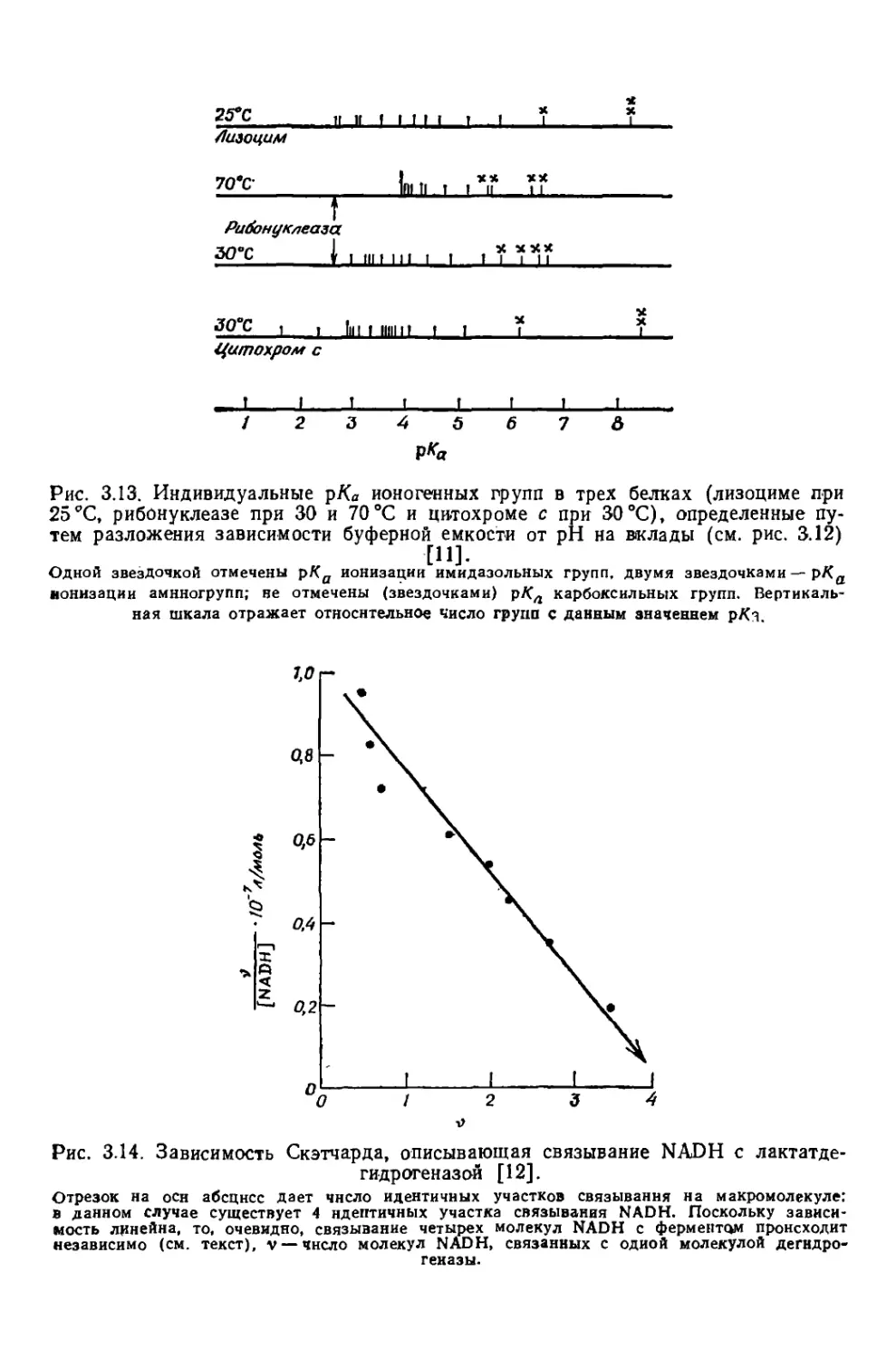
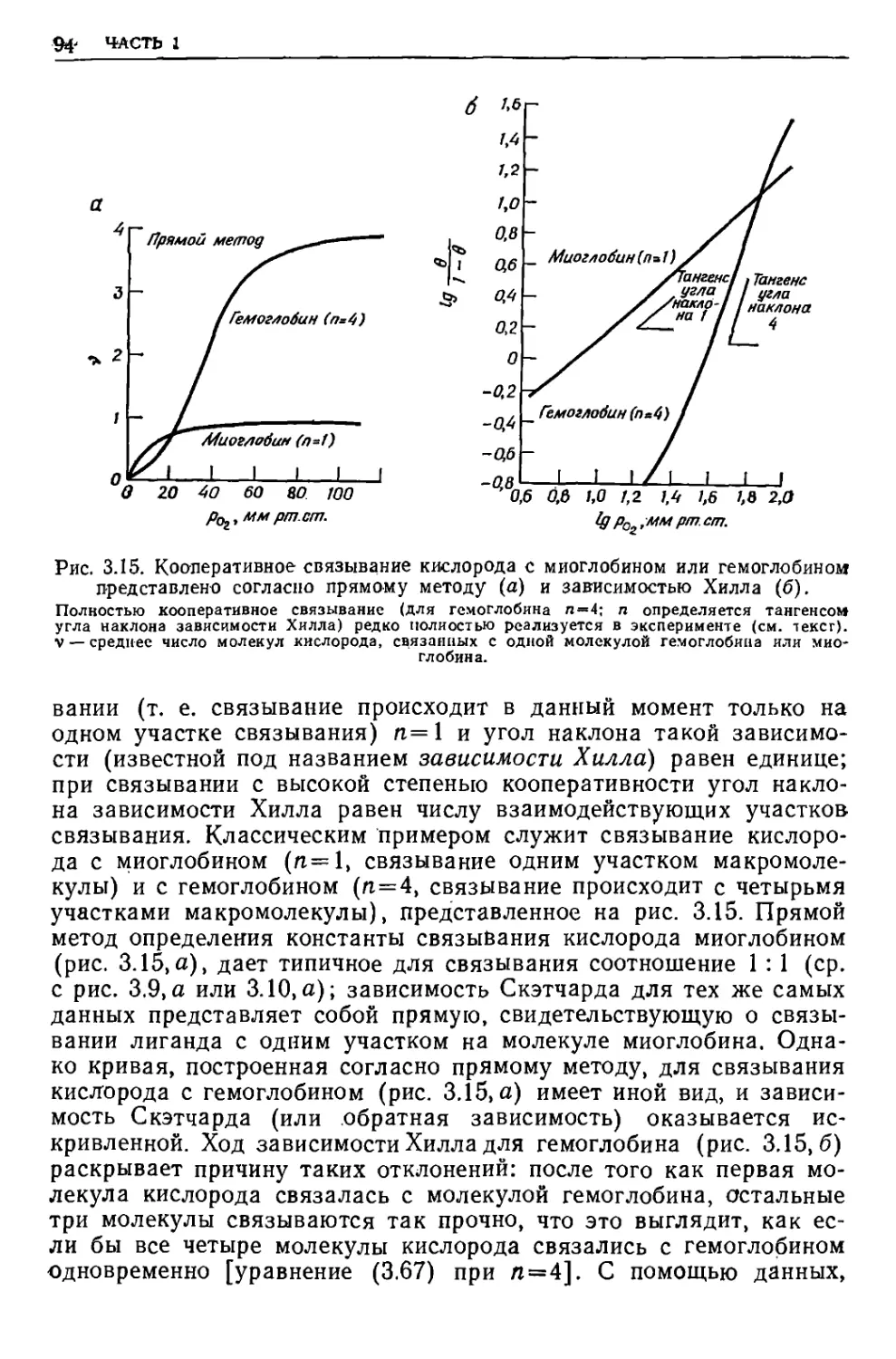
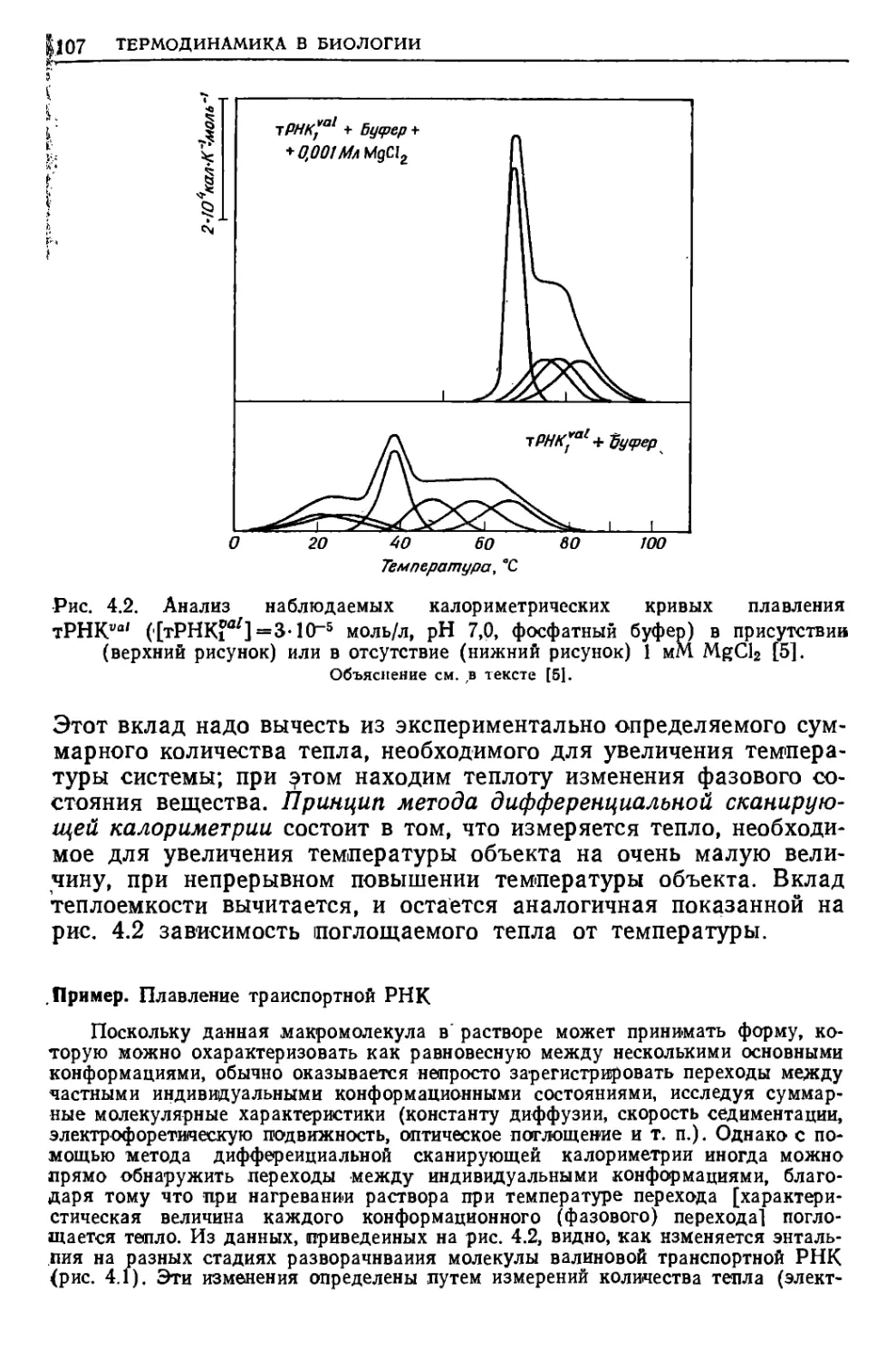
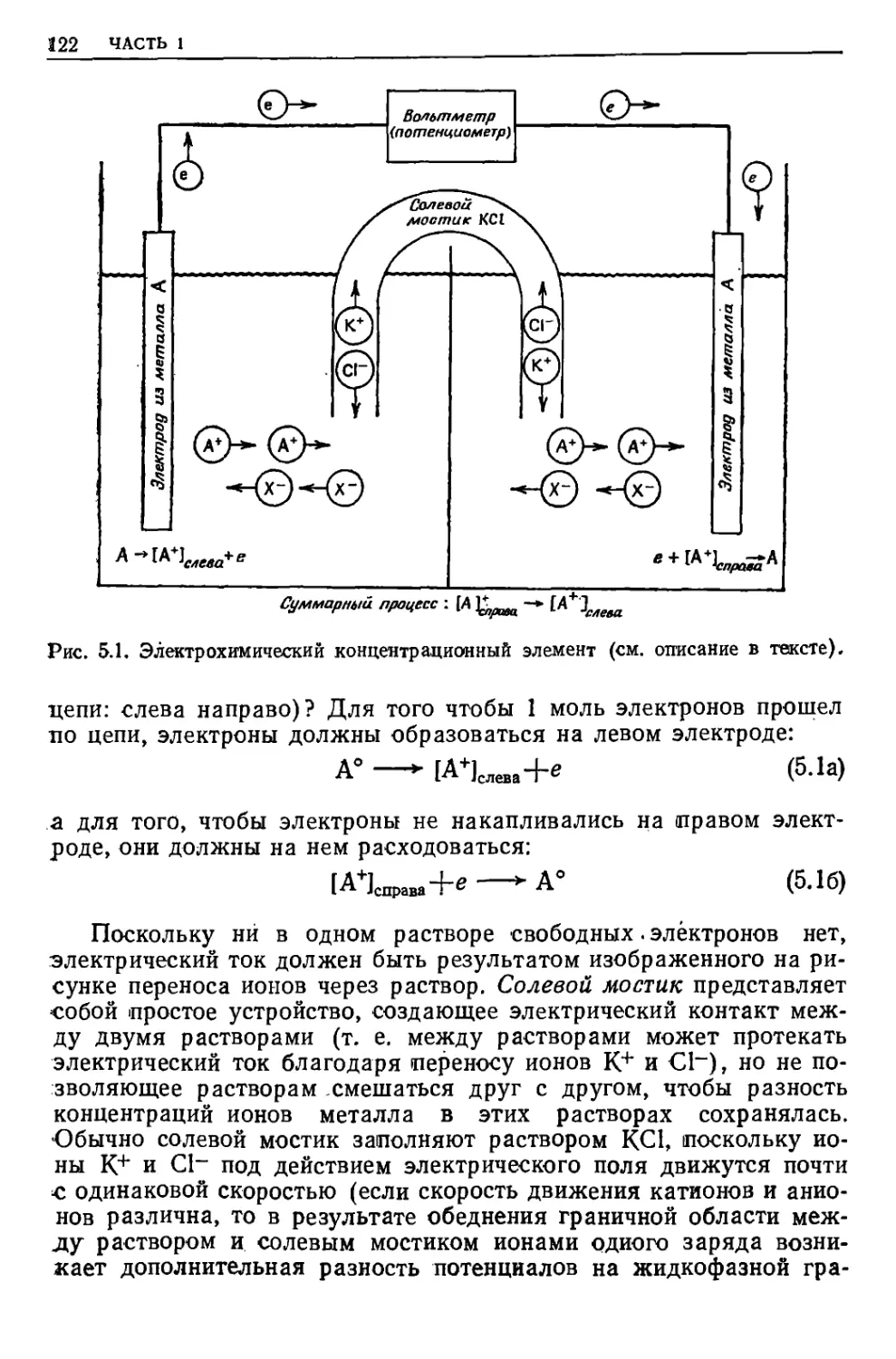
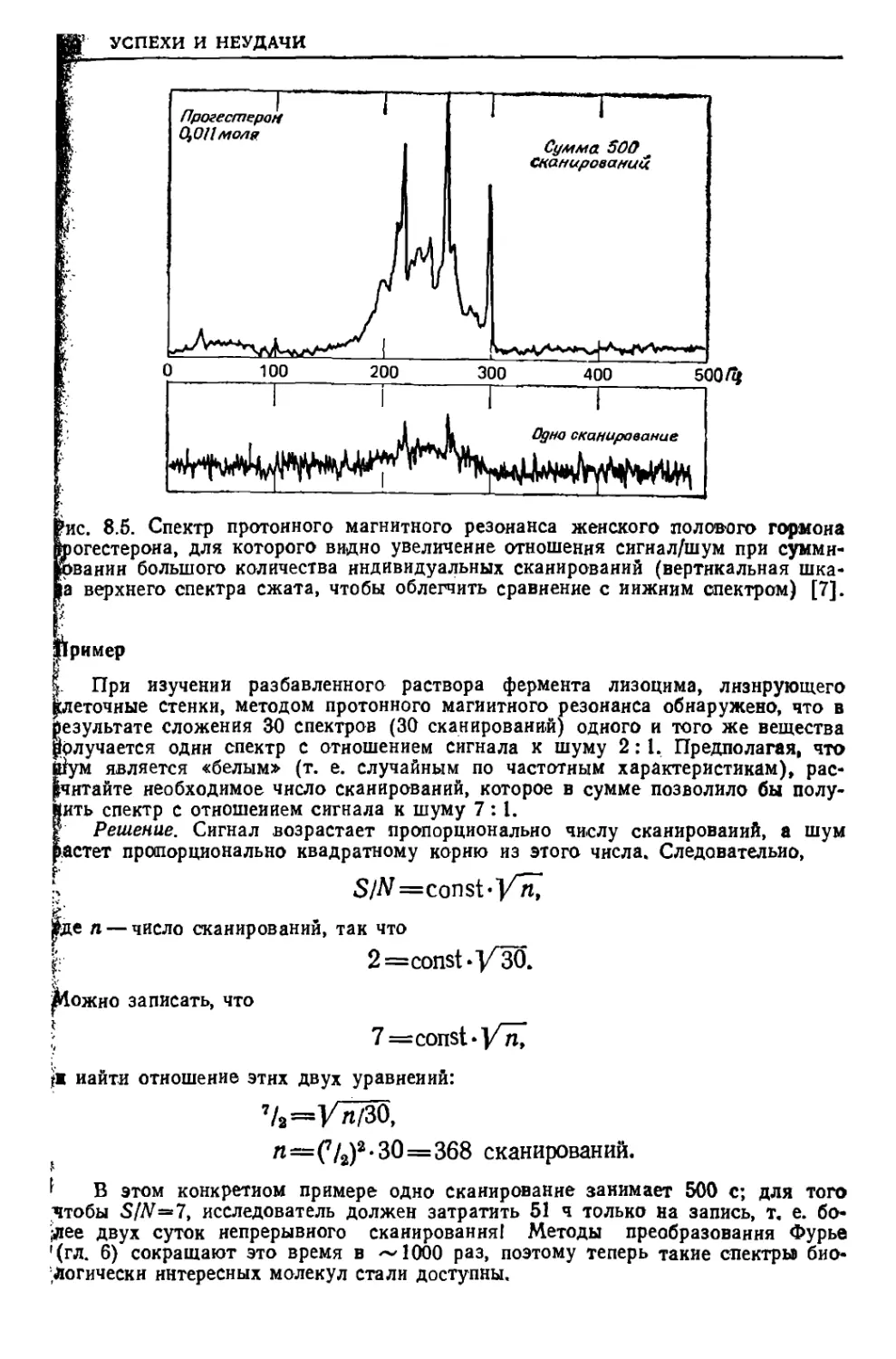
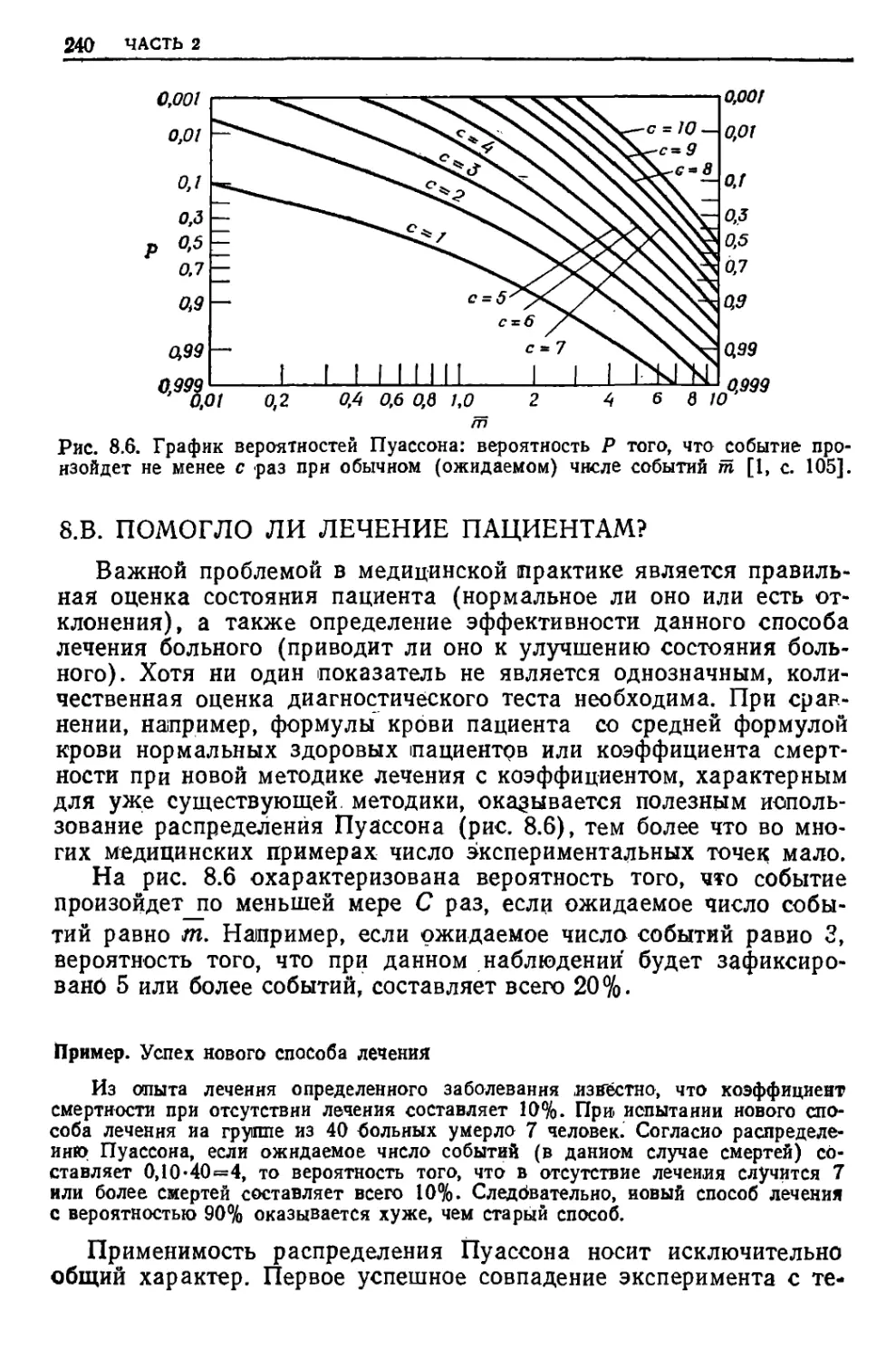
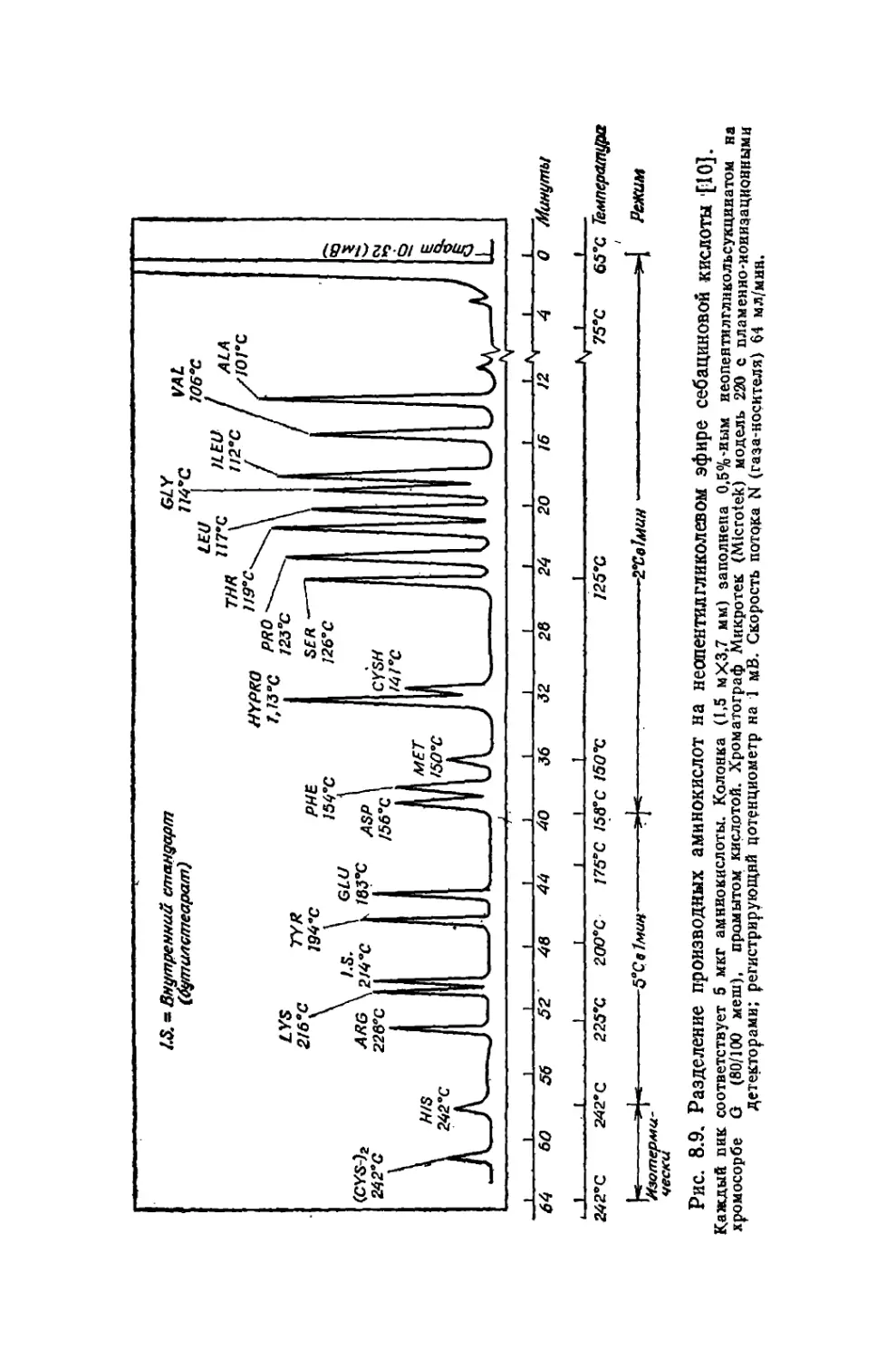
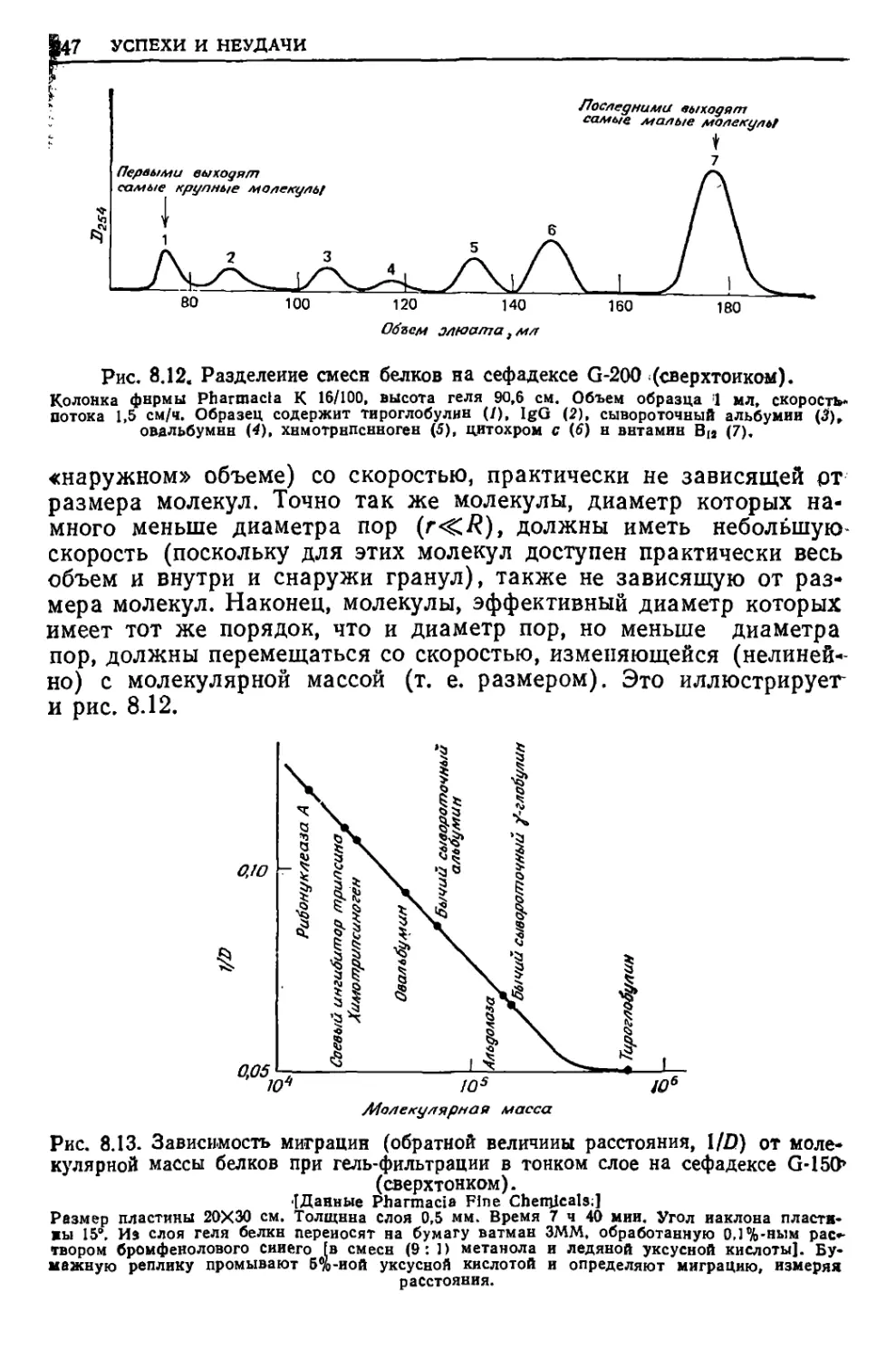
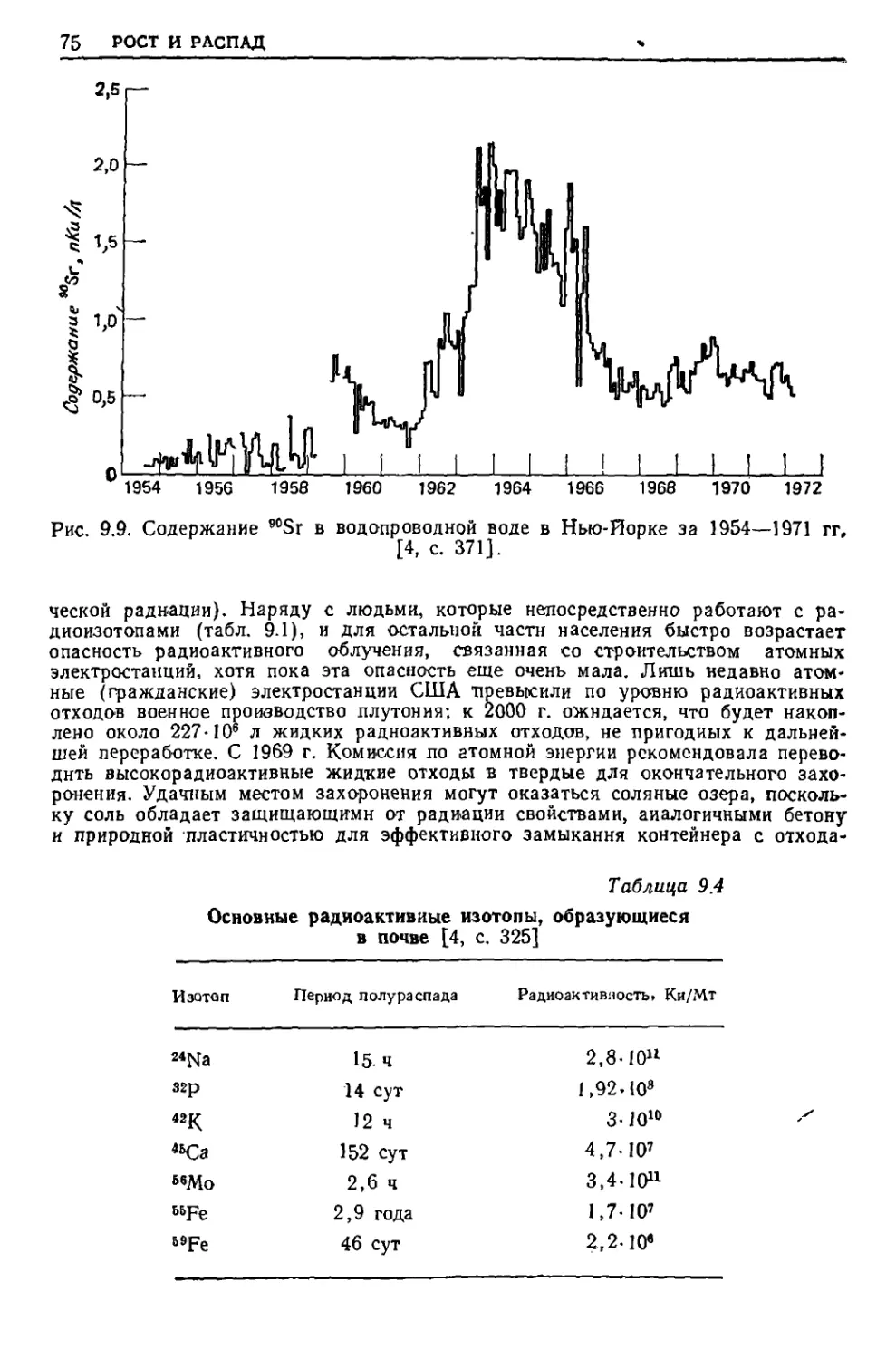
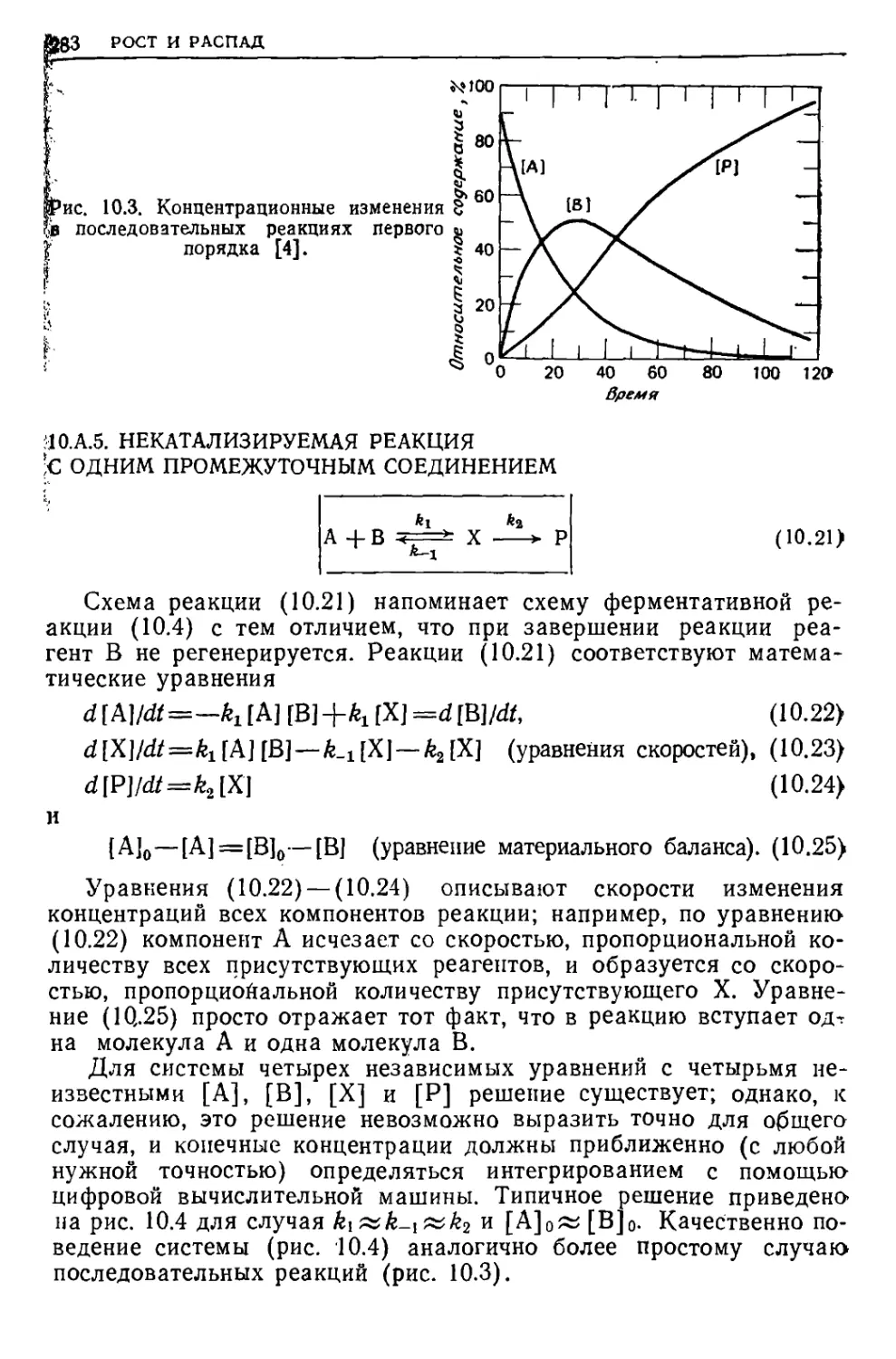
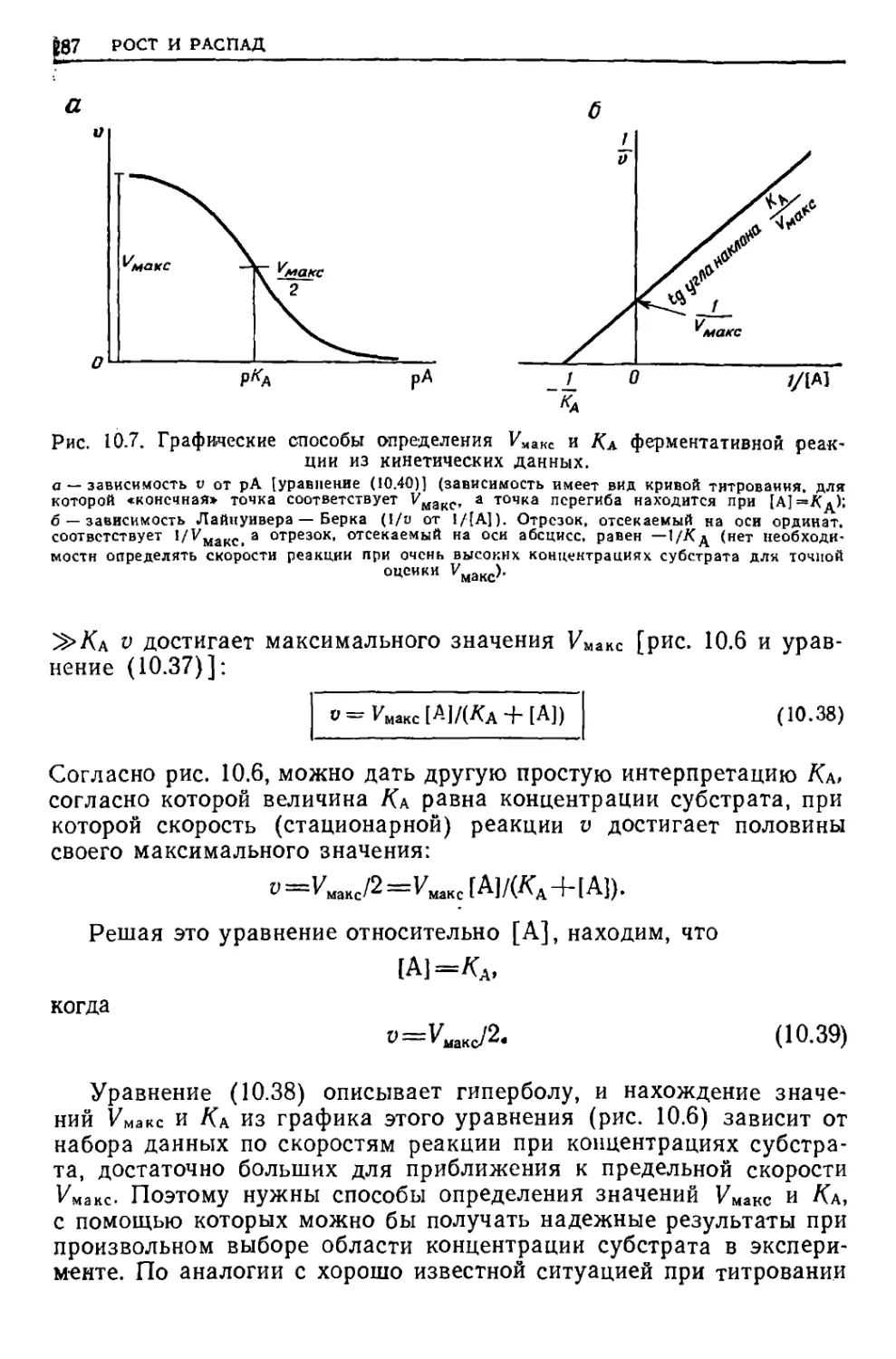
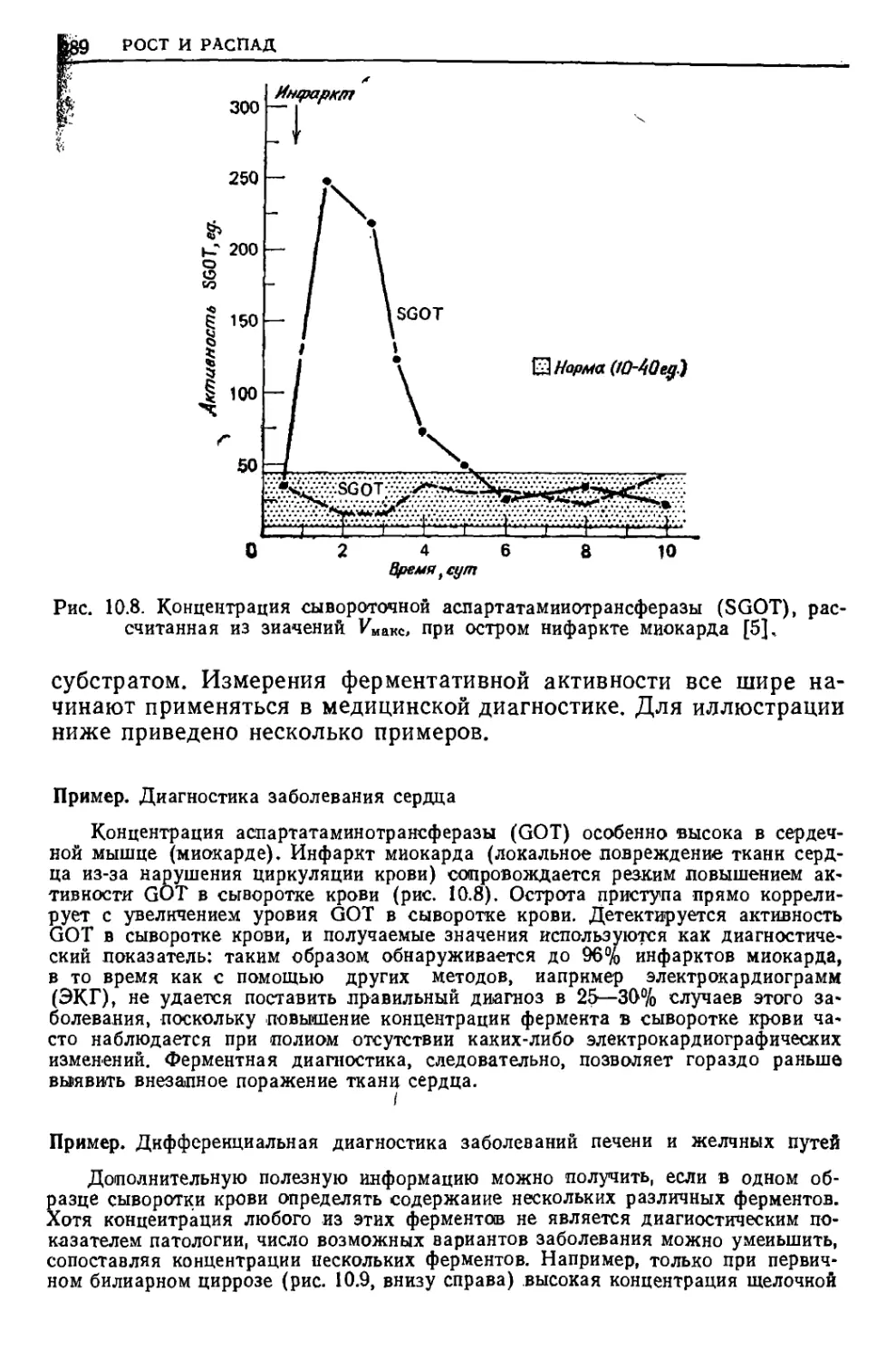
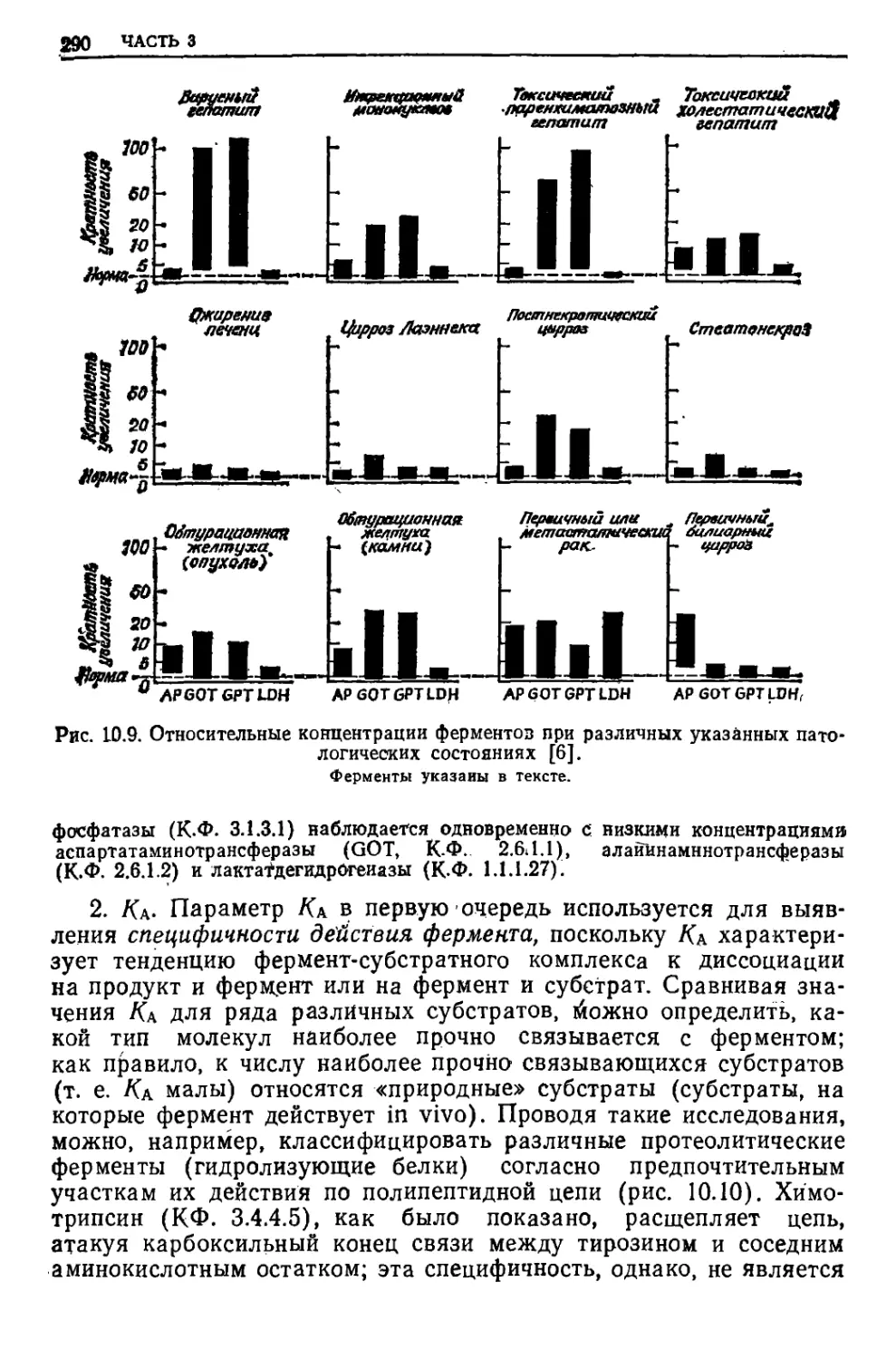
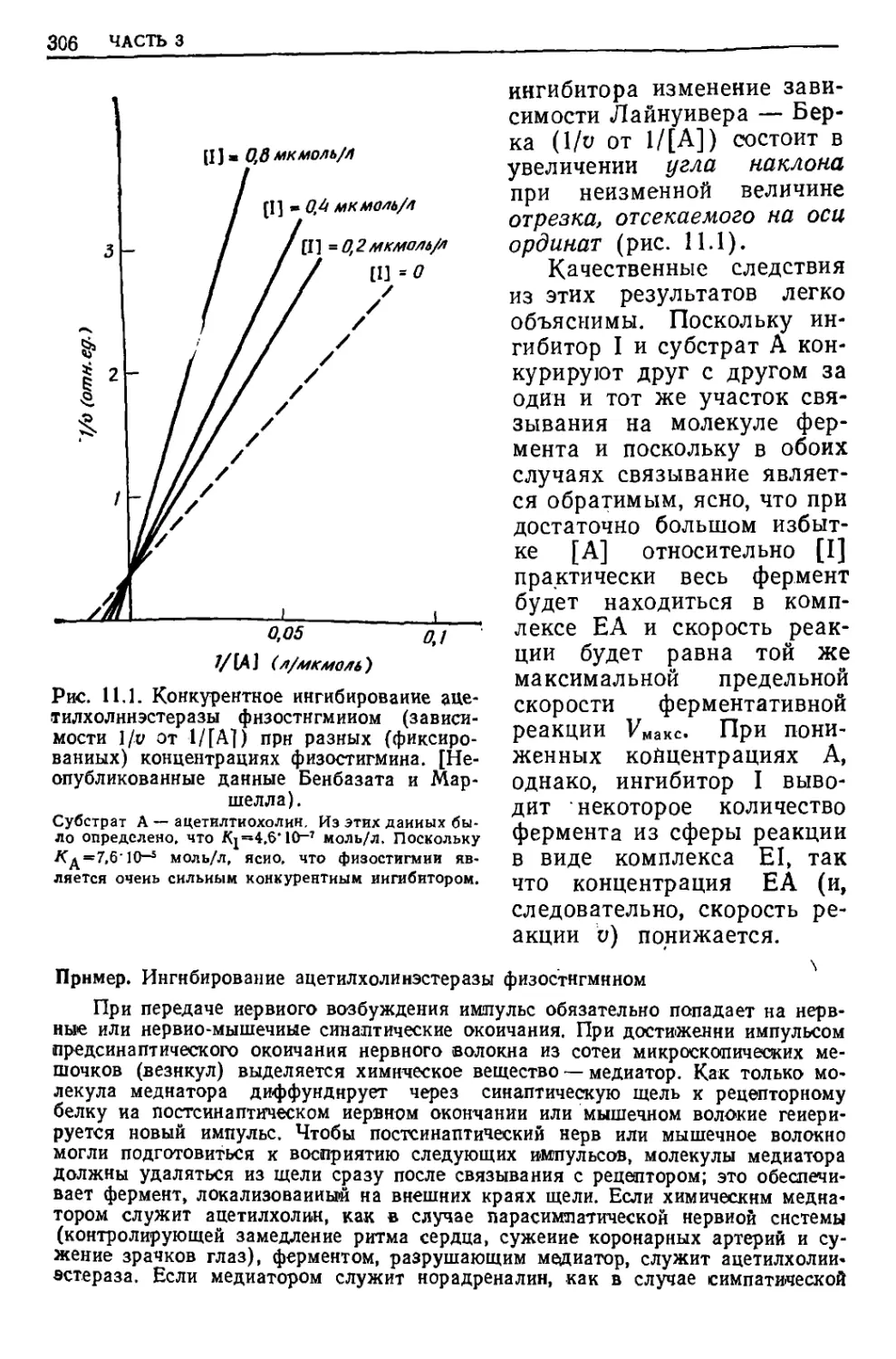
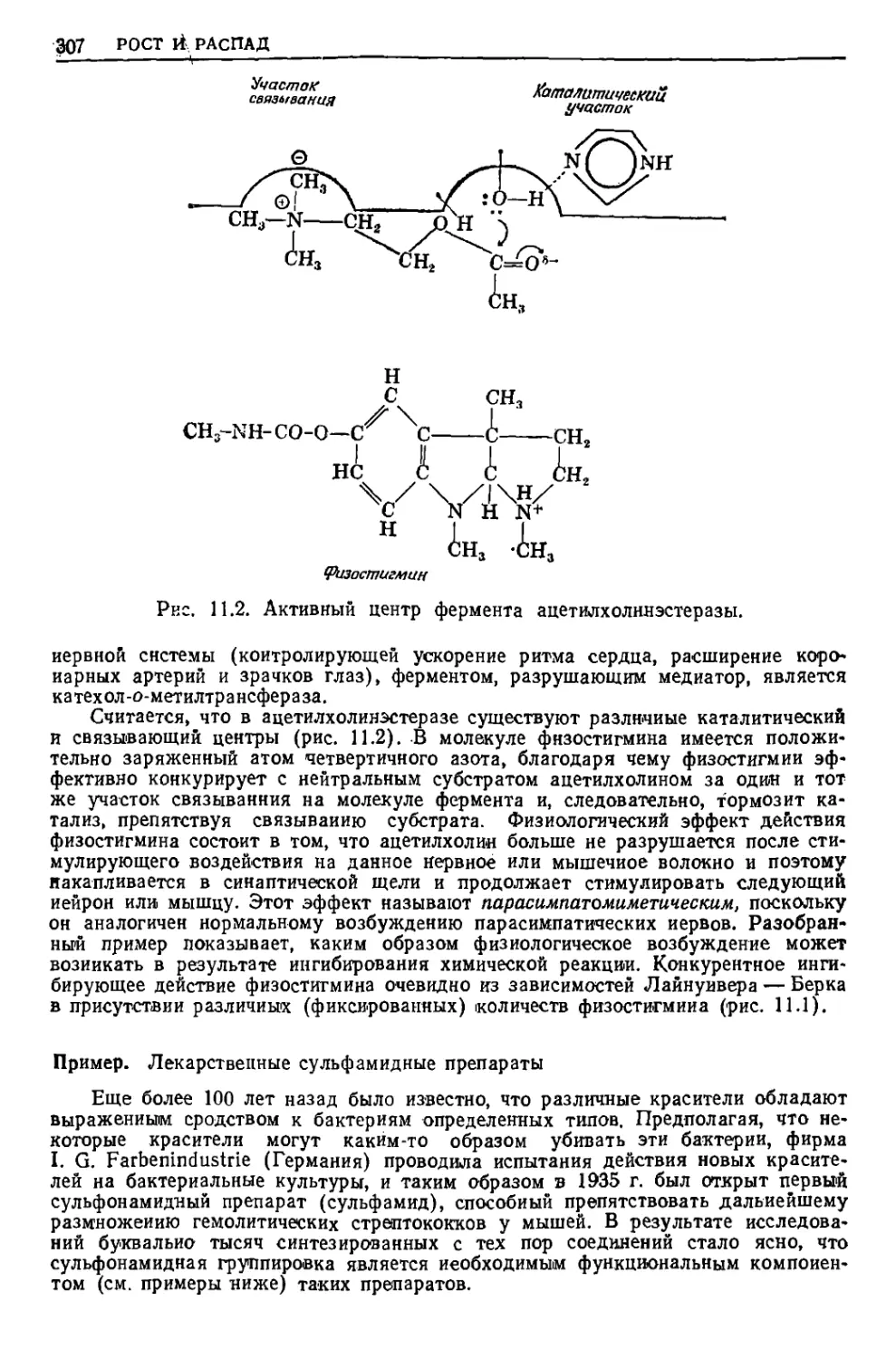
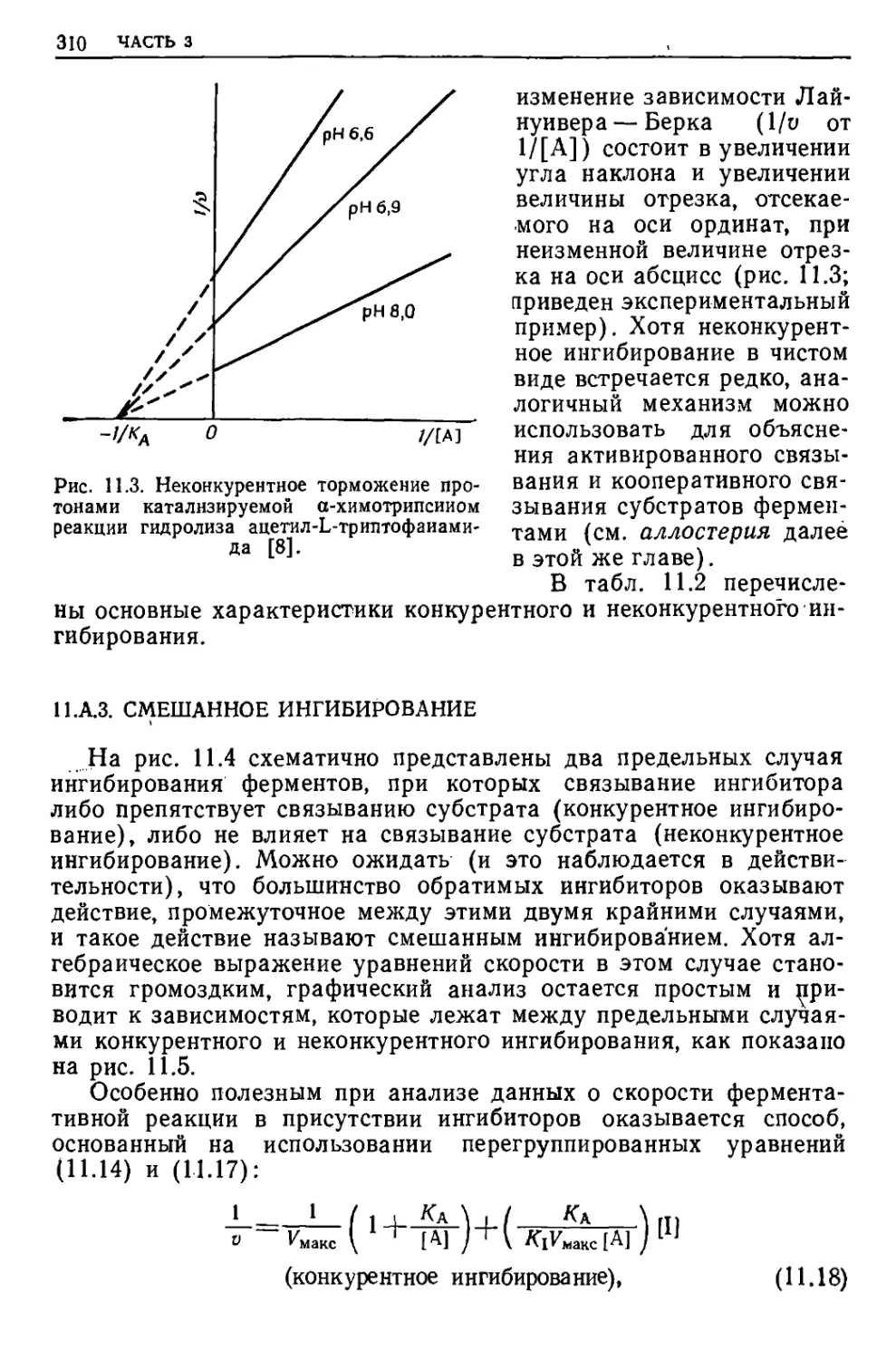
/


































































