/
































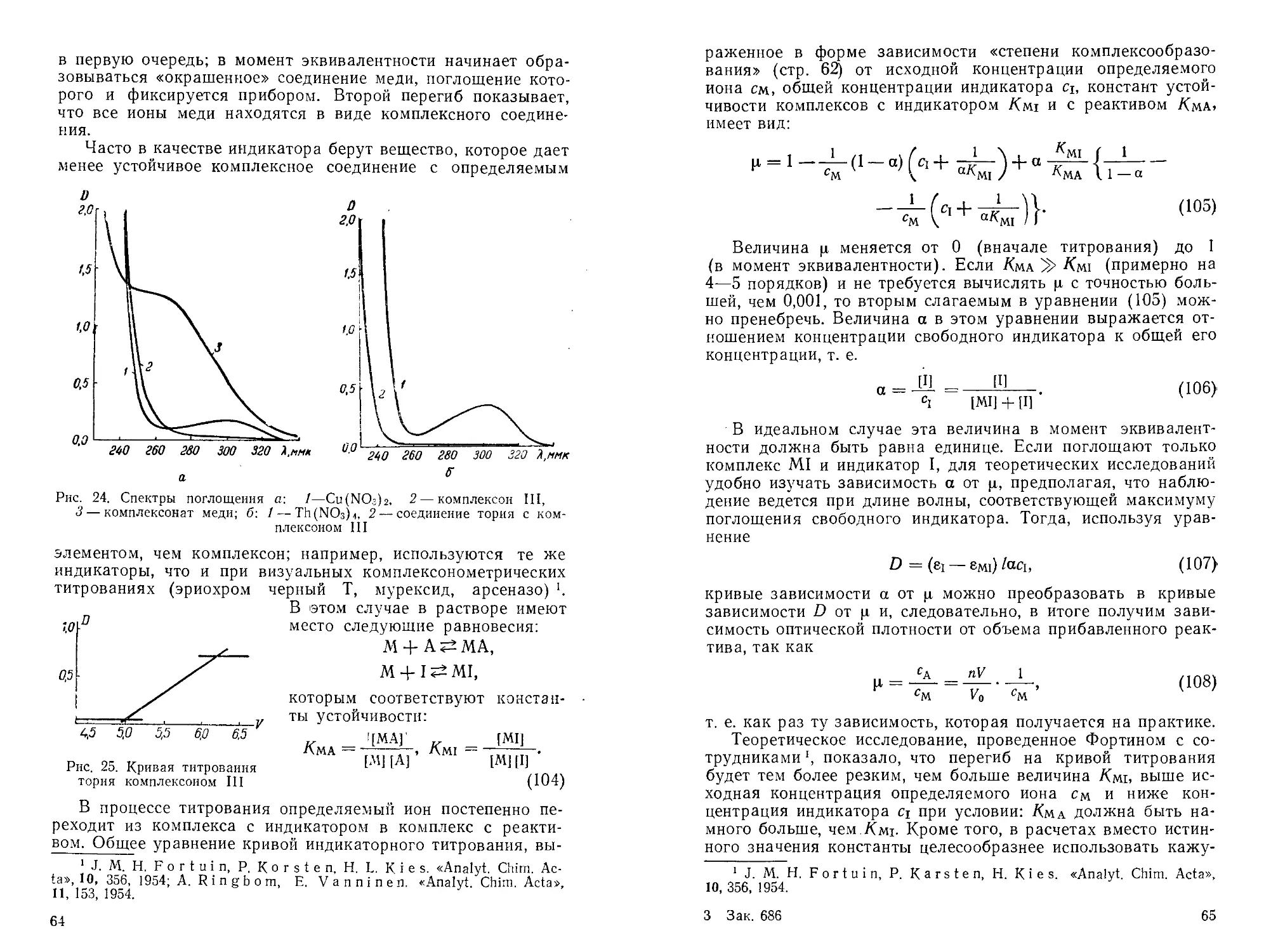






























































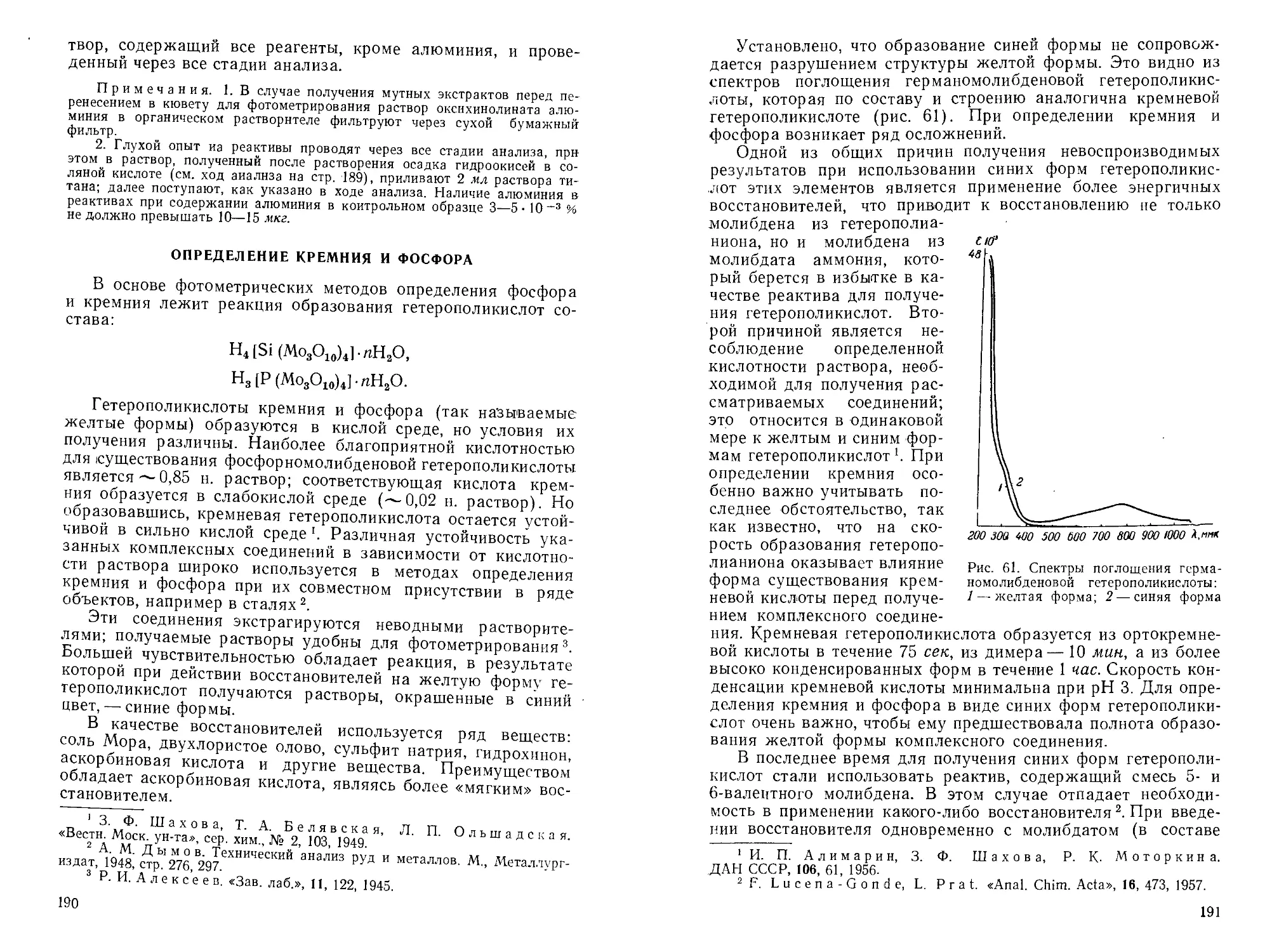








































Автор: Пешкова В.М. Громова М.И.
Теги: спектральные методы анализа оптические методы анализа химия
Год: 1965
Текст
В. М. ПЕШКОВА, М. И. ГРОМОВА
ПРАКТИЧЕСКОЕ
РУКОВОДСТВО
по
СПЕКТРОФОТОМЕТРИИ
И КОЛОРИМЕТРИИ
Издание второе,
переработанное и дополненное
Допущено Министерством высшего
и среднего специального образования
РСФСР в качестве учебного пособия
для университетов
ИЗДАТЕЛЬСТВО
МОСКОВСКОГО УНИВЕРСИТЕТА
19 6 5
УДК 543.42.062(076.5) 4-543.432(076.5)
Книга является учебным пособием для практи-
ческих занятий студентов в лабораториях фото-
метрических методов анализа химических факуль-
тетов вузов.
В ней изложены теоретические основы фотомет-
рического метода, современные направления его
развития, описана аппаратура, а также даны при-
меры использования метода для изучения систем
в растворах и в практике количественного опреде-
ления ряда элементов.
Книга может быть также использована работ-
никами исследовательских и производственных
лабораторий.
Отв. редактор чл.-корр. АН СССР
проф. И. П. АЛИМАРИН
Печатается по постановлению
Редакционно-издательского совета
Московского университета
ПРЕДИСЛОВИЕ
В практику химических лабораторий прочно вошли коло-
риметрические методы определения очень большого числа
элементов. В последнее время в связи с задачами, стоящими
перед работниками аналитических и ряда других химических
лабораторий, широкое применение находит спектрофотомет-
рический метод, преимущества которого по сравнению с коло-
риметрическим мы старались показать в предлагаемом руко-
водстве.
Ограниченное число практических руководств в учебных
лабораториях вузов и трудности, возникающие в связи с этим
при проведении практических занятий со студентами, специа-
лизирующимися в области аналитической химии, побудили
нас составить настоящее руководство.
Книга состоит из пяти разделов. В I — теоретическом
разделе — рассмотрен основной закон светопоглощения
и кратко перечислены задачи, которые можно решить в прак-
тике аналитических лабораторий, применяя спектрофотомет-
рический метод. При изложении этого материала обращено
внимание на преимущество в работе с растворами, которые
подчиняются основному закону светопоглощения, показаны
способы графического изображения процесса светопоглоще-
ния в растворе.
Во II разделе изложены методы измерения интенсив-
ности окраски растворов и методы расчета концентраций,
принятые при выполнении работы на визуальных и фото-
электроколориметрических приборах, позволяющих измерять
оптические плотности или коэффициенты пропускания рас-
творов.
В III разделе даны отдельные примеры применения
спектрофотометрического метода, чтобы показать его преиму-
щество по сравнению с колориметрическим. В качестве при-
меров взяты методы спектрофотометрического титрования
и дифференциальный спектрофотометрический, позволяющие
расширить возможности определения как очень малых, так
3
и больших концентраций веществ, значительно повысить точ-
ность определения, в частности, при работе с «окрашенными»
реагентами по дифференциальному спектрофотометрическому
методу. Даны также спектрофотометрические методы опреде-
ления констант кислотной диссоциации органических реаген-
тов, проявляющих слабую кислотную функцию. Все эти при-
меры ярко показывают преимущество спектрофотометров —
приборов, имеющих высокую степень монохроматизации.
В IV разделе описана аппаратура, даны дополнитель-
ные сведения по практическому использованию приборов.
Основное внимание уделено обсуждению возможностей каж-
дого прибора и целесообразности их применения в тех или
других случаях. Описание конструкции приборов дается нами
кратко, в расчете на то, что студент использует для ознаком-
ления заводское описание, прилагаемое к прибору.
В V разделе дано описание практических работ. В их
число входят определение константы диссоциации двух орга-
нических реактивов, примеры, в которых показано преимуще-
ство призменных спектрофотометров перед фильтровыми, ме-
тоды определения отдельных элементов как в растворах их
чистых солей, так и в различных объектах, включая чистые
металлы.
Быстрое развитие методов фотометрического анализа по-
требовало изложения некоторых вопросов теории и практики
на новом современном уровне, а также и некоторого перерас-
пределения материала по отдельным вопросам. Поэтому
во втором издании пособия значительно переработаны и до-
полнены отдельные разделы.
Так, в I раздел внесено обсуждение точности фотометри-
ческих методов и переработаны параграфы: причины откло-
нения от законов светопоглощения, возможности и преимуще-
ства спектрофотометрического метода и исследование фото-
метрической реакции. Существенные изменения внесены в
III раздел: расширен материал по теоретическим основам
дифференциального метода и метода СФ-титрования, приве-
дены методы расчета истинных значений молярных коэффи-
циентов погашения и дано изложение некоторых фотометри-
ческих методов определения состава и констант устойчивости
комплексных соединений. В V раздел включены практические
работы по применению дифференциального метода для опре-
деления ряда элементов: железа, марганца, меди. Дополнены
методы определения ультрамалых количеств примесей.
Авторы приносят благодарность чл.-корр. АН СССР
проф. И. П. Алимарину, акад. АН УССР А. К. Бабко,
проф. А. И. Бусеву и проф. Л. И. Адамовичу за ряд ценных
советов при переиздании книги.
ВВЕДЕНИЕ
Абсорбционный анализ основан на избирательном погло-
щении потока лучистой энергии различными однородными
средами. В зависимости от условий изучения светопоглоще-
ния, т,- е. от аппаратуры применяемой для этой цели, разли-
чают два метода данного анализа: спектрофотометрический и
колориметрическийОни основаны на общем принципе — су-
ществовании пропорциональной зависимости между светопо-
глощением какого-либо вещества, его концентрацией и тол-
щиной поглощающего слоя. Другими словами, в основу этих
методов положен общий объединенный закон светопоглоще-
ния: закон Бугера — Ламберта — Бера. Но названные ме-
тоды существенно отличаются по тем задачам, которые мо-
гут быть решены с их помощью.
В колориметрическом методе в качестве источника осве-
щения используется немонохроматизированный поток лучи-
стой энергии видимого участка спектра. Поэтому этот метод
применяется только в концентрационном анализе, т. е. при
определении концентрации вещества в растворе.
Задачи концентрационного анализа решаются также и
с помощью спектрофотометрического метода, но в отличие
от колориметрического метода в нем используется всегда мо-
нохроматический поток лучистой энергии различных участков
спектра (видимого, ультрафиолетового, инфракрасного). Это
значительно .расширяет возможности спектрофотометриче-
ского метода по сравнению с колориметрическим (стр. 18).
На взаимодействии потока лучистой энергии с веществом,
через которое он проходит, основан еще ряд методов ана-
лиза: нефелометрический, турбидиметрический, люминесцент-
ный. Нефелометрический, турбидиметрический и абсорбцион-
ный методы часто объединяют в группу фотометрических ме-
1 В настоящее время термин колориметрия обычно заменяют термином
фотометрия, так как первый, строго говоря, применим к оценке цветнос-
ти, а не относительной интенсивности потока лучистой энергии.
5
тодов, хотя они и не имеют общего принципа: первые два
основаны на взаимодействии потока лучистой энергии с дис-
персной системой, т. е. на рассеянии потока лучистой энергии
(нефелометрический метод — на измерении отраженного све-
та, турбидиметрический — проходящего), а спектрофотомет-
рический и колориметрический методы — на взаимодействии
света с однородными системами.
В последние годы к фотометрическим методам чаще всего
относят спектрофотометрический и колориметрический ме-
тоды.
В данном руководстве термин фотометрирование исполь-
зуется для определения степени поглощения потока лучи-
стой энергии веществами, находящимися в растворе. Измеряя
светопоглощение, нельзя непосредственно определить массу
вещества, как это имеет место в весовом и объемном ана-
лизах, но светопоглощение раствора или интенсивность его
«окраски» непосредственно связаны с концентрацией веще-
ства в растворе, т. е. с массой вещества. Таким образом, из-
меряют доступный параметр, косвенно связанный с массой.
Каждая однородная система обладает способностью изби-
рательно поглощать лучистую энергию определенной длины
волны. В видимой части спектра воспринимаемый цвет есть
результат избирательного поглощения определенного уча-
стка спектра падающего непрерывного потока лучистой энер-
гии (белого света). Мы видим цвет в зависимости от погло-
щения того или иного участка спектра. Цвет раствора всегда
является дополнительным к цвету поглощенного излучения
(табл. 1).
Таблица 1
Наблюдаемые цвета и соответствующие им поглощенные участки спектра
Интервал длин волн поглощенного излучения, ММ.К Цвет поглощенного излучения Наблюдаемый цвет (дополнительный)
400—450 Фиолетовый Желто-зеленый
450—480 Синий Желтый
400—550 Сине-зеленый Оранжевый
500—560 Зеленый Красно-пурпурный
400—610 Сине-зелено-желтый Красный
450—650 Зелено-желто-красный Пурпурный
625—750 Красный Сине-зеленый
Смещение полосы поглощения от фиолетового конца види-
мого спектра к красному дает такую последовательность вос-
принимаемых цветов: желтый оранжевый красный -*
пурпурный синий сине-зеленый. Если происходит сме-
щение полосы поглощения в сторону длинных волн, то это
6
явление называется батохромным эффектом, а в коротковол-
новую часть спектра — гипсохромным.
Остановимся на величинах, которые характеризуют спектр
поглощения. Спектр поглощения вещества в растворе выра-
жают обычно как зависимость оптической плотности D или
пропускания Т потока лучистой энергии от длины волны X.
Единицами измерения длин волн служат ангстремы (1А =
= Ю~8 см), микроны (1 мк = 10~4 см), миллимикроны
(1 ммк = 10-7 см), нанометры (1 ммк = 1 нм = 10-9 м). Дли-
ны волн в видимой и ультрафиолетовой (УФ) областях спект-
ра выражают обычно в ангстремах или в миллимикронах,
в инфракрасной (ПК) —в микронах. Длина волны зависит
от показателя преломления среды, в которой излучение рас-
пространяется. Поэтому для характеристики определенного
участка спектра часто используют частоты или волновые чис-
ла, которые не зависят от рефракции среды.
Частота излучения v выражается отношением скорости
распространения излучения (скорости света) с к длине вол-
ны %
(Величины скорости света и длины волны должны быть взя-
ты для одной и той же среды, в которой распространяется
излучение).
Частота измеряется в обратных секундах (сект1), герцах
(Hz) или Френелях (/)
1Hz = сек1 = 1012/.
Волновое число v показывает, какое число длин волн при-
ходится на 1 см пути излучения в вакууме и определяется
соотношением
где % — длина волны в пустоте. С частотой волновое число
связано соотношением
v = cv,
где с — скорость света в пустоте, равная 3-1010 см/сек.
Например, если % = 250 ммк, то v = 40 000 см-1 и v —
= 1200-1012 сек-1 = 1200/.
Поглощая лучистую энергию определенного участка спект-
ра, система переходит на более высокий энергетический уро-
вень. Частота связана с величиной запаса энергии в началь-
7
ном Et и конечном £2 состояниях через постоянную Планка
h уравнением
£2 —£х = hv.
При переходах между двумя уровнями энергии возникают
отдельные спектральные линии или характерные полосы по-
глощения. Если вещество будет находиться в жидком состоя-
нии или растворено в каком-либо растворителе, то вслед-
ствие межмолекулярного взаимодействия отдельные линии,
характерные для спектра вещества, находящегося в газооб-
разном состоянии, сильно расширяются и образуют полосы.
Природа полос поглощения в ультрафиолетовой (УФ) и ви-
димой областях спектра одинакова и связана главным обра-
зом с числом и расположением электронов в поглощающих
молекулах и в заряженных частицах вещества (электронные
переходы внешних валентных электронов), а в инфракрасной
(ИК) области — с колебаниями атомов в молекуле. Стро-
гого разграничения границ видимой, УФ и ИК областей
спектра нет. Обычно принято считать
УФ область 200 —400 ммк,
видимая область 400 — 700 ммк,
ИК область:
ближняя 700 ммк— 2 мк,
основных (фундаментальных)
частот 2 — 50 мк,
дальняя > 50 мк.
Монохроматические потоки лучистой энергии могут быть
получены различным путем. В приборах, которые могут счи-
таться упрощенными спектрофотометрами (фотометр ФМ,
фотоэлектроколориметры ФЭК-Н-52, ФЭК-Н-54, ФЭК-Н-57,
ФЭК-56 и др.), для получения монохроматических потоков
лучистой энергии используются светофильтры с достаточно
узкой полосой пропускания (стр. 116), в спектрофотометрах
СФ-4, СФ-4А, СФ-5, СФ-2М, СФ-10 — диспергирующие приз-
мы, в спектрофотометре СФД-2 — дифракционная решетка.
I. ТЕОРЕТИЧЕСКИЕ ОСНОВЫ ФОТОМЕТРИЧЕСКИХ
МЕТОДОВ АНАЛИЗА
1. ОСНОВНЫЕ ЗАКОНЫ СВЕТОПОГЛОЩЕНИЯ
Между поглощением монохроматического потока лучистой
энергии и количеством поглощающего вещества существует
определенная зависимость. Рассматривая эту зависимость,
предполагаем, что имеем дело с однородным раствором ве-
щества при толщине слоя / (рис. 1).
Рис. 1. Изменение интенсивности потока лучистой энергии
при прохождении через «окрашенный» раствор
Монохроматический поток лучистой энергии, падая на
объект, частично поглощается, отражается и проходит через
слой вещества. Интенсивность первоначального монохрома-
тического излучения при прохождении через кювету с погло-
щающим раствором разлагается на сумму интенсивностей из-
лучений:
1о — 11
где 70 — интенсивность первоначального монохроматического
излучения, падающего на объект; Л — интенсивность монохро-
матического излучения, прошедшего через объект; 1а — ин-
9
тенсивность поглощенного объектом монохроматического из-
лучения; 1Г—интенсивность монохроматического излучения,
отраженного стенками кюветы и растворителем. Величина /г
слагается из двух величин: Л, — интенсивности монохромати-
ческого излучения, отраженного стенками сосуда, и 1гг — от-
раженного растворителем.
Интенсивность потока лучистой энергии, прошедшего че-
рез исследуемый раствор, всегда измеряют относительно рас-
твора сравнения или «нулевого» раствора, при приготовлении
и исследовании которого используется растворитель и кю-
веты, аналогичные применяемым для приготовления и иссле-
дования испытуемого раствора. Таким образом, величина 1Г
может быть в целом исключена.
Представим, что слой вещества I состоит из бесконечно
тонких слоев dl, и в этот тонкий слой поступает поток моно-
хроматической лучистой энергии с длиной волны %. После
прохождения через поглощающий слой, имеющий толщину
dl, интенсивность потока лучистой энергии уменьшается в ре-
зультате поглощения на величину dr.
~ — (1)
dl
или
у =—ad/, (2)
где a — коэффициент поглощения среды.
Следовательно, наблюдается относительное ослабление
потока лучистой энергии при прохождении его через каждый
тонкий слой однородного раствора вещества.
Интегрируя уравнение (2) от /о до Л по всей толщине
слоя, получим
Л i
У -у = — a^dl, (3)
/о О
In 7,— In /0 = — а/. (4)
Или в экспоненциальной форме
Л = (5)
Переходя к десятичным логарифмам
Л = /ою-«, (6)
a = 2.3026&,
где коэффициент погашения k — величина, обратная толщине
слоя, необходимой для ослабления интенсивности падающего
10
света до одной десятой от величины первоначального излу-
чения (ослабление в 10 раз).
Зависимость между ослаблением интенсивности направ-
ленного параллельно монохроматического потока лучистой
энергии и толщиной поглощающего слоя, установленная Бу-
герой в 1729 г. и подтвержденная Ламбертом в 1760 г., со-
ставляет сущность первого закона светопоглощения:
Относительное количество поглощенного пропускающей
средой света не зависит от интенсивности падающего излуче-
ния. Каждый слой равной толщины поглощает равную
долю проходящего монохроматического потока лучистой
энергии.
Второй закон светопоглощения дан Бером в 1852 г. и вы-
ражает связь между интенсивностью монохроматического по-
тока лучистой энергии и концентрацией вещества в погло-
щающем растворе:
Поглощение потока лучистой энергии прямо пропорцио-
нально числу частиц поглощающего вещества, через которое
проходит поток лучистой энергии.
Объединенный закон Бугера — Ламберта — Бера выра-
жается уравнением
/z = /o10~^, (7)
или
1g = е/с, (8)
‘t
так как k = ес.
Величину, выраженную логарифмом отношения , на-
ы
зывают оптической плотностью поглощающего вещества и
обозначают буквой D.
D = \g~ = slc. (9)
Таким образом, закон Бугера-—Ламберта выражает про-
цесс светопоглощения при постоянной концентрации веще-
ства в растворе и различной толщине слоя, и закон Бера —
ту же зависимость при постоянной толщине слоя испытуе-
мого раствора и различной концентрации вещества в рас-
творе.
Отношение интенсивности монохроматического потока из-
лучения, прошедшего через исследуемый объект, к интенсив-
ности первоначального потока излучения называется про-
зрачностью, или пропусканием раствора, и обозначается бук-
вой Т.
Т = — = \0~е1с. (10)
11'
Оптическая плотность D и пропускание (прозрачность) Т
связаны уравнением
Z) = -lgT. (11)
Обычно величину Т выражают в процентах, тогда
D = 1g 1-100, (12)
или
£) = 2 — IgT. (13)
Величины D и Т зависят от длины волны и концентрации
вещества в растворе (рис. 2 и 3).
Прямолинейная зависимость оптической плотности и ло-
гарифма пропускания от концентрации вещества в растворе
имеет место при условии подчинения растворов закону све-
топоглощения (рис. 3). При этом кривые спектров поглоще-
Рис. 2. Зависимость светопоглоще-
ния D, Т от длины волны Л
Рис. 3. Зависимость оптиче-
ской плотности и логарифма
пропускания от концентрации с
вещества в растворе
ния (D — X) имеют одну и ту же форму независимо от тол-
щины слоя раствора или концентрации вещества в растворе
(рис. 4) и характеризуются сохранением положения макси-
мума при одной и той же длине волны X.
Прямо пропорциональная зависимость между величинами,
характеризующими процесс светопоглощения (D,lgT), и кон-
центрацией вещества в растворе или толщиной поглощаю-
щего слоя, может быть получена только при постоянном зна-
чении молярного коэффициента погашения е. Величина е яв-
ляется коэффициентом пропорциональности.
Если концентрация раствора выражена в моль/л, и тол-
щина слоя в см, то е называют молярным коэффициентом по-
12
гашения. При концентрации раствора, равной 1 моль/л, и тол-
щине слоя, равной 1 см, ъ = D. Молярный коэффициент пога-
шения характеризует индивидуальные свойства исследуемого
вещества в растворе и является функцией длины волны из-
лучения (е = f (X)); он не зависит от концентрации вещества
в растворе и толщины поглощающего слоя, если к исследуе-
мым растворам приложим основной закон светопоглощения.
Согласно основному закону светопоглощения
е>-==-^, (14)
cl
где е рассчитывается на основании измеренной опытным пу-
тем величины D, деленной на толщину слоя и молярную кон-
центрацию поглощающего компонента. Следовательно, для
получения истинной величины молярного коэффициента пога-
шения необходимо, чтобы:
1) из всех присутствующих
в растворе компонентов при
той длине волны, при которой
измеряется величина D, имел
поглощение только изучаемый
компонент;
2) была известна равно-
весная концентрация изучаемо-
го компонента.
На практике такие идеаль-
ные условия существуют не
всегда. К идеальным условиям
приближаются случаи образо-
вания очень устойчивых комп-
лексов, поглощающих в обла-
сти, где отсутствует поглоще-
ние реактива, или случаи, ко-
гда измеряют поглощение рас-
творов, приготовленных раство-
рением точной навески изуча-
емого соединения в органиче-
Рис. 4. Зависимость оптической
плотности D от длины волны X
для ряда растворов одного и то-
го же соединения различной кон-
центрации
ском растворителе, где диссоциация комплекса ничтожно
мала.
Чаще всего встречаются случаи, когда при данной длине
волны поглощают несколько компонентов, в том числе и ис-
пользуемый реагент. Измерять величину оптической плот-
ности по отношению к раствору реагента, взятому в той же
концентрации, что и для реакции, с целью выявления истин-
ного поглощения самого комплексного соединения можно
только в том случае, когда избыток реагента достаточно ве-
лик: во-первых, чтобы иметь возможность пренебречь той до-
лей реагента, которая вступила в реакцию, а во-вторых,
13
иметь гарантию, что весь ион-комплексообразователь связан
в комплекс. При ступенчатом комплексообразовании такой
расчет дает представление о поглощении конечного (п-го)
комплекса, но ничего не говорит о поглощении промежуточ-
ных комплексов.
Если имеется область спектра, где поглощает только сам
комплекс и не поглощают остальные компоненты реакции,
для получения истинной величины е необходимо, чтобы реак-
ция практически протекала до конца, тогда исходная концен-
трация иона-комплексообразователя фактически будет равна
концентрации комплекса.
Чаще всего величина е, полученная делением измеренной
оптической плотности растворов, приготовленных в реальных
условиях опыта, на исходную концентрацию иона-комплексо-
образователя, является лишь средней величиной моляр-
ного_коэффициента погашения. При этом постоянство значе-
ния е для растворов с различной концентрацией не является
доказательством того, что получено истинное значение мо-
лярного коэффициента погашения. В данном случае лишь
соблюдается соотношение: если ct = nc2, то Di = nD2, или в об-
Dt ct
щем виде , т. е. доля исходной концентрации опре-
ck
деляемого иона, переходящая в комплекс, всегда постоянна,
нет побочных реакций и поэтому оптическая плотность про-
порциональна концентрации.
Недостаточно исследовать реакцию при какой-то одной
длине волны. Необходимо изучить поглощение во всем интер-
вале длин волн. Постоянство величины е для одной какой-
либо длины волны не обязательно для всех остальных длин
волн.
Как было отмечено, при условии выполнения основного
закона светопоглощения положение максимума поглощения
на кривых для растворов с различной концентрацией сохра-
няет постоянство (рис. 4); при условии отклонений от закона
наблюдается смещение максимума поглощения. Если рассчи-
тать величины е и построить кривые зависимости е от X, то
при выполнении закона светопоглощения для всех концентра-
ций будет получена одна и та же кривая; в случае отклоне-
ний от закона кривые зависимости е от X совпадать не будут
(рис. 5).
Если при изменении концентраций реагирующих веществ
изменяется молярный коэффициент погашения, то это указы-
вает на возможность возникновения побочных процессов (из-
менение степени диссоциации комплекса, явление полимери-
зации, ступенчатое образование комплексов и др.) и дает
возможность спектрофотометрически исследовать состояние
веществ в растворах. Совпадение кривых зависимости е от X,
14
для растворов с различными концентрациями также не озна-
чает, что данные величины е есть истинные молярные коэф-
фициенты погашения какого-либо компонента, а показывает,
что спектрофотометрически невозможно исследовать данную
систему. В дальнейшем будут даны (стр. 40) некоторые при-
емы расчета величин истинных
молярных коэффициентов пога-
шения, знание которых необхо-
димо для вычисления равно-
весных концентраций при полу-
чении количественных характе-
ристик процессов комплексооб-
разования.
Абсолютная величина мо-
лярного коэффициента погаше-
ния служит хорошей характе-
ристикой чувствительности ре-
акции. Сравнивая величины е
в максимумах поглощения рас-
творов двух различных комп-
лексных соединений одного и
того же элемента, можно ска-
зать, использование какого из
этих соединений дает возмож-
Рис. 5. Зависимость е от Л, в слу-
чае подчинения (ci) н неподчине-
ния (с2, с3, с<) основному закону
светопоглощения
ность определить меньшие концентрации данного элемента.
Например, для аммиаката меди е~ 500, для дитизоната меди
8~ 50 000. Таким образом, последняя реакция по чувстви-
тельности превосходит первую примерно в 100 раз.
2. ПРИЧИНЫ ОТКЛОНЕНИЯ ОТ ОСНОВНОГО ЗАКОНА
СВЕТОПОГЛОЩЕНИЯ
Отклонения от основного закона светопоглощения свя-
заны, с одной стороны, с немонохроматичностью потока лучи-
стой энергии, с другой — с состоянием исследуемого веще-
ства в растворе. Первая из этих причин вызывает отклонение
от общего объединенного закона, вторая — приводит к откло-
нению от закона Бера, которое имеет место значительно
чаще.
Недостаточная монохроматичность потока лучистой энер-
гии вызывает обычно отрицательное отклонение от закона.
Рассмотрим два потока лучистой энергии, охватывающие ин-
тервалы длин волн Za — и — Кп (рис. 6, а). Предполо-
жим, что измерения в интервале длин волн %а— где по-
ток лучистой энергии приближается к идеально монохрома-
тическому излучению, дают величину оптической плотности,
равную Ймакс, а в интервале длин волн %] — кп— Перед из
суммы плотностей, полученных при измерениях с идеально
15
монохроматическими излучениями определенных длин воли,
входящими в этот интервал
— =£»сред. (15)
Совершенно очевидно, что £)макс > £сред> так как величины
плотностей во всех точках, отличных от Хмакс, будут иметь
меньшее значение, чем при этой длине волны. При малых
концентрациях ct кривая спектра поглощения имеет менее
крутой подъем, чем кривые спектров поглощения концентри-
рованных растворов с2. Следовательно, при малых концен-
трациях разность Омаке — О сред будет мала, а при больших —
Рис. 6. Влияние монохроматизации потока лучистой энергии на выполне-
ние основного закона светопоглощения
велика, вследствие чего наблюдаются отрицательные откло-
нения от основного закона светопоглощения.
Отклонения от закона, связанные с состоянием вещества
в растворе, могут быть вызваны:
а) причинами, не связанными с изменением концентра-
ции поглощающих частиц в растворе;
б) причинами, обусловленными изменением концентрации
поглощающих частиц в растворе.
К причинам, не связанным с изменением концентрации
поглощающих частиц в растворе, можно отнести следующие.
1. Присутствие посторонних электролитов вызывает де-
формацию молекул или комплексных ионов окрашенных ве-
ществ и, следовательно, влияет на поглощение лучистой
энергии; с разбавлением раствора светопоглощение изме-
няется.
2. Сольватация (гидратация) также сказывается на по-
глощении потока лучистой энергии раствором, так как с из-
менением концентрации раствора этот процесс протекает не-
равнозначно.
16
Причины, связанные с изменением концентрации погло-
щающих частиц в растворе, могут иметь место в следующих
случаях.
1. С изменением концентрации меняется сила взаимодей-
ствия частиц в растворе (например, наблюдаются явления
полимеризации или деполимеризации)1.
2. Изменение концентрации водородных ионов в растворе
определяемого вещества влияет на выполнение закона Бера
в нескольких направлениях:
а) если реактив обладает кислотными свойствами, то пол-
нота образования окрашенного соединения зависит от pH
раствора
H2R + Ме2+ MeR + 2Н+,
так как меняется диссоциация самого реактива; чем менее
устойчиво образующееся соединение, тем сильнее сказы-
вается pH раствора;
б) при изменении pH раствора может меняться состав
образующегося соединения; например, в зависимости от pH
раствора железо (III) с салициловой и сульфосалициловой
кислотами образует три комплексных соединения различного
состава (стр. 138);
в) изменение pH раствора (уменьшение кислотности) ча-
сто приводит к разрушению измеряемого комплексного со-
единения или неполноте его образования вследствие склон-
ности центрального иона-комплексообразователя присоеди-
нять гидроксильные группы, давая металл-гидрокси-ионы
[Me (ОН)х];г'х.
Из сказанного ясно, что во всех рассмотренных выше слу-
чаях для получения воспроизводимых результатов необхо-
димо контролировать pH раствора.
3. Изменение степени диссоциации вещества в растворе
при разбавлении приводит к отклонению растворов от закона
Бера.
Следовательно, имеется ряд причин, осложняющих при-
менение фотометрических методов и связанных главным об-
разом с химическими свойствами вещества.
Для уменьшения влияния факторов, которые приводят
к осложнениям в применении законов светопоглощения, боль-
шое значение имеют выбор реагента для определения элемен-
тов и способы приготовления испытуемого и эталонных рас-
творов. Если в результате реакции образуется устойчивое
комплексное соединение, то разбавление раствора не влияет
практически на состояние определяемого вещества в рас-
1 Абсорбционная спектроскопия. Сб. статей. М., ИЛ, 1953, стр. 15.
17
творе и при приготовлении испытуемого и эталонных раство-
ров можно не учитывать концентрацию реактива. Использо-
вание мало устойчивого комплексного соединения требует
особого внимания к способу приготовления исследуемого и
эталонных растворов. Этому вопросу отведено большое место
в работах А/ К. Бабко
Наиболее практически ценными являются два метода при-
готовления исследуемого и эталонных растворов. Первый за-
ключается в том, что избыток реагента берется в определен-
ном кратном отношении к определяемому элементу. Во вто-
ром методе соблюдается постоянная концентрация реагента,
взятого в определенном большом избытке по отношению к
определяемому элементу.
3. ВОЗМОЖНОСТИ И ПРЕИМУЩЕСТВА
СПЕКТРОФОТОМЕТРИЧЕСКОГО МЕТОДА
Спектрофотометрический и колориметрический методы
анализа основаны на одном общем законе светопоглощения.
Но ввиду особенностей аппаратуры, применяя спектрофото-
метрический метод, можно решить ряд задач, недоступных
колориметрическому методу. Использование спектрофотомет-
ров с кварцевой и стеклянной оптикой, обеспечивающих вы-
сокую (от 0,5 до 2 ммк в зависимости от участка спектра)
монохроматизацию потока лучистой энергии, позволяет изу-
чать спектры поглощения веществ. Это открывает большие
возможности как для повышения чувствительности, так и для
увеличения избирательности методов определения отдельных
элементов.
1. Только спектрофотометрический метод дает возмож-
ность определить истинное значение е в максимуме поглоще-
ния, используя монохроматический поток лучистой энергии;
недостаточная монохроматизация потока лучистой энергии
приводит к получению среднего значения молярного коэффи-
циента погашения. Например, использование потока лучи-
стой энергии с интервалом длин волн В—Г дает значение
молярного коэффициента погашения — 2700, с интервалом
Б—Д~2200, с интервалом А—Е ~ 1700 (рис. 7) 1 2. Таким
образом, используя монохроматические потоки лучистой
энергии, значительно повышают чувствительность реакции.
Различия в монохроматизации потока лучистой энергии
сказываются также на чувствительности фотометрических
определений следующим образом. Оптическая плотность, как
1 А. К. Бабко. А. Т. Пилипенко. Колориметрический анализ.
М. — Л., Госхимиздат, 1951; А. К. Б а б к о. Физико-химический анализ ком-
плексных соединений в растворах. Киев, Изд-во АН УССР, 1955.
2 Г. В. Юинг. Инструментальные методы химического анализа. М.^
Госатомиздат, 1963, стр. 30.
18
известно, есть разность логарифмов двух потоков лучистой
энергии
(16)
D = lg/0 — lg/z.
Если первоначальный поток лучистой энергии /0 является
суммой большого числа излучений различных длин волн, из
которых лишь некоторые поглощаются испытуемым раство-
ром, то эта разность будет иметь меньшую величину, чем
в случае использования потока лучистой энергии близкого
к идеальному монохроматическому излучению, поглощаемо-
му данным исследуемым раствором (рис. 6,а). Таким обра-
f кг3
зр-
ЗР-
Рис. 8. Спектры поглощения соедине-
ний Pd с диоксимами; 1 — фенил-
диоксимом; 2 — бензилдиоксимом;
3 — а-фурилдиоксимом; 4 — аиизил-
диоксимом
Рис. 7. Влияние монохроматиза-
ции потока лучистой энергии на
значение молярного коэффициен-
та погашения
зом, при переходе от концентрации к концентрации в послед-
нем случае наблюдается большее изменение оптической плот-
ности и соответственно кривая зависимости D от с будет
иметь более крутой подъем (рис. 6,6).
2. Возможность исследования связи между спектрами по-
глощения соединений и строением органических реагентов,
используемых для определения отдельных элементов, позво-
ляет выбирать практически ценные реактивы. Например, на
рис. 8 видно, что соединение палладия с а-фурилдиоксимом
обладает наибольшим поглощением.
19
3. Растворы солей редкоземельных элементов ввиду осо-
бенности строения их атомов имеют характерные «пики»
(узкие полосы) поглощения для каждого элемента в различ-
ных участках спектра (см. приложение), что позволяет иден-
тифицировать соли элементов по их спектрам поглощения.
Используя характерные полосы поглощения, можно количе-
ственно определить, например, следующие элементы:
Nd — 521,8 ммк Sm — 402,0 ммк
Ег — 379,2 ммк
Pr —444,5 ммк
Eu — 393,9 ммк Gd — 272,8 ммк
Tu — 682,5 ммк Yb — 975,0 ммк
Спектрофотометрический метод дает возможность просле-
дить изменения в процессе комплексообразования для солей
указанных элементов1, фиксируя смещение максимумов на
I
0J2V
О.н\
Рис. 9. Спектры поглощения соединений Sm с триоксиглутаровой кислотой
при различных pH: 7—1,8; 2—3,28; 2—9,4; 4—5т(С1О4)з
кривой светопоглощения под влиянием pH раствора и кон-
центрации реагента. Это можно наблюдать по сдвигу макси-
мумов на 2—4 ммк на кривых поглощения этих соединений
(рис. 9).
4. Спектрофотометрический метод применим как для ис-
следования систем, содержащих одно вещество, обладающее
поглощением в определенном участке спектра, так и для си-
стем, содержащих несколько поглощающих компонентов2.
В последнем случае оптическая плотность раствора смеси п
компонентов, измеренная при длине волны Ка, будет равна
1 В. М. Пешкова, М. И. Громова. ЖНХ, II, 1356, 1957;
М. И. Громова, Я. И. X и л ь м а и, В. М. Пешкова. «Вести. Моск,
ун-та», № 6, 41, 1961.
2 Н. П. Ко марь. «Уч. зап. Харьковск. ун-та», XXXVII, 43, 51, 1951.
20
сумме оптических плотностей (при условии их аддитивности)
растворов отдельных компонентов
Яобщ = Da, + D\ + ... + D^. (17)
Чтобы определить концентрацию каждого компонента со-
ставляют систему уравнений
а—п
D = l£ %са, (18)
а=1
измеряя оптические плотности раствора смеси компонентов
при п длинах волн (соответственно числу компонентов). Ре-
шить эту систему можно, зная величины молярных коэффи-
циентов погашения каждого компонента при всех этих дли-
нах волн.
5. Приборы с кварцевой оптикой дают возможность рабо-
тать в ультрафиолетовой и инфракрасной областях спектра,
что позволяет измерять поглощение бесцветных и окрашен-
ных в слабо-желтый цвет растворов. Как уже отмечалось
(см. пункт 1), возможность работать в максимумах на кри-
вых светопоглощения значительно увеличивает чувствитель-
ность применяемой химической реакции и позволяет опреде-
лять малые концентрации с большой точностью. Например,
для определения ультрамалых количеств никеля а-бензил-
диоксимом измерение проводят в области его максималь-
ного поглощения в ультрафиолетовой области при X 273 ммк
(стр. 157), используя спектрофотометр СФ-4. Определение ко-
бальта 2-нитрозо-1-нафтолом при X 307 ммк позволяет опре-
делять ультрамалые количества кобальта (стр. 172).
6. Спектрофотометрический метод дает возможность ис-
следовать процессы комплексообразования, изучать состоя-
ние вещества в растворе:
а) определять константы диссоциации органических реа-
гентов (стр. 45) и константы нестойкости комплексных со-
единений (стр. 53);
б) для определения состава комплексных соединений, при-
меняют метод физико-химического анализа, строя диаграммы
состав — свойство, где в качестве свойства берется оптиче-
ская плотность D или пропускание Т раствора (стр. 51).
7. На основе большой точности измерения величин опти-
ческой плотности развиваются новые спектрофотометриче-
ские методы — дифференциальный (стр. 66) и спектрофото-
метрическою титрования (стр. 57).
4. ИССЛЕДОВАНИЕ ФОТОМЕТРИЧЕСКОЙ РЕАКЦИИ
Одной из главных задач, которые могут быть решены
с помощью фотометрических методов, является определение
концентрации вещества в растворе.
21
Любое фотометрическое определение состоит из двух эта-
пов: 1) приготовление раствора для фотометрирования (пе-
реведение анализируемой пробы в раствор и проведение фо-
тометрической реакции — получение «окрашенного» соедине-
ния); 2) измерение величины поглощения испытуемого рас-
твора (фотометрирование). Очень редко фотометрирование
проводят сразу же после переведения анализируемой пробы
в раствор, так как величина поглощения в этом случае бы-
вает очень незначительна и невозможно определять малые
количества вещества. Поэтому на практике определяемый
компонент обычно переводят в соединение, обладающее зна-
чительным поглощением, и стремятся использовать аппара-
туру, которая дает возможность производить измерения в об-
ласти его максимума поглощения (большие величины опти-
ческих плотностей соответствуют большим величинам моляр-
ных коэффициентов погашения). Чаще всего определяемый
элемент переводят в комплексное соединение с различными
органическими реагентами.
При выборе реагента для определения какого-либо эле-
мента следует учитывать прежде всего его селективность,
а также чувствительность определения, которая может быть
при этом достигнута. Селективность реагента в фотометриче-
ском методе определяется в первую очередь возможностью
найти область спектра, в которой поглощает испытуемое со-
единение, свободную от наложения поглощения посторонних
компонентов, присутствующих в растворе. Кроме того, сле-
дует стремиться подобрать специфические условия проведе-
ния реакции, в которых образуется комплексное соединение
только определяемого элемента. Оптимальные условия опре-
деления требуют полного связывания определяемого эле-
мента в комплекс. Большинство органических реагентов об-
ладают кислотно-основными свойствами. В общем виде урав-
нение реакции образования комплексного соединения в этом
случае можно представить следующим образом:
Ме"+ + mHR -> MeR^~m)+ + mH+
Следовательно, оптимальные условия образования ком-
плексного соединения будут зависеть не только от избытка
реагента, но также от pH раствора, особенно в том случае,
когда используемый реагент является слабой кислотой.В тех
случаях, когда комплексное соединение отличается малой
прочностью, для сдвига равновесия в сторону более полного
образования комплексного соединения используют органиче-
ские растворители: спирт, ацетон, или экстрагируют его
в слой органического растворителя, несмешивающегося с во-
дой (экстракционно-фотометрический метод, стр. 71). Кроме
того, поглощение самого органического реагента очень ча-
сто меняется с изменением кислотности раствора, что сле-
22
дует учитывать при выборе оптимальной длины волны для
измерения поглощения комплекса.
Таким образом, использованию фотометрической реакции
для количественного определения элемента должно предше-
ствовать изучение ионного состояния компонентов, вступив-
ших в реакцию, определение их фотометрических характе-
ристик, выяснение оптимальных условий полноты образова-
ния комплексного соединения, а также предварительное изу-
чение кинетики реакции. Только после этого можно присту-
пить к выяснению приложимости основого закона светопо-
глощения к раствору, в котором находится определяемый эле-
мент, и к разработке условий количественного его опреде-
ления.
Если предполагается, что в реакцию вступает ион эле-
мента с органическим реагентом, являющимся одно-, двух- или
многоосновной кислотой, т. е. реакция протекает по типу за-
мещения протона кислоты ионом металла, то этапы указан-
ного исследования следующие.
Исследование органического реагента
1. Для фотометрического исследования реагента необхо-
димо снять спектр поглощения его водного раствора или рас-
твора в неводном растворителе, если таковой применяется
для экстракции исследуемого соединения (D = f(%) ). В по-
следнем случае необходимо определить значение коэффи-
циента распределения и выяснить зависимость этой величины
от значения pH раствора (q = f (pH) ). Также нужно выяс-
нить зависимость светопоглощения раствора реагента от его
кислотности при выбранной длине волны (D = /(pH)) и рас-
считать значения молярного коэффициента погашения е реа-
гента в молекулярной форме (НА) — кислая область и пол-
ностью диссоциированвдй (А-)—щелочная область.
Для недиссоциированной и диссоциированной форм реа-
гента значение е можно считать практически истинным, если
отсутствует поглощение посторонних веществ (например, ком-
понентов буферного раствора).
2. Для растворд реагента в неводном растворителе необ-
ходимо определить, не наблюдается ли в растворе явление
полимеризации. Для этого снимают спектры поглощения рас-
творов реагента различной концентрации и рассчитывают
значение е. Постоянство значения этой величины указывает
на отсутствие явления полимеризации в интервале исследо-
ванных концентраций.
3. Результаты фотометрического исследования реагента
можно использовать для изучения кислотно-основных свойств
органического реагента (стр. 45). Если имеется различие в
поглощении диссоциированной и недиссоциированной форм
23
реагента, то для определения константы его кислотной дис-
социации используют метод «изобестических точек» (стр. 47)
или метод Н. П. Комаря (стр. 49).
4. Если предполагается применить еще какие-либо вспо-
могательные реагенты, то исследуют поглощение их раство-
ров в условиях проведения реакции.
Исследование растворов определяемого элемента
1. В объеме учебного курса практикума по спектрофото-
метрии нет возможности изучать ионное состояние элемента,
но необходимые данные по гидролизу для большого числа
элементов можно найти в соответствующих справочниках1.
2. Для водного раствора соли определяемого элемента
также следует снять спектр поглощения и рассчитать значе-
ние е.
Всеми этими данными необходимо располагать, чтобы ре-
шить более сложную задачу определения состава образующе-
гося комплексного соединения и его устойчивости с примене-
нием фотометрических методов.
Исследование комплексного соединения
Для выяснения оптимальных условий образования ком-
плексного соединения и получения ориентировочных данных
по его устойчивости важно предварительно выяснить влияние
разбавления, величины pH, избытка реактива, времени, тем-
пературы, а также последовательность добавления реагентов
при приготовлении фотометрируемого раствора. Растворы
должны обладать устойчивой и воспроизводимой окраской.
При изучении фотометрической реакции также следует нахо-
дить предел для определения минимальных и максимальных
концентраций определяемого вещества. Это исследование ре-
комендуется проводить в таком порядке.
1. Для выяснения области максимального поглощения
раствора соединения снимают спектр поглощения его, ис-
пользуя имеющиеся приборы (ФТ, ФЭК-Н-52, ФЭК-Н-54,
ФЭК-Н-57, ФЭК-56 или СФ-5 и СФ-4), и строят график за-
висимости оптической плотности D или пропускания Т от
длины волны X (рис. 4), используя в качестве «нулевого»
раствора растворитель. Таким образом, определяется область
максимального поглощения раствора комплексного соедине-
ния, которая в дальнейшем используется при исследовании
этого соединения и количественном определении искомого
элемента.
1 J. Bjerrum, G. Schwarzenbach, L. Sielen. Stability con-
ctants. London, 1957; A. E. Martell, M. Calvin. Chemistry of the me-
tal chelate compounds. New York, 1953.
.24
2. Далее приступают к выяснению оптимальных условий
образования комплексного соединения.
а. Если комплексное соединение устойчиво, то при р а з-
бавлении раствора и увеличении толщины поглощающего-
слоя в п раз значение D не изменится. Например, если для
измерения оптической плотности испытуемого раствора была
использована кювета с 1 — 2 см, а при разбавлении вдвое —
с / = 4 см и при более резком изменении объема V и тол-
щины слоя I значение D практически остается постоянным,
то в этом случае можно говорить об относительной устойчи-
вости комплексного соединения. Однако при изучении влия-
ния разбавления нужно предвидеть, не наступит ли гидро-
лиз данного соединения. В этом случае для поддержания
постоянного значения pH разбавление проводят буферным
раствором.
б. Для выяснения влияния избытка реагента
на полноту образования комплексного соединения необхо-
димо построить график в координатах D——. Более пра-
R
вильно в таких исследованиях брать хлорнокислые соли эле-
ментов, так как будет отсутствовать постороннее комплексо-
образование и, кроме того, соли перхлоратов хорошо диссо-
циируют. При образовании устойчивого комплексного соеди-
нения наблюдается резкий излом (рис. 10), который опреде-
ляет необходимый избыток реагента, и вид кривой а позво-
ляет сделать заключение о соотношении компонентов в ком-
плексном соединении. Получение кривой б говорит о более
сложном процессе комплексообразования или о получении
малоустойчивого соединения.
в. На полноту образования комплексного соединения су-
щественно влияет значение pH раствора. Влияние pH мо-
жет сказываться различно.
Если в реакции, в качестве лиганда участвуют анионы
слабой органической кислоты, то от величины pH раствора
будет зависеть концентрация лиганда.
Малоустойчивые комплексные соединения при увеличении
значения pH раствора разрушаются или меняют состав
вследствие гидролиза иона-комплексообразователя. Поэтому
необходимо построить график D = f (pH) (рис. 11). В интер-
вале а—б значений pH комплексное соединение образуется
в максимальной степени. При этом следует использовать най-
денную концентрацию реагента (рис. 10, кривая а). Если
комплексное соединение извлекается неводным растворите-
лем, то необходимо определить интервал значений pH макси-
мальной экстракции соединения (стр. 71).
г. Необходимо также исследовать зависимость светопо-
глощения раствора комплексного соединения от времени и
построить график D = f (/). Фотометрирование испытуемого
25.
раствора следует проводить в течение промежутка времени,
соответствующего горизонтальному участку кривой.
3. Выяснив оптимальные условия образования комплекс-
ного соединения, устанавливают область выполнения основ-
ного закона светопогло-
Рис. 10. Влияние избытка реагента
на полноту образования комплекс-
ного соединения
0 1 2 За Ч 5 Б 7 8 9 10 1112613 pH
Рис. 11. Зависимость полноты осаждения
гептоксимата никеля от pH раствора
щёния.
а. Приготовив ряд эта-
лонных растворов, содер-
жащих определенное ко-
личество элемента, сни-
мают спектры поглощения
растворов с различной
концентрацией определяе-
мого элемента и строят
графики зависимости ве-
личин D и Т от длины вол-
ны X. Если на кривых по-
глощения наблюдается со-
хранение положения ма-
ксимума при одной и той
же длине волны (рис. 4),
то это свидетельствует о
выполнении основного за-
кона светопоглощения.
б. Затем рассчитывают
значения молярного коэф-
фициента погашения по
полученным данным (для ряда растворов с различной кон-
центрацией) и строят график в координатах е—к.
Такие графики дают возможность сравнивать спектры
поглощения независимо от концентрации растворов. Если
растворы подчиняются основному закону светопоглоще-
ния, то значение молярного коэффициента погашения
остается постоянным и на графике получается одна
кривая (рис. 5). Когда известны спектры поглощения реа-
гента и исследуемого комплексного соединения, очень полезно
учитывать величину ДХ, т. е. разность в положении максиму-
мов поглощения указанных веществ, а также разность в значе-
ниях молярных коэффициентов погашения Де при выборе
наиболее выгодной длины волньи для количественного опреде-
ления. Большое значение величин ДА. и Де повышает ценность
рассматриваемой реакции. Особенно важно учитывать эти
величины при сравнительном исследовании новых реагентов.
Преимущество следует отдать реагентам с большим значе-
нием в ДА и Де ’.
в. Находят зависимость оптической плотности D и пропу-
1 А. К. Б а б к о, П. П. К и ш. ЖАХ, 17, 692, 1962.
26
скания Т от концентрации, откладывая по оси ординат значе-
ния этих величин, взятые в области максимума поглощения.
Если при построении графиков D — с (рис. 3) наблю-
дается прямо пропорциональная зависимость оптической
плотности от концентрации, то растворы подчиняются закону
Бугера — Ламберта — Бера.
г. Если растворы не подчиняются общему основному за-
кону светопоглощения, то вначале проверяют подчиненность
растворов закону Бугера — Ламберта, устанавливая зависи-
мость величин D и 1g Т от толщины слоя раствора. Для это-
го окрашенный раствор наливают в кюветы различного диа-
метра и, определив значение D или lg Т, строят график зави-
симости D и 1g Т от / — толщины слоя раствора. Получение
прямолинейного графика, показывающего соблюдение закона
Бугера — Ламберта для данного раствора, позволяет изме-
рять величину D для растворов с различной интенсивностью
окраски, используя кюветы различного диаметра. Найденная
прямолинейная зависимость между D и / дает возможность
полученные после пересчёта данные нанести на один график
в координатах D — с (стр. 39).
Если было установлено, что закон Бугера — Ламберта вы-
полним, то необходимо изучить причины, вызывающие от-
клонение от закона Бера (стр. 16).
Для измерения светопоглощения растворов комплексных
соединений большое значение имеет приготовление «нуле-
вого» раствора. Если ранее было выяснено, что в области
максимального поглощения комплексного соединения имеет-
ся поглощение других веществ, участвующих в реакции, то
их необходимо вводить в «нулевой» раствор. В случае боль-
шого поглощения реагента следует рекомендовать в дальней-
шем дифференциальный метод (стр. 70).
Обычно при экстракции комплексного соединения реагент
также переходит в слой органического растворителя и, имея
собственное поглощение, не дает возможность с достаточной
точностью выяснить поглощение комплексного соединения.
В этом случае важно определить различие в зависимости
коэффициентов распределения реагента и исследуемого со-
единения от значений pH (стр. 72).
Спектрофотометрический метод может быть использован
для более детального изучения комплексных соединений. Ес-
ли предварительно известны данные по ионному состоянию
элемента, то состав комплексного соединения можно опреде-
лить по методу изомолярных серий (стр. 51). В данном руко-
водстве не рассматриваются более сложные 'Случаи ступенча-
того комплексообразования. В общем случае указанный ме-
тод применим для определения стехиометрических коэффи-
циентов в уравнении реакции образования комплексного со-
единения.
27
Для определения констант устойчивости комплексных со-
единений также существует ряд фотометрических методов.
Один из них рассматривается на стр. 53.
4. Фотометрическую реакцию всегда характеризует ее
чувствительность. Чувствительность обычно выражают в зна-
чениях молярного коэффициентна погашения (стр. 15) или в
мкг определяемого элемента. Если при определении исполь-
зован монохроматический поток лучистой энергии и соблю-
дены другие условия (стр. 13), то получают истинное значе-
ние молярного коэффициента погашения. Для характери-
стики чувствительности реакции можно использовать и зна-
чение е (среднее), которое получают, используя фильтровые
приборы (ФЭК-Н-57, ФТ, ФЭК-56), но не ФЭК-М, так как све-
тофильтры этого прибора имеют очень широкие полосы про-
пускания (100—120 ммк). В целях единого подхода к оценке
чувствительности реакции всегда необходимо указывать ус-
ловия определения молярного коэффициента погашения: для
спектрофотометров — ширину щели, для фильтровых прибо-
ров — их марку и лЭф светофильтра.
Чувствительность фотометрической реакции в мкг опреде-
ляют следующим образом. Бланк 1 для характеристики опре-
деляемого минимума элемента предлагает использовать фор-
мулу
т = -^-иЛ-103, (19)
е
где D — наименьшее допустимое значение оптической плот-
ности; п — число атомов элемента, входящих в молекулу по-
глощающего соединения; А — атомный вес определяемого
элемента; q — диаметр кюветы в см.
Е. Сендел2 дает открываемое минимальное количество
элемента в мкг, обнаруживаемое в слое раствора, имеющем
поперечное сечение, равное единице (1 см2), отнесенное к
0 = 0,001. Для сравнительной оценки фотометрических реак-
ций, рекомендуемых для определения одного элемента, этот
прием удобен.
5. ТОЧНОСТЬ СПЕКТРОФОТОМЕТРИЧЕСКОГО
МЕТОДА
Обычный метод построения градуировочного графика в
координатах D — с, удобен тем, что дает возможность уста-
новить область выполнения основного закона светопоглоще-
ния, но не позволяет, выбрать интервал значений D, в кото-
1 Методы анализа веществ особой чистоты и монокристаллов. «Мето-
ды определения следов элементов». Сб. Харьков, ВНИИмонокристаллов,
1962; А. Б. Б л а н к. ЖАХ, 17, 1040, 1962.
2 Е. Сендел. Колориметрические методы определения следов метал-
лов. М., «Мир», 1964, стр. 472.
28
ром можно измерить данную величину с достаточной точ-
ностью.
Из самого смысла величины D = lg-2- следует, что в об-
Л
щем случае эта величина может принимать значения от О
до со. Зависимость оптической плотности D от интенсивности
потока лучистой энергии Л, прошедшего через испытуемый
раствор, имеет вид, показанный на рис. 12. Из этого рисунка
следует, что область плотностей, имеющих практическое зна-
чение, очень ограничена. Как показывают расчеты наимень-
шая ошибка получается при измерении
D = —— = 0,4343, (20)
lge10 7
а оптимальный интервал оптических плотностей лежит в пре-
делах значений D от 0,2 до 0,8. Такой же вывод можно сде-
лать, если рассмотреть зависимость относительной ошибки от
величины пропускания Т
(предполагается, что при
фотометрическом измере-
нии допускается ошибка
в 1 %). Как видно из
рис. 13, наименьшая ошиб-
ка соответствует значе-
нию Т=36,8% и возрас-
тает незначительно в ин-
тервале пропусканий от 15
до 65% (что соответст-
вует интервалу оптиче-
ских плотностей от 0,8
до 0,2). Более наглядное
представление о том, ка-
Рис. 12. Зависимость оптической плот-
ности D от интенсивности потока лучис-
той энергии, прошедшего через погло-
щающий раствор
кова будет ошибка в оп-
ределении данной концен-
трации при минимальной
ошибке в измерении Т на
данном приборе, дает по-
строение кривой зависимости Т (%) — с (%) (рис. 14).
Таким образом, ограничение интервала значений D, Т,
в котором можно измерить эти величины с достаточной точ-
ностью, вызывает в свою очередь ограничение интервала
концентраций, которые могут определяться фотометрическим
методом. Поэтому спектрофотометрический метод нельзя ис-
пользовать для определения больших концентраций веществ,
хотя его применение в значительной степени ускорило бы
проведение анализа. В связи с этим в последнее время полу-
' Г. В. Юинг. Инструментальные методы анализа. М., Госатомиздат,
1963, стр. 32; С. Н i s к с е у. «Anal. Chem.», 21, 1440, 1949.
29
чает все более широкое развитие дифференциальный
спектрофотометрический метод, в котором в каче-
стве «нулевого» раствора вместо растворителя (как это име-
ет место в обычном колориметрическом методе) используется
один из растворов эталонного ряда1. В этом случае расши-
Рис. 13. Зависимость отиоситель- Рис. 14. Зависимость пропускания от
иой ошибки от пропускания рас- концеитрации раствора
твора
ряется область значений D, Т, измеряемых с допустимой
ошибкой, так как оптимальное значение/),измеряемое с наи-
меньшей ошибкой, в данном случае определяется соотноше-
нием
£)опт = 0,43 — £)0, (21)
где Do — оптическая плотность «нулевого» раствора, изме-
ренная по отношению к растворителю. Если Do 0,43, то
Dom = 0 во всех случаях.
Однако,как показали расчеты2, дифференциальный спек-
трофотометрический метод не только расширяет область
концентраций, доступных спектрофотометрическим определе-
ниям, но и повышает точность измерений. При этом было
установлено, что отношение интенсивностей двух потоков лу-
чистой энергии h и /ь прошедших через раствор с большей
концентрацией с2 и через раствор с меньшей концентрацией
’ R. Bastian. «Anal. Chem.», 21, 972, 1949; 22, 160, 1950; 23, 580,
1951; 25, 259, 1953; G. Kortum. «Angew. Chem.», 50, 193, 1937; С. H i s-
k e y. «Anal. Chem.», 21, 1440, 1949; C. Hiskey, J. Rabinowitz,
I. Young. «Anal. Chem.», 22, 1464, 1950; C. Hiskey, I. Young. «Anal.
Chem.», 23, 1196, 1951; C. Hiskey, D. Firestone. «Anal. Chem.», 24,
342, 1952.
2 C. Hiskey. «Anal. Chem.», 21, 1440, 1949.
30
Cj, зависит от оптической плотности раствора сравнения («ну-
левого» раствора) — Dt.
А. = Ю-е/с,(а—1) _ JQ—ОДа—1)
11
где
a = — = С1+ Ас .
С1 С1
(22)
(23)
Из рис. 15 видно, что с возрастанием Di можно опреде-
лять меньшие величины а, так как при одном и том же зна-
чении а больше будет чувствоваться различие в интенсивно-
стях двух потоков лучистой энергии /г и Л (отношение — -
11
уменьшается). Следователь-
но, в работе целесообразно
использовать «нулевой» рас-
твор, имеющий возможно
большее собственное погло-
щение. Однако это не всег-
да можно осуществить на
практике, так как использо-
вание раствора сравнения с
большим поглощением тре-
бует либо увеличить интен-
сивность источника освеще-
ния, либо повысить чувстви-
тельность приемника—фото-
элемента и его усилительной
схемы, что не всегда возмож-
но сделать при использова-
нии в работе приборов опре-
деленных конструкций. По
этим причинам упрощенные
визуальные спектрофотомет-
Рис. 15. Зависимость отношения “Г“
'I
от оптической плотности «нулевого»
с2
раствора при различных а= —
ры типа универсального фотометра использовать для диффе-
ренциальных измерений невозможно. При работе на спектро-
фотометрах СФ-4, СФ-5 интенсивность освещения можно уве-
личить за счет увеличения ширины щели. Это, однако, может
привести к значительным отклонениям от основного закона
светопоглощения.
Хиски и Юнг1 показали, что в случае невыполнения ос-
новного закона светопоглощения ошибка при дифферен-
циальных измерениях будет минимальной, когда в качестве
«нулевого» используется раствор, для которого величина про-
изведения e'Ct имеет наибольшее значение; в' = (| —есть
\ de J
1 С. Н i s к е у, J. Y о u n g. «Anal, Chem.», 23, 1196, 1951.
31
Рнс. 16. Выбор оптимальной
концентрации «нулевого» рас-
твора при дифференциальных
измерениях
тангенс угла наклона касательной к кривой зависимости
D от с в точке, соответствующей концентрации Ci (рис. 16).
Как следует выбирать на практике оптимальную концен-
трацию для «нулевого» раствора и находить интервал кон-
центраций, в котором можно производить дифференциальные
измерения, будет описано в специальной главе (стр. 66).
Помимо работ Хиски в последнее время появились ра-
боты1, в которых также рассматриваются вопросы точности
спектрофотометрического мето-
да: теоретически и эксперимен-
тально изучается зависимость
точности спектрофотометриче-
ских измерений от используе-
мой аппаратуры, невоспроизво-
димое™ установки кювет в
приборе, пропускаемости изме-
ряемых растворов и т. п.; при
этом обычный спектрофотомет-
рический метод рассматривает-
ся как частный случай диффе-
ренциального спектрофотомет-
рического метода. В данном
случае рассмотрены вопросы
точности спектрофотометриче-
ского метода, связанные толь-
ко с инструментальными ошиб-
ками. Безусловно, не меньшее
значение имеют ошибки, свя-
занные с условиями протека-
ния химических реакций в конкретных условиях проведения
данного фотометрического определения. Поэтому решить воп-
рос о точности какого-либо метода спектрофотометрического
определения можно лишь на основании статистической обра-
ботки целой серии результатов, полученных в конкретных усло-
виях проведения данного определения 2.
