/





























































































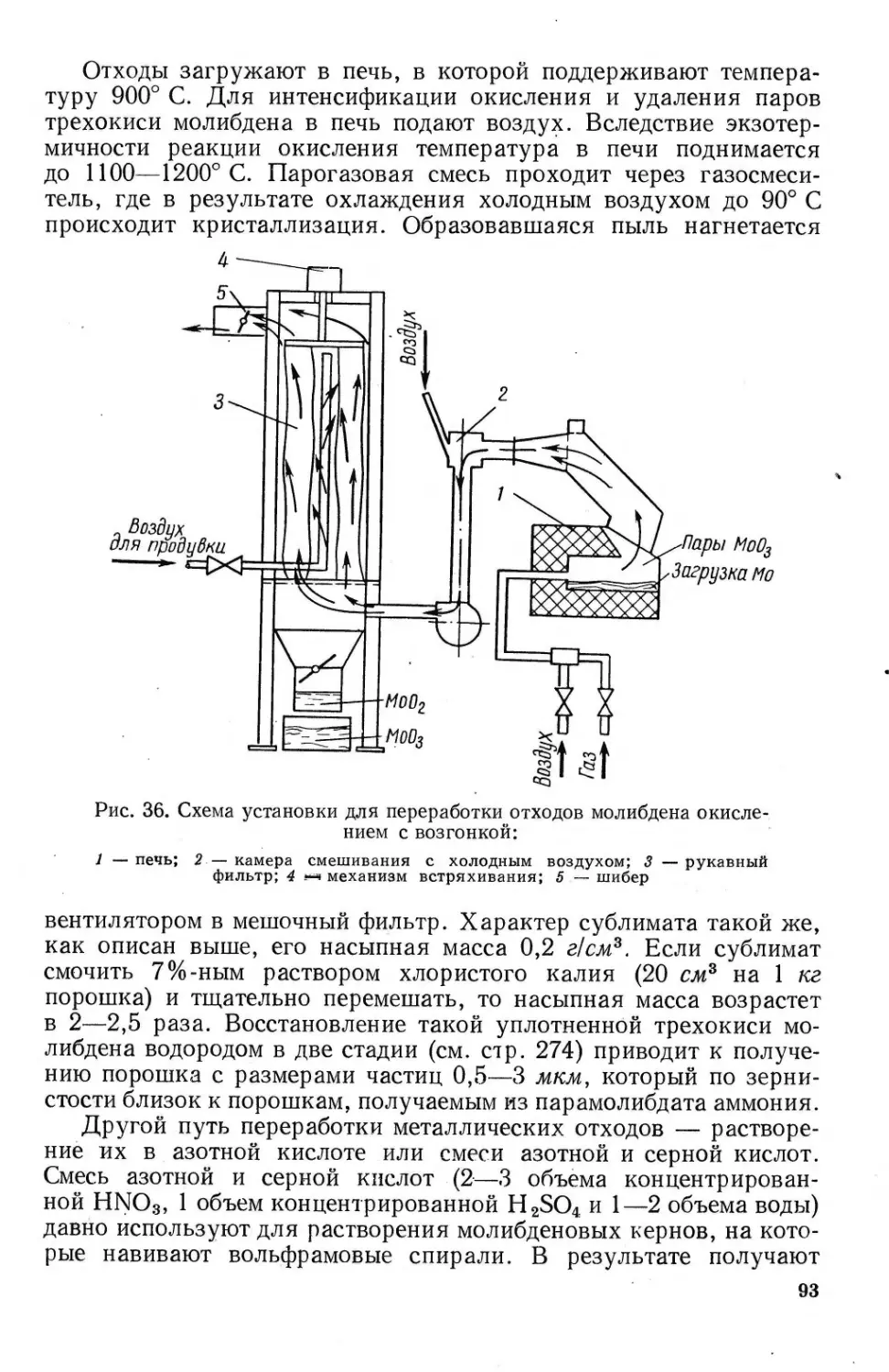






















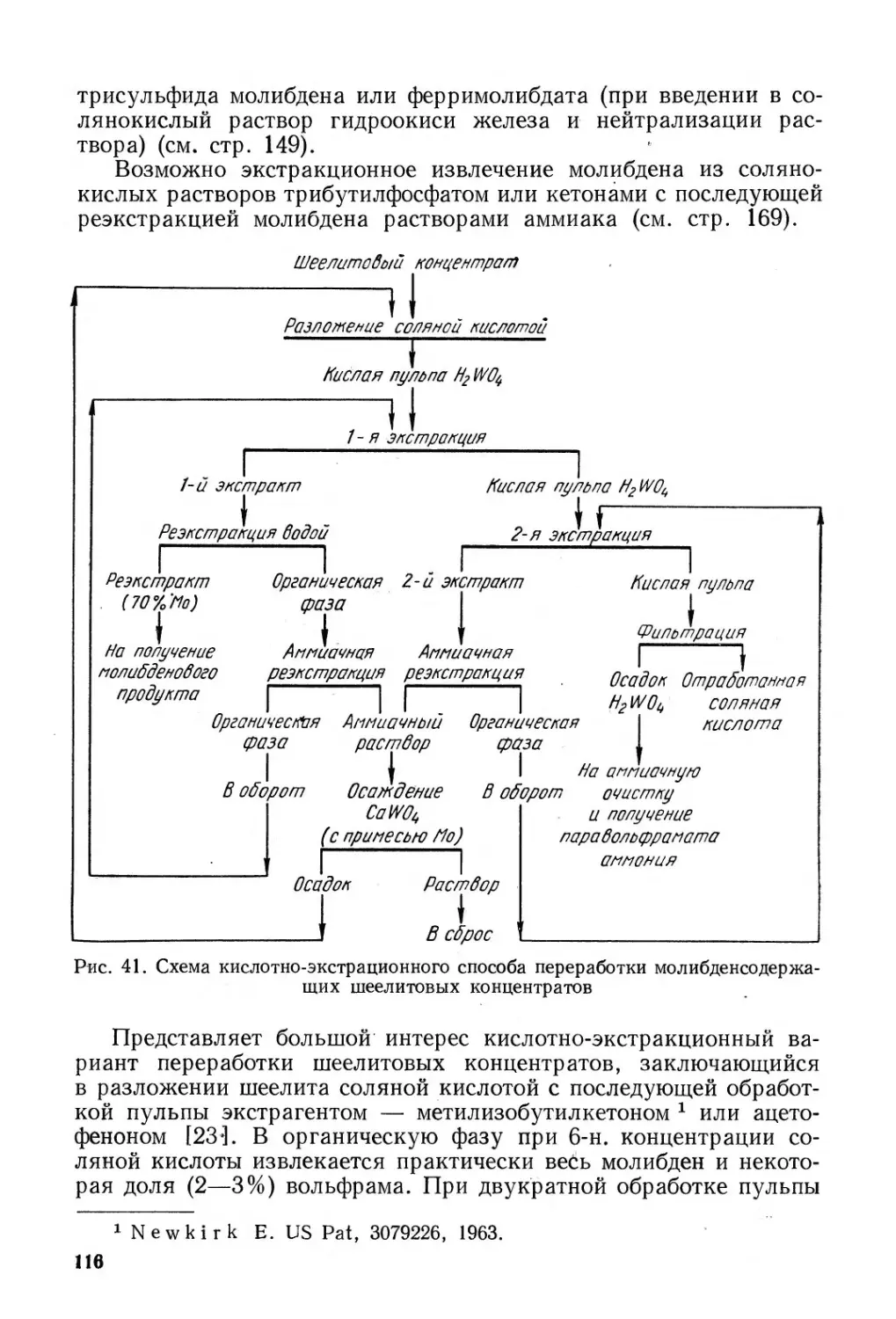




















































































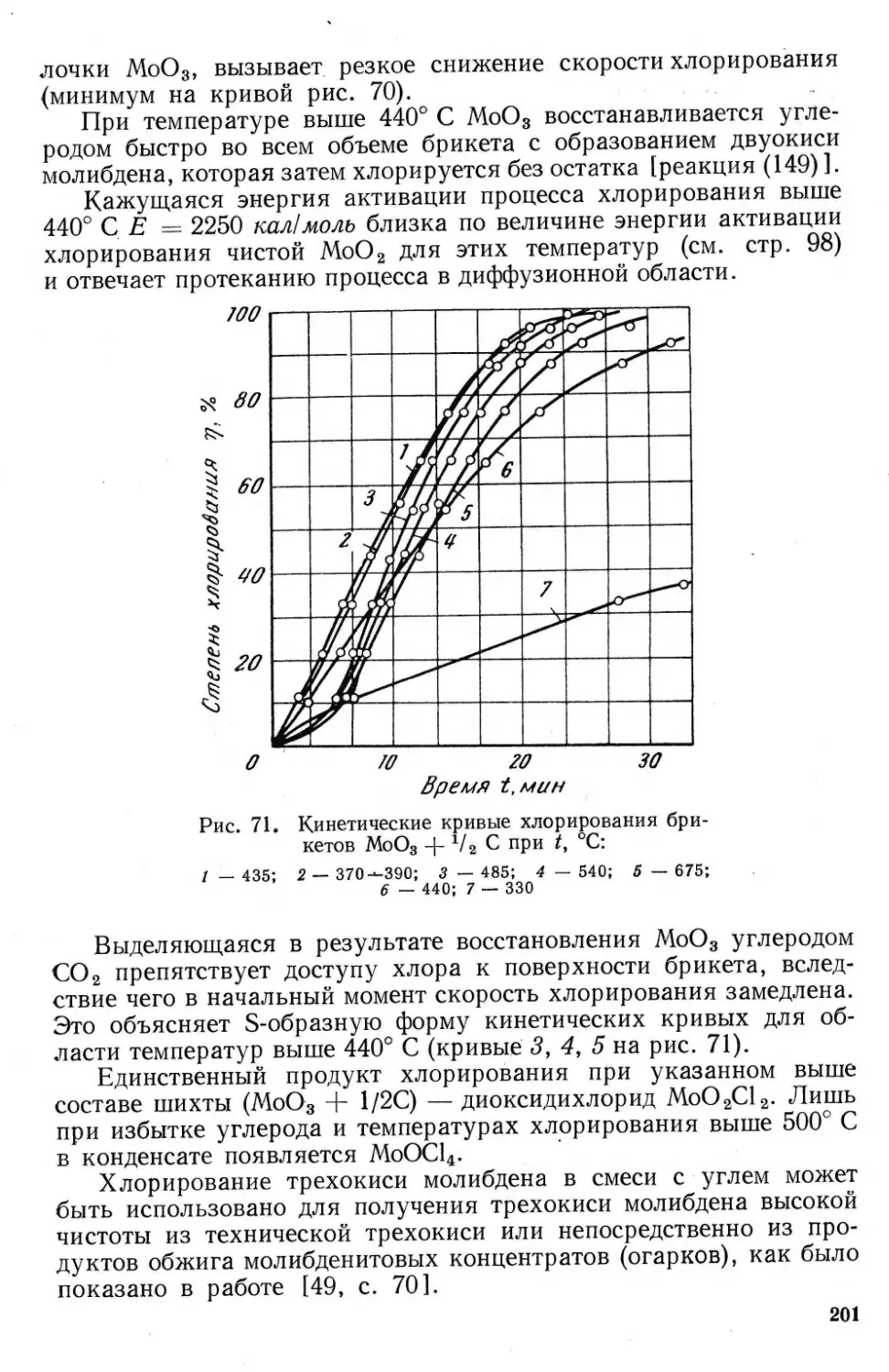











































































































































































































































Автор: Зеликман А.Н.
Теги: цветные металлы в целом металлургия гидрометаллургия порошковая металлургия молибден
Год: 1970




Текст
A. H. ЗЕЛИКМАН
МОЛИБДЕН
ИЗДАТЕЛЬСТВО „МЕТАЛЛУРГИЯ"
МОСКВА 1970
A. H. ЗЕЛИКМАН
МОЛИБДЕН
ИЗДАТЕЛЬСТВО „МЕТАЛЛУРГИЯ"
МОСКВА 1970
УДК 669.28
Молибден. Зеликман А. Н. Изд-во
«Металлургия», 1970, 440 с.
Рассмотрены физико-химические основы и
практика процессов производства молибдена
и его важнейших соединений (окислов, хло-
ристых соединений, сульфидов, силицидов,
боридов, карбонила).
Большое внимание уделено новым направ-
лениям в гидрометаллургии молибдена и про-
изводстве компактного металла методами по-
рошковой металлургии и плавки. Рассмотрены
свойства молибдена и области его применения.
В отдельной главе изложены вопросы попут-
ного извлечения рения в производстве молиб-
дена.
Книга содержит обширную библиографию
работ советских и зарубежных ученых.
Предназначается для инженерно-техниче-
ских и научных работников заводов и научно-
исследовательских институтов, преподавате-
лей вузов, аспирантов и студентов старших
курсов Илл. 184. Табл. 103. Библ. 827 назв.
ЗЕЛИКМАН АБРАМ НАУМОВИЧ
МОЛИБДЕН
Редактор издательства М. С. Архангельская
Технический редактор Л. В. Добужинская Переплет художника П. П. Перевалова
Сдано в производство 14/1II 1969 г. Подписано в печать 3/XII 1S69 г.
Бумага типографская № 1, 60х90*/1в — 13,75 бум. л. 27,50 печ. л.
Уч.-изд. л. 27,33 Изд. № 4733 Т-15289
Тираж 2200 экз. Заказ 123 Цена 2 р. 93 к.
Издательство «Металлургия», Москва, Г-34, 2-й Обыденский пер., 14
Ленинградская типография № 6 Главполиграфпрома
Комитета по печати при Совете Министров СССР
Ленинград, С-144, ул. Моисеенко, 10
3-10-3
Б5-69
ОГЛАВЛЕНИЕ
Предисловие.,,................................................... *6
Глава I. МИНЕРАЛЫ МОЛИБДЕНА, РУДЫ И ИХ ОБОГАЩЕНИЕ 7
1. Краткие исторические сведения............................. 7
2. Геохимия молибдена...................................... &
3. Минералы и руды молибдена .................... . . 10'
4. Обогащение молибденовых руд............................. 13.
5. Состав молибденовых концентратов и некоторых продуктов
их переработки ........................................ 18
Глава II. ОКИСЛИТЕЛЬНЫЙ ОБЖИГ МОЛИБДЕНИТОВЫХ
КОНЦЕНТРАТОВ.................................................. 21
6. Физико-химические основы окислительного обжига молиб-
денитовых концентратов ............. - . . 21
7. Практика окислительного обжига......................... 37
Глава III. ПРОИЗВОДСТВО ТРЕХОКИСИ МОЛИБДЕНА . . 50-
Производство трехокиси молибдена гидрометаллургическим способом . 50
8. Выщелачивание ........................................ 50
9. Очистка аммиачных растворов от примесей осаждением
сульфидов .................................... 57
10. Выделение молибдена из аммиачных растворов в виде по-
лимолибдатов .................................... 58
11. Распределение примесей и чистота трехокиси молибдена . 69
12. Извлечение молибдена из хвостов после выщелачивания
огарков................... ... . . 71
Производство трехокиси молибдена методом возгонки.... . 75
13. Физико-химические основы процесса возгонки трехокиси
молибдена из огарков .......................... . . 76
14. Технология возгонки трехокиси молибдена............... 87
15. Чистота и физические свойства возогнанной трехокиси
молибдена '...................................... 90
Получение трехокиси молибдена из отходов обработки и вторичного ме-
талла ...................................................... 91
Получение трехокиси молибдена из молибдата кальция................. 94
1* 3
Глава IV. ПРОИЗВОДСТВО ФЕРРОМОЛИБДЕНА ....................... 95
16. Силикотермический способ ........................ 96
17. Карботермический способ ......................... 97
Глава V. ПЕРЕРАБОТКА РАЗЛИЧНЫХ ПОЛУПРОДУКТОВ
ОБОГАЩЕНИЯ................................................... 99
18. Получение молибдата кальция из молибденитовых и повел-
литовых полупродуктов .............................. 99
19. Извлечение молибдена из бедных ферримолибдитовых руд
и концентратов ................................... 106
20. Извлечение молибдена из шеелитовых концентратов и полу-
продуктов ...................................... . . 113
21. Переработка вульфенитовых концентратов.......... 117
Глава VI. ГИДРОМЕТАЛЛУРГИЧЕСКИЕ СПОСОБЫ
РАЗЛОЖЕНИЯ МОЛИБДЕНИТОВЫХ КОНЦЕНТРАТОВ 119
22. Разложение азотной кислотой ................... 119
23. Окисление молибденита кислородом под давлением (авто-
клавный процесс) .................................. 122
24. Окисление молибденита растворами гипохлорита натрия 131
25. Другие способы окисления молибденита........... 136
Глава VII. СПОСОБЫ ВЫДЕЛЕНИЯ МОЛИБДЕНА ИЗ СЛОЖНЫХ
И БЕДНЫХ РАСТВОРОВ.......................................... 140
26. Осаждение двуокиси молибдена .................. 140
27. Выделение полимолибдата аммония из растворов молиб-
дата натрия ....................................... 146
28. Осаждение трисульфида молибдена 147
29. Осаждение ферримолибдатов ..................... 149
30. Извлечение молибдена из растворов сорбцией на ионооб-
менных смолах и углях ............................. 150
31. Извлечение молибдена из растворов экстракцией орга-
ническими растворителями ................. . . 158
Глава VIII. ХЛОРИСТЫЕ СОЕДИНЕНИЯ МОЛИБДЕНА ................. 175
32. Пятихлористый молибден . ............ 177
33. Низшие хлориды молибдена . . . . 186
34. Оксихлориды молибдена ........................ 191
35. Хлорирование кислородных соединений молибдена . . . 196
Глава IX. СУЛЬФИДЫ МОЛИБДЕНА ............................... 209
36. Полуторасульфид Mo2S3 ......................... 209
37. Трисульфид молибдена MoS3.................... 212
38. Дисульфид молибдена ........................... 214
Глава X. ДРУГИЕ СОЕДИНЕНИЯ МОЛИБДЕНА ....................... 233
39. Силициды молибдена ............................. 233
40. Бориды молибдена ............................. 240
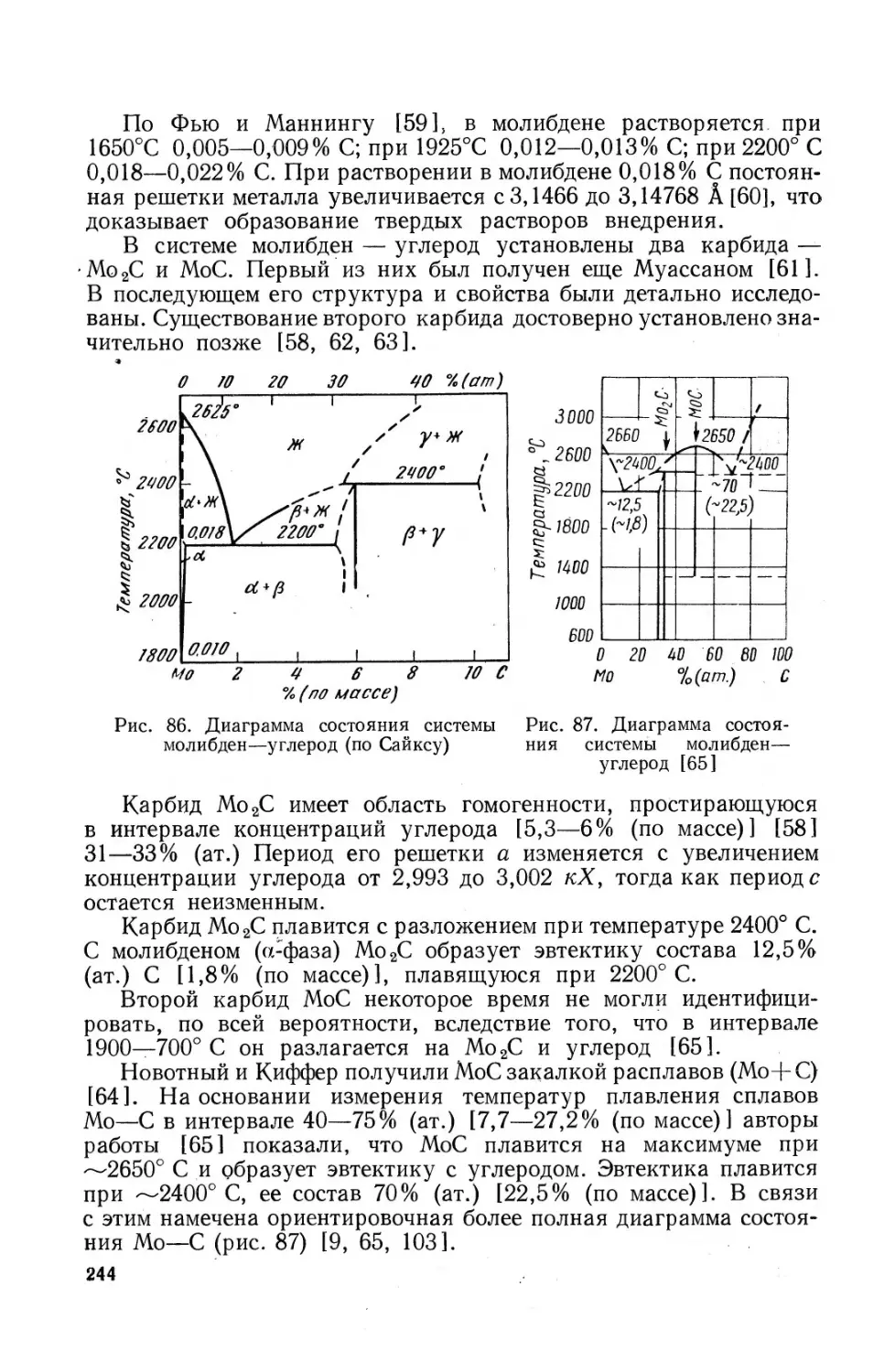
41. Карбиды молибдена..................... . . . 243
42. Карбонил молибдена Мо(СО)6 ................... 248
Глава XI. ПРОИЗВОДСТВО ПОРОШКООБРАЗНОГО МОЛИБДЕНА 254
43. Окислы молибдена ............................ 254
44. Физико-химические условия восстановления трехокиси
молибдена водородом ............................... 264
45. Производственная практика восстановления МоО3 водо-
родом ............................................. 268
4
46. Другие варианты восстановления трехокиси молибдена
водородом ............................................... 282
47. Карботермическое восстановление кислородных соедине-
ний молибдена ..................................... 285
48. Восстановление трехокиси молибдена кальцием и магнием 290
49. Получение молибдена из молибденита .... 292
50. Получение молибдена из карбонила . . 295
51. Получение молибдена из галогенидов . . .... 299
52. Получение молибдена электролизом .................... 306
Глава XII. ПОРОШКОВАЯ МЕТАЛЛУРГИЯ МОЛИБДЕНА ... 315
53. Прессование заготовок молибдена..................... 315
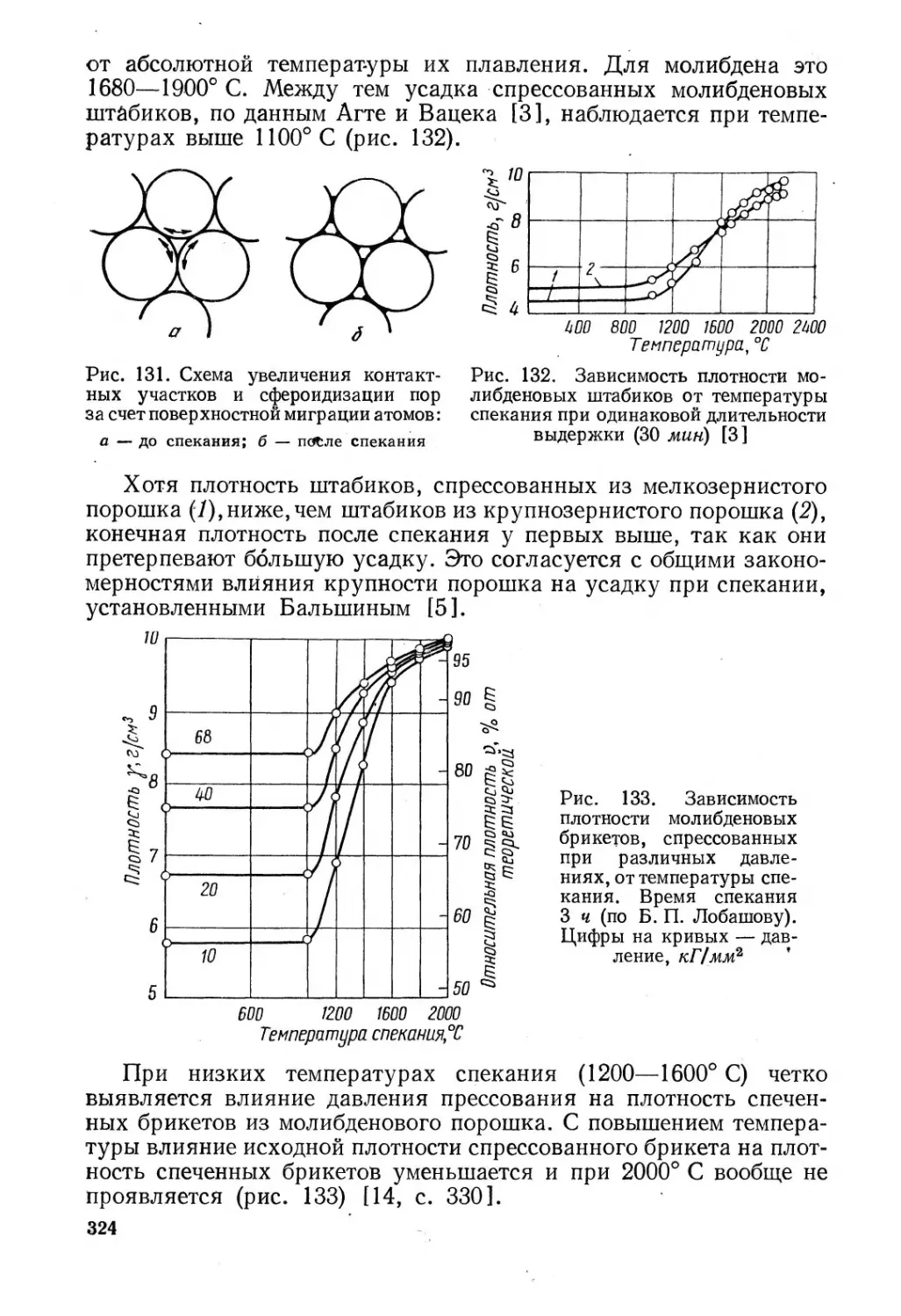
54. Спекание спрессованных заготовок молибдена.......... 322
Глава XIII. ПЛАВКА МОЛИБДЕНА .............. 338
55. Дуговая плавка ..................................... 338
56. Электронно-лучевая плавка .......................... 347
57. Получение монокристаллических слитков молибдена 352
Глава XIV. ОБРАБОТКА ДАВЛЕНИЕМ, МЕХАНИЧЕСКИЕ
СВОЙСТВА, МЕТАЛЛОГРАФИЯ МОЛИБДЕНА . . . . 362
58. Обработка давлением................................. 362
59. Механические свойства молибдена .................... 368
60. Металлографический контроль в производстве молибдена 384
Глава XV. СВОЙСТВА И ОБЛАСТИ ПРИМЕНЕНИЯ МОЛИБДЕНА 387
61. Физические свойства . . ..................... 387
62. Химические свойства ................................. 392
63. Защита молибдена от окисления........................ 395
64. Области применения молибдена .......... . . 397
Глава XVI. ПОПУТНОЕ ИЗВЛЕЧЕНИЕ РЕНИЯ
В ПРОИЗВОДСТВЕ МОЛИБДЕНА.......................................... 402
65. Общие сведения о рении ............................. 402
66. Сырьевые источники рения ........................... 407
67. Извлечение рения из различных отходов переработки
молибденитовых концентратов ...................... 408
68. Получение рения .................................. 413
Литература ...................................................... 420
ПРЕДИСЛОВИЕ
Молибден не новый металл в современной технике. До недав-
него времени его использовали преимущественно в производстве
легированных сталей и некоторых сплавов с цветными металлами.
Применение чистого металла ограничивалось производством элек-
троосветительных и электровакуумных приборов, а также нагре-
вателей высокотемпературных печей. Положение существенно из-
менилось в последнее десятилетие, когда молибден и сплавы на
его основе стали наиболее перспективными жаропрочными кон-
струкционными материалами в новых областях техники. В связи
с этим развились методы дуговой и электронно-лучевой плавки,
позволяющие получать заготовки и изделия крупных размеров
из чистого и легированного молибдена.
С увеличением производства и расширением областей исполь-
зования молибдена появились новые исследования, охватившие
разнообразные направления технологии молибдена — производ-
ство химических соединений высокой чистоты, металлических
порошков и компактных заготовок, монокристаллических слитков
и сплавов.
Автор поставил перед собой задачу — осветить современное
состояние технологии молибдена, уделяя главное внимание физико-
химическим основам процессов. Значительное место в книге отве-
дено новым технологическим направлениям в гидрометаллургии
молибдена, производстве порошков и компактного металла.
Книга, по возможности полно отражая наиболее существенные
работы, опубликованные в советской и зарубежной печати, одно-
временно является итогом многолетних исследований, проводимых
автором и его сотрудниками в области разработки процессов полу-
чения химических соединений молибдена из рудных концентратов.
Автор выражает глубокую благодарность профессору
Н. С. Грейверу за ценные замечания, сделанные при рецензирова-
нии рукописи книги.
6
ГЛАВА I
МИНЕРАЛЫ МОЛИБДЕНА,
РУДЫ И ИХ ОБОГАЩЕНИЕ
1. КРАТКИЕ ИСТОРИЧЕСКИЕ СВЕДЕНИЯ
Название «молибден» происходит от греческого слова «молиб-
дос», означающего в переводе «свинец». До конца XVIII в. этим сло-
вом называли не только свинец и свинцовый блеск, но и похожие
на свинец сульфидные минералы, а также графит. Молибденит
(MoS2) и графит в течение многих столетий считали идентичными.
Лишь в 1778 г. шведский химик К. В. Шееле установил неиден-
тичность графита и молибденита и открыл новый элемент, назван-
ный молибденом. Разлагая молибденит азотной кислотой, Шееле
выделил молибденовую кислоту, получил трехокись и некоторые
соли молибдена 11]. В 1782 г. соотечественник Шееле Гьельм
получил металлический порошок молибдена восстановлением трех-
окиси молибдена древесным углем [2]. Более чистый металл был
позже получен Берцелиусом восстановлением трехокиси молиб-
дена водородом.
Применение молибдена было известно задолго до его открытия.
Так, в XIV в. в Японии изготовляли сабли из стали, содержащей
молибден; в XVI в. молибденит употребляли, подобно графиту,
в качестве карандаша. Примерно во второй половине XIX в. в тех-
нике начали использовать некоторые соединения молибдена,
в частности молибдат аммония как реактив при определении фос-
фора и молибденовую синь как краситель.
К 1900 г. мировое производство молибдена составляло всего
лишь 11m. Стимулом к развитию добычи молибденовых руд послу-
жило открытие в конце XIX в. влияния присадок молибдена на
свойства сталей.
В 1896 г. молибденовая сталь с содержанием 3,72% Мо и
0,52% С была выплавлена на Путиловском заводе в Петербурге [3 ].
Было установлено, что влияние молибдена на свойства стали ана-
логично влиянию присадок вольфрама. В этот же период начали
выплавлять стали с присадками молибдена во Франции (заводы
7
Крезо, 1894 г.) и в США (1898 г.) при изготовлении броневых и ин-
струментальных сталей.
Для присадок молибдена в стали в 1900 г. был разработан про-
цесс выплавки ферромолибдена [4].
В годы, предшествовавшие первой мировой войне, в Германии
на заводах Круппа начали применять молибден в производстве ста-
лей для орудийных и ружейных стволов, броневых плит, броне-
бойных снарядов, гранат, котлов высокого давления в военном
флоте, частей подводных лодок. Это вызвало развитие производ-
ства молибденовых сталей в Англии и других странах.
В этот же период (1915—1917 гг.) в России в небольших коли-
чествах была начата добыча молибденовых руд Чикойского место-
рождения в Забайкалье.
Промышленное производство металлического молибдена и при-
менение его в электроосветительной технике началось примерно
в те же годы, что и производство вольфрама (1909—1910 гг.), когда
был разработан металлокерамический способ получения этих ме-
таллов в компактной форме.
До 1925 г. мировая добыча молибдена не превышала в среднем
нескольких тысяч тонн. Интенсивное развитие производства молиб-
дена началось с 1925 г. после открытия крупнейшего месторождения
Клаймакс в США. К этому времени были усовершенствованы спо-
собы обогащения бедных, в частности мелковкрапленных, молиб-
деновых руд, изучены свойства различных сталей, легированных
молибденом, установлена возможность замены вольфрама молибде-
ном в быстрорежущих сталях, наметились широкие возможности
его использования в развивающейся электровакуумной и электро-
осветительной технике, а также в сплавах с цветными металлами.
Производство молибденовой продукции резко возросло в трид-
цатых годах и достигло в капиталистических странах в период
второй мировой войны к 1943 г. 31,3 тыс. т металла в концентра-
тах. В последующие годы производство сохранялось в среднем на
уровне 25—30 тыс. т, в 1960—1965 гг. составляло 40—44 тыс. т
в год, а в 1967 г. достигло 56 тыс. т.
Значительным достижением в технологии производства молиб-
дена явилось освоение в начале 50-х годов промышленной выплавки
крупных слитков молибдена, что расширило возможности приме-
нения молибдена и сплавов на его основе в качестве жаропрочного
конструкционного материала.
Производство металлического молибдена создано в СССР
в 1928 г., а тремя годами позже — в 1931 г. — возникло произ-
водство ферромолибдена [5]. В эти же годы началась промышлен-
ная эксплуатация открытых отечественных месторождений молиб-
деновых руд — Чикойского, а затем Умальтинского. В последую-
щие годы в результате активной геологической разведки было от-
крыто большое Число месторождений, что полностью обеспечило
отечественную молибденовую промышленность рудным сырьем.
8
2. ГЕОХИМИЯ МОЛИБДЕНА
По последним оценкам Виноградова [6] среднее содержание
молибдена в земной коре 1,1 • 10~4 % (по массе). Данные о содержа-
нии молибдена в породах различного типа, морской и речной
воде, растениях и почвах приведены в табл. 1, заимствованной
из работы Михайлова [7].
РАСПРЕДЕЛЕНИЕ МОЛИБДЕНА В ПРИРОДЕ
ТАБЛИЦА 1
Объект исследования Среднее содержание молибдена, п-1*0~4, % (по массе) Литератур- ная ссылка
Земная кора Морская и океанская вода Речная вода Сланцы и глины Песчаники Карбонатные породы Кремнистые породы Растения Почвы 1,1 0,01 0,001 2,0 1,0 1,2 0,8 0,4 0,3 0,2 2,2 [6] [12] [8] 6 [8 9 8 [8 [8] [Ю] [7] *
* Усредненное значение.
Курода и Сенделл отмечают, что кислые и основные породы
мало отличаются по содержанию молибдена. Так, среднее содер-
жание молибдена в гранитных породах 1,1-10“4%, базальтах и
диабазах 1,1 • 10-4%, габбро 0,6-10"4%. Ультраосновные породы
содержат меньше молибдена, чем другие типы пород, — 0,4 X
X 10-4% [8].
В условиях первичных магматических процессов молибден
отличается высоким сродством к сере и значительно меньшим
к кислороду. Поэтому в первичных рудах эндогенных месторожде-
ний молибден представлен преимущественно сульфидом четырех-
валентного молибдена (минерал молибденит). В зонах окисления
преобладают кислородные соединения шестивалентного молиб-
дена (молибдаты), образовавшиеся в результате окисления молиб-
денита.
Хрущов дает следующую общую геохимическую характери-
стику молибдена [11].
В земной коре максимальные концентрации молибдена соз-
даются в результате гидротермальной деятельности интрузий
гранитоидной магмы, что приводит к образованию основных про-
мышленных месторождений эндогенного типа.
9
В экзогенных условиях молибден (в форме кислородных соеди-
нений) концентрируется в осадочных породах, в особенности
в кремнисто-углистых глинистых сланцах и углях, где он тесно
связан с органическим веществом. Значительные концентрации мо-
либдена известны также в нефти и твердых нефтебитумах. Гео-
химия молибдена в окисленной зоне рассмотрена в работе [7].
Характерны ассоциации молибдена в природных условиях со
следующими элементами [11]:
с оловом, висмутом и бериллием в высокотемпературных пег-
метитовых и пневматолитовых месторождениях. Молибден в них
большей частью присутствует как второстепенная примесь;
с вольфрамом, преимущественно в скарновых месторождениях,
где часто образуются крупные концентрации обоих металлов;
с медью в гидротермальных месторождениях, из которых мно-
гие имеют важное промышленное значение (медно-порфировые
РУДЫ);
со свинцом и цинком, а также с ураном (уран-молибденовая
рудная формация) в сравнительно низкотемпературных месторо-
ждениях;
с ванадием и ураном в экзогенных условиях образования
в углистых сланцах, углях и нефтебитумах.
3. МИНЕРАЛЫ И РУДЫ МОЛИБДЕНА
Известно около 20 минералов молибдена. Наиболее часто встре-
чающиеся из них: молибденит MoS2; повеллит СаМоО4; молибдит
(ферримолибдит) Fe2 (МоО4)3-пН2О; вульфенит РЬМоО4; молибдо-
шеелит Са (W,Mo)O4; чиллагитРЬ(Мо,\¥)О4; кёхлинитВ[2(МоО4)О2;
линдгренит Си3 (МоО4)2 (ОН)2; ильземанит МоО3- SO3- 5Н2О; ура-
номолибдат UO2-UO3-2MoO3.
Промышленное значение имеют лишь первые четыре минерала.
Молибденит MoS2 -— самый распространенный минерал молиб-
дена. Подавляющую часть молибдена добывают из недр в форме
молибденита.
Молибденит мягкий свинцово-серого цвета минерал с металли-
ческим блеском. Плотность MoS2 4,7—4,8, твердость по шкале
Мооса 1—1,5. Минерал имеет гексагональную кристаллическую
структуру слоистого типа (см. стр. 221). Слои ионов молибдена
расположены между двумя слоями ионов серы, образуя трехслой-
ные пакеты. Структура молибденита хорошо объясняет совершен-
ную спайность его кристаллов. Она обусловлена слабой связью
между тройными слоями S—Мо—S.
В последнее время найдена редко встречающаяся ромбоэдри-
ческая модификация молибденита в Армении [13], Альпах [14]
и в Канаде [15]. Найдено, что образованию ромбоэдрического
молибденита способствует повышенное содержание примеси ре-
ния [13]. При нагревании без доступа воздуха до 1300—1350° С
молибденит частично диссоциирует, при 1650—1700° С плавится
10
с разложением [16]. При нагревании на воздухе до 500° С минерал
легко окисляется до МоО3. Азотная кислота и царская водка окис-
ляют молибденит. Молибденит залегает большей частью в кварце-
вых жилах, часто ассоциируется с шеелитом, вольфрамитом, кас-
ситеритом, халькопиритом, арсенопиритом, висмутовым блеском.
В виде изоморфной примеси в молибденитах встречается элемент
рений (0,04—0,0001%), который попутно извлекается при пере-
работке молибденитовых концентратов.
В результате выветривания в верхних зонах рудных жил мо-
либденит медленно окисляется, образуя молибденовые охры, мо-
либдит, повеллит, вульфенит и другие вторичные минералы.
Повеллит СаМоО^ — наиболее распространенный минерал зон
окисления молибденовых месторождений. Обычно встречается
в форме пленок на молибдените (на поверхности и по плоскостям
спайности). Цвет грязно-серый, иногда зеленоватый. Кристалли-
зуется в тетрагональной сингонии, вид симметрии — бипирами-
дальный. Плотность 4,35—4,52, твердость 3,5. Вторичный повел-
лит образуется в результате окисления молибденита и взаимодей-
ствия образующейся трехокиси молибдена (или молибденовой
кислоты) с кальцийсодержащими поверхностными водами.
Встречается, хотя и редко, первичный (гидротермальный) по-
веллит в виде кристалликов желтовато-зеленого цвета. Первичный
повеллит может содержать изоморфную примесь вольфрама (до 8%).
Повеллит легко растворяется в соляной кислоте, чем поль-
зуются в химическом анализе для раздельного определения окис-
ленного и сульфидного молибдена.
Молибдит (ферримолибдит) — молибдат трехвалентного же-
леза Fe2 (МоО4)3-пН2О (п = 7 4-8). Цвет лимонно-желтый. Часто
встречается в зонах окисления молибденитовых руд в участках
с повышенным содержанием минералов железа. В отличие от по-
веллита, молибдит не образует псевдоморфоз по молибдениту,
а выносится из первичных мест залегания в пределах 0,5—1 м и
отлагается в трещинах и пустотах выщелачивания [11]. Плот-
ность молибдита 4,5. Минерал растворяется в соляной и серной
кислотах, разлагается растворами аммиака и щелочей. Скопления
нормального ферримолибдата, отвечающего написанной выше фор-
муле, редки. Известно, что из водных растворов ферримолибдат
стехиометрического состава получается в узких пределах pH =
= 3,5 4-3,6 [17]. Большей частью в зонах окисления имеются
скопления молибденсодержащих гидроокислов и окислов железа
с переменным соотношением Fe2O3 : МоО3, состав которых можно
выразить общей формулой xFe2O3-#MoO3-zH2O. Молибдит и мо-
либденсодержащие окислы железа представляют некоторый ин-
терес как источник молибдена при разработке месторождений
с развитыми зонами окисления. Так, в крупнейшем месторождении
США Клаймакс в верхних зонах месторождения около 25% Мо
присутствует в виде молибдита.
и
Вульфенит РЬМоО^ встречается в зоне окисления свинцовых
месторождений, содержащих молибденит, или в присутствии
свинца в молибденовых рудах. Цвет бурый, иногда желтый, желто-
зеленый, красный, что обусловлено содержанием примесей (меди,
вольфрама, хрома, ванадия). Кристаллизуется в тетрагональной
сингонии. Плотность 6,3—7, твердость 2,5—3. Промышленное
значение вульфенита в настоящее время весьма малое.
Основную массу молибдена добывают в настоящее время из эндо-
генных месторождений гидротермального происхождения. Концен-
трация молибдена в рудах низкая. Эксплуатируются руды, содер-
жащие десятые и сотые доли процента молибдена.
Генетические типы месторождений молибдена и их характери-
стика освещены в монографии Хрущова [11].
С точки зрения задач обогащения можно ограничиться подраз-
делением молибденовых руд на следующие три группы:
1. Простые кварцево-молибденитовые
р у д ы, в которых содержание других сульфидов (пирит, халько-
пирит) незначительное. Молибденит залегает в кварцевых жилах,
его содержание в рудах составляет десятые доли процента, иногда
до 1%. К этому типу относится крупнейшее в мире молибденовое
месторождение Клаймакс (Колорадо, США).
2. Медно-молибденовые руды, в которых содер-
жание сульфидов меди и пирита преобладает. К медно-молибдено-
вым относятся штокверкововкрапленные руды, связанные со вто-
ричными кварцитами, а также медные порфировые руды, содержа-
щие молибденит. Содержание молибдена в медно-молибденовых
рудах обычно низкое и выражается в сотых, а иногда тысячных
долях процента. В СССР к этому типу руд относятся руды таких
крупных медных месторождений, как Коунрадское и Бощекуль-
ское в Казахстане, Алмалык в Узбекистане, Каджаранское в За-
кавказье [11, 19]. Эти руды являются важным источником полу-
чения молибденовых концентратов.
3. Скарновые молибдено-вольфрамовые
р у д ы. В рудах этого типа молибденит вместе с шеелитом и суль-
фидами (пирит, халькопирит) находится в кварцевых прожилках
и гнездах, заполняющих трещины в скарнах (окремненных из-
вестняках, содержащих силикаты кальция, железа, алюминия).
Наряду с молибденитом обычно содержится повеллит.
К этому типу относится одно из крупнейших в мире Тырны-
Аузское месторождение молибдено-вольфрамовых руд на Север-
ном Кавказе [11, 19 ].
В рудах всех типов в той или иной мере могут быть развиты
зоны окисления, в которых содержатся вторичные минералы — по-
веллит, ферримолибдит, молибденсодержащие окислы железа. На
отдельных участках месторождений руды могут быть чисто окис-
ленными (почти не содержащими молибденита) или смешанными,
в которых наряду с молибденитом присутствуют окисленные ми-
12
нералы. Типичный пример — Сорское месторождение в Хакассии,
где в зоне окисления, простирающейся на глубину 25—50 м, на-
ходятся чисто окисленные и смешанные руды [22].
Разведанные запасы молибдена в капиталистических странах
приведены в табл. 2 [18].
ТАБЛИЦА 2
РАЗВЕДАННЫЕ ДОСТОВЕРНЫЕ ЗАПАСЫ
МОЛИБДЕНА В КАПИТАЛИСТИЧЕСКИХ СТРАНАХ, ТЫС. Т
(ПО СОСТОЯНИЮ НА 1967 г.)
Страна или территория Запасы Страна или территория Запасы
Всего 2870 Филиппины 1,5
В том числе: Канада 320
США 1800 Норвегия 3
Чили 550 Мексика 1
Перу 100 Япония 2
Гренландия 90 * Турция 2
* Оценка.,
Наиболее крупные производители молибденовых рудных кон-
центратов в капиталистических странах — США, Чили, Перу и
Канада.
4. ОБОГАЩЕНИЕ МОЛИБДЕНОВЫХ РУД
Основной способ обогащения молибденовых руд — флотация.
Лишь при доводке концентратов иногда используют методы магнит-
ного обогащения (для удаления минералов железа, отделения воль-
фрамита), а также применяют химические методы для удаления
примесей. Теория и практика флотации молибденовых руд различ-
ного типа рассмотрена в капитальных монографиях Митрофа-
нова [19], Полькина [20], Фишмана и Соболева [21]. В этих
книгах обобщен накопленный опыт флотации молибдена на оте-
чественных и зарубежных предприятиях.
Получение молибденитовых концентратов
Молибденит — легко флотируемый минерал, обладающий гид-
рофобностью и хорошо смачиваемый углеводородами. Эффектив-
ными собирателями при флотации молибденита служат обычно
керосин или трансформаторное масло, веретенное масло или дру-
гие нейтральные масла. Их добавляют частично при измельчении
и частично — при флотации вместе с пенообразователями — сос-
новым маслом или ксиленолом. Молибденит может флотировать
с одним лишь вспенивателем, однако собиратели (керосин, масла)
повышают флотируемость молибденита. Флотацию молибденита
ведут в содовой среде при pH = 7,5 ч-8 или несколько выше.
и
При обогащении простых кварцево-молибде-
нитовых РУД флотацию первоначально ведут при крупном
измельчении, получая грубые молибденовые концентраты, содер-
жащие большие количества кварца в сростках с молибденитом. Гру-
бые концентраты затем дополнительно измельчают (для разруше-
ния сростков) и проводят перечистную флотацию. Если необхо-
димо, проводят несколько стадий измельчения и перечистки,
в особенности при тонкой вкрапленности молибденита.
Для отделения примесей сульфидов (пирита, халькопирита)
перечистку проводят, добавляя депрессоры сульфидов меди и же-
леза — сернистый натрий или цианиды, а иногда — смеси этих
депрессоров. В случае присутствия в руде или образования при
измельчении тонких силикатных шламов для лучшего их отделе-
ния в перечистные операции добавляют жидкое стекло.
На рис. 1 в качестве примера приведена схема обогащения квар-
цево-молибденитовой руды на предприятии Клаймакс. Из руды,
содержащей 0,2—0,5% молибдена, 1—3% пирита, 0,02% меди
(халькопирит), получают молибденитовый концентрат, содержащий
90% MoS2 или 54% Мо. В концентрат извлекают 90% Мо от об-
щего его содержания и 98% от содержания сульфидного молибдена.
Более сложны схемы обогащения медно-молибдено-
вых руд. Наиболее распространены схемы, в которых перво-
начально получают коллективный медно-молибденовый концен-
трат, а затем проводят селективную флотацию с выделением молиб-
денового и медного концентратов.
При флотации коллективного медно-молибденового концен-
трата на фабриках СССР большей частью используют реагенты:
трансформаторное масло, ксантогенат (собиратель для сульфидов
меди), терпинеоль (вспениватель). Состав коллективных концен-
тратов зависит от соотношения меди и молибдена в исходной руде.
Так, если исходная руда включает 0,7% Си и 0,01% Мо, в’коллек-
тивном концентрате содержится —25—30% Си, —0,3—0,5% Мо.
Из медной порфировой отечественной руды, содержащей 1% Си
и 0,005% Мо, получают коллективный концентрат с содержанием
15—18% Си и 0,05% Мо.
Возможны два пути разделения коллективного медно-молибде-
нового концентрата:
1) флотация молибденита при депрессии сульфидов меди сер-
нистым натрием или цианидами;
2) флотация сульфидов меди при депрессии молибденита крах-
малом.
В промышленной практике распространен первый метод.
Коллективный концентрат сгущают (для удаления избытка реа-
гентов), добавляют сернистый натрий или цианиды для депрессии
сульфидов меди и пирита и флотируют молибденит с нейтральными
маслами и пенообразователями, проводя многократные его пере-
чистки.
14
PydaOfiKHo
| 12,5 мм
Измельчение
Классификация
Основная флотация
Очистная
Контрольная
Классификация
Измельчение Фло/Ь
'ация
Контрольная
Классификация
Флотация
Изнельчение
Сгущение
Слив
Флотация
Флотация
Классификация
ьчение
Флотация
Очистная
Перенашивание
Перемешивание
Перечистная
Ио концентрат
Хвосты
Рис. 1. Схема флотации кварцево-молибденитовой руды на фабрике Клаймакс
15
Для снижения расхода сернистого натрия пульпу коллектив-
ного концентрата пропаривают с известью при 85° С. Известь раз-
рушает пленки собирателя (ксантогената) на зернах окисленных
медных минералов, вызывающих повышенный расход депрессора.
На рис. 2 показана схема флотации медно-молибденовой руды
на Каджаранской фабрике [21]. В руде медь представлена в ос-
новном халькопиритом, молибден — молибденитом. Основная
пустая порода — кварц.
Слив классификатора
55-607. -0,07Ьнн
Перевешивание
Основная
коллективная флотация
Рис. 2. Схема флотации медно-молибденовой руды на Каджаранской фабрике
16
При разделении коллективного концентрата молибден фло-
тируют, используя в качестве депрессора халькопирита сернистый
натрий, причем для снижения его расхода применяют пропарку
с известью. Вспенивателем служит терпинеоль. На фабрике полу-
чают молибденитовый концентрат следующего примерного со-
става: 49% Мо, 0,8% Си, 5,74% SiO2, 1,68% Fe. Медный кон-
центрат содержит 14% Си, 22,6% SiO2 и 21,4% Fe.
При селективной флотации молибдена из коллективного кон-
центрата с низким содержанием молибдена (например, 0,05% Мо
и 15—18% Си) часто ограничиваются получением молибденового
полупродукта с концентрацией молибдена 15—20%, меди 0,5—2%,
высоким содержанием SiO2, Fe и других примесей. Такой полу-
продукт поступает на гидрометаллургическую переработку, в ре-
зультате которой получают молибдат кальция (см. гл. V) [20].
Комбинирование флотации с гидрометаллургическим обогащением
обеспечивает более высокое извлечение молибдена в концентрат.
При обогащении молибденито-шеелитовых
скарновых руд (типа тырны-аузских) первоначально выде-
ляют флотацией молибденит. Реагентами-собирателями в молиб-
деновой флотации служат керосин, сосновое или трансформатор-
ное масло и аэрофлот (дитиофосфат); для депрессии сульфидов
меди, железа, мышьяка используют цианиды или цианиды с сер-
нистым натрием.
В хвостах сульфидной флотации остаются шеелит и повеллит.
В тырны-аузских рудах значительная часть повеллита изоморфно
связана с шеелитом. Шеелит вместе с повеллитом флотируют, ис-
пользуя в качестве собирателя олеиновую кислоту в содовой
среде. Для депрессии минералов пустой породы вводят жидкое
стекло [20]. Получаемые шеелитовые концентраты содержат 5—
6% Мо. Извлечение из них молибдена может быть осуществлено
гидрометаллургическим способом с выделением вольфрамового
и молибденового химических концентратов (см. гл. V).
Флотация окисленных молибденовых руд
Значительные запасы молибдена заключены в рудах, содер-
жащих окисленные минералы молибдена, — повеллит, ферримо-
либдит, молибденсодержащие окислы и гидроокислы железа.
Вследствие трудностей обогащения извлечение молибдена из окис-
ленных руд осуществляется лишь в ограниченных масштабах.
Трудности обогащения окисленных молибденовых руд обуслов-
лены следующими обстоятельствами [20]:
1. Окисленные минералы молибдена находятся в тонкодисперс-
ной вкрапленности, часто в форме охристых образований, которые
легко шламуются при измельчении и теряются в хвостах при обо-
гащении.
2. Тесная связь повеллита с другими кальциевыми минералами
(кальцит, флорит, апатит), а ферримолибдита с окислами и гидро-
2 А. Н. Зеликман 17
окислами железа ограничивает возможности обогащения. Удается
получить лишь бедные концентраты.
Следует при этом учитывать низкое содержание молибдена в ру-
дах зон окисления (0,01—0,05%).
В СССР в промышленных масштабах осуществляют флотацию
некоторых смешанных руд, содержащих, наряду с молибденитом,
повеллит, который извлекают из хвостов сульфидной флотации.
Флотацию повеллита проводят в щелочной среде (pH 10,2)
с олеиновой кислотой или другими собирателями типа жирных
кислот. Для депрессии кальциевых и других минералов пустой
породы добавляют жидкое стекло. Пенообразователь — сосновое
масло. После ряда перечисток, перед которыми проводят пропарку
при 80—90° С с добавкой жидкого стекла, получают бедные повел-
литовые концентраты, содержащие 6—12% Мо, 2—3% Си, 30—
35% СаО, 3—8% Fe, 4—6% S. Их направляют на гидрометаллур-
гическую переработку.
Сложная проблема — извлечение молибдена из окисленных
ферримолибдитовых руд [20—22]. Ввиду тесной связи молиб-
дена в этих рудах с окислами железа обогащение сводится к извле-
чению в концентрат минералов железа.
Исследования ряда институтов по обогащению сорских ферри-
молибдитовых руд, содержащих 0,03—0,05% Мо, 55—60% SiO2,
2—3% Fe, показали возможность получения флотационных кон-
центратов с содержанием 0,3—0,5% Мо при общем извлечении
молибдена в концентрат примерно 65—70% [22].
Ферримолибдит и молибденсодержащие окислы железа можно
флотировать, используя в качестве собирателя олеиновую кислоту,
вспенивателя — сосновое масло. Дорогую олеиновую кислоту
можно заменять продуктами перегонки нефти (асидол, мылонафт,
окисленный керосин, окисленный петролатум) или продуктами
жирового производства — отходами щелочной рафинации расти-
тельных масел (соапсток и др.). Хорошие результаты получены при
использовании окисленного петролатума и соапстока [221.
Из получаемых бедных окисленных ферримолибдитовых кон-
центратов молибден, как показали исследования, может быть из-
влечен гидрометаллургическими способами (см. гл. V).
5. СОСТАВ МОЛИБДЕНОВЫХ КОНЦЕНТРАТОВ
И НЕКОТОРЫХ ПРОДУКТОВ ИХ ПЕРЕРАБОТКИ
Выпускаемые в СССР молибденитовые концентраты по содержа-
нию молибдена и примесей должны удовлетворять требованиям,
приведенным в табл. 3.
Кроме молибденитовых концентратов, выпускают химические
концентраты — продукты гидрометаллургической переработки
полупродуктов обогащения. К ним относятся молибдат кальция
(МДК) — продукт переработки медно-молибденовых и повелли-
18
ТАБЛИЦА 3
МАРКИ И СОСТАВ МОЛИБДЕНИТОВЫХ* КОНЦЕНТРАТОВ В СССР
Марка Молибден, %, не менее Примеси, %, не более
S1O2 As Sn Р Си
КМ1 50,0 5,0 0,07 0,07 0,07 0,5
КМ2 48,0 7,0 0,07 0,07 0,07 1,00
кмз 47,0 9,0 0,07 0,07 0,15 2,00
товых концентратов и концентраты типа КМГ, получаемые при
переработке шеелито-повеллитовых полупродуктов обогащения.
Требования к составу химических концентратов приведены
в табл. 4 и 5.
ТАБЛИЦА 4
ТЕХНИЧЕСКИЕ УСЛОВИЯ НА МОЛИБДАТ КАЛЬЦИЯ
Марка Мо, %, не менее Примеси, %, не более
Са Р S
МДК1 44 22 0,1 0,2
МДК2 40 24 0,2 0,3
ТАБЛИЦА 5
СОСТАВ КОНЦЕНТРАТОВ, ПОЛУЧАЕМЫХ ПРИ ГИДРОМЕТАЛЛУРГИЧЕСКОЙ
ПЕРЕРАБОТКЕ ШЕЕЛИТО-ПОВЕЛЛИТОВЫХ ПОЛУПРОДУКТОВ
Марка Мо, %, не менее Примеси, %, не более
Si О 2 As Sn Р Си wo3
КМГ1 50 3,0 0,10 0,05 0,05 0,15 3,0
КМГ2 48 5,0 0,15 0,07 0,07 0,25 4,0
Молибденовые концентраты служат исходным сырьем для вы-
плавки ферромолибдена, используемого для присадок
молибдена в стали, и для получения химическихсоеди-
нений молибдена высокой чистоты. Важнейшее из
них—трехокись молибдена, которая является исходным соеди-
нением для производства молибдена.
Лимитирование содержания ряда примесей в концентратах
диктуется требованиями производства ферромолибдена. Известно,
что сера вызывает красноломкость стали, мышьяк и фосфор —
хладноломкость, медь способствует увеличению зерна и делает
сталь хладноломкой, олово понижает режущие свойства инстру-
ментальных сталей.
2*
19
Концентраты марок КМ1 и КМ2 предназначены для выплавки
ферромолибдена марки Mol, а марки КМЗ —для ферромолибдена
марок Мо2 и МоЗ (табл. 6).
ТАБЛИЦА 6
СОДЕРЖАНИЕ ПРИМЕСЕЙ В ФЕРРОМОЛИБДЕНЕ (НЕ МЕНЕЕ 55% Мо)
«
Примеси, %, не более
Марка с Si S р Sb Си Sn
Mol 0,10 1,00 0,10 0,10 0,05 0,80 0,05
Мо2 0,15 1,50 0,15 0,15 0,08 1,5 0,08
МоЗ 0,20 2,00 0,20 0,20 0,10 2,5 0,10
Для производства чистой трехокиси молибдена пригодны молиб-
денитовые концентраты всех выпускаемых марок. Трехокись молиб-
дена для производства металла на заводах СССР получают терми-
ческим разложением парамолибдата аммония (NH4)6 -Мо7О24 X
X 4Н2О, чистоту которого характеризует табл. 7.
*
ТАБЛИЦА 7
СОСТАВ ПАРАМОЛИБДАТА АММОНИЯ (1-й СОРТ); СОДЕРЖАНИЕ
ПРИМЕСЕЙ ОТНЕСЕНО К КОЛИЧЕСТВУ МОЛИБДЕНА В СОЛИ
Примесь Содержание, %, не более Примесь Содержание, %, не более
Кремний (SiO2) .... Марганец Мышьяк Никель Сера Фосфор Цинк 0,03 0,01 0,005 0,005 0,05 0,002 0,1 Полуторные окислы . . В том числе Fe2O3 Щелочные металлы (в пе- ресчете на NaCl) . . Щелочноземельные ме- таллы (CaO+MgO) 0,03 0,01 0,01 £ 0,008
ГЛАВА II
ОКИСЛИТЕЛЬНЫЙ ОБЖИГ
МОЛИБДЕНИТОВЫХ КОНЦЕНТРАТОВ
Основной, широко применяемый в промышленности способ раз-
ложения молибденитовых концентратов, независимо от типа вы-
пускаемого продукта, — окислительный обжиг. Продукт обжига
(огарок), содержащий трехокись молибдена и ряд других соедине-
ний, служит исходным материалом для выплавки ферромолибдена
и получения химических соединений (трехокиси молибдена, мо-
либдата кальция и др.),
В производстве химических соединений разложение молибде-
нита можно осуществлять гидрометаллургическими методами,
исключающими предварительный окислительный обжиг. Эти про-
цессы будут рассмотрены в гл. VI.
6. ФИЗИКО-ХИМИЧЕСКИЕ основы окислительного
ОБЖИГА МОЛИБДЕНИТОВЫХ КОНЦЕНТРАТОВ
При обжиге молибденитовых концентратов протекает ряд хими-
ческих реакций, которые могут быть подразделены на четыре
группы [1; 2, с. 25]:
1. Окисление молибденита кислородом до трехокиси молиб-
дена.
2. Вторичное взаимодействие молибденита с трехокисью молиб-
дена с образованием низших окислов молибдена.
3. Окисление сульфидных минералов сопутствующих элемен-
тов (меди, железа, цинка, свинца) с образованием окислов и суль-
фатов.
4. Взаимодействие между трехокисью молибдена и кислород-
ными соединениями других металлов (окислами, сульфатами, кар-
бонатами) с образованием молибдатов.
21
Кинетика и химизм окисления молибденита
Дисульфид молибдена при температурах выше 500° С интен-
сивно взаимодействует с кислородом по суммарной реакции
MoS2 + З^Оз —> МоО3 + 2 SO2; Д//298 = — 228,5 ккал.
Изменение свободной энергии реакции в зависимости от темпера-
туры описывается уравнением 1
AZ0 = _ 265180 — 8,125Т lg Т + 83,35Т;
при 600°С
AZr = —213,3 ккал-, К = = Ю53.
Ро2
Реакции, сопровождающиеся столь большой убылью свободной
энергии, практически необратимые: окисление MoS2 должно про-
текать при сколь угодно малой концентрации кислорода в газовой
фазе. Для таких реакций решающее значение приобретает кинетика
процесса. По мере окисления частицы молибденита покрываются
оболочкой образующейся трехокиси молибдена. Поэтому скорость
окисления определяется структурой окисной оболочки, через ко-
торую кислород и сернистый газ должны диффундировать в про-
тивоположном направлении.
В результате изучения скорости реакции и продуктов окисле-
ния компактных образцов молибденита (полученных прессованием
порошка MoS2 в цилиндрической пресс-форме) автор и Беляевская
пришли к следующим заключениям о кинетике и химизме окисле-
ния молибденита [3]:
1. В интервале температур 400—600° С молибденит взаимодей-
ствует с кислородом с образованием МоО3, минуя стадию образо-
вания двуокиси молибдена. Наблюдаемая при температуре 600° С
промежуточная прослойка МоО2 обусловлена реакцией вторичного
взаимодействия MoS2 с МоО3 (см. стр. 29).
Таким образом, при 600° С протекают следующие реакции:
MoS2 + 3Х/2О -> МоО3 + 2SO2, (1)
MoS2 + 6МоО3 -> 7МоО2 + 2SO2, (2)
МоО2 + 1/2О2 —МоО3. (3)
При 400 и 500° С единственный обнаруживаемый продукт окисле-
ния — МоО3, так как реакция (2) протекает при этих температу-
рах со значительно меньшей скоростью, чем реакции (1) и (3).
1 При выводе уравнения принято:
Mo + S2ra3 MoS2; kZT = — 85870 + 37,33 Т [9];
Мо + 1Х/2О2 ^Z±MoO3; AZr = —177810 — 8,125 lg Т + 86,05 Т[ 10, 11];
S2 + 2О2 2SO2; AZr = — 173240 + 34,627 [11].
22
Структура спрессованных цилиндрических образцов молиб-
денита после их окисления в потоке воздуха при различных тем-
пературах характеризуется схемой, показанной на рис. 3. При 500
и 600° С наблюдается отчетливо выраженный фронт окисле-
600°С 500° С ООО С
Рис. 3. Схема окисления спрессованных образцов
молибденита при различных температурах
Продолжительность окисления,ч *
Рис. 4. Зависимость глубины окисле-
ния спрессованных образцов молибде-
нита от времени при 500 и 600° С
ния. Образцы покрыты оболочкой трехокиси молибдена (при
600° С между MoS2 и МоО3 имеется тонкий промежуточный слой
МоО2); при 400° С видимый фронт окисления отсутствует. Каждое
зерно минерала покрыто окисной оболочкой. Окисление одновре-
менно протекает по всей массе
образца.
2. Скорости и закономер-
ности окисления MoS2 при раз-
личных температурах зависят
от структуры оболочек твердых
продуктов. При 600° С оболочка
МоО3 рыхлая и не оказывает
существенного диффузионного
сопротивления. Зависимость
глубины окисления от времени
(рис. 4) носит линейный харак-
тер, т. е. скорость процесса
определяется скоростью химиче-
ской реакции (осуществляется
кинетический режим). Константа
скорость К600 ^ 0,0085 mmImuh.
При 500° С окисная оболочка
более плотная. В этом случае
по мере ее утолщения происходит переход от кинетического
режима к промежуточному, а затем к чисто диффузионному.
Зависимость глубины окисления X от времени выражается урав-
нением Хп = Лд, причем во времени п изменяется от 1 до 2.
Следует, однако, учитывать,' что диффузионное сопротивление
оболочки МоО8 при 500° С проявляется лишь при толщине ее
более 0,1—0,2 мм. Поэтому при окислении частиц MoS2 малого
размера (меньше 0,1 мм), из которых обычно состоит флотацион-
ный концентрат, окисление протекает в кинетической области.
23
При 400° С окисная оболочка плотная и механически отделяет
поверхность минерала от газовой фазы. Осуществляется чисто
диффузионный режим, характеризуемый параболической зависи-
мостью X2 = Дт (К = 1-4-5-10"12 мм2/мин). Это объясняет от-
сутствие фронта окисления при 400° С. Скорость диффузии газов
по межзеренным границам выше, чем скорость диффузии через
плотную окисную оболочку, окру-
жающую каждое зерно спрессо-
ванного образца.
3. Первоначальный акт взаимо-
действия кислорода с дисульфи-
дом молибдена — химическая ад-
сорбция молекул кислорода на по-
верхности минерала. Это вытекает
из сопоставления энергий связей
атомов в молекуле кислорода
(118 ккал) и связей MozzS в ди-
сульфиде (45,6 ккал). Следова-
тельно, чтобы молекула кислорода
вступила в реакцию с MoS2, потре-
бовалось бы преодолеть большой
активационный барьер (~73 ккал).
Между тем кажущаяся энергия
активации реакции окисления
значительно ниже (—43 ккал).
Рис. 5. Схема возникновения актив-
ных центров вследствие перехода
электрона в зону проводимости.
Закрашены атомы со свободными
валентностями. На рисунке пока-
заны только 3 из 6 атомов серы,
окружающих каждый атом молиб-
дена
Это объясняется реагированием с MoS2 лишь тех молекул кисло-
рода, которые адсорбированы на активных участках его поверх-
ности, что приводит к ослаблению и даже разрыву связей атомов
в молекуле кислорода. Высокая концентрация активных цен-
тров характерна для полупроводников, к которым относится
дисульфид молибдена. Энергия, необходимая для нарушения
валентной связи и перехода электрона в зону проводимости, у по-
лупроводников незначительная. Возбуждение одного электрона
в кристалле MoS2, которое может произойти, например, под дей-
ствием тепла, света, приводит к образованию двух свободных ва-
лентностей (рис. 5). Примеси и дефекты в кристаллической решетке
увеличивают число активных центров.
Молекула кислорода, попадая на активный центр поверхности,
вступает в реакцию с образованием промежуточного поверхност-
ного соединения и свободного атома кислорода.
При окислении молибденита переходными могут быть соедине-
ния типа оксисульфидов, образующихся в результате поверхност-
ных реакций на участках со свободной валентностью (MoS2):
MoS2 4- О2 — MoS2 [О2]адс,
MoS2 [О2]адс -> MoS2O 4- О,
(4)
(5)
24
или
MoS2 [02]адс -> MoSO2 + S.
(6)
На образование промежуточных соединений подобного типа
затрачивается энергия активации. Дальнейшее взаимодействие
кислорода с ними идет самопроизвольно с выделением энергии.
Реагирование облегчается генерацией поверхностью MoS2 свобод-
ных атомов кислорода и серы, а также бирадикалов SO. Можно
представить следующий ряд реакций, в результате которых обра-
зуются МоО3 и SO2:
MoS2O + О2 (аде) — МоО3 + 2S, (7)
MoOS2 4- О2 (аде) МоО3 + SO, (8)
S + O2 —SO + O, (9)
SO + O2->SO2 + O. (10)
Таким образом, частично осуществляется известный цепной
механизм окисления серы [4]. Свободные атомы кислорода могут
в адсорбционном слое вступить в поверхностную реакцию с не-
активными участками поверхности MoS2, образуя те же промежу-
точные соединения:
MoS2 + О -> MoS2O
и т. д.
Согласно изложенному представлению о механизме процесса,
молибденит непосредственно окисляется до МоО3, минуя стадию
образования МоО2, что соответствует экспериментальным данным.
Опубликованы работы, в которых изучалась кинетика окисле-
ния в кипящем слое частиц синтетического дисульфида молиб-
дена (размер зерен 0,15—0,2 мм) [5] и гранул молибденитового
концентрата крупностью от 0,5 до 2,6 мм, имевших пористость
—55% [6]. Использовались гранулы, полученные в заводских
условиях на чашевом грануляторе с применением в качестве связки
бентонитовой глины [43].
Навеску вводили в кипящий слой инертного материала — квар-
цевого песка, что обеспечивало изотермические условия снятия
кинетических кривых. Конструкция кварцевого реактора кипя-
щего слоя подобна описанному в работе [8]. Сернистый газ по-
глощался раствором перекиси водорода и фиксировалось измене-
ние электропроводности раствора.
На рис. 6 приведены кинетические кривые окисления гранул
в интервале температур 550—620° С. Величина кажущейся энер-
гии активации, рассчитанная из кинетических данных, составляет
43 000 кал, что указывает на протекание процесса в кинетической
области [6]. (Близкая величина —50 000 кал получена для реак-
ции окисления синтетического дисульфида молибдена [5]).
25
Кривые окисления для гранул различной крупности (от 0,5
до 2,6 мм) при одинаковой температуре оказались совершенно
идентичными. Из этого следует, что окисление идет одновременно
во всем объеме гранулы, т. е. одновременно окисляются все частицы
дисульфида молибдена, находящиеся в пористой грануле. Поэтому
кинетические кривые окисления негранулированного и гранулиро-
ванного концентрата должны быть идентичны (при размере гра-
нул до 2,5 мм).
Рис. 6. Кинетические кривые окисления гранул мо-
либденитового концентрата в кипящем слое в интер-
вале температур 550—620° С. Линии — эксперимен-
тальные, точками обозначены значения, рассчитанные
по уравнению (14). Температура окисления, °C:
/ — 620; 2 — 600; 3 — 580; 4 — 570; 5 — 560; 6 — 550
В интервале температур 550—620° С скорость окисления в ки-
пящем слое не зависит от концентрации кислорода в газовом по-
токе, т. е. реакция имеет нулевой порядок по кислороду [5, 6].
Для аналитического описания зависимости степени окисления
гранул концентрата от времени в работе [6] было использовано
известное положение, что время, необходимое для достижения оди-
наковой степени окисления а при различных температурах, об-
ратно пропорционально константам скорости реакций при этих
температурах:
^2 Кг Г / 1 1 \1 /11\
ехр '7\)j ’ 6 о
где т — время, необходимое для достижения одинаковой степени
окисления а;
К — константа скорости;
Т — абсолютная температура;
Е — энергия активации.
26
Если зависимость степени протекания реакции при темпера-
туре 7\ описать многочленом типа
a (txTx) = А (7\) та + В (Тг) т6 + С (7\) / + (12)
коэффициенты которого Л, В и С зависят от температуры, то из
зависимости между временем и константой скорости следует, что
для температуры Т2:
Л(Т2) = Л(Т1)ехр '
В(Тг) = В(Т1)ехр[-^-(^--А-)],
(13)
[С (Т2) = С (Л) ехр
Таким образом, если для какой-либо одной температуры най-
ден многочлен типа (12), описывающий кинетику окисления, то
с помощью соотношения (13) можно найти коэффициенты много-
члена при других температурах. Для температуры 570° С был
найден многочлен, с высокой точностью описывающий экспери-
ментальную кривую окисления гранулированного молибденито-
вого концентрата:
а (т)570 = 5,85-10~3т —9,37- Ю^т1’25 _р
4- 1,240т0’25 —0,938т0-125.
Учитывая соотношение (13) и подставляя Е = 43 000 кал, по-
лучим аналитическое выражение для расчета кривых окисления
при различных температурах:
а(т, Т) = 5,85-10 3 ехр
2-|6-10‘(-ет-+)]т-
- 9,37-Ю"3 ехр [2,70-4-^] т1,25 +
+ 1,240 ехр [о,54-104[4о-- 441 т0-25 —
-0,938ехр [о,27.1О-4(4з--4-)] т°’125- (14)
Как видно из рис. 6, значения а, рассчитанные по уравнению,
близки к экспериментальным.
Температура воспламенения молибденита
Температура воспламенения сульфида — важная кинетическая
характеристика реакции взаимодействия сульфида с кислородом.
Выше температуры воспламенения окисление сульфида может про-
текать самопроизвольно, за счет теплоты реакции.
27
Температура воспламенения определяется соотношением между
скоростью выделения тепла в результате экзотермической реакции
. Выше температуры
к ах /отв
. Это обусловливает возмож-
отв
Отв
зависит от удельной
пр
( и скоростью отвода тепла
\ “Т /Пр
( dQ \ . / dQ \
воспламенения ) >> ( )
\ ат /пр \ аТ /с;
ность самопроизвольного развития процесса. Окисление сульфида
начинается при температурах ниже температуры воспламенения,
( dQ \ / dQ \
но в этом случае ( ) < ( —~ )
\ аТ /пр \ аТ /отв
Температура воспламенения зависит от крупности частиц [ско-
рость реакции и, следовательно
поверхности материала] и от способа осуществления обжига, опре-
/ dQ \
деляющего скорость отвода тепла ( ~^г) •
В табл. 8 приведены температуры начала окисления и воспла-
менения порошков молибденита различной крупности, определен-
ные методом снятия дифференциальных кривых нагревания при
прососе воздуха через неподвижный слой порошка. Температуру
начала окисления определяли по появлению в газах следов (не
более 0,0003 г) сернистого газа.
отв
ТАБЛИЦА 8
ТЕМПЕРАТУРА НАЧАЛА ОКИСЛЕНИЯ И ВОСПЛАМЕНЕНИЯ
ПОРОШКОВ МОЛИБДЕНИТА
Крупность порошка, мм Температура начала окисления, °C Температура воспламенения, °C
<0,063 207 365
0,09—0,127 230 465
0,2—0,35 300 510
Сопоставление, примерно для одного класса крупности, тем-
ператур воспламенения молибденита и сульфидов других метал-
лов [39] показывает, что по возрастающей температуре воспла-
менения они располагаются в ряд:
FeS2 — MoS2 -> Cu2S — ZnS NiS PbS.
Температура воспламенения порошка молибденита в кипящем
слое выше, чем в неподвижном слое, через который просасывается
воздух, так как в кипящем слое больше скорость отвода тепла.
Так, для одного и того же молибденитового концентрата воспла-
менение в неподвижном слое наблюдалось при 360—360° С, в ки-
пящем слое при 490—500° С.
28
Взаимодействие между MoS2 и МоО3
Без доступа воздуха (например, внутри спекшихся кусков ма-
териала, образующихся при обжиге концентрата в случае пере-
грева материала) в огарке появляется двуокись молибдена в ре-
зультате взаимодействия трехокиси молибдена с дисульфидом
по реакции
MoS2 + 6МоО3 -> 7МоО2 + 2SO2, (15)
AZ°r = 44290 + 48,75 Т 1g Т — 218,52 Т*.
Возможно также реагирование с образованием промежуточного
окисла Мо4О1г:
MoS2 + 27МоО3 -> 7Мо4Оп + 2SO2. (16)
Из термодинамических расчетов следует, что реакция (15) мо-
жет протекать при температурах выше 250° С (табл. 9).
ТАБЛИЦА 9
УБЫЛЬ СВОБОДНОЙ ЭНЕРГИИ И РАВНОВЕСНОЕ ДАВЛЕНИЕ
РЕАКЦИИ МЕЖДУ MoS2 и МоО3 С ОБРАЗОВАНИЕМ МоО2
Температура, °C AZ0, ккал/моль Равновесное давление PSO2’ am
25 + 14 890 3,44-10~6
250 0,00 1,0
300 —3 710 5,15
600 —21 710 5,2-102
Автором [13] было изучено взаимодействие МоО3 с MoS2 в ин-
тервале температур 400—700° С в вакууме (при начальном давле-
нии 0,1—0,5 жж pm. cm.) и в атмосфере аргона. В ходе реакции
фиксировали количество выделившегося сернистого газа. О составе
продуктов судили по фазовому рентгенографическому и химиче-
скому анализу.
На рис. 7 показано нарастание во времени давления SO2 в ре-
акционном сосуде при нагревании смеси MoS2 + 6МоО3. Реакция
с заметной, хотя и незначительной, скоростью протекает уже при
400° С. Скорость взаимодействия резко увеличивается при темпе-
ратурах выше 550° С, что объясняется относительно высоким дав-
лением пара Л1оО3 и возможностью протекания реакции с участием
газовой фазы (давление пара при 600° С ~0,005 мм pm. cm., при
* При выводе уравнения принято: для МоО2 AZ^ =—133 600
+ 43,07 Т [12]; для остальных компонентов приняты значения AZ, приведенные
в сноске на стр. 22.
29
700° С ~0,05 мм). При 700° С реакция заканчивается примерно
за 10 мин. Продукты реакции при 600—700° С темно-коричневого
цвета, содержат двуокись молибдена.
Время, Mt/н
Рис. 7. Изменение давления /SO2 во времени при на-
гревании смеси MoS2 + 6МоО3 при различных тем-
пературах в манометрической установке
При температурах ниже 550 С, помимо МоО2, в продуктах со-
держится Мо4О1х. При 700° С на кривой давление SO2 — время
имеется отчетливый максимум
Рис. 8. Степень реагирования MoS2
с МоО3 в зависимости от времени
в атмосфере аргона. Состав смеси:
MoS2 -|- 6МоО3
(наблюдаемый в менее выраженной
степени и при 650° С). Некоторое
снижение давления SO2 связано
с реагированием МоО2 с SO2 по
реакции
2МоО2 + SO2 = 2МоО3 + X/2S2.
(17)
Как видно из рис. 8, высокая
скорость реакции между MoS2 и
МоО3 при температурах 600 и
700° С наблюдается также в атмо-
сфере аргона: за 60 мин всту-
пает в реакцию при 600° С —45%
смеси MoS2 + 6МоО3, а при 700° С
около 90 %.
Поскольку двуокись молибдена
мало растворима в растворах
аммиака или соды, используемых при выщелачивании молибдена
из огарков, необходимо проводить обжиг в условиях, исключаю-
щих возможность протекания реакции между MoS2 и МоО3.
зо
Образование молибдатов.
Системы молибдат — трехокись молибдена
При окислительном обжиге молибденитового концентрата при-
меси сульфидных минералов меди, железа, цинка, свинца превра-
щаются в окислы, частично в сульфаты х. В концентратах часто
содержится карбонат кальция (минерал кальцит), иногда карбо-
нат магния (магнезит). Окислы, сульфаты и карбонаты названных
элементов в интервале температур 500—700° С активно реагируют
с трехокисью молибдена, образуя молибдаты:
МеО + МоО3 -> Me МоО4, (18)
Me SO4 + МоО3 — Me МоО4 + SO3 (SO2, О2), (19)
Me СО3 + МоО3 — Me МоО4 + СО2. (20)
Термохимические свойства ряда молибдатов приведены в
табл. 10.
ТАБЛИЦА 10
ТЕРМОХИМИЧЕСКИЕ СВОЙСТВА НЕКОТОРЫХ МОЛИБДАТОВ
Молибдат
Температура
плавления,
°C
дЯ298’
ккал/моль
л 7°
AZ298’
ккал/молъ
S0
^298’
э. е.
СаМоО4
MgMoO4
РЬМоО4
FeMoO4
1'е2 (МоО4)8
ZnMoO4
CuMoO4
Na2MoO4
-1515
1063,5 [22]
1056 [23]
956 [23]
990 [30]
820 [15]
(с разложением)
687
—369,5+0,3 [26]
—334,8±0,2 [26]
—249,7±0,9 [25]
—257,2 [24]*
—258,1 [24]*
—708,2 [28]
—272,4 [24]*
—225,7 [24]*
—350,4 [41]
—344,0±0,3 [26]
—309,7 [26]
—236,4 [24]*
—235,3 [24]*
—249,4 [24]*
—202,7 [24]*
29,3±0,2 [27]
28,4+0,2 [27]
50,6 [24]*
30,9 [33]
37,6 [24]*
35,6 [24]*
38,1 [27]
* Оценочные значения.
Возможность образования молибдатов при нагревании ряда
окислов с трехокисью молибдена была установлена Тамманом и
Вастергольдом [14] по термограммам нагревания смесей окислов.
В последующем условия образования молибдатов при нагре-
вший и окислов или солей различных металлов с МоО3 изучали ав-
тор и Беляевская [13, 15], Гинстлинг и Фрадкина [16], Козма-
ИОВ 117], Рейнгольд и Смогунов [18]. В этих работах использо-
В1ЛИ дифференциальный термический анализ, рентгено-фазовый
и химический анализы, сопоставление химических свойств исход-
1 Катализаторами реакции SO2 -ф г/г О2 являются окислы меди,
железа и особенно трехокись молибдена. Образованию сульфатов благоприят-
ствуют низкие температуры обжига (550—580° С).
31
ных смесей и продуктов их нагревания. Высокая химическая ак-
тивность трехокиси молибдена в рассматриваемых реакциях объ-
ясняется двумя обстоятельствами [13]: а) низкой температурой
начала спекания МоО3 (550—560° С), при которой атомы приобре-
тают достаточную подвижность для обмена местами; б) сравни-
тельно высокой упругостью пара МоО3 при температурах выше
500° С, обусловливающей возможность протекания реакций с уча-
стием газовой фазы. При этом существенное Значение имеет поли-
мер изованность молекул МоО3 в парах [19, 20].
Поскольку в условиях обжига молибденитового концентрата
молибдаты образуются в присутствии избытка трехокиси молиб-
дена, важно рассмотреть изученные системы Л+МоО4—МоО3.
Ниже рассмотрены основные данные об условиях образования
молибдатов и их взаимодействии с МоО3.
Молибдат кальция. Взаимодействие МоО3 с СаО и СаСО3
с образованием СаМоО4 наблюдается при температурах 400 и
300° С соответственно и активно протекает при температурах
выше 500° С [13, 15, 18].
Реакция СаО + МоО3 = СаМоО4 сопровождается значитель-
ным тепловым эффектом (ДЯдев ~ —42Л ккал1моль), что отме-
чается на термограммах нагревания. Между тем при нагревании
смеси СаСО3 с МоО3 термические эффекты не наблюдаются, так
как реакция СаСО3 + МоО3 = СаМоО4 + СО2 протекает с очень
малым изменением энтальпии (Д77298 = +0,25 ккал). Однако
убыль свободной энергии реакции значительна. Она может быть
рассчитана по приближенному уравнению:
Д2° - 250 — 39,87+
AZ298 = — 1 1,65 ккал\
= — 30,55 ккал.
Взаимодействие СаСО3 и МоО3 наблюдается при температурах
значительно ниже температуры диссоциации карбоната кальция
(800—900° С).
При температурах интенсивного протекания реакции (550—
600° С) в начальный момент находящаяся в газовой смеси трех-
окись молибдена непосредственно реагирует с карбонатом каль-
ция. В дальнейшем процесс состоит из возгонки МоО3, диффузии
ее через оболочку СаМоО4, химического взаимодействия и диф-
фузии СО2 через оболочку твердого продукта.
В зависимости от соотношения компонентов в смеси и размеров
их частиц скорость процесса лимитируется либо диффузией, либо
скоростью возгонки. Так, по данным работы [16], в которой изу-
* При расчетах использовали термодинамические функции для молибдатов,
приведенные в табл. 9. Для окислов карбонатов и сульфатов других металлов —
данные работы [И].
.32
чалась кинетика реакции между СаСО3 и МоО3 в интервале тем-
ператур 580—620° С, при нагревании эквивалентной смеси отно-
сительно крупных частиц СаСО3 (г = 0,18 мм) и мелкозернистой
МоО3 (г = 0,036 мм) образуется толстая оболочка СаМоО4. В этом
случае процесс лимитируется диффузией и подчинается уравнению
1 — (1 - G)2/3 — 2/3G = Кдт, (21)
где G — степень превращения «покрываемого» компонента
(СаСО3), доли единицы;
т — время, мин;
Кд — константа скорости.
При t — 600° С Кд 11,7* 10~4. За 60 мин вступает в реакцию
60—65% МоО3.
При большом избытке СаСО3 (молярное отношение СаСО3 :
: МоО3 = 15), малой величине его частиц (г = 0,03 мм) и относи-
тельно крупных частицах МоО3 (0,06—0,13 мм) образуется тонкий
диффузионный слой СаМоО4 и сильно уменьшается поверхность
испарения МоО3. Процесс лимитирует возгонка, причем справед-
ливо уравнение
1-(1-G)2/3 = Kbt, (22)
где G — степень превращения МоО3, доли единицы. При t =
= 620° С Кв = 2,76-10'2. За 20 мин вступает в реакцию ~72%
МоО3. При обжиге концентрата часть кальцита в результате реак-
ции с содержащимся в газах серным ангидридом переходит в суль-
фат кальция, который взаимодействует с МоО3 с образованием
СаМоО4 лишь выше 650° С [2, с. 27; 18; 21].
В системе МоО3—СаМоО4 (рис. 9) имеется одна эвтектика при
содержании 24% (по массе) СаМоО4, плавящаяся при 727±
±3°С [22].
Молибдат магния. Окись и карбонат магния реагируют с МоО3
при температурах 500—700° С с образованием MgMoO4 [18]:
MgO + МоО3 — MgMoO4;
Д//298 = — 17,0 ккал/моль;
Д/298 = — 17,1 ккал/моль;
MgCO8 + МоО3 - MgMoO4 + СО2;
Д#298 = + 7,25 ккал/моль;
Д2298 = —5,5 ккал!моль;
Д2?73 = — 25,7 ккал/моль.
Первая реакция протекает со значительным выделением тепла,
вторая — эндотермическая, что обусловлено эндотермическим эф-
фектом диссоциации карбоната магния.
3 А. Н. Зеликман 33’
Молибдаты железа. Молибдат двухвалентного железа FeMoO4
образуется в результате реакции между FeO и МоО3 в интервале
температур 500—700° С при отсутствии воздуха (начало реакции
наблюдается при 300° С). Цвет продукта черный. Благоприятные
условия для возникновения FeMoO4 создаются при образовании
в процессе обжига спекшихся
кусков, куда затруднен доступ
воздуха. При нагревании на
воздухе при температурах выше
500° С FeMoO4 окисляется с об-
разованием Fe2O3 и МоО3 [13,
17]. FeMoO4 плавится при тем-
пературе 1056° С и образует
с МоО3 эвтектику при содержа-
нии 27,8% (мол.) [36,4% (по
массе)] FeMoO4 плавящуюся
при 705° С [23] (рис. 10). На
термограммах нагревания сме-
сей Fe2O3+MoO3 не наблюдается экзотермических эффектов,
отвечающих взаимодействию в твердом состоянии. На этом осно-
вании авторы работ [14] и [15] заключили, что между этими окис-
лами нет химического взаимодействия с образованием ферримо-
либдита Fe2 (МоО4)3. Однако Козманов [17] с помощью рентгено-
графии установил, что при
нагревании смеси Fe2O3+
+ МоО3, начиная с 700° С,
образуется Fe2(MoO4)3, при-
чем непременное усло-
вие для протекания реак-
ции — достаточно большая
концентрация паров МоО3
в окружающей атмосфере.
Цвет продукта светло-пес-
чаный. Тот же вывод о хи-
мическом взаимодействии
между Fe2O3 и МоО3 сде-
лан, в более поздней работе
[18], где был использован
Рис. 10. Система МоО3—FeO
метод высокотемпер ату р -
ной рентгенографии, причем Fe2(MoO4)3 образовался в интер-
вале температур 530—600° С. Ферримолибдат при нагревании
на воздухе диссоциирует на Fe2O3 и МоО3 и быстро разлагается
при 900° С.
По данным Ягера, Рамеля и Бекера [23], в системе Fe2O3—
МоО3 образуется ферримолибдат Fe2(MoO4)3, который плавится
при 956° С (рис. 11). Fe2(MoO4)3 образует эвтектику с МоО3,
плавящуюся при 722° С.
34
При нагревании Fe3O4 с МоО3 образуются смеси Fe2 (МоО4)3
к FeMoO4, содержащие в зависимости от соотношения исходных
компонентов избыточную МоО3 или Fe2O3 [18].
Псевдобинарная система Fe2(MoO4)3—FeMoO4 — эвтектиче-
ского типа (рис. 12). Другие молибдаты в ней не обнару-
жены [23].
Помимо молибдатов железа, установлено существование двух
молибдитов железа Fe2MoO4 и Fe4Mo6O16 — производных двуокиси
молибдена. Они образуются при
нагревании (без доступа воздуха)
FeO с МоО2. Оба молибдита
железа плавятся выше 1150° С.
Ге203 Мо03°/О(мол) моО3
Рис. 11. Система МоО3—Fe2O3
о го 40 во so юо
Fe2(MoOh)3 °/0(по массе} FefioO^
Рис. 12. Псевдобинарная система
Fe2 (МоО4)3—FeMoO4
На рис. 13 приведен ориентировочный изотермический разрез
системы железо — молибден —- кислород примерно для темпера-
тур 800—1100° С [23], который показывает, какие фазы могут
находиться в равновесии между собой.
Молибдат меди. Окись и сульфат меди реагируют с Мо03
в интервале температур 300—700° С образованием СиМоО4 — ве-
щества желто-зеленого цвета. По данным "работ [13, 15],
СпМоО4 плавится с разложением при 820° С и образует эвтек-
тику с МоО3, отвечающую составу 42,2% (по массе) СиМоО4,
Плавящуюся при 560° (рис. 14). Согласно более поздней публи-
кации, * в системе МоО3—СиО существуют два молибдата: СиМоО4
ИЗСиО • 2МоО3. Оба молибдата плавятся с разложением при 850
И 870е С соответственно. Авторы нашли, что эвтектика
МоО,—СиМоО4 плавится при 710° С, а найденная в работе [15]
температура эвтектики в действительности соответствует поли-
морфному превращению СиМоО4, наблюдаемому при 550—560° С.
* Koh 1 in й Her R. Faurie. I. C. R. Acad. Sc. Paris, 1967, p. 1751?
3* 35
Молибдат цинка. Реакция между окисью цинка и МоО3 с обра-
зованием ZnMoO4 наблюдается выше 400° С и интенсивно проте-
кает в интервале 600—700° С [15]. Продукт реакции белого цвета
с возовым оттенком
Рис. 13. Ориентировочный изотермический раз-
рез системы Fe—Мо—О для 800—1100° С
МоО3 СиО
7о(ло массе)
Рис. 14. Система МоО3—СиО
Систему Мо03—ZnO изучали авторы работ. [22] и [30], данные
которых несколько расходятся. На рис. 15 приведена система
МоО3—ZnO, построенная Зобниной и Кисляковым [30]. По их
данным, молибдат цинка плавится конгруэнтно при 990° С и обра-
Рис. 15. Система ZnO—МоО3
зует эвтектику с МоО3 состава, % (мол.): 81,3 МоО3 и 18,7 ZnO,
плавящуюся при 660° С. По данным работы [22], эвтектика
имеет состав 15,9% (мол.) ZnO и плавится при 707° С. Кроме того,
авторы считают, что ZnMoO4 плавится при 1000° С с разложением.
Эти расхождения, вероятно, объясняются близостью положений
точки плавления молибдата и температуры эвтектики, отличаю-
щихся лишь на 5 град (рис. 15).
36
Молибдат свинца. Окись свинца РЬО и трехокись молибдена
реагируют при температурах выше 340° С с образованием
РЬМоО4 [13]. Взаимодействие
сульфата свинца с МоО3 отме-
чается выше 550° С [18]:
PbSO4 + МоО3 =
= РЬМоО4 + SO3.
Система МоО3—РЬМоО4
имеет одну эвтектику при содер-
жании 49% (по массе) РЬМоО4,
плавящуюся при 670° С. Мо-
либдат свинца плавится без
разложения при 1063° С [22]
(рис. 16).
Рис. 16. Система МоО3—РЬМоО4
7. ПРАКТИКА ОКИСЛИТЕЛЬНОГО ОБЖИГА
В заводской практике молибденитовые концентраты обжигают
в пламенных или муфельных печах с ручным перегребанием мате-
риала, вращающихся трубчатых печах, многоподовых печах с ме-
ханическим перегребанием и в печах кипящего слоя.
Одноподовые печи (пламенные или муфельные) и барабанные
печи имеют существенные недостатки и мало пригодны для обжига
молибденитовых концентратов. В них трудно поддерживать опти-
мальную температуру обжига (—570—580° С), наблюдаются пере-
гревы, ведущие к спеканию материала и интенсивному протеканию
вторичных реакций (образование МоО2, молибдатов). Кроме того,
в этих печах не используется теплота реакций окисления: обжиг
ведут при непрерывном подводе тепла, так как нет полного про-
тивотока газов и обжигаемого материала. Более совершенны мно-
гоподовые печи и печи кипящего слоя, которые используют в на-
стоящее время на ряде заводов СССР [35, с. 251 и 265; 38].
Обжиг в многоподовых печах
Многоподовые печи с механическим перегребанием давно ис-
пользуют для обжига сульфидных концентратов железа, меди,
цинка. Полный противоток газов и обжигаемого материала, ин-
тенсивное окисление во взвешенном состоянии в моменты пере-
сыпания материала с пода на под обеспечивают в печах этого типа
возможность ведения процесса за счет теплоты реакции. Разогрев
форсунками, устанавливаемыми, например, на четных подах, не-
обходим только при запуске печи. Однако на нижних подах для
доведения содержания серы в огарках до установленных кондиций
(0,05—0,25% 80бщ) форсунки постоянно работают. Температура
37
на подах не должна превышать 580—590° С. Нагревание до 700—
800° С совершенно недопустимо, так как при этих температурах
трехокись молибдена интенсивно возгоняется. Кроме того, наблю-
даемое в этом случае спекание и оплавление огарка вызывает за-
растание отверстий и быстрый износ гребков. Поддержание тем-
Рис. 17. Распределение температуры по подам механической
многоподовой печи:
а — подвод воздуха на нижние поды, отвод газов сверху, давление
под сводом 1,25 мм вод. ст.; б — подвод воздуха на каждый под и
отвод газов с каждого пода в общий газоход; разрежение под сво-
дом от 0,75 до 1,25 мм вод. ст.
пературы на уровне 580—590° С в многоподовых печах обычной
конструкции, применяемых для обжига сульфидного сырья, за-
труднительно.
В связи с этим на заводах Клаймакс в США многоподовые печи
для обжига молибденитовых концентратов были реконструиро-
ваны [29]. Для регулирования температуры воздух подают от-
дельно на каждый под, а газы отводят с каждого пода через регу-
лируемую заслонку в общий газоход. Это позволяет поддерживать
необходимую температуру на подах. Распределение температуры
по подам при таком способе работы показано на рис. 17. Обжиг
ведут в печах с 8, 12 и 16 подами диаметром от 4 до 5,4 ж. Пылеунос
составляет 18%, причем пыль состоит в основном из необожжен-
38
кого концентрата, ее улавливают и возвращают в печь на обжиг.
Вал с гребками вращается со скоростью 2/3—1 об!мин, слой мате-
риала имеет толщину не более 60 мм. Производительность много-
подовых печей по концентрату составляет 60—70 кг на 1 м2 пода
печи в сутки.
Обжиг в кипящем слое
В химической и металлургической промышленности для техно-
логических операций, основанных на гетерогенных реакциях газ —
твердое тело, широко распространены процессы, протекающие
в слое зернистого материала, приведенного в легкоподвижное, так
называемое «псевдожидкое» состояние (или состояние «кипящего
слоя»), при пропускании через слой материала восходящего по-
тока газа. В такое состояние, ха-
рактеризуемое интенсивным хаоти-
ческим движением частиц, зерни-
стый материал переходит при
опредёленных скоростях газового
потока, превышающих некоторую
критическую величину (умин). Ее
экспериментально определяют из
зависимости между сопротивле-
нием слоя (ДР) и линейной ско-
ростью потока v, показанной на
рис. 18.
Сопротивление кипящего слоя
аналогично гидростатическому
давлению столба жидкости и может
быть рассчитано по уравнению
&р = Я(Утв- Тгаз)(1 - б), •
где
Н — высота слоя;
0 2 4 6 6 Ю
Скорость Воздуха., см/сек
Рис. 18. Зависимость между сопро-
тивлением слоя зернистого мате-
риала (ДР) и скоростью потока
газа (и) при различной высоте слоя
молибденового огарка, мм:
1 — 27; 2—51; 3 — 79; 4 — 107
утв, Тг?з — плотности твердых частиц и газа;
6 — относительный объем, занимаемый газом в ки-
пящем слое, доли единицы.
К наиболее важным свойствам кипящего слоя, определяющим
эффективность его использования, относятся:
а) подвижность слоя, подобная подвижности жидкости, что
позволяет легко осуществить непрерывную подачу и самопроиз-
вольную выгрузку материала из аппарата;
б) высокие теплопроводность и коэффициенты теплопередачи
кипящего слоя, обусловленные интенсивной циркуляцией частиц.
Это позволяет строго поддерживать во всей массе слоя заданную
температуру даже при реакциях со значительными тепловыми эф-
фектами, что в ряде случаев важно для предотвращения побочных
реакций. Избыточное тепло легко отводится с помощью холо-
дильников (например, труб, установленных в слое и охлаждае-
мых водой);
39
в) высокая скорость химических реакций, протекающих в слое,
благодаря большой поверхности контакта и омыванию каждой
частицы потоками газа.
Общие закономерности кипящего слоя освещены в специальных
монографиях [31, 32].
В применении к молибденитовым концентратам обжиг в кипя-
щем слое был впервые исследован и осуществлен в промышленных
масштабах в СССР [34, 35, 37, 38].
Обжиг стандартных негранулированных концентратов
в печах КС
Из рассмотренного выше химизма обжига молибденитовых кон
центратов следует, что для получения огарков с высоким содержа-
нием выщелачиваемого молибдена необходимо проводить обжиг
при строго определенной температуре, не допуская спекания ма-
териала, и при возможно меньшем контакте частиц между собой
(для сведения к минимуму образования молибдатов). В наиболь-
шей степени эти условия соблюдаются при обжиге в кипящем слое.
Исследования показали, что содержание выщелачиваемого мо-
либдена (извлечение в раствор аммиака) в огарках кипящего слоя
составляет 90—93%, что на 10—12% выше, чем в огарках подовых
печей. Это объясняется существенными отличиями в рациональном
составе огарков. Огарки, полученные при обжиге концентратов
в кипящем слое, не содержат двуокиси молибдена, тогда как в огар-
ках подовых печей содержание МоО2 составляет 4—4,6%. Боль-
шая часть кальцита при обжиге в подовых печах реагирует с МоО3,
образуя СаМоО4, что существенно снижает степень извлечения
молибдена в аммиачные растворы. Между тем при обжиге в кипя-
щем слое подавляющая часть кальцита реагирует с SO3, обра-
зуя CaSO4.
При обжиге в кипящем слое условия благоприятны для обра-
зования SO3: температуру обжига строго поддерживают в преде-
лах 560—570° С, взвешенные частицы МоО3 играют роль катали-
затора для реакции SO2 + 1/2O2^lSO3 [118]. Большая часть
меди в огарках подовых печей содержится в составе СпМоО4,
а в огарках КС — в составе CuSO4. При обжиге в подовых печах
значительная часть окислов железа реагирует с МоО3, образуя
молибдаты [FeMoO4, Fe2(MoO4)3], тогда как при обжиге в КС
вследствие отсутствия постоянного тесного контакта между ча-
стицами и исключения спекания материала нет условий для
образования молибдатов железа.
Для обжига молибденитовых концентратов в кипящем слое
используют печи различных конструкций [35,38]. На рис.. 19
показана одна из промышленных печей и общая схема установки.
Печь представляет собой цилиндрическую шахту, футерованную
жароупорным бетоном или фасонным шамотным кирпичом. В ниж-
40
ней части шахты расположена воздухораспределительная решетка
(подина). Решетка состоит из ряда сопел с грибовидными съем-
ными колпачками, что предотвращает просыпание материала под
решетку.
Равномерное питание печи концентратом — важнейшее усло-
вие поддержания заданного режима обжига. Концентрат подается
в печь с помощью автоматизированного узла загрузки, состоящего
из цилиндрического бункера \ под которым находится тарельча-
тый питатель с регулируемой скоростью оборотов. Нож питателя
Рис. 19. Схема установки для обжига молибденитовых концентратов в кипящем
слое:
1 — воздуходувка; 2 — вентиль; 3 — форсунка; 4 — выносная топка под давлением;
5 — система водяного охлаждения для отвода избыточного тепла; 6 — течка; 7 — бачок
для мазута; 8 — тарельчатый питатель; 9 — бункер; 10 — шлюзовый затвор; 11 — камера
печи; 12 — циклон; 13 — решетка (подина); 14 — разгрузочный порог; 15 — дымосос;
16 — электрофильтр
сбрасывает концентрат в бункер герметичного шлюзового затвора,
откуда он по трубе (течке) поступает в кипящий слой. На уровне
1000—1500 мм от подины находится разгрузочное отверстие (по-
рог выгрузки), через которое непрерывно выгружается огарок,
ссыпающийся по шлюзовому затвору в приемники.
Газы вместе с уносимыми с ними тонкими частицами материала
проходят пылеулавливающие устройства (циклоны, электро-
фильтр) и выбрасываются в атмосферу. Вследствие близости тем-
ператур возгорания молибденитового концентрата в кипящем слое
(500—510° С) и начала спекания огарков (580—590° С) обжиг кон-
центрата можно проводить лишь при относительно низкой темпера-
туре в слое, поддерживаемой в пределах 550—570° С. При более
высокой температуре на стенках печи в надслоевой зоне обра-
1 В конических бункерах слегка влажные молибденитовые концентраты
(1—2% влаги) зависают.
41
зуются плотные наросты (результат-окисления уносимых тонких
частиц концентрата), куски которых падают в слой и накапли-
ваются на подине, что приводит к нарушению процесса.
Заданная температура в слое поддерживается автоматически.
Система регулирования основана на изменении количества подз-
ываемого концентрата. При повышении или понижении темпера-
туры по сравнению с заданной соответственно автоматически умень-
шается или увеличивается количество подаваемого в печь в еди-
ницу времени концентрата путем изменения числа оборотов та-
рели питателя (рис. 19). Практика показада, что система обеспе-
чивает надежное регулирование температуры в пределах ± 2,5 град
от заданной [40, 42]. Если при средней установленной часовой за-
грузке концентрата выделяется много избыточного тепла, его
отводят, подавая воду в трубы, установленные в кипящем слое.
Для запуска печи первоначально создают в ней кипящий слой из
огарка, разогретый до температуры зажигания молибденитового
концентрата (500—510° С) подогретым воздухом, для чего преду-
смотрена выносная топка, работающая под давлением Ч Затем
включают систему питания печи концентратом (примерно 50—
55 кг!ч на 1 ж1 2 пода печи). Попадая в разогретый кипящий слой
огарка, концентрат возгорается, температура в слое за 15—30 мин
достигает оптимальной (560—570° С) и далее поддерживается авто-
матически системой регулирования.. Рабочие линейные скорости
воздуха в шахте печи для создания кипящего слоя при обжиге
молибденитовых концентратов обычно лежат в пределах 5—
7 см!сек (при нормальных условиях), что отвечает расходу воздуха
180—250 мЧч на 1 ж2 пода печи и коэффициенту избытка воздуха
а = 1,8 4-2,2. При описанных режимах обжига производительность
печи по концентрату составляет примерно 1200—1300 кг/м2, пода
печи в сутки, что в 15—20 раз выше производительности подовых
печей. Огарки содержат 0,6—0,8% сульфидной серы.
Часть тонких частиц концентрата (25—45% в зависимости от
гранулометрического состава концентрата) уносится с газами.
Система, состоящая из циклов и сухого электрофильтра, обеспе-
чивает хорошее (—>99%) улавливание пыли, причем в циклонах
оседает 85—90% пыли. Однако улавливаемая пыль окислена не-
полно (примерно на 70—80%) и содержит 8—10% сульфидной серы
(против 30—32% S в стандартных концентратах). Вследствие низ-
кой температуры обжига (560—570° С) средняя температура в над-
слоевой зоне печи равна 500—510° С. В этих условиях тонкие ча-
стицы MoS2, уносимые с газами, за время пребывания в печи (1—
2 мин при высоте печи 9 ж) не успевают окислиться.
1 В других вариантах выносная топка отсутствует. Камера печи и начальная
партия загрузки разогреваются пламенем форсунок. Однако в этом случае запуск
приходится проводить на инертном тугоплавком материале (хвостах выщелачи-
вания или кварцевом песке).
42
Проблема получения окисленной пыли при обжиге молибде-
нитового концентрата в кипящем слое оказалась сложной. По-
пытки повышения температуры в надслоевой зоне (например, улуч-
шением теплоизоляции печи) приводили к образованию наростов
на стенках печи. Возвращать пыль на дообжиг в слой без предвари-
тельного, хотя бы частичного, укрупнения частиц нельзя, так как
это приводит к прогрессирующему увеличению доли уносимого
с газами материала. Неполно окисленную пыль приходится до-
полнительно обжигать в других печах либо направлять на выще-
лачивание для извлечения трехокиси молибдена, а остаток перера-
батывать вместе с хвостами после выщелачивания огарков
(см. рис. 25 и 26).
Радикальное решение — введение предварительной грануля-
ции концентрата, что позволяет возвращать неполно окисленную
пыль на грануляцию, а также обжигать в кипящем слое высоко-
дисперсные концентраты, при обжиге которых пылевынос дости-
гает 60—70%.
Обжиг гранулированных концентратов в печах КС
Технология грануляции молибденитовых концентратов и об-
жига гранулированного материала описана в работе [43]. Гра-
нуляция осуществляется на чашевом грануляторе. В качестве
связки используют бентонит Г Примерный состав шихты для гра-
нуляции: 5—6% бентонита, 12—16% воды, остальное концентрат
и оборотная пыль (оптимальную влажность с точностью ±0,5%
определяют опытным путем для каждой партии концентрата). Для
получения качественных гранул необходимо строгое соблюдение
оптимального состава шихты, хорошее перемешивание компонен-
тов, растирание смеси в бегунах, разрыхление материала перед
поступлением в чашевый гранулятор.
Изучая скорости псевдоожижения фракций с гранулами раз-
личного размера, остановились на оптимальном наборе с круп-
ностью гранул от 0,2 до 2 мм, для которого минимальная скорость
псевдоожижения равна 12 см!сек. Скорость псевдоожижения смеси
гранул значительно ниже, чем крупных фракций смеси (40 см!сек
для фракций 1—2 мм\ 25 см!сек, для фракций 0,5—1 мм). При опти-
мальном режиме грануляции (хорошее перемешивание компонен-
тов и разрыхление шихты; наклон чаши 45° при окружной скорости
вращения чаши —0,9 м!сек) выход фракции гранул крупностью
до 2 мм составляет 80—90%. Фракцию +2 мм возвращают в бе-
гуны. Прочность на раздавливание гранул крупностью —2 мм
из смеси концентрата с 20—25% оборотной циклонной пыли
—370 г после сушки и —720 г после обжига.
1 Бентонит — один из видов глин, обладающих хорошими вяжущими свой-
ствами.
43
Производительность гранулятора с диаметром чаши 1000 мм
и высотой борта 100 мм равна 300 кг/ч.
При изучении обжига гранулированного концентрата на
опытно-промышленной печи с площадью пода 1 ж2 установлена
возможность непосредственной загрузки в слой сырых гранул,
т. е. исключения предварительной их сушки. Было обнаружено,
что характер пыли, образующейся при обжиге гранул, сильно от-
личается от пыли обжига гранулированного концентрата: она зна-
чительно сильнее окислена (средняя степень окисления —94%
против ~70% при обжиге негранулированного концентрата) и го-
раздо мельче: средний размер частиц циклонной пыли при обжиге
негранулированного концентрата 35 мкм, тогда как при обжиге
гранул средний размер частиц циклонной пыли 6,2 мкм, а частиц
тонкой пыли, собранной за циклоном, —2,5 мкм.
Высокая степень окисления пыли и очень, малый размер ее
частиц свидетельствуют о том, что большая часть пыли при обжиге
гранул вторичного происхождения, т. е. образуется в результате
взаимного истирания гранул в кипящем слое. Лишь около 60%
пыли, образующейся при обжиге гранул, улавливается циклоном.
Более тонкая пыль полно улавливается рукавным фильтром с ру-
кавами из стеклоткани при поддержании в фильтре температуры
—200° С (для устранения конденсации серной кислоты).
Основные показатели обжига гранулированного сорского кон-
центрата 1 во время длительной кампании (23 суток) в сравнении
со средними показателями обжига в кипящем слое негранулиро-
ванного тырны-аузского концентрата сопоставлены в табл. 11.
Гранулы обжигали при средней температуре 575—580° С. Расход
воздуха составлял 300—350 ж3/(ч-ж2), что примерно в два раза
ниже оптимального для гранул крупностью до 2 мм, и обусловлено
недостаточной пропускной способностью системы пылеулавлива-
ния.
Как видно из данных табл. 10, степень пылеуноса при обжиге
гранул высокая (~38%), однако тонкая пыль, улавливаемая ру-
кавным фильтром (более 40% от общего количества пыли), имеет
высокую степень окисления (~99%) и направляется вместе с огар-
ком на выщелачивание. Благодаря этому выход материала, не-
посредственно направляемого на выщелачивание, значительно
выше, чем при обжиге негранулированного концентрата. Циклон-
ная пыль, окисленная неполно (—90%), возвращается на грану-
ляцию, что позволяет замкнуть схему обжига. При увеличении
расхода воздуха до нормального [—-720 ж3/(ч- ж2) ] тепловой баланс
обжига изменится и производительность печи в этом случае воз-
1 Сорский концентрат тонко дисперсный (~82% частиц размером меньше
74 мкм), пылевынос при обжиге негранулированного концентрата составляет
60—70%. Тырны-аузский концентрат более крупнозернистый (50—60% частиц
размером меньше 74 мкм)]
44
ТАБЛИЦА 11
ОСНОВНЫЕ ПОКАЗАТЕЛИ РАБОТЫ ОПЫТНО-ПРОМЫШЛЕННОЙ
ПЕЧИ КС (ПЛОЩАДЬ ПОДА 1 ж2, ВЫСОТА СЛОЯ 1 ж) ПРИ ОБЖИГЕ
ГРАНУЛ СОРСКОГО КОНЦЕНТРАТА И НЕГРАНУЛИРОВАННОГО
ТЫРНЫ-АУЗСКОГО КОНЦЕНТРАТА [43]
Показатели Обжиг гранул из сор- ского концентрата (влажность 13%, содер- жание молибдена в сухом 47,8%) Обжиг неграну- лированного тырны-аузского концентрата
Суточная производительность по за- грузочному материалу, кг/сутки Выгрузка огарка, кг/сутки .... 1650 1160
810 652
Выгрузка циклонной пыли, кг/сутки 280 416
Выгрузка пыли рукавного фильтра, кг/сутки 210 -
Количество материала, непосредствен- но поступающего на выщелачивание, кг/сутки 1020 652
Пылевынос, % от суммы огарка и пы- ли 37,8 39
Прямой выход материала, непосред- ственно направляемого на выщела- чивание, % от суммы огарка и пы- ли 78,7 61
Степень окисления огарка, % ... 97,5—98,0 95,0
Степень окисления циклонной пыли, % 90,0 60—65
Степень окисления пыли рукавного фильтра 99,0 —
растет на 70—80%, т. е. составит 1,7 ml сутки на 1 м2 по материалу,
непосредственно направляемому на выщелачивание (или составит
по влажному гранулированному концентрату ^>2,8 т!сутки на
1 м2 пода печи).
Производительность печей КС
В кипящем слое твердые частицы интенсивно перемешиваются.
Среднее расчетное время пребывания материала в кипящем слое
равно отношению общей массы материала М} находящегося в ванне
печи, к загрузке в единицу времени т: хср = —. Однако вслед-
ствие быстрого перемешивания частиц материала в выгружаемом
огарке содержатся частицы, время пребывания которых в слое
колеблется от близкого к нулю до превышающего во много раз
среднее время пребывания. Следовательно, остаточное содержание
серы у частиц огарка будет различным в зависимости от времени
пребывания их в слое. Поэтому при расчете производительности
и качества огарков печей кипящего слоя, кроме кинетики про-
цесса окисления, необходимо учитывать распределение частиц
в огарке по времени их пребывания в слое.
45
Если обозначить F (т) — зависимость остаточного содержания
серы в огарке от продолжительности обжига [т. е. F (т) характери-
зует кинетику обжига], то содержание серы Cs в огарке, выгру-
жаемом из слоя, выразится уравнением [44—46]
т
Cs-р(т)Л? (т),
о
(23)
где dC (т)—функция времени, характеризующая распределение
частиц по времени в выгружаемом материале;
Т — время, достаточное для полного окисления материала.
Выражение (23) легко получить, исходя из следующих рассу-
ждений.
Разобьем отрезок времени Т на k участков. Очевидно, что если
в огарке доля частиц со временем пребывания от tz до tz + Дт
равна ACZ, а содержание сульфидной серы в частицах со временем
пребывания тг- равно F (тг), то полное содержание серы в огарке
для всех частиц со временем пребывания в слое tz < Т равно сумме
произведений ^CtF (tz):
i=k
Cs = s F(MACZ.
i—1
(24)
Переходя к пределу Дт —-> 0, получим уравнение (23).
При небольших размерах печи (с расстоянием от точки за-
грузки до точки выгрузки до 1,5 м) перемешивание частиц в ки-
пящем слое можно принять идеальным, т. е. принять, что загру-
женные в слой частицы мгновенно равномерно распределяются
по всему объему. В этом случае функция распределения dC (т)
имеет вид * 1
dC (т) = dr,
(25)
где М — общая масса материала в ванне печи;
m — загрузка в единицу времени (в пересчете на эквивалент-
ную массу получаемого огарка).
Подставляя выражение (25) в уравнение (23), получим
т
«dx-
6
(26)
tn
1 Точнее, dC (т) = e M dx. Однако без большой погрешности можно
принять [44] е м = 1,
46
Решая уравнение относительно т, получим уравнение для рас-
чета производительности печей небольшого размера при заданном
остаточном содержании серы в огарке:
Се М
а0_____
т
F (т) dx
о
(27)
т
Значение интеграла J F (т) dx может быть определено гра-
о
фически — это площадь, ограниченная осями координат в кинети-
ческой кривой окисления F (т), которая определяется на лабора-
торной установке кипящего слоя с периодической загрузкой.
Кроме того, значение этого интеграла можно определить по резуль-
татам обжига концентрата в лабораторной или опытно-промышлен-
ной печи непрерывного действия:
j F (т) dx = . (28)
О
В качестве примера приведем расчет производительности реаль-
ной печи для обжига молибденитового концентрата с площадью
пода 1,92 м2 и высотой слоя 1 м. Содержание сульфидной серы
в огарке Cs = 0,6%. Пылевынос составляет 30% от массы кон-
центрата. Объемная масса материала в ванне 1000 кг!м\ Умень-
шение массы частиц при обжиге 10%. Масса материала в печи в пе-
ресчете на массу концентрата
М=- 1,92-1000-0,9 — 2133 кг.
т
Значение интеграла J F (т) dx было определено из опыта обжига
о
концентрата в лабораторной печи с площадью пода 0,06 м2 *:
т
J F (x)dx = 18,57 %/ч.
о
Максимальное количество материала, которое может быть загру-
жено в ванну при предельном содержании в огарке 0,6% S,
0,6-2133
m = —— = 69 кг ч.
18,57
Производительность печи по концентрату (с учетом пылеуноса 30%)
69:0,7^98,5 кг/ч.
* Детальнее см. работу [45].
47
Реальная производительность печи составляла по концентрату
100 кг/ч, что хорошо согласуется с приведенным расчетом.
При расчете печей со значительным расстоянием между точ-
ками загрузки и выгрузки перемешивание нельзя считать мгно-
венным. Чтобы загруженные частицы равномерно распределились
по всему объему кипящего слоя, должно пройти некоторое время,
в течение которого концентрация частиц в точке выгрузки увели-
чивается от нуля до среднего по всему слою. Поскольку механизм
выравнивания состава слоя аналогичен диффузии, для вывода
уравнения, пригодного для расчета печей любого размера, может
быть использовано уравнение диффузии:
дС (х, т) __ п д2С (х, т) ,9СП
du D dx2 9
где х — расстояние от точки загрузки;
D — коэффициент перемешивания.
Определив функцию распределения dCx из уравнения диффузии
и подставив его в формулу (23), получим уточненное выражение
для концентрации серы в огарке [46] *:
(30)
где I — расстояние от точки загрузки до точки выгрузки.
Таким образом, для расчета печей любого размера, помимо
кинетики окисления F (т), должна быть известна величина коэф-
фициента перемешивания твердых частиц в кипящем слое, которую
можно определить по методике, описанной в работе [50]. Вели-
чина коэффициента перемешивания при обжиге гранулированного
молибденитового концентрата с размерами гранул 0,2—2 мм равна
22,5 см21сек.
Анализ уравнений (27) и (30) позволяет определить пути повы-
шения производительности печей КС без увеличения их площади
и при сохранении качества огарка [2, с. 33].
Увеличение высоты слоя. Производительность печи прямо про-
порциональна количеству материала в кипящем слое М. Следо-
вательно, увеличение высоты кипящего слоя (высоты порога вы-
грузки) позволяет повысить производительность печи без измене-
ния остаточного содержания серы в огарке.
Повышение температуры обжига. При повышении температуры
обжига ускоряется окисление концентрата [уменьшается значе-
* Детальный вывод см. в работе [46].
48
ние интеграла ]
6
F (r) dx , что позволяет повысить производитель-
ность печи без ухудшения качества огарка. Однако возможности
повышения температуры обжига весьма ограничены, так как пре-
дельная температура обжига негранулированного концентрата
равна —570° С.
Грануляция концентрата перед обжигом. Повысить произво-
дительность печей КС, работающих на негранулированном кон-
центрате, вследствие увеличения высоты слоя можно лишь на
40—50%, так как при дальнейшем росте производительности по-
даваемого в печь воздуха будет недостаточно для полного окисле-
ния концентрата (увеличить расход воздуха нельзя из-за возраста-,
ния пылевыноса). При обжиге гранул скорости воздуха в несколько*
раз больше, чем для негранулированного концентрата, что уве-
личивает возможности роста производительности в результате по-
вышения порога выгрузки.
Практика показала, что при обжиге гранул работая темпера-
тура в слое может быть повышена до 580—590° С, соответственно
этому возрастает также производительность печи.
Учитывая сказанное, можно считать, что при обжиге гранул
производительность печей КС можно увеличить в 2—2,5 раза по
сравнению с обжигом негранулированного концентрата.
4 А. Н. Зеликман
ГЛАВА III
ПРОИЗВОДСТВО ТРЕХОКИСИ МОЛИБДЕНА
Трехокись молибдена высокой чистоты (99,8—99,9% МоО3) —
основной исходный материал для получения молибдена. Сырьем
для производства чистой трехокиси молибдена служат продукты
окислительного обжига’стандартных молибденитовых концентра-
тов — молибденовые огарки.
В промышленной практике чистую трехокись молибдена полу-
чают гидрометаллургическим способом или способом возгонки.
В данной главе рассмотрены физико-химические основы и прак-
тика этих процессов.
ПРОИЗВОДСТВО ТРЕХОКИСИ МОЛИБДЕНА
ГИДРОМЕТАЛЛУРГИЧЕСКИМ СПОСОБОМ
Принципиальная схема получения чистой трехокиси молиб-
дена гидрометаллургическим способом показана на рис. 20.
Огарок выщелачивают раствором аммиака. Из аммиачного
раствора, содержащего молибдат аммония, после очистки его от
примесей выделяют молибден в форме полимолибдатов (парамолиб-
дат или тетрамолибдат аммония).
Трехокись молибдена получают термическим разложением
кристаллов молибдата аммония.
8. ВЫЩЕЛАЧИВАНИЕ
Трехокись молибдена быстро растворяется в растворах амми-
ака. Однако в огарках, как показано выше, кроме трехокиси мо-
либдена, могут присутствовать низшие окислы молибдена (МоО2,
Мо4ОХ1), молибдаты кальция, магния, меди, цинка, свинца, же-
леза (двух- и трехвалентного), сульфаты кальция, магния, меди,
неокислившийся молибденит, соли щелочных металлов, кремне-
50
зем и другие примеси. Поэтому извлечение молибдена из огарка
в аммиачный раствор зависит от рационального состава огарков
и поведения отдельных составляющих при действии раствора
аммиака.
на
Обожженный нолибденитовый концентрат
( oeaPw>РасгпворМН^
Выщелачивание
Лвосты Раствор . . .
। ( "ГЦ /2
На дополнительное | f 1
извлечение полибдено ।Очистка।
Очищенный раствор Сульфидный кек
вариант! | Варианте отвал
Нейтрализация выпорка
Кислый Осадок
поточный раствор полиполибдата
На регенерацию полибдено
Перекристаллизация Кристаллизация
Паточный Кристаллы Паточный
раствор паранолибдата ан и оная раствор
----------"“I Прокаливание ----
I ПГ
Периодически выводят Трехокись
на очистку от припесей нолибдена
Рис. 20. Принципиальная схема получения трехокиси молибдена из огарков
гидрометаллургическим способом
Из данных табл. 12, которая характеризует пове^^ние соеди-
нений молибдена по отношению к различным растворителям,
можно сделать следующие выводы:
1. Имеется сходство в поведении MoS2, МоО2 и Мо4ОХ1. Эти
соединения мало растворимы в воде, растворах щелочей и соляной
кислоте.
Некоторая растворимость обусловлена медленным окислением,
причем склонность к окислению в воде и водных растворах щело-
чей возрастает в ряду MoS2 —> МоО2 —> Мо4Охх.
4* 51
СП
to
ТАБЛИЦА 12
ПОВЕДЕНИЕ СОЕДИНЕНИЙ МОЛИБДЕНА ПРИ ДЕЙСТВИИ РАЗЛИЧНЫХ РАСТВОРИТЕЛЕЙ [52]
(РАСТВОРИМОСТИ ВЫРАЖЕНЫ В г/л СОДЕРЖАНИЯ МОЛИБДЕНА ПРИ 20° С)
Соединение Растворитель
вода растворы аммиака (5%-ные) растворы соды (5%-ные) соляная кислота (5%-ная)
MoS2 Нерастворим Весьма незначительная ра- створимость, связанная с окислением ъ Весьма малая раствори- мость, связанная с оки- слением Нерастворим
МоО2 Мало растворим (<С0,025) Незначительная мость, связанная нием (0,1—0,2) раствори- с окисле- Незначительная раствори- мость, связанная с оки- слением (0,06—0,08) Мало растворим (0,005—0,01)
-- Мо4Оц Мало растворим (^0,09) Незначительная мость, связанная нием (^2—4,5) раствори- с окисле- Незначительная раствори- мость, связанная с оки- слением (1—2,5) Мало растворим (^0,08)
МоО3 Мало растворим (0,4-2) Быстро растворяется Быстро растворяется Растворяется
СаМоО4 Мало растворим (0,03—0,06) Мало растворим (^ 0,09) Разлагается с переходом мо- либдена в раствор Растворяется
Продолжение табл. 12
Соединение Растворитель
вода растворы аммиака (5%-ные) растворы соды (5% -ные) соляная кислота (5%-ная)
СпМоО4 Мало растворим (-0,16) • Ёыстро растворяется Разлагается с переходом молибдена в раствор Растворяется
ZnMoO4 Мало растворим (-1,6) Растворяется Растворяется Растворяется
FeMoO4 Мало растворим (-0,08) Медленно разлагается с пере- ходом молибдена в раствор Разлагается с переходом мо- либдена в раствор Растворяется
PbMoO4 Мало растворим (-0,03) Мало растворим (~0,13) Растворяется Растворяется
Fe2 (MoO4)3 Мало растворим, гидролитически разлагается Легко разлагается с перехо- дом молибдена в раствор Легко разлагается с перехо- дом молибдена в раствор Растворяется
MgMoO4 Растворяется (15,9%) Разлагается с переходом мо- либдена в раствор Легко разлагается с пере- ходом молибдена в рас- твор Растворяется
2. Молибдаты кальция, меди, цинка, железа и свинца мало
растворимы в воде, растворяются в соляной кислоте и разла-
гаются растворами соды. Относительно высокой растворимостью
в воде отличается молибдат магния (—160 г!л при 25° С). По отно-
шению к растворам аммиака они подразделяются на три группы:
а) СиМоО4 и ZnMoO4 растворяются с образованием молибдата
.аммония и аммиачных комплексов Me (NH3)4 (ОН)2;
б) СаМоО4, РЬМоО4 практически не разлагаются;
в) FeMoO4 разлагается медленно, Fe2 (МоО4)3 разлагается
быстро с переходом молибдена в раствор и образованием гидро-
закиси и гидроокиси железа.
