/























































































































Автор: Малютина Т.М. Конькова О.В.
Теги: цветные металлы в целом анализ металлургия цветные металлы
ISBN: 5-229-00421-5
Год: 1988




Текст
Т, М. Малютина
О. В Коньков;
АНАЛИТИЧЕСКИЙ
КОНТРОЛЬ
! В МЕТАЛЛУРГИИ
ЦВЕТНЫХ
И РЕДКИХ
МЕТАЛЛОВ
Т. М. Малютина
О. В. Конькова
АНАЛИТИЧЕСКИЙ
КОНТРОЛЬ
В МЕТАЛЛУРГИИ
ЦВЕТНЫХ
И РЕДКИХ
МЕТАЛЛОВ
Издание второе,
переработанное
и дополненное
Допущено
Управлением кадров и учебных заведений
Министерства цветной металлургии СССР
в качестве учебника
для техникумов цветной металлургии
ft
МОСКВА «МЕТАЛЛУРГИЯ» 1988
УДК 669.2 + 669.85/.86 (543—547+075)
Рецензент: Каменск-Уральский алюминиевый техникум.
УД К 669.2 + 669.85/.86 (543—547 + 075)
Аналитический контроль в металлургии цветных и редких металлов.
Малютина Т. М., Конькова О. В.: Учебник для техникумов.—
2-е изд., перераб. и доп.— М.: Металлургия, 1988. 240 с.
Во втором издании (первое — в 1977 г.) описаны методы анализа
продуктов производства цветных и редких металлов от сырья до го-
товой продукции. Особое внимание уделено метрологической характе-
ристике методов и применяемой аппаратуре.
В практической части изложены методы, тщательно отработанные
в лабораториях научно-исследовательских институтов н предприятий
цветной металлургии, а также новые перспективные методы.
Для учащихся металлургических техникумов, обучающихся по
специальности «Анализ и контроль в цветной металлургии». Ил. 25.
Табл. 5. Библиогр. список 10 назв.
УЧЕБНИК ДЛЯ ТЕХНИКУМОВ
Тамара Михайловна Малютина
Ольга Викторовна Конькова
АНАЛИТИЧЕСКИЙ КОНТРОЛЬ
В МЕТАЛЛУРГИИ
ЦВЕТНЫХ И РЕДКИХ МЕТАЛЛОВ
2-е изд.
Редактор издательства Л. М. Элькинд
Художественный редактор Ю. И. Смурыгин
Технический редактор В. М. Курпяева
Корректоры Т. В. Морозова, В. М. Гриднева
ИБ № 2860
Сдано в набор 06.04.87. Подписано в печать 16.12.87. Т-24067. Формат бумаги
84Х 1О8'/з2- Бумага типографская № 2. Гарнитура литературная. Печать высокая.
Усл. печ. л. 12,6. Усл. кр.-отт. 12,6. Уч..-изд. л. 15,19. Тираж 3120 экз. За-
каз № 129. Цена 90 к. Изд. № 1206.
Ордена Трудового Красного Знамени издательство «Металлургия»,
119857, ГСП, Москва, Г-34, 2-й Обыденский пер., д. 14.
Областная книжная типография,
320091, Днепропетровск, ул. Горького, 20
2605000000—037
М -----------------60—87
—88
ISBN 5-229-00421-5 © Издательство «Металлургия», 1988
2
СОДЕРЖАНИЕ
Предисловие ........................................• • . 4
I. Значение контроля производства........................... 5
II. Правила отбора пробы и подготовка. ее к анализу ... 7
III. Оценка результатов анализа............................. 12
IV. Производственная классификация видов анализа .... 22
V. Научно-методическая классификация методов анализа . . 25
Химические методы.................................. 26
Физико-химические методы........................... 32
Физические методы.................................. 43
VI. Разделение и маскирование элементов. Вскрытие проб . . 45
Осаждение.......................................• • 45
Хроматография...................................... 46
Экстракция......................................• • '47
Маскирование..........................................• 48
Разложение материалов................................... 48
VII. Методы определения содержания цветных металлов ... 49
Алюминий..............................................• 49
Хром..........................................• • . 59
Кобальт...............................................• 66
Никель................................................• 76
Медь.............................................• • 83
Цинк.............................................• • 98
Кадмий................................................• 104
Олово............................................• • 107
Свинец........................................• . • ИЗ
VIII- Методы определения содержания редких металлов . • . . 118
Титан.........................................• . • 118
Цирконий и гафний • . •.............................. 131
Ниобий и тантал........................................ 146
Молибден н вольфрам ................................... 164
Рений . . • . •........................................ 180
Редкоземельные элементы................................ 190
Скандий .............................. 205
Галлий, индий, таллий ..................................211
IX- Методы определения содержания сопутствующих и при-
месных элементов . -.....................................222
Железо . . . . •...................................• 222
Кремний . •........................................... 226
Фосфор , . . . •...................................• 231
Азот н сера . .................................• . 234
Кальций и магний .......................................236
Рекомендуемый библиографический список . . . •..............241
Предметный указатель .........................................239
ПРЕДИСЛОВИЕ
Основными направлениями экономического и социального развития
СССР на 1986—1990 годы и на период до 2000 года предусмотрено1
в цветной металлургии опережающими темпами развивать алюмини-
евую, вольфрамо-молибденовую, редкометалльную и оловянную про-
мышленность.
Настоящая книга является вторым изданием (первое — вышло в
издательстве «Металлургия» в 1977 году под названием «Технический
анализ в металлургии цветных и редких металлов»1)- Материал, вклю-
ченный во второе издание, соответствует разделу «Аналитический
контроль в производстве цветных и редких металлов» программы по
предмету «Химические и физико-химические методы анализа». При
написании книги авторы учитывали, что изучению этого раздела пред-
шествует изучение курсов «Качественный анализ» и «Количественный
анализ». В связи с этим в настоящей книге рассмотрены лишь основы
химических и физико-химических методов анализа. Основное внимание
уделено особенностям применения каждого из методов в аналитиче-
ском контроле производства цветных и редких металлов. Рассмотрен
анализ разнообразных объектов: руд, концентратов, сплавов, раство-
ров и т. д. Большое внимание уделено способам разложения материа-
лов в сочетании с гравиметрическими, титриметрическими, электрохи-
мическими, фотометрическими, атомно-абсорбционными методами ана-
лиза.
Во второе издание книги внесены следующие изменения и допол-
нения: 1) согласно учебной программе, включены новые разделы:
«Кальций», «Магний» и «Фосфор»; 2) предусмотрено применение по-
суды из стеклоуглерода вместо дорогостоящей — платины; 3) приведена
методика определения меди в сплавах способом внутреннего электро-
лиза с использованием катодов в виде тигля из стеклоуглерода (мето-
дика разработана преподавателями МИСиС В. П. Гладышевым и
Л. 3. Козель); 4) приведен ряд новых методик (например, определе-
ния свинца, железа); некоторые методики исключены.
Практической части анализа предшествуют характеристика при-
родного сырья, рассмотрение применения цветных и редких металлов
в различных областях техники, свойств соединений, имеюших значение-
для аналитической химии элементов.
Порядок расположения материала, относящегося к цветным и
редким металлам, в основном соответствует порядковому номеру эле-
мента в Периодической системе элементов Д. И. Менделеева; элемен-
ты с близкими химическими свойствами (цирконий и гафний, ниобий
и тантал, молибден и вольфрам) рассмотрены в одной главе.
Более подробные сведения по всем рассмотренным в книге вопро-
сам учащиеся могут найти в литературе, список которой приведен в
конце книги, а также в серии монографий, вышедших в издательстве
«Наука» и посвященных аналитической химии отдельных элементов;
алюминия, кобальта, никеля, цинка, кадмия, олова, циркония, гаф-
ния, ниобия, тантала, вольфрама, молибдена, рения, редкоземельных
элементов, иттрия, индия, галлия, таллия, кремния, азота и серы.
Разделы I—IV и VIII книги (кроме главы «Скандий») написаны
Т. М. Малютиной, раздел VII — О. В. Коньковой и раздел IX иаписан
совместно.
Авторы выражают благодарность канд. хим. наук А. И. Лазареву^
А. П. Савостину, Н. Л. Климасенко и проф. Ю. М. Полежаеву за
ценные замечания, сделанные при просмотре рукописи.
1 Термин «технический анализ» выходит из употребления.
4
I. ЗНАЧЕНИЕ КОНТРОЛЯ ПРОИЗВОДСТВА
В задачу курса «Аналитический контроль в металлургии
цветных и редких металлов» входит изучение методов
определения химического состава сырья, промежуточных
продуктов производства, вспомогательных материалов и
готовой продукции.
Изучение химического состава исходного сырья (руд,
концентратов), промежуточных продуктов производства
(электролитов, пульп, растворов различного состава), го-
товой продукции (металлов, сплавов и т. д.), вспомога-
тельных материалов (воды, реактивов и т. п.) осуществля-
ется путем аналитического контроля произ-
водства. Анализ выполняется по утвержденным схемам,
в которых указаны объекты анализа, определяемые ком-
поненты, диапазон определения их содержаний, точки от-
бора пробы, периодичность анализа, методика анализа.
Значение аналитического контроля для производства
огромное, так как дает возможность судить о ходе техно-
логического процесса, его соответствии установленным ре-
жимам, о качестве используемого сырья и готовой про-
дукции. Без аналитического контроля предприятие не
может выпускать продукцию, соответствующую установ-
ленным нормам. Хорошо налаженный контроль способ-
ствует ритмичной работе предприятия, повышению каче-
ства продукции, снижению брака и уменьшению отходов
производства. На основании результатов анализа осуще-
ствляют классификацию продукции по сортам или маркам
и производят финансовые расчеты между поставщиком
продукции и ее потребителем.
Организация контроля производства. На предприятиях
технологический процесс контролируется специальной
службой, в которую входят лаборатории: ЦЗЛ (централь-
ная заводская), цеховые, экспрессного анализа, специаль-
ного назначения (анализ воды, промышленных стоков)
и санитарно-промышленные.
ЦЗЛ является научно-техническим центром предприя-
тия, в задачу которого входит организация п осуществле-
ние контроля процесса производства продукции на всех
стадиях. Кроме того, ЦЗЛ должна осваивать и использо-
вать новые, наиболее перспективные методы контроля,
участвовать в подготовке новых ГОСТов и ТУ, проводить
арбитражные и проверочные анализы, осуществлять мето-
дическое руководство цеховыми лабораториями и т. д.
5
Цеховые лаборатории систематически контролируют
процесс производства отдельных цехов, способствуя вы-
явлению причин технологических неполадок и разработке
предложений для их устранения. Экспресс-лаборатории
обслуживают отдельные стадии (переделы) технологиче-
ского производства, требующие наиболее быстрого конт-
роля. Деятельность заводских лабораторий тесно связана
с работой ОТК (отдела технического контроля), который
контролирует качество поступающего сырья и продукции
предприятия, а также соблюдение технологических режи-
мов. В функции ОТК входит также наблюдение за пра-
вильностью хранения готовой продукции, сырья и мате-
риалов; контроль качества тары, упаковки и маркировки.
Кроме того, ОТК осуществляет отбор средних проб, обе-
спечивает их хранение. Вопросы, связанные с проведением
арбитражных анализов, с предъявлением рекламаций так-
же решаются при участии ОТК-
Требования к методам анализа. Продукция предприя-
тий цветной металлургии весьма разнообразна.
Объектами анализа являются руды, концентра-
ты, оксиды, соли, металлы, сплавы, производственные раст-
воры и т. д. Важной задачей является анализ сточных
вод предприятий с целью определения содержания цен-
ных компонентов и токсичных веществ, а также анализ
атмосферы цехов и выбросов в атмосферу.
Отличительной особенностью большинства руд цветных
и редких металлов является низкая концентрация основно-
го металла—от тысячных и десятитысячных долей про-
цента до целых процентов. Лишь в некоторых рудах, на-
пример алюминиевых, титановых, содержание элементов
исчисляется десятками процентов. В продуктах переработ-
ки руд — концентратах — содержания цветных и редких
металлов также достигают десятков процентов.
Развитие новых отраслей промышленности — атомной
энергетики, ракетостроения, полупроводниковой техники —
связано с применением материалов особой чисто-
т ы. К ним относятся, например, элементные полупровод-
ники (германий, селен, теллур), полупроводниковые соеди-
нения (арсенид галлия, фосфид индия), высокочистые
цирконий, ниобий и др. В отдельных случаях содержание
примесей в этих материалах не должно превышать 10~6—
10-7%. Для определения различных содержаний элементов
необходимы соответствующие методы анализа. В одних
случаях для применяемых методов характерным является
низкий предел обнаружения, в других — в ы с о-
6
кая воспроизводимость результатов. При конт-
роле отдельных стадий технологических процессов, особен-
но быстропротекающих, высокие требования предъявля-
ются к скорости выполнения анализа.
Руды цветных и редких металлов являются комплекс-
н ы м сырьем. Например, в рудах меди, свинца и цинка
обычно содержатся кадмий, золото, серебро, селен, теллур,
молибден, мышьяк, висмут, сера и другие элементы. Такие
руды называют полиметаллическими. Задача аналитика —
определить содержание одного или нескольких элементов
в присутствии многих других. Методы, применяемые для
анализа многокомпонентных материалов, должны быть
избирательными.
Для выполнения анализа применяют химические, фи-
зико-химические и физические методы, характеризующие-
ся различными пределами обнаружения, воспроизводи-
мостью и скоростью выполнения. Определение содержаний
элементов в пределах 10-6—1О-8О/о стало возможным при
использовании физических методов анализа (масс-спект-
ральных, активационных). Повышению экспрессное™ ана-
лиза способствовало применение различных инструмен-
тальных методов.
Наиболее совершенной формой анализа технологическо-
го процесса является автоматический непрерывный конт-
роль. При этом используются квантометры, автоматиче-
ские хроматографы, масс-спектрометры, экспресс-анализа-
торы, автоматические колориметры и другие автоматические
приборы. Значение высокопроизводительных методов
контроля особенно велико в условиях производства, когда
необходимо выполнять массовые анализы.
II. ПРАВИЛА ОТБОРА ПРОБЫ
И ПОДГОТОВКА ЕЕ К АНАЛИЗУ
Материалы добываются или производятся обычно боль-
шими партиями, достигающими десятков или сотен тонн.
Анализируется же небольшое количество материала, как
правило навеска пробы не превышает 1 г. Как бы точно
и тщательно ни был выполнен анализ, все усилия бес-
полезны, если анализируемая проба не отражает средний
состав партии, т. е. не является представительной.
Небольшая часть материалов (концентрата, металла и
т. п.), состав которой соответствует среднему составу всей
партии, называется средней пробой данной партии.
7
Из-за неоднородности состава материала нельзя брать
для анализа первый попавшийся кусок или пробу в любом
месте партии. Это объясняется следующими причинами.
Под воздействием окружающей среды материалы могут
увлажняться или, наоборот, терять влагу, образовывать
химические соединения, например с кислородом воздуха.
В первую очередь отмеченные процессы происходят на
поверхности материала, и таким образом он становится
неоднородным. Твердые сыпучие материалы могут быть
в виде крупных и мелких частиц, различающихся химиче-
ским составом. При транспортировке частицы материала
перераспределяются: крупные куски перемещаются ближе
к поверхности слоя, а мелкие сосредоточиваются в нижней
его части.
Неоднородность химического состава сплавов (слитка
или отливки) обусловлена ликвацией. Кристаллиза-
ция сплава происходит не при определенной температуре
в отличие от чистых металлов, а в некотором интервале
температур. Химический состав закристаллизовавшихся в
разное время (т. е. при разной температуре) частей спла-
ва оказывается неодинаковым. Отдельные составляющие
сплава при охлаждении перемещаются в глубинные зоны
слитка, застывают в последнюю очередь. На поверхности,
таким образом, металл более чистый. Это явление ликва-
ции иногда обнаруживается визуально благодаря неодно-
родности окраски поверхности или излома слитка. На-
пример, в сплавах меди с оловом, цвет которых желтый
с красноватым оттенком, можно наблюдать белые пятна
олова. Причем таких пятен в глубине слоя больше, чем
на его поверхности. Значительная ликвация наблюдается
и в других сплавах цветных металлов, в частности сви-
нец— цинк, медь — свинец, цинк — олово, медь — серебро.
Для получения средней пробы исходный материал
измельчают, перемешивают и сокращают, соб-
людая определенные правила.
Перемешивание пробы осуществляют перекаты-
ванием, для чего ее помещают на большой лист глянцевой
бумаги или полиэтиленовую пленку. Каждый угол листа
или пленки попеременно приподнимают до тех пор, пока
проба не переместится к противоположному углу. После
ряда перекатываний по одной диагонали операцию повто-
ряют по другой диагонали. Для лучшего перемешивания
пробы приподнимают взаимно перпендикулярные края
пленки. Вместо перекатывания можно применять способ
перемешивания с помощью стального шпателя. После
8
перемешивания приступают к сокращению пробы, т. е. к
уменьшению ее массы.
Сокращение пробы предварительно производят
квартованием, для чего насыпают материал в виде кону-
са, затем дощечкой придают ему усеченную форму или
форму диска, который делят на четыре равные части дву-
мя взаимно перпендикулярными линиями. Две противо-
положные части объединяют, перемешивают, вновь обра-
зуют конус и сокращают.
Операции перемешивания и сокращения производят до
получения пробы определенной массы. По окончании по-
следних перемешивания и сокращения материал, разме-
щенный на ровной поверхности (глянцевой бумаге, пленке
или металлическом листе), распределяют с помощью шпа-
теля тонким слоем в виде прямоугольника.
По поверхности слоя шпателем проводят ряд горизон-
тальных и вертикальных линий и из середины полученных
квадратиков (из каждого или через один) отбирают при-
мерно одинаковые количества материала. Отобранный ма-
териал (10—50 г) измельчают в стальной, агатовой или
фарфоровой ступке. Пробу просеивают через сито опреде-
ленного номера. Остаток, не прошедший через сито, до-
измельчают и вновь просеивают. Полученную таким обра-
зом лабораторную среднюю пробу помещают в бюкс или
склянку с притертой пробкой.
Методика отбора средней пробы, установленная
ГОСТом, разработана для каждого материала, состав ко-
торого нужно установить. Основными этапами этого про-
цесса являются отбор первичной средней пробы и лабора-
торной средней пробы.
1. Отбор первичной средней пробы производится раз-
личными способами в зависимости от агрегатного состоя-
ния материала, подлежащего анализу. Правила отбора
проб составлены специально для материалов, которые
перевозятся в вагонах, баржах. При этом учитывается
способ транспортировки материала — в мешках, бочках,
навалом и т. д. Обычно в отборе первичной средней про-
бы аналитик не участвует, поэтому этот вопрос нами не
рассматривается.
2. Отбор лабораторной средней пробы (подготовка про-
бы к анализу) производится ОТК, тем не менее техник-
химик должен быть знаком с этой операцией.
Масса отобранной первичной средней пробы, как пра-
вило, велика (500 г). Часть первичной средней пробы, из
которой непосредственно берут навески для анализа, на-
9
зывается лабораторной средней пробой. Масса
ее (10—50 г) достаточна для выполнения анализов. Состав
лабораторной средней пробы должен соответствовать
среднему составу первичной средней пробы и составу всей
партии. Для этого дополнительно сокращают, перемеши-
вают и измельчают пробу.
а. Отбор лабораторной средней пробы сыпучих мате-
риалов (руд, концентратов и др.). Обычно первичная сред-
няя проба, из которой нужно отобрать лабораторную
среднюю пробу, уже достаточно измельчена. Крупность
пробы определяется путем просеивания вещества через
сито. Сита характеризуются размером сторон отверстий
в миллиметрах или числом отверстий, приходящихся на
1 см2. Иногда сита характеризуют в мешах (меш — число
отверстий на 1 линейный дюйм, или на 25,4 мм). Для из-
мельчения пробу необходимо брать небольшими порция-
ми. Степень измельчения имеет большое значение для
полноты вскрытия материала. Пробу считают достаточно
измельченной, если при растирании материала между
пальцами не ощущаются отдельные крупинки (частицы
диаметром 0,074 мм, или 200 меш).
б. Отбор лабораторной средней пробы металлов и
сплавов. Анализируемые материалы могут быть в
виде слитка, листа или губки. Поверхность слитка
металла или сплава зачищают от окалины и других за-
грязнений с помощью наждачного круга или стальной
щетки. Сверлом из специальной стали отбирают стружку,
просверливая пробу металла насквозь или до середины
слитка с противоположных сторон. Если взять стружку
только с поверхности, то из-за ликвации проба не будет
представительной. При отборе пробы металла, раскатан-
ного в виде листа, сверление производят в нескольких
местах по определенной схеме, например в шахматном по-
рядке. Процесс отбора лабораторной средней пробы ме-
талла или сплава является очень ответственной опера-
цией. Химический состав пробы при отборе не должен
измениться, стружка во время сверления или строгания
не должна подвергаться окислению при нагревании.
Для определения некоторых примесей, например кисло-
рода, пробу отбирают в виде кусочков, а не стружки (во
избежание окисления). Кусочки вырубают с помощью зу-
била.
Кроме рассмотренных общих приемов отбора проб для
некоторых материалов, а также для определения отдель-
ных примесей, существуют специальные приемы и прави-
10
ла, обеспечивающие правильность анализа. Например,
титан, полученный магниетермическим способом, на-
ходится в виде так называемой губки. Для определения
содержания металлических примесей — железа, никеля —
часть средней пробы перемешивают и делят на три части.
Из одной части прессуют электрод диаметром 40 мм и мас-
сой 1,3 кг. Остальные две части хранят в герметичной
упаковке для арбитражных анализов. Из спрессованного
электрода выплавляют в вакууме слиток, из которого от-
бирают пробу в виде стружки для определения примесей
железа и никеля. Для определения содержания хлора про-
бу не плавят во избежание его улетучивания, а прессуют
брикет, с торца которого снимают стружку.
в. Отбор пробы жидкостей. Если жидкость однородна,
то среднюю пробу можно брать в любом месте емкости.
Если жидкая или полужидкая масса неоднородна и ее
трудно перемешать, то среднюю пробу составляют из част-
ных проб, взятых на разных уровнях раствора или пульпы.
Для отбора проб пульпы существуют специальные пробо-
отборники.
3. Упаковка проб, хранение, документация. Первичную
среднюю пробу сыпучих материалов массой 500 г поме-
щают в две стеклянные или металлические банки, плотно
закрывают их пробками. Пробу, находящуюся в одной из
банок, используют для отбора лабораторной средней про-
бы и проведения анализа, другую сохраняют в качестве
арбитражной. Банку с арбитражной пробой заливают по-
верх пробки сургучом или мастикой, опечатывают и хра-
нят до тех пор, пока весь материал не израсходуют или
он не будет окончательно принят потребителем. Обычно
контрольные или арбитражные пробы хранят в течение
6 мес в специальных помещениях, где не работают с
кислотами и другими агрессивными веществами. Каждая
банка снабжается этикеткой с указанием материала, его
марки, массы партии, названия завода-поставщика, време-
ни отбора пробы и т. д.
В лаборатории пробу регистрируют порядковым номе-
ром, в журнале отмечают дату поступления пробы, номер
и дату сопроводительного документа, массу пробы.
Полученную лабораторную среднюю пробу, а также
ее остаток помещают в бюкс или склянку с притертой
пробкой. При проведении массовых анализов пробы мо-
гут находиться в пакетиках из глянцевой бумаги (если не
предполагают определять в ней содержание углерода)
или из полиэтилена, а арбитражные пробы хранят в бюк-
11
сах. Часть лабораторной средней пробы используют для
определения в ней содержания гигроскопической влаги.
4. Определение содержания гигроскопической влаги,
или воды, которую можно удалить из пробы в процессе
сушки при 105—110 °C.
Содержание гигроскопической влаги изменяется в за-
висимости от влажности воздуха. В одинаковых навесках
пробы одного и того же вещества процентное содержание
определяемых компонентов будет различным в зависимос-
ти от влажности пробы.
Содержание гигроскопической влаги определяют одно-
временно с анализом материала на другие компоненты.
Для этого берут отдельную навеску пробы. Содержание
основных компонентов определяют в воздушно-сухой про-
бе. Предварительно высушивать пробу перед анализом
нецелесообразно, так как тонкоизмельченные порошки
проб легко адсорбируют влагу из воздуха.
Для определения содержания гигроскопической влаги
обычно берут навеску (2—3 г) воздушно-сухой пробы и
помещают ее в бюкс с притертой крышкой. Навеску сушат
при температуре 100—110 °C в сушильном шкафу в тече-
ние 2 ч, при этом крышку бюкса приоткрывают. Затем
бюкс закрывают крышкой и помещают его на 30—45 мин
в эксикатор для охлаждения. Перед взвешиванием крыш-
ку бюкса на 1 с приподнимают для выравнивания давле-
ния в бюксе с атмосферным.
После первого высушивания и взвешивания пробу вновь
сушат. Операцию повторяют до получения постоянной
массы. Разность масс бюкса с пробой до и после высуши-
вания соответствует содержанию гигроскопической влаги.
Содержание (х, %) определяют по формуле x=[(Gi—
— G2)/G] 100, в которой G — навеска воздушно-сухой про-
бы, г; Gi — масса бюкса с пробой до высушивания, г;
G2 — то же, после высушивания, г.
Результаты анализа пересчитывают на сухое вещество
по формуле С = Ci 100/ (100—х), где С—содержание опре-
деляемого элемента в сухом веществе, %; С;— содержа-
ние определяемого элемента в воздушно-сухой пробе, %.
III. ОЦЕНКА РЕЗУЛЬТАТОВ АНАЛИЗА
Результат любого тщательно выполненного анализа
(х) отличается от истинного содержания элемента
в анализируемом материале (ц). Разность величин (х—
12
ц) называется ошибкой или погрешностью анали-
за. Анализ позволяет установить наиболее вероятное со-
держание определяемого компонента. Для оценки точности
анализа по данной методике предварительно проводят ее
метрологическую аттестацию, т. е. оценку пра-
вильности и воспроизводимости для всего интервала опре-
деляемых концентраций на достаточно большом статисти-
ческом материале.
Ошибки, или погрешности аналитических определений.
Ошибки аналитических определений могут быть система-
тическими (систематические погрешности) и случайными
(случайные отклонения). К третьему типу ошибок отно-
сятся грубые ошибки или промахи.
Систематические погрешности — это погреш-
ности, которые повторяются при повторных анализах без
изменения величины и знака и зависят от постоянно дей-
ствующих причин. Полученные результаты могут быть
близкими между собой и в то же время неправильными,
т. е. будут отличаться в большей или меньшей степени
от истинного содержания элемента в пробе. Наличие сис-
тематических погрешностей устанавливают с помощью
стандартных образцов, в которых содержание определя-
емого элемента известно. С этой же целью анализируют
различными методами один и тот же образец или варьи-
руют навески, или применяют «способ добавок» определя-
емого элемента к пробе.
Если величина и знак систематической погрешности
установлены, то в результаты анализа можно ввести со-
ответствующую поправку и тем устранить ошибку опре-
деления.
Правильность результатов анализа характе-
ризуется близостью полученного результата к истинному
содержанию определяемого элемента в пробе, т. е. систе-
матическая погрешность должна быть близкой к нулю.
Случайные отклонения — это погрешности, за-
висящие от неопределенных причин и не повторяющиеся
при повторных анализах. Другими словами, их величина
и знак непостоянны. Случайные отклонения результатов
анализа можно уменьшить увеличением числа параллель-
ных определений. Однако следует иметь в виду, что стан-
дартная погрешность среднего результата анализа умень-
шается пропорционально корню квадратному из числа
определений. Для уменьшения погрешности в 4 раза нуж-
но провести 16 определений, в 5 раз — уже 25 и т. д., т. е.
затраты времени увеличиваются.
13
Воспроизводимость характеризует степень бли-
зости друг к другу отдельных результатов определений
или измерений одной и той же величины в одном и том
же объекте по одной и той же методике анализа (измере-
ния), но в различных условиях: разные аналити-
ческие лаборатории, разные образцы прибора, разные ана-
литики, разное время. Воспроизводимость количест-
венно характеризуют стандартным отклонением
(Звоспр.). Воспроизводимость тем лучше, чем меньше зна-
чение 5воспр,-
Сходимость — характеризует степень близости друг
к другу отдельных результатов определений (или измере-
ний) одной и той же величины в одном и том же объекте
по одной и той же методике и в одинаковых усло-
виях: одна и та же аналитическая лаборатория, один и
тот же прибор, один и тот же аналитик, одно и то же
время. Сходимость количественно характеризуют стан-
дартным отклонением сходимости (Scx)• Схо-
димость тем лучше, чем меньше значение Scx.
Для характеристики результатов анализа, отражающей
близость к нулю ошибок всех видов, как систематических,
так и случайных, пользуются термином точность.
Грубыми ошибками, или промахами назы-
ваются ошибки, которые сильно искажают результаты ана-
лиза. Промахи, как правило, объясняются ошибочными
действиями аналитика. При вычислении среднего резуль-
тата ряда параллельных
определений результат, со-
держащий грубую ошибку,
отбрасывают.
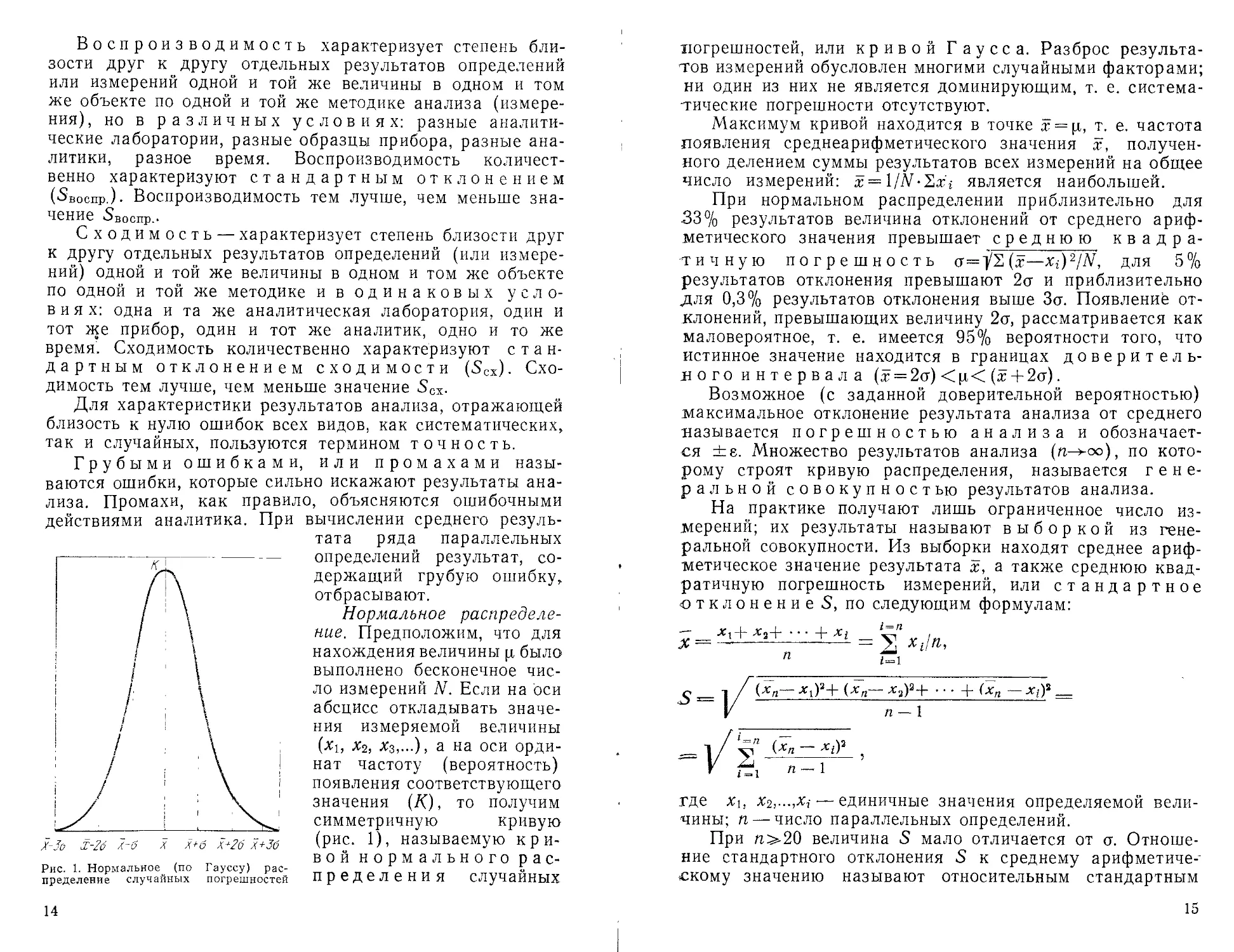
Нормальное распределе-
ние. Предположим, что для
нахождения величины р было
выполнено бесконечное чис-
ло измерений N. Если на оси
абсцисс откладывать значе-
ния измеряемой величины
(%1, х2, Хз,...), а на оси орди-
нат частоту (вероятность)
появления соответствующего
значения (Д), то получим
симметричную кривую
(рис. 1), называемую кри-
вой нормального рас-
пределения случайных
14
погрешностей, или кривой Гаусса. Разброс результа-
тов измерений обусловлен многими случайными факторами;
ни один из них не является доминирующим, т. е. система-
тические погрешности отсутствуют.
Максимум кривой находится в точке т = ц, т. е. частота
появления среднеарифметического значения х, получен-
ного делением суммы результатов всех измерений на общее
число измерений: x^l/N-'Xxi является наибольшей.
При нормальном распределении приблизительно для
33% результатов величина отклонений от среднего ариф-
метического значения превышает среднюю квадра-
тичную погрешность ст=/2(х—для 5%
результатов отклонения превышают 2ст и приблизительно
для 0,3% результатов отклонения выше Зст. Появление от-
клонений, превышающих величину 2ст, рассматривается как
маловероятное, т. е. имеется 95% вероятности того, что
истинное значение находится в границах доверитель-
но го и н т е р в а л а (г = 2ст) <ц< (х + 2ст).
Возможное (с заданной доверительной вероятностью)
максимальное отклонение результата анализа от среднего
называется погрешностью анализа и обозначает-
ся ±е. Множество результатов анализа (п->оо), по кото-
рому строят кривую распределения, называется гене-
ральной совокупностью результатов анализа.
На практике получают лишь ограниченное число из-
мерений; их результаты называют выборкой из гене-
ральной совокупности. Из выборки находят среднее ариф-
метическое значение результата х, а также среднюю квад-
ратичную погрешность измерений, или стандартное
отклонение S, по следующим формулам:
X - X 2 -р • • -р XI !
х = 1 = У Xtln,
п м.
£ = рЛ СМг— M)S+ (хп~~ М)2+ + (хп — xty =
где Xi, X2,...,Xi — единичные значения определяемой вели-
чины; п — число параллельных определений.
При и >20 величина S мало отличается от ст. Отноше-
ние стандартного отклонения S к среднему арифметиче-'
скому значению называют относительным стандартным
15
отклонением Sr = S/x и представляют в виде десятичной
дроби. Во избежание путаницы термин «коэффициент ва-
риации» V=100Sr, выраженный в процентах, применять
не рекомендуется в тех случаях, когда результат анализа
также выражен в процентах.
Случайную погрешность среднеарифметического резуль-
тата анализа х характеризуют доверительным интерва-
лом е. Для этого используют значение стандартного от-
клонения S, вычисленного заранее на основе большого
числа измерений (п>20). Значение е с надежностью а=
= 0,95 (вероятностью 95 и/о) составляет ±2S/Vn, а истин-
ное значение определяемой величины лежит в интервале
значений х — (2S/Vn) и х 4- [2S/]/п).
Распределение Стьюдента. Если число измерений не-
велико (п<20), то для вычисления доверительного интер-
вала вводят поправочный коэффициент t в величину S,
которую при этом обозначают символом Sn- Значения t
определяют из так называемого распределения
Стьюдента, или ^-распределения, которое несколько
шире нормального. В табл. 1 приведены значения коэф-
фициента Стьюдента для различных уровней надежностей
а (вероятностей) и числа параллельных определений п.
Таблица 1. Значения tan для различных степеней надежности
и числа определений
п а = 0,95 сс = 0,99 п а=0,95 а—0,99 п а=0,95 а=0,99
2 12,71 63,66 9 2,31 3,36 16 2,13 2,95
3 4,30 9,93 10 2,26 3,25 17 2,12 2,92
4 3,18 5,84 11 2,23 3,17 18 2,И 2,90
5 2,78 4,60 12 2,20 3,11 19 2,10 2,88
6 2,57 4,03 13 2,18 3,06 20 2,09 2,86
7 2,45 3,71 14 2,16 3,01 21 2,09 2,85
8 2,37 3,50 15 2,15 2,98
Погрешность Еап при заданной надежности и числе
определений п, выполненных при установлении Sn, вы-
числяют по формуле San = ±Sntan /Т'м. Истинное значение
определяемой величины находится в пределах от хп—еКя
до Если число измерений превышает 20, то счи-
тается, что распределение Стьюдента практически совпа-
дает с нормальным.
Допускаемые расхождения результатов определений.
Обычно при проведении анализа число параллельных опре-
16
делений не превышает трех-четырех. В методиках, стан-
дартных инструкциях, ГОСТах на методы анализа, как
правило, указываются значения величин допускаемых рас-
хождений результатов определений (анализа).
Допускаемым расхождением (7?тах) называют разность
наибольшего и наименьшего из п результатов параллель-
ных определений. Величина /?тэх при доверительной веро-
ятности а = 0,95 (т. е. в 95 случаях из 100) равна 2,77S при
n = 2, 3,315S при ц = 3 и 3,63S при п=4.
Допускаемые расхождения приводят для всего интер-
вала определяемых содержаний в виде таблицы или гра-
фика, по оси ординат которого откладывают значения
Лтах, а по оси абсцисс — значения соответствующих кон-
центраций. По найденному значению концентраций нахо-
дят допускаемые расхождения.
Предварительная аттестация методик, т. е. установле-
ние значения S, на основании которого рассчитывается ве-
личина допускаемых расхождений, проводится по большо-
му числу параллельных определений (п>20). Если в мето-
дике не указаны значения допускаемых расхождений, но
известно относительное стандартное отклонение Sr, то
находят значение S по формуле S — Srx и рассчитывают
значения допускаемых расхождений результатов анализа,
как указано выше. Значение Sr в методике обычно при-
водится для конкретных содержаний определяемого эле-
мента. Если дается только одно значение Sr, то это озна-
чает, что относительное стандартное отклонение сущест-
венно не зависит от определяемых содержаний.
Пример. Содержание меди в руде определено по ме-
тодике, относительное стандартное отклонение которой
установлено при /1 = 30, Sr=0,018. Получены следующие
результаты: xj = 4,40, %2 = 4,60. Среднее значение х= (4,40 +
+ 4,60) /2 = 4,50.
Допускаемые расхождения результатов (при а = 0,95)
находим по формуле 2,77S = 2,77S7+ = 2,77-0,018-4,50~
— 0,22. Разность результатов не превышает допуска: 4,60—
4,-40 = 0,20 <0,22. Результат анализа выдается как
X + г = X ± 2SlVn = 4,50 + 2-0,81 //2 = 4,50 ±0,12%.
Выявление промахов. Если приходится выполнять ана-
лиз по методике, для которой неизвестны ни допускаемые
расхождения, ни относительное (абсолютное) стандартное
отклонение, то результат анализа характеризуется средне-
арифметическим годных результатов. Предварительно
устанавливают пригодность полученных результатов, как
17
описано ниже. Эти же расчеты выполняют при определе-
нии Sr по методике с небольшим числом определений.
Для проверки пригодности сомнительного результата,
т. е. члена, максимально отличающегося от других членов
ряда, при м<8 используют формулу Q'=(x2—Xi)/(хп—
—xi), в которой числитель—разность сомнительного и
соседнего результатов, а знаменатель — разность крайних
результатов ряда.
Для исключения сомнительных результатов вычислен-
ное значение Q' сравнивают со следующими значения-
ми Q:
«... 3 4 5 6 7 8
Qa=0,95... 0,94 0,77 0,64 0,56 0,51 0,48
Qa = 0,99... 0,99 0,89 0,76 0,70 0,64 0,58
Если Q'>Q, то сомнительный результат исключают из
процесса обработки данных, а если Q'<Q, то принимают
его в расчет.
Пример. При определении содержания меди в пробе
руды по методике, для которой неизвестны S, Sr, е, было
получено шесть результатов, которые расположены в по-
рядке возрастания их численных значений: 4,45; 4,48; 4,52;
4,60; 4,65; 5,42%.
Из крайних членов ряда сомнение вызывает значение
5,42. Вычисляем значение Q':
Q'= (5,42 - 4,65)/(5,42 — 4,45) =0,79.
Для данного значения и = 6 и а = 0,95 табличное зна-
чение Q = 0,56. Так как Q'>Q, то значение 5,42 следует
исключить из последующих вычислений. Если сомнение
вызывают несколько значений, то указанные расчеты про-
водят последовательно для каждого из них. Допускается
исключение не более >/з всех результатов. В противном
•случае, результаты считают неудовлетворительными и ана-
лиз повторяют.
Вычисления значения х и погрешности анализа прово-
дят, как указано в табл. 2. ______________
Стандартное отклонение 5Л = ]//ЛЕ( х—xz)2/(« —1) =
]/0Д278/4~ 0,Со-
относительное стандартное отклонение S,-=Sn/x = 0,08/
/4,54 = 0,018. Поскольку число определений невелико (м =
= 5), погрешность результата анализа с надежностью а=
= 0,95 составляет ± 0,08-2,78 / уД = ± 0,10.
18
Таблица 2. Вычисления стандартного отклонения по пяти определе-
ниям
t xi X—Xi
1 4,48 4,54—4,48=+0,06 ( + 0,06)2=+0,0036
2 4,60 4,54—4,60=—0,06 (—0,06) 2=+0,0036
3 4,45 4,54—4,45=+0,09 ( + 0,09)2=+0,0081
4 4,65 4,54—4,65=—0,11 (—0,11)2= +0,0121
5 4,52 4,54—4,52=+0,02 ( + 0,02)2= + 0,0004
л=5; 2Xi = 22,70; _ ( = 5 х = 2л';//г = 1 = 1 = 22,70 : 5 = 4,54 2(7—X;)2 = 0,0278
Для выявления систематической погрешности средний
результат анализа х = 4,54 сравнивают с установленным
содержанием меди в стандартном образце (С). Если Д =
—х—С>еап, то систематические погрешности имеются.
Предположим, что при выполнении анализа по методи-
ке, для которой вычислено по небольшому числу опре-
делений (п=5), в производственных условиях получены
значения Xi = 4,42 и х2 = 4,58. Стандартное отклонение ме-
тодики S = 0,08; допускаемое расхождение результатов
двух параллельных определений 2,78S/y« = 0,31.
Разность результатов 4,58—4,42 = 0,16<0,31, следователь-
но, результаты пригодны для определения х, т. е.
- (4,58 + 4,42) . сп т-т
— х'_!—_—!—' =4,50. Погрешность результата анализа
е= ±0,08-2,78/У2. Итак, значения определяемой величи-
ны составят 4,50 ±0,16%.
Стандартные образцы применяют: 1) для проверки
правильности результатов анализа химического состава
вещества при проведении арбитражных анализов; 2) для
проверки правильности вновь разработанной, усовершен-
ствованной или внедряемой методики; 3) для градуировки
приборов при их освоении и проверке. Стандартные образ-
цы представляют собой материалы, в которых точно уста-
новлено содержание ряда элементов. Для установления
состава стандартных образцов применяют различные ме-
тоды.
19-
Найденное содержание элемента называют установ-
ленным (С). Установленное содержание вносят в сви-
детельство (паспорт), прилагаемое к стандартному образ-
цу, при одновременном указании погрешности е, с которой
установлено содержание элемента (погрешность ат-
тестации с заданной доверительной вероятностью).
Ниже в качестве примера приведены результаты аттеста-
ции стандартного образца лопаритового концентрата (а=
= 0,95):
СеО2 LasOa NdsOa Dy, Оз
Y2O,
С, %
8, %
. . . . 16,2 8,4 4,1 0,017 0,033
. . . . ±0,3 ±0,2 ±0,2 ±0,002 ±0,004
Eu2O5
0,053
±0,02
При использовании стандартного образца необходимо
учитывать, что его химический состав должен наиболее
близко соответствовать составу анализируемой пробы.
Стандартные образцы подразделяют на три основные
категории: государственные, отраслевые и стандартные
образцы предприятия. Их применение способствует улуч-
шению качества аналитического контроля, устранению
спорных моментов между поставщиками и потребителями
продукции.
Критерий необходимости арбитражного анализа. По ре-
зультатам анализа производят сортировку продукции, т. е.
относят ее к определенному сорту или марке. Продукции
более высокого сорта соответствует и более высокая цена.
Бывают случаи, когда продукция попадает не в установ-
ленные сорта, а в брак. Завод — поставщик продукции
терпит при этом ущерб.
Результаты анализа одного и того же продукта, выпол-
ненные в ЦЗЛ поставщика и потребителя по одной и той
же методике с известным относительным стандартным
отклонением, могут не совпадать. В этом случае необходим
арбитражный анализ. Прежде чем приступить к его выпол-
нению, необходимо найти значение е; — максимальную по-
грешность разности средних результатов анализа постав-
щика (ал) и потребителя (жг) и сопоставить ее с раз-
ностью этих средних результатов (xi—хъ).
Если (oh—x2)<Si, то арбитраж проводить не следует,
если (Ji—х2) >е15 то арбитражный анализ необходим, так
как расхождения не случайны. Значение ei вычисляют по
формуле
£1 = 35, К*? + = (3Sr! Vn) Vх^+ х22,
где n — ni = ti2 (а=0,99).
20
Пример. Требуется установить необходимость прове-
дения арбитражного анализа для случая, когда завод—
поставщик продукции получил при анализе значение 5-] =
= 4,8%, а завод-потребитель £2 = 4,5%.
Анализы на заводах выполняли по одной и той же ме-
тодике: Sr = 0,04; число определений nj = n2 = n = 2;
(3-0,04//2) j/(4“8^j + (4,5)2 0,6; х2 =
= 4,8 — 4,5 = 0,3 < 0,6.
В этом случае необходимости проведения арбитражно-
го анализа нет, так как оба результата л-] и 5% характе-
ризуют значение определяемой величины с учетом погреш-
ности методики.
Некоторые термины метрологических характеристик.
Такие термины, как «точность» и «чувствительность», раз-
ными авторами понимаются и используются по-разному.
Возможность качественного обнаружения искомого
компонента характеризуется так называемым пределом
обнаружения, под которым понимают наименьшее
содержание, при котором по данной методике можно об-
наружить определяемый компонент с заданной довери-
тельной вероятностью. За предел обнаружения принимают
значение 33 или 63, где S — стандартное отклонение ко-
лебаний контрольного («холостого») опыта.
Наименьшее значение определяемого содержания, огра-
ничивающее область значений (диапазон) определяемых
содержаний снизу, называется нижней границей или
нижним пределом определяемых содержаний и обо-
значается Сн. В практике анализа чистых веществ за ниж-
нюю границу принимают то минимальное содержание, ко-
торое можно определить данным методом с относитель-
ным стандартным отклонением 0,33. Нижний предел вы-
числяют как утроенное стандартное отклонение результа-
та определения при концентрации, близкой к предельной.
Если на достаточном числе определений установлено, что
стандартное отклонение величины 5-10-5% составляет
1-10~5%, то нижний предел определения не менее чем
3-10-5%.
Термин коэффициент чувствительности сле-
дует толковать как наклон (угловой коэффициент) прямой
градуировочного графика. Смысл терминов коэффи-
циент чувствительности и чувствительность
ГОСТ 16263—70) совпадает.
21
Следует различать понятия «метод анализа» и «мето-
дика анализа». Метод анализа — это краткое определение
принципов, положенных в основу анализа вещества, на-
пример титриметрический метод анализа или экстракцион-
но-фотометрический метод анализа. Методика анализа—•
это подробное описание всех условий и операций, которые
обеспечивают заданные характеристики правильности и
воспроизводимости.
Контрольный (холостой) опыт — выполнение тех же
операций, которые проводятся и при анализе пробы, с
теми же реактивами, в той же посуде, на тех же приборах.
При нахождении истинного содержания определяемого
компонента в объекте анализа находят поправку контроль-
ного опыта и вычитают ее из результатов анализа.
IV. ПРОИЗВОДСТВЕННАЯ КЛАССИФИКАЦИЯ
ВИДОВ АНАЛИЗА
Различают следующие виды технического анализа в зави-
симости от задач, решаемых с их помощью: 1) маркиро-
вочные; 2) арбитражные (проверочные или контрольные);
3) для текущего контроля технологического процесса.
Маркировочные анализы предназначены для сырья, по-
ступающего на предприятие, и готовой продукции данного
производства. Основным требованием к этим анализам
является достаточно высокая точность, так как результа-
ты, например, маркировочных анализов сырья являются
исходными для расчета технологического процесса. Резуль-
таты анализа готовой продукции позволяют судить о ее
качестве, соответствии нормам. Анализ дает возможность
классифицировать продукцию по сортам или маркам, ко-
торым соответствует определенная цена.
В табл. 3 в качестве примера приведены технические
требования к молибденовым флотационным концентратам
отдельных марок. Области применения концентратов раз-
личны, от этого зависят требования к содержанию как
основного вещества (молибдена), так и лимитируемых
примесей. В данном случае молибден является ценным
компонентом, поэтому чем больше его содержание в кон-
центрате, тем выше марка продукта. Кремний, алюминий,
олово, фосфор и медь, наоборот, являются «вредными»
примесями, т. е. концентрат с меньшим содержанием этих
примесей будет отнесен к более высокой марке. В техни-
ческих требованиях, например, к высшей марке КМ.Ф-1
22
Таблица 3. Технические требования к молибденовому флотацион-
ному концентрату
хМарка Сорт Mo, %, не менее Примесей, %, не более
SiO2 As Sn р Си
КМФ-1 1-Й 51 5,0 0,04 0,03 0,03 0,4
КМФ-2 2-й 48 7,0 0,06 0,05 0,04 0,8
КМФ-3 3-й 47 9,0 0,07 0,07 0,05 1,5
КМФ-4 4-й 45 12,0 0,07 0,07 0,05 2,5
Примечание. Концентрат используется в производстве ферромолибдена
1-го сорта и молибденита высокой чистоты (КМФ-1), ферромолибдена 2-го
сорта и молибденовокислого аммония (КМФ-2), ферромолибдена 3-го сорта,
молибденовокислого аммония и других солей (КМФ-3) и солей молибдена
.(КМ-Ф-4).
норма содержания Ценного компонента— молибдена долж-
на быть не менее 51% при содержании примесей, %, не
более: SiO2 5,0; As 0,04; Sn 0,03; Р 0,03; Си 0,4.
Для выполнения маркировочных анализов используют
различные химические, физико-химические и физические
методы. В связи с высокими требованиями, предъявляе-
мыми к точности маркировочных анализов, необходимо,
например, тщательное отделение мешающих элементов, что
приводит к увеличению продолжительности анализа. Иног-
да для этих целей используют достаточно длительный, но
точный гравиметрический анализ.
Арбитражные (проверочные или контрольные) анали-
зы проводят в тех случаях, когда результаты выполненного
ранее анализа вызывают сомнение. Если сомнение возник-
ло в лаборатории, выполнившей анализ, то контрольный
анализ проводит либо более квалифицированный, чем в
предыдущем случае, аналитик, либо два аналитика ана-
лизируют шифрованные (содержание компонентов не из-
вестно аналитику) пробы.
Арбитражный анализ необходим также тогда, когда
•одно из подразделений данного предприятия — цех или ла-
боратория—опротестовало результаты анализа. Наконец,
арбитражный анализ выполняется в случае, если резуль-
таты маркировочного анализа опротестованы предприя-
тием— потребителем данной продукции. В этом случае
проверочные анализы из одной и той же отобранной лабо-
раторной средней пробы могут проводиться либо силами
заинтересованных сторон (внутренний арбитраж), либо
с участием третьей, незаинтересованной организации
23
(внешний арбитраж). В качестве такой арбитражной ор-
ганизации привлекаются наиболее авторитетные (голов-
ные) лаборатории, имеющие большой опыт в выполнении
анализа данной продукции.
Если есть сомнения в представительности пробы, то
для арбитражного анализа вновь отбирают лабораторную
среднюю пробу. Для арбитражных анализов в основном
применяют те же высокоточные методы, что и для мар-
кировочных анализов.
Анализы для текущего контроля технологического про-
цесса должны быть ускоренными (или экспрессными).
Ускоренные (экспрессные) анализы предназначены для
регулирования технологического процесса, внесения необ-
ходимых корректив. Технолог, ведущий процесс, на основа-
нии результатов ускоренного анализа при необходимости
может менять некоторые параметры, например повышать
или понижать температуру процесса, регулировать кон-
центрацию реактивов. Ускоренность анализа возможна
благодаря исключению некоторых операций в ущерб точ-
ности применяемого метода.
В основном же для проведения ускоренных анализов
используют другие методы, которые являются, как прави-
ло, менее точными, чем маркировочные, но достаточно
быстрыми. Для ускоренных анализов применяют из хи-
мических методов титриметрические, из физико-химиче-
ских, например фотометрические, ионометрические и др.
Из физических методов наиболее пригодными являются
методы, с помощью которых легко осуществим Автомати-
ческий контроль. Например, в последние годы на пред-
приятиях цветной металлургии применяют рентгеноспект-
ральные методы анализа, позволяющие контролировать
содержание элементов непосредственно в потоке раствора
или пульпы (квантометры «Поток» КРФ-13).
Нормативно-техническая документация. Для осуществ-
ления аналитического контроля производства применяют
проверенные, наиболее надежные методики, согласованные
предприятиями — унифицированные методики.
Унифицированные методики обычно публикуются в соот-
ветствующих сборниках, которые служат первой стадией
унификации (сборники методов анализа титана, арсенида
галлия и др.).
Унифицированные методики, как правило, не привяза-
ны к определенным маркам продукции, содержат условия
проведения анализа для сравнительно широких пределов
изменения состава объектов анализа.
24
Следующей стадией унификации методик являются
технические условия (ТУ), отраслевые стандарты (ОСТы)
и государственные стандарты (ГОСТы)—все это различ-
ные категории нормативно-технической документации
(НТД). При составлении НТД на методы анализа исходят
из технических условий на марки продукции, т. е. исполь-
зуют методики определения содержания элементов, кото-
рое в данной НТД нормируется. НТД на методы анализа
содержит данные о допускаемых отклонениях результатов
анализа при определении концентрации на уровне норм.
В ряде случаев технические условия и методы анализа
включают в общий ГОСТ на данный материал.
V. НАУЧНО-МЕТОДИЧЕСКАЯ КЛАССИФИКАЦИЯ
МЕТОДОВ АНАЛИЗА
Методы технического анализа подразделяются на химиче-
ские, физико-химические и физические. К химическим от-
носятся гравиметрия и титриметрия, а также газовый
(газообъемный) анализ. К физико-химическим относятся
методы: 1) оптические — фотометрия (спектрофотометрия,
фотоколориметрия), нефелометрия, турбидиметрия, флуо-
риметрия, атомно-абсорбционная спектрофотометрия,
2) электрохимические — электрогравиметрия, электрохи-
мическая титриметрия (кондуктометрия, потенциометрия,
амперометрия, кулонометрия), полярография, ионометрия;
3) кинетические методы. Физические методы включают
спектральный анализ, фотометрию пламени, рентгеноспект-
ральный, масс-спектральный, активационный, радиометри-
ческий анализы.
Следует отметить, некоторую условность де юния ме-
тодов на химические, физико-химические и физические.
Существуют также другие классификации. В последние
годы получили развитие так называемые комбинирован-
ные методы анализа, к которым можно отнести, например,
химико-спектральный, экстракционно-атомно-абсорбцион-
ный, экстракционно-фотометрический методы. Эти методы
сочетают предварительную химическую подготовку пробы
(разделение, концентрирование) с последующим определе-
нием содержания элементов физическими или физико-хи-
мическими методами.
25
Химические методы
Гравиметрическим (весовым) называют метод количе-
ственного анализа, состоящий в точном измерении массы
определяемой составной части анализируемого вещества,
выделенной либо как соединение определенного состава,
либо в элементном виде.
Гравиметрия имеет ряд особенностей, определяющих
ее роль в техническом анализе. Она позволяет получать
результаты высокой точности, так как взвешивание явля-
ется одной из наиболее точных операций количественного
анализа. Погрешность взвешивания на аналитических ве-
сах составляет несколько десятых долей миллиграмма.
Фактор пересчета влияет на величину суммарной погреш-
ности определения. Большой фактор пересчета обеспечи-
вает уменьшение суммарной погрешности, если порядок
величин исходной навески и весовой формы одинаков
(обычно Gmax = 200 мг). Если весовая форма значительно
меньше исходной навески, то суммарная погрешность ана-
лиза возрастает.
Гравиметрию применяют в основном при достаточно
высоких содержаниях определяемого элемента (единицы
или десятки процентов), когда другими методами, напри-
мер физико-химическим, трудно достичь высокой точности
результатов. При определении основных компонентов гра-
виметрическим методом относительное стандартное откло-
нение Sr достигает значений 0,001—0,005.
Гравиметрия является длительным методом, так как
включает такие продолжительные операции, как фильтро-
вание, промывание, высушивание, прокаливание и доведе-
ние осадка до постоянной массы. Часто приходится пере-
осаждать осадок для удаления соосадившихся элементов.
В большинстве случаев результаты анализа можно полу-
чить через несколько часов, в сложных случаях — на вто-
рые или третьи сутки. По этой причине гравиметрии не
применяют для ускоренных (экспрессных) анализов, по-
зволяющих наблюдать за ходом технологических процес-
сов. Однако ее часто используют при выполнении высоко-
точных маркировочных и арбитражных анализов на пред-
приятиях. Нередко роль гравиметрического анализа сво-
дится к контролю результатов, полученных другими, более,
ускоренными инструментальными методами. Гравиметрия
используется для установления химического состава стан-
дартных образцов, титров растворов, анализа товарных
продуктов.
26
При определении редких элементов (тантала, ниобия,
вольфрама и др.) в материалах сложного химического
состава, например в сырье, часто требуются длительные
и сложные операции отделения определяемого элемента от
сопутствующих. В этом случае следует учитывать не толь-
ко длительность и трудоемкость, но и недостаточную на-
дежность гравиметрического анализа. При нахождении
суммарного содержания тантала и ниобия в некоторых
концентратах необходимо вводить поправку на соосадив-
шийся фосфор, для чего заранее определяют его содер-
жание. Однако нахождение содержания фосфора в этих
концентратах является весьма сложной задачей, решение
которой не всегда приводит к надежным результатам.
Гравиметрия относится к сравнительно дешевым мето-
дам анализа, так как не требует сложной и дорогостоящей
аппаратуры, проста в технике выполнения и доступна прак-
тически для любой аналитической лаборатории. Несмот-
ря на значительную продолжительность одного определе-
ния, возможность одновременного анализа целого ряда
проб и навесок дает этому методу преимущества при вы-
полнении массовых анализов.
Гравиметрические методы в настоящее время вытесня-
ются титриметрическими, физико-химическими и физиче-
скими методами анализа.
Расчеты в гравиметрии. 1. Искомое вещество определяют
в той же форме, в которой оно находится в пробе. Например, при
расчете содержания меди (х %) в медных концентратах на основании
данных электрогравиметрического анализа применяют формулу х=
= (g—gJ-lOO/Gn, в которой g— масса катода с выделившейся медью,
г; gi—масса катода, г; GK—навеска концентрата, г. 2. Искомое ве-
щество определяют не в той форме, в которой оно находится в про-
бе. Например, при определении содержания никеля в сплавах на
медной основе диметилглиоксимом его содержание (х %) вычисляют
по формуле х=Л-0,2032-100/Go, в которой А — масса просушенного
осадка диметилглиоксимата никеля, г; 0,2032 — коэффициент пересчета
диметилглиоксимата никеля на никель; Go — навеска образца, г.
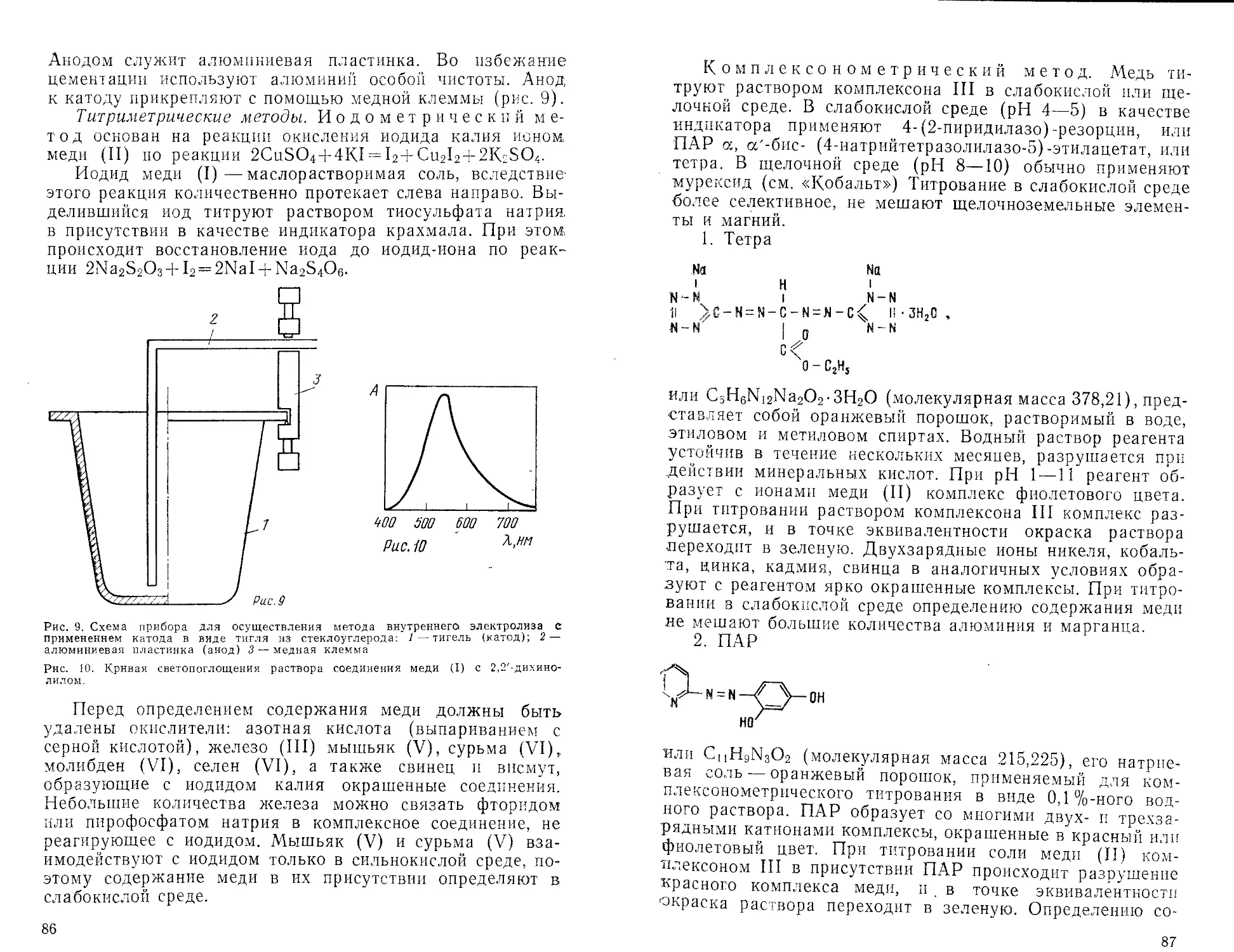
Титриметрический (объемный) метод анализа основан
на точном измерении количества раствора реагента (тит-
ранта) известной концентрации, который израсходован
на реакцию с определяемым элементом.
Операцию добавления рабочего раствора реагента точ-
но известной концентрации (титрованный раствор)
к исследуемому раствору называют титрованием.
Титрование производят до наступления момента окончания
реакции — точки эквивалентности, о котором судят
визуально по изменению окраски раствора, образованию
27
осадка, с помощью специальных индикаторов или с ис-
пользованием физико-химических методов индикации, на-
пример кондуктометрии, потенциометрии. В последнем
случае точка (или момент) эквивалентности фик-
сируется специальными приборами по изменениям электро-
проводности, потенциала электрода, происходящим в ре-
зультате реакции.
По характеру протекающих реакций в титриметриче-
ском анализе различают следующие методы: нейтрализа-
ции, окисления-восстановления, осаждения, комплексо-
образования.
К особенностям титр иметр и и при ее сравнении,
например, с гравиметрией относится ее ускоренность.
В основе титриметрии часто находятся избирательные ре-
акции в отличие от гравиметрии. Обычно титриметриче-
ский анализ используют для определения средних и вы-
соких содержаний элементов. Титриметрию широко при-
меняют на производстве, чему, в частности, способствует
простота используемого обрудования при визуальных опре-
делениях.
Комплексонометрия относится к методам тетраметри-
ческого анализа, являясь, по словам чешского ученого
Р. Пришибила, его ядром. В этом методе титрование про-
изводится раствором какого-либо комплексона. Комплек-
соны— органические соединения, представляющие собой
аминополикарбоновые кислоты или их соли.
Из исследованных комплексонов в наибольшей степени
применяется этилендиаминтетрауксусная кислота (EDTA),
или комплексон II (краткая форма написания Н4У, где
У — радикал этилендиаминтетрауксусной кислоты)
Н00СН2ф CHjCOOH
'n-CHj-CHj-N
HOOCHZQ/ CHjCOOH
Хорошо растворимая в воде двунатриевая соль этилен-
диаминтетрауксусной кислоты (Na—EDTA) получила на-
звание комплексона III или трилона Б (краткая форма
написания NaoEpy)
НООСН2С CHjCOONa
CH J- CH J-/ .2HjO.
NaOOCHjC ' CHjCOOH
28
Метод комплексонометрического титрования основан на
способности EDTA образовывать прочные внутрикомплекс-
ные соединения, с ионами металлов например Zn2+, Cd2+,
Са2+, Mg2+, Sn2+, In3+, Zr(IV), Ln+3.
Прочность комплексных соединений EDTA с металла-
ми так высока, что катионы этих металлов в растворах
не обнаруживаются с помощью обычных аналитических
реакций. Такая прочность объясняется образованием не-
скольких связанных между собой устойчивых пятичленных
клешнеобразных колец:
*^С0
Чем больше величина IgA в табл. 4, тем устойчивее
комплекс. Реакция взаимодействия EDTA с ионом опреде-
ляемого металла протекает в строго стехиометрическом
отношении: 1 молекула EDTA или Na-EDTA реагирует
с 1 атомом металла независимо от его степени окисления
при этом выделяются 2 иона водорода; лишь молибден
(V) и молибден (VI) образуют комплексы с EDTA соста-
ва: EDTA=Mo = l:2.
Т а б л и ц а 4. Логарифмы констант устойчивости (К) комплексов
EDTA
Ион IgX Ион IgK Ион I IgK Ион IgX Ион IgK
AF+ 16,1 Си2+ 18,8 1 Ga3+ 20,3 Ni2+ 18,6 T1O2+ 17,3
Cd2+ 16,6 Dy3+ 18,3 Но3+ 18,7 Pb2+ 18,3 TP+ 5,8
Cd3+ 17,4 Ег3+ 18,8 In3+ 24,9 Pr3+ 16,4 Tm3+ 19,3
Се3+ 15,8 Еи3+ 7,7 La3+ 15,4 Sc3+ 23,1 ‘ Y3+ 18,0
Со2+ 16,2 Еи2+ 17,4 Lu3+ 19,8 Sm3+ 17,1 Yb3+ 19,5
Со3+ 36,0 Fe2+ 14,3 Mn2+ 14,0 Tb3+ 17,9 Zn2+ 16,3
Сг2+ Сг3+ 13,0 24,0 Fe3+ 25,1 Nd3+ 16,6 Ti3+ 21.3 Z4(IV) 29,5
Уравнения реакции:
29
1) HOOCH2C CHjCOONa
- CH2- CH2- N4 + CaCl2-—
NaOOCH2C CH2COOH
i— OOCHjC CHjCOONa
— 'xN-CH2-CH2-N^ + 2HCI
NaQOCHjC^ X‘CH2GOO-i
--------------Ca---------------1
2) H00CH2C CH2COONa
^N-CH2-CH2-Nx + FeCl3---
NaOOCH2CX CH2C00H
r-OOCH2C CH2COONa
— - CHj- CH2- + 2HCI ;
Na00CH2C X‘CH2COO-|
--------------Fe---------------1
I
Cl
3) HOOCHjC CH2COONa
J^N-CHj-CHj-N^ + ZrOClj—-
NaOOCHjC GHjCOOH
i—OOCHjC CH2C00Na
— ^N-CH2-GHz-N^ + 2HCI
NaOOCHjC '' CHjCOO—।
-------------Zr Q-------------1
В качестве индикаторов при комплексонометрическом
титровании применяют органические реагенты, которые
образуют окрашенные комплексные соединения с ионами
металлов. Эти индикаторы называются металл-индикато-
рами. Металл-индикаторами являются, например, эрио-
хромчерный Т, мурексид, ксиленоловый оранжевый и др.
Металл-индикаторы с металлами образуют менее прочные
комплексы, чем комплексоны.
Комплексонометрическое титрование применяют в двух
вариантах: прямом и обратном.
Прямое титрование проводят раствором ком-
плексона III до изменения окраски раствора. Перед титро-
ванием к раствору соли определяемого металла, например
циркония, прибавляют в качестве индикатора какое-либо
органическое соединение (эриохромчерный Т, арсеназо III,
ксиленоловый оранжевый и др.), образующее с цирконием
интенсивно окрашенный комплекс. Прочность этого ком-
30
плекса, однако, значительно меньше прочности комплек-
соната циркония. Благодаря этому комплексон III вытесняет
ионы циркония из его комплекса с индикатором. В про-
цессе титрования по мере добавления комплексона III к
раствору, содержащему цирконий, последний постепенно
переходит в бесцветный комплексонат. В момент достиже-
ния точки эквивалентности, т. е. когда цирконий оказы-
вается полностью связанным с комплексоном, происходит
резкое изменение окраски раствора — возникает окраска
свободного индикатора.
Индикатор эриохромчерный Т, окрашенный в розовый
цвет в кислых средах, образует с ионами циркония (цир-
конила) в 1,5—2М солянокислом растворе комплекс си-
него цвета по реакции
При титровании комплексоном в момент достижения
точки эквивалентности комплекс циркония с эриохромчер-
ным Т разрушается и образуется бесцветный комплексонат
циркония:
NaOOC-CHj CHjCOONa
^n^-ch2-chz-n/
рООС-СН2 ХСНгСОО—1
1-------------ZrO------------J
Раствор приобретает окраску свободного индикатора,
т. е. становится розовым.
При обратном титровании к раствору соли опре-
деляемого катиона прибавляют отмеренное количество
титрованного раствора комплексона III-и титруют избыток
31
комплексона III стандартным раствором соли какого-либо
металла с металл-индикатором. При оттитровании избыт-
ка комплексона III наблюдается четкий переход окраски
индикатора в точке эквивалентности. Для титрования ис-
пользуют раствор соли такого металла, комплекс которого
с комплексоном III менее устойчив по сравнению с ана-
логичным комплексом определяемого металла.
Метод обратного титрования применяют в тех случаях,
когда нет подходящего металла-индикатора для определя-
емого металла, когда реакция образования комплекса про-
ходит слишком медленно или когда в условиях титрова-
ния образуется осадок.
Метод комплексонометрии прост, быстр и удобен, так
как позволяет в большинстве случаев проводить анализ
без дополнительных операций отделения сопутствующих
элементов. В то же время он является высокоточным ме-
тодом: относительное стандартное отклонение результатов
•определений S,= 0,002-=- 0,008. Комплексонометрия широко
применяется в практике лабораторий предприятий для
определения содержания индия, галлия, циркония, РЗЭ,
цинка и других элементов.
