциальная энергия иэ,я всех
электронов и ядер системы есть сумма
потенциальных энергий utj взаимодействия
между всеми возможными парами частиц. и9,я — однородная фун-
— (тг п V-) =
r% Ft.
(а)
Умножая обе части равенства (а) на dt, интегрируя по t от нуля до т и деля на ^т
получим средние по интервалу времени для обоих частей равенства (а):
где т. <у.2>есть удвоенное среднее кинетической энергии частицы, т. е. 2<7\ >.
Если*/*, и v. в интервале 0</<т конечны, то при х -* со получим
Аналогичные уравнения получаются для других частиц системы. Суммируя ио
всем частицам, получим (II 1.5). Доказательство теоремы о вириале для квантовых
систем имеется в кн. Д. Слэтэра (см. литературу к введению, ссылка [3]).
* Однородная функция и(гъ ..., г ) степени п от гъ ..., гп по определению
Удовлетворяет условию
и (krl9 ... , Xr,,) = hnu (ri, ... , гл),
где X— произвольный множитель. Если первые производные непрерывны, то6
Дифференцируя обе части уравнения по % и полагая X = 1, получим
ди
-z—
drt
= пи .
51
кция первой степени от коорлинат частиц. Пусть Ft —
равнодействующая всех электростатических сил, действующих на г-тую
частицу. Вириал этих сил
^i= S rtFi. (III.7)
Согласно (II 1.6) имеем для п = —1
Величина электростатической силы, с которой атом с действует на атом
d и атом d действует на атом с, равна—du3tn IdR. Если мы хотим, чтобы
атомные ядра покоились, то к ним надо приложить внешние силы,
противонаправленные внутренней электростатической силе межатомного
взаимодействия.
Перейдем теперь к квантовомеханическому описанию системы;
воспользуемся адиабатическим приближением. Пусть атомы находятся в
стационарных состояниях. Сила, с которой они действуют друг на
друга, равна квантовомеханическому среднему электростатической
силы—диэ,я/д#. Она связана с энергией межатомного взаимодействия
E(R) уравнением (В. 12).
Как уже говорилось, в адиабатическом приближении положение
атомных ядер фиксировано. Чтобы выполнить это условие, надо к
атомным ядрам приложить внешние силы—F, равные по величине и
противонаправленные силам взаимодействия атомов друг с другом.
Квантовомеханическое среднее вириала всех сил этой системы
представляет собой сумму среднего вириала внутренних сил, следующих
закону Кулона, <CW1>, и среднего вириала внешних сил <№г>,
который равен —rf = R . Применяя теорему о вириале и учи-
dR
тывая (II 1.8), имеем
Так как Е представляет собой полную энергию обоих атомов
>
то
/и ч-2Е4-7?-^- (HL9)
Если функция E(R) известна,то с помощью (III.9) легко найти
зависимость средней кинетической энергии электронов (ядра покоятся) и
средней потенциальной энергии электронов и ядер от расстояния
между ядрами.
Допустим, что между атомами действуют только силы отталкивания.
Примем, что потенциальная энергия отталкивания подчиняется
эмпирическому уравнению Борна—Мейера (II 1.2). Запишем это уравнение
52
в форме £" = е х , где Е' = £УС и безразмерная переменная л;
^ 7?/р. Тогда с помощью (III.9) получим
< мэ, *) = Се-х(2 — х). (ШЛО)
Графики, изображающие эти функции, представлены на рис. 5.
Диаметры атомов различных элементов отличаются не очень сильно; они
лежат в интервале примерно от 0,1 до 0,4
нм. Величины i?, при которых
отталкивание играет существенную роль, не
превышают этих значений.
Природа близкодействующего
отталкивания. Из рис. 5 следует, что резкое
возрастание энергии отталкивания Е при
соприкосновении атомов вызвано ростом
средней кинетической энергии электронов.
Причины этого можно объяснить с помощью
принципа неопределенности Гейзенберга.
Пусть электрон заключен в пространстве,
ограниченном стенками цилиндра и
поршнем. Расстояние между дном цилиндра и
поршнем равно X. Стенки цилиндра и
поршень для электрона непроницаемы.
Поэтому волновая функция электрона вне
указанного постранства равна нулю.
Обозначим среднее значение координаты
электрона через <Х>, среднее значение
компоненты его импульса, направленной вдоль
оси X, — через <рх>\ средние
квадратичные отклонения координаты и
импульса от <Х> и <рх>— через<(ДХ2)>=
= <(Х — <Х>)2 > и <(Др*)2> =
= <(рх— <рх)2>- По принципу
неопределенности имеем
-0,02 -
(III.11)
Рис. 5. Зависимость полной
энергии электронов и ядер
Е'у средней потенциальной
энергии электронов и ядер
<«э,я>и средней
кинетической энергии электронов
<ГЭ> от межъядерного
расстояния R при малых R
порядка R ^ 0,2 нм.
[Потенциальная энергия
отталкивания подчиняется уравнению Бор-
на — Мейера. Энергия выражена
в безразмерных величинах (см.
текст)]
Для рассматриваемого нами электрона <рх > = 0; <(ДХ)2>^ X2.
Средняя кинетическая энергия движения электрона вдоль оси X равна
<Тх> = <Рх >/2т, где m — масса электрона. Согласно (II 1.9)
8/72*2
(III.12)
При движении поршня по направлению к стенке цилиндра X будет
уменьшаться и средняя кинетическая энергия <Тх> электрона
увеличиваться. <Тх> будет расти за счет работы, требующейся для
движения поршня. Нечто подобное происходит и при сближении атомов.
53
Если расстояние между атомами очень мало, так что R <р,
средняя кинетическая энергия электронов, вычисленная с помощью
потенциала Е' = е~х, становится отрицательной. Такой результат не имеет
физического смысла, так как при очень малых R потенциал Борна—
Мейера неприменим. В этом случае можно пользоваться
экранированным кулоновским потенциалом (II 1.3).
Для Ry близких к сумме радиусов частиц, энергия отталкивания
между несколькими частицами с хорошей степенью приближения может
быть представлена как сумма энергий отталкивания между всеми
возможными парами из этих частиц. При этом каждое из парных
взаимодействий считается независимым от всех остальных.
§ 21. Химическая связь
Перекрывание электронных облаков молекул сопровождается
химическим взаимодействием. Происходит перераспределение
электронной плотности и образуется химическая связь, сильная или слабая.
В жидкостях соседние молекулы находятся в непосредственном
контакте друг с другом. Здесь при описании различных взаимодействш!
пренебрегать перекрыванием электронных облаков, как правило,
нельзя.
Чтобы вычислить энергию Е химического взаимодействия, надо
знать волновую функцию г|> системы, состоящей из химически
связанных друг с другом частиц:
£ = <Ф1 Я| ф>. (III. 13)
Л
Оператор Гамильтона Н есть сумма оператора потенциальной энергии
Л Л
и и оператора кинетической энергии Т:
*•'"."•-. (III.,4>
где m — масса электрона.
Согласно (III.13) энергия Е — есть квантовомеханическое среднее
оператора Гамильтона. Пусть ось Z направлена по линии химической
связи, которая образуется при сближении каких-либо двух атомов или
молекул. На примере иона Н2+ М. Д. Фейнберг и К. Рюденберг [121
показали, что помимо потенциальной энергии важную роль в'химичес-
кой связи играет компонента Tz оператора кинетической энергии.
В области пространства посередине между атомными ядрами иона
Нг+ вторая производная волновой функции ij) no Z близка к нулю и,
согласно (III. 14), вклад в компоненту кинетической энергии,
зависящую от этой производной, невелик. Уменьшение потенциальной
энергии при образовании химической связи обусловлено стягиванием
электронного облака к ядрам. Если бы химическая связь не возникала, то
стягивание электронного облака к ядрам было бы затруднено ростом
54
кинетической энергии электронов по причинам, рассмотренным в § 19.
При образовании химической связи потенциальная энергия снижается,
компоненты кинетической энергии, перпендикулярные линии
связи Тх и Ту , растут, и Tz ^ 0. В итоге и кинетическая и потенциальная
энергия системы могут принимать более низкие значения, чем в случае
химически несвязанных атомов (при том же расстоянии R). Несмотря
на общее возрастание кинетической энергии электронов за счет роста
Тх и Ту, полная энергия отрицательна и система приобретает
устойчивость.
Способы отыскания точной волновой функции молекул, за
исключением некоторых очень простых случаев, пока не найдены. Расчеты
свойств систем, в которых имеются химические связи, обычно носят
качественный характер. Главным источником сведений об энергиях
химических связей и других их свойствах служит опыт.
Отметим следующие важные особенности химических связей.
а. Энергия химических связей в системе, состоящей из N частиц,
не может быть представлена в виде суммы энергий попарных
взаимодействий этих частиц, зависящих только от расстояний между ними.
б. Совокупность химически связанных частиц имеет определенную
структуру. Оговоримся, что для очень слабых химических связей это
утверждение может оказаться неправильным. Здесь оно еще нуждается
в проверке.
Теория химических взаимодействий подробно рассматривается в
руководствах по квантовой химии, органической и неорганической
химии, теории твердых тел. Она далека от совершенства, но, опираясь на
химические и физические исследования молекул и методы квантовой
механики, шаг за шагом постепенно вскрывает сложную картину тех
процессов, которые протекают при образовании и разрушении молекул,
а также при изменениях их состояния.
Глубокий анализ теории строения молекул, а также путей и
перспектив развития этой теории имеется в книге В. М. Татевского [13].
Экспериментальные данные о сильных химических взаимодействиях
есть во многих учебниках, монографиях и справочниках по химии.
Экспериментальные исследования слабых химических взаимодействий
широко развернулись лишь в последнее десятилетие. Это вызвано
успехами оптических, рентгенографических, радиофизических,
термодинамических, акустических и многих других методов изучения вещества.
Информация о слабых химических взаимодействиях между
молекулами пока еще неполна и порой противоречива. Основная трудность
состоит в том, что ни один из экспериментальных методов не является
универсальным, т. е. дающим во всех случаях надежное и
исчерпывающее знание свойств слабых химических связей. Такие сведения могут
быть получены, как правило, лишь с помощью нескольких независимых
методов.
* Описание этих методов не входит в нашу задачу. Многие из них
рассматриваются в книге Ю. Я. Фиалкова, А. Н. Житомирского и
Ю. Н. Тарасенко [14]. Здесь мы рассмотрим добытые с их помощью
Данные о слабых химических связях. Лучше других изучены слабые
химические взаимодействия, протекающие с участием водорода.
55
§ 22. Водородная связь
Водород принято считать одновалентным, но это не полностью
отражает способность водорода соединяться с другими атомами. Атом
водорода, имеющий сильную химическую связь с каким-либо атомом X,
часто может образовывать вторую химическую связь с другим атомом
или группой атомов Y. Обычно эта вторая связь намного слабее первой.
Чтобы различить их, слабую связь называют водородной или Н-связью
и обозначают пунктиром (X—H...Y). Однако бывают случаи, когда обе
связи водорода различаются мало или даже одинаковы. Таковы,
например, связи в ионе [FHF]".
Способность атома водорода образовывать две химические связи
была впервые обнаружена Ильинским и Бекетовым в 80-х годах XIX в.
Позднее это свойство водорода рассматривалось Вернером и рядом
других исследователей. Понятие о водородной связи утвердилось в науке
лишь после работы Латимера и Родебуша (1920). Они воспользовались
этим представлением, чтобы объяснить ассоциацию молекул воды [15].
Постепенно стало очевидным, что Н-связь широко распространена и
сильно влияет на свойства не только воды, но и многих других веществ.
Водородная связь, подобно другим химическим связям, образуется
только при непосредственном контакте между молекулами. Она
локализована в пространстве. Положение водородной связи в молекуле и
направление ее действия фиксированы, хотя и не столь жестко, как для
сильных химических связей.
Водородная связь может образовываться не только легким изотопом
водорода Н, но и тяжелым водородом — дейтерием D [5].
Атомы X и Г, участвующие в Н-связяхмеждуХ—НиУ. С
улучшением методов исследования выяснилось, что круг атомов, способных
участвовать в образовании Н-связей, очень широк. В качестве атома X
может фигурировать любой атом, образующий с водородом обычную
химическую связь: фтор, кислород, азот, хлор, бром, сера, фосфор,
углерод и т. д. Наиболее ярко способность участвовать в Н-связях
проявляется обычно у тех групп X—Н, где атом X обнаруживает
сильное сродство к электрону. Таковы фтор, кислород и азот. Атомами Y,
вероятно, могут быть любые атомы, даже атомы инертных газов.
Например, А. В. Иогансен и Э. В. Броун показали, что НВг и НС1
образуют слабые водородные связи с аргоном и ксеноном. Изменение
энергии системы при образовании этих связей составляет около
4 кДж/моль*. РольУ могут выполнять и ароматические циклы
углерода (связи X—Н... я-орбитали), группы
\с = с/ , —С=С— , —C=N и т. д.
Молекулы четыреххлористого углерода, ранее считавшегося инертным
растворителем, способны принимать участие в образовании слабых
водородных связей X—Н...С1.
* Это означает, что при образовании Na связей изменение энергии равно
4 кДж/моль (Na— число Авогадро).
56
Если в системе имеются не только слабые, но и сильные Н-связи с
энергией порядка 20 кДж/моль,то слабые водородные связи обнаружить
труднее. Влияние сильных связей на свойства системы
относительно велико, и присутствие слабых Н-связей обычно остается
незамеченным. Поэтому создалось впечатление, что группы С—Н молекул алка-
нов и алкильных радикалов не могут образовывать Н-связи.
Например, когда обсуждают свойства растворов метилового спирта в воде,
то, как правило, упоминают лишь о связях вида О—Н...0 между
молекулами Н2О, СН3ОН и между молекулами воды и спирта:
/Оч СНз СН3
н н а оч
'•о хн. .и * хн
н н U °
Здесь связи О—Н...0 имеют энергию около 20 кДж/моль. Не
исключено, что в этих растворах имеются и связи вида С—Н...0 с энергией
порядка 4 кДж/моль:
I /
НО—G-H—О
Но выявление таких, значительно более слабых связей, в этих
условиях затруднительно.
Когда прочные Н-связи отсутствуют, существование водородных
связей С—Н...0 с участием групп С—Н молекул алканов и циклоалка-
нов в ряде случаев удается обнаружить.
Так, группы С—Н молекул я-гексана и циклогексана образуют
комплексы с триоксаном С3Н6О3 с помощью связей С—Н...О, энергия
которых составляет около 4 кДж/моль. В этих растворах имеются еще и
связи С—Н...0 между молекулами триоксана, но прочность их
примерно та же, что и у Н-связей я-гексана с триоксаном.
Способность атома Y принимать участие в Н-связях нередко
ассоциируется с присутствием у Y так называемых неподеленных пар
электронов. Атом азота в пиридине C5H5N имеет одну неподеленную пару
электронов и может принимать участие в образовании одной Н-связи.
Атом кислорода в воде имеет две неподеленные пары электронов и
соответственно способен участвовать в двух Н-связях. Каждый из
атомов хлора в СС14 имеет три неподеленные пары электронов; по-
видимому, каждая из них может принимать участие в одной связи.
Способность молекул RX—Н и YRX образовывать Н-связи
существенно зависит от вида молекул. Например, карбонильная группа /С =0
в кетонах образует менее прочные Н-связи с гидроксилом О—Н,
чем карбонильная группа в карбоновых кислотах. В спиртах кислород
гидроксила способен присоединять группы О—Н с образованием
57
ассоциатов
R R
-к -«к
4
В уксусной кислоте кислород гидроксила такой способностью не
обладает, так как в группе C\q ^ внешние электроны гидроксильного
кислорода, по-видимому, сравнительно прочно связаны не только с Н,
но и с группой С=О. Они не могут образовывать сильные Н-связи с
другими молекулами.
Способность молекул к образованию Н-связей иногда зависит от
конфигурации радикалов R и Rx. Радикалы могут частично или
полностью экранировать группы X—Н и Y и, следовательно,
препятствовать возникновению Н-связи. Например, в молекуле третичного
бутилового спирта группа О—Н отчасти экранирована группами СН3;
в молекуле 2,4,6-три-т/?ет-бутилфенола группа О—Н полностью
экранирована третичными бутильными группами.
Меж- и внутримолекулярные Н-связи. Водородная связь бывает
межмолекулярной и внутримолекулярной. В первом случае водород
связывает два атома, принадлежащие разным молекулам. Во втором
случае оба атома, связанные водородом, принадлежат одной и той же
молекуле. Например, молекулы о-хлорфенола в парах имеют
внутримолекулярную связь ОН...С1, Kj\ P • В жидком о-хлорфеноле
^^сУ
внутримолекулярная связь О—Н...С1 конкурирует с
межмолекулярной О—Н...О. Связь О—Н.. .О более прочна, поэтому в жидком
о-хлорфеноле преобладают ассоциаты (CeH^HCl)^, сходные по
структуре с ассоциатами фенола [16].
Межмолекулярные водородные связи обычно линейные. Это
означает, что ядра атомов X, Ни Y располагаются на одной прямой. По-
видимому, такая конфигурация Н-связей наиболее устойчива.
Внутримолекулярные Н-связи часто бывают нелинейными, как в ортохлорфе-
ноле. Нелинейные водородные связи могут быть в тех случаях, когда
другие химические связи препятствуют линейному расположению
атомов X—Н и Y.
Классификация Н-связей на межмолекулярные и
внутримолекулярные до известной степени условна. Например, связи О—Н...0 в
кольцевом димере уксусной кислоты
р-Н—Оч
ШС^/у \f" ртт
з— \ // "— з
являются межмолекулярными относительно мономеров СН3СООН.
Эти связи можно рассматривать и как внутримолекулярные относи-
58
тельно молекулы димера (СН3СООН)2. Внутримолекулярные
водородные связи мы подробно рассматривать не будем. Обзор свойств веществ
с внутримолекулярными Н-связями имеется в книге Д. Пиментела и
О. Мак-Клеллана [15].
§ 23. Энергия AU и энтальпия ДЯ водородной связи
д U и ДЯ есть соответственно изменение внутренней энергии и
энтальпии системы в результате реакции образования Н-связи:
RX-H + Y :£ RX-H--Y (111.15)
При возникновении Н-связи энергия и энтальпия системы
уменьшаются, поэтому AU и ДЯ отрицательны. (Энергия или энтальпия начального
состояния вычитается из энергии или энтальпии конечного
состояния). Для описания изменений состояния системы в результате
реакций образования Н-связей пользуются как величинами ДЯ, так и
величинами AU. Но в жидкой фазе различием между AU и ДЯ = AU -\-
-\-PAV можно пренебречь; так как изменение объема мало и PAV <t
д^7. Реакция (III. 15) может сопровождаться рядом других побочных
явлений, особенно в жидкой фазе, поэтому экспериментальные
значения Д U илиДЯ нередко представляют собой результат наложения
нескольких процессов. Чтобы пояснить, о чем идет речь, рассмотрим
реакцию образования кольцевых димеров уксусной кислоты:
2СН3СООН -> (СН8СООН)2 (II 1.16)
В газовой фазе ДЯ этой реакции составляет около—63 кДж/моль.
Следователь но, на одну водородную связь приходится около —31,5 кДж/моль.
В жидкой уксусной кислоте ДЯ реакции (III.16) равно примерно
—29,7 кДж/моль [17]. На одну Н-связь приходится приблизительно
—15 к Дж/моль. Расхождение между величинами ДЯ в жидкой и
парообразной фазах объясняется тем, что в жидкой фазе реакция (III. 16)
сопровождается рядом других процессов.
Молекулы мономера СН3СООН имеют две различные
конфигурации, различающиеся положением группы ОН, —цис- и траяоизомеры:
Н
0 0 О Оч
СН3 СН3
цис» транс»
В парах уксусной кислоты присутствуют в основном молекулы цис-
изомера, гдеони более устойчивы. Ихэнергия примерно на 6,ЗкДж/моль
ниже энергии mpa^-изомера. В жидкой фазе концентрация транс-
формы мономера СН3СООН выше, чем концентрация цис-форыы.
Повышение устойчивости шраяс-формы СН3СООН при переходе от пара
к жидкости вызвано'тем, что дипольный момент транс-формы [хтранс ^
^3,9D, а дипольный момент ^м>формы jnUHC~ 1,4 D. Если молекула,
имеющая в вакууме дипольный момент \xv, переносится из вакуума в
59
среду со статической диэлектрической проницаемостью е5, то ее потен-
циальная энергия уменьшается на величину, равную энергии
реактивного взаимодействия (11.36):
Р _ Bs — 1 8 со + 2 \%
Для уксусной кислоты при 20°С es = 6,2, e^ = 2,08, V = 57 мл,
а3 = 2,26-10~23 см3. Пользуясь этими значениями, получаем £#,цис- =
= 2,5 кДж/моль; £^,транс- = 20 кДж/моль. Таким образом,
потенциальная энергия молекул траяс-изомера в жидкой уксусной кислоте
примерно на 11,2 кДж/моль ниже, чем потенциальная энергия цис-
изомера (20—2,5—6,3 = 11,2). Концентрация цис- и транс-форм NT
и Nu, связаны с разностью их свободных энергий FT и Fu соотношением:
^Г (111.17)
В рассматриваемом случае разность свободных энергий приближенно
равна разности потенциальных энергий FT — Еп ~ 11,2 кДж/моль.
Условия применимости этого приближения рассмотрены в кн. [18],
стр. 468. При 20° С RT = 2,42 кДж/моль и, следовательно, NT ~
~ e4>6Nn са 102 Nn. Таким образом, почти все молекулы мономера
уксусной кислоты в жидкой фазе имеют транс-форму. Входе реакции
(III. 16) две полярные молекулыСН3СООН образуют неполярную
молекулу кольцевого димера. При этом молекулы траяс-изомера должны
переходить в цис-форму. Следовательно, разность потенциальных
энергий кольцевого димера и молекул мономера в среднем составит 63—
—2-11,2=40,6 кДж/моль. Эта величина на 11 кДж/моль больше
наблюдаемой. Расхождение может быть обусловлено более слабыми
химическими связями между молекулами уксусной кислоты, неточностью
значений jiu и |дт и приближениями, принятыми в ходе расчета.
В жидкой фазе экспериментальные величины Д{/ и АН могут
представлять собой некоторые средние изменения внутренней энергии и
энтальпии, происходящие при реакции образования Н-связи X—H...Y
в реакциях комплекса RXH...YRlt а также его составных частей с
окружающими молекулами S.:
(RX — Н — YRi) S* (111.18)
Молекулы S. могут образовывать связи между собой, с молекулами
RX—Н и RXY, с комплексами или ассоциатами*, содержащими связи
X—H...Y.
Усреднение в ходе эксперимента производится по всем возможным
взаимодействиям со всеми i видами молекул S., присутствующих в
жидкой фазе. Энергия Н-связей иногда может составлять всего лишь
* Здесь и далее «комплексами» мы будем называть химические соединения
различных молекул, ассоциатами — соединения одинаковых
молекул.
60
Таблица 3
Энтальпии водородных связей
/. Од покомпонентные системы
Вид связи
О—Н...0
С-Н...С,
F—H...F
(F-H...F)"
Вещество
Вода, лед
Спирты C^Hgrt+iOH
Кислоты CrtH2/2+iCOOH
Бензол
Фтористый водород
Бифторид калия
—АН, кДж/моль
24
23
30,5
5,0
29,2
96
2. Двухкомпонентные системы
Вид связи
0—Н...0
0—H...N
Система
Фенол — дифениловый эфир
Фенол — нитрометан
Фенол — диоксан
Фенол — диэтиловый эфир
Фенол — тетрагидрофуран
Фенол — этилацетат
Фенол — ацетон
Фенол — диметилформамид
Фенол — триэтилфосфат
Фенол — диметилсульфоксид
Tpem-бутиловый спирт — этилацетат
Трет-бутиловый спирт —
диметилсульфоксид
Фенол — ацетонитрил
Фенол — пиридин
Фенол — триэтиламин
Tpem-бутиловый спирт — трихлораце-
тонитрил
Tpem-бутиловый спирт—акрилони-
трил
Tpem-бутиловый спирт — бензони-
Tpem-бутиловый спирт — ацетонитрил
Ацетонитрил — изопропиловый спирт
Ацетонитрил — трихлор-2,2-метилэта-
нол
Ацетонитрил — 1 -трихлорэтанол
Ацетонитрил — 1,3-гексахлоризопро-
панол
—А Я, кДж/моль
9,2
12,1
20,5
22,6
23,8
19,2
19,6
25,9
27,6
28,0
11,7
19,2
16,3
30,0
37,6
2,9
9,2
6,7
8,3
9,6
11,7
18,0
22,6
61
Продолжение табл. 3
Вид связи
0—Н...С1
0—Н...Сг
N—H...0
N-H...C,
N-H...N
C-H...N
С-Н...Сте
Система
Фенол — хлорциклогексан
Трет -бути ловый спирт — трихлор -
ацетонитрил
Фенол — бензол
Дифениламин — диоксан
Дифениламин — диметилформамид
«-Метиланилин — бензол
я-Метиланилин — диметиланилин
я-Метиланилин — диметиланилин
«-Метиланилин — пиридин
Бензол — диоксан
Хлороформ — ацетон
Ацетилен — диоксан
Ацетилен — ацетон
Бензол — цианистый водород
Хлороформ — пиридин
Хлороформ — бензол
—Д#, кДж/моль
9,2
2,9
6,7
9,6
15,0
6,3
6,7
8,3
15,8
5,0
10,5
4,2
5,9
18,0
9,2
5,4
около 1 кДж/моль, что для одной молекулы сравнимо со средней
энергией теплового движения —кв Т, приходящейся на одну степень
свободы молекулы при 20°С. В других случаях она может достигать
величин порядка 40 кДж/моль и более (табл. 3). Энергия и другие свойства
Н-связи зависят от вида атомов X и Y, с которыми соединен водород,
от свойств молекул, в состав которых входят атомы X и Y, йот
структуры комплексов. Например, энергия одной связи О—Н...0 в воде
равна £^21 кДж/моль; в димерах уксусной кислоты ~34 кДж/моль; в
комплексах воды с ацетоном — около 8 кДж/моль.
62
Как уже говорилось, в жидкости изменение энергии при разрыве
или образовании связи существенно зависит от энергии реактивного
взаимодействия. Именно этим объясняется то, что изменение
энтальпии АН при разрыве одной Н-связи в кольцевомдимере(СН 3СООН)г
в жидкой уксусной кислоте составляет ~8,0 кДж/моль, а при
разрыве Н-связи в разомкнутом димере ~21 кДж/моль [17]. Различие
связано с тем, что в разомкнутом димере примерно 70% молекул СН3СООН
имеет траяс-конформацию. Иногда полагают, что «сопряженные» Н~
связи, например, в спиртах и воде
R
А,
i /н \
.о, о о
'•о н "
R об
.н
значительно прочнее, чем изолированные. При образовании
«сопряженных» Н-связей энергия одной водородной связи по мнению
некоторых исследователей иногда увеличивается на 4—6 кДж/моль.
Серьезные доказательства справедливости таких утверждений в
литературе отсутствуют.
Надо иметь в виду, что энтальпия и энергия образования какой-
либо химической связи могут считаться мерой прочности связи лишь
приближенной в сопоставимых условиях. Подробнее об этом говорится
в § 38.
Энергия водородной связи в воде, спиртах и ряде других вещеега
возрастает на величину до 400 Дж/моль при замене водорода в связи
X—H...Y на дейтерий. Этот интересный эффект детально исследовал
И. Б. Рабинович [5].
Точность определений энергий Н-связей, как правило, невелика.
Значения энергии и энтальпии одних и тех же Н-связей, найденные
различными методами и разными исследователями, нередко различаются на
2—4 кДж/моль и более.
§ 24. Межъядерные расстояния X—Н и Н ... Y
Как уже говорилось, обычно связи водорода в системе X—H...Y
неэквивалентны. Протон располагается ближе к атому X, с которым
он образует более сильную химическую связь. Если энергия Н-связи
достигает нескольких килокалорий на моль, то наблюдается небольшое
растяжение связи X—Н. Например, в мономерных молекулах воды
расстояние О—Н равно 0,096 нм, а в ассоциатах оно увеличивается до ~
^0,1 нм. С ростом энергии Н-связи расстояние между X и Y
уменьшается. Так, расстояние О...О меняется от 0,33 нм в кристаллах Са(0Н)2
До 0,250 нм в кристаллах КН2РО4. В табл. 4 приведены расстояния
63
X... Y для некоторых Н-связей, а также сведения о положении протона.
Эти данные носят отрывочный характер, в особенности для Н-связей
в жидкой фазе.
Таблица 4
Расстояния X—Н и X ... Y для некоторых связей X—H...Y(b 10"1 нм)
Соединение
н2о
D2O
НСООН
СНдСООН
СН3ОН
<СН2ОН)2
NaHCO3
Ni(OH)2
Mg(OH)2
х-н.
О—Н.
О—D.
О—Н.
О—Н.
О—Н.
О—Н.
О—Н.
О—Н.
О—Н.
. Y
. О
. О
. О
. О
.. О
. О
. О
. О
. О
х-н
1,00
1,01
1,04
1,03
0,96
0,96
0,92
1,01
1,06
X ... Y
2,76
2,76
2,58
2,61
2,70
2,96
2,55
3,1
3,2
§ 25. Внутримолекулярные колебания
и водородная связь
Нормальные и характеристические колебания. Водородная связь
X—Н...Y влияет на внутримолекулярные колебания и приводит к
появлению новых колебательных степеней свободы, что находит отражение в
инфракрасных (ИК) спектрах и спектрах комбинационного рассеяния
(КР) света. Как известно, молекула, состоящая из п атомов, имеет Зп
степеней свободы, из которых для нелинейных молекул 6, а для
линейных — 5 внешних степеней свободы связаны с поступательным и
вращательным движениями молекулы как целого. Остальные Зп—6
или Зп — 5 внутренние степени свободы связаны со всевозможными
колебаниями атомных ядер в молекулах. Колебательное движение
может быть описано с помощью естественных координат X.,
определяющих отклонения межъядерных расстояний и валентных углов
относительно равновесного положения. При равновесной конфигурации
атомных ядер все естественные координаты X. обращаются в нуль.
Колебания атомных ядер в молекулах взаимосвязаны, поэтому изменения
естественных координат атомных ядер также взаимосвязаны. Если
считать колебания гармоническими, то во многих случаях с помощью
методов, разработанных механикой малых колебаний молекул,
приближенно можно осуществить переход от естественных координат Хг к
нормальным координатам Q..
Каждая естественная координата представляет собой некоторую
линейную функцию всех нормальных координат. Число нормальных
координат равно числу внутренних степеней свободы молекул, т. е.
Зп — 6 или 3/г — 5. При колебательном движении молекулы каждая
нормальная координата изменяется независимо от других нормальных
координат. Каждой нормальной координате соответствует некоторое
64
нормальное колебание системы, имеющее определенную частоту v^ и
форму [19]. Нормальные колебания являются гармоническими. При
поглощении, испускании и комбинационном рассеянии света энергия i
нормального колебания изменяется на fro)f, где сог = 2тг^. Поэтому
для не очень сложных молекул частоты нормальных колебаний могут
быть найдены с помощью данных об ИК- и КР-спектрах. Хотя, в
принципе, каждое нормальное колебание молекулы сопровождается
смещением всех ат@мных ядер, во многих случаях основную роль в таком
колебании играют лишь несколько атомных ядер, а иногда только одно
атомное ядро. В этом случае в ИК- и КР-спектрах наблюдаются так
называемые «характеристические» частоты, а соответствующие
нормальные колебания называют характеристическими.
Если молекулы образуют ассоциаты или комплексы, то все
характеристические частоты и интенсивности соответствующих полос в ИК-
спектрах, как правило, меняются. При слабых химических
взаимодействиях эти изменения незначительны. Исключение представляет
водородная связь. Участвующий в Н-связи протон подвижен, потому
что его масса мала, поэтому спектроскопические проявления Н-свя-
зей обычно сравнительно ярко выражены.
Колебания комплексов с Н-связью. Пусть молекула, содержащая
группу атомов X—Н, имеет пг атомных
ядер, а молекула, содержащая атом или ys у ^ н—— у
группу атомов Y, имеет п2 атомных ядер. /
Будем считать, что обе молекулы нелиией- ^ ч^
ны. До образования Н-связи общее число
колебательных степеней свободы двух
молекул равно 3(/гх -f- n2). Из них 12
степеней свободы являются внешними,
связанными с поступательными и
вращательными движениями каждой из молекул.
Остальные 3(ях + п2) — 12 степеней
свободы — внутренние, колебательные. После
образования связи X—H...Y число
внешних степеней свободы системы
уменьшается на 6, а число колебательных степеней
свободы соответственно возрастает на 6. Из
них одна степень свободы с частотой
колебаний vT соответствует трансляционному
колебанию, т. е. такому, которое происходит
вдоль связи X...Y; пять других связаны с
деформационными колебаниями,
сопровождающимися изменением ориентации
молекул относительно направления Н-связи. В
Циклических димерах с двумя Н-связями
имеется два валентных колебания вдоль
связей X...Y и четыре деформационных.
Некоторые из нормальных колебаний
комплексов с Н-связью являются
характеристическими в ИК-и КР-спектрах. Типы
3-503
R
/
"У
Рис. 6. Некоторые типы
колебаний комплекса с
Н-связью [15]:
v — X—Н валентное колебание
(/,5 • 1013 — 1,05 • 101* Гц, 3000 —
4000 нм); ^ — R — X — Н
деформационное (плоское) колебание
(3- 1013 —5,1 • 1013 Гц; 6000 —
10 000 нм); v^ — R—X—Н
крутильное (неплоское) колебание (9 х
X Ю12—2,7 • 1013 Гц, 11 000-30 000
нм); колебательное движение
атома водорода здесь происходит
перпендикулярно плоскости R—X—Y;
va — X ... Y трансляционное
колебание (1,5 • 1012 — 7,5 • 1012 Гц;
4 . 10* — 2 • 105 нм); v<o — X —
Н... Y деформационное
колебание (< 1,5 • Ю12 Гц; > 0,2 мм)
65
этих колебаний представлены на рис. 6. Стрелками показаны глав»
ные смещения, происходящие при колебательном движении атомов.
Латинские индексы s, Ъ, t указывают на тип колебания молекулы
RXH до образования ею Н-связи с другой молекулой. Греческие
индексы а и (3 указывают на тип колебания, возникающего после
образования Н-связи. Для каждого типа колебаний приближенно
указаны частоты (в Гц) и длины волн (в нм), где обычно
наблюдаются соответствующие области поглощения в ИК-спектрах. При
возникновении Н-связей частота валентного колебания X—Н, т. е. vJf
обычно понижается, а интенсивность соответствующей полосы в ИК-
спектре обычно резко возрастает. Эти изменения, если они
наблюдаются, указывают на существование водородной связи с участием группы
X—Н. Наиболее чувствительным признаком водородной связи является
увеличение интенсивности полосы ИК-спектра, соответствующей
валентному колебанию X—Н. Интенсивность этой полосы может
возрастать в 10 и более раз. Изменение частоты иногда может и не
наблюдаться. Например, частота валентного колебания С—Н в молекуле
хлороформа не изменяется при образовании Н-связи С—Н...0 с ацетоном.
В принципе, допустимы случаи, когда интенсивность полосы
валентного колебания X—Н остается неизменной или даже убывает.
Далее будет показано (см. § 29), что теоретически такие случаи
возможны, хотя пока еще не наблюдались.
Частота деформационного колебания v^ группы X—Н при
образовании водородной связи возрастает, а ее интенсивность существенно
не изменяется.
§ 26. Водородная связь и резонанс Ферми
Если два колебательных уровня многоатомной молекулы,
принадлежащие различным колебаниям или комбинациям колебаний, имеют
почти одинаковую или одинаковую энергию (а значит, и частоту),
то между этими случайно совпадающими по частоте колебательными
уровнями возникает резонанс Ферми. Происходит «отталкивание»
уровней друг от друга и перераспределение энергии колебаний
между этими уровнями. Например, когда частота валентного колебания
v5 группы X—Н близка к частоте первого обертона деформационного
колебания vb и симметрия этих колебаний одинакова, то между
колебаниями 2v6 и v5 возникает резонанс Ферми. Колебательные уровни
v5 и 2v6 «отталкиваются». В ИК-спектрах вместо одной полосы,
соответствующей наложению колебаний v5 и 2v6, наблюдается две полосы,
интенсивность полосы обертона 2v6 возрастает за счет интенсивности
основной полосы Vy, которая ослабляется. Происходит перекачка
энергии от валентного колебания X—Н к первому обертону
деформационного колебания той же группы атомов. Это наблюдается, например,
в метане. Но обычно частота v5 существенно превышает частоту 2vft и
резонанс Ферми между v5 и 2vb в свободных молекулах, содержащих
группу X—Н, не возникает. Например, у молекул воды v^ =Л,095 X
X 1014 Гц (3650 см"1), a 2v6 —6,45-1013 Гц (2150 см"1); у аминов и амидов
v^ = (1,02—1,05). 1014 Гц (3400—3500 см""1), 2v6 =9-1013 Гц (ЗОООслГ1)
66
и т. д. При образовании связи X—H...Y, как уже было сказано,
v уменьшается, a v^ возрастает и уровни v5 и 2vfc сближаются тем
болыие,чем прочнее Н-связь, поэтому создаются благоприятные усло-
вия для возникновения ферми-резонанса. А. В. Иогансен [20] доказал,
что при образовании прочных Н-связей действительно происходит фер-
ми-резонанс в простейшем случае между колебательными уровнями
v5 и 2v6. Вместо одной полосы v5 наблюдаются две полосы. Одна из них
соответствует колебанию v5, а другая обертону 2v6. Вследствие
перекачки энергии интенсивность обертона усиливается уже на расстоянии
/^7,5-1012 Гц (250 см"1) от полосы v5. При остром резонансе, т.е. когда
частоты v5 и 2v6 очень близки друг к другу, возникает дублет с
расстоянием между максимумами ~3-1012 Гц (100 см"1). Это наиболее
простой вариант. Резонанс Ферми, обусловленный Н-связью, может
приводить и к возникновению значительно более сложной структуры
низкочастотного крыла полосы v5 в ИК-спектрах.
Учет резонанса Ферми необходим для правильной характеристики спектров
веществ с Н-связями и понимания их природы. По мнению А. И. Иогансена,
усложнения структуры полос валентных колебаний X—Н в ИК-спектрах веществ со
связями X—H...Y, вызванные ферми-резонансом, в ряде случаев
истолковывались неправильно. Предполагалось, что существуют разные типы Н-связей;
высказывались гипотезы о модуляции \$. межмолекулярными колебаниями, о
том, что потенциальные кривые для протона на линии связи Н—H...Y имеют два
минимума. Отсюда следовала гипотеза о туннельных переходах протона от
одного минимума на потенциальной кривой ко второму. С учетом резонанса Ферми, как
считает А. И. Иогансен, эти гипотезы нуждаются в дополнительном
обосновании. Следует также иметь в виду, что частоту колебания v5 нельзя
отождествлять с частотой какого-либо одного максимума сложной полосы. В приведенном
выше простейшем случае резонанса двух колебательных уровней невозмущенная
частота v^ совпадает с центром тяжести резонансного дублета. Для более
сложных резонансных картин частота v^ тоже определяется по положению центра
тяжести полосы. Ранее это обстоятельство не учитывалось, поэтому цитируемые
в литературе значения частот колебаний vs в относительно прочных Н-связях
(если сказанное здесь не было учтено) завышены тем более, чем прочнее Н-связь.
§ 27. Изотопные сдвиги частот колебаний [5]
Если в группе X—Н заменить водород на дейтерий, то при
образовании водородной связи X—D...Y происходят изменения частот v5 и
vA, аналогичные тем, которые наблюдаются для связи X—H...Y.
Отношения ks = v"/v? и kb = v"/v? для различных групп X—Н и X—D
и связей X—H...Y и X—D...Y почти не отличаются. Согласно
И. Б. Рабиновичу для воды, спиртов, кислот и анилина ks~kb =k;
обычно величина к лежит в интервале от 1,34 до 1,37. Уменьшение
частоты A^s валентных и увеличение частоты Д^ деформационных
колебаний сопровождаются изменением энергии этих колебаний.
В приближении гармонического осциллятора средняя энергия атомных
колебаний, имеющих частоту ш = 2jcv, равна
h W h о)
Е"— + -7^П{ (ШЛ9)
Для валентных и деформационных групп X—Н и X—D при Т ~
^ 300К hto > kb Т, поэтому энергия Е близка к нулевой энергии Ео:
67
Уменьшение нулевой энергии sub колебаний при образовании Н-свя-
зи и D-связи равно:
Д££с1яа(Д^ +Av£); AE$ = nh(&4? + Av£) = АЕ% / k. (III.21)
Таким образом, при переходе от Н- к D-связи энергия валентного и
деформационного колебаний уменьшается примерно на 25% и
соответственно уменьшается разность Д£о. Образование водородной связи
сопровождается изменением крутильных t колебаний групп X—Н или
X—D и возникновением валентных су- и деформационных (3-колебаний в
группах X—H...Y или X—D...Y. Замещение в группе X—Н водорода
дейтерием вызывает уменьшение частоты, а следовательно, и энергии
крутильных и деформационных колебаний примерно в 1,3—1,4 раза.
Расчеты И. Б. Рабиновича [5], основанные на экспериментальных
данных о частотах колебаний, показывают, что при 25°С замещение
водорода в гидроксильной группе метилового спирта на дейтерий в паре
вызывает уменьшение энергии s-, Ь- и /-колебаний на 8350 Дж/моль.
В жидкой фазе при таком замещении энергия s-, b-y а- и (3-колебаний
уменьшается на 8700 Дж/моль. Следовательно, разность энергий
указанных характеристических колебаний в паре и жидкой фазе при
замене Н на D возрастает на 343 Дж/моль. Эта величина в пределах ошибок
опыта равна разности энергий диссоциации D-связи и Н-связи в
метиловом спирте.
Разность энергии s-, b-, g- и (3-колебаний в жидкой D2O и Н2О при
25°С равна —16 220 Дж/моль. Разность энергий s-, b- и /-колебаний
в парах D2O и Н2О равна —14 800 Дж/моль. При замене водорода на
дейтерий разность энергий характеристических колебаний пара и
жидкости здесь возрастает на 1420 Дж/моль. Заметим, что (в отличие от
метилового спирта) основной вклад в эту разность вносит
изотопический сдвиг частот внеплоскостных деформационных (3-колебаний
водородной связи. Экспериментальная величина разности энтальпий
парообразования тяжелой и обычной воды составляет 1380 Дж/моль.
Таким образом, есть основание считать, что изотопные разности
энергии водородной связи при замещении водорода дейтерием вызваны
преимущественно изменениями энергии колебаний атомов,
принимающих участие в образовании водородной связи. Эти примеры указывают
на важность исследований изотопических эффектов для теории
межмолекулярных взаимодействий. Учет изменений энергии
характеристических колебаний атомов при образовании Н-связи полезен для
выяснения вопроса, куда расходуется энергия, требующаяся для разрыва
водородной связи.
Так, например, суммарная энергия s- и 6-колебаний молекул СН3ОН
при возникновении связи О—Н...О уменьшается на 1670 Дж/моль.
§ 28. Правила Иогансена
Обширные и тщательные исследования ИК-спектров позволили
А. В. Иогансену и его сотрудникам установить ряд важных
эмпирических закономерностей в свойствах Н-связей.
68
Правило интенсивностей [21]. Пусть АН обозначает изменение
энтальпии (в Дж/моль), аДЛ—изменение интегральной интенсивности*
полосы v5 валентного колебания X—Н в результате реакции
ХН + Y ^t XH - Y (II 1.22)
Правило интенсивностей указывает, что —АН пропорционально
квадратному корню из АЛ:
(III 23)
Замечательно, что постоянная К почти не зависит от вида Н-связи.
Для многих водородных связей О—H...Y (в воде, спиртах,
комплексах карбоновых кислот с основаниями), Н-связей N—H...Y, С—H...Y
(в ацетилене, хлороформе) численное значение константы К = 12.
(Если Д Н выражена в ккал/моль, то К = 2,90 [21])
Правило интенсивностей было установлено на основе данных об
инфракрасных спектрах молекул RXH, YRX и комплексов RX—H.-.YRi
в разбавленных растворах в «инертных» растворителях, таких как
я-гептан и четыреххлористый углерод.
В концентрированных растворах и чистых жидкостях, как уже
говорилось в §22, реакция (II 1.22) может сопровождаться рядом других
процессов такого же характера. Здесь экспериментальные значения
АН и АЛ являются некоторыми эффективными усредненными
величинами, поэтому правило интенсивностей выполняется хуже.
Правило интенсивностей справедливо во всем известном интервале
энергий водородных связей, включая взаимодействия с энергией
порядка ± 2 кДж/моль, где смысл термина «связь» при Т ~ 300 К уже
теряется.
Правило интенсивностей не выполняется для нелинейных
внутримолекулярных Н-связей, например, для пятичленных циклов типа
Правило частот [22]. Сдвиг частоты полосы v5 при образовании
Н-связи приближенно пропорционален квадрату изменения энтальпии:
Av5~(Atf)2, (II 1.24)
гдеД^ r=vg — v5; vj—частота валентного колебания группы Х—Н, ке
участвующей в Н-связи. Например, для комплексов фенол—основание
Интегральная интенсивность Л определяется соотношением
где С — концентрация вещества, содержащего группу X—Н; / и /0, имеющие
частоту v, — интенсивности падающего излучения и излучения, прешедшего
через поглощающий слой толщиной /.
69
при Д Vy, лежащих в интервале от 3-1012 до 2,4« 1013 Гц (от 100 до 800
см"1), соблюдается зависимость
А^ = v°— ^ = 20 + 0,515
где Vg обозначает частоту валентного колебания группы О—Н фенола
в разбавленном растворе в четыреххлористом углероде. При малых
энергиях Н-связи правило (II 1.24) соблюдается для газов лучше, чем
для жидкостей, так как в жидкой фазе сказывается влияние
окружающих молекул. Для прочных Н-связей правило частот выполняется
точнее. С усилением Н-связи чувствительность Д^ быстро растет и
обгоняет чувствительность |/ДЛ к изменению энергии взаимодействия,
поэтому влияние окружающих молекул на прочные комплексы можно
изучать по изменениям частот колебаний vs в связях X—H...Y.
Комбинируя уравнения (III.24) и (III.23), можно установить связь
между усилением и смещением полос v Так, например, для Н-связей
фенол — основание
\ \pp (111.25)
где vs> г — частота валентного колебания О—Н в парах фенола; vSt/,.p —
в разбавленных растворах фенола.
Правило растяжений Н-связей [23]. Колебания va — комплекса
X—H...Y представляют собой чередование растяжений и сжатий
водородной связи. При этих растяжениях и сжатиях возникает
квазиупругая сила/а, стремящаяся вернуть систему в состояние равновесия. Если
г0 — равновесное расстояние H...Y, а г — расстояние H...Y в момент
сжатия или растяжения, то
!*=-*«('-'*) = —К,Я* (II 1.26)
где коэффициент пропорциональности Кз называют силовым
коэффициентом; q = г — г0. Силовой коэффициент связан с потенциальной
энергией системы Е, которая зависит от расстояния H...Y. Для малых
значений q, разлагая Е в ряд, имеем:
В положении равновесия, где Е = Ео, потенциальная энергия
проходит через минимум, поэтому ( ) = о. Выбирая за начало отсче-
\ dq /o
\ q /q
та потенциальную энергию в положении равновесия, т. е. считая Ео =
= 0, получаем:
** ^(L (IIL28)
Согласно правилу растяжений, установленному Иогансеном и
Розен бергом,
/Са=а(—ЬН)Ь= 12,6-1013 D\f° =2,0-103(—ДЯ)1*20 , (II 1.29)
70
где Dh — энергия диссоциации одной молекулы комплекса
RX—H...YR!*, Ко выражена в дин/см; АН— в ккал/моль; Dh — в
эрг/молекула.
Рис. 7 показывает, что правило растяжений соблюдается и для
двухатомных гидридов элементов верхней части таблицы Менделеева.
Таким образом, эта зависимость охватывает практически весь интервал
Рис. 7. Зависимость силовой
постоянной Ко , дин/см*, от энергии связи—
А Я, ккал/моль, ряда двухатомных
молекул и Н-ассоциатов [23]:
/ — фенол — триэтиламин; 2 — фенол —
пиридин; 3 — хлористый водород — диметиловый
эфир; 4 — хлористый водород — диэтиловый
эфир; 5 — бромистый водород — диметиловый
эфир; 6—ассоциаты «-метилацетамида; 7 —
цепочечные ассоциаты муравьиной кислоты;
8 — ассоциаты молекул воды во льду; 9 —
цепочечные ассоциаты одкоатомных спиртов;
10— димеры муравьиной кислоты; // —ди-
меры хлористого Еодорода; X — дифторид
калия К (F — Н — F)£
ig Ын)
возможных значений энергий химической связи водорода от ~1 до 600
кДж/моль.
Поскольку коэффициент Ко не только слабых, ко и сильных
химических связей Еодорода следует уравнению (II 1.29), А. В. Иогансен и
М. Ш. Розенберг приняли, что и потенциальная энергия этих связей,
как функция межъядерного расстояния H...Y, описывается одним и
тем же уравнением. Они воспользовались потенциалом Липпинкотта:
Е (Я) = De {1 - ехр [- п (R - Re)2№]} ,
(111.30)
где R — межъядерное расстояние H...Y; Re— расстояние, при
котором E(R) минимальна. Для /?, мало отличающихся от Rey
экспоненту можно разложить в ряд по степеням n(R—Re)2/2R.
Ограничиваясь членом с первой степенью, имеем:
где К — силовой коэффициент растяжения Н-связи при R = Re.
Считая К равным Ко и пользуясь уравнением (II 1.29), имеем
-20 -0,20
10 D = 5,010-4(—
-0,20
(III.32)
* Здесь и далее в рисунках, заимствованных из работ других авторов,
использованы те единицы измерений, которые были приняты в оригинале. Перевод
в систему СИ см. стр. 3.
71
Для водородных связей О—Н... О А. В. Иогансен и М. Ш. Розенберг
приняли параметр п в потенциале Липпинкотта величиной
постоянной, такой же, что и для О—Н, т. е. п =7,1- Ю9 мм"1; они получили
^==0,57 . Ю-10 D-°t20 = 2,44 • 10-* (-A//)"0'20 . (111.33)
Результаты расчетов, приведенные в табл. 5, показывают, что
соотношение (III.33) действительно соблюдается во всем .интервале энергий
химических связей водорода с кислородом.
Таблица 5
Межъядерные
—Д#, кДж/моль
6
24
30
40
400
расстояния Н...0 при различных
(в 10-1 нм) [23]
R по уравнению (II 1.33)
2,25
1,72
1,63
1,53
0,97
энтальпиях связи
^эксп
2,13
1,76
1,59
1,48
0,97
Правило факторов [24]. Изменение свойстваф, где ф = АН; ]^ДЛ;Дапри
реакции образования Н-связи
ХгН + Yj ц£ X^H ... Yj + ^ij (111.34)
выражается произведением двух чисел — фактора i'-той кислоты Р. и фактора
/-того основания Ej:
--AyuPiEj . (111.35)
Символы Х.Н н Yj могут относиться и к разным, и к одинаковым молекулам.
Например, все кислоты, содержащие группы ОН и NH, могут выступать и в роли
оснований, т. е. фигурировать и как ХуН и как Yj. В уравнении (II 1.34) влияние
среды в явной форме не учитывается. По существу, это уравнение отражает
взаимодействие изолированных молекул в газе. Но обычно величины Дф.;- измеряют
в жидкостях, например в разбавленных растворах четыреххлористого углерода.
Чтобы не осложнять пока задачу учетом влияния среды, можно ограничиться
сначала разбавленными растворами Х^Н и Yj в четыреххлористом углероде при
комнатной температуре, считая такие условия реакции (II 1.34)
стандартными. Это ограничение не очень жесткое, так как различия Дф в таких «инертных»
растворителях, как C2CI4 и СС14, не превышают ошибок измерений.
Приведем примеры. Согласно правилу факторов (II 1.35) энтальпия каждой из
Н-связей выражается следующим соотношением:
= — 22PtEj кДж/моль,
(111.36)
где Д #11 — изменение энтальпии при образовании произвольного стандартного
комплекса XiH...Y, для партнеров которого положено Pi = Е\ = 1.
Растворителем является четыреххлористый углерод. Если в качестве стандарта принять
комплекс фенола^ с диэтиловым эфиром, то Д Ни = —22 кДж/моль.
Безразмерные Р. и Ej факторы характеризуют относительную
энергетическую способность х!Ни Yy к образованию Н-связей. В формуле (II 1.36)
факторы Р. и Ej — это константы, не зависимые друг от друга. Примененный здесь
термин «энергетическая способность» означает, что факторы Р. и Ej в этом урав-
72
нении характеризуют уменьшение энтальпии при образовании Н-связи, но не
вероятность протекания реакции (II 1.34), которая зависит от изменения
свободной энтальпии системы и оценивается величиной константы равновесия этой
реакции. Изменение свободной энтальпии Л G = АН—TAS зависит как от АН,
так и от А 5. Связь между АН и AS для некоторых классов Н-связей линейна,
так что А 5 = аАН + Ь, где а и Ъ постоянные. Тогда из (III.36) следует
(ASij-b) = (AS11-b)PiEj. (II 1.37)
Уравнение (III.37) применимо в гораздо более узких рамках, чем (III.36),
потому что линейная связь между АЯи А5 часто нарушается, например, когда
структура молекул RX.H или YjRi такова, что при протекании реакции (III.34)
возникают стерические препятствия.
Из правила интенсивностей (II 1.23) и уравнения (III.36) следует, что
jt (II 1.38)
где V АЛпимеет размерность л1/2- моль~]/2. м~1/2. Численное значение постоянной
К в уравнении (III.23) принято равным 12. Комбинируя (III.25) и (III.38), имеем
Aotj = AanPJo) Ej = 54Р<°> Ej см"1/* . (111.39)
В уравнении (111,39) сохраняются те же значения Ejt что и в (111.36). Факторы
Р? в ряде случаев несколько отличаются от /уфакторов уравнения (II 1.36),
поэтому здесь и фигурирует второй верхний индекс.
Соотношение (II 1.39) в отличие от (II 1.38) применимо лишь к не очень слабым
Н-связям (—АН) > 8—12 кДж/моль. Как уже говорилось, сдвиги частот ^
пропорционально (А Я)2 уменьшаются по мере ослабления Н-связи и
маскируются другими эффектами среды. Некоторые из Р. и Ej факторов
приведены в работе [24]. Общее число Р. факторов М равно чисйу различных групп Х.Н,
общее число Ej факторов N равно числу различных групп Y^. Зная М + N
чисел Р^ и Ej, с помощью формул (111.35)—(II 1.39) можно вычислить одно или
несколько свойств MN различных Н-связей в разбавленных растворах.
Факторы Р. и Ej, как следует из дальнейшего, характеризуют локальную
электронодонорную способность Х^Н и локальную электроноакцепторную
способность Yj. Правило факторов означает, что электроноакцепторная способность
каждой группы Х.Н одинакова по отношению ко всем донорам электронов Yj,
а электронодонорная способность одинакова по отношению ко всем акцепторам
электронов. Q
Описывая сдвиги частот, факторы Pi характеризуют способность связей
Х.Н к динамическому «взаимодействию» со связями H...Y. Pt и Ej факторы, как
было сказано, характеризуют локальные свойства активных центров Х^Н и Y;-
в молекулах. Из правила факторов следует, что взаимодействие этих центров,
приводящее к образованию водородной связи, лишь в малой мере затрагивает
строение молекулы в целом. Поэтому Pt и Ej факторы не коррелируют с диполь-
ными моментами, поляризуемостями, потенциалами ионизации и другими
молекулярными параметрами. Pi и Ej факторы не имеют простой общей связи с
другими химическими реакциями, более глубоко меняющими электронное
состояние молекул RXH и YRi, чем реакция (III.22). Вместе с тем, правило факторов не
является специфической особенностью Н-связей. Аналогичныеэмпирические соот-
отношения справедливы и для некоторых других типов слабых химических
взаимодействий между молекулами (см. гл. IV). Средняя энергия дипольных
взаимодействий и энергия лондоновских взаимодействий в сущности тоже следует
правилу факторов. В этом отношении водородная связь и ряд других типов
химических связей имеют общие черты с вандерваальсовыми взаимодействиями.
Влияние среды. Сравнительный метод расчета. Правило факторов
позволяет осуществлять приближенный пересчет характеристик Н-
связей, найденных в некотором растворителе S к стандартному раствору
в растворителе So (например, СС14) или к состоянию идеального газа.
Это расширение правила строится на допущении, что вДср^- сущест-
73
венный вклад вносят только взаимодействия молекул со средой. А они
сами могут быть Н-связями и следовать правилу факторов. Влияние
иных взаимодействий на связь X—H...Y не учитывается. Реакция
образования Н-связей в растворителе S имеет вид
Х,Н ...S + HS ... Y,- ^t Х;Н ... Y; + S ... HS + Д<р?;.. (III.40)
Применяя правило факторов, получаем:
Ч- = А?п Р°1^+ А?и P°s E°s ~ ^п P°i E°s ~ A*?i P°s Щ
(Ш.42)
Здесь индекс «О» указывает состояние идеального газа. Уравнение
(III.42) представляет собой, по существу, результат применения
сравнительного метода расчета свойств различных соединений. Общие
основы этого метода разработаны В. А. Киреевым, М. X. Карапетянцем,
В. М. Татевским и его сотр. [25].
§ 29. О природе водородных связей
Водородная связь, как и другие взаимодействия между
молекулами, в конечном счете обусловлена электрическими зарядами электронов
и атомных ядер. Вопрос заключается в том, происходит ли при
образовании Н-связи перераспределение электронной плотности в молекулах,
и если оно происходит, то какую роль играет. Ранее считали, что
перераспределением электронной плотности можно пренебречь. Тогда
Н-связь была бы в сущности вандерваальсовым взаимодействием, но на
малых расстояниях между молекулами, когда представление о
точечных диполях и мультиполях уже неприменимо. При этом свойства
Н-связи можно было бы вычислить с помощью параметров,
характеризующих распределение зарядов и другие свойства изолированных
молекул.
Н. Д. Соколов [26], а затем и ряд других исследователей доказали,
что при образовании Н-связи главную роль играет перераспределение
электронной плотности взаимодействующих молекул. Тем самым было
установлено, что водородная связь, по существу, представляет собой
химическое взаимодействие молекул. Хотя изменения в состояниях
молекул, образующих Н-связь, как правило, невелики, свойства
Н-связи не могут быть описаны с помощью свойств изолированных
молекул.
Вывод о перераспределении электронной плотности молекул при
образовании Н-связей вытекает из анализа ИК-спектров. Как уже
говорилось, при образовании водородной связи интенсивность
валентного колебания v5 группы X—Н возрастает. В то же время
интенсивность деформационного колебания v& группы X—Н не только не
возрастает, но даже несколько уменьшается. Простое объяснение этих
фактов можно дать, следуя работам [27].
74
Предположим, что молекула X взаимодействует с молекулой Y,
причем происходит перераспределение электронной плотности.
Молекула X приобретает некоторый отрицательный заряд —В, а молекула
Y — равный по величине положительный заряд +8. Таким образом,
межмолекулярное взаимодействие сопровождается возникновением ди-
польного момента jim. Пусть [i0 — дипольный момент свободной
молекулы X; Qoi — нормальная координата i нормального колебания
свободной молекулы X; Q. — координата i нормального колебания в
системе X...Y. Когда межмолекулярные силы много слабее
внутримолекулярных, различием между Qoi и Qt можно пренебречь при условии,
что частоты колебаний выше 9-1012 Гц (300 см"1), так как в этих
случаях различия между Qo. и Q. относительно малы.
Рис. 8. К расчету интенсивности „валентных и
деформационных колебаний группы X—Н в
связи X—H...Y
Если нормальные колебания считать гармоническими, то в этом
приближении интегральная интенсивность Аг полосы ИК-спектра,
соответствующей i нормальному колебанию, связана с производной
{л = |л0 -f. jim по нормальной координате Q. с помощью следующего
соотношения:-
я
IT
п
17
dQt
(II 1.43)
Для слабых химических связей X...Y с хорошей степенью
приближения можно принять, что производная d\bm/dQt независима от
производной d^JdQv Ориентация молекулы Y относительно молекулы X
фиксирована.
Пользуясь (III.43), получаем
ri)
(III .44)
Изменение интенсивности АА t зависит от направлений векторов d^
и d\xo/dQr Например, если d^0/dQi и dpJdQ. равнонаправлены, то
интенсивность растет; если они противонаправлены, интенсивность
падает.
Рассмотрим линейные Н-связи X—H...Y. Схема такой связи
представлена на рис. 8. Обозначения р0, jiOT, rl9 r2, аг и а2 ясны из
рисунка. Диполи направлены вдоль связей. Суммарный момент системы
Н бй
= jte -f
Нормальные координаты колебаний v5 и v^, как пра-
75
вило, примерно совпадают со смещениями dX и dY ядра Н при
закрепленных ядрах X и Y. Это объясняется тем, что масса протона обычно
много меньше массы ядра X.
Для v имеем
ДЛЯ
dY = 0; dQs -dX = dr1 = — dr2; As
dX = 0; dQb ~ dY = rxdax = — r2da2; Ab <
dX
dY
(111.45)
1
гл
я
а*Зс
а»!
дро
дХ
г2 д
• +
«я
d\im
2
2
IX
Зс
гл
Vm
г?,
Подставляя в (111.45) ji = ji0 -|- jiOT, пренебрегая членами
второго порядка малости и учитывая, что \ьОу == \i0 ax и \ьтУ = jAm а2, получим:
(111.46)
Опыт показывает, что при образовании Н-связи интенсивность
полосы v5 возрастает, a v6 обычно падает. Отсутствие роста
интенсивности полосы валентных колебаний X—Н не наблюдалось. Из этого факта
и уравнения (II 1.46) следует, что векторы d^JdX и дрт/дХ в случае
Н-связи обычно равнонаправлены. Направления дипольных моментов
|а0 и \хт тоже должны совпадать; иначе не было бы выполнено условие
&ь ^ Аьо- Зависимость \im от г2 может быть представлена степен-
ной функцией [im = аг2 п. Тогда
должен быть в п раз больше,
чем | \^т1гг\- Если п велико, то прирост интенсивности полосы
валентного колебания X—Н будет большим, в то время как изменение
интенсивности полосы деформационного колебания будет малым.
Итак, спектральные свойства Н-связей указывают на
перераспределение электронной плотности между X—Н и Y. Как правило,
происходит перенос отрицательного заряда от молекулы RXY к молекуле
RXH. Этот вывод подтверждается, например, исследованиями
диэлектрической проницаемости бензола [28]. Свободные молекулы
бензола неполярны, но при ассоциации в жидкой фазе возникают связи
между группами С—Н одной молекулы и я-орбиталями другой,
сопровождающиеся переносом заряда. При этом молекулы комплекса
приобретают дипольный момент порядка 0,2D.
О влиянии среды на Н-связь. Существует несколько точек зрения по
этому вопросу. Исследования А. В. Иогансена и ряда других авторов
показали, что на энергию и другие свойства Н-связей X—H.-.YRi
могут влиять химические взаимодействия молекул RXH и YRX с
соседними молекулами. Вандерваальсовы взаимодействия на Н-связь
практически не влияют [22, 27, 29].
76
§ 30. Другие слабые химические взаимодействия
между электрически нейтральными молекулами
Слабые химические взаимодействия, происходящие без участия
водорода, изучены не так систематически и полно, как водородные
связи. Слабые химические взаимодействия очень широко распространены и,
несомненно, еще более разнообразны, чем сильные химические связи.
В этом параграфе рассмотрим лишь некоторые из не относящихся к
водородной связи слабых химических взаимодействий между
электрически нейтральными атомами или молекулами.
Инертные газы и теперь еще нередко фигурируют как примеры
веществ без химического взаимодействия. Их атомы имеют наиболее
устойчивые электронные оболочки и приближенно могут быть
уподоблены шарам. Теплота испарения сжиженных инертных газов мала. При
нормальной температуре кипения у Не3 она составляет всего
24,7Дж/моль; у Не4—85 Дж/г-атом; у неона—174Дж/моль; у аргона—
6480 Дж/моль; у криптона — 9050 Дж/моль и у ксенона —
12 620 Дж/моль. Если между атомами инертных газов и действуют
химические силы, то они должны быть очень слабыми.
Вывод, что между атомами инертных газов существуют слабые
взаимодействия химического типа, следует из данных о свойствах паров
этих веществ и структуре кристаллов. В парах инертных газов
обнаружены димеры и в некоторых случаях — более сложные ассоциаты
[30]. Энергия диссоциации молекул неона Ne2 составляет около
355 Дж/моль; аргона Аг2 — около 1090 Дж/моль, ксенона — около
2920 Дж/моль, молекул HeNe — около 117 Дж/моль и т. д.
Тщательные оптические исследования, выполненные И. Танака и
К. Иошино [31], позволили не только уточнить энергию связи атомов в
молекулах ряда инертных газов, но и изучить спектры
внутримолекулярных колебаний. На рис. 9, а представлен график, изображающий
потенциальную энергию E(R) молекулы Аг*0 как функцию расстояния
между атомами. По оси ординат отложены энергии* в условных
единицах — см"1, по оси абсцисс — расстояния между центрами атомов
R в 10"1 нм. Глубина потенциальной ямы (энергия диссоциации) D =
~ 91,6 см"1 = 1090 Дж/моль. Основной колебательный уровень
молекулы лежит на глубине Dg = 76,9 см"1 = 885 Дж/моль.
На рис. 9, б изображены результаты исследований потенциальной
энергии взаимодействия между атомами аргона, когда R меньше 0,3 нм,
т. е. в области сильного отталкивания. При R = 0,22 нм
потенциальная энергия сил отталкивания E(R) достигает 11 000 см"1, что
соответствует около 117 кДж/моль. Молекулы инертных газов могут
возникать при взаимодействии атомов, находящихся в основных или
возбужденных квантовых состояниях. Исключением является гелий, ато-
* 100 см"1 = 1190 Дж/моль. Этот способ выражения энергии применяется
с
иногда в спектроскопии в связи с тем, что энергия кванта з = h& = h— =
А.
= 3- 1010/Ь', где частота V в см"1 есть число колебаний электромагнитной волны,
которые она испытывает, проходя путь в 1 см.
77
£ cm'L
4,0 5,0 6,0
a
Рис. 9. a — Кривая потенциальной
энергии взаимодействия двух атомов
аргона, находящихся в основном
состоянии
(кривая следует уравнению Морзе с Re =
= 3,81 A,De = 262 кал/моль, Do ~ 220
кал/моль, где DQ — потенциальная энергия
молекулы в колебательном состоянии v = 0);
б — кривая потенциальной энергии
при малых межъядерных
расстояниях
(В области R > 3 А кривая б эквивалентна
кривой а, но глубина потенциальной ямы
несколько преувеличена. Кривая смещена
влево по отношению к а так, что абсцисса
минимума располагается при Re «=» 3,60 А. Это
необходимо для «сшивания» с потенциалом в
области R< 3 А) [31]
Рис. 10. Схема плотнейшей шаровой
упаковки в одном слое [33]
мы которого в основном состоянии
димеров не образуют. Молекулы
Не2 могут возникать лишь при
условии, когда хотя бы один из
взаимодействующих атомов находится
в возбужденном состоянии [31].
При температуре 89 К и давлении
2,74-105 Па может быть
ассоциировано 2% атомов неона; при
давлении 5,07- 10б Па концентрация
ассоциированных атомов аргона,
криптона и ксенона достигает 3%
(при температурах 302 К, 419 К и
513 К соответственно) [32].
Известно немало химических соединений
инертных газов с атомами других
элементов.
Инертные газы обычло
кристаллизуются с образованием плотней-
ших упаковок атомов. (О
некоторых отклонениях от этого
правила см. стр. 79). Чтобы понять, на
чем основан упомянутый выше
вывод о слабых химических
взаимодействиях между их атомами,
надо сначала рассмотреть некоторые
из свойств плотнейших упаковок
шаров одинакового размера. Мы
воспользуемся описанием,
имеющимся в книге А. И.
Китайгородского [33] и работе Л. Иенсена ([4],
стр. 251). Наиболее плотная
упаковка шаров одинакового размера
достигается следующим образом.
Разместим несколько шаров так,
чтобы они плотно прилегали друг
к другу (рис. 10). Внутри такого
упакованного слоя каждый шар
имеет шесть соприкасающихся с ним
соседей. Это единственно
возможный способ создания наиболее
плотной упаковки в слое
одинаковых шаров. Между шарами
имеются лунки. В эти лунки сверху
можно положить шары. Тогда мы
получим второй плотно упакованный
слой. Отметим, что одни из лунок
нижнего слоя будут заняты, а
другие останутся свободными. Лун-
78
ки заполняются через одну, так как заполнить все лунки шарами того
же размера, что и шары нижнего слоя, невозможно. На рис. 10 лунки
первого слоя, остающиеся пустыми, зачернены. Если бы мы поместили
шары только в зачерненные лунки, то тогда незачерненные лунки
остались бы пустыми. Упаковка второго слоя не изменилась бы.
Таким образом, плотная упаковка, состоящая из двух слоев шаров
одинакового размера, может быть только одна.
Но если мы будем располагать в лунках второго слоя шары
третьего слоя, то для третьего слоя возникают две возможности. Одна из
них— центры шаров третьего слоя лежат над центрами шаров первого
слоя; положения шаров первого и третьего слоев полностью совпадают.
Другая — шары находятся над зачерненными лунками первого слоя.
Хотя обе трехслойные структуры и обладают одинаковой плотностью
упаковки, они различны. Обозначим нижний слой символом Л, второй
слой символом В. Если третий слой совпадает с первым, то мы опять
получаем слой А. Последовательность слоев А В ABA В... представляет
собой гексагональную плотнейшую упаковку (ПГУ) шаров одинакового
размера. Если третий слой не повторяет слой Л, то его можно
обозначить символом С, так как его положение отличается и от слоя Л и от
слоя В. Слой С можно получить из слоя А, повернув слой А на угол
60° вокруг оси, перпендикулярной к плоскости слоя.
Последовательность слоев ABC ABC ABC... представляет собой гранецентрированную
кубическую (ГКЦ) плотнейшую упаковку шаров одинакового размера.
Можно построить и множество других плотнейших упаковок,
отличающихся последовательностью слоев, например АВСВАВСВ.... Но нас
интересуют только первые две простейшие упаковки: гексагональная и
гранецентрированная кубическая. Неон, аргон, криптон и ксенон
кристаллизуются с образованием ГКЦ решетки. Жидкий Не4 при
температурах ниже 1 К и давлениях порядка 30-105 Па кристаллизуется с
образованием ПГУ структуры. В интервале от 1 до 2 К Не4
кристаллизуется в объемноцентрированной кубической (ОЦК) решетке (см. гл.
XI), которая при возрастании давления быстро переходит в
гексагональную плотноупакованную (ПГУ) структуру. Жидкий Не3 при
давлении порядка 30-105 Па и температурах ниже 3 К кристаллизуется с
образованием ОЦК структуры. При повышении давления до 1-107 Па
ОЦК модификация переходит в плотноупакованную гексагональную
(ПГУ) структуру.
Можно получить кристаллы аргона с гексагональной плотнейшей
упаковкой атомов, но они неустойчивы и превращаются в кристаллы с
гранецентрированной кубической решеткой.
И в гексагональной, и в гранецентрированной кубической решетке
каждый атом Ихмеет 12 непосредственно соприкасающихся с ним
соседних атомов (координационное число Z = 12). Но в решетке с
гексагональной упаковкой расстояния между соответствующими атомами в
первом и третьем слоях немного короче, чем в решетке с
гранецентрированной кубической упаковкой. Если бы между атомами было только
вандерваальсово (лондоновское) притяжение на больших расстояниях и
близкодействующее отталкивание на малых расстояниях, то инертные
газы должны были бы кристаллизоваться с образованием гексагональ-
79
ной плотной упаковки атомов. Разность вандерваальсовых энергий
обоих типов упаковок очень мала — она составляет всего лишь 0,01%
энергии решетки. Но важно то, что энергия ПГУ структуры ниже,
поэтому все попытки объяснить большую устойчивость гранецентрирован-
ной кубической структуры кристаллов инертных газов, не прибегая к
представлению о взаимодействиях, сопровождающихся
обобществлением электронов, оказались безуспешными. Объяснение наблюдаемой
структуры кристаллов инертных газов, как показал Иенсен, может быть
дано лишь с помощью представлений о слабых химических
взаимодействиях между атомами. В кристаллах инертных газов существуют
трехатомные фрагменты Х3 (например, Аг3), энергия которых, согласно
расчетам Иенсена, зависит от угла б между направлениями «связей»:
Как в кубической, так и в гексагональной решетках каждый атом
образует с какими-либо двумя из 12 ближайших соседей 66
треугольников. Из них 57 треугольников для обеих решеток одинаковы, а 9
различны [4].
Расчеты показали, что при 180° > 6 > 120° потенциальная
энергия групп Х3 атомов инертных газов почти не зависит от величины 6.
Но в области значений 6, меньших, чем 120°, потенциальная энергия
групп X з быстро возрастает, поэтому трехатомные группы с 0 <
<; 120° снижают устойчивость гексагональной плотной упаковки
атомов инертных газов. Более высокая устойчивость гранецентрирован-
ной кубической упаковки инертных газов, согласно Иенсену [4],
вызвана тем, что при переходе от гексагональной плотной упаковки к гра-
нецентрированной кубической три тримера с 6 = 110° превращаются
в три тримера с 6 = 120°.
Соответствующее этому вращение на 60° Л-слоя (гексагональной
решетки), в результате которого образуется С-слой (кубической
решетки), сопровождается небольшой потерей энергии попарных
взаимодействий между атомами. Эта потеря с избытком компенсируется
энергией трехчастичных взаимодействий химического типа,
сопровождающихся обобществлением электронов и образованием тройников Х3 с 6^
- 120°.
Итак, если бы между атомами инертных газов не было химических
взаимодействий, характеризующихся появлением некоторых
преимущественных «валентных углов» между связанными друг с другом
атомами, то твердые инертные газы должны были бы иметь ПГУ
структуру. 3hro, очевидно, справедливо и в других случаях, когда речь идет о
взаимодействии частиц сферической формы. Ближайшей к ПГУ
структуре по уровню вандерваальсовой энергии является ГЦК упаковка
сферических атомов. Ее потенциальная энергия при отсутствии
химических взаимодействий выше; она менее стабильна. ОЦК решетка
обладает еще более высокой потенциальной энергией вандерваальсовых
взаимодействий. При отсутствии химических сил она менее стабильна,,
чем ГЦК упаковка.
80
Вывод о слабом химическом взаимодействии между атомами
инертных газов качественно следует из экспериментальных данных о
структуре кристаллов и ее изменении с ростом давления. Иенсен дал
приближенное квантовомеханическое обоснование этого вывода. Но если
слабые связи химического типа между атомами инертных газов
наблюдаются в парах и в твердой фазе, то, надо полагать, они имеются и в
жидкости.
Итак, даже у атомов инертных газов при их контакте друг с другом
появляются слабые химические взаимодействия. Естественно ожидать,
что в большинстве других случаев контакты между атомами или
молекулами тоже будут приводить к возникновению слабых химических
связей. Опыт подтверждает это.