Разложение FeMoO4 раствором аммиака первоначально про-
текает быстро, однако после разложения ~30% скорость реакции
резко понижается, вероятно, вследствие образования пленок гидро-
закиси железа, а также гидроокиси, образующейся в результате
окисления Fe (ОН)2. Возобновление активного реагирования на-
блюдается после высушивания остатка при 100° С, при котором
нарушается структура пленок гидроокисей железа [52]. Из про-
каленного на воздухе при 600—700° С FeMoO4 весь молибден легко
выщелачивается аммиачной водой, что связано с его окислением
и образованием Fe2O3 и МоО3. Осадки гидроокисей железа, обра-
зующиеся при разложении FeMoO4 и Fe2 (МоО4)3 растворами ам-
миака, способны прочно сорбировать молибдат-ионы. Вследствие
этого остатки (хвосты) от выщелачивания огарков даже после
^многократной промывки содержат растворимый молибден.
Автор, Буровой и Никитина изучали кинетику выщелачивания
молибденовых огарков растворами аммиака в условиях, исклю-
чающих внешнедиффузионное сопротивление. Скорости выщела-
чивания высокие. Так, практически полное выщелачивание молиб-
дена из огарков печи КС (размер частиц 0,05—0,6 мм) 8%-ным NH3
достигается на 10—12-й минуте при 20° С и 1,5—2-й минуте при
50° С при большом избытке аммиака.
Было установлено, что степень выщелачивания в интервале
концентрации аммиака 1—8% и температурах 20—60° С изме-
няется во времени по экспоненциальному закону относительно
•содержания молибдена в твердой фазе. В частности, при концен-
трации аммиака 8% зависимость константы скорости от темпера-
туры описывается уравнением
1пЛ = —-^+31,6, (31)
1 dG
^Мо
где GMo — содержание молибдена в твердой фазе, доли единицы.
Энергия активации, равная 19 ккал!моль, остается постоянной
в интервале температур 20—60° С.
’1
54
Скорость реакции выщелачивания имеет нулевой порядок по
концентрации молибдена в растворе (в интервале концентраций
О—120 г/л Мо) и первый порядок по концентрации аммиака.
Огарки выщелачивают 8—10%-ным раствором аммиака на хо-
лоду или при нагревании до 50—60° С при отношении т : ж —
= 1 : 3-1 : 4. Расход аммиака на выщелачивание колеблется от
115 до 140% от теоретически необходимого количества. Следует
учитывать необходимость сохранения в конечных растворах из-
быточной концентрации аммиака (25—30 г/л) для предотвращения
образования полимолибдатов. В зависимости от состава огарков
извлечение молибдена в аммиачный раствор составляет 80—95%,
выход хвостов (масса хвостов по отношению к массе исходного
огарка) колеблется от 10 до 30%, а содержание молибдена в них —
от 5 до 25%. Поэтому, как правило, необходимо дополнительное
извлечение молибдена из хвостов (см. стр. 71).
Низкое прямое извлечение преимущественно связано с повышен-
ным содержанием в исходных огарках молибдата кальция и дву-
окиси молибдена, а также неокислившегося молибденита.
Как отмечалось, при обжиге в кипящем слое кальцит преиму-
щественно превращается в CaSO4, меньшая доля — в СаМоО4.
Однако при выщелачивании огарков аммиачной водой сульфат
кальция переходит в менее растворимый молибдат кальция х:
CaSO4 + МоОГ -> СаМоО4 + SO1-. (32>
Предотвратить эту реакцию и этим повысить общее извлечение
молибдена в аммиачный раствор можно добавлением карбоната
аммония, с которым CaSO4 реагирует, образуя СаСО3. Кроме того,
в присутствии карбоната аммония молибдат кальция частично
разлагается по реакции
СаМоО4 + (NH4)2 СО3 СаСО3 + (NH4)2 МоО4. (33)
Реакция, однако, быстро прекращается из-за образования на
частицах молибдата пленок карбоната кальция.
Благоприятное действие карбоната аммония состоит также и
в том, что сульфаты и молибдаты железа, реагируя с карбонатом,
образуют осадки основных карбонатов1 2 , обладающие меньшей
адсорбционной способностью, чем гидроокиси железа. Это сни-
жает содержание выщелачиваемого молибдена в хвостах.
В работах [2, с. 35; 56] показано, что добавки карбоната аммо-
ния в растворы аммиака повышают прямое извлечение молибдена
из огарков печей кипящего слоя с 83—85 до 95—96,5% и сни-
жают содержание молибдена в хвостах с 27—28 до 7—8%, причем
1 Растворимость CaSO4 в воде при 20° С составляет ^>2 г/л, СаМоО4;
0,028 г/л [49].
2 Для трехвалентного железа известны, например, основные карбонаты
состава: 3Fe(OH)3* FeOHCO3 и 7Fe(OH3)« FeOHCO3 [61 ].
влияние добавки сказывается тем сильнее, чем больше содержа-
ние кальция в исходном огарке. Результаты существенно зависят
от порядка добавления реагентов. Необходимо, чтобы на огарок
непосредственно воздействовал карбонатсодержащий раствор.
Если карбонат аммония добавляют после предварительной обра-
ботки огарка раствором аммиака (при которой весь сульфат каль-
ция превращается в СаМоО4, а соли железа разлагаются с образо-
ванием гидроокисей железа), положительный эффект мало прояв-
ляется, так как молибдат кальция лишь частично разлагается
карбонатом аммония, а осадки гидроокисей железа не переходят
в основные карбонаты. Для обеспечения достаточно высокого
извлечения молибдена из огарков рекомендуется добавлять в рас-
твор аммиака примерно 60—80 кг карбоната аммония на тонну
огарка. При выщелачивании аммонийно-карбонатным раствором
обожженных хвостов от выщелачивания огарков общее извлечение
молибдена в растворы, по данным цеховых опытов, достигает
97—99% в зависимости от происхождения огарка [56] *.
В производственной практике выщелачивание огарков аммиач-
ной водой ведут в реакторах с механическими мешалками или ба-
рабанных вращающихся выщелачивателях, применяя для более
полного извлечения молибдена трех-четырехкратную обработку.
Крепкие растворы первых двух выщелачиваний, содержащие
120—140 г/л Мо, объединяют и выводят на очистку от примесей.
Слабые растворы последующих стадий возвращают на первые
стадии. Хвосты выщелачивания после промывки направляют на
дополнительное извлечение молибдена (см. стр. 171) [55, 60].
Для понижения степени перехода примесей в аммиачный рас-
твор иногда перед выщелачиванием огарок промывают водой.
При этом в раствор переходят растворимые соли щелочных ме-
таллов, сульфаты меди, железа, магния, отчасти кальция. Так, при
промывке водой огарка, полученного при обжиге тырны-ауз-
ского молибденитового концентрата и содержавшего, %: 51,2 Мо,
0,76 Fe, 0,75 СаО, 0,14 СиО, 0,29 Na, 0,11 MgO, 0,86 SO?2, в про-
мывную воду перешло, %: 58,3 Na, 19,3 Са, 14,5 Mg, 23,8 Fe,
76,1 S и 8,9 Си от количества этих примесей в исходном огарке.
Однако потери молибдена с промывной водой составляют
3—4%. Концентрация МоО3 в водном растворе достигает 3—5 г/л.
Это объясняется, как нами было показано [52], повышенной рас-
творимостью молибдатов в воде в присутствии трехокиси молиб-
дена (табл. 13) вследствие образования растворимых полимолибда-
тов, в частности метамолибдатов Л4еМо4О13 (где Me — Са, Си и др.)
по реакции
Me МоО4 + ЗМоО3 Me Мо4О13. (34)
Вследствие этого промывка огарков водой нецелесообразна.
* ЗеликманА. Н., Тараканов Б. М. Авт. свид. СССР, № 165546,
10/1 1963 г. Бюлл. изобр., 1964, № 19.
56
ТАБЛИЦА 13*
ИЗВЛЕЧЕНИЕ МОЛИБДЕНА В РАСТВОР ПРИ ОБРАБОТКЕ ВОДОЙ
СМЕСИ МОЛИБДАТА С ТРЕХОКИСЬЮ МОЛИБДЕНА ПРИ 20° С [52]
Молибдат Концентрация молибдена, г/л, в
М е МоО4 смеси Me МоО4 + ЗМоО3
СаМоО4 0,06 2,45
СиМоО4 0,16 8,45
ZnMoO4 1,57 10,86
FeMoO4 0,034 2,13
PbMoO4 0,08 0,29
Можно интенсифицировать процесс выщелачивания, если про-
водить его в аппарате кипящего слоя непрерывного действия ана-
логично тому, как это описано для выщелачивания цинковых огар-
ков серной кислотой [36, 98].
В аппарат, представляющий собой колонку с изменяющимся
по высоте диаметром, непрерывно снизу поступает раствор ам-
миака, а сверху (в среднюю часть колонны) подается водная
пульпа огарка. Из верхней расширенной части выпускаются про-
дукты выщелачивания (осветленный раствор и шламы). .
Производительность достигала 30—35 т огарка на 1 м3 аппа-
рата в сутки. Существенными преимуществами являются поддер-
жание постоянной концентрации молибдена в растворах молиб-
дата аммония и возможность полной автоматизации процесса Ч
9. ОЧИСТКА АММИАЧНЫХ РАСТВОРОВ ОТ ПРИМЕСЕЙ
ОСАЖДЕНИЕМ СУЛЬФИДОВ
Аммиачные растворы содержат примеси меди, цинка, железа,
никеля, щелочноземельных и щелочных металлов, ионы SOl”
и др. Примеси меди и цинка находятся в составе прочных ком-
плексных анионов Ше (NH3)4]2+, константы нестойкости которых
равны 2,14-10"13 и 3,46-10"10 соответственно. Никель также
образует стойкий аммиакат Ni (NH3)|+ с константой нестойкости
1,86-10"9 [*96].
Аммиачные растворы обычно содержат также примеси железа:
Fe (II) образует неустойчивый аммиакат, тогда как примесь
Fe (III), по всей вероятности, находится в растворе в составе
коллоидных частиц гидроокиси. Если при выщелачивании огар-
ков применяют добавку карбоната аммония, трехвалентное железо
может присутствовать в составе аммиачно-карбонатного ком-
плекса (NH4)X [Fe (СО3) ]п (ОН)Щ] [61]. Для очистки от примесей
1 Л. С. Никитина. Диссертация. МНС и С, 1969.
57
меди и железа их сульфиды осаждают сернистым аммонием.
Добавляемый сернистый аммоний реагирует с молибдатом аммо-
ния с образованием оксисульфомолибдатов:
(NH4)2 МоО4 + х NH4HS (NH4)2 MoO4_xSx + х NH4OH. (35)
Это обусловливает низкую концентрацию ионов S2" в растворе.
Однако в растворе остается некоторая концентрация ионов S2“,
соответствующая равновесию реакции. Произведение раствори-
мости CuS очень мало (8,5-10“45). Это обеспечивает полное оса-
ждение меди, несмотря на то что она связана в прочный комплекс.
Железо также полно осаждается. Произведение растворимости
FeS (3,7* 10~19) значительно больше, чем CuS, но аммиачные ком-
плексы железа непрочные, концентрация ионов железа достаточна
для достижения произведения растворимости. По-иному обстоит
дело с примесями цинка и никеля. Произведения растворимости
ZnS и NiS достаточно малы (6,9-10~26 и 7,9- 10~26 соответственно).
Однако вследствие прочности аммиачных комплексов концентра-
ция ионов цинка и никеля столь низка, что произведение раство-
римости их‘сульфидов (с учетом того, что в растворе мало свобод-
ных ионов S2~) не достигается. Вследствие этого примеси цинка
и никеля остаются в растворе.
Раствор сернистого аммония вливают в аммиачный раствор
небольшими порциями, контролируя полноту осаждения. Избыток
сернистого аммония может- быть ликвидирован добавлением не-
которого количества свежего аммиачного раствора.
Объемистый осадок сульфидов меди и железа сорбирует неко-
торое количество молибдата аммония, что обусловливает потерю
примерно 0,3% Мо.
10. ВЫДЕЛЕНИЕ МОЛИБДЕНА ИЗ АММИАЧНЫХ РАСТВОРОВ
В ВИДЕ ПОЛИМОЛИБДАТОВ
Условия образования и состав полимолибдатов
Молибденовая кислота способна присоединять различное числе
молекул МоО3 с образованием поликислот, производными которые
являются полимолибдаты. В отличие от нормальных молибдатов
у полимолибдатов молярное отношение ТИе2О : МоО3 (где Me —
Na+, К+, NHt) меньше единицы и изменяется в широких преде
лах. Известны, например, следующие типы полимолибдатов:
ЛМ2О • 2МоО3 ..............
ЗЛ1е2О-7МоО3 1
5Л4е2О-12МоО3 J ...........
/Ие2О-ЗМоО3 ...............
ТИе2О • 4МоО3 .............
/Ие2О*10МоО3 .......
Димолибдаты
Парамолибдаты
Тримолибдаты
Метамолибдаты (или
тетр амолибдаты)
Декамолибдаты
58
и ряд других. Большей частью они выделяются из растворов в виде
кристаллогидратов с различным числом молекул воды. Полимо-
либдаты получаются при подкислении растворов нормальных
молибдатов щелочных металлов и аммония в определенном интер-
вале значений pH, например от 6,5 до 1. При этом ионы МоО1”
переходят в полимерные анионы по упрощенной схеме:
п МоОГ + 2 (п — х) Н+ М0„О^+х + (П — х) Н2О.
Так, например, образование парамолибдат-иона MoyO24G и мета-
молибдат-иона МО4О13- можно представить реакциями:
7МоО1~ + 8Н+ МоуОгГ + 4Н2О,
4МоО2Г + 6Н+ Мо4О1Г + ЗН2О.
В равновесии с этими полианионами
могут быть анионы типа НМО7О2Г,
Н2МО7О24 , Н3МО7О24 , НМО4О13ИДР.,
образующиеся в результате присое-
динения протонов к полиионам. Зна-
чения pH, при которых образуются
полиионы, зависят от концентрации
молибдена в растворе, что было уста-
новлено Карпени [65] и в более
поздних работах Яцимирским и Алек-
сеевой [66, 77] и Морачевским и
Лебедевой [67].
Рис. 21. Диаграмма состояния
молибденовой кислоты в водных
растворах [66]
Как видно из рис. 21, pH начала образования полимерных
ионов возрастает с увеличением концентрации молибдена в рас-
творе. При каждом значении pH в зависимости от концентрации
молибдена имеются три области [66]: область I — существования
только ионов МоО1~ (наиболее низкие концентрации молибдена);
область II, в которой ионы МоО1~ находятся в равновесии с по-
лимерными анионами, и область III, отвечающая существованию
только полимерных анионных форм. При концентрациях молиб-
дена, встречающихся в производственной практике (50—150 г/л),
уже при pH = 7,5 и ниже мы попадаем в область существования
различных полимерных форм, находящихся в равновесии между
собой и ионами МоО|Л
Изучению состава, природы и условий образования полимер-
ных ионов, содержащих молибден, посвящено много работ, вы-
воды которых существенно расходятся (преимущественно в отно-
шении состава сложных ионов).
Яндер с сотрудниками [68] из измерений коэффициентов диф-
фузии определял молекулярную массу полимерных ионов. Сопо-
ставляя полученные результаты с данными кондуктометрического'
титрования и термо-титрования
ствовании следующих анионов:
они пришли к выводу о суще-
pH
Ионы
>6,1 ............
-6,0 ............
-4,5—6,0...........
2,9—4,5............
0,9—2,9............
Только М.ОО4
и HMo3OjJ“
НМо6О*Г
Н3М°бО21 , Н7Мо24О78
^9^°24^78
После изоэлектрической точки (pH — 0,9) по мере повышения
кислотности разрушаются полимерные ионы и появляются ком-
плексные катионы. В последующей работе того же автора [69]
выражается сомнение в существовании тримолибдатионов Мо3Оп".
По данным Бринцингера с сотрудниками [71; 72], изучавших
состав анионов молибдена в слабокислой среде при концентрации
молибдена 0,05 моль!л с помощью диализа и потенциометрического
титрования, в растворах находятся ионы следующего состава:
pH Ионы
>6,4 ........... МоО|~
5,2—6,4...............Мо3О|р в равновесии с Мо6О2^“,
M°6O|j“ и МоО4“
1,5—5,0...................... Мо6О^7
Бийе [73—75], использовавший криоскопический метод ис-
следования, предполагает существование анионов. МО7О2Г,
Мо60^о , МО4О13 , НМОбО^о •
Линдквист [76] считает, что в различных интервалах pH
в равновесии существуют полиионы с семью атомами молибдена:
7МоОГ 4- 8Н+ МО7О2Г + 4Н2О,
МО7О2Г + Н+ НМО7О2Г,
нмо7о|г + н+ Н2МО7О2Г.
Константы равновесия этих реакций, поданным работ [63, 64, 70],
приведены в табл. 14.
Из данных табл. 14 следует, что наибольшей прочностью отли-
чаются парамолибдат-ионы МО7О2Г.
Чаньи [78, 79], применивший метод хроматографии на бумаге,
пришел к выводу, что в слабокислых растворах доминирующими
являются гексамолибдат-ионы МобОэд”-
1 При термо-титровании регистрируется изменение температуры в процессе
титрования.
во
ТАБЛИЦА 14
КОНСТАНТЫ РАВНОВЕСИЯ РЕАКЦИИ ОБРАЗОВАНИЯ ПОЛИМОЛИБДАТОВ
Реакция 1g А, по данным
[70] [64]
МоО1~ + Н+zi НМоОГ 1 if 4,08 3,9
7МоО|- + 8Н+ Мо7О|7 + 4Н2О 57,7 57,76
Мо7О^ + н+ НМо7О|7" 4,33 4,41
НМо7О|7 + н+ Н2Мо7О^ 3,7 3,5
Мартинес и Кабанес [84] криоскопическим методом нашли
следующие анионные формы в различных интервалах pH:
pH Формула
>6,1............ МоО4—
2,9—4,5............ МоХЙГ
~6,0.............. Мо4О^~
0,9—2,9............. Мо7О^
4,5—6,0............ Мо.О,Г
Ниже изоэлектрической точки, лежащей примерно при pH =
= 0,9 4-1,1 [68. 81, 82], полимерные анионы разрушаются и до-
минирующими становятся комплексные оксикатионы, например
молибденила МоО2+:
МоОГ + 4Н+
МоО2+ + 2Н2О.
(36)
В ряде работ показано, что в слабокислых растворах присутствуют
преимущественно полимерные оксикатионы, например Mo2Ol+,
Мо3Ов+ [82, 87, 90, 91].
При переходе от менее кислых растворов к более кислым проис-
ходит деполимеризация оксикатионов с образованием мономера:
2Мо30|+ + 2Н+ ЗМо2Од+ + Н2О, (37)
Мо2025+ + 2Н+ 2МоО2+ + Н2О. (38)
Катионы, находящиеся в равновесии с анионными формами,
появляются еще до изоэлектрической точки, начиная с pH =
= 2,5 4-2. Гохштейн [81] доказал это полярографическим мето-
дом, Бабко [83] — изучением сорбции молибдена на анионитах и
катионитах при различных значениях pH раствора, Романо
Рао [86] —осаждением при pH = 2,4 оксихинолином желтого
осадка оксихинолята молибденила МоО2 (C9N6NO)2.
61
Несмотря на имеющееся расхождение данных различных ис-
следователей о составе полимерных комплексных ионов молиб-
дена в растворе, можно считать установленным:
А) выше pH = 7,5-г-7 (в зависимости от концентрации молиб-
дена в растворе) существуют только молибдат-ионы МоО1~;
б) в интервале pH от 7,5—6,5 до —2,5 (при концентрации мо-
либдена более 0,2 моль/л) происходит полимеризация с образова-
нием сложных анионов: Mo4Oi^ (метамолибдат-ион), МО7О2Г (па-
рамолибдат-ион), Мо6О2(Г (гексамолибдат-ион) и некоторых дру-
гих. Эти ионы, по всей вероятности, гидратированы, т. е. пред-
ставляют собой акваполиионы;
в) при pH меньше 2,5 начинается образование катионных ком-
плексных ионов, находящихся в равновесии с анионными фор-
мами;
г) после изоэлектрической точки (pH меньше 1,0) молибден
находится в растворе преимущественно в составе комплексных
катионов МОО2+ Мо20|+, Мо3Ов+.
Механизм полимеризации и структура полианионов, образу-
ющихся при подкислении растворов молибдатов, недостаточно
выяснены.
Силлен [80] считает, что полимеризация протекает ступенчато,
в растворах в равновесии находятся различные полимеры, по-
строенные по типу «ядро—звенья» (т. е. содержащие «ядро» и
различное число связанных с ним однотипно построенных
«звеньев»).
По представлениям Чожнак [85], полимеризации предше-
ствует, как первая стадия, протонизация анионов МоО|”, приво-
дящая к образованию сложных катионов, способных к конден-
сации с анионными формами молибдена. Соответственно этому
в слабокислом растворе протекают три процесса:
1. Протонизация:
н+ н+ н+
МоО1~ [МоОз(ОН)]” [Мо02 (ОН)2]° [МоО (ОН)3]+.
2. Гидратация:
[МоО3 (ОН)]- + 2Н2О 77 [МоО (ОН)6Г,
[МоО2 (ОН)2]° + 2Н2О^Г [МоО (ОН)6]°,
[МоО (ОН)3]+ + 2Н2О^ [Мо (ОН)6 Н2О]+.
3. Конденсация:
2 [Мо (ОН)6]° 7" [(ОН)5 — Мо — О — Мо — (ОН)6]° + Н2О,
[МоО (ОН)5Г + [Мо (ОН)5 Н2О]+ 77
[(ОН)5 — Мо — О — Мо — (ОН)6]° + Н2О.
62
Таким образом, по мнению автора, в растворах в равновесии
могут находиться, наряду с полимерными анионами и катионами,
также полимерные нейтральные молекулы. Приведенная выше
схема, хотя и объясняет качественно полимеризацию, не позволяет
вывести конкретные полимерные формы — тетрамолибдат Мо4О1Т\
парамолибдат МО7О2Г и др.
Спицын [62] в результате ряда исследований считает, что
в образовании акваполисоединений участвуют ионы водорода,
связывающие между собой анионы, участвующие в построении
комплексного иона. Кроме того, предполагается наличие в струк-
туре акваполианионов оксониевых группировок. Так, например,
аниону паравольфрамата приписывается состав [ОН3 (WO4)3 X
X (Н3О)3 (Н2О)3]5“.
Способ выпарки
Парамолибдат аммония 3 (NH4)2O- 7МоО3* 4Н2О или
(NH4)6 Мо7О24- 4Н2О кристаллизуется из растворов молибдата ам-
мония, в которых молярное отношение NH3 : МоО3 равно 6 : 7
или несколько выше. Это соотношение достигается выпариванием
раствора, при котором удаляется часть аммиака:
7 (NH4)2 МоО4 -> (NH4)e Мо7О24 + 8NH3 + 4Н2О. (39)
Парамолибдат аммония устойчив на воздухе. Водные растворы
его имеют слабокислую реакцию. Растворимость парамолибдата
аммония в воде (в пересчете на содержание МоО3) составляет:
при 10° С 17,47%; при 35° С 34,6%; при 60° С 43,75%. Из раство-
ров выделяется кристаллогидрат с 4 молекулами воды. На выпа-
ривание поступают растворы, содержащие 120—140 г/л МоО3
(плотность растворов 1,09—1,12). Выпаривание проводят в аппа-
ратах из нержавеющей стали с паровой рубашкой в две стадии.
Предварительную выпарку ведут до плотности 1,20—1,23, после
чего растворы отстаивают и фильтруют для отделения осадка
сульфидов меди, железа и гидроокиси железа, которые выде-
ляются в результате коагуляции коллоидных частиц и тонкодис-
персных взвесей [60]. Затем проводят основную выпарку до плот-
ности 1,38—1,40 (примерно 400 г/л МоО3), горячий раствор филь-
труют и собирают в кристаллизаторе.
Во избежание образования молибдатов более кислых, чем
парамолибдат (т. е. с меньшим отношением NH3 : МоО3), необхо-
димо в процессе выпарки сохранять в растворе некоторый избыток
аммиака (5—10 г/л), а также перемешивать раствор, чтобы избе-
жать местных перегревов. Молибдаты более кислые, чем пара-
молибдат, выделяются в форме мелкокристаллических трудно-
растворимых осадков на стенках выпарного аппарата. Кристалли-
зацию проводят в кристаллизаторах с мешалкой и принудитель-
ным охлаждением в несколько последовательных стадий. При пер-
63
вой кристаллизации выделяется 50—60% соли, содержащейся
в растворе, при второй еще 15—20%, при третьей —10%. Хво-
стовой маточный раствор, в котором концентрируются примеси,
выпаривают досуха, а остаток прокаливают при 350—400° С и
полученную трехокись молибдена возвращают на выщелачивание
раствором аммиака вместе с огарком.
Мелкокристаллический осадок парамолибдата аммония, отде-
ленный от маточного раствора центрифугированием и промытый
холодной водой, является товарным продуктом или поступает на
термическое разложение для получения трехокиси молибдена.
Парамолибдат аммония, полученный описанным выше методом
выпарки, часто содержит примеси в количествах, превышающих
допустимые техническими условиями. Это относится к примесям
щелочных металлов (при повышенном содержании их в огарке),
примесям цинка и никеля (которые не отделяются при сульфидной
очистке), примесям вольфрама. При проведении фракционной
кристаллизации примеси вольфрама и цинка концентрируются
в первых фракциях кристаллов, что позволяет отделить большую
их часть. Однако это в значительной мере снижает выход чистых
кристаллов парамолибдата аммония. Отмеченные выше недостатки
обусловили применение на отечественных заводах первоначаль-
ного выделения молибдена из аммиачных растворов в форме
полимолибдата методом нейтрализации.
Выделение тетрамолибдата аммония из слабокислых
растворов
При подкислении производственных растворов молибдата ам-
мония (с концентрацией МоО3 от 140 до 300 г/л) до pH = 2—3 при
температуре 50—70° С и интенсивном перемешивании молибден
выделяется в составе мелкокристаллического осадка двухвод-
ного тетрамолибдата (метамолибдата) (NH4)2 Мо4О13* 2Н2О или
(NH4)2 О-4МоО3-2Н2О:
4(NH4)2MoO4 + 5H2O—2^(NH4)2Mo4O13-2H2O + 6NH4OH.
(40)
Условия выделения тетрамолибдата из производственных рас-
творов детально исследованы Золотаревым [58, 59], который изу-
чал растворимость и состав фаз в системе NH3—МоО3—НС1—Н2О.
В табл. 15 и на рис. 22 приведены данные для части изотермы си-
стемы [20% (по массе) NH3 + 80% (по массе) МоО3]—НС1—Н2О
при концентрации МоО3 29,6% (плотность раствора 1,25). Весовое
отношение NH3 : МоО3 = 20 : 80 соответствует обычным произ-
водственным растворам, содержащим 5—10 г/л свободного NH3.
Минимум растворимости наблюдается при pH = 2, когда вы-
деляется двухводный тетрамолибдат аммония. Двухводный тетра-
молибдат аммония — метастабильная фаза. При контакте с маточ-
64
ним раствором происходит его дегидратация с превращением
в безводный стабильный тетрамолибдат (NH4)2Mo4O13. Превраще-
ние сопровождается измельчением
частиц до размеров меньше 1 мкм.
Образующийся при этом объеми-
стый осадок трудно отделяется
от маточного раствора и адсорби-
рует примеси. На рис. 23 и 24
приведены кинетические кривые
дегидратации в зависимости от тем-
пературы (рис. 23) * и в зависи-
мости от pH раствора при 60° С
(рис. 24) *. При температурах
20—40° С водная соль относительно
устойчива (индукционный период
составляет 3—4 ч). При 50° С и
выше индукционный период резко
сокращается, фазовое превраще-
ние протекает быстро. Уменьше-
ние pH маточного раствора с 3
до 1 при температуре 60° С спо-
собствует ускорению дегидратации
соли (рис. 24).
Для сохранения кристалличе-
ского осадка водного тетрамолиб-
Рис. 22. Зависимость содержания
молибдена в маточном растворе
от pH при нейтрализации раствора
молибдена аммония соляной кисло-
той
дата рекомендуется быстрое отделение осадка от маточного рас-
твора фильтрацией с тем, чтобы свести до минимума контакт кри-
ТАБЛИЦА 15
СОСТАВ ТВЕРДЫХ ФАЗ И СОДЕРЖАНИЕ MoO3 В РАВНОВЕСНОМ
РАСТВОРЕ ПРИ 35° С ДЛЯ СИСТЕМЫ 20% (ПО МАССЕ) NH3 + 80%
(ПО МАССЕ) МоО3 — НС1 — Н2О; КОНЦЕНТРАЦИЯ МоО3 В ИСХОДНОМ
РАСТВОРЕ 29,6%, ПЛОТНОСТЬ РАСТВОРА 1,25
Содержание МоО3 в растворе, % pH раствора Состав твердой фазы (формула безводной соли)
2,28 1,92 1,71 0,81 0,61 0,43 0,27 0,10 0,05 0,07 0,09 0,13 5,25 ] 5,03 J 4,92 1 4,43 4,08 3,62 J 3,15 I 2,65 2,13 1,67 ] 1,22 J 0,83 > 3 (NH4)2O-8MoO3 (NH4)2O-3MoO3 (NH4)2O-4MoO3 (NH4)2O-6MoO3 (NH4)2O-7MoO3
* Золотарев Л. Л. «Гидролитический метод очистки от примесеи
в гидрометаллургии молибдена». Кандидатская диссертация, 1958.
5 А. Н. Зеликман
65
сталлов с раствором. С этой целью пульпу из реактора-осадителя
выпускают на нутч-фильтр, где маточный раствор отделяется
за несколько минут от кристаллов, которые затем отжимают
на центрифуге.
Рис. 23. Кинетика дегидратации дигидрата тетрамо-
либдата аммония при различных температурах при
pH = 2
При осаждении из растворов с концентрацией 280 г/л МоО3
(если необходимо, растворы предварительно концентрируют вы-
париванием) с осадком двухводного тетрамолибдата выделяется
97—98% Мо. В кислом маточном растворе остается 2—3% Мо
Рис. 24. Кинетика дегидратации дигидрата тетрамолибдата
в зависимости от pH раствора при 60° С (цифра на кри-
вых — pH)
(что отвечает концентрации 3—5 г/л Мо). После длительного стоя-
ния из маточного раствора дополнительно выделяются аморфные
осадки полимолибдатов различного состава, которые после раство-
рения в аммиачной воде возвращают на операцию очистки от при-
66
»
месей — осаждение сульфидов сернистым аммонием. Хвостовые
маточные растворы содержат ~1 г!л Мо (—0,5—0,7% от исход-
ного содержания молибдена в аммиачном растворе). Из них молиб-
ден может быть регенерирован сорбцией на ионообменных смолах
(см. стр. 150). Отфильтрованные на центрифуге и промытые хо-
лодной водой осадки тетрамолибдата содержат мало посторонних
примесей. Подавляющая часть примесей (ионы Na, К, Са, Mg,
Zn, Си, Ni, Fe, Sb, As, S) остается в слабокислом (pH ^2—3)
маточном растворе. Исключение составляет вольфрам, большая
часть которого соосаждается с полимолибдатом. Кроме того,
осадки содержат примесь ионов хлора (0,2—0,4%), трудно отмы-
ваемых водой. Для очистки тетрамолибдата от хлора и превраще-
ния его в обычный товарный продукт — парамолибдат аммония,
проводят перекристаллизацию осадка через аммиачный раствор.
С этой целью растворяют кристаллы тетрамолибдата в 3—5 %-ном
растворе аммиака при 70—80° С до насыщения (плотность рас-
твора 1,41—1,42). После охлаждения до 15—20° С из раствора
выделяется 50—60% Мо в составе парамолибдата 3(NH4)2O*
•7МоО3-4Н2О. Маточный раствор может служить для последова-
тельной перекристаллизации 8—10 партий осадков тетрамолиб-
дата. Постепенно в маточном растворе накапливаются примеси
и его выводят, присоединяя к растворам, поступающим на оса-
жден ие су л ьфидов.
Выделение тетрамолибдата из слабокислого раствора в сочета-
нии с последующей перекристаллизацией осадка через аммиачные
растворы обеспечивает получение парамолибдата аммония более
высокой чистоты, чем это достигается при использовании способа
выпарки и кристаллизации парасоли.
Термическое разложение парамолибдата аммония
Термическое разложение молибдатов аммония различного со-
става изучалось в работах [92—95] с использованием термогра-
фического и термогравиметрического методов.
Для парамолибдата состава 3(NH4)2O 7МоО3-4Н2О приводятся
следующие схемы разложения.
По Фунаки и Сегава [92]:
110° С 200° С
3 (NH4)2 О • 7МоО3 • 4Н2О-------* 3 (NH4)2 О • 7МоО3----->
280° С
(NH4)2 О • 4МоО3------* Мо03.
По Дювалю [93]:
50_спо р 140_200° С
3 (NH4)2 о• 7МоО3 • 4Н2О —2 (NH4)2O • 5Мо03------------------->
„ ... „ 270—310° С „ „
- (NH4)2 О 4Мо03-----------* Мо03.
5* ’ 67
По Золотареву:
3 (NH4)2 О - 7МоО3 • 4НаО (Nh4)2 О • ЗМоО3 ——
>260° С
—» (NH4)2 О • 6МоО3---->МоО3.
По Хегедюш с сотрудниками [94],. изучавшему разложе-
ние парамолибдата в восстановительной атмосфере (30% Н2 +
+ 70% N2):
3 (NH4)2 О • 7МоО3 • 4Н2О 2 (NH4)2 О • 5МоО3 -*
[210—255° С , г 300° С т т _ _ _ 350—400° С _
--------* (NH4)2O • 4МоО3---->Н2МоО4----------*МоО3.
Для парамолибдата состава 5(NH4)2O 12Мо03- 12Н2О (отли-
чающегося от обычного производственного продукта) Роде и
Твердохлебов приводят схему [95]:
5 (NH4)2 О • 12Мо03 • 12Н2О 5 (NH4)2 О • 12МоО3
г 250—265° С тг" 320—370° С
—[2 (NH4)2 О • 5МоО3--------> (NH4)2 О • 4МоО3--------> МоО3.
Данные различных авторов о термическом разложении
3(NH4)2O-7МоО3 отличаются преимущественно по составу про-
межуточных фаз и температурному интервалу их образования.
Это обусловлено различием примененных методов исследования.
Так, при термографическом анализе, когда скорость подъема
температуры относительно высока, могут быть обнаружены не-
стабильные фазы, которые при длительном изотермическом нагре-
вании перейдут в фазы другого состава. При длительных выдерж-
ках начало фазового превращения может быть обнаружено при
значительно более низких температурах, чем это отмечается по
термическому эффекту на термограммах.
Из большей части работ следует, что парамолибдат аммония
полно обезвоживается (теряет 4 молекулы воды) при температурах
90—110° С, а продуктом, непосредственно предшествующим обра-
зованию МоО3, является безводный тетрамолибдат (NH4)2O-
• 4МоО3, который в интервале температур 280—380° С разла-
гается до МоО3.
По Золотареву, дигидрат тетрамолибдата аммония разлагается
по схеме
ел_960° С' х>260° С
(NH4)2 0.4МоО3 • 2Н2О----------> (NH4)2 О - 4МоО3 ----> МоО3.
Роде и Твердохлебов отмечают образование промежуточного
гидрата [95]:
105_woo р
(NH4)2 О • 4МоО3 • 2Н2О----------* (NH4)2 О • 4МоО3 • Н2О —
145—155° С _ __ _
-------* (NH4)2 О-4МоО3
320—365° С , ,
---------> МоО3.
68
В производственной практике термическое разложение пара-
молибдата аммония с целью получения трехокиси молибдена про-
водят при температурах 450—500° С в электрических печах с труб-
чатым муфелем прямоугольной формы, куда помещаются лодочки
из нержавеющей стали или сплава никеля с молибденом, запол-
ненные солью. Они продвигаются с определенной скоростью
вдоль трубы печи. Кроме того, для термического разложения
парамолибдата применяют барабанные печи непрерывного дей-
ствия [60]. При соответствующем режиме работы полученный
порошок трехокиси молибдена однороден и имеет бледно-зелено-
желтый цвет. Белые включения свидетельствуют о неполном раз-
ложении парамолибдата, а черные включения — показатель ча-
стичного восстановления МоО3 выделяющимся аммиаком. Это воз-
можно при недостаточно быстром удалении газообразных продук-
тов разложения (при плохой тяге).
11. РАСПРЕДЕЛЕНИЕ ПРИМЕСЕЙ И ЧИСТОТА
ТРЕХОКИСИ МОЛИБДЕНА
Описанная выше технология получения трехокиси молибдена
(парамолибдата аммония), включающая выделение тетрамолиб-
дата аммония (см. рис. 20), обеспечивает получение продукта
высокой чистоты, характеризуемого следующим содержанием при-
месей, %: не более 0,0001 Sn, Pb, Bi, Sb, Cd; не более 0,001 Zn,
Си, Mg, As, Р, S, Ni, Cr, Ca, Na; не более 0,003 Si, Al; не более
0,005 Fe.
Содержание примесей значительно ниже предусмотренного
техническими условиями на парамолибдат аммония первого
сорта, %, не более1:
Полуторные окислы R2O3................ 0,03
В том числе Fe2O3 ................... 0,01
Никель................................. 0,005
Марганец.............................. 0,01
Мышьяк ............................... 0,005
Фосфор................................. 0,002
Сера ................................. 0,05
Остаток от гидрохлорирования.......... 0,15
В том числе:
двуокись кремния .............. 0,03
щелочноземельные металлы (СаО
+ MgO) .......................... 0,008
щелочные металлы в пересчете на NaCl 0,1
В табл. 16 приведено распределение некоторых примесей
между продуктами по стадиям технологии, которое было просле-
жено с использованием метода радиоактивных индикаторов
[97; 2, с. 91].
1 Содержание примесей отнесено к содержанию молибдена
69
о
ТАБЛИЦА 16
РАСПРЕДЕЛЕНИЕ ПРИМЕСЕЙ В ПРОЦЕССЕ ПРОИЗВОДСТВА ПАРАМОЛИБДАТА АММОНИЯ ПО СХЕМЕ,
ВКЛЮЧАЮЩЕЙ ОСАЖДЕНИЕ ТЕТРАМОЛИБДАТА АММОНИЯ [97]
Продукт • Содержание примеси, %
Zn Sn Sb* Fe ‘ Co p As s Pb Cu
Исходный аммиачный раствор (NHJ2 МоО4 100 100 100 100 100 100 100 100 100 100
Раствор^после очист- ки сернистым ам- монием 86 16—41 91—95 2—4 40—70 88—95 95—97 95 0,1- 0,3—0,5 *
Маточный раствор по- сле осаждения те- тр амо л ибдата ам- мония 85 12—36 80—91 2—4 35—70 77—94 86—93 93 0,85 *
Осажденный дигид- рат тетрамолибда- та аммония . . . 0,004— 0,5—2,6 0,3—2,0 0,2—0,4 2—8 0,1—10 0,14—1,6 0,82
Маточный раствор после перекристал-* лизации тетрамо- либдата 0,07 0,004— 0,2-1,7 0,2—0,9 0,1—1,0 1,4-1,6 0,4—5,0 0,06—1,0 0,5-0,6 •
Парамолибдат аммо- ния ........ 0,005 0,0016— 0,2—0,9 0,08—0,3 0,04— 0,12— 0,45— 0,04— 0,2—0,3
• 0,002 0,08 0,7 0,7 0,14
* Выделение примесей РЬ и Си осаждением их сульфидов столь полное, что на последующих стадиях они не обнаружены.
1
При очистке растворов сернистым аммонием наиболее полно
удаляются примеси РЬ, Си и Fe, тогда как Zn, Sn, Sb, Со оса-
ждаются лишь частично. Весьма существенная очистка от этих
примесей происходит при осаждении тетрамолибдата аммония,
в том числе Си и Fe, если они неполно выделены в виде сульфи-
дов. В кислом маточном растворе остается подавляющая часть
примесей. Дополнительная очистка происходит при перекристал-
лизации "гетрамолибдата с получением парамолибдата аммония.
Растворы молибдата аммония можно очистить от примесей
фосфора, мышьяка и отчасти вольфрама соосаждением с гидро-
окисью железа. С этой целью в исходный раствор молибдата
аммония добавляют нитрат железа [~2,5 г Fe (NO3)3 на 1 кг
(NH4)2MoO4 при содержании .<^0,01 % As] и эквивалентное коли-
чество NH4OH. После нагревания при 60° С и отстаивания осадка
содержание As и Р в растворе снижается в 10 раз. С гидроокисью
железа соосаждается до 50% W (при исходном его содержании
в молибдате аммония 0,16%) [2, с. 91].
12. ИЗВЛЕЧЕНИЕ МОЛИБДЕНА ИЗ ХВОСТОВ
ПОСЛЕ ВЫЩЕЛАЧИВАНИЯ ОГАРКОВ
Хвосты выщелачивания в зависимости от состава концентрата
и способа обжига содержат от 5 до 30% Мо (чаще 15—20%).
Он находится в составе молибдатов кальция и железа, дву-
окиси молибдена, неокислявшегося молибденита. Кроме того,
часть молибдена (иногда 1/3—1/4 часть от его содержания) при-
сутствует в виде ионов МоО|“\ сорбированных на гидроокиси
железа и трудно отмываемых водой. Другие компоненты хвостов:
окислы и гидроокиси железа, кремнезем, иногда вольфрам в форме
шеелита или вольфрамита.
Для извлечения молибдена из хвостов выщелачивания приме-
няют щелочные способы вскцытия (спекание с содой или выщела-
чивание растворами соды) и разложение хвостов соляной кис-
лотой.
Щелочные способы. При спекании хвостов с содой, проводимом
в окислительных условиях при температуре 700—750° С, все
молибденсодержащие соединения реагируют с содой с образова-
нием молибдата натрия:
СаМоО4 + Na2CO;
з 7^ Na2MoO4 4- СаСО3,
(41).
2FeMoO4 + 2Na2CO3 + 1 /2 О2 -»2Na2MoO4 + Fe2O3 + 2CO2) (42)
MoO2 + Na2CO3 +1/20
2 Na2MoO4 + 'CO2,
(43)
2MoS2 + 6Na2CO3 + 9O2 -> 2Na2MoO4 + 4Na2SO4 + 6CO2. (44)
•*
Затем продукт обжига выщелачивают водой.
71
Вместо спекания с содой может быть успешно” применена
обработка хвостов растворами соды. Для полного разложения
молибдатов при температурах 80—100° С требуется небольшой
избыток соды (~25—30% по отношению к теоретически необхо-
димому по реакции разложения наиболее прочного из них СаМоО4).
Это вытекает из значений констант равновесия реакций взаимо-
действия различных молибдатов с растворами соды [53, 54] *.
При значительном содержании в хвостах MoS2 и МоО2 обработке
растворами соды должен предшествовать окислительный обжиг
с целью окисления этих соединений до МоО3.
В работе [88] хвосты от выщелачивания огарков растворами
соды обрабатывали в автоклаве при температуре 200° С (давле-
ние ~15 ат) в течение 3—4 ч. Расход соды при выщелачивании
составлял 250% от теоретически необходимого. Извлечение мо-
либдена в раствор составляло 97—99%. Эти параметры процесса
и высокий расход соды недостаточно обоснованы. Несомненно,
достаточную скорость и полноту вскрытия хвостов можно обеспе-
чить при более низкой температуре в автоклаве (140—150° С)
и расходе 120—130% от теоретически необходимого.
При содовых способах вскрытия сложен переход от получаемых
растворов молибдата натрия к растворам молибдата аммония.
Обычно такой переход осуществляется через осаждение ферри-
молибдата хлорным железом при оптимальном pH = 3,5**. Под-
держание оптимального pH при осаждении затруднительно.
Поэтому осадки содержат переменное количество Fe2O3 и МоО3
и не отвечают по составу нормальному ферримолибдиту
Fe2 (МоО4)3-пН2О. При значении pH выше 3,7 осадки содержат
избыточную гидроокись железа, при более низком значении —
кислые молибдаты и молибденовую кислоту (см. также стр. 149).
Вместе с молибдатом железа осаждаются фосфорнокислые и
мышьяковокислые соли железа [55].
Отфильтрованные желто-коричневые осадки ферримолибдита
выщелачивают аммиачной водой. При этом часть выщелачиваемого
молибдена остается в хвостах вследствие адсорбционных свойств
объемистых осадков гидроокиси железа. Более полное извлечение
молибдена в раствор обеспечивается предварительным обжигом
осадков ферримолибдата при 800° С, так как при этом ферримо-
либдат разлагается на МоО3 и Fe2O3.
Необходимое для осаждения ферримолибдата хлорное железо
получается растворением осадков после аммиачного выщелачива-
ния, содержащих Fe (ОН)3 или Fe2O3 в соляной кислоте (см.
рис. 25).
* Более детально о взаимодействии молибдатов с растворами соды см.
в гл. V.
** О некоторых других путях получения парамолибдата аммония из рас-
творов Na2MoO4 см. в гл. VII.
72
Описанная выше технология переработки хвостов имеет недо-
статки:
1. При значительном содержании в хвостах минералов воль-
фрама (шеелит, вольфрамит) они разлагаются полно при спека-
Обо^менный концентрат
Рис. 25. Схема переработки молибденовых огарков, включающих спекание с содой
хвостов после выщелачивания
нии с содой и в большой степени при обработке растворами соды.
Во всех последующих операциях вольфрам сопутствует молиб-
дену и загрязняет конечный продукт — парамолибдат аммония.
2. Получаемые аммиачные растворы содержат повышенные
количества ионов натрия вследствие трудности отмывки осадков
73
Концентрат
Сушка и обжиг
Огарок
М/3।
Выщелачивание
Рис. 26. Схема переработки молибденовых огарков, включающая кислотное раз-
ложение хвостов выщелачивания
ферримолибдата от хлористого натрия. Это затрудняет получение
парамолибдата аммония, кондиционного по примеси натрия, в осо-
бенности при использовании метода выпарки и кристаллизации.
Кислотный способ. Отмеченные выше недостатки вскрытия
хвостов спеканием с содой (или обработки растворами соды) вы- •
звали поиски других путей их переработки. В результате был
разработан [60, 89] способ разложения хвостов соляной кислотой,
используемый в заводской практике (рис. 26).
При обработке хвостов 30%-ной соляной кислотой на холоду
(т : ж — 1 : 1) все молибдаты растворяются, тогда как минералы
вольфрама (вольфрамит, шеелит) в этих условиях не разлагаются.
Молибден находится в солянокислом растворе в форме МоО2С12
и оксихлормолибденовых комплексных кислот типа Н2 (МоО2С14)
или Н(МоО2С13) [90, 91].
Таким образом, растворение, например, молибдата кальция
протекает по приближенной реакции
СаМоО4 + 6НС1 -» Н2 [МоО2С14] + СаС12 + 2Н2О. (45)
Двуокись молибдена и молибденит не растворяются в соляной
кислоте и остаются в нерастворившейся части хвостов вместе
с SiO2 и минералами вольфрама.
При нейтрализации кислой пульпы, полученной в результате
обработки хвостов соляной кислотой, аммиачной водой до pH =
= 2,7ч-3,4, молибден полно (—98,90%) осаждается в составе
ферримолибдата в смеси с полимолибдатами аммония различного
состава. При этом весь кальций и часть избыточного железа, медь,
магний, примеси щелочных металлов остаются в кислом растворе.
После фильтрации и промывания 1%-ной НС1 (контролируется
отмывка от ионов кальция) осадок обжигают при 550—600° С
(для окисления МоО2 и MoS2 и разложения ферримолибдата)
и затем выщелачивают аммиачной водой. Степень выщелачива-
ния повышается при добавке карбоната аммония (см. стр. 55).
Содержание молибдена в отвалах в этом случае составляет 0,5—
0,8%, а потери молибдена с отвальными хвостами не превы-
шают 1 % от исходного содержания молибдена в перерабатываемом
концентрате. Общее извлечение молибдена в товарный продукт —
парамолибдат аммония достигает 94,5—95%.
ПРОИЗВОДСТВО ТРЕХОКИСИ МОЛИБДЕНА
МЕТОДОМ возгонки1
Относительно низкая температура кипения трехокиси молиб-
дена (1155° С) послужила основой для разработки промышленных
процессов получения чистой трехокиси из огарков. Метод приме-
1 Мы использовали общепринятое название «метод возгонки», однако в дей-
ствительности речь идет об испарении трехокиси молибдена из расплава.
75
няют на заводах США [100—102], Австрии [99], Польской
Народной Республики [103, 104], Его преимущества состоят
в возможности получения чистой трехокиси молибдена по корот-
кой технологической схеме, включающей возгонку и улавливание
возгонов.
В СССР физико-химические и технологические исследования
процесса возгонки проведены в Московском институте стали и
^плавов автором, Беляевской и др. [106, 114].
13. ФИЗИКО-ХИМИЧЕСКИЕ ОСНОВЫ ПРОЦЕССА ВОЗГОНКИ
ТРЕХОКИСИ МОЛИБДЕНА ИЗ ОГАРКОВ
Технологические показатели возгонки трехокиси молибдена
из огарков (производительность печи для возгонки и чистота
возогнанной трехокиси молибдена) определяются: а) давлением
пара и скоростью испарения МоО3 при температуре процесса;
б) характером взаимодействия трехокиси молибдена с примесями,
присутствующими в огарке, и их поведением при возгонке.
Давление пара трехокиси молибдена
Давление пара над твердой и жидкой трехокисью молибдена
впервые определено Файзером[105] в интервале температур 610—
1155° С. В этой работе скорость испарения трехокиси молибдена
из лодочки с поверхностью 100 мм2 сравнивалась со скоростью
испарения при температуре кипения 1155° С. Она была установ-
лена по остановке на кривой нагревания.
Давление пара рассчитывали по формуле
т
Р = Р°-^’
где р0 = 760 мм рт. ст.;
т и т0 — молярные количества испарившегося МоО3 при
температуре опыта и температуре кипения соот-
ветственно.
Такая методика была принята в связи с отсутствием данных
о молярном составе пара трехокиси молибдена, которая, как пред-
положил автор, находится в парах в виде полимеризованных мо-
лекул. Это было подтверждено в работе [106], в которой опреде-
лялось давление пара над жидкой МоО3 при 900—1100° С двумя
методами: по температуре кипения при заданном давлении (метод
точек кипения) и методом переноса (метод струи).
Первый метод не требует знания плотности пара или молеку-
лярной массы вещества в газовой фазе и позволяет непосредственно
определить истинное давление пара.
При расчете давления пара по второму методу необходимы дан-
ные о молекулярной-массе пара. Следовательно, сопоставление зна-
чений давления пара, полученных двумя методами, позволяет сде-
Л1ТЬ заключение о степени полимеризации молекул в газовой фазе.
Авторы показали, что степень полимеризации МоО3 в интер-
вале температур 810—1000° С изменяется от 3,7 до 3, при темпера-
турах 950—1000° С в парах находятся молекулы (МоО3)3 или
Мо3О9. Позже это подтвердили Берковиц с сотрудниками [107],
установившие, что теплота разложения тримера на мономер:
(МоО3)3 газ ЗМоО3 газ;
ЛЯ = 220,9 ± 9,5 ккал/моль тримера.
Значения давления пара над жидкой трехокисью молибдена,
полученные по методу точек кипения, близки к данным Файзера
(табл. 17) и хорошо описываются
уравнением [106]
7685
JgP(MoO3)3 7' '
ИЛИ
. __ 7685
1§/?(МоОз)3~_ т
Отсюда теплота испарения
АЯ-4,576-7685 -
— 35 200 кал!моль,
(47)
ТАБЛИЦА 17
ДАВЛЕНИЕ ПАРА НАД
ЖИДКОЙ ТРЕХОКИСЬЮ МОЛИБДЕНА
Темпе- ратура , °C Давление пара, мм pm. cm.,- по данным
[106] [Ю5]
800 12,6 10,1
900 51,3 53,9
1000 167,5 179,8
1100 457,0 476,2
1155 760,0 760,0
энтропия испарения
АЗ — 4,576-5,37 — 24,60 кал!моль.
Давление пара над твердой трехокисью молибдена изучалось
в работах [107—113 ]. Значительные расхождения данных обуслов-
лены различной точностью примененных методов (метод потока,
эффузионный метод Кнудсена) и различиями в оценке степени
полимеризации (табл. 18).
Берковиц и др. [107] определяли давление пара над твердой
МоО3 методом Кнудсена с масс-спектрометрическим анализом га-
зовой фазы. Ими установлено, что в газовой фазе находятся поли-
меры трехокиси молибдена со степенью полимеризации 3, 4 и 5.
Как видно из табл. 19, среднее число молекул в полимере из-
меняется от 3,24 при 600° С до 3,47 при 710° С. При температу-
рах выше 800° С пары содержат в основном полимеры (МоО3)3.
Эффузионный метод Кнудсена применяли также Блекборн
[108], Хорбе [109] и Гульбранзен [111] для определения давле-
ния пара над твердой трехокисью молибдена. Все они использо-
вали среднюю молекулярную массу в парах, рассчитанную на
основе данных работы [107] (табл. 19). Ариа, ГЦукарев и Глуш-
кова [112] и Аккерманн с сотрудниками [ПО] определяли давле-
ние пара Над твердой МоО3 методом потока. В табл. 18 сопостав-
77
ТАБЛИЦА 18
ТЕПЛОТА И ЭНТРОПИЯ СУБЛИМАЦИИ ТРЕХОКИСИ МОЛИБДЕНА
ПО ДАННЫМ РАЗЛИЧНЫХ АВТОРОВ
Интервал температур, °C АН, ккал/моль пара AS, э. е./молъ пара Метод определения Литератур- ный источник
635—675 62,5 42,8 Эффузионный (Кнудсе- [113]
на)
668—714 67,0 * 53,3 * Метод потока 112
537—727 85,4 71,7 Эффузионный [107]
535—685 78,8 66,6 » 108
707—787 87,8 73,0 Метод потока [ПО]
600—710 88,6 88,7 Эффузионный [109]
600—700 75,4 62,3 » [111]
* Данные скорректированы с учетом полимеризации молекул в газовой фазе.
ТАБЛИЦА 19
ДАВЛЕНИЕ ПАРА НАД ТВЕРДОЙ ТРЕХОКИСЬЮ МОЛИБДЕНА
Температура, °C Среднее число молекул п в поли- мере [107] Давление пара, мм рт. ст., по данным
[Ю9] [111]
600 3,238 1,67-10-3 4,62-10-3
610 3,253 3,20-10-8 —— —
620 3,269 4,81-10-3
625 3,277 1,12-Ю-2
630 3,286 7,16-10“3
640 3,303 1,28-10“2 ———
645 3,323 — 5,18-10-2
650 3,323 2,49-IO"2 —
660 3,343 4,21-10-2 —
670 3,364 6,86-10-2 —
675 3,375 8,22-10-2 1,54-IO’1
690 3,410 2,13-10-1 —
700 3,438 3,14-Ю-1 3,56-Ю-1
710 3,469 5,18-10-1 -
лены значения теплоты и энтропии сублимации трехокиси мо-
либдена, полученные различными авторами. Используя эти дан-
ные, можно рассчитать давление пара по уравнению
4,576 lgp(Moo3)„ =
—Н AS (ат).
(48)
На рис. 27 сопоставлены данные различных авторов по давле-
нию пара над твердой и жидкой трехокисью молибдена.
78
Более близкие значения давления над твердой трехокисью
молибдена получены в работах [107—109 и 111], однако и они
значительно отличаются. Данные Хбрбе [109] и Гульбранзена
[111] приведены в табл. 19.
Если принять ДЯсубл = 75,4 ккал [111], то разность между
теплотой сублимации и теплотой испарения (ДТ/ = 35,2 ккал)
Рис. 27. Зависимость логарифма давления пара
от обратной температуры над твердой и жидкой
трехокисью молибдена по данным различных
авторов:
/ — Файзер; 2 — Уено; 3 — Ариа, Щукарев (скорректи-
ровано); 4 — Беркович и др.; 5 — Гульбранзен и др.;
6 — Аккерманн и др.; 7 — Блекборн и др.; 8 — Зелик-
ман и др.; 9 — Хбрбе и др.
составит 40,2 ккал на 1 моль (МоО3)п. При п = 3 на каждый моль
МоО3 приходится 12,4 ккал. Теплота плавления МоО3, равная
12,6 ккал!моль [117], близка к этой величине. Это дополнительно
подтверждает преимущественное присутствие тримера в газовой^
фазе.
Хбрбе отмечает, что образование полимерных молекул можно
объяснить, исходя из особенностей структуры трехокиси молиб-
дена, которая показана на рис. 92 (см. стр. 259). Она состоит из
слоев октаэдров. Полимеризованным молекулам легче отделиться
от кристалла (в виде сочлененных октаэдров), чем одиночной
молекуле МоО3 [109].
79
Скорость испарения трехокиси молибдена
Скорость испарения МоО3 в большой мере зависит от скорости
потока газа над поверхностью расплава, т. е. от скорости отвода
тяжелых полимеризованных молекул от поверхности испарения.
В табл. 20 приведены скорости испарения МоО3 при различ-
ных температурах, полученные Файзером [105] при испарении
из плоской платиновой лодочки с поверхностью, равной 1 см2.
Скорость прососа воздуха была малой, порядка нескольких литров
в час (линейные скорости ~0,15—0.3 см!сек).
ТАБЛИЦА 20
СКОРОСТЬ ИСПАРЕНИЯ ТРЕХОКИСИ МОЛИБДЕНА, ПО ДАННЫМ
ФАЙЗЕРА [105]
Температура, °К -у-10» Скорость испарения г, г/(см2-сек) Л#"*1 lg i „ Скорость испарения, кг/(м2-ч)
1073 0,93 0,64 —6,1938 2,3
1123 0,89 1,48 —5,8297 5,33
1173 0,85 3,43 —5,4647 12,35
1223 0,817 5,66 —5,2472 20,4
1273 0,785 П,4 —4,9431 41,0
1323 0,755 18,3 —4,7375 65,9
1373 0,928 30,2 —4,5200 109
1423 0,702 58,3 —4,2343 210
Данные Файзера в координатах lg i — -у- хорошо уклады-
ваются на прямой (рис. 28). Энергия активации процесса испаре-
ния Е — 39 200 кал, что на 4000 кал больше теплоты испарения
(35 200 кал). Наблюдаемое различие говорит о существенной
роли диффузии полимеризованных молекул трехокиси молибдена
в процессе испарения. Даже при малых скоростях потока газа-
носителя скорости испарения чистой трехокиси молибдена, полу-
ченные Файзером, достаточно высоки и возрастают от 12,3 кг/(м2-ч)
при 900° С до 109 кг/(м2-ч) при 1100° С (см. табл. 20).
В работе [114] кинетику испарения изучали гравиметрическим
методом. Испарение проводили из кварцевого стаканчика высотой
8 мм и диаметром 14 мм (поверхность испарения 1,5 см2), запол-
ненного до верхнего края предварительно переплавленной и из-
мельченной трехокисью молибдена. Кварцевый стаканчик подве-
ршивали к коромыслу аналитических весов в кварцевой трубе
диаметром 60 мм. Опыты проводили при скорости потока воздуха
2,3 см!сек.
Как видно из рис. 29, кинетические кривые имеют нелинейный
характер, причем в начальный период темп убывания скорости
испарения больше, чем в последующие. Уменьшение скорости испа-
рения во времени, по всей вероятности, объясняется понижением
уровня жидкости, что приводит к падению фактической скорости
80
потока газа над ее поверхностью и увеличению толщины диффу-
зионного слоя, через который диффундируют молекулы (МоО3)3.
Зависимость скорости испарения от количества испарившейся
трехокиси хорошо описывается эмпирическим уравнением
ат + b ’
(49)
где i — скорость испарения, г/(см2-сек);
т — количество испарившейся МоО3, г!см2\
а и b — постоянные.
Экспериментальные значения скорости испарения для различ-
ных участков кинетических кривых (ix при значениях от 0,5 до
Рис. 28. Зависимость логарифма
скорости испарения от обратной
температуры, по данным Фай-
зера [105]
Время г, сек
Рис. 29. Зависимость количества
испарившейся трехокиси молиб-
дена с единицы поверхности от
времени при различных темпе-
ратурах [114]
0,7 г!см\ 12 при т от 1,0 до 1,17 г/сж2), а также экстраполирован-
ные значения /э для начального момента (т % 0), когда скорость
испарения максимальная, приведены в табл. 21. Реальные значе-
ния скорости испарения и f2), полученные в работе [114],
того же порядка, что определены Файзером (ср. табл. 20 и 21).
При одинаковых скоростях потока газа скорость испарения
трехокиси молибдена понижается с увеличением поверхности
испарения. Это является следствием зависимости скорости испаре-
ния от разности концентрации паров МоО3 на фазовой границе
и в газовом потоке:
D(nF~ nL>
L
(50)
где D — коэффициент диффузии;
nF— концентрация паров на фазовой границе;
nL — концентрация паров в ядре потока;
L — расстояние от фазовой границы, на котором концен-
трация паров равна nL.
6 А. Н. Зеликман
81
ТАБЛИЦА 21
ЭКСПЕРИМЕНТАЛЬНЫЕ И ЭКСТРАПОЛИРОВАННЫЕ ЗНАЧЕНИЯ
СКОРОСТИ ИСПАРЕНИЯ МрО3 ПРИ ТЕМПЕРАТУРАХ 830 —1000° С,
ПО ДАННЫМ [114]
Температура, °C Экспериментальные величины, г/(см2-сек)-10~4: Экстраполированная величина, , г/(см2-сек)-МГ*1
Ц Ц
835 1,165 0,803 3,18
865 2,19 1,42 5,75
888 3,44 2,32 8,85
930 6,3 4,51 17,75
1000 * 19,87 13,12 53,55
* Значения при 1000° С получены графической экстраполяцией зависимости lg i-—
При неизменной поверхности испарения с увеличением ско-
рости потока газа возрастает скорость испарения, так как увели-
чивается разность концентраций nF — nL и уменьшается L.
Так, например, при 830° С из чашечки с поверхностью 3,4 см2
испаряется за 60 мин при скорости потока воздуха 23 л/ч 0,3 г!см2,
а при потоке 58 л/ч 0,48 г/см2.
Кинетические кривые испарения МоО3 из молибденовых огар-
ков подобны приведенным выше для испарения чистой трехокиси
молибдена.
Однако абсолютные скорости ниже. Это обусловлено присут-
ствием в огарках окислов ряда металлов, растворяющихся
в жидкой МоО3 и снижающих давление пара трехокиси.
На рис. 30 приведены кривые изменения во времени скорости
испарения МоО3 из молибденового огарка состава, %: 57,3 Мо;
0,1 СаО; 2,47 Fe; 0,97 Си; 1,23 5общ; 0,51 8сульфидн; 6,5 SiO2.
Огарок засыпали на постель из чистого кварцевого песка. Испаре-
ние проводили из кварцевой чашечки с поверхностью 3,4 см2 при
линейной скорости потока воздуха 2,3 см/сек и непрерывной ре-
гистрации убыди в массе г.
Для сопоставления на график нанесены данные ю скорости
испарения чистой трехокиси молибдена в тех же условиях.
Как и в случае чистой трехокиси молибдена, изменение ско-
рости испарения во времени хорошо описывается уравнением
г “ ’ что позволяет экстраполяцией (при т 0) опреде-
лить максимальную скорость испарения.
1 Данные заимствованы из дипломной работы И. Нарамовского, выполнен-
ной под руководством автора и Л. В. Беляевской.
82
Табл. 22 дает представление о реальных скоростях испарения
трехокиси молибдена из огарков при скорости потока воздуха
2,3 см!сек. При температуре 1000° С они имеют величину порядка
от 10 до 20 кг/(ж2-ч). Скорость испарения можно увеличить, при-
менив большие скорости потока воздуха или повысив температуру
до 1050—И 00° С.
Дитц и Кластерманн [47 ] исследовали влияние ряда факторов
на возгонку трехокиси молибдена из огарков на лабораторной
Рис. 30. Зависимость скорости испарения МоО3 из огарка
от степени испарения:
1 — огарок; 2 — чистая трехокись молибдена
установке, моделирующей возгоночную печь (см. рис. 34). Воз-
гонку проводили из кварцевого тигля диаметром 65 мм, высотой
250 мм, установленного под углом 45° и вращающегося с регули-
руемой скоростью. Скорость возгонки изучали в зависимости от
температуры, а также скорости и температуры подаваемого в ти-
гель газа-носителя (воздуха).
ТАБЛИЦА 22
ЭКСПЕРИМЕНТАЛЬНЫЕ И ЭКСТРАПОЛИРОВАННЫЕ ЗНАЧЕНИЯ
СКОРОСТИ ИСПАРЕНИЯ МоО3 из огарка. СКОРОСТЬ потока
ВОЗДУХА 2,3 см/сек
Температура, °C Скорость испарения, кг/(м2-ч) * Экстраполированное значение А, кг/(м2-ч)
i1 /2
820 1.0 0,51 2,6
920 6,9 5,5 8,45
1000 14,1 9,66 30,2
* — участок кривой при т от 0,59 до 0,1 г/см2’, i2 — при т от 0,19 до 0,23 г/см,2.
6*
83
Было установлено, что при подаче в пространство над распла-
вом холодного воздуха скорость возгонки выше, чем при подо-
греве воздуха до температуры сублимации. Это объясняется тем,
что холодный воздух охлаждает на 200—300° пары сублимата,
что приводит к конденсации части паров над расплавом с образо-
ванием тонкой взвеси, уносимой воздухом. Вследствие этого по-
нижается парциальное давление паров над расплавом, что вызы-
вает ускорение испарения.
Существенное значение при возгонке из огарка имеет враще-
ние тигля, так как если поверхность испарения остается спокой-
ной, образуется корка. Имеется некоторая оптимальная скорость
вращения тигля, при которой скорость возгонки достигает макси-
мума.
Авторы работы [47] сопоставляют полученные ими экспери-
ментальные данные по скорости испарения с рассчитанными по
формуле Матца [100]:
Рс
= VT -75—2---- , (51)
•^о 1 о •Ьо р — ps х '
где Wso — скорость испарения, г/ч;
Vt0 — скорость подачи газа-носителя, л!ч\
ps — плотность паров испаряемого вещества, г!л\
Ps — равновесное давление паров при температуре испа-
рения;
Р — общее давление.
Это уравнение предполагает равновесное насыщение газа-
носителя испаряемым веществом.
При расчетах максимально возможных скоростей сублимации
по этому уравнению авторы работы [47] допустили ошибку, не
учитывая полимеризации молекул МоО3 в парах и приняв значе-
ние pSo = 6,4, тогда как действительное значение с учетом содер-
жания в парах (МоО3)3 равно 19,3. Следовательно, рассчитанные
значения скорости испарения втрое выше и, вопреки выводу авто-
ров, реальные значения намного ниже расчетных при всех ско-
ростях газа-носителя. Это вполне объяснимо, так как газ-носи-
телъ не насыщен парами трехокиси молибдена.
Следует, кроме того, учитывать, что давление пара над обож-
женным концентратом ниже значений над чистой трехокисью
молибдена, использовавшихся в расчетах.
Поведение примесей при возгонке трехокиси молибдена
В продуктах обжига молибденитовых концентратов (огарках)
содержится большое число примесей — окислы Fe, Са, Mg, Al,
Si, Си, Pb, Zn в количествах от сотых долей до нескольких про-
центов.
14
В работе [114] было изучено поведение примесей при возгонке
трехокиси молибдена, в состав которой вводили окислы перечис-
ленных выше элементов в количестве 1—3%. Возгонку проводили
в струе воздуха (6 см!сек), подо-
гретого до температуры опыта.
В лодочку загружали трехокись
молибдена в смеси с кварцевым
песком (крупностью —1,0 +0,2 лш)
в отношении 1:1. Содержание
примесей в возгонах определяли
спектральным методом.
Как видно из рис. 31 и табл. 23,
примеси по-разному влияют на
степень возгонки трехокиси молиб-
дена. Примеси окислов меди,
цинка и алюминия практически
не снижают скорости и степени
возгонки МоОз при 1000° С, но
несколько ухудшают показатели
возгонки при 850° С. Примеси
кальция, магния и железа суще-
ственно снижают скорость и сте-
пень возгонки.
Различное влияние примесей
окислов Zn, Си, А1 и Са, Mg, Fe
хорошо объясняется характером
диаграмм состояния их окислов
Рис. 31. Влияние примесей на сте-
пень возгонки трехокиси молибдена
при различных температурах (коли-
чество введенной примеси указано’
в табл. 22):
1 — А12О3; 2 — СиО; 3 — РЬО; 4 —
ZnO; 5 — Fe2O3; 6 — СаО; 7 — MgO
с трехокисью молибдена (см. стр. 34—37). Окись меди образует
молибдат, разлагающийся при плавлении (>900° С). С А12Оа
трехокись молибдена соединений не образует. Молибдаты каль-
ТА БЛИЦА 23
ПОВЕДЕНИЕ ПРИМЕСЕЙ ПРИ ВОЗГОНКЕ ТРЕХОКИСИ МОЛИБДЕНА.
ВО ВСЕХ ОПЫТАХ НАВЕСКА 4 г Мо03 + ПРИМЕСЬ И 4 г SiO2.
ВРЕМЯ ВОЗГОНКИ 20 мин
Исходный материал Возогнано МоО3, %, при Содержание примеси в возгонах, %, при
850° С 1000° С 850° С 1000° С
Чистая МоОо 78,6 99,7
МоО3- 1-1,0% РЬО 53,2 97,4 0,0015 0,016
МоО3- 1-1,25% ZnO 48,8 95,9 <0,008 <0,008
МоО3- -3% А12О3 73,8 98,2 <0,00025 <0,00025
МоО3- -4,3% Fe2O3 36,0 80,2 <0,001 <0,001
Мо03- -0,38% СиО 68,2 98,3 0,0002 0,0005
Мо03- F3,3%MgO 24,8 64,0 <0,00025 <0,00025
МоО3- 1-2,8% СаО 16,2 67,9 Не опр. Не опр.
85
ция, магния, железа, свинца и, вероятно, цинка при 1000° С
не разлагаются. Расчет показывает, что при связывании МоО3
в MgMoO4, СаМоО4 и Fe2 (МоО4)3 теоретически возможная сте-
пень возгонки МоО3 составляет для смесей с 3,3% MgO88%,
с 2,8% Сад 92%, с 4,3% Fe2O3 88%.
В проведенных опытах при 1000° С (когда за время опыта сво-
бодная трехокись полностью возгоняется) степень возгонки трех-
окиси молибдена на 10—25% ниже расчетной^ что объясняется
уменьшением скорости возгонки вследствие снижения упругости
пара МоО3 над расплавом, содержащим растворенные молибдаты.
РЬМоО4 также относится к числу молибдатов, плавящихся без
разложения; влияние примеси РЬО на степень возгонки вследствие
относительно малого ее количества (1% РЬО связывает ~0,5%
МоО3 в РЬМоО4) выявляется в. малой степени. л
Для всех изученных примесей, кроме свинца, отмечается очень
незначительная возгонка примеси даже при высоких степенях
возгонки трехокиси молибдена (до 97%). Содержание в возгонах
примесей Си, Mg и А1 составляет 0,0002—0,0005%, содержа-
ние Fe и Zn ниже чувствительности анализа, т. е. <0,001 % Fe,
<0,008% Zn. Содержание всех этих примесей в возгонах практи-
чески не меняется с изменением температуры в пределах 850—
1000° С и степени возгонки МоО3.
С увеличением температуры возгонки резко возрастает содер-
жание примеси свинца в возгонах от ~0,002% при 850—900° С
до 0,01% при 950° С и 0,015% при 1000° С, что вызвано значи-
тельным ростом давления пара РЬМоО4.
По данным работы [114], давление пара РЬМоО4 составляет
при 1016° С 0,08 мм, при 1036° С 0,13 мм, при 1060° С
0,23 мм рт. ст.
Влияние кварцевого песка на скорость возгонки
Известно, что ^в промышленной практике при возгонке трех-
окиси молибдена огарки смешивают с кварцевым песком.
Исследования показали [114], что добавление кварцевого
песка снижает среднюю скорость возгонки МоО3 при загрузке
огарка на кварцевую постель и при предварительном смешивании
огарка с песком. Однако без кварцевого песка остаток от возгонки
‘(содержащий обычно 20—30% невозогнанной трехокиси), засты-
вая, образует прочный монолит, что затрудняет разгрузку и по-
следующую переработку остатка. Из этого следует, что добавле-
ние песка необходимо для получения рыхлого, легко выгружае-
мого остатка от возгонки. Для обеспечения более высокой скорости
возгонки огарок рекомендуется загружать на кварцевую постель,
не перемешивая его с песком.
Поскольку кварц химически не взаимодействует с расплавлен-
ной трехокисью, влияние кварца на скорость возгонки в большой
мере определяется смачиваемостью кварца расплавом трехокиси.
«6
Качественная оценка смачиваемости кварца чистой расплав-
ленной трехокисью молибдена, а также трехокисью с примесями
2% СаО, 2% СпО и 3% Fe2O3 была проведена по растеканию
лежащей капли [114].
Чистая трехокись молибдена хорошо смачивает кварц при тем-
пературах 800—1000° С. В присутствии примесей СаО, СиО,
Fe2O3 смачивание ухудшается, капля растекается медленно,
наблюдается большой краевой угол смачивания. Соответственно
этому огарки с высоким содержанием примесей плохо смачивают
кварц.
Понижение скорости возгонки трехокиси молибдена в присут-
ствии кварцевого песка объясняется тем, что лишь в первоначаль-
ный момент испарение происходит с поверхности загрузки.
По истечении некоторого времени верхний слой обедняется трех-
окисью молибдена, так как скорость испарения' опережает ско-
рость капиллярного поднятия жидкой МоО3. Далее испарение
идет с транспортировкой паров трехокиси молибдена через пары
(капилляры) в слое кварца, что замедляет скорость испарения.
14. ТЕХНОЛОГИЯ ВОЗГОНКИ ТРЕХОКИСИ МОЛИБДЕНА
На предприятиях фирмы Клаймакс Молибденум в США [101]
возгонку трехокиси молибдена из молибденовых огарков проводят
в электрических печах с вра-
щающимся кольцевым подом,
покрытым слоем кварцевого
песка (рис. 32, 33) *. Для на-
грева используют силитовые
нагреватели (стержни из кар-
бида кремния), расположенные
радиально над подом и обеспе-
чивающие температуру внутри
•печи 1000—И 50° С. Техничес-
кую трехокись молибдена (ога-
рок) непрерывно загружают
на подину. Расплавленная МоО3
пропитывает слой песка, что
создает постоянную постель
пода. Над подом с определенной
скоростью просасывается воз-
дух, уносящий возогнанную
трехокись молибдена. Газы
Рис. 32. Схема печи с вращающимся
подом для возгонки трехокиси молиб-
дена:
1 — вращающийся под; 2 — силитовые
нагреватели; 3— воздух, подаваемый в печь;
4 — трехокись молибдена — в кулера и
рукавные фильтры
проходят через ряд отверстий, расположенных над подом,
по трубам в общий коллектор. Далее они проходят через систему
кулеров (где оседает часть МоО3) и затем поступают в мешочные
фильтры, где полностью улавливаются возгоны.
* N о у S. М. S m i t h D. C. US Pat., 2958588, 1960.
Печное пространство разделяется перегородками на питающую
и нагревательную зону (зону возгонки). В питающую зону непре-
рывно подается смесь огарка с песком. Поскольку в этой зоне воз-
можно пыление, что может снизить чистоту продукта, высасыва-
ющие трубы питающей зоны связаны с отдельным эксгаустером
и пылеуловителем.
Рис. 33. Внешний вид печи для возгонки с вращающимся
подом и под печи с силитовым нагревателем
Загруженный огарок проходит под нагревательными элемен-
тами один раз. За один оборот пода возгоняется примерно 60%
трехокиси молибдена. Оставшаяся масса удаляется с пода шне-
ковым скребком и перерабатывается гидрометаллургическим спо-
собом или поступает на выплавку ферромолибдена.
Производительность печи 3,75 т чистой МоО3 в сутки.
Для получения однородного продукта система отрегулирована
так, чтобы обеспечивались одинаковые условия во всех участках
печи (разрежение, скорость потока воздуха, температура расплава).
Исходным сырьем служат высокосортные молибденитовые кон-
центраты следующего состава, %: 56,0 Мо, 0,16 Си, 0,06 Са,
88
0,28 Диез, 37,6 S, 0,06 Zn, 0,08 Mg, 0,03 P, 0,04 Pb, 0,38 Fe,
4,5 SiO2, следы As. Огарки, полученные в результате обжига
концентратов в многоподовой печи, содержат 80—90% МоО3.
Возогнанная из огарков трехокись молибдена имеет высокую
чистоту (99,975% МоО3). Она весьма дисперсна (масса утряски
около 0,5 г/см3). Такие лег-
кие объемистые порошки
неудобны для транспорти-
ровки и использования.
Рис. 34. Схема и^внешний вид установки для возгонки
трехокиси молибдена на заводе в Австрии:
1 — кварцевый тигель; 2 — кожух; 3 — электронагрева-
тель; 4 — подача сжатого воздуха; 5 — вытяжной зонт;
6 — вентилятор; 7 — мешочный фильтр
Перед упаковкой трехокись увлажняют дистиллированной во-
дой, уплотняют и сушат. В результате насыпная масса порошка
увеличивается до 3,5 г!см3.
Печи описанной выше конструкции применяют также на заво-
дах фирмы Шаттук Хемикал в США. Здесь возгонку ведут при
более высоких температурах (1120—1260° С), что снижает степень
чистоты продукта. Содержание МоО3 в нем 99,5% или несколько
выше [102].
В Австрии на заводе Планзее масштабы производства чистой
трехокиси молибдена значительно меньше. Возгонку осуще.
89
ю оз <м о (М
"S СО 1 о ь- со
G0 с о о о с с о о о о о о
Чр 0х 00 ОЗ СО Ь-
Ьй О -Н о о сч о
Й тЧ £ о о о с о о
< И о о о о о о
Н и 03 К о и •
о
и со о Ю Г- 00 ОЗ Г- •
о 00 СЧ с”*)
я Рч О о о о о о
о =s о о о о о о
X си о
ГТ ф
5 S к
Р. ю ь- со со £"•4^
00 к > "Ч СЧ СО о о о
си о о о о о о
р. К о о о о о о
И
Й 03
О
Л СО
Р. Ct со ь- ю
о си г о 1 со о
и - Й о о О 1 о о
о о о о о о
00
К
X xf Оз ОЗ ю — oi
3 СО О о о СО 1—4
из и о о о о О О
я о с о о О о
X
ш
л s
X № Я
си К я ► о « о Ю 00 ю ю
о си я© Ю хг г- ю ю
ь СО о
4—1 О 0.2
ео
О 1
о я Я
£ (X со Я <ХЯ„ О О о о о о
X си с S — о о о —• о о о — о
*си со О
к
о
[ 1
00
О о *
й сЗ О сЗ О
Й О (З’ • г—1 о
ь- со о
Чр 00 , Оз 00 -
Ш О О4* Й о О ° 0,5 1
щ ей т—
g к Е- О О • * о • - о^о • •* • о О Цч X1 ...«К « ofc©
Рч с м о § ар £ § со с75
Г), (М ю
к 03 и. О 8,09 3, 0,2 ^оГсо" 6,35 2,1 14, я си X
ГС ю >>
<3^ (X
к а. к , аК сЗ S сЗ я \о о
Е-> эД 03 о И »s S гр НН] CU я
о о я & Ct Я О £ X jjf cj Я сЗ Оч сЗ м о Q- о СКИЙ ирова 1 Zn :
к о Я СЗ >ор ул]
к
ствляют в небольших печах
периодического действия \
Обожженный молибденовый
концентрат (вероятно, в смеси
с кварцевым песком) поме-
щают в кварцевый тигель
вращающейся- электрической
печи, установленной под уг-
лом 35?. Наклонное положе-
ние позволяет увеличить по-
верхность испарения трех-
окиси молибдена. Возгонку
ведут при температуре 900—
1000° С. В тигель подается
воздух, который уносит пары
трехокиси молибдена. Над
печью установлен зонт, через
который с помощью вентиля-
тора пары МоО3 подаются
в мешочный фильтр (рис.,34).
15. ЧИСТОТА И ФИЗИЧЕСКИЕ
СВОЙСТВА ВОЗОГНАННОЙ
ТРЕХОКИСИ МОЛИБДЕНА
Чистота возогнанной трех-
окиси молибдена зависит от
состава исходных огарков,
температуры процесса и сте-
пени возгонки. Это отчетливо
видно из табл. 24, где при-
ведены результаты опытов
возгонки в укрупненно-лабо-
раторных масштабах при за-
грузке огарка на слой песка
[114].
Продукт достаточно высо-
кой чистоты, отвечающий
техническим условиям на па-
рамолибдат первого сорта,
получается при возгонке из
богатого по молибдену огарка
(57% Мо или 85,5% МоО3) при
температуре 1000—-1100° С
и степени возгонки около
1 Schwarzkopf Р. DR Pat.,
480287, 1926; 566948, 1927; О. Pat.
116561, 1930.
90
60—70%. Более бедные по молибдену огарки с повышенным со-
держанием примесей дают возгоны с содержанием ряда примесей
в сотых долях процента. В этом случае повторная сублимация
позволяет повысить чистоту продукта.
Формы частиц и физические характеристики возогнанной трех-
окиси молибдена изучали Бенесовский и Видмайер [115, 116].
Воздушной сепарацией в спиральном сепараторе авторы выде-
лили две фракции частиц — легкую и тяжелую. Химически они
идентичны, однако они отличаются насыпной массой и формой
частиц.
Тяжелая фракция имеет насыпную массу утряски 1,43 г/см&
и основной размер частиц 3,5 мкм, а легкая — насыпную массу
0,3 г/см? и размер частиц 2,5 мкм.
Электронномикроскопические исследования показали, что тя-
желая фракция содержит преимущественно частицы округлой*
формы, представляющие собой агрегаты из субмикроскопических
кристаллов, тогда как легкая фракция содержит иглообразные-
(или пластинчатые) кристаллы (рис. 35). Дифракционная картина
в обоих случаях одинакова и, следовательно, обе формы имеют
одинаковую кристаллическую решетку.
Авторы предполагают, что шарообразные частицы образуются
в результате выноса мелких капелек из расплава или из хороша
оформившихся кристаллов, поверхность которых оплавилась при
прохождении вблизи горячих зон печи.
В работе [471 показано, что форму частиц и насыпную массу
конденсата можно регулировать скоростью и температурой газа-
носителя.
При использовании холодного газа-носителя получается суб-
лимат с частицами шарообразной формы и массой утряски 1,2 г!см3,
тогда как в случае подачи подогретого воздуха конденсат состоит
из игольчатых кристаллов, а его масса утряски равна 0,19 г!см\
ПОЛУЧЕНИЕ ТРЕХОКИСИ МОЛИБДЕНА
ИЗ ОТХОДОВ ОБРАБОТКИ
И ВТОРИЧНОГО МЕТАЛЛА
В производстве молибдена накапливаются обрубки и обрезки,,
получаемые при производстве изделий, а также брак проката и
протяжки проволоки. Кроме того, заводы-потребители собирают
и направляют в переработку молибденовые детали вышедших
из строя электровакуумных приборов.
Наиболее простой путь получения трехокиси молибдена из
металлических отходов — окисление с сублимацией трехокиси,
разработанный С. А. Викторианским, Р. Б. Маргулисом и
др. [119].
На рис. 36 показана установка для возгонки, используемая
на одном из заводов в СССР.
91!
Рис. 35. Формы частиц возогнанной трехокиси
молибдена. Электронная микроскопия:
а — легкая иглообразная форма. Х6800; б — тяжелая
шарообразная форма. X1600
92
Отходы загружают в печь, в которой поддерживают темпера-
туру 900° С. Для интенсификации окисления и удаления паров
трехокиси молибдена в печь подают воздух. Вследствие экзотер-
мичности реакции окисления температура в печи поднимается
до 1100—1200° С. Парогазовая смесь проходит через газосмеси-
тель, где в результате охлаждения холодным воздухом до 90° С
происходит кристаллизация. Образовавшаяся пыль нагнетается
Рис. 36. Схема установки для переработки отходов молибдена окисле-
нием с возгонкой:
1 — печь; 2— камера смешивания с холодным воздухом; 3 — рукавный
фильтр; 4 механизм встряхивания; 5 — шибер
вентилятором в мешочный фильтр. Характер сублимата такой же,
как описан выше, его насыпная масса 0,2 г!см3. Если сублимат
смочить 7%-ным раствором хлористого калия (20 см3 на 1 кг
порошка) и тщательно перемешать, то насыпная масса возрастет
в 2—2,5 раза. Восстановление такой уплотненной трехокиси мо-
либдена водородом в две стадии (см. cip. 274) приводит к получе-
нию порошка с размерами частиц 0,5—3 мкм, который по зерни-
стости близок к порошкам, получаемым из парамолибдата аммония.
Другой путь переработки металлических отходов — растворе-
ние их в азотной кислоте или смеси азотной и серной кислот.
Смесь азотной и серной кислот (2—3 объема концентрирован-
ной HNO3, 1 объем концентрированной H2SO4 и 1—2 объема воды)
давно используют для растворения молибденовых кернов, на кото-
рые навивают вольфрамовые спирали. В результате получают
93
растворы, содержащие 300—500 г!л H2SO4, 100—300 г!л HNO3
и до 180 г!л Мо. Для выделения молибдена из таких растворов
В. Н. Парусников [120] рекомендует их нейтрализацию аммиач-
ной водой с последующей выпаркой раствора и кристаллизацией
смеси нитрата, сульфата и молибдена аммония. Прокаливая эту
смесь при 500—600° С, получают трехокись молибдена, которая
может быть очищена растворением в растворе аммиака с выделе-
нием парамолибдата аммония.
ПОЛУЧЕНИЕ ТРЕХОКИСИ МОЛИБДЕНА
ИЗ МОЛИБДАТА КАЛЬЦИЯ
Молибдат кальция — один из товарных продуктов обогати-
тельных фабрик.
Для получения трехокиси молибдена из молибдата кальция
(если этого требуют условия производства) может быть исполь-
зован аммонийно-фторидный метод, предложенный в работе [57 ] *.
Метод основан на реакции обменного разложения между мо-
либдатом кальция и фтористым аммонием в аммиачной среде:
СаМоО4 + 2NH4F -> (NH4)2 МоО4 + СаЕ>. (52)
Константа равновесия этой реакции
[МоС>2-]
А25° С = [Г -]2
равна 7410. Столь высокое ее значение определяет практическую
необратимость реакции, которая полно протекает при температу-
рах 20—25° С и стехиометрическом расходе фторида аммония.
Для создания необходимой концентрации ионов фтора в раствор
аммиака можно вводить рассчитанное количество плавиковой
кислоты. Кроме того, можно использовать выпускаемый техни-
ческий бифторид аммония.
Для обеспечения высокого извлечения молибдена в раствор
(~99%) рекомендуется осуществлять противоточное двухстадий-
ное выщелачивание.
При обработке технического молибдата кальция с содержанием
42%. Мо аммонийно-фторидным раствором при 20—25° С полу-
чали растворы с концентрацией молибдена 100—130 г! л, содержа-
щие не более 1 г! л ионов фтора. Из таких растворов методом вы-
парки был выделен кондиционный парамолибдат аммония.
Осадки фтористого кальция, получаемые в результате обмен-
ной реакции, могут применяться в металлургии (флюсы), для
обмазки сварочных электродов или быть направлены на установку
для регенерации плавиковой кислоты.
* 3 е л и к м а н А. Н., Крейн О. Е., Ракова Н. Н. «Способ полз
чения парамолибдата и паравольфрамата аммония». Авт. свид. СССР № 17929С
Бюлл. изобр., 1966 № 5.
ГЛАВА IV
ПРОИЗВОДСТВО ФЕРРОМОЛИБДЕНА
Ферромолибден — лигатурный сплав, используемый для вве-
дения добавок молибдена в хромистые, никелевые, хромоникеле-
вые, вольфрамовые и другие специальные стали с целью улуч-
шения их механических, антикоррозионных или магнитных
свойств.
Ферромолибден обычно содержит 55—65% Мо. Из диаграммы
состояния системы Fe—Мо (см. рис. 171) можно видеть, что сплавы
такого состава в основном должны содержать 8-фазу (соединение
Fe7Mo6 переменного состава) и о-фазу (твердые растворы на основе
соединения FeMo) в различном соотношении.
Сплавы, отвечающие указанному выше составу, тугоплавки.
Полное их плавление происходит при 1700—1800° С. Поэтому
выпуск жидкого сплава из печи затруднителен.
Требования, предъявляемые к составу ферромолибдена раз-
личных марок в СССР, приведены в табл. 6.
Сырьем для получения ферромолибдена служат стандартные
молибденитовые концентраты, содержащие 47—50% Мо и строго
ограниченное количество примесей мышьяка, фосфора, олова и
меди — элементов, сильно влияющих на свойства стали.
В производственной практике ферромолибден плавят из обож-
женных молибденитовых концентратов (огарков) с содержанием
0,05—0,2% S. Обжиг проводят в многоподовых печах.
Теория и практика выплавки ферромолибдена обстоятельно
освещены в монографии [1].
Наиболее распространен силикотермический спо-
соб получения ферромолибдена, разработанный в СССР В. П. Елю-
тиным [2, 3]. Ограниченное применение (в тех случаях, когда до-
пустимо повышенное содержание углерода) имеет карботер-
мический способ выплавки ферромолибдена в электродуго-
вых печах.
95
16. СИЛИКОТЕРМИЧЕСКИЙ СПОСОБ
Трехокись молибдена восстанавливается кремнием по экзо-
термической реакции
2МоО3 + 3Si = 2Мо + 3SiO2. (53)
Для этой реакции при 1300° К
А^1зоо=—299,1 ккал, AZ1300 = —252,0 ккал.
Удельный тепловой эффект реакции ^1300 = —805 ккал на
1 кг смеси стехиометрического состава г.
Реакция восстановления окиси железа кремнием 2Fe2O3 +
+ 3Si = 4Fe + 3SiO2 характеризуется удельным тепловым эф-
фектом: <71300 = —508 ккал на 1 кг исходной смеси.
Практически в качестве восстановителя применяют не крем-
ний, а ферросилиций (70—75% Si).
С целью увеличения удельного теплового эффекта реальной
шихты до значения, обеспечивающего самопроизвольное разви-
тие процесса, часть ферросилиция (7—8%) заменяют алюминием,
восстанавливающим МоО3 и Fe2O3 по реакции
МоО3 —р 2А1 = Мо —р А12О3;
А#1300 =•—240,9 ккал', 71300 ~— 1210 ккал!кг смеси;
(54)
Fe2O3 + 2А1 = 2Fe + А12О3;
ДЯ1300 =—210,4 ккал', ?1300 =— Ю00 ккал/кг смеси.
В шихту для выплавки ферромолибдена входят следующие ма-
териалы: обожженный молибденовый концентрат (огарок), бога-
тая железная гематитовая руда или железная окалина, желез-
ная стружка, флюсы (известь, плавиковый шпат) и восстанови-
тели (ферросилиций, алюминиевая крупка).
Шихту составляют из расчета получения 60%-ного ферромо-
либдена и шлака примерно следующего состава, %: 62,5 SiO2;
9,6 А12О3; 23,1 FeO; 3,0 СаО; 1,8 CaF2.
Закись железа, известь и плавиковый шпат снижают темпе-
ратуру плавления и вязкость шлака.
Ниже приведен примерный состав шихты для выплавки ферро-
молибдена [1], %:
Обожженный концентрат (85% МоО3) . . 51,5
Ферросилиций (73,6% Si) ..............18,9
Железная руда (96,0% Fe2O3) ..........15,3
Железная стружка ...................... 9,8
Алюминиевая крупка .................. 1,94
Известь.............................. 1,54
Плавиковый шпат . . г........... . . 1,02
1 Все данные для расчетов тепловых эффектов взяты из справочного руко-
водства [5].
96
Удельный тепловой эффект шихты приведенного состава равен
525 ккал!кг шихты, что обеспечивает при масштабах 3—4 т кон-
центрата в одну плавку проведение процесса внепечным способом
за счет теплоты реакций. Измельченные компоненты шихты пере-
мешивают в смесительном барабане.
Ферромолибден выплавляютв металлическом цилиндре (горне),
футерованном шамотным кирпичом. Его устанавливают в песоч-
ное основание, в котором сделано углубление для выплавляемого
сплава. Над горном устанавливают вытяжной зонт, соединенный
с пылеуловителем. Плавку ведут с верхним или нижним запалом.
При плавке с верхним запалом в горн засыпают всю навеску
шихты. Затем шихту разравнивают, на ее поверхности делают
2—3 лунки, в которые засыпают запальную смесь. Она состоит
из смеси стружки магниевого сплава (электрон), алюминиевой
крупки и селитры в соотношении (по массе) 1 : 1 : 1,5. Запальную
смесь зажигают раскаленным железным прутом. Реакция быстро
распространяется по всей массе шихты и заканчивается за 40—
70 мин. После дополнительной выдержки в течение 20 мин (для
оседания мелких корольков сплава) открывают летку, находя-
щуюся на уровне сплава, и выпускают шлак. Затем цилиндр под-
нимают краном. Блок металла в течение нескольких часов осты-
вает в песке. Затем его замачивают в ванне с проточной водой и
дробят на дробилке на куски массой до 5 кг.
При плавке с нижним запалом на дно горна засыпают 10—
15% всей шихты и поджигают ее запалом. Затем постепенно за-
гружают из бункера с помощью шнекового питателя остальную
шихту, вначале медленно, а через 5—10 мин, когда реакция рас-
пространится по всему сечению горна, быстро загружают остав-
шуюся шихту. Окончание реакции определяют по прекращению
бурления шлака.
Прямое извлечение молибдена в сплав составляет 92—93%.
В шлаке содержание молибдена не должно превышать 0,3%. При-
мерно 2—4% Мо, содержащегося в исходной шихте, уносится
с газами. Уловленную пыль возвращают на шихтовку. Извлече-
ние молибдена с учетом оборотных материалов (пыль, отходы
разделки блоков, выплески шлака) достигает 98—99%.
Нцже приведен типичный состав выплавляемого ферромолиб-
дена, %: 60,5 Мо; 0,97 Si; 0,02 С; 0,035 S; 0,018 Р; 0,23 Си; 0,028 А1.
17. КАРБОТЕРМИЧЕСКИЙ СПОСОБ
Выплавку ферромолибдена с использованием углеродистого
восстановителя применяют на некоторых зарубежных заводах
и испытывали в СССР [1, 4]. Трехокись молибдена восстанавли-
вается углеродом в две стадии:
лол_с
МоО3 + 1 /2 С------> МоО2 + 1 /2 СО2, (55)
~>800° С
Мо02 + 2С—------> Мо + 2СО. (56)
7 А. Н. Зеликман 97
Первая из них экзотермическая, вторая эндотермическая (Д//
% + 46,8 ккал) *.
После полного удаления кислорода образуется карбид молиб-
дена Мо2С.
Карботермическую выплавку ферромолибдена проводят в одно-
фазных (300—500 ква) или трехфазных (500—1500 ква) электро-
дуговых печах, используя в качестве исходного материала обож-
женные молибденовые концентраты. Футеровка стен дуговых
печей набивная доломитовая, подина — угольная.
Шихту, состоящую из молибденового огарка, восстановителя
(древесный уголь, коксик) и флюсов (СаО, А12О3у CaF2), брикети-
руют. В качестве связки используют известковую муку, желез-
ный порошок.
Для получения ферромолибдена с относительно низким со-
держанием углерода (0,1—0,2% С) плавку ведут в две стадии.
Первоначально в печи расплавляют отходы разделки блока и обо-
ротный (богатый молибденом) шлак предыдущей плавки, затем
' постепенно загружают брикеты шихты (размер брикетов — при-
мерно половина кирпича) и железную стружку. На этой, первой
стадии получают сплав с повышенным содержанием углерода
(1—2%). На второй стадии плавки для снижения содержания
углерода в печь грузят брикеты с недостатком углерода, получая
конечный сплав и оборотный шлак. При этом шлак должен быть
основной. Примерный состав шлака, %: 40—45 SiOo; 50—55 СаО;
5—10 А12О3; 2—3 CaF2.
Ввиду высокой температуры плавления сплава плавку ведут
на блок.
Ниже приведен характерный состав ферромолибдена, выплав-
ляемого этим способом, %: 60—65 Мо; 0,03 S; 0,01 Р; 0,23 Si;
0,16 С.
Общая степень извлечения молибдена в сплав (с учетом обо-
ротов и улавливания пыли) составляет 95%. Удельный расход
электроэнергии 4450—6400 квт-ч/т ферромолибдена.
* Более детально о термодинамике и механизме восстановления см. гл. XI.
ГЛАВА V
ПЕРЕРАБОТКА РАЗЛИЧНЫХ ПОЛУПРОДУКТОВ
ОБОГАЩЕНИЯ
В результате обогащения полиметаллических сульфидных руд,
например медно-молибденовых, помимо стандартных концентра-
тов, получают промпродукты, содержащие 5—20% Мо, а при
обогащении окисленных руд — бедные концентраты с содержа-
нием от 5—6% Мо (повеллитовые концентраты) до 0,2—0,4% Мо
(при обогащении окисленных руд, содержащих ферримолибдит).
Значительные количества молибдёна (до 5—7% Мо) содержатся
в некоторых шеелитовых концентратах (например, тырны-аузских).
Молибден из перечисленных полупродуктов обогащения извле-
кают гидрометаллургическими методами, получая «химические
концентраты» — молибдат кальция или другие продукты с высо-
ким содержанием молибдена, которые могут быть использованы
для легирования сталей (молибдат кальция), выплавки ферромо-
либдена и получения чистых соединений молибдена.
18. ПОЛУЧЕНИЕ МОЛИБДАТА КАЛЬЦИЯ ИЗ МОЛИБДЕНИТОВЫХ
И ПОВЕЛЛИТОВЫХ ПОЛУПРОДУКТОВ
' Молибдат кальция широко используют в сталеплавильном
производстве для легирования стали молибденом преимущественно
в тех случаях, когда его содержание в стали не превышает 1%.
При введении молибдата кальция в смеси с кремнеземом в ванну
мартеновской печи или электропечи, а также в конвертер молиб-
ден полностью переходит в сталь, причем роль восстановителя
выполняет железо:
СаМоО4 + SiO2 + 2Fe = CaO.SiO2 + Fe2O3 + Mo.
Молибдат кальция, кроме того, может служить исходным мате-
риалом для выплавки ферромолибдена.
Технология переработки сульфидных молибденовых полу-
продуктов разработана в СССР Ленинградским горным инсти-
7* 99
тутом под руководством Н. С. Грейвера [1, 2]. В 1941 г. на одном
из отечественных предприятий вступил в эксплуатацию цех произ-
водства молибдата кальция 13].
Сульфидный концентрат
Обтиг
Огарок
Сода
7-е выщелачивание
Сода
2-е выщелачивание
Подготовка растворов
Сода
Фильтрация
3-е выщелачивание
СаС1г
Фильтрат Оборотный
кек
Сода
Осаждение
6-е выщелачивание
вода
Сода
вода
Нолибдат
кальция
Декантат
на извлечение
71 о и ве
5-е выщелачивание
Пронывка
Фильтрация
Отвальные
хвосты
Прон 6 а до
в отвал
Фильтрат
в отвал
Фильтрация
Полибдот
кальция
Рис. 37. Технологическая схема получения молибдата каль-
ция из молибденитовых полупродуктов
Сушка
Упаковка
Ниже приведен типичный
дукта обогащения [3], %:
состав молибденитового полупро-
Мообщ .... 15,0—22,0 SiO2
Моокисл.......... 0,2—0,4 А1Д
Fe . ........... 3,0—7,0 СаО
21,0—24,0
10,0—12,0
0,8—1,2
0,2—0,6
9,0—11,0
Си .............. 2,0—5,0 MgO
5общ ............15,0—23,0 Уголь
. ' В составе полупродукта обогащения содержатся минералы
молибденит (основная часть молибдена), повеллит, халькопирит
100
и пирит, карбонаты кальция и магния, кварц, алюмосиликаты.
Уголь попадает в концентрат из технического сернистого натрия,
применяемого при флотации, и отчасти из руды.
На рис. 37 приведена применяемая в производстве технологи-
ческая схема переработки молибденитовых промпродуктов. Она
включает окислительный обжиг, выщелачивание огарков раство-
рами соды, очистку растворов и осаждение технического молиб-
дата кальция. Другие технологические варианты переработки
концентратов (окисление кислородом в автоклавах, гипохлорит-
ный метод) рассмотрены в гл. VI.
Обжиг промпродукта
Основы и практика окислительного обжига молибденитовых
концентратов были детально рассмотрены выше (см. гл. II). Отли-
чительная особенность при обжиге молибденовых промпродук-
тов — высокое содержание в огарках молибдатов меди и железа,
в особенности в том случае, когда обжиг осуществляется в муфель-
ных или механических многоподовых печах. В настоящее время
в результате проведенных исследований обжиг молибденовых
промпродуктов проводят в печах кипящего слоя при температуре
в слое 650—660° С, что почти на 100 град выше рабочей темпера-
туры при обжиге в кипящем слое стандартных концентратов. Это
объясняется более высокой температурой спекания частиц обож-
женного молибденового полупродукта в связи с пониженным
содержанием в нем молибдена и значительным содержанием Fe2O3
и SiO2 и других тугоплавких компонентов [5].
Выщелачивание
Продукты обжига, кроме трехокиси молибдена, содержат мо-
либдаты [СаМоО4, CuMoO4, FeMoO4 и Fe2(MoO4)3], сульфаты
(CuSO4, CaSO4), кремнезем, алюмосиликаты и др. Для обеспече-
ния высокого извлечения молибдена выщелачивание огарков про-
водят растворами соды, которые, в отличие от аммиачной воды,
легко разлагают молибдаты.
В процессе выщелачивания, помимо растворения трехокиси
молибдена, протекает реакция обменного разложения молибда-
тов кальция, меди и железа с образованием молибдата натрия,
карбоната кальция, основных карбонатов переменного состава и
гидроокиси железа:
СаМоО4тв + Na 2СО3 раст Na2MoO4 раст + СаСО3 тв, (57)
(х + у) СиМоО4 тв + (х + у) Na2CO3 раст + 2у Н2О х СиСО3 х
х у Си (ОН)2 + (х + у) Na2MoO4 + 2у NaHCO3) (58)
(х + У) FeMoO4 тв -|- (х + 2у) Na2CO3 + 2у Н2О х” х FeCQ3 х
Ху Fe (ОН)2 + (х + у) Na2MoO4 + 2z/ NaHCO3> (59)
101
Fe2 (MoO4)3 + 3Na2CO3 + 3H2O ДГ 3Na2MoO4 + 2Fe (0H)3 + 3CO2,
(60)
Na2CO3 + CO2 + H2O = 2NaHCO3. (61)
Необходимый избыток соды зависит от.величин констант равно-
весия реакций между раствором соды и молибдатом.
Равновесие реакции СаМоО4 — раствор соды изучали в рабо-
тах [6, 8, 9]. Поданным работы [6], зависимость константы равно-
весия от температуры для этой реакции описывается уравнением 1
1g + 2,875, (62)
где
К _ [Na2MoO4]
Л1 [Na2CO3]
(концентрация выражена в моль!л).
При 25° С значение = 1,23, при 90° С = 3,05.
Из этого следует, что для полного разложения СаМоО4 и пере-
вода молибдена в раствор минимально необходимый избыток соды
при 25° С составляет 80% сверх стехиометрического количества,
а при 90° С 22%.
Полное разложение СиМоО4 достигается при расходе соды
1,12 моль на 1 моль СиМоО4. Состав осадка в этом случае примерно
отвечает формуле 1,5 CuCO3-Cu (ОН)2. При обработке FeMoO4
молибден полностью переходит в раствор при расходе соды
1,25 моль на 1 ;иолб’РеМоО4 [6].
Молибдат трехвалентного железа легко разлагается растворами
соды при количестве ее, близком к стехиометрическому.
Для обеспечения высокого извлечения молибдена в раствор
при минимальном расходе соды огарки следует выщелачивать
при температуре 80—90° С, что вытекает из температурной зави-
симости изменения констант равновесия реакций взаимодействия
молибдатов с растворами соды. Взаимодействие соды с молибда-
тами замедляется образованием пленок малорастворимых карбо-
натов и гидроокисей на частицах исходных соединений.
При выщелачивании огарков растворами соды вместе с молиб-
деном в раствор переходят примеси кремния, фосфора и мышьяка
в виде натриевых солей кремневой, фосфорной и мышьяковой
кислот, а также частично растворяется медь. Н. С. Грейвер пред-
полагает, что медь образует малоустойчивые растворимые ком-
плексы состава mCuCO3-nNaCO3 или mCuCO3-nNaHCO3 12].
Растворимость меди растет с концентрацией соды и снижается
с повышением температуры (рис. 38). Максимум растворимости
наблюдается при 20° С.
1 Приведенные значения /С реакции получены при работе, с СаМоО4, синте-
зированным из окислов при 600° С.
102
ЯР SO 50 О о 30 20 10 О
/емлерагпура раствора. °C
Рис. 38. Извлечение меди в раствор
при обработке CuSO4 растворами
соды различной концентрации при
различных температурах (цифры
у кривых — концентрация соды, %)
Приведенные на рис. 38 кривые получены при обработке суль-
фата меди растворами соды различной концентрации [2]. Они
показывают, что для снижения содержания меди в растворах
необходимо к концу выщелачивания иметь возможно более низ-
кую остаточную концентрацию соды. В нейтральных растворах
при нагревании карбонатные комплексы меди разрушаются и медь
полностью выделяется из раство-
ров в составе основных карбона-
тов.
Другая точка зрения на при-'
чины растворения меди выдвинута
В. Д. Пономаревым и К. Б. Лебе-
девым [4]. Авторы считают, что
переход меди в раствор вызван
образованием золя карбоната
меди в растворах углекислого
натрия [п СиСОз • т СОз~ • k Na+].
При этом ионом-стабилизатором
будет служить карбонат-ион, уда-
ление которого вызывает разру-
шение золя углекислой меди и
выделение меди из раствора.
Присутствующая в содовых
растворах примесь* железа также,
вероятно, находится в форме кол-
лоидных образований.
Огарки выщелачивают 8—10%-ным раствором соды в 4—5 ста-
дий по принципу прерывистого противотока (см. рис. 37). Это
позволяет лучше использовать соду и выводить растворы, нейтра-
лизованные свежей порцией выщелачиваемого огарка, до pH %
~ 8-^8,7. Выщелачивание ведут в стальных реакторах с мешал-
ками при подогреве глухим паром через рубашку или змеевики
до температуры 85—90° С. В корпус каждого реактора на различ-
ных уровнях вварены 5 кранов, через которые выпускается освет-
ленный раствор после отстаивания. Продолжительность каждого
выщелачивания и отстаивания — примерно 3 ч, а всего цикла опе-
раций в одном выщелачивателе (включая загрузку, перекачку,
подогрев) 8 ч. Отфильтрованные растворы, содержащие 50—
70 г Мо в 1 л раствора, поступают на осаждение молибдата каль-
ция. Извлечение молибдена в раствор составляет 97,5—98%.
Очистка растворов
Растворы, полученные в результате выщелачивания огарков,
содержат примеси меди, железа, кремния (Na2SiO3), иногда
фосфора и мышьяка (Na3AsO4, Na3PO4), ионы SO1“ (8—11 а/л),
Cl” (1—2 а/л), избыточную соду.
103
Для очистки от мед и, железа и кремнекислоты
растворы нейтрализуют до pH 7-^-8 и кипятят. При этом раз-
лагается избыточная сода (удаляется СО2), разрушаются медные
карбонатные комплексы или золи, гидролизуется силикат натрия
с выделением кремневой кислоты, коагулируют коллоидные при-
меси железа. Ранее нейтрализацию растворов проводили, добавляя
серную кислоту, что увеличивало концентрацию сульфат-ионов.
В последующем был найден более рациональный путь — нейтра-
лизацию растворов стали проводить добавлением в раствор, вы-
водимый из цикла выщелачивания, свежего молибденового огарка
до установления pH = 8ч-8,7 [4].
Как показала практика работы отечественных предприятий,
содержание в растворах фосфора и мышьяка не превышает сотых
долей процента, что исключает необходимость очистки от этих
примесей.
При повышении содержания примесей Р и S очистка может
быть осуществлена солями закиси или окиси железа, добавляе-
мыми в виде концентрированного раствора в нейтральный раствор
при интенсивном перемешивании и нагреве до 40—50° С. Полное
выделение мышьяка и фосфора обеспечивается окклюдированием
фосфата и арсената железа осадками гидроокиси и гидрозакиси
железа. Для осаждения примесей достаточно двух- трехкратного
избытка осадителя [2 ].
Предварительную очистку растворов от сульфат-ионов, яв-
ляющихся основной примесью в растворах, не проводят. Они
отделяются в процессе осаждения молибдата кальция и его про-
мывки.
Осаждение молибдата кальция
Растворы, поступающие на осаждение молибдата кальция,
имеют следующий примерный состав, г/л: 40—60 Мо; 0,25—
0,5 соды; 8—10 SO1”; 0,4—0,5 Си; 1,0—2,0С1; pH = 8ч-9. Влияние
различных факторов (pH раствора, количество осадителя, темпе-
ратура и др.) на состав и физическую структуру осадков молиб-
дата кальция детально изучено в работах [2, 3, 7].
По действующим техническим требованиям в молибдате каль-
ция марки МДК-1 должно быть не менее 44% Мо и не более 0,2% S,
в молибдате кальция марки МДК-2 допускается содержание
40—44% Мо и 0,2—0,3% S.
Молибдат кальция можно избирательно осаждать, если боль-
шую часть сульфат-ионов оставлять в растворе, регулируя коли-
чество добавленного осадителя (хлористого кальция) и оставляя
определенное количество молибдена в маточном растворе. Это
объясняется большим различием в растворимости СаМоО4 (до
0,35 г/л в реальных растворах) и CaSO4 (~2 г/л).
При остаточном содержании молибдена 0,8—1 г/л содержание
серы в осадках меньше 0,4%, что лишь в два раза выше технических
104
требований. При меньшей концентрации ЗО^-ионов в исходном
растворе содержание серы в молибдате кальция снижается [3, 7].
Содержание молибдена в осадках молибдата кальция в боль-
шой мере зависит от остаточной концентрации соды в растворе,
так как карбонат кальция с низкой растворимостью (0,0144 г/л)
осаждается вместе с. молибдатом кальция. Ввиду этого необходимо
контролировать содержание соды в исходных растворах, ее кон-
центрация не должна превышать 0,5 г/л.
Практически молибдат кальция осаждают, постепенно добав-
ляя в нагретый раствор, нейтрализованный до pH = 8,64-8,7,
раствор хлористого кальция (350—500 г/л) при перемешивании
до содержания в маточном растворе 0,6—0,7 г/л Мо. После оконча-
ния осаждения раствор кипятят при перемешивании в течение
1 ч %ля улучшения структуры осадка.
С целью доведения содержания серы до кондиций (<: 0,2%)
осадки промывают 3—4 раза декантацией холодной водой. При
промывке растворяется CaSO4. Промывные воды содержат 0,2—
0,25 г/л Мо. Промытый молибдат кальция фильтруют на вакуум-
фильтре, сушат и упаковывают.
При повышенном содержании серы в молибдате кальция для
ее удаления может быть использован следующий способ. Молиб-
дат кальция смачивают раствором соды, взятой в количестве 1 —
1,25-кратном к теоретически необходимому, для связывания серы
в сульфат натрия. Затем материал прокаливают при 600—650° С.
и выщелачивают водой. Содержание серы в молибдате кальция
может быть снижено до 0,01—0,06%, переход молибдена в растворы
не превышает десятых долей процента [2].
Извлечение молибдена из маточных растворов
и промывных вод
Маточные растворы от осаждения молибдата кальция содержат
0,6—0,8 г/л Мо, растворы после промывки молибдата кальция
0,2—0,3 г/л Мо. Раньше из таких растворов молибден осаждали
в виде трисульфида, как описано на стр. 147.
В настоящее время молибден из маточных и промывных рас-
творов регенерируют сорбцией на слабоосновной смоле АН1.
Условия сорбции и десорбции рассмотрены на стр. 150.
Описанная выше схема производства молибдата кальция из
молибденитовых полупродуктов обеспечивает извлечение молиб-
дена ~94,7%. С отвальными хвостами теряется —2,4%, механи-
ческие потери (включая обжиг) составляют —2,9% [2].
Получение молибдата кальция из повеллитовых
полупродуктов обогащения
При обогащении руд, содержащих повеллит и молибденит,
повеллит извлекают из хвостов сульфидной флотации. При фло-
тации хвостов получают повеллитовый полупродукт, в котором
105
концентрируются также кальцит, апатит, флюорит и другие мине-
ралы Ч
Примерный состав повеллитового концентрата, получаемого
на одной из отечественных обогатительных фабрик, следующий, %:
Мообщ .... 6—10 СаО............30—50
•Моокисл .... 4—7 F..............11—13
S.......... 5—7 Fe............ 2—4
SiO2.......8—10 MgO...........0,2—0,4
Помимо окисленных минералов молибдена — повеллита и неко-
торого количества молибдата, в концентрате содержится и неболь-
шое количество молибденита.
Исследования Лебедева, Апполонова и др. [3] показали воз-
можность применения описанной выше схемы производства молиб-
дата кальция из сульфидных полупродуктов для"' переработки по-
веллитового концентрата.
Присутствие в повеллитовом концентрате сульфидного молиб-
дена, а также флотореагентов и механической примеси угля
обусловило необходимость обжига концентратов перед выщела-
чиванием растворами соды.
Противоточное пятистадийное выщелачивание обеспечивает
высокое извлечение молибдена в раствор, однако расход соды
в этом случае значительно выше, а содержание в растворах мо-
либдена ниже, чем при переработке сульфидных промпродуктов.
Растворы, поступающие на осаждецие молибдата кальция, со-
держат много свободной соды (10—15 г/л) и лишь 10—15 г/л Мо.
Для их нейтрализации необходимо вводить в раствор значитель-
ное количество серной кислоты, что привело бы к загрязнению
серой молибдата кальция. В связи с этим целесообразно парал-
лельно перерабатывать сульфидные и окисленные полупродукты,
направляя первый раствор окисленной ветви на третью стадию
выщелачивания сульфидной ветви [3]. Это позволяет использо-
вать избыточную соду окисленной ветви и получить богатые по
молибдену растворы, из которых выделяется стандартный молиб-
дат кальция.
19. ИЗВЛЕЧЕНИЕ МОЛИБДЕНА ИЗ БЕДНЫХ
ФЕРРИМОЛИБДИТОВЫХ РУД И КОНЦЕНТРАТОВ
Многие месторождения молибдена отличаются значительным
развитием зон окисления, в которых молибден представлен
вторичными минералами, преимущественно ферримолибдитом
(xFe2O3-z/MoO3-zH2O) и повеллитом (СаМоО4). Значительные
запасы молибдена заключены в некоторых железистых кварцевых
рудах, содержащих ферримолибдит. Большая часть ферримо-
1 Повеллит флотируется с жирными кислотами сравнительно хорошо из
безжелезистых кварцевых руд.
106
либдита находится в дисперсном (субмикроскопическом) состоя-
нии в окислах и гидроокислах железа — лимоните, гидрогема-
тите, гетите и гидрогетите. Обогащение таких руд по существу
сводится к извлечению в концентрат молибденсодержащих окис-
лов и гидроокислов железа. Это делает принципиально невозмож-
ным получение богатых по молибдену концентратов. Из окислен-
ных руд, содержащих 0,05—0,07% Мо, удается флотацией полу-
чить концентраты с содержанием 0,2—0,3% Мо при извлечении
около 70—75% [10].
В связи с этим внимание исследователей было направлено на
разработку рентабельных способов химического извлечения мо-
либдена из окисленных руд и бедных концентратов. Ниже сделан
обзор результатов этих исследований, проводившихся преиму-
щественно с сорскими окисленными рудами и концентратами,
‘состав которых приведен в табл. 25 [13, 14].
ТАБЛИЦА 25
СОСТАВ ОКИСЛЕННЫХ РУД И КОНЦЕНТРАТОВ
Материал Содержание, %
Мо общ. Мо окисл. SiO2 Fe Al 2O3 СаО s
Окисленная руда 0,08 0,07 67,2 2,78 16,48 0,99 0,28
Концентрат . 0,29 0,21 51,4 7,53 14,67 —— 0,96
Изученные химические способы извлечения молибдена из
окисленных руд и концентратов можно подразделить на чисто
гидрометаллургические (выщелачивание молиб-
дена из руд или концентратов кислотами или растворами щелочей)
и комбинированные (сочетание пирометаллургических
операций вскрытия с гидрометаллургическими).
Гидрометаллургические способы
Выщелачивание серной кислотой. Известно, что все кислородные
соединения шестивалентного молибдена растворяются в серной
и соляной кислотах. На этом основаны аналитические методы
определения окисленного молибдена в рудах и концентратах.
Естественно, что изучению возможности технологического при-
менения обработки кислотами для извлечения молибдена из руд-
ного сырья было уделено значительное внимание. Исследования
проводили с серной кислотой как наиболее дешевой и легко
транспортируемой.
В результате обработки рудного материала, содержащего
0,07—0,2% окисленного молибдена, 5—10%-ной серной кислотой
при нагревании до 80—90° С в растворы извлекается 85—90%
107
Mo [13]. Высокое извлечение молибдена в кислые растворы
объясняется одновременным растворением окислов железа, с ко-
торыми молибден тесно связан. Однако этот способ оказался не-
рентабельным из-за высокого расхода серной кислоты (150—
180 кг купоросного масла на тонну руды). При осуществлении
оборотного выщелачивания в три-четыре стадии с промежуточным
подкреплением растворов расход кислоты можно снизить до 90—
100 кг на тонну руды [14]. Значительные трудности представляет
выделение из сернокислых растворов богатых молибденовых про-
дуктов, так как растворы сильно загрязнены железом и алюминием.
При нейтрализации растворов (с целью их очистки) с гидратными
кеками теряется 10—15% Мо.
Перечисленные недостатки делают кислотный метод мало
перспективным. '
Выщелачивание растворами щелочей. Извлечение молибдена
при выщелачивании окисленной молибденовой руды разбавлен-
ными растворами едкого натра или соды, как правило, значительно'
ниже, чем извлечение в сернокислые растворы. Это объясняется
тем, что щелочи, в отличие от серной кислоты, не вскрывают мине-
ралы железа, с которыми связан молибден.
В работе автора и Горовиц [13] было установлено, что удовле-
творительное извлечение молибдена (75—80%) достигается при
выщелачивании окисленной руды 2—2,5%-ными растворами соды
при весьма тонком мокром измельчении руды (или совмещении
измельчения с выщелачиванием) при температурах 120—150° С
(под давлением 2—5 ат). Рудный материал необходимо измельчать
в мельницах мокрого помола до содержания в нем 80—90% частиц
ТАБЛИЦА 2&
РЕЗУЛЬТАТЫ ПРОМЫШЛЕННЫХ ОПЫТОВ ОСАЖДЕНИЯ МОЛИБДЕНА
ИЗ БЕДНЫХ РАСТВОРОВ В ВИДЕ ТРИСУЛЬФИДА
Показатели Объем исходного раствора, л
300 240 65 5000
Концентрация молибдена в исходном растворе, г!л 0,21 0,775 0,48 0,40
Количество сернистого натрия, % к теоретическому 150 125 300 300
Содержание молибдена в осадке три- сульфида, % 32,7 33,04 28,7 18,0
Извлечение молибдена в осадок три- сульфида, % 99,6 99,7 99,1 99,(
Содержание SiO3 в осадке, % ... 9,54 1,16 5,0 11,0
Содержание молибдена в продукте окислительного обжига осадка три- сульфида, % 44,0 * I1 54,8 50,0 52,0
* Раствор был плохо осветлен.
108
меньше <<0,067 мм. В зависимости от состава обрабатываемой
руды растворы содержат 0,2—1 г!л Мо и в незначительной степени
загрязнены примесями. Извлекать молибден из таких бедных рас-
творов можно осаждением трисульфида молибдена или используя
ионообменное концентрирование.
Как видно из табл. 26, трисульфид осаждается сернистым нат-
рием (по режимам, описанным на стр. 147) полно даже при содер-
жании в растворе 0,2 г!л Мо. В результате окислительного обжига
осадка получаются химические концентраты с содержанием в сред-
нем 50% Мо [13].
Прямое выщелачивание растворами соды в автоклавах может
быть использовано для рудного материала, содержащего только
окисленный молибден. В случае присутствия, наряду с окислен-
ным, также и сульфидного молибцена (что характерно для бедных
концентратов, получаемых флотацией хвостов после обогащения
смешанной руды), обработке растворами соды должен предшество-
вать окислительный обжиг при 700—800° С.
Общее извлечение молибдена в химический концентрат по рас-
сматриваемой схеме составляет 70—75% при следующем расходе
реагентов, кг/т руды:
Сода кальцинированная . . . 50—60
Сернистый натрий............ 3—5
Серная кислота ............. 6—10
К недостаткам автоклавно-содовой схемы следует отнести необ-
ходимость весьма тонкого измельчения руды, которое является
дорогой операцией. Второй недостаток — наблюдаемое отличие
в степени извлечения молибдена в раствор для руд из различных
участков месторождения, что обусловлено неодинаковым характе-
ром ассоциации молибденовых минералов с окислами железа.
Комбинированные схемы
Чтобы повысить извлечение молибдена из окисленных руд
и концентратов, исследовали технологические варианты, в кото-
рых гидрометаллургическим операциям предшествуют пироме-
таллургические процессы вскрытия. К ним относятся обжиг
с известью, обжиг с хлористым натрием, хлорирование.
Обжиг с известью. При обжиге окисленной руды или концен-
трата с известью в интервале температур 700—850° С окислы же-
леза реагируют с окисью кальция с образованием феррита каль-
ция, молибден связывается в молибдат кальция. Таким образом,
полностью вскрываются рудные минералы. При выщелачивании
продукта обжига растворами соды молибден извлекается в рас-
твор, из которого выделяется осаждением трисульфида молибдена
или концентрируется с помощью ионообменной смолы. Эта простая
схема (рис. 39) была испытана в полупромышленных масшта-
бах [15].
109
Известь в количестве 3—4% смешивают с рудой или концен-
тратом. В барабанной печи шихту обжигают при 800—850° С.
Огарок выщелачивают 2—3%-ным раствором соды при т : ж —
— 1 : 2,5 и температуре 90° С. В раствор извлекается 80—85% Мо.
Примерный состав растворов при переработке флотационного
концентрата, содержащего 0,3% Мообщ и 0,19% Моокисл, г!л:
0,4 Мо; 0,0009 Р; 0,006 SiO2; 12,9 SO1“.
Соре кий окисленный концентрат 0,3 7О По)
Шихтовка с известью
Обти£ (600 В5(ГСЦ
Раствор 2- 25 %> ПагС03
Вы щели ч и ванне
Отстаивание и фильтрование
веки Растворы
В отвал Подкисление (до, рН-3) и нагревание
Л
Сорбция и десорбция
на колонках с анионитовой снолои
Т
Анниачные растворы (70-вОг/л Но )
Выпарка и кристаллизация параколиддата аинония
А мн ио к на регенерацию Прокалка Паточный раствор
Т 1
Ко03 I_________
Рис. 39. Технологическая схема извлечения молибдена из окисленных руд и кон-
центратов, включающая обжиг с известью
При сопоставлении двух возможных вариантов извлечения
молибдена из растворов приведенного выше состава — осаждения
в виде MoS3 и ионообменного способа — выявились экономиче-
ские преимущества второго способа, состоящие в пятикратном
снижении расхода серной кислоты при отсутствии затрат на сер-
нистый натрий [16].
Описанная схема обжига с известью наиболее разработана и
может обеспечить вполне экономичное извлечение молибдена из
окисленных руд и концентратов. Общее извлечение молибдена
В конечный продукт составляет 77—80%.