Расчеты в титриметрии. Титр, или нормальность рабочего
раствора, устанавливают по рассчитанной и точно взвешенной на-
веске химически чистого исходного вещества. Титр часто выражают
в граммах определяемого вещества, соответствующего 1 мл рабочего
раствора: если исходное вещество неустойчивое или недостаточно чис-
тое, то готовят его раствор приблизительной концентрации, а затем
находят поправочные коэффициенты для приведения нормальности
растворов к заданной.
1. Прямое титрование. Например, при комплексонометрическом
определении содержания цинка (х %) в медных концентратах (см.
-«Цинк») применяют формулу x=TV-l00/G, в которой Т — титр раст-
вора комплексона III, выраженный в граммах цинка в 1 мл раствора;
V — объем раствора комплексона III, израсходованный на титрование
цинка, мл; G — навеска концентрата, г.
2. Обратное титрование. Например, при титриметрическом опреде-
лении содержания хрома в хромовых рудах и концентратах, содер-
жащих >0,05 %V (см. «Хром»), содержание оксида хрома (х, %)
вычисляют по формуле Х|= (Г7<—!/|)7’| ЮО/'О, в которой V—объем
раствора соли Мора, взятый для анализа, мл; К — поправочный коэф-
фициент или соотношение объемов растворов перманганата калия и
соли Мора; Vi — объем раствора перманганата калия, израсходованный
яа титрование избытка раствора соли Мора, мл; 1\ — титр 0,1 н. раство-
ра перманганата калия, выраженный в граммах оксида хрома в 1 мл
раствора; G—навеска руды или концентрата, г.
Физико-химические методы
Фотометрические методы — спектрофотометрия и фото-
колориметрия, относящиеся к методам абсорбционной
спектроскопии, основаны на избирательном поглощении
32
(абсорбции) света анализируемым раствором. Эти методы
различаются применяемой аппаратурой.
Спектрофотометрия основана на поглощении
монохроматического излучения, или точнее света в очень
узком интервале длин волн (1—2 нм). Аппаратурой явля-
ются спектрофотометры, позволяющие работать как с
окрашенными, так и с неокрашенными растворами, по-
глощающими излучение в ультрафиолетовой, видимой,
или ближней инфракрасной областях спектра.
Фотоколориметрия основана на поглощении ана-
лизируемым раствором полихроматичного света. Аппара-
тура представляет собой фотоэлектроколориметры, снаб-
женные светофильтрами, выделяющими свет в достаточно
узком интервале длин волн. Для наиболее узкополосных
светофильтров современных фотоэлектроколориметров ши-
рина области пропускания составляет 20—30 нм.
Колориметрический анализ основан на определении
концентрации элемента по интенсивности окраски раство-
ра, оценку которой производят или визуально путем
сравнения с эталонным раствором, или с помощью прос-
тых оптических приборов — фотометров и колориметров.
Воспроизводимость результатов при визуальной колори-
метрии невысока (Хч=0,1 -е-0,2). Этот метод в первую
очередь представляет интерес при нахождении содержа-
ния микропримесей, так как возможно оценить интенсив-
ность окраски раствора малого объема (~1 мл) находя-
щегося в колориметрической пробирке.
В основе фотометрических методов лежит зависимость
интенсивности монохроматического светового потока I, про-
шедшего через слой окрашенного раствора, от интенсив-
ности падающего потока света /0, концентрации окрашен-
ного вещества С и длины оптического пути кюветы I. Эта
зависимость определяется уравнением /=/0 10~hcl, которое
является математическим выражением основного за-
кона светопоглощения, или объединенного зако-
на Бугера — Ламберта — Бера. В этом уравнении
k — коэффициент поглощения, величина которого зависит
от природы растворенного вещества, растворителя, темпе-
ратуры и длины света. Если концентрация С выражена в
молях на литр раствора, а I в сантиметрах, то коэффи-
циент k называют молярным коэффициентом по-
гашения и обозначают буквой е. В этом случае 1 =
= 10 10~ЕСг. Оптическая плотность раствора А = П011. При
соблюдении основного закона светопоглощения оптическая
плотность прямо пропорциональна молярному коэффициен-
2. 129.
33
ту погашения, концентрации вещества и длине оптического
пути, т. е. А — е,С1.
Если в последнем соотношении С = 1 мол/л, 1=1 см, то
А = е, т. е. молярный коэффициент погашения представ-
ляет собой оптическую плотность 1 М раствора, помещен-
ного в кювету длиной оптического пути 1 см. Для боль-
шинства окрашенных соединений измерение Л = е не пред-
ставляется возможным, так как оптические плотности 1 М
растворов слишком высоки. По этой причине при опреде-
лении е ,моль-1 см-1, по формуле г = А)С1 значение С<1 М.
Обычно вычисляют значение эффективного молярного
коэффициента погашения, так как для нахождения истин-
ного значения е необходимо соблюдение ряда условий,
трудно выполнимых на практике (строгая монохроматич-
ность света, поглощение одного типа частиц известной
концентрации, отсутствие влияния посторонних веществ
и ионной силы раствора).
Молярный коэффициент погашения является важной
характеристикой окрашенных соединений, отражающей их
индивидуальные свойства. Величина е зависит от длины
волны проходящего света, температуры раствора и приро-
ды растворенного вещества и не зависит от длины опти-
ческого пути и концентрации вещества.
Для различных цветных реакций значения е также раз-
личны. Например, молярные коэффициенты погашения окра-
шенных аква-ионов РЗЭ не превышают 10. Для большого
числа цветных реакций элементов с органическими соеди-
нениями значения е составляют п-103—п-104. Наиболее
чувствительные реакции характеризуются значениями е =
= п-105, например реакция образования соединения цир-
кония с арсеназо III достигает значения 1,2-105 которое,
согласно теоретическим расчетам, приближается к пре-
дельно возможному для цветных реакций.
Существует зависимость между значением молярного
коэффициента погашения и чувствительностью цветной
реакции. Чем выше значение молярного коэффициента по-
гашения, тем выше чувствительность, которая в этом слу-
чае характеризуется угловым коэффициентом (или танген-
сом угла наклона) градуировочного графика (рис. 2).
Значения оптической плотности А, измеряемые с доста-
точной воспроизводимостью, находятся в пределах от 0,1
до 1,0. Чем больше значение е, тем меньшая концентрация
раствора будет соответствовать указанному интервалу зна-
чений А.
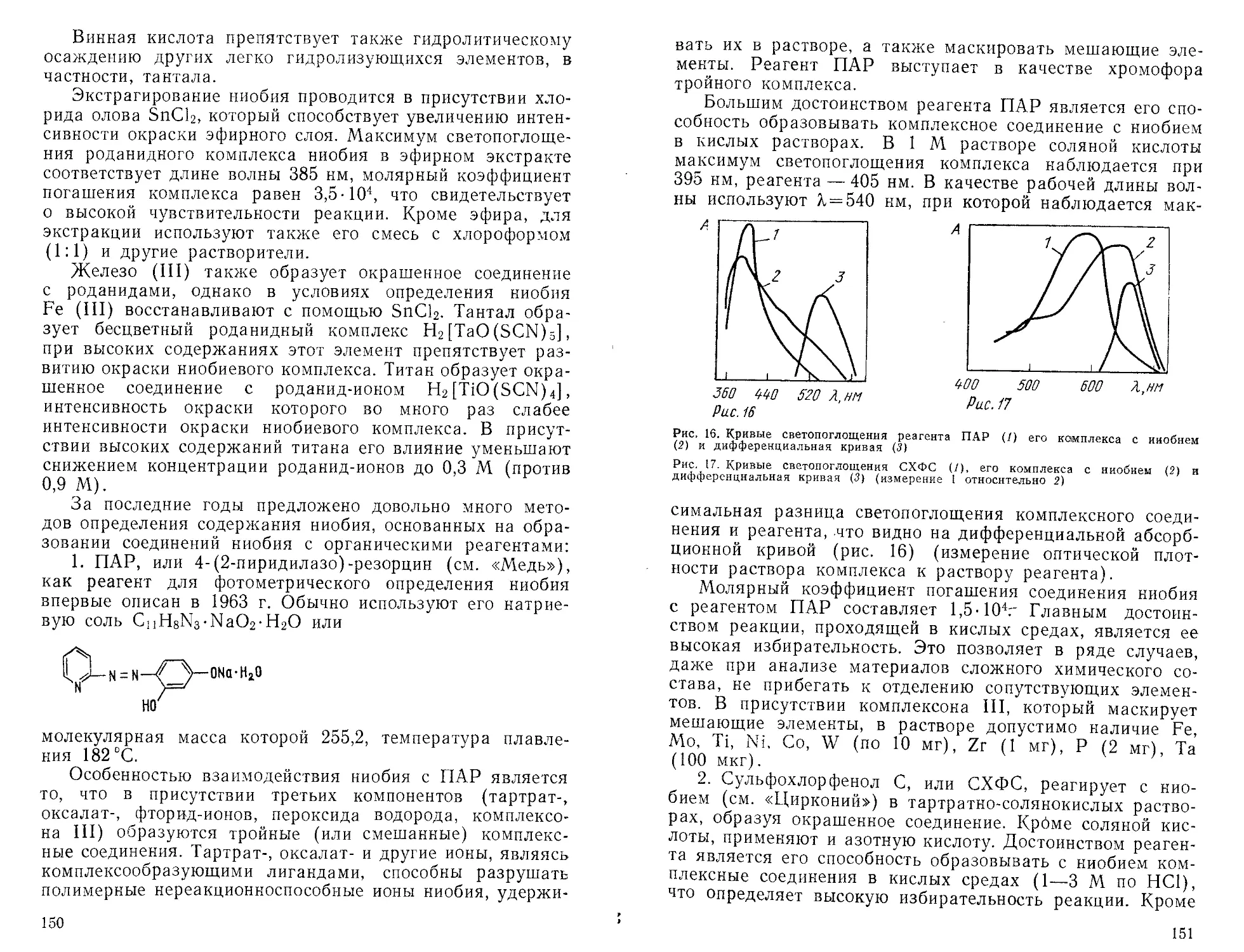
Предел обнаружения фотометрических реакций (по Сен-
34
делу) колеблется от 0,01 до 0,001 мкг/см2. Эти значения
относят к минимально измеряемой величине Л = 0,001. На
практике надежно можно измерить значения А = 0,05. Для
оценки предела обнаружения в реальных условиях учиты-
вают воспроизводимость результатов измерения оптиче-
ской плотности в области низких значений А и принимают
>lmin = 5SA, где Sa — стандартное отклонение оптической
плотности при значениях величины А до 0,1.
Интервал оптических плотностей, а, следовательно, и
определяемых концентраций, которые могут быть измере-
Ри.с.3
Рис. 2. Градуировочные графики для цветных реакций (1—3), характеризующих-
ся различными значениями молярных коэффициентов погашения 81>82>83
Рис. 3. Градуировочный график в методе полной дифференциальной спектро-
фотометрии
ны обычным (абсолютным) фотометрическим методом,
ограничен. Воспроизводимость измерений при обычном
методе невысока и характеризуется относительным стан-
дартным отклонением результатов определений 0,02—
—0,05 в зависимости от прецизионности прибора. Этот ме-
тод применяют для определения малых содержаний эле-
ментов (10~3—10~10%) или единиц процентов. Для рас-
ширения интервала определяемых концентраций (>10%)
и повышения воспроизводимости анализа применяют диф-
ференциальный фотометрический метод. Вос-
производимость дифференциального фотометрического ме-
тода характеризуется величиной относительного стан-
дартного отклонения 0,005—0,01 и приближается к
воспроизводимости классических гравиметрических и тит-
риметрнческих методов. В то же время метод является
более быстрым, чем гравиметрический.
Работы по дифференциальному спектрофотометрическо-
му методу у нас в стране впервые (в 1958 г.) выполнены
под руководством советского ученого Ю. А. Чернихова
его сотрудниками Т. М. Малютиной и Б. М. Добкиной. При
дифференциальном спектрофотометрическом методе опти-
ческая плотность исследуемого раствора измеряется не
2*
35
относительно растворителя или раствора реактивов, а
относительно раствора сравнения («нулевого» раствора),
содержащего известное количество определяемого веще-
ства.
Принцип дифференциальной фотометрии поясним на
следующем примере. Допустим, что концентрация 0,2 г
меди в 100 мл раствора может быть определена абсолют-
ным методом фотометрии с относительным стандартным
отклонением 0,05. При увеличении концентрации меди в
10 раз не получается в 16 раз большая воспроизводимость.
Если даже оптическая плотность, соответствующая такой
высокой концентрации меди, может быть измерена на при-
боре, то погрешность измерения оптической плотности бу-
дет выше, чем в рассмотренном выше случае. Это объясня-
ется тем, что измерения оптической плотности будут
произведены не в оптимальной области шкалы прибора.
Вышесказанное относится к случаю, когда раствором
сравнения является вода.
Предположим, что вместо воды используют стандарт-
ный раствор, содержащий 1,8 кг меди в 100 мл, а окраска
раствора подчиняется закону Бугера — Ламберта — Бера
и при высокой концентрации меди. Тогда для раствора,
содержащего 2,0 г меди, оптическая плотность должна
соответствовать такому же показанию шкалы прибора, как
и при измерении 0,2 г меди по отношению к воде. Если
возможно определять разницу концентраций и 0,2 г с
относительным стандартным отклонением 0,05, то общая
концентрация автоматически будет определена с относи-
тельным стандартным отклонением 0,005. Этот процесс
может быть продолжен. Если взять 19,8 г меди для уста-
новления прибора на нуль, то при соблюдении всех выше-
указанных условий можно получить относительное стан-
дартное отклонение 0,005 для раствора 20 г меди и т. д.
Таким образом, дифференциальный метод измерений
позволяет получить значительный выигрыш в воспроиз-
водимости результатов по сравнению с абсолютным ме-
тодом.
При дифференциальном методе измерений раствором
сравнения является один из растворов эталонного ряда,
оптическая плотность которого достаточно высока и в то
же время близка к оптической плотности исследуемого
раствора. Светопоглощенне раствора сравнения условно
принимается равным нулю. Чем выше оптическая плот-
ность раствора сравнения, тем выше воспроизводимость
измерений, так как погрешность измерений относится в
36
этом случае к большему значению концентрации. Макси-
мальная воспроизводимость измерений при данной кон-
центрации раствора сравнения наблюдается в случае ра-
венства концентраций растворов сравнения и испытуемого,
т. е. /2/Л = 1 (теоретические расчеты). Экспериментальным
путем установлено, что оптимальный интервал оптических
плотностей в дифференциальном методе равен 0,1—0,7.
Повышение оптической плотности раствора сравнения
связано с необходимостью увеличения светового потока.
Это объясняется тем, что поток света, прошедший через
сильно поглощающий раствор, должен обладать достаточ-
ной интенсивностью, пригодной для измерения фотоэле-
ментом. Увеличение потока света при измерениях на
спектрофотометрах осуществляется раскрытием щели.
Увеличение интенсивности потока света может вызвать
отклонения от закона светопоглощения, приводящие к
уменьшению воспроизводимости измерений. Нарушения за-
кона светопоглощения наблюдаются также при измере-
ниях на фотоэлектроколориметрах в связи с недостаточной
интенсивностью света, прошедшего через сильно окрашен-
ный раствор и попадающего на фотоэлемент. При нару-
шении пропорциональной зависимости между концентра-
цией и оптической плотностью необходимо установить
оптимальную концентрацию раствора сравнения.
Выбор оптимальной концентрации раствора сравнения
производится следующим образом. Готовят ряд эталонных
растворов с постоянной разницей концентрации АС и со-
ответствующей разницей ЛЛ в интервале значений от 0,3
до 0,4. Измеряют оптические плотности каждого после-
дующего раствора по отношению к предыдущему и вычис-
ляют произведение а— (ДЛ/АС)С0, где Со — концентра-
ция раствора сравнения. В качестве оптимальной выби-
рают ту концентрацию, при которой значение а макси-
мально. Относительно выбранного раствора сравнения из-
меряют оптические плотности серии растворов. По полу-
ченным значениям вычисляют значение фактора F или
строят градуировочный график зависимости ДЛ от АС.
Вычисление фактора производят по формуле F= (Ci—
—Со)/А, в которой С,— концентрация раствора, более
концентрированного, чем раствор сравнения; Со — концент-
рация раствора сравнения; Л — оптическая плотность более
концентрированного раствора, измеренного по отношению
к раствору сравнения.
Оптическую плотность испытуемого раствора измеряют
по отношению к раствору сравнения и определяют кон-
37
центрацию первого по градуировочному графику или по
формуле CX~CO + AF, где А—оптическая плотность испы-
туемого раствора, измеренного по отношению к раствору
сравнения, а Сх — концентрация испытуемого раствора.
Если оптическая плотность испытуемого раствора мень-
ше оптической плотности раствора сравнения, то произ-
водят измерение оптической плотности раствора сравне-
ния по отношению к испытуемому раствору и значение А
берут со знаком минус. Предварительно для тех же усло-
вий строят градуировочный график. Метод дифференци-
альной фотометрии с использованием как положительных,
так и отрицательных значений А называется методом пол-
ной (двусторонней) дифференциальной спектрофотометрии
(рис. 3). Он предложен советскими химиками В. Ф. Бар-
ковским и В. И. Ганопольским.
Для дифференциальных измерений используют как
спектрофотометры, так и фотоэлектроколориметры. При ис-
пользовании спектрофотометров можно достичь лучшей
воспроизводимости измерений по сравнению с фотоэлектро-
колориметрами, что объясняется более высокими значе-
ниями А для раствора сравнения в первом случае. (О вы-
боре Со при работе на фотоэлектроколориметрах см.
«Алюминий»).
В последние годы вариант высокоточной фотометрии —
дифференциальная (разностная) фотометрия дополнился
вариантом прямого измерения абсорбции, основанным на
использовании современной высокоточной микропроцес-
сорной аппаратуры. Показано, что стандартное отклонение
S измерения абсорбции вплоть до Л~2 на микропроцес-
сорном спектрофотометре Рп 8800 методом прямой фото-
метрии находится на уровне (1ч-2)-10—4 при времени
интегрирования 10 с и ширине щели 8 нм. При Л>2,2 S
увеличивается в 2—3 раза. Дифференциальный вариант
метода (абсорбция раствора сравнения Л~2) позволяет
при этом уменьшить S примерно в 2 раза.
Уменьшение погрешности с помощью дифференциалы-
ной фотометрии, а также реализация возможностей спек-
трофотометров типа Рп 8800 в варианте прямой фотомет-
рии достигаются, если погрешности предварительных ста-
дий анализа (взвешивания, разложения пробы, взятия
аликвотных частей, получения окрашенных фотометрируе-
мых форм и построения градуировочных графиков) не
превышает погрешности фотометрического измерения.
Нефелометрия основана на измерении света, рассеян-
ного дисперсной средой (взвешенными частицами).
38
Турбидиметрия предусматривает измерение поглощения
света, прошедшего через дисперсный раствор. Два послед-
них метода используют, например, при определении содер-
жания хлорид-ионов в виде соединения AgCl.
Флуориметрия (люминесцентный анализ) основан на
измерении вторичного излучения, возникающего в резуль-
тате взаимодействия ультрафиолетового излучения с опре-
деляемым компонентом. Содержание катионов, не обла-
дающих собственной люминесценцией, определяют с по-
мощью флуоресцентных реакций комплексов катионов с
органическими реагентами. Для определения содержания
индия, галлия, тантала и др, флуориметрическим методом
используют например, родаминовые красители. Флуори-
метрические методы характеризуются низким пределом
обнаружения (10 '6<7о), но они часто являются недостаточ-
но селективными. Используются в основном для опреде-
ления содержания микропримесей в материалах высокой
чистоты.
Атомно-абсорбционная спектрофотометрия основана на
измерении светопоглощения (абсорбции) определяемого
элемента, переведенного в атомный пар. Принцип метода
очень прост: излучение лампы с полым катодом проходит
через слой атомного пара определяемого элемента; затем
излучение резонансной длины волны выделяется монохро-
матором и направляется на фотодетектор.
Лампа с полым катодом представляет собой кругло-
донную колбу из молибденового стекла, в которую впаян
анод из молибденовой проволоки и катод в виде сквоз-
ного цилиндра из определяемого металла. Для атомизации
проб применяют пламя, высокотемпературные печи, на-
пример графитовую кювету Львова (по имени советского
ученого, предложившего ее конструкцию), а также лазер
и другие способы.
Атомное поглощение характеризуется экспоненциаль-
ным убыванием интенсивности проходящего света I в за-
висимости от толщины слоя поглощения I и концентрации
раствора по закону Бугера — Ламберта — Бера.
Метод получил наибольшее развитие с 1955 г. в связи
с предложением австралийского ученого А. Уолша просве-
чивать атомный пар светом лампы, которая излучает
спектр изучаемого элемента. В опытах исследователей
XIX в. атомный пар просвечивали источником света со
сплошным спектром, и для наблюдения ослабления излу-
чения требовались приборы высокой разрешающей спо-
39
собности. Благодаря предложению Уолша метод получил
современное аппаратурное оформление.
Наибольшее распространение имеют двухлучевые атом-
но-абсорбционные приборы американской фирмы «Пер-
кин-Эльмер», японской «Хитачи» и др. В нашей стране
выпущены двухлучевые приборы СФПА, «Сатурн», «Са-
турн-1» (рис. 4). Двухлучевую систему применяют в связи
с тем, что свечение пламени часто бывает интенсивнее,
чем излучение лампы с полым катодом. Прошедший через
атомный пар образца основной пучок света совмещают с
пучком сравнения и измеряют их соотношение с помощью
электронной схемы.
Рис. 4. Функциональная схема атомно-абсорбционного спектрофотометра:
1 — атомизатор; 2 — блок подготовки газа; 3— компрессор: 4 — лампа с по-
лым катодом; 5 —модулятор; 6 — датчик синхроимпульсов; 7 — конденсатор
неатомного поглощения при работе по однолучевой схеме; 8 — полупрозрачное
зеркало; 9 — монохроматор; 10 — фотоумножитель (преобразователь световых
импульсов в электрические); 11 — блок 'цифровой регистрации и обработки резуль-
татов; 12 — усилитель: 13 — усилитель синхроимпульсов; 14 — блок управления
графитовой печью: ЭВМ — электронно-вычислительная машина; ЦПУ — цифро-
печатающее устройство
В настоящее время метод атомной абсорбции позволяет
определять большинство элементов Периодической системы
Д. И. Менделеева. Метод является быстрым, достаточно
точным и в то же время характеризуется низким пределом
обнаружения (для некоторых элементов до 0,001 мкг/мл).
Метод атомной абсорбции успешно конкурирует с мето-
дами эмиссионного спектрального анализа, а также с по-
лярографическими п химическими методами анализа.
40
Электрогравиметрический метод основан на выделении
элемента из раствора в свободном состоянии (иногда в
виде оксидов) с помощью электролиза на взвешенном
электроде. По увеличению массы предварительно взвешен-
ного электрода вычисляют содержание определяемого эле-
мента в пробе. Этот метод относится, как показывает его
название, к гравиметрическим методам. При анализе про-
дукции цветной металлургии электрогравиметрический ме-
тод применяют для определения содержания меди в спла-
вах, черновой меди, для разделения и определения
содержания меди и никеля при совместном их присут-
ствии. При электролизе химическая реакция на электро-
дах протекает под действием электрического тока (внеш-
ний электролиз).
К электрогравиметрическим методам относится также
метод внутреннего электролиза. Метод назван так потому,
что электролиз проходит не под действием внешнего ис-
точника электрической энергии, а в результате окислитель-
но-восстановительных реакций, протекающих в анализи-
руемом растворе (например, в растворе соли меди) при
погружении в него двух металлов, например, платины и
цинка, составляющих гальванический элемент. Металличе-
ские платина и цинк образуют гальванический элемент при
соединении их с помощью металлического проводника. При
электролизе на менее активном металле (Pt-катод) проис-
ходит процесс восстановления с выделением из раствора
определяемого металла (меди): Cu2++2e->Cu. В качестве
анода используют менее благородный металл, чем тот, ко-
торый определяют. Например, при определении меди в
качестве анода берут пластинку металлического цинка.
При электролизе цинк растворяется и переходит в раствор:
Zn—2e->Zn2+.
Схема гальванической цепи в рассматриваемом случае:
- Zn/Zn2+ il Cu2+/Cu (Pt)+.
окисление восстановление
Титриметрические электрохимические методы. Помимо
установления точки эквивалентности по изменению окрас-
ки раствора визуальным путем, применяют различные фи-
зико-химические методы, основанные на измерении электро-
проводности (кондуктометрическое титрова-
ние), потенциала электрода (потенциометрическое
титрование), величины диффузионного тока (амперо-
метрическое титрование).
41
По сравнению с титрованием по изменению окраски
раствора метод потенциометрического титрования, дает
например более точные результаты и может быть приме-
нен для титрования таких веществ, для которых не подо-
браны индикаторы, или для анализа окрашенных раство-
ров.
Кулонометрическое титрование, основанное
на измерении количества электричества, расходуемого в
ходе электрохимической реакции, дает возможность опре-
делять с высокой точностью малые и большие количества
вещества.
Полярография (предложена в 1922 г. чешским ученым
Яросл. Гейровским). Этот метод является одним из важ-
нейших электрохимических методов, в основе которого
лежат процессы электроокисления или электровосстанов-
ления на ртутном капающем электроде. Ток электрохими-
ческой реакции зависит от потенциала и концентрации
определяемого вещества, а предельный ток пропорциона-
лен концентрации. В качестве электродов сравнения при-
меняют насыщенный каломельный электрод, металличе-
скую ртуть на дне сосуда (донная ртуть).
Методом полярографии можно определять содержание
элементов, способных окисляться на аноде или восстанав-
ливаться на катоде. Полярографический метод анализа
характеризуется достаточно низким пределом обнаруже-
ния, который составляет 10-5 моль/л. Метод является до-
статочно избирательным и позволяет определять содер-
жание нескольких элементов, одновременно присутствую-
щих в растворе. Его используют для определения меди,
цинка, кадмия, свинца, теллура и других элементов в раз-
личных продуктах производства цветной металлургии.
Для анализа материалов высокой чистоты применяют
полярографию переменного тока, которая поз-
воляет снизить предел обнаружения примерно на два по-
рядка (до 10-7 моль/л). Метод полярографии перемен-
ного тока основан на изучении зависимости переменной
составляющей тока ячейки от постоянного напряжения при
одновременной подаче на электроды постоянного и пере-
менного напряжения небольшой амплитуды.
Полярограмма переменного тока представляет собой
график зависимости величины переменного тока от по-
стоянного поляризующего напряжения. Величина макси-
мума на кривой пропорциональна концентрации определя-
емого вещества. Полярографией переменного тока возмож-
но определять содержание элементов с низким пределом
42
обнаружения. Она характеризуется большей разрешающей
способностью, чем классическая полярография, что позво-
ляет в ряде случаев определять содержание элементов без
разделения — упрощается ход анализа.
Ионометрия основана на использовании ионселективных
электродов для определения содержания отдельных ионов.
Ионселективным электродом называют индикаторный
или измерительный электрод с относительно высокой спе-
цифичностью к отдельному иону или типу ионов. Самым
первым ионселективным электродом, который получил ши-
рокое практическое применение, был стеклянный электрод,
используемый для измерения pH — активности ионов
водорода.
В настоящее время разработаны электроды для опреде-
ления хлорид-, фторид-, цианид-ионов, ионов меди, серебра,
натрия и др. По виду применяемых в электродах мембран
они делятся на стеклянные, кристаллические и жидкие.
Ионселективый электрод позволяет измерить активность
ионов а\. Для получения результатов в единицах концент-
рации (Ci) пользуются выражением а,- = Сгу, где у— коэф-
фициент активности.
Использование ионселективных электродов дает воз-
можность быстро анализировать производственные раство-
ры, вести непрерывный контроль производства. Большое
применение нашел фторселективный электрод для опреде-
ления фторид-иона в сточных водах предприятий, в про-
мышленных растворах в воздухе с предварительным по-
глощением газообразных фторидов щелочными растворами.
Кинетические методы, основаны на зависимости скорос-
ти реакции (v) от концентрации реагирующих веществ:
п = где Сд и Св — концентрации реагирующих ве-
ществ А и В соответственно. Кинетические методы харак-
теризуются низким пределом обнаружения (до 10-6 мкг/мл)
и низкой селективностью.
Физические методы
К физическим относятся методы анализа, в которых
не применяются химические реакции.
Эмиссионный спектральный (атомно-эмиссионный) ана-
лиз основан на регистрации линейчатых спектров, излу-
чаемых парами определяемого вещества в плазме электри-
ческой дуги или в искре. Эти спектры называют спектрами
излучения или эмиссионными спектрами, а анализ — эмис-
сионным спектральным. Спектральный анализ характери-
43
зуется пределом обнаружения 10~3—10“®%. Преимущест-
вом метода является возможность одновременного
определения содержания ряда элементов, спектральные
линии которых могут быть сняты на одну фотографическую
пластинку (свыше 70 элементов).
Сочетание спектрального анализа с предварительным
химическим концентрированием элементов (с помощью
экстракции и др.) положено в основу комбинированного
химико-спектрального метода. Этот метод
позволяет по сравнению со спектральным снизить пре-
делы обнаружения элементов.
' Метод фотометрии пламени заключается в распылении
анализируемого раствора в пламени и измерении интен-
сивности излучения с длиной волны, характерной для
определяемого элемента. Метод применяется, например,
для определения содержания щелочных и щелочноземель-
ных металлов.
Рентгеноспектральный анализ основан на возбуждении
атомов вещества при облучении его электронами больших
энергий или рентгеновскими лучами. В первом случае
процесс называют прямым возбуждением, во втором —
вторичным или флуоресцентным. Линии рентгеновского
излучения возникают вследствие перемещения электронов
в наиболее глубоких внутренних электронных слоях.
Рентгеноспектральный (рентгенофлуоресцентный) ана-
лиз пригоден для определения содержания всех элементов,
атомный номер которых >13, т. е. начиная с алюминия.
Особое преимущество метод имеет при анализе смесей
элементов, близких по свойствам, например редкоземель-
ных элементов, тантала и ниобия. Рентгеноспектральный
метод применяют для анализа руд, сплавов, металлов,
различных продуктов химической технологии. Диапазон
определяемых концентраций очень широк: можно опреде-
лять макро- (от 1 до 100%) и микро- (КН—КП3 %) ком-
поненты.
В последние годы разработаны многоканальные анали-
заторы, квантометры, которые позволяют определять со-
держание целого ряда элементов с высокой экспресс-
ностью. Метод применяется для анализа растворов
и пульп непосредственно в потоке (определение меди в
нескольких точках технологического процесса и др.). При-
менение современных рентгеновских квантометров сочета-
ется с. автоматизированной доставкой проб для анализа;
автоматизация процесса обработки данных осуществля-
ется с помощью электронно-вычислительных машин.
44
К недостаткам рентгеноспектрального анализа относит-
ся существенное влияние состава пробы, что приводит к
необходимости введения ряда поправок.
Масс-спектрометрия — способ исследования вещества
путем ионизации атомов и разделения ионов по величине
отношения массы (т) к заряду (е). Массы ионов изме-
ряют в атомных единицах массы, за которую принимают
712 часть массы изотопа углерода 12С. Масс-спектромет-
рию применяют для определения содержания всех эле-
ментов и соединений, которые можно перевести в паро-
образное состояние. При использовании искровой масс-
спектрометрии нижний предел определяемых концентраций
составляет 10~'4—10~8%. Метод используют в основном для
анализа вещества высокой чистоты.
Радиоактивационный анализ основан на использовании
различных ядерных реакций для определения содержания
элементов. Метод применяется для определения как основ-
ных компонентов (высоких содержаний), так и примесей.
Анализируемый объект предварительно подвергается облу-
чению какими-либо ядерными частицами или гамма-луча-
ми. В результате ядерных реакций образуются радиоактив-
ные изотопы, по активности которых проводят определение
содержания элементов.
В ряде случаев анализ можно выполнять без разруше-
ния образца. Метод находит широкое применение для ана-
лиза особо чистых веществ, используемых в полупровод-
никовой технике, а также металлов и сплавов.
Радиометрический метод применяется для определения
содержания урана, тория, радия и других естественно-ра-
диоактивных элементов по испускаемому ими а, р или у
излучению.
VI. РАЗДЕЛЕНИЕ И МАСКИРОВАНИЕ ЭЛЕМЕНТОВ.
ВСКРЫТИЕ ПРОБ
Осаждение
Метод разделения элементов, основанный на осаждении,
относится к наиболее старым; часто его стараются за-
менить более современными методами. К недостаткам
метода осаждения относится: 1) отсутствие полноты раз-
деления элементов из-за явления соосаждения; 2) зна-
чительная продолжительность операций его выполнения
(фильтрования, промывания осадков и т. д.).
45
Для разделения ионов используется различие раствори-
мостей их соединений. Например, разделение молибдена
и рения основано на малой растворимости молибдата каль-
ция в воде и на достаточно большой растворимости перре-
ната кальция.
Для осаждения элементов применяют органические ре-
агенты, например 8-оксихинолин, купферон, образующие с
ионами ряда металлов малорастворимые комплексные со-
единения.
Для разделения могут быть применены методы осаж-
дения гидроксидов аммиаком, едким натром при различ-
ных значениях pH. Например, при pH 5,3, который соз-
дается добавлением ацетатного буферного раствора,
осаждают титан (IV), цирконий (IV), торий (IV), желе-
зо (III), алюминий, хром (III). Два последних элемента
полностью осаждаются лишь в присутствии железа (III).
В растворе остаются кобальт (II), никель (II), марганец
(II), цинк, молибден (VI), вольфрам (VI) и медь.
Хроматография
Большое практическое значение для разделения эле-
ментов имеет хроматографический метод анализа, пред-
ложенный в 1903 г. русским ученым М. С. Цветом. Метод
основан на распределении разделяемых веществ между
неподвижной фазой, которой может быть жидкость или
твердое тело, и подвижной — газом или жидкостью. Хро-
матографию подразделяют на два вида: газовую и жид-
костную. Ниже рассмотрены подвиды жидкостной хрома-
тографии.
Жидко-жидкостная хроматография основана на
распределении вещества между двумя жидкими фазами.
В качестве неподвижной фазы используют воду или орга-
нический растворитель. В первом случае подвижной фазой
является органический растворитель, во втором — водный
раствор. Жидко-жидкостную хроматографию подразделяют
на колоночную распределительную и бумаж-
ную.
Твердо-жидкостная хроматография основана на
избирательной адсорбции веществ такими адсорбентами,
как силикагель, оксид алюминия, молекулярные сита, по-
ристое стекло. Разновидностью твердо-жидкостной хрома-
тографии является тонкослойная, в которой адсорбент
распределен тонким слоем на поверхности пластики.
Ионообменная хроматография основана на спо-
46
собности ионов анализируемого раствора вступать в реак-
ции обмена с подвижными ионами сорбента, который, на-
зывают ионитом. Иониты, способные к обмену катионов,
называют катионитами, а к обмену анионов — анионита-
ми. В ионитах с амфотерными свойствами происходит об-
мен анионов и катионов. Эксклюзионная хроматогра-
фия основана на различии размеров молекул разделяемых
веществ.
Экстракция
Экстракция — это процесс переноса растворенного ве-
щества из одной жидкой фазы (воды) в другую (органи-
ческий растворитель), с ней не смешивающуюся. Экстрак-
цию применяют как для извлечения в органическую фазу
определяемого элемента, так и для удаления мешающих
компонентов.
Широкое распространение получил способ экстракции
с использованием делительных воронок. Его осуществляют
путем встряхивания анализируемого раствора с органиче-
ским растворителем в течение определенного промежутка
времени (1—3 мин). Одновременно с извлечением опреде-
ляемого элемента в слой органического растворителя мо-
жет происходить концентрирование этого элемента, т. е.
переведение его из большого объема водного раствора в
меньший объем органического слоя (абсолютное концен-
трирование) .
Абсолютное концентрирование эффективно
при анализе сточных вод предприятий. При анализе чис-
тых веществ возникает задача отделения определяемого
элемента (микрокомпонента) от других элементов, присут-
ствующих в резко преобладающих количествах, т. е. от
макрокомпонентов. Экстракция позволяет уменьшить со-
отношение содержаний макро- и микрокомпонентов. Такое
концентрирование называется относительным.
Отношение концентрации вещества определенного сос-
тава в органической фазе к концентрации вещества того
же состава в водной фазе (в условиях равновесия) назы-
вается константой распределения: Кл= [А] о/ [А] в-
Отношение суммарной концентрации вещества в органи-
ческой фазе (Со) к суммарной концентрации его в вод-
ной фазе (Св) при установлении равновесия называется
коэффициентом распределения или коэффициен-
том экстракции: Z) = C0/CB. Форма нахождения элемента в
той или другой фазе во внимание не принимается.
6
47
Экстракцию часто применяют в сочетании с другими
методами. Например, экстракционно-фотометрический ме-
тод нашел широкое применение при определении содержа-
ния микропримесей в материалах высокой чистоты.
Маскирование
Реагенты, с помощью которых удается устранить дей-
ствие элементов, мешающих определению искомого ком-
понента, причем без их отделения, называются маскирую-
щими. Наиболее распространенным видом маскирования
является комплексообразование. Мешающий элемент обра-
зует с маскирующим реагентом комплекс более прочный,
чем с основным реагентом, применяемым для определения
содержания искомого элемента. Определяемый же эле-
мент не должен давать с маскирующим веществом ком-
плекс, более прочный, чем с реагентом, которым он опре-
деляется.
Если подобран маскирующий реагент, то способ устра-
нения его мешающего влияния на анализ следует пред-
почесть более сложным способам разделения элементов
(осаждению, ионному обмену, экстракции и др.). В каче-
стве маскирующих реагентов применяют винную, лимон-
ную, щавелевую кислоты или их соли, комплексон III и
т. д. Например, при определении содержания титана с
пероксидом водорода в присутствии больших количеств
железа последнее связывают в бесцветный комплекс фос-
форной кислотой [Ее(РО4)2]3^.
Большое число элементов Периодической системы
Д. И. Менделеева реагирует с комплексоном III, т. е. сам
комплексон как реагент для титрования не является се-
лективным по отношению к какому-либо одному или не-
скольким элементам. Селективность методов комплексоно-
метрического титрования повышается в присутствии ре-
агентов, маскирующих мешающие элементы. Например,
винная кислота в кислой среде маскирует олово (IV),
сурьму, титан, цирконий, хром (III), ниобий, вольфрам и
используется при комплексонометрическом определении
содержания меди, цинка, индия, галлия, свинца, висмута,
железа (III), никеля.
Разложение материалов
Разложение пробы и переведение в раствор определя-
емых компонентов, являющееся одним из важных этапов
анализа, должно протекать в условиях, обеспечивающих
48
вскрытие материала без потерь определяемого элемента.
Удачно выбранный способ разложения материала позво-
ляет не только перевести в раствор определяемый элемент,
но и облегчить его отделение от сопутствующих элементов
или создать благоприятные условия для конечного опреде-
ления содержания элемента без операций разделения.
Разложение пробы —часто более трудоемкая операция,,
чем само определение содержания, особенно это относит-
ся к анализу сложного минерального сырья.
При анализе материалов высокой чистоты способ раз-
ложения пробы должен обеспечить получение низких зна-
чений «контрольного» опыта, величина которых
зависит от количеств определяемого элемента, вноси-
мого в раствор реактивами и посудой, в которой проводят
разложение пробы. Низкие значения «контрольного» опыта
достигаются использованием малого числа реагентов и
операций, причем степень чистоты применяемых реагентов
должна быть высокой или очистка реагентов должна
легко осуществляться. Цель очистки состоит в уменьшении
содержания определяемого элемента в реагентах. Оно
должно быть малым по сравнению с содержанием элемен-
та в пробе.
Скорость растворения вещества зависит от его поверх-
ности: чем мельче проба руды, или металла, тем быстрее
завершится растворение. Однако не рекомендуется из-
мельчать пробу в большей степени, чем это необходимо,
из-за явлений окисления и адсорбции. Скорость раство-
рения, как правило, увеличивается с повышением темпера-
туры.
Способы разложения материалов растворением в кисло-
тах, в смесях кислот, щелочным или кислым, окислитель-
ным или восстановительным сплавлением, а также спека-
нием приведены подробно при рассмотрении методов опре-
деления отдельных элементов.
VII. МЕТОДЫ ОПРЕДЕЛЕНИЯ СОДЕРЖАНИЯ
ЦВЕТНЫХ МЕТАЛЛОВ
Алюминий
Алюминий открыт в 1825 г. датским ученым X. Эрстедом и назван
по латинскому названию квасцов (alumen).
Нахождение в природе. Алюминий, являясь одним из самых рас-
пространенных металлов в земной коре (8,8%), сосредоточен главным
49
образом в алюмосиликатах состава Al2O3-x1SiO2- t/H2O. Из них осо-
бенно распространены алюмосиликаты из группы полевых шпатов, вклю-
чающей такие минералы, как ортоклаз, альбит, анортит. Промышлен-
ным источником алюминия служит боксит — горная порода, состоящая
из гидратов глинозема А12О3-хН2О, оксидов железа, хрома, титана,
кальция, магния, галлия, скандия, кремнезема. Минерал, представлен-
ный одноводным гидратом глинозема АЮ(ОН), называется диаспором
или бемитом, а минерал, представленный трехводным гидратом глино-
зема А1(ОН)3,— гидраргиллитом или гиббситом. В зависимости от при-
роды основного породообразующего минерала различают три типа бок-
ситов: диаспоровые, гидр аргиллитовые и смешанные. Металлический
алюминий получают электролитическим путем.
Применение. Металлический алюминий используют для изготовле-
ния проводов, химической аппаратуры, конденсаторов, для защиты
отдельны?; металлов от коррозии. В виде порошка или мелкой струж-
ки алюминий служит восстановителем при получении многих цветных
и редких металлов из их кислородных соединений. Этот метод алю-
минотермии осуществляется по реакциям типа Сг2О3+2А1 = А12Оз+2Сг.
Широко применяются сплавы на основе алюминия с добавками (для
увеличения прочности) меди, кремния, титана, железа, никеля, мар-
ганца, цинка, магния. Кроме того, производят сплавы на основе пе-
речисленных элементов, в которые алюминий добавляют в качестве
легирующей добавки. Особенностью алюминиевых сплавов является
легкость. Их производство стимулировалось в основном развитием
авиации. В настоящее время они применяются во многих других
областях народного хозяйства.
Свойства алюминия и его соединений. Алюминий —
элемент Ш группы Периодической системы Д. И. Менде-
леева. Электронное строение атома в основном состоя-
нии— ls22s22p63s23p’. Устойчивой является степень окисле-
ния + 3, однако известны соединения алюминия, в степени
окисления +1, которые образуются при высоких темпера-
турах.
Алюминий — металл белого цвета; температура его
плавления 660 °C. Вследствие образования защитной оксид-
ной пленки алюминий устойчив против действия разбав-
ленных и концентрированных кислот (азотной и фосфор-
ной), слабо растворяется в разбавленной и концентриро-
ванной серной кислоте. Более агрессивна в отношении
алюминия соляная кислота: 2А1 + 6НС1 = 2А1С13 + ЗН2.
Металл также легко растворяется в щелочах с образова-
нием алюминатов и выделением водорода: 2Al + 2NaOH +
+ 10H2O = 2Na [А1(ОН)4(Н2О)2]4-ЗН2.
Гидроксид алюминия А1(ОН)3 выпадает в виде
белого студенистого осадка, содержащего основные соли,
при действии щелочи на растворы солей алюминия или при
действии солей неорганических кислот. При рН>5 А1(ОН)3
растворяется в избытке щелочи с образованием алюми-
натов.
Гидроксид алюминия — амфотерное соединение,
-50
атом водорода в котором может быть замещен на металл»,
а группа ОН-—на кислотный остаток. В результате этого
при взаимодействии гидроксида алюминия с сильными
кислотами или щелочными образуются соли:
А1(ОН)3+ 3HNO3= A1(NO3)34~3H2O; Al (ОН)3 +
+ NaOH4-2H2O = Na[Al(OH)4(H2O)2].
Оксид алюминия. Путем прокаливания гидрокси-
да или солей алюминия получают аморфный белый поро-
шок оксида алюминия AI2O3. Он существует в различных
модификациях. Весовой формой в гравиметрии является
негигроскопичный оксид кристаллической структуры, полу-
чаемый при прокаливании аморфного оксида выше 1000 °C.
Фторид алюминия A1F3 образуется при растворе-
нии гидроксида во фтористоводородной кислоте. С фто-
ридами щелочных металлов образует комплексные соеди-
нения. Na3AlF6 (криолит) имеет большое значение в метал-
лургии и аналитической химии алюминия.
Алюминиевые сплавы растворяются в щелочах,
в соляной кислоте или в смеси кислот, например соляной
и азотной.
Алюмосиликаты разлагаются сплавлением с пиро-
сульфатом калия или со смесью соды и буры. Бокситы
гидраргиллитового типа разлагаются смесью кислот (сер-
ной, азотной и соляной). Бокситы диаспорового и смешан-
ного типов разлагаются сплавлением с пиросульфатом
калия или Na2CO3.
Методы определения содержания алюминия
В аналитической химии элемента используют амфотер-
ность гидроксида, способность иона А13+ к комплексообра-
зованию с галогенид-ионами, оксикислотами и образова-
нию внутрикомплексных соединений. Для определения
содержания алюминия применяют титриметрию, гравимет-
рию (с использованием неорганических и органических
реагентов), фотометрию, люминесцентные методы с ис-
пользованием органических реагентов. Атомно-абсорбци-
онное определение алюминия до недавнего времени было
затруднительным вследствие образования в пламени термо-
стойких оксидов. С появлением более совершенных при-
боров, позволяющих использовать высокотемпературное
пламя оксида азота (I) — ацетилена, эти затруднения
исчезли. Разработаны методы определения содержания
51
алюминия в различных образцах, например в солях алю-
миния, в боксите.
Гравиметрические методы. Осаждение гидроксида алю-
миния в настоящее время применяется для отделения его
от других элементов. Гидроксид алюминия выделяют раз-
личными способами: с помощью солей неорганических
кислот, аммиаком, соединениями, выделяющими при нагре-
вании аммиак, слабыми органическими основаниями (на-
пример, пиридином). При этом удается отделить алю-
миний от никеля и марганца, щелочных и щелочноземель-
ных элементов.
Самым точным гравиметрическим методом определения
содержания алюминия считается оксихинолиновый
метод.
8-оксихинолин (оксин)
ОН
или C9H7ON (молекулярная масса 145,15). Оксихинолин
предложен русским ученым Р. Бергом в 1927 г. Из уксусно-
кислых и аммиачных растворов в области pH 4,2—9,8
реактив осаждает желто-зеленый кристаллический окси-
хинолинат алюминия состава А1(СдН6ОМ)з. Реагент по-
зволяет отделить алюминий от магния, щелочных и ще-
лочноземельных элементов, а также от бериллия.
Применение комплексообразующих веществ повышает
селективность метода, так как появляется возможность
разделить алюминий и железо, алюминий и титан.
Фторид натрия предложен советским ученым
И. В. Тананаевым в качестве осадителя алюминия в виде
труднорастворимого комплексного соединения — криолита:
AlCl3 + 6NaF=Na3AlF6+3NaCl. Введение в раствор хлори-
да натрия понижает растворимость криолита. Метод от-
личается высокой селективностью: присутствие м^эганца,
меди, кобальта, никеля, железа не мешает определению
содержания алюминия. В случае осаждения из сернокис-
лых растворов в присутствии комплексообразующих ве-
ществ допустимы ограниченные содержания титана, цирко-
ния, молибдена, ниобия, ванадия, вольфрама.
Титриметрические методы. Наибольшее распростране-
ние получили фторидный и комплексонометрический ме-
тоды определения содержания алюминия.
.52
В основе фторидного метода лежит образование
криолита ЫазА1Р6 при условии избытка фторида натрия
и хлорида натрия до насыщения. В результате гидролиза
растворы солей алюминия имеют кислую реакцию. Обра-
зование криолита приводит к изменению реакции раствора
до нейтральной.
Избыток раствора фторида натрия оттитровывают ра-
створом хлорида алюминия до изменения окраски кислот-
но-основного индикатора (метилового красного). Свобод-
ную кислоту, обычно содержащуюся в растворах солей
алюминия, после добавления фторида натрия и до титро-
вания хлоридом алюминия нейтрализуют щелочью (по
фенолфталеину).
Известны различные варианты фторидного метода, на-
пример вариант с применением индикаторного ион-селек-
тивного фторидного электрода.
Комплексонометрический метод. Прямое ти-
трование алюминия комплексоном III затруднено вслед-
ствие образования устойчивых аква- и оксикомплексов,
медленно реагирующих с раствором титранта. Титрование
проводят при нагревании в присутствии индикаторов: хром-
азурола S, эриохромцианина R, алюминона, ксиленоло-
вого оранжевого.
В большинстве случаев рекомендуется обратное титро-
вание растворами солей цинка, свинца, магния с индика-
торами ксиленоловым оранжевым и эриохромчерным Т.
1. Ксиленоловый оранжевый [ (3,3-бис)-N, N'-ди (кар-
боксиметил) -аминометил-о-крезол-сульфофталеин]
или C3iH32O13N2S (молекулярная масса 672,44) —это крас-
но-коричневый кристаллический порошок, легко раствори-
мый в воде, имеющий окраску раствора: желтую при pH
5—6 и красную при рН>6. Соединения реагента с иона-
ми металлов, в частности с ионами Zn2+, Pb2+, образуются
при pH 3—6 и также окрашены в красный цвет. Обратное
титрование избытка добавленного комплексона III раство-
53
ром соли цинка или свинца проводят при pH 5,5—6,0;
при этом желтая окраска раствора переходит в крас-
ную.
2. Эриохромчерный Т (натриевая соль) [1-(1-окси-2-
нафталазо)-6-нитро-2-нафтол-4-сульфокислоты]
ОН НО
или C20H12O7N3SNa (молекулярная масса 461, 41) пред-
ставляет собой порошок коричневого или черного цвета,
растворимый в воде и спирте с образованием окраски:
синей при pH 7—И, оранжевой при рН>11,5 и красной
при рН<6. При pH 9—10 многие катионы (Mg2+, Zn2+)
образуют с реагентом комплексное соединение красного,
цвета. При титровании комплексоном III это комплексное
соединение разрушается и в точке эквивалентности наблю-
дается синяя окраска реагента. При определении содержа-
ния алюминия обратным титрованием избытка добавлен-
ного комплексона III раствором соли магния или цинка
в точке эквивалентности синяя окраска раствора пере-
ходит в красную.
Растворы комплексона III готовят растворением препа-
рата в дистиллированной воде и хранят в полиэтиленовой
посуде. При хранении в стеклянной посуде титр раствора
со временем уменьшается в результате перехода в раст-
вор ионов металлов из стекла (Zn2+, Са2+). Исходным
веществом для определения титра служат чистые металлы,
например цинк, а также безводные соли, негигроскопич-
ные и устойчивые при нагревании до ПО °C, например
РЬС12, Pb(NO3)2. Целесообразно определять титр раство-
ра комплексона III в тех же условиях, в которых проводит-
ся титрование.
Фотометрические методы определения содержания алю-
миния, не обладающего хромофорными свойствами, осу-
ществляют с применением рассмотренных в разделах
«Алюминий», «Титриметрические методы» комплексономет-
рических индикаторов.
1. При фотометрическом определении содержания алю-
миния часто применяют алюминон.
54
Алюминон (аммонийная соль ауринтрикарбоновой кис-
11 coonh4
о
лоты) или СггНгзОдЫз (молекулярная масса 473,43), явля-
ется растворимым в воде реагентом, дающим красную
окраску. При выполнении реакции образования соедине-
ния алюминия с алюминоном требуется строгое соблюде-
ние pH 4,4—4,75, применение емкого ацетатного буферного
раствора. Максимальное светопоглощение комплекса на-
блюдается при длине волны 535 нм. Окраска комплекса
развивается во времени при нагревании. Свойства реаген-
та отдельных партий значительно различаются, в связи с
этим необходима частая проверка градуировочного гра-
фика.
2. Эриохромцианин 7? (тринатриевая соль диметилсуль-
фооксифуксондикарбоновой кислоты)
или CgsHisOgSNas (молекулярная масса 536,4), использу-
ется в качестве реагента при определении концентрации
алюминия в узком интервале pH 6,2—6,5. При рН>6,5
комплексное соединение алюминия разлагается. Молярный
коэффициент погашения комплекса при 2«тах=535 нм со-
ставляет 6,5-104. Это самый чувствительный реагент для
определения содержания алюминия. Л4ешающее определе-
нию железо восстанавливают аскорбиновой кислотой, медь
маскируют тиосульфатом. Определению содержания алю-
миния мешают также хром, бериллий, ванадий, цирконий,
и не мешают лантан, индий, цинк, свинец, никель, олово
и марганец.
3. Хромазурол S (тринатриевая соль дихлорсульфоди-
метилоксифуксондикарбоновой кислоты)
55
или С2зН1зОдС125Маз (молекулярная масса 546,0)—это
кристаллическое вещество красного цвета, хорошо раство-
римое в воде. Свободная кислота известна в литературе
под названием «альберон».
В качестве реагента при определении алюминия хрома-
зурол S, напоминая эриохромциамин R, обладает несколь-
ко меньшей чувствительностью, но комплексное соедине-
ние его с алюминием более устойчиво к действию восста-
новителей, чем в сравниваемом случае. Содержание алю-
мия определяют при pH 6,0 и Х=547 нм. Молярный
коэффициент погашения комплекса составляет 5-Ю4.
Практическая часть
Комплексонометрическое определение алюминия в боксите
Принцип метода. Алюминий связывают в комплекс с
комплексоном III, избыток последнего оттитровывают ра-
створом соли цинка в присутствии индикатора ксиленоло-
вого оранжевого. Предварительно отделяют гидроксиды
алюминия, титана, железа уротропином и растворяют их
в соляной кислоте. Титан и железо в полученном растворе
отделяют от алюминия едким натром. Относительное стан-
дартное отклонение равно 0,01 при содержании —45%
AI2O3.
Реактивы и растворы. 1) аммиак водный, р = 0,91 г/см3;
2) хлорид аммония; 3) ацетатный буферный раствор (для
его приготовления 250 г ацетата натрия помещают в мер-
ную колбу вместимостью 1000 мл, растворяют в воде, до-
бавляют 20 мл уксусной кислоты и доводят объем до метки
водой); 4) карбонат натрия, безводный; 5) кислота азот-
ная, р=1,40 г/см3; 6) кислота соляная, р=1,19 г/см3 и
разбавленная 1:1, 1:40; 7) кислота уксусная, р =
= 1,05 г/см3; 8) кислота серная, р=1,84 г/см3; 9) ксилено-
ловый оранжевый, 0,1%-ный раствор; 10) комплексон III,
или динатриевая соль этилендиаминтетрауксусной кисло-
ты 0,05 М раствор (для его приготовления 18,6 г комплек-
56
сона III растворяют в воде, фильтруют в мерную колбу
вместимостью 1000 мл, разбавляют водой до метки и тща-
тельно перемешивают); 11) гидроксид натрия, 20%-ный
раствор; 12) ацетат натрия; 13) уротропин, 25 и 0,5%-ные
растворы; 14) цинк металлический гранулированный;
15) раствор нитрата цинка 0,05 М (для его приготовле-
ния 3,269 г металлического цинка растворяют при нагре-
вании в смеси 100 мл воды и 15 мл азотной кислоты; ра-
створ выпаривают до 5—10 мл и разбавляют водой до
1000 мл).
Для определения поправочного коэффициента 0,05 М.
раствора комплексона III по раствору нитрата цинка
бюреткой отбирают в коническую колбу вместимостью
500 мл 20 мл раствора комплексона III и разбавляют его
водой до 200 мл. Вводят 5—6 капель ксиленолового оран-
жевого и добавляют по каплям аммиак до синей окраски,
которую устраняют добавлением по каплям разбавленной
соляной кислоты. Затем в колбу приливают 20 мл ацетат-
ного буферного раствора и титруют раствором нитрата
цинка до изменения желтой окраски раствора в розовую.
Поправочный коэффициент 0,05 М раствора комплек-
сона III находят из соотношения K=VJV, в котором V) —
количество 0,05 М раствора нитрата цинка, мл, а V —
количество раствора комплексона III, мл.
Выполнение определения. Навеску боксита 0,5 г поме-
щают в платиновый тигель, тщательно смешивают с 1 г
карбоната натрия и сплавляют при 1100 °C в течение
30 мин.
После охлаждения сплав помещают в стакан вмести-
мостью 200 мл и осторожно приливают 35 мл соляной
кислоты (1:1). Остаток сплава в тигле полностью раст-
воряют той же кислотой при нагревании и переносят раст-
вор из тигля в стакан, а затем в мерную колбу вмести-
мостью 500 мл. Аликвотную часть 100 мл помещают в
стакан вместимостью 250 мл, нейтрализуют аммиаком до
начала выпадения осадка гидроксидов. Осадок растворяют
в разбавленной соляной кислоте, приливаемой по каплям.
К раствору прибавляют 0,5 г хлорида аммония, 25 мл
25%-кого раствора уротропина; смесь нагревают в тече-
ние 20 мин для коагуляции осадка и фильтруют через
неплотный фильтр. Осадок промывают шесть-восемь раз
теплым 0,5%-ным раствором уротропина, затем смывают
с фильтра в стакан, в котором проводилось осаждение.
Фильтр промывают горячей разбавленной соляной кисло-
той (в количестве 25 мл) и три-четыре раза горячей во-
57
дой, собирая фильтрат в тот же стакан. Содержимое стака-
на нагревают до растворения осадка, после чего перево-
дят в мерную колбу вместимостью 250 мл, содержащую
50 мл нагретого до 80 °C раствора едкого натра. Стакан
ополаскивают три-четыре раза горячей водой.
Раствор охлаждают, разбавляют водой до метки и тща-
тельно перемешивают. Затем раствор фильтруют через
сухой фильтр в сухой стакан или колбу, отбрасывая первые
порции фильтрата. Отбирают 200 мл раствора, помещают
в коническую колбу вместимостью 500 мл, добавляют
50 мл 0,05 М раствора комплексона III, нагревают его
до кипения. Затем добавляют пять-шесть капель раствора
ксиленолового оранжевого и нейтрализуют раствор разбав-
ленной соляной кислотой, прибавляя ее по каплям до пере-
хода окраски раствора в желтый цвет. После нейтрализа-
ции к раствору прибавляют 20 мл ацетатного буферного
раствора. Смесь кипятят 2—3 мин, охлаждают, прибав-
ляют еще две-три капли раствора ксиленолового оранже-
вого и избыток комплексона III, оттитровывают раствором
нитрата цинка до первого изменения желтой окраски ра-
створа в оранжевую. Каждый миллилитр разности вели-
чин добавленного 0,05 М раствора комплексона III и из-
расходованного 0,05 М раствора нитрата цинка соответ-
ствует 1,3491 мг алюминия.
Определение алюминия в магниевых сплавах
дифференциальным фотометрическим методом
Принцип метода. Определение алюминия основано на
реакции образования комплексного соединения с хрома-
зуролом S. Оптическую плотность испытуемого раствора
измеряют по отношению к раствору сравнения с извест-
ным содержанием алюминия. Относительное стандартное
отклонение результатов определений составляет 0,01 при
содержаниях —10% алюминия.
Реактивы, растворы и аппаратура. 1) кислота соляная, р =
= 1,19 г/см3, разбавленная 1:1 и 0,1 М раствор; 2) аскорбиновая
кислота, 1%-ный раствор; 3) хромазурол S, 0,12%-ный раствор;
4) ацетат натрия, 2 М. раствор; 5) рабочий раствор алюминия (гото-
вят следующим образом: растворяют 0,0375 г металлического алюми-
ния в 2 мл соляной кислоты (1:1) и разбавляют водой в мерной
колбе вместимостью 500 мл; в 1 мл полученного раствора содержится
0,075 мг алюминия); 6) спектрофотометр СФ-4 или аналогичный
прибор.
При использовании фотоэлектроколориметров выбирают в каче-
стве оптимального такое значение Со, для которого величина а макси-
мальна (см. с. 37—38), а отсчеты Л0Тн удается произвести с точностью
58
абсолютного метода (отклонение стрелки гальванометра на всю шкалу
или максимальное раскрытие сектора индикаторной лампы).
Построение градуировочного графика. В шесть мерных
колб вместимостью по 100 мл вводят с помощью микро-
бюреткп от 1,0 до 1,5 мл стандартного раствора алюми-
ния. Приливают в каждую колбу по 2 мл раствора аскор-
биновой кислоты, 5 мл 0,1 М раствора соляной кислоты,
воду примерно до объема 50 мл, по 5 мл раствора хрома-
зурола S, 5 мл раствора ацетата натрия и доводят до
метки водой. Оптическую плотность растворов измеряют
при % = 545 нм в кюветах (/=10 мм) относительно раство-
ра, содержащего 0,075 мг алюминия в 100 мл. По резуль-
татам измерений строят градуировочный график зависи-
мости оптической плотности от концентрации или вычисля-
ют значение постоянного фактора, которым пользуются
при расчете содержания алюминия в пробе.
Выполнение определения. Навеску сплава 0,100 г ра-
створяют в 2 мл соляной кислоты (1:1), переводят раст-
вор в мерную колбу вместимостью 500 мл и разбавляют
водой до метки. Аликвотную часть раствора (5 мл) поме-
щают в мерную колбу вместимостью 100 мл. Вводят 2 мл
раствора аскорбиновой кислоты, воду примерно до объема
50 мл и далее поступают, как указано при построении гра-
дуировочного графика. Содержание алюминия находят,
используя градуировочный график, или вычисляют по со-
ответствующей формуле.
Хром
Хром открыт в 1797 г. французским ученым А. Вокленом в хро-
мате свинца, встречающемся в природе в виде минерала крокоита.
Название элемента произошло от греческого слова chroma — цвет
(из-за яркой окраски соединений).
Нахождение в природе. В земной коре содержатся значительные
количества хрома (0,02%). Основным сырьем для производства хрома
является хромистый железняк FeO-CrjOp или хромиты, содержание
хрома в которых составляет 15—60%. Хром изоморфно замещают мар-
генец, алюминий, магний, цинк.
Применение. Применение хрома разнообразно. Его используют для
покрытия металлоизделий в целях повышения износоустойчивости.
Добавка хрома улучшает качество сталей; стали с высоким содержа-
нием хрома (14—18%) получили название нержавеющих. Сплавы хро-
ма применяются в печах сопротивления как нагревательные элементы.
Свойства хрома и его соединений. Хром—элемент
VI группы Периодической системы Д. И. Менделеева.
Электронное строение атома в основном состоянии
ls22s22p63s23p63d54s1. Хром может иметь степень окисления
от +2 до +6, в образовании связей, помимо наружного
59
электрона, участвуют электроны предпоследнего недостро-
енного уровня. Наиболее устойчивыми являются степени
окисления +6 и +3. Растворы и твердые соли хрома (II),
как сильные восстановители, неустойчивы на воздухе.
Основные степени окисления хрома (0, +2, +3, +6) мож-
но проследить на схеме: металлический хром Cr^-ион хро-
ма (II) Сг2+->оксид хрома (II) Сг О (основной)-э-ион хрома
(III) Сг3+-^-оксид хрома (III) Сг2О3 (амфотерный), оксид
хрома (VI) СгО3 (кислотный), хромат-ион СгО42~, дихро-
мат-ион Сг2О72'.
Хром — очень прочный металл серебристо-белого цвета
с синеватым оттенком; температура его плавления высокая,
1890 °C. Под действием концентрированной азотной кисло-
ты и царской водки хром покрывается слоем защитной
оксидной пленки. Пассивированный хром в кислотах не
растворяется. В разбавленных серной и соляной кисло-
тах металл растворяется с выделением водорода и образо-
ванием солей хрома. Хром (II) является сильным восста-
новителем и окисляется воздухом до хрома (III): 4Сг +
+ 6H2SO4+О2 = 2Сг2(SO4)з + 2Н2О + 4Н2, 4Сг+12НС1 + О2 =
— 4СгС13+2Н2О + 4Н2.
Соли хрома ярко окрашены вследствие хромофор-
ных свойств самого атома хрома.
Руды хрома и концентраты разлагают смесью сер-
ной и фосфорной кислот, сплавляют с пероксидом натрия
в железных тиглях или с содой в присутствии окислителя.
Щелочное сплавление позволяет отделить хром от элемен-
тов, образующих нерастворимые гидроксиды и основные
соли: 2FeO • Сг2О3 + 7Na2O2 = Fe2O3 + 4Na2CrO4+3Na2O;
Cr2O3 + Na2CO3 = 2NaCrO2 + CO2; Cr2O3 + 2Na2CO3 +
+ 3KNO3 = 2Na2CrO4 + 3KNO2 + 2CO2.
Плав выщелачивают водой. В нерастворимом остатке
содержатся гидроксид железа, карбонаты щелочноземель-
ных элементов и магния. Элементы, образующие кислот-
ные и амфотерные оксиды, переходят в раствор (хром,
ванадий, марганец).
Методы определения содержания хрома
Наибольшее распространение получили титриметриче-
скне и фотометрические методы определения содержания
хрома, а в последнее время все шире применяется атомно-
абсорбционный анализ.
Титриметрические методы. Ведущими среди них явля-
ются методы с применением окислительно-
60
восстановительных реакций. Хром (VI) титруют
раствором соли железа (II) в присутствии окислительно-
восстановительного индикатора (дифениламина, орто-
фенантролина, фенилантраниловой кислоты, 4-арсоно-фе-
нилантраниловой кислоты): 2Na2CrO4+6FeSO4+8H2SO4 =
= Cr2(SO4)3 + 3Fe2(SO4)3 + 2Na2SO4+8H2O. Определению
содержания хрома мешает ванадий.
При анализе металлов, сплавов и руд применяют обрат-
ное титрование, т. е. осуществляют предварительное окис-
ление xpoiw (III) до хрома (VI) прибавлением избытка
титрованного раствора соли железа (II). Избыток добав-
ленного раствора соли железа (II) затем оттитровывают
раствором перманганата калия до появления слабо-розовой
окраски на зеленом фоне раствора: I0FeSO4 + 2KMnO4 +
+ 8H2SO4 = 5Fe2 (SO4) 3 + 2MnSO4 + K2SO4+ 8H2O. Присут-
ствие ванадия при обратном титровании определению со-
держания хрома не мешает. Варианты метода обратного
титрования различаются способами окисления хрома и
разрушения избытка окислителя.
Широко применяется на практике окисление хрома (III)
в сернокислой среде персульфатом аммония в присутствии
катализатора — нитрата серебра или смеси растворов суль-
фатов кобальта и никеля: Cr2(SO4)3 + 3 (NH4)2S2O8 +
+ 7H2O = H2Cr2O7 + 3 (NH4)2SO4 + 6H2SO4. Зеленый цвет
ионов Сг3+ переходит в оранжевый. В присутствии марган-
ца оранжевая окраска хромовой кислоты маскируется фио-
летово-красной окраской марганцовой кислоты. Если мар-
ганец отсутствует, то его специально вводят в раствор
анализируемого образца для контроля полноты окисления
хрома, так как марганец окисляется позже хрома и по-
явление окраски перманганат-иона указывает на заверше-
ние окисления хрома.
Далее хромовую кислоту титруют раствором соли же-
леза (II). Марганцовую кислоту перед титрованием раз-
рушают добавлением соляной кислоты или хлорида на-
трия: 2НМпО4+14НС1 = 2МпС12 + 5С12 + 8Н2О. Содержаще-
еся в растворе серебро связывается в виде труднораство-
римого AgCl. Раствор, имеющий оранжевую окраску,
титруют раствором соли железа (II) в присутствии окисли-
тельно-восстановительного индикатора — фенилантранило-
вой кислоты
/СООН
61
или C13H11D2N (молекулярная масса 213,23). Окисленная
форма индикатора красного цвета, восстановленная —
бесцветная при нормальном окислительном потенциале
ниже +1,08 В. В присутствии Н2СГ2О7 индикатор окисля-
ется и раствор окрашивается в красный цвет. При
титровании раствором соли железа (II) сначала восста-
навливается Н2СГ2О7, а затем индикатор; окраска раство-
ра при этом переходит в зеленую (прямое титрование).
Комплексонометрический метод Ионы хро-
ма (III) медленно взаимодействуют с комплексоном III
при кипячении в слабокислой среде (pH 5,3—6) с образо-
ванием интенсивно окрашенного фиолетового комплекса.
Окраска комплекса мешает определению точки эквивалент-
ности. Применение флуоресцирующих индикаторов позво-
ляет устранить эти затруднения и определить комплексо-
нометрически значительные количества хрома обратным
титрованием (прямое его титрование невозможно).
Фотометрические методы определения содержания хро-
ма основаны на собственной окраске хрома (VI) или реже
хрома (III), а также на образовании комплексных окра-
шенных соединений с комплексонами и органическими ве-
ществами других классов. Для большого числа материа-
лов, содержащих хром, разработаны методы определения
его содержания дифенилкарбазид'ом или (C6H5NHNH)2CO
О
NH-NH-C-NH-NH 1
(молекулярная масса 242,27) — розовато-белого кристал-
лического вещества. Его растворяют в ацетоне, водно-аце-
тоновой среде или в спирте. Реагент взаимодействует с
хромом (VI), окисляясь до дифенилкарбазона:
G6H5NHNH)2C0 + Crof + 8Н+ = Сг3++ 4Н20+
о
NH-NH-C-N = N-£2^ .
Окисленная форма реагента взаимодействует с катио-
ном Сг3+, образуя окрашенное соединение. Фиолетовое
комплексное соединение хрома III с дифенилкарбазоном
характеризуется молярным коэффициентом погашения при
Zmax = 546 нм (рис. 5), равным 4,17-104. Комплексное сое-
динение устойчиво в 0,025—0,1 М растворе серной кислоты
62
В более кислой среде оно разрушается. Мешающие опре-
делению содержания хрома железо (III), молибден, мар-
ганец, ртуть предварительно отделяют или маскируют
Рис. 5. Кривая светО'
поглощения раствора со*
единения дифенилкар-
базида с хромом
различными комплексообразующими веществами. В при-
сутствии ванадия (V:Cr< 10:1) измеряют А через 10 мин.
Практическая часть
Титриметрическое определение хрома
в хромовых рудах и концентратах
Принцип метода. Определение основано на окислении
хрома (Ш) в сернокислой среде персульфатом аммония
в присутствии катализатора — нитрата серебра или смеси
растворов сульфатов кобальта и никеля.
Хром (VI) титруют раствором соли Мора (NH4)2
[Fe(SO4)2] в присутствии в качестве индикатора фени-
лантраниловой кислоты. При содержании 0,05%V хром
восстанавливают раствором соли Мора и его избыток тит-
руют раствором перманганата калия. Метод применим при
содержании 10—65%Сг в пробе. Относительное стандарт-
ное отклонение результатов определений 0,015 при содер-
жании 10—65% хрома.