1 Г. С. Т е р е ш и н. ЖАХ, 13, 388, 1959; 14, 516, 1960; Н. П. К о м а р ь,
В, П. Самойлов. ЖАХ, XVIII, 1284, 1963; G. S v е h 1 а, А. Р а 11,
L. Е г d е у. «Taianta», 10, 719, 1963.
2 В. В. Налимов. Применение математической статистики при ана-
лизе вещества. М., Физматгиз, 1960; Н. П. К о м а р ь. ЖАХ, 7, 925, 1952.
II. МЕТОДЫ ОПРЕДЕЛЕНИЯ И РАСЧЕТА
КОНЦЕНТРАЦИИ ВЕЩЕСТВ В РАСТВОРАХ
Фотометрические методы определения концентрации ве-
ществ в растворах основаны на измерении ослабления интен-
сивности потока лучистой энергии, прошедшего через «окра-
шенный» раствор. Ввиду трудности измерения абсолютной
интенсивности потока лучис-той энергии во всех фотометриче-
ских методах, как уже отмечалось (стр. 10),используется от-
носительное измерение интенсивности потока лучистой
энергии, прошедшего через исследуемый раствор, т. е. срав-
нивают его с потоком лучистой энергии, прошедшим через
раствор, выбранный в данном случае за эталон.
Все фотометрические методы можно разделить на две
группы: визуальные и фотоэлектроколориметрические. Ме-
тоды, в которых интенсивность потока лучистой энергии оце-
нивается с помощью глаза, называются визуальными. В фо-
тоэлектроколориметрических методах в качестве приемника и
анализатора лучистой энергии используют фотоэлемент, си-
ла фототока в котором согласно законам фотоэффекта про-
порциональна интенсивности падающего на него света. По-
ток лучистой энергии, прошедший через поглощающий рас-
твор, попадает на фотоэлемент, который превращает лучистую
энергию в электрическую. Сила тока, возникающего при
этом, измеряется с помощью гальванометра.
Фотоэлектроколориметрические методы обладают некото-
рыми преимуществами по сравнению с визуальными.
1. Глаз наблюдателя не утомляется при проведении боль-
шого числа измерений, например, при массовых анализах.
2. В зависимости от природы фотоэлемента, материала,
из которого изготовлены оптические детали прибора, и каче-
ства кювет фотоэлектроколориметрические методы можно
использовать для определения интенсивности «окраски» рас-
творов не только в видимой, но и в других областях спектра.
Например, в приборах ФЭК-56 имеются кюветы из стекла,
пропускающего поток лучистой энергии с длиной волны от
326 ммк, и сурьмяно-цезиевые фотоэлементы, чувствительные
к ультрафиолетовым излучениям.
2 Зак, 686
33
3. Применение визуальных методов, даже в случае исполь-
зования фотометров, ограничено способностью глаза улавли-
вать незначительные различия интенсивности освещения со-
седних участков фотометрического поля. Фотоэлектроколори-
метрические методы позволяют это ограничение снижать на
несколько порядков.
Для сравнения интенсивности двух потоков лучистой энер-
гии необходимо добиться, чтобы первоначальная их интен-
сивность (до прохождения одного из них через поглощающее
вещество) была одинакова. В различных фотометрических
методах эта цель достигается по-разному.
В методах, не требующих использования каких-либо при-
боров (разбавления, колориметрического титрования, стан-
дартных серий), задача сводится к подбору пробирок и ци-
линдров по диаметру и пропускаемости. Подбор пробирок по
диаметру производят путем наполнения серии пробирок оди-
наковым объемом жидкости и сравнения уровня заполнения
их при этом. Подбор по пропускаемости производят при за-
полнении пробирок одинакового диаметра одним и тем же
окрашенным раствором. Если при этом интенсивность наблю-
даемой окраски раствора во всех пробирках (цилиндрах)
одинакова, то их можно использовать для количественных
определений. В приборах одинаковая интенсивность первона-
чальных потоков лучистой энергии достигается прежде всего
настройкой осветителя. Кроме того, кюветы также должны
быть одинаковыми по пропускаемости.
Рассмотрим ряд фотометрических методов, отличающихся
в принципе путями сравнения (уравнивания) интенсивностей
потоков лучистой энергии, прошедших через эталонный и ис-
пытуемый растворы. В каждом из этих методов существует
определенный путь расчета концентрации испытуемого рас-
твора. В зависимости от того, используется ли в этих расче-
тах формула математического выражения основного закона
светопоглощения, можно сделать вывод, требуется ли стро-
гое выполнение этого закона при использовании на практике
данного метода.
Рассмотрим визуальные фотометрические методы.
Метод разбавления. В двух одинаковых градуи-
рованных колориметрических цилиндрах в идентичных усло-
виях готовят два раствора — один испытуемый, второй эта-
лонный с известной концентрацией определяемого элемента.
Раствор, окрашенный более интенсивно, разбавляют до тех
пор, пока окраски испытуемого и эталонного растворов не бу-
дут одинаковыми при наблюдении их по горизонтальному на-
правлению.
В случае выполнения основного закона светопоглощения
(s имеет постоянное значение) в момент уравнивания интен-
сивностей окрасок
34
— s/gCg, •— c2, — /3,
так как диаметр цилиндров одинаков и концентрация веще-
ства в 1 мл обоих растворов одинакова. Но
тогда
m, т2
~йГ - V
(24)
Следовательно, весовое содержание вещества прямо пропор-
ционально объему раствора.
Отмечают объем раствора в обоих цилиндрах и делают
расчет по формуле
тх __ Кг
mt Еа
где тх и т.2 — весовое количество вещества в испытуемом
и эталонном растворах; Vx и V2— объем испытуемого и эта-
лонного растворов.
Концентрация может быть выражена в весовых единицах
(л1г) или процентах.
Этот метод не отличается большой точностью и может
быть рекомендован для сравнения растворов, близких по
окраске. Колориметрические градуированные цилиндры мож-
но заменять колориметрическими пробирками, которые для
удобства наблюдения помещают в штатив.
Метод колориметрического титрования. Бе-
рут два одинаковых колориметрических цилиндра. В одном
из них проводят реакцию с испытуемым раствором, приготов-
ленным растворением навески анализируемого образца или
составляющим аликвотную часть исходного раствора. Во вто-
рой цилиндр добавляют те же количества всех реактивов, ко-
торые брали для приготовления испытуемого раствора; объем
растворов в обоих цилиндрах должен быть примерно одина-
ков. Во второй цилиндр из бюретки прибавляют стандартный
раствор, содержащий определяемый элемент, до получения
растворов в обоих цилиндрах с окраской одинаковой интен-
сивности. Если объемы растворов значительно отличаются,
их выравнивают разбавлением испытуемого раствора водой.
Содержимое второго цилиндра перемешивают встряхиванием.
Окраски растворов удобнее сравнивать в отраженном или
проходящем свете, используя в качестве экрана лист филь-
тровальной бумаги. Содержание определяемого компонента
находят по формуле
„ __ сСтЕст100
Сисп
а
2*
35
где Сисп—количество определяемого элемента в процентах;
Ест—объем стандартного раствора в миллилитрах; сст —
титр стандартного раствора; а — навеска или ее аликвотная
часть.
Совершенно очевидно, что в данном методе не обяза-
тельно строгое выполнение основного закона светопоглоще-
ния.
Метод стандартных серий. Берут ряд (8—10) спе-
циально подобранных колориметрических пробирок одинако-
вого диаметра и цвета стекла, с притертыми пробками и ста-
вят их в штатив. В одну из пробирок наливают испытуемый
раствор, во все остальные — стандартный, постепенно увели-
чивая концентрацию определяемого элемента; во всех про-
бирках выравнивают объем и добавляют одинаковое коли-
чество реактива. Проводят все операции, необходимые для
.получения окрашенного соединения, и наблюдают, сравнивая
интенсивность окраски испытуемого раствора с эталоном.
Если окраска испытуемого раствора занимает среднее поло-
жение между окраской двух растворов эталонного ряда, то
принимают за искомое среднее значение их концентрации или
готовят новый эталон с концентрацией, равной предполагае-
мой концентрации испытуемого раствора, и еще раз сравни-
вают интенсивность окрасок этих двух растворов в отражен-
ном или проходящем свете (в зависимости от условий: цвета,
интенсивности окраски). Наблюдение можно проводить в го-
ризонтальном или вертикальном направлении.
В отдельных случаях, если окраска получаемых соедине-
ний сохраняется длительное время не меняя своей интенсив-
ности, готовят постоянный ряд эталонов, запаивая пробирки;
это особенно удобно для полевых лабораторий. Иногда про-
бирки эталонного ряда можно заменить набором специально
приготовленных цветных стекол. Растворы эталонного ряда
более правильно готовить, увеличивая концентрацию опреде-
ляемого элемента в геометрической прогрессии, если приме-
няемый реактив бесцветен. В случае применения окрашен-
ного реактива, когда в испытуемом и эталонных растворах
наблюдается изменение окраски, большое внимание нужно
уделить подбору концентраций в ряду эталонных растворов;
иногда более подходящими являются условия, в которых кон-
центрация определяемого элемента увеличивается в арифме-
' тической прогрессии.
Метод стандартных серий очень распространен. Рацио-
нально использовать его в однотипных массовых анализах
для установления квалификации реактивов. В этом методе
так же, как в методе колориметрического титрования, не
обязательно строгое выполнение основного закона светопо-
глощения. Единственным требованием является идентичность
условий приготовления испытуемого и эталонных растворов.
36
Преимущество данного метода перед методом колориметри-
ческого титрования состоит в том, что могут быть использо-
ваны реакции, протекающие во времени.
Метод изменения толщины поглощающего
слоя, (Этот метод ранее носил не совсем правильное назва-
ние метода «уравнивания».)
Если имеются два раствора одного и того же соединения,
но взятого в разных концентрациях, то, изменяя толщину
слоя окрашенных растворов, находят положение, при кото-
ром (при наблюдении по вертикальному направлению) ин-
тенсивности окраски растворов будут одинаковы. Согласно
основному закону светопоглощения
D1 = &llc1 (1-й раствор),
О2 = s/,c2 (2-й раствор).
В момент уравнивания интенсивностей окраски растворов
= D2. Значения молярных коэффициентов погашения бу-
дут также равны, если эти растворы подчиняются основному
закону светопоглощения. Следовательно,
/1Д = /2с2, (27)
т. е. между концентрацией раствора и толщиной его слоя
(высотой столба окрашенного раствора) существует обратно
пропорциональная зависимость.
Концентрацию испытуемого раствора находят по формуле
(28)
1Х
где сх и С\ — концентрация веществ в испытуемом и эталон-
ном растворах; 1Х и 1\ — высоты столбов окрашенных испы-
туемого и эталонного растворов.
Описанный метод может быть применен в случае подчи-
нения растворов объединенному основному закону светопо-
глощения. В этом методе применяется колориметр погруже-
ния. В настоящее время наиболее распространенным являет-
ся концентрационный колориметр марки КОЛ-1М (стр. 74).
Метод диафрагмирования. В наиболее широко
применяемых в настоящее время приборах для уравнивания
интенсивности потоков лучистой энергии используются диаф-
рагмы с переменной величиной отверстия. Диафрагма соеди-
нена с барабаном, который имеет шкалу, проградуированную
в величинах оптической плотности D или процентов пропу-
скания Т. К такому типу приборов относятся как визуаль-
ные: универсальный и горизонтальный фотометры, — так
и фотоэлектрические: фотоэлектроколориметры ФЭК-М,
ФЭК-Н-52, ФЭК-Н-54, ФЭК-Н-57, ФЭК-56.
Принцип измерения путем диафрагмирования заключает-
ся в том, что вначале прибор настраивают на 0 при уста-
37
новке раствора сравнения на пути потока лучистой энергии,
а затем на пути того же потока лучистой энергии устанавли-
вают исследуемый раствор; изменение в его интенсивности,
вызванное поглощением раствора, компенсируют раскрытием
щели диафрагмы. По отсчетному барабану снимают показа-
ние величин D или Т. Более детально с принципом измерения
на том или ином приборе можно ознакомиться в разделе
«Аппаратура».
Помимо описанных выше в приборах различных конструк-
ций используются также другие пути уравнивания интенсив-
ности потоков лучистой энергииНапример, в микроколори-
метре КОЛ-52 это достигается с помощью оптических
клиньев.
В наиболее совершенных приборах, используемых в на-
стоящее время в фотометрических исследованиях, — спектро-
фотометрах, осуществляется как оптическая компенсация
(с помощью щели), так и электрическая (с помощью потен-
циометров) .
Если растворы подчиняются основному закону светопогло-
щения и имеются приборы, при помощи которых можно изме-
рять ослабление интенсивности потока лучистой энергии при
прохождении через окрашенный раствор, снимая показания
значений оптической плотности D или пропускания Т, то рас-
считывают концентрацию раствора, пользуясь следующими
методами.
1. Измеряя D или Т для ряда эталонных растворов с раз-
личной, но известной концентрацией определяемого элемента,
строят график в координатах 1g Г, D — с. Концентрацию ве-
щества в исследуемом растворе находят по калибровочному
графику (рис. 3) —графический метод.
2. Приготовив два эталонных раствора, которые содер-
жат окрашенное вещество в известной концентрации, и, изме-
рив для них значения D, находят концентрацию вещества в
испытуемом растворе, используя прямо пропорциональную
зависимость между D и с (стр. 14).
3. Если в испытуемом растворе имеется «посторонняя»
окраска, то определяют значение Dt без добавления в него
реактива. При измерении оптической плотности D испытуе-
мого раствора, когда в нем проведена химическая реакция,
вследствие которой образуется окрашенный раствор, из зна-
чения D вычитается значение Z?i.
Концентрацию рассчитывают по формуле
(О-Вх) = (29>
Вэт сэт
1 А. К. Б а б к о, А. Т. П и л и п е н к о. Колориметрический анализ.
М. — Л., Госхимиздат, 1951; Абсорбционная спектроскопия. Сб. статей. М.,
ИЛ, 1953, стр. 51.
38
Отклонения от закона Бера (непрямолинейная зависи-
мость D от с) встречаются чаще, чем от закона Бугера —
Ламберта.
Определение концентрации графическим методом (по гра-
дуировочному графику, стр. 38, пункт 1) в принципе возмож-
ет закона Бера (если интенсив-
но и в случае отклонении
ность окраски растворов
"воспроизводима). Однако
в этом случае для точного
хода градуировочного гра-
фика необходимо приго-
товить большее число эта-
лонных растворов. Кроме
того, как видно из рис. 17,
прямолинейный график
повышает точность опре-
деления (пропорциональ-
ные изменения величин
оптических плотностей со-
ответствуют пропорцио-
нальным изменениям кон-
центраций растворов —
£1, С2, Сз в случае прямо-
линейного графика и не-
пропорциональным — с,
£2, с3 в случае криволи-
Рис. 17. Повышение точности определе-
ния концентрации раствора при прямо
линейной зависимости D от с
нейного).
Кюветы подбирают так, чтобы измеряемые оптиче-
ские плотности укладывались в интервал значений от 0,1 до
1,0 (о точности измерений величин D см. стр. 28). Если рас-
творы подчиняются закону Бугера — Ламберта, то выбор кю-
веты облегчается; например, если кювета с диаметром в 2 см
непригодна для измерений интенсивности окраски начальных
и конечных растворов эталонного ряда, то можно, измерив
оптические плотности с кюветой большего или меньшего диа-
метра, пересчитать на оптические плотности, соответ-
ствующие кювете в 2 см, используя прямую пропорциональ-
ность между D и I, и нанести их на один и тот же градуиро-
вочный график.
III. ПРИМЕРЫ ИСПОЛЬЗОВАНИЯ
СПЕКТРОФОТОМЕТРИЧЕСКОГО МЕТОДА
1. ВЫЧИСЛЕНИЕ ИСТИННЫХ ЗНАЧЕНИЙ
МОЛЯРНЫХ КОЭФФИЦИЕНТОВ ПОГАШЕНИЯ
РАСТВОРОВ КОМПЛЕКСНЫХ СОЕДИНЕНИЙ
Значение величины молярного коэффициента погашения,
вычисленного по формуле (14)
пХ
Cl
не всегда соответствует истинному его значению (стр. 13).
Для растворов, в которых имеют место процессы комплек-
сообразования, полимеризации, диссоциации и т. п., закон
светопоглощения, как уже отмечалось на стр. 14, выполним
лишь в том случае, если величины молярных коэффициентов
погашения всех компонентов раствора одинаковы. В против-
ном случае, наблюдаются отклонения от основного закона
светопоглощения и величины средних молярных коэффициен-
тов погашения, вычисленные по формуле (14), будут раз-
личны при переменных условиях. Подобные случаи ослож-
няют количественные определения, но очень удобны для изу-
чения процессов, протекающих в растворах.
Рассмотрим некоторые методы вычисления истинных ве-
личин молярных коэффициентов погашения.
1. Метод, предложенный Н. П. Комарем требует предва-
рительного определения состава комплексного соединения и
написания уравнения реакции его образования.
Пусть реакция протекает по уравнению
В"+ + <?НА ВА<"“")+ + 9Н+.
Проводят ряд опытов, которые отличаются исходными
концентрациями реагирующих компонентов, при необходимых
условиях:
а) начальные концентрации1 2 реагирующих компонентов
Сна и св во всех опытах должны сохранять постоянное соот-
ношение, т. е.
Сна = <?св; (30)
1 Н. П. Ко марь. «Уч. зап. Харьковск. ун-та», XXXVII (8), 57, 1951.
2 Для простоты заряды ионов опущены.
40
б) величины Т, pH, I и Л во всех опытах должны быть
постоянны.
Рассмотрим случай, когда окрашенными (т. е. обладают
поглощением в исследуемой области длин волн) являются
используемый реагент и образующийся комплекс.
Введем обозначения:
Св = с;
£на = qc\
[BAf] =- х;
[НА] = qc — qx = q(c — х);
[Н] = hx (используется буферный раствор);
ёна и еВлд — молярные коэффициенты погашения
реагента и комплекса соответственно.
[В] — с — х;
Для опыта i по закону действующих масс
______________________________Xjhq______
{с^ — xt)[q(Ci~ Xi)]4
откуда
X, = (с. — x.)'?+’.
r \ i if
Согласно основному закону светопоглощения
= q (ct. — ёна/ + хгЁВА9/,
откуда
Di qe^pCtl
1 (eBAq ~ ^Ha)
Объединяя формулы (32) и (34), получим
О,- — qe^Cjl (_q\q % Г c‘eBAgZ~-0» р+1
1 (8BAg ~?8нд) V А / I (еВА;?—<78на) J
Аналогичное уравнение получим для опыта k. Деля
этих уравнений на другое, получим
1
с‘1еВАд ~ Di _ (Dj— qenAlCj) 9+‘
ckl^BAa~Dk ‘
q ' (Dr—q^UA.lck)q+
В правую часть этого уравнения входят величины, которые
известны или могут быть определены на опыте, поэтому для
простоты дальнейших преобразований обозначим это выра-
жение через В.
(31)
(32)
(33)
(34)
(35)
одно из
(36)
41
Исходные концентрации комплексообразователя в опытах
i и k можно взять в соотношении с,- = пс^. Тогда, решая урав-
нение (36) относительно вва^, получим
8вА^ —
п (Dj — BDk)
lct (п — В)
(37>
Вычисленное таким путем значение £ва? дает возможность
вычислить Xi по уравнению (34), если предварительно было
найдено енд. Следовательно, по уравнению (31) можно найти
и величину константы равновесия реакции образования ком-
3,2 -
"l 28-
2,6-
Рис. 18. Зависимость величины
молярного коэффициента пога-
шения от концентрации при раз-
личных длинах волн в случае не-
выполнения основного закона све-
топоглощения
плексного соединения.
2. В методе, предложенном
Юнг-пен-Тонгом и Кингом1
при изучении явлений полиме-
ризации в растворах 6-валент-
ного хрома, рассматривается
равновесие
2НСгО4 СгаО?~ + НаО.
Исследование проводят при
такой кислотности, когда в за-
метных количествах существу-
ют только ионы НСгО Г и:
СГ2О7 , поэтому равновесие с
[Сг2Оу~]
[HCrOJT
зависит от с и не зависит от [Н+].
В области длин волн 380—400 ммк закон светопоглоще-
ния не выполняется. Это видно помимо графика обычного
спектра из кривых зависимости 1g е от 1g с (рис. 18). Удобнее
всего проводить исследование в области 390—400 ммк. Сред-
ний молярный коэффициент погашения равен:
8 = 61(1 — у) + еаг/, (39>
где Bi — молярный коэффициент погашения НСгО4;
е2— молярный коэффициент погашения Сг2О7~’;
у —доля Cr (VI) в димерной форме.
Соотношение между К, с и у выражается уравнением
2/<с =----у---. (40).
(1 - У)2
Таким образом, имеем два уравнения с тремя неизвестны-
ми— К, 8], 82, поэтому однозначно решить их невозможно.
В этом случае используется метод последовательных прибли-
1 J. Ying-pen-Tong, Е. King. «J. Am. Chem. Soc.», 75, 6180, 1953.
42
жений с предварительным предположением одной из неизве-
стных величин. Предположение делается относительно К, ко-
торой задают несколько значений и на основании этого вы-
числяют у. Для удобства расчетов вычисляют таблицу функ-
ций
F (у) =------- (41)
w (1-у)2
в интервале значений от 0,600 до 0,001. С помощью этой таб-
лицы можно получить путем интерполяции значение у, соот-
ветствующее любому F (у) (т. е. 2/<с).
Уравнения для е, число которых определяется числом рас-
творов с различной концентрацией, решаются по методу
усреднений. Мерой точности в определении величин К, ej, ег
является среднее процентное отклонение вычисленной по
уравнению величины е от наблюдаемой е в опыте.
3. В методе, предложенном Циленом и Конником* 1 2 для
вычисления молярного коэффициента погашения комплекса
циркония с теноилтрифтор ацетоном (ТТА), используется
константа распределения этого комплекса между двумя рас-
творителями.
Мак Ви2 установил, что цирконий образует с ТТА ком-
плекс в соотношении 1 : 1 (при условии, когда отсутствуют
явления гидролиза и полимеризации) по уравнению реакции:
Zr4+ + НК ZrK3+ + Н+
Комплекс имеет максимум светопоглощения при Z. 336 лшк.
Для вычисления молярного коэффициента погашения дан-
ного комплексного соединения при этой длине волны Цилен
и Конник использовали две серии растворов:
1. Водные растворы. Серия состоит из нескольких (на-
пример, шести) пар растворов и в каждой паре концентра-
ция ТТА одинакова, но цирконий содержит только один из
растворов.
2. Водно-бензольные растворы. Серия включает двухфаз-
ную систему вода — бензол и состоит из таких же пар рас-
творов, как и водная серия, но водная фаза во всех раство-
рах приведена в равновесие с равным объемом бензольной
фазы.
Предполагается, что поглощает только ТТА и комплекс
2’гК3+
Тогда разность оптических плотностей двух растворов
одной и той же пары водной серии будет равна
lZrK3+], (42)
1 A. Z i е 1 е n, R. С о n n i k. «J. Am. Chem.. Soc.», 78, 5786, 1956.
2 W. Н. Me Vey. United States Atomic Energy Commission HW-21487.
June, 1951.
43
так как концентрация циркония намного больше концентра-
ции реактива (ТТА) и, следовательно, [ZrK3+] = [НК]исходн-
Такое уравнение можно написать для каждой пары раство-
ров.
Кроме того, для всех шести пар растворов отношение АТ>
при К 366 ммк к Д£> при любой другой Z. будет величиной по-
стоянной:
дпЗбб „366 __„366
®ZrK3+ ®НК _ „}.
— .К „Л. Л1'
8ZrK3+ НК
В серии бензол — вода, учитывая зависимость
,, [НК]б енз
р_ [НК]Нг0 ’
где Р — константа распределения реагента НК, для
фазы получают следующее соотношение:
И66 = (Р+1)С6кз+-46£ = ,
ДО2Л (3+ 1)<4кз+-е*к 2’
К соотношению (45) приходят в результате следующих рас-
суждений.
Так как [Zr4+] )§> [ТТА], то в водной фазе
= eZrK3_)_/cZrK3_|„
(43)
(44)
водной
(45)
и
D’2 = 8нк/С[НК]нгО,
но
[ню исходи [НК]н2о+[НК]бе„=.
Учитывая зависимость (44), можно записать
[НК]Исходн = [НК]н2о + [НК]н2о? = [НК]н2о(1 + й»
откуда
rtJTZl [НК]иСХОДИ
[НК1н-° ’ + Р) -
но
[НК]_ = [ZrK3+],
следовательно,
= D, - D2 = I [(1+P)^+8hLJ [ZrK3+]
44
и
= „л
л°2 <₽ + »’ггк«- - «1к
Совместным решением уравнений для Ki и К 2 получим
к366 3-!-
EZrK3
Таким образом, для вычисления s по этому методу нуж-
но знать коэффициент распределения реактива в системе бен-
зол— вода, состав комплекса и уравнение реакции его обра-
зования. Кроме того, берется большой избыток комплексооб-
разователя, чтобы весь реактив перевести в комплекс. Ком-
плекс должен нести заряд, чтобы он не мог переходить в
слой органического растворителя. Метод непригоден для слу-
чая малоустойчивых комплексов.
2. ВЫЧИСЛЕНИЕ КОНСТАНТ ДИССОЦИАЦИИ
ОРГАНИЧЕСКИХ РЕАГЕНТОВ
Если максимумы поглощения недиссоциированной и дис-
социированной форм органического реагента, являющегося
слабой кислотой находятся в различных областях спектра, то,
изучая поглощение растворов этого реагента при различных
значениях pH, можно спектрофотометрически определить кон-
станту его диссоциации. Например, максимум светопоглоще-
ния недиссоциированной формы бензоилацетона находится
при К 250 ммк, а диссоциированной формы — Z320 ммк
(рис. 19) По мере увеличения значения pH раствора возра-
стает степень диссоциации реагента, т. е. концентрация его
недиссоциированной формы уменьшается, а диссоциирован-
ной увеличивается. Следовательно, поглощение растворов при
К 250 ммк будет уменьшаться, а при К 320 ммк — возрастать
до тех пор, пока не упадет до минимума и не достигнет мак-
симума соответственно в момент, когда реагент перейдет пол-
ностью в диссоциированную форму.
Если концентрация реагента, ионная сила и температура
растворов постоянны, то все кривые пересекутся в одной так
называемой «изобестической» точке (рис. 19, Z. — 270 ммк).
При длине волны, соответствующей этой точке, обе формы
реагента (недиссоциированная и диссоциированная) имеют
одинаковое поглощение и независимо от значения pH раство-
ры одной и той же концентрации данного реагента имеют по-
стоянное поглощение. Поэтому для определения концентра-
ций органических реагентов имеет смысл использовать имен-
но эту длину волны.
1 А. П. 3 о з у л я, Н. Н. М е з е и ц е в а, В. М. П е ш к о в а, Ю. К. Ю р ь-
ев. ЖАХ, XIV, вып. 1, 17, 1959.
45
Рассмотрим несколько методов вычисления констант дис-
социации органических реагентов, используемых в спектро-
фотометрическом методе. Во всех случаях имеется ряд об-
щих положений.
Рис. 19. Кривые спектров поглощения водных растворов бензоилацето-
на при различных значениях pH: 1 — 2,03—6,58; 2—7,8; 3—8,4; 4—8,9;
5 — 10,02—11,08
' Пусть органический реагент диссоциирует как однооснов-
ная кислота по уравнению
НА Н+ + А~,
тогда
[Н+] [А-]
[НА]
(46)
где К — константа диссоциации реагента. В общем случае по-
глощение раствора реагента, содержащего как недиссоцииро-
ванную, так и диссоциированную формы, при определенной
длине волны Z. равно (в уравнениях опущены заряды частиц):
46
DeM = ЁНА/С (!—*)+ EAlCX>
(47)
где Dcm — оптическая плотность раствора, содержащего смесь
А~ и НА; с — концентрация реагента, во всех опытах сохра-
няется постоянной; I — толщина поглощающего слоя раство-
ра; х = [А~] — доля диссоциированной формы; (1—х) =
='[НА] — доля недиссоциированной формы; е^А и —мо-
лярные коэффициенты погашения недиссоциированной и дис-
социированной форм реагента при длине волны Z..
Расчетный метод1. Подставив в выражение (46) ве-
личины с, х и (1—х), получим
[Н]сх = Кс(1— х). (48)
Решая это уравнение относительно х, находим
К
[Н] + К
Комбинируя (47) и (49), получим
£>см = енд/с (------+ Мс (
\ [Н] + к J А <
к
[Н] + К
(50)
где ёна^с = -Она и ёд/с = Па— оптические плотности раствора
реагента соответственно в недиссоциированной и диссоции-
рованной формах при длине волны К, концентрации с и тол-
щине поглощающего слоя I.
Решая уравнение (50) относительно К, получим
К =
Дем-Дна
Да —Дем
[Н].
(51)
Следовательно, для вычисления константы диссоциации
реагента данным методом необходимо определить Ина» Da
и Псм хотя бы при одном значении pH.
Чтобы определить константу диссоциации опытным путем,
готовят ряд растворов, сохраняя постоянной концентрацию
реагента и меняя значение pH в широких пределах. Снимают
спектры поглощения всех приготовленных растворов, исполь-
зуя кювету с постоянной толщиной, и строят кривые этих
спектров. На основании полученных графиков определяют
область длин волн, в которой наблюдаются наибольшие раз-
личия в величинах D растворов. Величины П^А и D\ опре-
деляют исходя из следующих рассуждений.
1 W. S t е п s t г о m, N. Goldsmith. «J. Phys. Chem.», 30, 1683,
1926; С. V. Banks, А. В. Carlson. «Analyt. Chim. Acta», 7, 291, 1952.
47
По мере возрастания кислотности растворов наступит та-
кой момент, когда весь реагент перейдет в недиссоциирован-
ную форму и при дальнейшем увеличении кислотности не
будет меняться концентрация этой формы реагента; следова-
тельно, с этого момента не будет также изменяться оптиче-
ская плотность растворов. Аналогично при уменьшении кис-
лотности наступает, момент, начиная с которого реагент в
растворе будет существовать только в диссоциированной
форме и дальнейшее увеличение pH раствора не вызовет из-
менения его оптической плотности. Величины оптической
плотности, достигшие постоянства по мере возрастания и
уменьшения кислотности раствора берутся как £>на и Da со-
ответственно. Эти величины измеряют при одной и той же
длине волны, которую выбирают в интервале длин волн, где
наблюдаются наибольшие изменения оптической плотности
-с изменением кислотности растворов. При этой же длине вол-
ны измеряют Da, раствора, имеющего концентрацию водо-
родных ионов [Н+], лежащую между значениями pH, которым
соответствуют величины Дна и D&- Все эти величины под-
ставляют в формулу (51) и вычисляют значение К-
Можно вычислить величину К на основании нескольких
значений DKCM и [Н+] и, статистически обработав результаты,
получить среднюю величину К-
Этот метод (его часто называют методом «изобестических
точек») может быть использован и для определения констант
устойчивости комплексных соединений ’.
Графический метод. Константа диссоциации орга-
нического реагента, функционирующего как кислота, может
быть найдена графически на основании изучения спектров
поглощения растворов этого реагента при различных значе-
ниях pH в условиях указанных в предыдущем разделе. В об-
ласти низких значений pH имеется интервал, в котором не
происходит диссоциации реагента, а в области высоких зна-
чений pH — интервал, в котором реагент существует практи-
чески в полностью диссоциированной форме. Кривая зависи-
мости оптической плотности (измеренной при той длине вол-
ны, где происходит наибольшее изменение оптической плот-
ности при диссоциации реагента) от значений pH растворов
дана на рис. 20. Участок крутого подъема ограничен двумя
горизонтальными прямолинейными участками, соответствую-
щими упомянутым выше интервалам значений pH. Пер-
пендикуляр, опущенный из середины участка крутого подъема
на ось абсцисс, дает численное значение обратного лога-
рифма константы диссоциации реагента. Это видно из
уравнения (51):
1 L. V а г е i 11 е. «Bull. Soc. Chim. France», п° 6, 870, 1955.
48
К =
Дем-Дна
Да-Д™
[Н],
если
тогда
DZM
Ра + Д^а
2
lg/C = lg[H] и —lg/(==pH.
Этот метод применим и для многоосновных кислот при
условии достаточно большого различия в значениях констант
диссоциации и в величинах молярных коэффициентов пога-
шения различных форм кислоты, соответствующих различ-
ным ступеням ее диссоциации при одной и той же длине вол-
ны, или при условии, что разные формы кислоты максималь-
но поглощают при раз-
личных длинах волн.
Метод Н. П. К о м а-
р я ’. Метод определения
константы кислотной дне-'
социации реактива, пред-
ложенный Н. П. Комарем,
так же как и расчетный
метод, основан на совме-
стном решении двух урав-
нений: уравнения основ-
ного закона светопогло-
щения и закона действую-
щих масс. Но в отличие
от предыдущих метод
Н. П. Комаря может быть
применен в тех случаях,
когда невозможно практи-
12 3 4 5b 7 8 S 10 // 12 13
PH
Рис. 20. Зависимость оптической плот-
ности растворов бензоилацетона от pH
при различных длинах волн
чески получить в чистом виде данные о поглощении реагента
в диссоциированной или недиссоциированной формах ввиду
наложения их спектров поглощения. Следовательно, метод не
требует предварительного знания величин внаи 8а (или со-
ответствующих величин D) в максимуме поглощения диссо-
циированной и недиссоциированной форм кислоты, а, напро-
тив, позволяет их вычислить для той длины волны, при кото-
рой измеряют величины D.
Предполагается, что реактив диссоциирует как однооснов-
ная кислота
НА Н+ + А~.
Готовят серию растворов, в которых исходная концентра-
ция кислоты берется одинаковой и равной с. Концентрация
водородных ионов непостоянна.
1 Н. П. Ко марь. «Уч. зап. Харьковск. ун-та», XXXVII (8), 57, 1951.
49
Пусть в опыте i
[Н] = ht, [А] = xz, [НА] = с —х(,
тогда по закону действующих масс в момент равновесия
= (52)
С — Xi
Если ёна—молярный коэффициент погашения НА; еА —
молярный коэффициент погашения А-, то
= [(с-х^ + х^]/, (53)
откуда
— SHAC^ Dl
Х‘~
Подставляя полученное выражение в уравнение (52) для.
константы равновесия, получим
— Кг\с1 = hfi^cl — hrf (55)
уравнение с тремя неизвестными К, ёна и еА. Для опыта k
аналогично получим
DlK - Кг\с1 = h^cl - hkDKk. (56)
Вычитая одно уравнение из другого, получим
(^ - DKk) К = ендс/ (h( - hk) - (D^i - DKkhk). (57)
Это уравнение содержит уже только два неизвестных ёна
и К- Комбинируя опыт i с каким-либо опытом п, мы снова
получим уравнение с теми же неизвестными ёна и К:
($ - D„) К = енас/ (^ - Л„) - (D^ - DKnhn). (58)
Решая два линейных уравнения (57) и (58) с двумя не-
известными, получим
X 1 ~ (ДЧ' - Dghn) - (ДгХ - РКп) (Д^- - Д^)
еНА cl (DK_DK)(h._hn)_(DK_ DK)(h._hk) ' ( >
= (hi - hk) (Pfyhj - DKnhn) - (hj - hk) (D^hj - PKkhk)
(D^-D^(ht-hn)-(P^-D^)(hl-hk) • *
Вычисленные значения ёна и К можно подставить в урав-
нение (55) и получить из него еА«
50
Если можно найти такой участок спектра, где поглощает
только одна из форм (диссоциированная или недиссоцииро-
ванная), то расчеты упрощаются.
Пусть при данной длине волны поглощает только А-, тогда
4k = О, D, = х,1г\,
(61)
/8а
Подставив полученное выражение в уравнение для кон-
станты равновесия, получим
Л^ = Д-(8*с/-^). (62)
Аналогичное выражение получим для опыта k-t решая два
линейных уравнения для опыта i и k с двумя неизвестными,
получим
' D}ht — D^hk
Dk — Di
еХ = 1 .
cl ‘
Эти формулы можно использовать и для спектрофотомет-
рического определения pH растворов, так как, зная К, 8 и
начальную концентрацию с, на основании измерений величи-
ны D всегда можно вычислить [Н].
Для практического освоения данных методов расчета кон-
станты кислотной диссоциации органического реагента мож-
но определить константы диссоциации диметилглиоксима,
нитрозо-Я-соли и тимолсульфофталеина, пользуясь методикой
приготовления растворов, стр. 125, 127 и 128.
3. ОПРЕДЕЛЕНИЕ СОСТАВА КОМПЛЕКСНОГО
СОЕДИНЕНИЯ
Известно, что состав комплексного соединения меняется
при переходе его из твердого состояния в раствор, а многие
комплексные соединения существуют только в растворе.
Метод физико-химического анализа, разработанный
И. В. Тананаевым, А. К. Бабко, Н. П. Комарем 1 и другими
учеными, широко применяется для определения стехиометри-
1 И. В. Тан ан а ев. ЖАХ, 4, 68, 136, 1949; 3, 276, 1948; 5, 82, 1950;
Н. П. К о м а р ь, В. Н. Толмачев. ЖАХ, 5, 21, 1950; Н. П. К о м а р ь.
ЖАХ, 5, 139, 1950; А. К. Бабко. Физико-химический анализ комплекс-
ных соединений в растворах. Киев, Изд-во АН УССР, 1955.
51
ческих коэффициентов в уравнениях реакции образования
комплексных соединений, находящихся в растворе. Этот ме-
тод позволяет определять и состав комплексных соединений,
если известно ионное состояние компонентов, участвующих
в образовании комплексного соединения, и в условиях его об-
разования не наблюдаются явления гидролиза и полимери-
зации.
Для определения состава комплексных соединений часто-
применяется метод Остромысленского—Жоба обычно на-
зываемый методом изомолярных серий. Сущность метода
Рис. 21. Определение соотноше-
ния реагирующих компонентов
методом изомолярных серий
заключается в том, что го-
товят растворы обоих ком-
понентов одинаковой мо-
лярной концентрации и сме-
шивают в определенном по-
следовательном соотноше-
нии; при этом сумма кон-
центраций (г~моль/л) во
взятом определенном объе-
ме всегда остается постоян-
ной.
Для образования ком
плексного соединения необ
ходимо соблюдать опреде-
ленное значение pH раство
ра, поэтому при приготовле-
нии растворов применяют буферные растворы. При выборе бу-
ферного раствора необходимо, чтобы отсутствовало комплексо-
образование у иона металла с веществами, составляющими
буферный раствор.
В качестве свойства изучаемой системы при фотометриче-
ских исследованиях состава соединений используют оптиче-
скую плотность D приготовленных растворов. Например, го-
товят растворы обоих компонентов одинаковой молярной кон:
центрации и смешивают в следующих соотношениях (мл):
раствор 1соли металла 123456789
раствор комплексующего вещества 987654321
Растворы перемешивают, добавляют в них одинаковое-
количество буферного раствора (если это необходимо), до-
водят водой (или другим растворителем) до определенного
объема, измеряют значения D и строят график зависимости
D от соотношений молярных концентраций реагирующих ве-
ществ (рис. 21). Для получения воспроизводимых результа-
тов ионная сила растворов должна быть постоянной.
Максимальное поглощение дает раствор, в котором обра-
1 Л. И. Остро мысленский. «Вег.», 44, 268, 1911; Р. Job. «Апп.
Chim.», 9, 113, 1928.
52
зующееся соединение преобладает. Рекомендуется готовить-
несколько серий растворов (2—3), отличающихся по концен-
трации Ci и С2 (рис. 21), и снимать светопоглощение приго-
товленных растворов при нескольких длинах волн (по край-
ней мере двух). Максимум на кривой состав — свойство-
определяет стехиометрические коэффициенты в уравнении
образования комплексного соединения. В случае образования
малоустойчивых комплексных соединений на кривых не бу-
дет наблюдаться резкого излома.
Если на графике состав — свойство положения максиму-
мов совпадают для растворов различной концентрации, то
это говорит о постоянстве состава комплексного соединения.
Смещение максимума на кривых обычно происходит при об-
разовании нескольких комплексных соединений. Рис. 21 ил-
люстрирует случай, когда в растворе образуется и поглощает
только одно соединение. Если же один из компонентов дает
окрашенные растворы, то предварительно измеряют светопо-
глощение этих растворов (различной концентрации) и вычи-
тают полученные значения из величин оптических плотностей
приготовленных смесей растворов. Этот метод детально рас-
смотрен в монографии А. К. Бабко Е
4. РАСЧЕТ КОНСТАНТ УСТОЙЧИВОСТИ
ПРИ СТУПЕНЧАТОМ КОМПЛЕКСООБРАЗОВАНИИ
Спектрофотометрические методы вычисления констант
равновесия реакций образования комплексных соединений и
констант устойчивости комплексных соединений тесно свя-
заны с методами вычисления величин истинных значений
молярных коэффициентов погашения (см. глава II, пункт 1).
Как те, так и другие основаны на совместном решении урав-
нений двух законов: закона действующих масс и закона све-
топоглощения. Поэтому при рассмотрении некоторых приемов
вычисления истинных величин молярных коэффициентов по-
гашения (стр. 40) мы одновременно касались и методов вы-
числения констант равновесия реакций комплексообразова-
ния.
Остановимся на одном методе вычисления констант
устойчивости при ступенчатом комплексообразовании, пред-
ложенном К. Б. Яцимирским1 2.
При ступенчатом образовании моноядерных комплексов
имеют место следующие равновесия:
МА„ MAn-1 + А МА„_2 + 2А ...ZW + «А.
1 А. К. Бабко. Физико-химический анализ комплексных соединении
в растворах. Киев, Изд-во АН УССР, 1955.
2 К. Б. Яцимирский. ЖНХ, 1, № 10, 2306, 1956.
53-
Для простоты в уравнениях опущены заряды частиц. При
постоянной ионной силе
_[МА]_ = Р _ГМА2] = JMA.] _ (
[М] [A] rl [М] [А]» [М] [А]л п
Суммарная концентрация металла см будет равна:
см = [М] 4" [МА] Д- [МАа] 4" • • • 4" [МА„]. (66)
Оптическая плотность раствора, содержащего все ком-
поненты, будет равна:
D = 80 [М] 14- 8i [МА] 14- е2 [МАа] 14- ... 4~ [MAn] I. (67)
Средний молярный коэффициент погашения равен:
D _ е0 4- 81Р1 [А] 4- е2р2 [А]2 * * 4- • • • 4~ s *nPn [А]п (68)
“ CMZ 1 + Pi [А] + р2 [А]а + ... + Р„ [Ар ’ 7
вычитая 80, получим
д~__ ~___е _ AeiPx [А] 4- Ае2р2 [А]2 4~ • • • 4- Аепрп [Ар /gg.
° 1 4- Pi [А] 4- Рз [А]2 4- • • • 4- Р» [АР
Это уравнение справедливо не только для 8, но и для D, ес-
ли все измерения производились в кювете с одной и той же
толщиной слоя.
В результате выполнения серийных определений можно
получить большое число значений Ае, и, следовательно,
большое число уравнений типа (69). В дальнейшем необхо-
димо найти коэффициент для этих уравнений.
Предлагаемый метод решения сводится к построению ря-
да вспомогательных функций и экстраполяции их на нулевое
значение.
Рассмотрим простую вспомогательную функцию
. __ Ае
71 “ W
Из уравнения (69) следует
j. __ ACjPi Ае2р2 [А] 4- Ае3Рз [А]2 4- • 4~ Аепрп[Ар 1
Г1~ 1 + Pi [А] 4- р2 [А]2 4- ... + р„ [А]л
Экстраполяция на нулевую концентрацию лиганда дает
lim Д = ах = As$v
[А]-»0
Эту экстраполяцию можно осуществить графическим мето-
дом, если по оси абсцисс откладывать равновесную концен-
трацию лиганда, а по оси ординат — значение fi. На оси ор-
динат при этом отсекается отрезок, численно равный Аефь
.54
(70)
(71)
(72)
Дифференцирование функции fi и экстраполяция производ-
ной на нулевую концентрацию лиганда дает
lim = а2 = Де2ра - Ae^f. (73>
[А]~>о d [А]
Значение а2 можно найти путем построения новой вспо-
могательной функции /2:
Эта функция при экстраполяции на нулевую концентра-
цию лиганда принимает следующие значения:
lim fz = a2 = Ае2р2 — Ае-^. (75).
Г А]-»0
Аналогично строится третья вспомогательная функция /з:
При экстраполяции f3 на нулевую концентрацию лиганда
получим
1 im /3 = а3 = Аезрз — Ae^f. (77).
[А]->0
Этот же результат может быть получен при дифференци-
ровании f2 и экстраполяции производной на нулевую концен-
трацию лиганда.
Совершенно аналогично могут быть построены функции
fi, fs, f& и т. д.
(78>
Экстраполяция такой функции на нулевую концентрацию
лиганда дает
lim /г = at = АеД. — Де^'. (79)
А]-»0
Общее число экстраполяционных уравнений оказывается
в 2 раза меньше, чем число подлежащих определению вели-
чин (р и Ае).
В связи с этим целесообразно ввести новую переменную
величину у, связанную с концентрацией лиганда простейшим
соотношением
55<
После деления числителя и знаменателя уравнения (69)
на [А]п, учитывая соотношение (80), получим
д—___ Лелрл + Аеп 1ря 1£/ + • • • + AriPiz/" 1
₽л + Рл-1 у + ... + Р1/“1 + уп
Экстраполяция значения Ае на нулевое значение
lim Ае = Ьг = Аел.
у-*о
Дифференцирование Ае по у и экстраполяция производ-
ной на нулевое значение у приводит ’к следующему резуль-
тату:
Иш = (Аел_! - Ае ) • (83)
ду Рл
Аналогичный результат может быть получен и путем по-
дстроения вспомогательной функции <pi:
(81)
у дает
(82)
в рас-
(86)
(87)
(88)
(89)
их оп-
(90)
У
При экстраполяции этой функции на нулевое значение у,
как видно из уравнения (81) и (82), находим
lim Ф1 = &2 = (Де„_1 — Де„)-^Д_. (85)
^->0 Рп
По этой же схеме строят функции ф2, фз,... Фп-i и экстра-
полируют их на нулевое значение у.
При сочетании значений, полученных экстраполяцией
функций Ае, Ф1, Ф2,... фп-ь со значениями, полученными экс-
траполяцией функций fi, fz, fs,—, fn, можно найти все коэффи-
циенты уравнения (69). Так, например, при наличии
творе только двух комплексов МА и МА2 получим
ai = AeiPi>
Я2 = АВ2Р2 AbiP],
\ = Ае2,
b2 = (Aej — Ае2) А
Ра
Отсюда находим константы устойчивости Pi и р2 и
тические характеристики Aei и Ае2.
р __ а1Ь1 — а2Ь2
aib2 + bf
.56
(92>
а\ + б?2&1
6Z1&2 + ь\
+ ^1^1
Aej = ———
fll&i — Q%b%
Aea = bx. (93}
В случае устойчивых комплексов некоторые затруднения
вызывает вычисление равновесных концентраций лиганда.
Если комплексы не отличаются высокой устойчивостью, то
общая концентрация лиганда не будет существенно отли-
чаться от равновесной.
5. МЕТОД СПЕКТРОФОТОМЕТРИЧЕСКОГО
ТИТРОВАНИЯ
Для определения момента эквивалентности в титриметри-
ческих методах в последнее время широко стал использовать-
ся спектрофотометрический метод. С изменением концентра-
ции какого-либо поглощающего компонента в процессе тит-
рования меняется оптическая плотность раствора.
Метод спектрофотометрического титрования основан на
последовательном измерении светопоглощения раствора, ме-
няющегося в процессе титрования. Непременным условием
для применения данного метода является приложимость,
к изучаемому раствору основного объединенного закона све-
топоглощения Бугера — Ламберта — Бера, справедливого для
монохроматического потока лучистой энергии.
Обычно графики строят в координатах D = f(V). По ор-
динате откладывается значение D, по абсциссе — объем F
реагента, концентрация которого известна. Процесс титрова-
ния графически выражается двумя пересекающимися прямы-
ми линиями.
Метод спектрофотометрического титрования по способу
определения момента эквивалентности сходен с кондуктомет-
рическим и особенно амперометрическим методом. Во всех
этих методах для нахождения конечной точки титрования
прибегают к экстраполяции. Для экстраполяции используют
такие участки кривой титрования, где в избытке находится
либо титруемый ион, либо реагент, т. е. участки, где диссо-
циация комплекса практически подавлена. Такие методы име-
ют преимущества перед методами, в которых момент эквива-
лентности определяется по отрезку кривой вблизи от точки
эквивалентности (например, в методе потенциометрического
титрования).
Ход кривой спектрофотометрического титрования зависит
от того, какой из компонентов, участвующих в реакции, по-
5Г
глощает при выбранной длине волны. Типы отдельных кри-
вых титрования приведены на рис. 22.
1. Поглощает раствор испытуемого вещества А; поглоще-
ние его уменьшается по мере добавления раствора реаген-
та Б. Продукт реакции В и реагент Б не поглощают
(рис. 22,а).
2. Поглощает реагент Б, испытуемое вещество А и про-
дукт реакции В не поглощают. Значение оптической плот-
ности возрастает после достижения точки эквивалентности
(рис. 22,б).
3. Поглощает продукт реакции В, испытуемое вещество
А, и реагент Б не поглощают. Светопоглощение раствора воз-
растает по мере накопления «окрашенного» продукта реак-
ции и становится постоянным после достижения точки экви-
валентности (рис. 22,в).
4. Поглощает испытуемое вещество А и реагент Б, про-
дукт реакции В не поглощает. Светопоглощение раствора
уменьшается по мере того, как испытуемое вещество А всту-
пает в реакцию и возрастает с увеличением избытка «окра-
шенного» реагента Б (рис. 22,г).
5. Поглощают продукт реакции В и реагент Б, испытуе-
мый раствор не поглощает (рис. 22, д). Возможны два слу-
чая:
а. Реагент Б поглощает в большей степени, чем продукт
реакции В. В процессе титрования светопоглощение увеличи-
вается до наступления момента эквивалентности, так как на-
капливается «окрашенный» продукт реакции В. Светопогло-
щение вновь возрастает при добавлении избытка «окрашен-
ного» реагента Б и после достижения точки эквивалентности
(кривая 1).
б. Продукт реакции В поглощает больше, чем реагент Б.
Светопоглощение увеличивается по мере накопления в рас-
творе «окрашенного» продукта реакции В. После достижения
точки эквивалентности поглощает избыток реагента Б (кри-
вая 2).
6. Поглощает испытуемое вещество А и продукт реак-
ции В, реагент Б не поглощает. Здесь также возможны два
случая (рис. 22, е):
а. Испытуемое вещество А поглощает больше, чем про-
дукт реакции В. Светопоглощение раствора падает по мере
уменьшения концентрации испытуемого вещества А и стано-
вится постоянным после достижения точки эквивалентности
(кривая 1).
б. Продукт реакции В поглощает больше, чем испытуе-
мое вещество А. Светопоглощение раствора увеличивается до
наступления момента эквивалентности, так как возрастает
концентрация «окрашенного» продукта реакции (кривая 2).
7. Поглощают все три компонента: испытуемое веще-
.58
2?
Рис. 22. Различные типы кривых спек-
трофотометрического титрования
ство А, реагент Б и продукт реакции В. Возможны три слу-
чая (рис. 22, ж):
а. Испытуемое вещество А и продукт реакции В погло-
щают эквивалентно, поэтому до наступления момента экви-
валентности светопоглощение раствора остается постоянным
и увеличивается только после добавления избытка реагента
Б (кривая 1).
б. Испытуемое вещество А поглощает больше, чем про-
дукт реакции В. Светопоглощение раствора уменьшается до
момента эквивалентности (кривая 2).
в. Продукт реакции В поглощает больше, чем испытуе-
мое вещество А. Светопоглощение раствора увеличивается
.до момента эквивалентности (кривая 3).
Во всех трех случаях после достижения момента эквива-
лентности светопоглощение раствора определяется избытком
реагента Б.
8. Все три компонента не поглощают. Добавленный чет-
вертый компонент вступает в реакцию с избытком реагента
Б и поглощает при определенной длине волны. Такое веще-
ство (четвертый компонент) является индикатором. Этот слу-
чай графически воспроизводится как первый или второй слу-
чай (рис. 22, а, б).
Излом на кривой титрования может не всегда совпадать
с моментом эквивалентности по стехиометрическому уравне-
нию реакции. В последнем случае необходимо ввести эмпири-
ческую поправку. Последнюю находят в процессе титрова-
ния ряда растворов с различной концентрацией и берут сред-
ние значения из полученных результатов.
В методе спектрофотометрического титрования могут быть
использованы все химические реакции, встречающиеся в дру-
гих титриметрических методах, т. е. практически необрати-
мые реакции.
Однако метод спектрофотометрического титрования имеет
ряд преимуществ перед визуальными методами титрования,
так как применим для титрования растворов с низкой кон-
центрацией и к реакциям, которые не заканчиваются в точке
эквивалентности (например, образование малоустойчивых
комплексов, нейтрализация слабых кислот и оснований, реак-
ции окисления-восстановления систем с потенциалами, не
сильно отличающимися по величине). Кроме того, метод бо-
лее специфичен и позволяет легко проводить последователь-
ное титрование двух компонентов.
Метод спектрофотометрического титрования имеет то пре-
имущество перед методом прямого спектрофотометрического
определения, что он позволяет проводить определение в при-
сутствии компонентов, частично поглощающих при выбран-
ной длине волны, так как важно изменение оптической плот-
ности в процессе титрования, а не абсолютное значение ее;
.60
при этом должно выполняться лишь одно условие: поглоще-
ние определяемых ионов должно быть значительно больше
поглощения присутствующих в растворе посторонних ве-
ществ. Этим объясняется большое внимание исследователей
к методу спектрофотометрического титрования1. Хорошо раз-
работаны основы фотометрического титрования кислот и ос-
нований 2.
Особенно широко в методе спектрофотометрического тит-
рования используются реакции комплексообразования. Имеет-
ся сравнительно небольшое число работ, в которых лиган-
дами являются неорганические соединения 3.
В большинстве работ в качестве реагентов используются
комплексоны и главным образом этилендиаминтетрауксусная
кислота или ее двунатриевая соль. В этом случае применя-
ются варианты безындикаторного и индикаторного титрова-
ния..
Вывод уравнения безындикаторного титрования был сде-
лан на основании следующих рассуждений4. Пусть в про-
цессе титрования образуется комплексное соединение по
уравнению
М + А МА,
т. е. компоненты реагируют в соотношении 1:1. Если при вы-
бранной длине волны поглощают все три компонента (ион-
комплексообразователь, лиганд и комплекс), то согласно ос-
новному закону светопоглощения
D = V е( [i] I, (94)
I
где D — суммарная оптическая плотность, 8г— молярный
коэффициент погашения каждого компонента, [/]— кон-
центрация каждого компонента, I — толщина поглощающего
слоя. Ввиду того что в процессе титрования толщина погло-
щающего слоя остается постоянной, произведение е^/ = ki
есть постоянная величина. С другой стороны,
D = [М] kK + [МА] feMA 4- [A] feA- (95)
1 R. Е. G о d d u, D. N. Hume. «Anal. Chem.», 26, 1740, 1954-
J. В. H e a d r i d g e. «Taianta», 1, 293, 1958.
2 R. Muller. «Ind. Eng. Chem., Anal. Ed.», 11, 1, 1939; Ю. IO. Лу-
рье, Э. Таль. «Зав. лаб.», 9, 702, 1940; J. Mika. «Z. anal. Chem.», 128,
159, 1948; A. Ringbom, F. S u n d m a n. «Z. anal. Chem.», 116, 104 1939-
R. F. God du, D. N. Hume. «Anal. Chem.», 26, 1679, 1954.
3 W. Boyer. «Ind. Eng. Chem., Anal. Ed.», 10, 475, 1938; P. West.
C. de Vries. «Anal. Chem.», 23, 334, 1951; O. Menis, D. Manning
R. Ball. «Anal. Chem.», 30, 1772, 1958.
4H. Flaschka. «Taianta», 8, 381, 1961.
61
В каждой фазе титрования должны выполняться следую-
щие соотношения:
см = [М] + [МА], (96)
са = [А] + [МА], (97)
где см — общая концентрация металла, са — общая концен-
трация лиганда. Для теоретических исследований удобнее
вместо объема прибавляемого реагента по абсциссе откла-
дывать «степень комплексообразования» ц, выражаемую со-
отношением:
р=-Х (98)
см
Комбинируя (95), (96), (97) и (98) получим
D = CM [&МА + &а (р-—1)] + (&м+&а—Ама) [М]. ‘ (99)
Это уравнение показывает, что оптическая плотность в
процессе титрования может быть выражена как функция ве-
личин р и [М]. Однако величина [М] может быть также дана
как функция ц путем комбинации (96), (97) и выражения
для константы устойчивости комплекса
к [МА]
[М] [А]
(В данном случае следует использовать кажущуюся кон-
станту комплекса, которая вычисляется из истинного значе-
ния константы с учетом реальных условий — величины pH,
посторонних лигандов и т. п.). Таким образом,
ГМ1 _ [АсмО-р)—1] + /[Ксм(1 —р)—1]2 + Шм
[_•* J 1 v *)!
(100)
2К
Комбинируя (99) и (101), получим общее уравнение кри-
вой безындикаторного спектрофотометрического титрования:
Д = - - {2Ксм [^ма+^а (р- — 1)] + (&м+&а—Ама) [Асм (1 —р) — .
2д
— 1 + /[Асм(1 -р]-1]2 + 4Ксм]}. (Ю2)
Если удается найти длину волны, при которой поглощает
только образующееся комплексное соединение, то уравнение
упрощается, так как йм = Аа = 0, и принимает вид
D = Ама (см—[АТ]). (103)
Из приведенных уравнений видно, что при заданных ве-
личинах молярных коэффициентов погашения ход кривой за-
висит от произведения Кем- На рис. 23 даны кривые титро-
62
вания, рассчитанные для одного и того же значения «ма
и различных значений /<см- При малых значениях Кем кри-
вая титрования раньше начинает отклоняться от прямой до
точки эквивалентности и позже выходит на прямую после
точки эквивалентности. Однако, как показывают эксперимен-
тальные исследования, при величине Кем = 50 момент экви-
валентности может быть установлен путем экстраполяции из
точек удаленных от точки эквивалентности с достаточной точ-
ностью. На том же рисунке даны кривые зависимости рМ
от ц. Из сравнения их с кри-
выми спектрофотометриче-
ского титрования становятся
ясны преимущества метода
экстраполяции по сравнению
с обычными методами опре-
деления момента эквива-
лентности. Из уравнения
кривой спектрофотометри-
ческого титрования (103)
также следует, что чувстви-
тельность метода можно по-
высить увеличением тол-
щины слоя используемой
кюветы и выбором соответ-
ствующей длины волны, так
как Йма = 6ма^.
Таким образом, метод
безындикаторного спектро-
фотометрического титрования
концентраций даже в случае
Рис. 23. Вид кривых титрования в ко-
ординатах D=f(p.) и рМ=/(ц):
К см Ксм ^МА=
1. 10s 10~2 1000 100
2. 5-Ю3 10-2 50 100
можно применять для малых
использования малоустойчивых
комплексов.
В тех случаях, когда в доступной области длин волн ни
один из компонентов реакции не поглощает, используется ме-
тод индикаторного спектрофотометрического титрования. В ка-
честве индикатора может быть использована соль такого
металла, который дает менее устойчивое комплексное соеди-
нение, чем определяемый элемент; причем комплексное со-
единение элемента-индикатора должно иметь поглощение в
доступной области спектра. Так, определение тория ком-
плексоном можно проводить в присутствии соли меди *.
На рис. 24,6 даны спектры поглощения нитрата тория и
его комплексного соединения; последнее не поглощает в об-
ласти 280—320 ммк. На рис. 24, а показано, что при 290 ммк
поглощает только соединение меди с комплексоном. При этой
длине волны и производится титрование тория (рис. 25).
Комплексное соединение тория более устойчиво и образуется
1 R. Malmstadt, Е. Gohrbandt. «Anal. Chem.», 26, 442, 1954.
63
в первую очередь; в момент эквивалентности начинает обра-
зовываться «окрашенное» соединение меди, поглощение кото-
рого и фиксируется прибором. Второй перегиб показывает,
что все ионы меди находятся в виде комплексного соедине-
ния.
Часто в качестве индикатора берут вещество, которое дает
менее устойчивое комплексное
Рис. 24. Спектры поглощения a: t—Си(МОз)2, 2— комплексон III,
3 — комплексонат меди; б: 1 — Th(NOs)4, 2— соединение тория с ком-
плексоном III
соединение с определяемым
элементом, чем комплексон; например, используются те же
индикаторы, что и при визуальных комплексонометрических
титрованиях (эриохром черный Т, мурексид, арсеназо)
В этом случае в растворе имеют
место следующие равновесия:
М + А МА,
М4-1^М1,
которым соответствуют констан- •
ты устойчивости:
|[МА] _ [МЦ
(104)
определяемый ион постепенно пе-
Рис. 25. Кривая титрования
тория комплексоном III
В процессе титрования
реходит из комплекса с индикатором в комплекс с реакти-
вом. Общее уравнение кривой индикаторного титрования, вы-
1 J. М. Н. F о г t u i n, Р. К о г s t е n, Н. L. Kies. «Analyt. Chim. Ac-
ta», Ю, 356, 1954; A. Ringbom, E. Vanninen. «Analyt. Chim. Acta»,
11, 153, 1954.
64
раженное в форме зависимости «степени комплексообразо-
вания» (стр. 62) от исходной концентрации определяемого
иона см, общей концентрации индикатора Ci, констант устой-
чивости комплексов с индикатором К mi и с реактивом Кма,
имеет вид:
ц = 1
-!_(1-а) (С1 +
см \
a^Mi / Kma I 1 — а
См ( 1 + “Км1 ) 1
(105)
Величина ц меняется от 0 (вначале титрования) до I
(в момент эквивалентности). Если Кма Kmi (примерно на
4—5 порядков) и не требуется вычислять ц с точностью боль-
шей, чем 0,001, то вторым слагаемым в уравнении (105) мож-
но пренебречь. Величина а в этом уравнении выражается от-
ношением концентрации свободного индикатора к общей его
концентрации, т. е.
= [I] [И
ci [MI] + [I] ’
(106)
В идеальном случае эта величина в момент эквивалент-
ности должна быть равна единице. Если поглощают только
комплекс MI и индикатор I, для теоретических исследований
удобно изучать зависимость а от ц, предполагая, что наблю-
дение ведется при длине волны, соответствующей максимуму
поглощения свободного индикатора. Тогда, используя урав-
нение
D = (si — eMi)/aci,
(107)
кривые зависимости а от ц можно преобразовать в кривые
зависимости D от ц и, следовательно, в итоге получим зави-
симость оптической плотности от объема прибавленного реак-
тива, так как
сА nV 1
см То см
(Ю8)
т. е. как раз ту зависимость, которая получается на практике.
Теоретическое исследование, проведенное Фортином с со-
трудниками ’, показало, что перегиб на кривой титрования
будет тем более резким, чем больше величина Kmi, выше ис-
ходная концентрация определяемого иона см и ниже кон-
центрация индикатора Ci при условии: Кма должна быть на-
много больше, чем.Хмь Кроме того, в расчетах вместо истин-
ного значения константы целесообразнее использовать кажу-
1 J. М. Н. Fort u in, Р. Karsten, Н. Kies. «Analyt. Chim. Acta»,
10, 356, 1954.
3 Зак. 686
65
щиеся константы (стр. 62). Однако при использовании очень
низкой концентрации индикатора сильно понижается абсо-
лютное значение оптической плотности, поэтому выбор оп-
тимальной концентрации индикатора определяется, с одной
стороны, выбором оптимальной длины волны, для которой
разность ei—ем! имеет максимальное значение, с другой —
максимально возможной толщиной слоя используемой кю-
веты. Для получения хороших результатов величина /<mi
должна быть порядка 104— 105, следовательно, необходимо
учитывать оптимальное значение pH образования комплекса
с индикатором.
Для проведения спектрофотометрического титрования
применяется сравнительно простое устройство (стр. 108), если
имеется источник монохроматического потока света. До
1950 г. для титрования применялись фотометры и фотоэлек-
троколориметры со светофильтрами, имеющими достаточно
узкие полосы пропускания. В последнее время используются
спектрофотометры различных марок (ДЦ В, СФ-4, СФ-5).
В ряде работ металлическую крышку кюветного отделения
заменяют хорошо пригнанной бакелитовой или деревянной
крышкой с двумя отверстиями, через которые проходит кон-
чик бюретки и мешалка. Мешалку всегда помещают в кю-
вете так, чтобы она не находилась на пути потока лучистой
энергии, проходящего через раствор и падающего на фото-
элемент. В отдельных случаях перемешивание производят
током СО2 и N2, опуская стеклянную трубочку, по которой
проходит газ, на дно кюветы для титрования. Для перемеши-
вания также применяют магнитную мешалку.
Для уменьшения влияния разбавления на светопоглоще-
ние рекомендуется использовать концентрированный рас-
твор реагента, объем которого обычно измеряют микробю-
реткой.
Спектрофотометрический метод титрования несомненно
найдет широкое применение в аналитических лабораториях.
6. ДИФФЕРЕНЦИАЛЬНЫЙ
СПЕКТРОФОТОМЕТРИЧЕСКИЙ МЕТОД
Дифференциальный спектрофотометрический метод разви-
вается в настоящее время главным образом в трех направ-
лениях: 1) спектрофотометрическое определение больших
концентраций; 2) устранение мешающего влияния посторон-
них компонентов; 3) исключение поглощения реактива.
Как было указано на стр. 30, дифференциальный спектро-
фотометрический метод обеспечивает также более высокую
точность измерения поглощения исследуемого раствора, чем
обычный спектрофотометрический метод.
66
Определение больших количеств веществ.
Для определения больших количеств веществ ранее обычно
использовались весовые, объемные и электрохимические
методы. С развитием спектрофотометрического метода
появилась возможность применить его для определения
больших концентраций «окрашенных» веществ в растворе
с точностью, не уступающей указанным методам. Напри-
мер, определение марганцовой кислоты, хромата или би-
хромата с концентрацией марганца и хрома порядка 1 г/л
можно провести спектрофотометрическим дифференциаль-
ным методом с точностью, не уступающей классическим объ-
емным методам '. Теоретические основы этого метода разра-
ботаны Хиски с сотрудниками (стр. 30). Сущность метода
состоит в том, что в качестве «нулевого» берется эталонный
раствор с несколько меньшей концентрацией определяемого
элемента, чем в испытуемом растворе. Точность метода по-
вышается, если соотношение интенсивностей потоков лучи-
стой энергии, прошедших через испытуемый и эталонный
растворы, близко к 1.