НО
Обжиг с хлористым натрием. Обжиг руды или концентрата
с хлористым натрием приводит к различным результатам в зави-
симости от условий его проведения. Трехокись молибдена (см.
также стр. 192) реагирует с NaCl с образованием Na2MoO4 и окси-
хлорида МоО2С12:
2МоО3 + 2NaCl = МоО2С12 + Na2MoO4. (63)
В окислительной среде и в присутствии небольших количеств
соды подавляющая часть молибдена переходит в молибдат натрия,
который затем извлекается в водный раствор. Наоборот, при об-
жиге в присутствии сернистого газа (например, при добавках
пирита в шихту) большая часть молибдена возгоняется в виде
оксихлорида. Смещение реакции обусловлено образованием суль-
фата натрия:
МоО3 + 2NaCl + SO2 + V2O2 = МоО2С12 + Na2SO4. (64)
Вариант обжига с хлористым натрием, исключающий возгонку
оксихлорида, исследован в лабораторном и полупромышленном
масштабах [1’3]. При обжиге окисленных руд, содержащих 0,06—
0,08% Мо, с 2% NaCl и 0,5% соды при 700—800° С в течение 1 ч
происходит достаточно полное вскрытие. При последующем выще-
лачивании обожженной руды 0,5—1%-ным раствором соды при
т : ж — 1 : 24-2,5 и температуре 80—90° С в раствор извлекается
85—90% Мо, который далее выделяется в виде трисульфида.
Возможно применение ионообменного его концентрирования.
При использовании этого процесса для вскрытия окисленных кон-
центратов необходимо увеличить дозировку хлористого натрия
и соды примерно пропорционально увеличению содержания в них
железа по сравнению с исходной рудой.
При высоком содержании сульфидной серы в рудном материале
(4—5% S) при обжиге наблюдается значительная возгонка молиб-
дена в виде оксихлорида, что требует увеличения дозировки соды
или проведения предварительного окислительного обжига мате-
риала.
Второй вариант обжига с NaCl — хлоридовозгонка — изучался
авторами работ [13 и 17]. Было установлено, что при обжиге окис-
ленного концентрата, содержащего 3—4% сульфидной серы (если
необходимо, добавляют пирит), с поваренной солью (~10% NaCl
в шихте) при температуре 800° С 80—90% Мо возгоняется в со-
ставе оксихлорида МоО2С12. Просос воздуха, содержащего сер-
нистый газ, способствует повышению степени возгонки (табл. 27).
При обеспечении окислительной атмосферы в печи железо воз-
гоняется в незначительной степени, так как в этих условиях хло-
рид железа неустойчив. Присутствие восстановителя в шихте
резко снижает степень возгонки молибдена.
Обжиг с возгонкой можно проводить в барабанных печах,
улавливая оксихлорид в барботере или скруббере с циркулирую-
111
ТАБЛИЦА 27
ВЛИЯНИЕ СОДЕРЖАНИЯ СУЛЬФИДНОЙ СЕРЫ В ШИХТЕ
(КОНЦЕНТРАТ + 10% NaCl) и SO2 В ГАЗОВОЙ ФАЗЕ НА СТЕПЕНЬ
ВОЗГОНКИ МОЛИБДЕНА
(ТЕМПЕРАТУРА ОБЖИГА 800° С, ВРЕМЯ 1 ч) [13]
Содержание сульфидной серы в концентрате, % Среднее извлечение молибдена в возгоны, % Газовая среда
0,96 3,71 4,36 46,3 70,8 80,2 Просос воздуха
0,96 85,4 Просос смеси:
3,71 94,7 83% воздух а+
4,36 90,1 + 17% SO2
3,62 * 82,7 Просос воздуха
* Содержание серы в шихте, к которой было добавлено 5% FeS2
щим раствором аммиака. Молибден из растворов может быть затем
извлечен осаждением СаМоО4 или выпаркой с выделением полимо-
либдатов. Поскольку при обжиге возгоняется 80—90% Мо, из-
влечение молибдена в химический концентрат должно быть не
ниже 75%. Способ хлоридовозгонки представляет большой инте-
рес, так как в этом случае исключаются операции гидрометаллур-
гической обработки больших масс бедной руды или концентрата.
Его эффективность может быть, однако, выявлена при осуществле-
нии непрерывного процесса в сравнительно крупных масштабах.
Метод хлорирования хлором. Прямое хлорирование окислен-
ных молибденовых руд и концентратов изучали в работах [17, 18].
В присутствии активаторов (угля или пирита) хлорирование про-
текает активно при температурах 400—450° С с извлечением в воз-
гоны 85—95% Мо в виде оксихлорида. Шихта должна содержать
5—7% угля или 2—5% пирита (с учетом содержания сульфидной
серы в руде или концентрате). Была показана возможность осу-
ществления процесса в шахтном хлораторе непрерывного дей-
ствия с загрузкой гранулированного материала. В качестве
связки при грануляции на чашевом грануляторе может быть ис-
пользован сульфитно-целлюлозный щелок [18].
Существенный недостаток прямого хлорирования рудного ма-
териала хлором заключается в переходе большей части железа
в хлорид железа, возгоняемый вместе с оксихлоридом молибдена.
Это вызывает высокий расход хлора и усложняет систему конден-
сации. Возможности раздельной конденсации оксихлорида мо-
либдена и хлорного железа не изучены. Возникает также проблема
119
использования хлорного железа, которая, как известно, не нашла
решения при организации производства четыреххлоридного ти-
тана.
В связи с указанными недостатками представляется более пер-
спективным метод хлоридовозгонки, основанный на обжиге с хло-
ристым натрием.
20. ИЗВЛЕЧЕНИЕ МОЛИБДЕНА ИЗ ШЕЕЛИТОВЫХ
КОНЦЕНТРАТОВ И ПОЛУПРОДУКТОВ
В некоторых месторождениях вольфрамо-молибденовых руд,
например Тырны-Аузском, шеелит содержит изоморфную примесь
повеллита. При обогащении таких руд получают стандартный мо-
либденитовый концентрат и шеелитовый концентрат с содержа-
нием 5—6% Мо. Большая часть окисленного молибдена нахо-
дится в решетке шеелита, меньшая доля представлена неизоморф-
ными ферримолибдитом и повеллитом. Для доводки по содержа-
нию примеси фосфора шеелитовый концентрат обрабатывают
на холоду соляной кислотой так, чтобы конечная кислотность
была 30—40 г!л. В раствор вместе с фосфором извлекается часть
молибдена (преимущественно неизоморфно связанного) и 2—4%
вольфрама. Из кислого раствора известью осаждают «известковый
продукт», содержащий смесь фосфата, вольфрамата и молибдата
кальция. В составе такого продукта 10—17% Мо, 8—12% W, 1 —
3% Р.
Таким образом, молибден содержится и в стандартных шеели-
товых концентратах, и в полупродуктах доводочных операций.
Для извлечения молибдена из шеелитовых концентратов и
полупродуктов их подвергают гидрометаллургической перера-
ботке. В промышленной практике наибольшее распространение
получил автоклавно-содовый способ переработки шеелитсодержа-
щегб сырья. Кроме того, перспективны схемы, предусматривающие
разложение шеелита соляной кислотой.
Автоклавно-содовый способ. Технология разработана совет-
скими исследователями И. Н. Масленицким, Н. Н. Масленицким,
Л. М. Перловым и др. и детально изложена в ряде публикаций
[19—21]. Исходный материал разлагают в автоклаве при темпе-
ратуре 200° С растворами соды, в которые вместе с вольфрамом
переходит весь окисленный молибден. Растворы очищают от при-
месей фтора, фосфора, мышьяка и кремния обычными методами,
после чего осаждают молибден в виде трисульфида сернистым
натрием при pH = 1,54-2. При соответствующей дозировке оса-
дителя (~125% от теоретически необходимого количества) оса-
ждается 98—99% Мо и небольшая доля вольфрама. При содержа-
нии вольфрама в осажденном продукте более 2,5% переосаждают
трисульфид молибдена. С этой целью осадок растворяют в растворе
соды 250 г!л при 80—90° С, добавляют в раствор Na2S и нейтрали-
зуют его соляной кислотой до pH = 1,54-2.
8 А. Н. Зеликман 113
Осадки MoS3 после сушки на воздухе обжигают при темпера-
туре 400—500° С, при этом происходит термическая диссоциа-
ция MoS3 с образованием MoS2, который частично окисляется.
Получаемые молибденовые гидрометаллургические концен-
траты (КМГ) содержат 48—50% Мо и 2—3% WO3 (см. табл: 4).
Общая схема процесса показана на рис. 40.
Кислотный способ. Распространенный способ переработки
шеелитовых концентратов — непосредственное разложение их
концентрированной соляной кислотой, в результате которого по-
лучают осадок технической вольфрамовой кислоты. При сохране-
нии высокой остаточной кислотности (150—200 г!л НС1) большая
часть содержащегося в шеелите, молибдена переходит в соляно-
кислый раствор, что объясняется высокой растворимостью молиб-
деновой кислоты в соляной кислоте (табл. 28).
ТАБЛИЦА 28
РАСТВОРИМОСТЬ МОЛИБДЕНОВОЙ И ВОЛЬФРАМОВОЙ КИСЛОТ В
СОЛЯНОЙ КИСЛОТЕ [22]
Растворимость, г/л, при температуре, °C
Концентра- ция НС1, г/л 20 50 70
Н2МоО4 H2WO4 Н2МоО4 H2WO4 Н2МоО4 H2WO4
400 380 7,34 477,3 25,9 506,3 19,9
270 182 0,032 241,6 0,27 260,0 0,097
200 91,8 0,01 117,3 0,16 135,5 0,011
130 24,0 0,01 35,6 0,011 41,1 0,013
80 11,1 0,01 13,6 0,011 13,9 0,019
40 2,34 0,01 4,8 0,011 4,65 0,016 Н
При достаточном избытке соляной кислоты (170—200% от
теоретически необходимого количества) в отработанную соляную
кислоту переходит от 75 до 90% Мо в зависимости от исходного
его содержания. Практически не удается получить осадки вольф-
рамовой кислоты с содержанием молибдена ниже 0,3—0,4%-
Более полное удаление молибдена достигается при разложении
шеелита соляной кислотой в присутствии восстановителя, восста-
навливающего шестивалентный молибден до молибденовой сини.
В качестве восстановителя Ферей предложил 1 [11] исполь-
зовать порошок ферросилиция, при растворении которого в соля-
ной кислоте выделяется водород, восстанавливающий Мо (VI)
до Мо (V). При разложении шеелитового концентрата, содержа-
щего 2,26% Мо, соляной кислотой в присутствии ферросилиция
(75% Si) может быть получена вольфрамовая кислота с содержа-
нием 0,1 % Мо.
Из солянокислых растворов молибден может быть выделен
в виде молибдата кальция (добавлением известкового молока),
1 Furey I. L. US Pat., 222510 1941.
114
сульфида нолибдена
I
Фильтрация и проныдка
СаС1г
Раствор
Упаривание
Осаждение
Осадок
Осадок C!oS<
Репульпация,
фильтрация
Раствор
Фильтрация
и пронывка
Осадок СаМ(Д
, Раствор
Сушка
и частичный
обжиг
Дополнительное
осаждение
Разложение
в канализацию
Полибденовый
концентрат
Осадок Раствор
в канализации}
Пронывка
и фильтрация
Раствор Осадок H2W0^
в канализацию |
Сушка
Прокаливание
вольфранобый ангидрид
Рис. 40. Схема автоклавно-содового способа переработки молибденсодержащих
шеелитовых концентратов
8*
115
трисульфида молибдена или ферримолибдата (при введении в со-
лянокислый раствор гидроокиси железа и нейтрализации рас-
твора) (см. стр. 149).
Возможно экстракционное извлечение молибдена из соляно-
кислых растворов трибутилфосфатом или кетонами с последующей
реэкстракцией молибдена растворами аммиака (см. стр. 169).
Шеелитовый концентрат
Разложение соляной кислотой
Кислая пуло па Н2 W0$
- я экстракция
Реэкстракт
(7070'Но)
1-й экстракт Кислая пульпа H2W0li
| |
Реэкстракция водой 2-я экстракция
---------)
Кислая пульпа
Фильтрация
Осадок Отработанная
Н2 W0$ соляная
। кислота
аммиачную
очистку
Органическая
фаза
2-и экстракт
Но получение
молибденового
продукта
Аммиачная
реэкстракция
Аммиачная
реэкстракция
Органическая
фаза
Аммиачный
раствор
В оборот
Осаждение
Органическая
фаза
I На
В оборот
(спримесью Но)
Осадок Раствор
__I В сброс
и получение
паравольфрамата
аммония
Рис. 41. Схема кислотно-экстрационного способа переработки молибденсодержа-
щих шеелитовых концентратов
Представляет большой интерес кислотно-экстракционный ва-
риант переработки шеелитовых концентратов, заключающийся
в разложении шеелита соляной кислотой с последующей обработ-
кой пульпы экстрагентом — метил изобутил кетоном 1 или ацето-
феноном [23L В органическую фазу при 6-н. концентрации со-
ляной кислоты извлекается практически весь молибден и некото-
рая доля (2—3%) вольфрама. При двукратной обработке пульпы
1 Newkirk Е. US Pat, 3079226, 1963.
116
кетоном содержание молибдена в вольфрамовой кислоте сни-
жается до 0,02—0,05%.
Автор и Калинина, изучавшие экстракцию молибдена из
пульп вольфрамовой кислоты ацетофеноном (метилфенйлкетоном)г
нашли, что примерно 70% Мо можно реэкстрагировать из органи-
ческой фазы водой вместе с небольшой долей вольфрама, содержа-
щегося в экстракте. Из полученного слабокислого раствора можно
выделить молибденовый химический концентрат (например, мо-
либдат кальция или трисульфид молибдена). Оставшиеся в орга-
нической фазе молибден и вольфрам реэкстрагируются аммиач-
ной водой. Из аммиачного реэкстракта осаждают смесь CaWO4
и СаМоО4, которую возвращают на разложение [23].
Предложенная авторами кислотно-экстракционная схема пе-
реработки молибденсодержащего шеелитового концентрата пока-
зана 1 на рис. 41.
Преимущества кислотно-экстракционной схемы состоят в пол-
ном отделении молибдена от вольфрама, ее недостаток — высокий
расход соляной кислоты.
21. переработка вульфенитовых концентратов
Вульфенит не имеет существенного значения в мировой до-
быче молибдена, так как промышленные руды этого типа встре-
чаются редко. Обычно вульфенит находится в зонах окисления
свинцовых месторождений и сочетается с другими минералами,
например ванадатами, арсенатами и др. Высокая плотность вуль-
фенита, равная 6,6—7, обусловливает успешное применение гра-
витационных методов обогащения руд. Концентраты содержат
от 15 до 25% МоО3 (табл. 29).
ТАБЛИЦА 29’
СОСТАВ ВУЛЬФЕНИТОВЫХ КОНЦЕНТРАТОВ
Компоненты Содержание компонентов, %
США, Колорадо Мексика Чили
МоО3 16,60 28,03 21,80
РЬ 50,00 49,70 54,20
SiO2 6,00 12,80 8,00
FeO 11,00 — —
СаО 2,00 — —
As 0,80 — —
Р 0,05 1,20 0,30
13еликман А. Н. и Калинина И. Г. Авт. свид. СССР № 186136,
1965 г. Бюлл. изобр., 1965, № 18.
117
При переработке вульфенитового концентрата необходимо
обеспечить извлечение не только молибдена, но и свинца. Проще
всего это достигается при сплавлении концентрата с содой и углем
или с сульфидом натрия [12].
Сплавление с содой и углем. При оплавлении вульфенита с со-
дой и углем выплавляется металлический свинец и образуется
шлак, содержащий молибдат натрия:
РЬМоО4 + Na2CO3 + С = Na2MoO4 + Pb + СО + СО2. (65)
В свинце растворяются примеси золота и серебра. Шлак обра-
батывают водой и из растворов выделяют соединения молибдена.
Сплавление с сульфидом натрия. При сплавлении вульфенита
с сернистым натрием или смесью сульфата натрия и угля молиб-
ден образует сульфомолибдат натрия, а свинец— сульфид:
РЬМоО4 + 5Na2S = Na2MoS4 + PbS + 4Na2O. (66)
Плав ликвирует по плотности и большая часть сульфида свинца
собирается на дне плавикового тигля вместе с сульфидами меди и
висмута.
При выщелачивании водой верхней части сплава, содержащей
сульфомолибдат натрия, PbS остается в нерастворимом осадке.
Из раствора молибден может быть выделен в виде MoS3.
Сплавление с Na2S может быть заменено обработкой вульфе-
нита растворами сульфида натрия.
Описанные в литературе методы ^кислотного разложения вуль-
фенита не имеют практического значения.
ГЛАВА VI
ГИДРОМЕТАЛЛУРГИЧЕСКИЕ СПОСОБЫ
РАЗЛОЖЕНИЯ МОЛИБДЕНИТОВЫХ КОНЦЕНТРАТОВ
* Разложение молибденитовых концентратов может быть осу-
ществлено чисто гидрометаллургическими методами, исключаю-
щими предварительный окислительный обжиг. К ним относится
разложение азотной кислотой, окисление молибденита кислоро-
дом под давлением в щелочном растворе, окисление растворами
гипохлорита натрия и другие. Некоторые из этих методов уже
нашли применение в промышленной практике.
22. РАЗЛОЖЕНИЕ АЗОТНОЙ КИСЛОТОЙ
Азотная кислота концентрации 25—50% при нагревании
активно окисляет молибденит. Это используется в химическом
анализе для разложения исходной навески рудного материала и
представляет определенный интерес также для промышленной
переработки молибденитовых концентратов. Одно из преимуществ
разложения стандартного концентрата азотной кислотой заклю-
чается в возможности обеспечения более полного извлечения
спутника молибдена — рения. Он концентрируется вместе с частью*
молибдена в азотно-сернокислых маточных растворах, из которых
оба элемента могут быть извлечены экстракцией органическими
растворителями [67]. Существенно также, что при использовании
азотной кислоты практически исключаются вредные сбросы, так
как из азотно-сернокислых маточных растворов после их выпарки
кристаллизуется смесь сульфата и нитрата аммония, используе-
мая в качестве удобрений. В настоящее время разложение молиб-
денита азотной кислотой применяют на одном из заводов СССР.
Разложение молибденита азотной кислотой изучалось в рабо-
тах [1; 2; 41, с. 86 и 95; 60, с. 122, 129, 170], опубликованных в по-
следние годы. Взаимодействие MoS2 с азотной кислотой в основном
описывается реакцией
MoS2 + 6HNO3 = Н2МоО4 + 2H2SO4 + 6NO. (67}
119
Это подтверждается близким соответствием между реальным рас-
ходом кислоты и ее количеством, рассчитанным по написанной
выше реакции при достигнутой степени окисления [1 ]. Бурые пары
.двуокиси азота появляются вследствие взаимодействия закиси
азота с кислородом воздуха. Часть образующейся молибденовой
кислоты (в зависимости от режима разложения) остается в азотно-
сернокислом маточном растворе преимущественно в форме суль-
<фато-комплексов, например типа [Мо2О5 (SO4)n]~<2«-2). Наличие
Рис. 42. Кинетические кривые
окисления молибденита 54%-ной
.азотной кислотой при 80° С (/)
и 20° С (5). Кривая 2 — доля, мо-
либдена, перешедшего в раствор
анионных комплексов установлено
в работе [60, с. 175] методом элек-
трофореза на бумаге. Кроме того,
по всей вероятности, некоторая доля
молибденовой кислоты находится
в растворе в коллоидной форме.
В работах.”[1;*60, с. 170] было
установлено, - что при температуре
разложения выше 60° С и концен-
трации HNO3 более 20% в началь-
ный период (30—40 мин) весь окис-
лившийся молибден переходит в рас-
твор, однако в последующем выде-
ляется молибденовая кислота и резко
падает концентрация молибдена в рас-
творе (рис. 42). После выделения
молибденовой кислоты скорость раз-
ложения молибденита снижается
в результате тормозящего действия
молибденовой кислоты,отлагающейся
на частицах (рис. 42, кривая 1). С уве-
личением концентрации азотной кис-
лоты и температуры скорость окисления молибденита возрастает,
причем оба фактора способствуют коагуляции молибденитовой
кислоты. Так, при температуре 80° С с увеличением исходной
концентрации кислоты с 15—420 % до 54% содержание молибдена
в растворе понижается с 13—15 до 5 г/л. При тех же концентра-
циях кислоты и температуре 90° С содержание молибдена в рас-
творе соответственно равно 10—11 и 2,5 г/л. Присутствие в азотно-
сернокислом растворе значительных количеств ионов трехвалент-
ного железа приводит к повышению концентрации молибдена
в маточном растворе.
Теоретический расход 60%-ной азотной кислоты для разло-
жения стандартного молибденитового концентрата (с содержа-
нием молибдена 48—50%) примерно 3,16 т на 1 т концентрата.
Однако для обеспечения полного разложения приходится затра-
чивать значительно больше кислоты. При этом следует учиты-
вать расход кислоты на взаимодействие с минералами-приме-
сями (сульфиды меди, железа, кальцит и др.), а также испа-
120
рение кислоты в процессе разложения, проходящем при 80-*-
90° С.
Высокая степень разложения может быть обеспечена при двух-
стадийной схеме разложения (рис. 43), когда свежая кислота
поступает на разложение хвостов после выщелачивания аммиаком
осадка, полученного после первой ступени разложения. При этом
расход кислоты ниже, чем при одностадийном разложении [2].
*
Молибденитовый концентрат
1-я стадия разложения
Газы N0tN02
Фильтрация и промывка
На регенерацию Кислый
HN03 маточный
раствор
( Mo,RetFetCa и др)
Осадок
Раствор НН^ОН
Выщелачивание
На доизвлечение
молибдена
и извлечение
рения
Раствор
(ПН^)2 MoOk
Хвосты
HN03
На выделение
2-я стадия
полимолибдато разложения
Фильтрация Гтзы
но регенерацию
Осадок Азотнокислый
NH^OH раствор
Рис. 43. Принципиальная
схема разложения молиб-
денитового концентрата
азотной кислотой
Выщелачивание
Растворе Отвальные
j хвосты
Получаемые растворы молибдата аммония можно перераба-
тывать по обычной схеме (описанной в гл. III) с тем отличием,,
что для подкисления раствора до pH % 2н-3 (с целью осаждения
полимолибдата) вместо соляной кислоты применяют азотную.
Обязательное условие экономичности разложения азотной
кислотой — регенерация кислоты из выделяющихся окислов
азота. При этом одновременно обеспечивается полное улавлива-
ние окислов азота и исключение их выброса в атмосферу, что<
совершенно необходимо по условиям техники безопасности.
Как показано в работе [1 ], расход азотной кислоты существенно-
снижается при циркуляции газов с одновременным введением
в систему кислорода. В этом случае двуокись азота, образующаяся
121:
в результате окисления NO кислородом, растворяется с образова-
нием HNO3 и HNO2, которые окисляют молибденит.
Участие кислорода в процессе окисления позволяет полностью
разлагать молибденитовый концентрат при затрате азотной кис-
лоты меньшей, чем теоретически необходимое количество [1].
Так, при разложении молибденита 27%-ной азотной кислотой,
взятой в количестве, равном 80% от стехиометрического, в слу-
чае циркуляции газов с впуском кислорода за 2 ч при температуре
80° С минерал полностью окисляется. Это отвечает расходу 2,8 т
54%-ной азотной кислоты на тонну концентрата.
Молибденитовый концентрат можно разлагать азотной кисло-
той в аппаратуре из нержавеющей стали. При расчете объема
аппаратов необходимо учитывать сильное вспенивание пульпы
в процессе разложения из-за выделения окислов азота и при-
сутствия в концентрате флотореагентов.
Осадки, содержащие молибденовую кислоту, должны быть
отмыты от ионов кальция, чтобы при выщелачивании их раство-
ром аммиака не образовывался молибдат кальция.
Разложение молибденитового концентрата азотной кислотой
применяют в промышленных масштабах.
23. ОКИСЛЕНИЕ МОЛИБДЕНИТА КИСЛОРОДОМ ПОД ДАВЛЕНИЕМ
(АВТОКЛАВНЫЙ ПРОЦЕСС)
Окисление молибденита кислородом под давлением в водных
щелочных средах и применение этого процесса для вскрытия мо-
либденитовых концентратов изучалось в ряде работ [3—6].
Кинетику и механизм окисления в растворах КОН исследовал
Дрешер с сотрудниками [3], в растворах NH4OH, Na2CO3, NaOH—
Соболь и Нелень [5, с. 94]. В этих работах изучали скорость окис-
ления молибденита в зависимости от ряда факторов (температуры,
давления кислорода, концентрации щелочи и др.), причем исполь-
зовали цилиндрические плотные таблетки молибденита, получен-
ные прессованием порошка чистого минерала.
Окисление молибденита может быть описано следующим сум-
марным уравнением:
MoS2 + 9/2О2 + 6ОН- -> МоОГ + 2SO1” + ЗН2О. (68)
Реакция протекает через стадию образования тиосульфата
S2Oh, обнаруживаемого в растворе при неполном окислении
минерала:
MoS2 + %О2 + 4ОН~ -»МоО1_ + S2oi“ + 2Н2О. (69)
Концентрация S2O3 выше в начальной стадии процесса и сни-
жается во времени (рис. 44).
122
Написанные сложные реакции протекают через ряд более про-
стых промежуточных ступеней.
На рис. 45 показана зависимость скорости реакции от давле-
ния кислорода в опытах, проведенных с растворами КОН
2,6 моль/л при интенсивном перемешивании, исключавшем влия-
ние внешней диффузии [3]. Скорость процесса возрастает с увели-
чением давления кислорода, причем форма кривых характерна
для кривых сорбции газа на твердой поверхности. Зависимость,
скорости реакции от концентрации ионов ОН- сложная (рис. 46).
В интервале низких концен-
траций КОН (до 1 моль/л)
скорость реакции растет
с увеличением концентрации
щелочи, при более высокой
концентрации щелочи зави-
симость обратная, что объ-
ясняется уменьшением рас-
творимости кислорода с рос-
том щелочности раствора
(рис. 47).
При низких концентра-
циях щелочи растворимость
кислорода достаточно высо-
кая и скорость реакции ли-
митируется притоком ионов
ОН “. Поэтому скорость реак-
Рис. 44. Изменение во времени концен-
трации ионов S2O|— при автоклавном
окислении молибденита. Условия опытов:
t— 156° С; давление кислорода 21 ат;
концентрация КОН 2,6 моль/л, 38,6 г MoS^
на 1 л КОН
ции возрастает с увеличением
концентрации ОН-. При концентрациях КОН больше 1 моль/л
растворимость кислорода уменьшается настолько, что скорость
реакции лимитируется поступлением кислорода к поверхности.
Дрешер, Вардсворс и Фассел предполагают, что реакция
окисления состоит из следующих стадий, протекающих с различ-
ной скоростью [34:
1. Адсорбция молекулы кислорода на поверхности MoS2
(быстро):
MoS2 + О2 - MoS2 [О2]адс. (70}
2. Адсорбция второй молекулы кислорода (медленно):
MoS2 [О2]адс + О2 7^ MoS2 [2О2]адс,
К2
(71}
где Ki и К2 — константы скорости прямой и обратной реакций.
3. Образование'активного комплекса, способного реагировать
с ОН “-ионами (медленно):
MoS2 [2О2]адс -* [MoS2 . . . 2О2]акт,
где К3 — константа скорости реакции.
(72)
125
Давление кислорода, am
Рис. 45. Влияние давления кислорода
на скорость окисления молибденита.
Условия:концентрация КОН 2, блюл ь/л,
скорость перемешивания 750 об/мин
(кривые — расчетные, точки — экспе-
риментальные)
Рис. 46. Влияние концентрации
КОН на скорость окисления
молибденита. Условия: избыточ-
ное давление кислорода 28 ат\
скорость перемешивания
750 об/мин
Рис. 47. Растворимость кисло-
рода в зависимости от концен-
трации КОН:
s — растворимость кислорода; р —
абсолютное давление кислорода
124
4. Взаимодействие активного комплекса с ОН с образова-
нием на поверхности промежуточных гидроксокатиона молибде-
нила и S2O2 (быстро):
[M0S2 • • . 2О2]акт + ОН" -> МоО2 (ОН)+ + S2O£~. (73)
5. Взаимодействие промежуточного гидроксокатиона с ОН-
с образованием МОО4", переходящего в раствор (быстро):
МоО2 (ОН)^ + ОН" — МОО4- + 2Н+. (74)
6. Н++ ОН-->Н2О (быстро). (75)
7. S2O1" + !/2О2 — S2O1" (быстро). (76)
Ионы тиосульфата затем окисляются в растворе кислородом
до SO1“:
S2O|~ + 2О2 + 2ОН" — 2SO4-
+ Н2О.
(77)
Если принять долю всех участков, покрытых кислородом
(MoS2 [2О2]адс), 0, то в условиях равновесия скорость образо-
вания таких участков равна скорости их исчезновения.
Скорость образования покрытых участков = К0Ki (1 — 6)-
Скорость исчезновения покрытых участков складывается из ско-
рости десорбции кислорода по реакции (71) (/Со/С20) и скорости
образования активных комплексов по реакции (72) (/СоДз0),
так как эти комплексы быстро превращаются в ионы МоО|~, пере-
ходящие в раствор.
Таким образом, скорость исчезновения участков
к0к2е + к0к3е,
где (1 —0)—доля поверхности, не покрытой кислородом;
и К2 — константы скорости сорбции и десорбции кисло-
рода [реакция (71)];
7<3 — константа скорости реакции (72) — образования
активного комплекса;
/<0 — константа перехода от кажущейся доли поверх-
ностного покрытия к истинной поверхностной
концентрации. Она содержит множители Ks$ —
фактор учета шероховатости поверхности; Как1 —
доля активных участков поверхности от общего
числа участков; К' — число молекул, приходя-
щихся на единицу поверхности молибденита.
При равновесии
№ (1-0) [О2] - к0к2е - аде - о. (78)
125
Отсюда
(79)
В работе [3] скорость реакции окисления молибденита кисло-
родом определяли по скорости повышения концентрации ионов
МоО1~ в растворе. Поскольку скорость реакции (73) велика,
скорость образования МоО^” определяется скоростью образова-
ния активного комплекса по реакции (72):
Скорость реакции =
d [МоО^
dt
(80)
Подставляя выражение для 0 из уравнения (79), получим
d [МоС>4 ] [О2]
(81)
TS
Обозначив Ка = и Кь = --р , можно выразить ско-
Л2 “Г A3
рость реакции более простым уравнением:
d [МоО^-]
dt
КаКъ [Оа]
1 + Кь[О2] •
(82)
Это уравнение удовлетворительно описывает кривые зависи-
мости скорости реакции от давления кислорода, представленные
на рис. 45.
Горизонтальные участки кривых (где скорость реакции мало
зависит от давления кислорода) отвечают полному покрытию по-
верхности кислородом, когда 6 —> 1. В этом случае скорость реак-
ции равна Ка, т. е. определяется скоростью образования активных
комплексов по реакции (72).
Анализируя экспериментальные данные, авторы работы [3]
приходят к заключению, что в процессе окисления равновесие
сорбции кислорода на поверхности [реакция (71)] «не дости-
гается, и предполагают, что К% > К2, откуда Вели-
АЗ
чина энергии активации сорбции кислорода на поверхности мо-
либденита [по реакции (71)] определена равной 11,88 ккал!моль,
а энергия активации образования активированного комплекса
[реакция (72)1 6,60 ккал!моль,
126
Приближенная оценка показала, что доля активных участков
поверхности, участвующих в процессе окисления, равна лишь
0,26% общей поверхности. Из этого следует, что поверхность
пластинок молибденита относительно неактивна и поверхностные
реакции протекают преимущественно на ребрах.
В работах Соболя и Неленя [5, 6, с. 392] были сопоставлены
скорости окисления кислородом молибденита в растворах аммиака,
соды и щелочи. При прочих равных условиях скорость процесса
возрастает в ряду аммиак—сода—каустическая щелочь, что свя-
зано с соответствующим повышением концентрации ОН-ионов.
Авторы обнаружили каталитическое действие соединений меди
на процесс окисления молибдена кислородом в аммиачных рас-
творах и в растворах соды и щелочи. При концентрации меди
всего 100 мг/л скорость перехода молибдена в раствор аммиака
или соды вдвое выше, чем при отсутствии меди. Это позволяет,
добавляя соли меди, снизить параметры процесса (давление кисло-
рода,, температура, время обработки) при окислительном авто-
клавном выщелачивании молибденитовых концентратов. Механизм
каталитического действия меди авторами не разъяснен. Возможно,
здесь имеется аналогия с механизмом каталитического действия
меди при окислении гипохлоритом натрия (см. стр. 133).
Окислительное автоклавное выщелачивание было детально
изучено применительно к переработке молибденово-медных пром-
продуктов в работах [4—6]. Сульфидные молибденитовые пром-
продукты содержали, %: 5,8—6,3 Мо; 0,4 WO3; 6—9 Си; 12—
17 Fe; 21—27 S; 1,8 С; 0,01—0,014 Re; 26 SiO2; 1,7 СаО; 0,05—
0,07 As; 0,02—0,05 Р.
Авторы рекомендуют проводить окислительное выщелачива-
ние концентрата в растворах едкого натра. Опыты показали, что
при выщелачивании в растворах аммиака или соды для достиже-
ния приемлемой скорости' процесса его необходимо проводить
при температуре не ниже 200° С и парциальном давлении кисло-
рода 10—15 ат, тогда как при выщелачивании в растворах едкого
натра можно снизить температуру до 130—140° С и давление
кислорода до 1—2 ат. Это позволяет при работе с растворами
едкого натра заменить кислород сжатым воздухом, поскольку
общее давление в автоклаве будет не выше 8—10 ат. Кроме того,
применение аммиака для выщелачивания промпродуктов с высо-
ким содержанием меди приведет к переходу ее вместе с молибде-
ном в раствор, что усложняет последующую переработку раство-
ров. При применении растворов едкого натра хорошее раз-
деление меди и молибдена происходит в процессе выщелачи-
вания.
Помимо отмеченного выше, существенный недостаток раство-
ров аммиака — взрывоопасность смесей в системе аммиак—кисло-
род—водяной пар при высоких температурах и давлениях кисло-
рода.
127
Образующийся при выщелачивании молибдат-ион при pH
ниже 7 может реагировать с гидроокисями железа, меди и каль-
ция с образованием малорастворимых молибдатов:
ЗМоОГ + 2Fe (ОН)з Fe2 (МоО4)з + 6ОН(83)
МоО4“ + Си (ОН)2 СиМоО4 + 2ОН“, (84)
МоОГ + Са (ОН)2 СаМоО4 + 2ОН~. (85)
Константа равновесия этих реакций прйближенно может быть
представлена уравнением
= [ОН-]2
Г МоО^-'
Из этого следует, что каждой концентрации молибдена в рас-
творе соответствует некоторая минимальная концентрация ОН-
ионов, предупреждающая образование малорастворимого молиб-
дата [5, с. 82].
При концентрации молибдена 12—15 г/л, характерной для
растворов после выщелачивания промпродуктов приведенного
выше состава, конечная величина pH должна быть не ниже 8,5—9.
При выщелачивании в растворах едкого натра для обеспечения
необходимого конечного pH раствора достаточно избыточной кон-
центрации щелочи 3—5 г/л. Избыток реагента при использовании
соды значительно выше вследствие образования бикарбоната по
реакции соды с углекислотой, выделяющейся в процессе окисле-
ния и накапливающейся в газовой фазе автоклава:
MoS2 + %О2 + 3Na2CO3—> Na2MoO4 + 2Na2SO4 + ЗСО2,|
Na2CO8 + CO2 + H2Ozt2NaHCO3. J (86)
В табл. 30 сопоставлены оптимальные параметры и показатели
окислительного выщелачивания промпродуктов, а также конди-
ционного молибденитового концентрата в различных средах [4].
После выщелачивания кондиционного концентрата в раство-
рах соды или едкого натра при отношении ж : т = (5 4-6) : 1
автоклавная пульпа должна быть разбавлена водой примерно
на 20% в соответствии с растворимостью молибдата натрия в си-
стеме Na2MoO4—Na2SO4—Н2О при комнатной температуре [30].
Растворимость молибдата аммония в присутствии сульфата
аммония выше, чем молибдата натрия в присутствии сульфата
натрия. Поэтому разбавлять пульпу не требуется. Отношение
ж : т = 5 : 1 определяется расходом реагента — 25%-ного рас-
твора аммиака.
Из данных табл. 30 можно видеть, что извлечение в раствор
при применении соды и едкого натра одинаково высокое (98—
99%), однако несомненны преимущества едкого натра. Общее
128
ТАБЛИЦА 30
ПАРАМЕТРЫ И ПОКАЗАТЕЛИ ОКИСЛИТЕЛЬНОГО АВТОКЛАВНОГО
ВЫЩЕЛАЧИВАНИЯ [4]
Показатели Промпродукт Кондиционный концентрат
Раст воры
Na2CO3 NaOH Na2CO3 nh4oh
Расход реагента, % от стех иометр ического 115—120 103—105 115—120 135
Температура, °C ... . 200 130 200 200
Давление кислорода, ат 10—15 2 10—15 10—15
Давление общее, ат . . 30—35 5—6 30—35 50
Время обработки, ч . . 5—6 • 7—8 5—6 12—13
Отношение ж : т ... 5 : 1 5 : 1 6 : 1 5 : 1
Прямое извлечение в ра- створ молибдена и ре- ния, % 98—99 98—99 99,5 95—98
давление в этом случае в 6—7 раз ниже и, следовательно, во
столько же раз можно снизить толщину стенок автоклава (и соот-
ветственно стоимость автоклава). Вместо специальных пульповых
насосов высокого давления могут быть использованы стандартные
центробежные насосы. Возможна замена кислорода сжатым воз-
духом от обычных компрессоров, поскольку общее давление не
превысит 8—10 ат.
Некоторое затруднение представляет выбор материала, устой-
чивого при работе с растворами едкой щелочи в присутствии
кислорода, поскольку в этих условиях нержавеющие стали типа
1Х18Н9Т подвержены растрескиванию. Была, однако, установлена
возможность использования в этих условиях некоторых низколе-
гированных сталей [5, с. 237].
Для окислительного выщелачивания молибденового промпро-
дукта могут быть использованы автоклавы с механическим пере-
мешиванием. Перемешивающее устройство должно обеспечивать
хорошее диспергирование кислорода (воздуха) и засасывание его
из газовой фазы в раствор.
На рис. 48 показана предложенная технологическая схема
переработки медно-молибденового промпродукта [5]. В резуль-
тате окислительного выщелачивания в автоклаве по режимам,
описанным выше, в раствор полностью переходят молибден, ре-
ний, сера в форме натриевых солей молибденовой, рениевой и
серной кислот. Растворы содержат 12—15 г/л Мо; 170—180 г/л
Na2SO4; 20—40 мг/л Re, а также примеси соединений меди, воль-
фрама, кремния, мышьяка, сурьмы и фосфора. Первоначально
нейтрализацией раствора серной кислотой до pH % 8,5 и добав-
кой в раствор сульфата окиси железа (1—2 г/л) при температура
90—95° С осаждают гидроокись меди, кремневую кислоту, мышьяк
9 А. Н. Зеликман 129
и сурьму (в составе арсената и антимоната железа). Затем под-
кисляют раствор до pH = 2,5 ч-З и после охлаждения его до
О -Щ-5)° С выделяют до 75% ионов SOl” в составе десятиводного
сульфата натрия.
Для выделения молибдена из растворов с высоким содержанием
сульфата натрия непригодно осаждение молибдата кальция (осадки
Автоклавное восстановление
Фильтрация и пропь/вка
Осадок МоОг
Получение
петаллического
полибдено
Технический полибден
Раствор
Доизвлечение Но
Извлечение Re
Соль рения
Рис. 48. Технологическая
схема переработки молиб-
денитового промпродукта
автоклавным методом
(порошок )
сильно загрязнены сульфатом кальция). Ввиду этого был приме-
нен новый процесс — осаждение двуокиси молибдена восстановле-
нием ионов МоОГ молибденовым порошком в автоклаве при
200° С (см. стр. 140). Последующее восстановление двуокиси мо-
либдена водородом приводит к получению технического молибде-
нового порошка с содержанием примесей в нем, %: 2,1—2,5 Fe;
0,016—0,048 S; 0,17—0,26 Си; менее 0,01 As; менее 0,01 РЬ; следы
Sb, Sn, Р, Bi. Такой порошок может быть использован для леги-
рования сталей и получения сплавов с цветными металлами. Часть
130
порошка возвращают в процесс для осаждения двуокиси молиб-
дена.
Из маточного раствора после осаждения двуокиси молибдена
выделяют рений ионитно-адсорбционным методом.
Технологическая схема обеспечивает извлечение в готовый
продукт не менее 96% Мо и 85—90% Re, что выше, чем при ис-
пользовании обычной технологии переработки бедных молибде-
новых промпродуктов с получением молибдата кальция.
24 ОКИСЛЕНИЕ МОЛИБДЕНИТА РАСТВОРАМИ
ГИПОХЛОРИТА НАТРИЯ
Гипохлорит натрия, обладающий высоким окислительным
потенциалом, окисляет в щелочных средах все сульфидные мине-
ралы, в частности молибденит. Изучению условий окислитель-
ного выщелачивания растворами гипохлорита натрия молибдени-
товых руд и концентратов, а также некоторых других материалов
посвящено значительное число исследований [7—15].
Окисление молибденита гипохлоритом натрия описывается
следующей суммарной реакцией:
M0S2 -f- 9С1О~ + 6ОН— — МоС>4_ + 2SO4- + ЗНаО + 9СГ. (87 >
Изменение свободной энергии реакции весьма велико (AZ2980 к ==
= —343,6 ккал) и, следовательно, эта гетерогенная реакция
практически необратима.
Кинетику окисления молибденита гипохлоритом натрия иссле-
довали Иорданов и автор [10] на плотных цилиндрических образ-
цах молибденита, полученных прессованием под давлением 12т/с;и2
порошка чистого минерала. Опыты проводили в интервале темпе-
ратур 20—80° С при концентрации раствора гипохлорита 15—
60 г/л, интенсивно перемешиваемого мешалкой (от 300 до
600 об!мин). Контролировали нарастание во времени концентра-
ции молибдена в растворе и убыль гипохлорита. Скорости выра-
жали в г - ион/(см2' сек).
Молибденит окисляется с постоянной (не изменяющейся во*
времени) скоростью, возрастающей с повышением температуры
(рис. 49 и 50). Энергия активации процесса Е — 5250 кал/моль.
Скорость реакции линейно возрастает с увеличением концентра-
ции гипохлорита натрия (рис. 51) и мало зависит от концентрации
свободной щелочи в растворе.
Расход гипохлорита при окислении молибденита приблизи-
тельно соответствует теоретически необходимому по написанной
выше реакции.
Окисление молибденита протекает через стадию образования
тиосульфата 520з“, концентрация которого во времени проходит
через максимум и к концу процесса снижается до нуля.
9* 13®
При использовании почти всего гипохлорита натрия на по-
верхности образца молибденита наблюдаются пленки фиолето-
вого или коричневого цвета с фиолетовым оттенком, вероятно,
Время, мин
Рис. 49. Зависимость скорости
окисления молибденита гипо-
хлоритом натрия от времени.
Условия: концентрация NaClO
40 г/л\ скорость перемешивания
300 об/мин
Рис. 50. Зависимость ло-
гарифма константы ско-
рости (Д) окисления мо-
либденита гипохлоритом
натрия от обратной тем-
пературы .
отвечающие низшим окислам Мо4Оп и МоО2, которые могут обра-
зоваться в результате вторичного взаимодействия ионов МоО^
с молибденитом:
ЗМоОГ + M0S2 + 2ОН— Мо4Оц + S2O1~ + 2Н+.
(88)
О 10 20 30 ио 50 60
Кон цент рация Na СЮ, г/л
Рис. 51. Зависимость ско-
рости окисления молибденита
от начальной концентрации
гипохлорита натрия. Усло-
вия: t = 20° С; скорость пе-
ремешивания ЗООоб/лшн
Из опытов с плотными образцами
была вычислена средняя линейная ско-
рость окисления молибденита при 20° С,
равная 0,00045 мм/мин. Рассчитанное
по этой величине время полного окисле-
ния частиц молибденита различной
крупности близко к определенному
экспериментально. В растворе гипохло-
рита натрия концентрации 30 г/л при
20° С установлено следующее время
окисления:
Размер частицы, мм . 0,84 0,25 0,11 0,07
Время, мин.......... 960 280 218 75
Поскольку окисление растворами
гипохлорита натрия представляет инте-
рес преимущественно для бедных молибденовых концентратов
или руд, обычно содержащих сульфиды меди и железа, необхо-
димо рассмотреть их взаимодействие с гипохлоритом натрия.
132
Эти сульфиды окисляются гипохлоритом в щелочном растворе
с образованием гидроокисей.
Известно, что в присутствии соединений меди, кобальта, же-
леза, в частности гидроокисей этих элементов, гипохлорит натрия
в щелочной среде каталитически разлагается в основном с выде-
лением кислорода по реакции [16—18]
NaClO -> NaCl + Х/2О2, (89)
тогда как реакция образования хлората
3NaC10 — 2NaCl + NaC103
(90)
протекает лишь в незначительной степени. У хлората натрия
окислительные свойства в щелочных растворах мало выражены.
Прокопчик [17] установил, что каталитическое разложение
гипохлорита в щелочных растворах в присутствии меди обуслов-
лено образованием и разложением соединений трехвалентной
меди — куприатов. Причем здесь происходит и гомогенный, и
гетерогенный катализ.
Механизм гомогенного каталитического действия можно пред-
ставить реакциями:
2Си (ОН)2- + СЮ- + Н2О 2Си (ОН)? + СГ + 2ОН-
2Си (ОН)? + 2ОН- 2Си (ОН)2- + Н2О + Х/2О2
сю- - СГ +
Обе реакции-стадии необратимы и в щелочной среде ведут к раз-
ложению гипохлорита с выделением кислорода. .
Гетерогенное каталитическое разложение гипохлорита свя-
зано с образованием неустойчивых гидроокисей меди высшей
валентности:
2Cu (ОН)2 + СЮ" + Н2О 2Cu (ОН)3 + СГ
2Си (ОН)3 — 2Сп (ОН)2 + Н2О + 1/2О2
сю- -.СГ + ^Оз,
Скорость каталитического разложения гипохлорита в присут-
ствии гидроокиси меди сравнительно медленна при 20° С; с темпе-
ратурой она растет и зависит от количества добавленной гидро-
окиси меди (рис. 52) [19].
При. 80° С каталитическое разложение гипохлорита протекает
быстро, поэтому его расход значительно выше теоретически необ-
ходимого для окисления сульфидов, так как часть выделяюще-
гося при разложении гипохлорита кислорода удаляется из рас-
твора и не участвует в процессе окисления. Поэтому обрабаты-
вать концентраты растворами гипохлорита натрия целесообразно
при температурах не выше 40° С, когда каталитическое разложе-
133
ние гипохлорита протекает медленно и его расход близок к тео-
ретически необходимому для окисления сульфидов.
Разложение растворами гипохлорита натрия в щелочных сре-
дах изучали применительно к труднообогатимым молибденитовым
рудам и медно-молибденовым промпродуктам.
Щелочные растворы гипохлорита для практических целей
приготовляют, пропуская хлор в концентрированный раствор
щелочи с последующим разбавлением раствора до нужной кон-
центрации по гипохлориту.
Кокс и Шелингер [8] исследовали условия извлечения молиб-
дена из бедной молибденовой руды (содержащей 0,015% MoS2)
Время, мин
Рис. 52. Разложение гипохло-
рита натрия во времени при
и флотационного медного концентрата
(1,05% MoS2). Авторы рекомендуют
проводить окислительное выщелачива-
ние 3%-ным раствором гипохлорита
натрия в слабощелочной среде (pH =
= 10) при комнатной ” температуре.
В лабораторных опытах выщелачивание
проводили методом перколяции. Через
слой 4000 г руды и 1000 г концентрата
пропускали 2000 мл 3%-ного раствора
гипохлорита со скоростью 70 мл!мин,
различных температурах что отвечает времени выщелачивания
в присутствии гидроокиси зд мин. при этом в раствор извле-
МеДИ ПСиО, ЗеНТРаЦИИ кали 93,3% Мо из руды и 90,6% Мо
1 — юс- 2 - 50 из концентрата. Полученные бедные
растворы пропускали через колонку
с сильноосновной смолой IRA-400 в С1"-форме. Емкость смолы
0,24 г МоО1~/г смолы. Со [смолы молибден [элюировали 8%-ным
раствором едкого натра. Общее извлечение молибдена в элюаты
составляло 93 и 90,6% из руды и концентрата соответственно.
В результате ионообменного концентрирования могут быть
получены растворы с содержанием 40—50 г/л Мо, из которых
выделяется молибдат кальция или молибденовая кислота (нейтра-
лизацией раствора азотной кислотой).
При обработке бедных по молибдену материалов расход гипо-
хлорита натрия против теоретически необходимого для окисления
молибденита очень велик (примерно 24-кратный избыток).
Авторы не приводят данных, в какой мере расходуется гипо-
хлорит на окисление сульфидов меди и железа, содержащихся
в обрабатываемом сырье, и о возможности снижения расхода
гипохлорита вследствие циркуляции раствора.
Монев и Мирева [7] приводят результаты лабораторных опы-
тов выщелачивания молибдена щелочными растворами гипохло-
рита натрия из медного концентрата следующего состава, %:
22,5 Си; 40,7 Fe; 0,35 Мо; 2,8 SiO2.
134
Авторы получили максимальное извлечение молибдена 96,8%
при 80° С и продолжительности выщелачивания 60 мин. Расход
гипохлорита значительно выше необходимого для окисления суль-
фидов. Это объясняется высокой температурой обработки, при
которой часть гипохлорита каталитически разлагалась, не всту-
пая в реакцию окисления.
Условия обработки медно-молибденовых промпродуктов рас-=
творами гипохлорита натрия изучены в работах [13, 14].
Концентраты содержали, %: 23,2 Мо; 8,9 Си; 14,5 Fe; 31,2 S;
8,3 SiO2; 1,0 СаО; 1,5 А12О3; 0,0056 Re. Около 86% частиц кон-
центрата имело размер меньше 0,06 мм.
Скорость извлечения молибдена в раствор сильно возрастает
с повышением температуры от 20 до 60° С. Однако автор реко-
мендует проводить процесс при 20° С для сокращения расхода
гипохлорита. Оптимальная концентрация гипохлорита 30 а/л,
концентрация свободной щелочи 20—30 г/л. С увеличением кон-
центрации свободной щелочи скорость окисления молибденита
понижается. Это объясняется уменьшением окислительного по-
тенциала гипохлорит-иона с возрастанием щелочности.
Рекомендуемое отношение т : ж = 1 : 100. Такое большое раз-
бавление обусловлено низкой концентрацией гипохлорита натрия
(при концентрации 30 г/л NaClO в указанном разбавлении расход
гипохлорита составляет 150—200% от теоретически необходимого
для окисления молибденита). При оптимальных режимах за 3 ч
достигается высокое извлечение молибдена в раствор (96—98%).
Степень извлечения молибдена в раствор из медно-молибде-
нового промпродукта выше степени окисления сульфидной серы,
содержащейся в концентрате. Из этого следует, что молибденит
окисляется гипохлоритом с большей скоростью, чем сульфиды
меди и железа. Это объясняется образованием на частицах суль-
фидов меди и железа пленок нерастворимых продуктов окисления
(гидроокисей), тормозящих протекание процесса.
Из получаемых бедных по молибдену растворов (2—3 г/л)
автор рекомендует Извлекать молибден сорбцией на смолах.
В работе [12] изучалось применение гипохлорита для перера-
ботки бедных медно-молибденовых промпродуктов, содержащих
4,6—5,8% Мо.
Для удаления флотореагентов, препятствующих смачиванию
минералов раствором, промпродукты предварительно высушивали
при 200° С. Концентраты обрабатывали содовым раствором гипо-
хлорита.
Авторы отмечают, что при увеличении концентрации гипохло-
рита с 30 до 275 г/л (при сохранении избытка гипохлорита 400%
от теоретического для окисления молибденита) степень разложе-'
ния молибденита понижается. Поэтому остановились на концен-
трации гипохлорита 30 г/л и расходе гипохлорита 400—500% от
необходимого для окисления MoS2. Это отвечает отношению
135
т : ж = 1 : (43 4-60). Высокая степень извлечения молибдена в рас-
твор получена при указанных выше условиях и температуре обра-
ботки 50° С. Растворы содержали 0,8—1,5 г!л Мо и 10—15 г!л
ионов SO4~- Для извлечения из них молибдена использована
сорбция на слабоосновных анионитах с последующим элюирова-
нием 10%-ным раствором аммиака.
В работах [15, 59] изучен гипохлоридный вариант выщела-
чивания молибдена из хвостов и промпродуктов обогащения,
содержащих 1,3—4,8% Мо и 8,9—9,2% Си, который заключается
в том, что хлор подают в содовую пульпу обрабатываемого мате-
риала. Пульпа (т : ж 1 : 10-г-1 j 15) должна содержать значи-
тельный избыток соды .(250% от стехиометрического количества),
обеспечивающий конечную ее концентрацию не ниже 3—5 г/л.
Выщелачивание проводят при 20° С с пропусканием хлора
в течение 2 ч, что обеспечивает извлечение в раствор 90—95% Мо»
Расход хлора лишь несколько выше теоретического, равного
6,66 кг хлора на 1 кг молибдена. Из растворов молибден извлекают
ионообменным способом. Этот вариант проверен в полупромыш-
ленных масштабах.
Преимущество применения гипохлорита натрия для выщела-
чивания молибдена из бедных сульфидных концентратов и руд —
высокая избирательная способность реагента к окислению молиб-
денита и достижение полного извлечения молибдена в раствор
при низких температурах обработки (20—40° С). Однако суще-
ственное препятствие — высокий расход гипохлорита. Теорети-
чески необходимо затрачивать 9 моль гипохлорита на 1 моль MoS2
(или 7 кг гипохлорита на каждый килограмм молибдена в мате-
риале). Практический расход в 1,5—2 раза выше. Между тем стои-
мость гипохлорита (определяемая стоимостью хлора) относи-
тельно высокая. По мнению авторов работы [11], экономика
гипохлоритного выщелачивания будет благоприятна при условии
регенерации гипохлорита. Отмечается возможность осуществления
регенерации электролитическим способом при наличии дешевой
электроэнергии.
25. ДРУГИЕ СПОСОБЫ ОКИСЛЕНИЯ МОЛИБДЕНИТА
Кроме рассмотренных выше, изучены другие варианты окис-
лительного выщелачивания молибденита в щелочных и кислых
растворах:
окисление в щелочных средах перманганатом калия [9],
кислородом в присутствии медно-аммиачного катализатора [22],
окисью меди в автоклаве при повышенных температурах [23, 24];
окисление в кислых средах (серная, соляная кислоты) — хлора-
том калия или натрия, перманганатом калия, пиролюзитом,
азотной кислотой, растворами нитрозы, гипохлоритом кальция
[9, И, 20, 21].
136
Действие различных окислителей сравнивается в работах
Бхаппу, Рейнольдса и др. [И], окисление в сернокислых раство-
рах детально изучено Собиняковой с сотрудниками [20, 21].
Окисление в сернокислых растворах. Извлечение молибдена из
бедных продуктов обогащения обработкой их сернокислыми рас-
творами с окислителями перспективно для промышленного ис-
пользования.
В табл. 31 сопоставлено окисление молибденита и извлечение
молибдена в раствор при действии различных окислителей в серно-
» 4,
ТАБЛИЦА 31
СРАВНЕНИЕ ПОКАЗАТЕЛЕЙ ВОЗДЕЙСТВИЯ РАЗЛИЧНЫХ
ОКИСЛИТЕЛЕЙ НА МОЛИБДЕНИТ В СЕРНОКИСЛЫХ РАСТВОРАХ
[21] (Z = 90-т-95о С, ВРЕМЯ 3 ч)
Показатель < КСЮз HNO3 Нитроза* СаС1 (ОС1) КМпО4 МпО2
Расход окислителя, % от теоретического . . . 150 105—130 - 200 150 730
Расход окислителя, г/г MoS2 4,14 2,5—3,12 11,2—13,4 28,6 5,0 8,88 36,0
Концентрация H2SO4, % 10,0 20—25 ** 25—30 ** 5,0 10—20
Отношение ж : т ... 350 50 350 1000 1000 350
Окисление без абразива, % 100 92—100 100 26 30,4
Окисление с абразивом (песком), % 100 92—100 100 100 100 4—6,4
Извлечение молибдена в раствор, % .... 100 92—100 100 100 13,3 0,2
* Использовали нитрозу состава 88% H2SO4 и 11% окислов азота.
** Указана концентрация смеси кислот H2SO4 : HNO3 = 75 : 25.
кислой среде в оптимальных условиях. По эффективности окисле-
ния молибденита и растворения молибдена окислители могут быть
расположены в следующий ряд:
КСЮз > HNO3 > нитроза > CaCl (ОС1) > КМпО4 > МпО2.
Окисление гипохлоритом кальция и перманганатом калия
тормозится твердыми продуктами реакции — CaSO4 (в случае
гипохлорита) и труднорастворимыми марганец-молибденитовыми
соединениями. Добавки в пульпу абразива (песка), механически
снимающего пленки продуктов реакции, способствуют окислению
(табл. 30). Однако и в этом случае степень перехода молибдена
в раствор при использовании КМпО4 остается низкой вследствие
связывания молибдена в нерастворимые соединения.
Степень окисления молибденита гипохлоритом кальция по-
нижается с возрастанием концентрации серной кислоты, что также
137
объясняется увеличением количества образующегося сульфата
кальция, тормозящего процесс окисления [25].
Таким образом, в сернокислых средах перспективные окисли-
тели — хлораты калия или натрия и азотная кислота.
Окисление хлоратом приближенно описывается уравнением:
MoS2 + ЗСЮГ + ЗН2О — НМоОГ + 2HSO? + ЗСГ + ЗН+. (91)
Расход хлората по сравнению с теоретически необходимым
увеличивается из-за присутствия в обрабатываемом материале
других сульфидов, а также
удаления частично обра-
зующихся продуктов вос-
становления хлората —
хлора и С1О2 [11 ].
концентрация NaCl03t г/50 мл
Рис. 54. Влияние концентрации NaC103
на скорость растворения молибденита
при концентрации H2SO4 2% (объемн.)
Рис. 53. Влияние концентра-
ции H2SO4 на скорость рас-
творения молибденита при
концентрации 5% NaClOj
Как видно из рис. 53 и 54, скорость выщелачивания молибде-
нита находится в линейной зависимости от концентрации серной
кислоты и хлората натрия.
При обработке руды или бедных промпродуктов молибден из
получаемого разбавленного раствора можно извлекать ионным
обменом, экстракцией органическими растворителями или сорб-
цией на углях.
Окисление в щелочных растворах. Среди вариантов окисления
молибденита в щелочных растворах несомненный интерес пред-
ставляет окисление кислородом в присутствии катализатора —
медно-аммиачного комплекса [Си (NH3)4]2+, предложенное Соби-
няковой с сотрудниками [22, 25]. Авторы обрабатывали силикат-
ные руды, содержащие 0,2% Мо (~50% сульфидного молибдена)
растворами едкого натра, в которые добавляли гидроокись аммо-
ния и сульфат меди.
13«
Было установлено, что в присутствии медно-аммиачного ком-
плекса ускоряется окисление молибденита кислородом воздуха,
барботирующим через пульпу при температуре 85—90° С. При
концентрации 40—60 г!л NaOH, расходе аммиака 4—8 кг/т руды,
CuSO4 3—4 кг/т руды при отношении т : ж = 1 : 1 за 6—12 ч
в раствор извлекается 91—95% Мо.
Преимущества процесса — возможность окисления кислоро-
дом воздуха при нормальном давлении (исключается обработка
в автоклаве). Существенный недостаток — использование дорогого
реагента (CuSO4) и низкая скорость окисления (продолжитель-
ность окисления 12 ч).
В работах [23, 24] предложено окислять молибденит в медно-
молибденовом промпродукте (15—20% Мо, 3—4% Си) окисью
меди в растворе NaOH.
При оптимальных условиях (температура 200—250° С, т : ж =
= 1:3 — 5, расход окиси меди 60% от массы концентрата;
время обработки в автоклаве 3—4 ч, концентрация NaOH 300 г/л)
в раствор извлекали до 80% Мо. Для столь богатого концентрата
такое извлечение нельзя признать удовлетворительным.
139
ГЛАВА VII
СПОСОБЫ ВЫДЕЛЕНИЯ МОЛИБДЕНА
ИЗ СЛОЖНЫХ И БЕДНЫХ РАСТВОРОВ
В предыдущих главах освещены способы выделения молибдена
из аммиачных растворов в форме полимолибдатов (тетрамолиб-
дат, парамолибдат) и осаждение молибдата кальция из растворов
молибдата натрия. Они применимы лишь к достаточно чистым
растворам с высокой концентрацией молибдена.
Ниже рассмотрены другие технологические варианты извле-
чения молибдена из различных растворов, а также ионообменные
и экстракционные методы извлечения и концентрирования мо-
либдена.
26. ОСАЖДЕНИЕ ДВУОКИСИ МОЛИБДЕНА
Из растворов молибдата натрия с относительно высокой кон-
центрацией молибдена (от 15 до 80 г/л) молибден большей частью
осаждают в виде молибдата кальция. Недостаток способа —
низкое содержание молибдена в техническом молибдате кальция
(44—45%) и значительное содержание в нем примесей (до0,5% S,
до 0,4% Р и As). Из растворов с высоким отношением SO4"? Мо
(например, получаемый при окислительном выщелачивании мо-
либденитовых концентратов в автоклавах или растворами гипо-
хлорита натрия) практически вообще невозможно получить мо-
либдат кальция с кондиционным содержанием примеси серы [29 L
Большую часть сульфат-иона можно отделить из нейтрального
раствора кристаллизацией Na2SO4- 10Н2О при температурах
—104-12° С [5]. Однако в системе Na2MoO4—Na2SO4—Н2О
имеются обширные области выделения изоморфных смесей [301,
что обусловливает сокристаллизацию части Na2MoO4 с сульфатом
натрия. Кроме того, соосаждается рений.
В поисках путей выделения технического молибденового про-
дукта достаточной чистоты и богатого по содержанию молибдена
был разработан способ осаждения двуокиси молибдена восстано-
140
влением раствора Na2MoO4 молибденовым порошком или водо-
родом при повышенных температурах и давлениях [6, с. 414;
27; 28].
Восстановление молибденовым порошком. В работах Соболя
[5, 6] было показано, что молибдат-ион легко восстанавливается
молибденовым порошком в слабокислой среде (pH = 2,5 4-3,0)
при температуре 200° С с выделением двуокиси молибдена по
реакции
2МоО1“ + Мо + 4Н + — ЗМоО21 + 2Н2О. (92)
Изменение свободной энергии реакции
AZ298* = — 67,7 ккал.
Отсюда константа равновесия
Л [н+]4[МоО2~]2 ‘
Таким образом, восстановление ионов МоО1“ порошком
молибдена термодинамически может протекать полно уже при ком-
натной температуре. Однако практически с достаточной ско-
ростью процесс идет при температурах выше 100° С, т. е. в авто-
клаве.
Для температур 200° С в работе [6, с. 4141 определены ориенти-
ровочные значения константы равновесия К от 1016 до 1020.
Реакция протекает полно при теоретическом расходе порошка
молибдена в интервале pH — 2 4-3.
При недостатке порошка молибдена конечные растворы окра-
шены в интенсивно синий цвет, обусловленный присутствием про-
межуточных окислов (Мо4Оп) и молибденовой сини в коллоидной
форме. Из этого можно заключить, что восстановление ионов
МоО4“ порошком молибдена проходит через промежуточные ста-
дии образования соединений с валентностью между +6 и +4.
Осаждение двуокиси молибдена из растворов с концентрацией
15—60 г!л Мо рекомендуется проводить при температуре 200° С
и начальном pH = 2,5 4-3,0. В этих условиях при теоретическом
расходе порошка молибдена за 1 —1,5 ч осаждается более 95,5% Мо.
Остаточное содержание молибдена в растворе 5—15 мг/л. При-
сутствие сульфата натрия вплоть до концентрации насыщения
при комнатной температуре (растворимость Na2SO4 34% при 25° С)
не влияет на показатели восстановления. Однако его предвари-
тельное выделение в составе десятиводного сульфата натрия
выгодно для сокращения объема растворов, поступающих на вы-
деление двуокиси молибдена в автоклаве.
* При расчете принято: для МоО2~ AZSJgs ” —204 ккал, для МоО2
— —120,77 ккал.
141
Поскольку в слабокислых растворах (pH = 2 ч-З) молибден
находится в составе ионов полимолибдатов, десятиводный сульфат
натрия в этих условиях кристаллизуется при охлаждении рас-
твора до 0 ч-(+5° С) без изоморфного соосаждения молибдена.
Осадки двуокиси молибдена, промытые противоточной репуль-
пацией в 1%-ном растворе NaCl (что обеспечивает хорошее от-
стаивание дисперсного осадка), отфильтрованные и высушенные
на воздухе, содержали, %: 64—68 Мо (или 85,5—91% МоО2);
и Ст. Присутствие последних
Время, у
Рис. 55. Степень осаждения дву-
окиси молибдена во времени при
различных значениях pH раствора:
Кривая
начальное конечное
1 2 * 5,76
2 3 6,0
3 5 6,24
4 7 7,87
Концентрация молибдена в исход-
ном растворе 40 а/л, t = 200° С,
Рн2 ~ 40 ат
трех компонентов обусловлено коррозией хромоникелевой стали,
из которой изготовлен автоклав. Покрытие автоклава и мешалки
материалом, устойчивым против действия серной кислоты, должно
привести к снижению содержания этих примесей.
Восстановление водородом под давлением. Условия выделения
двуокиси молибдена из растворов молибдата натрия восстановле-
нием водородом изучали автор и Ляпина [27, 28; 5, с. 105].
Реакция восстановления приближенно выражается уравне-
нием 1
Мос>4-
+ Н2 = МоО2 + 2ОН~.
(93)
Константа равновесия реакции
к = [ОН-]2
[МоО2-]рНг •
Естественно, что изменение концентрации (активности) водо-
родных ионов в растворе должно существенно влиять на проте-
1 В слабокислых растворах (pH < 5) молибден присутствует в растворах
также и в составе анионов изополикислот Мо4О^~, Мо7О2^“, МобОэд“и др. Однако
это не изменяет общего хода рассуждений.
142
кание процесса, причем в результате восстановления pH раствора
должно возрастать. Это подтверждают данные, приведенные на
рис. 55, где отчетливо видно резкое влияние начального значения
pH раствора на степень осаждения молибдена. По мере протека-
ния реакции pH раствора возрастает, что ведет к замедлению про-
цесса [27].
Было найдено, что небольшие
добавки порошка молибдена при
pH — 2 4-3 существенно увеличивают
скорость восстановления водородом
(табл. 32). Это объясняется быстро
протекающим восстановлением ионов
МоО1“ порошком металлического
молибдена с образованием частиц
двуокиси молибдена, являющихся
затравкой для выделения МоО2 по
основной реакции восстановления
Рис 56. Зависимость константы
скорости восстановления молиб-
дат-иона водородом от парци-
ального давления водорода.
(/С, a* ami (см3* мин). В исходном
растворе 40 г Мо/л, t = 200° С)
водородом. Для ускорения процесса
достаточно ввести в исходный рас-
твор 3 г!л порошка молибдена.
При изменении парциального
давления водорода с 10 до 60 ат
скорость восстановления возрастает
прямо пропорционально корню квадратному -из давления водо-
рода (рис. 56). Из этого следует, что водород участвует в кон-
тролирующей процесс стадии в атомарной форме.
С повышением температуры возрастает скорость осаждения
двуокиси молибдена и крупность ее частиц. При 100 и 150° С
осадки мелкокристаллические и плохо фильтруются, при 200° С
выделяются крупнозернистые осадки.
Ниже приведены средние значения константы скорости вос-
становления при различных температурах, определенные для
ТАБЛИЦА 32
СТЕПЕНЬ ВЫДЕЛЕНИЯ Мо02 ИЗ РАСТВОРА Na2MoO4 БЕЗ ЗАТРАВКИ
И В ПРИСУТСТВИИ ЗАТРАВКИ (КОНЦЕНТРАЦИЯ Мо В РАСТВОРЕ
43 г/л, ртт = 40 ат; pH = 2; t = 200° С)
Н2 нач
Время опыта, ч Степень осаждения, %
без затравки с затравкой ~3 г/л
1 17 57
2 36 71
4 51 80
6 65 01
143
начального периода протекания процесса (исходные растворы
содержали 45,08 г!л Мо, рНиач = 2, давление водорода 60 ат):
Температура, °C....... 100 150 175 200
К, г-ат Мо/(см3-мин) • 10“4 0,042 0,118 0,169 0,244
С повышением температуры от 100 до 200° С скорость процесса
возрастает в 5,75 раза. Рассчитанная величина кажущейся энер-
гии активации Е = 6150 кал!моль.
Скорость выделения МоО2 находится в линейной зависимости
от начальной концентрации молибдена в растворе.
Время, необходимое для полного выделения молибдена, возра-
стает с увеличением концентрации молибдена. Так, при прочих
одинаковых условиях (рНнач = 2; t = 200° С, рн = 50 ат, за-
травка 3 г!л порошка Мо) из раствора с концентрацией 5,33 г/л Мо
полное осаждение достигается за 30 мин, а для раствора с концен-
трацией молибдена 43,6 г/л 3,5 — 4 ч.
В первые минуты от начала восстановления раствор окра-
шивается в темно-синий цвет, затем по мере выделения двуокиси
молибдена обесцвечивается. По всей вероятности, восстановление
протекает через стадию образования молибденовой сини. Из-
вестно, что молибденовая синь образуется при действии восстано-
вителей (SO2, H2S, Zn и др.) на кислые растворы молибдатов.
Молибденовая синь представляет собой соединение, близко от-
вечающее формуле Мо5О14- х И 2О (или по другим данным Мо8О23 X
X хН2О). Молибденовая синь находится в растворе в коллоид-
ном состоянии и, обладая большой удельной поверхностью,
адсорбирует растворенный водород, который восстанавливает ее
до двуокиси молибдена.
Рекомендуемые в работах [27, 281 оптимальные условия оса-
ждения двуокиси молибдена: температура 2003 С, давление во-
дорода рНг = 60 ат, затравка 3 г/л порошка Мо, рНнач = 2 -ч-З.
Время восстановления 1—4 ч в зависимости от исходной концен-
трации молибдена в растворе.
В этих условиях в результате восстановления выделяется
крупнозернистый черный осадок с синевато-коричневым оттен-
ком. Кристаллическая структура осадка совершенно идентична
структуре двуокиси молибдена. По химическому составу осадки
также близки к двуокиси молибдена (содержат 74,6—74,7% Мо).
Осадки практически нерастворимы в концентрированных серной
и соляной кислотах, а также в растворах щелочи. Азотная кислота
быстро их окисляет до молибденовой кислоты.
Недостаток выделения двуокиси молибдена восстановлением
водородом, по сравнению с описанным выше восстановлением
молибденовым порошком, — необходимость' использования авто-
клава, рассчитанного на рабочее давление 75—80 ат (давление
водорода 60 ат + давление паров воды при 200° С 15 ат), тогда
М4
как при восстановлении молибденовым порошком общее давление
в автоклаве 15—20 ат. Однако во втором случае требуется вос-
становление двуокиси молибдена водородом при температуре
850—900° С для получения молибденового порошка.
Отделение молибдена от вольфрама селективным восстановле-
нием водородом. При переработке молибденовых и вольфрамовых
концентратов часто получают промежуточные продукты и хвосты,
содержащие оба металла в различных соотношениях. После
вскрытия таких материалов с использованием щелочных реаген-
тов получают растворы молибдата и вольфрамата натрия.
Исследования показали [28], что имеются существенные отли-
чия в химизме и условиях восстановления растворов Na2WO4
и Na2MoO4 водородом.
Вольфрамат натрия восстанавливается до малорастворимых
соединений типа вольфрамовых бронз по реакции
5Na2WO4 + Н2 + ЗН2О -> Na2O-4WO3- WO2 + 8NaOH, (94)
тогда как молибдат натрия, как описано выше, восстанавливается
с образованием двуокиси молибдена по реакции (93).
Вольфрам в виде вольфрамовой бронзы выделяется с высокой
скоростью без затравки при температуре 200° С и давлении водо-
рода 20 ат, что существенно ниже оптимального давления водо-
рода для выделения МоО2 (60 am).
Различия в условиях и скорости осаждения соединений воль-
фрама и молибдена из растворов при восстановлении водородом
позволяют селективно выделять первоначально вольфрам, а затем
молибден.
ТАБЛИЦА 33
ПОСЛЕДОВАТЕЛЬНОЕ ОСАЖДЕНИЕ ВОЛЬФРАМА И МОЛИБДЕНА
ИЗ РАСТВОРОВ, СОДЕРЖАЩИХ 41 г/л Мо и 5 г/л W
- Состав раствора Осаждено, % к исход- ному Состав осадка, %
Раствор после осаждения 1 + о £ £ о S
о £ О'* о £ о £
Вольфрама (1,5 ч\ р н2 = %® апг) • • Молибдена (^н2 = = 60 ат, 4 ч\ за- травка 3 г/л Мо) 38,91 0,3 0,5 0,3 1,27 50 90,0 4,0 5,0 93,6 40,0 0,63 46,75 73,8 46 —0,85
1 t = 200° С, pH = 2.
10 А. Н. Зеликман
145
В табл. 33 приведены условия и результаты последовательного
осаждения вольфрама, а затем молибдена. Из исходного раствора,
содержащего 41,0 г/л Мои 5,0 г/л W [W/(Mo + W) = 10,9%],
получена двуокись молибдена, в которой отношение W/(Mo +
+ W) 0,85% при извлечении молибдена в осадок ~94%.
Этот способ, хотя и не дает глубокой очистки молибдена от воль-
фрама, обеспечивает отделение большей его части и, несомненно,
заслуживает внимания, так как способы отделения вольфрама от
молибдена в случае присутствия вольфрама в растворах молибдата
натрия не разработаны.
27. ВЫДЕЛЕНИЕ ПОЛИМОЛИБДАТА АММОНИЯ ИЗ РАСТВОРОВ
МОЛИБДАТА НАТРИЯ
Как было показано Попильским \ из растворов молибдата
натрия при введении в них ионов аммония (добавление NH4C1
или NH4OH) в определенном интервале pH можно выделить мо-
либден в составе полимолибдата аммония с незначительной, при-
месью натрия. Это объясняется различиями в растворимости
аммонийных и натриевых полимолибдатов. В последующем способ
был более детально изучен Шапиро [33, 34]. Высокая степень
осаждения молибдена (более 99%) наблюдается в интервале
pH = 1,5 4-2 из растворов с концентрацией не ниже 60 г/л Мо
при введении в раствор хлористого аммония в молярном отно-
шении NH4C1 : МоО3 =1:1 или несколько больше (примерно
0,87 кг NH4C1 на 1 кг Мо). Кристаллические осадки близки по
составу к тетрамолибдату (NH4)2O-4MoO3-2H2O и содержат не-
большую (0,3—0,5%) примесь Na2O. По всей вероятности, примесь
натрия • находится в кристаллической решетке, что обусловлено
изоморфным соосаждением полимолибдата натрия с тетрамо-
либдатом аммония.
Осаждать тетрамолибдат аммония рекомендуется на холоду
при перемешивании раствора и введении затравочных кристаллов
тетрамолибдата аммония. В этих условиях полное осаждение
молибдата достигается примерно за 6 ч. Осадки хорошо промы-
ваются и фильтруются. Присутствие сульфата натрия в растворах
(до 200 г/л) не снижает степени выделения молибдена в виде тетра-
молибдата. Однако соль содержит 0,05—0,06% S (по отношении
к молибдену).
Для очистки от примесей натрия и серы осадки тетрамолибдат;
подвергают перекристаллизации через аммиачные растворы с по
лучением обычного товарного продукта — парамолибдата аммо-
ния. Другой путь очистки состоит в обработке осадка тетрамолиб-
дата аммония 7%-ным раствором хлористого аммония, пред-
варительно нейтрализованного соляной кислотой до pH = 2
1Попильский Я. М. и др. Авт. свид. СССР № 113630 и 113474,
1957.
146
В результате обменной реакции с хлористым аммонием примеси
Na+ и SOt- переходят в раствор, их содержание в тетрамолиб-
дате аммония не превышает 0,01 % (по отношению к молибдену).
Описанный способ представляет интерес для выделения молиб-
дена из растворов молибдата натрия, получаемых при переработке
низкосортных концентратов, а также других молибденсодержащих
промышленных продуктов и отходов.
28. ОСАЖДЕНИЕ ТРИСУЛЬФИДА МОЛИБДЕНА
Осаждение трисульфида молибдена — простой и надежный
способ его выделения из бедных растворов, получаемых, например,
при переработке промпродуктов обогащения или непосредственно
руд (например, окисленных руд и концентратов) и содержащих
0,2—1 г!л Мо (см. стр. 106). Наиболее благоприятны для выделе-
ния трисульфида молибдена щелочные растворы, содержащие от-
носительно мало примесей катионов тяжелых металлов.
В такие раствооы первоначально добавляют раствор серни-
стого натрия, а затем их подкисляют до pH = 2 4-3. После на-
гревания раствора при 80° С выделяются хлопьевидные темно-
коричневые осадки:
Na2MoO4 -j- 4NaHS XNa2MoS4 + 4NaOH,
Na2MoS4 + H2SO4^-MoS3| + Na2SO4 + H2S.
4 । O’* Ci i ct
(95)
(96)
Приведенные реакции описывают процесс приближенно. В дей-
ствительности в растворе, кроме ионов сульфомолибдата MoSl-,
присутствуют ионы оксисульфомолибдатов MoOxS^~ (х + у =
— 4). Соответственно этому выпадающие осадки представляют
собой оксисульфид молибдена MoOxS3_x.
Более правильно реакцию осаждения трисульфида молибдена
представлять как реакцию гидролиза сульфомолибдат-иона:
MoSl- + 2Н2О Д2?—* MoS3 4- 2О1-Г + H2S. (97)
При pH = 2 -?3 реакция полностью сдвинута вправо. Образую-
щиеся ионы ОН“ связываются добавляемой кислотой.
Высокая степень осаждения (99—99,5%) из растворов с кон-
центрацией 0,2—1,0 г!л Мо обеспечивается при соблюдении сле-
дующих условий: избытке сернистого натрия 25—50% по отно-
шению к требуемому по реакции (избыток зависит от концентрации
молибдена в растворе), подкислении раствора серной кислотой
до pH = 2ч-3 (что отвечает концентрации —2 4-2,5 г!л H2SO4)
и при нагревании до 70—80° С примерно в течение 1—2 ч (см.
табл. 25).
Более полное осаждение обеспечивается в герметичном ап-
парате. Осадки трисульфида хорошо оседают и легко фильтруются.
10* . 147
Содержание в них молибдена колеблется от 20 до 35%, что зави-
сит от степени осветления раствора (содержания тонких взвесей,
например кремнезема или кремневой кислоты), а также избытка
сернистого натрия. При значительных избытках Na2S в осадках
содержится элементарная сера. После обжига при температурах
450—500° С содержание молибдена в продукте повышается до
50—55%. При низкотемпературном обжиге в условиях ограничен-
ного доступа воздуха происходит термическая диссоциация три-
сульфида молибдена с образованием дисульфида. Выделяющаяся
при этом сера взаимодействует с кислородом, содержащимся в суль-
фидном осадке. Если представить осадок оксисульфида форму-
лой %MoS3-г/МоО3, то протекающие при обжиге процессы можно
выразить реакциями:
MoS3MoS2 + V2S2, ' (98)
2МоО3 + 3 V2S2 — 2MoS2 + 3SO2. (99)
Таким образом, в результате низкотемпературного обжига
получают концентрат, содержащий сульфид молибдена. Отчасти
продукт содержит МоО2 и МоО3.
Проведение обжига трисульфида молибдена при более высо-
ких температурах и достаточном доступе воздуха с получением
трехокиси молибдена затруднительно вследствие высокой экзо-
термичности процесса, что ведет к перегревам и значительному
улетучиванию трехокиси молибдена.
Осаждение трисульфида молибдена широко используют при
переработке вольфрамовых концентратов как метод отделения мо-
либдена от вольфрама [35]. Вольфрамовые концентраты часто
содержат 0,5—1% Мо, а в некоторых шеелитовых концентратах
содержание молибдена составляет 4,5—5%, причем он находится
в форме изоморфной примеси в решетке шеелита (тырны-аузские
шеелитовые концентраты). При щелочных методах вскрытия
вольфрамовых концентратов (спекание с содой, разложение рас-
творами соды в автоклавах) получают растворы вольфрамата
натрия с концентрацией 1—10 г!л Мо (см. стр. 113). Из них мо-
либден может быть селективно выделен в виде трисульфида, что
объясняется различиями в условиях образования сульфосолей
молибдена и вольфрама.
Для образования сульфосолей молибдена достаточно добавить
в растворы лишь небольшой избыток сернистого натрия, тогда
как для перехода ионов WO4” в ионы WS1“ или оксисульфо-
ионы WOXS^“ необходима высокая концентрация ионов S2“"
в растворе.
Выделяемые из растворов вольфрамата натрия осадки три-
сульфида молибдена содержат 2—5% WO3, а общая потеря воль-
фрама с. осадком не превышает ~1%.
148
С целью снижения содержания вольфрама в осадках трисуль-
фида молибдена их растворяют в растворе сернистого натрия
и нейтрализацией раствора до pH = 2 4-3 снова осаждают три-
сульфид молибдена [35].
29. ОСАЖДЕНИЕ ФЕРРИМОЛИБДАТОВ
Удобным полупродуктом для выплавки ферромолибдена могут
служить осадки ферримолибдатов, которые выделяются из рас-
творов в определенном интервале pH.
Как показано в работе [11], степень извлечения молибдена
в осадок ферримолибдата и состав осадков сильно зависят от pH
раствора. Данные, приведенные в табл. 34, получены при смеши-
ТАБЛИЦА 34
ЗАВИСИМОСТЬ СОСТАВА ОСАДКОВ ФЕРРИМОЛИБДАТОВ ОТ pH
РАСТВОРА
pH осаждения Молярное отношение Содержание МоОз, % (на сухой продукт) Извлечение Мо в осадок, %
Ге2О3 МоО3 Н2О
1,11 1,0 4,15 13,10 79,0 27,7
1,53 1,0 4,14 9,60 77,4 89,0
1,57 1,0 4,03 9,50 77,0 89,4
2,18 1,0 3,54 8,02 76,1 94,4
2,32 1,0 3,30 6,49 73,6 >99
2,35 1,0 3,23 5,94 73,3 >99
2,41 1,0 3,10 5,47 72,3 >99
2,82 1,0 2,95 5,22 70,0 >99
вании раствора, содержащего 8 г/л Мо, и раствора, содержащего
3,12 г/л трехвалентного железа (молярное отношение Fe : Мо =
= 1:3) при контролировании значений pH раствора. В интервале
pH = 2,32 4-2,8 выделяются желтые осадки, содержащие 70—
74% МоО3 (в пересчете на сухой продукт) при степени выделения
молибдена из раствора более 99%.
Осажденные соединения не отвечают составу минерала ферри-
молибдита [Fe2 (МоО4)3-71/2Н2О], но представляют собой смеси
ферримолибдатов. До pH = 1,6 и осадках молярное отношение
Fe2O3 : МоО3 =1:4, что примерно отвечает молибдату со-
става FeOH* (НМоО4)2. В интервале pH = 2,4 4-2,8 отношение
Fe2O3 : МоО3 =1:3, осадки приближенно соответствуют со-
ставу Fe (ОН)2-(НМоО4). Практически осадки содержат смесь
этих ферримолибдатов, соотношение между ними зависит от pH
раствора.
При избытке ионов Fe3+ и pH >2,4 вместе с ферримолибда-
тами осаждается гидроокись железа, что снижает содержание МоО3
в продукте.
149
Если ферримолибдиты осаждают из раствора молибдата на-
трия, наиболее целесообразный способ проведения операции, по
всей вероятности, состоит в одновременном вливании в реактор
раствора молибдата натрия и раствора Fe2 (SO4)3, исходные pH
которых должны быть подобраны из такого расчета, чтобы под-
держивать pH раствора в пределах 2,5—3,0.
Недостатки осаждения ферримолибдитов — трудности поддер-
жания оптимального pH раствора (так как pH изменяется по мере
осаждения) и глинистый характер осадков, обусловливающий
трудности их фильтрации.
30. ИЗВЛЕЧЕНИЕ МОЛИБДЕНА ИЗ РАСТВОРОВ СОРБЦИЕЙ
НА ИОНООБМЕННЫХ СМОЛАХ И УГЛЯХ
Сорбцию на ионообменных смолах широко применяют в про-
мышленной практике для извлечения молибдена из различных
маточных растворов производства молибдата аммония и молиб-
дата кальция, растворов, получаемых при переработке бедного
сырья, а также для извлечения молибдена из рудничных вод.
Маточные растворы и растворы, получаемые при выщелачива-
нии бедного сырья, обычно содержат от 0,5 до 5 г!л Мо и значи-
тельные количества анионов SOI-, С1“-ионов и иногда ионов NOJT
(например, при разложении молибденита азотной кислотой). Со-
держание молибдена в рудничных водах очень низкое и колеб-
лется от 1 до 10 г Мо на 1 ж3 воды (или от 1 до 10 мг!л) [43].
Исследования Лурье и Филипповой [51], Бабко [48], Рябчи-
кова [49], Алимарина [50] показали, что молибден сорбируется
из растворов катионитами и анионитами. Однако применение катио-
нитов малоэффективно, так как катионы (МоО1+ и др.) присут-
ствуют в сравнительно кислых растворах (pH < 1), когда велика
конкуренция Н+-ионов. Кроме того, ъ производственных растворах
в значительных концентрациях присутствуют другие конкури-
рующие катионы (Na+, NH^, Cu2+ и др.).
Из слабокислых растворов молибден лучше всего поглощается
наименее совершенным катионитом — сульфоуглем, тогда как
синтетические катиониты типа СБС, Ку-1 и др. имеют весьма ма-
лую емкость по молибдену. Из этого следует, что поглощение мо-
либдена сульфоуглем преимущественно обусловлено молекуляр-
ной сорбцией, а не ионным обменом. Ряд экспериментов, под-
тверждающих эту точку зрения, провел Попов [37].
Для сорбции молибдена из промышленных растворов эффек-
тивно использование только анионообменных смол.
В многочисленных публикациях освещаются результаты ис-
пытаний сорбции молибдена на смолах различной основности и
структуры [37—40, 42—45, 47]. Характеристика некоторых из
них приведена в табл. 35.
150
ТАБЛИЦА 35
ХАРАКТЕРИСТИКА НЕКОТОРЫХ АНИОНООБМЕННЫХ СМОЛ [52]
Марка Характеристика основности Активные группы Обменная емкость, мг*экв!г по 1-н. НС1
АВ-16 Полифункциональный сильноосновной (СССР) =NH, hen и 15—20% пиридиновых групп — У 9,8—10,5
АВ-17 Монофункциональный сильноосновной (СССР) —N (СН3)3 4,3
AM Сильноосновной (СССР) -N (СН8)8 —
АМ-П То же -ЙС_> —
Амберлит IRA-400 Сильноосновной (США) -N (СН8)3 2,5—3,9
Амберлит IRA-410 То же N—СН2—СН2ОН Н3С СН8 3—3,9
Вофатит SBW Сильноосновной (ГДР) —N (СН3)з 3,5
ЭДЭ-10П Полифункциональный, среднеосновной (СССР) Вторичные и третичные аминогруппы и чет- вертичные аммоние- вые группы в радика- ле алифатического ряда (10—12%) —9,0
АН-1 Полифункциональный, слабоосновной (СССР) Первичные и вторичные аминогруппы, а также триазиновые кольца 4,2 (0,1-hJhCI)
15!
Продолжение табл. 35
Марка Характеристика ОСНОВНОСТИ Активные группы Объемная емкость, мг-экв1г по 1-н. НС1
АН-2Ф Полуфукциональный слабоосновной (СССР) Вторичные и третичные аминогруппы алифа- тического ряда =N, =NH 10,6
АН-9 То же То же 4,5 '
НО То же г То же 4,1
И сильноосновные, и слабоосновные смолы больщей частью
имеют максимальную сорбционную емкость по молибдену в слабо-
кислых средах в интервале pH от 5 до 2, т. е. в области существо-
вания в растворах полимерных анионов типа МО7О2Г, МО4О1Г
и др. (см. стр. 60). При повышении концентрации кислоты ем-
кость смол по молибдену снижается вследствие конкурирующего
действия аниона кислоты. Кроме того, снижение емкости в этом
случае объясняется деполимеризацией полианионов молибдена
и частичным переходом анионных форм в катионные.
Из нейтральных и слабощелочных растворов слабоосновные
аниониты практически не сорбируют молибден, тогда как сильно-
основные аниониты имеют достаточно высокую емкость при pH =
= 10-12.
Сорбционная емкость смолы по молибдену в большой мере зави-
сит от структуры смолы.
Для некоторых смол поглощение крупных полимерных ионов
молибдена встречает стерические затруднения, вызванные силь-
ным экранированием ионообменных групп или малыми размерами
ячейки.
Это иллюстрируется сопоставлением структуры и емкости по
молибдену (при сорбции из слабокислых растворов) трех сильно-
основных анионитов (табл. 36).
Низкой емкостью обладает смола АМП, в которой активный
азот сильно экранирован, с одной стороны, пиридиновым коль-
цом, с другой — высокомолекулярным углеводородным ради-
калом.
Наиболее высокая емкость у смолы AM, в которой азот слабо
экранирован (его окружают три метильные группы); промежу-
точное положение занимает смола IRA-410. При сорбции из слабо-
щелочного раствора емкость смол AM и АМ-П по молибдену одина-
152
ТАБЛИЦА 36
ОБМЕННАЯ ЕМКОСТЬ ПО МОЛИБДЕНУ СИЛЬНООСНОВНЫХ СМОЛ
РАЗЛИЧНОЙ СТРУКТУРЫ [40, с. 127]
(Концентрация в исходном растворе 1,0 г/л Мо, 0,1 моль/'л H2SO4)
Марка
смолы
Строение смолы
Объемная
емкость,
мг/г
AM
IRA-410
АМ-П
R-CH2-N (СН3)з
СН3
R— СН2—N— СН2—СН2ОН
530
380
98
кова, так как в этом случае молибден сорбируется в виде простого
молибдат-иона МоО|“ и структурные особенности этих двух смол
не влияют на сорбируемость молибдена.
Лимитирующей стадией сорбции молибдена обычно является
диффузия ионов внутри зерен смолы, причем величины коэффи-
циентов диффузии и констант скорости обмена зависят от струк-
туры анионита (табл. 38).
Так, для среднеосновного анионита ЭДЭ-10П величина коэф-
фициента диффузии и константы скорости обмена на три порядка
выше, чем для смолы АН-1 (при одинаковых размерах набухших
частиц). Следовательно, при сорбции молибдена на смоле ЭДЭ-10П
ТАБЛИЦА 37
ОБМЕННАЯ ЕМКОСТЬ ПОЕ И КОЭФФИЦИЕНТ РАСПРЕДЕЛЕНИЯ (К )
ПРИ СОРБЦИИ МОЛИБДЕНА АНИОНИТАМИ
[20]
(Концентрация молибдена 1,6 г/л, рН = 3)
Анионит ПОЕ, мг/г
марка основность
АВ-16 Сильная 394 20 615
ЭДЭ-1 Оп Средняя 288 12 954
НО ] { 261 4 369
АН-1 У Слабая 233 3 598
АН-2Ф J 1 181 1 820
153
можно применить большие скорости фильтрации, а динамическая
емкость (емкость до проскока) на этой смоле будет в меньшей мере
зависеть от скорости потока, чем при сорбции на смоле АН-1.
Константа скорости обмена сильно зависит от размера частиц
смолы (табл. 38).,
ТАБЛИЦА 38
КИНЕТИЧЕСКИЕ КОНСТАНТЫ СОРБЦИИ МОЛИБДЕНА НА
АНИОНИТАХ (рН=3) [40, с. 132]
Анионит Средний радиус набухшей частицы г, см Константа скорости обмена В, сек"1 Коэффициент диффузии D, см2/сек
ЭДЭ-1 Оп АН-1 АН-1 0,044 0,01’8 0,037 2,2-Ю-з 2,9-10~6 6,82-10-® 4,35-10-’ 0,95-10-» 0,96-10-» 51
В работе Собиняковой с сотрудниками [201 определены значе-
ния полной обменной емкости и коэффициенты распределения мо-
либдена (Кр) между смолой и раствором для анионитов различного
типа (см. табл. 37)
К (Мо)тд
ЛР (Мо)р ’
где (Мо)тв — концентрация молибдена в смоле, мг!г (сух.);
(Мо)р — равновесная концентрация молибдена в растворе,
мг!мл.
Исходные растворы содержали 1,6 г!л Мо и имели pH = 3,
при котором значения /Ср и ПОЕ для всех смол максимальны.
Как видно из табл. 37, наиболее высокие параметры сорбции
молибдена получены для сильноосновной смолы АВ-16, несколько
меньше для среднеосновной смолы ЭДЭ-10П и еще ниже для слабо-
основных анионитов.
В промышленной практике часто предпочитают использование
слабоосновных или среднеосновных анионитов, хотя их емкость
ниже емкости сильноосновных смол. Это объясняется возмож-
ностью элюирования молибдена со слабоосновных смол 5—10 %-
ными растворами аммиака, тогда как сильноосновные смолы,
более прочно удерживающие сорбированные ионы молибдена,
требуют применения для элюации растворов щелочей. Кроме того,
некоторые слабоосновные смолы отличаются высокой избиратель-
ной способностью в отношении полианионов молибдена. К ним
относится, например, смола АН-1, которая имеет малую емкость
по отношению к сильноразбавленным серной и соляной кислотам
1М
и практически не поглощает железа из растворов с величиной
pH - 2,5-3,5.
Условия сорбции молибдена из маточных растворов производ-
ства молибдата аммония были изучены в работах [38, 40, 45].
Такие растворы содержат, г/л: 0,5—1,5Мо; до 0,3 Си; —2,0 Са;
~6,88R2O3; ~4,5Fe; 35—40СГ; ~7SO1-; pH = 2,5-3.
Маточный раствор, 0,5 -1,5г/л Мо, pH- 2,5-3
Рис. 57. Схема сорбционного извлечения молибдена из маточных растворов произ-
водства молибдата аммония
Для сорбции молибдена рекомендуются слабоосновные анио-
ниты АН-9 в С1_-форме [38] и АН-1 в С1“-форме'[40, с. 132].
Последний из слабокислого раствора (pH = 3) в незначительной
степени сорбирует железо и медь и обладает большей емкостью,
чем смола АН-9. При удельной нагрузке 2 и 3 объема раствора на
1 объем смолы в час емкость смолы АН-1 по молибдену составляла
312 и 270 кг!т соответственно. Молибден легко вымывается со
смолы 5—10%-ными растворами аммиака с получением растворов
с концентрацией 60—120 г!л Мо.
155
Технологическая схема процесса показана на рис. 57. Общее
извлечение молибдена при его сорбции на анионитах из маточных
растворов составляет 95—96%.
Автор с сотрудниками [39] исследовал сорбцию молибдена из
растворов, полученных при выщелачивании молибдена из бедных
окисленных концентратов растворами соды. Растворы содержали
ОД г/л Мо и 12,9 г/л SO1-. При сопоставлении емкости по молиб-
дену слабоосновных смол трех марок: АН-1, АН-9 и АН-2Ф
(табл. 39) было установлено, что в С1“-форме они имеют близкую
емкость по молибдену, равную 21—23%, тогда как в форме SO4~
высокой емкостью отличается только смола АН-1.
ТАБЛИЦА 39
ПОЛНАЯ ОБМЕННАЯ ЕМКОСТЬ ПО МОЛИБДЕНУ РАЗЛИЧНЫХ
СЛАБООСНОВНЫХ СМОЛ ПРИ ПОГЛОЩЕНИИ ИЗ РАСТВОРА Na2MoO4
С КОНЦЕНТРАЦИЕЙ Мо 0,4 г/л, рН=3 [39]
Марка смолы ПОЕ, % анионитов в форме
С1“
АН-1 21,2 21,7
АН-9 8,3 23,5
АН-2Ф 8,1 23,3
Из растворов указанного выше состава сорбцию на смоле АН-1
в форме SOr~ можно осуществлять при нагрузке 5 м3 раствора на
1 м3 смолы в час.
Вымывание легко осуществляется 5%-ным раствором аммиака
при удельной нагрузке 10 м3 вымывающего раствора на 1 м3
смолы в час. Содержание молибдена в элюатах составляет 50—
60 г/л.
Шамсиев и Сенявин исследовали возможность применения ион-
ного обмена для извлечения молибдена из рудничных вод, содержа-
щих от 1 до 10 г Мо в 1 м3 (от 1 до 10 мг/л) [43]. Для таких бедных
растворов удовлетворительные результаты были получены при
сорбции на смоле АВ-17 в форме SO1- при pH раствора 6—7.
Существенные затруднения возникли при элюировании молибдена
со смолы. Растворы КОН (от 3 до 18%), NaOH + NaCl, NH4OH
элюировали не более 50—60% Мо. Полное вымывание достигается
при использовании раствора КОН в смеси с перекисью водорода
(10% КОН + 3% Н2О2), что обусловлено переводом молибдена
в надмолибденовую кислоту. При полупромышленных испытаниях
степень концентрирования молибдена составила 104, т. е. получали
растворы с содержанием в среднем 50 г/л, из которых легко вы-
делить богатый молибденом продукт.
156
Высокой сорбционной способностью в отношении ионов мо-
либдена обладают некоторые сорта активированных углей [40,
с. 124; 44].
ТАБЛИЦА 40
СОРБЦИЯ МОЛИБДЕНА И СТРУКТУРА АКТИВИРОВАННЫХ УГЛЕЙ
РАЗЛИЧНЫХ МАРОК [44].
(концентрация молибдена в исходном растворе 0,144 г/л, концентрация серной
кислоты 0,1 м/л. Для сопоставления приведена емкость по молибдену
анионита ЭДЭ-10п)
Марка сорбента ДОЕ, мг/г ПОЕ, мг/г у 2 W's ^ма v п V ми
ЛАМ 4,3 64 1,011 0,323 0,685 0,135 0,191
БАУ 16,7 119 1,495 0,344 1,151 0,113 0,231
КАУ 14,4 145 0,010 0,503 0,507 0,178 0,325
скт 14,4 180 0,949 0,626 0,320 0,129 0,500
N 1 28,8 300 2,584 0,941 1,643 0,623 0,318
ЭДЭ-Юп 1.8,0 298 — — — — —
Примечание. ДОЕ и ПОЕ — динамическая и полная обменная емкости.
Г2 — суммарный объем пор; — предельный объем сорбционного пространства; Ума —
объем макропор; Еп — объем переходных пор; Уми — объем микропор.
ТАБЛИЦА 41
СОРБЦИЯ МОЛИБДЕНА АКТИВИРОВАННЫМИ УГЛЯМИ РАЗЛИЧНЫХ
МАРОК
(концентрация молибдена в исходном растворе 1 г/л, концентрация серной
кислоты 0,1 мол [44 1)
Марка угля £ Концентрация молибдена в равно- весном растворе, г/л ПОЕ, мг/г а S- ср
БАУ 0,579 107 185
КАД иодный ....... 0,455 156 343
МКАД 0,437 164 .376
Древесный уголь Б 0,320 210,5 659
АГ-3 0,300 215 716
скт 0,300 215 716
Древесный уголь А ..... 0,276 228 826
ОУК кислый 0,274 229 836
ОУК щелочной 0,266 232 870
скл •_ . . 0,223 249 1116
В табл. 40 и 41 сопоставлены сорбционные свойства активиро-
ванных углей различных марок. Максимальные емкости углей по
молибдену лежат в области pH = 1 ч-З, отвечающей максималь-
ной концентрации полимерных анионных комплексов молибдена.
157
Однако с возрастанием концентрации молибдена в растворе об-
ласть оптимальных значений кислотности раствора расширяется
[40].
В работе [44 ] показано, что скорость поглощения молибдена
углями существенно выше, чем ионобменной смолой при примерно
равной ионообменной емкости.
Динамическая и полная обменная емкость по молибдену за-
висят от структуры угля. ПОЕ растет с увеличением предельного
объема сорбционного пространства (табл. 40). Поскольку для
крупных полимерных анионов молибдена мало доступны мелкие
микропоры, определяют сорбционную емкость переходные поры
или крупные микропоры. Авторы работы [44] для сорбционного
извлечения молибдена из сернокислых растворов рекомендуют
угли с широко развитой пористостью в области переходных и
микропор типа СКТ и N1 (табл. 40).
Хорошими сорбционными свойствами и относительно высокой
механической прочностью отличаются также угли марки СКТ [40].
Молибден может быть десорбирован с активированного угля
растворами щелочей.
31. ИЗВЛЕЧЕНИЕ МОЛИБДЕНА ИЗ РАСТВОРОВ ЭКСТРАКЦИЕЙ
ОРГАНИЧЕСКИМИ РАСТВОРИТЕЛЯМИ
Многообразие форм нахождения молибдена в водных раство-
рах (простые и полианионы, оксикатионы, комплексные анионы)
обусловливает возможность экстракции молибдена почти всеми
типами органических экстрагентов:
анионообменными экстрагентами типа аминов и четвертичных
аммониевых оснований;
катионообменными экстрагентами типа алкилфосфорных кислот;
«нейтральными» экстрагентами, к которым относятся трибу-
тилфосфат, спирты, простые эфиры, кетоны.
Использование экстракции в технологии молибдена весьма
перспективно. Преимущества процесса в сравнении с ионообмен-
ной сорбцией на смолах состоят в большей избирательной способ-
ности (возможно селективное извлечение молибдена в присутствии
высоких концентраций анионов кислот), а также применимости
процесса для разбавленных и богатых по содержанию молибдена
растворов.
Ниже приведен обзор наиболее существенных из опубликован-
ных работ по экстракции молибдена из водных растворов.
Экстракция аминами
Амины представляют собой алкильные производные аммиака.
В зависимости от числа водородных атомов, замещенных в аммиаке
на алкильные радикалы, различают первичные, вторичные и тре-
158
тичные амины, а также соединения тетраалкиламмония (или соли
четвертичных аммониевых оснований):
7?
I
Н—N
I
Н
первичный
амин
вторичный
амин
третичный
амин
соль четвертичного
аммониевого
основания
Здесь R — углеродный радикал; X — анион кислоты.
В аминах, как и в аммиаке, азот имеет неподеленную пару
электронов, что обусловливает способность к образованию коор-
динационных соединений:
R
7?—N + HNO3
R
NO3“
Образующиеся комплексные соли амина способны обменивать
анион кислоты на анионы металлов. Поэтому амины часто называют
жидкими анионообменниками.
Первичные, вторичные и третичные амины — сравнительно
слабые основания. Они экстрагируют анионы металлов в кислых
средах.
Экстракционные свойства аминов в большей мере зависят от
длины углеводородной цепи радикала. Например, для триалкила-
минов с нормальной углеводородной цепью максимальное извле-
чение анионов металлов достигается при содержании в цепи
8—10 атомов углерода [53, 54]. На экстракционные свойства
аминов сильно влияют стерические факторы. Так, при эк-
стракции молибдена из сернокислых растворов 0,1-м. раствором
триоктиламина в керосине коэффициент распределения DMo =
= 200 (исходная концентрация молибдена 1 г/л), тогда как для
три-2-этилгексиламина коэффициент распределения меньше 1. На-
личие этильных групп в непосредственной близости к атому азота
вызывает стерические затрудения, особенно при экстракции круп-
ных комплексных анионов, таких как полимолибдаты.
Наиболее широко для экстракции неорганических веществ из
кислых растворов применяют триоктиламин ТОА — (C8H17)3N.
Триоктиламин, а также его соли — сульфаты, хлориды, нитраты —
растворяются в керосине. Для повышения растворимости и улуч-
шения расслоения фаз в керосин обычно добавляют спирты фрак-
ций С7—С9.
159
Молибден экстрагируется из сернокислых растворов различ-
ными аминами с длинной углеводородной цепью. Преимущественно
изучали экстракцию триоктиламином (ТОА) [53, 54, 58].
Экстрагируемость молибдена ТОА сильно зависит от концен-
трации водородных ионов. По данным Ласкорина с сотрудниками
[54], максимум экстракции наблюдается при рН = 2н-3, что
отвечает присутствию в растворе полимерных анионов. Причиной
снижения коэффициента распределения в интервале pH = Зч-6
авторы считают гидролиз соли амина. При увеличении кислотно-
сти (pH < 1) снижение параметров экстракции объясняется пре-
имущественным присутствием молибдена в составе катионов. На-
блюдаемая экстракция молибдена из растворов с высокой концен-
трацией молибдена, по всей вероятности, обусловлена образова-
нием анионных комплексов типа [МоО2 (SO4)2]2- и др.
В табл. 42, по данным работы автора и Дрэган \ приведены
результаты экстракции молибдена из сернокислых растворов
1%-ным раствором ТОА в керосине.
ТАБЛИЦА 42
ПОКАЗАТЕЛИ ЭКСТРАКЦИИ МОЛИБДЕНА В ЗАВИСИМОСТИ ОТ
КОНЦЕНТРАЦИИ СЕРНОЙ КИСЛОТЫ 1%-ным РАСТВОРОМ ТОА41 В КЕРОСИНЕ
(исходная концентрация Мо 0,48 г/л\ VODr: Гв0 = 1 : 1)
Ь' г\
[Мо]орг Извлечение Мо
Концентрация H2SO4, % D — ГМ°1воДн • в экстракт, %
5 16,5 94,2
10 8,85 90,5
20 6,51 86,7
30 1,63 62,2
40 1,06 51,6
50 0,48 32,6
60 0,085 7,82
При экстракции молибдена из сернокислых растворов при пол-
ном насыщении органической фазы молярное отношение в
ней Мо : ТОА = 2:1 [54, 58]. Из этого следует, что молиб-
ден экстрагируется в виде полимерных анионов. Так, соотно-
шению Мо : ТОА = 2:1 отвечает экстракция анионов Мо4О1Г :
•• (R3NH)2SO4 + Mo4O?3~^(R3NH)2Mo4O13 + soT~.
Авторы работы [54] считают, что в пределах pH = 1-н4 эк-
стракцию молибдена можно описать уравнением
(Я3 NH)2 SO4 + (Н2МоОД2 (£3 NH)m [H2n_m (MoO4)rt] +
JU
+ -f-so24- (100)
1 Л. Дрэган. Диссертация, МИСиС, 1964.
160
При этом упрощенно предполагается, что молибден находится
в слабокислых растворах в виде полимеризованных молекул
молибденовой кислоты.
В работе [54] установлено, что с увеличением концентрации
анионов SO1~, Cl- и NOjT (вводившихся в виде аммонийных со-
лей) от 0,1 до 10 моль!л снижается экстрагируемость молибдена.
Авторы показали, что значения коэффициентов распределения за-
висят от природы кислоты. Из азотнокислых растворов молибден
экстрагируется ТОА хуже, чем из сернокислых или солянокислых
растворов.
Реэкстракция молибдена из органической фазы возможна при
контакте с растворами аммиака, соды или щелочей.
Применение экстракции молибдена триоктиламином в техно-
логии переработки молибденсодержащего сырья неизвестно. К не-
достаткам экстракции молибдена ТОА относится малая избира-
тельность: вместе с молибденом в широком интервале кислотности
экстрагируется рений (анионы ReO?), а также анионы [фосфорной
и мышьяковой кислот.
Экстракция солями четвертичных аммониевых оснований
(ЧАО)
ЧАО — сравнительно сильные основания (сильнее гидроокиси
аммония) и образуют хорошо диссоциирующие соли. Поэтому,
в отличие от аминов, они способны экстрагировать металлосо-
держащие анионы не только из кислых, но и щелочных сред. Эк-
стракционные свойства солей ЧАО зависят от типа алкильных
групп.
В опубликованных работах * [61, 62] по изучению экстракции
молибдена солями" ЧАО использовали соли типа
"сн3 - nr2
сн2с6н5 _
С1 — диалкилметилбензиламмоний хлорид
(ДАМБАС), где R — углеводороды С8—С18;
а также соли типа
* Kelley S. Т. US Pat., 3083085, 1963.
11 А. Н. Зеликман
161
Соли ЧАО экстрагируют анионы из растворов по ‘механизму
межфазного анионного обмена:
п + тМеХп~ = m[R^N]nMeX + nYm~, (101)
где п — заряд металлосодержащего аниона;
т — заряд аниона соли ЧАО;
Y — анион кислоты.
Растворителями солей ЧАО обычно служат керосин, ксилол
или другие углеводородные растворители. В органическую фазу
добавляют алифатические спирты для улучшения расслоения фаз
и растворимости солей ЧАО.
Автором и Калининой установлено, что из солей ЧАО приве-
денного выше типа более высокие показатели экстракции молиб-
дена дает ДАМБАС в БО^-форме. Экстракцию 'проводили из рас-
творов, содержащих 12 г!л Мо. Органическая фаза содержала 5%
ДАМБАС и 3% (объемн.) изооктилового спирта, растворенных
в керосине [62].
Как видно из табл. 43, в интервале pH = 3=5 коэффициенты
распределения молиблена высокие, причем они сильно зависят
от pH, что указывает на изменение ионного состава раствора.
В интервале pH = 7=12 коэффициенты распределения низкие и
не зависят от pH среды. Это объясняется тем, что при pH > 7
в растворах присутствуют только анионы МоО1~.
Высокие значения £)Мо при pH = 2 =5 объясняются экстрак-
цией полимерных анионов молибдена.
В этой области pH экстракция упрощенно может быть описана
реакциями:
(7?4N)2SO4 + Мо4О?3“7^ (/?4N)2Mo4O13 + SO1“,
3 (7?4N)2 SO4 + МО7О2Г (7?4N)6 Mo7O24 + 3SO4~.
В щелочной среде
(£4N)2 SO4 + MoO4~i± (#4N)2 MoO4 + SO4-
Таким образом, в слабокислых растворах молярное отношение
Мо : ЧАО = 2 : 1 и 7 : 6, тогда как в щелочной среде Мо : ЧАО =
= 1:2.
Несмотря на низкие значения DMo = (2,2 =2,3), экстракцию
молибдена солями ЧАО из щелочных растворов можно проводить
достаточно эффективно (табл. 43).
Реэкстракцию молибдена из органической фазы можно прово-
дить растворами соды, щелочей.
Попытки разделения молибдена и вольфрама экстракцией
с'олями ЧАО не дали положительных результатов. В интервале pH
от 2 до 12 молибден и вольфрам при их совместном присутствии
в растворе экстрагируются с близкими коэффициентами распре-
деления [62].
162
ТАБЛИЦА 43
ЗАВИСИМОСТЬ КОЭФФИЦИЕНТА РАСПРЕДЕЛЕНИЯ D И СТЕПЕНИ
ЭКСТРАКЦИИ Е МОЛИБДЕНА ОТ pH РАСТВОРА
(экстрагент ДАМБАС (5% в керосине), концентрация Мо в исходном растворе 12 г/л,
V : V =1-1)
орг води •
Исходный pH пМо Е, % Исходный pH £>Мо Е, %
2 1020,4 99,9 8 2,2 68,64
3 1286,3 99,91 9 2,2 68,64
4 1142,0 99,9 10 2,3 68,65
5 592,3 99,8 11 2,2 68,94
6 2,3 70,1 12 1,6 62,30
7 2,3 .69,9 -
Значительный интерес представляет возможность разделения
молибдена и рения экстракцией солями ЧАО, установленная Чер-
чвудом ц Розенбаумом [61]. Исходные растворы содержали:
16 г/л Мо; 1 г/л Re. При экстракции 5%-ными растворами соли
[R3CH3N]C1 в интервале pH = 1ч-12 коэффициент распределе-
ния молибдена понижается от 160 до 0,03, тогда как коэффициент
распределения рения уменьшается с 480 до 140. Это позволяет
селективно экстрагировать рений из раствора с pH = 12, а затем
при более низком значении pH экстрагировать молибден или вы-
делить его другими методами.
Экстракция Ди-2-эти л гексилфосфорной кислотой (Д2ЭГФ К)
Д2ЭГФК — производное ортофосфорной кислоты, в которой
два водородных атома замещены на этилгексиловый радикал
С8Н17 (или
СН3 — СН2 — СН2 — СН2 — СН — СН2—).
С2Н5
Структурная формула Д2ЭГФК:
НО
RO—Р - О.
Распространенный способ синтеза Д2ЭГФК основан на реакции
между спиртом и фосфорным ангидридом в керосине [64]. Обычно
Д2ЭГФК применяют в виде 5—20%-ного раствора в керосине,
что связано с высокой вязкостью неразбавленного экстрагента.
11* 163
№
В керосине Д2ЭГФК находится в димеризованном состоянии, что
обусловлено водородными связями [651:
(ОЯ)2
II
НО —Р = о
I I
[ I
6=р—он
II
(0Я)а
В слабокислых и умереннокислых растворах Д2ЭГФК ведет
себя как катионообменный экстрагент (осуществляется обмен ме-
жду катионами металлов в водной фазе и ионами водорода алкил-
фосфорной кислоты).
Однако из-за активных фосфорильных групп образующиеся при
экстракции комплексы обычно не являются простыми солями типа
(Т?2РОГ)г, а содержат сольватированные молекулы кислоты.
Общая формула соединений Mez+ (RzPO^z' ^НТ^РО4. При
увеличении концентрации кислоты подавляется диссоциация ал-
килфосфорной кислоты и экстракционные ее свойства как катио-
нообменного экстрагента должны уменьшаться. Вместе с тем
при высокой кислотности начинает проявляться способность
Д2ЭГФК к экстракции по сольватационному механизму • [66,
68—71, 73].
Действительно, в структуре алкилфосфорной кислоты имеется
фосфорильная группа Р=О, способная (при высокой концентра-
ции водородных ионов) к образованию координационных соеди-
нений (сольваты или гидросольваты) по аналогии с трибутилфос-
фатом.
Сольватационный механизм проявляется, в частности, в спо-
собности Д2ЭГФК экстрагировать минеральные кислоты, как
показано в работе [66]. Двойственный механизм экстракции
Д2ЭГФК отчетливо выявляется на рис. 58, где, по данным автора
и Нерезова [68], построена зависимость значений коэффициента
распределения молибдена от pH раствора при экстракции из
сернокислого раствора, содержащего 1 г!л Мо. 0,136-м. раствором
Д2ЭГФК в керосине. Аналогичный характер носят зависимости,
полученные при экстракции из азотнокислых и солянокислых
растворов.
В слабокислых и умереннокислых растворах (pH = 0 4-5),
где экстракция проходит по катионообменному механизму, мак-
симум экстракции наблюдается в интервале pH = 1,5 4-2,5
(рис. 58). Оптимум экстракции соответствует значениям pH, при
которых в растворах присутствуют в значительных концентрациях
164
оксикатионы МоОг+ и др. в равновесии с анионными ^формами.
В менее кислой области при увеличении pH от 3 до 5 концентра-
ция катионов уменьшается и экстрагируемость молибдена падает.
В более кислой области (pH < 1,5) с увеличением концентрации
водородных ионов снижение экстрагируемое™ молибдена обуслов-
Рис. 58. Зависимость лога-
рифма коэффициента распре-
деления молибдена (IgZ^Mo)
от pH равновесной водной
фазы [68]. Органическая фа-
за: 0,136-м. раствор Д2ЭГФК
в керосине. Водная фаза (ис-
ходная): 1 г!л Мо H2SO4.
Отношение объемов УОгр •
• ^водн = 1 • 1
pH равновесной водной разы
лено конкурирующим влиянием ионов водорода (подавляется дис-
социация алкилфосфорной кислоты).
Экстрагируемый комплекс молибдена имеет состав МоО2
(Т?2РО4)2- 2Н7?2РО4 [46]. Вода в состав соединения не входит.
Наиболее вероятная структура комплекса:
(OR2)2 (ORz)z
О = р — О ZO —Р — он
zMO02\
НО — P = QZ О — Р = О
(or2)2 (or2)2
Таким образом, две избыточные молекулы Д2ЭГФК сольва-
тированы вследствие присоединения Р=0 групп к катиону мо-
либдена.
В слабокислых растворах (pH = 2 ч-4, участок 1 кривой,
рис. 58) экстракция молибдена определяется равновесием реак-
ций:
МО7О2Г + 2ОН“^7МоО2+ + 10Н2О,
2МО7О2Г + 26H+^±7Mo2Os+ + 13Н2О,
(Ю2)
(ЮЗ)
Мо2О|+ + 4 (Н7?2РО4)2 7^ 2МоО2 (Т?2РО4)2 • 2HJR2PO4 + 4Н+, (104)
МоО2+ + 2 (Ш?2РО4)2^МоО2 (Z?2PO4)2-2H7?2PO4 + 2Н+. (105)
165
Переход оксикатионов в органическую фазу смещает равно-
весие первых двух реакций в сторону образования оксикатионов.
Кроме МоО2+ и Мо2Об+, в азотнокислых и солянокислых раство-
рах присутствуют также тримеры МозО|+. При умеренной кислот-
ности [pH = 1 -т- (—0,5), участок 2 кривой рис. 58] в растворе
доминируют оксикатионы. Экстракция протекает по реакциям
(104) и (105).
При высокой концентрации кислот (участок 3 кривой рис. 58)
механизм экстракции сольватационный. Как показали иссле-
дования, в этом случае из сернокислых растворов (концентрация
Н 2SO4 > 720 г/л) экстрагируется тетрасольват состава Мо2О5
(SO4)-4Н7?2РО4, из азотнокислых растворов (HNO3 >>200 г/л) —
тетрасольват Мо2О5 (NO3)2-4Н7?2РО4 *.
Состав экстрагируемого соединения из солянокислых раство-
ров не изучали. Можно, однако, полагать, что в этом случае об-
разуются сольваты диоксидихлорида МоО2С12• nHR 2РО4 или
НМоО2С13-пН/?2РО4 по аналогии с экстракцией молибдена из со-
лянокислых растворов трибутилфосфатом [72].
Более детально изучены условия экстракции молибдена
Д2ЭГФК из слабокислых растворов в связи с использованием
этого процесса для извлечения молибдена из промышленных рас-
творов [54, 67].
Присутствие натриевых и аммонийных солей нитратов, хло-
ридов и сульфатов до их концентрации 200 г/л практически не
влияет на коэффициент распределения молибдена. Однако примеси
ионов РО4-, AsOl- и особенно вольфрамат-ионов снижают эк-
страгируемость молибдена, что видно из данных табл. 44, где
приведена зависимость остаточного содержания молибдена в вод-
ной фазе от числа стадий экстракции свежим растворителем.
ТАБЛИЦА 44
ВЛИЯНИЕ ПРИМЕСЕЙ ФОСФОРА, МЫШЬЯКА И ВОЛЬФРАМА НА
ЭКСТРАКЦИЮ МОЛИБДЕНА 0,1-м. Д2ЭГФК В КЕРОСИНЕ [67 ]
(отношение Горг : Гводн = 1:1)
Примесь в исходной водной фазе, а/л (1 г/л Мо, рН=2) Содержание Mo в равновесной водной фазе, а/л, по стадиям
1 2 3 4 5 6
0,3WO^~. IWO^- IP lAs 0,007 . 0Д ’ 0,36 0,22 0,21 Отс. 0,05 0,2 0,07 0,06 л 0,045 0,12 0,035 0,03 ж» 0,040 0,08 0,015 0,01 0,03 0,07 0,003 Отс. 0,05
* В. М. Перезов. Диссертация, МИСиС, 1967.
166
Известно, что вольфрам из слабокислых растворов Д2ЭГФК
практически не экстрагируется [54]. Однако в присутствии воль-
фрама полное извлечение молибдена не достигается даже за 6 ста-
дий экстракции, тогда как без него молибден полно извлекается
за две стадии. Аналогичное явление, но в меньшей степени, на-
блюдается в присутствии фосфора и мышьяка [67].
Потеря индивидуальных свойств молибдена в указанных слу-
чаях обусловлена образованием гетерополикомплексных анионов
молибдена с примесями анионов фосфорной, мышьяковой и воль-
фрамовой кислот.
Примеси катионов Са2+, Си2+ и Fe2+ при их концентрации
1—2 /гл не влияют на экстракцию молибдена Д2ЭГФК из слабо-
кислых растворов при pH 2. Однако приместь катионов Fe3+
сильно влияет на .извлечение молибдена в органическую фазу.
Катионы Fe3+ экстрагируются Д2ЭГФК из слабокислых раство-
ров с образованием соединения с отношением Ре:Д2ЭГФК =
= 1:6. Это соединение прочное. При реэкстракции молибдена
растворами аммиака железо остается в органической фазе. На-
копление железа в органической фазе приводит к резкому сниже-
нию извлечения молибдена. Поэтому до экстракции молибдена
необходима очистка растворов от железа нейтрализацией их
до pH = 8.
В случае накопления железа в органической фазе для его
удаления необходима обработка экстрагента растворами едкого
натра. При этом выпадает гидроокись железа.
Ласкорин с сотрудниками [54] предложили проводить экстрак-
цию молибдена из слабокислых растворов Д2ЭГФК с добавками
трибутил фосфата для лучшего расслоения фаз.
По данным автора и Нерезова, в условиях, далеких от насы-
щения органической фазы, добавки ТБФ в органическую фазу
снижают коэффициент распределения молибдена. Это объясняется
взаимодействием Д2ЭГФК с ТБФ с образованием сольвата:
(НЯ2РО4)2 + 2ТБФ X 2 (Н/?2РО4• ТБФ).
В результате уменьшаются активная концентрация Д2ЭГФК и
коэффициент распределения молибдена (табл. 45).
Реэкстракцию молибдена из Д2ЭГФК можно проводить, в за-
висимости от условий технологии, растворами соды, едкого натра
или аммиака. Применительно к молибденовому производству
выгодно осуществлять реэкстракцию аммиачной водой.
При изучении реэкстракции было установлено, что расход
аммиака в три раза выше эквивалентного в расчете на образование
алкилфосфата NH4/?2PO4. Это обусловлено образованием соедине-
ния NH47?2PO4-2NH4OFI, которое выпадает в виде белого осадка.
Добавки в водный раствор аммиака нитрата аммония (10 г/л)
предотвращают выделение осадка. Вероятно, присутствие NH4NO3
167
ТАБЛИЦА 45
ВЛИЯНИЕ ДОБАВОК ТБФ НА ЗНАЧЕНИЕ Г>Мо ПРИ ЭКСТРАКЦИИ
0,078-м. Д2ЭГФК В КЕРОСИНЕ
(исходная концентрация 1 г/л Мо, отношение объемов О : В = 1 : 1)
Концентрация ТБФ, моль/л pH = 1,65 pH =2 •
Мо, г/л, в вод- ной фазе £>мо Мо, г/л, в вод- ной фазе DMo
0 0,024 40,5 0,015 65
0,035 0,028 34,6 — —
0,070 0,035 ' 27,6 0,023 44
0,105 0,040 24,0 — —
0,14 — — 0,035 27,5
снижает pH водной фазы, что предотвращает образование соеди-
нения указанного выше состава [67].
Экстракцию Д2ЭГФК применяют в промышленной практике
для извлечения молибдена из маточных растворов, получаемых
при переработке молибденовых концентратов с использованием
разложения концентрата азотной кислотой [67]. Выбор "Д2ЭГФК
был обусловлен доступностью этого экстрагента, а также тем, что
Д2ЭГФК не экстрагирует рений. Это позволяет отделить основ-
ное количество молибдена от рения, если он присутствует в раство-
рах х.
Исходные маточные растворы содержали, г/л: 0,01—0,02Re;
0,5—3,0Мо; 150—200NQT; 25—50SO1-; 0.28СГ; 0,2РО1-; 0,2—
0,6Fe; 0,1Мп; 1—1,5Си. После подкисления азотной кислотой
до pH = 2 растворы проходили пятиступенчатую экстракцию
раствором 7—8% Д2ЭГФК в керосине в экстракторе типа смеси-
тель—отстойник.
Молибден реэкстрагировали раствором 7—10% NH3 в двух
последовательных ступенях при отношении объемов О : В —
= 10:1 (десятикратное обогащение). Для предотвращения образо-
вания осадков в раствор аммиака добавлялся исходный маточный
раствор из расчета 10% по объему, что обеспечивало необходимую
концентрацию NH4NO3. После регенерации экстрагента контак-
том с 30 %-ной HNO3 органическую фазу, возвращают в цикл
экстракции.
Извлечение молибдена в аммиачные реэкстракты, содержа-
щие 15—20 г/л Мо, составляет 93—95%. Рафинаты молибденовой
экстракции содержали 50—100 мг/л Мо. Оставшийся молибден
не удавалось доизвлечь даже при увеличении числа стадий экс-
1 Зеликман А. Н. и Дрэган Л. Авт. свид. СССР № 176684,
1964 г. Бюлл. изобр., 1965, № 23.
1в8
трагирования до 8—10. Это объясняется, вероятно, связыванием
молибдена в неэкстрагируемые гетерополисоединения (см. та-
блицу 44).
Процесс также успешно используют для извлечения молибдена
из маточных растворов после осаждения полимолибдатов. Пер-
спективно его применение для извлечения молибдена из сернокис-
лых растворов, получаемых в мокрых системах пылеулавливания
газов обжиговых печей.