Литература по вопросу о молекулярных комплексах и ассоциатах
стала столь обширна и растет так быстро, что полное описание всех
установленных фактов в рамках этой книги невозможно. Мы
рассмотрим лишь некоторые характерные случаи. Подробности и
библиографию можно найти в монографии Л. Эндрюса и Р. Кифера [34], обзорах
Г. А. Бента [35], Г. Б. Сергеева и И. А. Леенсона [36], Г. И. Биттриха
[37] и др.
Иод. Рентгенографический анализ кристаллов иода показал, что
каждый атом иода участвует в одной сильной и по меньшей мере двух
слабых химических связях. Расстояние между центрами атомов,
связанных сильной химической связью, равно 0,270 нм, что на 0,003 нм
больше, чем расстояние между атомами иода у молекул парообразного
12. Атомы, соединенные слабой химической связью, удалены друг от
друга на 0,354 нм, что примерно на 0,08 нм меньше их вандерваальсо-
ва расстояния. Вандерваальсово расстояние приближенно
характеризует то расстояние между центрами атомов, при котором энергия их
взаимодействия сравнима с &Б Т при 300 К-
Эта характеристика является условной, так
как расстояние, при котором
взаимодействие сравнимо с &б Т, для одного и того
же атома в различном окружении может
быть разным. Но, как показывает опыт,
существенное (порядка 3-10~2 — 5-10~2 нм
и более) уменьшение межъядерного
расстояния по сравнению со средним «вандер-
ваальсовым межъядерным расстоянием»
свидетельствует о химическом
взаимодействии.
Итак, слабые химические связи между
молекулами иода немного растягивают
сильную связь. Энергия слабых химических
связей между молекулами 12 пока не
определена. Слабые и сильные химические связи
между атомами иода приводят к
образованию плоского слоя, строение которого
изображено на рис. И. На этом рисунке
показано, ЧТО раССТОЯНИе Между ДРУГИМИ СОСеД- расстояния в А
Рис. 11. Строение плоского
слоя, образованного
молекулами h в кристалле иода
135]:
кружки обозначают атомы иода;
сплошные линии — сильные
химические связи между атомами;
пунктир — слабые химические связи;
цифры обозначают межъядерные
81
ними атомами иода в решетке равно 0,407 нм. Оно на 2,3-10~2 нм
меньше вандерваальсова расстояния. Кристаллическая структура
брома и хлора подобна структуре иода. Для С12, Вг2 и 12 вандерва-
альсовы расстояния больше расстояний между атомами, связанными
слабой химической связью на 3, 6 и 8-10~2 нм соответственно.
Структура кристаллов бромистого
иода 1Вг также близка к
строению 12.
Межмолекулярные расстояния Вг...1Вг и
I...BrI равны
соответственно 0,318 и 0,376 нм.
Хлористый иод IC1
кристаллизуется в двух формах а
и р, В обеих формах
молекулы IC1 образуют
зигзагообразные цепочки,
изображенные на рис. 12. Эти цепочки
содержат два типа' молекул
IC1. В молекулах одного
типа оба атома принимают
участие в слабых химических
связях вдоль цепочки. Эти
молекулы изображены на
рис. 12 незачерненными
кружками. В молекулах второго
типа атомы хлора образуют
ответвления. Атомы хлора и
иода второго типа молекул
на рис. 12 изображены
черными кружками. а-Форма IC1 состоит из цепочек, в которых
ответвляющиеся атомы хлора занимают ^шмтоложение, т. е.
направлены в одну сторону. В Р-форме цепочки построены так, что
ответвляющиеся атомы хлора занимают транс-положения по
отношению друг к другу, т. е. направлены в разные стороны. На рис. 12 углы
между связями С1*—I...I и I—C1...I для простоты представлены, как
прямые углы. (Здесь С1* —ответвляющийся атом хлора). В
действительности же они равны 87 и 116° для (3-формы IC1 и 94 и 102° для а-
формы IC1. Расстояние между ядрами I и С1 в свободной молекуле IC1
равно 0,23 нм. В цепочках оно возрастает, причем для молекул,
участвующих в двух слабых связях, сильная связь растягивается до 0,244 нм.
У молекул, образующих одну слабую связь, расстояние I—С1
равно 0,237 нм. Расстояние I...C1 и I...I примерно на 0,1 нм короче
соответствующих вандерваальсовых расстояний.
Взаимодействие галогенов с кислородом, азотом, серой и селеном.
Как показывают кристаллографические исследования между атомом
галогена Г и атомом X, где X =0, N, S, Se, очень часто
наблюдаются слабые химические связи. Они обнаруживаются по существенному
уменьшению расстояния Г...Х сравнительно с суммой
вандерваальсовых радиусов атомов Г и X. Атомы Г и X могут быть составными частя-
dlCl
Рис. 12. Молекулярные цепи в
кристаллическом монохлориде иода [35]:
большие кружки — атомы кода; малые—атомы
о
хлора; цифры — межъядерные расстояния в А;
>{<) — изолированная молекула монохлорида иода
82
Таблица 6
Межъядерные расстояния в соединениях со слабой химической связью
Г...Х [35] (Г = С1, Вг, I; X = О, N, S, Se)
Молекула, содержащая
атом X
1,4-Диоксан
1,4-Диоксан
1,4-Диоксан
Ацетон
Метанол
1,4-Диоксан
Циклогексан-1,4-дион
1 -4-Диоксан
Ацетонитрил
Молекула, содержащая
атом Г
С12
Оксалилхлорид
Вг2
Вг2
Вг2
Оксалилбромид
Дииодацетилен
Дииодацетилен
Вг2
Связи и межъядерные расстояния,
10-1 Нм
2,67
(3,20)
3,18
°- C1(1,72)C1
(3,20)
2,82
(3,35)
2,78
О...ВГ2'28 Вг
(2,28)
(3,35)
2,78
—tt,*
(3,35)
°-ВГ(1,89)С
2,93
°"'1 (1,99)°
(3,55)
2,65
°-1(1.99)С
(3,55)
2,82
N-Br(2,28)Br
(3,45)
83
Продолжение табл.6
Молекула, содержащая
атом X
Гексаметилентетраамин
Пиразин
Триметиламин
4-Пиколин
Триметиламин
Пиридин
Хинолин
1,4-Дитиан
1,4-Дитиан
Молекула, содержащая
атом Г
Вг2
Тетрабромэтилен
12
I.
IC1
IC1
Йодоформ
1а
Дииодацетилен
Связи и межъядерные расстояния,
Ю-1 Нм
2,26
N... Вг-^-Вг
(2,28)
(3,45)
3,02
(3,45)
2,27
2,83
N ... I—■ I
(2,67)
(3,65)
2,31
2,83
"-■.«г»1
(3,65)
2,30
2,52
N--! (2,30)C1
(3,65)
2,26
2,51
"-\2.*»Ql
(3,65)
3,05
"-'(...s,0
(3,65)
2,87
-■О
(4,00)
3,27
S-I(1.99,C
(4,00)
84
Продолжение табл.6
Молекула, содержащая
атом X
1-4-Дитиан
1,4-Дитиан
Бензилсульфид
Сера
Сера
1,4-Диселенан
1,4-Диселенан
1-4-Диселенан
Тетрагидроселенофен
Молекула, содержащая
атом Г
Йодоформ
Sbl3
I.
Йодоформ
Sbl3
Ii
Дииодацетилен
Тетраиодэтилен
1,
Связи и межъядерные расстояния,
Ю-1 Нм
3,32
s...i2'10 с
(2,13)
(4,00)
3,27
2 77
S...Sb I
(2,70)
(4,35)
2,78
s...i2-82 i
(2,67)
(4,00)
3,50
s...i 2Л0 с
(2,13)
(4,00)
3,60
s...i 2'75 sb
(2,70)
(4,00)
2,83
Se-I(2,66)1
(4,15)
3,34
"-'(..и,0
(4,15)
3,41
(4,15)
2,76
Se-I'IS)1
(4,15)
85
ою-
•ою-
ми каких-либо молекул. Некоторые из примеров слабых химических
связей Г и X приведены в табл. 6, заимствованной нами из обзорной
работы Г. А. Беыта [35]. В графе «связи и межъядерные расстояния»
этой таблицы цифры над символами связи указывают расстояния после
образования соответствующей слабой химической связи. Цифры под
символами связи указывают расстояния до образования слабой
химической связи. Например, в комплексе 1,4-диоксан С12 расстояние
между атомными ядрами О и С1 равно 0,267 нм; сумма вандерваальсовых
радиусов О и С1 равна 0,320 нм; межъядерное расстояние в свободной
молекуле С12 равно 0,199 нм; межъядерное расстояние в молекуле С12>
участвующей в комплексе, равно 0,202 нм. Автор обзора [35] полагает,
что во всех случаях, приведенных в табл. 6, атом Г играет роль
акцептора электронов, а молекула, содержащая атом X, является донором
электронов. 'Это утверждение
быть может и правильное, в
каждом конкретном случае
нуждается в обосновании. Слабая
химическая связь может возникнуть
не только вследствие переноса
заряда. Для ее образования
может быть достаточно и
перераспределения электронной
плотности в пространстве между
молекулами. Так, например, комплекс бензола с Вг2 состава 1 : 1
(его структура представлена на рис. 13) некоторое время
рассматривался как типичный пример комплекса с переносом заряда от
бензольных колец к молекулам брома. Но Хупер на основе анализа
спектров ядерного квадрупольного резонанса
(ядра 81Вг) пришел к выводу, что в этом комплексе
нет заметного переноса заряда от бензольных колец
к молекулам брома. То же было найдено по
отношению к комплексу /г-ксилола и СВг4 (рис. 14).
Нитрилы. Оптические исследования нитрилов
и анализ данных о статической диэлектрической
проницаемости привели к выводу, что в жидкой
фазе образуются ассоциаты (RCN)n, имеющие
цепочечную структуру:
Рис. 13. Строение молекулярного
комплекса С6НбВг2 в твердой фазе
115°
\
Таким образом, в ассоциатах (RCN)rt атом азота
образует связь с атомом углерода. Изменение
энтальпии А Я при образовании одной связи в жид-
Рис. 14. Строение
молекулярного
комплекса пара-
ксилола и четырех-
бромистого
углерода:
черные кружки
—атомы углерода
86
кой фазе составляет 4—6 кДж/моль. В парах изменение энтальпии
при образовании связи C...N составляет, по-видимому, несколько
большую величину. Уменьшение энтальпии связи C...N в жидкой фазе
объясняется так же, .как и для связи О—Н...0 уксусной кислоты (см.
гл. II). Молекулы нитрилов имеют большой дипольный момент. Для
ацетонитрила он равен 3,8Ш. При переходе мономерных молекул из
пара в жидкую фазу их потенциальная энергия уменьшается на
величину ER за счет поляризации окружения и реактивной поляризации
самой молекулы. При образовании ассоциатов эффективный
дипольный момент одной молекулы падает, так что ER, а следовательно, и
Д# уменьшается примерно на 4 кДж/моль.
Связи С...О. На рис. 15 в качестве примера изображена структура
комплексов, образующихся вследствие слабого химического
взаимодействия между кислородом
карбонильной группы одной из
молекул тетрахлор-я-бензохинона
и углеродом карбонильной
группы другой молекулы.
Четыреххлористый углерод.
Способность четыреххлористого
углерода к образованию
ассоциатов была обнаружена М. Г.
Воронковым, М. В. Поздняковой
и Л. А. Жагата [38]. Ими было
изучено понижение температуры
кристаллизации циклогексана
при растворении в нем
четыреххлористого углерода.
Результаты опытов указывают, что
четыреххлористый углерод,
растворенный в циклогексане,
образует ассоциаты (СС14).
М. И. Шахпаронов, Б. Г. Ка-
питкин и В. В. Левин [28] на
основании радиоспектроскопических исследований в микроволновом
диапазоне также пришли к заключению, что в жидком четыреххлористом
углероде имеются не только мономерные молекулы, но и димеры, триме-
ры, а также более сложные ассоциаты (CCIJ^. Мономерные молекулы
СС14 в основном квантовом состоянии не имеют электрического диполь-
ного момента. При образовании ассоциатов происходит
перераспределение электронной плотности молекул. Когда конфигурация ассоциатов не
имеет центра симметрии, молекула ассоциата приобретает
электрический момент. Оказалось, что в жидком четыреххлористом углероде
имеются полярные ассоциаты, так что эффективный момент, приходящийся
на одну молекулу четыреххлористого углерода, составляет около О,ID.
Ассоциация четыреххлористого углерода подтверждается
исследованиями ИК-спектров. В области частот около 1,2-1011 Гц (40 см"1),
т. е. в субмиллиметровом диапазоне длин волн, наблюдается полоса
Рис. 15. Строение ассоциатов в тетра-
хлорпарабензохиноне СбС14О2 [35]:
пунктиром указаны наиболее сильные связи О ... G
с межъядерным расстоянием 2,85 °к
87
поглощения, которая, по всей вероятности, вызвана низкочастотными
колебаниями молекул СС14 в ассоциатах. Ан-алогичные данные имеются
для сероуглерода. Энергия связи между молекулами СС14 составляет
около 4 кДж/моль.
В гл. V будут описаны ассоциаты бензола и комплексы бензола с
хлороформом за счет л;-орбиталей ароматического цикла. Аналогичные
явления обнаружены Г. Биттрихом и его сотрудниками в растворах
тетрагидрофуран—бензол и диэтиламин—хлорбензол [37].
§ 31. Слабые химические взаимодействия ионов
Взаимодействия между ионом и окружающими его частицами
(атомами, молекулами, ионами) можно подразделить на химические и
электростатические. Химические взаимодействия осуществляются только с
теми частицами, которые вступают в непосредственный контакт с
ионом. Они сопровождаются обобществлением электронов.
Электростатические взаимодействия вызваны электрическим зарядом иона.
Они распространяются на всю массу вещества, окружающего ион.
Химические взаимодействия ионов с другими частицами могут быть
сильными или слабыми. При взаимодействии протона Н+ с молекулой
водорода Н2 образуется ион Н+2 и выделяется энергия, равная 290
кДж/моль. Энергия, выделяющаяся при взаимодействии с Н+ атомов
гелия, равна 173 кДж/моль, кислорода — 468 кДж/моль, молекулы
воды — 710 кДж/моль, аммиака — 905 кДж/моль. Все это примеры
сильных химических взаимодействий, происходящих с участием
ионов [3].
Слабые химические взаимодействия наблюдаются, например, в тех
случаях, когда связанные друг с другом ионы изоэлектронны атомам
инертных газов и имеют заполненные электронные оболочки. Таковы
ионы галогенидов щелочных металлов. Рассмотрим этот случай
подробнее.
Галогениды щелочных металлов. Существование слабых химических
взаимодействий между ионами галогенидов щелочных металлов, как и
в случае атомов инертных газов, вытекает из результатов анализа
свойств кристаллов и паров этих веществ. Отличие от инертных газов
здесь состоит не только в том, что ионы заряжены, но и в том, что анион
и катион одного галогенида неодинаковы по размерам. Ранее
возможность химического типа взаимодействий между ионами в кристаллах
галогенидов щелочных металлов не учитывалась. Ионы
рассматривались как «химически насыщенные» частицы. Взаимодействие между
ионами в существенных чертах предполагалось парным, так что полная
энергия кристаллической решетки считалась суммой энергий
взаимодействия ионов, взятых попарно. При этом учитывались все возможные
парные комбинации ионов.
М. Борн и Д. Мейер предполагали, что энергия всех парных
взаимодействий между ионами в кристаллах галогенидов щелочных
металлов состоит из трех слагаемых:
а) электростатической энергии взаимодействия ионов в решетке,
88
если их рассматривать как точечные заряды. Отнесенная к одной
ячейке кристалла эта энергия имеет вид:
где ге — наименьший ионный заряд в решетке кристалла; /?0 —
наименьшее расстояние между двумя ионами; а' — постоянная Маделун-
га, зависящая от структуры кристаллической решетки;
б) энергии близкодействующего отталкивания. М. Борн и Д. Мейер
вычисляли ее с помощью эмпирического потенциала (II 1.2). У галоге-
нидов щелочных металлов полная энергия близкодействующего
отталкивания ионов во много раз превышает энергию межатомного
отталкивания в кристаллах инертных газов. Электростатическое притяжение
сжимает кристалл в такой степени, что ближайшие ионы
отталкивают друг друга;
в) энергии вандерваальсова притяжения между ионами.
По расчетам Борна и Мейера, при нормальных давлении и
температуре кристаллы всех галогенидов щелочных металлов должны иметь
структуру типа NaCl. Катионы и анионы образуют две вставленные
друг в друга гранецентрированные кубические решетки. Давно
известно, что большинство галогенидов щелочных металлов действительно
образуют кристаллы типа NaCl. Но в ряду Na, К, Rb, Cs
кристаллическая решетка типа NaCl постепенно теряет устойчивость. Хлористый,
бромистый и йодистый цезий кристаллизуются в решетке типа CsCl.
Катионы и анионы располагаются в узлах двух вставленных друг в
друга простых кубических решеток. В последние десятилетия
выяснилось, что при повышении давления у кристаллов многих из галогенидов
щелочных металлов, включая и NaCl [39], наблюдается переход от
решетки типа NaCl к решетке типа CsCl.
Удовлетворительное объяснение относительно высокой
стабильности решетки типа CsCl для кристаллов галогенидов щелочных металлов в
рамках теории Борна—Мейера не было найдено. По расчетам Л. Иен-
сена [4], помимо электростатического взаимодействия между ионами и
близкодействующего отталкивания, важную роль играют трехчастич-
ные взаимодействия, сопровождающиеся обменом электронами между
ионами. Но в отличие от инертных газов трехчастичные
взаимодействия между ионами в кристаллах галогенидов щелочных металлов
способствуют возникновению треугольников с малыми углами б. При
6 ^ 120° имеет место притяжение, при 6>120° — отталкивание.
Вклад в полную энергию решетки трехчастичных взаимодействий
химического характера (сопровождающихся обобществлением
электронов) составляет от 12 до 25 кДж/моль. Вандерваальсо-
вы взаимодействия Иенсеном не учитывались, так как их
влиянием на структуру решетки галогенидов щелочных
металлов можно пренебречь. Расчеты Иенсена позволили не
только объяснить стабильность решеток типа CsCl в кристаллах
хлорида, бромида и иодида цезия, но и вычислить величину
давлений, при которых происходят фазовые переходы между
кристаллами с решетками типа NaCl и CsCl. Рассчитанные величины давлений
хорошо согласуются с экспериментальными.
89
Итак, даже в тех случаях, когда ионы имеют заполненные
электронные оболочки, подобные атомам инертных газов, они проявляют
способность к слабым химическим взаимодействиям. Химическая
активность таких ионов еще более отчетливо выражена, когда они
взаимодействуют с частицами, не имеющими заполненных электронных оболочек.
Одним из характерных примеров могут служить взаимодействия
отрицательно заряженных одноатомных ионов галогенов и
электрически нейтральных молекул галогенов, приводящие к образованию
полиатомных ионов.
Полиатомные ионы галогенов. Известны, например, трехатомные
ионы галогенов. Они могут иметь линейную структуру и быть
асимметричными или симметричными. Строение иона зависит от катиона,
с которым ион связан. Так, ион ^асимметричен в трииодиде аммония
и цезия и симметричен в трииодиде
Q3.03 s-\ 183 г\ тетрафениларсония (C6H5)4AsI3. На
—'■ \^у— \^J рис. 16 более слабая связь в несиммет-
а ричном трииодидионе изображена
пунктиром. Цифры указывают
межатомные расстояния в 10"1 нм. Ион ио-
да может присоединять две молекулы
^ 12 с образованием пентаиодидиона,
Рис. 16. Строение иона I,- [35]: имеющего плоскую структуру.
а - асимметричный триодион в триодио- ОтМвТИМ, ЧТО В ПОЛИИОДИДИОНЗХ
дах аммония и цезия; б — симметричный ГГОИ ООраЗОВаНИИ СЛЭОЫХ ХИМИЧеСКИХ
SSTA^raS SSSSHSE?
связей происходит «растяжение» силь-
ядерные расстояния в А ных связей- Чем сильнее «слабая»
связь, тем короче соответствующее
межъядерное расстояние и
соответственно больше растяжение сильной связи (табл. 7).
Полиатомные ионы галогенидов наблюдаются в кристаллах солей, в расплавах
и растворах.
Могут существовать не только полиатомные анионы галогенидов,
но и полиатомные катионы. Так, например, некоторые растворы иода
в концентрированной серной кислоте, по-видимому, содержат катионы
1+, 1з\ Is". Существуют полиатомные ионы галогенидов смешанного
состава, например, CIF2", CIF4", 1С\Т и т. д.
Сольватация ионов. Взаимодействие ионов с растворителем
называют сольватацией ионов. Для водных растворов используется термин
гидратация. Согласно сказанному сольватация включает в себя
химическое и электрическое взаимодействие ионов с окружающими частицами.
На примере галогенидов щелочных металлов можно видеть, что оба
эти типа взаимодействий тесно взаимосвязаны, поэтому исследование
каждого из них в отдельности затруднительно. В разбавленных
растворах ионов на первый план нередко выступают электростатические
силы. В концентрированных растворах обычно увеличивается влияние
химических взаимодействий.
Химическое взаимодействие между ионами и растворителем в
жидкой фазе пока еще изучено недостаточно систематически. Тем не менее
90
Таблица 7
Расстояние между ядрами атомов иода в молекулах
и группах I ... I—I полииодидионов [35]
Частица
I» (г*з.)
!2 (крист.)
If" (крист.)
1|" (крист.)
If (крист.)
1^~ (крист. несимм.)
1|" (крист.)
1"^ (крист. симм.)
«Короткое»
расстояние
в Ю-1 нм
2,67
2,70
2,74
2,80
2,81
2,83
2,85
2,90
«Длинное»
расстояние
I ... I
в 10-1 нм
4,30*
3,54
3,44
3,42
3,17
3,03
3,00
2,90
* ВандерваальсоЕО расстояние.
многочисленные качественные, а в некоторых случаях и количественные
характеристики строения сольватов или гидратов имеются. Так,
например, рентгенографические исследования А. Ф. Скрышевского [40]
показали, что в водных растворах LЮН, содержащих 10 молекул воды
на одну молекулу гидрата окиси лития, каждый ион Li тетраэдрически
окружен двумя молекулами воды и двумя ионами ОН". Расстояния
между Li+ и О составляют около 0,2 нм. В водных растворах хлорида
магния, содержащих на одну молекулу MgCl2 25 молекул воды, ионы
Mg2+ гидратированы в среднем шестью молекулами воды, образующими
правильный октаэдр.
Расстояние между Mg2+ и О составляет 0,2 нм. Каждый ион хлора
С1~ также окружен шестью молекулами воды. Расстояние С1—О
составляет 0,325 нм. Сходную структуру имеет и первая гидратная оболочка
хлорида никеля NiCl2, растворенного в воде. Эти гидраты, по существу,
представляют собой координационные соединения, в которых
молекулы воды являются лигандами. Таким образом, проблема сольватации
ионов тесно связана с химией координационных соединений и теорией
поля лигандов. Здесь нет ни возможности, ни необходимости излагать
проблемы теории поля лигандов, химии координационных соединений и
другие вопросы, связанные с изучением сольватации ионов. Материалы
и библиографию о сольватации ионов можно найти в книгах О. Я.
Самойлова [41], К. П. Мищенко и Г. М. Полторацкого [42], Н. А.
Измайлова [43], Р. Робинсона и Р. Стокса [44], Л. Эндрюса и Р. Кифера
144], В. С. Шмидта [45], серии монографий «Современные проблемы
электрохимии» [46—49], сборнике «Вопросы физической химии
растворов электролитов» [50]. Проблемы теории поля лигандов и химии
координационных соединений рассматриваются в книгах Р. Коттона
и Дж. Уилкинсона [51], И. Б. Берсукера [52] и многих других изданиях.
91
ГЛАВА IV
ЭМПИРИЧЕСКИЕ МЕТОДЫ ОПИСАНИЯ
МЕЖМОЛЕКУЛЯРНЫХ ВЗАИМОДЕЙСТВИЙ
§ 32. Об эмпирических формулах
Теоретические расчеты межмолекулярных взаимодействий пока еще,
как правило, имеют значение для качественных выводов об их
особенностях. Количественные характеристики в подавляющем большинстве
случаев получаются с помощью эксперимента. Экспериментальные
данные об энергии межмолекулярного взаимодействия могут быть
описаны с помощью эмпирических формул. Некоторые из них будут
рассмотрены в этой главе. Почти все они основаны на анализе свойств
разреженных газов. Формулы, пригодные для эмпирического описания
межмолекулярных взаимодействий в разреженных газах, часто
применяют для тех же целей к жидким системам. Здесь порой упускают из
виду следующее. Во-первых, в разреженных газах среднее расстояние
между молекулами велико, поэтому сравнительно большой вклад во
взаимодействие вносят дальнодействующие силы. (Когда молекулы
электрически нейтральны, то это в основном дипольные и лондонов-
ские взаимодействия.) В жидкостях же, как мы видели,очень важна роль
близкодействующих сил. Во-вторых, энергия реактивного
взаимодействия полярных молекул с окружающей средой в газах мала, а в
жидкостях велика и может существенно изменять энергию образования связей
между молекулами. В этом отношении формулы, основанные на
свойствах газов, ведут к недооценке роли дальнодействующих сил.
В-третьих, при переходе от жидкой фазы к парам межмолекулярные силы
могут испытывать качественные изменения, обусловленные влиянием
коллективного взаимодействия большого числа частиц. Так происходит,
например, при испарении металлов. В-четвертых, эмпирические
формулы представляют собой усредненную эффективную характеристику
межмолекулярных сил. Способ усреднения обычно не ясен, но он должен
зависеть от метода исследования энергии взаимодействия и влиять на
математическую форму эмпирической потенциальной функции E(R) и
значения фигурирующих в этой функции параметров.
Методы, позволяющие связывать макроскопические свойства
вещества с межмолекулярными взаимодействиями, применимые для
разреженных газов, не всегда пригодны при описании свойств жидкостей.
По этим причинам эмпирические формулы, применимые для
характеристики межмолекулярных взаимодействий в газах, нельзя
безоговорочно применять в ходе исследования жидких систем.
Моделирование жидкостей с помощью эмпирических соотношений,
полученных для их паров, возможно. Однако выводы, основанные на
применении таких моделей, нуждаются в проверке.
В сущности, то же самое следует сказать и о моделях и
эмпирических соотношениях, основанных на свойствах твердых тел. Этот вопрос
подробно разбирается Харрисоном [53].
92
§ 33. Потенциалы Морзе и Липпинкотта
Взаимодействие двух атомов, сопровождающееся возникновением
сильных или слабых химических связей, во многих случаях хорошо
описывается потенциалом Морзе:
где De — энергия диссоциации двухатомной молекулы в состоянии,
когда ее потенциальная энергия минимальна; Re — расстояние, при
котором E(R) минимальна; (3 — эмпирическая постоянная. Потенциал
Морзе широко применяется при описании двухатомных молекул,
возникших в результате сильного химического взаимодействия, таких,
например, как НС1, НВг, О2 и т. д.
Потенциал Морзе лучше других эмпирических функций
характеризует взаимодействие двух атомов инертных газов на относительно
малых расстояниях, когда образуются молекулы Ne2, Ar2, NeHe [31] в
результате очень слабых химических взаимодействий (см. стр. 78).
Наряду с потенциалом Морзе применяется потенциал Липпинкотта:
Е (R) = De{ 1 - ехр [п (R - Re)* / 2Д]} , (IV.2)
где De — энергия диссоциации молекулы; п — эмпирическая
постоянная. Потенциал Липпинкотта описывает взаимодействие между
атомами Н и С1 в молекуле НС1 и взаимодействие между теми же атомами
различных молекул НС1, сопровождающееся образованием слабой
Н-связи Н—С1... Н—С1. То же относится к HF и F...HF, О—Н и
О—Н...О, а также к ряду других водородных связей [32]. Функции
(IV. 1) и (IV.2) пригодны для описания близкодействующих
межатомных взаимодействий как в газах, так и в жидкостях.
§ 34. Использование результатов исследований
свойств газов
Если газ разрежен, то его уравнение состояния может быть
представлено в виде ряда по степеням V'"1, где V — объем одного моля газа:
(ВС)
PV = RrT h +— + — + ... , (IV.3)
где Р — давление газа; Rr — газовая постоянная; Т —
температура, К; В — так называемый второй вириальный коэффициент, С —
третий вириальный коэффициент и т. д. Вириальные коэффициенты
являются функциями температуры газа.
Классическая статистическая механика дает следующее выражение
для второго вириального коэффициента:
оо
B=2nNA J {1— ехр(— E(R)/kBT)}R*dRt (IV.4)
о
где Na — число Авогадро. Формула (IV.4) применима в том случае,
93
если силы, действующие между молекулами, не зависят от ориентации
молекул по отношению друг к другу.
Выбирая различные эмпирические функции для E(R) и подставляя
их в уравнение (IV.4), стремятся подобрать эмпирические параметры
так, чтобы вычисленное значение В совпало с опытным в возможно
более широком интервале температур. При выборе эмпирических
функций E(R) стараются, чтобы эти функции по возможности были
простыми и не противоречили теоретическим представлениям о
межмолекулярном взаимодействии. В принципе также поступают при анализе
результатов измерений вязкости и других свойств разреженных газов.
Параметры функции E(R) для одних и тех же молекул, полученные
разными методами, как правило, различаются. Это объясняется тем,
что эмпирические функции E(R) и расчетные формулы неточны.
Степенной потенциал. Для простейших неполярных симметричных
молекул (атомы инертных газов, молекулы метана и т. п.) часто
пользуются степенной функцией E(R) вида
Е (R) = — aR-m + bR'n9 (IV.5)
где а, Ьу тип — эмпирические параметры. Первый член в правой
части этого уравнения учитывает потенциальную энергию притяжения.
Уравнение (IV.5) иногда называют формулой Ми и Грюнайзена.
Если т = 6, то (IV.5) переходит в уравнение
Е (/?)£= - ай-в + bR'n. (IV.6)
Эту формулу можно преобразовать, выразив эмпирические постоянные
а и Ь через параметры, имеющие ясный физический смысл.
Дифференцируя (IV.6), получим:
dE 6a пЬ
~dR = ~W ~~ Rn+l ' (IV'?)
Приравнивая (IV. 7) нулю, определяем расстояние/^, при котором
притяжение уравновешивается отталкиванием:
(Яо)""6 =-~-. (IV.8)
оа
Минимальная энергия при R = Ro
Подставляя (IV.9) в (IV.7), получим
^(Л <■*■'«
Если в (IV. 10) подставить то расстояние d0, при котором E(R) = 0,
то, решая это уравнение относительно dQy найдем
94
Вводя обозначение е* =
чим
—Ет{п и подставляя (IV. 11) в (IV. 10),
полуdQ = R0 — =0,8909tf0;
1 2 '
Е(Ю
График этой функции представлен на рис. 17. При /г =12 уравнения
(IV.8)—(IV. 12) приобретают особенно простой вид:
R60 = 2b/a; (IV.13)
(IV.14)
(IV.15)
(IV.16)
(IV.17)
Уравнения (IV. 16) и (IV. 17) часто называют уравнениями Леннарда—
Джонса или (12—б)-потенциалом.
В кн. [2] приведены параметры 8* и d0 (12—6)-потенциала
Леннарда—Джонса для некоторых атомов и простых молекул. Параметры
получены с помощью анализа
результатов измерений вязкости газов и
второго вириального коэффициента. Эти
параметры приближенно
характеризуют особенности взаимодействия
молекул, но значение их не следует
преувеличивать. Даже для атомов
инертных газов истинный парный
потенциал в области малых значений R
глубже и круче потенциала
Леннарда—Джонса. Только благодаря
случайной взаимной компенсации
ошибок у исследователей существовало
представление, что с помощью
парного (12—6)-потенциала можно
описывать свойства инертных газов во всей
области состояний от кристалла до
разреженного газа [54]. В действительности потенциал Леннарда—Джонса
дает заниженные значения энергии взаимодействия при малых /? и
завышенные значения при больших JR. Следует также иметь в виду, что
потенциал Леннарда—Джонса качественно непригоден для описания
взаимодействия между атомами аргона и большинства других
инертных газов в конденсированных фазах.
Как говорилось в § 30, в аргоне и других жидких инертных газах,
за исключением гелия (см. гл. XI), имеются ассоциаты, содержащие
три и более атомов. Взаимодействие между атомами в таких ассоциатах
зависит не только от межатомных расстояний, но и от «валентных уг-
Рис. 17. График потенциала,
следующего уравнению (IV. 12)
95
лов». Тем не менее знание параметров (12—6)-потенциала может
принести пользу, так как позволяет приближенно-охарактеризовать
межмолекулярные взаимодействия. Это относится и к веществам, состоящим
из несферических, в том числе полярных молекул, приведенных в кн.
[2], к которым не только (12—6)-потенциал, но и любой степенной
потенциал вида (IV.5) заведомо неприменим.
§ 35. Свойства постоянных уравнения
Ван-дер-Ваальса
Межмолекулярное взаимодействие в газах и жидкостях иногда
оценивают с помощью постоянных а и b уравнения Ван-дер-Ваальса:
4г
— b) = RrT, (IV. 18)
где V — объем одного моля газа; параметр а учитывает влияние
межмолекулярного притяжения, параметр b связан с силами отталкивания
между молекулами.
Уравнение (IV. 18) было предложено в 1873 г. Ван-дер-Ваальсом.
Оно может быть выведено теоретически [54, 55]. Эмпирические
параметры аи b представляют неменьший интерес, чем, например,
параметры е* и d0 потенциала Леннарда—Джонса. Приближенная
статистическая теория показывает, что b равно учетверенному собственному
объему молекул:
b = ^-nNA 4; (IV.19)
о
а = — 2nNA \ E(R)R*dR. (IV.20)
Параметры аи b можно определить, зная температуру Тк и давление Рк
или объем Укодного моля вещества в критической точке жидкость — пар:
27 RT ~
a = —RTKb; b = —f-; VK = 3b. (IV.21)
Опыт показывает, что параметры аи b обладают свойством
аддитивности. В гомологических рядах соединений атомам можно приближенно
приписывать некоторые значения параметров а. и 6.. Параметр b
химического соединения получается суммированием величин 6. всех
атомов, входящих в молекулу соединения (Бачинский):
Для параметров а и а. справедливо следующее соотношение (Ван-Лаар):
/!= 2 V^i- (IV-23>
96
Если вещество является двухкомпонентным газовым или жидким
раствором, то
а ~ацл? +2а1шх! х2 + а22л| ; (IV.24)
Ь =t Ьп х\ + 2Ь12 х1 х2 + Ь22 х\ , (IV.25)
где хг и х2 — мольные доли компонентов 1 и 2; ап, 6П, а22 и Ъ22 —
параметры уравнения Ван-дер-Ваальса чистых компонентов, а12 и Ь12
могут быть грубо оценены с помощью уравнений:
(IV.26)
\ [у/~hi + у Щ • (IV.27)
Уравнение (IV.26) было установлено Галициным и Вертело.
Подробное рассмотрение параметров aiJf btJ имеется в монографии М. П. Ву-
каловича и И. И. Новикова [55].
Второй вириальный коэффициент двухкомпонентных газовых
растворов Bv (см. § 34) тоже подчиняется приближенным эмпирическим
соотношениям вида (IV.24) и (IV.26) [56, 57]:
Bv — Bnx2l+ 2В12 хг х2 + В22 х\ ; (IV. 28)
Bi2 - У Вп В22 . (IV.29)
Здесь Вп и В22— вторые вириальные коэффициенты индивидуальных
компонентов 1 и 2, Bi2 — второй вириальный коэффициент,
учитывающий взаимодействие между компонентами 1 и 2.
Обзор и анализ экспериментальных данных о вторых вириальных
коэффициентах газов и их смесей имеется в монографии Э. Мейсона и
Т. Сперлинга [56], статьях Н. П. Маркузина [57], Г. Фрейданка и
М. Реч [58].
Соотношения (IV.26) и (IV.29) и аналогичное эмпирическое
уравнение
£i2 (Я) - VEvl (R) E22 (Ю (IV.30)
часто используются в теории растворов. Уравнение (IV.30) можно
сопоставить с уравнением (1.30) для усредненной по всем ориентациям
полярных молекул энергии дипольного взаимодействия < Е0А > и
уравнением (1.58) для энергии лондоновского взаимодействия Е0Лт <Я0Д> и
Еол следуют соотношениям вида^(1У.ЗО), но их сумма, которая в
основном представляет собой теоретическую энергию вандерваальсова
взаимодействия двух молекул
2 2
/Е \,Е — _ 2 , H-i Р-2 __ 3 . aia2 . /1/2 HV ЗП
<*«> + *«- г — т -*- 7ГТ7Г' ( }
<«> + « з^г т * 7ГТ7Г
в тех случаях, когда оба слагаемых по порядку величины одинаковы,
не может быть выражена в виде двух сомножителей, зависящих от
свойств молекул компонентов.
4-503 97
Уравнение (IV.30) имеет форму, аналогичную правилу факторов
Иогансена для энтальпий образования Н-связей(см. гл. III).
Поскольку основной вклад в энергию межмолекулярных взаимодействий вносят
химические связи, возможно, что закономерности, подобные правилу
факторов, применимы и для некоторых других видов слабых
химических взаимодействий. Правило факторов указывает на некоторое
сходство близкодействующих слабых химических взаимодействий с даль-
нодействующими диполькыми и лондоковскими взаимодействиями
молекул.
§ 36. Атом — атом потенциалы
А. И. Китайгородский предложил метод расчета энергии решетки
молекулярных кристаллов с помощью «атом — атОхМ потенциалов».
Каждый атом, входящий в молекулу, рассматривается как некоторый
силовой центр. Энергия взаимодействия молекул равна сумме энергий
парных взаимодействий атомов / и /, принадлежащих разным
молекулам. Энергии взаимодействия атомов Е.;- зависят лишь от сорта атомов.
Они не зависят от того, в какую молекулу и в каком валентном
состоянии атомы входят. Для E.j могут быть приняты различные
аналитические выражения, например потенциал Леннарда—Джонса и др.
Параметры эмпирических соотношений подбираются так, чтобы, зная все
межъядерные расстояния в кристалле, можно было получить
правильное значение энергии решетки кристалла. Подробное описание этого
метода и примеры его применения приведены в монографии А. И.
Китайгородского «Молекулярные кристаллы» [59] и обзоре П. М.
Зоркого и М. А. Порай-Кошица [60]. Метод атом—атом потенциалов дает
возможность подобрать межатомные потенциалы на основе
экспериментальных данных для нескольких представителей какого-либо
класса органических веществ, а затем применять полученные кривые для
вычисления свойств всех остальных веществ этого класса. Так,
например, зная потенциалы взаимодействия атомов Си С, СиН, НиН,
можно рассчитывать энергию и ряд других свойств множества кристаллов
углеводородов.
Схема атом — атом потенциалов, как показал А. И.
Китайгородский, представляет интерес и при расчетах энергии молекул. Хотя метод
атом — атом потенциалов применяет эмпирические соотношения для
Еф в целом его нельзя считать необоснованным теоретически. Из
работ В. М. Татевского [13], как нам представляется, следует, что
метод атом — атом потенциалов применим к любым химическим
связям, слабым или сильным. Там же указаны и условия
непротиворечивого применения этого подхода.
Мы рассмотрели близко- и дальнодействующие силы порознь, но
природа межмолекулярных взаимодействий едина. Все они
обусловлены зарядами атомных ядер и электронов, из которых состоят
молекулы. Надо полагать, что со временем будут созданы эффективные
методы расчета межмолекулярных взаимодействий с помощью теоретичес-
98
ких соотношений, справедливых при любых расстояниях между двумя
или большим числом молекул. Первая попытка построения такой
теории принадлежит О. Синаноглу [4]. Но полученные им формулы в
сущности дают лишь указание на принципиальную возможность
решения проблемы.
Мы видели, что при малых расстояниях межлу молекулами
притяжение в основном вызвано химическими взаимодействиями, т. е.
такими, которые сопровождаются обобществлением электронов. Именно
такие взаимодействия, как будет показано далее, определяют
особенности строения большинства жидкостей. Поэтому жидкости можно
рассматривать как макроскопические молекулы, строение которых
постоянно варьирует, испытывая под влиянием теплового движения
небольшие отклонения от среднего. Отсюда следует, что понятие о малых
молекулах как о некоторых химически изолированных частицах не вполне
строгое даже для газов, еще менее строгое для жидкостей [13]. Оно
сохраняет свой смысл, если химические связи между малыми
молекулами на порядок слабее, чем химические связи атомов в молекулах. Но
оно теряет обычный смысл, если малые молекулы образуют друг с
другом связи, почти столь же прочные, что и внутримолекулярные.
Понятие об ионах в конденсированной фазе тоже имеет приближенный
характер. Оно оправдано лишь в той мере, в какой можно пренебречь
химическим взаимодействием между ионами и их окружением.
Если химическое взаимодействие между молекулами (или между
ионами) велико, то в качестве структурных единиц жидкости иногда
могут фигурировать атомные остатки, т. е. атомные ядра и прочно
связанные с ними электроны внутренних оболочек. Такие жидкости могут
быть расплавленными металлами; они могут быть сходны с жидкими
галогенидами щелочных металлов и их структура может напоминать
строение жидкого аргона с той лишь разницей, что энергия
взаимодействия между атомными остатками на два — три порядка больше, чем
энергия связи атомов аргона.
В этой части книги не рассматривался вопрос о межчастичных
взаимодействиях в жидких металлах. Металлическая связь представляет
собой взаимодействие большого числа атомных остовов с
коллективизированными электронами. Она существенным образом зависит от
строения металлов, в особенности от способа распределения ближайших
частиц относительно друг друга. Вопрос о металлической связи
логически более уместно излагать после характеристики строения жидких
систем. Поэтому металлическая связь будет рассмотрена в начале
третьей части этой книги (см. гл. VIII).
Литература
1. А. А. Соколов, И. М. Тернов. Квантовая механика и атомная
Физика. М., «Просвещение», 1970.
2. Д ж. Гиршфельдер, Ч. Кертисс, Р. Берд. Молекулярная
теория газов и жидкостей. М., ИЛ., 1961.
3. В. И. Веденеев , Л. В. Гурвич, В. Н. Кондратьев,
В- А. Медведев, Е. Л. Франкевич. Энергии разрыва химических
4* 99
связей. Потенциалы ионизации и сродство к электрону. Справочник. М., Изд-во
АН СССР, 1962.
4. Современная квантовая химия. Под ред. О. Синаноглу. Т. 2. М., «Мир»,
1968.
5. И. Б. Рабинович. Влияние изотопии на физико-химические
свойства жидкостей. М., «Наука», 1968.
6. О. А. О с и п о в, В. И. М и н к и н, А. Д. Г а р н о в с к и й.
Справочник по дипольным моментам. М., «Высшая школа», 1971.
7. М. Д. Г а л а н и н. Резонансный перенос энергии возбуждения в люми-
несцирующих растворах. Тр. ФИАН СССР. М., Изд-во АН СССР, 1960,12,3.
8. А. Н. Теренин. Фотоника молекул красителей и родственных
органических соединений. Л., «Наука», 1967.
9. М. И. Ш а х п а р о н о в. О резонансной миграции внутримолекулярных
колебаний, Ж. Ф. X. 39, 2370, 1965.
10. Г. Ф р е л и х. Теория диэлектриков. М., ИЛ, 1960.
И. Я. Ю. А х а д о в. Диэлектрические свойства чистых жидкостей. М.,
Изд-во стандартов, 1972.
12. М. I. F e i n b е г g, К. R uedenberg. J. Chem. Phys., 54, 1495,
1971.
13. В. М. Т а т е в с к и й. Классическая теория строения молекул и
квантовая механика. М., «Химия», 1973.
14. Ю. Я. Ф и а л к о в, А. Н. Житомирский, Ю. А. Т а р а-
с е н к о. Физическая химия неводных растворов. Л., «Химия», 1973.
15. Д. Пименте л, О. Мак- Клеллан. Водородная связь, М.,
«Мир», 1964.
16. А. Е. Л у ц к и й, М. Ф. Ш а л и м о в, Б. А. Т р о м з а. Теор. и
экспер. химия, 5, 690, 1969.
17. М. И. Ш а х п а р о н о в, П. С. К а с ы м х о д ж а е в, В. В.
Левин, М. И. Л у п и н а. О строении жидкой уксусной кислоты и кинетике
процессов перестройки ее ассоциатов. Сб. «Физика и физико-химия жидкостей», вып.
2, Изд-во МГУ, 1973, стр. 11.
18. М. И. Ш а х п а р о н о в . Введение в молекулярную теорию
растворов. Гостехиздат, 1956.
19. Л. М. С в е р д л о в, М. А. К о в н е р, Е. П. К р а й н о в.
Колебательные спектры многоатомных молекул. М., «Наука», 1970.
20. А. В. И о г а н с е н. Оптика и спектроскопия. Сб. статей. Л., «Наука»
1967, Т. III, стр. 228—231.
21. А. В. И о г а н с е н. Докл. АН СССР, 164, 610, 1965;
А. В. И о г а н с е н, Б. В. Рассадин. Журн. прикл. спектр., 6, 493,
801, 1967;
А. В. И о г а н с е н, Г. А. К у р к ч и, Б. В. Р а с с а д и н. Журн.
прикл. спектр., 11, 1054, 1969.
22. А. В. И о г а н с е н, Б. В. Рассадин. Журн. прикл. спектр., 11,
828, 1969; С. Е. Одиноко в, А. В. Иогансеч, А. К. Дзизенко.
Журн. прикл. спектр., 14, 418, 1971.
23. А. В. И о г а н с е н, М. Ш. Р о з е н б е р г. Докл. АН СССР, 197,
117, 1971.
24. А. В. И о г а н с е н. Теорет. и экспер. химия, 7, 302, 312, 1971.
25. В. М. Т а т е в с к и й , В. А. Б е н д е р с к и й, С. С. Яровой.
Методы расчета физико-химических свойств парафиновых углеводородов. М.,
Гостоптехиздат, 1960.
26. Н. Д. Соколов. Докл. АН СССР, 58, 611, 1947; Усп. физ. наук, 57,
205, 1955.
27. А. В. И о г а н с е н и Г. А. К у р к ч и. Опт. и спектр., 13, 480, 1962;
А. В. И о г а н с е н. Докл. АН СССР, 184, 1350, 1969.
28. М. И. Ш а х п а р о н о в, Б. Т. К а п и т к и н, В. В. Левин.
Ж. Ф. X., 46, 498, 1972.
29. А. В. И о г а н с е н, Э. В. Б р о у н, Г. Д. Л и т о в ч е н к о. Опт.
и спектр., 18, 38, 1965.
30. Т. A. Milne, F. Т. G г е е n e. J. Chem. Phys., 47, 4095, 1967.
31. Y. ТапакаД. Y о s h i n o. J. Chem. Phys. 50, 3087, 1969; 53, 2012,
100
1970; 57, 2964, 1972.
32. В. Г. Ф а с т о в с к и й, А. Е. Р о в и н с к и й, Ю. Р.
Петровский. Инертные газы, изд. 2-е, М., Атомиздат, 1972.
33. А. И. Китайгородский. Порядок и беспорядок в мире атомов.
М., «Наука», 1966.
34. Л. Э н д р ю с, Р. К и ф е р. Межмолекулярные комплексы в
органической химии. М., «Мир», 1967.
35. Н. A. Bent. Структурная химия донорно-акцепторных
взаимодействий. Chem. Rev., № 5, 587—648, 1968.
36. Г. Б. С е р г е е в, И. А. Л е е н с о н. Образование свободных радикалов
из молекулярных комплексов. Усп. химии, 41, 1566, 1972.
37. Г. И. Б и т т р и х. Межмолекулярное взаимодействие и методы
селективного разделения. Zs. Phys. Chem, DDR, № 6, 201, 1970. См. также С h,
D e h m e 1 t, M. F i n k e, H. J. В i t t r i с h. Z. phys. Chem. 255, 251, 1974;
K. Geier, H. J. В i t t г i с h. Z. phys. Chem. 255, 305, 1974.
38. M. Г. В о р о н к о в, М. В. Позднякова, Л. А. Ж а г а т а.
Х 40, 1425, 1970.
39. В. В. Е в д о к и м о в а, Л. Ф. Верещагин. ФТТ, 4, 1965, 1963.
40. А. В. С к р ы ш е в с к и й. Структурный анализ жидкостей. М.,
«Высшая школа», 1971.
41.0. Я- Самойлов. Структура водных растворов электролитов. Изд-во
АН СССР, 1957.
42. К. П. М и щ е н к о, Г. М. Полторацкий. Вопросы
термодинамики и строения водных и неводных растворов электролитов. Л., «Химия», 1968.
43. Н. А. И з.м а й л о в. Электрохимия растворов. Изд-во Харьковского
гос. ун-та, 1959.
44. Р. Р о б и н с о н, Р. Стоке. Растворы электролитов. М., ИЛ, 1963.
45. В. С. Шмидт. Экстракция аминами. М., Атомиздат, 1970.
46. Некоторые проблемы современной электрохимии. Под ред. Дж. Бокриса.
М., ИЛ, 1958.
47. Новые проблемы современной электрохимии. Под ред. Дж. Бокриса, М.,
ИЛ, 1962.
48. Современные аспекты электрохимии. Под ред. Дж. Бокриса и Б. Конуэя.
М., «Мир», 1967.
49. Современные проблемы электрохимии. Под ред. Дж. Бокриса и Б.
Конуэя. М., «Мир», 1971.
50. Вопросы физической химии растворов электролитов. Сб. статей под ред.
Г. И. Микулина. Л., «Химия», 1968.
51. Ф. К о т т о н, Д ж. У и л к и н с о н. Современная неорганическая
химия. Т. I—III, M., «Мир», 1969.
52. И. Б. Б е р с у к е р. Строение и свойства координационных соединений.
Л., «Химия», 1971.
53. У. X а р р и с о н. Теория межатомного взаимодействия в твердых телах.
УФН, 108, 285, 1972.
54. Физика простых жидкостей. Статическая теория. Под ред. Г. Темперли,
Дж. Роулинсона, Дж. Рашбрука. М., «Мир», 1971.
55. М. П. В у к а л о в и ч и И. И. Н о в и к о в. Уравнение состояния
реальных газов. М., Госэнергоиздат, 1948.
56. Э. М е й с о н, Т. С п е р л и н г. Вириальное уравнение состояния. М.,
«Мир», 1972. стр. 212.
57. Н. П. М а р к у з и н. 2-е вириальные коэффициенты органических
соединений и их смесей. Сб. «Химия и термодинамика растворов», вып. 2. Изд-во
ЛГУ, 1968.
58. Н. F г е у d a n k, M. R a t z s с h. Вторые вириальные коэффициенты
бинарных газовых смесей с учетом явлений ассоциации. Zs. Phys. Chem. DDR,
248, 83, 1971; 249, 33, 1972.
59. А. И. Китайгородский. Молекулярные кристаллы. М.,
«Наука», 1971.
60. П. М. 3 о р к и й, М. А. П о р а й - К о ш и ц. Новые представления
в кристаллохимии молекулярных структур. Сб. «Современные проблемы
физической химии». 1, 98, 1968.
ЧАСТЬ ВТОРАЯ
СТРОЕНИЕ ЖИДКИХ ФАЗ
ГЛАВА V
АССОЦИАТЫ И КОМПЛЕКСЫ
Строение жидкостей — это картина распределения ее частиц в
пространстве. Существуют три способа приближенного описания
строения однокомпонентных жидкостей и растворов. Один из них опирается
на представление обассоциатах и комплексах, другой связан с
понятием о функциях распределения частиц, в частности о корреляционных
функциях, третий — использует понятие о флуктуациях. Способы эти
взаимосвязаны и в известной мере дополняют друг друга. Мы
рассмотрим каждый из них и приведем примеры.
§ 37. Ассоциаты и комплексы
Каждую жидкую фазу, как уже отмечалось, можно считать
гигантской макромолекулой. Но в таких огромных макромолекулах
встречаются однотипные малые фрагменты, содержащие небольшое число
атомных ядер, взаимное расположение которых более или менее фиксировано.
Упорядоченные образования, возникающие в результате
химического взаимодействия между частицами, мы будем называть ассоциата-
ми и комплексами. Ассоциаты и комплексы, по существу, однотипные
образования и различаются только своим составом. Мы будем называть
ассоциатами такие упорядоченные образования которые состоят из
одинаковых частиц (мономерных звеньев). Таков ассоциат (Н2О)П, в
котором имеется п молекул воды, ассоциат метилового спирта (СН3ОН)т
и т. д. Комплексы состоят из разнородных частиц. Например, в
растворах вода — метиловый спирт кроме одиночных молекул и ассоциа-
тов присутствуют комплексы (H2O)V (CH3OH)m. Деление на ассоциаты
и комплексы удобно, но многие авторы им не пользуются, называя
комплексами или ассоциатами любые упомянутые образования, вне
зависимости от их состава.
Как уже говорилось в гл. I—IV, взаимодействия между
молекулами сопровождаются возникновением слабых или сильных химических
связей и, следовательно, появлением ассоциатов и комплексов. Были
описаны некоторые из ассоциатов аргона, иода, четыреххлористого
102
углерода, ацетонитрила, галогенцианидов, комплексов брома с
ацетоном, иода с триметиламином и ряда других.Убедительных доказательств
того, что существует хотя бы одна жидкость, в которой нет
ассоциатов и хотя бы один раствор, где нет комплексов, не имеется
[за исключением жидкого гелия (см. гл. XI)].
Для характеристики комплексов и ассоциатов необходимо знать их
состав, структуру, а также энергии химических связей между
частицами, образующими комплекс или ассоциат. Когда состав и структура
комплексов и ассоциатов установлены, требуется найти их
концентрации. Если концентрации всех комплексов, ассоциатов и мономерных
частиц в жидкой фазе известны, то в рамках понятий об ассоциатах
и комплексах строение жидкости выяснено. Определив концентрации
мономерных молекул, ассоциатов и комплексов и, если возможно,
отыскав их коэффициенты активности, вычисляют константы химического
равновесия для реакций ассоциации и комплексообразования,
протекающих в жидкости. Если эти константы найдены при ряде температур
и постоянстве давления или объема системы, то с помощью уравнений
изобары или изохоры химической реакции определяется изменение
энтальпии АН или внутренней энергии AU жидкости, связанное с
ассоциацией или комплексообразованием. А величины АН и AU
позволяют судить о тех изменениях энергии жидкости, которые происходят при
образовании или разрушении соответствующих химических связей,
в. частности водородных связей.
Бензол. Рассмотрим, например, жидкий бензол. Анализ измерений
релеевского рассеяния света [1] показал, что в жидком бензоле есть
ассоциаты. Они образуются при
взаимодействии групп С—Н одной
молекулы с я-орбиталями другой
молекулы бензола (л-ассоциаты). Согласно
[1] при комнатной температуре
ассоциировано не менее 70% молекул
жидкого бензола. Молекула
бензола—симметричный волчок. Через
центр-молекулы перпендикулярно плоскости, в
которой лежат атомные ядра
углерода, проходит ось симметрии Сб. Если
положение оси симметрии Св
определено, то ориентация молекулы
бензола в пространстве задана. Две
молекулы бензола могут
взаимодействовать с образованием я-ассоциата, в
котором, по-видимому, имеются две
С—Н...Сс связи (рис. 18). Энтальпия образования одной С—Н...С*
связи составляет около 4 кДж/моль. Угол между осями С6 молекул
бензола в димере близок к 90°. Если каждую ось С6 изобразить чертой, то
строение димера можно представить* с помощью следующей схемы:
Рис. 18. Строение л;-ассоциатов
жидкого бензола:
• — протоны двух С — Н ... Ск связей
между двумя молекулами бензола;
пунктиром указано положение оси симметрии
Св молекулы бензола, расположенной в
плоскости, ортогональной к плоскости
рисунка; точка в центре правой
молекулы бензола — проекция оси Св на
плоскость рисунка
103
Тримеры бензола могут с равной степенью вероятности иметь одну из
следующих структур:
Л
1
Тетрамеры (СвН6)4 образуют 16 структурных изомеров и т. д. В
принципе, в жидкой фазе изредка могут встречаться и значительно более
сложные ассоциаты, содержащие десятки и даже сотни молекул,
которые состоят из связанных друг с другом слоев. Примерно такую же
структуру имеют и кристаллы бензола. На рис. 19 показано
расположение молекул в элементарной ячейке кристаллической решетки
бензола [2]. Структура рыхлая, ближайшее расстояние между атомами
углерода соседних молекул равно 0,38 нм. Заштрихованные молекулы
составляют один слой, а незаштрихованные — другой. Плоскости, в
которых лежат оба слоя, отстоят друг от друга на половину высоты
элементарной ячейки.
Ассоциаты жидкого бензола, в сущности, представляют собой очень
малые фрагменты кристаллов бензола. При температуре кристаллизации
они могут стать зародышами кристаллов. Этот пример еще раз говорит
Рис. 19. Расположение
молекул бензола в элементарной
ячейке кристаллической
решетки бензола [2].
(Заштрихованные молекулы
составляют один слой, а незаштрихованные —
другой)
Рис. 20. Строение цепочечных ассо-
циатов в кристаллах уксусной
кислоты [15]
о том, что при изучении строения ассоциатов и комплексов полезно иметь
сведения о строении соответствующих твердых фаз. Другие примеры,
подтверждающие сказанное, уже были приведены в гл. III.
Уксусная кислота. Ассоциаты и комплексы в жидких фазах далеко
не всегда имеют ту же структуру, что и кристаллы, которые получаются
104
при замерзании этих жидкостей. Например, твердая уксусная кислота
состоит из цепочечных полимерных ассоциатов, изображенных на рис.20.
В кристаллах уксусной кислоты нет кольцевых димеров, описанных
в гл. III. При плавлении уксусной кислоты большая часть цепочечных
ассоциатов разрушается с образованием кольцевых димеров. Пары
уксусной кислоты практически не содержат цепочечных ассоциатов. Они
почти полностью состоят из кольцевых димеров.
В жидкой фазе могут быть любые ассоциаты и комплексы, которые
способны возникать при химическом взаимодействии молекул.
Растворы бензола. Структура растворов бензола сложнее, чем
строение чистого бензола. Кроме ассоциатов бензола в растворах бензола
присутствуют его комплексы с молекулами иных компонентов и
ассоциаты молекул остальных компонентов. В качестве примера приведем
растворы бензол—хлороформ.
В табл. 8 представлены идеализированные схемы простейших
ассоциатов и комплексов, которые обнаруживаются в растворах
Таблица 8
Схемы простейших ассоциатов и комплексов
в растворах бензол — хлороформ (р-тип молекул)
1
2
J
и
5
Схема
_L
_~—
_J
Н
н
6
7
8
9
10
Схема
н-
—►
h-
-+-
1*
//
12
13
и
15
Схема
—~
Лпд'
_L_
к
бензол — хлороформ с помощью релеевского рассеяния света,
радиоспектроскопических измерений, инфракрасной и акустической
спектроскопии [3]. Чертой изображены молекулы бензола. Положение черты
соответствует ориентации оси симметрии С6 этих молекул. Стрелкой
указаны молекулы хлороформа. Положение стрелки соответствует
ориентации дипольного момента (л молекул СНС13 (дипольный момент
направлен вдоль оси симметрии С 3 от Н к С). По мере разбавления
бензола каким-либо другим растворителем концентрации сложных
ассоциатов (С6Н6)П уменьшаются. В разбавленных растворах обычно
остаются в основном мономерные молекулы С6Нб с примесью димеров
(СвНв)2.
Более сложные ассоциаты если и есть, то в столь малых количествах,
что ими можно пренебречь. Это наблюдается, когда ассоциаты
неустойчивы и легко распадаются. Молекулы стабильных ассоциатов,
например многих из полимеров, могут существовать в разбавленных
растворах сколь угодно долго.
105
§ 38. Устойчивость ассоциатов и комплексов
Следует отличать вопрос о стабильности одной молекулы ассо-
циата или комплекса (в вакууме) от вопроса о стабильности таких
молекул ввеществе, например в растворе [4]. Оба вопроса различны
как по своему содержанию, так и по способу решения. Ответ на первый
вопрос определяется видом потенциальной энергии, как функции
расстояний между атомными ядрами одной изолированной молекулы ас-
социата в вакууме. Если потенциальная функция при некоторых
значениях координат атомных ядер проходит через минимум, то
молекула соответствующего ассоциата или комплекса сама по себе
устойчива. Вопрос о существовании минимума потенциальной энергии
одной изолированной молекулы ассоциата или комплекса мог бы быть
решен, например, с помощью квантовомеханических расчетов. Но
решение этой задачи, как правило, пока невыполнимо.
Второй вопрос связан с поведением макроскопической системы—
раствора — при тех физических условиях, в которых раствор
пребывает. Точнее, в данном случае речь идет о поведении соответствующего
термодинамического потенциала. Когда раствор находится при
заданных значениях температуры Т (в термостате) и давления Р, то
устойчивость ассоциатов и комплексов в растворе определяется
свойствами свободной энтальпии G = Н — TS как функции Г, Р и концентраций
мономерных молекул, ассоциатов и комплексов хъ ..., хк. В состоянии
термодинамического равновесия свободная энтальпия минимальна при
некоторых, вполне определенных значениях концентраций хъ... хь ...,
хк (здесь хг — мольная доля молекул /-того сорта). Величины xlt...9
хи ..., хк в принципе можно было бы определить с помощью к условий.
Каждое из этих условий имеет вид
dG(T, Р, *!, ... , xi9 ... , ^)^Q у1)
Жидкая фаза не содержала бы ассоциатов или комплексов только в
том случае, если свободная энтальпия имела бы минимум при хг = 1,
х2, ..., хк =0. В этом случае при заданных внешних условиях
(определенных Т и Р) молекулы, обозначенные индексом «1», были бы в
жидкой фазе вполне устойчивы. Но в действительности жидкости всегда
содержат целый ряд различных химических частиц, ассоциатов,
комплексов и т. д., получающихся в ходе реакций с участием молекул
сорта «1». Поэтому термодинамически устойчивой жидкой фазой всегда
является раствор ассоциатов, состав которого определяется
значениями х.у обращающими G в минимум. Расчетфункцииб(.%,..., х.,...,хк) —
задача статистической механики. Трудности, связанные с решением
этой задачи, для жидких систем пока еще не преодолены.
Однозначной взаимозависимости между строением ассоциатов и
комплексов и их устойчивостью нет. Но обычно относительно
стабильнее в жидкой фазе при заданных ТиРте ассоциаты и
комплексы,энтальпия образования которых в данных условиях больше. Поясним
это с помощью следующих качественных аргументов. Пусть раствор,
в котором имеются молекулы А и В, находится при температуре Т и
106
давлении Р. С появлением химической связи между А и В число
внешних степеней свободы частиц А и В уменьшается. По законам
статистической механики это ведет к уменьшению энтропии S раствора, так что
Д5 <0. Энтальпия такого раствора обычно тоже падает А Я <0.
Если модуль |А Н\ больше модуля |А 5|, то свободная энтальпия
раствора снижается на величину AG =АН—TAS. Ясно, что большие
абсолютные величины А Н при прочих равных условиях
благоприятствуют стабильности комплекса АВ.
§ 39. Среднее время жизни ассоциатов и комплексов
Понятие о среднем времени жизни молекулы ассоциата или
комплекса имеет смысл для неизолированных молекул. Изолированная
стабильная молекула, если не протекают процессы радиоактивного
распада, должна была бы существовать сколь угодно долго. Среднее время
жизни ассоциата или комплекса i в жидкой фазе при постоянных
Т и Р зависит от свободной энтальпии активации процесса распада
молекул этого ассоциата или комплекса
т ~ е ь , (V.2)
где AG* — изменение свободной энтальпии жидкости при переходе
одной молекулы ассоциата или комплекса из активированного в
равновесное состояние. A G^ =Л Нф— TAS*, где А Н*—энтальпия и A S^—
энтропия активации реакции, ведущей к разрушению ассоциата или
комплекса. Средние времена жизни различных ассоциатов и
комплексов могут различаться на много порядков. Например, среднее время
жизни молекул окиси хлора СЦО, растворенной в четыреххлористом
углероде, по порядку величины равно 104 с [5], а среднее время жизни
ассоциатов (СС14)п в этом растворе составляет всего лишь с^10~12с.
Если свободная энтальпия активации реакции велика, то реакция
будет протекать медленно, хотя бы она и вела к образованию
устойчивого комплекса или ассоциата. Такова, например, реакция соединения
третичных алкиламинов с галоидными алкилами (Меншуткин, 1887).
§ 40. О методах изучения ассоциатов и комплексов
Универсальных способов определения состава, структуры и^
концентраций ассоциатов и комплексов нет. Для выяснения структуры
какой-либо жидкой фазы обычно требуются нестандартные
исследования. Если среднее время жизни молекул ассоциатов и комплексов
велико, то для их изучения применяются методы аналитической химии,
описанные, например, в монографии [6]. Когда речь идет об ассоциа-
тах или комплексах растворенного вещества в разбавленных
растворах, то задача в известной мере облегчается. Как уже было отмечено,
уменьшение концентрации растворенного вещества часто
сопровождается разрушением сложных структур. Сохраняются, в основном,
лишь более простые молекулы ассоциатов и комплексов. В этих усло-
107
виях эффективность инфракрасной спектроскопии, калориметрии,
криоскопии и ряда других методов [5—7] возрастает. Успешно
исследуются не только стабильные, но и неустойчивые простые ассоциаты и
комплексы с малыми средними временами жизни молекул т >- 10~4с.
Иначе обстоит дело, когда требуется выяснить строение быстро
разрушающихся ассоциатов и комплексов с участием молекул
компонента, концентрация которого в растворе велика. В пределе это может
быть однокомпонентная жидкость. В таких случаях картина
ассоциации и комплексообразования обычно усложняется. Анализ ее лучше
выполнять несколькими независимыми методами, дополняющими и
контролирующими друг друга. Когда среднее время жизни ассоциатов
или комплексов в концентрированных растворах меньше 10~3 — 10~4с,
применение ИК-спектроскопии или ЯМР обычно указывает лишь на
существование явлений ассоциации и комплексообразования.
Обнаруживаются изменения химических сдвигов, смещения в ИК-
спектре характеристических полос поглощения, аномальное
изменение их интенсивности, появление новых полос. Эти факты порой дают
косвенные основания для гипотез о структуре жидкой фазы. Но
теории, однозначно связывающей инфракрасные спектры или спектры
ЯМР со строением жидкостей, нет, поэтому гипотезы, основанные на
данных об этих спектрах для концентрированных растворов
нуждаются в проверке. Например, ИК-спектры жидкой уксусной кислоты
исследуются около 40 лет. Спектры показывают, что в жидкой
уксусной кислоте имеются водородные связи С—Н...О; но они не дают
сведений о строении ассоциатов (СН3СООН)П и их концентрациях. Одни
из авторов утверждают, что уксусная кислота состоит из кольцевых
димеров, другие находят цепочечные образования, третьи отмечают,
что спектр связей О—Н...0 цепочечных и кольцевых ассоциатов
одинаков и поэтому с помощью ИК-спектров эти структуры различать
невозможно. Другой пример — жидкий диметилформамид. Спектры
ЯМР дают основание считать, что в жидком диметилформамиде и его
растворах присутствуют ассоциаты (CH3)2NCHO. Было высказано
предположение, что молекулы диметилформамида в жидкой фазе
образуют кольцевые димеры. Но, как вскоре выяснилось, наблюдавшиеся
особенности спектров ЯМР главным образом обусловлены не
ассоциацией, а влиянием реактивного поля. Оказалось, что ассоциаты
(CH3NCHO) имеют в основном цепочечную структуру.
При изучении структуры индивидуальных жидкостей и
концентрированных растворов существенную пользу могут принести
рентгенография, радиоспектроскопические измерения и релеевское рассеяние
света. Преимущество этих методов состоит в том, что исследователи
располагают теорией, устанавливающей вполне определенную связь
между результатами измерений и строением жидких фаз. Вопросы
рентгенографии жидкостей обсуждаются в следующей главе. Здесь мы
дадим некоторое представление о возможностях анализа данных,
получаемых методами диэлектрической радиоспектроскопии и релеевского
рассеяния света.
Радиоспектроскопические измерения во многих случаях позволяют
вычислить среднее статистическое значение квадрата дипольного мо-
108
мента молекул жидкой фазы <ja2>. Разумеется, речь идет о таких
жидких фазах, в которых есть полярные молекулы. Теория,
позволяющая вычислить <Ср-2>, развита Онзагером. Хорошее изложение этой
теории имеется в книге Фрелиха [8]; ряд вопросов выяснен автором
[9]. Зная <н-2>, можно подобрать такие физически обоснованные
пространственные модели ассоциатов; которые удовлетворяли бы
уравнению
Np -2 (р= 1,2,3, ...); (V.3)
N
для чистой жидкости или несколько более сложным уравнениям для
растворов. Здесь Np — число молекул ассоциата вида р в единице
объема; N — общее число молекул жидкости в единице объема в
пересчете на мономер (т. е., как если бы ассоциации не было); \ьр— диполь-
ный момент ассоциата вида р.
Физически обоснованная структура ассоциатов и комплексов не
должна противоречить выводам о межмолекулярном взаимодействии,
если они надежно установлены спектроскопическими,
рентгенографическими, ЯМР или какими-либо другими методами.
Напомним, что ассоциаты и комплексы могут различаться не только
составом, но и строением. Поэтому ассоциаты, молекулы которых
содержат одинаковое число мономерных звеньев, но различаются
расположением этих звеньев, обозначаются различными номерами индекса
р. Когда структура молекул ассоциата или комплекса задана и диполь-
ные моменты мономерных молекул известны, то, пользуясь
правилами суммирования векторов, нетрудно вычислить р2р. При этом
следует учесть влияние внутреннего вращения полярных молекул в ассо-
циатах. Подробности таких расчетов описаны, например, в
монографии [10] и работе [И].
Метод, основанный на анализе измерений релеевского рассеяния
света, разработан автором [9,12]. Поляризуемость молекулы (см. гл. I)
вдали от полосы поглощения света представляет собой симметричный
тензор второго ранга aif. Один из инвариантов этого тензора —
анизотропия поляризуемости молекул *[**•
' л L\ А/С У У ) \ Ал ZZi / \ У Y ZZi / J * *
где аХх , aw и dzz — главные значения тензора поляризуемости
молекулы. Эллипсоид значений тензора поляризуемости** называют эллип-
* Иногда для обозначения анизотропии тензора поляризуемости
применяется символ у-
** Дипольный момент Д jx, индуцированный внешним полем 8, для слабых
полей связан с 8 линейными соотношениями:
Тензор ujj симметричен, т. е. а^ = а^, если поглощением энергии
электромагнитного поля при распространении света в жидкости можно пренебречь. Если
109
соидом поляризуемости (рис. 21). Величины &хх, &yy и o^z, а
следовательно, и if2 зависят от структуры молекул. Так, если аХх = <*yy =
= azzy то эллипсоид поляризуемости переходит в шар, анизотропия
поляризуемости равна нулю, ?2 = 0. Пользуясь результатами
измерений релеевского рассеяния света, можно вычислить среднюю
статистическую анизотропию поляризуемости молекул жидкости <72>>.
Затем подбирают такие физически обоснованные пространственные
модели ассоциатов, которые удовлетворяют уравнению
2
II
N
(V.5)
где Npu N имеют тот же смысл, что и в уравнении (V. 3); ^ —
анизотропия поляризуемости молекул ассоциата вида р (р = 1, 2, 3, ...).
Для растворов <л2> вычисляется
по несколько более сложной формуле.
Если полуоси эллипсоидов
поляризуемости мономерных молекул
известны и структура молекул ассоциата или
комплекса вида р задана, то вычис-
—X лить fp нетрудно.
Анализ строения жидкостей с
помощью уравнений (V. 3) и (V. 5)
опирается, в сущности, на тот же подход
к решению проблемы, что и
рентгенография кристаллов или
электронография молекул. Во всех этих методах
подбираются такие характеристики
структуры (тип и параметры кристаллической решетки, валентные
углы и межъядерные расстояния в молекулах, углы, определяющие
взаимную ориентацию молекул в ассоциатах и комплексах), которые
позволяют наиболее полно и корректно описать результаты
систематических экспериментальных исследований.
Методы, основанные на определении и анализе средних
статистических значений <р2> и <Ci2> и отыскании дипольных моментов
|Ар и анизотропии поляризуемости ?2 ассоциатов и комплексов,
похожи друг на друга еще и потому, что и \хр и ^ сильно зависят от
симметрии молекул ассоциатов. Эти методы дополняют друг друга и
осуществляют взаимный контроль.
Рис. 21. Эллипсоид значений
тензора поляризуемости || а^ у
вектор 8 имеет единичную длину и вращается вокруг начала координат, то конец,
вектора А ^и описывает поверхность, уравнение которой, приведенное к главным;
осям, имеет вид
= 1.
аХХ
Эта поверхность есть эллипсоид с полуосями сг
ПО
уу
и a
zz ,
Допустим, например, что в жидкости, состоящей из полярных
молекул, имеются ассоциаты вида р, содержащие п мономерных молекул.
Пусть структура этих /г-меров вида р такова, что выполняется
следующее соотношение:
p£ = ntf. (V.6)
где [J<|—дипольный момент мономера. Тогда соответствующий член
суммы в уравнении (V. 3) имеет вид:
Nо 9 flNn о
Ho nNp есть число мономерных молекул жидкости, входящих в состав
ассоциатов вида р, поэтому молекулы я-мерного ассоциата р,
удовлетворяющего условию (V. 7), вносят в <{а21> такой же вклад, какой
давали бы nNp свободных мономерных молекул. Таковы, например,
димеры диметилформамида. Дипольные моменты у мономерных звеньев
в молекулах димеров расположены под углом 90° друг к другу. В этом
случае расчет показывает, что
,,2 п.,2
Уравнение (V.3), а следовательно, и радиоспектроскопические методы,
позволяющие вычислить <С^2>, «не замечают» присутствия в
жидкости ассоциатов, удовлетворяющих условию (V. 6).
Точно так же релеевское рассеяние света «не обнаруживает»
присутствия я-мерных ассоциатов вида р, строение которых таково, что
7р = "7?, (V.8)
где 7? — анизотропия поляризуемости молекул мономера. Так,
например, анизотропия поляризуемости тех из тримеров бензола,
которые имеют одну из следующих структур (индекс р = 3):
равна 3^2 . Но ч2р других тримеров бензола (индекс р =4),
структура которых может быть представлена с помощью следующих схем:
-к
не удовлетворяют уравнению (V. 8). Их анизотропия поляризуемости
равна (1 — —-Л^ , поэтому они вносят вклад в <12>,
отличающийся от того, что дают неассоциированные молекулы СбНб [3].
Как правило, ассоциаты, дипольный момент которых следует
условию (V. 6), имеют анизотропию поляризуемости, не подчиняющуюся
ill
условию (V. 8), и, наоборот, ассоциаты, структура которых такова,
что уравнение (V. 8) выполняется, не подчиняются уравнению (V. 6).
Для комплексов условия, аналогичные (V. 6) и (V. 8), имеют более
сложный вид и выполняются реже, так как молекулы различных
компонентов имеют неодинаковые дипольные моменты и поляризуемости.
Таким образом, с помощью релеевского рассеяния света можно
обнаружить такие ассоциаты и комплексы, которые не фиксируются
радиоспектроскопическими измерениями. Последние в свою очередь
обогащают информацию о строении жидких фаз и дают возможность
повысить степень надежности выводов, полученных с помощью
релеевского рассеяния света.