Реактивы и растворы. 1). Кислота азотная, р = 1,40 г/см3; 2) ки-
слота ортофосфорная, р=1,70 г/см3; 3) кислота серная, р=1,84 г/см3
и разбавленная 1:1, 1:4, 1:100; 4) сульфат марганца, раствор 1 г/л;
5) нитрат серебра, раствор 1 г/л (для большей устойчивости раствора
к нему добавляют 0,5 мл азотной кислоты на каждый литр раствора
и хранят в склянке из темного стекла); 6) сульфат кобальта; 7) суль-
фат никеля; 8) кобальто-никелевый катализатор, раствор (для его
приготовления 15 г сульфата кобальта и 15 г сульфата никеля по-
мещают в стакан вместимостью 700—800 мл, наливают 500 мл воды
и перемешивают до растворения солей); 9) персульфат аммония, ра-
створ 25%-ный (годен к применению в течение 7—10 сут); 10) хло-
рид натрия, раствор 50 г/л; И) карбонат натрия безводный, раствор
0,2%-ный; 12) кислота фенилантраииловая, раствор (содержит 2 г, кнс-
63
лоты в 100 мл раствора карбоната натрия); 13) пероксид натрия;
14) соль Мора, 01 н, титрованный раствор (для его приготовления
39,5 г солн Мора растворяют в 250 мл разбавленной 1:4 серной
кислоты; раствор фильтруют в мерную колбу вместимостью 1000 мл,
доливают водой до метки и перемешивают); 15) калия дихромат,
0,1 н. титрованный раствор (для его приготовления 4,9032 г дихро-
мата калия, дважды перекристаллизованного и высушенного в течение
2—3 ч при 150—170 °C, растворяют в 500—600 мл воды в мерной
колбе вместимостью 1000 мл, доливают водой до метки и перемеши-
вают); 16) перманганат калия, 0,1 н, титрованный раствор (для его
приготовления 32 г перманганата калия растворяют в 1000 мл воды;
раствор переливают в бутыль вместимостью 10 л из темного стекла,
добавляют 9 л воды, перемешивают и оставляют на 7—10 сут); ра-
створ переливают в другую бутыль из темного стекла, пользуясь
сифоном, не доходящим до дна первой бутыли на 15 мм; вместо
сифонирования можно применять фильтрование через прокаленный
асбест); 17) оксалат натрия, перекристаллизованный и высушенный
при 105—НО °C до постоянной массы.
Титр раствора соли Мора устанавливают по дихромату калия;
25 мл 0,1 н раствора дихромата калия помещают в коническую колбу
вместимостью 500 мл, приливают 200 мл воды, 40 мл разбавлен'ной
1:1 серной кислоты. Раствор перемешивают, охлаждают, прибавляют
к нему 5—6 капель раствора фенилантраниловой кислоты. Раствор
снова перемешивают и медленно титруют раствором соли Мора до
перехода его вишневой окраски в зеленую.
Титр раствора соли Мора (7), выраженный в граммах оксида
хрома, вычисляют по формуле Т = 0,1226 -0,5166/V, в которой 0,1226 —
масса дихромата калия, содержащаяся в 25 мл раствора, взятого
для установления титра, г; 0,5166 — коэффициент пересчета дихромата
калия на оксид хрома; V—объем 0,1 н. раствора соли Мора, из-
расходованный на титрование, мл.
Титр раствора перманганата калия устанавливают по оксалату
натрия: 0,2 г оксалата натрия помещают в коническую колбу вмести-
мостью 250 мл и растворяют при слабом нагревании в 75 мл воды.
Приливают к раствору 15 мл разбавленной 1:1 серной кислоты, нагре-
вают его до 70—80 °C и титруют раствором перманганата калия до
появления розовой окраски, устойчивой в течение 1—2 мин. Титр
0,1 н. раствора перманганата калия (Ti), выраженный в граммах ок-
сида хрома, вычисляют по формуле Ti = G• 0,3781/V, в которой G — на-
веска оксалата натрия, г; V — объем раствора перманганата калия,
израсходованный на титрование, мл; 0,3781 — коэффициент пересчета
оксалата натрия на оксид хрома.
Поправочный коэффициент раствора соли Мора по раствору пер-
манганата калия устанавливают следующим образом: в коническую
колбу вместимостью 250 мл наливают из бюретки 20 мл раствора соли
Мора, приливают 50—60 мл воды и титруют 0,1 н. раствором
перманганата калия до появления слабо-розовой окраски, устойчивой в
течение 1 мин.
Выполнение определения. При анализе руд и концент-
ратов, содержащих <0,05%V, навеску разлагают в смеси
серной и ортофосфорной кислот; 0,05—0,2 г хромовой руды
или концентрата помещают в коническую колбу вмести-
мостью 500 мл, смачивают водой, приливают 10 мл орто-
фосфорной кислоты, перемешивают, приливают 20 мл
64
серной кислоты, вновь перемешивают и нагревают до раз-
ложения при периодическом перемешивании содержимого
колбы.
Полученный раствор охлаждают, приливают 300—
350 мл воды и перемешивают. Затем приливают 1 мл раст-
вора сульфата марганца, 10 мл раствора нитрата серебра
или 10 мл раствора кобальто-никелевого катализатора,
25 мл раствора персульфата аммония и нагревают до по-
явления малиновой окраски, что указывает на полное
окисление хрома. Раствор кипятят 12—15 мин (до пре-
кращения выделения пузырьков кислорода), прибавляют
к нему 10 мл раствора хлорида натрия и снова кипятят
до исчезновения малиновой окраски. После этого раствор
охлаждают, приливают к нему 25 мл разбавленной 1:1
серной кислоты. Раствор вновь охлаждают, вводят 5—6 ка-
пель фенилантраниловой кислоты и титруЮт раствором
соли Мора до перехода вишневой окраски раствора в зе-
леную.
Для руд и концентратов, содержащих >0,05%V, ана-
лиз проводят так же, как и в рассмотренном выше случае
до момента исчезновения малиновой окраски раствора. За-
тем раствор охлаждают и приливают к нему раствор соли
Мора до изменения желтой окраски на зеленую, после
чего вводят 5—6 мл избытка соли Мора, который оттитро-
вывают раствором перманганата калия до появления ро-
зовой окраски, устойчивой в течение 1—2 мин. 1 мл 0,1 н.
соли Мора соответствует 1,7332 мг хрома; 1 мл 0,1 н. пер-
манганата калия соответствует 1,7332 мг хрома.
Фотометрическое определение хрома
в сплавах на основе меди
Принцип метода. Определение основано на реакции
образования окрашенного комплексного соединения хрома
(III) с комплексоном III. Медь удаляют из раствора с
помощью электролиза. Определению не мешают титан (IV),
а также железо (III) (до 1 мг), никель (II), кобальт (II),
марганец (II), алюминий (до 10 мг).
Относительное стандартное отклонение результатов
определения составляет 0,05 при содержаниях хрома 0,5—
2 %.
Реактивы и растворы. 1) Аммиак, разбавленный 1:1; 2) комплек-
сон III (5%-ный); 3) кислота азотная, р = 1,40 г/см3, разбавленная
1:1; 4) кислота серная, р=1,84 г/см3, разбавленная J: 1; 5) кислота
уксусная, р = 1,05 г/см3, 2 М раствор; 6) фенолфталеин; 7) рабочий
раствор хрома (для его приготовления растворяют 1 г металлического
3- m 65
хрома в нескольких миллилитрах серной кислоты и доводят объем
раствора в мерной колбе водой до 1000 мл. В 1 мл раствора содер-
жится 1 мг хрома).
Построение градуировочного графика. В ряд стаканчи-
ков вместимостью по 50 мл вводят от 0,2 до 2,0 мг хрома
с интервалом в 0,2 мг, добавляют по 2 мл серной кислоты,
раствора комплексона III. Растворы нейтрализуют по
фенолфталеину аммиаком до появления бледно-розовой
окраски, после чего добавляют по 5 мл уксусной кислоты,
доводят водой примерно до 25 мл, нагревают до кипения
и кипятят 5 мин. По охлаждении растворы переводят в
мерные колбы вместимостью 50 мл и доводят до метки
водой. Измеряют оптическую плотность растворов при
Л=550 нм в кювете с / = 50 мм. В качестве раствора сравне-
ния применяют раствор контрольного опыта. По получен-
ным данным строят график зависимости оптической плот-
ности от концентрации хрома, отдельные точки которого
периодически проверяют.
Выполнение определения. Навеску сплава 0,5 г раство-
ряют при нагревании в 25 мл смеси кислот (60 мл азотной
кислоты и 40 мл серной кислоты); нагревание продолжают
до выделения паров серной кислоты. По охлаждении раст-
вор разбавляют водой до 70—75 мл, нагревают до 60—
70 °C и проводят выделение меди электролизом при силе
тока 2А и напряжении 2,5 В до обесцвечивания раствора.
По окончании электролиза раствор переводят в мерную
колбу вместимостью 100 мл и доводят до метки водой.
Отбирают аликвотную часть раствора, равную 20 мл, и
переводят ее в стаканчик вместимостью 50 мл. Туда же
добавляют 2 мл раствора комплексона III, 4—5 капель
раствора фенолфталеина и нейтрализуют аммиаком до по-
явления в растворе бледно-розовой окраски и далее по-
ступают так же, как описано при построении градуировоч-
ного графика. Содержание хрома определяют, используя
градуировочный график.
Кобальт
Металлический кобальт получен в 1735 г. шведским ученым
Г. Брандтом. Соединения кобальта с древних времен применяются
для окрашивания стекол в синий цвет. Назван элемент по имени ми-
фологического злого духа Кобальда, по-видимому, в связи с тем, что
при обжиге кобальтовых руд, являющихся в основном арсенидами,
выделяются ядовитые соединения мышьяка.
Нахождение в природе. В земной коре содержится 0,00-1% кобаль-
та. Важнейшими минералами кобальта являются: кобальтин (кобаль-
товый блеск) CoAsS, эритрин (кобальтовый цвет) Соз(А5О4)2-8Н2О,
66
асболан Со2Оз-СоО-рМпО2-дРе20-з'ГСН2О. В виде примеси кобальт
содержится в полиметаллических, марганцовых, железных, никелевых,
медных и серебряных рудах, которые служат источником его добы-
вания.
Применение. Устойчивость металлического кобальта против корро-
зии, высокая температура плавления позволяют применять его в ка-
честве добавки для улучшения свойств стали, для изготовления сплавов
различного назначения (космическая техника, атомная энергетика).
Широко применяются кобальтовые катализаторы.
Свойства кобальта и его соединений. Кобальт — эле-
мент VIII группы Периодической системы Д. И. Менделе-
ева, составляет наряду с железом и никелем первую триа-
ду этой группы.
Электронное строение атомов в основном состоянии
/s22s22p63s23p63d74s2. Наиболее устойчивой является степень
окисления +2, в некоторых соединениях, особенно в ком-
плексных, кобальт проявляет степень окисления +3 и +4.
Кобальт — металл стального цвета с синим отливом.
Температура его плавления 1493 °C.
В разбавленной азотной кислоте кобальт растворяется
с выделением оксидов азота (II): 3Co+8HNO3 =
= 3Co(NO3)2 + 2NO + 4H2O; в концентрированной азотной
кислоте выделяется оксид азота (IV): Co + 4HNO3 =
= Со (NO3) 2+ 2NO2+2Н2О.
Дымящая азотная и концентрированная серная кисло-
ты его пассивируют. В разбавленных серной и соляной
кислотах кобальт образует соли кобальта (II): Со +
-bH2SO4 = CoSO4+H2, Со+2НС1 = СоС12 + Н2. Фтористо-
водородная кислота и щелочь практически не действуют на
кобальт.
Гидроксид кобальта Со(ОН)2 образуется при
действии щелочи на растворы солей кобальта (II). Вна-
чале образуется синяя основная соль которая переходит в
розовый гидроксид. Последний растворим в кислотах и
аммиаке с образованием комплексных аммиакатов различ-
ного состава и окраски: CoSO4 + 2NaOH = Co(OH)2+
4-Na2SO4; Co(OH)2+6NH4OH= [Co(NH3)6] (OH)2 + 6H2O.
В большом избытке щелочи образуются кобальтиаты
синего цвета К2[Со(ОН)4].
Гидроксид кобальта Со(ОН)з—аморфное вещество
темно-коричневого цвета, образуется при действии окисли-
телей на растворы солей кобальта (II), является промежу-
точным продуктом при получении Со3О4 и солей кобаль-
та (III).
Оксид кобальта (II) СоО — порошок зеленого
цвета, образующийся при прокаливании гидроксида ко-
3* 67
бальта, а также при восстановлении оксидов кобальта (III)
в токе водорода. Как и гидроксид, оксид кобальта обла-
дает основными свойствами.
Оксид кобальта (III) Со2О3 существует в виде-
моногидрата коричневого или черного цвета, образуясь при
воздействии на оксид — кобальта (II) сильных окислите-
лей. Оксид кобальта (III) растворим в соляной кислоте с
образованием соли в степени окисления +2 и выделением,
хлора.
Оксид кобальта (II—III) Со3О4— черное крис-
таллическое вещество, получаемое из оксида кобальта (II)
насыщением кислородом или прокаливанием моногидрата
Со(Ш) Со2Оз-Н2О. Оксид кобальта (II—III) растворим
в соляной кислоте с образованием тех же продуктов реак-
ции, что и при растворении оксида кобальта (III). Оксид
кобальта (II—III) является конечным продуктом гидро-
металлургической стадии переработки кобальтсодержащих
руд, из него получают металлический кобальт.
Кобальтовые руды предварительно обжигают для уда-
ления основной массы мышьяка и серы. Не разлагаемые
кислотами руды сплавляют со щелочами в железных
тиглях.
Кобальтовые сплавы с цветными металлами и сульфид-
ные руды разлагают смесью азотной и соляной кислот:
3CoS + 6HC1 + 2HNO3 = 3CoC12 + 2NO + 3S + 4H2O.
Методы определения содержания кобальта
Ион кобальта (II) характеризуется способностью обра-
зовывать растворимые комплексные соединения в избытке
аммиака, экстрагирующиеся органическими растворителя-
ми комплексные соединения с роданид-ионом. Селективны-
ми реактивами, позволяющими определять кобальт в при-
сутствии других элементов (меди, никеля, железа), явля-
ются оксинитрозосоединения. В зависимости от содержания
кобальта в анализируемом объекте (оно колеблется от
десятых долей до десятков процентов) применяют титри-
метрические, фотометрические, полярографические и атом-
но-абсорбционные методы. Сравнительно редко прибегают
к гравиметрическим и люминесцентным методам опреде-
ления содержания кобальта.
Фотометрические методы получили наибольшее прак-
тическое применение для определения содержания кобаль-
та. При этом методы, основанные на измерении светопо-
глощения сульфатных, хлоридных, цианатных и оксалат-
68
ных комплексов, как малочувствительные, используются
мало. Более чувствителен и часто применяем роданидный
метод, однако и он уступает по чувствительности методам,
в которых используются органические реактивы. Высокой
чувствительностью и избирательностью обладают оксинит-
розосоединения. Оксинитрозосоединения в форме моноок-
сима дают с кобальтом соединения в кислой и щелочной
средах. Соединения, образованные в кислой среде, экстра-
гируются органическими растворителями. При этом избы-
ток реактива, перешедший в органическую фазу, переводят
снова в водную фазу действием 2 М щелочи. Наиболее
употребимы 1-нитрозо-2-нафтол (рис. 6, кривые 1,2), а так-
же более чувствительные 2-нитрозо-1-нафтол (рис. 7, кри-
вые 3,4) и нитрозо-И-соль (рис. 7).
1.1-Нитрозо-2-нафтол
NO
или C10H7O2N (молекулярная масса 173,17)—оранжево-
бурый порошок, растворимый в воде, легко растворимый
в спирте, растворах щелочей, эфире, бензоле, в ледяной
уксусной кислоте. Нитрозонафтолы окисляют кобальт (II)
нис. б. Кривые светопоглощения растворов I-нитрозо-2-нафтола (7) и его соеди-
нения с кобальтом (2); 2-нитрсзо-1-нафтола (<?) и его соединения с кобаль-
том (4)
Рис. 7. Кривые светопоглощения раствора нитрозо-Р-соли (/) и ее соединения
с кобальтом (2) (относительно раствора реагента)
до кобальта (III). 1-Нитрозо-2-нафтол применяют для вы-
деления и определения больших и малых количеств ко-
бальта.
69
Определение микроколичеств кобальта проводят, приме-
няя экстракцию внутрикомплексного соединения кобальта
в хлороформ, из уксуснокислой среды (pH 2,5ч-5). Мак-
симальное светопоглощение экстракта оранжевого цвета
наблюдается при длине волны 415 нм. Градуировочный
график в области концентраций кобальта 10—50 мкг в
объеме 25 мл при толщине поглощающего слоя 5 мм имеет
вид прямой линии. Молярный коэффициент погашения
комплекса при лтах = 415 нм составляет 2,9-104. Окрашен-
ные соединения с реагентом образуют медь, железо (II), ни-
кель (II), их разрушают кипячением с 1 М азотной кисло-
той после прибавления реагента. Собственную окраску же-
леза (III) маскируют фосфат-ионом.
Большие количества меди и никеля можно удалить аце-
татным гидролизом перед прибавлением реагента. Марга-
нец (II) окисляют азотной кислотой. Не мешают опреде-
лению содержания кобальта цинк, алюминий, титан.
В настоящее время перед определением кобальта нит-
розонафтолами часто отделение мешающих элементов про-
водят с помощью анионного обмена: кобальт вместе с
медью, цинком, железом, свинцом сорбируют на анионите
(АВ-17) из 8—9 М раствора соляной кислоты. При этом
происходит отделение никеля. Затем десорбируют ко-
бальт 4 М раствором соляной кислоты. Вместе с кобаль-
том извлекается 20—25% железа, которое при последую-
щем определении маскируют фосфат-ионом.
2. Нитрозо-И-соль (1-нитрозо-2-нафтол-3,6-дисульфокис-
лоты динатриевая соль), существует в виде двух изомер-
ных соединений:
N-ОН
или C!0H5O8NS2Na2 (молекулярная масса 377,3) — кристал-
лическое вещество желтого цвета, растворимо в воде, эти-
ловом и метиловом спиртах. Образует при pH 5 с кобаль-
том (Ш) комплексное соединение красного цвета, раство-
римое в воде, устойчивое при подкислении.
Светопоглощение растворов комплекса кобальта изме-
ряют пли при 7тах = 415 нм (молярный коэффициент по-
гашения 3,5-10*), или при ?. = 500 нм с меньшей чувстви-
тельностью (молярный коэффициент погашения 1,5-104).
Во втором случае влияние собственной окраски реагента
70
меньше, и в качестве раствора сравнения можно приме-
нять воду.
Гравиметрические методы определения. Красный оса-
док соединения кобальта (III) с 1-нитрозо-2-нафтолом при-
мерного состава Со(СюНб02П)з-пН20 образуется в слабо-
кислых (pH 3,8—4,0), нейтральных и аммиачных
растворах. Образовавшееся соединение при подкислении
не разрушается. Мешают осаждению кобальта серебро,
висмут и олово. Железо и вольфрам можно маскировать
фторид-ионом. Не мешают осаждению кобальта равные по
содержанию количества никеля, алюминия, кадмия, каль-
ция, магния, бериллия, хрома, свинца, марганца, цинка,
сурьмы, мышьяка, ртути. В присутствии больших количеств
никеля проводят переосаждение кобальта. После высушива-
ния при 115 °C состав соединения становится постоянным
(п = 2), и оно применимо для гравиметрического опреде-
ления содержания кобальта. В некоторых случаях отделе-
ние Со от сопутствующих элементов проводят осаждением
в виде кобальтинитрита (гексанитрокобальтата III) калия:
2Co(N02)3+6KN024-3H20 = 2K3[Co(N02)6]-3H20.
Желтый осадок кобальтинитрита калия образуется в
уксуснокислом растворе в присутствии больших количеств
никеля, железа, цинка, марганца и многих других эле-
ментов, имеет постоянный состав и применяется для гра-
виметрического определения содержания кобальта. Оса-
док легко растворим в минеральных кислотах. Получен-
ный раствор можно использовать для определения содер-
жания кобальта фотометрическим методом.
Титриметрические методы определения. Применяется
титрование кобальта неорганическими и органическими
реактивами. Кобальт можно титровать такими органически-
ми реактивами, как 1-нитрозо-2-нафтол, диметилглиоксим,
нитрозохромотроповая кислота, комплексон III и др. Точ-
ка эквивалентности устанавливается амперометрически,
фототурбидиметрически и визуально. В комплексонометри-
ческих методах индикаторами служат мурексид и ксиле-
ноловый оранжевый.
1. Мурексид (аммонийная соль пурпуровой кислоты)
ONH+ 0ч
/ %
NH-C C-NH
/ / \
0=С C-N=c С=0
NH-cf \-NH
71
или СвНзОбЫб-НгО (молекулярная масса 302,21) порошок
красно-фиолетового цвета. Водные растворы реагента не-
устойчивы, особенно в кислой среде. Его применяют при
комплексонометрических титрованиях в щелочной среде в
виде смеси реагента с хлоридом натрия (соотношение
1:100) в количестве 0,2—0,3 г на каждое определение.
С ионами кобальта (II), никеля (II), меди (II) в щелоч-
ной среде (pH 8—10) мурексид образует комплексные сое-
динения желтого цвета. При титровании комплексоном III
комплексы разрушаются, в точке эквивалентности окрас-
ка раствора переходит в фиолетовую.
Определению содержания кобальта мешает большое
количество аммиака, разлагающее комплекс с мурексидом
вследствие образования аммиаков.Поэтому при нейтрали-
зации кислых растворов следует проявлять осторожность.
Кроме того, в присутствии больших количеств аммиака
Со2+ легче окисляется до Со3+ кислородом воздуха. Ще-
лочноземельные элементы определению содержания ко-
бальта не мешают.
2. Ксиленоловый оранжевый (см. раздел «Алюминий»)
образует с ионами кобальта в уксуснокислой среде комп-
лекс фиолетового цвета. При титровании раствором комп-
лексона III комплекс разрушается, и в точке эквивалент-
ности окраска раствора переходит в оранжевую. Опреде-
лению кобальта мешают многие элементы.
При анализе металлургических образцов широкое при-
менение находит потенциометрический метод титрования
кобальта гексацианоферратом (III) калия (феррициани-
дом калия) в аммиачном растворе: [Fe(CN)6] 3- +
+ [Со (NH3)6] 2+= [Fe(CN)6]-4+ [Co(NH3)e]3+.
Аммиачный комплекс иона Со3+ окрашен в красно-бу-
рый цвет. Окисление Со2+ происходит вследствие различия
нормальных окислительных потенциалов систем:
для [Fe(CN)6]3-/[Fe(CN)6]4- Ео= - 0,36В;
для [Co(NH3)6]3+/[CO(NH3)g]2+ Ео=О,1ОВ.
Определению кобальта не мешают большие количества
никеля, цинка, меди (II), мышьяка (V); допустимы значи-
тельные количества железа (III).
Мешают определению кобальта марганец (его влияние
устраняют добавлением фторид-ионов), железо (II) и
большие количества железа (III). Гидроксид железа (III)
сорбирует ионы кобальта. Во избежание этого добавляют
комплексообразующие вещества — лимонную кислоту, свя-
зывающую железо в неосаждаемые аммиаком соединения.
72
Йодометрическое титрование кобальта выполняют, окис-
ляя Со (II) в нейтральном растворе, например, перокси-
дом водорода. При этом выпадает осадок Со(ОН)3. Избы-
ток окислителя разрушают, прибавляют иодид калия и
титруют выделившийся иод раствором тиосульфата натрия.
Атомно-абсорбционный метод определения содержания
кобальта применяют при анализе горных пород, минера-
лов, сплавов и чистых металлов. Оптимальной является
линия 240,7 нм, по которой возможно определение
>0,13 мкг/мл. Лампы повышенной яркости позволяют
обнаружить 0,01 мкг/мл. В воздушно-ацетиленовом пла-
мени определению содержания кобальта никель, хром,
медь и другие элементы практически не мешают.
Практическая часть
Фотоколориметрическое определение кобальта
в медных концентратах
Принцип метода. Метод основан на измерении свето-
поглощения окрашенного в красный цвет соединения ко-
бальта (III) с нитрозо-К-солью. Кобальт предварительно
отделяют от мешающих определению элементов осажде-
нием в виде кобальтинитрита. Метод применим при содер-
жании кобальта 0,03—0,15%. Относительное стандартное
отклонение результатов определения составляет 0,1.
Реактивы и растворы: 1) Кислота азотная, р=1,40 г/см3, разбав-
ленная 1:1 (перед разбавлением кислоту кипятят для удаления окси-
дов азота); 2) кислота серная, р=1,84 г/см3, разбавленная 1:99';
3) кислота соляная, р=1,19 г/см3, разбавленная 1:1; 4) кислота уксус-
ная, р=1,04 г/см3; 5) аммиак, р = 0,91 г/см3; 6) нитрит калия, 50 и
2%-ные растворы, подкисленные уксусной кислотой; 7) карбонат ка-
лия (поташ), насыщенный раствор; 8) нитрат калия; 9) сульфат ко-
бальта, перекристаллизованный и высушенный на воздухе; 10) ацетат
натрия, 50%-ный раствор; И) фторид натрия; 12) нитрозо-И-соль,
0,1%-ный раствор; 13) рабочий раствор кобальта, содержащий в 1 мл
0,1 мг кобальта (для его приготовления 0,4767 г перекристаллизован-
ного сульфата кобальта помещают в стакан, прибавляют 50 мл воды
и перемешивают до растворения соли, затем переливают раствор в
мерную колбу вместимостью 1000 мл, доливают разбавленной серной
кислотой до метки и перемешивают).
Построение градуировочного графика. В пять мерных
колб вместимостью 50 мл отмеривают 1; 2; 3; 4; 5 мл ра-
бочего раствора кобальта. Шестая колба служит для про-
ведения контрольного опыта. В нее помещают 5 мл раз-
бавленной серной кислоты. В колбы прибавляют аммиак
до pH 5—6 (по универсальной индикаторной бумаге),
приливают 5 мл раствора ацетата натрия и кипятят в те-
73
чение 2—3 мин. Затем к растворам прибавляют 10 мл
раствора нитрозо-Н-соли, кипятят 1—2 мин, после чего
приливают 5 мл разбавленной 1:1 азотной кислоты, снова
кипятят раствор в течение 1 мин, охлаждают, доливают до
метки водой и перемешивают.
Оптическую плотность растворов измеряют на фото-
электроколориметре, применяя светофильтр с областью
светопропускания 520—540 нм в кювете с / = 50 мм.
Выполнение определения. Навеску концентрата 0,5—
1 г помещают в стакан (250—300 мл), смешивают с 0,4—
0,5 г фторида натрия, приливают 25—30 мл соляной кис-
лоты п нагревают 20 мин. Затем к раствору порциями
приливают 13—15 мл азотной кислоты, нагревают раствор
до исчезновения темных частиц и выпаривают до получе-
ния влажных солей. Выпаривание повторяют и прибавляют
5 мл соляной кислоты. К остатку от выпаривания прили-
вают 5 мл соляной кислоты, 40 мл горячей воды, кипятят
13—15 мин, затем охлаждают и фильтруют раствор через
фильтр средней плотности в стакан вместимостью 250 мл.
Осадок на фильтре промывают 6 раз водой, собирая
фильтрат в тот же стакан.
К фильтрату, объем которого должен быть примерно
100 мл, прибавляют небольшими порциями раствор кар-
боната калия до появления слабого помутнения, не исче-
зающего при сильном перемешивании, затем приливают
15 мл уксусной кислоты и перемешивают до полного про-
светления раствора.
К раствору прибавляют 25 мл 50%-ного раствора нит-
рита калия, тщательно перемешивают, прибавляют 5 г
нитрата калия, снова перемешивают и оставляют раствор
с осадком кобальтинитрата калия на 1 ч. Затем осадок
отфильтровывают через плотный фильтр, содержащий
небольшое количество фильтробумажной массы, и промы-
вают 2%-ным раствором нитрита калия до полного обес-
цвечивания промывных вод.
Осадок на фильтре растворяют в 20—25 мл горячей
разбавленной 1:1 соляной кислоты, приливаемой порциями
по 4 5 мл. Раствор собирают в стакан, в котором произ-
водилось осаждение. Фильтр промывают 5 раз горячей
разбавленной 1:99 соляной кислотой. Затем раствор вы-
паривают до получения влажных солей, прибавляют 5 мл
соляной кислоты и нагревают до полного растворения со-
лей. После этого к раствору приливают 25—30 мл воды,
охлаждают, переливают в мерную колбу вместимостью
100 мл. доливают до метки водой и перемешивают.
74
Аликвотную часть раствора 5—10 мл переносят в мер-
ную колбу вместимостью 50 мл, прибавляют аммиак до
pH 5—6 (по универсальной индикаторной бумаге) п да-
лее поступают, как описано при построении градуировоч-
ного графика.
Потенциометрическое определение кобальта
в сплавах никеля
Принцип метода. Определение основано на титровании
кобальта (II) в аммиачном растворе феррицианидом ка-
лия. Определению содержания кобальта мешают марга-
нец (II), железо (Ш). Предварительно марганец (II) окис-
ляют перманганатом в присутствии фторид-ионов. Желе-
зо и другие сопутствующие элементы маскируют цитратом
калия. Относительное стандартное отклонение результатов
определений 0,03 при содержаниях — 3% кобальта.
Реактивы и растворы. 1) Аммиак р = 0,91 г/см3, разбавленный 1:1;
2) фторид аммония; 3) хлорид аммония; 4) нитрат калия; 5) кислота
азотная, р = 1,40 г/см3; 6) кислота серная, р = 1,84 г/см3, 2 М раствор;
7) кислота соляная, р=1,19 г/см3, разбавленная 1:1; 8) перманганат
калия, 0,01 н, титрованный раствор; 9) смесь азотной и соляной кис-
лот 1:3; 10) феррицианид калия, раствор 0,05 М.
Выполнение определения. Навеску сплава 2 г помещают
в стакан вместимостью 250 мл и растворяют при нагрева-
нии в 30—50 мл смеси кислот. Раствор выпаривают до
сиропообразного состояния, обмывают стенки стакана
15 мл воды и нагревают до полного растворения солей.
Раствор нейтрализуют разбавленным аммиаком до выпа-
дения осадка гидроксидов, который растворяют в разбав-
ленной соляной кислоте. К полученному раствору прибав-
ляют 7,5 мл 2 М серной кислоты, 60 мл воды, 3 г фторида
аммония, охлаждают до 20—22 °C и титруют марганец
0,01 М раствором перманганата калия с применением пары
электродов платина'—вольфрам до отклонения стрелки
гальванометра. (В случае отсутствия в образце марганца
операцию титрования перманганатом исключают). Затем
к раствору прибавляют 10 г цитрата калия, 25 г хлорида
аммония, 25—30 мл аммиака и титруют кобальт раство-
ром феррицианида калия с применением той же пары
электродов. 1 мл 0,05 М раствора феррицианида калия
соответствует 2,9467 мг кобальта.
75
Атомно-абсорбционное
определение кобальта в никеле
Принцип метода. Образец растворяют кислотным спо-
собом, раствор фотометрируют в ацетилено-воздушном
пламени по линии 240,72 нм. Градуировочный график
строят по водным растворам соли кобальта при содержа-
нии 4—40 мг/л кобальта. Относительное стандартное от-
клонение результатов определений 0,05—0,01 при содер-
жании кобальта 0,005—0,8%.
Реактивы и растворы. 1) Кислота серная, р= 1,84 г/см3; 2) кислота
фосфорная, р=1,70 г/см3; 3) кислота азотная, р = 1,40 г/см3; 4) раство-
ры соли кобальта, содержащие в 1 мл 4—40 мкг кобальта; 5) смесь
серной и фосфорной кислот 1:1.
Выполнение определения. Навеску никеля 0,5 г раст-
воряют в 20 мл смеси серной и фосфорной кислот, окис-
ляют раствор несколькими каплями азотной кислоты, до-
водят его, объем в мерной колбе до 100 мл. Фотометри-
руют полученный раствор, и содержание кобальта опреде-
ляют, используя градуировочный график.
Никель
Никель был открыт в 1751 г. шведским химиком А. Кронштедтом,
предложившим название элемента по имени мифологического злого
духа Ника. В XVII в. из руды красного цвета состава NiAs неудачно
пытались получить медь и назвали ее купферникель (негодная медь),
обвинив в неудаче духа Ника.
Нахождение в природе. Содержание никеля в земной коре состав-
ляет 0,01 %. Промышленное значение имеют сульфидные медно-никеле-
вые руды. Основным минералом сульфидных руд является пентлан-
дит (Fe, Ni)9S8. Наряду с никелем и медью из медно-никелевых руд
получают платиновые, редкие, рассеянные элементы, золото, серебро,
кобальт.
Применение. Никель используют в качестве защитного покрытия
(никелирование), для изготовления катализаторов и сплавов различ-
ного назначения — жаропрочных, магнитных, не расширяющихся при
нагревании.
Свойства никеля и его соединений. Никель — элемент
VIII группы Периодической системы Д. И. Менделеева,
входит наряду с железом и кобальтом в первую триаду
элементов этой группы. Электронное строение атомов в
основном состоянии—ls22s2p63s23p63d84s2. Устойчивой яв-
ляется степень окисления +2, но в комплексных соедине-
ниях проявляется степень окисления +3 и +4.
Металлический никель получают в виде порошка чер-
ного цвета или в виде слитков. Металл в слитках имеет
серебристо-белый цвет. Температура плавления никеля
76
1453 °C, он легко растворяется в разбавленной азотной и
соляной кислотах, а также в аммиаке в присутствии кар-
боната аммония: 3Ni + 8HNO3 = 3Ni(NO3)2 + 2NO + 4H2O;
Ni + 2HCl = NiCl2 + H2.
Щелочи на никель не действуют, концентрированная
азотная кислота его пассивирует.
Гидроксид никеля Ni(OH)2 выделяется при
pH 7 из растворов никелевых солей под действием щело-
чи. Гидроксид никеля имеет желтоватый цвет, переходя-
щий в зеленый со временем, легко растворяется в аммиа-
ке и солях аммония с образованием комплексного иона
[Ni(NH3)e]2+, окрашенного в синий цвет. Комплексный
аммиакат никеля обесцвечивается при действии цианида
калия вследствие образования цианистого комплекса ни-
келя: [Ni(NH3)62++4KCN = K2[Ni(CN)4] +6МНз + 2К+.
Эта реакция служит основой титриметрического цианидного
метода определения содержания никеля.
Действие щелочи на соли, комплексные аммиакаты и
цианиды никеля в присутствии сильных окислителей при-
водит к образованию черного осадка соответствующего
гидроксида никеля Ni(OH)3. Осаждение Ni(OH)3 едким
кали в присутствии бромной воды с последующим прока-
ливанием осадка до NiO применяется в аналитической
химии для гравиметрического определения содержания
никеля: NiCl2 + 2KOH = Ni(OH)2 + 2KCl; 2Ni(OH)2 + Br2 +
+ 2KOH = 2Ni(OH)3 + 2KBr.
Окси д н и к е л я NiO — зеленое кристаллическое ве-
щество, получаемое прокаливанием солей никеля. В метал-
лургии оксид никеля является конечным продуктом пере-
работки никелевого концентрата и исходным продуктом
для выплавки металла. Применяется оксид никеля также в
керамической промышленности и для получения катализа-
торов.
Сульфидные никелевые руды разлагают перед анализом
азотной кислотой или ее смесью с соляной кислотой (обыч-
но после предварительного удаления серы и мышьяка об-
жигом) : 3NiS + 6НС1 + 2HNO3 = 3N iCl2 + 2NO + 3S + 4Н2О.
Окисленные руды и силикаты разлагают соляной кислотой,
а руды, не разлагаемые кислотами, сплавляют со щело-
чами.
Методы определения содержания никеля
Растворимые комплексные аммиакаты, цианиды, внут-
рикомплексные соединения с органическими веществами,
в особенности типа диоксима, а также окрашенные соеди-
77
нения с серу- и азотсодержащими органическими вещест-
вами применяются в аналитической химии никеля с целью
выделения его из растворов и последующего определения.
Выделение никеля обычно проводят едкими щелочами,
диметилглиоксимом, а также электролизом. К наиболее
распространенным методам определения содержания нике-
ля относятся следующие: гравиметрический, титриметри-
ческий, фотоколориметрический, комплексонометрический
(в настоящее время вытеснивший цианидный метод) и
атомно-абсорбционный.
Гравиметрический метод с применением диметилглиок-
сима. Диметилглиоксим
СН,-С-С-СН5
5 п и
HON NOH ’
или C4H8O2N2 (молекулярная масса 116, 12), представляет
собой белый кристаллический порошок, малорастворимый
в воде, легко растворимый в растворах щелочей, диоксане,
спирте, эфире. Этот самый распространенный реактив для
выделения и определения никеля был предложен в 1905 го-
ду русским ученым Л. А. Чугаевым и часто называется
реактивом Чугаева. Применяется в виде 1%-ного спирто-
вого или аммиачного раствора. Диметилглиоксим выделяет
из аммиачных и нейтральных растворов солей никеля
кристаллический осадок красного цвета, который можно
отфильтровать через тигель с фильтрующим дном, вы-
сушить и взвесить в форме ИЦСДДИгОгД.
Большие количества меди, кобальта и цинка замедляют
осаждение никеля. Присутствие многих других элементов
не препятствует применению гравиметрического метода.
Перед осаждением диметилглиоксимата никеля к кисло-
му раствору добавляют винную кислоту для связывания в
прочные растворимые комплексные соединения элементов,
дающих гидроксиды в аммиачной среде. Ионы алюминия,
марганца, железа, титана, хрома и других металлов за-
мещают атомы водорода карбоксильных и гидроксильных
групп винной кислоты:
^0—।
с-он с^о
1 I \ !
H-G-OH г Н - С - о - Ге + ЗН+
I + Fe3+ = I /
Н-С-ОН н-С-0
I I
Сг-ОН С-ОН
о Xo-J
78
Аналогично реагирует лимон-
ная кислота Н2С(СООН)—
С(ОН) (СООН)—СН2(СООН).
Метод осаждения никеля диме-
тилглиоксимом очень точен, он при-
годен для определения больших и
малых количеств никеля, но длите-
лен. При небольших количествах
никеля (<0,5 мг), а также в при-
сутствии кобальта, меди и цинка
растворы с осадком оставляют на
ночь.
Фотометрический метод с при-
менение M диметилглиоксима. Диме- щения водного раствора ди-
ТИЛГЛИОКСИМЭТ никеля растворим “ХТенияСсМникелеМИ(2)еГ°
в неполярных органических раство-
рителях, а также в щелочных растворах. Молярный коэф-
фициент погашения диметилглиоксимата никеля в хлоро-
форме при Хтах = 360 нм равен 3,4-103, а при Х = 400 нм
составляет 1,8-103.
В щелочной среде в присутствии окислителя (персуль-
фата аммония, иода или бромной воды) диметилглиокси-
мат никеля растворяется и окрашивает раствор в красный
цвет. Молярный коэффициент погашения диметилглиокси-
мата НИКеЛЯ В ЭТИХ УСЛОВИЯХ Составляет 1,3- 104 При Хтах =
= 470 нм (рис. 8). Высокий молярный коэффициент пога-
шения растворов комплексного соединения никеля с ди-
метилглиоксимом позволяет применять при фотометриче-
ском определении небольшие навески. Допустимы значи-
тельные количества меди и кобальта.
Фотометрическое определение никеля осуществляют
также с другими реактивами, например с дитизоном, а
также с производными диметилглиоксима, например с
а-фурилдиоксимом:
HON NOH
Титриметрические методы. Содержание никеля можно
определять прямым титрованием растворами органических
реактивов: диметилглиоксима с внешним индикатором (бу-
мага, смоченная раствором диметилглиоксима), диэтил-
дитиокарбамата, дитизона. 8-оксихинолина и некоторых
других. Очень точные результаты дает цианидный метод,
79
но вследствие токсичности титранта метод мало применя-
ется.
Йодометрическое титрование выполняют, окисляя в ще-
лочном растворе никель (II), например, персульфатом ка-
лия. При этом выпадает осадок гидроксида никеля (III).
Прибавляют иодид калия, серную кислоту и титруют вы-
делившийся иод раствором тиосульфата натрия.
В настоящее время наибольшее распространение полу-
чил метод титрования комплексоном III в присутствии раз-
личных индикаторов. Следует применять разбавленные
растворы, содержащие никель, так как собственная окрас-
ка комплексоната никеля затрудняет установление точки
эквивалентности.
Вследствие замедленного образования комплексоната
никеля применяют нагревание растворов и метод обрат-
ного титрования. При обратном титровании отчетливые
переходы окраски получены для соли цинка с индикато-
рами эриохромчерным Т, ксиленоловым оранжевым (см.
«Алюминий»), а при прямом — с мурексидом (см. «Ко-
бальт»), а также с сульфарсазеном, предложенным со-
ветскими учеными А. М. Лукиным и Г. С. Петровой.
1. Сульфарсазен (4-нитро-2-арсонобензол-1,4/-диазоами-
ноазобензол-4-сульфокислоты натриевая соль)
AsO5H2
02N
N = N - NH
N='N
SOjNa
или CigHuOeNeSAsNa (молекулярная масса 572,32), пред-
ставляет собой порошок красно-коричневого цвета, раство-
римый в воде. Особенно хорошо он растворим в слабоще-
лочных растворах, мало растворим в спиртах и нерастворим
в неполярных органических растворителях. При титро-
вании соли никеля комплексоном III в присутствии суль-
фарсазена происходит разрушение розового комплекса
никеля с сульфарсазеном, и в точке эквивалентности ок-
раска раствора переходит в зеленую (цвет комплексоната
никеля; pH 9,5—10).
2. Мурексид с ионами никеля в щелочной среде обра-
зует комплексное соединение желтого цвета. При титро-
вании комплексоном III этот комплекс разрушается, и в
точке эквивалентности окраска переходит в фиолетовую.
Атомно-абсорбционный метод. Содержание никеля оп-
ределяют атомно-абсорбционным методом в горных поро-
дах, сульфидных минералах, сплавах и чистых металлах.
80
В качестве наиболее чувствительной линии рекомендуете®
линия 232 нм, по этой линии возможно определение со-
держания никеля >0,12 мкг/мл. Часто используется ли-
ния 341,5 нм. Средством атомизации обычно служит пламя
ацетилен — воздух, рекомендуется также пламя ацети-
лен— оксид азота (I), в котором меньше проявляются по-
мехи со стороны железа и хрома.
Практическая часть
Гравиметрическое определение никеля в сплавах
на медной основе, не содержащих олова
Принцип метода. Определение основано на осаждении-
никеля диметилглиоксимом в присутствии ацетата натрия.
Медь отделяют электролизом (см. «Медь»). Мешающие
определению Fe, Al маскируют лимонной кислотой. Осадок,
высушивают при НО °C и взвешивают. Относительное
стандартное отклонение результатов определений 0,015 при
содержании никеля 0,3—3%.
Реактивы и растворы. 1) Аммиак, р = 0,91 г/см3; 2) хлорид аммо-
ния, 25%-ный раствор; 3) диметилглиоксим, 1%-ный спиртовый ра
створ; 4) кислота азотная, р=1,40 г/см3; 5) кислота лимонная; 6) кис-
лота серная, р=1,84 г/см3; 7) кислота соляная; р = 1,19 г/см3 и раз-
бавленная 1:1; 8) ацетат натрия; 9) смесь азотной и серной кислот
(для ее получения к 600 мл воды прибавляют 160 мл азотной и
180 мл серной кислот); 10) смесь соляной и азотной кислот 3:1.
Выполнение определения. Навеску сплава 0,5—1 г по-
мещают в стакан вместимостью 300 мл, прибавляют 30—
50 мл смеси соляной и азотной кислот, растворяют при
нагревании и кипятят до удаления оксидов азота. Затем
раствор разбавляют горячей водой до 200 мл, нагревают
до кипения, кипятят 1 мин и выделяют основную массу
меди электролизом при силе тока 1 —1,5 А и напряжении
2—3 В в течение 2 ч. Электролит выпаривают до объема
100 мл. К раствору прибавляют 2 г лимонной кислоты,
15 мл раствора хлорида аммония, нейтрализуют аммиа-
ком и снова осторожно, по каплям, подкисляют соляной
кислотой. Прибавляют 5 мл спиртового раствора диметил-
глиоксима на каждые 10 мг никеля и 15 мл в избыток,
затем при непрерывном перемешивании раствора добав-
ляют ацетат натрия до появления осадка диметилглиок-
симата никеля и 2 г ацетата натрия дают в избыток.
Раствор оставляют стоять 3 ч. Выделившийся осадок от-
фильтровывают на фильтре средней плотности, промывают
8—10 раз теплой водой, осадок растворяют на фильтре
81,
горячей соляной кислотой (1:1), собирая фильтрат в тот
же стакан, в котором проводилось осаждение. Фильтр
промывают 8—10 раз теплой водой и отбрасывают. Повто-
ряют осаждение никеля, для этого к раствору добавляют
2 г лимонной кислоты и далее поступают, как описано вы-
ше. Через 20—30 мин фильтруют осадок во взвешенный
•стеклянный тигель-фильтр с пористым дном, промывают
8—10 раз теплой водой, высушивают в сушильном шкафу
при 110 °C до постоянной массы. Фактор пересчета на ни-
кель составляет 0,2032.
Комплексонометрическое определение никеля
в солях никеля
Принцип метода. Титрование никеля комплексоном III
проводят при pH 9,5—10 в присутствии сульфарсазена или
мурексида в качестве индикатора. Обратное титрование
проводят при pH 5,5—6,0 с индикатором ксиленоловым
оранжевым. Относительное стандартное отклонение резуль-
татов определений 0,01 при содержании никеля 40—50%.
Реактивы и растворы. 1) Аммиак, р=0,91 г/см5; 2) хлорид аммо-
ния; 3) бура, 0,05 М раствор; 4) буферный аммонийно-аммиачный ра-
створ, pH 9,5—10 (для его получения 54 г хлорида аммония раство-
ряют в 200 мл воды; к полученному раствору прибавляют 350 мл
раствора аммиака и доводят объем раствора до 1 л водой); 5) ком-
плексон III, 0,05 М раствор; 6) мурексид; 7) хлорид натрия; 8) инди-
каторная смесь (мурексид растирают с хлоридом натрия в соотноше-
нии 1:100); 9) сульфарсазен, 0,05°/о-ный раствор в растворе буры.
Выполнение определения. Слабокислый раствор, содер-
жащий 70—85 мг никеля, помещают в коническую колбу
вместимостью 250 мл, прибавляют 10 мл аммонийно-
аммиачного буферного раствора, разбавляют водой до
100 мл. затем прибавляют 0,1 г индикаторной смеси или
Ю,3 мл раствора сульфарсазена. Полученный раствор пе-
ремешивают и титруют комплексоном III до перехода ин-
тенсивной желтой окраски раствора в фиолетовую в случае
применения в качестве индикатора мурексида или до пе-
рехода розовато-фиолетовой окраски в зеленую в случае
применения в качестве индикатора сульфарсазена. При
этом I мл точно 0.05 М раствора комплексона III соответ-
ствует 2,9350 мг никеля
Атомно-абсорбционное определение никеля
в сплаве на основе кобальта
Принцип метода. Определение основано на измерении
интенсивности линии поглощения никеля 232 нм в присут-
32
ствии основы и сопутствующих элементов (марганца, же-
леза). Относительное стандартное отклонение результатов
определений 0,01 при содержании никеля 3%.
Реактивы и растворы. 1) Кислота азотная, р= 1,40 г/см5; 2) кисло-
та соляная, р=1,19 г/см3; 3) смесь азотной и соляной кислот 1:3;
4) раствор соли никеля (100 мкг/мл) и разбавленные растворы.
Выполнение определения. Навеску сплава 0,1 г раст-
воряют в 5 мл смеси кислот. Раствор упаривают досуха»
остаток растворяют в 3 мл соляной кислоты. Полученный
раствор переносят в мерную колбу вместимостью 100 мл
и доливают до метки водой. Из этого раствора ведут оп-
ределение содержания никеля, градуировочный график
строят по данным для раствора соли никеля.
Медь
Медь известна с глубокой древности. Латинское ее обозначение
cuprum происходит от названия острова Кипр, где древние греки до-
бывали медную руду. Медь и ее сплавы имели большое значение при
создании материальной культуры человечества.
Нахождение в природе. В земной коре содержится 0,01% меди,
в основном находящейся в виде сульфидных соединений. Промышлен-
ное значение имеют примерно 40 минералов меди, важнейшими из
которых являются халькопирит (медный колчедан) CuFeSs, халькозин
(медный блеск) Cu2S, ковеллин CuS, малахит CuCO3-Cu(OH)2.
В зависимости от содержания примесных элементов медные руды
делят на медно-молибденовые, медно-цинковые, медно-никелевые,
медно-кобальтовые. В этом комплексном сырье содержатся и другие
ценные элементы; благородные металлы, селен, теллур, индий, герма-
ний, таллий, рений, свинец, кадмий, висмут.
Наиболее распространены пирометаллургические методы получения
меди.
Применение. Высокая теплопроводность и малое электрическое
сопротивление меди позволяют применять ее в электротехнической
промышленности. Разнообразное применение находят такие сплавы,
как бронзы, латуни, мельхиор, томпак, нейзильбер, константан, сплав
Деварда, сплавы меди с серебром и золотом для изготовления монет
и ювелирных изделий, катализаторы на основе меди.
Свойства меди и ее соединений. Медь — элемент 1 груп-
пы В подгруппы Периодической системы Д. И. Менделе-
ева. Электронное строение атомов в основном состоянии
Is22s22p63s23p634Zl04s1. Наиболее устойчивая степень окисле-
ния атома +2, менее устойчивая +1. Известны соединения
Си (III), которые считаются сильными окислителями.
Медь — мягкий металл красного цвета. Температура
плавления меди 1083 °C. Металлическая медь растворяется
в кислотах-окислителях. В азотной кислоте образуются
оксиды азота, в горячей концентрированной.серной кисло-
83
те — диоксид серы и соответствующая соль меди (II):
3Cu + 8HNO3 = 3Cu(NO3)2+2NO + 4H2O; Cu + 2H2SO4 =
= CuSO4+SO2 + 2Н2О.
Сульфат меди (сернокислая медь) CuSO4— бес-
цветная соль, растворимая в воде. Из водных растворов
сульфата меди кристаллизуется медный купорос CuSO4-
5Н2О — служащий исходным веществом для получения
других соединений меди. Медный купорос является побоч-
ным продуктом рафинирования меди, кроме того, его по-
лучают в промышленных масштабах растворением меди
в нагретой разбавленной серной кислоте при продувании
воздухом: 2Cu + 2H2SO4+ О2 + 8Н2О == 2CuSO4 • 5Н2О.
При действии аммиака из растворов солей Си (II) вы-
падает основная соль, растворимая в избытке аммиака с
образованием комплексных ионов, окрашенных в синий
цвет различной интенсивности: [Cu(NH3)2(H2O)2]2+,
[Си (NH3)3H2O[2+, [Си (NH3)4[2+. Реакции солей меди (II)
с аммиаком имеют вид: 2Cu2++SO42~4-2NH4OH =
= (CuOH)2SO4 + 2NH+ ; (CuOH)2SO4 + 2NH + + 6NH3 =
= 2 [Cu (NH3) 4[ 2++ SO2“ + 2H2O.
В присутствии солей щелочных металлов соли меди (II)
образуют двойные соединения, содержащие медь в виде
комплексного аниона [СиС14[2~. Комплексные аммиакаты
и галогениды разрушаются при действии цианида калия
с образованием комплексного иона [Cu(CN)4[2~.
Сульфид меди (II) CuS — порошок черного цвета,
образующийся при кипячении растворов солей меди в
степени окисления +2 в присутствии серноватистокислого
натрия (тиосульфата натрия); CuSO4 + Na2S2O3 +Н2О =
= CuS + Na2SO4 + H2SO4.
При диссоциации тиосульфата натрия в водных раст-
ворах создается небольшое, но достаточное для осаждения
малорастворимого сульфида меди (II) количество ионов
S- . В осадок выпадает также сера и сульфид меди (I):
2CuSO4 + 2Na2S2O3 + 2H2O = Cu2S + S + 2Na2SO4+2H2SO4.
Свойство меди легко соединяться с серой используется
в пирометаллургическом методе получения меди, при очи-
стке свинца от меди, в аналитической химии меди для от-
деления ее от сопутствующих элементов.
Сульфидные минералы меди и ее сплавы, например
бронзы, растворяются в азотной кислоте, а латуни и спла-
вы с благородными металлами — в смеси азотной и соля-
ной кислот: 6Cu +2HNO3 + 6HC1 = 3Cu2C12 + 4H2O + 2NO;
ЗС и + 2HNO3 + 6НС1 = ЗСиС12 + 4Н2О + 2NO; ЗСи2С12 +
84
4-8НМОз = ЗСи(Ь1Оз)2 + ЗСиС12 + 2ЫО + 4Н2О. Сплавы на
алюминиевой основе растворяются в растворе 20%-ного
гидроксида натрия.
Методы определения содержания меди
Методы определения и отделения меди от сопутствую-
щих элементов основаны на способности ее образовывать
аммиачные, галогенидные комплексы, а также комплекс-
ные соединения со многими кислород-, серо-, азотсодер-
жащими органическими и неорганическими соединениями.
Наиболее распространенными методами определения со-
держания меди являются: электрогравиметрический метод,
пригодный для выделения больших количеств меди (до
5 г), гравиметрические методы с применением органических
осадителей, из которых наиболее часто используют а-бен-
зоиноксим, 8-оксихинолин, хинальдиновую кислоту, меркап-
тобензтиазол. Широко распространены титриметрические
комплексонометрические методы и очень точный йодомет-
рический метод. Для определения микроколичеств меди
наиболее часто применяют спектрофотометрические методы,
основанные на реакциях меди с чувствительными органи-
ческими реагентами, такими как диэтилдитиокарбамат
натрия, дитизон, дихинолил, вытеснившими старый метод
определения содержания меди, основанный на образовании
его окрашенного аммиаката. Применяются также инстру-
ментальные методы: атомно-абсорбционный и эмиссион-
ный спектральный.
Электрогравиметрический метод основан на выделении
меди электролизом на платиновом сетчатом катоде при
силе тока 1,5—2,5 А и напряжении 2—3 В из азотно-серно-
кислого раствора: Cu2++2e-->Cu.
Определению меди мешают большие количества желе-
за (III), окисляющего медь, а также хлорид- нитрит-, ро-
данид-ионы, олово (IV), сурьма (III), мышьяк (III), се-
лен (IV), теллур (IV), висмут, молибден, металлы плати-
новой группы.
При электрогравиметрическом определении меди мето-
дом внутреннего электролиза в различных сплавах, раст-
ворах солей меди платина может быть заменена на стекло-
углерод. Снятие катодных осадков меди с поверхности
изделий из стеклоуглерода азотной кислотой, как это при-
нято в известных методиках, происходит количественно.
Применяют катод из стеклоуглерода в виде чашки или
ти! я СУ-2000, ТУ 48-20-117—82, вместимостью 50 мл.
85
Анодом служит алюминиевая пластинка. Во избежание
цементации используют алюминии особой чистоты. Анод
к катоду прикрепляют с помощью медной клеммы (рис. 9).
Титриметрические методы. Йодометрический ме-
тод основан на реакции окисления иодида калия ионом
меди (II) по реакции 2CuSC>4 + 4KI = I2+CU2I2+2K.2SO4.
Иодид меди (I)—маслорастворимая соль, вследствие’
этого реакция количественно протекает слева направо. Вы-
делившийся иод титруют раствором тиосульфата натрия,
в присутствии в качестве индикатора крахмала. При этом,
происходит восстановление иода до иодид-иона по реак-
ции 2Na2S2O3 + l2 = 2NaI + Na2S4O6.
Рис. 9. Схема прибора для осуществления метода внутреннего электролиза с
применением катода в виде тигля из стеклоуглерода: 1 — тигель (катод); 2—
алюминиевая пластинка (анод) 3 — медная клемма
Рис. 10. Кривая светопоглощения раствора соединения меди (I) с 2,2'-дихино-
лилом.
Перед определением содержания меди должны быть
удалены окислители: азотная кислота (выпариванием с
серной кислотой), железо (III) мышьяк (V), сурьма (VI),
молибден (VI), селен (VI), а также свинец и висмут,
образующие с иодидом калия окрашенные соединения.
Небольшие количества железа можно связать фторидом
или пирофосфатом натрия в комплексное соединение, не
реагирующее с иодидом. Мышьяк (V) и сурьма (V) вза-
имодействуют с иодидом только в сильнокислой среде, по-
этому содержание меди в их присутствии определяют в
слабокислой среде.
86
Комплексонометрический метод. Медь ти-
труют раствором комплексона III в слабокислой или ще-
лочной среде. В слабокислой среде (pH 4—5) в качестве
индикатора применяют 4-(2-пиридилазо)-резорцин, или
ПАР а, а'-бис- (4-натрийтетразолилазо-5)-этилацетат, или
тетра. В щелочной среде (pH 8—10) обычно применяют
мурексид (см. «Кобальт») Титрование в слабокислой среде
более селективное, не мешают щелочноземельные элемен-
ты и магний.
1. Тетра
Nd Na
। Н ।
N-N I N-N
II >C -N = N- C - N = N - C< II 3H20 ,
N-N | о N-N
c<
0 — C2H5
или C3H6Ni2Na2O2-3H2O (молекулярная масса 378,21), пред-
ставляет собой оранжевый порошок, растворимый в воде,
этиловом и метиловом спиртах. Водный раствор реагента
устойчив в течение нескольких месяцев, разрушается при
действии минеральных кислот. При pH 1—И реагент об-
разует с ионами меди (II) комплекс фиолетового цвета.
При титровании раствором комплексона III комплекс раз-
рушается, и в точке эквивалентности окраска раствора
переходит в зеленую. Двухзарядные ионы никеля, кобаль-
та, цинка, кадмия, свинца в аналогичных условиях обра-
зуют с реагентом ярко окрашенные комплексы. При титро-
вании в слабокислой среде определению содержания меди
не мешают большие количества алюминия и марганца.
2. ПАР
или C11H9N3O2 (молекулярная масса 215,225), его натрие-
вая соль — оранжевый порошок, применяемый для ком-
плексонометрического титрования в виде 0,1%-ного вод-
ного раствора. ПАР образует со многими двух- и трехза-
рядными катионами комплексы, окрашенные в красный или
фиолетовый цвет. При титровании соли меди (II) ком-
плексоном III в присутствии ПАР происходит разрушение
красного комплекса меди, п . в точке эквивалентности
окраска раствора переходит в зеленую. Определению со-
87
держания меди не мешают магнии и щелочно-земельные
элементы. Комплексонометрическое титрование меди с ПАР
проводят при pH 4—5 или 10.
Атомно-абсорбционный метод. Медь — один из элемен-
тов, наиболее легко определяемых методом атомной аб-
сорбции. Малые количества меди, от 0,1 мкг/мл, опреде-
ляют по резонансной линии 324,75 нм; для больших коли-
честв меди рекомендуется линия 249,2 нм. Средством
атомизации служит пламя ацетилен — воздух, низкотемпе-
ратурное пламя природный газ — воздух, а также непла-
менные средства, например графитовая печь. Определение
содержания меди методом атомной абсорбции сочетают с
выделением ее экстракцией, электролизом и другими из-
вестными способами. Методом атомной абсорбции опреде-
ляют концентрацию меди в рудах, минералах, медных кон-
центратах, сплавах.
Фотометрические методы. Экстракционно-фотометриче-
ский метод с 2,2'-дихинолилом (купреином)
или Ci8Hi2N2 (молекулярная масса 256,31). Реагаент явля-
ется нерастворимым в воде белым порошком, хорошо ра-
створим в амиловом, изоамиловом спиртах. С ионами ме-
ди (I) образует комплекс, окрашенный в фиолетовый цвет.
При pH 6 и выше соединение извлекается амиловым или
изоамиловым спиртом. Обычно применяют его экстракцию
в изоамиловый спирт; растворы реагента в изоамиловом
спирте готовят 0,02—0,03 %-ными.
В области максимального светопоглощения комплекса:
при 2vmax=545 нм (рис. 10) реагент не поглощает свет.
Окраска комплекса прямо пропорциональна концентра-
ции меди при содержании ее до 40 мкг в 10 мл органиче-
ской фазы при толщине поглощающего слоя 10 мм. Опре-
делению мешают оксалат-, роданид-, цианид-ионы.
Экстракционно-фотометрический метод с диэтилдитио-
карбаминатом натрия
(C2H5)2N- СС
SNa-3Hz0 ,
или C5Hi0NS2Na-3H2O (молекулярная масса 225,31). Реа-
гент является белым кристаллическим порошком, легко раст-
воримым в воде, с ионами меди (II) образует трудно раст-
воримое в воде соединение желто-коричневого цвета, экст-
88
рагируемое в широком интервале pH (4,5—11) в хлоро-
форм. Область максимального светопоглощения комплекса
436 нм; его окраска прямо пропорциональна концентрации
меди при ее содержании 5—20 мкг в объеме органической
фазы 25 мл и толщине поглощающего слоя 30 мм. Опреде-
лению мешают железо, алюминий, никель, кобальт, мар-
ганец, титан. Влияние этих элементов можно устранить
маскированием различными комплексообразующими веще-
ствами. Не поддаются маскированию висмут, таллий (III),
теллур (IV), сурьма (III). Бесцветные соединения обра-
зуют свинец, цинк и кадмий. Избирательность метода по-
вышается при использовании в качестве реагента диэтил-
дитиокарбамината свинца (0,5 М. НС1).
Практическая часть
Электрогравиметрическое определение меди
в сплавах на основе железа, алюминия, меди
с применением стеклоуглерода1 *
Принцип метода. Мед.ъ выделяют методом внутреннего
электролиза. В качестве катода применяют стеклоуглерод
(в виде чашки или тигля), в качестве анода — алюминие-
вую пластинку. Относительное стандартное отклонение
0,01 при содержании меди от 0,1 до 100%.
Реактивы, растворы и материалы. 1) Кислота соляная, р =
= 1,19 г/см3; 2) кислота азотная, р=1,40 г/см3; 3) кислота серная,
разбавленная 1:1, 1:3 и 1:4; 4) гидразин сернокислый; 5) чашка или
тигель из стеклоуглерода марки СУ-2000; 6) алюминиевая пластинка
из алюминия ос. ч.
Выполнение определения. 1. Определение меди в раст-
воре сульфата меди. В предварительно взвешенную чаш-
ку или тигель из стеклоуглерода (катод) отбирают раст-
вор сульфата меди, добавляют 5 мл серной кислоты (1:4)
и 20 мл дистиллированной воды, нагревают раствор до
~60—70°C на песчаной бане. В горячий раствор погружа-
ют алюминиевый анод, прикрепленный к катоду с помощью
медной клеммы. Электролиз проводят при нагревании на
песчаной бане в течение 45 мин, затем доливают горячей
водой так, чтобы уровень жидкости в чашке повысился на
0,5 см, и приводят электролиз еще 15 мин. Затем из чаш-
ки выливают раствор, промывают ее дистиллированной
водой, спиртом и эфиром, высушивают в сушильном шка-
1 Методика разработана преподавателями МИСиС В. П. Глады-
шевым и Л. 3. Козель.
89
фу при температуре 80 °C до постоянной массы п взвеши-
вают. По разности находят массу выделившейся меди.
2. Определение меди в сплавах на основе железа и
алюминия. Навеску сплава (1 г при содержании меди
0,1—1% и 0,2 г при содержании меди 1 —10%) растворяют
в предварительно взвешенной чашке или тигле из стекло-
углерода в 20 мл концентрированной соляной кислоты при
нагревании на песчаной бане. Затем прибавляют по каплям
концентрированную азотную кислоту до прекращения
вспенивания, после чего прибавляют 4 мл серной кислоты
(1:1), выпаривают до паров SO3, охлаждают, прибавляют
20 мл дистиллироваанной воды, добавляют 0,5 г сернокис-
лого гидразина для восстановления Fe3+ до Fe2*3 и 4 мл
серной кислоты (1:4), нагревают до 60—70 °C и далее
проводят электролиз, как описано выше.
3. Определение меди в медных сплавах (сплавы на
основе меди с содержанием 50—98%). Навеску сплава
(0,4 г) растворяют в 10 мл концентрированной азотной
кислоты в стакане вместимостью 250 мл при нагревании.
Добавляют 10 мл серной кислоты (1:4), выпаривают до
паров SO3. Содержимое стакана 'переносят в мерную кол-
бу вместимостью 100 мл и разбавляют до метки дистилли-
рованной водой. Отбирают для электролиза 5 мл получен-
ного раствора в чашку или тигель из стеклоуглерода
(катод), добавляют 5 мл серной кислоты (1:3) и 20 мл
дистиллированной воды, нагревают до 60—70 °C и далее
проводят электролиз, как описано выше.
Электрогравиметрическое определение меди
в медных концентратах
Принцип метода. Определение основано на выделении
меди электролизом при содержании серебра в концентратах
не более 500 г/т. Мешающие определению меди, железо и
алюминий предварительно отделяют, осаждая их аммиа-
ком в виде гидроксидов. Относительное стандартное откло-
нение результатов определений 0,01 при содержании меди
20—40%.
Реактивы и растворы. 1) Кислота азотная, р = 1,40 г/см3, пред-
варительно прокипяченная и охлажденная; 2) кислота серная, р =
= 1,84 г/см3 п разбавленная 1:1, 5:95, 1:99; 3) кислота соляная, р =
= 1,19 г/см3; 4) аммиак, р = 0,91 г/см3 и разбавленный 1:99, 5) перок-
сид водорода; 6) мочевина; 7) спирт этиловый (гидролизный) ректи-
фикованный.
Выполнение определения. Навеску медного концентра-
та 1 г (при содержании меди до 20%) или 0,5 г (при со-
90
держании меди свыше 20%) помещают в колбу вмести-
мостью 250 мл, смачивают водой, приливают 15 мл соля-
ной кислоты и нагревают в течение 15—20 мин. Затем к
раствору приливают 20 мл азотной кислоты, накрывают
колбу часовым стеклом и нагревают до прекращения вы-
деления оксидов азота. Стекло снимают, обмывают его
водой над колбой и выпаривают раствор до состояния
влажных солей. Далее приливают 10 мл разбавленной
1:1 серной кислоты и выпаривают раствор до выделения
паров серной кислоты. При наличии в остатке от выпари-
вания темных включений приливают азотную кислоту пор-
циями по 1 мл до обесцвечения остатка и снова выпари-
вают раствор до выделения паров серной кислоты. Оста-
ток охлаждают, приливают к нему 5 мл воды, обмывая
стенки колбы, и снова выпаривают до выделения паров
серной кислоты. К охлажденному остатку приливают 50 мл
воды, раствор кипятят 3—5 мин и отфильтровывают оста-
ток на плотный фильтр. Колбу и фильтр промывают 7—
8 раз разбавленной 1:99 серной кислотой.
Фильтрат и промывные воды собирают в стакан вмес-
тимостью 400 мл, нагревают до 70 °C и приливают аммиак
до полного выпадения гидроксидов, а после проверки пол-
ноты осаждения добавляют 2—3 мл в избыток. Раствор
с осадком оставляют на 30 мин при 70—90 °C. Затем оса-
док отфильтровывают на фильтре средней плотности.
Фильтр с осадком промывают 3—4 раза горячим разбав-
ленным 1:99 аммиаком.
Осадок с фильтра смывают струей горячей воды в
стакан, в котором проводилось осаждение, прибавляют
30 мл разбавленной 5:95 серной кислоты, 10 мл пероксида
водорода и нагревают до полного растворения осадка.
В полученном растворе осаждение аммиаком проводят
еще два раза, как указано выше.
Все фильтраты и промывные воды собирают в стакан
вместимостью 400 мл и выпаривают до 200 мл. Затем ней-
трализуют раствор азотной кислотой и добавляют 8 мл
в избыток.
Предварительно взвешенный катод укрепляют в держа-
теле установки, затем погружают его в раствор так, чтобы
верхний край электрода находился на 5 мм выше поверх-
ности раствора, и подвергают раствор электролизу (не
перемешивая) при силе тока 2—2,5 А и напряжении 2,2—
2,5 В. Во время электролиза стакан должен быть накрыт
двумя половинками часового стекла с полукруглыми выем-
ками.
91
После обесцвечивания раствора прибавляют 0,1—0,2 г
мочевины и включают мешалку. Через 20 мин раствор до-
ливают водой до полного погружения электрода, обмывая
стекла и стенки стакана. Если через 20 мин на вновь по-
крытой раствором поверхности катода не появится медь,
электролиз считают законченным.
Катод освобождают от электролита, промывают над
стаканом водой и отключают ток. Далее катод снимают,
промывают погружением в спирт, высушивают при 100—
105 °C в течение 5 мин, охлаждают в эксикаторе и взвеши-
вают. Содержание меди в пробе рассчитывают, как ука-
зано на с. 27.
Иодометрическое определение меди
в цинковых сплавах, не содержащих олова
Принцип метода. Определение основано на реакции
восстановления меди (II) иодидом калия до меди (I).
Выделившийся по реакции элементный иод титруют раст-
вором тиосульфата натрия. Предварительно медь осаж-
дают тиосульфатом натрия. Относительное стандартное
отклонение результатов определений 0,01 при содержании
меди 5—20%.
Реактивы и растворы. 1) Кислота азотная, р=1,40 г/см3; 2) кисло-
та серная, р=1,84 г/см3 и разбавленная 1:1; 3) иодид калия; 4) крах-
мал растворимый, раствор 5 г/л; 5) карбонат натрия кристаллический;
6) тиосульфат натрия, растворы 500 г/л; 0,1 н. титрованный, установ-
ленный по дихромату калия.
Выполнение определения. Навеску образца 0,2 г поме-
щают в стакан вместимостью 500 мл, прибавляют 5 мл
азотной кислоты, 5 мл разбавленной серной кислоты и
растворяют при нагревании. Обмывают стенки стакана
водой и выпаривают до появления паров серной кислоты.
Последнюю операцию повторяют дважды. Охлаждают, со-
ли растворяют в воде и разбавляют водой до объема
200 мл. Нагревают раствор до кипения, прибавляют 60—
80 мл раствора тиосульфата натрия (500 г/л) и кипятят
до просветления раствора. Фильтруют на быстрофиль-
трующем фильтре и промывают 8—10 раз горячей водой.
Фильтр с осадком помещают в фарфоровый тигель, под-
сушивают и прокаливают при температуре 500 °C, остаток
в тигле растворяют в 5 мл азотной кислоты, прибавляют
5 мл разбавленной серной кислоты и дважды выпаривают
до паров серной кислоты.
Раствор переносят из тигля в коническую колбу вмести-
мостью 500 мл, прибавляют воды до объема 200 мл, 5 г
92
йодистого калия, выдерживают в темном месте в течение
3—5 мин и титруют выделившийся иод 0,1-н. раствором
тиосульфата натрия до светло-желтой окраски. Прибавляют
3—5 мл свежеприготовленного раствора крахмала (5 г/л)
и медленно при перемешивании титруют до исчезновения
синей окраски крахмала. 1 мл 0,1 н. раствора тиосульфа-
та натрия соответствует 6,354 мг меди.
Комплексонометрическое определение меди
в щелочных электролитах меднения и латунирования
Принцип метода. Прямое титрование меди комплексе'
ном III проводят при pH 8—10 в присутствии насыщенно-
го раствора мурексида в качестве индикатора. Относи-
тельное стандартное отклонение результатов определений
0,01 при содержании меди 5—10%.
Реактивы и растворы. 1) Аммиак, р = 0,91 г/см3; 2) буферный
аммонийно-аммиачный раствор, pH 9,5—10; комплексон III 0,05 М
раствор; 4) мурексид, насыщенный водный раствор; 5) пероксид во-
дорода.
Выполнение определения. К 10 мл разбавленного элек-
тролита прибавляют 5 мл аммиака и по каплям (чтобы
раствор не пенился) 2—3 мл пероксида водорода. Если
раствор синеет,' его слегка нагревают, кипятят, после
охлаждения добавляют мурексид и титруют раствором
комплексона III до перехода оранжевой окраски в фио-
летовую. 1 мл 0,05 М раствора соответствует 3,1770 мг
меди.
Комплексонометрическое
определение меди в латунях
Принцип метода. Прямое титрование меди комплексо-
ном III проводят при pH 10 в присутствии ПАР в качест-
ве индикатора. Относительное стандартное отклонение ре-
зультатов определений 0,01 при содержании меди — 60%.
Реактивы и растворы. 1) Аммиак, р = 0,91 г/см3; 2) буферный аце-
татный раствор, pH 5; 3) буферный аммонийно-аммиачный раствор,
pH 9,5—10; 4) комплексон III, 0,05 М раствор; 5) кислота азотная,
р=1,40 г/см’; 6) кислота соляная, р=1,19 г/см3; 7) кислота уксусная,
р=1,04 г/см3; 8) метиленовый голубой, 0,05%-ный раствор; 9) ПАР
0,1%-ный спиртовый раствор; 10) персульфат аммония, 10-%-ный раст-
вор; 11) тиосульфат натрия, 1%-ный раствор; 12) смесь азотной и
соляной кислот (1:3).