В разделе «Точность спектрофотометрического метода»
было указано, что практическое использование этого метода
в данном случае требует предварительного выяснения двух
основных моментов, определяющих точность дальнейших
спектрофотометрических измерений:
а) определение области концентраций, где наблюдаются
наименьшие отклонения от основного закона светопоглоще-
ния, вызванные недостаточной монохроматичностью потока
лучистой энергии;
б) выбор оптимальной концентрации «нулевого» раствора
(раствора сравнения), для которого е'со.т имеет максималь-
ное значение.
Область выполнения основного закона светопоглощения
определяют обычным путем (стр. 12), измерив оптические
плотности ряда эталонных растворов по отношению к рас-
твору с наименьшей концентрацией в данном эталонном ряду
и построив график зависимости D от с.
Для выбора оптимальной концентрации «нулевого» рас-
твора поступают следующим образом. В области концентра-
ций, в которой не наблюдается значительных отклонений от
основного закона светопоглощения, готовят ряд эталонных
растворов с такой Дс, чтобы интервалы ДД были порядка
0,3—0,4. Измеряют оптические плотности каждого последую-
щего раствора по отношению к предыдущему и вычисляют
е = и 8 с0)/ (Со,г — концентрация в растворе, кото-
рый используется в данном i-том измерении в качестве «ну-
1 В. Ф. Барковский, И. Н. В торы г ин а. ЖАХ, XVII, 865, 1962.
3* 67
левого»). Раствор, для которого е'с0,, имеет наибольшую ве-
личину, используют в качестве «нулевого».
Примечание. При выборе оптимальных условий дифференциаль-
ных измерений следует прежде всего найти ту предельную концентрацию,
при которой еще возможно используемый прибор установить на нуль.
Дальнейшее определение концентрации испытуемого рас-
твора можно провести графически1, измерив его оптическую
плотность по отношению к тому же «нулевому» раствору, ко-
торый был использован при измерении оптических плотностей
эталонных растворов.
Для расчета концентрации испытуемого раствора можно
также использовать следующее уравнение:2
cx=DxF-i-c1, (109)
где Ci —концентрация вещества в «нулевом» растворе; Dx—
оптическая плотность испытуемого раствора, измеренная по
отношению к «нулевому» раствору с концентрацией F—
фактор пересчета, предварительно вычисленный.
Это уравнение можно вывести следующим образом. Если
взять два раствора какого-либо «окрашенного» соединения
с известной концентрацией щ и с2 и третий испытуемый рас-
твор с неизвестной концентрацией сх и измерить интенсив-
ность светопоглощения всех этих растворов по отношению
к растворителю, то соответственно получим величины D\, D2
и Dx.
В случае подчинения основному закону светопоглощения
7^2 D1 ^2 С1 (110)
Dx — Dx сх —
Разности D2—Di и Dx — Dx, обозначенные через D’2 и D‘xf
представляют собой оптические плотности растворов с2 и сх,
измеренные по отношению к раствору сб
= (1Н)
Dx сх — Ci
D’2cx — D’2Ci =D'x(c2 — c^, (112)
= + (113)
U2
F
1 В. Ф. Барковский, И. H. Вторыгииа. «Зав. лаб.», XXVIII,
275, 1962; В. И. Ганопольский, В. Ф. Барковский, Т. А. Гано-
польская. «Зав. лаб.», XXX, 267, 1964.
2 С. D. S u s а п о, О. М е п i s, С. К. Talbott. «Anal. Chem.», 28, 1-072,
1956; Б. М. Добкина, Т. М. Малютина. «Зав. лаб.», XXIV, 1336;
1958; XXVII, 650, 1961.
68
Таким образом, фактор F представляет собой отношение
разности концентраций двух растворов к оптической плот-
ности более концентрированного раствора Сг, измеренной по
отношению к раствору с меньшей концентрацией сь
Практически для нахождения фактора F поступают сле-
дующим образом. Готовят ряд эталонных растворов, взяв
интервал концентраций, в котором выполняется основной за-
кон светопоглощения. Измеряют оптическую плотность каж-
дого из эталонных растворов по отношению к первому из
них; затем измеряют оптические плотности всех последующих
растворов по отношению ко второму и т. д. По формуле
F = Ci-~C1 (114)
вычисляют значения фактора для каждого измерения и на-
ходят среднее его значение. Измеряют оптическую плотность
испытуемого раствора по отношению к выбранному, как ука-
зано ранее, «нулевому» раствору и по формуле (109) вычис-
ляют его концентрацию, используя рассчитанный фактор.
Определение в присутствии мешающих
компонентов. Для определения в присутствии мешаю-
щих компонентов приведенный выше метод оказывается не-
пригодным, поэтому можно применить другой вариант диф-
ференциального спектрофотометрического метода, в котором
необходимым условием остается применимость основного за-
кона светопоглощения к анализируемым растворам.
В этом варианте как для приготовления «нулевого», так
и испытуемого растворов в качестве исходного используют
раствор образца и отбирают аликвотные части следующим
образом. Перед проведением фотометрической реакции в три
мерные колбы помещают определенные объемы исследуемого
раствора: в первую несколько меньший, в две следующие
одинаковый, но больший, чем в первую. Кроме того, в третью
колбу прибавляют еще определенное количество стандарт-
ного раствора определяемого компонента. После этого во
всех трех колбах проводят колориметрическую реакцию и из-
меряют оптическую плотность второго и третьего растворов
по отношению к первому. Если Со— содержание определяе-
мого компонента в объеме Vi испытуемого раствора, прибав-
ленного в первую колбу; сх— содержание определяемого ком-
понента в объеме V2 испытуемого раствора, прибавленного во
вторую и третью колбы; Dx— оптическая плотность раствора
с содержанием определяемого компонента сх, измеренная по
отношению к растворителю; Do — оптическая плотность рас-
твора с содержанием определяемого компонента Со, измерен-
ная по отношению к растворителю; са — количество опреде-
ляемого компонента, добавленного в третий раствор; Da —
оптическая плотность раствора с содержанием определяемого
69
компонента са, измеренная по отношению к растворителю, то
согласно закону светопоглощения
Рх — Ра _ сх~с0
Рх 4" ^0 Сх 4“ со
После простых преобразований получим
с a ~
(115)
(116)
(П7)
где D'x— оптическая плотность второго раствора (с содержа-
нием определяемого компонента сх), измеренная по отноше-
нию к первому раствору (с содержанием определяемого ком-
понента Со); с’х — содержание определяемого компонента в.
таком объеме испытуемого раствора, который равен раз-
ности объемов его, прибавленных во вторую и в первую
колбы.
Например, при определении никеля в стали (стр. 159) для
приготовления «нулевого» раствора со берут 2 мл контроль-
ного образца стали, а для второго раствора сх— 2,5 мл\ для
третьего раствора также 2,5 мл, но, кроме того, к нему до-
бавляют 0,05мг никеля (са). Таким образом, суммарная кон-
центрация в третьем растворе будет равна сх 4- са. Во всех
трех колбочках проводят реакцию с диметилглиоксимом в
одинаковых условиях и измеряют оптические плотности вто-
рого и третьего растворов по отношению к первому рас-
твору с0. По формуле (117) вычисляют содержание никеля в
объеме 0,5 мл контрольного раствора образца стали. Da мо-
жет быть найдена как разность оптических плотностей треть-
его и второго растворов.
Использование «окрашенных» реагентов.
Дифференциальный спектрофотометрический метод может
быть применен в тех случаях, когда имеется значительное
наложение в спектрах поглощения комплексного соединения
и реагента *. Оптические плотности растворов реагента D\ и
комплексного соединения измеряют по всей доступной
области спектра по отношению к растворителю. Если реа-
гент берется в большом избытке (1:50; 1:100), то концен-
трацией реагента, вошедшей в комплексное соединение, мож-
но пренебречь при работе с малыми концентрациями веще-
ства; тогда разность Д2—Di принимают за ДИст — поглоще-
ние раствора чистого комплексного соединения. Таким обра-
1 G. В а и е г j е е. «Anal. Chem.», 29, 55, 1957.
70
зом, получают кривую истинного спектра поглощения соеди-
нения Дист—X, где /)Ист есть разность D2—D\ для каждой
длины волны.
При работе с «окрашенными» реагентами можно исполь-
зовать также в качестве «нулевого» раствор реагента, кото-
рый готовится в условиях приготовления испытуемого рас-
твора. Этот прием возможен в том случае, если прибор на-
страивается на 0 при указанном «нулевом» растворе.
В дифференциальном методе кроме спектрофотометров
во многих случаях с таким же успехом могут быть использо-
ваны приборы, являющиеся упрощенными спектрофотометра-
ми: фотоэлектроколориметры типа ФЭК-Н-52, ФЭК-Н-54,
ФЭК-Н-57 и ФЭК-56.
7. ЭКСТРАКЦИОННО-ФОТОМЕТРИЧЕСКИЙ МЕТОД
В последнее время, несмотря на большое развитие це-
лого ряда физико-химических методов, отличающихся боль-
шой избирательностью по'сравнению с химическими мето-
дами, не всегда удается непосредственно определять многие
элементы в сложных смесях. Решение этой задачи во многом
зависит от предварительного разделения, которое с успехом
может быть проведено методом экстракции, основанном на
различном распределении компонентов в системе неводный
растворитель — вода. Распространению метода экстракции
способствовало появление ряда теоретических работ, посвя-
щенных физико-химическому исследованию этого процесса *.
Метод позволяет разделять вещества, сильно отличающиеся
по концентрации, поэтому в настоящее время экстракцион-
ные методы нашли широкое применение в практике аналити-
ческих лабораторий.
Особое значение приобретает метод экстракции ввиду не-
обходимости определения примесей в особо чистых веще-
ствах, широко применяемых в атомной и полупроводниковой
технике. Для определения малых и ультрамалых количеств
элементов, являющихся примесями, метод экстракции приме-
няется не только для выделения определяемого элемента, но
и для целей его концентрирования.
Очень удачным является сочетание метода экстракции
с последующим спектрофотометрическим определением эле-
ментов (экстракционно-фотометрический метод).
В настоящем практическом руководстве не затрагиваются
вопросы механизма экстракции и классификации экстрак-
ционных систем, которые освещены в ряде монографий и
1 Ю. А. Золотов, И. П. А л им ар ин. ДАН СССР, 136, 603, 1960;
Р. М. Д а й м о н д, А. Г. Так. Экстракция неорганических соединений. М.,
Госатомиздат, 1962; Н. П. К о м а р ь. «Тр. химфака и НИИхимии Харь-
ковск. ун-та», 19, 9, 1963.
71
сборников ', а рассматриваются возможности экстракционно-
фотометрического метода для определения ультрамалых ко-
личеств отдельных элементов. В большинстве случаев для
определения искомых элементов используют комплексные со-
единения этих элементов с органическими реагентами. В спек-
трах поглощения растворов комплексного соединения и реа-
гента наблюдается наложение максимумов, что осложняет
спектрофотометрическое определение. Для того чтобы исклю-
чить поглощение реагента, в отдельных случаях используется
Рис. 26. Зависимость процента экстракции хлороформом от значе-
ния pH водной фазы: 1 — гептоксимата никеля; 2 — гепоксима
дифференциальный метод (стр. 70). Например, при опреде-
лении цинка дитизоном (стр. 201) измеряют поглощение испы-
туемого раствора по смешанной окраске, используя в каче-
стве «нулевого» раствор дитизона.
Перспективным является прием, позволяющий путем ре-
экстракции перевести реагент из слоя органического раство-
рителя в водный слой. Этот прием основан на различной
зависимости коэффициента распределения комплексного со-
единения и реагента от величины pH раствора (рис. 26). Та-
ким образом, можно сравнительно простым приемом полу-
чить определяемое соединение в чистом виде и освободиться
от необходимости применять приемы работы и расчеты, ис-
пользуемые при работе с многокомпонентными системами.
1 Д. Моррисон, Г. Фрейзер. Экстракция в аналитической хи-
мии. Л., Госхимиздат, 1960; В. В. Фомин. Химия экстракционных процес-
сов. М., Атомиздат, 1960; В. И. Кузнецов. Химические основы эк-
стракционно-фотометрических методов анализа. М.., Госгеолтехиздат, 1963.
72
Однако следует учитывать, что для экстракции ультрама-
лых количеств элементов характерно смещение интервала
значений полной экстракции в более щелочную область. На-
пример, соединение никеля с диоксином 1,2-циклогександиона
количественно экстрагируется хлороформом при содержании
никеля ~10 мкг в интервале значений pH 4,2—11; при со-
держании никеля 5 мкг и меньше — при pH 5,4—12,75.
Помимо этого метод экстрагирования дает возможность
исследовать новые комплексные соединения с целью получе-
ния их количественных характеристик (констант устойчиво-
сти, истинных значений е).
IV. АППАРАТУРА
КОНЦЕНТРАЦИОННЫЙ КОЛОРИМЕТР КОЛ-1М
Концентрационный колориметр КОЛ-IM. применяется
для определения концентраций окрашенных растворов по
методу изменения толщины поглощающего слоя (метод
«уравнивания»).
Принципиальная схема прибора изобра-
жена на рис. 27, а; внешний вид прибора
показан на рис. 27,6.
Свет от источника 1 параллельным пуч-
ком проходит через две кюветы 2 с окра-
шенными растворами, в которые погружа-
ются два стеклянных столбика 3 одинако-
вой длины и плотности. Призма 4 сводит оба
Рис. 27. Принципиальная схема (а) и внешний вид (б) концентрацион-
ного колориметра К.ОЛ-1М
пучка света, прошедшие через каждую кювету, к одной оси.
Пройдя через светофильтр 11 и окуляр 5, свет попадает в глаз
наблюдателя. Поле зрения окуляра разделено на две поло-
вины, каждая из которых освещается пучком света, прошед-
шим через одну из двух кювет. Освещенность полей окуляра
74
уравнивают поднятием или опусканием столбиков, погружен-
ных в растворы. Поскольку пропускаемость этих двух стол-
биков совершенно одинакова, то интенсивность светового по-
тока изменяется только за счет поглощающих свойств раство-
ра, пропорциональных, как известно, концентрации и толщине
слоя, которая в данном случае ограничивается высотой под-
нятия столбика. Высоты столбиков отсчитывают по шкале,
снабженной нониусами 12. Для удобства наблюдения яркости
освещения полей окуляра поворотом кольца 6 делают резкой
линию раздела поля зрения на две половины.
Порядок работы на приборе. Для сравнения от-
носительной интенсивности окраски двух растворов, кото-
рыми наполнены кюветы, необходимо, чтобы пучки света,
попадающие в каждую из этих кювет, были одинаковой ин-
тенсивности.
Работу следует начинать с установки осве-
тителя. Для этого готовят два эталонных раствора с раз-
личными концентрациями, вставляют столбики в пазы, имею-
щиеся на пластинке 7, укрепленной перпендикулярно кювет-
ному столику, и вдвигают их до упора. Обе кюветы напол-
няют окрашенным раствором одной и той же концентрации
и, поставив столбики в самое верхнее положение, вставляют
кюветы в прибор, вдвигая их сбоку в пазы, имеющиеся на
кюветном столике прибора /3; при этом следует обращать
внимание на обозначения, имеющиеся на кюветах (левая и
правая). Опускают столбики в кюветы, вращая винты 8,
и устанавливают оба на одной и той же высоте (например,
на деление 20). Одев кожух 9 и включив лампу прибора че-
рез трансформатор, наблюдают освещенность полей окуляра,
уравнивая ее поворотом патрона лампы 10 вокруг оси. После
того как уравняют таким образом освещенность полей оку-
ляра по раствору одной концентрации, делают ту же самую
проверку установки лампы по раствору другой концентрации,
наполнив им обе кюветы и установив оба столбика опять на
одной и той же высоте. Смену раствора в кюветах
и любую другую операцию, требующую выдви-
жения кювет из прибора или столбиков из па-
зов, в которых они укреплены, необходимо
производить только при поднятых вверх до
отказа столбиках.
Концентрацию определяют после того, как
будет установлен осветитель. Вылив раствор изве-
стной концентрации из одной кюветы, наполняют ее раство-
ром неизвестной концентрации и, установив один из столби-
ков на какой-то определенной, фиксированной высоте, ме-
няют высоту другого столбика до уравнивания освещенности
полей окуляра. Уравнивание производят 5—6 раз и из схо-
дящихся результатов берут среднее арифметическое.
75
Концентрацию неизвестного раствора рассчитывают по
формуле
<118>
"х
Примечания. 1. Колориметр погружения КОЛ-1 М применяется
для определения концентрации окрашенного раствора, если к таковому
приложим закон Бугера — Ламберта — Бера.
2. Определить концентрацию раствора по этому методу можно и в
случае неподчинения окраски раствора закону Бугера — Ламберта—Бера,
но при условии, что концентрации исследуемого и стандартного растворов
достаточно близки.
3. Для того чтобы исключить возможные ошибки из-за неодинаковой
освещенности и пропускания кювет, можно предложить следующий метод
работы. Наполнив одну из кювет окрашенным раствором какой-то концен-
трации (не обязательно точно известной), уравнивают по отношению к
нему сначала стандартный, затем исследуемый растворы, помещая их по
очереди во вторую кювету.
Концентрацию исследуемого раствора рассчитывают по формуле (118).
Выбор светофильтра. Подбирают цвет, дополни-
тельный к цвету окрашенных растворов, или наливают в кю-
веты растворы с концентрациями, отличающимися друг от
друга не менее чем на 10%, и устанавливают столбики на
одной и той же высоте. Вращая диск со светофильтрами, на-
ходят светофильтр, для которого разница в яркостях освеще-
ния полей наиболее заметна.
МИКРОКОЛОРИМЕТР КОЛ-52
В основу конструкции микроколориметра КОЛ-52 поло-
жен принцип уравнивания двух световых потоков путем из-
менения интенсивности одного из них с помощью фотометри-
ческого клина.
Рис. 28. Оптическая схема микроколориметра КОЛ-52
Оптическая схема прибора дана на рис. 28. Свет от источ-
ника 1, нить которого расположена в фокусе конденсора 2,
попадает на диафрагму 3, пропускающую два узких парал-
лельных пучка света. Один из них проходит через кювету
76
с исследуемым раствором 4 и левый клин 5; другой — через
правый клин 5'. Призма 6 сводит оба световых пучка к одной
оси. Пройдя через светофильтр 7 и окуляр 8, пучки света по-
падают в глаз наблюдателя, зрачок которого совмещен со
зрачком выхода прибора. Наблюдатель видит круг, разделен-
ный на две половины, имеющие в общем случае различную
яркость.
Внешний вид прибора показан на рис. 29, а, б, где видно
расположение всех его деталей.
Порядок работы на приборе. Перед тем как
включить лампу, следует наладить водяное охлаждение, ко-
торое осуществляется путем подвода водопроводной воды че-
рез каучуковые шланги к штуцерам 9 кожуха лампы 10.
Лампу включают через трансформатор Т-2, рассчитанный на
включение в сеть переменного тока 127 или 220 в. При изме-
нении питающего напряжения со 127 на 220 в или наоборот
следует переставить винт в соответствующее гнездо транс-
форматора. После этого вилку осветителя прибора включают
в розетку трансформатора, а вилку трансформатора в питаю-
щую сеть. Выходное напряжение трансформатора поворотом
рукоятки можно менять от 10 до 12 в, меняя соответственно
и яркость освещения.
Прежде всего следует проверить правильно ли
установлен осветитель. Для этого на пути пучка
света устанавливают поворотом диска 12 светофильтр № 6.
По шкале нониусов 13 с помощью рукояток 14 правый клин
устанавливают на деление 0, левый — на деление 70. Кювету
наполняют растворителем (например, водой) и устанавлива-
ют ее на пути левого пучка света с помощью рукоятки дер-
жателя кювет 15. Следует помнить, что кювета и с раство-
ром и с растворителем всегда ставится только на пути пучка
света, проходящего через левый клин.
Перед окуляром устанавливают лупу 16 и наблюдают че-
рез нее зрачок выхода прибора. Если наблюдатель видит яр-
кий кружок и при перекрытом правом световом пучке остает-
ся видимым полный кружок без добавочных бликов снизу,
то следует считать установку нити лампы удовлетворитель-
ной. Слегка вдвигая лупу, фокусируют ее на изображение ни-
ти лампы. Наблюдаемая при этом картина должна соответ-
ствовать представленной на рис. 30.
После этого отодвигают лупу и приступают к выбору
среднего фотометрического равновесия, на-
пример, для левого клина (точно так же этот выбор про-
изводится и для правого клина). Наблюдаемое поле зрения
окуляра во время работы должно быть освещено равномерно.
Для того чтобы линия раздела поля зрения на две половины
была резкой, следует ее сфокусировать поворотом линзы оку-
ляра 17 (рис. 29). Левый клин устанавливают на деление 0.
77
lb -
Рис. 29. Внешний вид микроколориметра КОЛ-52
Рпс. 30. Наблюдаемое в
окуляр изображение нити
лампы микроколориметра
КОЛ-52 при правильной
установке осветителя
Уравнивают поля окуляра по яркости освещения, вращая
рукоятку правого клина. Ввиду того что клинья соединены
со шкалами в обратном порядке, т. е. нулевому делению ле-
вого клина соответствует наибольшая плотность, а правого
клина — наименьшая плотность, положение фотометрического
равновесия в данном случае должно приходиться на интер-
вал делений шкалы правого клина от 60 до 70. Соответствен-
но положение среднего фотометрического равновесия при ну-
левом положении правого клина должно находиться в интер-
вале делений шкалы левого клина от 60 до 70. Яркость
освещения полей окуляра уравнивают несколько раз (5—6)
и из всех сходящихся отсчетов берут среднее арифметиче-
ское. Если положение среднего фо-
тометрического равновесия не укла-
дывается в указанный предел зна-
чений шкалы, то устанавливают
лампу осветителя, как это рекомен-
дуется в описании, прилагаемом
к прибору.
После того как было установлено
положение среднего фотометриче-
ского равновесия, приступают
к измерениям. Измерения мож-
но производить как с правым, так
и с левым клином. При работе с ле-
вым клином правый клин устанавли-
вают в положение среднего фото-
метрического равновесия. В свето-
вой поток вводится кювета с анали-
зируемым раствором. Вращая ру-
коятку левого клина, добиваются равномерного освещения по-
лей окуляра. Освещенность полей окуляра уравнивают, как
обычно, несколько раз и из сходящихся отсчетов по шкале
нониусов берут среднее арифметическое. При работе с правым
клином левый клин устанавливают в положение среднего
фотометрического равновесия и яркость освещения полей оку-
ляра уравнивают правым клином.
Измерения с помощью левого клина производят при по-
стоянной яркости поля зрения, что является преимуществом
данного метода. Однако если не удается уравнять яркость
освещения полей окуляра ни левым, ни правым клином
(этот случай может иметь место при достаточно больших
значениях оптической плотности анализируемого раствора),
то при измерении с помощью правого клина можно восполь-
зоваться дополнительным нейтральным светофильтром, кото-
рый вводится в правый пучок света рукояткой 18. Тогда к от-
считанному по шкале правого клина числу прибавляют чис-
ло, соответствующее условному увеличению отсчета при вве-
70
дении дополнительного нейтрального светофильтра. Это число
определяется для каждого цветного светофильтра, с которым
производят измерение, следующим образом.
Правый клин устанавливают на деление 60 и левым кли-
ном определяют положение среднего фотометрического рав-
новесия. После этого устанавливают левый клин в положе-
ние среднего фотометрического равновесия и, вводя в правый
световой поток нейтральный светофильтр, уравнивают пра-
вым клином поля окуляра по яркости. Берут отсчет по шкале
правого кл 'на П\ и вычисляют число /гСф, соответствующее
плотности нейтрального светофильтра по формуле
пСф = 60 — пх. (119)
Прибор дает возможность снять спектральную характери-
стику, т. е. проследить изменение оптической плотности рас-
твора в зависимости от длины волны. Так как отсчеты по
шкале микроколориметра пропорциональны величинам опти-
ческих плотностей растворов, то для нахождения оптической
плотности исследуемого раствора необходимо отсчет по шка-
ле умножить на коэффициент пропорциональности. Коэффи-
циенты пропорциональности для каждого светофильтра даны
в паспорте прибора. Там же даны величины оптической плот-
ности нейтрального светофильтра для каждого цветного све-
тофильтра; при измерениях с дополнительным нейтральным
светофильтром к вычисленному значению оптической плот-
ности прибавляют оптическую плотность нейтрального свето-
фильтра.
Для количественных определений светофильтр выбирают,
как обычно, согласно наибольшей величине оптической плот-
ности (стр. 24, 117).
Определение и расчет концентраций производят одним из
двух принятых методов:
1. Если окраска подчиняется основному закону светопо-
глощения в каком-то интервале концентраций, то концентра-
ция исследуемого раствора сх может быть вычислена по фор-
муле
_ С!ПХ
—
П1
где С] — концентрация эталонного раствора; ni и пх соответ-
ствуют отсчетам по шкале микроколориметра для эталонного
и исследуемого растворов.
Подобный же расчет возможен и в случае неподчинения
окраски раствора основному закону светопоглощения, если
эталонный раствор имеет концентрацию, достаточно близкую
к концентрации исследуемого раствора, и, следовательно, их
оптические плотности будут располагаться на прямолинейном
80
отрезке кривой зависимости оптической плотности от кон-
центрации.
2. Можно приготовить ряд эталонных растворов и, по-
строив график зависимости отсчетов по шкале от концентра-
ции, согласно отсчету, сделанному для исследуемого рас-
твора, определить с помощью графика концентрацию этого
раствора.
Данный тип визуального колориметра удобно использо-
вать в тех случаях, когда имеются очень малые количества
анализируемого материала. Прибор позволяет- работать
с растворами достаточно малых концентраций, так как тол-
щина кюветы достаточно велика, а самое главное, — с ма-
лыми объемами растворов (минимальное количество раство-
ра, необходимое для измерения его концентрации, равно
0,5 мл).
УНИВЕРСАЛЬНЫЙ ФОТОМЕТР ФМ
Универсальный и горизонтальный фотометры представ-
ляют собой виды упрощенного визуального спектрофотомет-
ра, в котором монохроматизация света достигается с по-
мощью светофильтров. Универсальный фотометр позволяет
измерять коэффициенты пропускания и оптические плотности
жидких и твердых прозрачных (нерассеивающих) веществ.
Наличие в приборе ряда узкополосных монохроматиче-
ских светофильтров позволяет также снимать спектральную
характеристику раствора данного соединения. В основу
устройства прибора положен принцип уравнивания интенсив-
ности двух световых потоков путем изменения одного из них
с помощью диафрагмы с отверстием переменной величины.
В случае одинаковой интенсивности окраски растворов, на-
полняющих обе кюветы, при одинаково открытых диафраг-
мах обе половины поля зрения окуляра имеют одинаковую
освещенность. Если же интенсивность окраски растворов не-
одинакова, то для уравнивания освещенности полей окуляра
необходимо уменьшить отверстие диафрагмы, расположенной
на пути пучка света, прошедшего через менее интенсивно ок-
рашенный раствор.
Принципиальная схема прибора дана на рис. 31, внешний
вид — на рис. 32.
Два параллельных пучка света, пройдя через две кю-
веты 1, одна из которых наполнена раствором исследуемого
вещества, а другая — «нулевым» раствором, по отношению
к которому измеряется пропускание или плотность исследуе-
мого раствора, попадают на щели двух диафрагм 2, бара-
баны 11 которых имеют две шкалы: красную — оптических
плотностей и черную — коэффициентов пропускания. Оптиче-
ской системой прибора оба пучка сводятся к одной оси и на-
81
правляются через окуляр 3 в глаз наблюдателя, который ви-
дит круг, разделенный на две половины.
Порядок работы на приборе. Для определения
величины изменения интенсивности одного из попадающих в
Рис. 31. Принципиальная схема универсального фотометра ФА"!
Рис. 32. Внешний вид универсально-
го фотометра ФМ
окуляр световых потоков
(в результате ослабления
потока за счет поглоще-
ния света окрашенным
раствором) необходимо,
чтобы оба пучка
света, падающие
на кюветы с рас-
твором и раствори-
телем, были одина-
ковы по интенсив-
ности. Поэтому перед
началом измерений сле-
дует проверить установ-
ку осветителя и одинако-
вы ли параллельные пуч-
ки света, проходящие
через обе кюветы, по ин-
тенсивности.
Прежде всего следует
проверить, как установле-
ны спирали осветителя.
Включают лампу через
трансформатор. Наблю-
даемые в окуляр 3 спира-
матовых стеклах из пазов
резкое изображение. В том
окуляр, их изображение вво-
с помощью зеркала 5. Если
ли при вынутых рассеивающих
конденсаторов 4 должны! иметь
случае, если спирали не видны в
дится в поле зрения окуляра
спирали не видны в окуляр или изображение их недостаточно^
82
резко, то следует, освободив винт 6, поворотом головки осве-
тителя 7 вокруг оси или передвижением ее по вертикали
добиться отчетливой видимости спиралей в окуляр. Фокуси-
ровку нитей спирали следует произвести с помощью конден-
соров 4. Наблюдая в окуляр, добиваются четкого изображения
нитей спирали.
После этого следует вставить рассеивающие матовые стек-
ла в пазы конденсоров 4 и проверить одинаковы ли световые
потоки по интенсивности следующим образом. Поворотом
диска со светофильтрами 9 установить на пути светового по-
тока зеленый светофильтр. Линию раздела поля зрения на
две половины следует сделать резкой с помощью кольца 10.
(Зеленый светофильтр выбирают потому, что в этой области
спектра сильно повышается чувствительность человеческого
глаза). Барабан И, например, левой диафрагмы следует по-
ставить в положение, соответствующее 50% пропускания
(т. е. в области плотностей, где глаз наиболее легко улав-
ливает малейшие изменения интенсивности световых пото-
ков,— стр. 29). Наблюдая в окуляр, медленно вращают бара-
бан правой диафрагмы до уравнивания освещенности полей
окуляра. Повторяют эту операцию до получения сходящихся
показаний. Если среднее арифметическое из сходящихся по-
казаний шкалы правого барабана лежит в данном случае в
интервале значений 49,5 — 50,5%, то установку осветителя
следует считать удовлетворительной. Если показания выхо-
дят за предел этих значений, то следует добиться лучших ре-
зультатов одним из следующих приемов:
а) поменять матовые рассеиватели местами;
б) вставить в паз одного из конденсоров прозрачное
стекло, прилагаемое к прибору;
в) слегка перемещать один или оба конденсора в освети-
теле; вызываемая при этом дефокусировка нити лампы не
имеет значения для работы с прибором.
Если ни одним из этих приемов не удается достигнуть
желаемого результата, то снимают головку осветителя
с кронштейна и устанавливают лампу в осветителе заново,
как это рекомендуется в описании, прилагаемом к прибору.
К измерениям приступают после того; как
добьются равномерной освещенности полей окуляра. Изме-
ряют оптические плотности D или коэффициенты пропуска-
ния Т следующим образом.
Один из барабанов, например правый, устанавливают в
положение, соответствующее 100% пропускания или 0 опти-
ческой плотности. На пути пучка света, проходящего через
соответствующую ему диафрагму, на столик 8 ставится кю-
вета с исследуемым раствором (см. примечание). На пути
пучка света, проходящего через левую диафрагму, ставится
кювета, наполненная раствором, по отношению к которому
83
измеряют оптическую плотность или коэффициент пропуска-
ния исследуемого раствора («нулевой» раствор).
Поворотом левого барабана добиваются равномерной
освещенности обоих полей окуляра и по шкале этого же ба-
рабана снимают показания Т и D. Освещенность полей оку-
ляра уравнивают несколько раз; из сходящихся результатов
берут среднее арифметическое.
Чтобы снять спектральную характеристику какого-либо
окрашенного раствора, необходимо измерить оптическую
плотность этого раствора, как было указано ранее, после-
довательно со всеми светофильтрами. Затем откладывая по
оси абсцисс значения эффективной длины волны каждого
светофильтра, найденные в табл. 2 (|стр. 116) соответственно
его марке, а по оси ординат — полученные значения оптиче-
ской плотности, соответствующие измерениям при данном
светофильтре, можно построить график зависимости D от X,
или, иначе, представить графически спектр поглощения дан-
ного раствора, как, например, это дано на рис. 4.
Для определения концентрации раствора, как известно,
наиболее выгодно использовать область максимального по-
глощения данного окрашенного раствора. Поэтому, перед
тем как приступить к измерению светопоглощения, следует
на пути светового потока поставить тот светофильтр, для ко-
торого была найдена наибольшая оптическая плотность при
снятии спектральной характеристики. Концентрацию раство-
ра рассчитывают одним из обычных методов, указанных
в гл. II (стр. 38).
Примечания. 1. Кюветы универсального фотометра следует на-
полнять растворами так, чтобы при наблюдении по вертикали через крыш-
ку кюветы не было видно пузырьков воздуха, присутствие которых вызы-
вает сильное изменение оптической плотности измеряемых растворов.
2. Для повышения точности измерений (стр, 28) в тех случаях, когда
это возможно (измерение малых интенсивностей поглощения), барабан диа-
фрагмы, расположенной на пути светового потока, проходящего через испы-
туемый раствор, следует установить не на 100% пропускания, а на 20—
30%. В остальном порядок определения концентраций остается прежним.
Горизонтальный фотометр представляет собой
разновидность универсального фотометра с расположением
всех основных узлов на горизонтальной оси. Принципиальная
схема прибора и его возможности такие же, как и для уни-
версального фотометра, кроме возможности измерять
пропускание твердых образцов. Отличается он также устрой-
ством кювет, которые имеют прямоугольные грани, что удоб-
но при установке их в кюветодержателе, расположенном на
горизонтальной оси; наполняются они доверху. В остальном
методика работы с этим прибором и измерение оптических
плотностей ничем не отличаются от описанных выше для уни-
версального фотометра.
84
ФОТОЭЛЕКТРИЧЕСКИЙ КОЛОРИМЕТР ФЭК-М
Прибор ФЭК-М относится к типу приборов, в которых ин-
тенсивности световых потоков сравнивают с помощью селено-
вых фотоэлементов, благодаря чему исключаются субъектив-
ные ошибки, свойственные индивидуальным особенностям
Рис. 33. Принципиальная схема фотоэлектроколориметра ФЭК-М
глаза различных наблюдателей и имеющие место при работе
с визуальными приборами. Конструкция прибора основана на
принципе уравнивания интенсивностей двух световых потоков
при помощи щелевой диафрагмы. Принципиальная схема
прибора дана на рис. 33, общий вид прибора — на рис. 34, а, б.
Световые пучки, идущие от одного и того же источника 13,
отразившись от двух зеркал 14, проходят через светофильт-
85
Рис. 34. Внешний
вид фотоэлсктроколориметра ФЭК-М
ры 6 и кюветы 15 и попадают на два фотоэлемента 16 и 16'.
Фотоэлементы соединены между собой по дифференциальной
схеме; в качестве нулевого прибора служит стрелочный галь-
ванометр 1. На пути левого пучка, падающего на фотоэле-
мент 16, расположены нейтральные клинья 4 и 5; на пути
правого пучка, падающего на фотоэлемент 16',— щелевая
диафрагма 17, связанная с двумя отсчетными барабанами 2.
Отсчетные барабаны имеют две шкалы 3: красную — оптиче-
ских плотностей и черную — коэффициентов светопропуска-
ния.
Принцип измерений заключается в том, что вначале про-
изводят компенсацию фототоков, идущих от каждого фото-
элемента, устанавливая гальванометр на нуль с помощью
нейтральных клиньев 4, 5 при помещенных в оба пучка света
кюветах с растворителем и нулевом положении диафрагмы по
отсчетному барабану. Затем в правый световой поток вводят
кювету с поглощающим раствором. Это вызовет отклонение
стрелки гальванометра, так как на правый фотоэлемент бу-
дет падать меньше световой энергии, чем на левый. Уравнять
потоки света можно, меняя ширину щели диафрагмы. По
шкале барабана определяют оптическую плотность данного
раствора или процент пропускания.
Порядок работы на приборе. Работу сле-
дует начинать с проверки установки освети-
теля. Лампу осветителя включают через стабилизатор за
15—20 мин до начала работы, чтобы она успела приобрести
устойчивую эмиссию. Для того чтобы световой поток был
равномерным и полностью заполнял окно измерительной
диафрагмы и нейтральных клиньев, нить лампы осветителя
должна изображаться на поверхности одного из свето-
фильтров 6 в виде резкой и отчетливой спирали, распо-
ложенной в центре светофильтра. Изображение нити сле-
дует рассматривать, прикрыв светофильтры папиросной бу-
магой. Устанавливают осветитель с помощью регулировоч-
ных винтов 7.
После проверки установки осветителя можно присту-
пать к измерениям. Прежде всего освобождают арретир
гальванометра, повернув рукоятку 8 из положения «закрыто»
в положение «открыто». По окончании работы делается об-
ратное переключение. После перенесения прибора на новое
место следует с помощью винта 9 установить механический
нуль гальванометра (этого ни в коем случае не следует де-
лать при положении «закрыто»), но перед началом работы
каждый раз необходимо только проверить правильность уста-
новки механического нуля.
При работе с фотоэлектроколориметром ФЭК-М оптиче-
скую плотность (или коэффициент пропускания) можно из-
мерить двумя способами: по правому и левому барабану.
87
По правому барабану измерения проводят следующим об-
разом.
1. Правый барабан устанавливают на делении 0 (при
этом щель диафрагмы имеет наименьшую ширину).
2. И в правый и в левый пучок света вводят кюветы с рас-
творителем, по отношению к которому измеряют плотность
исследуемого раствора. Кюветы устанавливают в кюветодер-
жатели 11, каждый из которых имеет три гнезда для уста-
новки кювет и вращается на оси, имеющей три фиксирован-
ных положения. Правая и левая кюветы должны быть уста-
новлены в кюветодержателях на одинаковом расстоянии от
входного отверстия.
3. Компенсируют фототоки, устанавливая стрелку гальва-
нометра на нуль, пользуясь нейтральными клиньями 4, 5,
с помощью которых меняется освещенность левого фотоэле-
мента.
Включают гальванометр с помощью рукоятки 10, кото-
рая имеет три положения: 0 — гальванометр выключен, 1 — ма-
лая чувствительность, 2 — большая чувствительность. Начи-
нать работу всегда следует при положении рукоятки на ма-
лой чувствительности; большая чувствительность
включается тогда, когда произведено урав-
нивание световых потоков при малой чув-
ствительности. Ни в коем случае не следует ставить ру-
коятку в положение 0, расположенное за положением 2, так
как при обратном переключении сразу же будет включена
большая чувствительность! При положении выключателя 1
компенсация производится сначала грубым клином 4 и затем
тонким клином 5. Точно так же производится компенсация
при положении выключателя 2.
4. Поставив выключатель 10 в положении 0, поворотом
держателя вводят в правый световой поток кювету с испы-
туемым раствором. При этом освещенность правого фотоэле-
мента уменьшается вследствие поглощения света этим рас-
твором, и для уравнивания световых потоков необходимо
раскрыть щель диафрагмы, расположенной на пути правого
потока, поворотом барабана и по шкале правого барабана
произвести отсчет оптической плотности или коэффициента
пропускания.
Поставив поворотом кюветодержателя в правый световой
поток кювету с раствором другой концентрации, можно, не
меняя компенсации нейтральными клиньями, измерить плот-
ность второго раствора и т. д.
Следует только помнить, что первоначаль-
ная компенсация производится всегда при
положении 1 рукоятки 10 и лишь затем вклю-
чается положение 2. Смена кювет произво-
дится при положении 0 этой рукоятки.
S8
Шкала правого барабана имеет значения D от 0 до 0,52;
Г — от 100 до 30%.
Плотность более концентрированных растворов измеряют
по левому барабану.
1. На пути левого светового потока устанавливают кю-
вету с растворителем, на пути правого — с исследуемым рас-
твором. Таким образом, при любом способе измерений в ле-
вом световом потоке всегда находится только кювета с рас-
творителем.
2. Левый барабан устанавливают в положение, соответ-
ствующее 0 оптической плотности (или 100% светопропуска-
ния). При этом диафрагма оказывается полностью откры-
той.
3. Производят компенсацию стрелки гальванометра на 0
с помощью нейтральных клиньев.
4. На пути правого светового потока ставят кювету с рас-
творителем. Ввиду того что освещенность правого фотоэле-
мента после этого возросла, для уравнивания освещенности
необходимо закрывать Диафрагму, пока стрелка гальвано-
метра не придет в нулевое положение. Отсчет производят по
шкале левого барабана.
Следует заметить, что работа с правым барабаном
имеет ряд преимуществ: во-первых, точность отсчета по шка-
ле правого барабана значительно выше ввиду того, что на
полный оборот барабана приходится меньший интервал зна-
чений, например, оптической плотности от 0 до 0,52, в то вре-
мя как на тот же оборот левого барабана приходится интер-
вал значений оптической плотности от 0 до 2,0; во-вторых,
как уже отмечалось, при работе с правым барабаном воз-
можно, произведя однократную компенсацию нейтральными
клиньями, измерить плотность всего ряда эталонных раство-
ров, если светофильтр не меняется.
Определение концентрации испытуемого
раствора начинают с выбора светофильтра.
Поскольку фотоэлектроколориметр ФЭК-М снабжен ограни-
ченным набором из трех широкополосных светофильтров —
зеленого, красного и синего (спектральные характеристики
светофильтров представлены на рис. 46), то не представ-
ляется возможным, пользуясь этим прибором, снять спек-
тральную характеристику испытуемых растворов. Свето-
фильтры устанавливаются попарно на пути световых потоков
с помощью рукоятки 12.
С каждым из светофильтров измеряют оптическую плот-
ность D или пропускание Т одного из растворов эталонного
ряда и выбирают светофильтр согласно общим принципам
(стр. 24). Выбрав нужный светофильтр, концентрацию испы-
туемого раствора определяют, пользуясь одним из описанных
ранее способов, основанных на измерении D (стр. 38).
89
ФОТОЭЛЕКТРОКОЛОРИМЕТР-НЕФЕЛОМЕТР
ФЭК-Н-52, ФЭК-Н-54, ФЭК-Н-57
Данный прибор имеет ту же оптическую схему, что и опи-
санный ранее фотоэлектроколориметр ФЭК-М. Однако в его
конструкцию внесен ряд усовершенствований, расширяющих
возможности этого прибора (общий вид прибора дан на
рис. 35).
Прибор снабжен расширенным набором из девяти узко-
полосных светофильтров, поэтому он может быть использо-
ван как упрощенный спектрофотометр. Так как селеновые
фотоэлементы заменены в нем сурьмяно-цезиевыми, то имеет-
ся возможность использовать светофильтр с /.Эф 360 ммк,
захватывающий ближайшую ультрафиолетовую область. Фо-
тоэлементы включены по дифференциальной схеме через уси-
литель на стрелочный нуль-гальванометр 1. Схема включения
предусматривает предварительную компенсацию «темнового
тока» рукояткой 21, или так называемую установку электри-
ческого нуля, которая производится перед началом измере-
ния при перекрытых с помощью рукоятки 18 световых пото-
ках. В начале работы на приборе, перед тем как перекрыть
световые потоки для установки электрического нуля, следует
некоторое время (15—20 мин) осветить фотоэлементы, от-
крыв щель, чтобы привести их к нормальному режиму, когда
сила фототока становится пропорциональной интенсивности
освещения.
Установка электрического нуля производится, как обыч-
но, сначала на малой, а затем на большой чувствительности.
Усилитель повышает чувствительность прибора и включает-
ся одновременно с осветителем.
Прибор может быть использован как нефелометр. Для
этого необходимо в отверстия, где помещаются линзы, рас-
положенные непосредственно перед кюветным отделением,
вставить точечные диафрагмы 19, сильно суживающие све-
товые потоки, и поставить рукоятку 20 в положение «нефело-
метр». Для нефелометрических измерений имеются три све-
тофильтра—№ 9, 10 и 11. В остальном принцип работы на
этом приборе ничем не отличается от принципа работы на
фотоэлектроколориметре ФЭК-М. Следует только помнить,
что перед началом измерений нужно произвести установку
электрического нуля. (Обозначения деталей на рис. 34 и
рис. 35 тождественны.)
ФОТОЭЛЕКТРОКОЛОРИМЕТР-НЕФЕЛОМЕТР ФЭК-56
Фотоэлектрический колориметр-нефелометр ФЭК-56 име-
ет оптику, которая позволяет производить измерения в об-
ласти спектра от 315 до 630 лгл/к. Приемниками световой энер-
90
Рис. 35. Внешний вид фотоэлектроколориметров ФЭК-Н-52,
ФЭК-Н-54. ФЭК-Н-57
гии служат два сурьмяно-цезиевых фотоэлемента. В качестве
источников света в приборе используют две лампы: лампу
накаливания, дающую сплошной спектр испускания в види-
мой области, и ртутно-кварцевую лампу, дающую линейчатый
спектр испускания в ультрафиолетовой и видимой областях.
В качестве монохроматоров служат светофильтры с узкими
полосами пропускания. Следовательно, прибор может быть
использован для изучения спектров поглощения. Максимумы
пропускания большинства этих светофильтров практически
совпадают с рядом линий в эмиссионном спектре ртути. По-
этому с ртутно-кварцевой лампой можно производить изме-
рения в ультрафиолетовой и видимой областях спектра
с очень узкими монохроматическими пучками при следующих
длинах волн (ммк): 577,9; 546; 436; 405,8; 365; 313.
Оптическая схема прибора дана на рис. 36, общий вид —
на рис. 37. Свет от источника 1, пройдя через светофильтр 2,
попадает на призму 3, которая делит пучок на два: левый и
правый. Далее параллельные пучки света идут через кю-
веты 4, диафрагмы 5 и 5' и попадают на фотоэлементы 6,
включенные по дифференциальной схеме через усилитель на
индикаторную лампу 7. На пути левого светового потока по-
стоянно находится кювета с «нулевым» раствором (в ча-
стности, с растворителем). На пути правого светового потока
могут быть последовательно установлены кюветы с испытуе-
мым и «нулевым» растворами. Интенсивности световых пото-
ков, проходящих через правую и левую кюветы, уравнивают
с помощью диафрагм «кошачий глаз»: левая диафрагма 5'
92
Рис. 37. Внешний вид фотоэлектроколоримстра ФЭК-56
служит для компенсации световых потоков при установлен-
ном на нуль барабане правой диафрагмы 5; правая'диафраг-
ма является измерительной — отсчет относительной величины
поглощения испытуемого раствора по сравнению с «нулевым»
производится всегда по правому барабану (о порядке изме-
рений см. далее).
Неодинаковая освещенность фотоэлементов вызывает раз-
мыкание сектора индикаторной лампы, используемой в дан-
ном случае в качестве прибора-индикатора вместо галввано-
Рис. 38. Питающее устройство к
фотоэлектроколориметру ФЭК-56
метра. Интенсивность световых потоков, падающих на каж-
дый фотоэлемент, выравнивается изменением ширины щели
диафрагмы до тех пор, пока сектор индикаторной лампы не
сомкнется.
Лампа накаливания и ртутно-кварцевая лампа имеют
каждая собственную панель 8, с помощью которых их пооче-
редно устанавливают в осветителе 9, вдвигая по пазам 10 н
закрепляя винтом И. Лампу накаливания 12 включают в те-
лефонные гнезда 13, ртутно-кварцевую лампу 14 — в теле-
фонные гнезда 15.
Усилитель и источник света получают питание от сети
переменного тока через специальное питающее устройство
(рис. 38). Питающее устройство соединяют с прибором штеп-
сельным разъемом 16. Для включения в работу соответствую-
щей лампы (накаливания или ртутно-кварцевой) на панели
питающего устройства имеется переключатель 17.
94
Порядок работы. Питающее устройство соединяют
с прибором штепсельным разъемом 16 и включают в сеть
переменного тока с напряжением 220 в с помощью шланга 18.
Переключателем 17 включают в работу соответствующую
лампу. Ставят переключатель 19 в положение «включено» и
дают усилителю и лампе прогреться 20—30 мин. При работе
с ртутно-кварцевой лампой усилитель прогревают при вы-
ключенной лампе, включая ее за 5 мин до начала измерений.
Устанавливают лампу с помощью юстировочных
винтов 20. В трубки измерительных диафрагм вставляют
металлические пробки, имеющиеся в комплекте прибора. На-
блюдая положение кружков света на поверхности пробок,
с помощью юстировочных винтов 20 добиваются положения,
когда пучки света будут располагаться концентрично отно-
сительно трубок измерительных диафрагм.
К измерениям приступают после проверки уста-
новки осветителя. Устанавливают электрический нуль при-
бора; нажав в направлении оси и повернув в положение «3»
(закрыто) рукоятку 21, перекрывают световые пучки и ру-
кояткой 24 приводят сектор индикаторной лампы в сомкну-
тое положение. После этого вновь открывают шторку рукоят-
кой 21, нажав вдоль ее оси и повернув в положение «О» (от-
крыто). В левый кюветодержатель устанавливают кювету
с «нулевым» раствором, в правый — кювету с измеряемым
раствором. Шкалу правой диафрагмы рукояткой 5 устанав-
ливают в положение, соответствующее значению D = Q (крас-
ная шкала) или значению Т = 100 (черная шкала). Вращая
рукоятку 5' левой диафрагмы, добиваются сомкнутого поло-
жения сектора индикаторной лампы. После этого с помощью
рукоятки 23 заменяют в правом световом потоке кювету с ис-
следуемым раствором на кювету с «нулевым» раствором. Ме-
няя щель правой диафрагмы рукояткой 5, приводят вновь
сектор индикаторной лампы в сомкнутое состояние. По шка-
ле правой диафрагмы 22 снимают показания оптической
плотности D или пропускания Т.
Спектр поглощения изучают путем измерения
поглощения исследуемого раствора с различными свето-
фильтрами и построения зависимости D от К, используя в ка-
честве К табличные данные для максимума пропускания соот-
ветствующего светофильтра. Для количественных определе-
ний используют светофильтр, соответствующий наибольшей
величине D или наименьшей величине Т.
Светорассеяние, вызываемое взвесями, эмульсиями и
коллоидными частицами, измеряют турбидиметрически (в про-
ходящем свете). Светорассеяние растворов со слабой мут-
ностью на данном приборе измерить невозможно.
Примечание. Большое значение для получения надежных резуль-
татов измерений имеет чувствительность прибора, которая определяется
95
числом делений шкалы, на которое нужно повернуть барабан для полного
размыкания сектора индикаторной лампы (чем это число меньше, тем выше
чувствительность прибора). Чувствительность проверяют следующим об-
разом: 1) устанавливают электрический нуль прибора и открывают штор-
ку; 2) включают светофильтр № 6; 3) по шкале правого барабана уста-
навливают деление Т=80%; 4) поворотом левого барабана приводят сектор
индикаторной лампы в сомкнутое положение; 5) меняя щель правой диаф-
рагмы рукояткой барабана 5, приводят сектор в полностью разомкнутое
состояние. Отсчет по шкале правого барабана должен при этом находиться
в пределах значений 73—87. Если полученные значения выходят далеко
за пределы этого интервала, то это указывает на неисправность одного из
следующих электроузлов: индикаторной лампы, фотоэлемента или лампы
6Н8С усилителя.
СПЕКТРОФОТОМЕТРЫ СФ-4, СФ-4А и СФ-5
Нерегистрирующий фотоэлектрический кварцевый спектро-
фотометр СФ-4 служит для измерения коэффициентов пропу-
скания и оптических плотностей жидких и твердых веществ.
Монохроматизация света в данном приборе достигается
с помощью кварцевой диспергирующей призмы, от которой
пучок света попадает на щель, имеющую переменную вели-
чину и обеспечивающую дополнительную монохроматизацию.
Поскольку вся оптика в спектрофотометре сделана из кварца,
то возможно изучать спектры поглощения веществ не только
в видимой, но также в ультрафиолетовой и ближней инфра-
красной областях спектра. Эти измерения можно проводить
в интервале от 220 до 1100 ммк.
Монохроматизация светофильтрами в этом приборе отсут-
ствует. Имеющиеся в оптической схеме прибора два фильтра
10 (рис. 40) используются только для предотвращения попа’-
дания на щель рассеянного света, отраженного от различных
оптических деталей конструкции. Эти фильтры имеют очень
широкую область пропускания: с УФС-2 работают в области
от 320 до 400 ммк; с ОС-14 — от 580 до 620 ммк.
Общий вид прибора дан на рис. 39, а, б; оптическая схе-
ма— на рис. 40. Лучи света от источника 1 падают на зер-
кало-конденсор 2, которое собирает и направляет пучок лу-
чей на плоское зеркало 3, поворачивающее лучи на 90° и на-
правляющее их через защитную кварцевую пластинку 4 на.
входную щель монохроматора 5. Зеркальный объектив 6,
в фокусе которого расположена щель, направляет параллель-
ный пучок лучей на кварцевую диспергирующую призму 7
с отражающей задней гранью. Призма разлагает свет в
спектр и направляет его обратно на объектив. Луч, прошед-
ший призму под углом, близким к углу наименьшего откло-
нения, попадает на выходную щель 8, расположенную под
входной щелью. Поворачивая призму вокруг оси, можно по-
лучить на выходе монохроматора лучи различных длин волн.
Выходящий монохроматический пучок света проходит кварце-
вую линзу 9, фильтр 10, измеряемый образец 11, линзу 12 и
96
Рис. 39. Внешний вид спектрофотометра СФ-4
4 Зак. 686
падает на светочувствительный слой фотоэлемента 13. Фото-
ток, возникающий в фотоэлементе под влиянием падающей
световой энергии, усиливается электронными радиолампами
и передается на миллиамперметр (прибор-индикатор) 14. Ве-
личину поглощения света испытуемым объектом измеряют
на СФ-4 относительно какого-либо образца", поглощение ко-
торого принимается равным нулю. Поэтому сущность измере-
ний на спектрофотометре сводится к следующему.
Вначале, когда на пути светового потока стоит «нулевой»
образец, стрелка миллиамперметра приводится к нулю с по-
Рис. 40. Оптическая схема спектрофотометра СФ-4
мощью одного потенциометра. При установке на пути пучка
света измеряемого образца с иным поглощением, чем «нуле-
вое», изменение интенсивности светового потока, падающего
на фотоэлемент, и соответствующее этому изменение тока
вызовет отклонение стрелки миллиамперметра, которая мо-
жет быть снова приведена к нулю путем изменения сопротив-
ления на другом потенциометре, соединенном с отсчетными
шкалами. Более подробно порядок работы на приборе будет
изложен ниже.
Источники освещения. Для обеспечения работы в
широком интервале длин волн в приборе имеются два источ-
ника освещения со сплошным спектром излучения: водород-
ная лампа для измерений в области 220—350 ммк и лампа
накаливания для измерения в области 320—1100 ммк.
Лампы устанавливают в специальные осветители 31, ко-
торые попеременно укрепляют на кронштейне 15, приделан-
98
ном к корпусу прибора. Внутри каждого осветителя имеется
панель для укрепления лампы и зеркало-конденсор, которое
направляет пучок света, идущий от лампы, в сторону патруб-
ка, соединяющего осветитель с корпусом прибора. Панели
снабжены четырьмя винтами, которые используются при на-
стройке ламп: один поворачивает лампу вокруг оси, два дру-
гих двигают ее в продольном и поперечном направлениях,
четвертый винт передвигает лампу в вертикальном направ-
лении. Винты поворачивают специальным ключом, имею-
щимся в комплекте, прилагаемом к прибору. Помимо этого,
при настройке лампы можно пользоваться винтом 2, вращаю-
щим конденсорное зеркало; винт поворачивают с помощью
часовой отвертки.
Лампа накаливания питается от аккумулятора напряже-
нием 6 в. Во время работы следует проверить, насколько хо-
рошо припаяны к клеммам аккумулятора концы провода от
лампы, так как в случае плохих контактов невозможно обес-
печить постоянную устойчивую эмиссию лампы, что совер-
шенно необходимо в процессе измерений.
Питание водородной лампы от сети высокого напряжения
осуществляется через электронный стабилизатор, стабилизи-
рующий колебания напряжения в сети не более 30%. Если
колебания в сети высокого напряжения превышают 30%, то
необходима дополнительная стабилизация, которая может
быть осуществлена путем параллельного подключения еще
одного стабилизатора. Включают лампу через стабилизатор
согласно прилагаемой инструкции.
Следует помнить, что ток накала включают при пол-
ностью введенном потенциометре (рукоятка его находится
при этом в крайнем левом положении) и лишь постепенно
ток накала доводится поворотом рукоятки потенциометра на-
право до значения, указанного в паспорте лампы и контро-
лируемого амперметром, подключенным к стабилизатору. Вы-
сокое напряжение включают лишь через 10 мин после вклю-
чения тока накала. Выключают стабилизатор в обратном по-
рядке: сначала высокое напряжение, а затем ток накала.
Помимо ламп, используемых в процессе измерений в ка-
честве источников освещения, в комплекте прибора имеется
также ртутная лампа, обладающая линейчатым спектром,
которая используется для проверки градуировки шкалы длин
волн. Питание этой лампы осуществляется так же, как и во-
дородной, от сети переменного тока высокого напряжения,
и при работе с ней при включении и выключении следует со-
блюдать т^же предосторожности.
Фотоэлементы и питание усилительной
схемы. Так как данный прибор работает в достаточно боль-
шом интервале длин волн от 220 до 1100 ммк, нет возмож-
ности подобрать такой фотоэлемент, который был бы чув-
4*
99
падающеи на них световой
Рис. 41. Панель для подклю-
чения батарей БАС-Г-60 и кар-
манного фонаря в спектрофо-
тометре СФ-4
ствителен к излучениям в этом интервале, поэтому в приборе
имеются два фотоэлемента: сурьмяно-цезиевый фотоэлемент
для работы в интервале от 220 до 650 ммк и «ислородно-це-
зиевый — в интервале 600—1100 ммк. Переключение с од-
ного фотоэлемента на другой производится при 625 ммк
с помощью рукоятки 16-. при полностью вдвинутой рукоятке
на пути пучка света устанавливают сурьмяно-цезиевый фото-
элемент, при полностью выдвинутой — кислородно-цезиевый.
Фототоки, возникающие в фотоэлементах под влиянием
нергии, передаются на усилитель-
ное устройство, питание которого
осуществляется, с одной стороны,
от аккумулятора напряжением
6 в (от которого питается также
лампа накаливания), с другой
стороны, от сухой анодной бата-
реи БАС-Г-60 и батарей для кар-
манного фонаря. Напряжение от
этих источников, подаваемое на
усилитель, должно находиться
под постоянным ежедневным
контролем, так как в случае да-
же незначительного его отклоне-
ния от предусмотренного по схеме
невозможно наладить правильную
работу прибора. Поэтому каждый
раз перед началом работы следу-
ет проверять напряжение на акку-
муляторе (которое под нагрузкой, т. е. при включенной лампе
накаливания и включенном спектрофотометре, должно быть
не менее 6 в) и на концах анодной сухой батареи и батарей
для карманных фонарей. Анодные сухие батареи БАС-Г-60
и батареи для карманного фонаря помещены в ящцк, распо-
ложенный в задней части прибора. Подключение батарей
производится через панель (рис. 41), к которой с другой сто-
роны подключены провода, передающие напряжение от ба-
тарей на усилитель. На этой панели в положении 1 находится
общий минус для трех отводов от анодной сухой батареи.
Между этим минусом и клеммой 2, а также между клеммами
1 и 3 напряжение должно быть не менее 30 в; между клем-
мами 1 и 4 не менее 12 в. К клеммам 5 и 6 подключаются
соответственно + и — от двух батарей для карманного фо-
наря, соединенных между собой последовательно. Напряже-
ние между этими клеммами должно быть не менее 8 в. Если
напряжение на клеммах будет меньше указанных значений,
то следует сменить батареи. В случае отсутствия батарей
БАС-Г-60 можно заменить их последовательно соединенными
батарейками для карманного фонаря. Следует еще заметить,
100
что во избежание всяческих помех в работе и для обеспече-
ния точности и воспроизводимости результатов измерений на
приборе, особое внимание необходимо уделять качеству кон-
тактов при подсоединении аккумулятора и питающих бата-
рей. Концы шланга, идущие от усилительного устройства и
лампы накаливания, а также от сухих батарей к аккумуля-
тору, должны быть накрепко припаяны к клеммам аккумуля-
тора и панели.
Кюветы. В комплекте спектрофотометра имеются два
вида кювет: прямоугольные с толщиной слоя в 1 см и ци-
линдрические с различной толщиной слоя (рис. 42). Прямо-
угольные кюветы сделаны из оптического кварца и могут
быть использованы для работы в любой области спектра, до-
ступной данному прибору. Для них имеется специальный ме-
таллический держатель (рис. 43) с четырьмя гнездами.
В одном из этих гнезд устанавливают кювету с раствором,
поглощение которого в данной работе принимается равным
нулю (чаще всего это растворитель). В три остальных гнез-
да вставляют кюветы с' исследуемыми растворами. Таким
образом, можно одновременно измерять поглощение трех
растворов, если нас интересует их поглощающая способность
по отношению к одному и тому же растворителю. Металличе-
ский держатель с кюветами помещают в кюветное отделение
(рис. 39, а, 17) таким образом, чтобы белая точка-указатель
на каретке в кюветном отделении соответствовала точке-ука-
зателю на держателе кювет. На это следует обращать вни-
мание, так как каретка имеет четыре фиксированных поло-
жения (соответственно числу установленных в держателе
кювет), обозначенных на ее рукоятке (рис. 39, а, 18) и соот-
ветствующих только данному держателю и при данной его
установке в каретку. Поэтому ни в коем случае нельзя поль-
зоваться держателем, взятым из другого спектрофотометра.
Цилиндрические кюветы комплектуются попарно: по две
кюветы с толщиной слоя (л-ш): 4,05; 4,10; 4,20; 4,50; 5,0; 10; 20;
50 и 100. Соответственно имеются по два металлических дер-
жателя (рис. 43) для каждой из пяти последних пар кювет,
но кюветы с толщиной слоя от 4,0 до 4,5 мм вставляются в
держатели для кювет с толщиной в 5,0 мм. Точные данные
относительно толщины слоя кювет и их пропускаемости при-
ведены в аттестате соответствующего прибора. Эти кюветы
(рис. 42) представляют собой стеклянные стаканы, закры-
вающиеся с обоих сторон крышками, кварцевыми или стек-
лянными в зависимости от того, в какой спектральной об-
ласти предполагается вести работу. Стеклянные крышки обо-
значены буквой «С» и используются для работы в области
400—1000 ммк.
Наполняют кюветы следующим образом. Одна из кры-
шек помещается на чистую твердую подкладку и сбоку на
101
Рис. 42. Кюветы к спектрофотометру СФ-4: 1 — прямоугольные;
2 — цилиндрические; 3 — тефлоновые
Рис. 43. Металлические держатели для кювет к спектрофотометрам
СФ-4 и СФ-2М; 1 — держатель для прямоугольных кювет; 2 — дер-
жатель для цилиндрических кювет; 3 — держатель для твердых об-
разцов; 4 — держатель для кювет к прибору СФ-2М; 5 — держатель
для тефлоновых кювет
102
нее надвигают стакан кюветы. Затем наполняют кювету рас-
твором до образования выпуклого мениска. Вторую крышку
надвигают сверху так, чтобы в стакане не осталось пузырь-
ков воздуха. После этого осторожно снимают кювету с под-
кладки, придерживая крышки, и вставляют ее в отвинченную
крышку металлического держателя, в которую предваритель-
но помещают каучуковую прокладку. Сверху помещают вто-
рую каучуковую прокладку и навинчивают металлический
стакан вместе со второй металлической крышкой. Кюветы
следует держать в чистоте, не оставлять надолго наполнен-
ными исследуемым раствором и мыть сразу после окончания
работы во избежание разъедания стенок и адсорбции на них
веществ, изменяющих пропускаемость. Моют кюветы водой,
но в случае сильного загрязнения стенок их можно опустить
ненадолго в концентрированную азотную кислоту. Следует
еще указать на необходимость периодической проверки про-
пускаемое™ прямоугольных кювет, наполненных дистиллиро-
ванной водой, и сопоставления полученных данных с данны-
ми аттестата. Это необходимо делать ввиду того, что со вре-
менем на стенках кюветы могут адсорбироваться посторон-
ние вещества, которые вызовут значительные ошибки при
дальнейшей работе с этими кюветами.
Порядок работы на приборе. Прежде чем при-
ступить к измерению оптических плотностей исследуемого
объекта, необходимо проверить работу всех узлов прибора,
а именно: 1) правильность градуировки шкалы длин волн
прибора; 2) правильность показаний шкалы оптических плот-
ностей.
Проверяют градуировку шкалы длин волн
(рис. 39, а, 29) по линиям ртутного спектра. Для этого в
комплекте прибора, как уже говорилось выше (стр. 99),
имеется ртутная лампа. Длины волн эмиссионного спектра
ртути можно найти в любом атласе спектральных линий, но,
помимо этого, в аттестате каждого прибора имеется таблица
длин волн линий ртутной лампы, по которым проверяют гра-
дуировку шкалы при заводском выпуске прибора. Сущность
данной проверки заключается в том, что, наблюдая тем
или иным путем (визуально или фотоэлектрически) про-
хождение через щель определенной линии ртути при враще-
нии рукоятки (рис. 39, а, 19) шкалы длин волн, останавли-
вают это вращение.как раз в тот момент, когда через щель
проходит максимум линии, и сравнивают полученное показа-
ние шкалы с данными атласа или аттестата.
В комплекте прибора имеется зрительная труба для ви-
зуальной градуировки прибора.
Визуальную градуировку целесообразно произ-
водить только при первоначальной установке прибора или пе-
реносе его на новое место, т. е. в тех случаях, когда можно
103
ожидать значительный сдвиг шкалы. Делают это следующим
образом: отвернув стопорные винты, снимают камеру фото-
элементов (рис. 39, а, 20), выдвигая ее вверх; на ее место
устанавливают зрительную трубку (рис. 39, б, 21). На крон-
штейн для осветителя устанавливают ртутную лампу. Уста-
новку ртутной лампы проверяют визуально: поворотом ру-
коятки (рис. 39, а, 22) открывают щель на полную ширину —
2 мм. Шкалу длин волн устанавливают на Л 546,1 ммк (зе-
леная линия ртути). В кюветное отделение со стороны, про-
тивоположной щели, вставляют лист белой бумаги и наблю-
дают освещенность прямоугольника, являющегося изображе-
нием щели. При правильной установке лампы этот прямо-
угольник должен равномерно освещаться зеленым светом.
Если освещенность прямоугольника неравномерна или на-
блюдаются затемненные углы, то следует, пользуясь спе-
циальным ключом и часовой отверткой, настроить лампу
с помощью винтов и конденсорного зеркала, как это было
описано выше (стр. 99), до достижения равномерного запол-
нения прямоугольника щели зеленым светом.
После этого можно приступить непосредственно к градуи-
ровке. Визуальную градуировку производят по той же зеле-
ной линии ртути Л 546,1 ммк. Щель уменьшают настолько,
чтобы в трубку можно было видеть только резкую линию
(т. е. до 0,02 — 0,04 мм). Поворачивая объектив зрительной
трубки, фокусируют изображение щели, добиваясь рез-
кости. Передвигая шкалу длин волн с помощью рукоятки
(рис. 39, а, 19) и подводя линию 546,1 ммк со стороны корот-
ких длин волн (например, от 530 ммк), наблюдают момент
наиболее яркого освещения щели, что соответствует прохож-
дению через щель максимума интенсивности этой линии.