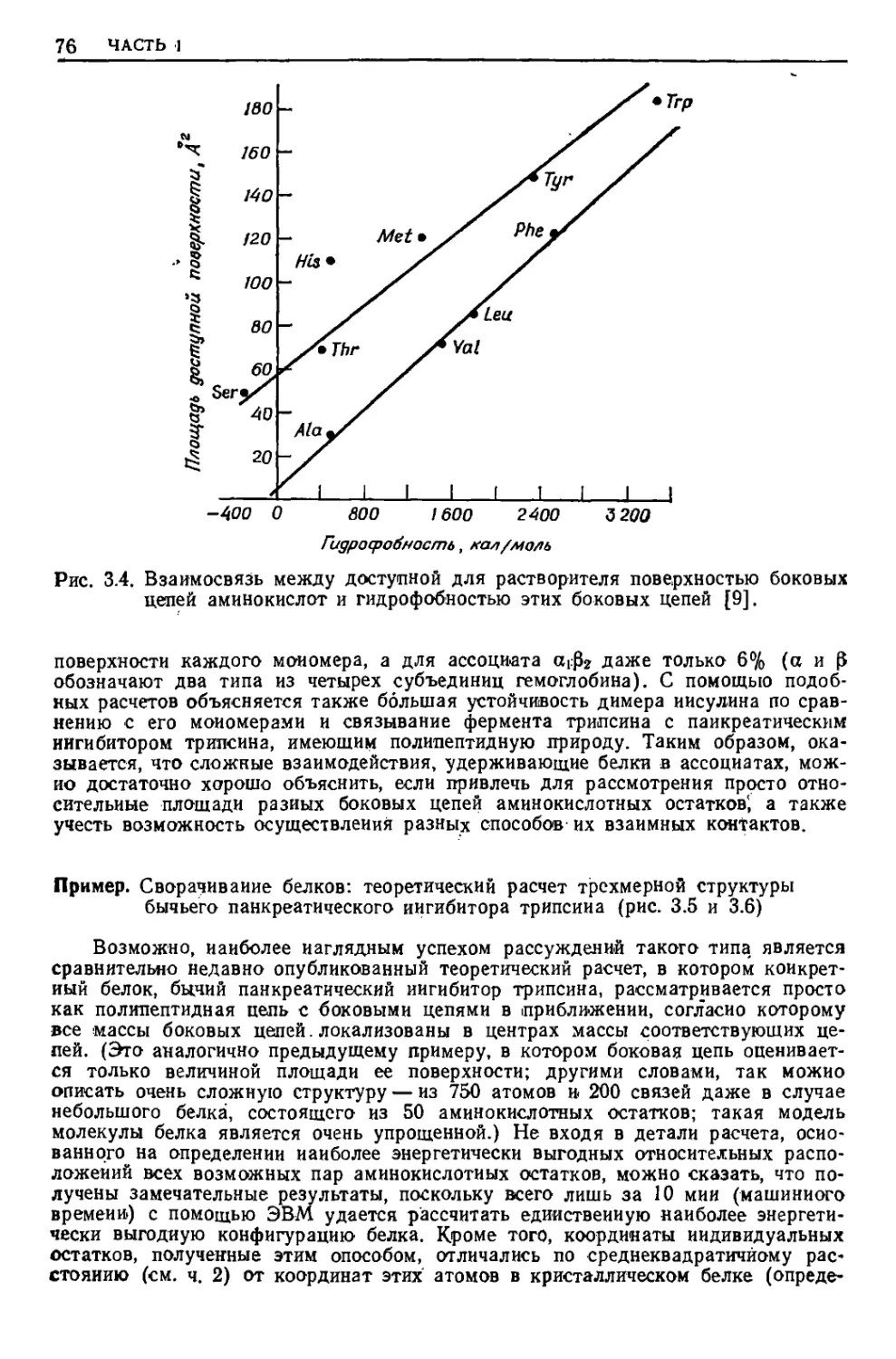
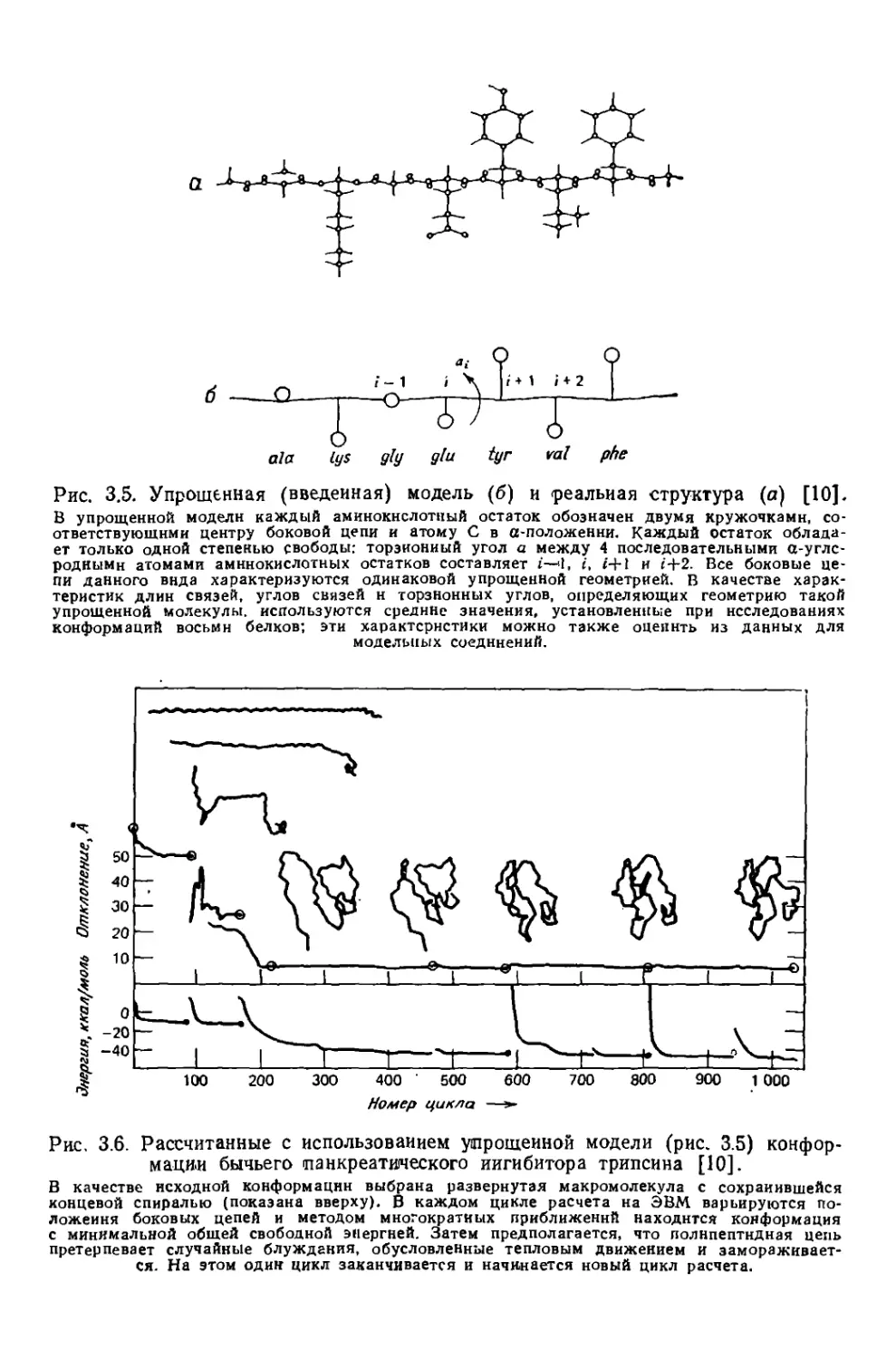
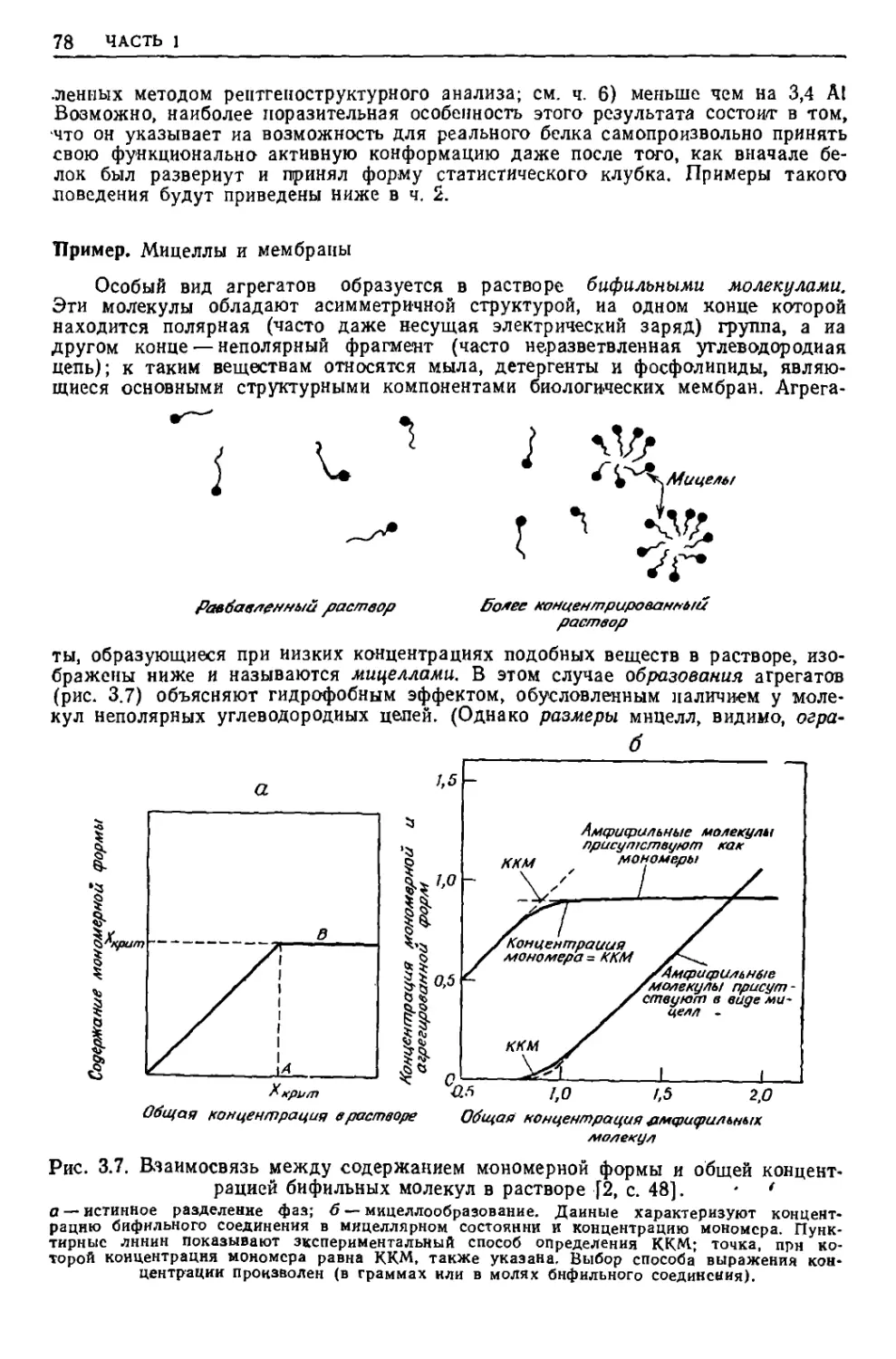
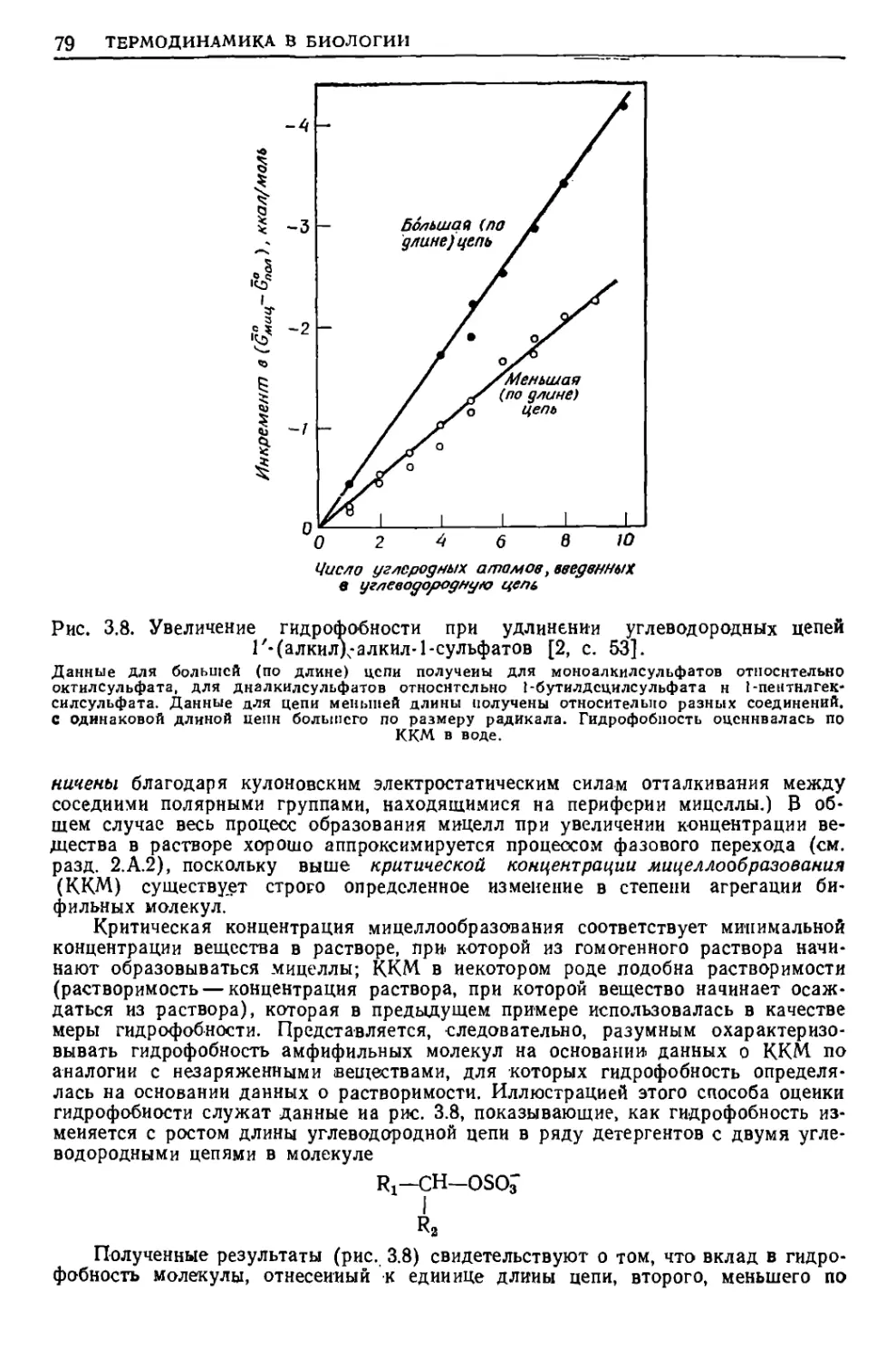











































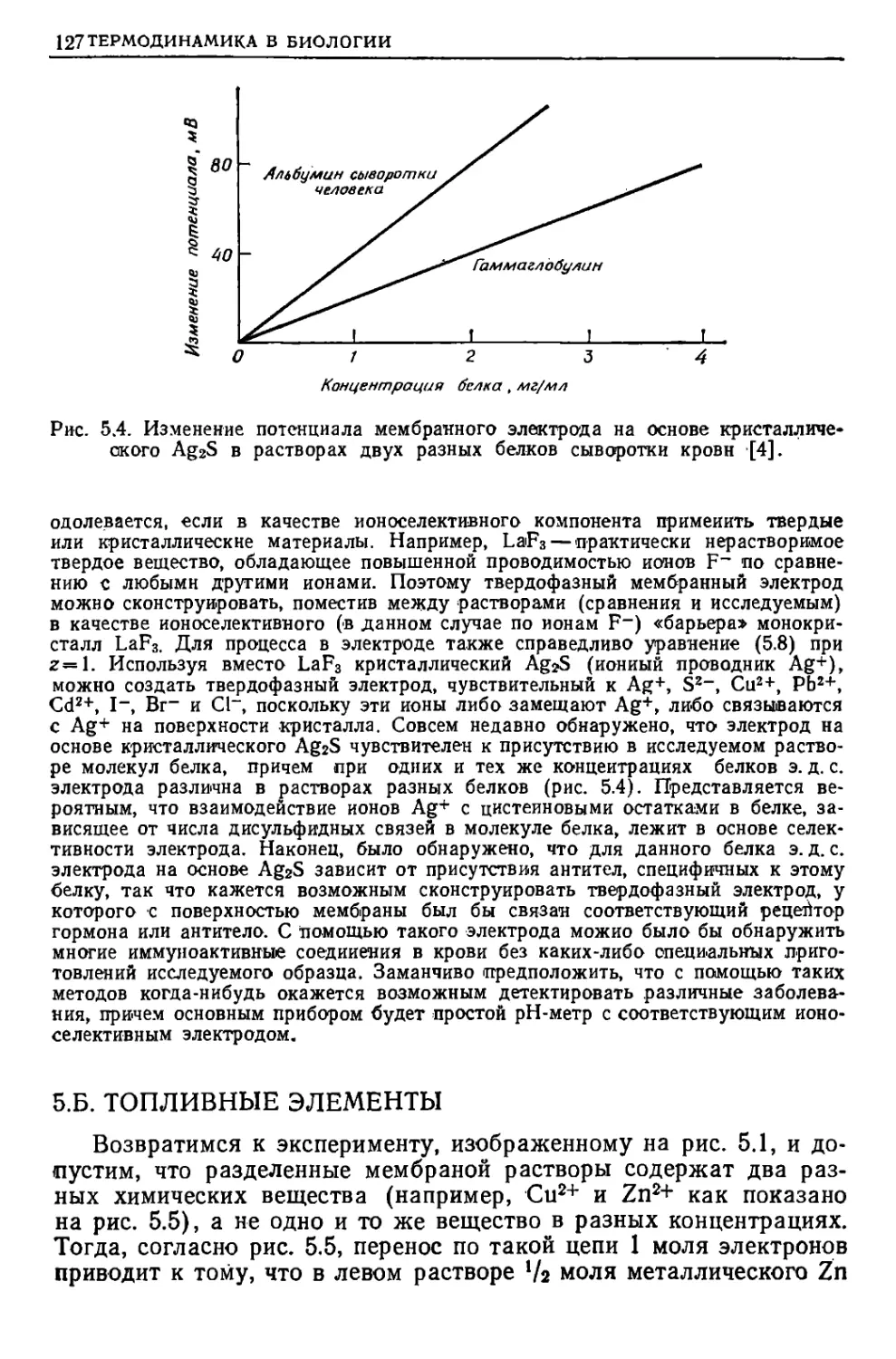
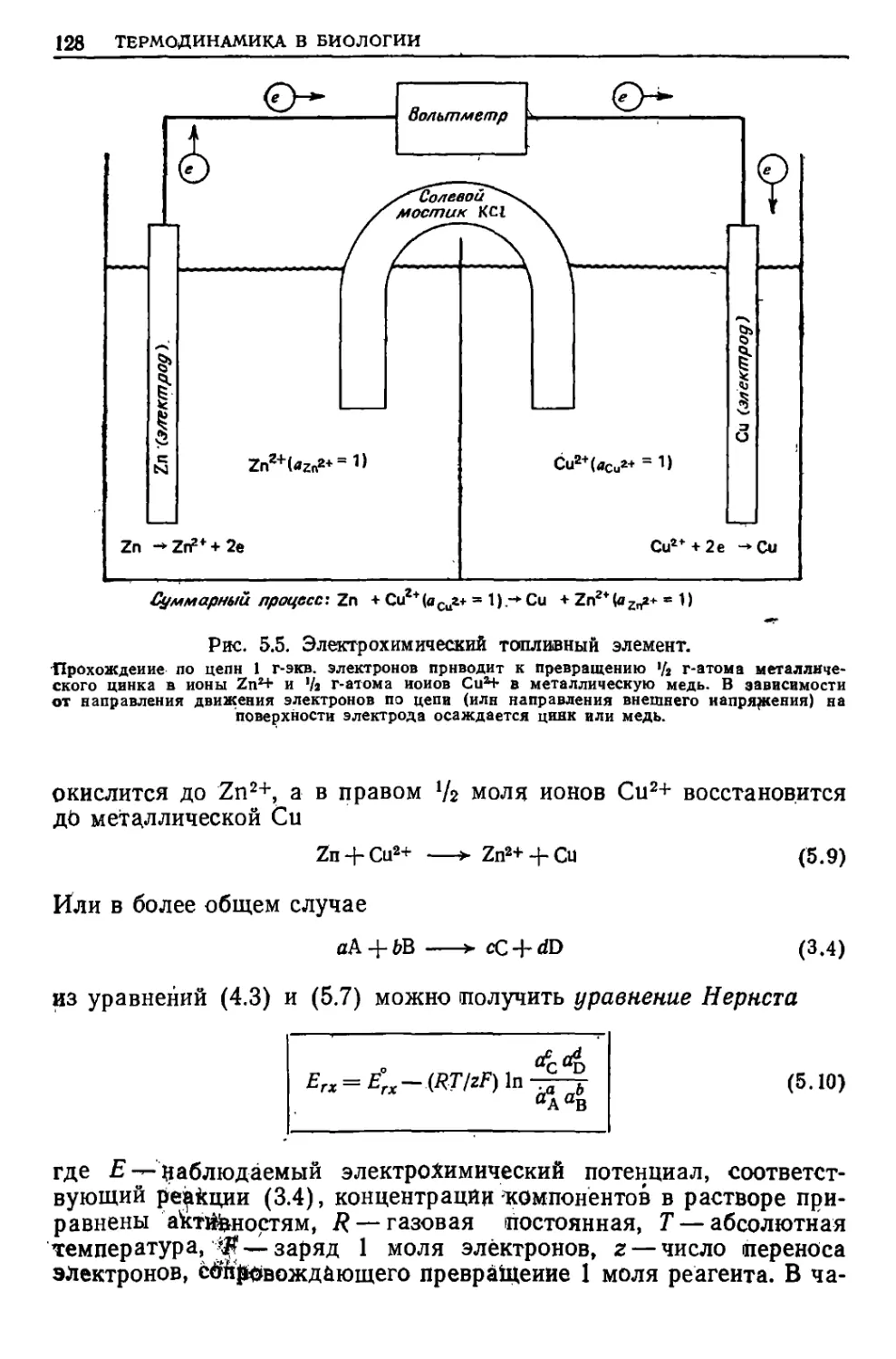

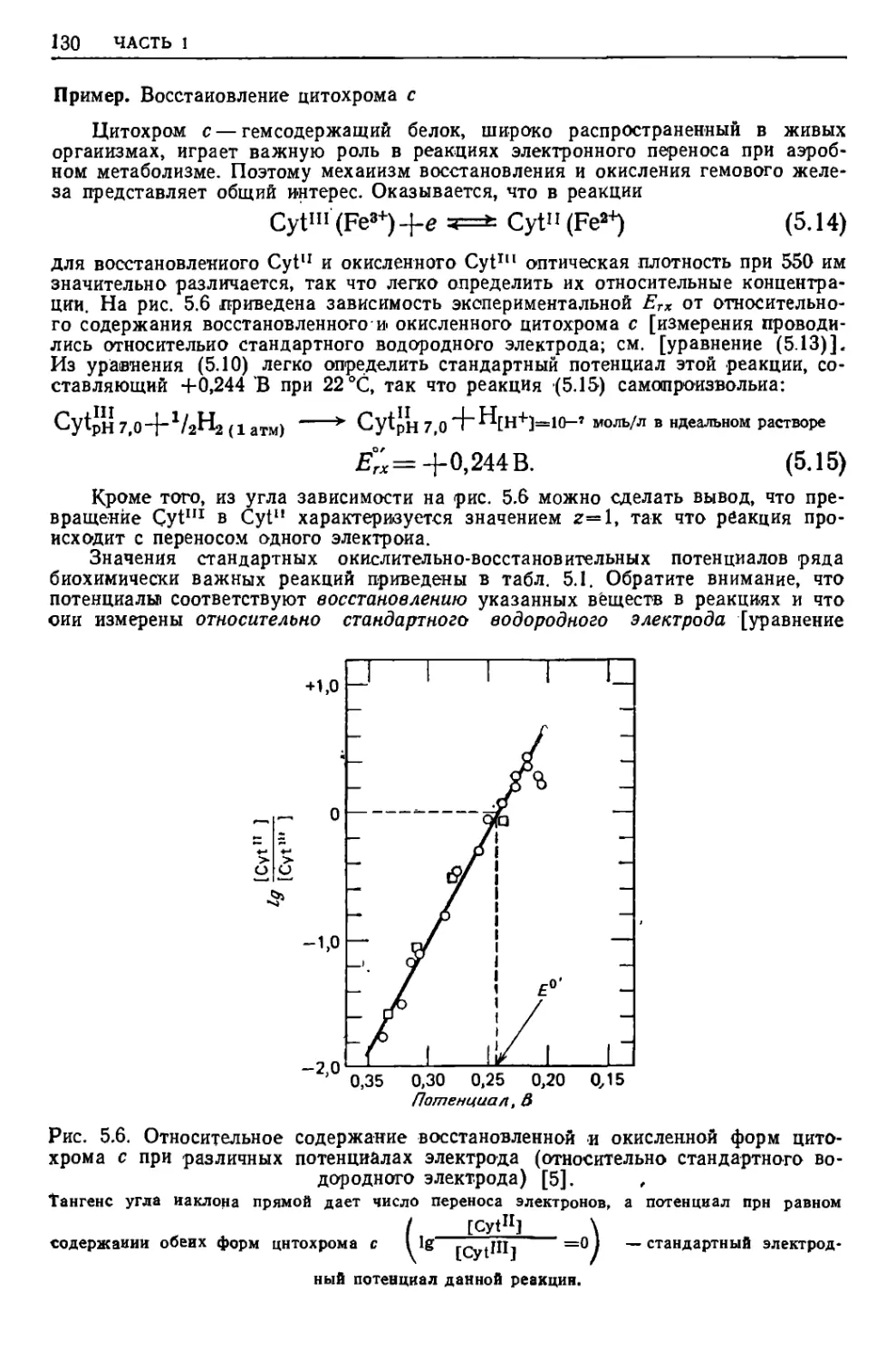







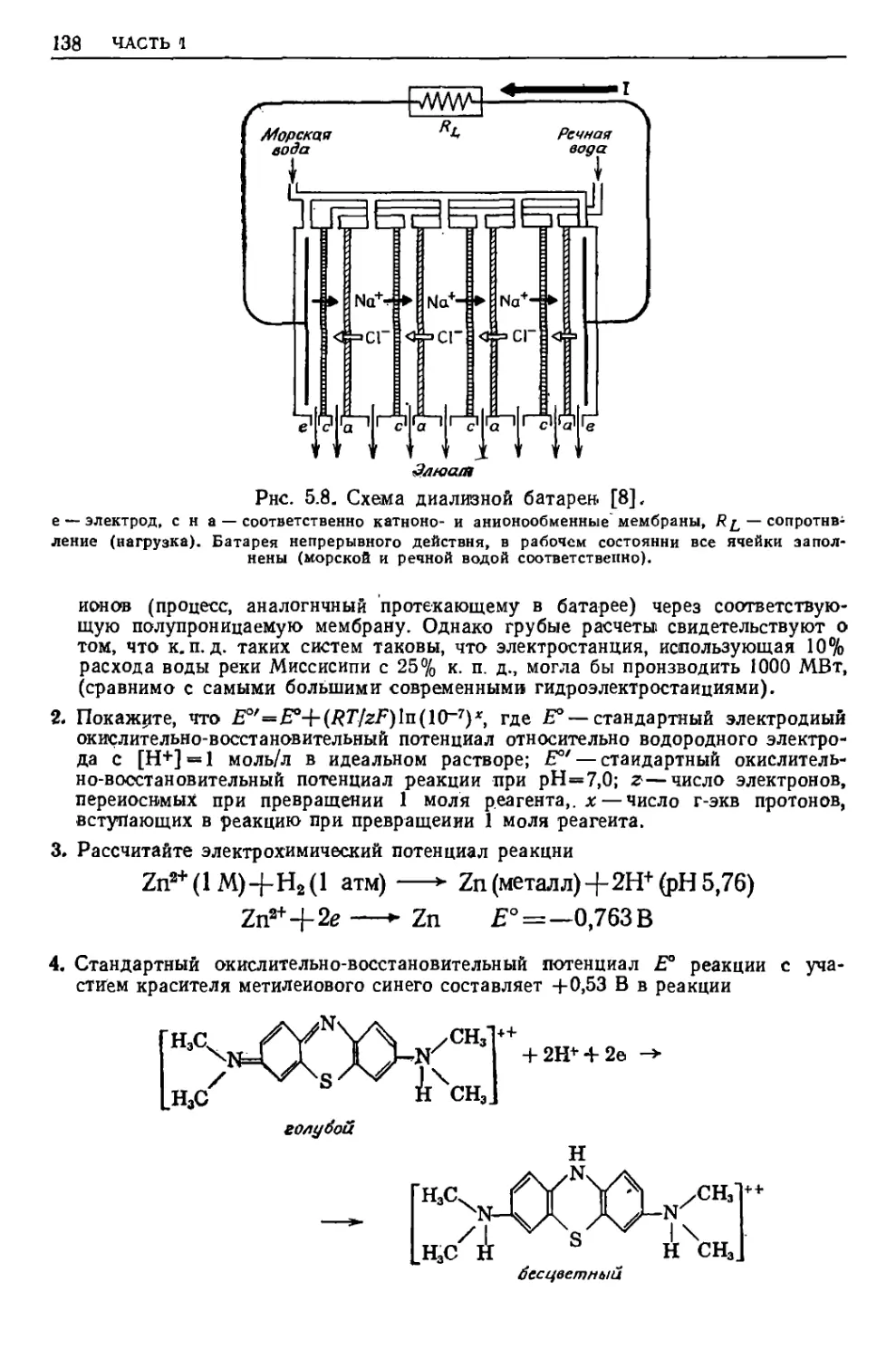















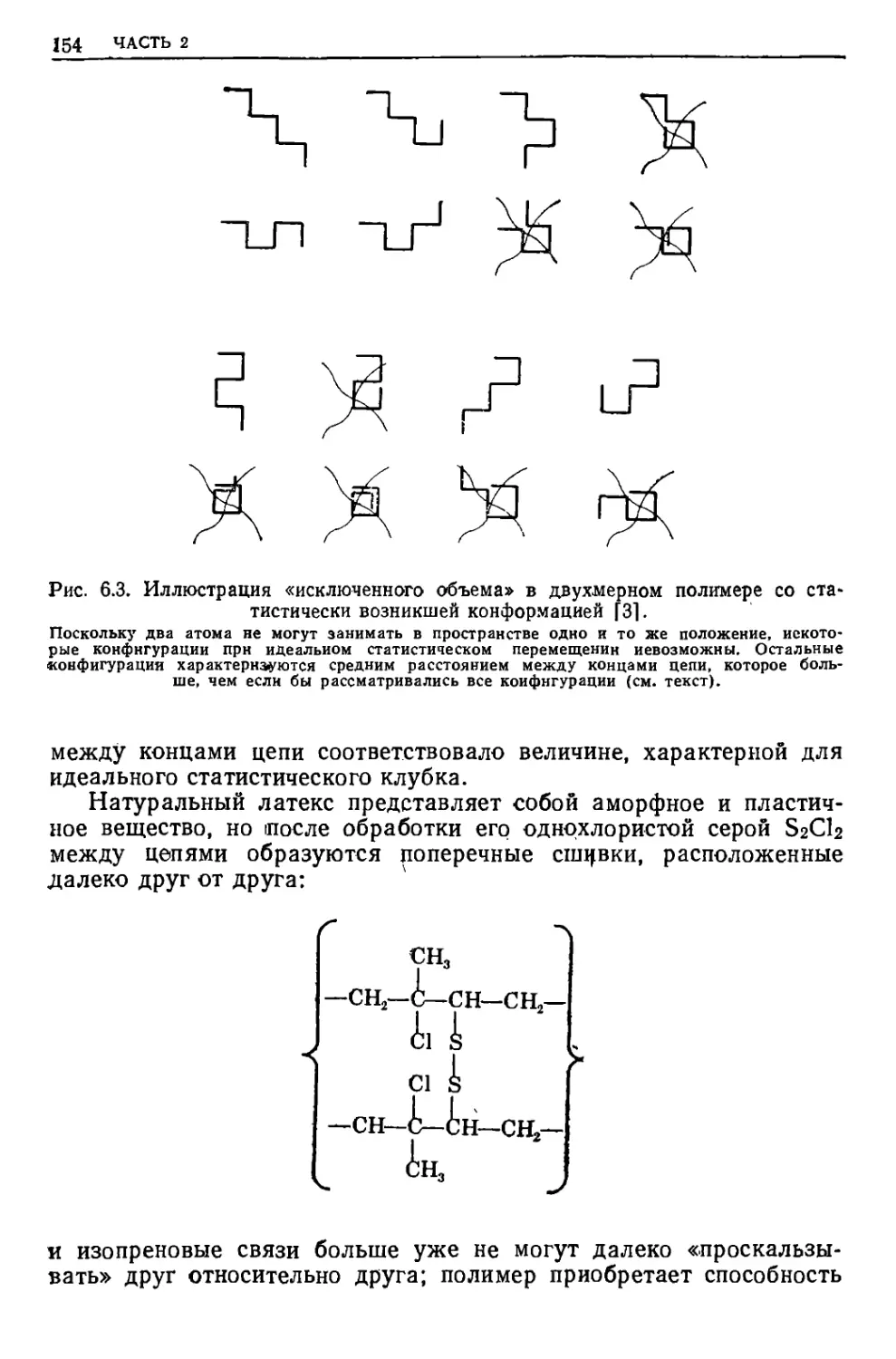






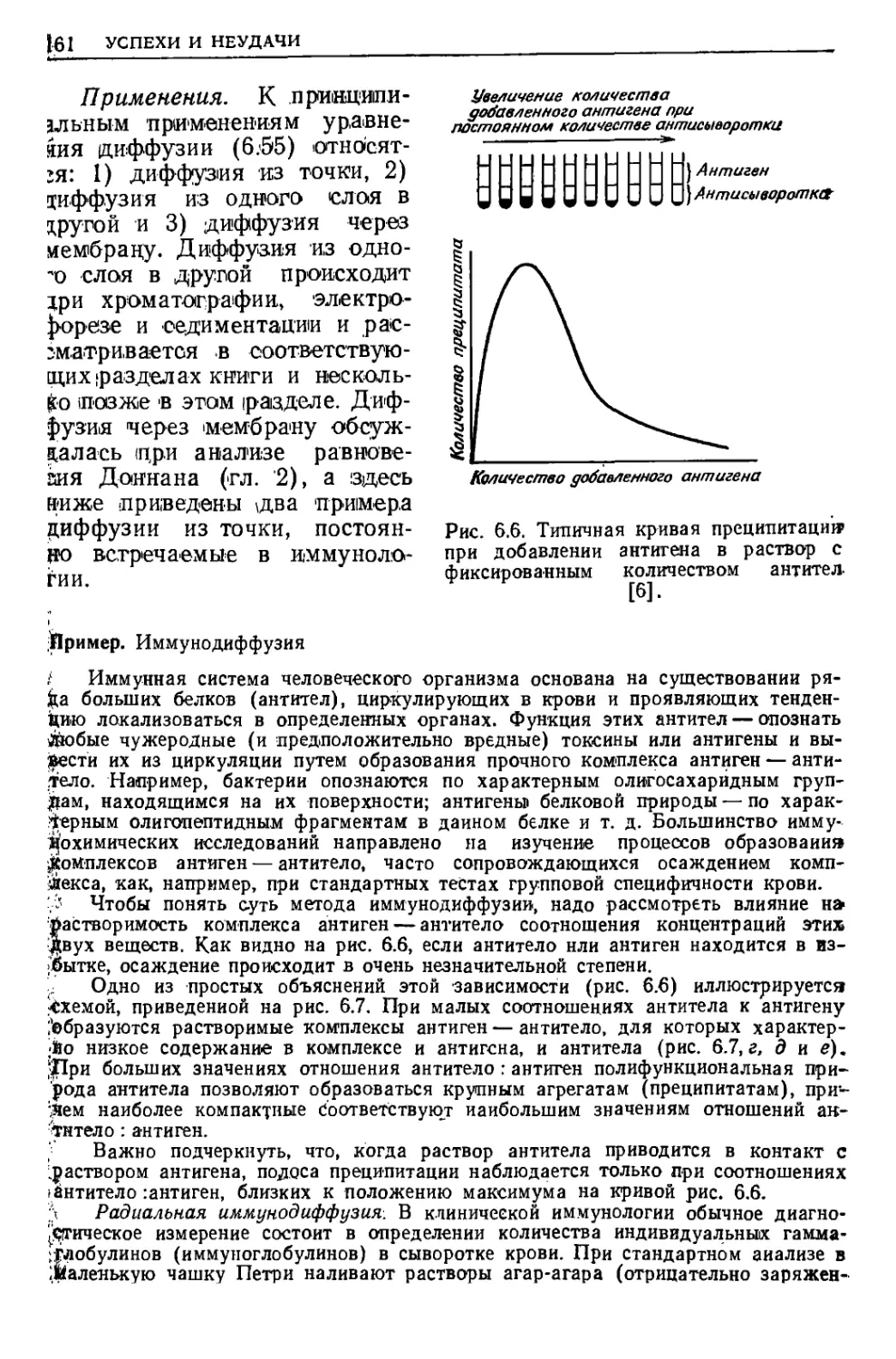


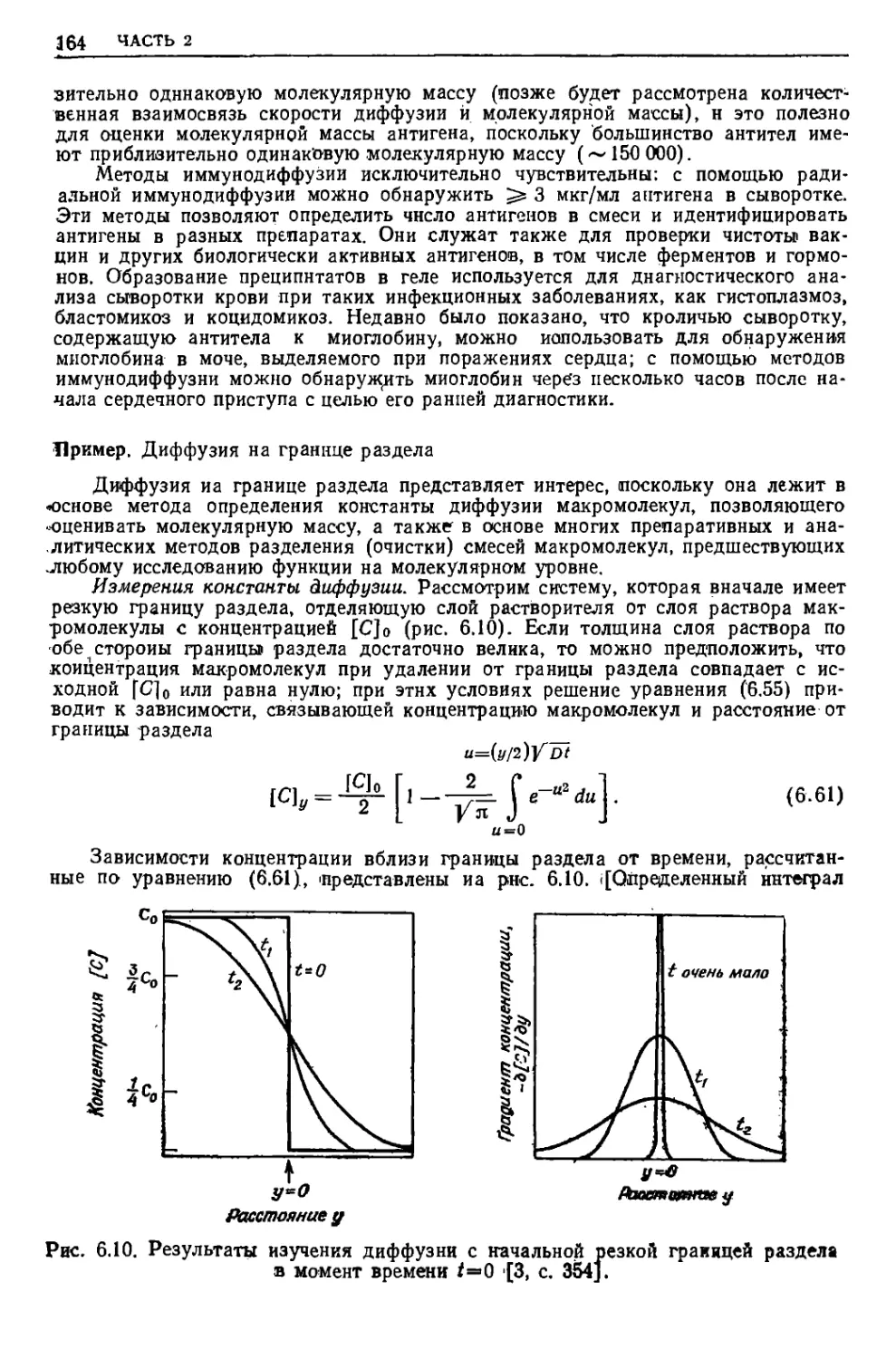










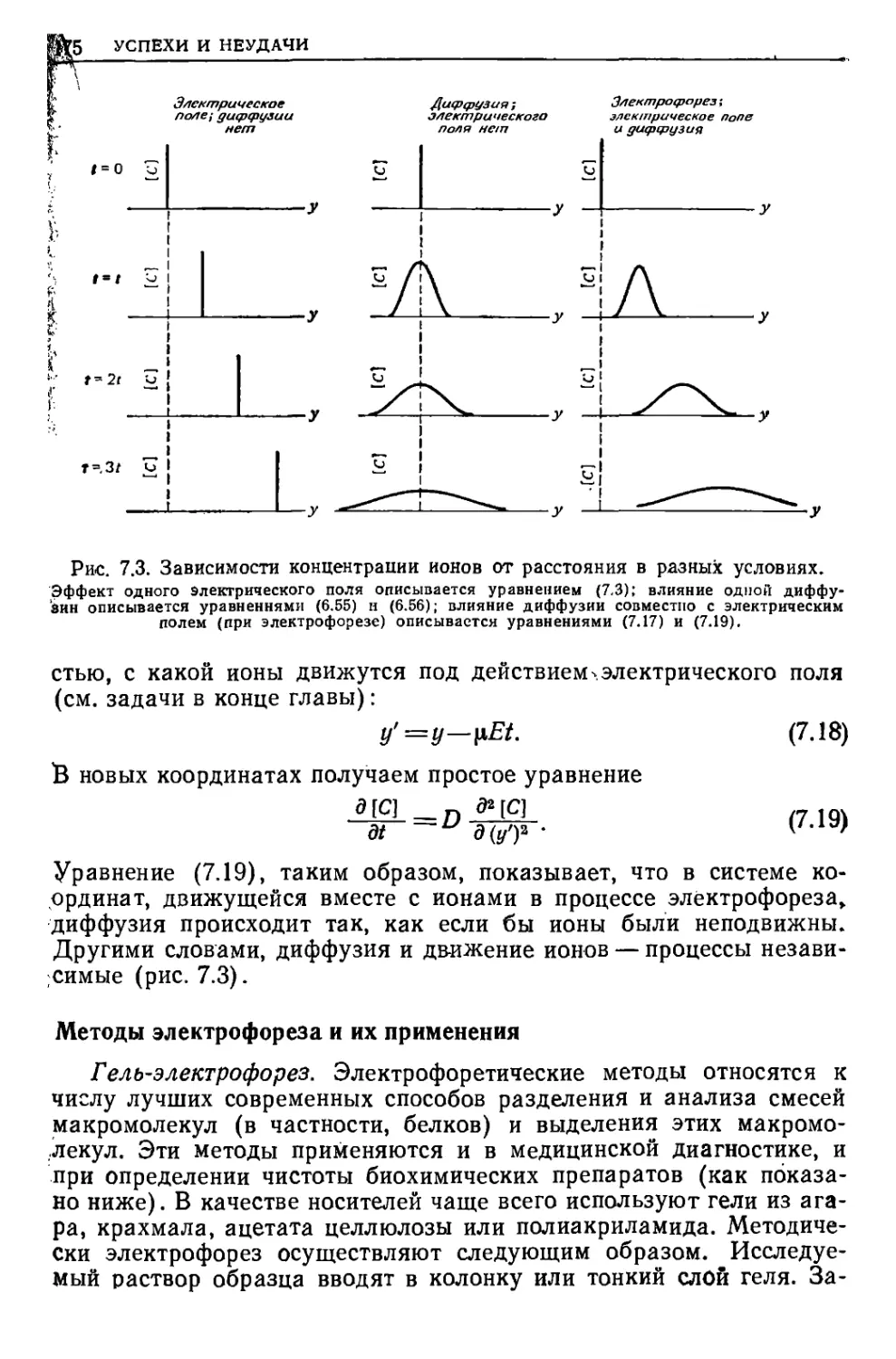

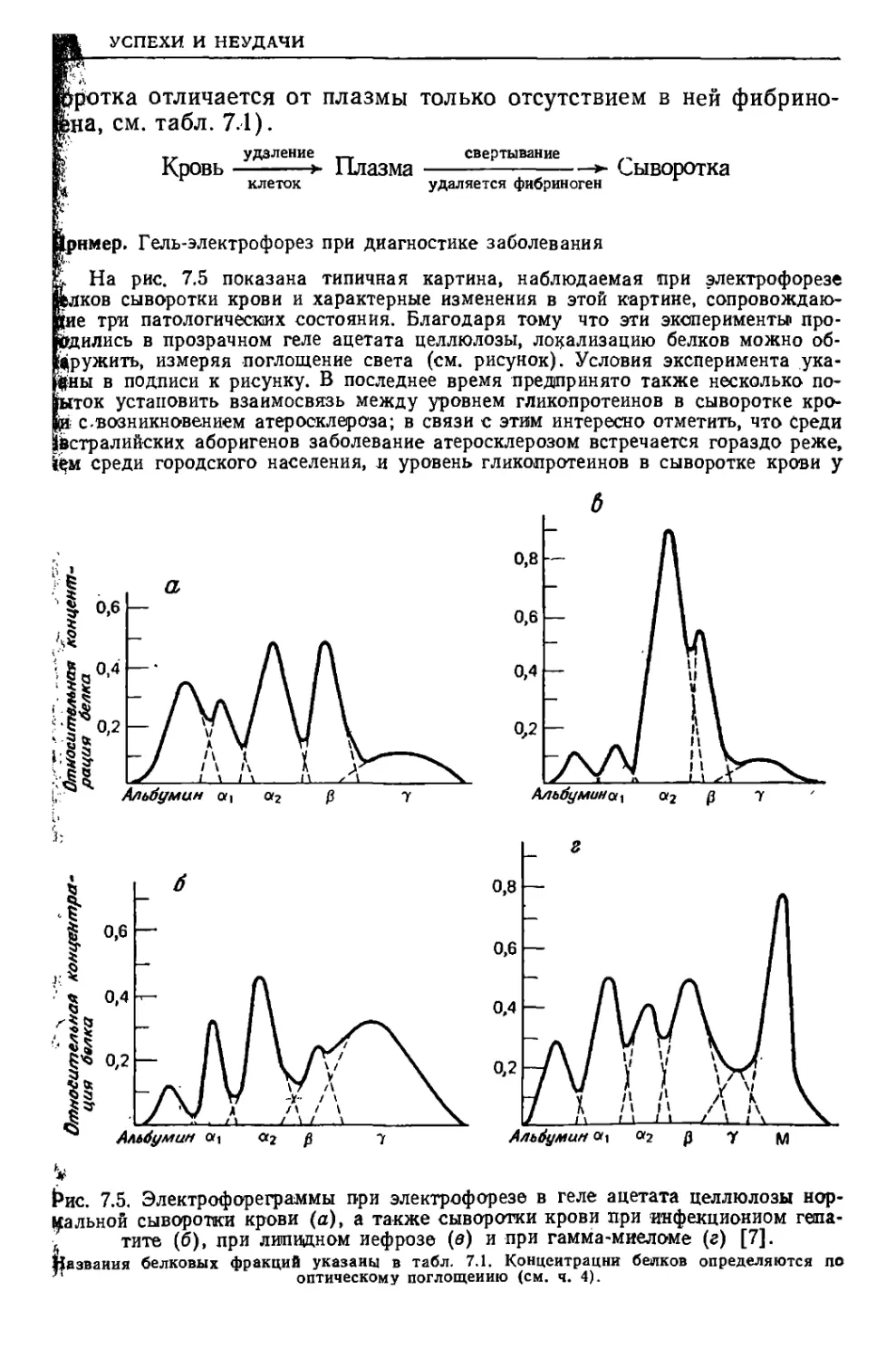
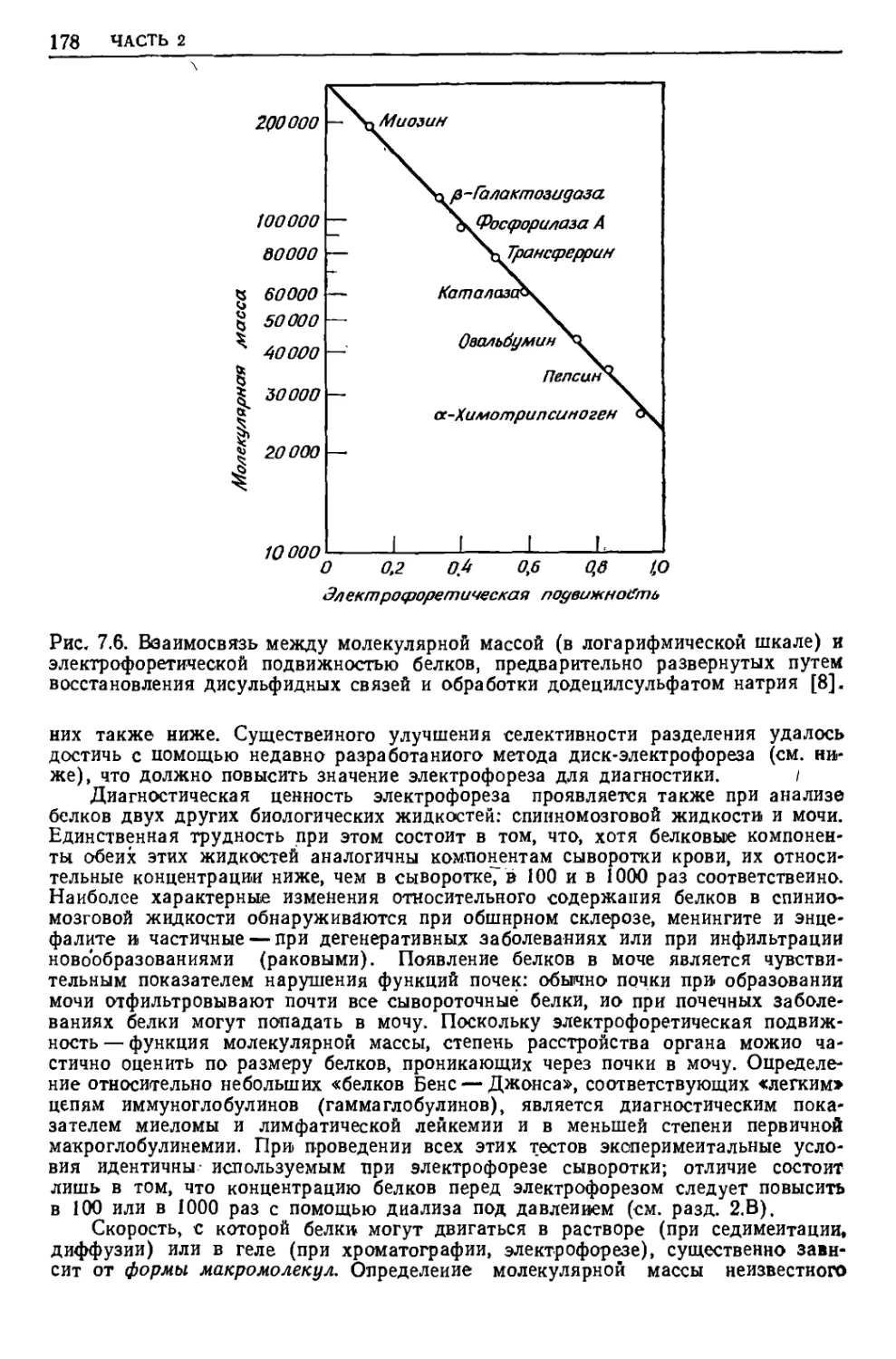