Выполнение определения. Навеску латуни 0,1 г раст-
воряют в смеси кислот. Раствор упаривают до удаления
93
оксидов азота, разбавляют водой, нейтрализуют аммиа-
ком до начала образования осадка, который растворяют
в уксусной кислоте. Затем прибавляют буферный раствор
pH 5,10 мл раствора тиосульфата натрия. Полученный
раствор разбавляют водой до 70—80 мл, нагревают до
70—75 °C, вводят 1 мл раствора ПАР, 0,5 мл раствора
метиленового голубого.
Титрование производят раствором комплексона III до
перехода синевато-красной окраски в желто-зеленую (та-
ким образом определяют содержание цинка). Затем вво-
дят аммиачный буферный раствор pH 10,5 мл раствора
персульфата аммония и тем же раствором комплексона
III титруют медь до перехода голубой окраски раствора
в желтоватую. »
Комплексонометрическое
определение меди в бронзах
Принцип метода. Прямое титрование меди комплексо-
ном III приводят при pH 4 в присутствии индикатора тет-
ра. Относительное стандартное отклонение результатов
определений 0,01 при содержании меди —90%.
Реактивы и растворы. 1) Буферный ацетатно-аммиачный раствор,
pH 4; 2) кислота азотная, р= 1,40 г/см3, разбавленная 1:1; 3) комплек-
сон III, 0,01 н. раствор; 4) фторид натрия; 5) тетра, 0,2%-ный ра-
створ.
Выполнение определения. Навеску бронзы 0,2 г раст-
воряют в 10 мл разбавленной! азотной кислоты, раствор
.кипятят до удаления оксидов азота, затем переносят в
мерную колбу вместимостью 200 мл и доводят объем до
метки водой. Аликвотную часть раствора 10—20 мл поме-
щают в коническую колбу, прибавляют 10 мл буферного
раствора pH 4, 0,5 фторида натрия (если присутствует
Fe, Sn), 1—2 капли раствора индикатора и титруют раст-
вором комплексона III до перехода фиолетовой окраски
раствора в зеленую.
Фотометрическое определение меди
в металлическом никеле
Принцип метода. Определение основано на образова-
нии ионами меди (II) окрашенного в желто-коричневый
цвет соединения с диэтилдитиокарбаматом натрия, экстра-
гирующегося хлороформом. Относительное стандартное от-
клонение результатов определения 0,1 при содержании
0.003—0,02% меди. Мешающие элементы маскируют ли-
монной кислотой и комплексоном III.
94
Реактивы и растворы. 1) Аммиак, р=0,91 г/см3; 2) ди эти;: лигно-
карбамат натрия, раствор 1 г/л; 3) кислота азотная р= 1,40 г/см3;.
4) кислота соляная, р= 1,19 г/см3 и разбавленная 1:1; 5) кислота ли-
монная; 6 комплексон III, раствор 100 г/л; 7) медь особой чисто-
ты— 99,95% Си (раствор, содержащий 0,001 мг/мл); 8) хлороформ;
9) раствор цитрата аммония (для его приготовления к 210 мл аммиа-
ка прибавляют 150 мл воды и 200 г лимонной кислоты небольшими
порциями, раствор перемешивают и охлаждают, разбавляют водой до
500 мл; в полученном растворе концентрация цитрата аммония состав-
ляет 40 г/л).
Построение градуировочного графика. В стаканы
вместимостью 100 мл помещают 0, 3, 6, 9, 12, 15, 18, 21,
24 мл раствора меди, что соответствует 0, 3, 6, 9, 12, 15,
18, 21, 24 мкг меди, прибавляют при перемешивании 5 мл
раствора цитрата аммония, 10 мл раствора комплексона
III, воды до 70 мл, затем нейтрализуют аммиаком до зна-
чения pH 8,5—9 по универсальной индикаторной бумаге
и далее поступают, как указано при описании выполнения,
определения.
Выполнение определения. Пробу образца 0,25—1 г по-
мещают в стакан вместимостью 300 мл, растворяют при
нагревании в 15—25 мл соляной кислоты. После раство-
рения окисляют железо и карбиды азотной кислотой, при-
ливая ее по каплям до прекращения вспенивания, кипя-
тят и выпаривают раствор досуха. Сухой остаток смачи-
вают 10 мл соляной кислоты, снова выпаривают досуха.
Выпаривание с 10 мл соляной кислоты повторяют. Сухой
остаток растворяют при нагревании в 20 мл разбавленной
соляной кислоты, переводят в мерную колбу вместимостью
100 мл, доводят до метки водой и перемешивают.
Аликвотную часть раствора помещают в делительную
воронку вместимостью 200 мл, прибавляют 5 мл раствора
лимоннокислого аммония, 10 мл раствора комплексона,
перемешивают, затем нейтрализуют аммиаком по универ-
сальной индикаторной бумаге до pH 8,5—9, разбавляют
водой до объема 70 мл, прибавляют 5 мл раствора ди-
этилдитиокарбамата натрия и экстрагируют диэтилдитио-
карбамат меди двумя порциями хлороформа по 10 и 5 мл,
энергично взбалтывая каждую порцию в течение 2 мин.
После разделения фаз сливают слой хлороформа в
сухую мерную колбочку вместимостью 25 мл, доливают
хлороформом до метки и перемешивают. Фильтруют часть
раствора через фильтр средней плотности в кювету с длиной
оптического пути 30 мм. Оптическую плотность раствора
измеряют на фотоэлектроколориметре с синим светофильт-
ром. В качестве раствора сравнения применяют хлоро-
95
форм. Через все стадии анализа проводят контрольный
«опыт.
По найденной разности оптических плотностей иссле-
дуемого раствора и контрольной пробы находят процент-
ное содержание меди, используя градуировочный график.
А томно-абсорбционное определение меди
Принцип метода. Образец растворяют в смеси азотной
и соляной кислот, затем разбавляют водой до содержания
меди 2—60 мг/л.
Метод применим для определения меди в медных кон-
центратах и огарках при содержании в них 6—30%Си.
При использовании прибора фирмы «Перкин—Эльмер»,
модель 303—4а, снабженном фотоэлектрической системой
регистрации с цифропечатающим выходным устройством,
относительное стандартное отклонение результатов анали-
за не превышает 0,02.
Реактивы и растворы. 1) Кислота азотная, р=1,40 г/см3; 2) бром;
3) фторид аммония; 4) кислота соляная, р = 1,19 г/см3; 5) растворы
меди (2—60 мг/л).
Выполнение определения. Навеску образца 0,5—1 г по-
мещают в колбу, приливают 10 мл азотной кислоты и 1 мл
брома, накрывают колбу часовым стеклом, оставляют на
2—3 ч. Затем часовое стекло снимают, колбу ставят на
нагретую плиту, избыток брома отгоняют. В смесь вво-
дят 0,3 г фторида аммония, 10 мл соляной кислоты, раст-
вор упаривают до небольшого объема. Раствор с нераст-
воримым остатком переносят в мерную колбу вмести-
мостью 250 мл, доводят до метки водой и перемешивают.
Отстоявшийся прозрачный раствор используют для опре-
деления меди. Содержание меди в растворе определяют,
используя градуировочный график.
Цинк
Название происходит от латинского слова zinkum, означающего
«белый налет», упоминается немецким естествоиспытателем и врачом
Парацельсом в 1530 г. Еще в древности были известны сплавы цинка
с медью — латуни. Металлический цинк в Европе научились получать
в конце средних веков.
Нахождение в природе. В земной коре содержится 0,0015% Zn в
основном в виде сульфида ZnS—цинковой обманки, или сфалерита.
В этом минерале цинковых руд цинку всегда сопутствует значитель-
ное количество железа, изоморфно его замещающего, а также десятые
доли процента кадмия и в меньших количествах германий, индий,
таллий, галлий. В разнообразных минералах, в железных и полиметал-
96
лических рудах цинк содержится в качестве примеси. При плавке
железных руд цинк накапливается в пыли колошниковых газов.
Цинковые концентраты, служащие основным источником цинка,
получают путем селективной флотации полиметаллических руд. Метал-
лический цинк получают электролитическим или пирометаллургическим
способами.
Применение. Металлический цинк на воздухе покрывается защит-
ной пленкой и не корродирует. Оцинкованное железо, листовой цинк
широко применяются в быту. Цинковая пыль служит восстановителем
в органическом синтезе, вытесняя металлы из растворов их соедине-
ний. Химический элемент Даниэля работает на основе реакции цинка
с ионами меди: Cti2+ + Zn=Zn2+ + Cu.
Применение в промышленности получили следующие сплавы цинка:
латуни, антифрикционные сплавы с алюминием, типографские.
Свойства цинка и его соединений. Цинк — элемент
II группы элементов Периодической системы Д. И. Мен-
делеева. Электронное строение атомов в основном состоя-
нии— ls22s22ps3s23p63rf104s2.
Устойчивая степень окисления цинка +2. Металличе-
ский цинк растворяется в разбавленных кислотах, особен-
но легко в соляной кислоте, с выделением водорода:
Zn + 2HCl = ZnCl2 + H2.
В концентрационной серной кислоте образуется оксид
серы (IV). Продуктами восстановления могут быть также
-сера и сероводород. В умеренно концентрированной азот-
ной кислоте — оксиды азота: Zn + 2H2SO4=ZnSO4+SO2+
+ 2Н2О; Zn + 5H2SO4 = 4Z.nSO4 + H2S + 4H2O; 3Zn + 4H2SO4==
= 3ZnSO4+S 4~ 4H2O; 4Zn 4- IOHNO3 = 4Zn (NO3) 2 -j- N?O +
+ 5H2O; 3Zn + 8HNO3 = 3Zn (NO3) 2 + 2NO + 4H4O.
В разбавленной азотной кислоте образуется нитрат
аммония: 4Zn+I0HNO3 = 4Zn(NO3)2 + NH4NO3 + 3H2O.
Цинк высокой степени чистоты растворяется медленно,
и для ускорения процесса растворения добавляют несколь-
ко капель сильно разбавленного раствора медного купо-
роса.
Цинк легко растворяется в щелочах с образованием
цинкатов, в растворах аммиака и хлорида аммония: Zn +
+ 2КОН = K2ZnO2 + Н2; Zn + 2NH4C1 + 2Н2О = ZnCl2 + Н2 +
+ 2NH4OH.
Амфотерные свойства соединений цинка позволяют от-
делить его при избытке едкого натра от гидроксидов же-
леза, хрома, марганца никеля, кобальта, р. з э., титана,
циркония. Явление соосаждения цинка с гидроксидами
металлов приводит к необходимости переосаждения. От-
деление цинка от гидроксида железа и алюминия аммиа-
ком также требует операции переосаждения вследствие
соосаждения цинка.
4, 129.
97
Оксид цинка ZnO образуется при сгорании метал-
ла и вследствие летучести находится в воздухе помеще-
ний там, где металл нагрет выше температуры плавле-
ния. Оксид цинка растворяется в кислотах, щелочах^
аммиаке.
Гидроксид цинка Zn(OH)2 выделяется из раст-
воров солей цинка действием щелочи при pH 6—8. При
рНП гидроксид цинка растворяется в избытке щелочи,
образуя цинкаты.
Белый сульфид цинка ZnS выпадает в осадок
в нейтральных растворах солей цинка под действием се-
роводорода или сульфида аммония, со временем пере-
ходя в труднорастворимую модификацию: Zn2++H2S^
4=tZnS + 2H+. В свежеосажденном виде легко растворяется
в сильных кислотах: ZnS + 2HCl=ZnC12 + H2S.
Из щелочной среды сульфид цинка можно выделить
с помощью тиоацетамида — заменителя сероводорода:
8
CHj-C-NH2 ,
или CH3CSNH2 (молекулярная масса, 75, 134). В кислой
или щелочной средах реагент подвергается гидролизу и
от него отщепляется H2S по реакции CHsCSNH2 + 2H2O-^
-*-CH3COONH4 + H2S. Одновременно осаждаются ионы ме-
ди (II), марганца (II), железа (III) и свинца (II) (в ще-
лочной среде).
Природные соединения цинка разлагаются кипячением
с соляной кислотой. Такой метод удобен тем, что весь
цинк переходит в раствор, а в нерастворимом остатке ос-
тается основная масса элементов мешающих определению
цинка. Для полного разложения материала добавляют
окислитель — азотную кислоту, которую затем удаляют
выпариванием с серной кислотой. Нерастворимый остаток
сплавляют с содой, растворяют в соляной кислоте и при-
соединяют к основному раствору. Полиметаллические ру-
ды обычно легко разлагаются смесью соляной и азотной
кислот.
Методы определения содержания цинка
В настоящее время основными методами определения
содержания цинка в объектах цветной металлургии и про-
дуктах их переработки являются: титриметрические, фото-
98
метрические, быстрый полярографический (широко приме-
няются в массовой работе), а также атомно-абсорбционный.
Наибольшее применение в анализе сложных систем нашло
предварительное выделение цинка в виде сульфида и
сорбция его ионитами.Анионные комплексы цинка [ZnCk]2-
извлекают из растворов анионитами, а ион Zn2+—катио-
нитами.
Титриметрические методы. Наибольшее распростране-
ние имеет комплексонометрический метод определения со-
держания цинка, вытеснивший классический ферроцианид-
ный метод. Г. Шварценбах с сотрудн. предложили в ка-
честве индикатора при определении цинка эриохромчер-
ный Т, который применяется и в настоящее время ввиду
очень четкого перехода окраски в точке эквивалентности.
Недостатком индикатора является неустойчивость его
водных растворов (см. «Алюминий»),
Цинк титруют комплексоном III в аммиачном буферном
растворе pH 10. Селективность метода невысокая, однако
применение маскирующих средств позволяет значи-
тельно ее повысить, и даже в сложных системах цинк
можно определять комплексонометрически без отделения
сопутствующих элементов. Индикаторы: ксиленоловый
оранжевый (см. «Алюминий»), сульфарсазен(см.«Никель»),
ПАР (см. «Медь»), обладающие четким переходом окрас-
ки в точке эквивалентности и, кроме того, позволяющие
титровать цинк в слабокислой среде (рН~5); применя-
ются наиболее часто в сочетании с приемом маскирова-
ния сопутствующих элементов.
Фотометрический метод. В качестве фотометрического
реагента наиболее часто применяют дитизон (дифенил-
тиокарбазон)
NH-NH-CjHj N-NH-C6HS
! ~
5 = С =HS-C
n = n-c6h5 N = N-C6Hs
или C13H12N4S (молекулярная масса 256, 39), который
представляет собой порошок черного цвета, растворимый
в хлороформе и четыреххлористом углероде с образова-
нием зеленой окраски. Растворяясь в щелочах, он окра-
шивает их в красный цвет.
Дитизонат цинка имеет строение
C.H5-N = N-CC'S^ZnrS~H-N = N~C^
'“N-NH 'NH-N
4*
99
Красный дитизона? цинка образуется при встряхива-
нии слабокислого или слабощелочного раствора цинка с
раствором дитизона в хлороформе или четыреххлористом
углероде. Экстракция в четыреххлористом углероде про-
текает быстрее. Максимальное светопоглощение (рис. 11)
в четыреххлористом углероде наблюдается при pH 6,5—
7,5 и а = 535 нм. Мешают определению цинка никель, ко-
бальт, кадмий, медь, серебро, ртуть (II), висмут, золото.
Применение маскирующих средств позволяет повысить се-
лективность определения цинка. Часто определение со-
держания цинка дитизоном проводят после удаления ме-
шающих элементов ионообменным методом.
400 500 600 Л,ни
Рис. 11. Кривые свето-
поглощения растворов
дитизона (/) и его со-
единения с цинком (2)
в четыреххлористом угле-
роде
Дитизон предложен в 1929 г. Позднее для фотометри-
ческого определения цинка предложены соединения из
класса формазанов (цинкон и другие), пиридилазосоеди-
нения (ПАН, ПАР), азоарсониевые соединения (сульфар-
сазен), антипириновые красители (хромпиразол), окси-
азосоединения. Наиболее перспективными из них являются
хромпиразол и оксиазосоединение — сокращенно назван-
ное НААН или НФАГН [5-нитрофенил (2-азо-Р) -2'-(р-аце-
тилгидразино) -нафталин]. В настоящее время этот реагент
применяется при анализе сплавов сложного состава.
Полярографический метод широко применяется при
анализе сложных материалов, содержащих цинк. На фоне
аммиачного раствора хлорида аммония цинк восстанав-
ливается на ртутнокапельном или платиновом электроде
с образованием ярко выраженной волны.
Атомно-абсорбционный метод применяется для опреде-
ления цинка в разнообразных материалах: рудах, мине-
ралах, солях, концентратах, сплавах, чистых металлах.
Определение проводят, применяя пламя воздух—ацети-
лен, а также непламенные средства атомизации (графи-
товую печь). Обычно используется резонансная линия
213,86 нм, рекомендована также линия 307,59 нм. По
резонансной линии определяют содержание цинка от
100
0,02 мкг/мл. Близко к резонансной линии цинка нахо-
дятся линии меди, поэтому при работе с соединениями,
содержащими медь, катод лампы не должен содержать ее
примеси.
Практическая часть
Комплексонометрическое определение цинка
в медных концентратах
Принцип метода. Определение основано на титровании
цинка в ацетатной буферной среде pH 5,6—5,8 раствором
комплексона III в присутствии в качестве индикатора кси-
ленолового оранжевого. Свинец отделяют в виде сульфа-
та, другие метающие элементы маскируют комплексообра-
зующими веществами. Метод применении при содержании
цинка более 3%. Относительное стандартное отклонение
результатов определений 0,01.
Реактивы и растворы. 1) Аммиак, р = 0,91 г/см3 и разбавленный
1:1; 2) роданид аммония, раствор 100 г/л; 3) фторид аммония, раст-
вор 100 г/л; 4) бифторид аммония; 5) буферный раствор, pH 5,6—5,8
(125 мл уксусной кислоты смешивают со 135 мл аммиака, разбавляют
водой до 1000 мл и перемешивают): 6) кислота азотная, р= 1,40 г/см3
и разбавленная 1:1; 7) кислота соляная, р=1,19 г/см3 и разбавленная
1:1; 8) кислота серная, р=1,84 г/см3 и разбавленная 1:1, 1:99;
9) ксиленоловый оранжевый (смешивают индикатор с нитратом ка-
лия 1:99); 10) комплексон III, 0,025 М. раствор (для его приготовле-
ния 9,305 г комплексона III растворяют в 200—250 мл воды, перели-
вают в мерную колбу вместимостью 1000 мл, разбавляют водой до
метки и перемешивают); И) тиосульфат натрия, раствор 200 г/л;
12) цинк металлический гранулированный; 13) раствор, содержащий
в 1 мл 0,01 г цинка (для приготовления раствора 1,0000 г цинка
растворяют в 25 мл соляной кислоты при нагревании; раствор вы-
паривают до 3—5 мл, затем прибавляют 25 мл воды, 20 мл соляной
кислоты; полученный раствор переносят в мерную колбу вместимостью
100 мл, разбавляют водой до метки и перемешивают).
Выполнение определения. Навеску медного концентра-
та 0,5—1 г помещают в колбу вместимостью 250 мл, сма-
чивают водой, приливают 10—20 мл соляной кислоты.
Раствор нагревают в течение 15—20 мин, затем выпари-
вают до 5—7 мл, охлаждают, приливают 10—15 мл азот-
ной кислоты и продолжают нагревание до прекращения
выделения оксидов азота. После этого раствор вновь ох-
лаждают, приливают 20 мл разбавленной 1:1 серной кис-
лоты и выпаривают до обильного выделения паров сер-
ной кислоты. Остаток охлаждают, обмывают стенки колбы
водой в количестве 5—7 мл. Повторяют выпаривание раст-
вора до появления паров серной кислоты. Остаток снова
101
охлаждают, приливают к нему 100 мл воды и кипятят
15—20 мин.
Колбу с содержимым помещают в ванночку с проточ-
ной водой для охлаждения в течение 2 ч. Осадок отфильт-
ровывают на тампоне из фильтробумажной массы и про-
мывают разбавленной 1:99 серной кислотой до исчезно-
вения реакции на железо с роданидом аммония, а затем
два раза водой.
Фильтрат и промывание воды собирают в мерную кол-
бу вместимостью 250 мл для последующего определения
цинка.
Аликвотную часть раствора, содержащую 20—30 мг
цинка, помещают в коническую колбу вместимостью
250 мл и разбавляют водой до 50—70 мл. К раствору при-
бавляют аммиак небольшими порциями до начала выпа-
дения нерастворимого осадка гидроксидов, а затем при-
бавляют 20 мл раствора фторида аммония. Полученный
раствор перемешивают до растворения осадка гидрокси-
дов, прибавляют 1—2 г бифторида аммония, 20 мл раст-
вора тиосульфата натрия, 15 мл буферного раствора, 0,1 —
0,2 г индикатора ксиленолового оранжевого. Затем цинк
титруют раствором комплексона III до перехода фиолето-
вой окраски раствора в желтую.
Для установления титра 0,025 М. раствора комплексо-
на III, выраженного в граммах цинка, микробюреткой от-
меривают в три колбы 2, 4, 6 мл раствора цинка, при-
бавляют аммиак небольшими порциями и далее проводят
анализ, как описано при подготовке аликвотной части
раствора цинка к анализу. 1 мл 0,025 М раствора комп-
лексона III соответствует 1,6345 мг цинка.
Ускоренное полярографическое определение цинка
в сплавах на алюминиевой основе
Принцип метода. Содержание цинка определяют на
приборе переменного тока на фоне 1 М раствора хлорида
алюминия без предварительного отделения основы. Медь
в количестве до 40 мг определению не мешает. При боль-
ших количествах сопутствующего никеля из суммарной
волны следует вычитать волну никеля. Относительное
стандартное отклонение результатов определений 0,05 при
содержании цинка 1—2%.
Реактивы и растворы. 1) Алюминий металлический (99,95% А1);
2) бром; 3) хлорид гидроксиламмония, раствор 100 г/л; 4) кислота
соляная, р=1,19 г/см3 и разбавленная 1:1; 5) растворы цинка, никеля,
содержащие по 100 мкг/мл.
102
Выполнение опред -.ения. Нароску сплава 0,7 г раство-
ряют в 20 мл разбавленной соляной кислоты. По оконча-
нии растворения добавляют две капли брома и выпари-
вают раствор до получения влажных солей, затем смывают
стенки стакана водой и снова упаривают до влажных со-
лей. Растворяют соли в 10—15 мл горячей воды, добавля-
ют 2 мл раствора хлорида гидроксиламмония и кипятят
полученный раствор 1—2 мин. Затем раствор охлаждают,
переводят в мерную колбу вместимостью 25 мл, доливают
до метки водой и перемешивают.
Волну цинка снимают от потенциала — 0,9 В. Расчет
содержания цинка выполняют по градуировочному графи-
ку, построенному по добавкам раствора цинка к чисто-
му алюминию или по стандартному образцу. Для введе-
ния поправки на содержание никеля к навеске чистого
алюминия добавляют соответствующее количество раство-
ра никеля, растворяют алюминий в 20 мл разбавленной
соляной кислоты и производят определение, как описано
выше.
Атомно-абсорбционное определение цинка,
кадмия, меди в сульфидных минералах
Принцип метода. Определение основано на кислотном
разложении образца и фотометрировании полученного
раствора в воздушно-ацетиленовом пламени по резонанс-
ным линиям элементов. Метод применим при содержании
цинка до 1•10—5 %, марганца, железа, кобальта, никеля
или хрома до 1 10 3%. Относительное стандартное откло-
нение результатов определений 0,1.
Реагенты и растворы. 1) Кислота соляная, р= 1,19 г/см3; 2) кисло-
та азотная, р = 1,40 г/см3; 3) кислота винная, 20%-ный раствор; 4) раст-
воры металлов. Для построения градуировочного графика применяют
растворы, содержащие, мг/мл; кадмия 0,04; меди и хрома по 0,1; же-
леза, марганца, никеля, кобальта по 0,2, цинка 0,01.
Выполнение определения. Навеску образца 0,1—0,5 г
разлагают в 10 мл соляной кислоты, добавляют к раст-
вору несколько капель азотной кислоты и упаривают его
до небольшого объема. Затем добавляют к раствору 5 мл
соляной кислоты и нагревают досуха. К сухому остатку
добавляют 2 мл раствора винной кислоты, охлаждают,
переводят в мерную колбу вместимостью 50 или 100 мл,
доливают до метки водой. Полученный раствор фотомет-
рируют и определяют содержание элементов, используя
метод градуировочного графика.
103
Кадмий
Кадмий открыт в 1817 г. немецким ученым Ф. Штромейером в
карбонате цинка и назван им по греческому названию руды цинка
«кадмейя» или «кадмиа», которую древние греки добавляли к меди
при выплавке латуни.
Нахождение в природе. Кадмий-рассеянный элемент, постоянно
сопутствующий цинку. Содержание его в земной коре составляет
0,000013%. Его собственные минералы встречаются редко, основной
минерал гринокит CdS лишь тонкой корочкой покрывает цинковые ми-
нералы, в особенности сфалерит. Кадмий получают из отходов цин-
кового, медного и свинцового производства гидроэлектрометаллурги-
ческим методом.
Применение. Металлический кадмий применяют для антикор-
розионных покрытий, более устойчивых, чем цинковые, никелевые и
полученные лужением, а также для изготовления различных сплавов
(антифрикционных, легкоплавких, припоев, ювелирных, типографских)
с такими элементами, как медь, платина, золото, свинец, олово, железо
и др.
Кадмий высокой степени чистоты применяется в атомной про-
мышленности как поглотитель тепловых нейтронов. Кадмиевые электро-
ды применяются в аккумуляторах.
Свойства кадмия и его соединений. Кадмий — эле-
мент II группы Периодической системы Д. И. Менделе-
ева. Электронное строение атомов в основном состоянии —
ls22s22p63s23p63dl04s24p64dl05s2. Устойчивая степень окисле-
ния кадмия +2.
Кадмий — серебристо-белый мягкий металл, имеет тем-
пературу плавления 321 °C, не растворяется в щелочах,
медленно растворяется в кислотах, легче всего растворя-
ется в азотной кислоте: 3Cd + 8HNO3 = 3Cd(NO3)2 + 2NO +
+ 4Н2О.
Оксид кадмия CdO получают прокаливанием ни-
трата или карбоната кадмия.
Гидроксид кадмия Cd(OH)2, обладающий ос-
новными свойствами, получают действием щелочи на раст-
воры солей кадмия: Cd(NO3)2 + 2KOH = Cd(OH)2 + 2KNO3.
Труднорастворимый сульфид кадмия осаждается
из кислых растворов солей кадмия сероводородом, окра-
шен в оранжево-желтый цвет: Cd(NO3)9 + H2S4=iCdS +
+ 2НКтОз.
Оксид, гидроксид и сульфид растворимы в аммиаке,
цианидах и в кислотах.
сульфид кадмия растворим также в растворах гало-
генидов с образованием комплексной соли.
В аммонийно-аммиачной среде в присутствии сульфат-
и хлорат-ионов кристаллизуются труднорастворимые
соли составов: [Cd(NH3)4] (СЮ4)2; [Cd(OH2)2(NH3)4] SO4.
Руды, минералы, сплавы, концентраты, содержащие кад-
104
мий, разлагают кислотным способом. Кадмий бокситов,
хромитов и касситеритов восстанавливают в токе водоро-
да и растворяют в смеси соляной и азотной кислот (1:3).
Методы определения содержания кадмия
Наибольшее распространение получили два метода оп-
ределения содержания кадмия: полярографический (широ-
ко применяемый при массовых анализах) и атомно-аб-
сорбционный. Применяются также гравиметрические, элек-
трогравиметрические, титриметрические методы, основан-
ные на выделении труднорастворимых соединений кадмия
с последующим переводом их в растворимые комплексы,
а также комплексонометрические методы (прямое и об-
ратное титрование). Большое значение имеют фотомет-
рические методы.
Полярографический метод широко применяется при
анализе руд и продуктов металлургического производст-
ва, содержащих кадмий. Ионы кадмия легко восстанав-
ливаются на ртутном электроде: Cd2++2e+Hg = Cd(Hg).
Наиболее часто используют хлоридно-аммиачный фон
(£v,=-0,85 В).
Атомно-абсорбционный метод. Определение содержа-
ния кадмия от 0,002 мкг/мл проводят в пламени кислород-
водород, кислород-ацетилен по резонансной линии 228,8 нм.
Применяют также непламенные средства атомизации кад-
мия (в графитовой печи). Состав образца практически не
влияет на определение.
Фотометрический метод. Комплексные галогениды и
цианиды кадмия образуют окрашенные ассоциаты с анти-
пирилметаном, уротропином, тетрафениларсонием. Орга-
нические серо- и азотсодержащие соединения взаимодейст-
вуют с кадмием, образуя окрашенные комплексы, приме-
няющиеся для фотометрического определения кадмия.
При анализе сложных материалов часто применяют
ионообменные и экстракционные методы отделения со-
путствующих элементов с последующим фотометрирова-
нием дитизоната.
Дитизон (см. «Цинк») образует с кадмием в щелочной
среде комплексное соединение красного цвета, экстраги-
руемое органическими растворителями. В аналогичных
условиях (pH 6—14, хлороформ) реагируют катионы цин-
ка, меди, никеля, кобальта. В их присутствии при отде-
лении кадмия используют . различие устойчивости дитизо-
натов в зависимости от pH среды.
105
Практическая часть
Атомно-абсорбционное определение кадмия
в цинке и цинковых рудах
Принцип метода. Образец переводят в раствор кислот-
ным разложением и разбавляют водой до содержания кад-
мия 2—5 мкг/мл. Растворы фотометрируют при длине вол-
ны 228,8 нм в пламени пропан-бутан. Для увеличения
чувствительности определения в анализируемые растворы
вводят 5% изобутанола. Содержание кадмия определяют
методом добавок. Метод применим при содержании 0,2—
0,005 % Cd. Относительное стандартное отклонение резуль-
татов определений 0.03—0,06.
Реактивы и растворы. 1) Кислота соляная, р=1,19 г/см3; 2) кисло-
та азотная, р=1,40 г/см3; 3) изобутанол; 4) раствор кадмия, содержа-
щий кадмия 10 мкг/мл; 5) смесь азотной и соляной кислот в отноше-
нии 3:1.
Выполнение определения. Навеску металлического
цинка 2,5—5,0 г растворяют в 15 мл соляной кислоты,
переводят раствор в мерную колбу вместимостью 50 мл,
доводят водой до метки и перемешивают. В три пробир-
ки берут аликвотные части раствора по 5 мл, в две из
них добавляют раствор соли кадмия в количестве, содер-
жащем 20 и 50 мкг кадмия, разбавляют до 10 мл водой
и фотометрируют.
При анализе руды навеску 2,5 г растворяют в смеси
кислот, выпаривают до 1—5 мл, разбавляют полученный
раствор до объема 50 мл в мерной колбе, в три пробирки
берут аликвотные части раствора по 5 мл и далее посту-
пают, как описано выше.
Полярографическое определение кадмия
в чистых солях
Принцип метода. Определение основано на полярогра-
фировании кадмия на хлоридно-аммиачном фоне при по-
тенциале восстановления в интервале 0,7—1,0 В относи-
тельно нормального каломельного элемента. Относитель-
ное стандартное отклонение результатов определений 0,03.
Реактивы и растворы. 1) Аммиак, р = 0,91 г/см3; 2) хлорид аммо-
ния; 3) сульфит натрия; 4) желатина пищевая, 1%-ный раствор;
5) растворы кадмия; 6) раствор фона (для его приготовления 100 г
хлорида аммония и 100 г сульфита натрия растворяют в 500 мл воды,
добавляют 150 мл раствора аммиака,' 50 мл раствора желатины, раз-
бавляют водой до 1 л).
106
Выполнение определения. К 5—10 мл нейтрального
раствора соли, содержащего 0,25—50,0 мг кадмия, поме-
щенного в мерную колбу вместимостью 50 мл, приливают
20 мл раствора фона, доводят объем водой по метки,
перемешивают. Через 30 мин (после полного восстанов-
ления растворенного в жидкости кислорода) полярогра-
фируют при 0,7—1,0 В (н. к. э) и определяют содержа-
ние кадмия, используя градуировочный график, построен-
ный в идентичных условиях.
Олово
Олово известно с глубокой древности. Его латинское название
stannum означает стойкий. Русское название произошло от общесла-
вянского названия о1 — белый или желтый.
Нахождение в природе. Содержание в земной коре составляет
~ 0,008 %. Из 20 известных минералов олова промышленное значение
имеют два: касситерит (оловянный камень) SnO2 и станнин (оловян-
ный колчедан) Cu2FeSnS4 В виде примеси олово входит в состав
полиметаллических руд, в минералы титана, ниобия, тантала. Оловян-
ные руды с низким содержанием элемента предварительно обогащают.
Концентраты содержат 40—70% олова.
Применение. Металлическое олово в виде белой жести применяет-
ся в консервной промышленности, которая потребляет 40% выплав-
ляемого металла. Лужение посуды, производство фольги, припоев и
других различных сплавов — важные области применения олова. Олово
входит в состав бронз (сплавы олова с медью), типографских спла-
вов (сплавы олова со свинцом и сурьмой), баббитов (сплавы для под-
шипников, состоящие из олова, свинца, сурьмы и меди), сплава для
атомной энергетики с цирконием. На производство сплавов расходуется
более 50% выплавляемого металла.
Свойства олова и его соединений. Олово — элемент
IV группы Периодической системы Д. И. Менделеева,
входит в главную подгруппу германия. Электронное строе-
ние атома в основном состоянии — ls22s22p63s23p63d104s24ps
4dI05s25p2. Олово имеет две степени окисления +2 и +4,
из них последняя более устойчивая.
Металлическое олово серебристо-белого цвета, тускнеет
на воздухе. Это мягкий легкоплавкий металл (темпера-
тура плавления 231,9 °C), существующий в двух аллотроп-
ных формах: а — серое олово, устойчивое ниже 13,2 °C,
и р — белое олово, устойчивое выше 13,2 °C. Выше 161 °C
и ниже 13,2 °C олово легко рассыпается в порошок.
Олово легко растворяется в концентрированной соля-
ной кислоте, щелочах, царской водке: Sn + 2HCl = SnCl2 +
+ Н2; Sn + 2NaOH + O2 = Na2SnO3 + H2O; 3Sn + 4HNO3 +
+ 12HCl = 3SnC14 + 4NO + 8H2O.
Медленно идет растворение олова в разбавленной
азотной и концентрированной серной кислотах: 4Sn+
107
+ 10HNO3 = 4Sn(NO3)2 + NH4NO3 + 3H2O; Sn + 4H2SO4=
= Sn(SO4)2 + 2SO2 + 4H2O. Концентрированная азотная
кислота при кипячении образует нерастворимую р-оловян-
ную кислоту: Sn + 4HNO3 = H2SnO3 + 4NO2 + H2O. Выделе-
ние нерастворимой [3-оловянной кислоты применяется в
аналитической химии для отделения олова от сопутствую-
щих элементов.
Оксид олова (IV) SnO2 (белого цвета) резко от-
личается по растворимости от оксида олова (II) SnO (чер-
ного цвета). Оксид олова (IV) очень стоек по отношению
к кислотам и щелочам, а оксид олова (II) легко раство-
рим в кислотах.
Хлорид олова (II) легко окисляется до SnCl4, а
в присутствии избытка хлорид-ионов до хлоростаннат (IV)-
иона [SnCle]2~. Гидроксиды олова Sn(OH)2 и
Sn(OH)4 образуются при действии аммиака и щелочей
на растворы солей олова. Они амфотерны и растворяются
в кислотах с образованием соответствующих солей; при
растворении в щелочах образуются соответственно стан-
ниты и станнаты [Sn(OH)3]_, [Sn(OH)6]2~.
Разложение материалов, содержащих олово, проводят
различными методами в зависимости от их составов и
состояния содержащегося в них олова. Наибольшие труд-
ности возникают при переведении в раствор касситерита,
который разлагается или сплавлением с Na2O2, со щело-
чами в присутствии пероксида натрия, металлического
цинка, натрия, калия, или же сплавлением с бурой. Иног-
да применяют для разложения касситерита длительный
метод нагревания в токе водорода, отличающийся от ще-
лочного сплавления тем, что в раствор не вводятся посто-
ронние металлы. Оловянные руды и концентраты, содер-
жащие касситерит, разлагают также с помощью иодида
аммония. При нагревании возгоняется тетраиодид олова,
растворимый в серной кислоте: SnO2 + 4NH4I = SnI4 +
4NH3 + 2H2O.
Методы определения содержания олова
Обычно определению олова предшествует отделение его
от сопутствующих элементов: меди, кремния мышьяка,
железа, марганца и т. д. При растворении оловянных спла-
вов в азотной кислоте удобно отделять олово в виде |3-оло-
вянной кислоты. Применяют также отгонку олова в виде
летучих галогенидов SnBr4, Snl4, восстановление олова до
металла цинком, алюминием, кадмием: [SnCI6] 2-+ 2Zn =
108
= Sn + 2ZnCl2 + 2Cb, осаждение или экстракцию купфе-
роната.
Наиболее распространенные титриметрические химиче-
ские методы определения олова основаны на окислитель-
но-восстановительных реакциях. Применяются фотометри-
ческие методы, например дитиоловый и фенилфлуороно-
вый. Перспективным методом определения олова является
рентгенорадиометрический.
Титриметрические методы. Растворимые станнаты, по-
лученные после сплавления оловосодержащих материалов
с Na2O2, при подкислении раствора переходят в соли оло-
ва (IV), которые восстанавливают до олова (II) метал-
лическим железом: [SnC16]2-+Fe = SnCl2 + FeCl24-2CI_, а
затем титруют иодометрически: Sn2++I2 = Sn4++2I~. В ка-
честве восстановителя, кроме железа, применяют алюми-
ний, никель, свинец и др.
Фотометрический метод. Фенилфлуорон
с6н5
или С19 Н12О5 (молекулярная масса 320, 30), образует при
рН1 с оловом (IV) труднорастворимый комплекс, который
можно удержать в растворе в дисперсном состоянии в
присутствии защитного коллоида. Молярный коэффициент
Рис. 12. Кривые свето-
поглощекия водных ра-
створов фенилфлуорона
</) и его соединения с
оловом (2)
'0 500 Л, НИ
погашения псевдораствора оранжево-красного цвета при
&тах=510 нм (рис. 12) равен 7,7-104. Определению олова
мешают окислители и многие катионы. Сурьму маскируют
лимонной кислотой; тантал, ниобий, молибден и титан —
109
пероксидом водорода. При выполнении определения тре-
буется строгое соблюдение pH (1,1—1,2). При его заниже-
нии реакция не протекает количественно, при завышении —
в осадок выпадает реагент желтого цвета.
Практическая часть
Спектрофотометрическое
определение олова в меди
Принцип метода. Метод основан на измерении свето-
поглощения окрашенного в красный цвет соединения оло-
ва с фенилфлуороном. Предварительно олово отделяют от
мешающих элементов осаждением его совместно с гидрок-
сидом железа (III); относительное стандартное отклоне-
ние результатов определения составляет 0,1 при содер-
жании олова 0,01—0,05%.
Реактивы и растворы. 1) Бромная вода, насыщенный раствор;
2) кислота соляная, р=1,19 г/см3; 3) кислота азотная, р=1,40 г/см3
и разбавленная 1:1; 4) кислота серная, р = 1,84 г/см3, разбавленная
1:1, 1:4, 1:9; 5) аммиак, р = 0,91 г/см3 и разбавленный 1:1, 1:50;
6) аммония нитрат; 7) кислота винная, 5%-ный раствор; 8) кислота
аскорбиновая, 5%-ный раствор; 9) сульфат железа (III), 1%-иый
раствор; 10) желатина, 0,5%-ный раствор; 11) индикаторная бумага
конго; 12) спирт этиловый; 13) фенилфлуорон, 0,03 %-ный спиртовой
раствор (для его получения 0,03 г фенилфлуорона растворяют при
нагревании на водяной бане в 100 мл этилового спирта, содержащего
1 мл серной кислоты, разбавленной 1:1); 14) олово марки 01; 15) за-
пасной раствор олова (для его получения 0,1 г измельченного олова
растворяют в 10 мл серной кислоты при нагревании; после охлаж-
дения приливают 180 мл серной кислоты, разбавленной 1:1, раствор
перемешивают и переносят в мерную колбу вместимостью 1000 мл,
доливают до метки водой и вновь перемешивают); в 1 мл раствора
содержится 0,1 мл олова; 16) рабочий раствор (для его получения
10 мл запасного раствора переносят пипеткой в мерную колбу вмести-
мостью 100 мл и доливают до метки раствором серной кислоты, раз-
бавленной 1:9); в 1 мл раствора содержится 0,01 мг олова.
Построение градуировочного графика. В семь мерных
колб вместимостью 25 мл прибавляют по 2 мл раствора
сульфата железа (III), 0,3 мл раствора винной кислоты
и затем последовательно 0; 0,5; 1,0; 1,5; 2,0; 3,0 мл рабо-
чего раствора олова, нейтрализуют растворы аммиаком,
разбавленным 1:1 до щелочной реакции по бумаге конго,
прибавляют 1,6 мл разбавленного 1:1 раствора серной
кислоты, доливают водой до объема 15 мл. По каплям
прибавляют бромную воду до тех пор, пока раствор не
приобретет желтую окраску, и оставляют на 5 мин. Избы-
ток брома удаляют прибавлением раствора аскорбиновой
110
9
кислоты до обесцвечивания и в избыток еще 1 мл, раствор
вновь оставляют на 5 мин. Затем прибавляют 2,5 мл рас-
твора желатины, 5 мл раствора фенилфлуорона, доливают
водой до метки и оставляют стоять на 30 мин. Измеряют
оптическую плотность растворов при длине волны 510 нм,
используя подходящую кювету по отношению к раствору
контрольного опыта, не содержащего олова. По найден-
ным значениям оптических плотностей строят градуиро-
вочный график.
Выполнение определения. Навеску меди 1 г помещают
в стакан вместимостью 400 мл и растворяют в 15 мл азот-
ной кислоты, разбавленной 1:1. После окончания бурной
реакции прибавляют 5 мл соляной кислоты. Раствор осто-
рожно нагревают до полного растворения меди и удаления
оксидов азота, затем доливают водой до 200 мл и при-
бавляют 2 мл раствора сульфата железа. Нагревают до
температуры 60—70 °C, гидроксиды осаждают аммиаком,
разбавленным 1:1, который прибавляют при перемешива-
нии в таком количестве, чтобы вся медь перешла в ком-
плексное соединение, и еще в избыток 1 мл.. Раствор с
осадком выдерживают на водяной бане в течение 10—
15 мин, а затем фильтруют на фильтре средней плотности.
Стакан дважды обмывают горячим раствором аммиака,
разбавленного 1:1. Фильтр с осадком промывают 5—6 раз
этим же раствором аммиака. Осадок смывают в стакан,
в котором производили осаждение гидроксидов, прибав-
ляют 10 мл серной кислоты, разбавленной' 1:1, доливают
водой до 150 мл и вновь осаждают гидроксиды. Осадок
фильтруют на том же фильтре и промывают два раза го-
рячим раствором аммиака, разбавленным в соотноше-
нии 1:50.
Осадок растворяют горячим раствором серной кислоты,
разбавленной 1:4. В стакан прибавляют 5 мл серной кис-
лоты, разбавленной 1:1. Раствор осторожно выпаривают
до появления паров серного ангидрида. В случае, если
раствор имеет темную окраску, прибавляют несколько
кристалликов соли нитрата аммония и затем снова выпа-
ривают до обесцвечивания раствора. Остаток охлаждают,
стакан ополаскивают 10 мл воды, раствор переливают в
мерную колбу вместимостью 250 мл и доливают до метки
раствором серной кислоты, разбавленной 1:4. Отбирают
аликвотную часть раствора 25—10 мл в стакан вмести-
мостью 100 мл и снова выпаривают до появления белых
паров SO3 (остаток должен быть влажным). Раствор
охлаждают, нейтрализуют раствором аммиака, разбавлен-
111
ным 1:1, по индикаторной бумаге конго и по каплям при-
бавляют раствор аммиака, разбавленный 1:1, до измене-
ния окраски бумаги от синего до красного цвета. Затем
в раствор прибавляют 1,6 мл серной кислоты, разбавлен-
ной 1:1 и далее поступают, как описано при построении
градуировочного графика.
Полярографическое
определение олова в меди
Принцип метода. Метод основан на восстановлении оло-
ва из оксалатного комплекса на ртутном электроде в при-
сутствии электролита, содержащего метиленовый голубой.
Медь осаждается в виде оксалата и не мешает определе-
нию олова. Введение метиленового голубого повышает чув-
ствительность определения олова вследствие адсорбции
на ртутном электроде.
Применяется полярограф с наложением переменного
напряжения. Относительное стандартное отклонение ре-
зультатов определения составляет 0,2—0,1 при содержа-
ниях олова 0,003—0,05%.
Реактивы и растворы. 1) Кислота .азотная, р = 1,40 г/см3, разбав-
ленная 1:1; 2) кислота серная, р = 1,84 г/см3, разбавленная 1:1 и 1:9;
3) кислота фтористоводородная, р=1,13 г/см3, 1М раствор; 4) метиле-
новый голубой (МГ), 0,37 %-ный водный раствор; 5) олово марки 01;
6) растворы олова (готовят так же, как указано в методике спектро-
фотометрического определения олова и меди); 7) щавелевая кислота,
1М раствор.
Выполнение определения. Навеску меди 0,5 г раство-
ряют в 10 мл азотной кислоты, разбавленной 1:1, прили-
вают 10 мл серной кислоты, разбавленной 1:1, нагревают
до выделения паров серной кислоты, охлаждают, обмы-
вают стенки стакана раствором серной кислоты, разбав-
ленной 1:9, снова нагревают до выделения паров серной
кислоты и обмывают стенки стакана серной кислотой,
разбавленной 1:9. Раствор выпаривают до прекращения
выделения паров серной кислоты (не пересушивая), при-
ливают 25 мл горячего раствора щавелевой кислоты.
Раствор кипятят в течение 3—5 мин и оставляют на
20 мин для осаждения оксалата меди. Затем раствор с
осадком переносят в мерную колбу вместимостью 50 мл
раствором щавелевой кислоты. Прибавляют 1,5 мл раство-
ра метиленового голубого, доливают до метки раствором
щавелевой кислоты и перемешивают. Часть раствора де-
кантируют на плотный фильтр и собирают в сосуд для
112
пропускания азота, продувают азотом и полярографируют
от —0,55 В. Потенциал пика —0,9 В (донная ртуть).
Расчет проводят методом добавок. Для этого отбирают
часть рабочего раствора олова, выпаривают до прекраще-
ния выделения паров серной кислоты (не пересушивая).
К остатку прибавляют часть анализируемого раствора,,
перемешивают и после отстаивания осадка раствор декан-
тируют на плотный фильтр, продувают азот и полярогра-
фируют, как описано выше.
Свинец
Металлический свинец был доступен людям с глубокой древнос-
ти. Происхождение названия элемента не установлено.
Нахождение в природе. В земной коре содержится 0,0016% свинца.
Основным минералом свинца является галенит (свинцовый блеск) PbS,.
которому сопутствуют сульфиды меди, железа, серебра, цинка. Суль-
фидное сырье перерабатывается комплексно. Источником промышлен-
ного добывания свинца является свинцово-цинковые полиметаллические'
РУДЫ.
Применение. Свинец применяется в производстве аккумуляторов,-
кабелей, в химической промышленности он служит материалом реак-
ционных приборов для защиты от радиоактивного излучения, из него
изготовляют легкоплавкие сплавы (типографские, припои).
Свойства свинца и его соединений. Свинец — эле-
мент IV группы Периодической системы Д. И. Менде-
леева, относится к подгруппе германия.
Электронное строение атомов в основном состоянии
/s22s22p63s23p63d104s24/?64d104P45s25p65d106s26/72. Наиоблее
устойчивая степень окисления свинца +2, степень окисле-
ния атома +4 неустойчивая, РЬО2— очень сильный окис-
литель.
Металлический свинец очень мягкий металл, имеет се-
рый цвет с синеватым оттенком. Поверхность свежеполи-
рованного свинца серо-белого цвета, но она быстро туск-
неет на воздухе, покрываясь защитной оксидной пленкой.
Металл легко растворяется в азотной кислоте, в горячей
концентрированной серной кислоте с образованием кис-
лой соли, в других кислотах практически нерастворим:
3Pb + 8HNO3 = 3Pb(NO3)2+2NO + 4H2O; PbSO4 + H2SO4=
= Pb(HSO4)2.
Оксид свинца (II) РЬО существует в виде двух
модификаций: красная — глет, желтая — массикот; обе
модификации растворимы в кислотах с образованием со-
лей свинца (II), а в щелочах — плюмбитов: РЬО +
+ 2СН3СООН = РЬ (СНзСОО) 2+ Н2О; РЬО + 2NaOH =
= Pb(OH)2 + Na2O; Pb(OH)2 + 2NaOH = Na2PbO2 + 2H2O.
па
Большинство солей свинца труднорастворимо, многие
из них используются для отделения свинца от других эле-
ментов. Например, осадок сульфата свинца PbSO4 раство-
ряют в ацетате или тартрате аммония с образованием
комплексных ионов или в щелочи с образованием плюмби-
тов, отделяя таким образом свинец от сульфатов щелоч-
ноземельных элементов:
GOONH4
।
PbSO4+CHOH =
I
снон
I
COONH4
COONH4
I
CH(L
I >b + Н2504
ОНО
I
сооын4
Методы определения содержания свинца
Широкое применение находят полярографический, атом-
но-абсорбционный и дитизоновый фотометрический методы
определения содержания свинца. Часто используется кос-
венный'хроматный титриметрический метод, а также ком-
плексонометрический метод.
Атомно-абсорбционный метод. В процессе анализа ре-
комендуется фотометрировать резонансную линию 283 нм,
по которой можно определить содержание свинца от
0,02 мкг/мл. В пламени воздух—ацетилен помехи со сто-
роны других элементов практически отсутствуют. В низко-
температурных пламенах мешают элементы образующие
со свинцом труднорастворимые соединения; их влияние
можно устранить добавлением комплексона III.
Комплексонометрический метод. Комплексонометриче-
ское титрование свинца проводят в аммонийно-аммиачной
буферной среде pH 10 или в ацетатной среде pH 6,5—7
соответственно с индикаторами эриохромчерным Т (см.
«Алюминий») или ксиленоловым оранжевым (см. «Алю-
миний»). В среде pH 8—9 применяют также сульфарсазен
(см. «Никель») в качестве индикатора, обладающий чет-
ким переходом окраски в эквивалентной точке. При ана-
лизе сложных систем требуется предварительное отделе-
ние мешающих элементов. Например, свинец отделяют
от сопутствующих элементов в виде сульфата, растворяют
осадок в избытке комплексона III и титруют свинец рас-
твором соли цинка способом обратного титрования.
Хроматный титриметрический метод. Свинец выделяют
в виде хромата, растворяют его в смеси соляной кислоты
и хлорида натрия и определяют иодометрически эквива-
114
лентное свинцу количество хромовой кислоты: РЬ2+-Ь
СгО 1~= РЬСгО4; 2РЬСгО4+ 2Н+=2РЬ2+ + Сг2О72- + Н2О;:
Cr2O7~ + 14H++6I- = 3I2 + 2Cr3++7H2O; I2 + 2S2C32- =
= S4O42- + 2I-
Малорастворимые хроматы образуются многими эле-
ментами, в связи с этим свинец предварительно осаждают
в виде сульфата, растворяют сульфат в ацетате аммония
и осаждают хромат в ацетатной среде. Барий мешает
определению содержания свинца.
Практическая часть
Полярографическое определение свинца
в цинковых концентратах
Принцип метода. Метод основан на восстановлении
иона свинца (II) на ртутном капельном электроде в соля-
нокислом растворе. Мешающие определению ионы желе-
за (III) и меди (II) устраняют железом, восстановленным
водородом.
Применяется полярограф с наложением постоянного
напряжения с ртутными электродами; относительное стан-
дартное отклонение результатов определения составляет
0,03 при содержаниях 3—5 % свинца.
Реактивы и растворы. 1) Кислота соляная, р=1,19 г/см3 и разбав-
ленная 1:1, 1:3; 2) кислота азотная, р=1,40 г/см3 и разбавленная
1:1; 3) железо хлорное, 2%-ный раствор; 4) желатина пищевая,
0,5%-ный свежеприготовленный раствор; 5) железо, восстановленное
водородом; 6) свинец марки СО; 7) раствор, содержащий в 1 мл 1 мг
свинца (для его получения 1,000 г свинца растворяют в 10 мл раз-
бавленной азотной кислоты и выпаривают раствор до объема 3 мл.
К остатку приливают 15 мл соляной кислоты и выпаривают досуха.
Выпаривание повторяют два раза, каждый раз добавляя 5 мл соляной
кислоты. К остатку приливают 250 мл разбавленной 1:1 соляной кисло-
ты, раствор нагревают до растворения хлорида свинца, переливают
в мерную колбу вместимостью 1000 мл, прибавляют еще 250 мл раз-
бавленной 1:1 соляной кислоты, охлаждают, доливают до метки водой
и перемешивают).
Выполнение' определения. Навеску концентрата 1—
0,25 г помещают в стакан вместимостью 250 мл, прили-
вают 20 мл соляной кислоты (р = 1,19), нагревают на пе-
сочной бане 30 мин, прибавляют 10 мл азотной кислоты
(р=1,40) и, накрыв стакан часовым стеклом, продолжают
нагревание до прекращения бурного выделения оксидов
азота. Стекло обмывают над стаканом небольшим количе-
ством воды и раствор выпаривают почти досуха. Выпари-
115
вание раствора повторяют еще дважды, прибавляя каждый
раз 5 мл разбавленной 1:1 соляной кислоты. К остатку
приливают 20 мл разбавленной 1:1 соляной кислоты и на-
гревают 15 мин. К раствору приливают 20 мл воды и
вместе с нерастворимым остатком переливают в мерную
колбу вместимостью 250 мл. Охлаждают раствор, прибав-
ляют 50 мл соляной кислоты (р = 1,19) и после охлажде-
ния до комнатной температуры раствор разбавляют до
метки водой, перемешивают и дают осесть нерастворимому
осадку на дно колбы. Далее отбирают часть раствора и
восстанавливают железо и медь порошком железа, восста-
новленного водородом. Раствор с порошком железа остав-
ляют на 25 мин, затем фильтруют через сухой фильтр,
собирая фильтрат в сухой стакан.
Часть фильтрата переносят в электролизер, прибавляют
0,5 мл раствора желатина и полярографируют в интервале
от — 0,2 до —0,6 В по отношению к ртутному аноду.
Для приготовления эталонных растворов свинца в мер-
ные колбы вместимостью 250 мл отмеривают бюреткой 5,
10, 20 и 25 мл раствора свинца, что соответствует кон-
центрациям свинца 20, 40, 80 и 100 мг/л, приливают по
5 мл раствора хлорного железа доливают растворы до
метки разбавленной 1:3 соляной кислотой, перемешивают
и полярографируют, как указано выше.
Содержание свинца (х) в процентах вычисляют по
•формуле
х —
~ КО 1000-1000 ’
где Н — высота волны свинца, полученная при полярогра-
фировании испытуемого раствора, мм; V — объем мерной
колбы, мл; К — средняя величина отношений волн, полу-
ченных при полярографировании эталонных растворов, к
концентрациям этих растворов, мм-л/мг; G — навеска кон-
центрата, г.
Комплексонометрическое определение свинца
в свинцовом сурике
Принцип метода. Свинцовый сурик РЬ3С>4 является
смесью оксидов свинца (II и IV) (2:1). При растворении
в горячей азотной кислоте в присутствии пероксида водо-
рода Pb (IV) восстанавливается до Pb (II). Метод осно-
ван на титровании Pb (II) раствором комплексона III в
присутствии индикатора ксиленолового оранжевого в сре-
.116
де уротропина. Относительное стандартное отклонение ре-
зультатов определения 0,02.
Реактивы и растворы. 1) Кислота азотная, р=1,40 г/см3, разбав-.
ленная 1:4; 2) ксиленоловый оранжевый, 0,1%-ный раствор; 3) метило-
вый оранжевый, 0,1%-ный раствор; 4) гидроксид натрия, 5 н. раствор;
5) комплексон III, 0,05 М. раствор; 6) пероксид водорода, 30%-ный
раствор; 7) уротропин.
Выполнение определения. Навеску свинцового сурика
0,2 г помещают в коническую колбу вместимостью 250 мл,
прибавляют 15 мл раствора азотной кислоты и две капли
пероксида водорода. Раствор перемешивают, разбавляют
водой до 50 мл и кипятят 2 мин. После полного растворе-
ния сурика раствор охлаждают, разбавляют водой до
100 мл, прибавляют две-три капли раствора метилового
оранжевого и нейтрализуют раствором гидроксида натрия
до желтой окраски. Прибавляют 0,5 мл раствора ксилено-
лового оранжевого и порциями на кончике шпателя при
перемешивании, уротропин до сине-фиолетовой окраски
раствора (pH 5). Титруют раствором комплексона III до
перехода окраски раствора в лимонно-желтую. 1 мл 0,05 М
раствора соответствует 0,01116 г РЬО.
Йодометрическое определение диоксида свинца
в свинцовом сурике
Принцип метода. Определение основано на селектив-
ном растворении РЬО2 смесью растворов уксусной кисло-
ты и уксуснокислого натрия, восстановлении Pb (IV) до
Pb (II) иодидом калия и титрования выделившегося иода
раствором тиосульфата натрия. Относительное стандарт-
ное отклонение результатов определения 0,006.
Реактивы и растворы. 1) Калий йодистый; 2) кислота уксусная,
разбавленная 1:1; 2) крахмал, 0,5%-ный раствор, свежеприготовлен-
ный; 3) натрий серноватисто-кислый, 0,1 н. раствор; 4) натрий уксусно-
кислый, насыщенный раствор.
Выполнение определения. Навеску свинцового сурика
0,2 г помещают в коническую колбу вместимостью 400—
500 мл с притертой пробкой, содержащую раствор, состоя-
щий из 20 мл раствора уксуснокислого натрия и 3 мл рас-
твора уксусной кислоты. Непосредственно перед введением
навески прибавляют 0,5 г йодистого калия. Раствор раз-
бавляют водой до 50—70 мл, закрывают колбу пробкой
и тщательно перемешивают до полного растворения. Опо-
ласкивают пробку водой, доводят объем раствора водой
до ,Щ0 мл, прибавляют 1 мл раствора крахмала и титруют
117
раствором серноватистокислого натрия до исчезновения
синей окраски раствора. 1 мл 0,1 н. раствора соответствует
0,01196 г РЬО2.
VIII. МЕТОДЫ ОПРЕДЕЛЕНИЯ СОДЕРЖАНИЯ
РЕДКИХ МЕТАЛЛОВ
Титан
Титан открыт в 1790 г. английским любителем-минералогом Грегером.
В 1795 г. немецкий химик и минералог Клапрот проанализировал не-
известный в то время минерал рутил и обнаружил в нем новый эле-
мент, которому он дал название «титан» (в греческой мифологии Ти-
тан—•сын богини Земли). Через несколько лет Клапрот подтвердил,
что элемент, открытый Грегором и им, является одним и тем же ве-
ществом.
Нахождение в природе. Титан относится к числу элементов, ши-
роко распространенных в природе. Содержание его в земной коре
составляет 0,61%, что соответствует девятому месту после кислорода,
кремния, алюминия, железа, кальция, натрия, калия, магния. Титан
обнаружен в метеоритах, в образцах Лунных пород, доставленных на
землю космическими кораблями. В природе существует много мине-
ралов с высоким содержанием этого элемента. Отнесение титана к
числу редких металлов связано с трудностями, которые возникали при
его получении из руд.
К основным минералам титана относятся рутил, ильменит, титано-
магнетит. В рутиле содержится до 97% ТЮ2 и в качестве примесей —
железо, ниобий, тантал, олово, хром, ванадий, марганец. Ильменит
(титанистый железняк) представляет собой титанат железа FeTiOj,
содержащий —32% титана и —37% железа, в качестве примесей
присутствуют магний и марганец. Титаномагнетит— смесь ильменита
и магнетита — имеет формулу mFeTiO3-/iFe2O3. Реже встречаются дру-
гие минералы: сфен (титанит) CaTiSiO5, содержащий 40,8% TiO2, и
примеси оксидов железа, марганца, ниобия и др.; перовскит СаТЮ3,
содержащий 58,9% Ti с изоморфной примесью ниобия, тантала, лан-
тана.
Титан также входит в состав минералов, включающих редкозе-
мельные элементы, тантал, ниобий, ванадий, например, в лопарит,
химическая формула которого (Са, Се, Na)2 (Nb, Ti)2O6; минерал со-
держит 32,22% TiO2 (полный химический состав см. «Ниобий и тан-
тал»). Титан присутствует в осадочных породах — бокситах, глинах.
Применение. Широкому применению титана способствует исключи-
тельная коррозионная стойкость металла и его сплавов в агрессивных
химических средах, а также в морской воде. При высоких температу-
рах сплавы титана превосходят по прочности алюминиевые сплавы
и даже нержавеющую сталь. Титан и его сплавы широко применяются
в авиационной технике, ракетостроении, судостроении, химическом ма-
шиностроении. Порошок металлического титана находит применение
как поглотитель газов (геттер) в электровакуумной технике. Диоксид
титана используется в качестве белого пигмента (титановые бе-
лила).
118
Свойства титана и его соединений. Титан-—• химиче-
ский элемент IV группы Периодической системы Д. И. Мен-
делеева, относится к первому ряду переходных элементов,
является элементом d-группы.
Электронная конфигурация атомов в основном состоя-
нии— ls22s22p63s23p63d24s2; в атоме имеются четыре ва-
лентных электрона (3d24s2). Устойчивость состояния окис-
ления уменьшается от Ti (IV) к Ti (III) и Ti (II).
Титан — тугоплавкий металл серебристого цвета. Тем-
пература плавления 1668±4°С. Как отмечалось, его кор-
розионная стойкость является одним из наиболее ценных
свойств. При комнатной температуре титан не- раство-
ряется в минеральных кислотах, водных растворах щело-
чей; он нерастворим и в горячих водных растворах щело-
чей. Растворяется при нагревании в разбавленных соляной
и серной кислотах с образованием соединений Ti (III),
окрашенных в фиолетовый цвет. Эти соединения являются
неустойчивыми: при взаимодействии с кислородом возду-
ха Ti (III) постепенно окисляется до Ti (IV), соединения
которого бесцветны: 2TiCl3 + 2HCl + 1/2O2 = 2TiCl4+H2O.
Для ускорения окисления титана к сернокислому или соля-
нокислому растворам, полученным после растворения ти-
тана, добавляют какой-либо окислитель, например азот-
ную кислоту.
Соединения титана при недостаточно высокой концен-
трации кислоты (<5% H2SO4) легко подвергаются гидро-
лизу, особенно при нагревании, в результате чего выпадает
в осадок метатитановая кислота: Ti(IV)+3H2O = H2TiO3| +
+ 4Н+.
Пероксид водорода способствует получению устойчивых
растворов титана благодаря образованию комплекса с
Ti (IV).
При действии фтористоводородной кислоты на титан
сначала образуется TiF3, который в присутствии окисли-
теля (кислорода воздуха или HNO3) переходит в TiF4 и
далее, при избытке HF, в комплексную фторотитановую
кислоту: 2Ti + 6HF = 2TiF3 + ЗН2, 2TiF3 + >/2О2 + 2HF = 2TiF4+
+ Н2О, TiF4+2HF = H2[TiF6],
Фторид титана TiF4 летуч; во избежание потерь
титана растворение ведут в присутствии серной кислоты,
при этом образуется устойчивая титанилсерная кислота:
TiF4 + 2H2SO4+H2O = H2[TiO(SO4)2]+4HF, где TiO2+ —
титанил-ион.
Перевод в раствор металлического титана осуще-
ствляется также сплавлением с пиросульфатом калия и
119
последующим растворением плава в растворе органиче-
ской кислоты, например щавелевой или винной, с образо-
ванием соответственно титанилщавелевой Н2 [TiO (€204)2!
или титанилвинной Н2 [TiO (С4Н4О6)2] кислоты. Кроме ука-
занного способа, для растворения плава применяют сер-
ную кислоту, часто в присутствии пероксида водорода.
Гидроксид титана Н2ТЮз, или дигидроксид оксо-
титана (IV) ТЮ(ОН)2 (метатитановая кислота)—белый
кристаллический осадок, нерастворимый в серной, азот-
ной и соляной кислотах. Гидроксид титана H4TiO4 или
Ti(OH)4 (ортотитановая кислота) получают осаждением
на холоду из водных растворов солей титана щелочью.
Это белый аморфный хлопьевидный осадок, растворимый
в минеральных кислотах. При кипячении переходит в бо-
лее стабильный гидроксид титана H2TiO3.
Диоксид титана TiO2 — порошок белого цвета, с
трудом растворимый в концентрированной серной кислоте
даже при длительном нагревании: TiO2 + 2H2SO4 =
= Н2 [TiO(SO4)2]+Н2О. В результате образуется титанил-
серная кислота; ускорению процесса растворения TiO2
способствует добавление сульфата аммония.
Более быстрое разложение TiO2 происходит при его
сплавлении с пиросульфатом калия при температуре
— 900 °C. Плав можно растворить в растворе органической
кислоты, например винной, или серной кислоты, с добавле-
нием пероксида водорода. В растворимое состояние оксид
титана (IV) можно перевести при нагревании с фтористо-
водородной кислотой: TiO2 + 6HF = H2 [TiF6]+2Н2О.
Разложение титансодержащих руд, минералов и кон-
центратов производят следующими способами: а) раство-
рением в серной кислоте или серной кислоте в присутствии
сульфата аммония; б) сплавлением пиросусльфатами ще-
лочных металлов.
При анализе ряда минералов сложного состава необхо-
димо предварительно обработать пробу смесью фтористо-
водородной и серной кислот (с последующим удалением
фтористоводородной кислоты) и дополнительно сплавить
пробу с пиросульфатом калия (или натрия).
Разложение сплавов титана. Сплавы титана раство-
ряют в разбавленных серной или соляной кислотах с по-
следующим добавлением азотной кислоты (для окисления
Ti (Ш) до Ti (IV). Применяют также смесь фтористо-
водородной и азотной или серной кислот. Недостатком
этого способа является необходимость удаления фтористо-
120
водородной кислоты, мешающей последующему определе-
нию титана.
Удобным и надежным способом разложения некоторых
сплавов титана, не растворяющихся в минеральных кисло-
тах, является сплавление с пиросульфатом калия (или
натрия). Этот способ успешно используется, например, при
анализе сплавов, содержащих ниобий.
Методы определения содержания титана
Фотометрические методы имеют наибольшее практиче-
ское применение для определения содержания титана. Они
основаны на избирательных цветных реакциях титана с
неорганическими и особенно органическими лигандами.
Эти методы применяют для определения как микро-, так
и макроконцентраций титана.
Ниже рассмотрены реагенты, имеющие наибольшее при-
менение для определения титана в различных материалах:
1. Пероксид водорода. Пероксидный метод определе-
ния содержания титана является наиболее старым и одним
из распространенных методов благодаря своей простоте и
доступности реагента. Метод основан на образовании в
сернокислом растворе в присутствии пероксида водорода
окрашенного в желтый или оранжевый (при высоких кон-
центрациях титана) цвет комплексного соединения титана.
По данным разных авторов, образуется пероксотитановая
[ТЮ(Н2О2)]2+ или пероксотитаносерная [TiO2 (SO4) 2]2—
кислота: Ti (ЗО^г + НгОг + ЗНгО^НДЮяН-гНгЭСЛ или
TiOSO4 + Н2О2 + H2SO4 = Н2 [TiO2 (SO4)2] + Н2О.
Метод достаточно избирателен, но обладает сравни-
тельно невысокой чувствительностью. Молярный коэффи-
циент погашения при длине волны Хтах = 410 нм равен
— 7,0-102. Оптимальной кислотностью образования ком-
плексного соединения является 1—2 М по серной кис-
ло'те. Окрашенные ионы Fe (III) при их высоких содер-
жаниях мешают определению титана. Для устранения
влияния железа применяют фосфорную кислоту, которая
связывает железо в бесцветный комплекс [Fe(PO4)2] 3~.
Фосфорная кислота ослабляет окраску также и комплекс-
ного соединения титана в связи с образованием бесцвет-
ного комплексного аниона, поэтому кислоту вводят в
стандартные растворы. К другим элементам, мешающим
определению титана окраской собственных ионов или об-
разующим с пероксидом водорода окрашенные соединения,
относятся никель (II), хром (III), ванадий (V), молиб-
ден (VI), ниобий (V).
121
Влияние указанных элементов устраняют либо пред-
варительным их отделением от титана, либо их введе-
нием в раствор сравнения. В последнем случае необхо-
димо первоначально установить содержание элементов в
анализируемой пробе. При использовании фотоэлектро-
колориметров и особенно спектрофотометров метод стано-
вится более избирательным по сравнению с визуальным
определением. Например, влияние никеля, хрома значи-
тельно уменьшается.
Фториды, образующие более прочные комплексные сое-
динения с титаном, ослабляют окраску, при высоких со-
держаниях фторидов растворов обесцвечивается.
Достоинством пероксидного метода является высокая
устойчивость соединения (окраска устойчива в течение
14 сут).
Строгое подчинение окраски закону Бугера—Ламбер-
та — Бера в широком диапазоне содержаний титана позво-
ляет использовать метод для определения не только низ-
ких, но и высоких концентраций титана (дифференциаль-
ный вариант метода).
Дифференциальные измерения на спектрофотометрах
рекомендуется проводить не при tanax = 410 нм, а при длине
волны 390 нм, когда действие рассеянного света можно
уменьшить с помощью светофильтра УФС-2. Обычно этот
светофильтр рекомендуется использовать при измерениях
в области 320—380 нм. Однако при дифференциальном
способе измерений велико влияние «рассеянного» света,
поэтому и при ?i = 390 нм необходимо работать с использо-
ванием указанного светофильтра.
2. Диантипирилметан (ДАМ) или C23H24O2N4 (молеку-
лярная
н3с-с=с-сн2-с=с-сн3
II II
H,C-N 0=0 0 = 0 N-CH3 ,
•* \ / \ /
N N
I 1
С6Н5 С6Н5
масса 388,47), образует с титаном в кислой среде окра-
шенное в желтый цвет комплексное соединение. По дан-
ным советского химика В. П. Живописцева, образуется
комплексный катион [П(ДАМ)з]4+, в котором каждая мо-
лекула диантипирилметана связана с титаном через кар-
бонильные кислороды. Максимум светопоглощения сое-
динения находится при 385 нм (рис. 13).
122
Метод обладает рядом преимуществ по сравнению с
другими фотометрическими методами определения титана.
Образование комплексного соединения происходит в кис-
лых средах — в 1—6 М солянокислом растворе. При до-
статочном избытке реагента (0,8%-ный раствор ДАМ)
максимальная окраска комплекса развивается практиче-
ски мгновенно и устойчива длительное время (по крайней
мере, в течение суток).
Реакция является достаточно чувствительной (моляр-
ный коэффициент погашения при %тах = 385 нм равен 16-Ю3)
и достаточно избирательной. Хотя с реагентом ДАМ, кро-
Рис. 13. Кривые свето-
поглощения реагента
ДАМ (/) и его ком-
плекса с титаном (2)
ме титана, реагируют и другие элементы, однако многие
из них образуют бесцветные комплексы, не мешающие
•определению содержания титана. Железо (III), образую-
щее с реагентом ДАМ окрашенное комплексное соедине-
ние, необходимо восстанавливать до железа (II), которое
окрашенного соединения с реагентом не образует. Мешаю-
щее влияние ряда элементов устраняют, применяя опре-
деленную кислотность раствора. Наиболее благоприятной
средой для определения содержания титана в материа-
лах, содержащих большие количества железа, является
2—3,5 М соляная кислота; определение титана возможно
в присутствии оксалатов, фосфатов, а также фторидов при
использовании 6 М соляной кислоты.