В этот момент останавливают вращение шкалы длин волн
и смотрят, насколько точно показание шкалы (рис. 39, а, 29)
совпадает с данными аттестата. Если отклонение невелико
(не более 3—5 ммк), то можно перейти к фотоэлектрической
проверке градуировки. Если же отклонение более значитель-
но, то следует его уменьшить поворотом зеркального объек-
тива 30 (как это будет указано при фотоэлектрической гра-
дуировке) .
Фотоэлектрическая, более точная, проверка градуи-
ровки производится (в отличие от визуальной) системати-
чески по прошествии небольшого срока работы на приборе
(через 1—2 месяца), так как даже при постоянном нахожде-
нии прибора на одном и том же рабочем месте шкала длин
волн может сместиться в процессе работы.
Ставят переключатель 23 в положение «Х1» и при закры-
той шторке-переключателе 24 (опущена вниз) устанавливают
стрелку миллиамперметра 14 на нуль поворотом рукоятки
«темнового тока» 25 (т. е. компенсируют темновой ток). За-
104
тем устанавливают потенциометр чувствительности в рабочее
положение, т. е. делают 4—5 оборотов рукоятки 26 от край-
него положения (безразлично, правого или левого) и измери-
тельный потенциометр 27 ставят на 3—5% пропускания. От-
крывают фотоэлемент (рукоятка 24 поднята вверх до отказа)
и, регулируя щель, приводят стрелку миллиамперметра 14 в
пределы шкалы. Медленно вращая шкалу длин волн, подво-
дят линию 546,1 ммк; при этом стрелка двигается влево. Ре-
гулируя щель, следует удерживать стрелку в пределах шка-
лы. В момент прохождения через щель максимума интенсив-
ности линии наблюдается также максимальное отклонение
стрелки миллиамперметра влево. Вращение шкалы длин волн
следует остановить как раз в этот момент: до начала обрат-
ного движения стрелки вправо.
Отчет по шкале 29 должен быть равным 546,1 ммк с от-
клонением, не превышающим указанного в аттестате для
данной длины волны. Таким же образом нужно проверить
градуировку во всем диапазоне от 220 до 1100 ммк по дли-
нам волн, указанным в аттестате. Если не наблюдается до-
статочно хорошего совпадения в показаниях, то необходимо
этого добиться поворотом зеркального объектива 30. Для
этого крышку с надписью «регулировка зеркала» снимают и
зеркальный объектив поворачивают с помощью специального
ключа, имеющегося в комплекте прибора.
Визуальную установку водородной лампы
производят прежде, чем приступают к измерениям. Ее уста-
навливают так же, как ртутную лампу, но по красной линии
водорода 656 ммк. Фотоэлектрическую установку
водородной лампы производят следующим образом:
переключатель 23 ставят в положение «включ.», и при закры-
том фотоэлементе стрелку миллиамперметра приводят к нулю
с помощью потенциометра «темнового тока» 25. Устанавли-
вают ширину щели 2 мм (максимальное раскрытие) и потен-
циометр чувствительности 26 ставят в крайнее левое поло-
жение (минимум чувствительности), открывают шторку фото-
элемента и вращают шкалу длин волн с помощью рукоятки
19 в сторону меньших длин волн. Значение длины волны, при
котором стрелка миллиамперметра приходит к нулю, срав-
нивают с паспортом данной лампы, в котором указана пре-
дельная граница излучения лампы в ультрафиолетовой об-
ласти. Если показания шкалы длин волн и данные паспорта
отличаются более чем на 2—3 ммк, то, следовательно, водо-
родная лампа установлена плохо и ее устанавливают с по-
мощью юстировочных винтов и конденсорного зеркала, о ко-
торых уже говорилось выше (стр. 99). Возможность работать
в крайней ультрафиолетовой области, допускаемой излуче-
нием данной водородной лампы, может быть достигнута
только в условиях оптимального фототока, возникающего в
105
фотоэлементе,!, е. при полностью открытой щели (2 мм) и ми-
нимальной чувствительности.
Лампу накаливания также можно установить визуально
и фотоэлектрически. Визуальная ее установка производится
аналогично установке ртутной лампы по зеленой линии
546,1 ммк. При фотоэлектрической установке учитывается,
что интенсивность излучения лампы накаливания неодина-
кова во всем рабочем интервале. Максимум интенсивности
приходится на область 520—550 ммк. Следовательно, в этой
области имеется возможность для работы с минимальной
щелью.
Исходя из этого установку лампы накаливания проверяют
следующим образом. Скомпенсировав, как обычно, темновой
ток при закрытом фотоэлементе, устанавливают по шкале
длин волн значение 546,1 ммк (находящееся как раз в об-
ласти максимальной интенсивности излучения этой лампы).
Поворачивают на 4—5 оборотов потенциометр чувствитель-
ности от одного из крайних положений, т. е. устанавливают
его в усредненное положение. Открыв шторку фотоэлемента,
приводят стрелку миллиамперметра к нулю, уменьшая щель.
Если стрелка миллиамперметра приводится к нулю при рас-
крытии щели не более чем на 0,02—0,03 мм, то установку
лампы накаливания можно считать удовлетворительной.
Большее раскрытие щели, требующееся в этом случае, за-
ставит работать при иных длинах волн с еще более широкой
щелью, что, понятно, не выгодно. Поэтому плохо установлен-
ную лампу следует настроить так же, как водородную, с по-
мощью юстировочных винтов (стр. 99).
Далее можно приступить к измерениям.
Прежде всего следует проверить правильность показания
шкалы оптических плотностей. Поворотом шкалы длин волн
устанавливают ту длину волны, при которой хотят измерить
оптическую плотность или процент пропускания.
Ставят переключатель 23 в положение «включ.» и при за-
крытой шторке-переключателе 24 устанавливают стрелку
миллиамперметра на нуль с помощью потенциометра «темно-
вого тока» 25. Против щели устанавливают кювету с рас-
твором, поглощение которого в данной работе принимается
равным нулю. Открыв шторку-переключатель, устанавливают
стрелку миллиамперметра на нуль, меняя ширину щели ру-
кояткой 22 или пользуясь потенциометром чувствитель-
ности 26. Таким образом добиваются установки нуля отсчета
по измерительному потенциометру. Однако при положении
переключателя на «включ.» измерительный потенциометр
оказывается отключенным и его положение в период только
что описанных операций в сущности безразлично. Если же-
лательно проверить правильность компенсации на «нулевой»
раствор, то необходимо поставить переключатель 23 в поло-
106
жение «X 1» и тем самым подключить измерительный потен-
циометр. Стрелка миллиамперметра должна приводиться
к нулю при установке измерительного потенциометра на нуль
оптической плотности или на 100% пропускания. Стрелка
миллиамперметра не приводится к нулю при недостаточно
точной компенсации на «нулевой» раствор и в случае боль-
шой ошибки в показаниях отсчетного потенциометра.
Совершенно очевидно, что подобную проверку не следует
производить перед каждым измерением и имеет смысл сде-
лать ее лишь при общей проверке правильности градуировки
шкалы отсчетного потенциометра. Для удобства же работы
конструкцией прибора предусмотрена компенсация на «нуле-
вой» раствор при отключенном отсчетном потенциометре.
После компенсации на «нулевой» раствор перед щелью
устанавливается кювета с исследуемым раствором, переклю-
чатель устанавливают в положение «X 1», отсчетным потен-
циометром приводят стрелку миллиамперметра к нулю и по
его шкале отсчитывают оптическую плотность или процент
пропускания. Измерять при новой длине волны начинают
также с установки длины волны и далее повторяют все вы-
шеописанные операции.
Для того чтобы монохроматизация света была достаточно
высокой, выгодно работать при возможно более узкой щели.
Кроме того, изменение ширины щели ведет хотя и незначи-
тельному, величины оптической плотности (соответствен-
но процента пропускания). Поэтому для нахождения точ-
ного положения максимума необходимо все измерения
производить при постоянной ширине щели. Однако при изу-
чении спектра поглощения в широком интервале длин волн
невозможно сохранить постоянной ширину щели (в силу
различной интенсивности источников света в различных уча-
стках спектра и различной чувствительности фотоэлемента
к различным длинам волн). Поэтому целесообразно при из-
мерениях в широкой спектральной области компенсацию на
«нулевой» раствор производить как с помощью щели (не фик-
сируя ее), так и потенциометром чувствительности.
При измерениях же в области обнаруженного максимума
щель следует сохранять постоянной, выбрав ее минимально
возможной в данном участке спектра, а для компенсации на
«нулевой» раствор пользоваться только потенциометром чув-
ствительности.
Правильность показаний отсчетного по-
тенциометра можно проверить по нейтральным фильт-
рам НС-6 и НС-8, имеющимся в комплекте прибора, вели-
чина пропускания которых при различных длинах волн дана
в аттестате прибора. Кроме того, можно использовать рас-
творы ряда веществ (например, KiCrCh, CuSO4 и т. п.), имею-
щих постоянные величины молярных коэффициентов погаше-
107
ния при определенных длинах волн. Рецепты приготовления
таких растворов даются в литературе
Приспособление для спектрофотометриче-
ского титрования к спектрофотометру СФ-4 довольно
простое. В крышке кюветного отделения проделывают два
отверстия, одно для микробюретки, второе для механической
мешалки. Кювета из оптического стекла должна иметь объем
около 25 мл (для титрования в видимой области спектра
можно использовать кювету от фотоэлектроколориметра
ФЭК-М). Стенки кюветы покрывают черным лаком, остав-
ляя отверстие диаметром в 1 см на пути светового потока.
Мешалка вводится через отверстие в крышке кюветного от-
деления так, чтобы ее конец находился против затемненного
участка кюветы. Раствор реактива (достаточно концентриро-
ванный) прибавляют из микробюретки, чтобы избежать зна-
чительного разбавления титруемого раствора (стр. 66).
Спектрофотометр СФ-4А отличается от спектрофо-
тометра СФ-4 только модернизированной электрической схе-
мой. Усилитель питается не от аккумулятора и сухих бата-
рей, а от сети высокого напряжения через единый блок пита-
ния, включающий стабилизатор и выпрямитель. Все лампы
питаются также от сети высокого напряжения через стаби-
лизатор. Лампы устанавливаются в одном и том же освети-
теле, который имеет два цоколя: один для установки лампы
накаливания, второй — для водородной и ртутной ламп, уста-
навливаемых попеременно. Для того чтобы направить в при-
бор через входное отверстие световой пучок от соответствую-
щей лампы, необходимо лишь повернуть зеркало-конденсор,,
которое помещается в осветителе между лампами. На ящике
блока питания имеются выключатели для включения усили-
теля и ламп; пользоваться выключателями следует строго
согласно инструкции, прилагаемой к прибору.
Для расширения границ точности измерения потенциомет-
ры «чувствительности» и «темнового тока» снабжены пере-
ключателем на четыре рабочих интервала. Рукоятки пере-
ключателей расположены на передней стенке прибора рядом
с рукоятками соответствующих потенциометров. Положение
1 рукоятки потенциометра «чувствительности» соответствует
самой высокой точности измерения, но требует работы с ши-
рокой щелью, положению 4 соответствует обратная зависи-
мость.
В целом порядок работы на приборее и его возможности
остаются такими же, как и для спектрофотометра СФ-4.
Спектрофотометр СФ-5 в принципе ничем не отли-
чается от спектрофотометра СФ-4. В отличие от последнего-
он имеет стеклянную оптику, поэтому область длин волн.
1 Абсорбционная спектроскопия. Сб. статей. М., ИЛ, 1953, стр. 128.
108
в которой возможно производить измерения, пользуясь этим
прибором, ограничена интервалом 380—1100 ммк. Прибор
снабжен теми же фотоэлементами и набором кювет, что и
СФ-4, с той лишь разницей, что сваренные прямоугольные
кюветы сделаны из оптического стекла, а не из кварца и
в комплекте отсутствуют кварцевые крышки к цилиндриче-
ским кюветам. Поскольку нет возможности работать в
ультрафиолетовой области спектра, в комплекте прибора име-
ются только два источника освещения: ртутная лампа — для
градуировки шкалы длин волн и лампа накаливания-—для
измерений. Лампа накаливания питается не от аккуму-
лятора, а через стабилизатор так же, как ртутная. Каждая
лампа имеет свой собственный держатель, который после
установки его на кронштейне закрывается кожухом, общим
для той и другой лампы.
В остальном конструкция прибора, его оптическая и элек-
трическая схемы, а также порядок работы на нем сохраня-
ются совершенно теми же, что и для спектрофотометра СФ-4.
РЕГИСТРИРУЮЩИЕ СПЕКТРОФОТОМЕТРЫ
СФ-2М, СФ-10
Регистрирующий спектрофотометр позволяет записывать
на специальном бланке спектры поглощения и пропускания,
а также измерять коэффициенты отражения различных об-
разцов. Запись по всей длине видимого спектра может быть
произведена несравненно в более короткое время, чем промер
этого же участка спектра на спектрофотометре СФ-4. Прибор
имеет двойной монохроматор, поэтому монохроматизация
света здесь достаточно высокая. Ширина входной и выход-
ной щелей того и другого монохроматора изменяется во вре-
мя работы прибора автоматически соответственно дисперсии
призм. Таким образом, при достаточно высокой монохрома-
тизации вырезается спектральный участок постоянного спек-
трального интервала. Однако рабочий диапазон прибора ох-
ватывает только видимую часть спектра от 400 до 700 ммк и,
следовательно, он обладает в этом отношении меньшими
возможностями, чем нерегистрирующий кварцевый спектро-
фотометр СФ-4.
Общий вид прибора дан на рис. 44, принципиальная опти-
ческая схема — на рис. 45. Свет от источника 1 падает на
входную щель 2, расположенную в фокальной плоскости
объектива коллиматора 3. Выходящий из него параллельный
пучок света проходит первую диспергирующую призму 4 и
претерпевает разложение в спектр.
Объектив дает изображение спектра в плоскости средней
щели (Л — Л), образованной ножом 5 и изображением его
109
3/. /S'
Рис. 44. Внешний вид спектрофотометра СФ-2М
в плоском зеркале 6. Требуемый участок спектра вырезается
перемещением зеркала и ножа вдоль спектра. Вырезанный
участок спектра проходит через второй монохроматор 4',
действие которого аналогично действию первого, и проекти-
руется в плоскость выходной щели 7. Пройдя далее ряд пло-
скополяризующих призм, свет в виде двух расходящихся пуч-
ков проходит кюветное отделение 8 и через передние окна 9
интегрирующей сферы 10 падает на плоскость ее задних
/О
Рис. 45. Принципиальная схема спектрофотометра СФ-2М
окон 11. В интегрирующей сфере происходит суммирование
двух световых пучков, и свет суммарной интенсивности попа-
дает затем на фотоэлемент 12. Освещенность сферы являет-
ся функцией суммарной мгновенной величины интенсивности
света, отраженного от двух эталонов (в случае измерения
оптических плотностей и процентов пропускания) или образ-
ца и эталона (в случае измерения коэффициентов отраже-
ния), помещенных соответственно в задних окнах интегри-
рующей сферы. Фототок, возникающий в фотоэлементе под
влиянием суммарного светового потока, передается через
усилитель на кинематическую систему прибора.
Источником освещения служит кинопроекционная лампа
К-ЗО, установленная в специальном кожухе 33. Юстировка
лампы производится с помощью винтов 13 и контролируется
по резкому изображению нити лампы в центре поверхности
коллиматорной линзы (для наблюдения следует отодвинуть
111
шторку левого окна 15 в кожухе прибора). Если необходимо
сменить лампу, освобождают винты 14 и, отодвинув в сто-
рону кожух лампы, вынимают лампу из гнезда патрона.
Перед началом записи следует выполнить
следующие операции.
1. Проверить правильность подключения шлангов 16
с проводами, соединяющими шкаф питания 17 с прибором,
согласно нумерации на шлангах.
2. Установить ширину входной и выходной щели. Для
этого отодвигают шторку правого и левого окон 15—15'
в кожухе прибора и устанавливают требуемую ширину
спектрального интервала согласно цифрам на барабанчи-
ках 18.
3. Рукояткой 19 установить в рабочее положение кулачок,
поворачивающий призму Рошона, для записи по оптическим
плотностям (в окошке рукоятки должна появиться буква D)
или коэффициентам отражения или пропускания (соответ-
ствует буква Т).
4. Вставить перо и завинтить гайку 20.
5. Завернув винт 21, установить по шкале длин волн 22
отсчет, соответствующий 400 ммк, с помощью рукоятки 23,
немного оттянув ее на себя. (Вращать шкалу длин волн с по-
мощью рукоятки 23 можно как против часовой стрелки в сто-
рону больших длин волн, так и по часовой стрелке. Однако
по часовой стрелке это вращение вручную ограничено пре-
делами шкалы.)
6. Установить бланк для записи на барабане 24. Для это-
го предварительно обрезают бланк, имеющийся в комплекте
прибора так, чтобы линия отреза прошла через два штриха,
нанесенные ниже шкалы длин волн на расстоянии 15 ± 0,2 мм.
Навернуть бланк на барабан так, чтобы линия отреза вплот-
ную прилегла к правому бортику барабана, а перо при опу-
скании его на бланк стояло на абсциссе, соответствующей
400 ммк. После этого прижать бланк металлическим пружин-
ным прижимом 25.
Далее можно приступить к включению электромеханиче-
ской системы прибора.
1. Включить сетевой шланг, идущий от шкафа питания,
в штепсельную розетку на 127 в.
2. Включить выключатель «сеть» на передней стенке шка-
фа питания 26.
3. Включить лампу выключателем, расположенным на
щитке прибора 27. Произвести юстировку лампы, как ука-
зано на стр. 111, поставив выключатель на цоколе лампы в
положение «юстировка».
4. Включить усилитель выключателем 28 на передней
стенке шкафа питания и дать прогреться 2—3 мин.
5. Включить мотор-модулятор выключателем на щитке
112
прибора и нажатием кнопки «пуск мотора-модулятора». Ес-
ли мотор не работает, включение повторить.
6. Переключить лампу на «измерение».
7. Включить мотор отработки выключателем на щитке
прибора и отрегулировать с помощью усилителя рукояткой
29 небольшие повороты муфты мотора отработки.
Если в кюветном отделении отсутствуют какие-либо по-
глощающие объекты и в окнах интегрирующей сферы уста-
новлены два одинаковых эталона, то при положении рукоятки
19 на D перо должно двигаться вправо, т. е. в сторону нуля
оптической плотности и соответственно влево при положении
Т, т. е. в сторону ста процентов пропускания. Если перо дви-
жется не в том направлении, в котором следует, то необхо-
димо перебросить рукоятку «переключение мотора отработ-
ки» на щитке прибора.
8. Если перо не доходит до прямых, соответствующих
D = 0 или Т = 100% (при отсутствии измеряемых образцов
на пути светового потока), или стремится выйти за их пре-
делы, то следует отрегулировать его положение поворотом
призмы Рошона с помощью винта 30 через правое окно ко-
жуха прибора. После этого необходимо произвести запись по
прямой, соответствующей Т = 100% или 0 = 0, включив мо-
тор длин волн и освободив перед этим винт 21.
9. Выключив мотор длин волн, а затем мотор отработки,
завинчивают стопорный винт 21 и, подняв перо, возвращают
шкалу к 400 ммк.
10. Сняв кожух 31 с кюветного отделения, устанавливают
на пути светового потока исследуемый образец. Кюветы
с растворами и твердые образцы укрепляются в специаль-
ных держателях (рис. 43, 4). «Нулевой» образец устанавли-
вают всегда в правом световом потоке, исследуемый — в ле-
вом. Закрывают кюветное отделение кожухом.
11. Включив мотор отработки, дают перу занять положе-
ние, соответствующее поглощению или пропусканию образца
при Z 400 ммк.
12. Освободив стопорный винт 21, включают мотор длин
волн и производят запись по спектру.
Проверяют градуировку шкалы длин волн
по дидимовому стеклу, имеющемуся в комплекте прибора,
контрольная запись спектра которого прилагается к пас-
порту прибора.
Скорость записи по спектру регулируется рукояткой 32.
Спектры поглощения веществ, обладающих широкими и ма-
ло интенсивными максимумами, можно записывать на боль-
шой скорости (при положении указателя 3—4). Спектры типа
спектров поглощения солей редкоземельных элементов сле-
дует регистрировать при минимальной скорости записи, ина-
че при большой скорости поворота барабана перо не успеет
113
пройти вдоль барабана и выписать большой и резкий мак-
симум.
Проверяют правильность показаний опти-
ческих плотностей и процентов пропускания
по светофильтрам № 1 и 2, контрольная запись которых так-
же прилагается к паспорту прибора.
Останавливать запись и выключать прибор рекомендуется
в следующем порядке: 1) выключить мотор длин волн;
2) выключить мотор отработки. Если нужно записать новый
образец, то как раз в этот момент можно поставить его в кю-
ветное отделение и, установив шкалу длин волн, как обычно,
на требуемое значение, произвести запись, включив сначала
мотор отработки и затем мотор длин волн. Если требуется
выключить прибор полностью, то отдельные его узлы выклю-
чают в следующем порядке: 1) мотор длин волн; 2) мотор
отработки; 3) мотор модулятор; 4) лампа; 5) усилитель;
6) сеть; 7) вынуть вилку из розетки тока высокого напря-
жения.
Спектрофотометр СФ-10 в принципе ничем не отли-
чается от спектрофотометра СФ-2М, но является более позд-
ней конструкцией. Оба прибора обладают одинаковыми воз-
можностями и приемы работы на них совершенно одинаковы.
Однако спектрофотометр СФ-10 имеет более совершенную
электромеханическую часть и поэтому удобнее и надежнее
в работе.
СВЕТОФИЛЬТРЫ
Одним из способов монохроматизации света является мо-
нохроматизация с помощью светофильтров.
Светофильтры представляют собой твердые (стеклянные,
желатиновые, целлофановые и т. п.) или жидкие среды, об-
ладающие избирательным поглощением лучистой энергии и
пропускающие свет в достаточно узком интервале длин волн.
Обычно цвет светофильтра соответствует тому участку спект-
ра, который этим светофильтром пропускается. Ширина про-
пускания определенного спектрального участка колеблется
от 100 ммк (рис. 46) (светофильтры в ФЭК-М) до 20—40 лси/с
(рис. 47) (светофильтры в ФЭК-Н-57 и ФТ). Вполне очевидно,
что в первом случае мы не можем говорить о какой-либо
спектральной характеристике, полученной при измерениях на
фотоэлектроколориметре ФЭК-М. В данном случае свето-
фильтр может лишь способствовать повышению точности и
чувствительности количественных определений.
Светофильтры, имеющиеся в фотометрах и фотоэлектро-
колориметре-нефелометре, позволяют получить достаточно
точную спектральную характеристику большинства веществ,
водные растворы которых имеют достаточно широкие макси-
114
мумы поглощения. Совершенно непригодны эти приборы
лишь при изучении спектров поглощения таких веществ, как
соли редкоземельных элементов, которые имеют большое чис-
ло острых максимумов поглощения, расположенных друг от
друга на расстоянии 20—50 ммк. Понятно, что разрешить эти
максимумы с помощью светофильтров с шириной пропуска-
ния в 40 ммк не представляется возможным.
Все светофильтры, имеющиеся в визуальных приборах,
характеризуются величиной эффективной длины волны лэ<к
которая вычисляется по
специальным формулам,
учитывающим чувстви-
тельность глаза. Величи-
на ЛЭф указывает, что
Рис. 47. Кривые пропускания све-
тофильтров к фотометрам
Рис. 46. Кривые пропускания
светофильтров к фотоэлектро-
колориметру ФЭК-М
данный светофильтр примерно эквивалентен по своему дей-
ствию идеально-монохроматическому светофильтру, пропу-
скающему только излучение с длиной волны, к которой наи-
более чувствителен глаз в области пропускания свето-
фильтра.
Для желтых и зеленых светофильтров 73ф приблизительно
совпадает с максимумом пропускания. Для красных свето-
фильтров это не так. Например, для красного светофильтра
максимум пропускания лежит в области 700—680 ммк,
а А,Эф — 659 ммк. Такое расхождение между максимумом све-
топропускания и величиной 7Эф является результатом умень-
шения чувствительности глаза на краю красной части
спектра.
В табл. 2 приведены данные для 73ф наиболее распростра-
ненных светофильтров.
115
Таблица 2
Максимумы пропускания различных светофильтров
Светофильтр А эф, ммк А максимума про- пускания, ммк Ширина области пропускания, ммк
М-43 436 416 + 10 41
М-47 465 455+10 43
М-50 496 500 + 10 40
М-53 533 540+10 36
М-55 550 — —
М-57 574 580 + 10 34
М-59 590 — —
М-61 619 610 + 10 41
М-66 665 660+10 64
М-72 726 — 64
К-1 659 — —
К-2 633 — 85
к-з 570 — .—
К-4 541 — 51
К-5 519 .— —
К-6 478 —. 77
К-7 461 —. —
Для приборов с фотоэлектрической регистрацией обычно
вместо лЭф даются X максимума пропускания соответствую-
щих светофильтров. ХЭф и X максимума пропускания свето-
фильтров обычно приводятся в аттестатах соответствующих
приборов.
В визуальных приборах (К0Л-1М, КОЛ-52, ФТ) свето-
фильтры укреплены в револьверном диске 1 (рис. 48), пово-
ротом этого диска на пути светового потока устанавливают
необходимый светофильтр. Рабочее положение светофильтра
определяется защелкиванием пружинного фиксатора, а но-
мер светофильтра, находящегося в рабочем положении, оп-
ределяется по цифре, появляющейся в окошке 2. Если же не-
обходимо проверить марку светофильтра, а также сменить
или почистить светофильтр, открывают крышку 3 и с по-
мощью прилагаемого к прибору ключа вынимают свето-
фильтр из отверстия.
На диске со светофильтрами расположен окуляр. Фоку-
сировка линии раздела поля зрения окуляра па две поло-
вины производится с помощью кольца 5. После фокусировки
для удобства дальнейших наблюдений на окуляр надевается
наглазник 4; второй глаз закрывается наглазником 4'.
Таким образом, для получения спектральной
характеристики исследуемого объекта необходимо про-
извести измерения последовательно со всеми светофильтрами
и, построив график зависимости О (оптическая плотность) от
116
(эффективная длина волны или Л максимума пропуска-
ния), получить спектр поглощения.
Выбор светофильтра для количественных определений
производится на основании следующих принципов:
1) при работе с окрашенными растворами выбирается
светофильтр, цвет которого является дополнительным к цве-
ту испытуемого раствора;
Рис. 48. Диск со светофильтрами к микроколориметру КОЛ-52
2) если известен спектр поглощения испытуемого объекта,
то в работе используется светофильтр с областью максималь-
ного пропускания, близкой к области максимального погло-
щения испытуемого объекта.
ФОТОЭЛЕМЕНТЫ
В фотоэлектроколориметрах, как уже отмечалось (стр. 33),
в качестве приемника лучистой энергии служит вместо глаза
фотоэлемент.
В основе использования фотоэлементов лежит явление фо-
тоэффекта, открытое А. Г. Столетовым в 1888 г. Оно заклю-
чается в том, что под действием света с поверхности различ-
на
иых тел вырываются электроны, вследствие чего данное тело
приобретает заряд. Причем это явление имеет место только
при условии, что энергия светового кванта больше работы,
необходимой для освобождения электрона с поверхности дан-
ного вещества и сообщения ему какой-то кинетической энер-
гии. Как известно, энергия светового кванта определяется
длиной волны света Z или частотой его колебаний v.
W = hv=h—. (121)
%
Таким образом, для каждого вещества существует опре-
деленная длина волны (или соответственно частота колеба-
ний) света, называемая порогом фотоэффекта, при которой
начинает наблюдаться фотоэффект.
Сила возникающего фототока также зависит от длины
волны падающего на фотоэлемент света, т. е. для каждого
фотоэлемента имеется спектральная характеристика чувстви-
Рис. 49. Спектральная характеристика фотоэлементов: Cs — сурьмя-
но-цезиевый; Se — селеновый; Cu2O (I)—купроксный с фронтовым
фотоэффектом; Cu2O (II)—купроксный с тыловым фотоэффектом
тельности его к различным участкам длин волн (рис. 49).
Спектральная характеристика фотоэлементов сильно зависит
от температуры. Кроме того, А. Г. Столетовым было установ-
лено, что сила фототока прямо пропорциональна интенсив-
ности падающего на фотоэлемент излучения, хотя строгая
пропорциональность имеет место только для монохроматиче-
ского пучка света. Таким образом, регистрируя гальвано-
метром силу тока, возникающую в фотоэлементе и пропор-
циональную интенсивности падающей на фотоэлемент свето-
вой энергии, можно установить, насколько ослабляется ин-
тенсивность светового потока при прохождении его через кю-
вету с поглощающим веществом.
При работе с фотоэлементами следует всегда иметь в
виду ряд факторов, влияющих на получение точных и вос-
производимых результатов.
118
Во-первых, спектральная чувствительность фотоэлементов
так же, как и интегральная (равная силе тока, возникающего
при освещении поверхности элемента световым потоком в
1 лм), может со временем меняться, т. е. наблюдается «ста-
рение» фотоэлемента, что вызывает необходимость его
замены.
Во-вторых, для фотоэлементов характерно явление «утом-
ления» (уменьшение силы фототока со временем), которое
наблюдается при длительном непрерывном освещении фото-
элемента достаточно ярким светом. Поэтому во время работы
для фотоэлемента необходим «отдых», т. е. временное пре-
кращение его облучения.
В-третьих, чувствительность фотоэлемента бывает неоди-
накова по всей его поверхности, поэтому большое внимание
следует уделять настройке осветителя, чтобы при параллель-
ных измерениях всегда освещался один и тот же участок по-
верхности фотоэлемента. Освещаемая площадка при этом не
должна быть очень маленькой (диаметр освещаемого пятна
должен быть равен примерно 1 см). Иногда равномерная
освещенность фотоэлементов достигается использованием ма-
товых рассеивателей.
Кроме того, следует также заметить, что на точность фо-
тоэлектроколориметрических измерений существенно влияет
качество гальванометра, с помощью которого измеряют силу
тока, возникающего в фотоэлементе. Для того чтобы наблю-
далась линейная зависимость между силой фототока во внеш-
ней цепи фотоэлемента и силой падающего света, сопротив-
ление гальванометра должно быть возможно малым и не
должно превышать внутреннего сопротивления фотоэлемента.
В фотоэлектроколориметрии обычно употребляются три
типа фотоэлементов 1) фотоэлементы с запирающим слоем
(вентильные), 2) фотосопротивления (фотоэлементы с внут-
ренним фотоэффектом) и 3) фотоэлементы с внешним фото-
эффектом (вакуумные или газонаполненные).
Из вентильных фотоэлементов с запирающим слоем наи-
большее распространение получил селеновый фотоэлемент1 2
(рис. 50); он представляет собой железную пластинку 1, по-
крытую слоем элементарного селена (полупроводника) 2.
Поверхность селена покрыта очень тонкой полупрозрачной
пленкой золота или платины 3, на которой помещено метал-
лическое контактное кольцо 4. Фотоэлемент с целью защиты
поверхностного слоя от повреждений и воздействия паров и
1 В. С. Асатиани. Биохимическая фотометрия. М., Изд-во
АН СССР, 1957.
2 И. П. Алимарии. Фотоэлектрические колориметры с селеновыми
фотоэлементами и применение их в химическом анализе. М., Госгеолиздат,
1944.
119
Рис. 50. Принципиальная схема селе-
нового фотоэлемента
ностью, вследствие чего после
фототок не «ползет». Кроме
тазов помещают в футляр из пластмассы с выводами от кон-
тактов; отверстие в футляре прикрывается тонкой стеклянной
пластинкой или целлулоидной пленкой.
Принцип действия фотоэлемента заключается в следую-
щем. При попадании света на фотоэлемент с поверхности
селена выбиваются электроны, которые попадают в слой зо-
лота, так как благодаря полупроводниковым свойствам се-
лена они не могут проникать через его слой и дойти до же-
лезной пластинки. В результате слой золота оказывается от-
рицательно заряженным по отношению к железной пластинке
и, если соединить эти два слоя через гальванометр 5, мож-
но измерить фототок, появляющийся во внешней цепи.
Именно такие фотоэлементы
имеются в фотоколоримет-
рах ФЭК-М.
Селеновьве фотоэлемен-
ты получили широкое рас-
пространение благодаря ря-
ду положительных качеств.
Интегральная чувствитель-
ность этих фотоэлементов
достаточно велика (350—
500 лм), что позволяет ис-
пользовать гальванометры
чувствительностью 10_6—
10-7 а без употребления
внешних источников пита-
ния. Селеновые фотоэлемен-
ты обладают малой инерт-
ключения источника света
го, доброкачественные фото-
элементы уменьшают свою чувствительность по прошествии
года не более чем на 1%. Спектральная чувствительность
глаза и селенового фотоэлемента очень близки (рис. 49).
Кроме описанных селеновых фотоэлементов с фронтальным
фотоэффектом имеются также селеновые фотоэлементы с ты-
ловым фотоэффектом, у которых электроны выбиваются под
действием света на границе между селеном и железом. Эти
фотоэлементы обладают большим внутренним сопротивле-
нием, вследствие чего дают меньшую силу тока во внешней
цепи. Однако они обладают более постоянными характери-
стиками. К тому же типу фотоэлементов с запирающим сло-
ем относятся и меднозакисные (запирающий слой — закись
меди), серносеребряные (запирающий слой — сернистое се-
ребро) и серноталлиевые (запирающий слой — сернистый
таллий). Первый из названных фотоэлементов не выдержи-
вает сравнения с селеновым ввиду большой чувствительности
к изменениям температуры и малой интегральной чувстви-
120
тельности. Два последних фотоэлемента обладают более вы-
сокой интегральной чувствительностью и, кроме того, они
могут быть использованы в более широкой спектральной об-
ласти, чем селеновый фотоэлемент. Они широко использу-
ются для измерений в ближней инфракрасной области
спектра.
В основе действия фотосопротивлений лежит уменьшение
сопротивления вещества при облучении его светом опреде-
ленной длины волны. Фотоактивными в этом случае могут
быть различные полупроводники, из которых чаще всего при-
меняют таллофид (сплав сульфида таллия с окисью таллия),
сернистый свинец и реже селен. Таллофидные и сернисто-
свинцовые сопротивления обладают большой чувствитель-
ностью в инфракрасной области спектра, но имеют ряд недо-
статков, поэтому их применение ограниченно.
Действие фотоэлементов с внешним фотоэффектом осно-
вано на выбивании электронов под действием света из све-
точувствительного слоя, являющегося катодом. Выбитые элек-
троны устремляются к аноду, в результате чего во внешней
цепи возникает электрический ток.
Наиболее распространенными фотоэлементами с внешним
фотоэффектом являются кислородно-цезиевые и сурьмяно-це-
зиевые. Первые используются в основном для работы в ин-
фракрасной области спектра, вторые—в ультрафиолетовой и
видимой областях спектра (см. спектрофотометр СФ-4,
стр. 99). Основными недостатками фотоэлементов с внеш-
ним фотоэффектом являются необходимость использовать
внешнее напряжение, а также их малая интегральная чув-
ствительность, вследствие чего они требуют применения лам-
повых усилителей. Однако благодаря их чувствительности
к более широкому интервалу длин волн они применяются в
наиболее совершенных приборах последних конструкций (на-
пример, СФ-4, СФ-2М и т. д.).
Более удобны вакуумные фотоэлементы, так как они хотя
и уступают газонаполненным в чувствительности, но отлича-
ются от них рядом положительных свойств: меньшей зависи-
мостью характеристики от колебаний приложенного напря-
жения, меньшей инерционностью (т. е. меньшим запаздыва-
нием изменения фототока при изменении светового воздей-
ствия) .
КЮВЕТЫ
Типы кювет. В зависимости от конструкции прибора
кюветы, в которые помещают испытуемые растворы, имеют
различную форму. Наиболее распространенными являются
прямоугольные кюветы. Наборами таких кювет снабжены фо-
тоэлектроколориметры ФЭК-М, ФЭК-56, ФЭК-Н-54 и
ФЭК-Н-57, а также горизонтальный фотометр и спектрофо-
121
тометр СФ-2М. В каждом наборе имеется несколько пар
кювет с различной толщиной слоя от 1 до 50 мм. Парные
кюветы должны иметь одинаковую пропускаемость при за-
полнении их одинаковыми растворами.
Горизонтальный фотометр и спектрофотометр СФ-2М
снабжены одним набором, так как одновременно в нем мо-
гут быть помещены всего две кюветы одной толщины.
К каждому фотоэлектроколориметру придается по два
набора, так как при измерении можно использовать четыре
Рис. 51. Кюветы для
универсального фотометра ФМ
кюветы: одну, заполненную растворителем,— в левом кювет-
ном отделении, и одну с растворителем и две с испытуемыми
растворами — в правом. Кюветы этого типа заполняются рас-
твором почти доверху и сверху прикрываются плоскими
крышками.
Кюветы универсального фотометра ФМ, ввиду того что
они должны ставиться на предметный столик, имеют расши-
ренное плоское основание (рис. 51). Световой поток проходит
через эти кюветы снизу вверх, поэтому толщина их слоя огра-
ничивается крышками. Кюветы должны быть заполнены рас-
твором таким образом, чтобы при рассматривании сверху
под крышкой не наблюдалось пузырьков воздуха.
Особое устройство имеют кюветы микроколориметра
КОЛ-52. Они имеют большую толщину измеряемого слоя I
и очень низкий уровень заполнения (до риски) при столь же
122
малой их ширине. Такое устройство кюветы позволяет
работать с малыми объемами (0,5 мл) разбавленных рас-
творов.
Внешний вид кювет концентрационного колориметра
КОЛ-IM и погруженных в них столбиков дан на рис. 27,6.
Следует заметить, что при работе с этим прибором абсолют-
ный объем'жидкости, который помещен в кювету, значения
не имеет, так как толщина слоя определяется расстоянием от
дна кюветы до конца столбика, погруженного в раствор. Но в
кювету должно быть налито такое количество раствора, чтобы
он не выливался через край при полном погружении стол-
бика и чтобы конец столбика при самом верхнем его поло-
жении не выходил из раствора.
В комплекте спектрофотометра СФ-4 имеются два вида
кювет: прямоугольные кварцевые кюветы и цилиндрические
со сменными кварцевыми и стеклянными крышками. О ра-
боте с этими кюветами и способе их наполнения см. в описа-
нии спектрофотометра (стр. 101).
Для измерений растворов малых концентраций и малых
объемов с целью повышения чувствительности определения
удобно использовать цилиндрические кюветы с большой тол-
щиной слоя, но малым диаметром. Представленные на рис. 42
кюветы с толщиной слоя 10 см сделаны из тефлона и имеют
кварцевые окна диаметром 5 мм. Для их заполнения тре-
буется лишь — 2,5 мм испытуемого раствора, но следует уде-
лять особое внимание тщательности их установки в кювет-
ном отделении в строго параллельном световому потоку по-
ложении. Это достигается с помощью специального держа-
теля (рис. 43).
Обращение с кюветами. Большое значение в коло-
риметрии имеет чистота рабочих граней кювет. Многие веще-
ства обладают способностью адсорбироваться на поверхности
и загрязнять стенки кювет. Это может вызвать большие
ошибки при измерении оптических плотностей исследуемых
растворов. Поэтому никогда не следует надолго оставлять
кюветы наполненные испытуемыми растворами. Их следует
сразу же по окончании работы вымыть, ополоснуть дистилли-
рованной водой и вытереть снаружи мягкой тряпочкой.
Во время мытья или вытирания нельзя скоблить стенки кю-
вет чем-либо жестким, чтобы не повредить их поверхность,
что вызовет изменение их пропускаемости. Не следует допу-
скать вытекания раствора из кюветы на внешнюю стенку или
в кюветное отделение, так как это, во-первых, сказывается на
прозрачности стенок кювет, а во-вторых, портит детали при-
бора.
Заполнение кювет растворами. Чисто вымытые
и вытертые снаружи кюветы никогда специально не сушатся
изнутри. Для того чтобы концентрация раствора не измени-
123
лась вследствие разбавления его водой, оставшейся в кювете,
перед заполнением кюветы раствором ее следует ополоснуть
небольшой порцией этого же раствора.
Если измеряют плотность ряда эталонных растворов, со-
держащих один и тот же определяемый компонент, то мыть
кювету водой перед заполнением ее раствором, отличающим-
ся только по концентрации, не следует. Необходимо перед
наполнением кюветы очередным эталонным раствором толь-
ко ополоснуть ее этим же раствором. Рекомендуется произ-
водить измерения вначале менее концентрированных раство-
ров, последовательно увеличивая их концентрацию.
V. ПРАКТИЧЕСКИЕ РАБОТЫ
1. ОПРЕДЕЛЕНИЕ КОНСТАНТ КИСЛОТНОЙ
ДИССОЦИАЦИИ ОРГАНИЧЕСКИХ РЕАГЕНТОВ
Теоретические основы методов расчета констант диссо-
циации реагентов, имеющих кислотно-основные функции, на
основании спектрофотометрического исследования системы
изложены на стр. 45. Любым из этих методов можно рассчи-
тать константу кислотной диссоциации реагента, если изу-
чить спектры поглощения серии растворов, имеющих различ-
ную кислотность, но постоянную концентрацию изучаемого
реагента и постоянную ионную силу.
В настоящем разделе даны рецепты приготовления рас-
творов некоторых реагентов перед изучением их спектров по-
глощения. Данные спектрофотометрических измерений можно
записать в приведенную ниже таблицу и использовать для
расчетов по одному из изложенных ранее (стр. 45) методов.
X ммк D
pH 0,7 pH 1,5 pH 2,3 и т. д.
230 240 и т. д.
а. Определение константы диссоциации диметилглиоксима
СН3—С—С—СН3
II II
НО—N N—ОН
Первая константа диссоциации диметилглиоксима соот-
ветствует уравнению
H2D Н+ + HD-
125
и может быть определена спектрофотометрически на основа-
нии изучения спектров поглощения растворов, имеющих раз-
личные значения pH в интервале 3 — 12. (По второй ступени
диметилглиоксим диссоциирует в более щелочной среде:
HD- Н+ + D2-).
Максимум светопоглощения недиссоциированной формы
лежит в области 220 — 230 ммк, диссоциированной — в об-
ласти 260—270 ммк. Поэтому для изучения изменений в
спектре поглощения диметилглиоксима в зависимости от
значения pH используют спектрофотометр, позволяющий ра-
ботать в ультрафиолетовой области спектра.
Необходимые реактивы:
1. Диметилглиоксим, насыщенный раствор, разбавленный
в 100 раз.
2. Хлорная кислота, 0,1 н. раствор.
3. Едкий натр, 0,1 н. раствор.
4. Буферный раствор, pH 9,24 (0,05 М раствор буры).
5. Перхлорат натрия, 1 М раствор.
Готовят серию растворов:
1. Раствор имеет pH 3. В мерную колбу на 100 мл берут
10 мл раствора диметилглиоксима, 1 мл раствора хлорной
кислоты, 10 мл раствора перхлората натрия. Объем доводят
водой до 100 мл. Диметилглиоксим находится в форме НА.
2. Раствор имеет pH 12. В мерную колбу на 100 мл берут
10 мл раствора диметилглиоксима, 10 мл раствора щелочи,
9 мл раствора перхлората натрия и объем доводят водой до
100 мл. Диметилглиоксим находится в форме А-.
3. Раствор имеет pH 11. В мерную колбу на 100 мл берут
10 мл раствора диметилглиоксима, 1 мл раствора щелочи
и 10 мл раствора перхлората натрия. Объем доводят водой
до 100 мл.
4. Раствор имеет pH 10. В мерную колбу на 100 мл берут
10 мл раствора диметилглиоксима, 1 мл раствора щелочи
0,01 н. (10 мл 0,1 н. щелочи разбавляют водой до 100 мл)
и 10 мл раствора перхлората натрия. Объем доводят водой
до 100 мл.
5. Раствор имеет pH 9. В колбу на 100 мл берут 10 мл
раствора диметилглиоксима, 66 мл раствора буры и объем
доводят водой до 100 мл.
Все растворы имеют постоянную ионную силу 0,1.
Точные значения pH растворов измеряют на ламповом по-
тенциометре со стеклянным электродом. Измеряют поглоще-
ние каждого раствора в области длин волн от 220 до 300 ммк
с интервалом в 10 ммк, строят кривые спектров поглощения
на основании результатов измерений и вычисляют первую
константу диссоциации диметилглиоксима (стр. 45).
126
б. Определение константы кислотной диссоциации
нитрозо-К-соли
В нитрозо-К-соли водород оксимной
к диссоциации:
N=O
группы способен
NOH
NaO3S—
—SO3Na
—SO3Na NaO3S—
Максимум поглощения недиссоциированной формы лежит
в области 260—265 ммк, диссоциированной — 420—430 ммк.
Поэтому определить константу диссоциации нитрозо-И-соли
можно на основании изучения поглощения растворов этой
соли в зависимости от значения pH в видимой области спект-
ра (при X > 400 ммк); следовательно, кроме спектрофото-
метра СФ-4 можно использовать спектрофотометры СФ-5,
СФ-2М, СФ-10.
Серия изучаемых растворов должна иметь переменные
значения pH в интервале 3—12. Все растворы должны иметь
ионную силу, примерно равную 0,5. Поэтому по формуле
и =12“'
вычисляют приближенное значение ионной силы, которая со-
здается в каждом растворе путем прибавления буферного
раствора, и какое количество перехлората натрия необходимо
добавить для того, чтобы создать ионную силу, равную ~ 0,5.
Необходимые реактивы:
1. Нитрозо-И-соль, 0,2г в 1 л раствора.
2. Буферный раствор, pH 6. Берут 500 мл 0,1 М бифтала-
та калия и 454,5 мл 0,1 М раствора щелочи; объем доводят
водой до 1 л.
3. Буферный раствор, pH 7. Берут 400 мл 0,15 М раствора
однозамещенного фосфата натрия и 600 мл 0,15 М двузаме-
щенного фосфата натрия.
4. Буферный раствор, pH 8. Берут 50 мл 0,15 М раствора
однозамещенного фосфата натрия и 950 мл 0,15 М рас-
твора двузамещенного фосфата натрия.
5. Тетраборат натрия, 0,05 М раствор.
6. Едкий натр, 0,1 н. раствор.
127
7. Хлорная кислота, 0,1 и. раствор.
Готовят серию растворов:
1. Раствор имеет pH 3. В мерную колбу на 50 мл берут
5 мл раствора нитрозо-К-соли, 1 мл раствора хлорной кисло-
ты и раствор перхлората натрия.
2. Раствор имеет pH 6. В мерную колбу на 50 мл берут
5 мл раствора нитрозо-К-соли, 20 мл буферного раствора,
имеющего pH 6, и раствор перхлората натрия.
3. Раствор имеет pH 7. В мерную колбу на 50 мл берут
5 мл раствора нитрозо-К-соли, 20 мл буферного раствора,
имеющего pH 7, и раствор перхлората натрия.
4. Раствор имеет pH 8. В мерную колбу на 50 мл берут
5 мл раствора нитрозо-К-соли, 20 мл буферного раствора,
имеющего pH 8, и раствор перхлората натрия.
5. Раствор имеет pH 9. В мерную колбу на 50 мл берут
5 мл раствора нитрозо-К-соли, 20 мл, 0,05 М раствора тетра-
бората натрия и раствор перхлората натрия.
6. Раствор имеет pH 12. В мерную колбу на 50 мл берут
5 мл раствора нитрозо-К-соли, 5 мл 0,1 н. раствора щелочи
и раствор перхлората натрия.
После добавления необходимого количества перхлората
натрия объем растворов доводят водой до 50 мл. Значения
pH приготовленных растворов проверяют на ламповом потен-
циометре со стеклянным электродом. Измеряют оптические
плотности полученных растворов по отношению к воде в об-
ласти длин волн от 400 до 700 ммк с интервалом в 10 ммк.
На основании полученных данных строят кривые спектров
поглощения всех растворов и вычисляют константу диссо-
циации нитрозо-К-соли (стр. 45).
в. Определение константы кислотной диссоциации
тимолсульфофталеина (тимолового синего)
Н3С сн3 Н3С сн3
128
Кислотно-основной индикатор тимолсульфофталеин яв-
ляется двухосновной кислотой. При изменении кислотности
его водных растворов в интервале значений pH от 1 до 10 их
окраска меняется дважды: из красной в желтую при рН~2,
что соответствует диссоциации его по первой ступени; из жел-
той в синюю при pH ~ 8,8, что соответствует диссоциации по
второй ступени:
H2R rt HR- + Н+ R1 2- + 2Н+.
красная желтая синяя
Максимумы поглощения различных форм — H2R, HR- и
R2~ находятся соответственно при А. 555, 425 и 600 ммк. Та-
ким образом, спектры поглощения тимолового синего изу-
чают в видимой области спектра. Изучая спектры поглоще-
ния растворов тимолового синего, имеющих переменные зна-
чения pH в интервале 1—7, можно определить первую кон-
станту его диссоциации Кь в интервале 7—10 — вторую Кг-
Необходимые реактивы:
1. Раствор тимолового синего — 0,5%-ный. 5 г тимоло-
вого синего растирают в ступке с 21,5 мл раствора едкого
натра (0,05 н.), переносят в колбу и разбавляют до объема
в 1 л; перед употреблением раствор разбавляют в 10 раз.
2. Две серии (соответственно двум константам диссоциа-
ции) буферных растворов, имеющих следующие значе-
ния pH
1-я серия 2-я серия
1,2 7,6
1,5 8,0
1,7 8,8
2,3 9,3
2,9 9,6
7,6 10,0
В мерные колбы на 25 мл помещают по 0,4 мл раствора
индикатора, доводят объем до метки колбы соответствую-
щими буферными растворами, согласно одному из приведен-
ных выше рядов, и тщательно перемешивают. Измеряют оп-
тические плотности приготовленных растворов по отношению
к воде в области длин волн от 400 до 700 ммк с интервалом
в 10 ммк. На основании полученных данных строят кривые
спектров поглощения всех растворов и вычисляют соответ-
ствующую константу диссоциации тимолсульфофталеина
(стр. 45).
Примечание. Точные значения pH растворов измеряют на лампо-
вом потенциометре со стеклянным электродом. Ввиду того что некоторые
различия в ионной силе растворов мало сказываются на поглощении в
видимой области спектра, иет необходимости специально регулировать ион-
ную силу растворов, приводя ее к одному значению во всей серии раство-
ров.______
1 Е. Н. Виноградова. Методы определения концентрации водород-
ных иоиов. Изд-во МГУ, 1956; Ю. Ю. Л у р ь е. Справочник по аналитиче-
ской химии. М„ Госхимиздат, 1962.
5 Зак. 686 129
2. ОПРЕДЕЛЕНИЕ СОСТАВА КОМПЛЕКСНЫХ
СОЕДИНЕНИЙ КОБАЛЬТА (II) С НИТРОЗОНАФТОЛАМИ
МЕТОДОМ ИЗОМОЛЯРНЫХ СЕРИЙ
Кобальт с 1-и 2-изомерами нитрозонафтолов образует два
соединения: одно — растворимое в воде, второе — раствори-
мое в неводных органических растворителях, но нераствори-
мое в воде. Первое соединение образуется в щелочной среде;
в кислой среде в присутствии окислителя выделяется нерас-
творимое в воде второе соединение. Это соединение для ма-
лых количеств кобальта может образоваться и без добавле-
ния окислителя, если в избытке присутствует реагент, окис-
ляющий кобальт до 3-валентного состояния, который и дает
соединение, нерастворимое в воде, но растворимое в невод-
ных органических растворителях.
а. Определение состава соединения кобальта
с 1 -нитрозо-2-нафтолом 1
Для определения состава комплексного соединения ко-
бальта (II), образующегося в щелочной среде, необходимы
реактивы:
1. Растворы соли кобальта (хлористого или сернокислого)
с концентрацией 5 • 10-в и 1 • 10-5 моль/л.
2. Растворы 1-нитрозо-2-нафтола с концентрацией 5-10-5
и 1 • 10-5 моль/л.
3. Сегнетова соль, 20%-ный раствор.
4. Едкое кали, 5%-ный раствор.
В пять мерных колб емкостью 50 мл вводят растворы
соли кобальта и реагента в следующих соотношениях:
число миллилитров реактива 10,0; 15,0; 20,0; 22,5; 25;
число миллилитров соли кобальта 20,0; 15,0; 10,0; 7,5; 5,0.
Добавляют 5 мл раствора сегнетовой соли и 5 мл раствора
едкого кали. Растворы перемешивают, доводят водой до мет-
ки колбы и измеряют светопоглощение при длинах волн 416
и 480 ммк. При снятии светопоглощения растворов (X 416 лице)
нужно внести поправку на поглощение реактива. Для этого
готовят растворы одного реагента в тех же условиях, кото-
рые указаны для приготовления смесей растворов, только не
вводят соль кобальта и измеряют D при X 416лцик. При по-
строении графика для смеси растворов, снятых при
X 416 ммк, вносят поправку на поглощение реагента для рас-
творов, в которых реагент присутствует в избытке. Графики
строят для двух серий растворов с различной концентрацией,
1 В. М. Пешкова, В. М. Бочкова. «Тр. комиссии по аналитичес-
кой химии». М., Изд-во АН СССР, VIII (XI), 125, 1958.
130
снятых при двух длинах волн — 416 и 480 ммк (стр. 51). На
основании полученных графиков делают заключение о со-
ставе соединения кобальта с 1-нитрозо-2-нафтолом.
б. Определение состава соединения кобальта
с 2-нитрозо-1-нафтолом
Для определения состава комплексного соединения ко-
бальта, образующегося в щелочной среде, необходимы реак-
тивы, кроме указанных на стр. 130:
1. Растворы соли кобальта (хлористого или сернокислого)
с концентрацией 5-10~7 и 1 • 10~6 моль/л.
2. Водные растворы натриевой соли 2-нитрозо-1-нафтола
той же концентрации: 5-10-5 и 1 • 10~6 моль/л.
В пять мерных колб емкостью 50 мл вводят растворы со-
ли кобальта и реагента в следующих соотношениях:
число миллилитров реактива 10,0; 15,0; 20,0; 22,5; 25;
число миллилитров соли кобальта 20,0; 15,0; 10,0; 7,5; 5,0.
Добавляют 5 мл раствора сегнетовой соли и 5 мл раство-
ра едкого кали. Растворы перемешивают, доводят водой до
метки колбы и измеряют светопоглощение при длинах волн
510 и 540 ммк.
Для приготовления смесей растворов используют исход-
ные растворы двух концентраций (5-10“7 и 1 • 10~6 моль/л)
и измерение каждой серии растворов производят при двух
длинах волн. На основании полученных графиков делают за-
ключение о составе соединения кобальта с 2-нитрозо-1-наф-
толом.
3. ПРИМЕРЫ, ПОКАЗЫВАЮЩИЕ ПРЕИМУЩЕСТВА
БОЛЕЕ ВЫСОКОЙ МОНОХРОМАТИЗАЦИИ ПОТОКА
ЛУЧИСТОЙ ЭНЕРГИИ
Особые преимущества призменных спектрофотометров
(СФ-4, СФ-5, СФ-2М, СФ-10) и спектрофотометров с дифрак-
ционной решеткой (СФД-2), обеспечивающих достаточно вы-
сокую монохроматизацию потока лучистой энергии, перед
фильтровыми упрощенными спектрофотометрами можно по-
казать на примере выполнения следующей эксперименталь-
ной работы.
а. Сравнительное изучение возможностей фотометрических
приборов различного типа
1. Готовят ряд эталонных растворов с переменной кон-
центрацией определяемого элемента, пользуясь методикой
определения этого элемента в растворе его чистой соли: мар-
5*
131
ганец в виде перманганата (стр. 150) и соединения с формаль-
доксимом (стр. 154); никель с диметилглиоксимом (стр. 159);
железо ic сульфосалициловой кислотой (стр. 138); фосфор в
виде гетерополисоединения (стр. 192) и т. п.
2. Готовят раствор соли редкоземельного элемента (р.з. э.)
с концентрацией порядка 0,05 М.
3. Измеряют оптические плотности одного из растворов
эталонного ряда, приготовленного согласно пункту 1, и рас-
твора соли р.з. э. (пункт 2), пользуясь фотоэлектроколори-
метром ФЭК-М с тремя имеющимися в нем светофильтрами.
На основании полученных результатов выбирают свето-
фильтр, для которого оптические плотности имеют наиболь-
шую величину, измеряют поглощение всех растворов эталон-
ного ряда с этим светофильтром и строят градуировочный
график, используемый в дальнейшем для количественных
определений.
4. Измеряют оптические плотности растворов, приготов-
ленных согласно пунктам 1 и 2, пользуясь фотоэлектроколо-
риметрами ФЭК-56, ФЭК-Н-57, ФЭК-Н-54 или фотометром
ФМ со всеми имеющимися в них светофильтрами. На осно-
вании полученных результатов строят кривые спектров по-
глощения всех исследуемых растворов. Вычисляют величины
молярных коэффициентов погашения в максимумах поглоще-
ния. На основании сохранения положения максимума в спект-
ре поглощения всех эталонных растворов и постоянства ве-
личин молярных коэффициентов погашения делают заключе-
ние о пределах выполнения основного закона светопоглощения.
Используя значения величин оптических плотностей в макси-
мумах поглощения всех растворов эталонного ряда строят
градуировочный график в координатах D — с и сравнивают
его наклон с наклоном градуировочного графика, получен-
ного в условиях, указанных в пункте 3. Он также может быть
использован для количественных определений.
5. Измеряют спектры поглощения одного из растворов
эталонного ряда( пункт 1) и раствора соли р. з. э. (пункт 2),
используя спектрофотометр (СФ-4, СФ-5, СФ-10, СФД-2
и СФ-4А). Измерения вначале производят грубо с интервалом
в 10 ммк, а затем в области максимального поглощения
более точно — с интервалом в 1—2 ммк. Строят кривые
спектров поглощения каждого раствора и вычисляют значе-
ния величин молярных коэффициентов погашения в макси-
муме поглощения.
Сравнивая характер спектров поглощения, а также вели-
чин молярных коэффициентов погашения, полученных при
работе на приборах двух типов (пункты 4 и 5), делают за-
ключение о возможностях этих приборов и целесообразности
использования их для различных целей.
132
б. Возможность изучения процессов комплексообразования
в растворах солей редкоземельных элементов
1. Снимают спектр поглощения раствора соли редкозе-
мельного элемента, пользуясь спектрофотометром СФ-4 и фо-
тоэлектроколориметрами ФЭК-56, ФЭК-Н-54, ФЭК-Н-57
(или фотометром ФТ), и строят кривые полученных спектров.
2. К тому же раствору соли редкоземельного элемента
прибавляют лимонную кислоту и щелочь. Например, к 1 мл
0,01 М раствора редкоземельного элемента (Pr, Nd, Er, Sm
и т. п.), помещенному в градуированную пробирку, прибав-
ляют 1 мл 0,1 М раствора лимонной кислоты (соотношение
р. з. э.: л. к. = 1: 10) и 0,2 мл насыщенного раствора КОН;
объем раствора доводят водой до 10 мл и измеряют значе-
ние pH.
3. Снимают спектр поглощения раствора редкоземельного
элемента в присутствии лимонной кислоты также на прибо-
рах: спектрофотометре СФ-4 и фотоэлектроколориметрах
ФЭК-56, ФЭК-Н-54, ФЭК-Н-57. Устанавливают, каким из
приборов возможно обнаружить сдвиг полосы максимального
поглощения в процессе комплексообразования.
в. Повышение чувствительности метода при работе
в области максимального поглощения соединения
1. Приготовить раствор трисульфосалицилата железа по
методике определения железа в растворе чистой его соли
(стр. 142).
2. Снять спектр поглощения этого раствора по отноше-
нию к воде на спектрофотометре СФ-4 в интервале длин волн
от 220 до 1100 ммк и фотоэлектроколориметрах ФЭК-56.
ФЭК-Н-54, ФЭК-Н-57 (или фотометре ФМ), используя все
имеющиеся светофильтры.
3. Вычислить молярные коэффициенты погашения в мак-
симумах поглощения, обнаруженных в том и другом случае,
и сравнить их величины.
4. МЕТОДЫ ОПРЕДЕЛЕНИЯ ОТДЕЛЬНЫХ ЭЛЕМЕНТОВ
ОПРЕДЕЛЕНИЕ ЖЕЛЕЗА
Для определения железа существует ряд колориметриче-
ских методов, позволяющих определять железо в 2- и 3-ва-
лентном состоянии. Наиболее распространенными являются
методы определения железа в виде роданида и соединений
с сульфосалициловой кислотой. Заслуживают внимания так-
же методы определения железа (II) с реактивами а, а'-дипи-
ридилом, о-фенантролином и диоксимом 1,2-циклогександио-
на. Выбор метода определяется наличием и влиянием сопут-
ствующих элементов (табл. 3).
133
Таблица 3
Чувствительность реакции и некоторые свойства соединений железа*
Реагент Чувствительность мкг Fe/си2, (X ммк) Устойчивость окраски раствора
а, а'-Дипиридил 1,10-Феиаитролин 4,7-Дифенил-1,10-фенантролин (спирт—вода) 2,6-б«с-(4-Фенил-2-пиридил)-4-фе- иилпиридин (СНС1з—спирт) Роданид Родаиид+ацетон Роданид и трибутиламмоний в амилацетате Сульфосалициловая кислота 0,007 (522) 0,005 (508) 0,0025 (523) 0,002 (583) 0,008 (480) 0,004 (480) 0,0025 (480) 0,01 (430) очень устойчива » > » неустойчива » устойчива в течение 1 дня »
*Е, Сен дел. Колориметрические методы определения следов металлов.:
М., «Мир», 1964, стр. 472.
Определение железа в виде роданида
Железо (III) в кислой среде образует с роданид-ионом в
зависимости от его концентрации ряд комплексных соедине-
ний различного состава, отличающихся сравнительно малой
устойчивостью.
Состав комплексного Константа диссоциации
иона комплексного соединения
[Fe (SCN)]2+ 3 • Ю" 3
[Fe(SCN)2]1+ 1,4 • 10~2
Fe (SCN)3 4 • 10~2
[FelSCNJd1- 1,6 • 10~1
[Fe(SCN)6]2~ 7 • 10“’
В растворе могут существовать1 и сосуществовать комплекс-
ные ионы с координационным числом от I до 6.
Комплексный ион [Fe(SCN]2+ в растворе образуется при
концентрации ионов родана, равной 5 10~3 М. При увеличении
концентрации роданид-иона происходит образование ионов
с большим координационным числом. Так, при концентрации
роданид-иона 1,2-10~2М в значительном количестве обра-
зуется комплексный ион [Fe (SCN)z]1+. В водном растворе
1 А. К. Б а б к о, А. Т. П и л и п е н к о. Колориметрический анализ.
М.— Л., Госхимиздат, 1951, стр. 168.
134
всегда имеется смесь комплексных соединений и невозможно
создать условия для существования какого-либо одного ком-
плексного соединения, что видно из рис. 52. Поэтому для по-
лучения воспроизводимых результатов очень важно произво-
дить определение железа, строго соблюдая предлагаемые
условия. Следует точно соблюдать равную концентрацию ро-
данид-иона в испытуемом и эталонных растворах, чтобы при
равной концентрации железа в этих растворах получить оди-
наковую интенсивность окраски. При колориметрическом оп-
ределении железа всегда нужно добавлять большой избыток
роданида. В этих условиях
раствор подчиняется закону
Бера в большом интервале
концентраций железа. В каче-
стве реактива можно исполь-
зовать роданистый аммоний
или калий.
Так же важно проводить
рассматриваемую реакцию, соз-
дав определенную кислотность
раствора, обеспечив существо-
Рис. 52. Образование железо-ро-
данидных комплексов в зависимо-
сти от концентрации роданида ам-
мония
вание в ионном состоянии же-
леза (III) и роданида. Кис-
лотность раствора должна быть
таковой, чтобы предотвратить
гидролиз Fe(III), не понизив
концентрацию роданид-иона, необходимого для образования
комплексного соединения. При большой кислотности возни-
кает опасность образования комплексных ионов [Fede]3-,
[Fe(SO4)3]3_. Оптимальной кислотностью является интервал
0,05—0,1 н.; подкисление следует проводить серной или соля-
ной кислотой.
Соединения роданового железа экстрагируются неводными
растворителями, что значительно повышает ценность реак-
ции, увеличивая ее чувствительность, а также расширяя воз-
можность ее применения для определения железа в окрашен-
ных растворах солей, например, в солях никеля.
Чувствительность реакции при визуальном наблюдении
0,005 мг в 10 мл неводного растворителя при экстрагирова-
нии из 50 мл водного раствора.
Для определения железа может быть рекомендован ме-
тод колориметрического титрования, а также графический
метод с использованием в этом случае фотометра или фото-
колориметра. Большое практическое применение находит ме-
тод колориметрического титрования.
Родановая реакция применяется только для определения
железа (III); 2-валентное железо не вступает в реакцию
с роданидами.
135
В качестве окислителя чаще всего используют азотную
кислоту, а также и другие окислители в зависимости от при-
роды объекта, в котором определяют содержание железа: пе-
рекись водорода, персульфат аммония, перманганат калия.
Из табл. 3 видно, что чувствительность метода определе-
ния железа роданидами повышается, если реакцию прово-
дить в присутствии ацетона; чувствительность метода еще
больше повышается, если определение железа проводить
смесью трибутиламмония и амилового спирта. Проведению
реакции мешает ряд веществ. Прежде всего должны отсут-
ствовать анионы ряда кислот, которые дают более прочные
комплексные соединения, чем роданид железа: фосфаты,
ацетаты, арсенаты, фториды, бораты, а также хлориды и
сульфаты, присутствующие в значительных количествах.
Также должны отсутствовать элементы, ионы которых дают
комплексные соединения с роданидом: кобальт, хром, висмут,
медь, молибден, вольфрам, титан в 3- и 4-валентном состоя-
нии, ниобий, палладий, кадмий, цинк, ртуть.
Предлагаемые ниже условия могут быть использованы
для определения железа (III) в водных растворах и в при-
сутствии посторонних веществ при условии, что таковые не
реагируют с роданидами или не образуют с железом более
устойчивых комплексных соединений, чем роданиды железа.
Необходимые реактивы:
1. Стандартный раствор соли железа (III). Навеску
0,864 г х. ч. железо-аммонийных квасцов (отбирают прозрач-
ные кристаллы) помещают в мерную колбу емкостью 1 л,
растворяют в воде, подкисленной 5 мл серной кислоты (уд.
в. 1,84), доводят водой до 1 л и тщательно перемешивают —
раствор А. 1 мл раствора А содержит 0,1 мг железа. 10 мл
раствора А разбавляют водой в мерной колбе на 100 мл —
раствор Б. 1 мл раствора Б содержит 0,01 мг железа. Рас-
твор Б готовят в день его употребления.
2. Роданистый аммоний, 4 н. раствор.
3. Серная кислота, 4 н. раствор.
4. Азотная кислота 1:1.