Сказанное относится и к другим методам.
Важные, хотя и косвенные сведения о строении жидких веществ,
дают методы, позволяющие судить о кинетике и механизме быстрых
физико-химических процессов, протекающих в жидкостях. Некоторые
из таких методов охарактеризованы в кн. Е. Н. Еремина [5]. В
последнее время больших успехов достигли сверхвысокочастотная
диэлектрическая радиоспектроскопия и акустическая спектроскопия. С помощью
акустических методов стали доступны исследованию процессы
перестройки ассоциатов и комплексов, протекающие за периоды времени
до 10"10с включительно. Методы диэлектрической радиоспектроскопии
позволяют наблюдать даже процессы, протекающие за 10 "13 с.
В заключение отметим, что встречающееся иногда в литературе
качественное подразделение свойств жидкостей на
«структурно-чувствительные» и «не чувствующие» структуру относительно. Свойства
жидкостей в конечном счете обычно сложным образом связаны с их
структурой. Для одних жидких систем такая связь бывает заметна и
соответствующее свойство «чувствует» структуру. Для других жидких систем
тоже самое свойство может оказаться «не чувствующим структуру».
Наиболее интересны для изучения структуры те свойства, для которых
имеется хорошо обоснованная теория, позволяющая производить
расчеты величин, или функций, характеризующих структуру жидких фаз»
ГЛАВА VI
ФУНКЦИИ РАСПРЕДЕЛЕНИЯ ПОЛОЖЕНИЙ ЧАСТИЦ
В ЖИДКОСТЯХ
§ 41. Распределение молекул жидкости
Рассмотрим сначала простую жидкость, например жидкий аргон*.
Если пренебречь слабым химическим взаимодействием между атомами
* «Простыми жидкостями» называют жидкие простые вещества. Иногда
простыми жидкостями именуют неассоциированные жидкости, где нет химически
связанных друг с другом атомов, образующих молекулы различной формы и
размера. Факты, приведенные в гл. I, показывают, что таких жидкостей в природе не
существует. Это абстрактная теоретическая модель, лишенная важной черты жид -
кого состояния вещества — взаимодействия между молекулами (или атомами)»
сопровождающегося обобществлением электронов.
112
аргона, то можно принять, что жидкий аргон состоит из атомов. В этом
приближении жидкий аргон относят к группе одноатомных простых
жидкостей. Пусть V обозначает объем некоторого количества жидкого
аргона. Выделим, мысленно, в пространстве, занятом жидким аргоном,
элемент объема dV = dXdYdZ, настолько малый, что в нем может
поместиться лишь один атом аргона (рис. 22). Элемент объема не должен
находиться около поверхности
жидкости, в остальном его место выбирается
внутри V произвольно. Атомы жидкости
в ходе теплового движения могут
переходить от одного элемента объема к
другому, т. е. будет происходить процесс
самодиффузии. При этом слабые
химические связи между ними будут то
возникать, то разрушаться. В ходе
самодиффузии стечением времени каждый из
атомов побывает в любой точке объема V,
занятого жидкостью. Найдем
вероятность dW того, что некоторый,
произвольно выбранный атом жидкого аргона
располагается в элементе объема dV, т. е.
его координаты лежат в интервалах (X,
X +dx); (У, Y +dY)\ (Z, Z +dZ).
В соответствии с теорией вероятности
dW определяется соотношением
dv,
Рис. 22. К расчету распреде
ления молекул в жидкой фазе-
= p (Xy Y, Z)dXdYdZ1
(VI. 1)
где р(Х, У, Z) — плотность вероятности указанного события, которая
определяется свойствами системы, их зависимостью координат X, Y, Z.
В отсутствие внешнего поля жипкий аргон изотропен, т. е. его
свойства* не зависят от координат, поэтому плотность вероятности
р(Х9 У, Z) = const = В.
Любой из атомов аргона, в том числе и тот, который нами выбран,
в соответствии с условиями задачи находится в объеме V. Это
достоверное событие, его вероятность равна единице:
=l= JJJ P(X,Y,Z)dXdYdZ =
Следовательно, если учесть, что dXdYdZ =dV>
(VI.2)
(VI.3)
Итак, плотность вероятности распределения атомов жидкого аргона в
объеме V равна F"1.
* Речь идет о величинах, усредненных по времени. Например, плотность
аргона в малом элементе объема в какой-либо момент времени может отличаться от
средней плотности. (Подробнее см. гл. VII.)
ИЗ
Допустим, что в жидкой фазе имеются сравнительно обособленные
структурные фрагменты, молекулы, содержащие несколько атомных
ядер и тесно связанных с ними электронов. Такие жидкости иногда
называют многоатомными. Положение молекулы, если она не имеет
сферической симметрии, в общем случае можно определить, задавая
координаты центра тяжести молекулы (X, Y, Z) и эйлеровы углы,
определяющие ориентацию молекулы в пространстве (8-, ср, <]>). Для
осесимметричных молекул достаточно задать углы & и ср, определяющие
положение оси симметрии. Напомним, что 0 < Ь <: тс; 0 <ср < 2тс;
О <: ф < 2т:. Повторяя те же рассуждения, что л для жидкого аргона,
мы найдем для несимметричных молекул плотности вероятности
распределения молекул р(Х, Y, Z, &, ср, ф) = (8т:2V)'1; для
осесимметричных р(Х, Y, Z, ft, ф) = (AnV)'1. Этот результат справедлив для
молекул любой изотропной жидкости.
Радиальная функция распределения. Вернемся к образцу
жидкого аргона в объеме V. Рассмотрим теперь два малых элемента объема
dViYL dV2, центры которых удалены друг от друга на расстояние R
(см. рис. 22). dVt и dV2 по порядку величины равны d%, где d0 —
диаметр атомов жидкости. Выберем два каких-либо атома аргона и
обозначим их индексами 1 и 2. Вероятность dWx того, что атом 1
находится в элементе объема dVv равна
dW1 = dV1/V. (VI. 4)
Вероятность того,' что атом 2 находится в элементе объема dV2i равна
dW2 = dV2/V. (VI. 5)
Найдем вероятность dWi2 такого события, когда атом 1 находится в
элементе объема dVly а атом 2 располагается в dV2- Если движения
атомов не зависят друг от друга, то вероятность dWi2 равна
произведению вероятностей dW{ и dW2:
Опыт показывает, что уравнение (VI. 6) справедливо, если расстояние
R между атомами изотропной жидкости велико. Лишь в тех случаях,
когда состояние жидкости соответствует окрестности критической точки,
уравнение ( VI. 6) не выполняется при довольно больших R ~ 102 нм
(подробнее см. гл. VII). Но если расстояние R по порядку величины
сравнимо с диаметром атомов, то из опыта следует, что перемещения
атомов 1 и 2 нельзя считать независимыми друг от друга. В этом
случае вероятность нахождения атома 1 в dVx и атома 2 в dV2 не равна
произведению dWi и dW2. Это обстоятельство можно учесть, введя
некоторый множитель g, зависящий от R:
Когда R ->■ оо, g(R) ~v 1 и уравнение (VI. 7) переходит в уравнение
(VI. 6). Если R <Zd0, т. е. элементы объема dV^ и dV2 пересекаются,
то dWi2 равно нулю, так как такое событие невероятно, поскольку
114
молекулы непроницаемы друг для друга. Следовательно, при R
g(R) = о.
Функция g(R) учитывает корреляцию, т. е взаимосвязь между
атомами жидкости. Так как жидкость изотропна, то она зависит
только от абсолютной величины R. Функция g(R) не зависит от
направления радиуса-вектора /?, величина которого отсчитывается от центра
молекулы У, поэтому ее называют радиальной функцией распределения
атомов.
Поскольку функция g(R) связана с понятием о вероятности dWi2y
она является усредненной статистической характеристикой строения
жидкости. Радиальная функция распределения g(R) позволяет
находить относительную частоту появления тех или иных межатомных
расстояний в жидкости при заданных средней плотности р и температуре Т.
Следовательно, радиальная функция распределения зависит от
плотности жидкости и ее температуры, как от параметров, g=g(R; p, T).
Радиальная функция распределения атомов, по существу,
представляет собой своеобразную термодинамическую характеристику строения
жидкости.
В теоретических расчетах свойств одноатомных жидкостей часто
пользуются функцией h(R) = g(R) — 1, называемой парной
корреляционной функцией. При увеличении R функция h(R) стремится к нулю.
§ 42. Расчет функции g{R)
Радиальная функция распределения атомов простых жидкостей
может быть найдена по данным о рассеянии рентгеновских лучей,
нейтронов или электронов [13—17]. Рентгеновские лучи рассеиваются
главным образом электронами атомов; нейтроны — преимущественно
атомными ядрами, за исключением магнитных веществ, где рассеяние элект-
троыами существенно. Электроны рассеиваются всеми частицами атома
в целом. Различие в физической картине рассеяния ведет к некоторым
отличиям в содержании получаемой информации [16]. Тем не менее
методы обработки и анализа результатов эксперимента имеют много
общего. В качестве примера дадим представление о расчетах
радиальной функции распределения на основании сведений о рассеянии
рентгеновских лучей.
Пусть параллельный пучок монохроматических рентгеновских
лучей, длина волны которых X, падает на слой одноатомной жидкости.
Рентгеновские лучи рассеиваются электронами атомов по всем
возможным направлениям. Рассеянное излучение подразделяется на
когерентное и некогерентное. Когерентно рассеянное рентгеновское
излучение имеет ту же длину волны, что и лучи, падающие на слой
жидкости. Когерентно рассеянные лучи, по определению, имеют постоянные
фазовые соотношения, зависящие от положений рассеивающих частиц
жидкости, поэтому они интерферируют.
Интенсивность рассеянного излучения измеряется на расстояниях,
во много раз превышающих длину волны рассеянного света, так что
рассеянные рентгеновские волны можно считать плоскими. Опыт
показывает, что благодаря интерференции на фотопластинке, регист-
115
рирующей излучение, обычно наблюдается система концентрических
колец—областей повышенной и пониженной интенсивности излучения.
Следовательно, интенсивность / когерентной части рассеянного
излучения не монотонно зависит от угла рассеяния 6. Некогерентное
рассеяние образует на фотопластинке сплошной фон, интенсивность
которого увеличивается с ростом 6. После учета поправок на этот фон,
на поглощение рассеянного излучения и некоторых других находят
зависимость интенсивности / от угла рассеяния 9 или от к= Я5Ш .
Отношение интенсивности / когерентного рассеяния рентгеновских
лучей от N атомов в жидкости к интенсивности рассеяния такого же
пучка рентгеновских лучей от N изолированных друг от друга атомов
(т. е. атомов идеального газа) называют структурным фактором S(k):
I (k)
S(^TFW" <VI8>
где f2(k) — интенсивность рассеяния рентгеновских лучей одним
изолированным атомом. Разумеется, речь идет о тех атомах, из которых
состоит данная простая жидкость. /2(&) можно рассчитать теоретически
или определить экспериментально по рассеянию рентгеновских лучей
в парах.
Структурный фактор S(k) связан с радиальной функцией
распределения атомов жидкости уравнением
Для квантовых жидкостей (см. гл. XI) при температурах Т ->0 К
структурный фактор S(k) выражается более простым соотношением:
), (VI. 10)
где E(k) — энергия фотона с волновым вектором k\ h — постоянная
Планка; m — масса одного атома жидкости. При малых значениях
модуля волнового вектора k> Е = hck> где с — скорость звука (см. гл.,
XI). Для неквантовых жидкостей при k -»■0, S(0) =р&Б71^> где &Б —
постоянная Больцмана; ^—изотермическая сжимаемость, р—плотность.
Применяя к (VI. 9) преобразование Фурье, получают
*. (VI. 11)
Это уравнение позволяет вычислить g(R), если структурный фактор
S(k) известен для достаточно широкого интервала значений k.
Расчеты по уравнению (VI. 11) не дают вполне точных значений
радиальной функции распределения, в особенности для малых R,
сравнимых с диаметром атомов d0. Объясняется это, в частности, тем, что
при расчетах значений g(R) для малых R важную роль играет точность
116
знания функции I(k) при больших k, т. е. больших углах рассеяния.
Но с ростом 0 интенсивность когерентно рассеянного излучения
быстро убывает; ее трудно отличить от фона и точно измерить. Некоторые
затруднения связаны с тем, что k не может превышать 2тс/Х, в то время
как верхний предел интегрирования в (VI. 11) бесконечен. При
выводе уравнения (VI. 11) пренебрегают вкладом в / (k) многократно
рассеянного излучения. Кроме того, предполагается, что каждый из
атомов жидкости рассеивает рентгеновские лучи независимо от своих
соседей. Такое предположение было бы справедливо, если бы химические
взаимодействия между соседними атомами, сопровождающиеся
обобществлением электронов, отсутствовали. Но, как было показано в
гл. III, даже между атомами инертных
газов действуют слабые силы химического
происхождения.
На рис. 23 изображена радиальная
функция распределения атомов жидкого аргона.
По оси абсцисс отложены отношения
межатомного расстояния R к диаметру атомов
d0. Радиальная функция распределения
непрерывна. В жидкости можно встретить
любые расстояния между атомами при
условии, что R > d0. Этим строение
жидкости отличается от строения кристалла. В
идеальном кристалле, атомы которого
находятся только в узлах решетки,
возможны лишь некоторые, избранные
межатомные расстояния, зависящие от параметров
решетки.
Из рис. 23 видно, что g(R) проходит через ряд максимумов и
минимумов, совершая затухающие колебания около значения g(R) = 1.
При R} равном ~4d0, отклонения радиальной функции распределения
от 1 обычно находятся в пределах ошибок измерений. Иначе говоря,
корреляция атомов жидкости обычно не распространяется далее, чем
на расстояния порядка 4—5 диаметров атомов. Отметим, что это как
раз такие расстояния, на которых нельзя пренебречь перекрыванием
волновых функций атомов при рассмотрении лондоновских
межмолекулярных взаимодействий. Максимумы g(R) соответствуют тем
значениям межатомных расстояний, которые относительно более
вероятны, т. е. чаще встречаются. Минимумы g(R) указывают на те
межатомные расстояния, которые встречаются реже, чем соседние с ними.
§ 43. Кривые атомного распределения.
Среднее координационное число
Часто вместо радиальной функции распределения атомов g(R)
пользуются радиальной функцией распределения плотности p(R). Связь
между p(R) и g(R) имеет вид:
0 12 3
R/d0
Рис. 23. Радиальная
функция распределения атомов
жидкого аргона
Р(Я) =—*(*).
(VI. 12)
117
где NIV есть среднее число атомов в единице объема жидкости, т. е.
средняя плотность жидкости, деленная на массу одного атома. Таким
образом, p(R) пропорциональна средней плотности жидкости. р(Щ
обозначает среднюю плотность на расстоянии R от произвольного
выбранного атома одноатомной жидкости.
Выберем в жидкой фазе за начало отсчета положение какого-либо
из атомов, удаленного от поверхности жидкости. Среднее число атомов,
Рис. 24. Кривые атомного
распределения аргона при различных
давлениях и температурах,
соответствующих равновесию жидкость —
насыщенный пар для точек U 2, 3, 4, 5 и
6 на рис.25 [17]
100 120 ПО 160 V'К
Рис. 25. Р—Т-диаграмма аргона:
цифры 1 — 6 — состояния аргона, которым
соответствуют кривые атомного
распределения на рис. 24
находящихся в сферическом слое радиусом R и толщиной dR,
окружающем выбранный нами центральный атом, равно inR2p(R)dR. Если
по оси ординат откладывать величины 4гс7?2р(7?), а по оси абсцисс
значения R, то получают так называемые кривые атомного
распределения.
На рис. 24 изображены кривые атомного распределения аргона при
различных давлениях и температурах, соответствующих равновесиям
жидкость — насыщенный пар, для состояний, определяемых точками
/, 2, Зу 4, 5 и 6 на рис. 25 [18]. Кривая 1 получена для жидкого аргона
вблизи тройной точки [температура и давление тройной точки Т =
118
= 83,99 К, Р= 6,84 • 104 Па (0,684 атм). Кривая 6 получена для паров
аргона, близких к насыщению. Тонкими линиями на рис. 24
изображены кривые атомного распределения, если бы атомы аргона
располагались неупорядоченно, хаотически, как в идеальном газе. Это параболы,
представляющие собой совокупность значений 4nR2— .
Экспериментальные кривые атомного распределения пересекают параболы,
проходя через ряд максимумов и минимумов. Это—отражение
корреляции атомов аргона. Наиболее сильно корреляция выражена у
соседних атомов, т. е. при Ry немного превышающем d0 (для
атомов аргона d0 = 0,34 нм). Рис. 24 показывает, что корреляция
атомов наблюдается как в жидкой фазе, так и в парах, хотя и менее
отчетливо. С уменьшением плотности пара корреляция ослабевает. У
аргона корреляция атомов обнаруживается даже при плотностях, в 15 раз
меньших, чем плотность аргона в критической точке. Корреляция
атомов аргона зависит от плотности обычно гораздо сильнее, чем от
температуры. При постоянной плотности вдали от критической
точки жидкость — пар корреляция практически не зависит от
температуры, даже если температура меняется на 20—30°.
Радиус корреляции. Наибольшее расстояние между частицами
(атомами, молекулами, ионами) жидкости, при котором еще можно
наблюдать корреляцию, называют радиусом корреляции. Обычно
эту величину обозначают символом L. Величина L зависит от метода и
точности наблюдения корреляции, поэтому L определена не строго.
Радиус корреляции зависит от температуры, давления и состава
жидкой фазы. Если состояние жидкости далеко от критической
точки, то L, как правило, не превышает 3—4 диаметров молекул. В
окрестности критической точки жидкость — пар радиус корреляции резко
Бозрастает, достигая значений порядка 10а нм и более.
Среднее координационное число. Первый максимум на кривых
атомного распределения характеризует положения соседних атомов.
Абсцисса максимума приблизительно равна наиболее вероятному
расстоянию между ближайшими атомами жидкости. В случае
одноатомных жидкостей максимумы на кривых атомного
распределения не изолированы друг от друга. Площадь под первым
максимумом представляет собой среднее первое координационное
число z, т. е. среднее число атомов, окружающих любой, произвольно
выбранный атом жидкости ( кроме атомов, находящихся около
поверхности)
г = 4я f R*p(R)dR9 (V.13)
dQ
где величина R± зависит от способа выделения площади под
максимумом.
На рис. 26 в качестве примера приведена кривая атомного
распределения жидкого золота при 1100°. Максимумы этой кривой налагаются
друг на друга. На рисунке площадь под первым максимумом выделена
путем продолжения кривой справа от максимума симметрично левой
119
части кривой до пересечения с осью абсцисс в точке Rv Ясно, что
определение площади под этим максимумом, а следовательно, и
координационного числа z не может быть вполне точным. Величина z зависит
от способа выделения площади, а таких способов насчитывают четыре
[14]. К сожалению, способ, с помощью которого вычислялось
координационное число, указывается далеко не во всех работах по
рентгенографии жидкостей. В этих случаях величины г, найденные
различными исследователями, трудно сопоставимы. Средние случайные ошибки
при определении z могут превышать 10%. Тем не менее ценность этой
Рис. 26. Кривая атомного
распределения жидкого золота при 1100
°С [23]:
пунктир — кривая атомного распределения
атомов золота при той же температуре,
если бы упорядочение отсутствовало
Рис. 27. Кривая атомного
распределения жидкого хлора при 25° С и 7,7
атм [13]
характеристики строения жидкостей не вызывает сомнений, в
особенности, когда речь идет о простых жидкостях.
Для золота z = 11,0, т. е. каждый атом золота в жидкой фазе в
среднем окружен примерно 11 атомами. Золото кристаллизуется с
образованием гранецентрированной кубической решетки. В кристаллах
золота z = 12. Таким образом, при плавлении золота координационное
число немного уменьшается.
Другой пример — кривая атомного распределения жидкого хлора
при 25° С и 7,8 • 105 Па (рис. 27). Первый максимум на этой кривой
изолирован. Абсцисса в точке первого максимума равна 0,201 нм,
d0 = 0,15 нм, 7?! = 0,24 нм. Расчет по формуле (VI. 13) показывает,
что среднее число атомов хлора, соответствующее первому максимуму,
в пределах ошибок опыта равно единице. Это согласуется с
представлением, что жидкий хлор состоит из двухатомных молекул С12.
Расстояние между атомными ядрами в изолированной молекуле С12 равно
0,199 нм. Молекулы С12, как уже говорилось в гл. III, взаимодействуют
120
друг с другом с образованием слабых химических связей. Можно
предполагать, что увеличение на 2 • 10~3 нм межъядерного расстояния
молекулы С12 вызвано слабым химическим взаимодействием между
соседними молекулами хлора. Так, например, при слабом химическом
взаимодействии молекул С12 с диоксаном это расстояние растягивается до
0,202 нм (см. табл. 6).
Вандерваальсов радиус атомов хлора составляет около 0,18 нм.
Следовательно, межъядерные расстояния между химически
несвязанными атомами хлора должны быть не менее 0,36 нм. Второй максимум на
кривой атомного распределения жидкого хлора имеет абсциссу,
равную 0,4 нм, и обусловлен расстоянием между примыкающими друг
к другу атомами хлора соседних молекул С12. Грубо приближенная
оценка площади под этим максимумом приводит к выводу, что каждая
молекула хлора окружена шестью ближайшими соседями (А. Ф. Скры-
шевский [ 13]). Отсюда можно заключить, что расположение соседних
молекул жидкого хлора даже при температурах выше точки кипения
(температура кипения равна — 34,05°С) мало отличается от расположения
молекул в кристаллах хлора. Каждый атом хлора молекул С12 в
кристаллической фазе имеет две слабые химические связи с двумя
соседними молекулами, расположенными в той же плоскости. Расстояние
между центрами слабо связанных друг с другом атомов хлора соседних
молекул равно 0,33 нм. Таким образом, молекула С12имеет4 соседние
молекулы, расположенные в одном плоском слое (так же, как и 12). Еще
две соседние молекулы хлора помещаются в плоских слоях,
находящихся выше и ниже того слоя, в котором имеется выбранная нами
центральная молекула С12. С ростом межатомного расстояния корреляция
положений молекул жидкого С12 быстро исчезает, как это следует из
рис. 27.
§ 44. Корреляция трех атомов.
Суперпозиционное приближение
Пусть dW123 есть вероятность того, что три произвольно выбранных
атома жидкости располагаются в малых элементах объема dVlydV2 и dV3,
каждый из которых по порядку величины равен d0. Если элементы
объема dVlt dV2 и dVз не очень сильно удалены друг от друга, то, как
показывает опыт, между атомами жидкости возникает корреляция.
В этом случае dW123 не будет равно произведению плотностей
вероятностей dVt/V (i = 1, 2, 3). Это обстоятельство можно учесть тем же
способом, что и при написании уравнения (VI. 7), вводя тернарную
функцию /(/?!, R2, R3) распределения
(VI. 14)
где Rly i?2 и R3 — расстояния между ядрами атомов /, 2, 3. Функция
/C^i» #2, R з) учитывает корреляцию трех атомов. Следуя Д. Кирквуду,
нередко предполагают, что для изотропной жидкости при отсутствии
внешних сил функция /(/?1э R2t R3) может быть представлена в виде
121
произведения трех радиальных функций распределения:
/(Ль Ла. R*) = g№ g(R*) g(K3)- (VI.15)
Это так называемое суперпозиционное приближение. Уравнение (VI. 15),
несомненно, выполняется, когда расстояния Rly R% и R3 стремятся
к бесконечности. В этом случае правая и левая части уравнения (VI. 15)
равны единице. Так как атомы непроницаемы друг для друга, то
вероятность сближения атомов на расстояния, меньшие, чем d0, равна
нулю. Поэтому при Rlt R2 или Rs, меньших, чем dOf обе части уравнения
(VI. 15) равны нулю. Основываясь на этих свойствах
суперпозиционного приближения, ранее интуитивно предполагали, что оно может
приводить к качественно правильным результатам при статистических
расчетах свойств жидкостей, когда требуется учитывать все
возможные расстояния между атомами, большие, чем диаметр атомов d0. Но
единственное основание для введения суперпозиционного
приближения — упрощение теоретических расчетов свойств жидкостей. Сомнения
в применимости формулы (VI. 15) при расчетах свойств жидкостей
ранее высказывались многими авторами. Анализ этого вопроса был
выполнен И. П. Павлоцким в работе [19]. Оказалось, что
суперпозиционное приближение (VI. 12) применимо лишь тогда, когда газ
разрежен настолько, что его уравнение состояния может быть представлено в
виде ряда по степеням У"1, где V — объем газа [см. ур. (IV.3)i.
Суперпозиционное приближение описывает свойства такого газа с
точностью до члена, содержащего третий вириальный коэффициент
включительно.
§ 45. Корреляция и ассоциация
Поскольку строение жидкостей определяется короткодействующими
силами, ясно, что и корреляция, т. е. взаимосвязь положений молекул,
также должна зависеть, в основном, от короткодействующих сил
химического типа. Эти силы определяют вероятные положения молекул
первой координационной сферы. Теми же силами устанавливаются
вероятные положения молекул второй координационной сферы по
отношению к молекулам первой координационной сферы и т. д. Таким
образом корреляция, по существу, есть статистическое описание
ассоциации и комплексообразования. Функции, описывающие корреляцию
молекул и атомов, имеют статистическую природу. Поэтому связь
между радиальной функцией распределения g(R\ P, Т) и
межмолекулярными взаимодействиями, а также строением ассоциатов и комплексов,
сложна и неоднозначна, В рамках суперпозиционного приближения
аналитическое выражение связи между радиальной функцией
распределения атомов и потенциальной энергией межатомного
взаимодействия было найдено рядом авторов. Наиболее последовательный и
математически совершенный вариант теории был развит Н. Н.
Боголюбовым [20]. Анализ интегрального уравнения Боголюбова и
вычисления радиальной функции распределения с помощью этого уравнения
выполнены И. 3. Фишером [21]. Расчет радиальной функции
распределения атомов для некоторых простых видов эмпирических функций
потенциальной энергии может быть осуществлен с помощью ЭВМ.
122
Описание методов расчета имеется в статье А. М. Евсеева и его сотр.
[22] и кн. И. 3. Фишера [21]. Некоторые исследователи находили
радиальные функции распределения с помощью механических моделей.
Обзор ранних работ такого рода имеется в статье И. В. Радченко [23].
О более поздних работах рассказано в обзоре Д. Бернала и С. Кинга
[24].
Так, например, в опытах Бернала и Кинга около 3000 стальных
шаров диаметром около 6 мм каждый засыпалось в баллон, который
затем встряхивался и сжимался. Оказалось, что в случайной плотноупа-
кованной структуре, которая возникает в баллоне, шары занимают
примерно 64% объема баллона. В гранецентрированной кубической
плотной упаковке шары занимали бы 74% объема. Отношение
плотностей при гранецентрированной кубической и случайной плотной
упаковках шаров равно 1,16. В тройной точке аргона, как показывает опыт,
отношение плотностей твердой и жидкой фаз равно 1,15.
Затем Д. Бернал и С. Кинг измеряли координаты каждого шара и
находили радиальную функцию распределения шаров. Получалось,
что эта функция почти совпадает с функцией радиального
распределения атомов жидкого аргона в окрестности тройной точки. Было
подсчитано среднее координационное число в случайном плотноу пакован -
. ном распределении стальных шаров. Для расстояний между шарами,
не превышающих их диаметр более чем на 5%, оно близко к 8. Но
встречаются и такие локальные координационные числа, как 4 и 12.
Потенциальная энергия взаимодействия между стальными шарами
диаметра d0 имеет вид:
Е (R) = + оо при R < ^
(VI. 16)
Е (R) = 0 при R > d0.
Но почти та же радиальная функция распределения получается и для
модели, в которой потенциальная энергия взаимодействия между
атомами подчиняется уравнению Леннарда—Джонса (IV. 17):
Е (Я) = + оо
Введение дальнодействующих сил притяжения, пропорциональных
R~7, как и можно было ожидать, практически не отражается на
функции радиального распределения атомов. Если же потенциал (IV. 17)
заменить уравнением Леннарда—Джонса для всех значений R как
больших, так и меньших диаметра атомов d0 (нежесткие сферы), то
радиальная функция распределения атомов для значений R, лежащих
в окрестности диаметра атомов dOi изменится; g(R] p, T) будет точнее
соответствовать экспериментальной радиальной функции
распределения атомов, поскольку в реальных системах возможны межатомные
расстояния, меньшие, чем диаметр одного атома.
Применение корреляционных функций. Как уже говорилось,
радиальная функция распределения атомов позволяет определять сред-
123
нее координационное число простых жидкостей. В ряде случаев можно
получать сведения о среднем координационном числе и структуре
сложных жидкостей.
Можно полагать, что в жидком аргоне соседние атомы
располагаются в среднем примерно так, как в случайной плотной упаковке
стальных шаров. Но мы ничего не можем сказать о фрагментах этой
упаковки. Являются ли они ассоциатами атомов аргона или случайными
скоплениями несвязанных частиц, — этот вопрос остается без ответа.
Радиальная функция распределения отражает среднюю корреляцию
между атомами, но природа этой корреляции обычно выясняется
другими методами. Так, анализ свойств паров аргона говорит о
присутствии в парах ассоциатов. Изучение свойств кристаллов аргона приводит
к выводу о важной роли многочастичных взаимодействий,
сопровождающихся обобществлением электронов.
Опираясь на результаты исследований g(R; р,7) и многих других
свойств веществ в жидкой, твердой и газовой фазах, можно подобрать
модели строения молекул жидкости, соответствующие
действительности, с некоторой, иногда, быть может, высокой степенью вероятности.
Но сами по себе функции радиального распределения атомов не дают
однозначных указаний о строении и свойствах жидкостей. Формула
(VI. 11), с помощью которой вычисляется^/?; р, 7), справедлива не
только для одноатомных жидкостей, но и для кристаллических порошков,
когда они состоят из одного сорта атомов. На этом основаны попытки
рассматривать жидкости как поликристаллические образования [25].
В последние годы такой подход получил развитие в работах Д. Бер-
нала [24]. По мнению Бернала, строение жидкостей можно представлять
себе как статистическую совокупность различных многогранников,
в вершинах которых помещаются атомы. В принципе, можно было бы
ассоциаты и комплексы рассматривать как такого рода многогранники
или их элементы.
Таким образом радиальная функция распределения не позволяет
получить исчерпывающую информацию о строении даже простых
жидкостей.
§ 46. Прямая корреляционная функция
Иногда пытаются использовать корреляционные функции для
вычисления энергии межатомных взаимодействий. С этой целью в ряде
работ вводится понятие о прямой корреляционной функции с (R, Т)
[24], которая связана с парной корреляционной функцией h(R; р, Т) =
= g(R\ P, Т)—\ уравнением:
h(R) = c(R)+p § c(R — R')h(R')dR', (VI. 18)
где R — вектор, соединяющий произвольно выбранные атомы 1 и 2;
R' — вектор, соединяющий атом 2 и некоторый произвольно
выбранный атом 3. R—R' — вектор, соединяющий атомы 2 и 3; р =N/V,
т. е. число атомов в единице объема.
Уравнение (VI. 18) не имеет строгого обоснования. Оно имеет
следующий физический смысл. Прямая корреляционная функция с (/?)=
124
^ c(R, T) учитывает непосредственное взаимодействие между атомами
I и 2. Подынтегральное выражение в (VI. 18) учитывает непрямую
корреляцию атомов 1 и 2 через некоторый третий атом. Интегрирование по
R' означает учет всех возможных непрямых корреляций между
атомами 1 и 2 через любой третий атом. Но корреляция между различными
атомами 3 не учитывается.
Предполагается, что прямая корреляционная функция c(R) более
тесно связана с потенциальной энергией взаимодействия атомов E(R)y
чем парная корреляционная функция h(R). На основе качественных
аналогий принимается, что связь между c(R) и E(R) имеет вид
= ехр{-
1. (VI.19)
Найденная с помощью уравнения (VI. 19) функция E(R), по мнению
некоторых авторов [24], позволяет охарактеризовать взаимодействия
между молекулами или ионами, из которых состоит жидкость. Для
жидких металлов функция E(R) проходит через ряд максимумов и
минимумов.
§ 47. Связь функций распределения
с термодинамическими функциями
Радиальная функция распределения и парная корреляционная
функция представляют собой, в сущности, своеобразные
термодинамические функции,потому что зависят от термодинамических переменных—
температуры и плотности. Естественно, что другие термодинамические
функции сравнительно просто могут быть связаны с функциями g(R;
р, Т) или h(R;py T). Наиболее проста связь между парной
корреляционной функцией и изотермической сжимаемостью одноатомных
жидкостей [14]:
/ 00 s
J*<*>*« <VI-20)
J
^ 0 '
где v = VIN — средний объем, приходящийся на один атом
жидкости; множитель v/kb T представляет собой сжимаемость, если бы она
следовала уравнению состояния идеального газа; второй член в
фигурных скобках учитывает влияние отклонений от свойств идеального
газа, обусловленных межатомными взаимодействиями.
Приведем еще выражение для внутренней энергии U изотропной
системы (жидкости или газа), состоящей из Л^а частиц сферической
формы в объеме V [20]:
3 2nN% r
U RT^ E(R)g(R)R*dR. (VI.21)
о
V *)
о
Первый член в правой части этого уравнения есть кинетическая
энергия теплового движения сферических частиц, вторая часть — вклад
125
во внутреннюю энергию U, обусловленный потенциальной энергией
взаимодействия E(R) всех пар частиц изотропной системы. В случае
вандерваальсовых сил, как было показано в гл. I, формулы для E(R)
имеют смысл только при тех расстояниях RB между молекулами,
которые на порядок превышают их диаметр d0. В таких условиях
радиальная функция распределения g (R) = 1. Подставляя выражение для
E(R) дипольных, лондоновских и поляризационных взаимодействий в
уравнение (VI. 17) и полагая g(R) = 1, после интегрирования по R от
RB до оо получаем уравнение (1.65), описывающее вклад во внутреннюю
энергию системы, обусловленный вандерваальсовыми силами.
Другие соотношения между корреляционными и
термодинамическими функциями приводятся, например, в [24]. Как и уравнение (VI. 17),
все они имеют односторонний характер в том смысле, что, зная функцию
h(R) при всех R , можно вычислить |3, и другие термодинамические
функции, но по (3/ и иным термодинамическим характеристикам
жидкости нельзя найти h(R).
Следует подчеркнуть, что корреляционные функции
малочувствительны даже к существенным изменениям не только дальнодействую-
щей, но и близкодействующей компоненты потенциальной энергии
межмолекулярного взаимодействия. Термодинамические свойства, в
частности внутренняя энергия, сильно зависят от E(R). Поэтому
малые различия между корреляционными функциями, в сообенности в
областях малых R порядка d0, приводят к сильно отличающимся
уравнениям состояния жидкости и большим расхождениям в других
термодинамических свойствах. Но именно в области малых R
рентгенографические и нейтронографические исследования корреляционных функций
дают наименее точные результаты.
§ 48. Функции распределения частиц в растворах
Если раствор двухкомпонентен, причем оба компонента простые
жидкости, то корреляции атомов компонентов 1 и 2 можно учесть,
вводя три радиальные функции распределения guiR), gz2(R) h^12(/?).
Эти функции могут быть определены с помощью рассеяния нейтронов,
если использовать изотопы компонентов 1 и 2. Интенсивность
рассеяния нейтронов атомными ядрами изотопов должна существенно
различаться. Предполагается, что замена атомов одного изотопа атомами
другого не меняет структуру раствора. Это предположение в
большинстве случаев справедливо. Варьируя изотопный состав раствора,
можно получить ряд различных кривых рассеяния нейтронов при одной
и той же концентрации компонентов. Чтобы определить три
перечисленные радиальные функции распределения, нужно получить не менее
трех различных нейтронограмм. Впервые такие опыты выполнили Дж.
Эндерби, Д. Норт и П. Эгельстафф в 1966 г. (см. [14], стр. 91). Они
исследовали жидкий сплав меди и олова, в котором на 6 атомов меди
приходилось 5 атомов олова. Были изучены три варианта такого сплава:
с естественной медью, с медью, обогащенной до 99% изотопом 63Си, и
медью, обогащенной до 99 % изотопом G5Cu. К сожалению, интенсивнос-
126
ти рассеяния нейтронов естественной медью и изотопом 63Си
отличаются очень слабэ, поэтому измерения были не достаточно точны, но
показали, что корректное определение функций gn(R)> giz(R) и g22(R)
раствора с помощью этого метода возможно.
ГЛАВА VII
ФЛУКТУАЦИИ В ЖИДКОСТЯХ
§ 49. Виды флуктуации
Флуктуации представляют собой локальные отклонения свойств
вещества от их среднего значения, случайно возникающие под
действием теплового движения и молекулярных сил. Для характеристики
строения жидкостей существенны флуктуации плотности,
анизотропные флуктуации и (когда речь идет о растворах) флуктуации
концентрации.
Флуктуации плотности — случайные локальные сгущения и
разрежения вещества. Анизотропные флуктуации — случайные
локальные отклонения свойств жидкости от изотропного поведения.
Флуктуации концентрации — случайные локальные отклонения от среднего
состава раствора. Флуктуации плотности, концентрации и
анизотропные флуктуации статистически независимы друг от друга. Флуктуации
плотности и флуктуации концентрации в среднем (т. е. статистически)
не связаны с локальными отклонениями жидкости от изотропного
поведения. Это позволяет отнести флуктуации плотности и
концентрации к группе так называемых изотропных флуктуации.
Иногда встречающиеся термины «флуктуации анизотропии» или
«флуктуации ориентации» не удачны. Речь идет не об изменении
(флуктуации) анизотропии жидкости или ориентации молекул, а о случайном
возникновении локальной анизотропии в жидкой среде, которая в
среднем изотропна, поэтому мы пользуемся термином «анизотропные
флуктуации».
Понятие о флуктуациях было введено раньше, чем представление о
радиальной функции распределения, и давно уже стало одним из
основных понятий статистической физики. Но в теории растворов оно
долгое время играло сравнительно небольшую роль. В течение многих
лет господствовало мнение, согласно которому флуктуации не являются
существенной характеристикой структуры жидкости и не оказывают
заметного влияния на свойства жидких систем, если жидкости не
находятся в непосредственной окрестности критической точки жидкость —
пар или критической точки расслаивания. Исследования ([9, 26] и др.),
проведенные в течение последних двух десятилетий, показали, что это
мнение во многих случаях не соответствует действительности.
Наиболее прямым экспериментальным подтверждением
присутствия флуктуации, как известно, являются измерения рассеяния света.
Если среда вполне однородна, то она свет не рассеивает. Между тем
наблюдения показывают, что твердые тела, жидкости и газы
рассеивают свет.
127
Допустим, что через кювету, заполненную бензолом, водой,
метиловым спиртом или какой-либо другой хорошо очищенной прозрачной
жидкостью, проходит яркий пучок естественного света. Тогда,
наблюдая этот пучок сбоку, например под углом 90°, можно видеть, что
жидкость рассеивает свет, причем рассеянный свет имеет синевато-
фиолетовый оттенок. В начале нашего века считали, что рассеяние
света прозрачными жидкостями и газами обусловлено посторонними
включениями, пылинками. Но затем выяснилось, что при полном удалении
пыли рассеяние света остается. Оно зависит от температуры и состава
рассеивающей среды и обладает рядом других особенностей, которые
не могли бы иметь места, если бы источником рассеяния были
посторонние включения — пылинки.
Рассеяние света обусловлено взаимодействием света с молекулами
среды. Если молекулы распределены вполне равномерно, то тогда в
результате интерференции свет, рассеянный от отдельных молекул,
взаимно погашается. В этом случае рассеяние отсутствует. Источником
рассеяния света в обеспыленных газах и жидкостях являются
отклонения от равномерного распределения молекул, или флуктуации.
Флуктуации тесно связаны с термодинамическими свойствами
жидких систем. Они влияют и на кинетику некоторых молекулярных
процессов, например на диффузию и на поглощение звука в растворах.
§ 50. О теории флуктуации
Пусть индивидуальная жидкость или раствор занимает объем V и
находится в состоянии термодинамического равновесия. Разделим V
на области v. (i =1,2,..., п). В ходе теплового движения в жидкости
будут возникать флуктуации. В различных областях vt температура,
плотность, концентрация и другие физические величины могут
принимать неодинаковые значения. Термодинамическая теория флуктуации
основана на допущении, что внутренняя энергия (или свободная
энергия для «системы в термостате» при заданной температуре) аддитивно
слагается из энергий областей vv т. е. U = 2£/4; G = 2G.. Энергия
взаимодействия области v. с остальной частью системы мала по
сравнению с энергией взаимодействия частиц, находящихся внутри области
v.. Это означает, что энергия области v. системы однозначно
определяется заданием макроскопических переменных, характеризующих
состояние системы в vr
Условие аддитивности энергии приводит к выводу о независимости
флуктуации, происходящих в соседних областях v.. Иначе говоря,
получается, что флуктуации распределены хаотически.
Термодинамическая теория флуктуации неприменима, если условие
аддитивности энергий теряет силу.
В некоторых случаях работа, которая необходима для
возникновения флуктуации, очень мала. Тогда лаже малые взаимодействия
между vt и остальной частью системы приводят к большим
отклонениям от состояния равновесия в области vr Таково, например,
поведение жидкости в критической точке жидкость — пар. В этих усло-
128
виях изменения термодинамических потенциалов областей v. при пояе-
лении флуктуации определяются не только отклонением плотности,
состава и других термодинамических переменных от равновесных
значений, но и градиентами этих переменных. По существу, это
означает учет взаимодействий между v. и остальной частью системы.
В принципе выводы термодинамической теории флуктуации точны,
если области vv в которых изучаются флуктуации, бесконечно велики.
На практике теория нередко приводит к хорошему количественному
совпадению с опытом даже тогда, когда линейные размеры области у.
жидкости или неидеального газа по порядку величины равны 10 нм,
т. е. малы по сравнению с длиной волны видимого света, но велики по
сравнению с диаметром молекул, порядок величины которого
составляет 0,1 нм — 1 нм. В таких областях жидкости может находиться около
104 молекул бензола, ацетона и других низкомолекулярных веществ.
Качественное согласие теории с опытом наблюдается и для более
мелких элементов объема.
Условие аддитивности, а вместе с ним и термодинамическая теория
флуктуации теряет силу в том случае, когда области vt настолько малы,
что содержат лишь небольшое число молекул. Энергия
взаимодействия столь малой области с окружающей средой по порядку величины
сравнима с энергией самой области, поэтому энергией взаимодействия
с окружающей средой здесь пренебрегать нельзя. Однозначной связи
между плотностью, составом и энергией в такой малой области
жидкости нет.
Теория флуктуации в больших областях vi жидкостей хорошо
разработана. Для малых областей, содержащих лишь десятки молекул,
теория развита недостаточно. Экспериментальные исследования
флуктуации в столь малых элементах объема жидкостей (микрофлуктуа-
ций) пока еще почти не производились. Не исключено, что поведение
флуктуации в малых элементах объема жидкостей напоминает то,
которое наблюдается для флуктуации в окрестности критической точки
жидкость — пар.
Рассмотрим замкнутую область vv пренебрегая ее
взаимодействием с окружающей средой. Состояние вещества в области vv где
происходит флуктуация, может быть охарактеризовано, если квантовой
неопределенностью энергии АЕ (для замкнутой области v.) также можно
пренебречь.
Пусть х обозначает какую-либо макроскопическую характеристику
состояния области v. (плотность, концентрацию, давление и т. д.).
Связь между неопределенностями Е и х имеет вид (см. [27], стр. 70):
&EAx~hxt (VII.1)
где х — скорость изменения величины х\ Ь — постоянная Планка
(ft = 1,05-10"34 Дж-с).
Понятие о скорости изменения л: приобретает смысл только в том
случае, если процесс изменения х протекает достаточно медленно,
чтобы в ходе изменения х максвелловское распределение скоростей не
нарушалось. Только в этом случае можно пользоваться понятиями о
5—503 129
температуре области vtno x как некоторой термодинамической
характеристике системы в области v..
Допустим, что в области v.'образовалась флуктуация, так что
величина х отличается от равновесного среднего статистического'зна-
чения <СО>. В замкнутой системе <Х> не зависит от времени. Если в
начальный момент времени свойство х замкнутой системы почему-либо
отличается от <Ж>, то с течением времени замкнутая система будет
стремиться к своему равновесному состоянию и х будет неограниченно
приближаться к <СС>. Этот процесс называется релаксацией. Ясно,
что производная по времени от разности Дх = х— <Х> равна х.
Скорость, с которой х стремится к равновесному значению <00, в
простейшем случае следует уравнению
х = — — (х — <*>), (VII.2)
где т — постоянная, имеющая размерность времени; т называют
временем релаксации. В рассматриваемом нами случае оно
характеризует по порядку величины среднюю продолжительность существования
флуктуации. Из сказанного следует, что т должно быть по крайней
мере на порядок больше периода времени At, необходимого для
установления максвелловского распределения скоростей в области vr Из
(VII. 1) и (VI 1.2) вытекает, что
| АЯ | | А* | ^ h | х | = -^ \х - (х) | . (VII.3)
Разность х — <Сх> может иметь смысл только тогда, когда квантовая
неопределенность |Дл:| <£\х — <%>|. Поэтому должно выполняться
неравенство
1 А£| » — . (VII.4}
т
Согласно одному из основных принципов статистической физики —
принципу Больцмана — плотность вероятности возникновения
флуктуации х в изолированной системе равна
W(x) = const eAS{x)/kB, (VI 1.5)
где AS — разность энтропии рассматриваемого и равновесного
состояний. Эта формула'имеет смысл только для таких изменений состояния,
когда квантовая неточность определения TAS = АЕ мала по
сравнению с &Б7\ т. е. AE<^kBT. Следовательно, неравенство (VII.4)
усилится, если АЕ заменить на кБ Т. Таким образом, среднее «время
жизни» термодинамических флуктуации должно удовлетворять
неравенству
.ч h 1,05. 10-34 ю-ii
Z>>1^T- 1,38. ю- т -—7~'с- (VIL6)
Это означает, что при комнатных температурах термодинамическое
описание возможно только для таких флуктуации, среднее время суще-
130
ствования которых на порядок больше, чем 10~13с. Неравенство (VI 1.6)
указывает порядок наименьших времен релаксации, в течение которых
можно не учитывать динамические квантовые эффекты. Учет
квантовых эффектов необходим для процессов, которые протекают с
характерными временами порядка Ыкъ Т и менее.
В (VI 1.6) нет указаний о величине периода времени А/,
требующегося для установления максвелловского распределения скоростей в
таких элементах объема, к которым применимо макроскопическое
описание. Величина At зависит от плотности и температуры системы. Для
очень разреженных газов период времени At может быть большим.
Экспериментальные и теоретические исследования показывают, что
для жидкостей при комнатных температурах Д^ по порядку величины
составляет около 10"13 с. Для классических (т. е. неквантовых)
жидкостей At ~ h/(kBT).
Иногда минимально возможное значение т, при котором поведение
жидкой системы можно описывать, применяя макроскопические
характеристики, определяют с помощью величин максвелловского
времени релаксации Ту. Это время сдвиговой релаксации в жидкостях,
т. е. релаксации напряжения при некоторой заданной сдвиговой
деформации. Максвелловское время релаксации определяют с помощью
отношения коэффициента вязкости к модулю сдвига жидкости.
Четкого способа обоснования такого подхода к определению минимальных
возможных значений т, по-видимому, нет. Да и модуль сдвига
жидкостей — величина, далеко не всегда известная. Для жидкого аргона
вблизи точки плавления xs имеет величину порядка 6-10"13 с. Но для
жидкого натрия получается слишком малая величина xs ~ 10~13 с,
не удовлетворяющая неравенству (VII.6). Для жидкого глицерина
имеется несколько максвелловских времен релаксации; одно из них
при 20°С равно—4-10~9 с, другое—4-10"10 с. Если среднее время жизни
флуктуации в области v. настолько мало, что неравенство (VII.6) не
выполняется, то такие флуктуации нельзя рассматривать с помощью
термодинамической теории.
§ 51. Флуктуации плотности
Рассмотрим некоторый замкнутый участок v, расположенный
внутри объема V жидкости, т. е. не около ее поверхности. Этот участок,
как уже говорилось, должен быть достаточно большим, чтобы для
характеристики его состояния были применимы термодинамические
переменные. Пусть <р> обозначает среднюю плотность жидкости. Время
от времени в области v случайно в ходе теплового движения возникают
отклонения плотности р от <р>, т. е. флуктуации плотности Др =
= р—<р>. ЕслиДр>0, то в v, очевидно, возникает сгущение
жидкости. Когда Др<0, происходит разрежение жидкости.
Вероятность dW возникновения флуктуации Др области v жидкости
равна
dr=/pwAp. (VII.7)
5* 131
Если состояние жидкости не соответствует непосредственной
окрестности критической точки жидкость — пар, то плотность вероятности
подчиняется нормальному распределению. Это утверждение является
следствием принципа Больцмана (см., например, [28]).
hv— тАоГТТаТТ^Г еХР О//Л.Л9А • (VI I. 8)
Статистическое среднее квадрата флуктуации плотности прямо
пропорционально изотермической сжимаемости fl, и обратно
пропорционально величине того объема о, в котором рассматривается
флуктуа*
ция*:
В правой части (VI 1.9) р есть средняя плотность жидкости (ради
упрощения здесь и далее мы не пользуемся скобками <...>, если нет
необходимости различать величины <Х> и х).
Уравнение (VI 1.9) имеет простой физический смысл. Для малых
областей v можно принять, что изменение объема ДУ большого
образца жидкости при изменении давления на APV равно v. Тогда
=kBT. (VII. 10)
Величина v* = v представляет собой то изменение
объема жидкой фазы, для осуществления которого требуется затратить
работу, равную кБ Т.
Когда состояние жидкости далеко от критической точки жидкость—
пар, то р, обычно близка к 10"11 Па"1. Легко подсчитать, что при Т =
= 300 К объем и* составляет всего около 4-10~24 мл. Молярный объем
низкомолекулярных жидкостей V ~ 102 мл, следовательно, объем,
приходящийся на одну молекулу в жидкой фазе, приблизительно равен
2-10"24 мл. В окрестности критической точки жидкость — пар |3,
возрастает на 2 порядка и более. Жидкость становится мутной.
Наблюдается критическая опалесценция. Так, например, для жидкого СО2
* Подчеркнем, что здесь и далее всюду речь идет об истинной изотермической
1 / dV \
сжимаемости $t = — -—I ) , когда интервал давлений А Р, в котором
измеряется сжатие жидкости, очень мал. Приводимые в литературе величины р t в
большинстве своем есть средние значения сжимаемости для больших интервалов
давлений А Р, равных десяткам и сотням атмосфер. Например, при 20° С истинное
значение изотермической сжимаемости бензола равно 8,41 • 10~12 Па"1 [12]. При
той же температуре, но в интервале давлений (100—300)- 105 Па средняя
сжимаемость "JT. равна 8,0- 10~12 Па"1, а в интервале давлений (300—500)- 105 Па"1^ =
= 6,85-10"12 Па"1. Прямые измерения истинной изотермической сжимаемости
возможны, но представляют собой нелегкую задачу ([12], стр. 42). Более просто и
вместе с тем достаточно надежно величины р^ можно получить на основе
анализа данных о рассеянии света, пользуясь методикой расчета, указанной в
[12].
132
поданным, приведенным в работах В. П. Скрипова и Ю. Д. Колпако-
ва [29, 30], вблизи критической точки fit= 10"9 Па"1. Имеется в виду
граница области состояний, где уравнение (VII.9) еще применимо.
В этом случае и* составляет около 4-10~22 мл, что приблизительно в 4
раза превышает объем, приходящийся на одну молекулу СО2 в жидкой
фазе около критической точки.
Уравнения (VII.8)—(VII.9) справедливы, когда флуктуации Др . и
др;- в любых двух областях v. и Vj жидкости статистически
независимы, т. е. когда
<APiAPy> = 0. (VII. 11)
В ближайшей окрестности критической точки жидкость — пар
условие (VII. 11) и уравнения (VII.8) и (VII.9) неприменимы. Это видно
уже из того, что когда температура Т приближается к температуре
критической точки Тк, а давление Р к давлению в критической точке Рк,
сжимаемость р/ стремится к бесконечности. Левая же часть равенства
(VII.9) всегда конечна. Область состояний жидкости, где
термодинамическая теория флуктуации плотности теряет силу, невелика. Так, по
данным В. П. Скрипова и Ю. Д. Колпакова [29], отклонения от
формулы (VI 1.9) для жидкого СО2 могут наблюдаться, лишь когда разность
j1 т
Тк— Т<0,1 К, т. е. когда —- ^гз . 10"4. Область состояний, где
наблюдается критическая опалесценция, не следует смешивать с
областью отклонений от уравнения (VI 1.9) и от закона Релея.
Отклонения от уравнения (VII.9) проявляются в интервале температур,
примерно в 10 раз более узком, чем для критической опалесценции.
§ 52. Флуктуации плотности
и парные корреляции молекул
Изотермическая сжимаемость простых жидкостей связана с парной
корреляционной функцией h(R) уравнением (VI.20), поэтому
{ОО
4nN С
1+~ J
<(Лр)2> = 7л^1+~ * h(R)R*dR\. (VII.12)
где (Nv) — среднее число молекул жидкости в объеме v. Деля обе
части равенства на величину m2/v2, где т — масса одного атома,
получим
00 ч
-\-~- Г h(R) R2dR\ , (VII. 13)
J I
о )
где ДЛ^—флуктуация числа молекул, т.е. Nv —{Nz)\ ((ANV)2} —
средняя квадратичная флуктуация числа молекул в области v простой
жидкости. Уравнение (VI 1.13) выведено для простых жидкостей, но
оно применялось и для анализа связи между флуктуациями плотности
и корреляционной функцией воды [31]. Расчеты с помощью уравнения
133
(VII. 13) для многоатомных жидкостей во многих случаях могут давать
качественно правильные результаты. При этом можно условиться,
что R обозначает расстояние между центрами тяжести молекул.
Возможны и другие способы выбора R. Уравнение (VII. 12) дает основу
для характеристики средних размеров флуктуации плотности.
Средний радиус флуктуации плотности — это радиус корреляции (см.
гл. VI), т. е. такая величина R = L, что при всех R > L парная
корреляционная функция h(R) может быть принята равной нулю.
Другой, независимый от этого способ оценки величины L дают
измерения интенсивности / рассеяния света при разных длинах
световых волн X для прозрачных жидкостей. Если % > L, то соблюдается
закон Релея, т. е. / ~ Х~4. В непосредственной окрестности
критической точки радиус корреляции L имеет
тот же порядок величины, что и X в
оптическом диапазоне длин волн, т. е.
102 нм. В этом случае закон Релея не
соблюдается: / — Кгп, где п < 4.
Представление о том, как меняется
строение жидкости при повышении
температуры и приближении к критической
точке, дает работа Миколаи и Пингса
[14]. На рис. 28 сопоставлены парные
корреляционные функции h(R) аргона
для состояний, сравнительно далеких от
критической точки, и состояния,
близкого к критической точке. Видно, что в
окрестности критической точки радиус
области, где при всех R, Л (/?) > О растет.
Это значит, что радиус сгущений
жидкости около произвольно выбранного
атома увеличивается. Ясно, что вместе с
этим растет и радиус областей
разрежения. Жидкость становится все более
пористой. Непосредственно около
критической точки флуктуации плотности не
следуют уравнению (VII. 11), т.е.
коррелируют друг с другом. Это означает,
что при Т ->- Тк и Р ->РК области сгущений все более и более
последовательно чередуются с областями разрежений.
Низкомолекулярная Жидкость становится похожей на губку с тем отличием, что
диаметр пор в этой губке в среднем имеет величину порядка 1 нм.
Рис. 28. Парные
корреляционные функции h(R)
жидкого аргона [14]
(Нижняя кривая соответствует
состоянию, близкому к критической
точке)
§ 53. Микрофлуктуации плотности
Попытаемся теперь охарактеризовать нетермодинамические
флуктуации плотности. Речь идет о флуктуацияхДр илиД А^,, которые
возникают в очень малых элементах объема v < 10~18 мл. В особенности
нас будут интересовать такие элементы объема, где область v имеет
радиус немного более d0. Такая область может, например, быть занята
134
молекулой в центре и ее непосредственным окружением (первая
координационная сфера)*.
Методов расчета флуктуации плотности в столь малых элементах
объема пока что нет. Но с помощью модельных опытов и по данным о
радиальной функции распределения атомов можно найти средний
квадрат флуктуации координационного числа <С(Д-г)2>. Он отличается от
<;(дЛ^)>. При расчетах средних квадратов флуктуации плотности и
числа молекул предполагается,что объему неподвижен. При
вычислении среднего квадрата флуктуации координационного числа
рассматривается движущийся объем v', неизменно связанный с каким-либо
атомом жидкости, находящимся в его центре. Для вычисления <(Д zf>
надо было бы знать тернарную функцию распределения (см. § 44). Но
трудности расчета тернарной функции очень велики, поэтому И. 3.
Фишер и В. К. Прохоренко [21, 26] воспользовались суперпозиционным
приближением (см. гл. VI). В этом приближении средний квадрат
флуктуации числа молекул в упомянутом перемещающемся объеме равен
^(\R\)(\R\)[(\RR\)l]dRdR (VII.14)
где /?2 и /?3 — радиусы-векторы, определяющие положение молекул
2 и 3 по отношению к молекуле ), находящейся в центре v'\
dR2 = dx2 dy2 dz2\ dR3 = dx3 dyB dz3
И. З. Фишер и В. К- Прохоренко подчеркивают, что расчеты по
формуле (VII. 14) должны приводить к заниженным величинам <(AAV )2>.
Сопоставим уравнение (VII. 14) с формулой, определяющей средний
квадрат флуктуации числа молекул в неподвижном объеме** v:
N2 С С
— JJ
(VII. 15)
Объемы v и v' одинаковы, <zNv>&<NV' >. В подынтегральном
выражении (VII. 14) радиальные функции распределения атомов g(\R21) и
g(\R 3|) можно заменить их средними значениями по области с/. По
порядку величины средние значения g(R) в интервале R от 0 до 1,5 d0 близки
к единице. Если принять значения g(R) равными единице, то величины
<(ДА^')2> и <(ДАу2> совпадут. Но как видно из рис. 23, среднее
значение g(R) в интервале R от 0 до 1,5 d0 в действительности несколько
меньше единицы, поэтому<(Д AV)2>, найденная по уравнению(УП. 14),
должна быть того же порядка, но несколько меньше, чем<(ДЛ^)2>.
Практически точная величина отношения <(ANV> )2>/< Л^>
известна пока что только в одном случае, а именно для модели аргона
при температуре и давлении, близким к тройной точке (84,4 К; 6,84 X
X 104Па). Опыты Д. Бернала и С. Кинга со стальными шарами (см. гл.
VI) показали, что их модель хорошо описывает строение жидкого ар-
* Принимаем, что молекула находится внутри области v, если ее центр
тяжести попадает в эту область.
** Если линейные размеры v велики по сравнению с d0, то, интегрируя
(VII. 15) по Rx или /?2, получим уравнение (VII.13) (подробности см. [20]).
135
гона около тройной точки. Пользуясь материалами, приведенными в
их работе [24], можно подсчитать, что при v' = v = — я(1,05 do)s =
= 1,9-10~22 мл величина б Nv> = V<(&Nv )2>f<Nv> > равна 0,14.
Расчеты И. 3. Фишера и В. К. Прохоренко по приближенной формуле (VII. 14)
при Г = 86,3 К дают dz ~ 6Nv' — 0,11. Используя завышенное значение
среднего координационного числа z = 10,5 при 7=84,4 К эти авторы получили для
жидкого аргона заниженные значения ог = }Л((Дг)2) / z = 0,06. Если
воспользоваться более правильным значением г = 8,5, то 6г = 0,075. Несомненно, что
относительные флуктуации bz — bNv' рассчитанные в работах И. 3. Фишера и
В. К- Прохоренко, по порядку величины правильны и близки к относительным
флуктуациямoiV^, = Y((kNv)2') / {Nv} в неподвижных элементах объема v,
радиус которых равен диаметру атомов ф>.
Сравним среднюю величину микрофлуктуаций плотности со
средним значением термодинамических флуктуации плотности, которые
должны были бы наблюдаться в объеме v, если бы термодинамическое
уравнение (VI 1.9) было применимо к объемам жидкости порядка
10~22 мл. Такое сопоставление позволит нам оценить характер и
величину отклонений от термодинамической теории, происходящих в
объемах порядка 10~22 мл.
Изотермическую сжимаемость жидкого аргона при 84 К можно
вычислить с помощью формулы
р,---!-+!"£-. (VII.16)
Вблизи тройной точки аргона плотность р = 1,416 г/мл; скорость
звука а = 875 м/с; теплоемкость Ср = 1,100 кДж/(кг-К); объемный
коэффициент расширения (вычисленный по зависимости р от Т) ар =
=3,8-10"3 К"1. Подставляя эти значения в уравнение (VI 1.6), получим
§t = 1,7-10"11 Па"1. Зная р„ по уравнению (VII.9) найдем бЛЛоерм =
= 0,084. Таким образом, микрофлуктуации плотности в модели
жидкого аргона при 84 К, исследованный Берналом и Кингом, почти в 1,7
раза больше, чем можно было бы ожидать на основе термодинамической
теории. Эта разница не столь уж велика. При радиусе области vt
равном 2dQy отношение величины б Nvf , рассчитанной по данным Бернала
и Кинга, к bNlepM для аргона тоже близко к 1,7. Для воды около
температуры замерзания б z =0,33, a 8NTvem равно 0,14. Здесь
получается расхождение, несколько большее, чем для модели аргона. Для
жидкой ртути вблизи точки замерзания (—38° С) расчет по уравнению
(VII. 14) даетб^Уу' =0,14, т. е. почти ту же величину, что и для
жидкого аргона. Но термодинамическая теория приводит к величине б Л^ерм,
равной 0,03, т. е. примерно в 5 раз меньшей. Отметим, что отношение
плотностей твердой и жидкой фаз в точке плавления ртути равно 1,04,
что значительно меньше, чем у аргона, для которого отношение равно
1,15. Сжимаемость ртути вблизи температуры затвердевания в 45 раз
меньше сжимаемости жидкого аргона около его тройной точки.
В табл. 9 приведены некоторые из величин уг((А2)2)/г,
вычисленных И. 3. Фишером и В. К. Прохоренко по рентгенографическим
136
Таблица 9
Относительные флуктуации координационного числа
в жидкостях [21, 31]
Жидкость
Гелий
Неон
Аргон
»
»
Ксенон
Натрий
Калий
Золото
Ртуть
Температура,
К
2,25
26,0
86,3
91,8
149,3
183
373
668
1273
235
Т//"<(Дг)2>/2
0,18
0,13
0,11
0,24
0,26
0,18
0,17
0,20
0,11
0,14
Жидкость
Ртуть
»
Германий
Висмут
»
»
Вода
»
»
»
Температура,
К
273
373
1373
458
673
823
274,5
303
335
356
У <(Л2)2>/2
0,21
0,23
0,15
0,19
0,22
0,22
0,33
0,33
0,28
0,26
данным о радиальной функции распределения. Из таблицы видно, что
флуктуации координационных чисел, а значит, и микрофлуктуации
плотности жидкостей, далеких от критической точки, слабо зависят от
вида жидкости и от температуры. Впрочем, этот вывод качественно
следует из графиков радиальной функции распределения для разных
жидкостей и различных температур. В интервале О < R < l,5d0 эти
графики, как правило, не очень
сильно различаются.
На рис. 29 изображены
зависимости отношений §Nv/%NvTevM
от радиуса сферической
области для жидкого аргона,
воды и ртути при состояниях,
близких к тройной точке. Здесь
8NV = /<(А^)а>/<^>
вычислено по уравнению (VII. 13),
абЛГгерм_ по уравнению (VII.9)
при той же величине объема
жидкости v. Кривые на рис. 29
представляют собой
качественную характеристику
наблюдаемых закономерностей. Как
следует из табл. 9 и рис. 29,
микрофлуктуации плотности
низкомолекулярных жидкостей в очень
малых объемах, линейные
размеры которых порядка 1 нм, в
основном определяются не
свойствами МОЛекуЛ, а СреДНИМ ИХ ^,-ради>с элемента объема, в котором рас-
числом в таком элементе объема, сматР»ваетс* фл-кТижо:ст1? ~ диаметр шлекул
3
LgRv/d0
Рис. 29. Сопоставление средних
относительных микрофлуктуаций плотности в
жидкой фазе, рассчитанных с помощью
статистических методов, 6NV и
термодинамической теории
137
т.е. плотностью системы. Для таких жидкостей, как аргон, где даль-
нодействующие силы не существенны и дальнодействующие
корреляции слабы, величины микрофлуктуаций плотности не очень сильно
превышают уровень термодинамических флуктуации плотности. С
ростом дальнодействующих корреляций и при усилении ассоциации
термодинамические флуктуации уменьшаются (падает сжимаемость (3^).
Расхождение между микрофлуктуациями плотности и
термодинамическими флуктуациями плотности возрастает.
Важной особенностью миркофлуктуаций плотности является их
корреляция. Малые области сжатия окружены столь же малыми
областями разрежения. Корреляция быстро затухает и на расстояниях
порядка 10 нм исчезает. Распределение микрофлуктуаций плотности
может быть не вполне симметричным. Естественно ожидать, что в
жидкостях, строение которых близко к плотной упаковке, математическое
ожидание микрофлуктуаций, ведущих к понижению плотности и
координационного числа, больше, чем для флуктуации противоположного
знака. В жидкостях с рыхлой структурой (вода) вероятность
появления малых сгущений больше, чем разрежений. Это подтверждается
модельными опытами Д. Бернала и С. Кинга [24] и расчетами
И. 3. Фишера и В.К- Прохоренко. Впрочем, согласно Берналу и
Кингу, в областях у, радиус которых равен dOi отклонение от
симметрии составляет всего около 6%.
§ 54. Флуктуации плотности и ассоциация
Как уже было сказано, связь между средним квадратом
флуктуации плотности <(Ар)2> и ассоциацией или комплексообразованием
существует. Но она не проста. В общем, появление устойчивых ассвци-
атов и комплексов, рост ассоциации сопровождаются уменьшением
изотермической сжимаемости 0, и, следовательно <(Ар)2>. Увеличиваются
критические температура Тк и давление Рк. Например, вода более
ассоциирована, чем метанол. Изотермическая сжимаемость воды при
20°С равна 4,7-10~12 Па"1, а метанола 12,1- 10~и Па"1, соответственно
критические температуры и давления воды Тк = 374,2° С, Рк =
= 222-105 Па, метанола Тк = 240° С, PK = 79J-105 Па. Другой"
пример — третичный бутанол значительно слабее ассоциирован, чем
нормальный бутанол. В соответствии с этим у третичного бутанола
Тк = 235°С, а у нормального Тк = 287,0°С. У органических
соединений и других молекулярных жидкостей энергия взаимодействия
молекул, как правило, не превышает 10—20 кДж/моль, поэтому и
сжимаемость таких жидкостей вдали от критического состояния при одинако-
Т Р
вых приведенных температурах 9 = и давлениях я = раз-
Тк Рк
личается не очень сильно. В жидких металлах энергия
взаимодействия между атомами на порядок больше. Соответственно этому и
сжимаемость жидких металлов на порядок меньше. Так, например,
изотермическая сжимаемость жидкой ртути при 20° С составляет всего
около 4,0-10"18 Па"1.
138
Вместе с тем отыскание количественной связи между средним
квадратом флуктуации плотности или сжимаемостью и ассоциацией
представляет собой сложную проблему. Это задача молекулярной теории
термодинамических свойств жидких фаз.
При постоянных Т и Р средняя флуктуация плотности в области v
любой изотропной жидкой фазы (однокомпонентной жидкости или
раствора) связана со спонтанными изменениями свободной энтальпии G
области v на величину, равную ks Т. Но изменение свободной энтальпии
G = АН—TAS. Изменения энтальпии АН и энтропии AS являются
сложными функциями ассоциации и комплексообразования.
§ 55. Адиабатические и изобарические флуктуации
плотности
До сих пор мы рассматривали флуктуации плотности как единое
целое. Но в действительности в жидкой фазе наблюдаются два типа
флуктуации плотности — адиабатические и изобарические.
Их свойства существенно различаются.
Плотность однокомпонентной жидкости р в области v есть функция
двух независимых переменных: давления Р и энтропии S; р = р(Р,
5), поэтому флуктуация плотности связана с флуктуациями Р и S
уравнением
дР/s ' \dS Jp
Так как S и Р независимые переменные, то их флуктуации
статистически независимы <APAS> = 0. Следовательно,
(VII.18)
Таким образом, средний квадрат флуктуации плотности слагается
из среднего квадрата адиабатических флуктуации плотности <(Ap)2>s
и среднего квадрата изобарических флуктуации плотности <сЛ(р)2>/>.
Можно показать [32], что
(VII.19)
(VII.20)
гдеа = — / J — термический коэффициент расширения жидкости;
СР — теплоемкость жидкости при постоянном давлении; р5 =—_.( J
— адиабатическая сжимаемость.
Когда жидкая фаза представляет собой раствор, то, чтобы
определить состояние системы, недостаточно задать значения переменных Р
и S. Необходимо определить концентрации независимых компонентов
съ •.., cv ..., сК. В этом случае флуктуация плотности Ар связана не
139
только с флуктуациями Р и 5, но и с флуктуациями концентрации
Aq, ..., Acit ..., Аск:
Индекс «с» в уравнении (VII.21) является сокращенным обозначением
условия постоянства концентраций всех независимых компонентов
с1у ..., ск. Так как флуктуации независимых переменных Р9 S и с.,
т. е. ДР, AS и Дс., статистически независимы:
то после возведения Др в квадрат и статистического усреднения, члены,
содержащие <ДРДс.>, <Д5Дс.> и <ДРД5>, исчезают. Поэтому
уравнения (VII.19) и (VII.20) справедливы и для растворов. Члены
вида | др ) <(Aq)2>, обусловленные флуктуациями концентрации,
будут рассмотрены позже.
Здесь следует сказать, что статистически независимы флуктуации
тех величин, которые выбраны в качестве независимых переменных,
характеризующих состояние системы. Обычно этот выбор диктуется
условиями эксперимента. Безотносительно к выбору независимых
переменных, характеризующих состояние системы, вопрос о
статистической независимости тех или иных флуктуации не имеет смысла. Так,
например, флуктуации плотности и флуктуации концентрации
статистически независимы, если в качестве независимых переменных,
определяющих состояние раствора, выбраны его плотность и концентрации
с. компонентов. Они статистически зависимы, если в качестве
независимых переменных, характеризующих состояние раствора, выбрать
его плотность и химические потенциалы ц. компонентов. Тогда
<ДрД^.>1А =^=0, где индекс ja означает, что флуктуации плотности и
концентрации рассматриваются при некоторых заданных значениях
химических потенциалов ц. независимых компонентов раствора. Такой
выбор независимых переменных может быть полезен, например, при
изучении влияния гравитационного поля на рассеяние света в растворах в
окрестности их критической области жидкость — пар. Некоторые
результаты подобных расчетов приведены в работе А. Д. Алехина,
А. 3. Голика, Н. П. Крупского, А. В. Чалого, Ю. И. Шиманского [33].
Расчеты выполнены с помощью термодинамического потенциала
F (Г, v, (j.) = jlxx Ч— Pv> предложенного М. А. Анисимовым [34].
Адиабатические флуктуации плотности по своей физической
природе эквивалентны адиабатическим сгущениям и разрежениям,
возникающим при распространении в жидкостях продольных звуковых волн.
В сущности, адиабатические флуктуации плотности есть затухающие
звуковые колебания, распространяющиеся в жидкости со скоростью
звука во всех направлениях от области возникновения флуктуации.
Ясно, что затухание этих волн происходит тем быстрее, чем больше
коэффициент их поглощения.
140
Известно, что затухающие звуковые колебания можно представить
как результат суперпозиции множества гармонических, т. е.
монохроматических, звуковых волн (фононов). Математически это можно
сформулировать, разлагая адиабатические флуктуации плотности
—) АР в трехмерный ряд Фурье:
dp /s
2
k
eikr , (VI1.22)
где к — волновое число, вектор, модуль которого равен 2л/Л; Л —
длина монохроматической звуковой волны; г — радиус-вектор,
начало которого отсчитывается от центра области v\ Др k — амплитуда
монохроматической звуковой волны. Она отыскивается с помощью
обратного преобразования Фурье:
A?k = — Г Ape~ikr dv. (V11.23)
Так как амплитуда Др* вещественна, то
Др*=Др*, (VI 1.24)
гдеДр %— комплексно-сопряженное значение Др и . Так как жидкость
изотропна, то
Др—к — Др*. (VI 1.25)
Итак,* благодаря адиабатическим флуктуациям плотности, в жидкости
распространяются звуковые волны, вызванные тепловым движением.
Они^могут быть обнаружены и изучены с помощью рассеяния света.
§ 56. Спектр света, рассеянного флуктуациями
плотности
Компоненты Мандельштама — Бриллюэна. Пусть на жидкость
падает монохроматический пучок света, волновой вектор которого равен
к0 (рис. 30). Будем рассматривать только ту часть рассеянного
излучения, которая обусловлена флуктуациями плотности. Предположим,
что рассеянное излучение (релеевское рассеяние), волновой вектор
которого равен к, наблюдается под углом б к направлению падающей
световой волны. Тогда в рассеянии принимают участие те
монохроматические звуковые волны, волновой вектор кф которых удовлетворяет
следующему соотношению:
кф = к0-Ь. (VII.26)
Физический смысл этого уравнения прост. Согласно квантовой
механике ^fe0, %k и %кф есть векторы количества движения фотоновр0, р
и фонона* рф. Умножая обе части уравнения (VII.26) на h, получим
квантовомеханическую формулировку закона сохранения количества
Движения. Волновые векторы к0 и А? образуют угол 9 (рис.31). По-
* Фононом называется квазичастица, соответствующая плоской
монохроматической звуковой волне, возникшей при тепловом возбуждении системы.
141
скольку модули k0 и k почти одинаковы, то
0
ИЛИ
а0 = 2/гЛ sin •
2 '
(VI 1.27)
где Л—длина волны фонона, рассеивающего свет.
Уравнение (VI 1.27) называют условием Брэгга. Оно может быть
найдено с помощью классической теории интерференции света,
рассеянного звуковыми волнами. Впервые на это обратил внимание французский
физик Л. Бриллюэн еще в 1914 г. Подробный анализ этого вопроса
был дан им в статье, опубликованной в 1922 г. В 1926 г. в несколько
иной форме проблема рассматривалась Л. И. Мандельштамом. Из
Рис. 30. Направления волновых
векторов падающего на жидкость
монохроматического света kQi рассеянного
света k и тепловой
монохроматической звуковой волны или фонона k^
[34] Ф
Рис. 31. К расчету
уравнения (VII.27)
этих работ следует, что в спектре света, рассеянного адиабатическими
флуктуациями плотности, должны наблюдаться две симметрично
расположенные компоненты. Максимумы этих компонент должны быть
смещены по отношению к частоте падающего излучения на величину,
равную
AV =
п аг . 6
±2/iv0— sin —
(VI 1.28)
где п — показатель преломления; с — скорость света в вакууме;
аг — скорость гиперзвуковой волны, длина которой Л определяется
уравнением (VII.27) (рис.32). Экспериментальное доказательство этого
теоретического результата принадлежит Е. Ф. Гроссу.