Определению содержания титана не мешают магний,
алюмиий, цинк, кадмий, марганец, РЗЭ, медь, цирконий,
церий, кобальт, молибден (V), ванадий (IV). Молибден (VI)
образует с реактивом окрашенное соединение и его мешаю-
щее влияние устраняют также, как и мешающее влияние
железа (III) и ванадия (V), восстановлением аскорбиновой
кислотой, гидроксиламином. Никель, хром (III) мешают
определению содержания титана собственной окраской.
Избирательным методом определения титана является
экстракционно-фотометрический, основанный на образова-
нии тройного соединения Ti—ДАМ — хлорид олова (II).
123
В качестве экстрагента используется хлороформ. Моляр-
ный коэффициент погашения комплексного соединения
невысок: е=18-103 при Л. = 20 нм (имеющиеся в литературе
сведения о величине е = 68-103 являются ошибочными). Од-
нако использование экстракционного варианта позволяет
сократить объем колориметрируемого раствора, а, следо-
вательно, и снизить передел обнаружения титана. Хлорид
олова способствует экстракции соединения титана с реа-
гентом ДАМ и одновременно устраняет мешающее влия-
ние железа. Определению мешают лишь вольфрам и нит-
рат-ионы.
3. Хромотроповая и дихлорхромотроповая кислоты.
Комплекс титана с хромотроповой кислотой образуется
при pH 2—3,3, максимальное светопоглощение комплекса
(е=17-103) наблюдается при Х = 470 нм. В связи с тем, что
реакция проводится при определенном значении pH, реак-
тив менее удобен в работе, чем ДАМ.
2,7-Дихлорхромотроповая кислота (используют динат-
риевую соль) взаимодействует с титаном в кислой области
при pH 1. Преимуществами реагента по сравнению с хро-
мотроповой кислотой являются его большая устойчивость
против окисления и избирательность реакции: определе-
ние содержания титана возможно в присутствии фосфа-
тов, фторидов, комплексона III, оксалатов; е = 11 • 103 при
^max = 490 НМ.
Титриметрические методы определения содержания ти-
тана имеют достаточно большое применение. Эти методы
используются для нахождения макроконцентраций титана
в сплавах, концентратах, шлаках и т. д. Из титриметри-
ческих методов наибольшее практическое применение по-
лучили комплексонометрический и оксидиметрический.
Комплексонометрическое определение.
Комплексон III в присутствии пероксида водорода взаимо-
действует с титаном (IV) в 0,3 М солянокислом растворе
с образованием устойчивого (lgK~20,4) соединения: [TiO
(Н2О2) ] 2++ Na2H2T^Na2 [TiO (Н2О2) У] + 2Н+.
Содержание титана определяют путем титрования из-
бытка комплексона III раствором соли висмута в присут-
ствии ксиленолового оранжевого. Определению не мешают
многие элементы: Mg, Мп, Zn, La, Се (III), A1,W (в коли-
честве до 50 мг каждого из элементов), Та (до 70 мг),
Мо (до 100 мг), Си (до 5 мг), Nb (до 60 мг).
Железо (III) и цирконий, образующие устойчивые в
кислой среде комплексонаты, мешают определению тита-
на, поэтому сначала находят их суммарное содержание
124
титрованием без пероксида водорода, а затем в присутст-
вии пероксида водорода определяют сумму трех элемен-
тов. Содержание титана определяют по разности этих оп-
ределений. Мешающее действие небольших количеств же-
леза можно устранить добавлением аскорбиновой кислоты..
Оксидиметрический метод определения со-
держания титана основан на восстановлении титана (IV)
до титана (III) и последующем титровании (окислении)
восстановленного титана раствором окислителя, например
перманганата калия или соли Fe (III) в присутствии ро-
данида аммония (или калия) в качестве индикатора. Роль
восстановителя обычно выполняют металлические цинк,
кадмий, их амальгамы, алюминий: 2TiOSO4+Zn + 2H2SO4=
= Ti2(SO4)3 + ZnSO4 + 2H2O; Ti2(SO4)3 + Fe2(SO4)3 + 2H2O =
= 2TiOSO4+ 2FeSO4+ 2H2SO4.
Нормальный окислительно-восстановительный потенци-
ал Eo равен для системы Ti (IV)/Ti (III)—0,04 В, для
Cd/Cd (И)—0,402 В, для Zn/Zn (II)—0,762 В и для
Al/Al (III) — 1,7 В.
Полученные в результате восстановления соединения
титана (III) являются весьма неустойчивыми и под дейст-
вием кислорода воздуха вновь окисляются до Ti (IV). По
этой причине раствор Ti (III) приходится изолировать во
избежание контакта с воздухом и титрование проводить
в атмосфере углекислого газа.
При введении в сернокислый раствор титана (III) раст-
вора сульфата аммония происходит образование комплекс-
ного аниона [Ti(SO4)2] ~:Ti2(SO4)s+ (NH4)2SO4-^-2NH4[Ti
(SO4)2], благодаря чему потенциал системы Ti (IV)/Ti (III)
возрастает на 0,3 В, раствор становится устойчивым по
отношению к кислороду воздуха, и титрование можно
вести без применения углекислого газа.
Следует учитывать возможное мешающее влияние на
определение титана оксидиметрическим титрованием дру-
гих элементов, характеризующихся переменной степенью
окисления. Наиболее частым спутником титана во многих
объектах анализа является железо. Простым, эффектив-
ным, позволяющим проводить определение титана в мате-
риалах, содержащих большие количества железа, явля-
ется способ, основанный на титровании восстановленного
титана раствором соли железа (III) в присутствии рода-
нида калия или аммония в качестве индикатора.
Гравиметрические методы. При гравиметрическом оп-
ределении содержания титана основной весовой формой
является оксид титана TiO2, который образуется в ре-
125.
зультате прокаливания гидроксида титана Ti(OH)< или
его соединений с органическими осадителями.
Один из наиболее старых гравиметрических методов
определения содержания титана основан на осаждении
его аммиаком в виде гидроксида с последующим прока-
ливанием выделенного осадка до оксида титана ТЮ2.
В данном случае определению мешают элементы, осаж-
даемые аммиаком (например, железо, цирконий, ниобий,
тантал и др.), а также фосфор, ванадий, мышьяк.
Осаждением гидроксида титана избытком едкого натра
при нагревании титана отделяют от ванадия (V), хро-
ма (VI), молибдена (VI), вольфрама (VI), фосфора,
.алюминия. Для более полного отделения необходимо
двойное осаждение.
В качестве органических соединений для осаждения
титана используют купферон, уротропин, таннин, 5,7-диб-
ромоксихинолин и др.
Купферон (аммонийная соль N-нитрозо-М-фенилгид-
роксиламина)
C4HS- N - ONH4
N = 0
(молекулярная масса 155,16) осаждает из сильнокислых
растворов титан в виде купфероната
/С6Н, - N - о \
1 „> Ti
\ N = 0 /4
Осадок прокаливают при 800—900 °C и взвешивают
в виде диоксида. Вместе с титаном осаждаются железо
(III), цирконий, тантал, ванадий, торий, частично РЗЭ и
вольфрам.
Более устойчивый аналог купферона — М-бензоил-N-
^фенилгидроксиламин (БФГА)
Z~V-C-N-/~A
Хаш/ || । \==/
0 ОН
или С1зНцО2Н (молекулярная масса 213,1). Наряду с ти-
таном он осаждает ниобий в виде NbO(Ci3Hio02N)3, тан-
тал, молибден, цирконий. В присутствии комплексона III
и пероксида водорода выделяются в осадок ниобий, тан-
тал и молибден, в то время как титан остается в растворе.
:12б
Таким образом, ни один из неорганических или орга-
нических осадителей не является достаточно специфичным
для осаждения титана. С помощью соответствующих комп-
лексантов, например комплексона III, винной, лимонной
кислот, возможно более избирательное выделение титана.
Практическая часть
Оксидиметрическое определение титана
в ильменитовых концентратах
Принцип метода. Определение основано на восстанов-
лении титана до титана (III) металлическим алюминием
и последующем титровании Ti (III) раствором железоам-
монийных квасцов с роданидом аммония в качестве инди-
катора. Методика рассчитана на определение содержания
ТЮ2 от 45 до 65%. Относительное стандартное отклонение
результатов определений 0,02—0,015.
Реактивы, и растворы., 1) Пиросульфат калия; 2) кислота соляная,.
р=1,19 г/см3 и разбавленная 1:4; 3) карбонат натрия безводный;
4) квасцы железоаммонийные (для приготовления 0,1 н. раствора
48,22 г квасцов растворяют в 400 мл дистиллированной воды, содер-
жащей 50 мл концентрированной серной кислоты; по охлаждении раст-
вор доводят водой до 1 л; титр раствора устанавливают по стан-
дартному образцу с известным содержанием титана); 5) алюминий
гранулированный металлический; 6) роданид аммония, 10%-ный раст-
вор (хранят в склянке темного стекла).
Выполнение определения. Навеску 0,2 г ильменитового
концентрата (с точностью до четвертого знака) тщатель-
но перемешивают в фарфоровом тигле стеклянной палоч-
кой с 5 г пиросульфата калия. Смесь сплавляют в муфель-
ной печи при 800—850 °C, выдерживают 10—20 мин, а за-
тем охлажденный плав выщелачивают разбавленной 1:4
соляной кислотой в конической колбе вместимостью 250 мл.
В колбу добавляют 2 г гранулированного алюминия, на-
крывают ее часовым стеклом и содержимое колбы уме-
ренно кипятят на электролите. Общий объем раствора до.
восстановления должен быть 150—200 мл.
После прекращения выделения водорода, т. е. после
полного растворения алюминия, колбу с йлиты снимают и
добавляют в нее 10 мг карбоната натрия. Обмыв дистил-
лированной водой стекло и горлышко колбы, ее закрывают
плотно резиновой пробкой (пробка должна быть со стек-
лянной трубкой для отвода газов во избежание взрыва)
и охлаждают, после чего раствор титруют 0,1 н. раство-
ром квасцов в присутствии 10 мл роданида аммония в.
среде углекислого газа до появления слабо-бурой окраски.
127
Температура титруемого раствора должна быть в пре-
делах 18—20 °C. Перед концом титрования каждую сле-
дующую каплю титранта добавляют после тщательного
перемешивания содержимого колбы.
Параллельно проводят контрольный опыт для опреде-
ления содержания в реактивах веществ, восстанавливаю-
щихся алюминием и титрующихся раствором квасцов. 1 мл
0,1 н. раствора квасцов соответствует 7,99 мг оксида ти-
тана.
Определение титана в ильменитовых концентратах
дифференциальным фотометрическим методом
Принцип метода. Определение титана основано на ре-
акции образования, окрашенного в желто-оранжевый цвет
комплексного соединения титана с пероксидом водорода
в кислой среде. Оптическую плотность испытуемого раст-
вора измеряют по отношению к раствору сравнения, ко-
торый представляет собой окрашенный раствор пероксид-
ного соединения титана определенной концентрации. Ме-
шающее влияние железа (III) устраняют добавлением
фосфорной кислоты.
Измерения оптической плотности производят на спек-
трофотометре СФ-4а при Л = 390 нм или применяют фото-
электроколориметр марок ФЭК-56, ФЭК-60 (А,щах~410 нм).
Относительное стандартное отклонение результатов оп-
ределений при спектрофотометрическом варианте метода
-составляет 0,005, при фотоколориметрическом 0,01—0,015.
Реактивы и растворы. 1) кислота серная, р= 1,12 г/см3 и 20%-ный
раствор (для его приготовления к 890 мл воды осторожно добавляют
ПО мл концентрированной серной кислоты и дают раствору остыть
при комнатной температуре или помещают стакан с кислотой в сосуд
с водой); 2) кислота фосфорная, р=1,70 г/см3; 3) пероксид водорода,
30- и 6%-ный растворы; 4) сульфат аммония; 5) пиросульфат калия;
6) оксид титана (IV); 7) рабочий раствор титана (для его приготов-
ления навеску 0,5000 г оксида титана (IV) помещают в коническую
колбу вместимостью ~250 мл, прибавляют 12,5 г сульфата аммония
и 27.5 мл серной кислоты (р= 1,84) и нагревают на электроплитке до
полного растворения ТЮг- После охлаждения раствор переводят в
мерную колбу вместимостью 250 мл, доводят водой до метки и пере-
мешивают. В 1 мл полученного раствора содержится 1,2 мг титаца.
Для приготовления раствора титана из оксида титана можно также
сплавить его навеску с 5 г пиросульфата калия при 800—900 °C и
затем растворить плав в серной кислоте (р=1,12) с добавлением
~1 мл 30%-ного пероксида водорода).
Построение градуировочного графика. В четыре мерные
колбы вместимостью по 100 мл вводят с помощью пипеток
или бюреткой 12, 14, 15 и 18 мл стандартного раствора
128
титана. Приливают в каждую колбу по 20 мл фосфорной
кислоты, 50 мл 20%-ного раствора серной кислоты, 5 мл
6%-ного раствора пероксида водорода, доводят объем до
метки тем же раствором серной кислоты и перемешивают.
Раствор, содержащий 12 мг титана в 100 мл, исполь-
зуют в качестве раствора сравнения. Оптическую плот-
ность остальных трех растворов измеряют по отношению
к раствору сравнения на спектрофотометре СФ-4а в кюве-
тах (/=10 мм) при 2^ = 390 нм с введением светофильтра
УФС = 2.
По полученным данным строят градуировочный график
зависимости оптической плотности от концентрации или
вычисляют значение постоянного фактора, которым поль-
зуются при расчете содержания титана в пробе.
При работе на фотоэлектроколориметрах в качестве
раствора сравнения используют раствор с несколько мень-
шим содержанием титана (6 мг) и относительно него из-
меряют оптическую плотность растворов с соответственно
более низкими концентрациями титана. Например, со-
держание титана в растворе сравнения равно 6 мг/100 мл,
а в измеряемых растворах содержится 8 и 10 мг титана
в 100 мл. В этом случае погрешность метода будет не-
сколько большей, чем при измерениях на спектрофото-
метре.
Выполнение определения. Навеску 0,3000 г концентра-
та помещают в фарфоровый или кварцевый тигель, до-
бавляют несколько капель серной кислоты (р=1,84) и
сплавляют при 800—850 °C с 5 г пиросульфата калия до
получения прозрачного плава. Полученный плав раство-
ряют в 50 мл серной кислоты (р=1,12), содержащей 1 мл
6 %-него раствора пероксида водорода. Раствор охлаж-
дают, переводят в мерную колбу вместимостью 100 мл,
доводят тем же раствором серной кислоты до метки и
перемешивают. Отбирают аликвотную часть раствора
(15 мл) в мерную колбу вместимостью 100 мл, приливают
20 мл фосфорной кислоты и далее все реактивы, как ука-
зано при построении градуировочного графика.
Относительную оптическую плотность анализируемого
раствора измеряют по отношению к раствору сравнения.
Содержание титана либо находят по градуировочному
графику, либо вычисляют, пользуясь соответствующей
формулой.
5. 129-
Фотометрическое определение титана
в сплавах с ниобием
Принцип метода. Метод основан на реакции образова-
ния окрашенного в желтый цвет соединения титана с ди-
антипирилметаном в кислой среде. Гидролиз ниобия пре-
дотвращают добавлением в раствор винной кислоты. Ме-
тодика рассчитана на определение содержаний титана до
10%. Относительное стандартное отклонение результатов
определений составляет 0,03.
Реактивы, растворы и аппаратура. 1) Кислота серная, р= 1,84 г/см3
и разбавленная 1:1; 2) пиросульфат калия; 3) кислота винная, 15%-
ный раствор; 4) кислота соляная, р=1,19 г/см3 и разбавленная 1:1;
5) диантипирилметан, 2%-ный раствор (для его приготовления 20 г
препарата растворяют в 300—400 мл воды, к которой предварительно
добавлено 60 мл разбавленной 1:1 серной кислоты, добавляют ~2 г
аскорбиновой кислоты для восстановления железа, обычно присутст-
вующего в качестве примеси в реактиве; раствор переводят в мерную
колбу вместимостью 1 л и разбавляют водой до метки);-6) запасной
раствор, содержащий в 1 мл 1,0 мг титана (для его приготовления
0,1000 г металлического титана сплавляют с 3 г пиросульфата калия
в муфеле при 800—900 °C; плав растворяют в 20—30 мл винной
кислоты и разбавляют водой до 100 мл; 7) рабочий раствор, содер-
жащий в 1 мл 100 мкг титана (готовят соответствующим разбавле-
нием запасного раствора); 8) фотоэлектроколориметр ФЭК-56 (свето-
фильтр № О, 2ьтах — 490 нм) или аналогичный прибор.
Построение градуировочного графика. В ряд мерных
колб вместимостью по 50 мл вводят от 1 до 5 мл стан-
дартного раствора титана с интервалом 1,0 мл. В колбы
добавляют по 10 мл винной кислоты и помещают их на
5 мин в кипящую воду. Не охлаждая раствора, приливают
по 15 мл раствора диантипирилметана и по 10 мл серной
кислоты. Затем растворам дают остыть до комнатной
температуры и доводят их объем до метки.
Оптическую плотность растворов измеряют при Х =
= 490 нм в кюветах (/ = 30 мм) по отношению к раствору
контрольного опыта, не содержащего титана. По полу-
ченным данным строят градуировочный график зависи-
мости оптической плотности от концентрации, отдельные
точки которого периодически проверяют.
Выполнение определения. Навеску, сплава 100 мг
сплавляют в кварцевом тигле с 3—5 г пиросульфата ка-
лия. Плав растворяют при слабом нагревании в 20—30 мл
раствора винной кислоты, затем раствор переводят в мер-
ную колбу вместимостью 100 мл, доводят объем до метки
водой и перемешивают (раствор А). Отбирают 20 мл
раствора А и переводят в мерную колбу вместимостью
100 мл, добавляют 20 мл винной кислоты и доводят объем
130
водой до метки (раствор Б). Для определения содержания
титана отбирают 5—10 мл раствора Б (в зависимости от
содержания титана) в мерную колбу вместимостью 50 мл,
добавляют 10 мл винной кислоты, помещают на 5 мин
в кипящую воду и далее поступают, как указано при
построении градуировочного графика. Содержание тита-
на находят, используя градуировочный график.
Цирконий и гафний
Цирконий открыт в 1789 г. немецким химиком и минералогом
М. Клапротом в минерале цирконе ZrSiOi. Название циркония проис-
ходит от арабского слова Zerk — драгоценный камень или персидско-
го Zargiin — золотой камень, так как минералы циркония в древности
были известны как драгоценные камни.
Гафний, являющийся постоянным спутником циркония, открыт
венгерским химиком Г. Хевеши и нидерландским физиком Д. Костером
значительно позже. Это связано с тем, что химические свойства цирко-
ния и гафния чрезвычайно близки, элементы было трудно разделить и
установить индивидуальность гафния. Гафний получил свое название
в честь города Копенгагена (по-латински Hafnia). Положение гафния
в Периодической системе элементов было предсказано Д. И. Менделе-
евым задолго до его открытия.
Нахождение в природе. Цирконий относится к числу элементов,
достаточно распространенных: в земной коре его содержание состав-
ляет 0,025%, что превышает содержание таких элементов, как медь,
никель, цинк и других цветных металлов. Однако минералов, в кото-
рые цирконий входит как основная составная часть, немного. Чаще
он находится в рассеянном состоянии в силикатных породах. Основ-
ными минералами циркония являются следующие.
Бадделеит — по составу почти чистый оксид циркония (IV) (до
98% ZrO2). В качестве примесей содержит НЮ2 (1,2%), а также
ThO2 (до 0,2%), SiO2 (0,06%), Fe2O3 (2,1%), MgO (0,64%), CaO
(0,80%), MnO (0,23%), TiO2 (0,65%); иногда присутствует оксид
урана (до 1%).
Циркон — ортосиликат циркония ZrSiO4 — содержит ZrO2 67,2%,
SiO2 32,8%. В качестве примесей в минерале находятся НЮ2 (от
0,5 до 4%), Fe2O3 (0,35%), СаО (0,05—4%), изредка РЗЭ, ниобий
и тантал.
Эвдиалит или силикат циркония, натрия, кальция, содержащий
хлор: (Na, Са)6 Zr(Si60ts) (ОН, С1]2-
Гафний — значительно менее распространенный, чем цирконий, эле-
мент; его содержание в земной коре составляет 3-10~4%. Это типич-
ный рассеянный элемент, являющийся постоянным спутником цирко-
ния во всех его минералах. Самостоятельных минералов гафния не
найдено.
Применение. Цирконий, его сплавы и соединения находят широкое
применение в различных областях техники. Благодаря высокой кор-
розионной стойкости, низкому сечению захвата тепловых нейтронов
цирконий применяется в атомной энергетике в качестве конструкцион-
ного материала атомных реакторов. Так как химический аналог и
постоянный спутник циркония — гафний обладает высоким сечением
захвата тепловых нейтронов, для использования циркония в атомной
технике его тщательно очищают от гафния.
5*
131
Кроме металлического циркония, в атомной энергетике использу-
ется жаропрочный сплав на основе циркония — циркалой-2, содержа-
щий легирующие добавки олова, железа, хрома, никеля.
В качестве легирующей добавки цирконий используют при произ-
водстве сталей для их раскисления, очистки от азота (деазотирова-
ния) и связывания серы. Сплавы цветных металлов, содержащие 0,1—
15% Zr, обладают высокой прочностью, термической устойчивостью,
электропроводностью. В производстве электровакуумных приборов цир-
коний используют как геттер (поглотитель газов), а порошкообразный
цирконий — в военной технике для приготовления бездымного пороха.
Соединения циркония находят широкое применение в производстве
огнеупоров, керамики, фарфора, эмалей и стекла. Сульфат циркония
применяют в кожевенной промышленности в качестве хорошего дуби-
теля.
Гафний в промышленности используется пока еще мало. Представ-
ляет интерес его применение в регулирующих и защитных устройствах
атомных реакторов благодаря высокому сечению захвата тепловых
нейтронов. Перспективно применение соединений и сплавов гафния
в производстве высокотемпературных и жаропрочных материалов. На-
пример, температура плавления карбида гафния 3890°С; сплав ниобия
и тантала, содержащий 2—10% гафния и 8—10% вольфрама, прочен
даже при 2000 °C.
Диоксид гафния применяется при изготовлении оптических стекол
с высоким показателем преломления и в качестве катализатора в орга-
ническом синтезе.
Свойства циркония и гафния и их соединений. Химиче-
ские свойства циркония, гафния и их соединений очень
близки. Сходство именно этих элементов наибольшее по
сравнению с другими родственными парами (тантал —
ниобий, вольфрам — молибден). Это объясняется тем, что
вследствие лантаноидного сжатия радиусы атомов Zr и
Hf (соответственно 0,145 и 0,144 нм), а также радиусы
ионов Zr4+ и Hf4+ (0,074 и 0,075 нм) практически одина-
ковы. Ниже рассмотрены свойства этих элементов и не-
которых их соединений.
Свойства гафния нами отдельно не рассматриваются,
а отмечаются лишь особенности свойств гафния или его
соединений по сравнению со свойствами циркония и его
соединений. Для циркония и гафния характерной явля-
ется степень окисления +4.
Температура плавления циркония 1852±10°С, гафний
более тугоплавок: его температура плавления 2222±30°С.
Цирконий и гафний — металлы серебристо-бело-
го цвета. Одним из наиболее ценных свойств является их
стойкость против коррозии. Азотная, соляная и разбав-
ленная (50%-ная) серная кислоты, а также растворы ще-
лочей практически не растворяют металлы.
Металлические цирконий и гафний растворяются во
фтористоводородной кислоте (разбавленной и концентри-
132
рованной) с образованием фторидных комплексов: Zr+
+ 6HF = H2 [ZrFe]+2Н2. Вследствие летучести фторидов
циркония и гафния упаривание фторидных растворов не-
обходимо проводить в присутствии серной кислоты. Для
растворения металлов применяют также сплавление с
фторидами щелочных металлов. Со щелочами и карбо-
натами щелочных металлов металлический цирконий не
сплавляется.
Цирконий и гафний растворимы в концентрированной
серной кислоте при нагревании: Zr + 4H2SO4 = Zr(SO4) 2 +
+ 2SO2 + 4H2O. При избытке серной кислоты образуется
комплексная цирконосерная кислота: Zr(SO4)2 + 2H2SO4 =
= Н4 [Zr (SO4) 4]. Растворение происходит легче в смеси
серной кислоты с сульфатом аммония или калия.
Для растворения металла можно применить сплавле-
ние его в виде мелкой стружки с пиросульфатом натрия
или калия. Так как двойные соли сульфата циркония и
натрия более растворимы, чем соответствующие соли
калия, то для сплавления лучше использовать пиросуль-
фат натрия. Однородный плав, полученный в результате
сплавления, можно перевести в раствор 1—2 М серной
кислотой или оксикислотой. Например, при растворении в
винной кислоте образуется комплексный анион цирконил-
винной кислоты [ZrO (С4Н4О6)2) 2“.
Использование для растворения рассматриваемых ме-
таллов серной кислоты или пиросульфатов щелочных
металлов иногда является предпочтительным по сравнению
с быстро протекающим растворением во фтористоводород-
ной кислоте, так как не требуется платиновая посуда и
исключается стадия упаривания раствора с серной кисло-
той для удаления фторид-ионов, обычно мешающих после-
дующему анализу.
Для растворения металлов можно применять царскую
водку, хотя этот способ является достаточно длительным:
3Zr + 4HNO3+ 12HCl = 3ZrCl4+4NO + 8H2O.
Диоксид циркония (IV) ZrO2 — кристалличе-
ское вещество, температура плавления которого 2700 °C.
В природе находится в виде минерала бадделеита, черный
цвет которого зависит от присутствующих примесей же-
леза и марганца. Растворимость диоксида циркония (IV)
зависит от кристаллического состояния соединения. Диок-
сид циркония, прокаленный при температуре выше 1050 °C,
практически нерастворим даже в концентрированной сер-
ной кислоте, медленно растворяется во фтористоводород-
ной кислоте и лучше всего разлагается при сплавлении с
133
пиросульфатами или бисульфатами натрия или калия:
ZrO2 + 4KHSO4 = Zr(SO4)2 + 2K2SO4 + 2H2O.
При сплавлении диоксида циркония с бифторидом ка-
лия образуется комплексный фторцирконат калия: ZrO2 +
+ 3KHF2 = K2[ZrF6] +КОН + Н2О, а со щелочами образу-
ются цирконаты: ZrO2 + 2KOH = K2ZrO3 + H2O, которые при
растворении плава в воде гидролизуются с выделением
гидроксида циркония: K2ZrO3 + 3HOH = Zr(OH)4 + 2KOH.
Для сплавления используют также пероксид натрия.
Диоксид циркония, прокаленный при более низкой, чем
1050 °C, температуре (800—900 °C), не растворяется в раз-
бавленных кислотах, но растворим при длительном кипя-
чении в концентрированной серной кислоте, особенно при
добавлении сульфата аммония.
Диоксид гафния (IV) HfO2 — тугоплавкое ве-
щество белого цвета, температура плавления которого
2800 + 25 °C. Получают его при прокаливании гидроксида
или солей гафния (нитрата, оксихлорида и др.).
Аморфный диоксид гафния, полученный при темпера-
туре прокаливания <400 °C, растворим в минеральных кис-
лотах. Диоксид гафния, прокаленный при более высоких
температурах, не растворяется в соляной и азотной кис-
лотах, но растворим в концентрированной фтористово-
дородной и серной кислотах; разлагается при спавлении
со щелочами, содой, пиросульфатами щелочных метал-
лов.
Гидроксид циркония получают осаждением из
растворов его солей аммиаком или щелочью: Zr(IV) +
+ 4OH_ = Zr(OH)4. Все соли циркония легко подвергаются
гидролизу, результатом которого является образование
солей цирконила ZrO2+ и конечного продукта — гидрати-
рованного диоксида циркония — белого студенистого ве-
щества. Осажденный на холоду гидроксид циркония (а-
модификация) Zr(OH)4xH2O растворяется в разбавленных
минеральных и органических (например, щавелевой) кис-
лотах, в карбонате аммония. При длительной выдержке
(старении) или нагревании a-модификация постепенно пе-
реходит в ^-модификацию ZrO(OH)2nH2O, которая пло-
хо растворяется в кислотах.
Из солянокислых растворов на холоду осаждение гид-
роксида циркония начинается при pH 1,9, из сернокис-
лых— при pH 1,7, из азотнокислых — при pH 2,3—2,4.
Гафний из солянокислых растворов начинает осаждаться
при несколько более высоком значении pH»2,3 ч- 2,4, а из
азотнокислых — при pH— 2,5. Если в растворе присутствуют
134
органические кислоты—винная, лимонная, щавелевая, то
они препятствуют осаждению гидроксида циркония (или
гафния), так как образуют с ионами этих элементов проч-
ные комплексные соединения. Взаимодействие со щавеле-
вой кислотой, например, проходит по реакции Zr(IV)4-
+ 2С2О42~ +НОН= [ZrO(C2O4)2]2“' + 2H+ с образованием
комплексного цирконилоксалата.
Двухзамещенный фосфат циркония
Zr(HPO4)2—белый хлопьевидный осадок, образуемый при
взаимодействии солей циркония с растворимыми фосфа-
тами: Zr(IV) +2HPO42“ = Zr(HPO4)2. Осадок труднораст-
ворим даже в довольно концентрированных растворах сер-
ной кислоты. После прокаливания при температуре
>1000 °C гидрофосфаты циркония переходят в пирофос-
фат ZrP2O7, нерастворимый во всех разбавленных кисло-
тах, кроме фтористоводородной. Пирофосфат циркония
используется для гравиметрического определения содержа-
ния циркония. Фосфат гафния еще менее растворим в кис-
лотах, чем соответствующая соль циркония, что исполь-
зуется для фракционного разделения этих элемен-
тов.
Хлорид циркония ZrCl4 — белый кристаллический гиг-
роскопичный порошок, гидролизуется во влажном воздухе
и в водных растворах с образованием оксихлорида цир-
кония (хлорида циркония).
Оксихлорид циркония кристаллизуется в виде соеди-
нения ZrOCl2-8H2O, хорошо растворимого в воде, но слабо
растворимого в соляной кислоте. При прокаливании ZrOCl2-
•8Н2О переходит в ZrO2.
Разложение циркониевых руд и минералов. Для раз-
ложения руд и минералов циркония предложено большое
число плавней. Удобным способом разложения является
сплавление их в соотношении 1:4 или 1:3 со смесью кар-
боната натрия (соды) и тетрабората натрия (буры). Для
полного вскрытия руды требуется высокая температура
(1000—1200 °C). Плав выщелачивают водой и осадок раст-
воряют в 10 %-ном растворе соляной кислоты.
При разложении циркона переход циркония в раствор
происходит по следующей схеме: ZrSiO4+3Na2CO3 +
+ Na2B4O7^-Na2ZrO3 + Na2SiO3 + 4NaBO2 + 3CO2. Образую-
щийся метацирконат натрия гидролизуется при растворе-
нии плава в воде: Na2ZrO3 + 3H2O^-Zr(OH)4 + 2NaOH. При
растворении осадка в кислоте происходит образование
оксихлорида циркония: Zr(OH)4 + 2HCl-*ZrOCl2 + 3H2O.
135
Для разложения циркониевых минералов применяют
также сплавление с едким кали или едким натром. Цир-
коний при последующем растворении плава в воде пере-
ходит в гидроксид, легко растворимый в кислотах. Смесь
щелочи и пероксида натрия применяют для вскрытия труд-
норазлагаемых руд.
Для разложения минерала циркона применяют также
сплавление с кислым фторидом калия. В результате сплав-
ления образуется фторцирконат калия K2ZrF6 и фторси-
ликат калия K2SiF6.
Бедделеит можно вскрыть сплавлением с пиросульфа-
том натрия с последующим растворением плава в 1 М.
растворе серной кислоты; при этом цирконий в растворе
будет находиться в виде сульфата.
Одним из методов перевода в раствор природных ма-
териалов, содержащих цирконий, является разложение
пробы смесью фтористоводородной и серной кислот. Од-
нако при этом не разлагается минерал циркон ZrSiO4, и
нерастворимый остаток дополнительно разлагают сплав-
лением его с бурой Na2B4O7, щелочами или кислым фто-
ридом калия.
Соляная кислота не разлагает большинство минера-
лов циркония и применяется для предварительной обра-
ботки руд и минералов с высоким содержанием железа.
При этом железо растворяется в кислоте и переходит в
раствор, а основная масса циркония остается в остатке
и разлагается при последующем сплавлении остатка с
содой. (Минерал эвдиалит разлагается соляной кисло-
той) .
Растворение сплавов циркония. Для растворения спла-
вов, содержащих цирконий, используют фтористоводород-
ную кислоту или ее смесь с азотной кислотой. Раз-
ложение сплавов удобно проводить также сплавлением с
пиросульфатом натрия или калия и последующим раство-
рением плава в растворе органической кислоты.
О состоянии циркония в растворах. В недостаточно
кислых средах (<0,5 М растворы соляной кислоты) опре-
деление содержания циркония, особенно в присутствии
других редких элементов, может быть ненадежным. Это
связано с возможностью образования в этих условиях не-
реакционноспособных соединений циркония, когда цветные
реакции с органическими реагентами не возникают или
сильно замедленны.
Вопрос о форме нахождения циркония в растворах яв-
ляется сложным, окончательно не решенным и в то же
136
время очень важным для аналитической химии этого эле-
мента, так как многие реагенты способны вступать в реак-
цию с цирконием, находящимся в виде негидролизованных
ионов Zr4+, образующихся лишь в сильнокислых раство-
рах, или с ионами цирконила ZrO2+. В средах с недоста-
точной кислотностью цирконий чрезвычайно склонен к
гидролизу с образованием не только ионов цирконила, но
и различных гидролизованных форм, полимерных молекул
или даже коллоидных частиц. Гидроксо-ионы циркония
образуются в результате реакции ZrO2+4-H2O->Zr(OH)22+,
которая в слабокислых растворах сдвинута вправо.
Гидролизованные формы имеют тенденцию к полиме-
ризации в результате наличия мостиковых (оловых) ОН-
групп:
„ОН.
Zr
он^
а также
имеющих
к образованию сложных полиядерных веществ,
цепочечные или циклические структуры, в кото-
рых атомы циркония соединены кислородными мостиками
или гидроксомостиками
। ।
—Zr--0 — Zr-
/ \ / \
НО он но он
\ / \ /
-Zr—0 —-Zr—
I I
Так как длина полиядерных частиц может быть разной,
реакционная способность их будет также неодинаковой,
и результаты определения содержания циркония получа-
ются плохо воспроизводимыми.
Деполимеризация, т. е. перевод циркония в
реакционноспособное состояние, достигается лишь кипяче-
нием достаточно кислых, подлежащих анализу растворов.
Этот способ дает хорошие результаты, например, при
определении циркония в растворах 1—2 М по НС1. При
проведении реакции в более кислых средах (>6 М НС1)
предварительное нагревание раствора не требуется, так
как такая среда обеспечивает нахождение циркония в
виде реакционно-способного катиона Zr4+. В связи с изло-
женным ниже будут рассмотрены некоторые органические
реагенты, дающие цветные реакции с цирконием в кислых
средах.
Методы определения содержания циркония (гафния)
Фотометрические методы определения содержания цир-
кония предусматривают использование реагентов, взаимо-
действующих с цирконием в кислых средах: арсеназо III,
сульфохлорфенол С, ксиленоловый оранжевый.
1. Арсеназо III (1,8-диоксинафталии-3,6-дисульфокис-
лота-2,7-бис <азо>-2-фениларсоновая кислота) относится
к бисазосоединениям на основе хромотроповой и о-амино-
фениларсоновой кислот. Структурная формула арсена-
зо III:
Обычно арсеназо III применяют в виде двунатриевой
соли. Реагент предложен советским ученым С. Б. Савви-
ным в 1959 г., используется для определения ряда эле-
ментов. Цирконий (гафний) образует с арсеназо III в 2—
10 М НС1 комплексное соединение, окрашенное в зеленый
цвет. Окраску комплекса стабилизируют желатиной. Пред-
полагаемая структура комплекса следующая:
Молекула арсеназо III сочетает в себе три группиров-
ки: AsO3H2, способствующая образованию прочного комп-
лекса в кислой среде; N = N-aso-rpynna, обеспечивающая
окраску комплекса; ОН-группы, образующие второй
шестичленный цикл при взаимодействии с катионом цир-
кония, что способствует упрочнению комплекса и углуб-
лению окраски.
На рис. 14 приведены кривые светопоглощения соеди-
нения циркония с арсеназо III и реагента. Максимум све-
топоглощения комплекса соответствует длине волны 665 нм;
поглощение света раствором реагента при этой длине
волны незначительно. Реакция является высококонтраст-
ной: разница максимального светопоглощения комплекса
и реагента составляет 125 нм.
138 £
Максимальная интенсивность окраски соединения цир-
кония с арсеназо III наблюдается в 9—10 М НС1; моляр-
ный коэффициент погашения комплекса при Х = 665 нм
равен 12-104, т. е. эта реакция является одной из наибо-
лее чувствительных.
В тех случаях, когда содержание циркония в материа-
лах не слишком мало (>10_2%), реакцию образования
комплексного соединения проводят в растворе 2 М по НС1.
В этих условиях определению содержания циркония (в
50 мл раствора) не мешают следующие элементы: Al, Mg,
Са (по 100 мг), Мп (50 мг), РЬ, Мо (по 30 мг), Sn
500 000 7ОО 500 soo
Рис. « А>т Рис15 А,НП
Рис. 14. Кривые светопоглощения арсеиазо III (/) и его комплекса (2) с цир-
конием
Рис. 15. Кривые светопоглощения ксиленолового оранжевого (/) и его ком-
плекса с цирконием (2)
(20 мг), Fe(II), V (по 15 мг), Ni, Со, Т1 (по 10 мг) Си,
Сг, РЗЭ Fe (III), Nb (по 5 мг). Мешают определению со-
держания циркония уран и торий.
Благодаря большому сходству свойств циркония и
гафния, которое определяется строением их атомов, ионы
этих элементов дают также сходные реакции с органиче-
скими реагентами. Однако с некоторыми из них в силь-
нокислых средах, например с арсеназо III в 4 М НС1,
комплексы гафния менее устойчивы, чем соответствующие
комплексы циркония, что используется для определения
циркония (>1%) в присутствии гафния.
2. Сульфохлорфенол С, или СХФС, синтезированный
в 1964 г. советскими учеными С. Б. Саввиным и Ю. М. Дед-
ковым, является бисазопроизводным хромотроповой кис-
лоты, структурным аналогом широко известного реагента
арсеназо III. Он относится к ОД'-диоксиазосоединениям,
содержащим характерную ОД'-диоксиазогруппировку
139
Реагент образует с цирконием (гафнием) в растворе
0,5—1 М по НС1 окрашенное комплексное соединение.
Максимальное светопоглощение циркониевого комплекса
наблюдается при длине волны 640 нм, а реагента — при
длине волны 560 нм. Молярный коэффициент погашения
комплекса соответствует е = 50-103. По сравнению с арсе-
назо III сульфохлорфенол С дает более избирательную
реакцию с цирконием в присутствии таких элементов, как
торий, уран, РЗЭ.
Сульфохлорфенол С является также реагентом для оп-
ределения ниобия и молибдена, поэтому определение цир-
кония в материалах, содержащих эти элементы, без их
отделения невозможно. При использовании экстракцион-
но-фотометрического варианта метода избирательность оп-
ределения несколько повышается.
3. Ксиленоловый оранжевый (см. «Алюминий») отно-
сится к группе трифенилметановых красителей. Он обра-
зует в кислой среде с ионами циркония (гафния) комплекс
красного цвета. Молярный коэффициент погашения комп-
лексного соединения циркония в 0,8 М хлорной кислоте
составляет 3,5-104 при %тах = 535 нм. Максимум светопогло-
щения ксиленолового оранжевого находится при длине
волны 440 нм (рис. 15).
В 0,8 М хлорной или 0,6 М соляной кислоте опреде-
лению содержания циркония не мешают Fe (II), Al [по
5 мг], Ti [1 мг], Th [0,5 мг]; Fe (III) необходимо восста-
навливать до Fe (II). Определению циркония мешают
большие количества молибдена и ниобия, а также фто-
риды, фосфаты и оксалаты. Снижают интенсивность окрас-
ки большие количества сульфатов.
Определение содержания циркония в сплавах ниобия
основано на том, что в 0,2 М серной кислоте комплексон III
полностью разрушает окрашенный комплекс циркония с
ксиленоловым оранжевым и не оказывает влияния на
окраску комплекса ниобия с этим реагентом. Определению
циркония не .мешают также Mo (VI), W (VI), А1, РЗЭ,
U (VI) и Fe (по 50 мкг).
Цирконий при добавлении пероксида водорода не взаи-
модействует с ксиленоловым оранжевым, в то время как
интенсивность окраски соединения гафния в этих условиях
140
ослабевает лишь частично. На этом основан метод опре-
деления элементов при совместном присутствии.
Титриметрические методы. Среди титриметрических ме-
тодов определения высоких содержаний циркония главен-
ствующее место принадлежит комплексонометрическому
титрованию. Цирконий по сравнению с другими элемен-
тами образует наиболее прочное комплексное соединение
с комплексоном III (lgK—ЗО). Титрование циркония мо-
жет проводиться в более кислых средах, чем титрование
других элементов, в связи с чем метод является достаточ-
но селективным. Для определения содержания циркония
используют два варианта комплексонометрического титро-
вания: 1) прямое титрование циркония раствором ком-
плексона III в присутствии металлохромного индикатора;
2) обратное титрование избытка комплексона III раство-
ром соли другого элемента, образующего устойчивые ком-
плексы с комплексоном III.
Расчетным путем было установлено, что четкое изме-
нение окраски при титровании циркония комплексоном III
достигается в том случае, если константы устойчивости
соединения циркония с индикатором, по крайней мере, в
104 раз меньше константы устойчивости комплексоната
циркония.
Прямое комплексонометрическое титрование циркония
возможно выполнять в кислых растворах, когда избира-
тельность метода повышается. При прямом титровании
циркония в 0,3 М, растворе серной кислоты в присутствии
лимонной кислоты определению циркония не мешают боль-
шие количества титана; в 3 М растворе соляной кислоты
титрованию не мешают большие количества титана даже
без введения дополнительных комплексообразующих ве-
ществ. Подходящим индикатором, реагирующим с цирко-
нием в кислых средах и резко изменяющим свою окраску
в этих условиях, является ксиленоловый оранжевый.
В методе обратного титрования чаще всего для титро-
вания избытка комплексона III применяют соли висмута
или тория, которые образуют устойчивые комплексонаты
в кислых растворах, а в качестве индикатора используют
ксиленоловый оранжевый.
Следует указать, что гафний реагирует с комплексо-
ном III в тех же условиях, что и цирконий, поэтому метод
комплексонометрического титрования дает возможность
определять либо один из этих элементов в отсутствии
другого, либо их суммарное количество.
141
Гравиметрические методы. В связи с появлением изби-
рательных фотометрических и комплексонометрических ме-
тодов определения содержания циркония и гафния грави-
метрические методы определения этих элементов утратили
свое значение. Однако из гравиметрических методов до сих
пор применяют следующие.
Метод, основанный на осаждении циркония в виде гид-
роксида аммиаком, дает хорошие результаты в приложе-
нии к растворам чистых солей циркония; осадок гидрокси-
да прокаливают до ZrO2. При использовании метода, осно-
ванного на осаждении циркония в виде фосфата, осажде-
ние проводят из кислого, нагретого до 40—50 °C раствора,
содержащего 10% по объему серной кислоты; осажденный
белый хлопьевидный осадок прокаливают при 1000 °C до
безводного пирофосфата циркония ZrP2O?; при осаждении
фосфатом аммония цирконий отделяют от вольфрама, мо-
либдена, алюминия, марганца, меди и других элементов.
В присутствии Н2О2 цирконий отделяют от Ti (IV) и
Nb (V). Для гравиметрического определения содержания
циркония используют также купферон, таннин, фениларсо-
новую кислоту, миндальную кислоту, иодат калия.
Практическая часть
Гравиметрическое определение циркония
в оксихлориде циркония
(установление титра стандартного раствора циркония)
Принцип метода. Цирконий осаждают в виде гидрокси-
да аммиаком и затем осадок прокаливают до диоксида
циркония. Относительное стандартное отклонение резуль-
татов определений 0,005 при содержании ~ 30% циркония.
Реактивы и растворы. 1) Оксихлорид циркония; 2) хлорид аммо-
ния; 3) аммиак водный, 25%-ный раствор; 4) нитрат аммония, 2%-ный
раствор; 5) кислота соляная, р = 1,19 г/см3.
Выполнение определения. Навеску оксихлорида цирко-
ния 0,88 г растворяют в 40 мл соляной кислоты, разбав-
ляют в мерной колбе вместимостью 250 мл водой до метки
и перемешивают. Аликвотную часть раствора (50 мл) по-
мещают в стакан вместимостью 250 мл, разбавляют водой
до 100 мл, добавляют к раствору 1 г хлорида аммония,
нагревают смесь до 70 °C и приливают по каплям раствор
аммиака при перемешивании до появления запаха. Раствор
с осадком нагревают до кипения, дают постоять 20 мин
в теплом месте, затем отфильтровывают на фильтре с бе-
лой лентой.
142
Осадок на фильтре промывают пять раз раствором нит-
рата аммония. Фильтр с осадком помещают во взвешен-
ный тигель (фарфоровый или платиновый), подсушивают,
осторожно озоляют и прокаливают в муфельной печи при
900—1000 °C до постоянной массы. Титр стандартного рас-
твора циркония Т\ вычисляют по формуле, г/мл: T\ = GX
X0,7403/V, в которой G — масса осадка после прокалива-
ния, г; 0,7403 — коэффициент пересчета оксида циркония
на цирконий; V — объем аликвотной части стандартного
раствора циркония, взятой для определения циркония, мл.
Фотометрическое
определение циркония в рудах
Принцип метода. Метод основан на образовании ком-
плексного соединения циркония с арсеназо III в ~2 М
солянокислой среде. Метод позволяет проводить определе-
ния циркония непосредственно после разложения пробы
без предварительного отделения от сопутствующих эле-
ментов. Предел определения циркония составляет 4-10~3%.
Относительное стандартное отклонение 0,15—0,20 при со-
держании циркония 10-2—10_ 3%.
Реактивы, растворы и аппаратура. 1) кислота соляная (р =
= 1,19 г/см3), 2 М раствор; 2) карбонат натрия безводный (сода);
3) тетраборат натрия (бура), 10-водный; 4) смесь для сплавления
(растирают в фарфоровой ступке соду и прокаленную буру, взятые в
соотношении 3:2); 5) желатина, 1%-ный раствор; 6) арсеназо III,
0,05%-ный раствор; 7) оксихлорид циркония; 8) запасной раствор,
содержащий в 1 мл 0,001 г циркония (для его приготовления 0,8832 г
оксихлорида циркония растворяют в 40 мл концентрированной соляной
кислоты, разбавляют в мерной колбе вместимостью 250 мл водой до
метки и перемешивают); 9) рабочий раствор, содержащий в 1 мл
5 мкг циркония (для приготовления разбавляют запасной раствор в
200 раз 2 М соляной кислотой); 10) фотоэлектроколориметр ФЭК-60
(светофильтр № 6, ХЭф = 670 нм) или аналогичный прибор.
Построение градуировочного графика. В ряд стаканов
вместимостью до 50—100 мл вводят от 1 до 5 мл (с интер-
валом 1 мл) стандартного рабочего раствора циркония и
разбавляют до объема 10 мл 2 М соляной кислотой. Рас-
творы нагревают до кипения на песчаной бане. После
охлаждения растворы переносят в мерные колбы вмести-
мостью по 50 мл, вводят по 3 мл раствора желатины, по
2 мл раствора арсеназо III и доводят до метки 2 М соля-
ной кислотой.
Оптическую плотность растворов измеряют при Х =
= 670 нм в кювете / = 20 мм. Раствор сравнения содержит
все реактивы.
143
Выполнение определения. Навеску руды 0,1—0,2 г по-
мещают в платиновую чашку, в которую предварительно
вводят 5 г смеси соды и буры, и перемешивают пробу
стеклянной палочкой. Чашку помещают в муфельную
печь, нагретую до —800 °C, и ведут сплавление при этой
температуре в течение 7—10 мин. Вращением чашки рас-
пределяют плав таким образом, чтобы он застыл по стен-
кам тонким слоем. После охлаждения к плаву прибавляют
— 50 мл воды и нагревают его на песчаной бане. Раствор
с осадком переводят в стакан вместимостью 250 мл, раз-
бавляют до 100—150 мл водой и нагревают для лучшей
коагуляции осадка. После отстаивания осадок отфильтро-
вывают, промывают горячей водой и растворяют 40—60 мл
2 М соляной кислоты. Раствор переводят в мерную колбу
вместимостью 50—100 мл, доводят объем раствора до мет-
ки той же кислотой.
Отбирают в стакан вместимостью 50 мл аликвотную
часть раствора, содержащую предположительно 5—25 мкг
циркония. Раствор нагревают до кипения. По охлаждении
раствор переводят в мерную колбу вместимостью 50 мл,
прибавляют 3 мл раствора желатины и 1 мл раствора арсе-
назо III. Доводят объем раствора до метки 2 М соляной
кислотой и измеряют оптическую плотность раствора, как
это описано при построении градуировочного графика.
В качестве раствора контрольного опыта применяют
раствор, полученный в тех же условиях, что и анализируе-
мый раствор.
Комплексонометрическое определение
диоксида циркония в бадделеитовом концентрате
Принцип метода. Определение основано на прямом тит-
ровании циркония комплексоном III в 2 М растворе соля-
ной кислоты в присутствии индикатора ксиленолового
оранжевого. Относительное стандартное отклонение резуль-
татов определения 0,01 при содержании —98% диоксида
циркония.
Реактивы и растворы. 1) Карбонат натрия безводный (сода);
2) тетраборат натрия (бура), 10-водный; 3) смесь для сплавления
(растирают в ступке соду и прокаленную буру, взятые в соотноше-
нии 1,5:1); 4) кислота соляная, р=1,19 г/см3 и разбавленная 1:5;
5) ксиленоловый оранжевый, 0,1%-ный раствор; 6) циркония окси-
хлорид; 7) рабочий раствор, содержащий в 1 мл —0,001 г циркония
(для его приготовления 0,88 г оксихлорида циркония растворяют в
40 мл концентрированной соляной кислоты, разбавляют в мерной колбе
вместимостью 250 мл водой до метки и перемешивают; титр раствора
циркония устанавливают гравиметрическим методом осаждением цир-
кония в виде гидроксида аммиаком); 8) комплексон III, 0,05 М раст-
144
вор, титр которого устанавливают в условиях выполнения определения
циркония в пробе (1 мл 0,05 М раствора комплексона соответствует
4,561 мг циркония).
Выполнение определения. Навеску тонкоизмельченного
концентрата 0,2 г помещают в платиновую чашку, в кото-
рую предварительно вводят 5 г смеси соды и буры. Пробу
перемешивают и сплавляют в муфельной печи в течение
— 10 мин при 800—900 °C до получения однородного плава.
По окончании сплавления вращением чашки распределяют
плав таким образом, чтобы он застыл тонким слоем по
стенкам чашки. В чашку приливают воду, покрывая ею
всю поверхность плава, и ставят для нагревания на песча-
ную баню. Раствор с осадком переводят в стакан вмести-
мостью 200 мл, разбавляют до 150 мл водой и нагревают
для лучшей коагуляции осадка. После отстаивания осадок
отфильтровывают, промывают два-три раза горячей водой.
Осадок смывают с фильтра в тот же стакан 60—70 мл
горячей соляной кислоты (1:5) и, если нужно, нагревают
до полного растворения гидроксидов. По охлаждении рас-
твор переводят в мерную колбу вместимостью 100 мл и до-
водят объем до метки раствором разбавленной 1:5 соля-
ной кислоты.
Для титрования отбирают в коническую колбу вмести-
мостью 150—200 мл аликвотную часть раствора, равную
50 мл. Раствор нагревают до кипения, затем добавляют
индикатор и титруют 0,05 М раствором комплексона III
до резкого перехода малиновой окраски раствора в жел-
тую. Вновь нагревают раствор до кипения; если при этом
розовая окраска его восстанавливается, то продолжают
титрование, приливая раствор комплексона III по каплям
до окончательного перехода малиновой окраски раствора
в желтую.
Фотометрическое определение циркония
в сплавах на основе ниобия
Принцип метода. Определение содержания циркония
основано на измерении суммарной абсорбции комплексов
циркония и ниобия в 0,2 М сернокислой среде с ксилено-
ловым оранжевым по отношению к аликвотной части рас-
твора пробы, содержащей, помимо тех же реактивов, ком-
плексон III, маскирующий только цирконий. Для предот-
вращения гидролиза ниобия его предварительно связывают
в пероксидный комплекс. Метод рассчитан на определение
содержания от 1 до 5% циркония. Относительное стан-
дартное отклонение результатов определений 0,10.
145
Реактивы и растворы. 1) Сульфат аммония; 2) кислота серная,
р=1,84 г/см3 и 1 М раствор; 3) пероксид водорода, 30%-ный раст-
вор; 4) комплексон III, 0,05 М раствор; 5) ксиленоловый оранжевый,
0,1%-ный раствор; 6) запасной раствор, содержащий в 1 мл 1 мг цир-
кония (для его приготовления 0,1351 г оксида циркония (IV) сплавляют
с 3—4 г сульфата калия; плав растворяют при нагревании в 1 н. сер-
ной кислоте; раствор переводят в мерную колбу вместимостью
100мл и доводят объем до метки той же кислотой); 7) рабочий
раствор, содержащий в 1 мл 0,01 мг циркония (готовят в день упот-
ребления соответствующим разбавлением запасного раствора 1 н.
серной кислотой).
Построение градуировочного графика. В ряд мерных
колб вместимостью по 50 мл вводят из микробюретки от
1 до 4 мл стандартного рабочего раствора циркония (с
интервалом в 0,5 мл). В колбы добавляют до 20 мл 0,5 М
серной кислоты, по 1 мл ксиленолового оранжевого, пере-
мешивая раствор после добавления каждого реактива. До-
водят содержимое колб до метки водой и вновь перемеши-
вают. Не менее чем через 30 мин измеряют оптическую
плотность раствора в кюветах (( = 30 мм) при /. = 536 нм
относительно раствора контрольного опыта. Точки градуи-
ровочного графика периодически проверяют.
Выполнение определения. Навеску сплава 0,1 г раство-
ряют при нагревании в 3 мл серной кислоты (р=1,84)
с добавлением 0,5 г сульфата аммония в жаростойком ста-
кане. По охлаждении плава добавляют 0,3 мл пероксида
водорода и разбавляют раствор водой до 50 мл, охлаж-
дают, переводят в мерную колбу вместимостью 100 мл и
доводят объем водой до метки. Для определения содер-
жания циркония отбирают в две мерные колбы вмести-
мостью по 50 мл равные аликвотные части раствора, со-
держащие 20—35 мкг циркония, и разбавляют их до 20 мл
0,5 М раствором серной кислоты. В одну из колб вводят
0,2 мл раствора комплексона III и тщательно перемеши-
вают. Затем в обе колбы добавляют по 1 мл раствора
ксиленолового оранжевого, доводят объем раствора до
метки водой и перемешивают. Не менее чем через 30 мин
•измеряют оптическую плотность раствора, не содержащего
комплексон III, относительно раствора, с комплексоном
при Х = 536 нм в кювете (/ = 30 мм). Содержание циркония
находят, используя градуировочный график.
Ниобий и тантал
Ниобий открыт в 1801 г. английским химиком Ч. Гатчеттом,
который назвал новый элемент в честь Колумба — колумбием,
а минерал, в котором он был обнаружен — колумбитом. Тантал открыт
шведским химиком А. Экебергом в 1802 г. в минералах, которые
146
впоследствии получили название танталита и иттротанталита. Новый
элемент был назван танталом по имени героя древнегреческой мифо-
логии Тантала, осужденного Зевсом на вечные муки («муки Тантала»),
так как растворить в кислотах оксиды этого элемента мучительно
трудно.
В 1844 г. немецкий химик Г. Розе сделал сообщение об откры-
тии двух новых элементов, один из которых был подобен танталу,
открытому Экебергом, а другой был назван им ниобием. Это наз-
вание было дано по причине сходства нового элемента с танталом
(в мифологии Ниоба — дочь лидийского царя Тантала). В литературе
долгое время употреблялись оба названия одного и того же эле-
мента— колумбий и ниобий, с 1952 г. общепринятым стало название
ниобий.
Нахождение в природе. По распространенности в земной коре
ниобий (1-1О“зо/о) и особенно тантал (2-10-4%) являются редкими
элементами. В минералах элементы всегда находятся вместе; они
образуют либо самостоятельные минералы, либо входят как примеси
в состав минералов титана, циркония и др. Наиболее важными ми-
нералами являются метаниоботанталаты железа и марганца (Fe, Мп)
(Nb, Та)2О6; при преобладании содержания ниобия минерал называют
колумбитом, а тантала — танталитом. Большое практическое значение
имеет минерал лопарит — титанониобат редких земель цериевой груп-
пы, кальция и натрия (Са, Се, Na)2 (Nb, Ti)2O6. Ниже приведен хи-
мический состав лопарита, %: Nb2O5+Ta2O5 8—10; TiO2 39,22—39,24;
SiO2 0,27—0,72; ThO2 0,53; 2Ce2Q3+2Y2O3 32,30—34,61; P2O5 0,2;
V2O5 0,01; CaO 4,22—5,76; Na2O 7,88—9,06; K2O 0,26—0,75; Fe2O,
0,06—0,72; A12O3 0,72; SrO 3,0.
Пирохлоф, или титанониобат натрия и кальция имеет формулу
(Na, Ca)2(Nb, Ti)2(O, F)z. Формула микролита, или танталата кальция
2СаО-(Та, Nb)2O5.
Применение. Наиболее широкое применение ниобий находит в виде-
сплава с железом (феррониобий) в черной металлургии. Металличес-
кие ниобий и тантал и их сплавы используют в тех случаях, когда
необходимо работать при высоких температурах. Ниобий и тантал
входят в состав жаропрочных сплавов, используемых для изготов-
ления газовых турбин реактивных двигателей; находят применение
в атомной промышленности, в химическом машиностроении благодаря
их высокой коррозионной стойкости в агрессивных средах.
Способность тантала и ниобия поглощать газы используется
в электровакуумной промышленности, из ниобия и тантала изготов-
ляются сетки и другие детали электронных ламп. Тантал в виде
проволоки и листов используют в костной и пластической хирургии,
так как он не раздражает живую ткань.
Свойства ниобия, тантала и их соединений. Ниобий и
тантал — элементы V группы Периодической системы
Д. И. Менделеева, очень сходные между собой по хими-
ческим свойствам, что объясняется близкими радиусами
атомов (соответственно 0,145 и 0,147 нм), равными ра-
диусами ионов (0,069 нм для Nb5+ и Та5+). Существуют
и некоторые различия свойств элементов: ниобий химиче-
ски более активен, сравнительно легче восстанавливается
в водных растворах и др. Это объясняется структурой
электронных оболочек атомов. Электронная конфигурация
147
атомов ниобия 4d45s’ (сверх структуры криптона), а тан-
тала 5d36s2 (сверх структуры ксенона). Ниобий относится
к элементам второй переходной группы Периодической си-
стемы элементов, энергетические уровни электронной обо-
лочки атомов заполнены полностью до уровня 4р. При
незаполненном подуровне 4 <7 имеется один электрон на
подуровне 5s. Тантал относится к третьей переходной
группе элементов с недостроенным подуровнем электронов
5d при наличии двух спаренных электронов на подуровне
6s. Наиболее характерны степень окисления ниобия и тан-
тала + 5. Известны соединения, в которых ниобий прояв-
ляет степени окисления +4, +3, +2, 4-1.
Ниобий — металл светло-серого цвета, температура его
плавления 2470±10°С. Устойчивость против действия кис-
лот у ниобия несколько меньше, чем у тантала. Фтористо-
водородная кислота медленно взаимодействует с ниобием.
Ниобий растворим в смеси HF+HNO3, в смеси 40% HF +
4-10% НгО2. Расплавленные щелочи растворяют оба ме-
талла, образуя ниобаты и танталаты.
Тантал—металл серого цвета с синеватым оттенком,
температура плавления тантала 3010±50°С. Тантал ха-
рактеризуется высокой коррозионной стойкостью против
действия кислот: азотной, соляной, серной, хлорной, цар-
ской водки, что объясняется присутствием на его поверх-
ности прочной оксидной пленки. Металл растворяется в
смеси фтористоводородной и азотной кислот: 3Ta + 21HF +
4-5HNO3 = 3H2 [TaF7] 4-5NO4-ЮН2О, в смеси 40% HF +
+ 15% Н2О2.
Оксиды тантала (V) Та2О5 и ниобия (V) Nb2Os раство-
римы в горячей фтористоводородной кислоте, а также раз-
лагаются при сплавлении с пиросульфатами щелочных ме-
таллов, карбонатами и едкими щелочами. При растворении
пиросульфатного плава в воде выделяются гидроксиды
Ta2Os-xH2O и Nb2Os-xH2O, которые называются земель-
ными кислотами. Если же растворить пиросульфитный
плав не в воде, а в растворах органических кислот (вин-
ной или щавелевой), то получится прозрачный раствор,
содержащий тартратные и оксалатные комплексы тан-
тала и ниобия: [ТаО(С4Н4О6)2]~, [ТаО(С2О4)2]-,
[NbO(C4H4O6)2]-, [NbO(C2O4)2]-.
При растворении плава карбоната калия или едкого
калия с оксидами тантала и ниобия в воде образуются
растворимые танталаты и ниобаты калия. Танталаты и
ниобаты натрия плохо растворимы в воде и выпадают в
осадок.
148
В нейтральных и кислых растворах соединения ниобия
и тантала в отсутствии комплексообразующих веществ
чрезвычайно склонны к гидролизу и образованию полимер-
ных соединений. Гидролизованные и полимерные формы
разрушаются вспомогательными активаторами. К таким
активаторам относятся ионы фтора, оксикислот, пероксид
водорода, комплексон III.
При растворении металлов или земельных кислот в
присутствии фтористоводородной кислоты образуются ком-
плексные кислоты, а в присутствии фторида калия — их
соли: KzfTaF?], К2 [NbF7]. Классический метод Мариньяка,
который до сих пор используют для разделения тантала
и ниобия в промышленности, основан на различной раство-
римости фтортанталата и оксифторниобата калия. В рас-
творах, содержащих —7% HF, фторниобат калия гидро-
лизуется до оксифторниобата К2 [NbOFg] -Н2О, раствори-
мость которого в воде в 10—12 раз выше растворимости
K2[TaF7].
Для разложения тантал- и ниобийсодержащих материа-
лов применяют фтористоводородную кислоту, смесь фто-
ристоводородной, серной или азотной кислот, смесь суль-
фата аммония (NH4)2SO4 и серной кислоты. Используют
также сплавление с пиросульфатами щелочных металлов
и щелочами, а также с бифторидом калия.
Растворение пиросульфатных или бифторидных плавов
проводят в растворах винной и щавелевой кислот, а так-
же в смеси пероксида водорода с серной кислотой.
Методы определения содержания тантала и ниобия
Фотометрические методы определения ниобия. Роданид-
ный метод — наиболее старый метод, но в настоящее вре-
мя еще не потерял своего значения, особенно при опреде-
лении микроконцентраций ниобия (10~2—1О-зо/о). Это
объясняется высокой чувствительностью, избирательностью
и доступностью реагента.
Метод основан на взаимодействии ниобия (V) с рода-
нистоводородной кислотой в солянокислом растворе с об-
разованием комплексного соединения Н [NbO(SCN)4],
окрашенного в желтый цвет. Образующийся комплекс
экстрагируют эфиром или определяют содержание ниобия
в гомогенной водно-ацетоновой среде. Реакцию образова-
ния комплексного соединения ниобия проводят в присут-
ствии винной кислоты, предотвращающей гидролитическое
выделение ниобия: Н4 [NbOH (С4Н2О6)2] + 4I<SCN-MHCI^
->H[NbO(SCN)4] +2Н2С4Н4О6+4КС1.
149
Винная кислота препятствует также гидролитическому
осаждению других легко гидролизующихся элементов, в
частности, тантала.
Экстрагирование ниобия проводится в присутствии хло-
рида олова SnCh, который способствует увеличению интен-
сивности окраски эфирного слоя. Максимум светопоглоще-
ния роданидного комплекса ниобия в эфирном экстракте
соответствует длине волны 385 нм, молярный коэффициент
погашения комплекса равен 3,5 • 104, что свидетельствует
о высокой чувствительности реакции. Кроме эфира, для
экстракции используют также его смесь с хлороформом
(1:1) и другие растворители.
Железо (III) также образует окрашенное соединение
с роданидами, однако в условиях определения ниобия
Fe (III) восстанавливают с помощью SnCl2. Тантал обра-
зует бесцветный роданидный комплекс Н2 [ТаО (SCN)3),
при высоких содержаниях этот элемент препятствует раз-
витию окраски ниобиевого комплекса. Титан образует окра-
шенное соединение с роданид-ионом Н2 [TiO(SCN)4],
интенсивность окраски которого во много раз слабее
интенсивности окраски ниобиевого комплекса. В присут-
ствии высоких содержаний титана его влияние уменьшают
снижением концентрации роданид-ионов до 0,3 М (против
0,9 М).
За последние годы предложено довольно много мето-
дов определения содержания ниобия, основанных на обра-
зовании соединений ниобия с органическими реагентами:
1. ПАР, или 4-(2-пиридилазо)-резорцин (см. «Медь»),
как реагент для фотометрического определения ниобия
впервые описан в 1963 г. Обычно используют его натрие-
вую соль CnH8N3-NaO2-Н2О или
молекулярная масса которой 255,2, температура плавле-
ния 182 °C.
Особенностью взаимодействия ниобия с ПАР является
то, что в присутствии третьих компонентов (тартрат-,
оксалат-, фторид-ионов, пероксида водорода, комплексо-
на III) образуются тройные (или смешанные) комплекс-
ные соединения. Тартрат-, оксалат- и другие ионы, являясь
комплексообразующими лигандами, способны разрушать
полимерные нереакционноспособные ионы ниобия, удержи-
150
вать их в растворе, а также маскировать мешающие эле-
менты. Реагент ПАР выступает в качестве хромофора
тройного комплекса.
Большим достоинством реагента ПАР является его спо-
собность образовывать комплексное соединение с ниобием
в кислых растворах. В 1 М растворе соляной кислоты
максимум светопоглощения комплекса наблюдается при
395 нм, реагента — 405 нм. В качестве рабочей длины вол-
ны используют ?v=540 нм, при которой наблюдается мак-
Рис. 16
Рис. 16. Кривые светопоглощения реагента ПАР (/) его комплекса с ииобнем
(2) и дифференциальная кривая (3}
Рис. 17. Кривые светопоглощения СХФС (/), его комплекса с ниобием (2) и
дифференциальная кривая (3) (измерение I относительно 2)
симальная разница светопоглощения комплексного соеди-
нения и реагента, что видно на дифференциальной абсорб-
ционной кривой (рис. 16) (измерение оптической плот-
ности раствора комплекса к раствору реагента).
Молярный коэффициент погашения соединения ниобия
с реагентом ПАР составляет 1,5-104г Главным достоин-
ством реакции, проходящей в кислых средах, является ее
высокая избирательность. Это позволяет в ряде случаев,
даже при анализе материалов сложного химического со-
става, не прибегать к отделению сопутствующих элемен-
тов. В присутствии комплексона III, который маскирует
мешающие элементы, в растворе допустимо наличие Fe,
Mo, Ti, Ni. Со, W (по 10 мг), Zr (1 мг), Р (2 мг), Та
(100 мкг).
2. Сульфохлор фенол С, или СХФС, реагирует с нио-
бием (см. «Цирконий») в тартратно-солянокислых раство-
рах, образуя окрашенное соединение. Крбме соляной кис-
лоты, применяют и азотную кислоту. Достоинством реаген-
та является его способность образовывать с ниобием ком-
плексные соединения в кислых средах (1—3 М по НС1),
что определяет высокую избирательность реакции. Кроме
151
ниобия, в кислой среде окрашенные комплексы образуют
только цирконий, гафний и скандий. Разница светопоглоще-
ния комплекса и реагента максимальна при Х = 660 нм
(дифференциальная кривая) (рис. 17). Молярный коэффи-
циент погашения соединения ниобия с СХФС 3,6-104.
В 1 М. растворе соляной кислоты определению ниобия
не мешают W, А1 (по 40 мг), Fe, Ti (по 1 мг). Допустим
100-кратный по отношению к ниобию избыток циркония
(в присутствии комплексона III), 10-кратный избыток мо-
либдена (в присутствии восстановителя гидроксиламина)
и 5-кратный избыток тантала.
В 3 М растворе соляной кислоты повышается избира-
тельность реакции: при этой кислотности можно допустить
присутствие 2 мг Ti. Определению также не мешает
20-кратный избыток молибдена (в присутствии гидрокси-
ламина и комплексона III).
Фотометрические методы определения тантала. П и р о-
галловый метод определения содержания тантала
основан на образовании окрашенного в желтый цвет ком-
плексного соединения тантала с пирогаллолом в соляно-
кислом или сернокислом растворе. Определению тантала
в оксалатно-сернокислой среде (pH 1,8—2,2) сильно ме-
шает титан. Окраска растворов пирогаллового комплекса
титана примерно в пять раз интенсивнее окраски раство-
ров пирогаллового комплекса тантала. При проведении
реакции в оксалатно-солянокислой среде (4 М по НС1)
и измерении светопоглощения в ультрафиолетовой области
спектра мешающее влияние титана уменьшается в 70 раз.
Пирогалловый метод, впервые предложенный еще в
1937 г. М. С. Платоновым и Н. Ф. Кривошлыковым, до
последнего времени был наиболее распространенным ме-
тодом фотометрического определения содержания тантала.
В настоящее время этот метод не всегда удовлетворяет
требованиям контроля производства тантала по причине
сравнительно невысокой чувствительности (в среде, 4 М
по НС1, 8 = 2,4-103 при Хщах=335 нм) и недостаточной
избирательности (определению мешают титан, ниобий, мо-
либден, вольфрам, фториды).
К наиболее чувствительным и избирательным методам
определения содержания тантала относятся методы, осно-
152
ванные на экстракции соединений (солей) гексафтор-
танталата с основными красителями. Из основных
красителей чаще всего используются трифенилметановые
и родаминовые (ксантеновые). Экстракция фтортантала-
тов основных красителей впервые описана советским уче-
ным Н. С. Полуэктовым с сотрудниками. Тантал в раз-
бавленных растворах фтористоводородной кислоты нахо-
дится в виде отрицательно заряженных комплексных ионов
[TaFs], которые с катионом основного красителя обра-
зуют ионную пару, экстрагируемую бензолом или другим
экстрагентом. В водном растворе соединение практически
полностью диссоциировано, образование комплексной соли
происходит лишь в процессе экстракции.
В качестве экстрагентов в основном используют бензол
или толуол. Толуол, являющийся гомологом бензола, ме-
нее летуч. Температура кипения бензола С6Н6 80 °C, то-
луола СбНбСНз 110,8°C. Возможно также применение бу-
тилацетата (при содержаниях>0,1 % тантала).
Из всех изученных трифенилметановых краси-
телей бриллиантовый зеленый обеспечивает наиболее
высокую чувствительность экстракционно-фотометрическо-
го определения тантала. Бриллиантовый зеленый имеет
(C2H5)2h|.
x>N+(C2Hj)2d'
молекулярную массу 420,5.
На рис. 18 приведены кривые
светопоглощения толуольноаце-
тового экстракта гексафтортанта-
лата бриллиантового зеленого и
экстракта красителя. Максимум
светопоглощения экстракта ком-
плексного соединения соответст-
вует длине волны Х = 640 нм. Мо-
лярный коэффициент светопогло-
щения соединения при этой дли-
не волны 8= 1,2-105. Свето-
поглощение экстракта бриллиан-
тового зеленого значительно
меньше.