Экстракция молибдена трибутилфосфатом
Трибутилфосфат ТБФ — сложный эфир фосфорной кислоты
(С4Н9О)3РО — широко используют как экстрагент в гидрометал-
лургии. Экстракционные свойства ТБФ обусловлены наличием
фосфорильной группы Р=О, способной к присоединению эк-
страгируемых молекул благодаря присутствию свободной пары
электронов на фосфорильном кислороде [74].
ТБФ экстрагирует из кислых сред недиссоциированные моле-
кулы, образуя сольваты. Сольватирование происходит либо в ре-
зультате образования водородной связи (присоединение к фосфо-
рильному кислороду ионов Н+, Н3О+ или иона гидроксония
НэО^),или в результате непосредственного взаимодействия с ка-
тионом, металла, входящим в состав экстрагируемой молекулы.
При этом большей частью координационное число катиона металла
равно шести, что определяет возможное количество сольватиро-
ванных молекул ТБФ.
Нелидов и Даймонд [55] установили, что ТБФ является хо-
рошим экстрагентом для извлечения молибдена из солянокислых
растворов. Высокие коэффициенты распределения молибдена на-
блюдаются даже при относительно низкой концентрации кислоты.
Так, при экстракции из растворов 1- и 2-м. НС1, содержащих
а/л Мо, значения DMo равны 4,5 и 65 соответственно. При
разбавлении ТБФ до 10—20% (объемн.) керосином или другими
растворителями коэффициенты распределения уменьшаются, но из-
влечение молибдена остается достаточно высоким. Экстрагируе-
мый комплекс представляет собой дисольват МоО2С12* 2ТБФ,- т. е.
экстрагируются недиссоциированные молекулы хлорида молиб-
денила. Координационное число молибдена в комплексе равно 6
[72].
Молибден может быть реэкстрагирован из органической фазы w
растворами аммиака или соды.
В работе [36] предложено использовать экстракцию молиб-
дена ТБФ из кислых растворов, получаемых при разложении
шеелитоповеллитовых концентратов соляной кислотой.
Изучалась экстракция молибдена ТБФ из сернокислых сред.
Поданным Ю. Б. Герлита [56], при экстракции ТБФ из раствора
2-н. H2SO4 с концентрацией молибдена ^10г/л Z)Mo = 21,6.
169
ТБФ экстрагирует молибден из сернокислых растворов также при
слабой кислотности (в интервале pH = 6,4 4-0,5), однако коэф-
фициенты распределения в этом случае низкие. По данным автора
и Дрэган, при экстракции
100%-ным ТБФ из растворов
с содержанием 0,2 г!л Мо макси-
мум экстракции отмечается при
О 1 23й56789 10 11
концентрация нсцн
Рис. 60. Зависимость коэффи-
циента распределения молибдена
од концентрации соляной кис-
лоты при экстракции кетонами:
/ — ацетофенон; 2 — метилизобу-
тилкетон; 3 — циклогексанон
Рис. 59. Зависимость коэф-
фициента распределения мо-
либдена от pH раствора при
экстракции 100%-ным ТБФ
из сернокислого раствора.
Исходная концентрация
Мо 0,2 г!л
pH = 2 (рис. 59), чему соответствует DMo= 2,3 и извлечение в рас-
твор 70% Мо [68]. Химизм экстракции молибдена ТБФ при столь
низких концентрациях кислоты не выяснен. По всей вероятности,
экстрагируются поликислоты, например Н2Мо4О13 и др.
Экстракция молибдена кетонами
В ряде работ показано, что мрлибден хорошо экстрагируется
из солянокислых растворов кетонами, в частности метилизобу-
тилкетоном [26, 55, 57, 63] и метилфенилкетоном (ацетофеноном)
[63]. Экстракция кетонами представляет значительный интерес
в связи с выявившейся возможностью применения кетонов для
отделения молибдена от вольфрама экстракцией из солянокислых
пульп вольфрамовой кислоты1 [26, 62].
На рис. 60, по данным работ [62, 63], сопоставлены показа-
тели экстракции молибдена из растворов с содержанием 10 г!/г Мо
для трех распространенных кетонов, харатеристика которых при-
ведена в табл. 46.
1 Brown В. W., Brit. Pat., 854027; N е w k i г k Е. US Pat., № 3079226,
1963.
ПО
ТАБЛИЦА 46
НЕКОТОРЫЕ СВОЙСТВА КЕТОНОВ
Кетон Формула Молеку- лярная масса Темпера- тура кипения, °C Плот- ность, г/ел«3 Раствори- мость в воде, % (20° С) Темпера- тура плав- ления, °C Темпера- тура вос- пламене- ния, °C
t Метилизобутилкетон МИБК О сн3—с-с4н9 100,156 115,65 0,805 1,7—2,2 —83,5 15,6
Метилфенилкетон (ацетофенон) о сн3-с-свн6 120,15 202,3 1,020 Нераство- рим +20,5 105
Циклогексанон сн2 сн2 Н2с/ >с = о сн2 сн2 98,14 155,65 0,951 2 4 (31° С) —16,4 33,9
Экстракция молибдена кетонами при низких кислотностях
незначительная. Начиная с концентрации кислоты выше 2-н.
для циклогексанона (ЦГ) и 5-н. для ацетофенона (АЦФ) и метил-
изобутилкетона (МИБК), значения £>Мо резко увеличиваются, до-
стигают максимума и затем с дальнейшим ростом кислотности
уменьшаются. Различная величина максимальных значений DMo
у сопоставляемых кетонов объясняется главным образом различи-
ем в их растворимости в солянокислых растворах.
С повышением концентрации соляной кислоты все возрастаю-
щие количества ЦГ переходят в водную фазу, сольватируя водород-
ный ион, что снижает переход молибдена в органическую фазу.
Высокая растворимость ЦГ в соляной кислоте приводит к тому,
что при 6-н. концентрации НС1 не наблюдается расслоение фаз.
При использовании МИБК, растворимость которого в соляной
кислоте меньше, максимум 7)Мо достигается при более высокой
концентрации кислоты, а его значение выше. Расслоение отсут-
ствует при ~9-н. концентрации НС1.
Растворимость АЦФ даже в концентрированных растворах
НС1 очень мала. Соответственно этому максимум Z)Mo наблюдается
при 9-н. НС1, фазы расслаиваются даже при 11-н. НС1. Пониже-
ние экстрагируемое™ молибдена при превышении оптимальной
кислотности для МИБК и АЦФ объясняется экстракцией соля-
ной кислоты кетонами, которая становится заметной выше 7-н.
концентрации HCL
Известно, что в растворах НС1 концентрации выше 4-н. Мо
в большей мере присутствует в форме недиссоциированных моле-
кул диоксидихлорида МоО2С12 [31, 55], а при более высокой кон-
центрации НС1 — в составе оксихлоромолибденовых кислот типа
Н(МоО2С13); Н2(МоО2С14); Н(Мо3О6С17) [32, 57]. Некоторые соли
этих кислот выделены из сильнокислых растворов.
На основании изучения спектров поглощения в инфракрасной
области и химического анализа насыщенного молибденом АЦФ’
и МИБК установлено, что экстракция молибдена кетонами из
солянокислых растворов протекает по гидратно-сольватному ме-
ханизму [63].
При экстракции молибдена из 6-н. НС1 ацетофеноном образуется
сольват состава
[Н3О (Н2О)3 п ket]+ [МоО2С13 (Н2О)Г,
который частично диссоциирует в органической фазе, обусловли-
вая значительную ее электропроводность. Число п = 3 или 4,
избыточное количество молекул кетона служит растворителем
сольвата.
Экстракция молибдена кетонами МИБК и АЦФ может быть
использована для отделения молибдена от вольфрама при перера-
ботке молибденсодержащих шеелитовых концентратов, в которых
содержание молибдена иногда достигает 5—6%.
172
При разложении шеелита концентрированной соляной кисло-
той большая часть молибдена переходит в солянокислый раствор.
Однако в осадках вольфрамовой кислоты содержание молибдена
остается не ниже 0,2—0,3%.
Было установлено, что при обработке солянокислой пульпы
вольфрамовой кислоты кетоном молибден полностью извлекается
в органическую фазу. В вольфрамовой кислоте содержание мо-
либдена снижается до 0,02—0,04%, что обеспечивает получение
паравольфрамата аммония с содержанием примеси —0,001 % Мо.
В отличие от других экстрагентов при обработке пульпы
кетонами (МИБК, АЦФ) происходит хорошее разделение фаз
и не наблюдается «зависания» вольфрамовой кислоты ни в одной
из фаз.
В работах [26, 62] изучены условия отделения молибдена от
вольфрамовой кислоты при экстракции из пульпы вольфрамовой
кислоты ацетофеноном. Преимущество АЦФ — его доступность
и низкая стоимость, малая растворимость в солянокислых раство-
рах, высокая температура вспышки (табл. 46) и более высокие коэф-
фициенты распределения по сравнению с МИБК. Недостатки
АЦФ — высокая вязкость и плотность, близкая к плотности воды.
Однако при его использовании фазы разделяются достаточно бы-
стро.
В табл. 47 приведены результаты опытов разложения шеели-
тового концентрата, содержащего 4,5% Мо, соляной кислотой
с последующей обработкой пульпы ацетофеноном. Концентрация
соляной кислоты в конце разложения изменялась от 5,8 до 9,4-н.
При остаточной кислотности выше 6-н. перед экстракцией пульпу
разбавляли до концентрации НС1 6—6,2-н. для снижения содер-
жания вольфрамовой кислоты в солянокислом растворе. Экстрак-
ТАБЛИЦА 47
РЕЗУЛЬТАТЫ РАЗЛОЖЕНИЯ ШЕЕЛИТОВОГО КОНЦЕНТРАТА
СОЛЯНОЙ КИСЛОТОЙ С ЭКСТРАКЦИЕЙ МОЛИБДЕНА АЦЕТОФЕНОНОМ [26 ]
(состав концентрата: 51,1% WO3. 4,5?4 Мо)
Остаточная Отношение объемов Извлечение в экстра- гент, % от исход- ного Содержание Мо в H2WO4, %
кислотность, н. водн WO3 после разложе-. НИЯ после эк-
V иорг Мо W Мо стракции
в экстракте кислотой из пульпы
5,8 1 ; 3,6 92,99 3,26 0,40 0,50 0,45
6,0 1 : 3,2 "96,96 3,90 0,46 0,47 0,31
6,5 1 : 2,2 1 : 1,9 97,88 2,54 0,30 0,35 0,19
7,1 7,4 98,07 3,58 2,99 0,41 0,30 0,10
1 : 1,3 96,22 0,35 0,30 0,07
8,2 1 : 1,2 94,75 2,34 0,28 0,29 0,05
9,4 1 : 0,8 96,68 4,14 0,49 0,29 0,03
173
цию АЦФ проводили в две стадии при температуре 80° С в течение
30 мин. Содержание молибдена в вольфрамовой кислоте до эк-
стракции составляло 0,3—0,5%, тогда как после обработки эк-
страгентом содержание молибдена (при остаточной кислотности
выше 8-н.) снизилось до 0,05—0,03%. Извлечение молибдена в ор-
ганическую фазу составляло 96—98%, вольфрама 2,5—3% от
исходного.
Изменение содержания молибдена в исходном шеелите от 0,5
до 4% практически не влияет на степень извлечения вольфрама
в органическую фазу, примеси мышьяка и фосфора увеличивают
переход вольфрама в органическую фазу.
Следует при этом отметить, что экстракция приводит к суще-
ственной очистке вольфрамовой кислоты от мышьяка и фосфора.
Извлечение большей части молибдена (—70%) из органиче-
ской фазы возможно при реэкстракции водой. При этом вольфрам
реэкстрагируется в малой степени. Из водного реэкстракта может
быть выделен молибденовый химический концентрат (например,
осаждением CaMoOJ. Оставшиеся в органической фазе молибден
и вольфрам реэкстрагируются вместе 10%-ным раствором • ам-
миака, из которого осаждается вольфрамо-молибденовый оборот-
ный продукт, поступающий на разложение соляной кислотой
(см. рис. 41).
Полупромышленные испытания описанной кислотно-экстрак-
ционной схемы переработки шеелитового концентрата, содержа-
щего 4,5% Мо, подтвердили возможность получения высокочи-
стого паравольфрамата аммония с содержанием 0,005% Мо.
Приведенная схема применима и для переработки вольфрамо-
вых полупродуктов — искусственного шеелита с примесью мо-
либдена.
Существенный недостаток схемы — высокий расход соляной
кислоты, достигающий 4,3 т НС1 (плотность 1,18) на 1 т концен-
трата, что связано с необходимостью обеспечения высокой остаточ-
ной кислотности после разложения концентрата.
ГЛАВ-А VIII
ХЛОРИСТЫЕ СОЕДИНЕНИЯ МОЛИБДЕНА
Изучены свойства ряда хлоридов и оксихлоридов молибдена,
отвечающих различным степеням окисления молибдена: МоС1б,
МоС14, МоС13, МоО 2, МоО2С12, МоОС14, МоОС13, МоОС12. Кроме
того, имеются сведения о существовании более сложных (много-
ядерных) оксихлоридов (Мо2О3С14, Мо2О3С1б, Мо2О3С15), инди-
видуальность которых строго не установлена.
Значительный интерес к изучению свойств и условий синтеза
хлористых соединений обусловлен рядом причин:
а) развиваются хлорные процессы вскрытия различных бед-
ных, трудно обогатимых типов рудного сырья, при которых мо-
либден полно извлекается, образуя легколетучие хлористые сое-
динения;
б) летучесть хлористых соединений высшей валентности
(МоО2С12, МоС15 и др.) позволяет легко осуществлять глубокую
их очистку методами сублимации или дистилляции;
в) хлористые соединения молибдена могут быть использованы
для получения чистого молибдена или молибденовых покрытий
методами восстановления водородом в газовой фазе, термической
диссоциацией или электролизом расплавленных сред.
В табл. 48 приведены некоторые термохимические характери-
стики хлористых соединений молибдена по данным, которые
мы считаем наиболее достоверными из опубликованных в литера-
туре.
Значения термодинамических функций для хлоридов молибдена
в справочной литературе [14, 15] основаны на оценке, данной
Брюэром и др. [16].
Как было показано в ряде работ С. А. Щукарева с сотруд^
никами [5—8, 10, 11], экспериментально определенные значения
энтальпий образования хлоридов молибдена выше приводимых
Брюэром, что объясняется неправильным значением принятого
175
м
ТАБЛИЦА 48
S ТЕРМОДИНАМИЧЕСКИЕ СВОЙСТВА ХЛОРИСТЫХ СОЕДИНЕНИЙ МОЛИБДЕНА
Хлорид Со- стоя- ние * Температура плавления, °C Температура кипения, °C Энтальпия образования А-^2985 ккал/моль Энтропия о S298’ кал/град** Теплоем- кость при 300° к с [8] ** кал/ молъх X град Энтальг ккалЦ сублима- ции ,ИЯ АЯ298, ноль [8] испаре- ния Энтроп э. t сублима- ции ИЯ AS298- [8] испаре- ния
МоС15 т г 194 [4] 268 [4] —126 [5, 6] —104 [6, ?: 56 [5— 101 [5- 7] -7] (35) (26) 23 14 45 25
МоС14 т т Разл. выше 130° С [39] ~320° С (субл.) —116 [8] —90 [8] 00 СП - ОО о -Ф СП - (28) (22) — — — —
МоС13 т Разл. выше 530° С [39] —94 [1, 10] (43) [8] 24 — — — —
МоС12 т Разл. выше 730°С [39] — —69+5 [11] (31) 18 — — — —
МоО2С12 т г 170 под да вл. 1,5 ат [6] 159 (субл.) [12] —173+0,7 [5] —155,0 [5] 26 [8] 73 [8] (25) (14) ' 18 — 36 —
МоОС14 т г 104 [6] 250 [6] —154,0 [5] —141 [8] (50) [1 (75) [1 и и (33) (22) 13 12 25 22
МоОС13 т г 295 [13] 352 [13] (—160) [8] (—130) [8] (30) [1 (64) [1 5] п (27) (18) — — — —
МоОС12 т Разл. выше 350° С — —126+4 [20] — — — — —
* Т — твердое, Г — газообразное.
** В скобках приведены ориентировочные значения.
им исходного значения А/7 образования МоС15 (—90,8 ккал!моль},
взятого из работы Ван Лимпа [17]. Действительное значение этой
величины —126 ккал!моль (табл. 48).
32. ПЯТИХЛОРИСТЫЙ МОЛИБДЕН
Высший хлорид молибдена МоС15 — твердое вещество, кри-
сталлизующееся в форме черных игл с металлическим блеском.
Незначительные примеси оксихлоридов придают им зеленый от-
тенок [2]. Пары МоС15 красно-бурого цвета. Плотность МоС15
при 25° С составляет 2,9275 [1].
Пентахлорид молибдена образует молекулярные кристаллы —
связи между молекулами ванн-дер-ваальсовского типа.
Методом электронной дифракции в парах установлено, что
в молекуле МоС15 атомы хлора расположены в вершинах триго-
нальной бипирамиды с атомом металла в центре [3, 19]. Расстоя-
ние между атомами Мо—С1 равно 2,27 А, между атомами С1—С1
ЗД2 А.
При конденсации МоС15 происходит его димеризация. Моле-
кула димера Мо2С110 построена из двух октаэдров, соединенных
ребрами. Два атома хлора осуществляют мостиковую связь между
атомами молибдена. Расстояния Мо—С1 (концевой атом хлора)
2,24 А, Мо—С1 (мостиковый атом хлора) 2,53 А. Расстояние Мо—
Мо 3,84 А. [56]. МоС15 парамагнитен. Магнитный момент р =
= 1,55 магнетонов Бора [40].
В атмосфере сухого воздуха МоС15 при комнатной температуре
окисляется с образованием оксихлоридов:
МоС15 + Х/2О2 = МоОС14 + V.CU, (106)
[МоС15 + О2 = МоО2С12 + 3/2С12. (107) •
Во влажном воздухе МоС15 гидролизуется (дымит) и расплы-
вается, образуя бурую жидкость.
Пятихлористый молибден плавится при 194° С и кипит при
268° С без заметного разложения, образуя бурые пары.
Критическая температура МоС15 /кр = 577,0° С, критическая
плотность ркр = 0,74 г!см\ плотность жидкого МоС15 рж =
= 2,196 г/см3, [41].
Зависимость вязкости паров МоС15 от температуры описы-
вается уравнением [41]:
т]МГ1 = 0,990 —9,24-10’3А/+ 6,47-10"5 А/2—1,97-10“7 А/3 спз;
5
А/ == I tnjl.
Уравнение справедливо в интервале 194,4—310° С.
Наиболее точные данные о давлении пара МоС15 получены в ра-
боте [7], в которой измерения проводили с использованием мем-
бранного нуль-манометра. ‘
12 А. Н. Зеликман 177
Для сублимации (до температур 200° С)
1g Рат = 9,50-----
(Ю8)
для испарения (выше 200° С)
IgPam = 5,536 —
(Ю9)
Значения энтальпий и энтропий процессов сублимации и ис-
парения приведены в табл. 48. Теплота плавления МоС15 равна
8 ккал!молъ\ давление пара при температуре плавления
~80 мм рт. ст.
Выше 210° С в газовой фазе в области ненасыщенного пара
МоС15 диссоциирует с образованием МоС14 [7]: .
МоОбгаз МоСЦгаз 1/2С12-
Для этой реакции
2779
(110)
(111)
Водород в зависимости от температуры и давления восстанав-
ливает МоС15 до низших хлоридов (МоС13, МоС12) или до металла.
Пентахлорид молибдена гидролитически разлагается в воде
с образованием оксихлорида МоОС13. Прозрачные коричневые
растворы на воздухе синеют (протекает окисление с образованием
молибденовой сини).
МоС1б хорошо растворяется в безводных органических раство-
рителях — эфирах, спиртах, четыреххлористом углероде, серо-
углероде, хлороформе и др. С эфиром образует молекулярное сое-
динение типа МоС15-2С2Н5О [18, 31].
Для получения пятихлористого молибдена наиболее просты
и пригодны для промышленных целей способы хлорирования ме-
таллического молибдена, ферромолибдена или молибденита.
Значительно сложней методу, основанные на хлорировании ки-
слородных соединений молибдена (молибдатов, трехокиси молиб-
дена) различными хлорирующими агентами, так как в этом слу-
чае трудно избежать образования оксихлоридов молибдена.
Среди этих методов следует отметить лишь распространенный
препаративный метод, основанный на взаимодействии МоО3
с четыреххлористым углеродом в запаянной стеклянной ампуле
при температуре 260—400° С [271:
МоО3 + ЗСС14 = МоС15 + ЗСОС12 + 1/2С12. (112)
Ампулы из стекла пирекс диаметром 12—15 мм, длиной 200—
250 мм выдерживают при указанных температурах под давлением
40—80 ат. При температурах 280—300° С реакция полно проте-
кает примерно за 6 ч, при температурах 350—400° С — за I—2 ч.
178
После окончания реакции ампулу вскрывают, из нее отгоняют
при 100°С под вакуумом СС14 и летучие продукты (СОС12, С12),
а также примеси оксихлоридов.
Ниже более детально рассмотрены способы получения МоС15,
представляющие технологический интерес.
Хлорирование молибдена. Молибден активно реагирует с хло-
ром при температурах 500—600° С. При избытке хлора основной
продукт реакции МоС15. Однако в качестве промежуточных про-
дуктов в результате вторичного взаимодействия образуются низ-
шие хлориды, например по реакции
МоС15 + Мо = МоС13 + МоС12, ' (113)
AZiooo°c = —23,67 ккал.
Этим объясняется отсутствие в течение некоторого времени
после начала хлорирования (особенно при хлорировании в слое
брикетов) пятихлористого молибдена в парогазовой фазе: в верх-
них горизонтах слоя в результате взаимодействия МоС1б с молиб-
деном образуются низшие хлориды.
Для снижения содержания примесей оксихлоридов необхо-
димо до хлорирования восстановить окисные пленки на частицах
молибденового порошка сухим водородом при температурах 900—
1000° С. После снижения температуры до 450—500° С водород
вытесняется.чистым азотом, а затем в систему подают хлор.
Источником загрязнения МоС1б примесями оксихлоридов может
быть также примесь кислорода в хлоре. Для очистки от кислорода
рекомендуется пропускать хлор через слой угля, нагретого до
700—800° С.
Для снижения содержания оксихлоридов (МоО2С12 и МоОС14)
в МоС15 в зоне конденсации пентахлорида необходимо поддержи-
вать температуру 150—160° С. В этих условиях оксихлориды
как более летучие вещества не конденсируются.
МоС15 получали на установке с загрузкой 1 кг молибденового
порошка [22]. Порошок молибдена брикетировали (под давлением
10—20 кПсм2). Брикеты дробили на кусочки 3—8 мм, которые
загружали в кварцевые лодочки (диам. 48 мм, длиной 300—
350 мм), помещаемые в кварцевую трубу (диам. 50 мм). Воздух
из системы вытесняли аргоном или азотом, по достижении темпе-
ратуры 600° С выпускали хлор. В первоначальный момент хло-
рирования отгонялось небольшое количество оксихлоридов молиб-
дена, оседавших на стенке приемника в виде тонкой пленки,
а затем начинали поступать темно-бурые пары МоС15, конденси-
рующиеся в приемнике в форме черных игольчатых кристаллов,
которые по мере их накопления на стенках приемника и ухуд-
шения теплопередачи образуют оплавленную массу. С целью
очистки МоС15 от примесей оксихлоридов проводили простую
12* 179
перегонку в атмосфере аргона. Более глубокая очистка может
быть осуществлена ректификацией [22].
В более крупных масштабах наиболее удобно хлорировать
молибден (в форме кусковых отходов штабиков, отходов прокатки,
проволоки, а также брикетов из порошка) в расплаве NaCl, смеси
хлоридов NaCl—КО (при молярном отношении 1 : 1 температура
плавления солевой смеси ~660° С) или КО—MgO2. Материал
периодически загружается в расплав и хлорируется при темпе-
ратуре 750—800° С. Большая часть хлоридов примесей остается
в расплаве, в том числе и хлориды железа и алюминия.
Хлорирование ферромолибдена. Ферромолибден — наиболее
удобный и доступный материал для получения пятихлористого
молибдена в промышленных масштабах. Выпускаемый для леги-
рования сталей ферромолибден в среднем содержит 60% Мо.
Главные примеси, %: 1—2 Si; 0,5—2,5 Си; 0,1—0,15 S, Р, С
(каждой); по 0,05 Sb, Sn. Основная составляющая ферромолиб-
дена 8-фаза системы Fe—Мо (см. рис. 171) — соединение Fe7Mo&
переменного состава. Ферромолибден хрупок и может быть раз-
дроблен до кусков нужного размера на щековой дробилке. В ра-
боте [21 ] изучалась кинетика хлорирования ферромолибдена
в различных условиях.
Как видно из рис. 61, ферромолибден хлорируется с более вы-
сокой скоростью, чем молибден и железо. При хлорировании ку-
скового материала (размеры кусков 5—10 мм) вследствие экзо-
термичности реакции реальная температура кусков на 200—
300° С выше исходной (например, при начальной температуре
400—500° С реальная температура ~650—750° С). При хлори-
ровании одиночного куска ферромолибдена реакция протекает
беспрепятственно с постоянной скоростью. Однако при хлориро-
вании слоя кускового материала возникает торможение процесса,
обусловленное протеканием вторичного взаимодействия МоС1б
и FeCl3 с молибденом и железом в верхних слоях брикетов с об-
разованием нелетучих низших хлоридов по реакции (113), а также
по реакциям:
МоС15 + Fe МоС13 + FeCl2,
^Fwoo° к = —37,1 ккал',
(П4)
2FeCl3 + Fe
3FeCl2,
(115)
AFiooo° к = —36,7 ккал',
2FeCl3 + Мо
2FeCL + MoCL,
4i I £7
(116)
AFiooo° к == —23,3 ккал.
Низшие хлориды покрывают поверхность кусков ферросплава,
препятствуя доступу хлора.
180
1дНн,см/сек
_1------1-----1-----1------1------/ ,пз
1,0 1.8 1.8 2.0 2.2 -f 'O
'i । ।_____j_____J______I
1
830 7J5 625 555 500 055
Температура, °С
Рис. 61. Зависимость логарифма скорости
хлорирования молибдена (/), железа (2)
и ферромолибдена (5) от обратной темпе-
ратуры
В связи с этим целесообразно хлорировать ферромолибден
в расплаве хлористого натрия или его смеси с КС1 [21]. В этом
случае низшие хлориды растворяются в расплаве.
Дополнительное важное преимущество хлорирования ферро- #
молибдена в расплаве состоит в том, что большая часть железа
остается в расплаве в виде FeCl2 и соединения NaFeCl4 (см.
стр. 183). В конденсате пятихлористого молибдена содержится
лишь 0,5—1,5% Fe.
Хлорирование в расплаве
идет с весьма высокой ско-
ростью (линейная скорость
хлорирования при 850° С
равна 2,4 • 10-4 см!сек).
При столь высокой скорости
процесс лимитируется подво-
дом хлора, т. е. протекает
во внешнедиффузионной об-
ласти.
Принципиальная схема
аппарата для хлорирования
ферромолибдена в расплаве
показана на рис. 62. Хлори-
руемый кусковой материал
располагается на графитовой
решетке (или постели из ку-
сков графита) в хлораторе,
представляющем собой шах-
ту, футерованную плотным
шамотным кирпичом. Хлор подают в нижнюю часть хлоратора.
Хлорирование в случае ванны из NaCl первоначально про-
водят при температуре 850° С. По мере накопления в ванне хло-
ристого и хлорного железа и снижения температуры плавления
расплава температура процесса может быть снижена до 600—
650° С.
Поскольку в процессе хлорирования объем расплава возра-
стает (накапливаются хлориды железа), непрерывно (или периоди-
чески) часть расплава сливается через летку в сборник. Перио-
дически в хлоратор догружают хлористый натрий (для корректи-
ровки состава ванны) и кусковой ферромолибден. Выходящая из
расплава парогазовая смесь практически не содержит избыточного
хлора (весь хлор используется для хлорирования ферромолибдена
в ванне). Вследствие этого в газовой фазе над ванной возможен
термический распад МсС15 с осаждением низших хлоридов на
стенках газохода. Для предотвращения этого необходимо
вводить в парогазовую смесь, выходящую из расплава, некоторое
количество хлора (в расчете на парциальное давление хлора
~100 мм рт. ст.).
181
Пятихлористый молибден собирается в никелевом конденсаторе.
Хлорирование молибденита. Исследования Глухова и Бехтле
показали, что до температуры 400° С молибденит взаимодействует
с хлором с относительно малой скоростью, хотя заметное хлориро-
вание наблюдается уже при 200—220° С.
Выше 400° С скорость хлорирования резко возрастает. Однако
при хлорировании слоя порошка или брикетов из MoS2 длитель-
ное время в конденсаторе нет паров МоС15, несмотря на полное
использование хлора. Вначале появляются пары свободной серы,
затем хлористой серы и лишь после подачи около половины хлора
Рис. 62. Принципиальная схема
хлоратора для хлорирования фер-
ромолибдена в солевом расплаве:
1— хлоратор, футерованный шамотным
кирпичом; 2 — постель из кусков гра-
фита; 3 — фурмы для подачи хлора;
4 — слой из кусков ферромолибдена;
5 — летка для слива расплава; 6—бун-
кер с питателем; 7 — газоход; 8 — кон-
денсатор; 9 — воздушная рубашка;
10— сборник хлорида; 11 —сборник
расплава
от теоретически необходимого количества появляется пятихло-
ристый молибден [54]. Это объясняется вторичными реакциями
взаимодействия пятихлористого молибдена с молибденитом. Наи-
более вероятно протекание следующих реакций:
MoS2 + - МоС15 + S2C12, (117)
МоС15 + MoS2 — МоС13 + МоС12 + 2S, (118)
МоС15 + 2S—>МоС13 + S2C12. (119)
Таким образом, в начальной стадии процесса хлорируемый ма-
териал покрывается низшими нелетучими хлоридами, а парога-
зовая смесь содержит лишь серу и хлорид серы. В последующем
поступающий хлор реагирует с низшими хлоридами, переводя
их в пятихлористый молибден. Авторы работы [54] рекомендуют
хлорировать MoS2 при температурах 450—500° С при большом
избытке хлора.
При хлорировании молибденита необходимо обеспечить раз-
дельную конденсацию МоС15 (в первом конденсаторе) и хлористой
•серы 1 (во втором, охлаждаемом водой). В случае хлорирования
1 S2C12 — жидкость с точкой замерзания минус 77° С. Давление пара при
-40° С равно 28 мм рт. ст.
182
стандартных концентратов (содержащих ~85%~MoS2) основные
примеси в пятихлористом молибдене -— хлорное железо и хлори-
стый алюминий. Очистка от них рассмотрена ниже. Один из не-
достатков хлорирования молибденита — необходимость утилиза-
ции получаемой хлористой серы, а также повышенный расход
хлора.
Очистка МоС15
Чистота пятихлористого молибдена зависит от принятого спо-
соба его получения. Если исходным материалом служит ферро-
молибден или молибденитовый концентрат, МоС15 может содер-
жать значительные количества FeCl3,- а также примеси хлоридов,
других элементов.
Солевая очистка. Эффективным способом очистки пятихлори-
стого молибдена от FeCl3 (температура кипения 319° С), а также
от А1С13 (температура кипения 194° С) может служить пропуска-
ние паров МоС15 через колонку, заполненную хлористым натрием
или хлористым калием [23]. Хлориды железа и алюминия обра-
зуют с NaCl или КС1 легкоплавкие и малолетучие соединения
7VfeFeCl4 и МеА1С14, тогда как МоС15 с хлоридами щелочных метал-
лов химически не взаимодействуют. Соединение NaFeCl4 имеет
точку плавления 156° С, a NaAlCl4 108° С. При изучении систем
NaCl—FeCl3 и NaCl—А1С13 термическим анализом соединения
NaFeCl4 и NaAlCl4 обнаружить не удается (им практически от-
вечают эвтектические составы). Однако резкое понижение давле-
ния пара в системе FeCl3—NaCl над расплавами, содержащими
более 50% (мол.) NaCl по сравнению с чистым хлоридом железа,,
а также то, что смесь компонентов, отвечающая составу NaFeCl4,
перегоняется без разложения, указывает на образование соеди-
нения [24].
Давление пара NaFeCl4 определяется уравнением
\gpMM рт. ст. = - + 5,80. (120)
Оно составляет:
Температура, °C....... 502 550 602 650 720 800
Давление пара NaFeCl4,
мм рт. ст ............ 5,3 6,9 30,5 45,0 79,1 156
Системы МоС15—FeCl3 и МоС15—А1С13 — эвтектического типа
(рис. 63 и 64), химические соединения в них не обнаружены [25].
Изучена также диаграмма плавкости системы МоС15—А1С13—
FeCl3 [26].
В работе [23] описана солевая очистка пятихлористого молиб-
дена от хлоридов железа и алюминия при составе исходной
смеси хлоридов: 50% МоС15, 25% FeCl3 и 25% А1С13. Смесь хло-
ридов испарялась в испарителе и током сухого хлора увлекалась
183.
в кварцевую колонку, заполненную кусочками (10—20 мм) NaCl
или смесью NaCl + КО. Температуру в солевой колонке под-
держивали в интервале 300—400° С.
В сконденсированном МоС15 содержалось менее 0,005% Fe
и 0,003% А1. Повышение температуры в солевой колонке до
500—550° С ухудшает качество очистки из-за заметного давления
паров NaFeCl4 и NaAlCl4 при
этих температурах.
Очистка ректификацией и
возгонкой. Возможность очистки
пятихлористого молибдена рек-
Рис. 64. Диаграмма плавкости системы
МоС15—FeCl3
Рис. 63. Диаграмма плавкости си-
стемы МоС15—А1С13
тификацией показана в работе [22]. Ректификацию проводили
в стеклянной колонке с 15 ситчатыми тарелками. Диаметр
колонки 30 мм, общая высота 600 мм, расстояние между тарел-
ками 30 мм, диаметр отверстий на тарелке 0,8 мм, число отвер-
стий 45. Объем перегонного куба 1000 мл (рис. 65). Колонка
и дефлегматор обогревались электроспиралью. Куб присоединяли
к колонке с помощью шлифа, смазываемого графитом. Скорость
отбора фракций регулировали с помощью игольчатого клапана.
Было показано, что ректификация — эффективный способ раз-
деления молибдена и вольфрама. При ректификации смеси 95%
МоС15 и 5% WC16 была получена основная фракция МоС15 (отбор
в интервале 272—292° С), содержащая лишь следы вольфрама при
выходе чистого продукта 65,8%.
В случае отсутствия примеси оксихлоридов гексахлорид
вольфрама с точкой кипения 337° С (что на 69 град выше точки
кипения МоС15) концентрируется в кубовом остатке. Практически
МоС15 всегда содержит примеси оксихлоридов, например МоОС13,
образующихся при кратковременном контакте хлорида с возду-
хом. В этом случае значительная доля вольфрама вместе с частью
молибдена удаляется в первые низкокипящие фракции. Судя по
184
оранжевому цвету этих фрак-
ций и температуре кипения,
вольфрам присутствует в них
в виде окситетрахлорида
WOC14 (/кип = 232° С). По
всей вероятности, его образо-
вание происходит в резуль-
тате реакции
WC16 + МоОС13 -> WOCL +
+ МоС15. (121)
Хорошее отделение мо-
либдена от вольфрама проис-
ходит и в том случае, когда
в смеси преобладает WC16.
Так, при ректификации смеси
95% WC16 и 5% МоС15 полу-
чена фракция чистого WC16
(отбор при 336—337° С) с со-
держанием 0,01 % Мо при
выходе фракции 70—85% и
богатая молибденом фракция
(выход 82 % от исходного
количества молибдена) с со-
держанием Мо/Мо + W, рав-
ным 53—54*% [22].
Поведение малых коли-
честв примесей при ректифи-
кации МоС1б еще недоста-
точно изучено. По данным
автора, возможна глубокая
очистка от примесей Na, Mg,
Са, Си, Zn, Pb, Al, Fe.
Бадиали и др. [61 ] сопо-
ставили чистоту МоС15, полу-
ченного дистилляцией при
270—285° С и сублимацией
хлорида при .температуре
несколько ниже" точки плав-
ления (194° С) в токе хлора.
В конденсаторах? поддержи-
вали температуру 210—220°С.
Авторы отмечают, что при
дистилляции в аппарате из
стекла пирекс хлорид молиб-
дена был загрязнен приме-
сями элементов Na, В, Si
Рис. 65. Схема установки для ректифика-
ции МоС15:
/ — склянка с поглотителем влаги; 2 —"деф-
легматор; 3 — теплоизоляция; 4 — приемник
хлорида; 5 — ректификационная колонка;
6 — шлиф; 7 — куб-испаритель; 8 — электро-
плитка; 5 — термометр; 10 — штатив
185
и Al, содержащихся в стекле. Хлорид высокой чистоты был по-
лучен при сублимации в токе хлора. Из исходного пятихлористого
молибдена с содержанием, %: <0,0105 Fe; <0,0092 Al; 0,0014 Си
получали конденсат с содержанием, %: <0,001 Fe, Al, Si (каж-
дой), <5-10-бСг, остальные примеси (В, Мп, Mg, Ni, Си, Са,
Со) ниже предела чувствительности аналитических опоеделений.
33. НИЗШИЕ ХЛОРИДЫ МОЛИБДЕНА
Четыреххлористый молибден
МоС14 — мелкокристаллический порошок коричневого цвета,
который выше 130° С испаряется и разлагается (диспропорцио-
нирует) по реакции
2МоС14газ <1 МоОзтв “F МоС15Газ. (122)
В парах в интервале температур 330—1600° С равновесие реакции
сдвинуто влево — основная составляющая газовой фазы МоС14
[39]. В парах тетрахлорид мономерен.
Обратимость написанной выше реакции используют иногда
для синтеза тетрахлорида молибдена. С этой целью эквимолярную
смесь МоС13 + МоС15 с небольшим избытком МоС15 длительное
время (до 25 ч) выдерживают в запаянной ампуле при температуре
250° С. После охлаждения и вскрытия ампулы избыток МоС15
отгоняется под вакуумом при 120° С [28].
Распространенный метод синтеза тетрахлорида — хлориро-
вание двуокиси молибдена в смеси с углем (отношение по массе
5 : 1) парами четыреххлористого углерода в потоке чистого инерт-
ного газа при 300° С:
МоО2 + 2CCL = МоС14тв + 2СОС12, (123)
МоО, + CCL = МоС14тв + СО,. (124)
Азот барботирует через жидкий СС14 при температуре 20—
25° С и далее проходит над лодочкой с двуокисью молибдена. По-
лученный МоС14 очищается от летучих примесей нагреванием в ва-
кууме при 60—70° С [11].
Другие методы синтеза — взаимодействие МоО2 с СС14 в запаян-
ной ампуле при 250° С [27] и метод диспропорционирования три-
хлорида молибдена при 500° С (2МрС13 = МоС12 + МоС14) [1]
не приводят к получению чистых препаратов МоС14.
Авторы работы [39] утверждают, что ни один из описанных
выше способов синтеза не обеспечивает получения чистого МоС14.
Они разработали методику синтеза кристаллического тетрахлорида
молибдена высокой чистоты, основанную на упомянутой выше
реакции взаимодействия МоС13 с МоС15. Синтез проводят в за-
паянной ампуле (длина 130 мм, диаметр 16 мм), в которой нахо-
183
дится молибденовая проволока или фольга и пятихлористый мо-
либден.
Первоначально ампулу выдерживают 12 ч в печи с температур-
ным перепадом 450 —> 375° С, причем молибденовую проволоку
помещают в зону 450° С. В результате в зоне 375° С конденсируется
некоторое количество смеси МоС13 и МоС14. Кроме того, в ней
остается значительное количество МоС15. Затем ампулу выдер-
живают в печи с температурным перепадом 255 —> 235° С, причем
конденсат МоС13 и МоС14 находится в зоне 255° С. Трубку распо-
лагают слегка наклонно с таким расчетом, чтобы находящийся
в избытке МоС15, конденсирующийся в зоне 235° С в жидком виде,
стекал в зону 255° С, там испарялся и снова поступал в зону
235° С. Это обеспечивает хорошую циркуляцию газов. В процессе
выдержки МоС13 превращается в МоС14, который накапливается
в зоне с 235° С. В конце избыток МоС15 отгоняется при температур-
ном перепаде 150 —> 20° С.
Этим способом были получены черно-коричневые кристаллы
в форме гексагональных призм. Они без остатка растворяются
в воде, 6-н. НО, этаноле и эфире. Полное растворение МоС14
в воде служит критерием его чистоты [39]. Хорошие’растворители:
тетрахлорида молибдена СС14, CS2 и СНС13 [30].
Тетрахлорид молибдена кристаллизуется в тригональной син-
гонии, пространственная группа РЗ\С — Did [39]. Периоды ре-
о
шетки а = 6,058, с = 11,674 A, da = 1,927. Каждый атом молиб-
дена окружен 6 атомами хлора, расположенными в вершинах
октаэдра. Расстояния Мо — С1 равны 2,42 А, кратчайшие
расстояния С1—С1 равны 3,46; 3,47 и 3,69 А. Рентгенографиче-
ская плотность МоС14 равна 3,19 г!см3 [39].
*
Треххлористый молибден
МоС13 — мелкокристаллический порошок, по внешнему виду
подобный свежевосстановленной меди.
Трихлорид молибдена кристаллизуется в решетке моноклин-
ного типа [38, 39]. Пространственная группа С21т. Постоянные
решетки: а = 6,065,, b = 9,760, с = 7,250 А, [3 = 124°. Числа
атомов в элементарной ячейке z = 4.
Структура слоистая, нового типа, со слоями, параллельными
плоскости ху. Анионы расположены по закону плотной гексаго-
нальной упаковки. В слое катионов атомы молибдена расположены
подобно атомам углерода в структуре графита, но симметрия по-
нижена с 6 до 2 из-за тенденции атомов молибдена группироваться
парами. Расстояние между парными атомами Мо <--* Мо равно
о о “
2,77 А, а между парами 3,70 А. Каждый атом молибдена окружен 6
атомами хлора, расположенными в вершинах искаженного окта-
187
эдра. Расстояния Мо—С1 будут 2,40; 2,45 и 2,55 А. Расстояния
С1—С1 равны 3,23 А. Плотность dpeHT = 3,77 г! см*.
Трихлорид при нормальной температуре устойчив на воздухе,
при нагревании окисляется до оксихлорида МоО2С12.
При отсутствии кислорода при температуре выше 450° С дис-
пропорционирует с образованием дихлорида и тетрахлорида:
2МоС13тв = МоС12тв + МоС14Газ. (125)
Трихлорид нерастворим в холодной воде, но медленно гидро-
лизуется при нагревании с образованием гидратированных окси-
хлоридов, постепенно окисляющихся до молибденовой кислоты.
В серной кислоте МоС13 растворяется, давая зеленые растворы,
содержащие катионы Мо3+ и МоО+. Трихлорид мало растворим
в разбавленной соляной кислоте, но растворяется в концентри-
рованной соляной кислоте. Растворы окрашены в красный цвет,
что обусловлено образованием анионов (МоС16)3’. Трихлорид
молибдена не растворяется в этиловом спирте, этиловом эфире,
ацетоне. Хорошие растворители МоС13, пиридин (C5H5N) и нитро-
бензол (C6H5NO2). С первым он образует комплекс MoC13‘3C5H5N
[17, 31]. При сплавлении МоС13 с хлоридами натрия и калия
образуются комплексные соли 7Ие3МоС16, которые используются
при электролитическом получении молибдена.
Трихлорид молибдена получают хлорированием молибдена или
восстановлением МоС15 водородом.
Для получения МоС13 действием хлора на молибден*молибде-
новый порошок хлорируют при температурах 600—700° С хлором,
разбавленным азотом, так как при избытке хлора образуется
МоС15.
Полученный этим методом МоС13 содержит примесь МоС12, ко-
торый может быть удален растворением в абсолютном этиловом
спирте.
Лучший способ получения МоС13 — восстановление пентахло-
рида водородом.
В работе [28] рекомендуется проводить восстановление в авто-
клаве из нержавеющей стали, защищенном стеклянной облицов-
кой. Пятихлористый молибден восстанавливается водородом под
давлением ~7 ат при температуре 125° С и молярном отношении
Н2 : МоС15 =5:1. За 16 ч выход МоС13 составляет 98%. Для
очистки от МоС15 и НС1 продукт нагревают в вакууме при 250° С.
Чистый препарат может быть получен также действием паров
брома на КзМоС16 [31]:
К3МоС16 + 3/2Вг2 -> ЗКВг + МоС13 + 3/2С12. (126)
Пары брома пропускают над исходной солью при температуре
450° С. Бромистый калий отделяют от трихлорида обработкой
водой и затем этиловым спиртом.
188
В работе [39] МоС13 был синтезирован с использованием метода
химического транспорта на основе реакции (122). Были получены
черные кристаллы, которые при растирании образуют красный
порошок.
Дихлорид молибдена
МоО 2 — порошок желтого цвета, приобретающий при нагре-
вании выше 600° С зеленоватый оттенок. Плотность дихлорида
Df = 3,714 [1].
Давно установлено, что молекулы дихлорида молибдена поли-
меризованы, им приписывали состав Мо3С16. Два из шести атомов
хлора отличаются от остальных (они осаждаются нитратом се-
ребра, вступают в реакцию двойного обмена [34]). Это послужило
основанием рассматривать полимер как координационное соеди-
нение типа (Мо3С14) С12. Два внешнесферных иона хлора способны
замещаться на ионы брома или иода.
Другая особенность Мо3С16 состоит в способности присоединять
молекулы галогенидов или галогеноводородных кислот с образо-
ванием комплексных, солей или кислот. Известны, например,
соли К 2 (Мо3С18) -2Н2О; (NH4)2 (Мо3С18)-2Н2О, а также кислота
Н(Мо3С17)-Н2О.
Рентгеноструктурные исследования показали, что дихлориду
правильней приписать формулу гексамера состава (Мо6С18) С14.
Дихлорид молибдена Мо6С112 кристаллизуется в орторомбиче-
ской решетке с периодами а = 11,249, b = 11,280, с = 14,067 А,
пространственная группа ВЬаш—D^h. Элементарная ячейка со-
держит 4Мо6С112 [39].
В основе структуры лежит многоядерный ион (Мо6С18)4+,
строение которого показано на рис. 66, а [32—34]. Восемь ато-
мов хлора образуют куб с длиной ребра 3,48 А, несколько выше
центра граней которого расположены шесть атомов молибдена
(в вершинах октаэдра). Атомы молибдена лежат выше центра грани
куба на 0,11 А, расстояния Мо—Мо равны 2,613 А.
Остальные 4 атома хлора, лежащие вне иона (Мо6С18)4+, не-
равнозначны. В решетке можно выделить группы (MoeCls) CI2,
которые связаны в бесконечные цепи четырьмя атомами хлора
(С1а“а) (из которых два относятся к каждой группе). В целом
структура Мо6С112 может быть охарактеризована как «молеку-
2
лярная слоистая» и описана общей формулой сю [МобСЦ] Cl^Cl^
[39]. Взаиморасположение всех атомов показано на рис. 66, б.
Каждый СН-атом связан с тремя атомами Мо, каждый СГ~а-атом —
с двумя атомами Мо, каждый С1а-атом — с одним атомом Мо.
Расстояния между атомами Мо—С1 равны 2,47 А, Мо—С1а“а
равны 2,50 А, Мо—С1а равны 2,38 А. Углы между связями (Мо—
—Мо) равны 130,2 и (Мо—СР—Мо) равны 63,9 [39].
189
Дихлорид молибдена не растворяется в воде и разбавленных
кислотах, но растворяется в концентрированных галогеноводо-
родных кислотах. Из солянокислых растворов выделен кристал-
логидрат (Мо6С18) С14-4Н2О. При добавлении щелочи осаждается
гидроокись желтого цвета (Мо6С18) (ОН)4-14Н2О.
Таким образом, было установлено, что комплекс (Мо6С18)4+
сохраняется и в растворах [35].
Дихлорид молибдена хорошо растворим в этиловом спирте,
что используется при разделении низших хлоридов молибдена.
Рис. 66:
а — структура многоядерного иона ^MogClg]4-^; б — структура ^MogClg ClpCl^^
атомы Мо — меньшего размера, атомы С1 — большего размера
МоС12 устойчив в твердом состоянии до температуры при-
мерно 730° С. Выше этой температуры он диспропорционирует^по
реакции
2МоС12тв 7~* Мо + МоС14газ.
Давление пара МоС14, равное 1 ат, достигается для этой ре-
акции при 1080° С [39]. На воздухе МоС12 окисляется до МоО3.
Описаны следующие способы получения дихлорида молибдена:
1. Термическое разложение МоС13 [реакция (125)]. Методику
синтеза описали Сендерофф и Бреннер [36]. В запаянной эвакуи-
рованной трубке из стекла пирекс (диаметр 1,5 см, длина 30 см)
нагревают при 350° С в течение 48 ч смесь МоС15 и порошка мо-
либдена с избытком МоС15 ~ 14% от необходимого по реакции.
При этом образуется МоС13:
ЗМоС15 + 2Мо - 5МоС13. (127)
При синтезе трубку располагают наклонно, чтобы продукты реак-
ции были в нижнем ее конце. После охлаждения вскрывают верх-
ний конец трубки, припаивают к нему согнутую под углом 120° С
трубку-конденсатор. Проводят откачку и отпайку, после чего от-
100
гоняют в конденсатор при 300° С избыток пятихлористого молиб-
дена. Оставшийся чистый МоС13 нагревают примерно 24 ч при
650° С. Образующийся МоС14 летит и конденсируется в холодной
части, а в горячем конце остается дихлорид молибдена. Он содер-
жит незначительную примесь молибдена. Более чистый МоС12
можно получить, если проводить диспропорционирование МоС13
при 600° С, что требует большего времени выдержки. По всей
вероятности, вторую стадию синтеза можно проводить при непре-
рывной откачке, не запаивая трубку-конденсатор. Выход ди-
хлорида примерно 65%.
2. Хлорирование молибдена парами четыреххлористого угле-
рода, разбавленного азотом, при 600° С [17]:
Мо + 2СС14 - МоС12 + С2С1б. (128)
Этим методом не удается получить дихлорид, свободный от при-
меси молибдена, так как плотный слой МоС12 на частицах металла
препятствует протеканию процесса.
3. Взаимодействие трибромида молибдена с сухим хлористым
водородом [37]:
MoBr3 + 2НС1 = МоС12 + 2НВг + 1/2Вг2. (129)
Реакция протекает при 500° С. Для удаления летучих компонен-
тов продукт выдерживают в вакууме при 500° С. Этот способ поз-
воляет получить чистый препарат с хорошим выходом.
34. ОКСИХЛОРИДЫ МОЛИБДЕНА
Диоксидихлорид молибдена
Мо02С12 — наиболее устойчивый из оксихлоридов. Это ос-
новной продукт хлорирования кислородных соединений молиб-
дена (МоО2, МоО3, молибдатов). МоО2С12 конденсируется в форме
легко возгоняющихся светло-желтых чешуйчатых кристаллов.
Давление пара над твердым оксихлоридом достигает одной атмо-
сферы при температуре 159° С [12]. Оксихлорид плавится при
170° С под давлением 1,5 ат [6].
Зависимости давления пара от температуры, полученные раз-
личными авторами, существенно отличаются.
Так, по данным работы Шарупина*:
lgpQm = 9,84-^-; (130)
АЯсУбл =19,5 ккал\
^^субл 45 5. в.
* Б. П. Ш а р у п и н. Диссертация, ЛГУ, 1960.
191
По данным работы Суворова*:
lgPam = 7,98 —
(131)
ДЯсубл — 16,3 ккал\
Д^субл 36,5 э» с.
Согласно первому уравнению, давление 1 ат над МоО2С12
достигается при 161° С (что близко к данным работы [12]), тогда
как по второму уравнению оно достигается при 173° С.
МоО2С12 устойчив в парах до 500° С. При более высоких тем-
пературах, вероятно, протекает частичное разложение соответ-
ственно реакции
2МоО2С12газ = МоО3тв + МоОС14газ.
(132)
В сухом воздухе МоО2С12 не изменяется, в присутствии влаги
образуется гидрат МоО2С12-Н2О. В воде диоксихлорид раство-
ряется с образованием кислых растворов, содержащих катионные
и анионные комплексы молибдена.
Диоксидихлорид молибдена хорошо растворяется в органиче-
ских растворителях: кетонах, спиртах, СС14, СНС13 и др.
Диоксидихлорид молибдена может быть легко получен одним
из следующих способов, которые далее будут детальней рассмо-
трены:
1) хлорированием двуокиси молибдена при температуре 200° С
[49, с. 70];
2) хлорированием трехокиси молибдена в смеси с углем при
температурах 350—400° С [49, с. 70]:
МоО3 + С12 + 1/2С = МоО2С12 + 1/2СО2; (133)
3) хлорированием смеси МоО3 с MoS2, взятых в молярном от-
ношении 6: 1 при температурах 350—400° С [55, 57]:
6МоО3 + MoS2 + 7С12 = 7МоО2С12 + 2SO2; (134)
4) нагреванием смеси МоО3 с NaCl при температурах 700—
800° С [52]:
2МоО3 + 2NaCl = МоО2С12 + Na2MoO4. (135)
С целью очистки диоксидихлорид перегоняют при 160—180° С
в токе сухого воздуха или азота.
Моногидрат диоксидихлорида («хлорид Дебре») — соединение,
которому приписывают состав МоО2С12-Н2О или МоО3-2НС1.
Оно получается при действии на МоО3 сухим хлористым водоро-
дом при 170° С в форме легколетучего белого с зеленоватым от-
тенком порошка. В парах при 160° С диссоциирует.
* А. В. Суворов. Диссертация. ЛГУ, 1961.
192
Химическая природа моногидрата не установлена. Сиджвик
[42] приписывает ему формулу О—Мо(ОН)2С12.
Моногидрат диоксидихлорида легко растворяется в воде, спир-
те, ацетоне и других органических растворителях.
Окситетрахлорид молибдена
Индивидуальность МоОС14 была достоверно установлена лишь
недавно в работах Щукарева с сотрудниками [6, 11] и Глухова
с сотрудниками [43, 44].
Окситетрахлорид кристаллизуется в виде темно-зеленых куби-
ческих кристаллов, обладающих дихроизмом: в очень тонком слое
в отраженном свете МоОС14 зеленый, а в проходящем — красный.
Температура плавления МоОС14 равна 102° С [44].
При отсутствии хлора МоОС14 несколько диссоциирует уже при
комнатной температуре и быстро при нагревании с образованием
МоОС13:
Для этой реакции
МоОС14^МоОС13+ 1/2С12.
(136)
[7]
1g Кр = 3,152-
(137)
АЯ = 11,4 ккал;
AS- 14,4 э. е.
Давление пара МоОС14 можно измерить только в атмосфере
хлора.
В этих условиях зависимость давления пара от температуры
описывается уравнением [7]
lgpam = 4,783(138>
•АЯИС11 — 11,7 ккал;
Д5ИСГ1 —21,9 э. е.
Расчетное значение температуры кипения 260,5° С.
Во влажном воздухе МоОС14 расплывается, образуя синюю
жидкость.
Окситетрахлорид легко растворяется в различных органиче-
ских растворителях, таких как этиловый спирт, эфир, Дихлор-
этан, четыреххлористый углерод, хлороформ. С бензолом и аце-
тоном образует металлоорганические соединения [45].
13 А. Н. Зеликман 1^3
Описано несколько методов синтеза МоОС14:
1. Хлорирование МоО3 четыреххлористым углеродом:
МоО3 + 2CCL = MoOCl, + 2COCL.
° 1 5b Ц 1 Л
(139)
Синтез проводят в запаянных стеклянных ампулах при темпера-
туре 210—180° С в течение 5—7 ч.
Полученный продукт прогревают в вакууме при 50—70° С
для удаления СОС12 и СС14, а затем двукратно перегоняют под
вакуумом при 100—120° С.
2. Нагревание равномолярной смеси МоС1б и МоО2С12 в за-
паянных ампулах при 240° С [46, 44]:
МоС1б + МоО2С12 = МоОС14 + МоОС13. (140)
Для отделения МоОС14 от МоОС13 отгоняют окситетрахлорид
при температурах 180—200° С в холодную часть ампулы.
3. Хлорирование диоксихлорида молибдена смесью хлора
с однохлористой серой [43]:
4МоО2С12 + S2C12 + ЗС12 = 4МоОС14 + 2SO2. (141)
Над диоксидихлоридом при температуре 100—120° С медленно
пропускается струя хлора, насыщенного парами хлористой серы.
При этом возгоняется и конденсируется окситетрахлорид. Он очи-
щается сублимацией в токе хлора.
4. Хлорирование трехокиси молибдена и молибдатов смесью
хлористой серы и хлора [47].
Этот способ наиболее удобен для получения МоОС14 в относи-
тельно крупных количествах. Исходный материал обрабатывают
в стеклянной колбе жидкой хлористой серой при 100—120° С
при пропускании хлора. Протекающие реакции описываются урав-
нениями:
2МоО3 + S2C12 + ЗС12 = 2МоОС14 + 2SO2, (142)
4СаМоО4 + 3S2C12 + 9С12 = 4МоОС14 + 4СаС12 + 6SO2. (143)
Согласно работе [47], для получения МоОС14 в стеклянную
колбу с обратным холодильником загружают смесь МоО3 (или
СаМоО4) с серой в отношении 1,5—2 г-атом S на 1 г-атом Мо.
В колбу подают хлор для перевода всей загруженной серы в хло-
ристую серу, затем температуру поднимают до 100—120° С, при
которых бурно протекает реакция хлорирования. По окончании
реакции (прекращение выделения SO2 и появление избыточного
хлора) обратный холодильник заменяют трубкой и отгоняют
хлористую серу. Затем из колбы в токе хлора при температуре
200—220° С отгоняется окситетрахлорид, конденсирующийся
в приемнике, в котором поддерживают температуру 80—90° С.
Для очистки МоОС14 вторично перегоняют.
194
Окситрихлорид молибдена
МоОС13 синтезирован и изучен Глуховым и Елисеевым. Ав-
торы показали, что МоОС13 может быть получен взаимодействием
МоС1б с МоО2С12 [реакция (140)] или с МоО3 по реакции [44]:
МоО3 + 2МоС15 = 2МоОС13 + MoOCL.
(144)
Окситетрахлорид отделяется от менее летучего МоОС13 отгон-
кой.
Реакцию проводят в длинной эвакуированной запаянной ам-
пуле, в один из концов которой загружают стехиометрическую
смесь компонентов. Реакция начинается при 100° С. По окончании
реакции из полученной смеси оксихлоридов при температуре 180—
200° С отгоняется МоОСЦ, конденсирующийся в холодной части
ампулы. В нижней части ампулы остается почти чистый МоОС13,
который для очистки перегоняют при 280—300° С.
Другой метод отделения МоОСЦ от МоОС13 состоит в обработке
массы абсолютным четыреххлористым углеродом в атмосфере азота,
в котором растворяется окситетрахлорид. Полученные кристаллы
МоОС13 затем выдерживают в вакууме при 150—180° С.
МоОС13 представляет собой черные игольчатые кристаллы.
Плотность МоОС13 при 20° С равна 3,149 г! см3. Температура плав-
ления 295° С.
Давление пара в зависимости от температуры описывается
уравнением
lgpam = 8,764--^;
А//Субл = 25 ккал}
Д^субл ~ 40 э. е.
Расчетное значение температуры кипения 352° С [13].
Выше 200° С МоОС13 заметно диссоциирует по схеме
ЗМоОС13 - МоС13 + MoOCL + МоО2С12. (145)
О О 1 ху л \ Z
Во влажном воздухе МоОС13 расплывается в буро-коричневую
жидкость, которая затем синеет. В воде окситрихлорид быстро
гидролизуется, коричневый раствор постепенно синеет и затем
осаждается молибденовая синь.
МоОС13 растворяется в соляной и азотной кислотах с образо-
ванием зелено-желтых растворов. Оксихлорид растворим в абсо-
лютированном спирте и ацетоне и практически нерастворим на
холоду в четыреххлористом углероде, хлороформе, дихлорэтане,
бензоле, эфире, толуоле.
13* 195
Оксидихлорид молибдена
МоОС12 недавно синтезирован и изучен Шеффером и Тилла-
ком [48]. Возможны два метода его получения, в основе которых
лежат реакции:
МоО3 + 2МоС13 = ЗМоОС12,
5МоО3 + 6МоС15 + 4Мо = 15МоОС12.
(146)
(147)
Синтез проводят в длинных запаянных ампулах в атмосфере
хлора, что позволяет одновременно осуществлять химическую
‘транспортировку с получением крупных кристаллов МоОС12.
При синтезе по первой реакции взаимодействие компонентов
и химическая транспортировка осуществляются при градиенте
температур 400 300° С, при синтезе по второй реакции — при
градиенте 475 —> 350° С.
Оксидихлорид можно получить также по реакции
МоО2 + МоС13 + 1 /2С12 = 2МоОС12.
(148)
Исходные продукты нагревают в запаянной ампуле при гра-
диенте температуры 400 —* 300° С.
Оксидихлорид кристаллизуется в форме игл бронзово-корич-
невого цвета. Тип решетки — моноклинный с параметрами: а =
= 12,77 А, b = 3,759 А, с = 6,54 А; 0 = 104,8°. Рентгенов-
•ская плотность d = 3,98 г/см3, опытное значение d = 3,90 г!см3.
Ориентировочное значение энтальпии образования
ЛЯ = —125 ± 4 ккал!моль.
При нагревании в высоком вакууме (~10“б мм рт. ст.) выше
350° С МоОС12 разлагается с образованием МоС12 и МоО2С12.
При нагревании в сухом воздухе происходит окисление до МоО2С12,
МоОС13 и МоО3.
Оксидихлорид не растворяется в воде и концентрированной
соляной кислоте ни на холоду, ни при нагревании. Горячая кон-
центрированная серная кислота медленно с ним взаимодействует,
горячая азотная кислота окисляет его до молибденовой кислоты.
Серная кислота в присутствии КМпО4 или Н2О2 растворяет
МоОС12.
При действии нагретых растворов концентрированного аммиака
или 2-н. NaOH образуются черные суспензии.
35. ХЛОРИРОВАНИЕ КИСЛОРОДНЫХ СОЕДИНЕНИЙ
МОЛИБДЕНА
Окислы молибдена (МоО2, МоО3) и молибдаты кальция, свинца,
меди, железа и др. взаимодействуют с различными хлорирующими
агентами (хлор, четыреххлористый углерод, хлориды серы) пре-
имущественно с образованием оксихлоридов — МоО2С12 и МоОСЦ*
196
В определенных условиях получаются также и хлориды (МоС15,
МоС14), например при хлорировании окислов молибдена хлором
при высоких температурах в присутствии избытка угля или взаи-
модействии СС14 с МоО3 под давлением (в запаянных ампулах).
Однако они всегда содержат значительные количества оксихлори-
дов. Термодинамические характеристики некоторых реакций хло-
рирования окислов молибдена приведены в табл. 49.
ТАБЛИЦА 49
ТЕРМОДИНАМИЧЕСКИЕ ФУНКЦИИ НЕКОТОРЫХ РЕАКЦИЙ
ХЛОРИРОВАНИЯ ОКИСЛОВ МОЛИБДЕНА 1
№ реак- ции Реакция A w® дп298* ккал! моль ккал! моль
1 МоОзтв + С12 = МоО2С12Газ + 1/2О2 +23,2 +25,63
2 МоОзтв + С12 + 1/2С = —23,825 —25,95
= МоО2С12газ + 1/2СО2
3 1/2МоОетв +С12 — 1/2С = —28,425 —0,33
= 1/2МоОС14Газ — 1/2СО2
4 МоОзтв + 1/2С = МоО2тв + 1/2СО2 —9,625 + 17,67
5 МоОгтв CI2 == МоОгС^газ —14,2 +8,3
1 При расчетах для оксихлоридов использованы данные табл. 48.
Двуокись молибдена
Двуокись молибдена с заметной скоростью реагирует с хлором
при температурах 100—120° С, а при 200° С и выше реакция про-
текает активно с образованием диоксидихлорида:
МоО2 тв + С12 = МоО2С12 газ; (149)
А//298 = —14,2 ккал]молъ\
AZ°r = —14 200 — 8,ЗТ;
AZ230 = —18,35 ккал!моль.
Это наиболее простой способ получения диоксидихлорида молиб-
дена.
Двуокись молибдена может быть получена восстановлением
МоО3 увлажненным водородом при температуре 550° С.
Отсутствие остатка после хлорирования двуокиси молибдена
при температурах 180—300° С свидетельствует о соответствии ее
теоретическому составу. Если в двуокиси присутствуют примеси
промежуточных окислов, например Мо4Оп, образуется остаток
МоО3, так как при указанных температурах Мо4Ои взаимодей-
ствует с хлором по реакции
Мо4Ои + С12 = МоО2С12 + ЗМоО3.
(150)
197
Кинетика хлорирования двуокиси молибдена изучалась [49,
с. 70] методом непрерывного взвешивания цилиндрического бри-
кета МоО 2, спрессованного под давлением 2500 кПсм2.
Рис. 67. Зависимость степени
хлорирования брикета МоО2
от температуры. Время хлори-
рования 18 мин (/) и 22 мин (2)
На рис. 67 видно, что хлориро-
вание МоО2 наблюдается несколько
выше 100° С, скорость процесса резко
возрастает в интервале температур
170—200° С и затем мало зависит от
температуры.
При всех температурах реакция
протекает лишь на поверхности бри-
кета, который в процессе хлорирова-
ния сохраняет свою форму и перво-
начальную плотность. Это позволяет
по степени хлорирования рассчиты-
вать глубину хлорирования (X, см)
и линейную скорость хлорирования
К = — см/сек.
т
Из значений кажущихся энергий
активации (табл. 50) следует, что
в интервале температур до 185° С скорость хлорирования дву-
окиси молибдена лимитируется скоростью химического взаимо-
действия (кинетическая область), тогда как выше 185° С скорость
хлорирования определяется диффузией — подводом хлора к ре-
акционной поверхности.
ТАБЛИЦА 50
СКОРОСТЬ И КАЖУЩАЯСЯ ЭНЕРГИЯ АКТИВАЦИИ ХЛОРИРОВАНИЯ
МоО 2 ХЛОРОМ [49, с. 70]
Температура, °C Константа скорости А = ~Y“. см/мин Энергия активации Е, кал/моль
140 0,0014
175 0,0044 21 650
185 0,0102
200 230 0,0142 0,0172 2350
290 0,0210 »
370 0,0227 1400
540 0,0254
188
Трехокись молибдена
Хлорирование хлором в присутствии углерода
или четыреххлористым углеродом
Взаимодействие трехокиси молибдена с хлором наблюдается,
начиная примерно с 400° С; активно реакция протекает в интервале
600—700° С с образованием диоксидихлорида.
Это находится в хорошем соответствии с данными о зависимости
изобарно-изотермического потенциала реакции от температуры
Рис. 68. Зависимость AZ
от температуры для неко-
торых реакций хлориро-
вания окислов молиб-
дена:
1—5 — номера реакций (см.
табл,. 49)
(рис. 68); AZ < 0 при температу-
рах выше 630° С.
Температура активного хлори-
рования МоО3 резко снижается
°? м
I 50
30
Qj
£ 10
Б
200 300 • М 500 800 700
Температура, °C
Рис. 69. Степень хлорирова-
ния МоО3 различными хло-
рирующими агентами в зави-
симости от температуры.
Навеска МоО3 1 а, время
хлорирования 1 ч:
1 — хлор; 2 — смесь СО + С12;
3 — СС14; 4 — С12 —Н уголь
в присутствии угля, окиси углерода или при использовании че-
тыреххлористого углерода, что видно на рис. 69, построенном по
данным работы [50]. Хлорирование хлором в присутствии угля или
четыреххлористым углеродом протекает с высокой скоростью при
350—400° С, смесью хлора с окисью углерода — в интервале 450—
550° С.
Реакция с хлором в присутствии угля протекает с более вы-
сокой скоростью (особенно при низких температурах), чем с че-
тыреххлористым углеродом.
При хлорировании в присутствии угля с наибольшей убылью
свободной энергии протекает реакция (133) (см. рис. 68).
Возможно, хотя и менее термодинамически вероятно, также
образование МоОС14 по реакции
МоО3 + 2С12 + С = MoOCL + СОа.
(151)
199
В работе [49, с. 70] исследована кинетика хлорирования МоО3
в присутствии углерода гравиметрическим методом. Смесь ком-
понентов (МоО3 и ламповая сажа), взятых в соотношении, отве-
чающем реакции (133), брикетировали под давлением 2500 кПсм2.
Кинетику хлорирования изучали в интервале температур 300—
675° С при скорости потока хлора 140 см/мин. С течением времени
брикет сокращался в размерах, сохраняя форму, прочность и
исходный состав.
Механизм хлорирования в большой степени зависит от интер-
вала температур, в котором протекает процесс. Это хорошо выяв-
Рис. 70. Зависимость степени хлори-
рования брикетов из смеси трехокиси
молибдена с сажей от температуры.
Время хлорирования 16 мин (/) и
19 мин (2)
ляется при построении зависи-
мости степени хлорирования от
температуры (рис. 70), где можно
выделить несколько областей.
В интервале 280—370° С ско-
рость хлорирования резко рас-
тет с увеличением температуры
от 290° С (начало заметного
хлорирования) до максимума
при 370° С. В этой области тем-
ператур хлорирование проте-
кает в кинетической области
с высокой энергией активации
Е = 32 400 кал!моль.
В интервале температур
370—440° С отмечается резкое
торможение реакции (площадка
и минимум на кривой рис. 70).
Это объясняется тем, что выше
370° С, наряду с реакцией хло-
рирования, наблюдается восста-
новление МоО3 сажей, вероятно,
до промежуточного окисла Мо4Оп. Реакция восстановления
МоО3 углеродом в интервале температур 370—420° С идет пре-
имущественно на поверхности брикета, чему способствует при-
сутствие хлора, снижающего температуру начала восстановле-
ния окислов углеродом [49, с. 61].
Промежуточный окисел Мо4Оп взаимодействует с хлором с об-
разованием МоО3 по реакции (150). Это приводит к возникновению
на поверхности брикета оболочки трехокиси молибдена, тормозя-
щей реакцию хлорирования.
Возрастание скорости хлорирования с повышением температуры
компенсируется торможением реакции оболочкой МоО3, что объ-
ясняет наличие горизонтального участка на кривой рис. 70 в ин-
тервале 370—420° С. При дальнейшем увеличении температуры
(интервал 420—440° С) скорость восстановления возрастает. Ин-
тенсивное выделение СО2, наряду с тормозящим влиянием обо-
200
лочки МоО3, вызывает резкое снижение скорости хлорирования
(минимум на кривой рис. 70).
При температуре выше 440° С МоО3 восстанавливается угле-
родом быстро во всем объеме брикета с образованием двуокиси
молибдена, которая затем хлорируется без остатка [реакция (149)].
Кажущаяся энергия активации процесса хлорирования выше
440° С Е = 2250 кал!моль близка по величине энергии активации
хлорирования чистой МоО2 для этих температур (см. стр. 98)
и отвечает протеканию процесса в диффузионной области.
Время t.MUH
Рис. 71. Кинетические кривые хлорирования бри-
кетов МоО3 + V2 С при /, °C:
1 _ 435; 2 — 370—390; 3 — 485; 4 — 540; 5 — 675;
6 — 440; 7 — 330
Выделяющаяся в результате восстановления МоО3 углеродом
СО2 препятствует доступу хлора к поверхности брикета, вслед-
ствие чего в начальный момент скорость хлорирования замедлена.
Это объясняет S-образную форму кинетических кривых для об-
ласти температур выше 440° С (кривые 5, 4, 5 на рис. 71).
Единственный продукт хлорирования при указанном выше
составе шихты (МоО3 + 1/2С) —диоксидихлорид МоО2С12. Лишь
при избытке углерода и температурах хлорирования выше 500° С
в конденсате появляется МоОС14.
Хлорирование трехокиси молибдена в смеси с углем может
быть использовано для получения трехокиси молибдена высокой
чистоты из технической трехокиси или непосредственно из про-
дуктов обжига молибденитовых концентратов (огарков), как было
показано в работе [49, с. 70].
201
Брикеты из смеси огарка с нефтяным коксом (соотношение
МоО3 + 1/2С) рекомендуется хлорировать в интервале темпера-
тур 350—450° С. Вследствие присутствия нехлорируемых при-
месей (SiO2, А12О3) после полного хлорирования брикет сохра-
нял свою форму и прочность, однако существенного торможения
процесса при этом не наблюдалось. Степень хлорирования в за-
висимости от состава огарков колеблется от 98 до 99,9%. При зна-
чительном содержании молибдатов (СаМоО4 и др.) необходимо для
обеспечения полного хлорирования проводить процесс при 550—
600° С.
В табл. 51 приведено содержание некоторых примесей в про-
дуктах хлорирования, а также в диоксихлориде после его очистки
возгонкой при 180° С.
ТАБЛИЦА 51
ПОВЕДЕНИЕ ПРИМЕСЕЙ ПРИ ХЛОРИРОВАНИИ ОГАРКА И
ВОЗГОНКЕ КОНДЕНСАТА
Продукт Масса, г Содержание примесей, %
Fe Си SiO2 СаО А1 W
Эгарок Остаток после хлориро- 214,3 3,18 2,23 16,72 1,33 1,26 0,23
вания 108,2 3,65 4,43 33,3 2,63 2,52 0,43
Конденсат МоО2С12 • . . Остаток после возгонки 154,0 1,88 0,0002 0,003 0,001 0,001 <0,02
конденсата Сублимат МоО2С12 после 28,4 9,0 0,0002 0,003 0,001 0,001 *<0,02
возгонки 125,5 0,24 0,0002 0,003 0,001 0,001 <0,02
При хлорировании происходит достаточно полная очистка от
примесей меди, кремния, кальция, алюминия и вольфрама. В про-
цессе хлорирования и возгонки МоО2С12 железо отделяется не-
полно. Однако при последующем переводе оксихлорида молибдена
в трехокись достигается глубокая очистка от этой примеси.
Для получения МоО3 дйоксидихлорид растворяют в чистой
воде на холоду при т : ж = 1 : 3 (что отвечает концентрации мо-
либдена в растворе 160 г/л). После фильтрации от незначительной
взвеси раствор нейтрализуют до pH =2^-3 раствором аммиака
(8%-ным) при 50—60° С. Выпавший мелкокристаллический осадок
тетрамолибдата (NH4)2O-4МоО3-2Н2О фильтруют и после про-
мывки холодной водой прокаливают при 500—550° С. В табл. 52
сопоставлен состав исходного огарка и полученной трехокиси
молибдена, имеющей высокую чистоту.
202
ТАБЛИЦА 52
СОДЕРЖАНИЕ ПРИМЕСЕЙ В ИСХОДНОМ ОГАРКЕ И ТРЕХОКИСИ
МОЛИБДЕНА, ПОЛУЧЕННОЙ ЧЕРЕЗ ОКСИХЛОРИД
Примесь Исходный огарок, % МоО3, % Примесь Исходный огарок, % МоО3, %
SiO2 16,72 0,0024 As 0,07 0,001
Fe 3,16 0,0009 Сг He опр. 0,001
Al 1,02. 0,0007 Р 0,01 0,001
Са 0,95 0,0035 Sb 0,08 0,0001
Mg 0,40 0,00023 Zn He опр. 0,0004
‘ ! Na Не опр. 0,005 Bi » » 0,0001
К » » 0,007 Sn 0,7 0,0001
Мп 0,12 0,001 Pb 0,015 0,00006
Ni Не опр. 0,0002 Cd He опр. 0,0001
W 0,23 <0,02 Cu 2,23 0,0002
Хлорирование МоО3 в присутствии молибденита
Как уже отмечалось, трехокись молибдена активно взаимо-
действует с хлором лишь при температурах выше 600° С. В при-
сутствии молибденита хлорирование МоО3 протекает с высокой
скоростью при значительно более
низких температурах (350—400° С)
[50, 55].
Исследования кинетики хлори-
рования смесей МоО3 с MoS2, взя-
тых в разных соотношениях
показали, что максимальные ско-
рость и степень реагирования соот-
ветствуют составу смеси с моляр-
ным отношением MoS2 : МоО3 = *
= 1:6 (рис. 72) [55].
В интервале температур 400—
450° С протекает реакция
MoS2 + 6МоО3 + 7С12 =
= 7МоО2С12 + 2SO2. (152)
Рис. 72. Скорости хлорирования
смеси MoS2 + МоО3 в зависимости
от состава при различных темпера-
турах, °C:
/ _ зоО; 2 — 400; 3 — 450
, Если состав смеси отклоняется от стехиометрического для на-
писанной реакции, то в остатке содержится тот компонент, кото-
рый находится в избытке.
Поскольку MoS2 реагирует с МоО3 при температурах выше
400° С с образованием МоО2 (см. стр. 29), реакцию хлорирования
смеси молибденита с трехокисью молибдена можно представить
протекающей в две стадии:
MoS2 4- 6МоО3 = 7МоО2 + 2SO2,
7МоО2 + 7С12 = 7МоО2С12.
203
‘ Хлорирование смеси МоО3 с MoS2 может быть использовано как
простой способ получения чистой трехокиси молибдена из молиб-
денитовых концентратов и полупродуктов [57] *.
Шихту, состоящую из огарка (продукта обжига молибденито-
вого концентрата) и сырого концентрата (молярное отношение
MoS2 : МоО3 =1:6) брикетируют или гранулируют на чашевом
грануляторе. Брикеты или гранулы хлорируют хлором в шахт-
ной печи при температуре 400—450° С. Уловленный в конденса-
торе диоксидихлорид молибдена очищают возгонкой при 170—
180° С и затем превращают в трехокись молибдена.
Гранулы после хлорирования смеси огарка с молибденитовым
концентратом сохраняют свой первоначальный размер и имеют
достаточно высокую прочность из-за остающегося кремнезема и
других нехлорируемых минералов. Это благоприятствует осуще-
ствлению процесса в шахтном хлораторе непрерывного действия.
Рассмотренный вариант проверен в применении к различным
материалам: огаркам обжига стандартных концентратов, полу-
продуктам обогащения, циклонной пыли обжиговых печей. При
температуре хлорирования 400° С средняя степень извлечения мо-
либдена в возгоны составляла 97—98,4%.
Полученный из очищенного возгонкой при 160—170° С диокси-
дихлорида парамолибдат аммония имел высокую степень чистоты.
Содержание примесей в парамолибдате, %: 0,006 Na; 0,002 Fe;
0,0015 Al; 0,0015 Mg; 0,00015 Са; 0,001 As; 0,001 SiO2 [57].
Хлорирование MoO3 хлоридами серы
Хлориды серы SCI2 и S2C12 — активные хлорирующие агенты.
Они получаются при пропускании хлора над серой, с которой хлор
взаимодействует при комнатной температуре. Состав образую-
щейся смеси жидких хлоридов серы зависит от температуры и
избытка хлора.
Монохлорид S2C12 — жидкость с резким удушливым
запахом. Температура замерзания минус 77° С, кипения 138° С.
Плотность монохлорида 1,68.
Дихлорид серы SCI2 — жидкость красного цвета, за-
твердевает при минус 122° С, кипит при 60° С. Плотность 1,629.
Для хлорирования кислородных соединений хлоридами серы
предварительно пропускают хлор через смесь жидких хлоридов
серы при температуре 38—40° С [58]. Газовая фаза содержит
смесь хлоридов S2C12, SCI2 и частично SC14 (соединение малоустой-
чивое).
Равновесное атомное отношение сера : хлор в газовой фазе
при температуре 38—40° С примерно равно 1 : 4 [59].
* См. также Зеликман А. И., Тараканов Б. М. Авт. свид.
СССР № 186687. Бюлл. изобр., 1966, № 19,
В смеси хлоридов преобладающим и более устойчивым является
монохлорид серы S2C12.
Реакция хлорирования трехокиси молибдена хлоридами серы
в потоке хлора наблюдаются при температурах 160—170° С и
протекает с высокой скоростью при 250—300° С с образованием
диоксидихлорида молибдена:
4МоО3 + S2C12 + 3CI2 = 4МоО2С12 + 2SO2.
(153)
Роль серы состоит прежде всего в связывании кислорода в SO2>
что увеличивает убыль свободной энергии процесса. Кроме того,
по всей вероятности, каталитически ускоряет процесс дихлорид
серы, который легко диссоциирует с выделением атомного хлора:
S2C14
^±S2C12 4- 2С1.
Хлорирование трехокиси молибдена хлоридами серы не имеет
существенных преимуществ перед хлорированием трехокиси в при-
сутствии угля или в смеси с молибденитом. В последних двух слу-
чаях практическая температура хлорирования лишь на 100 град
выше, чем при использовании хлоридов серы. Между тем работать
с хлоридами серы сложней, необходимо улавливать хлориды серы
из парогазовой смеси в системе конденсации.
Хлорирование МоО3 хлористым натрием
Исследования автора и Горовиц [52] показали, что трехокись
молибдена активно реагирует с хлористым натрием в интервале
температур 500—700° С с образованием МоО2С12 и молибдата
натрия. Реакция протекает примерно с одинаковой скоростью
и выходом МоО2С12 в токе воздуха и в аргоне. Однако в воздухе
наблюдалось некоторое выделение хлора, тогда как в атмосфере
аргона хлор не был обнаружен. В соответствии с этим взаимо-
действие МоО3 с NaCl в присутствии кислорода можно предста-
вить реакциями:
МоО3 + 2NaCl + !/2О2 = Na2MoO4 + С12
МоО3 + С12 = МоО2С12 + Х/2О2
2МоО3 + 2NaCl = Na2MoO4 + МоО2С12.
Небольшая доля хлора при этом удаляется, не вступив в ре-
акцию.
В атмосфере инертного газа механизм процесса иной: перво-
начально образуется оксихлорид молибдена и окись натрия, ко-
торая затем вступает в реакцию с МоО3:
МоО3 + 2NaCl = МоО2С12 + Na2O
МоО3 + Na2O = Na2MoO4
2МоО3 + 2NaCl = Na2MoO4 + МоО2С12.
205
Согласно написанным выше реакциям, до 50% Мо должно воз-
гоняться в форме МоО2С12. Практически степень возгонки состав-
_ * ляет 20—30%, так как часть
0 20 йО 60 80 100
Иа2МоО^ °/о(М0Л) Мо03
Рис. 73. Система Na2MoO4—МоО3
образующегося молибдата нат-
рия вступает в реакцию с МоО3
с образованием полимолибдатов
(Na 2О • 2МоО3; Na 2О • 4МоО3)
(рис. 73).
Хлорирование МоО3 хлори-
стым натрием может служить
простым препаративным мето-
дом глубокой очистки трех-
окиси молибдена от приме-
сей [53].
В частности, примесь воль-
фрама полностью отделяется
даже при высоком его содер-
жании (табл. 53). Други? при-
* меси содержатся в количе-
ствах ниже тысячных и деся-
титысячных долей процента
(табл. 54).
• . ТАБЛИЦА 53
СТЕПЕНЬ ОЧИСТКИ МоО3 ОТ ВОЛЬФРАМА ПРИ ТЕМПЕРАТУРЕ
600° С, ПРОДОЛЖИТЕЛЬНОСТЬ НАГРЕВАНИЯ с NaCI 30 мин [53]
Содержание WOS в смеси МоО3 + NaCI, % Отношение W/(Mo + W), % Извлечение моли- бдена в возгойы, %
в исходной смеси в оксихлориде
1 1,19 1,7-10~3 21,54
5 5,90 0,93-КГ3 21,38
25 28,80 1-Ю-3 20,04
ТАБЛИЦА 54
СОДЕРЖАНИЕ ПРИМЕСЕЙ в МоО3, ОЧИЩЕННОЙ ХЛОРИРОВАНИЕМ
ХЛОРИСТЫМ НАТРИЕМ [53]
Примесь Содержание, %
в исходной MoO3 в МоО3, очищенной через оксихлорид
•
А1 0,015 0,0007
Si <0,0005 0,0008
Mg 0,001 <0,0005
Pb 0,00006 <0,00005
Sn <0,0001 <0,0001
Bi <0,0001 <0,0001
Cu 0,0002 <0,0002
Zn >0,01 <0,0004
306
Продолжение табл. 54
Примесь Содержание, %
в исходной МоО3 в МоО3, очищенной через оксихлорид
Cd <0,0001 <0,0001
Sb 0,00015 <0,0001
Cr <0,001 <0,001
As 0,0012 <0,001
Ni <0,001 <0,001
Ti <0,001 <0,001
Fe 0,0006 0,0005
W 0,03 0,0006
Молибдаты
Представляют интерес данные об условиях хлорирования мо-
либдатов кальция, меди, железа и свинца, которые могут содер-
жаться в продуктах обжига молибденитовых концентратов, а
также в окисленных рудах и концентратах (минералы повеллит,
вульфенит, ферримолибдит).
Хлорирование молибдатов хлором и другими хлорирующими
агентами в различных условиях изучал Синакевич [50, 51], хло-
ридами серы — Глухов с сотрудниками [58].
Молибдаты кальция, меди и свинца получали нагреванием
стехиометрической смеси окислов или осаждением из растворов.
Их состав соответствовал формуле Л4еМоО4. Молибдаты трехва-
лентного железа, осажденные из раствора, не отвечали составу
нормального ферримолибдата (Fe2O3 -ЗМоО3). Использованный
в работе [50] препарат примерно соответствовал формуле Fe2O3 X
X 5МоО3-Н2О, а в работе [58] — Fe2O3-4MoO3-7H2O.
Исследования показали, что по убывающей активности взаи-
модействия с хлором молибдаты располагаются в следующем ряду:
CuMoO4 > Fe2O3 • пМоО3 > РЬМоО4 > СаМоО4.
В этом же ряду возрастает термическая прочность молибдатов
(см. табл. 9). Молибдаты меди и железа начинают реагировать
с хлором около 400° С и быстро хлорируются при 650—700° С.
Молибдат свинца активно реагирует с хлором при 750—800° С,
а СаМоО4 — при 900—1000° С. Основная реакция хлорирования:
/ИеМоО4 + 2CL = МеС\2 + МоО2С12 + О2. (154)
Частично образуется также МоОС14.
При хлорировании хлором в присутствии углерода или приме-
нении четыреххлористого углерода рабочая температура хлори-
рования молибдатов снижается до 350—450° С, причем и в этом
случае основной продукт хлорирования МоО2С12. При хлориро-
207
вании ферромолибдата в присутствии восстановителей значитель
ная доля железа переходит в FeCl3, конденсирующийся вместе
с МоО2С12.
Хлорирование молибдатов хлором сильно активируется в при-
сутствии сульфидов — пирига или молибденита. Это объясняется
образованием хлоридов серы, вступающих в реакцию с кислород-
ными соединениями [50, 51 ].
Исследования [51 и 58] показали, что молибдаты кальция,
свинца и трехвалентного железа хлорируются при действии смесью
хлора с хлоридами серы, начиная с температур 100—170° С,
с образованием МоО2С12.
СаМоОд хлорируется интенсивно при температурах 250—300° С,
РЬМоО4 — при 200° С. В конце процесса реакция несколько тор-
мозится накапливающимися хлоридами кальция и свинца.
Ферримолибдат активно хлорируется в интервале температур
140—170° С, при этом большая часть хлорного железа остается
в остатке.
ГЛАВА IX
СУЛЬФИДЫ МОЛИБДЕНА
В системе молибден—сера достоверно установлено существо-
вание трех сульфидов: Mo2S3, MoS2 и MoS3. Существование Mo2S5
сомнительно. По данным работы [1 ], из кислых растворов молиб-
датов.. восстановленных цинком, при их насыщении сероводородом
выделяется гидратированный сульфид Mo2S5-3H2O. Однако это
соединение строго не идентифицировано.
При изучении системы молибден—сера сульфидов, отвечающих
степени окисления молибдена ниже 3+, не обнаружено [2, 3].
Растворимость серы в молибдене составляет 1,49% (ат.) [0,5%
(по массе) ].
Ниже описаны методы получения и свойства упомянутых трех
сульфидов молибдена, причем основное внимание уделено важ-
нейшему из них и наиболее устойчивому — дисульфиду молибдена.
36. ПОЛУТОРАСУЛЬФИД Mo2S3
До недавнего времени о существовании и условиях образования
полуторасульфида молибдена были противоречивые данные. Пер-
вые сведения о полуторасульфиде опубликованы Гишаром [4],
который получил серо-стальные иглы Mo2S3 при кратковремен-
ном нагревании дисульфида молибдена без доступа воздуха в элек-
тродуговой печи (следовательно, при температурах выше 1600—
1700° С). Это подтверждено также в более поздней работе автора
и Беляевской [10], показавших, что при температурах 1650—
1700° С MoS2 быстро разлагается с образованием низшего суль-
фида. Длительная выдержка при высоких температурах приводит
к полной диссоциации с получением молибдена [4]. Между тем
исследователи, изучавшие термическую диссоциацию MoS2 в ва-
кууме при 1000—1200° С [5—7], а также равновесие реакции
восстановления дисульфида молибдена водородом [8, 9], не
обнаруживали в качестве промежуточной фазы Mo2S3.
14 А. Н. Зеликман
209
Однако работами последнего времени достоверно установлено,
что термическая диссоциация дисульфида молибдена в вакууме
и восстановление его водородом сопровождаются образованием
Mo2S3 как промежуточного продукта.
Мак Кеб, изучая давление диссоциации MoS2 методом Кнуд-
сена, показал, что в равновесии с молибденом и парами серы
в интервале температур 1025—1150° С находится не MoS2, a Mo2S3
[11]. Это подтверждено также Опаловским и Федоровым [13J,
которые синтезировали Mo2S3, нагревая MoS2 в высоком вакууме
при 1280° С. Для реакции
4/3Мотв + $2газ
2/3Mo2S3tb
(155)
Мак Кеб вывел следующее уравнение зависимости AZ0 от темпе-
ратуры [11]:
AZ°r = —96800 4- 41,8т кал.
Стаблес и Ричардсон [12] исследовали равновесие в системе
Мо—S—Н циркуляционным методом при температурах 850—
1200° С. Водород циркулировал над смесями Мо + Mo2S3 и
Mo2S3 + MoS2 до- установления равновесия. Равновесную кон-
центрацию H2S в газовой фазе определяли радиохимическим ме-
тодом (при синтезе сульфидов добавляли радиоактивную серу).
Было установлено, что MoS2 восстанавливается водородом в две
стадии, для каждой из которых выведены уравнения зависимости,
изобарно-изотермического потенциала от температуры:
при 1000—1200° С
2MoS2tb Н2газ<—Mo2S3tb 4“ H2Sra3; * (156)
AZ° = 21610—8,237" кал(± 100 кал)\
при 850—1200° С
/З1 г^2^3тв I А12газ ** / З1 1<лгв I А2^газ
AZ° = 21270 — 6,47" кал(± 100 кал).
(157)
Комбинируя последнее уравнение с известными термодинами-
ческими данными для реакции диссоциации сероводорода, авторы
работы [12] вывели уравнение свободной энергии образования
полуторного сульфата по реакции (155):
Д2°г = —85700 4- 36,41 Г кал.
Значения AZ? вычисленные из этого уравнения, отличаются
от результатов Мак Кеба и, несомненно, более точны.
По данным Елинека [14], Mo2S3 обладает моноклинной синго-
нией с параметрами ячейки: а — 8,6335 А, b = 3,208 А, с —
= 6,092 А,Р = 102°43 , Z == 2, пространственная группа
РЪ^т^е. Рассчитанная из рентгеноструктурных данных плотность
равна 5.806 г! см*.
210
В структуре Mo2S3 атомы молибдена расположены внутри окта-
эдров из атомов серы, но они смещены на 0,32 А от центров окта-
эдров в направлении оси &, что приводит к образованию зигзаго-
образных цепочек с расстоянием Мо—Мо 2,85 А. Каждый атом мо-
либдена окружен шестью атомами серы, три атома серы располо-
жены на расстоянии 2,36 А, три других — на расстоянии 2,57 А
Рис. 74. Структура Mo2S3,
спроектированная по
оси Ь. Пунктиром обведе-
на элементарная ячейка.
Атомы при у = 1/4 нари-
сованы тонкими линиями,
при у — 3/4 — толстыми
линиями:
1 — атомы молибдена; 2 —
атомы серы
жены на расстоянии 2,36 А, три других —
(рис. 74).
Атомы серы образуют плотноупако-
ванную решетку с последовательностью
слоев chh. Все октаэдрические промежутки
между прилежащими слоями h заняты
атомами молибдена, тогда как между
соседними слоями с ь h только половина
октаэдрических промежутков заселена
атомами молибдена. Таким образом, по-
следовательность слоев в направлении
оси a: SMoSMoi/2SMoS. . .
Горох с сотрудниками [3] на основа-
нии морфологических исследований кри-
сталлов приписывают Mo2S3 структуру
ромбической сингонии ромбодипирами-
дального класса симметрии 312ЗРС, подоб-
ную решетке Sb2S3. Кристаллы Mo2S3
удлиненно-призматические, имеющие в по-
перечном сечении форму ромба с углами
между смежными гранями 40 и 140°. Та-
ким образом, структура Mo2S3 еще одно-
значно не расшифрована.
Свойства Mo2S3 мало изучены. Изве-
стно [3—5], что при обычной температуре
полуторасульфид не реагирует с соляной
а также смесью разбавленных соляной и
нагревании азотная кислота окисляет Mo2S
молибденовой и серной кислот. При нагревании на воздухе Mo2S3
окисляется до МоО3.
Твердость Mo2S3 по шкале Мооса 5, микротвердость колеб-
лется в пределах 350—510 кПмм2. Плотность 5,75 [3].
Полуторный сульфид, как уже отмечалось, можно получить
термической диссоциацией дисульфида молибдена в вакууме или
синтезом из элементов [15, 17]. .
По данным работы [17], термическую диссоциацию MoS2
проводят в эвакуированных (до остаточного давления 10 ~5 мм
рт. ст.) запаянных кварцевых длинных ампулах. Один конец
ампулы, где находится MoS2, нагревается до 1280—1300° С,
а другой конец выходит из печи, в нем конденсируется отгоняемая
сера. После длительной выдержки получается кристаллический
порошок серо-стального цвета, отвечающий составу Mo2S3.
и серной кислотами,
азотной кислот. При
з с образованием
14*
211
В работе [3] рекомендуется следующая методика синтеза Mo2S3
из элементов. Стехиометрические количества молибдена и серы
нагревают в эвакуированной кварцевой ампуле при 500° С в те-
чение 6 ч, затем при 900° С в течение 14 ч, после чего проводят
пятнадцатиминутную выдержку при 1400° С.
По всей вероятности, после выдержки при 500° С получается
смесь Мо 4т MoS2. При 900° С Мо и MoS2 реагируют с образова-
нием MosS3. В работе [16] показано, что Mo2S3 как стабильная
фаза появляется лишь при температурах выше 610° С. Это сле-
дует также из данных Стаблеса и Ричардсона [12], которые после
выдержки смеси молибдена с серой при 500° С в течение несколь-
ких суток получали смесь молибдена с MoS2. Последующее на-
гревание Мо + MoS2 в течение 16 ч в водороде при 1200° С привело
к образованию Mo2S3.
37. ТРИСУЛЬФИД МОЛИБДЕНА MoS3
Для получения трисульфида молибдена известны следующие
способы [18]:
1. Осаждение из слабокислых (pH = 2 4-3) растворов молиб-
датов сероводородом.
2. Подкисление растворов тиомолибдатов щелочных металлов
или аммония. Растворы тиомолибдатов окрашены в желтый цвет
и устойчивы в интервале pH = 7 4-10. При их подкислении соля-
ной или серной кислотой до pH = 24-3 осаждается темно-корич-
невый хлопьевидный осадок:
(NH4)2MoS4 + 2НС1 = MoS3 + H2S + 2NH4C1. (158)
В интервале pH = 44-7 растворы приобретают темно-красную
окраску, затем из них медленно выделяется осадок MoS3.
Исходными растворами могут служить растворы молибдатов
натрия, калия или аммония, а также органических оснований
типа пиперидина (C5H10-NH) или пиперазина (C4H10N2), которые
насыщают сероводородом, а затем подкисляют.
3. Разложение твердых тиомолибдатов аммония, пиперидина
или пиперазина разбавленными растворами серной, соляной или
уксусной кислот [19, 20]. При разложении твердого тиомолибдата
пиперидина или пиперазина авторы работы [19] получили MoS3
в виде мелких блестящих черных листочков, названных ими «кри-
сталлическим» трисульфидом молибдена.
4. Термическое разложение тиомолибдата аммония или тио-
молибдатов пиперидина или пиперазина в вакууме или инертной
среде при температуре 190—200° С [21, 22]:
(NH4)2MoS4 -> MoS3 + H2S + 2NH3. (159)
Препараты трисульфида молибдена, полученные всеми пере-
численными выше методами, рентгеноаморфны [18]. Обычно со-
держат избыток серы по сравнению со стехиометрическим соста-
212
bom MoS3, а также воду (в случае осаждения MoS3 из водных рас-
творов) [18, 21]. Лишь часть избыточной серы находится в сво-
бодном состоянии и может быть удалена растворением в сероуг-
лероде. Остальная часть серы, вероятно, входит в состав фазы пере-
менного состава на основе MoS3 [21].
Таким образом, препараты отвечают составу MoS34^ • z/H2O.
Значение х при осаждении MoS3 из растворов достигает 0,7 [21 L
Вся влага и избыточная сера (в том числе связанная) удаляется
в процессе нагревания в вакууме при 200° С [18].
При соблюдении определенного режима осаждения можно,
как показано в работах [23—25], выделить гидратированный три-1
сульфид (MoS3-2H2O) без избытка серы. Для этого рекомендуется
предварительно приготовить 15-н. раствор сульфида аммония или
20-н. раствор сульфида натрия, насыщая растворы соответствую-
щей гидроокиси сероводородом при охлаждении до 0° или ниже.
Полученный высококонцентрированный раствор сульфида ам-
мония или натрия добавляют в раствор молибдата аммония, затем
подкисляют 2-нб-н. НС1 образовавшийся раствор (NH4)2MoS4 до
обесцвечивания. После кипячения осадок фильтруют, промывают
водой, спиртом, эфиром и сушат в вакуумном эксикаторе. При
170—290° С дигидрат полностью обезвоживается [25].
Поведение трисульфида молибдена при его нагревании детально
исследовано в работах [18, 21].
Роде и Лебедев [21 ] методом дифференциально-термического
анализа и термогравиметрии установили, что при нагревании пре-
паратов MoS3_|_x-г/Н2О, полученных различными методами, в ат-
мосфере азота в интервале температур от 25 до ^250° С удаляется
вода и часть избыточной серы. В интервале от 250 до ~ 400° С
первоначально удаляется избыточная сера, а затем (примерно
с 350° С) начинается термическая диссоциация MoS3 с образова-
нием аморфного дисульфида молибдена. Быстрая кристаллизация
его наблюдается при 380—400° С, что отмечается на термограммах
резко выраженным эндотермическим эффектом.
После нагревания при температурах 400—750° С продукты
разложения большей частью не отвечают стехиометрическому
составу MoS2 и содержат избыточную серу. Полное удаление избы-
точной серы достигается при 900—1000° С.
Давление паров серы при диссоциации трисульфида молибдена
по реакции
MoS3 MoS2 + Sn газ,
где п = 2, 6 и8, изучали Парравано и Малькуори методом по-
тока в интервале температур 355—418° С [32], а также Преннер и
Брокмеллер [92] в интервале 475—500° С. Их данные приведены
ниже:
Температура, °C.........• . 355 390 418 475 480 500
Общее давление серы, мм рт. ст. 4,0 28,8 178,6 250 313 980
213
Написанная выше реакция, по всей вероятности, необратима,
что было отмечено Бильц и Кохер [93]. Высказывается, однако,
мнение, что образование MoS3 возможно при длительном нагрева-
нии дисульфида молибдена с парами серы при температуре 400° С
112].
Трисульфид молибдена растворяется в растворах сульфидов
аммония и щелочных металлов, а также в растворах щелочей.
В последнем случае образуются растворы оксисульфомолибдатов,
например Na2MoO2S2 и др. Соответственно этому известны окси-
сульфиды молибдена MoOxS^ (х + у = 3). Так, оксисульфид
состава MoO2S получен взаимодействием диоксидихлорида МоО2С12
с Na2S в абсолютном спирте [28]:
Мо02С12 + Na2S = MoO2S + 2NaCl. (160)
Гидратированные оксисульфиды MoOxS3_x -zH2O различного
состава выделены при подкислении растворов тиомолибдатов [26]
или насыщении сероводородом растворов молибдатов [27]. Без
доступа воздуха оксисульфиды при нагревании выше 300—350° С
разлагаются с образованием смеси MoS2 с МоО2 в различных соот-
ношениях, что согласуется с данными [29] об образовании MoS2
и МоО2 при термическом разложении окситиосульфата аммония.
При нагревании на воздухе MoS3 и оксисульфиды переходят
в МоО3.
38. ДИСУЛЬФИД МОЛИБДЕНА
Большое число работ о свойствах, структуре и методах полу-
чения дисульфида молибдена, опубликованных в литературе,
объясняется расширяющимся его использованием в качестве сма-
зывающего материала и катализатора, а также перспективами его
применения как полупроводникового материала.
Получение чистого молибденита
Высокосортные молибденитовые концентраты обычно содержат
примерно 85—95% MoS2 (51—57% Мо). Основная примесь —
кварц, другие часто присутствующие примеси — кальцит, суль-
фиды меди, железа (см. стр. 14). Для использования молибденита
в качестве смазывающего материала необходим продукт достаточно
высокой чистоты с содержанием 99,5—99,9% MoS2. В связи с этим
разработаны технологические схемы очистки молибденитовых
концентратов, в которых сочетаются тонкое измельчение с фло-
тацией и обработкой материала различными химическими реаген-
тами [34, 35].
В результате тонкого измельчения и ряда флотационных пере-
чисток получают концентрат с содержанием 97—98% MoS2, 0,3—
0,6%. SiO2, остальное — примеси минералов железа, кальция,
магния и др. Для дальнейшей очистки концентрат обрабатывают
214
на холоду концентрированной соляной кислотой (для удаления
примесей Ее, Са, Mg и др.), а затем 40%-ной плавиковой кислотой
с добавкой 10%-ной концентрированной серной кислоты (для уда-
ления SiO2). После промывки водой продукт обрабатывают раство-
ром соды или аммиака, снова промывают водой, затем спиртом
и сушат на воздухе с облучением инфракрасными лучами или
в вакууме. Такая обработка обеспечивает получение • продукта
примерно следующего состава, %: 99,5—99,85 MoS2; 0,01—
0,06 SiO2; 0,03 MgO; 0,05 СаО; 0,04 Fe2O3 + А12О3 1341.
Размер частиц полученного порошка 10—250 мкм. Для приме-
нения в качестве смазывающего материала его дополнительно
измельчают до крупности 0,5—3 мкм в струйных мельницах [36].
Возможно применение ультразвукового измельчения.
Для защиты тонкодисперсного порошка молибденита от окис-
ления предложено покрывать частицы минерала тонкой масляной
пленкой. Описано несколько способов нанесения масляных по-
крытий Ч Один из эффективных способов состоит в смешивании
определенного количества порошка, не содержащего масел, с по-
рошком, содержащим до 8% масел, и измельчении такой смеси
в струйной мельнице. В ней тонкое измельчение осуществляется
в результате удара и взаимного трения частиц при высоких ско-
ростях в потоке циркулирующей газовой среды (воздуха, азота
и др.). При этом получается однородное покрытие частиц порошка
маслом.
Для защиты от окисления достаточно содержания в порошке
0,2—0,3% масел. Применяют рафинированные нефтяные масла
и некоторые другие. Желательно, чтобы в составе масел были масла
с высокими точками кипения, порядка 250° С.
Синтетический MoS2
Известные методы получения дисульфида молибдена можно
подразделить на следующие группы:
1. Синтез из элементов.
2. Термическая диссоциация трисульфида молибдена.
3. Взаимодействие серы с окислами или молибдатами в рас-
плавах.
4. Взаимодействие серы или сероводорода с окислами, хлори-
дами или оксихлоридами молибдена.
5. Выделение MoS2 из растворов сульфомолибдата натрия при
высоких температурах.
Практически используют более изученные три первые группы
методов. Четвертая группа методов находит применение в послед-
нее время преимущественно для получения пленок дисульфида
молибдена на различных подложках.
1 Патенты (США) 3073779, 1963; 3082065, 1963; 3125413, 1964; 3101252, 1963.
215
Синтез MoS2 из элементов. Кинетику взаимодействия молиб-
дена с парами серы исследовали гравиметрическим методом автор
и Теслицкая [37]. Исходным материалом служили молибденовый
порошок (средний размер частиц 70 мкм) и молибденовая лента
высокой чистоты. Из кинетических кривых сульфидирования мо-
либденового порошка (рис. 75) следует, что молибден взаимо-
действует с серой с высокой скоростью при температурах выше
750° С. Полное сульфидирование порошка с размерами 70 мкм
с образованием MoS2 проходит (при 750° С) за ~35 мин. Вначале
Время г, мин
Рис. 75. Кинетические кривые суль-
фидирования молибденового порошка
парами серы при температуре, °C:
Рис. 76. Кинетические кривые
сульфидирования молибденовой
ленты при температуре, °C:
1 — 990; 2 — 930; 3 — 897; 4—830; 5 — J — 875; 2 — 750; 3 — 700; 4 —
800; 6 — 758; 7 — 715; 8 — 680; 9 — 660; 1020; 5 — 995; 6 — 650; 7 — 950
10 — 640
наблюдается инкубационный период, вероятно, обусловленный
наличием тонких окисных пленок на частицах молибдена. Скорость
ликвидации пленки растет с увеличением температуры (рис. 75).
В интервале температур 640—715° С реакция протекает в проме-
жуточной области (энергия активации Е = 15,6 ккал! моль), при
более высоких температурах (750—990° С) лимитирующей стадией
становится диффузия — подвод паров серы к реагирующей по-
верхности (£ = 9,2 ккал!моль). В интервале температур 750—
990° С кинетические кривые описываются уравнением параболы
сх2 = Кт, где а — степень превращения. Такая закономерность
характерна для гетерогенных реакций, лимитируемых диффузией
через твердые оболочки продуктов реакции.
В интервале температур 715—640° С зависимость скорости про-
цесса от времени выражается уравнением ап = Кт, причем зна-
чение п изменяется от 1,87 при 715° С до 1,43 при 640° С.
Иной характер имеют кинетические кривые сульфидирования
молибденовой ленты (толщина ленты 0,5 мм). В этом случае ско-
рость взаимодействия в интервале температур 700—875° С значи-
тельно превосходит скорость процесса при более высоких темпера-
216
турах 950—1020° С (рис. 76). Это объясняется образованием при
температурах 950—1020° С тонкой плотной пленки MoS2 на по-
верхности молибденовой ленты, резко тормозящей процесс. При
механическом удалении пленки взаимодействие возобновляется,
но затем снова резко тормозится. По всей вероятности, при сульфи-
дировании частиц порошка, имеющих развитую, неровную поверх-
ность, столь плотная пленка не образуется.
При сульфидировании порошка и компактного молибдена про-
дукт реакции соответствовал по химическому составу формуле
MoS2. Полуторасульфид Mo2S3 в продуктах сульфидирования не
был обнаружен.
Синтез MoS2 из элементов обычно проводят в эвакуированных
запаянных кварцевых ампулах/куда помещают порошок молиб-
дена и серу, взятую с небольшим избытком из такого расчета,
чтобы давление в ампулё при максимальной температуре не пре-
вышало ~10 ат. Рекомендуется использовать грубозернистые
(~70 мкм) молибденовые порошки (чтобы избежать бурного про-
текания реакции и перегрева). Могут быть использованы также
короткие отрезки тонкой молибденовой проволоки. Для снижения
содержания кислорода исходный молибденовый порошок реко-
мендуется предварительно выдержать в токе сухого водорода при
900—1000° С.
При синтезе применяют ступенчатый режим нагрева. Первона-
чально ампулу выдерживают некоторое время при 550—650° С
с тем, чтобы часть серы и молибдена вступила в реакцию. Затем
поднимают температуру до 800—900° С, выдерживая ампулу не-
сколько часов при этой температуре. Для получения упорядочен-
ной гексагональной структуры необходима длительная выдержка
при 1000—1100° С (см. стр. 225).
Недостаток метода синтеза из элементов в ампулах — ограни-
ченность масштабов. Обычно диаметр ампул 20—25 мм, длина
150—120 мм. Увеличение диаметра ампул опасно из-за возможно-
сти их взрыва.
Сульфидирование молибдена с получением MoS2 можно также
осуществить действием сероводорода или сероуглерода на по-
рошок молибдена при температурах 700—800° С [42].
Термическая диссоциация MoS3. Трисульфид молибдена, по-
лученный одним из рассмотренных выше способов, разлагают
нагреванием в потоке инертного газа (аргон, азот). На основании
работ [18, 21 ], в которых изучали термическую диссоциацию MoS3,
можно рекомендовать примерно такой режим разложения.
Лодочку с исходным материалом помещают в трубчатую печь,
нагретую до 400—450° С. В этом интервале температур материал
выдерживают 2—3 ч, после чего температуру поднимают до 900—
1000° С. При высокой температуре проводят продолжительный на-
грев (5—8 ч) для полного удаления избыточной серы и гомогени-
зации структуры.
217
Взаимодействие серы с окислами молибдена и молибдатами
в расплавах. Окисли и молибдаты реагируют с серой при темпера-
турах 500—700° С с образованием дисульфида молибдена. Это
положено в основу наиболее распространенного метода синтеза
MoS2, в котором сера и кислороцное соединение молибдена взаимо-
действуют в расплаве поташа или соды [4, 43—45, 18]. Расплав
служит растворителем серы и защищает образующийся продукт
от окисления.
Как показано автором и Крейн [44], исходным материалом
для синтеза может служить не только трехокись молибдена,
но также и молибдат кальция.
В любом случае в расплаве соды или поташа присутствует
молибдат щелочного металла 7ИеМоО4 (Me — Na или К) как
результат взаимодействия щелочного карбоната с МоО3 или
СаМоО4.
Согласно работе [44], при использовании МоО3 процесс в ос-
новном протекает по суммарной реакции
Na2MoO4 + 3S = MoS2 + Na2SO4.
(161)
Присутствие сульфата натрия в количестве, примерно экви-
валентном MoS2, доказано фазовым анализом затвердевшего рас-
плава.
При использовании молибдата кальция, помимо этой реак-
ции, протекает вторичное взаимодействие с образованием CaSO4’
СаСО3 + N a2SO4 Т± CaSO4 + Na2CO3.
В связи с присутствием в получаемом MoS2 примеси МоО2
Белл и Херферт (45 ] считают, что реакция проходит в две стадии:
2МоО3 + S = 2МоО2 + SO2)
МоО2 + 3S = MoS2 + SO2.
Эта схема, однако, не учитывает влияния карбонатного рас-
плава, в котором молибден находится в составе анионов МоО4~, а
не МоО3, и образуется сульфат натрия. В связи с этим правильней
отражает реальный процесс следующий ряд реакций, протекаю-
щих в расплаве:
3Na2MoO4 + S = ЗМоО2 + Na2SO4 + 2Na2O, (162)
ЗМоО2 + 6S = 3MoS2 + ЗО2, (163)
2S + ЗО2 + 2Na2O = 2Na2SO4. (164)
He исключено, что двуокись молибдена может образоваться
при недостатке серы (в конце процесса) также в результате взаимо-
действия MoS2 с Na2MoO4:
6Na2MoO4 + MoS2 = 7МоО2 + 2Na2SO3 + 4Na2O. (165)
218
В литературе приводятся различные режимы получения MoS2
по рассматриваемому методу.
В работах [44, 51 ] рекомендуется при использовании в ка-
честве исходного материала МоО3 и серы следующий режим. Соду,
МоО3 и измельченную серу перемешивают в весовом соотношении
40,7 : 33,5 : 25,8. Смесь помещают в фарфоровый тигель, закры-
тый крышкой. Тигель выдерживают в печи при 700—800° С в те-
чение 1 ч. Охлажденную массу измельчают, обрабатывают по-
следовательно горячей водой, раствором аммиака, 5%-ной соля-
ной кислотой и снова водой. Черный осадок промывают спиртем
и сушат в вакууме при 50—60° С. Извлечение в готовый продукт
составляет 80—85%. Состав дисульфида молибдена, полученного
в опытах с шихтой, содержащей 100 г МоО3, приведен в табл. 55.
ТАБЛИЦА 55
ХИМИЧЕСКИЙ И ФАЗОВЫЙ СОСТАВ ПАРТИЙ ДИСУЛЬФИДА МОЛИБДЕНА
[44]
Химический состав, % Фазовый состав, %
Элемент опыт № 1 опыт № 2 соединение опыт № 1 опыт № 2
-МОобщ ^°окисл Зобщ Зэлем 59,65 0,71 39,94 0,64 59,53 0,32 39,49 1,69 MoS2 МоО3 Зэлем 98,24 1,07 0,64 98,69 0,48 1,69
Сумма 99,95 100,86
Эти данные в последующем были подтверждены в опытах с загруз-
кой шихты, рассчитанной на получение 2000 г дисульфида мо-
либдена. Дисульфид содержит незначительное количество при-
месей окислов молибдена и элементарной серы, которая может
быть удалена нагреванием в вакууме при 700—800° С или обра-
боткой сероуглеродом. Удаление примеси МоО2 до содержания
<0,01% возможно, как показано в работе [45], при выдержке
продукта в расплавленном KCNS.
При использовании молибдата кальция процесс проводят так
же, как описано выше. Состав исходной смеси, %:*37,8 СаМоО4;
33,1 Na2CO3; 29,1 S [44, 51].
Авторы работы [18] для получения MoS2 с ромбоэдрической
структурой (см. ниже) рекомендуют следующий путь: 6 г Na2CO3
и 4 г серы нагревают в течение нескольких часов в кварцевом
тигле. После охлаждения в тигель добавляют 1/3 приготовленной
смеси, состоящей из 5 г МоО3, 1 г серы и 2 г Na2CO3. Смесь нагре-
вают в течение полутора часов при 900° С, после охлаждения
добавляют следующую часть смеси и т. д. Продукт реакции обра-
батывают раствором аммиака, водой, соляной кислотой, затем
промывают CS2 (растворение элементарной серы), спиртом и эфи-
ром. Выход MoS2 близок к теоретическому.
219
Метод синтеза MoS2 в расплаве солей прост в осуществлении
и может быть использован в промышленной практике.
Взаимодействие серы или сероводорода с окислами или хлори-
стыми соединениями молибдена. Эта группа методов мало изучена.
Известно, что пары серы и сероводород взаимодействуют с окис-
лами молибдена (МоО3, МоО2), пятихлористым молибденом и
оксихлоридами молибдена (МоО2С12, МоОС14) в интервале темпера-
тур 500—700° С с образованием MoS2.
Авторы работы [51 ] проводили реакцию между МоО3 и парами
серы в вертикально установленной трубе из нержавеющей стали
с пористой перегородкой, на которую засыпали слой порошка
трехокиси молибдена. Через пористую перегородку из испари-
теля поступали пары серы в смеси с аргоном. Реакция наблюда-
лась выше 300° С, однако лучшие результаты получены при 700° С.
Синтезированный MoS2 содержал 59,9% Мо и 40,2% S, что совпа-
дает с теоретическим составом.
Взаимодействие МоО3 с серой и H2S, по всей вероятности,
протекает с промежуточным образованием МоО2 [45, 47]:
2МоО3 + S = 2МоО2 + SO2, (166)
МоО2 + 3S = MoS2 + SO2,
(167)
MoO, + H,S = MoO, + H2O + S,
MoO, + 2H,S = MoS, + 2H,O.
£ I 4 I 4
(168)
(169)
В работе [47] отмечается, что при взаимодействии H2S с МоО3
в конечном продукте всегда содержится примесь двуокиси мо-
либдена.
Можно полагать, что МоС15 и МоО2С12 реагируют с серой
и сероводородом в газовой фазе при 500—600° С по следующим
суммарным реакциям:
МоС15 + 7S = MoS2 + 21/2S2C12, (170)
2МоС15 + 5H2S = 2MoS2 + S + 10HC1, (171)
MoO2Cl2 + 5S = MoS2 + SO2 + S2C12, (172)
MoO2Cl2 + 3H2S = MoS2 + S + 2HC1 + 2H2O. (173)
В работе [51] синтез MoS2, основанный на реакции между
МоС15 и H2S, проводили в кварцевой трубке, через которую
пропускали газовую смесь аргона, сероводорода и пентахлорида
молибдена. В нагретой зоне длиной 201) мм поддерживали темпера-
туру 700—800° С. В результате реакции на стенках трубки осаж-
дался дисульфид молибдена. Продукт содержал значительное
количество элементарной серы, которую удаляли обработкой
порошка раствором сернистого натрия. Полученный дисульфид
содержал 58,3% Мо, 40,1 % S. Выход не превышал 20%.
220
Некоторые из приведенных выше реакций предложено исполь-
зовать для получения пленок дисульфида молибдена. Так, опи-
сано получение пленок MoS2 на кварце, стекле, солях (например,
NaCI) [48*]. На поверхность подложки первоначально нано-
сят тонкую пленку МоО3 (например, напылением). Затем при 650—
700° С проводят ее сульфидирование действием паров серы или
сероводорода. Другой путь состоит в пропускании над нагретой
до 650—700° С поверхностью подложки смеси паров хлорида или
оксихлорида с парами серы или сероводородом *.
Описан метод получения пленок MoS2 взаимодействием серы
с молибденом [50]. На оптическое стекло Пирекс первоначально
напыляют пленку молибдена. Затем покрываемое стекло вместе
с небольшим количеством чистой серы выдерживают в эвакуиро-
ванной запаянной кварцевой трубке при 500° С. При пленке мо-
либдена толщиной 620 А получали кристаллы MoS2 размером 30 А.
Выделение MoS2 из растворов сульфомолибдата натрия. Ару-
тюнян и Хуршудян [94] показали, что из растворов сульфомо-
либдата натрия при их нагревании в автоклаве в интервале тем-
ператур 530—640° С тиосоль разлагается с выделением кристал-
лического дисульфида молибдена. Авторы не рассматривают
химизм процесса. По всей вероятности, его можно представить
следующими реакциями:
Na2MoS4 —>MoS3 + Na2S,
MoS3 —* MoS2 + S.
Выделяющаяся сера растворяется в растворе сульфида натрия.
Исходные растворы молибдата натрия (с концентрацией 10—
50 г!л Мо) насыщали сероводородом для образования Na2MoS4.
Затем раствор помещали в автоклав. При температурах 550—600° С
разложение соли с выделением кристаллического MoS2 проис-
ходит за 3—4 ч. Изменение pH от 7 до 11 не влияет на структуру
осадков и скорость выделения. Полученный дисульфид имел
•структуру, близкую к структуре ромбоэдрического MoS2.
Структура MoS2
До недавнего времени считали, что дисульфид молибдена имеет
.лишь одну кристаллическую форму — отвечающую структуре
минерала молибденита, обладающего гексагональной симметрией,
которую мы будем далее обозначать 2Я-Мо52.
Развитая в последние годы теория политипии молибденита,
обусловленной слоистым характером его структуры, позволила
установить возможность существования 8 политипных модифи-
каций, из которых 5—независимые (имеют неэквивалентные дифрак-
* Langaudie I. Fr. Pat., 1087879, 1955; 66764, 1957.
221
ционные признаки) [54]. В природе преобладает гексагональная
двухслойная модификация 2Н и недавно обнаружена менее рас-
пространенная трехслойная форма 3T?-MoS2 [61—63, 49].
Ромбоэдрический дисульфид молибдена (3Z?-MoS2) был впер-
вые получен Беллом и Херферигом [45] взаимодействием серы
с МоО3 в расплаве поташа по методу Шультена [43].
Образцы MoS2, полученные другими методами синтеза (син-
тез из элементов, термическая диссоциация MoS3, взаимодей-
ствие МоС15 с H2Sh др.), имеют структуру, отличающуюся от 2Н
и 3/?-форм [51]. Авторы работы [18] считают, что в этом случае
образуется неупорядоченная гексагональная форма с дефектами
упаковки слоев S — Мо — S. По всей вероятности, более пра-
вильно ее рассматривать как структуру/в которой чередуются
тонкие зоны с взаиморасположением слоев S — Мо — S по типу
2Н и 3R [55]*.
Структура гексагонального молибденита [2//-MoS2l. Струк-
тура молибденита исследована и расшифрована Дикинсоном и Пау-
лингом [52] и несколько позже Хасселем [53]. Решетка—гек-
сагональная, слоистая (пространственная группа Р 63/ттс).
Слои атомов молибдена расположены между двумя слоями
атомов серы, образуя трехслбйные пакеты S — Мо — S (рис. 77).
Эти пакеты располагаются один над другим таким образом,
что каждый третий повторяет первый, каждый четвертый — вто-
рой и т. д.
Ионы молибдена и серы внутри слоя прочно связаны (гомеопо-
лярная связь), тогда как между тройными слоями связи слабые,
ван-дер-ваальсовского типа. Это обусловливает совершенную
спайность по (0001) и пластинчатую форму кристаллов.
Особенность структуры, впервые найденной при изучении
молибденита, — расположение шести атомов серы, окружающих
атом молибдена, в вершинах правильной равнобедренной триго-
нальной призмы (вместо обычного расположения в вершинах
октаэдра) с углами S — Мо — S, равными 82 и 136°. Такая ко-
ординация обусловлена й4$р-гибридизацией атомных волновых
функций [57, 58].
Ионы молибдена в MoS2 имеют электронную конфигурацию
4d45s'5p, образующуюся в результате возбуждения одного d-элек-
трона и перехода его на 5р-уровень (в нормальном состоянии
в атоме молибдена электроны занимают 4d55s' орбиты).
Теоретически было показано [57], что б/4$р-конфигурации
соответствует шестиковалентная связь с тригонально-призмати-
ческой координацией, причем относительная прочность такой
связи в 2,98 раза выше, чем связи, осуществляемой s-электронами.
* Авторы работы [94] утверждают, что MoS2, выделенный из тиомолибдат-
ных растворов при 550—600° С, имеет структуру 3R. Однако и в этом случае
получается смесь 2Н 4- 3R [55].
222
Тригонально-призматическая координация не найдена в каких-
либо других соединениях молибдена и специфична для MoS2.
Более наглядно структуру 2//-MoS2 можно представить в виде
чередующихся слоев тригональных призм с ионами Мо4* в центрах
и пустых слоев из тетраэдров (рис. 78).
Элементарная ячейка содержит две молекулы MoS2. Пара-
метры: <2 = 3,1062 А, с = 12,294, da = 3,890. Расстояние Мо—S
равно 2,35 А, расстояния
S—S в тригональной приз-
ме 2,98 А, расстояние S—S
между соседними слоями
3,66 А 156].
QMD
Рис. 77. Структура
молибденита гексаго-
нальной модификации
Рис. 78. Структура гексагонального
молибденита MoS2 в виде координа-
ционных полиэдров (тригональных
призм). Видно, что основания призм
примыкают к пустым тетраэдрам
Ромбоэдрический MoS2- [ЗР-форма]. Белл и Херферт [45],
исследуя синтезированный ими по методу Шультена [43] дисуль-
фид молибдена, обнаружили существенные различия между дифрак-
ционной картиной молибденита и синтезированного MoS2. Вместо
отражений (103), (105) и (107), характерных для молибденита,
синтетический MoS2 давал дублеты, центры которых распола-
гались вблизи указанных «нормальных» отражений (рис. 79).
Индицирование рентгенограмм показало, что для синтетического
MoS2 выполняется ромбоэдрическое условие погасаний. Белл и
Херферт предположили, что структура 3/?-MoS2 отвечает типу
CdCl2 с октаэдрическим расположением атомов серы вокруг
223
атомов молибдена. Такая координация возникает в результате
поворота одного из слоев серы (например, верхнего) внутри пакета
по отношению к другому слою серы на 60°.
Однако Елинек с сотрудниками [59] и, независимо от них,
Семилетов [60] показали ошибочность такой интерпретации струк-
туры 3/?-MoS2 ввиду больших расхождений между теоретическими
и экспериментальными интенсивностями отражений. В действи-
тельности решетка 3T?-MoS2 построена из тех же слоев, что и 2Н-
MoS2 (т. е. сохраняется тригонально-призматическая коорди-
Рис. 79. Дифрактограммы гексагонального 2Н (а) и ромбоэдри-
ческого 37? (б) дисульфида молибдена
нация атомов молибдена с ближайшими атомами серы). Ромбо-
эдрическая модификация получается из гексагональной при пово-
роте на 60° целых трехслойных пакетов, что приводит к структуре
с пространственной группой R3 т.
Параметры решетки ромбоэдрического MoS2 [18]: а = 3,163 А,
с = 18,37 A, da = 5,81. Таким образом, параметры а у обеих
модификаций одинаковы, тогда как параметр с 3R-MoS2 в 1,5 раза
больше, чем 2/7-MoS2. Из этого следует, что в элементарной ячейке
2/7-MoS2 два трехслойных пакета, а в 3/?-MoS2 — три пакета
(рис. 80).