Изобарические флуктуации плотности. Гросс обнаружил, что кроме
компонент, предсказанных Бриллюэком и Мандельштамом, в
спектре рассеянного излучения имеется несмещенная (центральная)
компонента. Ее максимум соответствует частоте возбуждающего излучения
(рис. 32). Чтобы объяснить этот экспериментальный факт, Л. Д.
Ландау и Г. Плачек ввели представление об изобарических флуктуациях
142
плотности, вызванных флуктуациями энтропии (см. уравнение VII.20).
В отличие от адиабатических изобарические флуктуации плотности с
течением времени не перемещаются. Они в основном вызваны
локальными изменениями температуры (температурными волнами,
возникающими при появлении флуктуации плотности) [35]. Локальные
изменения температуры связаны с местными изменениями структуры — флук-
туацпями концентрации ассоциатов в однокомпонентных жидкостях,
ассоциатов и комплексов в растворах. В растворах эти флуктуации
не сопровождаются изменением концентраций независимых
компонентов системы, поэтому их не следует смешивать с флуктуация-
ми концентрации независимых компонентов.
Среднее время жизни изобарических флуктуации плотности
определяется коэффициентом температуропроводности %:
т ~ii/x = ££Lf (VII.29)
где х — коэффициент теплопроводности. Минимальные средние
времена жизни адиабатических и изобарических флуктуации плотности,
при которых термодинамическая
теория флуктуации еще
пригодна, можно оценить с помощью
неравенства (VI 1.6).
Рассмотрим в качестве
примера флуктуации плотности в
жидком бензоле при 300 К.
Полагая, что адиабатические
флуктуации плотности перемещаются
со скоростью звука, для бензола
равной 1,3-105 м/с, получим, что
время т пребывания
адиабатической флуктуации плотности в
области v на порядок превышает
10~13 с, если радиус области v не
меньше 1 нм.
Найдем теперь среднее время жизни изобарических флуктуации в
бензоле.
Коэффициент температуропроводности бензола составляет около
10~4 м2/с. Если радиус области v равен 1 им, то ее поверхность будет
порядка 4я/?2, т. е. около 10~17 м2. Из уравнения (VII.29) следует, что
среднее время жизни изобарической флуктуации плотности бензола в
такой области будет всего 10~13 с. Неравенство (VII.6) не выполняется.
Чтобы оно было удовлетворено, область v должна иметь радиус порядка
5 нм. Таким образом, в очень малых областях жидкости, меньших, чем
10~18 мл, изобарические флуктуации плотности исчезают быстрее, чем
адиабатические. Термодинамическая теория флуктуации плотности
применима к участкам жидкости, если их радиус составляет величину
порядка 10~8 м и более. Отметим, что при длине гиперзвуковых волн
А~Ю~8 м скорость теплообмена имеет тот же порядок, что и скорость
распространения звука. Поэтому в бензоле гиперзвуксвые волны, имею-
143
Рис. 32. Спектр релеевского триплета:
в центре компонента Гросса (или несмещенная
компонента); по обе стороны от нее симметрично
расположены компоненты Мандельштама — Брил-
люэна
щие частоту выше 1011 Гц, представляют собой не адиабатические, а
изотермические звуковые колебания.
Релеевский триплет. Итак, спектр тонкой структуры релеевского
рассеяния света (релеевский триплет) в чистых жидкостях обусловлен
адиабатическими и изобарическими флуктуациями плотности. В
растворах центральная компонента релеевского триплета, будем называть
ее компонентой Гросса (по имени открывшего ее в 1930 г. Е. Ф.
Гросса), зависит не только от изобарических флуктуации плотности, но и
от флуктуации концентрации. Изучая спектр центральной компоненты
релеевского триплета, изображенного на рис. 32, можно определить
коэффициент температуропроводности х и, если известно Ср,
—коэффициент теплопроводности х. Изучая спектр компонент
Мандельштама—Бриллюэна, получают сведения о скорости распространения и
коэффициенте поглощения звуковых волн [36]. Точность этих измерений
резко возросла с появлением газовых лазеров. Измерения проводятся
при углах рассеяния б, обычно превышающих 20—30°. В этих
условиях спектр компонент Мандельштама — Бриллюэна позволяет изучать
лишь гиперзвуковые волны, имеющие частоту порядка 1010 Гц. При
очень малых углах рассеяния в принципе можно было бы исследовать
скорость и поглощение звука в более широком диапазоне частот и
оптическим методом получать сведения о дисперсии скорости звука, т. е.
о зависимости скорости звука от частоты колебаний звуковых волн [37].
Отношение интегральной интенсивности компоненты Гросса
релеевского триплета к интегральной интенсивности двух компонент
Мандельштама—Бриллюэна согласно Л. Д. Ландау и Г. Плачеку равно:
2'м.б
■ —1. (VII.30)
М. С. Тунин в 1962 г. показал, что в тех случаях, когда при частотах
до 1010 Гц наблюдается дисперсия скорости звука (скорость звука
растет с частотой, а сжимаемости ^, ^ и теплоемкости Ср и Су
уменьшаются), уравнение (VII.30) дает правильные результаты, если р^, р„
Ср и Су уменьшить на величины, соответствующие наблюдаемой
дисперсии.
Многие из упомянутых здесь вопросов подробно излагаются в
работах [12, 36].
§ 57. Анизотропные флуктуации
Анизотропные флуктуации имеются в однокомпонентных
жидкостях и растворах, если в жидкой фазе есть анизотропные молекулы, или
анизотропные ассоциаты (что, в сущности, то же самое). Анизотропия
обычно наблюдается, если молекулы (или ассоциаты) не имеют шаровой
симметрии. Тогда их поляризуемость — тензор второго ранга (см.
гл. V). Нередко такие молекулы полярны, т. е. имеют постоянный ди-
польный момент. Связь между симметрией и ее дипольным моментом
подробно рассмотрена в монографии В. И. Минкина, О. А. Осипова,
Ю. А. Жданова [10]. Если молекула имеет центр симметрии, то это
144
достаточное (но не необходимое ) условие отсутствия у нее дипольно-
го момента. В общем же необходимо, чтобы векторная сумма диполь-
ных элементов всех полярных групп, присутствующих в молекуле,
была равна нулю. Это условие выполняется, например, для метана,
четыреххлористого углерода и других молекул, имеющих форму
правильного тетраэдра, хотя у этих молекул среди элементов симметрии
нет центра инверсии.
Спонтанный электрический момент жидкости. Рассмотрим кратко
виды анизотропных флуктуации, процессы их образования и раепада,
методы их изучения. Пусть имеется некоторый сферический участок v,
находящийся внутри большого объема V изотропной жидкости.
Размеры v примерно те же, что и при описании флуктуации плотности.
Жидкость может содержать любое число независимых компонентов.
Допустим, что в жидкой фазе некоторые из молекул или же все молекулы
полярны. Дипольные моменты полярных молекул могут быть
неодинаковы. Если жидкость изотропна, то средний электрический момент
области v должен быть равен нулю, М = 0. При возникновении в
области v анизотропной флуктуации электрический момент Mv этой
области будет отличаться от нуля. Распределение Mv является
нормальным. Плотность вероятности fMv появления в области v электрического
момента, модуль которого равен Mv, в соответствии с теорией
флуктуации, следует нормальному распределению:
fM,v = expj—--Д-1. (VH.31)
Таким образом, с ростом Mv вероятность его возникновения быстро
падает. Средний квадрат флуктуации электрического момента (УИ^) для
области v, находящейся внутри объема V низкомолекулярной
изотропной жидкости, определяется уравнением, вывод которого имеется
в монографии Г. Фрелиха [8]:
<Лр-*!»1. <*« + Ц('«-1> , (VII.32)
4я e
где ss — статическая диэлектрическая проницаемость. Уравнение
(VII.32) показывает, что при &s > I (Ml) прямо
пропорционально es. Физический смысл уравнения (VII.32) нетрудно понять. С этой
целью введем обозначение:
</п2> = <4яе,м2/(2е,+ 1)(е,-1)>. (VII.33)
Подставляя (VII.33) в (VII.32), получим:
i^l! = <m?> = *B7\ (VII.34)
v
Уравнение (VI 1.34) определяет величину статистического среднего
квадрата эффективного электрического дипольного момента единицы
объема жидкости {т\)у для создания которого надо затратить
работу, равную къ Т.
Mv в уравнении (VI 1.32) включает в себя не только тот
электрический момент, который непосредственно связан с анизотропным
распределение?*! постоянных дипольных моментов молекул, но и тот, который
индуцируется во всей жидкости под влиянием анизотропного
распределения полярных молекул в области v. Поляризация молекул,
расположенных вне области v, весьма существенно влияет на величину
Mv. Если бы сферическая область жидкости была окружена не такой
же жидкостью, а вакуумом, то связь между средним квадратом
спонтанного электрического момента (Л^)вакуум и г8 была бы совсем иной:
\ v /вакуум __ 9&Б Т ^ bs 1 (VI 1.35)
4~~ ' i7+~2 '
Формула (VI 1.35) показывает, что (Л^)вакуум в отличие от {M2v)
почти не зависит от sst если е5 > 1.
Теперь вернемся к сферическому участку жидкости v,
расположенному внутри большого объема V той же жидкости. Можно исключить
вклад в MV9 связанный с электронной и атомной поляризацией
молекул, находящихся в области v. Тогда вместо уравнения (VII. 32)
получится
где 8оо — вклад в es , связанный с электронной и атомной
поляризацией (см. гл. II); M'v обозначает только ту часть спонтанно
образующегося в v электрического момента, которая связана с
анизотропным распределением ориентации полярных молекул внутри v. Так же,
как и для Mv, здесь существенную роль играет среда, в которую
погружена интересующая нас область v. Если в жидкости нет полярных
молекул, то es =гоо и M'v обращается в нуль.
Величина <сМу> зависит от дипольного момента [х молекул,
имеющихся в жидкости. Если жидкость однокомпонентна, т. е. все
молекулы одинаковы, то <M^> = Nv <jx2>, где Nv — число молекул
в объеме v. Среднее статистическое значение квадрата дипольного
момента <|^2> связано с дипольными моментами ассоциатов и их
концентрациями уравнением (V.3).
Измеряя статическую диэлектрическую проницаемость жидкостей,
можно определять <Л^>, <ia2> и получать сведения о структуре
жидкостей, как об этом уже было сказано в гл. V. Изучая зависимость
диэлектрической проницаемости и диэлектрических потерь от
частоты электромагнитного поля (диэлектрическая радиоспектроскопия),
получают информацию о кинетике и механизме процессов образования
и разрушения тех анизотропных флуктуации, которые
сопровождаются появлением в области v электрического момента.
146
§ 58. Флуктуации диэлектрической проницаемости
Рассмотрим теперь более общий случай. Пусть жидкость содержит
анизотропные молекулы. Они могут быть и неполярными, как,
например, молекулы бензола или фенантрена. Если в области v появляется
анизотропная флуктуация, то диэлектрическая проницаемость этой
области представляет собой симметричный тензор второго ранга ег-.,.
Этот тензор состоит из скалярной части ebiK и симметричного
тензора Ае^:
е* = е^ + Де*, (VI 1.37)
где biK — единичный тензор; /, к = X,Y, Z, т. е. декартовы
координаты.
В уравнении (VII. 37) скаляр е слагается из средней
диэлектрической проницаемости жидкости г0 и флуктуации As скалярной
части тензора:
(VII.38)
Флуктуация скалярной части тензора As в свою очередь состоит из
флуктуации Аепл, обусловленных локальными изменениями
плотности, о которых мы говорили ранее, и флуктуации AsK,
вызванных случайными локальными изменениями концентраций
компонентов, если жидкая фаза состоит из двух или большего "числа
независимых компонентов:
Де = Дзпл + Дгк. (VI 1.39)
Флуктуации плотности и концентрации в среднем не нарушают
изотропии среды. Анизотропные флуктуации сопровождаются
появлением анизотропии диэлектрической проницаемости. В этом случае
компоненты симметричного тензора AstK не равны нулю:
(VI 1.40)
След тензора AeiK равен нулю. Так как жидкость ]в среднем
изотропна, т. е. все направления в ней равноправны, то < AeiK > = 0;
<(As^)2> одинаковы для всех 1фк\ <(Asa)2> одинаковы для
всех i. Величины < (AsfJ2 > можно . определить с помощью
рассеяния света. Пусть в направлении оси х, выбранной нами системы
декартовых координат распространяется плоская неполяризованная
монохроматическая световая волна. Длина волны X, интенсивность
потока света равна /0. Поток света рассеивается жидкостью, находящейся
в области v. Введем обозначения: i?aH — коэффициент рассеяния света
на анизотропных флуктуациях: Rx — коэффициент рассеяния света*,
* Коэффициентом рассеяния R называют величину, пропорциональную
отношению интенсивности / рассеянного света к /0:
147
электрический вектор которого колеблется параллельно оси X (рис. 33)
Если жидкость не содержит молекул полимеров и не находится в
состоянии, очень близком к критической точке, то при угле рассеяния 0 =
= 90°, т. е. при рассеянии в направлении оси Yу имеем [36]:
-
Рассеяние света в направлении оси Y обусловлено флуктуациями
ДAeYz и Aszx, но благодаря равенству всех < (Де^)2>
Падающий свет
Рассеянный
фет
чг
Рис. 33. Падающее излучение и рассеянное
излучение Eoz и Eqy компоненты электрического вектора
падающей монохроматической неполяризованной
световой волны:
Е% и Ех — компоненты электрического вектора рассеянной
световой волны; угол рассеяния 8 равен 90°
при i Ф к, уравнение (VII. 42) справедливо для всех i фк.
Статистическое среднее квадрата компонентов Ае^. тензора Аг{к связано с
7?ан уравнением
(VII.43)
Если рассеяние наблюдается в направлении оси У, то / =Z и, как уже
говорилось, все <(Двп)2> одинаковы.
§ 59. Поперечный звук.
Эффект Леонтовича—Фабелинского
При появлении анизотропных флуктуации в некоторых
сравнительно вязких жидкостях возникают упругие напряжения и деформации.
В жидкости распространяются быстро затухающие поперечные
колебания (поперечный звук). Частицы жидкости колеблются в плос-
где V — объем рассеивающей среды; г — расстояние от центра рассеивающего
объема до точки наблюдения рассеянного излучения. Для #ан в (VI 1.41) надо /
заменить на / —интенсивность света, рассеянного анизотропными флуктуациями.
Для Rx надо в (VI 1.41) заменить / на Iх.
148
кости, перпендикулярной направлению распространения деформации.
В 1941 г. М. А. Леонтович показал, что это должно вызывать
расщепление х-компоненты релеевского рассеяния света на два максимума,
симметрично расположенных по отношению к частоте v0
возбуждающей световой волны. Свет, рассеянный на флуктуациях анизотропии,
может менять свою частоту, если эти флуктуации вызывают
распространение поперечных звуковых волн.
Затухающие поперечные звуковые волны можно
разложить в спектр Фурье. Каждая компонента
такого спектра представляет собой
монохроматическую волну поперечного звука. Если
падающее излучение монохроматизировано и
его волновой вектор равен k0, то волновой
вектор рассеянного излучения должен
удовлетворять условию (VII. 26). При угле
рассеяния 6 вектор света, рассеянного
поперечной звуковой волной так же, как и при
рассеянии на продольных звуковых волнах (см.
§ 56), должен состоять из двух линий,
смещенных относительно частоты
возбуждающего излучения v0 на величину ±Av:
42 Of
ОЛ
а± • °
Д v = 2n v0 — sin — ,
(VI 1.44)
О 0,1
V, см'1
Рис. 34. Эффект Леонто-
вича — Фабелинского в
хинолине при 20° С
(расщепление х- компоненты
спектра Релея)
где а±— скорость распространения
поперечной звуковой волны; с — скорость света в
вакууме. Впервые расщепление спектра
л;-компоненты линии Релея, рассеянной в нитробензоле, хинолине и анилине,
наблюдалось И. Л. Фабелинскими его сотр. [38] (рис. 34). Это явление
можно именовать эффектом Леонтовича — Фабелинского.
§ 60. Процессы, приводящие к появлению
анизотропных флуктуации
Чтобы анизотропные флуктуации существенно повлияли на
свойства жидкости и могли, следовательно, быть замеченными, достаточно
изменить ориентацию лишь небольшого числа частиц. Например,
если только одна из каждой 10 000 полярных молекул жидкости
ориентирована в некотором определенном направлении, а остальные
распределены изотропно, то и в этом случае возникает электрический
момент My поле которого будет иметь напряженность, равную примерно
100 В/см. Для переориентации одной' молекулы в среднем требуется
время не менее 10"12 с. Неравенство (VII. 6) выполняется. Сведения о
механизме процессов образования анизотропных флуктуации были
получены в основном с помощью диэлектрической
радиоспектроскопии и рассеяния света. Анизотропные флуктуации могут возникать
(или исчезать) в ходе следующих процессов.
а. Изменение конформации молекулы, сопровождающееся
изменением величины или переориентацией дипольного момента молекулы
149
[х или изменением анизотропии тензора поляризуемости молекулы
72. Таковы, например, превращения цис-формы молекул уксусной
кислоты в транс-форму вследствие переориентации группы ОН.
Процессы перехода молекул поворотных изомеров друг в друга
находятся в динамическом равновесии. Если в каком-либо изотропном
элементе объема жидкости одна из молекул под влиянием теплового
движения случайно меняет свою конформацию, так что меняется [х или
72 (или (х и f2), то в этом элементе объема возникает анизотропная
флуктуация. Через некоторое время динамическое равновесие
восстановится благодаря изменению конформаций той же или других
молекул. Исчезает и анизотропная флуктуация в данном элементе
объема. Но появятся анизотропные флуктуации в других участках
жидкости. Эти процессы протекают непрерывно, если состояние жидкости
таково, что они могут происходить.
Одним из превращений того же типа являются инверсионные
переходы в молекулах аммиака, диметиламина и некоторых других.
Молекула аммиака имеет форму симметричной пирамиды. При
инверсионном переходе атом азота проходит через плоскость, в которой
находятся атомы водорода, и располагается по другую сторону от
этой плоскости. Молекула NH3 как бы выворачивается наизнанку.
Ее дипольный момент меняет направление на противоположное. Если
инверсионный переход молекулы NH3 произошел в изотропном
элементе объема жидкости, то возникает анизотропная флуктуация.
Обратный процесс приведет к ее исчезновению.
б. Изменение ориентации молекул в ходе теплового движения.
Сюда относятся два вида процессов: вращательные качания и
броуновские переориентации молекул. Вращательные качания есть
неупорядоченные малые изменения ориентации молекулы около
положения ее равновесия. Они не сопровождаются перераспределением
положений соседних молекул и не связаны с преодолением потенциального
барьера. Броуновские переориентации — скачкообразные, резкие
изменения ориентации молекулы. Обычно они сопровождаются
перераспределением положений соседних частиц и поэтому связаны с
преодолением некоторого потенциального барьера.
в. Процессы образования и разрушения ассоциатов и
комплексов, сопровождающиеся изменением электрического момента
жидкости или анизотропии ее поляризуемости. Таковы, например,
процессы образования и разрушения ассоциатов бензола, спиртов,
уксусной кислоты и т. д. Как правило, эти процессы сопровождаются
изменением ориентации молекул или их частей.
Изменения конформаций, броуновские переориентации,
процессы образования и разрушения ассоциатов и комплексов протекают с
преодолением некоторого потенциального барьера. Для этого нужно
чтобы молекулы, непосредственно участвующие в соответствующем
элементарном акте (обычно это одна-две молекулы), приобрели
энергию, достаточную для перехода через энергетический барьер.
Обычно этот барьер в несколько раз превышает кв Т.
Механизм преодоления энергетического барьера в ходе указанных
процессов еще слабо изучен. Здесь только отметим следующее. Если
150
энергетический барьер высок по сравнению с кв Т, то в накоплении
энергии принимает участие большое число молекул. В этом случае каждый
элементарный акт (изменение конформации, разрыв Н—связи и т. д.)
связан с изменением состояния многих молекул. Такие процессы
носят кооперативный характер. Если же энергетический барьер не очень
велик по сравнению с къ Т, то число молекул, меняющих свое состояние
б ходе отдельного элементарного акта, невелико. Такие процессы не
имеют кооперативного характера и подчиняются хорошо известным
законам химической кинетики. Конечно, оба типа процессов сосуществуют
друг с другом и между ними нет резкой границы. При более высоких
температурах преобладают некооперативные превращения. При
низких температурах на первый план выходят кооперативные процессы.
Термин «высокая» и «низкая» температуры здесь имеют смысл только
применительно к определенной жидкой фазе.
§ 61. Флуктуации концентрации
Впервые существование флуктуации концентрации было
установлено М. Смолуховским на основании теоретического анализа работ
Д. И. Коновалова о давлении насыщенного пара растворов. Работа
М. Смолуховского была исходным пунктом исследований А.
Эйнштейна и многих других авторов по вопросам теории флуктуации и
рассеяния света флуктуациями. Пусть N±n N2 — числа молекул
компонентов 1 и 2 в замкнутой области v раствора. Состав двухкомпо-
нентного раствора будем характеризовать отношением с = NJN^
Область v содержит достаточно большое число молекул, чтобы ее
состояние можно было описать с помощью термодинамических функций.
Флуктуации концентрации Д с = с — <С£> подчиняются
нормальному распределению. Пусть Р2 означает парциальное давление
насыщенных паров компонента 2, т. е. паров, находящихся в
термодинамическом равновесии с раствором. Согласно термодинамической
теории флуктуации
д1пР*\ (VII.45)
дс )pt т
где Nx — среднее число молекул компонента / в области v. Если
концентрации выражать в мольных долях х2 компонента 2, то уравнение
(VII. 45) примет вид:
«^'Ь- ' (VIM6)
A
дх2 Jpt т
где N = N± + Л^2 — среднее число молекул раствора в области v.
Для многокомпонентных растворов обобщение уравнения (VII.45)
имеется в монографии Хилла [39]. Работа образования флуктуации
концентрации Ас или Дл:. пропорциональна производной д\пР2/дс
или dlnPi/dx.. Чем меньше эта производная, тем больше средний квад-
151
рат флуктуации. Для идеального раствора, парциальное давление
насыщенных паров компонентов которого следует закону Рауля
Pi = Poi*i> 0"= 1,2) (VII.47)
производная д\пР2/дх2 = Vx2 и
ид
N
(VI 1.48)
Если парциальное давление насыщенных паров компонентов
раствора Р. меньше, чем Poixt (отрицательные отклонения от идеальности)
(рис. 35), то д1пР2/дх2 > х~1 и <(Д*.)2> меньше <(Дл:.)2 >„.
В тех случаях, когда парциальные давления компонентов раствора
больше, чем Poi х. (положительные отклонения от идеальности), статисти-
Р,АУ мм рт. ст. Р?Ь мм рт. ст. в ммрпгспг^
U1200°C I I t=35,17°C
110
Ag 0,2 Ofi 0,6 Ofi РЪ Сщ 02 о,4 0,6 0,8(CH3)2C0
а Хрь б *(си3)2со
Рис. 35. Парциальные давления насыщенных паров,
находящихся в равновесии с растворами:
а — серебро — свинец при 1200°С [40] (положительные отклонения от
идеальности); б — хлороформ — ацетон при 35,17°С [41] (отрицательные
отклонения от идеальности); концентрация х выражена в молярных долях
ческое среднее квадрата флуктуации концентрации больше, чем у
идеального раствора.
Сопоставим значения средних относительных флуктуации
концентрации Ъхг = Y((KxiJz) I xty рассчитанные по формуле (VII. 46), с ве-
личинами средних относительных флуктуации плотности ор = |/"((Ар)2)/р,
вычисленными по формуле (VII. 9). С этой целью воспользуемся
данными для растворов ацетон—нитробензол и ацетон — четыреххло-
ристый углерод, приведенными в монографии [9]. В этих растворах
наблюдаются положительные отклонения от идеальности, но критичес-
152
<(Axf>W*
10
кой точки расслаивания нет. При объеме v == 10~18 мл и 25°С имеем
для ацетона 5р = 2,3 • 10~3, для нитробензола Ьр = 1,3 • 10~3 и для
четыреххлористого углерода Ьр =2,1 • 10"3. При х = 0,5 и 20°С для
растворов ацетон — нитробензол &р = 1,7 • 10~3 и 8я = 1,7 • 10~2; для
растворов ацетон—четыреххлористый углерод 5р =2,1 • 10~3 и Ьх =
= 1,7- Ю'2.
В области концентраций х ~ 0,5, где ((Дх.)2) для указанных
растворов наиболее велико (рис. 36), средний квадрат относительных
флуктуации концентрации больше,
чем средний квадрат
относительных флуктуации плотности в 65
раз для растворов ацетон —
четыреххлористый углерод и в 40 раз
для растворов ацетон —
нитробензол. Относительные флуктуации
концентрации в этих растворах при
20°С имеют примерно ту же
величину, что и относительные
флуктуации плотности в чистых жидкостях
при состояниях, близких к
критической точке. Например, для воды
вблизи критической точки (Тк =
= 374,2°С, Рк = 222 . 105 Па) при
Т - 360°С и /> = 2 • 107 Па
изотермическая сжимаемость (3, ==
(СН5)2С0 0,25
При v = 10 18мл
Рис. 36. Зависимость среднего
квадрата флуктуации концентрации
<(Ах)21> от состава растворов,
выраженного в мольных долях х:
1 — растворы ацетон — нитробензол; 2 —
растворы ацетон — четыреххлористый углерод;
объем у, в котором рассматриваются
флуктуации концентрации, равен 10~16 см3 [91
-=3,76-10"8 Па"1
получаем Ьр = кв Гр^ = 1,4 X
X 10~2.
Отметим, что если бы растворы
ацетона в четыреххлористом
углероде и нитробензоле были бы
идеальными, порядок величины
средних флуктуации концентрации остался бы тем же. В обоих случаях
при х = 0,5 мы получили бы 8л; = 1,2 • 10~2.
Средние относительные флуктуации концентрации в нерасслаива-
ющихся растворах велики в окрестности х == 0,5. Они наиболее велики
при значительных положительных отклонениях от идеальности, т. е.
Ясно, что присутствие устойчивых и больших
когда
dxt
флуктуации концентрации качественно отличает строение многих
растворов от структуры однокомпонентных жидкостей.
§ 62. Флуктуации концентрации в окрестности
критической точки расслаивания
В некоторых растворах при понижении температуры происходит
расслаивание. Иногда оно возникает при повышении температуры
(рис. 37). Точка на кривой равновесия двух жидких фаз, в которой обе
фазы становятся тождественными по своим свойствам и расслаивание
153
исчезает, называется критической точкой расслаивания. Она
характеризуется критической температурой Тк и критической концентрацией
хк. Различают верхнюю критическую точку расслаивания (ВКТ) и
нижнюю точку (НКТ) в зависимости от того, когда появляется
расслаивание, при понижении температуры (ВКТ) или при ее повышении
(НКТ). Верхняя и нижняя критические точки
расслаивания меняются с давлением, хотя и слабо.
При изменении давления на 105 Па критическая
температура расслаивания обычно смещается на
сотые доли градуса. В одних случаях наблюдается
повышение 7К, в других — понижение. В
критической точке расслаивания (ВКТ и НКТ) на
кривых, отображающих зависимость парциального
давления насыщенного пара /-того компонента Р.
от его концентрации х.у возникают точки перегиба!
Следовательно, в критической точке расслаивания
дРг д*Рг
= 0; -ТТ" = О; (' = 1,2). (VII.49)
'Н20 0,1 0,2Х
dxt
В окрестности критической точки расслаивания
раствора работа, требующаяся для образования
Рис. 37. Диаграм-
н"вес^я°в°жидаГх флуктуации концентрации, очень мала. Статисти-
растворах вода — ческое среднее квадрата флуктуации концентрации
метил-4-шшеридин возрастает. Даже малые локальные изменения со-
с верхней и ниж- СТояния раствора оказывают заметное влияние на
ней кпитическими
точками расслаи- состав сравнительно больших его участков. Иначе
вания (ВКТ и говоря, возрастает радиус корреляции флуктуации
НКТ) концентрации. В окрестности критической точки
при (Т—Тк) ~ 1—2° флуктуации концентрации
встречаются так часто, что лучи света,
попадающие в раствор, нередко испытывают многократное рассеяние, прежде
чем выйти наружу. Поэтому раствор становится мутным.
Наблюдается критическая опалесценция. Постепенно радиус корреляции
флуктуации концентрации L достигает величин порядка 10"7.м,
сравнимых с длиной волны света. Тогда при рассеянии света
возникают отклонения от закона Релея. При устранении помех,
связанных с многократным рассеянием, и тщательном термостатировании
отклонения от закона Релея нередко наблюдаются лишь в узком
интервале температур, при \Т—7'к|<0,1 [42]. Растворы с развитыми
флуктуациями концентрации похожи на дисперсные системы с очень
малыми неоднородностями. Отличие от обычных дисперсных систем
состоит в том, что флуктуации концентрации неустойчивы. Они
случайно возникают и быстро исчезают. Среднее время их существования ^
обратно пропорционально коэффициенту диффузии. Исследования,
выполненные автором и его сотр. [43], показали, что в растворах с
положительными отклонениями от идеальности, состояние которых
далеко от критической точки расслаивания, т может лежать в интервале
10~5 — Ю~и с. Время 10~б с само по себе очень малое, в молекулярных
154
временных масштабах весьма велико. В окрестности критической точки
расслаивания х быстро растет. При Т-* Тк среднее время жизни
флуктуации стремится к бесконечности.
§ 63. Микрофлуктуации концентрации
Когда температура раствора отличается от Тк более чем на ~ 0,2 К,
длина радиуса корреляции L обычно не превышает 10~8 м. Флуктуации
концентрации представляют собой неоднородности в среднем малых
размеров, поэтому рассеяние света следует закону Релея.
Интенсивность рассеянного света / ~ >г4. При возрастании \Т—Тк\ средние
размеры флуктуации концентрации постепенно уменьшаются. Точнее
сказать, радиус корреляции постепенно сокращается до величин
порядка 1 нм.
Попытаемся охарактеризовать статистическое среднее квадрата
флуктуации концентрации ((Д#.)2) в очень малых элементах
объема v ~ 10~21 мл, радиус которого равен радиусу первой
координационной сферы.
Среднее время т жизни флуктуации концентрации в столь малых
элементах объема, очевидно, должно зависеть от скорости диффузии.
Оно не может быть меньше среднего времени, требующегося для
перескока молекулы из одного положения равновесия в другое.
Экспериментальные данные показывают, что среднее время, проходящее между
скачками молекулы из одного места в другое (соседнее), при Т = 300 К
для низкомолекулярных жидкостей равно 10"11 — 10~12 с.
Следовательно, даже для флуктуации в объемах порядка 10~21 мл, т. е. микрофлук-
туаций концентрации, условие (VII. 6) соблюдается. Следовательно,
может наблюдаться заметное влияние микрофлуктуаций концентрации
на термодинамические свойства вещества. Время, требующееся для
поляризации низкомолекулярных маловязких жидкостей при
наложении внешнего поля, обычно не превышает 10"11 с. Поэтому, когда
раствор с развитыми флуктуациями концентрации находится в
электрическом поле, его поляризация, а следовательно, и диэлектрическая
проницаемость ведут себя так, как если бы раствор представлял
собой обычную дисперсную систему с неоднородностями очень малых
размеров. Диэлектрическая проницаемость zs такой системы
уменьшается. Автором показано, что уменьшение диэлектрической
проницаемости Ass зависит от статистического среднего квадрата
микрофлуктуаций концентрации:
<'*-">'<(**)■> . (VII.50)
Здесь состав двухкомпонентного раствора выражен в объемных долях
ip r^ V2!{V1 + V2)y где V± и V2 — объемы компонентов до их
смешения; esl и es2 — статические диэлектрические проницаемости
индивидуальных жидкостей; г3 — статическая диэлектрическая
проницаемость раствора. Температура, при которой определяются
величины es, esl и ej2, одна и та же. Снижение es достигает 20%.
Подробный анализ этого явления имеется в монографии [9]. Пользу-
155
ясь уравнением (VII. 50), можно изучать микрофлуктуации, линейные
размеры которых близки к диаметру первой координационной сферы.
Статистическое среднее квадрата микрофлуктуаций концентрации
((Дл:)2) может достигать значений порядка 0,15. Средняя
абсолютная величина микрофлуктуаций концентрации |/<(Дл-)2)^ 0,4. Она
близка к предельному значению |Дх| == 0,5, при котором микрофлуктуации
концентрации представляют собой области чистых компонентов 1 и 2.
Для идеальных растворов статистическое среднее квадрата
микрофлуктуаций концентрации можно рассчитать по уравнению (VII. 48).
При х = 0,5 оно максимально и равно си 0,02, т. е. на порядок
меньше, чем в растворах с положительными отклонениями от идеальности.
Подобно микрофлуктуациям плотности микрофлуктуации
концентрации слабо зависят от температуры. Они немного увеличиваются в
окрестности критической точки расслаивания. Для растворов
неэлектролитов, имеющих критическую точку расслаивания или большие
положительные отклонения от идеальности, микрофлуктуации
концентрации в области их максимума близки по величине {(Ах)2) ~
~ 0,15. Влияние микрофлуктуаций концентрации на
диэлектрическую проницаемость существенно осложняет анализ строения
растворов с помощью данных об es. Некоторые аспекты этого вопроса
рассматриваются Ю. Я- Фиалковым [44]. Приведенные здесь оценки
средней величины микрофлуктуаций концентрации являются
приближенными. Они имеют, в основном, качественный характер.
§ 64. Флуктуации концентрации и строение
жидких растворов эвтектического состава
Как известно, жидкий раствор
эвтектического состава (жидкая
эвтектика) может находиться в
равновесии с твердыми фазами, число
которых равно числу компонентов
системы. На рис. 38 изображена
диаграмма плавкости с
эвтектической точкой двухкомпонентной
системы. При затвердевании жидкой
эвтектики образуется твердая
эвтектика. Она имеет своеобразную
пластинчатую или зернистую
микрогетерогенную структуру.
Области (твердые растворы), содержащие
избыток компонента У,
перемежаются с областями (твердыми
растворами), имеющими избыток
компонента 2. Когда компоненты 1 и 2
не образуют твердых растворов, то
твердая эвтектика состоит из
областей твердого компонента / и
твердого компонента 2. Размеры плас-
1100
60
80 Си
Вес. % Си
700
Рис. 38. Диаграмма плавкости
двухкомпонентной системы серебро —
медь [45]:
Аи В— точки плавления чистых
компонентов; Е — эвтектическая точка; С и D —
равновесные концентрации твердых растворов
меди в серебре и серебра в меди,
образующихся в эвтектическом сплаве (заштрихованы
области существования твердых растворов)
156
тинок или зерен твердой эвтектики зависят от скорости
замораживания жидкой эвтектики. Быстрое охлаждение ведет к уменьшению их
размеров до величин порядка 10"7 м.
Подробное описание свойств и строения эвтектических сплавов
имеется в кн. В. Я- Аносова и С. А. Погодина [45]. Иногда
высказывается предположение, что жидкие растворы эвтектического
состава имеют особую «микрогетерогенную» или «квазиэвтектическую»
структуру. В действительности же нет прямой связи между средней
величиной флуктуации концентрации и эвтектической структурой
(М. И. Шахпаронов [46]). Жидкий эвтектический раствор может
подчиняться закону Рауля, в нем могут наблюдаться положительные
или даже отрицательные отклонения от идеальности. Твердая
эвтектика во всех этих случаях будет иметь описанную выше структуру.
Термодинамические свойства жидкого раствора эвтектического
состава не имеют никаких особенностей. Производная д\пР2!дх2 не
претерпевает никаких существенных изменений. Флуктуации концентрации
в эвтектическом растворе могут быть большими или малыми и
существенно не отличаются от флуктуации в обычных растворах. С этим
согласуются результаты исследований В. М. Глазова [47, 48]. Это было
экспериментально подтверждено Г. П. Рощиной и Э. Д. Ищенко,
которые исследовали рассеяние света в расплавах эвтектического
состава нафталин — дифенил, фенол — монохлоруксусная кислота и
другие [49] и также в работе [50], где строение жидкой эвтектики
нафталин — бензойная кислота определялось рентгенографически
(В. В. Шилов, Н. Н. Миненко, А. К. Дорош, А. Ф. Скрышевский,
Г. И. Баталии). При изучении растворов, в особенности
металлических сплавов, рентгенографическими и другими методами иногда
выдвигается гипотеза о существовании «квазиэвтектической структуры».
В этих жидких системах, видимо, имеются положительные отклонения
от идеальности. Они сопровождаются большими микрофлуктуациями
концентрации, что влияет на результаты рентгеновских и других
измерений.
Характерная структура твердых эвтектических сплавов
обусловлена не тем, что в жидкости имеется уже готовая квазиэвтектическая
смесь, а особыми условиями кристаллизации в эвтектической точке,
так как здесь одновременно кристаллизуются две или несколько
твердых фаз. При этом большое значение имеют явления пересыщения.
Благодаря задержке в кристаллизации и возникновению локальных
областей пересыщенных растворов то одного, то другого компонента,
образуется твердая эвтектическая структура как в случае, когда
флуктуации концентрации в жидкой фазе велики, так и когда они очень
малы.
Первое и, возможно, до сих пор единственное систематическое
исследование механизма и кинетики кристаллизации сплавов
эвтектического типа было выполнено около 40 лет назад А. А. Бочваром [51].
Согласно А. А. Бочвару, эвтектическая кристаллизация происходит
только в тех областях, где обе твердые фазы соприкасаются друг
с другом и с раствором.
157
К сожалению, возможности эксперимента в те годы были невелики.
А. А. Бочвар в течение 8 лет искал бинарную систему, процесс
кристаллизации которой можно было бы детально изучать фотографическим
методом. Для этого оба компонента должны были быть прозрачны,
давать эвтектические сплавы, иметь разную окраску, образовывать
эвтектику с достаточно малой линейной скоростью кристаллизации,
чтобы можно было производить фотографирование растущих
кристаллов. Оба жидких компонента должны были бы легко переохлаждаться
без кристаллизации, плавились бы при невысокой температуре и,
наконец, не разлагались при нагревании. Лучше других этим условиям
удовлетворяла система азобензол (СбНб — N = N — С6Нб) — пипе-
( ° ^
( /ч \
ронал I СН2<^ р>С6Н3СНО I . Температура плавления азобензола 68° С,
а пиперонала 37° С. Эта система и была изучена А. А. Бочваром.
Описание других объектов основано в значительной мере на
экстраполяции выводов, полученных в ходе изучения этой системы,
поэтому механизм кристаллизации эвтектических сплавов, вероятно,
требует дальнейших исследований. Как нам представляется, можно
высказать следующие предположения.
Если вязкость жидкого сплава мала, ближний порядок жидкой
фазы близок к ближнему порядку твердой фазы, способность к
образованию пересыщенных растворов невелика — твердая эвтектика
даже при малых скоростях охлаждения может иметь мелкозернистую
структуру. Если одно или несколько перечисленных выше условий не
выполняется, твердая эвтектика при малых скоростях охлаждения
может иметь крупнозернистую структуру, обусловленную повышенной
способностью жидкого сплава к образованию пересыщенного
состояния.
Литература
1. М. И. Шахпаронов, Н. Г. Шленкина. Вестн. Моск. ун-та.
Сер. хим., 12, 398, 1971. Н. Г. Шленкина, М. И. Шахпаронов
Вестн. Моск. ун-та. Сер.хим., 12, 522, 1971.
2. Е. G. Cox., D. W. J. С г a i с k s h а п к, J. А. С. Smith. Ргос.
Roy. Soc, A247, 1, 1958. Е. G. Cox. Rev. Mod. Phys. 30, 159, 1958.
3. М. И. Шахпаронов. Сб. «Физическая адсорбция из
многокомпонентных фаз». М., «Наука», 1972, стр. 66.
4. В. М. Татевский. Квантовая механика и теория строения молекул.
Изд-во МГУ, 1965.
5. Е.Н. Еремин. Основы химической кинетики в газах и растворах. Изд-
во МГУ, 1971.
6. Ф. Росотти, X. Росотти. Определение констант устойчивости и
других констант равновесия в растворах. М., «Мир», 1965.
7. Г. Б. Сергеев, Н. Ф. К а з а н с к а я, Б. М. Ужинов,
В. И. П а п и с о в а, В. С. Г у р м а н, Г. Б. М е л у з о в а, С. В. 3 е н и н,
В. В. Романов. Экспериментальные методы химической кинетики. Под ред.
Н. М. Эмануэля. М., «Высшая школа», 1971.
8. Г. Ф р е л и х. Теория диэлектриков. М., ИЛ, 1960.
9. М. И. Ш а х п а р о н о в. Методы исследования теплового движения
молекул и строения жидкостей. Изд-во МГУ, 1963.
158
10. В. И. М и н к и н, О. А. О с и п о в, Ю. А. Жданов. Дипольыые
моменты в органической химии. Л., «Химия», 1968.
П. М. И. Ш а х п а р о н о в, П. К. К а с ы м х о д ж а е в, В. В. Ле-
п и н М. И. Л у п и н а. Сб. «Физика и физико-химия жидкостей», Вып. 2.
Изд-во МГУ, 1973, стр. 11.
12. М. И. Ш а х п а р о н о в. Об основных вопросах практики и теории
речеевского рассеяния света в жидкостях. Сб. «Современные проблемы
физической химии». Т. 5. Изд-во МГУ, 1970, стр. 3.
13. А. Ф. С к р ы ш е в с к и й. Структурный анализ жидкостей. М.,
«Высшая школа», 1971.
14. Сб. «Физика простых жидкостей. Экспериментальные исследования»
Под ред. Г. Темперли, Дж. Роулинсона, Дж. Рашбрука. М., «Мир», 1973. 1
15. Н. Г. М а р ч. Жидкие металлы. Под ред. В. М. Глазова.
«Металлургия», М., 1972.
16. Б. И. Хруще в. Структура жидких металлов. Изд-во ФАН. Уз. ССР,
1970.
17. А. М. Е в с е е в, Г. Ф. Воронин. Термодинамика и структура
жидких металлических сплавов. Изд-во МГУ, 1966.
18. A. Eisenstein, N. S. Gingrich. Phys. Rev. 62, 261, 1942.
19. И. П. Павлоцкий. Вестн. МГУ. Сер. хим., 13, 306, 1972.
20. Н. Н. Боголюбов. Проблемы динамической теории в статической
физике. ОГИЗ, Гостехиздат, М.—Л., 1946; Н. Н. Боголюбов. Избранные
труды в трех томах. Т. 2, Киев, «Наукова думка», 1970.
21. И. 3. Фишер. Статистическая теория жидкостей. М., Физматгиз, 1961.
22. А. М. Е в с е е в, М. Я. Ф р е н к е л ь, А. Н. Ш и н к а р е в. Сб.
«Физика и физико-химия жидкостей». Вып. 1. Изд-во МГУ, 1972, стр. 125.
23. И. В. Радченко. УФН, 17, 318, 1937.
24. Сб. «Физика простых жидкостей». Статистическая теория. Под ред.
Г. Темперли, Дж. Роулинсона, Дж. Рашбрука. М., «Мир», 1971.
25. Л. М. Б р е х о в с к и х. ЖЭТФ, 12, 287, 311, 1942.
26. Критические явления и флуктуации в растворах. Труды совещания
(январь 1960 г.). М., Изд-во АН СССР, 1960.
27. Л. Д. Ландау, Е. М. Л и ф ш и ц. Квантовая механика.
Нерелятивистская теория. М., Физматгиз, 1963.
28. М. А. Л е о н т о в и ч. Статистическая физика. ОГИЗ, Гостехиздат,
1944.
29. В. П. С к р и п о в, Ю. Д. Колпаков. Критическая опалесденция
в системе жидкость — пар. «Современные проблемы физической химии». Изд-во
МГУ, 1970, т. 5, стр. 295.
30. В. П. Скрипов. Метастабильная жидкость. М., «Наука», 1972.
31. И. 3. Ф и ш е р, В. К. Прохоренко. Сб. «Критические явления
и флуктуации в растворах». Под ред. М. И. Шахпаронова. М., Изд-во АН СССР,
1960; В.К. Прохоренко, О. Я. Самойлов, И. 3. Фишер. Докл.
АН СССР, 125, 396, 1959.
32. Я- П. Т е р л е ц к и й. «Статистическая физика». М., «Высшая школа»,
1966.
33. А. Д. Алехин, А. 3. Голик, Н. П. К р у п с к и й, А. В.
Ч ал ы й, Ю. И. Ш и м а н с к и й. Сб. «Физика жидкого состояния». Киев,
«Высща школа», 1973, стр. 65.
34. М. А. А ни симов. ЖФХ, 45, 1551, 1971.
35. Л. Д. Л а н д а у, Е. М. Л и ф ш и ц. Электродинамика сплошных
сред. М., Физматгиз, 1959, стр. 502.
36. И. Л. Ф а б е л и н с к и й. Молекулярное рассеяние света. М.,
«Наука», 1965.
37. М. И. Ш а х п а р о н о в. Докл. АН СССР, 174, 69, 1967.
38. В. С. Старунов, Е. В. Т и г а н о в, И. Л. Ф а б е л и н с к и й.
Письма ЖЭТФ, 5, 317, 1967; В. С. С т а р у н о в, И. Л. Ф а б е л и н с к и и.
Тепловое и вынужденное молекулярное рассеяние света. Сб. «Современные
проблемы физической химии». Изд-во МГУ, 1970, т. 5, стр. 175.
39. Т. Хил л. Статистическая механика. Принципы и избранные
положения. М., ИЛ, 1960.
159
вып
М.
М.
. 4
И.
43
Г.
44
45
40. А. А. Г р а н о в с к а я, А. П. Любимов. ЖФХ, 27, 1437, 1953
41. J. v о п - Z a we d i s k i. Zs. ph. Chem., 35, 129 (1900).
42. Д. К- Б е р и д з е, М. И. Шахпаронов. Уч. зап. МОПИ, 92,
' 49, 1960; Укр. физ. журн. 7, 771, 1962; М. А. Анисимов
Шахпаронов. ЖФХ, 40, 2331, 1966.
М. И. Шахпаронов, Ю. Г. Шорошев, С. С. Алиев
Халиулин, П. К. Хабибуллаев. ЖФХ, 43, 2543, 1969.
Ю. Я. Фиалков. Двойные жидкие системы. Киев, «Техника», 1969.
В. Я. Аносов, С. А. П о г о д и н. Основные начала
физико-химического анализа. М. — Л., Изд-во АН СССР, 1947.
46. М. И. Шахпаронов. Изв. АН СССР «Металлургия и топливо»
№ 3, 118, 1961.
47. В. М. Г л а з о в, А. А. В е р т м а н, Е. Г. Ш в и д к о в с к и й.
Изв. АН СССР, «Металлургия и топливо», № 3, 104, 1961.
48. В. М. Г л а з о в, А. А. В е р т м а н. Сб. «Исследование сплавов
цветных металлов», вып. IV. Изд-во АН СССР, 1963, стр. 85.
49. Г. П. Р о щ и н а, Э. Д. И щ е н к о. Укр. Физ. журн., 9, № 3, 1964.
50. В. В. Шилов, Н. Н. М и н е н к о, А. К- Д о р о ш,
А. Ф. С к р ы ш е в с к и й, Г. И. Баталии. Журн. структ. хим., 11, 923,
1970.
51. А. А. Б о ч в а р. Механизм и кинетика кристаллизации сплавов
эвтектического типа. М. — Л., ОНТИ, 1935.
ЧАСТЬ ТРЕТЬЯ
СТРОЕНИЕ ПРОСТЫХ ЖИДКОСТЕЙ
В предыдущих главах были рассмотрены основные понятия,
представления и методы, применяемые при описании строения жидких
фаз. Теперь, опираясь на эти понятия и методы, можно рассмотреть
структуру жидких систем. Множество различных жидких систем
бесконечно. В этой книге мы ограничимся простыми жидкостями.
Строение простых жидкостей и строение более сложных жидких систем в
основных чертах однотипно. С позиций молекулярной теории резкого
различия между простыми жидкостями и более сложными жидкими
системами нет. Систематический обзор строения и свойств простых
жидкостей позволяет в сжатой форме охарактеризовать особенности,
присущие строению и свойствам громадного числа более сложных
жидких фаз. Он важен и сам по себе, поскольку значение простых
жидкостей в науке и практике велико. Простые жидкости и более
сложные двух- и многокомпонентные жидкие фазы до последнего времени
было принято изучать раздельно. Эта традиция вызвана отставанием
молекулярной теории жидких систем, многолетним господством
феноменологических представлений и методов. Теперь, когда
исследования строения жидких систем, и в том числе простых жидкостей,
развернулись широко, указанная традиция потеряла смысл. Она уже
давно оставлена в теории твердых тел и газов. В монографиях и
учебной литературе строение и свойства твердых сплавов излагаются после
описания строения простых твердых тел. Так же поступают в
молекулярной теории газов. Пришла пора пойти по этому пути и в теории
жидких систем.
ГЛАВА VIII
ЖИДКИЕ МЕТАЛЛЫ, ПОЛУПРОВОДНИКИ
И ДИЭЛЕКТРИКИ
§ 65. Классификация. Диэлектрики. Полупроводники
Как уже говорилось, простыми жидкостями мы называем жидкие
простые вещества: водород, гелий, неон, аргон, азот, кислород, ртуть,
железо, золото и т. д.
Простые жидкости можно подразделить на квантовые и
классические, а также на жидкие диэлектрики и жидкие металлы, полуме-
1/27-503 161
таллы и полупроводники. Конечно, эти понятия неточны. Резкие
границы между классическими и квантовыми жидкостями отсутствуют.
Точно так же нет и резкой границы между жидкими металлами, полу,
металлами, полупроводниками и диэлектриками. Известны вещества и
состояния с промежуточными свойствами.
Существуют только две простые квантовые жидкости — жидкие
стабильные изотопы гелия 4Не и 3Не. Некоторыми свойствами
квантовой жидкости обладают электроны проводимости в металлах. Строе-
ние и свойства простых квантовых жидкостей 4Не и 3Не будет
рассмотрено в гл. XI.
Так как все жидкости состоят из частиц, имеющих электрический
заряд, — атомных ядер и электронов, •— то деление жидкостей на
группы в зависимости от их поведения во внешнем
электромагнитном поле вполне естественно. Оно диктуется природой жидкостей и
получило широкое распространение.
Как уже было сказано, различают жидкие диэлектрики,
полупроводники, полуметаллы и металлы. Аналогичная классификация
применяется и для твердых тел. Непосредственно эту классификацию обычно
связывают с электропроводностью вещества о:
с, Ом"1 • м"1
металлы — 104 —102
полуметаллы -- 102—1
полупроводники ~- 1 —10~8
диэлектрики — 10~8 и менее
Хорошие диэлектрики, применяемые для изоляции, имеют
электропроводность порядка 10~14 Ом"1 • м"1 и менее.
Различия в электропроводности представляют собой лишь одно
из характерных внешних проявлений тех особенностей вещества,
которые привели к указанной классификации, имеющей глубокие
физические основания. Здесь мы дадим краткое качественное
описание этих оснований, опираясь вначале на относительно более простую
картину, полученную в результате исследования кристаллов.
Диэлектрики. В идеальном диэлектрике нет зарядов, способных
свободно перемещаться под влиянием внешнего электрического поля.
Волновые функции электронов в диэлектрике почти полностью
локализованы около атомных ядер. При наложении внешнего
электростатического поля происходит поляризация, т. е. перераспределение
положений электрических зарядов. Возникает поле поляризации,
которое противонаправлено внешнему полю 80, но меньше его по
абсолютной величине. В объеме занимаемом диэлектриком, под влиянием
внешнего поля 80 возникает среднее макроскопическое электрическое
поле 8. В однородном изотропном диэлектрике оно равнонаправлеио
с полем 80. О поле 8 говорилось ранее в гл. П.
В реальных диэлектриках свободные заряды всегда есть, только их
концентрация очень мала. Поэтому при наложении
электростатического внешнего поля свободные заряды будут перемещаться под
влиянием внешнего поля, создавая обратное поле, компенсирующее внеш-
162
лее поле. Следовательно, в реальных диэлектриках величина среднего
макроскопического поля 8 с течением времени, хотя бы и крайне
медленно, должна уменьшаться.
Полупроводники. Известны два вида полупроводников —
электронные и протонные полупроводники. В электронных полупроводниках
истинными носителями тока являются электроны; в протонных
полупроводниках — протоны. Среди простых жидкостей и их растворов
протонных полупроводников нет, поэтому ограничимся описанием
свойств только жидких электронных полупроводников.
Электропроводность вещества зависит от концентрации носителей
тока и их подвижности. Характерная особенность чистых
электронных полупроводников — быстрое увеличение концентрации
проводящих электронов с ростом температуры. Причина этого состоит в
следующем. Спектр значений энергии изолированных атомов дискретен.
Если есть N изолированных атомов, то все они могут иметь одинаковую
энергию. Электроны, принадлежащие разным изолированным атомам;
могут находиться в одинаковых квантовых состояниях. В жидкости или
кристалле атомы взаимодействуют друг с другом. В результате этого
взаимодействия состояния движения электронов, одинаковые в
изолированных атомах, становятся различными. Дискретные
энергетические уровни изолированных атомов преобразуются в
энергетические зоны. Между зонами находятся области запрещенных
состояний. Это запрещенные зоны. Если все уровни в зоне полностью
заняты электронами, зона называется валентной. Зона, где не все уровни
заняты, называется зоной проводимости. Так как уровни энергии в
зоне очень близки, электроны зоны проводимости, получая даже
небольшие порции энергии, могут изменять свои состояния, в том
числе скорость и направление движения. При наложении внешнего
электромагнитного поля электроны зоны проводимости, взаимодействуя с
этим полем, будут менять состояние своего движения. Возникает
электрический ток.
Пусть в конденсированной фазе (например, кристалле) имеются
только заполненные (валентные) и незаполненные (пустые) зоны. Если
ширина запрещенных зон Аи велика (порядка 10 эВ), эта фаза —
диэлектрик. Но если Аи мала (порядка 1 эВ), то в результате теплового
возбуждения часть электронов валентной зоны будет преодолевать
энергетический барьер. Поэтому валентная зона станет
незаполненной, а пустая зона получит некоторое число электронов. Обе зоны
станут зонами проводимости. При повышении температуры число
электронов, переходящих из нижней зоны в верхнюю, будет быстро расти.
Концентрация носителей тока будет увеличиваться пропорционально
б Б . Такая фаза — полупроводник. Теория жидких
полупроводников изложена в кн. А. И. Губанова [1].
§ 66. Состояние электронов в энергетических зонах.
Металлы и полуметаллы
Теория, описывающая состояние электронов в энергетических
зонах, проще формулируется для кристаллов. Ее понятия с
несущественными изменениями сохраняют силу и для жидких систем. При
рассмотрении кристаллов обычно исходят из того, что потенциальная
энергия и(г) электронов проводимости в поле, создаваемом ионной
решеткой, является периодической функцией радиуса-вектора г, т. е.
и(г +1) = и{г). Здесь / —вектор решетки, определяемый ее
структурой:
/ = 1г а± + 12а2 + 13 а3, (VII 1.1)
где аъ а2, #з — базисные векторы решетки; ll9 /2, 13 — целые числа.
Волновая функция электронов в зоне проводимости приближенно
имеет следующий вид:
'h = Ak(r)eikr , (VIII.2)
гдеЛд(г) —функция Блоха, названная так по имени физика —
теоретика, впервые применившего ее. Ak (г), как и потенциальная энергия
u(r)t удовлетворяет условию трансляционной инвариантности, т. е.
Ак(г) = Ak(r +/); k — волновой вектор, определяемый равенством
k = ki b± + k2 b2 + k3 b3> (VI11.3)
где &lf &2, b3—векторы обратной решетки, определяемые через
векторы прямой решетки аъ а%, а3 с помощью следующих равенств:
[а2а3] [asai] . и Wia2] /WTTT ..
0i = —'г :; &2 = —i^ ;» &з=—; г- (VIII. 4)
аг [а2 а3\ аА [а2 а3\ а± [а2 а3\
Символ [ахпу] — обозначает векторное произведение ах и ау. Если
кристалл имеет размеры, равные по трем базисным направлениям
ЛГ\Г\(ЛГ\ГЧ
6 %
Рис. 39. Схематическое
изображение электронных волновых
функций в кристалле [2]:
а — потенциал вдоль цепочки атомов;
б — пример собственной функции (сама
функция комплексна; здесь показана
только ее действительная часть); эту
функцию можно представить в виде
произведений функции Блоха Ь, имеющей
периодичность решетки и плоской волны s
(здесь показана действительная часть
последней)
164
решетки Lxаъ L2a2 и L3a3y где Ll9 L2 и L3 — большие целые числа, то
2пт1 2пт2 2пт*
ki=-~; k2=—-*-; къ=—±. (VIII.5)
Lx L2 L3
где ml9 rn2 и т3 — целые числа. Произведение L1L2L3 равно числу
j\j элементарных ячеек в решетке. Вывод уравнения (VIII. 2) для фй
имеется в кн. У. Харрисона [2], Д. Займана [3] и Г. Джонса [4].
Схематическое изображение пространственных волновых функций электронов
проводимости в кристалле дано на рис. 39. Около ионов эти волновые
функции испытывают сильные осцилляции под влиянием электронных
состояний внутренних оболочек ионов. Приближенный учет этого
влияния осуществляется с помощью метода псевдо потенциал а (см. § 67).
В качестве самого грубого приближения можно принять, что
потенциал Е(г) постоянен. Тогда волновые функции электронов
проводимости будут плоскими волнами:
- <l>k = eikr . (VIII.6)
Энергия этих электронов равна
h2 k2
EW=—^> (VIIL7)
где т — масса электрона; k= | к \ = —— —- волновое число; X —
длина волны.
Пусть в образце кристалла имеется N элементарных ячеек. Тогда
существует ровно W различных значений волнового вектора к, для
которых
0<mi<Li — 1, (/=1,2,3). (VIII.8)
Если образец кристалла имеет макроскопические размеры, т. е.
число элементарных ячеек iV велико, то соседние значения векторов k мало
отличаются друг от друга. Таким образом, энергия электронов E(k)
макроскопического кристалла есть квазинепрерывная функция
волнового вектора &. Квазинепрерывность этой функции вызвана тем, что
волновые векторы могут иметь хотя и очень близкие друг другу, но
все же дискретные значения.
Рассмотрим ^-пространство, т. е. пространство, точки которого
соответствуют различным значениям волнового вектора к. В
^-пространстве область векторов k замкнута, т. е. точки, соответствующие т. = О
и т. = L. — 1, расположены рядом [4]. Области ^-пространства,
внутри которых E(k) является квазинепрерывной функцией k,
называют зонами Бриллюэна. Анализ движения электронов проводимости
показывает, что %к напоминает импульс. Чтобы отличать %k от
истинного импульса электрона, произведение ^& принято называть
квазиимпульсом.
Полуметаллы. В отличие от полупроводников энергетические зоны
полуметаллов слегка перекрываются (рис. 40) Область вблизи дна
второй энергетической зоны, например вблизи точки Л, может
оказаться ниже потолка первой зоны. Поэтому электроны как бы перелива-
6—503 165
ются через край первой зоны около точки А и углы зоны остаются
незанятыми [3]. В результате нижняя зона почти заполнена, а верхняя
почти пустая. Между этими зонами имеется область запрещенных
состояний. Концентрация носителей тока и проводимость
существенно зависят от температуры. Проводимость значительно выше, чем
у полупроводников.
Металлы. Если какая-либо из зон Бриллюэна заполнена не до
конца, то при наложении внешнего поля появляется ток, причем
проводимость будет велика. Изменение температуры лишь
перераспределяет энергии электронов в зоне, не влияя существенно на
концентрацию электронов проводимости. Зависимость электропроводности от
температуры сравнительно слаба. Она определяется в основном
процессами рассеяния электронов в жидкости или кристалле. Такие
вещества являются металлами.
Как уже было сказано, если кристалл содержит N элементарных
ячеек, то в зоне Бриллюэна имеется N разрешенных к векторов и,
следовательно, N возможных пространственных волновых функций
tyk(r). Но каждой из этих N пространственных волновых функций может
соответствовать два электрона с противоположно направленными
спинами. Поэтому в кристалле, содержащем N элементарных ячеек, на
каждую энергетическую зону приходится 2N электронных состояний.
Если на одну элементарную ячейку в кристалле приходится нечетное
Рис. 40. Перекрытие зон в
двухвалентном металле [3].
(Энергия Е как функция вектора k в
приведенной зоне)
Рис. 41. Зона
Бриллюэна и поверхность Ферми
одновалентных металлов
(меди, серебра, золота)
(сфера с центром в точке
k = 0):
пунктир — ребра зоны
Бриллюэна; сплошные линии —
поверхность Ферми
число свободных электронов, то кристалл всегда будет металлом.
Таковы, например, одновалентные щелочные металлы Li, Na, К, Rb,
Cs и благородные металлы Си, Ag, Аи. У этих металлов энергетическая
зона и зона Бриллюэна заполнены наполовину.
Наиболее существенной характеристикой энергетического спектра
электронов в зоне Бриллюэна является ферми-поверхность постоян-
166
ной энергии E(k) = EF. Эта поверхность при абсолютном нуле
температуры отделяет в k пространстве заполненные электронные состояния
от свободных. EF — уровень Ферми. Вид повер хности зависит от
структуры кристалла и числа электронов, которые могут распределяться по
состояниям в зоне Бриллюэна. При температурах, отличных от О К,
необходимо учитывать состояния, расположенные выше поверхности
Ферми. Некоторое представление о зонах Бриллюэна и уровнях
Ферми дает рис. 41. На этом рисунке изображены зона Бриллюэна и
поверхность Ферми меди, золота и серебра в пространстве обратной
решетки. Прямая решетка этих металлов — гранецентрированная
кубическая.
Поверхность Ферми строится на основании экспериментальных
исследований поведения металлов в электромагнитных полях при
низких температурах. Сведения о поверхности Ферми могут быть
получены с помощью измерений периодических колебаний магнитной
восприимчивости тонкой полоски металла в магнитном поле.
Поверхность Ферми может быть изучена методом циклотронного резонанса,
т. е. резонансного уменьшения поглощения электромагнитной
энергии заданной частоты металлом при определенной напряженности
магнитного поля. Описание принципиальных основ таких
исследований имеется в кн. У. Харрисона [2].
При температурах, отличных от О К, поверхность Ферми в
металлах немного размывается. В жидких металлах это различие несколько
больше, чем в твердых. Электроны рассеиваются на беспорядочно
расположенных ионах сильнее, чем когда они упорядочены. Поэтому
некоторая (небольшая) часть состояний в пространстве внутри сферы
Ферми не заполнена. Соответственно часть состояний вне сферы
Ферми занята электронами [4].
§ 67. Метод псевдопотенциала
В настоящее время основным методом электронной теории
непереходных, твердых и жидких металлов является метод
псевдопотенциала [5].
Волновые функции электронов проводимости ортогональны
волновым функциям внутренних электронных оболочек атомов. Поэтому
электроны проводимости испытывают отталкивание от ионов. Но
вместе с тем электроны притягиваются положительным зарядом ионов.
Отталкивание почти полностью компенсирует притяжение. В итоге
эффективный потенциал, изменяющий состояние движения электрона
(рассеивающий потенциал, или «псевдопотенциал»), мал по сравнению
с энергией электронов проводимости.
Метод псевдопотенциала используют следующие три
фундаментальных физических приближения. Во-первых, применяется
приближение самосогласованного поля. Взаимодействие между электронами
описывается некоторым средним потенциалом. Этот потенциал зависит
от состояний электронов, а состояния электронов определяются
упомянутым средним потенциалом.
6* 167
Во-вторых, все электронные состояния в металле подразделяются
на состояния зоны проводимости (почти свободные электроны) и
состояния внутренних электронных оболочек атомов. Число электронов
проводимости на атом металла нередко равно номеру группы
периодической системы, в которой находится рассматриваемый элемент. Это
валентные электроны. Волновые функции невалентных электронов
(электронов внутренних оболочек) сильно локализованы около
атомных ядер. Эти волновые функции не перекрываются и, следовательно,
взаимодействие между «ионами» металла в решетке сводится к
отталкиванию их положительных зарядов. Влияние электронов
проводимости и соседних «ионов» на волновые функции невалентных
электронов не учитывается*. Таким образом, считается, что волновые
функции оболочек «ионов» такие же, как у изолированных ионов, хотя
собственные энергии, соответствующие этим волновым функциям, в
решетке отличаются от собственных энергий изолированных ионов.
В-третьих, для описания свойств металла, зависящих от
электронов в зоне проводимости, применяется метод возмущений. Его
применение оправдано тем, что псевдопотенциал мал по сравнению с
энергией электронов в зоне проводимости.
В теории псевдопотенциала твердый или жидкий образец металла
рассматривается, по существу, как макромолекула. Псевдопотенциал
подбирается так, чтобы с его помощью можно было правильно описать
поверхность Ферми.
Если псевдопотенциал выбран удачно, то с его помощью можно
рассчитать электрическое сопротивление металлов, температуру их
перехода в сверхпроводящее состояние, фононные спектры. Задав
расположение атомных остовов, можно провести расчеты полной энергии
металла при температуре Т = 0°С. Таким образом, теория
псевдопотенциала позволяет установить взаимосвязь между многими
важными свойствами металлов. Наибольшие успехи достигнуты пока для тех
металлов, изолированные атомы которых имеют незаполненные s или р
электронные оболочки. Таков, например, алюминий.
Формула для псевдопотенциала сравнительно сложна. Для
исчерпывающей ее характеристики потребовалось бы предварительно
рассмотреть ряд вопросов квантовой теории твердого тела. Здесь
следует отметить только, что псевдопотенциал представляет линейный
нелокальный оператор**,напоминающий оператор возмущения Н' в
уравнении (1.15) (см. [5]). Матричные элементы псевдопотенциала за-
* В металлах, как правило, нет ионов в обычном смысле слова. Валентные
электроны у металлов обобществлены, поэтому термин «ион» применяется здесь
условно, чтобы подчеркнуть это, мы берем его в кавычки.
Л
** Пусть некоторый оператор Q, действуя на функцию ф (г) переводит ее в
другую функцию Ф(г) того же аргумента г:
Если оператор л и н е й н ы й, то функция ф никогда не должна возводиться
в какую-либо степень, отличную от единицы, и к ней не должны добавляться по-
168
висят от четырех независимых переменных. В этом смысле
псевдопотенциал напоминает эмпирический степенной потенциал (IV.5), в
котором фигурируют четыре произвольные постоянные: а, 6, т и п.
Эксперимент не дает достаточной информации для исчерпывающего
определения всех четырех независимых переменных, входящих в
псевдопотенциал. В связи с этим подбираются такие выражения
псевдопотенциала, чтобы с их помощью можно было описать поверхность
Ферми исследуемого металла.
Расчеты с помощью метода псевдопотенциала не связаны с
допущением о периодическом характере межионного потенциала.
Могут рассматриваться и такие случаи, когда «ионы» неупорядочены.
Поэтому метод псевдопотенциала широко применяется в теории
жидких металлов [6]. При этом используются экспериментальные данные
о радиальной функции распределения атомов жидких металлов.
Как уже было сказано в предисловии, описание расчетов свойств
жидких систем с помощью их моделей не входит в задачу этой книги.
Мы ограничиваемся здесь лишь качественной характеристикой
метода псевдопотенциала. Подробное изложение расчетов свойств
металлов с применением теории псевдопотенциала дано в монографиях
У. Харрисона [2] и В. Хейне, М. Коэна, Д. Уэйра [5]. Обзор
применений к жидким металлам имеется в статье Ю. П. Красного и
Н. П. Коваленко [5].
§ 68. Жидкие металлы. Состояние электронов
Атомы металлов в твердой и жидкой фазах образуют в основном
плотноупакованные структуры. При плавлении металлов
электропроводность а обычно падает примерно в 1,5—2 раза. При повышении
температуры жидкого металла электропроводность уменьшается, но
медленнее, чем у твердых металлов. В жидких свинце и висмуте
электропроводность почти не зависит от температуры, а у жидких цинка, кадмия
и ртути она даже растет с увеличением температуры. Число электронов
проводимости в единице объема жидких металлов часто почти совпадает
с числом валентных электронов. Подвижность электронов в металлах,
как было показано А. Р. Регелем [7], при плавлении меняется мало.
Плотность жидких металлов меняется при их затвердевании
незначительно. Сжимаемость жидких металлов, как и твердых, мала. Она
примерно на порядок меньше сжимаемости жидких диэлектриков.
Чтобы изучить строение жидких металлов, необходимо выяснить,
как распределены ионы металла и электроны проводимости. Распреде-
стоянные, не зависящие от <р. Если оператор локальный, то его действие
сводится к умножению функции ф(г) на некоторую заданную функцию того же
аргумента г. Все другие операторы нелокальны. Например, оператор Q
нелокален, если
Ф (г) - Q 9 (г) = J Q (г, г') ср (/•') dr'.
Квантовомеханические матричные элементы оператора Q имеют в этом случае
вид <ф|§| Ф> = S<}*(r)Q(r, r')cp(r')a'r'.
169
ление ионов в жидких металлах определяют с помощью рентгено- или
нейтронографии. Распределение электронов проводимости
исследуется на основе данных об оптических свойствах металлов и
электронных явлений переноса.
Электроны проводимости в основном скапливаются вокруг
положительных ионов и экранируют их [1]. Для характеристики состояния
электронов проводимости важно знать функцию распределения
энергии Е этих электронов. Эту функцию называют плотностью состояний
п(Е). Вид этой функции пока что еще не вполне ясен. Существует
мнение, что у жидких металлов поверхности Ферми имеют сферическую
форму. У большинства твердых
металлов в видимой области
спектра обнаруживаются
области поглощения света вследствие
возбуждения электронов с фер-
ми-уровня в пустые зоны над
ним. В жидких металлах
границы поглощения не наблюдаются
до тех пор, пока частота не
оказывается достаточно высокой,
чтобы возбудить невалентные
электроны ионных оболочек. Это
означает, что над ферми-уровнем
нет каких-либо признаков
зонной структуры [7]. Н. Мотт [6]
придерживается несколько иной
точки зрения. Он полагает, что
Рис. 42. Предполагаемый вид плотности
вероятности состояний электронов п(Е)
как функции их энергии в трехмерной
решетке [6]:
а — приближение почти свободных электронов
(имеется «хвост» плотности состояний,
простирающийся в область низких энергий); б—слабое
взаимодействие; в — сильное взаимодействие; г—
жидкий полупроводник
у ряда металлов плотность
состояний п(Е) около поверхности
Ферми мало изменяется при
плавлении. Согласно Мотту, в
неупорядоченных трехмерных
структурах существуют два типа
состояний электронов:
локализованные и нелокализованные. Внутри заданного интервала энергии
состояния либо локализованы, либо не локализованы. Между ними имеется
качественное различие. Если состояния локализованы, то электрон не
диффундирует из заданной области; проводимость при этом равна
нулю. Имеется критическая энергия Ес, разделяющая области с
локализованными и нелокализованными состояниями. Для локализованных
состояний пространственная волновая функция г|) убывает как sin(kr)e~^ry
причем ^ стремится к нулю, когда Е -+Е с. Для нелокализованных
состояний средняя длина свободного пробега электронов L
приближается к длине волны электрона X, когда Е -+Е с. Величина L не
может быть меньше X. Функция п(Е) электронов может иметь вид,
представленный на рис. 42. Области энергии, в которых могут
появиться локализованные состояния, заштрихованы. Стрелки указывают
положение уровня Ферми в двухвалентных металлах. Если уровень
Ферми лежит в незаштрихованной области, то вещество представляет
170
собой металл. «Если уровень Ферми при низких температурах лежит
в заштрихованной области, то материал представляет собой
полупроводник или изолятор. Здесь возможны два типа проводимости.
а. Возбуждение электронов в незаштрихованную область. При
этом перенос носителей заряда происходит так же, как и в
кристаллических полупроводниках
б. Перескок из одного локализованного состояния в другое. Для этого
всегда требуется конечная энергия активации, ибо каждому
локализованному состоянию отвечают свои собственные дискретные значения
энергии» ([6], стр. 38).
§ 69. О структуре твердых простых веществ.
Правило 8—N
Перейдем теперь к общей характеристике распределения атомов
в простых веществах. Мы будем говорить не только о жидкостях, но и о
кристаллах, потому что при изучении строения жидкостей полезно
знать, как располагаются атомы ниже температуры плавления.
Расположение атомов при заданных температуре Т и давлении Р в конечном
счете определяется условием минимума свободной энтальпии системы.
Факторы, влияющие на это условие, пока еще исследованы
недостаточно. Одним из них являются силы связи между атомами.
Правило 8—N. Влияние химических связей между атомами на
строение твердых и жидких фаз находит отражение в известном «правиле
8— N». Согласно этому правилу координационное число z атомов
данного элемента в конденсированной фазе (жидкости или кристалле) равно
8—N, где N — число электронов, находящихся на внешней оболочке
атома.
Это правило действует, когда между атомами образуются прочные
химические связи. Например, атомы галогенов имеют 7 валентных
электронов. Они образуют молекулы F2, Cl2, Вг2 и 12. Следовательно,
координационное число z этих атомов (т. е. число ближайших соседних
атомов) равно единице. Это справедливо не только для твердых и
жидких, но и газообразных галогенов до тех пор, пока под влиянием
высокой температуры молекулы не распадаются на атомы. Атомы кислорода,
серы, селена и теллура имеют 6 валентных электронов. Согласно
правилу 8—N эти вещества должны иметь структуру с координационным
числом 2. И, действительно, сера, селен и теллур образуют
цепочечные или кольцевые молекулы, в которых каждый атом имеет два
соседа, 2=2.
Далее этому правилу следуют фосфор, мышьяк, сурьма и висмут,
в кристаллах которых z = 3. Углерод, германий, кремний и олово
(при низких температурах) тоже подчиняются правилу 8—N. Они
могут иметь структуру типа алмаза. Тогда каждый атом окружен
четырьмя другими атомами, которые размещены симметрично в Еершинах
правильного тетраэдра.
Но правило 8—N выполняется далеко не всегда. Ему не
подчиняются, например, инертные газы. В табл. 10, которая с некоторыми
изменениями заимствована из кн. В. К. Григоровича [8], сопоставлены
171
Кристаллические структуры простых веществ (по Григоровичу [81]) [Темпера
их абсолютных температур разными спосо
гцк
Ш Предполагаемые ОЦК
структуры
8-N и ковалентно-металлические
структуры
структуры кристаллов простых веществ при атмосферном давлении.
Правило 8—N выполняется для 20 веществ, расположенных в правой
части этой таблицы. В табл. 10 структуры твердых простых веществ
закодированы различной штриховкой. Когда существуют две или более
полиморфных модификаций, площадь, отведенная каждой из них,
приблизительно соответствует температурной области ее существования,
выраженной в долях абсолютной температуры плавления (см. шкалу
температур у магния).
Отметим следующие закономерности. В правой части таблицы,
где господствуют неметаллы, чаще всего встречаются сравнительно
рыхлые молекулярные структуры. Многие из них следуют правилу
8—N. В левой части таблицы и ее центре, где находятся металлы, в
172
Таблица 10
хурные области существования различных структур элементов указаны в долях
бами штриховки (см. шкалу у Mg) ]
II
Gd
1316°
llfl
Tb
i392't|
Hill
Dy
U42f
Illllll
Ho
Sit
Er
ж
ТГГПТТ
Am
Cm
Bk
Cf
Es
Fm
No
Lv/
основном осуществляются плотноупакованные и объемноцентриро-
ванная кубическая структуры.