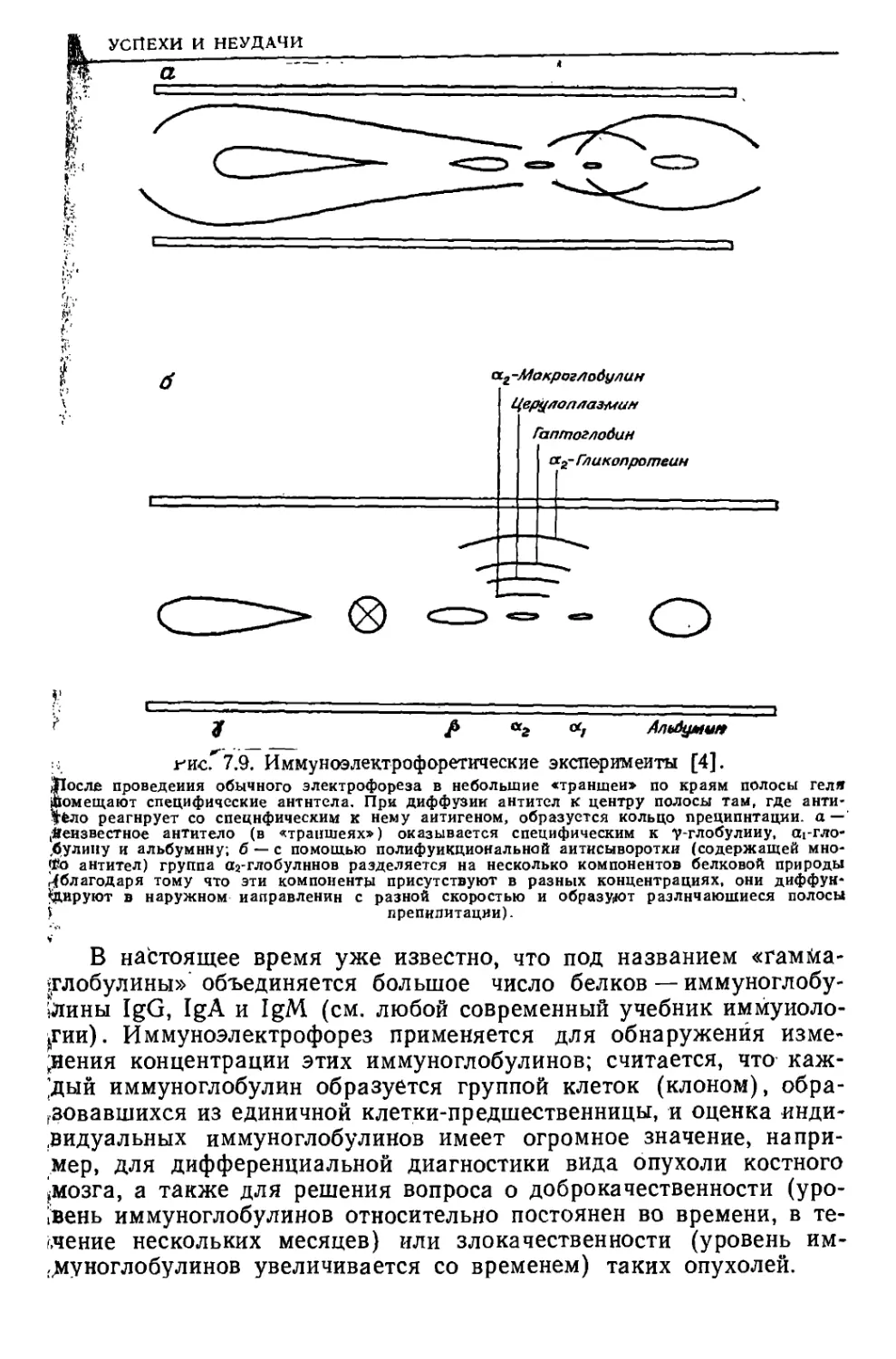




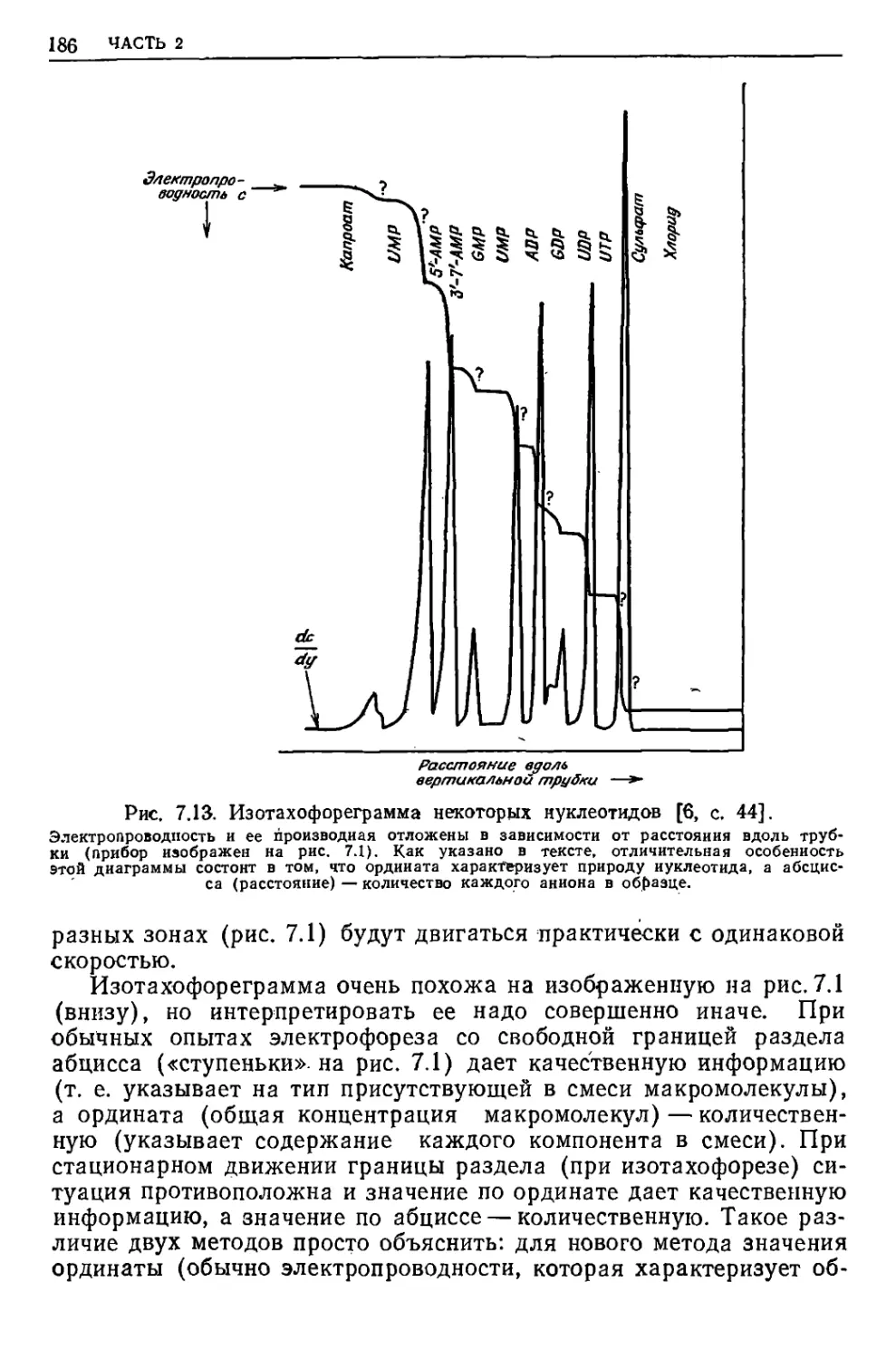
























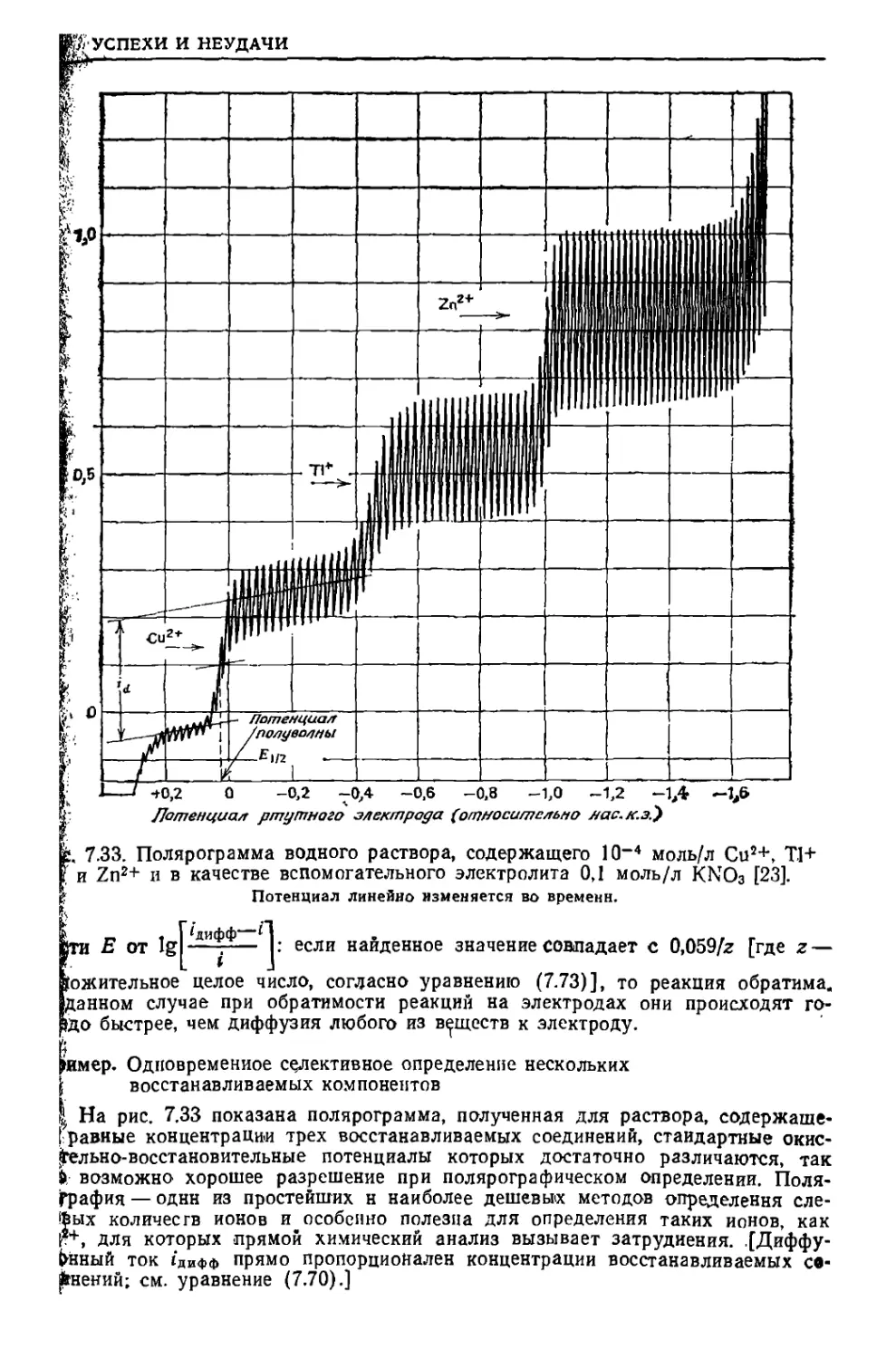







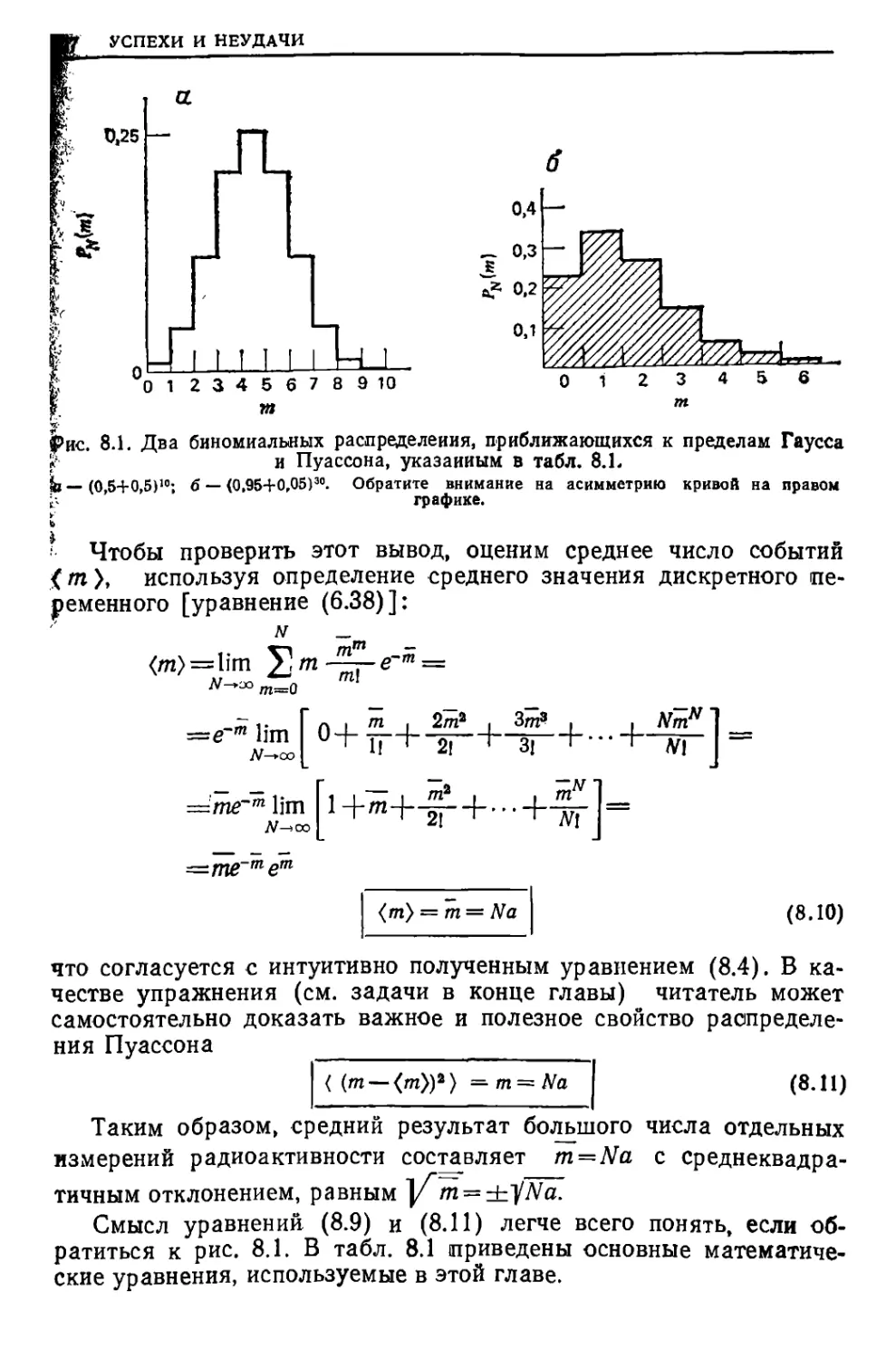








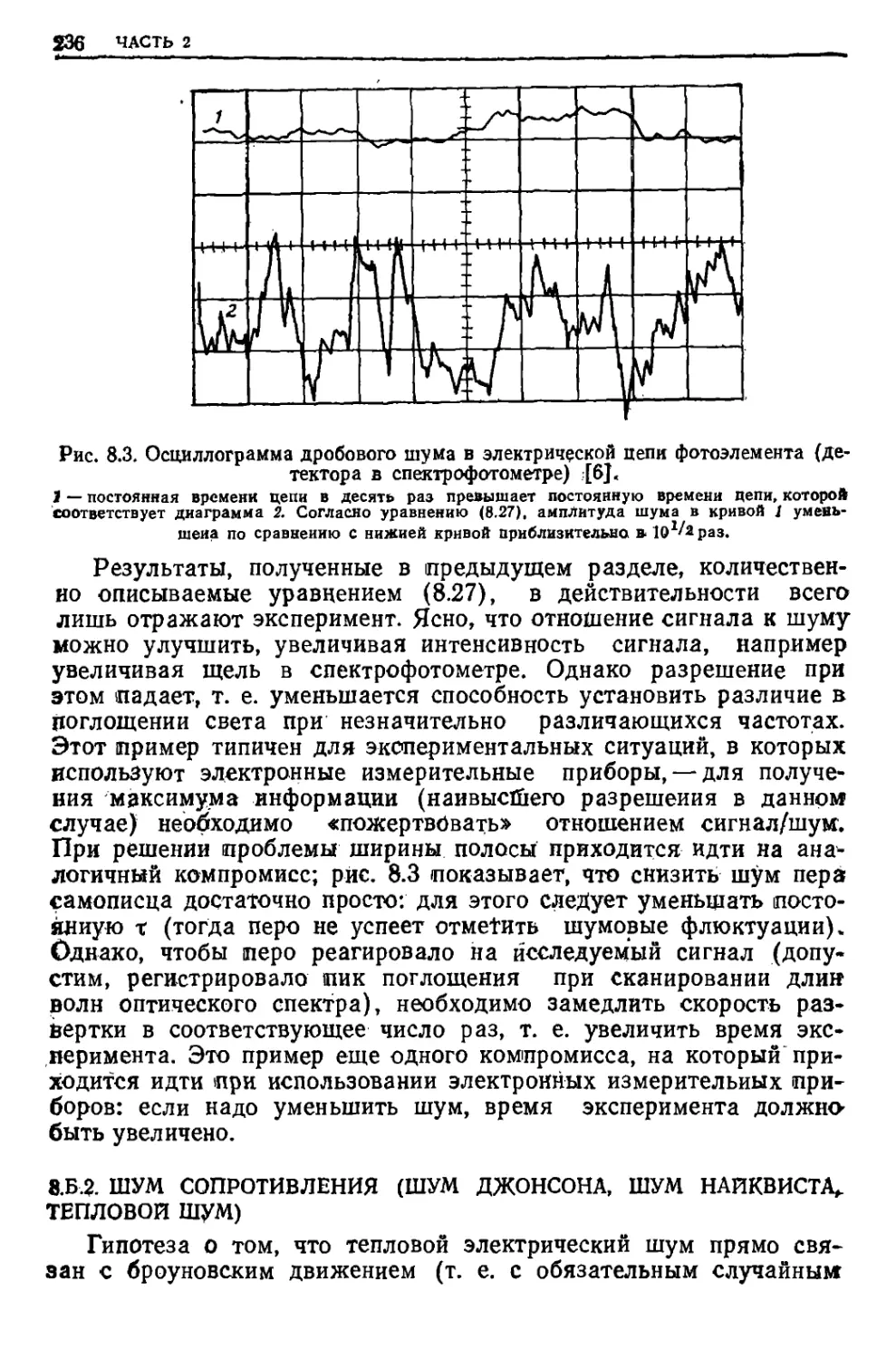

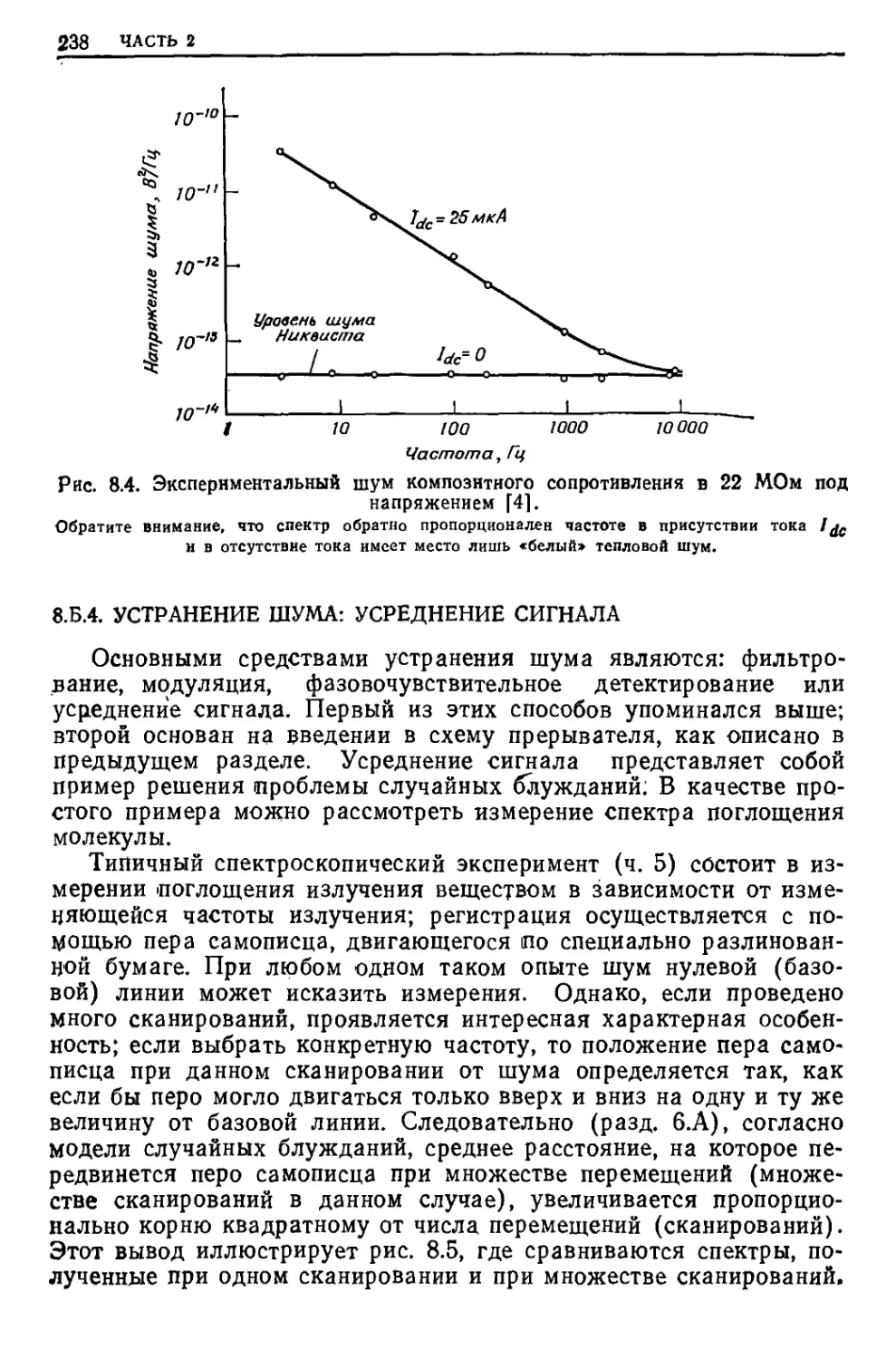






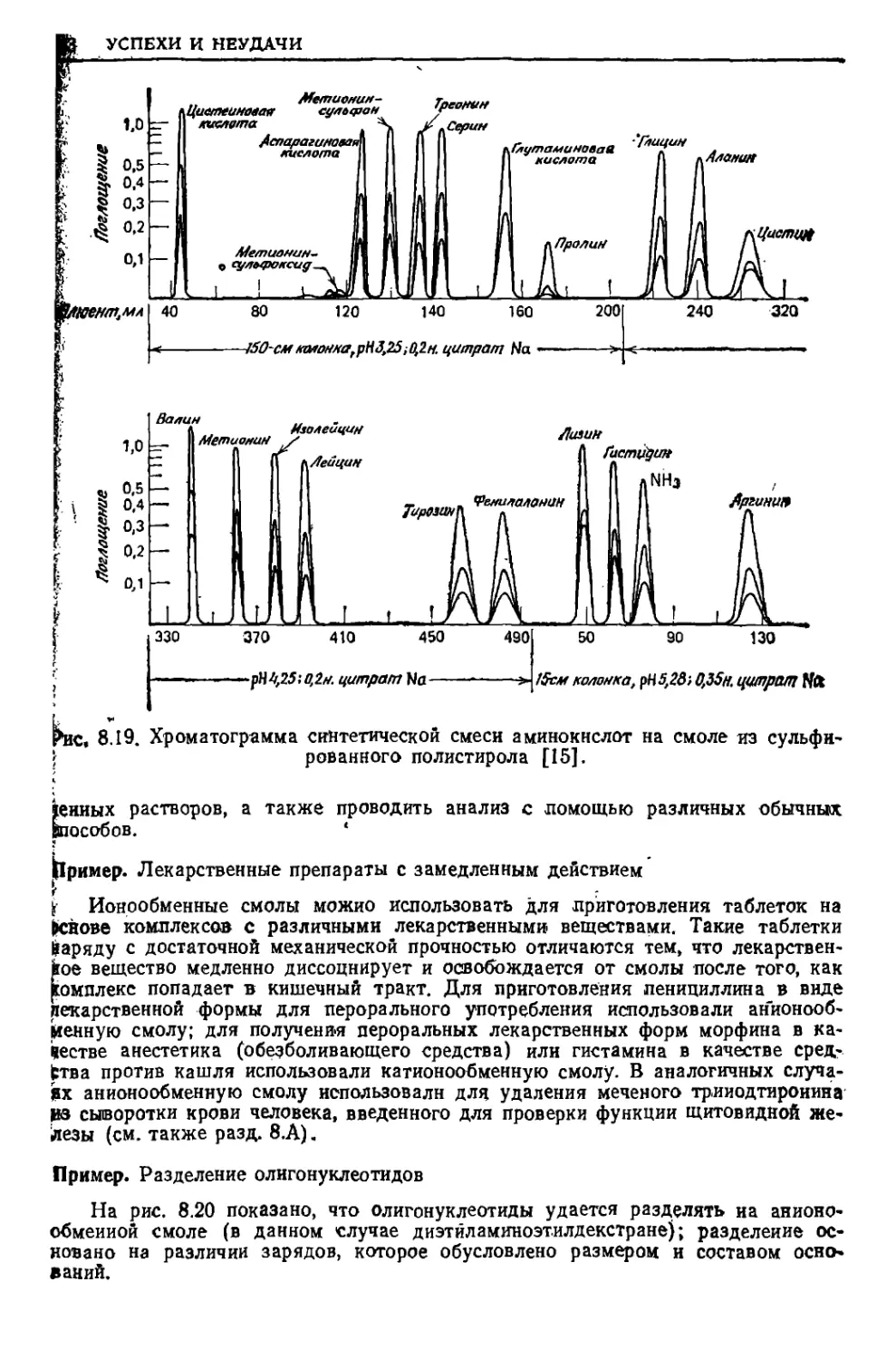
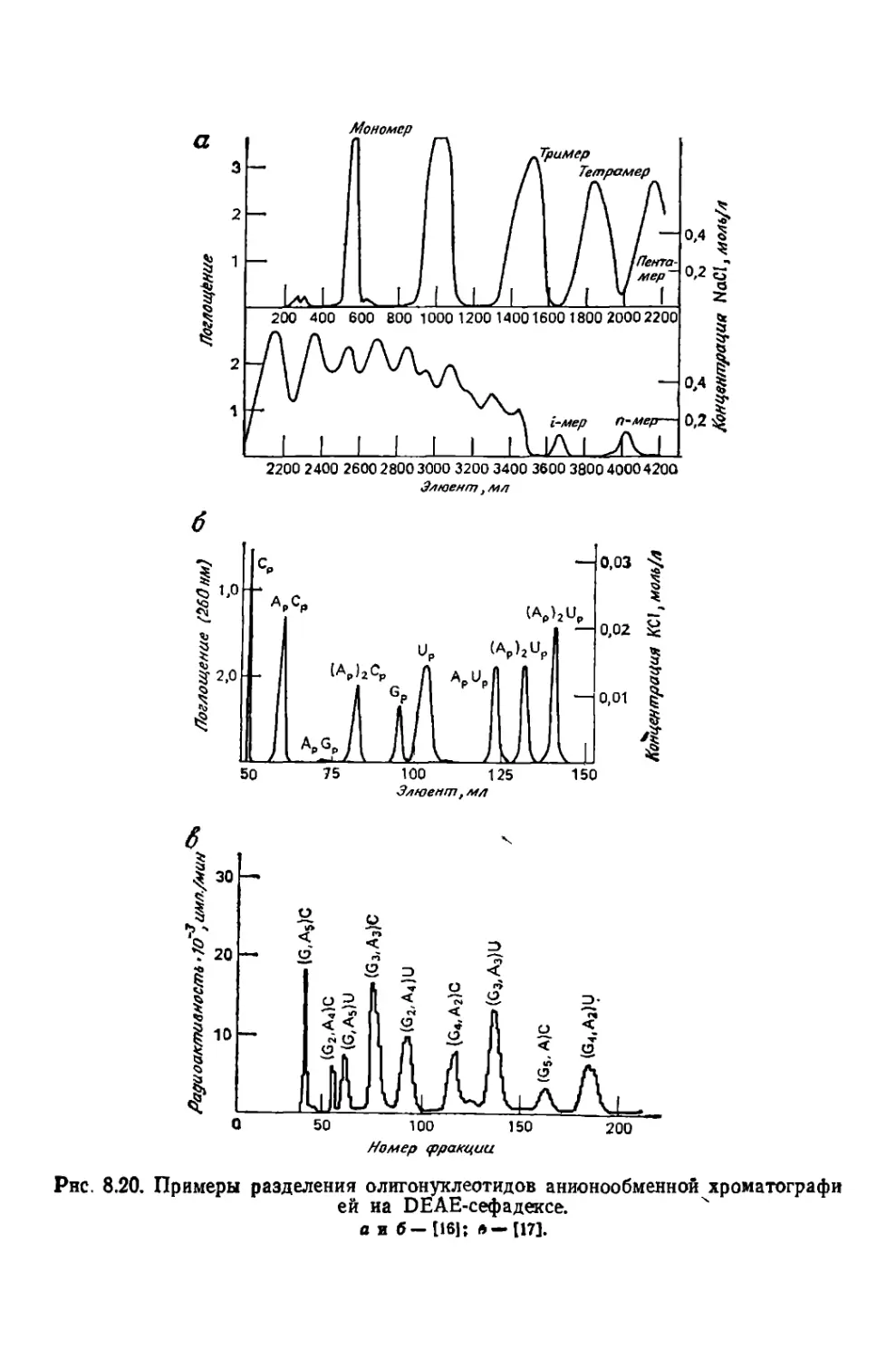






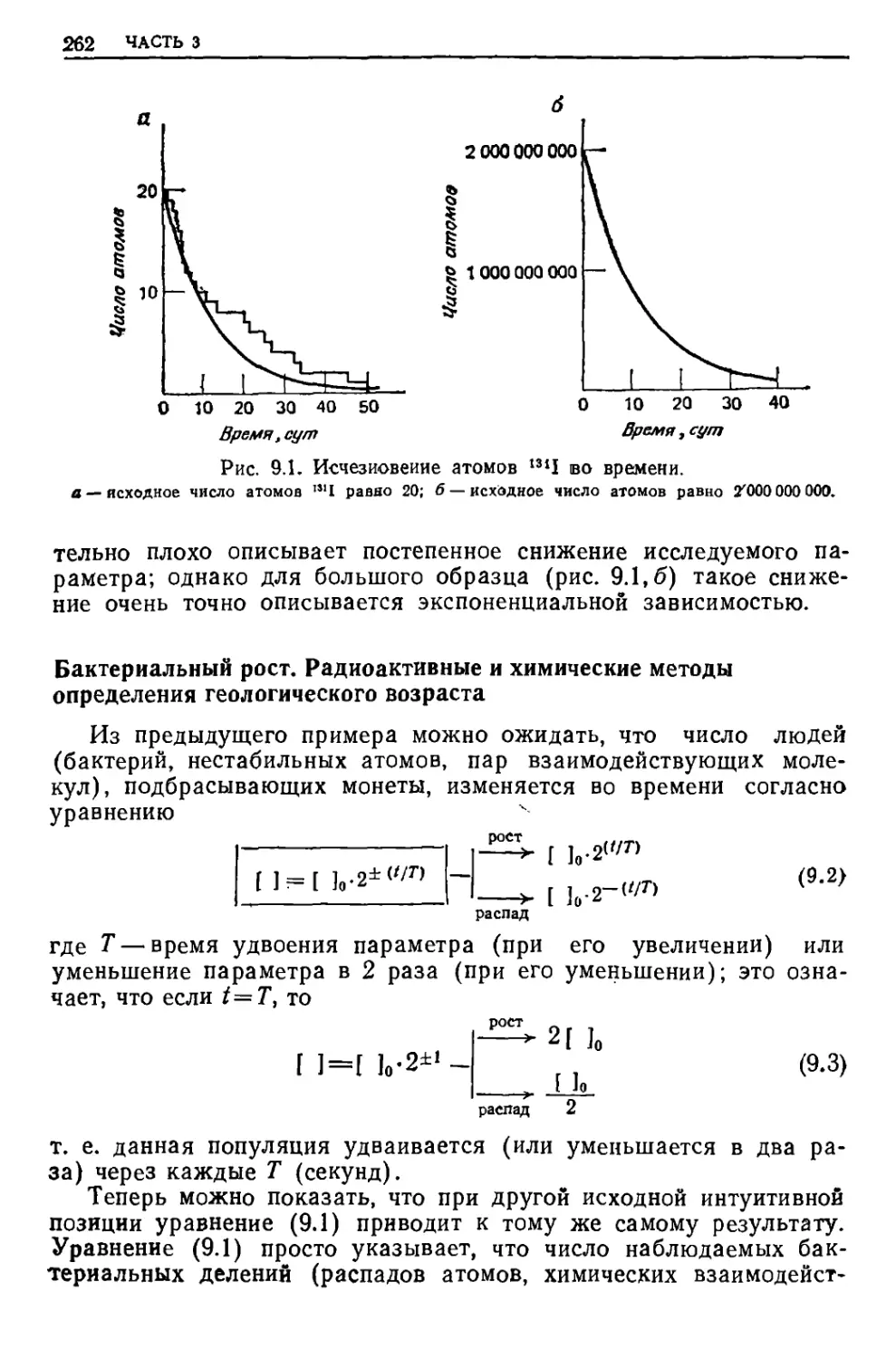

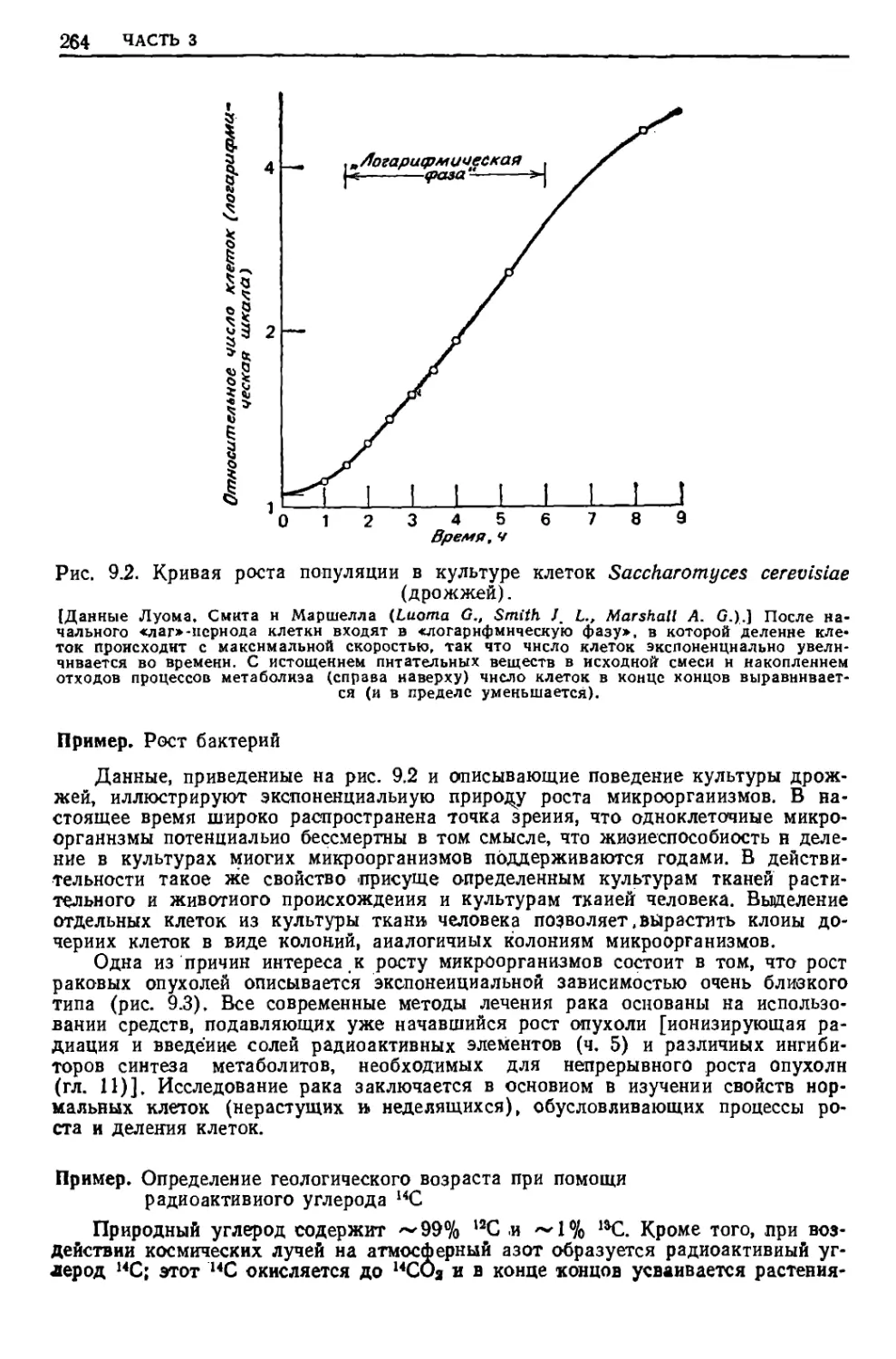

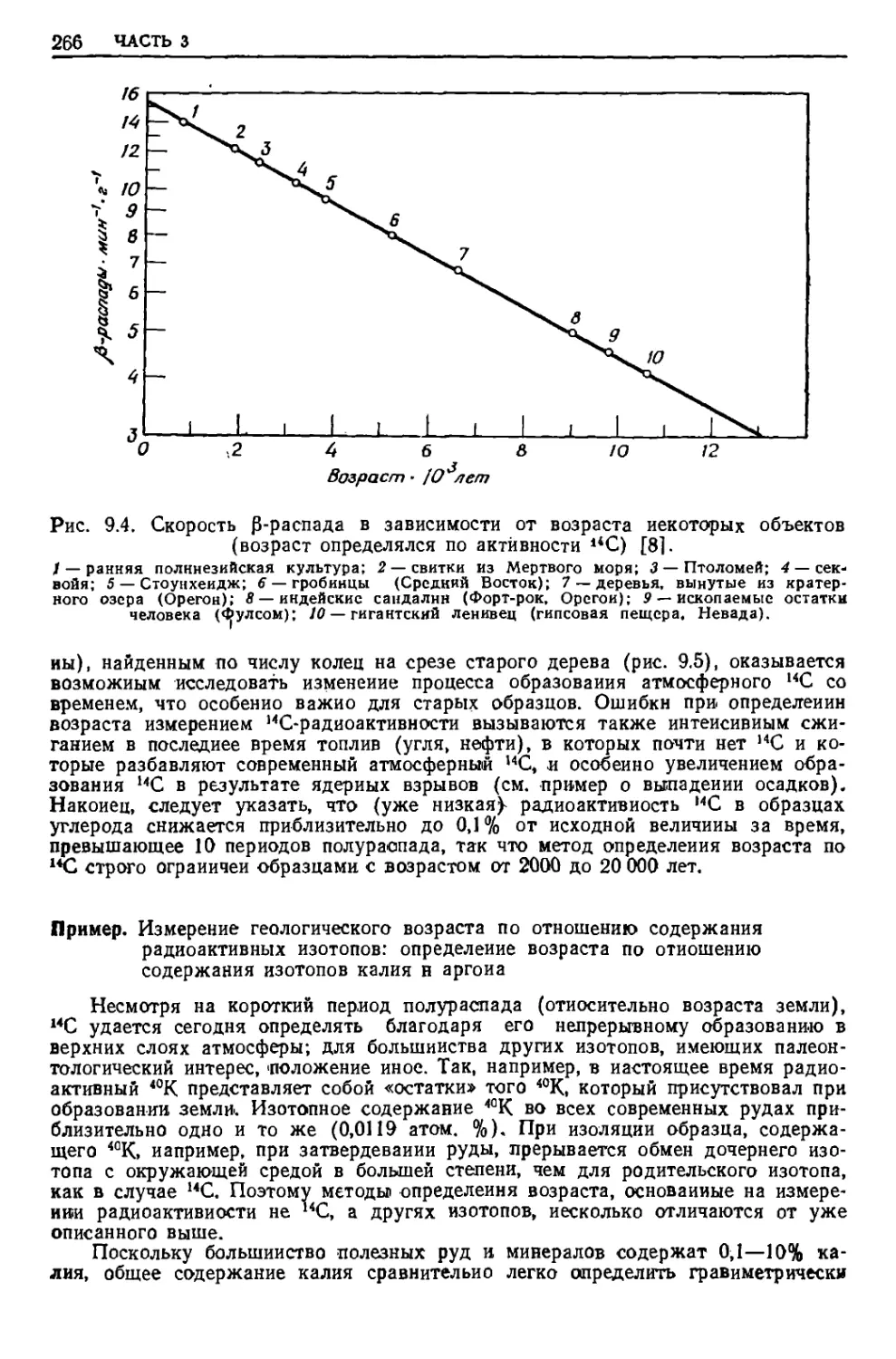
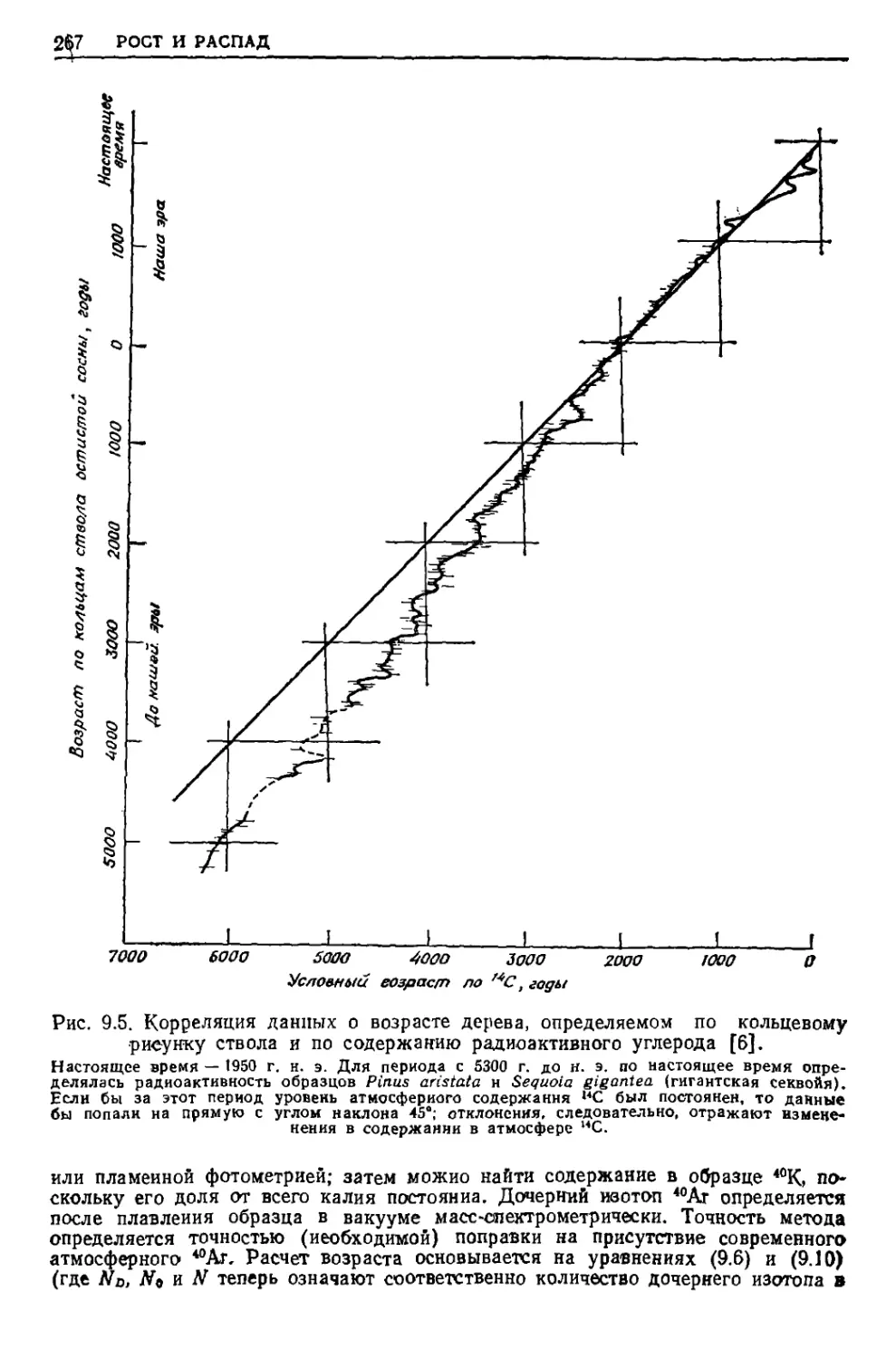



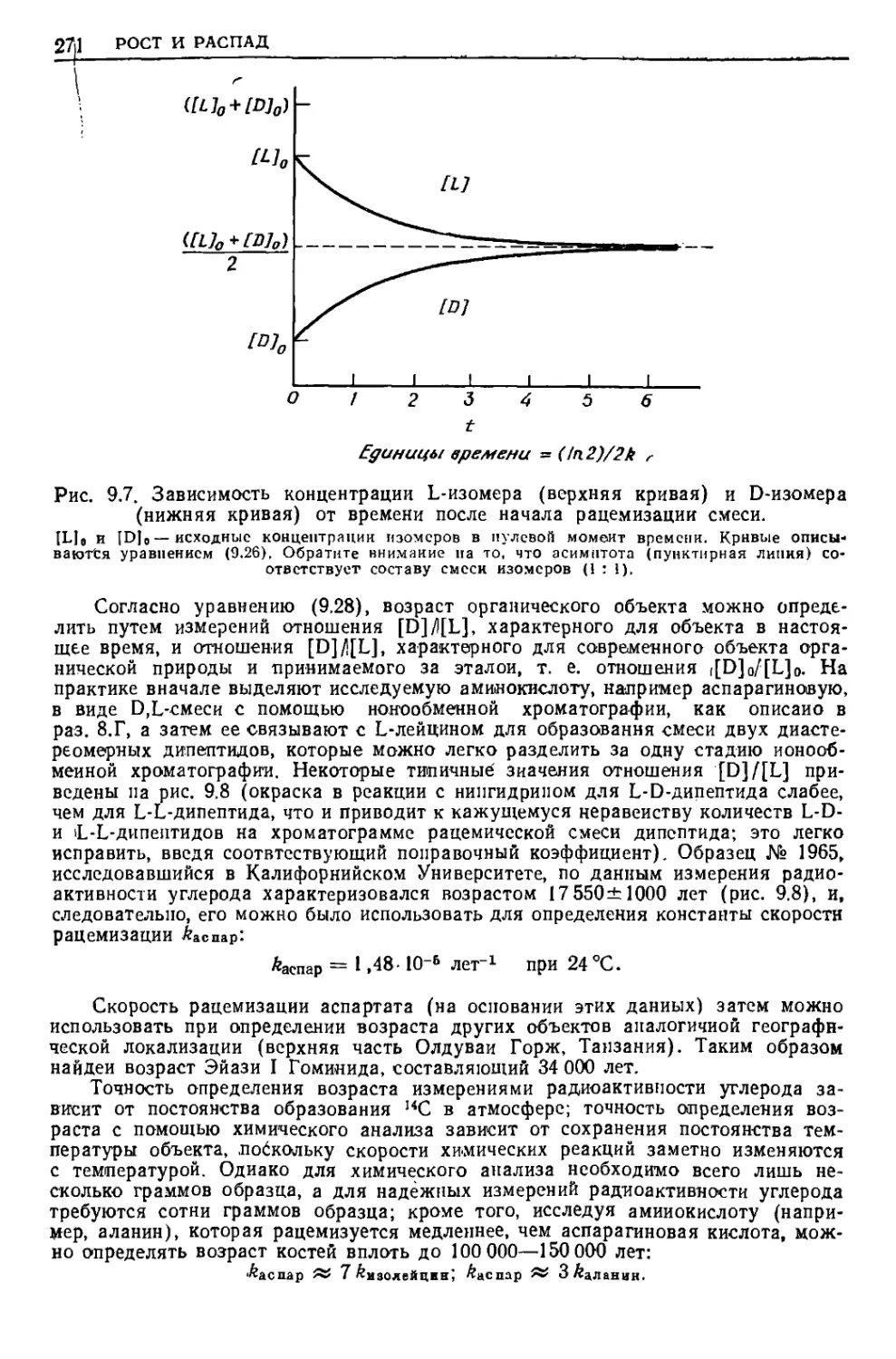
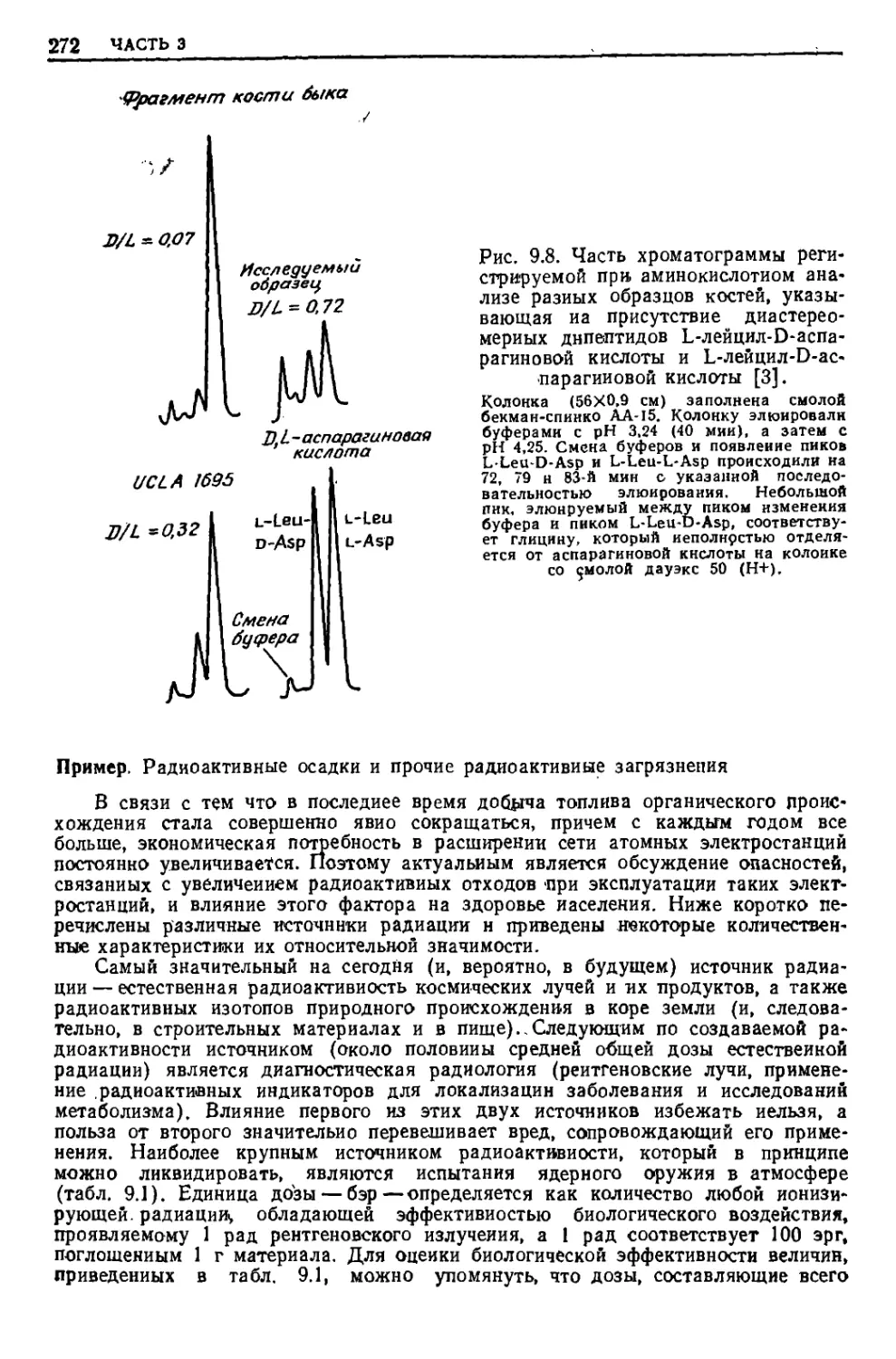








































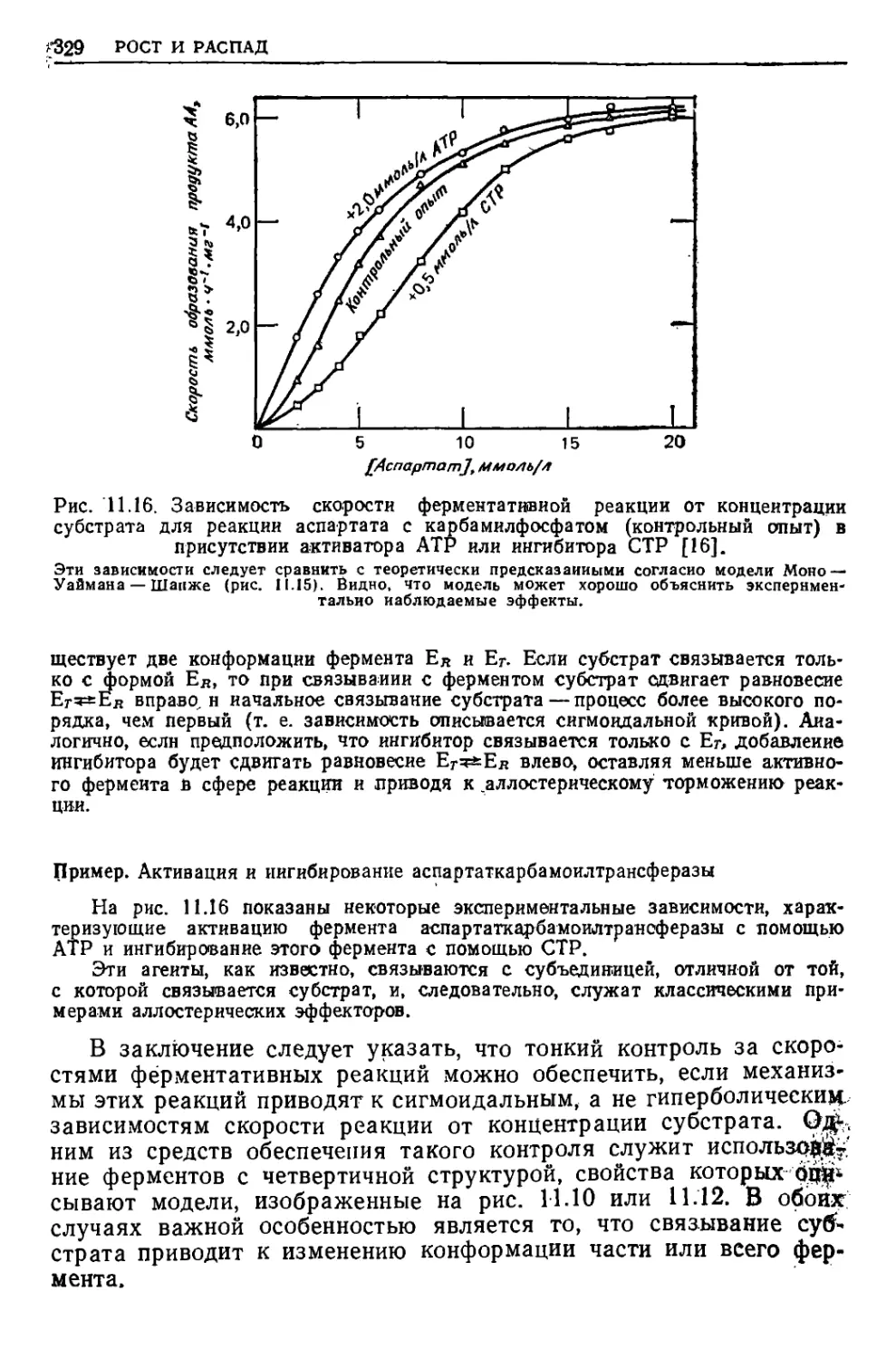





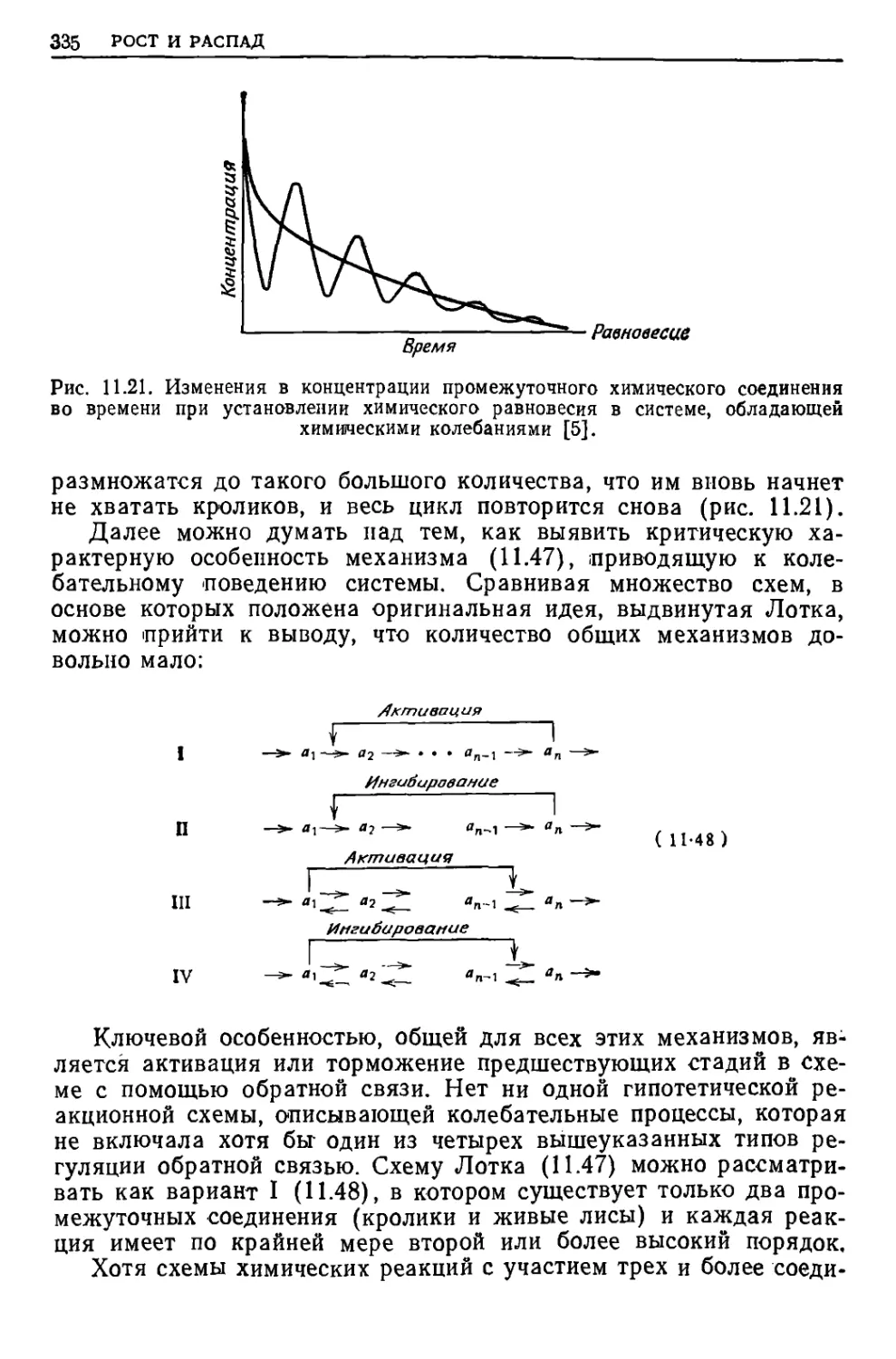

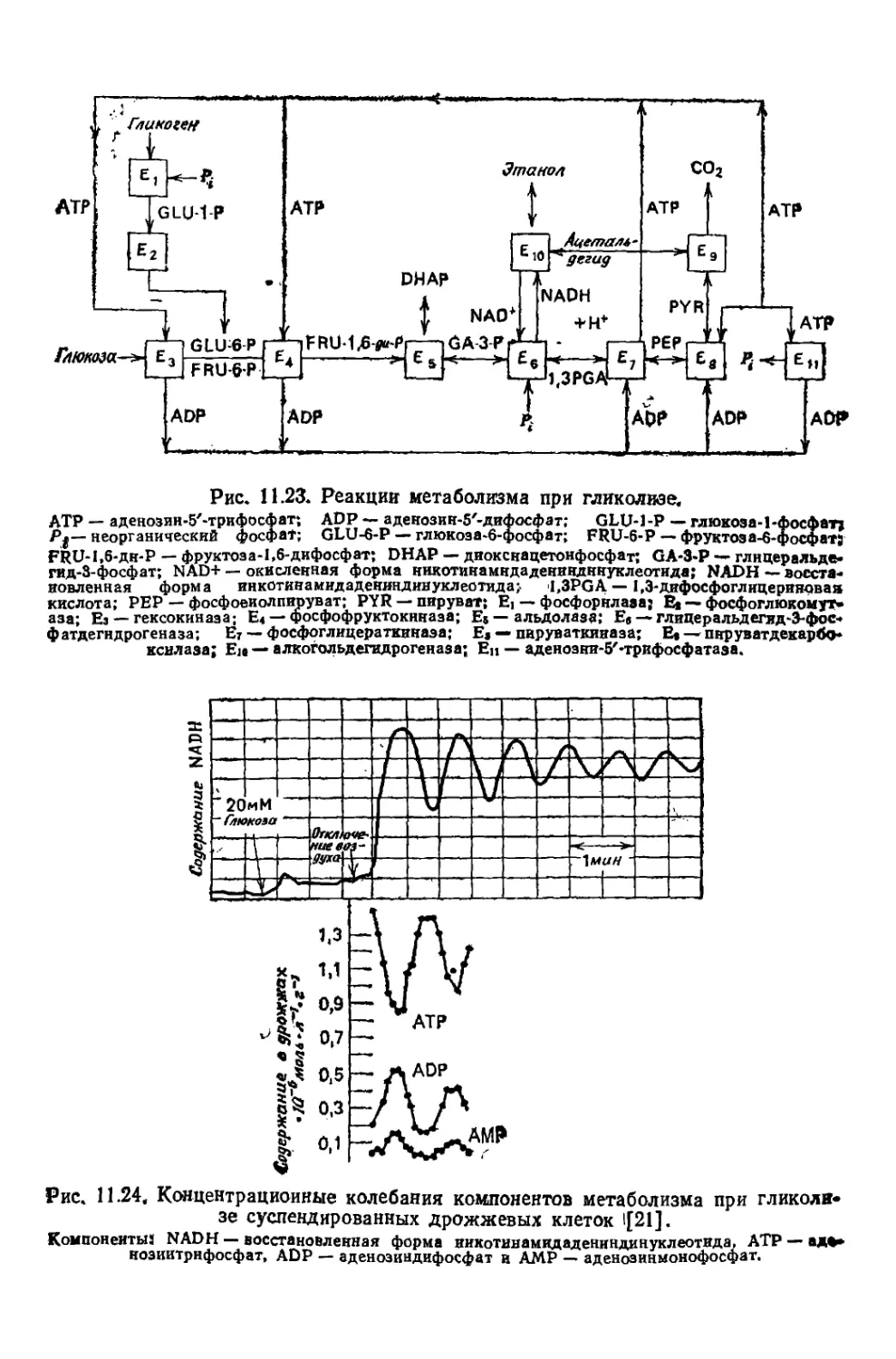

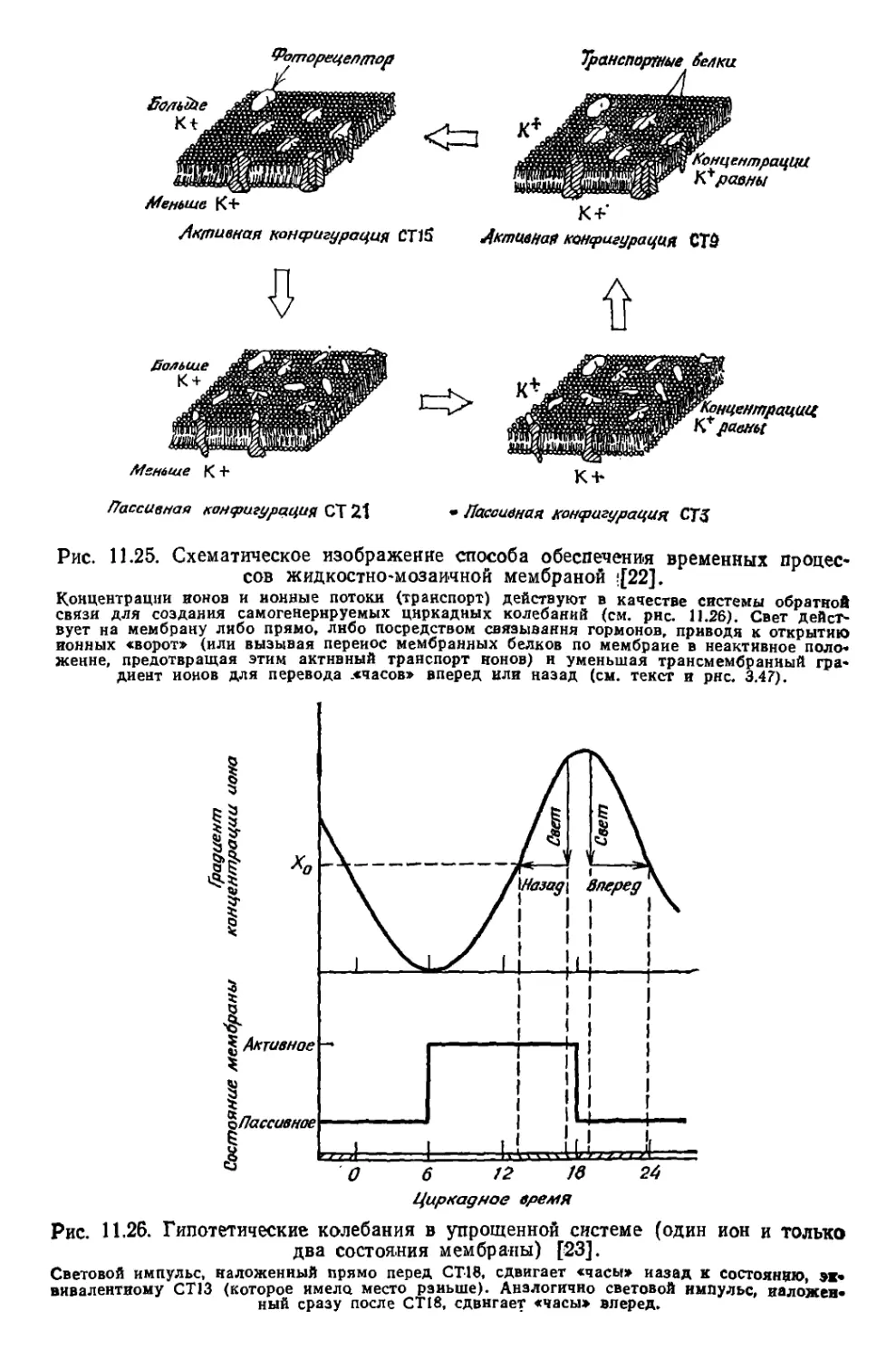





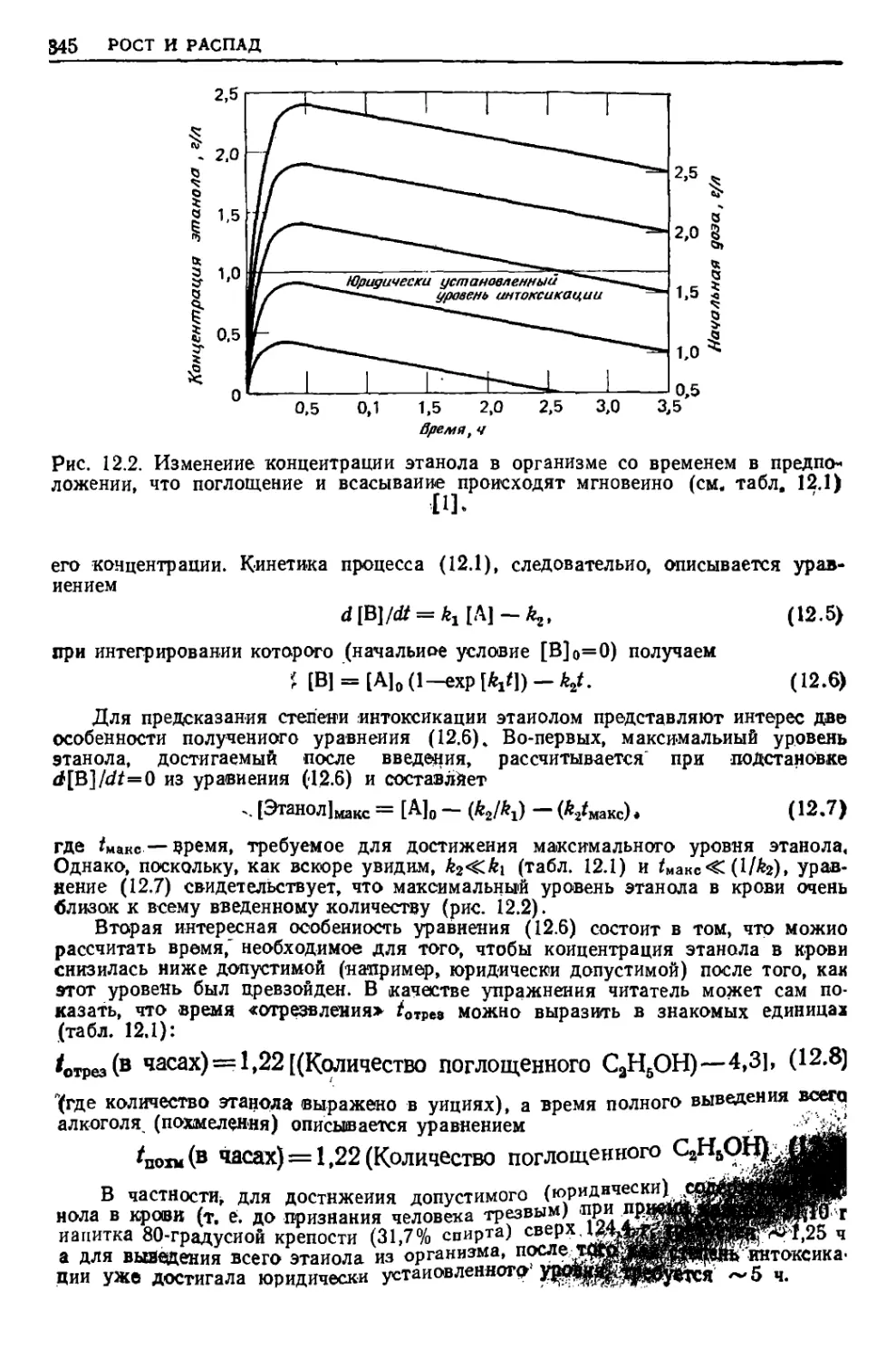
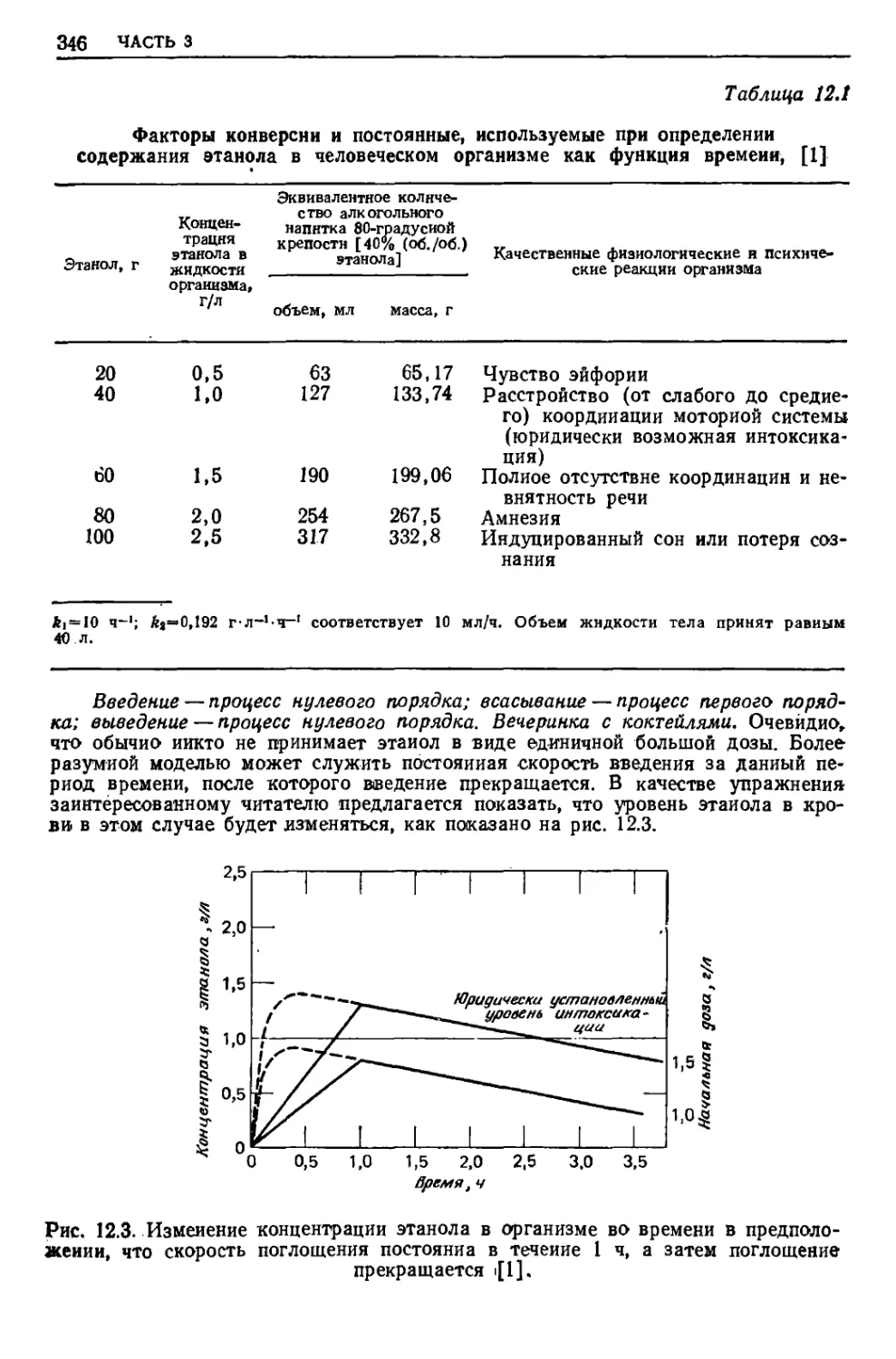