5. Изоамиловый спирт.
а. Определение железа в растворе его чистой соли
К Ю или 20 мл испытуемого водного раствора, содержа-
щего железо, добавляют 0,5 мл азотной кислоты, нагревают
раствор до начала кипения и после охлаждения переносят
его в тщательно вымытый с притертой пробкой цилиндр ем-
костью на 50 или 100 мл, прибавляют 5 мл раствора рода-
нистого аммония, 0,5 мл серной кислоты и 10 или 20 мл изо-
амилового спирта; доводят водой объем раствора до 50 мл,
взбалтывают и сравнивают появившуюся окраску спиртового
слоя с окраской спиртового слоя эталонного раствора, содер-
136
жащего то же количество воды, азотной и серной кислот, изо-
амилового спирта. К этому раствору из бюретки прибавляют
по капле стандартный раствор железа, содержащий в 1 мл
0,01 мг железа (раствор Б); после прибавления каждой пор-
ции и взбалтывания сравнивают интенсивность окраски слоев
спирта. Раствор железа прибавляют до уравнивания интен-
сивности окраски спиртовой фазы. Зная объем и концентра-
цию стандартного раствора железа находят содержание же-
леза в испытуемом растворе по формуле (стр. 35).
б. Определение железа в солях никеля
Для определения железа нужно иметь соль никеля, кото-
рая не содержит железа. Если такая соль отсутствует, то ее
необходимо приготовить следующим образом. Берут 3 г соли
никеля (сульфата, нитрата или хлорида) растворяют в 30 мл
воды, добавляют 1,5 мл серной кислоты и 1,5 мл азотной кис-
лоты. Раствор нагревают, осторожно кипятят в течение 2—
3 мин и разбавляют водой до объема 50—70 мл, прибавляют
1 г хлористого аммония, снова нагревают до начала кипения
и прибавляют аммиак до появления слабого запаха. Раствор
фильтруют, фильтрат нейтрализуют серной кислотой (по лак-
мусу), доводят объем до 90 мл и для приготовления эталон-
ного раствора берут 30 мл полученного раствора соли никеля,
не содержащей железо.
Примечание. В случае употребления азотнокислой соли никеля
операцию окисления железа следует опустить.
Для определения железа в солях никеля берут навеску
соли 1 г (с точностью до 0,01 г), растворяют в 30 мл воды,
прибавляют 0,5 мл серной кислоты, добавляют 0,5 мл азот-
ной кислоты, раствор нагревают и осторожно кипятят 1 —
2 мин, по охлаждении переносят в колориметрический ци-
линдр на 100 мл, доводят объем до 50 мл, прибавляют к нему
5 мл раствора роданистого аммония и 10 мл изоамилового
спирта, после чего содержимое цилиндра хорошо перемеши-
вают. В другой цилиндр переносят 30 мл раствора соли ни-
келя, не содержащей железо, вводят те же количества азот-
ной и серной кислот, роданистого аммония и изоамилового
спирта, что и в цилиндр с испытуемым раствором, и из бю-
ретки прибавляют по капле стандартный раствор железа.
Определение заканчивают в условиях, указанных для опреде-
ления железа в водном растворе.
в. Определение железа в алюминии и легких сплавах
на основе алюминия
Кроме указанных на стр. 136 необходимы реактивы:
1. Едкий натр, 10%-ный раствор (без железа).
2. Азотная кислота 1:1.
137
3. Аммиак, 10%-ный раствор.
Навеску измельченного образца от 0,5 до 2 г (в зависи-
мости от содержания железа) помещают в стакан емкостью
200—300 мл, обрабатывают раствором едкого натра, не со-
держащим железа, вводя его постепенно и прикрыв стакан
часовым стеклом. По растворении главной массы вещества
добавляют в стакан 50 мл горячей воды. На навеску 0,5 г бе-
рут 10—20 мл раствора едкого натра. Полученный остаток,
содержащий медь, железо и магний, отфильтровывают и про-
мывают его на фильтре горячей водой. Остаток на фильтре
растворяют в 5—10 мл подогретого раствора азотной кисло-
ты; фильтр промывают, наполняя его три раза горячей водой.
В фильтрате, предварительно подогретом, железо осаж-
дают раствором аммиака, добавляя его до щелочной реакции
на лакмус или до ощущения запаха. Осадок на фильтре
промывают горячей водой и растворяют в 2—3 мл азотной
кислоты; промывают фильтр три раза, наполнив его горячей
водой. В дальнейшем определение железа производят, как
указано на стр. 136.
Определение железа сульфосалициловой кислотой
Железо с сульфосалициловой кислотой образует ряд ком-
плексных соединений в зависимости от кислотности опреде-
ляемого раствора *.
В кислой среде, в интервале pH 1,8 — 2,5, образуется ком-
плексный катион моносульфосалицилата железа
/°—
[Fe(HSO3.CeH3< )]•+
\с-о—
II
О
В интервале pH 4—8 образуется комплексный анион ди-
сульфосалицилата железа
.О—
[Fe(HSO3.C,H3< )аГ-
ХС—О-
II
о
1 В. И. К у з и е ц о в. «Зав. лаб.», 12, 278, 1946.
138
В щелочной среде, в интервале pH 8—11, образуется так-
же комплексный анион, но трисульфосалицилата железа
/О—
[Fe(HSO3-CeHZ )3]3~
ХС—О—
Образующиеся соединения моно- и трисульфосалицилата
железа имеют спектры поглощения (рис. 53) с максималь-
ной областью поглощения соответственно при X 510 и
Рис. 53. Спектры поглощения: 1 — моно- и 2 — трисульфосалицилата же-
леза
416 ммк и молярными коэффициентами погашения 1600
и 4000.
Чувствительность реакции при визуальном наблюдении
0,005 мг в 50 мл водного раствора при наблюдении в верти-
кальном направлении и по сравнению с идентичной колори-
139
метрической пробиркой, содержащей 50 мл дистиллирован-
ной воды. Рассматриваемая реакция может быть использо-
вана для определения железа (III) в кислой среде и суммы
железа (II и III) в щелочной среде.
Комплексные соединения сульфосалицилата железа явля-
ются более устойчивыми, чем роданиды железа, что позво-
ляет применить рассматриваемый метод для определения
железа в присутствии фосфатов, ацетатов, боратов.
При проведении реакции в кислой среде определению же-
леза не мешают значительные количества меди и алюминия,
так как комплексные соединения этих элементов менее устой-
чивы, чем комплексное соединение сульфосалицилата желе-
за (Ш). Но соединение моносульфосалицилата железа менее
устойчиво, чем трисульфосалицилата железа, поэтому в кис-
лой среде исключается возможность определения железа в
присутствии фтор-иона.
Необходимые реактивы:
1. Стандартный раствор железа, содержащий в 1 мл
0,1 мг (стр. 136).
2. Серная кислота, 2 н. раствор.
3. Сульфосалициловая кислота, 10%-ный раствор.
4. NH4OH, 10% -ный раствор.
ОПРЕДЕЛЕНИЕ ЖЕЛЕЗА В ВИДЕ МОНОСУЛЬФОСАЛИЦИЛАТА
а. Определение железа в водном растворе
в отсутствие посторонних ионов
К Ю мл раствора, помещенного в колбу на 50 мл, прибав-
ляют 1 мл азотной кислоты, осторожно нагревают в течение
1—2 мин, охлаждают раствор и добавляют в него 1 мл сер-
ной кислоты, 5 мл раствора сульфосалициловой кислоты и
объем раствора доводят водой до метки колбы. Дальнейшее
определение производят в колориметре системы КОЛ-1М,
ФТ или ФЭК-М. В случае использования колориметра
КОЛ-1М готовят два эталонных раствора, содержащих от 0,1
до 0,3 мг железа в объеме 50 мл-, настраивают прибор, меняя
фокусное расстояние лампы осветителя последовательно по
двум приготовленным растворам, добиваясь одинаковой ок-
раски полей окуляра колориметра при одинаковой высоте
столбов окрашенных растворов.
Для определения железа в испытуемом растворе в каче-
стве раствора сравнения используют один из приготовленных
эталонных, который по окраске ближе подходит к испы-
туемому раствору. Содержание железа рассчитывают по фор-
муле (118) (стр. 76). Можно также использовать графичес-
кий метод определения концентрации окрашенного раствора
(стр. 38). При использовании приборов ФТ и ФЭК-М предва-
140
рительно подбирают светофильтр, область пропускания кото-
рого совпадает с максимумом на кривой светопоглощения
(рис. 53). Готовят пять эталонных растворов с содержанием
железа от 0,05 до 0,30 мг, увеличивая содержание железа в
каждой колбе на 0,05 мг. Условия приготовления эталонных
растворов аналогичны указанным выше для определения же-
леза в испытуемом растворе.
б. Определение железа в алюминии и легких сплавах
на основе алюминия
Кроме указанных необходимы реактивы:
1. Соляная кислота 1:1.
2. Азотная кислота 1:1.
Навеску измельченного образца от 0,5 до 2 г (в зависи-
мости от содержания железа) помещают в стакан или кони-
ческую колбу емкостью 250 мл и постепенно приливают из
пипетки 25—30 мл соляной кислоты. Если металл раство-
ряется медленно, раствор с остатком образца осторожно по-
догревают на песчаной бане. После растворения образца
раствор упаривают до начала кристаллизации и охлаждают.
Охлажденный остаток растворяют при нагревании в мини-
мальном количестве воды, добавляют 1 мл азотной кислоты;
раствор нагревают и осторожно кипятят 1—2 мин, нейтрали-
зуют раствором аммиака по индикатору тропеолин 00 или
по универсальной индикаторной бумаге до pH 3, переводят
в мерную колбу на 100 мл и разбавляют водой до метки
колбы. Для испытания берут 10 или 20 мл раствора в мер-
ную колбу на 50 мл, добавляют 1 мл серной кислоты, 5 мл
раствора сульфосалициловой кислоты и объем раствора дово-
дят водой до метки колбы. Дальнейшее определение произ-
водят в условиях, указанных для определения железа в вод-
ном растворе в виде моносульфосалицилата в отсутствие по-
сторонних ионов (стр. 140).
Интенсивность окраски измеряют на фотометре ФМ или
фотоколориметре ФЭК-М.
в. Определение больших количеств железа дифференциальным
спектрофотометрическим методом
10 мл испытуемого раствора, содержащего от 0,5 до 2,5 мг
железа, помещают в мерную колбу на 100 мл и прибавляют
20 мл сульфосалициловой кислоты, 1 мл H2SO4 и доводят
объем раствора до метки колбы водой. В тех же условиях
готовят 5 эталонных растворов с содержанием железа в
указанных пределах. Измеряют оптическую плотность рас-
творов на фотоэлектроколориметрах ФЭК-М, ФЭК-Н-52,
ФЭК-Н-54, ФЭК-Н-57, ФЭК-56 или спектрофотометре, исполь-
141
зуя светофильтр с Хмакс пропускания — 510 ммк и кювету
с толщиной слоя в 1 см. В качестве «нулевого» раствора при
измерении как оптических плотностей эталонных растворов,
так и контрольного, используют раствор, содержащий 0,5 мг
железа в 100 мл. Содержание железа в контрольном рас-
творе рассчитывают по градуировочному графику или по урав-
нению (109) (стр. 68), вычислив предварительно фактор F
как указано на стр. 69.
Примечание. При использовании кюветы с толщиной слоя в
0,5 см содержание железа в эталонных и контрольном растворах можно
увеличить вдвое.
ОПРЕДЕЛЕНИЕ ЖЕЛЕЗА В ВИДЕ ТРИСУЛЬФОСАЛИЦИЛАТА
а. Определение железа в водном растворе в отсутствие
посторонних ионов
К 10 мл испытуемого раствора, помещенного в мерную
колбу на 50 мл, прибавляют 5 мл раствора сульфосалицило-
вой кислоты, 5 мл раствора аммиака и объем раствора до-
водят водой до метки колбы. Дальнейшее определение про-
изводят в фотоколориметрах ФЭК-М., ФЭК-56, ФЭК-Н-54,
ФЭК-Н-57 или фотометре ФМ. Для этого готовят 6 эталон-
ных растворов, содержащих от 0,05 до 0,3 мг железа, строят
калибровочную кривую и графически находят количество же-
леза в испытуемом растворе.
Примечание. Определение железа в присутствии посторонних ве-
ществ, если концентрация их не превышает 0,3—0,5 г в 50 мл и они не реа-
гируют с сульфосалициловой кислотой, производят в условиях, указанных
выше.
б. Определение железа в растворимых в воде
фосфорнокислых солях1
Навеску анализируемого препарата 1—2 г (в зависи-
мости от содержания железа) помещают в коническую кол-
бу емкостью 50 мл, добавляют 10 мл воды и 5 мл раствора
сульфосалициловой кислоты и растворяют при слабом подо-
гревании (60—70°). При неполном растворении для получе-
ния прозрачного раствора добавляют несколько капель азот-
ной или соляной кислот. К полученному раствору добавляют
5 мл раствора аммиака. Образовавшийся окрашенный рас-
твор переводят в мерную колбу на 50 мл, добавляют водой
до метки колбы и измеряют интенсивность окраски в усло-
виях, указанных выше для определения железа в виде три-
сульфасалицилата.
1 В. М. П е ш к о в а, А. Д. Е г о р о в. «Зав. лаб.», 4, 885, 1935.
142
в. Определение железа в соляной кислоте
К 8,4 мл (10 г) испытуемой соляной кислоты (уд. в. 1,19),
помещенной в мерную колбу на 50 мл прибавляют 10 мл во-
ды, нейтрализуют раствором аммиака по индикаторной бу-
маге примерно до значения pH 6—8, добавляют 5 мл суль-
фосалициловой кислоты, 5 мл аммиака и объем раствора до-
водят водой до метки колбы. Дальнейшее определение про-
изводят в условиях, указанных на стр. 142 для трисульфоса-
лицилата железа.
г. Определение железа в азотной кислоте
К 7,1 мл (Ю г) испытуемой азотной кислоты (уд. в. 1,4),
помещенной в мерную колбу на 50 мл, прибавляют 10 мл во-
ды, нейтрализуют раствором аммиака по индикаторной бу-
маге примерно до значения pH 6—8, добавляют 5 мл раство-
ра сульфосалициловой кислоты, 5 мл аммиака и объем дово-
дят водой до метки колбы. Дальнейшее определение произ-
водят в условиях, указанных на стр. 142 для трисульфо-
салицилата железа.
д. Определение больших количеств железа
дифференциальным спектрофотометрическим методом
10—15 мл испытуемого раствора, содержащего от 0,5 до
1,5 мг железа, помещают в мерную колбу на 100 мл, прибав-
ляют 20 мл сульфосалициловой кислоты, 20 мл NH4OH и до-
водят объем раствора до метки колбы водой. В тех же усло-
виях готовят 5—6 эталонных растворов с содержанием же-
леза в указанных пределах. В дальнейшем определение
производят так же, как дано в методике определения железа
дифференциальным методом в виде моносульфосалицилата
(стр. 141), но измеряя оптические плотности при К 416 ммк.
Определение железа (II) о-фенантролином 1
Соли железа (II) с фенантролином дают комплексные со-
единения, в которых ион Fe2+ присоединяет три молекулы
реактива, образуя комплексный катион
1 Е. Б. С е н д е л. Колориметрические методы определения следов ме-
таллов. М„ «Мир», 1964, стр. 480.
143
Рис. 54. Спектры пропускания соеди-
нения железа с о-фенантролином при
различных концентрациях железа
Соединение образуется в широком диапазоне pH 2,0—9,0,
давая растворы, окрашенные в оранжево-красный цвет при
значительном содержании железа. Растворы комплексного
соединения отличаются большой устойчивостью, к ним при-
ложим закон Бугера — Ламберта — Бера. Железо можно вос-
становить до 2-валентного гидроксиламином или гидразином.
Спектр пропускания соеди-
нения дан на рис. 54 с мини-
мумом при А 508 ммк. Значе-
ние молярного коэффициен-
та погашения 11 000. Чувст-
вительность реакции при ви-
зуальном наблюдении 0,01 мг
в 25 мл раствора.
Реакция железа с о-фе-
нантролином применяется
только для определения же-
леза (II). Проведению реак-
ции мешает ряд веществ.
Серебро, висмут, кадмий,
цинк, ртуть образуют с ре-
активом малорастворимые
комплексы и уменьшают,
таким образом, интенсив-
ность окраски растворов же-
леза. При незначительном
содержании указанных эле-
ментов их влияние мож-
но устранить добавлением
большого избытка реактива.
Фосфаты, фториды, хлори-
ды и сульфаты не оказывают влияния, если присутствуют
в умеренных количествах.
Необходимые реактивы:
1. Стандартный раствор железа (II) с содержанием 0,1 мг
в 1 мл. 0,702 г х. ч. соли Мора помещают в мерную колбу
емкостью 1 л, растворяют в воде, подкисленной 4 мл серной
кислоты (уд. в. 1,11), доводят водой объем раствора до 1 л
и тщательно перемешивают.
2. Гидроксиламин солянокислый, 1О°/о-ный раствор.
3. Уксуснокислый натрий, 2М и 0,2М растворы.
4. о-Фенантролин, 0,5%-ный водный раствор.
5. Бромфенол синий, 0,1 %-ный раствор.
В мерную колбу на 25 мл вводят 10 мл испытуемого рас-
твора. В другую колбу берут также 10 мл испытуемого рас-
твора (аликвотную часть) и в присутствии индикатора бром-
фенолсинего добавляют такое количество ацетата натрия,
чтобы pH раствора было 3—6,0; найденный объем ацетата
144
натрия добавляют в первую колбу с испытуемым раствором,
вводят 1 мл раствора гидроксиламина и 1 мл раствора о-фен-
антролина, разбавляют водой до метки колбы и измеряют
интенсивность окраски раствора через 10 мин. В присутствии
фосфат-иона наблюдение следует производить через 1 час.
Интенсивность окраски измеряют на фотометре ФТ, фото-
колориметре ФЭК-М или ФЭК-56, ФЭК-Н-54, ФЭК-Н-57, вы-
бирая соответствующий светофильтр. Готовят 6 эталонов
в условиях, указанных для испытуемого раствора и содержа-
щих железо (II) в количествах (мг): 0,025; 0,05; 0,075; 0,1;
0,15; 0,20. Концентрацию железа в растворе определяют гра-
фическим методом.
Определение железа (II) 4,7-дифенил-1,10-фенантролином
в водном растворе 1
4,7-Дифенил-1,10-фенантролин, подобно о-фенантролину,
дает с железом (II) соединение аналогичного состава, но яв-
ляется более чувствительным реагентом для определения же-
леза (табл. 3, стр. 134).’
Кроме указанных на стр. 144 необходимы реактивы:
1. 4,7-Дифенил-1,10-фенантролин, 0,001 М раствор. 33 мг
реактива растворяют в смеси 1 : 1 изоамилового и этилового
спиртов.
2. Уксуснокислый натрий, 10%-ный раствор.
3. Этиловый и изоамиловый спирты.
К Ю мл водного раствора, содержащего железо, добав-
ляют 2 мл раствора гидроксиламина, 4 мл раствора ацетата
натрия и 4 мл раствора 4,7-дифенил-1,10-фенантролина. После
этого добавляют 6 мл изоамилового спирта и хорошо переме-
шивают. Через 5 мин разделяют фазы и переносят слой спир-
та в мерную колбу или цилиндр на 10 мл. Промывают дели-
тельную воронку 2—3 мл этилового спирта, добавляют его
к основному спиртовому раствору и доводят объем до 10 мл
этиловым спиртом. Поглощение измеряют при 533 ммк, при-
меняя спектрофотометры СФ-4, СФ-5, СФ-2М, СФ-10. Гото-
вят 5 эталонных растворов, содержащих железо в количе-
ствах (мг): 0,01; 0,025; 0,05; 0,075; 0,1 в условиях, указанных
для испытуемого раствора. Для определения железа исполь-
зуют графический метод, который рационально применять при
определении ультрамалых количеств железа.
Определение железа (II) диоксимом 1,2-циклогександиона2
2-валентное железо образует с а-диоксимами (H2D) со-
единения состава Fe (HD)2l подобно никелю, палладию, 2-ва-
1 Е. Сендел. Колориметрические методы определения следов ме-
таллов. М., «Мир», 1964, стр. 487.
2 В. М. Пешкова, В. М. Бочкова. «Методы анализа редких и
цветных металлов». Сб. статей. Изд-во МГУ, 1956.
145
лентной платине и меди. Железо (II) с диметилглиоксимом
образует в щелочной среде (pH — 10) растворы, окрашенные
в интенсивно красный цвет. В присутствии пиридина это со-
единение извлекается неводными растворителями.
В отличие от соединений Fe (II) с диметилглиоксимом
окрашенное соединение с диоксимом 1,2-циклогександиона
образуется в широком интервале кислотности раствора. В ам-
миачной среде окраска раствора сильно зависит от концен-
трации аммиака. Наиболее интенсивная окраска получается
при pH 9—10. Максимум поглощения растворов диоксиматов
железа с увеличением pH раствора смещается в длинновол-
новую область спектра.
2-валентное железо дает с диоксимом 1,2-циклогександио-
на два соединения: максимум поглощения первого соедине-
ния находится при 440 ммк, а второго — при 520 ммк. Моляр-
ные коэффициенты погашения равны соответственно 3300 и
7200. Первое соединение устойчиво в кислой среде при pH 3,
а второе — при pH 9—10. Чувствительность реакции повышает-
ся в щелочном растворе. Весьма вероятно, что в более щелоч-
ной среде образуется соединение типа [Fe D2]2+, а в кислой
среде Fe(HD2)2; спектры поглощения растворов при pH от 5
до 9, возможно, отвечают смеси двух соединений. При дли-
тельном стоянии растворов диоксиматов железа, полученных
в щелочной среде, максимум поглощения сдвигается в корот-
коволновую часть спектра, что видимо, связано с потерей ам-
миака раствором и переходом одного соединения в другое
(рис. 55).
Окраска раствора соединения 2-валентного железа с диок-
симом 1,2-циклогександиона и пиридином оказалась устойчи-
вой, не зависящей от избытка пиридина и более интенсивной,
чем в случае других диоксимов; молярный коэффициент по-
гашения 9800. Максимум поглощения находится при 520 ммк.
Соединение извлекается бензолом и хлороформом. В бензоль-
ном слое находится то же соединение, что и в водном рас-
творе. Окрашенные растворы подчиняются закону Бугера —
Ламберта — Бера.
Для определения железа (II) в присутствии железа (III)
в качестве комплексообразующих веществ берут соли лимон-
ной или винной кислот.
Кроме указанных на стр. 144 необходимы реактивы:
1. Пиридин, 10°/0-ный раствор.
2. Диоксим 1,2-циклогександиона, водный раствор.
3. Сегнетова соль, 20%-ный раствор.
Определение производят в следующих условиях. К вод-
ному раствору в делительной воронке, содержащему железо,
добавляют 0,5 мл водного раствора диоксима 1,2-циклогек-
сандиона, 3 мл раствора пиридина, 1,5 мл раствора сегнето-
вой соли, аммиак до щелочной реакции по фенолфталеину
146
и раствор доводят водой до 20 мл. Окрашенное соединение
извлекают из водного раствора двумя порциями бензола по
5 мл каждая.
Интенсивность окраски можно измерить, применяя фото-
метр ФТ и фотоколориметры ФЭК-М., ФЭК-56, ФЭК-Н-54,
ФЭК-Н-57. Готовят 5 эталонов в условиях, указанных для ис-
пытуемого раствора и содержащих железо (II) в количествах
485 478 ~496 ~~~533 541 574
А, ппк
Рис. 55. Спектры поглощения Fe (II) с диоксимом 1,2-циклогексан-
диона при различных значениях pH
(мг): 0,025; 0,05; 0,1; 0,15; 0,2. Определение производят гра-
фическим методом.
Определение железа (II) можно производить также в от-
сутствие пиридина, соблюдая в остальном условия, указан-
ные выше.
ОПРЕДЕЛЕНИЕ МАРГАНЦА
Определение марганца в виде перманганата
Для определения марганца используют ряд методов. Из
них наиболее широко известны методы, основанные на окис-
лении марганца (II) в марганцовую кислоту. Эти методы
обычно применяют для определения марганца в сталях, чу-
гунах, горных породах.
147
В качестве окислителей могут быть использованы пер-
сульфат аммония в присутствии катализаторов азотнокис-
лого серебра или солей кобальта, висмутат натрия и перйо-
дат калия. Реакцию окисления проводят в кислой среде; оп-
ределению мешают восстановители, поэтому хлор-ион дол-
жен отсутствовать. Если хлориды присутствуют в исходном
материале, то их предварительно удаляют выпариванием
с азотной и серной кислотами.
При окислении персульфатом аммония могут образовать-
ся соединения 4-валентного марганца, окрашивающие рас-
твор в винно-красный цвет. Для предотвращения образова-
ния этих соединений необходимо: 1) окисление марганца
производить в растворе, имеющем достаточно высокую кис-
лотность; 2) вводить фосфат-ион в виде фосфорной кислоты
или ее солей для образования промежуточного соединения
марганца (III); 3) к образовавшемуся раствору винно-крас-
ного цвета прибавить несколько крупинок муравьинокис-
лого натрия для восстановления 4-валентного марганца до
2-валентного, который затем снова окисляют в марганцовую
кислоту. Однако не следует брать избыток муравьинокислого
натрия во избежание восстановления самой марганцовой кис-
лоты.
При определении сравнительно больших количеств мар-
ганца полнота окисления достигается в присутствии перйо-
дата калия, который добавляют после окисления персуль-
фатом.
При успешном течении реакции окисления растворы
должны иметь чистый малиново-розовый цвет. Образующееся
соединение марганцовой кислоты имеет спектр поглощения,
приведенный на рис. 56 с максимумом поглощения при
525 ммк; молярный коэффициент погашения 2300.
Реакция чувствительна; определяемое минимальное коли-
чество марганца 0,05 мг в 50 мл раствора. Подчинение основ-
ному закону светопоглощения наблюдается до концентрации
150 мг/л.
ОПРЕДЕЛЕНИЕ МАРГАНЦА ПЕРСУЛЬФАТНЫМ МЕТОДОМ
Необходимые реактивы:
1. Стандартный раствор соли марганца, содержащий
0,1 мг в 1 мл, готовят двумя способами:
а) в мерную колбу емкостью 1 л помещают навеску
0,273 г х. ч. или ч. д. а. препарата сульфата марганца, пред-
варительно прокаленного до постоянного веса в воздушной
бане, растворяют соль в воде и водой доводят объем раство-
ра до 1 л;
б) 0,287 г перманганата калия, квалификации ч. д. а. по-
мещают в мерную колбу емкостью 1 л, растворяют в воде,
148
перегнанной в присутствии перманганата, доводят водой объем
раствора до 1 л и тщательно перемешивают.
2. Серная кислота, 6 н. раствор.
3. Азотнокислое серебро, 0,1 н. раствор.
4. Персульфат аммония, 20%-ный свежеприготовленный
' раствор.
5. Перйодат калия (или натрия), сухой препарат.
Рис. 56. Спектры поглощения: / — перманганат калия; 2—формаль-
доксимат марганца
6. Фосфорнокислый натрий двузамещенный, 10%-ный рас-
твор.
7. Раствор, состоящий из смеси кислот (серной, фосфор-
ной и азотной). Смесь кислот готовят следующим образом:
100 мл серной кислоты (уд. в. 1,84), 125 мл фосфорной кис-
лоты (уд. в. 1,7) и 250мл азотной кислоты (уд. в. 1,4) раз-
бавляют дистиллированной водой до 1 л. Для приготовления
указанной смеси кислот берут 500 мл воды, вводят туда по-
степенно серную кислоту при помешивании раствора, а затем
другие кислоты и доводят общий объем водой до 1 л.
7. Перекись водорода, 0,1 н. раствор.
149
а. Определение марганца в водном растворе,
не содержащем хлоридов
20 мл испытуемого водного раствора, не содержащего
хлоридов, помещают в колбу на 50 мл, прибавляют 20 мл
6 н. серной кислоты, 1 мл азотнокислого серебра, 1 мл пер-
сульфата аммония и 5 мл раствора фосфорнокислого натрия.
Колбу с раствором помещают в кипящую водяную баню или
на песчаную баню и выдерживают до появления устойчивой
розовой окраски. После этого раствор охлаждают, доводят
его объем до метки колбы водой и фотометрируют.
Ряд эталонных растворов с содержанием в них марганца
от 0,05 до 0,5 мг готовят в тех же самых условиях, предвари-
тельно вводя стандартный раствор марганца в 20 мл дистил-
лированной воды.
б. Определение марганца в сталях1
При определении марганца в сталях в эталонные раство-
ры вводят сталь, не содержащую марганец, или с известным
его содержанием. Эти образцы должны иметь состав, близ-
кий к определяемому образцу стали.
Для растворения стали применяют смесь серной, фосфор-
ной и азотной кислот. Серная и азотная кислоты используют-
ся в качестве растворителей. Фосфорная же кислота добав-
ляется с целью связать образующееся в процессе растворе-
ния стали 3-валентное железо в бесцветный комплекс, чтобы
исключить мешающее влияние окраски его солей в процессе
колориметрирования. О влиянии фосфат-ионов на процесс
окисления 2-валентного марганца до перманганат-ионов бы-
ло сказано выше (стр. 148).
В качестве стандартного раствора марганца, используемо-
го для приготовления эталонных растворов, применяют рас-
твор перманганата калия, который после введения его в мер-
ные колбы обесцвечивают перекисью водорода.
Приготовление эталонных растворов и по-
строение калибровочной кривой. 0,5 г стали с из-
вестным содержанием марганца помещают в коническую кол-
бу и растворяют при нагревании в 50 мл смеси кислот. Рас-
твор нагревают до прекращения выделения хорошо заметных
паров окислов азота, охлаждают его, переносят в мерную
колбу на 100 мл, доводят объем раствора дистиллированной
водой до метки колбы — раствор А. Затем для приготовления
эталонных растворов отбирают пипеткой по 20 мл раствора
А и переносят в мерные колбы на 100 мл. Приливают рас-
1 А. М. Дымов. Технический анализ руд и металлов. М., Металлург-
издат, 1948, стр. 280.
150
твор перманганата калия с таким расчетом, чтобы общее со-
держание марганца в каждом из эталонных растворов было
соответственно равно (лг):
1-й ряд 2-й ряд
0,5 1,1
0,7 1,3
0,9 1,5
1,1 1,7
К полученным растворам для обесцвечивания окраски
перманганата добавляют 3—4 капли перекиси водорода, из-
быток последней разрушают кипячением. Содержимое колб
охлаждают и добавляют по 0,5 мл азотнокислого серебра и
10 мл персульфата аммония. Растворы нагревают на водяной
бане (60—70°), пока окраска не перестанет усиливаться, за-
тем охлаждают, разбавляют до метки колбы дистиллирован-
ной водой, тщательно перемешивают и измеряют интенсив-
ность окраски, пользуясь фотометром и фотоколориметром.
По полученным данным строят калибровочную кривую.
Определение марганца в контрольном
образце стали. 0,1 г стали с известным содержанием
марганца растворяют в 10 мл смеси кислот в конической кол-
бе на 50—100 мл, нагревают раствор до прекращения выде-
ления окислов азота, охлаждают и переносят в мерную колбу
на 100 мл, приливают 0,5 мл раствора азотнокислого серебра
и 10 мл раствора персульфата аммония. Раствор нагревают
на песчаной бане или в кипящей водяной бане, пока интен-
сивность окраски не перестанет увеличиваться, охлаждают,
доводят объем раствора до метки колбы водой и измеряют
оптическую плотность. По калибровочной кривой, построен-
ной ранее по эталонным растворам, определяют содержание
марганца в испытуемом образце стали.
Примечание. Если содержание марганца в стали превышает 1%,
то в раствор после окисления персульфатом и охлаждения его добавляют
0,1—0.2 г перйодата калия, снова нагревают почти до кипения, выдержива-
ют при этой температуре 5 мин, охлаждают и доводят объем раствора до
метки колбы.
ОПРЕДЕЛЕНИЕ МАРГАНЦА ПЕРЙОДАТНЫМ МЕТОДОМ1
При окислении перйодатом растворы марганцовой кисло-
ты получаются более устойчивыми, чем при окислении пер-
сульфатом. Добавление фосфорной кислоты в данном случае
необходимо как в присутствии железа (для связывания его
в комплекс), так и в его отсутствие, так как она предотвра-
щает возможность осаждения перйодата или йодата мар-
ганца.
‘К. А. Бакланова, О. П. Бояршинова. Сб. «Анализ мине-
рального сырья». М., Госхимиздат, 1956, стр. 369.
151
а. Определение в растворах чистых солей
Необходимые реактивы:
1. Стандартный раствор, содержащий 0,1 мг Мп в 1 мл.
2. Перйодат калия (или натрия), сухой препарат.
3. Фосфорная кислота, концентрированная.
4. Серная кислота 1:1.
К исследуемому раствору в объеме около 30 мл, поме-
щенному в коническую колбу, прибавляют 6 мл серной кис-
лоты, 2—5 мл фосфорной кислоты и 0,3—0,5 г перйодата ка-
лия. Раствор нагревают до кипения и выдерживают при тем-
пературе, близкой к кипению, 5 мин, затем охлаждают и пе-
реносят раствор в мерную колбу на 50 мл. Доводят объем
раствора до 50 мл, перемешивают и измеряют интенсивность
его окраски.
Эталонные растворы с содержанием в них марганца от 0,1
до 0,5 мг готовят аналогичным образом, вводя предвари-
тельно стандартный раствор марганца в коническую колбу,
содержащую около 30 мл воды.
б. Определение марганца в стали
0,1 г стали с неизвестным содержанием марганца рас-
творяют в 10 мл смеси кислот в конической колбе на 50—
100 мл, раствор нагревают до прекращения выделения окис-
лов азота. Содержимое колбы охлаждают, переносят в мер-
ную колбу на 100 мл и производят окисление марганца в ус-
ловиях, указанных для определения марганца в водном рас-
творе перйодатным методом. Измеряют интенсивность окрас-
ки растворов и определяют содержание марганца по калибро-
вочной кривой в условиях, указанных при определении мар-
ганца персульфатным методом (стр. 150), заменив персуль-
фат на перйодат.
ОПРЕДЕЛЕНИЕ БОЛЬШИХ КОЛИЧЕСТВ МАРГАНЦА
ДИФФЕРЕНЦИАЛЬНЫМ СПЕКТРОФОТОМЕТРИЧЕСКИМ МЕТОДОМ
а. Определение марганца в отсутствие больших
количеств хрома
Большие количества марганца (5—10 мг в 100 лм; 5—10%
соответственно) можно определять, используя приведенную
выше методику приготовления растворов для случая перйо-
датного окисления марганца, но увеличив до 0,6 г количе-
ство перйодата.
В этом случае измеряют оптическую плотность контроль-
ного раствора (как и эталонных растворов) по отношению
к первому из эталонных растворов, используя кювету с тол-
152
щиной слоя в 0,5 см. Содержание железа в контрольном об-
разце рассчитывают по градуировочному графику или по
уравнению (109), вычислив предварительно фактор F, как
указано на стр. 69.
б. Определение марганца в присутствии больших
количеств хрома
Приведенные выше методики дают завышенные резуль-
таты при определении марганца в сталях, содержащих значи-
тельные количества хрома. В этом случае можно использо-
вать вариант дифференциального спектрофотометрического
метода, описанный на стр. 69.
0,5 г стали с неизвестным содержанием марганца раство-
ряют в 50 мл смеси кислот в конической колбе при нагрева-
нии до полного удаления окислов азота. Раствор переносят
в мерную колбу на 100 мл и доводят объем раствора водой
до метки колбы. Используя данный раствор стали, готовят
3 новых раствора:
1-й раствор — в коническую колбу на 100 мл помещают
20 мл раствора контрольного образца стали;
2-й раствор — в коническую колбу на 100 мл помещают
3.0 мл раствора контрольного образца стали;
3-й раствор — в коническую колбу на 100 мл помещают
30 мл раствора контрольного образца стали и прибавляют
0.5 мг марганца, используя его стандартный раствор.
Во все колбы прибавляют по 0,6 г перйодата калия, нагре-
вают почти до кипения и выдерживают при этой температуре
5 мин. После прекращения нарастания интенсивности окраски
перманганата растворы переносят в мерные колбы на 100 мл
и доводят объем раствора водой до метки колбы.
Измеряют оптические плотности второго Dx и третьего
Вх+а растворов по отношению к первому, используя кювету
с толщиной слоя 0,5 см, и вычисляют содержание марганца
в 10 мл раствора контрольного образца стали (что соответ-
ствует разности объемов, прибавленных во второй и первый
растворы) по формуле (117):
. ^хса
Сх^—-
иа
На основании полученного результата вычисляют про-
центное содержание марганца в контрольном образце стали.
Определение марганца формальдоксимом * 1
Для определения марганца в воде, в золе растений, когда
железо содержится в небольших количествах, можно исполь-
1 В. М. Пешкова, А. А. Овсянникова. «Зав. лаб.», 6, 800, 1937;
Е. С е н д е л. Колориметрические методы определения следов металлов. М.,
«Мир», 1964, стр. 550.
153
зовать в качестве реактива формальдоксим. Марганец 2-ва-
лентный реагирует с формальдоксимом в щелочной среде
с образованием окрашенного в красно-бурый цвет соедине-
ния, спектр поглощения которого имеет максимум при
А 455 ммк (рис. 56), е — 11 000. Состав соединения, выделен-
ного в этих условиях в виде сухого препарата, соответствует
формуле Mn(H2C = NO)3. Следовательно, марганец окис-
ляется до 3-валентного в щелочной среде кислородом воз-
духа.
Формальдоксим пригоден также для определения пер-
манганат-иона. Здесь формальдоксим вначале играет роль
восстановителя. В дальнейшем механизм реакции образова-
ния формальдоксимата марганца аналогичен указанному
выше.
Необходимые реактивы:
1. Стандартный раствор марганца, содержащий в 1 мл
0,1 мг Мп. Стандартный раствор марганца готовят так же,
как при определении марганца персульфатным методом в
растворах чистых солей.
2. Формальдоксим, раствор. Раствор формальдоксима го-
товят, прибавляя к 1 мл 40%-ного (не ниже!) раствора фор-
малина 2 г солянокислого гидроксиламина и 50 мл дистил-
лированной воды.
3. Едкий натр, 1 н. раствор.
а. Определение марганца в растворе чистой соли
10—15 мл испытуемого раствора, содержащего марганец,
помещают в мерную колбу на 50 мл и прибавляют 0,5 мл
раствора формальдоксима, 2—3 мл раствора щелочи и смесь
перемешивают; через 5 мин добавляют 4—5 капель раствора
формальдоксима и 0,5 мл раствора щелочи и наблюдают, не
увеличивается ли интенсивность окраски раствора. Если
окраска раствора не изменяется, то объем его доводят водой
до метки колбы. Если же интенсивность окраски возрастает
при вторичном добавлении реактива, то добавляют еще не-
сколько капель формальдоксима и щелочи, пока заметное
нарастание окраски не прекратится. Избыток формальдокси-
ма и щелочи не мешает.
Для приготовления эталонных растворов берут мерные
колбы того же объема, вводят в них раствор марганца, рас-
творы формальдоксима и щелочи в тех же количествах и при
условиях, которые были указаны для определения марганца
в испытуемом растворе.
Эталонные растворы, приготовленные в колбах на 50 мл,
должны содержать марганец в количествах от 0,05 до 0,40 мг;
интервал взятых концентраций марганца 0,05 мг. Измеряют
154
оптическую плотность на фотоколориметре (ФЭК-М, ФЭК-56,
ФЭК-Н-54, ФЭК-Н-57), фотометре или микроколори-
метре.
ОПРЕДЕЛЕНИЕ БОЛЬШИХ КОЛИЧЕСТВ МАРГАНЦА
ДИФФЕРЕНЦИАЛЬНЫМ СПЕКТРОФОТОМЕТРИЧЕСКИМ МЕТОДОМ
а. Определение марганца в отсутствие мешающих ионов
10—15 мл испытуемого раствора, содержащего от 0,2 до
0,7 мг марганца, помещают в мерную колбу на 50 мл, при-
бавляют 1,5 мл свежеприготовленного раствора формальдок-
сима, 10 мл щелочи, оставляют стоять 5 мин, вновь прибав-
ляют такое же количество формальдоксима и щелочи, после
чего доводят объем раствора до метки колбы водой и изме-
ряют оптическую плотность. В тех же условиях готовят
6—7 эталонных растворов с содержанием в них марганца в
указанных пределах. Измерение оптической плотности рас-
творов производят на любом фотоэлектроколориметре, ис-
пользуя светофильтр с ХМакс пропускания—450 ммк и кювету
с толщиной слоя в 0,3 — 0,5 см. В качестве «нулевого» рас-
твора как при измерении оптической плотности эталонных
растворов, так и контрольного используют раствор, содержа-
щий 0,2 мг марганца. Содержание железа в контрольном рас-
творе рассчитывают по градуировочному графику или по
формуле (109), вычислив предварительно фактор F, как ука-
зано на стр. 69.
б. Определение марганца в присутствии железа
Для определения марганца в присутствии железа может
быть использован вариант дифференциального спектрофото-
метрического метода, сущность которого изложена на стр. 69.
Раствор контрольного образца, содержащий марганец и
железо в количестве 0,5—3 мг, помещают в мерную колбу на
100 мл и доводят объем раствора до метки колбы водой.
Готовят три раствора:
1-й раствор — в мерную колбу на 100 мл помещают 10 мл
испытуемого раствора;
2-й раствор — в мерную колбу на 100 мл помещают 20 мл
того же испытуемого раствора;
3-й раствор — в мерную колбу на 100 мл помещают 20 мл
испытуемого раствора и прибавляют 0,1 мг марганца, исполь-
зуя его стандартный раствор.
Все растворы обрабатывают по приведенной выше мето-
дике для определения марганца дифференциальным спектро-
фотометрическим методом в отсутствие железа.
155
Измеряют оптические плотности второго Dx и третьего
Dx+a растворов, используя в качестве «нулевого» первый
раствор и кювету с толщиной слоя в 0,3—0,5 см, и вычисля-
ют содержание марганца в 10 мл испытуемого раствора (что
соответствует разности объемов, прибавленных во второй и
первый растворы) по формуле (117) (стр. 70).
Примечание. Этим методом возможно произвести определение
марганца в присутствии других элементов (медь, никель), реагирующих
так же, как и железо с формальдоксимом.
ОПРЕДЕЛЕНИЕ НИКЕЛЯ
Наиболее распространенными реагентами для определе-
ния никеля являются а-диоксимы. Впервые предложенные
Л. А. Чугаевым 1 для открытия и определения никеля реаген-
ты указанной группы не потеряли своего значения до настоя-
щего времени. Распространение получили две группы мето-
дов определения никеля. В первой — используются окрашен-
ные соединения с а-диоксимами, получаемые при действии
ряда окислителей в щелочной среде, во второй — окрашенные
растворы, получаемые при растворении в неводных раствори-
телях соединений диоксиматов никеля. Известна довольно
большая группа а-диоксимов, кроме диметилдиоксима, кото-
рые нашли широкое применение для фотометрического опре-
деления никеля с использованием метода экстракции.
Интерес к другим а-диоксимам возник в связи с необхо-
димостью определения ультрамалых количеств никеля в ряде
объектов. Для решения последней задачи преимуществом об-
ладают органические реагенты, образующие очень устойчи-
вые комплексные соединения с большими значениями моляр-
ных коэффициентов погашения. Для определения ультрама-
лых количеств никеля применяют: а-бензилдиоксим, а-фу-
рилдиоксим и диоксим 1,2-циклогептандиона (гептоксим).
Комплексные соединения никеля со всеми а-диоксимами
имеют три полосы поглощения; более интенсивная из них
лежит в ультрафиолетовой области, где поглощают и сами
реагенты. В табл. 4 приведены некоторые характеристики
комплексных соединений никеля. Из этой таблицы видно, что
более устойчиво соединение никеля с а-бензилдиоксимом;
это же соединение имеет большее значение е. Близким по
устойчивости является соединение никеля с гептоксимом,
комплексные соединения никеля с этими двумя реагентами
не разрушаются при обработке экстрактов раствором ще-
лочи, применяемой для разрушения соответствующих ком-
плексных соединений кобальта и меди, часто сопутствующих
никелю и образующихся при действии а-бензилдиоксима и
1 Л. А. Чугаев. Избр. труды, т. I. М., Изд-во АН СССР, 1954.
J56
гептоксима'. Кроме того, действием щелочи избыток этих
двух реагентов удаляется из слоя органического раствори-
теля при экстракции комплексного соединения и открывается
возможность измерять поглощение в ультрафиолетовой об-
ласти при Х,макс только комплексного соединения (стр. 72,
рис. 26). Комплексное соединение никеля с а-фурилдиокси-
мом менее устойчиво; при удалении избытка реактива ком-
плексное соединение разрушается, поэтому в ультрафиолето-
вой области производить измерения невозможно. Тем не ме-
нее указанный реактив является ценным для фотометриче-
ских определений никеля, так как имеется возможность из-
мерять D в видимой области спектра (X 438 ммк), при е =
= 19 000; реактив в этой области спектра не поглощает.
Значения 1g Куст для комплексных соединений получены
методом экстракции; для а-бензилдиоксима — методом рас-
творимости. Для определения ультрамалых количеств никеля
и кобальта необходимо применять реактивы квалификации
«особой чистоты» или в отсутствие таковых подвергать реак-
тивы специальной очистке..
Таблица 4
Значение и 1g Куст комплексных соединений никеля
Реагент \<акс> ммк емакс 1g Куст (общая)
Диметилдиоксим 262 2,45-10* 17,61 + 0,14
327 4,58-103
374 3,43-103
Диоксим 1,2-циклогептан- 263 2,49-10* 21,90+0,15
диона (гептоксим) 330 5,05-103
377 4,05-Ю3
а-Бензилдиоксим* 273 5,0-10* 25,20
361 1,0-10*
406 1,2-104
а-Фурилдиокснм* 293 5,0-10* 14,85+0,02
384 1,0-10*
435 1,9-10*
* Данные наших исследований. Остальные значения взяты из статьи
С. Banks, D. Barnum. «J. Amer. Chem. Soc», 80, 4767, 1958.
Определение никеля диметилглиоксимом
в присутствии окислителей
Реакция определения никеля диметилглиоксимом в ще-
лочной среде в присутствии окислителей получила большое
распространение. В результате реакции образуются растворы,
1 В. М. Пешкова, Н. Г. Игнатьева. ЖАХ, XVII, 1086, 1962.
157
окрашенные в бурый цвет. Максимальное поглощение наблю-
дается при 470 ммк; значение молярного коэффициента по-
гашения 13 000. В результате ряда исследований, в которых
авторы применяли различные окислителиможно предпола-
гать, что в условиях проведения реакции происходит окисле-
ние никеля. Наши исследования1 2 показали, что в присутствии
аммиака и щелочи образуются два комплексных соединения,
так как спектры их
поглощения различны
(рис. 57).
Определение состава
соединений, полученных в
присутствии аммиака и
щелочи, показывает, что
в этих соединениях отно-
шение Ni: DH2= 1 : 3. Наи-
более вероятно, что ва-
лентность никеля в этих
соединениях равна трем.
Более устойчивые ком-
плексные соединения об-
разуются в присутствии
щелочи. В качестве оки-
слителя лучше использо-
вать раствор иода. Ни-
кель может быть опреде-
лен указанной реакцией
в сталях в присутствии
кобальта, ванадия, мо-
либдена. Вольфрам, хром
и титан могут присут-
ствовать до 18%. Ме-
шают медь и все эле-
Рис. 57. Спектры поглощения: 1 —
диметилглиоксим; 2 — диоксимат ни-
келя в присутствии NH3; 3 — диокси-
мат никеля в присутствии NaOH
менты, которые дают осадки гидроокисей при действии
растворов аммиака и едкой щелочи.
Необходимые реактивы:
1. Стандартный раствор соли никеля с содержанием 0,1 мг
в 1 мл. Для приготовления стандартного раствора навеску'
сернокислого никеля, квалификация х. ч. с известным содер-
жанием никеля, соответствующую 0,1 г никеля, помещают в
мерную колбу на 1 л, растворяют в воде, подкисляют 2 мл
концентрированной серной кислоты квалификации х. ч., дово-
дят объем водой до 1 д и тщательно перемешивают — рас-
твор А. Перед употреблением раствор А разбавляют в 10 раз.
1 А. Ок а с, М. Si те к. «Chem. Listy», 52, 1903, 1958; А. С. Андре-
ев, О. П. Азрельян, Е. С. Поспелова. ЖАХ, 6, 375, 1951.
2 В. М. Пешкова, Н. В. Мельчакова. Сб. «Методы анализа ред-
ких и цветных металлов». Изд-во МГУ, 1956, стр. 101.
158
2. Иод, 0,05 М. раствор.
3. Диметилглиоксим, 0,05 М. спиртовый раствор.
4. Едкий натр, 1 н. раствор.
5. Азотная кислота 1 : 5.
6. Сегнетова соль, 20%-ный раствор.
7. Соляная кислота, 1 н. раствор.
а. Определение никеля в растворе чистой соли
К 20 мл испытуемого раствора, помещенного в мерную
колбу на 50 мл, добавляют 1—2 капли соляной кислоты,
0,5 мл раствора иода, 0,5 мл раствора диметилглиоксима,
2,5 мл раствора щелочи и объем доводят водой до 50 мл.
После перемешивания измеряют интенсивность окраски рас-
твора, используя фотоколориметры ФЭК-М., ФЭК-56,
ФЭК-Н-54, ФЭК-Н-57 или фотометр. Эталонные растворы го-
товят в аналогичных условиях, обязательно соблюдая исход-
ный объем раствора, содержащего никель, и указанный поря-
док прибавления реактивов. Содержание никеля в эталонных
растворах может быть от 0,005 до 0,1 мг в 50 мл раствора.
б. Определение никеля в сталях, не содержащих меди
Приготовление эталонных растворов и по-
строение калибровочной кривой. Навеску 0,1 г
стали, не содержащей никеля, переносят в коническую колбу
на 50 мл, растворяют в 10 мл азотной кислоты, нагревают на
песчаной бане до удаления окислов азота, раствор охлаж-
дают, переносят его в мерную колбу на 100 мл и доводят во-
дой до метки — раствор А. В мерные колбы на 50 мл отби-
рают пипеткой по 2,5 мл раствора А и вводят стандартный
раствор соли никеля в таких количествах, чтобы содержание
никеля (мг) в эталонных растворах соответствовало:
1-й ряд 2-й ряд
0,005 0,06
0,01 0,07
0,02 0,08
0,03 0,09
0,04 0,1
0,05
К растворам добавляют по 5 мл раствора сегнетовой со-
ли, объем доводят водой до 20—25 мл, после этого добав-
ляют 0,5 мл раствора иода, 0,5 мл раствора диметилглиок-
сима, 3 мл раствора едкой щелочи и водой доводят объем до
50 мл. По приготовленным эталонным растворам строят ка-
либровочный график.
Определение никеля в контрольном образ-
це стали. Для определения никеля в сталях 0,1 г исследуе-
мого объекта переносят в коническую колбу на 50 мл, добав-
159
Рис. 58. Спектр поглощения соедине-
ния N1 с а-фурилдиоксимом в СНС1з
ляют 10 мл азотной кислоты, нагревают раствор на песчаной
бане до удаления окислов азота, охлаждают его, переносят
в мерную колбу на 100 мл и доводят водой объем раствора
до метки — раствор А. В мерные колбы на 50 мл отбирают
пипеткой по 2,5 мл раствора А, добавляют те же количества
реактивов в той же последовательности, как это указано для
приготовления эталонных растворов, не вводя стандартные
растворы соли никеля. Измерение интенсивности окраски
растворов производят в тех же условиях, в которых построе-
на калибровочная кривая.
Примечания. 1. При содержании Ni в стали около 0,20% и мень-
ше берут для испытания 5 мл раствора А.
2. При определении никеля в стали можно использовать эталонные
растворы, приготовленные в условиях, указанных для определения никеля
в растворе чистой соли (стр. 159). Но при измерении величины оптической
плотности испытуемого раствора в этом случае в качестве «нулевого» сле-
дует использовать также испы-
туемый раствор стали, в котором
не проведена колориметрическая
реакция (без добавления к нему
диметилглиоксима, щелочи и окис-
лителя) .
Определение никеля
а-фурилдиоксимом методом
экстрагирования 1
В методе экстрагирова-
ния используется раствори-
мость диоксиматов никеля
в неводных растворителях.
Из диоксимов лучше всего
использовать а-фурилдиок-
сим, так как его соединение
с Ni имеет максимум по-
глощения в видимой части
спектра (рис. 58) при Хмакс
438 ммк, значение молярно-
го коэффициента погашения
при этой же длине волны —
19 000. Соединение получа-
ют в результате реакции
Ni с а-фурилдиоксимом в
присутствии фосфатного буферного раствора (pH 8,5—9,5).
В качестве неводного растворителя используют хлороформ
или бензол. Метод можно рекомендовать для определения
никеля в сталях, в легких сплавах, в алюминии.
1 В. М. Пешкова, Е. А. Грибова, Г. А. Гончарова,
И. В. П у з д р е и к о в а. ЖАХ, 8, 114, 1953; В. М. П е ш к о в а, В. М. Б о ч-
к о в а, Л. И. Лазарева. ЖАХ, 15, 610, 1960.
160
Необходимые реактивы:
1. Стандартный раствор соли никеля с содержанием
0,001 мг готовят разбавлением 10 мл раствора А (стр. 158)
до 1 л. Раствор пригоден в день его приготовления.
2. а-Фурилдиоксим, 0,2%-ный водный раствор (пригоден
в течение недели).
3. Na2HPO4, 0,2 н. раствор.
4. Хлороформ или бензол.
5. Сегнетова соль, 20%-ный раствор.
6. Азотная кислота 1 : 5.
7. Раствор NaOH, 10%-ный.
8. NH4OH 1 : 50.
а. Определение никеля в растворе чистой соли
К раствору соли никеля в объеме 2—20 мл (в зависимости
от его содержания), помещенному в делительную воронку на
50 мл, добавляют 1 мл раствора а-фурилдиоксима, 20 мл
раствора соли двузамещенного натрийфосфата и регулируют
pH раствора до значения'8,5—9,5 по универсальной индика-
торной бумаге, прибавляя по каплям раствор NaOH, дают
постоять содержимому 15 мин и образовавшееся соединение
диоксимата никеля экстрагируют в течение 5—10 мин двумя
порциями хлороформа или бензола по 5 мл каждая. Для уда-
ления следов влаги в хлороформе или бензоле после отделе-
ния слоев его можно или профильтровать через сухой бумаж-
ный фильтр, или перелить в сухую колбочку; в последнем
случае влага удерживается на стенках стеклянного сосуда.
Практически с хлороформом работать удобнее, так как раз-
деление слоев с этим растворителем происходит быстрее.
При содержании никеля от 0,05 мг и выше экстрагирова-
ние следует проводить из 15—20 мл водного раствора, беря
первую порцию растворителя 10 мл, и делать повторную об-
работку водного раствора еще двумя порциями (по 5 мл)
растворителя. Все порции растворителя сливают в мерную
колбочку на 25 мл или градуированную пробирку на 10 мл
и доводят объем до метки применяемым растворителем (хло-
роформом или бензолом).
Содержание никеля в рядах эталонных растворов зави-
сит от процентного состава анализируемого объекта:
1-й ряд 2-й ряд 3-й ряд 4-й ряд
Кювета 5 см СФ-4 Кювета 2 см и СФ-4 Любые измерительные приборы
0,05 мкг 0,5 мкг 0,005 мг 0,025 мг
0,1 » 0,7 » 0,007 » 0,05 »
0,2 » 0,9 » 0,01 » 0,075 »
0,3 » 1,1 » 0,025 » 0,1 »
0,4 » 1,3 » 0,05 » 0,15 »
0,5 » 1,5 » 0,075 » 0,2 »
61/4 Зак. 686
161
Эталонные растворы готовят в аналогичных условиях, обя-
зательно соблюдая исходный объем раствора, содержащего
никель, постоянным; порядок прибавления реактивов и усло-
вия экстрагирования соединения указаны выше.
Интенсивность окраски раствора измеряют, используя фо-
тометр, фотоколориметры ФЭК-56, ФЭК-Н-54, ФЭК-Н-57
или спектрофотометры СФ-4, СФ-5 и СФ-2М, если опреде-
ляют малые концентрации никеля. Раствором сравнения яв-
ляется хлороформ.
б. Определение никеля в сталях1
Навеску 0,1 г стали переносят в коническую колбу на
50 мл, растворяют ее в 10 мл азотной кислоты, нагревают
раствор на песчаной бане до удаления окислов азота, выпа-
ривают его до объема 2—3 мл, переводят в мерную колбу на
50 мл и доводят водой объем до метки. К 1—2 мл приготов-
ленного раствора (в зависимости от содержания никеля), по-
мещенного в делительную воронку, добавляют 10 мл раство-
ра сегнетовой соли, 1 мл раствора а-фурилдиоксима, 10 мл
раствора фосфата натрия и регулируют pH раствора до зна-
чения 8,5—9,5 по универсальной индикаторной бумаге, до-
бавляя по каплям 25%-ный раствор NaOH. Дают постоять
содержимому воронки 5 мин и экстракцию образовавшегося
соединения диоксимата никеля производят точно в условиях,
предложенных для определения никеля в растворе чистой
соли.
Все хлороформенные или бензольные экстракты собирают
в делительную воронку и энергично перемешивают с 10 мл
аммиака. Сливают хлороформенные экстракты в мерную
колбу на 25 мл или градуированную пробирку на 10 мл и
доводят объем до метки применяемым растворителем (хлоро-
формом или бензолом). Для приготовления эталонных рас-
творов используют водные растворы никеля. Никель вводят
в количествах, соответствующих одному из рядов эталонных
растворов, указанных выше, в зависимости от его процент-
ного содержания в испытуемом образце. Далее измеряют D,
как указано в методике определения никеля в растворе чи-
стой соли.
в. Определение никеля в алюминии 2
Кроме указанных на стр. 161 необходимы реактивы:
1. Стандартный раствор соли никеля с концентрацией 10
и 1 мкг/мл готовят разбавлением раствора А (стр. 158) в
день употребления.
1 A. G a h I е г, A. A. Mitchell, М. Mellon. «Anal. Chem.», 23, 500,
1951.
2 В. М. Пешкова, В. М. Бочкова, Л. И. Лазарева. ЖАХ,
15, 610, 1960.
162
2. Кислота азотная по ГОСТу 4461—48, концентрирован-
ная, перегнанная в кварцевом приборе.
3. Аммиак 1 :40. Готовят из аммиака по ГОСТу 3760—47
насыщением дважды перегнанной воды при стоянии в экси-
каторе.
4. Едкий натр, 4 н. раствор.
5. Соляная кислота по ГОСТу 3118—46, концентрирован-
ная, очищенная перегонкой.
6. Хлороформ по ГОСТу 3160—51, очищенный от карбо-
нилхлорида и спирта промыванием 5%-ным раствором ще-
лочи и несколькими порциями воды и затем после осушки
перегнанный. Хлороформ хранят в склянке темного стекла.
7. Винная кислота особой чистоты, 20%-ный водный рас-
твор.
8. Фосфатный буферный раствор, pH 9,6.
9. а-Фурилдиоксим, 0,2%-ный спиртовый раствор.
10. Дистиллированная вода, дважды перегнанная в квар-
цевом приборе, или полученная из обычной дистиллирован-
ной воды на специальной ионнообменной колонке пропуска-
нием через смесь катионита и анионита.
Вся работа проводится с водой, полученной указанным
способом.
11. Универсальная индикаторная бумага с интервалом
pH от 8,6 до 9,8.
Все перечисленные реагенты должны быть испытаны спек-
трофотометрическим методом на отсутствие в них никеля по-
реакции с а-фурилдиоксимом в условиях, изложенных Ниже.
Измерения производят на спектрофотометре СФ-4 или
СФ-5. Кюветы стеклянные с рабочей длиной 5 см\ ввиду ма-
лого объема растворов используют ограничитель с шириной
щели 5 мм. Кюветы тефлоновые с кварцевыми стеклами; ра-
бочая длина 10 см, емкость 2,5—3 мл.
Приготовление эталонных растворов. В де-
лительные воронки конической формы емкостью 50 мл поме-
щают раствор никеля с различным его содержанием, 5 мл
раствора а-фурилдиоксима, 10 мл буферной смеси; если pH
раствора меньше 9,5, то добавляют раствор щелочи до ука-
занного значения pH. Через 15 мин образовавшееся соедине-
ние экстрагируют двумя порциями (по 2,5 мл каждая) хло-
роформа; содержимое воронки встряхивают в течение 15 мин
после прибавления каждой порции хлороформа, используя
механический вибратор. Перед второй экстракцией добав-
ляют в воронку еще 5 мл раствора а-фурилдиоксима. Хлоро-
форменные растворы сливают в мерные градуированные ци-
линдры и доводят объем хлороформом до 5 мл. Затем рас-
творы помещают в кювету и измеряют светопоглощение.
61/а Зак. 686
163-
В зависимости от содержания никеля (мкг) готовят три ряда
эталонных растворов:
1-й ряд 2-й ряд 3-й ряд
0,005 0,05 0,30
0,01 0,10 0,40
0,015 0,15 0,50
0,02 0,20 0,60
0,05 0,25 0,70
0,30 0,80
0,90
1,00
По данным измерений строят калибровочный график.
В качестве раствора сравнения («нулевого» раствора) ис-
пользуют раствор а-фурилдиоксима в хлороформе, которым
обработан раствор, содержащий все реагенты, кроме соли
никеля, и прошедший все стадии, описанные выше.
Определение никеля в контрольном об-
разце алюминия. 1 г алюминия помещают в тефлоно-
вую или кварцевую чашку емкостью 70—100 мл и растворяют
в 10—15 мл соляной кислоты, добавляя последнюю неболь-
шими порциями. Раствор выпаривают почти досуха, добав-
ляют 20 мл 20%-ного раствора винной кислоты и переносят
в делительную воронку, смывая чашку двумя порциями по
5 мл. К раствору в делительную воронку добавляют 10 мл
буферной смеси, 5 мл раствора а-фурилдиоксима. После это-
го измеряют значение pH раствора по универсальному инди-
катору.
Если pH раствора будет меньше 9,5, то добавляют раствор
щелочи до указанного значения pH. Через 15 мин получен-
ное соединение никеля экстрагируют двумя порциями (по
2,5 мл) хлороформа. Содержимое воронки встряхивают в те-
чение 15 мин после прибавления каждой порции хлороформа,
используя механический вибратор. Перед второй экстракцией
еще добавляют в воронку 5 мл раствора а-фурилдиоксима.
Объединенные экстракты промывают 10 мл раствора аммиа-
ка (1 :40). Далее поступают согласно описанию определения
никеля в водном растворе. В зависимости от содержания ни-
келя используют первый, второй или третий ряд эталонных
растворов, измеряя D как указано в методике приготовления
эталонных растворов.
г. Определение никеля в галлии
Приготовление эталонных растворов. В де-
лительные воронки конической формы емкостью 50 мл поме-
щают раствор никеля с различным его содержанием, 5 мл
164
раствора а-фурилдиоксима и 10 мл буферной смеси. Через
15 мин экстрагируют образовавшееся соединение двумя пор-
циями (по 2,5 мл каждая) хлороформа. Содержимое воронки
встряхивают в течение 15 мин после прибавления каждой
порции хлороформа, используя механический вибратор. Пе-
ред второй экстракцией добавляют в воронку еще 5 мл рас-
твора а-фурилдиоксима. Хлороформенные растворы сливают
в мерные градуированные цилиндры и доводят объем хлоро-
формом до 5 мл. Затем растворы помещают в кювету и из-
меряют их светопоглощение. В зависимости от содержания
никеля готовят эталонный ряд растворов (стр. 164). В каче-
стве «нулевого» используют раствор а-фурилдиоксима в хло-
роформе, которым обработан раствор, содержащий все реа-
генты, кроме соли никеля, и прошедший все стадии, описан-
ные выше.
Определение никеля в контрольном образ-
це галлия. 1 г металлического галлия, помещают в теф-
лоновую или кварцевую чашку и растворяют в смеси 2 мл со-
ляной и 6 мл азотной кислот. После растворения полученный
раствор упаривают почти досуха на водяной бане. Остаток
растворяют в 10 мл винной кислоты и нейтрализуют раство-
ром щелочи, прибавляя его постепенно до pH 9,5 (по инди-
каторной бумаге). Раствор переносят в делительную воронку,
добавляют 5 мл раствора а-фурилдиоксима и оставляют
стоять 15 мин. После этого проводят экстракцию двумя пор-
циями (по 2,5 мл каждая). Содержимое воронки встря-
хивают в течение 15 мин после прибавления каждой пор-
ции хлороформа, используя механический вибратор. Пе-
ред второй экстракцией добавляют еще 5 мл раствора а-фу-
рилдиоксима. Объединенные экстракты промывают 10 мл
раствора аммиака (1 :40). Хлороформенный раствор сливают
в мерный цилиндр и доводят его объем хлороформом до
5 мл. Затем раствор помещают в кювету и измеряют его све-
топоглощение. Далее поступают согласно описанию приготов-
ления эталонных растворов.
д. Определение никеля в двуокиси кремния
При определении никеля в двуокиси кремния основная
масса кремния удаляется с плавиковой кислотой в виде
H2SiF6 и SiF4.
Кроме указанных на стр. 163 необходимы реактивы:
1. Плавиковая кислота, концентрированная, квалификации
ч. д. а., очищается перегонкой в тефлоновом приборе.
2. NH4OH 1 :40.
3. Серная кислота 1:1.
Определение никеля в контрольном образ-
це двуокиси кремния. Навеску 2 г двуокиси кремния
6Ч2* 165
помещают в тефлоновую чашку и обрабатывают плавиковой
кислотой (около 25 мл) с несколькими каплями серной кис-
лоты. Затем раствор выпаривают до объема одной капли на
воздушной бане при ~ 80°. После выпаривания остаток рас-
творяют в 5 мл винной кислоты и раствор нейтрализуют в
чашке раствором щелочи до pH ~ 9,5 по индикаторной бу-
маге. Полученный раствор переносят в делительную воронку
объемом 50 мл, промывая чашку 5—10 мл воды. Затем до-
бавляют 5 мл раствора а-фурилдиоксима и оставляют стоять
15 мин. После этого экстрагируют соединение никеля двумя
порциями (по 2,5 мл каждая) хлороформа. Содержимое во-
ронки встряхивают в течение 15 мин после прибавления каж-
дой порции хлороформа, используя механический вибратор.
Хлороформенные экстракты объединяют и промывают 10 мл
аммиака.
Полученные хлороформенные растворы сливают в мер-
ные цилиндры на 5 мл и доводят хлороформом объем до
5 мл. Полученный раствор помещают в кювету и измеряют
его светопоглощение. В качестве «нулевого» используют рас-
твор а-фурилдиоксима в хлороформе, которым обработан
раствор, содержащий все реагенты, кроме соли никеля, и
прошедший все стадии, описанные выше. Далее поступают
согласно описанию приготовления эталонных растворов
(стр. 164). В зависимости от содержания никеля готовят эта-
лонный ряд растворов.