При повышении температуры а-олово, представляющее собой
полупроводник со структурой алмаза, переходит в металлическое белое
олово, обладающее объемноцентрированной тетрагональной структурой.
Сложные кубические структуры а- и р-марганца, сложные структуры
а- и (3-урана и нептуния, а-, р- и f-плутония, имеющие отчетливо
выраженные локализованные химические связи между атомами,
переходят вОЦК структуру, типичную для металлов; причем у марганца
и плутония этому переходу предшествует превращение в гранецент-
рированную кубическую (ГЦК) модификацию. У большинства
полиморфных металлов низкотемпературная а-модификация имеет плотную
173
гексагональную (ПГУ) или ГЦК структуру, которые с повышением
температуры переходят .в ОЦК структуру (литий, натрий, кальций,
стронций, барий, скандий, иттрий, лантан, актиний и большинство
лантаноидов, а также титан, цирконий, гафний, таллий и другие—всего более
30 металлов). «У лантана и церия между гексагональной
плотноупакованной и ОЦК структурами располагается область устойчивости ГЦК
упаковки. В ряде случаев с повышением температуры происходит
переход ГЦК — ОЦК (торий, т — S-превращения марганца и железа)
или гексагональной в гранецентрированную кубическую (кобальт)»
[8]. Последовательность полиморфных превращений и устойчивость
модификаций определяются их термодинамическими свойствами.
Структура устойчива, когда при заданных температуре и давлении ей
соответствует минимальное значение свободной энтальпии G по
сравнению с ее величиной для других возможных видов структуры той же
системы.
§ 70. О возможных распределениях атомов в простых,
твердых и жидких веществах
Прежде чем перейти к описанию распределения атомов (т. е.
структуры) жидких простых веществ, рассмотрим вкратце те теоретические
представления по этому вопросу, которые имеются в литературе. Когда
речь идет о тех простых веществах, между атомами которых отчетливо
фиксируются направленные связи за счет валентных электронов
(например, так называемые ковалентные структуры, следующие правилу
8 — N), то особых разногласий о причинах возникновения структуры
не возникает. Ясно, что решающая роль (при атмосферном давлении)
принадлежит химическим связям между атомами. Иное дело металлы,
где валентные электроны более или менее коллективизированы.
Здесь единой точки зрения пока еще нет.
Обычно ограничиваются описанием возможных способов связи
между атомами металлов за счет валентных или невалентных электронов.
Состояние валентных электронов характеризуют с помощью метода
самосогласованного поля (метода Хартри—Фока) [9, 10, И].
Метод Хартри—Фока является приближенным. Каждый электрон
в атоме взаимодействует по закону Кулона со всеми остальными
электронами, поэтому его движение зависит от движения всех остальных
электронов. В методе Хартри—Фока взаимодействие между
электронами в атоме непосредственно не учитывается. Вместо этого
предполагается, что электрон движется в некотором эффективном
электрическом поле, получаемом в результате соответствующего усреднения
положений всех остальных электронов. Считается, что эта процедура
позволяет охарактеризовать распределение электронов в атомах с хорошей
степенью точности. Напомним, что метод псевдопотенциала,
применяемый для описания свойств почти свободных электронов в металлах,
тоже опирается на приближение самосогласованного поля.
Расчеты уровней энергии электронов с помощью метода
псевдопотенциалов осуществляются с точностью около 0,5 ридберга [5] (1 рид-
берг = 13,60 эВ = 1,31 МДж). Это намного превышает те изменения
174
энергии, которые происходят при фазовых переходах в металлах, в
том числе при их плавлении. Напомним также, что пространственная
волновая функция электронов проводимости, рассчитанная по методу
псевдопотенциала, испытывает резкие колебания около атомных
остовов. Эти колебания сложным образом зависят от волновых функций
внутренних электронных оболочек атомов металла и от потенциала,
действующего на электроны проводимости. Любопытно, что потенциал
косвенного взаимодействия ионов друг с другом через почти свободные
электроны проводимости оказывается короткодействующим. Радиус
его действия фактически не превышает расстояния между соседними
ионами. Вместе с тем надо иметь в виду, что любые объяснения
структуры металлов, исходящие из описания межатомных связей,
неполны. Знание энергии межатомных связей позволяет судить лишь
об относительной устойчивости фаз при О К- Для изолированной
системы необходимы сведения об ее энтропии. Для фаз,
находящихся при постоянных давлении и температуре, нужны данные о
свободной энтальпии.
Гипотеза Григоровича. По мнению В. К. Григоровича,
расположение атомов в твердых и жидких простых веществах определяется,
в основном, их электронным строением [8]. «В металлической решетке,
где внешние электроны положительных ионов сильно возбуждены
вследствие возмущающего действия соседних атомов, сравнительно
небольшой прирост температуры может быть достаточным для наступления
перекрытия и обменного взаимодействия внешних р6 оболочек ионов,
не перекрывающихся при низких температурах» ([81, стр. 202). Так,
например, объемноцентрированная кубическая структура натрия,
область существования которой простирается от 30 К до температуры
плавления, по Григоровичу, может быть объяснена с помощью
следующих соображений. Из экспериментальных данных (об оптических
свойствах, эффекте Холла и т. д.) известно, что натрий в твердом и жидком
состоянии имеет один электрон проводимости на атом. Это означает,
что его валентный электрон с 3s уровня переходит в электронный газ.
Атомы натрия в конденсированном состоянии имеют внешнюю 2s22p6
оболочку. Взаимодействие ионов Na+ с электронным газом приводит
к сближению и перекрыванию р-орбиталей внешних р6 оболочек ионов,
в результате чего возникают обменные /^вухэлектронные а-связи,
направленные по трем осям прямоугольных координат. Образование
шести связей каждым атомом со своими соседями приводит к простой
кубической ячейке со свободным объемом в центре, который может
быть заполнен таким же ионом. Так, из двух простых кубических под-
решеток, энергетически невыгодных, а потому редко реализующихся
в металлах, образуется ОЦК структура, одна из трех типичных
металлических структур. Гипотеза Григоровича иллюстрируется рис. 43.
Точно так же обосновывается возникновение ОЦК структур и у
других щелочных металлов. Для лития, ионы которого имеют Is2оболочку,
возникновение ОЦК структуры связывается с предположением о
переходе s электронов на р уровни.
Григорович отмечает, что при переходе ГЦК и ПГУ в ОЦК
модификации происходит уменьшение межатомных расстояний. Сжимае-
175
мость металлов, как уже говорилось, много меньше сжимаемости
диэлектриков. Эти факты говорят в пользу представления о
перекрывании атомных орбиталей соседних атомных остовов в металлах. Одна
из слабых сторон гипотезы Григоровича состоит в том, что
возможность непосредственного влияния обобществленных валентных
электронов на структуру металлов не учитывается.
Правило Юм-Розери. Совсем иное объяснение структуры
металлических кристаллов было предложено Юм-Розери (см., например,
ОЦК(р6-р6)
Рис. 43. Взаимодействие р-электронов, возникающее по гипотезе
Григоровича при перекрывании электронных облаков ионов [8]
(Изолированные ионы имеют р* конфигурацию электронных орбиталей. Взаимодействие р-
u электронов приводит к образованию ОЦК структур металлов и структур типа NaCl)
[12, 13, 14]). Оно основано на предложенном им понятии электронной
концентрации. Эта последняя равна среднему числу валентных
электронов, приходящихся на один атом в кристалле. Каждый атом в
металле окружен в среднем числом электронов, равным электронной
концентрации. По мнению Юм-Розери, положение каждого атома в металле
определяется его притяжением к почти свободным электронам,
расположенным около него. Поэтому в разных металлах и сплавах,
имеющих одинаковую электронную концентрацию, структуры должны быть
подобны. В некоторых случаях правило Юм-Розери оправдывается.
Но оно имеет множество отклонений. С его помощью нельзя объяснить
полиморфные переходы в металлах и то, что многие из них плавятся
без изменения электронной концентрации. «Ясно, что электронная
концентрация — не единственный фактор, определяющий структуру
чистого металла. Это видно также из анализа строения сплавов, где
появляются исключения почти при любой попытке объяснения структуры,
основанной на понятии электронной концентрации» [13].
Правило Энгеля. По Н. Энгелю, если валентное состояние атомов
металла соответствует одному s- и одному р-электронам, должна
возникать гексагональная, плотноупакованная структура. Когда атомы
176
металла имеют три валентных электрона, причем один находится в s-
состоянии, а два—в /^-состоянии, то ожидается гранецентрированная
кубическая решетка. Наконец, когда атом имеет один валентный s-
электрон или же когда внешняя оболочка атома имеет структуру,
подобную dn~x s-конфигурации, возникает объемноцентрированная
кубическая решетка. Попытка объяснения этих правил принадлежит
Л. Брюэру. По его мнению, химические связи между атомами
металлов, формирующие решетку, образуются в результате взаимодействия
валентных электронов ([14], стр. 72).
Теория строения простых веществ пока еще делает лишь первые
шаги. Последовательный учет всех факторов, определяющих строение
твердых и жидких фаз, — дело будущего.
ГЛАВА IX
ПОДГРУППЫ ЛИТИЯ, БЕРИЛЛИЯ
И ПЕРЕХОДНЫХ МЕТАЛЛОВ
Перейдем теперь к описанию структуры простых жидкостей. Мы
будем рассматривать простые жидкости, группируя их в соответствии
с периодическим законом Менделеева.
§ 71. Подгруппа лития (щелочные металлы)
(Li, Na, К, Rb, Cs)
В периодической системе элементов имеется правило, согласно
которому «только у второго или третьего элемента главной подгруппы
полностью проявляются характерные свойства группы, в то время как
первый и в меньшей степени также второй элемент обнаруживают
отклонения от этих характерных свойств. Первый элемент часто
является при этом переходным по своему поведению к следующей главной
подгруппе.
Второй элемент напоминает иногда соединения побочной подгруппы,
принадлежащей той же группе» ([15], стр. 184).
Щелочные металлы образуют главную подгруппу первой группы
периодической системы. Первый элемент в ряду щелочных металлов
литий в соответствии с упомянутым правилом по своим химическим
и некоторым физическим свойствам больше напоминает
щелочноземельные металлы. Натрий тоже считается не вполне характерным
представителем щелочных металлов. Свойства, вполне типичные для этой
подгруппы, проявляются только начиная с калия.
Пары щелочных металлов частично ассоциированы. Ассоциация
растет с увеличением молярной массы элемента. В парах присутствуют
димеры и более сложные ассоциаты. Энергии диссоциации димеров
равны для Li2 107; Na2 72,3; К2 49,3; Rb2 45,2; Cs2 43,5 кДж/моль [16].
При комнатной температуре все щелочные металлы
кристаллизуются с образованием объемноцентрированной кубической решетки.
177
tt
К
CO
H
,иия
UONNIO
h- СЧ 00 i—■ Ь-
CO j^ 00 •—« OO
00 t^> CO C^- cO
Tj« О t^- C5 Ю
CO CX> l>- CO CO
>S
1
t-0l-
CO CO CO CO CO
ooooo
Ю 00
-Ю Г--
о —ооо
со со со сэ
Ю (N О О 00
CO t^C^Tf О
CO CO CO t-- Г^
СО (N CSJ (N (М
см
Ю Ю
со ю ю ю со
*-« (NCN СМ <М
О !>■
(^ <Л Tf О 00
СО О СО <N -н*
ю ^ со —- о
tJh СО СО СО СО
СО СО "Ф TflO
о oocsj со со
—. г-^ г— о>со
00 00 00 00 00
гга^аа
ооосо
00 ООО-н 00
О 00 О С<1 00
COOCJ5 00N
S ее ТО >, О»
•- со
7 н
£3|
?&|
л СП ^*-
R5
« мя
178
Цо при низких температурах у лития и натрия устойчивы более
плотные упаковки. Некоторые свойства щелочных металлов приведены в
табл. И. Из этой таблицы следует*, что плавление не сопровождается
заметным изменением координационного числа г. Расхождения между
величинами z в твердой и жидкой фазах не выходят за пределы
ошибок опыта. Проводимость уменьшается на 30—40%. Постоянная Холла
почти не меняется [17]. Следовательно, состояние почти свободных
электронов при плавлении не претерпевает существенных изменений.
Замечательны оптические свойства щелочных металлов. Обладая
большим коэффициентом поглощения света в видимой области спектра,
они прозрачны для ультрафиолетовых лучей. Показатель преломления
в ультрафиолетовом диапазоне меньше единицы. При увеличении
атомного номера щелочного металла область длин волн, для которых
металл прозрачен, расширяется в сторону видимого спектра. Эти
свойства щелочных металлов полуколичественно объясняются теорией,
основанной на представлении о почти свободных валентных электронах
в металлах.
Обзор рентгенографических исследований строения жидких
щелочных металлов имеется в статье И. В. Радченко [19] и в кн. Б. И.
Хрущева [20], где описаны и результаты более поздних рентгенографических
и нейтронографических измерений. Рентгенографические
исследования показали, что координационное число натрия и калия мало
меняется даже при температуре, более чем на 300° превышающей
температуру плавления. Правда, экстремумы на кривой атомного
распределения несколько различаются. Таким образом, есть
основания считать, что в жидких щелочных металлах при не очень высоких
температурах и давлениях часто встречаются фрагменты объемноцен-
трированной кристаллической решетки.
Очень интересен вопрос о строении жидких щелочных металлов
при высоких давлениях. Косвенные сведения об их структуре дают
исследования фазовых равновесий. Производная dTldP для кривой,
описывающей состояние двух одно компонентных, сосуществующих,
термодинамически равновесных фаз, могущих взаимно превращаться
друг в друга, следует уравнению
£=-ЬЬ
где Vm и VT3 — молярные объемы; Sm и 5ТВ — энтропии одного моля
сосуществующих жидкой и твердой фаз. Это хорошо известное
уравнение Клапейрона—Клаузиуса. За исключением гелия (при очень
низких температурах), энтропия жидкости при температуре плавления
больше, чем энтропия сосуществующей с жидкостью твердой фазы.
Поэтому, когда производная dTldP положительна, плавление
сопровождается ростом объема. Если же dTldP отрицательна, плавление
приводит к уменьшению объема системы. На рис. 44 изображена фазовая
* При составлении табл. 11 и табл. 12—27 были использованы материалы,
имеющиеся в работах [1, 8, 17—23].
179
ш
wo
о
4/7 80
120
160
Р}к5
Рис. 44. Фазовая
диаграмма цезия [24]
диаграмма цезия [24]. При давлениях до с^2,0 ГПа производная dTldP
положительна, но с ростом давления до 2,25 ГПа постепенно падает
до нуля. При этом давлении объемы жидкой и твердой фаз одинаковы.
Температура плавления повышается от 28,6 до 197°С. При Р >
>2,25 ГПа производная dTldP становится отрицательной, т. е. объем
моля жидкого цезия меньше, чем твердого. При 2,54 ГПа и 19ГС
наблюдается тройная точка. В этой точке
жидкий цезий сосуществует с двумя
модификациями твердого цезия Csl и CsII.
Производная dTldP вдоль кривой
сосуществования кристаллов Csl и CsII
положительна. Кривая плавления CsII при давлении
около 3,05 ГПа снова имеет производную
dTldP, равную нулю. Затем эта
производная становится отрицательной. Кривая
плавления CsII заканчивается во второй
тройной точке, где жидкий цезий
сосуществует с CsII и CsIV. Производная dTldP
вдоль кривой сосуществования CsII и CsIV
положительна и сравнительно велика.
Кривая плавления CsIV в изученном до сих пор
небольшом интервале давлений имеет
положительную производную dTldP,
Фазовая диаграмма рубидия похожа^на
фазовую диаграмму цезия. Кривая
плавления рубидия проходит через максимум при более высоком
давлении около 4,0 ГПа. Здесь тоже имеется, по-видимому, не менее
трех модификаций рубидия. Но в отличие от цезия тройная точка,
в которой жидкая фаза сосуществует с Rbl и Rbll, расположена
при ~8,0ГПа,т. е. правее максимума на кривой плавления. Переход
Rbll и Rbl И, видимо, может происходить при давлениях порядка
20,0 ГПа.
Согласно табл, 11, разность атомных объемов жидкой и твердой
фаз щелочных металлов невелика, но с ростом молярной массы металла
возрастает. При плавлении координационное число почти не
меняется и энтропия плавления мала. Следовательно, можно полагать, что
плавление щелочных металлов связано с ростом числа вакансий.
Связи между атомами лития в решетке прочнее. Поэтому литий плавится
при более высокой температуре и концентрация вакансий в точке
плавления лития меньше, чем у других элементов его подгруппы.
Образование вакансий при плавлении ведет к росту объема. Повышение
давления содействует тому, что в жидкой фазе наряду с фрагментами
ОЦК структуры могут появляться фрагменты более плотноупакован-
ных структур, например гранецентрированной кубической или
плотной гексагональной. Это может приводить к уменьшению объема
системы. Когда при высоких давлениях доля плотно упакованных
образований в жидкой фазе становится большой, плавление кристаллов с
ОЦК структурой сопровождается уменьшением объема. Фазовые
диаграммы рубидия и цезия дают основания считать, что жидкая фаза
180
переходит к более плотной упаковке при меньших давлениях, чем
твердая фаза. Когда твердая фаза тоже плотно упакована, плавление
возможно в основном благодаря появлению вакансий. Здесь вновь
объем жидкости будет больше объема твердой фазы и производная
dT/dP станет положительной.
На рис. 45 изображены зависимости от давления удельных объемов
сосуществующих жидкого и твердого цезия. При относительно малых
давлениях, когда плавление
происходит почти без изменения
координационного числа, объем
жидкости немного больше объема твердой
фазы. С повышением давления
положение меняется. При давлениях
2,0—4,7 ГПа рост объема жидкости
за счет вакансий подавляется
уплотнением упаковки атомов. После
превращения CsII в плотноупако-
ванный CsIV, т. е. при давлениях
выше 4,72 Гпа, изменение объема
при плавлении определяется,
вероятно, лишь ростом концентрации
вакансий. Здесь удельный объем
жидкой фазы существенно
превышает удельный объем кристаллов
CsIV. Заметим все же, что при еще
более высоких давлениях в
принципе не исключена возможность
возникновения менее плотноупако-
ванных структур. Теория этого
вопроса отсутствует.
Экспериментально подобные переходы
наблюдались, например,у таллия, олова и
висмута.
Рис. 45. Удельные объемы "со
существующих твердого и жидкого цезия
[24]:
сплошная линия — удельный объем
кристаллического цезия; пунктир — удельный объем
жидкого цезия
§ 72. Подгруппа бериллия (щелочноземельные металлы)
(Be, Mg, Ca, Sr, Ba, Ra)
В парах легких щелочноземельных металлов найдены
двухатомные молекулы. Их концентрации невелики. Не исключено присутствие
и более сложных структур. Энергии диссоциации двухатомных
молекул в кДж/моль при 25°С равны: Ве2 около 71; Mg2 33,4; Са2 26,86
[11]. Двухатомные молекулы щелочноземельных элементов менее
устойчивы, чем аналогичные молекулы элементов подгруппы лития.
На элементарную ячейку структуры щелочноземельных металлов
приходится два электрона. Поэтому валентная зона могла бы быть
полностью заполненной, но она перекрывается с ближайшей пустой
зоной. В результате бериллий и его аналоги являются металлами.
Исследования подвижности электронов бериллия указывают на
сильную связь его валентных электронов с атомными остовами в крис-
181
s
оерил
ппа
гру
еС
О
Я^ОИ/жЦ"*
4if ow • М/ж}
■шои-ЯМ,
«w^
гОГ
ч
WH
03
и
N
I/
Е1Г1твхэис1м ecfX
чнюю/ж^м
Элемент
"'-С
1Hj Hdii
я -ии^
а1Е/ж-
'■■*
ц
SV
ахл
килч
Sag
0
О С7> ОО ОО О-
Ю00ОСЛ—"
О CM LQ CO lO
СМ •—•* ~-* у—> '—<
о olo о о о
IQ Q5 СО Tf —н О
t^ CO t^- СО С7> 00
СМ ^ <—' —« —« ^
0,56
0,62
3,66
0,75—0,33
"^ СО СО •>
О) CM <J> СО
СМ СО СМ »—'
-н СО СМ 00 ^~ч
IvOt.K^j^
11,7
8,960
8,667
(9,21)
7,66
(14,2)
8
СО
1556
923
1123
1043
983
973
CDO "Ф О СМ О
СМ СМ СГ> СО ^ 1>-
СМ СО СО т^ т^ tJ<
со см оо оо оо оо
осоооо
СО LQ CD
—н LQ t^ CD l>- Ю
Бериллий
Магний
Кальций
Стронций
Барий
Радий
таллической решетке. Магнии
и кальций проводят ток
примерно так же, как рубидий и
цезий. Электропроводность
бария и стронция (а также
бериллия) приблизительно в 5
раз меньше. Предполагается,
что у бария и стронция
перекрывание зон мало («плохие
металлы»). Удвоение числа
валентных электронов по
сравнению с щелочными
металлами сопровождается резким
увеличением температур
плавления и кипения.
Проводимость, плотность и энтропия
при плавлении изменяются
не очень сильно (табл. 12).
Кривые плавления
щелочноземельных металлов похожи
на кривые плавления цезия и
рубидия. Полнее других
изучена фазовая диаграмма
бария. Сжимаемость металлов
подгруппы бериллия
возрастает от бериллия к барию.
Кристаллы магния вблизи
температуры плавления
имеют ПГУ структуру.
Остальные металлы подгруппы
бериллия вблизи температуры
плавления имеют ОЦК
структуру. Можно предполагать,
что в жидких металлах
подгруппы бериллия (включая
магний) вблизи точки
плавления часто встречаются
фрагменты ОЦК структуры.
Рентгенографические
исследования строения этих металлов,
по-видимому, еще не
производились.
§ 73. Подгруппа скандия.
Лантаноиды и актиноиды
Подгруппа скандия (Sc, Y,
La, Ac) (табл. 13). Кристаллы
а-скандия при комнатной тем-
182
пературе имеют гексагональную
плотную упаковку атомов. Отношение
параметров решетки с/а = 1,5917. Оно
меньше, чем для гексагональной плот-
нОй упаковки шаров, где с/а = 1,6333.
При 1335°С а-скандий переходит в
3-скандий, имеющий объемноцентри-
рованную кубическую структуру. Та
же последовательность
кристаллических структур наблюдается у иттрия
с тем отличием, что в плотной
гексагональной упаковке а-иттрия с/а =
= 1,572. Превращение а-иттрия в р-ит-
трий, имеющий ОЦК структуру,
происходит при 1495° С. Твердый лантан
имеет три полиморфные
модификации— а, р и ]. а-Лантан
характеризуется плотной гексагональной
упаковкой атомов с отношением с/а =
= 1,604. Плотные слои в кристаллах
а-лантана упакованы в
последовательности АВАСАВАС. При 310°С
а-лантан переходит в [3-лантан,
имеющий гранецентрированную плотную
кубическую упаковку атомов. При
868°С ^-лантан переходит в ^-лантан с
ОЦК структурой. Плавление скандия,
иттрия и лантана, по-видимому, не
сопровождается существенным
изменением ближней координации атомов.
В этом отношении металлы подгруппы
скандия напоминают металлы
подгруппы лития. Энтропии плавления
металлов подгруппы скандия и лития
почти одинаковы. Энтропии испарения
различаются. Но сведения о теплотах
и энтропиях плавления, приведенные
в табл. 13, недостаточно точны, и
данные о составе паров в литературе
отсутствуют, поэтому анализ
упомянутых различий пока невозможен.
Лантаноиды (Се, Рг, Nd, Pm, Sm,
Eu, Gd, Tb, Dy, Ho, Er, Tm, Yb,
Lu). Лантаноиды очень интересны
Для изучения связей между
структурой простых веществ и электронным
строением их атомов. Атомы всех
лантаноидов имеют 5р6 и 6s2
электронные конфигурации. Изолирован-
ЕГ
S
Ч
\о
X
ска
/ппа
о.
и
О
ч с
в8 §
о.к.
<P^q,
£*с
л в
1-1^
1^
с
t?
*1
° j
л
§1
<|
ч
л
^"f
<ч
Ьй
"^
с
к
L
- о
N
NS
Bx4ll
Ч1Г0п/ж)^Я
0
ч
с^
С5 С^ t^- ^-^
**^ _ГСО
—н »- ОЩ
^^
(305,01
(393,5)
399,8
(402)
ооо о
(N СО СОСО^
Ю (N СО
CSJ-н СО
rj^CO rf
Tt< "4f CO
ClOO^s
t4^. r^i »—h CO
t^ CDIOIO
^ __
S55s
csjco со о
Ю -и CD CD
со "tf<co со"
ssss
oooooo^
ОЦК
ОЦК
ОЦК
(0ЦК)
i-hNNCO
00 Ol CD CT^
CO ^ CO CO
Sl^^
^-•QQ- J—
»s '»s
s »s я s
X Cu H S
те н К h
^ н те ^
C^ |^4 t^ *^
183
ные атомы лантаноидов различаются числами электронов,
находящихся в 4/ и 5d состояниях. Сопоставляя числа 4/ и bd
электронов в изолированных атомах со структурой твердых и жидких фаз,
можно попытаться проследить влияние строения электронных
оболочек атомов на строение конденсированных фаз лантаноидов.
К сожалению, химически чистые лантаноиды труднодоступны,
поэтому они слабо изучены. В табл. 14 сопоставлены в основном
термодинамические характеристики расплавов лантаноидов и некоторые
особенности кристаллических фаз. Рассмотрим эти последние
подробнее, потому что знание строения твердых фаз помогает изучать
структуру их расплавов.
Закономерности строения кристаллов лантаноидов удобно
проследить с помощью табл. 10. Все лантаноиды, изученные при температурах,
близких к плавлению, имеют ОЦК структуру. Для прометия, эрбия
и тулия надежных данных пока еще нет. У европия, расположенного
в центре группы лантаноидов, ОЦК структура устойчива, по-видимому,
во всей области существования твердой фазы. У остальных
лантаноидов при низких температурах устойчивы фазы, имеющие плотные
упаковки атомов с координационным числом 12. Лантаноиды подгруппы
церия, за исключением самария и европия, при низких температурах
имеют плотные упаковки атомов типа а-лантана (АВАСАВ) (Се, Pr, Nd,
Pm). У церия, подобно лантану, переход от гексагональной плотной к
ОЦК упаковке происходит через ГЦК упаковку атомов. а-Самарий
имеет специфическую ромбоэдрическую упаковку с расположением
слоев АВАВСАСВС. У лантаноидов подгруппы иттрия (Gd, Tb, Dy, Но,
Er, Tm и Lu) низкотемпературная модификация имеет плотную
гексагональную упаковку типа магния (АВАВ). Только у иттербия
низкотемпературная фаза обладает гранецентрированной кубической
упаковкой.
Таким образом, повышение температуры для всех лантаноидов,
за исключением (по-видимому) европия, сопровождается переходом
к упаковкам с более отчетливо выраженным химическим
взаимодействием между атомами. Напомним, что при отсутствии направленных
межатомных взаимодействий простые вещества должны были бы иметь
плотную гексагональную упаковку сфер; ГЦК упаковка указывает
на существование взаимодействий химического характера, ОЦК
упаковка представляет собой еще более отчетливое проявление
химических связей между атомами (см. гл. III). Низкотемпературные
кристаллические модификации лантаноидов, естественно, лучше изучены,
чем высокотемпературные. Обратим внимание на следующие
интересные особенности низкотемпературных фаз (см. табл. 14). Своеобразно
изменение межъядерных расстояний d0. Сначала в подгруппе церия
наблюдается уменьшение d'o с ростом порядкового числа атомов
лантаноида. Затем в конце подгруппы церия, у европия, атомы которого
имеют ровно наполовину заполненную систему /-состояний электронов,
d0 вдруг резко возрастает. Потом в подгруппе иттрия d'o снова
падает. Но у иттрия, атомы которого имеют как раз полностью заполненную
систему /-состояний, d0 опять внезапно резко увеличивается.
184
о
X
ctf
н
X
ев
«5
/ iid Li qirow • ^т/жУт* tLl^nsW
jr ndu 4itow/yKi$fyi * //V
о
с
<
Ю
H
™-X/-tf '"sv
-—
wh i_or^P
MtIO я Btfoxadau udAiEdauwai
"Лйи-.пг.хэйн^х^хо
0
eg
о
3
314
о
со
in
CM
I
fe
5,099
464
LQ
о
099
CO
998
*
О
8
CO
«
к
CD
Hf
CO
101
332,9
8
CO
§
CO
5,69
891
CO
т|«
656
CO
1071
О
CO
CO
1
CO
с
CO
s
283,9
о
CO
oo
00
5,53
147
t>
85
641
CO
1141
О
CO
CO
QQ-
s
g
°
Д
to4
(97
(293)
s
CO
(6,28)
^.
CO
о
о
С
о
(103
(191,7)
о
24)
6,61
00
ю
с^
604
со
1190
О
СО
со
СО-
sS
Он
(Я
и
оГ
(100
(293)
о
см
ю
8
9,21
Я
О
084
О
C7i
§
ПИЙ
о
ю
W
оо"
(103
(311,9)
о
е
6,36.
8
О
Ю
S
604
00
1535
О
^?
со
rV
1ИНИ
б
га"
U
(105)
(293)
00)
(28
ю
6,66
о
00
о
о
со
со
564
со
1589
О
оо
со
со
со
со.
VO
trim)
(251)
СО
"^
8
S
6,57
8
(N
446
со
1665
О
СО
СО
GQ-
га
рози
с
s
tz{
(96)
(251)
00)
(26
96)
(6,99)
,18)
ГО
t>-
452
со
1715
О
335
гг.
S
г
е ^
(154
(293)
о
8
11,09
S
2
514
со
о
со
со
S
^о
ГГ)
ОО4
88)
(213)
8
(24
5
9,29
СО
00
со
оо
оо
492
со
я
о
см
S
^.
155,3
о
ю
ю
оо
»
6,99
СО
СО
^
О5
О
880
со
1071
О
Ю
СО
аз-
1
6"
!^
см"
5^
(247)
00)
(22
т?
00
5Г
(16
§
СМ
468
СО
1953
О
ю
$
со
СО-
ЦИЙ
S
1=^
185
Любопытно также, что скачки межъядерного расстояния d'o в
кристаллах европия и иттрия не влияют на последующее
закономерное снижение величины d'o. Оно продолжается с ростом порядкового
числа атомов так, как если бы упомянутых скачков не было.
Можно полагать, что при высоких температурах, когда плотноупа-
кованные структуры переходят в ОЦК модификации,
последовательность межатомных расстояний остается той же, что и при низких
температурах. Косвенным указанием на это служит связь между d'o
твердых лантаноидов и температурой плавления. В тех случаях, когда
а'о в ряду лантаноидов проходит через максимум, Тпл проходит через
минимум. Последовательное снижение d0 сопровождается столь же
последовательным ростом Тпл.
Не лишен интереса и тот факт, что температуры перехода в ОЦК
модификации, приведенные в табл. 14, тоже обнаруживают тесную
связь с dq, похожую на ту, что наблюдается у температуры плавления.
Наименьшее значение температур перехода имеют церий, европий и
иттербий, т. е. как раз те лантаноиды, где d0 максимально. У европия,
как уже говорилось, модификация с иной, чем ОЦК, структурой,
по-видимому, вообще отсутствует. Последовательное уменьшение d\
сопровождается ростом температуры перехода в ОЦК модификацию.
Дифракционные исследования строения жидких лантаноидов не
производились. По аналогии со щелочными металлами и другими
металлами, имеющими ОЦК структуру вблизи температуры плавления,
можно полагать, что ближняя упорядоченность типа ОЦК упаковки
у многих жидких лантаноидов сохраняется. Косвенно об этом
говорят малые величины энтропии плавления, как правило, примерно
такие же, как у щелочных металлов, ASnjI^6,7 Дж/К-моль.
Теплоемкость Ср жидких лантаноидов вблизи температуры плавления
определена неточно. Тем не менее все же отметим, что величины Ср
относительно малы именно у жидких европия и
иттербия, где межатомные расстояния в ряду
лантаноидов максимальны. Температуры
кипения многих из лантаноидов определены не
очень точно, но в целом прослеживается
постепенное, хотя и не регулярное понижение
Ткш с ростом порядкового номера
лантаноида. Поэтому температурный интервал, в
котором существует жидкая фаза, постепенно
сужается. Если у церия разность А Тж = Ткш—
— Тпл превышает 1100°, то у эрбия &Тт
лишь немногим более 100°, а у лютеция около
200°. Вместе с тем у иттербия Д Тж ^ 700°, а у
европия Д7Ж составляет около 1800° —
максимальная величина среди лантаноидов.
Отсюда можно заключить, что свойства простых
жидкостей и, в частности, их устойчивость
весьма существенно зависят от деталей
строения электронных оболочек их атомов.
0 Ю 20 30 40 50 60 70
Р,кб
Рис. 46. Фазовая
диаграмма церия [87]
186
Большой интерес представляют сведения о фазовых равновесиях
лантаноидов при высоких давлениях. На рис. 46 изображена
диаграмма фазовых равновесий церия. С увеличением давления температура
плавления церия падает, проходит через минимум и затем возрастает.
Энтропия плавления при атмосферном давлении равна 5,10 Дж/К- моль.
Из уравнения Клапейрона—Клаузиуса (IX. 1) следует, что
плавление церия сопровождается уменьшением объема и составляет около
2,5 об.% твердой фазы. Вероятно, при плавлении координационное
число растет, возникают фрагменты плотноупакованных структур.
В точке минимума кривая плавления церия встречается с кривой
равновесия 8- и f-церия, как это показано на рис. 46. С исчезновением
фазы, имеющей ОЦК упаковку атомов, кривая плавления
приобретает «нормальный» вид. Для плавления плотноупакованных твердых
фаз требуется образование вакансий, т. е. рост объема. Как только
плотность жидкой фазы в точке плавления становится меньше
плотности твердой фазы, рост давления сопровождается увеличением
температуры плавления церия.
Энтропия плавления церия, празеодима и неодима значительно
меньше энтропии плавления других лантаноидов. Если это
обусловлено уплотнением структуры при плавлении, то фазовые диаграммы
празеодима и неодима должны быть похожи на фазовую диаграмму
церия.
Фазовые диаграммы европия и иттербия похожи друг на друга и
на фазовые диаграммы бария и стронция — соседей лантана в
периодической системе.
Диаграммы плавкости европия и иттербия проходят через
максимум, что дает основание предполагать следующее. При плавлении этих
металлов возрастает число вакансий, но наряду с фрагментами ОЦК
структуры появляются фрагменты плотных упаковок. В результате,
среднее координационное число растет. С ростом давления
концентрация фрагментов плотных упаковок в расплавленном металле
увеличивается, поэтому плотности жидкой и твердой фаз постепенно
сближаются. Они становятся одинаковыми в точке максимума кривой
плавления. При более высоких давлениях фрагменты плотных упаковок
в жидкой фазе играют основную роль. Среднее координационное число
возрастает до ~10—11. Плавление сопровождается сжатием системы,
поэтому температура плавления с ростом давления уменьшается. Так
продолжается до тех пор, пока твердая фаза сохраняет ОЦК
структуру. При еще более высоких давлениях следует ожидать, что в твердой
фазе произойдет переход от ОЦК упаковки к плотной упаковке. Тогда
плавление вновь будет сопровождаться увеличением объема и
температура плавления опять начнет расти.
Энтропия плавления большинства лантаноидов мало отличается
от энтропии плавления европия и иттербия. Отсюда можно
предполагать, что зависимости температуры плавления от давления
лантаноидов, как правило, похожи на диаграммы плавкости европия и
иттербия.
Актиноиды (Th, Pa, U, Np, Pu, Am, Cm, Bk). Строение и свойства
актиноидов столь же интересны, как и лантаноидов.
187
И"
к
5! '"
I-01 • >
BirifeiDHcto
.8вг,
О ^- ,О> ^.О О
"СГ> - -
£2S
о ^ О ® ® ^
LO О о О СО "^
со (М г^ Ю Tf Ю
^ "^ СО СО <М СО
s- a
Юго^Пс£>С
- -to о - -
ЮО t -COOOtN
CM
00
00OCDCCNOOO
(NOO^COt-HO
О CO ^t C5 (N rt* (D 00
CN Л
oocS<§< ^
со* со со со со со* со
дааЯа^гда
oooooooo
CO --H CO
ю to со
Имеющиеся в литературе
сведения об актиноидах
неполны.В табл. 15 содержатся
данные о структуре
актиноидов в точке плавления и ряде
других термодинамических
свойств, полезных для
характеристики жидких фаз [25-—
—26].
Следуя принятому
порядку, рассмотрим вкратце
строение твердых фаз. Торий
образует две аллотропные
модификации, ос-торий имеет
ГЦК структуру. При 1380°С
сс-торий переходит в Р-торий
с ОЦК структурой.
Протактиний при 20°С обладает объ-
емноцентрированной
тетрагональной решеткой. Сведения
о его структуре вблизи
температуры плавления
отсутствуют. Уран известен в трех
аллотропных модификациях.
Кристаллы а- и {J-урана имеют
сложную слоистую
структуру. В слоях основную роль
играют ковалентные
межатомные связи, между слоями —
металлические. При 770°С
образуется ^-модификация с
ОЦК структурой и плотность
урана немного
уменьшается — примерно на 0,02%. В
точке плавления плотность
падает на 8%.
Нептуний подобно урану
имеет а- и р-формы со
слоистой структурой ковалентно-
металлического типа и
высокотемпературную ОЦК 7Ф
зу. Переход в ОЦК фазу
сопровождается увеличением
объема на ~ 7%.
Плутоний образует шесть
аллотропных форм. Три
низкотемпературные формы (а, р
и ^) имеют сложные кова-
лентно-металлические струк-
188
туры. Переходы а — р и р — f сопровождаются падением
плотности на 9,62 и 2,67% соответственно. При 319° С образуется ГЦК
3_плутоний и плотность падает еще на 7%. При 451°С появляется
тетрагональный S'-плутоний; плотность увеличивается на 0,36%. Наконец,
при 476° С возникает ОЦК е-форма плутония, причем ее плотность на
2,16% ниже плотности В'-фазы. Плавление s-плутония опять
сопровождается падением плотности, но на этот раз на 0,82%.
Америций существует в трех аллотропных модификациях.
Структура а-америция подобна гексагональной плотной структуре а-лантана.
При 600° С происходит превращение в [3-форму, имеющую ГЦК
структуру. Строение '(-америция пока не установлено. Кристаллическая
структура, температура плавления и плотность металлического
америция близки к этим свойствам лантаноидов и существенно отличаются
от свойств нептуния и плутония. Металлические кюрий и берклий
близки по своим свойствам к америцию. а-Кюрий и а-берклий имеют
гексагональную плотную упаковку типа а-лантана. [З-Кюрий и р-бер-
клий аналогичны ^-америцию. ^-Кюрий и ^-берклий пока не
обнаружены, но их существование вероятно.
Таким образом, все хорошо изученные вблизи температуры
плавления актиноиды подобно лантаноидам имеют ОЦК структуру. Но в
отличие от лантаноидов четкой связи межатомных расстояний в твердой
фазе с температурами плавления и кипения не наблюдается. При
плавлении существенных изменений координационного числа, видимо, не
происходит. Об этом говорят такие же, как у лантаноидов, малые
энтропии плавления, не превышающие (там, где они надежно определены)
8 Дж/К • моль. Изменение энтропии при испарении жидкости имеет
тот же порядок, что и у лантаноидов.
§ 74. Подгруппы титана, ванадия,
хрома и марганца
Подгруппа титана (Ti, Zr, Hf). Кристаллы каждого из этих
элементов существуют в двух модификациях. Низкотемпературная а-мо-
дификация характеризуется гексагональной плотной упаковкой
атомов; ^-модификация обладает ОЦК структурой. Отношение
параметров решетки с/а у гексагональных упаковок составляет около 1,59;
оно несколько меньше, чем при гексагональной плотной упаковке
шаров. Энтропия плавления титана, циркония и гафния мала. Хотя
дифракционные исследования строения жидких фаз еще не производились,
можно думать, что в окрестности температуры плавления среднее
координационное число атомов жидкости остается почти таким же, как в
ОЦК кристаллах (см. табл. 16). Температурный интервал
существования жидкой фазы очень велик. У циркония он составляет более 2500 К,
а у гафния — более 3000 К. Можно предполагать, что в жидкой фазе
четыре валентных s- и d-электрона обобществлены и таким образом
концентрация электронного газа (или «электронной жидкости») велика.
Поэтому жидкая фаза сохраняет устойчивость до температур 4—5 тыс.
градусов. С этих позиций можно пытаться истолковать и аномально
большие энтропии испарения. Они могут быть обусловлены иониза-
189
итана
и
«j
с
Подгруп
—|<§
™?v
™#%
—я*
«m^tf»
"и
"VlV
*"™Z
WHj_0I • V
-,
Hdii Biri/'BX3Hd>i
BdAxHiCdxQ
4If0^;
Элемент
O(N(M
СЧ CN CM
429,1
582
(661)
3550
4650
(5500)
00O?_v
8,15
(7,8)
(8,8)
Щ
CO
1940
2125
2493
CO O5 h~
CN CO" CO
sss
oooo oo
OOO
^ CD CD
Титан
Цирконий
Гафний
Я
Я
о
ч^ои-^/ж1^
"""sv
Ч1гом/ж^/м
'иэи//у
>I 'UH}Ii
sx*
ffU^ ndu
T_wT.wo
'*-01-Ж*
qirow-^/жЬ1
«Жс/^
qirow-M/^YT1
'""sv
qirowy^tf м
■""яу
»1WJ
жг
8Хг
""j Hdu
BlflTBXDHdM
BdAxMAd xq
qIfOIt/жfи
■86SHV
Элемент
CD OCO
Ю ^ <M
<M CO CO
O5 O)
Ю CT> Ю
rf CD t"»
3650
5200
5700
o>o>
o"o"
Ю (M
oo o>
(NCO1^
о оГсо
*o со 52,
7,74
(9,6)
(9,6)
16,96
(26,8)
(31,8)
2193
2740
3250
~H 4f Tj<
О- О> О5
ofcsfcs
oo'oo'oo4
00 00 00
а'^гя'
OOO
00 CM~
to СЧ CSJ
«(N00
юьь
Ванадий
Ниобий
Тантал
190
c\3
s
4
Ю
ed
о
о.
X
a
с
I
ri*
иш£ Hdu
™'m
»-,
«./^
Hdu ^ ^
*.-oi-ж«
4JU«
-**
ин T_oi • °p
жг
ахг
BIflfFXDHdH
Bd/CxM^dxQ
ВДОК/Ж№
Элемент
ою t^
см со со
о о
"^G О
СОЮ 00
2915
5100
5800
00 О>
сГо"
СО 00
(39,3)
34,3
38
6,36
9,54
(9,6)
со
00 СОЮ
со r-^rt*
^ СМ СО
2176
2890
3650
ю
см см" см"
ОО'оо'оО*
оооо оо
оцк*
оцк
оцк
00 СЛ ^t4
СО CD 00
Хром
Молибден
Вольфрам
S
ей
U
Q.
сЗ
ев
С
С
>>
о.
и
ее
О
С
-&«
--«
•V-.
.Z Hdu
ч«л^
™'Ч
-^
М-,
«н01. 0р
жг
TS^J
Элемент
95,4
(117,0)
120,2
00
СМ Ю О-
2314
(4900)
5900
СО CD
—Г о
ю с^-
см" о"
СО СО СО
со со со
(14,6)
(23,0)
(33,1)
*-< ^ ю
1П Tf Tf
i-< CM CO
(£)-н1О
CO t^. O
CM CM <N
00 CM CM
ОС С
OO^N
CM CO t^
Марганец
Технеций
Рений
191
цией паров и относительно резкой перестройкой электронной
структуры системы при испарении жидкой фазы.
Подгруппа ванадия (V, Nb, Та). Ванадий, ниобий и тантал имеют
только одну устойчивую кристаллическую фазу с ОЦК структурой.
Свойства жидких ванадия, ниобия и тантала мало изучены.
Приведенные в табл. 17 данные показывают, что эти жидкости по своему
строению и свойствам, видимо, во многом подобны простым жидкостям
подгруппы титана. При плавлении концентрация электронов
проводимости почти не меняется, потому что электропроводность остается
почти такой же, как в твердой фазе. Концентрация обобществленных
электронов в жидкой фазе должна быть несколько выше, чем у металлов
подгруппы титана, так как атомы имеют пять валентных электронов.
Соответственно сказанному ранее, температуры плавления и кипения,
а также энтропии испарения металлов подгруппы ванадия больше,
чем у металлов подгруппы титана. Энтропии плавления имеют
величины, обычно наблюдаемые при плавлении кристаллов с ОЦК
структурой.
Подгруппа хрома (Cr, Mo, W). В последовательности хром •—
молибден — вольфрам температуры плавления и кипения сильно
возрастают (см. табл. 18). В три раза увеличивается интервал
температур, в котором устойчива жидкая фаза. В конденсированных
фазах подгруппы хрома происходит обобществление не только s-элек-
тронов, но и части d-электронов с образованием d-зон. В
последовательности хром — вольфрам доля d-электронов, участвующих в
образовании d-зон, растет. По современным представлениям именно этот
фактор повышает устойчивость твердой и жидкой фаз в металлах
подгрупп хрома и соседних подгрупп [7]. Электропроводность при
плавлении уменьшается незначительно, в основном за счет разупорядочения
решетки. В точке плавления металлы имеют ОЦК структуру.
(Относительно хрома сведения разноречивы; возможно существование
устойчивой высокотемпературной ГЦК модификации). Малая энтропия
плавления указывает на вероятность сохранения фрагментов ОЦК
структуры в жидкой фазе.
Подгруппа марганца (Mn, Tc, Re). Марганец имеет четыре
устойчивые кристаллические модификации. Низкотемпературные а- и р-мо-
дификации марганца имеют сложную ковалентно-металлическую
структуру. При 1100°С образуется ^-модификация с ГЦК плотной упаковкой.
При 1130°С 7"маРганеЦ переходит в 8-марганец с ОЦК
упаковкой атомов. Температуры плавления и кипения марганца значительно
ниже, чем технеция и рения. Концентрация коллективизированных
(почти свободных) электронов в конденсированных фазах марганца
меньше, чем у рения и технеция. Полагают, что в марганце
обобществляются в основном лишь s-электроны. У рения и технеция доля
обобществленных d-электронов возрастает. Малые изменения энтропии и
проводимости при плавлении дают основание считать, что среднее
координационное число жидких металлов подгруппы марганца мало
отличаются от координационного числа соответствующих твердых фаз
(см. табл. 19).
192
§ 75. Подгруппы железа
и платины
Как известно, элементы
подгруппы железа и платины
в свою очередь
подразделяются на металлы подгруппы
железа (Fe, Co, Ni) и металлы
подгруппы платины (Ru, Rh,
Rd, Os, Ir, Pt), которые по
сходству их свойств делятся
на три диады: рутений и ось-
мий, родий и иридий,
палладий и платина. Структуры
внешних электронных
оболочек атомов металлов
подгруппы железа и платины
приведены в табл. 20. Там же
содержится ряд других данных,
представляющих интерес для
качественной характеристики
строения расплавленных
металлов этой подгруппы. Так
как все эти металлы
тугоплавки, строение и свойства их
расплавов пока еще слабо
изучены.
Твердое железо
существует в трех устойчивых
полиморфных модификациях. До
911°С устойчива ОЦК
модификация. Эта модификация фер-
ромагнитна до 768°С (а-жел езо).
Выше 768°С а-железо
переходит в парамагнитное ^-железо
без изменения структуры. При
91 ГС происходит
превращение ОЦК структуры в ГЦК
модификацию ^-железа.
Наконец, при 1392°С снова
появляется ОЦК модификация
(8-железо). Эта модификация,
по-видимому, плотнее, чем
а-железо. Во всяком случае,
в точке плавления 8-железа
расстояние между соседними
атомными ядрами в жидкой
фазе по результатам
рентгенографических измерений
Я"
К
•=2
VO
о
'9-01 -
"sv
zOV
wh ,_0Г°Р
Н V
< см • o
о
COCOCOti
cs t^. ю
о" о" о"
СО OOCN ^^
СО Tj< »-H QQ QQ СО
Ю СО CD
со ю t^fc S <о -Гсо^
— ^ -ч ^^L^ CO C^ CS
со со ^f
ОЮООСОСОЮЮО-СЧ
OCOCSOOCOCM^fCM'^
19a
равно 0,251 нм, а у низкотемпературной фазы это расстояние
d'0=0,254 нм. После плавления среднее координационное число 8 в пре.
делах ошибок рентгенографических измерений не меняется. Рост
температуры плавления железа с давлением указывает на то, что
фрагменты ОЦК структуры сохраняют устойчивость и при повышенных
давлениях. Энтропия плавления железа такая же, как у большинства
металлов с ОЦК структурой.
Кобальт имеет две кристаллические модификации. Ниже 420° С
устойчив а-кобальт, имеющий гексагональную плотную упаковку
атомов. Выше 420°С и до температуры плавления устойчив ^-кобальт,
обладающий ГЦК структурой. Кристаллический никель имеет только
одну термодинамически устойчивую фазу с ГЦК упаковкой атомов.
Платиновые металлы не имеют устойчивых полиморфных
модификаций. Структура кристаллов платиновых металлов указана в табл. 20.
В каждой триаде (Fe, Co, Ni), (Ru, Rh, Pd), (Os, Ir, Pt) наблюдается
понижение температуры плавления и кипения с ростом порядкового
номера элемента. Энтропии плавления кобальта и последующих
металлов подгруппы железа и платины выше энтропии плавления железа и
в пределах ошибок опыта почти одинаковы. Во всех этих случаях
плавление связано с возрастанием числа вакансий в гранецентрированной
или гексагональной плотных упаковках атомов. Рост числа вакансий
сопровождается увеличением объема на 3—4% для металлов
подгруппы железа. Для платиновых металлов экспериментальные данные
отсутствуют. Можно полагать, что их поведение такое же.
В последовательностях Fe, Ru, Os, затем Со, Rh, Ir и Ni, Pd, Pt
температуры плавления и кипения резко возрастают. Видимо,
концентрации обобществленных электронов в конденсированных фазах
металлов растут в этих последовательностях. Как показывают
имеющиеся экспериментальные данные для железа, кобальта, никеля и
платины, электропроводность расплава вблизи точки плавления мало
отличается от электропроводности твердой фазы. Электронные
состояния металлов подгруппы железа и платины при плавлении не
претерпевают существенных изменений.
Пары металлов подгруппы железа и платины при относительно
низких температурах вблизи точки плавления практически
одноатомны. Так, например, при 1450° С суммарное давление паров молекул
палладия Pd2, Pd3 и Pd4 составляет менее 10~3 Па. При 1700° G
концентрация атомов Pd в 3-Ю5 раз выше концентрации молекул
Pd2. Но при повышении температуры с ростом общего давления паров
концентрация молекул палладия увеличивается.
§ 76. Подгруппы меди и цинка
Подгруппа меди (Си, Ag, Аи). В кристаллах и расплаве меди,
серебра и золота концентрация свободных электронов приблизительно
одинакова и составляет около одного электрона на атом. Обобществлены
электроны s-состояний. Предполагается, что й10-электронные оболочки
атомных остовов перекрываются слабо, поэтому взаимодействия
между атомными остовами в кристаллах и жидкой фазе не характеризуются
194
Отчетливо выраженными преимущественными направлениями. Атомы
металлов подгруппы меди в твердой фазе образуют плотную гранецент-
рированную кубическую упаковку.
При плавлении эта структура не претерпевает существенных
изменений. Средний радиус первой координационной сферы во всех
случаях остается тем же, что и в твердой фазе. Координационное
число уменьшается незначительно, по-видимому, за счет возрастания
концентрации вакансий. Температура плавления при повышении
давления растет.
Относительный рост объема металла AV7FTB при плавлении для всех
трех металлов близок. Энтропии плавления и испарения,
электропроводность а и отношения аж/атв различаются мало (табл. 21). Любопытно,
что они и ДV71/TB имеют в точности ту же величину, что и у алюминия
(см. табл. 23). Структура алюминия такая же, как у металлов
подгруппы меди, концентрация почти свободных электронов значительно
выше. По расчетам Т. Фабера [7], отношение длины свободного
пробега электронов в жидкой фазе к среднему межатомному расстоянию
у алюминия равно 6, у меди 13, серебра 18 и золота 10. Пары металлов
подгруппы меди, подобно парам алюминия, в основном одноатомны,
но, содержат небольшие (порядка 1%) примеси двухатомных и,
возможно полимерных молекул. Энергии диссоциации равны (в кДж/моль)
201 для Си2, 173,5 для Ag2 и 216 для Аи2.
Отметим, что энергия диссоциации А12 равна 192,5 кДж/моль,
и, следовательно, здесь тоже наблюдается сходство свойств.
Подгруппа цинка (Zn, Cd, Hg). Кристаллы цинка и кадмия имеют
гексагональную упаковку атомов. Но в отличие от плотнейшей
гексагональной упаковки сферических атомов решетки цинка и кадмия
вытянуты в одном направлении. Каждый атом окружен шестью другими
атомами, лежащими в одной плоскости или слое. Расстояние между
центрами соседних атомов в этом плоском слое а равно 0,26649 нм для
цинка и 0,29788 нм для кадмия. Назовем этот слой первым слоем. Выше
и ниже произвольно выбранного центрального атома в первом слое
находится еще по три атома, удаленных на несколько большее
расстояние, равное 0,2907 нм для цинка и 0,3287 нм для кадмия. Эти шесть
атомов представляют собой фрагменты слоев, расположенных над
первым слоем или под ним. Следующие за ними верхний и нижний
слои имеют точно такое же расположение атомов, как в первом слое.
Расстояние между центрами одинаково расположенных атомов в
первом и третьем слоях с равно 0,49468 нмдля цинка и 0,56167 нм для
кадмия. Отношения с/а равны соответственно 1,8563 и 1,8856. Таким
образом, решетки цинка и кадмия являются слоистыми. Число
атомов, составляющих ближайшее окружение любого атома решетки в
слое, равно шести. Следовательно, здесь наблюдается тенденция
к выполнению правила 8—N, где N — число валентных электронов,
равное 2 для цинка и кадмия.
Кристаллы ртути имеют решетку несколько иного типа. Каждый
атом в этой решетке окружен шестью другими атомами на расстоянии
3,3005 нм (при —46° С) и еще шестью атомами на расстоянии 0,3477 нм.
Гип решетки относится к гексагональной сингонии с параметрами эле-
195
cd
Cf
я
ЯХ /Ж
с/ с
1
«J
с
I
с
■01-
ЛУ
1:ч 5
У-
II&
СО ^ СО
о о о
ЮЮОО
00 ^О5
СЧ (М <N
S8S
00 00 ^Н
о* о" о"
сосооъ
со со см
S28
СО ^Ф СМ
(CON
О СО СО
со*4 _Tci
ЮСО —
•^сою
СО тР CD
tO СО СО
со см со
> СО СО
> 00 00
СО 00 00
LO 00 00
ЗПГЗ
UUU
Oi CD СО
СО 00 СО
СО СМ СО
II
г л ~\
о
a
X
S
с
с
О,
и
С
ишХ udu
qiro*;*«V
иинг Hdu
чи'ом/ж^/м
<иэи//у
иил
ж„
QI ,Ж
ITU j *
Jj HQU
'СоТ-^
чьто^'^/ж^т1
♦ Же/ п
ЧГГОМ'^/ж^/
SV
чггом/ж^/м
iIfII//V
ex
*>т Л
ЛУ
М i]rul
ж
V
о
N
JnT
as g
&3кв
£5к
ом
чи-ои/жЬ'н
о
о
S
0>
СО
S7,
СО
ю
00
см
%
о
t^-
см
СО
о
R
•^f
i>T
см
^jT
t>-
я
СО
35
СМ
S
СП
СП
8
о
см
о
о
СП
см
t>.
29,
§
СП
F^
о
со
^.
ю
8
со
ON00N
СО О
СО СО
Г^ 00
СП СП
см
$
СП ~~
СП
ю
СП
СО
см
^о
о°
см
СО
"-4
СП
Tt«
а
СП
8:
см
см
со
СП
см
S
со
о о
см см см со со со
00
о
со
+
со
и*
с
см
130
Цинк
со
00
со
+
со
с
111
»к
S
СО
t^
ui"9-
ю.оо
t^
со
+
со
>5
и.
с
,, ,
61,
Ртуть
196
ментарной ячейки а = 0,3463 нм; с =0,6706 нм и углом наклона между
а и д а=70о32'. Таким образом, структура ртути более рыхлая, чем
цинка и кадмия: с/а = 1,94. Координационное число г ртути тоже
следует правилу 8—N. С ростом отношения с/а температура плавления
металлов подгруппы цинка падает. Среднее координационное число
атомов этих металлов в жидкой фазе определено с помощью
рентгенографических и нейтронографических методов не очень точно.
По-видимому, существенного изменения типа их структуры при плавлении
не происходит. И. В. Радченко [19] полагает, что в точке плавления
строение жидкой ртути почти такое же, как у твердой фазы. При более
высоких температурах среднее координационное число атомов жидкой
ртути растет и структура приближается к плотноупакованной. Это
сопровождается увеличением средних межатомных расстояний за счет
теплового расширения.
Энтропии плавления всех трех металлов невелики (табл. 22). Это
согласуется с выводом об отсутствии коренной перестройки структуры
при плавлении металлов подгруппы цинка. Повышение давления
сопровождается ростом температуры плавления ртути. При давлении
6,0 ГПа ртуть плавится около 200° С.
В литературе имеются указания о возможности существования в
расплавах цинка, кадмия и ртути остатков
твердой фазы, имеющих размеры порядка 1 нм[27].
Хотя среднее число почти свободных
электронов на один атом у всех металлов под- п(Е)
группы цинка близко к двум, поведение
электронов ртути во многом аномально. Ее
электропроводность почти в три раза меньше,
чем у цинка и кадмия, и быстро растет с
увеличением давления. Термоэлектродвижущая
сила ртути тоже аномально велика.
Как показал Н. Мотт [28], аномальное по- п([)
ведение ртути объясняется тем, что валентная
зона, электронные состояния которой
полностью заполнены, слабо перекрывается со
следующей зоной. В результате, как видно из
графика на рис. 47, а, описывающего
зависимость плотности вероятности электронных ^(Г
состояний от их энергии, образуется провал
«псевдощель». При снижении плотности
ртути провал становится глубже (рис. 47, б).
Когда плотность ртути мала, «псевдощель»
переходит в обычную зону запрещенных
состояний и металлические свойства ртути исчезают
(рис. 47, в).
Пары этих металлов в основном
одноатомны, присутствуют лишь следы димеров.
Энергии диссоциации димеров: 25,1; 12,55 и
9,6 кДж/моль для Zn2, Cd2 и Hg2
соответственно.
6 и
Рис. 47. Плотность
вероятности электронных
состояний ртути по Мот-
ту [28]:
а — жидкая ртуть; б — ртуть,
когда ее плотность (в кГ/м3)
такова, что электропроводность
<у находится в интервале 3—20
Ом-^-м"1; в—плотность ртути
очень мала
197
ГЛАВА X
ПОДГРУППЫ БОРА, УГЛЕРОДА, АЗОТА,
КИСЛОРОДА, ФТОРА И ИНЕРТНЫХ ГАЗОВ
§ 77. Подгруппа бора
(В, Al, Ga, Inf Tl)
В соответствии с правилом, упомянутым при описании щелочных
металлов, свойства бора напоминают свойства углерода, а алюминий
сходен с элементами подгруппы скандия. Пары в основном одноатомны,
но содержат и двухатомные молекулы. Энергии их диссоциации в
кДж/моль при 25° С равны: В2 264,5; А12192; Ga2150; In297,5; Т1261,5
[16]. Эти молекулы более прочные, чем двухатомные молекулы
элементов подгруппы лития и бериллия, расположенных в тех же периодах
системы Менделеева.
Строение жидкого бора пока не исследовано, так как бор плавится
при высокой температуре — около 2300° С. Кристаллы бора тверды и
слабо проводят ток. Они имеют металлический блеск. Их
электропроводность с ростом температуры быстро увеличивается, поэтому
предполагается, что вблизи точки плавления бор становится металлом [8].
Хотя бор вдали от точки плавления неметалл, его строение не следует
правилу z =8—ЛЛ Поэтому правилу атомы бора должны быть
пятивалентными. Но в решетке бора, имеющей сложную тетрагональную
структуру, половина атомов образует шесть связей, а другая
половина — четыре связи, так что в среднем получается пять связей на атом.
Строение других простых жидкостей подгруппы бора изучалось
рентгенографически [19]. Положение главного максимума на
рентгенограмме жидкого алюминия вблизи точки плавления (700° С) совпадает
с расстоянием между соседними атомами в кристаллах. Алюминий крис*
таллизуется с образованием гранецентрированной кубической решетки.
В расплаве фрагменты ГЦК структуры сохраняются. Небольшое
снижение координационного числа после плавления, по всей вероятности,
связано с ростом концентрации дефектов (пустот) в ГЦК структуре
ближнего порядка, что ведет к увеличению объема в точке плавления
на 6%. Рост давления сопровождается увеличением температуры
плавления алюминия. В кристаллах алюминия на один атом
приходится 2 электрона проводимости. Плавление не сопровождается
существенным изменением концентрации этих электронов.
Кристаллы галлия имеют ромбоэдрическую структуру. Его атомы
образуют неплоские сетчатые слои, ячейки которых — неправильные
шестиугольники. В слое каждый атом связан с тремя другими атомами
галлия. Из них один находится на расстоянии 0,244 нм и два на
расстоянии 0,271 нм. Кроме того, два ближайших атома двух соседних слоев
(по одному от каждого слоя) расположены на расстоянии 0,274 нм.
В итоге общее число соседних атомов равно пяти, только размещены
они на разных расстояниях, поэтому правило 8—N не выполняется.
198
Структуру кристаллов
галлия можно описать и с
несколько иных позиций,
приняв, что в твердом галлии
имеются молекулы Ga2, которые
соединены друг с другом
несколькими более слабыми
химическими связями. На
каждый атом галлия приходится
один электрон проводимости.
Проводимость галлия такая
же, как у щелочных
металлов.
При плавлении
распределение атомов галлия
существенно изменяется — молекулы
Ga2 распадаются. Это
сопровождается аномальным ростом
энтропии, ASnjl равна 18,5
Дж/К • моль. И в результате
образуется плотноупакован-
иая структура с
координационным числом, близким к 12.
Ближайшее расстояние между
атомами в среднем равно 0,27/
нм. Отметим, что около точки
плавления в жидком галлии
обнаруживаются фрагменты
кристаллической структуры.
Они исчезают при нагревании
жидкости на 15—20° выше
температуры плавления [19]. Так
как при плавлении галлия
координационное число
увеличивается и плотность
растет, то температура плавления
галлия падает с повышением
давления. При 1,2 ГПа
появляется более плотная
модификация Gall. Кривая
плавления этой модификации имеет
положительную производную
dT/dP.
Индий кристаллизуется в
гранецентрированной
тетрагональной решетке, близкой
к ГЦК (v^1'075)- B
кристаллах индия атомы имеют
со
СМ
СЗ
Sf
Я
2.
«8
руппа
I
X Hdu
[Ч1Г01М ■}[/ Ж|/
' UDHsv
IIH'Mj, ndu
'UDh//V
, 11ИМ
Ж,,
iru ncju
U J Ж
с/ r>
vu£ ndu
W • NO
*£-01-Ж*
ЧИ'ОИ-^/Ж^
. ЖсГ9
4L-ow-M/>Ktr
41* ii _.
ПТ Л
гО I uil
м ДГ"^
i-ОГ V
imh x_oi P
CQ
H
&Я
б|=
Ч1ГОМ/Ж<С>1
э
g
I
CM
CM
CO
стГ
CO
Ю
о
о
CM
,—-.
t»-
(32,
CO
l^.
en
CM
CM
300
CM
CM
00
j;
4-
^ с
CO
cu
t-
<v
g-
tQ
oo
00 CO
о о
СП ~
CO t4-
cn >o
(>>! CM
oo
CM —«
Г— LO
CM CM
CO
Ю
см
СО
см^°
OCM |
— en
^Ф СО
о" о"
OO ^*
t^°o^
СО СМ
t^. »-.
TftO
z:S
СО "4f
1>- СП
СОЮ
ою
OCvj
со с
см со
СО О
СП СО
сп
г~-
-00 -
СМ 1- СМ
оГ
СО оо
00 Г-
см см
со сп
1
со
ч^ *2
исх
rf со
со см
S 9S
о "я ч
£sq
^ CU
< U
CD
(s^
СП
ю
СО
см
С-4
о
сч
со
см
со
5?
о"
о
со
о"
см
LO
сп
сч
о-
со^
г—
сч
со"
см
см
со
СП
см
i СМ
СП
сч
СП
см
см
со
»—<
о
см
СП
°
о"
*~1
о"
со
г-
см
^
г-
см
^л
см
со*
577
1 со о
со -со
—. со
* 1
00 1
см
СО
со
о
"7
оо
00
н-
Он
н
н
й
t^
со"
см
92
[нди.
S
со"
см
'-*F
со
00
00
о
о
00
алл!
н
199
четырех ближайших соседей на расстоянии 0,324 нм и восемь ца
расстоянии 0,337 нм. Координационное число атомов жидкого
индия немного выше точки плавления составляет около 10,5.
Следовательно, жидкий индий обладает довольно плотной упаковкой. При
повышении температуры до 650° С среднее координационное число снижается
до 9,4[29]. Малые изменения энтропии и объема при плавлении индия
(табл. 23) также говорят об отсутствии глубоких изменений ближнего
порядка.
Твердый таллий вблизи точки плавления имеет ОЦК структуру.
AV
Прирост энтропии Д5ПЛ = 7,5 Дж/К • моль и объема ~Г/—==3»2% при
У тв
плавлении близки к изменениям, которые испытывают другие металлы,
имеющие ОЦК структуру и плавящиеся с сохранением
координационного числа 8. Производная dTldP кривых плавления индия и таллия
положительна, но уменьшается с ростом давления. Интересны
результаты рентгенографического изучения переохлажденного жидкого таллия
при температурах ниже 300° С [30]. При 290° С координационное число
составляет около И. Повышение температуры сопровождается
постепенным уменьшением координационного числа. Первый максимум на
кривых радиального распределения атомов жидкого таллия
асимметричен. Его абсцисса составляет около 0,33 нм. Анализ кривых
радиального распределения и данных о плотности жидкого таллия,
выполненный авторами работ [30], показывает, что в первой координационной
сфере атомов таллия, вероятно, содержится 8 атомов. Это указывает
на сохранение фрагментов ОЦК структуры. О. В. Романова и
Б. А. Мельник [31] полагают, что в жидком таллии кроме ОЦК
структуры имеются фрагменты ПГУ структуры(плотноупакованиый
гексагональной). При повышении температуры доля ПГУ структур возрастает.
§ 78. Подгруппа углерода
(С, Si, Ge, Sn, Pb)
Атомы этих элементов проявляют большую склонность к
ассоциации. Прочность ассоциатов убывает с увеличением молярной массы
элемента. Энергия диссоциации двухатомных молекул в кДж/моль
при 25° С равна: С2603; Si2 314; Ge2 272; Sn2196; Pb2 100 [16].
Особенно велика способность к ассоциации у атомов углерода и кремния.
Присоединение к молекуле С2 еще одного атома углерода
сопровождается уменьшением энергии системы на 747 кДж/моль (при 25° С);
следовательно, С3 устойчивее, чем С2. Как показывают расчеты, энтальпия
реакции Сп = С^ + С для нечетных п составляет около 753 кДж/моль,
а для четных п — около 540 кДж/моль, причем п может быть сколь
угодно большим. Многоатомные ассоциаты кремния Si^ и германия Qen
тоже устойчивы. Энтальпия реакции Si4 — Si3 + Si составляет около
420 кДж/моль (при 1550° С); энтальпия реакции Ge4 = Ge3 -j-Ge
приблизительно равна 376 кДж/моль; энтальпия реакции Ge4 = 2Ge2
равна 468 кДж/моль [16].
Углерод. Широко известны две модификации твердого углерода:
алмаз и графит.
200
ж
Алмаз — изолятор, ширина его запрещенной зоны 5,6 эВ; его
удельное сопротивление при 25° С равно 5 • 1016 Ом • м. Алмаз
плавится при ~ 3800° С под давлением 1,0—2,0 ГПа. При
температурах выше 1000° С алмаз переходит в графит; теплота превращения
lf88 кДж/моль. Структуры алмаза и графита широко известны и мы
их описывать не будем. При 3700° С графит возгоняется. Он
плавится под давлением 10,3 ГПа около 3700° С. Вдоль оси а
(перпендикулярной слоям графита, образованным правильными шестиугольниками
из атомов углерода) графит слабо проводит ток, его
электропроводность составляет около 10~2 Ом"1 • м"1 при 25° С. Вдоль оси с
(расположенной в плоскости
упомянутых слоев) проводимость
графита составляет около
КЮм-^м"1, т. е. того же
порядка, что и у полуметаллов.
Строение жидкого
углерода пока еще не изучено.
Предполагается, что плавление
углерода сопровождается
ростом среднего
координационного числа, причем жидкий
углерод — металл. Очень
интересные косвенные указания
о строении жидкого углерода
можно получить из его
фазовой диаграммы,
представленной на рис. 48.
Графит имеет
куполообразную кривую плавления.
На левой ветви этой кривой, где производная dTldP положительна,
плавление, по-видимому, связано, в основном, с ростом концентрации
вакансий. Производная dTldP обращается в нуль при ~ 4600° С и
давлении около 6,0 ГПА (Н. С. Фатеева, Л. Ф. Верещагин [32]). На
правой ветви производная dTldP отрицательна и, надо полагать, что
плавление сопровождается ростом координационного числа. Для
алмаза вплоть до давлений порядка 6,0 ГПА производная dTldP
остается отрицательной.
Кремний кристаллизуется в решетке типа алмаза. В отличие от
углерода твердый кремний не имеет устойчивой графитоподобной
модификации. Кремний — полупроводник. При 20° С его проводимость
составляет около 10"8 Ом"1 • м"1. Кремний плавится при 1410° С,
поэтому изучение свойств жидкого кремния—нелегкая задача. Плавление
кремния сопровождается частичным разрушением алмазоподоб-
Ной структуры и уменьшением объема на 9,6%. При повышении
давления до 4,5 ГПа температура плавления кремния снижается до 1157° С.
Интересные и разносторонние исследования жидкого кремния высокой
степени чистоты, содержащего менее чем 10~6 мольн. % примесей, были
выполнены В. М. Глазовым и его сотр. [33]. Основным элементом
строения жидкого кремния, по-видимому, является размытый тепловым дви-
Алмаз+ \ Алмаз
метастад. \
графит. \
Рис. 48. Фазовая диаграмма углерода
[8]
8—503
201
жением объемноцентрированный куб (см. § 107). Разупорядочение
атомов при плавлении кремния сопровождается уменьшением
подвижности электронов примерно в 50 раз. Но концентрация электронов
проводимости возрастает более чем в 103 раз. В итоге электропроводность
при плавлении кремния резко увеличивается, так что кремний
переходит в металлическое состояние. Кремний диамагнитен—его магнитная
восприимчивость х отрицательна. Плавление сопровождается резким
уменьшением абсолютной величины |х| . Оно может быть объяснено
ростом концентрации электронов проводимости [33]. После плавления
абсолютная величина магнитной восприимчивости жидкого кремния
немного увеличивается с ростом температуры до значений,
превышающих температуру плавления примерно на 100 К. Дальнейшее
нагревание до температур порядка 1700° С не сопровождается изменением
магнитной восприимчивости. Вязкость жидкого кремния в интервале
температур AT ~ 120° выше точки плавления снижается с ростом
температуры сильнее, чем при более высоких температурах. Эти и ряд
других фактов приведенных в монографии В. М. Глазова, С. Н.
Чижевской и Н. Н. Глаголевой [33] дают основание полагать, что после
плавления кремния происходит постепенное разрушение фрагментов
алмазоподобной структуры с переходом к размытой тепловым
движением ОЦК структуре, характерной для многих металлов.
Германий тоже кристаллизуется в решетке типа алмаза. Каждый
его атом окружен четырьмя другими, находящимися на расстоянии
0,243 нм. Кристалл хорошо очищенного германия — полупроводник.
Ширина запрещенной зоны при комнатной температуре 0,72 эВ.
Электропроводность порядка 10~4 Ом"1- м"1 растет с температурой.
Плавление германия сопровождается увеличением координационного числа
от 4 до ~7. Одновременно возрастает и межатомное расстояние до
0,28 нм [19, 33, 34]. Резкое изменение структуры при плавлении
сопровождается очень большим приростом энтропии, А5ПЛ=28,85 Дж/КХ
Хмоль, и скачкообразным увеличением электропроводности. Жидкий
германий — металл (подробнее см. [21, 33]). Фазовые диаграммы
германия и кремния похожи. Кривые плавления имеют отрицательные
производные dT/dP.
Отметим, что энтропия плавления кремния примерно такая же,
как германия. Это дает основание предполагать, что при плавлении
среднее координационное число атомов кремния тоже возрастает от
4 до 8.
Результаты исследований свойств жидкого германия до температур
примерно на 350 К выше точки его плавления рассмотрены в кн.
В. М. Глазова, С. Н. Чижевской и Н. Н. Глаголевой [33]. Вблизи
точки плавления жидкий германий подобно кремнию содержит
фрагменты алмазоподобной структуры. Но концентрация таких
фрагментов меньше, чем у жидкого кремния, и при нагревании их влияние на
свойства расплавленного германия наблюдается в более узком
интервале температур AT ~ 80 К.
Олово имеет две модификации. а-Олово (серое) устойчиво ниже
13,2° С с алмазоподобной структурой. Кристаллы Р-олова (белое
олово) имеют объемноцентрированную тетрагональную структуру.
202
При превращении р-олова в
серое объем возрастает на
25,6%. При комнатной темпе-
ратуРе проводимость белого
олова равна ~103 Ом^-м"1.
Электропроводность серого
олова значительно ниже.
Энтропия плавления олова
в два раза меньше, чем
германия, но все-таки довольно
велика. Можно предполагать,
что плавление
сопровождается переходом ближнего
порядка к ОЦК структуре. По
рентгенографическим данным (см.
[21]) среднее
координационное число атомов олова в
жидкой фазе в пределах
ошибок опыта равно 8 и остается
таким при нагревании
жидкости даже на 900 К выше
точки плавления.
Свинец имеет гранецентри-
рованную плотноупакован-
ную кубическую структуру.
После плавления фрагменты
этой структуры сохраняются.
На это указывает сохранение
высокого координационного
числа и нормальные для
металлов, имеющих ГЦК
структуру, изменения энтропии и
объема при плавлении (табл.
24). Нагревание до 450° С
сопровождается
уменьшением координационного числа
до 9 (Я. И. Дутчак [35]). Это
может быть связано с ростом
концентрации вакансий в
жидкой фазе.
§ 79. Подгруппа азота
(N, Р, As, Sb, Bi)
Когда атомы простых
веществ подгруппы азота
образуют три химические связи
друг с другом, то валентные
углы между связями немно-
8*
03
Н
5
I
исШ
и<Ш
/IV
CO OS
эю s 00 со"
Э О О О) 00
CO Cl r^ О oT
°2^3 со С21^.