Как уже отмечалось, ромбоэдрический MoS2 получается только
при синтезе дисульфида по методу взаимодействия МоО3 с серой
в расплаве соды или поташа. Авторы работы [18] объясняют
это изоляцией одного первоначально образующего зародыша MoS2
от другого и последующим ростом их с параллельной ориентацией
слоев. Все другие методы синтеза приводят к получению продукта
со структурой 2Я-Мо52 либо со смешанной структурой 2Н + 3R.
224
Ус л о в и я структурных превращений М о S2.
Данные о влиянии генезиса рудных месторождений на появле-
ние ромбоэдрического молибденита неоднозначны. Авторы ра-
боты [61, 90] предполагают, что образованию 3/?-MoS2 способ-
ствует примесь рения (в образце молибденита с ромбоэдрической
структурой содержалось 1,88% рения), а также пониженные
температуры. Чухров, Звягин и др. [55], обстоятельно иссле-
довавшие 380 образцов молибденита из 90 месторождений, пришли
к выводу, что температура, внешнее давление и содержание ре-
ния не являются факторами,
определяющими возникнове-
ние 2Н и 37?-модификаций. Сле-
дует, однако, отметить, что
содержание рения во всех 380
образцах было низким (не более
сотых долей процента), что де-
лает выводы авторов об отсут-
ствии влияния примеси рения
на структуру молибденита мало
убедительными.
Более определенны данные
об устойчивости 2Н и 37?-форм
синтетического дисульфида мо-
либдена.
Рис. 80. Схема расположения слоев
MoS2 при их плотной упаковке в ре-
шетке:
• Автор, Теслицкая и Инден-
баум синтезировали дисульфид
молибдена с ромбоэдрической
а — гексагональная двухслойная упаков
ка АВ' АВ'-, б — ромбоэдрическая трех
слойная упаковка АВС АВС
(37?) и смешанной (2Н + 3R)
структурой и изучили влияние условий отжига, а также примеси
рения на изменение структуры M0S2. Отжиг M0S2 проводили
в запаянных кварцевых ампулах в вакууме и в парах серы под
давлением 4 и 18 атм в интервале температур 500—1100° С.
Продолжительность выдержки в вакууме была до 100 ч, отжига
под давлением паров серы — от 20 до 350 ч. Было установлено,
что ромбоэдрический MoS2, а также образцы со смешанной 2Н +
+ 3R исходной структурой устойчивы при длительном отжиге
в вакууме вплоть до 1100° С. Переход к гексагональной модифи-
кации происходит только при отжиге в парах серы при давлениях,
превышающих равновесное над MoS2.
По-видимому, дисульфид молибдена имеет область гомогенности,
и при недостатке атомов серы в решетке (отжиг в вакууме) ромбо-
эдрическая ориентация тригональных призм MoS6 достаточно
устойчива до температур 1100° С. Растворение серы в MoS2 (за-
полнение ею вакантных мест в решетке при отжиге под давлением
паров серы) делает возможным превращение 3R-^2H при повышен-
*А. Н. Зеликман и др. Кристаллография, 1969, № 5.
15 А. Н. Зеликман
225
ных температурах. Гексагональная модификация наиболее устой-
чива и не переходит в ромбоэдрическую при любых условиях
отжига.
В этой же работе было отчетливо выявлено влияние содержа-
ния рения (более 1,2%) в MoS2 на структуру дисульфида молиб-
дена.
Твердые растворы (Mo1_xReJS2 с содержанием до 12%
Re по отношению к MoS2 получили действием паров серы на сплавы
молибдена с рением при температуре 850—900° С.
Все образцы имели смешанную структуру 2Н + 37?, но с уве-
личением содержания рения с 1,2 до 12% количество 37?-формы
резко возрастает. При содержании рения выше 1,2% переход
37? —> 2Н не наблюдался после длительного отжига в парах серы
при 1000—1100° С.
В результате сульфидирования сплава Мо + 5% Re под дав-
лением паров серы ~50 ат (в стальном патроне) был получен
дисульфид (Mo^ReJ S2 с совершенной ромбоэдрической струк-
турой. ' Очевидно, что при замещении части атомов молибдена
в решетке MoS2 рением при высоком давлении серы трехслой-
ная 37?-упаковка^ становится более устойчивой, чем двухслой-
ная 2Н.
Термохимические свойства MoS2
Вопросы термической устойчивости MoS2 частично уже рас-
сматривались выше (стр. 210). Не вызывает сомнений, что -до
800° С в инертной атмосфере и в вакууме MoS2 практически не
диссоциирует. Заметная термическая диссоциация с образова-
нием Mo2S3 наблюдается при нагревании в вакууме при темпера-
турах 1000—1200° С [11, 17]. Длительное нагревание в вакууме
при 1400° С приводит к полному разложению MoS2 до молибдена
198].
При нагревании образца MoS2 в атмосфере аргона с плав-
ным повышением температуры от 20 до 1350° С в течение 3 ч от-
мечена заметная диссоциация молибденита (содержание S сни-
жается с 39 до 36%) [10].
.-.Эти качественные наблюдения позволяют заключить, что
дисульфид молибдена разлагается до плавления. При кратковре-
менном нагревании (2—3 мин) спрессованных образцов MoS2
наблюдалось [10], начиная с температур 1650—1700° С, появ-
ление жидкой фазы при одновременном интенсивном разложении
дисульфида молибдена. По всей вероятности, неравновесная
жидкая фаза представляла собой эвтектику Mo2S3 — MoS2 (эвтек-
тика наблюдалась на микрошлифах).
' Вероятно, образованием эвтектик объясняется спекание MoS2
при его нагревании выше 1000° С и покрытие поверхности мате-
риала труднопроницаемой коркой Mo2S3, затрудняющей диссо-
циацию [17/ 64 ].
226
Ввиду быстрого протекания термического разложения MoS2
при температурах выше 1600° С вызывают сомнение выводы ра-
боты [65], в которой утверждается, что точка плавления MoS2
находится около 2375° С. Понятие «точка плавления MoS2» не
имеет физического смысла. Можно лишь утверждать, что в опре-
деленных условиях (быстрый подъем температуры, кратковремен-
ная выдержка) появляются жидкие фазы.
Впервые давление диссоциации дисульфида молибдена было
экспериментально исследовано Парравано и Малькуори [8],
которые изучали методом потока равновесие реакции восстанов-
ления MoS2 водородом в интервале температур 800—1100° С. Из
сохранения постоянства газовой фазы в процессе восстановления
при 1100° С (вплоть до почти полного удаления серы) авторы
заключили, что восстановление протекает без промежуточного
образования Mo2S3. Используя экспериментальные данные Пар-
равано и Малькуо'ри и данные о теплоемкости MoS2, Келли вывел
уравнение зависимости AZ — f (7) для реакции Мо r|- SpoM6 7^ MoS2:
AZ0 — —57 640 — 15,787 lg7 + 5,6• 10“372 — 0,252т"1 + 49,247 кал\
AZ298 = —54 190 кал\
ДЯ298 — 57 270 кал.
Позже автор и Крейн [9], применив циркуляционный метод
изучения равновесия реакции восстановления MoS2 водородом,
также не обнаружили промежуточного образования Mo2S3. Рас-
считанные «в этой работе значения давления диссоциации MoS2
оказались близкими к вычисленным по уравнению Келли.
Стеблес и Ричардсон, однако, убедительно показали, что
восстановление MoS2 водородом протекает через стадию образо-
вания Mo2S3, и определили равновесный состав газовой фазы
(H2+H2S) над сосуществующими твердыми фазами: MoS2+
4- Mo2S3 и Mo2Ss 4- Мо [12].
Для реакции Мо 4~ S2ra3 = MoS2 ими выведено следую-
щее уравнение зависимости AZ0 от температуры:
AZ0 = —85 870 — 37,337 (точность ± 2000 кал).
Из этого уравнения и данных о теплоемкости MoS2 (Ср =
= 11,2 4- 13,50-10-37 [95]) рассчитана энтропия MoS2 : S298 —
= 16,9 ± 2 кал/град.
Исходя из этого значения энтропии и с учетом зависимости
теплоемкости от температуры найдены стандартные термодинами-
ческие величины для реакции образования дисульфида из эле-
ментов Мо 4~ 2Spom6 = MoS2:
AZ°98 = — 78 420 кал\
Д/^298 = — 91 340 кал.
15*
227
Физические свойства MoS2
Рис.^,81. Зависимость электро-
проводности (о, Ш!'1’ СМ'1)
синтетического Мо32(2/Лфор-
ма) от температуры. Отжиг
при 800° С под давлением
паров серы, ат:
1 — 15; 2 — 5; 3 — 0,5; 4 —
в вакууме
Электрические, магнитные, оптические и другие физические
свойства изучали преимущественно на образцах молибденита
и отчасти синтетического дисульфида молибдена.
Электропроводность. Дисульфид молибдена обладает полу-
проводниковыми свойствами. Об этом свидетельствует большой
отрицательный коэффициент электросопротивления [67]. При
нагревании проводимость MoS2 сильно повышается. При темпера-
турах 200—400° С и выше характер
проводимости электронный, подобный
проводимости металлов, о чем, в част-
ности, свидетельствует соблюдение за-
кона Ома, тогда как при комнатной
температуре закон Ома не соблюдается
[67,68]. Вследствие сильной анизотро-
пии кристаллов MoS2 удельная элек-
тропроводность в направлении, перпен-
дикулярном оси С, примерно в 1000 раз
больше удельной электропроводности
в направлении, параллельном оси С [68].
Электросопротивление сильно зави-
сит от содержания примесей и харак-
тера образцов (монокристаллические
пластинки, прессованные поликристал-
лические образцы).
По данным работы [71], удельная
электропроводность молибденита нахо-
дится в пределах 10~2—10-4ом'1 •см~1.
В работе [72] показано, что электропроводность различных
образцов молибденита (вдоль основной плоскости кристалла)
колеблется от ‘4,2 до 0,009
Для синтетического дисульфида молибдена Оганесян и Рудь
[75 ] определили величину удельной электропроводности 16,1 ом"1 X
X см-1. Столь высокое значение, вероятно, обусловлено откло-
нением состава образцов от стехиометрического. Поданным автора
и Теслицкой1, удельная электропроводность синтетического MoS2
высокой чистоты (гексагональной модификации) имеет значение
(1,2—5,3) -10"5 ом~1‘см~1 (25° С). Электропроводность возра-
стает с температурой и зависит от давления паров серы при от-
жиге синтетического MoS2 (рис. 81).
Электросопротивление уменьшается с увеличением приложен-
ного напряжения. При некотором максимальном напряжении
наступает «пробой», после которого электросопротивление резко
понижается. По всей вероятности, это обусловлено преимущест-
1 М. В. Теслицкая Диссертация. МИСиС, 1968.
228
венно тепловым действием тока [69]. Электросопротивление мо-
либденита сильно уменьшается с повышением внешнего давления
[70].
Термо-э. д. с. Дисульфид молибдена как полупроводник обла-
дает высокими значениями термо-э. д. с.
Для молибденита различных месторождений получены [71]
сильно отличающиеся значения коэффициента т. э. д. с. а. Так,
для молибденита высокотемпературных формаций установлены
положительная и отрицательная термо-э. д. с. со значениями
а — + 100 мкв!град и а = — 100 мкв!град, тогда как для молиб-
денита среднетемпературных месторождений найдена только отри-
цательная термо-э. д. с. (а —75 мкв). Из этого следует, что
в молибдените возможны и электронный, и дырочный типы про-
водимости, что связано с наличием примесей. По данным работы
[73], в интервале 20—300°С термо-э. д. с. молибденита линейно
возрастает с температурой. Для синтетического дисульфида
молибдена в работе [74] определили а — +120 мкв!град. По
данным автора и Теслйцкой1, для MoS 2, отожженного в парах
серы, а = 450ч-750 мкв!град.
Эффект Холла и другие электрические характеристики.
Вследствие различий в содержании примесей данные различных
авторов о величине постоянной Холла, концентрации и подвиж-
ности носителей тока, энергии возбуждения существенно отли-
чаются. Для образцов синтетического MoS2 авторы работы [75]
определили постоянную Холла R — —1,7-10“2 см31кул, концен-
трацию носителей тока п — 3,7- 1020 см~3; подвижность носителей
тока и 0,28 см2/(в-сек). Однако авторы не приводят характе-
ристики чистоты и структуры образцов синтетического дисуль-
фида, с которыми проводилась работа.
Измерения [72] показали, что у различных образцов природ-
ного молибденита значения коэффициента Холла колеблются
от 35 до 3000 см3/кул.
По данным работы [77], у молибденита концентрация носи-
телей тока п-5,9-1018 см~3. Для сколотой пленки молибде-
нита толщиной несколько микронов в работе [76] определено
п — 1016 см~3, а подвижность и = 100 см21(в-сек). Из измерений
электропроводности и постоянной Холла Самедова—Бейбутова
определила [78] энергию возбуждения молибденита АЕ — 0,75 эв.
По другим данным [72], энергия возбуждения для различных
образцов молибденита равна 0,55; 0,14; 0,12; 0,05 эв.
Магнитные свойства. Молибденит диамагнитен. Магнитная
восприимчивость в направлении, параллельном гексагональной
оси С, % к 87,1-10"6, а в направлении, перпендикулярном
гексагональной оси, = —44,3-10~6 [79]. Увеличение диа-
магнетизма в направлении, перпендикулярном оси С кристалла,
1 См. сноску па стр. 228.
229
обусловлено наличием свободных электронов с высокой подвиж-
ностью в базисной плоскости и ограниченной подвижностью
в перпендикулярном к ней направлении.
Фотоэлектрические и оптические свойства. Электросопротивле-
ние дисульфида молибдена понижается при его освещении. Макси-
мальный фотоэффект наблюдается при длине волны X = 0,8 мкму
а фоточувствительность простирается до X % 2 мкм [80]. Фото-
электрическая чувствительность возрастает примерно втрое при
понижении температуры с +70 до —70° С [81 ]. Оптические свой-
ства MoS2 изучали на тонких монокристаллических пленках
молибденита [50, 82, 84]. Для пленок толщиной 100 А при темпе-
ратуре 290° К в области 1—4 эв полосы поглощения найдены при
длинах волн, А: 2700, 3950; 4480; 6050; 6660 [50].
В спектре отражения монокристаллов молибденита при ком-
натной температуре в области 1—6 эв пики отражения располо-
жены при длинах волн 260; 400; 460; 600; 680 и 1100 микрометров
[83].
Другие физические свойства MoS%. Удельная тепло-
проводность дисульфида молибдена, х =
= 4,75- 10“4 кал!(см-град-сек) (при 80° С) [85].
У полупроводников теплопроводность определяется перено-
сом тепла электронами (хэл) и колебаниями молекул решетки хф
(фононная теплопроводность).
Установлено, что для MoS2 хф/хэл = 84, т. е. основной вклад
в механизм электропроводности вносят колебания решетки.
Дебаевская температура для MoS2 0 — 209° К [85].
Диэлектрическая проницаемость 8 = 5,6 при 80° С [85], при
— 196° С 8 = 7,6 [86].
Смазывающие свойства MoS2
Дисульфид молибдена — один из весьма эффективных твердых
смазочных материалов. Его широко применяют в виде сухой
смазки, в составе консистентных смазок, в виде суспензии в масле
в машиностроении, авиации, металлообработке, электрооборудо-
вании [36]. В практике преимущественно используют чистый
природный дисульфид молибдена — минерал молибденит.
Высокие смазывающие свойства молибденита обусловлены его
структурой — совершенной спайностью, обусловленной слабой
связью между слоями S—Мо—S. Хорошие смазывающие свойства
молибденита сохраняются в широком интервале температур
от —45 до +400° С (выше 400° С происходит окисление с образо-
ванием МоОз).
Как видно из табл. 56 и 57, при обычной температуре коэф-
фициент трения MoS2 ниже, чем графита, а долговечность работы
трущихся поверхностей значительно выше.
230
ТАБЛИЦА 56
КОЭФФИЦИЕНТ ТРЕНИЯ И ИЗНОС МОЛИБДЕНИТА И ГРАФИТА
(скорость скольжения 2,9 см/сек', осевая нагрузка 18 кГ, время опыта 30 мин.)
[96]
Материал Коэффициент трения за время, мин Площадь пятна износа, см2
1 30
Графит 0,06 0,11 0,02
MoS2 (молибденит) 0,017 0,047 0,010
ТАБЛИЦА 57
КОЭФФИЦИЕНТ ТРЕНИЯ И ДОЛГОВЕЧНОСТЬ ГРАФИТА И МОЛИБДЕНИТА
(удельная нагрузка 6000 кГ/см2, скорость скольжения 0,13 см/сек, предварительная
обработка —фосфатирование, толщина покрытия 8 —13 мкм) [97]
• Материал Коэффициент трения Долговечность, циклы
Графит 0,080 8 640
MoS2 . . 0,034 103 680
В отличие от молибденита, у которого низкий предел прочности
на сдвиг обусловлен его структурой, у графита малый предел
прочности на сдвиг является следствием присутствия адсорбиро-
ванных молекул паров воды на плоскостях сдвига [38]. Поэтому
смазывающие свойства графита резко ухудшаются в , вакууме,
при повышенных температурах (удаляются молекулы воды)
или при температурах ниже нуля.
По способности образовывать поверхностную пленку (нали-
пать на трущуюся поверхность) молибденит также превосходит
графит [39]. Механизм взаимодействия молибденита с поверхно-
стями металлов рассмотрен в работе [40], а теория его действия
как смазывающего материала — в работе [91].
Пленка дисульфида молибдена имеет высокое сопротивление
продавливанию в направлении, нормальном плоскости чешуек
молибденита. Предел текучести ее достигает 35000 кПмм\ что выше
предела текучести некоторых сталей. При столь высоких давле-
ниях возможна деформация (сглаживание шероховатости) поверх-
ностей металлов под пленкой, что приводит к резкому увеличе-
нию фактической поверхности контакта. Графит не выдерживает
столь высоких нагрузок [41, 46].
Относительно пригодности в качестве смазывающего материала
синтетического дисульфида молибдена в литературе имеются
разноречивые мнения [36]. Это объясняется различным каче-
231
ством применявшегося в испытаниях MoS2. При соответствии
состава синтетического дисульфида формуле MoS2, содержании
в нем не более 0,2—0,5% примесей смазывающие свойства синте-
зированного продукта не уступают свойствам молибденита. Это
вполне согласуется с сохранением у синтетического дисульфида
молибдена решетки слоистого типа [44].
Химические свойства MoS2
Поведение дисульфида молибдена при нагревании уже рас-
смотрено выше. Детально освещены были также кинетика окис-
ления (гл. II) и взаимодействие MoS2 с хлором (гл. VIII). Фтор
активно (с воспламенением) реагирует с MoS2 уже при слабом
нагревании [4].
В воде, соляной кислоте любой концентрации и разбавленной
серной кислоте, а также растворах соды на холоду и при нагре-
вании до 100° С дисульфид молибдена практически устойчив
[88]. Азотная кислота и царская водка при нагревании быстро
окисляют молибденит.
* В щелочных средах (pH 10) при доступе воздуха тонко-
дисперсные порошки MoS2 очень медленно окисляются. Скорость
окисления меньше в нейтральных и слабокислых растворах [87].
Выше 300° С MoS2 реагирует с водяным паром, вероятно, по
реакции [89]:
MoS2 + 6Н2О = МоО2 + ЗН2 + 2SO2. (174)
MoS2 начинает взаимодействовать с СО2 и SO2 выше 500° С,
реакция активно протекает при 800—1000° С [98]:
MoS2 + 6СО2 = МоО2 + 2SO2 + 6СО, (175)
MoS2 + SO2 = МоО2 + l1/^.
(176)
Выше 500° С при отсутствии кислорода MoS2 активно реаги-
рует с МоО3 с образованием МоО2 (см. стр. 29).
При нагревании MoS2 в инертной атмосфере с сульфидами
металлов группы железа образуются соединения TWeMoS^ (где
Me—Fe, Со, Ni, у — 2,5 ч-З). Сложный сульфид FeMoS2i5
обладает ферромагнитными свойствами [13].
ГЛАВА X
ДРУГИЕ СОЕДИНЕНИЯ МОЛИБДЕНА
39. СИЛИЦИДЫ МОЛИБДЕНА
Свойства силицидов
В системе Мо—Si установлено три силицида: Mo3Si (плавится
с разложением), Mo3Si2 и MoSi2 [1—5]. На рис. 82 приведена
ориентировочная диаграмма плавкости по данным работ [4, 6, 9].
70 20 30 00 7о(ат.)
2800
2600
2000
^2200
S 2000
С
| 7800
Аз
Z600
7000
Мо 2 О 6 8 70 72 70 76 78 81
7о (по массе)
Рис. 82. Диаграмма состояния системы молибден—
кремний [4, 9]
Соединение Mo3Si образуется по перитектической реакции между
Mo3Si2 и расплавом, тогда как два других соединения плавятся
конгруэнтно. Состав и точка плавления эвтектики MoSi2 — Mo3Si2
точно не установлены.
В справочной литературе [7, 8] приводится диаграмма состоя-
ния Мо—Si, построенная Киффером и Червенка [5], отличаю-
щаяся от приведенной на рис. 82 тем, что Mo3Si образуется по пери-
тектической реакции расплава с молибденом, а не с Mo3Si2. Од-
233
нако более поздняя работа [6] подтвердила данные, опублико-
ванные в работах [4].
Растворимость кремния в молибдене составляет 0,27% (по
массе) (при 1315° С), 0,8% (при 1425° С), 1,65% (при 2070° С)
О Si
Рис. 83. Структура MoSi2
[4, 10]. Молибден в кремнии практически
нерастворим.
В табл. 58 приведены свойства сили-
цидов молибдена. Наиболее изучен и
практически важен дисилицид молибдена.
Дисилицид молибдена — металлоподобное
соединение, обладающее металлической
электропроводностью. Кристаллизуется
в сложной -структуре тетрагонального
типа. Атомы Si образуют каркас, в пу-
стотах которого расположены атомы мо-
либдена. В направлении оси Z чередуются
двойные плотноупакованные слои из ато-
мов кремния со слоями из атомов молиб-
дена (рис. 83). О значительной доле метал-
лических связей у MoSi2 говорит рост
его электросопротивления с температу-
рой.
Ниже приведены свойства дисилицида молибдена.
Кристаллическая структура ....................
Периоды решетки, А[1] . . ....................
Плотность ....................................
Микротвердость при нагрузке 100 Г, кГ/мм2 [7]
Температура плавления, °C [5] ................
Теплота образования (298° К), ккал!моль [13, 14]
Теплоемкость (298—1150° С), кал!(г-град) [14]
Теплопроводность, кал!(см • град • сек) [7] . . . .
Коэффициент линейного расширения, град~х [7]
Предел прочности при сжатии, кГ/мм2 [8] . . . .
Предел прочности при растяжении, кГ!мм2 [16, 7]
Удлинение при растяжении, % [16, 7]...........
Кратковременная прочность, кПмм2 [7] . . . .
Модуль упругости, кПмм2 [8]...................
Температура перехода в пластичное состояние,
°t [7] ...................................
Удельное электросопротивление, мком-см [7] . .
Тетрагональная (тип
а = 3,206; с — 7,877;
с/а— 2,457
5,9—6,3
1290
2030± 50
— 31,4
15,75 + 3,24-10“3Т—
—1,08-10бТ'2
0,129 (150° С); 0,093 (540° С)
5,1 ИО"6
113
28 (980° С); 29,4 (1200° С)
<0,5 (27—1320° С)
60,5 (1100° С); 50,5 (1200° С)
43 000
>1600
21,5—25 (22° С);
75—80 (1600° С)
Дисилицид молибдена отличается высокой окалиностойкостью
(сопротивление окислению) при температурах 1300—1700° С.
Это объясняется образованием на поверхности MoSi2 газонепро-
ницаемой стеклообразной пленки, богатой SiO2 [19]. При темпера-
турах 600—1000° С стойкость против окисления низка, так как
234
ТАБЛИЦА 58
СВОЙСТВА СИЛИЦИДОВ Mo3Si И Mo3Si2
Свойство Mo3Si Mo3S 2
Кристаллическая структура . . . .
Периоды решетки, А ...............
Плотность, г/см3..................
Микротвердость при нагрузке 100 г,
кГ/мм2 ...........................
Температура плавления, °C . . . .
Теплота образования (298° К),
ккал/моль ........................
Кубическая (тип P-W) а = 4,90 [3] Тетрагональная, слоистая [12] а ~ 9,66; с = 4,99; с/а = 0,51
8,4+0,3 [3]
1310 —2120 [4] 2050 ±50 [5] 1170 —2190 [4] 2100 + 50 [5]
-23,5 [14] — 46,8 [13, 14]
образуются рыхлые слои окислов SiO2, МоО2 и МоО3, из которых
последний заметно возгоняется выше 750° С [17].
Толщина стекловидной защитной пленки на спеченных образ-
цах MoSi2 после выдержки в течение 2000 ч при 1400° С состав-
ляет 0,02—0,03 мм, а после 3000 ч при 1700° С—0,05—0,1 мм [17 ].
Изменение массы спеченных образцов MoSi2 после выдержки
при 1200° С в течение 4 ч составляло +0,3 мг!см2 [18, 19].
Самсонов и Портной для горячепрессованных образцов MoSi2
при той же температуре получили более высокие значения прибыли
массы: за 5 ч 6 мг!см\ за 40 ч 8 мг!см\ за 100 ч 24 мг!см<21 [7]..
По данным работы [34], окисление MoSi2 в интервале темпера-
тур 900—1300° С подчиняется временному закону вида W = Клп,
U/ — изменение массы; К — константа скорости. Показатель
п 0,72 (900° С); 0,42 (1200° С).
Дисилицид молибдена практически не растворяется в мине-
ральных кислотах (H2SO4, HF, НС1, HNO3) и в царской водке,
однако легко растворяется в смеси азотной и плавиковой кислот
17, 33].
Предварительное окисление MoSi2 с образованием защитного
слоя приводит к полной устойчивости силицида в азотной и соля-
ной кислотах любой концентрации.
При температуре 1000° С MoSi2 устойчив против действия SO2,
СОа, NO2. После защиты предварительным окислением он устой-
чив в хлористом водороде. Однако хлор быстро реагирует cMoSi2
даже после предварительной защиты окислением [7].
Дисилицид устойчив в расплавленных натрии, свинце, висмуте,
олове, ртути и других металлах, не образующих силицидов.
Расплавленный цинк взаимодействует cMoSi2 в малой степени.
При 800° С в силициде растворяется до 1 % Zn,который выделяется
при охлаждении [17].
235
Крипоустойчивость (сопротивление ползучести) у дисилицида
молибдена низкая, что видно из табл. 59 [20].
ТАБЛИЦА 59
КРИПОУСТОЙЧИВОСТЬ MoSi2 ПРИ ВЫСОКИХ ТЕМПЕРАТУРАХ
Температура, °C Напряжение, кГ/мм2 Время до разрушения, ч Скорость крипа, %л
870 24,5 107 0,0024
980 14,0 224 0,0028
1040 8,4 ПО 0,073
1095 . 7,0 85 0,18
К недостаткам MoSi2 как жаростойкого материала относится
низкая теплостойкость (т. е. устойчивость в условиях частых
смен температуры) [16].
Получение MoSi2
Синтез из элементов. Наиболее распространенный способ
получения чистого MoSi2 — нагревание смеси порошков молибдена
и кремния. Смесь порошков кремния и молибдена прессуют в бри-
кеты и выдерживают при температурах 1000—1100° С в атмосфере
инертного газа в течение 0,5—1 ч. Полученный силицид содержит
примерно 35% SiCBH3 и — 0,4% SiCBO6 [22].
Можно нагревать брикеты в графитово-трубчатых печах в ат-
мосфере водорода. В этом случаеMoSi2 содержит 0,1 % С, что вполне
допустимо [21]. Графитово-трубчатые печи удобны для промышлен-
ного получения дисилицида молибдена.
Брохин с сотрудниками [35] получил MoSi2 горячим прессова-
нием (спекание под давлением) смесей порошков молибдена и крем-
ния в графитовых пресс-формах. Горячее прессование проводили
при 1400—1450° С под давлением 150 кг!см2 с выдержкой в тече-
ние 3—5 мин. Размеры образцов 50x25 мм. Авторы отмечают,
что получению плотных образцов с хорошими характеристиками
окалиностойкости благоприятствует избыток кремния в шихте
до 5% по отношению к стехиометрическому количеству. Образцы
имели кажущуюся плотность 5,9 г!см3.
Алюмо- и силикотермические способы. Дисилицид молибдена
может быть получен восстановлением алюминием смеси МоО3 +
+ SiO2 в присутствии серы для образования легкоплавкого суль-
фидного шлака. Продукт восстановления попеременно обрабаты-
вают растворами NaOH, НО и HNO3 для удаления окиси алю-
миния [24, 1].
В промышленных масштабах, по данным Норткотта [23],
технический силицид молибдена получают алюмотермическим
восстановлением трехокиси молибдена в присутствии ферросили-
236
ция. Процесс проводят аналогично получению ферромолибдена.
Шихту, состав которой приведен в табл. 60, засыпают в стальной
цилиндр, футерованный огнеупорным кирпичом, установленный
в небольшом углублении на влажном песке. Шихту поджигают
смесью порошка алюминия и перекиси натрия («запал»), после
чего реакция протекает самопроизвольно. В нижней части цилиндра
собирается слиток силицида молибдена. Состав силицида приведен
в табл. 60.
ТАБЛИЦА 60
СОСТАВ ШИХТЫ И СИЛИЦИДА МОЛИБДЕНА, ПОЛУЧАЕМОГО
ИЗ НЕЕ АЛЮМОТЕРМИЧЕСКИМ МЕТОДОМ
Шихта Силицид молибдена
составляющие количество, кг составляющие содержание, %
Молибден (в . виде трех-
окиси) 450 Молибден 60
Алюминий (крупка) . . 117,5 Кремний ........ 34
Ферросилиций (90% Si) 345 Железо 4
Ферросилиций (50% Si) 75 Сера 0,04
Плавиковый шпат . . . 22,5 Медь 0,42
Известняк 72,5 Алюминий 0,37
Медно-силицидный метод. Метод основан на взаимодействии
молибдена с кремнием в расплаве меди [25, 26]. В расплаве обра-
зуются кристаллики силицида молибдена, состав которых за-
висит от концентрации кремния в ванне. Этим способом могут
быть получены все силициды молибдена. После измельчения сплав
обрабатывают попеременно растворами кислот и щелочей для от-
деления меди и избытка кремния от силицидов молибдена. Медно-
силицидный способ используют для нанесения защитного покры-
тия MoSi2 на изделия из молибдена.
Молибден погружают в расплавленную медь, содержащую
10—30% Si при температуре выше 1200—1300°С. Максимальная
скорость наращивания наблюдается, когда ванна содержит 16,5% Si
при 1300° С. За 20 мин толщина покрытия составляет — 100 мкм,
за 60 мин —200 мкм [27, 28].
Карботермический метод. Марковский и Векшина разработали
условия получения MoSi2 восстановлением углеродом смеси окис-
лов МоО3 и SiO2:
МоО3 + 2SiO2 + 7С -> MoSi2 + 7СО. (177)
Авторы показали, что при некотором избытке кремнезема в шихте
и кратковременной выдержке брикетированной смеси при 1900° С
(—15 мин) можно получить MoSi2 без примеси карбида кремния.
Содержание углерода в силициде колеблется от 0,2 до 0,7%, азота
1,5% (если процесс ведется без защитной атмосферы).
237
поверхности молибдена (ленте,
Рис. 84. Зависимость толщины покры-
тия молибденовой проволоки диамет-
ром 2 жж силицидом молибдена от вре-
мени и температуры, °C:
1 — 1800; 2 — 1600; 3 — 1400; 4 — 1200
Рекомендуемый состав шихты, %: 33,4 МоО3; 47,2 SiO2;
19,4 С. Брикеты можно нагревать в угольно-трубчатой печи.
Осаждение из газовой фазы. Способ состоит в восстановлении
четыреххлористого кремния водородом на нагретой до 1100—1800° С
проволоке) [29—32].
На поверхности молибдена
постепенно наращивается слой
MoSi2. Этот метод преимущест-
венно применяют для получения
защитных покрытий на молиб-
дене. В случае покрытия молиб-
деновой проволоки ее нагревают
прямым пропусканием электри-
ческого тока. При покрытии
массивных изделий из молиб-
дена целесообразно применять
индукционный нагрев.
Для полного использова-
ния SiCl4 предложено осуществ-
лять процесс в замкнутой си-
стеме с циркулированием паро-
газовой смеси SiCl4+H2 [31].
При температурах покрытия ниже 1420° С под наружным слоем,
содержащим MoSi2 + Si, находится слой низшего силицида
Mo3Si2, непосредственно примыкающий к молибдену. При темпера-
турах выше 1420° С промежуточный слой отсутствует [30].
На рис. 84 приведена зависимость толщины покрытия сили-
цидом молибдена от времени и температуры.
Получение изделий из MoSi2
Окалиностойкость дисилицида молибдена, коррозионная стой-
кость, металлическая проводимость и другие свойства послужили
основанием для его использования в качестве материала нагре-
вателей высокотемпературных печей (подобных силитовым нагре-
вателям), эксплуатируемых в атмосфере воздуха и агрессивных
газов. Недостаток MoSi2 как нагревателя — относительно высокая
электропроводность. С целью ее снижения при изготовлении нагре-
вательных стержней вводят присадки окислов (например, SiO2,
А12О3), а также бора и углерода [21].
Так, в Швеции выпускают нагреватели для печей «Супер-
Кантал», содержащие MoSi2, SiO2 с добавками других окислов
и металлов. Удельное сопротивление этих нагревателей изме-
няется от 40 мком-см при 20° С до 350 мком-см при 1600° С [37].
В отличие от нагревателей из карбида кремния, работающих
устойчиво только до 1400° С, нагреватели из MoSi2 с оксидными
добавками работают длительное время при 1600—1700° С.
238
Применяют защитные покрытия MoSi2 молибденовых нагрева-
телей (проволоки, ленты, стержней), наносимые из газовой фазы
(SiCl4 + Н2) или другими способами. Такие молибденовые нагре-
ватели могут работать в окислительной атмосфере при 1700° С.
Обсуждается возможность использования MoSi2 для изготовле-
ния огнеупоров и материалов теплообменников атомных котлов
(в связи с устойчивостью силицида в расплавленных металлах-
теплоносителях) [17, 37], а также его применения в качестве мате-
риала для изготовления химической аппаратуры.
Изделия из дисилицида молибдена могут быть получены го-
рячим прессованием или спеканием предварительно спрессован-
ных заготовок. Горячее прессование, в котором объединяются
операции прессования и спекания, обеспечивает получение наиболее
плотных изделий. Горячее прессование изделий проводят в пресс-
формах из плотного графита на прессах различной конструкции [38]
Высокоплотные изделия получаются при температуре —1900° С,
давлении прессования 250 кПсм? и времени выдержки 10лшн.
Спеченные образцы после горячего прессования для снятия
внутренних напряжений отжигают при температурах на 150—200° С
ниже температуры спекания.
При использовании метода спекания предварительно спрессо-
ванных заготовок порошок MoSi2 прессуют под давлением 2,8 Т1см\
а спекают при 1400—1700° С в вакууме. Качество таких изделий
ниже, чем горячепрессованных. Лучшие результаты получаются
при введении в исходный порошок перед прессованием пластифика-
торов (раствор каучука в бензине, парафина в бензине, бакелита
в спирте и др.), что облегчает формование. В процессе спе-
кания большая часть пластификатора удаляется, оставшаяся
примесь углерода способствует получению более прочных изде-
лий [8].
Для производства длинных трубок и стержней из дисилицида
молибдена (часто с оксидными добавками) применяют метод мунд-
штучного прессования массы порошка с пластификаторами. Кроме
перечисленных выше пластификаторов, можно использовать крах-
мальный клейстер [8]. Спрессованные заготовки сушат на воздухе
1—2 суток (или более короткое время в вакуум-сушилках) до ко-
нечной влажности 4—6%.* Спекание проводят в электропечах
сопротивления с молибденовыми нагревателями или угольно-
трубчатых печах в атмосфере водорода или инертного газа.
Стержневые трубчатые заготовки спекают в засыпках из А12О3
или ВеО. Контейнер со спекаемыми изделиями помещают в печь,
разогретую до 600—700° С, затем температуру за 1—3 ч поднимают
до максимальной (~1750° С), при которой изделия выдерживают
15—20 мин и охлаждают вместе с печью до 800—900° С. Пористость
изделий 4—6%. Из пластифицированных смесей можно также
изготовлять листы и пластины прокаткой в валках и последую-
щим спеканием [8].
239
40. БОРИДЫ МОЛИБДЕНА
Систему молибден—бор изучали многие исследователи [39—
46]. На рис. 85 приведена наиболее достоверная диаграмма со-
стояния, построенная Портным с сотрудниками [45]. Авторы
критически рассмотрели предшествующие данные и показали,
что источником ошибок было присутствие примеси углерода в ис-
следуемых образцах системы бор.— молибден. Чтобы исключить
примеси углерода, спрессованные образцы смесей бор—молибден
Рис. 85. Диаграмма состояния системы
молибден — бор
нагревались в печи с вольфрамовым нагревателем в условиях,
исключающих контакт образца с посторонним материалом. Диа-
грамма построена по данным термического, металлографического
и рентгеноструктурного анализов.
Растворимость бора в молибдене при эвтектической температуре
(2200° С) равна — 1%.
В системе установлено пять боридов молибдена: Мо2В, МоВ,
МоВ2, Мо2В5, МоВ~12. Из них МоВ и MoB~i2 плавятся кон-
груэнтно, остальные фазы плавятся с разложением по перитекти-
ческим реакциям. Все бориды, кроме Мо2В, имеют области гомо-
генности различного простирания. Борид Мо3В2 как стабильная
фаза в системе не обнаружен. Это согласуется также с данными
работы [44]. По всей вероятности, эта фаза стабилизируется лишь
в присутствии углерода.
В табл. 61 приведены данные о структуре и некоторых свой-
ствах боридов молибдена [8, 50].
240
ТАБЛИЦА 61
О
Зеликман
СТРУКТУРА И НЕКОТОРЫЕ СВОЙСТВА БОРИДОВ МОЛИБДЕНА
ьэ
Борид Структура и периоды решетки Теплота образования, —ккал/моль [W3] Температура плавления, °C [45] Микротвер- дость, кГ/мм2 (нагрузка 50 Г) [45] Плотность расчетная, z{cmz Удельное сопротивле- ние, мком-см [47] Температура перехода в сверх- проводящее состояние, °К
Мо2В Тетрагональная, тип СиА12 [39, 40] а = 5,543, с =
а-МоВ = 4,735 А, с/а = 0,854 . . Тетрагональная, а = 3,110, с = 16,95 A, da — 5,45 25,5 2270—2300 (перитек- тика) 1700—1900 9,31 40
Р-МоВ [39—41] Ромбическая, а = 3,16, b — 16,3 2100—2500 8,77 45 4,40 [49] <1,8 [48]
МоВ2 = 8,61, с = 3,08 А [40] . . Гексагональная, а = 3,06, с = — 2550 — — 25 —
Мо2В5 = 3,10 А, с!а = 1,01 [40] Ромбоэдрическая, а= 3,011, с = 20,93 А, с!а = 6,95 [39, 23,0 2350 (перитек- тика) 2100—2500 7,78 45
40] 50,0 2200 (перитек- тика) 2300—2800 7,48 25 <1,3 [491
Некоторые сведения о других свойствах, помимо приведенных
в табл. 61, имеются только для Мо2В5. Они приведены ниже [8].
Теплоемкость Ср, кал (г-град) ................. 0,11
Теплопроводность при 20° С, кал (см • сек • град) . . §4
Работа выхода электронов ф, эв ........ 3,38
Предел прочности при изгибе, кГ/м2............... 35
Бориды молибдена не отличаются высокой окалиностойкостью.
При температурах 1000—1100° С они быстро окисляются с обра-
зованием МоО3 и В2О3. Наиболее высокая скорость окисления
у Мо2В, значительно меньшая — у Мо2В5. Моноборид занимает
промежуточное положение [40].
Борид Мо2В5 устойчив на холоду против действия концентри-
рованных соляной и серной кислот, при нагревании он быстро
разлагается. В фосфорной, хлорной,щавелевой и плавиковой кисло-
тах, а также в 30 %-ном растворе едкого натра на холоду и при
нагревании Мо2В5 не устойчив [8].
Известны следующие способы получения боридов молибдена:
1. Синтез из элементов. Наиболее чистые бориды полу-
чают нагреванием брикетов из смеси порошков молибдена и бора
при 1200° С в вакууме [39] или атмосфере чистого водорода при
1500—1700° С [40, 51]. Может быть использовано также горячее
прессование.
2. Восстановление смеси окислов МоО3 и В2О3
углеродом или МоО3 смесью карбида бора и сажи по реакциям:
МоО3 + В2О3 + 6С = МоВ2 + 6СО, (178)
2МоО3 + В4С + 5С = 2МоВ2 + 6СО.
О * I 4J I
(179)
Борокарбидный метод имеет преимущества перед карботерми-
ческим, так как в первом случае В2О3, содержащийся в шихте,
частично летит, что затрудняет получение борида заданного состава
и приводит к повышенному содержанию углерода в бориде [52].
Смеси МоО3 с В4С и сажей нагревают в вакууме или в атмо-
сфере водорода при 1200—1300° С. Реакция протекает быстро —
за 0,5—1 ч. В вакууме получают более чистые бориды. Для повы-
шения производительности шихту брикетируют под давление
0,5—1 Т/см2.
При получении борида молибдена в графитово-трубчатой печи
в атмосфере водорода в шихту вводят, кроме В4С, борный ангид-
рид (3—7% от массы шихты) для удаления в процессе нагрева
свободного углерода в форме. СО.
При' получении диборида молибдена восстановлением углеродом
смеси МоО3 с В2О3, по данным работы [102], рекомендуется
вводить в шихту борный ангидрид в количестве 125% по отно-
шению к требуемому по написанной выше реакции (для вос-
полнения его потери при нагревании).
242
При выдержке шихты в угольно-трубчатой печи в течение 40 мин
при 1800° С получается диборид, близкий по составу МоВ 2. Однако
в нем содержится до 0,5% С, а также примесь В2О3. В связи
с этим предложен метод рафинирования диборида, состоящий
в прокаливании при 1600—1800° С в вакууме (5-10“4—5* 10"6мм
рт. ст.) брикетированной смеси диборида с добавками МоО3 и бора.
При этом было установлено, что химическое взаимодействие
диборида с указанными добавками резко активизировало процесс
усадки брикетов при спекании.
3. Электролиз расплавленных *сред. Электролиз
расплава, содержащего МоО3, Na2B4O7 и NaF, при 1000° С позво-
ляет выделить на катоде кристаллические осадки боридов молиб-
дена. Состав их зависит от соотношения молибдена и бора в ванне.
При начальном молярном отношении МоО3: В2О3: NaF=0,2:2: 1
на катоде выделялись кристаллические осадки, близкие по составу
Мо2В (5,33% В). Уменьшая содержание МоО3 в электролите
до 0,12—0,1, можно получить осадки МоВ (10,13% В) [57 .
Разработан электролитический способ покрытия молибдена
боридом молибдена. Борирование проводят из расплава, содержа-
щего 33% НВО2и67% NaF. При катодной плотности тока 9,6 а!см\
напряжении 11 в, температуре950—1200°С на молибденовом катоде
через определенное время (1—2 ч) образуются два слоя: внешний
(микротвердость 2650 кПмм2) и внутренний, граничащий с мо-
либденом (микротвердость 1680 кПмм2). По всей вероятности,
внешний слой соответствует бориду с более высоким содержанием
бора (МоВ 2 или Мо2В5) [53].
4. Осаждение из газовой фазы. При пропускании
смеси газов ВС13 + Н2 над поверхностью молибдена, нагретой до
1800—2000° С, образуются боридные покрытия.
Бориды молибдена обладают высокой твердостью, износо-
стойкостью, а также крипоустойчивостью, что позволяет их
использовать для изготовления режущего и бурового инструмента,
жаропрочных сплавов.
В работах [54, 55] установлено, что борид молибдена, цементи-
рованный никелем, дает сплав с режущими свойствами и стойкостью,
не уступающими сплавам на основе карбида вольфрама.
Для повышения стойкости боридов молибдена против окисле-
ния на воздухе предложено легирование их кремнием или сили-
цидом молибдена. Присутствие кремния или силицида приводит
в образованию стеклообразной пленки окиси кремния, подобно
тому как это имеет место при окислении силицида кремния [56].
41. КАРБИДЫ МОЛИБДЕНА
Система молибден — углерод, построенная Сайксом с сотр. [58]
на основе термического, рентгеноструктурного и металлографи-
ческого анализов, приведена на рис. 86. Растворимость углерода
в молибдене низкая и понижается с температурой.
16* 243
По Фью и Маннингу [59], в молибдене растворяется при
1650°С 0,005—0,009% С; при 1925°С 0,012—0,013% С; при 2200° С
0,018—0,022% С. При растворении в молибдене 0,018% С постоян-
ная решетки металла увеличивается с 3,1466 до 3,14768 Л [60], что
доказывает образование твердых растворов внедрения.
В системе молибден — углерод установлены два карбида —
Мо2С и МоС. Первый из них был получен еще Муассаном [61].
В последующем его структура и свойства были детально исследо-
ваны. Существование второго карбида достоверно установлено зна-
чительно позже [58, 62, 63].
Рис. 86. Диаграмма состояния системы Рис. 87. Диаграмма состоя-
молибден—углерод (по Сайксу) ния системы молибден—
углерод [65]
Карбид Мо2С имеет область гомогенности, простирающуюся
в интервале концентраций углерода [5,3—6% (по массе)] [58]
31—33% (ат.) Период его решетки а изменяется с увеличением
концентрации углерода от 2,993 до 3,002 кХ, тогда как периоде
остается неизменным.
Карбид Мо2С плавится с разложением при температуре 2400° С.
С молибденом (a-фаза) Мо2С образует эвтектику состава 12,5%
(ат.) С [1,8% (по массе)], плавящуюся при 2200° С.
Второй карбид МоС некоторое время не могли идентифици-
ровать, по всей вероятности, вследствие того, что в интервале
1900—700° С он разлагается на Мо2С и углерод [65].
Новотный и Киффер получили МоС закалкой расплавов (Мо+С)
[64]. На основании измерения температур плавления сплавов
Мо—С в интервале 40—75% (ат.) [7,7—27,2% (по массе)] авторы
работы [65] показали, что МоС плавится на максимуме при
—2650° С и образует эвтектику с углеродом. Эвтектика плавится
при —2400° С, ее состав 70% (ат.) [22,5% (по массе)]. В связи
с этим намечена ориентировочная более полная диаграмма состоя-
ния Мо—С (рис. 87) [9, 65, 103].
244
ТАБЛИЦА 62
СВОЙСТВА КАРБИДОВ МОЛИБДЕНА
Свойства Мо2С МоС
Максимальное содержание углерода,
% (по массе) 5,91 11,13
Область гомогенности, % (ат.) . . . 31—33,7
Кристаллическая структура .... Гексагональная Гексагональная
Периоды решетки, А [63] (структура вне- дрения) (тип NiAs)
а = 3,002, с = а — 2,898, с —
= 4,724, da — = 2,809, с/а =
Плотность, г!см3’. = 1,574 = 0,969
расчетная 9,06 8,88
экспериментальная 9,18 8,4
Микротвердость (100 г), кПмм2 . . . 1480 [66] —1500
Температура плавления, °C .... Теплота образования — Д/7298 [14], —2400 (перитек- тика) [58] —2650 [65]
ккал!моль —4,2 0,072 —2,0
Теплоемкость, кал1(г*град) Термический коэффициент линейного
расширения сс-106 Удельное электросопротивление, 9,3 (по осям а и с) .
мком • см (20° С) 133 [104] —
Модуль упругости, кПмм2 [68] . . . Точка перехода в состояние сверхпро- 22 100 —
водимости, °К 2,78 [67] 9,28 [69]
В табл. 62 приведены некоторые свойства карбидов молибдена.
Карбид Мо2С — порошок темно-серого цвета. Его кристалли-
ческая структура построена по типу фаз внедрения.
Атомы молибдена образуют плотную гексагональную упаковку.
Атомы углерода размещены в октаэдрических промежутках
решетки с координационным числом 6. Геометрическая возмож-
ность образования структуры внедрения определяется отношением
радиусов атомов углерода и молибдена, равным 0,55 (по Хэггу,
фазы внедрения образуются в пределах отношений гх : гМе =
- 0,41 ч-0;59).
Монокарбид МоС, подобно WC, кристаллизуется в гексаго-
нальной решетке типа NiAs, не относящейся к фазам внедрения.
Карбид Мо2С устойчив против действия соляной и плавиковой
кислот любой концентрации на холоду и при нагревании, а также
в разбавленной серной кислоте. В азотной кислоте, царской водке,
смесях H2SO4 + HNO3 или HNO3 + HF карбид растворяется.
Выше 500° С карбид активно окисляется на воздухе и взаимо-
действует с хлором. Фтор реагирует с Мо2С при комнатной темпера-
туре.
245
Наиболее простой путь промышленного получения карбидов
молибдена — нагревание смеси молибденового порошка с сажей
в графитово-трубчатой печи в атмосфере водорода.
Для получения Мо2С стехиометрическую смесь компонентов
нагревают при 1400—1500° С, для получения МоС — при 2000° С
с быстрым охлаждением [70].
' Полученный этим методом Мо2С содержит 6,05—6,10% С,
в том числе 0,15% свободного углерода.
Другой путь — восстановление трехокиси молибдена углеро-
дом по реакции
2МоО3 + 7С — Мо2С + 6СО.
Эта реакция протекает при температурах 1100—1300° С. Для
получения карбида стехиометрического состава шихту лучше
.нагревать в вакууме. При осуществлении процесса в водороде он
участвует в восстановлении трехокиси, что затрудняет дозировку
углерода в шихте и вызывает повышенное содержание свободного
углерода в карбиде.
По данным работ [71, 72], крупнокристаллические порошки
карбидов молибдена могут быть получены электролизом из рас-
плавленных сред, содержащих Na2CO3 + Na4B2O7 + LiF +МоО3.
Электролиз проводят в графитовом тигле (анод) с графитовым
катодом при температуре 800° С и напряжении на ванне 2,5—3 в.
Состав полученного карбида определяется молярным отношением
МоО3 : Na2CO3 в ванне. Так, при электролизе из расплава
Na2B4O7 + 2Na2CO3 + 4,5LiF + (0,28 4-0,35) МоО3 получаются
серебристые кристаллы Мо2С. Из ванны с более низким содер-
жанием МоО3 [Na2B4O7 + 3Na2CO3 + 6LiF + (0,12-4-0,14) МоО3]
на катоде осаждается монокарбид МоС. Из ванн промежуточного
состава выделяется смесь карбидов.
Описано получение отложений карбидов молибдена при дей-
ствии СО + Н2 или СН4 на молибден при температурах 800—
1000° С [73, 74]:
2Мо + СО + Н2 -> Мо2С + Н2О,
2Мо + ЗСН4 Мо2С + ОгНб + ЗН2.
По данным работы [79], плотные, хорошо связанные с под-
ложкой отложения Мо2С получаются при пропускании над нагре-
той до ~1400° С поверхностью молибденовых изделий (проволока,
лист и др.) гелия, содержащего ~1,2% пентана.
В работе [75] Мо2С получали термическим разложением
хлоридов (МоС14, МоС15) при давлении 20 мм рт. ст. или ниже
на нагретой до 1480—1600° С поверхности графитовых стержней.
Можно выделить карбид молибдена из газовой фазы термиче-
ским разложением карбонила Мо (СО)б при пониженном давлении
и температурах 300—800° С [76, 77].
* Образуются также другие продукты разложения СН4.
246
При температурах 350—475° С и давлении СО (результат раз-
ложения карбонила) до 0,2 мм рт. ст. была получена кубическая
модификация Мо2С, тогда как при более высоких температурах
и давлениях — обычный гексагональный Мо2С или смесь двух
модификаций. При разложении карбонила одновременно осаж-
дается и поропюк молибдена. .
Карбид молибдена Мо2С применяют в ограниченной степени
в некоторых твердых сплавах [78]. Твердые сплавы на основе
одного лишь Мо2С имеют недостаточную твердость и хрупки.
Например, сплав состава 90% Мо2С + 10% Ni имеет твердость
81 HRCqq и сопротивление на изгиб 60—80 кПмм2.
Несколько лучшими свойствами обладают сплавы, в которых
Мо2С сочетается с WC и цементирующим металлом — кобальтом
или никелем. Однако и эти сплавы уступают по свойствам сплавам
на основе WC.
Практическое значение имеют сплавы, в которых Мо2С со-
четается с карбидом титана. В процессе спекания образуются
твердые растворы карбидов, причем растворимость Мо2С в TiC.
достигает 50%, a TiC в Мо2С — 20%.
Сплавы Мо2С—TiC были первыми промышленно важными
титансодержащими твердыми сплавами. Цементирующей фазой
служил никель с добавками хрома. Они обладают более высокой
твердостью, чем сплавы WC—Со, но более низким сопротивлением
изгибу, и могут использоваться для чистовой обработки стали.
Однако их превосходят сплавы типа WC—TiC—Со. Добавки
Мо2С в сплавы WC—TiC—Со повышают их твердость, но снижают
их вязкость.
При повышенном содержании Мо2С вязкость возрастает, если
вместо кобальта в качестве цементирующего металла применить
никель [78]. В табл. 63 приведен состав и свойства твердых спла-
Т АБЛ ИЦА 63
СВОЙСТВА СПЛАВОВ WC—Мо2С —TiC С РАЗЛИЧНОЙ
ЦЕМЕНТИРУЮЩЕЙ СВЯЗКОЙ [78]
Состав, % HRA Прочность при изгибе, кГ/мм2
WC Мо2С TiC “ Ni Со
77 1 16 6 91 120
76 2 16 1 6 91 115
73 5 16 3 3 91 100
60 16 16 8 — 91 100
60 16 16 — 8 91,5 85 *
30 30 25 15 91 85
15 30 45 5 5 91 90
15 30 40 10 5 91 100
15 15 55 10 5 91 100
20 10 65 5 92 95
247
bob WC—Mo2C—TiC. Их можно успешно применять для резания
сталей, однако они более хрупки, чем соответствующие сплавы
без карбида титана.
42. КАРБОНИЛ МОЛИБДЕНА Мо[СО]6
Свойства карбонила молибдена
Гексакарбонил молибдена — белое кристаллическое вещество.
Решетка — ромбическая молекулярная, пространственная группа
— Рпа [80]. В молекуле карбонила атом молибдена окружен
Рис. 88. Структура молекулы
шестикарбонила молибдена
шестью группами СО, расположен-
ными в вершинах октаэдра. Атомы
Мо—С—О лежат на одной прямой
(рис. 88).
ТАБЛИЦА 64
ДАВЛЕНИЕ ПАРА Мо (СО)в
Темпе- ратура, °C Давление пара, мм рт ст., по уравнению
[76] [83]
25 0,011 0,01
100 41,2 36,9
155 828 760,0
200 55,6-10s 50,9-103
Расстояние
чески [81], А:
между атомами, определенное электронографи-
Мо—С .... 2,08±0,04
Мо—О .... 3,23±0,05
С—О .... 1,15±0,05
В элементарной ячейке 4 молекулы Мо (СО)6. Периоды ре-
шетки, А : а = 12,02; b = 6,48; с = 11,23. Плотность карбонила
молибдена при 25° С 1,96 г!см3. Высокая симметричность моле-
кулы карбонила обусловливает летучесть соединения — возгонка
наблюдается уже при 30—40° С.
Давление пара над твердым карбонилом, по данным различных
авторов, описывается уравнениями:
1gр = —-|- 11,795 мм рт. ст. [76],
1g р =-----— + 11,7274 мм рт. ст [83].
Вычисленные по этим уравнениям значения давления пара
близки между собой (табл. 64).
248
Некоторые термодинамические свойства карбонила молибдена
приведены ниже.
Температура сублимации, °К [83] ..... 428,2
Теплоемкость, 298° К, кал/град\
по [84]............................... 57,92
по [85]........................... 49,08
Теплота образования из элементов, ккал!моль
[86] ............................... 233,12
Теплота образования из металла и окиси уг-
лерода, ккал/моль [87] .............. 74,0
Свободная энергия образования из эле-
ментов, ккал/моль [88].................211,29
Теплота сублимации, ккал/моль [83, 76] 17,3
Свободная энергия сублимации, ккал/моль
[88]....................................+4,99
Энтропия сублимации, кал/моль [83] . . . +37,93
Энтропия при 298° К, э. е.:
твердый [84]......................... 78,17
газообразный [85].............117,71
До 150° С Мо (СО)6 термически устойчив, полное разложение
на молибден и СО наблюдается при 400° С.
Карбонил молибдена диамагнитен. Молярная магнитная вос-
приимчивость равна минус 80-10-6. В абсорбционном спектре
паров карбонила максимумы поглощения наблюдаются при вол-
новых числах 31 000, 34 500 и 43 500 см~г [101 ]. Гексакарбонил
обладает относительно высокой химической стойкостью. Соеди-
нение нерастворимо в воде и при комнатной температуре, не взаи-
модействует с концентрированными соляной и серной кислотами,
а также растворами щелочей.
Концентрированная азотная кислота быстро разлагает карбо-
нил молибдена с окислением молибдена до молибденовой кислоты.
Бром и иод взаимодействуют с карбонилом с образованием
низших галогенидов. Так, при действии брома идет реакция [98Т
Мо (СО)6 + 2Вг2 = MoBr4 + 6СО.
В спиртовом растворе едкого кали образуется карбонилгидрщц
По данным О. Д. Кричевской,*, растворимость карбонила мо-
либдена в органических растворителях ограниченная, несколько*
выше его растворимость в карбонилах железа и никеля.
Ниже приведена растворимость Мо (СО)6 в органических
растворителях при 20° С, г! 100 г:
Бензол . . *..........1,50
Толуол................1,29
о-Ксилол..............1,17
Ацетон................1,97
Этиловый эфир .... 2,00
н-Бутиловый спирт . . . 0,40
Изоэтиловый спирт . . . 0,64
Четыреххлористый уг-
лерод ..............1,46
Этиловый спирт .... 0,25
Fe (СО)6 4,94
Ni (СО)б ................3,95
*0. Д. Кричевская. Диссертация. ЛГИ, 1964.
249
Методы синтеза карбонила молибдена
•1
Многие исследователи пытались синтезировать карбонил мо-
либдена прямым взаимодействием окиси углерода с молибденовым
порошком различной степени дисперсности и активности при
давлениях СО 200 ат и выше и температурах 200—300° С. Однако
положительные результаты не были получены. Выход карбонила
не превышал 1—2% [91]. В последующем Несмеянов с сотруд-
никами в результате ряда работ пришли к заключению, что при
синтезе карбонила молибдена (как и карбонилов хрома и воль-
фрама) следует исходить не из металла, а из его соединений, на-
пример хлоридов, которые в процессе синтеза могут легко восста-
навливаться [92, 93].
Развивая эти взгляды, Белозерский и Кричевская отмечают,
что перестройка наружных электронных оболочек атома металла,
необходимая для формирования молекулы карбонила молибдена,
требует большой затраты энергии. Однако в процессе восстанов-
ления хлорида молибдена при отрыве ионов хлора временно
появляется возбужденный атом металла, внешняя электронная
оболочка которого оказывается подготовленной для образования
шестикарбонила (атом металла присоединяет молекулы окиси
углерода, не переходя в основное состояние нейтрального атома)
[95].
Разработаны «мокрые» (с применением органических раство-
рителей) и «сухие» методы синтеза карбонила молибдена из его
хлорида в присутствии восстановителей. Ниже рассмотрены важ-
нейшие из этих методов.
Синтез карбонила молибдена из хлорида «мокрым» способом.
Впервые карбонилы хрома, молибдена и вольфрама были полу-
чены методом, предложенным Джобом [96], и затем несколько
улучшенным Хибером [97, 98]. Согласно этому методу, суспен-
зия МоС15 в абсолютном эфире подвергается одновременному
воздействию реактива Гриньяра (например, фенилмагнийбромида
C6H5MgBr) и окиси углерода. Примерный режим синтеза: в реак-
ционный сосуд с быстроходной мешалкой, из которого воздух
вытеснен окисью углерода, вводят навеску безводного МоС15 и
заливают равные объемы абсолютного эфира и бензола (бензол
для растворения образующихся MgBr2 и MgCl2) примерно в ко-
личестве по 250 мл на 100 г хлорида. Затем при непрерывном
перемешивании небольшими порциями вводят фенилмагнийбромид
(около 3 моль на 100 г хлорида), растворенный в 1250 мл эфира.
Реакционный сосуд присоединён к ресиверу с окисью углерода.
Реакция продолжается около 6 ч, когда прекращается абсорбция
окиси углерода. Полученный коричнево-красный раствор гидро-
лизуют вливанием в 6-н. серную кислоту со льдом. Продукт
гидролиза отгоняют в течение 3—4 ч с водяным паром. Конец
отгонки определяют по прекращению выделения белых кристал-
250
лов в стекающем конденсате. После отгонки эфира под вакуумом
(ниже 60° С) и охлаждения до 0° С отделяют белые кристаллы
карбонила молибдена. Они могут быть очищены возгонкой при
40—50° С под вакуумом или перекристаллизованы из эфира.
Выход карбонила молибдена по описанному способу низкий
(порядка 6%).
О механизме реакции Джоба нет единого мнения [91]. Пред-
полагалось, что в присутствии реактива Гриньяра образуются
промежуточные органокарбонильные соединения, затем разла-
гающиеся с образованием карбонила металла. Однако Анисимов
и Несмеянов показали, что реактив Гриньяра играет лишь роль
восстановителя хлорида и может быть успешно заменен метал-
лами— цинком, железом и др. [92].
Возможно, что в результате восстановления и карбонилирова-
ния первоначально образуются соли карбонилгидрида — соеди-
нения типа 7?Мо (СО)5, в результате гидролиза и диспропорцио-
нирования которых получается карбонил [100]:
67?Мо (СО)5 + 12НС1 -> 6Н2Мо (СО)5 4- 6/?С12, (180)
6Н2Мо (СО)5 5Мо (СО)6 + 6Н2 + Мо. (181)
В работах [92, 93] при синтезе карбонила молибдена вместо
реактива Гриньяра был применен цинковый порошок, а в качестве
реакционной среды — безводный ацетон.
В реакционный сосуд с суспензией МоС15 в ацетоне в атмосфере
окиси углерода при —10° С постепенно вводили цинковую пыль.
Сосуд охлаждали, поддерживая температуру не выше 0° С. Далее
реакционную смесь разлагали серной кислотой со льдом и пере-
гоняли карбонил с паром. Выход составлял 10—11%.
При осуществлении описанного синтеза под давлением окиси
углерода 100 ат при комнатной температуре выход карбонила
возрос до 46%. В этом случае цинковую пыль можно вводить
в один прием.
Авторы работы [99], применяя особо чистые эфир и хлорид,
этим методом получили карбонил молибдена с выходом 83—91%.
В ряде патентов 1 указывается, что при проведении синтеза
в абсолютном эфире с использованием для восстановления хло-
рида или окисхлорида молибдена порошка алюминия или его
сплава с медью и цинком (50% Си, 45% А1, 5% Zn) при 70—100° С
и давлении СО 145—220 ат выход карбонила составляет 90% в слу-
чае хлорида и 75% в случае оксихлорида молибдена.
Реакция карбонилирования активируется содержанием в реак-
ционном газе до 2% карбонилов железа, никеля и кобальта.
Синтез карбонила «сухим» способом. Описанные выше мокрые
способы синтеза карбонила молибдена мало пригодны для про-
1 D. Т. Hurd US Pat., 2554194, 1951; 2557744, 1952; 2601022, 1952..
Brit. Pat., 678712, 1951; 678897, 1951; 703437, 1954.
25 i
мышленных целей. Они связаны с необходимостью использовать
легко воспламеняющиеся органические растворители высокой
чистоты (эфир, ацетон), потери которых в процессе синтеза и
отделения карбонила из реакционной массы значительны.
Более перспективны сухие методы, в которых восстановитель
и окись углерода воздействуют на соединения молибдена (МоС15,
оксихлориды) при отсутствии растворителя.
Первые исследования в этом направлении применительно
к хлоридам молибдена опубликованы Хибером [90], который полу-
Рис. 89. Схема установки для получения карбонила мо-
либдена из пентахлорида:
1 — ввод газа из раствора; 2 — вентили; 3 маслоотделитель;
4 — манометр; 5 — циркуляционный насос; 6 — предохрани-
тельный клапан; 7 — сборник карбонила; 8 — вентиль сброса
газа; 9 — реактор
чал Мо (СО)6, воздействуя на МоС15 или МоО2С12 окисью углерода
(рабочее давлёние 345—380 ат) при температуре 230° С и добав-
ляя в качестве восстановителя медь. Выход карбонила из МоС15
достигал 83%.
При использовании МоО2С12 лишь г/3 его превращалась в кар-
бонил, что, по мнению автора, объясняется распадом оксихло-
рида с образованием неустойчивого гексахлорида:
ЗМоО2С12 2МоО3 + МоС16.
Детальное исследование условий синтеза карбонила из МоС15
сухим способом проведено Белозерским и Кричевской [94, 95].
Авторы получали исходный МоС15 хлорированием молибденито-
вого концентрата при 500° С.
Для обеспечения максимального выхода карбонила необхо-
димо, чтобы скорость восстановления хлорида была близка ско-
рости присоединения молекул СО к возбужденному атому металла.
Этого можно достигнуть выбором восстановителя и варьирова-
нием температуры и давления окиси углерода. В качестве восста-
новителя авторы рекомендовали железную стружку крупностью
252
2 — 3 мм, количество которой берут с избытком 100% от теорети-
чески необходимого по реакции железа:
2MoCL + 5Fe = 2Мо + 5FeCl2.
М 1 I Ы
1 — сосуд для очистки раствором ще-
лочи; 2 — холодильник; 3 — вакуум-
фильтр; 4 — сосуд для перегонки
с водяным паром; 5—парообразователь
железа вследствие его покры-
Оптимальный режим процесса: температура 200° С, давление оки-
си углерода 280 ат, продолжительность операции 48—72 ч\ скорость
циркуляции реакционного газа 3—5 объемов в час. В этих усло-
виях среднее извлечение молибдена в карбонил составляет 67,5%.
При температуре 200° С и давле-
нии СО 280 ат равновесная кон-
центрация паров карбонила молиб-
дена составляет всего 2,7%. Вслед-
ствие этого необходим непрерыв-
ный отвод реакционного газа из
аппарата — его циркуляция.
На рис. 89 показана схема
укрупненно-лабораторной уста-
новки для получения карбонила
молибдена из хлорида. Все аппа-
раты выполнены из стали. В си-
стему включен циркуляционный
насос, перекачивающий газ из
реактора последовательно в охла-
ждаемый сборник карбонила, ко-
лонку с твердой щелочью (для
поглощения хлора), маслоотдели-
тель и осушитель с хлористым
кальцием.
Низкий прямой выход карбо-
нила (67,5%) обусловлен умень-
шением реагирующей поверхности
тия пленками FeCl2. Не исключено также превращение части
МоС15 в МоС13, который непригоден для получения карбонила
молибдена. Остаток от синтеза должен быть возвращен на опера-
цию повторного хлорирования и карбонилирования.
Сырой карбонил молибдена содержит примеси карбонилов
железа и меди, соли свинца и цинка и др. Кристаллы карбонила
очищают, перемешивая с 5%-ным раствором щелочи (соединения
примесей гидролизуются), и перегоняя затем карбонил молибдена
с водяным паром. Для более глубокой очистки проводят повторную
обработку 5—20%-ным раствором щелочи и перегонку с паром.
Это обеспечивает получение карбонила высокой чистоты. Схема
установки для очистки показана на рис. 90.
Описанная выше технология имеет существенные недостатки.
Процесс малопроизводителен — одна операция продолжается
50—70 ч. Прямой выход низкий (67,5%), что требует возвращения
остатка на операцию хлорирования.
253
ГЛАВА XI
ПРОИЗВОДСТВО ПОРОШКООБРАЗНОГО МОЛИБДЕНА
Основной промышленный способ производства молибдена —
восстановление чистой трехокиси молибдена водородом, в резуль-
тате которого получают порошкообразный металл, превращаемый
затем в компактные заготовки методом порошковой металлургии
или плавкой.
Известны и другие способы получения молибдена; восстановле-
ние МоО3 углеродом или металлами (кальцием, магнием); терми-
ческая диссоциация дисульфида молибдена; термическая диссо-
циация или восстановление галогенидов молибдена; электролиз
расплавленных сред. Некоторые из них уже находят применение.
43. ОКИСЛЫ МОЛИБДЕНА
В результате исследований Хегга и Магнели [1—3, 11],
Глемзера и Лютца [4] и более поздних работ Кильборга [5]„
Роде и Лысановой [6] в системе молибден—кислород в настоящее
время достоверно установлено существование следующих фаз г
МоО3 (a-фаза); моноклинной Мо9О26 (|3'-фаза) [1—4]; триклин-
ной Мо9О26 (£-фаза) [5, 6]; моноклинной Мо8О23 ф-фрза) [1—4];
орторомбической Мо4Ои (у-фаза) [1—4]; моноклинной Мо4Оп
(трфаза) [5, 61; тетрагональной МоО2(80 (Ф-фаза) [5, 6] и орто-
ромбической Мо17О47 (х-фаза) [5] и МоО2 (6-фаза). Большинство
из перечисленных выше промежуточных окислов между МоО3
и МоО2 соответствует по составу гомологическому ряду MonO3n_t
(окислы Мо9О26, Мо8О23, Мо4Оп) [4, И].
Все исследователи, изучавшие систему молибден—кислород,,
получали промежуточные окислы длительным нагреванием сме-
сей МоО3 с МоО2 или МоО3 с молибденом в инертной среде или
в вакууме в запаянных ампулах в интервале температур 500—800°С.
Промежуточные окислы термически неустойчивы. При температу-
рах выше 800° С они разлагаются с образованием МоО3 и МоО2 [4 ].
254
По утверждению некоторых авторов,
МоО2—Мо существуют окислы Мо2О3 [7,
[9, 10]. В других работах, однако, это не
ния [1—4, 6, 12].
в интервале составов
8], МоО [8] и Мо3О
получило подтвержде-
Рис. 91. Температур-
ные ^области существо -
вания промежуточных
окислов в системе
МоО3—МоО 2
На рис. 91 показаны температурные области, в которых обра-
зуются различные промежуточные окислы при нагревании в ваку-
уме смесей МоО3 с Мо или МоО2 по Кильборгу [5].
Данные о структуре в условиях образования и некоторых
свойствах окислов молибдена приведены в табл. 65.
Структура окислов молибдена
В большинстве окислов молибден окружен шестью атомами
кислорода, расположенными в вершинах октаэдра. Лишь в не-
которых окислах (Мо4Оп) шестерная координация существует
вместе с четверной (тетраэдрической). Таким образом, структуру
окислов молибдена можно наглядно описать, рассматривая ее
как состоящую из различно сочлененных координационных поли-
эдров—октаэдров, сочлененных либо только ребрами, либо вер-
шинами, либо и теми и другими. При соединении только ребрами
образуется плотнейшая кубическая упаковка. В этом случае
каждый атом кислорода принадлежит одновременно шести окта-
эдрам. Следовательно, на один атом металла приходится 1/6 X
X 6 = 1 атом кислорода (пМе : по = 1 : 1).
Если октаэдры соединены только вершинами, каждый атом
кислорода принадлежит двум октаэдрам: пМе : п0 = 1 : 3.
Если октаэдры соединены и ребрами и вершинами, могут
быть получены окислы с соотношением пМе ; по = от 1:1 до
1 : 3, т. е. со средней валентностью от 2 до 6 [13].
Трехокись молибдена Л4оО3. Высшей окисел молибдена кри-
сталлизуется в ромбической решетке слоистого типа [1, 16, 25]
(рис. 92). Каждый атом молибдена окружен шестью атомами кис-
лорода, расположенными в вершинах сильно деформированного
255
256
ТАБЛИЦА 65
СТРУКТУРА. УСЛОВИЯ ОБРАЗОВАНИЯ И НЕКОТОРЫЕ СВОЙСТВА ОКИСЛОВ МОЛИБДЕНА
Окисел 4 Кристаллическая структура, период решетки Плот- ность, г/смА Поведение при нагревании Условия образования Цвет и другие свойства Удельное электро- сопротивле- ние, ом-см, при 20° С [5]
МоО3 (а-фаза) Ромбоэдрическая ре- шетка слоистого типа, а = 3,954; b — 13,825; с = = 3,69 А 4,69 Плавится при 795° С, кипит при 1155° С. Поли- морфные превра- щения отсутству- ют. Заметно суб- лимирует выше 600° С * л Белая с зеленова- тым оттенком в растворах щело- чей, аммиака, ки- слотах. В парах полимеризована: (МоО3)з и др. —
MOgO^ ф-фаза) Моноклинная ре- шетка, а — 16,80; b = 4,04; с = = 13,4 А; р = = 106°5' — Выше 785° С разла- гается на МоО3 и 7-Мо4Ои [5]. Относительно ста- билен ниже тем- пературы образо- вания 650° С г При нагревании смесей МоО3 с Мо или МоО2 при 650—780° С [5] Сине-черный с ме- таллическим бле- ском 1,2
М.О9О26 (р' -фаза) Моноклинная ре- шетка, а — 16,76; b — 4,03; с = = 14,45 А; В = = 96° Плавится при 780—790° С с раз- ложением на МоО3 и у = Мо4Оп [6] При нагревании смесей МоО3 или МоО2 при 700— 780° С без суще- ственного терми- ческого эффекта . [5, 6] Темно-синие кон- гломераты с фио- летовым оттен- ком. В отражен- ном поляризован- ном свете изменяет окраску от ярко- розовой до голу- бой 3,7
Зеликман
Окисел Кристаллическая структура, период решетки Плот- ность, г {см* Поведение при нагревании
MOgO2e (g-фаза) Триклинная решет- ка [5], а = 8,145; b = 4,843; с = = 19,66 А; а = = 95,47°; р = = 90,39°; у = = 109,97° 4,74 При 760—780° С превращается в Р# = МодО2б. При 550° С медлен- но разлагается на МоО3 и т]-Мо4О11 [5]. По данным работы [6], плавится при 773—780° С с раз- ложением на МоО3 и у-Мо4Оп
MO17O47 (х-фаза) Орторомбическая ре- шетка [5], а = = 21,61б; Ь = — 19,632; с — = 3.95U А 4,72 При 630° С медлен- но разлагается на у-Мо4Ои и р-Мо8О23 [5]
МО4О11 (у-фаза) Ромбическая решет- ка, а = 24,49; Ь= = 5,457; с= = —6,75 А 4,15 Плавится при 815— 820° С с разложе- нием на МоО3 и МоО2 [6]. При 550° С медленно превращается в 'n-Mo4On
Продолжение табл. 65
Условия образования Цвет и другие свойства Удельное электро- сопротивле- ние, 0М‘СМ, при 20° С [5]
При нагревании смесей МоО3 с МоО2 или Мо в интервале 600— 750° С [5, 6], со- провождается эн- дотермическим эф- фектом [6] Сине-черный, не- правильные че- шуйчатые крис- таллы 4,74
При длительном на- гревании смесей МоО3 с МоО2 или Мо при 560° С Черные длинные тонкие иглы, при растирании в по- рошок черно-ко- ричневые [5] 0,05
При нагревании смесей МоО3 с МоО2 или Мо при 610—800° С с эн- дотермическим эф- фектом [5, 6]. В неравновесных ус- ловиях присут- ствует в смесях, нагретых при 500° С Т емно-фиолетовый кристаллический порошок, являет- ся полупроводни- ком 0,25
258
Окисел Кристаллическая структура, период решетки Плот- ность, г 1см? Поведение при нагревании
Мо4Оц (трфаза) Моноклинная ре- шетка [5], а — = 24,54; b = = 5,439; с = = 6,701 А; р = = 94,28° 4,17 Медленно превра- щается в у-Мо4Оп при 655° С. Выше 800° С разлагает- ся на МоО3 и МоО2
МоО2 (6-фаза) Моноклинная ре- шетка; а — 5,610; b = 4,843; с = = 5,526 А; В = = 119°62' 6,34 При нагревании в токе азота до 1200° С не претер- певает изменений. Выше 1800° С раз- лагается на МоО3 и Мо. Слегка сублимирует выше 1000° С
Продолжение табл. 65
Условия образования Цвет и другие свойства Удельное электро- сопротивле- ние, ОМ’СМ, при 20° С [5]
При нагревании смесей МоО3 с МоО2 или Мо при 500—600° С [5, 6], а также при тер- мической диссо- циации МоО3 в вакууме при 550° С Фиолетовый кри- сталлический по- • рошок 0,2
При нагревании , CMeqeft МоО3 с МоО2 или Мо при 650—700° С Темно-коричневый с фиолетовым от- тенком, иногда металлическим блеском —
октаэдра. Способ сочленения октаэдров показан на рис. 92, б
[14]. Структура состоит из трехслойных пакетов октаэдров, из
которых каждый третий слой «пустой» (не заселен молибденом),
что приводит к слоистой структуре. Внутри каждого слоя окта-
эдры соединены вершинами. Октаэдры соседних слоев соединены
ребрами и, следовательно, второй слой в пакете сдвинут по отно-
шению к первому на пол-октаэдра по оси а.
Рис. 92. Структура трехокиси молибдена:
а — элементарная ячейка; б — структура трехокиси молибдена в виде
координационных многогранников — октаэдров (по Н. В. Белову)
Поскольку при сочленении октаэдров последующих двух
слоев происходят сдвиги по осям с, то первый слой второго па-
кета не совпадает с начальным слоем первого пакета. С ним сов-
падает лишь первый слой третьего пакета. Следовательно, эле-
ментарная ячейка должна по высоте вмещать два пакета (шесть
слоев октаэдров), т. е. параметр b ячейки должен быть примерно
равен 3d (где d — высота октаэдра). Два других параметра а
и с близки (см. табл. 67).
Расстояния Мо—Мо в октаэдрах, соединенных вершинами,
3,66—3,92 А, а в соединенных ребрами 3,37 Л. Расстояния Мо—О
равны 1,90—2,34 Л, расстояния О—О равны 2,75—3,75 Л.
Трехокись молибдена обладает слабой, независимой от темпе-
ратуры парамагнитной восприимчивостью. Анализируя это
свойство, обнаруженное у ряда простых соединений переходных
элементов, Дорфман [17] приходит к заключению, что у таких соеди-
нений взаимная деформация электронных оболочек столь велика,
17*
259
что орбиты соседних ионов перекрываются и образуются непо-
лярные связи.
Окислы Мо9О26 (|3') и ТИо8О23 (|3) [2]. Структуры окислов сходны.
Основа структуры — октаэдры [МоО6], которые сочленяются
вершинами и образуют двумерные блоки с бесконечной протяжен-
ностью вдоль оси Ь. В перпендикулярном направлении ширина
блоков конечна (9 октаэдров для Мо9О26 и 8 октаэдров для Мо8О23).
Крайние октаэдры обоих концов блока имеют общие ребра с соот-
ветствующими октаэдрами других блоков. Кроме того, блоки
Рис. 93. Сочленение октаэдров в структуре:
I — Мо9О2в (3')» А — ширина блока 9 октаэдров; II — Мо8О23 (3). А — ши- .
рина блока 8 октаэдров
соединяются *между собой через общие вершины других окта-
эдров. Вследствие этого в каждой элементарной ячейке содер-
жатся две характерные группы из четырех октаэдров, сочленен-
ных исключительно ребрами (рис. 93).
Атомы молибдена образуют слои, параллельные грани ас.
Эти слои гофрированные, так как атомы Мо смещены один отно-
сительно другого с выходом из плоскости слоя на ±0,36 А. Пара-
метры решетки см. в табл. 67. Ниже приведены расстояния между
атомами, А:
Мо8О29 (30 Мо8О2з (3)
Мо—Мо:
в октаэдрах, соединенных
вершинами.................• 3,66—3,74 3,74—3,82
в октаэдрах, соединенных
ребрами ....................3,23 и 3,28 3,23 и 3,25
Мо—О ...........................1,80—2,1 1,8—2,2
0—0 ........................... 2,5—3,0 2,5—2,9
Мо9О26 обладает полупроводниковыми свойствами.
( Окисел Мо4Оп (у-фаза). В отличие от других окислов, струк-
тура Мо4Оп (у) состоит из октаэдров (МоО6) в сочетании с тетра-
эдрами (МоО4). В структуре имеются блоки октаэдров (ширина
блока — шесть октаэдров), сочлененных вершинами. Блоки со-
единены между собой искаженными тетраэдрами.
В элементарной ячейке содержится 4Мо4Ои. Из общего числа
16 атомов молибдена 12 (или 3/4) окружены кислородом октаэдри-
260
Рис. 94. Кристаллическая структура:
а — рутила; б — двуокиси молибдена
чески, а 4 — тетраэдрически. Если принять, что в первых распо-
ложены шестивалентные атомы, а во вторых — четырехвалентные,
то средняя валентность молибдена в окисле окажется равной
12 У64-4 У 4
—— ——— = 5,50. Это совпадает со средней валентностью мо-
либдена в Мо4Ои. В структуре Мо4Оп имеются каналы (пустоты)
вдоль оси с [13]. Постоянные решетки см. в табл. 67. Мо4Оп
обладают полупроводниковыми свойствами. Отмечается существо-
вание узкой области гомогенности — от МоО2>75 до MoO2i70 [4].
Двуокись молибдена (8-фа-
за). Структура МоО 2 детально
изучена Магнели [19]. Она
сходна со структурой рутила,
но в отличие от рутила эле-
ментарная ячейка не тетра-
гональная, а моноклинная.
Шесть атомов кислорода рас-
полагаются в вершинах де-
формированного октаэдра,
причем только половина окта-
эдрических пустот занята
атомами молибдена. Окта-
эдры [МоО6] соединены между
собой ребрами, образуя бес-
конечные цепи. Октаэдры
соседних цепей сочленены между собой вершинами (рис. 94).
В МоО 2 из-за двух лишних валентных электронов возникают
дополнительные связи, приводящие к искажению правильной
структуры, присущей рутилу. Атомы молибдена в соседних окта-
эдрах попарно сближаются (смещаются от центров октаэдров),
образуя зигзагообразные линии. При этом смещаются также и
атомы кислорода, что ведет к искажению октаэдра. Общие ребра
октаэдров (расстояние О—О) увеличины.
Ниже приведены расстояния между атомами в решетке МоО2, А:
Мо—Мо:
в октаэдрах, соединенных верши-
нами .............................3,69—3,71
в октаэдрах, соединенных ребрами 2,51—3,11
Мо—О............,..................... 1,9—2,1
О—О .................................. 2,7—2,9
Термохимические свойства окислов молибдена
Двуокись молибдена. Термохимические свойства МоО2 иссле-
дованы недостаточно.
Блекборн с сотрудниками [20], изучавшие давление пара
окислов молибдена эффузионным методом Кнудсена, показали, что
МоО2 при температурах выше 1000° С сублимирует с одновремен-
261
ным частичным диспропорционированием на МоО3 и Мо. По их
данным, теплота диспропорционирования по реакции
ЗМоО2 2МоО3газ + Мо
АЯ = 33,6 ккал/моль МоО3.
При температуре выше 1800° С диспропорционирование МоО2 на
Мо и МоО3 протекает быстро [20].
О температуре плавления двуокиси молибдена нет сведений.
По всей вероятности, МоО2 разлагается до плавления.
Опубликованные данные о теплоте образования МоО2 сильно
различаются (табл. 66). По всей вероятности, более точное зна-
ТАБЛИЦА 66
ЗНАЧЕНИЯ СТАНДАРТНОЙ ТЕПЛОТЫ ОБРАЗОВАНИЯ МоО2
ПО ДАННЫМ РАЗЛИЧНЫХ АВТОРОВ
Автор Год “ Д//298’ ккал/моль Литератур- ный источник
Микстер 1910 142,8 [21]
Шадрон . 1921 134,0 [22]
Тонасаки . . 1940 131,7 23
Штаскиевич и др 1955 140,9 25]
Мах 1957 140,8 [24]
чение Д//298 =—140,9 ккал [25, 24]. Сильно расходятся также
значения абсолютной энтропии, что обусловлено трудностями
измерения теплоемкости МоО2 из-за ее неустойчивости при нагре-
вании.
Так, по Кингу [26], $298 = 11,06 ± 0,05 э. е., по Тонасаки
S298 = 19,3 э. е. [23]. По оценке Кубашевского и Эванса S298 =
= 13,6 ± 2,0 э. е. [27].
Для изобарного потенциала образования МоО2 Томпсон [28]
по экспериментальным данным Тонасаки [23] вывел уравнение для
интервала температур 918—1096° К:
AZ = —130 884 — 9,9171g Т + 2,2 Ь 10-372 — 2,515 • Ю^’1 +
+ 67,577 кал/моль. (182)
Ричардсон и Джеффес [29] для интервала температур 298—
1200° С предложили приближенное уравнение:
AZ = — 133 600 + 43,077(точность ± 10 000 кал). (183)
Кубашевский и Эванс [27] рекомендуют для температур
289—1300° К уравнение
AZ = — 140 100 — 4,671g 7 + 55,87 (точность ± 6000 кал). (184)
262
Значения AZMoO2, рассчитанные по уравнению (184), на 6—
8 ккал больше рассчитанных по двум другим уравнениям (табл. 67).
При его выводе принято более высокое значение Д//298 ==
= —140,9 ккал!моль.
ТАБЛИЦА 67
ЗНАЧЕНИЯ ИЗОБАРНОГО ПОТЕНЦИАЛА ОБРАЗОВАНИЯ МоО2,
РАССЧИТАННЫЕ ПО УРАВНЕНИЯМ РАЗЛИЧНЫХ АВТОРОВ
Температура, °К л . — AZ, ккал/моль, по данным
[28] [29] * [27]
298 .. 120,8 126,8
673 109,3 102,0' 111,3
773 99,5 100,3 107,2
873 94,0 96,0 103,4
1073 88,2 87,3 95,0
1173 83,5 83,0 91,1
1373 76,4 74,4 83,3
Трехокись молибдена. Трехокись молибдена плавится при
795° С без разложения [30] и кипит при 1155° С [31, 32]. Теплота
плавления 12,55 ± 0,4 ккал!моль [33], теплота испарения
35,2 ккал!моль [32].
Трехокись молибдена заметно сублимирует в потоке воздуха
при 635° С, а в потоке воздуха с 3% паров воды — при 600° С
[39]. Данные об упругости пара рассмотрены на стр. 76.
Результаты различных авторов, измерявших теплоту образо-
вания МоО3, близки. Ниже приведены значения —А #298, ккал!моль:
Муз и Пар [34].............. 176,5
Нейман и др. [35] ........ 180,4
Штаскиевич и др. [25] . . . 178,0
Среднее из приведенных выше значений, равное —178,3 ккал!моль,
может быть принято как наиболее вероятное.
По данным Госгрова и Снайдера [33], зависимость молярных
теплоемкостей твердой и жидкой МоО3 от температуры подчи-
няется уравнениям:
жидкая МоО3:
Ср = — 219,7 + 0,2956Т — 7,0 • Ю"5?2 (выше 1068° К);
твердая МоО3:
Ср = — 20,07 + 5,90 • 10~3Г — 3,68 • 105Г'2.
Для Т = 2684-1068° К точность ±1%.
Для энтропии ими получено значение S298 = 18,65 кал!град,
что находится в хорошем согласии со значением S298 = 18,7 ±
± 0,3 кал!град, определенным Сельцем с сотрудниками [36].
263
Сельц и др. [36], приняв для теплоемкости МоО3 выведенное
ими уравнение (Ср = 14,1 + 12,1 • 10“3 Т) и значение АЯж =
= —176 500, получили для А//3 и AZ3 образования МоОз уравне-
ния:
ЛНт = — 174 600 — 4,ОТ + 4,9 • 10~3Т2 — 3,30 • ГО5?”1,
AZy = — 174 600 + 9,2 IT lg Т — 4,9 • 10“3Т2 — 1,65 - 105Т-1 +
+ 35,89Т. (185)
Кубашевский и Катеролл [38] приводят для изобарного
потенциала образования Мо03 уравнение
AZ® =— 178900 —4,6Т lg 7+ 75,37. (186)
Во всем интервале температур до плавления МоО3 значения,
полученные по выведенным уравнениям, базирующимся на уточ-
ненных данных о теплоемкости и теплоте образования МоО3,
близки к рассчитанным по уравнениям Сельца с сотрудниками.
Они отличаются примерно на +2 ккал, что объясняется приня-
тым Сельцем более низким значением А/7 образования МоО3.
44. ФИЗИКО-ХИМИЧЕСКИЕ УСЛОВИЯ ВОССТАНОВЛЕНИЯ
ТРЕХОКИСИ МОЛИБДЕНА ВОДОРОДОМ
Как показали исследования Хегедюша с сотрудниками [39],
при восстановлении трехокиси молибдена водородом отчетливо
выявляются две ступени восстановления, соответствующие реак-
циям:
МоО3 + Н2^±МоО2 + Н2О, (187)
МоО2 + 2Н2^±Мо + 2Н2О. (188)
Авторы установили, что на термогравиметрических кривых,
выражающих зависимость изменения массы от температуры
(«7И—Т»-кривые) наблюдается остановка, отвечающая составу МоО2
(колебания состава от МоО1>99 до МоО2>05), причем площадка больше
в случае присутствия влаги в водороде (рис. 95).
ТАБЛИЦА 68
ДАННЫЕ ТЕРМОГРАВИМЕТРИЧЕСКОГО ИЗУЧЕНИЯ
^ВОССТАНОВЛЕНИЯ МоО3 [39]
Состав газа Температура начала восстановле- ния, °C Состав продукта первой стадии Температура, °C
первой стадии полного восстановле- ния
Сухой водород .... Водород с 3% паров во- ды 390 510 МоО1>96 МоО2,05 590 610—710 670 795
264
Из табл. 68 видно, что температуры начала восстановления
МоО3 и температуры, отвечающие завершению первой стадии
восстановления и полному восстановлению в потоке сухого водо-
рода, примерно на 120 грай ниже, чем в водороде с влажностьюЗ%.
В продуктах неполного восстановления, состав которых лежит
в интервале МоО3—МоО2, рентгеновским анализом не были обна-
ружены промежуточные окислы типа МоО2,75 (Мо4Оп), МоО2,89
(Мо9О2в) и др.
Эти окислы, преимущественно Mo,tOn (у-фаза), появляются
в небольших количествах в продуктах восстановления лишь
после того, как почти вся трехокись молибдена восстановилась
Рис. 95. Кривые масса —*
температура для восста-
новления МоО3:
а — в потоке сухого водо-
рода; б — в потоке водорода
с 3% пгГров воды. Скорость
подъема температуры 150° С
в час (цифры на кривых —
температура, °C)
до двуокиси. Высказано предположение, что промежуточный
окисел у-Мо4Оп образуется в результате вторичного взаимодей-
ствия между МоО2 и МоО3:
ЗМоО3 + МоО2 = Мо4Оп.
(189)
Как показано в работе [6], эта реакция протекает быстро
с эндотермическим эффектом при температуре 610—640° С.
Между МоО2 и Мо не обнаружено каких-либо промежуточных
фаз. Так, при восстановлении до состава МоО1>8 в продукте най-
дены МоО2, Мо4Оп и а-молибден.
Роде и Лысанова [55], изучавшие термографическим, термо-
гравиметрическим и рентгенофазовым методами восстановление
трехокиси молибдена водородом, подтвердили основные выводы
работы Хегедюша с сотрудниками. Они обнаружили лишь две
стадии восстановления, отвечающие образованию МоО2 и Мо.
При температурах ниже 500° С продукты восстановления пред-
ставляют собой смеси МоО3 с МоО2, а выше 500° С — смеси МоО2
и Мо.
При составе, отвечающем МоО, продукт содержит лишь МоО2
и а-молибден [39].
Торможение процесса вблизи завершения первой ступени
восстановления МоО3 МоО2 (рис. 95), вероятно, объясняется
эндотермичностью второй стадии восстановления МоО2 до Мо.
Авторы работы [39] предлагают более сложное объяснение.
Они считают, что уже вблизи торможения возможно образование,
наряду с МоО2 и Мо4Оп, также и молибдена (аморфного или
265
Р-Mo). В этом случае торможение может быть вызвано протека-
нием превращения аморфного молибдена в кристаллический или
твердофазными реакциями между МодОп и Мо или MoqO26 и Мо
с образованием Мо02.
При дальнейшем повышении температуры Мо02 и промежуточ-
ные окислы восстанавливаются до металла, что отвечает второй
стадии восстановления:
МоО2 + 2Н2 = Мо + 2Н2О, (190)
МоО2,75Ч- 0,75Н2 = Мо02 + 0,75Н2О, (191)
МоО2 7Б + 2,75Н2 = Мо + 2,75Н2О.
(192)
Реакция, отвечающая первой стадии восстановления, экзо-
термическая, тогда как восстановление МоО2 до Мо сопровож-
дается поглощением тепла.
Точная оценка тепловых эффектов реакций первой и второй
стадий затрудняется расхождениями в значениях теплоты образо-
вания МоО2 и отсутствием данных о теплоемкости двуокиси
молибдена.
При расчете теплоты реакций (187) — первой ступени восста-
новления — по стандартным значениям теплот образования ком-
понентов получим:
* Д//298 = — 11,2 ккал
(если принять для МоО2 == —131,7 ккал), и
Д//298 = — 20,3 ккал
(если принять для МоО2 ДЯ298 = —140,8 ккал).
По оценке авторов работы [41 ] при температуре 510° С теп-
ловой эффект реакции равен 22,2 ккал, или 154 ккал на 1 кг МоО3.
Первая ступень восстановления сопровождается большой
убылью изобарного потенциала. Значения констант равновесия
столь велики (табл. 69), что реакция практически необратима
и может протекать при весьма низких (например, 0,1%) содер-
жаниях водорода в смеси Н2 + Н2О.
ТАБЛИЦА 69
ПРИБЛИЖЕННЫЕ ЗНАЧЕНИЯ ИЗОБАРНЫХ ПОТЕНЦИАЛОВ
И КОНСТАНТ РАВНОВЕСИЯ РЕАКЦИИ МоО„ + Н9 —МоО9 TR + Н9Огя_
О 1 О А < - 4 1 О & 1 do
Температура, °C AZ0, ккал/моль МоО3 Рц2
25 —21 279 3,98-10“
400 —23 730 5,0-Ю7
600 —24 805 1,7-106
800 —25 564 1,58-106
п римечание. При расчетах использованы уравнения AZ: для МоО2 (184),
для МоО3 (186), для Н2О : AZ = —58 900 + 13,1 Т.
266
Реакция (188) второй ступени восстановления эндотермиче-
ская. Для этой реакции в зависимости от принятого значения
ДЯ298 Для МоО2 стандартный тепловой эффект реакции варьирует
от +16,1 до +25,2 ккал.
При 815° С на второй стадии восста-
новления поглощается ~21,0 ккал на
1 моль МоО2, или 164,5 ккал на 1 кг
МоО2 1411.
Равновесие реакции восстановления
МоО2 водородом изучали Шадрон [40]
и Тонасаки [23], используя статиче-
ский метод.
Ниже приведены значения констант
Р Т_Т 0
равновесия Кр = р 2 -реакции восста-
новления МоО 2 водородом по Шад-
рону [401:
Температура
°C ... . 700 760 850 950 985 1040 1100
К„ .... 0,37 0,45 0,56 0,72 0,86 0,97 1,12
Значения, полученные Тонасаки,
существенно ниже, и вероятно, точней
(табл. 70). На рис. 96 приведена зави-
симость равновесных парциальных
давлений паров воды для рассматривае-
мой реакции по данным Тонасаки. Пунктиром показаны экстра-
полированные значения, полученные из линейной зависимости
1g [42].
ТАБЛИЦА 70
Рис. 96. Равновесное давле-
ние паров воды для реакции
восстановления МоО2 водо-
родом. Суммарное давление
1 ат. Сплошная линия —
данные Тонасаки, пунктир-
ная — экстраполированные
значения
КОНСТАНТЫ РАВНОВЕСИЯ И РАВНОВЕСНЫЕ ПАРЦИАЛЬНЫЕ
ДАВЛЕНИЯ ПАРОВ ВОДЫ ДЛЯ РЕАКЦИИ ВОССТАНОВЛЕНИЯ
МоО2 ВОДОРОДОМ, ПО ДАННЫМ ТОНАСАКИ [23]
(ниже 645 и выше 823° С значения экстраполированы по зависимости 1g К-],
общее давление 1 ат)
Температура, °C р РН2 Парциальное давление паров воды, мм рт. ст. Температура, °C РНзО Р Рн2 Парциальное давление паров воды, мм рт. ст.
227 0,013 9,5 754 0,343 194,1
327 0,038 27,8 778 0,370 204,9
427 0,076 . 53,7 797 0,389 213,2
527 0,138 92,9 823 0,421 224,8
645 0,234 143,6 927 0,550 269,7
665 0,252 153,0 1027 0,646 298,3
689 0,275 163,9 1127 0,759 327,9
717 0,302 176,7 1227 0,912 362,5
740 0,327 187,7
267
Из рис. 96 следует, что восстановление МоО2 до молибдена
термодинамически возможно в интервале температур 665—900° С
в том случае, если содержание паров воды в газовой смеси не
превышает 18 и 30% соответственно.
Практически для обеспечения высокой скорости восстановле-
ния нобходимо использовать хорошо осушенный водород и вести
процесс в потоке газа, непрерывно удаляя выделяющиеся пары
воды.
45. ПРОИЗВОДСТВЕННАЯ ПРАКТИКА ВОССТАНОВЛЕНИЯ
МоО3 ВОДОРОДОМ
Печи для восстановления
Трехокись молибдена восстанавливают водородом в много-
трубных печах с непрерывным или периодическим передвижением
лодочек с восстанавливаемым материалом вдоль трубы или
в трубчатых вращающихся печах непрерывного действия. Сухой
водород подают.в печи противоточно движению материала. Про-
цесс можно проводить в одну или две стадии. Предпочтение от-
дается двухстадийному восстановлению, в особенности в том слу-
чае, когда необходимо получить тонкодисперсный порошок опре-
деленной зернистости, предназначаемый для превращения в ком-
пактный металл методом порошковой металлургии.
При одностадийном процессе получаются грубозернистые по-
рошки вследствие того, что водород, содержащий пары воды,
в этом случае находится в длительном контакте с восстанавливае-
мыми окислами молибдена. Это благоприятствует росту зерен [42].
Кроме того, при одностадийном восстановлении низкая произво-
дительность печей обусловлена большим различием в насыпной
массе трехокиси молибдена (0,4—0,5 г! см3) и молибденового
порошка (~2,5 г/сж3).
На рис. 97 показана многотрубная печь для восстановления.
Печь состоит из горизонтально расположенных в два ряда 9 или
И труб из нержавеющей стали. Диаметр труб 50—70 мм, длина
5—7 м. Трубы заключены в стальной кожух, выложенный внутри
слоями теплоизоляционных материалов (листовой асбест, диато-
митовый и шамотный кирпич, асбоцементные плиты). Непосред-
ственно под трубами и над ними расположены плиты фасонной
керамики, в п^зах которой размещены электронагреватели из
спиралей нихромовой проволоки диаметром 4,5—5 мм. Для
увеличения срока службы труб и нагревателей целесообразно
у печей второй стадии восстановления герметизировать кожух
печи и заполнять его водородом.
Печь имеет несколько температурных зон (от 3 до 5) с общей •
длиной до 4 м, что обеспечивает заданный температурный режим
восстановления. Мощность печей колеблется в зависимости от
температуры и размеров труб в пределах 30—50 кет.
268
Рис. 97. Многотрубная печь для восстановления трехокиси молибдена водородом:
/ _рабочие трубы; 2 — нагреватели; 3 — толкатели лодочек; 4 и 5 — трубы для подачи и вывода водорода; 6 — теплоизоляция
Лодочки с восстанавливаемым материалом перемещаются вдоль
трубы с помощью металлических штанг механическим толкателем.
Скорость перемещения изменяется в пределах от 5 до 30 мм/мин.
Разгрузочные концы труб снабжены холодильниками. С обеих
сторон трубы герметично закрыты пробками. Каждая труба имеет
патрубки для подвода свежего водорода (со стороны выгрузки)
и отвода отработанного водорода (со стороны загрузки).
Наряду с описанными многотрубными печами, на заводах
СССР применяют барабанные печи непрерывного действия, кон-
струкция которых разработана инж. Бабичем (рис. 98, см. [44]).
Труба печи (нержавеющая сталь, диаметр 400 мм, длина 4 м)
опирается на два опорных ролика и вращается со скоростью
примерно 2 об!мин с помощью. системы передач. Корпус печи
установлен на станине с уклоном 2—4° в сторону разгрузки.
Внутри трубы расположены диафрагмы 5 и продольные лопасти 6.
Диафрагмы служат для предотвращения скольжения материала
вдоль трубы, лопасти способствуют пересыпанию материала и луч-
шему его контакту с водородом.
Трехокись молибдена подается в печь из бункера 11, снабжен-
ного рыхлителем 12, шнековым питателем 13. Разгрузка осуще-
ствляется через бункер 14 со шнековым разгружателем 15. По-
движное уплотнение трубы (в местах ввода шнека и ввода трубы
в разгрузочной бункер) обеспечивается графитовыми кольцами.
Обогрев печи — электрический, по длине печи имеются четыре
тепловые зоны, в каждой из которых температура автоматически
регулируется.
Барабанные печи применяют лишь для первого восстановле-
ния МоО3 до МоО2, так как в этом случае максимальная темпера-
тура процесса не превышает 650° С. При более высоких темпера-
турах, необходимых при втором восстановлении (900—1000° С),
возникают трудности выбора достаточно жаропрочного и жаро-
стойкого при рабочих температурах процесса материала для
изготовления трубы барабанной печи.
В барабанной печи описанной выше конструкции также терми-
чески разлагают парамолибдат аммония для получения трехокиси
молибдена. В случае последующего восстановления МоО3 до
МоО2 в барабанной печи обе печи устанавливают рядом: в первую
печь поступает парамолибдат аммония, выгружаемую из нее
трехокись молибдена элеватором подают в бункер печи для вос-
становления.
Питание печей водородом и его регенерация. В производстве
порошков молибдена предпочитают применять более чистый
электролитический водород, который должен быть хорошо осушен
и очищен от примеси кислорода. Очистка от кислорода дости-
гается пропусканием газа через контактные печи, содержащие
слой нагретого катализатора. В качестве катализаторов исполь-
зуют медную стружку или железомедноникелевый катализатор,
270
Рис. 98. Печь с вращающейся трубой для восстановления МоО3 водородом:
1— кожух печи; 2 — огнеупорная кладка; 3 — фасонная керамика с электронагревателями; 4 — стальная труба; 5 — диафрагмы;
6 — продольные лопасти; 7 — мотор; 8 — шкив; 9 — редуктор; 10 — цепь; 11 — загрузочный бункер; 12 рыхлитель,
13 —шнековый питатель; 14 —разгрузочный бункер; 15 —разгружатель; 16 —электромотор; 17 соединительная муфта,
18 — редуктор; 19 — станина
нагретый до 600—650°С, а также палладированный асбест. В послед-
нем случае достаточен нагрев катализатора до 250—300° С. На
поверхности катализатора кислород соединяется с водородом.
Для осушки водород пропускают через колонки, наполненные
влагопоглотителями — едким натром, едким кали, хлористым
кальцием, силикагелем, фосфорным ангидридом. Равновесные
осушителями в зависимости от
давления пара над различными
Широкие
Рис. 99. Равновесное давление паров
воды над некоторыми осушителями
в зависимости от содержания в них
воды при 20° С. Пунктирные линии
указывают практически достижимую
степень осушки
содержания в них воды приве-
дены на рис. 99 [73].
Гранулированный хлори-
стый кальций может быть ис-
пользован лишь для предвари-
тельной осушки газа, так как
после пропускания потока газа
через колонку с СаС12 содер-
жание влаги обычно не ниже
200 мг!м? (не достигается равно-
весное давление паров воды над
осушителем).
Для осушки удобно исполь-
зовать силикагель (гранулиро-
ванный и высушенный гель
кремневой кислоты). Этот вла-
гопоглотитель легко регенери-
руется нагреванием при 160—
180° С. С этой целью нагрева-
тель обычно располагают не-
посредственно в осушительной
колонке с силикагелем. При
использовании силикагеля сте-
пень обезвоживания достигает
20 жа/ти3 газа при условии, если поглотитель не насыщен водой
(содержит меньше 21% воды). Низкое содержание остаточной
воды при сушке силикагелем обусловлено быстрым установле-
нием сорбционного равновесия.
Весьма активный поглотитель влаги — гранулированное едкое
кали. При его использовании достигается остаточное содержание
влаги в осушаемом газе 2—4 мг!м3. Хороший, хотя и менее
эффективный осушитель—гранулированный едкий натр. Кроме
влаги, едкие щелочи поглощают углекислоту.
Емкий и быстро действующий осушитель—концентрирован-
ная серная кислота. При пропускании газа через промыватели
с серной кислотой можно снизить содержание воды до 3 мг/м3.
Наиболее острая осушка газа достигается при использовании
фосфорного ангидрида. При достижении равновесия содержание
воды в газе может быть доведено до <10,02 жг/ж3. Однако из-за
того, что равновесие устанавливается медленно, при практиче-
272
ских скоростях газа достигается содержание влаги примерно
0,5—1 мг/м3.
Для снижения расхода водорода отработанный газ, выходя-
щий из печей (содержащий пары воды), поступает в регенерацион-
ную установку, схема которой приведена на рис. 100.
По выходе из печи 1 отработанный водород из каждой трубы
печи поступает в общий коллектор 2, откуда через водяной затвор 3
проходит в трубчатый холодильник 4, где отделяется большая
часть паров воды. Далее газ смешивают со свежим водородом
Ьодяная магистраль
Рис. 100. Схема регенерации водорода
(очищенным от кислорода), поступающим из сети, и направляют
в осушительную башню 5. Увлекаемые потоком газа частицы
влагопоглотителя улавливаются рукавным фильтром 6. Осу-
шенный водород затем поступает в компрессор 7, который
создает необходимое избыточное давление в системе (давление до
компрессора не менее 100 мм вод., ст., после него — не выше
1000 мм вод. ст.). Перед поступлением в печь для более глубокой
осушки газ пропускают через еще одну осушительную колонку 8
с силикагелем или фосфорным ангидридом. Для предотвращения
взрывов служат гасители 9 — стальные баллоны, заполненные
тонкой вольфрамовой проволокой.
Подобные регенерационные установки монтируются у каждой
печи восстановления. Содержание паров воды в поступающем
в печи водороде контролируют по точке росы. В хорошо осушен-
ном водороде содержание паров воды не должны превышать
2—3 мг/м3 [76].
Количество свежего водорода, поступающего в печи, должно
восполнять расход его на реакции восстановления и потери при
загрузке и выгрузке лодочек.
При длительно действующей циркуляционной системе в во-
дороде появляются следы сероводорода, фосфористого и мышьяко-
18 А. Н. Зеликман 273
вистого водорода. В частности, источником РН3 может быть
технический фосфорный ангидрид, если он содержит низшие
окислы фосфора. Для очистки водорода от этих примесей газ
пропускают через колонку с активированным углем [76].
Режимы восстановления
Как уже отмечалось, большей частью восстановление МоО3
водородом проводят в две, а иногда и в три стадии. Первую ста-
дию (МоО3 —> МоО2) осуществляют при постепенном подъеме
температуры вдоль трубы печи с 450 до 650° С, причем образова-
ние двуокиси молибдена в основном должно закончиться до
температуры 550° С. При быстром продвижении материала в зону
максимальной температуры вследствие значительного выделения
тепла на первой стадии восстановления возможно оплавление
частично восстановленной трехокиси с образованием непроницае-
мой для газа корочки.Оплавление,вероятно, обусловлено наличием
легкоплавкой эвтектики в системе МоО3—МоО2.
В качестве примера ниже приводится режим первого восста-
новления в трубчатой печи с длиной трубы 3 м в никелевых ло-
дочках размером 250x44x25 мм. В лодочку загружают 320 г
МоО3. Максимальная температура восстановления 550° С. Ло-
дочки продвигаются в печи со скоростью ~20 мм/мин. Каждый
час из печи выгружается 4 лодочки. В каждую трубу диаметром
51 мм поступает 0,5—0,7 м3/ч водорода. Полученная двуокись
молибдена после просева поступает на второе восстановление.
Второе восстановление проводят при максимальной темпера-
туре 850—950 или 1000—1100° С. При температурах 850—950° С
достаточно полное удаление кислорода (до содержания 0,1 —
0,2%) возможно лишь при длительной выдержке материала
в печи (медленное продвижение лодочек) и высоком расходе
хорошо осушенного водорода.
Если эти условия не соблюдаются, порошки после второго
восстановления содержат 0,4—2% кислорода (в зависимости от
принятого режима). Для снижения содержания кислорода до
0,05—0,2% в этом случае проводят третье восстановление при
1000—1100° С. Естественно, что необходимость в третьем восста-
новлении отпадает, если при втором восстановлении поддержи-
вается максимальная температура 1000—1100° С. При темпера-
турах 1000—1100° С стойкость труб из хромоникелевой стали
и нихромовых нагревателей при соприкосновении с воздухом
заметно снижается. В этом случае обязательно использование
печей с герметизированным кожухом, заполненным водородом
для защиты труб и нагревателей от окисления.
Скорость продвижки лодочек на второй стадии восстановления
в 2—2,5 раза ниже, чем на первой, а расход водорода на одну
трубу примерно в 1,5—2 раза выше.
274
Восстановленные порошки для отделения крупных фракций
просеивают на виброситах с шелковой сеткой. Для получения
однородных партий смешивают 500—1000 кг восстановленных по-
рошков в смесителе лопастного типа или барабанном смесителе
и вновь просеивают всю партию через сетку с размером ячейки
0,08—0,071 мм.
Свойства молибденовых порошков
Размеры частиц порошков. В зависимости от режимов восста-
новления получают порошки с разной крупностью частиц и на-
бором зерен. Мелкозернистые порошки, предназначенные для
производства молибденовых штабиков методом порошковой ме-
Рис. 101. Отдельные частицы и агломераты
частиц восстановленного молибденового по-
рошка. Х2000
Рис. 102. Частицы молиб-
денового порошка откаэд-
рической формы. Х2000
таллургии, получаются по описанным выше режимам двустадий-
ного восстановления. Они содержат частицы размером от 0,1
до 6 мкм. Некоторые крупные зерна представляют собой агло-
мераты частиц (рис. 101) [42]. Образование агломератов объяс-
няется частичным спеканием частиц при высоких температурах.
Отдельные частицы имеют правильную октаэдрическую форму
(рис. 102). Они свидетельствуют о росте частиц в результате пере-
носа металла (окислов) через газовую фазу.
Присутствие агломератов затрудняет точное определение гра-
нулометрического состава порошка. Большей частью используют
микроскопический и седиментационный способы определения
гранулометрических характеристик порошка.
18* 275
Статистический микроскопический анализ состоит в следую-
щем: пробу порошка смешивают с раствором терпентина в скипи-
даре и растирают шпателем для разрушения слабых агломератов.
Затем каплю смеси наносят на предметное стекло. С помощью
микроскопа, снабженного окулярной сеткой, подсчитывают число
зерен различного размера. В результате замера 200—400 частиц
вычисляют распределение (в процентах) частиц порошка по их
величине в микронах.
Другой способ подготовки
порошка к микроскопическому
%
Рис. 103. Схема микропроектора:
1 — экран; 2 — зачерненный корпус;
3 — линза; 4 — полупрозрачная призма;
5 — микроскоп; 6 — кювета с водой; 7 —
осветитель; 8— столик микроскопа с изме-
ряемым образцом
Рис. 104. Типичные кривые распре-
деления частиц молибденовых по-
рошков по их размерам:
/ — тонкодисперсный порошок, микро-
скопический метод; 2—тот же порошок,
седиментационный метод; 3 — грубо-
зернистый порошок для дуговой плавки,
седиментационный метод
измерению размеров частиц состоит в смешивании порошка
с органическим цементом типа АКР или с бакелитом. Смесь прес-
суют в брикет и выдерживают при 140° С. При этом смола поли-
меризуется и прочно сцепляется с частицами порошка. Далее
один из торцов брикета шлифуют и полируют. Под микроскопом
в отраженном свете подсчитывают размеры зерен. Поскольку
на шлифе видны различные сечения зерен, замеры не отражают
истинную величину частиц. В связи с этим вводят поправку,
рассчитывая размеры зерен по формуле [52]
D — “тг^ср — l>265dcp,
J и
(193)
где dcp — средний размер зерен данной фракции, определенный
из измерений.
Существенно упрощает подсчеты размеров зерен использова-
ние микропроекционных аппаратов. Один из выпускаемых в СССР
приборов этого типа МСАН-2 состоит из универсального микро-
276
скопа, микропроекционной головки (призма, линза), зачерненного*
конуса с матовым экраном 400x400 мм и осветителя с тепловым
фильтром (рис. 103) При подсчете используют метод сравнения
с нормированными кругами или с помощью сетки, наклеенной
на экран.
Седиментационный анализ основан на определении скорости
оседания частиц порошка, суспендированных в какой-либо жид-
кой среде. Например, для порошков молибдена могут быть исполь-
зованы этиловый спирт, ацетон, смесь воды с глицерином.
По закону Стокса, скорость оседания частиц пропорциональна,
квадрату их диаметра, что позволяет рассчитать средние размеры
частиц суспензии.
Для определения скорости оседания отбирают пробы суспен-
зии в процессе оседания (пипеточный метод), взвешивают осадок,
выпавший из суспензии (седиментационные весы) и применяют
фотоседиментацию, основанную на измерении поглощения (абсорб-
ции) или рассеяния света, являющихся функциями степени дис-
персности частиц. Приборы, используемые для этих целей, и
методы обработки результатов описаны в работах [46—49].
На рис. 104 приведены типичные кривые распределения тон-
кодисперсного и грубозернистого молибденовых порошков по
их крупности, определенные двумя методами [42].
Различие кривых распределения, полученных микроскопи-
ческим и седиментационным методами, объясняется агрегирова-
нием частиц, которые трудно полностью исключить в седимен-
тационном методе.
Порошки молибдена, получаемые на заводах СССР, характе-
ризуются следующим гранулометрическим составом (по данным
микроскопического анализа):
Диаметр зерен, мкм Содержание фракции, %
<0,8 0,8—1,7 1,7-2,5 2,5—3,4 3,4-4,2 4,2—5,1 40—60 20—30 10—20 2—10 0—5 0—3
Допускаются лишь отдельные зерна величиной 8—9 мкм.
Косвенной характеристикой гранулометрического состава по-
рошков служит насыпная масса (унас) — масса единицы объема
свободно насыпанного порошка. Это важная характеристика для
технологии получения штабиков молибдена методом порошковой
металлургии. Величина насыпной массы зависит не только от
крупности и «набора» частиц порошка, но и от их формы, а также
степени шероховатости их поверхности. Однако размер частиц
имеет определяющее значение. Как правило, чем.крупнее частицы
порошка, тем больше его насыпная масса. Насыпная масса мо-
277
либденовых порошков при свободной насыпке колеблется в пре-
делах 0,9—1,4 г!см3, а масса утряски — от 1,6 до 2,2 г!см3.
Важная характеристика порошка — его удельная поверхность,
которая в большой мере определяет поведение порошка при
прессовании и спекании заготовок.
Для определения удельной поверхности порошков широко
используют метод газопроницаемости и адсорбции газов. Метод
газопроницаемости основан на фильтрации воздуха через слой
порошка (при давлениях, близких к атмосферному, или при вы-
соких разрежениях) и измерении сопротивления (разности давле-
ний). Различные варианты метода и соответствующие приборы
описаны в работах [48.. 50]. Метод газопроницаемости позволяет
определить удельную поверхность порошков и рассчитать сред-
ний (эквивалентный) диаметр частиц (число Фишера) по формуле
d _ 60000 -1 / ~ ' П941
где ц — вязкость воздуха, из;
с — проводимость (т. е. величина, обратная сопротивлению),
см31сек на единицу давления (Г1см2)\
р — плотность материала, г/сти3;
L — длина пробы, см\
w — масса образца; г;
V — кажущийся объем уплотненной пробы, см3',
р — избыточное давление, Г 1см*}
F — разность давлений, Г! см*.
Для массового контроля порошков по удельной поверхности
широко используют приборы, основанные на измерении про-
межутка времени, необходимого для прохождения через образец
определенного объема воздуха. В СССР к приборам этого типа
относится Т-3 (прибор Товарова), схема которого приведена на
рис. 105. Пробу порошка помещают в металлическую кювету на
пористый фильтр и уплотняют до определенного объема. При
помощи резинового аспиратора создают разрежение под слоем
материала. Под действием разности уровней жидкость начинает
перетекать из закрытого колена в открытое, вызывая просачива-
ние воздуха через слой материала. При постоянстве объема по-
рошка и объема просасываемого воздуха продолжительность про-
сасывания зависит от величины удельной поверхности порошка,
определяемой по формуле
So = -^, (195)
где t — время просасывания;
р — плотность порошка;
Л — константа прибора, определяемая по порошку с извест-
ной удельной поверхностью.
278
Метод газопроницаемости позволяет измерить лишь внешнюю
поверхность частиц порошков, которая значительно меньше пол-
ной поверхности.
Удельная поверхность производственных молибденовых по-
рошков, определенная с помощью прибора Т-3, колеблется в пре-
делах 0,15—0,20 ж2/г, тогда как полная поверхность равна
~1—1,5 мЧг.
Другой, более совершенный, прибор разработан Дерягиным.
Удельная поверхность в этом приборе определяется по сопротив-
лению фильтрации разреженного воздуха — ниже 0,1 мм рт: ст.
Рис. 105. Схема прибора
Т-3 для определения
удельной поверхности по
методу проницаемости:
1 — гильза с порошком;
2 — манометр-аспиратор;
3 — кран; 4 — регулятор
разрежения; 5 — резиновая
груша-аспиратор
При таком разрежении осуществляется кнудсеновский режим
(длина свободного пробега молекул больше размера пор). В этом
случае зависимость между поверхностью и кинетическим сопро-
тивлением выражается соотношением
<19б>
где Q — поток газа, прошедшего через слой материала;
8 — коэффициент пористости;
k — численный коэффициент;
dp
— градиент давления в слое материала в направлении
потока воздуха.
Описание прибора и практика измерений детально освещены
в работе [50].
Среди адсорбционных методов определения удельной поверх-
ности порошков высокой точностью отличается метод низкотем-
пературной адсорбции азота-—метод БЭТ (по имени авторов
Брунаэра, Эммета и Теллера), позволяющий измерять истинную
удельную поверхность порошков в широком интервале значений
от 0,1 до 2000 мЧг с относительной погрешностью 2—5%.
Адсорбцию азота навеской порошка измеряют объемным ме-
тодом в высоковакуумных сорбционных установках. После дега-
279
зации системы и навески порошка при температурах 200—300° С
в высоком вакууме в систему впускают некоторый объем азота
и определяют количество его, сорбированное порошком при
температуре кипения жидкого азота (—195,8° С). В методе БЭТ
определяют количество азота, адсорбированного мономолекуляр-
ным слоем. В этих условиях площадь, занимаемая 1 см3 азота,
а = 4,4 м2. Отсюда полную поверхность порошка можно рассчи-
тать по формуле
(197)
где q — навеска порошка;
Vm — объем поглощенного азота.
В связи со сложностью аппаратуры, а также длительностью
измерений (5—6 ч) метод БЭТ ранее использовали только в иссле-
довательной практике. Однако в настоящее время разработаны
упрощенные приборы и методы расчета. Так, прибор, созданный
во ВНИИНСМ [51 ], позволяет производить 12 определений в день,
что делает возможным его применение для серийных испытаний
порошков. Прибор предназначен для измерения удельной поверх-
ности от 0,3 до 1000 мЧг с точностью ±5%.
Для контроля порошков используют также сорбционные ме-
тоды, в которых определяют относительную удельную поверх-
ность. Эти методы сравнительно простые. Так, Креймер и др. [67]
разработали способ определения удельной поверхности по коли-
честву сорбированного порошком красящего вещества из рас-
твора (например, метиленовой сини). Другой метод, разработан-
ный во ВНИИТС, состоит в определении количества сорбирован-
ных порошком паров метанола (рис. 106). Количество сорбиро-
ванного метанола (СН3ОН) определяют по разности давлений
паров метанола до и после адсорбции. При равных навесках по-
рошка количество адсорбированного метанола служит мерой
относительной удельной поверхности.
Химический состав порошков. Исходные материалы для полу-
чения молибденовых порошков — трехокись молибдена или пара-
молибдат аммония — имеют высокую степень чистоты, что опре-
деляет низкое содержание примесей в получаемых порошках.
Типичное содержание примесей в молибденовых порошках харак-
теризуется следующими данными спектрального анализа, %:
0,001 К; 0,001 Na; 0,001 W; 0,001 Al; 0,001 Са; 0,004 Fe; 0,001 Ni;
0,002 Cr; 0,001 Si; 0,001 Cu; 0,001 Mg. Содержание углерода
колеблется от 0,01 до 0,001%, азота от 0,002 до 0,003% [42].
Основная примесь в порошке — кислород, содержание которого
в зависимости от режима восстановления составляет 0,05—0,2%.
Бурцева и Клячко [101], используя метод радиоактивных
изотопов, показали, что при восстановлении водородом парамо-
280
либдата аммония примеси калия и натрия остаются в молибдено-
вом порошке при температуре второго восстановления 1000° С.
Мышьяк начинает удаляться при первом восстановлении, но
большая часть (80—85%) улетучивается при втором восстанов-
Рис. 106. Схема прибора
для определения относи-
тельной удельной поверх-
ности порошка по сорбции
метанола:
1 — колба; 2 — манометр
для измерения давления ме-
танола; 3 — манометр для
точного измерения давления
в Системе с помощью микро-
катетометра; 4 — пробирка
с порошком; 5— стеклянный
баллон — ресивер
лении. Очистка от фосфора протекает менее полно — в процессе
второго восстановления удаляется 30—35%.
Свежевосстановленные молибденовые порошки активно сор-
бируют кислород и влагу при хранении их на воздухе. Как видно
из рис. 107 [81, с. 53], привес порошка на воздухе за 30 мин.
Продолжительность прерывания на
воздухе, ч
Рис. 107. Привес молиб-
деновых порошков (при
кратковременном их хра-
нении на воздухе) с вели-
чиной частиц, мкм:
1 — 3,2; ?. — 2,5; 3 — 4,2
4g
достигает 0,02—0,03%, после чего продолжается более медленное
поглощение. При выдержке в течение месяца привес в среднем
составлял 0,1%, причем х/8—*/4 привеса приходится на погло-
щенную влагу. По ориентировочной оценке Гульбранзена, тол-
щина окисных оболочек в порошке молибдена со средней величи-
ной зерна 2,5 мкм, выдержанного на воздухе в течение месяца,,
равна 30—40 А [81, с. 76]. Из этого следует, что молибденовые
порошки необходимо хранить в герметичной таре в атмосфере
инертного газа.
281
46. ДРУГИЕ ВАРИАНТЫ ВОССТАНОВЛЕНИЯ ТРЕХОКИСИ
МОЛИБДЕНА ВОДОРОДОМ
В связи с увеличением масштабов производства молибдена и
развитием плавки молибдена (дуговая и электронно-лучевая
плавка) ведутся исследования, направленные на разработку более
производительных процессов восстановления трехокиси молиб-
дена водородом с получением продуктов, удобных для питания
плавильных печей. Среди них представляют интерес непрерывные
процессы восстановления трехокиси молибдена водородом в кипя-
щем слое и в движущемся слое из брикетов (таблеток) восстанав-
ливаемых окислов.
Восстановление в кипящем слое
Мишель и Ханвей [45] показали, что при введении тонкодис-
персной трехокиси молибдена в кипящий слой частиц молибдена
при температуре 900—1000° С восстановление водородом в основ-
ном происходит на поверхности частиц, размеры которых увели-
чиваются по мере восстановления. Это обеспечивает возможность
Рис. 108. Схема установки для
восстановления трехокиси мо-
либдена водородом в кипящем
слое:
1 — кипящий слой; 2 — печь; 3 —
реактор; 4 — электрод; 5 — источ-
ник высокого напряжения; 6—цик-
лон; 7 — сборник пыли; 8 — элек-
тростатический осадитель пыли;
9—загрузочные бункера; 10— водо-
охлаждаемая труба для подачи
МоО3; 11 — шнековый питатель;
12 — подогреватель водорода; 13 —
ротаметры; 14 — разгрузочная
труба
проведения процесса в кипящем слое. Если бы восстановление
проходило в объеме, а не на поверхности частиц, образующиеся
тонкодисперсные порошки металла уносились бы из слоя пото-
ком газа.
Восстановление на поверхности частиц (а не в объеме), по мне-
нию авторов [45], объясняется тем, что внесенная в слой МоО3
расплавляется и смачивает частицы молибдена, что приводит
к ее восстановлению на поверхности частиц слоя. Исходя из этого,
восстановление в кипящем слое следует проводить в одну ста-
дию. Другое соображение в пользу одностадийного восстановле-
282
ния — рациональное использование теплоты, выделяющейся на
первой стадии восстановления, что снижает общее количества
тепла, которое нужно подвести в слой при предварительном нагре-
вании поступающего в слой водорода.