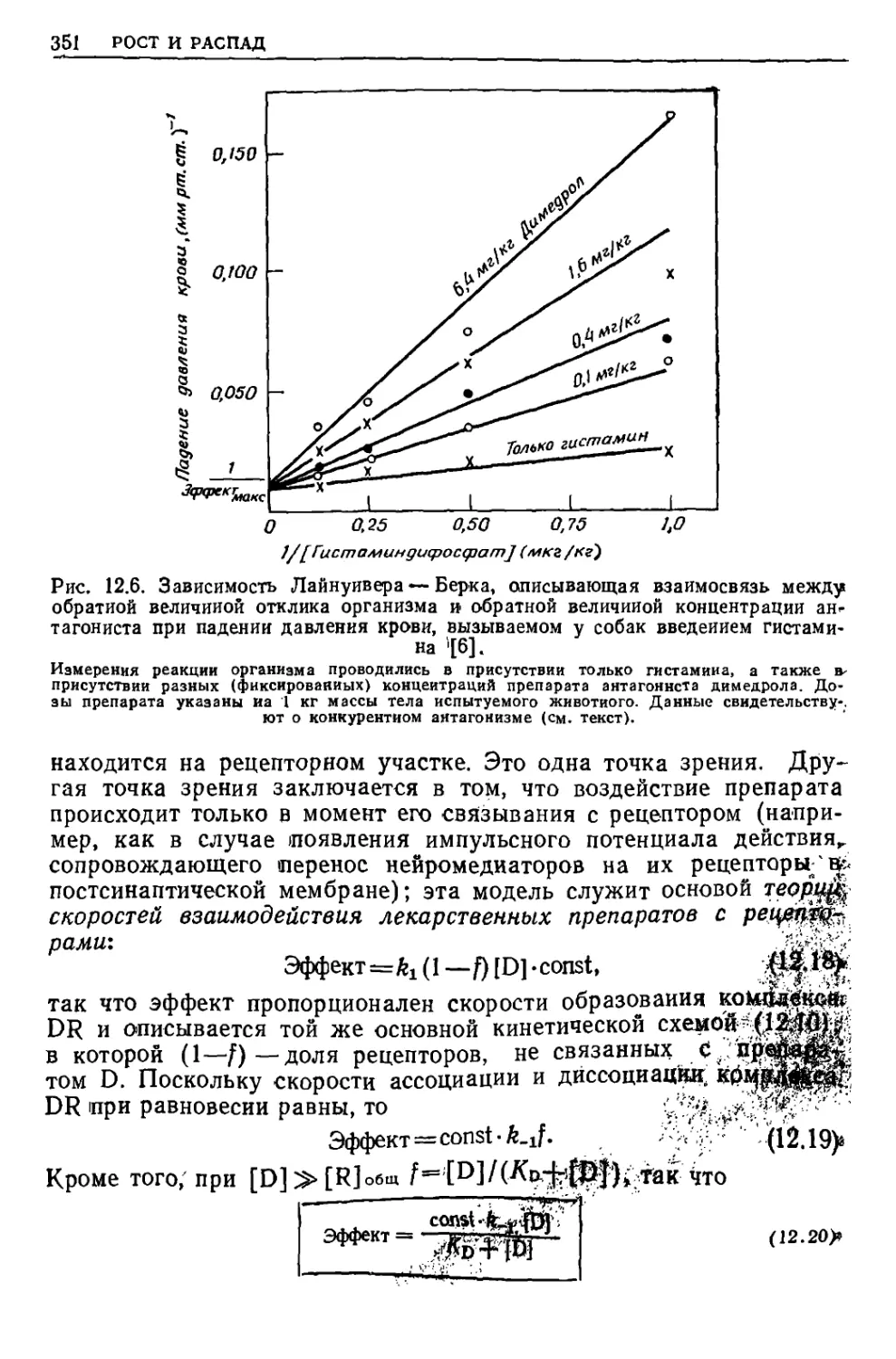

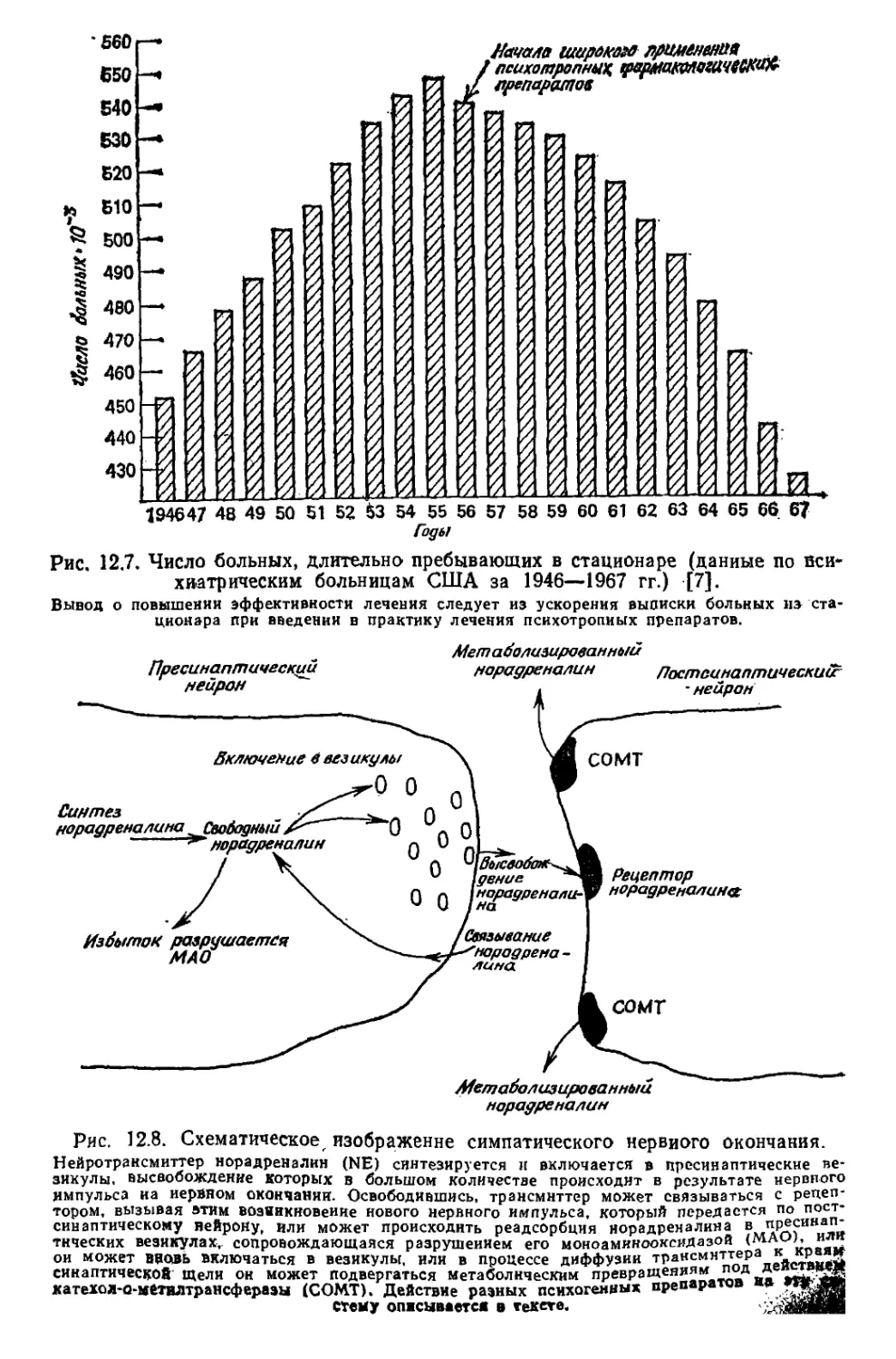













Текст
Э. Маршелл
Б опизическая
химия
i
ИЗДАТЕЛЬСТВО
..МИР"
BIOPHYSICAL CHEMISTRY
Principles, Techniques, and Applications
ALAN G. MARSHALL
University of British Columbia
John Wiley & Sons
New York Santa Barbara Chichester
Brisbane Toronto
Э. МАРШЕЛЛ
БИОФИЗИЧЕСКАЯ
ХИМИЯ
ПРИНЦИПЫ, ТЕХНИКА Ц ПРИЛОЖЕНИЯ
В 2-Х ТОМАХ
1
Перевод с английского
канд. хим. наук Б. Ю. ЗАСЛАВСКОГО
под редакцией доктора хим. наук,
проф. С. В. РОГОЖИНА
ИЗДАТЕЛЬСТВО «МИР» МОСКВА 1981
УДК 578.088
Книга американского автора представляет собой курс
физический химии применительно к биологическим объектам. В русском
переводе выходит в двух томах.
В томе 1 рассмотрены термодинамика и кинетика
биофизических систем, а также модели вероятностных биофизических
процессов. Каждый из разделов проиллюстрирован примерами, что
облегчает восприятие материала.
Предназначена для преподавателей, студентов биологических
вузов и для научных работников биохимических специальностей.
Редакция литературы по химии
© 1978, by John Wiley and Sons, Inc. AH Rights Re-
1805000000 served
Authorized translation from English language edi-
20503-083 tion published by Johm Wiley and Sons, Inc.
041(01)-81 © Перевод на русский язык, «Мир», 1981
ПРЕДИСЛОВИЕ РЕДАКТОРОВ ПЕРЕВОДА
Научно-техническая революция середины нашего столетия
вызвала к жизни много разнообразных физико-химических методов
исследования простых и сложных веществ, а также их смесей и
обеспечила быстрое развитие этих методов. Различные виды
хроматографии (газо-жидкостная, ионообменная, бумажная,
тонкослойная, гель-проникающая, аффинная, лигандообменная и т. д.),
электрофоретические методы, масс-спектрометрия, УФ-, ИК- и
радиоспектроскопия, ультрафильтрация и седиментационные методы
позволяют решать биохимические задачи практически любой
сложности. Эти методы обеспечили переход от чисто описательной
методологии к исследованиям, основанным на точных
количественных измерениях.
Наблюдающееся в последние десятилетия бурное развитие
некоторых естественных наук, особенно биологии, молекулярной
биологии, биофизики, биохимии и биофизической химии, в
значительной мере связано с разработкой новых методов.
Биофизическая химия, развивающаяся на стыке биохимии и биофизики,
относится к наиболее молодом направлениям естественных наук и
изучает физико-химические основы процессов жизнедеятельности,
протекающих, как правило, в высокоорганизованной среде.
Высокая структурная организация большинства
биокомпонентов проявляется в характере химического поведения при
включении их в субклеточные структурные ансамбли и различные орга-
неллы клеток. Так, например, фосфолипиды пищевых
растительных масел и фосфолипиды, включенные в состав клеточных
мембран, при одной и той же химической природе мало похожи по
функциональным свойствам.
Следовательно, физическое состояние биокомпонентов и
структурная организация по крайней мере ближайшего окружения
наряду с химическими параметрами этих биокомпонентоз могут
определять результаты биохимических процессов. Полное
понимание физико-химических процессов в живом организме практически
невозможно без учета взаимосвязи физических функций состояния
и химической природы веществ, участвующих в биохимических
процессах.
Книга Э. Маршелла написана как учебное пособие для
студентов биологических специальностей. Однако по характеру
изложения материала и его объему она заметно отличается от обычных
учебников.
В книгу включены разделы, которые входят в классические
курсы физической химии (термодинамика, химическое равновесие,
кинетика и т. п.), и разделы, посвященные практически всем
новейшим методам исследования, используемым в современной
биологии, таким, как квантовая механика, все виды спектроскопии,
хроматографии, фурье-анализ, светорассеяние и т. д.
Однако, естественно, при подобной широте охвата материала
разделы довольно кратки, что оказалось приемлемым благодаря
6 ПРЕДИСЛОВИЕ РЕДАКТОРОВ ПЕРЕВОДА
интересной схеме изложения и расположения материала. Большое
число сложных физических явлений сведено к рассмотрению
немногих простых математических моделей, что позволяет
сохранить строгость изложения, максимально сократив применение
математического аппарата. Каждая модель положена в основу
одной из шести частей книги. Заменяя в этих моделях отдельные
переменные и коэффициенты соответствующими физическими
параметрами можно, как оказалось, описать широкий круг явлений
и процессов. Например, уравнения движения груза на пружине
позволяют описать такие процессы, как поглощение и преломление
света, оптическое вращение, круговой дихроизм, ядерный
магнитный резонансг поглощение ультразвука, диэлектрическая
релаксация и др.
В каждой части имеется много удачно подобранных рисунков
и практических примеров, благодаря чему рассматриваемые
проблемы становятся более понятными и лучше воспринимаются,
несмотря на краткость изложения и сложность тематики. В конце
каждой главы предлагается несколько задач, самостоятельное
решение которых должно помочь закреплению излагаемого
материала.
Автор успешно справился со своей задачей — дать биологу и
биохимику «рабочие» знания основ современной физической
химии и раскрыть возможности использования их в практической
работе. Из-за сложности и разнообразия рассматриваемых
проблем отдельные разделы изложены почти конспективно, однако в
конце главы автор помещает библиографический список для
более глубокого изучения.
Книга написана ясно, живо, и даже, можно сказать,
занимательно. Она несомненно будет интересна и полезна не только для
студентов и аспирантов, но и для широкого круга научных
работников биологических и химических специальностей.
С. Рогожин
Б. Локшин.
Посвящается Мерилин, Уэнди, Брайан
ПРЕДИСЛОВИЕ
Цель этой книги — вооружить студентов, основные интересы
которых лежат в области биологических наук, в том числе в
области медицины и стоматологии, знаниями физической химии в
объеме, полезном при последующей работе. Большинство
существующих учебников для студентов, слушающих полу- или
одногодичный курс «физической химии для биологов», представляют собой
либо математически и теоретически разбавленный конспект
традиционных разделов физической химии (обычно с акцентом на
поведение малых молекул в газовой фазе, иногда с сокращенным
разделом о макромолекулах), либо ряд мало связанных между
собой глав по конкретным биофизическим методам с подробным
описанием методических и практических деталей при очень скупом
изложении принципов, лежащих в основе методов.
В настоящей книге предпринята попытка минимально
использовать математический аппарат в физической химии и при этом
помочь студентам применить полученные знания для решения
соответствующих биохимических и клинических проблем.
Для достижения указанной цели понадобилось разработать
новый принцип подачи материала. Поскольку большинство физико-
химических методов можно объяснить на немногих простых
математических моделях, в начале каждой части книги вводится своя
математическая модель, например модель груза на пружине.
Существенное достоинство такого подхода состоит в том, что
последовательное применение одной и той же модели к нескольким
разным биофизическим методам заключается просто в замене
обозначений в математических выражениях. Общность такого подхода
позволяет естественным образом рассмотреть также многие
современные проблемы и методы (которые обычно не обсуждаются в
элементарных учебниках), в том числе электронную микроскопию,
кинетику переходных состояний в химических реакциях и явления
релаксации, электрический шум, методы Фурье в дифракции и
спектроскопии, а также некоторые проблемы, связанные с
поглощением и дисперсией. Поскольку каждая из шести частей в
большой степени самостоятельна и связана с другими частями
многочисленными перекрестными ссылками, лектор, читающий этот курс,
может выбирать порядок подачи материала по собственному
вкусу.
Предлагаемый курс был опробован автором в течение восьми
лет преподавания физической химии студентам, изучающим науки,
связанные с проблемой жизни, в Университете Британской
Колумбии. Студенты третьего курса, предварительно прослушавшие
годичный курс математического анализа и годичный курс общей
химии, проявили высокий уровень восприятия материала,
содержащегося в гл. 1—17. (Программа курса была рассчитана на два
семестра.) Если же при чтении лекций опустить некоторые приме-
8 ПРЕДИСЛОВИЕ
ры, можно даже расширить круг охватываемых вопросов.
Конечно, если лектор располагает всего одним семестром, то при
составлении программы на основе этой книги ряд тем можно не
включать. Задачи, помещенные в конце каждой главы, должны
стимулировать творческую работу студентов, а не механическую
подстановку чисел в вызубренные формулы. (Дополнением к
данной книге мог бы служить сборник, включающий гораздо более
полный список задач с решениями.)
Во второй половине книги предпринята попытка описать самые
современные биофизические методы: «диск»-электрофорез,
аффинную хроматографию, рентгеноструктурный и рентгенографический
анализ при исследованиях макромолекул, использование ионосе-
лективных электродов, круговой дихроизм, лазерное
светорассеяние, флуоресценцию, ультразвуковой анализ и использование
магнитного резонанса «спин»-меченых соединений. Во всех случаях
рассматриваемые методы излагаются в прямой связи с
биологическими исследованиями, и заинтересованность студентов
поддерживается с помощью частных примеров, иллюстрирующих
применимость физического подхода для решения проблем в современной
биохимии или/и в клинической практике.
Автор выражает признательность членам своей семьи за
терпимость и поддержку в период написания этой книги. Автор
считает своим приятным долгом принести благодарность своим
коллегам за их советы, предложения, поправки и помощь. Это
Д: Кларк, М. Комисаров, А. Эддисон, Ц. Мэре, Э. Барнелл»
Л. Вербелов, Д. Рое, Дж. Бенбасат и Д. Кумб.
Э. Маршелл
|ЧАСТЬ 1
ГГЕРМОДИНАМИКА
fe БИОЛОГИИ
if
Полное описание поведения вещества, вообще говоря, следует
•начинать с квантовомеханического анализа движения и энергии
отдельных атомов и молекул; затем необходимо изложить некоторые
Ътатистико-механические (усредняющие) методы, позволяющие
;описать поведение системы с большим числом молекул с помощью
■термодинамики. (Скорость изменения свойств такой
макроскопической системы описывает кинетика.) Далее полезно ближе
познакомиться с математическими элементами разных расчетных методов:
математический аппарат квантовой механики можно развить из об-
яцей геометрии (ч. 5); для расчета среднестатистических величин
Применяется математический аппарат вероятностных процессов
'(ч. 2); кинетический анализ базируется на алгебраических
вычислениях (ч, 3).
*;■ В основе термодинамики лежат понятия работы и теплоты. Хо-
ря работа или теплота, затрачиваемые или выделяемые в
процессе, зависят от конкретного выбора его пути, соотношение между
|шми от пути процесса не зависит. Следовательно, для
термодинамического описания макроскопической системы используются
уравнения полных дифференциалов, не зависящие от выбора конкретно-
|*о пути. В общем случае значение термодинамического подхода
состоит в том, что отсутствует необходимость в знании механизма
■перехода из одного состояния в другое, поскольку конечный
результат не зависит от пути этого перехода. Большое практическое
значение термодинамики вытекает из того, что изменения таких
функций состояния, как энергия, не зависят от пути перехода, и поэтому
)при переходе системы из одного состояния в другое можно
приравнивать конечные результаты переходов, происходящих по разному
пути. Таким образом часто удается получить информацию о
процессах, прямое исследование которых невозможно, например, о
реакциях в живых клетках.
Термодинамика особенно полезна для предположительной
оценки энергетических изменений, происходящих в результате
химических реакций, часто в тех случаях, когда конкретная исследуемая
реакция плохо поддается прямому изучению. Термодинамический
подход позволяет предсказывать, можно ли ожидать самопроиз-
10 ЧАСТЬ I
вольного протекания данной реакции; если нет (реакция
несамопроизвольна), то какое количество энергии необходимо затратить
для проведения реакции. Эта информация особенно важна для
определения путей реакций различных метаболических процессов.
Можно рассчитать также энергии разрыва конкретных химических
связей; найденные величины служат в качестве характеристики
прочности связей при сопоставлении разных молекул.
Термодинамика объясняет существование констант равновесия и их
зависимость от температуры. С помощью термодинамики получают
детальную информацию о механизме ферментативных реакций,
позволяющую установить существование определенных промежуточных
соединений и характеризующую их энергетическую устойчивость.
С позиций термодинамики объясняется возникновение разности
давлений (осмотического давления) по обе стороны
полунепроницаемой мембраны, отделяющей раствор, в котором находятся
незаряженные макромолекулы, от раствора, в котором нет
макромолекул, а также падение потенциала (потенциал Доннана) между
растворами, разделенными такой мембраной, в присутствии
(заряженных частиц) ионов. С помощью термодинамики объясняется и
влияние соли, находящейся в растворе (солевой эффект), на
растворимость макромолекул; это явление имеет большое
практическое значение при выделении и очистке белков. Термодинамика
применяется при описании процессов в электрохимических ячейках, а
также для теоретических прогнозов при создании новых типов
электродов с избирательной чувствительностью к одному типу
низкомолекулярных соединений в растворе. Возникновение различных
«фаз» (твердой, жидкой, газовой и других) легко анализируется с
помощью термодинамики, и в настоящее время представляет
интерес привлечение термодинамики для объяснения физиологических
функций биологических мембран при разных температурах, а также
для развития теорий возникновения и эволюции жизни. Наконец,
термодинамика предсказывает эффективность той или иной
системы (превращающей теплоту в работу) и обеспечивает разумную
основу для обсуждения путей энергетического обмена в растениях.
Все формальное изящество термодинамики становится-
очевидным, если строго и последовательно развить первый и второй
законы термодинамики; однако такое изложение находится за
пределами задач, стоящих перед данной книгой, поскольку в основном
все необходимые преобразования выполняются на модели
идеального газа и могут быть лишь ограниченно применены
непосредственно к биологическим системам. Поэтому вначале в данной книге
приводится краткий обзор основных свойств термодинамических
функций состояния, а затем рассматриваются непосредственно
биохимические приложения. Более детальное обоснование
основных термодинамических соотношений и законов читатель сможет
найти в любой из отличных монографий (см. литературу) или в
большинстве современных учебников по основам химии.
ГЛАВА 1
РАБОТА, ТЕПЛО И ЭНЕРГИЯ
Термодинамика — это наука, изучающая взаимные
превращения различных видов энергии в макроскопических системах,
связанных с переходами энергии между системами в форме теплоты
и работы. Однако, поскольку предполагается, что общая энергия
вселенной как единой системы постоянна ( первый закон
термодинамики), обсуждение необходимо ограничить некоторой^ особой
изолированной частью пространства, системой. Тогда под
окружающей средой подразумевается остальная часть вселенной.
Система называется равновесной, если макроскопические свойства
(объем, температура, давление, концентрация, энергия и т. д.)
системы постоянны во времени. Термодинамическое состояние
системы определяется совокупностью изучаемых термодинамикой
свойств системы. С помощью термодинамики можно сделать ряд
полезных предсказаний о том, что должно произойти при
переходе системы из одного состояния в другое, например при
протекании химической реакции, фазовом переходе, переносе
растворителя и/или ионов через полунепроницаемую мембрану,
прохождении электрического тока, изменении температуры и т. п. Таким
образом, естественным путем приходим к рассмотрению работы и
теплоты, которые представляют собой «транзитные» формы
энергии в процессе изменений состояний системы.
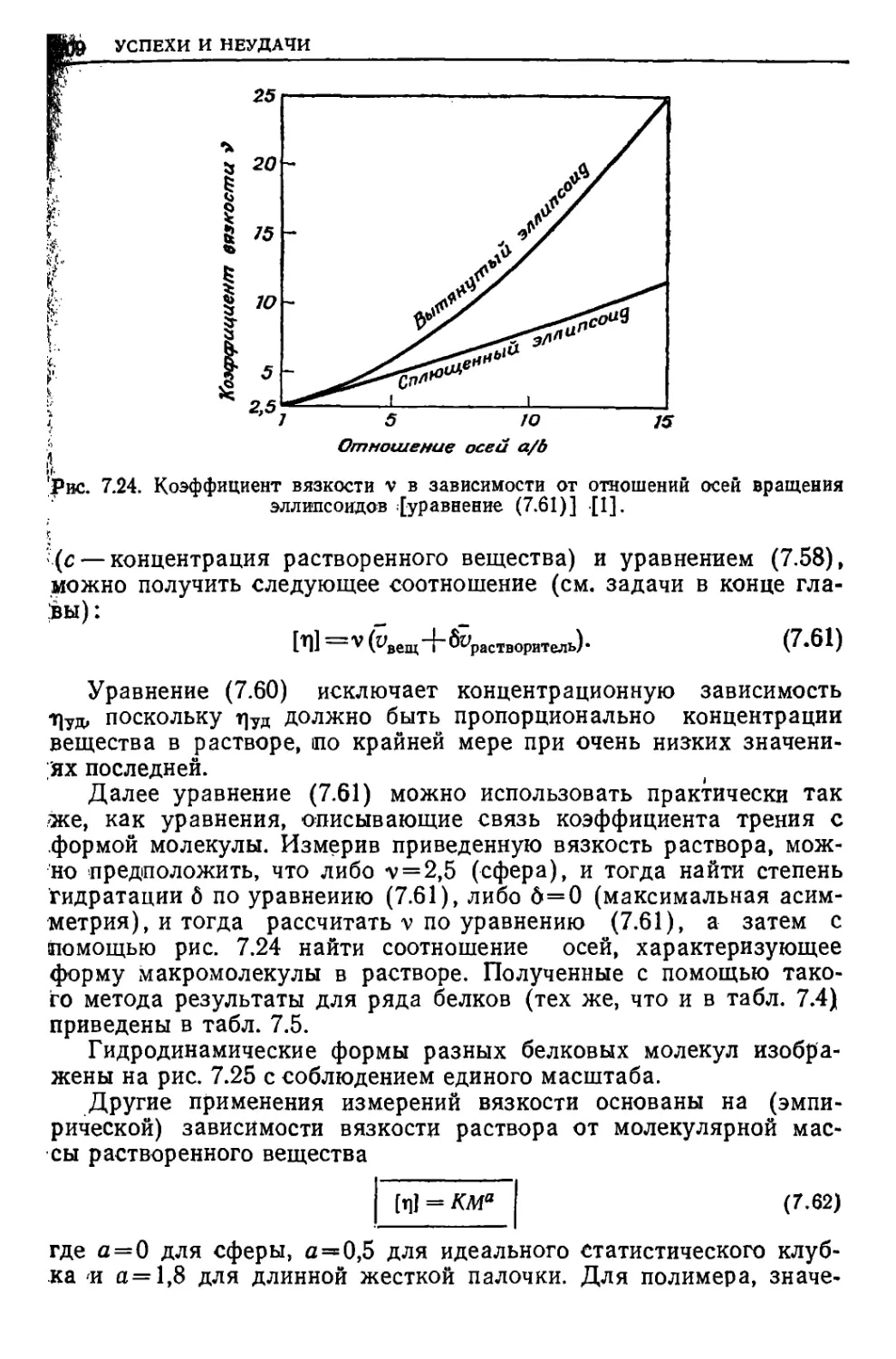
Как показано в табл. 1.1, работу можно выразить как
произведение обобщенной силы («интенсивный» параметр) на
обобщенное перемещение («экстенсивный» параметр). Поскольку
обобщенную силу можно считать постоянной, если
обобщенное перемещение достаточно мало, наиболее удобно
определять работу (как это сделано в табл. 1.1) при бесконечно малом
перемещении. Работа положительна, если она производится
системой, и отрицательна, если она совершается окружающей средой
над системой. Работа, совершаемая при переходе системы из
одного состояния в другое, зависит от пути перехода (т. е. от
последовательности бесконечно малых перемещений); особый вид пути,
на каждом бесконечно малом отрезке которого система близка к
равновесию, называется обратимым, и изменение любого
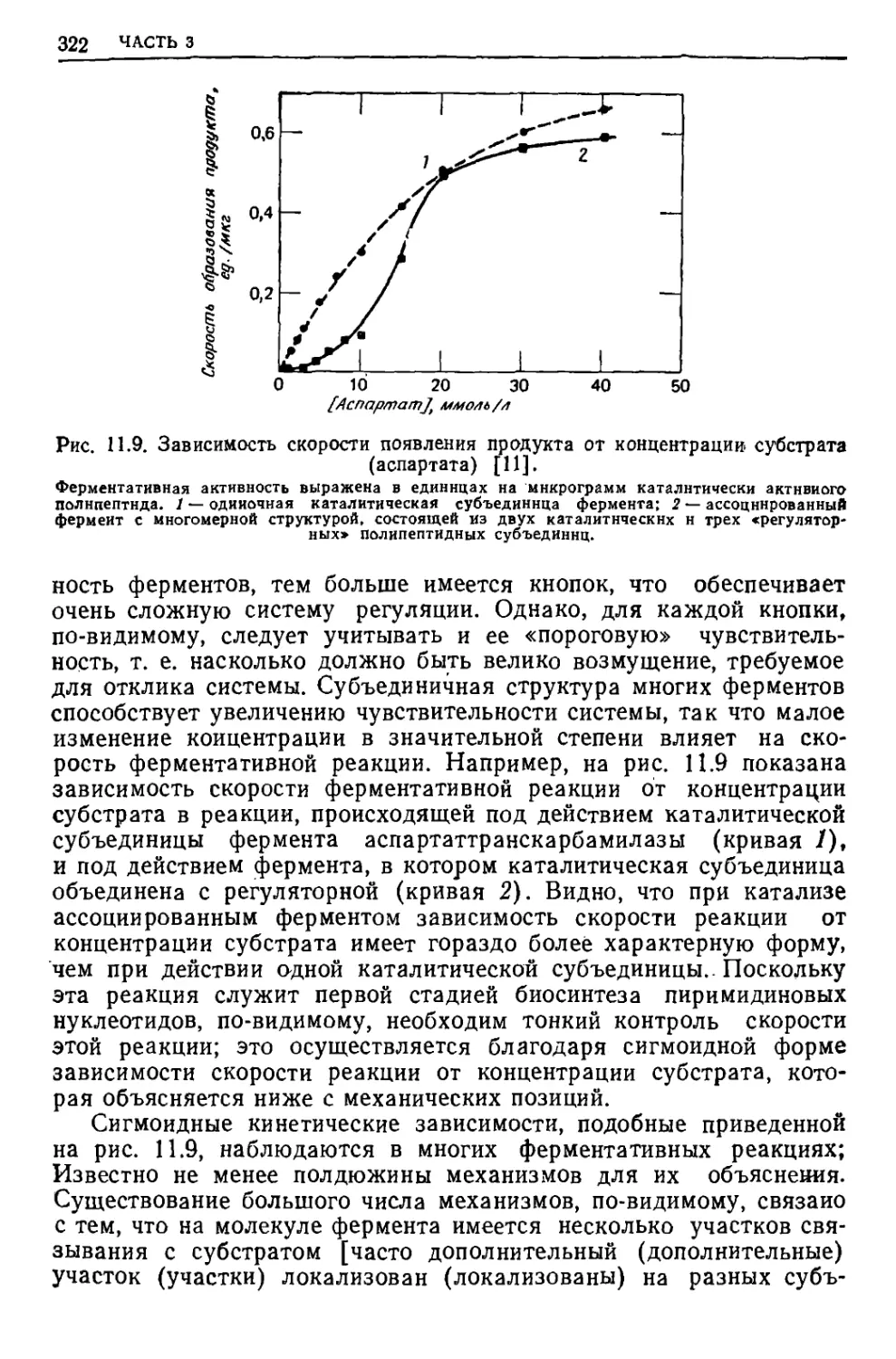
параметра этого процесса обозначается буквой d (например, dw —
обратимая работа); для обозначения необратимого изменения при лю-
12 ЧАСТЬ 1
Таблица 1.1
Виды работы (обратимая работа dw определяется как произведение
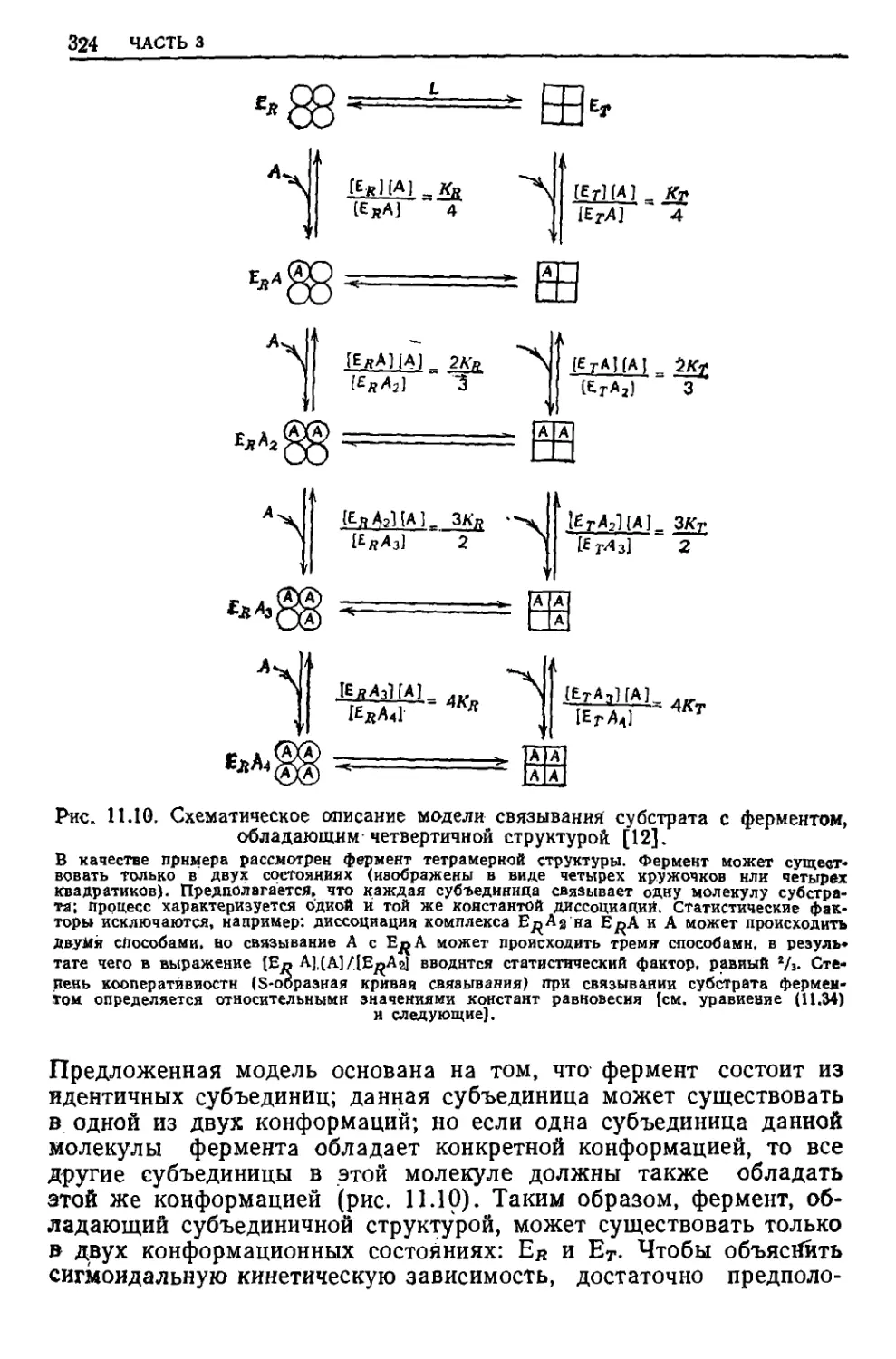
обобщенной силы на обобщенное перемещение)
dw = Обобщенная сила Обобщенное перемещение
PdV
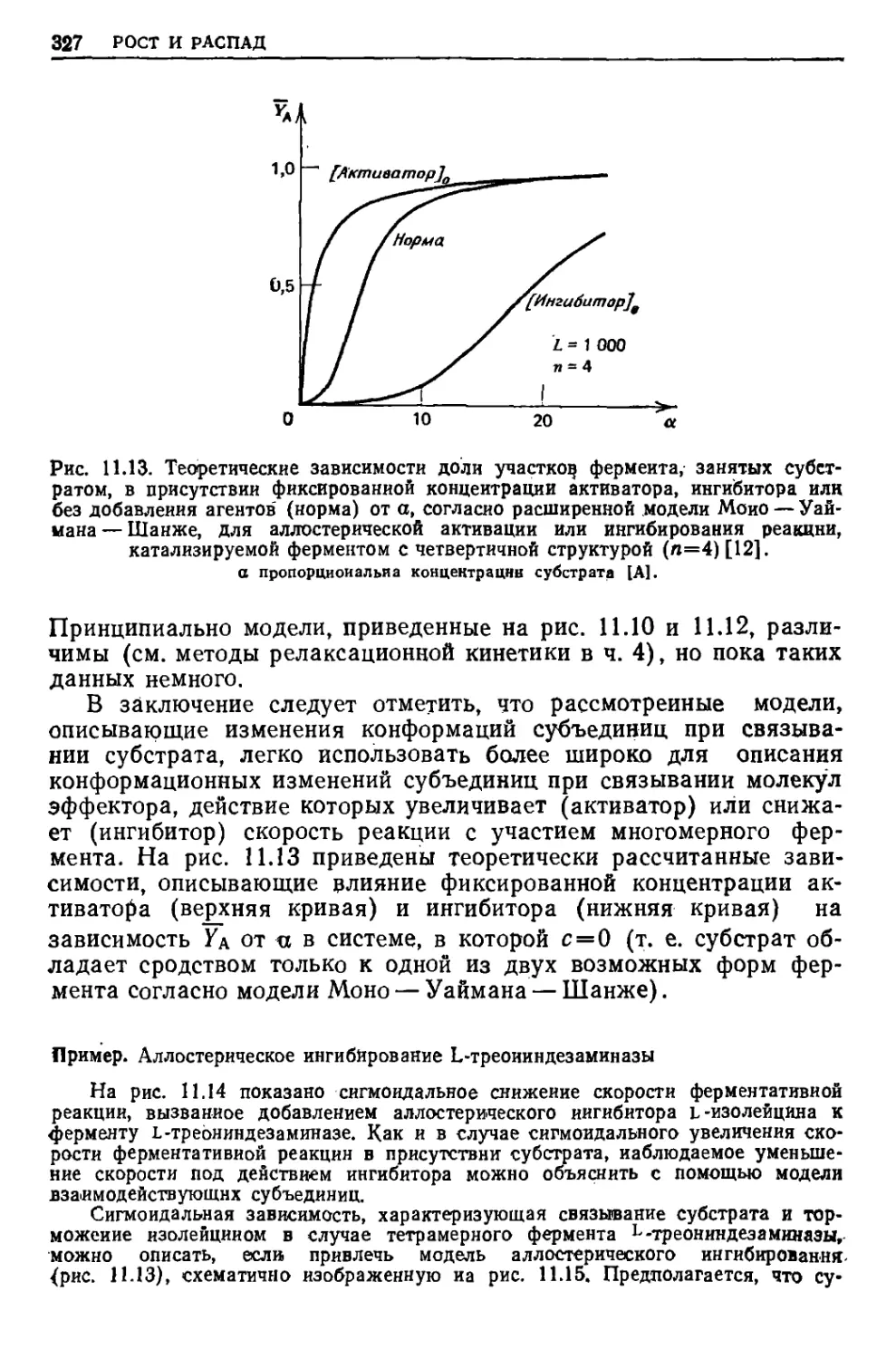
ydA
Edq
Hd\l
Fdx
P — давление (сила, деленная на
площадь)
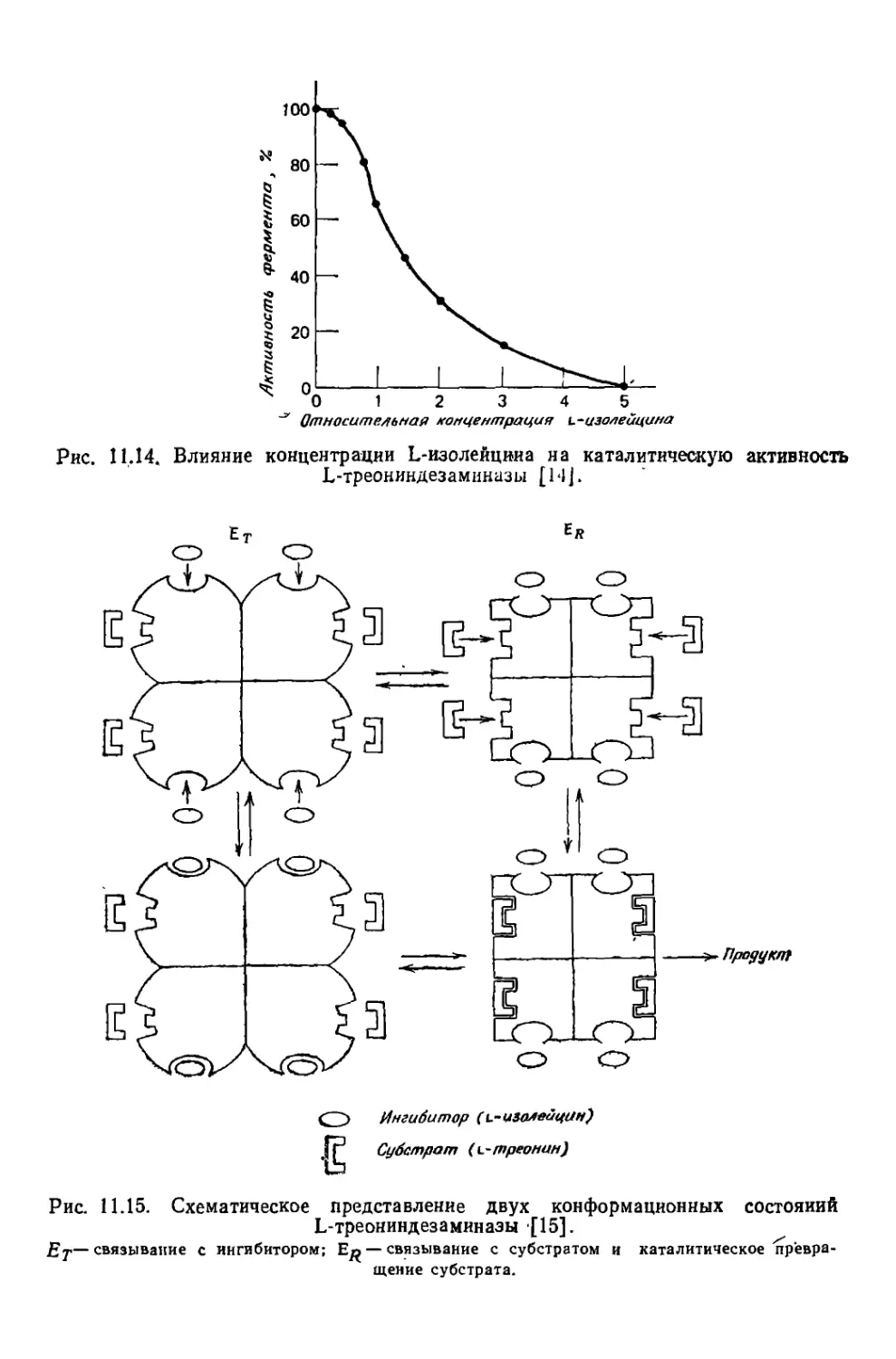
у—поверхностное натяжение (сила,
деленная на расстояние)
Е — э. д. с.
Н — напряженность магнитного поля
F—сила
V — объем
А — площадь
q — заряд
ц — магнитный момент
х — расстояние
бом другом пути перехода системы используется символ 6
(например, 6w — необратимая работа).
Считается, что два тела находятся в тепловом контакте, если
между ними может происходить передача энергии, но невозможен
перенос вещества. Теплота — это такой вид энергии, который
переходит от более нагретого тела к более холодному телу при
наличии между телами теплового контакта. В табл. 1.2 приведены
разные (по происхождению) виды теплоты. Теплота, выделяемая
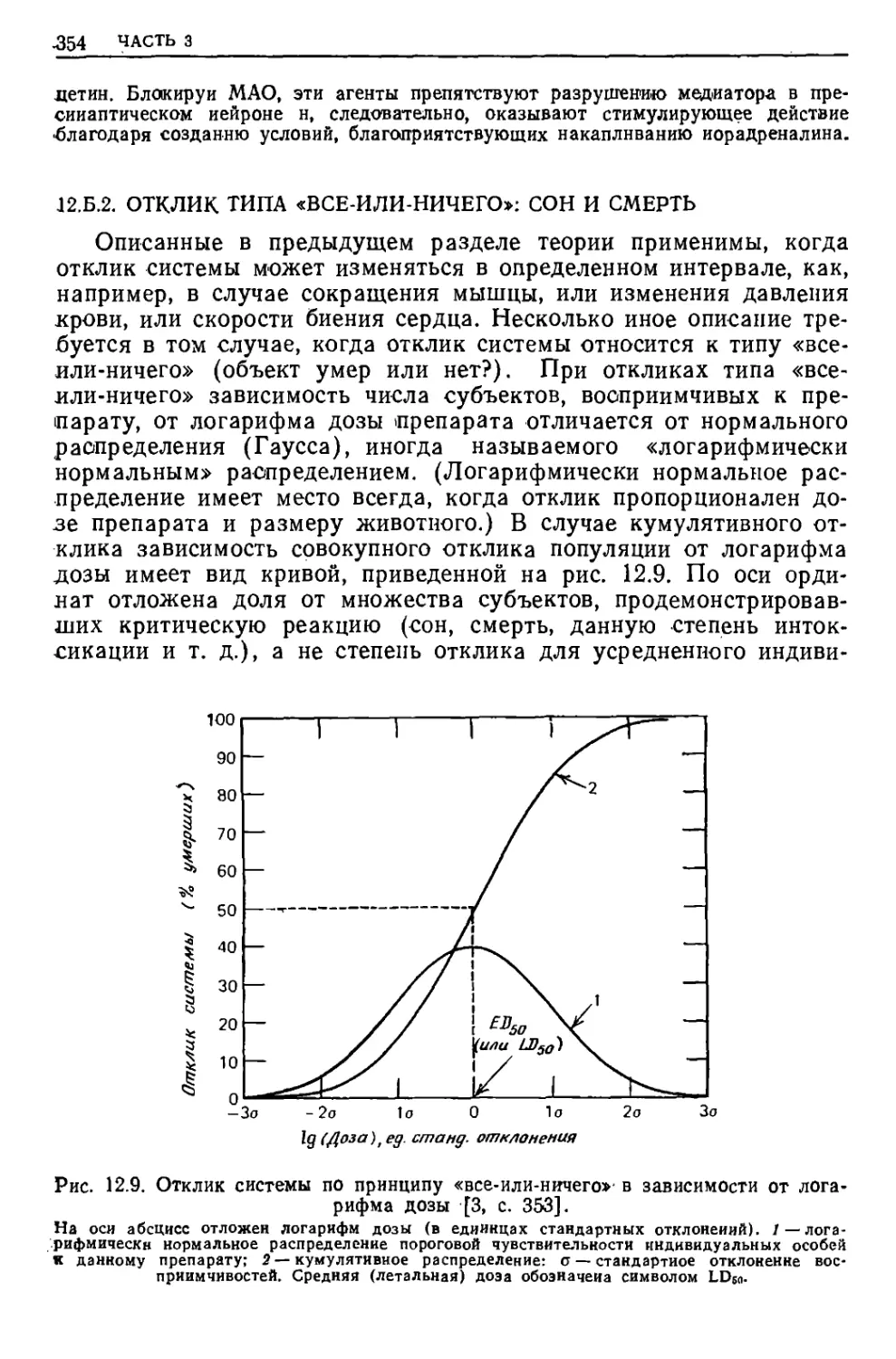
или поглощаемая при химической реакции, часто служит для
оценки прочности химических связей, которые разрываются или
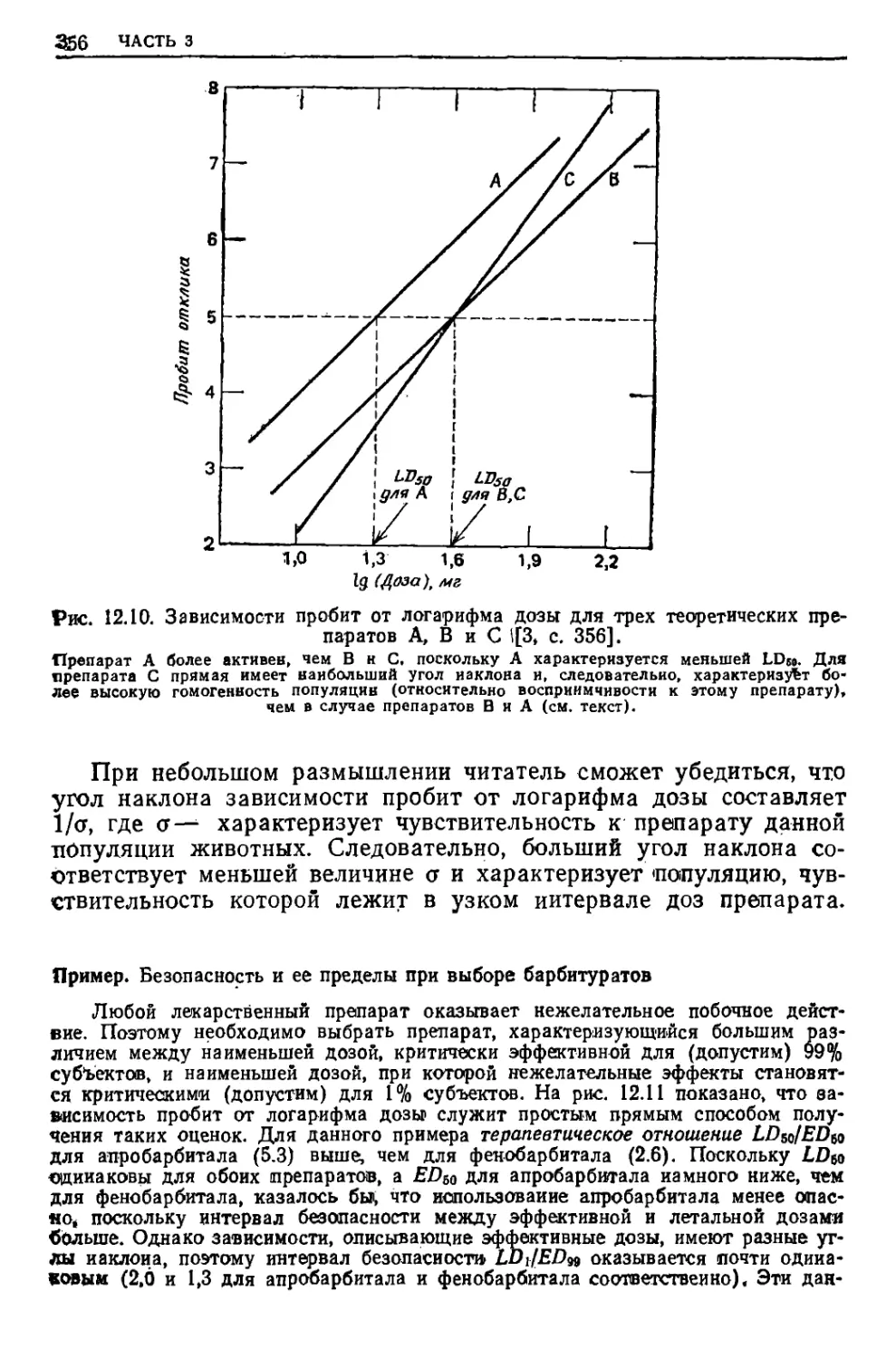
образуются в данном процессе. Количество тепла, требуемое для
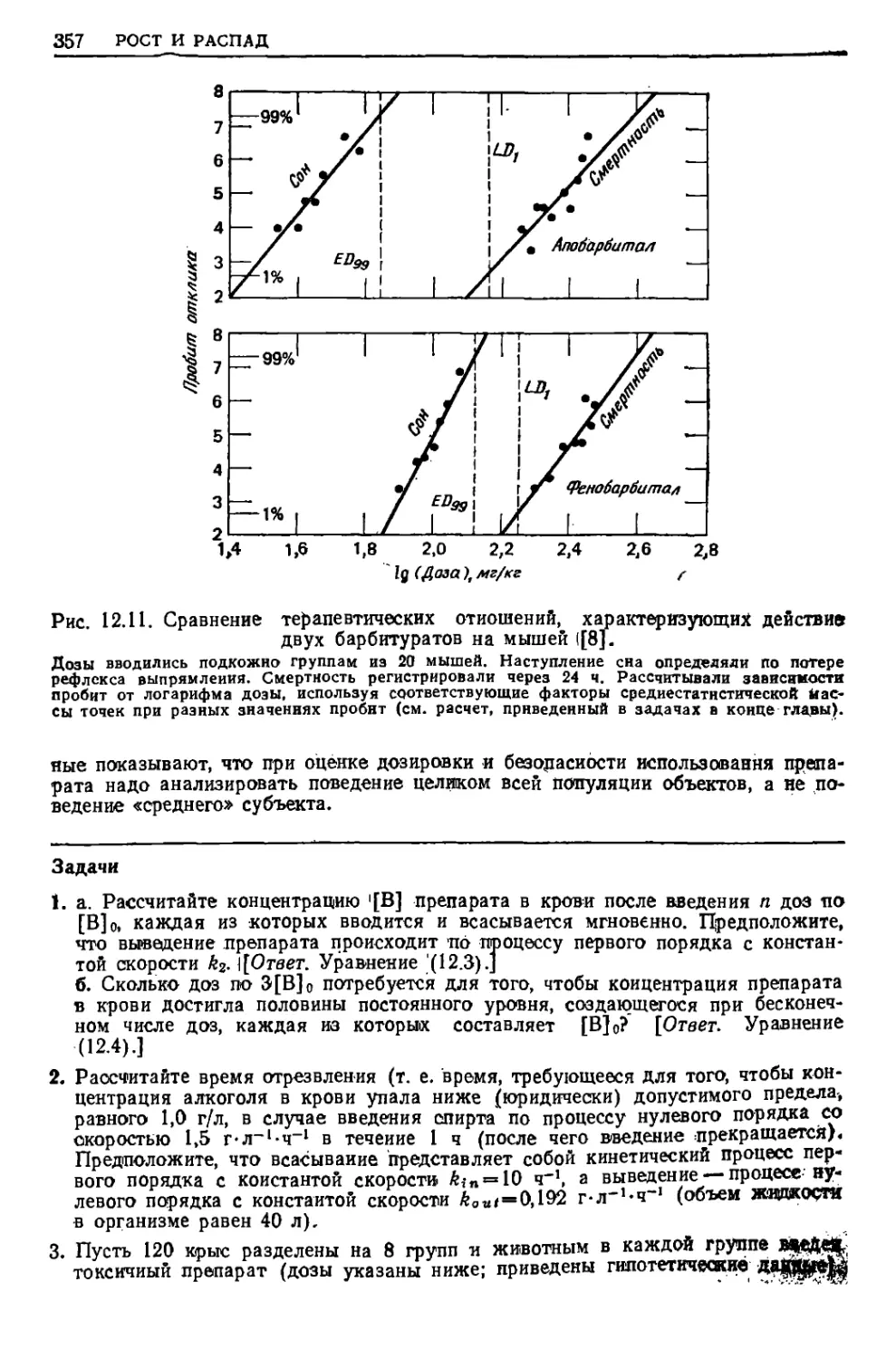
увеличения температуры вещества, позволяет предсказывать
результат процесса, проводимого при данной температуре
(отличной от той, при которой процесс изучен); это особенно важно в
связи с тем, что большинство термодинамических характеристик
химических реакций изучено при температуре, которая может
значительно отличаться от интересующей нас температуры,
например от температуры физиологического процесса. Теплота, выде?
ляемая при прохождении электрического тока (табл. 1.2),
является функцией квадрата силы тока; поэтому, поскольку мощность
равна произведению V-I (где V и / — напряжение и сила тока
соответственно), становится ясным, почему для снижения
тепловых потерь передачу электроэнергии на большие расстояния
нужно производить при максимально высоком возможном
напряжении. Принято считать положительной теплоту, которая
поглощается системой. Процесс называется адиабатическим, если система
не поглощает и не выделяет тепло (в окружающую среду).
Действие паровых двигателей, остающихся и сегодня одним из
главных источников механической энергии, основано на превращении
теплоты в механическую работу. Количество тепла,
выработанного в процессе, также зависит, как показано ниже, от пути
перехода ив начального состояния в конечное.
i' 13 ТЕРМОДИНАМИКА В БИОЛОГИИ
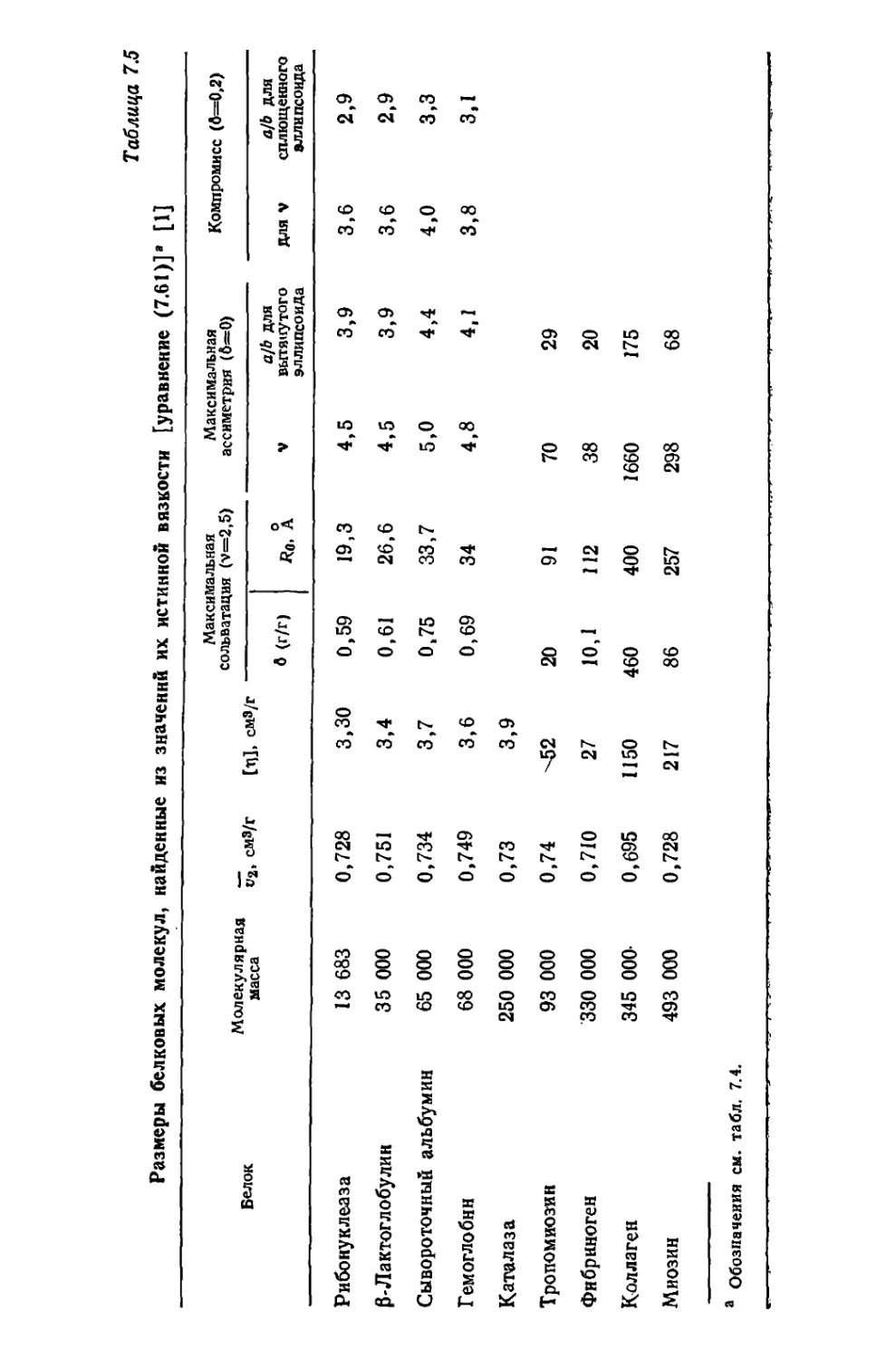
Таблица 1.2
Источники тепла
I
Исследуемый процесс
(,q — теплота, поглощаемая или выделяемая в процессе
Нагревание вещества
Электрический ток в
проводнике
Изменение физического
(фазового) состояния
вещества (например,
■плавление твердого
вещества, испарение
жидкости)
Химическая реакция
Трение
6<7 = CmdT, где С — теплоемкость, т — масса
вещества, Т — температура
6q = I2Rdt, где /—сила тока, t — время, R —
сопротивление проводника
б<7 = Mm, где dm — масса вещества, фазовое
состояние которого изменяется,
h — теплота, требуемая для
превращения единицы массы вещества
(удельная теплота плавления,
удельная теплота испарения)
bq = hdm, где dm—масса (допустим) реагента,
превращаемого в продукт, h —
теплота, требуемая для превращении
единицы массы реагента (удельная
теплота реакции)
&7 = Fdx, где F — противодействующая сила
трения, х—расстояние
Для данного процесса теплота и работа зависят от его пути;
однако, согласно первому закону термодинамики, их сумма
(полная внутренняя энергия системы) не зависит от пути процесса.
Используя принятые выше обозначения, можно записать
dE—8q-\-6w (первый закон термодинамики), (1.1)
где Е — энергия системы.
Физический смысл уравнения (1.1) состоит в том, что
невозможно существование какого-либо устройства, способного
производить эйергию, если отсутствуют другие взаимосвязанные
изменения в системе. Если предположить, что изменения суммарной,
энергии системы зависят от пути ее перехода из одного состояния
в другое, то можно было бы осуществить циклический процесс,,
при котором возвращение системы в начальное состояние могло
бы приводить к увеличению, ее энергии. Поскольку ни один такой
процесс не был обнаружен, предполагается, что он в принципе
невозможен. Физический смысл уравнения (1.1) математически
выражается так:
*
dE^O,
(1.2)
где ф обозначает некоторый (циклический) путь, возвращающий
систему в начальное состояние.
14 ЧАСТЬ I
Уравнение (1.2) является уравнением полного дифференциала
-dE. Точно так же любой полный дифференциал dF можно
выразить уравнением в частных производных:
-(-£L*+(-£L*+(Tf-L
{частные производные рассчитываются так же, как обычные
производные; при этом полагают, что переменные, указанные в
нижнем индексе, считаются постоянными: например, для функции
F(x, у, z)=x2y3ez частная производная по х равна (dF/dx)VtZ=
= 2xyzez]. Наконец, поскольку в конкретном случае часто
оказывается удобным оценить изменения теплоты и работы в процессе,
происходящем по обратимому пути, и поскольку уравнение (1.1)
выполняется для любого пути, можно рассчитать изменение
энергии следующим образом:
dE^=dq-{-dw (обратимый путь), (1.4)
Для любого выбранного пути изменение энергии в системе
равно dE.
Клаузиус (1850 г.) предложил еще одну функцию состояния
(т. е. полный дифференциал). Если выбранный замкнутый путь
разделить на бесконечно малые обратимые отрезки и для
простоты расчетов использовать модель идеального газа, то можно
показать, что
<Y)(dqlT)—0 (второй закон термодинамики). (1.5)
Принятые обозначения (dq) указывают на то, что любой
выбранный бесконечно малый участок пути является обратимым; Т —
абсолютная температура.
Таким образом, можно определить новую функцию состояния—
энтропию S, которая удовлетворяет определению полного
дифференциала
dS=dqjT: (1.6)
г
Энергия — количественный параметр, физический смысл которого должен
быть знаком большинству читателей из классической и> квантовой механики;
интуитивный смысл энтропнн, вероятно, менее понятен. Энтропия характеризует
степень неупорядоченности, или беспорядоченности, в системе. Для большей
наглядности воспользуемся схемой, приведенной на рис. 1Л.
Даны две сообщающиеся камеры, заполненные газом (для простоты приня*
то, что объем н давление в камерах одинаковы), и два идеальных газа. Первс*
начально в первой камере находится одни газ А (например, гелий), а во
второй— другой В (например, неон). Между камерами имеется кран. Если его от*
1& ТЕРМОДИНАМИКА В БИОЛОГИИ
крыть, то газы будут переходить из одной камеры в другую. При достижении
равновесия оба газа равномерно распределяются в обеих камерах. Поскольку
общее число молей газа п—пх+пъ, общий объем V и общее давление Р при
смешивании газов не изменяются, то, согласно закону идеального газа:
PV = nRT (где R = 8,314 Дж • моль-1 • К"1),
(1.7).
абсолютная температура Т также не изменяется. Далее, энергия идеального газа»
зависит только от температуры (для идеального одноатомного газа кинетическая
Начальное
состояние
рк-р
Ktav
Т
^-v
(XJ
к_У
РВ=Р
^нач
Т
Конечное
состояние
р =— р
*А 2^
2Унач
Г
Р =-Р
ГЬ 2
Рис. 1.1. Энтропия и «порядок» в системе: энтропия смешивания.
В начальный момент камеры разделены и в каждой находится один нз идеальных газов А
и В; затем камеры соединяются и система достигает нового равновесного состояния. Хотя
температура (и энергия; см. текст) обоих газов не меняется, доступный объем для газов
удваивается, и в результате (процесса смешивания) парциальное давление обоих газов
уменьшается в два раза. Согласно уравнениям (1.7)—(1.12), процесс «разупорядочивания»
сопровождается увеличением энтропии системы, несмотря иа то что общий объем,
температура, давление и энергия остаются неизменными.
энергия равна г]^ЯТ\ потенциальная энергия равна нулю, т. е. между молекулам»
идеального газа отсутствуют межатомные силы притяжения или отталкивания)
и^ следовательно, энергия при смешивании остается постоянной (dE=0). Из
уравнений (1.4) и (1.6) можно теперь рассчитать изменение энтропии в процессе
смешения:
dS = dq/T = (dE — dw)/T = —dw/T,
Поскольку для идеального газа
dw = — PdV,
уравнение (1.8) можно переписать в виде
dS=PdV/T = nR(dV/V).
(1.8)
(1.9)
(1.10)
Следовательно, процесс смешивания можно рассматривать как простое
расширение газа от начального объема Vn&4 до конечного объема VKonsx2V1,BT,. Тогда
суммарное изменение энтропии можно рассчитать как сумму индивидуальных
16 ЧАСТЬ 1
изменений энтропии двух газов А и В:
Д5 = SK0H — SHaq = ASa + Л<$в =
V V
vкон v кон
= nAR {dV/V + nBR ^dV/V = (1.11)
V V
vнач нач
= nAtf [In (21/нач) - In (1/нач)] + nBR [In (2VHa4) - In (VRa4)] =
= («A + "В) R ^ (21/нач/Инач) = (nA +nB) R\n2. (1.12)
В результате процесса смешивания общее давление, объем, температура и
энергия в рассматриваемой системе остаются постоянными, однако происходит
изменение энтропии; это изменение энтропии связано с увеличением
неупорядоченности, в системе: молекулы газов А и В теперь случайным образом
перемешаны друг с. другом, а не разделены на две группы. Говоря статистическим
языком, существует лишь один способ существования молекул А и В отдельно друг
ют друга, но существует бесконечное число способов расположения полов.ины
молекул А в каждой камере; следовательно, как только открыт кран, гораздо
•более вероятно равномерное распределение молекул А и В, чем их полное
разделение по камерам. '[Мы еще встретимся с таким же доводом в ч. 5 при
решении вопроса о том, как распределить общую энергию (а не число молекул)
системы разными способами.]
Связь между энтропией и неупорядоченностью является гораздо более
общей, нежели в данном примере, и часто привлекается для объяснения
наблюдаемого самопроизвольного протекания энергетически невыгодных химических
реакций (гл. 4).
Для обратимого пути процесса наиболее просто рассчитать
изменения энтропии по уравнению (1.6), причем конечное
суммарное, изменение энтропии также определяется только параметрами
начального и конечного состояний системы и не зависит от пути
между ними.
Первый и второй законы термодинамики вводят две функции
состояния — энергию Е и энтропию S, изменения которых не
зависят от пути, выбранного для перехода между конкретными
начальным и конечным состояниями. Существуют некоторые другие
очевидные функции состояния: температура Т, давление Р и объем
У. Уже указывалось, что теплота и работа не являются функция-
ми состояния, поскольку зависят от пути. Действительно,
последнее положено в основу конструкции тепловых двигателей и
холодильников: при переносе рабочего вещества (в данных случаях,
пара или фреона) в замкнутом циклическом процессе может
происходить переход работы в теплоту (или теплоты в работу).
Существуют еще три полезные функции состояния, образуемые
комбинациями предыдущих функций: энтальпия Н, свободная энергия
Гиббса G и свободная энергия Гельмгольца А
н =
G =
А =
= £+ PV
Н-
■Е-
-TS
-TS
(1.13)
(1.14)
(1.15)
Функции
состояния
Таблица 1.3
Термодинамические функции состояния и основные термодинамические уравнения
Название
Основное уравнение
Замечания
Р
V
т
Е
Н
Давление
Объем
(Абсолютная)
температура
Энергия
Энтропия
Энтальпия
dw= —PdV + dw'
PV = nRT
dE = bq-\- 8w
dE = dq -\- dw
dE = dq — PdV+ dw'
dS = dq/T
(второй закон термодинамики)
dE = TdS-PdV+dw'
H = E + PV
dH = dE + PdV+VdP
dH = TdS + VdP + dw'
dS = dH/T
G
A
Свободная энергия
Гиббса
Свободная энергия
Гельмгольца
а = н — ts
dG = dH — TdS — SdT
dG = VdP — SdT + dw'
A = E — TS
dA = dE — TdS — SdT
dA = dw = —PdV + dw'
Поскольку работа, производимая системой над
окружающей средой, отрицательна, а при расширении
системы Р и dV положительны, то перед первым
слагаемым необходим знак минус. dw'-~^другая
(обратимая) работа, отличная от PV
Уравнение состояния для (только) идеального газа
Первый закол термодинамики (основное уравнение)
Для (только) обратимых процессов
Для (только) обратимых процессов; dw — работа,
отличная от PV
Это уравнение определяет изменения энтропии 5 при
обратимом пути. Рассчитанное значение изменения
энтропии применимо для любого пути между теми
же начальным и конечным состояниями (хотя dS уже
не равно тогда bq/T)
Для (только) обратимых процессов
По определению
В общем случае
Для (только) обратимых процессов
При постоянном Р; dw' — Q (совершается только
работа PV)
По определению
В общем случае
Для (только ) обратимых процессов
По определению
В общем случае
Для (только) обратимых процессов, постоянная,
температура; dA — максимальная работа, совершаемая при
переходе системы из данного начального состояния
в конечное
18 ЧАСТЬ I
Таблица 1.4
Критерии существования равновесия в системах,
некоторые специфические свойства которых постоянны
Предварительно определенные условия
Поведение указанной функции состояния
Любой обратимый процесс
Теплоизолированная бомба
[постоянный V; адиабатический процесс
(теплообмена нет); dw=0
(совершается только работа PV)]
Любой обратимый процесс
Любой обратимый процесс
Изолированная система
(постоянные Е н V; dw'=0)
Любой обратимый процесс
Адиабатический процесс в
калориметре (постоянное Р; dq=0; dw=0)
dE=dq—PdV+dw
\
d£=0 (Е минимальна при
равновесны)
dS=dqfT
dE=TdS—PdV+dw
I
dS=0 (S максимальна при
равновесии)
dH=dq+VdP+dw
\
dff=0 (Н минимальна при
равновесии)
Любой обратимый процесс
Типичные условия химической
реакции (постоянные Т и Р; dw=0)
dG=VdP—SdT+dw
\
dG = 0 (G минимальна при
равновесии)
Любой обратимый процесс
Замкнутая реакционная кювета с
контролируемой температурой
(постоянные Т н V; dw'=0)
dA=dE—TdS—SdT
i
dA=0 (А минимальна при
равновесии)
Соотношения между основными функциями состояния (Р, V, Т,
Е, S, Я, G, А) приведены в табл. 1.3; эти соотношения
используются в данном разделе при рассмотрении приложений
термодинамики.
Одна из главных причин привлечения некоторых
перечисленных в табл. 1.3 функций состояния обусловлена тем, что они
служат в качестве критериев равновесия. Равновесие существует, если
все макроскопические свойства системы постоянны; однако если
предварительно указывается, что некоторые свойства постоянны
(пусть Т и У), то часто для выяснения вопроса, достигнуто ли в
системе равновесие, надо проанализировать лишь еще одну
функцию состояния (в данном случае А). В табл. 1.4 указаны
критерии равновесного состояния при различном выборе свойств,
считающихся постоянными.
Данные, приведенные в табл. 1.3 и 1.4, помогают объяснить
роль разных термодинамических функций состояния. Роль
функций Р, V и Т очевидна, однако их значение не столь уж велико.
Функции Е и S следуют непосредственно из первого и второго за-
19 ТЕРМОДИНАМИКА В БИОЛОГИИ
конов термодинамики, однако также не находят очень большого
практического применения. Изменения энтальпии И можно прямо
связать с процессами, происходящими в калориметре; изменения
этой функции применяются для характеристики прочности связей,
когда суммарное изменение энтальпий связей, трансформируемых
в химической реакции, проявляется в виде теплоты, выделяемой
в калориметре. Свободная энергия Гиббса (далее называемая
просто свободной энергией) при постоянных температуре и
давлении лежит в основе простого критерия равновесия, а также
показывает, что равновесие в таких условиях достигается благодаря
взаимосвязанным изменениям: увеличению 5 и уменьшению Е.
Свободная энергия Гельмгольца А характеризует максимальную
работу, которую можно получить в данном процессе; большинство
процессов проводится йе в оптимальных условиях, поэтому
действительная работа, получаемая при переходе из одного и того же
начального состояния в конечное, всегда меньше, чем dAb
указанная в табл. 1.3, но для сравнения все же полезно знать
максимально возможную работу.
Термохимия: применение полных дифференциалов
Напомним, что изменения термодинамических функций
состояния (Р, V, Г, Е, Н, S, G, А) зависят только от начального и
конечного состояний системы и не зависят от пути между ними. По-
видимому, очевидно, что температура, объем и давление обладают
этим свойством, т. е. являются функциями, удовлетворяющими
уравнению (1.3). Экспериментально установлено это и для
энергии Е (сущность первого закона термодинамики). Второй закон
термодинамики эквивалентен заявлению, что энтропия 5 также
обладает этим свойством. Поскольку остальные
термодинамические функции Я, G и А являются просто линейными
комбинациями перечисленных выше, то они также должны описываться
уравнениями полных дифференциалов.
Перейдем к рассмотрению химических примеров. Поскольку
Я — функция состояния (т. е. dH — полный дифференциал), то в
основу классификации прочности связей можно положить теплоту,
выделяемую или поглощаемую в химических реакциях. Кроме
того, можно вычислить теплоту реакции (из данных прочности
связей) в тех случаях, когда ее нельзя найти непосредственно из
опыта.
Прежде всего можно экспериментально определить изменения
энтальпии АН. Согласно определению (табл. 1.3), очевидно, что
значение АН можно измерять как теплоту, выделяемую (или
поглощаемую) при проведении процесса обратимо при постоянном
давлении в отсутствие электрического или магнитного полей:
H=E+PV (1.13)
20 часть i
или
Однако
так что
dH=dE-\-PdV+VdP. (1.16)
dE=dqo6p-^dwo6p-{-dWo6p, (1.1)
dH=dq06p— PdV+dw'o6P-{-PdV-{-VdP. (1.17)
Следовательно, при day обр =0
dH=dqo6p (при постоянном Р) (1.18а)
или
ДЯ = <7обр (ПРИ постоянном Р)
(1.186)
Поскольку Я—функция состояния, значение ЛЯ,
экспериментально определенное для обратимого пути процесса при
постоянном давлении, справедливо и для любого другого пути
независимо от того, является ли этот путь обратимым или нет.
Экспериментально измерения проводятся просто. Например, при изучении
эндотермического процесса (т. е. происходящего с поглощением
тепла) можно измерять мощность электроэнергии (рассеиваемую
в виде тепла в реакционной ячейке), необходимую для
поддержания постоянной температуры в реакционной смеси; затем,
интегрируя (суммируя) мощность по времени реакции
(мощность=энергия/время), с помощью такого «калориметрического»
эксперимента находят энергию (теплоту), поглощенную в ходе реакции, т. е.
суммарное количество тепла, которое требовалось для того, чтобы
температура в процессе реакции не снижалась.
В общем случае ЛЯ зависит от температуры, давления и
фазового состояния (жидкого, твердого, газообразного) реакционной
смеси, поэтому при сопоставлении нескольких реакций
необходимо выбрать некое стандартное состояние (стандартные условия).
За стандартное состояние чистого химического элемента принято
состояние этого элемента в его естественном виде (твердое
вещество, жидкость, газ) при 25 °С ( — 298 К) и I атм (101,325 Н/м2).
Это определение подходит для газов при низком давлении, но для
описания реакций в растворах используются более сложные
определения (см. гл. 3). Стандартная энтальпия (или- свободная
энергия) реакции — изменение энтальпии (или свободной энергии),
сопровождающее превращение фиксированного числа молей
реагентов в продукты при условии, что все реагенты и продукты
находятся в стандартных состояниях. Стандартную энтальпию (или
свободную энергию) обычно обозначают соответствующей буквой с
.21 ТЕРМОДИНАМИКА В БИОЛОГИИ
верхним индексом °; например, для простой химической реакции,
протекающей по уравнению
А-\-В *■ C-\-D (АЬВ,С и D находятся в стандартных
состояниях)
Н;х=Н9с+Нр~Н°А~Н0в. (1.19)
Наконец, наиболее удобно для сопоставления состояние, при
котором энтальпия Я по определению считается равной нулю.
Так, стандартную энтальпию индивидуального химического
элемента при 25°С и 1 атм обычно считают равной нулю и
используют в качестве точки отсчета. Проиллюстрируем вышеизложенное
на следующих примерах.
Пример. Определение энергий химических связей из теплот образования
Изменение энтальпии при образовании 1 моля химического соединения из
элементов при стандартных условиях (25 °С, 1 атм) называется стандартной
теплотой образования данного вещества ДЯгэв • Рассмотрим, например, образование
cc-d-глюкозы:
бСграфит -j- *К-)2 газ ~~г ^"2 газ *" СвН12Ов Кристалл
О
Д#29з (образование глюкозы) =
= Д#298 (продукты) — ДЙ298 (Реагенты) =
= Д#298 (глюкОза) — А6#298 (графит) — ДЗ^зэв (02)газ — Д6#298 (Н2)Газ =
= Д//298 (глюкоза)* = —215,8 ккал/моль — —902,9 Дж/моль.
Если Д#2м — отрицательная величина (как в обсуждаемом примере
теплота образования глюкозы), это означает, что тепло выделяется в результате
превращения реагентов в продукты реакции. Следовательно, энтальпия продукта
(или сумма энтальпий продуктов) должна быть меньше энтальпии реагентов, а
теплота образования характеризует прочность химических связей,
образовавшихся в результате реакции, т. е. —&Н%Ъ9 выражает количество тепла (энтальпию),
которое надо «ввести» в соединение, чтобы разорвать его связи и образовать
индивидуальные элементы.
Для глюкозы ДЯгав равна сумме энтальпий образования всех химических
связей в молекуле, а именно связей С—Н, С—О, С—С и О—Н. Для определения
энтальпии образования конкретной связи надо обратиться к более простой
молекуле. Например, для определения ДЯ§вв связи С—Н рассмотрим молекулу
метана СН*. Сравнивая энтальпии образования метана СН< и этана СгНв, можно
найти Д#2вз связи С—С (см. задачи в конце главы). Энтальпию связи О—Н
можно определить из энтальпии образования воды. Таким способом можно
составить таблицу средних значений энергий для большинства химических связей
(выраженных стандартной энтальпией образования связи). Эти значения затем
можно использовать при расчете ожидаемой энтальпии образования в реакциях,
для которых не существует доступных экспериментальных данных; рассчитанная
таким образом энтальпия образования может также сравниваться со значениями,
найденными методами квантовой механики.
Для определения средней энергии связи С—Н в молекуле СН4 энтальпию
образования четырех связей С—Н делят на 4. При этом получают стандартную
энтальпию образования • одной средней связи С—Н. Для этого надо
определить Д#198 процесса образования метана СН4.
* Для каждого элемента #§9в=0.
22 часть i
Путь № 1.
газ
+ 4Н-
газ
->- СН^газ
О О
АНА = Д#298 (СН4) = 4ДЯ298 (среднее значение для связи С—Н)
Экспериментально измерить АНА этой реакции невозможно. Однако,
поскольку Н — функция состояния, АН а можно получить косвенно, если- использовать
любой путь, приводящий от данных реагентов к данному продукту. Поэтому,
записав данные для трех следующих экспериментально исследуемых реакций
Q-рафит
2Н-
Ографит "г 2Н2 газ
~*~ С'газ
->- н
2 газ
сн,
4 газ
АНВ = Л#298 = 171,7 ккал/моль
АНС = ДЯ298 = —104,2 ккал/моль
АНр— АН29В = —17,9 ккал/моль
можно составить другой путь, приводящий от тех же реагентов к тому же самому
продукту.
Путь № 2.
V/ газ + 4Н-*о *СН4а„
АН*
'графит
IMJc
+ 4Н-гш *С
АН*
градшт
+ 2Н
г га»
Пользуясь тем, что Н — функция состояния, можно тут же записать
АНА = — АНВ + 2Д#С + AHD =
= —171,7 + 2 (—104,2) + (—17,9) = —398,0 ккал/моль,
так что средняя величина энтальпии связи С—Н в CPU составляет —'(398,0 : 4) =
——99,5 ккал/моль.
Здесь уместно отметить несколько особенностей. Во-первых,
полученная величина непосредственно характеризует энергию
химической связи, причем были использованы экспериментально
измеренные изменения энтальпий в некоторых химических реакциях.
Во-вторых, благодаря тому что Я — функция состояния, найдено
изменение энтальпии в (недоступной для исследования)
изучаемой реакции анализом другого (более длинного) пути,
составленного из процессов, изменения энтальпий в которых можно
измерить экспериментально. И наконец, в-третьих, рассчитанное
значение энтальпии связи представляет собой среднее значение
индивидуальных энтальпий, соответствующих последовательному
удалению атомов водорода из молекулы метана:
СН4 ► СН3. + Н. АН1
СН3. * СН2. + Н. ДЯ,
сн2. —>- сн. +н. д#3
сн. —•- с- +н. дя4
23 ТЕРМОДИНАМИКА В БИОЛОГИИ
■f
(Последние данные относительно указанных индивидуальных
энтальпий свидетельствуют о том, что A#i>A#2>A#3>"A#4.)
Энергию связи можно определять спектроскопически, измеряя
минимальную энергию фотона, достаточную для полной
диссоциации двух связанных атомов. Энергии связей можно также
получить из экспериментов, в которых молекулу подвергают
бомбардировке электронами высокой энергии, определяя минимальную
энергию электронов, достаточную для разрыва исследуемой связи,
и проводя масс-спектрометрическую идентификацию
образующихся при бомбардировке фрагментов молекулы. Наконец,
исследование колебательных спектров (ИК-область) позволяет найти
силовую постоянную химической связи (см. ч. 5). При этом
химическую связь рассматривают как механическую пружину,
связывающую два исследуемых атома.
Из всех этих характеристик энергии химической связи первые
и наиболее полные значения были получены измерениями
энтальпий реакций методом калориметрии (как указано выше) или из
данных.о температурной зависимости констант равновесия (гл. 4)
или э. д. с. (гл. 5). Сравнение энтальпий связей для одного и того
же атома в разных молекулах (см. задачи в конце главы)
значительно дополняет представления о природе химических связей в
молекулах и комплексах и о роли растворителей в химических
реакциях.
Пример. Теплоты химических реакций, недоступных для прямого исследования
Многие .интересные химические реакции, в частности большинство реакций,
происходящих в живых клетках, часто недоступны для прямого исследования.
Однако с помощью способа, описанного в предыдущем примере, можно
определить изменение любой функции состояния при переходе от реагентов к продуктам,
рассматривая другой (обычно более длинный, но более удобный) путь реакции.
Если известны теплоты образования из элементов для всех соединений (это
обычно справедливо для большинства промежуточных соединений в
биохимических процессах), то можно определить теплоту реакции, например разлагая все
реагенты до элементов, а затем образуя все продукты из элементов, входящих
в состав их молекул, как проиллюстрировано в следующем примере.
Рассмотрим гидрогенолизную систему образования муравьиной кислоты в
бактерии Е. coli. В этой системе фермент (см. ч. 3) катализирует (ускоряет)
реакцию взаимодействия водорода с бикарбонат-ионом, приводящую к
образованию формиат-иона и воды. Можно легко показать [1], что присутствие любого
истинного катализатора не может влиять на равновесные свойства (т. е.
состояние) системы, хотя катализатор может очень сильно увеличить скорость
достижения равновесия. Следовательно, при анализе исследуемой реакции присутствие
фермента в системе можно не принимать во внимание.
Путь № 1
__ фермент
Н2 газ + НСОз водн 25°С( i аТм НС00^дн + Н20жидк
Для того чтобы определить ЛЯ§98 реакции, происходящей по пути № 1,
начнем с того, что выпишем все доступные значения энтальпий, имеющие
отношение к этой реакции:
Сграфит + н2 газ + 02 газ »» НСООНЖидк Д#л = AtfLo = —99 750 кал/моль-
24 ЧАСТЬ I
Q-рафит "г 2 газ —' *" С02 газ
Д//в = Д#298 = —94 240 кал/моль
"а газ 4" 11чРг газ *" "г^Ькидк
Д#с = Д#298 ~ —68,310 кал/моль
С02 газ + Вода »- С02 насыщн.водн
ДЯд = Д#298 = —4844 кал/моль
С02 насыщ.води ~г "г^жидк *" ^ЦСОз води
Д#£ = ДЯддз = 0 (одни и те же
вещества)
Н2С03 водн *" ^водн ~Ь НСОз водн
Д#р = ДЯ^8 = +2075 кал/моль
НСООНЖидк + Вода *- НСООНводц
ДЯ0 = Д#298 = —1(*0 кал/моль
НСООН
води
■^н+дн + нсоо-дн
Д#я = Д#298 = —^ кал/моль
Замечание. Определение стандартного состояния растворенного вещества
(ионогенного или нейтрального) несколько более сложно, чем такое
определение для элементов ил» простых веществ, о которых шла речь до сих
пор. Это определение дано ниже в гл. 3; на данном этапе достаточно
указать, что за стандартное состояние вещества в растворе можно
приближенно принять его I M водный раствор.
Далее реконструируем исследуемый путь № 1, подбирая подходящую
последовательность реакций, теплоты которых известны.
Путь № 2
^г(а+ НСОз щн + Н щц
Наш + NCOOflHw + Н20
водн
Н2С03 в0дН
НСООН
водн
C0W+H2°«"ff*
НСООН
жидк
н2^ст + 0гщ+н2аа* \о2ш
С фцрц,* Н2ад,+ 02ав+ Н2ди+ ^°2да
Для удобства в левую и правую части исходного уравнения реакции
введены ионы Нводн, Сохраняя знаки при энтальпии ДЯ298 соответственно
последовательности реакции в схеме пути № 2, можно легко рассчитать искомую теплоту
реакции, проходящей по пути № 1
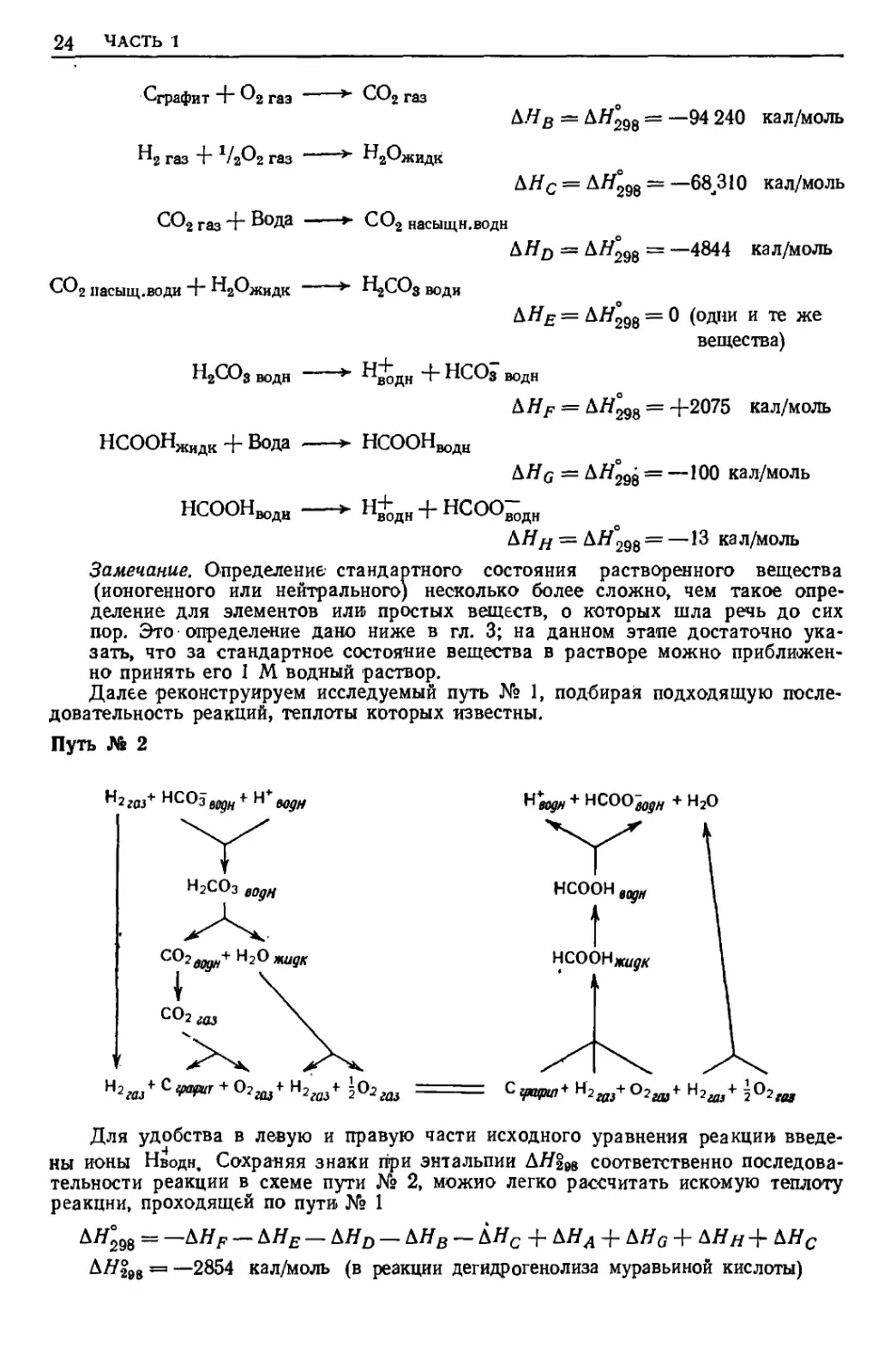
ДЯ°98 = —ЬНР — ДЯ£ — Д#0 — ДЯВ — ДЯС + ДЯЛ + ДЯ0 + ДЯЯ + ДЯС
Д#2в8 =3 —2854 кал/моль (в реакции дегидрогенолиза муравьиной кислоты)
!'25 ТЕРМОДИНАМИКА В БИОЛОГИИ
If Записанная реакция является экзотермической, т. е. при
протекании реакции слева направо при указанных концентрациях,
температуре и давлении тепло выделяется. Позже будет показано, как
;эта информация может быть использована для решения вопроса
;;0 возможности самопроизвольного протекания данной реакции на
^основании данных об изменении свободной энергии G, а не
энтальпии в качестве исследуемой функции состояния. Значение
Д#2°98 позволяет также предсказать качественное изменение
константы равновесия реакции с температурой: если, согласно
расчетам, следует ожидать (как имеет место в действительности), что
.записанная реакция сопровождается поглощением тепла, то при
;нагревании равновесие должно сдвигаться вправо. Степень
смещения равновесия (см. гл. 4) зависит от АН.
Таким образом, в данной главе рассмотрены следующие
практически важные вопросы: получение количественной
характеристики энергии химических связей из теплот образования и расчет
изменений термодинамической функции состояния (в данном
случае энтальпии) для процессов, которые не удается исследовать
прямо. Позже будет показано, как на основании данных о тепло-
тах реакций можно предсказывать зависимость константы
равновесия от температуры.
Задачи
1. Зная теплоты реакций при стандартных условиях, рассчитайте средние
значения энтальпий углерод-углеродных связей в этане С2Нб, этилене С2Н4 и
ацетилене СгН2. Предполагается, что во всех этих соединениях энтальпия
связи С—Н характеризуется одним и тем же значением. Какие выводы об
относительной прочности простой (одинарной), двойной ъ тройной
углерод-углеродных связей можно сделать нз полученных в результате расчета значений?
2Сграфит + ЗН2 газ »- С2Нв газ Д^298 = ~20190 кал/моль]
2Сграфит + 2Н2 газ > С2Н4 газ Д#2вв= 12555 кал/моль
2Сграфит + Н2 газ *- С2Н2Газ ДЯ298 = 54230 кал/моль
Н2Газ ** 2Н.гаэ Д#298= 104200 кал/моль
О-рафит »- С «газ ДЯ^д = 171700 кал/моль
Сграфит + 2Н2 газ *• СН4Гаэ ^^29&~—17865 кал/моль
2. Энтальпии образования НгОжидк н HjOraa составляют —68,32 и
—57,80 ккал/моль соответственно. Рассчитайте энтальпию испарения воды при
298 К
2Б °С, 1 атм
Нг^жидк *" "г^гаэ
(Теплота испарения соответствует энергии, необходимой для разрыва
межмолекулярных связей в жидкости.)