!§§ is
lO^f CO O) OJ
о
N
со SB
г
88
Ю00 Is-
5- 8£
СОЮ 00 СО
О Ю CN СО
СГ)\Л
I I
^ Iе?
С4 SCO
SS
qOoO b» 00 —«
S
«s
if
111 si
203
го превышают 90°. Молекулы, состоящие из 4 атомов, имеют форму
тетраэдра.
Пары элементов этой подгруппы содержат димеры, тримеры, тет-
рамеры и более сложные ассоциаты. Димеры азота очень прочны,
Энергия их диссоциации равна 945 кДж/моль. Тримеры значительно
менее устойчивы. Энтальпия реакции N 3 = N2 + N составляет всего
около 2,5 кДж/моль. В парах азота наблюдаются и молекулы N4,
С увеличением относительной молекулярной массы элементов под-
группы прочность димеров падает. Энергии диссоциации в кДж/моль
равны: Р2 488; As2 383; Sb2 295; Bi2 163. Но зато устойчивость более
сложных ассоциатов в ряду азот—сурьма возрастает (для висмута
данных нет). Энтальпия реакции Р4=2Р2 равна 228 кДж/моль;
As4 = 2As2 248 кДж/моль; Sb4 = 2Sb2 263 кДж/моль [16]. Примерно
в той же последовательности повышается устойчивость твердых и
жидких фаз. Растут температуры плавления и кипения.
Азот. Твердый азот существует в двух модификациях. Кубическая
а-форма при температуре — 237,5° С переходит в менее плотную
гексагональную (З-форму. Кривые атомного распределения жидкого азота
при 64 и 89 К, полученные Шарра в 1942 г., представлены на рис. 49.
Первый максимум при R ~ 0,13 нм изолирован. Площадь под ним
приблизительно равна одному атому. На этом основании полагают,
/
50
20
10
1
У
i
й
f
-/■
/.
/S
1
/ i
л
//
Ut
г
20
т
/ 2 J к 5 6 7
Я, А
Рис. 49. Кривые атомного рас- Рис. 50. Схема строения одного из слоев серого
пределения жидкого азота [37]: мышьяка или черного фосфора [36]
кривая (/) при 64 К; кривая (2) при (Атомы лежат на двух уровнях, отмеченных + и —. По-
89 К следовательные слои располагаются так, что нижние
атомы одного слоя лежат вертикально над «дырками» в
другом слое)
что жидкий азот состоит из молекул N2. На наш взгляд, этот
результат нуждается в проверке. Возможно, что в ранних работах по
рентгеновскому рассеянию площадь под первым максимумом была
определена недостаточно точно. Отметим, что расстояние между атомными
ядрами молекул N2 в парах азота равно 0,11 нм. Если абсцисса первого
максимума кривой атомного распределения Rml = 0,13 нм определена
204
правильно, то связь между атомами
азота сильно растянута за счет взаи-
модействия с другими соседними
атомами.
Далее кривая атомного
распределения имеет максимумы при 0,26; 0,4
и 0,48 нм. С нашей точки зрения, это
указывает на существование в
жидком азоте тримеров и более сложных
ассоциатов азота. Косвенным
указанием на присутствие таких
ассоциатов в жидком азоте и его парах
служит слишком малая величина
энтропии испарения; она равна 35,2 Дж/к-
• моль (табл. 25). Упомянутая
величина вычислена при допущении, что
испаряются не молекулы, а атомы
азота. Это, конечно, не так. Пусть
средняя молярная масса пара
соответствует N3, тогда ASHCn составит около
104 Дж/К-моль, т. е. будет такой же,
что и для других простых жидкостей
подгруппы азота с учетом их
ассоциации.
Фосфор имеет целый ряд
аллотропных модификаций. Основные:
черный, красный и белый фосфор.
При нормальных условиях наиболее
устойчив черный фосфор. Иначе
говоря, его свободная энтальпия
минимальна. Но потенциальный барьер,
препятствующий переходу метаста-
бильных модификаций в черный
фосфор, велик, поэтому при обычных
условиях черный фосфор не образуется.
Здесь тоже сказывается то интересное
правило периодической системы
элементов, о котором говорилось в гл.
IX при описании строения щелочных
металлов. Подобно графиту, черный
фосфор состоит из слоев Рп (рис. 50).
Атомы фосфора в слое группируются
в шестиугольники. Каждый атом
химически связан с тремя соседними
атомами фосфора.
Расстояние между соседними
атомами в слое равно 0,218 нм, а между
слоями 0,368 нм. В графите слои
плоские, в черном же фосфоре атомы
ю
сч
cr
s
ч
\о
cd
с
с
;>>
о.
I
41f°W'"D"sv
иин
Hdu чнчж/ж^н
-и
«.Л
4 ^ с/
ахл
,01 „„^^
Л'"1
ин T_oi • жр
ин :_oi • °Р
жг
ех2
HdU BlflfEJLOHdH
Tees^J
о
Элемент
35,2
25,5
(29)
35,63
82,71
2,721
14,07
(43,96)
67,95
151,6
77,3
548,1
(1500)
1910
1832
о о to ю
и г г
1,64
2,9—2.1
00 t-«.
о о
30,56
СОО5 ^Г О О>
со со t-* оо »-ч
^ LO
(NU0 СП СО
со со" о со
1 1
6,31
317,3
1096*
903
544,5
2,25-2,28
3,12
3,35
CN СО<МчГ
^ со en cs со
-н CN СМ СО СО
ol°?
— СО СО СО СО
гекс.
куб.
ромб,
ромб,
ромб.
473
334,1
289
262,5
208
Азот
Фосфор
Мышьяк
Сурьма
Висмут
S
205
слоя находятся на двух уровнях. Одни из атомов расположены над
плоскостью, проходящей через слои, а другие — на таком же
расстоянии под этой плоскостью.
Красный фосфор неоднороден. Его можно рассматривать как
твердый раствор ряда модификаций. Белый фосфор состоит из молекул Р4.
Существуют две модификации белого фосфора: ромбическая (3-форма
при —76,9° С переходит в немного менее плотную кубическую а-фор-
му. Ее плотность равна 1,828 г/мл при 20° С.
Белый и красный фосфор — диэлектрики; черный фосфор —
полупроводник. При 25° С ширина его запрещенной зоны составляет около
0,33 эВ. Электропроводность равна 7-10~3 Ом^-м"1.
Плавление черного фосфора происходит около 1000° С при
давлении 1,8 ГПа. Если же черный фосфор нагревать, не повышая
давления, то при 560—580° С он превращается в красный фосфор.
Температура плавления красного фосфора в зависимости от условий его
получения лежит в интервале 585—610° С. Белый фосфор плавится
при 44,1°С.
Кривые атомного распределения жидкого белого фосфора были
получены для температур 48° С и 226° С. Максимумы на кривых
распределения расположены при 0,225; 0,39; 0,6 нм [37].
Первый максимум изолирован и площадь под ним соответствует
примерно трем атомам [37]. Это дает основание считать, что белый
фосфор состоит из молекул Р4, имеющих форму правильного
тетраэдра. В парах расстояние между атомами фосфора в молекулах равно
0,221 нм (электронографические измерения). Небольшое увеличение
межатомных расстояний Р—Р у молекул Р4 в жидкой фазе можно
объяснить ассоциацией молекул Р4 (подобно увеличению расстояний I—I
в ассоциатах 12, описанных в гл. III).
Пары жидкого белого фосфора тоже состоят из молекул. Об этом
говорят не только измерения молекулярной массы паров, но и
нормальная величина энтропии испарения, если ее выразить в Дж/К-моль Р4
(см. табл. 25). Выше 800° С молекулы Р 4 диссоциируют с образованием
молекул Р2.
Мышьяк. Пары мышьяка так же, как и фосфора, до 800° С состоят
из молекул As 4. Выше 800° С они диссоциируют в заметных
количествах на молекулы As2. При 1700° С диссоциация на молекулы As2
заканчивается. Если пары мышьяка конденсируются на поверхности,
охлаждаемой жидким воздухом, то образуется желтый мышьяк. Его
свойства похожи на свойства белого фосфора, плотность равна 1,97 г/мл,
решетка кубическая, как у кристаллов а-формы белого фосфора. Жел.-
тый мышьяк неустойчив. Он легко переходит в металлический или
серый мышьяк. Это наиболее устойчивая и наиболее плотная
модификация мышьяка. Его плотность при 20° С равна 5,20 г/мл.
Серый мышьяк можно считать аналогом черного фосфора.
Структура их однотипна, но электропроводность гораздо выше. Она
составляет около 102 Ом"1 • м"1, т. е. серый мышьяк — металл. Серый
мышьяк, как и черный фосфор, плавится под давлением, но значительно
легче. При плавлении происходит резкое изменение структуры. Об
206
этом говорит высокая энтропия плавления, равная 25,4 Дж/К • моль.
Строение жидкого мышьяка не исследовано.
Зависимость температуры плавления серого мышьяка от давления
представлена на рис. 51, а. При не очень больших давлениях
плавление сопровождается ростом объема (dT/dP положительна). При
давлении порядка 60 ГПа удельные объемы жидкой и твердой фаз
становятся равными. В этой области давлений переход к более плотной
структуре при плавлении уменьшает объем в той же мере, в какой он растет
за счет увеличения концентрации вакансий.
80 100
Р,кбар
оии
600
Ш'
200
БЬЖ
1 "—""-—----^у
\
8ЪТ6
/
V 5Ь Тб
\ ПГУ
\
20 40 60 80 100
Р, кбао
600
400
100
О 20 40 60 80
б Р, кбар
Рис. 51. Фазовые диаграммы [8]:
— мышьяка; > — сурьмы; в — висмута
Bi/
ТВ.
В1Ж
гр
Bi
1
У
5^38^-
ЗЩ
\ПГУ
Bi У
О ЦК
i
207
С повышением молярной массы элементов подгруппы азота
потенциальный барьер, препятствующий переходу в наиболее устойчивую
аллотропную модификацию, падает. В этой подгруппе наиболее
устойчивая модификация отличается и наибольшей проводимостью.
Сурьма. Кристаллическая сурьма — металл; ее проводимость
при 20° С составляет около 106 Ом^-м"1. Структура того же типа,
что у черного фосфора и серого мышьяка. Как показали исследования
дифракции нейтронов, плавление сопровождается увеличением
среднего координационного числа от 3 для твердой фазы до 8,7—8,8 для
жидкой [38]. Одновременно растет электропроводность. Среднее
расстояние между соседними атомами в жидкой сурьме при 660° С
составляет 0,333 нм. При 800° С среднее координационное число*
уменьшается до 8,4—8,7. Зависимость температуры плавления сурьмы от
давления представлена на рис. 51,6. При температуре кипения пары
сурьмы содержат молекулы Sb2 и Sb4. Понижение температуры паров
сопровождается ростом концентрации Sb4.
Висмут — металл, структура его кристаллов того же типа, что у
сурьмы, серого мышьяка и черного форфора. При плавлении висмута,
по данным рентгенографических исследований [19], среднее
координационное число возрастает до 8, электропроводность увеличивается,
плотность растет. Фазовая диаграмма висмута приведена на рис. 51,в.
При нормальной температуре кипения висмута его пары в основном
одноатомны.
W)
50
30
20
10
2
ry
1
f?
^/j
/
/
/
/
<f
г/
0 12
§ 80. Подгруппа кислорода
(О, S, Se, Те, Ро)
Кислород. Атомы подгруппы
кислорода обладают способностью к
образованию цепочечных, а также в ряде
случаев (сера, селен) и кольцевых ас-
социатов. Менее, чем у других
элементов подгруппы, эта способность
выражена у кислорода. Его атомы образуют
прочные димеры О2, энергия
диссоциации которых равна 498,8 кДж/моль.
Энергия диссоциации тримеров О3 на О2
и О равна 105 кДж/моль (при 25° С). В
парах кислорода имеются тетрамеры О4
Рис. 52. Кривые атомного распределения жид»
кого кислорода:
кривая (/) при 62 К; кривая (2) при 89 К
4/7
30
10
10
7 8
R.A
* Вопрос о структуре жидкой сурьмы в известной мере дискуссионен.
Рентгенографические исследования приводят к несколько иным выводам. Согласно
[38] расхождение между результатами нейтронографических и
рентгенографических измерений обусловлено несовершенствами рентгенографических
исследований.
208
[39]. В жидкой фазе присутствие
О 4 было установлено в
результате тщательных, тонких и
разносторонних исследований
Л. И. Некрасова [40]. Молекулы
О 4 в жидком озоне О 3 имеют
полосу поглощения, максимум
которой соответствует частоте
3,78 - 1013 Гц (1260 см"1).
Атомная функция
распределения жидкого кислорода при
62 и 89 К представлена на рис.
52. На графике имеются два
изолированных максимума.
Площадь под первым максимумом
при 62 К равна 1,18, а при 89К
—1,08 атомов кислорода. По
мнению Н. Е. Гингрича [36],
первый максимум обусловлен
молекулами О2 и отчасти О3, а
второй—молекулами О3. В
молекулах О3 межатомные расстояния
равны 0,13 и 0,22 нм.
Расстояние 0,22 нм совпадает с
положением второго изолированного
максимума. Третий и четвертый
максимумы на кривых атомного
распределения, с нашей точки
зрения и в соответствии с
результатами исследований Л. И.
Некрасова, свидетельствуют о
существовании в жидком кислороде
более сложных и менее
устойчивых ассоциатов. Сведения о
ряде свойств жидкого кислорода и
других простых жидкостей
подгруппы кислорода приведены в
табл. 26.
Сера. У следующего члена
подгруппы— серы— способность
к ассоциации атомов выражена
значительно сильнее. Твердая
сера имеет много модификаций,
а жидкая обладает необычными
свойствами, поэтому строение
серы вызывает большой интерес и
изучалось неоднократно. Ниже
95,6° С наиболее устойчива
ромбическая сера. При 95,6° С она
8
VO
ГС»
Н
UHHj Hdu
UHHi Hdu
* 'nrai
ШХ Hdu яхо / жс
FUI Hdu
T_w • T_wo VOl • Жс
qra..X/«tflf"*1S
qifOW'^i/ж^' * 5"V
Qirnm/wV/M *^"^t7V7
1UU«^ »]j Л Г7 V
яхл
a'"»*
инх_^)1.жр
ж2
ахг
—. ^sus
о
Элемент
38,1
12,85
27,51
40,24
48,86
3,421
9,21
26,33
50,7
60,29
91,17
717,7
958
1263
1235
щ
8
„^gjw
0,222
1,591
5,44
17.50
(5,0)
со «ooot>T
54.45
385,95
490
723
519
112
CO CO*
il.e
Tf4f
Геке.
Ромб.
Геке.
Простая
куб.
fe
оГсою соIs»
Кислород
Сера
Селен
Теллур
Полоний
209
превращается в моноклинную серу. Если ромбическая сера не успела
перейти в моноклинную, то чистая ромбическая сера плавится при
112,8° С. Чистая моноклинная сера плавится при 119,3° С с
образованием маловязкой желтой жидкости.
Одна из интересных особенностей жидкой серы — аномальная
зависимость вязкости от температуры. При нагревании жидкой серы
ее вязкость сначала падает до 6,5 • 10"3 Па-с при 155° С. Затем с
повышением температуры вязкость возрастает. Цвет жидкости
меняется—она становится коричневой. Около 187° С сера превращается в
темно-коричневую массу. Вязкость серы достигает 93,3 Па • с. Затем
вязкость постепенно падает и при 300° С сера опять становится
подвижной. Жидкая сера кипит при 444,60° С, ее вязкость в точке кипения
равна 8,3-10"2 Па- с.
Пары серы при 150—200° С имеют оранжево-желтую окраску.
Нагревание сопровождается изменением цвета паров: они становятся
красными, потом при 600° С приобретают соломенно-желтый цвет.
В парах серы имеются молекулы S8, S6, S4, S2 и атомы S. Оранжево-
желтая окраска характерна для паров, состоящих в основном из
молекул S8. Соломенно-желтая наблюдается для паров, в которых
высока концентрация молекул S2. От 800 до 1400° С пары серы состоят в
основном из S2. Затем молекулы S2
диссоциируют. Выше 1700° С почти все молекулы S2
распадаются.
Молекулы S8 представляют собой
зигзагообразное кольцо (рис. 53). Выберем
положение какого-либо из атомов молекулы S8
Рис. 53. Строение молеку- в качестве начала координат. Тогда в коль-
лы 8 цевом ассоциате S8, изображенном на рис.
53, два атома будут находиться на
расстоянии 0,204 нм от начала координат; два атома будут удалены на 0,330 нм,
два атома ^— на 0,442 нм и один атом — на 0,466 нм. Атомный
радиус серы считают равным 0,104 нм.
Молекулы S8 обнаружены в парах, кристаллах и разбавленных
растворах серы.
Молекулы S6 и S4 — цепочечные фрагменты молекул S8. Энергия
диссоциации S8 на S6 и S2 равна 123,5 кДж/моль, Se на S4 и S2
152 кДж/моль, S4 на 2S2 133,8 кДж/моль. Энергия диссоциации S 6 на
S5 +S равна 280 кДж/моль, S4 на S3 + S 270 кДж/моль, S3 на S2 +
+ S 280,4 кДж/моль и S2 на 2S равна 416 кДж/моль (всюду при 25° С)
[16]. Поэтому распад молекул серы протекает преимущественно с
образованием ассоциатов, содержащих четное число атомов.
Ромбическая и моноклинная серы состоят из молекул S8.
Ближайшее расстояние между атомами соседних молекул S8 в кристаллах
равно 0,33 им.
Наиболее полные рентгенографические и нейтронографические
исследования строения жидкой серы были выполнены К. Томпсоном и
Н. Гингричем [41]. На рис. 54 представлены кривые атомного
распределения жидкой серы при 80° С (переохлажденная жидкость),
120°, 165°, 200°, 240°, 300° С. Кроме того, на этом рисунке имеются кри-
210
вые атомного распределения аморфной серы при 4° С и порошка
ромбической серы при 23° С. Для ромбической серы кривая атомного
распределения сглажена с помощью множителя ехр[—0,00558£2], чтобы
устранить колебания кривой, связанные с ошибками опыта. Здесь
fc=4nsin — /X; G — угол рассеяния; X — длина волны
монохроматического излучения (см. гл. VI).
Кривые атомного распределения жидкой, аморфной и ромбической
серы похожи друг на друга. Первый максимум изолирован, его
абсцисса на всех кривых одинакова — она равна 0,207 нм. Площадь под
30
20
10
о
Жидкость
Т=300°С
Т=240°С
Т=200°С
ТЧ65°Г
Т=120°Г
Т-80°С
Аморфная
сера
Ромбццес
пая сера
0 1 2 3 4 5 6 7 8 R,A
Рис. 54. Кривые атомного распределения серы [41]
первым максимумом всюду составляет около двух атомов. Этот
максимум обусловлен ближайшими соседними атомами S в молекулах S8
или их фрагментах S6 и S4. Второй максимум смещается от 0,346 нм
при 80° С до 0,334 нм при 300° С. У аморфной и ромбической серы он
находится примерно при 0,34 нм. Площадь под вторым максимумом
для жидкой серы меняется от 4,1 атома при 80° С до 3,1 атомов при
300°С. Для аморфной серы площадь под вторым максимумом
составляет около 3,7 атомов. Для ромбической серы эту площадь оценить
трудно, так как второй максимум плохо разрешен. Второй максимум
характеризует расстояния между теми атомами серы, которые расположены
через один атом в ассоциатах. Кроме того, примерно на том же расстоя-
211
нии находятся атомы S двух соседних молекул. При температурах
выше 165° С этот максимум постепенно расплывается. Его сглаживание
вызвано не только влиянием соседних молекул, но и ростом
амплитуды колебаний атомов S в цепочечных фрагментах серы. Третий
максимум располагается на расстоянии около 0,45 нм. Площадь под ним
определить трудно. Поэтому ее величина в работе [41] не приведена.
Этот максимум связан с положениями остальных трех атомов в
молекуле S8, а также атомов соседних молекул серы. При 0,55 нм у
ромбической, аморфной и переохлажденной жидкой серы наблюдается еще
один максимум. С повышением температуры он быстро сглаживается.
Этот максимум дает основания считать, что в жидкой и аморфоной сере
молекулы S8 имеют тенденцию к упорядоченному расположению,
образуя фрагменты структуры ромбической серы. Можно полагать, что
между ближайшими атомами соседних молекул S8, находящимися на
расстоянии около 0,3 нм друг от друга, имеются слабые химические
связи.
Разрыв колец S8 сопровождается образованием цепочечных ассоци-
атов Sn. По имеющимся в литературе оценкам [42], в области, где
вязкость жидкой серы велика (160—215° С), цепочечные ассоциаты при
разных температурах содержат в среднем от 5500 до 12 000 атомов
серы. Быстрое охлаждение вязкой жидкой серы приводит к
образованию метастабильной пластичной серы, которая имеет волокнооб-
разную структуру. В цепочечных ассоциатах возможность слабого
химического контакта между атомами серы, принадлежащими соседним
цепочкам, возрастает. Число слабых химических связей между
соседними молекулами резко увеличивается, поэтому вязкость серы быстро
растет и, как уже говорилось, достигает максимума около 187° С.
Дальнейшее нагревание сопровождается разрушением цепочек,
уменьшением средней степени ассоциации серы.
Исследования строения серы дифракционными методами
демонстрируют как достижения, так и слабые стороны этих методов. Если бы
сведения о строении молекул S8 в парах, кристаллах и разбавленных
растворах отсутствовали, то правильный анализ кривых атомного
распределения едва ли был бы возможен. На основании положения и
площади первого изолированного максимума легко можно было бы прийти
к неверному выводу о том, что жидкая сера состоит из молекул S3.
Твердая сера — диэлектрик. При плавлении проводимость
возрастает примерно в 10б раз, так что жидкая сера приближается к
полупроводникам. Согласно измерениям А. Р. Регеля и его сотр. [43], а
также Г. Веззоли [44], при нагревании жидкой серы проводимость
возрастает, проходит через максимум около 164,5° С, затем
понижается до температуры 186° С. Дальнейшее нагревание снова
сопровождается ростом электропроводности. При 212-214° С наблюдается
резкое ее увеличение. Затем она опять уменьшается, проходит через
минимум при 230° С и вновь медленно возрастает с дальнейшим
повышением температуры.
Малое значение энтропии испарения при температуре кипения ASHcn=
= 12,8 Дж/К . моль объясняется тем, что пары серы ассоциированы.
При 450° С они имеют следующий состав: 53,9% атомов серы образуют
212
ассоциаты S8; 37,0% —S6; 4,9%—S4 и 4,2% —S2. Таким образом,
средняя степень ассоциации паров равна 6,8. Следовательно, ASH£n =
= 12,8 • 6,08 = 87,0 Дж/К • моль. Этот пример показывает, как
энтропия испарения связана с ассоциацией молекул в парах.
Селен, как и сера, имеет ряд полиморфных модификаций,
образованных цепочками атомов селена. При обычной температуре устойчив серый
селен. Эта модификация состоит из очень длинных спиралевидных цепей.
Расстояние между соседними атомами в цепи равно 0,232 нм.
Расстояние между ближайшими атомами селена в соседних цепочках равно
0,346 нм. В отличие от серы, модификации селена, состоящие из
кольцеобразных молекул Se8, метастабильны.
Жидкий селен тоже состоит из длинных цепочек (Se)n. Вследствие
внутреннего вращения при тепловом движении эти цепочки имеют
множество различных конформаций, поэтому упаковка их не так
плотна, как в кристаллах серого селена. Среднее расстояние между
соседними атомами в цепочках жидкого селена равно 0,346 нм, т. е.
примерно то же, что и в твердой фазе. Но среднее расстояние между
ближайшими атомами селена в соседних цепочках значительно больше, чем
в кристаллах, оно равно 0,37 нм [21]. Проводимость селена в отличие
от серы при плавлении резко падает, уменьшаясь примерно на 3
порядка [43]. Нагревание до 700°С сопровождается увеличением
проводимости приблизительно на 5 порядков. Проводимость жидкого селена зависит
от величины напряжения электрического поля, прилагаемого к слою
жидкости. При повышении напряжения происходит переход из
состояния с малой проводимостью в состояние с высокой проводимостью.
Дальнейшее повышение напряжения ведет к новому переключению в
состояние с малой проводимостью [43, 45].
Если выливать расплавленный
селен в холодную воду, то
образуется стекловидный селен. Его
строение подобно жидкости. Для
превращения стекловидного селена в
серый селен требуется
перераспределить связи между атомами так,
чтобы цепи селена приобрели одну
и ту же спиральную конформацию
и выстроились параллельно друг
другу. Такое превращение связано
с преодолением большого
потенциального барьера, поэтому
стекловидный селен устойчив, хотя и ме-
тастабилен по отношению к серому
селену.
Пары селена состоят из
цепочечных и кольцевых молекул Se8,
а также молекул Se6, Se4, Se2n
следов атомов Se. От 1000° С до
1500°С пары состоят в основном из
молекул Se2.
Рис. 55. Фрагменты
спиралевидных цепочечных ассоциатов
теллура в его кристаллах [46]:
dlt d2, ^з»и^4—межъядерные расстояния.
Эллипсы символизируют относительно
слабые химические связи между соседними
цепочками
213
Теллур. Кристаллический теллур — аналог серого селена. Он
тоже состоит из очень длинных спиралевидных цепочечных ассоциа-
тов (Те)п, фрагменты которых изображены на рис. 55. Энтропия
плавления теллура более чем в два раза превышает энтропию плавления
селена и почти в 6 раз выше энтропии плавления серы (см. табл. 26).
Твердый теллур-полуметалл, при плавлении его
электропроводность возрастает примерно на порядок. Это дает основания полагать,
6 8 10R,A ' О 1
/0Я,А
Рис. 56. Радиальные функции распределения атомов g(R)
жидкого теллура при 575° и 1600° С [46]
что при плавлении теллура его строение меняется. Такой вывод
подтверждается экспериментально. Большой интерес в связи с этим
представляют нейтронографические исследования жидкого теллура,
выполненные К. Туаран, Б. Кабен и М. Брюиль. Обзор и анализ
результатов исследований имеется в работе [46]. Измерения проведены
при одиннадцати различных температурах, лежащих в интервале от
575° до 1700° С и давлениях до 6,0 МПа. Были найдены радиальные
функции распределения атомов для каждой из температур. На рис. 56
представлены эти функции для 575 и 1600° С. Кривые имеют три
узких отчетливых пика. Положения пиков при 575° С соответствуют
межатомным расстояниям 0,296 нм; 0,381 нм и 0,452 нм. При более высоких
температурах первый пик постепенно смещается до 0,307 нм при 1700°С.
Второй и третий пики в пределах ошибок опыта не меняют положения.
Координационное число z возрастает от 2 атомов в твердой фазе до-
Рис. 57. Структура теллура в различных
термодинамических состояниях [46]:
а—типический фрагмент струючфы жидкого телл/ра при 1700°С
и 60 атм (6Ы05 Па); б — фрагмент стр/ктуры кристаллического
теллура, в— типический фрагмент структуры жидкого теллура
214
3 в жидкой. Координационные числа, по мнению авторов работы [46],
определены со степенью точности порядка 10—15%; z падает, а г2 и z3
растут с повышением температуры. В пределах ошибок опыта сумма
z + 2ч равна 6 и не зависит от температуры.
Положения первых трех пиков радиальной функции
распределения близки к межатомным расстояниям dly d2 и d3 структуры
кристаллического теллура, изображенной на рис. 57.
К. Туаран, Б. Кабен и М. Брюиль [46] предлагают
правдоподобную картину распределения атомов жидкого теллура в первой и второй
координационной сферах. Рис. 57 помогает понять те изменения,
которые происходят в теллуре при его плавлении и нагревании
расплава. На рис. 57, б изображен фрагмент структуры кристаллического
теллура. Жирные линии указывают химические связи между атомами
в цепочках. Каждый атом теллура имеет две такие связи, образующие
угол в 102,6°. Пунктиром указаны более слабые химические связи
между атомами теллура, принадлежащими соседним цепочкам.
Каждый атом теллура имеет четыре такие связи. При плавлении строение
теллура изменяется так, как показано на рис. 57, в.
Фрагмент структуры жидкого теллура отличается следующим.
Каждый атом в среднем имеет три более прочные химические связи,
изображенные жирными линиями, и три более слабые, представленные
пунктиром. Расстояния d3n d4 одинаковы. Валентный угол между
более прочными связями уменьшается до 97°. При нагревании до 1700° и
повышении давления до 61 • 10б Па ближний порядок жидкого
теллура принимает структуру, характерную для простой кубической
решетки, изображенной на рис. 57, а. Такая же структура наблюдается
у кристаллов а-полония, следующего элемента подгруппы кислорода.
Различие между более сильными и более слабыми химическими
связями исчезает. Валентный угол между связями снижается до 90° .
По имеющимся в литературе данным [15], плотность жидкого
теллура при температуре, немного превышающей температуру плавления,
проходит через максимум.
Как уже отмечалось, электропроводность теллура при плавлении
возрастает примерно в 20 раз. С повышением температуры она растет
до 700° С. Выше 700° С (при атмосферном давлении)
электропроводность жидкого теллура не зависит от температуры. Температура
плавления теллура при повышении давления проходит через максимум при
482° С [8]. Перестройка структуры жидкого теллура, очевидно,
сопровождается изменениями его потенциальной энергии и энтропии.
Рост энтропии стабилизирует относительно более изотропную
конфигурацию атомов типа простой кубической решетки. Этому
противодействует возрастание потенциальной энергии системы. Потенциальная
энергия минимальна при образовании цепочечной структуры,
наблюдаемой у твердого теллура.
Таким образом, плавление теллура сопровождается ветвлением его
цепочек с образованием локальных плоских образований, быстро
исчезающих и возникающих вновь в ходе теплового движения. При более
высоких температурах (и давлениях) распределение становится еще
более изотропным.
215
Несколько слов о парах теллура. А. А. Кудрявцева и Г. П. Устю-
гов нашли, что при температурах 750—ИЗО К в парах теллура имеются
молекулы Те4, Те2 и атомы Те [18]. Этим объясняется низкая энтропия
испарения жидкого теллура.
Полоний — металл, его кристаллы, как уже говорилось, имеют
простую кубическую решетку с координационным числом, равным
шести. Строение жидкого полония не изучено. Приведенная в табл. 26
величина энтропии плавления полония определена не точно. Можно
предполагать, что среднее координационное число атомов полония в
жидкой фазе немного больше, чем в твердой. Пары полония при
нормальной температуре кипения, по всей вероятности, состоят из ассоци-
атов. Имеющееся в табл. 26 значение энтропии испарения дает
основания полагать, что средняя степень ассоциации паров близка к двум.
Итак, атомы подгруппы кислорода способны к образованию
цепочечных ассоциатов. Если каждый последующий атом занимает цис-
положение, то цепочки образуют кольца. Если же атомы занимают
траяс-положения, то колец не возникает. Число ковалентных связей
в кольце на единицу больше, чем в разомкнутой цепочке, имеющей такое
же число атомов, как и в кольце, поэтому потенциальная энергия
колец ниже потенциальной энергии разомкнутых цепочек.
Гексагональная упаковка цепочек плотнее, чем ромбоэдрическая или моноклинная
упаковка колец. С повышением молярной массы, усложнением
электронной структуры и удалением внешних электронов от атомного ядра
способность атомов элементов подгруппы кислорода к образованию
дополнительных химических связей увеличивается и такие связи
становятся прочнее.
В сере, где химические связи между атомами соседних цепочек
сравнительно слабы, в твердой фазе устойчивы кольцеобразные
цепочки. В селене кольцеобразные цепочки уже не возникают. В
теллуре связи между атомами соседних цепочек почти столь же сильны,
как и в самих цепочках. В полонии все связи эквивалентны и цепочек
не образуется.
§ 81. Водород
Этот элемент в периодической системе занимает особое место. Атом
водорода—простейший из атомов. Молекула водорода Н2 — самая
простая из электрических нейтральных молекул. Она состоит из двух
протонов и двух электронов. Для водорода более отчетливо, чем во
многих других случаях, проявляется связь между состоянием атомных
ядер в молекулах и свойствами вещества.
Рассмотрим некоторые свойства молекул, состоящих из одинаковых
ядер, таких, как Н2, D2 и др. Согласно квантовой механике при обмене
одинаковых ядер местами волновая функция ф молекулы или остается
без изменения, или же меняет свой знак. Если ф не меняется, то
состояние молекулы называется симметричным по отношению к этой
перестановке. Если же ф меняет знак, то состояние молекулы называется
антисимметричным. Когда атомные ядра имеют целый спин (s. = 0, 1, 2,...),
то волновая функция молекулы симметрична по отношению к переста-
216
новке двух таких одинаковых ядер*. Но если частицы имеют
полуцелый спин (s. = 1/2, 3/2, ...), то волновая функция молекулы
антисимметрична по отношению к перестановке этих частиц [47]. Такими
частицами являются электроны и некоторые из атомных ядер, например
протоны. Спин атомных ядер дейтерия равен 1.
Спины ядер — векторы. Сложение спинов дает полный ядерный
спин молекулы. Если спины каждого из ядер s. = 1/2, то ядерный спин
двухатомной молекулы может быть равен 0 или 1. В магнитном поле
5 может иметь только те направления, для которых компонента в
направлении поля равна s, s — 1, s—2,...,— s. Если s = 0, то возможна
лишь одна ориентация полного ядерного спина молекулы. Если s = 1,
то возможны три ориентации этого спина. Поэтому статистический вес
состояний молекул с s = 1 в три раза больше, чем статистический вес
состояний молекул с s = 0.
Далее надо учесть, что полная волновая функция молекулы ф с
хорошей степенью точности (но все же приближенно) может быть
представлена как произведение волновой функции фк, зависящей только
от координат частиц, и спиновой волновой функции ф5, зависящей от
ориентации спинов. И фк, и ф^ зависят от обмена одинаковых ядер
местами.
Координатная волновая функция в первом приближении имеет
вид
где фэл, фкол и фвр — электронные, колебательные и вращательные
собственные функции молекулы. Колебательная волновая функция
молекулы зависит только от межъядерного расстояния и не меняет знака при
обмене одинаковыми ядрами. Влияние обмена ядер на фк зависит от
изменений электронной и вращательной собственной функции. Если
электронная собственная функция не меняется, то при четных
вращательных уровнях молекулы (вращательные квантовые числа J = 0, 2,
4, ...) обмен ядрами не влияет на знак фвр, а следовательно, и на знак
Фж-
При нечетных вращательных состояниях (/ =1,3, 5,...) обмен
одинаковыми ядрами меняет знак фвр, а следовательно, и знак фк (если
знак фэл сохраняется). Что касается спиновой функции <|>5, то она
симметрична, когда s = 2s., (2s. — 2), (2s.—4), ... и антисимметрична,
когда s =(2st — 1), (2s. — 3), ....
Таким образом, для молекулы водорода Н2, находящейся в
основном квантовом состоянии, ф5 симметрична, когда s = 2s. = 1, так
как спины их протонов равны 1/2. В этом случае спины протонов
параллельны. Если же спины протонов в молекуле Нг антипараллельны,
т. е. s = 2s. — 1 = 0, то ф5 антисимметрична.
* s. здесь означает спиновое квантовое число. Абсолютная величина
собственного момента количества движения частицы st = hYsi (si + 1) » гдей=Ь/2л.
217
Как уже было сказано, для ядер, имеющих полуцелый спин,
полная волновая функция молекулы антисимметрична по отношению к
обмену ядер местами, Так как для молекулы водорода Н2 <|>эл
симметрична по отношению к этому обмену, то ф может быть
антисимметричной, когда s = 0 и вращательные уровни четные или же когда s = 1
и вращательные уровни нечетные. Молекулы Н2 с параллельными
спинами протонов называют молекулами ортоводорода. Молекулы с
антипараллельной ориентацией ядерных спинов называют молекулами
параводорода. Ортомодификацией молекул обычно называют ту,
которая имеет больший статистический вес* [48].
У дейтерия спин атомного ядра равен 1. Согласно сказанному
спиновая собственная функция молекул дейтерия D2 симметрична по
отношению к перестановке ядер, если полный ядерный спин
молекулы s равен 2 или 0, и антисимметрична, если s^l-При
s — 2 спины ядер параллельны, при s = 0 они антипараллельны, при
s = 1 направлены под углом 60° один к другому. Собственная
электронная функция основного состояния молекулы Ь2 симметрична по
отношению к обмену ядрами. При s. = 1 полная волновая функция
молекулы симметрична. Следовательно,' состояния с s =2иО соответствуют
четным вращательным уровням, а состояние с s =1—нечетным
вращательным уровням. Легко показать, что статистический вес состояний с
четными вращательными уровнями (равный 5 + 1 = 6) в два раза
больше статистического веса состояний с нечетными вращательными
уровнями. Вообще, если одинаковые ядра какой-либо двухатомной
молекулы имеют спин s., то отношение статистических весов орто-и
парамодификаций равно (s. + l)/sr
У дейтерия молекулы ортомодификации имеют четные
вращательные уровни (в отличие от молекулы Н2).
Квантовые переходы между симметричными и антисимметричными
состояниями, вообще говоря, запрещены. Но если спин ядра s.
отличен от нуля, то этот запрет не является вполне строгим. Поэтому
ортомодификации водорода и дейтерия могут превращаться в
парамодификаций и, наоборот, парамодификаций в ортомодификации. Равновесие
между орто- и парамодификациями зависит от температуры.
Отношение концентрации параводорода [пН2] и ортоводорода [оН2]
определяется уравнением [51]:
[ПН2] .7=0,2,
[оН2] ~
% e 2
J=l,3, ... ./=1,3,
* У атомов гелия тоже различают орто- и парамодификаций, но они
отличаются ориентацией спинов электронов. Спины электронов ортогелия параллельны;
парагелия — антипараллельны.
218
? +_9е +13е Н
3 [Зе +7е + Не + ••• )
где s. — спин ядра, равный 1/2; / — вращательный уровень; ej—
энергия вращения молекулы, находящейся на J вращательном уровне.
Для водорода Н2 при низких температурах более устойчива пара-
модификация; у дейтерия D2 — ортодейтерий. При очень низких
температурах водород состоит почти полностью из параводорода, а
дейтерий из ортодейтерия. Большая устойчивость параводорода и орто-
дейтерия при низких температурах связана с тем, что эти молекулы
могут занимать наинизший вращательный уровень с вращательным
квантовым числом, равным нулю. У самого низкого вращательного
уровня молекул ортоводорода и парадейтерия J = 1. Максимально
возможное содержание ортоводорода равно 75 %, т. е. когда на одну
молекулу параводорода в соответствии с указанными выше
статистическими весами приходится три молекулы ортоводорода. Такое
содержание для равновесного водорода почти достигается уже при комнатной
температуре. Максимально возможное содержание парадейтерия
равно 33,3%.
Переход ортоводорода в параводород и парадейтерия в
ортодейтерий при понижении температуры происходит очень медленно и может
длиться многими неделями и даже годами. Скорость
самопроизвольной бимолекулярной реакции превращения, обусловленного
магнитным взаимодействием молекул ортоводорода, пропорциональна
квадрату концентрации о-Нг- Константа скорости (при отсутствии
катализаторов этой реакции) равна 3,34 • 10~6 с"1 х мд"1 (мд — молярная
доля). Теплота орто — параконверсии твердого или жидкого
водорода равна 1,417 кДж/моль [49].
Если водород, равновесный при комнатной температуре, т. е.
содержащий около 75% молекул ортоводорода, охладить, то
концентрация ортомодификации в жидкой фазе в течение длительного времени
будет близка к 75%. Такой жидкий водород принято называть
нормальным. Равновесный жидкий водород почти нацело состоит из
параводорода. При нормальной температуре кипения жидкого водорода,
равной 20,4 К, в равновесном водороде содержится всего 0,21%
ортоводорода*.
Свойства нормального и равновесного жидкого водорода несколько
различаются. В табл. 27 приведены некоторые из свойств природного
водорода (99,97 %Нг и около 0,03% HD) в нормальном и равновесном
при 20,4 К состояниях. Различия между свойствами жидкого, нормаль-
* Другие двухатомные молекулы, состоящие из одинаковых атомов,
существуют в орто- и парасостояниях, если спин атомных ядер отличен от нуля. Так,
например, молекулы N1^, Cl3^, Вг1^ существуют в орто- и парасостояниях.
Молекулы С1! и О1^ существуют только в одной форме, так как спины дер С12 и О16
равны нулю. Молекулы О^О18, С135С137 и др., состоящие из атомов разных
изотопов, орто- и парасостояний не имеют.
219
Таблица 27
Некоторые свойства нормального и равновесного при 20,4 К водорода [49]
Свойство
В тройной точке:
температура
давление
теплота плавления
энтропия плавления
плотность жидкой фазы
плотность твердой фазы
изменение объема при плавлении
В нормальной точке кипения:
температура
молярный объем
теплота испарения
энтропия испарения
В критической точке:
температура
давление
молярный объем
Единица
измерения
К
Па
кДж/кг
Дж/К-моль
кг/м3
кг/м3
%
К
мл/моль
кДж/кг
Дж/К-моль
К
Па
мл/моль
Данные
нормальном
13,947
7,22-103
58,24
8,08
77,2060
86,67*
12,2
20,384
28,397
454,3
44,59
33,24
1,3.10е
67,0
о водороде
равновесном
при 20,4 К
13,803
7,05-103
58,24
8,12
77,0063
86,63**
12,5
20,267
28,481
446,10
43,96
32,99
1,295-10»
64,14
Дейтерий
18,72
—
98,4
10,5
—
—
—
23,57
—
461
38,9
38,35
12350
61(?)
* При давлении 7,2-108 Па; ** при давлении 7,03-10» Па.
ного и равновесного водорода очень интересны. Они не только
показывают, что свойства жидкостей зависят от состояния атомов ядер,
но и демонстрируют характер наблюдаемых зависимостей. Из
приведенных в табл. 27 данных следует, что параводород кипит при более
низких температурах, чем нормальный водород. Теплота испарения
жидкого параводорода меньше, а молярный объем больше, чем у
нормального водорода. Хотя различия и невелики, они дают основания
считать, что взаимосвязь между молекулами параводорода в жидкой
фазе слабее, чем между молекулами ортоводорода. По всей
вероятности, это вызвано различиями в магнитных взаимодействиях молекул.
Магнитные моменты молекул орто- и параводорода отличаются за счет
различий суммарных ядерных спинов и вращательных квантовых
чисел. Спины протонов в молекулах параводорода антипараллельны.
Они компенсируют друг друга и не вносят вклад в магнитный момент
молекулы. При низких температурах почти все молекулы
параводорода находятся на самом низком вращательном уровне, J = 0, поэтому
магнитный момент молекул параводорода равен нулю, т. е. они
немагнитны. Магнитный момент молекул ортоводорода всегда отличен от
нуля, потому что ядерные спины параллельны и самый низкий
вращательный уровень J — 1.
Отметим, что вязкость жидкого ортоводорода приблизительно на
5% выше, чем параводорода. Твердый водород имеет гексагональную
220
плотную упаковку. При повышении давления температура его
плавления растет. Параводород плавится при 80 К, когда давление достигает
5,47 • 108 Па, нормальный водород плавится при 80 К, если давление
равно 5,45 • 108 Па. С ростом давления производная dTldP
постепенно уменьшается. Плавление сопровождается резким увеличением
концентрации вакансий и объем параводорода возрастает на 12,5%. При
повышении давления концентрация вакансий в жидкой фазе падает.
Уменьшение производной dTldP, вероятно, вызвано этим фактором.
Сжимаемость нормального жидкого водорода, рассчитанная с помощью
данных о плотности при различных давлениях, равна 1,263 • 10~8 Па"1
при 19,1 К и 1,64 • 10~8 Па"1 при 20,4 К. Интервал давлений, в котором
определялась сжимаемость, составляет около 1,0 МПа. Нижняя
граница этого интервала — давление насыщенных паров водорода.
Диэлектрическая проницаемость жидкого нормального водорода,
находящегося в равновесии с парами водорода, равна 1,2533 при
14,10 К и 1,2305 при 20,49 К- Для жидкого дейтерия в области
равновесия жидкость — пар диэлектрическая проницаемость изменяется
от 1,282 при 18,80 К до 1,272 при 21,18 К [49]. Сведения о некоторых
из термодинамических свойств жидкого дейтерия приведены в
табл. 27. Энтропия плавления водорода и дейтерия составляет окало
8,1 Дж/К • моль. Такая величина ASnJ1 наблюдается в тех случаях,
когда плавление не сопровождается существенным изменением структуры
(например, у щелочных металлов). Можно предполагать, что
координационное число молекул Н2 в жидкой фазе около температуры
плавления близко к 12. Плотность жидкого водорода равна 70,8 кг/м3,
следовательно, среднее расстояние между молекулами Н2 в жидкой
фазе R составляет около 0,45 нм. Температура замерзания жидкого
водорода 14,05 К при Р = 1,01325* 105 Па. При столь низкой
температуре свойства жидкого водорода, вообще говоря, должны были
бы следовать законам квантовой статистики. Как известно,
отклонения свойств макроскопических систем от классической
статистической механики могут наблюдаться в тех случаях когда «тепловая
длина волны»
\т = (2nh2/mkBT)ln (X. 3)
сравнима со средним расстоянием между частицами [50]. Хг по
порядку величины равна длине волны де-Бройля частицы, имеющей
массу m и кинетическую энергию къ Т.
\ Тепловая длина волны молекул Н2, равная при 15 К 0,18 нм, по
порядку величины сравнима с /?, поэтому для жидкого водорода мож~
но наблюдать отклонения от законов классической статистической
механики. Но так как молекулы водорода связаны друг с другом
химическими и вандерваальсовыми взаимодействиями, то жидкий водород
затвердевает около 14 К, т. е. при такой температуре, когда влияние
квантовостатистических эффектов невелико. Отметим, что молекулы
Н2 следуют квантовой статистике Ферми, а молекулы D2 — статистике
Бозе.
221
sr
к
4
\o
чиоп/ж)^н 'L'u#
Л V
CO О
l>- 00
ю
юсчсо ю
- со i>^
О -^
NCOO —• С
t-- 00 C5 <П >
8
""З"1 О"! ^tf* ^н
Ю00О) О ю
со'оГоГ —Г^'
т—i CM rt4
OOCOCO LOv
C^- CO i-H ON
rf со'сГ со "^
o „ ч
i-H t—i О CO rt1
Ю CO 00 —i ~
e-
as
я I
^fX
— ooo oo
I
§ 82. Подгруппа фтора
(F, Cl, Br, I, At)
Сведения о
некоторых свойствах жидких
элементов подгруппы
фтора или галогенов
приведены в табл. 28.
Атомы галогенов
образуют димеры F2, Cl2, Вг2
и 12. Энергии их
диссоциации в кДж/моль при
25°С равны: 158,5 для
фтора, 242 для хлора,
192,5 для брома и 151
для иода [16].
Кристаллы фтора и жидкий фтор
состоят из молекул F2.
Строение кристаллов
фтора пока не
установлено, но известно, что
имеются две
аллотропные модификации фтора
аи?. При —227,6°С
р-фтор переходит в
а-фтор, а энтальпия
возрастает на 0,727
кДж/моль. а-Фтор
плавится при—219,46°С.
Энтропия плавления
невелика. Можно полагать,
что плавление фтора не
сопровождается
существенным изменением
среднего
координационного числа молекул F2.
Происходит рост
концентрации вакансий.
Жидкий фтор
окрашен в ярко-желтый цвет.
Его относительная
молекулярная масса [38]
близка к относительной
молекулярной массе
аргона [39,94]. Свойства
жидкого фтора —
температура плавления,
кипения, критическая
температура и критическое
222
давление, плотность, теплота и энтропия плавления, теплота и
энтропия испарения, вязкость газа — также близки к соответствующим
свойствам аргона [52]. Диаметры молекул F2 и атома аргона
составляют около 0,3 нм. Поляризуемость молекул фтора немного выше
поляризуемости атомов аргона. Статическая диэлектрическая
проницаемость жидкого фтора заменяется от 1,567 при 57,40 К до 1,517 при
83,21 К. Молярная поляризация?' = п^~—V в этом интервале
температур практически постоянна и равна 5,01 мл. Это немного
превышает величину молярной поляризации жидкого аргона, которая
равна 4,20 мл [53]. Таким образом, есть основания думать, что
между молекулами фтора действуют слабые химические силы того же
порядка, что и между атомами аргона.
Кристаллы хлора имеют тетрагональную структуру с параметрами
решетки а = 0,856 нм, с = 0,612 нм. Энтропия плавления
сравнительно велика. Плавление хлора сопровождается существенным
увеличением числа степеней свободы молекулы С12.
Результаты рентгенографических исследований строения жидкого
хлора рассмотрены в § 43. Жидкий и твердый хлор — диэлектрики,
их электропроводность около 10~8 Ом"1- м"1.
Бром кристаллизуется с образованием базоцентрированной
ромбической решетки, параметры элементарной ячейки которой а = 0,448 нм,
b = 0,667 нм и с = 0,827 нм. Электропроводность твердого брома
а = 10~6 Ом"1 • м"1. Энтропия плавления брома выше, чем хлора.
Диэлектрическая проницаемость
жидкого брома довольно велика — при
комнатной температуре она равна
3,2. Нейтронографические
исследования жидкого брома [54] при 23°С
дают основание считать, что
ближний порядок в расположении
молекул жидкого Вг2 вблизи
температуры плавления сохраняется. На рис.
58 приведена атомная функция
распределения жидкого брома
согласно данным, имеющимся в работе
[54]. В нижней части рисунка
приведены данные о радиальной
плотности атомов брома в идеальном
кристалле. Пунктирные линии
указывают положения и числа атомов
брома. На кривой имеются
отчетливые максимумы при 0,23; 0,39; 0,45;
0,55 и 0,62 нм. Максимум при 0,23 нм
хорошо разрешен и площадь под
ним соответствует одному атому Рис. 58. Кривая атомного распреде-
брома. Это согласуется с представ- ления жидкого брома при 23° С и
лением, что жидкий бром состоит ^J^l 1ЖШТЮМ указан0
ИЗ ДВухаТОМНЫХ МОЛекуЛ. МеЖМО- распределение атомов в кристаллах брома)
223
лекулярное расстояние Вг—Вг равно 0,230 ± 0,005 нм, что хорошо
гармонирует со значением 0,229 нм для молекул Вг2 в парах и
значением 0,227 нм для молекул Вг2 в кристаллах. Расстояние 0,296 нм,
при котором атомная функция распределения проходит через
минимум, согласно работе [54], есть минимальная дистанция между
двумя атомами брома, принадлежащими соседним молекулам Вг2.
Кривая атомного распределения, соответствующая расстояниям более
0,3 нм, отражает распределение молекул Вг2.
При свободном вращении молекул каждая из них должна была бы
занимать объем -^- |——^—^—| . кг3нм = 7,б5 • 10~29 м3. Если
свободно вращающиеся молекулы Вг2 были бы плотноупакованы, то
молярный объем брома был бы равен 62 мл/моль, в то время как в
действительности молярный объем жидкого брома при 23°С равен 54,4 мл/моль.
Поэтому молекулы брома в жидкой фазе, по всей вероятности, не
имеют свободного вращения. Они колеблются около временных
положений равновесия, будучи связаны друг с другом слабыми химическими
взаимодействиями. Расположение соседних молекул Вг2 при
плавлении существенно не меняется — возрастает число вакансий в
ближайшем окружении. При расстояниях порядка 0,6 нм наблюдаются
большие изменения в распределении молекул.
Иод в твердом состоянии имеет подобно брому ромбическую
решетку с периодами а = 0,725 нм; Ъ = 0,9772 нм и с = 0,4774 нм.
Строение кристаллов иода отчасти уже описано в гл. III.
Электропроводность кристаллов при 25° С равна 1,7 • 10~9 Ом"1 • м"1;
диэлектрическая проницаемость 10,3 при 23° С. Энтропия плавления иода
немного выше, чем у брома. Можно полагать, что строение жидкого иода
в основных чертах подобно строению жидкого брома.
Электропроводность жидкого иода при 138,2° С равна 4,48 • 10~7 Ом~* • м"1.
Диэлектрическая проницаемость при 118° С равна 11.
§ 83. Инертные газы
(Не, Ne, Аг, Кг, Хе, Rn)
Сводка некоторых из свойств инертных газов приведена в табл. 29
[55]. Гелий — квантовая жидкость (ему посвящена следующая глава).
Строение других жидких инертных газов изучалось дифракционными
методами неоднократно. Особенно подробно был исследован жидкий
аргон. О результатах этих работ говорилось в гл. VI.
Координационные числа атомов инертных газов, приводимые в литературе,
различаются на 20—30%. Расхождения объясняются неточностями
эксперимента и неоднозначностью способа расчета координационных чисел.
Наиболее достоверные значения 2 жидких инертных газов около температуры
плавления, по-видимому, близки к 8. Это значение
координационного числа в сочетании с данными о росте объема при плавлении,
приведенными в табл. 29, может быть истолковано с помощью модели
хаотически распределенных сфер, изученной Д. Берналом и С.
Кингом. Вместе с тем вопрос о строении жидких инертных газов пока еще
224
X
ч
qifow-м/жй1 ^"s1 V
llfU// у
"Л V
см см
CM —■
as
Ю CO
Ю
CO l^
2^
CM О
Is* ОЭ
CM rj«
о
Ю
rt«
CM
Ю 00
Ю Ю
Ю ^
CO t>-
C75
t^.
ю о
I I CSCCIIO'*
2SSSSS
«—< о} i4-» t4» со >-0
CM 00^ CO
О О CM
r^ CM 00 Ю
Ю Ю Ю CO
CO f^ CO C7>
CO ^ CO CM
о —«-н*см
ЮЮМОЮ
cx> lo" —Г —Г —Г ^-Г
Ю СО СО »—'
СМ 00—« СО
CM CO CO CO ^
X
к
л
ю ю ю
<л оо оо оо
00
к оо см см см см
СМ ^"1^*-1
8 х
* § Ё §
о u. s о
си a. a. a
Е<^^
225
не вполне ясен. Рентгенографические и нейтронографические измерения
дают информацию о среднем статистическом распределении атомов
инертных газов. Но в жидких инертных газах, несомненно,
присутствуют фрагменты упорядоченных структур, где атомы связаны слабым
химическим взаимодействием. Об этом свидетельствуют результаты
исследований твердых фаз и паров этих веществ, рассмотренные в гл. III.
ГЛАВА XI
КВАНТОВЫЕ ПРОСТЫЕ ЖИДКОСТИ 4Не И 3Не
§ 84. Введение
Как уже говорилось, квантовыми называют такие жидкости, для
описания свойств которых необходимо применять квантовую статистику.
Особенности, присущие квантовым жидкостям, наиболее отчетливо
обнаруживаются у жидких стабильных изотопов гелия 4Не и 3Не.
Это вызвано тем, что атомы гелия в основном состоянии не способны
к химическому взаимодействию друг с другом. При атмосферном
давлении они притягиваются друг к другу, видимо, лишь с помощью
дисперсионных сил, которые очень слабы и не могут упорядочить атомы даже
при О К- Упорядочению препятствует нетепловая (нулевая) энергия
движения гелия, поэтому 4Не и 3Не при атмосферном давлении не
затвердевают. Точка сосуществования жидкости, пара и твердой фазы у
4Не и 3Не отсутствует. Этот факт, необъяснимый с позиций докванто-
вой физики, есть одно из проявлений квантовых свойств жидкого
гелия. Представление о нулевой энергии движения вытекает из
квантовой теории.
Свойства жидкого гелия уникальны, поэтому они тщательно
изучаются. Жидкий гелий исследован примерно так же детально, как вода,
если не лучше. Так как его особенности очень интересны, мы опишем
свойства жидкого гелия подробнее, чем свойства других простых
жидкостей. Во многих случаях мы будем пользоваться материалами,
имеющимися в монографии В. Кеезома [56] и обзорных статьях К.
Мендельсона [57], В. П. Пешкова и К. Н. Зиновьевой [58, 59]. Наше
рассмотрение здесь, как и в других случаях, разумеется, никоим образом
не претендует на полноту. Мы обратим внимание читателя только на
те факты и представления, которые наиболее существенны.
Атомы 4Не и 3Не имеют заполненные электронные оболочки Is2.
s-Орбитали обладают шаровой симметрией, поэтому атомы гелия
можно уподобить шарам. Их диаметр равен примерно 0,24 — 0,25 нм.
Атомы 4Не имеют целочисленный спин. Из квантовой теории поля
(см. [47]) следует, что такие частицы представляют собой бозоны, т. е.
подчиняются квантовой статистике Бозе—Эйнштейна. Спин атомов
8Не равен 1/2, поэтому атомы 3Не являются фермионами, т. е. следует
статистике Ферми—Дирака.
226
Как уже было сказано при описании свойств жидкого водорода
(см. § 82), квантовостатистические эффекты начинают играть
существенную роль^когда тепловая длина волны Хт сравнима со средним
расстоянием Н между частицами. Некоторые из свойств 4Не и 3Не
охарактеризованы в табл. 29.
Рассмотрим сначала имеющиеся в литературе сведения о строении и
свойствах 4Не.
§ 85. Изотоп 4Не
Кристаллы 4Не прозрачны и получаются только при давлениях,
превышающих ~2,5-106 Па. 4Не кристаллизуется с образованием
плотной гексагональной упаковки ПГУ. В интервале температур
1,45—1,78 К имеется небольшая область существования кубической
объемноцентрированной фазы (ОЦК). Теплота перехода из
гексагональной в менее плотную кубическую фазу 4Не при 1,7 К составляет около
0,4 кДж/моль (т. е. 0,095 кал/моль). При более высоких давлениях
гексагональная плотноупакованная структура переходит в гранецен-
трированную кубическую.
Плавление твердого 4Не сопровождается рядом очень интересных
особенностей, которые подробно описаны в обзорах В. П. Пешкова [58]
и К. Мендельсона [57] (см. также [56] и [27]). На рис. 59 изображена
диаграмма фазовых равновесий 4Не. При температуре 0,77 К и
давлении 2,53 • 106 Па на кривой плавления 4Не наблюдается минимум.
В этой точке производная dPIdT обращается в нуль. При температуре
10*
ю2
10
10
r1
10
-2
10
,-з
10
,-4
Г5
10
10'
Г7
Где
--.
Жи
рдй
..-
дкс
\я ф
Не-
7С/ПЬ
J
аза
Ж-
а
F—
/
-■/--
-1 -
1
1
ОТ/'
ЫГ
f
у
у
'Гл^
Г(
-
* IT
/,7^5
?,/7Z
13
у
°/
г-З
2Я^£
7,8л/
1ГПМ
мм рт.ст.
W3
ю2
м рт. ст.
10
1
ю-'
да"2
«Г3
//»-♦
Ув"
0,1 0,2 0,5 1 2 3 57Ю W 50 100
I
Рис. 59. Р—Т-диаграмма фазовых равновесий 4Не [58]
227
1,765 К и давлении 3,14 • 1Об Па график -^г=
аТ
имеет излом.
На фазовой диаграмме рис. 59 в этой точке кривая плавления
соединяется с кривой, описывающей зависимость от давления и температуры
фазового перехода второго рода в жидком гелии. Этот фазовый переход
второго рода будет рассмотрен далее.
При 3,6 • 108 Па температура плавления 4Не равна 30,770 К и
разность молярных объемов АУПЛ =0,430 мл/моль [27]. При 7,370 • 108 Па
температура плавления 4Не равна 50 К [55]. При 2 • 109 Па АКП1
0,2 мл/моль и мольный объем твердого 4Не составляет всего 6,37 мл/моль
[58].
Так как разность молярных объемов сосуществующих жидкой и
твердой фаз конечна, то по уравнению Клапейрона — Клаузиуса
(IX. 1) обращение в нуль производной dPIdT означает, что энтальпия
и энтропия плавления 4Не при 0,77 К равны нулю:
TLSnJL = ЬНпл = Шпл+РЬУпл^й. (XI. 1)
Следовательно, в этих условиях изменение внутренней энергии при
затвердевании жидкого гелия Шш ~ —РАУПЛ. Как показали опыты
Г. К. Стрэти и Е. Д. Адамса, при температурах ниже 0,77 К
кристаллизация 4Не с помощью его сжатия приводит к поглощению теплоты,
т. е. сопровождается охлаждением системы. Правда, этот тепловой
эффект очень мал; при 0,7 К он составляет всего лишь около 3,5 • 10~4
Дж/моль, а при температурах, лежащих ниже 0,77 К, он еще меньше.
На рис. 60 изображена зависимость энтальпии плавления АНПЛ,
величин Р AVnjl и разности внутренних энергий Д{/пл жидкого и
твердого гелия от температуры. Разность энтальпий при температуре 0,77 К
обращается в нуль. При 0 К разность внутренних энергий Д£/ состав-
С, калIг град
кал/моль
6
5
J
2
0
1
\/
4-
q
-V
/
у
/
s*
/
/
'АН
PAV
AU
о / 2 j VOi
Рис. 60. Зависимость энтальпии
плавления А Нпл, величины PA VUJl и
разности внутренних энергий A UnJl, жидкого
и твердого 4Не от температуры |57]
1=^
V
Н
I]
F
р^/
0J46
0,1 Ь5
OJbl
0JU
0,140
0,139
0,138
0,137
о 1 г з i к
Рис. 61. Зависимость плотности р
и теплоемкости С жидкого 4Не от
температуры [55]
228
ляет около — 5,0 Дж/моль. Как отмечает К- Мендельсон, уже ниже
1,5 К процессы плавления и затвердевания гелия не похожи на
обычно наблюдаемые процессы плавления и кристаллизации. Гелий не
затвердевает при одном лишь охлаждении. Чтобы перевести гелий в
твердую фазу, необходимо приложить к нему некоторое давление,
фазовое превращение при этом становится практически механическим
процессом, не сопровождающимся тепловыми изменениями. Однако
плавление гелия происходит самопроизвольно при определенных
величинах давления и температуры.
Согласно третьему закону термодинамики энтропия жидкой фазы,
так же как и твердой, при абсолютном нуле температуры должна
обращаться в нуль. В связи с этим приобретает большой интерес вопрос о
распределении атомов в жидком гелии, особенно при наиболее низких
температурах. Плотность жидкого гелия мала, под давлением
насыщенных паров она составляет всего около 0,14 г/мл, что в значительной
мере объясняется малой молекулярной массой гелия (заметим, что
плотность жидкого водорода примерно в два раза меньше плотности жидкого
гелия). Необычна зависимость плотности 4Не от температуры (рис. 61).
Там же представлена температурная зависимость теплоемкости С
вдоль линии равновесия жидкость — пар. При температуре 2,173 К и
49,80 • 102 Па плотность жидкого 4Не проходит через максимум,
после чего функция р = f(T) резко меняет свое направление,
плотность быстро уменьшается. Теплоемкость тоже аномально зависит от
температуры. Кривая теплоемкости похожа на греческую букву X.
При 2,182 К теплоемкость является разрывной функцией. Здесь в
жидком 4Не происходит фазовый переход второго рода*. Температура этого
фазового перехода («Х-точки») немного снижается при увеличении
давления. Жидкую фазу при температурах, соответствующих Х-точкам и
ниже, принято называть «гелий II». Жидкая фаза при температурах,
лежащих выше Х-точек, названа «гелий I».
§ 86. О строении жидкого 4Не
Замечательной особенностью фазового перехода второго рода в
жидком 4Не является отсутствие изменений структуры жидкости,
т. е. изменений распределения атомов гелия в пространстве. Этот факт,
отмеченный в ранних рентгенографических исследованиях Кеезома и
других авторов, был подтвержден нейтронографическими измерениями
Д. Харста и Д. Хеншоу [61]. Они изучили рассеяние медленных
нейтронов (средняя дебройлевская длина волны равна 0,104 нм) жидким
4Не в интервале температур от 1,65 до 5,04 К, т. е. от температур,
лежащих ниже Х-точки, до температур, близких к критической точке.
Как известно, при заданной температуре частицы не могут быть
локализованы в области пространства, имеющей размеры порядка
средней длины волны де-Бройля. Средняя дебройлевская длина
определяется уравнением
* Понятие о фазовых переходах второго рода излагается в курсах
термодинамики. О фазовых переходах второго рода в жидкостях см. также [60].
229
хБ =
(XI.2)
Для атомов 4Не при 1,65 К ^б равна 9 • 10~2 нм, а при 5,0 }{
5 • 10~2 нм. Видимо, поэтому функция атомного распределения
4nR2p(R)f согласно работе [61], не имеет отчетливо выраженных
максимумов. Д. Харст и Д. Хеншоу подчеркивают, что в области
фазового перехода второго рода, т. е. в интервале температур от 1,65 до 2,25 К,
кривая атомного распределения жидкого 4Не практически не
меняется.
Некоторые особенности эта функция все-таки имеет. Д. Харст и
Д. Хеншоу приводят значения радиуса первой координационной
сферы Rz и дают оценки среднего координационного числа г. В интервале
температур 1,65—2,25 К Rz равно 0,37 нм, а г = 9; при 4,24 К Rz =
= 0,37 нм и z =8; при 5,04 К Rz = 0,39 и z = 7. Диаметр атомов
гелия составляет около 0,24 нм (по другим данным около 0,26 нм), а
радиус первой координацион-
№
/,5-
Рис. 62. Радиальные функции
распределения [62]:
а — атомов газообразного *Не при 4,2 К и 0,98 атм;
б — атомов жидкого 4Не при 0,56 К и жидкого 8Не
при 0,79 К
230
ной сферы равен или
превышает 0,37 нм. Исходя из
данных о плотности, легко
подсчитать, что среднее
расстояние между соседними атомами
— / q i7 V/3
жидкого 4Не, r = 2 6 v 1
V 4nNA )
при 3 К составляет около
0,45 нм. Следовательно,
средний объем, приходящийся на
один атом в жидком гелии,
приблизительно в 6 раз
превышает объем самого атома.
Это можно объяснить тем, что
энергия притяжения атомов
гелия друг к другу мала, а
энергия их отталкивания
сравнительно велика. Основное
квантовое состояние
характеризуется отталкиванием
атомов гелия друг от друга,
которое сравнительно велико
даже при больших расстояниях
между ними (см. гл. III).
Поэтому плотность гелия мала, а
сжимаемость велика. Тем не
менее среднее
координационное число атомов 4Не
довольно велико. Оно такое же, как
у плотных жидкостей,
например у жидких щелочных ме-
таллов. Такое же координационное число около температуры
плавления наблюдается и у других жидких простых веществ подгруппы
гелия.
Интересны измерения рассеяния рентгеновских лучей в жидких
*Не и 3Не и парах 4Не, выполненные Е. К. Ахтером и Л. Мейером
[62]. На рис. 62 представлены радиальные функции распределения
g(R). Для жидкого 3Не при 0,79 К и газообразного 4Не при 4,2 К и
9,93 • 104 Па радиальные функции распределения имеют два
максимума. Для жидкого гелия первый максимум соответствует Rz~
& 0,35 нм, для газообразного Rz ~ 0,4 нм. Первый максимум в
жидкости при 0,79 К выражен менее отчетливо, чем в парах при 4,2 К и
9,93 • 104 Па. Другие результаты этой работы мы рассмотрим далее.
§ 87. Сжимаемость, скорость звука,
молярная поляризация жидкого 4Не
Изотермическая сжимаемость жидкого гелия р, очень велика:
при 2,71 К она составляет около 11,85 • 10"8 Па"1 и приблизительно
в десять раз превышает изотермическую сжимаемость жидкого
водорода при 16 К- Адиабатическая сжимаемость $s жидкого гелия II
практически совпадает с изотермической сжимаемостью. Отношение
р, /р^ = CplCy у гелия II отличается от единицы всего на 0,1—0,5%.
У гелия I отношение теплоемкостей Ср /Су растет с повышением
температуры. Распространение звука в гелии I — адиабатический
процесс. В гелии II звуковые волны тоже адиабатические, но
расхождение между адиабатическими и изотермическими условиями
распространения звука здесь несущественно ввиду малого отличия
теплоемкости при постоянном давлении Ср от теплоемкости при
постоянном объеме Су . Скорость звука растет от ~ 180 м/с при 4,2 К до 237 ±
± 2 м/с при 0 К (экстраполяция). Скорость звука в окрестности X-
точки резко снижается. Объем моля жидкого 4Не при 3 К составляет
28,5 мл. При давлении около 80 • 106 Па и той же температуре он
уменьшается до 19,7 мл. При давлении 3,54 • 108 Па объем моля равен 10,1 мл
[27]. Статическая диэлектрическая проницаемость жидкого 4Не в
точке кипения (под давлением 1,006 • 105 Па)е5 = 1,049 ± 0,006.
Молярная поляризация р = . Bs~ . — равна 0,123 мл. В пределах
4я bs + 2 р
ошибок опыта Р не зависит от температуры и имеет те же значения для
гелия I, что и для гелия II. Молярная поляризация газообразного гелия,
полученная по уравнению р = . —-—— ' » равна 0,1234 мл.
4я /£, + 2 Р
Здесь /Zoo — показатель преломления парообразного гелия при 0° С и
1,01 • 105 Па, экстраполированный к бесконечной длине волны; /г» =
= 1,000346. Плотность паров гелия при этих условиях равна 0,17846
кг/м3. Если из вычисленного значения поляризации 0,1234,
полученного на основании оптических данных, подсчитать диэлектрическую
постоянную жидкого гелия в точке кипения при нормальном давлении,
231
ДНисп> кал/г
7
/
/
/
г
•bgfi
о
о
\
\
то получится 1,0491, что в точности совпадает с величиной, найденной
экспериментально [63]. Это дает основание считать, что атом гелия
почти не меняется при переходе гелия из газообразного состояния в
жидкое.
§ 88. Энтальпия испарения. Межъядерное взаимодействие
Энтальпия испарения жидкого 4Не представлена на рис. 63 ([56]
стр. 265). В X-точке производная энтальпии испарения по
температуре испытывает скачок, обусловленный резким изменением
теплоемкости жидкого 4Не. Линейная
экстраполяция к 0 К дает для энтальпии
испарения величину, равную примерно 6,07
Дж/моль (14, 5кал/моль). Заметим, что
при 0 К, по всей вероятности, давление
насыщенных паров гелия обращается в
нуль. При 0,6 К давление насыщенных
паров 4Не равно 2,41 • 10~2 Па и очень
быстро уменьшается с понижением
температуры. При 0,5 К давление
насыщенных паров 4Не уже равно 2,18 • 10"8 Па.
Снижение температуры на 0,1 К
сопровождается более чем десятикратным
уменьшением давления насыщенного
пара [55]. При температуре кипения
энтальпия испарения одного моля жидкого
гелия Д#исп составляет около 84 Дж/моль.
Изменение внутренней энергии при
испарении 1 моля в нормальной
точке кипения Д£/Исп = Д#исп —
—ЯДКИСп, где Я = 1,013- 105 Па и АКИСП — разность мольных
объемов жидкости и пара при температуре кипения. У 4Не
А1/исп = 210 мл, следовательно, P&V исп = 21 Дж/моль. Таким
образом, Д#исп составляет около 63 Дж/моль, что близко к приведенной
выше величине теплоты испарения 4Не, найденной линейной
экстраполяцией к 0 К-
Теперь оценим порядок величины энергии лондоновского (или
дисперсионного) притяжения между атомами 4Не в одном моле жидкого гелия.
По расчетам Д. Слэтера и Д. Кирквуда [64], энергия лондоновского
притяжения двух атомов 4Не равна 3,18 (ao/R)e ридберг (см. § 70). Здесь
а0 = 2,59 • 10"2 нм. В соответствии с измерениями Д. Харста и
Д. Хеншоу примем, что радиус первой координационной сферы в
жидком гелии R2 = 0,39 нм. Положим, что при всех расстояниях R> Rz
радиальная функция распределения g(R) равна единице. Тогда энергия
лондоновского взаимодействия для одного моля жидкого гелия равна*
* Расчет по приближенной формуле Лондона дает при тех же
представлениях £Ол = —14,2 к Дж/моль.
232
5 Т,К
Рис. 63. Энтальпия испарения
жидкого 4Не [56]
[см. гл. I, уравнение (1.65) и гл. VI, уравнение (VI. 21)]
(0,529)e • 10~24 • 3,18 . 1,3 • 106
F —
ZV
~ 67 Дж/моль.
Значение Е0Л1 конечно, сильно зависит от выбора величины Rzy
которая определена неточно, так как нулевое движение в жидком гелии
существенно влияет на результаты дифракционных исследований.
Если принять Rz == 0,28 нм, что немного превышает диаметр атомов
гелия и является нижним пределом величины £>в в уравнении (1.65),
то Еол будет составлять около 167 Дж/моль (см. также работу [65]).
Следовательно, предположение, что межатомное притяжение в
жидком гелии обусловлено лишь лондоновскими силами, не лишено
оснований. Гелий — единственная жидкость, атомы которой, по-видимому,
не испытывают короткодействующего притяжения,
сопровождающегося обобществлением электронов. Они притягиваются друг к другу лишь
с помощью хотя и очень слабых, но дальнодействующих лондоновских
сил. В твердом гелии, имеющем гексагональную плотную упаковку,
химические взаимодействия между атомами, вероятно, тоже
отсутствуют. При повышении давления они возникают и, возможно, являются
главной причиной упомянутых изменений структуры твердого гелия.
§ 89. Энтропия и теплоемкость жидкого 4Не
Большой интерес для суждения о строении жидкого 4Не
представляют исследования его энтропии. На рис. 64 представлена
энтропийная диаграмма гелия. Энтропия S вычислена с помощью
экспериментальных данных о теплоемкости выше 1 К с использованием сведений
об энтропии плавления и
испарения. Выше Х-точки
энтропия жидкого 4Не под
давлением насыщенных
паров меняется
приблизительно линейно с
температурой. Экстраполяция этих
линий к 0 К показывает,
что при абсолютном нуле
энтропия жидкого гелия I
была бы равна нулю.
Энтропия жидкого гелия I
уменьшается с ростом давления.
В области устойчивости
гелия II энтропия очень
быстро уменьшается при
понижении температуры.
Здесь рост давления
сопровождается увеличением
энтропии жидкости.
S, кал/г-град
0,8
0,2
Для понимания
природы жидкого гелия очень
9—503
О j 1 3 47, "К
Рис. 64. Энтропийная диаграмма 4Не [57]
233
важны измерения его теплоемкости. На рис. 65 представлены
результаты прецизионных измерений теплоемкости С, выполненных Г. А. Кра-
мерсом, Д. Васшером и К- Д. Гортером в интервале температур от 1,9
до 0,2 К. По оси ординат отложены величины теплоемкости в
логарифмическом масштабе. Наиболее интересной особенностью зависимости
теплоемкости от температуры является довольно резкое изменение
наклона кривой вблизи 0,7 К- Ниже этой температуры результаты можно
представить в виде С == 23,573 Дж/(кг • К).
Таким образом, в этой области температур теплоемкость жидкого
гелия пропорциональна Г3. Она зависит от температуры так же,
С, Дж/г град
0,2 0,3 0,h Ofi 0,8 1,0 1,1 1,5 2fl
Т,К
Рис. 65. Теплоемкость жидкого 4Не при низких температурах
[57]
как теплоемкость дебаевской модели изотропного твердого тела.
Теоретический анализ этой модели, выполненный П. Дебаем, показал,
что кубическая зависимость теплоемкости от температуры, часто
наблюдаемая при низких температурах, обусловлена фононами.
Следовательно, в этом отношении поведение жидкого 4Не подобно твердым
телам.
При температурах выше 0,7 К зависимость теплоемкости от
температуры нельзя представить в форме простой степенной функции Т.