Схема опытной установки для восстановления в кипящем слое
показана на рис. 108. Реактор кипящего слоя имел диаметр
125 мм и высоту 1200 мм. Водород, подогретый в камере, поступал
в нижнюю коническую часть реактора. Тонкодисперсная трех- ’
окись молибдена (—0,063 мм) вдувалась в нижнюю часть реактора
потоком холодного водорода. В верхней части кипящего слоя
(на высоте 250—300 мм) расположена разгрузочная труба.
Для улавливания тонких частиц, уносимых из слоя, служили
электрофильтр и циклон. Для реактора данного размера уста-
новлены следующие оптимальные условия восстановления:
Температура в слое, °C........................955
Скорость подачи МоО3-а/ч .....................400
Расход водорода, л!ч * ...................... 2530
Линейная скорость водорода (при нормальной
температуре), см/сек .....................11,2
Температура подогрева водорода, °C .... 600
Температура в печи, °C .......................975
Степень восстановления МоО3, % ................99
* 19-кратное количество к теоретически необходимому.
Авторы работы, однако, не указывают, какая доля поступа-
ющего в слой материала остается в слое, т. е. восстанавливается
на частицах.
В табл. 71 сопоставлен гранулометрический состав исходного
порошка, загруженного в слой, и продукта восстановления.
ТАБЛИЦА 71
ГРАНУЛОМЕТРИЧЕСКИЙ СОСТАВ ПОРОШКА МОЛИБДЕНА,
ВОССТАНОВЛЕННОГО В КИПЯЩЕМ СЛОЕ
Фракция, мм Доля фракции, %
исходный порошок продукт восстановления
0,83—0,59 0 4,7
0,59—0,42 0 29,6
0,42—0,29 50 34,3
Меньше 0,29 50 31,4
Средний диаметр частиц растет линейно со временем восста-
новления. Естественно, что при длительном ведении процесса
частицы укрупнятся в такой степени, что возникнет необходи-
мость обновить ванну.
283
В качестве исходной загрузки могут быть использованы от-
сеянные мелкие фракции порошков. Кроме того, возможно измель-
чение некоторого количества крупного порошка, например в вих-
ревой или струйной мельницах, где исключено загрязнение по-
рошка измельчающими телами.
Восстановленные в кипящем слое порошки молибдена содер-
жали 0,015% О. Ниже приведено содержание примесей в металле
после плавки порошка в электронно-лучевой печи, % (по массе):
'0,004 С; 0,0005 N; <0,01 Fe; <0,001 S; <0,001 Сг; <0,01 W;
0,008 О.
Восстановление в слое гранулированных окислов
Для питания плавильных печей удобен металл в форме гра-
нул, получаемых в результате восстановления гранулированной
•трехокиси молибдена. Возможность осуществления такого про-
цесса показана в работе [41].
Восстановление проводили в шахтной печи, позволявшей осу-
ществлять процесс в периодическом или непрерывном режиме.
Диаметр шахты в нижней части 225 мм, в верхней 150 мм. Высота
шахты 2900 мм. Внутренняя часть шихты выполнена из нержаве-
ющей стали. В печь поступала гранулированная трехокись мо-
либдена с размерами гранул 3—8 мм *, которая восстанавлива-
лась в две стадии: МоО3 —> МоО2 и МоО2 —> Мо.
На первой стадии выделяется значительное количество тепла
*(154 ккал на 1 кг МоО3), что требует обеспечения его отвода с целью
избежания локального перегрева и спекания шихты. На второй
стадии необходим подвод тепла (164,5 ккал на 1 кг МоО2).
Тепловой режим на каждой стадии регулировали рециркули-
рующим потоком водорода. Система рециркуляции включала
циркуляционный насос, теплообменник, подогреватель газа и
водяной скруббер.
Оптимальная температура первой стадии восстановления 500—
555° С. Такая температура процесса обеспечивается при условии
разбавления циркулирующего газа азотом до концентрации 30—
35% Н2.
Оптимальная температура второй стадии восстановления 830—
890° С при подаче в печь чистого неразбавленного водорода.
Основные режимы и показатели восстановления в шахтной печи
приведены в табл. 72.
Потеря в массе порошка молибдена после восстановления су-
лим водородом (0,16—0,44%) характеризует в основном содер-
жание в нем кислорода. По-видимому возможно снижение его со-
держания до 0,05—0,1% при повышении температуры второго
восстановления до 950—1000° С и более острой осушке водорода.
* Технология грануляции в работе [41 ] не описана. Однако известно, что
увлажненная трехокись молибдена хорошо гранулируется на чашевых грануля-
торах. Высушенные гранулы обладают достаточной прочностью.
284
ТАБЛИЦА 72
РЕЖИМЫ И ПОКАЗАТЕЛИ ВОССТАНОВЛЕНИЯ ГРАНУЛИРОВАННОЙ
ТРЕХОКИСИ МОЛИБДЕНА В ШАХТНОЙ ПЕЧИ
Показатели I стадия МоО3 -> МоО2 II стадия МоО2 -» Мо
Скорость подачи материала (в пересчете на МоО2 или Мо), кг!ч 8,35 8,85
Температура в реакционной зоне печи, °C ... . 550—555 850—890
Состав рециркулирующего газа Н2, % Скорость рециркуляции газа, мЧмин 32 100
—1,0 —0,85
Температура нагрева газа, °C 545 1025
Потеря в массе при восстановлении навески про- дукта водородом при 1000° С, % 25,0 0,16—0,44
47. КАРБОТЕРМИЧЕСКОЕ ВОССТАНОВЛЕНИЕ КИСЛОРОДНЫХ
СОЕДИНЕНИЙ МОЛИБДЕНА
Восстановление Мо03 углеродом
Хегедюш и Нейгебауер, изучавшие механизм восстановления
трехокиси молибдена углеродом (газовой сажей), показали, что
восстановление протекает в две отчетливо выявленные стадии —
МоО3
*--г—^<503
300035
600
МоОг
700gl0
МоО3
502
305002
Мо02
600 700 803
~мо*МоО2
Мо»мо2С
9151900
100.
Рис. 109. Термогравиметрическая
кривая восстановления МоО3 сажей
в атмосфере аргона:
а — в атмосфере аргона; б — в атмо-
сфере СО2 (цифры на кривых — тем-
пература, °C)
Рис. ПО. Дифференциальная
кривая нагревания смеси МоО3
с сажей в атмосфере азота (экзо-
термический эффект при 610° С—
первая стадия восстановления;
эндотермический эффект при
910°—вторая стадия)
восстановление МоО3 до МоО2, а затем МоО2 до молибдена. Это хо-
рошо иллюстрируется термогравиметрической кривой (рис. 109),
снятой при скорости подъема температуры 2,5 град/мин, а также
дифференциальной кривой нагревания (рис. ПО) [106].
Первая стадия
МоО3 + V2 СО - МоО2 + х/2 СО2
(198)
285
протекает в интервале температур 420—630° С. Реакция экзотер-
мическая, ДЯ % —9,6 ккал!моль МоО2.
Елютин и Павлов [107], определявшие температуру начала
восстановления трехокиси молибдена углеродом методом газовой
хроматографии, наблюдали начало восстановления при 415° С,
что хорошо согласуется с данными Хегедюша. Промежуточные
окислы между МоО3 и МоО2 появляются лишь при медленном
восстановлении МоО3 при низких температурах (~420—500° С).
Вторая стадия протекает в интервале
~1DD Г 800—900° С по эндотермическим реакциям:
МоО2 + С — Мо + СО2;
ДЯ % + 46,8 ккал,
МоО2 + 2С -> Мо + 2СО;
ДЯ = 4-88,1 ккал.
(199)
(200}
да /да J500 2000 Для последней реакции зависимость AZ
Температура,°К от температуры описывается уравнением
Рис. 111. Зависимость
изобарно-изотермического
потенциала от темпера-
туры:
1 — реакция МоО2 + 2С"<^-
Мо + 2СО; 2 — реакция
СаМо<Э4 —{- ЗС < Мо -j- СаО+
+ ЗСО
= 4-86 700 4-4,674g Т —97,77. (201}
Подсчет по этому уравнению дает следу-
ющие значения AZ реакции (рис. 111):
Температура, ° К 300 1000 1500
AZ, ккал ...» 60,2 -|-2,8 —13,5
Значение AZ <С 0 при температурах выше 780° С. Это согла-
суется с реально наблюдаемой температурой начала реакции вос-
становления МоО2 до Мо примерно при 800° С (см. рис. 109).
Авторы работы [106] указывают, что карбид Мо2С образуется
лишь после полного удаления кислорода. При соответствующем
составе шихты и режиме восстановления можно получить молибде-
новый порошок с небольшим содержанием примеси углерода.
В работах [106, 107] показано, что в процессе восстановления
происходит сублимация окислов молибдена и перенос их к по-
верхности восстановителя. Сублимация окислов ускоряется в при-
сутствии восстановителя, даже если нет прямого контакта его
с окислом [107]. Высказано предположение [109], что возгоняе-
мая трехокись молибдена находится в парах в активированном
(электронно-возбужденном) состоянии. Первую стадию восста-
новления можно представить следующей схемой:
1. Сублимация трехокиси молибдена с образованием парооб-
разных частиц, находящихся в активном состоянии.
2. Перенос активных частиц МоО3 к восстановителю и адсорб-
ция их на его поверхности.
286
3. Химическое взаимодействие окисла и углерода с образова-
нием низшего окисла молибдена и десорбция СО2 с поверхности
восстановителя.
Вторая стадия — восстановление МоО2 до Мо, протекающая
в интервале температур 800—900° С, может быть описана реак-
циями:
моо2тв^моо2газ>
МоО2газ + С^Мо + СО2>
СО2 4- с 7^ 2СО.
По мнению авторов работы [107], прямое взаимодействие
МоО2тв + С не играет существенной роли.
Рис. 112. Схема угольно-трубчатой печи сопротивления:
1 — кожух; 2 — сажевая теплоизолирующая засыпка; 3 — люк для загрузки сажи;
4 — подводящие электрический ток охлаждаемые головки; 5 — холодильник; 6 — кон-
тактные конусы; 7 — шины, подводящие ток; 8 — экранирующая угольная труба; 9 —
угольная или графитовая труба накала .
Трехокись молибдена может восстанавливаться углеродом
в угольно-трубчатых печах сопротивления так же, как при полу-
чении вольфрамовых порошков (рис. 112) [108].
В качестве восстановителя используют ламповую сажу, содер-
жащую не более 0,1% золы. В шихту вводят теоретическое коли-
чество углерода. Смесь набйвают в угольные патроны, которые
продвигаются вдоль трубы печи, проходя горячую зону печи
(с температурой 1200—1300° С) за 30—40 мин. Из печи непрерывно
выделяется окись углерода, которая поджигается на выходе
из печи. В результате восстановления получают рыхлые блоки
темно-серого цвета.
Как уже отмечалось, карбид молибдена образуется лишь после
удаления кислорода. Регулируя время восстановления, можно
получить порошки с содержанием не более 0,5% С и 0,2—
0,5% О.
В настоящее время в производственной практике трехокись
молибдена восстанавливают не углеродом, а водородом, что обес-
287
печивает получение более чистых порошков, не содержащих угле-
рода. Процесс может быть использован для получения техниче-
ских молибденовых порошков.
Восстановление молибдата кальция
В работах [109, 110] исследована термодинамика и кинетика
восстановления молибдата кальция ламповой сажей.
Из рис. 111 следует, что восстановление СаМоО4 углеродом
по реакции
СаМоО4 + ЗС
77 Мо + СаО + ЗСО
(202)
термодинамически возможно при температурах выше 830° С.
С достаточной скоростью реакция протекает при температуре
1000° С.
Состав твердой (разы
Рис. 113. Зависимость констант равновесия восста-
новления СаМоО4 окисью углерода от состава твер-
дых фаз при различных температурах
Восстановление идет в две стадии с промежуточным образова-
нием СаМоО3 [110], что отчетливо видно на рис. 113:
СаМоО4 + С СаМоОз + СО, (203)
CaMoOg + 2С Мо + СаО + 2СО.
(204)
В работе [109] изучали кинетику восстановления брикетиро-
ванной смеси СаМоО4 с ламповой сажей. Шихту готовили с 5%-ным
избытком восстановления.
Как видно из рис. 114, при 1000° С реакция заканчивается
за 80 мин, при 1200° С — за 25 мин.
До степеней превращения 0,6—0,7 реакция протекает
с постоянной скоростью, затем скорость монотонно убывает
(рис. 115).
288
Кинетические кривые восстановления СаМоО4 в брикетах хо-
рошо описываются уравнением
) — (1—а)’/з] =/Ст, (205)
где а — степень восстановления в долях единицы;
т — время;
К — константа скорости реакции.
До 1000° С восстановление протекает равномерно во всем
объеме брикета, выше 1000° С наблюдается фронтальное восста-
Рис. 115. Зависимость ско-
рости восстановления СаМоО4
углеродом от степени восста-
новления при различных
температурах, °C:
/—900; 2 — 950; <5 — 1000; 4 — 1050;
5 — 1080; 6 — 1110; 7 — 1210
ние. 114. Зависимость степени восста-
новления СаМоО4 углеродом от вре-
мени при различных температурах, °C:
J — 900; 2 — 950; 3 — 1000; 4 — 1050;
5 — 1080; 6 — 1100; 7 — 1210
новление брикета от периферии к центру. Энергия активации
реакции восстановления при 900—1000° С £ = 62,0 ккал, тогда
как при 1000—1200° С Е = 25 ккал. Следовательно, в этих обла-
стях температур различны лимитирующие стадии процесса.
В низкотемпературной области, по всей вероятности, лимити-
рующей стадией реакции является десорбция окиси углерода
с поверхности угля. Для процесса десорбции СО, по литера-
турным данным, кажущаяся энергия активации равна 43—
68 ккал!моль [111]. Поскольку при 900—1000° С скорость восста-
новления определяется десорбцией окиси углерода, а общая ско-
рость процесса мала, создаются благоприятные условия для
одновременного протекания реакции во всем объеме брикета.
В высокотемпературной области лимитирует процесс первая
стадия газификации угля:
СОз + СтГСО + СО^с.
а 1 - 1 d ДС
!1/- - 1
Для этой реакции энергия активации Е 254-27 ккал!моль.
19 а. н. Зеликман 289
Известно, что с повышением концентрации СО снижается ско-
рость взаимодействия СО2 с углеродом. Можно полагать, что
во внутренних слоях брикета давление СО повышенное. Это ведет
к торможению реакции газификации СО2, лимитирующей скорость
восстановления СаМоО4, и вызывает фронтальное восстановление
брикета.
Добавка в шихту ~1% СаС12 сильно ускоряет восстановле-
ние СаМоО4, особенно при низких температурах. Влияние СаС12,
вероятно, объясняется частичным его испарением при темпера-
туре восстановления и взаимодействием с углем с образованием
карбида кальция и хлора, сорбирующегося на поверхности угля.
Это способствует десорбции с угля СОадс и ускорению процесса
восстановления.
Восстановление молибдата кальция углеродом (в качестве ко-
торого может быть использована ламповая сажа или измельчен-
ный нефтяной кокс) может проводиться, как описано выше,
в угольно-трубчатых печах при температуре 1200—1300° С. Воз-
можно восстановление в графитовых или шамотных тиглях, куда
шихту плотно набивают или загружают в форме брикетов, по-
добно тому как это описано для восстановления CaWO4 [108].
Тигель плотно закрывают крышкой (неплотности замазываются
шамотом). Оставляют лишь отверстие для выделения СО. Тигель
помещают в газовый горн или электропечь с температурой 1200—
1300° С. После кратковременного мокрого измельчения продукта
восстановления основная масса окиси кальция может быть отде-
лена в гидроциклоне. Оставшуюся часть окиси кальция отделяют
при обработке разбавленной соляной кислотой. После промывки
водой и сушки получается технический молибденовый порошок,
в котором основными примесями являются углерод и кислород.
48. ВОССТАНОВЛЕНИЕ ТРЕХОКИСИ МОЛИБДЕНА
КАЛЬЦИЕМ И МАГНИЕМ
В 1960—1962 гг. появились сообщения, в которых описываются
результаты восстановления трехокиси молибдена или ее смеси
с двуокисью молибдена кальцием или магнием. Применение столь
сильных восстановителей как кальций и магний, для восстанов-
ления сравнительно малопрочных окислов молибдена обусловлено,
по всей вероятности, поисками методов выплавки слитков ле-
гированного молибдена (с легирующими присадками Ti, Zr, V,
Cr, Al и др.). Получение слитков легированного молибдена с
равномерным распределением легирующих элементов встречает
значительные трудности, вызывает необходимость повторных пла-
вок.
Между тем при восстановлении МоО3 в присутствии окислов
легирующих элементов прочные окислы титана, циркония, вана-
дия, хрома и др. будут восстановлены кальцием или магнием и
290
перейдут в сплав, который может быть затем подвергнут пере-
плавке в дуговой или электронно-лучевой печах.
Из данных табл. 73 видно, что тепловые эффекты реакций вос-
становления окислов молибдена кальцием и магнием весьма
высокие.
ТАБЛИЦА 73
СТАНДАРТНЫЕ ТЕПЛОВЫЕ ЭФФЕКТЫ ВОССТАНОВЛЕНИЯ МоО3 И
МоО2 КАЛЬЦИЕМ И МАГНИЕМ
Реакция Тепловой эффект, ккал/моль Удельный тепловой эффект, ккал/кг Теоретиче- ская температура,
МоО3- ЬЗСа = МоЧ -ЗСаО —277,4 —1050 —7500
МоО2“ -2Са = МоЧ -2СаО —163,0 —782 —5600
МоО3- h3Mg = Мо- 43MgO —252,8 —1165 —8300
МоО2- |-2Mg = Мо- j-2MgO —146,5 —830 —5100
В табл. 73 приведены теоретически возможные температуры
для стехиометрических смесей компонентов, рассчитанные в пред-
положении отсутствия тепловых потерь. Реальные температуры
должны быть порядка 3000° С, что достаточно для выплавки слит-
ков молибдена.
При столь высоких тепловых эффектах металлотермические
реакции протекают с большой скоростью, часто с выбросами.
Поэтому в шихту вводят инертные добавки, например окись
кальция.
Процесс проводят в герметичном стальном сосуде («бомбе») г
футерованном огнеупором (СаО и др.)- Поскольку при быстром
распространении экзотермической реакции по массе шихты раз-
виваются высокие давления, бомбы изготовляют из цельнотяну-
тых труб небольшого диаметра (150—200 мм) длиной 900—1100 мму
к которым приваривают дно и массивный фланец для герметич-
ного присоединения крышки. Бомбу можно футеровать окисью
магния или смесью СаО и MgO, получаемой из электроплавленого
доломита.
В лабораториях фирмы Ва-Чанг (США) проводили опыты вос-
становления трехокиси молибдена в смеси с кальциевой стружкой
в бомбе, рассчитанной на загрузку шихты, содержащей 22,7 кг Мо.
Были получены слитки с суммарным содержанием примесей 0,05%
(в основном азот и кислород) и твердостью 140—150 НВ [112].
Описано получение пластичного молибдена восстановлением МоОа
магнием "В бомбе по методу Кемпбелла й др. [113].
При восстановлении МоО3 магнием развиваются высокие дав-
ления — выше 340 ат. Поэтому рекомендуется восстанавливать
смеси МоО3 + МоО 2 (от 10 до 30% МоО2) и вводить в шихту
19* 291
инертную добавку — окись кальция. Это сильно снижает темпе-
ратуру и соответственно давление.
Улучшают качество металла небольшие количества углерода
в шихте, который служит раскислителем при восстановлении и
дуговой переплавке металла. Выплавленные в бомбе слитки были
сварены в электроды для дуговой плавки. После дуговой пере-
плавки получен металл с содержанием 99,2—99,8% Мо. В ряде
полученных слитков содержание кислорода было меньше
5-10~4%.
Испытания показали хорошую обрабатываемость молибдена,
в частности возможно обжатие на 50%.
49. ПОЛУЧЕНИЕ МОЛИБДЕНА ИЗ МОЛИБДЕНИТА
Разработаны два технологических варианта получения молиб-
дена непосредственно из молибденита — способ термической дис-
социации в вакууме и восстановление молибденита оловом.
Интерес к разработке способов прямого получения молибдена
из молибденита возник в связи с тем, что обогатительные фабрики
освоили в промышленных масштабах производство чистого молиб-
денита (99,5% MoS2) для использования в качестве твердого сма-
зывающего материала.
Как исходный материал для получения металла молибденит
имеет следующие потенциальные преимущества:
1. Поскольку исходный материал почти не содержит кисло-
рода, может быть получен молибден с низким содержанием кисло-
рода, а также (при соблюдении определенных условий) с низким
содержанием азота.
2. Металл получается по короткой схеме непосредственно
из минерала, исключаются операции обычной переработки молиб-
денита с целью получения чистой трехокиси молибдена.
Способ термической диссоциации
Известно, что дисульфид молибдена термически разлагается
на молибден й серу при нагревании в вакууме при температурах
выше 1370° С. Диссоциация протекает в две стадии:
4MoS2->2Mo2S3 + S2ra3, (206)
2/3Mo2S3 —>4/3Мо + S2ra3. (207)
Для реакции (207), по Стаблесу и Ричардсону (см. стр. 227),
AZ = 85 700 — 36,4 IT.
Расчет по этому уравнению дает следующие равновесные дав-
ления паров серы для реакции диссоциации Mo2S3:
Температура, °C 1400 1600 1800
ц$2, мм Рт- ст- 0,44 6,95 61,0
292
Из этих данных следует, что при внешнем давлении ниже
0,1 мм рт. ст. уже при 1400° С можно достигнуть полного разло-
жения Mo2S3.
Практически диссоциация протекает с достаточной скоростью
при 1650° С в условиях непрерывной откачки паров серы. Техно-
логия получения молибдена термической диссоциацией изучена
и проверена на полупромышленной установке в США исследова-
телями фирмы Клеймакс [56].
Рис. 116. Схема установки для получения молибдена термической диссоциацией
молибденита:
1 — печь с молибденовым нагревателем; 2 — конденсатор серы; 3 — охлаждаемая ло-
вушка; 4 — диффузионный насос; 5 — бустерный насос; 6 — механический вакуумный
насос; 7 — герметичная камера; 8 — дробилка; 9 — подъемник печи; 10 — мельница;
11 — система очистки аргона
На рис. 116 приведена схема полупромышленной установки,
рассчитанной на единовременную загрузку 72,5 кг чистого молиб-
денита, что отвечает 45,5 кг Мо.
Чистый молибденит в форме брикетов загружали в контейнер,
изготовленный из перфорированного молибденового листа. Кон-
тейнер помещали в печь с нагревателем из молибденовой ленты.
Печь монтирована в герметичной камере, которую до включения
нагревателя печи эвакуируют и заполняют чистым аргоном.
Атмосфера агрона, окружающая вакуумную печь в процессе
диссоциации, исключает подсосы воздуха. После окончания про-
цесса охлажденный продукт извлекают из печи в атмосферу чи-
стого аргона. Печь присоединена к конденсатору серы и вакуум-
ной системе. Конденсатор разделен перегородками на четыре
зоны, в которых последовательно охлаждаются и конденсируются
пары серы. Для конденсации тонкодисперсных частиц (серный
дым) им сообщали заряд, для чего в камеры конденсатора поме-
щены электроды, к которым подведено напряжение 12 000 в.
293
Между конденсатором и вакуумными насосами находится ло-
вушка, охлаждаемая смесью сухого льда со спиртом или жидким
азотом.
Вакуум создается системой, состоящей из бустерного масля-
ного насоса, диффузионного насоса и механического насоса пред-
варительного вакуума.
Термическую диссоциацию молибденита проводили при 1650° С
при давлении от 1 до 5 мкм. Полученные спекшиеся брикеты мо-
либдена дробили и измельчали в мельнице до крупности частиц
порошка 1,65 мм. Порошки молибдена содержали не более 0,02% S
и 0,004—0,008% С. Из порошков были изготовлены расходуемые
электроды, которые плавили в дуговой печи.
Содержание металлических примесей в слитках после дуговой
плавки молибдена, полученного диссоциацией MoS2, и обычного
производственного порошка близки (табл. 74). В первых выше
содержание алюминия (в печи использовали алундовый огнеупор)
и кобальта.
ТАБЛИЦА 74
СПЕКТРАЛЬНЫЙ АНАЛИЗ СЛИТКОВ МОЛИБДЕНА
Элемент Обычный производственный молибден, %-IO”4 Молибден, - полученный диссоциацией MoS2, %«10~4
Mg 6 7
Al 1 10
Si 32 32
Ti 68 45
V 1 2
Cr 6 8
Fe 63 65
Co 1 10
Ni 1 2
Cu 1 2
Ag 0,6 2
Pb 6 7
Фрактографическое изучение изломов слитков из порошков,
полученных термической диссоциацией, показало отсутствие про-
слоек окислов на границах зерен, причем раскисляющую добавку
углерода не добавляли. Из этого следует, что содержание кисло-
рода в порошке, полученном диссоциацией, ниже, чем в обычных
производственных молибденовых порошках.
Содержание примесей внедрения (О, N, Н) в слитках, опреде-
ленное методом вакуумной плавки, колебалось в следующих пре-
делах: кислород — от 6-10"4 до 5-10“3% (в зависимости от со-
держания углерода в порошке); водород — от <2-10“б до 7х
X 10"б%; азот — от <3* 10“4 до 4-10“4%. Содержание О, N и Н
того же порядка, что в слитках из обычного порошка молибдена.
294
Авторы работы [56] считают, что более низкое содержание
примесей внедрения, особенно кислорода, которое можно было
ожидать, не было реализовано по той причине, что не достигалось
достаточно низкое давление в печи на последних стадиях выдержки
продукта. Отмечается, что основные трудности при конструирова-
нии промышленных установок связаны с необходимостью конден-
сации больших количеств серы, а также с подбором огнеупорных
материалов для печи, устойчивых против действия паров серы.
Восстановление молибденита оловом
Процесс предложен Нахтманом и Пулом [57], которые пока-
зали, что молибденит можно восстановить оловом в потоке водо-
рода при 1250—1300° С. Пары образующегося SnS удаляются
с потоком газа (точка кипения SnS 1200° С):
MoS2 + 2Sn — Мо + 2SnS | (газ). (208)
Поток очищенного водорода нагревается, проходя через мо-
либденовые нагреватели сопротивления, и поступает в керамиче-
скую трубу — реторту, где помещаются брикеты шихты (MoS2 +
+ Sn). Водород — эффективный теплоноситель. Он сообщает бри-
кетам необходимое тепло для протекания реакции и испарения
сульфида олова. Газовый поток уносит пары SnS, который кон-
денсируется в холодной части реторты. Водород проходит систему
очистки и возвращается в реторту.
Слитки из молибдена, восстановленного оловом, по чистоте
сопоставимы со слитками, выплавленными из металла, получен-
ного термической диссоциацией молибденита [58].
Преимущества способа по сравнению с термической, диссоциа-
цией — более низкая температура процесса и более простая аппа-
ратура. Недостаток — необходимость использования олова и его
регенерации из сульфида олова.
50. ПОЛУЧЕНИЕ МОЛИБДЕНА ИЗ КАРБОНИЛА
Выше были описаны свойства и способы синтеза карбонила
молибдена (см. стр. 248). Поскольку карбонил молибдена терми-
чески разлагается при температурах выше 150° С на металл и
окись углерода, многие исследователи изучали возможность ис-
пользования карбонила для получения порошка молибдена или
молибденовых покрытий.
Условия разложения карбонила с целью получения молибде-
нового порошка или покрытий исследовали Хард с сотрудни-
ками [59], Джермер и Ландер [60], Белозерский и Кричев-
ская [61], Розен [62, 63] и др.
По данным работы [61], разложение карбонила по реакции
Мо (СО)6-> Мо + 6СО
295
протекает с достаточной скоростью при температурах 300—400° С
с выделением тонкодисперсного молибденового порошка. Однако
получаемый порошок содержит примерно 2,5% С и 4—5% О.
Это объясняется разложением окиси углерода с выделением сажи
по реакции
2СО^СО2 + С,
а также вторичными реакциями взаимодействия активного по-
рошка молибдена с СО2 и СО с образованием окислов и карбидов
молибдена, например по реакции
3 Мо СО2 = Мо2С + МоО2.
Розен [62] изучал зависимость скорости разложения карбо-
нила от температуры, пропуская смесь водорода с карбонилом
(при массовом потоке карбонила от 0,6-10~2 до 1,9-10"2 молъ/ч)
через нагретую стальную трубу. Автор отмечает, что на поверх-
ности разложение начинается при 150—160° С, тогда как реак-
ция гомогенного разложения, требующая более высоких энергий
активации, наблюдается при более высокой температуре.
Максимальная скорость осаждения молибдена на поверхности
трубки наблюдается при температурах 720—750° С, независимо
от давления водорода и массового потока карбонила. В металли-
ческом порошке содержалось 3,6—3,8% С.
Если термическое разложение карбонила проводят при пони-
женных давлениях (0,1—0,01 мм рт. ст.) (что допустимо лишь
в случае получения покрытий), то содержание углерода в осажден-
ном металле понижается с повышением температуры от —4%
при 250° С до —0,1% при 650—750° С [62].
Однако работа при низких давлениях неприемлема, когда тре-
буется получить порошок молибдена и необходимо обеспечить вы-
сокую производительность процесса. При нормальном давлении
и использовании в качестве газа-носителя СО, СО2 или водорода,
содержание углерода в порошке сохраняется примерно на уровне
2—2,5% в интервале температур 400—1000° С (табл. 75). Осу-
ществляя разложение в слабоокислительной среде (—2% О2),
можно снизить содержание углерода до нескольких десятых долей
процента, при этом неизбежно повышение содержания кислорода
в порошке [61]. Поскольку содержание углерода и кислорода
порядка 2—3% недопустимо в порошках молибдена, предназна-
ченных для превращения в компактные заготовки методом порош-
ковой металлургии или плавки, необходима очистка карбониль-
ного порошка молибдена, что сильно усложняет технологию.
Для очистки от углерода авторы работы [61 ] рекомендуют
обработку порошка в потоке влажного водорода при 800—1200° С.
Как видно из табл. 76, достаточно полно углерод удаляется в ре-
зультате длительной обработки порошка (—48 ч) при 1100° С.
296
ТАБЛИЦА 75
СОДЕРЖАНИЕ УГЛЕРОДА И КИСЛОРОДА В ПОРОШКЕ МОЛИБДЕНА,
ПОЛУЧЕННОМ РАЗЛОЖЕНИЕМ КАРБОНИЛА С РАЗНЫМИ
ГАЗАМИ-НОСИТЕЛЯМИ КАРБОНИЛА [б 1 ]
Температура, °C Скорость газового потока, л/мин Содержание в металле, %
С О
Окись углерода
400 0,38 2,24 —
600 0,52 2,37 4,97
800 0,52 2,43 4,63
1000 0,50 2,53 4,24
1200 0,56 2,64
Двуокись углерода
400 1 0 2,12
600 0,95 2,23 —
800 0,95 2,23 ' ——"*
1200 0,95 0,063 —
Водород
400 0,50 2,43 —
800 0,50 3,01 2,42
1000 0,52 2,76 3,89
1200 0,54 2,76 1,06
Азот -j-2,2% О2
400 1,3 0,27 —
800 1,2 0,24 -
1000 1,2 0,10 —
1200 1,8 0,12 1
ТАБЛИЦА 76
ОБЕЗУГЛЕРОЖИВАНИЕ КАРБОНИЛЬНОГО МОЛИБДЕНОВОГО
ПОРОШКА В СМЕСИ ВОДОРОДА С ПАРАМИ ВОДЫ [611
Температура, °C Время обработки, ч Содержание углерода, %
до обработки после обработки
400 3 0,108 0,0
950 3 3,08 2,18
1000 2 3,52 1,07
1100 24 0,35 0,05
1100 48 0,35 0,003
1200 8 0,062 0,021
297
Одновременно с этим возрастает содержание кислорода, который
рекомендуется удалять восстановлением порошка сухим водородом
при 1100° С, как это делается в производственном способе получе-
ния молибдена.
Сульфит - целлюлозная барда
Молибденитовый концентрат
или полупродукт
игольный порошок
Смешение, брикетирование
Хлор
Сушка, кальцинация (600 С;2Ьч)
Хлорирование
I конденсация (220°С) Остаток
И конденсация (120°С)
Ш конденсация (10 °C)
Cl
Улавливание
хлора раствором
щелочи в отвал
Технический
МоС15
Азот
Продукт,
содержащий
Cu,Pb,Zn и др.
Окись углерода
Смешение,
брикетирование
Карбонилирование (280ат; 72ч)
Тё(С0)5
Мо(СО)6
Сырой Мо(С0)6
Ректификация,
термическое
разложение
Пар
Окись Карбонильное
углерода железо
Остаток
Раствор I
NaOH 1
Трехстодийная
очистка от примесей
металлов
Отсев железа
Остаток Железная
от стружка
У истый Мо(С0)6
Термическое разложение
Оборотная Очистка молибденового порошка
окись углерода от углерода и кислорода
Уистый карбонильный
молибденовый порошок
Рис. 117. Схема получения молибденового порошка карбонильным способом
208
Из сказанного следует, что разложением карбонила молибдена
вряд ли могут быть получены порошки молибдена более чистые,
чем порошки, производимые восстановлением чистой трехокиси
молибдена водородом.
Между тем в целом технологическая схема карбонильного спо-
соба, как видно из рис. 117, весьма сложна и многостадийна [104].
Основные звенья схемы:
1. Подготовка молибденита к хлорированию, хлорирование и
стадийная конденсация хлоридов.
2. Брикетирование пентахлорида молибдена с железной струж-
кой и процесс карбонилирования при 200° С и 280 ат СО (см.
стр. 252).
3. Очистка карбонила от примесей двойной перегонкой.
4. Термическое разложение карбонила с получением молибде-
нового порошка.
5. Очистка порошка от углерода и кислорода.
Операция карбонилирования требует длительной выдержки
(50—60 ч), а прямой выход в карбонил составляет 60^—65%.
Весьма продолжительна операция обезуглероживания.
Усложняющее обстоятельство — применение в карбонильной
технологии четырех газов — хлора, азота, окиси углерода и во-
дорода. Вследствие этого карбонильный метод может найти лишь
ограниченное применение для производства порошка молибдена.
51. ПОЛУЧЕНИЕ МОЛИБДЕНА ИЗ ГАЛОГЕНИДОВ
Высшие галогениды молибдена МоС15 и MoF6 — летучие соеди-
нения, легко очищаются перегонкой и могут служить исходными
соединениями для получения молибденовых порошков высокой
чистоты и молибденовых покрытий осаждением из газовой фазы
[74, 78, 85]. Для этих целей необходимы галогениды высокой чи-
стоты, не содержащие примеси кислорода в форме оксигалоге-
нидов.
Наибольший интерес представляет восстановление галогени-
дов водородом. Галогениды можно восстанавливать водородом
в трех вариантах — в «факеле», в кипящем слое и осаждением
на нагретых поверхностях.
Восстановление в факеле
Этот процесс описан в литературе применительно к восстанов-
лению водородом WF6 и MoF6 [64]. Гексафторид молибдена
MoF6 — белоснежное кристаллическое вещество. Фторид плавится
при 17,5° и кипит при 35° С. Плотность MoF6 в точке плавле-
ния 2,551.
Давление пара над жидким MoF6 подчиняется уравнению [65]
^^Рммрт.ст* — 'г F 7,407.
299
Приближенные значения теплоты образования газообразного
MoF6 АЯ298 % —405 ккал!моль, энтропии S298 = 79,0 кал!моль.
Гексафторид молибдена наиболее просто может быть получен
прямым действием фтора на порошкообразный молибден или от-
ходы молибдена (брак штабиков, отходы проката и др.). Реакция
фторирования сильно экзотермическая и начинается при комнат-
ной температуре. Фторирование можно проводить в аппаратах
из сплава никеля с медью (монель). Для обеспечения полной кон-
денсации необходимо охлаждать конденсаторы до минус 60—
70° С [65].
Для фторирования могут быть использованы реакторы с не-
подвижным слоем фторируемого материала и реакторы кипящего
слоя.
Для очистки из сконденсированного MoF6 отгоняют низкоки-
пящие примеси (главным образом HF). При последующем испа-
рении MoF6 для подачи его в реактор восстановления водородом
гексафторид очищается от всех менее летучих примесей.
Процесс восстановления в факеле, описанный в работе [64],
предполагает самоподдерживаемую реакцию (за счет выделяюще-
гося тепла), которая протекает между газообразными компонен-
тами, подаваемыми в сопло, установленное в реакционной
камере.
Точная оценка теплового эффекта реакции восстановления
MoF6 водородом невозможна из-за весьма ориентировочного зна-
чения теплоты образования гексафторида. Однако эксперимен-
тально установлено, что для самопроизвольного протекания реак-
ции теплоты ее недостаточно. Чтобы сообщить тепло, необходимое
для проведения реакции «в факеле», в форсунку, кроме MoF6,
подают фтор и избыточный водород, которые взаимодействуют
с образованием HF. При этом на каждый моль HF выделяется
64,2 ккал.
На рис. 118 показана схема полупромышленного реактора,
который изготовлен из двух труб диаметром 100 мм и высотой
1,2 м из сплава монель, устойчивого против действия сухого HF
до 600° С.
В первую трубу, где протекает реакция восстановления через
две концентрично установленные трубки (сопло), вводят газооб-
разный MoF6, водород и фтор. Внутренний диаметр наружной
трубки сопла равен 11 мм, а центральной трубки 6,25 мм (внеш-
ний) и 3,75 мм (внутренний). Предпочтительно подавать смесь
MoF6 + Н2 во внешнюю трубку сопла, а фтор в центральную
трубку.
Стенки реактора охлаждают воздухом или водой, протека-
ющей по внешнему змеевиковому холодильнику. Вторая труба
реактора служит фильтром. Внутри трубы установлены пористые
фильтрующие патроны, изготовленные спеканием из порошка
сплава монель.
•00
Твердые продукты реакции собирали в сборниках, присоеди-
ненных к донной части реактора и фильтра. От 70 до 85% по-
рошка молибдена собиралось в сборнике фильтра. В сборнике
реактора содержался более крупный порошок (агломераты), от-
части загрязненный продуктами коррозии стенки трубы. Этот
материал фторировали и полученный MoF6 возвращали в процесс.
Для стряхивания порошка и предотвращения его спекания
реактору и фильтру сообщали вибрацию.
Восстановление проводили при подаче 3,2 кг!ч MoF6, расходе
фтора 0,25 кг на 1 кг MoF6. Водород подавали в смеси с гексафто-
ридом в количестве 200% от теоретически необходимого для вос-
Рис. 118. Схема реактора
для восстановления гек-
сафторида молибдена в
факеле:
1 — реактор; 2 — сопло для
подачи MoFe, Н2 и F2 (Л и
В — варианты подачи); 3 —
фильтр; 4 — фильтрующий
патрон; 5 — сборники по-
рошка
становления. Температуру в реакторе поддерживали в пределах
525—555° С. Восстановленный молибденовый порошок содержал
примесь 0,03—0,05% фторида. Для снижения содержания фтора
полученный порошок восстанавливали водородом в течение 24 ч.
После такой обработки содержание фторида снизилось до 0,0002%.
Общее содержание металлических примесей в порошке составляло
всего 0,0025%. Содержание углерода в порошке 0,015%. Источ-
ник примеси углерода — фтор, в котором были найдены следы CF4.
Средний размер частиц порошка, определенный микроскопи-
ческим методом, 2,4 мкм.
Порошки молибдена и вольфрама, восстановленные из фто-
ридов в факеле, имеют чрезвычайно большую поверхность. Удель-
ная поверхность полученного молибденового порошка равна
6 м2/г, что примерно в 6 раз больше удельной поверхности обычных
молибденовых порошков с тем же средним размером частиц.
Это может найти специальное применение в случае необходимости
использовать порошок с большой активной поверхностью.
Установлена возможность получения порошков вольфрамо-
молибденового сплава при совместном восстановлении смеси
W.F6 + MoF6 водородом в факеле. В результате восстановления
при 525—555° С получается однородный порошок, частицы кото-
рого представляют собой сплав вольфрама и молибдена, что
подтверждено рентгенографическим исследованием.
301
Восстановление галогенидов в кипящем слое
Принципиальная схема восстановления галогенидов в кипящем
слое показана на рис. 119. Очищенный дистилляцией галогенид
из испарителя поступает в кипящий слой из частиц порошка
металла, создаваемый восходящим потоком водорода (или смеси
водорода с инертным газом).
Галогенид восстанавливается водородом в кипящем слое и
металл осаждается на поверхности частиц, что приводит к посте-
пенному увеличению их размеров. Периодически крупнозернистый
металл выгружают из слоя, часть выгруженного продукта из-
СВежий Н2
НС1 или HF
Рис. 119. Принципиаль-
ная схема установки для
восстановления галогени-
дов молибдена водородом
в кипящем слое:
1 — испаритель галогенида;
2 — реактор кипящего слоя;
<3 — труба для выгрузки по-
рошка металла; 4 — фильтр;
5 — теплообменник; 6— осу-
шительные колонки; 7 —
скруббер; 8 — холодильник;
9 — насосы
мельчают (например, в вихревой мельнице) и возвращают в реак-
тор. Избыточный водород, содержащий галогеноводород (HF,
НС1), поступает в систему очистки (скруббер, осушительные ко-
лонки) и после подогрева возвращается в реактор [66].
Исходные порошки, загружаемые в реактор, должны иметь
некоторую оптимальную крупность частиц. Мелкозернистые по-
рошки (средйий размер 2—3 мкм) трудно перевести в состояние
однородного кипения вследствие их склонности к агломерирова-
нию. Однородное псевдоожижение в этом случае может быть до-
стигнуто применением вибрации, сообщаемой реактору.
Молибденовые порошки со средним размером частиц 20—
30 мкм легко переводятся в состояние однородного кипящего слоя.
Однако выше определенной температуры (750—800° С) кипе-
ние прекращается (оседает слой). По всей вероятности, это обус-
ловлено началом спекания металлических частиц, ведущим к агло-
мерации. Однако это явление, наблюдаемое и при работе с другими
металлическими порошками, еще недостаточно изучено [69].
Для осуществления процесса основное условие — установле-
ние режимов, при которых подавляющая часть галогенида вос-
станавливается на поверхности частиц с осаждением металла.
Если протекает гомогенное восстановление хлорида в газовой
фазе, образующиеся тонкодисперсные частицы металла выносятся
с потоком газа, причем они обычно содержат значительную при-
месь невосстановленных низших галогенидов.
на
Как показали исследования автора и Егорычева, при низких
температурах (550—600° С) пентахлорид восстанавливается водо-
родом в кипящем слое преимущественно на поверхности частиц,
тогда как с повышением температуры увеличивается доля тонко-
дисперсных частиц, выносимых из слоя.
Такая закономерность объясняется более низкими значе-
ниями энергии активации реакции, протекающей на поверхности,
по сравнению с энергией активации реакции гомогенного вос-
становления.
При восстановлении пентахлорида водородом температуру
в испарителе поддерживают в интервале 180—200° С. При более
высокой температуре наблюдается термическая диссоциация МоС15
с образованием малолетучего МоС13.
Обычно в испаритель подают газ-носитель (водород или ар-
гон), вместе с которыми галогенид поступает в кипящий слой.
Восстановлением галогенидов водородом в кипящем слое
можно получить порошки молибдена заданной крупности, которые
трудно получить другими способами. Крупнозернистые порошки*
молибдена необходимы, например, для изготовления синтериро-
ванных катодов электровакуумных приборов, пористых фильт-
ров.
Опубликованы данные о восстановлении гексафторида воль-
фрама водородом в кипящелМ слое, которые по близкой аналогии
металлов можно отнести к молибдену.
Гексафторид восстанавливается в кипящем слое частиц воль-
фрама с начальной крупностью от 5 до 50—100 мкм. Температура
восстановления 555—580° С. Продолжительность процесса зави-
сит от требуемой крупности порошка. Частицы имеют сфериче-
скую форму, кристаллиты ориентированы в радиальном направле-
нии. Максимальный размер частиц 420—590 мкм. Полученный
крупнозернистый порошок хорошо прессуется в заготовки. После
активированного спекания в атмосфере водорода они имели плот-
ность около 99% от теоретической 1 [68].
Восстановление МоС15 водородом в кипящем слое позволяет
покрывать молибденом частицы тугоплавких окислов (UO2, SiO2,
А12О3, ZrO2, MgO) и других тугоплавких соединений (SiC, ZrC,
бориды и др.) [69—72]. Прослойки металла улучшают физические
и механические свойства спеченных изделий из этих материалов.
Известно, что из двуокиси урана изготовляют тепловыделя-
ющие элементы ядерных реакторов. Использование для этой цели
частиц двуокиси урана, предварительно покрытых молибденом,
приводит к улучшению свойств спеченных заготовок. Повы-
шается их прочность и теплопроводность, устойчивость против
радиационного разрушения. Технология покрытия двуокиси урана
молибденом описана в работах [69, 71].
1 В 1 о с h е г I. М., Pearson L Н. US Pat., № 3234007, 1966.
303
Восстановление МоС15 на нагретой подложке
Восстановление на нагретых поверхностях используют преиму-
щественно для получения молибденовых покрытий. Однако в огра-
ниченной степени метод можно применять также для получения
толстых слоев осажденного чистого металла, который может быть
затем переплавлен. Принципиальная схема аппарата для получе-
ния молибденовых покрытий показана на рис. 120 [77].
Рис. 120. Схема
установки для по,
вых покрытий:
либдено-
7 — конденсатор; 2 — индуктор; 3 — покрываемое изделие;
4 — держатель; 5 — плита; 6 — асбестовая изоляция; 7 —
нагреватель; 8 — теплоизоляция печи; 9 — пятихлористый
молибден; 10 — подача водорода; 11 — термопара
Обстоятельное исследование условий осаждения молибдена
восстановлением МоС15 на нагретых поверхностях проведено
Чайльдсом с сотрудниками [74]. Авторы пропускали смесь высо-
кочистых МоС15 и водорода через кварцевую трубку с покрывае-
мым образцом металла, который нагревали с помощью индуктора
до 800—1100° С. Было установлено, что качество осадка зависит
от общего давления в системе, парциального давления паров МоС15
и температуры подложки. Лучшими физическими свойствами
(высокая прочность, хорошее сцепление с подложкой) обладают
грубозернистые осадки со столбчатой структурой. Они полу-
304
чаются при температуре подложки 850—900° С при общем давле-
нии в системе не выше 25 мм рт. ст. и парциальном давлении
MoCJ5 около 1/10 от давления водорода. При общем давлении
5 мм рт. ст. скорость осаждения при 900° С составляла 0,5 мм/ч.
При более высоких давлениях и низких температурах полу-
чаются рыхлые осадки или тонкодисперсные порошки, содержащие
низший хлорид МоС13.
В зависимости от требуемого парциального давления водорода
температуру в испарителе поддерживают в интервале 120—200° С.
Присутствие оксихлорида в газовой смеси нарушает сцепле-
ние молибдена с покрываемым металлом, что объясняется присут-
ствием двуокиси молибдена, образующейся в результате восста-
новления оксихлорида [75].
В работе [79] была оценена чистота молибдена, получаемого
осаждением на нагретой поверхности. Авторы установили, что
более чистый пентахлорид молибдена получается при очистке
сублимацией по сравнению с его дистилляцией.
Сублимацию проводили в потоке чистого хлора при темпера-
туре 194° С (несколько ниже температуры плавления МоС15),
пары конденсировали в стеклянном конденсаторе. Для восста-
новления МоС15 пропускали смесь МоС15 и водорода через нагре-
тую до 850° С викоровую трубку. Высокий выход молибдена до-
стигался при общем давлении в системе 5—20 мм рт. ст. В про-
веденном укрупненном опыте из 550 г МоС15 за 36 ч было получено
185 г металла (извлечение 96%).
В сублиматоре МоС15 поддерживали температуру 280° С, газом-
носителем служил чистый аргон, водород впускали отдельно в зону
восстановления.
Молибден был осажден в форме трубки и переплавлен в элек-
тронно-лучевой печи. Содержание примесей в осажденном и пере-
плавленном металле приведено в табл. 77.
ТАБЛИЦА 77
СОДЕРЖАНИЕ ПРИМЕСЕЙ В МОЛИБДЕНЕ, ПОЛУЧЕННОМ
ВОССТАНОВЛЕНИЕМ МоС1б ВОДОРОДОМ, %• 10“4
Примесь В металле, восстановленном из МоС1б водородом В металле после электронно-лучевой плавки
С н N О Mg Fe, Al, Si Ti, V, Cr, Co, Ni Mn Cu 8,9 3,0 9,0 6,5 1 (0,5—2) каждого <Z 1 каждого «1 <0,5 -0,2 9,0 5,0 Не опр.
20 А. Н. Зеликман
305
52. ПОЛУЧЕНИЕ МОЛИБДЕНА ЭЛЕКТРОЛИЗОМ
Многочисленные попытки электролитически выделить молибден
из водных растворов не дали результатов [84, 86]. Даже весьма
тонкие осадки (доли микрона) представляют собой низшие
окислы молибдена [84].
По данным работ [87—89], при электролизе нейтральных или
слабощелочных растворов молибдатов осаждающиеся на катоде
черные осадки отвечают по составу гидратированной полутора-
окиси Мо2О3-ЗН2О. В работе [89] рекомендуется использовать
электролиз для выделения молибдена из водных растворов молиб-
датов в виде полутораокиси, имеющей малую растворимость
в воде.
Установлена возможность получать электролитически осадки
сплавов молибдена (до 50% Мо) с железом, никелем или кобаль-
том из кислых или слабощелочных растворов лимоннокислых
солей [80] и осадков сплавов молибдена с железом или кобальтом
из концентрированных щелочных растворов х. Однако в слоях
осадков толщиной 0,025—0,05 мм, в которых содержалось до 25—
30% Мо, было много кислорода, что указывает на одновременное
осаждение низших окислов молибдена.
Не получены положительные результаты и в работах по выде-
лению молибдена электролизом из органических растворителей
и других неводных сред [82, 83, 86]. Лишь при электролизе рас-
плавленных сред выделяются осадки молибдена.
Наибольшее число работ проведено с расплавами электроли-
тов, содержащими фосфаты щелочных металлов (иногда с добав-
ками галогенидов щелочных или щелочноземельных металлов),
а также с ваннами на основе галогенидов щелочных и щелочнозе-
мельных металлов (фториды и хлориды натрия, калия, лития,
кальция). В первом случае исходные соединения молибдена, вво-
димые в расплав, — МоО3, СаМоО4 и другие молибдаты. В слу-
чае галогенидных ванн, кроме кислородных соединений, исполь-
зуют хлористые соли (К3МоС16, МоС15)..
Электролиз из фосфатных и боратных ванн
Мета- и пирофосфат натрия (NaPO3, Na4P2O7), а также тетра-
борат натрия Na2B4O7 — хорошие растворители кислородных
соединений молибдена.
Фосфаты NaPO3 и Na4P2O7 плавятся при температурах 625 и
990° С соответственно. Точка плавления эвтектики (70% NaPO3 +.
+ 30% Na4P2O7) 572° С. Поэтому часто применяют смеси фосфа-
тов. Борат Na2B4O7 плавится при 742° С.
По данным Андреева [90], потенциал разложения МоО3 в рас-
плаве метафосфата при 1000° С и концентрации 5% (мол.) МоО3
1 Y п t е m a L. F. US Pat., ’2428—404, 1947.
306
Е = 0,86 в, а в пирофосфате при тех же температуре и концен-
трации МоО3 Е = 0,94 в.
По Делимарскому и Назаренко [91], в расплаве тетрабората’
натрия при 840° С Е = 1,39 в.
В электрохимических рядах напряжений в расплавах фосфа-
тов и тетрабората натрия молибден занимает существенно различ-
ное место. Ряды напряжений [90, 91]:
Na4P2O7 (1000° С): Na, Zn, Wvi, Cd, Co, Ni, Cu”, Pb, Fe111, MoVI
Sbni, Nbv.
NaPO3 (720° C): Na, WVI, Zn, Cd, Fem, Pb, Co, Ni, Си», MoVI>
Bi, Sbni.
Na2B4O7 (840° C): Na, СгШ, MoVI, Fem, Mn1”, WVI, Zn, Sbni, Ni>
SnIV, Cd, Bi, TP, Co, Pb, Си”, Ag.
Интересно отметить, что из фосфатных ванн молибден должен
выделяться при более низких напряжениях, чем вольфрам, тогда
как в боратных ваннах потенциал разложения ниже у вольфрама.
Часто используют двойные фосфатно-боратные ванны или
тройные фосфатно-боратно-хлоридные ванны.
Электролиз МоО3 в пирофосфатной и боратной ваннах изучал
Григорьян [92]. Полученные им данные о физико-химических
свойствах систем МоО3—Na4P2O7 и МоО3—Na2B4O7 приведены
на рис. 121 и 122.
Первоначально электролиз проводили в небольших графито-
вых тиглях, служащих анодом, с графитовым катодом в центре,
а затем в укрупненно-лабораторном электролизере с амперной
нагрузкой до 600 а, эскиз которого показан на рис. 123. В нем
графитовый тигель служил анодом в центре — катод из молибдена
или графита. Электролиз проводили в атмосфере аргона. Катод
периодически извлекали и специальным приспособлением очи-
щали. Одновременно в электролиз опускали второй катод. В ванну
полунепрерывно или непрерывно подавали с помощью вибропита-
теля техническую трехокись молибдена.
В работе [92] рекомендованы следующие оптимальные ре-
жимы электролиза.
Для боратной и фосфатной ванн оптимальная концентрация
МоО3 6% (по массе), начальная катодная плотность тока DK =
= 8--15 а!см? (для ванны с центральным катодом), анодная плот-
ность Da = 0,1-4-0,15 а!см\ напряжение на ванне ~3,1 в. Темпе-
ратура электролиза для боратной ванны 850° С, фосфатной 920—
960° С. При указанных режимах выход по току равен 80—85%.
Катодный продукт подвергали мокрому измельчению, фильтро-
вали, промывали на фильтре и сушили.
20* 307
По данным работы [92], в среднем порошки содержали
99,5% Мо, однако полный анализ порошков не проводился, в част-
ности не определено содержание кислорода.
* 300-
200\-------1---1____I___
Мо03 25 50 75 Ыа^Рг07
% (аа массе)
% [по массе)
Рис. 121. Физико-химические свой-
ства системы МоО3—Na4P2O7:
а — диаграмма состоянйя; б — плот-
ность; в — вязкость; г — удельная
электропроводность; д — поверхност-
ное натяжение
Рис. 122. Физико-химические свой-
ства системы МоО3—Na2B4O7:
а — диаграмма плавкости; б — плот-
ность; в — вязкость; г — удельная
электропроводность; д — поверхност-
ное натяжение
Полученные порошки содержали 80—85% частиц размером
до 0,01—0,014 мм, 15—20% от 0,01 до 0,070 мм.
Ряд исследований по электролитическому получению молиб-
дена из боратных и фосфатных ванн выполнен в лабораториях
308
Горного бюро США. Так, изучался электролиз МоО3 в расплаве
из смеси галоидных и борнокислых солей [93]. Состав электро-
лита, %: 23 NaCl; 15 NaF; до 57 Na2B4O7; до 8 МоО3. Анод —
графитовый тигель, катод—графитовый стержень. Электролиз про-
водили при температуре 1000°C,
начальном DK^80 а!дм\ Da~
= 15 al дм2 без защитной атмо-
сферы. Расстояние между като-
дом и анодом до дна тигля 50 мм
и до стенки 25 мм. При элек-
тролизе чистой МоО3 ^и техни-
ческой (содержавшей до 10%
примесей, в том числе 5,5% W)
был получен молибденовый по-
рошок чистотой 99,5%.
Содержание примесей, %:
С.......... 0,003
О .... 0,025
А1 .... 0,01—0,05
В .... 0,001—0,0025
Са .... 0,005—0,01
Fe .... 0,0075—0,02
Mg, Ni, Си . По <0,001
Na .... <0,0025
Р.......... 0,0018
W.......... <з-ю-5
As .... 0,0005
Осадок представлял собой ден-
дриты, хорошо пристающие
к катоду.
После прессования в элек-
троды и переплавки в дуговой
печи суммарное содержание
примесей было меньше 0,1%.
Среди них, %:
С.......... 0,024
О .... 0,01
Fe .... 0,0075—0,025
Н .... 0,017—0,02
As, В, Cd,
Mg, Ni,
Si, Си . . По <0,001
Рис. 123. Схема укрупненно-лабора-
торного электролизера со сменным
катодом:
1 — пневмоподъемник; 2 — пневмоочисти-
тель катода; 3—вибропитатель; 4—прием-
ник катодного осадка; 5 — трубка для
отвода газов; 6— анод (графит); 7 — катод,
(графит или молибден); 8 — термопара;
9 — электронагреватели; 10 — тележка;
11 — подвод инертного газа
Примененный в рассматриваемой работе тетраборатно-хло-
ридно-фторидный электролит для электролиза МоО3 лучше, чем
испытывавшийся электролит из тетрабората, пирофосфата натрия
и хлорида натрия. Преимущества состоят в возможности более
широкого варьирования температуры и состава ванны, меньшем
осаждении МоО2 и фосфора на катоде. Кроме того, полученный
309
Опыты, проведенные в лабораторном электролизере в атмо-
сфере аргона при предварительной тщательной осушке электро-
лизера и исходных солей, показали, что молибден выделяется при
изменении катодной плотности от 3 до 100 а!дм1 2. При плотностях
тока выше 3 а!дм2 и температуре 600—900° С из ванн обоих соста-
вов на катоде выделяются рыхлые осадки, однако из второй ванны
(КС1—LiCl) при температуре 600° С и плотности тока 3 а!дм2
выделяются плотные, хорошо сцепленные с катодом покрытия
вплоть до толщины 0,5 мм, что может быть использовано для полу-
чения покрытий.
Катодные осадки кипятили в 10 %-ной соляной кислоте, промы-
вали водой, фильтровали, промывали ацетоном и сушили. Они
содержали 0,026% кислорода, другие примеси в . количествах от
-0,01 до 0,0001 %. Общее содержание металла не ниже 99,9%.
Электролиты из хлоридов щелочных металлов и К3МоС16 при-
годны также для электролитического рафинирования молибдена.
Электролиз кислородных соединений
из галогенидных ванн
В английском патенте 1 описано получение молибдена электро-
лизом в расплавленном электролите: 30—80% СаС12, 10—35% СаО.
В расплав вводят МоО3 из расчета молярного отношения СаО :
: Мо03 =1:1. Окись кальция растворяют в СаС12 до введения
J4oO3, что способствует быстрому растворению и распределению
трехокиси молибдена в расплаве. Вместо МоО3 в расплав СаС12
можно вводить СаМоО4, в этом случае добавка окиси кальция не
нужна.
По данным Хлебникова и Надольского [102], система СаС12 —
СаМоО4 на участке до содержания СаМоО4 25% (по массе) имеет
эвтектическую точку состава, % (по массе): 11,5 СаМоО4; 88,5СаС12.
Температура плавления эвтектики 738° С. Потенциал разложе-
ния Са МоО4 в расплаве СаС12 при 1000° С равен —0,89 в [ЮЗ].
Согласно патенту \ электролиз проводится без защитной атмо-
сферы при температуре 1050—1200° С. DK = 1,6=11,2 а!см2,
Da = 0,8=1,12 а!см2. Анод графитовый, катод молибденовый
или стальной. Выход по току близок к 100%, извлечение мо-
либдена ~88%. Осадок хорошо держится на катоде, чистота
полученного порошка (после отмывки электролита водой, сла-
бой НС1) 99,8—99,9%.
Описан 2 электролиз трехокиси молибдена в ванне, состоящей
из смеси фтороборатов (N^BF4, KBF4) и оксифтороборатов Na2O
(BF3)4 или К2О (BF3)4.
Состав ванны: NaBF4 : Na2O (BF3)4 : МоО3 = 500 : 10 : 20 ч.
(по массе). Электролиз ведут при температуре 420 ± 10° С;
1 Патент (англ.), 918167, 1963.
2 Патент (японск.), 6707, 1956.
512
Степень очистки зависит от потенциала электрода по отноше-
нию к данной примеси и ее концентрации в электролите
(табл. 78).
В работе [105] изучали условия рафинирования молибдена от
примесей Sn, Fe, Si, Си и Ni, вводившихся в количествах от 0,1
до 10% в электролит состава, г!л\ 330 КО; 270 и 200 КзМоСЛ6.
Электролиз проводили в защитной атмосфере аргона при измене-
нии катодной плотности тока от 3 до 100 а!дм2 и температуре 600
и 900° С.
Примеси вводили в виде солей SnCl2, FeCl2, Na2SiF6r CuCl
и NiCl2.
Было установлено, что при определенных условиях (плот-
ность тока, температура) можно обеспечить осаждение на катоде
молибдена с содержанием примеси не выше 0,01% (по массе).
Эти условия приведены в табл. 78. Данные таблицы показывают
существенную зависимость условий рафинирования от темпера-
туры. При 600° С нельзя получить достаточно чистый молибден
по примесям Sn, Си, Ni, тогда как при 900° С они хорошо отде-
ляются при определенных плотностях тока.
DK = 0,25 а!см\ напряжение на ванне 2,3 в. Анод — графит,. .
катод медный. Чистота осадка молибдена выше 99%.
Электролитическое рафинирование молибдена
Электролитическое рафинирование молибдена можно прово-
дить в электролитах из расплава галогенидов щелочных металлов
(NaCI—КС1, КС1—LiCl), в котором растворены МоС13 или КзМоС16.
Электролитическое рафинирование происходит при анодном рас-
творении молибдена (более благородные примеси остаются в анод-
ном шламе), а также при последующем осаждении, молибдена на
катоде (менее благородные примеси остаются в электролите).
Поскольку молибден занимает относительно благородное место
в электрохимическом ряду в расплаве щелочных галогенидов (см..
стр. 307), наиболее существенная очистка от примесей менее бла-
городных металлов должна наблюдаться при осаждении на катоде..
ТАБЛИЦА 78
УСЛОВИЯ ЭЛЕКТРОЛИЗА, ОБЕСПЕЧИВАЮЩИЕ СОДЕРЖАНИЕ
ПРИМЕСИ В ОСАЖДЕННОМ НА КАТОДЕ МОЛИБДЕНЕ НЕ ВЫШЕ 0,01% [105J
Соль, добавленная в электролит Содержание металла-примеси в ванне, % Отношение 3 + Мо к металлу примеси в ванне Максимально допустимая плотность тока, а/дм2, при температуре, °C
600 900
0,2 31 Нет * 100
SnCl2 1,5 6 » 100
6,7 0,6 » 100
0,04 12 30 100
FeCl2 0,5 4 10 100
6,0. 0,3 10 100
0,03 40 100 100
Na2SiF6 • 0,2 10 100 100
•> 1,6 0,3 100 100
0,1 34 Нет 100
CuCl 0,7 5 » 100
9,1 0,2 » 3
0,06 35 Нет 100
NiCl2 0,5 6 » 10
4,8 0,5 » Нет
* «Нет» — означает, что в пределах изученных плотностей тока (от 3 до 100 а/дм2}
нельзя получить осадка молибдена с содержанием примеси 0,01% или ниже.
313
поэтому тонкие порошки занимают больший объем (имеют меньшую
насыпную массу), чем грубозернистые. Первые обладают большей
удельной поверхностью, силы сцепления проявляются в них
в большей степени, препятствуя уплотнению порошка.
При прессовании порошка получают тело определенной формы,
обладающее некоторой прочностью. Это достигается в результате
увеличения контактных поверхностей между частицами и, сле-
довательно, возрастания сил сцепления между ними (величина
которых обратно пропорциональна расстоянию между частицами),
а также сил межчастичного механического зацепления.
При прессовании контактная поверхность первоначально уве-
личивается в результате заполнения частицами свободных про-
межутков. Дальнейшее уплотнение возможно лишь вследствие
пластической деформации частиц, приводящей к выравниванию
поверхности их соприкосновения. Однако в случае прессования
молибденовых порошков на холоду пластическая деформация
частиц незначительная, поэтому плотность прессовок обычно не
превышает 65% от плотности молибдена.
Плотность прессовок зависит от крупности частиц порошка.
При одинаковом удельном давлении плотность спрессованной за-
готовки тем ниже, чем меньше величина частиц порошка. С повы-
шением удельного давления возрастает плотность прессовок. Од-
нако для каждого порошка (в зависимости от крупности его* частиц
и формы) имеется критическое-удельное давление, выше которого
прессуемое тело начинает вести себя как сплошное — наблю-
дается скалывание или расслой. Для молибденовых порошков
предельное удельное давление равно 4—6 т1см2. Общие законо-
мерности прессования порошков детально освещены в специальной
литературе [4—6, 21 ].
Ниже рассмотрены вопросы технологии прессования молибде-
новых порошков.
Прессование штабиков
Для производства проволоки и листов небольшого размера мо-
либденовые порошки прессуют в стальных разъемных пресс-фор-
мах в штабики квадратного (для протяжки проволоки) или прямо-
угольного (для прокатки) сечения, имеющие длину 500—650 мм.
Примерные размеры прессуемых заготовок и условия прессования
приведены в табл. 79. Конструкция пресс-формы показана на
рис. 124. Пресс-формы изготовляют из твердых и износостойких
сталей.
Для повышения срока их службы практикуют покрытие вну-
тренней поверхности слоем стеллита. Внутренние поверхности
должны быть хорошо отшлифованы.
Прессование проводят на гидравлических прессах с верти-
кальным и горизонтальным цилиндрами. Горизонтальный ци-
линдр осуществляет боковое давление, с помощью которого за-
316
ГЛАВА XII
ПОРОШКОВАЯ МЕТАЛЛУРГИЯ МОЛИБДЕНА
До 1943—1945 гг. единственным способом производства ком-
пактных заготовок молибдена, поддающихся обработке давле-
нием, служил способ порошковой металлургии (или металлоке-
рамики), разработанный в 1909—1910 гг. Кулиджем примени-
тельно к изготовлению изделий из вольфрама1 [1].
Основные стадии технологии:
а) прессование порошка в заготовку (штабик, брикет);
б) спекание спрессованной заготовки (нагревание ее при опре-
деленной температуре, лежащей ниже точки плавления металла);
в) 'обработка спеченной заготовки с целью получения изделий
(ковка, протяжка, прокатка).
Способ Кулиджа, особенностью которого является нагревание
сравнительно небольших спрессованных заготовок (штабиков)
прямым пропусканием электрического тока, не . утратил своего-
значения и в настоящее время. Вместе с тем развиты, преимуще-
ственно для получения крупных заготовок, а также изделий слож-
ной формы, другие варианты технологии порошковой металлур-
гии молибдена.
53. ПРЕССОВАНИЕ ЗАГОТОВОК МОЛИБДЕНА
Порошок металла, свободно насыпанный в какую-либо емкость
и подвергнутый, утряске, не находится в состоянии максимального
уплотнения. Этому препятствуют силы трения между частицами,
а также чисто механическое взаимное их зацепление. Таким обра-
зом, порошок занимает объем, отвечающий промежуточному
равновесию между силами сцепления и весом его частиц. Именно
1 Способ порошковой металлургии для получения компактных металлов из
порошков впервые предложен русским инженером Соболевским в 1826 г., который
применил его для изготовления изделий из платины [2].
315
0,32 мм и в герметичной таре передают на прессование. Существен-
ное значение имеет равномерное распределение навески порошка
в пресс-форме. Кроме ручного разравнивания с помощью пла-
стины-шаблона, для этого может быть использовано заполнение
пресс-формы на вибрационных столах [3].
Плотность спрессованных штабиков зависит от удельного дав-
ления и размеров зерен порошка. Как видно из табл. 80, штабики,
спрессованные из мелкозернистых порошков, имеют меньшую
плотность.
ТАБЛИЦА 80
ПЛОТНОСТЬ ШТАБИКОВ МОЛИБДЕНА, СПРЕССОВАННЫХ ИЗ
ПОРОШКОВ С РАЗЛИЧНОЙ ВЕЛИЧИНОЙ ЗЕРЕН [3]
Удельное давление прессования, Т/см2 Плотность штабиков, г/см3
мелкозернистый порошок крупнозернистый порошок
1,0 4,3 5,5
1,5 5,2 6,0
2,5 5,8 6,4
3,5 6,4 6,6
4,5 6,6 6,8
Брикеты, спрессованные из молибденовых порошков (частицы
которых мало деформируются), имеют малую прочность на растя-
жение по сравнению с заготовками из мягких металлов. Это объ-
ясняется упругим расширением (упругим последействием) заго-
товки после снятия давления прессования, преимущественно в на-
правлении прессования, которое составляет примерно 0,5—0,6%
линейного размера. Оно ведет к нарушению и ослаблению меж-
частичных контактов в заготовках из твердых металлов, тогда
как в случае мягких металлов эти нарушения менее существенны.
При некотором критическом для данного порошка давлении
упругое последействие может быть причиной появления расслой-
ных трещин.
Наиболее часто встречающиеся виды брака спрессованных шта-
биков — трещины (поперечные и продольные). Они могут быть вы-
званы рядом причин: некачественным изготовлением пресс-формы,
сдвигом ее стенки при прессовании или извлечении штабика, не-
большим смещением пресс-формы под нагрузкой, неравномерным
распределением порошка в пресс-форме, а также отклонением
характеристик порошка от оптимальных по зернистости и содер-
жанию кислорода. Повышенное содержание кислорода (и, следо-
вательно, пленок окислов на частицах) может быть результатом
длительного хранения порошка при доступе воздуха. Поэтому
необходимо использовать свежевосстановленные порошки или
хранить их в атмосфере инертного газа.
318
жимается пресс-форма. Боковое давление должно быть несколько
выше вертикального давления на пуансон пресс-формы во избежа-
ние сдвига стенок разборной пресс-формы. Применяют гидравли-
ческие прессы мощностью 270—2000 т. На заводах СССР наиболее
распространен 520-т пресс П-801.
Известно, что при одностороннем прес-
совании порошка в пресс-форме давление
распределяется неравномерно по объему
прессуемой заготовки, что объясняется
трением порошка о стенки пресс-формы.
Вследствие этого в любом вертикальном
разрезе заготовки верхние слои плотней
нижних. В горизонтальных сечениях
в верхних слоях плотность заготовки уве-
личивается от центра к периферийным
областям, а в нижних слоях — от пери-
ферии к центру.
Для обеспечения более равномерного
распределения давления к порошку перед
прессованием добавляют смазывающие
жидкости — раствор глицерина в спирте
(в отношении 1,5 : 1) или раствор пара-
фина в бензине (4—5% парафина). Рас-
творы, выдавливаясь при прессовании
к стенкам пресс-формы и смазывая их,
Рис. 124. Пресс-форма для
прессования молибдено-
вых штабиков:
1 —- щеки; 2 — торцовые
пластины; 3 — нижняя под-
кладка; 4 — пуансон; 5 —
соединительные штифты
уменьшают трение частиц порошка о стенки. На каждый кило-
грамм порошка расходуется ~3—5 см3 раствора, который тща-
тельно перемешивается с порошком в смесителе в течение 20—
60 мин. Затем порошок просеивают через сетку с отверстиями
ТАБЛИЦА 79
ПРИМЕРНЫЕ РАЗМЕРЫ ШТАБИКОВ И РЕЖИМЫ ПРЕССОВАНИЯ
ПОРОШКОВ ЧИСТОГО МОЛИБДЕНА
Размер штабиков, мм Примерная навеска, г Удельное давление, Т /см2 Изготовитель
14X14X600 740 3,0 Заводы СССР [33]
18X18X650 1400 2,5
30X30X650 4000 3,0
40X40X650 6000 2,5
60X60X650 9500 2,0
20X20X300 800 4—5 Metallwerke Plansee
25X25X400 1600 3—4
40 X 40 X 600 5000 1,0—1,5 (Австрия) [3]
40X70X600 8000 1,0—1,5
317
ком. С целью утряски и равномерного распределения порошка
форму устанавливают на вибрационную плиту. Заполненную
форму закрывают резиновой крышкой и помещают в камеру
Рис. 126. Схематический
разрез формы для гидро-
статического прессования
цилиндрических загото-
вок:
1 — резиновый колпак («бе-
рет»); 2 — резиновые ман-
жеты; 3 — металлическая
гильза; 4 — резиновая обо-
лочка; 5 — прессуемый поро-
шок; 6—штуцер; 7—пробка;
а — заполненная форма;
б — форма в процессе прес-
сования
гидростатического прессования [9, с. 187]. Формы для прессования
цилиндрических заготовок и трубок показаны на рис. 127 и 128.
Наиболее важный узел рабочей камеры гидростатического прес-
сования — затвор. Различные конструкции камер и затворов
описаны в работе [И, с. 32].
Рис. 128. Формы для прессования
трубок
Рис. 127. Внешний вид деталей форм
гидростатического прессования ци-
линдрических брикетов:
/—резиновая оболочка; 2 — перфориро-
ванная гильза; 3—«берет»; 4—манжеты
На рис. 129 приведен схематический разрез одной из кон-
струкций рабочей камеры с затвором, особенностью которого
является упруго расширяющееся кольцо, которое в процессе уве-
личения давления в камере надежно обеспечивает ее герметич-
ность.
320
Прессование крупных заготовок
В связи с расширением использования молибдена как конструк-
ционного материала потребовались крупные заготовки металла
массой 100—200 кг и более для изготовления листов больших раз-
меров, труб и изделий других профилей. Для получения таких
заготовок прессование порошков в стальных пресс-формах прак-
тически непригодно из-за невозможности обеспечить их равномер-
ную плотность. Кроме того, в этом случае необходимы прессы
большой мощности (5000 т и более), что неэкономично.
Для прессования крупных заготовок однородной плотности ис-
пользуют метод гидр
о статического
Рис. 125. Принципи-
альная схема аппарата
для гидростатического
прессования порошка:
1 —насос высокого давле-
ния; 2 — рабочая камера;
3—манометр; 4—затвор;
5— вентиль для спуска
давления; 6 — прессуе-
мый порошок в эластич-
ной оболочке
прессова-
ния, при котором порошок металла, помещенный в эластичную
оболочку (из резины или другого цолимерного материала), под-
вергается равномерному всестороннему сжатию с помощью жид-
кости, подаваемой под давлением в рабочую камеру (рис. 125).
При гидростатическом прессовании нет трения порошка
о стенки пресс-формы, что обеспечивает равномерную плотность
спрессованной заготовки, отсутствие трещин и другие дефекты,
наблюдаемые при прессовании в стальных пресс-формах. Обору-
дование и промышленная технология гидростатического прессова-
ния разработаны в СССР в институте ЦНИИЧМ в начале 50-х
годов [9—И]. Процесс получил развитие также в США [15, 16]
и других странах.
Методом гидростатического прессования могут быть получены
заготовки цилиндрической или прямоугольной формы, трубки
и изделия более сложной формы. Нужную геометрическую форму
получают, помещая эластичную оболочку в перфорированные
стальные формы. Так, для получения цилиндрических или пря-
моугольных заготовок стальным формам придают соответственно
вид трубы или коробки (рис. 126) [9, 10]. Эластичная оболочка
растягивается и плотно прилегает к внутренней поверхности
формы. На нижний конец формы натягивают резиновую крышку
(«берет») и стяжную резину, после чего форму заполняют порош-
319
келя обеспечивалась при гидростатическом прессовании при давле-
ниях на 30—50% меньших, чем в случае обычного прессования.
Зависимость относительной плотности брикетов от удельного
давления гидростатического прессования описывается уравне-
нием [17]
1
=-А—>или !g =4“ igр—Alg (209)
'max
где Ф — относительная плотность, %;
р — удельное давление прессования;
Ртах — удельное давление, необходимое для достижения от-
носительной плотности, равной 100%;
т — постоянная (для интервала давлений, обеспечивающих
относительную плотность брикетов ^85%).
При изучении гидростатического прессования порошков мо-
либдена в интервале давлений 5—30 кПмм2 Лобашов определил
следующие значения параметров приведенного выше уравнения:
ртах = 169,0 — 170,2; — = 0,202 ч- 0,208.
54. СПЕКАНИЕ СПРЕССОВАННЫХ ЗАГОТОВОК МОЛИБДЕНА
Общие основы процесса
Основная технологическая задача спекания — получение проч-
ного брикета, поддающегося механической обработке давлением.
Исходя из этого, Меерсон определяет спекание, как «процесс
увеличения прочности межчастичного (контактного) сцепления,
приводящий к увеличению прочности всего брикета в результате
воздействия повышенных температур» [9, с. 11].
В результате спекания брикетов из чистых металлических
порошков, помимо увеличения прочности, всегда наблюдается
усадка (уменьшение размеров и, следовательно, увеличение плот-
ности) и изменение структуры тела (изменение размеров и формы
зерен, формы и числа пор).
Процессы, протекающие при спекании (упрочнение, усадка,
рост зерен), обусловлены повышенной подвижностью атомов при
температурах спекания. Особой подвижностью обладают атомы,
расположенные на свободных поверхностях, вследствие ненасы-
щенности их силовых полей. Избыток свободной энергии у по-
верхностных атомов проявляется в поверхностном натяжении,
стремящемся сократить свободную поверхность. При достижении
определенной температуры атомы, расположенные на свободных
поверхностях, приобретают достаточную подвижность для пере-
мещения (миграции) с выступов (менее устойчивые состояния)
322
В работе [11, с. 32] дано примерное описание порядка работы
на установке гидростатического прессования ЦНИИЧМ (рис. 130).
Перед прессованием рабочая камера заполняется водой, посту-
пающей из водопровода через фильтр 7. Избыток воды, возникаю-
щий при погружении формы в камеру, откачивают водокольцевым
насосом Р. После погружения формы опускают затвор 2, пере-
Рис. 129. Схема рабочей камеры
для гидростатического прессования:
1 — гайка для подъема стержня; 2 —
пробка; 3 — стержень; 4 — корпус
камеры; 5 — упруго расширяющееся
кольцо; 6 — резиновая прокладка;
7 — штуцер для подачи жидкости
крывающий цилиндр камеры прес-
сования. Оставшийся в рабочем
пространстве воздух вытесняется
водой. После удаления воздуха,
на что указывает вытекание воды
через вентиль 3, его перекрывают
и включают двигатель насоса вы-
сокого давления 5. После дости-
Рис. 130. Схема установки гидростатиче-
ского прессования:
1 — камера прессования; 2 — затвор (в подня-
том положении); 3 — вентиль высокого давле-
ния; 4— манометр высокого давления; 5 — на-
сос высокого давления; 6 —манометр водопро-
водной магистрали; 7 — фильтр очистки воды;
8 —сливная воронка; 9 — водокольцевой насос
жения заданного давления насос выключают, давление-сбрасывают
с помощью дистанционного вентиля, включенного в трубопровод
высокого давления.
Рабочее давление эксплуатируемых установок 1—2,2 Т1см\
оно обеспечивает плотность спрессованных заготовок 60—70% от
плотности компактного металла.
Исследования Борока и Лобашева показали, что заготовки
диаметром 200 мм, спрессованные из порошков различных ме-
таллов, в том числе молибдена, имеют одинаковую плотность по
всему сечению [9]. Балыпин и Дубровский [17], изучавшие гидро-
статическое прессование порошков меди, железа, молибдена и
никеля при различных давлениях (от 1 до 6 Т1см2), установили,
что одинаковая плотность брикетов из порошков молибдена и ни-
21 А. Н. Зеликман 321
от абсолютной температуры их плавления. Для молибдена это
1680—1900° С. Между тем усадка спрессованных молибденовых
штйбиков, по данным Агте и Вацека [3], наблюдается при темпе-
ратурах выше 1100° С (рис. 132).
Рис. 132. Зависимость плотности мо-
либденовых штабиков от температуры
спекания при одинаковой длительности
выдержки (30 мин) [3]
Рис. 131. Схема увеличения контакт-
ных участков и сфероидизации пор
за счет поверхностной миграции атомов:
а — до спекания; б — пЛле спекания
Хотя плотность штабиков, спрессованных из мелкозернистого
порошка (/), ниже, чем штабиков из крупнозернистого порошка (2),
конечная плотность после спекания у первых выше, так как они
претерпевают большую усадку. Это согласуется с общими законо-
мерностями влияния крупности порошка на усадку при спекании,
установленными Бальшиным [5 ].
Рис. 133. Зависимость
плотности молибденовых
брикетов, спрессованных
при различных давле-
ниях, от температуры спе-
кания. Время спекания
3 ч (по Б. П. Лобашову).
Цифры на кривых — дав-
ление, кГ/мм2,
При низких температурах спекания (1200—1600° С) четко
выявляется влияние давления прессования на плотность спечен-
ных брикетов из молибденового порошка. С повышением темпера-
туры влияние исходной плотности спрессованного брикета на плот-
ность спеченных брикетов уменьшается и при 2000° С вообще не
проявляется (рис. 133) [14, с. 330].
324
к впадинам и узким местам контакта частиц, где запас свободной
энергии меньше и, следовательно, состояние атомов более устой-
чиво. Процесс поверхностной миграции атомов приводит к сгла-
живанию свободных поверхностей и сфероидизации пор. При
этом возрастает поверхность контакта между частицами и в ре-
зультате этого увеличивается прочность всего брикета (рис. 131).
Однако, как ясно из рис. 131, поверхностная миграция атомов
не может привести к усадке спекаемой заготовки, так как общий
объем пор в этом случае остается неизменным.
Усадка тела возможна лишь в результате объемной деформации
частиц и «затекания» материала в поры под действием сил поверх-
ностного натяжения. С ростом температуры увеличивается по-
движность атомов внутри объема спекаемых частиц, что ведет
к понижению их прочности, повышению пластичности и способно-
сти к объемному течению под действием сил поверхностного натя-
жения. Следует при этом учитывать, что с повышением температуры
прочность падает быстрее, чем силы поверхностного натяжения.
Известно, что в процессе спекания усадка первоначально про-
исходит с большой скоростью, а затем замедляется. Можно пола-
гать, что в начальной стадии спекания вследствие громадного пре-
обладания свободных участков над контактными силы поверх-
ностного натяжения, сосредотачивающие свое действие на кон-
тактных участках, способны вызвать быструю пластическую де-
формацию сдвигового типа. Этому также способствует повышенная
концентрация дефектов кристаллической решетки в начале спе-
кания. По мере развития процесса спекания увеличивается пло-
щадь контактных участков, уменьшается свободная поверхность,
повышается прочность кристаллов вследствие уменьшения де-
фектов решетки. В результате этого удельное напряжение на 1 маг
площади контактных участков становится меньше критического,
необходимого для сдвиговой деформации, и быстрая усадка пре-
кращается. Дальнейшая медленная усадка происходит в резуль-
тате диффузионной ползучести (т. е. связана с диффузией атомов
по вакансиям).
Изложенные выше представления о механизме спекания раз-
виты и более детально рассмотрены в работах Меерсона [6, 9,
с. 11].
Собирательная рекристаллизация (рост более крупных кри-
сталлов за счет мелких), начинающаяся выше определенной темпе-
ратуры, связана с интенсивной перегруппировкой атомов. Это
активизирует действие сил поверхностного натяжения, вызываю-
щих усадку.
Однако рекристаллизация не является причиной усадки. Как
правило, она наступает лишь после начала усадки при достаточном
количестве контактных участков для ее протекания [21].
Известно, что температура начала рекристаллизации у тел,
спрессованных из металлических порошков, составляет 2/3—3/4
21* 323
Для проверки однородности штабика может быть использован
описанный в работе [3] прием. До спекания в середине штабика
керном наносят небольшую метку. Штабик после первого спекания
помещают накерненной меткой на острие ножа. Определяется раз-
личие в массе обоих концов, которое не должно превышать 1 %
от массы штабика.
Высокотемпературное спекание при прямом нагревании током
(«сварка»). Молибденовые штабики спекают при максимальной
Рис. 134. Схема аппарата для спе-
кания молибденовых штабиков:
I — стальная плита; 2 — колпак;
3 — токоподвод; 4—верхний непод-
вижный контакт; 5 — нижний под-
вижный контакт; 6 — щипцы; 7 —
шины, подводящие ток; 8 — груз;
9 — молибденовый штабик
Рис. 135. Аппарат для одновремен-
ного спекания шестнадцати молибде-
новых штабиков
температуре 2200—2400° С, которая достигается прямым про-
пусканием электрического тока через штабик. Эта операция в про-
изводстве называется сваркой.
На рис. 134 показана схема аппарата для спекания штабиков.
Концы штабика закрепляют между подводящими ток медными
контактами, охлаждаемыми водой. Для крепления используют
щипцы из вольфрамовых пластин.
Медная трубка, подводящая ток к нижнему контакту, проходит
через отверстие в стальной плите, имеющей по периферии кольце-
вой паз. В паз на резиновое кольцо устанавливают охлаждаемый
водой медный колпак, в который в процессе сварки непрерывно по-
ступает сухой водород. Нижний контакт — подвижный, так как
в процессе сварки происходит значительная линейная усадка шта-
326
Выше рассмотрен идеализированный процесс спекания, предпо-
лагающий отсутствие примесей, пленок окислов и адсорбированных
газов на частицах металла, а также отсутствие активной газовой
атмосферы.
Как уже отмечалось, порошки молибдена содержат 0,05—
0,2% кислорода и, следовательно, частицы порошка и спрессован-
ных заготовок покрыты пленками окислов молибдена. При прове-
дении спекания в водороде пленки окислов восстанавливаются, что
приводит к увеличению поверхности металлических контактов.
Активирующее действие на спекание молибденовых заготовок
оказывает присутствие влаги в водороде.
Спекание штабиков молибдена
Молибденовые штабики спекают большей частью в две стадии:
первая стадия — предварительное низкотемпературное спекание
с целью упрочнения штабика и повышения его электропроводности;
вторая стадия — высокотемпературное спекание, при котором на-
гревание большей частью осуществляется прямым пропусканием
тока через штабйк. Кроме того, штабики спекают в вакуумных
печах с косвенным нагревом.
Для молибденовых штабиков малых сечений — достаточно
прочных, предварительное спекание может не проводиться.
Предварительное спекание. Предварительное спекание прово-
дят при температуре 1000—1100° С в атмосфере водорода с вы-
держкой штабиков в горячей зоне печи от 30 до 120 мин (в зависи-
мости от размеров штабиков). Спекание проводят в муфельных
электропечах с алундовой трубой прямоугольной формы. Нагрева-
телем служит молибденовая проволока или лента. Для защиты
нагревателя от окисления кожух печи герметизирован и заполнен
водородом. Спрессованные штабики укладывают в никелевые
лодочки на молибденовые пластины или на подстилку из крупно-
зернистого молибденового порошка. В лодочку помещают от 3
до 30 штабиков в зависимости от их размеров.
Спрессованные штабики обладают открытой пористостью, во-
дород в процессе спекания диффундирует в поры, восстанавливая
пленки окислов, что способствует образованию межчастичных
металлических контактов.
Усадка при первом спекании незначительная (2—3%), однако
штабики заметно упрочняются из-за увеличения контактных уча-
стков в результате поверхностной миграции атомов. Штабики после
предварительного спекания проходят браковку, в которой выяв-
ляются различные дефекты прессования. Наиболее распространен-
ные виды брака: выкрашивание, расслой, брак
по навеске (допустимое отклонение от заданной навески не
более ±1,5%), брак по размерам (конусность шта-
бика) [33].
325
ных приборов АРШ-210 представляет собой трехпозиционный
программный регулятор тока с электронной следящей системой.
Программа задается сменными профилированными кулачками.
Режим сварки автоматически записывается на ленту [33].
В результате сварки пористость молибденовых штабиков сни-
жается с 40% (для спрессованных штабиков) до 6—9,7%, что
отвечает плотности 9,7—9,2 г!см*. Меньшая плотность характерна
для штабиков больших сече-
ний.
Трудности получения од-
нородных по плотности шта-
биков больших сечений объяс-
няются значительным темпе-
ратурным градиентом по се-
чению штабика и по длине.
В центре штабика темпера-
тура может быть на 100—
150° С выше, чем на поверх-
ности, интенсивно излучаю-
щей тепло. Поэтому перифе-
рийные части штабика оказы-
ваются недостаточно уплот-
ненными. При увеличении
силы тока в центральной
Рис. 136. Типичная структура спеченного части штабика достигается
молибденового штабика. Х250 температура плавления —
получается штабик с кана-
лом. Ввиду этого штабики сечением 60x60 мм приходится спе-
кать при пониженной температуре (~80% от тока переплавки).
Их используют для изготовления расходуемых электродов элек-
тродуговых печей).
Структура качественных спеченных штабиков мелкозернистая
однородная с числом зерен 25.00—5000 на 1 мм2. Типичная струк-
тура спеченного штабика показана на рис. 136.
Помимо определения плотности штабиков, для оценки их ка-
чества практикуют проверку их пористости по «чернильной пробе»..
Достаточно плотный штабик не впитывает чернил, нанесенных на
его поверхности. Кроме того, проводят технологическое опробова-
ние (7—10 штабиков от партии) обрабатываемости спеченных шта-
биков (ковка, протяжка).
Поведение примесей при «сварке» штабиков. При сварке шта-
бики очищаются от ряда примесей в результате испарения лету-
чих окислов, а также металлов. Однако поведение примесей изу-
чено недостаточно. В работе Бурцевой и Клячко [12] установлено,,
что в процессе сварки (при максимальной температуре сварки
2350° С) удаляется 70—75%- К идо 70% Са. От мышьяка и фосфора
происходит полная очистка.
328
бика (примерно на 12—15%). Подвижность нижнего контакта
обеспечивается подводом тока с помощью гибких шин. Для натя-
жения штабика служит груз, присоединяемый к нижнему кон-
такту через систему блоков.
Поскольку электросопротивление молибденовых штабиков от-
носительно низкое, нагрев их до 2300—2400° С требует значитель-
ной силы тока при низком напряжении. Так, для штабиков 18X
X 18x600 мм максимальная сила тока около 4500 а, а для штаби-
ков больших сечений (40x40 и 60x60 мм) 8000—12 000 а при
напряжении на концах штабика от 10 до 20 в. В соответствии с этим
сварочные аппараты питают от понижающего трансформатора,
а плавную регулировку напряжения осуществляют автотрансфор-
матором.
Подводимая электроэнергия расходуется в основном на излу-
чение нагретой поверхностью штабика и зажимных щипцов. Ин-
тенсивность излучения энергии поверхностью молибдена, нагре-
той до 2327° С, равна ~70 emlcM2 (см. табл. 96). Отсюда энергия,
излучаемая штабиками размером 25 X 25 X 600 мм и 40 X 40 X 600 мм,
равна 42 и 67,2 кет соответственно. На практике применяют
аппараты с установочной мощностью (в расчете на один штабик)
100 ква (для штабиков сечением 25 X 25 мм) и 200 ква (%ля штабиков
сечением 40x40 мм) [33].
Кроме описанного выше, в производственной практике приме-
няют аппараты для групповой сварки штабиков (рис. 135). В этом
случае сокращается расход электроэнергии, так как штабики
взаимно экранируют один другой. Последовательное соединение
штабиков позволяет работать при более высоком напряжении,
подводимом к аппарату, и соответственно меньшей силе тока [3].
Температуру сварки устанавливают и регулируют по силе
тока. Для этого на нескольких пробных штабиках отданной партии
определяют силу тока, необходимую для переплавки штабика.
Максимальная температура сварки (2200—2400° С) соответствует
силе тока, составляющей 85—92% от тока переплавки.
Режимы сварки зависят от размеров штабиков и их назначения.
При сварке штабиков сечением от 20 X 20 мм за 8—15 мин увели-
чивают силу тока до максимальной, выдерживают при максималь-
ной силе тока 6—20 мин и выключают ток. При сварке штабиков
сечением 40x40 и 60x60 мм силу тока плавно поднимают за 60—
80 мин с последующей выдержкой при максимальной силе тока
50—70 мин. По данным работы [3], на заводе Metallwerke Plan-
see (Австрия) в одном сварочном аппарате сваривают 14 молибде-
новых штабиков размером 20x20x300 мм. В течение 25 мин
силу тока поднимают до максимальной (4200 а при 56 з) с последую-
щей выдержкой в течение 30 мин при максимальной температуре.
В последние годы на заводах СССР введено автоматическое
управление режимом сварки штабиков с помощью программного
регулирования проходящего через них тока. Один из разработан-
327
Спекание в вакуумных печах с косвенным нагревом
Способ спекания штабиков в описанных выше контактных
(«сварочных») аппаратах имеет существенные недостатки [28]:
1. Высокий расход электроэнергии, обусловленный необходи-
мостью применения регулировочных трансформаторов, низких на-
пряжений и больших токов, а также значительными теплопоте-
рями, связанными с отсутствием теплоизоляции и присутствием
в аппарате водорода, обладающего высоким коэффициентом тепло-
проводности.
2. Необходимость отрезать недостаточно спеченные концы
штабиков, зажатые в водоохлаждаемых контактах (отходы со-
ставляют 3—5%).
3. Наличие градиента температуры по сечению и по длине
штабика.
4. Низкая производительность, невозможность осуществления
автоматизированного непрерывного процесса.
5. Невозможность спекания изделий любой формы.
В связи с этим советсткие исследователи разработали техноло-
гию спекания молибденовых штабиков в вакуумных печах косвен-
ного нагрева с графитовыми нагревателями [28, 29].
Первоначально исследование спекания молибденовых штаби-
ков было проведено в опытной вакуумной печи, конструкция кото-
рой описана в работе [29] (рис. 137), В печи применен графитовый
нагреватель (диаметр 200 мм, высота 880 мм) с питанием трехфаз-
ным током. Нагреватель подвешен на контактных башмаках, к ко-
торым прижимается стальными пластинами. Нижняя часть на-
гревателя может свободно расширяться при нагреве. Теплоизо-
ляция состоит из слоя ^графитовой крупки, засыпанной между
двумя цилиндрами: внутренним — графитовым и внешним —
стальным. Другие детали конструкции ясны из рис. 137. Вакуум
в печи порядка 10~3—10"4 мм рт. ст. создается форвакуумнььм
насосом ВН-4 с газобалластным устройством и бустерным на-
сосом БН-1500.
Спекаемые молибденовые штабики устанавливали в графито-
вом контейнере на подложку из нескольких листов молибдена и
поддерживали в вертикальном положении при помощи молибде-
новых проволок, образующих ячейки.
Штабики размером 18x18x450 мм вполне удовлетворитель-
ного качества получались при спекании в течение 1 ч при 1800° С
или 2 ч при 1650° С. Их обрабатывали по обычным режимам с про-
тяжкой проволоки диаметром до 0,03—0,05 мм.
Отмечено, что после спекания в вакууме штабики несколько
пластичней, чем обычные, спеченные в водороде в аппаратах кон-
тактного типа.
В дальнейшем была сконструирована высокопроизводительная
механизированная методическая вакуумная электропечь для спе-
329
кания штабиков и пластин. Принцип ее действия ясен из рис. 138,
Печь состоит из камер загрузки, предварительного спекания, охла-
ждения и разгрузки. Перед камерами загрузки и разгрузки
имеются шлюзовые камеры [14, с. 204].
Камера спекания футерована графитовыми блоками и тепло-
изолирована графитовой крупкой.
Графитовые контейнеры с уложенными в них молибденовыми
штабиками периодически передвигаются с помощью механического
Рис. 137. Опытная вакуумная печь с графитовыми нагрева-
телями:
1 — кожух; 2 — экраны; 3 — графитовая засыпка; 4 — нагрева-
тели; 5 — пробка с экранами; 6 — смотровое отверстие; 7 — токо-
подвод; 8 — верхняя крышка; 9 — контейнер со штабиками; 10 —
подставка; 11 — бустерный насос; 12 — форвакуумный насос
толкателя вдоль печи на ширину одного контейнера, перемещен-
ного в камеру разгрузки. На освободившееся место из камеры
загрузки вводится новый контейнер. Для нагревания используют
графитовые стержни, расположенные над и под контейнерами
со штабиками.
Для предохранения молибдена от науглероживания внутренние
стенки контейнера обмазывают смесью 60% молибденового по-
рошка и 40% бакелитового лака. После сушки и выдержки в ва-
кууме при 1850—1950° С на графите образуется плотный слой
молибдена в смеси с карбидом, устраняющий науглероживание
штабиков в процессе спекания. Штабики свободно укладывают на
дно контейнера в несколько рядов, причем верхние ряды уклады-
330
вают на графитовые пластины, покрытые предохранительным слоем
науглероженного молибдена. При спекании крупных штабиков
(35x35 или 40x40 мм), спрессованных в стальных пресс-формах
одностороннего прессования, иногда наблюдаются искривления и
поперечные разрывы, обусловленные неоднородностью плотности
по высоте. Эти явления отсутствуют у крупных штабиков, полу-
ченных методом гидростатического прессования.
Рис. 138. Схема устройства вакуумной методической §печи
для спекания молибденовых заготовок:
/ — камера загрузки; 2 — предварительная , камера; 3 — камера
спекания; 4 — камера охлаждения; 5 — камера разгрузки; 6 —
контейнеры со штабиками; 7 — шлюзы
Для спекания штабиков принят следующий режим: темпера-
тура 1900—1950° С, продвижка контейнера на 200 мм через каж-
дые 1,5 ч. В. этом случае штабики находятся в рабочей зоне в те-
чение 6—8 ч [14, с. 204].
Штабики имеют однородную структуру по сечению и длине.
Число зерен на 1 мм2 в среднем равно 3000. Плотность штабиков
9,7—9,85 г!см3. Среднее содержание углерода в штабиках 0,001%,
поверхностный слой толщиной около 1 мм содержит 0,005—
0,008% С.
Содержание газов N + О + Н в штабиках 0,0009%, тогда как
при спекании в водороде их содержание составляет 0,002%.
В описанной печи, кроме штабиков, спекают пластины для про-
катки молибденовой ленты.
Мощность печи 380 кет, удельный расход электроэнергии
14] квт-ч на 1 кг молибдена. Печь занимает площадь 80 м2.
Спекание крупных спрессованных заготовок
Крупные заготовки массой 50—200 кг, "полученные гидроста-
тическим прессованием, спекают в печах с косвенным нагревом.
Для этой цели используют муфельные печи с нагревателями со-
331
противления или индукционные печи. Создание таких печей для
рабочих температур 2200—2300° С встречает существенные за-
труднения. Ввиду этого спекание крупных заготовок проводят
при температурах 1700—1850° С, что влечет за собой увеличение
времени выдержки при температуре спекания. В зависимости от
размеров спекаемой молибденовой заготовки и температуры (в пре-
делах температур 1700—1850° С) продолжительность спекания
в водороде до максимального уплотнения составляет 3—15 ч
[14, 27].
В патентах фирмы «Вестингауз» с целью активирования про-
цесса предложено спекать заготовки молибдена в атмосфере влаж-
ного водорода [22]. По утверждению авторов спекание во влажном
водороде (насыщенном парами воды при 20—40° С) при темпера-
турах 1600—1700° С позволяет получить заготовки плотностью
10 г!см? при времени спекания 1,5—3 ч. Высказывается предполо-
жение, что активирование спекания во влажном водороде объяс-
няется протеканием обратимой реакции
Мо + 2Н2О^Г.МоО2 + 2Н2.
На поверхности мелких зерен (обладающих большим запасом
свободной поверхностной энергии) эта реакция может протекать
в сторону окисления, тогда как на поверхности более крупных зе-
рен пары МоО2 будут восстанавливаться. В результате процессов
восстановления — окисления ускоряется перенос атомов с по-
верхности мелких частиц к крупным, что активизирует спекание.
Снижение температуры спекания приводит к получению мелко-
зернистых заготовок с числом зерен около 28 000 на 1 мм2.
Некоторые исследователи не получили столь высокого эффекта
действия атмосферы влажного водорода [3] или вовсе его не обна-
ружили [27]. Кроме того, авторы работы [27], изучавшие спе-
кание молибденовых штабиков при низких температурах, нашлщ
что наибольшие степени уплотнения достигаются при спекании
в осушенном водороде (влажность по точке росы 0°), тогда как
с увеличением содержания влаги в водороде степень уплотнения
снижалась. Этот вопрос нуждается в дальнейшем изучении.
На рис. 139 показана муфельная печь для спекания крупных
заготовок молибдена размером 100х 100х 1200 и 200Х50Х 1200мм*
применяемая на заводе в Австрии [23, 26]. В качестве огнеупоров,
используют кирпич из А12О3 и ZrO2.
Молибденовый нагреватель расположен под сводом печи. Спрес-
сованные заготовки укладывают в толстостенные молибденовые
лодочки на слой подсыпки из окиси алюминия. Процесс спека-
ния длится в течение «многих часов» (точные данные не сообща-
ются) [26].
При спекании особое внимание обращают на подкладки под
заготовки, которые должны обеспечить свободную циркуляцию
332
газов вокруг заготовки, а также ее перемещение по подкладке
в процессе усадки во избежание появления поперечных трещин.
Этим требованиям удовлетворяет подкладка из тонкого молибде-
нового листа, гофрированного поперек направления усадки [22].
Созданы индукционные печи для спекания спеченных загото-
вок молибдена в вакууме [24] или в атмосфере водорода [13].
Для сопоставления спекания в вакууме и в водороде нет достаточ-
ных данных.
При одинаковой температуре спекания заготовки, спеченные
в вакууме, имеют меньшую плотность, чем спеченные в водороде.
Рис. 139. Схематический чертеж
муфельной печи для спекания круп-
ных заготовок молибдена:
1 — молибденовые крючки; 2 — тепло-
изоляция; 3 — огнеупорная футеровка
(А12О3); 4 — отверстие для измерения
температуры; 5 — молибденовые нагре-
ватели; 6 — молибденовая заготовка;
7 — водяная рубашка
Для достижения плотности 9,6—9,7 г!см3 в водородной атмосфере
можно проводить спекание при температурах 1800—1850° С,
тогда как в вакууме 10"3—10“4 мм рт, ст, температура спекания
должна быть на 200—300 град выше. По всей вероятности, при
спекании в вакууме полнее удаляется примесь кислорода. Так, по
данным работы [7], после спекания прессовок из молибденового
порошка в водороде и в вакууме содержание кислорода в образцах
составляло 0,014 и 0,007% соответственно.
На рис. 140 показана одна из конструкций индукционных печей
для спекания молибденовых заготовок в атмосфере водорода.
Кварцевая труба 16 (диаметром 425 мм, высотой 1100 мм) опи-
рается на резиновую прокладку в выточке нижней крышки 9,
Сверху труба закрыта кольцом 3 и крышкой 2. Крышки и кольцо
изготовлены из меди и охлаждаются водой. Вокруг кварцевой
трубы расположен индуктор 13, который нагревает до нужной
температуры молибденовый цилиндр 15, установленный на под-
333
Рис. 140. Схема индукционной печи для спекания заготовок в водороде:
1 смотровой патрубок; 2 — верхняя крышка; 3 — прижимное кольцо; 4 — кожух
индуктора; 5 — кирпичи из двуокиси циркония; 6 — шамотные подставки; 7 — стол
печи; 8 поддон; 9 — нижняя крышка; 10 — трубчатые стяжки; 11 — теплоизоляция;
** ^алундовая трубка; 13 — индуктор; 14 — молибденовая заготовка; 15 — молибде-
новый цилиндр (нагреватель); 16 — кварцевая трубка; 17 — молибденовая крышка;
18 — прихват; 19 — струбцинка
ставке из двуокиси циркония. Спрессованную заготовку устанав-
ливают на столик из молибдена. Теплоизолятором служит засыпка
из крупки плавленой окиси алюминия (корекс).
В индукционной печи спекали заготовки молибдена массой
25—30 кг при температуре 1800—1850° С. Общее время нагрева
составило 30 ч, из них 15 ч при максимальной температуре.
Было установлено, что при содержании в исходных порошках
0,017—0,02% щелочных металлов наблюдается неоднородность
плотности спеченной заготовки, вызывающая образование трещин
во внутренних зонах заготовки при ковке.
Примеси щелочных металлов, удаляющиеся в процессе нагрева
заготовки, интенсифицируют спекание наружных слоев заготовки
при низких температурах. Это затрудняет удаление примесей
из внутренних слоев. Возникающее внутреннее давление препятст-
вует усадке, иногда наблюдается растрескивание заготовки.
Снижение содержания щелочных металлов в молибденовом по-
рошке до 0,005—0,007% устраняет неоднородность в плотности
спеченных заготовок, которая колеблется в пределах 9,6—9,9 г!см3
[14, с. 330].
Описанная выше индукционная печь имеет существенные не-
достатки. Масса спекаемых заготовок не превышает 25—30 кг,
так как габариты печи определяются диаметром выпускаемых
кварцевых труб (425 мм). При работе в атмосфере водорода квар-
цевые трубы нагреваются до 900—1000° С, что приводит к быстрому
выводу их из строя (растрескивание, расстеклование). В связи
с этим разработаны индукционные печи без кварцевой трубы,
в которых можно спекать заготовки массой до 300 кг. Для герме-
тичности рабочего пространства в такой печи наружную поверх-
ность индуктора покрывают слоем пластика [13 \
Спекание молибденовых заготовок из порошков молибдена
с добавками легирующих элементов
Изучалось влияние ряда легирующих элементов на свойства
спеченных молибденовых заготовок (температуру рекристаллиза-
ции и перехода из вязкого состояния в хрупкое, длительную проч-
ность и др.) [7; 31; 14, с. 323]. .
В работте Скотта и др. [7] легирующие добавки порошков
титана, циркония, тантала и ванадия в количестве от 0,1 до 2%
перемешивали с порошком молибдена в защитной атмосфере ар-
гона. Отмечается важность использования свежевосстановлен-
ного порошка молибдена и его транспортировки и хранения под
аргоном для снижения содержания в легированном молибдене
примесей кислорода и азота. Порошки прессовали в заготовки
размером 19x38x200 мм под давлением 1,6—3,2 Т1см* и спекали
в вакууме в течение 4 ч при 2000—2300° С.
Основное количество газов выделялось в процессе подъема
температуры (время подъема до 2 ч). Заготовки имели относитель-
335
ную плотность не ниже 95%. Их прокатывали в горячем состоянии
(нагрев под прокатку 1100—1000° С). Общее обжатие после ряда
пропусков достигало 85—90%.
Борок отмечает, что введение легирующих добавок высоко-
реакционных металлов (титан, цирконий, ванадий и др.) ме-
ханическим смешиванием порошков не дает удовлетворительных
результатов. В связи с этим в ЦНИИЧМ был разработан способ
получения порошков сплавов на основе молибдена, сущность ко-
торого заключается в том, что смесь порошка молибдена с окис-
лами (TiO2, ZrO2 и др.), взятыми в необходимых соотношениях,
восстанавливают гидридом кальция при температуре 1150—1175° С.
При этом образуются частицы сплава. Полученный порошок отде-
ляют от окиси кальция, растворяя ее в слабых растворах кислот,
и затем высушивают. Содержание кислорода в таком порошке низ-
кое и он хорошо прессуется. Гранулометрический состав порош-
ков регулируют, подбирая состав шихты и условия восстановле-
ния [13; 11, с. 69] *. Состав порошков и другие их характеристики
приведены в работе [14, с. 323]. Порошки прессовали в заго-
товки в аппарате гидростатического прессования под давлением
0,9 Т/см2. Спекание проводили в вакууме 1—5-10“3 мм рт. ст.
После выдержки при 2000° С в течение 5 ч спеченные заготовки
имели плотность 9,5—9,8 г!см3.
В процессе спекания в вакууме удаляются кислород, азот и
водород, но в разной степени, зависящей от введенного легирую-
щего элемента.
Поскольку растворимость кислорода в молибдене весьма низ-
кая (~4-10“4%), большая часть кислорода находится в порошке
в виде окисных пленок МоО2. В процессе спекания в вакууме при
температурах выше 800° С кислород может удаляться из спекае-
мой заготовки в результате реакции диспропорционирования МоО2
с образованием летучей МоО3 и взаимодействия двуокиси с угле-
родом:
ЗМо02тв —» Мотв + 2МоО3газ,
МоО2тв + 2С Мо + 2СО.
Часть кислорода при температурах выше 1200° С может свя-
заться в прочные окислы легирующего элемента (титана, цирко-
ния и др.). Посторонние окислы в отличие от МоО2 располагаются
не в виде сплошной пленки по границам зерен, препятствующей
скольжению и деформации кристалла, а в виде разрозненных
мелких или крупных шаровидных включений, которые не оказы-
вают существенного влияния на пластические свойства металла.
Поэтому такие сильные раскислители, как титан и цирконий,
хотя и не способствуют удалению кислорода в процессе спекания
* Б. А. Борок, и др. Авт. свид. СССР № 127029, 1957.
336
молибденовой заготовки, обезвреживают его действие из-за лик-
видации сплошных окисных пленок по границам зерен.
Водород наиболее полно удаляется при спекании в вакууме.
Степень удаления азота зависит от введенного легирующего эле-
мента. Например, в присутствии лантана и титана часть азота
удерживается, вероятно, в виде нитридов.
Авторы работы [14, с. 323] для более полной дегазации реко-
мендуют медленный подъем температуры спекаемой заготовки
с тем, чтобы примеси кислорода, азота и водорода удалить при
сохранении открытой пористости и при температурах, когда диф-
фузионная подвижность легирующих элементов мала.
Влияние различных легирующих элементов на механические
свойства молибдена рассмотрено в гл. XIV.
Для повышения прочности и температуры рекристаллизации
в некоторые сорта молибдена, используемого в электровакуумной
технике, вводят кремнещелочную присадку (молибден марки МК)
в количестве 0,2—0,3%. Присадку вводят в виде силиката калия.
Для равномерного распределения присадки двуокись молибдена
пропитывают раствором силиката калия, полученную пасту сушат
и затем восстанавливают МоО2 водородом. В процессе спекания
при температурах 2000—2200° С большая часть кремнещелочной
присадки удаляется. Однако оставшиеся на границах зерен пленки
кремнещелочной присадки влияют на структуру рекристаллизо-
ванной молибденовой проволоки.
22 а. н. 3ел.икман
ГЛАВА XIII
ПЛАВКА МОЛИБДЕНА
Для получения крупных заготовок молибдена массой 500—
2000 кг, необходимых для прокатки листов большого размера,
вытяжки труб и других изделий, к началу 50-х годов была раз-
работана и получила распространение в промышленной практике
дуговая плавка молибдена с расходуемым электродом, и примерно
10 лет спустя — электронно-лучевая плавка.
Теория дуговой и электронно-лучевой плавок и конструктив-
ные варианты плавильных агрегатов освещены в специальных
монографиях [25, 36, 38].
55. ДУГОВАЯ ПЛАВКА
Конструкция печей и режимы плавки
В современных дуговых печах металл плавится в дуге, возни-
кающей между верхним (расходуемым) электродом и нижним
электродом — расплавленным металлом, находящимся в охла-
ждаемой медной изложнице (кристаллизаторе) (рис. 141). Вслед-
ствие высокой теплопроводности меди и быстрого отвода тепла
водой, поступающей в рубашку кристаллизатора, жидкий металл,
соприкасаясь со стенкой, быстро.затвердевает и плавка фактически
идет в гарниссаже из молибдена. Это исключает загрязнение мо-
либдена медью. Плавку молибдена большей частью ведут на по-
стоянном токе при полярности: расходуемый электрод — катод,
расплав — анод. Для выпрямления тока служат мотор-генера-
торы или мощные селеновые, германиевые или кремниевые вы-
прямители. Обратная полярность (положительный верхний элек-
трод) связана с большей вероятностью переброса дуги на стенку
кристаллизатора. Это может вызвать прожог стенки или ее ча-
стичное разъедание.
338
Молибден можно плавить также и в' ’дуге переменного тока.
При работе на постоянном токе температура катода существенно
не меняется во времени, что обеспечивает постоянство эмиссии
электронов. Между тем из-за синусоидальной формы крирой пере-
менного тока температура катода во времени изменяется. Однако
при плавке наиболее тугоплавких металлов (W, Та, Мо) при опре-
деленной плотности тока дуга достаточно стабильна, что объяс-
няется сохранением на электродах высокой температуры, необхо-
димой для поддержания дугового разряда [25, 40].
Рис. 141. Схема дуговой вакуумной
печи с расходуемым электродом:
1 — вакуумное уплотнение штока; 2 —
вместилище расходуемого электрода; 3 —
подвижной шток, несущий электрод; 4 —
электроде держатель; 5 — корпус печи;
6 — патрубок к вакуумному насосу; 7 —
расходуемый электрод; 8 — медный охла-
ждаемый кристаллизатор; 9 — водяная
рубашка; 10 — токоподвод к кристаллиза-
тору; И — смотровое отверстие; 12—токо-
подвод к электроду
Вода
Вода
t I
Вода
Наиболее распространены печи, в которых электрод предвари-
тельно изготовляют из спеченных молибденовых штабиков. Сты-
ковой сваркой штабики соединяют в прутки длиной 1—2,5 м.
Затем их связывают в пакет (4—16 прутков и больше в зависимости
от сечения штабиков и диаметра кристаллизатора) [34; 29, с. 65].
Электроды могут быть также изготовлены спеканием спрессо-
ванных цилиндрических брикетов небольшой высоты в печах
с использованием индукционного нагрева. Затем брикеты сое-
диняют в электрод.
Расходуемый электрод перемещают при плавке и подводят
к нему ток с помощью охлаждаемого штока, на конце которого
закреплен электрод (рис. 141), для подвода тока и перемещения
электрода применяют также ролики (рис. 142).
Плавку ведут с глухим кристаллизатором (рис. 141) или с вытя-
гиванием слитка, путем опускания поддона (рис. 142). Плавка
22* 339
с вытягиванием слитка обеспечивает лучшую откачку выделяющих-
ся газов, так как зона дуги в этом случае все время находится
в верхней части кристаллизатора, тогда как при плавке с глухим
кристаллизатором газы откачиваются из глубокого кольцевого
зазора между электродом и стенками кристаллизатора.
Перед плавкой на дно кристалли-
затора кладут молибденовый диск.
После создания в печи вакуума
(10-3—10-4 мм рт. ст.) электрод
опускают для зажигания дуги.
Рис. 142. Схема (а) и внешний вид (6) дуговой печи на предприя-
тии в Австрии (Планзее, Реутте):
/ — патрубок, подключаемый к вакуумной~системе; 2 — слиток; 3 — водо-
охлаждаемый кристаллизатор; 4 — механизм опускания слитка; 5 — пода-
ющие электрод ролики; 6 — приспособление для разгрузки; 7 — расхо-
дуемый электрод
В США в 1950 г. в результате работ, проводившихся с 1943 г.,
создана сложная плавильная установка, в которой электрод из
молибденового порошка прессуют и спекают непосредственно
в плавильной камере печи (рис. 143). Конструкция и работа уста-
новки УПСП 1 детально описаны в работах [39, 41].
Электрод шестигранной формы прессуют слоями высотой от 6
до 30 мм с помощью гидравлической системы в пресс-форме слож-
ной конструкции. Спрессованный электрод проходит между сколь-
зящими контактами (один из них расположен непосредственно
1 УПСП — установка прессования, спекания и плавки.
340
под пресс-формой) и спекается прямым пропусканием тока, при-
обретая достаточную прочность, чтобы выдержать массу электрода
длиной до 1,5 м.
Наиболее сложная часть установки — механизм формования
электрода. Шестигранная пресс-форма состоит из шести пластин,
из которых три могут реверсивно перемещаться в стальном блоке
на расстояние до 1,5 мм, что регулируется горизонтальным ги-
дравлическим цилиндром. Когда в процессе прессования давление
в вертикальном цилиндре повышается (вследствие сопротивления
прессуемого столбика) до заданного значения, давление в гори-
зонтальном цилиндре снижается и брикет перемещается вниз
до полного хода плунжера. Таким образом, скорость подачи элек-
трода на плавление непосредственно связана с толщиной прессуе-
мого брикета. Полный цикл прессования брикета длится 3 сек*
за минуту прессуется 20 брикетов.
Дуга установки УПСП работает на переменном токе. Для
зажигания дуги на дно кристаллизатора укладывают в форме
конуса молибденовую стружку. При соприкосновении электрода
со стружкой возникает короткое замыкание, нагревающее верхний
слой стружки и конец электрода до температуры плавления. В об-
разующемся зазоре возникает дуга, которая далее горит непре-
рывно на протяжении всей плавки.
В плавильном агрегате подобного типа выплавляют слитки
молибдена диаметром до 305 мм и массой 910 кг со скоростью 7,3—
9,1 кг! мин.
Описанн'ая установка весьма сложна, так как все ее части
должны быть смонтированы в вакуумной камере и все операции
(прессование, спекание, плавка) должны быть строго синхронизиро-
ваны. Так как в УПСП предусмотрено движение электрода только
вниз, подъем электрода, если дуга не загорелась, невозможен.
Это требует сложной системы автоматического управления, обес-
печивающей зажигание дуги и непрерывность ее горения в про-
цессе плавки.
Молибден в дуговых печах плавят при давлении остаточных
газов в камере печи 10”2—10 ”4 мм рт. ст. Давление в кристалли-
заторе над расплавом примерно на порядок выше, чем в камере.
Дуга горит в основном в результате присутствия в узком за-
зоре между расплавом и расходуемым электродом ионизированных
паров молибдена. Это следует из значительно меньшей величины
потенциала ионизации молибдена и молекул газов, а также малой
зависимости напряжения на дуге от остаточного давления в печи
(в пределах 10”1—10”4 мм рт. ст.) [25, с. 12].
Ниже приведены потенциалы первой ионизации, эв:
Молибден ...........7,2
Азот................12,4
Кислород ...........13,5
Водород............13,54
341
В процессе дуговой плавки необходимо обеспечить условия
стабильного горения дуги и исключить образование побочных
дуг между электродом и стенкой кристаллизатора.
Одно из таких условий — работа на короткой дуге (в пределах
20—35 мм). Длина дуги не должна превышать величину зазора
между катодом и стенкой тигля во избежание переброса дуги на
стенку. В зависимости от диаметра электрода зазор между элек-
тродом и кристаллизатором поддерживают примерно в пределах
30—40 мм [25].
При длине дуги 20—35 мм и разрежении 10“2—10"4 мм рт. ст.
напряжение на дуге составляет 30—40 в.
Автоматическая регулировка работы печи основана на зависи-
мости между напряжением и длиной дуги: при повышении на-
пряжения увеличивают скорость подачи электрода, при пони-
жении — уменьшают скорость подачи.
Для перемешивания жидкого металла, способствующего полу-
чению слитка однородного состава, применяют магнитную ка-
тушку (соленоид), устанавливаемую аксиально по отношению
к кристаллизатору. Кроме того, взаимодействие поля соленоида
с дугой позволяет предотвратить смещение столба дуги и стабили-
зировать дуговой разряд.
При плавке в дуговой печи с расходуемым электродом пере-
грев металла, находящегося в жидком состоянии (лунка), по от-
ношению к температуре его плавления невелик. Действительно,
перегрев молибдена на катоде не может быть значительным, так
как по достижении температуры, лишь несколько превышающей
точку плавления (реально при 2675° С), капли расплавленного
металла стекают с катода на расплав в кристаллизаторе — анод.
Температура анода несколько выше, чем катода, вследствие бом-
бардировки его электронами. По данным работы [25], температура
жидкого молибдена на аноде достигает 2800° С, что лишь на 180 град
выше точки его плавления. Глубина жидкой ванны возрастает
с увеличением плотности тока. Большей частью при плавке мо-
либдена отношение глубины жидкой лунки к диаметру кристал-
лизатора равно примерно 0,5—0,7.
Раскисление и легирование молибдена
Механические свойства плавленого молибдена, его обрабаты-
ваемость давлением в большой степени зависят от содержания при-
месей, в особенности примеси кислорода. Поскольку слитки мо-
либдена имеют крупнокристаллическую структуру (следова-
тельно, суммарная поверхность зерен относительно мала), уже
при весьма низком содержании кислорода, равном 0,0006 % , окислы
молибдена образуют сплошную пленку на границах зерен, что
обусловливает плохую обрабатываемость слитков. Снижение со-
держания кислорода достигается добавками раскислителей.
342
Из испытанных раскислителей (Al, Ti, Zr, С) наиболее эффек-
тивным при вакуумной дуговой плавке молибдена оказался уг-
лерод [37, с. 5; 42].
Для успешного раскисления рекомендуется использовать вы-
сокочистый измельченный графит, который вводят в небольшом
избытке по отношению
к необходимому для свя-
зывания кислорода. Избы-
точный углерод остается
в виде мелкодисперсных
включений карбида, не
оказывающих существен-
ного влияния на обрабаты-
ваемость слитка.
Более равномерное рас-
пределение раскислите-
ля и эффективное рас-
кисление достигается при
введении углерода в состав
р асходуемого электрода.
Такой способ практикуется
на заводе фирмы «Клай-
макс», использующей печь,
в которой электрод фор-
муется в плавильной ка-
мере (рис. 143).
Исходный молибдено-
вый порошок, содержащий
0,02—0,04% О и 0,002—
0,004% С, смешивают с из-
мельченным графитовым
порошком, вводимым в
шихту в количестве 75—
125 г на 450 кг порошка.
Кроме того, в шихту доба-
вляют до 30% молибдено-
вого скрапа (стружка
после обдирки слитков),
что способствует упрочне-
нию расходуемого элек-
трода и позволяет исполь-
зовать отходы [39].
Рис. 143. Схема дуговой печи фирмы «Клай-
*макс» для плавки молибдена:
1 — гидравлическая система для перемещения
вертикального и горизонтального цилиндров;
2 — сегментная пресс-форма для прессования
шестигранного электрода; 8 — вертикальный ци-
линдр; 4 — горизонтальный цилиндр; 5 — верх-
ний контакт; 6 — нижний контакт; 7 — бункер;
8 — питатель; 9 — спеченный электрод; 10 — кри-
сталлизатор; 11 — электроизоляция; 12 — элек-
троцепь
При выплавке сплавов в шихту вводят также высокосортные
порошки легирующих элементов — титана, циркония, ниобия,
ванадия и др.
Если ввести графит в состав расходуемого электрода по усло-
виям производства невозможно, раскислитель добавляют непо:
343
средственно в жидкую ванну. Однако при введении в жидкую ванну
измельченного графита трудно обеспечить равномерное его рас-
пределение — часть графита всплывает. В связи с этим рекомен-
дуется вводить углерод в виде лигатурного сплава высокой плот-
ности, например сплава молибдена с карбидом молибдена. Сте-
пень раскисления в большой мере зависит от парциального давле-
ния СО над расплавом, следовательно, от остаточного давления
в камере печи. Для эффективного раскисления углеродом необ-
ходимо проводить плавку при остаточном давлении 10“3—
1-0“4 мм рт. ст.
При получении сплавов молибдена легирующие добавки вво-
дят, подобно углероду, либо в состав электрода при его приго-
товлении, либо непосредственно в ванну. Последний способ полу-
чил наибольшее распространение.
В ванну вводят с помощью дозатора смесь порошков легиру-
ющих элементов или гомогенные лигатурные сплавы. В последнем
случае устраняется опасность сегрегации вводимых элементов
по плотности. При выплавке слитков большого диаметра можно вво-
дить в расплав легирующие добавки, а также раскислители в виде
таблеток или небольших кусков.
Однако при любом способе введения легирующих добавок
в жидкую ванну не удается получить достаточно равномерного
распределения легирующей добавки. Как правило, проводят по-
вторную переплавку с получением слитка большего диаметра.
Структура слитков и контроль их чистоты
Как видно из рис. 144, макроструктура слитков плавленого
молибдена столбчатая. Поскольку вертикальная стенка кристал-
лизатора охлаждается, а наиболее горячая часть ванны находится
сверху (под электродом), температурный градиент в направлении,
перпендикулярном к поверхности роста слитка, больше, чем в на-
правлении, параллельном ей. Такие условия благоприятны для
роста столбчатых кристаллов [42].
Для контроля качества слитков проводят химический анализ
на содержание газовых примесей (кислород, азот, водород), угле-
рода и легирующих добавок, а также металлографическое -иссле-
дование изломов (фрактография) и микрошлифов.
Ниже приведено типичное содержание
ках молибдена [39], %:
Нелегированный
молибден (0,02% С)
Кислород............ 0,0003
Водород .............. 0,00002
Азот ............... 0,0001
примесеи газов в слит-
Легированный
молибден (0,46% Ti
и 0,23% С)
0,0001
0,00001
0,001
Содержание углерода при строгой его дозировке на раскисле-
ние колеблется от 0,015 до 0,03%.
344
Удобный и чувствительный способ контроля степени раскисле-
ния слитков молибдена и содержания в них карбидов — исследо-
вание изломов (фрактография) \
На рис. 145 сопоставлены фото-
графии фрактографических образцов
и микрошлифов плавленого молиб-
дена с различной степенью раскисле-
ния и содержания карбидов.
Если при дуговой плавке не вво-
дят углерод для раскисления, то на
границах зерен выявляются пластин-
ки двуокиси молибдена (рис. 145, а).
При введении углерода пластинки
исчезают, двуокись молибдена при-
сутствует в виде мелких включений,
наряду с этим появляются перьеоб-
разные включения карбида молибдена
(рис. 145, б). При более глубоком
раскислении включения двуокиси
исчезают, остаются лишь вклю-
чения карбида молибдена (рис.
145, в).
Рис. 144. Макроструктура слит-
ка молибдена:
а — продольный разрез; б —* попе-
речный разрез
Гарниссажная дуговая плавка с разливкой металла
в изложницу
В последние годы разработан метод гарниссажной плавки^
позволяющей получать слитки с равномерным составом и мелким
зерном [19, 30]. Этот метод состоит в следующем.