500 620 Л,нм
Рис. 18. Кривые светопог-
лощения экстрактов брил-
лиантового зеленого (-0 и
его комплекса с танталом
(2)
153
Определению содержания тантала с использованием
бриллиантового зеленого не мешают большие количества
ниобия — постоянного спутника тантала. Ниобий образует
с реагентом экстрагируемые ассоциаты, прочность которых
значительно ниже прочности соответствующих .соединений
тантала. Коэффициент экстракции ниобиевых соединений
возрастают с повышением концентрации фторид-ионов.
Так, определению тантала при концентрации F- 0,22 М
не мешают 5 мг Nb, а при 0,5 М F- мешают 200 мкг Nb.
При концентрации фторид-ионов, равной 0,22 М, опре-
делению тантала не мешают Ti, Al, Zr, Fe, V, Ca, Mg, Cu
(no 10 мг). При увеличении концентрации фторид-ионов
до 0,65 М определению тантала не мешают Ti (50 мг), А1
(70 мг), Zr (100 мг), однако при этом, как уже отмеча-
лось будет несколько больше, чем при концентрации F-
0,22 М, сказываться влияние ниобия.
Рассмотренный метод применяется для определения
содержания тантала в различных материалах без отделе-
ния его от сопутствующих элементов.
Кроме высокой чувствительности реакции, достоинством
бриллиантового зеленого в качестве реагента на тантал
является возможность проведения экстракции из кислых
(2 М) растворов. При этом колебания кислотности, рав-
ные 0,2 М, практически не сказываются на результатах
определения тантала. В этом преимущество реагента по
сравнению с некоторыми другими трифенилметановыми
красителями, например с кристаллическим фиолетовым.
В случае использования кристаллического фио-
летового необходимо проводить реакцию при строго
определенном значении pH раствора, равном 1—1,3.
К родаминовым (ксантеновым) красите-
лям относятся родамин 6Ж, бутилродамин С, родамин С,
родамин ЗБ. Соединения родаминовых красителей с фтор-
танталатом используют для экстракционно-фотометриче-
ских (абсорбционнометрических) и экстракционно-флуори-
метрических методов. Последние являются более чувстви-
тельными, например нижний предел определения тантала
с бутилродамином С экстракционно-фотометрическим ме-
тодом равен 2 мкг/5 мл, а экстракционно-флуориметриче-
ским с тем же реагентом 0,002 мкг/5 мл.
Гравиметрические методы определения содержания
тантала и ниобия основаны на осаждении элементов в виде
труднорастворимых соединений с органическими или не-
органическими реагентами. При этом образуются соедине-
ния переменного состава, которые трудно использовать в
154
качестве весовой формы. В связи с этим осадки прокали-
вают при высокой температуре (1000 °C) до оксидов.
Гидролитическое осаждение. Для гравимет-
рического определения тантала и ниобия используют осаж-
дение их гидроксидов из тартратных растворов (тартрат-
ный гидролиз) или разложение щелочных ниобатов и тан-
талитов минеральными кислотами.
Осаждение тан н ином. Тантал из слабокислых
растворов осаждается таннином легче, чем ниобий. Ком-
плекс тантала с таннином (светло-желтого цвета) осаж-
дается первым, осадок титанового комплекса (красного
цвета) — вторым и комплекс ниобия с таннином (ярко-
красного цвета) —третьим. Танниновый метод применяют
для разделения тантала и ниобия. В ходе анализа раство-
ра, содержащих эти элементы, ниобий, частично соосадив-
шийся с танталом, определяют колориметрическим мето-
дом и вносят поправку в результаты определения тантала.
Танниновый метод применяют также и для осаждения и
определения суммарного количества тантала и ниобия.
В слабокислой оксалатной среде в присутствии комплексо-
на III ниобий и тантал количественно осаждают таннином
и отделяют от многих сопутствующих элементов.
Для осаждения тантала и ниобия применяют и другие
реактивы, например купферон, 8-оксихинолин, фениларсо-
новую кислоту и т. д. Гравиметрические методы опреде-
ления ниобия и тантала в материалах сложного химиче-
ского состава являются длительными и трудоемкими, так
как связаны с операциями отделения ниобия и тантала от
сопутствующих элементов. Эти методы часто заменяются
другими, например фотометрическими методами.
Титриметрические методы. Оксидиметрическое опреде-
ление содержания ниобия основано на восстановлении
Nb (V) до Nb (III) и последующем титровании соедине-
ния Nb (III) окислителями. Этот метод не всегда является
надежным, так как трудно получить ниобий в одной опре-
деленной степени окисления.
Для определения ниобия советский ученый Бабко А. К-
с сотрудниками предложили метод, основанный на образо-
вании пероксидного комплекса ниобия и окислении избыт-
ка пероксида водорода в 0,25—1,5 М серной кислоте пер-
манганатом калия.
Косвенное комплексонометрическое определение содер-
жания ниобия основано на образовании тройного комплек-
са ниобия с пероксидом водорода и нитрилотриуксусной
кислотой. Избыток нитрилотриуксусной кислоты оттитро-
155
вывают раствором сульфата меди с индикатором метил-
кальцеином при pH 5—5,5.
Титриметрические методы не получили широкого при-
менения для определения содержания ниобия, так как в
присутствии других элементов, как правило, они не яв-
ляются надежными.
Методы разделения. Кроме методов отделения тантала
и ниобия от других элементов, основанных на осаждении
(см. гравиметрические методы), используют экстракцион-
ные и хроматографические. Экстракцию тантала из
фторидных растворов циклогексаноном или метилизобутил-
кетоном в виде соединения HzTaF? используют для отде-
ления его от ниобия, который в растворах с малым содер-
жанием свободной HF склонен к образованию комплексов
HzNbOFs, которые не экстрагируются.
Для разделения тантала и ниобия применяют ионо-
обменную хроматографию, основанную на селек-
тивном вымывании ниобия смесью 1 М соляной и 0,5 М
щавелевой, а также смесью 1 М соляной и 0,05 М фтори-
стоводородной кислот.
Практическая часть
Определение ниобия в рудах
Принцип метода. Определение основано на образова-
нии и экстракции роданидного комплекса ниобия. Методи-
ка применяется при содержании ниобия в рудах до 0,05%.
Относительное стандартное отклонение результатов опре-
делений 0,2.
Реактивы и растворы. 1) Кислота фтористоводородная, р= 1,15 г/см3;
2) кислота серная, р = 1,84 г/см3 и разбавленная 1:1 и 1:9; 3) кис-
лота азотная, р=1,43 г/см3; 4) пиросульфат калия; 5) кислота вин-
ная, 15- и 3°/о-ные растворы; 6) пероксид водорода, 30%-ный раствор,
разбавленный в 10 раз водой; 7) роданид калия, 20%-ный раствор;
8) хлорид олова (II), раствор (для его получения 15г SnCl2-2H2O
растворяют в 38 мл соляной кислоты (р=1,19 г/см3) и доводят объем
водой до 100 мл); 9) эфир этиловый; 10) рабочий раствор ниобия,
содержащий в 1мл 0,05 мг Nb2O5 (для его приготовления 0,0125 г
Nb2O5 сплавляют с 2г K2S2O7; плав выщелачивают 50мл 15%-ного
раствора винной кислоты при нагревании и объем раствора доводят
водой до 250 мл); 11) рабочий раствор титана, содержащий в 1мл
1мг Т1О2 (для его приготовления 0,5 г ТЮ2 сплавляют с K2S2O7,
плав растворяют в 100 мл 15%-ного раствора винной кислоты;
объем раствора доводят водой до 500 мл).
Приготовление шкалы растворов сравнения или по-
строение градуировочного графика. В ряд пробирок вводят
от 0,05 до 0,4 мл рабочего раствора ниобия с интервалом
156
в 0,05 мл. Каждую пробирку доливают до 4 мл 3%-ным
раствором винной кислоты, прибавляют 3 мл раствора
KSCN, по 3 мл раствора SnCl2 и по 6 мл НС1 (1:1). После
добавления каждого реактива пробирки встряхивают. За-
тем приливают по 5 мл этилового эфира и раствор взбал-
тывают в течение 30 с.
Выполнение определения. Навеску 0,2—0,5 г руды сма-
чивают 0,5 мл воды в платиновом тигле, прибавляют
— 2,5 мл фтористоводородной кислоты, 0,5 мл серной кис-
лоты (1:1), выпаривают до появления паров серного ангид-
рида. Прибавляют две капли HNO3, выпаривают досуха
и прокаливают в течение 2—3 мин в муфеле. Остаток
сплавляют с 2—3 г пиросульфата калия. Плав растворяют
в 10 мл 15%-ный винной кислоты при нагревании, раствор
переносят в колбу вместимостью 50 мл и разбавляют во-
дой до метки.
Две аликвотные части раствора объемом по 1—4 мл
помещают в колориметрические пробирки с притертыми
пробками. В одной из них определяют содержание титана.
Для этого к анализируемому раствору приливают 1 мл
серной кислоты (1:9), 1 мл 3%-ной пероксида водорода
и сравнивают окраску с окраской раствора, содержащего
те же реактивы и 0,5 мг TiO2 (раствора с предельно до-
пустимым количеством титана). Если содержание титана
не превышает 0,5 мг, то определяют содержание ниобия
в другой аликвотной части раствора. Для этого испытуе-
мый раствор доводят до 4 мл 3%-ным раствором винной
кислоты и прибавляют все реактивы, как указано при при-
готовлении шкалы стандартных растворов. Окрашенный
эфирный слой сравнивают с эфирными слоями шкалы стан-
дартов. Сравнение окрасок можно производить визуально
или с помощью фотоэлектроколориметра (А. —385 нм).
Примечание. Для получения более правильных результатов
рекомендуется при достаточно высоких содержаниях титана вводить
в растворы сравнения такие количества титана, которые соответствуют
содержанию его в аликвотной части испытуемого раствора.
При анализе титановых руд или руд, содержащих < 0,05% Nb,
ниобий необходимо предварительно выделить путем однократного
осаждения таннином.
Определение ниобия в лопаритовом концентрате
фотометрическим методом
Принцип метода. Определение основано на реакции
образования комплексного соединения ниобия с 4-(2-пири-
дилазо)-резорцином в тартратно-солянокислом (1 М НС1)
растворе.
157
Измерение оптической плотности производят абсолют-
ным методом на фотоэлектроколориметре или с использо-
ванием дифференциальной фотометрии на спектрофото-
метре. Относительное стандартное отклонение результатов
определений при абсолютном методе равно 0,03, при диф-
ференциальном — 0,01.
Реактивы и аппаратура. 1) Кислота фтористоводородная,
,0=1,13 г/см3; 2) кислота серная, р=1,84 г/см3 и разбавленная 1:1;
3) пиросульфат натрия, 4%-ный раствор; 4) тартрат аммония, 10- и
4%-ные растворы; 5) аммиак водный, разбавленный 1:9; 6) кислота
соляная (р= 1,19 г/см3), 6М раствор; 7) комплексон III, 1%-ный
раствор; 8) 4-2-пиридилазо-резорцин, или ПАР, 0,15%-ный раствор
(для его приготовления навеску препарата 0,15 г, смоченную 1—2 мл
воды, тщательно растирают стеклянной палочкой в стакане, раство-
ряют в —50 мл воды, переводят в мерную колбу вместимостью 100 мл
и доводят объем водой до метки. Раствор пригоден в течение не более
1 мес.); 9) запасной раствор, содержащий в 1мл 1мг ниобия (для
его приготовления 0,1430 г оксида ниобия (V), содержащего не менее
99.5% NbjOs, сплавляют с 4г пиросульфата натрия; плав растворяют
при кипячении в 40 мл 10%-ного раствора тартрата аммония с до-
бавлением 10 мл аммиака (1:9); раствор разбавляют водой до метки
в мерной колбе вместимостью 100 мл; 10) рабочий раствор, содер-
жащий в 1 мл 100 мкг ниобия (готовят разбавлением запасного
раствора в 10 раз 4%-ным раствором тартрата аммония); 11) спект-
рофотометр СФ-4а или аналогичный прибор; 12) фотоэлектроколори-
метр ФЭК-56 или аналогичный прибор.
Построение градуировочного графика при абсолютном
методе анализа. В мерные колбы вместимостью по 50 мл
приливают стандартный рабочий раствор ниобия в количе-
ствах, соответствующих 200; 300; 400 и 450 мкг ниобия.
В одну из колб стандартный раствор не добавляют (рас-
твор контрольного опыта). Концентрацию тартрата аммо-
ния доводят до 400 мг, а концентрацию пиросульфата
натрия — до 200 мг соответствующими 4%-ными раство-
рами их солей. Добавляют в колбы по 5—7 мл воды, по
2 мл раствора комплексона III, по 8,5 мл 6 М раствора
соляной кислоты, по 6 мл раствора ПАР и доводят объем
раствора водой до метки. Растворы перемешивают и через
1 ч измеряют оптическую плотность по отношению к рас-
твору контрольного опыта при Х = 540 нм в кювете с 1 =
= 5 мм. Затем строят градуировочный график зависимости
оптической плотности от концентрации ниобия, отдельные
точки которого периодически проверяют. Графиком поль-
зуются при содержаниях <6°/о оксида ниобия в пробе.
Выполнение определения при абсолютном методе ана-
лиза. Навеску тонкоизмельченной пробы 0,150 г помещают
в платиновую чашку, смачивают 1 мл воды, приливают
6—7 мл фтористоводородной кислоты и осторожно, нагре-
158
вают в течение — 15 мин. К раствору приливают 5—6 мл
разбавленной 1:1 серной кислоты и нагревают до появ-
ления густых паров серной кислоты. Чашку охлаждают,
приливают еще 2—3 мл разбавленной 1:1 серной кислоты
и снова нагревают до появления паров серной кислоты.
Обработку серной кислотой повторяют, после чего содер-
жимое чашки упаривают на горячей плите досуха.
К остатку добавляют 2 г пиросульфата натрия, чашку
помещают в муфель и ведут сплавление при 850—900 °C.
до получения однородного плава. В процессе сплавления
добавляют 1—1,5 мл серной кислоты (р=1,84), которую
затем удаляют, оставляя тигель в муфеле до прекращения
выделения ее паров.
Плав растворяют в 40 мл 10%-ного раствора тартрата
аммония при кипячении. Раствор охлаждают и переводят
в мерную колбу вместимостью 100 мл, объем раствора до-
водят до метки водой. Раствор может быть использован
для определения тантала.
При определении содержания ниобия абсолютным ме-
тодом отбирают аликвотную часть прозрачного раствора
(5 мл), содержащую 300—450 мкг ниобия, в мерную колбу
вместимостью 50 мл, содержание тартрата аммония дово-
дят его 4%-ным раствором до 400 мг, пиросульфата нат-
рия— 4%-ным раствором его соли до 200 мг, затем при-
бавляют — 15 мл воды, 5 мл комплексона III, 8,5 мл 6 Л1
раствора соляной кислоты, 6 мл раствора ПАР. Объем
раствора доводят водой до метки, перемешивают и через
1 ч измеряют оптическую плотность раствора по отноше-
нию к раствору контрольного опыта в кювете толщиной
слоя 5 мм на фотоэлектроколориметре (Хтах~540 нм).
Концентрацию ниобия в аликвотной части раствора на-
ходят по градуировочному графику или расчетным путем
по пропорции, при составлении которой используют опти-
ческую плотность эталонного раствора, близкого по кон-
центрации к испытуемому.
Вычисление постоянного фактора при дифференциаль-
ном спектрофотометрическом методе, который применяется
для анализа концентратов, содержащих не менее 6% окси-
да ниобия (V).
В мерные колбы вместимостью по 100 мл вводят стан-
дартный рабочий раствор ниобия в количествах от 700 до
850 мкг ниобия с интервалом в 50 мкг, добавляют 10%-ный
раствор тартрата аммония до 800 мкг, 4%-ный раствор
пиросульфата натрия до 200 мг. Далее приливают 5 мл
раствора комплексона III, 15—20 мл воды, 17 мл 6 М
159
раствора соляной кислоты, 12 мл ПАР и воду до метки.
Растворы перемешивают и через 2 ч измеряют оптическую
плотность на спектрофотометре в кювете / = 20мм (Х,=
= 540 нм) по отношению к раствору сравнения, содержа-
щему 700 мкг ниобия. Значение фактора F вычисляют по
формуле, приведенной на с. 37.
Выполнение определения при дифференциальном спек-
трофотометрическом методе. Из растворов проб, приготов-
ленных как указано выше, отбирают в мерную колбу
вместимостью 100 мл аликвотную часть раствора, содер-
жащую 750—850 мкг ниобия. Затем приливают все реак-
тивы и измеряют оптическую плотность растворов, как
указано при вычислении постоянного фактора по отноше-
нию к раствору, содержащему 700 мкг ниобия.
Содержание ниобия в аликвотной части раствора вы-
числяют по формуле CX-CO + AF. Если в испытуемом раст-
воре содержится менее 700 мкг ниобия, то раствором срав-
нения служит этот испытуемый раствор, и по отношению
к нему измеряют оптическую плотность раствора сравне-
ния, содержащего 700 мкг ниобия. В этом случае произве-
дение AF при вычислении содержания ниобия в аликвот-
ной части вычитают из Со (см. с. 38).
Фотометрическое определение тантала
в сплавах с ниобием
Принцип метода. Определение основано на реакции об-
разования в сернокисло-оксалатном растворе соединения
тантала с пирогаллолом, окрашенного в желтый цевт. Влия-
ние ниобия устраняют связыванием его в оксалатный
комплекс. Методика рекомендуется для определения от
1 до 10% тантала. Относительное стандартное отклонение
результатов определений 0,03.
Реактивы и растворы. 1) Пиросульфат калия; 2) оксалат аммония,
4%-ный раствор; 3) кислота серная, р= 1,84 г/см3 и разбавленная
1:1, 1:9; 4) кислота соляная, р = 1,19 г/см3; 5) пирогаллол: раствор
20г/100мм (для его приготовления 20 г пирогаллола растворяют в сме-
си: 90 мл воды, 9 мл разбавленной 1:1 серной кислоты и 1мл 2М
раствора хлорида олова в соляной кислоте; если раствор опалесцирует,
то его фильтруют через два плотных фильтра; раствор пригоден для
работы в течение 3—4 сут. при условии хранения его в темной
склянке); 6) запасной раствор тантала, содержащий в 1 мл 1 мг тан-
тала (для его приготовления 0,1г металлического тантала сплавляют
с 5—6 г пиросульфата калия при 800—850 °C; плав растворяют при
нагревании в 50 мл 4%-ного раствора оксалата аммония, охлаждают,
переводят в мерную колбу вместимостью 100 мл и доводят объем
до метки раствором оксалата аммония); 7) рабочий раствор, содер-
жащий в 1 мл 100 мкг тантала (готовят соответствующим разбавле-
нием запасного раствора 4%-ным раствором оксалата аммония).
160
Построение градуировочного графика. В ряд мерных
колб вместимостью по 25 мл вводят из микробюретки от
0,0 до 6,0 мл рабочего раствора тантала с интервалом в
1 мл. В каждую колбу добавляют 2,5 мл серной кислоты
(1:9), 12 мл раствора пирогаллола, разбавляют до метки
раствором оксалата аммония и перемешивают. Измеряют
оптическую плотность раствора при Х,=400 нм в кюветах
с толщиной слоя 20 мл по отношению к раствору, не со-
держащему тантала. По полученным значениям строят гра-
фик зависимости оптической плотности от концентрации.
Выполнение определения. Навеску сплава 0,1 г сплав-
ляют в кварцевом или фарфоровом тигле с 3—5 г пиро-
сульфата калия. Плав растворяют при нагревании в 50—
70 мл раствора оксалата аммония, раствору дают остыть,
переводят в мерную колбу вместимостью 100 мл, доводят
его объем до метки раствором оксалата аммония и пере-
мешивают.
Для определения содержания тантала отбирают али-
квотную часть раствора (5—10 мл) в мерную колбу вме-
стимостью 25 мл. Добавляют в колбу 2,5 мл серной кис-
лоты и далее все реактивы, как указано при построении
градуировочного графика. Содержание тантала определя-
ют, используя градуировочный график.
Гравиметрическое определение
суммарного содержания ниобия и тантала
в технической смеси
гидроксидов редкоземельных элементов
и оксифторниобате калия
Принцип метода. Определение производят, используя
осаждение таннином из солянокислой среды после уда-
ления фторид-ионов упариванием раствора с серной кис-
лотой. Методика рассчитана на определение суммарного
содержания ниобия и тантала более 25%. Относительное
стандартное отклонение результатов анализа 0,01.
Реактивы и растворы. 1) Кислота фтористоводородная (р= 1,16 г/см3),
40%-нын раствор; 2) кислота серная (р = 1,84 г/см3), разбавленная 1:1;
3) танннн, 1%-ный раствор в разбавленной 1:9 соляной кислоте (све-
жеприготовленный) и 0,5%-ный раствор в разбавленной 1:99 соля-
ной кислоте; 4) пиросульфат калия; 5) оксалат аммония, 4%-ный
раствор; 6) кислота соляная (р = 1,19 г/см3) и разбавленная 1:9, 1:99.
Выполнение определения. Навеску пробы 0,3 г помеща-
ют в платиновую чашку, добавляют 3—5 мл фтористово-
дородной кислоты и 8—10 мл разбавленной 1:1 серной
кислоты.
/26. 129.
161
Смесь нагревают на песчаной бане и упаривают почти
досуха (до состояния влажных солей). Содержимое чашки
смывают в стакан вместимостью 250 мл с помощью 150 мл
1%-ного раствора таннина и чашку тщательно вытирают
фильтровальной бумагой, которую затем бросают в стакан.
Раствор с осадком кипятят в течение 2 мин и выдержи-
вают 2 ч или оставляют на ночь. Осадок отфильт-
ровывают, промывают 0,5%-ным раствором таннина и
переносят во взвешенный тигель. Фильтр с осадком озо-
ляют, прокаливают до постоянной массы при 900 °C, ох-
лаждают и взвешивают.
Для введения поправки на титан, присутствующий в
техническом гидроксиде ниобия, осадок суммы земельных
кислот сплавляют с 2 г пиросульфата калия при 900 °C
до получения прозрачного плава. По охлаждении плав
растворяют в 25—50 мл 4%-ного раствора оксалата ам-
мония и определяют содержание титана по реакции с
пероксидом водорода визуальным путем или с использо-
ванием фотоэлектроколирметра (см. «Титан»). Из найден-
ного содержания суммы земельных кислот вычитают ср-
держание титана.
Экстракционно-фотометрическое определение тантала
в рудах и тетрахлориде титана
Принцип метода. Метод основан на экстракции фтор-
танталата бриллиантового зеленого смесью толуола с аце-
тоном (9:1) или бутилацетатом из тартратно-сернокислого
раствора (0,5 М по серной кислоте). Определение тантала
проводят непосредственно после разложения материала.
Относительное стандартное отклонение результатов опре-
деления при содержаниях 1-Ю-3—0,7% тантала состав-
ляет 0,2—0,05 соответственно.
Реактивы, растворы и посуда. 1) Кислота фтористоводородная
(р= 1,13 г/см3); 2) кислота серная (р= 1,84 г/см3) и разбавленная
1:1 и 1:4; 3) тартрат аммония, 16- и 8%-ные растворы; 4) натрия
пиросульфат; 5) бриллиантовый зеленый, 1%-ный раствор; 6) толуол
ос. ч.; 7) ацетон ос. ч.; 8) бутилацетат (бутиловый эфир уксусной
кислоты); 9) фторид натрия ос. ч., насыщенный раствор; 10) запас-
ной раствор тантала, содержащий в 1 мл 500 мкг тантала (для его
приготовления 61 мг оксида тантала (V) сплавляют с 2 г пиросуль-
фата калия при 900° С с добавлением нескольких капель серной кис-
лоты р= 1,84 г/см3 до получения прозрачного плава; плав растворяют
при кипячении в 40 мл 10%-ного раствора тартрата аммония, добав-
ляют 10 мл аммиака (1:9) до получения прозрачного раствора, дают
раствору остыть при комнатной температуре и доводят водой до мет-
ки в колбе вместимостью 100мл); 11) рабочий раствор тантала,
содержащий в 1 мл 5 мкг тантала (для его получения 1 мл запасного
162
раствора разбавляют в мерной колбе вместимостью 100мл 8%-ным
раствором тартрата аммония; готовят в день применения); 12) спе-
циальная посуда: кварцевые цилиндры с притертыми пробками вмес-
тимостью 80 мл, стеклянные пробирки с притертыми пробками вмес-
тимостью 15 мл, пипетки из полиэтилена.
Построение градуировочных графиков. В кварцевые ци-
линдры с притертыми пробками приливают от 0,2 до 1,0 мл
стандартного рабочего раствора тантала с интервалом в
0,2 мл и от 1 до 5 мл того же раствора с интервалом в
1 мл и разбавляют до 5 мл 8%-ным раствором тартрата
аммония. К полученным растворам приливают по 4 мл
серной кислоты (1:4), 9 мл толуола, 1 мл ацетона, 6 мл
раствора фторида натрия (полиэтиленовой пипеткой) и
0,5 мл раствора бриллиантового зеленого. Цилиндры за-
крывают пробками и встряхивают в течение 2 мин. После
разделения слоев (спустя ~3 мин) отбирают сухой пи-
петкой с поршнем или грушей 7,0 мл экстракта и перено-
сят в сухую пробирку с притертой пробкой, в которую
предварительно добавлено 3,0 мл ацетона.
Содержимое пробирки перемешивают и измеряют оп-
тическую плотность раствора на спектрофотометре СФ-4А
(Х = 640 нм) в кювете 1 = 20 мм или фотоэлектроколориметре
ФЭК-56 со светофильтром № 8 (Хтах~607 нм). В качестве
раствора сравнения применяют воду. Раствор, не содер-
жащий тантала, используют в качестве раствора контроль-
ного опыта. Строят градуировочные графики зависимости
оптической плотности от концентрации тантала. При по-
строении градуировочного графика для содержания танта-
ла от 5 до 25 мкг в качестве экстрагента можно использо-
вать 10 мл бутилацетата.
Выполнение определения. При анализе руд 0,5 г пробы
помещают в платиновую чашку и растворяют при нагре-
вании в 10 мл фтористоводородной кислоты (р = 1,13) и
10 мл серной кислоты, разбавленной 1:1, а затем упари-
вают досуха. Остаток сплавляют при температуре 800 °C
с 2 г пиросульфата натрия. Плав растворяют в 25 мл
16%-ного раствора тартрата аммония при нагревании, ра-
створ охлаждают, переводят в мерную колбу вместимостью
500 мл и доводят водой до метки. Аликвотную часть раст-
вора, содержащую от 2 до 25 мкг тантала, помещают в
кварцевый цилиндр, объем доводят до 5 мл 8%-ным раст-
вором тартрата аммония, добавляют 2 мл серной кислоты
(1:4) и далее поступают, как указано при построении гра-
дуировочного графика.
При анализе тетрахлорида титана в мерную колбу
вместимостью 50 мл, содержащую 20 мл серной кислоты
»/а«* 163
(1:1), осторожно вливают с помощью пипетки с грушей
или поршнем 1,5 мл тетрахлорида титана. Содержание
колбы перемешивают до полного растворения рыхлой
вспучивающейся массы желтого цвета и поглощения бе-
лого дыма, разбавляют водой до метки и перемешивают.
Аликвотную часть раствора 2 мл, содержащую от 1 до
5 мкг тантала, переносят в кварцевый цилиндр с притер-
той пробкой, добавляют 5 мл 8%-ного раствора тартрата
аммония и далее поступают, как указано при построении
градуировочного графика. При расчете содержания тан-
тала в пробе тетрахлорида титана исходят из того, что его
плотность составляет 1,73 г/см3.
Молибден и вольфрам
Молибден в виде молибденовой кислоты открыт в 1778 г. швед-
ским химиком К. Шееле. Вольфрам открыт в 1781 г. К- Шееле
в минерале тунгстене — «тяжелом камне», который впоследствии по-
лучил название шеелит.
Нахождение в природе. Молибден и вольфрам относятся к мало-
распространенным элементам: в земной коре содержание молибдена
составляет 3-10-4, вольфрама 1-10~4%. Основными минералами мо-
либдена являются: молибденит, или молибденовый блеск MoS2 (суль-
фид молибдена), по внешнему виду напоминающий графит; молибде-
нит часто содержит в виде изоморфной примеси рений (10~5—10-2%);
повеллит СаМо04 (молибдат кальция); нередко часть молибдена
( — 10%) в повеллите замещена вольфрамом Са(Мо, W)O4. Меньшее
значение имеют минералы: вульфенит РЬМоО4 (молибдат свинца)
и молибдит хРе2О3-г/МоОз-пН2О. Молибден содержится также в медных
и медно-свинцовых рудах (до 0,01%), которые используются для его
извлечения при комплексной переработке сырья.
К основным минералам вольфрама относятся: вольфрамит
(Fe, Mn)WO4 (вольфрамат железа н марганца), представляющий со-
бой изоморфную смесь ферберита FeWO4 (вольфрамата Fe (II)) и
гюбнерита MnWO4 (вольфрамата Мп (II)); шеелит CaWO4 (вольфра-
мат кальция), содержащий часто до 10% оксида молибдена (VI).
Меньшее значение имеет штольцит PbWO4 (вольфрамат свинца).
Применение. Основная часть (75—80%) производимого молибдена
используется в черной металлургии для легированных сталей. Молиб-
ден находит применение для получения жаростойких и кислотостой-
ких сплавов. Благодаря высокой температуре плавления (262()±10°С),
прочности при высоких температурах, хорошей электропроводности
молибден используют в производстве электроламп и электронных
приборов. Из молибденовой проволоки в паре с вольфрамовой делают
термопары для измерения температур в интервале 1200—2000° С.
Молибден и его сплавы используют для изготовления лопаток тур-
бин и других деталей реактивных двигателей.
В качестве конструкционного материала молибден находит приме-
нение в энергетических ядерных реакторах, так как имеет сравнительно
малое сечение захвата тепловых нейтронов и прочен при высоких
температурах.
Дисульфид молибдена применяют как смазочный материал для
трущихся частей механизмов (молибденовая смазка). Молибдат нат-
164
рия используют в производстве красок и лаков, а оксиды молибдена —
в качестве катализаторов в химической промышленности. В последние
годы растворимые молибдаты стали применять как стимуляторы роста
растений в виде микродобавок к удобрениям.
Вольфрам промышленное значение приобрел почти через сто лет
после открытия. В конце XIX в. его начали применять в качестве
легирующих добавок к сталям (например, быстрорежущая сталь);
в начале XX в.— для изготовления нитей электрических ламп нака-
ливания, для получения твердых сплавов, основным компонентом
которых является карбид вольфрама.
В настоящее время вольфрам широко применяется в технике для
производства: специальных сталей, используемых для изготовления
пил, фрез, твердых сплавов на основе карбида вольфрама (85—95%
WC и 5—14%Со); жаропрочных и износостойких сплавов вольфрама
с другими тугоплавкими металлами — танталом, ниобием, молибденом,
рением — в авиационной и ракетной технике; контактных сплавов,
в состав которых входит 10—40% меди или серебра; этн сплавы ис-
пользуют для изготовления рабочих частей рубильников и выключа-
телей. Сплавы вольфрам — никель — медь иашли применение в ра-
диотерапии для защиты от у-лучей и изготовления контейнеров,
в которых хранятся радиоактивные изотопы. Чистый вольфрам при-
меняют в радиоэлектронике и рентгенотехнике.
Вольфрамат натрия используют в производстве лаков и пигментов,
для изготовления огнестойких и водоустойчивых тканей.
Свойства молибдена, вольфрама и их соединений,
Вольфрам является самым тугоплавким металлом,
его температура плавления 3395± 15 °C. Молибден так-
же относится к тугоплавким металлам, температура его
плавления 2620±10°С.
Молибден и вольфрам — элементы VI группы Перио-
дической системы Д. И. Менделеева, очень сходные меж-
ду собой по свойствам. Молибден относится ко второму
ряду главной или d-группы переходных элементов с ча-
стично заполненной 4 d-оболочкой. Наиболее характерная
степень окисления молибдена +6. Для аналитической хи-
мии имеют значения также степени окисления молибдена
+ 5 и +3. Вольфрам относится к третьему ряду главной,
или d-группы переходных элементов. В обычном состоя-
нии окисления, а также у нейтральных атомов d-оболочка
заполнена частично (5d46s2). Вольфрам в соединениях мо-
жет находиться в различных степенях окисления: +6, +5,
+ 4, +3, реже +2. Наиболее устойчивой является степень
окисления +6.
Молибден при комнатной температуре устойчив к дейст-
вию соляной и серной кислот. При нагревании с серной
кислотой образуется растворимый сульфат молибдена
(или молибденила): 2Mo + 3H2SO4 = Mo2(SO4)3 + 3H2. Мо-
либден уже на холоду медленно растворяется в азотной
кислоте: (Mo+2HNO3 = H2MoO4+2NO), царской водке и
165
в виде порошка — в пероксиде водорода. При нагревании
растворение ускоряется. Молибден растворяется в смеси
азотной, серной кислот и воды (соответственно 5:3:2),
В этой смеси вольфрам не растворяется. Молибден хорошо
растворим в смеси фтористоводородной и азотной кислот;
переходит в растворимое состояние при сплавлении с нит-
ритом калия; легко разлагается также при сплавлении в
щелочи или соде в присутствии окислителя нитрата натрия
(калия): Mo 4- 3NaNO3 4- Na2CO3 — NaaMoC^-p 3NaNO2 4-
+ CO2f.
В качестве окислителя применяют также Na2O2. Вод-
ные растворы щелочей на молибден не действуют.
Вольфрам на холоду практически нерастворим в ми-
неральных кислотах (НС1, H2SO4, HNO3). Растворяется
в смеси азотной и фтористоводородной кислот. При на-
гревании с соляной кислотой с последующей обработкой
азотной кислотой образуется вольфрамовая кислота
H2WO4. В расплавленных щелочах вольфрам устойчив в
отсутствие окислителей, но в присутствии, например,
KClOg, NaNO3 или при доступе воздуха образуются воль-
фраматы щелочных металлов: W+KC103 + Na2C03 =
= Na2WO4+KCl + CO2f. Порошкообразный вольфрам раст-
воряется в растворе пероксида водорода.
Оксид молибдена (VI) МоО3 — кристаллы свет-
ло-зеленого цвета, которые выше 600 °C начинают возго-
няться, поэтому при прокаливании этого соединения при
высоких температурах наблюдаются потери молибдена.
Сведения о растворимости оксида МоО3 в воде противо-
речивы: по данным разных авторов, при комнатной темпе-
ратуре растворяется от 0,4 до 2 г/л МоО3. В водных раст-
ворах щелочей и аммиака МоО3 растворяется с образо-
ванием соответствующих молибдатов. Оксид молибдена
(VI) растворим также в минеральных кислотах.
Оксид вольфрама (VI) WO3 — порошок желто-
зеленого цвета, получаемый при прокаливании металла
(выше 400 °C) или вольфрамовой кислоты. Выше 800 °C
WO3 улетучивается, не разлагаясь. Оксид вольфрама (VI)
практически нерастворим в минеральных кислотах, за
исключением фтористоводородной, растворяясь в которой,
образует оксифторид WOF4, легко растворим в щелочах,
карбонатах щелочных металлов с образованием раствори-
мых вольфраматов:
WO3-j-2NaOH = Na2WO4 + Н2О, WO3 + Na2CO3 =
== Na2’.vOs CO2.
166
Оксид вольфрама (VI) после прокаливания с трудом
растворяется в аммиаке. Благодаря амфотерным свойст-
вам он сплавляется также с бисульфатом калия: WO3 +
+ 2KHSO4= (WO2) SO4+ K2SO4+Н2О. При выщелачивании
плава водой и подкислении раствора кислотой в осадок
выпадает вольфрамовая кислота.
В растворах наиболее устойчивы соединения молибде-
на (VI). Соединения молибдена (III) и молибдена (V)
устойчивы только в растворах соляной кислоты высокой
концентрации (соответственно 9 и 4 М НС1). При меньшей
кислотности эти соединения легко окисляются кислородом
воздуха до молибдена (VI). Характерной особенностью мо-
либдена является его способность к образованию поли-
меризованных и гидратированных соединений. Следует от-
метить, что у вольфрама эти свойства выражены в большей
степени, чем у молибдена.
Молибденовая кислота Н2МОО4, или моно-
гидрат,— кристаллы белого цвета. Молибденовая кислота
образуется при длительной выдержке сильно подкислен-
ного горячей азотной кислотой раствора молибдатов.
В растворе молибденовой кислоты имеет место равновесие
Мо О4~+ 2Н+ МоО2(ОН)2^ МоО2+ + 2ОН-.
Кислая среда Щелочная среда
Вольфрамовая кислота H2WO4 или моногид-
рат (WO3-H2O),— порошок желтого цвета. Вольфрамо-
вая кислота осаждается из растворов вольфраматов го-
рячими минеральными кислотами, кроме фтористоводород-
ной, в виде моногидрата WO3-H2O (кислый гидролиз:
Na2WO4+2HCl = H2WO4+2NaCl). Из холодного раствора
вольфраматов осаждается дигидрат WO3-2H2O.
В отличие от вольфрамовой кислоты, которая не раст-
воряется в соляной, серной и азотной кислотах, молиб-
деновая кислота Н2МоО4 растворима в избытке
минеральных кислот: например, при 20 °C растворяется
Н2МоО4 в количестве 11,1 г/л, a H2WO4 — в количестве
0,01 г/л при концентрации НС1 80 г/л. Растворение молиб-
деновой кислоты в кислотах можно объяснить образова-
нием катиона МоО22+. Например, в серной кислоте обра-
зуется сульфат молибденила: H2MoO4+H2SO4=
= (MoO2)Sd4+2H2O.
Различие растворимостей молибденовой и вольфрамо-
вой кислот (или их оксидов) в минеральных кислотах ис-
пользуется для разделения молибдена и вольфрама. Воль-
фрамовая кислота легко растворима (особенно свеже-
167
осажденная) в аммиаке, а также в растворах едких ще-
лочей, карбонатов аммония, натрия. При этом образуются
растворимые вольфраматы аммония (NH^WOj или нат-
рия Na2WC>4.
Молибден и вольфрам образуют комплексные гетеро-
поликислоты с фосфором, мышьяком, кремнием и други-
ми элементами. Например, состав фосфорновольфрамовой
гетерополикислоты: Н7 [P(W2O7)6]-л:Н2О, фосфорномолиб-
деновой гетерополикислоты: Н7[Р(Мо2О7)6] -Н2О. Реакции
образования гетерополикислот с фосфорной, мышьяковой
и кремниевой кислотами являются основой фотометри-
ческих методов определения фосфора, мышьяка и крем-
ния. Вольфрам образует комплексные анионы состава
[W(C2H2O4)4]2~, [W(C4H4O6)4]2-.
Сплавы молибдена растворяются в кислотах:
в концентрированной соляной или серной (1:3) при добав-
лении азотной''кислоты. Удобным способом растворения
молибдена и его сплавов с вольфрамом является приме-
нение пероксида водорода или насыщенного раствора ща-
велевой кислоты в присутствии пероксида водорода; раст-
ворение происходит при умеренном нагревании раст-
вора.
Иногда для растворения сплавов применяют фтористо-
водородную кислоту или ее смесь с азотной кислотой.
Для разложения сплавов вольфрама приме-
няют сплавление их с пиросульфатом калия и последую-
щее растворение сплава в растворах органических кислот
(лимонной, винной, щавелевой). При растворении воль-
фрамсодержащих сплавов в кислотах, например серной,
.происходит выпадение вольфрамовой кислоты в осадок,
который растворяют в растворах аммиака, щелочи или
карбоната натрия. Для растворения сплавов вольфрама
с танталом, ниобием и другими элементами применяют
смесь фтористоводородной и азотной кислот. Полученный
раствор упаривают почти досуха и остаток растворяют в
растворах карбоната натрия или оксикислот.
Сплавы вольфрама с рением растворимы в 30%-ном пе-
роксиде водорода.
Для р а з л о ж е н и я руд и минералов молиб-
дена: молибденита MoS2, а также окисленных природных
соединений — вульфенита РЬМо04, повеллита Са (Mo, W) О4,
молибдита хРе20з-f/Mo03-лН2О применяют азотную кис-
лоту при нагревании с последующим добавлением соля-
ной или серной кислот.
168
Для разложения молибденовых руд и минералов при
определении в них содержания рения (см. «Рений») при-
меняют спекание проб с оксидом кальция в присутствии
окислителя (перманганата калия) или окисляют рений,
пероксидом водорода в растворе.
Для разложения руд молибдена применяют сплавление
проб с гидроксидами щелочных металлов в присутствии
окислителей (Na2O2, KNO3). При этом образуются раст-
воримые в воде молибдаты. Одновременно в раствор в
виде перрената переходят рений, вольфрам (в виде воль-
фрамата), а также кремний (в виде растворимого сили-
ката). Элементы, образующие нерастворимые гидрокси-
ды, остаются в осадке. Разложение минералов молибде-
на легко происходит и при их'сплавлении с пероксидом
натрия или с содой в присутствии окислителя: 2MoS2 +
+ 6Na2CO3 + 9O2 = 2Na2MoO4+4Na2SO4+6CO2.
Окисленные минералы молибдена переходят в раство-
ры карбоната аммония, в то время, как молибденит в этих
условиях не растворяется. Сульфидные руды часто пред-
варительно обжигают: 2MoS2 + 7O2 = 2MoO3 + 4SO2.
Для разложения вольфрамовых руд, мине-
ралов концентратов существует несколько способов,
но наиболее часто для этого используют концентрирован-
ные соляную и азотную кислоты.
При разложении в соляной кислоте раствор выпари-=
вают до определенного объема, при этом выделяется жел-
тая модификация вольфрамовой кислоты. Наряду с ней
образуются растворимые хлоридные комплексы вольфра-
ма, т. е. выделение вольфрама происходит неполностью..
Для разрушения хлоридных комплексов и предотвраще-
ния образования вольфрамовой кислоты в коллоидной,
форме прибавляют концентрированную азотную кислоту.
При таком способе разложения элементы, соединения ко-
торых растворимы в кислотах, остаются в растворе, а
в осадке вместе с вольфрамовой кислотой находятся сили-
каты, оксиды ниобия и тантала и касситерит SnO2. Воль-
фрамовую кислоту затем переводят в раствор аммиака
или щелочи, отделяя таким образом ее от неразложив-
шейся части руды.
Если в руде присутствует молибден, то часть его бу-
дет находиться в осадке с вольфрамовой кислотой и за-
тем с вольфрамом перейдет в раствор аммиака или ще-
лочи.
При разложении шеелита или вольфрамита в соляной
кислоте происходит выделение вольфрамовой кислоты в
169
•осадок: для шеелита CaWO4-F2HCl = H2WO4+CaCI2, а для
вольфрамита (Fe, Мп)WO4-F2HC1 = H2WO4-F (Fe, Мп)С12.
Шеелит также хорошо разлагается азотной кислотой при
нагревании.
Для разложения вольфрамовых руд с низким содер-
жанием вольфрама, а также при определении вольфрама
в касситерите применяют сплавление пробы с едким нат-
ром и последующее выщелачивание плава водой. При этом
вольфрам в виде растворимого вольфрамата натрия пере-
ходит в раствор, в котором можно определить концентра-
цию вольфрама фотометрическим методом. Для минера-
лов с высоким содержанием вольфрама такой способ раз-
ложения обычно не применяют, так как ионы щелочных
металлов препятствуют последующему гидролитическому
выделению вольфрамовой кислоты.
Быстрое разложение вольфрамовых руд происходит
при сплавлении их с пероксидом натрия или смесью его с
едкой щелочью.
Методы определения содержания
молибдена и вольфрама
Из фотометрических методов определения содержаний
молибдена и вольфрама наиболее широкое практическое
применение получили роданидный и дитиоловый:
1. Роданидный метод основан на взаимодействии ионов
молибдена (V) и вольфрама (V) с роданидом, в резуль-
тате которого образуются окрашенные в желтый или
оранжевый цвет комплексные соединения молибдена
(MoO(SCN)5]2~ или окрашенные в желтый цвет комплек-
сы вольфрама [WO(SCN)5] 2~. При определении содержа-
ния молибдена в качестве восстановителя молибдена (VI)
применяют хлорид олова (II) SnCl2, тиомочевину в при-
сутствии ионов меди (II) и другие вещества. При исполь-
зовании в качестве восстановителя SnCl2, окраска ком-
плексного соединения получается недостаточно устойчивой.
Предполагают, что SnCl2, являясь сильным восстановите-
лем, может восстанавливать молибден (VI) до степени
окисления ниже пяти.
При использовании в качестве восстановителя тиомо-
чевины реакцию проводят в присутствии катализатора —
соли меди (II), которая образует бесцветный комплекс с
тиомочевпной [Cu(SCN2H4)3]+, не мешающий определе-
нию содержания молибдена. Оптимальной кислотностью
обладают 1,5—2 М. растворы серной или соляной кислот;
170
при более низкой кислотности окраска раствора имеет по-
сторонний розовый оттенок, при более высокой — устой-
чивость окраски уменьшается. Большое значение имеет
последовательность прибавления реактивов (соли меди,
тиомочевины, роданида). Определению молибдена мешают
фосфор, железо (III), висмут; железо (III) восстанавли-
вают до железа (II). Вольфрам' в этих условиях находится
в виде W (VI) и не образует окрашенного комплекса с
роданидом. Вольфрам связывают в комплекс винной или
лимонной кислотой для предотвращения осаждения воль-
фрамовой кислоты в кислой среде.
Окраску комплексного соединения молибдена с рода-
нидом стабилизируют ацетоном, при этом повышается и
чувствительность метода. Предполагается, что в водно-
ацетоновом растворе образуется нейтральный комплекс
Рис. 19. Кривые свето-
поглощения роданидных
комплексов молибдена
(/) и вольфрама (2) в
изоамиловом спирте
состава МоО(8СМ)з. Окрашенное соединение можно эк-
страгировать органическими растворителями (изоамило-
вым спиртом и др.). При использовании экстракционного
варианта метода влияния никеля, хрома, ванадия устра-
няется.
Максимум светопоглощения комплексного соединения
молибдена с роданидом наблюдается в водных растворах
при 460 нм (е= 10,5-103), в растворах изоамилового спир-
та— при 470 нм (рис. 19, кривая 1).
При определении содержания вольфрама вольфрам
(VI) восстанавливают в солянокислом или сернокислом
растворах с помощью хлорида олова. Восстановление
необходимо проводить после прибавления роданида, в
противном случае образующиеся низшие соединения воль-
фрама могут находиться в коллоидном, менее реакцион-
носпособном состоянии. Оптимальная кислотность для
образования комплексного соединения создается 8,5—9,5 М
раствором соляной кислоты. Кроме хлорида олова, в ка-
честве восстановителя применяют Ti (III). Оптимальная
кислотность при использовании этого восстановителя соз-
171
дается 3,1—3,3 М раствором соляной и 2,2 М раствором
серной кислоты. Титан (III) является более сильным вос-
становителем, чем хлорид олова: нормальные окислитель-
но-восстановительные потенциалы составляют для систем
W6+/W5+ £=+0,26 В, Sn<+/Sn2+ £=+0,15 В, Ti4+/Ti3+ £ =
= + Ю В. При использовании хлорида титана (III) окраска
комплексного соединения развивается быстрее (через 15—
20 мин), чем при использовании хлорида олова (через
— 60 мин). Молярный коэффициент погашения комплексно-
го соединения в водном растворе равен 17-Ю3 при Хтах =
= 400 нм.
Роданидный комплекс вольфрама можно экстрагиро-
вать с помощью органических растворителей. Кривая
светопоглощения комплексного соединения вольфрама с ро-
данидом в изоамиловом спирте приведена на рис. 19, кри-
вая 2. В присутствии такого сильного восстановителя,
каким является хлорид титана (III), молибден (VI) вос-
станавливается до Mo (III), который с роданидом образует
лишь слабо окрашенное соединение: 1 мг Мо соответст-
вует 0,02 мг вольфрама. При определении вольфрама в
присутствии молибдена можно вводить поправку на со-
держание последнего.
Таким образом, хотя вольфрам и молибден образуют
окрашенные роданидные комплексы, роданидный метод
позволяет определять содержание вольфрама в присутст-
вии молибдена, а содержание молибдена — в присутствии
вольфрама без предварительного их разделения.
2. Определение содержания молибдена и вольфрама
дитиоловым методом. Молибден (VI) и вольфрам (V) об-
разуют в сильнокислой среде 4—12 М НС1 или 6—14 н.
H2SO4, окрашенные в сине-зеленый цвет комплексные сое-
динения с дитиолом (3,4-метил-1, 2-димеркаптобензолом)
Комплекс молибдена с дитиолом имеет формулу:
Соединение, трудно растворимое в воде, хорошо раство-
ряется в органических растворителях (амилацетате, бу-
тилацетате, хлороформе).
172
В качестве восстановителя вольфрама (VI) до воль-
фрама (V) применяют хлорид олова (II). Максимум све-
топоглощения дитиолата вольфрама в амилацетате находит-
ся при 640 мн (рис. 20, молярный коэффициент погашения
комплекса при этой длине волны равен 1,9-104. Макси-
мумы светопоглощения дитиолата молибдена наблюдаются
при 440 и 675 нм. При 675 нм молярный коэффициент
погашения комплекса равен 2,1 • 104.
Дитиоловый метод позволяет проводить анализ молиб-
дена и вольфрама при их совместном присутствии. В опре-
деленных условиях вначале получают дитиолатный комп-
лекс молибдена (VI), экстрагируют его, а затем в водном
Рис. 20. Кривые свете-
поглощения комплексов
дитиола с молибденом
(/) и вольфрамом (2) в
амилацетате
растворе после введения сильного восстановителя обра-
зуют комплекс вольфрама (V) с дитиолом.
На практике вместо дитиола применяют более устой-
чивый цинк-дитиол в виде взвеси в спирте или щелочи.
Гравиметрические методы применяют для определения
высоких содержаний молибдена и вольфрама (>0,5%).
1. Определение содержания молибдена в виде молиб-
дата свинца, или плюмбатный метод, является наиболее
старым методом, который применяется с 1871 г. и до на-
стоящего времени для определения макроконцентраций
молибдена. Благодаря большой молекулярной массе мо-
либдата свинца получаются более точные результаты, чем
при использовании других гравиметрических методов, в
которых весовой формой является оксид молибдена (VI).
При анализе материалов сложного состава в присутст-
вии других элементов (Cr, V, Sb, W, Si, Р, As, Fe) требу-
ются длительные и сложные операции отделения молибде-
на или введение поправки на содержания указанных эле-
ментов. Молибден осаждают в виде молибдата свинца:
РЬ2++МоО42- = РЬМоО4. Осаждение проводят из горячего
слабоуксуснокислого раствора, содержащего ацетат аммо-
ния, добавлением ацетата или нитрата свинца. Выделен-
ный осадок отфильтровывают, промывают, прокаливают и
взвешивают в виде РЬМоО4, При высоких содержаниях
вольфрама в исследуемом материале вводят соответствую-
173
щую поправку, для чего определяют содержание вольфра-
ма в осадке молибдата свинца фотометрическим методом
после отделения молибдена путем осаждения его в виде
сульфида.
Для гравиметрического определения содержания мо-
либдена осаждают ионы Mo (VI) из уксуснокислых
растворов с помощью а-бензоиноксима (купрона). Оса-
док высушивают при 105 °C и взвешивают в виде
МоО2(СпНi2O2N)2 или прокаливают при 500—550 °C и
взвешивают в виде МоО3. Подобное осаждение является
более селективным, чем осаждение в плюмбатном методе.
Определению мешают ниобий, кремний, вольфрам, тантал.
Влияние ванадия (V) и хрома (VI) устраняют восстанов-
лением их до низших степеней окисления.
2. Для гравиметрического определения содержания
вольфрама чаще всего используют метод, основанный на
выделении вольфрамовой кислоты при действии на раст-
воримые вольфраматы минеральных кислот (кислый гид-
ролиз WO42-+2HC1 = H2WO4 + 2C1_). Выделенный осадок
вольфрамовой кислоты отфильтровывают, промывают и
прокаливают при температуре не выше 800 °C (во избежа-
ние улетучивания вольфрамового ангидрида WO3) и взве-
шивают. Часть вольфрама может остаться в виде его
растворимых соединений в растворе, особенно в присутст-
вии фосфора, мышьяка, ванадия, фтора и щелочных метал-
лов. Для более полного выделения в осадок вольфрамовой
кислоты осаждение проводят в присутствии алкалоида
цинхонина Ci9H22ON2, который образует с вольфрамо-
вой кислотой труднорастворимый вольфрамат цинхони-
на. Наряду с цинхонином применяют бетанафтохинолин и
другие органические соединения. Осадок вольфрамовой
кислоты может быть загрязнен многими элементами. На-
пример, ванадий, молибден, фосфор образуют -комплекс-
ные соединения с вольфрамом; ниобий, тантал, титан пе-
реходят в осадок в результате кислого гидролиза; ионы
железа, хрома, марганца адсорбируются осадком H2WO4;
осадок содержит также кремниевую кислоту H2SiO3-nH2O.
Обычно вольфрамовую кислоту прокаливают, взвешива-
ют в полученном осадке WO3, определяют содержание
примесей и вводят соответствующую поправку при опре-
делении содержания вольфрама. Удаление кремниевой
кислоты производят обработкой осадка фтористоводород-
ной и серной кислотами до улетучивания кремния в ви-
де SiF-i-
Гравиметрический метод определения содержания воль-
174
фрама путем выделения вольфрамовой кислоты при ана-
лизе материалов сложного состава является достаточно
длительным и трудоемким.
Титриметрические методы, определения содержаний мо-
либдена и вольфрама включают;
1. Комплексонометрическое титрование молибдена.
Комплексон III образует с ионами молибдена (V) проч-
ное комплексное соединение. Предварительно молибден
(VI) восстанавливают до молибдена (V) с помощью суль-
фата или хлорида гидразиния в присутствии избытка-
комплексона III, который оттитровывают стандартным
раствором ацетата цинка по эриохромчерному Т в амми-
ачном растворе, ксиленоловому оранжевому при pH 5,0—
5,6 или раствором сульфата циркония в присутствии кси-
ленового оранжевого в качестве индикатора в кислой
среде (0,15 М H2SO4).
При определении в аммиачной среде в присутствии вин-
ной кислоты и фторида калия титан, ниобий, тантал,
вольфрам, алюминий, лантан анализу не мешают. При
определении в кислой среде анализу не мешают алюми-
ний, магний, цинк, кадмий, кобальт, свинец, РЗЭ при
отношении их количеств к количеству молибдена не бо-
лее 1:1. Ионы железа (III), циркония и гафния, образую-
щие устойчивые комплексонаты в кислой среде, определе-
нию содержания молибдена мешают.
2. Титриметрическое определение содержания вольфра-
ма используется сравнительно редко. Наибольшее приме-
нение получил метод, основанный на выделении вольфра-
мовой кислоты кислым гидролизом, растворении осадка в
отмеренном объеме титрованного раствора едкого натра,
взятого в избытке (H2WO4+2NaOH = Na2WO4 + 2H2O), и
оттитровывании избыточных количеств раствора NaOH
раствором кислоты (HNO3, H2SC>4). В качестве индикато-
ра применяют фенолфталеин.
Практическая часть
Комплексонометрическое определение молибдена
в молибденовых концентратах
Принцип метода. Определение основано на образова-
нии комплексного соединения молибдена (V) с комплек-
соном (Ш), избыток которого оттитровывают раствором
0,01 М. ацетата цинка в присутствии индикатора ксилено-
лового оранжевого при pH 5,0—5,6. В качестве восстано-
175
витсля Mo (VI) применяют хлорид гидразиния. Вольфрам
предварительно связывают в комплекс с винной кислотой,
другие мешающие определению элементы отделяют осаж-
дением едким натром. Методика рассчитана на опреде-
ление содержания молибдена от 45 до 60%. Относитель-
ное стандартное отклонение результатов определений ~0,01.
Реактивы и растворы. 1) Кислота азотная, р= 1,43 г/см3 и раз-
бавленная 1:3; 2) кислота соляная р= 1,19 г/см3 и разбавленная 1:1,
1:3; 3) кислота серная, р= 1,84 г/см3 и разбавленная 1:1; 4) кислота
винная, 20%-ный раствор; 5) кислота уксусная, р= 1,05 г/см3; 6) ам-
миак водный, 25%-ный раствор; 7) гидроксид натрия или натр едкий,
25%-ный раствор; 8) хлорид гидразиния, 20%-ный раствор, свеже-
приготовленный; 9) раствор, содержащий в 1 мл 1 мг молибдена (для
его получения 0,500 г металлического молибдена помещают в стакан
вместимостью 500 мл, приливают 50—60 мл разбавленной 1:3 азотной
кислоты. После растворения молибдена приливают 30 мл разбавлен-
ной 1:1 серной кислоты и нагревают раствор до выделения паров
серного ангидрида. Раствор охлаждают, приливают 2—3 мл воды
и снова нагревают до выделения паров серного ангидрида; снова
охлаждают и приливают 100 мл воды. Раствор кипятят 2—3 мин,
охлаждают, переливают в мерную колбу вместимостью 500 мл, до-
ливают водой до метки и перемешивают); 10) ацетат аммония;
11) ацетатный буферный раствор, pH 5,8 (для его получения 500 г
ацетата аммония растворяют в воде, переводят в мерную колбу
вместимостью 1 л, добавляют 20 мл уксусной кислоты, доводят до
метки водой и перемешивают); 12) ацетат цинка, 0,01 М раствор
(для его получения 2,195 г соли помещают в мерную колбу вмести-
мостью 1 л, прибавляют 25 мл ацетатной буферной смеси, доливают
водой до метки, перемешивают); 13) уротропин, 30%-ный раствор;
14) бромтимоловый синий (индикатор), 0,1%-ный раствор; 15) ксиле-
ноловый оранжевый (индикатор), 0,2%-ный раствор; 16) комплексон III,
0,01 М раствор.
Установка титра 0,01 М раствора комплексона III.
Отбирают пипеткой 50 мл стандартного раствора молибдена в кони-
ческую колбу вместимостью 500 мл, приливают 50 мл воды, 2—3 ка-
пли раствора бромтимолового синего и раствор едкого натра до появ-
ления синей окраски раствора. Затем к раствору приливают 2 мл
раствора винной кислоты, перемешивают и приливают 40 мл раствора
комплексона III, разбавленную 1:1 соляную кислоту до появления
желтой окраски и сверх этого количества в избыток 10 мл, 10 мл ра-
створа хлорида гидразиния; раствор кипятят в течение 4 мин, охлаж-
дают, прибавляют одну каплю бромтимолового синего и раствор ам-
миака до появления зеленого оттенка, затем вводят разбавленную 1:3
соляную кислоту до появления желтой окраски раствора и сверх
этого количества в избыток 2 мл, 15 мл раствора уротропина и через
10 мин 0,6—2 мл раствора ксиленолового оранжевого. После пере-
мешивания раствор титруют раствором ацетата цинка до изменения
окраски раствора до винно-красной.
Соотношение объемов растворов комплексона III и ацетата цинка
устанавливают следующим образом. В колбу вместимостью 500 мл
приливают 100 мл воды, 3—4 капли раствора бромтимолового синего,
40 мл раствора комплексона III из бюретки и разбавленную 1:3 соля-
ную кислоту до изменения окраски раствора и сверх этого количества
2 мл. Затем в колбу приливают 15 мл раствора уротропина, через
176
10 мин 10—12 капель раствора ксиленолового оранжевого и титруют
раствор 0,01 М раствором ацетата цинка до появления винно-красной
окраски раствора.
Выполнение определения. Навеску концентрата 0,25 г
помещают в коническую колбу вместимостью 250 мл, при-
ливают 40 мл азотной кислоты и нагревают в течение
10 мин. Затем приливают 10 мл соляной кислоты и про-
должают нагревание до прекращения выделения оксидов
азота, приливают 10 мл разбавленной 1:1 серной кислоты
и нагревают до выделения паров серного ангидрида. Раст-
вор охлаждают, стенки колбы обмывают 2—3 мл воды и
вновь нагревают до выделения паров серного ангидрида.
Раствор охлаждают, приливают 100—120 мл воды и на-
гревают до растворения солей. Затем добавляют 2—3 кап-
ли раствора бромтимолового синего и осторожно неболь-
шими порциями приливают раствор едкого натра до по-
явления синего окрашивания и, кроме того, 5 мл в
избыток. Раствор кипятят в течение 5 мин, охлаждают и
вместе с осадком переносят в мерную колбу вмести-
мостью 250 мл, доливают до метки водой, перемешивают
и фильтруют через сухой плотный фильтр в сухую кони-
ческую колбу, отбрасывая первые порции фильтрата.
В коническую колбу вместимостью 500 мл вводят
50 мл фильтрата, приливают 2 мл раствора винной кис-
лоты, 40 мл раствора комплексона III, 3—4 капли бром-
тимолового синего и небольшими порциями разбавленную
1:1 соляную кислоту до перехода окраски раствора в жел-
тую и сверх этого количества 10 мл в избыток. К раствору
приливают 10 мл хлорида гидразиния. После кипячения
в течение 5 мин полученный раствор охлаждают, прибав-
ляют одну каплю бромтимолового синего и далее все
реактивы, как указано при установлении титра раствора
комплексона III. Каждый миллилитр разности величин до-
бавленного 0,01 М раствора комплексона III и раствора
ацетата цинка соответствует 1,92 мг молибдена.
Фотометрическое определение молибдена
в сплавах с ниобием и вольфрамом (роданидный метод)
Принцип метода. Определение основано на реакции об-
разования соединения молибдена (V) с роданидом аммо-
ния в солянокислом растворе. В качестве восстановителя
используют тиомочевину в присутствии сульфата меди.
Влияние ниобия устраняют введением оксалата аммония.
Вольфрам определению не мешает.
7. 129-
177
Метод рассчитан на определение содержания молибде-
на от 1 до 5%. Относительное стандартное отклонение ре-
зультатов определений 0,05.
Реактивы и растворы. 1) Пиросульфат калия; 2) оксалат аммо-
ния, 4%-ный раствор; 3) кислота соляная, р=1,19 г/см3 и разбавленная
1:1; 4) сульфат аммония; 5) роданид аммония, 50%-ный раствор;
6) сульфата меди раствор (для его получения 4 г CuSO4-5H2O раст-
воряют в 100 мл воды); 7) тиомочевины раствор (для его получения
10 г тиомочевины растворяют в 100 мл воды при постоянном пере-
мешивании при комнатной температуре); 8) кислота серная, р =
= 1,84 г/см3; 9) запасной раствор молибдена, содержащий в 1 мл
1,0 мг молибдена (для получения такого раствора 1,840 г молибдата
аммония (NH4)eMo7O24-4H2O растворяют в воде, переводят в мерную
колбу вместимостью 1 л и разбавляют водой до метки); 10) рабочий
раствор, содержащий в 1 мл 50 мкг молибдена (раствор готовят
разбавлением запасного раствора в 20 раз).
Построение градуировочного графика. В ряд мерных
колб вместимостью по 50 мл вводят из микробюретки от
1,0 до 4,0 мл раствора молибдена, содержащего 50 мкг
молибдена в 1 мл, с интервалом в 0,5. мл. В колбы до-
бавляют по 25 мл разбавленной 1:1 соляной кислоты,
1,5 мл раствора сульфата меди; раствор перемешивают,
охлаждают примерно до 18 °C, добавляют к нему по 15 мл
раствора тиомочевины (при этом может образоваться оса-
док, который растворяется через несколько минут) и вновь
перемешивают. Через 10 мин в колбы вводят 1 мл раст-
вора роданида аммония, разбавляют до метки водой и
тщательно перемешивают. Через 10 мин измеряют опти-
ческую плотность растворов при Х = 460 нм в кювете с /=
= 20 мм по сравнению с водой. (Окрашенные растворы
устойчивы в течение 45 мин.)
По полученным значениям строят график зависимости
оптической плотности от концентрации молибдена, точки
которого периодически проверяют.
Выполнение определения. Навеску сплава 0,1 г сплав-
ляют в кварцевом тигле с 2—3 г пиросульфата калия при
температуре 900 °C. Остывший плав растворяют в 150 мл
оксалата аммония, переводят раствор в мерную колбу
вместимостью 250 мл, доводят до метки тем же раство-
ром оксалата аммония и перемешивают. Вместо сплавления
можно растворить сплав при нагревании на электроплитке
в жаростойком стакане в 3 мл серной кислоты с добавле-
нием 1 г сульфата аммония; по растворении добавить раст-
вор оксалата аммония.
Для определения содержания молибдена 10—15 мл
раствора отбирают в мерную колбу вместимостью 50 мл„
добавляют 12,5 мл соляной кислоты (1,19), 1,5 мл раство-
178
ра сульфата меди, перемешивают и охлаждают до ~18°С.
Далее прибавляют все реактивы, как указано при построе-
нии градуировочного графика. Содержание молибдена в
пробе рассчитывают по градуировочному графику.
Фотометрическое определение молибдена
в оксиде ниобия (дитиоловый метод)
Принцип метода. Определение основано на образовании
окрашенного комплекса молибдена (VI) с цинк-дитиолом
в 6 М солянокислом растворе. Дитиолат молибдена экстра-
гируют амилацетатом, бутилацетатом или хлороформом.
Основу отделяют путем сплавления пробы со щелочью.
Предел определяемых содержаний составляет 5-10”3% при
навеске 0,5 г. Относительное стандартное отклонение ре-
зультатов определения 0,2—0,1 при содержаниях молиб-
дена 5-10“3—5-10~ ’%.
Реактивы и растворы. 1) Натр едкий кристаллический, 2%-ный
раствор; 2) кислота соляная, р=1,19 г/см3; 3) цинк-дитиол (для при-
готовления суспензии 0,1 г цинк-дитиола растирают в фарфоровой
ступке с минимальным количеством спирта и разбавляют им до 25 мл;
перед применением суспензию взбалтывают. Можно приготовить сус-
пензию цинк-дитиола также путем нагревания' навески препарата при
температуре не выше 80 °C в 2%-ном растворе едкого натра; раствор
должен быть свежеприготовленным); 4) амилацетат (амиловый эфир
уксусной кислоты), или бутилацетат (бутиловый эфир уксусной кисло-
ты), или хлороформ; 5) запасной раствор, содержащий в 1 мл 0,1 мг
молибдена (для его приготовления 0,1 г металлического молибдена
растворяют в 15%ном растворе пероксида водорода, приливают избы-
ток аммиака; раствор кипятят до разрушения пероксида водорода и
разбавляют водой до 1000 мл); 6) рабочий раствор, содержащий в
1 мл 10 мкг молибдена (для его приготовления 10 мл запасного ра-
створа разбавляют в мерной колбе вместимостью 100 мл водой до
метки).
Построение градуировочного графика. В пять стакан-
чиков вместимостью по 50 мл приливают от 0,2 до 2 мл
стандартного рабочего раствора молибдена с интервалом
0,2—0,4 мл. В стаканчики приливают воду до объема 15 мл
и по 15 мл соляной кислоты. После прибавления каждого
реактива раствор тщательно перемешивают, приливают
по 2 мл суспензии цинк-дитиола и выдерживают растворы
в течение 15 мин, изредка помешивая. Затем растворы
переводят в делительные воронки вместимостью по 50 мл,
приливают по 5 мл экстрагента (бутилацетата, амилаце-
тата или хлороформа) и встряхивают в течение 1 мин. По
расслаивании водный слой отбрасывают, а экстракт пере-
водят в сухую кювету с длиной оптического пути 10 мм,
накрывают ее крышкой и дают экстракту отстояться до
7*
179
исчезновения пузырьков (~1 мин). Затем измеряют опти-
ческую плотность при длине волны Х = 675 нм по отноше-
нию к воде. Одновременно проводят контрольный опыт на
реактивы. По полученным значениям строят график зави-
симости оптической плотности раствора от концентрации
молибдена.
Выполнение определения. Навеску 0,5 г оксида ниобия
(V) помещают в никелевый тигель, где находится 1 г пред-
варительно расплавленного едкого натра, добавляют еще
3 г едкого натра и ведут сплавление в муфеле при 800—
850 °C до получения однородного плава. Плав выщелачи-
вают примерно в 60 мл воды в стакане вместимостью
150—200 мл при кипячении. По охлаждении раствор с
осадком переводят в мерную колбу вместимостью 100 мл,
разбавляют водой до метки, тщательно перемешивают и
выдерживают до тех пор, пока раствор над осадком не
станет прозрачным. Отбирают аликвотную часть раство-
ра в зависимости от содержания молибдена, помещают ее
в стакан вместимостью 50—70 мл и разбавляют водой до
15 мл. К раствору приливают 15 мл соляной кислоты и
перемешивают. Затем приливают 2 мл суспензии цинк-
дитиола и раствор переносят в делительную воронку вме-
стимостью 50 мл. Далее поступают, как указано при
построении градуировочного графика. Измеряют опти-
ческую плотность экстракта при Х = 675 нм. Контрольный
опыт на реактивы проводят через все стадии анализа.
Оптическая плотность раствора контрольного опыта не
должна превышать 0,01 при определении содержания мо-
либдена порядка 10~3%.
Рений
Задолго до открытия рения его существование было предсказа-
но Д. И. Менделеевым как аналога марганца (двимарганца). Открыт
рений был лишь много лет спустя, в 1925 г. немецкими химиками
В. Ноддаком, И. Таке и О. Бергом. Почти одновременно с ними
сообщения об открытии этого элемента сделаны английским исследо-
вателем Ф. Лорингом и чешскими учеными Г. Друце, Я. Гейровским
и В. Долейжеком. Название рения происходит от названия Рейнской
области в Германии.
Нахождение в природе. Рений является одним из самых редких
элементов; его содержание в земной коре составляет ~ I -10—7%• Это-
типичный рассеянный элемент. Он встречается в основном в виде
следов в рудах и минералах различных элементов. Наиболее бога-
тым источником рения является молибденит M0S2, содержащий
10-1—10-so/o рения. Предполагают, что рений в молибдените находит-
ся в виде сульфида ReS2 или оксида Re2O7.
Сырьевым источником рения являются также медные руды. Так,
в медных рудах Джезказганского месторождения в 1961 г. обнаружен
180
минерал, названный джезказганитом. Его формулу можно указать
лишь предположительно: (Re, MojSa—сульфид молибдена и рения
или CuReS4 — сульфоренат меди. Медные и молибденовые концентра-
ты, получаемые в результате переработки соответствующих руд, со-
держат от 2-Ю-3 до 2-10" 1о/о рения.
Применение. Благодаря высокой температуре плавления и хоро-
шим механическим свойствам при повышенных температурах рений
находит применение в производстве жаропрочных сплавов. Особенно
большое значение имеют сплавы рения с другими тугоплавкими ме-
таллами— вольфрамом и молибденом. Они используются при изготов-
лении термопар, работающих при температурах >2000 °C, электро- и
электронных ламп, электроконтактов, а также в авиационной и косми-
ческой технике. Рений и его сплавы применяются в приборостроении
при изготовлении деталей точных приборов, в качестве катализато-
ра при крекинге нефти вместо более дорогостоящих металлов плати-
новой группы.
Свойства рения и его соединений. Температура плав-
ления рения 3180 °C. После вольфрама рений является
наиболее тугоплавким из всех металлов. По внешнему
виду это серебристо-белый металл, напоминающий пла-
тину.
Рений является элементом VII группы Периодической
системы Д. И. Менделеева, он относится к третьему ряду
переходных элементов главной, или d-группы. Рений имеет
семь валентных электронов 5d56s2. В соединениях встре-
чается в восьми состояниях окисления: от +7 до — 1.
Главным образом, это степени окисления +7, +5 и +4.
Максимальная степень окисления +7 является наиболее
характерной для рения.
В водных растворах рений чаще всего находится в ви-
де оксоанионов ReOV (перренат-ионов), которые устойчи-
вы как в кислых, так и в щелочных средах. При обычных
температурах металл устойчив, его окисление начинается
при 300°С, а выше 600 °C окисление протекает интенсивно
с образованием рениевого ангидрида Re2O7.