§ 90. Аномалии в явлениях переноса
Аномалии термодинамических свойств гелия II сопровождаются
еще более удивительными особенностями в явлениях переноса. Под-
234
робный обзор этих особенностей выходит за рамки задач нашей книги.
Ограничимся краткой характеристикой упомянутых аномалий.
В 1935-36 гг. В. Кеезом и мисс А. Кеезом обнаружили, что гелий
II обладает огромной теплопроводностью, более чем в миллион раз
превышающей теплопроводность гелия I, и в 200 раз большей, чем
теплопроводность меди при комнатной температуре. Открытие
сверхтеплопроводности гелия II вызвало большой интерес и заставило
исследователей обратить пристальное внимание на процессы,
протекающие в этой жидкости.
Вскоре Д. Аллен, Р. Пейерлс и М. Аддин в Кембридже
установили, что понятие теплопроводности в обычном смысле как отношения
теплового потока к градиенту температуры в гелии II теряет смысл.
Оказалось, что величина теплового потока зависит не только от
градиента температуры, но и от размеров прибора, с помощью которого
производятся измерения. Для капилляра заданного диаметра, в котором
находится гелий, при постоянном градиенте температуры
теплопроводность гелия при охлаждении ниже Х-точки резко возрастает,
достигая максимума при 2 К, и затем снова падает при дальнейшем
понижении температуры.
В 1938 г. Д. Аллен и Г. Джонс обнаружили, что при появлении
градиента температуры в гелии II возникает градиент давления. Это
явление было впоследствии названо термомеханическим эффектом.
Можно было полагать, что создание в гелии II разности давлений будет
вызывать появление разности температур.
В 1939 г. Д. Доунт и К- Мендельсон установили, что течение
гелия II от более высокого уровня к более низкому действительно
сопровождается появлением градиента температуры.
Уже в ранних исследованиях, выполненных В. Кеезомом и его
сотр. в 1930 г., было обнаружено, что гелий II обладает
необыкновенной способностью легко проходить даже через очень малые,
капиллярные течи в сосудах. Это явление впоследствии детально исследовалось
П. Л. Капицей и одновременно Д. Алленом и А. Майсенером (1938).
П. Л. Капица показал, что гелий II свободно проходит через зазор
между двумя стеклянными дисками даже тогда, когда ширина зазора
равна 5 • 10~7 м, Д. Аллен и А. Майсенер обнаружили этот эффект
при течении гелия II через узкие капилляры, сечение которых
составляло около 10"5 м. Скорость течения жидкости почти не зависела от
напора и диаметра капилляра. В последующих опытах Д. Аллена и
А. Майсенера выяснилось, что скорость течения даже увеличивается
при уменьшении радиуса капилляра до 10"7м.
По оценкам П. Л. Капицы вязкость гелия при переходе от гелия I
к гелию II уменьшается nd крайней мере в 1500 раз. Это явление было
названо им сверхтекучестью. Следует отметить, что понятие о вязкости
для гелия II нуждается в уточнениях, которые будут даны далее.
Понятие о вязкости сохраняет свой обычный смысл для гелия I;
она в интервале температур от 5 до 2,7 К почти постоянна. При
дальнейшем понижении температуры вязкость гелия I в отличие от
обычных жидкостей уменьшается от 3 • 10~е до 2,2 • 10~6 Па • с в Х-точке,
причем в самой Х-точке не наблюдается какого-либо скачка вязкости.
9* 235
Абсолютная величина вязкости жидкого гелия I очень мала — она
всего лишь втрое превышает вязкость газа. Это можно объяснить
теми же причинами, которые обуславливают малую плотность жидкого
гелия.
Еще одной удивительной особенностью 4Не является эффект переноса
жидкого гелия II по пленке, возникающей на поверхности сосуда с
этой жидкостью (Д. ДоунтиК. Мендельсон; А. К- Кикоин и В.
Г.Лазарев, 1938). В опытах Д. Доунта и К. Мендельсона маленький
цилиндрический стеклянный сосуд,
подвешенный на тонкой стеклянной
нити, можно было опускать в ванну
с жидким гелием II и поднимать над
этой ванной (см. рис. 66). «При
частичном погружении пустого сосуда
в жидкость наблюдалось
постепенное заполнение его гелием, пока
уровень в нем не оказывался на
одной высоте с уровнем в ванне. При
небольшом подъеме сосуда процесс
шел в другую сторону и снова
приводил к полному выравниванию
X
X
Рис. 66. Схема опытов Доунта и
Мендельсона по изучению переноса
жидкого 4Не по пленке на поверхности
сосуда [57]
уровней.
Наконец, когда сосуд полностью
вынимался из ванны, было видно,
как на его наружной поверхности появились капли жидкости,
которые постепенно росли и через равные интервалы времени падали в
ванну» [57].
Оказалось, что при погружении сосуда в жидкий гелий II на
поверхности сосуда возникает пленка толщиной около 30 нм. Перенос
гелия II по пленке не зависит от разности уровней жидкости, длины
пути и высоты промежуточного барьера. При постоянной температуре
скорость переноса на единицу периметра поверхности, соединяющей
уровни жидкого гелия II, постоянна. Природа поверхности,
по-видимому, не влияет на скорость переноса. Для медных и алюминиевых
сосудов были получены те же значения скоростей переноса, что и для
стеклянных сосудов. Скорость переноса очень сильно зависит от
температуры. От нулевого значения в Х-точке скорость увеличивается
до практически не зависящей далее от температуры величины
7,5- 10~5 мл/с на 1 см периметра соединительной поверхности.
Последующие исследования аномальных свойств жидкого гелия II
тесно связаны с теоретическими работами в этой области.
§ 91. О теории жидкого гелия П.
Квантовая когерентность жидкого гелия при 0 К
История развития теории сверхтекучести и других особенностей
жидкого гелия очень интересна. Далеко не все вопросы выяснены, а
тем временем, как обычно, эксперимент открывает новые проблемы.
Мы ограничимся лишь кратким изложением главных результа-
236
тов теории. Она развивается в двух аспектах—феноменологическом и
молекулярном и оба эти подхода влияют друг на друга. Важной вехой
на пути как феноменологической, так и молекулярной теории явились
работы Ф. Лондона. Большую роль в создании феноменологической
теории свойств гелия II сыграли работы Л. Д. Ландау [66] и его
учеников. Молекулярная теория сверхтекучести была построена Н. Н.
Боголюбовым [67]. Нас интересует здесь, главным образом, молекулярная
природа тех явлений, которые происходят в жидких системах. Поэтому
вопросов феноменологической теории мы почти не будем касаться.
Читатель, интересующийся этой стороной дела, может получить
основные сведения в книге Л. Д. Ландау и Е. М. Лифшица «Механика
сплошных сред» [68], где есть глава «Гидродинамика сверхтекучей
жидкости», обзорных статьях И. М. Халатникова [69] и книге К. Ху-
анга [50].
Рассмотрим состояние некоторого макроскопического объема V
жидкого 4Не. Будем считать этот объем замкнутой системой, т. е.
независимой от окружения. Примем, что импульс и механический
момент этого образца жидкого 4Не, как целого, равны нулю. Иначе
говоря, в лабораторной системе координат образец находится в
состоянии покоя. В этом случае единственным интегралом движения
образца является его энергия. Возможны лишь такие комбинации
импульсов и координат атомов 4Не, при которых энергия образца остается
неизменной. Статистические свойства макроскопического образца
определяются его энергией.
Допустим теперь, что температура жидкого 4Не равна ОК.
Назовем это состояние основным. Пусть Uo обозначает энергию этого
состояния. В соответствии с принципами статистической механики
энтропия системы
S=f(U, V)=kblnT(U), (XI.3)
где T(U) — обозначает число состояний системы, энергия которых
равна U или лежит в некотором узком интервале значений,
включающем U. При 0К
S = fclnr0, (XI.4)
где Го — вырождение основного состояния, т. е. число состояний,
имеющих одну и ту же энергию Uo. Состояние квантовой системы,
как известно, определяется ее волновой функцией ф.
Из опыта следует, что энтропия жидкого гелия 4Не при 0К равна
нулю. Это означает, что при 0К существует только одно состояние
макроскопического объема жидкого гелия; мы будем называть его
основным состоянием. Оно определяется одной единственной волновой
функцией ф0. Эта волновая функция является собственной функцией
гамильтониана Н макроскопической системы, имеющего одно-едикст-
венное собственное значение энергии, равное Uo. При 0К жидкий 4Не
является квантовой когерентной системой. Поскольку его состояние
единственно, движения всех частей этой макроскопической системы
однозначно взаимосвязаны (когерентны).
237
Сверхтекучесть сравнительно легко наблюдается только в жидком
изотопе гелия 4Не. В изотопе гелия 3Не сверхтекучесть, по-видимому,
обнаружена при температурах ниже 0,0026 К [70]. Естественно
предположить, что это различие связано с тем, что 4Не следует статистике
Бозе, а 3Не — статистике Ферми.
§ 92. Конденсация Бозе—Эйнштейна.
Теория Н. Н. Боголюбова
Еще в 1925 г. А. Эйнштейн, исследуя свойства газа,
состоящего из бозонов, показал, что в этом газе ниже некоторой критической
температуры должна наблюдаться конденсация в пространстве
импульсов. Это означает, что при температурах ниже Тк некоторая конечная
доля бозонов в покоящемся бозе-газе должна иметь импульс /?, равный
нулю. С понижением температуры доля таких частиц или, как теперь
принято говорить, доля этого конденсата должна расти. Расчеты А.
Эйнштейна были раскритикованы Д. Уленбеком и представление о
конденсации бозе-газа было, по существу, забыто. В 1938 г. Ф. Лондон
выдвинул предположение, что 4Не как раз и есть такой объект, где бозе—
эйнштейновская конденсация происходит и именно она и является
причиной сверхтекучести и других необычных свойств гелия II.
Как отмечал Ф. Лондон, критическая температура Тк
конденсации идеального бозе-газа, имеющего плотность жидкого гелия и
состоящего из атомов такой же массы, как у 4Не, должна быть равна 3,14 К-
Эта температура отличается от температуры перехода гелия I в гелий II
лишь на 0,96 К. Лондон предположил, что расхождение обусловлено
взаимодействием между атомами жидкого 4Не. Идеи Лондона
развивались далее в работах Д. Тисса. По его представлениям, гелий II —
раствор конденсата, атомы которого имеют импульс р~ 0, и
«нормальной» жидкости, атомы которой имеют импульсы р ф 0. По мнению
Тисса, конденсат не может участвовать в каких-либо диссипативных
процессах и поэтому является сверхтекучим. При 0 К весь жидкий
гелий представляет собой конденсат. Представления Тисса
подверглись справедливой критике Л. Д. Ландау и других исследователей.
Частицы конденсата должны были бы обмениваться импульсом при
столкновениях с частицами нормальной жидкости, поэтому при
движении в жидком гелии атомы конденсата испытывали бы трение и
сверхтекучести не было бы. Далее, если бы при 0 К все атомы гелия
покоились, то гелий под влиянием сил межатомного притяжения должен был
бы кристаллизоваться, а этого не происходит.
Распределение импульсов в идеальном газе Бозе — Эйнштейна.
Опишем теперь результаты, полученные Боголюбовым, следуя, в
основном, содержанию работ [67], замечательных по своей глубине и
тонкости анализа.
Допустим, что газ, состоящий из бесспиновых молекул, идеален.
Взаимодействие между молекулами отсутствует и полная энергия этого
бозе-газа сводится к кинетической энергии. Рассмотрим
распределение по импульсам отдельных молекул. Воспользуемся для этого
функцией распределения со(р), определив ее так, чтобы N(u(p) было средним
238
числом молекул с импульсами из элементарного объема dp пространства
импульсов, в котором находится точка р. Эта функция нормирована так,
что fсоф = 1.
Функция распределения импульсов молекул идеального газа,
покоящегося как целое, имеет вид
з/2 ^ v 1
+Z' (Х1-5)
где m — масса атома газа; 8(р)—дельта функция, которая равна
нулю при р^О и 1 при р = 0.
Из формулы (XI. 5) вытекает следующее. Если Т> Тк, то со(р)
непрерывна относительно аргумента р, и, наоборот, при Т <С Тк со(р)
становится разрывной. При Т < Тк заметная, т. е. конечная часть,
молекул покоится. Эта часть молекул как бы «замерзла» в
импульсном пространстве, имея один и тот же импульс р = 0. Относительное
число этих «замерзших» молекул
(*Г
Следовательно, оно мало при Т близком к Тк, и постепенно
увеличивается при понижении температуры. При абсолютном нуле температуры
все молекулы «замерзают», ш(р) = 1 при р = 0. В интервале
температур 0 < Т < Тк существуют одновременно и «конденсат» из
замерзших молекул, и «нормальная компонента», которая содержит всю
энтропию системы. Таким образом, можно говорить отдельно о плотности
конденсата Р^ = — \ 1 — и нормальной компоненты рп = — х
7Г
Необходимо подчеркнуть, что бозе — эйнштейновская конденсация
наблюдается только в пространстве импульсов. В обычном же
координатном пространстве и конденсат, и нормальная компонента полностью
«размешаны» и в состоянии статистического равновесия распределены
в пространстве вполне однородно, если не учитывать поле тяготения.
В поле тяготения в принципе можно было бы отделить конденсат от
нормальной компоненты, как это происходит при обычном разделении
фаз.
Конденсация Бозе — Эйнштейна возникает в результате
особенностей симметрии волновой функции системы, а не благодаря
взаимодействию между бозонами. Далее мы увидим, что взаимодействие между
бозонами влияет на концентрацию конденсата, хотя и не служит
причиной его появления.
Явление бозе-эйнштейновской конденсации идеального газа было
использовано Д. Тисса для объяснения сверхтекучести жидкого
гелия. Но теория Боголюбова ясно показывает, что между
сверхтекучестью и бозе—эйнштейновской конденсацией идеального
газа нет связи. Из формулы (XI. 5) для функции распределения
импульсов следует, что средние скорости нормальной компоненты и
239
конденсата одинаковы. В рассматриваемом случае газа, покоящегося
как целое, эти скорости равны нулю. В общем же случае, когда полный
импульс отличен от нуля, следовательно, когда система как целое
движется с определенной скоростью а, вместо (XI.5) справедливо
равенство [67]:
(xi-7>
2mkbT
Первый член в правой части равенства характеризует функцию
распределения импульсов р конденсата. Дельта функция Цр — та) равна
единице, когда/? = mv = та. Она обращается в нуль при/? Ф та.
Следовательно, конденсат как целое движется со скоростью а. Второй
I \р — та? 1
член в правой части равенства содержит функцию ехр — — — •
Величина р — та представляет собой импульс любого произвольно
выбранного атома нормальной компоненты. Если нормальная
компонента как целое покоится, то а = 0 и импульс равен/;. Если же все атомы
нормальной компоненты движутся в некотором общем для них
направлении со скоростью а, то импульс произвольно выбранного атома
будет равен р — та, где — со <: р < + со.
«Наоборот, в случае сверхтекучести необходимо, было бы, чтобы в
состоянии статистического равновесия конденсат мог двигаться
относительно нормальной компоненты с определенным спектром
скоростей. Ясно, что для идеального газа это невозможно, так как тогда
атомы конденсата не должны были бы обмениваться импульсами с
атомами нормальной компоненты, а в идеальном газе соударения
происходят между парами индивидуальных атомов и, следовательно, ничто
не препятствует атому конденсата отдавать свой импульс атомам
нормальной компоненты» [67].
Статистическая теория идеального бозе-газа показывает, что
флуктуации плотности этого газа неограниченно возрастают, когда
температура, понижаясь, стремится к Тк. Эти флуктуации бесконечно
велики при всех температурах, лежащих в интервале 0 < 71 < Тк.
Следовательно, при переходе через критическую температуру
интенсивность релеевского рассеяния света должна была бы очень сильно
возрастать, а этого у жидкого гелия не происходит. Упомянутые
трудности заставили Л. Д. Ландау в 1941 г. построить совершенно
другую, правда, не молекулярную, а полуфеноменологическую теорию
сверхтекучести.
Неидеальный газ Бозе — Эйнштейна — модель жидкого гелия.
Перейдем теперь к свойствам неидеального газа Бозе—Эйнштейна.
Н. Н. Боголюбов воспользовался упрощенным выражением
потенциальной энергии взаимодействия молекул газа
E(R) = eE0(R), (XI. 8)
где е — параметр.
240
Молекулы имеют сферическую форму. Если параметр г мал, то
взаимодействие слабое и отклонения неидеального газа от идеального
невелики. Когда е =0, газ идеален. В уравнении (XI. 8) не учтено
быстрое возрастание E(R) при малых R, т. е. короткодействующее
отталкивание, обеспечивающее непроницаемость молекул. Мы еще вернемся
к этому вопросу и увидим, что учет короткодействующего
отталкивания в данном случае не играет большой роли.
При абсолютном нуле найденная Боголюбовым функция
распределения импульсов газа, состоящего из бозонов, имеет вид
(XI.9)
где No/N — доля молекул, импульс которых равен нулю; V — объем
системы; v (р) с точностью до множителя 1/V — амплитуда Фурье —
компоненты при разложении потенциала E(R) в ряд Фурье по
переменным p. v (р)-Амплитуда «волны импульса» или «волны количества
движения», возникающей благодаря межмолекулярному
взаимодействию. Разложение E(R) в ряд Фурье имеет вид
р
Вследствие радиальной симметрии потенциальной функции,
амплитуды
,(P) = $E(R)e-iipR)/hdR. (XI. 11)
В этом разложении v (р) зависят лишь от модуля \р\ вектора импульса
р. Е(р) в уравнении (XI. 9) — энергия элементарных возбуждений,
т. е. коллективных состояний жидкости, которые условно можно
охарактеризовать как квазичастицы с импульсом р. Квазичастицы
следуют статистике Бозе—Эйнштейна. Связь между Е и р имеет вид
Из уравнения (XI.9) следует, что у неидеального газа Бозе —
Эйнштейна при 0К лишь часть молекул имеет в точности нулевой импульс.
Остальные же непрерывно распределены по всему спектру импульсов.
Доля последних равна:
No V С
N e (2nh)9N ]
. (XI. 13)
2Е(р)\В(р)+-±£— + ^.(р)\
241
Уравнения (XI. 9) и (XI. 23) показывают, что неидеальный газ
Бозе — Эйнштейна даже при ОК — сложная система.
В работах Н. Н. Боголюбова нет численных оценок доли
молекул конденсата. Они были получены О. Пенроузом и Л. Онзагером
[71]. Для жидкого гелия при ОК по их расчетам концентрация
конденсата составляет около 8% атомов гелия. Другие авторы нашли, что
концентрация конденсата при ОК может достигать 25% (см. [72]).
Экспериментальная проверка этих расчетов производилась рядом
исследователей, изучавших рассеяние быстрых монохроматических нейтронов в
жидком гелии. Таким нейтронам соответствует короткая волна де-
Бройля. Поэтому каждый нейтрон взаимодействует лишь с одним
атомом жидкого гелия. Распределение импульсов рассеянных нейтронов в
этом случае несет информацию о распределении импульсов атомов
жидкого гелия. Достаточно тщательные измерения пока что были
выполнены О. Харлингом [72] с нейтронами, наименьшая де-бройлевская
длина волны которых составляла около 3 • 10~2 нм. Изучен интервал
температур от 4,2 до 1,27 К- При 1,27 К концентрация конденсата
No/N =8,8 ±1,3%. С ростом температуры No/N падает. Измерения
No/N при Т > 2,0 К недостаточно надежны, поэтому опыты Харлинга
не дают возможности определить критическую температуру
бозе—эйнштейновской конденсации в жидком гелии. О. Харлингом найдена
средняя кинетическая энергия гелия при ОК. Она составляет около
108,8 Дж/моль, если потенциальную энергию Е(0) при ОК считать
равной—180Дж/моль.Подругимданным[65], средняя кинетическая
энергия гелия при ОК равна 129 Дж/моль, а Е(0) = 176—171 Дж/моль.
Вернемся к теории Н. Н. Боголюбова. Как уже было сказано,
эта теория предполагает, что взаимодействия между молекулами
слабы. Под малостью взаимодействия понимается следующее. Пусть
E(R) = Emt (XI. 14)
когда R по порядку величины равно d0. Тогда, как показал Боголюбов,
его теория применима, если удовлетворяется неравенство
Для жидкого гелия неравенство (XI. 15) выполняется, следовательно,
к жидкому гелию теория Боголюбова применима*. Физический смысл
уравнений (XI. 9) — (XI. 13) состоит в следующем. В основном
состоянии, т. е. при 0 К, система когерентна. Это значит, что в ней
нельзя выделить нулевое движение отдельных молекул. В системе
распространяются «волны движения». Они могут быть представлены как
суперпозиция «гармонических колебаний движения», амплитуда которых
* При 0 К вследствие нулевого движения средняя энергия атомов гелия, как
мы видели, не равна нулю. Если принять, чтос10 -0,3 нм, N а £т~40 Дж и NJN^
=0,1, то для гелия х\ ^ 5- КГ2. Другое условие применимости Теории Боголюбова
сводится к требованию, чтобы Т < 7\ , где 7\ —температура перехода гелия П
в гелий I.
242
равна v(p)/l/ [см. уравнения (XI.10) и (XI. 11)]. С гармоническими
колебаниями движения связаны элементарные коллективные
возбужденные состояния, имеющие энергию Е(р). Эти состояния считаются
независимыми друг от друга, что, конечно, является упрощением. Они
могут рассматриваться как идеальный газ из квазичастиц, у которых
связь между энергией и импульсом определяется формулой
(XI. 12). При малых импульсах уравнение (XI. 12) для Е(р) легко
преобразовать к следующему выражению:
Д(р) = *оЫ(1 + ---). (XI.16)
Здесь а0 — скорость звука при 0 К. Наоборот, когда импульсы
достаточно велики, можно разложить Е(р) в ряд по степеням малого
параметра s и написать:
"^Г + у У(р)- (Х1Л7)
Так как, согласно (XI. 11), v (p) стремится к нулю ири увеличении )р|,
то для достаточно больших импульсов Е(р) стремится к кинетической
энергии одной молекулы i£-L==^i!_. Следовательно, отношение
Е(р)1\р\ является непрерывной положительной функцией |р|,
принимающей значение а0 > 0 при Ы = 0 и возрастающей как -L^J
т
при р -voo. Поэтому оно имеет существенно положительное
минимальное значение:
(XI. 18)
Неравенство (XI. 18) представляет собой условие существования
сверхтекучести в неидеальном газе Бозе—Эйнштейна. Это условие
впервые было указано Л. Д. Ландау [66].
§ 93. Причины сверхтекучести жидкого гелия [67]
Не приводя соответствующих математических выкладок,
охарактеризуем, следуя [67], молекулярную картину сверхтекучести.
Сверхтекучесть связана с присутствием неидеального бозе-конденсата.
Покоящийся конденсат имеет энергию Ео. Энергетический уровень
конденсата, движущегося со скоростью а:
Nma2
ниже уровней слабо возбужденных состояний, которые в нем могут
возникнуть, если \а\ < а*. Когда абсолютная величина скорости потока
неидеального бозе-конденсата меньше а*, элементарные возбуждения
в нем не могут появляться. Невозможность возникновения
возбужденных состояний означает, что кинетическая энергия потока жидкости не
может превращаться в тепло, иначе говоря, диссипация кинетической
энергии движения потока отсутствует, вязкости у потока нет. Поэтому
243
движение всей массы гелия при О К совершается без трения. При
температурах выше О К сверхтекуча та «компонента» гелия, которая
находится в основном квантовом состоянии. Сверхтекучесть вызвана
существованием скорости а*, связанной с особенностями спектра
возбужденных состояний Е(р). А эти последние вызваны взаимодействияхми
между молекулами. При отсутствии взаимодействий между молекулами
спектр коллективных возбуждений в основном состоянии бозе-газа
исчезнет. Все молекулы в основном состоянии будут покоиться, но
сверхтекучести не будет.
Кроме потенциала (XI. 8), не учитывающего
короткодействующего отталкивания, Н. Н. Боголюбовым рассмотрены случаи, когда
молекулы можно считать идеально упругими шарами, или упругими
шарами со слабым притяжением между ними. Показано, что
появление сверхтекучести обусловлено соотношением между силами
отталкивания и притяжения. Силы отталкивания «благоприятствуют»
сверхтекучести, силы притяжения — «препятствуют». Кроме
того, когда потенциал не имеет радиальной симметрии, теория тоже
остается верной.
Таким образом, гелий II при температурах выше О К состоит из
гелия в основном квантовом состоянии или сверхтекучей
«компоненты» и гелия в возбужденных квантовых состояниях — нормально й
«компоненты». Сверхтекучая и нормальная «компонента» в
пространстве неразделимы. Энтропия сверхтекучей «компоненты» равна
нулю. Энтропия нормальной «компоненты» растет с повышением
температуры. Вязкость сверхтекучей «компоненты» отсутствует. Нормаль-
ная «компонента» имеет вязкость хоть и малую, но измеримую.
§ 94. О проявлениях квантовой когерентности
жидкого гелия
Итак, сверхтекучесть есть проявление квантовой когерентности
жидкого гелия в его основном состоянии. Другим проявлением квантовой
когерентности является упорядоченность пространственного
распределения его плотности. Н. Н. Боголюбов получил выражение (1947) для
флуктуации плотности в элементе объема v, в среднем содержащем
Л/ молекул неидеального газа Бозе — Эйнштейна [67]:
l (XI.19)
При О К
(Nv _ <Л^»2> = <Л^> "~ Q < (Nv) (XI .20)
и флуктуации становятся очень малыми, что указывает на
упорядочение системы, сходное с упорядочением электронной плазмы. Эта
аналогия отмечалась еще в 1938 г. Ф. Лондоном.
Квантовая когерентность феноменологически проявляется в том,
что все слои сверхтекучей компоненты движутся согласованно.
Течение этой компоненты потенциальное. Скорость зависит от потенциа-
244
ла, который описывает поведение всей массы гелия, находящегося
в основном состоянии. Этим объясняются рассмотренные выше
опыты Доунта и Мендельсона, изучавших перенос гелия II по пленке,
возникающей на поверхности сосуда.
Очень наглядны в этом отношении опыты Бендта. В кольцевом
канале им создавался непрерывно циркулирующий поток
сверхтекучей жидкости. Стоило затормозить верхний слой гелия,
находящегося в основном состоянии, погрузив полоску фольги, как
сверхтекучая компонента мгновенно останавливалась во всем кольце [73].
Онзагер показал, что вихри в сверхтекучей компоненте гелия II
квантованы. «Момент количества движения атома гелия вокруг ствола
вихря оказывается кратным постоянной Планка:
masr = nh. (XI.21)
Это выражение, на первый взгляд аналогичное постулату Бора,
отличается от него тем, что радиус г орбиты гелиевого атома вокруг оси
вихря может принимать любые размеры, вплоть до макроскопических
размеров сосуда порядка 1 см. Таким образом, масштабы квантования
возрастают в сотни миллионов раз» [73]. Как видно из уравнения
(XI. 21), распределение скорости вокруг данного вихря определяется
выражением as = rih/mr.
§ 95. Возбужденные состояния гелия II
Молекулярная теория возбужденных состояний гелия II пока что
мало разработана. Развитие получила феноменологическая теория.
Экспериментальные сведения о теплоемкости гелия II и ряд других
данных позволили Л. Д. Ландау построить функцию,
описывающую энергетический спектр возбужденных состояний Е(р) гелия II
[66]. График этой функции, полученной в результате исследований
рассеяния тепловых нейтронов в жидком 4Не, представлен на
рис. 67, заимствованном из статьи Р. Коули и А. Вудса [74] (см.
также [62]).
По оси абсцисс отложены значения волнового вектора \к | = |p|/h,
по оси ординат величина энергии в единицах Е(р)/кв , где кв —
постоянная Больцмана. Модуль волнового вектора Щ имеет размерность
(ангстрем)"1, а Е/кв выражена в градусах шкалы Кельвина. На этом
графике кривая Ландау уточнена экспериментальными данными
Коули и Вудса. Поскольку гелий II при Т > О К представляет собой в
значительной мере упорядоченную, квантово-когерентную систему,
неудивительно, что тепловое движение в гелии II во многом
напоминает тепловое движение твердых тел при температурах, близких к О К.
Тепловое движение в твердых телах при низких температурах можно
представить как совокупность гармонических колебаний звуковых
квантов, или фононов. Такая совокупность упрощенно может
рассматриваться как идеальный фононный газ.
Энергия этих квазичастиц, т. е. фононов, связана с частотой звука
соотношением
E = ho>, (XI.22)
245
где со — круговая частота звуковых колебаний. Каждый фонон
характеризуется квазиимпульсом — вектором /?, аналогичным по
своим свойствам импульсу частицы и равным
(XI .23)
2я .
где кф — волновой вектор звуковой волны, его модуль равен — »
Л — длина звуковой волны.
При низких температурах в соответствии с теорией Боголюбова
[уравнение (XI. Щ]Е(р) линейно зависит от |р|:
Е (р) = ао\р\, (XI.24)
где а0 — скорость звука при О К-
Производная Е по \р\ в области малых |р| дает скорость звука в
гелии в окрестности О К; она равна 240 м/с.
Кривая Ландау (см. рис. 67) изображает энергетический спектр
одноквантовых возбуждений. Та часть этого спектра, которая лежит
около минимума, в соответствии с первоначальной терминологией,
предложенной Таммом и Ландау в 1941 г., носит название Гротонов.
Ркс. 67. Энергетический спектр однофононных
возбужденных состояний в жидком 4Не при 1,1 К [74]:
по оси абсцисс отложены значения волнового вектора k; по оси
ординат — энергия возбуждения £* в условных единицах —
градусах Кельвина; зачерненные точки, кружки и квадраты
относятся к трем сериям измерений, выполненных в различных условиях
опыта
246
В действительности, разделение квазичастиц на фононы и роторы не
имеет физического смысла, как это было показано вначале Н. Н.
Боголюбовым и затем Л. Д. Ландау в 1947 г. Более правильно говорить
о длинноволновых (малые квазиимпульсы) и коротковолновых
(большие квазиимпульсы) типах элементарных возбуждений. Тем не менее
термин ротоны широко применяется в литературе, хотя смысл его
неоднозначен. В жидком гелии II элементарные коллективные
возбуждения могут представлять собой продольные колебания (обычно
звуковые волны) и квантовые вихри.
§ 96. Другие аномалии гелия II
Рассматривая гидродинамику гелия II как системы, состоящей из
двух взаимно проникающих жидкостей, нормальной и сверхтекучей,
Д. Тисса (1938) и Л. Д. Ландау (1941) предсказали, что в этой
системе наряду с обычными звуковыми волнами должны существовать
температурные волны, распространяющиеся с некоторой скоростью а2.
Эти волны были названы вторым звуком. По Тисса, при О К скорость
второго звука должна обращаться в нуль, а по Ландау, она должна
сохранять конечное значение. Экспериментальное доказательство
существования второго звука было получено В. П. Пешковым в 1946 г.
[75]. Результаты измерений скорости второго звука, выполненных
В. П. Пешковым и другими исследователями, находятся в
качественном согласии с теорией Ландау. При
температуре, близкой к О К, скорость
второго звука составляет около 200 9п19
м/с и в соответствии с теорией Лан- '
дау растет с увеличением давления.
В. П. Пешков предположил, что
второй звук может существовать и в
твердом гелии. Через 20 лет это
предположение было подтверждено.
Как уже отмечалось, правильное
описание ряда необычных явлений,
наблюдаемых в гелии II, впервые было
дано Л. Д. Ландау. Он подчеркнул, °>50
что «рассмотрение гелия как «смеси»
двух жидкостей является не больше,
чем способом выражения, способом,
удобным для описания явлений,
происходящих в гелии II. Как и всякое
описание квантовых явлений в
классических терминах, оно не является
вполне адекватным. В
действительности надо говорить, что в кванто- 0 опт и
вой жидкости, каковой является ге- '
ЛИЙ II, могут существовать ОДНОВре- Рис- 68- Зависимость относитель-
менно два движения, каждое из 1о- Гмпон^ГДГох теГр'^Й
торых связано со своей «эффективной [75]
*****
***
ч$
У
г/
у
f
J
/
.7
/
7
/
1 /
\1
\!
\?
/
J
/
/
t
247
массой» (так что сумма обоих этих масс равна полной истинной массе
жидкости). Одно из этих движений «нормально», т. е. обладает теми
же свойствами, что и движение обычной жидкости, другое же
«сверхтекуче». Оба эти движения происходят без передачи импульса от
одного к другому. Особенно подчеркиваем, что здесь нет никакого
разделения реальных частиц жидкости на «сверхтекучие» и «нормальные».
В определенном смысле можно говорить о «сверхтекучей» и
«нормальной» массах жидкости как о массах, связанных с обоими
одновременно возможными движениями, но это отнюдь не означает
возможности реального разделения жидкости на две части» [66].
По предложению Л. Д. Ландау, Э. Л. Андроникашвили [76]
провел эксперимент, позволивший изучить зависимость эффективной
массы нормальной компоненты гелия II от температуры. Результаты
опытов Э. Л. Андроникашвили представлены на рис. 68.
§ 97. Сверхтекучесть и сверхпроводимость
Родственность явлений сверхтекучести и сверхпроводимости
отмечалась уже в ранних работах Л. Д. Ландау и многих других
авторов. Принципиальное сходство этих явлений было установлено
Д. Бардиным, Л. Купером и Д.' Шриффером и независимо от них
Н. Н. Боголюбовым. Находящиеся вблизи поверхности Ферми
электроны в металлах могут образовывать попарно связанные состояния.
Эти пары при низких температурах претерпевают конденсацию в
пространстве импульсов, что ведет к возникновению сверхтекучести.
Но сверхтекучесть таких систем проявляется как сверхпроводимость,
так как частицы системы имеют электрический заряд. Очень
интересен вопрос, являются ли сверхтекучесть гелия и и
сверхпроводимость электронной плазмы в металлах единственными квантово-ко-
герентными состояниями жидкостей. (Электронная плазма в металлах
напоминает жидкость.) Вполне вероятно, что сверхтекучесть и
сверхпроводимость во вселенной распространены более широко.
Они могут встречаться в больших сгустках ядерной материи,
которые, в сущности, тоже представляют собой жидкие системы.
Например, в нейтронных звездах. Этим проблемам посвящены статьи
В. Л. Гинзбурга [77] и Д. А. Киржница [78], монография Ф. Дай-
сона и Д. Тер-Хаара [79].
§ 98. Изотоп 3Не [58]
На каждые 107 атомов 4Не в природе встречается всего лишь от
одного до десяти атомов 3Не. Возможность исследований свойств
жидкого 3Не связана с накоплением трития3Н в ядерных реакторах.
В ходе медленно протекающего [3-распада трития образуется 3Не:
Атомы 3Не в отличие от 4Не имеют ядерный спин, равный 1/2, и
магнитный момент, равный 1,07 • 10~26 Дж • м2/Вб. Как уже говорилось,
248
атомы 3Не —фермионы, поэтому при низких температурах свойства
зНе существенно отличаются от свойств 4Не. Ядро 3Не легче, чем
ядра других газов, за исключением водорода. Взаимодействие между
атомами 3Не слабее, чем у любых других атомов, и 3Не сжижается
последним из газов. Число атомов в 1 мл у жидкого 3Не меньше, чем у
какой-либо другой жидкости, например, в 3 раза меньше, чем у
жидкого водорода, и в 1,3 раза меньше, чем у жидкого 4Не. Диаграмма,
описывающая связь между
плотностью 8Не и температу- р,г/см3
рой, представлена на рис. 69
[58].
Нормальная температура
кипения жидкого 3Не равна
3,19 К, температура,
давление и объем 3Не в
критической точке равны: Тк = 3,38
К; рк -1,24- 10б Па,ик =
=72,8 мл. 3Не, как и 4Не,
остается жидким при самых
низких достигнутых до сих пор
температурах. Твердый 8Не
образуется при давлениях
выше 2,9- 106 Па. Известны три
модификации твердого 3Не:
кубическая объемноцентриро-
ванная (ОЦК), плотноупако-
ванная гексагональная (ПГУ)
и кубическая гранецентриро-
ванная (ГЦК). Области
существования каждой из
модификаций представлены на рис.
69 и 70. На рис. 70
изображена Р—Т диаграмма 3Не.
При 1,9 К и давлении 9,8- 106
Па длина ребра куба ОЦК
модификации составляет
около 0,4 нм. Тройная точка
сосуществования ОЦК и ПГУ
модификаций с жидкой фазой
находится при 3,15 К и
1,42 • 107 Па. Другая
тройная точка, где жидкость
сосуществует с ПГУ и ГЦК
модификациями, наблюдается
при 15,98 К и 1,36- 107Па.
Рентгеногр афические ис-
следования строения и
спектра элементарных
возбуждений в жидком 3Не при 0,79 К
Твердая фаза Гексагональная ПГУ
0,04
Рис. 69. Зависимость плотности
жидкого 3Не от температуры [58]
Р. атм
10*
10~'
10'
г 5
10
10"
10
10"
т
I
Куб
7дая
Чет
jnech
ОЦК
Жид
каст
—i
i
/
I
фа
VdL
гая
■—■*
—f-
ь!
У~
ty
f—
за
г— 1
1на/1ьна
f
/
Га
/
3
(убичес
ГЦК.
ix"
:кая
1
0,1 0,1 0,5/2 5 10 20 50Т,К
ммрт.ст.
W3
Ю2
10
1
да-2
w3
ю'*
ю-5
Рис. 70. Р—Т-диаграмма фазовых
равновесий в 3Не [58]
249
были выполнены Е. К- Ахтером и Л. Мейером [62]. Полученная ими
радиальная функция распределения представлена на рис. 62. Нейтро-
нографические измерения структурного фактора жидкого 3Не при 0,4 К
и спектра элементарных возбуждений были выполнены Р. Б. Халло-
ком [80]. Из рис. 62 следует, что распределение атомов в жидком
3Не менее упорядочено, чем в жидком 4Не.
В жидком 3Не при 0,5 К и давлении насыщенных паров плотность
проходит через максимум, так что др/дТ = 0. Повышение давления
смещает эту особенность в область более высоких температур. Там,
где др/дТ ->0, тепловая конвекция резко падает. Это ухудшает
перенос тепла и осложняет исследования свойств при низких температурах.
Кривая плавления 3Не при~ 0,32 К имеет минимум. Когда температура
плавления находится ниже 0,32 К, производная dPIdT
отрицательна, и в соответствии с уравнением Клаузиуса — Клапейрона
плавление должно сопровождаться выделением тепла. Причины этой
замечательной особенности в свойствах 3Не будут рассмотрены далее.
Энтальпия испарения жидкого 3Не при Г-^0 почти на 37,6 Дж/моль
меньше, чем энтальпия испарения жидкого 4Не. Как показал
Е. С. Рудаков [81], большая часть этой разницы обусловлена тем,
что при испарении 4Не образуется бозе-газ, а 3Не — ферми-газ.
Расчет внутренних энергий жидких 3Не и 4Не при Т ->0 с учетом
упомянутого обстоятельства показывает, что внутренняя энергия 4Не лишь
на ~12,5 Дж/моль больше, чем внутренняя энергия 3Не.
Кинетическая энергия движения фермионов 3Не при^Г =0в среднем выше, чем
бозонов 4Не. Это ведет к большему объему жидкого 3Не и,
соответственно, меньшей энергии взаимодействия между его атомами.
Жидкий 3Не, как и 4Не, следует уравнению Лоренца — Лоренца.
Интересно то, что молярная поляризация обоих жидкостей одинакова
Р ~Nx<x =0,123 мл, хотя энергия взаимодействия атомов различна*.
Молярный объем V жидкого 3Не, полученный экстраполяцией к 0 К,
равен 36,8346 мл/моль (экстраполировалась функция V = f(T) при
давлении насыщенных паров).
Объемный коэффициент расширения аг=— ( ) зависит от
^ r r т V \ дТ ]р
температуры сложным образом. При 0 К *т = 0, между 0 К и
0,5 К %т отрицательно и проходит через минимум, расположенный
около 0,2 К. В точке минимума ат = —10,38 • 10~3 К"1. При температуре,
близкой к 0,5 К %т , равно нулю. Выше 0,5 К %т положительно и быстро
увеличивается с температурой до значения 480,86 • 10"3 К"1 при
3,20 К- Таким образом, при температурах ниже 0,5 К нагревание
жидкого 3Не сопровождается уменьшением объема жидкости.
Сжимаемость жидкого 3Не очень велика. При 1,59 К сжимаемость
Р; равна 4,4 • 10"7 Па"1. В окрестности критической точки при 3,1 К
{3, = 4,17 • 10~4 Па"1, что всего лишь в 2,4 раза меньше сжимаемости
идеального газа. Поэтому жидкий 3Не отчасти напоминает газовую
* Здесь а — поляризуемость одного атома 3Не.
250
фазу. Повышение давления сопровождается резким уменьшением
объема и сжимаемости.
Поверхностное натяжение жидкого 3Не при температурах ниже
1,0 К, когда оно достигает наибольшей величины и уже практически
не зависит от температуры, равно всего 1,52 • 10~2 Н • м"1. Это в 500
раз меньше, чем у воды, почти в 100 раз меньше, чем у жидкого азота,
в 20 раз меньше, чем у водорода, и в 2,3 раза меньше, чем у жидкого
4Не. Любопытно, что если нанести на график значения поверхностного
натяжения а3Не и 4Не в масштабе с/а0 как функции Т/Тк, где
а0 — значение поверхностного натяжения при Т = 0К и Тк —
критическая температура, то кривые совпадают [58].
§ 99. О теплоемкости жидкого 3Не
Особый интерес представляют измерения теплоемкости жидкого
3Не. Результаты этих измерений в логарифмическом масштабе
изображены на рис. 71, где они сопоставлены с теплоемкостью жидкого 4Не
и растворов 3Не и 4Не. Теплоемкость жидкого 3Не, находящегося под
давлением собственных паров, при температурах ниже 0,1 К прямо
пропорциональна температуре. Значение этого результата для теории
жидкого 8Не обсуждается далее.
*С,Дж/моль -град
80
60
50
40
30
20
15
10
8,0
60
Ко
4fi
2,0
15
W
0,8
0,6
0,3
0,2
11s)
01
/
/
/
29атм/
i r
He3
+*-
0,0.
—•
e»
0J5
-
005
; Hei
\0J5^
i
до|Щ
и'
'Ж-
If
1-Щ
BE
u
I
Ш
-H
t
0,01 0,02 щи о,ооц}о,ш
0,81$ 15 2 3 4 5Т, К
Рис. 71. Теплоемкость 3Не, 4Не и растворов 3Не в
4Не [58]
(Цифры на графиках указывают концентрацию 3Не в мольных
долях)
251
Интересно сопоставить теплоемкость Cs жидкого 8Не вдоль кривой
равновесия жидкость — пар с теплоемкостью при постоянном
объеме Су и постоянном давлении Ср . На рис. 72 приведены графики
зависимости С, Су, Ср и Ср /Су от температуры. При температурах ниже
1,0К все три функции «практически
совпадают и отношение Ср /Су
становится постоянным.
Теплоемкости двумерных слоев
3Не и 4Не, покрывающих
сорбирующую поверхность почти полным
одноатомным слоем (74% заполнения
поверхности для 3Не и 80% для 4Не),
от 0,25 до 4К пропорциональны
квадрату температуры. Теплоемкость
твердого 3Не в различных его
модификациях, как и теплоемкость
твердого 4Не, при низких температурах
пропорциональна Т3 (с некоторыми
уточнениями, о которых будет
говориться ниже).
Теперь надо сказать несколько
слов о теории, связывающей
теплоемкость со строением вещества и
природой его теплового движения.
Если вещество—идеальный газ,
состоящий из фермионов, то,
пользуясь статистикой Ферми —
Дирака, нетрудно показать (см., например, [50]), что при низких
температурах
1,0
Рис. 72. Теплоемкости при
постоянном объеме Су, давлении Ср и
при состояниях, соответствующих
равновесию с паром С5, жидкого
Не а также отношение Ср 1Су
[58]
(Теплоемкости выражены в безразмерных
единицах C/R)
С =
*
Nk\T
= const 7\
(XI .25)
где кБ — постоянная Больцмана; sF — энергия Ферми, т. е.
наивысший уровень энергии фермионов этой системы при Т = 0.
Допустим, что система двумерна, например, мономолекулярный
слой, адсорбированный какой-либо плоской поверхностью. Точный
расчет теплоемкости упрощенной модели классической (т. е.
неквантовой) двумерной упорядоченной системы (модели Изинга) выполнен
Л. Онзагером. Зависимость С от Т сложна. Но при температурах,
значительно меньших критической температуры двумерной фазы, С
приблизительно пропорционально Т2.
Для трехмерных кристаллов в ряде простых случаев выполняется
теория теплоемкости, развитая Дебаем. Согласно этой теории
тепловые колебания N атомов кристалла можно приближенно представить
как совокупность 3N независимых друг от друга звуковых волн, или
фононов (продольных и поперечных). Связь между их энергией г и
импульсом р имеет виде = ар, где а—скорость звука. Фононы можно
рассматривать как квазичастицы, подчиняющиеся статистике Бозе-—
252
Эйнштейна. Расчеты, основанные на применении этой статистики при
низких температурах, ведут к выражению
~ 12тт:4 / Т \»
Cv = —— Rr -к-- > (XI .26)
где 0д — так называемая температура Дебая. В теории Дебая 0д
определяется уравнением &Б ®д = ^,п, где ют — максимальная
круговая частота звуковых колебаний в кристалле. Для двумерных
решеток аналогичный подход ведет к формуле
е i (Х1-27)
Дебаевская температура упомянутой выше адсорбированной
мономолекулярной пленки 3Не равна 28 К, а 4Не 31 К. Теплоемкость
твердого 3Не при температурах от 6,3 до 14 К и при молярных объемах
от 24,4 до 12,57 мл/моль выражается следующей эмпирической
формулой:
3
Дж/к-моль. (XI. 28)
Член AT в (XI. 28) очень мал и, по-видимому, вызван примесями,
вакансиями и дислокациями, т. е. нарушениями идеальной
упорядоченной структуры кристалла. ^
Зависимость дебаевской температуры Эд от молярного объема V
как для гексагональных кристаллов (ПГУ), так и для кубических
(ОЦК) определяется формулой
йп
— = — 2,5. (XI .29)
din V
В точке перехода кубических кристаллов в гексагональные при V =
=20 мл/моль для гексагональных кристаллов @д= 35 К, а для
кубических вд = 29 К- Теплоемкость твердых гексагональных кристаллов
4Не также определяется формулами (XI. 28) и (XI. 29). Значения де-
баевских температур при этом совпадают со значениями 0Д для
кубических кристаллов 3Не [58]. Любопытно, что дебаевская
температура ОЦК кристаллов 3Не и ПГУ кристаллов 4Не очень близка
к дебаевской температуре адсорбированных мономолекулярных
пленок этих веществ в интервале температур от 0,25 до 4 К-
§ 100. Теплоты фазовых переходов
и энтропия 3Не. Магнитные свойства
Энтальпии испарения и плавления 3Не приведены на рис. 73.
У 8Не энтальпия перехода ОЦК модификации в гексагональную при
3,15 К составляет около 1,8 Дж/моль. Внизу слева на рис. 73 изоб-
253
ражена в десятикратном масштабе энтальпия плавления при
температурах ниже 1 К- Если температура ниже 0,32 К, то как уже отмечалосьу
плавление 3Не сопровождается выделением тепла (отрицательная
энтальпия плавления). При 0,15 К уменьшение энтальпии в
результате плавления сравнительно велико — достигает 0,25 Дж/моль*.
Энтропийная диаграмма 3Не представлена на рис. 74. Кривая А —
график функции S = f(T) жидкого 3Не, находящегося в равновесии
с паром. Кривая В при Т > 0,33 К описывает зависимость 5 от Г
гмоль-град
/У, Дж/моль
100
1 2 3
-1Ди<1'моль
5ГК
Рис. 73. Энтальпия фазовых
переходов 3Не [58]:
А — энтальпия испарения: В — энтальпия
плавления; С — продолжение кривой В в
десятикратном масштабе
20
18
16
74
11
10
8
В
0 0у5 1 1,5 2 2,5 3 3,5 ЦК
Рис. 74. Энтропийная диаграмма 3Не
[58]:
А — энтропия жидкого 8Не, находящегося в
равновесии с паром; В — энтропия жидкого 3Не,
находящегося в равновесии с твердой фазой;
выше линии R 1п2 располагаются значения
энтропии ОЦК и ПГУ модификаций 8Не
У
г
1
jf
/
/
за
ОЦК
/
/
а—»—
Г£=
/
/
—1 —
ПГУ
/
———
в
■■ -
жидкости, находящейся в равновесии с твердым 3Не. Ниже 0,32 К
кривая В отображает функцию S = /(Г) при давлении 29,4 • 105 Па
(29 атм).
Вертикальная линия при 3,15 К соответствует тройной точке, где
сосуществуют жидкая, гексагональная и кубическая твердые фазы
3Не. Линии со знаками ОЦК и ПГУ соответствуют значениям
энтропии кубических и гексагональных кристаллов, находящихся в
равновесии между собой. Верхняя линия этой части диаграммы соответствует
энтропии кубических кристаллов, находящихся в равновесии с
жидкостью. Область резкого спада энтропии твердого 3Не в области низ-
* Это позволяет применять адиабатическую кристаллизацию жидкого 3Не
для получения очень низких температур. Отметим, что кристаллизация жидкого
4Не путем сжатия при температурах ниже 0,77 К и давления 2,5-106 Па тоже
приводит к возрастанию энтальпии (поглощению теплоты). Но при 0,7 К изменение
энтальпии составляет всего 3,5-10~4 Дж/моль, а при других температурах оно
еще меньше [58].
254
ких температур обозначена произвольно [58]. Надежные
экспериментальные данные в этой области отсутствуют.
Таким образом, энтропийная диаграмма 3Не аномальна. В
отличие от всех других веществ плавление 3Не ниже 0,32 К сопровождается
уменьшением его энтропии.
Магнитные свойства. Магнитный момент атомов 3Не равен
магнитному моменту их атомного ядра и составляет 0,7618 |jio|, где jip —
магнитный момент протона. Магнитный момент ядра 3Не в отличие
от момента протона отрицателен. Он является наибольшим по
абсолютной величине отрицательным магнитным моментом атомного ядра.
Сведения о магнитных свойствах 3Не важны для выяснения причин
аномалий, наблюдаемых при низких температурах.
Изотоп 3Непарамагнитен, его магнитная восприимчивость
положительна. Рассматривая магнитные свойства 3Не, полезно сопоставить их
со свойствами парамагнитного ферми-газа, т. е. газа, состоящего из
фермионов. При наложении внешнего магнитного поля в таком газе
возникает магнитный момент, обусловленный перераспределением
частиц между состояниями с различными ориентациями их спина.
Пусть температура ферми-газа достаточно высока, чтобы
выполнялось условие
kBT^>e*fi (XI.30)
где е^ — магнитная энергия Ферми. Здесь символ ejj. означает
наивысший уровень энергии системы в магнитном поле при Т = 0 , когда
система полностью вырождена, т. е. нижележащие уровни ее энергии
в магнитном поле заполнены.
Если условие (XI. 30) выполнено, то ориентации спинов частиц
полностью разупорядочены. При наложении внешнего магнитного поля
каждая частица вносит одинаковый вклад в магнитную поляризацию
газа. В итоге магнитная восприимчивость х единицы объема газа
следует уравнению
MiLf {Х1.зп
1
1 3VkBT Т
где В = N\><2/3VkB . Когда объем газа задан, В постоянна и газ следует
уравнению Кюри:
уТ = в (XI.32)
{В носит название постоянной Кюри).
При понижении температуры взаимодействие между молекулами
газа приводит к упорядочению спинов. В некоторых случаях это
может сопровождаться возникновением намагниченности. Так или
иначе, магнитная восприимчивость уменьшается. Падение % с
температурой становится заметным в окрестности магнитной температуры
Ферми T*F, которая определяется соотношением
къ1*Р = *г (XI.33)
Когда Т ~T*F, число частиц, спины которых упорядочены, сравни-
255
тельно велико. Вырождение системы становится значительным,
поэтому температуру Ферми называют также температурой вырождения.
Если спонтанной намагниченности нет, то при О К магнитная
восприимчивость неидеального ферми-газа /о равна
(XI. 34)
В ходе вывода этого уравнения используется допущение, что ферми-
газ разрежен, т. е. его отклонения от идеальности малы [50].
Магнитная восприимчивость жидкого 3Не крайне мала. При 2,0К
она составляет около 2,2 • 10"4, что в 210 раз меньше магнитной
восприимчивости жидкого 4Не и в сто с лишним тысяч раз меньше
магнитной восприимчивости жидкого кислорода. (Заметим, что 4Не
диамагнитен, а кислород — парамагнитен.) Тем не менее надежные
измерения х жидкого 3Не были выполнены рядом исследователей. Наиболее
полные данные имеются в работе В. Б. Била и Д. Хэттона при
давлениях от 7 • 104 до 27,8 • 105 Па. Основные результаты их работы
сводятся к следующему. Выше 2Kb жидком 3Не выполняется уравнение
Кюри (XI. 32). При более низких температурах начинается
упорядочение ядерных моментов, связанное с их взаимодействием. При очень
низких температурах % не зависит от Т.
Затвердевание жидкого 3Не при низких температурах
сопровождается разупорядочением ориентации ядерных спинов. Этот
экспериментальный факт, вытекающий из анализа магнитных свойств
3Не, объясняет рост энтропии при затвердевании 3Не и связанную с
этим отрицательную теплоту плавления. Энтропия при температурах
порядка 0,05 К и ниже определяется, в основном, степенью
упорядоченности спинов атомных ядер, т. е. квантовыми особенностями этого
вещества. Впервые возможность упорядочения спинов при плавлении
3Не и связанных с этим аномалий энтропии и энтальпии была указана
И. Померанчуком еще в 1950 г. Поэтому отрицательная теплота
плавления 3Не иногда именуется эффектом Померанчука.
§ 101. О теории жидкого 3Не
Жидкий 3Не — изотропная ферми-жидкость, состоящая из частиц,
не имеющих заряда. Теория такой квантовой ферми-жидкости была
развита Л. Д. Ландау [82], его учениками Л. П. Питаевским,
И. М. Халатниковым, А. А. Абрикосовым [83] и рядом других
теоретиков [84]. Согласно этой теории спектр квантовых возбужденных
состояний изотропной ферми-жидкости подобен спектру квантовых
состояний идеального ферми-газа.
Как известно, при Т = 0, т. е. в основном квантовом состоянии
идеального ферми-газа, его молекулы заполняют все состояния с
импульсами, меньшими некоторого граничного значения pF. Состояния
с большими, чем pF, импульсами не заполнены. Выберем декартову
систему координат. Пусть по осям X, Y\\ Z будут отложены рх, ру и
pz компоненты импульсов р молекул газа. Тогда мы получим импульс -
256
ное пространство, в котором роль координат будут играть импульсы.
При О К заполненные состояния в импульсном пространстве образуют
сферу с радиусом pF. По Ландау, величина pF ферми-жидкости
определяется той же формулой, что и для идеального ферми-газа:
pF=:h(3n2/9)W t (XI.35)
где р — число частиц жидкости в единице объема, т. е. N1V.
Рассмотрим теперь слабовозбужденные квантовые состояния
изотропной ферми-жидкости. Их энергия должна мало отличаться от
энергии основного состояния. В возбужденных состояниях
распределение частиц по импульсам не такое, как при О К. Всякое возбужденное
состояние может быть получено из основного путем
последовательного перевода частиц из внутренней части ферми-сферы наружу. При
каждом таком элементарном акте, или, иначе говоря, элементарном
возбуждении, получается состояние, отличающееся от исходного
появлением частицы, имеющей импульс р > pF, и возникновением «дырки»
в ферми-сфере, где р < pF. Каждое элементарное возбуждение имеет
спин 1/2. Элементарные возбуждения всегда образуются парами. У
одного из них импульс больше pF\ импульс другого меньше pF.
Хотя спектр квантовых возбужденных состояний изотропной
ферми-жидкости, как уже было сказано, подобен спектру квантовых
состояний идеального газа, эти спектры все же несколько различаются.
Элементарные возбуждения в ферми-газе не взаимодействуют друг
с другом. В ферми-жидкости каждая частица взаимодействует со всеми
остальными, поэтому частица, находящаяся вне ферми-сферы,
движется вместе с тем возмущением остальных частиц, которое возникло
вследствие взаимодействия. Это, по существу, уже не частица, а некоторое
коллективное состояние ферми-жидкости, зависящее от движения
многих частиц. А так как элементарное возбуждение в ферми-жидкости
вблизи ферми-поверхности в некоторых отношениях все же подобно
частице, то оно носит название «квазичастицы». Квазичастицам
можно приписать определенный импульс и эффективную массу т*.
Квазичастицы взаимодействуют друг с другом. Теория Ландау
учитывает это взаимодействие с помощью ряда безразмерных
параметров F1 и F?,(l=0, 1,2, ...).
Эти параметры дают представление о величине энергии
взаимодействия квазичастиц по сравнению с их кинетической энергией.
Параметры F] обозначают те взаимодействия, которые не зависят
от симметрии волновой функции, описывающей движение двух
квазичастиц. Это симметричные по спинам части взаимодействия;
отсюда и индекс «s» у символа Ff. Параметры Ff зависят от симметрии
волновой функции. Они описывают ту часть взаимодействия, которая
связана с антисимметричным характером волновой функции,
описывающей движение двух фермионов, что отражается с помощью индекса
«а» у символа Ff. В теории играют основную роль фактически только
три параметра, характеризующих взаимодействие между
квазичастицами, — это Fq, F$ и F\.
257
Число квазичастиц пропорционально 71, а их средняя энергия
пропорциональная кБТ, поэтому связанная с тепловыми возбуждениями
разность энергий жидкости в возбужденном, т. е. при Т > 0 и
основном (при 71 = 0) состояниях пропорциональна Т2. Следовательно,
теплоемкость Су пропорциональна Т:
С помощью формулы (XI. 36), зная из опыта величину Су ферми-
жидкости в области, где Су пропорционально Т, можно найти
эффективную массу квазичастиц т*. А зная т*, по формуле
легко вычислить параметр взаимодействия F\. Формула (XI. 37)
позволяет охарактеризовать физический смысл этого параметра.
Ft — та часть симметричного по спинам взаимодействия между
квазичастицами, которая определяет их эффективную массу. Если
F* < —3, то система неустойчива. Такая система не может
существовать.
Магнитная восприимчивость слабо возбужденной ферми-жидкости,
обусловленная ориентацией спинов («спиновая восприимчивость»),
имеет вид
m*pF «a
(XI.38)
где Р = eh/2mc — магнетон Бора; е — заряд электрона; с — скорость
света.
Если Fq ->■—I, то х ->■ ^ и в ферми-жидкости возникает
спонтанная намагниченность. Тогда ферми-жидкость будет
ферромагнитной. Найдя pF и т* по уравнениям (XI.35) и (XI.36) можно по
данным о х с помощью уравнения (XI.38) вычислить Fq .
Физический смысл параметра F% следующий. Волновая функция,
описывающая относительное движение двух фермионов с
параллельными спинами, в соответствии с принципом Паули должна быть
антисимметричной по отношению к перестановке их координат.
Поэтому такие фермионы имеют малую вероятность находиться вблизи
друг друга. Они стремятся расположиться как можно дальше друг
от друга. FeiKaK раз и описывает то стремление к отталкиванию квг*
зичастиц ферми-жидкости, которое обусловлено принципом Паули,
т. е. особенностями симметрии волновых функций.
Скорость звука а следует уравнению
a2=i^(1 + Fo>' <XI-39)
258
Если Fo -*—1, то, согласно (XI.39), скорость звука стремится к
нулю. При Ff <—1 ферми-жидкость будет неустойчива. Зная
плотность р, скорость звука а и теплоемкость Су, можно по
уравнениям (XI.35), (XI.36) и (XI.39) найти Fo. Параметр Fs0
характеризует отталкивательное взаимодействие само по себе, т. е. не
зависящее от спина квазичастиц.
Мы видели, что, согласно теории Ландау, магнитная
восприимчивость и скорость звука ферми-жидкости не зависят от температуры.
Опыты с жидким 3Не при температурах ниже 0,05 К
подтверждают это. Измеряя Су и х жидкого 3Не при очень низких температурах,
когда х и а становятся постоянными, можно найти параметры т*,
Fо, Fa0 и Fi жидкого 3Не.
Из теории Ландау далее следует, что теплопроводность
слабовозбужденной изотропной ферми-жидкости должна быть
пропорциональна Т"1, а вязкость Т~2. Эти результаты тоже согласуются со
свойствами жидкого 3Не. Так, при температурах, меньших 0,-1 К, и
давлении насыщенных паров вязкость жидкого 3Не следует эмпирической
формуле [58]:
0,32-10~8
1 = -^ . Па-с. (XI.40)
Длина L свободного пробега квазичастиц в ферми-жидкости, по Ландау,
пропорциональна Г"2. Квазичастицы могут принимать участие в
распространении звука, если его длина волны X много больше L. При
достаточно низких температурах величина L сравнима с X и звук
распространяться не может. Но, как показал Л. Д. Ландау, в этих
случаях возникает особый вид движения, обусловленный квантово-
когерентными свойствами жидкости в окрестности 0 К. Это движение
было названо нулевым звуком. Оно сопровождается периодическими
деформациями ферми-поверхности в пространстве импульсов. В ходе
этих деформаций ферми-поверхность перестает быть сферой и
вытягивается в направлении распространения нулевого звука. Скорость
нулевого звука немного превышает скорость обычного звука. Нулевой
звук в жидком 3Не был обнаружен и изучен В. Р. Абелем, А. К.
Андерсоном и Д..К. Уитли [85].
Таким образом, теория слабовозбужденной ферми-жидкости,
основы которой были развиты Л. Д. Ландау и его учениками,
качественно, а в некоторых случаях и количественно описывает многие
свойства жидкого 3Не при температурах ниже 0,1 К. Весьма интересна
проблема существования у жидкого 3Не области сверхтекучего
состояния при очень низких температурах. Теория считает, что
сверхтекучее состояние может быть. Экспериментальное подтверждение этого
вывода, как уже отмечалось, по-видимому, получено [70]. Теория,
позволяющая объяснить поведение жидкого 3Не при температурах
выше 0,1 К, отсутствует.
ГЛАВА XII
СТРОЕНИЕ ПРОСТЫХ ЖИДКОСТЕЙ
И ПЕРИОДИЧЕСКАЯ СИСТЕМА МЕНДЕЛЕЕВА
§ 102. Введение
До сих пор мы сопоставляли свойства и строение простых
жидкостей в рамках какой-либо подгруппы периодической системы.
Переходя от одной подгруппы к другой, мы описывали строение
простых жидкостей в той мере, насколько оно изучено. Теперь сведем этот
материал воедино. Расположим простые жидкости в
последовательности, диктуемой порядковыми номерами соответствующих элементов,
и опишем некоторые их свойства, тесно связанные со строением.
Обозревая всю картину в целом, отметим ее наиболее характерные
черты.
Металлы и неметаллы. Из табл. 30 видно, что подавляющее
большинство простых жидкостей — металлы. (Речь идет о жидкостях,
находящихся под давлением своих паров.) Плавление простых
металлов ни в одном случае не приводит к потере металлических свойств.
Наоборот, при плавлении некоторые неметаллы (германий, кремний и
др.) становятся металлами. Таблица демонстрирует хорошо
известный факт: неметаллы концентрируются в правом верхнем углу
таблицы Менделеева. У жидких простых веществ эта особенность
выражена еще более отчетливо, чем у твердых.
§ 103. Молярные объемы
Молярные объемы и атомные радиусы. При характеристике
размеров атомов нередко пользуются атомными радиусами, определяемыми
с помощью рентгенографического анализа кристаллических структур.
Выбор атомных радиусов часто связан с введением некоторых
упрощений, поскольку атомы обычно не имеют сферической формы. Кроме
того, атомные радиусы зависят от аллотропных модификаций, как
правило, сильнее, чем молярные объемы. Поэтому для характеристики
размеров атомов мы будем пользоваться молярными объемами,
определяя их как частное от деления молярной массы на плотность
твердой или жидкой фазы. Объем моля простого вещества V, или молярный
объем, очевидно, есть термодинамическая функция температуры Т и
давления Р. При заданных Рп Г величина V определяется,
следовательно, минимумом свободной энтальпии G. Величина молярного объема
строго говоря, имеет смысл только тогда, когда указаны физические
условия, при которых она определена. Плотность электронного
облака вокруг любого изолированного атома в принципе отлична от
нуля даже на больших расстояниях от атомного ядра, хотя и резко
падает с расстоянием. Чтобы охарактеризовать объем атома,
необходимо указать физические условия существования атома, метод изме-
260
о
со
сз
1
60 §
« й
0Q О
а"8§
Ч S с
ч
g з S
<u 4 et
S ч s
s*
§ 1 g"
4
I!
| g
^ Си
0?
ё
И о и d э
рения объема или какие-либо теоретические допущения относительно
граничной плотности электронного заряда.
Молярные объемы простых веществ в принципе следовало бы
сопоставлять при эквивалентных физических условиях. Например,
интересно было бы сравнить молярные объемы всех простых
жидкостей при температурах затвердевания, температурах
кипения и температурах критической точки. Однако такие данные для
большинства простых веществ отсутствуют. На рис. 75 сопоставлены
молярные объемы простых веществ при "неэквивалентных физических услови-
90 г
80
70
60
50
30
20
10
20
30
50
60
70
80
90
Рис. 75. Зависимосгь молярных объемов твердых простых веществ от
порядкового номера п соответствующих элементов в периодической системе
Менделеева
ях. Для3Неи4Не выбраны молярные объемы жидких фаз, найденные
экстраполяцией экспериментальных данных к О К. Для водорода,
аргона и других простых веществ с температурой плавления ниже 0°С
приведены молярные объемы твердых фаз около температур их
плавления. Для других элементов даны молярные объемы
соответствующих фаз при комнатной температуре.
Молярные объемы и периодический закон. Несмотря на неполную
сопоставимость молярных объемов диаграмма, представленная на
рис. 75, пригодна для качественной и даже полуколичественной ха-
262
рактеристики зависимости молярных объемов твердых и жидких
простых веществ от порядкового номера соответствующих элементов.
Дело в том, что изменение объема простых веществ при нагревании
от комнатной температуры до температуры плавления обычно не
превышает 5—6%. Исключений немного. Например, марганец
увеличивает свой объем на 20%. Плавление тоже сопровождается небольшими
изменениями объема. Обычно они составляют 4—5% и лишь для
водорода, инертных газов и иода достигают 11—13%, что немного по сравнению
с различиями в молярных объемах простых веществ. Молярный объем
цезия равен 70 мл — это наибольший из молярных объемов, если не-
считать франция, для которого V неизвестен, но может быть еще
большим. Наименьший молярный объем имеет углерод. Для него V =
= 3,4 мл. Таким образом, различие в молярных объемах и,
следовательно, в числах атомов в 1 мл для твердых и жидких простых веществ
может быть 20-кратным. В 1 мл жидкого цезия вблизи точки плавления
находится примерно 8,0 • 1021 атомов, а в 1 мл жидкого углерода в
аналогичных условиях около 1,5 • 1023 атомов. Еще разительнее
различия в общем числе электронов, находящихся в 1 мл жидкой и
твердой фазы. Молярные объемы твердого или жидкого водорода и нептуния
одинаковы. Но у твердого водорода в 1 мл находится 5,2 • 1022
электронов, а в 1 мл нептуния в 93 раза больше, т. е. 4,9 • 1024 электронов.
Молярные объемы обнаруживают, в общем, периодическую
зависимость от порядкового числа я, но такая зависимость от п отчетливо
проявляется лишь начиная с третьего периода. Максимумы функции
V =f(ri), начиная с третьего периода, соответствуют щелочным
металлам. Минимумы этой функции в малых периодах соответствуют
элементам подгруппы углерода, а в больших периодах — элементам
подгруппы железа и платины*.
Хотя функция V = f(n) периодична, однозначной связи между
строением электронных оболочек атомов и молярными объемами нет.
Одной из важнейших характеристик состояния электронов в атомах
являются ионизационные потенциалы атомов. Они определяют
энергию связи электронов в атомах и тесно коррелируют со структурой
атомной оболочки. Первые ионизационные потенциалы атомов, т. е.
характеристики энергии, необходимой для отрыва одного электрона от
электрически нейтрального атома, представлены как функции
порядкового номера п на рис. 76. Из рисунка видно, что периодическая
зависимость первых ионизационных потенциалов 1г от п выражена ярко.
Максимумы 1г соответствуют атомам элементов подгруппы гелия, а
минимумы — атомам элементов подгруппы лития.
Сопоставим графики 75 и 76. Сходство графиков заключается в том,
что максимумам У для натрия, калия, рубидия, цезия и франция соот-
* Функция v = f(n), где п — порядковое число элементов периодической
системы, разумеется, разрывна. Она имеет смысл только для целых чисел п.
Изображение на рис. 75 этой функции в виде непрерывного графика — прием,
применяемый в целях удобства ее качественного описания. То же самое
относится и к другим функциям порядкового числа п, рассматриваемым в этой главе.
263
ветствуют минимумы соответствующих первых ионизационных по-
тенциалов. В остальном прямую связь между функциями Vи
^усмотреть трудно — различия между ними велики. За исключением гелия
для атомов элементов подгруппы гелия на графике 75 никаких
особенностей функции V(n) нет, а потенциалы ионизации атомов этих
элементов всюду максимальны. Функция V(n) проходит через ряд
минимумов, о которых говорилось выше. А функция I^ri) в соответствую-
Периоды
О 5 10 15 20 25 30 35 40 45 50 55 60 65 Ю~ 75 80 85 90
Порядковый номер п
Рис. 76. Ионизационные потенциалы атомов
щих интервалах значений п отчетливо выраженных особенностей не
имеет. ^ „
Природа функции V(n) более сложна, чем у /х(я). Функция V(n)
зависит от распределения и состояния электронов и атомных ядер в
пространстве твердых и жидких фаз. Упомянутое распределение в свою
очередь связано со свободной энтальпией и другими
термодинамическими функциями этих макроскопических систем, а следовательно,
зависит от условий, в которых находится жидкость или твердое тело.
Количественные методы описания распределения электронов и
атомных ядер пока недостаточно хорошо развиты. Еще слабее изучена
взаимосвязь между термодинамическими функциями и распределением
электронов и атомных ядер в конденсированных средах. Но опыт
позволяет выявить ряд важных факторов, качественно характеризующих
особенности этих связей, Молярные объемы твердых и жидких фаз
зависят от числа электронов во внешних оболочках атомов, от концентра-
264
ции обобществленных «почти свободных» электронов и от
распределения между соседними ядрами, так называемых необобществленных
электронов, нередко образующих связи ковалентного типа. Рост
концентрации «почти свободных» электронов и образование ковалентных
связей обычно уменьшают молярный объем твердых и жидких фаз. При
этом устойчивость конденсированных, т. е. твердых и жидких, фаз
возрастает. Подробнее об этом будет сказано в § 106. Рост числа
электронов во внешних оболочках атомов обычно повышает молярный
объем твердой и жидкой фаз. Следует, заметить, что увеличение
числа электронов сопровождается ростом заряда атомных ядер. А
этот фактор противодействует увеличению объема, так как с
увеличением заряда атомных ядер электронное облако сильнее стягивается к ядру.
Устойчивость конденсированных фаз с увеличением молярного
объема при прочих близких условиях, как правило, падает.
Подчеркнем еще раз, что связь между V и распределением электронов и ядер
не прямая, а опосредованная. При заданных Р и Т молярный объем
есть функция свободной энтальпии, которая, в свою очередь, есть
сложная, пока еще плохо изученная функция распределения
электронов и атомных ядер, каким-то образом связанная со средней
электронной плотностью на границах соседних атомов в жидкости или твердом
теле. „
Рассмотрим теперь некоторые особенности графика V =f(ri).
Молярные объемы подавляющего большинства твердых и жидких
простых веществ лежат в интервале от 7 до 20 мл/моль. Молярные
объемы металлов и неметаллов мало различаются. Так, например, почти
одинаковы V водорода и лития, неона и магния, серы, скандия,
германия, ртути, протактиния. Молярный объем твердого фтора,
по-видимому, меньше, чем водорода, лития и азота. В большинстве случаев
для элементов одной подгруппы периодической системы молярный
объем растет с увеличением порядкового числа. Это правило соблюдается
почти всегда, за немногими исключениями. Так, молярный объем гелия
больше, чем неона и аргона; у азота он больше, чем у водорода.
Молярные объемы серебра и золота, циркония и гафния, технеция и
рения почти одинаковы. Учитывая упомянутую выше условность
принятых нами значений V, нет оснований утверждать, что правило здесь
нарушается. Отметим постепенное снижение молярного объема с
ростом порядкового номера у лантаноидов. В ряду лантаноидов эту
закономерность резко нарушают европий и иттербий, молярные объемы
которых существенно превышают молярные объемы соседних с ними
лантаноидов. В этом случае рост молярного объема, вероятно,
вызван уменьшением концентрации обобществленных электронов.
При сжатии простых веществ взаимодействие между атомами
усиливается и молярные объемы уменьшаются. Чем больше
молярный объем, тем выше сжимаемость. Эта закономерность
иллюстрируется на рис. 77, где по оси абсцисс отложены величины давления
Р в мегабарах (1 бар = 10б Па), а по оси ординат относительный объем
вещества V/Vo, где V и Vo — объемы вещества при давлениях Р и
1,013 • 10б Па соответственно.
10—503 265
Как и следовало ожидать, прямой корреляции между сжимаемостью
и энергией связи атомов в твердой или жидкой фазах нет. Это вытекает
из данных, приведенных на рис. 77, и других факторов (см., например,
[86]). Так, энергия связи между атомами вольфрама составляет около
880 кДж/моль, а между атомами иридия 502 кДж/моль, т. е. почти в
два раза меньше. Но молярный объем вольфрама больше, чем иридия,
и его сжимаемость выше. Родий имеет меньшую сжимаемость, чем
молибден, хотя энергия связи атомов у молибдена равна 670 кДж/моль,
v>
3,0
2,0
1,5
CS
4
A
\
r
/^
"■"
^*
**r~
S£^
• La
■—
г=ё
^L
Re
eO
Ir
12 3 4 5 6 7R>MBl
Рис. 77. Зависимость относительного объема V/Vo от
давления у цезия, бария, лантана, таллия,,
вольфрама, рения и иридия [86] (1Мбар = 1011 Па)
а у родия 460 кДж/моль. Таким образом, при повышении давления
вещества с большими молярными объемами сжимаются больше и
максимумы функции V =f(n) сглаживаются (рис. 78). Когда давление
достигает 1012 Па (107 бар), максимумы исчезают. Молярный объем
лития снижается до ~ 1 мл, калия — до 3 мл, цезия и свинца — до 5 мл.
Функция V =f(n) становится почти монотонной. Электронные
оболочки разрушаются. На смену им, по современным представлениям,
приходит статистическое распределение электронов в поле атомных ядер.
la
Р=ЮМбар
РЪ
Рис. 78. Зависимость молярных объемов простых веществ от порядкового номера
п соответствующих элементов [86] при высоких давлениях
266
§ 104. Строение простых жидкостей
и твердых фаз
Перейдем теперь к общему обзору строения жидких и твердых
простых веществ. Строение твердых простых веществ представляет
интерес для изучения структуры жидкостей по крайней мере в двух
отношениях. Во-первых, все твердые фазы в определенных
термодинамических условиях находятся в равновесии с жидкими.