Первоначально в медном кристаллизаторе, вокруг которого
расположен соленоид, получают слиток молибдена обычной плав-
кой с расходуемым электродом. Затем вместо расходуемого уста-
навливают нерасходуемый (вольфрамовый) электрод, с помощью
которого расплавляется некоторое количество металла таким обра-
зом, что у стенок изложницы остается слой твердого металла —
гарниссаж. Полученную ванну жидкого металла выдерживают
необходимое время для полного протекания процессов раскисле-
ния, ликвации и дегазации. Затем проводят проплавление и дон-
ную разливку. С этой целью повышают ток соленоида, фокусируя
дугу и устремляя ее тепловое действие в осевом направлении.
Одновременно повышают до максимума ток дуги. В результате
этого глубина жидкой ванны увеличивается и достигает сливного
отверстия, предварительно высверленного в донной части гарнис-
сажа. Металл сливается в толстостенную медную изложницу.
После слива в заготовке образуется полость в виде полусферы,
1 Техника фрактографического исследования описана на стр. 385.
34 &
Рис. 145. Формы присутствия двуокиси и карбида молибдена в литом молибдене.
Х2000:
а, б — сплошные пластинки двуокиси молибдена; в, г — включения двуокиси и карбида
д, е — включения двуокиси молибдена отсутствуют; а, в, д — фрактография; б, г, е
микрошлифы, электролитическая полировка
которая снова заполняется с помощью расходуемого электрода для
повторения процесса.
При сливе в медную изложницу создаются благоприятные усло-
вия для объемной кристаллизации, что приводит к получению рав-
ноосной мелкозернистой структуры слитка. Однородность его
состава обеспечивается выдержкой жидкого металла в течение не-
обходимого времени до проведения проплава. Полученные слитки
хрупки вследствие образования напряженной структуры при бы-
стром охлаждении. Однако высокотемпературный отжиг сообщает
слитку технологическую пластичность. Обработка слитков проще,
чем крупнозернистых слитков дуговой плавки.
Разработаны также методы гарниссажной плавки с поворотным
тиглем со сливом металла в стационарную или вращающуюся
форму. Это позволяет получать фасонные слитки, а также центро-
бежные отливки с мелкой равноосной структурой.
56. ЭЛЕКТРОННО-ЛУЧЕВАЯ ПЛАВКА
Конструкция печей
Нагревание и плавка металлов электронным пучком основаны
на превращении большей части кинетической энергии электронов
при их соударении с поверхностью металла в тепловую энергию.
Некоторая доля электронов при столкновении с нагреваемым ме-
таллом отражается, однако большая их часть поглощается.
Кинетическая энергия поглощенных электронов превращается
в тепловую энергию и энергию рентгеновского излучения, а также
затрачивается на выбивание электронов (вторичная эмиссия).
При разгоняющих напряжениях до 20 кв подавляющая часть
энергии поглощенных электронов превращается в тепловую и лишь
малая доля в энергию рентгеновского излучения [38].
При напряжениях выше 20 кв (и соответственном повышении
скорости электронов) доля энергии, выделяющейся в форме рентге-
новского излучения, возрастает, так как лучи становятся более
жесткими (уменьшается длина волны). Необходимо принимать
меры для защиты персонала от излучения. Поэтому в установках
для электронно-лучевой плавки ограничиваются скоростями элек-
тронов, которые создаются разгоняющими напряжениями не выше
30—35 кв, большей частью применяют напряжения 20 кв.
Плавку электронным лучом ведут в глубоком вакууме (10-4—-
10"6 мм рт. ст.). Высокое разрежение необходимо для того, чтобы
пучок электронов на пути к нагреваемому телу потерял возможно
меньше энергии на столкновения с атомами и молекулами газов,
а также для удаления из расплавленного металла примесей, испа-
ряемых в условиях плавки.
В отличие от дуговых вакуумных печей при плавке электрон-
ным лучом можно осуществить значительный (контролируемый)
347
перегрев жидкого металла и поддерживать металл в жидком со-
стоянии любое заданное время. Это, а также возможность проведе-
ния плавки при весьма низком остаточном давлении, создает
условия для более полной дегазации металла, чем при дуговой
плавке. Дополнительное преимущество состоит в возможности
переплавлять металл в любом виде (штабики, порошок, стружка),
тогда как при дуговой плавке необходимо приготовление расходуе-
мого электрода [25, 38].
Известно несколько конструктивных типов установок для
электронно-лучевой плавки металлов [25, 36, 38]. Опыт их экс-'
плуатации показал преимущества плавки с помощью электронных
пушек.
Плавильные установки этого типа состоят из следующих эле-
ментов:
1) электронной пушки, в которой создается управляемый поток
электронов;
2) плавильной камеры;
3) системы для создания высокого вакуума;
4) источника постоянного тока высокого напряжения.
На рис. 146 показана принципиальная схема плавильной уста-
новки с одной электронной пушкой. Электронная пушка, явля-
ющаяся источником мощного пучка электронов, состоит из катода,
анода, фокусирующей электромагнитной катушки (линзы) и диа-
фрагмы. Катодом может служить накаленная вольфрамовая спи-
раль. Однако обычно применяют более надежные катоды с косвен-
ным подогревом, изготовленные из массивного вольфрамового или
танталового диска. Катоду сообщается высокий отрицательный
потенциал. Поток электронов, эмитируемый катодом, проходит
через полый ускоряющий анод (который заземлен) и фокусиру-
ющую электромагнитную линзу (или систему линз). Пучок вы-
водится в плавильную камеру через сложные диафрагмы, созда-
ющие значительное газодинамическое сопротивление, что позво-
ляет поддерживать в пушке более высокое разрежение, чем в ра-
бочей камере, с помощью независимой вакуумной системы. Диа-
фрагма предотвращает попадание в электронную пушку выделя-
ющихся из металла при плавке газов. В более мощных печах,
предназначенных для плавки крупных слитков, применяют не-
сколько электронных пушек (три, четыре и больше). На рис. 147
показана схема установки с четырьмя электронными пушками.
Материал может поступать на плавку в форме порошка, гранул
(рис. 146) или в виде предварительно приготовленных спрессован-
ных и спеченных заготовок (рис. 147).
Ниже приведены ориентировочные мощности электронно-лу-
чевых печей для выплавки слитков молибдена различного диа-
метра [43]:
Диаметр слитка, мм ... 65 90 130 185 270
Мощность, кет........ 60 120 240 480 900
348
Успешная плавка в электронно-лучевых печах возможна при
обеспечении высокой скорости откачки и остаточном давлении
в плавильной камере порядка 10~5 мм рт. ст.
Для расчетов вакуумных систем при плавке молибдена, по дан-
ным Смелянского и Гуттермана, можно принять количество газов,
Рис. 146. Схема электронно-лучевой
печи с одной аксиальной пушкой:
1 — катод пушки; 2 — анод пушки; 3 —
линия высоковакуумной откачки; 4—элек-
тромагнитная фокусирующая линза; 5 —
диафрагма, разделяющая пушку и плавиль-
ную камеру; 6 — шибер; 7 — плавильная
камера печи; 8 — пучок электронов; 9 —
к вакуумной системе; 10 — выплавляемый
слиток; 11—механизм вытягивания, слитка;
12 -— водоохлаждаемый кристаллизатор;
13 — дозатор подаваемого на плавку
порошка
Рис. 147. Схема плавильной установки
с четырьмя электронными пушками:
1 — электронная пушка; 2 — механизм
подачи заготовки на плавку; 3 — расхо-
дуемая заготовка; 4 — электронный луч;
5 — выплавляемый слиток; 6 — водоохла-
ждаемый кристаллизатор; 7 — плавильная
камера; 8 — механизм вытягивания слитка
выделяющихся из исходного молибдена (спеченные штабики),
равным 0,2—0,25 дм3/кг [40]. Если на электронно-лучевой пере-
плав поступает металл после дуговой плавки, газосодержание
в молибдене значительно ниже — 0,05 дм3/кг. Эго позволяет суще-
ственно увеличить производительность электронно-лучевой печи.
Чистота электронноплавленого молибдена
При температурах 2900—3000° С и давлении 10"6 мм рт. ст.,
которые поддерживают при электронно-лучевой плавке, из жид-
кого молибдена могут быть удалены примеси, давление паров кото-
349
рых выше давления пара молибдена. Среди металлов лишь у ре-
ния, тантала и вольфрама давление пара существенно ниже, чем
давление паров молибдена при температурах 2800—3000° С
(рис. 148).
Если принять, что металлические компоненты образуют в жид-
ком молибдене растворы, близкие к идеальным, можно ориентиро-
вочно рассчитать конечное содержание примеси после плавки при
определенном остаточном давлении и температуре по формуле
WTaHfHbMo Tl Fe
3000 2000 1500 7000
Температура, °C
Рис. 148. Давление пара металлов
(мкм рт. ст.) в зависимости от
температуры
где рост — остаточное давление
газов;
— давление чистого ком-
понента (примеси) при
температуре плавки;
Мп — конечная молярная
доля примеси в рас-
плаве.
В качестве примера можно при-
вести расчет предельного содержа-
ния примеси железа в молибдене
после плавки при 2900° С и оста-
точном давлении — 10"7 ат. Дав-
ление пара железа при температуре 2900° С равно примерно
2 ат. Отсюда остаточное содержание железа:
10“7
jVFe — —-— = 0,5-10"7 мол. доли, или ~3-10"6% (по массе).
23
Химический анализ показывает, что после электронной плавки
содержание железа находится в пределах чувствительности опре-
делений [порядка < 10"4% (по массе)], что согласуется с рас-
четом.
В работе [44] показано, что в результате электронно-лучевой
плавки молибден очищается от подавляющей части примесей,
в частности примесей О, N, С, Fe, Си, Ni, Мп, Со (рис. 149). После
15-жш выдержки в расплавленном состоянии чистота молибдена
повышается с 99,8 до 99,99%.
В процессе плавки кислород и углерод удаляются одновре-
менно, однако содержание кислорода убывает быстрей. Из этого
следует, что кислород может удаляться, не только в виде СО.
Действительно, даже при отсутствии углерода содержание кисло-
рода в молибдене при плавке в вакууме снижается вследствие
испарения низшего окисла [451. Для глубокой очистки от кисло-
рода (до содержаний ^0,0001 %) необходимо вводить раскисли-
тели.
350
Раскисление углеродом происходит, по всей вероятности,
в результате взаимодействия низшего окисла молибдена с карбидом
молибдена
по реакции
MoOow 4
Изменение
ж 2СОгаз
свободной энергии этой реакции при 3000° К AZ
ккал. Отсюда
(210)
К =_______—_______1011’5
Л [МоО2] [Мо2ср 1и ’
Это приближенное значение, так как не учтены значения сво-
бодной энергии растворения двуокиси и карбида в расплавленном
Рис. 149. Изменение концентрации некоторых примесей
в зависимости от выдержки молибдена в расплавлен-
ном состоянии при электронно-лучевой плавке
молибдене. Однако даже в том случае, если значение константы
равновесия на несколько порядков ниже, равновесная концентра-
ция кислорода при раскислении молибдена углеродом ничтожно
малая при остаточном давлении СО, равном 10"5—10~7 ат.
Реально при применении углерода для раскисления содержание
кислорода в молибдене после электронной плавки ^0,0001—
0,0003%. В случае строгой дозировки углерода его содержание
составляет 0,002—0,004%. Между тем при дуговой плавке для
обеспечения глубокого раскисления вводят больше углерода и его
содержание в слитках не ниже 0,015—0,02%.
Изучали раскисление молибдена в процессе электронной плавки
рядом активных металлов. Эффективные раскислители молибдена—-
титан и цирконий. При использовании этих раскислителей кисло-
род удаляется в результате испарения низших окислов TiO и,
возможно, ZrO. Наряду с испарением TiO в процессе плавки испа-
ряется титан, давление пара которого при температурах плавки
молибдена значительно (при 3000° К давление пара титана равно
351
47,3 мм рт. ст.). Вследствие этого титан может испариться прежде,
чем будет достигнуто достаточно глубокое раскисление молибдена.
Давление пара циркония значительно ниже, чем титана (при
3000° К pzr — 0,19 мм рт. ст.), что делает его более эффективным
раскислителем молибдена [45].
Раскисление алюминием при электронной плавке неэффективно
из-за быстрого его испарения. Отмечается перспективность ис-
пользования добавок бора для раскисления молибдена [46].
Содержание углерода, кислорода и водорода в слитках молиб-
дена, полученных электронно-лучевой плавкой, характеризует
табл. 81 по данным Семчишена и Бэрра [37, с. 149]. Авторы отме-
ТА БЛИЦА 8]
СОДЕРЖАНИЕ ПРИМЕСЕЙ ВНЕДРЕНИЯ И ТВЕРДОСТЬ СЛИТКОВ
МОЛИБДЕНА, ПОЛУЧЕННЫХ ЭЛЕКТРОННО-ЛУЧЕВОЙ ПЛАВКОЙ [37]
№ слитка Исходный материал * Содержание в исходном материале., % Число пере- плавок Твердость HV, кГ/мм2 Содержание примесей в слитке, %
С прочие С о н прочие
1 СП 0,003 3 179 0,002 0,0004 0,00018
2 СП 0,038 — 2 187 0,002 0,0003 0,00003 —
3 Деф. 0,023 — 3 176 0,017 0,0008 0,00008 —
4 СП 0,003 0,58 Ti 3 173 0,002 0,0002 0,00001 0,008 Ti **
5 СП 0,016 0,58 Ti 3 179 0,003 0,0002 0,00003 0,008 Ti **
6 СП 0,038 0,58 Ti 2 192 0,006 0,0002 0,00001 0,030 Ti
7 Деф. 0,003 0,15 Al 3 183 0,004 0,0002 0,00001 0,000 Al
8 СП 0,003 0,25 Al 3 189 0,003 0,0003 0,00004 0,000 Al
Молибден вакуумной дуго- 180 0,15 0,0002 0,00001 —
вон плавки <
* СП — спеченный электрод; Деф. — кованый пруток из слитка дуговой плавки.
** Предел чувствительности определения.
чают, что введение алюминия или титана при отсутствии углерода
не приводит к глубокому раскислению. Из табл. 81 можно видеть,
что большая часть введенных титана и алюминия удаляется в про-
цессе плавки. Ниже приведены результаты спектрального анализа
на содержание металлических примесей нелегированного моли-
бдена после электронной плавки (слиток №3, табл. 81), %:
0,0008 А1; 0,0002 Са; <0,0001 Сг; 0,0008 Mg; 0,0001 Fe; <0,0001
Си; <0,001 Pb; 0,003 Si; <0,001 Sn; <0,0001 Ti; <0,0001 Zn;
<0,0001 Ni; <0,01 W.
57. ПОЛУЧЕНИЕ МОНОКРИСТАЛЛИЧЕСКИХ СЛИТКОВ МОЛИБДЕНА
Монокристаллические слитки молибдена высокой чистоты отли-
чаются от поликристаллического молибдена технической чистоты
особыми физическими свойствами. Они пластичны вплоть до ге-
лиевых температур (4,2° К), тогда как обычному молибдену при-
352
суща хладноломкость. Высокая технологическая пластичность
позволяет получать из монокристаллических слитков полуфабри-
каты (проволока, листы и др.) для изготовления деталей электрон-
ных приборов, способных работать при высоких температурах и
сохраняющих пластичность при низких температурах. Монокри-
сталлы молибдена устойчивы при работе в плазме цезия, калия
и других щелочных металлов, что позволяет их использовать для
изготовления катодов термоэлектронных преобразователей. На-
мечаются и другие области применения монокристаллов моли-
бдена. В связи с этим разработаны способы получения монокри-
сталлических слитков молибдена. Наибольшее значение для
промышленного использования имеют два метода получения моно-
кристаллов молибдена — метод зонной плавки и метод вытягива-
ния из расплава (по Чохральскому). Методы получения и свойства
монокристаллов тугоплавких металлов детально освещены в моно-
графии [47].
Зонная плавка
Метод зонной плавки, или зонной перекристаллизации, широко
используют в промышленной практике для глубокой очистки и вы-
ращивания монокристаллов различных материалов. Теория и прак-
тика метода изложены в монографиях [48, 49]. Метод состоит
в перемещении с определенной скоростью узкой расплавленной
зоны вдоль сравнительно длинного слитка (или спрессованного из
порошка прутка). При этом происходит перекристаллизация ме-
талла, сопровождающаяся очисткой выделяющейся из расплава
твердой фазы, обусловленной различием растворимости примеси
в твердой и жидкой фазах и малой скоростью диффузии в твердой
фазе. Кроме того, при проведении процесса в вакууме из жидкой
зоны удаляются примеси, обладающие высоким давлением пара.
Степень возможной очистки от примеси зависит от величины
коэффициента распределения /С = ^тв , представляющего собой
отношение концентраций примеси в твердой и жидкой фазах.
При средней концентрации примеси в исходном материале Со
в первом бесконечно малом объеме выделившейся твердой фазы
концентрация примеси будет равна /СС0. При К < 1 (наиболее
распространенный случай систем, рис. 150, а) КС0 <С0 и, следо-
вательно, выпадающие кристаллы будут содержать меньше при-
меси, чем жидкость. Если К > 1 (менее распространенный слу-
чай, рис. 150, б) КС0 > Со и первые выделившиеся кристаллы
будут обогащены примесью. При К < 1, когда примесь понижает
точку плавления металла, по мере передвижения зоны концентра-
ция примеси будет возрастать вдоль слитка в направлении переме-
щения зоны. При /С >* 1 концентрация примеси будет возрастать
в направлении, противоположном движению зоны. Распределе-
23 А. Н. Зеликман 35»
ние примеси по длине слитка в результате зонной плавки (за один-
проход) приближенно описывается уравнением
Сх = Со [1 -(1 — (211)
где Сх — концентрация примеси на расстоянии х от начала слитка;
Со — начальная концентрация примеси;
К — коэффициент распределения;
L — ширина жидкой зоны.
Уравнение выведено при допущении, что К — величина по-
стоянная, диффузия в твердой фазе отсутствует, а в жидкой фазе
протекает столь быстро, что концентрация примесей одинакова
во всем объеме расплава.
Рис. 150. Части
диаграммы состоя-
ния в области твер-
дых растворов:
а— примесь понижает
температуру плавле-
ния металла; б—при-
месь повышает темпе-
ратуру плавления
При малых значениях К (К <0,1) высокая степень очистки
происходит даже при однократном прохождении зоны. Более
глубокая очистка достигается проведением нескольких последо-
вательных проходов жидкой зоны.
Как видно из рис. 151, зонная плавка менее эффективна для
очистки от примесей, повышающих точку плавления металла
(Х>1).
Зонная цлавка молибдена и других тугоплавких металлов осу-
ществляется по методу «плавающей зоны» с вертикальным распо-
ложением слитка (бестигельная зонная плавка), как показано на
рис. 152. Расплавленная зона удерживается между двумя частями
слитка силами поверхностного натяжения. Максимальная длина
зоны, которая может удерживаться- силами поверхностного натя-
жения, зависит от радиуса заготовки, а также от критического
параметра, определяемого величиной ]Ат/р, где ст — поверхност-
ное натяжение, р — плотность материала [50]. Отношение а/р
при бестигельной зонной плавке должно быть не ниже 100 : 1 [51 ].
Максимальный диаметр получаемых молибденовых монокристал-
лических слитков 18—22 мм при длине 300—350 мм. Длина зоны
обычно не превышает диаметра прутка.
Успешная очистка происходит при оптимальной скорости пе-
ремещения жидкой зоны, которая для молибдена лежит в пределах
0,5—4 mmImuh. Для обеспечения отвода в расплав примесей, кон-
центрирующихся у фронта кристаллизации, перемешивают жид-
кость, вращая заготовку. .
354
Для создания расплавленной зоны используют индукционный
нагрев или нагрев электронным пучком. Последний способ наи-
более распространен. Большей частью используют кольцевые
катоды из вольфрамовой проволоки, перемещаемые вдоль заго-
товки." Для максимального использования эмиссии электронов,
необходимо располагать катод возможно ближе к слитку. При
Рис. 151. Изменение концентрации
примеси при зонной плавке вдоль
слитка при различных значениях К.
Начальная концентрация Со = 1.
х
—-----относительная длина затвер-
жу
девшей части слитка (цифры на кри-
вых;— значения /С)
Рис, 152, Принципиальная схема
зонной плавки с плавающей
зоной:
1 — вакуумная камера; 2—верхний
изолятор; 3 — верхний держатель
образца; 4 — образец (очищаемый
металл); 5—кольцевой электронно-
лучевой нагреватель (вольфрамовый
катод); 6 — расплавленная зона;
7 —нижний держатель образца;
8 — нижний изолятор; 9 — экраны,
на которые подается регулируемый
потенциал для фокусировки пучка
электронов
этом на катоде и фокусирующем электроде (экране) могут конден-
сироваться пары и брызги металла (рис. 153). Это приводит к бы-
строму выходу катода из строя (часто срок службы ограничивается
несколькими часами), особенно в случае, если на зонную плавку
поступает металл со значительным содержанием газовых при-
месей.
Для быстрой замены катода без перерыва плавки служат ка-
тодные шлюзы, позволяющие заменить катод примерно за 15 мин\
или револьверное катодное устройство, с помощью которого катод
заменяют за 1 мин [36]. Разработаны установки, в которых рас-
плавленная зона создается тремя или более электронными пуш-
ками, расположенными симметрично в горизонтальной плоскости.
23* 355
В этом случае пушки закреплены стационарно. Заготовка пере-
мещается по вертикали и вокруг своей оси. Такая система устра-
няет загрязнение металла материалом катода и исключает конден-
сацию паров металла на катоде [52, с. 111].
Для приблизительной оценки потребляе-
v мой мощности на создание расплавленной
ж "ж зоны при плавке прутка диаметром d можно
1 > пользоваться уравнением [32]
fcli®' jF P = Ad + Bd2, (212)
ОCZ где —константа, пропорциональная чет-
। вертой степени температуры плав-
I II ления;
I J В—константа, пропорциональная тепло-
• проводности вблизи точки плавле-
L ния металла.
Рис. 153. Внешний вид
вольфрамового катода
после плавки
Если исходный материал достаточно
чист, в результате прохождения жидкой
зоны вдоль заготовки получается монокри-
сталлический слиток.
На рис 154 показан внешний вид установки для зонной очистки
и выращивания монокристаллов тугоплавких металлов, разработан-
ной в Институте металлургии АН СССР им. А. А. Байкова [53, 54 ].
Рис. 154. Внешний вид установки для зонной очи-
стки и получения монокристаллов
Образец закреплен в молибденовых зажимах с танталовыми
пружинами, позволяющими образцу свободно удлиняться при
нагреве. Кольцевой катод из вольфрамовой проволоки закреплен
в стальных держателях, к которым с помощью гибких шин подво-
дится ток для нагревания катода примерно до 2000° С. Кронштейн
со стальными держателями вертикально перемещается по ходо-
356
вому винту, связанному с электродвигателем. Для фокусировки
электронов, испускаемых катодом, служат две параллельные пла-
стины, расположенные на расстоянии 4—5 мм одна от другой.
Анодная сеть питается выпрямленным током с напряжением до
8 кв и максимальным током 0,8 а.
Вакуумная система установки, состоящая из форвакуумного
насоса, диффузионного паромасляного насоса, ловушки с жидким
азотом и заслонки, обеспечивают разрежение до 8-10"6 мм рт. ст.
Применение паромасляного насоса обусловливает возможность
загрязнения очищаемого металла углеродом вследствие попадания
в рабочую камеру паров масел и продуктов их крекинга. В неко-
торой степени это происходит даже при наличии азотной ловушки.
В связи с этим разработаны установки электронно-лучевой
зонной плавки с системой откачки, включающей цеолитовый сорб-
ционный насос и гетероионный насос [55].
Ниже приведен примерный электрический режим зонной
плавки молибдена при начальном вакууме 5-10"5—6х
Х10"5 мм рт. ст. [47]:
Диаметр заготовки, мм............... 12—15
Диаметр катода, мм ................. '24—27
Число проходов...................... 2—3
Напряжение на катоде, в............. 4,5—8
Разгоняющее напряжение, е .......... 4500—8000
Сила тока накала катода, а ......... 50—80
Сила тока на аноде, а .............. 0,7—1,0
Скорость перемещения зоны, мм/мин . . 0,5—3,0
В процессе плавки происходит значительное испарение моли-
бденовой заготовки, зависящее от скорости перемещения зоны.
Так, стержень молибдена диаметром 10 мм при скорости движения
зоны 0,42 мм/мин, теряет 25% своей массы [56, с. 303]. Следова-
тельно, с повышением скорости до 2 мм!мм потеря уменьшится
примерно до 5%. В этой связи стремятся работать при максимально
возможной скорости перемещения зоны (примерно 2—4 мм!мин}.
Однако при таких скоростях большинство примесей не успевает
оттесниться в расплав, что резко снижает эффективность зонной
очистки и требует увеличения числа проходов. Это недостаток
метода зонной очистки.
По данным Белка [32], при скорости плавки 3 mmImuh только
углерод перераспределяется по длине образца. Очистка от других
примесей происходит преимущественно в результате их испарения
из жидкой зоны.
Высокие пластические свойства монокристаллов молибдена
иллюстрирует рис. 155.
Монокристальность полученного прутка проверяют методом
поверхностного электролитического травления в 5—10%-ном
растворе едкого натра в воде. Травление выявляет на поверхности
357
монокристалла чередующиеся продольные матовые и блестящие
полосы. Кроме того, для контроля монокристальности снимают
лауэграммы. Рентгенографические исследования показали, что
в монокристаллических слитках молибдена (как и других металлов
с объемноцентрированной решеткой) кристалл при электронно-
Рис. 155. Иллюстрация высоких пла-
стических свойств монокристаллов
молибдена при 20° С
лучевой зонной плавке само-
произвольно растет в направле-
нии, близком к (100).
В случае необходимости по-
лучить слиток с заданной ориен-
тировкой используют затравку.
Затравочный монокристалл
с определенной кристаллографи-
ческой плоскостью закрепляют
строго вертикально в нижнем
зажиме, а заготовку — в верх-
нем зажиме. Между ними остав-
ляют небольшой зазор (до 1 мм).
В месте стыка затравки с заго-
товкой создается расплавленная
зона и начинается продвижение
зоны вверх. Растущий кристалл
приобретает кристаллографиче-
скую ориентировку затравки
[47].
Для оценки степени чистоты
монокристаллических слитков
молибдена современные методы химического < анализа недоста-
точно точны и дополняются изучением физических свойств.
. В1 табл. 82 приведено содержание примесей по длине слитка
ТАБЛИЦА 82
СПЕКТРАЛЬНЫЙ АНАЛИЗ МОЛИБДЕНА ПОСЛЕ ЗОННОЙ ПЛАВКИ
Примесь Исходный материал, %-ю-4 Анализ по зонам, %-10 4 Чувстви- тельность анилиза, %-10-4
I II III конечная зона
Углерод .... 80 35 20 55 “Ц 85 2
Кремний .... 20 25 23 10 20 2
Железо ..... 40 <^ <^ 1 <^ 1 0,5
Медь 40 11 8 11 1 0,4
Хром 10 <20 20 <20 .<20 10
Марганец .... <С^2 <^2 <^2 7 2
Никель . . . . . 70 <^ 1 <^ 1 <^ <^ 1 1
Кобальт .... 10 2 2 3 1
Кислород . . . . — ~1 ~1 ~1 -1 -1
358
(по четырем зонам) после зонной очистки заготовки металла дуго-
вой плавки при скорости перемещения зоны 3 mmImuh [52, с. 181 L
Содержание углерода в конце образца выше, чем в исходном,
что указывает на его перераспределение по длине образца. Однако
•общее уменьшение содержания углерода независимо от скорости
зонной плавки указывает на удаление части углерода вследствие
реакции с кислородом.
Содержание железа, меди и никеля уменьшается преимуще-
ственно в результате испарения этих элементов.
Марганец и кобальт испаряются и перераспределяются при
передвижении зоны по длине образца. Содержание кислорода,
азота и водорода в образцах после зонной плавки обычно ниже
10-40/о.
Одним из косвенных методов оценки чистоты металла служит
измерение остаточного электросопротивления — отношения удель-
ного электросопротивления при 300 и 4,2° К (жидкий гелий) х.
Электросопротивление металла слагается из двух составляющих —
первая определяется рассеянием электронов тепловыми колеба-
ниями атомов, вторая — рассеянием электронов на примесях и '
дефектах кристаллов. При температуре жидкого гелия электро-
сопротивление монокристалла в основном определяется приме-
сями, так как тепловое рассеяние электронов незначительно.
Измерения величины К = Рзоо°— для молибдена до и после
Р4,2° К
зонной очистки показали ее возрастание от 360 до 1400 за один
проход и от 360 до 2500 за четыре прохода [48, с. 126].
Показателем низкого содержания примесей внедрения в моно-
кристаллическом слитке молибдена может служить технологиче-
ская проба на нзгиб или гиб с перегибом на 90 или 180° при ком-
натной температуре, сужение площади поперечного сечения при
растяжении и температура перехода из пластичного состояния .
в хрупкое (температура перехода снижается при уменьшении со-
держания примесей внедрения) [47]. Данные о механических
свойствах монокристаллического молибдена приведены в гл. XIV.
Получение монокристаллов методом Чохральского
Метод Чохральского — наиболее распространенный способ
получения монокристаллов германия, кремния и ряда металлов
и соединений, состоит в «вытягивании» монокристаллического
слитка из расплава, поддерживаемого при температуре, близкой
к температуре затвердевания. В начале процесса в расплав опу-
1 Следует, однако, подходить критически к оценке чистоты по этому пока-
зателю. Так, примесь карбида МоС (если он не распался полно на Мо2С и С),
температура перехода которого 9,3° К, может обусловить высокое значение отно-
сительного сопротивления молибдена.
359
скают затравочный монокристалл, закрепленный в держателе.
Затем затравочный кристалл с определенной скоростью вытяги-
вают из расплава с помощью подъемного механизма. Выращивае-
мый монокристаллический слиток имеет ориентацию затравочного
кристалла.
Преимущества получения монокристаллов по методу Чохраль-
ского, по сравнению с описанным выше методом зонной плавки
с плавающей зоной, заключаются в возможности выращивания
слитков большого диаметра и более совершенной структуры.
Рис. 156. Схема установки для вы-
ращивания монокристаллов туго-
плавких металлов по Чохраль-
скому:
1 — катод; 2 — фокусирующий элек-
трод; 3 — держатель фокусирующего»
электрода; 4 — водоохлаждаемый ти-
гель; 5 — поддон; 6 — слиток; 7 —
ванна жидкого металла; 8 — вытяги-
ваемый монокристалл; 9 — магнитная
линза
Метод позволяет, в отличие от зонной плавки, применять сколь
угодно низкие скорости вытягивания, так как металл испаряется
из ванны, что не влияет на диаметр монокристаллического слитка.
Для получения монокристаллов тугоплавких металлов метод
Чохральского ранее не применяли вследствие трудностей создания
ванны жидкого тугоплавкого металла. Применение электронного
нагрева и охлаждаемого медного тигля позволило решить эту
задачу.
На рис. 156 показана принципиальная схема осуществления
процесса [57].
Для получения монокристалла используют предварительно
дегазированный молибден, что позволяет применить кольцевой
катод, обеспечивающий симметричное распределение энергии по
ванне. Слиток исходного материала помещают на молибденовый
поддон, который может вращаться и перемещаться возвратно-
поступательно. Поддон находится в водоохлаждаемой медной
изложнице.
В работе [35] описано получение .монокристаллов молибдена
на небольшой установке с медным тиглем диаметром 80 мм. Исход-
ную загрузку после расплавления электронным нагревателем
выдерживали в жидком состоянии некоторое время до стабилиза-
ции теплового и вакуумного режима. Затем устанавливали темпе-
ратуру ванны несколько выше точки плавления молибдена и
360
в расплав опускали затравку на глубину 1—2 мм. Далее включали
механизм вытяжки. Диаметр кристалла зависит от мощности и диа-
метра электронного пучка, величина которого регулируется с по-
мощью магнитной линзы. Были получены монокристаллы моли-
бдена диаметром 15—20 мм и длиной до 300 мм, которые имели
несколько более высокие характеристики пластичности, чем слитки
бестигельной зонной плавки.
По методу Чохральского можно получать не только цилиндри-
ческие слитки, но также вытягивать из расплава монокристалли-
ческие трубки, пластины и другие профили, что представляет
большой интерес для ряда областей техники.
ГЛАВА XIV
ОБРАБОТКА ДАВЛЕНИЕМ, МЕХАНИЧЕСКИЕ
СВОЙСТВА, МЕТАЛЛОГРАФИЯ МОЛИБДЕНА
58. ОБРАБОТКА ДАВЛЕНИЕМ
Молибден, подобно ряду других металлов с объемноцентри-
рованной кубической решеткой, имеет температурный порог пе-
рехода из пластичного состояния в хрупкое. В зависимости от
скорости деформирования (методики механического испытания),,
величины зерна заготовки металла, наличия и распределения при-
месей (кислород, азот, углерод), присутствия легирующих эле-
ментов температура перехода молибдена в хрупкое состояние
колеблется от 95 до 400° С [2—4,5].
Бехтольд показал, что при обработке молибдена в зависимости
от температуры можно выделить четыре зоны [4]:
1) хрупкую зону, примерно в области температуры ниже
—75° С, в которой металл не пластичен;
2) переходную зону, в интервале от ~150 до —75° С, которая
характеризуется быстрым увеличением предела прочности с од-
новременным падением пластичности при понижении температуры;
3) пластичную зону, в интервале от ~ 150 до —900° С, в кото-
рой молибден имеет относительно низкий предел прочности, вы-
сокую пластичность и волокнистую структуру после обработки;
4) нестабильную зону, выше 900° С, где происходит частичная
рекристаллизация металла.
Чтобы осуществить истинную горячую деформацию молибдена,
при которой рекристаллизация происходит одновременно с дефор-
мацией и в процессе повторных нагреваний, обработку молибдена
следовало бы проводить при температурах 1650—2000° С. Однако*
такие условия обработки трудно достижимы. На практике заго-
товки нагревают под ковку или прокатку при 1200—1400° С.
Это позволяет использовать для подогрева заготовок водородные
печи с молибденовой обмоткой, а также газовые печи. Для этих
целей применяют также соляные ванны и (в случае крупных за-
готовок) индукционный нагрев [1].
362
Для обеспечения необходимой прочности изделий из молибдена
и сплавов на его основе конечные стадии ковки или прокатки необ-
ходимо проводить при температурах ниже температуры рекри- .
сталлизации. При этом степень конечного наклепа должна обеспе-
чивать высокую прочность изделия в условиях эксплуатации.
Обработка спеченных заготовок
Спеченные штабики квадратного сечения со стороной 25 мм
и меньше, предназначенные для производства проволоки, перво-
начально куют на ротационных машинах для получения круглых
прутков, поступающих на протяжку проволоки. Спеченные за-
готовки, из которых получают пластины и ленты, подвергают пло-
Рис. 157. Схема ротационной ковочной машины:
/ — станина; 2 — вал; 3 — ролики; 4 — стальная обойма;
5 — ковочные плашки; 6 — стальные кольца для крепления
обоймы
ской ковке в пластины, поступающие затем на прокатку. Обработка
спеченных штабиков детально рассмотрена в монографии Агте
и Вацека [6].
Ротационная ковка. Принцип действия ротационной ковочной
машины состоит в том, что штабик получает большое число ударов
(10 000—12 000 ударов в минуту) двумя ковочными плашками,
вращающимися с большой скоростью вокруг молибденового прутка
(рис. 157). Ковочная машина состоит из чугунной неподвижной
станины, внутри которой на обойме находятся расположенные
по окружности ролики. В пространстве между роликами вра-
щается вал, имеющий центральный канал, через который прохо-
дит штабик, и прорезь, в которой свободно сидят ковочные плашки.
При вращении вала плашки попеременно отбрасываются от центра
к периферии (центробежной силой) и от периферии к центру (вы-
ступающим роликом), ударяя по штабику. На рабочей части ко-
вочных плашек из быстрорежущей стали сделаны выемки, соответ-
ствующие диаметру обрабатываемого прутка.
Штабик перед ковкой нагревают до 1350—1400° С в установлен-
ной вблизи ковочной машины печи с водородной атмосферой.
Обжатие при ковке составляет 10—12% за проход. По мере обжа-
363
тия заготовок температура ковки снижается от ^1300° С при диа-
метре прутка 20—25 мм до 950—1000° С при диаметре 2,5—3 мм.
Первоначально прутки подают в ковочную машину вручную.
Начиная с определенного диаметра, когда длина прутков увели-
чивается, переходят на механическую подачу и непрерывную
ковку. Пруток проходит через газовую или электрическую печь
и подается транспортером на ковку. Для предотвращения окисле-
ния молибдена и уменьшения износа плашек пруток перед поступ-
лением в печь покрывают аквадагом (смесь тонкого графита с ам-
миачной водой и добавкой сахара).
Рис. 158. Схема установки для волочения молибденовой
проволоки:
1 — ведущий барабан; 2 — фильера; 3 — газовая печь; 4 —
смазочная коробка; 5 — спускной барабан
Трудоемкость операции ротационной ковки и неровность по-
верхности кованых прутков вызвали поиски других путей перво-
начальной обработки спеченных штабиков. Исследования пока-
зали возможность эффективной замены ротационной ковки ги-
дроэкструзией — выдавливанием спеченной заготовки под
воздействием давления жидкости [8]. Оптимальная степень де-
формации при гидроэкструзии спеченных штабиков 80%. Было
установлено, что при гидроэкструзий существенно повышаются
прочностные характеристики молибдена. Так, предел прочности
при степени деформации 90% возрастает в 3,8 раза, тогда как при
ротационной ковке в 2 раза. Пластичное состояние при гидро-
экструзии достигается уже при степени деформации 40—50%,
а в случае ротационной ковки при деформации 80—90%. Из экс-
трудированных образцов была получена качественная молибде-
новая проволока.
Волочение проволоки. Проволоку подвергают волочению при
нагревании на механизированных станах. «Однако тонкое волоче-
ние (диаметры меньше 0,1 мм) можно проводить на холоду, что
обеспечивает более гладкую поверхность проволоки.
Проволоку диаметром 1—0,3 мм протягивают при 600—500° С,
меньших диаметров — при 500—300° С.
При диаметрах проволоки до 0,3 мм используют твердосплав-
ные фильеры, а на тонком волочении — алмазные фильтры.
364
Принципиальная схема протяжки проволоки приведена на
рис. 158. Исходная катанка, намотанная на спускной барабан,
проходит через смазочную коробку с аквадагом, газовую (или
электрическую) печь, фильеру
и наматывается на ведущий
барабан.
• Применяют волочильные
станки однократного и много-
кратного волочения, в кото-
рых проволока последова-
тельно протягивается через
ряд фильер, отличающихся
диаметром очка. Производи-
тельность таких станков вы-
сока, что сокращает произ-
водственные затраты.
Проволоку из чистого мо-
либдена выпускают диамет-
ром от 2,5 до 0,02 мм.
Для очистки поверхности
молибденовой проволоки от
графита и окалины приме-
няют различные способы:
непрерывное пропускание
проволоки через трубчатую
печь, в которой проволока
кратковременно отжигается
при 1300—1400° С в атмо-
сфере влажного водорода,
химическое травление (после-
довательно в растворе горя-
чей 20 %-ной щелочи и соля-
ной кислоты) и электрохими-
ческое травление [6].
На рис. 159 сопоставлена
микроструктура спеченного
молибденового штабика, де-
формированного штабика и
проволоки, у которой ясно
Рис. 159. Структура спеченного шта-
бика (а), прокованного и слабо отожжен-
ного штабика (б) и неотожженной прово-
локи диаметром 0,2 мм (в). Х200
ковке на пневматическом молоте
По мере деформации температура
с — 1500 до 1000° С. Если исход-
выражена волокнистая струк-
тура.
Плоская ковка и про-
катка. Для получения пла-
стин и ленты молибденовые
штабики подвергают горячей
до Vз—х/4 исходной толщины,
нагрева под ковку снижается
365
Рис. 160. Микроструктура молибденового листа после различ-
ных режимов отжига:
л — 950° С, 9 ч, неизменившаяся структура прокатки; б — 1200° С,
15 мин, частичная рекристаллизация; в — 1200° С, 30 мин, полная
рекристаллизация; г — 1200° С, 120 мин, вторичный рост зерен
366
ные спеченные штабики молибдена имеют прямоугольную форму,
их можно прокатывать без предварительной ковки.
Пластины прокатывают на двухвалковых станах и станах боль-
шей мощности. Обычно применяют поперечную прокатку, что
обеспечивает одинаковую пластичность листа во всех направле-
ниях. До толщины листа —1,5 мм операцию проводят при на-
гревании заготовок до 1200—1350° С в начале прокатки с после-
дующим снижением температуры до 800—900° С.
Молибден прокатывают до толщины от 1—1,5 до 0,2 мм при
комнатной температуре с промежуточными отжигами после каж-
дого обжатия в пределах 5—25%. По данным работы [6], при хо-
лодной прокатке молибдена от ленты толщиной 1 до 0,2 мм необ-
ходимо 15—20 проходов. Температура отжига должна быть ниже
температуры рекристаллизации молибдена. Верхний предел тем-
пературы отжига 800—850° С. После горячей прокатки ленты
очищают от слоя окалины погружением их в расплав NaOH +
+ NaNO3 или травлением растворами аммиака и перекиси водо-
рода или раствором бихромата калия.
На рис. 160 приведены фотографии микроструктуры прокатан-
ного молибденового листа после различных режимов отжига.
Обработка плавленого молибдена
Слитки плавленого молибдена, обладающие грубой крупно-
зернистой структурой, деформируются значительно трудней, чем
спеченные заготовки.
Недостаточная пластичность литого молибдена обусловлена
ослаблением связей между кристаллитами из-за присутствия на
границах примесей окислов. Поэтому лучше деформируются
хорошо раскисленные слитки с низким содержанием кислорода [7].
Для слитков малых диаметров (до 100 мм) может быть приме-
нена горячая ковка (при 1400—1450° С), однако заготовки диа-
метром 150 мм и больше проковать не удалось из-за образования
трещин. В связи с этим для разрушения первоначальной грубо-
зернистой литой структуры приходится выдавливать слиток мето-
дом прессования [1, с. 95].
Выдавливание проводят по методу Южина—Сежурне (рис. 161)
при температуре 1260—1300° С с применением для смазки стекла,
которым покрывают слиток. Процедура подготовки слитка и ре-
жимы выдавливания описаны Брукартом [1, с. 95].
Исходный молибденовый слиток обрабатывают точно под раз-
меры камеры выдавливания с учетом припуска на стеклянную-
смазку. Первоначально слиток нагревают в газовой печи до 760° С
и покрывают специальной эмалью, поверх которой накатывают
тонкоизмельченное стекло. Затем металл нагревают до 1260° С
индуктором в закрытом контейнере (или нагревают погружением:
в расплавленный хлористый барий). Нагретый слиток вновь по-
крывают стеклом и помещают в камеру выдавливания.
367
При контейнере диаметром 775 мм, предназначенном для вы-
давливания заготовок диаметром 250 мм, применяют 2500-Т пресс
с ходом плунжера 2,5 м при скорости 1,5 м!мин. Коэффициент
обжатия колеблется от 1,7 до 2,5. Выдавленные слитки после горя-
чей ковки при 1425° С и обрезки концов обтачивают на глубину
до 25 мм для удаления стекла и слоя окалины.
Выдавленные заготовки можно далее деформировать ковкой
с последующей прокаткой в листы подобно тому, как это выше
кратко описано для спеченных заготовок.
Рис. 161. Выдавливание молиб-
денового слитка, покрытого
стеклом, по способу Юджина —
Сежурне (французский патент):
/ — контейнер; 2 — плунжер; 3 —
прессуемая заготовка; 4 —стеклян-
ная вата; 5 — стеклянная пластинка;
6 — очко; 7 — выдавленный пруток
Изделия из спеченных и плавленых заготовок молибдена
(лента, проволока и др.) не отличаются по свойствам [14].
Потери молибдена при нагреве под ковку и прокатку составляют
значительную величину (12—15% и более). Для снижения потерь
и улучшения пластических свойств молибдена разработаны процес-
сы ковки в атмосфере инертного газа [9] и прокатки в вакууме
[11 —12]. Это сильно усложняет оборудование, но обеспечивает
снижение потерь молибдена вследствие окисления до 0,5% и ниже.
Изготовление изделий из молибдена. Для изготовления из
молибдена и сплавов на его основе изделий различных профилей
можно применять почти все известные способы обработки: вы-
давливание на токарно-давильном станке, ковку, прокатку круг-
лых и листовых профилей, штамповку, глубокую вытяжку, вы-
давливание и механическую обработку резанием [1, с. 95; 13].
•Сварка, пайка и механические способы соединения молибдена
детально описаны Норткоттом [13, с. 190—219], а также в ра-
боте [44].
59. МЕХАНИЧЕСКИЕ СВОЙСТВА МОЛИБДЕНА
Механические свойства молибдена в большой мере зависят от
предшествующей механической и термической обработки и чистоты
металла. Особенно сильно на механические свойства влияют при-
меси кислорода, углерода и азота.
В настоящее время достоверно установлено, что молибден вы-
сокой чистоты с низким содержанием примесей внедрения пла-
стичен при комнатной температуре и в литом, и в рекристаллизо-
ванном состоянии. Это объясняется смещением порога перехода из
вязкого состояния в хрупкое в область минусовых температур.
Обстоятельные обзоры механических свойств молибдена со-
держатся в работах [13, 17, 21].
368
NO
МЕХАНИЧЕСКИЕ СВОЙСТВА НЕОБРАБОТАННОГО МОЛИБДЕНА
РАЗНОЙ ЧИСТОТЫ И МОНОКРИСТАЛЛИЧЕСКОГО СЛИТКА [17]
Зеликман
ТАБЛИЦА 83
Материал Примеси, % (по массе) Механические свойства при растяжении НВ, кГ/мм2 Температура начала ре- кристалли- зации, °C Температура перехода в хрупкое состояние, °C
О н N кГ/мм2 б. % %
Спеченный штабик . . Слиток дуговой плавки 0,02 ’ — 0,04 50—100 — — 150—200 1250—1390 300
(два переплава) . . . Монокристаллический 0,003 0,0003 0,001 . 57,1 28,96 0,1 170—200 950—1050 25
слиток 0,0008 0,0004 0,0006 31 10—15 100 160—170 650 —196
МЕХАНИЧЕСКИЕ СВОЙСТВА МЕТАЛЛОКЕРАМИЧЕСКОГО
МОЛИБДЕНА В ЗАВИСИМОСТИ ОТ СТЕПЕНИ ДЕФОРМАЦИИ [15]
(а — неотожженный; б — отожженный для снятия напряжений)
ТАБЛИЦА 84
Материал Диаметр, мм о^, кГ/мм- б, % Материал Диаметр, мм кГ/мм2 6, %
а б а б а б а б
0,4 140 — —
Пруток [18] 6,25 2,50 52,5 70,0 5 5 Проволока Г1 о] 0,2 0,1 146 165 . 1 II —
1,25 84,0 63 5 15 НУ] 0,04 200 — — —
0,02 250 — — —
Проволока Г1 Я1 0,625 • 0,250 91 119- 70 87,1 5 1 15 15 Лист [18] 1,0 0,5 84 115,5 59,5 87,5 1 1 4 7
1 ю 1 0,125 133 98 1 15 0,25 122,5 91 1 7
Прочность при растяжении и твердость
В табл. 83 приведены некоторые прочностные характеристики
необработанного молибдена (спеченные штабики, слитки дуговой
плавки) и монокристаллического молибдена, сопоставление кото-
рых показывает большое влияние содержания примесей в техни-
ческом молибдене на его пластичность и температуру перехода
в хрупкое состояние.
ТАБЛИЦА 85
МЕХАНИЧЕСКИЕ СВОЙСТВА ГОРЯЧЕПРОКАТАННОГО МОЛИБДЕНА
ДУГОВОЙ ПЛАВКИ
(пруток диаметром 12,7 мм) [20 ]
Условия термообработки Gb> кГ/мм2 as’ кГ/мм2 6, % Ф, % НВ, кГ/мм2
После горячей прокатки 70,4 70,1 32 60,0 255
Отжиг при /, °C: 925 65,5 39 59,2 *» 1 —
1040 63,7 59,5 41 65,7
1050 51,4 38,0 53 64,2 187
1200 51,7 35,2 55 55,6 187
ТАБЛИЦА 86
МЕХАНИЧЕСКИЕ СВОЙСТВА МОЛИБДЕНА ПРИ ПОВЫШЕННЫХ
ТЕМПЕРАТУРАХ
(дуговая плавка, горячая прокатка на круг 22 мм) [20 ]
Температура испытания, °C Состояние д О ье 04 to ье oi е> « 6, (Z = =50 мм), % Чр я»
27 Катаный 64,4 — 1 3 2
Сняты напряжения 64,0 59,7 • - 10 9
Рекристаллизован 47,5 44,6 46 36
870 Катаный 43,2 35,3 18,3 18 72
Сняты напряжения 35,6 31,2 15,1 22 81
Рекристаллизован 24,2 8,1 2,4 46 84
982 Катаный 35,1 28,4 16,2 19 81
Сняты напряжения 28,4 25,0 15,8 32 81
Рекристаллизован 20,4 7,3 4,0 49 75
—
*
1066 Катаный 26,7 22,2 16,9 29 83
Сняты напряжения 22,2 16,1 11,5 36 84
Рекристаллизован 15,2 6,0 2,9 35 52
370
Прочность молибдена при растяжении увеличивается с возра-
станием степени деформации от 52,5 кПмм2 для прутка диаметром
6,2 мм до 250 кПмм2 для проволоки диаметром 0,02 мм
(табл. 84).
Механические свойства катаного молибдена дуговой плавки
в зависимости от температуры отжига приведены в табл. 85 [20, 13].
С повышением температуры отжига с 925 до 1200° С снижаются
прочность и твердость и увеличивается удлинение. Механические
свойства молибдена при повышенных «температурах приведены
в табл. 86.
Низкая прочность рекристаллизованного молибдена требует
легирования его элементами, повышающими температуру рекри-
сталлизации.
Зависимость твердости отожженного поликристаллического
молибдена от температуры в широком интервале от +1857 до
—189,5° С исследована Энглом с сотрудниками [22, 23]. Их дан-
ные показывают быстрое возрастание твердости при охлаждении
молибдена от 159,2 кПмм2 при 20° С до 373 кПмм2 при —189,5° С.
Изменение твердости в интервале 20—1857° С приведено в табл. 87.
ТАБЛИЦА 87
ТВЕРДОСТЬ МОЛИБДЕНА В ЗАВИСИМОСТИ ОТ ТЕМПЕРАТУРЫ [22, 231
Температура, °C НВ, кГ/мм2 Температура, °C НВ, кГ/мм2
20 159,2 1089 50,9
80 135,8 1243 40,6
142 124,0 1371 33,9
205 113,6 1610 22,1
347 100,8 1663 19,5
520 89,1 1727 16,9
681 77,0 1802 14,1
881 987 65,2 58,5 1857 12,6
Упругие характеристики
Молибден обладает высоким модулем упругости, уступая
в этом отношении лишь осмию, иридию, рению, рутению и вольф-
раму. Значения модуля упругости и модуля жесткости (модуль
сдвига) металла, полученного методом порошковой металлургии
и плавкой, близки (табл. 88). Модуль упругости понижается от
32 300 кПмм2 при комнатной температуре до 28 040 кПмм2 при
870° С [20].
Коэффициент сжимаемости молибдена, служащий мерой сил
сопротивления изменению объема, равен 0,347-10° см^/кг.
24* 371
о
ТАБЛИЦА 88
УПРУГИЕ КОНСТАНТЫ МОЛИБДЕНА [20 ]
Температура испытания, °C Модуль упругости (Юнга), кГ/мм2 Модуль жесткости (сдвига), кГ/мм2 Отношение Пуассона Динамический модуль упру- гости*, кГ/мм2
Молибден, полученный методом порошковой металлургии
26 31 800 12 160 0,307 33 530
120 31 600 12 090 0,308 33 250
315 30 700 11 810 0,307 32 340
480 30 150 11 460 0,307 31 400
590 29 500 11 250 0,312 30 770
Молибден, плавленный в дуговой печи
26 32 300 12 230 0,324 —
140 31 400 11 880 0,330 —
385 30 000 11 460 0,317 *'
635 28 820 11 000 0,314 —
870 28 040 10 600 0,321 —
* Данные Костера [24 ] получены по методу Камертона.
Ползучесть, длительная и усталостная прочность
Молибден обладает высоким сопротивлением ползучести при
повышенных температурах. По данным работы [25], напряжение,
вызывающее 1%-ную деформа-
цию за 24 ч при 1000° С, соста-
вляет, кПмм2\
Рис. 162. Длительная прочность мо-
либдена в сопоставлении с прочностью
других тугоплавких металлов при
1085° С (все металлы вакуумной плавки)
Pt................. 0,14
Rh ................. 4,6
Мо .............. 4,6—6,16
Та............. 4,4—6,3
1г.................. 9,2
W................... 9,2
Как видно из рис. 162, дли-
тельная прочность молибдена
при 1085° С примерно вдвое
выше, чем у тантала и ниобия.
Длительная прочность молиб-
дена при 1000° С равна длительной прочности жаропрочного
сплава типа стеллит (сплав на основе кобальта, содержащий
хром и вольфрам). При времени выдержки 100 ч напряжение
разрушения равно 7 кПмм2 [3].
Важная характеристика конструкционных металлов — предел
усталости — напряжение разрушения металла после большого
числа циклов приложения переменной нагрузки.
372
По данным Брукарта и Хайлера [27], предел усталости мо-
либдена дуговой плавки колеблется от 50,6 до 56,2 кПмм2 (при
испытаниях на базе 5-Ю7 циклов переменной нагрузки). Для мо-
либдена, полученного методом порошковой металлургии, значения
предела усталости в тех же условиях испытаний 47,8—52,7 кГ!мм\
Отношение предела усталости к пределу прочности молибдена
0,66—0,81, что выше, чем у стали (0,4—0,6).
Поскольку значения предела усталости сильно зависят от ка-
чества поверхности образца, изучали влияние надреза на эту
величину. Отношение пределов усталости ненадрезанных и над-
резанных образцов (коэффициент чувствительности к надрезу)
оказалось равным 2,05 (для молибдена дуговой плавки) и 1,36
(для молибдена, полученного методом порошковой металлургии).
Влияние примесей внедрения на пластичность молибдена
Как отмечалось выше, молибден имеет температурный порог
перехода из вязкого состояния в хрупкое. Температура перехода
является функцией ряда факторов, однако решающее влияние на
положение порога хрупкости
оказывают примеси внедре-
ния — кислород, углерод,
азот. Растворимость этих
примесей в молибдене весьма
низкая, что видно из диа-
грамм (рис. 163), построен-
ных Спайсилом и Вульфом
[1, с. 176] по данным раз-
личных авторов [28—31].
Эвтектические температуры
для систем Мо—Мо2С и Мо—
МоО2 взяты из работ Сайкса
с сотрудниками [32] и Перри
с сотрудниками [33]. Если
содержание примесей внедре-
ния не превышает предела
их растворимости, молибден
О ОО/0,020,030 0.008 0,0/6О ОООО 0,0/2
о
7О (по массе)
пластичен при низких темпе- Рис. 163. Диаграмма растворимости кисло-
ратурах. Превышение пре- рода, углерода и азота в молибдене
дела растворимости приво-
дит к выделению МоО2, Мо2С, нитридов на границах зерен„
что резко снижает пластичность и повышает температурный
порог хрупкости.
В табл. 89 приведены данные о влиянии кислорода, угле-
рода и азота на температуру перехода молибдена из пластичного
состояния в хрупкое. Наиболее сильно влияет на порог перехода
из вязкого состояния в хрупкое кислород.
37&
Из табл. 89 можно видеть, что для снижения температуры пере-
хода в область температур ниже 0° содержание кислорода должно
быть порядка 0,0001 %. Азот и углерод повышают температуру пе-
рехода при их содержании выше 0,001 %. Содержание азота обычно
ниже критического. Однако снижение содержания углерода до
0,001 % трудно выполнимо, особенно в том случае, если его исполь-
зуют как раскислитель при плавке молибдена.
ТАБЛИЦА 89
ВЛИЯНИЕ ПРИМЕСЕЙ ВНЕДРЕНИЯ НА ТЕМПЕРАТУРНЫЙ ПОРОГ
ХРУПКОСТИ МОЛИБДЕНА [28]
Слиток Содержание, % Температура пере- хода по испытанию на изгиб (угол изгиба < 4°), °C
О N с
Высокой чистоты 0,0001 — —80 —60
0,0002 - —- + 200
С содержанием кислорода 0,0006 — — +200
——... 0,0008 —40
0,0014 —40
С содержанием азота 0,0037 +40
-- — 0,033 — + 140
—, .. — 0,003 —40
— — 0,006 —30
0,008 —10
С содержанием углерода 0,01 0
— 0,02 + 40
— — 0,024 +50
Технический молибден с -4-90
0,003% с хм
В этой связи авторы работы [17] считают углерод наиболее
вредной примесью с точки зрения его влияния на пластические
свойства. Повышение содержания углерода с 0,004 до 0,017%
вызывает возрастание температуры перехода от —50 до- +120° С
[34]. Чтобы порог хрупкости наступал при комнатной температуре,
содержание углерода не должно превышать 0,005% [35]. При
данном содержании примеси температура перехода из вязкого
состояния в хрупкое у рекристаллизованного молибдена на 100—
300 град выше, чем в деформированном состоянии. Это объясняется
скоплением по границам зерен в рекристаллизованном металле
двуокиси и карбида молибдена [17].
374
Влияние легирующих элементов на механические свойства
молибдена
Некоторые двойные системы. Важнейшие двойные системы
приведены на рис. 164—175 по данным справочников [36] и [13],
Рис. 164. Система молиб-
ден—вольфрам
Рис. 165. Система молиб-
ден—хром
Рис. 166. Система молибден—
ванадий
в которых можно найти более детальное их описание и ссылки на
первоисточники. Системы Мо—V и Мо—Re исследованы позже
Савицким с сотрудниками [37, 38]. Системы Мо—С, Мо—Si
и Мо—В были детально рассмотрены в гл. X.
375
7о(ат.)
70(ло массе)
Рис. 168. Система молибден—рений
% (по пассе)
Рис. 167. Система молибден—титан
%(сш)
О 20 О О 60 60 100
2800
2000
0^> 2000
S Г600
S'
1200
800
ООО
Мо 20 00 60 80 Zr
% ( по массе)
Рис. 169. Система молибден—цирконий
376
/7 20 00 00 80 7о(ат)
2600
2000
2200
oL-Mo
2000
•° 7800
7600
Ъ 7000
| 1200
$
Й 7000
800
600
ООО
Мо 20 00 60 80 AL
% (по массе)
Рис. 170. Система молибден—алюминий
Л”
t
-\16
J76O
Mo3AL+MoAL2\
'ОС ~Мо+Мо3А7 |
Мо3А1
Ж
660°
7730
735
\Мо{12
моА15 | ^МоА15
20
7с (ат.)
80
00
7600
7000
7200
600
7000
800
О
7800
Мо 70 20 30 00 50 60 70 80 90 Fe
7с (по массе)
Рис. 171. Система молибден—железо
37Т
378
о 20 СО • 60 80< %
Мо 20 00 60 80 Со
°/о(по массе)
Рис. 172. Система молибден—кобальт
Рис. 173. Система молибден—никель
Рис. 174. Система молибден--уран
Рис. 175; Система молибден—бериллий
Норткотт подразделяет ис-
следованные системы на три
класса [13]:
Класс I. Системы с пол-
ной растворимостью
(при всех температурах):
Мо—Сг Мо—Ti
Мо—Та (выше 885° С)
Мо—V " Мо—W
Класс II. Системы с пе
промежуточная фаза):
ритектикой (в скобках —
Мо—А1 (Мо3А1)
Мо—Fe (FeMo)
Мо—Со (Со2Мо3)
Мо—Ni (NiMo)
Класс III. Системы с
п р омежуточн ая фаз а):
Мо—Be (МоВе2)
Мо—В (Мо2В)
Мо—U
(отсутствует)
Мо—Zr (ZrMo2)
Мо—Re (Re3Mo2)
эвтектикой (Мотв. р_р
Мо—С (Мо2С)
Мо—Si (Mo3Si)
379
Как видно из табл. 90, составленной Моргуновой [5], системы
с полной растворимостью образуют элементы, имеющие решетку,
однотипную с молибденом, за исключением циркония, у которого
атомный диаметр сильно отличается от атомного диаметра молиб-
дена и железа.
ТАБЛИЦА 90
ТИП РЕШЕТКИ И АТОМНЫЕ ДИАМЕТРЫ ЛЕГИРУЮЩИХ
ЭЛЕМЕНТОВ [5]
Характеристика легирующего элемента
Элемент тип решетки атомный диа- метр для координа- ционного числа 12, о А Разница в атомных диаметрах с Мо, % * Тип системы
W Nb Та V p-Ti Сг ₽-Zr Al ос-Fe P-Со Ni Be Объемноцентрирован- ный куб Гранецентрированный куб Объемноцентрирован- ный куб Гранецентрированный куб То же Гексагональная ком- пактная упаковка 2,82 2,94 2,94 2,71 2,96 2,57 3,22 2,85 2,55 2,507 2,49 2,551 +0,71 —5,0 +5,0 —3,2 +5,7 -8,2 + 11,1 + 1,78 —8,9 — 10,3 —11,1 Т вердый раствор Перитекти- ческая Эвтектиче- ская
* Тип решетки молибдена — обьемноцентрированный куб. Атомный диаметр 2,80 А.
Твердость, прочность и обрабатываемость сплавов. При леги-
ровании молибдена преследуют две цели: а) увеличение деформа-
ционной прочности в результате повышения температуры рекри-
сталлизации; б) снижение температуры перехода из вязкого со-
стояния в хрупкое.
Проведены многочисленные исследования влияния различных
легирующих элементов на свойства молибдена, полученного ме-
тодом порошковой металлургии [39; 42; 1, с. 372 и 226] и дуговой
плавкой [40; 1, с. 189; 41].
Общие закономерности для сплавов, полученных обоими ме-
тодами, одинаковы, однако метод порошковой металлургии позво-
ляет ввести несколько большее количество легирующего элемента
при сохранении ковкости сплава.
На рис. 176, по данным работы [40], показано изменение твер-
дости литых молибденовых сплавов в зависимости от количества
380
легирующей добавки. В наибольшей степени повышают твердость
и ухудшают ковкость молибдена элементы, которые обладают наи-
меньшей растворимостью в нем (Be, Ni, В, Si, Со). Наоборот,
элементы, обладающие неограниченной растворимостью в мо-
либдене, в малой мере повышают его твердость (W, Та, Nb, V,
Ti). Элементы Al, Zr, Сг занимают промежуточное положение х.
При повышении температуры порядок изменения твердости
остается тем же.
Деформируемыми являются лишь те сплавы, которые представ-
ляют собой твердые растворы. Это определяет возможное коли-
Содержание элемента, 7О (по массе)
с»
Рис. 176. Влияние различных легирующих элементов
на твердость литого молибдена при комнатной темпе-
ратуре
Так, в плавленом двойном сплаве на основе молибдена для со-
хранения его ковкости содержание легирующего элемента, по
данным Парке [20], не должно превышать, %: 0,8 А1; 2,0 Сг;
0,3 Со; 0,3 Fe; 0,05 Ni; 0,03 Si. Для сплавов, полученных методом
порошковой металлургии, в литературе [39, 43] приводятся сле-
дующие предельные содержания легирующих добавок, %: 0,25—
0,5 Be; >4 Ti; >1,5 Zr; 0,25—0,5 Th; 1—3 Nb; 1—10 Ta; >1,5 Cr;
до 100 W; 0,5—2 Mn; 1—2 Al; 0,3 Co; 0,4 Fe; 0,4 Ni.
Температура рекристаллизации и длительная прочность.
Как уже отмечалось, деформационное упрочнение (наклеп) —
практически единственный путь придания необходимой прочности
изделиям из молибдена и сплавов на его основе. Поскольку обра-
батываемые сплавы гомогенны (представляют собой твердые
растворы), они не поддаются дисперсионному упрочнению (старе-
1 Семчишен отмечает, что при добавках небольших количеств Al, Si, Сг,
Fe, Со, Ni твердость молибдена сначала снижается, проходит минимум, лишь
затем растет. Вероятно, это объясняется их воздействием на содержание и форму
нахождения некоторых примесей. В частности, Al, Si, Сг могут раскислять металл
[1, с. 189].
381
нию). Температура рекристаллизации технически чистого мо-
либдена при максимальных степенях деформации 900—1000° С.
Температура начала рекристаллизации и величина зерен после
рекристаллизационного отжига сильно зависят от степени предва-
рительной холодной деформации. Эта зависимость описывается
диаграммой рекристаллизации (рис. 177)., построенной Савицким
Степень деформации, %
Рис. 177. Диаграмма рекристаллизации молибдена
с сотрудниками [45] для плавленого молибдена. Температура
начала рекристаллизации показана пунктирной линией.
Легирование молибдена элементами, образующими твердые
растворы, приводит к росту температуры рекристаллизации, что
позволяет повысить рабочие температуры эксплуатации изделий
с сохранением их прочности. Эффективные легирующие добавки,
повышающие температуру рекристаллизации, — титан, цирко-
ний, ниобий.
ТАБЛИЦА 91
ТЕМПЕРАТУРА РЕКРИСТАЛЛИЗАЦИИ МОЛИБДЕНОВЫХ СПЛАВОВ1
Легирующие в молибденовом сплаве Температура рекристалли- зации, °C
добавка содержание, %
Нелегированный Мо (0,01% С) 1180
Ti 0,5 1340
Ti+Zr 0,5+0,08 1430
Zr 0,15 1450
Zr 0,05 1340
Ti+Zr+C 1,25+0,15+0,15 1540
1 Методом измерения твердости определяли минимальную температуру рекристалли
зации за 1 ч после деформации на 75—97% при различных температурах и режимах дефор
мации.
382
В табл. 91 приведены температуры рекристаллизации некоторых
сплавов с титаном и цирконием [21, с. 187].
На рис. 178 сопоставлены кривые 100-^ длительной прочности
сплавов молибдена и 13 лучших жаропрочных сплавов (на основе
никеля, кобальта, хрома и др.). В интервале температур 870—
1093° С молибден и его сплавы значительно превосходят другие
жаропрочные сплавы.
Порог хрупкости. По влиянию на температуру перехода из
вязкого состояния в хрупкое Олдс и Рендсторф [461 подразделяют
легирующие элементы на три
группы:
1. Элементы, введение кото-
рых в малых количествах вна-
чале снижает порог хрупкости,
а затем повышает: Ti, Се, V,
В, Y.
2. Элементы, повышающие
порог хрупкости: Zr; Al.
3. Элементы, снижающие
порог хрупкости: Th, Re.
Вероятно, элементы второй
группы (Zr и А1), так же как
и первой, снижают порог хруп-
кости при достаточно малых
содержаниях.
Понижение температуры пе-
рехода небольшими добавками
активных элементов (титан,
церий и др.) связано с раскис-
Рис. 178. Длительная прочность сплава
на основе молибдена и 13 жаропроч-
ных сплавов на основе никеля, хрома,
кобальта и железа
ляющим и деазотизирующим их
воздействием. При добавлении легирующего элемента в количестве,
превышающем необходимое для связывания кислорода и азота,
порог хрупкости возрастает, так как увеличивается концентра-
ция элемента в твердом растворе [10, 48].
Температура перехода деформированных и отожженных (сня-
тие напряжений) прутков сплава Мо — 0,5% Ti равна 50° С,
а листов 77° С. У рекристаллизованного сплава температура пере-
хода возрастает до 127—250° С, что мало отличается от порога
хрупкости технически чистого молибдена [21].
По данным Клоппа и др. [21, с. 190], температура перехода
рекристаллизованных сплавов молибдена с содержанием рения
10, 20, 25, 30 и 35% (ат.) равна соответственно —40, —100, —150,
—Д80 и —250° С (температура перехода исходного рекристал-
лизованного молибдена была равной 60° С). Аналогичные резуль-
таты получены Савицким с сотрудниками [50].
Механизм влияния рения, а также тория на пластичность мо-
либдена еще недостаточно изучен.
383
60. МЕТАЛЛОГРАФИЧЕСКИЙ КОНТРОЛЬ В ПРОИЗВОДСТВЕ
МОЛИБДЕНА
Металлографические методы используют в производстве мо-
либдена для определения зернистости порошков и величины зерен
заготовок, контроля чистоты металла и протекания процессов
рекристаллизации.
Методика контроля зернистости порошков, в том числе металло-
графическим методом, уже рассматривалась (см. стр. 275). Допол-
нительные сведения можно найти в работе [51].
Ниже дано краткое описание металлографического контроля
компактного молибдена.
Приготовление шлифов. Техника приготовления микрошлифов
спеченных штабиков, образцов плавленого молибдена или кованых
заготовок не отличается от обычно используемой. Образцы режут
тонкими корундовыми или алмазными кругами толщиной около
0,3 мм при интенсивном охлаждении водой [17]. Большей частью
образцы закрепляют впрессовыванием в подогретые пластмассы
(полистирол, плексиглас, бакелит, оргцемент типа АКР) или за-
ливают оловянным припоем (третник). Приемы закрепления тон-
кой проволоки, спиралей детально описаны Смителлсом [53].
Образцы шлифуют на шлифовальной бумаге с последовательно
уменьшающейся крупностью (Э32—Э25, Э20, Э4, ЭЗ, М20, М14) *.
Полировку осуществляют на вращающемся диске, покрытом вой-
локом или сукном, с суспензией из тонкодисперсной окиси алюми-
ния в щелочном растворе красной кровяной соли (травящая поли-
ровка). Используют также электролитический способ полировки.
В качестве электролита может быть использован 2—5%-ный
раствор NaOH [17] или раствор H2SO4 в метиловом спирте (до-
бавляют 25 мл концентрированной H2SO4 и 175 мл спирта) [52].
Электролитическая полировка позволяет получить шлифы более
высокого качества, чем при механической полировке. Кроме
того, при электролитической полировке (в отличие от механиче-
ской) исключается наклеп полируемой поверхности, искажающий
структуру образца.
Электрополировку проводят при напряжении около 6 в при
плотности тока 2—3 а!см2.
Травители. Для выявления границ зерен в молибдене и струк-
турных составляющих в сплавах на его основе применяют различ-
ные травители (табл. 92).
Травителй 1—7 (табл. 92) используют в лабораториях фирмы
«Клаймакс» [52], 8—10 — на заводах СССР [16], реактивы 11 —
13 — на ряде европейских предприятий [6].
Для травления нелегированного молибдена наиболее широко
распространен разбавленный щелочной раствор K3Fe (CN)6 и
2—10%-ный раствор NaOH. Хорошо выявляет границы зерен
* ГОСТ 5009—62 и 6456—62.
384
аммиачный раствор сульфата меди. Раствор персульфата аммония
используют для выявления включений карбида молибдена [16].
Травители, рекомендуемые для травления сплавов, приведены
в табл. 93.
ТАБЛИЦА 92
СОСТАВ ТРАВИТЕЛЕЙ ДЛЯ МОЛИБДЕНА И СПЛАВОВ НА ЕГО ОСНОВЕ
№ пп. Состав травителя Способ употребления Время травления
1 10 г NaOH, 20 сек
30 г K3Fe (CN)6,
100 мл Н2О
2 1 г NaOH, 35 г K3Fe (CN)e, Погружение на холоду 4—6 мин
150 мл Н2О
3 20—30 мл раствора 2, 12—16 »
100 мл Н2О
4 0,5 г щавелевой кисло- \ г
5 ты, 100 мл Н2О 10 мл НС1, Электролитическое тра- 10—20 » <
4 мл H2SO4, вление 10—20 сек
120 мл СН3ОН
6 3 мл HNO3, 1 100 мл Н2О > Погружение в горячий 3—5 мин or-
7 2 мл HF, 100 мл Н2О J раствор 3—5 » к
8 60 мл 5%-ного раствора Погружение на холоду *
KsFe (CN)e, 30 мл 50% - или при 40—60° С
ного раствора NaOH
9 10%-ный раствор Погружение на холоду —
(NH4)2S2O8
10 10%-ный раствор NaOH Электролитическое тра- —
вление
11 3%-ный раствор Н2О2 Погружение в нагретый 20—30 сек
раствор
12 25 мл 1-н. NaOH и Электролитическое тра- —
20 мл Н2О2 вление —
13 Аммиачный раствор Погружение на холоду ——.
сульфата меди
Типичные структуры микрошлифов спеченных штабиков мо-
либдена, литого молибдена, деформированных и рекристаллизован-
ных образцов приведены выше (рис. 136, 159, 160).
Фрактография (изучение изломов). Фрактографический метод,
примененный для исследования литого молибдена Вудсайдом
[49], —чувствительный метод обнаружения фаз, выделяющихся
на границах зерен (окислов, карбидов и др.) [52].
Излом молибдена литого или деформированного и рекристалли-
зованного может быть получен резким ударом молотком. В литом
материале размеры зерен столь велики, что можно легко разли-
чить изломы по кристаллу и по межзеренным границам.
25 А. Н. Зеликман 385
ТАБЛИЦА* 93
ТРАВИТЕЛИ, РЕКОМЕНДУЕМЫЕ ДЛЯ ТРАВЛЕНИЯ СПЛАВОВ [52]
Сплавы системы Номер травителя (по табл. 92) Примечание
Мо—А1 2 X " '
Мо—В 3 —
Мо—Be 1 —
Мо—С 3 —
Мо—Со 4 Структура в целом
Мо—Со 1 Выявляется различие между трфазой (коричневая) и 8-фазой (не травится)
Мо—Fe 5 Структура в целом
Мо—Fe 1 Выявляется туфаза — темная (твердый раствор Fe в молибдене)
Мо—N 3 —
Мо—Ni 5 Выявляется вторая фаза
Мо—Si 1
Мо—Ti 2 или 3 | Выявляется тонкая кружевоподобная фаза, ве-
Мо—Zr 2 или 3 j роятно, сложный карбид
Мо—Zr 6 Травится структура, не затрачиваются включения окислов
Мо—Zr 7 Травятся только включения окислов
Мо—U 3 —
Фрактографический метод состоит в изучении под микроскопом
при больших увеличениях межзеренных изломов. Исследуемый
образец надежно закрепляют в пластилине, помещенном в углуб-
ление предметного стекла, причем поверхность излома должна быть
параллельна стеклу. Затем предметное стекло с образцом перево-
рачивают и устанавливают над отверстием металлографического
микроскопа. Предметное стекло нельзя прикреплять зажимами
к столику во избежание повреждения объектива при его сопри-
косновении с образцом.
Рекомендуется рассматривать излом при увеличении 1500
с иммерсионным объективом, что позволяет лучше выявить харак-
тер фаз на границе. Сухие объективы с увеличением 1000 непри-
годны вследствие чрезвычайно коротких фокусных расстояний.
Эффективность применения фрактографического метода для
обнаружения окисных и карбидных фаз видна на рис. 145. Включе-
ния окислов не выявлены на обычном микрошлифе, тогда как на
фотографии излома они четко видны.
ГЛАВА XV
СВОЙСТВА И ОБЛАСТИ ПРИМЕНЕНИЯ МОЛИБДЕНА
61. ФИЗИЧЕСКИЕ СВОЙСТВА
Атомные свойства
Атомная масса молибдена 95,4.
Изотопы. В природном молибдене семь изотопов [ 1 ]:
Массовое число .......... 92 94 95 96 97 98 100
Содержание в природной смеси, % 15,86 9,12 15,7 16,5 9,45 23,75 9,62
Искусственно получено 10 радиоактивных изотопов (табл. 94).
ТАБЛИЦА 94
РАДИОАКТИВНЫЕ ИЗОТОПЫ МОЛИБДЕНА
Массовое
число
Период полураспада Тi
Характер излучения
90
91
91
93
93
93
99
100
102
105
5,7±0,2 ч
15,5±0,5 мин
1,25 + 0,1 »
6,95 ч
17 мин
>2
66 ч
14,6 мин
12 »
—5 »
Р+> У
р+ (Nb91)
р+(Nb91), у
В- (Nb93), И. П*.
6+ (Nb93) у
й- (Тс99) v, е~
6- (Тс101) у
(Тс102)
р- (Тс108)
* И. П. — изомерный переход; е — электрон внутренней конверсии.
** К — захват.
Поперечное сечение захвата медленных нейтронов равно
2,7 барн!атом.
Электронная конфигурация атома молибдена в нормальном
состоянии:
25*
387
Энергия ионизации, эв, Мо° —> Мо+ —> Мо2+ —> Мо3+ —>
—> Мо4+ Моб+ —» Мо6+ соответственно равна 7,10; 16,15; 27,13;
40,53; 55,6; 71,7.
Кристаллическая структура. Молибден имеет
объемноцентрированную кубическую решетку, структурный
тип А2.
Период решетки а = 3,1403 А [4]. Этот тип решетки
сохраняется вплоть до плавления.
Плотность, рассчитанная из рентгенографических дан-
ных, равна 10,23 г!см3 [5].
Атомный объем 9,38 см? I г-атом.
Атомный радиус молибдена для координационного
числа 8, по Полингу [6], 1,36 А.
Ионный радиус Мо4+ равен 0,68 А [7], Мо6+ 0,62 А [8].
Работа выхода электрон о в. Данные различных
авторов существенно расходятся, что может быть обусловлено раз-
личиями в степени дегазации металла. Для хорошо дегазирован-
ного молибдена определено ср = 4,15 эв [9]. В более поздних ра-
ботах найдены более высокие значения: ф — 4,37 [10] и 4,29 [11 ].
Термические свойства
Температура плавления. Общепринятое значение
температуры плавления молибдена 2622 ± 10° С [12]. Оно яв-
ляется средним из 97 измерений с использованием трех различных
методов.
Теплота плавления 6,6 ± 0,7 ккал/г-атом или
70 кал!г [13].
Температура кипения. Надежных данных нет.
Вероятная величина, по оценочным расчетам [14—16], 4830° С.
Давление пара твердого молибдена. Да-
вление пара твердого молибдена впервые было определено Ленг-
мюром с сотрудниками [18, 19] по скорости испарения металла
с нагретой нити в вакууме. Этим же методом давление пара изучал
Цвиккер [20]. Более точные данные получены Эдвардсом и др. [17]
методом испарения с поверхности цилиндра в вакууме. Эти данные
по давлению пара твердого молибдена приведены ниже.
Температура, °C . . 1878 1912 1958 1967 1987 2027 2124 2165 2189
Давление, мм рт. ст.
Х106 ........... 3,362 5,703 11,45 13,49 15,87 28,58 109,9 168,5 220,6
Зависимость давления пара от температуры описывается урав-
нением [17, 16]
1g Рмм рт. ст. = 23,68 - - 1,726 • 10-3 Т.
Теплота сублимации. По данным работы [17],
средняя величина теплоты субликации при 298° К равна
388
155,55 ккал!г-атом. Для 298° К в работах [14, 15] приводится
значение 157,5 кк,ал!г-атом.
Давление пара жидкого моли б д е н а. Экс-
периментальные значения отсутствуют. Имеются оценочные дан-
ные, которые приводятся ниже [21 ].
Температура, °C......... 3375 3666 3808 3963 4132 4237 4382 4595
Давление, мм рт. ст. . f 1 5 10 20 40 60 100 200
Ориентировочное значение теплоты испарения 1625 кал!г.
Скорость испарения. В табл. 95 приведены значе-
ния скорости испарения молибдена в вакууме по данным работ
[19, го:.
ТАБЛИЦА 95
СКОРОСТЬ ИСПАРЕНИЯ МОЛИБДЕНА В ВАКУУМЕ
Температура, °C Скорость испарения, г/{см2'сек)
по Джонесу и др. [19] по Цвиккеру [20] •
927 2,44-Ю’19 1,23-10“20
1127 1,29-10-15 1,20-10-16
1327 7,60-10-13 1,23-IO'13
1527 1,06-10-10 2,82-10~п
1727 5,34-Ю-9 2,07-10-э
1927 1,30-10*7 7,30-10-8
2227 5,62-10-6 5,03-10-в
2527 1,04-10-4 —
Теплоемкость. В интервале температур 298—1800° К
атомная теплоемкость молибдена описывается уравнением [22]
Ср = 5,48+ 1,30-10~37\
Зависимость теплоемкости от температуры следующая:
Температура, ° К............ 298 1000 1500
Атомная теплоемкость,
кал!(г-атом- град)........... 5,867 6,78 7,43
Удельная теплоемкость,
калЦг* град) ......... 0,061 0,070 0,077
Термическое расширение. Коэффициент терми-
ческого расширения молибдена зависит от степени деформации.
Демарке для неотожженной молибденовой проволоки получил
следующие значения а [23]:
Температура, °C ...... . 25—100 500 1000 1500 2000
Коэффициент линейного рас-
ширения сс-106 .......... 4,9 5,1 5,5 6,2 7,2
123
389
По данным Уайта, средний коэффициент линейного расширения
отожженного молибдена в интервале 0° С до температуры плавле-
ния подчиняется уравнению [24]:
ctcp. 106 = 5,05 + 0,31 • 10“3/ + 0,36-10"6 /2,
где t — температура, °C.
Теплопроводность. Ниже приведены значения тепло-
проводности молибдена по данным Осборна в интервале температур
927—1627° С [25]:
Температура, °C............ 927 1027 1127 1227 1327 1427 1527 1627
Теплопроводность,
кал/(см-сек-град) .... 0,259 0,244 0,230 0,216 0,202 0,188 0,173 0,159
При 17° С теплопроводность молибдена равна 0,346 кал!(сек-см-
•гряд).При температуре —483° С теплопроводность молибдена воз-
растает до 0,438 кал!(см-сек-град) [27].
Данные различных авторов о теплопроводности молибдена
сопоставлены в работе [26].
Оптические свойства
В табл. 96 приведены важнейшие оптические характеристики
молибдена — интенсивность радиации, лучеиспускательная спо-
собность, соотношение между яркостной и истинной температу-
рами.
ТАБЛИЦА 96
ОПТИЧЕСКИЕ ХАРАКТЕРИСТИКИ МОЛИБДЕНА [27—29]
Истинная температура, °К Яркостная температура * Коэффициент моно- хроматического излучения при К = 6650А Интенсивность радиации, вт/см2
°К °C
. 1000 958 685 0,390 0,55
1200 1139 866 0,382 1,43
1400 1816 1043 0,375 3,18
1600 1489 1216 0,367 6,30
1800 1658 1385 0,360 11,3
2000 . 1824 1551 0,353 19,2
2200 1986 1713 0,347 30,7
2400 2143 1870 0,341 47,0
2600 2297 2024 0,336 69,5
2800 2448 2275 0,331 98,0 .
2895 ** 2519 2246 0,328 116,0 ‘
о
* Измерено для X == 6650 А.
** Температура плавления.
390
Электрические и магнитные свойства
Электросопротивление. Удельное электросопро-
тивление молибдена при различных температурах приведено
в табл. 97 [28, 32].
ТАБЛИЦА 97
УДЕЛЬНОЕ ЭЛЕКТРОСОПРОТИВЛЕНИЕ МОЛИБДЕНА
Температура, Удельное электро- сопротивление, МКОМ-см Температура, °C Удельное электро- сопротивление, МКОМ-см
20 5,20 1527 47,3
127 7,6 1727 53,5
327 13,4 1927 59,2
527 18,6 2127 66,0
727 23,9 2327 71,8
927 29,2 2527 78,2
1127 35,2 2622 * 81,4
1327 41,2
* Температура плавления.
Температурный коэффициент электросопротивления в интер-
вале 20—2620° С равен 0,00479. Электросопротивление несколько
зависит от степени деформации [31].
Сверхпроводимость. Температура перехода моли-
бдена в сверхпроводящее состояние лежит в пределах 0,9—т
0,98° К.
Термоэлектрические свой-
ства. Молибден и вольфрам обладают
наиболее высокими термо-э. д. с. среди
тугоплавких металлов. По измерениям
Рудницкого [34], термо-э. д. с. молибдена
в паре с платиной имеет следующие зна-
чения:
Температура^
Рис. 179. Термо-э. д. с.
термопары вольфрам —
молибден
Температура, °C . . 480 780 1030
Термо-э. д. ,с., мв 10 20 30
На рис. 179 приведена зависимость
термо-э. д. с. термопары W—Мо от тем-
пературы. В интервале 1200—2000° С зависимость линейная,
что позволяет использовать термопару для измерения высоких
температур.
Магнитная восприимчивость. При 18° С
удельная магнитная восприимчивость молибдена равна 0,04 X
Х10-6 ед. CGS. [3]. Молибден парамагнитен.
39 Г
62. ХИМИЧЕСКИЕ СВОЙСТВА
В предшествующих главах были детально описаны свойства
ряда химических соединений молибдена (окислов, молибдатов,
хлоридов, сульфидов, силицидов, карбидов, боридов и др.). В дан-
ном разделе мы ограничимся рассмотрением взаимодействия метал-
лического молибдена с газами, коррозии молибдена в жидких
средах, вопросов окисления молибдена и путей его защиты.
Взаимодействие с газами
Кислород. При нагревании на воздухе порошкообразный
молибден окисляется примерно с 300° С, компактный молибден —
с 400° С. Активное окисление наблюдается выше 600° С.
Водород. Водород химически не взаимодействует с мо-
либденом вплоть до точки плавления. Однако водород растворяется
в молибдене. Растворимость водорода растет пропорционально
квадратному корню из величины давления газа и увеличивается
с повышением температуры [35]. Ниже приведена растворимость
водорода (р = 1 ат) в молибденовой проволоке:
Температура, °C............ 400 500 600 700 800 900 1000
Растворимость, сж3Н2 в 100 а ме-
талла ................... 0,165 0,175 0,185 0,21 0,25 0,29 0,50
При нагревании в вакууме выше 1000° С водород может быть
полно удален из молибдена.
Азот и аммиак. В системе молибден—азот установлены
три нитридных фазы [39, 40]: P-фаза MoN0)40 (Mo5N2); у-фаз а
M°N0,5 (Mo2N); б-фаза MoN. Они получены азотированием порошка
молибдена аммиаком в интервале температур 400—745° С.
Хегедюш с сотрудниками [41 ], изучавшие нитрирование
порошка молибдена азотом и аммиаком термогравиметрическим
методом, установили, что чистый азот не взаимодействует с по-
рошком молибдена, чему препятствуют пленки окислов. Однако
в присутствии водорода при температуре 760° С происходит быстрое
нитрирование до состава MoN0>23.
Аммиак нитрирует порошок уже при 355° С. При 510° С обра-
зуется нитрид MoN0,47, при 660° С MoN0)84- С повышением темпе-
ратуры до 910° С нитрирование протекает в малой степени. При
температурах выше 900° С в вакууме порошок можно полно дени-
трировать.
Компактный молибден при атмосферном давлении и темпера-
турах выше 1200° С поглощает азот, выше 1500° С образуются
нитридные фазы.
Ниже приведены данные Нортона и Маршала [36] о поглоще-
нии азота молибденовой проволокой (р == 1 ат):
Температура, °C . 1200 1600 2000 2400
Поглощение азота, см3 в 100 г ме-
талла ............ 0,84 3,44 8,4 16,0
392
Адсорбцию азота молибденом изучали в ряде других работ [35,
37, 38], данные которых сильно расходятся между собой, что тре-
бует дальнейшего уточнения.
Пары воды при 600—700° С быстро взаимодействуют с мо-
либденом с образованием МоО2 и МоО3.
Галогены. Фтор активно реагирует с молибденом при
комнатной температуре с образованием гексафторида MoF6 (тем-
пература плавления 17,5° С, точка кипения 35° С).
Хлор реагирует с молибденом при температурах выше 500° С
с образованием МоС15 (детальней см. стр. 179).
Пары брома при нагревании (700—800° С) вступают в реакцию
с молибденовым порошком, образуя летучий тетрабромид МоВг4.
Иод при атмосферном давлении практически не взаимодействует
с молибденом вследствие неустойчивости иодистых соединений мо-
либдена. При низких температурах (300° С) под давлением паров
иода порошок молибдена вступает в реакцию с образованием три-
иодида Мо13 (черные кристаллы, точка плавления 900° С) [42].
Окись углерода и углеводороды при температурах
выше 800° С взаимодействуют с молибденом, образуя карбид.
Двуокись углерода и сернистый газ оки-
сляют металл, примерно начиная с 800—900° С.
Сера при температурах выше 400° С вступает в реакцию
с образованием дисульфида.
Сероводород реагирует выше 700—800° С.
Окислы азота N2O, NO, NO2 при 700—800° С окисляют
молибден до МоО3.
Поведение в водных растворах различных реагентов
и расплавах
Молибден устойчив против действия неокисляющих минераль-
ных кислот на холоду и при нагревании: соляной, серной (до
50%-ной концентрации), фосфорной, плавиковой. Это иллюстри-
рует табл. 98, составленная по данным работы [43]. Концентри-
рованная серная кислота (выше 75%) при нагревании до кипения
активно окисляет молибден.
Азотная кислота и царская водка быстро растворяют молибден.
Хорошие растворители молибдена — смеси азотной и серной ки-
слот, используемые для растворения молибденовых кернов, на
которые навивают вольфрамовые спирали.
В перекиси водорода молибден растворяется ср значительной
скоростью, что используют в химическом, анализе для растворения
проб проволоки или тонкого листа [44].
Водные растворы гидроокисей калия и натрия различной кон-
центрации не действуют на молибден на холоду и в очень малой
степени — при нагревании без окислителей. Щелочной раствор
феррицианида окисляет молибден, что используют для травления
металлографических шлифов.
393
ТАБЛИЦА 98
КОРРОЗИЯ МОЛИБДЕНА (ОТОЖЖЕННЫЕ ПЛАСТИНЫ)
В РАЗЛИЧНЫХ МИНЕРАЛЬНЫХ КИСЛОТАХ [431
(размер образцов 1,6X25X50 мм; объем раствора 30 мл/см2', время испытаний 38 ч; аэрация
воздухом; коррозия, мм/год)
Температура, °C НС1 HCl-f-0,5FeCl3
20% 37% 20% 37%
20 71 Кипения 0,00025 0,0035 0,0023 * 0,0005 0,747 4,064 6,35 0,003
Температура, °C H2SO4 н3ро4 HF
20% 50% ю% 50% 25% 49%
20 71 Кипения 0,0025 0,00076 0,00635 * 0,00068 0,0033 * 0,00063 0,0038 * 0,0005 0,508 0,00025 0,406
* Без аэрации воздухом.
В растворе аммиака молибден медленно корродирует, покры-
ваясь темным налетом [45].
Расплавленные щелочи и сода при доступе воздуха окисляют
молибден с образованием молибдатов. Добавки окислителей (ни-
трат или хлорат калия, перекиси) сильно ускоряют окисление.
Коррозия в жидких металлах
Некоторые сведения о стойкости молибдена в жидких металлах
при различных температурах приведены в табл. 99 [46]. Молибден
ТАБЛИЦА 99
СТОЙКОСТЬ МОЛИБДЕНА ПРОТИВ РАЗЪЕДАНИЯ ЖИДКИМИ
МЕТАЛЛАМИ [46]
Металл Точка плавления, °C Стойкость 1 при 600° С
. Hg —38,9 Хорошая
Na, К или их сплав От —12,5 до 98 »
Ga 29,8 Неудовлетворительная
Bi 271 Хорошая
Pb 327,4 »
Li 186 Неизвестна
Mg 650 Хорошая
1 Стойкость молибдена в жидких металлах при 300° С хорошая.
394
устойчив в расплавленных свинце и висмуте при 980° С и сопротив-
ляется действию этих металлов под нагрузкой при 815° С. При сов-
местном погружении нержавеющей стали и молибдена в жидкий
натрий при 1000° С в течение 40 ч не было обнаружено коррозии.
63. ЗАЩИТА МОЛИБДЕНА ОТ ОКИСЛЕНИЯ
Возможности использования молибдена как конструкционного
жаропрочного материала ограничиваются его легкой окисляе-
мостью. Начало окисления молибдена (синевато-стальные цвета
побежалости) отмечается примерно при 300° С, а при 600° С обра-
зуется плотно прилегающий к металлу окисный слой.
При низких температурах (до 400° С) основной составляющей
окисного слоя является трехокись молибдена, тогда как в интер-
вале 400—650° С она состоит из двух слоев: внутреннего, прилега-
ющего к металлу слоя МоО2 и наружного МоО3. Окалина растет
в основном вследствие диффузии кислорода внутрь металла [51].
При 700° С и выше окисление сопровождается улетучиванием трех-
окиси молибдена, что приводит к убыли в массе образца (рис. 180)
[47, 48].
При температурах до 700° С скорость окисления в начальный
период подчиняется параболической зависимости (следовательно,
лимитируется диффузией), а затем зависимость становится линей-
ной. При 700° С и выше происходит быстрый переход к линейному
закону окисления.
Трехокись молибдена плавится при 795° С, а эвтектика МоО3—
МоО2 — при 778° С. Следовательно, выше 780° С окисление сопро-
вождается образованием жидкой фазы, что приводит к резкому
ускорению процесса [55].
При температурах 850—900° С и выше скорость испарения столь
велика, что жидкая фаза на образцах отсутствует, окисление
идет с постоянной скоростью и мало зависит от температуры
(рис. 181). Механизм окисления при температурах 900—1300° С
изучали Архаров и Козманов [50] и ряд других исследователей [51].
По данным работы [50], скорость окисления в значительной
мере зависит от парциального давления кислорода в атмосфере.
Проблему защиты молибдена от окисления можно решать:
1) легированием молибдена с целью придания ему жаростой-
кости при одновременном сохранении жаропрочности;
2) нанесением защитных покрытии.
Многочисленные исследования первого пути до настоящего
времени не привели к положительному результату [43, 53]. За-
щитные покрытия оказались пригодными в определенных условиях
эксплуатации молибденовых деталей. Различные способы нане-
сения покрытий освещены в работе Борисенко [51]. Их можно
подразделить на четыре группы: 1) диффузионное насыщение;
2) плакировка; 3) электролитические покрытия; 4) напыление
жаростойкими сплавами.
395
Диффузионное насыщение заключается в нагре-
вании покрываемой детали при 900—1200° С в порошке наносимого
металла. Для активизации процесса нагревание проводят в атмо-
сфере, содержащей хлористый водород, что приводит к образова-
нию газообразного хлорида диффундирующего металла, взаимодей-
ствующего с покрываемым металлом. Этим методом покрывают
молибден силицидом MoSi2, который обладает высокой жаростой-
костью. Однако его недостаток — хрупкость, недостаточное со-
противление тепловому и баллистическому удару. Диффузион-
ным методом можно также покрывать молибден хромом.
Метод плакировки состоит в совместной горячей
Рис. 180. Изотермы окисления молибдена
.в атмосфере воздуха [47 ]
Рис. 181. Скорость окисления мо-
либдена в атмосфере воздуха при
различных температурах [48]
прокатке при 900—’1100° С молибдена с материалом, стойким
к окислению, например сплавом инконель \ никелем или нержа-
веющей сталью. Этот метод перспективен для защиты листового
молибдена, но для деталей сложной формы не пригоден.
Электролитическим методом получали на
молибдене никелевые, хромовые или хромоникелевые покрытия из
водных растворов. Лучшие результаты дает комбинированное по-
крытие — хром в качестве подслоя, а сверху никель. Изучали
также возможность покрытия алюминием, кремнием и бором из
расплавленных сред. Преимущества электролитического метода
состоят в его применимости для изделий любой формы.
Хромоникелевые покрытия обладают высокой жаростойкостью,
однако они не выдерживают циклического температурного воз-
действия.
Напыление жаростойкими сплавами (ме-
таллизация) осуществляют расплавлением наносимого материала
в пламени горелки и подачей расплавленных частиц на покрывае-
мую поверхность. Перспективными оказались нанесенные напы-
лением покрытия из сплавов А1—Сг—Si, Ni—Сг—В и Ni—Si—В.
1 Сплав 72,7% Ni, 15% Сг, 7% Fe, 1% Nb, 2,5% Ti.
396
Первый из них пригоден для работы при 1430° С, вторые — при
1200° С.
Сплав с алюминием устойчив против окисления и теплового
удара, но плохо противостоит баллистическому удару.
Сплавы на основе никеля жаростойки и противостоят баллисти-
ческому удару, но слабо сопротивляются эрозии и тепловому
удару [43].
В литературе отмечается также возможность защиты молибдена
жаростойкими эмалями и погружением молибдена в ванну из жид-
ких металлов с последующим окислением с образованием покрытия
из тугоплавких окислов (например, Al2O3-SiO2, 3Al2O3-2SiO2,
2MgO-SiO2 и др.) [51, 52].
В настоящее время еще не созданы покрытия, способные обеспе-
чить защиту молибдена в любых условиях эксплуатации. В связи
с этим приходится выбирать тип покрытия с учетом условий, в ко-
торых оно будет применяться.
64. ОБЛАСТИ ПРИМЕНЕНИЯ МОЛИБДЕНА
Молибден, его сплавы и соединения широко используют
в современной технике. Возрастание потребления молибдена
косвенно характеризует табл. 100, в которой приведены данные
об изменении масштабов производства молибденовых концентратов
за период с 1931 по 1967 г.
По оценке фирмы «Клаймакс» [59], структура потребления
молибдена в США характеризуется следующими данными, %:
Легированные стали всех типов............73
Легированный чугун и прокатные валки . . 9
Коррозионностойкие сплавы на никелевой ос-
нове .................................. 4
Жаропрочные сплавы на основе молибдена 6
Металлический молибден (нелегированный) 1
Химикаты и смазки ....................... 7
Итого.............100
Молибден в сталях и чугунах
Около 75% Мо используют в черной металлургии для легиро-
вания сталей и чугунов. В стали молибден входит в состав твер-
дого раствора и сложных карбидов молибдена и железа. Обычно
молибден вводят в сталь вместе с другими легирующими добав-
ками — хромом, никелем, ванадием, марганцем. По данным ра-
боты [61 ], около 60% легированных сталей, выпускаемых в США,,
содержат молибден. “
Конструкционные стали содержат до 0,5% Мо. Молибден
существенно повышает свойства конструкционной стали, улучшая
ее структуру, которая становится более однородной и мелкозер-
нистой. Понижая температуру эвтектоидного распада стали, мо-
либден расширяет температурный интервал закалки и отпуска
397
таблица ibo
МАСШТАБЫ ПРОИЗВОДСТВА МОЛИБДЕНОВЫХ КОНЦЁНТРАТОВ в КАПИТАЛИСТИЧЕСКИХ СТРАНАХ [56—60,66]
Страны Металл в концентратах, т, за годы
1931— 1940 * 1941— 1950 * 1952 1954 1956 1958 1960 1963 1964 1965 1966 £ 1967
Всего 10 000 19 182 24 000 30 252 28 662 25 624 34 116 34 294 35 635 44 549 56 814 ’55 968
В том числе: США .... 8 958 15 961 21 629 26 400 26 060 18 625 30 952 29 488 29 748 35 095 41 065 39500
Чили .... 31 642 1 644 1 208 1 415 1 814 2 014 3 046 4 228 3 603 4 735 5 129
Канада . . • 1.3 151 ' 138 205 381 257 348 454 580 4 335 9 342 9 627
Мексика . . . 368 465 — 72 15 26 60 41 30 49 104 120
Перу .... 84 49 3 1 — — —- 633 455 673 757 794
Австралия . . 15 51 — — — 1 2 . 1 2 3 3
Япония . . . — 12,5 89 204 242 310 381 322 277 277 245 245
Южная Корея . 119 185 7 10 14 31 44 — — 202 299 295
Филиппины . — — — — — 68 107 105 77 50 45
Марокко . . . 122 11,3 —- — — — — — — — - —
Норвегия . . ' 298 148 128 152 166 218 248 ’ 201 201 226 214 210
Финляндия 4,7 75,5 — — — — — —
Прочие страны . . . — —• — — — — — — 1 —
* Среднегодовая добыча.
и влияет на глубину прокаливаемое™ стали. Молибден повышает
механические свойства стали — предел упругости, сопротивление
износу и удару. Одно из ценных свойств молибдена — его спо-
собность устранять отпускную хрупкость аустенитной стали.
Молибден входит в состав многих марок инструментальных ста-
лей (для штампов, быстрорежущих и др.). В сталях для штампов
его содержание колеблется от 1 до 1,5%, а в быстрорежущих ста-
лях от 5 до 7,5—8,5% (в случаях, когда молибден заменяет вольф-
рам). Молибден повышает красностойкость инструментальных
сталей, их твердость и прочность, сопротивление образованию
закалочных трещин, износу.
Присадки от 2 до 4% Мо в нержавеющие хромоникелевые стали
улучшают их антикоррозионные свойства.
Молибден повышает жаропрочность и снижает хрупкость хро-
мистых и хромоникелевых сталей в условиях длительной работы.
Небольшие присадки молибдена вводят в сталь, используя
молибдат кальция или брикеты смеси технической трехокиси мо-
либдена с окисью кальция. В высоколегированные стали моли-
бден вводят в форме ферромолибдена.
Введение в чугун 0,2—0,5% Мо уменьшает склонность к росту
зерен, повышает вязкость, сопротивление износу и улучшает
свойства при повышенных температурах. Из кремнемолибдено-
вого чугуна изготовляют кислотостойкую аппаратуру.
Молибден повышает прочность и вязкость литых стальных
и чугунных прокатных валков, удлиняя срок их службы.
Антикоррозионные и жаропрочные сплавы
с цветными металлами
Молибден входит в состав ряда кислотостойких и жаропрочных
сплавов, в которых он сочетается главным образом с никелем,
кобальтом и хромом. Основные составляющие жаропрочных спла-
вов — никель и кобальт, содержание которых достигает 50—60%.
Большинство жаропрочных сплавов этого типа (одновременно и
коррозионностойких) содержат 20—28% Сг и 3—10% Мо.
Составы и прочностные характеристики подобных сплавов при-
ведены в работе [61].
Например, сплав состава, %: 18 Сг; 37 Ni; 20,0 Со; 3,0 Мо;
2,8 Ti; 17,0 Fe имеет длительную прочность за 1000 ч при 650° С
43,8 кГ1мм\ а при 820° С 12,6 кПмм2. Сплавы такого типа приме-
няют для изготовления лопаток и дисков роторов газовых турбин.
В наиболее кислотостойких сплавах на основе никеля, сопро-
тивляющихся действию всех минеральных кислот, кроме плави-
ковой, содержится 17—28% Мо, остальные компоненты — хром,
вольфрам, железо — в разных количествах. Например, сплав
28% Мо, 5% Fe, остальное никель стоек против действия соляной
и серной кислот любой концентрации на холоду и при нагревании
до 100° С.
399
Применение молибдена и сплавов на его основе
Производство электроламп и электровакуумная техника. Вы-
сокая температура плавления, малая скорость испарения, жаро-
прочность, относительно высокая электропроводность обусловили
применение молибдена в производстве электроламп и электро-
вакуумных приборов. Одно из наиболее ранних применений чи-
стого молибдена — производство электроламп, в которых из мо-
либденовой проволоки изготовляют крючки, поддерживающие
вольфрамовую нить накала, В этой же области молибден исполь-
зуют в качестве керна для навивки («спирализация») вольфрамо-
вой проволоки. После отжига спиралей молибденовый керн уда-
ляют растворением в смеси серной и азотной кислот. Молибдено-
вые прутки, впаиваемые в специальное стекло с коэффициентом
расширения, близким к коэффициенту расширения молибдена,
служат для ввода тока в различные электровакуумные приборы
и колбы мощных источников света.
Молибден широко используют для производства различных
деталей «горячей арматуры» электровакуумных приборов — сеток
приемно-усилительных ламп, анодов и вспомогательных электро-
дов генераторных ламп, катодов газоразрядных трубок. Аноды
изготовляют из молибденового листа, причем аноды больших раз-
меров часто состоят из нескольких частей, соединенных заклеп-
ками или с помощью точечной сварки [31, 32].
Из молибдена изготовляют также ряд деталей рентгеновских
трубок, в частности фокусирующие электроды.
Нагревательные элементы высокотемпературных печей. Мо-
либденовую проволоку, ленту и прутки используют в качестве
нагревателей в электрических печах с температурой до 1700° С.
Различные конструкции таких печей описаны в работах [31, 32].
Нагреватели могут работать в защитной атмосфере водорода,
в аргоне или в вакууме.
В связи с освоением производства крупных слитков молибден
стали применять в качестве электродов в печах для плавки стекла.
Электроды, к которым подводят электроток, представляют собой
молибденовые прутки диаметром от 25 до 150 мм длиной до 1,8 м.
Большей частью их вводят через стенки печи ниже уровня жидкого
стекла. Электроды большого диаметра размещают иногда на подине
печи. Применяют также электроды в форме молибденовых пла-
стин, опускаемых в ванну. Молибден практически не реагирует
с расплавленным стеклом. Мешалки и другие детали стеклопла-
ь вильных печей также изготовляют из молибдена [62].
Авиация и ракетная техника. В этой области применяют ле-
гированный молибден с присадками титана, циркония, ванадия,
ниобия и др., повышающих его жаропрочность, для изготовления
авиационных газовых турбин и ракетных двигателей: рабочие
лопатки и сопловые лопатки турбин, выхлопные сопла и камеры
400
сгорания прямоточных реактивных двигателей, передние кромки
ракет, носовые конусы ракет и другие детали [62—64].
В некоторых деталях ракет срок службы столь мал, что окисле-
ние молибдена не имеет значения. Однако рабочие и сопловые
лопатки газовых турбин и другие детали требуют защиты от окисле-
ния с использованием методов, рассмотренных выше.
Атомная техника. Молибден относится к материалам с отно-
сительно низким сечением захвата тепловых нейтронов (~2,6 бар-
на) и может быть использован в энергетических ядерных реакто-
рах'как конструкционный материал, сохраняющий прочность при
температурах 800° С. Из молибдена можно изготовлять детали
активной зоны реактора — трубы, оболочки, контейнеры и др.
Молибден достаточно устойчив против действия жидких ме-
таллических теплоносителей типа свинцововисмутового сплава
и лития [62].
Обработка металлов давлением. Тугоплавкость, высокая тепло-
проводность, низкий коэффициент расширения в сочетании с жаро-
прочностью делают перспективным применение сплавов на основе
молибдена в области горячей обработки давлением. Так, по дан-
ным работы [62], хорошие результаты получены при использо-
вании прошивных пуансонов для прошивки заготовок из нержа-
веющей стали. Обычные пуансоны из легированной стали выходят
из строя после прошивки одной трубы, тогда как пуансон из
сплава молибдена с 0,5% Ti прошивает около 100 заготовок.
Применение химических соединений молибдена
Дисульфид молибдена (чистый минерал молиб-
денит) — весьма эффективный смазочный материал для трущихся
частей механизмов. Молибденитовая смазка превосходит графит
и может применяться в интервале температур от —40 до +350° С.
Молибдат натрия используют в производстве красок
и лаков. В зависимости от требуемого цвета его применяют один
или в смеси с вольфраматом натрия.
Так как молибдаты восстанавливаются с образованием молиб-
деновой сини, они служат для окраски шелка, шерсти, хлопчато-
бумажных тканей и мехов.
Трисульфид молибдена и окислы МоО3,
МоО2 применяют в качестве катализаторов в химической и неф-
тяной промышленности, в частности при гидрировании углей
и нефти. Детальный обзор использования соединений молибдена
в качестве катализаторов сделан в работе [67].
Молибдат аммония широко используют в лаборатор-
ной практике в качестве реактива.
В последние годы в связи с установленным влиянием микро-
количеств молибдена в почве на стимулирование роста растений,
особенно бобовых культур, расширяется применение молибдата
аммония впроизводстве микроудобрений [65 ].
26 А. Н. 3еликман 401
ГЛАВА XVI
ПОПУТНОЕ ИЗВЛЕЧЕНИЕ РЕНИЯ В ПРОИЗВОДСТВЕ
МОЛИБДЕНА
Д. И. Менделеев в 1869 г. предсказал существование двух
элементов VII группы, аналогов марганца, которым предвари-
тельно дал названия «эка-марганец» и «дви-марганец». Они соот-
ветствуют известным в настоящее время элементам — технецию
(порядковый номер 43) и рению (порядковый номер 75). В после-
дующие 53 года многие исследователи сообщали об открытии
аналогов марганца, но без достаточно убедительных оснований.
Теперь мы знаем, что поиски элемента № 43 в природных соеди-
нениях не могли увенчаться успехом, так как он неустойчив.
Лишь в 1937 г. Сегре и Перье искусственно получили этот эле-
мент бомбардировкой ядер молибдена дейтонами и назвали его
технецием, так как это был первый элемент, полученный искус-
ственно (от греческого слова техне — искусственный).
В 1922 г. немецкие химики Вальтер и Ида Ноддак начали си-
стематические поиски аналогов марганца в различных минералах.
Из 1 кг колумбита они выделили 0,2 г продукта, обогащенного
молибденом, вольфрамом, рутением и осмием. В этом продукте
был обнаружен новый элемент с порядковым номером 75, что
подтвердили характеристические рентеновские спектры. О своем
открытии Ноддаки сообщили в 1925 г. Они назвали элемент
рением. Несколько месяцев спустя последовали сообщения чеш-
ских ученых Друце и Лоринга, а также Гейровского и Долейжака
об открытии ими элемента № 75 в пиролюзите. В последующем
было установлено, что в наибольших концентрациях рений на-
ходится в молибденитах [1].
65. ОБЩИЕ СВЕДЕНИЯ О РЕНИИ
. Физические свойства
Рений — тугоплавкий тяжелый металл, по внешнему виду
похожий на сталь. Некоторые физические свойства рения [3—8,
11] приведены ниже.
402
Атомный номер.................................. 75
Атомная масса ..............................* 186,31
Кристаллическая структура ................... Гексагональная ,
о « плотноупакованная
Периоды решетки, А ....................... а — 2,76; с = 4,45*
Плотность, г/см3 ........................... 21,01
Температура плавления, °C .................., 3180±20
Температура кипения, °C ..................... 5900
Давление пара при 2225° С, мм рт. ст......... 1,18-10“6
Удельная теплоемкость при 0—1200° С (средняя),
кал!(г-град) . . . ......................... 0,03653
Коэффициент линейного расширения:
вдоль оси С . . ................................ 12,45 • 10“б
перпендикулярно оси с . ;..................... 4,67 • 10"6
Удельное электросопротивление, ом-см*1№, при
температуре, °C:
20.................................................. 19,8
2220 ................................... 125
2710................................ 134
Работа выхода, эв............................ 4,8
Теплопроводность, кал! (см* сек-град) .............. 0,17
Твердость по НВ, кПмм2, ........... —200
Предел прочности при растяжении (кованые и затем
отожженные прутки), кГ/мм2 ...................... 115,5
Модуль упругости, кГ/мм2 . ......................... 47 000
Сечение захвата тепловых нейтронов, барн ... 85
По температуре плавления рений занимает второе место среди3
металлов, уступая лишь вольфраму, а по плотности стоит на
четвертом месте (после осмия, иридия и платины). Удельное
электросопротивление рения почти в четыре раза выше, чем*
у вольфрама и молибдёна.
В отличие от вольфрама рений пластичен в литом и рекристал-
лизованном состоянии и может быть деформирован на холоду.
Рений обладает очень высоким модулем упругости. После
небольшой степени деформации его твердость сильно возрастает
(проявляется сильный наклеп), но после отжига в защитной среде
(водороде) или в вакууме он вновь приобретает пластичность.
Изделия из рения (в отличие от изделий из вольфрама) выдержи-
вают многократные нагревы и охлаждения без потери прочности.
Сварные швы не хрупкие.
Причиной высокой пластичности рения является то, что ме-
талл имеет гексагональную структуру и механическая деформа-
ция осуществляется скольжением и двойникованием, в то время7
как у вольфрама — только скольжением.
Прочность рения при температурах до 1200° С выше, чем.
вольфрама, и значительно превышает прочность молибдена.
Химические свойства
Рений устойчив на воздухе при обычной температуре. Окисле-
ние металла с образованием рениевого ангидрида Re2O7 наблю-
дается выше 300° С и интенсивно протекает при температурах
выше 600° С. Тонкие порошки рения при хранении на воздухе1
26*
403
увлажняются, что объясняется частичным окислением до Re2O7,
поглощающего влагу.
С водородом рений не реагирует вплоть до температуры плав-
ления. Металл значительно устойчивей вольфрама в атмосфере,
содержащей следы влаги, при высоких температурах.
С азотом не взаимодействует. Известны нитриды рения, но
они получаются не прямым действием азота на металл, а по реак-
ции между хлоридом рения и аммиаком.
Рений не образует карбидов. Эвтектика в системе рений —
углерод плавится при 2480° С. Несмотря на отсутствие химиче-
ского взаимодействия с углеродом, не рекомендуется нагревать
рений в контакте с графитом, так как углерод диффундирует
в металл, что снижает его механическую прочность.
С фтором и хлором рений реагирует при нагревании, с бромом
и йодом металл практически не взаимодействует.
Рений не корродирует в соляной и плавиковой кислотах на
холоду и при нагревании. В азотной кислоте, горячей концентри-
рованной серной кислоте и перекиси водорода он растворяется.
Рений устойчив против действия расплавленных олова, цинка,
серебра и меди, слегка разъедается алюминием и легко раство-
ряется в расплавленных никеле и железе.
Рений в значительно меньшей степени, чем вольфрам,взаимодей-
ствует с окисью алюминия при высоких температурах в вакууме,
в очень малой степени химически взаимодействует с окисью
стронция при высоких температурах в вакууме.
Наиболее устойчивы химические соединения семивалентного
рения. Кроме того, известны соединения, отвечающие положи-
тельной валентности VI, V, IV, III, II, I, а также соединения
с валентностью минус 1. Для технологии рения представляют
интерес лишь соединения с семи-, шести-, четырех- и трехвалент-
ным рением [1—3].
К важнейшим кислородным соединениям рения относятся
рениевый ангидрид Re2O7 (плавится при 297° С, ки-
пит при 363° С), рениевая кислота и ее соли п е р -
ренаты. Данные по растворимости некоторых перренатов
в воде приведены в табл. 101.
Среди низших окислов следует упомянуть двуокись ReO2 —
промежуточный продукт при восстановлении перрената аммония
водородом.
Среди хлористых соединений представляют интерес для техно-
логии рения высший хлорид ReCl5 (точка плавления 260° С,
точка кипения 330° С), трихлорид ReCl3 (плавится около 730° С)
и легколетучие оксихлориды ReOCl4 (температура кипения 228° С)
и ReO3Cl (кипит при 130° С) [24].
Известны два сульфида рения: Re2S7 (количественно осаж-
дается из кислых растворов сероводородом) и дисульфид ReS2,
изоморфный MoS2.
404
ТАБЛИЦА 101
НЕКОТОРЫЕ СВОЙСТВА ПЕРРЕНАТОВ
Перренат Растворимость, г/100 г Н2О, при температуре, °C Температура плавления, °C
0 30 50
LiReO4-2H2O 293,25 410 410 87,5
NaReO4 103 145 173 414
KReO4 0,36 ' 1,47 3,2 555
NH4ReO4 2,76 8,7 16,01 Разлагается
выше 200° С
RbReO4 0,39 1,57 3,52 598
CsReO4 0,38 1,11 2,47 616
AgReO4 0,43 1,39 2,71 455
TlReO4 0,115 0,298 0,555 525
Mg (ReO4)2 — 283,6 — 930
Ca (ReO4)2 — 187,0 — 934
Zn (ReO4)2 — 313,6 701
Cu (ReO4)2 — 210,4 — Разлагается
Pb (ReO4)2 4,14 14,81 29,63 562
Ni (ReO4)2 — 310 * — Разлагается
Fe (ReO4)2 — 233 * • »
Перренат нитрона 0,018 ** — — —
А При 27° С.
При 15° С.
Области применения рения [4—6, 11]
В настоящее время уже определились области эффективного
применения рения, причем спрос на металл превышает масштабы
его производства.
Производство электроламп и электровакуумных приборов.
В этой области уже применяют в ряде ответственных случаев
вместо вольфрама рений или сплавы рения с вольфрамом и молиб-
деном. Преимущества рения перед вольфрамом состоят в лучших
прочностных характеристиках и сохранении пластичности в ре-
кристаллизованном состоянии; меньшей склонности к испарению
в присутствии следов влаги (сопротивление водородноводяному
циклу); более высоком электросопротивлении; отсутствии устой-
чивых карбидов.
Перечисленные преимущества рения выявляются особенно
в тех случаях, когда необходима долговечность работы ламп и
электронных приборов, в особенности в условиях динамической
нагрузки (например, в электронных лампах для радарных уста-
новок и авиации, в фотопроекционных лампах, лампах для же-
лезнодорожного транспорта и др.). Рений и сплазы вольфрама
с рением применяют для изготовления нитей накала, в качестве
керна катодов и поддогревателей, а также материала навивки
405
сеток радиоламп. В отличие от вольфрама рекристаллизованный
сплав W + 30% Re достаточно пластичен при комнатной темпе-
ратуре.
В электронных приборах используют также сплав Мо +
+ 50% Re. В сплаве этого состава сочетается высокая пластич-
ность (обрабатывается на холоду) с прочностью.
В связи с меньшей, чем у вольфрама, склонностью к испарению
в присутствии следов влаги рений применяют в разрядных труб-
ках, заполненных водородом с примесью паров воды.
Термопары для высоких температур. Термопары из рения
и его сплавов с вольфрамом и молибденом обладают высокой
и стабильной термо-э. д. с.
Большое достижение последнего времени — разработка в СССР
термопары из сплавов W + 5% Re, W + 20% Re. Термо-э. д. с.
этой термопары в пределах температур 0—2500° С находится
в линейной зависимости от температуры. При 2000° С термо-
э. д. с. = 30 мв. Важное преимущество термопары — сохранение
пластичности термоэлектродов после длительного нагревания при
высокой температуре [4, с. 162, 5, с. 218].
Электроконтакты. Рений и его сплавы с вольфрамом отли-
чаются высокой износостойкостью, сопротивлением электроэро-
зии в условиях образования электрической дуги. Исследования
показали, что рений и его сплавы с вольфрамом имеют преимуще-
ства перед вольфрамом как материал для контактов, обладающий
большей стойкостью к атмосферной и тропической коррозии,
а также постоянством контактной проводимости при воздействии
повышенных температур и контактных дуг. Имеются сообщения
о большой эффективности применения рениевых контактов в ко-
рабельных магнето. Испытания контактов из сплава W + 15% Re
в регуляторах напряжения и в приборах зажигания показали
их преимущества перед вольфрамовыми контактами [4, с. 180].
Жаропрочные и тугоплавкие сплавы. Сплавы рения с другими
тугоплавкими металлами (вольфрамом, молибденом, танталом)
отличаются высокой жаропрочностью и тугоплавкостью. Одно-
временно с этим добавки рения в вольфрам и молибден улучшают
их пластические свойства. Жаропрочные сплавы рения применяют
для изготовления ответственных деталей сверхзвуковых самоле-
тов и ракет [11].
Приборостроение. Вследствие высокой твердости и износо-
стойкости рения и его сплавов весьма перспективно их применение
в приборах, например, для изготовления опор для весов, осей
геодезической аппаратуры (компасы и др.), шарнирных опор и т. п.
Большой интерес рений представляет как материал для пру-
жин, работающих при высоких температурах. Испытания работы
плоских пружин из рения при температуре 800° С при много-
кратных циклах нагрева показали отсутствие деформации и сохра-
нение начальной твердости.
406
Покрытия. Для защиты от коррозии и износа применяют
покрытия рением других металлов (меди, серебра, никеля). При-
меняют также покрытия рением деталей из тугоплавких металлов
(вольфрама, молибдена) в радиоэлектронике.
66. СЫРЬЕВЫЕ ИСТОЧНИКИ РЕНИЯ
Рений — типичный рассеянный элемент. Весовое содержание
рения в земной коре низкое 10"7%.
Собственных минералов рения не найдено г. Более высокое
содержание рения (по сравнению со средним его содержанием
в земной коре) наблюдается в гранитных пегматитах и пневмато-
литических образованиях. Повышенные концентрации рения,
имеющие промышленное значение, отмечаются в сульфидах меди
и особенно в молибдените, который в настоящее время служит
основным источником получения рения.
Связь рения с молибденом обусловлена изоморфизмом MoS2
и ReS2 (радиус иона Мо4+ равен 0,68 A, Re4+ 0,56 А).
Содержание рения в молибденитах различных месторождений
лежит в пределах 10"2—10 "5%. Более богаты рением молибде-
ниты медно-молибденовых месторождений. Так, молибдениты
Мансфельдского месторождения медистых сланцев (ГДР) содер-
жит 0,01 % Re. Молибденитовые концентраты, получаемые из
медно-молибденовых месторождений СССР, содержат от 0,025
до 0,04% Re [4, 10].
Молибденитовые концентраты обычно подвергают окислитель-
ному обжигу при температуре 550—600° С, в результате которого
получают огарок, содержащий трехокись молибдена и ряд при-
месей. При этом содержащийся в молибдените рений образует
семиокись Re2O7, которая уносится с газовым потоком (точки
кипения Re2O7 363° С). Степень возгонки рения зависит от усло^
вий проведения обжига и минералогического состава концентрата.
Так, например, при обжиге некондиционных молибденитовых
концентратов на поду муфельной печи с ручным перегребанием
материала на одном из заводов СССР степень возгонки рения
составляла 65—75%. В огарке оставалось 35—25% Re.
Неполный возгон рения при обжиге на поду муфельной печи
может быть обусловлен частичным взаимодействием Re2O7 с каль-
цитом, а также окислами железа и меди с образованием перре-
натов. Так, возможно образование перрената кальция по реакции
СаСО3 + Re2O7 -> Са (ReO4)2 + СО2.
В случае недостатка воздуха, например внутри спекшихся
кусков, может образоваться двуокись рения в результате взаимо-
действия Re2O7 с молибденитом по реакции [13, 14]
2Re2O7 + MoS2 —> 4ReO2 4 МоО2 4 2SO2.
1 Появились сообщения о существовании трех минералов рения: окисла,
сульфида и сульфорената. Однако ни один из них не выделен.
407
Советские исследователи установили, что наиболее полно
рений возгоняется при обжиге молибденитовых концентратов
в кипящем слое [12]. Степень возгона рения в этом случае со-
ставляет 92—96%. Это объясняется тем, что в условиях обжига
в кипящем слое в меньшей степени протекают побочные реакции
образования перренатов и невозможно образование низших
окислов рения.
При обычной системе пылеулавливания, состоящей из пыле-
вых камер и электрофильтров (при обжиге в муфельных печах)
или циклонов и электрофильтров (при обжиге в печах кипящего
слоя), не обеспечивается удовлетворительное улавливание рения.
В зависимости от режима работы электрофильтров 70—80% Re
теряется с отходящими газами.
В случае обжига в кипящем слое запыленность газов весьма
высокая, так как 25—45% Мо уносится с газами. Основная часть
уносимого молибдена (до 85%) улавливается циклонами. Чтобы
рений не улавливался в циклонах, необходимо поддерживать
в них температуру порядка 400° С. В пыли, оседающей в газо-
ходах за циклонами и шламах сухих электрофильтров, содержа-
ние рения колеблется от десятых долей процента до 1,5%. Однако
степень улавливания рения в этом случае низкая.
Эффективное улавливание рения достигается при установке
за циклонами мокрых пылеуловителей (скрубберы, барботеры,
мокрые электрофильтры) [15].
При мокром пылеулавливании большая часть рения содержится
в кислых растворах. Чтобы увеличить концентрацию рения,
растворы многократно возвращают в мокрый пылеуловитель
(циркуляция растворов). Растворы, выводимые из системы мою
рого пылеулавливания, содержат, г!л: 0,2—0,8Re; 2—5 Мо и
50—80 H2SO4. Часть рения содержится в шламах.
В случае неполного возгона рения при обжиге молибденито-
вого концентрата рений, оставшийся в огарке, переходит вместе
с молибденом в растворы выщелачивания и после осаждения соеди-
нений молибдена остается в маточных растворах.
Таким образом, источниками рения при переработке молибде-
нитовых концентратов могут служить пыли обжига концентратов,
кислые растворы мокрых пылеуловителей и маточные (сбросные)
растворы после гидрометаллургической переработки молибдено-
вых огарков.
67. ИЗВЛЕЧЕНИЕ РЕНИЯ ИЗ РАЗЛИЧНЫХ ОТХОДОВ
ПЕРЕРАБОТКИ МОЛИБДЕНИТОВЫХ КОНЦЕНТРАТОВ
В зависимости о г природы ренийсодержащих материалов
(пыль, шламы, растворы) и их состава применяют различные
схемы извлечения из них рения.
В пылях обжига молибденитовых концентратов рений содер-
жится преимущественно в составе Re2O7. Это позволяет приме-
408
нить для извлечения рения выщелачивание пыли водой. Так как
часть рения может находиться в пыли в виде малорастворимых
в воде низших окислов, выщелачивание ведут с добавкой окисли-
телей, например пиролюзита (МпО2).
Выщелачиванию пыли иногда предшествуют предварительные
пирометаллургические операции: обжиг с возгонкой (с целью
повышения концентрации рения в пыли) или обжиг пыли с из-
вестью для связывания МоО3 в молибдат кальция СаМоО4. При
последующем выщелачивании водой малорастворимый молибдат
кальция отделяется от извлекаемого в раствор перрената кальция.
Из различных производственных растворов рений извлекают
следующими способами:
1) осаждением малорастворимых соединений (перрената ка-
лия KReO4, сульфида Re2S7);
2) сорбцией на ионообменных смолах или угле;
3) экстракцией органическими растворителями.
Последние два способа применяют для получения более кон-
центрированных растворов, из которых затем выделяют перренат
калия или перренат аммония — соли, являющиеся исходными,
для производства рения.