Определение никеля в индии а-бензилдиоксимом
Кроме указанных на стр. 163 необходимы реактивы:
1. Едкий натр, 1 н. раствор.
2. Лимонная кислота квалификации «особой чистоты»,
20%-ный раствор.
3. Этиловый спирт, перегнанный над твердым едким нат-
ром.
4. а-Бензилдиоксим, дважды перекристаллизованный из
диоксана, 0,02%-ный спиртовый раствор.
Все перечисленные реагенты должны быть испытаны спек-
трофотометрическим методом: а) на отсутствие в них никеля
по реакции с а-бензилдиоксимом в условиях, изложенных ни-
же; б) на отсутствие других примесей, экстрагируемых хло-
роформом и поглощающих при % 275 ммк.
Приготовление эталонных растворов. В де-
лительные воронки конической формы емкостью 50 мл поме-
щают различные количества соли никеля, добавляют по 2 мл
раствора сегнетовой соли, 2 мл раствора а-бензилдиоксима
и спирт до концентрации его в растворе~20% (по объему).
Через 15 мин образовавшееся соединение экстрагируют
Двумя порциями (по 2,5 мл каждая) хлороформа. Содержи-
мое воронки встряхивают в течение 15 мин после добавления
166
каждой порции хлороформа, используя механический вибра-
тор. После расслаивания фаз, осторожно отделяют водный
слой пипеткой и проверяют полноту экстракции по величине
pH водного слоя (значение pH должно соответствовать ин-
тервалу 8,5—11,3).
Хлороформенный слой промывают двумя порциями (по
5 мл каждая) 1 н. раствора едкого натра. Содержимое во-
ронки встряхивают в течение 2 мин после добавления каж-
дой порции. Хлороформенную фазу переносят в мерный ци-
линдр и доводят хлороформом объем раствора до 5 мл. По-
лученный раствор хлороформа переводят в кювету спектро-
фотометра и измеряют его светопоглощение при % 275 ммк.
В качестве «нулевого» используют раствор хлороформа, ко-
торым обработан раствор, содержащий все реагенты, кроме
соли никеля, и прошедший все стадии, описанные выше. В за-
висимости от содержания никеля готовят эталонные растворы
(стр. 164).
Определение никеля в контрольном об-
разце индия. 1г металлического индия помещают в квар-
цевую или тефлоновую чашку и растворяют в 10 мл концен-
трированной азотной кислоты, упаривают почти досуха на
песчаной бане или электрической плитке. Полученную каше-
образную массу растворяют в 20 мл лимонной кислоты, до-
бавляют 4 н. раствор гидроокиси натрия до начала образова-
ния осадка, вводят сначала 10 мл, а затем еще 5 мл раство-
ра сегнетовой соли и доводят pH раствора до значения
9—10 раствором щелочи (по универсальной индикаторной
бумаге). Полученный раствор упаривают до объема 20 мл,
переносят в делительную воронку с помощью 5 мл раствора
сегнетовой соли и далее определяют никель а-бензилдиокси-
мом в условиях, описанных при приготовлении эталонных
растворов и построении калибровочного графика.
Определение никеля гептоксимом в металлическом индии
Кроме указанных на стр. 163 необходимы реактивы:
1. Азотная кислота х. ч., концентрированная, перегнанная
в кварцевом приборе.
2. Винная кислота, 20%-ный водный раствор.
3. Гептоксим, 0,2%-ный водный раствор.
4. Едкий натр, 6 н. раствор.
Приготовление эталонных растворов. В де-
лительные конические воронки емкостью 50 мл помещают
раствор никеля с различным его содержанием, добавляют
3 мл раствора гептоксима и доводят pH раствора до значе-
ния 8,2—8,5 раствором щелочи (6 н.) и оставляют стоять
б течение часа. После этого производят экстракцию тремя
порциями (по 2,5 мл каждая) хлороформа. Содержимое во-
167
ронки встряхивают в течение 15 мин на механическом виб-
раторе после прибавления каждой порции хлороформа. Пос-
ле отделения соединяют хлороформенные экстракты и про-
мывают раствор хлороформа 10 мл раствора щелочи (1 н.)
в течение 15 мин и водой (3—Ь мин), используя механический
вибратор. Сливают слой хлороформа в сухую пробирку на
10 мл и измеряют поглощение органического экстракта при
% 263 ммк в тефлоновых кюветах с I = 10 см. В качестве «ну-
левого» используют раствор хлороформа, которым обрабаты-
вают раствор, содержащий гептоксим и все реагенты, кроме
соли никеля, и прошедший все стадии, описанные выше.
Определение никеля в контрольном образ-
це индия. 0,5 г металлического индия помещают в тефло-
новую или кварцевую чашку емкостью 70—100 мл и раство-
ряют в 5 мл азотной кислоты. Раствор выпаривают почти до-
суха, добавляют к остатку 10 мл раствора винной кислоты
при нагревании. После охлаждения добавляют 3 мл раствора
гептоксима и доводят pH раствора до значения 8,2—8,5 рас-
твором щелочи (6 н.). Полученный раствор переносят в кони-
ческую делительную воронку емкостью 50 мл и оставляют
стоять в течение часа. После этого производят экстракцию
тремя порциями (по 2,5 мл каждая) хлороформа. Содержимое
воронки встряхивают на механическом вибраторе в течение
15 мин после прибавления каждой порции хлороформа. Пос-
ле отделения соединяют хлороформенные экстракты и про-
мывают раствор хлороформа 10 мл раствора щелочи (1 н.)
в течение 15 мин и водой (3—5 мин), используя механиче-
ский вибратор. Сливают слой хлороформа в сухую пробирку
на 10 мл и измеряют поглощение органического экстракта
при % 263 ммк в тефлоновых кюветах с I — 10 см. Раствор
сравнения должен быть приготовлен в условиях, указанных
в прописи для приготовления эталонных растворов.
ОПРЕДЕЛЕНИЕ КОБАЛЬТА
Для определения кобальта применяется ряд колориметри-
ческих методов. Наибольшее распространение получили ме-
тоды определения кобальта в виде роданида 1 и группа мето-
дов, в которых используются органические реагенты — произ-
водные нитрозо-нафтолов.
Определение кобальта в виде роданида
Кобальт с роданид-ионом в нейтральном или слабокислом
водном растворе в зависимости от условий образует ряд ком-
плексных соединений следующего состава: [Co(SCN)]+, %Макс
1 А. К. Бабко, О. Ф. Драко. ЖОХ. 19, 1809, 1949; «Зав. лаб.», 16,
1162, 1950.
168
530 ммк; Со (SCN)2, ^макс 570 ммк; [Со (5СМ)з]1_ и
[Co(SCN)4]1 2-, ^макс 620 ммк. Предполагают, что при большом
избытке роданид-иона и в присутствии спирта, или лучше
ацетона (50—60%-ного раствора), кобальт находится глав-
ным образом в виде комплексного аниона [Co(SCN)4]2-; рас-
творы соединения окрашены в синий цвет. Ввиду малой устой-
чивости рассматриваемого комплексного соединения следует
строго придерживаться идентичных условий в приготовлении
эталонных и испытуемого растворов. Определению мешают
железо (III), медь, висмут, а также элементы, дающие труд-
но растворимые роданиды *. Никель мешает зеленой окрас-
кой, если он значительно превышает содержание кобальта.
В этом случае рекомендуется отделить комплексное соедине-
ние кобальта, экстрагируя его изоамиловым спиртом; окраски
раствора изоамилового спирта сравнивают с таковой эталон-
ных растворов. Влияние меди (II) и железа (III) устраняют
добавлением раствора хлорида олова (II); железо связывают
также в комплексные соединения с фосфат- и пирофосфат-
ионами.
Необходимые реактивы:
1. Стандартный раствор соли кобальта, содержащий
0,1 мг Со в 1 мл. Навеску сернокислого кобальта без никеля,
квалификации х. ч. или ч. д. а. с известным содержанием ко-
бальта, соответствующую 0,1 г Со, помещают в мерную кол-
бу емкостью 1 л, растворяют в воде, добавляют 4 мл кон-
центрированной серной кислоты, доводят водой объем рас-
твора до 1 л и тщательно перемешивают. Более разбавлен-
ные растворы с содержанием 0,01 и 0,001 мг/мл готовят со-
ответствующим разбавлением; эти растворы пригодны только
в день приготовления.
2. Роданид аммония (или калия), сухой препарат.
3. Ацетон, перегнанный, т. кип. 56°.
4. Пирофосфат натрия, 10%-ный раствор.
5. Фенолфталеин, 0,1%-ный спиртовый раствор.
6. Соляная кислота х. ч. или ч. д. а., концентрированная.
7. Соляная кислота 1 :4.
8. Азотная кислота х. ч. или ч. д. а., концентрированная.
9. Аммиак 1 : 1.
10. Хлористый аммоний, сухой препарат.
11. Сульфит натрия, 20%-ный раствор.
а. Определение кобальта в растворе чистой соли
К Ю—12 мл испытуемого раствора, помещенного в мер-
ную колбу на 25 мл, добавляют 1 г сухого препарата рода-
нида аммония (или калия) и прибавляют ацетон до объема
1 Е. Б. Сендел. Колориметрические методы определения следов
металлов. М., «Мир», 1964, стр. 368.
169
25 мл. После перемешивания измеряют интенсивность окрас-
ки, используя фотоколориметры, фотометр и микроколори-
метр. Эталонные растворы готовят в аналогичных условиях,
обязательно соблюдая объем, в котором был взят испытуе-
мый раствор. Содержание кобальта в эталонных растворах
может быть от 0,005 до 0,1 мг кобальта.
б. Определение кобальта методом колориметрического
титрования в водном растворе его соли и в присутствии
железа1
К Ю мл испытуемого раствора, помещенному в градуи-
рованный колориметрический цилиндр с притертой пробкой,
прибавляют 1 г сухого препарата роданида аммония (или ка-
лия), 10 мл ацетона, раствор перемешивают и цилиндр за-
крывают пробкой. В другой цилиндр вводят 10 мл воды, 1 г
роданида, 10 мл ацетона и из бюретки постепенно при пере-
мешивании добавляют стандартный раствор соли кобальта.
При окончательном сравнении интенсивности окраски рас-
творов в обоих цилиндрах объем раствора в первом цилиндре
(добавлением воды) делают равным объему второго ци-
линдра. Содержание кобальта в исследуемом растворе равно
содержанию кобальта во введенном стандартном растворе.
Для определения кобальта указанным методом в присут-
ствии железа (в железо-никелевых рудах) поступают сле-
дующим образом. Навеску руды 0,1 г помещают в стакан из
жаростойкого стекла емкостью 50—100 мл, добавляют 5 мл
соляной кислоты (концентрированной) и 2 мл азотной кис-
лоты, накрывают стакан часовым стеклом и нагревают рас-
твор на песчаной бане до появления кристаллов солей. За-
тем сухой остаток дважды обрабатывают раствором по
2—3 мл соляной кислоты (концентрированной), выпаривая
каждый раз раствор досуха. Сухой остаток смачивают 0,5 мл
(около 10 капель) соляной кислоты (концентрированной), до-
бавляют 3—4 мл воды, накрывают часовым стеклом и выпа-
ривают до 1—2 мл. После охлаждения, не отфильтровывая
нерастворимый остаток, добавляют 1 г хлорида аммония и
нейтрализуют аммиаком (осторожно) до выпадения осадка,
который растворяют в 3—4 каплях соляной кислоты (кон-
центрированной). К полученному раствору добавляют 0,1 г
роданида аммония (или калия) и наблюдают образование
красного раствора роданида железа. Из бюретки при непре-
рывном помешивании добавляют раствор пирофосфата нат-
рия до исчезновения красного цвета. По каплям добавляют
аммиак в присутствии фенолфталеина до появления устой-
чивой розовой окраски, затем раствор нейтрализуют соляной
1 В. М. Звенигородская. «Зав. лаб.», 11, 1022, 1945.
170
кислотой (1:4) до исчезновения розовой окраски, переводят
в мерную колбу на 25 мл и разбавляют водой до метки. От-
бирают 10 мл раствора в колориметрический цилиндр с при-
тертой пробкой, добавляют 1 г соли роданида, 10 мл ацетона,
тщательно перемешивают и закрывают пробкой. В другой
цилиндр вводят все те же реактивы (за исключением пиро-
фосфата) и готовят раствор сравнения. Определение в даль-
нейшем производят в условиях, указанных для определения
кобальта в растворе чистой соли.
Окончание определения может быть проведено также
с использованием приборов — фотоколориметров. В этом
случае эталонные растворы готовят так же, как и при опре-
делении кобальта в растворе его чистой соли, но при изме-
рении интенсивности окраски испытуемого раствора в каче-
стве «нулевого» используют раствор анализируемого образ-
ца, предварительно обработанный как испытуемый раствор,
но без добавления в него (вторичного) 1 г соли роданида.
Содержание кобальта (дга) в эталонных растворах зави-
сит от процентного содержания кобальта в испытуемом об-
разце:
1-й ряд
0,005
0,01
0,03
0,05
0,07
2-й ряд
0,06
0,08
0,1
0,12
Примечания. 1. Если образец руды содержит медь, то после свя-
зывания железа в комплексное соединение добавляют к раствору 0,1 —
0,2 мл сульфита натрия и переводят его в мерную колбу на 25 мл.
2. Если раствор в колбе не будет прозрачным, его перед фотометриро-
ванием фильтруют через сухой фильтр.
Определение кобальта 1- и 2-нитрозонафтолами
N=O NOH
I I!
Z\/4_он
1 -нитрозо-2-нафтол;
OH
—N=O
II
/\/\=NOH
2-нитрозо-1 -нафтол;
171
N=0
NOH
NaO3S—
—SO3Na NaO3S—I
—SO3Na
нитрозо-И-соль
M. А. Ильинский 1 впервые наблюдал образование окра-
шенных соединений кобальта с 1,2- и 2,1-изомерами нитрозо-
нафтолов. При образовании комплексных соединений с ме-
таллами нитрозонафтолы находятся в форме монооксима.
Кобальт с указанными реа-
Рис. 59. Спектры поглощения: I — сое-
динения кобальта с 1-нитрозо-2-наф-
толом; II— соединения кобальта с 2-
нитрозо-1-нафтолом; I' — 1-нитрозо-
2-нафтол; II' — 2-нитрозо-1-нафтол
Н-57 с 2,1-изомером можно
трации кобальта.
Кроме 1,2- и 2,1-изомеров нитрозонафтолов для опреде-
ления кобальта широко используется сульфопроизводное
1-нитрозо-2-нафтола, получившее название нитрозо-К-соли.
Соединения кобальта с 1,2- и 2,1-изомерами, получаемые в
кислой среде, обладают преимуществом перед соединениями,
тентами в зависимости от
условий проведения реакции
дает 2 типа соединений. В
щелочной среде образуется
соединение, легко раствори-
мое в воде; отношение ко-
бальта к реагенту в нем
равно 1:2 (CoR2). В кис-
лой среде образуется соеди-
нение состава C0R3; соедине-
ние мало растворимо в воде
и хорошо растворимо в не-
водных растворителях, не
смешивающихся с водой. Из
1,2- и 2,1-изомеров нитрозо-
нафтолов предпочитают 2,1-
изомер. Преимущество это-
го реагента видно из срав-
нения спектров поглощения
этих реагентов (рис. 59).
2,1-изомер имеет елзо7 ммк ='
=' 53 000, 1,2-изомер —
е мбоклк = 30 000. При на-
личии приборов СФ-4,
ФЭК-56, ФЭК-Н-54, ФЭК-
определять меньшие концен-
выделяемыми в щелочной среде, так как первые экстрагиру-
ются неводными растворителями. Сам реагент также экстра-
1 М. А. Ильинский. «Вег.», 117, 2592, 1884.
172
гируется, но может быть легко переведен в водную фазу об-
работкой раствором 2 н. щелочи. Таким образом, при изме-
рении интенсивности окраски полученных растворов избав-
ляются от влияния реагента.
Присутствие меди, железа и никеля в значительных коли-
чествах не мешает определению кобальта ’, так как указан-
ные элементы дают менее устойчивые комплексные соедине-
ния с 1,2- и 2,1-изомерами нитрозонафтолов, легко разру-
шаемые раствором 2 н. соляной кислоты.
Кроме указанных на стр. 169 необходимы реактивы:
1. Нитрозонафтолы (1,2- и 2,1-изомеры), 0,5%-ный рас-
твор в ледяной уксусной кислоте.
2. Едкий натр, 2 н. раствор.
3. Соляная кислота 1 :4.
4. Бензол или хлороформ.
5. Перекись водорода, 30%-ный раствор.
6. Ацетатный буферный раствор, pH 5,2.
7. Смесь концентрированных соляной и азотной кислот,
3:1.
8. Уксусная кислота, концентрированная.
а. Определение кобальта в растворе чистой соли
в присутствии солей никеля и железа 2-нитрозо-1 -нафтолом
К помещенному в делительную воронку испытуемому рас-
твору (3—5 мл), содержащему кобальт, прибавляют 5 мл
ацетатного буфера и 5 мл раствора реактива; в течение 5 мин
экстрагируют двумя порциями бензола по 5 мл каждая. Слой
бензола промывают раствором щелочи двумя порциями по
5 мл каждая. Полученный раствор бензола можно фотомет-
рировать.
Эталонные растворы готовят в указанных условиях и вво-
дят стандартный раствор соли кобальта в таких количествах,
чтобы содержание кобальта в эталонных растворах соответ-
ствовало (мг):
1-й ряд 2-й ряд
0,001 0,01
0,003 0,03
0,005 0,05
0,007 0,07
0,01 0,1
По приготовленным эталонным растворам строят калибро-
вочный график. Если испытуемый раствор кроме кобальта
содержит никель и железо, то после экстракции испытуемого
раствора бензолом в указанных выше условиях бензольный
1 В. М. Пешкова, В. М. Бочкова. «Тр. комиссии по аналитиче-
ской химии». М„ Изд-во АН СССР, VIII(XI), 125, 1958.
7 Зак. 686 173
слой промывают раствором соляной кислоты двумя порция-
ми по 10 мл каждая, один раз водой и затем двумя порциями
щелочи по 5 мл каждая. Полученный слой бензольной вы-
тяжки фотометрируют. Определение концентрации кобальта
производят, используя калибровочный график.
б. Определение кобальта в стали 2-нитрозо-1-нафтолом
Навеску стали 0,1 г переносят в стакан, добавляют 10 мл
раствора смеси соляной и азотной кислот и нагревают на
песчаной бане. Раствор упаривают почти досуха; остаток
растворяют в воде и переносят раствор в мерную колбу на
250 мл. Для определения берут 2—5 мл раствора в зависи-
мости от содержания кобальта в стали. В дальнейшем опре-
деление производят в условиях, указанных для определения
кобальта в присутствии никеля и железа.
в. Определение кобальта в азотной кислоте
2-нитрозо-1 -нафтолом
Кроме указанных на стр. 169 и 173 необходимы реактивы:
1. Соляная кислота, 2 н. раствор.
2. 2-Нитрозо-1-нафтол, 0,02%-ный водный раствор.
3. Фосфатный буферный раствор, pH 6,1.
Приготовление эталонных растворов. В ряд
стеклянных стаканчиков емкостью 20 мл помещают раствор
кобальта с различным его содержанием, 10 мл буферного
раствора и 2 мл раствора 2-нитрозо-1-нафтола. Растворы на-
гревают почти до кипения, охлаждают и переносят в кони-
ческую делительную воронку на 50 мл. К полученному рас-
твору приливают 5 мл хлороформа и экстрагируют соедине-
ние кобальта в течение 10 мин, используя механический виб-
ратор. Водный слой отбирают пипеткой, используя для втя-
гивания раствора резиновую грушу. Для удаления избытка
реактива хлороформенный слой обрабатывают раствором
щелочи в течение 20 мин, используя механический вибратор
и повторяя эту операцию дважды. В качестве «нулевого» ис-
пользуют раствор хлороформа, которым обработан раствор,
содержащий все реагенты, кроме соли кобальта, и прошед-
ший все стадии, описанные выше. В зависимости от содер-
жания кобальта (мкг) готовят 2 ряда эталонных растворов:
1-й ряд 2-й ряд
0,01 0,05
0,02 0,075
0,03 0,1
0,05 0,2
0,5
174
Определение кобальта в контрольном об-
разце азотной кислоты (особой чистоты). 10 г
испытуемого раствора азотной кислоты упаривают в кварце-
вой или тефлоновой чашке на водяной бане почти досуха.
К остатку добавляют 10 мл буферного раствора и 2 мл рас-
твора 2-йитрозо-1-нафтола. Раствор нагревают почти до ки-
пения, охлаждают и переносят в делительную воронку на
50 мл. К полученному раствору приливают 5 мл хлорофор-
ма и экстрагируют соединение кобальта в течение 10 мин,
используя механический вибратор. Избыток реактива (2-нит-
розо-1-нафтола) удаляют из слоя хлороформа в условиях,,
описанных при приготовлении эталонных растворов.
Железо также образует комплексное соединение с 2-нит-
розо-1-нафтолом, но это соединение разрушается при удалении
избытка реактива из слоя хлороформа раствором щелочи.
Затем для разрушения комплексных соединений меди и ни-
келя (которые могут сопутствовать кобальту) раствор хло-
роформа промывают 5 мл соляной кислоты в течение 10 мин,
используя механический вибратор. Отбирают пипеткой вод-
ный слой, промывают его водой и, так как при этой операции
освобождается некоторое количество реактива, которое вхо-
дило в комплексные соединения меди и никеля, еще раз рас-
твор хлороформа промывают последовательно щелочью и во-
дой. Раствор хлороформа переводят в мерный цилиндр или
градуированную пробирку, добавляют хлороформ до объема
5 мл и измеряют светопоглощение раствора. Раствор сравне-
ния («нулевой» раствор) готовят в условиях, указанных при
приготовлении эталонных растворов. В зависимое™ от содер-
жания кобальта используют эталонные растворы 1-го или
2-го ряда (стр. 174).
г. Определение кобальта в щелочи (особой чистоты)
2-нитрозо-1 -нафтолом
Эталонные растворы готовят в условиях, указанных при
определении кобальта в азотной кислоте (стр. 174). Навеску
щелочи 10 г растворяют в 25 мл воды и раствор нейтрали-
зуют рассчитанным количеством азотной кислоты, беря не-
большой избыток ее (2—3 мл). Сухой остаток растворяют в
20 мл воды, добавляют 10 мл буферного раствора и далее
поступают, как описано при анализе азотной кислоты.
Примечание. Содержание кобальта в азотной кислоте, используе-
мой для нейтрализации щелочи, должно быть известно и сделана соответ-
ствующая поправка на его содержание.
д. Определение кобальта 2-нитрозо-1 -нафтолом
в чистом алюминии
Кроме указанных на стр. 174 необходимы реактивы-
1. Едкий натр, 4 н. раствор.
7*
175
2. Лимонная кислота, 25%-ный водный раствор.
Приготовление эталонных растворов. Эта-
лонные растворы готовят точно в условиях, указанных при
определении кобальта в азотной кислоте (стр. 174). Так как
в алюминии (чистом) предусматривается незначительное ко-
личество никеля, то готовят один эталонный ряд с содержа-
нием кобальта (мкг):
0,01
0,02
0,03
0,05
0,075
Определение кобальта в контрольном об-
разце алюминия. 1 г металлического алюминия раство-
ряют в 20 мл едкого натра (4 н. раствор), прибавляют по-
степенно раствор лимонной кислоты до pH 8. Раствор пере-
носят в мерную колбу емкостью 50 мл и доводят водой до
метки. В стаканчик на 50 мл переносят 10 мл приготовленно-
го раствора, добавляют 2 мл раствора 2-нитрозо-1-нафтола
и нагревают почти до кипения, охлаждают и переносят в ко-
ническую делительную воронку на 50 мл. К этому раствору
приливают 5 мл хлороформа и экстрагируют соединение ко-
бальта в течение 20 мин на механическом вибраторе. Водный
слой отбирают пипеткой (используя для втягивания раствора
резиновую грушу). Для удаления избытка реактива хлоро-
форменный слой обрабатывают раствором щелочи в течение
20 мин, используя механический вибратор. Затем промы-
вают водой. Комплексное соединение железа, если железо
присутствует, разрушается при удалении избытка реактива
из слоя хлороформа раствором щелочи. Для разрушения ком-
плексных соединений никеля и меди (которые могут также
находиться в качестве примесей) раствор хлороформа про-
мывают 5 мл соляной кислоты в течение 5 мин и снова во-
дой, используя механический вибратор. Так как при этой опе-
рации освобождается некоторое количество реактива, кото-
рое входило в комплексные соединения меди и никеля, то
еще раз раствор хлороформа промывают последовательно
щелочью и водой. Раствор хлороформа переводят в мерный
цилиндр или градуированную пробирку, добавляют хлоро-
форм до объема 5 мл и измеряют светопоглощение раствора.
Раствор сравнения («нулевой» раствор) готовят в условиях,
указанных для приготовления эталонных растворов при оп-
ределении кобальта в азотной кислоте.
е. Определение кобальта в растворе чистой соли
1 -нитрозо-2-нафтолом
5 мл испытуемого раствора соли кобальта помещают в
стаканчик на 30—40 мл, добавляют 0,2 мл раствора перекиси
176
водорода и раствора щелочи до полноты образования осадка
(~ 1 мл). Выпавший осадок гидроокисей растворяют при
слабом подогревании в 2 мл уксусной кислоты. К горячему
раствору добавляют 1 мл раствора 1-нитрозо-2-нафтола, пе-
реносят содержимое стаканчика в делительную воронку, раз-
бавляют водой до 20 мл и экстрагируют соединение кобальта
двумя порциями бензола по 5 мл каждая. Слой бензола про-
мывают раствором щелочи двумя порциями по 5 мл каждая
и фотометр ируют.
Эталонные растворы готовят в условиях, указанных при
использовании реагента 2-нитрозо-1-нафтола, но в качестве
реагента берут 1-нитрозо-2-нафтол. По приготовленным эта-
лонным растворам строят калибровочный график.
ж. Определение кобальта в стали 1 -нитрозо-2-нафтолом
Для определения кобальта в стали используют методику
растворения образца, указанную выше при использовании
2-нитрозю-1-нафтола. Приготовленный раствор переносят в
колбу на 250 мл и разбавляют водой до метки. Испытуемый
раствор в объеме 2, 5, 10 мл (в зависимости от содержания
кобальта в образце) подвергают всем операциям, указанным
для определения кобальта 1-нитрозо-2-нафтолом в растворе
чистой соли. Если для анализа было взято 10 мл испытуемого
раствора, то объем реактива увеличивают до 2 мл. Приго-
товление эталонных растворов производят в условиях, ука-
занных для определения кобальта в растворе чистой соли.
Определение кобальта нитрозо-И-солью в растворе
чистой соли 1
Кроме указанных на стр. 169 необходимы реактивы:
1. Нитрозо-К-соль, 0,1%-ный водный раствор.
2. Соляная кислота 1 : 1.
3. Азотная кислота 1:1.
4. Ацетат натрия, 40%-ный раствор.
Испытуемый раствор соли кобальта в объеме 10—15 мл
помещают в стакан емкостью 40—50 мл, добавляют 0,5 мл
соляной кислоты и несколько капель (3—5) азотной кислоты,
прибавляют 3 мл раствора нитрозо-К-соли и 5 мл раствора
ацетата натрия. Раствор нагревают до кипения, прибавляют
2 мл азотной кислоты и осторожно кипятят в течение 1 —
2 мин. После этого раствор охлаждают, переводят в мерную
колбу на 50 мл, доводят водой до метки, перемешивают и
фотометрируют. Эталонные растворы готовят в указанных
условиях и вводят стандартный раствор соли кобальта в та-
1 Д. Н. М а л ю г а. ЖАХ, 1, 176, 1946.
177
ких количествах, чтобы содержание кобальта в эталонных
растворах соответствовало (лг):
0,005
0,0075
0,01
0,0125
0,015
ОПРЕДЕЛЕНИЕ МОЛИБДЕНА
Молибден дает реакции окрашивания с рядом реактивов:
роданидом, фенилгидразином, этилксантогенатом, таннином,
перекисью водорода, сероводородом, тиосульфатом натрия.
Но в практике наибольшее распространение получил рода-
нидный метод в силу его большой чувствительности и воз-
можности в большинстве случаев проводить определение без
предварительного отделения молибдена, особенно если в про-
цессе определения проводится экстракция роданидного ком-
плексного соединения молибдена, а также пероксидный ме-
тод.
Определение молибдена в виде роданида
Соли молибдена с роданидом аммония или калия в зави-
симости от концентрации последнего в присутствии восста-
новителя образуют несколько роданидных комплексных со-
единений *. Наиболее интенсивно окрашенным из них являет-
ся роданид 5-валентного молибдена Mo (SCN)s. Поэтому при
колориметрическом определении молибдена нужно обеспе-
чить достаточную концентрацию роданида. С другой стороны,
очень большая концентрация роданида снижает чувствитель-
ность реакции, так как может образоваться менее интенсивно
окрашенный комплексный ион [Mo (SCN)e]1-.
В качестве восстановителя в большинстве случаев приме-
няется хлорид олова (II); большой избыток последнего вре-
ден, так как может произойти восстановление молибдена до
низших степеней валентности с образованием слабо окрашен-
ных роданидных комплексов. Значительную роль играет так-
же кислотность раствора. Оптимальной считается кислот-
ность в 5% по соляной кислоте; как при более низкой, так
и при более высокой кислотности интенсивность окраски па-
дает. При низкой кислотности также замедляется восстанов-
ление железа, что имеет существенное значение, если опреде-
ление ведется в его присутствии. С другой стороны, присут-
ствие железа в небольших количествах (примерно равных
определяемым количествам молибдена) способствует образо-
1 А. К. Б а б к о. ЖОХ, 17, 642, 1947.
178
ванию более устойчивой окраски *. В качестве восстановителя
можно также использовать тиомочевину1 2.
При экстрагировании органическими растворителями ро-
данид молибдена становится более устойчивым, чем в вод-
ных растворах. В качестве органических растворителей могут
быть использованы бутиловый и амиловый спирты, этиловый
эфир, этилацетат, амилацетат и др.
Определению молибдена в виде роданида не мешают алю-
миний, кобальт, уран, тантал. Мешающее влияние вольфра-
ма можно устранить, связывая вольфрам в виннокислый ком-
плекс, который препятствует реакции вольфрама с родани-
дами. Основными мешающими элементами являются хром и
ванадий, хотя эти помехи мало сказываются, если приме-
няется метод экстрагирования соединения роданида молиб-
дена.
Необходимые реактивы:
1. Стандартный раствор молибдена, содержащий в 1 мл
0,1 мг Мо, готовится следующим образом: 1,85 г молибдата
аммония квалификации х. ч. растворяют в дистиллированной
воде, переносят раствор в мерную колбу емкостью 1 л и до-
ливают водой до метки колбы. 100 мл этого раствора перед
использованием разбавляют водой до 1 л.
2. Роданид калия, 5%-ный раствор.
3. Смесь серной и азотной кислот: 350 мл азотной кисло-
ты (уд. в. 1,4) смешивают с 750 мл воды, после чего осто-
рожно приливают 225 мл серной кислоты (уд. в. 1,84).
4. Смесь серной и соляной кислот: 450 мл серной кислоты
(уд. в. 1,84) осторожно приливают при помешивании к
1450 мл воды. Смесь охлаждают и приливают к ней частями
100 мл соляной кислоты (уд. в. 1,19).
5. Раствор 2-валентного олова: 250 г соли SnCl2-2H2O рас-
творяют при нагревании в 200 мл соляной кислоты (уд. в. 1,10)
до полного посветления раствора, разбавляют его 800 мл во-
ды и прибавляют несколько кусочков металлического олова.
6. Этиловый эфир.
7. Соляная кислота, концентрированная.
8. Железо-аммонийные квасцы, 1%-ный раствор. 1 г же-
лезо-аммонийных квасцов растворяют в воде, добавляют 1 мл
концентрированной H21SO4 и доводят объем раствора до
100 мл водой.
9. Фосфорная кислота, концентрированная.
1 Анализ минерального сырья. Сб. под ред. Ю. Н. Книпович и
Ю. В. Морачевского. Л., Госнаучтехиздат, 1956, стр. 805.
2 W. R. S с h о е 11 е г, A. R. Powell, С. Jahn. «Analyst», 60, 506,
1935; 52, 504, 1927.
179
а. Определение молибдена в растворе чистой соли,
не содержащей посторонних ионов
К анализируемому раствору объемом 25 мл добавляют
4 мл концентрированной соляной кислоты, 1 мл железо-аммо-
нийных квасцов и разбавляют в делительной воронке до
45—50 мл. Раствор охлаждают до 15°, добавляют 6—7 мл
роданида калия, перемешивают и добавляют 2 мл (или, если
требуется, больше) раствора хлорида олова. Раствор встря-
хивают и через 1 мин добавляют 15 мл эфира. Встряхивают
с обычными предосторожностями 1—2 мин и дают жидко-
стям разделиться. Выпускают водный слой в стакан, обсуши-
вают внутри трубку воронки свернутой полоской фильтро-
вальной бумаги и сливают эфир в сухую мерную колбу на
25 мл. Водный раствор переводят обратно в делительную во-
ронку и экстрагируют еще 10 мл эфира. Вторую эфирную
вытяжку соединяют с первой и разбавляют эфиром до 25 мл.
После перемешивания измеряют оптическую плотность в ин-
тервале длин волн 460—480 ммк. Предварительно должен
быть снят спектр поглощения одного из эталонных растворов.
Эталонные растворы, содержащие от 0,01 до 0,05 мг Мо, го-
товят так же, как анализируемый.
б. Определение молибдена в стали
Приготовление эталонных растворов и по-
строение калибровочной кривой. 0,4 г стали, не
содержащей Мо, помещают в стакан или коническую колбу
емкостью 100 мл и растворяют навеску в 20 мл серно-азотной
смеси при нагревании на песчаной бане. Раствор выпаривают
до появления паров SO3. Охладив стакан, приливают к остат-
ку 60 мл серно-соляной смеси, нагревают до кипения для рас-
творения образовавшихся солей и, если необходимо, от-
фильтровывают раствор от осадка кремнекислоты и графита.
Раствор переводят в мерную колбу на 200 мл и объем дово-
дят водой до метки колбы — раствор А. Отбирают пипеткой
по 25 мл раствора А, переносят в делительную воронку, вво- .
дят стандартный раствор молибдена, содержащий в 1 мл
0,1 мг, с таким расчетом, чтобы общее содержание его (мг)
в каждом из эталонных растворов соответствовало:
0,1
0,2
0,3
0,4
0,5
Примечание. Если отсутствует образец стали, не содержащей Мо,
при приготовлении эталонных растворов учитывают содержание Мо во
взягом образце.
180
К полученному раствору приливают 4 мл раствора рода-
нида калия и 4 мл двухлористого олова, хорошо перемеши-
вают раствор, приливают к нему 20 мл эфира и плотно за-
крывают воронку: содержимое воронки энергично перемеши-
вают в течение 30 сек. и дают отстояться до образования
двух слоев. Нижний кислотный слой осторожно сливают в
чистый стакан, обсушивают внутри трубку воронки сверну-
той полоской фильтровальной бумаги и сливают эфирный
слой в сухую мерную колбу на 50 мл. Водный раствор пере-
водят обратно в делительную воронку и производят повтор-
ное экстрагирование 20 мл эфира. После разделения слоев
вторая вытяжка присоединяется к первой. Если при вторич-
ном экстрагировании эфирный слой заметно окрашен, то еще
раз производят экстрагирование 10 мл эфира. После экстра-
гирования доводят объем раствора эфиром до метки колбы
и в течение 15 мин производят измерение оптической плот-
ности. По полученным данным строят калибровочную кри-
вую.
Примечания. 1. При определении молибдена в вольфрамовых ста-
лях растворение пробы следует производить в присутствии 2—3 мл фос-
форной кислоты.
2. Выпаривание раствора до образования паров серной кислоты необ-
ходимо как для разрушения карбидов, оставшихся неразложившимися, так
и для удаления азотной кислоты, которая впоследствии может препятство-
вать восстановлению молибдена.
Определение молибдена в контрольном об-
разце стали. 0,2 г стали с неизвестным содержанием мо-
либдена растворяют в 10 мл серно-азотной смеси при нагре-
вании на песчаной бане. По окончании растворения выпари-
вают раствор до появления паров серной кислоты (см. приме-
чания 1 и 2).
Охладив стакан, к остатку приливают 30 мл серно-соля-
ной смеси. Полученный раствор переводят в мерную колбу
на 100 мл и доводят до метки колбы водой. 25 мл этого рас-
твора пипеткой переносят в делительную воронку и произво-
дят определение в условиях, описанных выше для построения
калибровочной кривой. Содержание молибдена в образце
стали находят по калибровочному графику, построенному по
эталонным растворам, приготовленным на образце стали, не
содержащей молибдена.
Определение молибдена перекисью водорода 1
Молибден в кислой среде при действии перекиси водо-
рода образует соединение, растворы которого окрашены в
желтый цвет; область максимального поглощения лежит при
1 G. Т е 1 е р, D. Boltz. «Anal. Chem.», 22, 1030, 1950.
181
К 330 ммк. Метод может быть использован для определения
молибдена в присутствии ванадия и титана, которые также
дают соединения с перекисью водорода, но максимумы погло-
щения этих соединений значительно сдвинуты в видимую
область L
а. Определение молибдена в растворе его чистой соли
Необходимые реактивы:
1. Стандартный раствор, содержащий 0,1 мг Мо в 1 мл
(стр. 179).
2. Смесь кислот (хлорной и фосфорной), 1:1.
3. Перекись водорода, 3%-ный раствор.
25 мл испытуемого раствора, содержащего молибден,
вносят в мерную колбу на 50 мл, добавляют 10 мл смеси кис-
лот и 1 мл раствора Н2О2, доводят водой до метки. Интенсив-
ность окраски раствора измеряют, используя спектрофото-
метр СФ-4, при длине волны 330 ммк. Если для опреде-
ления оптической плотности используются фотоколориметры
ФЭК-56, ФЭК-Н-54, ФЭК-Н-57, то работают со светофильт-
ром ХЭф 360 ммк. При приготовлении эталонных растворов
стандартный раствор соли молибдена вводят в 20 мл воды
и в остальном соблюдают условия, указанные для приготов-
ления испытуемого раствора. В эталонные растворы вводят
молибден в количествах (мг):
1,0
2,0
3,0
4,0
5,0
Определение молибдена по поглощению молибдата
в ультрафиолетовой области спектра 1 2
В растворах щелочных молибдатов в области спектра, до-
ступной при работе на спектрофотометрах СФ-2, СФ-5 (400—
1 100 ммк), не обнаруживается каких-либо максимумов по-
глощения. Однако поглощение этих растворов сильно возра-
стает в ультрафиолетовой области, что может быть использо-
вано для количественного определения содержания молиб-
дена.
Необходимые реактивы:
1. Молибдат аммония, содержащий 0,1 мг Мо в 1 мл рас-
твора (стр. 179).
2. Едкий натр, 20%-ный раствор.
1 A. W е i s s 1 е г. «Ind. Eng. Chem., Anal. Ed.», 17, 695, 1945.
2 К. Б. Я ци мирский, И. И. Алексеева. «Зав. лаб.», 24, 1428,
1958.
182
В мерные колбы на 100 мл вносят раствор молибдата ам-
мония с таким расчетом, чтобы содержание молибдена в эта-
лонных растворах было равно (мг):
0,06
0,10
0,14
0,18
0,22
В каждую колбу добавляют по 4 мл раствора NaOH и до-
водят водой до метки колбы.
Оптическую плотность приготовленных растворов измеряют
на спектрофотометре СФ-4 при X 230 ммк. По полученным
данным строят калибровочную кривую. В контрольный рас-
твор молибдата добавляют такое же количество щелочи и из-
меряют оптическую плотность в тех же условиях, что и для
эталонных растворов.
ОПРЕДЕЛЕНИЕ АЛЮМИНИЯ
Для определения алюминия используют ряд методов. Наи-
более распространенными являются методы определения али-
зариновым красным, алюминоном и 8-оксихинолином. Окрас-
ка реагента и соединения зависит в большой мере от pH рас-
твора, что осложняет использование указанных реагентов.
Поэтому очень важно при определении алюминия и других
металлов контролировать pH раствора. Рассмотрим метод
определения алюминия 8-оксихинолином.
Определение алюминия 8-оксихинолином
|N |N /<осн = 1 - 10-i°
НО н+ 0~
(кислая среда)
(щелочная среда)
8-Оксихинолин является амфотерным соединением и в за-
висимости от условий реагирует как слабая кислота или как
слабое основание1. Реагируя как основание, 8-окоихинолин
дает продукты присоединения, например, с гетерополикисло-
тами фосфора, кремния, германия.
Оксихинолин проявляет свойства слабой кислоты, вступая
в реакцию с ионами металлов. В результате реакции образу-
1 Р. Берг. Применение 8-оксихинолина в аналитической химии. М.,
ОНТИ, 1937, стр. 96.
183
0500
0400
0,300
350
поглощения:
Al в CHC13;
Рис. 60. Спектры
’ — оксихинолинат
2 — оксихинолин в СНС1з
400 Л.нмк
0,600
ются малорастворимые в воде внутрикомплексные соедине-
ния. Эти соединения растворяются в неводных растворите-
лях (бензол, хлороформ) с образованием растворов, которые
можно фотометрировать (рис. 60); алюминий количественно
экстрагируется 1 в интервале pH 4,3 ~ 6,6. При фотомет-
рировании в качестве «нулевого» необходимо брать рас-
творитель, в который проэкстрагирован 8-оксихинолин, так
как область максимального по-
глощения реагента и соедине-
ния близки2 %яакс ОКСИХИНОЛИ-
ната алюминия в хлорофор-
ме — 395 ммк, оксихинолина
—320 ммк.
а. Определение алюминия
в водных растворах его
чистой соли
Необходимые реактивы:
1. Стандартный раствор со-
ли алюминия с содержанием
1 мг А1 в 1 мл. Для его приго-
товления 1,000 г металлическо-
го алюминия (проволока или
тонкая пластинка) помещают в
мерную колбу емкостью 1 л,
растворяют в 10 мл НС1 (уд.
в. 1,12), доводят объем до
1 л и тщательно перемеши-
вают— раствор А. 10 мл раствора А разбавляют водой до
1 л с предварительным добавлением в него 5 мл НС1 той же
концентрации — раствор Б. 1 мл раствора Б содержит
0,01 мг А1; раствор пригоден в день его приготовления.
2. о-Оксихинолин, 5%-ный раствор в 2 н. уксусной кис-
лоте.
3. Ацетат аммония, 2 н. раствор.
4. Бензол.
5. Уксусная кислота, 2 н. раствор.
6. Едкий натр, 5%-ный раствор.
7. Соляная кислота, уд. в. 1,12.
8. Соляная кислота, 1 н. раствор.
К 20 мл испытуемого водного раствора, помещенного в
делительную воронку, прибавляют 0,5 мл 1 н. раствора соля-
ной кислоты, 0,5 мл раствора оксихинолина, перемеши-
вают, добавляют 1 мл раствора ацетата аммония, снова
1 Е. Б. С е н д е л. Колориметрические методы определения следов ме-
таллов. М., «Мир», 1964, стр. 203.
2 Th. Moeller, J. Cohen. «J. Amer. Chem. Soc.», 72, 3546, 1950.
184
перемешивают и еще добавляют 2 мл того же раствора аце-
тата аммония. Экстрагирование производят двумя порциями
бензола по 6 мл каждая, перемешивая раствор в течение
1—2 мин. При проведении экстрагирования второй порцией
бензола следят, чтобы водный раствор оставался бесцветным;
в противном случае продлевают время перемешивания. Для
удаления следов влаги в слое бензола его можно профильтро-
вать через сухой бумажный фильтр или перелить в сухую
колбочку; в последнем случае влага удерживается на стен-
ках стеклянного сосуда. Бензольный раствор переносят в су-
хую мерную колбочку на 25 мл и доводят объем раствора
бензолом до метки колбы.
Интенсивность окраски раствора измеряют, используя фо-
токолориметры ФЭК-56, ФЭК-Н-54, ФЭК-Н-57 или спектро-
фотометры СФ-4, СФ-5, если определяют малые концентра-
ции алюминия. Окраску бензольного слоя сравнивают с се-
рией эталонных растворов, приготовленных в аналогичных
условиях и содержащих алюминий в количествах (.иг):
' 0,005
0,01
0,02
0,03
0,05
В качестве «нулевого» раствора используют бензол, полу-
ченный при экстракции раствора, содержащего оксихинолин,
и ацетат аммония. Экстрагирование производится в усло-
виях, указанных при работе с испытуемым раствором.
б. Определение алюминия в солях бериллия
При определении алюминия в присутствии бериллия ис-
пользуют свойство оксихинолината алюминия экстрагиро-
ваться в более кислой среде по сравнению с оксихинолинатом
бериллия. По Сенделу 1 50 мкг алюминия определяют в при-
сутствии 0,1 г бериллия при pH 5 (соединение бериллия экс-
трагируется при pH 9).
Кроме указанных ранее (стр. 184) необходимы реактивы:
1. 8-Оксихинолин, 1%-ный раствор в хлороформе.
2. 8-Оксихинолин, 5%-ный раствор в спирте.
3. Хлороформ, очищенный2.
4. Аммиак, концентрированный.
5. Аммиак, 6М раствор.
6. Соляная кислота х. ч., 10 н. раствор.
7. Соляная кислота х. ч., 2 М раствор.
1 Е. Б. С е н д е л. Колориметрические методы определения следов ме-
таллов. М., «Мир», 1964, стр. 203.
2 А. Вайсберге р, Э. Проскауэр, Дж. Риддик, Э. Т у п с.
Органические растворители. М., ИЛ, 1958, стр. 390.
185
8. Раствор бисульфита аммония. Его получают пропуска-
нием сернистого газа в 6М раствор аммиака, не содержащий
алюминия, до кислой реакции по лакмусовой бумаге.
9. Перекись водорода, 3%-ный раствор.
10. Буферный раствор, pH 5. Для его приготовления берут
70 мл 0,2 М. раствора ацетата натрия в смеси с 30 мл
0,2 М. уксусной кислоты.
11. Комплексон III, сухой препарат.
Приготовление эталонных растворов и по-
строение градуировочной кривой. Для приготовле-
ния эталонных растворов берут 5 делительных воронок ем-
костью ~ 100 мл, вводят 40 мл воды и стандартный раствор,
содержащий алюминий в количествах (мкг):
10
20
30
50
Затем добавляют 5 мл буферного раствора, воду до объема
50 мл, 10 мл 1%-ного раствора 8-оксихинолина в хлороформе
и содержимое воронки встряхивают в течение 3 мин. Хлоро-
форменный экстракт переносят в мерную колбу на 25 мл и
доводят объем до метки хлороформом. Полученный раствор
фильтруют через сухой бумажный фильтр для удаления ка-
пель воды, отбрасывая первые 5 мл, и переносят в кювету
для фотометрирования. Оптические плотности хлороформен-
ных экстрактов измеряют при X 395 ммк (если используется
спектрофотометр) или используют светофильтр с Хмакс Про-
пускания в области 385—410 ммк (если измеряют на фото-
электроколориметрах) . В качестве «нулевого» используют
раствор оксихинолина в хлороформе, которым обработан рас-
твор, содержащий все реактивы, кроме алюминия. По полу-
ченным данным строят градуировочный график.
Определение алюминия в контрольном
препарате соли бериллия. 0,1 г соли бериллия рас-
творяют в 30—40 мл воды, добавляют соответственно 3—4 мл
10 н. раствора соляной кислоты (чтобы концентрация кис-
лоты равнялась 1 н.), 2 мл раствора бисульфита аммония,
кипятят в течение 2 мин и охлаждают. К полученному рас-
твору добавляют 1 г комплексона, перемешивают и добав-
ляют аммиак до щелочной реакции по лакмусовой бумаге.
Раствор переносят в делительную воронку, добавляют посте-
пенно 5%-ный спиртовый раствор 8-оксихинолина и выдер-
живают в течение 1 час. После этого добавляют 10 мл хло-
роформа и встряхивают содержимое воронки в течение 1 мин.
Хлороформенный слой сливают в новую делительную воронку
и проводят повторную обработку водного слоя 5 мл хлоро-
форма. Такую обработку продолжают до получения бесцвет-
186
кого экстракта (обычно бывает достаточно 3—4 порций хло-
роформа). Объединенные экстракты промывают 25 мл воды,
слой хлороформа отделяют, а водный слой встряхивают
с 5 мл хлороформа, который потом присоединяют к основно-
му раствору хлороформа. Для переведения алюминия в вод-
ную фазу к хлороформу добавляют 25 мл 2 М. соляной кис-
лоты. Если в слое хлороформа содержится много оксихино-
лината алюминия, то его дополнительно обрабатывают 10 мл
кислоты. К водному раствору прибавляют несколько капель
метилоранжа и аммиака (концентрированного) до получения
желтой окраски раствора, затем 5 мл буферного раствора,
10 мл хлороформа и встряхивают содержимое воронки в те-
чение 3 мин. Экстракцию повторяют, обрабатывая водный
раствор еще двумя порциями хлороформа. Объединенные
экстракты, помещенные в делительную воронку, встряхивают
в течение 1 мин с 25 мл 2М соляной кислоты. К водному
раствору добавляют 20 мл раствора перекиси водорода и не-
сколько капель фенолового красного. Через 1 мин добавляют
6М аммиак до получения раствора красного цвета, 10 мл
хлороформа и встряхивают содержимое воронки в течение
3мин. Слой хлороформа сливают в мерную колбу на 25 мл и
доводят объем до метки хлороформом. Полученный раствор
фильтруют через маленький сухой бумажный фильтр для
удаления капель воды, отбрасывая первые 5 мл, и переносят
в кювету для фотометрирования. Измеряют поглощение полу-
ченного раствора так же, как указано в прописи для приго-
товления эталонных растворов. В качестве «нулевого» рас-
твора используют хлороформ, которым обработан раствор,
содержащий все реактивы, кроме алюминия, и проведенный
через все стадии анализа.
в. Определение алюминия в металлическом титане 1
Алюминий определяют колориметрическим путем по реак-
ции с оксихинолином. От главной массы титана алюминий
отделяют соосаждением с гидроокисью бериллия в аммиач-
ной среде в присутствии перекиси водорода, титан при этом
удерживается в растворе в пероксидной форме. Удаление
остатков титана, а также железа производится экстракцией
их оксихинолинатов хлороформом при pH 2.
Чувствительность метода 5-10-3%.
Необходимые реактивы:
1. Стандартный раствор алюминия. 1 г алюминиевой про-
волоки или фольги растворяют в 10 мл перегнанной соляной
кислоты и разбавляют в мерной колбе емкостью 1 л водой до
метки; 1 мл содержит 1 мг алюминия. Более разбавленные
1 Разработано в Гиредмете.
187
растворы получают разбавлением исходного раствора водой.
Рабочий раствор содержит 20 мкг алюминия в 1 мл.
2. Соляная кислота, перегнанная и разбавленная 1:4.
3. Перекись водорода, 30%-ный раствор.
4. Аммиак, свежеперегнанный, хранится в парафиниро-
ванной склянке.
5. Нитрат аммония, 2%-ный раствор, содержащий не-
сколько капель аммиака и пергидроля.
6. Хлоридный буферный раствор pH 1,6. К 50 мл 0,2 М рас-
твора хлористого калия добавляют 26,3 мл 0,2 н. соляной кис-
лоты и разбавляют в мерной колбе емкостью 200 мл водой
до метки.
7. Оксихинолин, 0,5%-ный раствор в перегнанном хлоро-
форме.
8. Ацетат натрия, 2М раствор. 136,1 г ацетата натрия
(CH3CO2Na • ЗН2О) растворяют в 500 мл воды.
9. Оксихинолин, 0,7%-ный раствор в перегнанном четы-
реххлористом углероде.
10. Раствор бериллия, содержащий 4 мг бериллия в 1 мл.
0,4 г металлического бериллия осторожно растворяют в ста-
кане, покрытом часовым стеклом в перегнанной соляной кис-
лоте, сначала на холоду, затем при нагревании. Раствор раз-
бавляют водой, если нужно фильтруют, переводят в мерную
колбу емкостью 100 мл и разбавляют водой до метки.
11. Раствор титана, содержащий 2,5 мг титана в 1 мл.
0,25 г титана растворяют в 10 мл перегнанной соляной кис-
лоты. Раствор окисляют 2—3 каплями раствора перекиси во-
дорода и разбавляют в мерной колбе емкостью 100 мл соля-
ной кислотой (1:10) до метки.
Приготовление эталонных растворов и по-
строение градуировочной кривой. В делительные
воронки емкостью 100 мл вводят 20—25 мл воды, 15 мл хло-
ридного буферного раствора, 2 мл 2М раствора уксуснокис-
лого натрия, 3—5 мл 0,7%-ного раствора оксихинолина в че-
тыреххлористом углероде, стандартный раствор алюминия в
количествах (мкг):
5
10
15
20
Содержание воронок встряхивают в течение 2 мин. После
расслаивания органический слой переносят в колориметриче-
ские пробирки емкостью 10 мл и диаметром 1 см с притер-
той пробкой и приливают четыреххлористый углерод до* опре-
деленного объема (5—6 мл). Оптические плотности получен-
ных растворов измеряют при /. 395 ммк (если используется
спектрофотометр) или используют светофильтр с ХмаКс про-
188
пускания в области 385—410 ммк. В качестве «нулевого» ис-
пользуют раствор 8-оксихинолина в четыреххлористом угле-
роде, которым обработан раствор, содержащий все реактивы,
кроме алюминия. По полученным данным строят градуиро-
вочный график.
Определение алюминия в контрольном об-
разце металлического титана. 0,3 г металла рас-
творяют при умеренном нагревании в 5—10 мл соляной кис-
лоты (перегнанной) *. Приливают при помешивании 1 мл рас-
твора бериллия, 80 мл воды, 10 мл раствора перекиси водоро-
да и аммиак до pH 8 (по универсальному индикатору). Через
несколько минут осадок отфильтровывают через неплот-
ный фильтр2 и промывают 3—4 раза раствором азотнокис-
лого аммония. Осадок смывают с фильтра в стакан емкостью
50 мл сначала горячей водой, затем 10 мл горячей перегнан-
ной соляной кислоты (1:4) и затем снова горячей водой. Об-
щий объем раствора должен составлять 25 мл3. Раствор
нейтрализуют аммиаком (по каплям) до pH 1,5 (по универ-
сальному индикатору), переводят в делительную воронку ем-
костью 100 мл и приливают 15 мл хлоридного буферного
раствора. Добавляют 10мл 0,5%-ного раствора оксихинолина
в хлороформе и воронку встряхивают в течение 1—2 мин.
Слой хлороформа сливают и экстракцию подобным путем по-
вторяют 6—8 раз до тех пор, пока хлороформенный слой не
будет бесцветен. Водный слой тщательно отделяют от хлоро-
форменного, приливают 5 мл 2 М раствора уксуснокислого
натрия и раствор перемешивают. Приливают 3—5 мл4
0,7%-ного раствора оксихинолина в четыреххлористом угле-
роде и воронку встряхивают 2 мин. Алюминий при этом пере-
ходит в органический слой, который переводят в колоримет-
рическую пробирку и далее поступают так же, как указано
в методике приготовления эталонных растворов. В качестве
«нулевого» используют раствор 8-оксихинолина в четыреххло-
ристом углероде, которым предварительно обрабатывают рас-
1 Если металл неоднороден, растворяют 1—1,5 г в 25 мл перегнанной
соляной кислоты и отбирают для анализа соответствующую аликвотную
часть раствора. Если остается нерастворимый остаток, его отфильтровыва-
ют и отбрасывают. Иодидный или другой компактный металл должен
быть в виде очень тонкой стружки; 0,3—1,5 г стружки растворяют в 20 мл
соляной кислоты х. ч. (уд. в. 1,19) при умеренном нагревании, доливая
в процессе растворения ту же кислоту до 20 мл. В отсутствие соляной кис-
лоты, квалификации х. ч., металл растворяют в перегнанной соляной кисло-
те вначале на холоду в течение продолжительного времени, а затем при
нагревании.
2 Иногда в фильтратах через некоторое время после начала фильтро-
вания появляется осадок, на что не обращают внимания.
3 При содержании алюминия более 0,015% для дальнейших операций
отбирают аликвотную часть раствора.
4 При наличии алюминия в 10—15 мкг приливают 3 мл, при большем —
5 мл. Содержание алюминия не должно превышать 45 мкг.
189
твор, содержащий все реагенты, кроме алюминия, и прове-
денный через все стадии анализа.
Примечания. 1. В случае получения мутных экстрактов перед пе-
ренесением в кювету для фотометрирования раствор оксихинолината алю-
миния в органическом растворителе фильтруют через сухой бумажный
фильтр.
2. Глухой опыт иа реактивы проводят через все стадии анализа, прн
этом в раствор, полученный после растворения осадка гидроокисей в со-
ляной кислоте (см. ход анализа на стр. 189), приливают 2 мл раствора ти-
тана; далее поступают, как указано в ходе анализа. Наличие алюминия в
реактивах при содержании алюминия в контрольном образце 3—5 • 10 ~* 3 %
не должно превышать 10—15 мкг.
ОПРЕДЕЛЕНИЕ КРЕМНИЯ И ФОСФОРА
В основе фотометрических методов определения фосфора
и кремния лежит реакция образования гетерополикислот со-
става:
Н4 [Si (Мо3О10)41 • пН2О,
Н3 [Р (Мо3О10)4] иН2О.
Гетерополикислоты кремния и фосфора (так называемые
желтые формы) образуются в кислой среде, но условия их
получения различны. Наиболее благоприятной кислотностью
для существования фосфорномолибденовой гетерополикислоты
является ~ 0,85 н. раствор; соответствующая кислота крем-
ния образуется в слабокислой среде (~-0,02 н. раствор). Но
образовавшись, кремневая гетерополикислота остается устой-
чивой в сильно кислой среде'. Различная устойчивость ука-
занных комплексных соединений в зависимости от кислотно-
сти раствора широко используется в методах определения
кремния и фосфора при их совместном присутствии в ряде
объектов, например в сталях2.
Эти соединения экстрагируются неводными растворите-
лями; получаемые растворы удобны для фотометрирования3.
Большей чувствительностью обладает реакция, в результате
которой при действии восстановителей на желтую форму ге-
терополикислот получаются растворы, окрашенные в синий
цвет, — синие формы.
В качестве восстановителей используется ряд веществ:
соль Мора, двухлористое олово, сульфит натрия, гидрохинон,
аскорбиновая кислота и другие вещества. Преимуществом
обладает аскорбиновая кислота, являясь более «мягким» вос-
становителем.
'3. Ф. Шахова, Т. А. Белявская, Л. П. Олыпадская
«Вести. Моск, ун-та», сер. хим., № 2, 103, 1949.
А. М. Дымов. Технический анализ руд и металлов М Meraiivnr-
издат, 1948, стр. 276, 297.
3 Р. И. А л е к с е е в. «Зав. лаб.», 11, 122, 1945.
190
Установлено, что образование синей формы не сопровож-
дается разрушением структуры желтой формы. Это видно из
спектров поглощения германомолибденовой гетерополикис-
лоты, которая по составу и строению аналогична кремневой
гетерополикислоте (рис. 61). При определении кремния и
фосфора возникает ряд осложнений.
Одной из общих причин получения невоспроизводимых
результатов при использовании синих форм гетерополикис-
лот этих элементов является применение более энергичных
восстановителен, что приводит к восстановлению не только
молибдена из гетерополиа-
ниона, но и молибдена из
молибдата аммония, кото-
рый берется в избытке в ка-
честве реактива для получе-
ния гетерополикислот. Вто-
рой причиной является не-
соблюдение определенной
кислотности раствора, необ-
ходимой для получения рас-
сматриваемых соединений;
это относится в одинаковой
мере к желтым и синим фор-
мам гетерополикислот *. При
определении кремния осо-
бенно важно учитывать по-
следнее обстоятельство, так
как известно, что на ско-
рость образования гетеропо-
лианиона оказывает влияние
форма существования крем-
невой кислоты перед получе-
Рис. 61. Спектры поглощения герма-
номолибденовой гетерополикислоты:
1 — желтая форма; 2 — синяя форма
нием комплексного соедине-
ния. Кремневая гетерополикислота образуется из ортокремне-
вой кислоты в течение 75 сек, из димера— 10 мин, а из более
высоко конденсированных форм в течение 1 час. Скорость кон-
денсации кремневой кислоты минимальна при pH 3. Для опре-
деления кремния и фосфора в виде синих форм гетерополики-
слот очень важно, чтобы ему предшествовала полнота образо-
вания желтой формы комплексного соединения.
В последнее время для получения синих форм гетерополи-
кислот стали использовать реактив, содержащий смесь 5- и
6-валептного молибдена. В этом случае отпадает необходи-
мость в применении какого-либо восстановителя2. При введе-
нии восстановителя одновременно с молибдатом (в составе
1 И. П. А л и м а р и н, 3. Ф. Шахова, Р. К- Мотор кин а.
ДАН СССР, 106, 61, 1956.
2F. Lucena-Gonde, L. Prat. «Anal. Chim. Acta», 16, 473, 1957.
191
так называемого смешанного реагента) возможно непосред-
ственное образование синего комплекса без видимой стадии
образования желтого комплекса. В связи с этим кажется
вполне вероятной возможность использования 2-валентного
железа, образующегося при растворении железосодержащего
анализируемого материала (чугун, сталь), для восстановле-
ния кремнемолибденового комплекса, в частном случае для
определения кремния в таких материалах.
В результате исследования *, проведенного с целью прак-
тического осуществления указанной возможности, установ-
лено, что при смешении растворов силиката натрия и соли
Мора при кислотности 0,12—0,25 н. по H2SO4 возникает мак-
симальная и устойчивая окраска; присутствие 1000-кратного
избытка 2-валентного железа в аналитической системе прак-
тически не влияет на светопоглощение растворов синего крем-
немолибденового комплекса.
Все это подтверждает возможность получения синей фор-
мы кремнемолибденового комплекса в присутствии 2-валент-
ного железа без предварительного образования желтой
формы.
ОПРЕДЕЛЕНИЕ ФОСФОРА
В приведенных ниже методиках определение фосфора про-
изводят в виде комплексного соединения синей формы фос-
форномолибденового гетерополианиона. При этом исполь-
зуют как методы с применением восстановителей, так и без
них. В качестве восстановителей берут двухлористое олово
и гидразинсульфат.
а. Определение фосфора в растворе его чистой соли
В качестве реагента применяют раствор, содержащий
смесь 5- и 6-валентного молибдена (стр. 191). Растворы под-
чиняются основному закону светопоглощения в интервале
0,01—2 мкг.
Необходимые реактивы:
1. Стандартный раствор соли фосфора, содержащий 0,1 мг
Р в 1 мл. Для получения этого раствора навеску 0,439 г х. ч.
однозамещенного фосфата калия (КН2РО4) помещают в мер-
ную колбу емкостью 1 л, растворяют в воде, доводят водой
до метки колбы и тщательно перемешивают.
2. Смешанный реактив: смесь Мо (V) и Мо (VI) в отно-
шении 2:3. Для приготовления этого реактива навеску 8,15 д
молибдата аммония квалификации ч. д. а. растворяют в 60 мл
воды. К 25 мл этого раствора добавляют 12,5 мл 12 н. соля-
ной кислоты и разбавляют водой до объема 50 мл, добав-
1 3. Ф. Шахова, Е. Н. Дорохова. «Вести. Моск, ун-та» вып 2,
77, 1965.
192
ляют в этот раствор 30 г металлической ртути, энергично
смешивают 10 мин и фильтруют раствор. Полученный рас-
твор должен иметь красную окраску. К 30 мл первоначаль-
ного раствора молибдата аммония добавляют 50 мл соляной
кислоты (уд. в. 1,19), 56 мл серной кислоты (уд. в. 1,84) (по-
степенно) и при постоянном помешивании вводят 40 мл рас-
твора, содержащего предварительно восстановленный молиб-
ден (V). Этот раствор разбавляют водой до объема 200 мл.
Полученный указанным путем реактив пригоден для употреб-
ления в течение 6 месяцев; он имеет изумрудно-зеленый цвет.
3. Серная кислота, 2 н. раствор.
К 8—10 мл помещенного в мерную колбу емкостью 25 мл
испытуемого водного раствора, содержащего фосфор, добав-
ляют 1,8 мл серной кислоты, 0,3 мл смешанного реактива,
разбавляют водой до объема 15 мл, перемешивают и нагре-
вают в течение 15 мин, погрузив в кипящую водяную баню.
Быстро охлаждают раствор, доводят объем водой до метки
колбы, перемешивают и фотометрируют при ХмаКс 815 ммк,
используя спектрофотометры СФ-4, СФ-5. Раствором сравне-
ния («нулевой» раствор) является дистиллированная вода.
Определяют концентрацию фосфора в испытуемом растворе
графическим методом, строя предварительно калибровочный
график по ряду эталонных растворов, приготовленных точно
в тех же условиях, которые были указаны для определения
фосфора в испытуемом растворе. Эталонные растворы содер-
жат фосфор в количествах (мкг):
1-й ряд 2-й ряд
0,05 0,5
0,15 0,75
0,25 .1,00
0,35 1,5
0,5 2,0
Если содержание фосфора отвечает количествам его, указан-
ным во 2-м ряду эталонных растворов, фотометрирование
можно производить на фотоэлектроколориметрах марки
ФЭК-56, ФЭК-Н-54, ФЭК-Н-57.
б. Определение фосфора в металлическом титане 1
Фосфор определяется колориметрическим путем по реак-
ции образования фосфорно-молибденовой гетерополикислоты,
экстрагируемой эфиром с последующим восстановлением
хлористым оловом до молибденовой сини. Определение про-
изводят после отделения титана сплавлением с едкой ще-
лочью.
1 Разработано в Гнредмете.
193
Чувствительность метода в условиях анализа 1 •10-3%.
Необходимые реактивы:
1. Стандартный раствор фосфора (запасной). 0,426 г дву-
замещенного фосфата аммония растворяют в воде, переводят
раствор в мерную колбу емкостью 1 л и разбавляют до
метки. 1 мл этого раствора содержит 100 мкг фосфора. Стан-
дартный раствор для колориметрирования готовят путем раз-
бавления запасного раствора водой. 1 мл полученного рас-
твора должен содержать 1 мкг фосфора.
2. Едкий натр х. ч. Содержание фосфора в нем не должно
превышать 0,0001 %.
3. Винная кислота ч. д. а., 50%-ный раствор.
4. Азотная кислота, концентрированная и 7%-ный раствор.
5. Молибденовая жидкость. 150 г молибденовокислого
аммония растворяют в 1 л воды и осторожно при помешива-
нии вливают в 1 л азотной кислоты (1:1). Раствору дают
отстояться в течение нескольких дней и перед употреблением
фильтруют. В случае, если при хранении раствора выделяет-
ся много молибденовой кислоты, раствор к употреблению не
пригоден.