Генетическая связь между структурами находящихся в термодинамическом
равновесии фаз интересна. Во-вторых, некоторые из флуктуации
структуры в жидкостях могут представлять собой фрагменты тех
аллотропных модификаций, которые в окрестности температуры плавления
почему-либо неустойчивы.
Рассмотрим сначала строение твердых и жидких фаз около
температуры плавления при давлении насыщенных паров. В табл. 31
штриховкой указаны структуры твердых простых веществ вблизи точки
плавления. Перед плавлением твердые фазы 50 элементов имеют ОЦК
структуру. Правда, в 10 случаях, отмеченных пунктиром, это
утверждение опирается не на прямые экспериментальные данные, а на
косвенные доказательства. Речь идет о прометии, эрбии, тулии, франции,
радии, актинии, протактинии, америции, кюри и берклии.
Предположение об ОЦК структуре их кристаллов в точке плавления основано на
сопоставлении со структурой кристаллов элементов-аналогов в
периодической системе (см. табл. 10). У 17 элементов твердые фазы в точке
плавления обладают ГЦК структурой. Здесь пока еще нет прямых
экспериментальных доказательств только для радона. У 8 элементов
соответствующие твердые фазы имеют ПГУ решетку.
Кристаллическим фазам 22 элементов присущи такие ковалентные и ковалентноме-
таллические структуры, для которых координационное число следует
правилу 8—N, где TV — номер группы периодической системы (см. гл.
VIII).
Многие элементы имеют ряд аллотропных модификаций, среди
которых одна (а у железа две) обладает ОЦК структурой. Речь идет о
равновесных модификациях при малых давлениях. Небезынтересно
следующее правило. При повышении температуры в условиях
термодинамического равновесия аллотропные превращения твердых фаз
упомянутых элементов завершаются модификацией с ОЦК
структурой. Иначе говоря, если есть аллотропная
модификация с ОЦК структурой, то плавится
именно эта модификация. Например, у лития есть
две аллотропные модификации — а-литий с ПГУ решеткой и fS-ли-
тий с ОЦК решеткой. В точке плавления устойчив Р-литий. У железа
в точке плавления устойчиво ?-железо, имеющее ОЦК структуру.
Из шести аллотропных форм плутония одна имеет ОЦК структуру.
Эта форма и является устойчивой в точке плавления. Пока что нам не
известно ни одного исключения из этого правила. Подчеркнем, что
речь идет об аллотропии при малых давлениях. Высокие давления
меняют структуру твердых фаз и указанное правило при высоких
давлениях теряет силу. Случаи, когда ОЦК модификации предшествуют
10* 267
СЗ
а*
g
плав.
3
«г*
и
ев
О.
О)
С
н
S
м
1
CQ
СО
Н
£Г
са
х
2
н
8
О.
Б
ДЫХ
о.
о
Си
н
^
Ои
щол
0)
&
1
pi t
я
н
я
я
я
1
я
1=3
03
fr-
я
я
О)
я
о
СП
о
я
1
1
я
ю
о
X
)укту
штри
я
о
U
W о и d 9 ц
при более низких температурах другие аллотропные формы, указаны
в табл. 10.
Итак, около 50% простых твердых веществ в точке плавления
характеризуются ОЦК структурой и 30% обладает плотно упакованными
расположениями атомов. Посмотрим теперь, что происходит в
результате плавления. Как уже говорилось, важнейшей характеристикой
структуры простых жидкостей является среднее координационное
число г. Экспериментальные данные о координационных числах
известны приблизительно для 40 простых жидкостей. Изучены все
жидкие неметаллы, за исключением астата и радона (инертные газы,
водород, азот, кислород, галогены, фосфор, сера, селен, теллур)
Атомы жидких инертных газов имеют среднее координационное число,
лежащее в интервале 8—9. (Здесь и далее мы пользуемся более
поздними результатами дифракционных методов. Ранние измерения в ряде
случаев приводили к завышенным значениям координационных чисел.)
Остальные неметаллы подчиняются правилу 8—N.
Исследовано 22 жидких металла. У 16 металлов вблизи точки
плавления z находится в интервале от 8 до 9 (металлы подгруппы лития,
алюминий, галлий, индий, таллий, железо, кадмий, ртуть, висмут,
сурьма, германий, олово). Надо полагать, что в этих простых
жидкостях относительно широко распространены фрагменты ОЦК
структуры. В пяти случаях (медь, серебро, золото, свинец, цинк) 2 = 11.
В этих жидких металлах, видимо, преобладают фрагменты плотно-
упакованных структур. Если твердая фаза имеет ОЦК структуру, то
после плавления координационное число, как правило, сохраняется
близким к 8 и нередко остается почти без изменений в большом
интервале температур, достигающем несколько сот градусов (щелочные
металлы, алюминий). Когда твердая фаза в точке плавления не имеет
ОЦК структуры, во многих случаях после плавления z ~ 8.
Следовательно, строение жидкостей и в этих случаях можно
охарактеризовать как ОЦК решетку, содержащую столь большое число дефектов,
что дальняя упорядоченность атомов отсутствует. Таковы жидкие
инертные газы, олово, алюминий, никель, висмут, германий, сурьма,
галлий, индий, кадмий, ртуть.
При плавлении металлов с ОЦК структурой энтропия возрастает
всегда на 5—10 кДж/К • моль. В тех случаях, когда твердая фаза
имеет ОЦК структуру и ее энтропия плавления лежит в указанном
интервале, можно с большой вероятностью полагать, что среднее
координационное число атомов в расплаве будет близко к 8.
В табл. 32 сопоставлены прямые и косвенные сведения о строении
простых жидкостей вблизи точки плавления при невысоких
давлениях. Сплошной штриховкой указаны результаты экспериментальных
исследований, пунктирной штриховкой — выводы, основанные на
данных об энтропии плавления, строении твердой фазы при
температуре плавления и строении простых жидкостей соответствующих
элементов-аналогов в периодической системе Менделеева. Жидкие
углерод, кремний и мышьяк, как уже отмечалось ранее, — металлы.
Плавление твердых фаз углерода, кремния и мышьяка, имеющих ко-
валентные решетки, сопровождается сравнительно большим увеличе-
269
in
HI
§fj
ill
m
ill
8S
Щ
Щ,
111
s*
I-
% ш
SG
III
1Ш
Ш
li
CO sf Ю
tf о и d э и
нием энтропии порядка 25—30 Дж/К • моль. Такого же порядка А5ПЛ
у их ближайших аналогов в периодической системе—германия и
сурьмы. Среднее координационное число атомов Ge и Sb в жидкой фазе
близко к 8. Можно предполагать, что жидкие углерод, кремний и
мышьяк имеют структуру, подобную строению жидких Ge и Sb, т. е. тоже
относятся к ОЦК типу.
Итак, около температуры плавления при малых давлениях
примерно 70 простых жидкостей имеют структуру ОЦК типа; 17 простых
жидкостей, видимо, имеют структуру плотных упаковок. Сюда же
включен жидкий бор, поскольку атомы в кристаллах бора расположены
так, что имеют в среднем 6 атомов ближайшего окружения и еще 6,
немного более удаленных. Энтропия плавления бора невелика,
следовательно, больших изменений в координации атомов ожидать нет
оснований. Вероятнее всего, что после плавления среднее
координационное число атомов бора близко к И. Ковалентные структуры,
следующие правилу 8—N, наблюдаются у 11 простых жидкостей.
С повышением температуры при малых давлениях среднее
координационное число атомов жидкости в конечном счете должно снижаться.
Но возможны случаи и роста координационного числа в некотором
интервале температур, как это наблюдается, например, у серы и индия.
Повышение давления до нескольких десятков гигапаскаль (сотни
килобар) в одних случаях ведет к росту координационного числа,
а в других к его уменьшению. Но при очень высоких давлениях
порядка 1012 Паскаль можно полагать, что реализуется плотнейшая
упаковка. Здесь различия между твердой и жидкой фазами исчезают.
При столь высоких давлениях, видимо, должна существовать
критическая точка, в которой жидкая и твердая фазы становятся
тождественными по своим свойствам. Эта критическая точка — своеобразный
аналог критической точки жидкость — пар, в которой становятся
одинаковыми свойства жидкой фазы и пара. Критическая точка на
кривой фазового равновесия жидкость — твердое тело может
возникать при очень высоких далениях в результате изменения структуры
электронных оболочек атомов. Можно полагать, что эта критическая
точка подобна критической точке равновесия а- и ^-модификаций
церия. Согласно имеющимся экспериментальным данным эта точка
находится при температуре 350—400° С и давлении порядка 2,0 ГПа
[87]. а — 7 - Превращение церия замечательно, в частности, тем,
что оно сопровождается скачкообразным изменением объема без
изменения ГЦК структуры. Интересно отметить, что экстраполяция
фазовой границы в этом случае приводит к широкому минимуму на
кривой плавления.
§ 105. Объемно-центрированная
кубическая структура
Как мы видели, у простых твердых веществ вблизи температуры
плавления и в еще большей степени у простых жидкостей ОЦК
структура наиболее распространена и характерна. Количественный анализ
этой закономерности — дело будущего. Для качественного ее понима-
271
ния надо подробнее описать особенности, присущие ОЦК структурам.
Допустим, что атомы шарообразны. Как бы плотно шары ни
прилегали друг к другу, между шарами всегда остается незанятое
пространство. Если распределение шаров известно, то можно подсчитать ту
долю пространства q, которая заполнена шарами, т. е. коэффициент
компактности структуры. При гранецентрированной кубической
решетке ГЦК элементарная ячейка структуры — куб, длину ребра
которого (параметр решетки) обозначим ах. На каждый такой куб
приходится 4 атома. Следовательно, на один атом приходится объем vx =
= а?/4. Из геометрии элементарной ячейки структуры ГЦК легко
вывести, что радиус шара Rx связан с параметром решетки ах уравне-
лНГ
нием R±=-— ал. Имеем
4
А 4 з
—~ <7i = "T я/Ч- (ХИЛ)
4 о
Отсюда следует, что </1== я/3 |/*2"= 0,74. Такой же коэффициент
компактности имеют и все другие плотнейшие структуры, в
частности гексагональная плотная структура ПГУ.
У объемно-центрированной кубической решетки ОЦК
элементарная ячейка структуры есть куб, длина ребра которого равна а2. На
каждый такой куб приходится 2 атома, следовательно, один атом занимает
о
объем v = 2 . Из геометрии элементарной ячейки ОЦК стру-
2 2
ктуры следует, что радиус шара R2 связан с параметром решетки
УТ
а2 уравнением R2=-— a2, поэтому
4 4 «
ТЯш=Т«$. (XII-2)
Выражая а2 через R2y получаем q2 =—^— = 0,68. Предположим те-
о
перь, что превращение гранецентрированной кубической структуры
в объемно-центрированную кубическую происходит без изменения
объема системы, т. е. атомный объем vx = v2 или af/4 = а|/2. Пользуясь
приведенными выше выражениями для Rx и R2, можно показать, что
r± = 21/3 V2/3 = 1 ,оз/?2. (XI 1.3)
Таким образом, если в ходе превращения гранецентрированной
кубической аллотропной формы в объемно-центрированную кубическую
молярный объем не меняется, то атомный радиус должен
уменьшаться наЗ%, несмотря на снижение плотности заполнения
пространства на ^ 6%. Если же ГЦК <=* ОЦК превращение происходит
без изменения атомного радиуса, т. е. Rx = R2, то ax = i
и vx=— l/ -£- y2 = 0,925tv (XII.4)
272
Такое ГЦК =^= ОЦК превращение должно, следовательно,
сопровождаться увеличением молярного объема на 7,5%. Те же самые
соотношения атомных радиусов и молярных объемов справедливы (если
атомы считать шарами) для гексагональной плотной упаковки типа
магния (АВАВ) и типа а-лантана (ABAC), плотной упаковки типа
самария (АВАВСАСВС) и любых других плотных упаковок.
Приведем еще аналогичные соотношения при превращении
аллотропной формы со структурой типа алмаза А4 в
объемно-центрированную кубическую структуру ОЦК. Элементарная ячейка структуры
типа алмаза — куб с ребром а4, на который приходится 8 атомов;
VT
атомный радиус /?4 = -— а4; коэффициент компактности q 4 = 0,34.
8
Таблица 33
Изменение объема при аллотропных превращениях с образованием
объемно-центрированной кубической структуры (атмосферное давление)
Элемент и
аллотропные превращения
Кальций а -> р
Марганец 7 -* &
Железо 7 -* а
» y -*• &
Лантан р -> 7
Церий y — &
Иттербий а -* р
Торий а -» р
Уран р -> y
Нептуний р -> 7
Плутоний 5' -> е
Литий а -> р
Натрий а -> Р
Бериллий а ^ р
Стронций Р -> 7
Титан а -> р
Цирконий а -* р
Гафний а -> р
Таллий а -> р
Самарий а -> р
Тип
структуры исходной
аллотропной
формы
ГЦК
~гцк
ПГУ
3,14
0,8
1,06
0,48
1,3
0,1
-1,2
—0,1
0,71
2,8
—2
—0,4
-0,4
—3,58
-2,3
—0,55
—0,66
—1,05
—0,15
—0,05
превр'
737
1410
1183
1673
1140
1000
1071
1675
1050
850
750
78
5
1527
862
1155*
1138
2260
508
992
Примечания
В таблице использованы значения
А V/V', приведенные в работе
Рудмана [88]. Греческие
символы а, р, 7 и т. д.
соответствуют принятым в литературе
наименованиям соответствующих
аллотропных форм
Уран, нептуний и плутоний
имеют структуры, которые
можно рассматривать, как структуры
ГЦК с ромбоэдрическим
искажением
Самарий имеет плотноупакованную
структуру, близкую к ПГУ
273
Таблица 34
Изменение объема и электропроводности при плавлении с образованием
простых жидкостей, имеющих структуру типа объемно-центрированной
кубической ОЦК
Элемент
Неон
Аргон
Криптон
Ксенон
Алюминий
Никель
Индий
Кадмий
Ртуть
Литий
Натрий
Калий
Рубидий
Цезий
Таллий
Европий
Иттербий
Уран
Железо
Галлий
Германий
Тип структуры
тв. фазы
гцк
^гцк
^ ПГУ
ОЦК
Орторомбиче-
ская
Алмазная
AV кг
VTB
11,5
11,7
11,5
11,5
6,0
4,2
2,7
4,7
3,7
1,65
2,5
2,552
2,5
2,6
3,2
5,0
5,5
8
3,06
—3,2
——-О, О
Гпл> К
25,55
83,75
116,45
161,25
932
1728
429,32
594
234,9
453,7
370,97
336,4
312,0
301,8
577
1090
1097
1406
1809
303
1210,5
Ж
*ТВ
0,45
0,95
0,46
0,51
0,26—0,2
0,61
0,69
0,64
0,63
0,605
0,485
0,92
2,2
19
*ж-ю-в,
Ом-^м-1
4,14
1,18
3,02
2,97
1,32
3,96
10,45
7,71
4,55
2,78
1,4
0,723
3,88
1,59
Примечания**
с/а= 1,077
с/а = 1,89
с/а = 1,94
274
Продолжение табл. 34
Элемент
Олово
Сурьма
Висмут
Тип структуры
тв. фазы
Ковалентно-
металличе-
ская
Ковалентно-
металлическая
V-™
ктв
2,8
—0,94
—3,35
г„л.К
505
903
544,5
а *
Ж
атв
0,48
1,64
2,86
•ж.иг«.
Ом-*м"1
2,08
0,883
0,782
Примечания**
— электропроводности в точке плавления соответственно жидкой и твердой
фаз.
♦* В примечаниях указаны отношения параметров решеток с/а в структурах твердых фаз типа
ПГУ и ГЦК. У ртути ромбическая решетка тождественна ПГУ решетке с с/а — 1,94.
Если аллотропное превращение А4 ч=* ОЦК происходит без
изменения молярного объема, то /?2 = 2V3/?4 = 1,26/?4. Атомный
радиус увеличивается на 26%. Если же атомный радиус не меняется,
то v2 = 1/2 t>4. Молярный объем уменьшается в два раза, потому что
ОЦК структура вдвое более компактна, чем структура типа алмаза.
Посмотрим теперь, что наблюдается в действительности при
аллотропных превращениях или плавлении с участием ОЦК структуры.
В табл. 33 указаны изменения объема аллотропных форм при их
превращении с образованием объемно-центрированной кубической
структуры. В табл. 34 приведены данные об изменении объема и
электропроводности при плавлении с образованием простых жидкостей,
имеющих структуру типа ОЦК. Для сравнения в табл. 35 фигурируют
данные о тех же характеристиках при плавлении с образованием
простых жидкостей, имеющих структуру плотных упаковок ГЦК или ПГУ.
Экспериментальные данные, имеющиеся в этих таблицах, приводят к
следующим заключениям.
1. При аллотропных превращениях в условиях атмосферного
давления образование ОЦК структуры наблюдается только у металлов.
Причем ОЦК структура получается из шютноупакованных
аллотропных форм типа ГЦК и ПГУ (или слабо отличающихся от них упаковок
в случае протактиния, урана, самария и нептуния). Если исходная
фаза имеет гексагональную плотную упаковку атомов, то ео Бсех
изученных случаях образование объемно-центрированной кубической
упаковки сопровождается небольшим уменьшением молярного
объема, в среднем равным около 1%. Наиболее велико оно у бериллия
(—3,58%). Если же исходная фаза обладает кристаллической решеткой
типа ГЦК, то при образовании фазы с ОЦК структурой в одних
случаях наблюдается небольшое сжатие, в других незначительное рас-
275
ширение. В среднем изменение объема составляет +0,6% Наиболее
велико оно у кальция—тоже элемента подгруппы бериллия (-[- 3,14%).
Но даже в этом случае увеличение объема много меньше 7,5%.
Как мы отмечали ранее (см. гл. III), влияние химических
процессов на строение кристалла наименее выражено в плотноупакован-
ных структурах ПГУ. Превращение ПГУ ^ ГЦК есть следствие
химических взаимодействий между атомами. В ОЦК структуре
влияние химических взаимодействий еще сильнее, чем в ГЦК. Ясно, что
аллотропные превращения вида ПГУ ^±= ОЦК должны
сопровождаться в среднем большими изменениями интенсивности химических
взаимодействий между атомами, чем аллотропные превращения вида
ГЦК =•* ОЦК.
Тот факт, что аллотропные превращения ПГУ ^±= ОЦК
сопровождаются уменьшением объема, причем во всех случаях
высокотемпературная фаза имеет ОЦК структуру, вероятно, не случаен.
Аллотропные превращения вида ГЦК =** ОЦК и ПГУ ** ОЦК
связаны с небольшими изменениями объема. В рамках принятой выше
модели шарообразных атомов это означает, что указанные
аллотропные превращения всегда сопровождаются уменьшением
атомных радиусов. Обычно атомные радиусы уменьшаются более чем на
3%, что приводит к снижению молярного объема простого вещества.
Мы пренебрегаем здесь неточностями расчетов, связанными с неучетом
отклонений атомов от сферической формы.
Из того факта, что ГЦК-^ ОЦК и ПГУ -> ОЦК превращения
сопровождаются уменьшением атомных радиусов, вытекает
интересное и важное следствие. Переход плотноупакованных аллотропных
Таблица 35
Изменение объема и электропроводности
при плавлении простых веществ структуры ГЦК и ПГУ
с образованием простых жидкостей,
имеющих структуру типа плотных упаковок
Элемент
Медь
Серебро
Кобальт
Золото
Свинец
Магний
Цинк
Тип
структуры
тв. фазы
ГЦК
ПГУ
Л V
v™ 102
4,5
3,3
3,2
5,1
3,6
3,05
4,2
пл»
1356
1234
1765
1336
600,6
923
692,7
атв
0,48
0,48
0,77
0,44
0,51
0,56
0,46
Ом"1 см"1
5,0—4,75
5,82
11,8
3,22
1,053
3,66
2,68
276
форм в формы, имеющие объемно-центрированную кубическую
структуру, нельзя рассматривать как переход от более плотных к более
рыхлой структуре. Широко распространенное приближение, в котором
атомы считаются сферами с постоянным радиусом, здесь неверно не
только в количественном, но даже в качественном отношении.
Уменьшение атомных радиусов при аллотропных превращениях вида ГЦК ^
ч=± ОЦК и ПГУ +* ОЦК имеет только один физический смысл. Оно
означает перераспределение электронной плотности в пространстве
между атомными ядрами. В сущности, каждый кристалл можно
рассматривать как гигантскую макромолекулу. Так как превращения
вида ГЦК =*=*= ОЦК и ПГУ ^ ОЦК сопровождаются
перераспределением электронной плотности, они в принципе имеют ту же физическую
природу, что и химические превращения обычных молекул. Различие
состоит в том, что фазовые превращения ГЦК ^ ОЦК и ПГУ =е±=ОЦК —
один из видов коллективных процессов. В этих превращениях
одновременно принимает участие очень большое число частиц. А обычные
химические процессы — неколлективные*. Они состоят из множества
элементарных актов, в которых принимают участие одновременно
лишь одна, две, редко три частицы. В упомянутых элементарных
актах действует закон простых кратных отношений. При коллективных
химических процессах этот закон неприменим.
2. При атмосферном давлении плавление простых веществ,
имеющих плотноупакованные структуры ПГУ, ГЦК или близкие к ним
(как у индия или ртути), во всех известных случаях сопровождается
увеличением объема независимо от строения расплава. Расплав может
иметь дефектную структуру типа ОЦК или типа ПГУ, ГЦК (см. табл.34).
У металлов величина AV/VTB в обоих случаях лежит в интервале
от ~ 3 до 6%. Для неметаллов аналогичное сопоставление
невозможно. При давлениях порядка 105 Па простые жидкие неметаллы со
структурой типа ОЦК и ПГУ в окрестности точки плавления не существуют.
Межатомные силы притяжения у инертных газов при атмосферном
давлении намного слабее, чем у металлов. Форма атомов инертных
газов близка к сферической. Здесь при характеристике упаковок ПГУ,
ГЦК и ОЦК модель жестких шаров вполне применима. Плавление
неона, аргона, криптона и ксенона сопровождается ростом объема на
одну и ту же величину (11,5%), причем жидкая фаза имеет структуру,
напоминающую ОЦК (г ^ 8), а твердая имеет гранецентрированную
кубическую упаковку. Увеличение объема при плавлении этих
веществ на 4% больше, чем при «аллотропном» превращении плотноупа-
кованного распределения жестких шаров одинакового размера в более
рыхлое распределение ОЦК (см. стр. 272). Избыточные 4% можно
отнести за счет возникновения в структуре ОЦК большого числа
дефектов, благодаря чему дальняя упорядоченность расположения
атомов инертных газов после плавления исчезает. Среднее координаци-
* Мы не рассматриваем здесь вопрос о глубокой связи между коллективными
и неколлективными процессами при химических реакциях, протекающих в
жидких фазах. В природе нет резкой грани между этими двумя типами процессов.
277
онное число жидких инертных газов вблизи точки плавления намного
выше 8 (скорее всего за счет примеси фрагментов плотноупакованных
структур), поэтому общий объем, занимаемый дефектами ОЦК
структуры в жидких инертных газах, видимо, составляет немногим более 4%
от всего объема жидкости.
Что касается металлов, аллотропные превращения вида ГЦК ^
^ ОЦК и ПГУ =*=* ОЦК, как мы видели, обычно происходят почти без
изменения объема. Превращения вида ГЦК^ (ОЦК)Ж и ПГУ =р* (ОЦК)Ж
при плавлении металлов сопровождаются ростом объема в среднем
на 4,3%. Каждый из процессов плавления, происходящий без
изменения типа структуры, т. е. ГЦК ** (ЩК)Ж, ПГУ *± (ПГУ)Ж и
ОЦК^МОЦЮж» ведет к увеличению объема в среднем на 4%. Эти
изменения объема надо в основном отнести за счет образования
вакансий. Как можно полагать, для разрушения дальнего порядка в плотно-
упакованных структурах при атмосферном давлении необходимо,
чтобы вакансии составляли около 4% от общего объема вещества.
В структурах типа ОЦК, где химическое взаимодействие выражено
сильнее, объем вакансий должен быть несколько выше (около 4—5%
от общего объема). Это справедливо для всех простых веществ —
металлов и неметаллов.
Концентрация электронов проводимости здесь не играет большой
роли. Отметим, что при процессах плавления твердых простых веществ,
имеющих структуры типа ПГУ, ГЦК и ОЦК, изменение
электропроводности мало зависит от структуры твердой фазы и получающегося
из нее расплава (ПГУ)Ж, (ГЦК)Ж или (ОЦК)Ж. В этих случаях у всех
изученных металлов отношение электропроводностей жидкой и
твердой фаз, аж/<зтв, лежит в интервале 0,45—0,95; исключение
составляет лишь ртуть, где <зж/атв ~ 0,2. Как указано в табл. 34, у твердой
ртути отношение параметров решетки типа ПГУ очень велико: с/а =
= 1,94. Это намного превышает отношение с/а = 1,633 решетки типа
ПГУ, состоящей из шарообразных атомов. Можно думать, что падение
электропроводности в перечисленных выше процессах плавления в
основном вызвано резким возрастанием концентрации дефектов.
Уменьшение объема при плавлении галлия, германия, сурьмы и
висмута можно объяснить перераспределением электронной плотности,
сопровождающимся ростом концентрации электронов проводимости и
увеличением координационного числа. Во всех этих случаях
электропроводность расплава выше электропроводности твердой фазы.
Белое олово имеет тетрагональную объемно-центрированную
структуру, менее плотную, чем ОЦК. При плавлении белого олова
уменьшение объема, связанное с вероятным образованием фрагментов
структуры ОЦК, видимо, маскируется ростом объема за счет увеличения
числа вакансий.
У церия и плутония плавление твердой фазы, имеющей ОЦК
структуру, происходит с уменьшением объема. Строение жидких церия
и плутония пока еще не изучено. Можно думать, что плавление здесь
сопровождается образованием плотноупакованной структуры (ГЦК)Ж
или (ПГУ)Ж.
278
§ 106. Плавление простых веществ
Температура плавления является одной из характеристик
термодинамического равновесия твердой и жидкой фаз. При температуре
плавления (если давление задано) свободные энтальпии твердой и жидкой
фаз одинаковы: ОТВ = ОЖ. Свободные энтальпии — сложные и пока еще
слабо изученные функции температуры, давления, межатомных
взаимодействий и строения фаз. Аналитическая форма функций GTB и G*
даже для фаз, соответствующих одному элементу, например аргону,
ртути и т. д., по имеющимся данным различна. Еще более
существенны различия функций GTB и пж фаз, соответствующих разным
элементам. Температура плавления, следовательно, является сложной
функцией строения и межатомных взаимодействий двух фаз — жидкой и
твердой. А межатомные взаимодействия и структура фаз в свою
очередь сложным образом зависят от строения и свойств атомов. Ясно,
что связь между температурой плавления и порядковым номером п
соответствующего элемента в периодической системе Менделеева не проста.
Тем не менее значение функции Тш = / (п) полезно для изучения
перечисленных выше взаимосвязей и отыскания, на первых порах, хотя
бы качественных их особенностей.
3000
2000
1000
ю
20
30
50
60
70
80
90
Рис. 79. Зависимость температуры плавления простых веществ от
порядкового номера п соответствующих элементов в периодической
системе Менделеева
279
Функция Тпл = / (п) при давлении насыщенных паров
представлена на рис. 79. Для элементов главных подгрупп периодической
системы связь Тпл с периодическим законом отчетлива. Все инертные газы
имеют малые температуры плавления. Во всех случаях
соответствующие им точки располагаются в минимумах графика Тпл =f(n).
Температура плавления инертных газов последовательно растет с
увеличением их атомного веса. Тоже наблюдается у галогенов — ближайших
соседей инертных газов в таблице Менделеева. Но в подгруппе
кислорода последний элемент (полоний) выпадает из упомянутой
закономерности. Он плавится при более низкой температуре, чем его
предшественник— теллур. В подгруппе азота максимум температуры
плавления передвигается еще на один элемент влево, в область меньших п.
Здесь температура плавления максимальна у мышьяка. А в подгруппе
углерода самой высокой температурой плавления отличается углерод.
Переходя к другим элементам этой подгруппы, мы обнаруживаем
минимум Гпл у олова. Последний член этой подгруппы — свинец —
плавится почти на 100 К выше, чем предпоследний — олово. Заметим,
что в подгруппе бора экстремум, как ив подгруппе азота, сдвигается на
один элемент влево и наблюдается у галлия. Но в подгруппе азота речь
шла о максимуме 7ПЛ, а здесь наблюдается минимум. В подгруппе
бериллия минимум соответствует магнию — второму элементу
подгруппы. Температура плавления кальция выше, чем у магния. В ряду
кальций, стронций, барий, радий температура плавления вновь
падает. Наконец, в подгруппе лития температура плавления
последовательно падает от лития к цезию.
Сопоставляя температуры плавления с данными о строении
плавящихся фаз, можно отметить следующие закономерности. ТПЛ
неметаллов одной подгруппы и однотипной структуры растут с
увеличением порядкового номера п. Это наблюдается у инертных газов,
галогенов, кислорода и его аналогов, азота и фосфора. В подгруппе
кислорода полоний — металл, в подгруппе азота — мышьяк, сурьма
и висмут тоже металлы, поэтому они не следуют указанному правилу.
У металлов главных подгрупп наблюдается противоположная
закономерность. Температуры плавления металлов одной подгруппы и
однотипной структуры уменьшаются с ростом порядкового номера
п соответствующего элемента. Так происходит в подгруппах лития,
бериллия, бора и углерода. В подгруппе бериллия все металлы имеют
в точке плавления ОЦК структуру, а магний — ПГУ структуру.
Его температура плавления выпадает из последовательности. В
группе бора структура в точке плавления почти одинакова у алюминия и
индия. Остальные члены подгруппы имеют другую структуру и не
следуют указанной закономерности. В подгруппе углерода сходную
структуру в точке плавления имеют все члены подгруппы, за исключением
свинца, который отклоняется от упомянутой закономерности. В
подгруппе азота металлы — мышьяк, сурьма и висмут — обладают
однотипной структурой. Температура их плавления снижается от мышьяка
к висмуту.
Спрашивается, в чем причина столь различного поведения
неметаллов и металлов главных подгрупп периодической системы? Можно
280
предложить следующее качественное истолкование. Если структура
твердой фазы однотипна и элемент принадлежит одной подгруппе, то
температура плавления определяется энергией связи между частицами
твердой фазы: атомами у металлов и инертных газов, молекулами у
неметаллов. В рамках указанных ограничений температура плавления
тем выше, чем больше энергия связи. Мерой энергии связи является
изменение энтальпии АЯ, необходимое для превращения твердой
фазы при постоянных Т и Р в газ, состоящий из тех же частиц, т. е.
атомов или молекул, что и твердая фаза. Эта мера энергии неточна,
потому что упомянутые частицы не всегда однозначно определяются.
Кроме того, энтальпии фаз вблизи плавления во многих случаях
неизвестны. Поэтому мы пользовались стандартными энтальпиями
образования твердых фаз АНт . Влияние аллотропных превращений тем
самым не учитывалось. Но, как показывает опыт, эти неточности не
очень существенны. Качественно, закономерность одна и та же для всех
.подгрупп периодической системы: главных и побочных. Величины
стандартных энтальпий образования приведены в справочнике М. X. Ка-
рапетьянца и М. Л. Карапетьянц [23].
Когда энергия связи падает с ростом порядкового номера элемента
в подгруппе, то температура плавления фаз, имеющих однотипную
структуру, уменьшается. И, наоборот, если энергия связи возрастает,
температура плавления однотипных фаз увеличивается. С этих
позиций, например, в подгруппе кислорода температуру плавления
полония и теллура сопоставлять не имеет смысла, так как их структура
резко различается. Мы смогли обнаружить, в сущности, лишь два
отклонения от указанной связи между Тпл и АНт- Температура
плавления бария на 60 К ниже температуры плавления стронция, а
стандартная энтальпия бария AHhs на 10,1 кДж больше, чем стронция. Но
у стронция в отличие от бария при 862 К происходит аллотропное
превращение с уменьшением объема на 2,3%. Это означает, что вблизи
температуры плавления энтальпия образования твердой фазы и энергия
связи стронция могут быть выше, чем у бария. То же самое наблюдается
для лантана и актиния. Лантан плавится при более высокой
температуре, чем актиний, хотя стандартная энтальпия лантана на 26,7 кДж/моль
ниже, чем у актиния. У лантана подобно стронцию при 595°С
происходит аллотропное превращение с уменьшением объема на 0,5%.
Таким образом, отклонение бария и лантана от упомянутой
закономерности, по-видимому, кажущееся.
Энтальпия образования лантана и редкоземельных металлов
определена не очень точно. Расхождения между величинами AHhs ,
найденными разными исследователями, обычно составляют 40 кДж
и более. Резкое снижение температур плавления у европия и
иттербия по сравнению с температурами плавления соседних
редкоземельных элементов в соответствии с указанным правилом сопровождается
столь же резким уменьшением энтальпии образования твердых фаз
(см. табл. 14). Подчеркнем, что правило, связывающее АН и Гпл,
достаточно строго действует в рамках подгрупп периодической системы.
Если простые вещества обладают структурой одного типа, ко
принадлежат разным подгруппам, правило может нарушаться. Например,
281
таллий имеет более высокую энтальпию образования, чем стронций,
но в два раза более низкую температуру плавления. Аллотропные
превращения здесь не меняют картины. Энтальпия образования железа
выше, чем хрома, но температура плавления на 370 К ниже. Железо,
хром, таллий и стронций в точке плавления имеют
объемно-центрированную кубическую структуру. Металлы подгруппы железа и платины
следует располагать в триады Fe, Ru, Os; затем Со, Rh, Ir и Ni, Pd, Pt.
В каждой из триад правило действует. Для разных триад оно может
нарушаться (например, кобальт и платина).
Энтальпия плавления ДНПЛ. В рамках подгрупп для простых
веществ с однотипной структурой А#пл почти всегда меняется в той
АНпл,кДж-моль~1
50г
С(125)
35
30
25
20
15
10
0 10 20 30- 40 50 60 70 80 90 п
Рис. 80. Зависимость *молярной энтальпии плавления простых веществ
от порядкового номера п соответствующих элементов в периодической
системе Менделеева
282
же последовательности, что и Тпл. Небольшие отклонения от правила
наблюдаются у лантаноидов и у палладия в триаде Ni, Pd, и Pt.
Они могут появиться в некоторых случаях за счет ошибок при
измерениях энтальпий плавления. Расхождения между величинами АЯПЛ,
полученными в разных лабораториях, нередко превышают 4 кДж/моль,
в особенности, если вещества тугоплавки. Энтальпия плавления,
как и следовало ожидать, существенно зависит от энергии связи
между частицами. Что касается разных подгрупп, то здесь наблюдаются
отклонения от указанной зависимости между Д#пл и Тпл. Функция
Д//пл = f(n) приведена на рис. 80.
Энтропия плавления ASnJI. Связь между^ ASnJI при абсолютном
давлении и периодическим законом Менделеева обсуждалась
неоднократно. Наиболее подробно она рассмотрена в работах В. М. Глазова [89] и
В. К. Григоровича [8]. На рис. 81 по оси абсцисс отложены порядковые
числа элементов, а по оси ординат—энтропии плавления в Дж/К-моль
независимо от атомного или молекулярного строения соответ-
/•к/
ствующих простых веществ (иначе величины ASnjI были бы
несопоставимы). Энтропия плавления, как и другие термодинамические
функции, описывающие равновесие двух фаз, зависит от строения обеих
фаз — жидкой и твердой. При плавлении строение вещества меняется
неизбежно. Здесь можно выделить два крайних случая.
1. Строение почти не меняется. Среднее координационное число
атомов в жидкой фазе остается почти тем же. Плавление
сопровождается лишь ростом числа вакантных мест в решетке, концентрация
которых становится достаточно большой, чтобы дальни^порядок
расположения атомов или молекул исчез. Ближе всего этой картине
соответствует, пожалуй, плавление металлов подгруппы лития.
2. Строение претерпевает коренные изменения. Меняются характер
и число химических связей между атомами, резко возрастает или
уменьшается концентрация электронов проводимости и т. д. Таковы
процессы плавления углерода (алмаза), кремния и германия.
В первом случае число конфигураций, в которых может
находиться система, при плавлении увеличивается ставнительно мало.
Соответственно и AS™ должна быть невелика. Во втором случае
плавление сопровождается резким возрастанием числа возможных
микросостояний системы. Энтропия плавления, очевидно, должна быть
существенно больше, чем в первом случае.
«*»
Обратимся теперь к графику функции Д5ПЛ = /(я)- Для
подавляющего большинства простых веществ величины ASnJl располагаются
в интервале от 5,8 до 11,3 Дж/К • моль, т. е. от /?1п2 до #1п4. За
немногими исключениями эти вещества — металлы. Энтропия плавления
изотопов гелия, как уже говорилось в главе о квантовых жидкостях,
близка к нулю, и в некотором интервале температур даже
отрицательна. Напомним, что изотопы гелия затвердевают только при давлениях
порядка 30 • 105 Па.
283
О)
ч
<D
X
К
2
О
■ S
n
В ряде случаев наблюдаются
малые величины ASnJI. Как
правило, это неметаллы: водород,
азот, кислород, фтор, фосфор,
сера. Сюда относятся и металл
церий. Для неметаллов вопрос
решается просто. Основными
структурными единицами у
водорода, азота, кислорода и
фтора при температуре
плавления являются молекулы-димеры,
а у фосфора — тетрамеры.
Плавление не меняет этой особенности
структуры. Димеры и тетрамеры
здесь играют примерно ту роль,
которая присуща атомам в
металлах. Если рассчитать
энтропию плавления на 1 моль
упомянутых молекул, то ASn;l во-
- ^ дорода, азота, кислорода, фто-
о S ра и фосфора будет составлять
около 8,4 Дж/К • моль, т. е. при-
^ мет значение, характерное для
S большинства простых жидкостей.
Относительно низкая величина
энтропии плавления в расчете на
1 моль означает, что состояние
движения атомов при плавлении
меняется слабее, чем у
большинства других простых
веществ. Это вполне понятно. Ведь
атомы здесь входят в состав
молекул, сохраняющихся и после
плавления.
С этих позиций можно
качественно объяснить и малую
величину энтропии плавления
серы. Сера состоит из
циклических молекул S8. Если бы
молекулы S8 были вполне прочными и
жесткими, т. е. не разрушались
и не испытывали конформацион-
ных превращений, то ASm в
расчете на 1 моль серы S8, вероятно,
была бы приблизительно в 2 раза
меньше, чем ASnJI фосфора (Р4).
Однако при плавлении серы
некоторые из кольцеобразных моле-
cd
CQ
О
«
ОцО)
о ч
в <и
Iй
<и
М
xj
а*
«5
S О
X S
ч
В
Я
к
в
о
I"
О)
о
к
I
I
§
к
о
и
со
о
а,
284
кул разрушаются с образованием цепочек, происходят конформаци-
онные превращения молекул, некоторая доля энергии тратится на
разрыв слабых химических связей между молекулами в твердой фазе. В
итоге энтропия плавления серы в расчете на 1 моль молекул довольно
велика. Но при расчете на 1 моль одноатомной серы она меньше
«средней нормы», поскольку движение атомов серы в расплавах и твердой
фазе различается не очень сильно.
У церия плавление сопровождается переходом к более плотной
упаковке атомов и увеличением плотности на 2,5%. Плавление углерода,
кремния, галлия, германия, мышьяка, сурьмы, теллура, висмута
связано с большими изменениями их строения и свойств. Описание
этих изменений имеется в гл. X. С ними связаны высокие значения
А5ПЛ. У галогенов плавление сопровождается разрушением ряда
слабых химических связей между димерами. Эти связи ослабевают с
уменьшением порядкового номера галогена, поэтому энтропия
плавления фтора в расчете на 1 моль F2 составляет всего 9,55 Дж/К • моль,
а у других галогенов увеличивается с ростом порядкового номера п.
Относительно высокие значения энтропии плавления инертных газов
обусловлены резкой перестройкой их структуры от ГЦК в твердой фазе
к структуре, напоминающей ОЦК, в жидкой фазе, а также снижением
плотности упаковки атомов. Аналогичны причины повышенных
энтропии плавления алюминия и ртути.
Небольшие колебания А5ПЛ у лантаноидов и некоторых других
тугоплавких металлов могут в ряде случаев быть вызваны малой
точностью измерений теплот плавления при высоких температурах. В
итоге мы видим, что связь между энтропией плавления и положением
соответствующего элемента в периодической системе хотя и существует,
но не в ярко выраженной форме. Это и естественно, поскольку речь
идет о величине, зависящей в основном от различий в структурах двух
сосуществующих фаз — жидкой и твердой. Связь между структурой
фаз и положением соответствующего элемента в периодической системе
не проста и не однозначна. Эта зависимость еще менее однозначна для
А5ПЛ.
Кривые плавления. Эти кривые представляют собой часть более
общих Р—Т диаграмм фазовых равновесий. Содержательный и
интересный обзор этих диаграмм выполнен В. В. Евдокимовой [87]. Ею
показано, что фазовые Р—Т диаграммы элементов одной и той же
группы похожи, причем по мере роста порядкового номера элемента, как
правило, наблюдается их закономерное изменение. Те типы фазовых
превращений, которые у первых членов подгруппы происходят в
области высоких давлений, у следующих членов той же подгруппы
наблюдаются при меньших давлениях. Диаграммы как бы
«стягиваются» в область меньших давлений.
§ 107. Кипение простых жидкостей
Температуры кипения. Ткш при атмосферном давлеции зависят от
энергии связи примерно так же, как и температуры плавления. В рам-
285
кнл>
7500
5000
2500
Рис. 82. Зависимость нормальной температуры кипения
простых веществ от порядкового номера п соответствующих
элементов в периодической системе Менделеева
10
20
30
50
60
70
■Rn,
80
90
3000
2000
1000
О
80
90
won
Рис. 83. Зависимость температурного интервала устойчивости простых
веществ при 105 Па от порядкового номера п соответствующих элементов
в периодической системе Менделеева
ках подгрупп периодической системы последовательность температур
кипения обычно совпадает с последовательностью значений
стандартных энтальпий образования АЯгэв . Это правило не выполняется в
подгруппе скандия и у лантаноидов.
На рис. 82 изображена функция Ткш =zf(n). Температуры
кипения тугоплавких металлов известны с точностью, зачастую не
превышающей ± 100 К-
Разность нормальных температур кипения и плавления. Гкип —
— Тпл = АГЖ характеризует температурный интервал, в котором
жидкая фаза устойчива при давлении порядка одной атмосферы. АГЖ
зависит от строения и свойств трех фаз: жидкой и твердой фаз в точке
плавления и пара в точке кипения.
Функция АТЖ = f(n) представлена на рис. 83. Общая тенденция
такова: большие температурные интервалы устойчивости жидкой
фазы наблюдаются у тех простых веществ, где энергия межатомных
связей велика. Однако здесь наблюдается немало отклонений даже в
подгруппе лантаноидов. Факторы, управляющие термодинамической
устойчивостью жидкой фазы, пока еще слабо изучены.
Литература
1. А. И. Губанов. Квантово-электронная теория аморфных
полупроводников. М., АН СССР, 1963.
2. У. X а р р и с о н. Теория твердого тела. М., «Мир», 1972; У. X а р р и-
с о н. Псевдопотенциалы в теории металлов. М., «Мир», 1966.
3. Д. 3 а й м а н. Принципы теории твердого тела. М., «Мир», 1966.
4. Г. Джонс. Теория зон Бриллюэна и электронные состояния в
кристаллах. М., «Мир», 1968.
5. В. X е й н е, М. К о э н, Д. У э й р. Теория псевдопотенциала. М.,
«Мир», 1973; Н. П. К р а с н ы й, Н. П. Коваленко. «Метод
псевдопотенциала в теории равновесных и кинетических свойств жидких металлов» в сб.
«Физика жидкого состояния», Киев, «Вища школа», 1973, стр. 17.
6. Н. М о т т. Электроны в неупорядоченных структурах. М., «Мир», 1969.
7. А. Р. Регель. Укр. физ. журн., 7, 833, 1962.
8. В. К- Григорович. Периодический закон Менделеева и электронное
строение металлов. М., «Наука», 1966.
9. Ч. Коулсон. Валентность. М., «Мир», 1965.
10. Д. X а р т р и. Расчеты атомных структур. М., ИЛ, 1960.
11. Физика простых жидкостей. Статистическая теория. М., «Мир», 1971.
12. В. Юм-Розери. Введение в физическое металловедение. М.,
«Металлургия», 1965.
13. М. К. Смит. Основы физики металлов. М., Металлургиздат, 1959.
14. Устойчивость фаз в металлах и сплавах. Под ред. П. С. Рудмана,
Дж. Стрингера, Р.И. Иоффе. М., «Мир», 1970.
15. Г. Р е м и. Курс неорганической химии. Т. I. M., ИЛ, 1963.
16. В. И. В е д е н е е в, Л. В. Г у р в и ч, В. Н. Кондратьев,
В. А. Медведев, Е. Л. Франкевич. Энергия разрыва химических
связей. Потенциал ионизации и сродство к электрону. Справочник. М., Изд-во
АН СССР, 1962.
•— 17. В. А. Алексеев, А. А. Андреев, В. Я. Прохоренко.
Электрические свойства жидких металлов и полупроводников. УФН, 106, 393,
1972.
18. В. В. Михайлов. Давление пара металлов. Итоги науки и техники.
Сер. «Термодинамика и равновесия». Научн. редактор М. X. Карапетянц. М.,
1972, т. 2, стр. 366.
287
19. И. В. Р а д ч е н к о. Строение жидких металлов. УФН, 41, 249, 1957.
20. Б. И. Хруще в. Структура жидких металлов. Изд-во ФАН
Узб.ССР, Ташкент, 1970.
21. Д. К. Б е л а щ е н к о. Явления переноса в жидких металлах и
полупроводниках, М., Атомиздат, 1970.
22. J. L. Margrave. High Temperaatures — High Pressures, 2,583,
1970.
23. M. X. Карапетянц, М. Л. Карапетянц. Основные
термодинамические константы неорганических и органических веществ. М., «Химия»,
1968.
24. G. С. Kennedy, A. J а у а г a m a n, R. С. Newton. Phys.
Rev., 126, 1363, 1962.
25. В. М. Г о р б а ч е в, Ю. С. 3 а м я т и н, А. А. Лбов. Основные
характеристики изотопов тяжелых элементов. Справочник. М., «Атомиздат», 1970.
26. Б. Ф. Мясоедов; Л. И. Г у с е в а, И. А. Л е б е д е в, М. С.
Милюкова, М. К. Чмутова. Аналитическая химия трансплутониевых
элементов. М., «Наука», 1972.
27. А. У б б е л о д е. Плавление и кристаллическая структура. М., «Мир»,
1969.
28. N. F. Mott. Phyl. Mag., 26, 505, 1972.
29. Н. Ocken, С. N. J. W a g n e r. Phys. Rev., 149, 122, 1966.
30. N. С. Н о 1 d e г, С. N. J. W a g n e r. J. Chem. Phys., 45, 482, 1966.
31. О. В. Р о м а н о в а, Б. А. Мельник. Укр. физ. журн., 17, 79,
1972.
32. Н. С. Ф а т е е в а, Л. Ф. Верещагин. Письма в ЖЭТФ, 13, 157,
1971.
33. В. М. Г л а з о в, С. Н. Ч и ж е в с к а я, Н. Н. Г л а г о л е в а.
Жидкие полупроводники. М., «Наука», 1967.
34. S. P. I s h е г w о о d, В. R. О г t о n, R. М а п a i I a. J. Non-Cryst
Solids ., № 8—10, 691, 1972.
35. Я. И. Д у т ч а к. Укр. физ. журн., 5, 94, 1960.
36. Р. Э в а н с. Введение в кристаллохимию. М., Госхимиздат, 1948, стр. 95.
37. Н. Е. Г и н г р и ч. Успехи химии, 15, 297, 1946.
38. J. Waseda, К. Suzuke. Phys. Status solid. (В) 47, 581, 1971.
39. R. E. Leckenby and E. J. Robbins. Proc. Roy. Soc,
London, A.291, 389, 1966.
40. Л. И. Некрасов. Исследование в области синтеза
физико-химических свойств и структуры замороженных водородно-кислородных систем (перекис-
но-радиальных конденсатов). Дис. МГУ, 1972, стр. 210, 293.
41. С. W. Т о m p s о п, N. S. G i n g r i с h. J. Chem. Phys., 31, 1598,1959.
42. С. Г л е с с т о н, К. Л е й д л е р, Г. Э й р и н г. Теория абсолютных
скоростей реакций. М., ИЛ, 1948.
43. А. А. Андреев, М. Мамадалиев, А. Р. Регель. Физ. и
техн. полупроводников, 5, 2187, 1971; А. А. Андреев, В. А. Алексеев,
Э. А. Л е б е д е в, М. Мамадалиев, Б, Т. М е л е х, А. Р. Регель,
Ю. Ф. Рыжков. Физ. и техн. полупроводников. 6, 661, 1972.
44. G. С. Vezzol i. J. Am. Ceram. Soc, 55, 65, 1972.
45. H. G о b r e d e t, F. Mahagjuri. J. Phys. Soc. Solid, state.
Phys., 5, 366, 1972.
46. G. T о u r a n d, В. С a b a n e, M. В r e u i 1 e. J. Non. Cryst. Solids,
№ 8—10, 676, 1972.
47. А. А. Соколов. Введение в квантовую электродинамику. М., Физ-
матгиз, 1958, стр. 32.
48. А. А. Соколов, И. М. Тернов. Квантовая механика и атомная
физика. М., «Просвещение», 1970.
49. Б. Н. Е с е л ь с о н, Ю. П. Б л а г о й, В. Н. Г р и г о р ь е в, В. Г.
М а н ж е л и й, С. А. М и х а й л е н к о, Н. П. Неклюдов. Свойства
жидкого и твердого водорода. М., Изд-во стандартов, 1969.
50. К. Хуан г. Статистическая механика. М., «Мир», 1966.
51. С. Г л е с с т о н. Успехи общей химии. М.-Л., Госхимиздат, 1941, стр.
84.
288
52. Д. У. К э д и, Л. Л. Б а р д ж е р. Физические свойства фтора. В сб
«Фтор и его соединения». Т. I. M., ИЛ, 1953, стр. 267.
53. С. A. A b b i s s, С. M. Knobler, R.K. T e a g u e, C. J. P i n g s
J. Chem. Phys., 42, 4145, 1965.
54. G. С a g 1 i о t i and F. P. Ricci. Nuovo Cimento, 24, 103, 1962.
55. В. Г. Ф а с т о в с к и й , А. Е. Р о в и н с к и й, Ю. В.
Петровский. Инертные газы, 2-е изд. М., Атомиздат, 1972.
56. В. Кеезом. Гелий. М., ИЛ, 1949.
57. М. Мендельсон. Жидкий гелий. В сб. «Физика низких
температур». М., ИЛ., 1959, стр. 783.
58. В. П. Пешков. УФН, 94, 607, 1968.
59. В. П. П е ш к о в, К. Н. Зиновьев. УФН, 67, 193, 1959.
60. М. И. Шахпаронов. Введение в молекулярную теорию
растворов. М., Гостехиздат, 1956.
61. D. G. Hurst, D. G. Hens haw. J. Chem., Phys., 100, 994, 1955.
62. E. K. Achter, L. M e у е r. Phys. Rev., 188, 291, 1969.
63. Я. Ю. A x а д о в. Диэлектрические свойства чистых жидкостей. М.,
Изд-во стандартов, 1972.
64. Д. Слэтер. Диэлектрики, полупроводники, металлы. М., «Мир»,
1969.
65. A. G. Gibbs, О. К. Н а г 1 i n g. Phys. Rev., A3, 1713, 1971.
66. Л. Д. Ландау. ЖЭТФ, 11, 592, 1941; J. Phys (USSR) 11, 91, 1946.
67. Н. Н. Боголюбов. Изв. АН СССР. Сер. физ., И, № 1, 77, 1947;
Вестн. МГУ, № 7, 43, 1947; Избранные труды в трех томах. Киев, «Наукова
думка», 1970, т. II.
68. Л. Д. Ландау и Е. М. Л и ф ш и ц. Механика сплошных сред. М.,
Гостехиздат, 1953.
69. И. М. Халатников. УФН, 59, 673, 1956; 60, 69, 1956.
70. Ju. D. A n u f r i e v, Т. A. Alvesado, Н. К. С о 1 1 а п, N. Т.
О р h e i m, P. Wennerstrom. Phys. Lett., 43-A, 175, 1973; T. A. Alvesado,
J u. D. A n u f r i e v, H. К. С о 1 1 a n, O. L. L о u n a s m a a, P.
Wennerstrom. Phys. Rev. Lett., 30, 962, 1973.
71. O. P e n г о s e, L. О n s a g e r. Phys. Rev., 104, 576, 1956.
72. О. К. Harling. Phys. Rev., A3, 1073, 1971.
73. Э. Л. Андроникашвили. Квантовая когерентность и
проблема сверхтекучести. М., «Природа», 1973, № 1, стр. 9.
74. R. A. Cou ley, A. D. В. W о о d s. Can. J. Phys., 49, 177, 1971.
75. В. П. Пешков. ЖЭТФ, 16, 1000, 1946.
76. Э. Л. Андроникашвили. ЖЭТФ, 16, 78, 1946; 10, 201, 1946.
77. В. Л. Гинзбург. Сверхтекучесть и сверхпроводимость во
вселенной. УФН, 97, 601, 1969.
78. Д. А. К и р ж н и ц. Экстремальные состояния вещества
(сверхвысокие давления и температуры). УФН, 104, 489, 1971.
79. Ф. Д а й с о н, Д. Т е р - X а а р. Нейтронные звезды и пульсары.
М., «Мир», 1973.
80. R. В. Н а 1 1 о с k. J. Low. Temp. Phys., 9, 109, 1972.
81. Е. С. Рудаков. Термодинамика межмолекулярного взаимодействия.
Новосибирск. «Наука», Сибирское отделение, 1968.
82. Л. Д. Л а н д а у. ЖЭТФ, 30, 1058, 1956; 32, 59, 1957.
83. А. А. Абрикосов, Л. П. Г о р ь к о в, И. Е. Дзялошинс-
к и й. Методы квантовой теории поля в статистической физике. М., Физматгиз,
1962.
84. Д. П а й н с, Ф. Н о з ь е р. Теория квантовых жидкостей. М., «Мир»,
1967.
85. В. Р. Абель, А. К- Андерсон и Д ж. К- У и т л и. УФН, 91,
311, 1967.
86. Л. В. А л ь т ш у л е р и А. А. Б а к а н о в а. УФН, 96, 193, 1968.
87. В. В. Евдокимова. УФН, 88, 93, 1966.
88. P. S. Rudma n. Trans. Metallurg. Soc. A. I. ME, 233, 864, 1965.
89. В. М. Глазов. ЖФХ, 66, 606, 1972.
289
Общая литература
1. А. А. С о к о л о в, И. М. Тернов. Квантовая механика и атомная
физика. М., «Просвещение», 1970.
2. В. М. Татевский. Классическая теория строения молекул и
квантовая механика. М., «Химия», 1973.
3. Г. Фрелих. «Теория диэлектриков», М., ИЛ, 1960.
4. Ю. Я. Ф и а л к о в, А. Н. Ж и т о м и р с к и й, Ю. А. Т а р а с е н-
к о. Физическая химия неводных растворов. Л., «Химия», 1973.
5. А. В. Скрышевский. Структурный анализ жидкостей. М.,
«Высшая школа», 1971.
6. М. И. Шахпаронов. Методы исследования теплового движения
молекул и строения жидкостей. М., Изд-во МГУ, 1963.
7. В. К. Григорович. Периодический закон Менделеева и
электронное строение металлов. М., «Наука», 1966.
8. Н. М о т т . Электроны в неупорядоченных структурах. М., «Мир», 1969.
9. В. П. Пешков. Свойства 3Не и его растворов в 4Не. Усп. физ. наук,
94, 607, 1968.
10. Б. И. Хруще в. Структура жидких металлов. Ташкент, изд-во «ФАН»
Узбекской ССР, 1970.
11. В. М. Г л а з о в, С. Н. Ч и ж е в с к а я, Н. Н. Глаголева.
Жидкие полупроводники. М., «Наука», 1967.
12. Сб. «Физика простых жидкостей». Т. I. Статическая теория. Под ред.
Г. Темперли, Дж. Роулинсона, Дж. Рашбрука. М., «Мир», 1966; т. II
Экспериментальные исследования. М., «Мир», 1973.
13. О. Я. Самойлов. Структура водных растворов электролитов и
гидратация ионов. М., Изд-во АН СССР, 1957.
14. К. П. М и щ е н к о, Г. М. Полторацкий* Вопросы
термодинамики и строения водных и неводных растворов электролитов. Л., «Химия», 1968.
15. В. П. Скрипов. Метастабильная жидкость. М., «Наука», 1972.
16. И. Б. Рабинович. Влияние изотопии на физико-химические
свойства жидкостей. М., «Наука», 1968.
17. Д ж. Пименте л, О. Мак-Клеллан. Водородная связь.
М., «Мир», 1964.
18. И. Р. К р и ч е в с к и й. Фазовые равновесия в растворах при
высоких давлениях. М.—Л., Госхимиздат, 1952.
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Адиабатическое приближение 11
Адиабатические флуктуации 139,
140
Азот 204
Актиноиды 187—189
Акустическая спектроскопия 5, 112
Алмаза структура 2/3
Аллотропные превращения 275—
278
Амиды 66
Амины 66
Анизотропные флуктуации
127, 144—151
Анилин 67
Аргон 8, 10, 30, 77—80, 117 — 119,
124, 224, 225
Аррениуса теория 5
Ассоциаты, определение 60
Ацетилен 69
Атом — атом потенциалы 98
Галогениды щелочных металлов 88,
89
Галогенов ионы 90
Гекеаи 8
Гелий 6, 30, 77—79, 224—259
Гексагональная упаковка атомов,
плотнейшая (ПГУ) 79
Германий 202
Гранецентрированная кубическая
упаковка атомов,
плотнейшая (ГЦК) 79
Григоровича гипотеза 175, 176
Д
Дейтерий 67, 68, 218—221
Дифракционные методы 5, 91, 108,
109, ПО, 115, 116, 117
Диэлектрики 6, 37, 162, 260
Диэлектрическая
радиоспектроскопия 108—112
Бензол 35, 36, 38, 76, 103—105
Бериллия подгруппа 181, 182
Близкодействующие силы 10, 49 —
54
Бор 198—200
Бром 49, 223, 224
В
Ванадия подгруппа 192
Вандерваальсово взаимодействие
19—32, 36, 97
Вириал 50
Вириальное уравнение состояния 93
Висмут 208
Внешнее поле 39
Внутреннее поле 37, 38
среднее 40—42
Внутренняя энергия 125
Вода 10, 30, 31, 56, 57, 61—64,
66-69, 102
Водород 6, 15, 16, 35, 54, 216—221
Водородная связь 6, 56—76
— — внутримолекулярная 58
Возбужденные молекулы 8, 34, 35
Волновая функция 11, 15, 25, 34,
237
Ж
Железа и платины подгруппа 193,
194
Жидкости
— простые 6
— квантовые 6
Закон
— Генри 5
— Вант-Гоффа 5
— Рауля 5
Золото 119, 120
Зона
— Бриллюэна 166
— энергетическая 164
И
Иод 81, 224
Иода монохлорид, структура 32
Ионы полиатомные 90, 91
Калий 49, 178, 180
Кислород 208, 209
291
Кислоты карбоновые 61, 69
Колебания
— нормальные 64, 65
— характеристические 65
Коллективные процессы 227
Комплексы, определение 60
Компонента
— Гросса 142, 143
— Леонтовича—Фабелинского 148,
149
— Мандельштама—Бриллюэна 141,
143
Координационное числ 119
Координаты
— естественные 64
— нормальные 64, 75
Корреляционная функция
парная 115, 133, 134
— — прямая 124
Коэффициент вириальный второй 93,
97
Кремний 201, 202
Критическая точка
— — жидкость — пар 134
— — расслаивания 153, 154
— — жидкость — твердая фаза 271
Л
Лантаноиды 183, 184
Литий 6, 178 — 180
М
Марганца подгруппа 192
Меди подгруппа 194
Металлическая связь 11, 99
Металлы 6, 164—167, 260
Метод
— возмущений 18, 24, 25
— псевдопотенциала 167 — 169
— ядерного магнитного резонанса
108
Микрофлуктуации
— концентрации 155, 156
— плотности 134, 135
Момент дипольный 20, 30
— — вакуумный 44
— внешний 44, 46
— внутренний 44, 45
— макроскопический 41, 43
— перехода 20, 25, 35
Мышьяка подгруппа 206
Н
Натрий 49, 178, 180
Неон 77, 78, 224, 225
Неколлективные процессы 277
Неэлектролиты 6, 260
О
Объемноцентрированная
кубическая упаковка (ОЦК) 80
272
Окись хлора 107
Олово 202, 203
Оператор
— ан.тисимметризации 33
— возмущения 12, 15
— Гамильтона 11, 12, 15, 19, 54
— дипольного момента 20
— кинетической энергии 54
— локальный 168, 169
— нелокальный 168, 169
Ортоводород 218—220
П
Пароводород 218 —220
Парная корреляционная функция
115, 133, 134
Пиридин 22, 28, 29, 30, 31, 36, 38,
57
Плотности
— радиальная функция
распределения 117 — 119
— микрофлуктуации 134, 135
— флуктуации 127, 131 — 134
— — адиабатические 139, 140
— — изобарические 139, 140
Поле
— внешнее 39
— внутреннее 37, 38
— — среднее 40—42
— Лоренца 37—42
— макроскопическое среднее 37—43
— полости 43
— реактивное 29, 37, 43—47
Поляризация среды 29, 30, 37, 41 —
45
Поляризуемость
— макроскопическая 39, 41
— молекул анизотропия тензора
109, 110
— — дисперсия 27
: средняя 27, 30
тензор 27, 109, ПО
Показатель преломления 40
— — предельное значение 40, 47
Полоний 216
Полуметаллы 165, 166
Полупроводники
— электронные 6, 163
— протонные 163
Померанчука эффект 256
Потенциал
— Леннарда—Джонса 95
— степенной 94
Правила
292
— Иогансена 68
— Энгеля 176, 177
Правило
— Бачинского 96
— Ван-Лаара 96
— 8-N, 171-174
— Галицина—Вертело 97
— интенсивностей 69
— растяжений водородных связей
70—72
— факторов 72, 73, 98
— частот 176, 177
— Юм-Розери 176
Процессы
— коллективные 277
— неколлективные 277
Радиальная функция распределения
атомов 115, 117, 122, 124,
135
— — — плотности 117, 119
Радиус вандерваальсов 81, 86
Радиоспектроскопия 5, 108—112
Радиус корреляции 119
Рассеяние света релеевское 5, 103,
108—112, 128
Реактивное поле 29, 37, 43—47
Релеевская спектроскопия 141, 144,
147—149
Резонанс индуктивный
— — электронный 32—35
— — колебательный 33
Резонанс
— Ферми 66
— ядерный магнитный 108
Релеевский триплет 144
Рубидий 178-180
Света рассеяние 5, 103, 108—112,
128
Свинца подгруппа 203
Связь
— металлическая 11, 99
— химическая сильная 6, 9, 54,
55, 99
слабая 6, 9, 10, 54, 55, 99
Сера 203-213
Селен 213
Сжимаемость
— адиабатическая 139
— изотермическая 125
Силы
— близкодействующие 10, 49—54
— вандерваальсовы 19—32, 36
— — аддитивность 29
— дальнодействующие 10
— дипольные 6, 19—22,30, 31, 36
— дисперсионные или лондонов-
ские 6, 24—28, 30, 31, 36
— отталкивания 10
— химические 6, 54, 55
Скандия подгруппа 182, 183
Состояния газа уравнение 93, 96, 97
Спектроскопические методы (И. К.;
К. Р. и др.) 5, 108, 109, ПО
Спектроскопия
— акустическая 5, 112
— диэлектрическая 108 — 112
— релеевская 141, 144, 147 —149
Спирты 57, 58, 61, 63, 64, 67—69,
102
Сравнительный метод расчета
свойств 73
Степенной потенциал 94
Сурьма 208
Теллур 214, 215
Тепловая длина волны 221
Термодинамические методы 5, 106
Тернарная функция распределения
атомов 121
Титана подгруппа 189
Триплет релеевский 144
Углерод 200, 201
— четыреххлористый 8, 30, 36,
57, 69, 72, 73, 87, 107
Уксусная кислота 58—60, 62—64?
104, 105
Уравнение
— Борна—Майера 50
— Ван-дер-Ваальса 96, 9*7
— Клапейрона—Клаузиуса 179,
228
— Клаузиуса—Мосотти
макроскопическое 39
— Клаузиуса—Мосотти
молекулярное 40
— Ландау—Плачека 144
— Леннарда—Джонса 95
— Липпинкотта 71, 72, 93
— Лоренца—Лоренца 27, 40
— Лондона 27, 28
— Морзе 93
— Шредингера И, 12
Ф
Ферми поверхность 166 — 168, 170
Флуктуации
— адиабатические 139, 140
— анизотропные 127, 144 — 151
— диэлектрической
проницаемости 147, 148
— изобарические 139, 140
293
— концентрации 151 — 154
— теория 128—131
Фосфор 205
Фтор 222
Функция распределения
атомов 115, 117, 122, 124, 135
— — плотности 117—119
тернарная 121
Хлор 49, 120, 121, 223
Хлороформ 69, 105
Хрома подгруппа 192
Ц
Цезий 49, 178-181
Цинк подгруппа 195
Ч
Частот правило 69, 70
Эвтектического состава растворы
156, 157
Электропроводность 162, 169
Энергия
— близкодействующего
отталкивания 49, 53, 89
— взаимодействия двух атомов 13,
88, 98, 99
— — — — влияние среды 30, 73
— внутренняя 125
— диполь-дипольного
взаимодействия 20, 21, 30, 31
— кулоновская 13
— лондоновского (или
дисперсионного) взаимодействия 24—28,
30, 48
— образовалия химических связей
54, 55
— полная 12
— поляризация молекул 28, 30
Энергия
— потенциальная 13
— реактивного взаимодействия 37,
46—49
— Ферми 252, 255
Эффект
— Леонтовича—Фабелинского 148,
149
— Померанчука 256
ОГЛАВЛЕНИЕ
Cm p.
Связь между используемыми в книге единицами измерений в СИ и
других системах 3
Основные обозначения 3
Предисловие 5
Введение 8
§ 1. Растворы (8). § 2. Строение жидкостей (9). § 3. Взаимодействия между
молекулами (10). Литература (14)
Часть первая
МЕЖМОЛЕКУЛЯРНЫЕ ВЗАИМОДЕЙСТВИЯ
Глава 1. Дальнодействующие молекулярные силы 15
§ 4. Оператор возмущения Н' (15). § 5. Метод возмущений (18). § 6. Энергия
взаимодействия в первом приближении метода возмущений (19). § 7. Лондо-
новские (или дисперсионные) взаимодействия (24). § 8. Поляризационное
взаимодействие (28). § 9. Об аддитивности вандерваальсовых сил (29). § 10. Ди-
польное, лондоновское и поляризационное взаимодействия молекул в жидкой
фазе (30). § 11. Резонансное взаимодействие между молекулами (32).
§ 12. Влияние изотопных замещений (35).
Глава II. Реактивное поле и его влияние на межмолекулярные
взаимодействия 36
§ 13. Реактивное взаимодействие (36). § 14. Среднее макроскопическое поле и
внутреннее поле (37). § 15. Поле Лоренца (39). § 16. Реактивное поле и поле
полости (43). § 17. Отличие дипольного момента полярных молекул в жидкой
среде от дипольного момента в вакууме (44). §18. Энергия реактивного
взаимодействия (46):
Глава III. Близкодействующие силы 49
§ 19. Близкодействующее отталкивание (49). § 20. Теорема о вириале и ее
применение для анализа близкодействующего отталкивания (50). § 21.
Химическая связь (54). § 22. Водородная связь (56). § 23. Энергия AU и энтальпия
АН водородной связи (59). § 24. Межъядерные расстояния X—Н и Н...У (63).
§ 25. Внутримолекулярные колебания и водородная связь (64). § 26.
Водородная связь и резонанс Ферми (66). § 27. Изотопные сдвиги частот колебаний
[5] (67). § 28. Правила Иогансена (68). § 29. О природе водородных связей
(74). § 30. Другие слабые химические взаимодействия между электрически
нейтральными молекулами (77). § 31. Слабые химические взаимодействия
ионов (88).
Глава IV. Эмпирические методы описания межмолекулярных
взаимодействий 92
§ 32. Об эмпирических формулах (92). § 33. Потенциалы Морзе Липпинкотта
(93). § 34. Использование результатов исследований свойств газов (93).
§ 35. Свойства постоянных уравнения Ван-дер-Ваальса (96). § 36. Атом — атом
потенциалы (98). Литература (99).
Часть вторая
СТРОЕНИЕ ЖИДКИХ ФАЗ
Глава V. Ассоциаты и комплексы Ю2
§ 37. Ассоциаты и комплексы (102). § 38. Устойчивость ассоциатов и
комплексов (106). § 39. Среднее время жизни ассоциатов и комплексов (107). § 40.
О методах изучения ассоциатов и комплексов (107).
Глава VI. Функции распределения положений частиц в жидкостях . . ^^
§ 41. Распределение молекул жидкости (112). § 42. Расчет функции g(R)
(115). § 43. Кривые атомного распределения. Среднее координационное число
(117). § 44. Корреляция трех атомов. Суперпозиционное приближение (121).
§ 45. Корреляция и ассоциация (122). § 46. Прямая корреляционная функция
(124). § 47. Связь функций распределения с термодинамическими функциями
(125). § 48. Функции распределения частиц в растворах (126).
197
Глава VII. Флуктуации в жидкостях 1<6/
§ 49. Виды флуктуации (127). § 50. О теории флуктуации (128). § 51.
Флуктуации плотности (131). § 52. Флуктуации плотности и парные корреляции
молекул (133). § 53. Микрофлуктуации плотности (134). § 54. Флуктуации
плотности и ассоциация (138). § 55. Адиабатические и изобарические флуктуации
плотности (139). § 56. Спектр света, рассеянного флуктуациями плотности
(141). § 57. Анизотропные флуктуации (144). § 58. Флуктуации диэлектриче-
295
С trip.
ской проницаемости (147). § 59. Поперечный звук. Эффект Леонтовича — Фа-
белинского (148). § 60. Процессы, приводящие к появлению анизотропных
флуктуации (149). § 61. Флуктуации концентрации (151). § 62. Флуктуации
концентрации в окрестности критической точки расслаивания (153). § 63.
Микрофлуктуации концентрации (155). § 64. Флуктуации концентрации и строение
жидких растворов эвтектического состава (156). Литература (158)
Часть третья
СТРОЕНИЕ ПРОСТЫХ ЖИДКОСТЕЙ
Глава VIII. Жидкие металлы, полупроводники и диэлектрики .... 161
§ 65. Классификация. Диэлектрики. Полупроводники (161). § 66. Состояния
электронов в энергетических зонах. Металлы и полуметаллы (164). § 67.
Метод псевдопотенциала (167). § 68. Жидкие металлы. Состояния электронов
(169). § 69. О структуре твердых простых веществ. Правило 8—N (171). § 70.
О возможных распределениях атомов в простых твердых и жидких веществах
(174)
Глава IX. Подгруппы лития, бериллия и переходных металлов .... \jj
§ 71. Подгруппа лития (щелочные металлы) (Li, Na, К, Rb, Cs) (177). § 72.
Подгруппа бериллия (щелочноземельные металлы) (Be, Mg, Ca, Sr, Ba, Ra)
(181). § 73. Подгруппа скандия. Лантаноиды и актиноиды (182). § 74.
Подгруппы титана, ванадия, хрома и марганца (189). § 75. Подгруппы железа и
платины (193). § 76. Подгруппы меди и цинка (194)
Глава X. Подгруппы бора, углерода, азота, кислорода, фтора и
инертных газов 198
§ 77. Подгруппа бора (В, Al, Ga, In, Tl) (198). § 78. Подгруппа углерода (С,
Si, Ge, Sn, Pb) (200). § 79. Подгруппа азота (N, P, As, Sb, Bi) (203). § 80.
Подгруппа кислорода (О, S, Se, Те, Ро) (208). § 81. Водород (216). § 82. Подгруппа
фтора (F, Cl, Br, I, At) (222). § 83. Инертные газы (Не, Ne, Ar, Кг, Хе, Rn)
(224)
Глава XI. Квантовые простые жидкости 4Не и 3Не 226
§ 84. Введение (226). § 85. Изотоп 4Не (227). § 86. О строении жидкого 4Не
(229). § 87. Сжимаемость, скорость звука, молярная поляризация жидкого 4Не
(231). § 88. Энтальпия испарения. Межъядерное взаимодействие (232). § 89.
Энтропия и теплоемкость жидкого 4Не (233). § 90. Аномалии в явлениях
переноса (234). § 91. О теории жидкого гелия II. Квантовая когерентность
жидкого гелия при 0 К (236). § 92. Конденсация Бозе — Эйнштейна. Теория Н. Н.
Боголюбова (238). § 93. Причины сверхтекучести жидкого гелия [67] (243). § 94. О
проявлениях квантовой когерентности жидкого гелия (244). § 95 Возбужденные
состояния гелия II (245). § 96. Другие аномалии гелия II (247). § 97.
Сверхтекучесть и сверхпроводимость (248). § 98. Изотоп 3Не [58] (248). § 99. О
теплоемкости жидкого 3Не (251). § 100. Теплоты фазовых переходов и энтропия
3Не. Магнитные свойства (253). § 101. О теории жидкого 3Не (256)
Глава XII. Строение простых жидкостей и периодическая система
Менделеева 260
§ 102. Введение (260). § 103. Молярные объемы (260). § 104. Строение простых
жидкостей и твердых фаз (267). § 105. Объемно-центрированная кубическая
структура (271). § 106. Плавление простых веществ (279). § 107 Кипение
простых жидкостей (286). Литература (287)
Общая литература
Предметный указатель 291
Михаил Иванович Шахпаронов
ВВЕДЕНИЕ В СОВРЕМЕННУЮ ТЕОРИЮ РАСТВОРОВ
(Межмолекулярные взаимодействия. Строение. Простые жидкости)
Редактор М. М. Поплавск-ая. Художник Ю. Д. Федичкин. Художественный редактор
Т. М. Скворцова. Технический редактор А. К. Нестерова. Корректор С. К. Марченко.
Сдано в набор 8/VII-75r. Подп. к печати 1/III-76 г. Формат бОХЭО1/™- Бум. тип. №2
Объём 18,5 печ. л. Усл. п. л. 18,5 Уч.-изд. л. 19,35 Изд. № Хим.—525. Тираж
10 000 экз. Цена 84 коп. Зак. 503 План выпуска литературы для вузов и техникумов
издательства «Высшая школа» на 1976 г. Позиция № 80.
Москва, К-51, Неглинная ул., д. 29/14, издателоство «Высшая школа»
Ярославский полш*рафкэм5инат Союзполиграфпрома при Государственном комитете Совета
Министров СССР по делам издательств, полиграфии и книжной торговли. 150 014 Ярославль.
ул. Свободы, 97.