Человек в результате происходящих в его организме процессов метаболизма
выделяет ~2500 ккал/сут. Если поглощение 1 кал (тепла) повышает темпе*
26 ЧАСТЬ 1
ратуру воды на 1 К, рассчитайте увеличение температуры тела человека при
условии, что все выделяемое им тепло поглощается его телом
(предполагается, что тело человека в основном состоит из воды н поглощает тепла столько
же, сколько равное количество воды). Принять массу человека равной 70 кг.
В действительности организм человека не изолированная система, н
основным путем теплоотдачи является испарение воды (например, с выделением
пота). Рассчитайте количество воды, которое должно было бы испариться за
счет тепла, вырабатываемого в результате процессов метаболизма за одни
сутки.
Литература
1. Klotz M., Energy Changes in Biochemical Reactions, Academic Press, New
York, 1967. Прекрасное краткое рассмотрение.
2. Waser J., Basic Chemical Thermodynamics, W. A. Benjamin, New York, 1966.
Хорошее изложение основного материала.
!\ЛАВА 2
[ИМИЧЕСКИЙ ПОТЕНЦИАЛ
>■
i
Данные, приведенные в табл. 1.4, указывают на то, что
существование и характеристики любого химического равновесия луч-
ре всего можно установить детальным анализом общей свободной
энергии Гиббса G вблизи состояния равновесия. Однако,
поскольку в большинстве исследуемых равновесных процессов участвуют
ю меньшей мере два химически неидентичных вещества
(компонента), прежде всего необходимо выяснить, каким образом общая
Свободная энергия складывается из соответствующих свободных
мергий соединений. К сожалению, в общем случае свободная
Энергия смеси не равна простой сумме свободных энергий
отдельных компонентов, так как процесс смешивания может приводить
j изменению свободной энергии каждого компонента. Это
положено проще усвоить по аналогии, если рассмотреть другое экстен-
'ивное свойство (т. е. свойство, зависящее от количества вещест-
а, присутствующего в системе; интенсивное свойство, например
емпература или давление, не зависит от количества вещества в
истеме) —объем смеси двух компонентов.
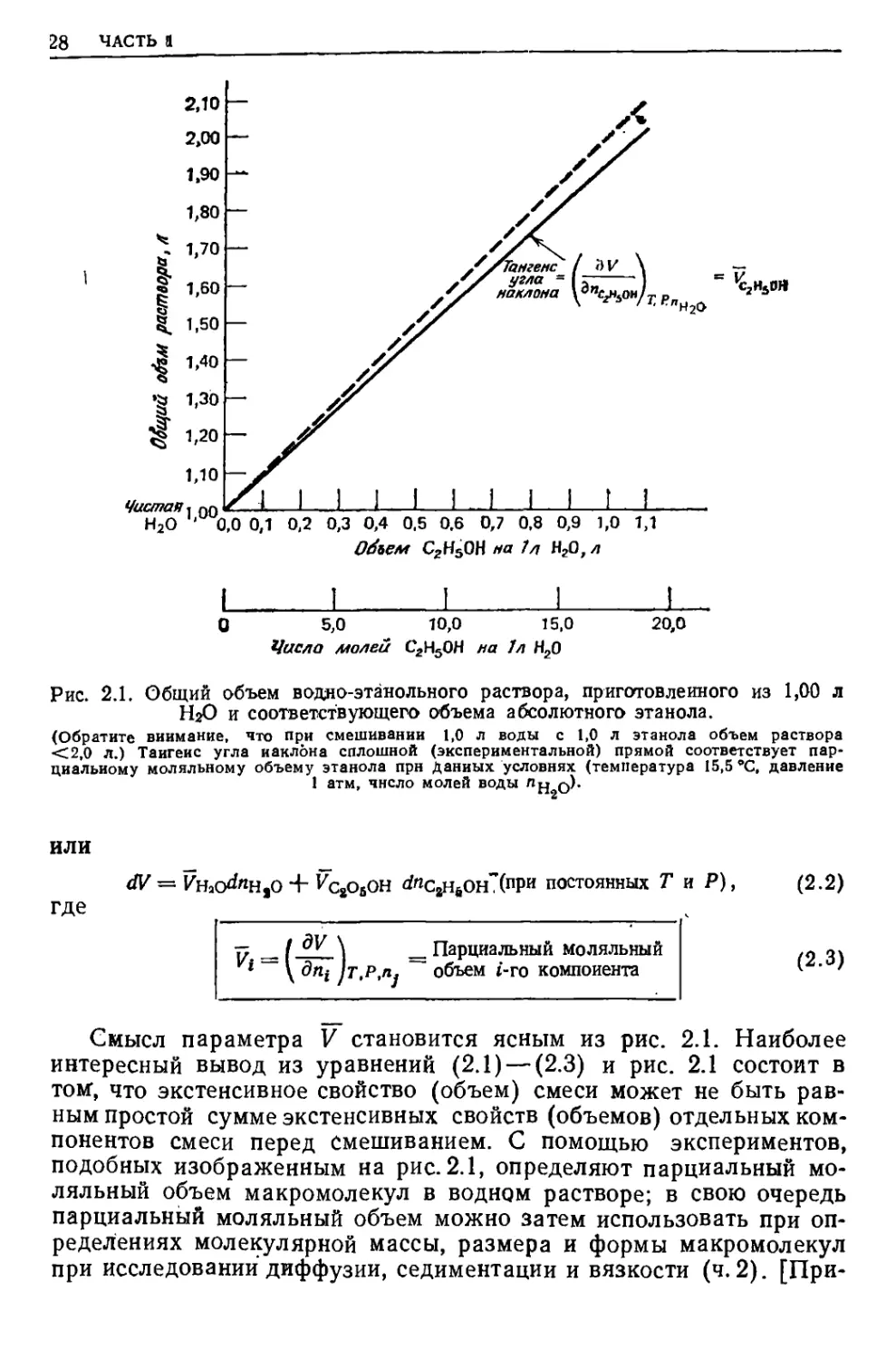
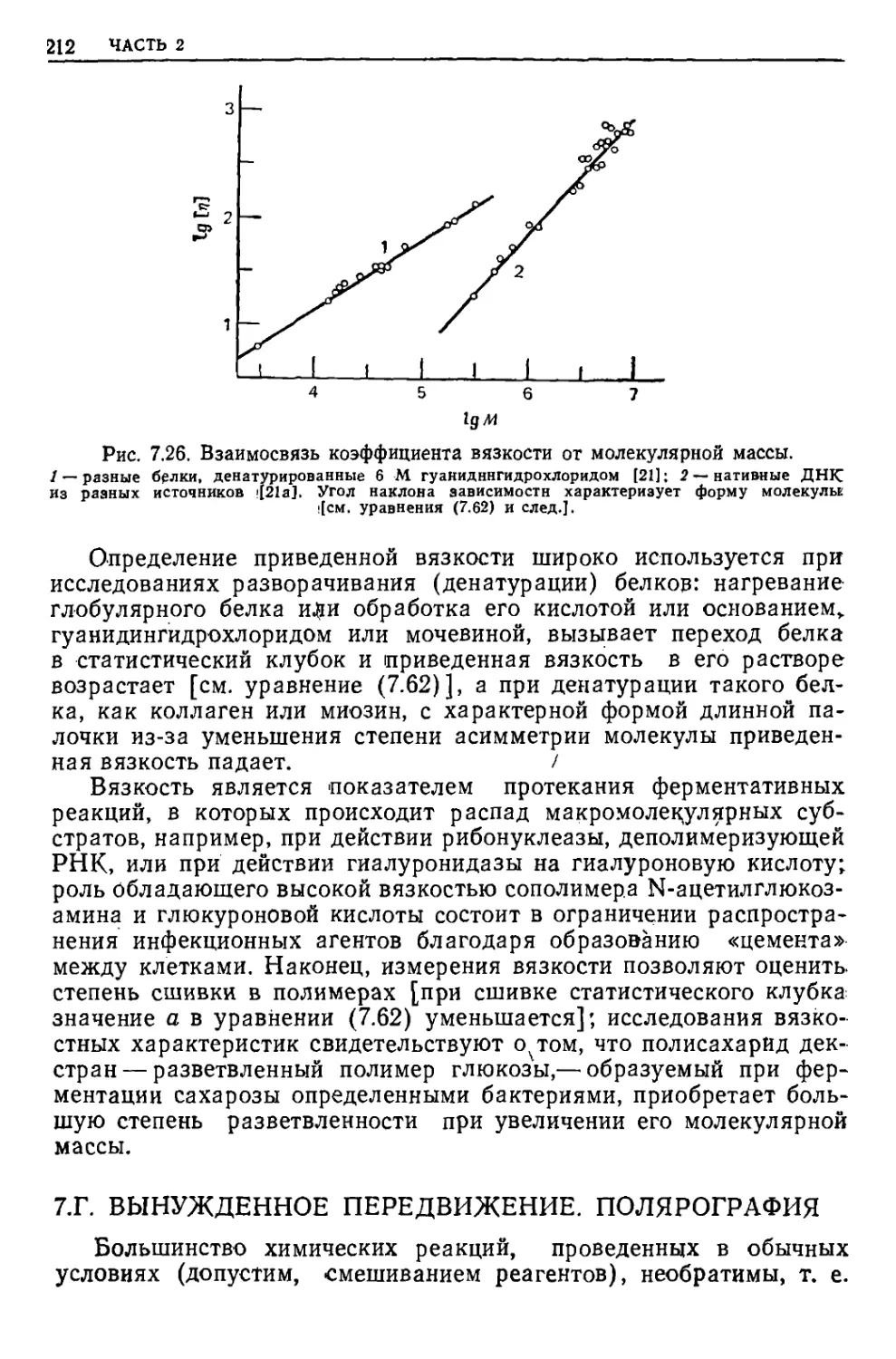
На рис. 2.1 показано, что объем смеси (раствора) после смеши-
зания 1,00 л воды и 1,00 л этанола не равен 2,00 л, а в
действительности немного меньше. (При смешивании воды и этанола в
любом соотношении суммарный объем раствора всегда меньше
£уммы начальных объемов воды и этанола, внесенных в
смеситель.) Этот результат просто отражает тот факт, что силы притя-
кения между молекулами воды и этанола отличаются от сил
притяжения, действующих между молекулами в каждом
растворителе. В общем случае, поскольку общий объем смеси V зависит от
Температуры Т, давления Р и количества каждого компонента
(числа молей п) и является функцией состояния, можно выразить
4V в виде полного дифференциала:
!' / dV \
\ дТ /р'лс2нвон.лн2о
£ +
( * )г."сан6он."н,о dP + (dZo )т.р.пС2ЩОН dn»*° +
fr / dV \
I +l^c8H6OHjr,P.„Hg0^2H6OH (2.1)
28 ЧАСТЬ 1
раствора, /г
i
1
2,10
2,00
1,90
1,80
1,70
1,60
1,50
1,40
1,30
1,20
1,10 —
Чистая
Н20
1.00
е2*ь0н/Т, Рл
*СгНй0И
н2о
0,0 0,1 0,2 0,3 0,4 0.5 0.6 0,7 0,8 0,9 1,0 Т,1
Объем СгН50Н на ?л Нг0, л
L
1
I
1
5,0 10,0 15,0
Числа молей С2Н50Н на 7л Н20
20,0
Рис. 2.1. Общий объем водно-этанольного раствора, приготовленного из 1,00 л
HjO и соответствующего объема абсолютного этанола.
(Обратите внимание, что при смешивании 1,0 л воды с 1,0 л этанола объем раствора
<2,0 л.) Таигеис угла наклона сплошной (экспериментальной) прямой соответствует
парциальному моляльному объему этанола прн Данных условиях (температура 15,5 *С, давление
1 атм, число молей воды л^ q).
ИЛИ
где
М = Инас^«н,0 + ^с8овон йяздонХпри постоянных Т и Р), (2.2)
Vi-[ дщ )т.Р,П; ~~
Парциальный моляльный
объем t-го компонента
(2.3)
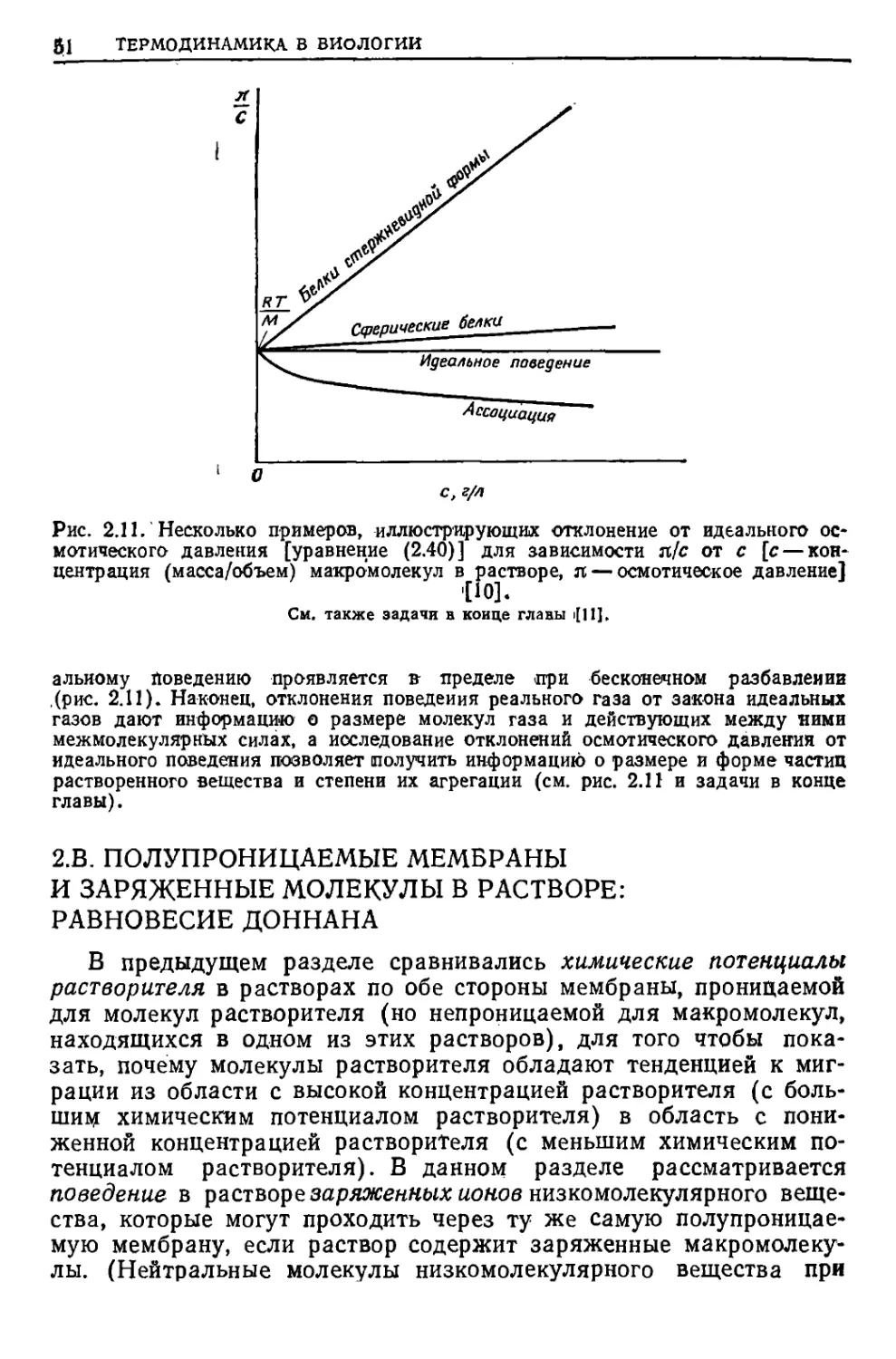
Смысл параметра V становится ясным из рис. 2.1. Наиболее
интересный вывод из уравнений (2.1) — (2.3) и рис. 2.1 состоит в
том1, что экстенсивное свойство (объем) смеси может не быть
равным простой сумме экстенсивных свойств (объемов) отдельных
компонентов смеси перед смешиванием. С помощью экспериментов,
подобных изображенным на рис. 2.1, определяют парциальный
моляльный объем макромолекул в водном растворе; в свою очередь
парциальный моляльный объем можно затем использовать при
определениях молекулярной массы, размера и формы макромолекул
при исследовании диффузии, седиментации и вязкости (ч. 2). [При-
t29 ТЕРМОДИНАМИКА В БИОЛОГИИ
мер, приведенный на рис. 2.1, представляет некоторый интерес для
расчета крепости алкогольных напитков; в США крепость (в
градуса]^) определяют как удвоенную объемную концентрацию спирта
(объем этанола, поделенный на общий объем смеси).]
Таким же способом, как описанный выше для экстенсивной
функции состояния V, можно исследовать другие экстенсивные
функции состояния (например, Е, S, Я, G и А, табл. 1.3). В
частности, можно записать полный дифференциал свободной энергии
Гиббса G, выразив его через независимые переменные Т, Р и тц
^=(ао/ар)7.,Л1(Ла>...л^р+(ао/аг)Р(Л1(Л2,...л^г+
(CQ/C/Zjj7ip> постоянные количества Ш1\» \^"^)
всех компонентов, кроме f-ro
Частные производные по давлению и температуре в уравнении
(2.4) легко идентифицировать, произведя некоторые
преобразования основных определений G, Н, Е и S для системы постоянного
состава (все m постоянны, т. е. drii=0).
Во-первых,
G=H—TS (по определению), (1.14)
так что
dG=dH—TdS —SdT. (2.5)
Однако
H=E-{-PV (по определению), (1-13)
так что
dH=dE+PdV+VdP (2.6)
и, следовательно,
dG=dE+PdV-irVdP— TdS— SdT. (2.7)
Теперь для обратимого процесса
dS=dq/T (по определению) (1.6)
и
dE=dq—PdV+dw', (1.4)
так что
dG=dq— PdV+dw' +PdV+VdP—T (dq/T) — SdT
или просто
dG=VdP— SdTA-dw' (2.8)
Наконец, для обратимого процесса в системе постоянного состава
при условии, что разрешенной является только работа PV (отсут-
+£
30 ЧАСТЬ. Л
ствуют электрические или магнитные поля), получаем
dG—VdP—SdT (постоянный состав, обратимый
процесс, только работа PV).
(2.9)
Из уравнения (2.9) можно легко найти искомые частные
производные
(2.10)
(2.П)
Теперь уравнение (2.4) запишем в более краткой форме, которая
часто используется в дальнейшем изложении
(dG/dP)Ttnitn2t.
и
(дС/дТ)РгП1,п2Г
..nt=V
..Гц = S
dG = VdP — SdT + 2 Gidni>
i
где
Gt = (dG/drii)^ П0СТ0ЯННЬ1е количества^г=Химический
всех компонентов кроме t'-ro потенциал
(2.12)
(2-13)
Символ суммы в уравнении (2.12) указывает на суммирование по
всем компонентам смеси. Из уравнений (2.10) и (2.11) вытекает
важное следствие: зависимость свободной энергии от давления
й\ли температуры. Позже будет показано, что свободная энергия
и константы равновесия взаимосвязаны, поэтому эти уравнения
применимы для предсказания изменений констант равновесия с
давлением или с температурой. В данный момент, однако,
принципиальный интерес представляет парциальная моляльная сво-
Таблица 2.1
Химический потенциал как критерий химического равновесия;
сопоставление с соответствующими критериями теплового или
механического"равновесия
Вид равновесия
По
определению при
равновесии
отсутствует
суммарный поток
Экстенсивное
свойство3
Интенсивное
свойство3
Условие равновесия
между фазами4 А и В
Тепловое Теплоты 5
Механическое Работы V
Химическое Вещества G или rit
Т
Р
0|
dS=0 или Та=Тв
(Р и щ постоянны)
dV=0 нлн РА=Рв
(Т н щ постоянны)
dG=Q или G? = Gf
(Т н Р постоянны)
а Экстенсивное свойство зависит от общего количества вещества;
зависит.
31 ТЕРМОДИНАМИКА В БИОЛОГИИ
V
Фиксированная граница
Фаза А
Граница ~>
разде/ia фаз
Фаза В
dS~dSA+dSu=
_ ~dq dq
" Та та
ds=o<z=>rA*rR
Моральная граница
Фаза а
Граница
раздела ->
/раз
Фаза В
dV
т
t?wA = PAdV
du>u = -P5dV
dw = dwr + diVfy
div = {-PA)dVA + <-PBWKB«
= PAdV-PBdV
dw = 0'
Фиксированная драница
.Фаза А
Граница
раздела ->
Фаз
Фаза В
dcA *-<?>,
4
dGB = G*dn.
dC - dCA + JCg »
. /~B.j
«-G^. + Gjtoi,
^с*о<=^>с2=<;4
a — теплового;
Рис. 2.2. Поведение двухфазной системы вблизи равновесия:
б — механического; в — химического.
При выводе критерия любого равновесия анализируется результат бесконечно малого
удаления системы от существующего условия равновесия. Видно, что при химическом
равновесии химический потенциал играет ту же роль, что температура или давление при
тепловом или механическом равновесиях (см. текст и табл. 2.1). В качестве границы раздела
(см. текст) двух фаз служит граница раздела жидкость — пар (а), непроницаемая
мембрана (б) или полунепроиицаемая мембрана (в).
бодная энергия Gi, которую иногда называют химическим
потенциалом*.
* Во многих учебниках для обозначения химического потенциала
используется более общий символ рн. Таким образом подчеркивается, что химический
потенциал (в подходящих условиях) можно определить как изменение любого
вида энергии, сопровождающее изменение количества вещества в системе
(dG\ (дИ\ -(—]
w - \ дщ )т,р,П] в { дщ )8,р,П) ~ [ дщ )s,v,nj
В данной книге целесообразнее использовать обозначение St, так как для
32 ЧАСТЬ 1
Роль химического потенциала Gt при описании химического
равновесия лучше всего, по-видимому, проиллюстрировать,
обсудив аналогичные критерии теплового или механического
равновесия, как показано на рис. 2.2 и в табл. 2.1.
Рассмотрим систему, состоящую из двух фаз (рис. 2.2),
например жидкости и пара, или двух несмешивающихся жидкостей, или
жидкости и твердого вещества. [Фазу можно определить как
систему, характеризующуюся макроскопической однородностью
(термодинамика рассматривает свойства макроскопических систем,
т. е. систем, состоящих из большого числа частиц, а не свойства
индивидуальных атомов) и по химическому составу, и по
физическому состоянию.] Можно говорить, что фазы А и В находятся в
тепловом равновесии, если от одной фазы к другой не существует
теплового потока, т. е. обе фазы находятся при одной и той же
температуре. В то же время, рассматривая (бесконечно малое)
изменение энтропии, сопровождающее перенос от одной фазы к
другой бесконечно малого количества теплоты при тепловом
равновесии (рис. 2.2,а), можно утверждать, что критерием теплового
равновесия является условие dS = 0 (табл. 2.1).
Аналогично две фазы находятся в механическом равновесии,
если между ними не происходит обмена работой. На рис. 2.2,6
показано, что условием такого равновесия является равенство
давлений обеих фаз.
Наконец, определим химическое равновесие как состояние, при
котором отсутствуют суммарные изменения в количествах всех
химических компонентов, присутствующих в системе. Тогда в
соответствии с диаграммой на рис. 2.2, в условием этого равновесия
(температура и давление постоянны; совершается только работа
PV) является rfG=0, поэтому Химический потенциал любого
компонента в одной фазе Gf главен химическому потеницалу этого
компонента в другой фазе Gi- Этот вывод можно сделать,
рассмотрев перенос бесконечно малого количества .i-го компонента
dni из фазы А в фазу В (рис. 2.2), по аналогии с примерами,
описывающими механическое и тепловое равновесия, для кодорых
рассматривались бесконечно малые изменения объема или
теплоты соответственно. В результате получаем
-~А ~fb при химическом равновесии
"' i (Т и Р постоянны, dw' = 0)
С помощью замечательного правила фаз Гиббса (см.
следующий раздел), просто используя уравнение (2.14), можно предска-
большинства процессов будут обсуждаться именно свободная энергия G, а ие
внутренняя энергия Е или энтальпия Н.
33 ТЕРМОДИНАМИКА В БИОЛОГИИ
зать, сколько фаз может существовать в системе при широком
варьировании условий.
Наиболее принципиальные выводы помещены в табл. 2.1.
Читателю следует хорошо усвоить эти выводы, так как на них
главным образом базируется изложение материала в данной главе.
2.А. ФАЗЫ И ФАЗОВЫЕ ПЕРЕХОДЫ
Фазовые переходы (плавление, переход в твердое состояние,
кипение) относятся к числу простейших «реакций», поскольку
часто в них участвует лишь один компонент. Прежде всего
необходимо решить вопрос о том, сколько фаз может присутствовать в
системе при различных условиях, и ответ на этот вопрос лежит
в основе всего обсуждаемого в этом разделе материала. Интерес
к фазовым переходам все более возрастает среди биологов,
поскольку биологические функции мембран можно скоррелировать
с фазовым составом фосфолипидного бислоя (разд. 2.А.2.).
2.А.1. СКОЛЬКО СУЩЕСТВУЕТ ФАЗ? (ПРАВИЛО ФАЗ ГИББСА)
При обсуждении данных, приведенных в табл. 2.1,
утверждалось, что состояние системы, находящейся в равновесии, можно
охарактеризовать температурой, давлением и концентрацией
(которая определяет химический потенциал) каждого химически
индивидуального составляющего (компонента) системы*. Для
описания системы, состоящей из р фаз и с компонентов, требуется
исследовать переменные параметры, общее число которых
составляет рс+2, поскольку в каждой фазе содержится с компонентов (и,
следовательно, с концентраций), в то время как температура и
давление одинаковы.для всех фаз, находящихся в равновесии.
Однако не все эти переменные независимы. Например, если
выразить концентрации различных компонентов через мольные
доли (для i-ro компонента мольная доля Х{)
*|в^-. (2.15>
i
то в любой выбранной фазе (пусть в фазе В) сумма мольных
долей всех компонентов, находящихся в этой фазе, равна единице:
Xf+*f+Xf+...+Xf=l. (2.16)
* На практике число компонентов можно определять как разность между
числом всех химических веществ (например, СаС03, СаО и СО*) и числом
химических реакций, которые связывают эти вещества (например, для данного при-
мера существует единственная реакция СаО-т-СОа=СаСОз, поэтому число
компонентов равно 3—1=2),
34 ЧАСТЬ !
Следовательно, если известны концентрации всех компонентов,
кроме одного, то его концентрацию можно рассчитать из
уравнения (2.16). То ж£ самое распространяется на каждую из р фаз
системы. Поэтому общее число параметров, необходимых для
описания состояния системы, можно уменьшить на р:
<рс+2-р)=р(с-1)+2.
Далее, согласно уравнению (2.14), в состоянии равновесия
химический потенциал, допустим, i-ro компонента в некоторой фазе
системы равен химическому потенциалу этого компонента в
любой другой фазе:
В системе с компонентов
Gc =GC — • • • =*(РС
(2.17)
(p—1) уравнений для каждого компонента
Анализ системы уравнений (2.17) показывает, что
накладывается еще с(р—1) ограничений, и это приводит к тому, что общее
число независимых интенсивных переменных (иногда называемых
степенями свободы) составляет
рли *
f = c — р + 2 (2.18)
Смысл правила фаз Гиббса, выражаемого уравнением (2.18),
легче всего понять на примере простой однокомпонентной
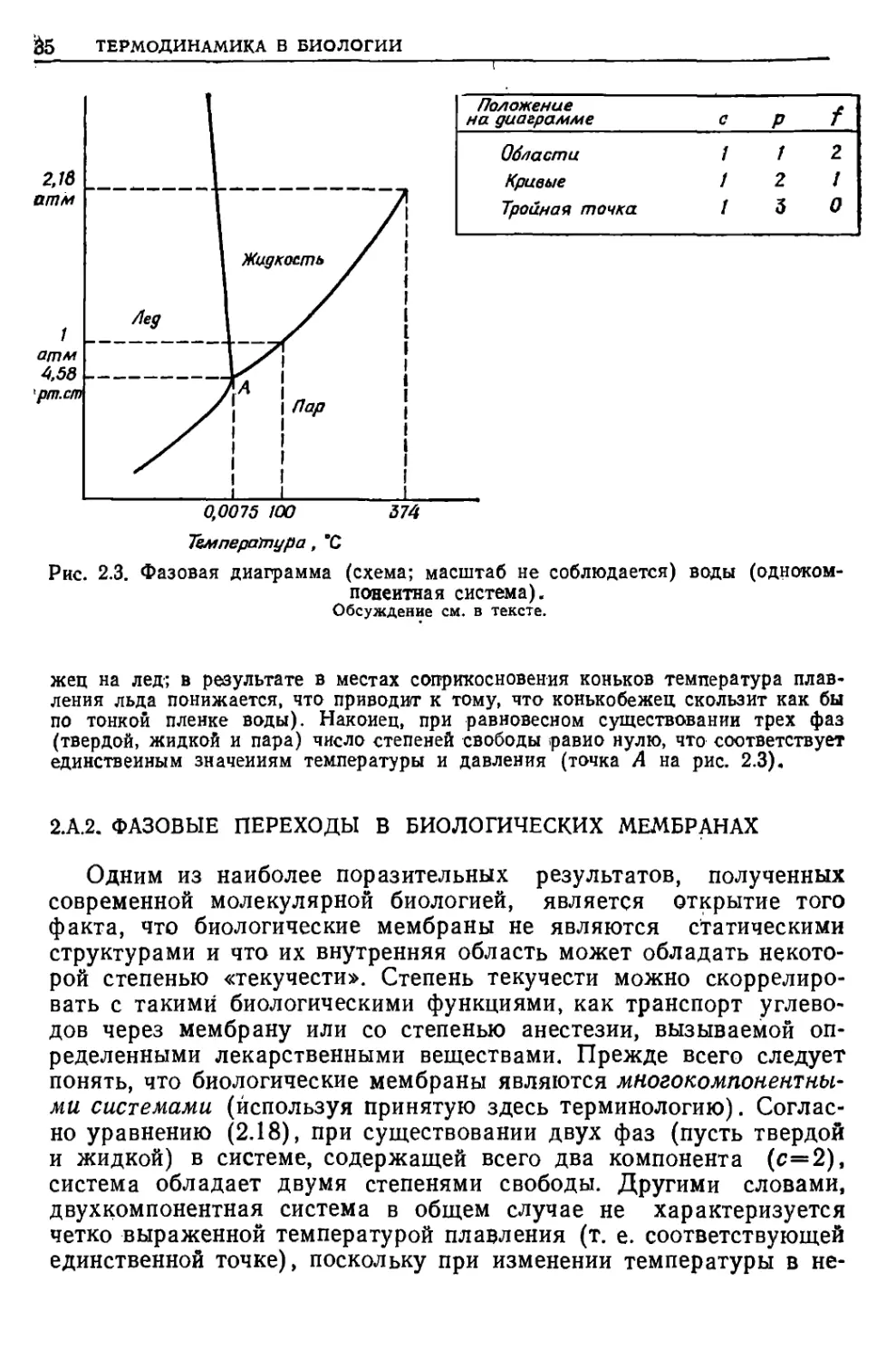
системы (рис. 2.3).
Примера Фазовая диаграмма воды (однокомпонентная система)
Уравнение (2.18) свидетельствует о том, что для однокомпонентной системы
(с=1) в зависимости от числа фаз р (1, 2 илн 3) существует f (2, 1 нлн 0)
степеней свободы, как показано в таблице (рнс. 2.3). Например, в интервале
температур и давлений, при которых вода существует в виде чистого пара, можно
изменять температуру и давление независимо друг от друга и в системе всегда
будет существовать только одна фаза (т. е. водяной пар). Точки на кривых
фазовой диаграммы, однако, соответствуют состоянию равновесия между двумя
фазами (например, между жидкостью и паром) в данной системе.
Следовательно, при фиксированном значении давления другая переменная (температура)
определяется значением на пограничной линии. Другими словами, прн данном
давлении вода характеризуется строго определенной температурой кипения.
Например, прн давлении 1 атм температура кипения воды равна 100 °С, в то время
как при уменьшении давления температура кипения воды заметно снижается.
Температура плавления льда также зависит от давлений (легкое скольжение на
коиьках отчасти объясняется высоким давлением, которое оказывает конькобе-
&5 ТЕРМОДИНАМИКА В БИОЛОГИИ
2,1 в
атм
Положение
на диаграмме
Области
Кривые
Тройная точка
1
2
3
2
/
О
0,0075 100 574
Температура, °С
Рис. 2.3. Фазовая диаграмма (схема; масштаб не соблюдается) воды (одноком-
поиеитная система).
Обсуждение см. в тексте.
жец на лед; в результате в местах соприкосновения коньков температура
плавления льда понижается, что приводит к тому, что конькобежец скользит как бы
по тонкой пленке воды). Наконец, при равновесном существовании трех фаз
(твердой, жидкой и пара) число степеней свободы >равио нулю, что соответствует
единственным значениям температуры и давления (точка А на рис. 2.3).
2.А.2. ФАЗОВЫЕ ПЕРЕХОДЫ В БИОЛОГИЧЕСКИХ МЕМБРАНАХ
Одним из наиболее поразительных результатов, полученных
современной молекулярной биологией, является открытие того
факта, что биологические мембраны не являются статическими
структурами и что их внутренняя область может обладать
некоторой степенью «текучести». Степень текучести можно скоррелиро-
вать с такими биологическими функциями, как транспорт
углеводов через мембрану или со степенью анестезии, вызываемой
определенными лекарственными веществами. Прежде всего следует
понять, что биологические мембраны являются
многокомпонентными системами (используя принятую здесь терминологию).
Согласно уравнению (2.18), при существовании двух фаз (пусть твердой
и жидкой) в системе, содержащей всего два компонента (с=2),
система обладает двумя степенями свободы. Другими словами,
двухкомпонентная система в общем случае не характеризуется
четко выраженной температурой плавления (т. е. соответствующей
единственной точке), поскольку при изменении температуры в не-
36 ЧАСТЬ 1
SO
40-
30 -
20 -
10 -
Жидкая
- (раза у
В Л А
у** *
Ж Жидкая фаза jL
/ Твердая фаза /
-**^
Тс
Твердая (раза
1 1 1 1 1 1 .Г F
\ .
~ о loo
DEL JJPL
Порержание jppj, л мол. %
Рис. 2.4. Фазовая диаграмма, описывающая фазовое разделение в водной смеси
днэлаидоилфосфатидилхолина (DEL) и дилальмитоилфосфатидилхолина (DPL)
на основании данных ЭПР [6].
Выше линии ликвидуса находится жидкая фаза, ниже линии солидуса — твердая фаза.
В области между кривыми, например в точке А, наблюдается сосуществование «жидкой»
и «твердой» фаз, и соответствующий этим фазам состав можно определить из пересечения
горизонтальной прямой, проходящей через точку А, с верхней (точка В) или нижней
(точка С) кривыми (см. текст). Точки D, Е и F соответствуют составу смесей, которые были
быстро заморожены и исследованы методом электронной микроскопии (см. рис. 2.5).
которых пределах в системе находят две фазы*. Биологическое
значение этой особенности теперь очевидно: если бы
биологические мембраны были построены из липидов только одного типа,
мембрана бы обладала резкой температурой плавления, и
организм мог бы отвечать на колебания температуры изменением
состояния и, следовательно, функции ' мембраны только по типу
«все-или-ничего». Однако благодаря многокомпонентному составу
мембрана может претерпевать постепенные изменения степени
текучести в широком интервале температур, обеспечивая гораздо
более «тонкий» контроль за функцией клетки в соответствии с
температурой окружающей среды.
Пример. Фазовые переходы в смесях синтетических фосфолипндов
Позже мы увидим (ч. 4),, что в фосфолипидную среду можно ввести
парамагнитную (нитроксидсодержащую) «спин-меченую» молекулу я что сигнал
ЭПР-спектра чувствителен к вращательной подвижности опнн-меченой молекулы.
Это свойство использовалось Мак-Коннеллом и сотр. для детектирования фазо-
* Правило фаз Гнббса таким образом объясняет, почему точка плавления
может использоваться в качестве критерия чистоты впервые выделенных или
синтезированных органических веществ: чистое соединение представляет собой
однокомпонентную систему и должно иметь резкую температуру плавления
(точку), в то 'Время как любая примесь изменяет ситуацию, создавая двухкомпонеит-
ную систему, температура плавления которой находится в некотором интервале.
38 ЧАСТЬ 1
п
6
1 I I I ( I I I L
О 50
Содержание ВРРЕ , мол. %
100%
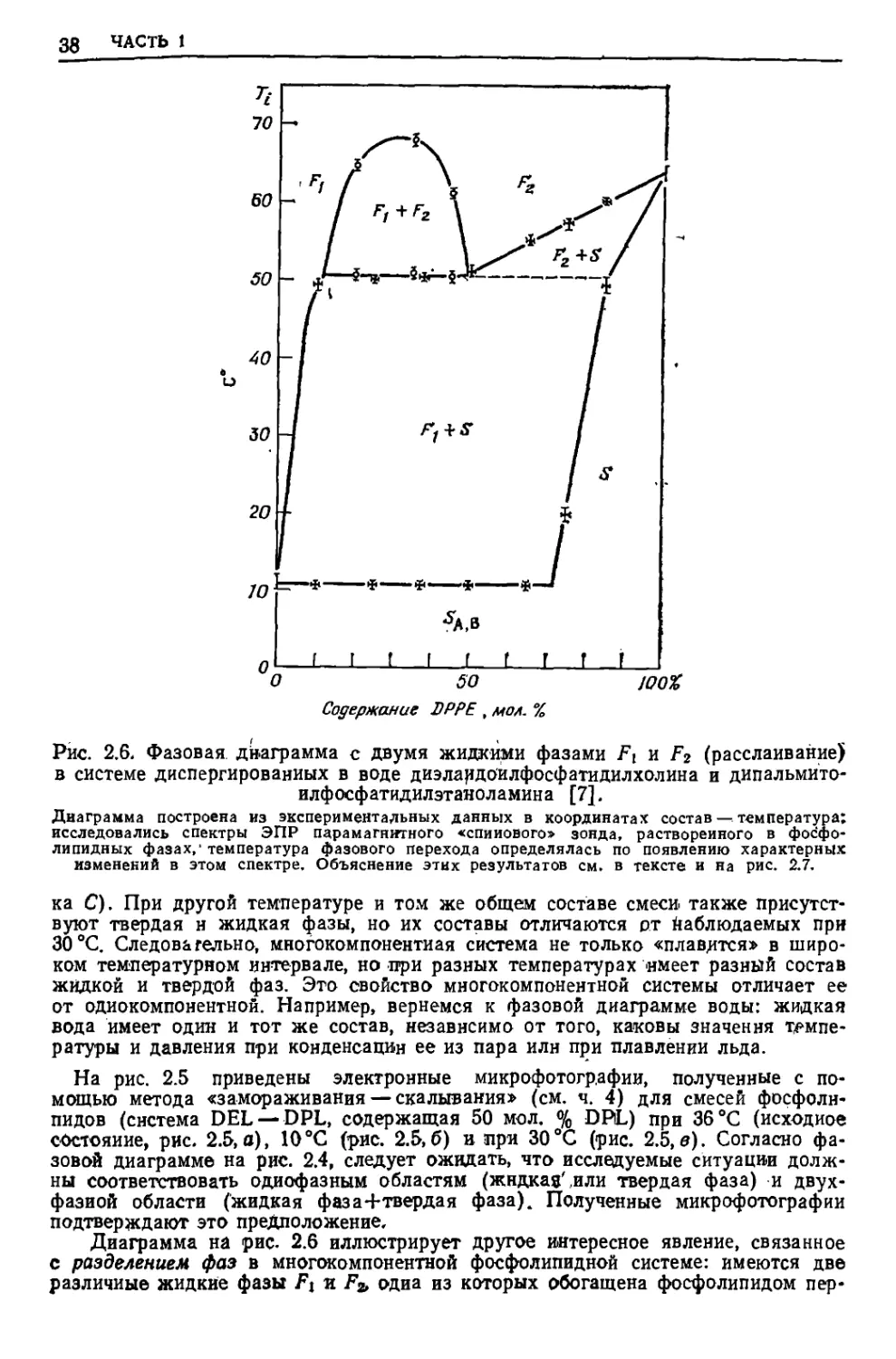
Рис. 2.6, Фазовая диаграмма с двумя жидкими фазами Fx и F2 (расслаивание)
в системе диспергированных в воде диэлаидоилфосфатидилхолина и дипальмито-
илфосфатидилэтаноламина [7].
Диаграмма построена из экспериментальных данных в координатах состав — температура;
исследовались спектры ЭПР парамагнитного «спинового» зонда, растворенного в фос'фо-
липидных фазах,' температура фазового перехода определялась по появлению характерных
изменений в этом спектре. Объяснение этих результатов см. в тексте и на рис. 2.7.
ка С). При другой температуре и том же общем составе смеси также
присутствуют твердая н жидкая фазы, но их составы отличаются рт наблюдаемых при
30 °С. Следовательно, многокомпонентная система не только «плавится» в
широком температурном интервале, но при разных температурах имеет разный состав
жидкой и твердой фаз. Это свойство многокомпонентной системы отличает ее
от одиокомпонентной. Например, вернемся к фазовой диаграмме воды: жидкая
вода имеет один и тот же состав, независимо от того, каковы значения
температуры и давления при конденсации ее из пара илн при плавлении льда.
На рис. 2.5 приведены электронные микрофотографии, полученные с
помощью метода «замораживания — скалывания» (см. ч. 4) для смесей фосфолн-
пидов (система DEL —DPL, содержащая 50 мол. % DPL) при 36 °С (исходное
состояние, рис. 2.5,0), 10°С (рис. 2.5,6) » при 30°С (рис. 2.5,б). Согласно
фазовой диаграмме на рис. 2.4, следует ожидать, что исследуемые ситуации
должны соответствовать однофазным областям (жидкая',или твердая фаза) и
двухфазной области (жидкая фаза+твердая фаза). Полученные микрофотографии
подтверждают это предположение.
Диаграмма на рис. 2.6 иллюстрирует Другое интересное явление, связанное
с разделением фаз в многокомпонентной фосфолипидной системе: имеются две
различные жидкие фазы F\ я Fa одна из которых обогащена фосфолипидом пер*
jg. ТЕРМОДИНАМИКА В БИОЛОГИИ
- ТТ?Т?ТТТТТ?ТТ?ТТ?Т Гомогенный
I
или
Разделение фаз1
в поперечном '
сечении
тттттттгтпт
г я
Рис. 2.7. Схематические представления возможных жидких фаз в смесях фосфо-
липндов [7].
■а — гомогенный жидкий раствор «черных» и «белых» липидов; б — частичная
несмешиваемость жидкость — жидкость, появляющаяся в результате латеральных разделений фаз на
области, сравнительно обогащенные белыми липидами, и области, сравнительно
обогащенные черными липидами (такой вид разделения фаз возникает между жидкой н твердой
фазами типа, рассмотренного на рис. 2.4 и 2.5): в—разделение фаз в поперечном сечении
соответствует обогащению одной половины бислоя черными липидами, а другой
половины — белыми липидами (можно ожидать, что мембрана более устойчива при изгибе по
типу г, чем при изгибе по типу д).
вого типа, а вторая — второго. Разделение фаз в липидном бислое может
происходить по латеральному типу (рис. 2.7, б); к такому типу относятся системы,
изображенные на рис. 2.4 и 2.5, в области жидкая фаза 4-тверда я фаза. Однако
существует и другая любопытная возможность разделения фаз: например путем
обогащения (допустим, что наружный слой мембраны обогащается одним фос-
фолипндом, а внутренний слой — другим фосфолипидом) |(рнс. 2.7, в). Тогда,
поскольку следует ожидать, что взаимодействие фосфолипидов с водой отличается»
такой тип разделения фаз может вызывать искривление мембраны.
Пример. Взаимосвязь между функцией мембраны
и фазовым состоянием мембраны
Одной нз биологических функций клеточных мембран является перенос
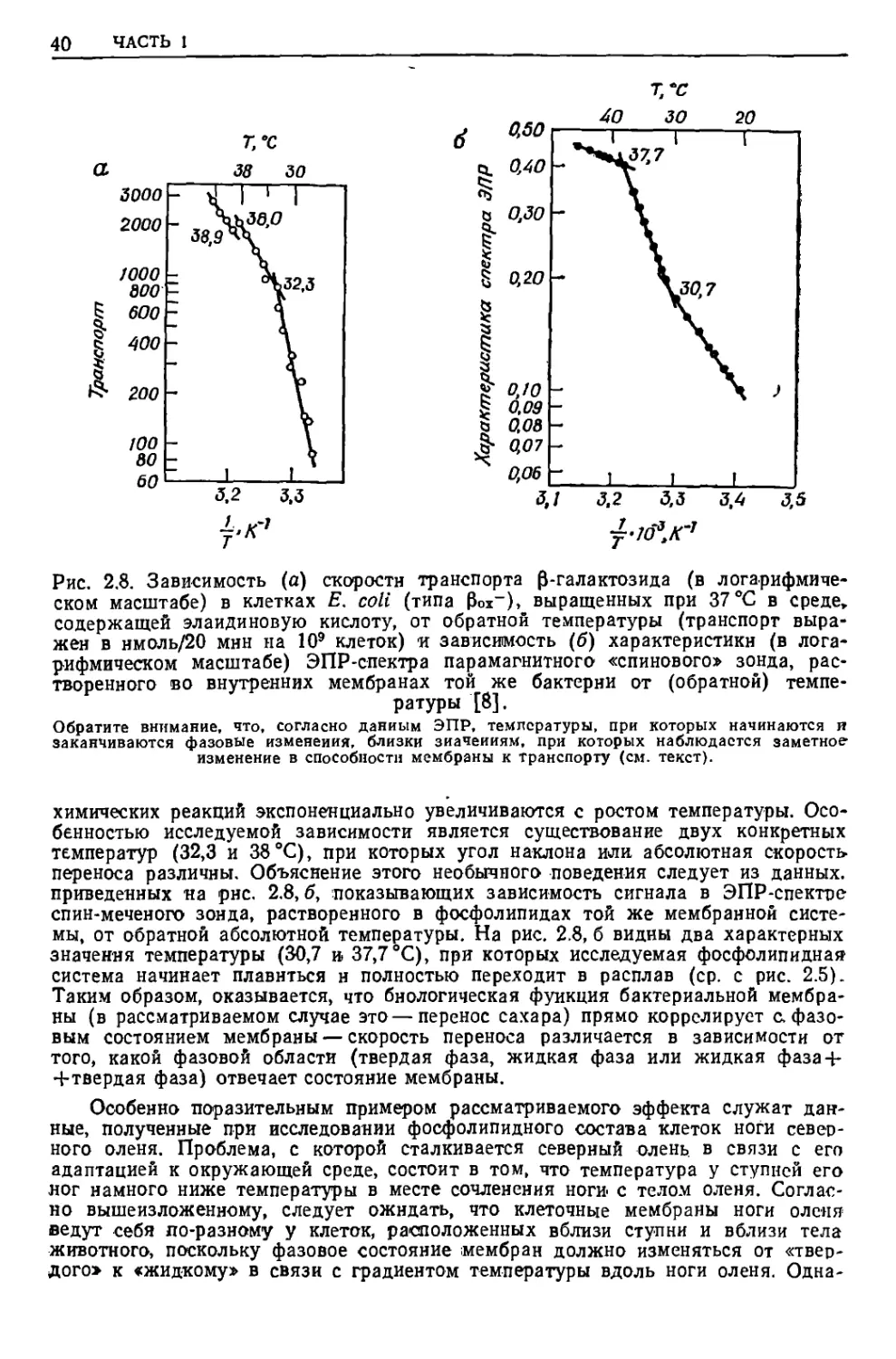
различных метаболитов внутрь клетки или из клетки. На рис. 2.8, а приведена
температурная зависимость специфической функции клеток Е. соЫ, а именно траис-
локации Р-галактозида с одной стороны мембраны иа другую. (,В качестве
хромофорной группы к р-галактозиду присоединена иитрофенильная группа для
того, чтобы исследовать процесс спектрофотометр'ически в УФ- и видимой
области.) Ниже показано (гл. 4), что графическая зависимость логарифма скорости
переноса от обратной абсолютной температуры должна быть прямой с
отрицательным тангенсом угла наклона, поскольку константы скоростей большинства
40 ЧАСТЬ 1
Т. "С
а
3000
2000
/000
800
I 600
р 400
I
Й- 200-
5.2 3,3
%
t
I
0,50
ОАО
0,30
0,20
0,10
0,09
0,08
0,07
0,06
*
-
-
-
—
—
40
1
1
30
1
37,7
у0,7
I
20
'
\ )
■
3,1 3.2 3,3 3,4
l-ioV
3,5
Рис. 2.8. Зависимость (а) скорости транспорта Р-галактозида (в
логарифмическом масштабе) в клетках Е. coli (типа Рох~), выращенных при 37 °С в среде,
содержащей элаидиновую кислоту, от обратной температуры (транспорт
выражен в нмоль/20 мнн на 109 клеток) я зависимость (б) характеристики (в
логарифмическом масштабе) ЭПР-спектра парамагнитного «спинового» зонда,
растворенного во внутренних мембранах той же бактерии от (обратной)
температуры [8].
Обратите внимание, что, согласно данным ЭПР, температуры, при которых начинаются и
заканчиваются фазовые изменения, близки значениям, при которых наблюдается заметное-
изменение в способности мембраны к транспорту (си. текст).
химических реакций экспоненциально увеличиваются с ростом температуры.
Особенностью исследуемой зависимости является существование двух конкретных
температур (32,3 и 38 °С), при которых угол наклона или, абсолютная скорость-
переноса различны. Объяснение этого необычного поведения следует из данных,
приведенных на рис. 2.8, б, показывающих зависимость сигнала в ЭПР-спектие
спин-меченого зонда, растворенного в фосфолипидах той же мембранной
системы, от обратной абсолютной температуры. На рис. 2.8, б видны два характерных
значения температуры (30,7 и 37,7 °С), при которых исследуемая фосфолипидная
система начинает плавиться и полностью переходит в расплав (ср. с рис. 2.5).
Таким образом, оказывается, что биологическая функция бактериальной
мембраны (в рассматриваемом случае это—перенос сахара) прямо коррелирует а
фазовым состоянием мембраны — скорость переноса различается в зависимости от
того, какой фазовой области (твердая фаза, жидкая фаза или жидкая фаза-Ь
+твердая фаза) отвечает состояние мембраны.
Особенно поразительным примером рассматриваемого эффекта служат
данные, полученные при исследовании фосфолипидного состава клеток ноги
северного оленя. Проблема, с которой сталкивается северный олень, в связи с его
адаптацией к окружающей среде, состоит в том, что температура у ступней его
ног намного ниже температуры в месте сочленения ног» с телом оленя.
Согласно вышеизложенному, следует ожидать, что клеточные мембраны ноги оленя
ведут себя по-разному у клеток, расположенных вблизи ступни и вблизи тела
животного, поскольку фазовое состояние мембран должно изменяться от
«твердого» к «жидкому» в связи с градиентом температуры вдоль ноги оленя. Одна-
I ТЕРМОДИНАМИКА В БИОЛОГИИ
?ист. 2.9. Схематическое представление поперечного сечения биологической мем
браны, содержащей белки и липиды [9].
Липиды изображены в виде светлых (граничные липиды, находятся в непосредственной
близости к белкам) и темных (другие липиды) кружочков с «хвостиками», а белки —
геометрически неправильными телами большего' размера. На схеме показано, что некоторые
белки прикреплены к структуре цитоскелета. Свойства граинчиых липидов отличаются от
свойств других липидов в мембране (ч. 4). с — все липиды в мембране находятся, в
жидком состоянии (волнистые «хвосты»); белки, не связанные с цитоскелетом, могут свободно
диффундировать в плоскости мембраны; б — мембрана находится при температуре
промежуточной относительно мембран а и в: некоторые липиды «заморожены» (прямые
«хвосты»), белки группируются в областях, содержащих «жидкие» лнпиды (в результате
появляются агрегаты белков); в-—температура мембраны соответствует «замороженному»
состоянию всех липидов; агрегация белков максимальна.
ко температура фазового перехода фосфолипида зависит от степени
ненасыщенности углеводородных цепей в данной фосфолипидной молекуле (чем больше
число двойных связей в цепи, тем ниже температура плавления). Поэтому в
организме северного оленя проблема адаптации решается так: клеточные мембраны,
локализованные вблизи ступни оленя, обогащены ненасыщенными фосфолипида-
ми по сравнению с мембранами клеток, расположенных вблизи тела животного.
Таким образом, в клетках ноги оленя фазовое состояние мембран (и.
следовательно, функция мембран) относительно одинаково, даже несмотря на то. что
температура клеток вдоль ноги изменяется значительно.
На рис. 2.9 приведена схематическая диаграмма, иллюстрирующая основные
представления о фазовом состоянии, структуре и функциях биологических
мембран.
Наконец, следует отметить, что разделение жидкой фазы на две (жидкость—
жидкость) не ограничивается фосфолипидными системами. Это явление давно
известно для определенных заряженных полисахаридов при смешивании их в
соответствующих соотношениях. Согласно некоторым теориям происхождения
жизни, предполагается, что процессы, 'подобные разделению фаз, происходили в
абиогенном «супе», состоящем нз смеси сахаров, аминокислот и других
молекул; при этом могла образоваться некоторого вида «клеточная» граница
раздела, на которой протекание химических реакций происходило бы иначе, чем в
остальном растворе. Для более глубокого рассмотрения этой и ранее затронутых
проблем читателю следует обратиться к литературе, указанной в конце этой
главы.
42 ЧАСТЬ I
2.Б. ПОЛУПРОНИЦАЕМЫЕ МЕМБРАНЫ
И НЕЙТРАЛЬНЫЕ МОЛЕКУЛЫ В РАСТВОРЕ:
ОСМОТИЧЕСКОЕ ДАВЛЕНИЕ
Для того чтобы понять, как возникает осмотическое давление»
прежде всего необходимо знать, что давление пара любого
растворителя Ра понижается при растворении в нем нелетучего
соединения (например, белка), как показано на верхней диаграмме
рис. 2.10. Интуитивно это явление можно объяснить следующим
образом: поскольку тот же самый объем теперь занимает только
Ха молекул исходного растворителя (где Хх— мольная доля
молекул растворителя), давление пара раствора Рд понижается:
Рд = ^а^А (закон Рауля)
(2.19>
Если поместить по обе стороны непроницаемой мембраны
(рис. 2.10,а) чистый растворитель (слева) и раствор (справа),
то разность давлений между ними составит (Ра —Ра)- Если
мембрана непроницаема, то давление в растворах можно выравнять
(т. е. привести систему в равновесие), наложив дополнительное
давление (Ра—Ра) к раствору (справа от мембраны), как
показано на рис. 2.10,6. Это уравнивающее давление является не
осмотическим, а обычным гидравлическим давлением.
а
1
t
—
*
Чистый
раствори'
гпель А
>* ^
рк
Растворитель
А *■ (не/гё-
тучее)раство-
р.ренное ее ■
<£_ щеавоо
"
■
Гидравлическое давление ,
■рана:
механическое равновесие
Непроницаемая мембрана
уве —
*-*-*
*«
я%гч
тлева пгпрааа *К
tp.+P
РА
т
^ч "^л'**
Полупроми цаемая
мембрана
Осмотическое давление
N\ Химическое равновесие
'. Х\, "° 6Д слева ~ бдсправа
IB
7
К
НШЯЮ—
Чистый
растворитель А
Раствори
а. тель А +
+(нелетучее)
-tfpae/лворен-
Т-ное
вещество &
L
v
Т
v
Непроницаемая мембрана
б
I
т
Ра**
У7Жт777\
Чистый
ра2???рл1~ \раствореннс
тель А {вещество В
Растворитель
А +(нелетучее)
л-
zPa+"
■i
РА*<
т
ра*я-
(Полупроницаемая) мембрана
проницаема только для растворители
Рис. 2.10. Схема, иллюстрирующая различие между гидравлическим и
осмотическим давлением.
Обсуждение см. в тексте.
43 ТЕРМОДИНАМИКА В БИОЛОГИИ
Однако если мембрана является проницаемой для молекул
растворителя и непроницаемой для молекул растворенного вещества
(полупроницаемая мембрана), то система при такой
неравновесной ситуации будет пытаться уменьшить разность давлений путем
переноса растворителя слева направо через полупроницаемую
мембрану. Перенос растворителя можно предотвратить и привести
систему к равновесию, наложив внешнее осмотическое давление л
(рис. 2.10,в).. На этом этапе важно отметить, что яф(РА—^а),
так что данный случай в некоторой степени отличается от того,
который рассматривался выше (рис. 2.10,6, простое
гидравлическое равновесие). Здесь речь идет скорее о химическом, а не о же"
ханическом равновесии; критерием равновесия (табл. 2.1)
является равенство химических потенциалов GA растворителя по обе
стороны мембраны (Ga,слева = GA,справа), а не равенство давлений по
обе стороны мембраны (при гидравлическом равновесии РСлева=
— гсправа).
Для расчета осмотического давления прежде всего надо
выразить взаимосвязь между химическим потенциалом и давлением.
Напомним, что для двухкомпонентной системы
dG=VdP—SdT+GAdnA+GbdnB (2.12)
(dGldP)T,nA,nB = V. (2.Ю)
Теперь, поскольку парциальный моляльный объем растворителя А
{обозначим его VA)
(-£Л,^ (2-3)
то, объединяя уравнения (2.3) и (2.10), получим
где второе равенство следует из того, что порядок
дифференцирования не имеет значения. Наконец, поскольку химический
потенциал определен как
(0G/drtA)r<PtnB=GA, (2.13>
можно переписать уравнение (2.20) в более удобной форме
Будем использовать два вида уравнения (2.21): для жидкости
А (при постоянной температуре и составе)
dGA = (VaWk dP
t
Жидкость
(2.21а>
44 ЧАСТЬ 1
и для пара Л (согласно закону идеальных газов Va—RTIPa)
dPk
dG^RT
Pa
dGk = RTd In PA
t
Пар
(2.21б>
Хотя уравнения (2.21а) и (2.216) правильно описывают
бесконечно малые различия в химическом потенциале через бесконечна
малые различия в давлении для идеального раствора [уравнение
(2.19)] и для идеального пара [ уравнение (1.7)], для того чтобы
выразить макроскопическое изменение химического потенциала
через макроскопическое изменение давления, необходимо найти?
сумму множества таких малых изменений. Рассмотрим сначала
жидкий растворитель; проинтегрируем уравнение (2.21а)
эксп.ссст эксп.давл
Ga=3
А (станд.сОст)
+ (X=A + (VAdP
станд сост
станд.давл
(2.22)
где G\ — константа интегрирования (химический потенциал для
произвольно выбранного стандартного состояния растворителя).
За стандартное состояние растворителя считают состояние чистого1
растворителя, находящегося в равновесии с его паром, так что»
стандартное давление соответствует давлению пара Ра чистого
растворителя. Кроме того, если предположить, что растворитель
практически несжимаем [т. е. что V\ практически не зависит от
давления и, следовательно, может быть вынесена из-под знака
интеграла в уравнении (2.22)], получаем
р
dP
G, = G
+ч
или
GA — GA -f- Va (P — РЛ (ДдЯ несжимаемой жидкости)
(2.23>
Аналогично в случае макроскопических измененийчхимического»
потенциала пара (идеального газа) уже принято условие
постоянства температуры [так что Т в уравнении (2.216) можно вынести
из-под знака интеграла]:
эксп.давл эксп.давл
(2.24)
GA=GeA+f/?77nnP=GA+/?r \dlnP
станд.дан
станд.давл
ТЕРМОДИНАМИКА В БИОЛОГИИ
: Наконец, определим в качестве стандартного состояния
состояние идеального газа при давлении 1 атм (обычное условие), так
что уравнение (2.23) можно записать в более сжатом виде
Р атм
'бА=(ГА-г-#Г <!d\nP=GOA + RT(lnP-\nl)=G0A-\-RT\n(Pn)
I атм
ИЛИ
(постоянная температура, идеаль-
UA-UAT К/ Ш У ный газ> р в атмосферах*)
(2.25)
Возвратимся к системе, изображенной на рис. 2.10. Очевидно,
что любой раствор находится в равновесии с своим паром, т. е.
(^л)пар,слева = (ЧОжндк.. слева (2.Zba)
И
\^А,)пар,справа~\^А,)жидк..справа, (Z..400J
так что
U^A/жндк.,слева \V А,) жидк.,справа \ЧА/пар,слева (УАлгар .справа. (.•*••*' )
Подставляя в уравнение (2.27) выражение для ((/а)пар
[уравнение (2.25)], получаем
[ёА)да.<сл«-(еА)жвдс.1С11Рава=
=GA+RT In Р°А - [GA +RT In РА] =
=#Г In (P°A/PA) =-ЯГ In (Pa/Pa). (2.28)
Но для идеального раствора, согласно уравнению (2.19),
(Ра/Ра) =^а, так что уравнение (2.28) принимает вид
(Ч\)жидк..слена (^А)жидк.,спрана== "1 *П (^л)справа- (л.2У)
Важность уравнения (2.29) состоит в том, что, поскольку
(^А)справа<1, 1п(Хд) справа — отрицательная величина, на
основании этого можно сделать вывод, что химический потенциал
растворителя.А (слева от мембраны) больше химического
потенциала раствора (справа от мембраны). Следовательно, точно так же,
как камень катится с горы вниз для уменьшения его
(механического) потенциала, молекулы растворителя проявляют тенденцию
к миграции через полупроницаемую мембрану для уменьшения
своего химического цотенциала.
* Обратите внимание, что, хотя Р в уравнении (2.25) выражается в
атмосферах, аргумент логарифма — безразмерная величина, так как отражает отношение
давлений (см. следующую главу).
46 ЧАСТЬ I
Однако, поскольку химический потенциал жидкого
растворителя можно увеличить, наложив внешнее давление [уравнение
(2.23)], можно полностью компенсировать стремление молекул
растворителя к переходу через полупроницаемую мембрану,
Приложив дополнительное внешнее давление л к раствору (справа1 от
мембраны), так чтобы его химический потенциал увеличился на
величину, точно равную начальной разности химических
потенциалов растворителя.и раствора.
Увеличение (ёА)жидк.справа, (VA)W (РА+п — Ра) +
обусловленное дополнитель- , ^ /р пв\— д> \ /о Ш
ным внешним давлением, ~г\уа)жщк\^а ^а)—^{Уа)жяак, \/-Щ
составляет
где внешнее давление я характеризуется тем, что
_,/Т7 ч _ D7.i./y v (осмотическое давление
я I v а}жидк ~—Ki in ^лА)справа идеального раствора)
(2.31)
Осмотическое давление — это такое дополнительное давление,
которое необходимо для того, чтобы- воспрепятствовать переносу
растворителя через полупроницаемую Мембрану. Кроме того,
осмотическое давление характеризует тенденцию к переносу
растворителя, наблюдаемую в отсутствие внешних воздействий.
Поскольку из практических соображений неудобно выражать
состав раствора через мольную долю растворителя [уравнение
(2.31)], преобразуем уравнение (2.31), введя концентрацию
(нелетучего) растворенного вещества В. Во-первых, поскольку
ХА+ХВ=1, <2-32)
можно переписать уравнение (2.31):
* (PaW=-ЯТ In [ 1 - (XbWuJ (2.33)
Кроме того, для очень разбавленных растворов
Хв « 1 (2.34)
и можно использовать приближение (см. приложение)
In (1 -Хв)» —Хв (2.35)
для того, чтобы получить
* &Лшт ~ЯГХв=ЯГ^Й_ жда-£, (2.36)
где /гв и /гА — число молей растворенного вещества В и
растворителя А в правом растворе на рис. 2.10, е. Теперь выразим число
молей растворителя пА через массу растворителя шА и его
молекулярную массу МА
nA=wA/MA. (2.37)
А ТЕРМОДИНАМИКА В БИОЛОГИИ
\ Тогда уравнение (2.36) преобразуется:
\ я=ДГЗ^а_. (2.38)
\ ^А^А
(Яа^онец, поскольку УА — объем моля жидкости (растворителя) А
VA=vAMA, (2.39)
(где vA называется парциальным удельнцм объемом,
приходящимся на 1 г растворителя А), получаем
я=#Г-^=#Г
_ "в
или
п = mRT (для идеального раствора)
(2.40)
где #(л«атм.моль_1-К)—универсальная газовая постоянная;
до а (воды) = 1 кг, тогда Vx=\ л и, следовательно, т—моляльная
концентрация (т. е. число молей растворенного вещества в 1 л
растворителя).
Формально уравнение (2.40) подобно закону идеальных газов
P=(n(V)RT. Тем не менее оно выполняется только для
разбавленных идеальных растворов, для которых применимы
приближения (2.19), (2.35) и (2.36)., Для биологических систем основное
значение уравнения (2.40) состоит в том, что оно показывает, как
в случае аккумулирования по одну сторону полупроницаемой
мембраны относительно малых концентраций крупных молекул
может возникать весьма значительная разность давлений;
различие концентраций на ~0,1 моль/л приводит к возникновению
осмотического давления ~2,5 атм! Осмотическое давление активно
участвует в процессе разрыва оболочки прорастающего семени.
Осмотическое давление играет большую роль в процессах
всасывания влаги из почвы корнями и стеблем растений. Для разрыва
(лизиса) клеток крови человека (или других клеток) обычно
используется способ, состоящий в помещении клеток в раствор с
пониженным осмотическим давлением (гипотонический раствор), в
котором клетки поглощают воду до тех пор, пока они не
лопаются. Теперь должно стать ясным, почему концентрация веществ в
растворах для медицинских инъекций должна быть такой, чтобы
осмотическое давление этого раствора было равно осмотическому
давлению самой крови (изотонический раствор): тогда не
происходит набухания или сжатия клеток крови in vivo. Далее
рассматриваются некоторые методы аналитического использования
осмотического давления при изучении размера макромолекул.
48 ЧАСТЬ I
Определения молекулярной массы из измерений
осмотического давления
Из уравнения (2.40) ясно, что измерение осмотического
Давления л при известной температуре позволяет определить моляль-
ную концентрацию нелетучего вещества в растворе, находящегося
по одну сторону полупроницаемой мембраны, откуда можно
определить молекулярную массу данного вещества.
Пример
В 1,00 мл воды растворено 80 мг исследуемого белка. Осмотическое
давление этого раствора при 25 °С оказалось равным 12 мм рт. ст. Какова
молекулярная масса исследуемого белка?
n = mRT (2.40)
(12/760 атм) = (80 г/л) (1/М моль/г) (0,08206 л-атм-моль^-Кг^ЭвК.
Решение приводит к
М = 124 000.
Благодаря тому что трудно осуществить точные измерения давлений много
меньших, чем в данном примере, ясно, что для определения молекулярной массы
вещества с помощью осмотического давления его раствора необходимы довольно
большие количества исследуемого вещества, особенно по сравнению с
количествами, требуемыми при использовании новых электрофоретических методов
исследования (см. ч. 2). Определение молекулярной массы ив измерений
осмотического давления, однако, важно с исторической точки зрения — именно этим
методом в 1925 г. было показано, что молекулярная масса гемоглобина
составляет 67 000, т. е. она в четыре раза превышает соответствующее значение,
отнесенное к одному атому железа (молекулярную массу мономера). Как
рассматривается ниже, измерения осмотического давления могут использоваться для
оценки полидисперсности смеси макромолекул разной молекулярной массы.
Понятия о средней молекулярной массе
в полидисперсных системах
Пусть надо измерить осмотическое давление смеси трех
высокомолекулярных веществ, содержащей ct\ г вещества с
молекулярной массой Mi, а2 г вещества с молекулярной массой М2 и а3 г
вещества с молекулярной массой Мз. Наблюдаемое осмотическое
давление равно сумме вкладов каждого из этих соединений:
п=т£Т-УщНТ+тъНТ=Ш~1~+-±?^+3^lW (2.41)
Однако, если неизвестно, что смесь гетерогенна,
экспериментально находят единственное (среднее) значение молекулярной
массы Мп для общего количества (^1+^2 + 03) макромолекул в
растворе. Запишем уравнение, связывающее среднюю молекуляр-
49 ТЕРМОДИНАМИКА В БИОЛОГИИ
ную массу с количествами и молекулярными массами
компонентов данной смеси:
я — (fli + аг + <h)IMn ^т _ {ахтг) + (а2/Мг) + fo/M,) ^
м„=
gi + а2 + %
« (ei/Al1) + (a1/Afa) + (e,/^«)
(frK+(ftW(&)*
(«iW + (<УМ2) + (%№ ~"
_ ATf Af д + iV»Af» + Ar,Af8
~ ^ + ^ + ^3 '
«„*■
2 ^Mi
f
2*i
(среднечисленная молекулярная масса)
(2.42)
где Ni — число молей i-ro компонента в смеси.
Важно отметить, что кажущаяся (средняя) молекулярная масса,
определяемая из измерений осмотического давления [уравнение
(2.42)], не равна величине кажущейся (средней) молекулярной
массы, определяемой для той же самой смеси при исследовании
диффузии [уравнение (2.43)] или седиментации [уравнение
(2.44)] (см. 2.2).
М„,—
ч&
NU =
_J
_/
2 N*M*
(средневесовая молекулярная масса)
(z-средняя молекулярная масса)
(2.43)
(2.44]
В табл. 2.2 перечислены различные экспериментальные методь
определения средней молекулярной массы.
Пример
Смесь макромолекул содержит 375 мг вещества с молекулярной массой
48 000, 500 мг вещества с молекулярной массой 67 000 и 220 мг вещества с моле
кулярной массой 300 000. Сравните значения (средних) молекулярных масс, оп
50 ЧАСТЬ 1
Таблица 2.2
Виды (средней) молекулярной массы, получаемые
с помощью различных экспериментальных методов
Экспериментальный метод или соответствующий
изучаемый параметр
Осмотическое давление
Химический анализ
Константа поступательной диффузии
Скорость седиментации и диффузии
Светорассеяние
Седиментационное равновесие
Вязкость
Mn<Mv*£Mw<Mz
Средняя
молекулярная масса
[уравнения
(2.42)-(2.44)]
мя
Мп
Mw
Mw
Mw
Mw, Mz
Mv
ределяемых при исследовании осмотического давления, диффузии и
седиментации.
/ 375 \ / 500 \ / 220 >
(-48Щ-] (48 00°) + Ь? 0007 (67 000> + ЫоОЙГJ <300 °°°)
**""* / 375 \ ( 500 \ / 220_ \ «=68400,
\ 48 000 J + [ 67 000 J + \ 300 000 J
375 "\ о / 50° ) „, „,«. /220
48 000 ) ( °00^3 ^~ V 67 000
-KW 000)» H-^-^ogg-1(300 000)»
Л*м= : ы \ \ тал ч ^-ойл—ч = 107000,
/ 375 "\ / 500 \
ll8M0-J(48000>a+\-6700CTJ<
1ш — / 375 \ / 500 \ / 220 \ л
( 48 000 J (48 000^ + I Т7"000 ) (67000) + ЬооШИ300000>
/ 375 \ / 500 "\ / 220 \
ЫШ) <48 С00>3 + Ыш) (67 °°0)3 + boOObTj (3Q0 °°0)а
/ 375 \ / 500 \ v / 220 \ '
(iSooo-J <48 000)а + (l>7wj<67 000)* + (-3Woo6J(300 00°)?
ЛК =
= 194 009.
Этот пример иллюстрирует, что обычно Afz>Afw>Af„. Данные три вида cpei*»
ней молекулярной массы имеют одно н то же значение, только если смесь моно-
дисперсна, т. е. состоит из одного компонента, имеющего заданную молекулярную
массу. Во всех случаях средние значения молекулярной массы чувствительны к
присутствию малых количеств примесей, молекулярная масса которых
значительно отличается от молекулярной массы исследуемого растворенного соединения
(см. задачи в конце главы).
Неидеальные растворы. Точно так же, как для реальных газов зависимость
Р — V отклоняется от закона идеальных газов, для реальных растворов
давление пара (и, следовательно, осмотическое давление) отклоняется от
предсказываемой уравнением идеального раствора {уравнение (2.19)}. Кроме того,
поведение реального газа в пределе при нулевом давлении приближается к
поведению идеального газа, аналогично тому как для растворителя тенденция к иде-
51 ТЕРМОДИНАМИКА В БИОЛОГИИ
/Г
С
I
1 о
с, г/л
Рис. 2.11. Несколько примеров, иллюстрирующих отклонение от идеального
осмотического давления [уравнение (2.40)] для зависимости п[с от с [с —
концентрация (масса/объем) макромолекул в растворе, я — осмотическое давление]
[Ю].
Си. также задачи в конце главы i[ll].
альиому поведению проявляется в пределе при бесконечном разбавлении
(рис. 2.11). Наконец, отклонения поведения реального газа от закона идеальных
газов дают информацию о размере молекул газа и действующих между ними
межмолекулярных силах, а исследование отклонений осмотического давления от
идеального поведения позволяет получить информацию о размере и форме частиц
растворенного вещества и степени их агрегации (см. рис. 2.11 и задачи в конце
главы).
2.В. ПОЛУПРОНИЦАЕМЫЕ МЕМБРАНЫ
И ЗАРЯЖЕННЫЕ МОЛЕКУЛЫ В РАСТВОРЕ:
РАВНОВЕСИЕ ДОННАНА
В предыдущем разделе сравнивались химические потенциалы
растворителя в растворах по обе стороны мембраны, проницаемой
для молекул растворителя (но непроницаемой для макромолекул,
находящихся в одном из этих растворов), для того чтобы
показать, почему молекулы растворителя обладают тенденцией к
миграции из области с высокой концентрацией растворителя (с
большим химическим потенциалом растворителя) в область с
пониженной концентрацией растворителя (с меньшим химическим
потенциалом растворителя). В данном разделе рассматривается
поведение в растворе заряженных ионов низкомолекулярного
вещества, которые могут проходить через ту же самую
полупроницаемую мембрану, если раствор содержит заряженные
макромолекулы. (Нейтральные молекулы низкомолекулярного вещества при
переходе через мембрану просто равномерно распределяются по
обе стороны мембраны; см. примеры, приведенные ниже:
«Равновесный диализ».)
Уравнение (2.29) (разд. 2.Б) показывает, что химический
потенциал Ga идеального растворителя увеличивается с ростом
концентрации растворителя согласно уравнению
dGk—RTd\n{Kk) (для идеального растворителя), (2.29)
где ХА — мольная доля (концентрация) растворителя. Аналогично
на основании зависимости давления пара разбавленного раствора
от концентрации вещества В в растворе можно описать
зависимость химического потенциала вещества в растворе от его
концентрации уравнением
dGB=RTdlnmB (для идеального раствора) (2.45)
где Шв — моляльная концентрация вещества (число молей
вещества в 1 кг растворителя) в растворе. Интегрируя уравнение (2.45),
чтобы описать макроскопическое изменение GB, получаем
ЭКСП.МОЛЯЛЬНОСТЬ
эксп.сост вещества
5B=GBrcTalw.c0cT)+jdGB=a+j/?rrfIn.mB==
станд.сост станд.молилыюсть
вещества
=<?в+RT In [ (тв)ЭКсп/(/лв)станд] (при постоянной температуре)
или
стандартное состояние* (/пв)станд —
= 1 моль/кг, постоянная температу- (2.46)
ра, идеальный раствор
Уравнение (2.29) легло в основу количественного описания
осмотического давления, а уравнение (2.46) непосредственно