6. Этиловый эфир.
7. Азотнокислая медь — раствор, содержащий 100 г соли
в 85 мл воды и 10 мл концентрированной азотной кислоты.
8. Азотнокислый кобальт, насыщенный раствор, разбав-
ленный водой 1:10.
9. Хлористое олово, 2%-ный раствор в соляной кислоте
(1:2).
10. Углекислый натрий, 2%-ный раствор.
Приготовление эталонных растворов. К 4—
5 мл раствора, содержащего от 0,2 до 1,2 мкг фосфора и по-
мещенного в делительную воронку емкостью 25 мл, добав-
ляют 1 мл молибденовой жидкости, перемешивают и через
5 мин прибавляют из бюретки 5,5 мл эфира1 и тщательно
взбалтывают в течение 2 мин. По расслаивании нижний вод-
ный слой сливают, а эфирный переводят в цилиндр для коло-
риметрирования и добавляют 10—11 капель раствора хлори-
стого олова, перемешивают и используют для дальнейшего
колориметрирования.
Можно использовать также серию постоянных эталонов,
имитирующих окраску фосфорномолибденовой гетерополи-
сини в эфирном слое. Серия постоянных растворов, имити-
рующих окраску эфирного слоя, готовится по табл. 5 в ци-
линдрах для колориметрирования с притертой пробкой диа-
метром около 13—14 мм.
1 После взбалтывания и последующего перевода в цилиндр для ко-
лориметрирования обычно остается только 5 мл.
194
Для уравнивания окраски раствора азотнокислой меди
с окраской фосфатного комплекса в эфирном слое к каждому
раствору шкалы добавляют по каплям раствор азотнокислого
кобальта. Уравнивание производят сравнением окраски шка-
лы с окраской эфирного слоя, содержащего соответствующие
количества фосфора в 5 мл эфира, приготовленного по про-
писи, указанной выше.
Таблица 5
Приготовление растворов, имитирующих окраску молибденовой сини
в эфирном слое, соответствующих различным количествам фосфора
Количество раствора азотнокислой меди, МЛ Количество 7%-ного раствора азотной кислоты, мл Содержание фосфора, мкг
в 25 мл эфира в 5 мл эфира
0,15 24,85 1 0,2
0,22 24,78 2 0,4
0,30 24,70 3 0,6
0,45 24,55 4 0,8
0,60 24,40 5 1,0
0,75 24,25 6 1,2
Шкала устойчива в течение нескольких месяцев.
Определение фосфора в контрольном об-
разце металлического титана. 0,3 г металла сплав-
ляют в никелевом тигле с 3 г едкого натра. Сплавление сна-
чала ведут на электроплитке для обезвоживания едкого нат-
ра, затем тигель переносят в муфельную печь, температуру
которой постепенно повышают от 200—300° до 600—700°.
Сплавление заканчивают по получении прозрачного плава.
По охлаждении тигля плав выщелачивают 25—30 мл 2%-ко-
го раствора углекислого натрия. Раствор с осадком переносят
в стакан емкостью 100 мл, кипятят 2—3 мин, осадок отфильт-
ровывают с отсасыванием через воронку Бюхнера с двойным
фильтром, стакан и фильтр промывают 5—6 мл 2 %-кого рас-
твора углекислого натрия, фильтрат переносят в стакан, со-
держащий 2 мл винной кислоты и 10,5 мл концентрированной
азотной кислоты *. По охлаждении раствор переводят в мер-
ную колбу емкостью 50 мл и доводят водой до метки.
Для определения фосфора отбирают пипеткой от 2 до
5 мл раствора, переводят в делительную воронку емкостью
25 мл и обрабатывают далее так, как указано в прописи для
приготовления эталонных растворов. Появившуюся окраску
сравнивают с серией эталонных растворов, находящихся в.
таких же цилиндрах, как и испытуемый раствор.
1 10,5 мл азотной кислоты необходимы для нейтрализации щелочи и
создания 10%-ной свободной кислоты в объеме 50 мл.
195
Можно использовать градуировочный график, построен-
ный на основании оптических плотностей эталонных раство-
ров, измеренных на одном из фотоэлектроколориметров
(ФЭК-М, ФЭК-Н-52, ФЭК-Н-54, ФЭК-Н-57, ФЭК-56).
Одновременно с проведением анализа необходимо прово-
дить глухой опыт по реактивам через все стадии анализа.
в. Определение фосфора в стали 1
При определении фосфора в стали в качестве восстано-
вителя для получения синей формы гетерополикислоты ис-
пользуют гидразин-сульфат.
Кроме указанных на стр. 192 необходимы реактивы:
1. Разбавленная азотная кислота. Добавляют 380 мл кон-
центрированной азотной кислоты к 620 мл воды.
2. Разбавленная бромистоводородная кислота. 20 мл
48 %-ной НВг добавляют к 80 мл воды.
3. Сульфит натрия. Растворяют 100 г безводного сульфи-
та натрия в 500 мл воды, разбавляют раствор до 1 л и филь-
труют.
4. Молибдат аммония. Растворяют 20 г молибдата аммо-
ния (NH4)6Mo7O24-4H2O в 10 н. серной кислоте и разбавляют
раствор до 1 л 10 н. серной кислотой.
5. Гидразин-сульфат. Растворяют 1,5 г гидразин-сульфа-
та и разбавляют до 1 л.
6. Смесь молибдата аммония и гидразин-сульфата. 25 мл
раствора молибдата разбавляют до 80 мл дистиллированной
водой. Добавляют 10 мл раствора гидразин-сульфата и раз-
бавляют до 100 мл. Этот раствор неустойчив, поэтому его
следует готовить непосредственно перед употреблением.
Приготовление эталонных растворов.
В 25 мл анализируемого раствора должно содержаться не
более 0,06 мг фосфора в виде ортофосфата. Раствор должен
быть нейтральным по лакмусу. К 25 мл анализируемого рас-
твора в мерной колбе на 50 мл добавляют 5 мл раствора мо-
либдата и 2 мл раствора гидразин-сульфата. Разбавляют до
метки дистиллированной водой и перемешивают. Погружают
колбу в кипящую водяную баню на 10 мин, вынимают и за-
тем быстро охлаждают. Снова перемешивают содержимое
колбы и, если необходимо, доводят объем до метки добавле-
нием нескольких капель воды. Измеряют оптическую плот-
ность раствора при X 830 ммк относительно раствора реа-
гентов в кювете с толщиной слоя 1 см. Калибровочная кри-
вая проходит через начало координат только при применении
в качестве раствора сравнения раствора реагентов. Необхо-
1 Д. Ф. Больтц. Колориметрические (фотометрические) методы опре-
деления неметаллов. М„ ИЛ, 1963, стр. 24.
196
димо следить, чтобы на стенках кювет не образовывались
пузырьки газа. Пузырьки удается устранить легким постуки-
ванием по стенке кюветы.
Определение фосфора в контрольном об-
разце стали. Навеску 50 мг углеродистой или низколеги-
рованной стали помещают в коническую колбу емкостью
125 мл, добавляют 5 мл разбавленной азотной кислоты и на-
гревают до полного растворения пробы. (При анализе неко-
торых хромовых сталей, кроме азотной кислоты, добавляют
еще 3 мл соляной кислоты 1:1.) Пипеткой добавляют 3 мл
хлорной кислоты. Осторожно выпаривают до появления па-
ров и после этого продолжают нагревание 3—5 мин до уда-
ления азотной кислоты. Охлаждают и, если сталь содержит
более 0,05% мышьяка, добавляют 5 мл разбавленной броми-
стоводородной кислоты и осторожно выпаривают до удале-
ния бромистого водорода. К охлажденному хлорнокислому
раствору добавляют 10 мл воды и 15 мл раствора сульфита.
Раствор нагревают до кипения и продолжают кипятить около
30 сек, охлаждают и переносят в мерную колбу на 50 мл,
применяя минимальное количество воды для споласкивания.
Добавляют 20 мл реагента молибдат аммония — гидразин-
сульфат. Разбавляют до метки дистиллированной водой и
перемешивают. Погружают колбу на 10 мин в кипящую во-
дяную баню, вынимают и быстро охлаждают. Если необхо-
димо, прибавляют несколько капель воды до метки. Изме-
ряют оптическую плотность при % 830 ммк в кювете I = 1 см
относительно аналогично приготовленного раствора из оди-
наковой навески и тех же количеств реагентов, но с прибав-
лением 5 мл 10 н. серной кислоты вместо 5 мл молибдата.
При определении фосфора в чугуне берут
навеску 100 мг, добавляют 25 мл разбавленной азотной кис-
лоты, удаляют азотную кислоту длительным кипячением, раз-
бавляют раствор точно до 100 мл, дают осесть графиту и от-
бирают 5 мл полученного раствора для определения. Далее
определение производят как описано выше, начиная с выпа-
ривания с хлорной кислотой и обработки бромистоводород-
ной кислотой.
ОПРЕДЕЛЕНИЕ КРЕМНИЯ
Определение кремния производят так же, как и фосфора,
в виде синей формы кремнемолибденового гетерополианиона.
В качестве восстановителя часто применяют двухлористое
олово. Аналогично определению фосфора можно проводить
определение кремния, не применяя восстановителя.
197
а. Определение кремния в сталях
(восстановитель — двухлористое олово)
Описываемый метод пригоден для определения кремния
в присутствии фосфора в сталях. Устойчивость гетерополи-
кислот кремния и фосфора, как было выше отмечено, зави-
сит от кислотности раствора. Образовавшаяся при низкой
кислотности (~ 0,02 н.) кремнемолибденовая гетерополикис-
лбта остается устойчивой при повышении кислотности рас-
твора, в то время как фосфорномолибденовая гетерополикис-
лота в этих условиях разрушается.
Необходимые реактивы:
1. Стандартный раствор кремния, содержащий 0,1 мг Si
в 1 мл. Для приготовления стандартного раствора берут на-
веску кремнекислого натрия (Na2,SiO3 • 9Н2О) с известным со-
держанием кремния (0,1 г), помещают в мерную колбу ем-
костью 1 л, растворяют в воде, доводят водой объем раствора
до 1 л и тщательно перемешивают—раствор А. 1 мл рас-
твора А содержит 0,1 мг Si. 10 мл раствора А разбавляют
до 200 мл — раствор Б. 1 мл раствора Б содержит 0,005 мг
кремния.
2. Серная кислота 1 :8.
3. Азотная кислота, 2,5 н. раствор.
4. Серная кислота, 0,15 н. и 8 н. растворы.
5. Молибдат аммония, 5%-ный раствор.
6. Двухлористое олово, 0,5%-ный раствор.
Для приготовления раствора в стакан на 100 мл поме-
щают 0,5 г соли двухлористого олова, добавляют 4 мл НС1
(уд. в. 1,19), 10 мл воды и нагревают до полного растворения
соли. После этого раствор переносят в мерную колбу на 100 мл
и доводят водой до метки. Раствор пригоден в день его при-
готовления.
Приготовление эталонных растворов. 0,1 г
стали, не содержащей кремния (или, если таковая отсутству-
ет, с известным заранее содержанием кремния, которое учи-
тывают при приготовлении эталонных растворов), помещают
в коническую колбу, добавляют 10 мл серной кислоты (1:8)
и нагревают на водяной бане при 85° до прекращения выде-
ления пузырьков. После этого приливают 5 мл азотной кис-
лоты и продолжают нагревание раствора в течение 2 мин.
Небольшой осадок графита дальнейшему ходу анализа не
мешает. Охладив раствор, переносят его в мерную колбу на
250 мл, доводят объем водой до метки, взбалтывают и дают
отстояться. Для приготовления эталонных растворов в мер-
ные колбы на 100 мл берут по 5 мл приготовленного раствора,
вводят в каждую колбу соответствующее количество стандарт-
ного раствора кремния, приливают 15 мл 0,15 н. раствора
198
серной кислоты, 5 мл раствора молибдата аммония и остав-
ляют стоять 5 мин. Прибавляют 25 мл 8 н. раствора серной
кислоты и после перемешивания добавляют 4 мл раствора
двухлористого олова. Растворы фотометрируют и строят ка-
либровочный график.
Содержание кремния в эталонных растворах должно соот-
ветствовать (мг):
0,005
0,01
0,015
0,02
0,025
Определение кремния в контрольном об-
разце стали. Навеску 0,1 г анализируемого образца ста-
ли растворяют при тех же условиях, которые были указаны
для приготовления эталонных растворов. После соответствую-
щего разбавления для испытания берут 5 мл раствора и про-
водят реакцию образования синей формы кремнемолибдено-
вой гетерополикислоты в условиях, описанных выше для при-
готовления эталонных растворов.
б. Определение кремния в растворе чистой, соли 1
без применения восстановителя
Метод основан на получении синей формы кремнемолиб-
деновой гетерополикислоты при использовании в качестве
реактивов солей молибдена (VI) и молибдена (V), прибав-
ляемых последовательно.
Кроме указанных на стр. 198 необходимы реактивы:
1. Молибдат аммония (молибден VI), 10%-ный водный
раствор.
2. Раствор восстановленной соли молибдена (молибден V)
(стр. 192). 57мл восстановленного молибдата аммония раз-
бавляют водой двойной дистилляции до 100 мл.
3. Вода двойной дистилляции.
4. Уксусная кислота I : 3.
5. Соляная кислота, 2 н. раствор.
6. Серная кислота, 12 н. раствор.
7. Серная кислота, 0,2 н. раствор.
В конические колбы емкостью 50 мл вводят стандартный
раствор соли кремния, разбавляют водой двойной дистилля-
ции до 5 мл, приливают 0,4 мл уксусной кислоты, 0,6 мл 0,2 н.
раствора серной кислоты, 0,2 мл раствора молибдата, остав-
ляют стоять 5 мин при комнатной температуре, добавляют
примерно 20 мл воды двойной дистилляции, 3 мл 2 н. рас-
1 Разработано 3. Ф. Шаховой, Р. К. Моторкнной, Л. И. Ващенко на
кафедре аналитической химии МГУ.
199.
твора соляной кислоты, 1,2 мл 12 н. раствора серной кис-
лоты, 0,2 мл раствора молибдена (V), разбавляют до 30 мл
и нагревают в кипящей бане в течение 15 .мин. Быстро охлаж-
дают растворы и фотометрируют при X 805 ммк по отноше-
нию к воде, взятой в качестве «нулевого» раствора. Содержа-
ние кремния в эталонных растворах соответствует (мкг):
1-й ряд 2-й ряд
1 7
3 10
5 15
7 20
10
Для определения кремния в испытуемом водном растворе
соблюдают все условия, указанные для приготовления эта-
лонных растворов.
в. Определение кремния в чугунах и сталях
без применения постороннего восстановителя ’
При определении кремния в чугунах и сталях не нужно
предварительно устранять 2-валентное железо путем окисле-
ния, а наоборот, целесообразнее использовать его в качестве
восстановителя (стр. 192), причем в тех случаях, когда же-
леза в анализируемом материале недостаточно для полного
восстановления комплекса, его следует вводить дополнитель-
но в качестве разбавляющего раствора. Необходимо отме-
тить, что при указанной выше кислотности образования крем-
немолибденового комплекса (0,12—0,25 н. по H2SO4) в рас-
творах может также присутствовать продукт восстановления
простого молибдата, который в отличие от гетерополиком-
плекса неустойчив при высокой кислотности. Поэтому после
образования гетерополисини при выбранной низкой кислот-
ности в растворе следует создать высокую кислотность (5 н.
по H2SO4). При изучении влияния концентрации молибдата
установлено, что 300-кратный избыток его является опти-
МЯЛЬНЫМ.
Кроме указанных на стр. 198 необходимы реактивы:
1. Серная кислота 1 : 10 и 1:3.
2. Соль Мора, 12%-ный раствор. 120 г соли Мора раство-
ряют в дистиллированной воде, добавляют 10 мл 1 н. серной
кислоты.
При содержании кремния 0,8—3,0% 0,1 г чугуна
с известным содержанием кремния растворяют в 10 мл H2SO4
(1:10) при нагревании на водяной бане до прекращения вы-
деления пузырьков газа. Раствор охлаждают и переносят в
1 Методика применима к образцам стали, растворимым в серной кис-
лоте.
200
мерную колбу емкостью 200 мл и разбавляют до метки рас-
твором соли Мора. 1,5; 2,0; 2,5; 3,0 и 3,5 мл полученного рас-
твора отбирают в колориметрические пробирки или мерные
колбы емкостью 25 мл, добавляют 2,4 мл раствора молиб-
дата аммония, выдерживают 5 мин, разбавляют до метки сер-
ной кислотой (1:3), перемешивают и через 15—20 мин изме-
ряют оптическую плотность на фотоэлектроколориметре
ФЭК-М с красным светофильтром в кюветах 0,5—1 см по от-
ношению к воде или раствору, содержащему несколько мень-
шее количество кремния и приготовленному в тех же условиях
(см. дифференциальный метод, стр. 66). Раствор чугуна с не-
известным содержанием кремния готовят в тех же условиях
и находят искомую концентрацию кремния по калибровоч-
ному графику.
При содержании кремния меньше 0,8% анализ
производят по той же методике, но навеску увеличивают до
0,3 г и для растворения ее берут 5 мл H2SO4 (1 : 10), разбав-
ляют солью Мора до 50 жл; в мерные колбы на 25 мл отби-
рают от 3 до 10 мл раствора.
ОПРЕДЕЛЕНИЕ ЦИНКА
Для определения цинка применяют ряд органических реа-
гентов. Из их числа большого внимания заслуживает дити-
зон ', который в зависимости от кислотности раствора может
реагировать в кето- или энольной форме
/NH—NH—СвН5
S=C\
XN=N-CeH5
Дитизон является одним из распространенных реагентов,
применяемых для определения тяжелых металлов. Реакции
эти отличаются большой чувствительностью; значения моляр-
ных коэффициентов погашения порядка 50 000. Дитизонаты
металлов хорошо растворимы в неводных растворителях, ко-
торые обычно используются для их экстракции. Дитизон реа-
гирует с большим числом элементов, но найдены условия
определения многих отдельных элементов (Zn, Cd, Си, Pb
и др.) при определенной кислотности раствора (pH) и при-
менении различных маскирующих веществ для устранения
влияния посторонних элементов. Раствор дитизоната цинка в
СС14 максимально поглощает при X 520 ммк (рис. 62).
а. Определение цинка в металлическом титане 2
В предлагаемой методике дан метод определения цинка
с предварительным отделением его от титана экстракцией
1 Н. Fischer. «Z. angew. Chem.», 42, 1025, 1929; 47, 685, 1934; 50
219, 1937.
2 Разработано в Гиредмете.
201
Рис. 62. Спектры поглощения:
1 — дитизон в СС14; 2 — дитизо-
нат циика в СС14
Диэтилдитиокарбамината цинка этйлацетатом с последую--
щей реэкстракцией в водный слой. Чувствительность метода
1 • ю-3%.
Необходимые реактивы:
1. Стандартный раствор цинка. 0,1 г металлического цин-
ка растворяют в соляной кислоте и раствор разбавляют в
мерной колбе емкостью 100 мл водой до метки — раствор А.
1 мл раствора А содержит 1 мг цинка. 1 мл раствора А раз-
бавляют в мерной колбе емко-
стью 100 мл водой до метки —
раствор Б. 1 мл раствора Б со-
держит 0,01 мг цинка; годен в.
день приготовления.
2. Азотная кислота, концен-
трированная, разбавленная 1 :3
и 0,01 н. раствор.
3. Борная кислота, 5%-ный
раствор.
4. Винная кислота, 15%-ный
раствор.
5. Плавиковая кислота, раз-
бавленная 1 : 5.
6. Аммиак водный.
7. Этилацетат перегнанный.
8. Диэтилдитиокарбаминат
натрия, 2%-ный раствор.
9. Уксуснокислый буферный
раствор, pH 3.
10. Уксуснокислый натрий, 50%-ный раствор.
11. Серноватистокислый натрий, 50%-ный раствор.
12. Четыреххлористый углерод, перегнанный, бесцветный.
13. Дитизон, 0,02%-ный раствор в четыреххлористом уг-
лероде (запасной).
0,020 г дитизона помещают в делительную воронку ем-
костью 500 мл и растворяют в 50 мл четыреххлористого угле-
рода; дитизон, растворяясь, окрашивает раствор в зеленый
цвет. По растворении дитизона добавляют в воронку 100 мл
аммиака (1 : 100) и воронку энергично встряхивают; при этом
дитизон переходит в аммиачный раствор, окрашивая его в
оранжевый цвет, а продукты окисления дитизона остаются
в слое органического растворителя. Последний сливают, в во-
ронку добавляют 100 мл чистого четыреххлористого угле-
рода, подкисляют аммиачный раствор несколькими каплями
разбавленной (1:1) серной кислоты и воронку встряхивают
для перевода дитизона обратно в слой четыреххлористого уг-
лерода. Отделив раствор дитизона от водного слоя, дважды
промывают его водой путем встряхивания в делительной во-
202
ронке, а затем фильтруют (для просушки) через бумажный
фильтр и хранят в склянке из темного стекла.
0,005%-ный раствор дитизона готовят разбавлением в че-
тыре раза запасного 0,02%-ного раствора дитизона четырех-
хлористым углеродом, т. е. на 25 мл 0,02%-ного раствора бе-
рут 75 мл четыреххлористого углерода. Раствор готовят в
день употребления.
Приготовление эталонных растворов. В мер-
ную колбу на 50 мл берут 5 или 10 мл в зависимости от ко-
личества испытуемого раствора 0,01 н. азотной кислоты и
стандартный раствор цинка в количествах (мг):
0,003
0,006
0,01
0,02
В остальном эталонные растворы готовят в условиях, ука-
занных ниже при определении цинка в испытуемом растворе.
Определение цинка в контрольном образ-
це металлического титана. 0,3 г металла помещают
в платиновую чашку и обрабатывают на холоду 10 мл пла-
виковой кислоты, прибавляя ее небольшими порциями; оче-
редную порцию плавиковой кислоты прибавляют по прекра-
щении бурной реакции. Помешивают раствор легким покачи-
ванием платиновой чашки. Растворение заканчивают при
слабом нагревании на водяной бане. После полного раство-
рения добавляют 5-—6 капель концентрированной азотной
кислоты и выпаривают на водяной бане до объема 1 —1,5 мл,
не допуская выпаривания до меньшего объема. Приливают
3 мл винной кислоты, 10 мл борной кислоты и разбавляют
водой до объема 25 мл. Раствор нейтрализуют аммиаком до
переходной окраски по бумаге конго, охлаждают и переводят
раствор в делительную воронку емкостью 100 мл. Платино-
вую чашку дважды промывают небольшими порциями воды
из промывалки, сливая ее в воронку. Туда же приливают
5—8 мл этилацетата, 1 мл раствора диэтилдитиокарбамината
натрия и энергично встряхивают воронку в течение 10—
20 сек. После отстаивания тщательно отделяют слой этилаце-
тата от водного слоя, переводя последний во вторую дели-
тельную воронку. К водному слою снова приливают этилаце-
зат и диэтилдитиокарбаминат натрия в тех же количествах
и воронку встряхивают. Экстракцию повторяют еще 2—3 ра-
за, пока окраска слоя этилацетата не останется без измене-
ния. Объединенный экстракт, собранный в одну делительную
воронку, отделяют от отстоявшихся капелек водного слоя,
промывают 2 раза по 5 мл уксуснокислым буферным раство-
ром и водный слой отбрасывают.
203
Экстракт фильтруют через плотный сухой складчатый
фильтр диаметром 11 см, собирая его в другую делительную
воронку емкостью 50 мл. Туда же приливают 10 мл азотной
кислоты (1:3) и воронку энергично встряхивают для извле-
чения цинка в водный слой. После отстаивания тщательно
отделяют водный слой, собирая его в стакан емкостью
50 мл. Обработку экстракта 5—10 мл той же кислоты повто-
ряют еще 2 раза. Объединенную азотнокислую вытяжку упа-
ривают на песчаной бане досуха. Сухой остаток смачивают
несколькими каплями концентрированной азотной кислоты
и слегка подсушивают на водяной бане. Затем остаток рас-
творяют в 0,01 н. азотной кислоте, раствор переводят в мер-
ную колбу емкостью 50 мл и разбавляют до метки той же
кислотой. Отбирают пипеткой 5—10 мл раствора и перено-
сят в воронку для экстрагирования (в растворе не должно
быть более 10 мкг цинка), приливают 1 мл раствора уксус-
нокислого натрия и 1 мл раствора серноватистокислого нат-
рия (растворы перед употреблением очишают взбалтыванием
в делительной воронке с дитизоном до тех пор, пока вновь
добавленная порция дитизона не будет оставаться зеленой)
и после перемешивания 15 мл 0,005%-ного раствора дитизона
в четыреххлористом углероде. Полученные растворы фотомет-
рируют, используя в качестве «нулевого» раствор дитизона,
применяемый для экстракции и которым обработаны все
реактивы, согласно примечанию.
Примечание. Необходимо проводить контрольный опыт с реак-
тивами через все стадии анализа, остерегаться применения химической по-
суды, содержащей цинк, и работать с перегнанными (желательно в кварце-
вой посуде) кислотами и водой.
ОПРЕДЕЛЕНИЕ РЕНИЯ
Для определения рения имеется ряд фотометрических ме-
тодов, в которых используется главным образом роданидная
реакция '. В последнее время появилось несколько сообщений
о применении а-фурилдиоксима для спектрофотометрического
определения рения в кислой среде1 2 (НС1 или H2SO4) в при-
сутствии хлорида олова (II) и ацетона3 или спирта. Приво-
дим метод определения рения, разработанный в нашей лабо-
ратории.
1 С. Hiskey, V. М е 1 о с h. «Ind. Eng. Chem., Anal. Ed.», 12, 503,
1940; S. Trib a la t. «Anal. Chim. Acta», 3, 113, 1949; 5, 115, 1951.
2 В. M. Пешкова, M. И. Громова. «Вести. Моск, ун-та», сер.
хим., № 10, 85, 1952; V. М е 1 о с п, К- Martin, W. Webb «Anal
Chem.», 29, 524, 1957.
3 В. M. Пешкова, Чон Ун Ам. «Вести. Моск, ун-та», сер. хнм.,
№ 4, 59, 1960.
204
Определение рения а-фурилдиоксимом
Процесс образования соединения рения с а-фурилдиокси-
мом в системе вода — неводный растворитель (ацетон и
спирт) является, по-видимому, сложным. В зависимости от
кислотности раствора, соотношения рения и реагента, а так-
же от концентрации неводного растворителя наблюдается
различная окраска растворов: желтая, желтовато-оранжевая,
коричневатая, фиолетовая и малиновая. В интервале кислот-
ности 0,6—1,0 н. по соляной
кислоте в присутствии хлорида
олова (II) и ацетона (24—26%
по объему) образуется окра-
шенное комплексное соедине-
ние рения, имеющее максимум
на кривой спектра поглощения
при X 530 ммк, е = 43 ООО±200
(рис. 63). При отдельном изу-
чении спектров поглощения чи-
стого ацетона и а-фурилдиок-
сима в ацетоне установлено,
что раствор реактива в ацетоне
поглощает в коротковолновой
части спектра (220—330 ммк) и
поглощение этих веществ не
сказывается на спектре комп-
Рис. 63. Спектры поглощения сое
динений рения с а-фурилднокси
мом в присутствии различных ко
личеств ацетона
лексного соединения рения с
а-фурилдиоксимом. Комплекс-
ное соединение рения с а-фу-
рилдиоксимом, полученное в
кислой среде, хорошо экстра-
гируется хлороформом и изоамиловым спиртом. Полнота
экстракции достигается при двукратном извлечении, если ис-
пользуется хлороформ, и трехкратном с изоамиловым спир-
том (объем растворителя 10—15.мл).
При выполнении экстрагирования растворы хлороформа
очень часто получаются мутными. Для получения прозрач-
ных растворов их дважды фильтруют через сухой фильтр
или, что значительно удобнее, к раствору хлороформа, полу-
ченному после экстракции, добавляют изоамиловый спирт.
Растворы, полученные после экстракции хлороформом и раз-
бавленные изоамиловым спиртом, подчиняются закону Буге-
ра— Ламберта — Бера в области концентраций от 0,04 до
7,2 мкг/мл. Производя фотометрирование с кюветой, имею-
щей диаметр 1 см, можно определять 0,04 мкг/мл Re. При
использовании кюветы диаметром 5 см и 10 см соответствен-
но может быть снижен предел для определения рения до
0,08 мкг/мл и 0,004 мкг/мл Re.
8 Зак. 686
205
Из литературных данных известно1, что оксикислоты да-
ют комплексные соединения с рением, имеющим валентность
ниже семи. При изучении влияния оксикислот на реакцию об-
разования комплексного соединения рения с а-фурилдиокси-
мом было установлено2, что максимальная окраска соедине-
ния развивается в течение 15 мин, без добавления оксикислот
для этого необходимо 45 мин. Видимо оксикислоты (щавеле-
вая или винная), фиксируя определенное валентное состояние
рения (IV), способствуют более быстрому образованию
соединения рения с а-фурилдиоксимом. Следует отметить, что
при определении рения в присутствии больших количеств мо-
либдена с добавлением оксикислоты (винной) оптическая
плотность комплексного соединения рения понижается, дости-
гая постоянного значения так же через 15 мин. Но если ис-
пользовать вариант дифференциального метода (стр. 69), то
большие количества молибдена не мешают определению ре-
ния.
Необходимые реактивы:
1. Стандартный раствор соли рения. 0,155 г х. ч. препарата
перрената калия (KReCh) растворяют в воде в мерной колбе
на 250 мл — раствор А. 1 мл раствора А содержит 0,1 мг Re.
10 мл раствора А разбавляют водой до 100 мл — раствор Б.
1 мл раствора Б содержит 0,01 мг Re; годен к употреблению
в течение 1—2 дней.
2. а-Фурилдиоксим, 0,015М раствор в ацетоне.
3. Соляная кислота, 5 н. раствор.
4. Изоамиловый спирт, ацетон, хлороформ, свежепере-
гнанные.
5. Двухлористое олово, 10%-ный раствор в 5 н. НС1.
6. Винная кислота, 20%-ный водный раствор.
7. Барий хлористый, 10%-ный водный раствор.
а. Определение рения в растворе чистой соли
К водному раствору, содержащему рений в объеме 3—
5 мл и помещенному в делительную воронку, добавляют 3 мл
соляной кислоты, 10 мл винной кислоты, 6,5 мл раствора
а-фурилдиоксима в ацетоне, 2,5 мл раствора двухлористого
олова, разбавляют раствор до объема 25 мл водой и через
45 мин экстрагируют комплексное соединение рения двумя
порциями хлороформа (по 6 мл). Полнота экстракции полу-
чается при встряхивании в течение 1 мин каждой порции рас-
твора.
Дают слоям разделиться, слой хлороформа переносят в
мерную колбу на 25 мл, в которую предварительно добав-
1 S. Т г i b а 1 a t. «Ann. d. Chemie», 4, 289, 1949.
2 В. М. Пешкова, Н. Г. Игнатьева. «Тр. Второго Всесоюзного
совещания по рению». М., «Наука», 1964, стр. 239.
206
лено 10 мл изоамилового спирта, доводят объем до метки
изоамиловым спиртом и фотометрируют при X 530 ммк\ «ну-
левым» раствором является смесь используемых растворите-
лей. Эталонные растворы, содержащие от 0,5 мкг до 0,06 мг
рения, готовят так же, как и анализируемый раствор.
б. Определение рения в присутствии молибдена1
Рений в природных соединениях обычно сопутствует мо-
либдену. Изложенный выше метод определения рения может
быть применен для определения этого элемента в присут-
ствии молибдена. Молибден в малых концентрациях в пред-
лагаемых условиях не мешает определению рения. Для соот-
ношений Re: Мо, равных 1:1, 1:5, 1:10, 1:15, рений опреде-
ляется количественно в интервале концентраций 6—30 мкг.
Определение производят в следующих условиях. К 5—7 мл
водного раствора, содержащего рений (VII) и молибден (VI),
помещенного в делительную воронку, добавляют 3 мл соля-
ной кислоты, 10 мл винной кислоты, 6,5 мл раствора а-фурил-
диоксима в ацетоне, 2,5 мл раствора двухлористого олова,
разбавляют раствор до объема 25 мл водой, экстрагируют
комплексное соединение рения двумя порциями хлороформа
(по 6 мл), добавляют изоамиловый спирт и фотометрируют
в условиях, указанных ранее для определения рения в рас-
творе его чистой соли.
Концентрацию рения в растворе находят графическим ме-
тодом. Эталонные растворы готовят в условиях, приведенных
для определения рения в растворе его чистой соли. В эта-
лонные растворы вводят рений в количествах (мг):
1-й ряд 2-й ряд
0,005 0,0075 0,01 0,015 0,015 0,02 0,025 0,03
в. Определение рения в присутствии больших количеств
молибдена дифференциальным спектрофотометрическим
методом 1
При определении рения в присутствии больших количеств
молибдена всегда получаются заниженные результаты. При
соотношении Re: Мо = 1 :40 ошибка достигает 25—30% уже
при определении 5 мкг рения.
Для определения рения в присутствии больших количеств
xvJiBiQ^'iQrI4eU,K0Ba’ И Г Игнатьева’ Г. П. Озерова. ЖАХ,
V 111 j хУО, 1 у00,
8* 207
молибдена следует рекомендовать вариант дифференциаль-
ного метода (стр. 69). Оптическую плотность D испытуемого
раствора измеряют относительно раствора сравнения, кото-
рый представляет собой раствор испытуемого объекта, взятый
в меньшем объеме.
В излагаемом ниже методе обработки руды большая часть
молибдена отделяется '.
Навеску 2 г руды (молибденового или медного концентра-
та) помещают в фарфоровый тигель, добавляют 0,2 г КСЮз,
3 г СаО и содержимое тигля осторожно перемешивают. Ти-
гель помещают в тигельную печь (температуру которой посте-
пенно доводят до 650°С) и нагревают 2 час при t = 650° ±
± 10°С. Затем тигель вынимают и охлаждают. При охлажде-
нии спек выщелачивают 150 мл горячей воды в стакане ем-
костью 300 мл при постоянном и тщательном перемешивании
раствора. Затем содержимое стакана упаривают на песчаной
бане до объема 80 мл, осадок отфильтровывают в горячем
состоянии на небольшой воронке Бюхнера. Осадок молиб-
дата кальция промывают двумя небольшими порциями воды.
Фильтрат переносят в стакан емкостью 150 мл, приливают
5 мл 10%-ного раствора хлорида бария для осаждения ионов
сульфата, который может присутствовать в растворе. Раствор
упаривают на песчаной бане до объема 20—30 мл, фильтруют
через фильтр с синей лентой в мерную колбу на 50 мл, ох-
лаждают и доводят водой до метки. Для определения рения
берут 2 порции по 10 мл (Рж) приготовленного раствора, од-
на из них имеет концентрацию сх и вторая с той же концен-
трацией искомого элемента (Re), но к ней добавляют неболь-
шое известное количество стандартного раствора Re (са), на-
пример 0,01—0,02 мг Re. Для приготовления раствора срав-
нения («нулевого» раствора) берут 9 мл (Vo) анализируемого
раствора. Ко всем трем растворам добавляют по 10 мл вин-
ной кислоты, 6,5 мл раствора а-фурилдиоксима в ацетоне,
4 мл раствора SnC^ (3;75 н. по НС1) и воды (из бюретки) до
объема 25 мл. Через 15 мин проводят экстракцию окрашен-
ного комплексного соединения двумя порциями хлороформа
(8 + 4 мл) в течение 1—2 мин. Хлороформенные экстракты
собирают в мерные колбы объемом 25 мл (носик воронки пе-
ред сливанием хлороформенного слоя следует осушить филь-
тровальной бумагой) и доводят объем раствора до метки кол-
бы изоамиловым спиртом (небольшое количество изоамилово-
го спирта следует прилить в колбы перед сливанием хлоро-
форменного слоя). Измеряют светопоглощение двух растворов
Dx и Dx+a относительно раствора сравнения при 530 ммк. На-
ходят значение иа и по формуле сх =----
Ва
V
-pe-
1 Б. Н. Р а и с к и й. «Зав, лаб,», 24, 803, 105S.
sos
деляют содержание рения в аликвотной части. Для примера
приведены данные в табл. 6 по определению рения в молиб-
деновом концентрате описанным методом.
Таблица 6
Определение рения в молибденовом концентрате
(содержание Re п • 10—2 %; V0=9 мл; 1Д=10 мл; 1=1 см)
Добавлзно Re (са), мкг Dx Dx+a Найдено Re, мкг Найдено Re, %
10 0,066 0,150 393 1,8-10—2
20 0,067 0,226 421 2,1-10-2
ср. 2,0-10-2
г. Определение рения в сплавах рений — вольфрам
Кроме указанных на стр. 206 необходимы реактивы:
1. Азотная кислота х. ч., концентрированная.
2. Плавиковая кислота х. ч., концентрированная.
3. Едкий натр х. ч., 50%-ный водный раствор.
Навеску сплава 0,2 г (содержание рения около 20%) или
0,8 г (при содержании ~5 %) растворяют в тефлоновой чашке
в смеси 10 мл HF и 5 мл HNO3 (азотную кислоту добавляют
по каплям). После растворения раствор упаривают на водя-
ной бане досуха, осадок растворяют в воде с добавлением
2,5 мл щелочи, 10 мл раствора винной кислоты (из расчета
1 мл на 20 мг W) и разбавляют до 200 мл. Этот раствор до-
полнительно разбавляют в 10 раз и в нем уже проводят опре-
деление рения. Для этого берут 2 раза по 3 мл испытуемого
раствора, к одному из них добавляют 0,01 мг рения. Для при-
готовления раствора сравнения берут 2 мл испытуемого рас-
твора. Далее определение ведут так же, как указано выше
для определения рения в присутствии молибдена, с тем
лишь различием, что экстрагируют соединение рения через
20—30 мин после прибавления всех реактивов.
д. Определение рения в присутствии ванадия
Кроме указанных на стр. 206 необходимы реактивы:
1. Соляная кислота, 4 н. раствор.
Присутствие 10 000-кратного избытка ванадия не мешает
прямому определению рения, поэтому готовят эталонные рас-
творы в условиях, указанных (стр. 206) для определения ре-
ния в растворе чистой соли. Определение рения в присутствии
ванадия проводят в следующих условиях.
К раствору, содержащему рений и ванадий в мерной кол-
бе на 25 мл, добавляют 2,5 мл соляной кислоты, 10 мл рас-
209
твора винной кислоты, 6,5 мл раствора а-фурилдиоксима,
4 мл раствора двухлористого олова и доводят водой до метки
колбы. Через 15 мин проводят экстракцию в условиях, ука-
занных для определения рения в растворе чистой соли
(стр. 206).
ОПРЕДЕЛЕНИЕ ЦИРКОНИЯ
За последнее время появилось сравнительно большое чис-
ло методов определения циркония, но применение их иногда
приводит к получению невоспроизводимых результатов. При-
чиной этому является состояние иона циркония в водных рас-
творах. Ионы циркония благодаря высокому заряду и неболь-
шому радиусу (0,47 А) легко гидролизуются и дают полиме-
ризованные частицы в водных растворах. Исследованию ион-
ного состояния циркония в растворах положила начало ра-
бота Конника и Мак Ви'. Общий вид процесса комплексооб-
разования выражается уравнением
IV
Zr + п (HA)‘~Z ZrA*-z/I + пН+.
Гидролиз можно рассматривать как процесс комплексооб-
разования иона циркония с ОН-ионами: Az~— ионьи гидрокси-
ла, a HA1-Z— молекулы воды. По данным, полученным Кон-
ником и Мак Ви, А. С. Соловкин1 2 вычислил общие константы
комплексообразования и константы гидролиза Ki. Нами было
рассчитано процентное распределение циркония между гидро-
ксокомплексами по формуле
МА _ 1°°Р» [д1г
1 1+2P/W ’
В результате расчета было показано, что цирконий в вод-
ном растворе дает ряд оксикатионов, которые и могут давать
соединения различного состава.
Определение циркония 2-(п-сульфонилазо)-1,8-
диоксинафталин-3,6-дисульфокислотой («спаднс»)
дифференциальным методом
С учетом ионного состояния циркония в растворе изучена3
колориметрическая реакция этого элемента с реагентом
«спаднс», предложенным Банерджи 4 для определения тория
1 R. С о п п i k, W. Me. V е у. «J. Am. Chem. Soc.», 71, 3182, 1949.
2 А. С. С о л о в к и и. ЖНХ, 2, 611, 1957.
3 В. М. Пешкова, Н. В. М е л ь ч а к о в а, Е. Д. Синицына.
«Изв. высш, учеби. зав.», химия и химическая технология, III, вып. 1, 72,
1960.
4G. Banerjee. «Z. anal. Chem.», 147, 105, 1955; «Anal. Chem. Acta»,
16, 56, 62, 1957.
210
и циркония. В работе использован один из вариантов диф-
ференциального спектрофотометрического метода. Установ-
лено большое влияние кислотности раствора на условия об-
разования соединения. Значение оптической плотности рас-
творов комплексного соединения достигает максимального
значения в том случае, если исходный раствор соли циркония
имеет кислотность, равную 5 н. (по НС1О4). После добавле-
ния раствора реагента кислотность
понижена до pH 0,9—1,1.
Раствор комплексного сое-
динения циркония со
«спаднс» подчиняется закону
Бера в интервале концент-
раций циркония от 4 до
40 мкг в 25 мл конечного
объема. Чувствительность
метода 0,02 мкг в 1 мл,
e5soMMK = 18 900 (определено
по дифференциальному ме-
тоду) . Соотношение компо-
нентов в комплексном сое-
динении Zr: «спаднс» = 1 : 2.
Необходимые реактивы:
1. Стандартный раствор
соли перхлората циркония,
содержащий 10 мкг Zr в
1 мл. Сульфат циркония очи-
щают осаждением гидрооки-
си циркония, растворением
ее в разбавленной соляной
кислоте и после упарива-
ния раствора осаждением
при прибавлении концент-
раствора должна быть
1 — «спаднс» по отношению к воде;
2 — соединение циркония со
«спаднс» — к воде; 3 — соединение
циркоиия со «спаднс» — к реактиву
рированной НС1 кристалли-
ческого осадка хлорокиси
циркония ZrOCb • 8Н2О. Для приготовления раствора перхло-
рата циркония навеску хлорокиси циркония 0,36 г растворяют
в фарфоровой чашке при нагревании в рассчитанном количест-
ве хлорной кислоты и упаривают раствор до удаления паров
соляной кислоты.
Соль перхлората циркония растворяют в 2 н. хлорной кис-
лоте, переносят в мерную колбу емкостью 1 л и доводят объ-
ем до 1 л той же кислотой — раствор А. Полученный раствор
содержит 0,1 мг Zr в 1 мл. 10 мл раствора А разбавляют 2 н.
НСЮ4 в мерной колбе на 100 мл — раствор Б. 1 мл раствора
Б содержит 10 мкг циркония.
2. «Спаднс», 0,1%-ный водный раствор.
3. Хлорная кислота, 2 н. раствор.
211
4. Едкий натр, 1 н. раствор.
К 3,5 мл испытуемого раствора циркония, помещенного
в мерную колбу на 25 мл, добавляют 2 мл 0,1%-ного раство-
ра реактива «спаднс», 6 мл раствора щелочи и доводят объ-
ем раствора до метки. Через 10—15 мин измеряют светопо-
глощение на СФ-4, СФ-5 при X 580 мк (рис. 64). В качестве
раствора сравнения берут раствор, содержащий 2 мл 0,1 %-ко-
го раствора «спаднс» в 25 .мл конечного объема (поглощение
реактива не зависит от кислотности раствора). Для построения
калибровочной кривой готовят 7 эталонных растворов с со-
держанием Zr от 5 до 35 мкг, добавляют в первые 6 колб
раствор НСЮ4 до объема 3,5 мл и проводят реакцию полу-
чения соединения циркония со «спаднс» в условиях, указан-
ных выше для определения циркония в испытуемом растворе.
Полученные растворы фотометрируют, строят график в
координатах D—с и по нему определяют концентрацию цир-
кония в испытуемом растворе.
Определение циркония методом спектрофотометрического
титрования 1
Цирконий с комплексоном III и п-нитробензолазопирока-
техином образует комплексные соединения различной устой-
чивости, обладающие поглощением в разных областях спект-
ра. Область максимального поглощения комплексного соеди-
нения циркония с n-нитробензолазопирокатехином лежит при
510 льи/с, комплексного соединения циркония с комплексо-
ном — при 375 лъмк:. Поэтому п-нитробензолазопирокатехин
может быть использован в качестве индикатора.
Если индикатор добавлен в количестве, недостаточном
для связывания всего циркония в комплексное соединение, то
при титровании раствором комплексона сначала титруются
свободные ионы циркония, а затем связанные в комплексное
соединение с индикатором. Получается кривая титрования,
соответствующая рис. 22, а (стр. 59). Установлено, что в пре-
деле концентраций циркония 10 мкг—1 мг растворы подчи-
няются основному закону светопоглощения.
Кроме указанных на стр. 211, необходимы реактивы:
1. Раствор комплексона III. Готовят раствор из препа-
рата квалификации х. ч. по точной навеске. Определение кон-
центрации его проводят методом спектрофотометрического
титрования по соли циркония известной концентрации.
2. п-Нитробензолазопирокатехин, 0,01 % -ный спиртовый
раствор.
3. Соляная кислота 1 : 1.
1 Разработано В. М. Пешковой, Н. В. Мельчаковой, Л. С. Тараруш-
кииой на кафедре аналитической химии МГУ.
212
а. Определение циркония в растворе чистой соли
В мерной колбе на 25 мл готовят испытуемый раствор со-
ли перхлората циркония с содержанием циркония от 0,1 до
5 мг. В кювету (в которой проводят титрование) переносят
5 мл испытуемого раствора, добавляют раствор хлорной кис-
лоты до объема 18—19 мл и 0,2 мл раствора индикатора и
помещают кювету в спектрофотометры СФ-4, СФ-5. Раствор
комплексона прибавляют из микробюретки отдельными пор-
циями по 0,1 мл, перемешивают и измеряют оптическую плот-
ность при 510 лиик после прибавления каждой порции реак-
тива. По полученным данным строят кривую титрования, на-
ходят точку эквивалентности графическим путем и вычис-
ляют содержание циркония.
б. Определение циркония в магниевом сплаве
Точную навеску сплава (0,1—0,15 г) растворяют в 10 мл
соляной кислоты и раствор упаривают до небольшого объема
(— 1 мл), затем добавляют 30 мл хлорной кислоты и вновь
упаривают до удаления паров соляной кислоты. Раствор пе-
реносят в мерную колбу на 50 мл и доводят объем до метки
раствором хлорной кислоты. Переносят в кювету аликвотную
часть (15—20 мл) и проводят титрование точно в условиях,
указанных ранее для определения циркония в растворе его
чистой соли.
Примечание. Настраивают спектрофотометр перед титрованием
следующим образом:
1) компенсируют темновой ток;
2) наполняют кювету дистиллированной водой и компенсируют фото-
ток (при открытой шторке-переключателе) потенциометром чувствитель-
ности и щелью при выбранной для титрования длине волны;
3) наполняют кювету анализируемым раствором и фиксируют показа-
ние оптической плотности после прибавления очередной порции реагента.
Компенсация на «нулевой» раствор (дистиллированную воду) в про-
цессе всего титрования остается постоянной.
Определение циркония реагентом арсеназо III (бензол-2'-
арсоновая кислота-) 1'-азо-2(-бензол-2"-арсоновая кислота-)
1-азо-7 (-1,8-диоксинафталин-3,6-дисульфокислота) методом
спектрофотометрического титрования 1
AsO3Ha НО ОН HaO3As
z____I I I I___
N=N— N=N—У 4,
HO3S SO3H
1 H. В. Мельчакова, M. H. Станиспавская, В. M. Пешко-
в а. ЖАХ, 19, вып. 6, 701, 1964.
213
При определении циркония реагентом арсеназо III ис-
пользовали метод прямого безындикаторного титрования
(стр. 61). Реагент арсеназо III, предложенный С. Б. Савви-
ным 1 в 1959 г. и успешно примененный для прямого спектро-
фотометрического определения циркония2, может быть ис-
пользован также и в методе спектрофотометрического титро-
вания. Из рис. 65 видно, что реагент максимально поглощает
при X 535 ммк; у комплексного соединения циркония с арсе-
Рис. 65. Спектры поглощения
в 6 и. НСЮг. 1 — арсеиазо III;
2 — комплексного соединения
циркония с арсеиазо III
Рис. 66. Зависимость объема арсеиа-
зо III, который пошел иа титрование,
от концентрации циркония в 2 и. (7)
ибн. (2) НС1О4; Л 670 ммк
назо III имеются две максимальные полосы поглощения при
X 620 ммк и X 670 ммк. Чувствительность и селективность,
метода увеличивается, если титрование проводится при
X 670 ммк и кислотности 6 н. по НС1О4.
Кроме указанных на стр. 211 и 212 необходимы реактивы:
1. Арсеназо III, водный раствор с концентрацией!
— 10~3 моль/л (добавляют 1—2 мл 2 н. НС1О4 на 100 мл
раствора). В работе используют эмпирический титр раствора
арсеназо III, определяемый титрованием соли циркония из-
вестной концентрации.
2. Хлорная кислота, 6 н. раствор.
1 С. Б. С а в в и и. ДАН СССР, 127, 1231, 1959.
2 В. Г. Горюшииа, Е. В. Романова. «Зав. лаб.», 26, 415, I960;
В. Г. Горюшина, Е. В. Романова, Т. А. Арчакова. «Зав. лаб.»,
27, 795, 1961; С. В. Е л и н с о и, Н. А. Мирзоян. «Зав. лаб.» 27, 798.
1961. .
214
При использовании арсеназо III для титрования необхо-
димо провести предварительное исследование для выяснения
степени чистоты реактива, т. е. установить, есть ли примеси,
имеющие поглощение при используемой длине волны X. Для
этого готовят раствор соли циркония, содержащий в объеме
25 мл 6 н. НС1О4 около 100 мкг циркония. В кювету спек-
трофотометра вводят последовательно по 0,5; 1,0; 1,5; 2,0 мл
раствора циркония, добавляют 6 н. НСЮ4 до закрытия око-
шечка кюветы (стр. 108) и титруют из бюретки раствором арсе-
назо III, добавляя по 0,1 мл и учитывая наблюдаемые зна-
чения оптической плотности D раствора. По эксперименталь-
ным данным строят кривые титрования в координатах
D — Vapc in и находят экстраполяцией точку эквивалентности.
Найденные значения наносят на график в координатах
Vapcш = f ([Zr]) (рис. 66). Все точки должны лежать на пря-
мой; при экстраполяции на ось ординат получают отрезок,
который дает поправку, учитывающую объем арсеназо III,
идущий на титрование посторонних поглощающих веществ.
Эту поправку вносят при титровании раствора с неизвестным
содержанием циркония. Использованный прием можно реко-
мендовать и в других случаях титрования, если не получают
пропорциональной зависимости в координатах D—V.
а. Определение циркония в растворе чистой соли
В мерной колбе на 25 мл готовят испытуемый раствор со-
ли перхлората циркония с содержанием от 0,1 до 1 мг цир-
кония. В кювету для титрования переносят 0,5 мл испытуе-
мого раствора, добавляют раствор хлорной кислоты до объ-
ема 18—20 мл и помещают кювету в спектрофотометр (СФ-4
или СФ-5). Раствор арсеназо III добавляют из микробюретки
отдельными порциями по 0,1 мл, перемешивают и измеряют D
при X 670 ммк после прибавления каждой порции реагента.
По полученным данным строят кривую титрования, находят
точку эквивалентности графическим путем и вычисляют содер-
жание циркония.
б. Определение циркония в сплавах на основе магния
и алюминия
Точную навеску сплава (01—0,15 г) растворяют в 10 мл со-
ляной кислоты и раствор упаривают до небольшого объема
( 1 мл), добавляют 30 мл 6 н. хлорной кислоты и вновь упа-
ривают до удаления паров соляной кислоты. Раствор перено-
сят в мерную колбу на 50 мл и доводят объем до метки рас-
твором хлорной кислоты. Переносят в кювету аликвотную
часть и проводят титрование точно в условиях, указанных для
определения циркония в растворе его чистой соли.
215
ОПРЕДЕЛЕНИЕ РЕДКОЗЕМЕЛЬНЫХ ЭЛЕМЕНТОВ
Спектрофотометрическое определение индивидуальных
редкоземельных элементов (р. з. э.) возможно в растворах, со-
держащих в качестве посторонних ионов только неорганиче-
ские анионы.
Спектры поглощения растворов солей редкоземельных
элементов ввиду особенностей строения 4 f-оболочки состоят
из определенного числа довольно узких максимумов, распо-
ложенных при определенных длинах волн.
Ввиду того что спектр поглощения каждого элемента этой
группы имеет строго индивидуальный характер, можно про-
вести качественный и количественный анализ смеси солей
этих элементов, имея спектрофотометр с достаточно высокой
монохроматизацией потока лучистой энергии, иначе невоз-
можно будет разрешить очень близко расположенные (на
расстоянии 5—10 ммк) узкие максимумы поглощения. Спек-
трофотометрическому анализу (качественному и количествен-
ному) смеси редкоземельных элементов обычно предшествует
выделение суммы этих элементов из анализируемого объекта
в виде фторидов или оксалатов.
Использование органических реагентов, хотя и повышает
чувствительность определения в тысячи и даже десятки ты-
сяч раз, приводит, однако, к потере специфичности, так как
благодаря сходству в химических свойствах элементов этой
группы они вступают в реакции с одними и теми же реаген-
тами и в одинаковых условиях.
Отдельные элементы, например лантан, церий и европий,
можно определить в присутствии других элементов этой груп-
пы, используя более основной характер первого и способ-
ность последних легко давать соединения с переменной ва-
лентностью.
Молярные коэффициенты погашения соединений редкозе-
мельных элементов с какими-либо органическими реагентами
также очень близки. Поэтому определение одного элемента
в присутствии других элементов данной группы с использо-
ванием органических реагентов удается провести очень редко
и требует специальной математической обработки данных
спектрофотометрических измерений. В большинстве случаев
органические реагенты используются для определения суммы
элементов этой группы. Такими реагентами являются ализа-
рин красный S', алюминон1 2, салицилфлуорон3, нафтазарин4.
1 R. W. Rin echard. «Analyt. Chem.», 26, 1820, 1954; Л. С. Сер-
дюк, Г. Ф. Федорова. ЖНХ, 4, 88, 1959.
2 L. Holleck, L. Hartinger, D. Eckard t. «Angew, Chem.», 65,
347, 1953; L. Holleck, D. E c k a r d t. «Z. anal. Chem.», 146, 103, 1955.
3 Ф. В. 3 а й к о в с к и й, Г. Ф. Садова. ЖАХ, 16, 29, 1961.
4 Th. Moeller, М. Teckotzky. «J. Am. Chem. Soc.», 77, 2649, 1955-
216
В последнее время широко используют реагенты группы
о'-арсоно-о'-оксиазосоединений (арсеназо I1 и арсеназо III2)
и реагенты, производные трифенилметана (ксиленоловый оран-
жевый3 и его аналоги). Эти же реагенты используют как
индикаторы при комплексонометрическом титровании редко-
земельных элементов (визуальном и спектрофотометриче-
ском) .
В настоящем руководстве даны лишь примеры, иллюстри-
рующие основные принципы спектрофотометрического анали-
за смеси на содержание индивидуальных редкоземельных
элементов, и некоторые методы определения суммы редкозе-
мельных элементов. Особое внимание уделено методам спек-
трофотометрического титрования редкоземельных элементов,
которые, обладая такой же высокой чувствительностью, дают
более высокую точность определения, чем прямые спектрофо-
тометрические методы.
Спектрофотометрическое определение индивидуальных
редкоземельных элементов в смеси неорганических солей
этих элементов
а. Качественный анализ
Для проведения качественного анализа раствора солей
редкоземельных элементов снимают спектр поглощения ана-
лизируемого раствора и определяют наличие характерных
максимумов для тех или других элементов.
Контрольный раствор смеси солей редкоземельных эле-
ментов помещают в кювету (прямоугольную или цилиндри-
ческую). Снимают спектр поглощения этого раствора по от-
ношению к дистиллированной воде в интервале длин волн от
220 до 1100 ммк\ первоначально измерения производят че-
рез 5—10 ммк и в области намечающихся максимумов по-
вторяют исследование спектра через 0,5—1 ммк. Пользуясь
кривыми поглощения растворов солей редкоземельных эле-
ментов (стр. 229). определяют, какие из них присутствуют в
смеси.
1 В. И. к У 3 н е ц о в. ЖАХ, 7, 226, 1952; В. И. К У з н е ц о в, Л. М. Б у-
данова, Т. В. Матросова. «Зав. лаб.», 22, 406, 1956; А. Ф. Кутей-
ников. «Бюл. ВИМС», № 5 (169), 1, 1957; Y. Fritz, М. Richard,
W. J. L а п е. «Analyt Chem.», 30, 1776, 1958.
2 S. В. Savvin. «Taianta», 8, № 9, 673, 1961; В. Г. Г о р ю ш и н а,
С. Б. С а в в и н, Е. В. Р о м а н о в а. ЖАХ, 18, 1340, 1963.
3 Л. С. Сердюк, В. С. Смирная. ЖАХ, 19, 451, 1964; Д. И. Ряб-
чиков, В. А. Рябухин. Методы определения и анализа р. з. э. М., Изд-
во АН СССР, 1961, стр. 159.
217
б. Определение количественного содержания редкоземельных
элементов
Рядом исследований * было установлено, что величины
молярных коэффициентов погашения в максимумах поглоще-
ния растворов неорганических солей редкоземельных элемен-
Таблица 7
Молярные коэффициенты погашения растворов солей редкоземельных
элементов (по Моллеру и Брантли)*
Соль \макс* ммк e Ширина щели, мм Минималь- ная кон- центрация, г/л
PrCl3 444,5 9,85 0,025 0,143
Pr(NO3)3 444,5 9,20 0,025 0,153
NdCl3 521,8 4,33 0,013 0,333
Nd(C104)3 521,8 4,30 0,013 0,336
Nd(NO3)3 521,8 3,88 0,013 0,372
Nd(C2H3O2)3 523,0 3,80 0,013 0,380
SmCl3 402,0 3,14 0,044 0,480
Sm(NO3)3 402,0 3,28 0,044 0,458
Sm(C2H3O2)3 402,5 3,68 0,044 0,410
EuCl3 393,9 2,92 0,029 0,520
Eu(NO3)3 394,5 2,05 0,053 0,743
Eu(C2H3O2)3 394,7 1,92 0,053 0,793
GdCl3 272,8 2,34 0,205 0,640
Gd(C2H3O2)3 272,9 3,28 0,205 0,479
Er(C104)3 379,2 4,00 0,030 0,419
TmCl3 682,5 2,58 0,018 0,659
Tm(C104)3 682,5 2,49 0,018 0,680
YbCI3 975,0 1,94 0,026 0,901
Yb(NO3)3 975,0 1,91 0,026 0,912
Yb(CIO4)3 975,0 1,96 0,026 0,885
Yb(C2H3O2)3 972,0 1,99 0,026 0,864
* Th. Moeller, J. Brantley. «Anal. Chem.», 22, 433, 1950.
tob могут быть использованы для определения количествен-
ного содержания отдельного элемента в их смеси. Выбор ха-
рактерных максимумов определяется отсутствием интерфе-
ренции при этой длине волны со стороны сопутствующих
элементов. В табл. 7 и 8 приведены длины волн и соответ-
ствующие им величины молярных коэффициентов погаше-
ния, которые могут быть рекомендованы (по данным двух
1 Th. М о е 11 е г, J, В г a n 11 е у. «Anal. Chem.», 22, 433, 1950' F S ped-
rf i n g, А. V о i g h t, Е. G 1 a d г о w, N. S 1 е i g h t, J. Р о w е 11, J. М г i g h t,
T. Butler, P. F i g a r d. «J. Amer. Chem. Soc.», 69, 2786, 1947; F. S p e d-
d i n g, E. Fulmer, T. Butler, E. G 1 a d г о w, M. G о b u s’h, P. P о r-
ter, J. Powe 1, J. Wright. «J. Amer. Chem Soc», 69, 2812 1947-
D. S t e v a r t, D. Kato. «Anal. Chem.», 30, 164, 1958; L. H о 11 e c k, L. Ha-
rt i n g e r. «Angew. Chem.», 67, 648, 1955
218
Таблица 8
Молярные коэффициенты погашения в характерных максимумах
поглощения растворов неорганических солей редкоземельных
элементов (по Холлек и Хартингер) *
Соль Хмакс- MMK e Минимальная концентрация, г/л Мешающие элементы
PrCl3 444,0 10,50 0,78
NdCl3 575,2 7,11 0,78 Рг
NdCl3 521,9 4,51 1,56
SmCl3 401,6 3,49 1,56 Ег
EuCl3 394,3 2,7 1,56 Sm
GdCl3 272,9 3,53 2,9 Sm, Dy, Но, Ег
DyCl3 907,5 2,15 3,13 Но, Er, Yb
DyCl3 350,0 2,44 6,25 Но
HoCl3 536,8 4,26 0,78 —
HoCl3 640,7 2,81 3,13 Ег
ErCl3 523,0 3,04 3,13 Но
ErCl3 487,1 1,92 3,13 Но
TuCl3 682,5 ' 2,52 2,70 Ег
TuCl3 777,5 1,07 2,70 —(
YbCl3 972,5 1,90 1,56 Ег
‘L. Holleck, L. Hartinger. «Angew. Chem.», 67, 648, 1955.
авторов) для количественного анализа смеси солей редкозе-
мельных элементов. Однако следует помнить, что значения
молярных коэффициентов погашения несколько меняются в
зависимости от присутствующего аниона (табл. 7). В при-
сутствии лигандов смещается также положение максимума
поглощения 1 и возрастает величина молярного коэффициента
погашения, что используется в ряде работ для повышения
чувствительности данного метода 2.
Контрольный раствор соли редкоземельного элемента илр:
смеси элементов этой группы, качественный состав которой
известен, помещают в кювету (прямоугольную или цилиндри-
ческую, предварительно высушенную). Пользуясь кривыми
спектров поглощения растворов солей редкоземельных эле-
ментов (стр. 229), определяют область длин волн, в которой
имеются характерные для данного элемента полосы поглоще-
ния. Снимают спектр поглощения контрольного раствора в
1 В. М. Пешкова, М. И. Громова. ЖНХ, 11, 1356, 1957;
В. М. Пешкова, М. И. Громова, Н. А. Канаев, И. П. Ефи-
мов. Сб. «Редкоземельные элементы». М., Изд-во АН СССР, 1958, стр. 277.
2 Н. С. Полуэктов, В. Т. Мищенко. ЖАХ, 17, 825, 1962;
К. В. Церкасевич, Н. С. Полуэктов. ЖАХ, 19, 1309, 1964;
М. И. Громова, Т. И. Романцева, В. М. Пешкова. «Вести. Моск,
ун-та», сер. хим., № 4, 57, 1964; Th. Moeller, D. Jackson. «Analvt.
Chem.», 22, 1393, 1950.
219
найденном интервале длин волн по отношению к дистиллиро-
ванной воде, производя измерения через 0,5—1 ммк. Поль-
зуясь табл. 7, вычисляют концентрацию исследуемого рас-
твора на основании измеренной величины оптической плот-
ности в максимуме поглощения.
Определение редкоземельных элементов
арсеназо III
Комплексные соединения редкоземельных элементов с ар-
сеназо III имеют два максимума поглощения: при X 600 и
650 ммк (рис. 67). Максимумы поглощения при этих же дли-
Рис. 67. Спектры поглощения растворов комплексного соедине-
ния р. з. э. с арсеназо III при pH: 1 — 2,35; 2а—1,5—-3,3; 26—
4,35; 2в—-8,37
нах волн имеет сам реагент в области значений pH > 3. Од-
нако оптимальным для образования комплексного соединения
является интервал значений pH 2—4. Кроме того, для самого
реагента оба эти максимума поглощения являются в равной
мере характерными, в то время как для комплексного соеди-
нения более характерен максимум поглощения при X 650 ммк;
поглощение при этой длине волны значительно больше, чем
при X 600 ммк. Поэтому определение редкоземельных элемен-
220
тов арсеназо III проводят, измеряя оптическую плотность
растворов, имеющих pH 2,5—3 при X 650 ммк. (В данном ме-
тоде не рекомендуется использовать буферные растворы для
создания необходимого значения pH, так как это снижает
оптическую плотность растворов).
Необходимые реактивы:
1. Раствор соли редкоземельного элемента, содержащий
0,1 мг в 1 мл.
Точную навеску свежепрокаленной окиси р. з. э. раство-
ряют в разбавленной соляной кислоте, взятой с небольшим
избытком. Раствор упаривают досуха на водяной бане, при-
ливают небольшое количество воды и слегка нагревают до
полного растворения полученной соли; переносят в мерную
колбу и доводят объем раствора до метки колбы водой. Раз-
бавлением этого раствора готовят раствор с содержанием
10 мкг р. з. э. в 1 мл.
2. Арсеназо III, 0,015%-ный водный раствор.
3. Хлорная кислота, 0,08 н. раствор.
К испытуемому раствору, содержащему 4—40 мкг редко-
земельного элемента и помещенному в мерную колбу на
25 мл, прибавляют 6—7 мл раствора арсеназо III, 1 мл хлор-
ной кислоты и доводят объем до метки колбы водой. Изме-
ряют оптическую плотность полученного раствора по отноше-
нию к воде при X 650 ммк, используя спектрофотометр. Со-
держание р. з. э. в испытуемом растворе определяют по гра-
дуировочной кривой, построенной по данным измерений опти-
ческих плотностей 5 эталонных растворов, содержащих р. з. э.
в количествах (мкг)-. 5, 10, 20, 30, 40 и приготовленных в
тех же условиях, что и испытуемый.
Определение редкоземельных элементов 1 методом
спектрофотометрического титрования
Как сумма редкоземельных элементов, так и отдельные
элементы этой группы могут быть определены методом спек-
трофотометрического титрования. В качестве реактивов мо-
гут быть использованы такие комплексообразующие лиганды,
как этилендиаминтетрауксусная кислота (обычно использует-
ся ее двунатриевая соль —комплексон III) или триоксиглута-
ровая кислота.
Индикаторами в этом случае могут служить комплексооб-
разующие реагенты, дающие с редкоземельными элементами
комплексные соединения, имеющие характерное поглощение
при определенной длине волны, например арсеназо I или эри-
1 В. М. Пешкова, М. И. Громова, И. П. Ефимов, А. В. Иса-
ченко. «Вести. Моск, ун-та», сер. хим., № 4, 59, 1961.
221
охром черный Т. Однако арсеназо I как индикатор имеет це-
лый ряд преимуществ, так как его растворы в воде устой-
чивы довольно длительное время, не подвергаются окисле-
нию кислородом воздуха, как это имеет место в случае инди-
катора эриохром черный Т.