Рений не растворяется ни на холоду, ни при нагрева-
нии в соляной и фтористоводородной кислотах. В серной
кислоте при нагревании рений растворяется, образуя ре-
ниевую кислоту HReCR. В азотной кислоте, пероксиде
водорода рений также растворим с образованием рение-
вой кислоты: 3Re + 7HNO3 = 3HReO4 + 2H2O + 7NO 2Re +
+ 7Н2О2 = 2HReO4 + 6Н2О.
В растворе аммиака в присутствии пероксида водорода
рений растворяется с образованием соответствующей аммо-
нийной соли — перрената аммония NH4ReO4. Рений раство-
ряется при слабом нагревании в бромной воде с образо-
ванием HReO4. Рений, находящийся в виде порошка или
мелкой стружки, можно сплавить со щелочами. Более
1 81
быстрому разложению способствуют такие окислители, как
пероксид натрия Na2O2, нитрат калия KNO3; при этом
образуются соответствующие перренаты натрия и калия.
Оксид рения (VII) Re2O7, или рениевый ангидрид,
является высшим и наиболее устойчивым кислородным
соединением рения. Это кристаллы светло-желтого цвета,
хорошо растворимые в воде с образованием рениевой
кислоты HReO4:Re2O7 + H2O = 2HReO4.
Рениевая кислота HReO4— сильная однооснов-
ная кислота, являющаяся сравнительно слабым в отличие
от марганцовой кислоты окислителем: окислительно-вос-
становительный потенциал системы МпОГ/МпО2 £=1,68 В,
а системы ReO£/ReO2 £ = 0,51 В.
В промышленности рений получают путем восстанов-
ления соли рениевой кислоты — перрената аммония
NH4ReO4 водородом. О растворимости перренатов в воде
(при 30 °C) можно судить по следующим данным:
Перренат ..............Ca(ReO4)2 NaReO4 NH4ReO4 KReO4
Растворимость, г/100 мл 187 145 8,70 1,47
Таким образом, наиболее растворим перренат кальция,
наименее — перренат калия.
Известны перренаты органических катионов, например
перренат нитрона С20Н1бИ4-HReO4, который благодаря ма-
лой растворимости применяется для гравиметрического
определения содержания рения.
Разложение руд и концентратов, содер-
жащих рений, производят в азотной или в смеси
азотной и соляной кислот при нагревании. Если в мине-
ралах или рудах содержатся тантал, ниобий, вольфрам,
титан, кремний, то для разложения применяют смесь азот-
ной и фтористоводородной кислот.
Для минералов, не растворяющихся в кислотах, при-
меняют сплавление с карбонатом натрия или едкими ще-
лочами в присутствии окислителей — пероксида натрия,
нитрата натрия. Применяется также сплавление с одним
пероксидом натрия.
Для разложения молибденовых концентратов их спе-
кают с оксидом кальция в присутствии окислителей KNO3,
КМпО4 при температуре 650—700 °C. В процессе после-
дующего растворения спека в воде основная часть мо-
либдена остается в осадке в виде малорастворимого мо-
либдата кальция СаМоО4 (растворимость 0,0056 г/100 мл).
Рений в виде перрената кальция Ca(ReO4)2, хорошо раст-
воримого в воде, переходит в раствор: 4ReS2 + 2CaO +
182
+ 15O2 = 2Ca(ReO4)2 + 8SO2. В осадке с основной частью
молибдена находятся малорастворимые соли вольфрама
(VI), гидроксиды железа (III), меди (II), марганца (IV)
и других элементов. В раствор, кроме рения, лишь в не-
больших количествах переходят молибдат-, вольфрамат-
и сульфат-ионы. Хлорид бария образует с указанными ио-
нами малорастворимые соединения и способствует их бо-
лее полному осаждению.
Сплавы рения растворяют в смеси азотной и со-
ляной, соляной и серной кислот с добавлением пероксида
водорода, а также в смеси фтористоводородной и азотной
кислот. ‘Сплавы рения с вольфрамом и молибденом раст-
воримы в пероксиде водорода. Для разложения некото-
рых сплавов, содержащих рений, применяют сплавление
со щелочами в присутствии окислителей, со смесью соды и
хлората калия.
Используя для разложения ренийсодержащих матери-
алов кислотное растворение, необходимо помнить, что сое-
динения рения (VII) характеризуются высокой лету-
честью при нагревании. При выпаривании солянокислых
растворов наблюдаются потери рения. Выпаривание сер-
нокислых растворов рения (VII) в присутствии или в от-
сутствие соляной и азотной кислот ведет к улетучиванию
рения. В присутствии фтористоводородной кислоты рений
не улетучивается. На летучести соединений рения осно-
ваны методы его отделения от других элементов. Из раст-
воров концентрированной серной кислоты рений отгоняют
при температуре 260—280 °C.
Методы определения содержания рения
Фотометрические методы определения содержания ре-
ния включают роданидный, тиомочевинный, диметилглиок-
симный методы и, кроме того, методы с основными краси-
телями.
1. Роданидный метод относится к наиболее старым
методам определения рения, но он еще имеет достаточно
широкое применение, особенно при определении малых
содержаний элемента. Метод основан на образовании в
1—4 М солянокислом растворе окрашенного в оранжевый
цвет комплексного соединения рения с роданид-ионом в
присутствии восстановителя хлорида олова (II). Предпо-
лагают, что состав соединения рения (V) с роданидом
выражается формулой Кз[ReO2(SCN)4].
Максимум светопоглощения комплекса находится при
430 нм (рис. 21); реакция является достаточно чувстви-
183
тельной: молярный коэффициент погашения е = 26-103.
Определению рения мешают молибден (VI), вольфрам
(VI), медь (II), ванадий (V), железо (III). Молибден с
роданидом образует также окрашенный комплекс
[MoO(SCN)5]2~. Влияние небольших количеств молибдена
устраняется проведением реакции в 4,5—5 М соляной кис-
лоте, комплекс молибдена при этом разрушается.
При определении рения в материалах с высоким содер-
жанием молибдена последний отделяют путем спекания
пробы с оксидом кальция или экстракцией молибденового
комплекса диэтиловым эфиром.
Рис. 21. Кривая свето-
поглощения роданидного
комплекса рения в изо-
амиловом спирте
Для снижения предела обнаружения рения применяют
экстракцию комплексного соединения рения с роданидом.
При экстракции комплекса бутилацетатом из 3—5 М соля-
нокислого раствора молярный коэффициент погашения
комплекса увеличивается до 41,5-103. При экстракционном
варианте метода избирательность определения повышается:
допустимо присутствие по 1 мг железа, вольфрама, меди,
хрома, никеля и некоторых других элементов.
2. Тиомочевинный метод является также одним из рас-
пространенных для определения содержания рения. Он
основан на образовании окрашенного в желто-зеленый
цвет комплексного соединения рения с тиомочевиной в
3—5 М солянокислых растворах в присутствии восстано-
вителя хлорида. Формула соединения имеет вид
zNH2
[ReO2(S = C( )4] +
Молярный коэффициент погашения комплекса равен
10,5-103 при Z = 390 нм и 6,96-103 при 450 нм.
Определению рения не мешают 1 мг молибдена, по
10 мг железа и алюминия, 2 мг вольфрама. Мешает опре-
делению рения медь. Ниобий и тантал маскируют с по-
мощью оксалат-ионов.
184
3. Диметилглиоксим образует с рением в солянокислом
или сернокислом растворах после восстановления рения
(VII) хлоридом олова (II) до низших степеней окисле-
ния комплексное соединение, состав которого, по-видимому,
[Re(OH)2H2Z)m] С12, где H2Z)m—молекула диметилглиокси-
ма. Молярный коэффициент погашения комплексного со-
единения равен 6,9-103 при Z = 440 нм. Этот метод находит
широкое применение благодаря доступности реагента, про-
стоте выполнения реакции (водная среда), устойчивости
окраски комплексного соединения.
Так как молярный коэффициент погашения комплекса
невысок, то метод с использованием диметилглиоксима
применяют для определения макросодержаний рения, на-
пример при анализе сплавов.
При определении содержания рения в сплавах с редки-
ми металлами — вольфрамом, ниобием анализ возможен
без отделения от указанных элементов, так как они не
образуют окрашенных соединений с диметилглиоксимом.
Для удержания указанных редких элементов в растворе
вводят различные комплексанты — фторид-ионы, пероксид
водорода, винную кислоту.
Атомно-абсорбционное определение содержания рения.
В этом случае используется аналитическая линия рения,
соответствующая длине волны 346,1 нм. Для атомизации
применяется пламя оксид азота (I) — ацетилен.
Метод имеет преимущества перед другими методами
при анализе сплавов, особенно бинарных. В этом случае
легко приготовить эталонные растворы путем смешивания
растворов компонентов сплавов.
Гравиметрические методы. При определении содержа-
ния рения в виде перрената нитрона рений осаж-
дают нитроном (1,4-дифенил-3,5-(эндоанилино)-4-5-дигид-
ро-1,2,4-триазол)
с6|н5
N
н - с/чс
V
С6Н5
При этом образуется соединение C20Hi6N4- HReO4.
Осаждение проводят из раствора 0,3—0,5%-ного уксусно-
кислого нитрона, в котором перренат нитрона практически
185
нерастворим (в воде при О °C растворяется —0,0017 г/л
перрената нитрона). Определению содержания рения с
нитроном мешают вольфрам, ванадий, молибден, а также
окислители (MnO4-, NO?7, Cr20z"), которые необходимо
удалять, в связи с чем метод является длительным.
Определение содержания рения в виде перрената
тетрафен иларсония основано на том, что перре-
нат-ион образует с хлоридом тетрафениларсония
(CeHs^AsCl кристаллический осадок: ReO 4“+ (C6Hs) 4As+ =
= (C6H5)4AsReO4. Количественное осаждение рения проис-
ходит как из сильнокислых (5 М) так и из сильноаммиач-
ных (6 М NH4OH) растворов. Осадок устойчив при нагре-
вании от 106 до 185 °C. Этот метод является более избира-
тельным по сравнению с нитронным.
Электрохимические методы. Метод потенциометри-
ческого титрования основан на восстановлении Re
(VII) до Re (V) в солянокислых растворах хлоридом
олова. В качестве индикаторного электрода применяют
платину.
При потенциометрическом определении содержания
рения восстановлением Re (VII) солями хрома (II) рений
восстанавливается до Re (IV): ReO.r + 4H++3e—>-ReO2 +
+ 2Н2О. Титрование рения (VII) проводят в горячем (60—
70 °C) сернокислом (2 М) растворе в присутствии катали-
затора (иодида калия). Точки эквивалентности устанавли-
вают с помощью индикаторного электрода — платиновой
пластинки; в качестве электрода сравнения применяют на-
сыщенный каломельный полуэлемент.
Полярографические методы определения со-
держания рения основаны на возможности получения волн,
соответствующих переходу Re (VII) в низшие степени
окисления.
Практическая часть
Фотометрическое определение содержания рения
в концентратах и рудах
Принцип метода. Определение основано на реакции
образования соединений рения с тиомочевиной (при содер-
жаниях рения выше 1 -10—2%) или роданид-ионом (при со-
держаниях рения ниже 1 • 10~2%). Относительное стандарт-
ное отклонение результатов определений составляет 0,25
при содержаниях рения от 2-1О~зо/о До 5-10—2% и 0,15 при
содержаниях рения >5-10"2%.
186
Реактивы и растворы. 1) Кислота соляная, р= 1,19 г/см3; 2) каль-
ция оксид; 3) хлорид бария, 20%-ный раствор; 4) роданид калия
(аммония); 5) перманганат калия; 6) хлорид олова, растворы (для их
получения соответственно 20 и 35 г соли растворяют при нагрева-
нии в 20 мл соляной кислоты, охлаждают растворы и разбавляют их
до метки водой в колбах вместимостью 100 мл); 7) хлорид железа
(III), раствор (для его получения 10 г соли растворяют в 100 мл
воды, подкисленной соляной кислотой); 8) тиомочевина, 10%-ный ра-
створ; 9) запасной раствор, содержащий в 1 мл 1 мг рения (для его
получения 0,1553 г перрената калия растворяют в воде; раствор пере-
водят в мерную колбу вместимостью 100 мл, доводят его объем водой
до метки и перемешивают); 10) рабочий раствор, содержащий в 1 мл
0,1 мг рения (раствор А, который готовят разбавлением запасного
раствора в 10 раз); И) рабочий раствор, содержащий в 1 мл 0,01 мг
рения (раствор Б, который готовят разбавлением запасного раствора
в 100 раз).
Определение содержания рения по реакции с тиомоче-
виной. Построение градуировочного графика.
В мерные колбы вместимостью по 25 мл вводят соответ-
ственно от 50 до 200 или от 10 до 50 мкг рения в виде
раствора. К растворам добавляют по 5 мл соляной кис-
лоты, разбавляют до 20 мл водой и затем вводят по
2,5 мл раствора тиомочевины, по 1 мл раствора хлорида
олова (20 г/100 мл). Объемы растворов доводят до метки
водой и перемешивают. Оптическую плотность растворов
измеряют через 30 мин на фотоэлектроколориметре,
ИСПОЛЬЗуЯ Светофильтр, ДЛЯ которого 2-гпах = 390 нм. Кю-
вета должна иметь Z = 50 мм при содержании рения от 10
до 50 мкг и / = 20 мм при содержании рения от 50 до
200 мкг. В качестве раствора сравнения применяют рас-
твор, содержащий все реактивы.
Графики зависимости оптической плотности от содер-
жания рения строят для содержания рения в 25 мл рас-
твора соответственно 10—50 и 50—200 мкг. Эти кривые
используют в зависимости от содержания рения в пробе.
Выполнение определения. Навеску концентра-
та 0,3—0,2 г помещают в фарфоровый тигель и переме-
шивают с 3—4 г оксида кальция и 0,1—0,2 г перманганата
калия, покрывают сверху слоем оксида кальция (~2 г)
и помещают в муфельную печь, температуру которой по-
степенно повышают до 650 °C. Спекание производят при
650—700 °C в течение 2 ч. Спек растворяют в воде
(—150 мл), находящейся в стакане вместимостью 250 мл
при нагревании и помешивании в течение 20 мин.
Раствор фильтруют с помощью водоструйного насоса
через воронку Бюхнера. Осадок промывают пять-шесть раз
горячей водой и отбрасывают. Фильтрат и промывные
воды помещают в стакан вместимостью 500—600 мл и упа-
187
ривают до объема 50 мл, затем в стакан приливают 5—6 мл
раствора хлорида бария и нагревают раствор до кипения.
Осадок отфильтровывают через плотный фильтр и промы-
вают два-три раза горячей водой. Фильтрат и промывные
воды собирают в мерную колбу вместимостью 100 мл, до-
водят объем раствора до метки водой и перемешивают.
Аликвотную часть раствора, содержащую 10—200 мкг
рения, переводят в мерную колбу вместимостью 25 мл,
приливают 5 мл соляной кислоты, доводят объем раствора
приблизительно до 20 мл, вводят в колбу 2,5 мл раствора
тиомочевины, 1 мл раствора хлорида олова (20 г/100 мл).
Объем раствора доводят до метки водой.
Оптическую плотность раствора измеряют в кюветах
толщиной слоя 50 мм при содержаниях рения от 10 до
50 мкг и в кюветах с 7 = 20 мм при содержаниях рения
от 50 до 200 мкг (Л = 390 нм) по отношению к раствору,
содержащему все реактивы. Содержание рения в пробе
находят, пользуясь градуировочным графиком в зависи-
мости от содержания рения в пробе.
Определение содержания рения по реакции с роданидом.
Построение градуировочного графика. В мер-
ные колбы вместимостью по 25 мл вводят соответственно
от 5 до 25 мкг рения (с интервалом 5 мкг). В колбы до-
бавляют по 10 мл соляной кислоты. Разбавляют растворы
до 20 мл модой и затем вводят 1 мл раствора хлорида
железа, 2 мл раствора роданида аммония (калия), 1 мл
раствора хлорида олова (35 г/100 мл). Объемы растворов
до метки доводят водой и через 30 мин измеряют их опти-
ческие плотности на фотоэлектроколориметре или спектро-
фотометре (Х = 430 нм) в кювете толщиной слоя 50 мм.
В качестве раствора сравнения используют раствор,
содержащий все реактивы. По полученным данным строят
градуировочный график зависимости оптической плотности
от концентрации.
Выполнение определения. Подготовку раство-
ра пробы для анализа проводят так же, как указано при
определении содержания рения по реакции с тиомочевиной,
с той лишь разницей, что навеска пробы составляет 2 г.
Для фотометрирования отбирают аликвотную часть, содер-
жащую 5—25 мкг рения, и далее поступают, как при по-
строении градуировочного графика. Раствором сравнения
служит раствор контрольного опыта на реактивы, прове-
денный через все стадии анализа.
Фотометрическое определение рения
в сплавах с молибденом
Принцип метода. Определение основано на реакции
образования соединения рения с диметилглиоксимом в сер-
нокислом растворе. Методика рекомендуется для опреде-
ления рения при его содержании в сплаве ~10%. Относи-
тельное стандартное отклонение результатов определе-
ний 0,05.
Реактивы и растворы. 1) пероксид водорода, 30%-ный раствор;
2) кислота серная, р=1,84 г/см3, разбавленная 1:1; 3) диметилглиок-
сим, 1%-ный спиртовой раствор; 4) хлорид олова (II), 10%-ный ра-
створ; 5) раствор, содержащий в 1 мл 100 мкг рения (см. с. 187).
Построение градуировочного графика. В пять мерных
колб вместимостью по 50 мл вводят соответственно 100;
200; 300; 400 и 500 мкг рения, добавляют по 3 мл серной
кислоты, 15 мл воды, 5 мл раствора диметилглиоксима,
4 мл раствора хлорида олова. Объем растворов доводят
до метки водой. Через 2 ч измеряют оптическую плотность
растворов на спектрофотометре или фотоэлектроколори-
метре при Х = 400 нм в кюветах с /=10 мм по отношению
к раствору контрольного опыта. По полученным значе-
ниям строят график зависимости оптической плотности от
концентрации.
Выполнение определения. Навеску сплава 0,1 г раство-
ряют в 2 мл пероксида водорода. Раствор переводят в мер-
ную колбу вместимостью 100 мл и доводят его объем
до метки водой. Аликвотную часть раствора, в которой
ориентировочно содержится 50—500 мкг рения, переводят
в мерную колбу вместимостью 50 мл, добавляют 3 мл сер-
ной кислоты и далее и поступают так же, как указано при
построении градуировочного графика. Содержание рения
в пробе находят, пользуясь градуировочным графиком.
Гравиметрическое определение рения
s перренате аммония с использованием нитрона
Принцип метода. Рений осаждают в виде соединения
Г/еОГ с нитроном из сернокислого раствора. Осадок сушат
и взвешивают. Методика рассчитана на определение содер-
жания рения в пробе —70%. Относительное стандартное
отклонение результатов определений 0,01.
Реактивы и растворы. 1) Кислота серная, р=1,84 г/см3 и 2 М
раствор; 2) кислота уксусная, р = 1,05 г/см3; 3) нитрон, уксуснокис-
лый раствор (для его получения 5 г нитрона смешивают с 3 мл
уксусной кислоты и растворяют смесь в 100 мл воды; перед употреб-
6
189
лением раствор фильтруют через тигель с пористым дном); разбавле-
нием полученного раствора в 20 раз получают 0,25 %-ный раствор).
Выполнение определения. К нейтральному раствору,
содержащему не более 100 мг рения в 50 мл, осторожно
добавляют 1 мл 2 М серной кислоты и нагревают раствор
до 80 °C. К раствору прибавляют 7—8 мл раствора аце-
тата нитрона (5 г в 100 мл). Полученный раствор пере-
мешивают и выдерживают в течение 2 ч в ледяной воде,
периодически перемешивая.
Осадок фильтруют через высушенный при НО °C и
взвешенный тигель с пористым дном, промывают 10—20 мл
ледяного 0,25%-ного раствора ацетата нитрона (порциями
по 3—5 мл), а затем два-три раза промывают ледяной во-
дой, насыщенной ацетатом нитрона (порциями по 2—
3 мл) для удаления захваченного осадком нитрона. Оса-
док сушат 2—3 ч при 110 °C и взвешивают. Фактор пере-
счета с перрената нитрона на содержание рения равен
0,3306.
Редкоземельные элементы
(лантаноиды и иттрий)
В группу редкоземельных элементов (РЗЭ) входят 15 элементов
с порядковыми номерами от 57-го до 71-го, начиная с лантана и
кончатая лютецием (лантаноиды), а также элемент иттрий с поряд-
ковым номером 39. Иногда к этой группе относят скандий (порядко-
вый номер 21).
Редкоземельные элементы открыты в 1794 г. академиком Петер-
бургской академии наук И. Я. Гадолином в минерале иттербите, ко-
торый в честь ученого был переименован в гадолинит. Для РЗЭ харак-
терно исключительное сходство основных химических и физических
свойств (кроме свойств их ядер), в связи с чем в Периодической
системе Д. И. Менделеева они помещены в одну клетку, которая
ранее была отведена лантану.
РЗЭ делят на две подгруппы — цериевую, в которую входят лан-
тан, церий, празеодим, неодим, прометий, самарий и европий; и иттри-
евую, которая включает иттрий, гадолиний, тербий, диспрозий, голь-
мий, эрбий, тулий, иттербий, лютеций (а также скандий). Свойства
элементов этих подгрупп несколько различны.
Нахождение в природе. По содержанию в земной коре (10~2%)
РЗЭ, особенно элементы цериевой подгруппы, являются достаточно
распространенными. Элементы нечетных номеров менее распростране-
ны, чем четных. Содержание наиболее редкого из РЗЭ — тулия (8Х
X10~5%) соизмеримо с содержанием таких широко применяемых в
практике элементов, как сурьма (4-10~5%) и кадмий (5-10-5%).
В земной коре наиболее распространенного из РЗЭ церия содержится
почти столько же (4,5-10~3%), сколько олова (4-10-3%) или цинка
(5- 10~з0/о). Вопрос о нахождении прометия в земной коре окончатель-
но не решен. РЗЭ в значительной степени являются рассеянными
элементами.
К основным минералам РЗЭ относятся: монацит (фосфаты РЗЭ)
190
(Ce, La,...)P04, содержащий торий, цирконий и др., ксенотим (фос-
фат иттриевых земель) УРО4, содержащий небольшие количества то-
рия; бастнезит (фторкарбонат церия и лантана) (Се, La) (COS)F и
паризит (фторкарбонат цериевых земель и кальция)
(Се, La)гСа (СО^)зр2-
РЗЭ входят в состав минералов, представляющих собой сложные
оксиды различных элементов. Некоторые минералы, например, лопа-
рит, пирохлор, содержат ниобий, тантал, титан. РЗЭ входят также
в состав циркониевых и урановых минералов.
Применение. РЗЭ применяются в черной и цветной металлургии,
стекольной, керамической химической промышленности, медицине, сель-
ском хозяйстве. В качестве присадок их применяют при производстве
чугуна, стали и сплавов цветных металлов. Присадки вводят в виде
сплава смеси лантаноидов, называемого мишметаллом.
Добавки оксидов РЗЭ в процессе стекловарения позволяют полу-
чать стекла различных цветов: ярко-красные (оксид неодима), зеле-
ные (оксид празеодима). Оксид церия (IV) применяют в стекольной
промышленности для полировки стекла. Экраны кинескопов для цвет-
ного телевидения изготовляют из ванадата иттрия, активированного
оксидом европия. РЗЭ используются в производстве оптических кван-
товых генераторов — лазеров (иттрий-гадолиниевые, а также гадоли-
ний-таллиевые гранаты, легированные неодимом).
Свойства РЗЭ и их соединений. Электронные конфи-
гурации атомов лантаноидов можно представить общей
формулой ls22s22p63s23p63dl04s24p64dlo4fn5s25pe5dm6s2, где
п изменяется от 2 до 14, a m принимает только два значе-
ния (0 или 1). В связи с тем, что число остальных элек-
тронов остается постоянным, для описания электронной
конфигурации лантаноида достаточно указать число 4f- и
5б/-электронов. Именно заполнением глубоколежащего
электронного уровня 4f объясняется исключительная бли-
зость химических свойств элементов этой группы.
Заполнение уровня 4f происходит таким образом, что
для первых семи элементов (от церия до гадолиния) спины
электронов параллельны, а для последующих элементов
^от тербия до лютеция) заполнение уровня электронов
происходит таким образом, что спины электронов антипа-
раллельны относительно первых. Такой порядок заполне-
ния электронами уровня 4f и является физической основой
деления элементов на цериевую и иттриевую подгруппы.
Эти подгруппы элементов различаются между собой хи-
мическими свойствами, в частности, растворимостью их
соединений, способностью к комплексообразованию.
Все РЗЭ в нормальных условиях имеют степень окис-
ления + 3, однако в особых условиях некоторые РЗЭ спо-
собны проявлять другую, «аномальную» степень окисления:
+ 4 для церия, празеодима и тербия; +2 для самария,
европия и иттербия. В состоянии «аномальной» степени
окисления элементы приобретают химические свойства,
191
резко отличающиеся от свойств остальных РЗЭ. Напри-
мер, церий (IV) напоминает элементы подгруппы титана,
а самарий (II), европий (II) и иттербий (II) приобретают
сходство с щелочноземельными металлами.
С увеличением порядкового номера ионные радиусы
РЗЭ в степени окисления +3 изменяются лишь незначи-
тельно, причем при переходе от La3+ к Lu3+ происходит
уменьшение ионных радиусов от 0,006 до 0,085 нм. Это
явление, называемое «лантаноидным сжатием», объяс-
няется особенностями строения оболочки 4f, сжимающейся
по мере заполнения. Иттрий по величине ионного радиуса
попадает в группу РЗЭ. Ионные радиусы иттрия и голь-
мия почти равны. Этим и объясняется сходство свойств
иттрия с РЗЭ.
РЗЭ — серебристо-белые металлы, напоминающие по
внешнему виду железо. Они очень реакционноспособны:
на воздухе быстро тускнеют, выше 200 °C реагируют с кис-
лородом, образуя оксиды. Иттрий устойчив на воздухе
вследствие образования на его поверхности защитной
оксидной пленки. Точки плавления элементов цериевой
группы ниже, чем элементов иттриевой группы (для лан-
тана /пл = 920 °C, для лютеция /Пл=1675°С). РЗЭ разла-
гают воду (на холоду медленнее, чем при нагревании),
легко растворяются в соляной, азотной и серной кислотах.
Во фтористоводородной кислоте металлы устойчивы, что
объясняется образованием защитных пленок малораство-
римых фторидов.
Оксиды РЗЭ, или «редкие земли»1 в основном
имеют состав ЬпгОз (где Ln — лантоноид). Исключение
составляют: церий (который при окислении образует оксид
СеО2), празеодим (при прокаливании в токе кислорода
или на воздухе образуется Рг6Оц), тербий (при тех же
условиях ТЬ4О7). Если нагревание вести в токе водорода,
то при 500—600 °C празеодим и тербий дают оксиды типа
Ln2O3.
Оксиды РЗЭ обладают основными свойствами, мало-
растворимы в воде. Оксид лантана, обладающий наиболее
основными свойствами, показывает щелочную реакцию во-
ды. Оксиды РЗЭ, за исключением оксида церия (IV) СеО2,
хорошо растворяются в кислотах, даже в слабой уксусной
кислоте. Оксид церия (IV) растворяется в горячей серной,
кислоте с образованием оранжево-красного сульфата це-
рия (IV).
1 Этот термин применяют и для редкоземельных элементов.
192
Гидроксиды РЗЭ Ln(OH)3 являются сильными
основаниями. Основные свойства их уменьшаются по мере-
возрастания порядкового номера элемента, т. е. при пере-
ходе от лантана к лютецию. Исключение составляет
гидроксид У(ОН)з, который является более слабым осно-
ванием, чем La(OH)3.
При прибавлении к растворам солей РЗЭ раствора
едкой щелочи или аммиака гидроксиды (или основные
соли) осаждаются в виде аморфных осадков. С уменьше-
нием основности в ряду лантаноидов значения pH начала-
осаждения понижаются от 7,8 для La до 6,3 для Lu.
Гидроксид Се (IV) осаждается из растворов при pH 0,9—1,
что позволяет отделить церий от других лантаноидов
после окисления его до Се (IV).
Гидроксиды РЗЭ растворимы в кислотах, нерастворимы,
в избытке аммиака, в растворах едких щелочей и в воде.
Растворимость солей РЗЭ в воде неодинакова.
К плохо растворимым солям относятся оксалаты
Ln2(C2O4)3-10Н2О, фториды LnF3-0,5H2O (где Ln—La, Се
и т. д.), фосфаты LnPO4-ftH2O и карбонаты. Хорошо рас-
творимы в воде нитраты, хлориды, перхлораты и сульфаты.
Сульфаты Ln2(SO4)3-8H2O хорошо растворимы в ледяной
воде и отличаются по растворимости от двойных сульфа-
тов щелочных металлов и РЗЭ Ln2(SO4)3-Na2SO4-HH2O.
РЗЭ могут быть разделены на три группы методом,,
основанным на различной растворимости двойных сульфа-
тов РЗЭ и щелочных металлов Ln2(SO4)3-Na2SO4-12Н2О!
в холодном насыщенном растворе сульфата натрия. Це-
риевая группа (лантан, церий, празеодим, неодим, сама-
рий) , а также скандий дают нерастворимые двойные суль-
фаты. Тербиевая группа (европий, гадолиний, тербий)
образуют умеренно растворимые сульфаты. Иттриевая
группа (иттрий, диспрозий, гольмий, эрбий, туллий, иттер-
бий, лютеций) характеризуется образованием растворимых
двойных сульфатов. В аналитической химии в настоящее
время этот метод применяется исключительно редко, так
как дает возможность лишь с небольшой точностью раз-
делить РЗЭ на группы. Он имеет скорее исторический
интерес, так как от него происходит деление на цериевые
и иттриевые земли.
Разложение руд и минералов РЗЭ. Минералы группы
фосфатов — монацит [(Се, La)PO4] и др.— разлагают при
нагревании с концентрированной серной кислотой. Для
вскрытия проб их сплавляют также с содой, пероксидом
193,
натрия, фторидом натрия Na2F2 или с кислыми фторида-
ми NaHF2.
Минералы группы фторкарбонатов, например паризит
(Се, Ьа)2Са(СО3)3Р2, бастнезит (Се, La)CO3F, разлагают
концентрированной серной кислотой и прокаливают при
650 °C. РЗЭ в виде сульфатов извлекают в водную вытяж-
ку, а кальций и железо переводят в труднорастворимые
осадки.
Сложные минералы, содержащие титан, тантал и нио-
бий, вскрываются достаточно трудно. Например, лопарит
и пирохлор разлагают фтористоводородной кислотой при
нагревании, при этом удаляется кремний (в виде SiF.*),
а титан, тантал, ниобий, железо, алюминий переходят в
раствор (в виде растворимых фторидов). В остатке остают-
ся фториды тория и РЗЭ; после выпаривания с серной
или соляной кислотами РЗЭ переходят в раствор, из кото-
рого их можно осаждать в виде оксалатов.
Для разложения минералов применяют также раство-
рение при нагревании в смеси (NH4)2SO4+H2SO4. При
этом в раствор переходит большое число элементов. Для
отделения от них РЗЭ последние осаждают в виде фто-
ридов.
Разложение сплавов, содержащих РЗЭ. Сплавы на
железной основе обычно растворяют в соляной кислоте
или царской водке. Если стали или сплавы содержат
вольфрам, ниобий, тантал, цирконий, то разложение ведут
в присутствии фтористоводородной кислоты с последую-
щей обработкой пробы серной кислотой.
Методы определения содержания РЗЭ
Фотометрические методы. 1. Определение содержания
РЗЭ с использованием органических реагентов основано
на образовании комплексных соединений. При этом воз-
можно определять как суммарное содержание РЗЭ, так
и содержание отдельных РЗЭ, в том числе Се (III) в от-
сутствии других РЗЭ.
Арсеназо III (формулу реагента см. «Цирконий и гаф-
ний») в слабокислом растворе взаимодействует как с сум-
мой РЗЭ, так и с каждым из них, образуя комплексное
соединение, окрашенное в зеленый цвет. Сам реагент в
слабокислых растворах имеет красно-розовую окраску.
При максимальном светопоглощении комплекса ХШах =
= 650 нм реагент практически не поглощает света (А,тах=
= 515 нм). Молярный коэффициент погашения (е) ком-
194
плекса арсеназо III с церием составляет 6,65-104, опти-
мальный pH раствора 2,3—2,7. Для других РЗЭ значе-
ние £ несколько отличаются: так, для комплекса арсе-
назо III с лантаном £ = 6,63-104, с гадолинием £=?6,92-10\
с иттрием £ = 7,96-104 и т. д. Арсеназо III применен совет-
скими химиками В. Г. Горюшиной, С. Б. Саввиным и
Е. В. Романовой для определения РЗЭ в рудах.
Для повышения избирательности определения анализ
ведут в растворе, более кислом (pH — 1,8), чем рассмотре-
но выше (pH 2,3—2,7). В этих условиях допустимо при-
сутствие 15 мг Са (при введении равных количеств в рас-
твор контрольного опыта), 50 мг Mg, 0,2 мг Си, 0,01 мг
Fe (III), 5 мг Fe (II) (восстановитель хлорид гидроксил-
аммония), 0,1 мг Ti, 0,5 мг Zr (в присутствии щавелевой
кислоты), 0,01 мг Nb, 0,2 мг Мо, 3 мг F-, 100 мг SOf, 40 мг
комплексона III, 5 мг А1 (в присутствии сульфосалицило-
вой кислоты), 0,5 мг NOT, Ю мг РО4’-.
2. Спектрофотометрические методы определения содер-
жания отдельных РЗЭ основаны на использовании спект-
ров поглощения растворов солей РЗЭ — хлоридов, нитра-
тов, перхлоратов. Из всех элементов Периодической си-
стемы Д. И. Менделеева только у солей РЗЭ (и солей
актинидов) наблюдаются довольно узкие полосы погло-
щений с острыми максимумами в инфракрасной, видимой
и ультрафиолетовой областях спектра. Узкополосные спект-
ры поглощения аква-ионов лантаноидов объясняются осо-
бенностями строения их оболочек, причем спектр погло-
щения каждого РЗЭ имеет характерный, только ему при-
сущий вид (рис. 22), так как отражает электронные
переходы на оболочке 4f. Исключение составляют ионы
иттрия, лантана и лютеция, которые не обладают соб-
ственным поглощением в растворах их
солей. Спектры поглощения РЗЭ исполь-
зуют для определения содержания от-
дельных РЗЭ с помощью спектрофото-
метров или фотоэлектроколориметров,
снабженных ртутной лампой СВД-120А
(ФЭК-56), дающей линейчатый спектр.
В табл. 5 приведены сведения об окраске
аква-ионов РЗЭ.
Молярные коэффициенты поглощения
окрашенных растворов солей РЗЭ неве-
лики (е=1-И0), поэтому спектрофото-
метрические методы определения их со-
400 500
К,нм
Рис. 22. Кривые све-
топоглощения хлори-
да празеодима
195
Таблица 5. Атомные номера элементов и окраска аква-ионов Ме3+
Атом- ный номер Элемент Цвет Атом- ный номер Элемент
57 La Бесцветный 71 Lu
58 Се 70 Yb
59 Рг Зеленый 69 Tu
60 Nd Красновато-фиолетовый 68 Er
61 Pm Ярко-розовый, желтый 67 Но
62 Sm Желтый 66 Dy
63 Eu Бледно-розовый 65 Tb
64 Gd Бесцветный 64 Cd
держаний по собственному поглощению используют для
определения достаточно высоких концентраций РЗЭ (не-
сколько ’миллиграммов в 1 мл).
Наиболее интенсивным поглощением обладают раство-
ры солей неодима и празеодима екаа3:=:7,5 при Х=575 нм
и 8Prci3 = 11 >2 при %=444 нм.
3. Фотометрические методы определения содержания
церия. Методы определения содержания церия в присут-
ствии других РЗЭ основаны на использовании его «ано-
мальной» степени окисления, равной +4.
Церий (IV) способен окислять окрашенные органиче-
ские соединения — метиловый голубой, о-толидин и др.
При этом происходит изменение окраски раствора.
Для определения содержания церия применяют также
метод, основанный на образовании окрашенного в желтый
цвет соединения церия (IV) с пероксидом водорода в ще-
лочном растворе в присутствии карбонат-, цитрат-ионов
:или нитрилтриуксусной кислоты. Максимальное поглоще-
ние соединения наблюдается в ультрафиолетовой части
спектра (%Шах = 305 нм).
Титриметрические методы. 1. Комплексонометрическое
-определение РЗЭ производят путем титрования раствора,
содержащего РЗЭ, комплексоном III при pH от 4 до 6,
когда другие элементы оказывают наименьшее влияние.
В качестве индикатора применяют ксиленоловый оран-
жевый, арсеназо III, смесь реактивов (метиленового си-
него и ализарина S) и др.
Метод комплексонометрического титрования является
методом группового определения РЗЭ (III): титруется
сумма РЗЭ, а также отдельный РЗЭ, если в растворе от-
сутствуют другие РЗЭ. Для предотвращения окисления
196
церия добавляют восстановитель — аскорбиновую кислоту
или гидроксиламин.
2. Окислительно-восстановительные реакции исполь-
зуют для определения содержания церия. Се (IV) являет-
ся сильным окислителем. Церий (III) предварительно
окисляют персульфатом аммония до Се (IV) и затем тит-
руют раствором восстановителя, например растворами ща-
велевой кислоты (с индикатором ферроином), растворами
железа (II) (в присутствии фенилантраниловой кислоты
или ферроина). При использовании в качестве восстано-
вителя соли Fe (II), или соли Мора, в растворе допустимо
присутствие больших количеств железа.
Отделение от сопутствующих элементов и гравиметри-
ческие методы определения содержания РЗЭ основаны на
предварительном осаждении РЗЭ в виде окса-
латов /.^(СгО^з-пНгО и последующем их прокалива-
нии до оксидов. Осаждение оксалатов РЗЭ проводят из
солянокислого или азотнокислого растворов при pH 0—2
путем добавления равного объема насыщенного раствора
щавелевой кислоты при температуре 60—80 °C и выдерж-
ки в течение нескольких часов. Растворимость оксалатов
в этих условиях незначительна и уменьшается с увеличе-
нием атомного номера элемента (оксалаты РЗЭ иттриевой
группы заметно растворимы в оксалате аммония). Окса-
латы прокаливают при 900 °C. Весовой формой является
смесь оксидов РЗЭ.
В виде оксалатов осаждается также торий. Однако в
присутствии комплексона III торий остается в растворе
(pH 3,2). К соосаждению с оксалатами РЗЭ склонны
цирконий и скандий, которые также образуют плохо рас-
творимые в воде и кислотах оксалаты. При добавлении
избытка щавелевой кислоты оба элемента образуют хоро-
шо растворимые комплексные соединения и почти пол-
ностью переходят в раствор. При большом избытке по
сравнению с РЗЭ алюминия, железа, ванадия, хрома, мо-
либдена, вольфрама, циркония эти и некоторые другие эле-
менты могут соосаждаться с РЗЭ. Кроме того, они могут
препятствовать полноте осаждения оксалатов РЗЭ в связи
с образованием смешанных комплексных оксалатов, напри-
мер Ln [Ре(С2О4)з], обладающих достаточно высокой ра-
створимостью.
При осаждении оксалатов РЗЭ в растворе должны от-
сутствовать фтор-ионы, особенно при малых количествах
РЗЭ.
197
Для выделения малых количеств РЗЭ в виде оксала-
тов их осаждают совместно с оксалатом кальция СаС2О4-
• Н2О, используя его в качестве коллектора.
Перед осаждением РЗЭ в виде оксалатов предва-
рительно осаждают их в виде гидрокси-
дов. Эта операция позволяет отделить РЗЭ от больших
количеств щелочноземельных металлов, а также некото-
рых металлов в степени окисления +4 при соответствую-
щем регулировании pH. Гидроксиды РЗЭ выделяются в
виде аморфных объемистых осадков при действии раст-
воров аммиака или щелочи. Цвет осадков соответствует
цвету осаждаемого иона.
Осаждение РЗЭ в виде фторидов исполь-
зуется для их отделения от многих элементов. При осаж-
дении РЗЭ из водного раствора их солей действием раст-
вора фтористоводородной кислоты образуется аморфный
слизистый, труднофильтруемый и промываемый осадок.
Фторидный метод, как и оксалатный, позволяет отделить
РЗЭ от железа, алюминия, титана, циркония, урана (VI),
ниобия, тантала и некоторых других элементов. В ходе
анализа обычно отделяют все РЗЭ от сопутствующих
элементов путем осаждения в виде фторидов с последую-
щего их осаждения в виде гидроксидов или оксалатов.
Выделенное суммарное количество РЗЭ анализируют на
содержание отдельных РЗЭ, используя, например, фо-
тометрическое определение церия (IV), спектрофотомет-
рические методы определения неодима, празеодима и т. д.
(по собственному поглощению их солей), а также спект-
ральное определение отдельных РЗЭ в их сумме.
Метод отделения и гравиметрического определения со-
держания церия. Предложенный в 1940 г. Ю. А. Черниховым
и Т. А. Успенской гравиметрический метод определения со-
держания церия в виде йодата еще не потерял своего зна-
чения. Из всех РЗЭ только Се (IV) осаждается из силь-
но азотнокислых растворов йодатом калия. Образую-
щийся нерастворимый осадок 2Се(Ю3)4-К1О3-8Н2О после
промывания спиртом или эфиром можно высушить и
взвесить. Для определения содержания церия в осадке
можно использовать его йодометрическое титрование.
Методы разделения РЗЭ. В современной аналитической
химии методы фракционного осаждения часто заменяют
методами ионного обмена и экстракции.
Ионообменная хроматография является эф-
фективным методом разделения РЗЭ, основанным на том,
что по мере уменьшения ионного радиуса РЗЭ (понижения
198
основности) прочность их комплексов с комплексообразую-
щими веществами увеличивается. Если медленно пропу-
скать раствор, содержащий ионы РЗЭ, через колонку с
катионообменной смолой, то ионы тяжелых элементов бу-
дут проходить через нее первыми. Разделение улучша-
ется при использовании комплексообразующих веществ,
например комплексона III. РЗЭ вводят в колонку в виде
раствора хлоридов. Ионы более тяжелых элементов сла-
бее удерживаются смолой, но дают более прочные комп-
лексы с комплексоном III и поэтому легче переходят в
водную фазу.
Смолу применяют в форме Си2+, десорбцию ведут при
pH 7,5—8.
РЗЭ можно отделить от других элементов экстрак-
цией трибутилфосфатом, или ТБФ—(С4Н9О)3РО, из
азотнокислых растворов, который образует с нитратами
РЗЭ комплексы состава £«(МО3)3-ЗТБФ.
Практическая часть
Комплексонометрическое определение
суммарного содержания РЗЭ
& технической смеси хлоридов РЗЭ
Принцип метода. Определение основано на комплексо-
нометрическом титровании суммарного количества РЗЭ в
присутствии индикатора — смеси ализарина S с метилено-
вым синим или ксиленолового оранжевого.
Метод рассчитан на определение содержания —35%
РЗМ (в пересчете на металл). Относительное стандартное
•отклонение результатов определений —0,01.
Реактивы и растворы. 1) Комплексон III, 0,05 М. раствор (титр
этого раствора устанавливают по 0,05 М. раствору хлорида цинка с
индикатором эриохромчерным Т при pH 9); 2) хлорид цинка, 0,05 М.
^раствор (для его получения 1,6345 г гранулированного цинка марки
ч.д.а. растворяют в 10—15 мл разбавленной 1:1 соляной кислоты; ра-
створ переводят в мерную колбу вместимостью 500 мл и разбавляют
•водой до метки); 3) кислота аскорбиновая, 1%-ный раствор; 4) бу-
ферный раствор, pH 9 (для его получения 10 мл 20%-ного раствора
хлорида аммония смешивают с 10 мл концентрированного раствора
•аммиака и разбавляют водой до 100 мл); 5) буферный раствор,
pH» 4,5 (для его получения 7,7 г ацетата аммония растворяют в
100 мл 6%-ного (по объему) раствора уксусной кислоты; значение
pH контролируют с помощью рН-метра); 6) кислота соляная (р =
= 1,19 г/см3), разбавленная 1:1; 7) аммиак водный, 25%-ный раствор
и разбавленный 1:1; 8) смешанный индикатор (для его получения к
-45 мл 0,5%-ного водного раствора ализарина S прибавляют 15 мл
спиртового раствора метиленового синего); 9) ксиленоловый оранже-
199
вый, 1%-ный раствор; 10) эриохром черный Т (для его получения
0,12 г индикатора смешивают с 25 г хлорида натрия; смесь тщательно
растирают в ступке).
Выполнение определения. Навеску 2,5 г хлоридов РЗЭ
помещают в стакан вместимостью 200 мл, прибавляют
10 мл разбавленной 1:1 соляной кислоты и нагревают в
течение нескольких минут на песчаной бане. Приливают
— 50 мл воды и продолжают нагревание до растворения
навески. Раствор охлаждают, переводят в мерную колбу
вместимостью 100 мл и разбавляют водой до метки. От-
бирают пипеткой 10 мл раствора и переводят в кониче-
скую колбу вместимостью 500 мл. Приливают в нее
— 150 мл воды, Гмл аскорбиновой кислоты, нейтрализуют
аммиаком до pH —4 (по универсальному индикатору).
Затем прибавляют к раствору 25 мл буферного раствора
(pH —4,5) и 15 капель индикатора (смешанного или кси-
ленолового оранжевого). Раствор нагревают до кипения и
титруют раствором комплексона III до перехода окраски
раствора из малиново-красной в зеленую (смешанный ин-
дикатор) или из малиновой в желтую (ксиленоловый оран-
жевый). При этом 1 мл 0,05 М раствора комплексона III
соответствует 0,00703 г РЗМ в пересчете на мишметалл
или 0,00823 г в пересчете на оксид РЗЭ.
Гравиметрическое определение
суммарного содержания РЗЭ
в технической смеси их хлоридов
Принцип метода. РЗЭ осаждают в виде гидроксидов,
а затем производят осаждение оксалатов РЗЭ, которые
прокаливают и взвешивают в виде оксидов. При содер-
жании в смеси —35% РЗЭ относительное стандартное от-
клонение результатов определения —0,01.
Реактивы и растворы. 1) кислота соляная, разбавленная 1:1;
2) кислота щавелевая, насыщенный и 1%-ный растворы; 3) аммиак
водный, 25%-ный и разбавленный 1:1.
Выполнение определения. Навеску пробы 0,5 г поме-
щают в стакан вместимостью 100 мл, прибавляют несколь-
ко миллилитров воды и 1—2 мл соляной кислоты. Смесь
нагревают до растворения пробы. Полученный раствор
разбавляют водой до 50—70 мл и в случае помутнения
фильтруют. К нагретому почти до кипения раствору при-
бавляют до слабого запаха раствор аммиака, содержи-
мое стакана перемешивают, выдерживают —5 мин и
фильтруют через фильтр средней плотности. Осадок на
200
фильтре растворяют в 10 мл горячей соляной кислоты,
собирая фильтрат в тот же стакан, в котором велось
осаждение гидроксидов. Раствор разбавляют водой до
100 мл, нагревают до кипения, приливают равный объем
горячего насыщенного раствора щавелевой кислоты, кипя-
тят 4—5 мин и оставляют стоять на теплой плите в тече-
ние 2 ч или на ночь. Затем осадок отфильтровывают, про-
мывают холодным 1%-ным раствором щавелевой кислоты.
Фильтр с осадком помещают во взвешенный фарфоро-
вый тигель, высушивают и прокаливают до постоянной
массы при 800—900 °C.
Фотометрическое определение содержания РЗЭ
в рудах (монаците, цирконе, паризите, ксенотиме, лопарите)
Принцип метода. Определение основано на образовании
комплексного соединения РЗЭ с арсеназо III. РЗЭ пред-
варительно выделяют в виде оксалатов или гидратов.
Методика рассчитана на определение содержания РЗЭ
в рудах от 0,01 % при навеске 0,1 г. Относительное стан-
дартное отклонение результатов определений 0,2—0,1 при
содержаниях РЗЭ от 0,02—1%.
Реактивы и растворы. 1) Кислота фтористоводородная, р=
= 1,13 г/см3; 2) кислота серная (р=1,84 г/см3), разбавленная 1:1;
3) пероксид водорода, 30%-иый раствор; 4) кислота соляная, р =
= 1,19 г/см3, а также 6; 0,2 и 0,01 М растворы; 5) хлорид кальция,
2,5%-ный раствор; 6) кислота щавелевая кристаллическая, 1%-ный
раствор; 7) аммиак, 25%-ный водный раствор; 8) буферный раствор,
pH 1,8 (для его приготовления 83 мл 0,2 М соляной кислоты смеши-
вают в мерной колбе вместимостью 1 л с 250 мл 0,2 М раствора
хлорида калия и доводят объем до метки водой); 9) арсеназо III,
0,1%-ный раствор (навеску 0,1 г реагента растворяют в воде, добав-
ляют несколько капель 10%-ного раствора карбоната натрия и на-
гревают при 50—60°C (синяя окраска); после растворения реагента
раствор подкисляют 6 М соляной кислотой до перехода окраски из
синей в фиолетово-красную, охлаждают и доливают до метки водой в
колбе вместимостью 100 мл); 10) запасной раствор РЗЭ, содержащий
в 1 мл 0,5 мг Ра20з (в зависимости от состава руд готовят различ-
ные растворы РЗЭ; для руд, содержащих минералы цериевой группы,
50 мг оксида лантана растворяют в 10 мл разбавленной 1:1 соляной
кислоты при нагревании и доводят объем до метки водой в колбе
вместимостью 100 мл); 11) рабочий раствор, содержащий в 1 мл
100 мкг La2O3; получают разбавлением запасного раствора в 50 раз.
Для руд, содержащих минералы цериевой и иттриевой подгрупп,
запасной раствор РЗЭ готовят растворением смеси оксидов лантана
и иттрия в соляной кислоте. Смесь составляют в таком соотношении,
которое установлено для руд данного месторождения. При анализе
любых руд для приготовления стандартного раствора РЗЭ можно
исходить из суммы оксидов РЗЭ, выделенных оксалатным методом
из руды, которая анализируется.
201
Построение градуировочного графика. В мерные колбы
вместимостью по 50 мл вводят от 5 до 30 мкг РЗЭ (с ин-
тервалом 5 мкг) или в виде раствора хлорида лантана
для руд, содержащих минералы цериевой подгруппы, или
в виде смеси растворов лантана и иттрия для руд, содер-
жащих минералы цериевой и иттриевой подгрупп, или
раствора суммы РЗЭ. В колбы добавляют по 25—30 мл
0,01 М. раствора соляной кислоты, 5 мл буферного раст-
вора, 2 мл арсеназо III. Объем растворов доводят до
метки 0,01 М раствором соляной кислоты и тщательно пе-
ремешивают. Оптическую плотность растворов измеряют
на фотоэлектроколориметре или спектрофотометре при
Х = 650 нм в кюветах с /=30 мм. В качестве раствора срав-
нения используют раствор контрольного опыта, содержа-
щего все реактивы. По полученным данным строят график
зависимости оптической плотности растворов от концент-
рации.
Выполнение определения. Навеску руды 0,1—0,2 г раз-
лагают при нагревании в платиновой чашке в 10—15 мл
фтористоводородной кислоты, приливают 5 мл серной кис-
лоты (1:1) и упаривают раствор до состояния влажных
солей. Сульфаты растворяют при охлаждении в 80—
100 мл воды с добавлением двух-трех капель пероксида
водорода. Затем к раствору добавляют 10 мл 0,01 М. со-
ляной кислоты и кипятят до его просветления. В раствор
вводят 1 мл раствора хлорида кальция, нагревают до ки-
пения, добавляют 1,5 г щавелевой кислоты, а затем амми-
ак до рН~5 (контроль по универсальной индикаторной
бумаге) и выдерживают в течение 2 ч или оставляют на
ночь. При осаждении руд с высоким или неизвестным со-
держанием кальция перед осаждением оксалатов произ-
водят осаждение гидроксидов аммиаком, не содержащим
СО2 по фенолфталеину, причем аммиак берут с избытком
в 10 мл. Осадок оксалатов отфильтровывают и промывают
три раза 1%-ным раствором щавелевой кислоты. Фильтр
с осадком высушивают в фарфоровом тигле и прокали-
вают при 600 °C до разложения оксалатов.
Прокаленный осадок растворяют в малом объеме кон-
центрированной соляной кислоты с добавлением одной-
двух капель пероксида водорода: раствор упаривают досу-
ха. Остаток растворяют в 0,01 М. растворе соляной кис-
лоты, полученный раствор переводят в мерную колбу
вместимостью 50—100 мл и доводят его объем до метки
той же кислотой.
Для определения содержания РЗЭ отбирают аликвот-
202
ную часть раствора, содержащую ориентировочно 5—
30 мкг РЗЭ. Раствор переводят в мерную колбу вмести-
мостью 50 мл, добавляют 5 мл буферного раствора и
2 мл раствора арсеназо III. Объем раствора в колбе доводят
до метки 0,01 М раствором соляной кислоты и тщательно
перемешивают. Измеряют оптическую плотность раство-
ров при 2^ = 650 нм в кювете с 7 = 30 мм по отношению к
раствору контрольного опыта, содержащему все реакти-
вы. Содержание РЗЭ находят, используя градуировочный
график.
При анализе лопаритовых руд после разложения на-
вески 0,1 г как указано в п. «Выполнение определения»,
осаждение гидроксидов ведут аммиаком. Осадок раство-
ряют в соляной кислоте и содержание РЗЭ определяют по
реакции с арсеназо III.
Дифференциальное фотометрическое определение неодима
в сплавах магний — неодим и в оксиде неодима
Принцип метода. Определение основано на измерении
светопоглощения азотнокислых растворов неодима при
Х = 575 нм по сравнению с раствором, содержащим извест-
ное количество нитрата неодима. Метод рассчитан на
определение содержания неодима от 40 до 45%. Магний
определению не мешает. Относительное стандартное от-
клонение результатов определений 0,01.
Реактивы, растворы и аппаратура. 1) Кислота азотная (р =
= 1,40 г/см3), разбавленная 1:1; 2) рабочий раствор неодима, содер-
жащий в 1 мл 50 мг NdsOj (для его получения 5,00 г прокаленного
при 800 °C оксида неодима растворяют в азотной кислоте при на-
гревании; раствор охлаждают, переводят в мерную колбу вмести-
мостью 100 мл и доводят объем до метки азотной кислотой); 3) спек-
трофотометры СФ-26 или аналогичные приборы; 4) фотоэлектроколо-
риметр ФЭК-56 (ртутная лампа, светофильтр № 7).
Примечание. Стандартные растворы нитрата неодима устой-
чивы длительное время. В связи с этим раствор сравнения, содер-
жащий в 25 мл 150 мг Nd2O3, а также растворы, содержащие в
25 мл 200—250 мг Nd2O3, можно использовать для целого ряда изме-
рений. Чтобы концентрация растворов сохранялась постоянной, не-
обходимо пользоваться сухими кюветами.
Построение градуировочного графика. В мерные колбы
вместимостью по 25 мл вводят из микробюретки 3,0; 3,5;
4,0; 4,5; 5,0 мл стандартного рабочего раствора неодима,
разбавляют водой до метки и перемешивают. Оптическую
плотность растворов измеряют при 2^=575 нм в кюветах с
/ = 50 мм. Пользуясь спектрофотометром СФ-26, необхо-
димо работать со светофильтром ОС-14 для уменьшения
203
влияния рассеянного света. В качестве раствора сравне-
ния используют раствор, содержащий в 50 мл 150 мг
Nd2O3. По результатам измерений строят график зависи-
мости оптической плотности раствора от концентрации
или вычисляют значение постоянного фактора.
Выполнение определения. Навеску сплава 2,500 г раст-
воряют при нагревании в 20 мл азотной кислоты, охлаж-
дают, переводят раствор в мерную колбу вместимостью
50 мл и разбавляют водой до метки. В мерную колбу
вместимостью 25 мл вводят 10 мл раствора пробы, раз-
бавляют водой до метки, перемешивают и измеряют оп-
тическую плотность раствора, как указано при построе-
нии градуировочного графика.
При анализе образца оксида неодима 0,200—0,250 г
пробы растворяют при нагревании в 5—6 мл азотной
кислоты (1:1) и далее продолжают анализ, как указано
при построении градуировочного графика.
Фотометрическое определение неодима
в сплавах магний—неодим (абсолютный метод)
Принцип метода. Определение основано на измерении
светопоглощения азотнокислых растворов неодима при
% = 575 нм. Магний определению не мешает. Метод рассчи-
тан на определение содержания неодима в сплаве до
15%. Относительное стандартное отклонение результатов
определений 0,05.
Реактивы, растворы и аппаратура. Те же, что при дифференциаль-
ном фотометрическом определении содержания неодима в сплавах
магний — неодим и в оксиде неодима.
Построение градуировочного графика проводят для со-
держания 10—100 мг Nd2O3, сравнивая оптическую плот-
ность растворов и воды.
Выполнение определения. Берут аликвотную часть рас-
твора, соответствующего 10—100 мг Nd2O3, и измеряют
оптические плотности по отношению к воде.
Т итриметрическое
определение церия в мишметалле
Принцип метода. Определение основано на титровании
(восстановлении) церия (IV) раствором соли Мора в при-
сутствии ферроина или фенилантраниловой кислоты в ка-
честве индикатора. Церий (III) предварительно окисляют
до церия (IV) персульфатом аммония. Метод рассчитан
на содержания церия в мишметалле от 40 до 75%. Отно-
204
сительное стандартное отклонение результатов определе-
ний 0,02.
Реактивы и растворы. 1) Кислота серная, р = 1,84 г/см3 и 2,5 М
раствор; 2) персульфат аммония, 25%-ный раствор (свежеприготов-
ленный); 3) сульфат железа (II)—аммония (соль Мора), 0,017 М
раствор (для его приготовления 10 г соли растворяют в 200—300 мл
воды, к раствору добавляют 50 мл концентрированной серной кисло-
ты; полученный раствор переводят в мерную колбу вместимостью
1 л, доводят его объем до метки водой и перемешивают; титр раст-
вора соли Мора устанавливают по раствору сульфата церия, концент-
рация которого предварительно определена гравиметрическим оксалат-
ным методом, после окисления церия (III) до церия (IV) персульфатом
аммония в условиях, описанных ниже в этой методике); 4) ортофе-
нантролин; 5) индикатор (ферроин или фенилантраниловая кислота);
6) ферроин (для приготовления его раствора 1,5 г ортофенантролина
и 0,7 г соли Мора растворяют в 100 мл воды); 7) фенилантраниловая
кислота, 0,2%-ный раствор (для его приготовления 0,2 г фенилантра-
ниловой кислоты растворяют в 100 мл 0,2%-ного горячего раствора
углекислого натрия).
Выполнение определения. Навеску мишметалла 0,1 г в
конической колбе растворяют в 20 мл 4 н. серной кисло-
ты. Раствор разбавляют водой приблизительно до 75 мл,
добавляют 25 мл раствора персульфата аммония. Кис-
лотность полученного раствора должна быть ~1 н. по
H2SO4. Раствор нагревают до кипения, кипятят в течение
20 мин для разложения избытка окислителя, поддерживая
первоначальный объем добавлением воды. Затем раствор
охлаждают до комнатной температуры, добавляют 1 кап-
лю ферроина и титруют раствором соли Мора до перехо-
да окраски в розовую.
При использовании в качестве индикатора фенилантра-
ниловой кислоты охлажденный раствор титруют раство-
ром соли Мора до бледно-желтой окраски, затем при-
бавляют две капли раствора фенилантраниловой кислоты
и продолжают титрование до обесцвечивания раствора.
При расчете исходят из того, что 1 мл 0,025 н. раство-
ра соли Мора соответствует 4,303 мг СеО2.
Скандий
Скандий открыт шведским ученым Л. Нильсоном в 1879 г. в про-
цессе разделения элементов эрбиевой подгруппы, полученной из мине-
рала гадолинита со Скандинавского полуострова, по имени которого
и назван. Скандий предсказан Д. И. Менделеевым в 1871 г.
Нахождение в природе. Содержание скандия в земной коре оце-
нивается равным 0,0006%. В природе скандий рассеян и встречается
лишь в виде незначительной примеси в минералах редкоземельных эле-
ментов, бериллия, тантала, ниобия, олова, вольфрама, циркония, ти-
тана, алюминия, а также в золах углей, природных водах и окамене-
лых остатках рыб. Для получения 1 г оксида скандия нужно перера-
ботать 3—4 кг гадолинита.
205
Собственные минералы скандия: тортвейтит Sc2(Si2O7), бефанамит
(Sc, Zr)2 [Si2O7], стереттит Sc(PO4)-2H2O встречаются очень редко.
Сырьем для добывания скандия служат отходы производства тех
металлов, которым скандий сопутствует в их минералах.
Применение. Практическое применение скандия пока ограничено;
его используют для изготовления ферритов; искусственный радио-
активный изотоп 4eSc широко применяется в химических, геологиче-
ских и металлургических исследованиях.
Свойства скандия и его соединений. С к а н д и й— эле-
мент III группы Периодической системы Д. И. Менделе-
ева. Электронное строение атома в основном состоянии —
ls22s22p63s23p63d'4s2. Скандий обладает устойчивой сте-
пенью окисления +3, известна неустойчивая степень
окисления +1.
В свободном состоянии скандий — металл с серебрис-
тым блеском на свежем срезе, быстро тускнеющий на
воздухе, не реагирующий с водой. Металл обладает не-
большой плотностью и высокой температурой плавления
(1539 °C).
Металлический скандий получают кальций-термическим
восстановлением хлорида или фторида скандия.
Оксид скандия Sc2O3 получают прокаливанием
гидроксида или солей скандия. Гидроксид скандия Sc (ОН) 3
образуется при взаимодействии растворов соли скандия и
щелочи или аммиака. В водных растворах соединения скан-
дия гидролизуются и склонны к образованию основных
солей. Ионы Sc3+ обладают высокой комплексообразую-
щей способностью и легко полимеризуются. Металлический
скандий, его оксид и гидроксид растворяются в разбавлен-
ных кислотах.
Методы определения содержания скандия
Основными методами количественного определения
скандия являются .спектральный, комплексонометриче-
ский, фотометрический. Эмиссионный пламенно-фотометри-
ческий и атомно-абсорбционный методы обладают в отно-
шении скандия низким пределом обнаружения. Ввиду
разнообразия скандийсодержащих объектов и недоста-
точной избирательности органических реагентов, предло-
женных для определения скандия, применению фотомет-
рических методов предшествует отделение скандия от
сопутствующих элементов. Практически часто при анали-
зе технических и природных материалов применяется до-
вольно специфичное осаждение скандия тартратом аммо-
206
ния в виде скандий-аммоний-тартрата или же в виде
скандий-иттрий-аммоний-тартрата.
Комплексонометрический метод определения содержа-
ния скандия основан на том, что скандий образует проч-
ное комплексное соединение состава 1:1 с динатриевой
солью этилендиаминтетрауксусной кислоты (комплексо-
ном III) в интервале pH 2,5—9,5. Константа нестойкости
этого соединения составляет величину 1,56-10-22. Прямое
или обратное титрование скандия проводят в присутствии
различных металлиндикаторов. Индикаторами, обладаю-
щими наибольшей избирательностью и четкостью перехо-
да, признаны ксиленоловый оранжевый и мурексид.
Комплексонометрическое определение содержания скан-
дия с ксиленоловым оранжевым применяют при анализе
600 ПВО 560 А,.нм
Рис. 23
Рис. 23. Кривые светопоглощения ксиленолового оранжевого (/) и его соединен
ння со скандием (2)
Рис. 24. Кривые светопоглощения растворов ДОТРИХАФ (/) и его соединения
со скандием (2) в этилацетате
антимонида и арсенида скандия, силикатов и шлаков, а
также стекол, содержащих скандий.
Из фотометрических методов определения содержания
скандия широкое распространение получил метод опреде-
ления с ксиленоловым оранжевым. Скандий образует
прочное комплексное соединение состава 1:1 при pH 1,5—
5,0. Нижний предел определения равен 0,1 мкг/мл; не-
большие количества редкоземельных элементов определе-
нию не мешают; ионы железа (III) и церия (IV) восста-
навливают аскорбиновой кислотой. Мешают определению
скандия торий, галлий, индий, цирконий. Кривые свето-
поглощения растворов ксиленолового оранжевого и его
соединения со скандием показаны на рис. 23. С помощью
ксиленолового оранжевого скандий определяют в металли-
ческом магнии и его сплавах, в медных сплавах, в воль-
фрамите.
207
Чувствительным и более специфичным является пред-
.ложенный советскими учеными В. М. Островской и
В. М. Дзиомко реагент ДОТРИХАФ (1,5-ди(2/-окси-4/-5/-
-б'-трихлорфенил) -3-ацетилформазан)
I
с=о
I
сн3
Реагент образует со скандием комплексное соедине-
ние состава 2:1 зеленого цвета, экстрагируемое из раст-
воров с pH 4,5—5,5 в органические кислородсодержащие,
растворители, например в этилацетат. Кривые светопогло-
щения ДОТРИХАФ и его комплекса со скандием приведе-
ны на рис. 24. С данным реагентом содержание скандия
определяют в технических образцах на основе циркония,
титана, алюминия. Основные компоненты образцов пред-
варительно удаляют купфероновой или трибутилфосфат-
ной экстракцией. В настоящее время ДОТРИХАФ явля-
ется единственным реагентом, позволяющим отличить
скандий от других элементов по образованию комплексно-
го соединения характерного зеленого цвета.
Скандий легко извлекается из шлаков и некоторых
минералов при кипячении с концентрированной соляной
кислотой и его можно быстро обнаружить в солянокислой
вытяжке в присутствии всех редкоземельных элементов.
Гравиметрические методы определения скандия основа-
ны на осаждении двойного тартрата аммония-скандия
NH4OOC— (СНОН)2—COOSc(OH)2, переходящего в оксид
скандия при 850—900 °C. Осаждение проводят из прозрач-
ного раствора соли скандия, содержащего избыток тар-
трата аммония, при прибавлении аммиака в интервале
pH 6,8—11,0. Практически удобно кислый раствор соли
скандия нейтрализовать до перехода окраски нейтрально-
го красного и затем добавлять такое количество-раствора
аммиака, чтобы его избыток соответствовал 0,1 М при
осаждении скандия без носителя или 0,5 М в случае при-
менения иттрия в качестве носителя. Концентрация тарт-
рата аммония поддерживается в пределах 5—20%. Полно-
208
му осаждению тартратов иттрия и в несколько меньшей
степени скандия препятствуют большие количества же-
леза, алюминия, титана, циркония, гафния, ниобия. По
этой причине эти элементы предварительно должны быть
удалены.
Практическая часть
Качественное обнаружение скандия
в растворах солей
Принцип метода. Метод основан на образовании в сла-
бокислой среде характерно окрашенного комплексного
соединения с реагентом из класса формазанов ДОТРИХАФ.
Реактивы и растворы. 1) Буферный ацетатный раствор, pH 4,9;
2) ДОТРИХАФ, 0,005%-ный раствор в этилацетате; 3) солянокислый
раствор скандия; 4) кислота уксусная, р = 1,05 г/см3; 5) соляная кисло-
та 12 и 2 М растворы; 6) ацетат натрия; 7) спирт этиловый (гидро-
лизный) ректификат; 8) этилацетат.
Выполнение определения. Каплю 0,05—0,001 М раствора
соли скандия помещают в пробирку с притертой пробкой,
добавляют 2 мл ацетатного буферного раствора, 1 мл
этилового спирта, 2 мл раствора ДОТРИХАФ. Раствор
встряхивают 3 мин; после расслаивания фаз появляется
зеленая окраска органической фазы. Обнаруживаемый
минимум равен 2 мкг Sc, предельное разбавление состав-
ляет 1:6 000 000.
Фотометрическое определение скандия
в сплавах магния
Принцип метода. Метод основан на образовании окра-
шенного в красный цвет комплексного соединения скандия
с ксиленоловым оранжевым. Относительное стандартное
отклонение результатов определения 0,05 при содержа-
ниях — 0,1% скандия.
Реактивы и растворы. 1) Бумага конго; 2) буферный раствор,
pH 2 (для его получения смешивают 263 мл 0,2 М раствора соляной
кислоты и 500 мл 0,2 М раствора хлорида калия); 3) кислота соляная,
р=1,19 г/см3, разбавленная 1:1 и 0,2 М раствор; 4) кислота аскор-
биновая, 2%-ный раствор; 5) хлорид калия, 0,2 М раствор; 6) ксиле-
ноловый оранжевый, 0,1%-ный раствор; 7) ацетат натрия, 50%-ный ра-
створ; 8) запасной раствор скандия, содержащий в 1 мл 1 мг
скандия (для его получения 0,153 г оксида скандия растворяют при
нагревании в 5 мл 6 М раствора азотной кислоты, охлаждают и до-
водят водой до 100 мл); 9) рабочий раствор скандия, в 1 мл кото-
рого содержится 0,01 мг скандия (готовят разбавлением запасного
раствора в 100 раз).
8. 129.
209
Построение градуировочного графика. В мерные колбы
вместимостью по 100 мл вводят 10—20 мл соляной кисло-
ты 1:1, стандартный раствор скандия, 5 мл раствора ас-
корбиновой кислоты, по каплям раствор ацетата натрия
до перехода индикаторной бумаги конго в сиреневый цвет,
5 мл буферного раствора, 5 мл раствора ксиленолового
оранжевого и доводят объем до метки водой. Через 20 мин
измеряют оптическую плотность раствора на фотоэлектро-
колориметре ФЭК-М с зеленым светофильтром в кювете
с Z=30 мм.
Выполнение определения. Навеску сплава 0,1—0,2 г
растворяют в 10—20 мл соляной кислоты (1:1) и перено-
сят раствор в мерную колбу вместимостью 100 мл. При-
бавляют в колбу 5 мл раствора аскорбиновой кислоты,
далее поступают, как описано при построении градуиро-
вочного графика. Содержание скандия определяют, исполь-
зуя градуировочный график.
Комплексонометрическое
определение скандия в стекле
Принцип метода. Титрование скандия комплексоном III
проводят при pH 2—3 в присутствии ксиленолового оран-
жевого в качестве индикатора. Относительное стандартное
отклонение результатов определения 0,01 при содержаниях
~ 1 % скандия.
Реактивы и растворы. 1) Аммиак, р = 0,91 г/см3; 2) кислота фто-
ристоводородная, р=1,15 г/см3; 3) кислота серная, р=1,84 г/см3;
4) кислота соляная, р=1,19 г/см3 и разбавленная 1:4; 5) комплексон
III, 0,05 М раствор.
Выполнение определения. Для определения содержания
скандия в стекле тонкоизмельченную пробу растворяют в
смеси H2SO2 + HF при нагревании и раствор упаривают.
Остаток растворяют в 25 мл соляной кислоты (1:4), пере-
водят в мерную колбу и доливают до 25 мл водой. К али-
квотной части добавляют 8—10 капель водного раствора
ксиленолового оранжевого, прибавляют по каплям 25%-
ный раствор аммиака до перехода желтой окраски раство-
ра в оранжевую устанавливают pH 2—3 (малиновая ок-
раска) и титруют 0,05 М раствором комплексона III до
перехода малиновой окраски раствора в лимонно-желтую.
При этом 1 мл точно 0,05 М раствора комплексона соот-
ветствует 2,248 мг скандия.
210
Фотометрическое определение скандия
в силикатных породах
Принцип метода. Метод основан на осаждении скан-
дия в виде тартрата скандия — иттрия — аммония и по-
следующем прокаливании осадка до оксидов; скандий
определяют фотометрически с ДОТРИХАФ. Относитель-
ное стандартное отклонение результатов определения 0,1
при содержаниях 0,01% скандия.