приводит к объяснению равновесия Доннана.
* Относительно уравления (2.25) заметим, что, хотя (тв)жсв выражено мо-
ляльной концентрацией, аргумент логарифма в уравнении (2.46) — безразмерная
величина, поскольку представляет собой отношение экспериментально
определяемой концентрации (моляльности) раствора к моляльности стандартного раствора
вещества. Стандартным состоянием вещества в растворе, согласно уравнению
(2.46), является 1 Мл раствор этого вещества (окружение каждой молекулы
растворенного вещества соответствует бесконечно разбавленному раствору, а
именно каждая молекула растворенного вещества окружена только молекулами
растворителя, среди которых отсутствуют молекулы растворенного вещества).
Детальное обоснование этого условия очень сложно (его можно найти в более
подробных курсах термодинамики (например, [1]) и здесь опущено.
Использование уравнения (2.46) не связано с определением условий стандартного
состояния; в этой и следующих главах сделана попытка показать это. Для целей
изложения материала в данном разделе достаточно считать, что идеальный раствор —
это раствор, подчиняющийся уравнению (2.46); иеидеальное поведение раствора
обсуждается в следующей главе.
GB = GB+^7,,n(/"B)3Kcn
53 ТЕРМОДИНАМИКА В БИОЛОГИИ
Начальное состояние
Конечное состояний
(равновесие)
[Nal
справа
[CI-1
справа
Мембрана проницаема для воды и
ионов соли, но непроницаема для
молекул белка
Рис. 2.12. Распределение ионов при равновесии Доинана (б) при. помещении»
растворов неорганической соли и (заряженного) белка по обе стороны
полупроницаемой мембраны (а).
При равновесии концситрацнн ионов натрия и хлора должны удовлетворять уравнении!»
(2.49). (Миграцией растворителя из-за осмотического давления здесь пренебрегают.).
Пусть небольшое количество соли (натрия) заряженного
(отрицательно) белка растворено в растворе, находящемся по одну
сторону полупроницаемой мембраны, и небольшое количество
хлорида натрия внесено в раствор по другую сторону мембраны.
Поскольку первоначально раствор слева не содержал ионы С1~
(рис. 2.12), а раствор справа содержал некоторое конечное
количество этих ионов, то в системе проявляется тенденция к
миграции ионов С1~ через мембрану справа налево [см. уравнение-
(2.46)]. Однако, для того чтобы при переносе через мембрану
раствор оставался электронейтральным, переносу каждого аниона С1~
должно соответствовать перенос катиона Na+. Поэтому
равновесие будет достигнуто, когда химический потенциал
(растворенного) NaCl станет одинаковым по обе стороны полупроницаемой-
мембраны
лслева /^справа
uNaCl — "м-''»
водн
Naci (B СОСтоянии равновесия)
(2.47>
Поскольку ЫаС1в0дн полностью диссоциирован на ионы Na+ иг
С1~, а также
'NaCl
водн
:(w4-tfcr.
уравнение (2.47) следует переписать:
слева | лолева ^справа i ^справа
Na+ H-Gcr =GNa+ "Г°СГ
Подставляя уравнение (2.46) в (2.48), получаем
G°Na++RT\nmcNT +5сг+ЯПптссГ-ва =
RT (1птсклае+ва+1п^Га)=^Г (lnm^+lnmcr^),
(2.48>
54 ЧАСТЬ I
ИЛИ
i_ /„слева I слева\ i_/ справа | справа\
In(mNa+ -Г "Кг j==In('"Na+ -hrrtcr J
„слева слева ^справа справа
mNa+ Шсг — mNa+ ШСГ
Обычно используется запись
[№+1слева [С1~]сц>ава , .
[Na+]cnpaBa - Icrle^T (B С0СТ°ЯНИИ РавНовесия>
(2.49)
Уравнение (2.49) описывает одну из форм равновесия Донна-
«а. В присутствии многозарядных ионов соответствующие
рассуждения и преобразования приводят к более общему выражению
V [М3+]справа ) ~\ [Хмелева J (2'50'
и т. д. (см. задачи в конце главы)
-Пример. Простой расчет концентраций при равновесии Доннана
Концентрации исходных растворов слева и оправа от полупроницаемой
мембраны равны соответственно ([Na^4-]0—10~* моль/л и [NaClJ°=10~2 моль/л.
Найти концентрации всех ионов в конечном растворе.
Прежде всего следует убедтъея в том, что в данных условиях NaCl будет
■перемещаться справа налево [уравнение (2.49)]. Пусть объемы растворов по обе
^стороны мембраны равны; тогда, если обозначить количество NaCl,
перемещающегося оправа налево, х, то равновесные концентрации ионов должны
удовлетворять уравнениям (растворы должны оставаться электронейтральными)
[Na+JMeBa=4.10-3-}-*,
[Na+^^IO-*-*,
V^* ]слева==^»
[Cl-UpaBa^lO-2-*
Подставляем эти выражения в уравнение (2.49), описывающее условие
равновесия Доннана.
4.10"* + * Ю-* — х
"Ю-2—-х = х ' д: = 4,17-Ю-9 моль/л.
Тогда получаем концентрации в конечных растворах
[Na-Чслева = 8,17 • I О"3 Моль/Л,
[Ма+]справа = 5,83.10"* МОЛЬ/Л,
[С1"]слева = 4.17- Ю~3 МОЛЬ/Л,
[СГЬправа^ 5,83. Ю-* моЛь/Л
55 ТЕРМОДИНАМИКА В БИОЛОГИИ
Пример. Влияние эффекта Доинана на осмотическое определение
молекулярной массы
Если внимательно проанализировать предыдущий пример, можно заметить,,
что в результате переноса некоторого количества NaCl через мембрану к
осмотическому давлению исходного раствора белка (относительно солевого раствора по
другую сторону мембраны) прибавляется вклад, соответствующий
дополнительному количеству соли. Прямые (но утомительные) алгебраические
преобразования приводят к следующему выражению наблюдаемого осмотического давления
раствора белка, измеряемого относительного давления солевого раствора по
другую сторону мембраны:
R1 Сбелок
я =•
VAM6l
елок
2 2
^белок^белок
1 + ~~*
4ц
(2.51>
где Сбелок — концентрация белка (в граммах белка в 1 кг воды), V& — объем-
раствора, содержащего 1 кг воды, Мбелок — молекулярная масса белка, 2белок—
заряд белка, Шбелок — моляльность белка (число молей белка в 1 кг. воды) и
|А=у22т/2* — ионная сила раствора (см. разд. З.Б.1). Надо лишь отметить, что
поскольку в уравнение (2.51) входит второе слагаемое, то зависимость я/с от
с характеризуется положительными отклонениями от прямой (см. рис. 2.11).
Таким образом, для любой концентрации белка в конечном растворе
наблюдаемое осмотическое давление превышает ожидаемое значение, так как
(заряженный) белок эффективно «перетягивает» в свой раствор дополнительное
количество соли. Следовательно, если бы не было известно, что белок заряжен,
найденное значение осмотического давления относилось бы исключительно к
свойствам белковых молекул и рассчитанная величина среднечислеииой молекулярной?
массы [уравнение (2.42)] была бы намного меньше истинной молекулярной
массы белка. Согласно уравнению (2.51), при этом ошибка увеличивается
пропорционально квадрату заряда белка и обратно пропорционально концентрации соли*
в исходном растворе. Поэтому для уменьшения таких ошибок желательно
проводить измерения при сравнительно высокой концентрации соли (см. задачи в-
конце главы) и при условиях, как можно более близких к изоэлектрической
точке белка (т. е. рН, при котором суммарный заряд молекулы белка равен нулю).
Подобные поправки надо вводить и при других экспериментальных
исследованиях в растворах, например методами седиментации и электрофореза, в
противном случае кажущиеся значения молекулярных масс могут быть ниже
истинных более чем и а порядок.
Пример. Диализ и его применения
С раствором макромолекул, помещенных в мешок с полупроницаемым»
стенками (т. е. непроницаемыми для макромолекул, но проницаемыми для воды»
и низкомолекулярных нейтральных или заряженных соединений), можно
провести широкий круг различных исследований. Простейший эксперимент, диализ,
заключается в изменении состава раствора (концентрация макромолекул не
меняется) внутри диализного мешка путем промывания мешка в часто (или даже
непрерывно) сменяемом внешнем растворе. Раствор внутри мешка будет иметь
практически тот же состав (исключая эффекты Доннана для заряженных
соединений), что и внешний раствор, но макромолекулы будут находиться только во
внутреннем растворе. Поскольку многие методы выделения белков включают
стадию осаждения солью (такой, как сульфат аммония), диализ является
удобным способом удаления соли из раствора ресуспендированного белка. Диализ
также является удобным способом изменения рН, замены буфера или изменения-
концентрации соли в растворе белка. В клинической практике описанная форма
диализа иопользуется при удалении продуктов обмена из крови больных с
заболеваниями почек. Для этого кровь больного пропускают через аппарат, где она
456 часть i
О Л
00-
to.y
Начальное состояние Конечное состояние
(равновесие)
Рис. 2.13. Схема простого опыта по диализу.
Фаствор белка и концентрированной солн помещается в мешок, стеики которого
непроницаемы для белка, ио проницаемы для растворителя и ннзкомолекулярных соединении (а).
■При равновесии (б) молекулы низкомолекулирного соединения практически равномерно рас-
-ярсделеиы во всей системе (внутри и снаружи мешка), а белок остается внутри. Конечный
эффект состоит в том, что основная часть соли удаляется из раствора белка внутри мешка,
используя большой (и/или часто заменяемый) объем внешнего раствора, можно удалить
»нли ввести любое низкомолекулярное соединение, которое проходит через мембрану.
Измеряя коицеитрацин низкомолекулярного соединения или иоиоа внутри н снаружи мешка,
■можно определять количественные характеристики связывания этих соединений с белком
(см. текст).
непрерывно циркулирует по одну сторону полупроницаемой мембраны, с другой
стороны который непрерывно подается свежий изотонический раствор. (Одна из
нормальных функций почек — обеспечить пассивный перенос выводимых
продуктов метаболизма из крови в мочу через полупроницаемую мембрану почечных
канальцев.)
Равновесный диализ: связывание молекул или ионов низкомолекулярного
вещества с макромолекулами. Допустим, поставлена задача измерить константу
образования комплекса, например, аспирина с сывороточным альбумином, и
имеется аспирин, меченный радиоактивным изотопом. Можно ввести меченый аспирин
в раствор (внешний раствор), в который погружается мешок, содержащий
раствор альбумина (внутренний раствор), и позволить системе прийти к равновесию
(рис. 2.13). Игнорируя существование заряда на молекуле аспирина, можно легко
-определить общую концентрацию аспирина во внешнем и внутреннем растворе,
измеряя радиоактивность обоих растворов. Поскольку при этом измеряется об-
лцее содержание свободного (в растворе) и связанного с альбумином аспирина,
общая концентрация аспирина в диализном мешке представляет собой сумму '
[Аспирин]ВЯуТр>>общ = [Аспирин] внутр. .свобод + [Аспирин • Альоумин]виутр (а)
[Аспирин]внеШи. ,общ— [Аспирин]внешИр>СВОбоД' (б)
Игнорируя заряд на молекуле аспирина, в первом приближении можно
приравнять концентрации свободного аспирина по обе стороны мембраны
[Аспирин]внуТр,>СВобод =» [Аспирин]внешНмСВОбод, (в)
поскольку молекулы аспирина равномерно распределены внутри и снаружи
диализного мешка. Приведенные уравнения можно решить, определив
концентрации свободного и связанного аспирина во внутреннем растворе (т. е. в
растворе белка), а также использовав независимые данные о суммарной концентрации
-белка (например, полученные путем измерения оптической плотности при 280 им)
[Альбумин]об1ц = [Альбумии]своб0д + [Альбумин ■ Аспирин].- (г)
В уравнениях (а), (б) и (г) концентрации являются экспериментально
измеряемыми величинами.
jj7 ТЕРМОДИНАМИКА В БИОЛОГИИ
Зная концентрацию связанного аспирина, можно определить остальные
концентрации, необходимые для определения константы комплексообразования
аспирина с альбумимом
[Альбумин • Аспирин]
Аобраз = [Аспирии]свобод [АльбуМИН]свобод'
Описанный вариант метода равновесного диализа позволяет исследовать
комплексообразование любой нейтральной молекулы низкомолекулярного
вещества с макромолекулой при условии, что доступен способ измерения общей
концентрации низкомолекулярного вещества внутри диализного мешка (в данном
случае измерялась радиоактивность). С учетом эффекта Доннана описанный
метод применим и для изучения комплексообразования ионов (см. задачи в конце
главы).
Задачи
1. Перегонка хорошо иллюстрирует применение на практике фазовых диаграмм.
Фазовая диаграмма, описывающая двухкомпонеиткую систему толуол —
бензол (рис. 2.14,а), аналогична фазовой диаграмме, характеризующей фосфоли-
пидную систему (рис. 2.4).
а. Рассчитайте число степеней свободы внутри каждой области на кривых.
б. Начиная с жидкого раствора, состав которого соответствует точке А иа
диаграмме а, допустим, что собираемый пар конденсируется путем понижения
температуры, полученная жидкость доводится до кипения и собираемый пар"
вновь тем же способом конденсируется. Изобразите эти действия и а фазовой
диаграмме и покажите, что в результате многократного повторения таких
операций будет получен чистый бензол. Этот процесс называется фракционной
дистилляцией; применяется в промышленности при разделении нефти» иа
компоненты.
ПО
«J МО
I
I
80-
70.
*кип
-(СбНдСНз)
Жидкость***
АУ
—
1
4*4v. Пар
! i
1
*кип
(С6Н6)
1
0,0 0,2 0,4 0,6 0,6 1,0
ХсФь
а
бб
_ 64-
62-
и 60
I
t
54
Пар
/ *
- /А
-7 \
*кип I
(СН3СОСНз|)
/
/Жидкостб\Жидш(т\
^3
I I
! '«/л
I (снег,)
1
\ У
! лазеотроп
! i
0,2 0,4 0,6 Q0 КО
Ясна,
Рис 214. Фазовые диаграммы бензол — толуол (а) и ацетон — хлороформ (6*)
[11].
58 часть а
в. Начиная с точки В на диаграмме б, изобразите описанный выше процесс,
если на каждой стадии собирать жидкость. Покажите, что конечным
результатом этого процесса будет жидкость постоянного состава, которая не
представляет собой чистый ацетон или чистый хлороформ. Такая смесь называется
азеотропной, и при последующих перегонках ее состав не изменится. Вода и
этиловый спирт образуют азеотропную смесь, в состав которой входит 95,6%
этанола; следовательно, при многократной перегонке разбавленных водио-
спиртовых смесей невозможно добиться повышения концентрации этанола
более, чем до 95,6%.
2. Смесь содержит 2,5 мг белка с молекулярной массой 20 000, 1,5 кг белка с
молекулярной массой 50 000 и 1,0 мг белка с молекулярной массой 100 000.
Найдите (среднюю) молекулярную массу для этой смеси различными
методами:
а. измерениями осмотического давления;
б. измерениями коэффициента диффузии;
в. измерениями седиментационного равновесия.
3. Осмотическое давление крови человека при 40 °С составляет 7,7 атм.
Предполагая, что раствор ведет себя как идеальный найдите:
а. Какова общая концентрация веществ, растворенных в крови?
б. Каково осмотическое давление крови при 4 °С?
4. Закон идеальных газов описывает поведение молекул газа как точечных масс,
между которыми отсутствуют межмолекуляриые силы притяжения. Для
осмотического давления аналогичное по форме уравнение (2.40), n=mRT, также
применимо к растворенным веществам, молекулы которых занимают
пренебрежимо малый объем и для которых можно не учитывать межмолекуляриые
взаимодействия. Поэтому по аналогии с уравнением Ван-дер-Ваальса для не-
идеальных газов
а \
Р + ~yf) (V — b)—RT (для одного моля газа)
;[где Ь — исключенный (недоступный) объем, обусловленный конечным
размером молекул газа, а—,постоянная, связанная с силами притяжения,
действующими между молекулами газа] выведите выражение осмотического давле-
-ния для «неидеального» раствора
я _1 Вс
~Wc = Ж + м* •
а
{где В=* {Ь—рт), с — концентрация макромолекул в граммах на 1 л].
Используйте полученный результат для объяснения отклонения от идеального
поведения осмотического давления, показанного на рис. 2.11.
15. Пусть препарат содержит 40 г полимера с молекулярной массой 100 000 и
0,04 г этилакриловой кислоты с молекулярной массой 100 (примесь).
а. Рассчитайте среднюю молекулярную массу этой смеси, определяемую из
измерений осмотического давления.
б. Принимая, что данный препарат содержит 99,9 масс. % полимера,
объясните, почему придается такое большое значение высокой степени очистки
препаратов при определениях молекулярной массы макромолекул (для
объяснения привлеките полученный выше результат).
S. Имеет место равновесие Доииана Дописанное выше уравнение (2.49)], в
качестве соли присутствует NaCl.
а. Проведите расчет равновесия Дониана для случая, когда присутствуют
ионы: Na+, K+, С1~ и NOs. (Проанализируйте также миграцию всех солей,
которые могут образоваться из этих ионов.)
б. Предполагая, что в оистемё присутствуют многозарядные ионы «[например,
такие, какие дает при диссоциации соль La2(S04)s], выведите уравнение,
описывающее связь между равновесными концентрациями катионов и анионов по
обе стороны полупроницаемой мембраны (на рис. 2.12, а) в условиях равно*
весия Доннана и в присутствии соли, имеющей общую формулу МГХ,.
ТЕРМОДИНАМИКА В БИОЛОГИИ
I. а. В нулевой момент времени два раствора, показанные ниже, помещаются
с двух сторон полупроницаемой мембраны (Prot~ — молекулы белка, несущие
единичный заряд). Пренебрегая переносом растворителя, рассчитайте
равновесные концентрации Mg2+ и С1~ по обе стороны мембраны.
0,00дМлЩС\г
0,ООШлЩ{?го\)г
б. Рассчитайте изменение (увеличение или уменьшение?) осмотического
давления в правой ячейке по сравнению с тем осмотическим давлением, которое бы
наблюдалось для нейтрального белка при той же концентрации.
в. Наконец, если истинная молекулярная масса белка равна 70 000, найти
кажущуюся молекулярную массу из измерений осмотического давления в
растворе справа от мембраны, если считается, что миграция соли через мембрану
отсутствует?
8. При описании метода равновесного диализа для определения констант комп-
лексообразования молекул низкомолекулярных веществ с макромолекулой
предполагалось, что молекулы низкомолекулярных веществ нейтральны, так
что при равновесии их концентрация одинакова по обе стороны
полупроницаемой мембраны. Теперь пусть необходимо найти коистаиту комплексообразова-
ния иодид-иона с альбумином:
а - . , . ^ г, [Альбумин-1]
Альбумин+-Ы- *=* Альбумин-1; Кобраз- [Альбумии+] [1Т
Для простоты стехиометрия комплекса принята равной 1:1. Пусть имеется
меченый иодид и можно измерять рН внутреннего (внутри полупроницаемого
мешка) и внешнего растворов. Альбумин находится во внутреннем растворе.
Разработайте схему эксперимента для определения /Собраз (какие измерения
следует сделать?), при этом используйте условие равновесия Доннана.
Литература
1. Морге W. /., Physical Chemistry, any edition, Prentice-Hall, Englewood Cliffs»
N. J. Наиболее легкодоступное детальное рассмотрение химического
потенциала.
2. Morris J. G„ A Biologist's Physical Chemistry, 2nd ed., Edward Arnold,
London, 1974. Традиционное рассмотрение термодинамики растворов.
3. Holde van К. Е., Physical Biochemistry, Prentice-Hall, Englewood Cliffs,
N. J. 1971. Хорошее описание осмотического эффекта и эффекта Доннана.
4. Мартин Р. Введение в биофизическую химию. М.: Мир, 1966. Хороший
раздел, посвященный осмотическому давлению.
5. Мак-Коннелл Г. Молекулярное движение в биологических мембранах, в кн.
Метод спиновых меток/Ред. Берлинер Л., М.: Мир, 1979, с. 570—607.
Хороший современный обзор явлений фазовых переходов в биологических мем*
браках.
6. Grant С. W. М., Wu S. H. W., McConnell H. M., Biochim. Biophys. Acta, 363,
151 (1974).
7. Wu S. H. W., McConnell H. M„ Biochemistry, 14, 847 (1974).
8. Linden C, Wright K., McConnell H. M„ Fox С F., Proc. Natl. Acad. Sci.
U. S. A., 70, 2271 (1973).
9. Linden C. D., Fox C. F., Accts. Chem. Res., 8, 321 (1975).
10. International Critical Tables, McGraw-Hill Book Co., New York, 1926—1930.
ГЛАВА 3
ХИМИЧЕСКИЕ РЕАКЦИИ
И КОНСТАНТЫ РАВНОВЕСИЯ
Прежде всего в этой главе, опираясь на определение
химического потенциала, введем константы равновесия. Затем объясним,
почему кажущиеся константы равновесия (т. е. получаемые при
использовании молярных или моляльных концентраций
реагентов и продуктов реакции) зависят от концентрации, и как эта
зависимость может оказаться полезной при осуществлении
выделения и очистки макромолекул прямым и обратным «высаливанием».
Наконец, более или менее детально проанализируем особенно
часто встречающееся и интересное равновесие, описывающее комплек-
сообразование (связывание) одной или нескольких молекул
низкомолекулярного вещества (нейтральных метаболитов, ионов или
протонов) с макромолекулами, и некоторые полезные способы (по
кривым титрования, способ Скэтчарда и т. д.) определения числа
участков связывания и констант комплексообразования при
связывании на разных участках.
З.А. КОНСТАНТЫ РАВНОВЕСИЯ
Из ранее полученного общего выражения для малых
изменений свободной энергии Гиббса
dG^VdP—SdT+% Gtdnt (2.12)
i
ясно, что при постоянном давлении и температуре
dG = 'VGidfit (постоянные Т и Р)
i
и, следовательно, при постоянных Т и Р
G = 2 ntGi
Уравнение (3.2) просто утверждает, что общая свободная
энергия смеси представляет собой сумму числа молей каждого ком-
(ЗЛ)
(3.2)
Ц ТЕРМОДИНАМИКА В БИОЛОГИИ
рнента, умноженных на свободную энергию моля компонента
(т. е. химический потенциал) при указанной концентрации. Теперь
1адо лишь знать, как d зависит от концентрации i-ro компонента:
Gj = C?J -f- RT InXi i — (идеальный) растворитель (З.За)
Gf = (?! -f- RT In rtii i — (идеальное) растворенное вещество (3.36)
* (в жидком растворе)
Gj = G, + RT \TiPi i — (идеальный) газ (3 .Зв)
G« «= (ь + RT In Я* ' — (идеальное) растворенное вещество (3.Зг)
(в жидком растворе)
где Xi — мольная доля, /щ—моляльная концентрация и Pi—
(парциальное) давление.
Уравнение (3.3а) выведено, как и уравнение (2.29) в
предыдущей главе, в приближении, что поведение растворителя
соответствует идеальному раствору, т. е. раствору,, для которого
применимо уравнение (2.19). Уравнения (3.36), (З.Зв) и (З.Зг)
действительны также в условиях бесконечно разбавленных растворов или
газов (см. обсуждение понятия «активность» в разд. З.А.1); при
таком ограничении уравнения выполняются и определяют
«идеальное» поведение. G? во всех случаях соответствует химическому
потенциалу идеального растворителя, растворенного вещества или
газа, который должен был бы наблюдаться в растворах с
единичной концентрацией (Xi, rtii, Pi).
Для равновесной типичной химической реакции
оА+6Вч=±сС+Д) (3.4)
в которой а молей вещества А реагируют с Ь молями вещества В
с образованием с молей вещества С и d молей вещества D,
можно утверждать, что в состоянии равновесия (при постоянной
температуре и давлении) суммарная свободная энергия продуктов
реакции равна суммарной свободной энергии реагентов [табл. 2.1
и уравнение (2.14)], поскольку в противном случае равновесие
сдвигается в ту или иную сторону:
GA+GB=GC+GD (3.5)
Подставляя уравнение (3.2) в уравнение (3.5), находим, что
aGA+bGB==cGc+dGD (3.6)
а после подстановки уравнения (3.3) в уравнение (3.6) получаем:
а [ОаН-ЯПпЮ] +6 [0°в+ЯГ1п(тв)] =
=с [Gc+RT In (mcj\ +d [й> +ДГ In (mD)]
62 ЧАСТЬ 1
ИЛИ
cGq-\- dGu—aGx—Ы}&—
=RT [—с In (mc)—d In (mD) -fa In (/nA) +b In (mB)]
(Gc+G°d)-(G'a+Gb)=-RT [In (mtf+ln (/nD)d-ln (/nA)«-ln(/nB)']
(tf°)
продукты
-(^реагенты =AG;x = -^Г In Г^|1. (3-7)
Напомним, что каждый реагент и продукт рассматривались как
идеальные вещества в растворе [уравнение (3.36)].
Основной смысл уравнения (3.7) состоит в том, что поскольку
левая часть уравнения постоянна (т. е. не зависит от
концентрации), то и правая часть должна также не зависеть от общих
концентраций всех веществ, участвующих в реакции. Следовательно,
для обозначения выражения в скобках в уравнении (3.7) можно
ввести новую величину — константу равновесия КР*
эавн.
Лоавн —
_с d
_с Р_ (постоянные Т и Р, идеальные
ра*н пЛть вещества в растворе)
(3.8)
Кроме того, должно быть ясно, что если один из реагентов —
растворитель, газ или твердое вещество в растворе, то необходимо
обязательно выражать его концентрацию при подстановке в
уравнение (3.8) в мольных долях (для растворителя или
растворенного вещества) или через парциальное давление (для газа). В
частности, поскольку растворитель обычно присутствует в большом
избытке, его мольная доля обычно очень близка к единице, и
поэтому в выражении константы равновесия часто опускается.
Аналогично мольная доля твердого вещества в растворе обычно равна
единице, исключение представляют смесн твердых веществ типа
амальгам.
Допустим, что система, описанная уравнением (3.4), находится
в химическом равновесии (постоянные Т и Р). Тогда
уравнение (3.8) указывает, что при введении в равновесную смесь
дополнительного количества одного из компонентов, например В,
равновесие реакции (3.4) сдвигается вправо или влево (в данном
случае вправо) до тех пор, пока новые равновесные концентрации
всех компонентов не будут удовлетворять уравнению (3.8).
Множество расчетов такого рода приводится в элементарных
учебниках по химии, и поэтому здесь эти расчеты опущены.
AG?X —изменение сбободной энергии, происходящее-в
результате превращения а молей вещества А и Ь молей вещества В в с
молей вещества G и d молей вещества D при условии, что все
компоненты находятся в стандартном состоянии, т. е. при единич-
63 ТЕРМОДИНАМИКА В БИОЛОГИИ
ной концентрации (выраженной в мольных долях, моляльностях
«ли парциальным давлением) для идеального растворителя,
растворенного вещества или газа. Реальные растворы и газы, однако,
могут значительно отклоняться от идеального поведения, и ниже
рассмотрено, каким образом и с помощью каких преобразований
необходимо модифицировать вышеприведенные уравнения, чтобы
объяснить и описать неидеальное поведение.
-З.А.1. АКТИВНОСТЬ: ТЕРМОДИНАМИЧЕСКАЯ КОНЦЕНТРАЦИЯ
Хотя константа равновесия /Сравн, выраженная уравнением (3.8),
пригодна для описания равновесия в разбавленных растворах
(особенно растворов незаряженных веществ), оказывается, что для
более концентрированных растворов (особенно растворов
электролитов) правая часть уравнения (3.8) зависит от общей
концентрации компонентов в равновесной системе. Другими словами,
константа равновесия не всегда является постоянной величиной.
Сделаем попытку объяснить это явление, а затем покажем, как его
можно использовать при разделении и очистке макромолекул в
растворе.
Возвращаясь к определению константы равновесия [уравнение
(3.8)], находим, что мольная доля растворителя Ха была введена
с некоторым приближением [уравнения (2.19) и (2.29)] для
описания отношения давлений пара Ра/Ра в уравнении (2.28) (где
Ра — давление пара, раствора, Ра—давление пара чистого
растворителя). Хотя это приближение выполняется при условии
бесконечно разбавленного раствора, оно нарушается в более
концентрированных растворах, для которых следует непосредственно
использовать отношение Ра[Р°а. Мольную долю растворителя Ха
можно, следовательно,^рассматривать в качестве приближения к
истинной «термодинамической» концентрации (активности ад),
которую можно найти из Ха с помощью соответствующего
коэффициента, известного под названием коэффициента активности ул'-
аА =* ^а/^а *= Va-^a (для растворителя А)
где
VA > 1 при ^а *■ 1 (бесконечно
разбавленный раствор)
(3.9а)
(3.10а)
Таким образом, константа равновесия действительно постоянна
при условии, что в правой части уравнения (3.8) используются не
концентрации, а активности (см. ниже).
Аналогичные рассуждения приводят к определению активности
как истинной термодинамической характеристики содержания
вещества (газа или твердого) в растворе аъ, если концентрация
выражена через моляльность раствора (или парциальное давление.
64 ЧАСТЬ 1
или мольную долю). При этом вводят соответствующий коэффи-
циент активности, как показано ниже*:
ав — Тв'ив [Для твердого вещества (раствор в жидкости)]
Yb *■ 1 ПРИ тв *" О
<*в = Yb^b (для газов)
Yb *- 1 при Рв *" О
аВ — Тв^в [Для твердых веществ (твердый раствор)]
Yb *- 1 при Яв *■ 1
(3.96)
(3.106)
(3.9в)
(З.Юв)
(3.9г)
(З.Юг)
В уравнении (3.9) все активности — безразмерные величины,
поскольку они соответствуют отношениям концентраций к
концентрациям в некотором стандартном состоянии (см. примеры в
гл. 4). Используя введенные активности, теперь можно переписать
уравнения (3.7) и (3.8) в виде, независимом от общей
концентрации веществ в системе
AGrJC = —RT In Ка (при постоянных Т и Р),
где
*с4
<4
(Ка не зависит от общей
концентрации веществ в системе)
(3.11)
(3.12)
Важно понять, почему приближенное определение константы
равновесия [уравнение (3.8)] часто вполне пригодно для расчетов
действительных равновесных концентраций. Во-первых, если речь
идет о разбавленном растворе, то все коэффициенты активности
[уравнения (3.10)] приближаются к единице и /Сравн=/(а:
(3.13)
lim/Ca==/CpaBH.
бесконечное
разбавление
Следовательно, уравнение (3.8) обычно подходит для описания
разбавленных (^0,01 моль/л) растворов. Во-вторых, поскольку
* Активность определена как произведение некоторым образом выраженной
концентрации на соответствующий коэффициент активности. Хотя выбранные в
уравнении (3.9) единицы концентрации, а именно мольные доли (для
растворителя или твердого вещества в растворе), и молярность, широко применяются,
можно так же легко определить активность вещества в растворе, например, как
произведение моляльности илн мольной доли на соответствующий коэффициент
активности. Во всех случаях в идеальном растворе (обычно гипотетическом)
0=1, если действительная концентрация растворенного вещества (моляльность,
молярность, мольная доля) равна единице, т. е. 7=1. Поэтому дл"я практического
расчета констант равновесия важным моментом является выбор подходящих
условий при определении активностей растворенного вещества и растворителя.
К счастью, эту трудность можно часто обойти, проводя все эксперименты при
постоянной ионной силе или в разбавленном растворе, как рассматривается выше.
65 ТЕРМОДИНАМИКА В БИОЛОГИИ
(см. разд. З.В) коэффициенты активности в первую очередь
зависят от ионной силы раствора (по крайней мере, для заряженных
частиц в растворе), коэффициенты активности в уравнении (3.14)
можно сохранять неизменными, проводи измерения при
постоянной ионной силе так, что множитель /Сравн, в который входят
концентрации, также останется постоянным:
п° Л „с d ..с .,d • ..с „с/
к _°сдр _ т&пъ УсУр __к УсУр п 1л\
аАав тАтв 7АУв УаУв
Таким образом, поскольку Ка не зависит от концентраций,
/Сравн также не будет зависеть от концентраций при условии, что
уг постоянны при постоянной ионной силе.
Поскольку активность и концентрация могут различаться очень
существенно (в 10 раз и более в растворах некоторых солей; см,
рис. 3.1), необходимо найти способ определения активности
каждого компонента в равновесной смеси для того, чтобы рассчитать
истинную константу равновесия Ка. Активность растворителя
можно определить, измерив давление пара раствора [см. уравнение
(3.9а) и табл. 3.1]. Активность растворенного вещества получают
из уравнения Гиббса — Дюгема, вывод которого помещен ниже.
Общую свободную энергию G системы, состоящей из
растворителя А и растворенного вещества В, можно выразить через
свободные энергии обоих компонентов системы GA и GB:
G=GA-\-GB=nAGA-\-nBGB (при постоянных Т и Р) (3.2)
Для бесконечно малого изменения свободной энергии G,
следовательно, получаем
dG=nAdGA -{- GAdn A -j- nBdGB -j- GBdnB. (3.15)
Однако из уравнения (3.1) известно, что
dG^GAdnA+GBdnB, (3.1)
так что, сравнивая уравнения (3.15) и (3.1), приходим к выводу,
что
npAGk -\- nBdGB — 0 (уравнение Гиббса—Дюгема)
(3.16)
Поскольку Gi = G°i ~\-PT\n(ai), в общем виде уравнение (3.3)
можно выразить следующим образом:
dG^RTdlnfa), (3.I7)
а используя новые обозначения, можно переписать уравнение
(3.16) в виде
nxd\naA+nBd\naB=0. (3.18)
66 ЧАСТЬ 1
Таблица 3.1
Мольные доли и активности растворителя и растворенного вещества
в растворах сахарозы3 при 50 °С [6]
Вода (растворитель) Сахароза (растворенное вещество)
>ные доли Ад
0,9940
Ю,9864
0,9826
0,9762
0,9665
0,9559
0,9439
0,9323
0,9098
0,8911
активность
0,9939
0,9834
0,9799
0,9697
0,9617
0,9477
0,9299
0,9043
0,8758
0,8140
мольные доли Л"д
0,0060
0,0136
0,0174
0,0238
0,0335
0,0441
0,0561
0,0677
0,0902
0,1089
активность
°в=?в х в
0,0060
0,0136
0,0197
0,0302
0,0481
0,0716
0,1037
0,1390
0,2190
0,3045
"Активность воды рассчитана из давления пара раствора .[уравнение (3.9а) 1 и
использована для расчета активности сахарозы путем интегрирования уравнения Гиббса — Дюгсма
'[уравнение (3.18)]. См. также задачи в конце главы. Обратите внимание, что в
разбавленных растворах концентрация (растворителя или растворенного вещества), выраженная в
мольных долях незначительно отличается от действительной термодинамической
концентрации (активности); различия между этими величинами увеличиваются при переходе к
более высококонцентрированным растворам (и по мере увеличения концентрации вещества
в растворе).
Наконец, поскольку состав данного раствора известен (яд и
Пв) и активность растворителя можно измерить, пользуясь
уравнением (3.9а), то с помощью уравнения (3.18) (см. табл. 3.1)
можно найти активность растворенного вещества. Полученные таким
образом активности растворенных веществ затем используются
для расчета коэффициентов активности [уравнения (3.96) — (3.9г)],
которые, можно считать, представляют собой отношение
«термодинамической» концентрации ав к измеряемой концентрации тв,
что иллюстрирует рис. 3.1 для нескольких растворенных' веществ.
Для растворов электролитов ситуация аналогична, но требует
некоторых дополнительных замечаний. Поскольку в водном
растворе многие ионогенные соединения полностью диссоциируют
MX ч—+ М+ + Х" (3.19)
химический потенциал растворенной соли Омх(водн) р'авен сумме
химических потенциалов ионов, входящих в ее состав:
^МХ водн =^М+ води ~f-Gx~води (3.20)
7 ТЕРМОДИНАМИКА В БИОЛОГИИ
Концентрация, ллоль/г
Рис. 3.1. Коэффициенты активности [уравнения (3.9) и (3.246)] различных
веществ в водных растворах при 25 °С [5].
«Неидеальное» поведение (т. е. у^.ф1) электролитов (сплошные кривые) проявляется при
более низких концентрациях, чем для неэлектролитов (пунктирные линии).
Проинтегрировав уравнение (3.17) и подставив результат в
уравнение (3.20), получаем
GMX водн + RT lnjZMX В0ДН =
=GM+ BOnn+RT In aM+ B0AH+G°X- водн+^Г lnax- в0дн. (3.21)
Однако, поскольку уравнение (3.20), в частности, справедливо для
(гипотетического) стандартного состояния одномоляльного
водного раствора соли MX при идеальных условиях
Gmx водн*=(/№*■ водн-{~бх~ водн (3.22)
то совместное решение уравнений (3.21) и (3.22) приводит к
простому выводу:
аМХ водн = (аМ+ водн) i^X" водн) • (3 • 23)
Однако, поскольку невозможно получить раствор, содержащий
только положительно или только отрицательно заряженные ионы,
а оба вида ионов всегда должны присутствовать одновременно, не
существует пути для определения активности для Мводн или ХГодц
раздельно. Максимально возможным является определение
средней активности а+ вещества:
(а±)« = ом+ах-.
(a±)J = (?±)*mM+mx-
(3.24а>
(3.25б>
68 ЧАСТЬ 1
где у+ — средний коэффициент активности ионов; примеры
приведены на рис. ЗЛ. Для более сложных по составу электролитов
MrXs +=*= rM+'-f-sX-r (3.25)
среднюю активность и средний коэффициент активности можно
определить следующим образом (см. задачи в конце главы):
(«±Yr+s) = №)' (*хУ - (y±Yr+s) №Г КТ
(3.26)
З.Б. РАСТВОРИМОСТЬ МАКРОМОЛЕКУЛ
З.Б.1. «ВСАЛИВАНИЕ» И «ВЫСАЛИВАНИЕ»
Между растворимостью макромолекул в солевых растворах и
зависимостью у± от концентрации соли (рис. 3.1) имеется прямая
взаимосвязь. В основе лежит гипотеза, что ион с данным знаком
заряда окружен ионами с противоположным знаком заряда
© © ее
©ее »-" е е е
ее ее
В этом случае между ионами существует электростатическое
взаимодействие, энергия которого должна учитываться в выражении
свободной энергии раствора соли:
^МХ водн == (^МХ водн)хим I ^электр»
^МХ «оди =<?МХ водн +RT In (mMX в0дн)8+бэлектр- (3.27)
Поскольку для идеального раствора 7мх(водн) = 1,/ все
отклонения от этого условия естественно отнести за счет
дополнительного потенциала электростатического ион-ионного взаимодействия
{уравнения (3.27)]:
Электр — ВТ 1П YMX водн- (3- 28)
Энергия электростатического взаимодействия должна
увеличиваться с увеличением заряда иона и с увеличением концентрации
соли (в более концентрированном растворе соли среднее
расстояние между иойами меньше, а потенциал обратно пропорционален
ТЕРМОДИНАМИКА В БИОЛОГИИ
Ьадрату расстояния между ионами), потому представляется
разумным, что 1п7± выражается формулой
IgY
где
/ =
±~
1
2
~А|
2j m
1
г+г
а
1*1
_|/V.
ионная
(уравнение
сила
Дебая-
-Хюккеля),
(3.29)
(3.30)
к А — коэффициент пропорциональности; для водных растворов
при 25°С А =0,509. Уравнение (3.29) получено Дебаем и Хюкке-
лем путем длинного прямого расчета с использованием
потенциала ион-ионного взаимодействия при среднем расстоянии между
ионами, определяемым концентрацией соли. 2+ и 2-— заряды
положительного и отрицательного ионов. Ионную силу / можно
рассматривать, как средневзвешенную концентрацию иона, при этом
показателем веса является квадрат заряда иона. Теперь можно
перейти к обсуждению вопроса о растворимости.
Введем константу — произведение растворимости —
(заряженного) белка Рг+ в воде
>Х, ^* P*+4-zX" (3.31)
*«=ДРгдрхГ)г =Дрг+ (а*-)г=ПР <3"32>
(архг=1). Используя определение коэффициента активности для
ионогенных твердых веществ в растворе [уравнение (3.26)], из
уравнения (3.32) получаем
ПР=(mp*-) {mx-Y у%+1). (3.33)
Если теперь в том же растворе растворить какую-либо другую
соль (например, M+Y~), то, согласно уравнению (3.29), 1пу±
станет более отрицательным, т. е. у± уменьшится. Однако ПР
определено через активности и, следовательно, является истинной
константой равновесия процесса растворения. Поэтому, если ПР
имеет постоянное значение, а у± при добавлении' в раствор
другой соли уменьшается, то концентрации mPz* и тх— в уравнении
(3.33) (т. е. растворимость белка) должны пропорционально
увеличиться. Такое увеличение растворимости макромолекулярного
вещества при увеличении содержания соли в растворе известно
как эффект всаливания (рис. 3.2). Таким образом, эффект всали-
вания удается объяснить действием сил притяжения между
ионами в солевом растворе. Уравнения (3.29) и (3.30) получаются
только при рассмотрении системы, характеризующейся
бесконечно малой ионной силой; в условиях более высоких концентраций
солей требуется дальнейший анализ, который помещен ниже.
70 ЧАСТЬ I
NaCl
Всаливание
(MH4)2S04
Высаливание
[Преобладание ион-\
\ионнб/х взаимодеа-Т
ствий '
Преобладание
взаимодействий между
ионом ираст
eopumefleM
2 <3
Ионнай сила
Рис. 3.2. Относительная растворимость карбоксигемоглобина в зависимости от
ионной силы в растворах разных неорганических солей [6].
Участки кривых при ионной силе «XI линейны и описываются уравнением (3.29).
Качественное объяснение кривых, когда становятся заметными эффекты всаливання и
высаливания, см. в тексте.
Данные, приведенные на рис. 3.2, указывают на
дополнительное влияние добавок соли при более высоких концентрациях соли,
а именно на противоположный эффект, приводящий к уменьшению
растворимости белка с увеличением количества соли (эффект
высаливания). Эффект высаливания можно объяснить связыванием
молекул воды благодаря электростатическому притяжению между
заряженным ионом и молекулой воды, обладающей большим ди-
польным моментом
о0ъ
<§ о
О ^
Таким образом, происходит как бы эффективное «удаление»
молекул воды и они становятся не доступными для сольватации
молекул белка. При высокой концентрации соли с ионами соли
«связано» столько воды, что эффективная концентрация белка
увеличивается и белок осаждается из раствора (рис. 3.2).
Хотя, анализируя ион-дипольные взаимодействия, не удается
получить реального количественного описания эффекта
высаливания, некоторые качественные выводы из получаемых результатов-
сделать можно. Данные, представленные на рис. 3.2,
свидетельствуют о том, что эффект высаливания в гораздо большей, степени
определяется природой аниона (SO?~ илиСГ-), чем природой
катиона (Na+ или К+); этот вывод является общим. При сравнений
ряда анионов или катионов следовало бы ожидать, что степень-
1\ ТЕРМОДИНАМИКА В БИОЛОГИИ
увязывания молекул воды данным ионом должна зависеть от двух
(взаимозависимых) факторов: размера иона (ионы большего
размера должны характеризоваться способностью к связыванию
большего числа молекул воды) и плотности заряда па его
поверхности (поверхность с большей плотностью заряда должна сильнее
притягивать молекулы воды и, следовательно, ион связывает
большее число молекул воды). Данные табл. 3.2 для разных катионов
и анионов иллюстрируют влияние этих двух факторов.
Поскольку между радиусом иона и плотностью заряда на его поверхности
существует обратная зависимость, нельзя сразу сказать, влияние
какого фактора на активность иона и, следовательно, на
растворимость белка будет превалировать. Согласно экспериментальным
данным (например, на рис. 3.2), отрицательно заряженные ионы
оказывают большее высаливающее действие благодаря тому, что
они (как правило) крупнее положительно заряженных ионов и,
следовательно, могут удерживать на периферии иона больше
молекул растворителя. Хотя объяснения уже известных
экспериментальных данных гораздо менее полезны, чем предсказания,
полученные результаты все же представляют интерес. Сравнивая
относительную растворимость данного белка в присутствии очень
многих солевых добавок, можно установить эмпирические
последовательности эффективности высаливающего действия различных
катионов и анионов [уравнения (3.34а) и (3.346)]; эти последова-
Таблица 3.2
Ионные радиусы и (относительная) напряженность электрического поля
иа поверхности иона3 для различных катионов и анионов
«о Напряженность (относительная)
Ион Ионный радиус , А электрического поля на поверхности иона
Высокая
Li+
Na+
К+.
Nrtf
Cs+
Mg2+
Zn2+
Mn2+
Ca2+
F"
ci-
Br-
I-
0,60
0,95
1,33
1,48
1,69
0,65
0,74
0,80
0,99
1,36
1,81
1,95
2,16
Низкая
Высокая
t
Низкая
Высокая
t
Низкая
aC большим приближением можно считать, что между этими двумя параметрами
существует обратная зависимость.
иЦля сравнения: средний диаметр молекулы воды составляет 2 А.
72 ЧАСТЬ 1
s
-2»
-0,50
2 4 6
Ионная сила
в
Ю
Рис. 3.3. Селективное осаждение белков путем добавления сульфата аммония [7].
Ионная сила рассчитывалась по уравнению (3.30).
т — растворимость белка. 1 — фибриноген; 2 — гемоглобин; 3 — псевдоглобулин; 4 —
сывороточный альбумин; 5 — многлобнк.
тельности известны под названиями лиофобных и лиофильных
рядов или рядов Гофмейстера:
Анионы: Цитрат3" > Тартрат2- > SO2;- > Ацетат" >CI" > NC£ >
>Br->r>CNS' (3.34a)
Катионы: Th*+>AI8+>H+>Ba2+>Sr2+>Ca2+>K+>Na+>NHj (3.346)
Пользуясь этими последовательностями, можно выбрать такую
комбинацию катион — анион, которая должна оказаться наиболее
эффективной для осаждения белков из раствора*. Наиболее
широко используемым высаливающим агентом является сульфат
аммония, поскольку в его присутствии происходит селективное
осаждение некоторых белков, в то время как другие белки остаются
в растворе (рис. 3.3). Например, гемоглобин .высаживается из
раствора сульфата аммония при ионной силе ~6, а миоглобин
при той же концентрации (белка) остается в растворе. Таким
образом, добавление сульфата аммония — один из способов
селективного выделения (осаждения) некоторых белков из смеси; этот
способ находит широкое применение при очистке белков.
Один из аспектов практической ценности рядов Гофмейстера
состоит в том, что, хотя эти последовательности невозможно
предсказать заранее, аналогичные ряды наблюдаются при изучении
многих других явлений (кроме отмеченной здесь растворимости),
например электрофореза, вязкости, седиментации и др. Причина
* В действительности соль LiCNS, соответствующая самым крайним
(справа) ионам, практически не проявляет высаливающего действия, а его всаляваю-
щее действие приводит даже х растворению шелковых чулок (натуральный шелк
состоит из молекул белка) в концентрированных растворах LiCNS.
fj(3 ТЕРМОДИНАМИКА В БИОЛОГИИ
■'
:.
Этого, возможно, связана с влиянием соли на химический потен-
\циал ионов (т. е. на растворимость белков); следовательно,
последовательность типа рядов Гофмейстера можно ожидать для
любого явления в растворах, содержащих растворенную соль. Многие
^константы равновесия и константы скорости химических реакций
^будучи отложенными на графике в логарифмическом масштабе)
[находятся в пропорциональной зависимости от квадратного корня
шз ионной силы, и это представляется разумным, как следует из
[уравнений (3.29) и (3.33) для частного случая равновесия при
'растворении.
?З.Б.2. ГИДРОФОБНОСТЬ: НЕКОВАЛЕНТНАЯ АССОЦИАЦИЯ
НЕПОЛЯРНЫХ МОЛЕКУЛ
Большинство молекул и их комплексов, представляющих интерес
для химиков, существуют благодаря водородным ковален/гным
связям или силам электростатического кулоновского притяжения
Между заряженными молекулами. В молекулярной биологии,
однако, известно множество примеров очень прочной ассоциации между
практически неполярными молекулами (т. е. между
незаряженными молекулами с очень малым дипольиым моментом) в водной
среде. Примерами таких взаимодействий являются: белок-белковые
(например, ассоциация субъединиц фермента, приводящая к
образованию белка с четвертичной структурой; см. гл. .11.Б.2),
сворачивание белка в более функциональноспособную форму;
образование липидных мицелл и бислойных мембран и др. Для того чтобы
понять происхождение и природу таких гидрофобных
взаимодействий, прежде всего необходимо ввести характеристику прочности
гидрофобных связей.
Для обычных ковалентных химических связей мерой прочности
обычно служит изменение энтальпии АН или энергии АЕ при
образовании данной связи из ^несвязанных атомов. Основная причина
выбора данных параметров состоит в том, что их легко измерить
экспериментально методами спектроскопии (АЕ) или
калориметрии (ДЯ) или найти из температурной зависимости констант
равновесия (АН). В случае исследуемых в биохимии крупных молекул
{часто обладающих ограниченной летучестью или растворимостью)
тепловые эффекты слишком малы для измерений в разбавленном
растворе, а спектры обычно слишком сложны для извлечения
информации о прочности связей. Поэтому прочность биохимических
связей обычно оценивают по изменению свободной энергии AG или
(что эквивалентно; см. гл. 5) по изменению электрохимического
потенциала, так как AG легко получить из измерений константы
равновесия, как было показано в предыдущем разделе.
Поэтому логически прочность гидрофобных связей можно
характеризовать через изменение (стандартной) свободной энергии,
обнесенной к 1 молю, AG0 для переноса растворенного вещества из
74 ЧАСТЬ 1
веды (растворителя, в растворе которого молекулы растворенного
вещества распределены равномерно) в джйгсан (растворитель, в
котором существуют гидрофобные взаимодействия, которые
обусловливают, например агрегацию молекул растворенного вещества)
Гидрофобность, отнесенная к 1 молю растворенного вещества =
== ^диоксан ^вода. (^ ■ &£>)
Далее, поскольку химический потенциал данного растворенного
вещества в его насыщенном растворе имеет одно и то же значение
в любом растворителе (т. е. при достижении концентрации
насыщения растворенное вещество проявляет одну и ту же тенденцию к
осаждению независимо от выбранного растворителя), то
^диоксаи—^вода (насыщенный раствор в любом растворителе),
^диоксаК'Г'А-' 1П 1^иасЫщ.,диоксаи_Г^-' ^П7иасЫщ.,диоксан ~
—О ^^ ^^
= £вода-ГА/ 'п [£]насыщ.,вОда~Г"-ЭДП ^П7иасыщ.,вода (3.36)
Поскольку концентрации в уравнении (3.36) представляют
собой просто растворимости данного вещества в обоих
растворителях, а коэффициенты активности для малорастворимых веществ
(так как их растворы очень разбавлены) близки к единице,
можно объединить уравнения (3.35) и (3.36)
Гидрофобность, отнесенная к 1 молю __ рт , (Растворимость в воде)
растворенного вещества * (Растворимость в диокеане)
(3.37)
Гидрофобность, определенную уравнением (3.37),
следовательно, легко найти экспериментально, измеряя растворимость. Ниже
рассмотрены некоторые применения на практике относительной
гидрофобности, определенной по уравнению (3.37).
Пример. Гидрофобность боковых цепей аминокислот в белковых молекулах
Уравнение (3.37) можно использовать для расчета гидрофобности любых
аминокислот, встречающихся в нативных белках и ферментах. Поскольку глицин
является основной единицей звеньев с пептидными связями, образующими основу
белков, сворачивание полипептидной цепи для принятия частной конфигурации,
характерной для данного белка, должно определяться боковыми цепями
остальных аминокислот (эти аминокислоты, кроме пролина, отличаются от глицииа
просто структурой боковой цепи, ответвляющейся от а-углеродного атома).