а. Спектрофотометрическое титрование редкоземельных
элементов в растворах их чистых солей комплексоном III
с арсеназо I (бензол-2'-арсоновая кислота-(Г-азо-2)-1,8-
диоксинафталин-3,6-дисульфокислота) в качестве индикатора
Возможность спектрофотометрического титрования редко-
земельных элементов комплексоном III с арсеназо I в каче-
стве индикатора обусловлена меньшей устойчивостью ком-
плексных соединений этих элементов с индикатором, чем
с комплексоном III. Комплексное соединение редкоземельного
элемента с арсеназо I имеет максимум поглощения в области
л 570—575 ммк, в то время как сам реактив максимально по-
глощает при X 510 ммк. Титруя редкоземельные элементы
раствором комплексона III в присутствии арсеназо I, наблю-
дают при X 575 ммк постепенное уменьшение оптической
плотности в результате перехода редкоземельных элементов
из комплексного соединения с арсеназо I в соединение с ком-
плексоном III. (Комплексное соединение редкоземельного
элемента с комплексоном III в данной области длин волн не
поглощает.) Таким образом, в процессе титрования получают
ход кривой, представленный на рис. 22, а (стр. 59).
Необходимые реактивы:
1. Комплексон III, 1 -10“4М раствор. Раствор готовят по
точной навеске и концентрацию его проверяют титриметри-
чески по раствору цинка с эриохром черным Т в качестве ин-
дикатора. В случае необходимости комплексон III можно
очистить перекристаллизацией из спирта.
2. Арсеназо I, 1 • 10-5М раствор.
3. Уротропин, 25%-ный раствор.
4. Соляная кислота, 0,1 н. раствор.
5. Аскорбиновая кислота, 1%-ный раствор (свежеприго
товленный!).
К раствору соли редкоземельного элемента (отдельному
или смеси), помещенному в мерную колбу на 50 мл и содер-
жащему редкоземельный элемент в количестве от 0,5 мг до
5 мкг, прибавляют 5 мл раствора арсеназо I, 0,5 мл соляной
кислоты и 1,5 мл уротропина (для создания pH 6,5) и дово-
дят объем раствора водой до метки колбы. После перемеши-
вания отбирают пипеткой 20 мл этого раствора и переносят в
кювету для титрования. Настройку спектрофотометра произ-
водят так, как указано на стр. 213 при титровании циркония.
222
Раствор комплексона III прибавляют из микробюретки
отдельными порциями по 0,1 мл, перемешивают и измеряют
оптическую плотность при X 575 ммк после прибавления каж-
дой порции реактива. По полученным данным строят кривую
зависимости D от V, графически определяют момент эквива-
лентности и вычисляют количество редкоземельного элемен-
та, учитывая, что редкоземельные элементы образуют с ком-
плексоном III соединение в соотношении 1:1.
Примечания. 1. При титровании суммы редкоземельных элемен-
тов в присутствии церия необходимо добавлять 3 мл аскорбиновой кислоты.
2. Следует помнить, что для получения надежных результатов концен-
трация реагента (комплексона III) не должна превышать концентрацию
определяемого элемента более чем в 100 раз, а концентрация индикатора
должна быть одного и того же порядка илн в крайнем случае ниже на
порядок, чем концентрация определяемого элемента.
3. При определении 2 р. з. э. расчет производят по среднему атом-
ному весу.
б. Определение редкоземельных, элементов в присутствии
тория 1
Метод спектрофотометрического титрования редкоземель-
ных элементов комплексоном III в присутствии арсеназо I
как индикатора может быть также использован для опреде-
ления этих элементов в присутствии тория. Возможность эта
вытекает из того факта, что комплексное соединение тория
с арсеназо I, которое максимально поглощает в той же об-
ласти, что и соединение с редкоземельными элементами, об-
разуется при pH 2, в то время как соединение редкоземель-
ного элемента с этим реагентом образуется при pH ~ 6—7.
Кроме того, торий образует с комплексоном III более устой-
чивое соединение, чем с р. з. э. Таким образом, имеется воз-
можность провести последовательное титрование сначала то-
рия при pH 2, а затем редкоземельного элемента при pH — 6,5.
При этом добавления индикатора перед титрованием не тре-
буется, так как индикатор «регенерируется» в процессе тит-
рования тория. Однако этот метод дает удовлетворительные
результаты только при титровании смесей, содержащих экви-
валентные или меньшие количества тория в сравнении с ко-
личествами редкоземельного элемента (отношение Th: р. з. э. =
= 1:1, 1 : 10, 1 : 100), а не наоборот.
Методика определения следующая. К раствору, содержа-
щему 5 мкг — 0,5 мг редкоземельного элемента и ~5 мкг
тория, помещенному в мерную колбу на 50 мл, прибавляют
5 мл арсеназо I, 1 мл соляной кислоты для создания pH 2
и доводят объем раствора до метки колбы дистиллированной
водой. По,еле перемешивания 20 мл этого раствора отбирают
пипеткой, переносят в кювету и титруют раствором комплек-
1 В. М. Пешкова, М. И. Громова, Н М. Александрова.
ЖАХ, XVII, 218, 1962.
223
сона III, прибавляя его по 0,1 мл из микробюретки и измеряя
оптическую плотность после каждой прибавленной порции
реактива. Когда значения оптической плотности станут по-
стоянными, создают в растворе pH 6,3, прибавив 1 мл уро-
тропина, и продолжают титрование. Кривая зависимости D
от V будет иметь два излома: один,— соответствующий коли-
честву тория, второй — редкоземельного элемента. Графиче-
ски определяют момент эквивалентности и вычисляют содер-
жание тория и редкоземельного элемента.
Если содержание тория превосходит содержание р. з. э.
более чем в 10 раз, то совместное титрование также возмож-
но. Но в этом случае содержание р. з. э. вычисляется по раз-
ности результатов титрования двух отдельных проб, прове-
денных при pH 2 и pH 6,5.
ОПРЕДЕЛЕНИЕ МЕДИ
Определение больших количеств меди
дифференциальным спектрофотометрическим
методом 1
Определение меди производят в виде аммиачного ком-
плексного соединения. Светопоглощение растворов измеряют
со светофильтром, имеющим ХЭф 610 ммк. Работа может быть
выполнена на фотоэлектроколориметрах ФЭК-56, ФЭК-Н-57.
Необходимые реактивы:
1. Стандартный раствор соли меди с содержанием 2 мг в
1 мл. Для приготовления стандартного раствора навеску
сульфата меди, квалификации х. ч. с известным содержанием
меди, соответствующую 0,5 г меди, помещают в мерную кол-
бу на 250 мл растворяют в воде, подкисленной 1 мл концен-
трированной серной кислоты, доводят водой до объема 250 мл
и тщательно перемешивают; 1 мл раствора содержит 2 мг
меди.
2. Аммиак, водный раствор, концентрированный.
а. Определение меди в водном растворе его чистой соли
10 мл испытуемого раствора, содержащего от 5 до 15 мг
или от 10 до 20 мг, помещают в мерную колбу на 50 мл, до-
бавляют 5 мл аммиака, доводят объем раствора до метки
колбы водой и измеряют оптическую плотность. В тех же
условиях готовят 5—6 эталонных растворов с содержанием
меди в указанных выше пределах, с интервалом в 2 мг меж-
ду соседними растворами. В качестве «нулевого», как при
измерении оптической плотности эталонных растворов, так и
1 Т. Г. Малкина, В. Н. Подчайнова. ЖАХ, XIX, вып. 6, 668,
1964.
224
контрольного, используют раствор, содержащий 5 или 10 мг
меди в зависимости от того, в каком интервале концентраций
производится работа (5—15 мг или 10—20 мг).
Установку прибора на нуль производят, поместив в оба
световых потока кюветы с одним и тем же «нулевым» рас-
твором. Содержание меди в контрольном растворе рассчиты-
вают по предварительно построенному калибровочному гра-
фику или по формуле (109)
= DxP + Q,
вычислив предварительно фактор F следующим образом. Из-
меряют оптическую плотность Dt каждого эталонного рас-
твора по отношению к первому из них; затем те же измере-
ния производят по отношению ко второму раствору всех по-
следующих растворов, по отношению к третьему раствору
и т. д.
По формуле (114)
р — Ci ~ С1
D{
вычисляют значения фактора для каждого измерения и из
полученных результатов находят его среднее значение. Изме-
ряют оптическую плотность испытуемого раствора Dx по от-
ношению к одному из растворов эталонного ряда Ci и вычис-
ляют его концентрацию по формуле (109).
б. Определение меди в бронзе и томпаке
Кроме указанных на стр. 204 необходимы реактивы:
1. Азотная кислота х. ч. 1:1.
Навеску 0,5 г исследуемого образца бронзы и томпака
растворяют при легком нагревании в 25 мл азотной кислоты,
раствор упаривают до небольшого объема, переносят в мер-
ную колбу емкостью 50 мл и разбавляют водой до метки
колбы. 1—2 мл (в зависимости от содержания меди) пере-
носят в мерную колбу на 50 мл, добавляют 5 мл аммиака,
разбавляют водой до метки колбы и тщательно перемеши-
вают.
Содержание меди в контрольном образце определяют по
предварительно построенному калибровочному графику или
по формуле (109), вычислив предварительно фактор F, как
указано выше при определении меди в растворе чистой соли.
ЗАДА ЧИ
При определении марганца в виде перманганата оп-
тическая плотность раствора (X 525 ммк), содержащего
0,12 мг Мп в 100 мл, при толщине слоя в 3 см оказалась
равной 0,152. Вычислить молярный коэффициент погаше-
ния 8.
225
2. Оптическая плотность раствора формальдоксимата
Марганца при начальной концентрации Мп, равной 0,07 мг в
J50 мл, измеренная при X 455 ммк и толщине слоя кюветы
в 1 см, оказалась равной 0,280. Вычислить значение кажуще-
гося молярного коэффициента погашения.
3. Оптическая плотность раствора трисульфосалицилата
железа, измеренная при X 433 ммк, в кювете с толщиной слоя
в 2 см равна 0,149. Для реакции было взято 4 мл 0,0005820 М
раствора железа и колориметрическая реакция была прове-
дена в колбе на 50 мл. Вычислить значение кажущегося мо-
лярного коэффициента погашения для данной реакции при
X 433 ммк.
4. При спектрофотометрическом определении никеля в ви-
де соединения с диметилглиоксимом в присутствии окислителя
в щелочной среде для раствора с концентрацией никеля
0,025 мг в 50 мл было получено значение оптической плот-
ности, равное 0,324 (при измерении в кювете с толщиной
слоя 2 см; X 470 ммк). Вычислить значение кажущегося мо-
лярного коэффициента погашения.
5. При определении железа в виде моносульфосалици-
лата оптическая плотность раствора, содержащего 0,23 мг Fe
в 50 мл, оказалась равной 0,264. (Толщина слоя кюветы была
равна при этом 2 см.) Вычислить значение кажущегося мо-
лярного коэффициента погашения.
6. Пропускание испытуемого раствора равно 62,3%. Ка-
кова оптическая плотность данного раствора?
7. Оптическая плотность исследуемого раствора равна
0,205. Вычислить, каково пропускание этого раствора в про-
центах.
8. Для определения железа методом колориметрического
титрования было взято 15 мл испытуемого раствора. На ти-
трование пошло 4,3 мл стандартного раствора железа, содер-
жащего 0,01 мг в 1 мл. Вычислить молярную концентрацию
испытуемого раствора.
9. Для определения марганца в стали методом колоримет-
рического титрования было взято 15 мл раствора, приготовлен-
ного растворением 0,1 г образца стали в 50 мл, и проведено
окисление марганца до перманганат-иона. На титрование по-
шло 4,5 мл стандартного раствора перманганата, содержа-
щего 0,01 мг Мп в 1 мл. Вычислить процентное содержание
марганца в испытуемом образце стали.
10. Содержание перманганата в эталонном растворе в мо-
мент уравнивания интенсивностей окраски растворов по ме-
тоду разбавления равно 1,55 мг. Вычислить содержание мар-
ганца в граммах в контрольном растворе, если объемы эта-
лонного и контрольного растворов в момент совпадения ин-
тенсивности их окраски были соответственно равны 32 5 мл.
и 23,4 мл.
226
11. При колориметрическом определении железа по ме-
тоду изменения толщины поглощающего слоя было вычис-
лено, что содержание железа в контрольном растворе равно
0,173 мг. Каково процентное содержание железа в анализи-
руемом образце алюминия, если навеска этого образца в 0,2 г
была растворена в объеме 100 мл, а для колориметрической
реакции было взято 0,2 мл?
12. При колориметрическом определении марганца с фор-
мальдоксимом был использован метод изменения толщины
поглощающего слоя. При этом высота столбов эталонного и
контрольного растворов в момент уравнивания интенсивности
окраски полей окуляра в колориметре были соответственно
равны 20,5 и 17,3 при содержании марганца в эталонном рас-
творе 0,05 мг. Какова молярная концентрация испытуемого
раствора, если для колориметрической реакции было взято
5 мл этого раствора?
13. Содержание молибдена в стали составляет при-
мерно 0,3%. Какова должна быть навеска этой стали, рас-
творенная в 100 мл, чтобы содержание молибдена в испы-
туемом растворе, приготовленном перенесением 20 мл исход-
ного раствора стали для проведения колориметрической реак-
ции в колбу объемом в 50 мл, не превышало 0,5 мг?
14. Навеску стали, соответствующую 0,1 г, растворили в
100 мл смеси кислот. Для колориметрической реакции на ни-
кель было взято 15 мл полученного раствора. Реакция была
проведена в колбе объемом 50 мл. По калибровочному графи-
ку было найдено, что содержание определяемого компонента
в 50 мл равно 0,123 мг. Вычислить процентное содержание
никеля в стали.
15. Значение кажущегося молярного коэффициента пога-
шения раствора моносульфосалицилата железа равно
1,6- 103. Рассчитать, каково должно быть содержание железа
(в мг) в эталонных растворах, приготовленных в мерных кол-
бах на 100 мл, чтобы оптические плотности при измерении
в кюветах с толщиной слоя в 1 см укладывались в интервал
значений от 0,1 до 1,0.
16. Величина кажущегося молярного коэффициента пога-
шения раствора комплексного соединения равна 15 000. Ка-
кова минимальная концентрация компонента (в г), которую-
можно определить с помощью данной колориметрической
реакции, если эта реакция проводится в мерной колбе на
25 мл и оптическая плотность раствора имеет величину не
ниже 0,1 при толщине слоя кюветы 3 см?
17. Для определения Мп в стали навеску ее в 0,1 г рас-
творили в смеси серной, фосфорной и азотной кислот. В даль-
нейшем раствор разбавили до 100 мл. Для колориметриро-
вания отбирали по 20 мл этого раствора. По калибровочному
графику найдено, что содержание Мп в колориметрируемом
227
растворе равно 0,71 мг. Каково процентное содержание Мп
в стали?
18. Известно, что образец стали содержит около 0,5%
кремния. Какую навеску стали следует растворить в 100 мл,
чтобы, отбирая 25 мл этого раствора в колбу на 50 мл, по-
лучить окраску, по интенсивности соответствующую окраске
раствора, содержащего 0,25 мг в 50 мл раствора?
19. Навеску стали в 0,1 г, содержащей никель, растворили
в азотной кислоте и довели объем раствора до 100 мл. Для
колориметрического определения никеля диметилдиоксимом
было взято 2,5 мл этого раствора. По калибровочному графи-
ку найдено, что количество никеля, содержащееся в этом
растворе, равно 0,014 мг. Каково процентное содержание ни-
келя в стали?
20. Найти процентное содержание Мо в образце стали,
если известно, что навеска стали в 0,2 г была растворена в
100 мл смеси кислот, для определения отбиралось по 25 мл
раствора и найденное по калибровочному графику количе-
ство Мо составляло 0,3 мг.
21. Содержание фосфора в анализируемых силикатах ле-
жит в пределах 1,0—0,5%. Какую навеску силиката следует
брать для анализа, если конечный объем раствора силиката
составляет 100 мл, на анализ фосфора идет 25 мл этого рас-
твора, а эталонный ряд содержит от 2 до 10 мг фосфора в
40 мл раствора?
22. Коэффициент молярного погашения а-фурилдиоксима-
та никеля в хлороформе составляет 19 000. Какое минималь-
ное процентное содержание никеля в чистом алюминии мо-
жет быть определено этим реактивом, если навеска А1 не
должна превышать 1 г, максимальный объем хлороформен-
ного экстракта составляет 10 мл, рабочая длина кювет равна
5 см, минимальная оптическая плотность раствора, при кото-
рой ошибка измерения не превышает 10%, равна 0,020?
23. Необходимо определять Си в полупроводниковых ма-
териалах в количествах 1 -10—6%. Каким минимальным коэф-
фициентом молярного погашения должно обладать комплекс-
ное соединение меди, в виде которого ее определяют спектро-
фотометрически, если навеска образца не должна превышать
1 г, конечный объем измеряемого раствора не должен быть
меньше 5 мл, кювета имеет 1 = 5 см, минимальное значение
оптической плотности, при которой возможна точность не ни-
же 10%, 0,020?
Приложение
СПЕКТРЫ ПОГЛОЩЕНИЯ
РАСТВОРОВ СОЛЕЙ
РЕДКОЗЕМЕЛЬНЫХ ЭЛЕМЕНТОВ
РгС13
6-
4-
2-
220 260 300 340 3S0 420 460 500 540 500
620 660 700 740 780 820 860 300 Л, ммк
Nd С13
2-
* 7 П—J—I—I—I—I—i—I—Г-1—I—Г“1—I—I—Г -Г*‘1-
610 650 690 730 770 810 850 890 930 \ 970 7, ммк
м _________________________________________
ГТ I I I ( I I ""Тл ! I | | I 7 I I
620 660 700 740 780 820 860 900 940 98QA.MMK
Ег(С104)з
-j—r ,т/'<\х,г ‘ Г' T^T~~~i Г i—г-i r-i n
620 660 700 740 780 820 860 800 340 A,MMK
TuClj
IX
2-
820 860 300 340 380 A, MMK
YbCL3
ОГЛАВЛЕНИЕ
Предисловие...................................................... 3
Введение......................................................... 5
I. Теоретические основы фотометрических методов анализа 9
1. Основные законы светопоглощения........................... 9
2. Причины отклонения от основного закона светопоглощения 15
3. Возможности и преимущества спектрофотометрического
метода . 18
4. Исследование фотометрической реакции......................21
5. Точность спектрофотометрического метода...................28
II. Методы определения и расчета концентрации веществ в рас-
творах ..........................................................33
III. Примеры использования спектрофотометрического метода 40
1. Вычисление истинных значений молярных коэффициентов
погашения растворов комплексных соединений .... 40
2. Вычисление констант диссоциации органических реагентов . 45
3. Определение состава комплексного соединения .... 51
4. Расчет констант устойчивости при ступенчатом комплексо-
образовании .................................................53
5. Метод спектрофотометрического титрования..................57
6. Дифференциальный спектрофотометрический метод ... 66
7. Экстракционно-фотометрический метод.......................71
IV. Аппаратура..............................................74
Концентрационный колориметр КОЛ-1М...........................74
Микроколориметр КОЛ-52...................................76
Универсальный фотометр ФМ....................................81
Фотоэлектрический колориметр ФЭК-М...........................85
Фотоэлектроколориметр-нефелометр ФЭК-Н-52, ФЭК-Н-54,
ФЭК-Н-57...................................................90
Фотоэлектроколориметр-нефелометр ФЭК-56..................90
Спектрофотометры СФ-4, СФ-4А и СФ-5..........................96
Регистрирующие спектрофотометры СФ-2М, СФ-10 .... Ю9
Светофильтры................................................114
Фотоэлементы . . ................................ 117
Кюветы.................................................... 121
V. Практические работы.........................................125
1. Определение констант кислотной диссоциации органических
реагентов...................................................125
а. Определение константы диссоциации диметилглиоксима . 125
б. Определение константы кислотной диссоциации нитро-
зо-К-соли................................................127
в. Определение константы кислотной диссоциации тимолсуль-
фофталеина (тимолового синего)...........................128
2. Определение состава комплексных соединений кобальта (II)
с нитрозонафтолами методом изомолярных серий .... 130
3. Примеры, показывающие преимущества более высокой моно-
хроматизации потока лучистой энергии ...................... 131
а. Сравнительное изучение возможностей фотометрических
приборов различного типа ............................. 131
б. Возможность изучения процессов комплексообразования
в растворах солей редкоземельных элементов . . . . 133
в. Повышение чувствительности метода при работе в области
максимального поглощения соединения.......................133
4. Методы определения отдельных элементов..................133
Определение железа........................................133
Определение марганца......................................147
Определение никеля ...................................... 156
Определение кобальта .................................... 168
Определение молибдена.....................................178
Определение алюминия......................................183
Определение кремния и фосфора ........................... 190
Определение цинка ....................................... 201
Определение рения ....................................... 204
Определение циркония .................................... 210
Определение редкоземельных элементов .................... 216
Определение меди..........................................224
Задачи....................................................225
Приложение................................................229
Спектры поглощения растворов солей редкоземельных эле-
ментов ............................................ ... 229
Замеченные опечатки
Страница
Строка
Напечатано
Должно быть
9'
130
131
143
3 снизу
4 снизу
21 сверху
10 и 22
сверху
формула
колориметр-нефелометр ФЭК-56
фотоэлектроколориметр-нефе-
лометр ФЭК-56
5-10-в
5-10-7
колориметр ФЭК-56
фотоэлектроколориметр
ФЭК-56
5-ИГ5
5-10~5
146 20 сверху
21 сверху
154 20 сверху
203 8 и 9 сверху
[FeD2]2-*,
Fe (HD3)2;
Формальдоксим, раствор.
В мерную колбу
[FeD2]2—,
Fe (HD),;
Формальдоксим, свеже-
приготовленный раствор.
В делительную вороику
Зак. 686
в. Определение константы кислотной диссоциации тимолсуль-
фофталеина (тимолового синего)........................128
2. Определение состава комплексных соединений кобальта (II)
с нитрозонафтолами методом изомоляриых серий . . . . 130
3. Примеры, показывающие преимущества более высокой моно-
хроматизации потока лучистой энергии ................ 131
я Спяннительное изучение возможностей фотометрических
Валентина Моисеевна Пешкова
Маргарита Ивановна Громова
ПРАКТИЧЕСКОЕ РУКОВОДСТВО
ПО СПЕКТРОФОТОМЕТРИИ
И КОЛОРИМЕТРИИ
Тематический план 1965 г. № 17
Переплет художн. Е. А. Михельсона
Редактор Е. Г. Дергачева
Технический редактор К. С. Чистякова
Корректоры Г. И. Чугунова,
Л. С. Клочкова
Сдано в набор 13/V 1965 г.
Подписано к печати 29/XI 1965 г.
Л-49717 Формат 60 x 90/J6
Физ. печ. л. 14,75 Уч.-изд. л. 14,87
Изд. № 798 Зак. 686 Тираж 6500
Цена 62 коп.
Издательство Московского
университета
Москва, Ленинские горы
Административный корпус.
Типография Изд-ва МГУ.
Москва, Ленинские горы
Московский Государственный университет
имени М.В.Ломоносова
Химический факультет
Кафедра аналитической химии
СБОРНИК МЕТОДИК
Д2Я ПРАКТИКУМА ПО СПЕКТРОФОТОМЕТРИИ
Под общей редакцией
профессора В.М.Пемковой
Москва - 1970
Настоящий сборник является дополнением ко второму изда-
нию учебного пособия В.М.Пешковой и М.и.Громовой "Практичес-
кое- руководство по спектрофотометрии и колориметрии” /Изд.
МГУ. 1965/. Поэтому в оглавлении сборника указано, к какому
разделу этого учебного пособия относится данное дополнение»и
в тексте имеется ряд ссылок на соответствующие разделы, для
краткости имеющие следующую форму: "Руководство”, стр. ...
Сборник составлен сотрудниками кафедры аналитической хи-
мии МГУ - профессором В.М.Пенковой, доцентами М.И.Громовой,
И.Ф.Долмановой, Н.В.Мельчековой и В.М.Савоотиной.
ДОПОЛНЕНИЕ К РАЗДЕЛУ Ш
I. ОПРЕДЕЛЕНИЕ СОСТАВА И КОНСТАНТ УСТОЙЧИВОСТИ
КОМПЛЕКСНЫХ СОЕДИНЕНИЙ.
Для определения состава комплексных соединений или со -
отноиения компонентов в яих имеется ряд методов. Приводим два
из них, пригодных в случае образования только одно-
го моноядерного комплексного соединения и ступенчатым компле-
ксообразованием можно пренебречь.
В общем виде уравнение образования этого комплекса мо -
жет бить записано так:
М~ nR — МК‘Ж"’'+ (П
I/ Метод молярных отношений (метод насыиения1.
Для определения оостава соединения этин методом строят
кривую зависимости какого-либо свойства (например, оптичес -
кой плотности) от концентрации лиганда; при этом концентрация
комплексообраэователя остается постоянной. Вид получающейся в
этом случае кривой (кривая насыщения) приведен на рио. I.
При образовании в системе достаточно прочного комплекса
на кривой насыщения получается резкий излом в течке, где мо-
лярное отношение иона-комплексообразователя и лиганда соот -
зетствует таковому в комплексе.
При получении кривей насыщения, не имеющей резкого изло-
ма о составе или соотношении компонентов в комплексном соеди-
нении можно судить на основании экстраполяции из прямолиней-
ных участков на кривой насыщения.
2/ метод предельного логарифмирования. Бента и френча.
При образовании малспрочных комплексов, когда получаются
кривые насыщения без резкого излома (с плавным переходом),бы-
вает затруднительно определить искомые состав или соотношение
компонентов. В этом случае может быть применен метод Бента и
Френча, который одновременно позволяет определить и константу
устойчивости образующегося соединения.
Общая константа устойчивости комплекса равна:
5
R.
Рис. I. Вид кривых насыщения
I/ устойчивое комплексное соединение.
Ue : R. = I : г
2J неустойчивое комплексное соединение
Ue : R, =1:3
6
. [HR,1
A (2)
Логарифмируя это выражение) получаем:
= П fy[Rj (3)
где, n - число лигандов, участвующих в комплекоообраэо -
вании о одним ионом металла.
Преобразуя уравнение (3), получаем
(4)
В случае, если мы за свойство комплекса берём его оптическую
плотность и можем выбрать такую длину волны, при которой по-
глощает только один комплекс » то [ГН RnJ
пропорциональна J) ; а концентрация металла . [MJ- , не
связанного в комплекс, пропорциональна J) , где
- оптическая плотность раствора, в котором весь металл
связан в комплекс MR.n .
Тогда уравнение (4) может быть преобразовано:
= %А + nt$N
Награбите - f ([Rj) п Судет
являться тангенсом угла‘наклона прямой к оси абсцисс, а
- отрезком,отсекаемым этой прямой на оси ординат.
В случае, когда комплекс MR^ экстрагируется несме-
шивающимся в водой растворителем уравнение (5) несколько ви-
доизменяется.
Константа распределения MR.n между водной и органи-
ческой фазами, т\п может быть представлена уравнением:
\ _ [Mftnjppr.
7
Комбинируя (6) и (2)* будем иметь:
, 1 _ [MR] орг
п "МЛ
С7)
После логарифмирования уравнения (7) и замены [' !Knjopr.
И [Mlg 'через пропорциональные им £орг и Рорг “
- J) L получаем уравнение аналогичное (5), из которого ле-
гко найти п и . Ej^n^n
+ п^ (8)
Может быть предложен другой метод определения
Как легко видеть из уравнения С7), для момента 50%-ной экст-
ракции» когда
[нкХг_ <
[м] -1
справедливо соотношение .
" И" Го1
Таким образом» определив из.кривой насыщения [К]
стветствушщую тсй точке» где ])1=4"]?0 . > и зная
по уравнению СЮ) можно вычислить
(9)
(Ю)
, со-
П .
2. ДОПОЛНЕНИЕ К ОПИСАНИЮ МЕТОДА ДИФФЕРЕНЦИАЛЬНОЙ
СПЕКТРОФОТОМЕТРИИ.
В дифференциальном методе измерения высоких плотностей
для расчёта определяемых концентраций в случае выполнения за-
кона поглощения лучистой энергии можно пользоваться как граду-
ировочным графиком, так и применяя фактор F /"Руководство",
стр. 69/.
Однако, если закон не выполняется, то необходимо для по-
строения, градуировочного графика предварительно выбрать,"ну-
левой" раствор. Оптимальным в зтом случае будет раствор", для
которого произведение будет иметь максимальное
значение /"Руководство", стр. 67/.
8
Практически работа в этой случае должна состоять из сле-
дующих этапов:
I. Готовят серию эталонных растворов, отличающихся на
одну и ту хе концентрацию.
2» Измеряют оптическую плотность каждого последующего
из них по отношению к предыдущему и рассчитывают текущее зна-
чение ' ‘РД— /во всех рассчвтах дС = пест. , со-
гласно условиям приготовления растворов/ и , где
С;- концентрация тоге раствора» который был взят в качестве
"нулевого” в данном измерении.
3. Измеряют оптические плотности всей серии растворов
пс отноиению к тому» для которого произведение име-
ет максимальное значение.
При этом совершенно очевидно, что ряд величин J) будет
иметь отрицательное значение. Так как обычно спектрофотометри
ческие приборы не приспособлены для измерения отрицательных
значений J) » они могут быть получены» если настроить вна -
чале "0я шкалы прибора по испытуемому раствору, а затем из-
мерить по отношению к нему оптическую плотность выбранного
"нулевого” раствора и взять её со знаком /-/.
k. По полученным данным строят градуировочный график.
используя как положительную, так и отрицательную зетвь ,
и определяют по нему концентрацию контрольного раствора.
Примечание: Зля приготовления эталонных растворов ис -
пользуют соответствующую пропись методики в "Руководстве".
Интервал используемых концентраций должен быть равен:
I. Для определения Fe в виде моносульфосалицилата от
2.8 до 7,0 через 0,7 мг (стр. 141).
2» Для определения Fe в виде трисульфосалицилата от
0,5 мг до 0,3 мг через 0,5 мг (стр. I43A.
3. Для определения Пп в виде перманганата от 2 мг до
12 мг через 2,мг (стр. 151).
9
3. КИНЕТИЧЕСКИЕ ЖГОДЫ ОПРЕДЕЛЕНИЯ
МИКРОКОИПОНЕНТОВ
Кинетические методы анализа основаны на использовании
зависимости между скоростью реакции и концентрацией реагиру-
ющих -веществ:
V( = kCaCb (II)
Y - скорость реакции; ,
К - константа скорости;
Сд и Q- соответственно концентрации реагирующих веществ
А и В.
Длн определения малых количеств вещества обычно исполь-
зуют каталитические реакции, в которых катализатором являет-
ся определяемый элемент
Ч=э€^ДСвСкат.
- скорость каталитической реакции;
К - каталитический коэффициент;
Скат. “ ковдентрация катализатора.
Реакция, скорость которой определяется концентрацией ка-
тализатора, называется индикаторной: а вещества, по изменению
концентрации которых судят о скорости реакции - индикаторными
веществами. В том случае, когда индикаторным веществом явля -
етсн продукт реакции, концентрацию его обозначим через X и
уравнение 02) принимает вид:
ЗГ ^в^кат.
Если в процессе реакции концентрации реагирующих ве-
ществ А и В существенно не меняются, то заменив в уравнении
(13) их произведение СдСв на постоянную Пс , по-
лучаем
='Х ПсСкаги (14)
или Й"К’Скат. (15>
10
К* - условная константа скорости» в величину которой входят
концентрации реагирующих веществ, взнтых в избытке.
Между концентрацией индикаторного вещества и временем
протекания реакции, в этом случае» существует прямопропорци -
овальная зависимость /дифференциальный вариант кинетических
методов анализу/;
х = К (16)
Если концентрация-хотя бы одного из реагирующих веществ, на -
пример вещества А, в процессе реакции меняется существенно.то
уравнение (13) можно записать в следующем виде:
(Q_‘3cJ^B^Kani.
а - начальная концентрация вещества А. (а-х) его концентра-
ция в каждый данный момент времени.
Преобразуй уравнение (17) и интегрируя его, получаем:
. м —^'СК0П1 dt (18)
W «Л*
Когда концентрация одного или нескольких, реагирующих ве-
ществ в процессе времени меняется существенно, применяется
интегральный вариант кинетических методов анализа. В этом
случае прямопропорциональная зависимость наблюдается между
какой-либо функцией концентрации индикаторного вещества и
временем реакции. Например, между функцией ~ и
временем реакции или функцией и временем, если
величина Q во всех опытах сохраняется постоянной.
Если каталитическан реакция протекает с изменением окрас-
ки раствора /или её интенсивности/, то измеряя величину J)
/значение оптической плотности раствора/ можно следить за из-
менением концентрации индикаторного вещества во времени.
Для определения неизвестной концентрации по данным кине-
тических измерений чаще всего используют способ тангенсов.При
работе по способу тангенсов оптическую плотность растворов, со-
- II -
Рис. 2. Зависимость J) от времени
при различных концентраци-
ях определяемого вещества.
12
держащих различное количество элемента - катализатора) изме-
ряют через определенные промежутки времени. Строят затем гра-
фики в координатах оптическая плотность-время /дифференци -
альный вариант кинетического метода анализа. рис. 2/ или фун-
кция оптической плотности-время /интегральный вариант/. По
графику определяют тангенсы наклона угла полученных прямых и
строят калибровочный график в координатах - концентра-
ция катализатора (рис. 3). Для определения неизвестной кон -
центрации по данным кинетических измерений используют также
способы фиксированного времени« фиксированной концентрации и
ряд других способов /I/.
ДОПОЛНЕНИЕ К РАЗДЕЛУ 1У
I. ИНСТРУКЦИЯ ДЛЯ ИСПОЛЬЗОВАНИЯ ФЭК-56 В МЕТОДЕ
СПЕКТРОФОТОМЕТРИЧЕСКОГО ТИТРОВАНИЯ
Спектрофотометрическое титрование может быть выполнено
как на спектрофотометрах СФ-4. СФ-4А. СФ-5» так и фотозлект-
гю колориметрах типа ФЭК-56. ФЭК-Н-57 и т.п. Краткое описание
приставки к спектрофотометру) используемой для титрования
дано в "Руководстве" /стр. 108/.
В последнее время нашей промышленностью выпускаются спе-
циальные приставки для титрования к фотозлектроколориметру
ФЭК-56.
В настоящей инструкции даны указания по использованию
приспособления для титрования к фотозлектроколориметру ФЗК-56.
выполд**:ой механической мастерской химического факультета
МГУ.
В зависимости от первоначальной настройки осветителя
фотозлектроколориметра ФЭК-56 более интенсивным может оказа-
ться или правый) или левый световой поток. В связи с этим мо-
гут быть использованы два варианта измерений оптической плот-
ности. меняющейся в процессе титрования.
I. Левый поток более интенсивен.
V В оба потока устанавливают кюветы с растворителем
/можно установку производить и по воздуху/;
W К.Б.Яцимирский. Кинетические методы анализа. М..Изд."Хи-
мия") 1967г.
2/ Правый барабан устанавливают на значение Т=100Й
/Р =0/.
3/ Уравнивают потоки» меняя ширину щели левой диафра-
гмы.
Записывают отсчёт по барабану левой диафрагмы / PQ /•
4/ Устанавливают в правом потоке кювету с титруемым
веществом.
5/ Компенсируют фототоки.» меняя ширину щели левой ди-
афрагмы перед началом титрования» отсчёт по шкале левого ба -
рабана равен Рв ,
6/ В процессе титрования после прибавления очередной
порции титранта и перемешивания компенсацию фототоков произ-
водят такхе» меняя ширину щели левой диафрагмы и отсчитывая
показания оптической плотности по шкале левого барабана.
Действительное значение оптической плотности» которую
имеет раствор в каждый момент титрования» равно
П. Правый поток более интенсивен,
I/ Так хе, как в варианте I.
2/ Левый барабан устанавливают на значение Т=1002б /J) =0/.
3/ Уравнивают потоки /компенсируют фототоки/» меняя ши-
рину щели правой диафрагмы. Записать отсчёт по шкале правого
барабана / /.
4/, 5/> 6/ То же» что и в варианте I. Действительное зна-
чение оптической плотности в этом случае будет равно
График зависимости оптической плотности от объема приба-
вленного титранта может быть построен как в координатах =
= |(v) , так и в координатах /или
/. Момент эквивалентности определяют как
обычно графически по пересечению двух прямолинейных участков
на кривой титрования; можно применять экстраполяцию /"Руковод-
ство", стр. 57/.
14
ДОПОЛНЕНИЕ К РАЗДЕЛУ У
I. ДОПОЛНЕНИЕ К ОПИСАНИЮ МЕТОДОВ ОПРЕДЕЛЕНИЯ КОНСТАНТ
ДИССОЦИАЦИИ ОРГАНИЧЕСКИХ РЕАГЕНТОВ
В методики» приведённые в "Руководстве" /стр.45» 125-
-129/» необходимо внести некоторые изменения. Порядок выпол-
нения работы по определению К,„„ реагентов должен быть еле-
дмии»
ДУ»ч»“- серию
I. Приготовляют растворов» имеющих одинаковую концент-
рацию реагентов и различные значения pH» используя соответст-
вующую пропиоь в "Руководстве"» но изменив интервал значений
pH» которые для различных реагентов должны быть соответствен-
но равны указанным в таблице I.
Таблица I.
Значения pH растворов для определения КЛИА. реагентов
tefc Г Нитрозо- Тимоловый синий/стр.128/I Диметилди-
пп ; -соль i /стр. 127/ ! К1 ! к2 •; оксим i /стр. 125/
I. /Змл 0,1Н НИО4/ Т/разб.до метки колбы 2N HCIOj/ 7 2/10мл 0,Ih| НС104/
2. 3,0 1.2 7,6 3/1мл 0,1М НСЮ^
3. 6.0 1,5 8,0 6
4. 7,0 1,7 8,8 9
5. 8,0 2.3 . 9,3 10
6. 9,0 2,9 9,6 II
7. 12,0 4,5 10,0 12
8.>12/10мл 0.IN ЫаОН/ 7,6 12/5МЛ
Значения pH растворов после их приготовления измеряют на
потенциометре.
2. Снимают спектры поглощения трех растворов реагента»
значительно отличающихся значениями! pH. Из серии растворов»
указанных в прописи для их приготовления» целесообразно вы-
15
брать два» имеющих граничные значения pH» и один с промежу-
точным.
3. На основании изучения полученных кривых спектров по-
глощения выбирают длину волны» при которой наблюдаются наи -
больиие изменения в величинах оптических плотностей растворов
с изменением их значений pH.
4. Измеряют при выбранной длине волны значения оптичес -
ких плотноотей остальных растворов приготовленной серии и вы-
числяют значения кди0С.» используя расчётный и графический
методы обработки результатов /“Руководство11, стр. 45/.
2. ОПРЕДЕЛЕНИЕ СОСТАВА И КОНСТАНТЫ УСТОЙЧИВОСТИ
АССОЦИАТА ФЕРРОИНА С ИОДИД-ИОНАМИ
С помощью описанных ранее методов /огр. 5 / можно опре-
делить состав и устойчивость ассоциата ферроина с иодид-иона-+
ми. Ферроин- довольно прочный комплексный катион [Fe(Phen*),f
который образует о J t Ct О* и некоторыми другими
анионами солеобразные продукты/аоооциатц/, экстрагируемые
органическими растворителями. Эти" аоооциаты обладают невысокой
прочностью. К определению их состава и констант устойчивости
может Сытьприменен описанный на стр. 7 метод, если
[FefPhenLT* принять за практически не диссоциированные
частицы М1+.
Необходимые реактивы.
I. Раствор соли Мора, содержащий I мг железа /П/e I мл.
Способ приготовлении см. “Руководство", стр. 144.
2. Гидрокоиламин солянокислый, 10^-ный раствор.
3. О—фенантролин, I^-ный водный раотвор.
4. Раотвор ферроина: готовят смешиванием 5,6 мл раствора
соли Мора, 30 мл раствора фенантролина, 20 мл раствора гидро-
ксиламина и разбавляют водой до I л.
5. Хлороформ.
6. Ацетат натрия, I М раствор.
7. Иодид калия, О,1М раствор.
* Phen - о-фенантролин.
16
В деояп делительных воронок или сосудов с притертыми
пробками помещают по I мл раотвора ферроина. 10 мл ацетата
натрия и от I до ю мл раотвора иодида калия. После
перемешивания добавляют во все раотворы по 10 мл хлороформа и
вотряхивают их содержимое в течение 2-3 минут. Через 15 мин
пооле расслаивания жидкостей хлороформные экстракты отделяют
и измеряют их оптичеокую плотность в однооентиметровой кюве-
те при А 520 нм на СФ-4. СФ-4А или СФ-5. По полученные
данным отроят график завиоимооти J)opr.—f ([3'])
По участку кривой насыщения, где достигается постоянство оп -
тичеокой плотности, определяют РорГ. и строят график зави-
симости
*3 тЛ? = МР1) т
х#рг.
Состав комплекса /п ./ определяют по графику /8/. как
тангено угла наклона полученной прямой к оси абсцисс.-По кри-
вой насыщения вычисляют J$n^n : из точки кривой, где
/5О2б-ная экстракция/ опускают перпен -
дикуляр на ось [3’] 'и вычисляют /?ПА = "[ J j" •
3. ИЗУЧЕНИЕ РАЗЛИЧИЙ В РАБОТЕ С ИУ1НОЙ ЛАМПОЙ И ЛАМПОЙ
НАКАЛИВАНИЯ НА ФОТОЭЛЕКТРОКОЛОРИМЕТРЕ ФЭК-56.
Фотозлектроколометр ФЭК-56 имеет два иоточника излучений:
лампу накаливания и ртутную лампу. Производить измерения мож-
но как о той, так и о другой лампой. Однако, ввиду того, что
соответствующий светофильтр выделяет из спектра испускания
ртутной лампы отдельные линии, а из спектра-испускания лампы
накаливания опиеделенный интервал длин волн, то монохроматич-
ность падающего на измеряемый объект излучения будет в первом
олучае выше.
Чтобы в этом убедиться необходимо провеоти следующую ра-
боту:
I. Снимают спектры поглощения иоследуемого раотвора,
пользуясь ртутной лампой и лампой накаливания, делая измере-
17
ния при следующих светофильтрах и соответствующих им длинах
волн» указанных в табл. 2.
Таблица 2.
Длины волн максимумов пропускания светофильтров
и линий испускания ртутной лампы
№ светофильтра !Л(нм) макс, пропуска- |Л(нм) линий испу-
! ния светофильтра !скания ртутной
! ! лампы
- » - _
I 315 313
2 364 365
3 400 405/8
4 440 436
5 490 -
6 540 546
7 582 577 /9/
8 610 -
9 630 -
2. Используя длину волны» соответствующую полосе макси-
мального поглощения» измеряют оптические плотности эталонных
растворов с той и другой лампой и строят соответствующие гра-
дуировочные графики.
3. Определяют в каком из зтих случаев tg угла на -
клона / £ / будет больше. _
4- . Вычисляют значения £ на основе измерений о каждой
из ламп. Данные обрабатывают статистически и определяют по -
грешность.
При выполнении данной работы можно использовать в качес-
тве испытуемых растворы соединений:
а/ кобальта с нитрозо- R -солью
/“Руководство11» стр. 177/,
б/ трисульфосалицилата железа
"Руководство", стр. 14-2/5
18
з/ перманганат калия
/"Руководство", стр. 152/»
г/ диоксимата никеля в присутствии окислителей
/“Руководство", стр. 159/»
Я/ сульфата церия
Примечания. I. Для приготовления 5 эталонных растворов
сульфата церия готовят ряд с содержанием церия ст 0,5 мг дс
5 мг в объёме 50 мл.
2. Для приготовления эталонных растворов кобальта с ни-
трозо- R -солью берут 1,5 мл реагента вместе 3 мл, указанных
в "Руководстве", стр. 177.
3. Аля приготовления растворов диоксимата никеля в при-
сутствии окислителей добавлять в каждый эталонный раствор
вместо щелочи /"Руководство", стр. 159/ 3 мл концентрирован-
ного аммиака.
4. ОПРЕДЕЛЕНИЕ ЖЕЛЕЗА /П/ В ВИДЕ ФЕРРОИН-ИОДИДА,
Для определения микроколичеств железа рационально ис -
пользовать образование соединения ферроин-иодида [Fe(Phen)3]7^
которое хорошо экстрагируется хлороформом.
Необходимые реактивы:
I. Стандартный раствор железа /П/ с содержанием 0,1 мг
в I мл./"Руководство", стр. 144/. Ьаствор с содержанием же -
леза 0,5 мкг в I мл готовят разбавлением в день употребления.
2. Гидроксиламин солянокислый "х.ч.", 10^-ный раствор.
3. О-фенантролин, солянокислый, 0,1^-ный водный раствор.
4. калий иодистый "х.ч.“, 20^-ный раствор-
5. Т рил он Б, 20^-ный раствор.
6. Хлороформ.
7. Аммиак (i:p.
Гидроксиламин солянокиолый очищают перекристаллизацией
из воды.
Хлороформ очищают по методике Вайсбергера (I).
А.Вайсбергер и др. Органические растворители, М.,ИЛ,1958г.
19
а/ Определение железа в растворе его чистой соли.
5 мл испытуемого раствора, содержащего железо» помещают в
делительную воронку и прибавляют растворы: 5 мл трилона Б» по
I мл гидраксиламина о-фенантролин и йодистого калия. Раотвор
нейтрализуют аммиаком до pH 3-4 по универсальной индикаторной
бумаге и оставляют стоять 40 минут. Далее добавляют 10 мл хю~
роформа и экстрагируют образовавшееся соединение в течение I
минуты. В указанных, условиях готовят пять эталонных растворов,
содержащих 0,3? 0,5; 0,7; 1,0 и 2,0 мкг железа. Для измерения
оптической плотности растворов используют СФ-5, СФ-4 или СФ-4А.
В качестве раствора сравнения используют хлороформ. Калиброво-
чный график рассчитывают методом наименьших квадратов. По это-
му графику находят содержание железа в испытуемом растворе.
б/ Определение железа в гипофосфите натрия,
Навеоку объекта /1г/ растворяют в 5 мл раствора трилона
Б, прибавляют по I мл растворов солянокислого гидроксиламина,
о-фенантродина и иодистсго калия, нейтрализуют аммиаком по
индикаторной бумаге до рН=3-4 и доводят водой объем раотвора
до 10 мл. Через 40 минут приливают 10 мл хлороформа и экстра-
гируют ферроин-иодид в течение I минуты. Оптическую плотнооть
растворов измеряют на приборах СФ-5, СФ-4, СФ-4А по отношению
к холоотому опыту. Для получения растворов сравнения /"холос-
того опыта"/ проводят экстракцию из указанных количеств реак-
тивов в тех же условиях и полученный экстракт в хлороформе ис-
пользуют в качестве раствора оравнения.
Содержание железа определяют, используя калибровочный
расчётный график (п. а/).
ъ/ Определение железа в нитрате кобальта.
Для определения микроколичеств железа в солях кобальта
имеется ограниченное число работ Для отделения железа
1а П.И.Артюхин,'З.Н.Гильберг, В.А.Пронин, В.И.Иоралев.Заводск.
лаборатория, 21» 926 /1967/.
2. F.Vyche,fU4>ht. Tafanta, 3
20
от макрокомпонента /нитрата кобальта/ и его концентрирования
рекомендуется предварительная экотракция хлоро комплексов же-
леза кислород содержащими растворителями Опыты пока -
вали» что незначительное количество кобальта при этой опера-
ции» созкстрагируется о железом. Для устранения мешающего
действия кобальта при определении железа был применен один из
вариантов дифференциального метода» для которого нужен только
раствор испытуемого образца /"Руководство"» стр. 69/.
Кроме указанных на стр. 19 необходимы реактивы:
I. Соляная кислота особой чистоты б й
2. Дибутиловый эфир.
3. Раствор аммиака /1:10/.
Навеску 5 г испытуемого образца нитрата кобальта поме-
щают в мерную колбу на 25 мл и доводят до метки соляной кис-
лотвй. Для приготовления первого /"нулевого"/, раствора /Со/
берут аликвотную часть 2<5 мл» /0,5 г образца; для второго
/Сх/~7»5 мл. раствора (1»5 г образца) и для третьего /Сх+Са/»
также 7»5 мл раствора и добавляют в него 0»5 мкг железа /Са/.
Вое эти растворы помещают в три делительные воронки и добавля-
ют соляной кислоты до общего объёма 10 мл. В каждую воронку
добавляют 10 мл дибутилового эфира и экстрагируют образовав-
шееся комплексное соединение железа в течение 3-х минут. ир -
паническую фазу промывают последовательно двумя порциями соля-
ной кислоты /по 10 мл каждац/» Из органической фазы комплекс-
ное соединение железа ре экстрагируют в водную фазу добавлени-
ем 20 мл раствора аммиака и 5 мл трилона Б и отделяют водную
фазу. В полученный раствор добавляют по I мл растворов гидро-
ксиламина и о-фенантролина. Через 20-30 минут добавляют I мл
иодида калин и экстрагируют образовавшееся соединение ферро -
ин-иодида 5 мл хлороформа в течение I минуты. После разделе -
ния фаз сливают органическую фазу в градуированную пробирку
на Ю мл» добавляют хлороформ до объёма Ю мл и проводят фо
тометрирование» как описано в "Руководстве” на отр. 69. Расчёт
содержания железа производят по |уравнению
______________С* = ^^К (2(П
L t. Bankman, H.Spekei. Z. anal. Chem., 162,18/1358/
tA CU$sen,L.Ba$ttK9S.Z. anal. Chem., 1П, -453/1958/
где - оптическая плотность второго раствора
/ Сх / п0 отношению к “нулевому" раствора
( /Со/.
Сх - содержание железа в количестве образца,пред-
ставляющее разность "нулевого" и второго рас-
твора /1г/
-разность оптических плотностей второго и тре-
тьего растворов, измеренных по отношению к
первому.
Содержание железа в контрольном образце нитрата кобаль-
та рассчитывают по формуле:
Ге % - • 100
где Q - навеска.
5. ОПРЕДЕЛЕНИЕ МИКРОКОЛИЧЕСТВ КОБАЛЬТА
КИНЕТИЧЕСКИМ МЕТОДОМ
Индикаторной реакцией для определения иикроколичеств ко-
бальта является катализируемая им реакция окисления ализари-
на перекисью водорода в боратном буферном растворе при pH
12,4. Определение проводится способом тангенсов интегрально-
го варианта кинетических методов анализа.
Необходимые реактивы:
I. Стандартный раствор соли кобальта /"Руководство",стр. 169/.
2. Ализарин /1,2 диоксиантрахинон/, 4.1О“^М водный раствор.
При растворении 0,0247 г ализарина очищенного двойной пе-
рекристаллизацией из воды добавляют 8 мл 0,01 tl раст -
вора едкого натра и доводят объём до 250 мл.
3. Перекись водорода "ос.ч?« 3,ЗМ раствор.
4. Боратный буферный раствор /pH - 12,37/. Для пииготовления
I л буферного раствора смешивают 400 мл 0,05М раствора
тетрабората натрия и 600 мл 0,1 раствора едкого натра.
Используют едкий натр "ос.ч." и тетраборат натрия ква-
лификации "х.ч." /ГОСТ 4199-48/, дважды перекристаллизован-
ный из воды. Все растворы готовят на деионизированной воде.
22
Построение калибровочного графика
способом тангенсов
В кварцевый смеситель с тремя отростками вводят последо-
вательно - в первый отросток 0,6 мл перекиси водорода, во вто-
рой - раствор, содержащий соль кобальта, в третий - 0,5 мл
раствора ализарина и такое количество боратно-щелочного буфе-
рного раствора, чтобы общий объём раствора был равен 10 мл .
Растворы термостатируют в течение 25-30 минут при 25°С,в мо-
мент смешения реагирующих веществ включают секундомер и из -
меряют-оптическую плотность раствора каждую минуту, начиная о
первой, в течение 10 минут на приборе ФЭК-Н-52 с термостати-
руемым корпусом или ФЭК-М, ФЭК-54 или ФЭК-57 с термостатиру-
емой камерой для кювет / I - 3 см; ЛЭф - 530 нм/.
По полученным данным строят графики в координатах tg
оптической плотности - время. Углы наклона этих прямых про -
порциональны концентрации кобальта. Затем строят график в ко-
ординатах tg еС -концентрация кобальта.
Содержание кобальта в эталонных растворах может быть от
5 10“’ - 5 Ю“2 мкг в 10 мл раствора.
Определение кобальта в растворе чистой соли
Для определения неизвестной концентрации кобальта I мл
испытуемого раствора помещают в один из отростков смесителя,
в другие два отростка добавляют указанные выше реактивы и
после тегрмоотатирования и смешения как указано выше фиксиру-
ют изменение оптической плотности во времени. По калибровоч-
ному графику находят концентрацию кобальта, соответствующую
полученному значению tg d */.
6. ОШЕДЕДЕНИЕ МИКРОКОДИЧЕСТВ МЕДИ КИНЕТИЧЕСКИМ
МЕТОДОМ
Индикаторной реакцией для определения микроколичеств ме-
ли является катализируемая медью реакция окисления гидрохино-
на перекисью водорода в присутствии пиридина в боратном бу -
^В.М.Пешкова, И.Ф.Долманова, Н.М.Семёнова. Х.аналит.химии,
18, 1228 /1963/.
23
ферном растворе при pH 7,8.
Определение меди проводится по способу тангенсов диффе-
ренциального варианта кинетических методов анализа.
Необходимые реактивы:
Т. Стандартный раствор меди, содержащий 2 мг/мл./"Руко-
водство" стр. 224*1 Растворы с меньшим содержанием меди (0,1
мкг/мл и 0,01 мкг/мл) готовят ежедневно разбавлением исход-
ных растворов.
2. Гидрохинон, 5,4 10*^М водный раствор. Технический ги-
дрохинон /ГОСТ 2549-60/ очищают возгонкой. Свежие растворы
готовят каждые 2-3 дня.
3. Пиридин ГОСТ 2747-44. высушивают над едким кали и пе-
регоняют в кварцевом приборе; готовят водный раствор концен-
трации 3
4. Перекись водорода "о.ч.", 1,6 М раствор. Точную кон-
центрацию перекиси водорода устанавливают титриметрически
каждые 5-7 дней.
5. Боратный буферный раствор - pH 7,8. Для приготовле -
ния I л буферного раствора смешивают 800 мл 0,2 N раство-
ра борной кислоты и 200 мл 0,5 М раствора тетрабората натрия.
Тетраборат натрия квалификации х.ч. и борную кислоту квали -
фикации х.ч. Дважды перекристаллизовывают из воды.
Построение калибровочного графика
способом тангенсов
В кварцевый смеситель с тремя отростками вводят: в пер-
вый отросток 0,5 мл гидрохинона, во второй - I мл перекиси
водорода, в третий - последовательно I мл пиридина, раствор
соли меди и такое количество боратного буферного раствора,
чтобы общий объём раствора был равен 10 мл. Растворы термо -
статируют 25-30 минут при температуре 25°С. В момент смеше -
ния раотворов включают секундомер и измеряют оптическую пло-
тность растворов, через I минуту в течение 10 минут на прибо-
ре ФЭК-М, ФЭК-54, ФЭК-57 с термостатируемой камерой для кю -
вет или ФЭК-Н-52 с термостатируемым корпусом / С= 3 см;АЭм-
- 420 нм/. По данным измерений строят графики в координатах
оптическая плотность - время, и тангенс угла наклона получеи-
- 24 -
ных прямых /^3°^/ - концентрация меди.
Содержание меди в эталонных растворах может быть 5 10~^_
- 5 10“^ мкг в 10 мл раствора.
а/ Определение меди в растворе чистой соли.
Для определения неизвестной концентрации меди I мл ис -
лытуемого раствора помещают в один из отростков смесителя, в
другие два отростка добавляют указанные выше реактивы и пос-
ле термостатирования и смешения как указано выше фиксируют
изменение оптической плотности во.времени. По калибровочному
графику находят концентрацию меди, соответствующую получен -
ному значению У*
б/ Определение меди в азотной и соляной кислотах.
Кроме указанных на стр. 24 требуются реактивы:
I. Едкий натр., 2 hl раствор.
2. Серная кислота ос.ч” конц.
10-15 мл испытуемой кислоты помещают в тефлоновые чашки
упаривают раствор в боксе на плитке, покрытой аобестом ди
объёма ~ 5 мл, добавляют 1-2 капли концентрированной сер -
ной нислоты, и далее упаривают раствор до объёма 0,5 мл, до-
бавляя I мл деионизированной воды. Раствор нейтрализуют ед-
ким натром до pH - 7 по универсальной индикаторной бумаге,
переносят в отросток кварцевого смесителя, споласкивая тефло-
новую чашку известным объёмом буферного раствора,-добавляют в
другие два.отростка кварцевого смесителя реактивы, указанные
в методике, и проводят определение меди так, как это указано
при определении меди в растворе чистой соли.
7. ОПРЕДЕЛЕНИЕ МЕДИ МЕТОДОМ СПЕКТРОФОТОМЕТРИЧЕСКОГО
ТИТРОВАНИЯ НА СПЕКТРОФОТОМЕТРЕ СФ-4.
Медь с комплексоном Ш образует устойчивое комплексное
соединение / tyK = 18,3/, обладающее максимальным поглоще -
нием потока лучистой энергии при А 745 нм. 4
И.Ф.Долманова, В.М.Пешкова. Ж. аналит. химии, 19, 297
(1964).
Р. Sweetsex, Р. Anatyt. Chem., 25, 253/1953/
25
Комплексонат железа в этой области не поглощает» поэто-
му возможно последовательное титрование железа и меди при
их совместном присутствии^. Вначале титруется.железо» обра-
зующее с комплексоном более прочное соединение» чем медь.
Необходимые реактивы:
I. Стандартный раствср соли меди» содержащий 2 мг Сы
в I мл ("Руководство?» стр. 224)
2. Комплексон Ш, 10 М раствор.
Раствор готовят из препарата квалификации "х. ч." по точ-
ной навеске и концентрацию его проверяют методом спектрофото-
метрического титрования по соли меди известной концентрации.
3. Ацетатный буферный раствор pH = 3.
.К 900 мл раствора 0»2 ь! уксусной кислоты добавляют 100
мл 0»2 tj уксуснокислого натрия.
Определение меди в растворе чистой соли
в отсутствии железа.
В мерную колбу на 50 мл помещают раствор» содержащий от
10 до 20 мг меди» и доводят объём ацетатным буферным раство-
ром до метки колбы.
После перемешивания отбирают пипеткой 20 мл этого раст-
вора и переносят в кювету для титрования. Раствор.комплексо-
на Ш прибавляют из микробюретки порциями по ~ 0»! мл, пе -
ремешивают и измеряют оптическую плотность при 745 нм после
прибавления каждой псрции реактива. По полученным данным стро-
ят кривую титрования, графически определяют, точку эквивалент-
ности и вычисляют количество меди, учитывая, что медь образу-
ет с комплексоном Ш соединение в соотношении 1:1
A.Lindenwood, Anatgi. Chem.. 25, 1910/lS53/
26
ОБРАБОТКА РЕЗУЛЬТАТОВ
ИЗМЕРЕНИЙ
I. ПРИМЕНЕНИЕ МЕТОДА НАИМЕНЬШИХ КВАДРАТОВ ДЛЯ
ПОСТРОЕНИЯ КАЛИБРОВОЧНОГО ГРАФИКА.
При построении калибровочного графика по эталонным
растворам может иметь меото значительный разброс результа-
тов измерения особенно при работе с малыми концентрациями
определяемых веществ. Это затрудняет определение правильно-
го хода градуировочного графика» который может быть в таком
случае построен только по методу наименьших квадратов.
Основное положение метода наименьших квадратов заклю -
чается в утверждении: "Для лучшей кривой сумма квадратов от-
клонений минимальна". На рис. I буквами Oj 0g 0^ 0^ 05
обозначены точки» соответствующие экспериментально получен-
ным данным измерений. Если из каждой точки опустить перпен-
дикуляр на кривую» то сумма квадратов отклонений ( д' ) дол-
жна быть минимальной» т.е.
i=n „
(21}
ь=1
Рассмотрим применение метода наименьших квадратов для
построения прямолинейного калибровочного графика.
Уравнение прямой можно записать в следующем виде:
у = |(х) = Вв+В,Х (22)
где BQ и Bj - постоянные /Во соответствует отрезку» отсека-
емому этой прямой на оси ординат, Bj - тангенсу угла накло-
на прямой к оси абсциос, X- переменная величина/.
Если мы определим постоянные величины BQ и Bj с учётом
основного требования метода наименьших квадратов /уравнение
21/» то по экспериментальным точкам 3^,3^,...3J, сможем
построить прямую» наилучшим образом удовлетворяющую полу -
чённым данным.
Известно» что прямую можно построить по двум зкспери-
- 27 -
Рис. 4. Разброс экспериментальных ре-
зультатов относительно истин-
ной кривой.
ментальным точкам (х,^) и (Х^у^) • Тогда» согласно
условию метода наименьших квадратов:
Bo+B.Xi-yr й *1
Вв+В(хгу8=<Ц (23)
F=V**s(B0+B.x-y,)‘+
+(B0+Bla^-ya)*=mi,n.
(24)
Известно, что значение Функции минимально, когда её част-
ные производные равны 0. Поэтому продифференцировав уравнение
/24/ по переменным Во и и приравняв частные производные к
0, получим следующие уравнения:
28
3g -yl)+2(Be+B,x%~yt)
^•-^(Bo+BtXfyJa^ 2(B0+ В1х,-уя)ха
. B0+B|X’y,-t-Bo+-Blxa-yjL=0
Box,+B1x*-ylx)+Be^+BlxJ~a*yt«O
Сгруппировав члены уравнения (26) по Bo и Bj, получим
2В,+8,(х,+*«)=У.-*-у»
В,(х,+х,)+В,(я?+х1*)=а,^+^,
>
Мы получили систему уравнений (27)» рассмотрели случай
построения графика по двум экспериментальным точкам. Если
калибровочный график строится по " " экспериментальным то-
чкам, то система уравнений (27) примет следующий вид)
nBs-l-B,(x1 + x2+...+xn)=lJ,+^+-.+a„
В.(х, +xs+.. +XJ+В,(х( + х‘+„ ,+х»)=
= х,у,+х4у4+...+хпул
или
1=И 1=п
пВ.+ в,£х;=
1=1 1=1
I’M i,=n i,=n
б£х’+ B0£x; =
l»l 6=1 1=1
(29)
Решая эту систему относительно BQ и Bj получим:
29
и Bj и строят по полученный данным прямую, используя уравне-
ние (22>«>
Пример: Расчёт калибровочного графика для определения меди
кинетическим методом по способу тангенсов.
Таблица 3.
Экспериментальные данные ( И = 4)
X ; Кол-во Си „ i (мкг/мл х 10s) 1 1 1 416 ! 8 ! 10 ! ! ! । । ।
У 1 tf* 7,7 j 10,4 j 15,0 j 20,3 i . •' !
Подставляем полученные данные в. уравнение (28) или (29)
4 Во + В2 /4+6+8+Ю/ = 7,7 + 10,4 + 15,0 + 20,3
Во /4+6+8+10/ + Вг /(42) + (6)2 + (8)2 + (Ю)2/ =
= 4 х 7,7 + 6 х 10,4 + 8 х 15,0 + 10 х 20,3.
Откуда рассчитываем Во = 2,05 и В = -I и получаем уравнение
для прямой:
У= 2,05 X- I.
Используя полученную зависимость, находим точки для нострсе-
ижя градуировочного графика по методу наименьших квадратов:
Таблица 4.
Данные для построения градуировочного графика
X ! ! Кол-во вСиа мкг/мл X 10s г * } 6. ! 8, 1 10
У ! —и- tgoL | ,7,2 ! 11,3 | 15,4 J 19,5
30
2. ОБРАБОТКА ЭКСПЕРИМЕНТАЛЬНЫХ РЕЗУЛЬТАТОВ
Результаты измерений должны быть обработаны с применени-
ем методов математической статистики^ » так как оценка до -
стоверности получаемых результатов имеет первостепенное зна-
чение» особенно при работе с микроколичествами веществ. Мето-
ды математической статистики применяются для оценки случайных
ошибок измерения.
Если было оделано "Л" измерений» то мпжно вычислить:
X - среднее арифметическое значение для выборки " п " наблю -
даемых результатов:
- Х,4-Хя+Х3+...+Хп
JC- п 1
О
$ - выборочную дисперсию для " Ц" наблюдений:
s - п-1
S- среднее квадратичное отклонение /или средне-квадратичную
ошибку/ определения: _____________
Дисперсия и среднеквадратичное отклонение характеризуют вое -
про из водимо сть метода» т.е. рассеяние отдельных измерений от-
носительно их среднеарифметического х.
При условии отсутствия систематических ошибон оценка то-
чности определения проводится путем вычисления доверительного
интервала» внутри которого с заданной степенью надежности " "
лежит иотинное значение определяемой величины
Г 34)
В.В.Налимов. Применение математической статистики при ана-
лизе веществ, м., Физматгжз, I960.
31
tg oC - табличная величина распределения Стьюдента, ко-
торая табулирована для определенного значения степени надеж-
ности а о(." я числа степеней свободы К /или f / = Л -I.
Степень надежности задается самим экспериментатором. /Обычно
сС принимают равнш 0,95 или 0,90/.
Таким образом, полученные экспериментальные данные сво -
дят в следующую таблицу:
32
ТАБЛИЦА РЕЗУЛЬТАТОВ ИССЛЕДОВАНИЯ ФОТОМЕТРИЧЕСКОЙ РЕАКЦИИ
33
СОДЕРЖАНИЕ
стр.
Дополнение к разделу Ш,
I. Определение состава и констант устойчивости
комплексных соединений................................ 5
2. Дополнение к описанию метода дифференциаль-
ной спектрофотометрии.......................... 8
3. Кинетические методы определения микрокомпо-
нентов ..................................... 10
Дополнение к разделу 1У.
I. Инструкция для использования ФЭК-56 в мето-
де спектрофотометрического титрования ............ 12
Дополнение к разделу У.
I. Дополнение к описанию методов определения
К--... органических реагентов........................ 15
2. Определение состава и нэястант устойчивое -
ти ассоциата ферроина с иодид-ионами ................ 16
3. Изучение различий в работе с ртутной лампой
и лампой накаливания на фотоэлектроколори -
метре ФЭК-56......................................... 17
4. Определение железа /П/ в виде ферроин-иоди-
да................................................... 19
5. Определение микро количеств кобальта кинети-
ческим методом....................................... 22
6. Определение микроколичеств меди кинетичес -
ким методом...................................... 23
?. Определение меди методом спектрофотометри -
ческого титрования на спектрофотометре СФ-4 25
Обработка результатов измерений.
I. Применение метода наименьших квадратов для
построения калибровочного графика.......... 27
2. Обработка экспериментальных результатов ... 31
Таблица результатов исследования фотометри-
ческой реакции................................... 33
Заказ 147 Тираж 130 Без цены
Лфлп. Химический фак-т МТУ