Реактивы и растворы. 1) Тартрат аммония, 40 и 3,5%-ные раст-
воры; 2) аммиак, р = 0,91 г/см3; 3) раствор оксида иттрия (для его
получения 1 г оксида растворяют в 10 мл соляной кислоты и раз-
бавляют до 200 мл водой); 4) кислота соляная, р= 1,19 г/см3 и раз-
бавленная 1:4; 5) кислота винная, 40%-ный раствор; 6) нейтральный
красный, 0,1%-ный раствор.
Выполнение определения. К раствору, упаренному до
объема 5—10 мл, после отделения железа и алюминия,
добавляют 50 мл воды, 5 мл раствора оксида иттрия, что
соответствует 25 мг У20з. Раствор осторожно нейтрализуют
аммиаком до появления мути; ее растворяют, прибавляя
по каплям разбавленную 1:1 соляную кислоту. К полу-
ченному раствору добавляют 50 мл 40%-ного раствора
тартрата аммония или винной кислоты и нагревают раст-
вор до кипения, после чего прибавляют несколько капель
раствора нейтрального красного и аммиака (при помеши-
вании) до резкого перехода окраски индикатора. Затем
к раствору прибавляют еще 1,5 мл аммиака и помещают
его на 30 мин в водяную баню, после чего нагревание
прекращают.
Если при кипячении или нагревании в водяной бане
выпавший вначале осадок тартратов растворяется, то к
раствору добавляют еще 0,5—1,0 мл аммиака. Спустя 12—
18 ч выпавший осадок тартратов переносят на беззольный
фильтр и промывают 100 мл 3,5%-ного раствора тартрата
аммония, к которому добавлено 3 мл аммиака. Осадок
вместе с фильтром осторожно озоляют в платиновом тигле
и прокаливают до образования оксидов. Скандий опреде-
ляют с ДОТРИХАФ (см. с. 209).
Галлий, индий, таллий
Существование галлия (экаалюминия) было предсказано в 1870 г.
Д. И. Менделеевым. Элемент в 1875 г. открыл французский химик
Лекок де Буабодран. Галлий получил свой символ в честь латинского
названия Франции (Gallium).
Индий открыт в 1863 г. немецкими химиками Ф. Рейхом и Т. Рих-
тером, обнаружившими при исследовании цинковой обманки новые
синие спектральные линии (цвета индиго).
8*
211
Таллий открыт в 1861 г. английским ученым У. Круксом при спе-
ктроскопическом исследовании шламов сернокислотного производства.
Название элемент получил по характерной зеленой линии спектра
Х = 535,046 нм (от латинского слова thallus — зеленая ветка).
Нахождение в природе. Галлий, индий, таллий относятся к числу
рассеянных элементов. Содержание их в земной коре невелико:
Ga 1,5-10-3%, In 1 -10—5% и Т1 3-1О-«/о.
Галлий накапливается в виде изоморфной примеси в сфалеритах.
Попутно с алюминием его извлекают из бокситов, нефелинов. Един-
ственный минерал галлия — галдит CuGaS2 встречается очень редко,,
богатым источником галлия является германит Cu3(FeGe)S4-
Минералы индия чрезвычайно редки; он содержится в виде изо-
морфной примеси в минералах цинка, свинца, олова. Таллий содержит-
ся в ничтожно малых количествах в различных горных породах, почве.
Собственные минералы таллия являются, по выражению В. И. Вернад-
ского, большой минералогической редкостью и не имеют практического-
значения. Таллий встречается главным образом, в минералах вместе-
со свинцом, цинком, медью и др.
Применение. Из рассеянных редких металлов меньше всего ис-
пользуется галлий. Вследствие низкой температуры плавления (29,8 °C)-
и высокой температуры кипения (2230 °C) металл предложено исполь-
зовать для изготовления высокотемпературных термометров. Легко-
плавкие (<60°С) сплавы галлия с рядом металлов (висмутом, кад-
мием, свинцом, цинком, индием, таллием) могут быть использованы
в сигнальных устройствах. В последнее время галлий находит при-
менение для получения полупроводниковых соединений — арсенида,
фосфида, антимонида галлия. Таллиевые оптические стекла характе-
ризуются высокой отражательной способностью. Сплавы, содержащие
галлий, предложено применять в зубоврачебной практике.
Индий находит более широкое, чем галлий, практическое приме-
нение. Легкоплавкие сплавы индия (In—Sn—Cd—Bi, In—Pb—Sn,
In—Pb) употребляют в качестве припоев для соединения металлов,
стекла, керамики, а также в системе пожарной сигнализации. Индий
широко используется в полупроводниковой технике для изготовления
германиевых выпрямителей. Большое применение индий находит также
для антикоррозионных покрытий (вместо серебряных в рефлекторах
и т. д.).
Таллий и его соединения имеют разнообразное применение: в ма-
териалах для инфракрасной оптики, в производстве селеновых выпря-
мителей, для изготовления антикоррозионных подшипниковых спла-
вов, в люминесцентных лампах. Токсичный сульфат таллия T12SCU
применяется в сельском хозяйстве для борьбы с грызунами. Моно-
кристаллы твердых растворов бромида и иодида таллия применяют в
оптических приборах, работающих в инфракрасной области спектра.
Некоторые соединения таллия используются для изготовления опти-
ческих стекол с высокой преломляющей способностью. Амальгамы тал-
лия, затвердевающие при 60 °C, применяют для измерения низких
температур.
Свойства элементов и их соединений. Галлий, индий,
таллий, как и лантаниды, относятся к III группе Периоди-
ческой системы Д. И. Менделеева. Они находятся в глав-
ной подгруппе и являются аналогами алюминия. В основ-
ном электронном состоянии атомы этих элементов имеют
строение внешних электронных оболочек s2pl. Три электро-
212
на определяют их степень окисления +3; один неспарен-
ный электрон обусловливает степень окисления +1. При
переходе от галлия к таллию устойчивость элементов в
степени окисления +3 уменьшается, а в степени окисле-
ния + 1—увеличивается. Галлий в растворах находится
всегда в степени окисления +3, таллий — в основном в
+ 1. Кроме того, для таллия характерны соединения, в ко-
торых он находится одновременно в степени окисления
+ 2 и +3.
Таллий (I) сходен со щелочными металлами: образует
хорошо растворимые в воде гидроксиды, растворимые нит-
раты, карбонаты. В отличие от щелочных металлов тал-
лий (I) образует плохо растворимые в воде галогениды
(растворим лишь T1F), сульфид T12S, хромат Т12СгО<.
Галлий — серебристо-белый металл сравнительно
МЯГКИИ И ХрупКИЙ /пл = 29,8 °C, £кип=2230°С. Галлий, так же
как и алюминий, проявляет амфотерные свойства; медлен-
но растворяется в азотной, быстрее — в серной, соляной и
хлорной кислотах при нагревании с образованием ионов
галлия (III). Галлий хорошо растворяется в царской вод-
ке; медленно реагирует с раствором NaOH с образованием
галлата натрия Na[Ga(OH)4].
Гидроксид галлия Ga(OH)3 осаждается в слабокислом
растворе (pH 3—4) и растворяется в слабощелочной среде
(pH 8—9) с образованием галлатов. Гидроксид галлия
растворим также в кислотах. В отличие от А1(ОН)з гид-
роксид галлия растворим в растворах аммиака; при ки-
пячении вновь выпадает гидроксид галлия. Кислотные
свойства у Ga(OH)3 выражены сильнее, чем основные.
Индий — серебристо-белый металл, мягкий и ковкий;
/пл = 157 °C, /кип=2020 °C. Образует соединения в степени
окисления +1, +2, +3, из которых последние наиболее
устойчивы. Индий растворяется в азотной, серной и со-
ляной кислотах. Гидроксид индия 1п(ОН)з осаждается при
pH 3—4, обладает слабыми амфотерными свойствами. Кис-
лотные свойства 1п(ОН)з выражены слабее, чем Ga(OH)3.
В растворах едкого натра (рН~14) 1п(ОН)3 растворяется
незначительно, образуя индат натрия Na3 [In(ОН)6]-2Н2О.
Это соединение неустойчиво и разлагается при нагрева-
нии или длительном стоянии. В растворах аммиака гид-
роксид индия нерастворим (в отличие от гидроксида гал-
лия), но хорошо растворим в кислотах.
Таллий — серебристо-белый металл с сероватым
оттенком, легко режется ножом; /лл = 304°С, /ОТШ=1475°С.
Металлический таллий хорошо растворим при нагревании
213
в азотной, серной и хлорной кислотах с образованием со-
лей Т1(1). В соляной кислоте таллий растворяется с тру-
дом вследствие образования плохо растворимого хлорида
таллия; со щелочами таллий практически не взаимодейст-
вует. В присутствии кислорода таллий медленно раство-
ряется в воде, образуя гидроксид таллия ТЮН.
В ряду напряжений таллий стоит между индием и ко-
бальтом: £0(т1/т1+)= —0,336 В; £'o(ti3+/ti+) = +1,221 В. Поэто-
му окисление таллия (I) в таллий (III) возможно только
сильными окислителями — хлором, бромом, персульфата-
ми, перманганатом, царской водкой и др. Соединения тал-
лия (III) легко восстанавливаются в кислых растворах
иодидами и другими восстановителями.
Гидроксид таллия ТЮН является растворимым в воде
сильным основанием, которое осаждается щелочью при
pH 1; в щелочах ТЮН нерастворим. Бесцветные ионы
ТР+ существуют только в сильнокислой среде; при pH— 0,3
выпадает коричневый осадок гидроксида Т1(ОН)3, не об-
ладающего амфотерными свойствами.
Окраска оксидов: белая для Ga2O3, желтая для 1п20з
и коричневая для Т12О3. Оксиды нерастворимы в воде, но
растворимы в кислотах. В ряду Ga2O3—In2O3—Т12О3 раст-
воримость увеличивается. Оксид Ga2O3 получают обезво-
живанием его гидроксида. Сильно прокаленный оксид
Ga2O3 плохо растворяется в кислотах и щелочах.
Ионы Ga3+, In3+ бесцветна, а ион Т13+ имеет желтова-
тую окраску. Большинство солей этих ионов хорошо рас-
творимо в воде, но склонно к гидролизу.
Рассматриваемые элементы не обладают большой
склонностью к комплексообразованию. Известными комп-
лексными соединениями галлия, индия и таллия, ус-
тойчивыми в водных растворах, является хлорид-
ные комплексы, комплексы с оксалатами, тартратами:
[Ga(C2O4)3|3-, [Ga (С4Н4О6)3] 3~, а также с комплексо-
ном III.
Экстракция. Хлоридный комплекс галлия экстрагиру-
ется эфирами, кетонами, спиртами из солянокислых раст-
воров. Из 7—8 М раствора соляной кислоты изопропило-
вым эфиром экстрагируется 99% галлия в виде HGaCl4.
Из 6 М раствора соляной кислоты диэтиловый эфир экс-
трагирует лишь следы индия. Индий экстрагируется ди-
этиловым эфиром в виде иодидного комплекса Н1п14-
• пНгО из 0,5—2,5 М иодистоводородной кислоты. В этих
условиях галлий не экстрагируется, а таллий экстраги-
руется. Алюминий и железо (III) не экстрагируются.
214
Индий хорошо экстрагируется в виде бромидного
комплекса, например изопропиловым эфиром из 6 М бро-
мистоводородной кислоты. Отделение индия в виде бро-
мида менее селективно, чем в виде иодида. Вместе с ин-
дием в экстракт переходят галлий (III), железо (III),
таллий (III) и др., цинк остается в водной фазе. (От ме-
таллов, образующих растворимые аммиачные комплексы —
серебра, меди, никеля, кобальта, цинка, кадмия, индий
можно отделить путем осаждения его аммиаком в виде
1п(ОН)з).
Из 1 М бромистоводородной кислоты диэтиловым эфи-
ром количественно извлекают таллий, при этом в незна-
чительных количествах экстрагируются железо (III), гал-
лий, индий. Хлорид таллия (III) экстрагируют эфиром из
2 М раствора соляной кислоты и отделяют от железа (Ш),
галлия (III) и индия (III).
Другие методы отделения. Для отделения галлия от
сопутствующих элементов используют также ионообмен-
ную хроматографию. Аммиачный раствор (pH 9—10), со-
держащий галлий, цинк и винную кислоту, пропускают
через катионит СБС в NH^ -форме. Цинк в виде проч-
ного аммиачного комплекса [Zn(NH3)4]2+ задерживается
катионитом, а галлий в виде тартратного комплекса
[Оа^ДЦОбЦ]3- переходит в раствор.
Ионный обмен используется также для отделения ин-
дия. Из растворов хлоридов (0,1 М НС1) на катионите
Дауэкс-50 в Н+-форме сорбируются индий и галлий. Ин-
дий вымывают из катионита 0,4 М, а галлий—1,3 М
раствором соляной кислоты.
Из растворов, содержащих медь, железо, цинк, таллий
(I), при pH 4 в присутствии комплексона III на колонке
с сильно кислотным катионитом удерживается только
таллий (I). Затем его вымывают 2 М раствором соляной
кислоты.
Разложение минералов, содержащих галлий, индий,
таллий. Галлий, индий и таллий встречаются в основном
в полиметаллических рудах, содержащих цинк, свинец,
олово, а также в бокситах, германитах и т. д. Методы
разложения этих минералов при определении содержания
галлия, индия и таллия в принципе те же, что и при ана-
лизе цветных металлов. Так, цинковые обманки разла-
гают обработкой соляной кислотой при нагревании с по-
следующим добавлением азотной кислоты. Для разложе-
ния оловянных руд используют сплавление с пероксидом
натрия или со смесью NaOH + Na2O2. При анализе бок-
215
ситов пробу разлагают сплавлением с пиросульфатом ка-
лия или другим путем. Подробнее разложение полиметал-
лических руд см. «Методы определения содержания цвет-
ных металлов».
Соединения таллия склонны к летучести; потери его
наблюдаются при прокаливании анализируемых материа-
лов, сплавлении с содой, бурой или едким натром. При
разложении проб кислотами (НС1, HNO3, H2SO4, НСЮ4)
потерь таллия не происходит.
Для разложения сплавов, содержащих галлий, индий,
таллий, применяют растворение в кислотах: HC1 + HNO3;
HNO3; HC1 + HNO3 + H2SO4 и др.
Методы определения содержания галлия
Спектральные методы имеют большое значение для оп-
ределения галлия (а также индия и таллия) при содержа-
нии < 1 %.
Титриметрические методы. Комплексонометрические ме-
тоды получили наибольшее практическое применение для
определения содержания галлия.
Комплексонометрическое определение выполняют в сла-
бокислых, нейтральных или аммиачно-тартратных раство-
рах методом прямого или чаще обратного титрования. Об-
ратное титрование проводят при pH 10 и температуре 70—
80 °C растворами солей цинка с индикатором эриохром-
черным Т в присутствии тартрат-ионов, предотвращающих
гидролиз.
Индикаторы 4-(2-пиридилазо)-резорцин или ПАР и 1-
(2-пиридилазо) -2-нафтол или ПАН позволяют проводить
прямое титрование в кислых средах (pH 2,0—2.6). Селек-
тивность и точность определения галлия может быть по-
вышена при использовании индикаторной системы: комп-
лексонат меди (II)—ПАН или ПАР (pH ~2).
Фотометрические методы. Галлий не обладает хромо-
форными свойствами и дает цветные реакции только с
окрашенными реагентами или реагентами, поглощающими
свет в ближней ультрафиолетовой области спектра. Одним
из лучших и наиболее распространенных методов опреде-
ления галлия является метод, основанный на образовании
ионной пары хлоридным комплексом галлия с основным
ксантеновым красителем родамином Б. Ионный ассо-
циат экстрагируют бензолом из 6 М раствора соляной
кислоты.
Бензольный экстракт используется как для фотометри-
216
ческого, так и для флуоресцентного определения содержа-
ния галлия. Молярный коэффициент погашения комплекса
в бензольном экстракте е = 20-103 при Zmax = 560 нм. При
использовании бутилродамина Б молярный коэффициент
погашения выше: е = 90-103 при Zmax= 565 нм.
Из 6 М раствора соляной кислоты с родамином Б, кро-
ме галлия, бензолом экстрагируются соединения железа
(III), золота (III), сурьмы (V), таллия (III). Однако в
присутствии восстановителей (Т1С13, аскорбиновой кисло-
ты и др.) умеренные количества этих металлов не мешают
определению содержания галлия. С использованием рода-
мина Б содержание галлия определяют в горных породах,
минералах, бокситах, свинце, цинке, алюминии и др. При
условии больших содержаний алюминия галлий выделяют
экстракцией амилацетатом из 6 М раствора соляной кис-
лоты.
Для определения галлия применяют также методы, ос-
нованные на реакции образования комплексного соедине-
ния с ксиленоловым оранжевым, ПАР и др.
Гравиметрические методы. При осаждении галлия в ви-
де гидроксида, соединений с таннином, купфероном осад-
ки прокаливают при 1000 °C и взвешивают в виде Оа2Оз.
При осаждении галлия с помощью 8-оксихинолина весовой
формой является оксихинолинат галлия Оа(С9Н6ОК)з-
Предварительно осадок сушат при £=105—120 °C.
Как было указано выше, в настоящее время гравимет-
рические методы определения содержания галлия приме-
няются редко.
Полярографические методы. Наиболее удобным фоном
для полярографического определения галлия является
5%-ный раствор роданида калия (0,03 М раствор по со-
ляной кислоте). Четкая полярографическая волна галлия
получается также на фоне салициловой кислоты (pH 2,8—
3,8). Потенциал полуволны (относительно насыщенного
каломельного электрода) в первом случае равен—0,80 В,
во втором — 0,85 В.
Методы определения содержания индия
Титриметрические методы. Из титриметрических мето-
дов определения индия интерес представляет комплексо-
нометрическое титрование. В качестве индикаторов при-
меняют ксиленоловый оранжевый (pH 2—3), а также ПАН
[1-(2-пиридилазо)-2 нафтол] (pH 2,3—2,5), пирокатехи-
новый фиолетовый (pH 2—3), эриохромчерный Т (pH 8—
217
Фотометрические методы. Для определения содержания
индия широко применяют родамины. Определение индия
с родамином С производят в 2,0—2,5 М растворе бромисто-
водородной кислоты фотометрическим или флуориметри-
ческим методами. Методы не являются избирательными и
требуется предварительное отделение индия с помощью
экстракции, ионного обмена и др. Применяют также ме-
тоды, основанные на реакции образования комплексных
соединений с ПАР, ксиленоловым оранжевым, салицил-
флуороном.
Гравиметрические методы, основанные на взвешивании
оксида 1п20з или оксихинолината (С9НбОЫ)з1п, широкого
применения не имеют.
Электрохимические методы. Для определения содержа-
ния индия применяют титрование его раствором комплек-
сона III при рН~1 с установлением точки эквивалентности
амперометрическим путем. Полярографическое
определение содержания проводят в среде 3 М со-
ляной кислоты при потенциале от —0,4 до —0,8 В относи-
тельно насыщенного каломельного электрода. При анали-
зе сульфидных руд сопутствующие элементы (медь, сви-
нец, кадмий, олово) отделяют.
Методы определения содержания таллия
Титриметрические методы. При комплексонометрии тит-
рование таллия (III) комплексоном III проводят при
рН~2. Индикатором служит реактив ПАР или ПАН.
Определению содержания таллия цинк, свинец и кадмий
не мешают.
Применяют амперометрическое и потенциометрическое
титрование, основанное на использовании окислительно-
восстановительных реакций таллия.
Фотометрические методы, основанные на использовании
родаминовых и трифенилметановых красителей, в ряде
случаев позволяют определять содержание таллия без
предварительного его отделения от сопутствующих эле-
ментов.
Таллий (I) в виде бромида образует комплекс с рода-
мином 6Ж, который экстрагируют и используют для оп-
ределения содержания таллия флуориметрическим мето-
дом. Этот метод представляет интерес с той точки зрения,
что не требует окисления таллия (I), соединения которого
являются наиболее устойчивыми. Реакция образования
соединения таллия (I) проводится в 5 М растворе бро-
218 й
мистоводородной кислоты или 15 М растворе серной кис-
лоты. Для фотометрического определения содержания тал-
лия используют реагенты ПАР или ПАН.
Гравиметрические методы. Осаждение таллия (III)
можно проводить электролизом на аноде. В качестве ано-
да используют платиновую чашку, которую взвешивают
вместе с выделившимся оксидом Т12О3, а затем массу чаш-
ки вычитают. Для гравиметрического определения содер-
жания таллия используют осаждение его в виде Т12СгО4 из
щелочного раствора.
Полярографические методы используют для определе-
ния содержания таллия в рудах и минералах. Полярогра-
фирование производят на фоне аммиачного раствора суль-
фата аммония и сульфата натрия.
Практическая часть
Комплексонометрическое определение индия
в сплавах индий—олово
Принцип метода. Метод основан на комплексонометри-
ческом титровании индия в присутствии индикатора ксиле-
нолового оранжевого. Метод рассчитан на определение
содержания индия до 50%; относительное стандартное от-
клонение 0,01.
Реактивы и растворы. 1) Кислота азотная, р=1,43 г/см3; 2) кисло-
та соляная, р=1,19 г/см3; 3) кислота винная, 20%-ный раствор;
4) смесь кислот (готовят следующим образом: к 150 мл раствора
винной кислоты прибавляют 45 мл азотной и 5 мл соляной кислот
и перемешивают); 5) кислота муравьиная; 6) аммиак водный, 25%-ный
раствор; 7) формиатный буферный раствор pH 3 (готовят следующим
образом: в мерную колбу вместимостью 1 л приливают 50 мл муравьи-
ной кислоты, 25 мл аммиака и разбавляют водой до метки); 8) натр
едкий, 10%-ный раствор; 9) комплексон III, 0,05 М раствор; 10) кси-
леноловый оранжевый, 0,5%-ный раствор.
Выполнение определения. Навеску 0,2 г сплава поме-
щают в стакан вместимостью 50 мл, приливают 10 мл
смеси кислот и растворяют при умеренном нагревании. По
окончании растворения раствор переводят в мерную кол-
бу вместимостью 100 мл, объем в колбе доводят до метки
водой и содержимое колбы перемешивают. Аликвотную
часть раствора, равную 20 мл, помещают в коническую
колбу вместимостью 250 мл, приливают 70'мл воды, бро-
сают кусочек бумаги конго и нейтрализуют раствором
щелочи до изменения окраски индикаторной бумаги из
синей в фиолетовую (pH 3). Приливают 5 мл буферного
219
раствора 0,5 мл ксиленолового оранжевого и титруют
индий 0,05 М раствором комплексона до перехода малино-
вой окраски раствора в лимонно-желтую. В конце титро-
вания раствор комплексона вводят медленно, с интерва-
лом между каплями 10—12 с; 1 мл 0,05 М раствора комп-
лексона III соответствует 5,7410 мг индия.
Фотометрическое определение индия
с 4-(2-пиридилазо)-резорцином
в производственных растворах
Принцип метода. Определение основано на взаимодей-
ствии индия с 4-(2-пиридилазо)-резорцином при рН~3.
Определению не мешают 20 мкг цинка, 40 мкг свинца и
300 мкг алюминия. Относительное стандартное отклонение
результатов определений 0,05 при содержаниях индия
20 мг/л.
Реактивы и растворы. 1) 4-(2-пиридилазо)-резорцин, 0,01%-ный
раствор; 2) ацетатный буферный раствор (рН~3); 3) кислота соля-
ная, р=1,19 г/см3 и разбавленная 1:1; 4) запасной раствор, содержа-
щий в 1 мл 1 мг индия (для его приготовления растворяют 0,1 г
металлического индия в 5 мл соляной кислоты; раствор разбавляют
водой в колбе вместимостью 100 мл); 5) рабочий раствор, содержа-
щий в 1 мл 20 мкг иидия (для его приготовления разбавляют запасной
раствор 0,01 М соляной кислотой).
Построение градуировочного графика. В ряд мерных
колб вместимостью по 25 мл вводят от 1 до 5 мл стан-
дартного раствора индия с интервалом в 1 мл. В колбы
прибавляют по 5 мл раствора 4-(2-пиридилазо)-резорцина
и 10 мл буферного раствора. Доливают растворы водой
до метки и измеряют их оптическую плотность при Х=
= 510 нм, в кюветах с 1 = 20 мм, используя в качестве раст-
вора сравнения раствор, содержащий все реактивы.
Выполнение определения. К слабокислому раствору, со-
держащему не более 100 мкг индия, добавляют 5 мл раст-
вора 4-(2-пиридилазо)-резорцина и далее все реактивы,
указанные при построении градуировочного графика.
Амперометрическое
определение индия в концентратах
Принцип метода. Определение основано на титровании
индия при pH 1,0 раствором комплексона III. Точку эк-
вивалентности устанавливают по исчезновению предель-
ного диффузионного тока восстановления 1п3+ на ртут-
ном капельном электроде при потенциале от —0,7 до
220
—0,8 В относительно насыщенного каломельного электро-
да. Определению не мешают цинк, марганец, кадмий,
кобальт, алюминий, 0,5 мг свинца, 10мгжелеза (II). Влия-
ние олова (до 5 мг) и сурьмы (до 2 мг) устраняют вве-
дением винной кислоты; медь (<0,5 мг) маскируют тио-
мочевиной. Основную массу свинца и мышьяк отделяют
от индия в ходе анализа, а именно, при разложении про-
<бы смесью соляной и серной кислот и упаривании раство-
ра до появления паров серной кислоты. Большие коли-
чества меди (>0,5 мг) устраняют при осаждении гидрок-
еида индия и других элементов аммиаком. Относительное
стандартное отклонение результатов определений 0,05 при
содержании индия 10~2%.
Реактивы и растворы. 1) Кислота соляная, р = 1,19 г/см3; 2) кисло-
та сериая р= 1,84 г/см3; 3) раствор аммиака, р = 0,91 г/см5; 4) кислота
яинная, кристаллическая; 5) кислота аскорбиновая, 4%-ный раствор;
6) тиомочевина, 5%-ный раствор; 7) тропеолин 00, 0,1%-ный раствор;
8) буферный раствор, pH 1,0 (для его приготовления 50 мл 0,2 М
раствора хлорида калия и 97 мл 0,2 М соляной кислоты разбавляют
водой до объема 200 мл); 9) комплексон III, 0,005 или 0,02 М раст-
воры.
Выполнение определения. Навеску концентрата 0,5 г
разлагают смесью соляной и серной кислот (по 5 мл).
Раствор упаривают до появления паров серной кислоты.
Остаток разбавляют водой. до 50 мл, осадок отфильтро-
вывают. Из фильтрата осаждают гидроксиды избытком
раствора аммиака. Осадок отфильтровывают через 2—Зч
и растворяют в соляной кислоте. Раствор переводят в
мерную колбу вместимостью 50—100 мл и разбавляют во-
дой до метки.
В стакан для титрования отбирают аликвотную часть
раствора (10—20 мл), вводят 0,5—1 г винной кислоты,
1—2 мл раствора аскорбиновой кислоты и 0,2 мл раствора
тиомочевины. Полученный раствор нейтрализуют раство-
ром аммиака по тропеолину 00 до изменения красной ок-
раски раствора в желтую. Затем к раствору прибавляют
15—20 мл буферного раствора и далее титруют раство-
ром комплексона III из полумикробюретки вместимостью
5 мл. Точку эквивалентности находят графически по изме-
нению диффузионного тока индия в зависимости от коли-
чества прибавленного раствора комплексона III.
IX. МЕТОДЫ ОПРЕДЕЛЕНИЯ СОДЕРЖАНИЯ
СОПУТСТВУЮЩИХ И ПРИМЕСНЫХ ЭЛЕМЕНТОВ
Железо
Для определения содержания железа наиболее часто при-
меняют фотометрические и титриметрические методы.
Фотометрическое определение содержания .железа про-
водят в основном с роданидом, сульфосалициловой кисло-
той и ортофенантролином.
Роданидный метод основан на образовании в кислой
среде окрашенного в красный цвет комплекса железа (III)
с роданид-ионами. Роданидный комплекс [Fe(SCN)n]3-”
(где п = 1, ..., 6) недостаточно устойчив в водных растворах,
поэтому для его стабилизации и повышения интенсивности
окраски применяют водно-ацетоновые растворы. Моляр-
ный коэффициент погашения роданидного комплекса при
Z,=480 нм в водном растворе равен 7-103, в водно-ацетоно-
вом -15-Ю3.
Медь (II), кобальт, висмут, титан и молибден образуют
с роданид-ионами окрашенные комплексы. Роданидный
комплекс железа экстрагируют эфиром, изоамиловым спир-
том или другими растворителями.
Для определения содержания железа применяют ме-
тод, основанный на образовании комплекса железа (II) с
1,10-фенантролином (ортофенантролином).
Этот комплекс (ферроин) очень устойчив.- Молярный
коэффициент его погашения при Х = 490 нм равен 10,6-103.
Железо (III) предварительно восстанавливают хлоридом
гидроксиламмония, реакцию проводят при рН~Зн-9 в
присутствии тартрата аммония. При анализе допустимо
присутствие, мг/л: Mo, W по 5; Со, Си по 10, Ni 2, Сг (III)
20. Наряду с ортофенантролином применяют его произ-
водное батофенантролин.
Для фотометрического определения содержания железа -
применяют также сульфосалициловую кислоту
222
Молярный коэффициент погашения комплекса в амми-
ачной среде при Z, = 430 нм равен 6-Ю3. Преимуществом
метода является то, что в аммиачном растворе сульфо-
салициловой кислоты железо (II) переходит в железо (III)
и образует комплекс желтого цвета. Таким образом, в ам-
миачной среде с помощью этого реагента определяют
суммарное содержание железа. В кислых растворах суль-
фосалициловая кислота реагирует только с железом (III),
образуя комплекс красного цвета. По разности двух опре-
делений находят содержание железа (II). Мешают опреде-
лению окрашенные ионы.
Титриметрические методы определения содержания же-
леза основаны на восстановлении железа (III) до железа
(II) хлоридом олова (II), удалении избытка восстанови-
теля и титровании восстановленного железа титрованным
раствором окислителя (КМпС>4 или К2СГ2О7).
Восстановление железа (III)': 2Fe3++Sn2++6Cl_=
= 2Fe2++ [SnCl8]2-.
Титрование K2Cr2O7:6Fe2++Cr2O2-7 + 14H+=6Fes++
+ 2Сг3++7Н2О.
Избыток восстановителя SnCl2 окисляют сулемой-хло-
ридом ртути (II). К раствору прибавляют фосфорную кис-
лоту для связывания в комплекс ионов железа (III), об-
разующихся при титровании. Исчезновение желтой окрас-
ки ионов железа (III) облегчает наблюдение конечной
точки титрования. Снижение окислительного потенциала
системы Fe3+/Fe2+ способствует окислению железа (II).
Используют также титрование железа (III) раствором
восстановителя после предварительного окисления желе-
за (II).
Для определения содержания железа применяют комп-
лексонометрическое титрование. В качестве индикатора
используют сульфосалициловую кислоту или роданид ам-
мония. Титрование проводят при pH 2—3. Медь, цинк,
магний, марганец, определению не мешают.
Для гравиметрического определения содержания желе-
за используют осаждение его гидроксида аммиаком в при-
сутствии солей аммония. Многие элементы при этом выде-
ляются в осадок вместе с железом, т. е. метод не
является избирательным.
В последние годы для определения содержания желе-
за применяют атомно-абсорбционный метод. При опреде-
лении группы примесей содержание железа вместе с дру-
гими элементами определяют эмиссионным спектральным
методом.
223
Фотоколориметрическое определение железа
с применением сульфосалициловой кислоты
в сплавах на основе ниобия
Принцип метода. Железо образует с сульфосалицило-
вой кислотой в аммиачной среде комплексное соединение
желтого цвета. Мешающее действие ниобия устраняют,
добавляя оксалат аммония.
Метод позволяет определять 0,05—5% железа с отно-
сительным стандартным отклонением 0,1—0,05.
Реактивы и растворы. 1) Кислота фтористоводородная, р =
= 1,15 г/см3; 2) кислота азотная, р=1,40 г/см3; 3) кислота серная,
р = 1,84 г/см3 и разбавленная 1:4; 4) оксалат аммония, 4%-ный раст-
вор 5) кислота сульфосалициловая, 20%-ный раствор; 6) аммиак,
25%-ный раствор; 7) раствор, содержащий в 1 мл 0,1 мг железа (для
его получения в 30 мл соляной кислоты, р=1,12 растворяют 1 г чистого
железа типа Армко; раствор переводят в мерную колбу вместимостью
1 л и доводят объем до метки водой; в мерную колбу вместимостью
100 мл переносят 10 мл полученного раствора, доводят объем до мет-
ки водой, перемешивают).
Построение градуировочного графика. В мерные колбы
вместимостью по 100 мл вводят по 20 мл воды, от 0,5 мл
до 2 мл раствора железа из микробюретки, по 5 мл сер-
ной кислоты (1:4), 20 мл раствора сульфосалициловой
кислоты, 15 мл раствора аммиака. Объем раствора дово-
дят водой до метки и тщательно перемешивают. Интен-
сивность окраски растворов измеряют, как указано в ходе
анализа. По полученным данным строят градуировочный
график.
Выполнение определения. Навеску сплава 0,1 г поме-
щают в платиновую чашку и растворяют при нагревании
в 7—10 мл фтористоводородной кислоты, добавляя по кап-
лям азотную кислоту. После охлаждения к раствору ос-
торожно приливают 15 мл серной кислоты (р=1,84) и
упаривают до паров серной кислоты.
Пробу охлаждают, переносят с помощью 10 мл раст-
вора оксалата аммония в мерную колбу вместимостью
100 мл и доводят объем до метки водой.
Отбирают аликвотную часть (10 мл) испытуемого раст-
вора в мерную колбу вместимостью 100 мл, приливают
20 мл воды, 20 мл раствора сульфосалициловой кислоты
и 15 мл раствора аммиака. Доводят объем раствора до
метки водой и тщательно перемешивают. Интенсивность
окраски раствора измеряют на фотоэлектроколориметре
ФЭК-М с синим светофильтром в кювете с Z = 50 мм на
фоне раствора контрольного опыта (раствор реактивов.,
проведенный через весь ход анализа).
224
Содержание железа в сплаве находят, используя гра-
дуировочный график.
Атомно-абсорбционное определение железа
в хромоникелевом сплаве
Принцип метода. Определение основано на измерении:
поглощения железа по линии 248,3 нм без отделения ос-
новы и других сопутствующих элементов. Относительное
стандартное отклонение результатов определения 0,03 при
содержании 0,5% железа.
Реактивы и растворы. 1) кислота азотная, р=1,40 г/см3; 2) кисло-
та соляная, р= 1,19 г/см3.
Выполнение определения. Навеску сплава 0,1 г раство-
ряют в 10 мл смеси соляной и азотной кислот (3:1), раст-
вор упаривают досуха. Сухой остаток растворяют в 3 мл
соляной кислоты, раствор переводят в мерную колбу
вместимостью 100 мл и доводят объем до метки водой.
Содержание железа определяют по аналитической линии.
248,3 нм в пламени ацетилен-воздух. Градуировочный гра-
фик строят, используя раствор соли железа.
Комплексонометрическое определение железа
в материалах на основе меди
Принцип метода. Метод основан на образовании комп-
лексного соединения железа (III) с комплексоном III.
В качестве индикатора применяют сульфосалициловую
кислоту (при содержании железа выше 5 мг) или роданид,
аммония (ниже 5 мг). Железо предварительно отделяют
от меди путем его осаждения аммиаком. Относительное
стандартное отклонение результатов определений 0,005—
0,01 при содержаниях железа соответственно 10—1%.
Реактивы и растворы. 1) Комплексон III, 0,025 М раствор;
2) сульфосалициловая кислота, 10%-ный раствор; 3) роданид аммония,
10%-ный раствор; 4) натр едкий, 1 М раствор; 5) кислота соляная,,
р = 1,19 г/см3 и 1 М раствор.
Выполнение определения. Навеску 1—0,1 г (в зависи-
мости от содержания железа) разлагают обычно приня-
тым способом (см. «Медь»); железо осаждают аммиаком.
Осадок гидроксида растворяют в минимальном количестве
соляной кислоты, добавляют постепенно едкий натр до
образования неисчезающей мути, затем добавляют 9 мл
1 М соляной кислоты (рН~1). Раствор разбавляют водой
до 100 мл, нагревают до 70—80 °C, прибавляют 1 мл суль-
фосалициловой кислоты или роданида аммония и титруют
225.
раствором комплексона III до перехода лиловой окраски
раствора в желтую. 1 мл 0,025 М раствора комплексона III
соответствует 1,3962 мг Fe.
Титриметрическое определение железа в рудах
Принцип метода. Определение основано на титровании
(окислении) железа (II) раствором дихромата калия в
присутствии дифениламинсульфоната в качестве индика-
тора. Железо (III) предварительно восстанавливают рас-
твором хлорида олова (II) до железа (II). Метод рассчи-
тан на определение 1—50% железа. Мешают определению
сурьма, мышьяк, и большие количества меди. Относитель-
ное стандартное отклонение результатов определения 0,05.
Реактивы и растворы. 1) Дихромат калия, 0,1 н. титрованный
раствор; 2) индикатор дифениламиносульфонат; 0,2%-ный раствор;
3) кислота серная, 3 М раствор; 5) кислота фосфорная, р = 1,7 г/см3;
4) кислота соляная, 6 М. раствор; 6) хлорид олова (II) (для его
приготовления 150 г днгидрата хлорида олова (II) растворяют в
1000 мл 6 М раствора соляной кислоты); 7) хлорид ртути (II), 5%-иый
раствор.
Выполнение определения. Образец руды разлагают кис-
лотой или сплавляют с пероксидом натрия. Полученный
раствор помещают в стакан, прибавляют равный объем
6 М соляной кислоты, нагревают почти до кипения. При-
бавляют горячий раствор хлорида олова (II) до получения
бледно-желтой окраски, быстро охлаждают стакан с рас-
твором в струе водопроводной воды, приливают 10 мл
раствора хлорида ртути (II) и оставляют в покое на 2—
3 мин до появления слабого помутнения раствора. Прили-
вают 200 мл воды, 10 мл 3 М серной кислоты, 5 мл раство-
ра фосфорной кислоты, 6—8 капель раствора индикатора
и титруют раствором дихромата калия до первого измене-
ния зеленой окраски раствора в фиолетовую. Один милли-
литр точно 0,1 н. раствора дихромата калия соответствует
5,58 мг железа.
Кремний
Фотометрические методы определения содержания крем-
ния основаны на образовании желтого кремнемолибдено-
вого комплекса Si(Moi204o)4- (pH 1—2) или восстановлен-
ного комплекса синего цвета (I М H2SO4). Молярный
коэффициент погашения желтого комплекса е = 20-103 при
А = 300 нм и е = 7-103 при ^. = 350 нм. Молярный коэффи-
циент погашения восстановленной кремнемолибденовой
226
гетерополикислоты равен 20-103 при ^=810^-820 нм. Вос-
становленный кремнемолибденовый комплекс можно экс-
трагировать бутиловым или изоамиловым спиртом. Рсак-
ция образования комплекса желтого цвета: SiO| +
+ 12МоО2- + 22Н+=Si (М012О40) 4~+ 11Н2О.
Гравиметрические методы. Для определения содержа-
ния кремния часто используют выделение в кислой среде
кремнекислоты в виде коллоидного раствора с последую-
щим ее обезвоживанием (выпариванием с соляной или
серной кислотами): Na2SiO3 + 2HCl = H2SiOs + 2NaCl. Для
выделения кремнекислоты добавляют желатину, что спо-
собствует более полной коагуляции. Кремнекислоту, выде-
ленную одним из указанных способов, прокаливают при
1000—1200°C и взвешивают в виде оксида кремния (IV)
SiO2. Полученный таким образом оксид кремния (IV) за-
грязнен соосадившимися элементами. Для внесения по-
правки кремний отгоняют в виде SiF4 или H2SiFe:SiO2 +
+ 4HF = SiF4 + 2H2O. Остаток вновь прокаливают и взве-
шивают. Потеря массы соответствует содержанию SiO2 в
пробе.
Гравиметрическое определение кремния
в боксите гидраргиллитового минералогического состава
Принцип метода. Определение основано на разложении
боксита смесью серной и соляной кислот, выделении крем-
ниевой кислоты из солянокислого раствора в присутствии
желатина и определении диоксида кремния по разнице в
массе до и после обработки фтористоводородной кислотой.
Методика рассчитана на определение содержания крем-
ния от 1 до 15%. Относительное стандартное отклонение
результатов определений 0,02.
Реактивы и растворы. 1) Кислота соляная, р=1,19 г/см3 и раз-
бавленная 1:1 и 1:49; 2) кислота серная, р=1,84 г/см3 и разбавлен-
ная 1:1; 3) кислота фтористоводородная, р=1,15 г/см3; 4) смесь сер-
ной и соляной кислот для растворения боксита (готовят, осторожно
вливая в 1000 мл воды 250 мл серной кислоты и после охлаждения
приливают 625 мл соляной кислоты); 5) желатин, 1%-ный раствор
(для приготовления его раствора 1 г желатина помещают в коническую
колбу вместимостью 250 мл со 100 мл нагретой до 70 °C воды и ра-
створяют желатин при нагревании, не доводя раствор до кипения).
Выполнение определения. Навеску боксита 1 г поме-
щают в стакан вместимостью 400 мл, приливают 60 мл
смеси кислот, накрывают часовым стеклом и выпаривают
содержимое стакана на песочной бане до выделения бе-
лых паров. После охлаждения приливают в стакан 10 мл
227
1%-ного раствора желатина, 10 мл концентрированной со-
ляной кислоты, 150 мл горячей воды, перемешивают и на-
гревают (не доводя до кипения) до полного растворения
сульфатов. Через 1—2 мин раствор фильтруют через не-
плотный беззольный фильтр, обмывают стенки стакана и
промывают осадок на фильтре 3 раза горячей соляной
кислотой, разбавленной 1:49, и 6 раз горячей водой.
•Фильтр с осадком помещают в платиновый тигель, озоляют
и прокаливают при 950—1000 °C в течение 40 мин, затем
охлаждают в эксикаторе и взвешивают. После достижения
постоянства массы осадок диоксида кремния в тигле сма-
чивают водой, прибавляют 3 капли концентрированной сер-
ной кислоты, 5 мл фтористоводородной кислоты и выпари-
вают досуха на песочной бане. Остаток прокаливают при
950 °C в течение 10 мин, охлаждают в эксикаторе и взве-
шивают. Коэффициент пересчета оксида кремния (IV) на
кремний равен 0,4674.
Дифференциальное фотометрическое определение кремния
в боксите
Принцип метода. Определение основано на образова-
нии кремнемолибденовой гетерополикислоты, восстановлен-
ной до синего комплекса аскорбиновой кислотой. Методи-
ка рассчитана на определение содержания кремния 1—
15%. Относительное стандартное отклонение результатов
определений 0,02.
Реактивы и растворы. 1) Кислота аскорбиновая, 1%-ный раствор;
2) кислота серная, р=1,84 г/см3 и 0,07 М, 4 М растворы; 3) кислота
соляная, р=1,19 г/см3 и разбавленная 1:3; 5) молибдат аммония,
5%-ный раствор. 5. Карбонат натрия безводный; 6) натрий тетраборно-
кислый 10-водный, обезвоженный при 400 °C; 6) оксид кремния (IV);
7) смесь для сплавления (для ее получения смешивают карбонат
натрия и тетраборат натрия в соотношении 3:1; 8) растворы оксида
кремния (IV) (Для приготовления раствора А 0,2 г растертого и
предварительно прокаленного в течение 1 ч при 1000 °C оксида крем-
ния (IV) сплавляют в платиновом тигле с 5 г безводного карбоната
натрия при 900 °C в течение 10—15 мин до получения прозрачного
плава, который затем растворяют в воде при нагревании в платиновой
чашке; после охлаждения раствор помещают в мерную колбу вмести-
мостью 1 л и доводят водой до метки); 1 мл раствора А содержит
0,2 мг оксида кремния (IV). (Раствор Б готовят перед применением,
для его получения 100 мл раствора А помещают в мерную колбу
вместимостью 500 мл и доводят водой до метки); 1 мл раствора Б
содержит 0,04 мг оксида кремния (IV).
Построение градуировочного графика. В ряд мерных
колб вместимостью по 100 мл вводят 0—12,0 мл раство-
ра Б с интервалом 2,0 мл. Раствор в каждой колбе дово-
.228
,дят до 50 мл 0,14 н. раствором серной кислоты, приливают
5 мл раствора молибдата аммония, через 10 мин при пере-
мешивании приливают 25 мл 8 н. раствора серной кисло-
ты обмывая ею стенки колбы, через 2—3 мин добавляют
10 мл раствора аскорбиновой кислоты, доливают до метки
водой и перемешивают. Через 30 мин измеряют оптиче-
скую плотность растворов относительно раствора, содер-
жащего 5,0 мл раствора Б, на фотоэлектроколориметре
со светофильтром с максимумом светопропускания 630 нм
или на спектрофотометре при длине волны 815 нм. При
этом следует пользоваться постоянными кюветами, из ко-
торых одна предназначена для испытуемого раствора,
а другая — для раствора сравнения. Строят график зави-
симости оптической плотности от концентрации кремния.
Выполнение определения. Навеску боксита 0,5 г поме-
щают в платиновую чашку, смешивают с 3 г смеси для
сплавления и сплавляют в муфельной печи, нагретой до
900—950 °C в течение 20 мин. Одновременно проводят кон-
трольный опыт: 3 г смеси для сплавления помещают в пла-
тиновую чашку и выдерживают в муфельной печи 5 мин
до расплавления смеси, после охлаждения плавы выщела-
чивают 50 мл горячей воды при нагревании. Полученные
растворы переносят в стаканы вместимостью 300 мл, со-
держащие 60 мл соляной кислоты, разбавленной 1:3. Тща-
тельно обмывают стенки платиновой чашки водой, раствор
присоединяют к основному раствору, разбавляют водой до
250 мл, перемешивают и нагревают содержимое стакана
до полного растворения осадка. Охлажденные растворы
переносят в мерные колбы вместимостью 500 мл, доливают
до метки водой и перемешивают. Аликвотную часть раство-
ра 5 мл помещают в мерную колбу вместимостью 100 мл
и далее поступают, как указано при построении градуиро-
вочного графика. Содержание оксида кремния (IV) опре-
деляют по градуировочному графику.
Фотометрическое определение кремния
в ильменитовых концентратах
Принцип метода. Определение основано на образова-
нии кремнемолибденовой гетерополикислоты, восстанов-
ленной до синего комплекса аскорбиновой кислотой. Опре-
делению кремния предшествует отделение титана путем
сплавления навески с едким натром.
Методика рассчитана на определение содержания крем-
ния 1—2%. Относительное стандартное отклонение резуль-
татов определений 0,05.
229
Реактивы и растворы. 1) Натр едкий; 2) кислота борная; 3) кис-
лота серная, р=1,84 г/см3 и 8 н. раствор; 4) кислота соляная, р =
= 1,19 г/см3; 6 М и 0,15 М растворы; 5) молибдат аммония, 5%-ный
раствор; 6) кислота аскорбиновая, 1%-ный раствор; 7) стандартный
образец ферротитана с известным содержанием кремния в 1 мл раст-
вора (38,4 мкг SiOs). Для получения раствора 0,1 г образца сплавляют
в никелевом тигле с 3 г едкого натра. Плав выщелачивают при на-
гревании водой в платиновой чашке и далее поступают, как при вы-
полнении определения, начиная со слов: «Раствор с осадком филь-
труют»... и т. д. Раствор должен быть 0,15 М по соляной кислоте.
Его хранят в полиэтиленовом сосуде.
Построение градуировочного графика. В ряд мерных
колб вместимостью по 100 мл из микробюретки вводят
раствор ферротитана в количестве 0,5—6,0 мл с интерва-
лом 0,5 мл. Растворы разбавляют до 50 мл 0,15 М соля-
ной кислотой, приливают 5 мл молибдата аммония. Рас-
творы перемешивают и выдерживают 10 мин. Затем до-
бавляют по 30 мл 8 н. серной кислоты, 10 мл аскорбиновой
кислоты. Объем растворов доводят до метки водой. Через
20—30 мин измеряют оптическую плотность растворов
при длине волны 630 нм в кювете с 1 = 20 мм. Строят гра-
фик зависимости оптической плотности от концентрации
кремния.
Выполнение определения. Навеску тонкоизмельченного
концентрата 0,1 г помещают в никелевый тигель и сплав-
ляют с 3 г едкого натра в муфельной печи, нагретой до
400 °C, постепенно повышая температуру до 600—700 °C.
Сплавление производят при этой температуре в течение
10—15 мин до получения однородного плава. Плав выще-
лачивают 100 мл воды в платиновой чашке при кипячении
на песчаной бане. Раствор с осадком фильтруют в поли-
этиленовый сосуд через парафинированную воронку а
фильтр белая лента.
Осадок на фильтре промывают несколько раз водой,
затем к фильтрату прибавляют 17,5 мл 6 М соляной кис-
лоты, переводят раствор в мерную колбу вместимостью
250 мл, доводят объем до метки водой и вновь переводят
в полиэтиленовый сосуд. Кислотность раствора должна
быть равной 0,15 М (необходимое количество кислоты на-
ходят следующим образом: взвешивают 3 г едкого натра,
растворяют его в 50 мл воды и титруют раствор 6 М со-
ляной кислотой по индикатору метилоранжу до перехода
окраски; в избыток прибавляют 6,25 мл 6 М соляной кис-
лоты) .
Аликвотную часть раствора (5—10 мл) отбирают в мер-
ную колбу вместимостью 100 мл, разбавляют примерно
до 50 мл 0,15 М соляной кислотой, прибавляют 5 мл мо-
230
либдата аммония и далее поступают, как указано при по-
строении градуировочного графика.
В качестве раствора сравнения используют раствор
контрольного опыта, проведенный через все стадии ана-
лиза. Содержание оксида кремния (IV) определяют по
градуировочному графику.
Фосфор
Аналитической формой при определении фосфора яв-
ляется фосфат-ион. Поэтому в процессе разложения ана-
лизируемого образца выполняют окисление различных
форм существования фосфора в РО^ -ион, применяя силь-
ные окислители: азотную кислоту, царскую водку, перман-
ганат калия. При действии неокисляющих соляной или
серной кислот фосфор может теряться в виде летучего
фосфина (фосфористого водорода).
В аналитической химии элемента используют способ-
ность иона РО? к образованию труднорастворимых со-
лей, гетерополисолей, гетерополикислот. На основании
данных реакций разработаны гравиметрические, титримет-
рические и спектрофотометрические методы определения
фосфора.
Гравиметрические методы. Осаждение магний-аммоний-
фосфата MgNH4PO4-6H2O медленно происходит в раство-
ре, содержащем фосфаты и магнезиальную смесь: раствор
хлорида или сульфата магния и гидроксида аммония.
Осадок промывают, сушат, нагревают до 600—700 °C и
взвешивают в виде пирофосфата магния Mg2P2O7.
Осаждению магний-аммоний-фосфата не мешают неко-
торые элементы например кальций, алюминий, железо.
Недостатком метода являются медленное осаждение и не-
обходимость переосаждения. В целях отделения от мешаю-
щих элементов применяют также предварительное осажде-
ние фосфат-ионов из кислой среды в виде аммониевой соли
молибденофосфорной гетерополикислоты желтого цвета:
Н3РО4+ 12(NH4)2MoO4+ 21HNO3 =
= (NH4)3[PO4- 12МоО3] + 21NH4NO3 + 12H2O.
Труднорастворимые гетероцолисоли образуют также
мышьяк, кремний, ванадий, вольфрам.
Завершают определение в виде различных весовых
форм: в виде пирофосфата магния, желтой соли, синей
соли РгО5-24МоОз, получаемой прокаливанием желтой со-
231
ли; в виде молибдата свинца РЬМоО4 следующим образом:
желтую соль растворяют в аммиаке, а затем осаждают
молибдат-ион, действуя раствором ацетата свинца:
(NH4)3[PO4-12МоО3] +23NH4OH = 12(NH4)2MoO4 +
+ (NH4)2HPO4+ 11Н2О,
(NH4)2Mo04+ Pb(CH3COO)2 = PbMoO4+ 2NH4CH8COO.
Многообразие вариантов завершения метода вызвано
стремлением исследователей устранить трудности, вызван-
ные непостоянством состава осадка гетерополисоли.
Быстрым и точным методом определения содержания
фосфатов является хинолиновый метод.
Хинолин
или C9H7N' (молекулярная масса 129,17). Из кислого рас-
твора, содержащего фосфаты, реактив осаждает соль жел-
того цвета. При осаждении в присутствии цитрат-ионов
фосфат-ион отделяется от многих элементов, образующих
труднорастворимые фосфаты и гетерополисоли (V, W, Та,
Nb, Zr, La и др.):
Н3РО4 + 12Na2MoO4 + 3C9H7N + 24HNO3=
= (C9H7N)3H3[PO4- 12MoO3] + 12H2O + 24NaNO3.
Можно применять также 8-оксихинолин (см. «Алюминий»).
Косвенные титриметрические методы. Ацидиметрический
метод основан на растворении гетерополисолей в избытке
титрованного раствора щелочи, которую оттитровывают
раствором азотной кислоты в присутствии фенолфталеина:
(C9H7N)3H JPO4 • 12МоО3] + 26NaOH =
= Na,HPO4 + 12Na2MoO4 + 3C9H7N + 14H2O;
(NH4)3[PO4-12MoO3] -P 26NaOH =
= Na2HPO4 4- 12Na2Mo04 + 3NH3 + 14H2O.
Метод с хинолином отличается более четким переходом
окраски в конечной точке титрования.
Коплексонометрический метод. Титрованным раствором
соли металла: висмута магния или цинка осаждают фос-
фат-ион. Избыток соли металла оттитровывают раствором
комплексона III.
232
Спектрофотометрические методы определения содержа-
ния фосфатов основаны на образовании гетерополикислот:
молибдено-фосфорной и молибдено-ванадиево-фосфорной:
Н3РО4-VOs-11МоО3.
Гравиметрическое определение фосфора
в метафосфатах алюминия и редкоземельных металлов
Принцип метода. Определение основано на образовании
гетерополисоли хинолиния и фосфорной кислоты в присут-
ствии цитрат-ионов. Предварительно метафосфаты сплав-
ляют со щелочью в стеклоуглеродных тиглях, а затем плав
обрабатывают кислотой.
Относительное стандартное отклонение результатов
определения 0,01—0,005 при содержании фосфора в пере-
счете на Р2О5 20—80%.
Реактивы и растворы. 1) Кислота азотная, р = 1,40 г/см3; 2) кис-
лота соляная, р = 1,19 г/см3; 3) кислота лимонная; 4) калия гидроксид
в гранулах; 5) натрия молибдат; 6) хинолин (можно заменить 8-окси-
хинолином); 7) реакционная смесь молибдата натрия н-лимонной кис-
лоты. (Для приготовления этой смеси 94,00 г молибдата натрия
растворяют в 350 мл воды, полученный раствор вливают в смесь,
содержащую 60 г лимонной кислоты, растворенной в 250 мл воды и
140 мл соляной кислоты; разбавляют водой до 1000 мл. Раствор
годен к употреблению в течение 1 мес; 8) раствор хинолина (готовят
следующим образом: в колбу вместимостью 1000 мл отмеривают ци-
линдром 300 мл воды, 60 мл соляной кислоты и 50 мл хинолина,
перемешивают, разводят водой до 1000 мл н снова перемешивают).
Выполнение определения. В стеклоуглеродный тигель
помещают 7—8 гранул гидроксида калия, смачивают 1 —
2 каплями воды, расплавляют на электроплитке и охлаж-
дают.
В тигель помещают 0,1000 г продукта прибавляют еще
4—5 гранул гидроксида калия, смачивают 1—2 каплями
воды и расплавляют на электроплитке, слегка вращая ти-
гель до полного разложения продукта в течение 10—15 мин.
По охлаждении содержимое тигля переносят водой в ста-
кан с меткой на 150 мл, к полученной суспензии прили-
вают пипеткой 10 мл соляной кислоты, доводят объем
полученного раствора водой до метки и кипятят на элек-
троплитке в течение 1 ч. Во избежание выброса при кипе-
нии в стакан помещают стеклянную палочку. По охлажде-
нии раствор переносят в колбу вместимостью 100 мл и
доводят объем водой до метки; 25 мл полученного раствора
пипеткой отмеривают в стакан вместимостью 250 мл, при-
ливают цилиндром 75 мл воды, 35 мл реакционной смеси,
нагревают на электроплите почти до кипения, к горячему
233
раствору медленно по каплям при перемешивании прибав-
ляют 5—7 мл раствора хинолина до прекращения образо-
вания осадка. Перемешивание продолжают до перехода
белой мути в желтый осадок. Через 10 мин осадок пере-
носят на тигель ТФ ПОР-16, предварительно высушенный
до постоянной массы при 150 °C и взвешенный, осадок про-
мывают водой и сушат при 150 °C в течение 1,5 ч до по-
стоянной массы и взвешивают на тех же весах. Коэффи-
циент пересчета гетерополисоли на оксид фосфора (V)
равен 0,03209. После анализа осадок из тигля удаляют,
тигель промывают аммиаком, водой и сушат.
В случае применения 8-оксихинолина осадок промы-
вают фильтратом и 1%-ным раствором нитрата натрия,
сушат при 105—ПО °C. Коэффициент пересчета гетерополи-
соли на оксид фосфора (V) равен 0,03128.
Азот и сера
Дистилляционно-ацидиметрический метод
определения азота в металлах и сплавах
Принцип метода. Азот в металлах содержится преиму-
щественно в виде нитридов и карбонитридов (например
TIN и TisCN^. При растворении металлов-в кислотах хи-
мически связанный азот переходит в соответствующие
аммонийные соли.
Определение заканчивается ацидиметрически в присут-
ствии смешанного индикатора (метиленового синего и ме-
тилового красного) после отгонки аммиака из щелочного
раствора (по методу Кьельдаля). Правильность проведе-
ния анализа контролируется стандартным образцом, на-
пример титана.
Определение содержания азота (растворение пробы,
отгонку аммиака) необходимо проводить в таком помеще-
нии, где не ведутся работы с аммиаком и его солями.
Одновременно через все стадии анализа проводится кон-
трольный опыт на реактивы. При этом реактивы исполь-
зуются в тех же количествах, что и при анализе испытуе-
мого образца. Определяемое количество азота (аммиака)
не должно быть меньше количества азота в контрольном
опыте. Нижняя граница определяемых содержаний азота
3-10-3%.
Гравиметрическое определение серы в боксите
Принцип метода. Определение основано на окислении
серы до сульфатной спеканием с перманганатом калия и
содой. После выщелачивания спека водой сульфат-ионы
осаждают хлоридом бария: Ba2++SO%-^BaSO4. Относи-
тельное стандартное отклонение результатов определения
серы составляет 0,02 при ее содержании до 0,5% и 0,01
при более высоких содержаниях.
Реактивы и растворы. 1) Кислота соляная, р = 1,19 г/см3 и раз-
бавленная 1:1; 2) хлорид бария, раствор 100 г/л; 3) пероксид водо-
рода, 3%-ный раствор; 4) перманганат калия; 5) карбонат натрия
безводный, 0,5%-ный раствор; 6) нитрат серебра, 1%-ный раствор;
7) метиловый красный, 0,1%-ный спиртовой раствор; 8) смесь для
спекания (для ее приготовления смешивают растертые перманганат
калия и безводный карбонат натрия в соотношении масс 1:1).
Выполнение определения. Навеску боксита 1 г поме-
щают в фарфоровый тигель, тщательно перемешивают с
5 г смеси для спекания и сверху засыпают еще 2 г указан-
ной смеси. Тигель закрывают крышкой, помещают в хо-
лодный муфель, постепенно нагревают до 700—800 °C и
выдерживают при этой температуре в течение 30 мин. Не
допускается проводить спекание в муфеле, в котором про-
водилось сплавление с бисульфатами или разложение
сульфатов. Тигель с содержимым охлаждают, помещают
в стакан вместимостью 400 мл, прибавляют 150 мл горя-
чей воды и нагревают до полного выщелачивания спек-
шейся массы, время от времени помешивая и раздавливая
комочки стеклянной палочкой. Затем тигель вынимают из
раствора и обмывают водой. Раствор фильтруют через
плотный беззольный фильтр, содержащий небольшое ко-
личество фильтробумажной массы. Фильтрат собирают в
стакан вместимостью 250 мл. После этого осадок промы-
вают на фильтре четыре-пять раз горячим раствором кар-
боната натрия и пять-шесть раз горячей водой.
Полученный фильтрат нагревают, добавляют 5—6 мл
пероксида водорода и кипятят 3—4 мин. Затем раствор
нейтрализуют по метиловому красному разбавленной со-
ляной кислотой, прибавляют 2 мл избытка той же кисло-
ты на каждые 100 мл раствора и снова нагревают до ки-
пения. К кипящему раствору (около 200 мл) прибавляют
при перемешивании 10 мл горячего раствора хлорида ба-
рия. Раствор с осадком продолжают нагревать в течение
30 мин, после чего выдерживают 12 ч.
Раствор над отстоявшимся осадком сливают на плот-
ный беззольный фильтр, содержащий небольшое количе-
235
ство беззольной фильтробумажной массы. Осадок в ста-
кане промывают три раза декантацией теплой водой, затем
его переносят на фильтр и промывают до тех пор, пока
20 мл промывных вод дадут очень слабое помутнение с
раствором нитрата серебра.
Влажный фильтр с осадком помещают во взвешенный
платиновый тигель, осторожно обугливают, не давая вос-
пламениться, и сжигают при достаточном доступе воздуха.
Остаток прокаливают при 800 °C в течение 40 мин,,
охлаждают в эксикаторе и взвешивают. Коэффициент пе-
ресчета сульфата бария на серу равен 0,1374.
Кальций и магний
Кальций получен Деви в 1909 г. Металлический кальций в
основном получают электролизом хлорида кальция. Кальций является
одним из распространенных металлов. Содержание его в земной коре
составляет 3,25%. Наиболее часто кальций встречается в природе в-
виде известняка и мела.
Известняк состоит в основном из минерала кальцита СаСО3, со-
держащего примеси магния, железа, марганца и др. Мел содержит
99% чистого кальцита. Реже встречается кристаллическая форма кар-
боната кальция-— мрамор.
Кальций входит в состав многих осадочных пород, водных алюмо-
силикатов, рудных минералов. Кальций содержится в металлургиче-
ских шлаках в форме различных химических соединений — силикатов,
фосфатов и др.
Магний принадлежит к числу широко распространенных эле-
ментов. Общее содержание его в земной коре составляет 2,35%. В при-
роде встречаются огромные залежи карбонатов — доломита (СаСО3-
•MgCCfc) и значительно реже магнезита (MgCO3).
Методы определения кальция
Гравиметрический метод определения кальция основан
на осаждении его в виде оксалата кальция щавелевой
кислотой или щавелевокислым аммонием: СаС12 +
+ (NH4)2C2O4-^-CaC2O4 + 2NH4Cl. Выделившийся в осадок
оксалат кальция отфильтровывают и прокаливают до окси-
да кальция СаС2О4-э-СаСО3-э-СаО.
Титриметрический метод определения кальция основан
на осаждении кальция в виде оксалата, обработке осадка
разбавленной серной кислотой и титровании освободив-
шейся щавелевой кислоты раствором перманганата калия:
СаС2О4+ H2SO4 = CaSO4 + Н2С3О4;
5Н2С2О4 + 2KMnO4+ 3H2SO4 ==
= 2MnSO4+ K2SO4+ 10 СО2+ 8Н2О .
236
Наиболее часто для определения кальция применяют
комплексонометрический метод (См. «Химические мето-
ды», «Комплексонометрия»). В качестве индикатора
используют эриохром черный Т или другие индикаторы.
Часто комплексонометрическое титрование применяют для
определения жесткости воды (определение суммарного
содержания кальция и магния, т. е. так называемой «каль-
циевой» и «магниевой» жесткости воды).
Методы определения магния
Гравиметрический метод определения магния основан
на образовании осадка фосфата магния и аммония соста-
ва MgH4PO4-6H2O. Отфильтрованный и промытый осадок
прокаливают при 1000 °C и взвешивают в виде пирофос-
фата : 2MgNH4PO4 • 6H2O^Mg2P2O7 + 2NH3 + 7Н2О.
Второй гравиметрический метод определения магния
основан на образовании осадка оксихинолята магния
MgCl2+ 2H(C9H6ON) + 2NH4OH =
= Mg (C9H6ON)2 • 2H2O + 2NH4C1.
Наиболее широкое применение для определения магния
находит комплексонометрический метод.
Комплексонометрическое определение
кальция и магния
в известняках и карбонатных породах 1
Принцип метода. Суммарное содержание кальция и
магния определяют титрованием раствора комплексо-
ном III. В другой части раствора после отделения кальция
в виде оксалата, определяют магний. Относительное стан-
дартное отклонение при определении кальция — 0,05, при
определении магния — 0,07.
Реактивы и растворы. 1) Кислота соляная, р=1,19 г/см3, и разбав-
ленная 1:1; 2) аммиак, р = 0,91 г/см3 и разбавленный 1:1; 3) индикатор
метиловый красный; 4) индикатор кислотный хромовый темно-синий;
5) хлоридно-аммиачный буферный раствор; 6) нитрат аммония, 2%-ный
раствор; 7) комплексон III, 0,1 М раствор; 8) оксалат аммония.
Выполнение определения. Навеску пробы 0,5 г раство-
ряют при умеренном нагревании в 20—30 мл соляной кис-
лоты (1:1). После растворения пробы раствор охлаждают,
переводят в коническую колбу вместимостью 250 мл, до-
1 Например, в доломите MgCO3-CaCO3.
237
бавляют до 100 мл воды, нейтрализуют большую часть
кислоты аммиаком, не содержащим карбонатов, и слабо-
кислый раствор нагревают до кипения. Затем осторожно
(из капельницы) прибавляют раствор аммиака до пере-
мены цвета индикатора метилового красного. Осадку гид-
роксидов дают отстояться в течение нескольких минут
и раствор фильтруют горячим через неплотный беззоль-
ный фильтр. Затем осадок промывают 3—4 раза горячим
2%-ным раствором нитрата аммония, нейтрализованным
аммиаком по метиловому красному. Фильтрат и промыв-
ные воды от осадка гидроксидов собирают в мерную кол-
бу вместимостью 250 мл, разбавляют водой до метки колбы
и перемешивают.
При определении суммы кальция и магния отбирают
из колбы 50 мл раствора, разбавляют водой в два раза,
добавляют 10 мл хлоридно-аммиачного буферного раство-
ра, 5—8 капель индикатора кислотного хромового темно-
синего и титруют 0,1 М раствором комплексона III до пе-
рехода окраски из красной в синюю.
Для раздельного определения магния 100 мл раствора
подкисляют уксусной кислотой до розовой окраски мети-
лового красного, нагревают до кипения и осаждают каль-
ций оксалатом аммония. Осадок оксалата кальция от-
фильтровывают через 1—2 ч и промывают горячей водой,
содержащей небольшое количество оксалата аммония.
Фильтрат и промывные воды собирают в коническую кол-
бу вместимостью 500 мл, добавляют аммиак (до появле-
ния запаха) и 15—20 мл хлоридно-аммиачной буферной
смеси. Затем добавляют 15—20 капель индикатора и тит-
руют 0,1 М раствором комплексона III.
БИБЛИОГРАФИЧЕСКИЙ
РЕКОМЕНДУЕМЫЙ СПИСОК
Алексеев Р. И., Коровин Ю. И. Руководство по вычислению и обра-
ботке результатов количественного анализа. М.: Атомиздат, 1972.
72 с.
Бусев А. И., Типцова В. Г., Иванов В. М. Руководство по аналитиче-
ской химии редких элементов. М.: «Химия», 1978. 431 с.
Канаев И. А. Ускоренное определение редкоземельных элементов. М.:
«Металлургия», 1971. 224 с.
Коростелев П. П. Реактивы и растворы в металлургическом анализе.
М.: Металлургия, 1977. 350 с.
Лурье Ю. Ю. Справочник по аналитической химии. М.: «Химия», 1979.
480 с.
Мусакин А. П. Таблицы и схемы аналитической химии. Л.: «Химия»,
1971. 128 с.
Практическое руководство по неорганическому анализу / В. Гиллебрандт,
Г. Лендель, Г. Брайт, Д. Гофман. М.: «Химия», 1966. 1111 с.
Сусленникова В. М., Киселева Е. К- Руководство по приготовлению
титрованных растворов. Л., «Химия», 1978. 184 с.
Филиппова Н. А., Шкробот Э. П., Васильева Л. Н. Анализ руд цвет-
ных металлов. М.: Металлургия, 1980. 150 с.
Харламов И. П. Спектрофотометрический анализ в черной металлургии
М.: Металлургия, 1980. 208 с.
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Анализ 22, 23, 24
Определение
— атомно-абсорбционное:
алюминия 51
железа 223, 225
кадмия 105
кобальта 73, 76
меди 88, 96
никеля 80, 82
рения 185
свинца 114
— гравиметрическое:
алюминия оксихинолином
(оксином) 52
— фторидом натрия 52
вольфрама 174
галлия таннином 217
— купфероном 217
железа 223
индия 218
кальция 238
кобальта 71
кремния 227
магния 239
меди 85, 89, 90, 91, 92
молибдена 173
— осаждением
а-бензоиноксимом 174
никеля 78, 81
ниобия в виде оксида 155
— осаждением таннином 155
РЗЭ осаждением
оксалатов 197, 200
---- фторидов 198
рения осаждением
нитроном 185, 189
---- хлоридом
тетрафениларсония 186
серы 237
скандия 206, 208
фосфора 231, 233
церия 198
циркония 142
— спектральное галлия 216
— титриметрическое:
азота 234
алюминия
комплексонометрическое 53, 56
вольфрама 175
галлия 216
железа
комплексонометрическое 225
239
— окислительно-
восстановительное 223, 226
индия 217, 219
кальция 238, 239
кобальта
йодометрическое 73
— комплексонометрическое 71
магния 239
меди
йодометрическое 86, 92
— комплексонометрическое
'93, 94
молибдена 175
никеля йодометрическое 80
— комплексонометрическое
79, 80, 82
РЗЭ 196, 199
свинца йодометрическое 117
— комплексонометрическое
114, 116
— хроматное 114
скандия 207, 210
таллия 218
титана
комплексонометрическое 124
— окислительно-
восстановительное 125, 127
•фосфора 232
.хрома
комплексонометрическое 62
— окислительно-
восстановительное 60, 61, 63
церия 204
цинка 99, 101
циркония 141
— фотометрическое:
алюминия алюминоном 55
— хромазуролом S 56
— дифференциальное 58
— эриохромцианином R 55
вольфрама дитиолом 172
— роданидом 170
галлия
ксиленоловым оранжевым 217
— родамином Б 216
железа роданидом 222
— сульфосалициловой
кислотой 222, 223, 224
— 1, 10-фенантролином 222
индия 218
кадмия 105
кобальта
1-нитрозо-2-нафтолом 69
— нитрозо-К-солью 70, 73
кремния 226, 228, 229
меди 2,2'-дихинолилом 88
— диэтилдитиокарбаматом
88, 94
молибдена дитиолом 172, 179
— роданидом 170, 177
неодима
дифференциальное 203
ниобия дифференциальное
159, 160
— ПАР 150, 157
— роданидом 149, 156
— сульфохлорфенолом С 151
олова 109, ПО
РЗЭ арсеназо III 194, 201
— по окраске солей 195
рения диметилглиоксимом
185, 189
— роданидом 183, 188
— тиомочевиной 184, 186 1
скандия
ДОТРИХАФ 208, 211
— ксиленоловым оранжевым
207, 209
таллия 218
тантала бриллиантовым
зеленым 153, 162
— кристаллическим
фиолетовым 154
— пирогаллолом 152
титана диантипирилметаном
122, 123, 130
— дифференциальное 122, 128
— хроматроповой кислотой 124
хрома дифенилкарбазидом 62
— комплексоном III 65
церия 196
циркония арсеназо III
138, 143
— ксиленоловым
оранжевым 140, 143
— сульфохлорфенолом С 139
электрохимическое:
галлия 217
индия 218, 220
кобальта 72, 75
меди 85, 89, 90
олова 112
рения 186
свинца 115
таллия 219
цинка 100, 102