Следовательно, если определить гидрофобность каждой аминокислоты, а затем в каждом
случае вычесть гидрофобность глицина, то получим характеристику
гидрофобности каждой из боковых цепей в аминокислотах. Эти величины собраны в
табл. 3.3.
Данные, приведенные в табл. 3.3, показывают, что перенос неполяриого
аминокислотного остатка из водной среды (на поверхности белка) в относительно
иеполярную внутреннюю область белковой макромолекулы может
сопровождаться большим выигрышем свободной энергии (А&°<£.0). Механизм этого эффекта
трудноуловим. Ои не вызван притяжением (по типу «подобное к подобному:»)
ТЕРМОДИНАМИКА В БИОЛОГИИ
Таблица 3.3
Шкала гидрофобности боковых цепей аминокислот,
полученная путем вычитания гидрофобности глицина
из гидрофобности каждой из указанных
аминокислот3 [8]
Аминокислота, боковая цепь которой Гидрофобность
рассматривается (кал/моль) при 25 °С
Триптофан
Норлейцин
Фенилаланин
Тирозин
Диоксифенилаланин
Лейцин
Валин
Метиоиин
Гистидин
Алании
Треонии
Серии
—3400
—2600
—2500
—2300
—1800
—1800
—1500
—1300
—500
—500
—400
+300
Большинство приведенных значений средние (обычно из
близких); использованы данные, полученные относительно «не-
полярных> растворителей (диоксаиа или этанола).
самих гидрофобных (т. е. углеводородных алифатических или ароматических)
групп; он также не вызван каким-либо существенным отталкиванием между
гидрофобной группой и растворителем. Причина состоит просто в том, что между
молекулами воды существуют мощные взаимодействия, которые должны
нарушаться при растворении вещества в воде. При растворении ионогенных
соединений эти* нарушения в основном компенсируются заменой взаимодействий вода —
вода иа взаимодействия иои — вода. Однако при растворении неполяриых
(гидрофобных) веществ такой компенсации нет и растворение вещества в воде
энергетически невыгодно. Более энергетически выгодной является ассоциация
молекул вещества друг с другом, при которой взаимодействия между молекулами
воды нарушаются в наименьшей степени. Этот эффект легко понятен из
корреляции, показанной на рис. 3.4, демонстрирующей линейную зависимость
гидрофобности от площади неэкранированной молекулами растворителя поверхности
боковой цепи аминокислоты. Гидрофобность аминокислотных остатков для
разных белков, таким образом, одинакова и составляет около —25 кал
(принимается во внимание только иеэкранированная поверхность).
Взаимосвязь между гидрофобиостью и площадью поверхности боковой цепи
можно использовать для расчета свободной энергии, выигрываемой при
ассоциации двух белков или белковых цепей в данной макромолекуле с известным
аминокислотным составом в области контакта цепей. Согласно расчетам, для
объяснения наблюдаемой устойчивости таких агрегатов можно считать, что контакт
осуществляется иа сравнительно малых площадях. Например, субъединицы
гемоглобина (см. ч. 3) удерживаются друг с другом в аосоциате благодаря
контактам: для ассоциата aiB, такие контакты затрагивают всего лишь 12% доступной
76 ЧАСТЬ ]
-400 0 800 1600 2400 3 200
Гидрофобность, кал/моль
Рис. 3.4. Взаимосвязь между доступной для растворителя поверхностью боковых
цепей аминокислот и гидрофобностью этих боковых цепей [9].
поверхности каждого мономера, а для ассоциата atfo даже только 6% (а и Р
обозначают два типа из четырех субъединиц гемоглобина). С помощью
подобных расчетов объясняется также большая устойчивость димера инсулина по
сравнению с его мономерами и связывание фермента трипсина с панкреатическим
ингибитором трипсина, имеющим полипептидную природу. Таким образом,
оказывается, что сложные взаимодействия, удерживающие белки в ассоциатах,
можно достаточно хорошо объяснить, если привлечь для рассмотрения просто
относительные площади разных боковых цепей аминокислотных остатков', а также
учесть возможность осуществления разных способов их взаимных контактов.
Пример. Сворачивание белков: теоретический расчет трехмерной структуры
бычьего панкреатического ингибитора трипсина (рис. 3.5 и 3.6)
Возможно, наиболее наглядным успехом рассуждений такого типа является
сравнительно недавно опубликованный теоретический расчет, в котором
конкретный белок, бычий панкреатический ингибитор трипсина, рассматривается просто
как полипептидная цель с боковыми цепями в приближении, согласно которому
все массы боковых цепей, локализованы в центрах массы соответствующих
цепей. (Это аналогично предыдущему примеру, в котором боковая цепь
оценивается только величиной площади ее поверхности; другими словами, так можно
описать очень сложную структуру — из 750 атомов и 200 связей даже в случае
небольшого белка, состоящего из 50 аминокислотных остатков; такая модель
молекулы белка является очень упрощенной.) Не входя в детали расчета,
основанного на определении наиболее энергетически выгодных относительных
расположений всех возможных пар аминокислотных остатков, можно сказать, что
получены замечательные результаты, поскольку всего лишь за 10 мии (машинного
времени») с помощью ЭВМ удается рассчитать единственную наиболее
энергетически выгодную конфигурацию белка. Кроме того, координаты индивидуальных
остатков, полученные этим опособом, отличались по среднеквадратичному
расстоянию (см. ч. 2) от координат этих атомов в кристаллическом белке (опреде-
a
a.
Tr_8~~f'
*-r4~
a/a ^ fffy glu tyr val phe
Рис. 3,5. Упрощенная (введенная) модель (б) и реальная структура (а) [10].
В упрощенной модели каждый аминокислотный остаток обозначен двумя кружочками,
соответствующими центру боковой цепи и атому С в a-положенни. Каждый остаток
обладает только одной степенью свободы: торэиониый угол а между 4 последовательными а-углс-
родкымн атомами аминокислотных остатков составляет i—Л, i, l+l и i+2. Все боковые
цепи данного вида характеризуются одинаковой упрощенной геометрией. В качестве
характеристик длин связей, углов связей н торзнонных углов, определяющих геометрию такой
упрощенной молекулы, используются средине значения, установленные при исследованиях
конформаций восьми белков; эти характеристики можно также оценить из данных для
модельных соединений.
Ц^$ $
1 L
i—г
1 "Г
400 500
Номер цикла
900 1 000
Рис. 3.6. Рассчитанные с использованием упрощенной модели (рис. 3.5)
конформаций бычьего панкреатического ингибитора трипсина [10].
В качестве исходной конформацнн выбрана развернутая макромолекула с сохранившейся
концевой спиралью (показана вверху). В каждом цикле расчета на ЭВМ варьируются
положения боковых цепей и методом многократных приближений находится конформация
с минимальной обшей свободной энергией. Затем предполагается, что полнпептндная цепь
претерпевает случайные блуждания, обусловленные тепловым движением и
замораживается. На этом один цикл заканчивается и начинается новый цикл расчета.
78 ЧАСТЬ 1
.ленных методом рентгеноструктурного анализа; см. ч. 6) меньше чем на 3,4 Al
Возможно, наиболее поразительная особенность этого результата состоит в том,
-что он указывает иа возможность для реального белка самопроизвольно принять
свою функционально активную конформацию даже после того, как вначале
белок был развернут и принял форму статистического клубка. Примеры такого
доведения будут приведены ниже в ч. 2.
Пример. Мицеллы и мембраны
Особый вид агрегатов образуется в растворе бифильными молекулами.
Эти молекулы обладают асимметричной структурой, иа одном конце которой
находится полярная (часто даже несущая электрический заряд) группа, а иа
другом конце — неполярный фрагмент (часто неразветвленная углеводородная
цепь); к таким веществам относятся мыла, детергенты и фосфолипиды,
являющиеся основными структурными компонентами биологических мембран. Агрега-
i ^
1
Равйав/генный раствор
Более концентрированный
раствор
ты, образующиеся при низких концентрациях подобных веществ в растворе,
изображены ниже и называются мицеллами. В этом случае образования агрегатов
(рис. 3.7) объясняют гидрофобным эффектом, обусловленным наличием у
молекул неполярных углеводородных целей. (Однако размеры мнцелл, видимо, огра-
6
Хкрнт
Общая концентрация в растворе
1,0 1,5 2.0
Общая концентрация /г/ифидги/ннш
молекул
Рис. 3.7. Взаимосвязь между содержанием мономерной формы и общей
концентрацией бифильных молекул в растворе |2, с. 48]. '
а — истинное разделение фаз; б — мицеллообразование. Данные характеризуют
концентрацию бифильного соединения в мицеллярном состоянии и концентрацию мономера.
Пунктирные лннин показывают экспериментальный способ определения ККМ.; точка, прн
которой концентрация мономера равна ККМ, также указана. Выбор способа выражения
концентрации произволен (в граммах или в молях бнфильного соедиксиия).
79 ТЕРМОДИНАМИКА В БИОЛОГИИ
!
-4 -
-з
■•5
I
я-
3
-2
-1
Большая (по
длине) цепь
Меньшая
(по длине)
цепь
Ю
Число углеродных атомов, введенных
в углеводородную цепь
Рис. 3.8. Увеличение гидрофобное™ при удлинении углеводородных цепей
Г- (алкил)ч-алкил-1 -сульфатов [2, с. 53].
Данные для большей (по длине) цепи получены для моноалкилсульфатов относительно
октилсульфата, для дналкилсульфатов относительно 1-бутилдсцилсульфата н 1-пентнлгек-
силсульфата. Данные для цепи меньшей длины получены относительно разных соединений.
с одинаковой длиной цепн большего по размеру радикала. Гидрофобпость оценивалась по
ККМ в воде.
чинены благодаря кулоновским электростатическим силам отталкивания между
соседними полярными группами, находящимися на периферии мицеллы.) В
общем случае весь процесс образования мицелл при увеличении концентрации
вещества в растворе хорошо аппроксимируется процессом фазового перехода (см.
разд. 2.А.2), поскольку выше критической концентрации мицеллообразования
(ККМ) существует строго определенное изменение в степени агрегации би-
фильных молекул.
Критическая концентрация мицеллообразования соответствует минимальной
концентрации вещества в растворе, при которой из гомогенного раствора
начинают образовываться мицеллы; ККМ в некотором роде подобна растворимости
(растворимость — концентрация раствора, при которой вещество начинает
осаждаться из раствора), которая в предыдущем примере использовалась в качестве
меры гидрофобности. Представляется, следовательно, разумным охарактеризо-
вывать гидрофобностъ амфифильных молекул на основании данных о ККМ по
аналогии с незаряженными веществами, для которых гидрофобность
определялась на основании данных о растворимости. Иллюстрацией этого способа оценки
гидрофобности служат данные на рис. 3.8, показывающие, как гидрофобность
изменяется с ростом длины углеводородной цепи в ряду детергентов с двумя
углеводородными цепями в молекуле
Ri— CH-OSO3
Полученные результаты (рис. 3.8) свидетельствуют о том, что вклад в
гидрофобность молекулы, отнесеииый к единице длины цепи, второго, меньшего по
U0 ЧАСТЬ i
■размеру, гидрофобного «хвоста», введенного в молекулу детергента, уже
облагающую одним более длинным хвостом, меньше, чем вклад, привносимый более
длинным хвостом. Однако вклад в гидрофобность, приходящийся на один
углеводородный атом длинного радикала, ие зависит от наличия или отсутствия в
молекуле второго хвоста. Другими словами, цепи в одной молекуле, видимо,
взаимодействуют друг с другом. Это предположение совсем недавно было
подтверждено данными о корреляции координат и подвижности атомов в обеих
углеводородных цепях природных фосфолипидиых молекул в агрегатах,
моделирующих структуру бислоя мембраны; результаты были получены методом
ЯМР-спектроскопии (см. ч. 4).
З.В. СВЯЗЫВАНИЕ НЕБОЛЬШИХ МОЛЕКУЛ
ИЛИ ИОНОВ С МАКРОМОЛЕКУЛАМИ
Большая часть принятых в настоящее время представлений о
механизмах химических и биохимических процессов основана на
измерениях констант равновесия при связывании одной молекулы
с другой. В следующем разделе данные такого рода будут
использованы для объяснения действия ферментов, лекарственных
веществ, ядов и гормонов. Как правило, в основе действия
перечисленных веществ лежит связывание одной или большего числа
небольших молекул I с эквивалентным числом участков на данной
макромолекуле Е
E-fnl ^=fc Е1я (3.38)
где равновесные концентрации удовлетворяют соотношению*
^TIT- (3'39)
В простейшем возможном случае, когда п=\ (т. е. на*
макромолекуле существует всего лишь один участок связывания), для
определения искомой константы равновесия (диссоциации) /Ci из
экспериментальных данных существует несколько графических
методов. Графическое представление данных всегда предпочтительнее
по сравнению с табличным, поскольку на графике легче обнару-.
жить случайные или систематические тенденции (особенно когда
графическое построение выбрано так, что теоретически ожидается
прямолинейная зависимость), которые могут быть не .столь
очевидны из табличных данных. Вначале более или менее детально об^
суждается связывание с одним участком, а затем с помощью
выявленных закономерностей рассматривается анализ процессов
связывания с большим числом участков. Для определения одной и той
же константы равновесия описано несколько разных способов
построения графиков и каждый способ используется при изучении
* В' этом разделе для простоты используется приближенное выражение
константы равновесия [через концентрации, а не активности, т. е. уравнение (3.8), а
не (3.12)]. Условия, при которых применимо это приближение [см. обсуждение
уравнения (3.12)], часто выполняются для биохимических процессов.
ll ТЕРМОДИНАМИКА В БИОЛОГИИ
шстемы конкретным экспериментальным методом. Например, с по-
ющью рН-метра непосредственно измеряется логарифм
концентрации Н+, так что получаемые данные (кривые титрования; см. ни-
gike) лучше всего представить в виде графической зависимости от
логарифма [Н+].
^Способы графического представления данных
при одноместном связывании
I
1- Начнем с рассмотрения одностадийного связывания с одним
участком
=* EI
E+I
(3.40)
приближенно характеризуемого константой равновесия
(диссоциации) /Ci
(3.41)
Для рассмотрения оказывается удобным использовать параметр
v, характеризующий долю общего числа макромолекул,
образовавших комплекс
v =
[EI]
[EI] + [Е]
(3.42)
Методы определения К\ из экспериментальных данных основаны
на разных способах объединения уравнений (3.41) и (3.42); часто
можно прямо экспериментально найти величину v. Например, если
поглощение в ультрафиолетовой или видимой области спектра
(ч. 4) свободных макромолекул Е в растворе известно и отлично
от поглощения макромолекул в комплексе EI, то долю молекул в
комплексе EI легко рассчитать из спектральных измерений.
Также v можно найти из результатов измерений методом равновесного
диализа, как описано в разд. 2.В.
Прямой метод. Если [EI] из уравнения (3.41) подставить в
уравнение (3.42), то
(3.43)
Для определения /Ci можно построить зависимость v от [I] при
постоянной [Е]общ. Эта зависимость имеет вид гиперболы; согласно
уравнению (3.43), если в комплексе связана ровно половина
макромолекул (т. е. v=0,5), то [1]=/С[. Построение такой зависимо-
82 часть i
Рис. 3.9. Сопоставление графических способов определения константы
диссоциации К\ при, одностадийном связывании низкомолекулярного соединения (или
иона) I с одним участком на макромолекуле.
а—прямой метод '[уравнение (3.43)]; б — кривые Бьеррума или кривые титрования
[уравнение (3.48)]; в—метод Бенези — Хильдебранда (обратная зависимость) [уравнение (3.51)];
г — кривые Скэтчарда .[уравнение (3.52)]. Хотя кривая на рис. б, вероятно, наиболее
знакома для большинства читателей, прн существовании на макромолекуле нескольких
участков связывания с разными константами связывания для разных участков предпочтительным
является построение кривых Скэтчарда (г) (см. текст). Уравнения, применяемые при
графическом определении AV если связывание происходит с п эквивалентными участками
макромолекулы, приведены в табл. 3.4.
I
сти, следовательно, позволяет определить Ki из экспериментальных
значений v, измеренных при разных концентрациях [I] (рис. 3.9,а).
Сложность использования прямого метода состоит в том, что
получаемая зависимость нелинейна, и поэтому для нахождения точки
перегиба кривой, для которой [1]=/Сь необходимо получать данные
в широкой области концентраций [I]. Этот вопрос еще будет
обсуждаться далее.
Построение кривых Бьеррума (кривых титрования). Для частно--
го случая, когда 1 = Н+, наиболее удобным способом определение
[Н+] служит рН-метрия
рН—lgaH+^ -lg [H+]. (3.44)
Тогда известное уравнение Хендерсона — Хассельбалха
получается логарифмированием обеих частей уравнения (3.41)
PH = p/ta-Hg([E]/lEH+]), (3.45)
где
p/Ca=-lg/Ca=-lg([E][H+I/[EH+]). (3.46)
S3 ТЕРМОДИНАМИКА В БИОЛОГИИ
В более общем случае решение уравнения (3.43) относительно /G
запишется следующим образом:
/C, = [I][(l-v)/vI (3.47)
и затем, прологарифмировав обе части этого уравнения
lg(/CI) = lg([I])+lg((l-v)/v),
или
-lg/C,=-lg[I]-lg((l -v)/v),
или
-lg[U=-lg/C,+lg((l-v)/v),
получаем
PI = p/Ci + lg((l-
где
pl = -lg[l]
p/tl = -lg/ti
-V)/v),
Уравнение (3.45) [и более общее уравнение (3.48)], таким образом,
приводит к построению зависимости v от рН (или в более общем
виде от pi), показанной на рис. 3.9,6 (кривая титрования). Точно
так же, как константа диссоциации обычной кислоты Ка
определяется значением рН, при котором кислота наполовину
нейтрализована
рН=р/Са при [Е] = [ЕН+] (3.50а)
можно определить константу диссоциации при равновесном
одноместном связывании Ki по концентрации небольших молекул (или
ионов), при которой комплекс образуется с половиной участков
связывания макромолекул.
pl=p/d при v=0,5. (3.506)
Условие (3.506) выполняется в точке перегиба на кривой Бьер-
рума (или кривой титрования) на рис. 3.9, б.
Наконец, согласно уравнению (3.48), экспериментальные данные
можно представить в виде прямолинейной зависимости,
откладывая, lg((l—v)/v) от pi. Эта зависимость (см. обсуждение кривой
Хилла в конце этого раздела) особенно полезна для определения
кооперативного типа связывания.
Метод обратной зависимости (кривые Бенези — Хильдебранда).
Существует несколько способов преобразования уравнения
гиперболы (3.43) в уравнение прямой. Один из способов состоит просто
84 часть i
в том, чтобы взять обратные величины от обеих частей уравнения
(3.43). При этом получаем '
l/v=l + Ki(l/[l\)
(3.51)
Как показано на рис. 3.9, в, искомую константу диссоциации
К\ получают прямо из угла наклона прямой (1/v) от (1/[1]).
Преимущества использования прямолинейной зависимости состоят в
том, что, во-первых, легко получить количественную оценку
точности результата по отклонению точек от прямой и, во-вторых, К\
можно найти, проводя измерения в. менее широкой
концентрационной области, чем при использовании прямого метода или метода,
основанного на построении кривой титрования (кривой Бьеррума).
Формально идентичная уравнению (3.51) ситуация встречается
также при построении кривых Лайнуивера — Берка, описывающих
кинетику ферментативных реакций (ч. 3).
Построение прямолинейной зависимости (кривые Скэтчарда).
Как легко можно убедиться, другой способ превращения
уравнения гиперболы (3.43) в уравнение прямой состоит в следующем
(v/[I])=(l//C,)-(l//C,)v. (3.52)
Таким образом, кривая Скэтчарда (v/[IJ) от v (см. рис. 3.9,г)
выражается прямой с углом наклона, равным (—1/Ki),
пересекающей ось ординат в точке (l/'/Ci), где 1//Ci=/Cb — константа
связывания (а не константа диссоциации) для одноместного связывания
по уравнению (3.40). Спрсоб Скэтчарда оказывается особенно
полезным при определении констант связывания с большим числом
участков (см. ниже).
Вышеописанные примеры показывают, что существует
несколько способов определения Ki (или, что то же самое, /Cb=(1//Ci)) из
экспериментальных значений v и [I]. Прямой метод определения
требует наименьших преобразований экспериментальных данных,
но приводит к неудобной форме кривой (гиперболе), для
построения которой необходимо, чтобы данные охватывали широкую
область концентраций [I]. Кривая титрования также нелинейна, но
часто используется в тех случаях, когда концентрации измеряются
прямо в логарифмическом масштабе (как, например, рН) или
когда экспериментальные значения [I] охватывают очень
широкую область (например, это имеет место при исследованиях
связывания лекарственных веществ) (см. ч. 3). Построение обратной
зависимости обычно применяется при исследовании одноместного
связывания, особенно при изучении кинетики ферментативных
реакций (см. ч. 3), в то врем,я как кривая Скэтчарда особенно
полезна при анализе связывания с большим числом участков (см.
ниже). Наконец, даже в случае одноместного связывания зави-
85 ТЕРМОДИНАМИКА В БИОЛОГИИ
симость Скэтчарда (рис. 3.9, г) оказывается наиболее удобным
способом графического представления результатов, поскольку она
обеспечивает оптимальное удельное влияние данных, полученных
при разных концентрациях [I]; (Короткое, но полезное
обсуждение достоинств и недостатков описанных способов графического
представления результатов имеется в работе [13].)
Анализ данных при связывании с большим числом участков:
идентичные независимые друг от друга участки связывания.
Когда данная небольшая молекула (ион) может связываться с
любым из нескольких независимых эквивалентных участков
связывания на макромолекуле с константой Кг
/г — tEl П1 (для каждого из п независимых /3 53)
1 (EI] участков на макромолекуле)
то доля занятых участков относительно общего их числа
описывается уравнением (3.42). Если каждый участок связывания на
любой макромолекуле характеризуется одной и той же
константой связывания лиганда, то, чтобы получить число молекул лиган-
да, связанных с одной макромолекулой, надо просто найти среднее
число молекул, связанных на каждом участке [уравнение (3.42)]:
(л—число идентичных независи- ,д е4\
мых участков связывания) * ' '
Под независимыми участками связывания подразумеваются
такие, для которых связывание молекулы с любым из них не влияет
на связывание другой молекулы с другим участком той же
макромолекулы. Уравнение (3.54) для связывания с большим числом
участков напоминает уравнение (3.43) для одноместного
связывания; для этих двух случаев похожи и формы кривых (рис. 3.9 и
■ЗЛО) при условии, что все участки связывания на любой данной
макромолекуле идентичны (см. табл. 3.4).
В реальных макромолекулах разные участки связывания
лиганда характеризуются разными константами связывания.
Например, различные аминокислоты в белке характеризуются
неодинаковым сродством к протонам. Хотя уравнение (3.54) легко
обобщить для описания щ участков t-ro типа с разными константами
равновесия (диссоциации) К\
Sti\ [1] (среднее число занятых молекулами
(/(■л. л_ m 1 участков в макромолекуле)
(3.55)
однако определение разных л* и К\ графическим анализом
усложняется. Конечно, можно приспособить экспериментальные данные
Таблица 3
Уравнения, применяемые при графических способах определения кднстаиты равновесия (диссоциации) К\ (рис. 3.9)
из экспериментальных измерений доли занятых участков связывания v в зависимости от концентрации
свободных молекул лиганда [1]а
Способ
Уравнение
Зависимость
Определение пн^
Прямой
Построение кривых Бьерру-
ма (кривых титрования)
Метод Бенези — Хильдеб-
ранда (обратная
зависимость)
Построение кривых Скэт-
чарда
v
п[Ц
*i+[U
In — v\
Pl=p/Ci + Igf——J
v против [I] (гипербола)
v против Ig [I] (сигмоидная
кривая)
1/v = 1/л+ (Ki/n) (l/[IJ) l/v против l/fll (прямая)
v/[l] = n/Ki-v/Ki
v/[I] против v (прямая)
n = limv
[I]-co
Ki = {I] при v = (л/2)
n = limv
lg[Ij-*0O
pKi — pi при v = n/2
n = (отрезок на оси у)-1
x~Q
Кi = —(отрезок на оси х)~г
п = (отрезок на оси х)~1
</=о
=о \
*1
х-
отрезок на оси у
Г
а Уравнения обобщены для случая связывания низхомолекулярных соединений (или ионов) с п независимыми эквивалентными участками на макромш
куле
tEj[I]
Е+я1 '< ■ >• EI ; /Cj= идентичное независимое связывание с каждым из п участков макромолекулы
••"»ло молекул лиганда, связанных с одной макромолекулой
87 термодинамика в биологии
(v как функция [I]) к уравнению (3.55) с помощью
вычислительных методов и определить искомые п{ и (Ki)i расчетом на ЭВМ.
(В действительности, даже когда данные можно согласовать с
выбранной графической зависимостью, желательно найти конечные
параметры в цифровом выражении, поскольку такие расчеты
легче воспроизвести другим исследователям.) Тем не менее
построение графической зависимости всегда желательно в качестве
диагностического качественного средства—«случайно выброшенные»
экспериментальные точки можно легко найти на графической
зависимости и повторить эксперимент, так как несколько таких
«плохих» точек может повлиять на результаты самого тщательного
расчета. Также использование доверительных интервалов,
характеризующих точки на графике, упрощает решение вопроса о том,
какие точки и в какой степени надо учитывать для конечной
«подгонки» зависимости. Ниже разные графические методы получения
констант связывания (диссоциации) и числа участков связывания
каждого типа на макромолекуле сопоставлены друг с другом.
Рис. 3.10 показывает, как выглядят обычные графические
зависимости, описывающие процесс связывания, в случае двух типов
участков связывания [(/Ci)i = 0,001 моль/л; (/(1)2 = 0,04 моль/л,
если число участков каждого типа «i = 10 и я2 = 30]. Очевидно, что
из четырех обычно применяемых методов для определения П\, п2,
(/Ci)i и (/Ci) 2 наиболее подходящим является построение кривой
Скэтчарда.
Несмотря на достоинства метода1 кривых Скэтчарда, очевидные
из 3.10, существует один тип связывания, для которого удобнее
воспользоваться методом кривых титрования, поскольку
экспериментальные данные измеряются в логарифмическом масштабе (а
именно измерение рН). Преобразуем зависимость Бьеррума
(кривые титрования) в вид, более пригодный для анализа данных,
получаемых при титровании многоосновных макромолекул.
Буферная емкость. Для упрощения проблемы представим
кривые титрования в виде индивидуальных вкладов функциональных
групп, для которых известны константы рКа, откладывая
зависимость угла наклона кривой титрования от рН, а не строя саму
кривую титрования. Таким образом, вводится понятие буферной
емкости р как величины, обратной изменению рН раствора при
добавлении в раствор сильного основания:
о dB
v=~jm> (3-56)
где d(pH)—инкремент рН, вызванный добавлением dB
основания. Уравнение (3.56) согласуется с обычным представлением о
буферных растворах: большую буферную емкость имеет раствор,
при титровании которого, для того чтобы хотя бы немного
изменить рН, надо добавить большое количество основания.
188 ЧАСТЬ 1
■АО
35
30
_ Кривые бьеррума
(кривые тшпроватг
25-
-5,0-4,0 -3,0 -2,0 -1,0 О
Кривые Снэтчарда
10 /5 20 25 30 35 АО
7 t
Рис. ЗЛО. Сопоставление графических способов определения констант связывания
и числа связывающих участков макромолекулы (константа связывания данного
низкомолекулярного соединения известна) [3, с. 614—618].
Пунктирные кривые характеризуют связывание на макромолекуле с 10 идентичными
независимыми (между собой) участками связывания, каждый из которых характеризуется
константой связывания К\ =1(М моль/л"; сплошные кривые — связывание на макромолекулах
с теми же 10 участками связывания </Cj =I0-3 моль/л) н дополнительными 30
идентичными независимыми (между собой) участками связывания (Ki »=0,04 моль/л). Очевидно, что
А2
«з экспериментальных данных легче всего определить Кл , К* < nt н Пз с помощью завн-
симости Скэтчарда. v—среднее число связанных с макромолекулой молекул низкомолеку-
ляриого соединения '[уравнение (3.55)], а Щ —концентрация ннзкомолекулярного соедине-
«ия в равновесных условиях. Прн [1] =*1Н+] кривая титрования легко получается из
измерений рН (см. раздел «Буферная емкость»).
Буферная емкость воды легко выводится из константы
равновесия диссоциации воды
К*= [Н+ПОН-]
(3.57)
89 ТЕРМОДИНАМИКА В БИОЛОГИИ
откуда
ig/C=ig[H+]-Hg[OH-]
или
-\g[R+]=pH=\g[OH-]-\gKw. (3.58)
В растворе сильного основания, например NaOH, £ = [ОН~]
в уравнении (3.56), и, поскольку d (рН) — d\g[OH~], из уравнения
(3.58) находим
АТ= d-^L=i/2.303i^L=1/2;3031oH-)
или проще
dB/(pH) = $ = 2,303 [ОН~] (для сильного основания, добавленного
к воде). (3.59а)
Аналогично при добавлении к воде сильной кислоты
-JJ|_=2,303 [Щ (3.596)
Объединяя уравнения (3.59а) и (3.596), найдем буферную
емкость водного раствора сильной кислоты или основания:
dB/d (рН) = ft = 2,303 ([Н+] + [ОН"]) (для воды)
(3.59в)
Наконец, рассмотрим буферную емкость водного раствора слабой
кислоты НА
НА т~у Н+ + А-
/(,=-№• <3-60>
В классическом эксперименте по титрованию, в котором В—
количество добавленного основания (ОН-), получаем один А- на
каждый добавленный ОН-
НА + ОН- *■ А- + Н20
/
так что уравнение (3.60) можно переписать в виде
*«=w- <3'61>
Кроме того, [НА] = [НА]о — В, где [НА]0 — количество слабой
кислоты, присутствовавшей перед введением в раствор основания
В. Уравнение (3.61), следовательно, принимает вид
Кй [НА],-Д» <3'62>
90 ЧАСТЬ 1
А
OJ5
0,10
0,05
Г
\^4/-
/Ло,2мсн3соон
J \,0,/МСНлСООН
1 >0^ 1 1 _
/
он-/
1
6 8
рН
10
12
Рис. З.П. Зависимость буферной емкости р от рН в растворах сильной (Н+) и
слабой (уксусной) кислоты и сильного основания (ОН-) [4, с. 112].
Для * растворов уксусной кислоты высота пика пропорциональна концентрации растворов
при (рН=р/Са) н соответствует максимальной буферной емкости р.
последнее решается относительно В и приводит к
[Н+]+/Са •
Дифференцируя уравнение (3.63)
dB/d[H+]=-
*а[НА]0
([H+J+/Ca)a
(3.63)
(3.64)
и используя известное правило дифференцирования, получаем
dB _ dB d\ti+\ t /Cfl[HAi0 \ 0 «n +
d pH ~" d [H+] d (PH) ~l ([H+] + Ka)V{ * l J;
_dS__ 2,303/Cg [H+] [HAj0
dpH -P- ([H+]+/Ca)2
(3.65)
На рис. 3.11 показана зависимость от рН буферной емкости
для уксусной (слабой) кислоты. Читатель легко сможет
убедиться (см. задачи в конце главы), что для данной (слабой) кислоты
буферная емкость р максимальна при рН = рКа- Кроме того,
согласно уравнению (3.65), максимальная буферная емкость
пропорциональна общему количеству слабой кислоты, присутствующей в
исходном растворе. Для правильного представления о значении
таких зависимостей следует подробнее остановиться на кривых
титрования многоосновной молекулы, например, аспарагиновой
кислоты (рис. 3.12). Хотя для этой молекулы рКа трудно точно
определить непосредственно из кривой титрования, достаточно
просто разложить зависимость буферной емкости от рН на вклады
от трех способных к диссоциации протонов аспарагиновой
кислоты.
Важной особенностью зависимости буферной емкости от рН
является сохранение ею одинаковой формы. Следовательно, кривые
91 ТЕРМОДИНАМИКА В БИОЛОГИИ
1
0,3
0,2
0,1
рлу*-
—
I '
р/Гт-»-*
' I '
X
ркз -\
о.ю —
0,05
10
о ? 4в 8 ю 12
рН
2
t
р/С,
4
t
Р*2
t
рК3
Рис.
3.12. Сопоставление двух способов анализа кривых титрования для
определения рКа отдельно титруемых групп в молекуле.
а — обычная кривая титрования (кривая Бьеррума) 0,1 М аспарагнновой кислоты сильным
основанием (например, NaOH); различие между двумя первыми рК выражено не очень
четко; б— зависимость буферной емкости Р от рН для той же системы; эту зависимость-
легко разбить на (как показано) вклады каждого из трех диссоциирующих протонов ас-
парагииовой кислоты для определения трех искомых рКа Аналогичные методы можно
использовать для определений индивидуальных констант диссоциации и оно генных групп в
молекулах рКа белков и числа групп с данным значением рКа в этих молекулах. (Как
показано на рис. 3.13, число титруемых групп в молекулах белков намного больше.)
титрования белков, содержащих большое число способных к
диссоциации протонов, можно разложить на соответствующие вклады
всех этих протонов, согласуя наблюдаемую зависимость буферной
емкости от рН с уравнением вида
е=£
2,303/С;[Н+]П;[НА]С
{Кг + [Н+])2
(3.66>
где rii — число способных к диссоциации протонов в молекуле
белка для которых Ka—Ki. Поскольку для каждого вклада любой
группы форма зависимости буферной емкости (от рН) одна и та
же, необходимо лишь указать щ и Ki для каждого типа
диссоциируемых протонов, чтобы соответствующим образом количественно-
описать экспериментальные данные. Примеры такого рода
приведены на рис. 3.13.
Анализ данных, описывающих связывание
с большим числом участков:
взаимосвязанные участки связывания
Выше рассмотрена макромолекула с большим количеством
возможных участков связывания небольшой молекулы I; участки
связывания могут иметь разные равновесные константы диссоциации
Ки однако связывание I с одним участком не влияет на связыва-
25°C
/1изоцим
70'C
II 11 ! 1 I 1 1 T 1 *
Initi i i и ii
X
1
T
Ридонуклеаэа
■V)°r J x ас эе <
•ы ь * i in ii 11 i I f i i 11
30"C ,
Цитохром
_l „ 1
i lull il f I *
с
1 t t 1 1
at
X
I
1
A 5
&
Рис. 3.13. Индивидуальные рКа ионогенных групп в трех белках (лизоциме при
25 °С, рибонуклеазе при 30 и 70 °С и цитохроме с при 30 °С), определенные
путем разложения зависимости буферной емкости от рН на вклады (см. рис. 3.12)
[11].
Одной звездочкой отмечены рКа ионизации имидазольных групп, двумя звездочками — рКд
ионизации аминогрупп; не отмечены (звездочками) рКл карбоксильных групп.
Вертикальная шкала отражает относительное число групп с данным значением р/Ст.
Рис. 3.14. Зависимость Скэтчарда, описывающая связывание NADH с лактатде-
гидрогеназой [12].
Отрезок на осн абсцисс дает число идентичных участков связывания на макромолекуле:
в данном случае существует 4 идентичных участка связывания NADH. Поскольку
зависимость линейна, то, очевидно, связывание четырех молекул NADH с ферментом происходит
независимо (см. текст), v —число молекул NADH, связанных с одной молекулой дегндро-
генаэы.
93 ТЕРМОДИНАМИКА В БИОЛОГИИ
ние другой молекулы I с любым другим участком на той же
макромолекуле. Экспериментальный пример такой ситуации,
представленный зависимостью Скэтчарда, показан на рис. 3.14,
характеризующей связывание четырех молекул NADH с идентичными
независимыми участками в молекуле лактатдегидрогеназы.
В этом разделе рассмотрим противоположный крайний случай
равновесного связывания [уравнение (3.67)], т. е. связывание с
такой высокой степенью кооперативности, что связывание первой
молекулы I способствует связыванию последующих молекул I, в
результате чего оставшиеся участки на макромолекуле
заполняются немедленно (т. е. как если бы связывание всех п молекул I
происходило сразу за одну стадию)
Е + л1 г-ь^ЕГ,,; /Ci =
IE] [I]"
JEW
(одностадийное связывание
на п участках)
(3.67)
Если вновь определить v как число молекул I, связанных с
одной молекулой Е,
п [EI„]
V—■
IEI„I + [El '
(3.68)
и, решив уравнение (3.67) относительно [Е1п], подставить
полученное выражение в уравнение (3.68), то после соответствующих
преобразований получим
.._ п[1)п
и
V
Шл + К i
п —v
tfi
<
(3.69)
Уравнение (3.69) обычно упрощают введением обозначения 9;
определяемого как доля всех занятых участков; тогда число
занятых на макромолекуле участков v равно
—
(3.70)
(3.71)
Наконец, логарифмируя обе части уравнения (3.71) [обратите
внимание, что оно напоминает уравнение (3.48)], находим
V = П0,
так что
v 9
я —v ~~ 1—9 ~~
1б(-ГГ9~)==я,б^11~,б/С1
(3.72)
Зависимость lg(9/[l—9]) от lg[I] должна представлять собой
прямую с углом наклона п. В результате при независимом связы-
9ф ЧАСТЬ 1
О 20 40 60 80 100
р^мм рт.ст.
анаше/ , Тангенс
г У^а I / уг/т
"наУ'! Iна1<Лона
0,6 0,6 1,0 1,2 1,4 1,6 1,д 2,0
?в Poz,mm рт.ст.
Рис. 3.15. Кооперативное связывание кислорода с миоглобином или гемоглобином
представлено согласно прямому методу (а) и зависимостью Хилла (б).
Полностью кооперативное связывание (для гемоглобина п=4; п определяется тангенсом
угла наклона зависимости Хилла) редко полностью реализуется в эксперименте (см. текст),
v — среднее число молекул кислорода, связанных с одной молекулой гемоглобина или мио-
глобина.
вании (т. е. связывание происходит в данный момент только на
одном участке связывания) п= 1 и угол наклона такой
зависимости (известной под названием зависимости Хилла) равен единице;
при связывании с высокой степенью кооперативности угол
наклона зависимости Хилла равен числу взаимодействующих участков
связывания. Классическим примером служит связывание
кислорода с миоглобином (п=1, связывание одним участком
макромолекулы) и с гемоглобином (гс=4, связывание происходит с четырьмя
участками макромолекулы), представленное на рис. 3.15. Прямой
метод определения константы связывания кислорода миоглобином
(рис. 3.15,а), дает типичное для связывания соотношение 1 : 1 (ср.
с рис. 3.9,а или 3.10,а); зависимость Скэтчарда для тех же самых
данных представляет собой прямую, свидетельствующую о
связывании лиганда с одним участком на молекуле миоглобина.
Однако кривая, построенная согласно прямому методу, для связывания
кислорода с гемоглобином (рис. 3.15, а) имеет иной вид, и
зависимость Скэтчарда (или обратная зависимость) оказывается
искривленной. Ход зависимости Хилла для гемоглобина (рис. 3.15,6)
раскрывает причину таких отклонений: после того как первая
молекула кислорода связалась с молекулой гемоглобина, остальные
три молекулы связываются так прочно, что это выглядит, как
если бы все четыре молекулы кислорода связались с гемоглобином
одновременно [уравнение (3.67) при я=4]. С помощью данных,
95 ТЕРМОДИНАМИКА В БИОЛОГИИ
приведенных на рис. 3.15,6, можно выявить еще одну общую
закономерность: связывание редко происходит с высокой степенью
кооперативное™ во всей области концентраций лиганда (угол
зависимости Хилла равен четырем только в определенном интервале
рСЬ, а при очень высоких или при очень низких значениях рОг
связывание происходит менее кооперативно). Таким образом, коопе-
ративность (т. е. взаимодействие между участками связывания)
минимальна в условиях, когда очень мало участков или когда
почти все участки связывания заняты молекулами лиганда.
Задачи
1. Дайте качественное объяснение того, как надо использовать уравнение Гибб-
са — Дютема для расчета зависимости активности сахарозы от ее
концентрации, используя приведенные в табл. 3.1 данные (мольные доли и давления
пара раствора).
Замечание. Начните с рассмотрения зависимости In cA от Ха-
2. Подтвердите, что активность (т. е. термодинамическая концентрация
)индивидуальных ионов оа растворах для сложного электролита МГХ8 связана с
активностью соли в исходном растворе уравнением
aMADOflH = (%s+)r(V-)s
Это уравнение приводит к определению средней активности а± и среднего
коэффициента активности у±:
(v+Г (v-)* - C±Y+S=(v±)'+s cv+)' Cv-)s
3. Выведите уравнения, а также дайте обоснования графических построений и
методов определения числа эквивалентных независимых участков связывания
п и константы равновесия К\ (диссоциации), характеризующей участок
связывания, для прямого метода, методов кривых Бьсррума, обратных
зависимостей и зависимостей Скэтчарда (табл. 3.4).
4. Сигнал ЭПР, характеризующий ион Мп2+ в водной среде, заметно изменяется
при связывании иона Мп2+ с макромолекулой. На основании данных ЭПР
для растворов, содержащих разные (известные) соотношения иона Мп2+ и
трипсина (протеолитический фермент с молекулярной массой ~ 25 000), были
получены следующие данные*:
Гмп2+]связ
[Трипсин]общ [Мп5
2950
2100
1500
700
450
250
'своб
[Мп2+]связ
1ТРипсин]общ
0,3
0,5
1,0
2,6
3,6
7,0
* Marshall А. С, Kang S. Y., unpublished data.
96 ЧАСТЬ 1
Из зависимости Скэтчарда, построенной по этим данным, в
предположении о существовании двух типов участков связывания оцените величины
констант связывания и число участков каждого типа.
5. Изоэлектрическую точку молекулы можно определить как величину рН, при
которой суммарный заряд молекулы равен нулю. Покажите, что изоэлектри-
ческая точка молекулы с двумя способными к диссоциации протонами (и,
следовательно, двумя значениями рКа) определяется следующим образом;
РНизоэле"ктр = У2 (рК*1 + РК*2). где VKa = —lg Ka,
(Ка — константа кислотной диссоциации исследуемой группы).
Н
I
6. Аминокислота лизин H2N(CH2b—С—СООН характеризуется тремя констан-
NH2
тами диссоциации p/C = 2,I8, pKn=S,95 и р/С3= 10,53.
а. Какому значению рН соответствует изоэлектрическая точка лизина?
б. Какова наиболее вероятная формула цвиотер-иона?
7. Покажите, что для слабой кислоты буферная емкость
dB/d (рН) = В = ([Н+]+/Са)2
достигает своего максимального значения при pH=pi(a.
Литература
1. Moore W. !., Physical Chemistry, Prentice-Hall, Englewood Cliffs, N. J.,
любое издание. Хорошее рассмотрение стандартных состояний и активностей,
включая теорию всалнвания Дебая — Хюккеля.
2. Tanford С, The Hydrophobic Effect, John Wiley & Sons, New York, 1973.
Хорошее обсуждение вопросов гидрофобности и аспектов применения этого
явления.
.3. Edsall J. Т., Wyman^ J., Biophysical Chemistry, Vol. I, Academic Press, New
York, 1958. Хороший анализ способов определения констант связывания и
числа участков связывания в реальных случаях7связывания с большим
числом участков. '
4. Bull Н. В., An Introduction to Physical Biochemistry, 2nd ed.t F. A. Davis
Co., Philadelphia, 1971. Хорошее краткое рассмотрение буферной емкости.
5. Barrow G. M., Physical Chemistry for the Life Sciences, McGraw-Hill, New
York, 1974, p. 238.
6. Cohn E. J., Chem. Rev., 1», 241 (1936).
7. Cohn E. J., Edsall I. T., Proteins, Amino Acids, and Peptides, Reinhold, New
York, 1943, p. 602.
8. Nozaki Y., Tanford C, J. Biol. Chem., 246, 2211 (1971).
9. Chothta C, Nature, 248, 338 (1974).
10. Levitt M„ Warshel A., Nature, 253, 694 (1975).
11. Bull H. В., Breese K-, Arch. Biochem. Biophys., 117, 106 (1966).
12. Anderson S., Weber G., Biochemistry, 4, 1948 (1965).
13. Deranleau D. A., J. Am. Chem. Soc, 91, 4044 (1969).
ГЛАВА 4
САМОПРОИЗВОЛЬНОЕ ПРОТЕКАНИЕ
ХИМИЧЕСКИХ РЕАКЦИЙ:
ТЕМПЕРАТУРНАЯ ЗАВИСИМОСТЬ
КОНСТАНТ РАВНОВЕСИЯ
И СКОРОСТЕЙ РЕАКЦИЙ
При рассмотрении любой химической реакции прежде всего
надо выяснить, а произойдет ли она. Согласно предыдущему
рассмотрению химических равновесий при обычных условиях, т. е. при
постоянной температуре и давлении (табл. 2.1), свободная энергия
(Гиббса) G минимальна при равновесии. Следовательно, в общем
случае химическая реакция
аА+6В —* cC+dD (4.1)
происходит самопроизвольно, если суммарное изменение
свободной энергии, характеризующее превращение а молей вещества А
и Ь молей вещества В в с молей вещества С и d молей вещества
D, — отрицательная величина:
Если (/продукты — Ореагенгы = AGrje < 0 (реакция происходит
самопроизвольно)
(4.2)
Более простой критерий самопроизвольного протекания
реакции следует из ранее выведенного соотношения
№гх = —ЦТ\пКд
(3.11)
где Ка — константа равновесного процесса (4.1), записанного
уравнением обратимой реакции (3.4), hG°rx — изменение свободной
энергии при превращении а молей вещества А и Ь молей
вещества В в с молей -вещества С и d молей вещества D при условии,
что все компоненты, находятся в стандартном состоянии (см.
разд. З.А.2). Следовательно, при условии стандартного состояния
компонентов уравнения (4.2) и (3.11) можно объединить и полу-»
чить простой критерий равновесия
Ка ]> I -*—*■ реакция протекает самопроизвольно при
превращении реагентрэ в стандартном
состоянии в продукты в стандартном
состоянии
(4.3)
Аналогично, если /Са<1, реакция не происходит
самопроизвольно;, при Ка=\ реакция не-происходит ни в том, ни в другом на-
98 часть 1
правлении при условии, что реагенты и продукты находятся в
соответствующих стандартных состояниях.
Из уравнения (4.3) следует, что, если равновесие сдвинуто
вправо (/Со>1), реакция при условии стандартных состояний
происходит самопроизвольно, а если равновесие сдвинуто влево
(Ка<\), реакция является несамопроизвольной. Естественно,
сразу же возникает проблема выбора способа выражения
концентраций компонентов реакции, от которого зависит значение Ка\ это
усложняет оценку самопроизвольности реакции. Кажущаяся
трудность решена здесь прежде всего (в начале этой главы), причем
следует отметить, что вопрос о самопроизвольности реакций
является, в частности, важным для установления последовательности
химических реакций в процессах биологического обмена (путей
метаболизма). Затем рассмотрены зависимость константы
равновесия (и, следовательно, самопроизвольности и
несамопроизвольности химических реакций) от температуры и способы
предсказания этой зависимости. В конце главы обсуждаются значения и
температурные зависимости констант скоростей реакций,
отношение которых определяет константу равновесия, поскольку многие
самопроизвольные, согласно термодинамическим критериям
(уравнение (4.2) или (4.3)], реакции столь медленно достигают
равновесия, что термодинамический критерий оказывается
несостоятельным для предварительной оценки направления и
самопроизвольности протекания реакции.
4.А. КРИТЕРИИ САМОПРОИЗВОЛЬНОСТИ ХИМИЧЕСКОЙ
РЕАКЦИИ
Трудность выбора способа выражения концентраций при
расчете константы равновесия сразу же отпадает, если вспомнить, что
активность — это всегда отношение двух концентраций. Например,
активность растворителя аА можно выразить как отношение
давления пара исследуемого раствора РА к давлению пара чистого
растворителя Р\:
аА = (Ра'/Ра) = Активность растворителя А, (3.9а)
аА —*■ 1 при ХА *■ 1
[Ра соответствует стандартному состоянию растворителя (т. е.
чистому растворителю)]. Подобным образом активность
растворенного вещества ав можно выразить как отношение молярных
концентраций (или моляльных концентраций, или мольных долей)
вещества в исследуемом растворе к концентрации, выраженной в
тех же единицах (молярности, моляльности или мольных долях)
вещества в идеальном растворе:
<*в^Ъ(тв[гпв), (4.1)
99 ТЕРМОДИНАМИКА В БИОЛОГИИ
где в идеальном растворе (уЪ = 1)гпв равно единице (при
соответствующем выражении концентрации); это условие стандартного
состояния вещества в растворе.
Например, если в качестве стандартного состояния вещества в
растворе выбрать идеальный раствор с концентрацией 1 моль/л,
то активность этого вещества в реальном 2,3 М растворе составит
Ов=ув(2,3) (где yb — коэффициент активности, соответствующий
2,3 М раствору) и (тв/т|)=2,3. Таким образом, несмотря на то
что значение 2,3 — молярная концентрация вещества В в растворе,
в действительности это число выражает отношение молярной
концентрации вещества в растворе к молярной концентрации этого
вещества в стандартном состоянии.
В качестве наиболее важного с практической точки зрения
примера рассмотрим раствор, содержащий ионы водорода. Можно
сравнить действительную концентрацию ионов водорода в
исследуемом растворе тн+ с концентрацией ионов водорода в
стандартном состоянии, соответствующей содержанию 1 моля
вещества в идеальном растворе, однако тогда рН раствора в
стандартном состоянии соответствовало бы значению, близкому к нулю,
которое слишком далеко от условий протекания физиологических
(или даже обычных химических) реакций. Поэтому при изучении
биохимических процессов обычно определяют в качестве
стандартного состояния ионов водорода в водной среде состояние,
соответствующее идеальному раствору с концентрацией 10~7 моль/л Н+
(рН 7,0). Таким образом, в зависимости от определения
стандартного состояния в выражение для активности ионов водорода в
водной среде вводится множитель 107:
%+=Yh+ (/"н+/^н+) = YH+mH+. (4-2а)
где тн+ =1 моль/л (условие протекания химических реакций) или
%+=Yh+ (^н+/^н+) = Ю7 Ун+тн+, (4.26)
где тн+=Ю~7 моль/л (условие протекания биохимических
процессов).
Переход от одного способа выражения концентрации к другому
соответствует переходу от одного выбранного стандартного
состояния (т. е. концентрация выражена в определенных единицах) к
другому стандартному состоянию (т. е. концентрация выражена в
других единицах). Следовательно, поскольку Ка и AG?X
действительно зависят от способа выражения концентрации, уравнения
(3.11) и (4.3) удовлетворяют любому выбору единиц концентрации
при условии, что эти единицы одинаковы при измерениях К& и
определении AGrx-
Разобравшись с выбором способа выражения концентраций,
можно перейти к рассмотрению критерия самопроизвольности
реакции (4.1), в которой реагенты и продукты присутствуют в про*
100 часть i
извольных (а не в стандартных или равновесных) концентрациях.
Подставляя уравнения (3.2) и (3.3) в уравнение (4.2):
Qi^nfii- (общая свободная энергия п молей
t-ro компонента) (3.2)
Gi==G\-\rRT\n(ai) (химический потенциал t-ro
компонента с активностью аг) (3.3)