Ниже рассмотрены примеры технологических схем, в которых,
используют упомянутые способы извлечения рения из отходов
производства.
Извлечение рения из пыли газоходов и шламов
электрофильтров
Пыль, улавливаемая в газоходах и шламах электрофильтров^
содержит 0,4—1,5% Re. Основная составляющая пыли — соеди-
нения молибдена (МоО3, MoS2). В пыли обычно много серной:
кислоты, так как в обжиговых газах содержится SO3 и пары
воды. Схема переработки пыли и шламов, применяемая на одном
из заводов СССР, показана на рис. 182 [4, с. 51].
Пыль двукратно выщелачивают горячей водой при отношении,
т : ж = 1 : 2,5 4-3, добавляя в раствор тонкоизмельченный пи-
ролюзит (МпО2) для окисления низших соединений рения. Не-
растворимый остаток отделяют фильтрацией. Объединенные рас-
творы и промывные воды содержат, г!л\ 0,5—0,6 Re; 8—10 Мо
и 20—30 H2SO4, а также сульфаты меди и железа.
Для выделения большей части молибдена, а также меди и:
железа растворы нейтрализуют известковым молоком до pH —
= 11-4-12. При нагревании до 60—70° С из раствора выделяются1
СаМоО4, CaSO4, гидроокиси меди и железа. Потери рения с осад-
ком после его промывки незначительны. Растворы упаривают
в аппарате из нержавеющей стали до концентрации рения 15—
20 г!л и затем осаждают перренат калия добавлением в нагретый
раствор хлористого калия (из расчета 50—60 г КС1 на 1 л рас-
123 , 40В
твора). После охлаждения отделяют кристаллический осадок
технического перрената калия. Степень осаждения рения, состав-
ляет 98—99%. Маточные растворы содержат 0,02—0,05 г!л Re.
Из них рений можно извлечь сорбцией на смолах. Технический
перренат калия очищают, проводя несколько последовательных
перекристаллизаций.
Растворимость KReO4 в воде при 90—100° С — около 100 г!л,
а при 20° С 10 г/л. Следовательно, выход кристаллов- при пере-
Пыло и шламы
Вода I Пиролюзит
Выщелачивание
Фильтрация
Остаток Раствор Л .
| I Са(0Н)2
В производство молибдена | |
Нейтрализация
Раствор Осадок
| СаНоОь, CaSOk, Ре (ОН) 3, Си(0Н)2
Выпаривание |
- I В производство
1 I соединений молибдена
Осаждение перрената калия
Осадок KReOk Раствор
Очистка перекристаллизацией Но регенерацию рения
Чистый кВеОц
Рис. 182. Схема извлечения рения из пыли газоходов и шламов электрофильтров
кристаллизации составит примерно 90%. При охлаждении рас-
твора до 5° С выход можно увеличить до 95%. Общее извлечение
рения из пыли в перекристаллизованный перренат калия состав-
ляет 85 %.
Другой вариант переработки пыли состоит в предварительном
«обжиге пыли с известью при температуре 570—670° С. Пыль сме-
шивают с гашеной известью в отношении 3 : 2 (по массе) и обжи-
гают на поду муфельной печи 2—4 ч. Затем выщелачивают про-
дукт обжига водой при т : ж = 1 : 3 и температуре 70—80° С.
Чтобы увеличить концентрацию рения, последовательно выщела-
чивают одним и тем же раствором до шести порций спека. Про-
мывные воды — оборотные. Растворы содержат до 0,3 г/л Re.
Их упаривают и выделяют перренат калия, как описано выше [3]
410
Сорбционное извлечение рения из маточных растворов
после выделения молибдата кальция
При переработке медно-молибденовых концентратов на молиб-
дат кальция по схеме, описанной в гл. V, в маточных растворах
остается весь рений. Примерный состав растворов, г!л [3]: 0,015—-
0,04 Re; 0,4—0,9 Мо; 27—30 СГ; 15—35 Sol-; 0,7—1,3 С1ОГ;
28—35 Na+; pH = 8,54-8,7.
Для извлечения молибдена и рения из таких растворов при-
меняют избирательную сорбцию ионов молибдена на анионито-
вой смоле, а затем сорбцию рения из раствора на активированном
угле [3, 23].
Раствор подкисляют до рН= 3, нагревают для удаления угле-
кислого газа (в исходном растворе содержится сода). Затем хо-
лодный отфильтрованный раствор пропускают через ионитовую
колонку, заполненную анионитом «Эспатит АН-1» в сернокислой
форме. Величина зерен смолы 0,15—0,6 мм. В слабокислом рас-
творе молибден находится в составе полианионов, которые сорби-
руются на смоле.
Из слабокислого раствора анионитом АН-1 ионы ReOT почти
не сорбируются и остаются в фильтрате, который содержит 15—-
40 мг!л Re и 10—20 мг!л Мо. Такие растворы поступают на адсорб-
ционные фильтры, заполненные активированным углем (марки
«КАД») крупностью 0,1—0,8 мм, который полно адсорбирует
из раствора рений и молибден [23].
Емкость угля по рению (при его содержании в растворе до*
20 мг!л) низкая (примерно до 2%). С угля можно избирательна
десорбировать вначале молибден (холодным 1%-ным раствором
Na2CO2), а затем рений нагретым до 90° С раствором соды. Рас-
творы содержат 200—400 мг!л Re. С целью концентрирования:
рения повторяют операцию сорбции на угле. Из полученных,
растворов осаждают перренат калия.
Возможно разделение молибдена и рения сорбцией на сильно-
основных смолах [18].
Извлечение рения из кислых растворов экстракцией
органическими растворителями
Рений экстрагируется из кислых растворов различными орга-
ническими растворителями: спиртами [2, 27], трибутил фосфатом
[5, с. 66; 28], аминами [17, 21, 28].
Как показано автором и Дрэган [17], из перечисленных типов
экстрагентов наиболее эффективен для экстракции рения из.
сернокислых растворов триоктиламин (ТОА) (рис. 183). В отли-
чие от ТБФ и изоамилового спирта (ПАС) его можно использовать
при любом разбавлении, что важно при экстракции из бедных,
растворов (табл. 102).
411!
Триоктиламин хорошо экстрагирует рениевую кислоту в ши-
роком интервале концентраций серной кислоты (от 1 до 50%).
Состав экстрагируемого соединения (R3N)2 HReO4. В интервале
кислотности 0,3—2,5 моль!л H2SO4 и концентрации ионов ReO4X
х5-10~4 моль!л среднее значение константы равновесия экстрак-
Равновесная яонцентрп1(ия[н2ЗО1}]6 гмол
Рис. 183. Зависимость коэффициента рас-
пределения рения D от концентрации
серной кислоты:
1 — ИАС, 50% в керосине; 2 — ДОА, 0,75%;
3 — ТБФ, 50%; 4 — ТОА, 0,75%
ции равно ~2-106.
Присутствие в сернокис-
лом растворе ионов С1“ и
NOjT в широком интервале
концентраций (от 20 до 150 г/л)
не снижает степени экстрак-
ции рения ТОА. Реэкстрак-
ция рения из ТОА легко
осуществляется 5—10 % -ным
раствором аммиака.
Для предотвращения вы-
падения солей аминов, срав-
н ител ьно малор аствор имых
в разбавителе — керосине,
в органическую фазу обычно
добавляют спирты (фракции
С7—С9). Однако они несколь-
ко снижают коэффициент
распределения рения. В работе [29] была показана возможность
замены спирта добавками ТБФ, который, в отличие от спиртов,
не снижая экстрагируемое™ рения, устраняет выпадение солей
амина.
По данным работы [26], экстракцию ТОА используют для
извлечения рения из азотносернокислых маточных растворов,
ТАБЛИЦА 102
КОЭФФИЦИЕНТ РАСПРЕДЕЛЕНИЯ РЕНИЯ В ЗАВИСИМОСТИ ОТ
КОНЦЕНТРАЦИИ ЭКСТРАГЕНТА В КЕРОСИНЕ
(концентрация Re 100 мг/л, отношение объема фаз 1:1)
Концентра- ции экстра- гента в керо- сине, % ТБФ ИАС ТОА
Концентрация H2SO4, %
10 30 5 25 10 30
0,05 0,5 1,0 5,0 10,0 30,0 50,0 75,0 100,0 0,06 0,06 3,05 8,33 21,4 0,256 1,26 39,0 38,5 39,6 0,29 7,43 2,99 60,8 45,17 49,8 53,5 323,3 66,5 186,5 498
412
получаемых при разложении молибденитового концентрата азот-
ной кислотой. Исходные растворы содержат, г/л: 0,5—3 Мо;
0,015—0,030 Re; 100—150 NO^J 50—100 SO4~. Первоначально
из раствора при pH = 2 ди-2-этилгексилфосфорной кислотой
извлекается молибден, а затем ТОА рений. Экстракция рения
осуществляется 7—8% раствором ТОА в керосине с добавкой ТБФ
(до полного растворения нитрата амина) при pH исходного рас-
твора 1—2 и отношении объемов О : В = 1 : 1.
Для концентрирования рения при реэкстракции 10%-ным раст-
вором аммиака поддерживают отношение объемов О : В = 10 : 1.
В результате двукратного концентрирования рения в цикле
экстракция — реэкстракция содержание рения в аммиачном
реэкстракте достигает 1,5—2 г/л. Высокое содержание нитрат-
ионов и примесь органических веществ затрудняют переработку
реэкстрактов на соли рения. Для получения перрената калия
из рениевого реэкстракта принята схема, включающая выпарку
раствора, смешивание его с известью, сушку и прокалку извест-
ковой пасты при 600° С (для удаления нитрат-ионов и органиче-
ских веществ), выщелачивание водой перрената кальция из
продукта прокаливания, упарку фильтрата и осаждение из него
перрената калия. Из полученного раствора вместо выделения
перрената калия можно непосредственно получать перренат аммо-
ния методом ионного обмена на слабоосновной смоле АН-2Ф.
Описанная технология экстракции ТОА может быть исполь-
зована для извлечения рения из сернокислых растворов мокрых
систем пылеулавливания.
Для экстракции рения предложено также использовать че-
твертичные аммониевые соли (см. стр. 161) [19].
68. ПОЛУЧЕНИЕ РЕНИЯ
Известные способы получения рения можно подразделить на
следующие группы [5, с. 71; 8].
1. Восстановление перрената калия или перрената аммония
водородом.
2. Восстановление двуокиси рения водородом.
3. Электролитическое выделение рения из водных растворов.
4. Термическая диссоциация галогенидов рения.
Последние два способа применяют преимущественно для
получения рениевых покрытий.
Восстановление перрената калия водородом
Извлечение рения из различных обогащенных им продуктов
часто заканчивается получением перрената калия. Естественно,
что рений первоначально получали восстановлением этой соли
водородом по реакции
KReO4 + Зг/2Н2 -> Re + КОН + ЗН2О.
413
Восстановление проводят в две стадии. После первого восста-
новления при температуре 500—550° С продукт восстановления
многократно промывают водой для удаления щелочи. Затем его
повторно восстанавливают при 900—1000° С. Рениевый порошок
промывают разбавленной соляной кислотой, водой и сушат в ва-
кууме или в токе водорода.
Порошки рения, полученные восстановлением перрената ка-
лия водородом, содержат не ниже сотых долей процента калия.
Содержание примеси калия не удается снизить даже после дву-
кратного восстановления и промывки порошка кислотой..
Между тем примесь калия при ее содержании выше 0,006% тор-
мозит спекание рениевых штабиков (не удается -получить шта-
бики с плотностью более 60—70% от теоретической), что обуслов-
ливает хрупкость металла, невозможность его обработки давле-
нием. В связи с этим в настоящее время восстановление перрената
калия водородом применяют только для получения технических
рениевых порошков.
Восстановление перрената аммония водородом
Восстановление перрената аммония водородом — наиболее
распространенный способ производства чистого рениевого по-
рошка, который затем превращают в плотные штабики, поддаю-
щиеся обработке давлением.
Получение перрената аммония. Перренат аммония большей
частью получают из перрената калия, для чего используют раз-
личные способы, кратко рассмотренные ниже.
Способ возгонки семиокиси рения Re2O7*
Первоначально перренат калия восстанавливают водородом при
500° С, получая после промывки водой технический рениевый
порошок, который окисляют кислородом при 800° С в кварцевой
трубке. Семиокись рения возгоняется и конденсируется на хо-
лодном конце трубки. Ее растворяют в небольшом количестве
воды и раствор, нейтрализуют аммиаком для выделения перрената
аммония. Маточный раствор выпаривают для доизвлечения рения..
Осажденный перренат аммония содержит примесь гидроокиси
железа. Для очистки от железа соль растворяют в разбавленном?
растворе аммиака. Затем раствор выпаривают и кристаллизуют
перренат аммония [30].
Извлечение рения в перренат аммония по этому методу состав-
ляет 85%, остальное количество рения дополнительно извлекается:
из маточных растворов и промывных вод. Общая сумма примесей
в полученном этим способом перренате аммония около 0,02%.
Как показали исследования Павловой [5, с. 117], метод воз-
гонки обеспечивает эффективную очистку при относительно низ-
ком содержании калия (0,02—0,04%).
414
Способ ионного обмена. Предложен в СССР
Ковыршиным [16]. Способ основан на использовании реакций
ионного обмена.
При пропускании раствора перрената калия через катионит
в Н+-форме или ИН^-форме протекают реакции обмена с полу-
чением раствора рениевой кислоты или перрената аммония:
. [Кат.] Н + KReO4 -» [Кат.] К + HReO4,
[Кат.] NH4 + KReO4 - [Кат.] К + NH4ReO4.
Таким образом, в результате обменной сорбции на катионите
можно получить либо раствор рениевой кислоты, либо непосред-
ственно раствор перрената аммония. Для этой цели может быть
использован советский катионит КУ-2.
Другой вариант метода состоит в применении анионообменных
смол. В .этом случае на смоле поглощаются анионы ReOJ”, кото-
рые затем вымываются гидроокисью аммония.
Этим способом можно получить перренат аммония с содержа-
нием примеси калия 0,01—0,005%.
Способ экстракции. Способ разработан Бибиковой
и др. [5, с. 66]. Он состоит в экстракции рения из раствора перре-
ната калия, содержащего 0,3-н. НС1, трибутилфосфатом с после-
дующей реэкстракцией 2—2,5-н. раствором аммиака. Способ
позволяет получить перренат аммония высокой чистоты с содержа-
нием всех примесей от* 0,001 до 0,0001%.
Сульфидный способ. Перренат калия при нагре-
вании растворяют в 10%-ной соляной кислоте. Из раствора
пропусканием сероводорода осаждают сульфид рения Re2S7>
который суспендируют в 10%-ном растворе аммиака и окисляют
перекисью водорода. При этом получают раствор, содержащий
перренат и сульфат аммония. Затем выпаривают раствор и кри-
сталлизуют перренат аммония.
.Растворимость перрената аммония в присутствии сульфата
аммония сильно понижается и составляет около 5 г/л, что позво-
ляет выделить из раствора большую часть рения.
Восстановление. Полученный одним из рассмотренных выше
способов перренат аммония обычно содержит 0,01—0,005% ка-
лия и небольшое количество примесей железа, кремния, алюми-
ния и других элементов.
Для получения мелкозернистого рениевого порошка перренат
аммония перед восстановлением измельчают в покрытых рези-
ной мельничных барабанах с измельчающими телами из облом-
ков рениевых штабиков. Измельченный перренат аммония вос-
станавливают водородом в трубчатых печах. Соль засыпают тон-
ким слоем (6—8 мм) в лодочки из молибдена или сплава никеля
с молибденом. Первоначально соль сушат в токе водорода при
415
300° С в течение 1 ч. Затем ведут восстановление при 800°С 1—2 ч.
Размер частиц получаемого порошка от 1 до 25 мкм.
Примерный химический состав порошков приведен в табл. 103.
ТАБЛИЦА 10$
СОСТАВ ПОРОШКОВ РЕНИЯ, ПОЛУЧЕННЫХ РАЗЛИЧНЫМИ СПОСОБАМИ
Состав порошков, полученных разными способами, %
Элемент из KReO4 из NH4ReO4, полученного экстракцией [5, с. 66] из ReO2, полу- ченной разло- жением ReCl5 [30] очисткой через: оксихлорид ReOCl4 [31]
Fe 0,02 0,0003 0,012 0,00004
Al 0,002 0,0003 0,008 0,0003
Мо 0,001 <0,0002 Не обнаружен 0,0002
Ni 0,028 0,00004 » » 0,00004
Си 0,0035 0,0003 0,0002 0,0003
к 0,04 0,004 Не обнаружен 0,0007
Na 0,06 0,0005 » » 0,0001
Са 0,1 <0,001 0,002 0,0014
РЬ 0,002 — Не обнаружен —
Sn 0,0006 —1 — —
Cd 0,001 . —— — —
Mg — 0,0002 0,005 0,00006
Si — 0,003 0,015 0,0002
Очистка рения через летучие хлористые соединения
Очистить рений от примесей, в особенности щелочных и щелоч-
ноземельных металлов, можно хлорированием технических рение-
вых порошков (например, полученных восстановлением перре-
ната калия) с возгонкой пятихлористого рения. Разработаны два
варианта последующего получения очищенного порошка рения
из ReCl6.
Способ института им. Батале (США) [30]. Исходный рений
хлорируют при температуре 750° С. Образующийся пятихлори-
стый рений дистиллируют. Полученный хлорид разлагают водой.
При этом около 70% Re осаждается в виде гидратированной
двуокиси рения:
3ReCl5 + (8 + х) Н2О — 2ReO2-x Н2О + HReO4 + 15НС1.
Отфильтрованный и высушенный осадок двуокиси рения вос-
станавливают водородом в две стадии — вначале при 400—600° С,
а затем (после растирания порошка) при 800° С. Из маточного
раствора после окисления соединений низшей валентности пере-
кисью водорода выделяют перренат аммония, поступающий на.
восстановление.
Способ обеспечивает хорошую очистку от примесей щелочных
и щелочноземельных металлов. Однако содержание железа и
416
кремния остается таким же, как в исходных порошках (табл. 103).
Общее извлечение рения по данному методу с учетом оборотов 95%.
Очистка через окситетрахлорид ReOCl^. Для более глубокой
очистки рения советские исследователи разработали метод очистки,
состоящий в следующем [31]. Рениевый порошок хлорируют,
как описано выше, с получением конденсата пятихлористого
рения. Затем без перегрузки из конденсатора ReCl5 окисляют
при 160—180° С кислородом, превращая его в окситетрахлорид:
ReCl5 + - ReOCI4 + V2C12.
Окситетрахлорид (температура плавления 30° С, кипения 228° С)
очищают ректификацией в стеклянной колонке, затем разлагают
водой высокой чистоты, при этом частично образуется осадок
двуокиси рения:
3ReOCl4 + 7Н2О -> 2HReO4 + ReO2 + 12НС1.
Для окисления двуокиси рения добавляют перекись водорода,
затем раствор нейтрализуют аммиачной водой, выпаривают и
кристаллизуют перренат аммония. Порошки рения, полученные
из перрената аммония, имеют высокую чистоту по всем примесям,
включая железо и кремний (табл. 103).
Получение порошкообразного рения электролизом [4, с. 100]
Электролитический способ выделения рения из растворов
применяют в основном для нанесения рениевых покрытий. Однако
этим способом можно получать и порошки рения.
Для электролиза могут быть использованы перренаты калия
и аммония; в зависимости от применяемой соли рекомендовано
два состава электролита, г!л\
1. Перренат калия .... 50
Серная кислота .... 75
Сернокислый аммоний 40
2. Перренат аммония ... 100
Серная кислота .... 100
Сернокислый аммоний 60
Режим электролиза при использовании обоих растворов сле-
дующий: катодная плотность тока 100—200 а!дм\ температура
электролита 75° С. Электролиз проводят при постоянной кон-
центрации рения в электролите и непрерывной - циркуляции
электролита.
При проведении электролиза в небольших масштабах при-
меняют платиновые аноды, катоды из танталовых пластин. Ка-
тоды периодически вынимают и очищают с них осадки рения.
Их промывают водой, слабой соляной кислотой, снова водой
и сушат.
При высокой чистоте исходных солей порошки имеют низкое
содержание примесей. В отличие от порошков, полученных вос-
становлением перрената аммония водородом, электролитические
порошки состоят из более крупных зерен.
27 А. Н. Зеликман 417
Термическая диссоциация галогенидов
Рений высокой чистоты можно получить термической диссо-
циацией ReCl5 или ReOCl4 на нагретой вольфрамовой или рение-
вой нити [25]. Способ применяют главным образом для получения
рениевых покрытий на
изделиях из вольфрама
и молибдена. Термическую
диссоциацию проводят
либо в вакууме, либо в ат-
мосфере инертного газа,
служащего носителем па-
ров хлорида или окси-
хлорида. Поверхность
нити нагревают до 1200—
1300° С. Рениевые покры-
тия на вольфраме и молиб-
дене, полученные этим
способом, прочно связаны
с покрываемым металлом.
Рис. 184. Схема установки для
непрерывного ренирования воль-
фрамовой проволоки:
1 —вольфрамовая проволока; 2 —
стеклянная трубка; 3 — кварцевый
экран; 4 —испаритель оксихлорида
рения; 5 — нагреватель испарителя;
6 — спираль для нагрева стенок
трубки; 7 — скользящие контакты
для нагрева проволоки; 8 — капил-
ляры; 9 — термопара; 10 — гидра-
влический затвор; 11—расходомер
аргона; 12—14—перемоточные ка-
тушки; 15 — редуктор; 16 — дви-
гатель; 17 — стойка
Как показали автор и Барышников [25], при использовании
ReOCl4 обеспечиваются значительно более высокие скорости
отложения, чем при ReCl6. В отличие от ReCl6, который в твердом
состоянии диссоциирует при 200° С на хлор и малолетучий ReCl3,
окситетрахлорид рения не разлагается до 600° С. Это позволяет
поддерживать высокую концентрацию его в парах.
Оксихлорид разлагается с выделением рения по реакции
19П0_1400° С
3ReOCl4———2Re + ReO3Cl + 5Х/2С12.
Легколетучий ReO3Cl удаляется вместе с хлором. Выход рения
по этой реакции 66%, практический выход 50—60%.
На рис. 184 показана схема установки для непрерывного рени-
рования вольфрамовой проволоки.
418
Получение компактного рения
Компактный рений получают методом порошковой металлургии.
Порошки рения имеют размеры зерен от 1 до 5 мкм (средняя
величина зерен ~2 мкм), их насыпная масса примерно 2,25 г!см3.
Для производства компактного металла порошки прессуют в сталь-
ных пресс-формах под давлением 4—5 Т1см2 в прямоугольные
штабики (12X12 мм), плотность которых около 9,5 г!см3 (~45%
от теоретической плотности). Для некоторого упрочнения и уда-
ления части летучих примесей штабики предварительно спекают
при 1200° С в вакууме (давление 0,5—1 мкм) или в водороде
в течение 2 ч. Высокотемпературное спекание («сварку») ведут,
в водороде в аппаратах, применяемых для сварки вольфрамовых
штабиков. Максимальная температура сварки 2800—2850° С.
Степень усадки при спекании зависит от содержания летучих
(в условиях спекания) примесей. К ним прежде всего относятся
щелочные металлы. Влияние примеси калия на усадку видно
из данных, приведенных ниже [6].
Содержание калия, % . . . 0,006 0,2—0,05 0,06—0,1 0,1
Плотность спеченного шта-
бика, % от теоретической
плотности ............ 90 80—88 70—-80 65—75
Вредная примесь — медь, которая испаряется в условиях
сварки, хотя допустимый предел ее содержания в порошке рения
выше, чем калия. Влияние других примесей на усадку штабиков
при спекании еще недостаточно изучено.
Спеченные штабики, которые должны иметь плотность не ниже
18,9 е/сж3 (~90% от теоретической плотности рения), уплотняют
ковкой или прокаткой на холоду с промежуточными отжигами.
Горячую обработку, как и в случае вольфрама и молибдена,
для рения не применяют из-за его красноломкости, которая обус-
ловлена образованием легкоплавкой семиокиси рения по гра-
ницам зерен.
Сложность механической обработки рения состоит в том, что
рений отличается высоким сопротивлением деформации. Даже
при малой степени обжатия на холоду (5—10%) его твердость
резко возрастает от 250—300 до 800 HV. Поэтому после холодной
деформации ведут отжиг при 1700—1800° С в течение 30—60 мин
и далее продолжают механическую обработку на холоду.
Рений относительно легко поддается прокатке. Возможно
получение фольги толщиной до 25 мкм. Протяжка металла труд-
ней. Проволоку протягивают до диаметра 75 мкм.
Кроме способа порошковой металлургии, для получения ком-
пактного рения применяют плавку. Дуговая плавка с расходуе-
мым электродом в охлаждаемом медном тигле дает слитки с круп-
нозернистой структурой, что усложняет его механическую обра-
ботку. Глубокая очистка рения может быть осуществлена зонной
плавкой в высоком вакууме с применением электронного пучка.
27*
ЛИТЕРАТУРА
I
К главе I
1. S с h е е 1 е С. W. Kgl. Svenska Vetensk. Akad. Handl., 1778, v. 39, p. 247;
1779, v. 40, p. 233.
2. H j e 1 m P. J. Kgl. Svenska Vetensk. Akad. Handl., 1788, v. 49, p. 288.
3. Л и п и н В. H. Горный журнал, 1897, № 4; Stahl und Eisen, 1897, № 3,
S. 571.
4. Грегг Д. А. Сплавы железа с молибденом. ГОНТИ, 1938.
5. Елютин В. П. и др. Производство ферросплавов. Металлургиздат, 1957,
6. Виноградов А. П. Геохимия, 1962, № 7, с. 555.
7. М и х а й л о в А. С. Геохимия, 1964, № 11, с. 1171.
8. Курода П., Сен дел л Э. Веб. «Геохимия редких элементов» (перев,
с англ.). ИЛ, 1959, с. 209.
9. Рабинович А. В. и др. Геохимия, 1958, № 2, с. 118.
10. Виноградов А. П. Геохимия редких и рассеянных элементов в почвах.
Изд. АН СССР, 1957.
11. X рущов Н. А. Оценка месторождений при поисках и разведках, вып. 19,
«Молибден». Госгеолиздат, 1961.
12. Гольдберг Э. Д. Геохимия литогенеза. ИЛ, 1963.
13. Ф а р м а з я н А. С., X у р ш у д я н Э. X. ДАН Арм. ССР, 1963, т. 37, №4Г
с. 211.
14. S. G г а е s е г. Schweiz. Mineral. Petrogr. mitt.,1964, В. 1, S.44.
15. R. J. T г e i 1. Canad. mineralogist, 1963, v. 7, № 3, p. 524.
16. 3 e л и к м а н A. H., Беляевская Л. В. ЖНХ, 1956, № 10, с. 2239,
17. Karobbi Bull. Soc. Nat. Napoli, 1930, v. 41, p. 169.
18. Минеральные ресурсы капиталистических стран. Под ред. Н. А. Быховера,
Госгеолтехиздат, 1968.
19. Митрофанов] С. И. Селективная флотация. Металлургцздат, 1958,.
с. 542—579.
20. Полькин С.И. Флотация руд редких металлов и олова. Госгортехиздат,.
1960, с. 591—619.
21. Фишман М. А., С о б о л е в Д. С. Практика обогащения руд цветных
металлов, т. IV. Госгортехиздат, 1963, с. 11—65.
22. Технология извлечения окисленного молибдена из руд. ОБТИ, Главзолото,.
1957.
К главам II и III
1. Зеликман А. Н.,Беляевская Л. В. В сборнике научных трудов
(МИЦМиЗ), № 25. Металлургиздат, 1955, с. 226—243.
2. Сб. «Металлургия вольфрама, молибдена и ниобия». Изд-во «Наука», 1967,
420
3. Зеликман А. Н. иБ ел я евск ая Л. В. ЖНХ, 1956, № 10, с. 2245.
4. Семенов Н. Н. и Р я б и н и н Ю. Н. ЖРФХО. Часть физич, 1928, т. 60,
с. 361.
5. Д е е в В. И., Смирнов В. И. Изв. вузов. Цветная металлургия, 1961.
№ 3, с. 44—49.
6. Зеликман А. Н. иВольдман Г. М. Изв. вузов. Цветная металлур-
гия, 1968, № 4, с. 71.
7. Зеликман А. Н. и др. Цветные металлы, 1966, № 2, с. 63.
8. П и с к у н о в И. Н. Изв. вузов. Цветная металлургия, 1960, № 1, с. 133.
9. Stubbles S. R.,Richardson F. D. Trans. Faraday Soc., 1960, v. 56,
№ 10, p. 1460.
10. S e 11 z H. a. o. Journ. Amer. Chem. Soc., 1943, v. 65, p. 600.
11. Kubaschewski O., Evans E. Metallurgical Thermochemistry.
London, 1958.
12. Richardson F. D., J e f f e s I. H. E. Journ. Iron Steel Inst., 1948,
v. 160, p. 261.
13. 3 e л и к м а н A. H. ЖНХ, 1956, т. I, № 12, c. 2778.
14. T a m m a n G., West ercho 1 d F. Z. Anorg. Allg. Chem., 1925, Bd. 149,
S. 21—46.
15. 3 e л и к м а н A. H., Беляевская Л. В. ЖПХ, 1954, т. 37, № 11,
с. 1151—1162.
16. Гинстлинг А. М. и Фрадкина Т. П. ЖПХ, 1952, т. 25, № 11,
с. 1134.
17. К о з м а н о в Ю. Д., Угольникова Т. А. ЖНХ, 1958, т. 3, № 5,
с. 1267.
18. Р е й н г о л ь д Б. М., См а гунов В. Н. В сб. «Научные труды»
(ИРГИРЕДМЕТ). Изд-во «Недра», 1965, вып. 12, с. 330—343.
19. Зеликман А. Н. и др. ЖНХ, 1956, № 1, с. 632.
20. Berkowitz J. а. о. Journ. of Chem. Phisics, 1957, v. 26, p. 842.
21. Пономарев В. Д., Букетов Е. А. В сб. трудов Казахского горно-
металлургического ин-та, 1958, № 16, с. 377.
22. К у н е в Д. и др. ЖНХ, 1966, № 8, с. 1989.
23. J a g е г W. u. a. Arch. Eisenhiittenwesen, 1959, Bd. 30, S. 435.
24. Ж a p к о в а М. А., Герасимов Я. И. ЖФХ, 1961, т. 35, с. 2291.
25. Muldrow С. N., Hepler L. G. Journ. phys. chem., 1958, v. 62, p. 982.
26. В a r a n у R. US Bur. Min. Rept. Invest., 1962, № 6143.
27. Weller W. W., King E. G. US Bur. Min. Rept. Invest., 1963, № 6147.
28. Rossini F. D. at.al. Selected values oj chemical thermodynamic properties.
Circ. Nat. Bur. Stand., 1952, p. 500.
29. Wheeler E. S. Metals Technology, 1944, VIII, p. 1—11. AIMME Techn.
publ., 1944, № 1718.
30. Зобнина A. H.,Кисляков И. П. Неорганические материалы. Изв.
АН СССР, 1966, № 3, с. 511.
31. Гельперин Н. И. и др. Основы техники псевдоожижения. Изд-во
«Химия», 1967.
32. Забродский С. С. Гидродинамика и теплообмен в псевдоожиженном
слое. Госэнергоиздат, 1963.
33. Weller W., US Bur. of Mines. Rept. Invest., 1966, № 6782.
34. 3 e л и к м а н A. H. и др. Цветные металлы, 1956, № 8, с. 41.
35. Сб. «Применение в СССР процессов обжига в кипящем слое». ЦИИН ЦМ, 1960.
36. Б у р о в о й И. А. и др. Цветная металлургия. Цветметинформация, 1965,
№ 14, с. 25.
37. П е т р о в В. М., Зеликман А. Н. Изв. вузов. Цветная металлургия,
1960, № 2, с. 126.
38. Зеликман А. Н. и др. С сб. «Применение кипящего слоя в народном
хозяйстве СССР». ЦИИН ЦМ, 1965, с. 122.
39. С м и р н о в В. И. и др. Цветные металлы, 1953, № 6, с. 24.
40. Буровой И. А., Б е р и ш т е й н И. М. Бюллетень ЦИИН ЦМ, 1956,
№ 11—12, с. 12.
421
41. Graham R.L.,H ер 1 er L. G. J. Amer. Chem. Soc., 1956, № 78* p. 4846.
42. .Л од ысев a M. С., Б у p о в о й И. А. Журнал Всесоюзного Химич,
общества им. Д. И. Менделеева, 1961, т. VI, № 5.
43. Зелик-м ан А. Н. и др. Цветные металлы, 1966, № 2, с. 63.
44. Вольдман Г. М. иЗеликман А. Н. Изв. вузов. Цветная металлур-
гия, 1962, № 4, с. 73.
•45. Зеликман А. Н.,Вольдман Г. М. Цветные металлы, 1964, № 5, с. 23.
46. Вольдман Г. М.,Зеликман А. Н. Изв. вузов. Цветная металлур-
гия, 1969, № 2 с. 108.
47. Von Н. Dietz, W. Klostermann. Abhandl. Deutsche Academ. Wiss.
KI. Math. Phys. Techn., 1966, № 1, S. 221.
48. Зеликман A. H. и др. Труды Совещания по обжигу материалов в кипя-
щем слое. Металлургиздат, 1956 с. 75.
49. Спицын В. И., Савич И. А. ЖОХ, 1952, т. 22, с. 1278.
*50. Вольдман Г. М., 3 е'л и км а н А. И. Изв. вузов. Цветная металлур-
гия, 1969, № 3 с. ПО.
•51. Башилов И. Я., Ки ндя ков П. С. Цветные металлы, 1931, № 7, с. 879.
52. Зеликман А. Н., Пр осей кова Т. Е. ЖНХ, 1961, т. 6, № 1, с. 212.
53. Зеликман А. Н.,Беляевская Л. В. ЖПХ, 1956, т. 28, № 1, с. 11.
•54. Рейнгольд Б. М. В сб. «Научные труды» (Иргиредмет). Изд-во
«Недра»., вып. 13, 1965, с. 398.
55. Зеликман А. Н. Металлургия вольфрама и молибдена.Металлургиздат,
1949.
•56. Зеликман А. Н.идр. Бюллетень ЦИИН ЦМ, 1965, № 12, с. 37.
57. Зеликман А. Н., Р а к о в а Н. Н. Цветные металлы, 1966, № 3, с. 57.
58. Золотарев Л. Л. Цветные металлы, 1958, № 1, с. 57.
59. Золотарев Л. Л. Изв. вузов. Цветная металлургия, 1958, № 2, с. 107.
60. Абашин Г. И. иПогосян Г. М. Технология молибдена и вольфрама.
Металлургиздат, 1960.
€1. Зайцев Л. М. ЖНХ, 1956, т. 1, № 10, с. 2425.
62. Спицын В. И. ЖНХ, 1957, т. 2, с. 502. .
53. Силлен Л. Г. О полианионах в растворах. Вестник ЛГУ, Сер. физики
и химии, 1964, вып. 1, № 4, с. 82.
64. Sasaki Y. а. о. Journ. inorg. nucl. chem., 1959, v. 9, p. 93.
65. С a г p e n i G. Bull. soc. chim. France, 1947, v. 14, p. 484.
66. Яцимирский К. Б., Алексеева И. И. ЖНХ, 1959, № 4, с. 818.
67. Морачевский Ю. В., Лебедева Л. И. ЖНХ, 1960, № 5, с. 2238.
68. Jander G., Witzman Н. Ueber amphotere Oxyhydrate. Z. anorg.
Chem., 1933, Bd. 215, S. 310.
69. Jander G., Spandau F. Z. phys. chem., 1941, Bd. 188, S. 65.
70. I n g r i N., Sill e n L. G. Acta chem. Scand., 1962, v. 16, p. 173.
71. Brinzinger H., R a t amarat C. Z. Anorg Chem., 1931, Bd. 196,
S. 55; 1935, Bd. 224, S. 97.
72. Brinzinger H. Z. phys. Chem., 1940, Bd. 187, S. 317.
73. В у e J. Bull. Chim. France, 1942 (5), v 9, p. 626.
74. В у e J. Bull. Soc. chim. France, 1942 (5), v. 9, p. 517.
75. В у ё J. Ann. chim. (France), 1945, v. 20, p. 463.
76. L i n d q i s t J. Nova Acta Reg. Soc. Sci. Ups., 1950, v. 15, № 1.
77. Яцимирский К- Б., Алексеева И. И. ЖНХ, 1963, т. 8, с. 2513.
78. С s а п у L. S. Acta chim. Acad; Sci. Hung, 1958, v. 15, p. 257.
79. C s a n у L. S. Mgyar kem. [olyoirat, 1955, Bd. 61, S. 54.
*80. S i 1 1 e n L. G. Acta chem. Scand., 1954, v. 8, p. 299.
*81. Гохштейн Я- П. Восстановление ионов молибдена на капельном ртут-
ном катоде. Труды комиссии по аналитической химии. Отдел хим. наук АН
СССР, 1949, № 2 (5), с. 54.
62. Бабко А. К., Набиванёц Б. И. ЖНХ, 1957, т. 2, с. 2085.
83. Бабко А. К., Н а б и в а н е ц Б. И. ЖНХ, 1957, т. 2, с. 2096.
84. Martinez J. В., С a b a n е s F...P. Anales Real Soc. Espan. Fis. Quim
(Madrid), 1961, v. 57 (Ref. Chem. Abstr. v. 57, 6850b).
422
85. Chojnacka J. Rocziki chemii, 1963, v. 37, p. 259.
86. Ramano Rao. Analytical chemical Acta 1957, v. 17, p. 538.
87. Jones M. Journ. Amer Chen. Soc., 1954, v. 76, p. 4233.
88. Б о й к о В. А. Бюллетень ЦИИН ЦМ, 1959, № 15, с. 25.
89. А й р а п е т я н Г. М. Промышленность Армении, 1958, № 3,
90. N е 1 i d о v L.,Diamond R. М. Journ. Phys. Chem., 1955, v. 59, p. 710.
91. S а с с о n i L., C i n i R. Journ. Amer. chem. Soc., 1954, v. 76, p. 4239.
92. F u n a k i K., S e g a v a T. Journ. Electrochem. Soc. Japan, 1950, v. 152.
93. Duval C. Inorganic Thermogravimetric Analysis, Amsterdam, 1953, p. 332.
94. A. Hegediis, K. S a s v a r i, S. Neugebauer Z. Anorg. Chem., 1957, Bd.
293 S 56
95. P о д e E. Я., Твердохлебов В. H. ЖНХ, 1958, № 3, c. 2343.
96. Я Ц и м и p с к и й. К. Б., Васильев В. П. Константы нестойкости
комплексных соединений. Изд. АН СССР. 1959.
97. Конева К. Г.,Савельский С. Л. Распределение примесей в произ-
водстве молибдена и фольфрамата аммония. В кн. «Металлургия и металло-
ведение». Изд. АН СССР, 1958, с. 189—195.
98. Буровой И. А.,Драчева Т. В. Цветные металлы, 1967, № 9, с. 54.
99. Kieffer R.,Hotop W. Pulvermetallurgie und Sinterwerkstoffe. Berlin
1948, 2 Aufg. S. 245.
100. Matz W., Chem. Ing. Techn., 1958, v. 30, p. 319.
101. Murphy W. W., W h e e 1 e r E. S., Linz A. Mining and Metallurgy, 1946»,
v. 27, p. 350—352.
102. Chemical and Engineering News. 1959, v. 37, p. 40.
103. Chmielkowski J. Rudi i MetalleNiezelazne, 1961, Rok. 3 , St. 411.
104. Mularczyk T. Rudi i Metalle Niezelazne, 1962, Rok. 7, St. 414.
105. F e i s e r J. Metall und Erz., 1931, B. 28, S. 297—302.
106. Зеликман A. H. и др. ЖНХ, 1956, т. 1, № 4, с. 332.
107. Berkowitz J. а. о. Journ. chem. phys., 1957, v. 26, p. 842.
108. Blackburn P., Hoch M., Johnston H. L. Journ. phys. Chem.»
1958, v. 62, S. 769.
109. H 6 r b e R. u. a. Z. Erzbergbau und Melallhuttenwesen, 1961, Bd. 14, № 5»
S. 232.
110. Ackermann R. G. a. o. Journ. phys. Chem., 1960, v. 64, p. 350.
111. G u 1 b r a n s e n E. A. a. o. Journ. Electrochem. Soc., 1963, v. 110, p. 242.
112. A p и а С. M. и др. ЖОХ, 1953, т. 23, с. 2063:
113. Ueno К., Journ. Chem. Soc. Japan, 1941, v. 62, p. 990.
114. Беляевская Л. В. и др. В сб. «Тугоплавкие металлы». Изд-во «Метал-
лургия», 1967, с. 46.
115. Benesovsky F., VidmajerA. Osterreichische chemiker Zeitung»
1952, v. 53, № 13—14, S. 158.
116. V i d m a j e r A. Kolloid Zeitschr., 1953, Bd. 130, S. 69.
117. С о s g г о v e L. A., S n у d e r P. E. J. Amer. Chem. Soc., 1953, Bd. 75»
S. 1227.
118. К i 1 1 e f f e r D. H. L i n z A. Molybdenum Compounds. New-York—Lon-
don, 1952.
119. Викторианский С. А. и др. Цветные металлы, 1961, № 7, с. 87..
120. Парусников В. Н. Цветные металлы, 1960, № 12, с. 61.
' К главе IV
1. Елютин В. П. и др. Производство ферросплавов. Металлургиздат, 1957,.
гл. VI.
2. Елютин В. П. Новости техники, 1938, т. 7, № 4, с. 26—28.
3. Е л ю т и н В. П. В кн. «Электрометаллургия ферросплавов». ОНТИ, 1937.
4. Р о б и т т А. Дж. Е. Практика электроплавки (пер. с англ.). Металлургиздат,.
1960, с. 371.
5. О. К u b a s с h е w s k i, Е. Evans Metallurgical Thermochemistry. New-
York—London, 1958.
42$
К главе V
1. Г рейвер Н. С. Хибинские апатиты, 1933, т. VI, с. 147—153.
2. Грейвер Н. С. В кн. «Основы металлургии». Изд-во «Металлургия»,
1967, т. IV, с. 31—51.
3. Лебедев К- Б. Производство молибдата кальция. Изд. АН Каз. ССР,
Алма-Ата, 1962.
4. «Л е б е д е в К. Б.,По ном а р ев В. Д. Изв АН Каз. ССР. Серия метал-
лургическая, 1957, № 1, с. 12.
5. Петров В. М.,Зеликман А. Н. Изв. вузов. Цветная металлургия.
1960, № 2, с. 126.
6. Зеликман А. Н., Беляевская Л. В. ЖПХ, 1956, № 21, с. 11—17.
, 7. Лебедев К- Б. Изв. АН Каз. ССР. Серия металлургическая, 1959, № 1,
56.
- Киндяков П. С. Редкие металлы, 1934, № 4, с. 48.
9. Рейнгольд Б. М. «Добыча и обработка руд редких, цветных и благо-
родных металлов». Сб. трудов Иргиредмета. Изд-во «Недра», 1965, вып. 13,
с. 398.
10. Технология извлечения окисленного молибдена. ОБТИ. Главзолото. 1957.
11. К. С. L i, Chung Y i Wang. Tungsten. New-York—London, Third Edi-
tion, 1955
12. В о n a r d y. Chem. Metallurg. Ing., 1919, v. 21, p. 364.
13. Зеликман A. H., Г о p о в и ц Н. Н. В сб. «Металлургия и технология
цветных металлов» (МИЦМиЗ). Металлургиздат, 1960. с. 186—201.
’4. Зырянов М. Н. и др. Труды Иркутского политехи, ин-та. Серия
металлургическая, 1963, вып. 18, с. 123.
15. 3 е л и н с к и й В. И. Бюллетень ЦИИН ЦМ, 1957, № 9, с. 10.
16. Зеликман А. Н. и др. Бюллетень ЦИИН ЦМ, 1962, № 7, с. 32.
17. С и н а к е в и ч А. С. В сб. трудов Иргиредмета. Металлургиздат, 1958,
вып. 7, с. 171.
18. С и н а к е в и ч А. С., 3 ы р я н о в М. Н. В сб. трудов Иргиредмета. Ме-
таллургиздат, 1961, вып. 9, с. 184.
19. М а с л е н и ц к и й И. К. Цветные металлы, 1939, № 4—5, с. 140.
20. Масленицкий Н.Н. Сборник материалов по применению автоклавных
процессов в металлургии цветных и драгоценных металлов. ЦИИН ЦМ, 1960,
с. 42.
21. Масленицкий Н. Н. Веб. «Обогащение руд» (Механобр), 1957, № 4,
с. 3.
22. Никитина И. С., В е л л е р Р. Л. «Обогащение и металлургия цветных
металлов». Сборник трудов Гинцветмета, Металлургиздат, 1957, № 13, с. 129.
23. Зеликман А. Н., Калинина И. Г. Веб. «Тугоплавкие металлы».
Изд-во «Металлургия», 1968, с. 26.
К главам VI и VII
1. Зеликман А. Н. идр. Известия вузов. Цветная металлургия, 1969, № 6.
2. Ю р к е в и ч Ю. Н., Шапиро К. Я- В сб. «Металлургия вольфрама,
молибдена и ниобия». Изд-во «Наука», 1967, с. 53.
3. D г е s h е г W. Н. а. о. Journ. of Metals, 1956, v. 8, № 6, p. 7)4.
4. С о б о л ь С. И. и др. Бюллетень ЦИИН ЦМ, 1959, № 12, с. 27.
5. Сборник материалов по применению автоклавных процессов в металлургии
цветных и драгоценных металлов. ЦИИН ЦМ, 1960.
6. Сб. трудов Гинцветмета, Металлургиздат, 1961. № 18.
7. М о н е в Г., М и р е в а С. Химия и Индустрия (Болг.), 1958, № 5, с. 136.
8. Сох Н., С h е 1 linger А. К. Eng. Min. Journ., 1958, v. 159, № 10, p. 101.
9. Юхтанов Д. M., Леонтьева К- Д. Цветные металлы, 1953, № 3,
с. 43.
424
10. Иорданов Хр. В., 3 ел и км ан А. Н. Химия и Индустрия (Болг.),
1961, № 6, с. 171.
И. В h а р р u R. В. а. о. Journ. of Metals, 1965, v. 17, № 11, S. 1199.
12. Ш а п и p о К. Я., К у л а к о в а В. В. Цветные металлы, 1963, № 9, с. 88.
13. Иорданов Хр. В. Минное дело и Металлургия (Болг.), 1961, т. 16, № 9,
с. 25.
14. Васильев Хр., Добрев Ив. Годишник на химикотехнологическия
институт (Болг.), 1962, т. 9, кн. I, с. 231.
15. Хрящев С. В., Козловская Э. М. Цветные металлы, 1967, № 2, с. 1*3.
16. М е 1 1 о г s L. W. Comprehensive Treatise on inorganic and Theoretical Che-
mistry. Lond.-N.-York—Toronto, 1946, v. 2, p. 250.
17. И p о к о п ч и к А. Ю. Труды АН Латв. ССР, Сер. Б, 1964, № 3, с. 31—48
18. Гликман Г. С., Д а й н Б. Я- ЖОХ, 1941, т. 11, № 3.
19. Иорданов Хр. В. Химия и индустрия, 1962, № 1, с. 7—9.
20. С о б и н я к о в а Н. М. и др. Минеральное сырье. Госгеолиздат, 1961,
вып. 2, с. 206.
21. Собинякова Н. М., Иванцова Г. А. Минеральное сырье. Изд во
«Недра», 1966, вып. 13, с. 70.
22. С о б и н я к о в а Н. М. и др. Минеральное сырье. Госгеолиздат, 1963,
вып. 9, с. 49.
23. Уднасынова 3. Д.,Пономарева Е.Н. Изв. АН Каз. ССР. Серия
химич. и технологич. наук, 1963, вып. 2.
24. Пон омарева Е. И., Лебедев К. Б. Труды Института металлургии
и обогащения АН Каз. ССР, 1962, т. 4, с. 28.
25. Собинякова Н. М. идр. Способ извлечения молибдена из руд. Авт.
свид. СССР, № 152738. Бюлл. изобретений, 1963, № 2.
26. Зеликман А. Н., К а л и н и н а И. Г. В сб. «Тугоплавкие металлы».
Изд-во «Металлургия», 1968, с. 26.
27. Л я п и н а 3. М., Зеликман А. Н. Изв. вузов. Цветная металлургия,
1959, № 3, с. 93—98.
28. 3 е л и к м а н А. Н., Л я п и н а 3. М. Изв. вузов. Цветная металлургия,
1960, № 2, с. 119.
29. Соболь С. И. «Металлургия цветных металлов». Сб. трудов Гинцветмета,
№ 15. Металлургиздат, 1959, с. 481.
30. С a d b и г у W. Е. Journ. Phys, chemistry, 1955, v. 59, № 3, p. 257.
31. Jones M. M. Journ. Amer. Chem. Soc., 1954, v. 76, p. 4232.
32. W e i n 1 a n d E., К и о 1 1 W. Z. Anorg. Chem. 1905, v. 44, S. 81.
33. Щ а п и p о К. Я. ЖПХ, 1962, т. 35, № 3, с. 486.
34. Ш а п и р о К- Я. Цветные металлы, 1961, № 7, с. 57—60.
35. М а с л е н и ц к и й Н. Н. Обогащение руд, 1957, № 4. с. 3.
36. Волк-Корачевская И. В. и др. Цветные металлы, 1967, № 8, с. 74.
37. Попов И. Ф. В сб. «Материалы совещания по применению ионного обмена
в цветной металлургии». ЦИИН ЦМ, 1957, с. 61.
38. Богомильская Е. М. Бюллетень ЦИИН ЦМ, 1958, № 20, с. 30.
39. Зеликман А. Н. и др. Бюллетень ЦИИН ЦМ, 1962, № 7, с. 32.
40. Сб. «Ионообменные сорбенты в промышленности». Изд. АН СССР, 1963.
41. «Металлургия и обогащение» (сб. работ аспирантов и соискателей), т. 11. Алма-
Ата, 1966.
42. Сох Н., Schellinger А. К. Eng. and Mining Journ., 1958, v. 159, № 10,
p. 101.
43. Шамсиев С. M., Сенявин M. M. Цветные металлы, 1963, № 10, с. 8.
44. К у з и н И. А., П л а ч е н о в Т. Г. и др. ЖПХ, 1961, т. 34, № 11.
45. Ч ер нобров С. М., Богатырева Е. М. «Обогащение руд», 1963,
№ 6, с. 24.
46. 3 е л и к м а н А. Н., Н е р е з о в В. М. ЖНХ, 1969, № 5, с. 206.
47. Плаксин И. Н., Т э т а р у С. А. Гидрометаллургия с применением ио-
нитов. Изд-во «Металлургия», 1964.
48. Б а б к о А. К-, Н абиванец Б. И. ЖНХ, 1957, т. 2, с. 2083, 2096.
49. Р я б ч и к о в Д. И., Борисова Л. В. ЖАХ, 1958, т. 12, с. 153.
425
50. Алимарин И. П., Медведева А. М. Труды Комиссии по аналит.
хим.; 1955, т. 6, с. 351.
51. Лурье Ю. Ю. и Филиппова Н. А. Заводская лаборатория, 1947г
т. 13, № 5.
52. С а л д а д з е К. М. и др. Ионообменные высокомолекулярные соединения.
Госхимиздат, 1960.
53. Колман и др. Вторая конференция по мирному использованию атомной
* энергии (доклад № 510). Избранные доклады иностр, ученых. Атомиздат, т. 7,
1959, с. 352.
54. Ласкорин Б.Н. и др. ЖПХ, 1962, т. 45, с. 2409.
55. N е 1 i d о v L., D i а ш о и d R. М. Journ. Phys. Chem., 1955, v. 59, p. 710.
56. Гер л и т Ю. Б. 2-я Женевская конференция. Доклады Советских ученых,
т. 4, Атомиздат, 1959, с. 181.
57. D i a m о n d R. М. Journ. Phys. Chem., 1957, v. 61, p. 75.
58. 3 а й ц e в А. А. и др. ЖНХ, 1963, т. 8, с. 2184.
59. X р ящев С. В.; Коч етков аЭ. А. Изв. вузов. Цветная металлургия,
1968, № 1, с. 56.
60. Федулов О. В., П о н ом а р е в В. Д. и др. «Металлургия и обогащение»
(сб. статей аспирантов), т. III. Алма-Ата, 1967, с. 170.
61. Churchwood Р. Е., Rosenbaum S. В. Journ. of Metals, 1963, v. 15,
№ 9, S. 648.
62. Зеликман A. H.,Калинина И. Г. Цветная металлургия, Цветмет-
информация, 1966, № 16, с. 52.
63. 3 е л и к м а н А. Н., Калинина И. Г. ЖНХ, 1968, № 10, с. 2778.
64. Ласкорин Б.Н. идр. Экстракционные свойства ал кил фосфорных кислот.
В сб. «Экстракция», вып. 1. Атомиздат, 1962, с. 171.
65. Д а й м о н д Р. М., Т а к Д. Г. Экстракция неорганических соединений.
Атомиздат, 1962. с. 32.
66. Зеликман А. Н., Нерезов В.М. ЖНХ, 1967, № 3, с. 769.
67. Зеликман А. Н., Н е р е з о в В. М. Цветные металлы, 1968, № 1, с. 65.
68. 3 е л и к м а н А. Н. Изв. вузов. Цветная металлургия, 1968, № 3, с. 85.
69. Н а г d у С. J. а. о. J. Inorg. Nucl. Chem., 1958, v. 7, p. 257.
70. П p и в а л о в M. М., Коновалов В. Ф. ЖНХ, 1965, т. 1, с. 251.
71. Peppard D. F., F е г г а г о I. R. J. Inorg. Chem., 1960, v. 3/4, Р. 365.
72. Arend К. Н., Specker H.Z. Anorg. Allg. Chem., 1964, Bd. 333, № 1—3,.
S. 18.
73. Карпачева С. M. и др. ЖНХ, 1967, № 7, с. 1925.
74. Николаев А. В. и др. В сб. «Экстракция», вып. 2. Атомиздат, 1962, с. 63.
К главе VIII
1. Biltz W.,Fendins С. Zeitschr. Anorg. Chem., 1928, v. 172, p. 385.
2. Глухов И. А., Б e x т л e Г. А. Труды АН Тадж. ССР, 1958, т. 84, с. 35.
3. Evans R., Lister M. Trans. Farad. Soc., 1938, v. 34, p. 1358.
4. D e b г а у H. Compt. Rend. Acad. Sci., 1868, v. 66, p. 732.
5. Щ у к a p e в С. А. и др. ЖНХ, 1960, № 5, t. 1650.
6. Щ у к a p e в С. А. и др. Вестник ЛГУ, Сер. физ. хим., 1959, № 4, с. 73.
7. Щ у карев С. А., Суворов А. В. ЖНХ, 1961, № 6, с. 1488.
8. С у в о р о в А. В., Новиков Г. И. и др. В сб. «Химия редких элемен-
тов». Изд-во ЛГУ, 1964, с. 26—32.
9. Hultgren N., Brewer L. Journ. phys. chem., 1956, v. 60, p. 947.
10. Щ у к a p e в С. А. и др. В сб. «Чистые металлы и полупроводники».
«Металлургиздат», 1959, с. 140.
И. Щукарев С. А. и др. Вестник ЛГУ, 1960, Сер. физ-хим., № 10.
12. Б а с и т о в а С. М. и др. Изв. АН Тадж. ССР. Отд. естеств. наук, 1958, т. 84,
с. 17.
13. Глухов И. А., Елисеев С. С. ЖНХ, 1962, т. VII, № 1, с. 81.
14. К u b a s с h е w s k i О., Evans E. L. Metallurgical Thermochemistry.
Pergamon Press. New-York, 1958, 3d ed.
426
15. Уилкс К. Е., Блок Ф. Е. Термодинамические свойства 65 элементов, их
окислов, галогенидов, карбидов и нитридов. Изд-во «Металлургия», 1965.
16. В г е w е г L. а. о. Chemistry and Metallurgy of Miscellaneous Materials:
Thermodinamics, by L. L. Quiel. New-York, 1950, p. 276.
17. Gmelins Handbuch der anorganischen Chemie, Syst. № 53 (Mo), Verlag Chemie,
Berlin, 1935.
18. W о r d 1 о u W., W e b b K. J. Chem. Soc., 1930, p. 2100.
19. Спиридонов В. П., Романов Т. В. Вестник МГУ, Сер. химич.,
1967, № 1, с. 98—99.
20. Schater Н.,Tillack J. Journ. Less-Common Metals, 1964, v. 6, № 2,
p. 152.
21. Зеликман A. H. и др. Получение пятихлористого молибдена хлорирова-
нием ферромолибдена. Цветные металлы, 1969, № 1, с. 67.
22. 3 е л и к м а н А. Н. и др. ЖПХ, 1962, т. 35, № 7, с. 1467.
23. К о р ш у н о в Б. Г. и др. Изв. вузов. Цветная металлургия, 1962, № 1,
с. 101.
24. К о р ш у н о в Б. Г. и др. Изв. вузов. Цветная металлургия, 1960, № 5,
с. 67.
25. Коршунов Б. Г., Г о л ь д и н В. И. ЖНХ, 1961, т. 6, № 7, с. 1642.
26. Коршунов Б. Г. и др. ЖНХ, 1963, т. 8, № 6, с. 1531.
27. Michael A., Morphy A. Amer. Chem. Journ., 1910, v. 44, p. 371.
28. Couch E., Brenner A. Journ. Res. Nat. Bur. Standards, 1959, A 63,
№ 2, p. 185—188.
29. Klemm R. Z. Anorg. Chem., 1936, Bd. 227, S. 193.
30. В i 1 t z W., F e n d i u s C. Z. Anorg. Chem., 1928, Bd. 176, № 1—3, S. 49.
31. C a m p b e 1 1 T. T. Journ. Electrochem. Soc., 1959, v. 106, № 2, p. 119.
32. В г о s e t C. Ark. Kern. Mineralog. Geol., 1945, v. A 20, № 7.
33. В г о s s e t C. Arkiv Kemi. Mineral. Geol., 1946, v. A22, № 1.
34. Г p и н б e p г А. А. Введение в химию комплексных соединений. Изд. «Хи-
мия», 1966, с. 501.
35. V a u g h a n Р. A., S t u d i v a n t I. H., P а и 1 i n g L. S. J. Amer. chem.
Soc., 1950, v. 72, p. 5477.
36. Senderoff S.,Brenner A., J. Electrochem. Soc., 1954, v. 101, № 1,
p. 29.
37. E m e 1 e n s H. E., G и t m a n V. J. Chem. Soc., 1949, p. 2979.
38. Schnering H. G., Wohrle H. Naturwissenschaften, 1963, Bd. 50,
№ 3, S. 91.
39. Schaffer H. u. a. Z. Anorg. Allg. Chem., 1967, B. 353, Heft 5/6, S. 281.
40. Klemm W., Steinberg H. Z. Anorg. Allg. Chem., 1936, Bd. 227,
S 193 201
41. Нисельсон Л. А., С о к о л о в а Т. Д. ЖНХ, 1965, т. 10, № 1, с. 18.
42. S i d g w i c k N. V. The Chemical Elements and their Compounds. Oxford, 1952
43. Глухов И. А., Родионова P. А. ДАН Тадж. ССР, 1959, т. 2, № 5,’
с. 15.
44. Г л у х о в И. А., Е л и с е е в С. С. Изв. отдел, геолого-химич. и техн,
наук АН Тадж. ССР, 1959, вып. 1, с. 79.
45. Глухов И. А., Тихомиров Л. А. ДАН Тадж. ССР, 1961, т. IV, № 1,
с. 7.
46. Puttbach W. Ann. Chim., 1880, Bd. 201, S. 128.
47. Глухов И. А., Тихомиров Л. А. ДАН Тадж. ССР, 1960, т. III, № 2,
с. 15.
48. Schaffer Н.,Tillack J. Journ. Less-Common Metalls, 1964, v. 6, № 2,
p. 152.
49. Сб. «Тугоплавкие металлы». Изд-во «Металлургия», 1968.
50. С и н а к е в и ч А. С. Сб. научн. трудов Иргиредмета, 1958, № 7, с. 170.
51. С и н а к е в и ч А. С., 3 ы р я н о в М. Н. В сб. трудов Иргиредмета.
Металлургиздат, 1959, № 8, с. 220.
52. 3 е л и к м а н А. Н., Г о р о в и ц Н. Н. ЖОХ, 1954, т. 34, с. 1916.
53. Зеликман А. Н., К р е й н О. Е. ЖПХ, 1961, т. 34, № 3, с. 679.
427
54. Г л ух о в И. А., Б ехтл е Г. А. В сб. «Исследования в области химии
редких металлов и солей». Тр. Инет, химии АН Тадж. ССР, 1958, вып. 2,
с. 17.
55. Т а р а к а н о в Б. М. и др. ЖНХ, 1968, № 3, с. 871.
56. Z а 1 k i n A., S a n d s D. Е. Acta Cristallogr. (Copenhagen), 1958, v. 11,
p. 615.
57. 3 e л и к м а н A. H. и др. Изв. вузов. Цветная металлургия, 1968, № 2,
с. 71.
58. Глухов И. А., Ш а л у х и н а. Л. М. ДАН Тадж. ССР, 1960, т. III, № 2;
1961, т. IV, № 1; 1961, т. IV, № 3; 1961, т. IV, № 4.
59. Rozebuum Н., Aten A. Proc. Acad. Amsterdam, 1904, v. 6, p. 559.
60. A. A t e n Z. phys. chem., 1906, Bd. 54, S. 55.
61. В a d i a 1 i M. A., Kirshenbaum N. W., В a k i s h R. Trans. Metal-
lurg. Soc. AIME, 1963, v. 227, № 1, p. 32—36.
К главе IX
1. Mawrow F.,Nikolow M. Zeitschr. Anorg. Chem., 1916, Bd. 95, S. 188.
2. Schaeffer S. C. u. a. Trans. Met. Soc. of AIME, 1964, v. 230, № 3, p. 594.
3. Г о p о x А. В. и др. ДАН СССР, 1964, т. 156, № 3, с. 541.
4. Guichard М. Ann. Chim. phys., 1901 (7), v. 23 (4), v. 51, p. 552.
5. Guichard M. Compt. Rend. Bull. Soc. Chim., 1932 (4), v. 51, p. 569.
6. Picon M. Compt. Rend., 1929, v. 189, p. 96.
7. Montoro K. Atti. Acad. Lincei, (6), 1929, v. 9, p. 331.
8. Parravano N., M a 1 q u о r i G. Atti Acad. Lincei, 1928 (6), v. 7, p. 109.
9. 3 e л и к м а н A. H., Крейн О. E. ЖФХ, 1965, т. 29, № 11, с. 2081.
10. 3 e л и к м а н А. Н., Беляевская Л. В. ЖНХ, 1956, т. I, № 10,
с 2239.
11. М с Cade С. L. Journ. of Metals, 1955, v. 7, № 1, p. 61.
12. Stubbles J. R.,Richardson F. D. Trans. Faraday Soc., 1960, v. 56,
№ 10, p. 1460.
13. Опаловский А. А., Федоров В. E. Халькогениды молибдена.
Успехи химии, 1966, т. 35, № 3, с. 427.
14. J е 1 1 i n е k F. Nature (London), 1961, v. 192, р. 1065.
15. О п а л о в с к и й А. А., Ф е д о р о в В. Е. В сб. «Халькогениды». Изд-во
«Наукова думка», 1967, с. 79.
16. М о г i m о t о N., К u 1 1 е г и d G. Carnegie Institution of Washington.
Yearbook 61, 1961—1962.
17. Опаловский А. А.,Федоров B.E. ДАН СССР, 1966, т. 163, с. 900.
18. Wildervanck J. С., Jellinek F. Z. Anorg. Allg. Chem., 1964,
B. 328, № 5—6, S. 309.
19. D e b u c q u e t L., V e 1 1 u z L. Bull. Soc. Chim. France, 1932 (4), v. 51,
p. 1571.
20. G 1 e m s e r O. u. a. Z. anorg. Chem., 1948, Bd. 257, S. 241.
21. Роде E. Я., Лебедев Б. А. ЖНХ, 1961, т. 6, № 5, с. 1189.
22, G i о v a n n i Р. Ricerca Scient., 1963, v. АЗ, р. 1137.
23. Т a i m n i I. К., A g о r w a 1 R. P. Proc. Nat. Acad. Sci. India, 1951, v. 20A,
P, 48.
24. T a i m n i I. K-, Agarwal R. P. Analyt. Chim. Acta, 1953, v. 9, p. 16;
1953, v. 9, p. 203.
25. T a i m n i I. K., Raksphal R. Analyt. Chim. Acta. 1960, v. 22, p. 34.
26. 3 в о p ы к и н А. Я- и др. ЖНХ, 1961, т. 6, с. 1994.
27. R a m а п о Rao D. V. J. Sci. and Industr. Res., 1954, v. 14B, p. 38.
28. Ter M u e 1 e n H. Chem. Weekbl., 1925, v. 22, p. 219.
29. Spengler G., Weber A. Chem. Ber., 1959, v. 92, p. 2163.
30. В i 1 t z W.,K6cher A. Z. Anorg. Chem., 1941, Bd. 248, S. 172.
31. P r e n n e r A. G., Brock moller I. Z. phys. Chem., 1913, Bd. 81, S. 151.
32. Parravano N., M a 1 q u о r i G. Atti. Accad. Lincei, 1928 (6), v. 7, p. 19.
428
33. Strivastava M. H. W., G h о s h S. Proceedings Nat. Acad. Sci. India*
1960, v. A29, № 2, p. 178—180, 181—186.
34. С к о p о в В. А., Кулешов В. А. Цветные металлы, 1960, № 3, с. 1—4.
35. Bor bash Е. Canad. Min-Journ., 1961, v. 82, № 8, р. 66.
36. Сентерюхина Л. Н., Опарина Е. М. Твердые дисульфидмолиб-
деновые смазки. Изд-во «Химия», 1966.
37. Зеликман А. Н.иТеслицкая М. В. Изв. вузов. Цветная металлур-
гия, 1968, № 2, с. 76.
38. .В г у а и t Р. J. а. о. Wear, 1964, v. 7, № 1, р. 118.
39. J о s t Н. Р. Aircraft Prod., 1960, v. 22, № 2, р. 5761.
40. L a n k a s t е г I. К. Wear, 1966, v. 9, № 3.
41. А л ь ш и цИ. Я. и др. Труды 3-й конференции по трению и износу в машинах.
Изд-во АН СССР, 1960, т. III, с. 172.
42. Bowden F. Р., R owe G. W. Engineer, 1957, v. 204, р. 667.
43. D е S с h u 1 t е n A. Bull. Min. France, 1889, v. 12, p. 545.
44. Зеликман A. H., К p e й н О. E. ЖПХ, 1960, № 1, с. 49.
45. Bell R. E., H e r f e r t R. E. J. Am. Chem. Soc., 1957, v. 79, № 13, p. 3351-
46. Braitwhaite E. R. Solid Lubricants and Surfaces. Pergamon Press, Ox-
ford—Lond.—New-York—Paris. 1964.
47. Romanowski W. Roczn. Chem., 1963, v. 37, p. 1077.
48. L a n g a u d i e I. Bull. Soc. France Miner. Cristall, 1956, v. 79, p. 576.
49. Круглова В. Г. и др. «Новые данные о минералах СССР». Тр. Минерало-
гического музея. Изд-во «Наука», 1965, № 16, с. 233.
50. F г i n d t R. F., У о f f е A. D. Proc. Roy. Soc. London, Sec. A, 1963, v. 273,
p. 69—83.
51. Зеликман А. Н.и др. Кристаллография, 1961, т. 6, № 3, с. 289.
52. Dickinson R. G., Р a u 1 i n g L. J. Am. Chem. Soc., 1923, v. 45, p. 1466.
53. Hassel O., Z. Kristallog Mineralog., Petrogr., 1925, Bd. 61, S. 92.
54. 3 в я г и н Б. Б., С о б о л е в а С. В. Кристаллография, 1967, т. 12, № 1,
с. 57.
55. Ч у х р о в Ф. В., 3 в я г и н Б. Б. и др. Геология рудных месторождений,.
1968, т. Ю, № 2, с. 12.
56. W у с к о f f W. G. Crystal Structures. Second. Ed. Vol. I, Intersci. PubL.
New-York—London, 1963, p. 280.
57. К i m b a 1 G. Journ. Chem. Phys., 1940, v. 8, p. 188.
58. Б a p и н с к и й P. Л., Вайнштейн Э. E. Изв. АН СССР, Сер. физ.,
1957 т. 21 с. 1387.
59. J е 1 1 i п е с k F. а. о. Nature, 1960, v. 185, № 4710, р. 376.
60. С емилетов С. А. Кристаллография, 1961, т. 6, с. 536.
61. Фармазян А. С., X у р ш у р я н Э. X. ДАН Арм. ССР, 1963, т. 37, № 4,
с. 211.
62. Graeser S. Schweiz. Minerak. Petrogr. Mitt., 1964, v. 1, p. 44.
63. T r a i 1 R. I. Canad. Mineralogist., 1963, v. 7, № 3, p. 524.
64. J о n s t о n R. R. M., Moore A. I. W. J. Phys. Chem., 1964, v. 68, p. 3399.
65. С a n n о n P. Nature, 1959, v. 183, p. 1612.
66. Исакова P. А., Изв. АН Каз. ССР, Сер. металлургия, обогащение, огне-
упоры, 1961, № 3, с. 3.
67. Pierce G. W. Physic. Rev., 1909, v. 28, № 3, p. 175.
68. D u t t a A. K. Nature, 1947, v. 159, p. 477.
69. Waterman A. T. Phyl. Mag., 1917, v. 33, p. 237, Physic. Rev. 1923, v. 21,
№ 5, p. 540.
70. Minomura S., Drickamer H. G. J. Appl. Phys, 1963, v. 34, № 10,
p. 3043.
71. Горбатов Г. А. и др. Минеральное сырье, 1962, вып. 5, с. 96—109.
72. М a n s f i 1 d R., S а 1 a m S. A. Proc. Phys. Soc., 1953, v. 66B, p. 377.
73. Раджабян Ф. M. Ученые записки Азербайджанского университета,
1957, № 3.
74. С а м с о н о в Г. В., О г а н е с я н В. X. ДАН УССР, 1965, № 10, с. 1317.
75. Оганесян В. X., Рудь Б. М. Порошковая металлургия, 1965, № 12, с. 54.
429
76. N a к a d a I. J. Phys. Soc. Jap., 1956, v. 11, p. 1122, 1958, v. 13, p. 1547.
77. S с a 1 e s W. W. Bull. Amer. Phys. Soc., 1957, v. 2, p. 97.
78. Самедова-Бейбутова 3. A. Tp. Азерб. политехи, инет., 1963,
вып. 9, с. 44—48.
79. D u t t а А. К. Indian Journ. Phys., 1944, v. 18, p. 249.
80. С о b 1 a n t z W. W. Scient. pap. Bur. Hand. Washington, 1920, № 398; 1924,
№ 486.
81. Heaps C. W., Leach W. F. Bull. Amer. phys. Soc., 1956, v. 1, p. 87.
82. В r e n b e r S. L. J. Phys, and Chem. Solids, 1964, v. 25, p. 1427.
83. Соболев В. В. Оптика и спектроскопия, 1965, т. 18, с. 334.
84. Evans В. L., Young Р. A. Proc. Roy. Soc. bond., 1965, v. A284, p. 402.
85. S о 1 a m S. A. Proc. Math, and Phys. Soc. U. A. R., 1960, v. 24, p. 41.
86. L a g r e n a u d i e I. J. Phys, radium, 1954, v. 15, p. 229.
87. Усатая E. С. Зап. Всес. Минерал. Общ., 1952, т. 81, с. 298.
88. Христофоров Б. С. и др. Зап. Всес. Минерал. Общ., 1954, т. 83, с. 58.
89. Самедов М. А., Р за - 3 аде П. Ф. Тр. Ин-та химии АН Азерб. ССР,
1954 т. 13 с. 5.
90. X у р ш у д я н Э. X. ДАН СССР, 1966, т. 171, № 1, с. 186.
91. Брейтуэйт Е. П. Твердые смазочные материалы и антифрикционные
покрытия. Изд-во «Химия», 1967, с. 92—108.
92. Р г е n n е г A. G., В г о с k m б 1 1 е г I. Z. Phys. Chem., 1913, Bd. 81, S. 151.
93. В i 1 t z W., К ocher A. Z. Anorg. Chem., 1941, Bd. 248, S. 172.
94. A p у т ю н я н Л. А., Хуршудян Э. X. Геохимия, 1966, № 6, с. 650.
95. Kelley К. К., Bur U. S. Mines Bull., 1949, № 476, p. 210.
96. Peterson M.B.,J ohnspn R. L. Lubr. Eng., 1955, v. 11, № 5, p. 325.
97. S t u p p В. C. Lubr. Eng., 1953, v. 9, № 4, p. 195.
98. S c h о 1 t z W. G. a. o. Trans. Metallurg. Soc. AIME, 1961, v. 221, p. 356.
К главе X
l. Zachariasen W. H. Z. phys. Chem., 1927, Bd. 128, S. 39.
2. Brewer L. a. o. J. Am. Ceram. Soc., 1950, v. 33, p. 291.
3. Templton D. H., D a u b e n С. H. Acta cryst., 1950, v. 3, p. 261.
4. Climax Molybdenum Co. (USA) Arc-cast Molybdenum-base Allays. First Annual
Report, 1950, Second. Report, 1951.
5. Kieffer R., Cerwenka E. Z. Metallkunde, 1952, Bd. 43, S. 101.
6. Nowotny H. a. о. Monatsh. chem., 1954, v. 85, p. 255.
7. С а с о н о в Г. В. Силициды и их использование в технике. Изд. АН Укр.
ССР, 1959.
8. Самсонов Г. В., Портной К- И. Сплавы на основе тугоплавких
соединений. Оборонгиз, 1961.
9. Хансен М., Ан дер ко К. Структуры биндрных сплавов. Металлург-
издат, 1962.
10. Ham I. L. Trans. ASME, 1951, v. 73, p. 723.
11. G e a c h G. A., J о n e s F. O. Plansee Proc. 1955, p. 80—90; Met. Abstr.,
1957, v. 24, p. 366.
12. P a r t h ё E. a. o. Monatsh. Chem., 1955, v. 86, p. 182.
13. Kubaschewski O., Catterall S. A. Thermochemical data of
Alloys. Pergamon Press. London, 1956.
14. В e p я т и н У. Д. и др. Термодинамические свойства неорганических ве-
ществ. Атомиздат, 1965.
15. Р г е 1 1 е г Н. VDJ— Zeitschrift, 1956, Bd. 98, S. 1611.
16. Materials and Methods, 1956, v. 43, p. 131, 133.
17. F i t z e r E., S c h w a b J. Metall, 1955, Bd. 9, S. 1062.
18. Kieffer R. u. a. 1952, Bd. 43, S. 284.
19. Kieffer R., Benesovsky F. Symposium of powder Metallurg. Iron
and Steel Inst., 1953, prep. gr. IV, p. 40.
20. M a x w e e 1 W. US Nat. advis. Cite. Aeronautics, Research. Meto. 1952
(E52, D09), p. 19. Цит. no Met. Abstr. 1954.
430
21. Самсонов Г. В., Н е ш п о р В. С. Огнеупоры, 1958, № 1, с. 28.
22. С а м с о н о в Г. В. и др. ЖНХ, 1954, т. 4, с. 2759.
23. Northcott L. Molybdenum. London. Butterworths Scient. Publ. 1956
[см. перев. в сб. «Молибден», ИЛ, 1959].
24. Н б n i g s с h m i d O. Monatshefte Chemie, 1907, Bd. 28, S. 1017.
25. H 6 n i g s c h m i d O. Karbide und Silizide Halle/Saale, 1914.
26. D e f a c q z E. Compt. Rend. (Paris), 1907, v. 144, p. 1424.
27. Sedlatschek K.,Stadler H. I. Planseeberichte fur Pulvermetallur-
gie, 1961, Bd. 9, S. 40.
28. Самсонов Г. В., Э п и к А. П. Покрытия из тугоплавких соединений.
Изд-во «Металлургия», 1964.
29. С a m р b е 1 1 I. а. о. Journ. Electrochem. Soc., 1949, v. 96, p. 318.
30. В e i d 1 e r E. a. o. Journ. Electrochem. Soc., 1951, v. 98, p. 21.
31. Прокошин Д. A., A p з а м а с о в Б. H. В сб. «Исследования по жаро-
прочным сплавам». Изд-во АН СССР, 1962, т. IX, с. 177.
32. Kieffer R., Nachtigal Е. Heraues-Festschrift, Hanau, 1950, S. 186.
33. К о с о л а п о в а Т. Я., К о т л я р Е. Е. ЖНХ, 1958, т. 3, № 5, с. 1241.
34. Глушко П. И. и др. Физика металлов и металловедение, 1962. т. 13, № 6.
35. Б р о х и н И. С. и др. Цветные металлы, 1958, № 9, с. 61.
36. Марковский Л. Я-, Векшина Н.В. ЖНХ, 1957 т. 2, № 7, с. 1693.
37. Metallurgia, 1956, № 318, р. 175; 1957, v. 55, р. 239.
38. Семенов Г. В., Ковальченко М. С. Горячее прессование. Гостех-
издат УССР, 1961.
39. К i е s s 1 i n g R. Acta Chem. Scand., 1947, v. 1, p. 893, 1949, v. 3, pp. 90,
595, 603; 1950, v. 4, pp. 146, 209.
40. S t e i n i t z R. a. o. Journ. of Metals, 1952, v. 4, № 9, p. 983.
41. R a u t a k a P., S t e i n i t z R. Journ. of Metals, 1953, v. 5, № 5, p. 747.
42. Gilles P. W., P о 1 1 о с k В. D. Journ. of Metals, 1953, v. 5, № 11, p.1537.
43. К i e f f e r R. u. a. Z. Anorg. Allg. Chem., 1952, v. 268, № 3, p. 191.
44. R u d у E. u. a. Z. fur Metallrunde, 1963, Bd. 54, № 6, S. 345.
45. П о p т н о й К. И. и др. Изв. АН СССР, Металлы, 1967, № 4.
46. Climax molybdenum Company of Michigan. Arc cast Molybdenum base allays.
Second, annual Reports, 1951.
47. S t e i n i t z R. J. of Metals, 1952, v. 4, p. 148.
48. Z i e g 1 e r W. T., Y о u n g R. A. Oxford Confer, on low temper, physics.
Oxford, 1951.
49. H u 1 m I. K., Mathias В. T. Phys. Rev., 1951, v. 82, p. 273. .
50. Огнеупоры для космоса (справочник). Изд-во «Металлургия», 1967.
51. Tobias С. W. Napay-Szabo J. Am. Chem. Soc., 1949, v. 71, p. 1882.
52. M e e p с о н Г. А., Самсонов Г. В. ЖПХ, 1954, т. 27, с. 1505.
53. В е с k W. Metal Industry, 1955, v. 86, р. 43.
54. S t е i n i t z R., Binder I. Powder Met. Bull., 1953, v. 6, p. 123.
55. Schwarzcopf P. The Machinist, Juni 1953, p. 927.
56. П о p т н о й К. И. и др. Изв. АН СССР, сер. «Металлургия и топливо», 1959,
№2, с. 117.
57. Weiss G., A n d г i е u х L. Bull. Soc. Chim., France, 1948, v. 15, p. 598.
58. S у к e s W. P. a. o. Inst. Min. Met. Eng. Techn. Publ., 1935, № 647.
59. Few W. E., Manning G. K. J. of Metals, 1952, v. 4, p. 271.
60. S p e i s e r R. a. o. J. of Metals, 1952, v. 4, p~. 275.
61. Mo i s s a n H. Compt. Rend., 1893, v. 116, p. 1225, 1895. v. 120, p. 1320;
1897, v. 125, p. 839.
62. T и t i у a H. Sci. Papers Inst. Phys. Chem. Research, Tokyo, 1932, v. 19,
p. 384.
63. Kuo К., H a g g G. Nature, 1952, v. 170, p. 245.
64. Novotny H., Kieffer R. Z. Anorg. Chem., 1952, v. 267, S. 261.
65. Novotny H. a. o. Monatsh. Chem., 1954, v. 85, p. 255.
66. Ковальский A. E., Макаренко T. Г. В еб. «Микротвердость».
Изд-во АН СССР, 1951, с. 170.
67. Mattias В. Т., Н u 1 m J. К. Phys. Rev., 1952, v. 87, р. 799.
431
68. Koster W.,Rauscher W. Z. Metallkunde, 1948, v. 39, p. 111.
69. Hardy G. F., Hulm J. K- Phys. Rev., 1954, v. 93, p. 1004.
70. D a w i h 1 W. Z. Anorg. Allg. Chem., 1950, Bd. 262, S. 212.
71. A n d r i e u x L., W e i s s G. Bull. Soc. Chim. France, 1948, v. 15, p. 598.
72. Andrieux L., Lebeaux P. Les hautes temperatures et leurs utilisa-
tions en Chimie. Paris. 1950, v. I, p. 375.
73. H i 1 p e r t S., Ornst’ein M. Ber., 1913, v. 46, p. 1669.
74. Schenck R. u. a. Z. Anorg. Allg. chem., 1931, Bd. 203, S. 159.
75. P r i n g I. N., F i e 1 d i n g W. J. Chem. Soc., 1909, v. 95, p. 1497.
76. Lander I. L, G e r m e r L. H. An. Inst. Min. Met. Eng. Techn. Publ., 1947,
№ 2259.
77. Campbell I. E. a. o. J. Electrochem. Soc., 1949, v. 96, p. 318.
78. Киффер P.,Шварцкопф А. Твердые сплавы. Металлургиздат, 1957.
79. P о w e 1 1 C. F. a. o. Vapor-Planting. New-York—London, 1955.
80. Rudolf W., H о f m a n n U. Z. Phys. Chem., 1935, Bd. 28, S. 351, 370.
81. Brockway S. a. o. Trans. Farad. Soc., 1938, v. 34, p. 1350.
82. M о n d L. u. a. Z. Anorg. Chem., 1910, Bd. 68, S. 217.
83. Резухина T. H.,Швы рев В. В. Вестник МГУ (химия), 1952, № 6,
с. 41—45.
84. А с т р о в Д. Н. и др. ЖФХ, 1955, т. 29, № 3, с. 424.
85. К a w a i К., М u г a t а Н. Bull. Chem. Soc. Japan, 1960, v. 33, № 7,
p. 1008—1013.
86. Шарифов К-А.,Резухина T. H. Труды Института физики и мате-
матики АН Азерб. ССР, Сер. физич., 1953, т. 6, с. 53.
87. Luks R., Denke О. Gmelins Handbuch Anorg. Chem. Syst., 1935, № 53,
p. 199.
88. С о t t о n F. A., a. o. Journ. Am. Chem. Soc., 1950, v. 78, p. 5168.
89. H i e b e r W., R о m b e r g E. Z. Anorg. Chem., 1935, Bd. 221, S. 332.
90. H i e b e r W. Naturf. und Medizin in Deutschland, 1939—1946, B. 24, TI.
2 (1948), 108—145.
91. Белозерский H. А. Карбонилы металлов. Металлургиздат, 1958.
92. Анисимов К. Н.,Несмеянов А. Н. ДАН СССР, 1940, т. 26, № 1,
с. 57—58.
93. Кочетков К. А. и др. ДАН СССР, 1940, т. 26, № 1, с. 53—56.
94. Белозерский Н. А.,Кричевская О. Д. В сб. «Чистые металлы и
полупроводники». Металлургиздат, 1957, с. 328.
95. Белозерский Н. А., Кричевская О. Д. Труды ин-та «Гипро-
никель», Л., 1958, вып. 3, с. 284.
96. Job А., С a s s а 1 A. Bull. Soc. Chim., 1927 (4), v. 41, p. 1041.
97. Hieber W.,Romberg E. Z. Anorg. Chem., 1935, Bd. 221, p. 321.
98. H i b e г W. u. a. Berichte, 1932, Bd. 65, S. 1090.
99. Волков В. Л. и др. ЖНХ, 1958, т. 3, № 11, с. 2433.
100. Белозерский Н. А., Кричевская О. Д. В сб. «Металлургия
вольфрама, молибдена и ниобия». Изд-во «Наука», 1967, с. 109.
101. Скуратов С. М. Коллоидный журнал, 1947, т. 9, с. 133.
102. Горбунов А. Е. Порошковая металлургия, 1966, № 11, с. 52.
103. Киффер Р., Бенесовский Ф. Твердые материалы (пер. с нем.).
Изд-во «Металлургия», 1968,
104. Самсонов Г. В. ЖТХ j 1956, т. 26, с. 716.
К главе XI
1. Hagg G.,Magneli A. Arkiv Kemi, Mineral GeoL, 1944, A 19, № 2, p. 1.
2. M a g n e 1 i A. Acta chem. scand., 1948, v. 2, pp. 501, 861.
3. M a g n e 1 i A. Acta crystallogr. (Copenhagen), 1953, v. 6, p. 495.
4. G 1 e m s e r O., Lutz G. Z. Anorg. Allg. chem., 1951, 263, 1—3, p. 1—14.
5. К i h 1 b о r g L. Acta Chem. Scand., 1959, v. 13, № 4, p. 954.
6. P q д e E. Я., Лыса нова Г. В. ДАН СССР, 1962, т. 144, № 2, с. 351.
7. Watt G. W., D a v i s D. D. J. Amer. Chem. Soc., 1948, v. 70, p. 3751.
432
8. Funaki К.,Ogawa T. J. Electrochem. Soc. Japan, 1950, v. 18, pp. 198,
220.
9. Schonberg N. Acta Chem. Scand., 1954, v. 8, p. 617.
10. Hatterer A., Harold A., Rerat С. C. R. Hebd. Seances Acad. Sci,
1955, v. 241, p. 750.
ll. Magneli А., В 1 о m b e r g - H a n s s о n B., Kihlborg L. J
Lindqvist Acta chem. Scand., 1955, v. 9, p. 1382.
12. H e g e d u s A. I., S a s v a r i K-, Neugebauer I. Z. Anorg. Allg.
chem., 1957, B. 293, № 1—2, S. 56.
13. Озеров P. П. Кристаллохимия кислородных соединений ванадия, вольф-
рама и молибдена. Успехи химии, 1955, т. 24, № 8, с. 951.
14. Белов Н. В. Структура ионных кристаллов и металлических фаз. Изд.
АН СССР, 1947, с. 102.
15. В г а к к е n Н. Z. Kristal, 1931, Bd. 78, S. 484.
16. Wooster N. Z. Kristal, 1931, Bd. 80, S. 504.
17. Дорфман Я. Г. Магнитные свойства и строение вещества. Техтеорет-
издат, 1955, с. 239.
18. G 1 е m s е г О., Lutz G. Naturwiss., 1947, Bd. 34, S. 215.
19. M a g n e 1 i A. Arkiv Kemi Mineral Geol., 1946, v. 24A, № 2.
20. В 1 а с к b u r n P. E. a. o. Journ. Phys. Chem., 1958, v. 62, p. 769.
21. M i x t e r W. G. Amer. Inst. Sci., 1910, v. 29, p. 488.
22. C h a u d г о n G. Ann. Chim., 1921, v. 16, p. 221.
23. T о n a s a к i K. Bull. Inst. phys. Chem. Res. (Tokyo), 1940, v. 19, p. 126—132.
24. M a h A. D. J. Phys. Chem., 1957, v. 61, p. 1572.
25. S t a s к i e w i c z B. A. a. o. J. Amer. Chem. Soc., 1955, v. 77, № 11, p. 2987.
26. King E. G. J. Amer. Chem. Soc., 1958, v. 80, p. 1799.
27. Kubaschewski O., Evans E. Metallurgical Thermochemistry, Lon-
don, 1958.
28. К a у Thompson M. D. Total and Free Energy of formation of the oxides
of 32 Metals. Electrochem. Soc. New-York, 1942.
29. R i c h a r d s о n F. D., J e f f e s I. H. F. Journ. Iron Steel Inst., 1948, v. 160,.
p. 261.
30. Iaeger F., G e r m s H. Z. Anorg. Chem., 1921, Bd. 119, S. 145.
31. F e i s e r I. Metall und Erz., 1931, Bd. 19, Heft 12, S. 297.
32. Зеликман A. H. и др. ЖНХ, 1956, № 4, с. 332.
33. G о s g г о v е L. A., Snyder Р. Е. J. Am. Chem. Soc., 1953, v. 75, № 5,
р. 1227.
34. М о о s е J. Е., Parr S. W. Journ. chem. Soc., 1924, v. 46, p. 2660.
35. N e u m a n n B. u. a. Z. Anorg. Chem., 1934, Bd. 218, S. 379.
36. S e 1 t z H. a. o. J о u r n. Amer. Chem. Soc., 1943, v. 65, p. 600.
37. Spenser H. M. J. Amer. Chem. Soc., 1945, v. 67, p. 1859.
38. Kybaschewski O.,Catterall S. A. Thermodinamic date of alloys.
Pergamon, Lond., 1956.
39. H ege d u s A. I. u. a. Z. Anorg. Chem., 1957, Bd. 293, № 1—2, p. 56—83.
40. C h a u d г о n G. Compt. Rend., 1920, v. 170, p. 182, Ann. de chimie et de
physique, 1921, v. 16, p. 182.
41. Kay H.,Langston B.G. Journ. of Metals, 1964, v. 16, № 11, p. 877.
42. T о й с и н г С. X. В сб. «Молибден». ИЛ, 1962, с. 28.
43. Меерсон Г. А., Зеликман А. Н. Металлургия редких металлов..
Металлургиздат, 1955.
44. А б а ш и н Г. И., П о г о с я н Г. М. Технология получения вольфрама и
молибдена. Металлургиздат, 1960.
45. М i с h а е 1 А. В., Н a n w а у I. Е. Journ. of Metals, 1964, v. 16, № 11,
p. 881.
46. О r r C., D e 1 1 a v a 1 1 e Y. M. Fine particle measurement size, Surface and
pore volume, New-York, 1959.
47. Фигуровский H. А. Седиментационный анализ. Изд. АН СССР, 1948..
48. Карелин Б.А. иЛуцкий В. К. Методы и аппаратура для измерения
размеров частиц порошков. Цветная металлургия. Цветметинформация, 1966.
28 а. н. Зеликман 433
49. Авдеева Н. Я. Об аналитическом методе расчета сидементометрического
дисперсного анализа. Изд. Ростовского Университета, 1964.
50. Дерягин Б. В. и др. Определение удельной поверхности порошков по
. сопротивлению фильтрации разреженного воздуха. Изд. АН СССР, 1957.
51. Соминский Д. С., Ходаков Г. С. Прибор для определения поверх-
ности дисперсных материалов методом БЭТ. Промстройиздат, 1957.
52. Смителлс К. Дж. Вольфрам. Изд-во «Металлургия», 1952.
53. Третьяков В. И. Металлокерамические твердые сплавы. Металлург-
издат, 1962.
54. Федорченко И. М.,Андриевский Р. А. Порошковая металлур-
гия. Изд. АН УССР, Киев, 1961, с. 98—121.
55. Роде Е. Я., Л ы с а н о в а Г. В. ДАН СССР, 1962, т. 145, № 3, с. 573.
56. Scholz W. G. а. о. Trans. Met. Soc. of AIME, 1961, v. 221, № 2, p. 356.
57. N a c h t a m I. S., P о о 1 H. G. Pat. office, 1958, v. 730, № 2, p. 459; 1962,
№ 1, p. 212.
58. Abraham A. D. a. o. Journ. of Metals, 1960, v. 12, № 1, p. 26.
59. Hurd D. T., M с E n t e e H. R., Brisbin P. H. Ind. Eng. Cem., 1952,
v. 44, p. 2432.
50. German L. H., Lander LI. Metals Technology, 1947, № 2, p. 559.
61. Белозерский H. А.,Кричевская О. Д. «Гипроникель», вып. 3,
1958 с. 284.
62. Розен А. А., Укр. хим. журн. 1959, т. 25, с. 735; 1960, т. 26, с. 151.
*63. Степанова Т. И., Розен А. А. Физика металлов и металловедение,
1960, т. 10, № 5, с. 650.
64. Smiley S. Н., а. о. Journ. of Metals, 1965, v. 17, № 6, p. 605.
«65. Ruff O., A s c h e r E. Z. Anorg. Chem., 1931, Bd. 196, S. 418.
56. О x 1 e у G. H., C a m p b e 1 1 I. E. Journ. of Metals, 1954, № 2, p. 45.
67. Kp e й м e p T. С. и др. Заводская лаборатория, 1949, № 15, с. 159.
•68. Steel, 1953, v. 153, № 19, р. 102, 105.
69. Blocher I. М. а. о. Batalle Memor. Inst. I, 1960, p. 1440.
70. Cline I. E., W u 1 f f I. Journ. Electrochem. Soc., 1951, v'. 38, № 10, p. 385.
71. Balachandra L, Gadiyar H. S. Trans. Indian Inst. Metals, 1962,
v. 15, p. 168.
72. Lansberg A. a. o. Journ. of Metals, 1965, v. 17, № 6, p. 850.
73. Лукс Г. Экспериментальные методы в неорганической химии. Изд-во «Мир»,
1965 с. 330.
74. Childs W. I. а. о. Trans. Amer. Soc. Met., 1951, v. 43, p. 105—121. (Cm.
перев. в книге «Методы получения чистых металлов». ИЛ, 1957, с. 155).
75. W 1 о d е k S. Т., W u 1 f f I. Journ. Electrochem. Soc., 1960, v. 107, № 6,
p. 565.
76. А г т e К., Вацек H. Вольфрам и молибден. Изд-во «Энергия», 1964.
77. Р о w е е 1 С. F. а. о. Vapor Plating New-York, 1955.
78. С e н д e p о в С. В сб. «Молибден». ИЛ, 1962, с. 129.
79. В a d i а 1 i М. A., Kirshenbaum N. W., В a k i s h R. Trans. Metal-
lurg. Soc. AIME, 1963, v. 227, № 1, p. 32—36.
80. Seim H. L, H о 1 f M. L. Trans. Electrochem. Soc., 1949, v. 96, p. 205.
81. The Metal Molybdenum Publ. Amer. Soc. Metals, Cleavelend, Ohio, 1958, Ed
, I. I. Harwood.
82. G m e 1 i n s Handbuch der Anorganischem Chemie. Syst, 1935, № 53, Molyb-
den, S. 155.
83. Booth H. S.,Mer 1 ub S ob о 1 M. J. Phys. Chem., 1931, v. 35, p. 3319.
84. K's ycki M. J.,Yntema L. F. Trans. Electrochem. Soc., 1949, v. 96, p. 48.
85. Л e oht ь e в Г. А. В сб. «Металлургия и металловедение чистых металлов».
1959, вып. 1, с. 70.
86. L a n d е г I. I., G е г m е г L. Н. Metals Technology, 14 (6), Techn. Publ.
№ 2259 (Sept. 1947).
87. Kollock L.,Smith E. J. Am. Chem. Soc., 1901, v. 23, p. 669.
88. Самарцев А. Г., Л e в и т и н а Э. И. ЖФХ, 1968, т. 32, с. 1023.
89. Гончаренко А. С. Цветные металлы, 1963, № 10, с. 60.
434
90. А н д р е е в В. Н. Укр. хим. жури., 1953, № 21, с. 569.
91. Делим арский Ю. К., Назаренко Г. Д. ЖНХ, 1957, № 2, с. 890.
92. Григорьян А. Л. В сб. «Порошковая металлургия и металлообработка».
НТО Машпром, Ереван, 1963, с. 33.
93. Н е i n е n Н. J., Z a d г а I. В. Reports Investig. Bur. Min. U. S. Dept.
Interior, 1964, v. 17, № 6444.
94. Backer Don H. J. of Metals, 1964, v. 16, № 11, p. 873.
95. S e n d e г о f f S. The Journal of the Institute of Metals, July 1966, Metallurg.
Reviews, v. 11, № 106, p. 97—112.
96. Z a d r a I. B., Gomes I. M. Bur. Mines Reports Investig., 1959, N 5554,
p. 23.
97. J. of Metals, 1960, № 8, p. 627.
98. Senderoff S., Brenner A. J. Electroch. Soc., 1954, v. 101, № 1,
pp. 28, 31, 29.
99. Рыжик О. А., С м и p н о в M. В. Изв. вузов. Цветная металлургия, 1963,
№ 6, с. 102.
100. Смирнов М. В., Р ы ж и к О. А. Труды Института электрохимии
Уральск, филиала АН СССР. Свердловск, 1965, вып. 6, с. 11—17.
101. Бурцева К. Г., К л я ч к о Л. И. В сб. «Металлургия вольфрама, мо-
либдена и ниобия». Изд-во «Наука», 1957, с. 91.
102. Хлебников Б. И., Надольский А. П. Труды Иркутского поли-
технического института, вып. 27, Сер. химич., 1966, с. 101.
103. Хлебников Б. И.,Надольский А. П. Изв. вузов. Цветная ме-
таллургия, 1968, № 1, с. 82.
104. Г рейвер Н.С.Вкн. «Основы металлургии». Изд-во «Металлургия», 1967,
т. IV с. 31_____35.
105. Couch D. Е., Senderoff S. Trans. Met. Soc. of AIME, 1958, v. 212,
№ 3, p. 320.
106. Hegediis A. J., Neugebauer J. Zeit. Anorg. Allg. Chem., 1960,
Bd. 305, № 3—4, S. 216.
107. Елютин В. П. и др. Изв. вузов. Черная металлургия, 1964, № 7, с. 5.
108. 3 е л и к м а н А. Н. Металлургия вольфрама и молибдена. Металлург-
издат, 1949.
109. Зеликман А. Н. и др. Изв. АН СССР, Неорганические материалы,
1966, т. 2, № 12, с. 2204.
110. Зеликман. А. Н. и др. Изв. АН СССР, Металлы, 1968, № 3, с. 80.
111. Есин О. А., Г е л ь д П. В. Физическая химия металлургических про-
цессов, ч. 1, Металлургиздат, 1962.
112. Y i р S. а. о. Wah Chang Corp. Albany, Oregon, 1960, 29 pp. U. S. Govern-
ment Research Reports 37, 1962, № 10, p. 541.
113. Metal Industry, 1962, 22 apr., p. 490.
К главам XII и XIII
1. С о о 1 i d g e W. D. J. Amer. Inst. Electr. Eng., 1910, v. 29, p. 953.
2. Соболевский П. Г. Горный журнал, 1827, ч. I, кн. 1; ч. II, кн. 6.
3. Агте К., В ацек К. Вольфрам и молибден. Изд-во «Энергия», 1964.
4. S е е 1 i g R. Р. Fundamentals of Pressing of Metals Powders in «The Physics of
Powder Metallurgy», New-York, Chap. 21, 1951, p. 344.
5. Бальшин M. Ю. Порошковая металлургия. Машгиз, 1948.
6. M e e p с о н Г. А. В сб. «Вопросы порошковой металлургии». Изв. АН УССР,.
1955, с. 16—51.
7. Скотт Г. и др. В сб. «Молибден». ИЛ, 1962, с. 43.
8. The Metal Molybdenum published dy Amer. Soc. for Metals, Cleaveland ohioF
1958, Ed. I. I. Harwood.
9. Сб. «Порошковая металлургия», НТО Машпром, Ярославль, 1956.
10. Борок Б.А.и др. Технология и оборудование для гидростатического прес-
сования. Филиал ВИНИТИ, 1959.
28*
435
И. Сб. «Порошковая металлургия» (ЦИИН ЦМ). Изд-во «Металлургия», 1965,
вып. 43.
12. Бурцева К- Г., К л я ч к о Л. И. В сб. «Металлургия вольфрама, молиб-
дена и ниобия». Изд-во «Наука», 1967, с. 91.
13. Борок Б. А. и др. Порошковая металлургия, 1967, № 11, с. 29.
14. Сб. «Порошковая металлургия». Изд-во «Высшая школа», Минск, 1966.
15. L е b а с h I. Steel, 1960, v. 146, № 15, р. 93.
16. L а с h ш а и J. С., Industr. Eng. Chem., 1961, v. 53, № 12, A56—A59.
17. Бальшин M. Ю.,Дубровский А. П. ДАН СССР, 1961, т. 136, № 2,
с. 332. * '
18. Steel, ’1959, v. 145, № 19, р. 158—160.
19. Гуляев Б. Б. и др. Литье из тугоплавких металлов. Изд-во «Машино-
строение», 1964.
20. Самсонов Г. В.,Плоткин С. Я. Производство железного порошка.
Металлургиздат, 1957, с. 190.
21. Федорченко И. М., Андриевский Р. А. Основы порошковой
. металлургии. Изд-во АН Укр. ССР, 1961.
22. Miller G. L. Metal Industry Lnd., 1949, v. 75, № 20, p. 411, №21, p. 439.
23. Kieffer R.,Benesovsky F. Powder Metallurgy. Bulletin, 1956, v. 7,
№ 3—6, p. 113.
24. Гаврилин В. И.,Лобашев Б.П. Индукционные вакуумные высоко-
температурные печи. Филиал ВИНИТИ, 1957.
25. Смелянский М. Я. и др. Дуговые вакуумные печи и электронные пла-
вильные установки. Металлургиздат, 1962.
26. Kieffer R., В eneso vsky F. Metall, 1959, № 5, p. 379—385.
27. Савин А. В., Э й д у к Ю. А. Цветные металлы, 1959, № 5, с. 81.
28. Мармер Э. Н. и др. В сб. «Применение вакуума в металлургии».Изд-во
АН СССР, 1963, с. 213.
29. Сб. «Применение вакуума в металлургии». Изд. АН СССР, 1960, с. 290.
30. Г и р ш о в В. Л., Постнов Л. Н. В сб. «Вакуумная металлургия»,
вып. 2. Л., Изд. ДНТП, 1964, с. 33.
31. Киффер Р., Пипитц Э. В сб. «Молибден». ИЛ, 1962, с. 373.
32. Belk J. A. Less-Common Metals, 1959, v. 1, № 1, р. 50.
33. Абашин Г. И., Погосян Г. М. Технология получения вольфрама и
молибдена. Металлургиздат, 1955.
34. Строев А. С., Иванов А. М.,Овсепян Е. С. Веб. «Применение
вакуума в металлургии». Изд. АН СССР, 1958, с. 62.
35. Сушкин Н. Г. и др. Изв. АН СССР, Металлы, 1967, № 3, с. 94.
36. Грубер. Плавка металлов электронным лучом. ИЛ, 1963.
37. Сб. «Тугоплавкие металлы и сплавы» (пер. с англ.). Изд-во «Металлургия»,
1965.
38. Заборонок Г. Ф., Зеленцов Т. И. и др. Электронная плавка
металлов. Изд-во «Металлургия», 1965.
39. Тиммонс Дж. А., Ийнглинг Р. Дж. Дуговая плавка молибдена.
В сб. «Молибден», ИЛ, 1962, с. 74.
40. Смелянский М. Я-, Гуттерман К-Д- Рабочий процесс и расчет
вакуумных дуговых печей. Госэнергоиздат, 1962.
41. Parke R. М., Н a m I. L. Trans. Am. Inst. Min. Met. Eng., v. 171, 1947, p. 16.
42. N о r t h с о t t L., Molybdenum. London, Butterworths Scient. Publ. 1956.
43. О g i e r m a n n G. Vacuum, 1960, v. 10, № 6, p. 454.
44. V e г о t J. L., Forestier H. Comp. Rend., 1961, v. 253, № 4, p. 662.
45. Смит X. P. и др. В сб. «Электронная плавка металлов». Изд-во «Мир», 1964.
46. S е a g 1 е S. R., М а г t i n R. L., В е г t е а О. Journ. of Metals, 1962, v. 14,
№ 11, p. 812.
47. Савицкий E. M. и Бурханов Г. С. Металловедение тугоплавких
металлов и сплавов. Изд-во «Наука», 1967.
48. П ф а н н В. Дж. Зонная плавка. Металлургиздат, 1960.
49. Парр Норман. Зонная очистка и ее техника. Металлургиздат. 1963.
50. Heywang W. Z. Naturforschung, 1956, Bd. На, S. 238.
436
51. Донской А. Д., Б а ш е н к о В. В. Применение электронно-лучевого
нагрева в промышленности. Изд-во «Знание», 1962.
52. Введение в технологию электронно-лучевых процессов под ред. Р. Бешин
(пер. с англ.). Изд-во «Металлургия», 1965.
53. Савицкий Е. М. и др. Заводская лаборатория, 1961, № 8, с. 879.
54. С а в и ц к и й Е. М., Бурханов Т. С. и др. Труды совещания по росту
кристаллов. Изд-во «Наука», 1965, № 6, с. 308.
55. Ивановский Г. Ф., Загорская Т. Н. Изв. АН СССР, Металлы,
1965, № 3, с. 65.
56. Калверин А.Всб. «Физические и химические свойства металлов высо-
кой чистоты». Изд-во «Металлургия», 1964.
57. Сушкин Н. Г. Теплофизика высоких температур, 1963, № 2, с. 313.
К главе XIV
1. Сб. «Молибден», ИЛ, 1962.
2. Ham J. L. Trans. ASME, 1951, v. 73, № 6, p. 731.
3. Bechtold J., S с о t t H. J. Electrochem. Soc., 1951, v. 98, p. 495.
4. Bechtold J.J. of Metals, 1953, № 5, p. 1469.
5. Моргунова H. H. Жаропрочные сплавы на основе молибдена. ВИНИТИ,
1959.
6. Агте К., В ацек И. Вольфрам и молибден. Изд-во «Энергия», 1964.
7. Н a m J. L. Steel, 1951, v. 128, № 3, р. 106.
8. М о ч а л о в Г. А. и др. Металлургия и металловедение чистых металлов,
вып. 6. Атомиздат, 1967, с. 155.
9. Kolbe С. L. J. Electrochem. Soc., 1954, v. 101, № 12, р. 61.
10. Hahn G. T. a. о. Proc. Refractory Metals Simpos. Chicago, April, 1962, p. 23.
11. Крупин А. В. и др. В сб. трудов МИСиС (XLVI) «Прокатка металлов и
биметаллов в вакууме». Изд-во «Металлургия», 1968, с. 15 и 65.
12. Крупин А. В., П а в л о в И. М. и др. Цветные металлы, 1966, № 5, с. 93.
13. Northcott L. Molybdenum. London. Betterworths, Sc. Publ. 1956 (cm.
перев. в сб. «Молибден», ИЛ, 1959).
14. В е е h t о 1 d J. Н., S с о t t Н. Metal Progress, 1952, v. 61, p. 82—88.
15. Э с п e В. Технология электровакуумных материалов (пер. с нем), т. I.
Металлы и материалы с металлической проводимостью. Госэнергоиздат, 1962.
16. Металлы и сплавы для электровакуумных приборов. Под ред. проф. Р. А. Ни-
лендера. Изд-во «Энергия», 1965.
17. Савицкий Е. М., Бурханов Г. С. Металловедение тугоплавких
металлов и сплавов. Изд-во «Наука», 1967.
18. F a n s t е е 1. Met. Corp. Tungsten and Molybdenum. Tech, inform. North.
Chicago, 1950.
19. V a n A r k e 1 A. E. Reine Metalle, Berlin, 1939.
20. P a r k e R. M. Metal Progr., 1951, v. 59, p. 81—96.
21. T i t z T. E., Wilson J. W. Behavior and Properties of Refractory Metals,
London, 1965.
22. Engl J., F 6 r m e r J. Z. phys., 1935—36, Bd. 98, S. 702.
23. Engl J., К a t z J. Z. phys., 1937, Bd. 106, S. 1.
24. Koster W. Z. Metallkunde, 1948, Bd. 39, S. 1.
25. Allen N. P., Carrington W. E., J. Jnst. Metals, 1953—1954, v. 82,
p. 525.
26. P u g h J. W. Trans. ASM, 1955, v. 47, p. 47.
27. В r u c k a r t W. L., H у 1 e r W. S. J. Metals, 1955, Bd. 7, № 2. Sec. 2.
28. О 1 d s L. E., R e n g s t о r f f G. W. J. Metals, 1956, v. 8, p. 150.
29. Marigner R. E., Schwope A. D. Trans. AIMME, 1954, v. 200, p. 65.
30. Few W., Manning G. K- J. Metals, 1952, v. 2, p. 271; 1955; v. 2, p. 343.
31. S p e i s e r R. W. a. o. Trans. AIMME, 1952, v. 194, p. 276.
32. CaseS. L. Metallurgy of Molybdenum, Summery Report to the Office of Naval
Research Battelle. Memorial Inst. Oct. 1954.
32a. Sy kes W. D. a. o. Trans. AIMME, 1935, v. 117, p. 173.
437
33. Р е г г у Т. а. о. J. Metall Progress, 1954, v. 65, р. 75.
34. J a f f е е R. I. J. Metals, 1964, № 5, p. 410.
35. Braun H. a. o. Metallwerk Plansee AG Reutte/Tyrol, 1965, p. 35.
36. Хансен M., Ан дер ко К. Структура двойных сплавов. Металлург-
издат, 1962, т. I и II.
37. С а в и ц к и й М. Е. и др. ЖНХ, 1959, т. 4, № 2.
38. Б а р о н В. В. и др. Изв. АН СССР, ОТН, Металлургия и топливо, 1958,
№ 4, с. 36.
39. В г и с k а г t W. L. а. о. Trans. Amer. Soc. Metals, 1953, v. 45, p. 286.
40. Ham J. L. Trans. Amer. Soc. Meeh. Engrs, 1951, v. 73, p. 723.
41. Савицкий E. M., Барон В. В. В сб. «Прочность металлов». Изд-во
АН СССР 1956 с. 144.
42. Р i р i t z’ Е., К i’e f f e r R. Z. Metallkunde, 1955, Bd. 46, S. 187.
43. С а в и ц к и й Е. М., Т ы л к и н а М. А. В сб. «Исследования по жаро-
прочным сплавам». Изд-во АН СССР, 1956.
44. Refractory metals and Allays III. Applied Aspects. Los Angelos. December,
9—10, 1963, Edited by R. I. Jaffee.
45. С а в и ц к и й М. Е. и др. ДАН СССР, 1957, т. 113, № 4, с. 1070.
46. О 1 d s L. Е., R е n d s t о г f f G. W. J. Metals, 1957, v. 9, № 4, p. 468.
47. H e r z i g A. J. Tool Engr, 40 Febr., 1958, p. 212.
. 48. С а в и ц к и й Е. М. и др. В сб. «Вопросы теории и применения редкоземель-
ных металлов». Изд-во «Наука», 1964.
49. Woodside G. С. Iron Age Dec., 1947, v. 4, p. 78.
50. С а в и ц к и й Е. М. и др. Физика металлов и металловедение, 1962, т. 14,
№ 1, с. 148.
51. Agte G.,PetrdTikM. Vlastnosti kovovych prasku a jejich mereni. Techni-
ckovedecke vydavatelstvi, Praha, 1951.
52. С о о n s W. C. Fractographic and Metallographic techniques for Molybdenum
and Molybdenum-base Alloys. The Metal Molybdenum. Publ. by Amer. Soc.
Metals, Cleveland, Ohio, 1958, p. 394.
53. С м и т e л л с К. Дж. Вольфрам. Металлургиздат, 1958, с. 164.
К главе XV
1. W i 11 i a m s D., Y u s t e r P. Phys. Rev., 1946, v. 69, p. 556.
2. Несмеянов Ан. H. и др. Получение радиоактивных изотопов. Госхим-
издат, 1954.
3. Northcott L. Molybdenum. London. Betterworths. Sc. Publ. 1956 (nep.
с англ, в сб. «Молибден», ИЛ, 1959).
4. Lu S. S., С h a n g Y. L. Proc. phys. soc. Lond., 1941, v. 53, p. 517.
5. Owen, Iball. Phil. Mag. J. Sci., 1932, v. 6, p. 13.
6. П о л и н г Л. Природа химической связи. Госхимиздат, 1947.
7. Goldschmidt V. М. Вег. deutsch. Chem. Ges., 1927, Bd. 60, S. 1270.
8. P a u 1 i n g L. J. Amer. Chem. Soc., 1927, v. 49, p. p. 765.
9. D u В r i g e L. A., R о her W. W. Phys. Rev., 1932, v. 42, p. 52.
10. Herring C.,N ichols M. H. Rev. Mod. Phys., 1949, v. 21, p. 185.
11. Ц a p e в Б. M. Радиотехника и электроника, 1957, т. 2, № 6, с. 675.
12. W о г t i n g A. G., J. Franklin Inst., 1925, v. 199, p. 549.
13. Kubaschewski O., Evans E. Metallurgical Thermochemistry. Lon-
don, 1958.
14. H о n i g R. Review a. technical Journ., 1955, v. 28, p. 400.
15. S t u 1 1 D. K., S i n k e С. C. Thermodinamic properties of the Elements.
Advances in chemistry. Ser. 18. Am. Chem. Soc., Washington, 1956.
16. Несмеянов Ан. H. Давление пара химических элементов. Изд. АН
СССР, 1961.
17. Edwards J. W. а. о. J. Amer. Chem. Soc., 1952, v. 74, p. 1539.
18. Lan g'm u i r I. Phys. Zeitschr., 1913, Bd. 14, S. 1273.
438
19. J о n e s H, A., Langm u ir L, G. M. Me. Key Phys. Rev., 1927, v. 30,
p. 201.
20. Z w i к к e r C. Physica (Niderland), 1927, v. 7, p. 71.
21. С т э л л Д. P. Таблицы давления пара индивидуальных веществ. ИЛ, 1949.
22. Kelley К- К. Bull. U. S. Bur. Mines, 1949, № 476.
23. Demar quay J. Compt. Rend., 1945, v. 220, p. 81—83.
24. White J. L. «The Suitability of Platinum, Molybdenum, Tantalum zand
Tungsten as Hightemperature Length Standards». Naval Research. Laboratory
Reports, 1958, № 5153, June 17.
25. О s b о r n R. H. J. Opt. Soc. Sm., 1941, v. 31, p. 428.
26. Tietz T. E., Wilson J. W. Behavior and Properties of Refractory Me-
tals, London, 1965.
27. К a n n u 1 u i к W. G. Proc. Roy. Soc., 1933, v. 141, p. 159.
28. Witney L. V. Phys. Rev., 1935, v. 48, p. 458.
29. Worthing A. G. Phys. Rev., 1925, v. 25, p. 853; 1926, v. 28, p. 190.
30. Z w i к к e r C. Physika, 1927, Bd. 7, S. 71.
31. Э с п e В. Технология электровакуумных материалов. Госэнергоиздат, 1962»
32. Агте К-, Вацек И. Вольфрам и молибден. Изд-во «Энергия», 1964.
33. Линтон Э. А. Сверхпроводимость. ИЛ, 1964.
34. Рудницкий А.А. Термоэлектрические свойства благородных металлов
и их сплавов. Изд-во «Наука», 1965.
35. Sieverts A.,Bruning К. Arch, fur Eisenhiittenwesen, 1933—1934, Bd.
7, S. 641.
36. Norton F. J., Marshall A. L. Trans. Amer. Inst. Met. Eng., 1935,
v. 116, p. 179.
37. Жуков И. И. Изв. .ин-та физ.-хим. анализа, 1926, т. 3, с. 14—15.
38. М а е t i n Е. Arch. Eisenhiittenw., 1924—1930, Bd. 3, S. 407.
39. H a g g G. Z. phys. Chem., 1930, Bd. 7, S. 339.
40. S c h 6 n b e r g N. Acta Chem. Scand., 1954, v. 8, p. 204.
41. Heged iis A. I. u. a. Z. Anorg. Allg. Chem., 1957, v. 293, № 1—2, S. 56.
42. R о 1 s t e n R. F. Iodide metals and Metal iodides New-York—London, 1961.
43. A p ч e p P. С. Молибден. Справочник по редким металлам (пер. с англ.).
ИЛ, 1965, с. 400.
44. Analytical Chemistry, 1961, v. 33, № 8, р. 1125.
45. М а 1 о w a n S. L. Z. Metallkunde, 1931, Bd. 23, S. 69.
46. Ядерные реакторы. Материалы для ядерных реакторов, ИЛ, 1956, т. III,
с 159
47, N а с h t i g а 1 E. Z. Metallkunde, 1952, Bd. 43, № 1, S. 23.
48. Lu s t m a n B. Metal Progress, 1950, v. 57, № 5, p. 629.
49. К ub aschewsk i O., Schneider A. J. Inst. Metals, 1949, v. 75,
p. 403.
50. A p x a p о в В. И., К а з м а н о в Ю. Д. Физика металлов и металловеде-
ние, АН СССР, 1956, т. 2, № 8, с. 361.
51. Борисенко А. И. Защита молибдена от высокотемпературной коррозии.
Изд. АН СССР, 1962.
52. Защита от окисления тугоплавких металлов. ИЛ, 1962, с. 11, 42 и 131.
53. М о р г у н о в а Н. Н. Жаропрочные сплавы на основе молибдена. ВИНИТИ,
1962.
54. Couch D. Е. а. о. Electrochem. Soc., 1958, v. 105, р. 450.
55, J о n е s Е. S. а. о. Corrosion 14, January, 1958.
56. Minerals Yearbook, 1963, 1964. Chapter Molybdenum.
57. Кочергина Д. Г. Молибденовая промышленность капиталистических
стран*. Цветная металлургия. Цветметинформация, 1966, № 5, с. 53.
58. Хрущов Н.А. Оценка месторождений при поисках и разведках, «Молиб-
ден». Госгеолиздат, 1961, вып. 19.
59. S a w у е г Н. A. Engng and Mining Journ., 1967, № 2, p. 155—157.
60. W i g 1 e С. P., Canad. Min. Journ., 1967, v. 88, № 2, p. 174.
61. M a n t e 1 1 C. L. Engineering Materials Handbook, 1958, Sect. 13, p. 29—37.
62. Фримен P. P. В сб. «Молибден», ИЛ, 1962, с. 11.
439
63. M c Cloud J. L. Metal Progr., august 1958, v. 74 (2), p. 75.
64. Jet Propuls, 1956, v. 26, № 11, p. 1042.
65. Ратнер Е.И., Б у p н и н И. А. Молибден и урожай. Изд. АН СССР, 1959.
66. Canadian Mining J. 1968, v. 89, № 2, p. 133.
67. К il letter D. H., Linz A. Molybdenum Compounds. New-York — Lond.,
1952.
К главе XVI
1. Д p у ц е Н. Рений, ИЛ, 1951.
2. Т г i b а 1 a t S. Rhenium et Technetium. Paris, 1957.
3. Лебедев К. Б. Рений. Металлургиздат, 1963.
4. Рений. Труды 1-го Всесоюзного совещания по проблеме рения. Изд. АН СССР,
1961.
5. Рений. Труды 2-й Всесоюзной конференции по проблеме рения. Изд. АН СССР,
1964. *
6. Rhenium. Ed. by В. W. Gonser. Amsterdam—New-York, 1962.
7. С и м с X. T., Д ж а ф ф p и P. И. Свойства рения (пер. с англ.). Проблемы
современной металлургии, 1957, № 3.
8. 3 е л и к м а н А. Н. Производство рения, его свойства и области приме-
нения. «Вопросы радиоэлектроники», сер. 1, Электроника, 1962, вып. 7, с. 23.
9. Тронев В. Г. Рений. Химическая наука и промышленность, 1956, № 5,
с. 539.
10. Бибикова В. И. Цветные металлы, 1946, № 4, с. 44.
11. Савицкий Е. М. идр. Сплавы рения. Изд. АН СССР. 1965.
12. Зеликман А. Н. и др. Цветные металлы, 1958, № 11, с. 47.
13. З’еликман А. Н. и др. Изв. АН СССР. Металлургия и горное дело, 1963,
№ 6, с. 113.
14. Д е е в В. И., С м и р н о в В. И. Цветные металлы, 1964, № 4, с. 63.
15. С а в р а е в В. П., С а м н о в Е. А. Цветные металлы, 1960, № 12, с. 53.
16. К о в ы р ш и н В. Т. Цветные металлы, 1958, № 10, с. 43.
17. 3 е л и к м а н А. Н., Д р э г а н Л. ЖНХ, 1967, № 1, с. 261.
18. Зеликман А. Н., Мейснер Г. ЖПХ, 1966, т. 39, с. 224.
19. С h и г с h w о о d Р. Е., R о s е n b a u m J. В. J. of Metals, 1963, v. 15,
р. 648.
20. Петров А. В. и др. ЖНХ, 1965, № 10, с. 986.
21. Зайцев А. А. и др. ЖНХ, 1963, № 9, с. 2184.
22. Погорел ый А. Д.,Малюгин А. С. Изв. вузов. Цветная металлургия,
1964, № 5.
23. Гуляева Е. И. и др. Цветные металлы, 1959, № 5, с. 73.
24. Барышников Н. В.,Зеликман А. Н. Изв. вузов. Цветная метал-
лургия, 1962, № 6, с. 98.
25. 3 е л и к м а н А. Н. и др. Изв. АН СССР. Металлургия и горное дело,
1963, № 4, с. 161.
26. Зеликман А. Н., Н е р е з о в В. М. Цветные металлы, 1968, № 1, с. 65.
27. G о i s h i W., Libby W. F. J. Am. Chem. Soc., 1952, v. 74, p. 6109.
28. К e r t es A. S., В e c k A. J. Chem. Soc., 1961, № 5, p. 1921; 1961, № 11,
p. 5046.
29. Нерезов В. M., Зеликман А. Н. Изв. вузов. Цветная металлургия,
1967, № 5, с. 113.
30. Rosenbaum D. а. о. J. Electrochem. Soc., 1956, v. 103, № 9, р. 518.
31. Зеликман А. Н. и др. В сб. «Тугоплавкие металлы». Изд-во «Металлур-
гия», 1968, с. 95.
63. M c Cloud J. L. Metal Progr., august 1958, v. 74 (2), p. 75.
64. Jet Propuls, 1956, v. 26, № 11, p. 1042.
65. Ратнер E. И., Б у p н и н И. А. Молибден и урожай. Изд. АН СССР, 1959.
66. Canadian Mining J. 1968, v. 89, № 2, p. 133.
67. К il letter D. H., Linz A. Molybdenum Compounds. New-York — Lond.,
1952.
К главе XVI
1. Д p у ц е Н. Рений, ИЛ, 1951.
2. Т г i b а 1 a t S. Rhenium et Technetium. Paris, 1957.
3. Лебедев К. Б. Рений. Металлургиздат, 1963.
4. Рений. Труды 1-го Всесоюзного совещания по проблеме рения. Изд. АН СССР,
1961.
5. Рений. Труды 2-й Всесоюзной конференции по проблеме рения. Изд. АН СССР,
1964. »
6. Rhenium. Ed. by В. W. Gonser. Amsterdam—New-York, 1962.
7. Симс X. T., Д ж а ф ф p и P. И. Свойства рения (пер. с англ.). Проблемы
современной металлургии, 1957, № 3.
8. 3 е л и к м а н А. Н. Производство рения, его свойства и области приме-
нения. «Вопросы радиоэлектроники», сер. 1, Электроника, 1962, вып. 7, с. 23.
9. Тронев В. Г. Рений. Химическая наука и промышленность, 1956, № 5,
с. 539.
10. Бибикова В. И. Цветные металлы, 1946, № 4, с. 44.
11. Савицкий Е. М. и др. Сплавы рения. Изд. АН СССР. 1965.
12. 3 е л и к м а н А. Н. и др. Цветные металлы, 1958, № 11, с. 47.
13. З’еликман А. Н. и др. Изв. АН СССР. Металлургия и горное дело, 1963,
№ 6, с. 113.
14. Деев В. И., С м и р н о в В. И. Цветные металлы, 1964, № 4, с. 63.
15. С а в р а е в В. П., С а м н о в Е. А. Цветные металлы, 1960, № 12, с. 53.
16. К о в ы р ш и н В. Т. Цветные металлы, 1958, № 10, с. 43.
17. 3 е л и к м а н А. Н., Д р э г а н Л. ЖНХ, 1967, № 1, с. 261.
18. 3 е л и к м а н А. Н., Мейснер Г. ЖПХ, 1966, т. 39, с. 224.
19. Churchwood Р. Е., Rosenbaum J. В. J. of Metals, 1963, v. 15,
р. 648.
20. Петров А. В. и др. ЖНХ, 1965, № 10, с. 986.
21. 3 а й ц е в А. А. и др. ЖНХ, 1963, № 9, с. 2184.
22. Погорел ый А. Д.,Малюгин А. С. Изв. вузов. Цветная металлургия,
1964, № 5.
23. Гуляева Е. И. и др. Цветные металлы, 1959, № 5, с. 73.
24. Барышников Н. В.,Зеликман А. Н. Изв. вузов. Цветная метал-
лургия, 1962, № 6, с. 98.
25. 3 е л и к м а н А. Н. и др. Изв. АН СССР. Металлургия и горное дело,
1963, № 4, с. 161.
26. Зеликман А. Н., Н е р е з о в В. М. Цветные металлы, 1968, № 1, с. 65.
27. G о i s h i W., Libby W. F. J. Am. Chem. Soc., 1952, v. 74, p. 6109.
28. К e r t e s A. S., В e c k A. J. Chem. Soc., 1961, № 5, p. 1921; 1961, № 11,
p. 5046.
29. Нерезов В. M., Зеликман А. Н. Изв. вузов. Цветная металлургия,
1967, № 5, с. 113.
30. Rosenbaum D. а. о. J. Electrochem. Soc., 1956, v. 103, № 9, р. 518.
31. Зеликман А. Н. и др. В сб. «Тугоплавкие металлы». Изд-во «Металлур-
гия», 1968, с. 95.