и
и _
Д</гх=</Пр0дукты—(We..™ (изменение свободной энергии
при превращении а молей
вещества А с активностью аА
и Ь молей вещества В с
активностью ав в с молей
вещества С с активностью ас и d
молей вещества D с
активностью aD),
получаем
Д Grx=cGc -j- dGD—aGA—bGB=
=cGl+&T In ac+d(jD-\-dRT]naD—oG\—aRTlnaA—&% —
—bRT\naB=G°c+GaD—GA—G°B+RT (in acc+lnadD—1паА —\nbB)
AGrx = Д<?" +ДГ1п
°Сар
4 <
Все реагенты и продукты в
стандартных концентрациях
(ад = ав = ас = o-d = 1)
(реагенты и продукты в
произвольных концентрациях)
(4.3)
Равновесие при постоянных
Т и Р (все реагенты и
продукты в равновесных
концентрациях)
AG = ДО
rx
AGrjc = 0
AGrx = — RT In
flCaD
A
lb
= —RT In Ka
Уравнение (4.3) указывает схему расчета AGrx в реакции
(4.1), когда реагенты и продукты присутствуют в произвольных
концентрациях, а соответствующие предельные случаи для
стандартных или равновесных концентраций показаны под
уравнением (4.3). Рассчитав AGrx из уравнения (4.3), можно определить,
будет ли реакция происходить самопроизвольно (AGrx<:0:
реакция происходит самопроизвольно слева направо), или несамопро-
рзвольно (Д(7Гдс>0: реакция происходит самопроизвольно справа
101 ТЕРМОДИНАМИКА В БИОЛОГИИ
налево) или состояние равновесия (AGrx—0: реакция не
происходит). Важно отметить, что даже при A(jr*>Q
(несамопроизвольный процесс) реакцию можно провести, подводя к системе
энергию, равную по меньшей мере AGr* (см. пример, приведенный
Ниже). Использование уравнений (4.3) и (3.11) хорошо
иллюстрируется конкретной реакцией, связанной с сотнями различных
биохимических процессов, а именно, на примере взаимного
превращения аденозинтрифосфата (АТР) и аденозиндифосфата (ADP).
Пример. АТР — «разменная монета» биоэнергетической экономики
Множество жизненно важных химических реакций клеточного метаболизма,
согласно уравнению (4.3), оказываются несамопроизвольными. Энергия,
необходимая для осуществления этил реакций, обеспечивается путем ^сопряжения
такой реакции с другой (самопроизвольной) так, чтобы изменение свободной
энергии в обеих сопряженных друг с другом реакциях было отрицательным и
суммарный (сопряженный) процесс являлся самопроизвольным. Наиболее широко
распространенной «движущей» реакцией является гидролиз АТР до ADP и
неорганического фосфата Pi. Поэтому в живой клетке непрерывно ведется синтез
АТР из ADP, чтобы обеспечить постоянный запас АТР, необходимый для
осуществления несамопроизвольных реакций метаболизма. Реакция, схема которой
записана уравнением (4.4), является, возможно, наиболее важной химической
реакцией в живых клетках (она происходит в нескольких сотнях почти из тысячи
ферментативных процессов в клетках животных); проанализируем изменение
свободной энергии в.этом процессе при разных условиях:
ADP + Р, + Н+ т—»■ АТР + Н20 (4.4)
для которой
flATPflH20
Ка = аАвроРлн+ (ПРИ Равновесии> (,4-5>
и
ДС° =— RTlnKa [изменение свободной энергии при превра- (4.6)
гх щении 1 моля ADP в АТР, где все
реагенты и продукты находятся в стандартных
состояниях (концентрация равна единице
в идеальном растворе)]
Хотя действительное значение Д<3°* зависит также от концентраций Mg2+ и
Са2+, обычно принимают, что AG°rx составляет
о
AGrJC = —2,2 ккал/моль при условии, что активности всех
реагентов и продуктов равны единице
(4.7)
Таким образом, реакция, по-видимому, должна происходить
самопроизвольно слева направо. Стандартное состояние Н+ (ан+=1) соответствует [Н+]=»
= 1 моль/л в идеальном растворе (т. е. рН раствора около нуля!), однако эта
постоянная далеко сдвинута относительно обычного значения рН в живых
клетках., Поэтому в биохимии обычно определяют в качестве стандартного состояния
Н+, такое, при котором ан+=1, т. е. >[Н+1 = Ю-7 моль/л (рН 7,0); тогда
получают значение AG0', более соответствующее действительным условиям биологических
процессов. Теперь вернемся к уравнению (4.2) и покажем (см. задачи в конце
главы), что
д5н+ = АО^ + Я!Г1п10-7, (4.8)
102 ЧАСТЬ 1
где Д(гд+ —свободная энергия Н+в стандартном состоянии (тн + =1 моль/л в
__ идеальном растворе);
AGtf4- — свободная энергия Н+ в стандартном состоянии (тн+=10~7 моль/л
в идеальном растворе с рН=7,0).
Отсюда можно рассчитать изменение стандартной свободной энергии AGrx для
нового стандартного состояния протонов в водной среде [уравнение (4.26)]:
ДС/х = ^АТР + ^Н20 — ^Н+ — GADP — Ц>1 =
=С;тР+ён2о'СвН++^Г 1п 10-'-GADP-G°Pl= Д^-ЛПп 10-' = -2,2 + 9,5
AG°' =+7 3 ккал/моль гДе стандартное состояние Н+ соответ-
гх ' ствует концентрации 10~7 моль/л,
стандартное состояние других веществ в
растворе—I моль/л в идеальном
растворе и стандартное состояние
растворителя—чистая вода)
(4.9)
Уравнение (4.9) показывает, что при снижении концентрации ионов
водорода до 10~7 моль/л, реакция происходит самопроизвольно в противоположном
направлений [т. е. оправа налево; см. схему (4.4)]. Этот расчет должен
продемонстрировать, что энергия, необходимая для проведения данной химической
реакции, не постоянная величина, а заметно зависит от действительных
концентраций различных компонентов реакции.
Читатель должен сам убедиться в том, что при еще более близких к
реальным физиологическим условиям (пусть [ATP]H[ADP]='[P,-] = 10~4 моль/л)
изменение свободной энергии в реакции (4.4) возрастает до ~ 12,7 ккал/моль.
[Лучшие на сегодня оценки концентраций всех компонентов этой реакции в
типичной клетке свидетельствуют о том, что действительное значение AGrx в
реакции (4.4) составляет около +12 ккал/моль.]
Одним из основных «питательных» веществ клетки является глюкоза
CeHjaOe. При распаде глюкозы в клетке на С03 и HjO выделяется большое
количество (свободной) энергии и часть этой энергии используется для образования
АТР из ADP. Используя только что полученное значение AGTX, характеризующее
образование АТР из ADP, можно рассчитать эффективность клетки при захвате
и хранении энергии (в виде высокоэнергоемкой фосфатной связи АТР),
высвобождаемой при распаде глюкозы (см. задачи в конце главы). Этот расчет
показывает, что клетка является исключительно эффективным преобразователем
энергии.
О Of
^Сотя, как показано выше, AGrx или даже AGrx, характеризующие
превращение реагентов в продукты в стандартных состояниях, позволяют сравнительно
грубо оценить действительное значение AGTX при физиологических концентрациях
реагентов и продуктов, тем не менее оказывается лолезным сравнить ряд
гомологичных реакций при одном и том же стандартном состоянии, как показано в
табл. 4.1. Данные, приведенные в таблице, характеризуют изменение свободной
энергии при гидролизе (удалении фосфатной группы) нескольких биологически
активных соединений. Очевидно, что фосфатные связи в некоторых из этих
соединений являются еще более энергоемкими, чем в АТР. Например, при
сопряжении верхней реакции с превращением ADP в АТР фосфоенолпируват мог бы
использоваться для переноса фосфата к ADP при образовании АТР — в
действительности это один из путей биологического образования АТР в процессе
распада углеводов. Для дальнейших упражнений в расчетах зависимости AGTX от
концентраций реагентов и продуктов читателю следует обратиться к задачам в
конце главы.
103 ТЕРМОДИНАМИКА В БИОЛОГИИ
Таблица 4.1
Свободные энергии гидролиза
(удаления фосфатной группы)
некоторых фосфорилированных
соединений3
Соединение
Фосфоеноллируват
Карбамоилфосфат
Ацетилфосфат
Креатинфосфат
Пирофосфат
ATiP (до ADP)
Глюкоза-1 -фосфат
Глюкоза-6-фосфат
Глюкоза-3-фосфат
о/
AGrJC, ккал/моль
— 14,8
— 12,3
—10,3
—10,3
—8,0
-7,3
—5,0
—3,3
—2,2
а °'
AG гхопределена для стандартных состояний:
[Н+] = 10~7 моль/л; идеальные растворы всех
других веществ, кроме Н+, с концентрацией I моль/л;
растворитель — чистая вода НгО.
4.Б. ИЗМЕНЕНИЯ КОНСТАНТ РАВНОВЕСИЯ
С ТЕМПЕРАТУРОЙ
Из исходного определения свободной энергии Гиббса
G=H—TS (1.14)
и общего соотношения между AG?X и константой равновесия Ка
AGarx=-RT In К4 (З.П)
легко вывести, что при любой данной (постоянной) температуре Т
AG°rx=-RT In Ka=AH°rx-TAS°rx, (4.10)
где AHfX и AS°rx — соответствующие изменения энтальпии и
энтропии при превращении 1 моля реагента в продукт при условии, что
все реагенты и продукты находятся в стандартном состоянии.
Переписав уравнение (4.10)
,пК _ АЯ~ , *»«
(4.11)
104 часть l
видим, что зависимость \пКа от (1/Г) выражается прямой с углом
наклона — (AH?X/R), пересекающей ось ординат в точке (AS?xfR)*.
Уравнение (4.11) имеет двойное значение: для предсказания по
известным АНгх и AS?X изменения константы равновесия с
температурой и, следовательно, для определения константы равновесия
при любой температуре, если известны AH0rXiASr'x и Ка при какой-
то одной температуре; кроце того, для определения АН°гХ и AS°rx
в данной реакции из температурной зависимости константы
равновесия Ка- Поскольку степень «самопроизвольности» протекания
химической реакции зависит как от АН°ТХ, так и от AS?X, решим
вопрос, что является основной причиной протекания данной
химической реакции: суммарное освобождение энергии в результате
разрыва и образования новых химических связей в реакции
(А//?х<0) или суммарное возрастание степени упорядоченности
при переходе от реагентов к продуктам реакции (AS^>0). Часто
особенно полезно сопоставлять тенденции, проявляющиеся в
изменении АНгх или AS% для гомологического ряда соединений; такие
тенденции составляют основу при обсуждении «стерических»,
«индукционных» и «резонансных» эффектов в механизмах
органических реакций.
Пример. Константы равновесия при стандартной температуре (25 °С)
и при температуре человеческого тела (37 °С)
Взаимодействие воды с фумарат-ионом с образованием аниона яблочной
кислоты
фумарат2-+ Н20 <■ * Малат2-
является ключевой стадией метаболизма в цикле трикарбоновых кислот.
Используя следующие данные, найдем и сопоставим константы равновесия этой реакци»
при 25 °С (обычная стандартная температура, при которой исследуются
химические реакции) с константой равновесия при 37 °С (температура человеческого
тела):
AG"rx = — 880 кал/моль при 25 "С
&Н\Х = +3560 кал/моль, причем можно считать А#°л постоянной в интервале
температур 25—40 °С.
Прежде всего из уравнения (3.11) найдем 1п/С0 при более низкой
температуре:
1
\nKa*=—AG0rx/RT=—(—880)/(1,99 кал-моль"1, град"1) (298 °К) —1,49;
^,(25»с)=^49=4,42 при 25°С.
Тогда,, поскольку, согласно уравнению (4.11),
dtoKjdft} =-AH\xjR (4.12)
* Строго говоря, это верно лищь когда &Н?Х и AS« не зависят от
температуры. Для большинства обычных биологических реакций, происходящих в узкой
области температур, можно считать, что это приблизительно справедливо.
.105 ТЕРМОДИНАМИКА В БИОЛОГИИ
прн независимости Д#£я и A-S*^-- от температуры (предположение, которое
обычно выполняется в очень узком температурном интервале, например для данного
примера в интервале 15°С) выполняется:
(1пКа)7-2 — (1пКа)7-г= — —Ц \Т~ ~" ~Т~ J
(4.13)
или
<ln#0„,= l,49-(J[W»:) (-4—sir) =1,72,
0Qwc=el'n=616 при 37 еС.
В этом случае увеличение температуры заметно сдвигает равновесие вправо.
Согласно принципу Ле-Шателье, удается предсказать качественные изменения:
для реакции, которая протекает слева направо с поглощением тепла (kH°rX>0t
как в данном случае), увеличение температуры должно сдвигать равновесие в
таком направлении, чтобы скомпенсировать наложенное воздействие (в данном
случае — подвод тепла). Таким образом, уравнение (4.13) поясняет смысл
принципа Ле-Шателье.
В разд. 4.А и 4.Б сделана попытка показать, что
самопроизвольность химической реакции предсказуемым образом зависит от
концентраций реагентов и продуктов реакции и от температуры,
при которой проводится реакция. Читатель теперь может
рассчитать AGrx или Ка при любых стандартных концентрациях
компонентов реакции и при любой .темйературе. На практике самым
удобным методом определения А(}гх (и, следовательно, fta) из
экспериментальных данных часто являются электрохимические
измерения (см. гл. 5).
Дифференциальная сканирующая калориметрия
Ранее указывалось (гл. 1), что значения теплоты, выделяемой
или поглощаемой в процессе химической реакции при постоянном
давлении, т. е. АНГХ, можно использовать для создания полезной
шкалы прочности химических связей. Кроме того, в той..же главе
предложен способ оценки АНГХ из температурной зависимости
константы равновесия реакции. Изменение физического (фазового)
состояния вещества при постоянном давлении (разд. 2.А.2) также
может сопровождаться -поглощением или выделением тепла:
например, при плавлении 1 г льда надо затратить ~80 кал, а при
испарении 1 г воды (жидкой) при давлении 1 атм необходимо
~ 540 кал. Чем больше АН, характеризующая изменение фазового
состояния, тем более значительна молекулярная реорганизация
в материале при этом изменении. Поэтому изменения конформа-
ции макромолекулы можно детектировать, регистрируя тепло,
выделяемое или поглощаемое при конформационных переходах.
Поскольку типичный фазовый переход происходит в сравнительно
узком температурном интервале, его регистрируют, измеряя коли-
106 ЧАСТЬ 1
E.coli tPHKj c
С
А
pG- с
G ; С
G С70
U А
G С
А U 60
U ACUGCC c U
4tU I I I I ' *
С G д ..Ю А J И I G
л С U С G GGCGGt г
О ||,, п 50 Т V» °
G
G
МИ
G A G С *7mG
G A A G G
20 С G
С G
U А
ЗОС G40
С G
С А
VA C
Рис. 4.1. Нуклеотидная последовательность в тРНК*"1' из Е. coli [4].
чество тепла, необходимого для увеличения температуры системы
в пределах этого интервала. Основная экспериментальная
сложность при этом состоит в том, как подвести определенное
количество тепла для увеличения температуры любого материала на еди*
ницу (градус) при постоянном давлении (типичные условия). Это
количество тепла называется теплоемкостью при постоянном
давлении СР:
CP=(dH/dT)P=const. (4.14)
Для данного вещества СР, как правило, постоянна, по крайней
мере, это справедливо в сравнительно узком температурном
интервале (условие, выполнимое для реакций, описывающих
физиологические процессы в биологии), Следовательно, вклад
теплоемкости составляет
?*
АН=\ CpdT (тепло, необходимое для увеличения
f температуры системы от 7\ до Т2 в (4.15)
отсутствие каких-либо фазовых
переходов).
107 ТЕРМОДИНАМИКА В БИОЛОГИИ
{
1.
к
.
1
(>'
!;■
*
h
Р-.
•»
•в
S
CJ
-?
4=
|
**
CS
*!:
см
уРНК,а' + Bytpep +
+о,оогмлщс\г
yPHKfal+ дурер
20 АО 60
Температура, "С
во
100
Рис. 4.2. Анализ наблюдаемых калориметрических кривых плавления
тРНК1"" ('|>PHKie/]=3-10-5 моль/л, рН 7,0, фосфатный буфер) в присутствии
(верхний рисунок) или в отсутствие (нижний рисунок) 1 мМ MgCl2 [5].
Объяснение см. ,в тексте [51.
Этот вклад надо вычесть из экспериментально определяемого
суммарного количества тепла, необходимого для увеличения
температуры системы; при этом находим теплоту изменения фазового
состояния вещества. Принцип метода дифференциальной
сканирующей калориметрии состоит в том, что измеряется тепло,
необходимое для увеличения температуры объекта на очень малую
величину, при непрерывном повышении температуры объекта. Вклад
теплоемкости вычитается, и остается аналогичная показанной на
рис. 4.2 зависимость поглощаемого тепла от температуры.
.Пример. Плавление транспортной РНК
Поскольку данная макромолекула в' растворе может принимать форму,
которую можно охарактеризовать как равновесную между несколькими основными
конформациями, обычно оказывается непросто зарегистрировать переходы между
частными индивидуальными конформационными состояниями, исследуя
суммарные молекулярные характеристики (константу диффузии, скорость седиментации,
электрофоретическую подвижность, оптическое поглощение и т. п.). Однако с
помощью метода дифференциальной сканирующей калориметрии иногда можно
прямо обнаружить переходы между индивидуальными конформациями,
благодаря тому что при нагревании раствора при температуре перехода
[характеристическая величина каждого конформационного (фазового) перехода]
поглощается тепло. Из данных, приведенных на рис. 4.2, видно, как изменяется
энтальпия на разных стадиях разворачивания молекулы валиновой транспортной РНК
(рис. 4.1). Эти изменения определены путем измерений количества тепла (элект-
108 часть l
рический обогрев), необходимого для поддерживания одной и той же
температуры у образца, содержащего тРНКиа/* и у образца сравнения при медленном
увеличении температуры обоих образцов (благодаря присутствию образца
сравнения вносится поправка иа вклады теплоемкостей в АН). Кривые на рис. 4.2
свидетельствуют о том, что разворачивание молекулы происходит за шесть
стадий и этот процесс характеризуется суммарной теплотой плавления
370 ккал/моль. Из данных, приведенных на рисунке, ясно, что структура
макромолекулы стабилизируется в присутствии иона Mg2+ i(t. е. тРНК плавится при
более высокой температуре), который связывает между собой какие-то области
молекулы (поскольку три стадии разворачивания, различающиеся друг от другая
в отсутствие Mg2+, в его присутствии сливаются в одну стадию).
Последовательность и парность оснований в данной нуклеиновой кислоте должна
совпадать с характерными для фенилаланиновой транспортной РНК, структура
которой, известна по данным рентгеноструктурного анализа (см. ч. 6), поэтому
представляется разумным, что данные трн высокотемпературных перехода
соответствуют «плавлению» трех областей спаренных оснований, которые в молекуле
связаны водородными связями, а переходы при более низких температурах —
разрушению гораздо более слабых взаимодействий между основаниями, с
помощью которых удерживаются друг возле друга различные «ручки» структуры
«кленового листа». Данные такого рода позволяют проводить исследования
участков взаимодействия различных химических агентов, связывающихся с
молекулой тРНК, наблюдая их влияние (как в случае Mg2+) на прочность
внутримолекулярных связей в разных частях исследуемой макромолекулы.
4.В. ТЕМПЕРАТУРНАЯ ЗАВИСИМОСТЬ ИНДИВИДУАЛЬНЫХ
КОНСТАНТ СКОРОСТЕЙ РЕАКЦИЙ*
В состоянии равновесия скорости прямой и обратной
химических реакций равны (см. ч. 3). Например, для реакции
А + В *==±: С-f-D (4.16)
*-i
скорость прямой реакции равна £i[A][B],- а скорость обратной
реакции составляет &_i[C][D], поэтому kj[A] [В] =&_i[C][D] N
или
*•/**=-№• (4Л7)
В правой части уравнения (4.17) записано как раз выражение
для константы равновесия реакции (4.16), следовательно
(4.18)
Хотя формула (4.18) в данном случае выведена для частного
случая равновесия (4.16), она является общей.
Вспомним, что константа равновесия изменяется с
температурой согласно уравнению
dln/CpaBH/d(l/T)=-A/4/tf, (4.12)
* Читатели, совершенно незнакомые с кинетикой химических реакций,
могут, прежде чем читать этот раздел, ознакомиться с ч. 3.
109 термодинамика в биологии
а уравнение (4.18) перепишем в логарифмическом виде
ln^paBH=ln^-ln^1( (4.18а)
тогда можно ожидать, Что температурная зависимость
индивидуальной константы скорости прямой или обратной реакции (k\ или
k-\) также должна удовлетворять уравнению
d\nk/d(UT)=—EajR, (4.19)
где
^а 'прямаягх) **а (обратнаяrx) ==&"rx (4.2U)
для того чтобы уравнения (4.19), (4.12) и (4.18а) согласовались
друг с другом. Экспериментально найдено, что константы скоростей
реакций, описывающих одностадийное равновесие^ действительно
удовлетворяют эмпирическому уравнению (4.19), обычно
записываемому в интегральной форме:
k = А ехр (—EJRT) (уравнение Аррениуса)
(4.21)
где предэкопоненциальный множитель А не зависит от
температуры, а Еа имеет размерность энергии и называется
(экспериментальной) энергией активации Аррениуса для данной реакции. В
течение последних 60 лет предпринимались невероятные усилия для
разработки теоретических основ предсказания абсолютных значег
ний параметров А и Еа для разных реакций. Хотя, за исключением
простейших реакций в газовой фазе количественные оценки А и
Еа оказались невозможными, были разработаны две теории,
объясняющие физический смысл параметров. А и Еа. Обе эти теории
позволили создать некоторые, механические представления о
химических реакциях. Они называются «теорией столкновений» (1917 г.)
и «теорией переходного состояния» (1935 г.).
4.В.1. ТЕОРИЯ СТОЛКНОВЕНИЙ И КОНСТАНТЫ СКОРОСТИ РЕАКЦИИ
Первый теоретический подход основан на гипотезе о том, что
скорость реакции между двумя веществами пропорциональна
числу столкновений между их молекулами за единицу времени —
поэтому эта теория называется теорией столкновений. Поскольку не
все реагирующие молекулы могут обладать достаточной энергией,
необходимой для осуществления реакции, следует ожидать, что
Скорость химической реакции (взаимодействия в секунду) =
=(Число столкновений в секунду между А и В)Х
Х(Доля реагентов с достаточной энергией).[А]»[В] (4.22)
ПО ЧАСТЬ 1
В случае реакции между молекулами идеальных газов можно
рассчитать частоту столкновений (в реакции между идеальными
газами):
Частота столкновений =Гя^еАВ j/ ^~ [А] [В], (4.23)
(реагенты—иделыше газы)
где й?ав — средний диаметр реагирующих молекул [й?ав=Г^а +
+^в)/2)], NQ— число Авогадро, k — постоянная Больцмана, Т —
абсолютная температура и \х — средняя масса молекул :[р.=
= тАтв/(гпА+тв)]. Можно показать, что доля молекул с
достаточной для реакции энергией составляет ехр[—Е*ЩТ], где Е*—
минимальная энергия, требуемая для того, чтобы реакция
произошла (см. гл. 19). Следовательно, поскольку
d [A]/dt=—k [А] [В], (4.24)
сравнение уравнений (4.22) — (4.24) приводит к следующему
результату:
#о г лГ SkT (теория столкновений,
k = 1Ш~ я^ав V ~^Г ехР {—Е*1%т) реакция между нде-
' р альными газами)
(4.25)
Согласно уравнению (4.25), скорость взаимодействия более
легких молекул (т. е. с меньшей jx), имеющих также большую
площадь поперечного сечения (яс?ав), выше; это же относится и к
скоростям реакций с меньшей минимальной энергией Е*. Эти
качественные предсказания были подтверждены при исследованиях
некоторых реакций между простыми молекулами в газовой фазе.
Значение k, рассчитываемое из уравнения (4.25), обычно плохо
сходится с экспериментальным в основном из-за того, что при
расчете не учитывались требования взаимной ориентации
сталкивающихся молекул, необходимой для осуществления реакции; ясно,
что, чем крупнее взаимодействующие молекулы, тем меньшую
долю от общей площади поверхности молекул составляет реакцион-
ноактивный участок и тем важнее становится это ориентацион-
ное (или стерическое) требование, и поэтому в общем случае
экспериментальное значение константы скорости гораздо меньше
того, которое предсказывается уравнением (4.25). Наконец,
благодаря тому что для обычных условий изменение температуры в
некотором интервале соответствует очень незначительным
изменениям Т и более значительным для ехр[—Е*ЩТ], уравнение (4.25)
предсказывает температурную зависимость констант скоростей
реакций, вид которой практически совпадает с описываемым
эмпирическим уравнением Аррениуса (4.21). Далее обсуждается
применение теории столкновений для качественного объяснения
механизма реакций первого порядка. Этот пример предвосхищает
Ill ТЕРМОДИНАМИКА В БИОЛОГИИ
помещенный несколько позже анализ стационарной кинетики
ферментативных реакций; к нему можно еще раз вернуться для
извлечения болЫшей пользы после изучения ч. 3.
Пример. Вывод выражения константы скорости химической реакции
первого порядка
Один из вопросов, который сразу же возникает при обсуждении скоростей
химических реакций следующий: почему некоторые реакции подчиняются
кинетическому уравнению первого порядка d[A]/dt= ±Я[А], когда1 практически все
элементарные химические реакции инициируются путем столкновения двух
реагирующих молекул? (Исключим из рассмотрения фотохимические реакции,
инициируемые поглощением света с последующей химической реакцией.) Линдемани
(1922 г.) смог решить этот вопрос, используя теорию столкновений, в
соответствии с механизмом, описываемым ниже.
Предполагается, что при большом числе столкновений в какой-то момент
одна из молекул реагента приобретает большую энергию:
*i
А + А +==* А+А* (4.26а)
«-1
Затем можно показать, что энергия возбужденной молекулы А* быстро
разделится на составляющие: энергию поступательного движения, энергию
вращательного движения и энергию колебательного движения молекулы. Если энергия
внутримолекулярных колебаний молекулы А* достаточно велика, амплитуда
колебаний по одной (или большему числу) связи настолько увеличивается, что
связь (связи) рвется:
А* »- Продукты (4.266)
Если лишь малая часть молекул А обладает достаточной энергией для
распада по такому механизму (фактор ехр [—E*/RT] в модели теории столкновений),
то механизм реакции (4.26) формально аналогичен схеме Михаэлиса—Ментен в
кинетике ферментативных реакций (ч. 3) и его можно описать, используя
аналогичное приближение условия стационарности (ом. детальное обоснование в
ч. 3):
dlA*]!dtezO*=kllA.}[A]—k_1lk}[k*]--kt[A*} (4.27)
или
Наконец, скорость образования продуктов описывается уравнением
Скорость образования продуктов = k2 [А*] = -т—пп _i_ ^' - (4.29)
Физический смысл уравнения (4.29) легче всего понять, рассмотрев
соответствующие граничные условия.
Нижний предел концентрации £2^>&i [А]. При этом условии А* обычно
превращается в продукты в большей степени, чем в реагенты [убедитесь из уравнения
(4.27)1 и выражение скорости [уравнение (4.29)] упрощается:
Скорость образования продукта = (k^/kj [A]2 = kx [A]2. (4.30)
В этом случае общий процесс описывается кинетикой второго порядка, как и
следовало бы интуитивно ожидать из того, что реакция инициируется столкновением
двух молекул А.
112 часть l
Б&рхний предел концентрации &-i[[A]»fc2. В этом случае А* обычно чаще
распадается вновь на реагенты, а не превращается в продукты и уравнение (4.29)
упрощается
Скорость образования продукта ^(kjijk^) [A].. (4.31)
Уравнение (4.31), таким образом, показывает, как реакция, начинающаяся
в результате столкновения двух молекул, может тем не менее:описываться
уравнением первого порядка по концентрации реагента. Очевидно,' кинетика первого
и второго порядков —крайние случай для таких реакций, так нто вообще следует
ожидать: порядок,реакции относительно 1[А]—дробная величина между 1 и 2
и приближение" к любому из целых значений зависит от концентрации реагента;
это было 'Подтверждено в исследованиях ряда реакций в газовой фазе. Наконец,
согласно, предложенному механизму реакции, можно качественно объяснить
низкие скорости большинства химических реакций по сравнению со скоростями
"столкновений' молекул друг с другом: лишь незначительная часть из общего
числа столкновений приводит к образованию продуктов с энергией, достаточной
для разрыва химических связей и осуществления реакции.
Теория столкновений, таким образом, позволяет объяснить
физическую природу наблюдаемой температурной зависимости
ехр[—EJRT] экспериментальных констант скоростей реакций и
зависимости констант скоростей от размера и массы реагентов.
Хотя действительные значения экспериментальных констант скоростей
занижены (на некий стерический фактор, р<С1) по сравнению с
•теоретическими, рассчитанными по уравнению (4.25),
сопоставление стерических факторов ряда реагентов-гомологов позволяет
обнаружить важную роль ориентации при исследовании данного
ряда реакций. И наконец, согласно теории столкновений,
предложена модель происхождения реакций первого и дробного
порядков. Такие схемы обычно типичны для реакций с участием
свободных радикалов, которые в последнее время привлекают
большое внимание в связи с изучением атмосферных загрязнений, а
также с возможным ингибирующим свободные радикалы
действием радиопротекторов.
4.В.2. ТЕОРИЯ ПЕРЕХОДНОГО СОСТОЯНИЯ И СКОРОСТИ
ХИМИЧЕСКИХ РЕАКЦИИ
Теория переходного состояния основывается на анализе
изменения потенциальной энергии системы в процессе реакции,
схематично изображенного на рис. 4.3. На рисунке изображена
диаграмма, на которой сплошными линиями отмечены уровни равной
энергии — следовательно, при протекании реакции реагенты,
находящиеся в исходном состоянии в «долине» потенциальной
энергии (область внизу справа на рис 4.3, в), преодолевают «вершину
барьера», или «седловину» (обозначенный как активированный
комплекс .на рис. 4.3, в), и после превращения в продукты
реакции спускаются в другую «долину» (область наверху слева на
рис. 4.3,в). [Если сделать «сечение» поверхности потенциальной
энергии при фиксированном значении г2 на рисунке, становится яс-
113 ТЕРМОДИНАМИКА В БИОЛОГИИ
Сечение при
Расстояние (вдаль пути реакции)
D + H + H
Активированный комплекс
(переходное состояние}
+
Профиль
реакции
(
Рис. 4.3. Поверхность потенциальной энергии для реакции D+H^—»-DH+H
[Л, с. 294].
а — поперечное сечение при постоянном гг; б — профиль изменения потециальной энергии
в реакции, происходящей по пути с минимальной энергией; в — контуры постоянной
энергии.' зависимость от межъядерного расстояния Г\ н г*
но, что продукты лежат в «долине» потенциальной энергии
(рис. 4.3,а).] Наконец, описывая наиболее энергетически
выгодный профиль изменения потенциальной энергии в системе в
процессе реакции, получаем кривую на рис. 4.3, б, свидетельствующую
о том, что система в этом процессе проходит через энергетический
«барьер».
' В теории столкновений при описании скоростей реакций
введен стерический множитель, для того чтобы объяснить причину, по
которой наблюдаемая скорость протекания реакций часто
намного ниже скорости, которую можно было бы ожидать при учете
реальной доли молекул, энергия которых достаточна для вступле-
П4 ЧАСТЬ I
ния в реакцию. В обычных равновесных ситуациях изменения
степени «согласованности» взаимодействия молекул (т. е. изменения
их ориентации или степени влияния стерического фактора)
можно описать также изменением степени неупорядоченности три
переходе от одного равновесия к другому, т. е. изменением энтропии
AG°rx=— RT \ri(Ka) =AH°rx—TbS°rx (при постоянной Г). (3.11)
Следовательно, можно надеяться, что смысл констант скоростей
химических реакций станет более понятным, если бы удалось
как-то согласовать предыдущее описание химического равновесия
с картиной пути реакции на рис. 4.3. Теория переходного
состояния основана на этой связи.
По теории переходного состояния предполагается, что реагенты
А и В находятся в динамическом равновесии* с активированным
комплексом (переходным состоянием) АВ^ (рис. 4.3)
** А*
А+В ч=* АВ* —*- Продукты (4.32)
так что можно определить константу равновесия /С* уравнением
**--&■' (433)
Поскольку скорость образования продуктов по схеме реакции
(4.32) предполагается пропорциональной концентрации
активированного комплекса АВ*, уравнение (4.33) можно решить отйоси-
тельно [AB+J
—d[A]/dt=[AB*]* (Скорость перехода через барьер) —
=[АВ*]£* =
=£*К*[А][В]. (4.34>
Из обычного выражения скорости прямой реакции
—d [A]/dt==kf [A] [BJ (4.24)
и уравнения (4.34) получаем
Константа скорости прямой реакции =tkf=k*K*. (4.35)
Вспомнив знакомое соотношение между константой
равновесия и свободной энергией (3.11), можно переписать уравнение
(4.35):
Константа скорости прямой реакции=&*ехр[—AG°*/RT],
kf =k* exp [bS°*jR] exp [—AH°*!RT], (4.36)
* Очевидно, что это равновесие не может быть истинным, так как
концентрации А и В изменяются со временем. Детальные расчеты, однако, указывают,
что эта модель тем не менее приводит к полезным результатам, кроме случаев
с очень низкими энергиями активации (т. е. ниже величины 5RT—3000 кал при
комнатной температуре).
115 ТЕРМОДИНАМИКА В БИОЛОГИИ
где Д(/0*, АН0* и AS0*—соответствующие изменения
стандартной свободной энергии, энтальпии и энтропии, сопровождающие
переход от реагентов к активированному комплексу.
И наконец, скорость перехода через энергетический барьер
можно приравнять к частотен колебаний связи (ей), которая (ые)
должна (ы) разорваться; другими словами, если атомы колеблются
с частотой v колебаний в секунду, то существует v возможностей
в секунду для ее разрыва. Поскольку, согласно классическим
представлениям, энергия колебаний связи составляет hv, где h —
постоянная Планка (Л=6,62-10~27 эрг-с), и поскольку тепловая
энергия колебания составляет kT (k — постоянная Больцмана),
имеем
hv=kT, (4.37)
так что
k*=v=kT/h (4.38)
и, следовательно,
(4.39)
Константа скорости прямой реакции — kf~
= (kT/h) exp (aS°*/#) exp (—ЬН°*[Ят)
Теперь можно сравнить уравнение переходного состояния (4.39)
с эмпирическим уравнением Аррениуса (4.21):
dlakf _ Eg _АЯ°* ■ 1 _£aH°* + RT
dT ~ RT*~ RT* "т" Т ~ RT*
откуда Ea=AH°*+RT. (4.40)
Уравнение (4.40) описывает связь между (теоретическим)
переходным состоянием и (экспериментальным) уравнением
Аррениуса, описывающим температурную зависимость констант
скоростей. Поскольку при комнатной температуре #Г=~600 кал, в то
время как для большинства химических реакций Еа составляет
много килокалорий, разность эмпирической энергии активации Еа
и энтальпии активации Яоф в теории переходного состояния
невелика. Уравнение (4.39), согласно теории переходного
состояния, позволяет объяснять различия в константах скоростей
химических реакций на термодинамическом языке энтальпии и
энтропии, уже знакомом нам из анализа обычных равновесий. Ниже
приведено несколько примеров.
Пример. Денатурация белков
Согласно теории переходного состояния, смысл уравнения (4.39),
описывающего константу скорости химической реакции, состоит в том, что это уравнение
позволяет найти как величину константы скорости, так и ее зависимость от
температуры, а это дает возможность охарактеризовать энергию, необходимую для
разрыва связей в реакции (АНФ), и изменение неупорядоченности в системе
116
ЧАСТЬ 1
а
ft
Переходное состояние
Agi* ^обратная реакция)
(прямая
реакция)
____!—. %
Субстрат k • \
Продукт
6
\
%ъ
Щ
Цъ
а*
^
Некатализцруемая
реакция
/" — N
t \
1 \
1 Катали-\
jjupj/емая у
. реакция i
If "N^V
\-_Jr У\
Субстат \
Продукт
ПутЪ, реакции
Путь реакции
Рнс. 4.4. Свободная энергия в химическая реакция.
а — некатализируемая химическая реакция. Переходное состояние определяется как
промежуточное между реагентами и продуктами состояние с максимальной свободной энергией
(аналогично активированный комплекс — промежуточное между реагеитами и продуктами
состояние с максимальной энтальпией). АО*Ф определяется из температуриой зависимости
1 > . константы скорости реакции [уравнение (4.39)1.
б—сравнение некатализируемой и ферментативной химических реакций. Действие
катализатора состоит в снижении свободной энергии активации реагентов.
(AS*). Таким образом, разность свободных энергий .реагентов и продуктов
реакции—лучший критерий самопроизвольности протекания всей реакции в целом,
н по аналогии разность свободных энергий реагентов и переходного состояния
выполняет такую же роль при решении вопроса о протекания реакции по
данному пути.
Такой путь анализа особенно хорош при описании скорости реакций
денатурации- белков. В этих реакциях (см. задачи в конце главы) энергия активации
может составлять sg; 100 000 кал/моль, так как для разворачивания белка из
компактной нативиой конформацин в статистически свернутую цель необходимо
разрушить много (вероятно, ионных) связей. Из уравнений (4.39) н (4.40) следует,
что, если Д//0^ большая положительная величина, константа скорости реакции
должна быть очень мала. Действующая сила в- такой реакции обеспечивается
очень значительным увеличением AS0*,, т. е. изменением неупорядоченности при
переходе от реагентов к /переходному состоянию, поскольку в действительности
при очень небольшом числе иативных конформаций бедка существует огромное
число возможных конформаций для белка денатурированного. Величина Д^^
компенсирует величину АН°* в уравнении (4.39), и в результате константа
скорости может быть большой. Однако, если энтальпия реакции AH°t велика,
скорость реакции значительно.изменяется в температурном интервале (см. задачу о
денатурации яичного альбумина), узком по сравнению с большинством
химических реакций.
Пример. Термодинамические характеристики ферментативной реакции
На рис. (4.5) показано, как можно осуществить термодинамический анализ
ферментативной реакции, если предположить, что реакция протекает по
механизму
Е + А *=± ЕА *z
=fc ЕР 4=fc Е + Р (см. ч. 3)
(4.41)
Необходимо исследовать температурную зависимость равновесной константы
суммарной реакции (Гчтобы получить ДЯ,* по уравнению (4.12)], температурную
зависимость (псевдоравновесной) константы Михаэлиса Ка для прямой или
/Сравя обратной реакции >[чтобы получить ДЯ связывания А или Р по уравнению
(4.12)] и температурную зависимость константы скорости прямой нли обратной
реакции [чтобы определить энергию активации Еа из уравнения (4.21)].
Найденные таким образом значения Д# дают полную количественную характеристику
П7 ТЕРМОДИНАМИКА В БИОЛОГИИ
(к
I
I
Переходное состояние
{активированный комплекс £2)
Реагент
+
Каталаза/пор
^Х
Продукт
+
Катализатор ■
З^Е + Р
Адсорбированный реагент
(комплекс ЕА)
Адсорбированный продукт
(комплекс £Р)
iVpaeHi
вант
Уравнение Аррёниуса (4.2f)
L
(У А ^реакции • вычисленная из А^ад приТ{иТг
Г5\ йНобразования,кои/>лекса ЕА.вычисленнай изK.Ownmun |1А.
<S> реакция) при Т, и Тг *АГ \Ураононив
Ся) йН образования комплекса LP,вычисленная из'/* (odpam-\ оантчодкра
*% наяЬеакция>при T.U ,z . /кр г J (4-Г2)
(4) Еа (.прямая реакцияхыгчисленная изЬ \
>< (Прямая реакция) при Т, иТ, \
(5) Еа (обратная реакция), вычисленная из к с
у>у (обратная реакция) при Tf uTs )
- - — - • .
Путь реакции—*■
Рис. 4.5. Эндотермическая ферментативная реакция, в качестве характеристик»
которой используется энтальпия [б].
Механизм, на котором основана диаграмма, рассматривается в тексте.
энергии, поглощаемой нлн выделяемой прн разрьвве илн образовании связей на
каждой стадии данной реакции. Сравнивая значения АН с теми^ которые
получены для некаталитических реакций,, можно сделать одре делённые
предположения об относительных достоинствах разных механизмов при ферментативном
катализе. Для более подробного ознакомления с .разными механизмами
ферментативного катализа, основанных на данных об энтальпии, читателю следует
обратиться к любому из современных учебников биохимии.
Задачи
1. Изменение свободной энергии при по/лном окислении 1 моля глюкозы до
углекислого газа и воды
602(1 атм, идеальный газ) + СвН1а06 (1 М, идеальный раствор)
ц—■.»- 6С02(1 атм, идеальный газ) + 6Н20 (чистая вода)
составляет
AGrx= —675 ккал/моль; стандартные состояния компонентов соответствуют
указанным выше.
Изменение свободной энергии прн превращении 1 моля ADP в ] моль
АТР при тех же стандартных состояниях всех компонентов реакции составляет/
7,3 ккал/моль (рН 7,0).
а. Рассчитайте, какое максимальное число молей АТР могло бы быть
образовано из ADP и фосфата, если бы в этой реакции можно было утилизировать,
всю энергию сгорания 1 моля глюкозы.
б. Живой клеткой из 1 моля глюкозы образуется ~38 молей АТР.
Используя полученный выше результат, рассчитайте к. п. д. превращения энергии в
клетке.
118 часть i
в. Теперь рассмотрите условия, более близкие к типичным физиологическим:
Рсо2 = 40 мм рт. ст.,
PQi = 100 мм рт. ст.,
[Глюкоза! =1 мг/см3,
[ATP] = [ADP] = [HP01-J = 0,0001 моль/л,
рН 7,0.
Выше уже было показано, что энергия превращения I моля ADP в АТ-Р при
таких условиях составляет ~ 12 ккал/моль. Рассчитайте изменение свободной
энергии при окислении в этих условиях I моля глюкозы.
т. Используя условия, указанные выше (пункт в), вновь рассчитайте к. п. д.
•превращения энергии в клетке.
д. Максимально возможная к. п. д. обычного теплового двигателя (т. е.
устройства, превращающего тепловую энергию — в данном случае теплоту
сгорания— в механическую работу) составляет (Т2—T{]jT2 [где Т\ н
Т2—(абсолютные) температуры рабочего вещества в разных частях двигателя].
К. п. Д. двигателя внутреннего сгорания достигает около 75% от
теоретического значения. Если рабочие температуры в двигателе составляют 1650°С и
600 °С, рассчитайте ожидаемую величину к. п. д. такого двигателя. Сравните с
результатом, полученным выше (пункт г). Попытайтесь объяснить причины,
почему биологическое преобразование энергии (которое представляет собой
изотермический процесс) оказывается настолько более эффективным.
2. а. Для большинства химических реакций установлено эмпирическое правило:
скорость реакции возрастает в два раза при увеличении температуры иа 10°С.
Если отношение констант скоростей прн двух температурах, отличающихся
на 10 °С, <2к>, рассчитайте энергию активации Еа, соответствующую Qu>=2 в
интервале от 25 до 35 °С.
б. Процесс оплодотворения яйцеклетки происходит со скоростью, значение Q\o
которой составляет ~600. Рассчитайте энергию активации з этом процессе.
в. Процесс денатурации яичного альбумина происходит с энергией активации
порядка 100 000 кал/моль. Рассчитайте, насколько больше времени
потребовалось бы на то, чтобы сварить яйцо на высоте 1220 м (давление 656 мм рт. ст.,
так что температура кипения воды 96 °С), чем на уровне моря.
3. Если- Sty— (стандартный) химический потенциал нонов Н+ в водной среде
в стандартном состоянии (ан+ = 1) при концентрации 1,00 моль/л в идеальном
растворе н если &ц+—стандартный) химический потенциал нонов Н+ в
водной среде в (другом) стандартном состоянии (ан+=1) при концентрации
10~7 моль/л в идеальном растворе, покажите, что
(?!н- = gIh- (2,303)-7-RT, гДе R—газовая постоянная, Т—температура в градусах
н ' ' Кельвина.
4. Из совместного анализа уравнений Аррениуса k=A expf[—EfRT] н Вант-Гоффа
dln*o AH°rx
д(\/Т) = —п— покажите, что
A#rje = Еа (для прямой реакции) — Еа (для обратной реакции).
5. Из данных, приведенных на рис. 4.6, рассчитайте &Н°ГХ денатурации химотрип-
синогена при рН 2,0 и рН 3,0. Рассчитайте также константу равновесия и
AGrx этого процесса при 25 и 37 °С (в значительной пи степени"
денатурирован зимоген при этих температурах?). И наконец, рассчитайте AS°X при двух
указанных значениях рН и двух температурах. Какой вывод можно сделать
о температурной зависимости параметров АН°ГХ н AS^X ?
119 ТЕРМОДИНАМИКА В БИОЛОГИИ
+ 0,8 F
О -
-1,0-
Рис. 4.6. Зависимость lg К от ЦТ для денатурации хнмотрнпснногена прн рН 2,0
пя денатург
и 3,0 [7].
6. Хорошо известно, что большинство ферментов денатурирует в присутствии
концентрированной (положим, 8 М) мочевины. Также хорошо известно, чта
этот процесо обычно ускоряется прн увеличении температуры. Недавно [8]
показано, что в присутствии разбавленной мочевины (0,05 моль/л) денатурация
а-химотрнпсина ускоряется прн снижении температуры < О °С! Из известного-
правила фаз папыггайтесь объяснить кажущееся отрицательное значение
энергии» активации для этого процесса.
7. При исследовании ферментативной реакции превращения фумаровой кислоты в
L-яблочную кислоту
С ООН
Hi
II
сн
I
соон
фумаровая
кислота
фумараза
+ НОН +
вода
кофактор
ч ■—
СООД
НСН
носн
i,
:оон
L-яблочиая
кислота
найдены температурные зависимости константы равновесия при связывании
Ко, константы скоростей прямой и обратной реакций kf н kr и константы Ми-
хаэлиса Кл и Кр обеих (прямой н обратной) реакций:
ain/ca/a<i/r) = +i8i2 к,
d\nkf/d(UT) = — 3070 К,
dlnkr/d(l/T)=~7599 К,
а1пКА/д(1/Г) =—2114 К.
a in Яр/а (1/л = +604 к.
Предполагая, что константу Мнхаэлиса можно рассматривать как
константу равновесия (диссоциации) при связывании субстрата (Ка) или
продукта (Кр) с ферментом, нарисуйте схему изменения энтальпии этой реакции
как функцию от пути реакции. Укажите: реагенты, продукты,
фермент-субстратный комплекс, комплекс фермента с продуктом и активированный комплекс
(переходное состояние).
Л20 ЧАСТЬ 1
Литература
1. Moore W. J., Physical Chemistry, any edition, Prentice-Hall, Englewood Cliffs,
N. J. Хорошее рассмотрение равновесия и самопроизвольности химических
реакций.
2. Kitzinger С, Benzinger Т. Н„ Methods of Biochemical Analysis, 8, 309 (1960);
Brown H. D., Biochemical Microcalorimetry, Academic Press, New York, 1969.
Обсуждение метода дифференциальной сканирующей микрокалориметрин н
его применений для изучения биологических макромолекул.
3. Wentworih W. E., Ladner S. J., Fundamentals of Physical Chemistry, Wads-
worth Publ. Co., Belmont, California, 1972. Хорошее рассмотрение
температурной зависимости констант скоростей химических реакций.
4. Barrell В. G., Clark B. F. С, Handbook of Nucleic Acid Sequences, Oxford,
Joynson-Bruvvers, Ltd., 1974, p. 55.
5. Privaldv P. L., Filimonov V. V., Benkstern T. V., Bayev A. A;, J. Mol. Biol.,
97, 279 (1975).
-6. Williams V. R, Williams H. В., Basic Physical Chemistry for the Life Sciences,
2nd ed., W. H. Freeman, San Fraflcisco, 1973, p. 320.
7. Eisenberg M. A., Schwert G. W., J. Gen. Physiol., 34, 583 (1951).
-8. Pincock R. E., Lin W.S., J. Agric. & Food Chem., 21, 2 (1973).
ГЛАВА 5
ЭЛЕКТРОХИМИЧЕСКИЙ ПОТЕНЦИАЛ
с
Выше показано, что разность химических потенциалов
возникает на границе двух растворов, отличающихся по концентрации
растворителя (осмотическое давление), то концентрации
заряженного вещества (равновесие Доннана), по реакционной
способности растворенных соединений или по температуре. В
соответствующих экспериментальных условиях (см. ниже) можно свести-
эти различия в химическом потенциале к разности электрических
потенциалов, которую можно затем измерить с помощью
вольтметра (потенциометра). Рассмотрим электрохимические
элементы, основанные на различиях в концентрации растворенного
вещества (концентрационные элементы) и на различиях в
реакционной способности (химические). Концентрационные элементы
получают все более широкое распространение в качестве устройств для
измерения разности концентраций двух растворов, в одном из
которых концентрация известна. Топливные элементы уже давно
используются для определения термодинамических параметров
AGrx, АНГХ и ASTX химических реакций, и их начинают все более
серьезно рассматривать в качестве перспективных практических
устройств для хранения энергии в химической форме с целью
дальнейшего превращения ее в электрическую энергию. В начале этой
главы будут рассмотрены устройства, способные преобразовывать
разность химических потенциалов (обусловленную различием
концентраций растворов) в измеряемую разность электрических
потенциалов или напряжение.
5.А. КОНЦЕНТРАЦИОННЫЕ ЭЛЕМЕНТЫ
Рассмотрим два раствора на рис. 5.1. Металлический электрод;
(например, металлическая проволока) погружен в раствор слева,
содержащий соль металла А+Х- с концентрацией [А+]'. Такой же-
электрод погружен в раствор оправа, в котором эта соль
находится в концентрации [А+] справа. Что произойдет ори прохождении
по цвпи.1 моЛя электронов в указанном направлении (на рис. 5.1
показано типичное направление движения электронов по внешней'
122 часть i
£у*
h
Вольтметр
(потенциометр)
&*
(£Ъ(£Ъ
A"*IA+Wi+e
?
« + rA+L^A
•'слева
Суммарный процесс : [А Xh/ява ~* ^А+%,
Рис. 5.1. Электрохимический концентрационный элемент (см. описание в тексте).
депи: слева направо)? Для того чтобы 1 моль электронов прошел
до цепи, электроны должны образоваться на левом электроде:
А° —* [А+]слева+е (5.1а)
з. для того, чтобы электроны не накапливались на правом
электроде, они должны на нем расходоваться:
1-А Справа "Г" ^ •"•
(5.16)
Поскольку ни в одном растворе свободных. электронов нет,
электрический ток должен быть результатом изображенного на
рисунке переноса ионов через раствор. Солевой мостик представляет
собой простое устройство, создающее электрический контакт
между двумя растворами (т. е. между растворами может протекать
электрический ток благодаря (переносу ионов К+ и С1~), но не
позволяющее растворам смешаться друг с другом, чтобы разность
концентраций ионов металла в этих растворах сохранялась.
Обычно солевой мостик заполняют раствором КС1, поскольку
ионы К+ и С1- под действием электрического поля движутся почти
с одинаковой скоростью (если скорость движения катионов и
анионов различна, то в результате обеднения граничной области
между раствором и солевым мостиком ионами одного заряда
возникает дополнительная разность потенциалов на жидкофазной гра-
123 ТЕРМОДИНАМИКА В БИОЛОГИИ
нице (на этом эффекте основан эксперимент, рассматриваемый
при обсуждении метода диск-электрофореза в ч. 2).
Электрическая работа переноса п молей электронов с левого
электрода на правый равна
Электрическая работа=(Заряд)«(Разность электрических
потенциалов)=nFE, (5.2)
где F — заряд 1 моля электронов (96 500 Кл), Е — разность
электрических потенциалов между электродами, измеряемая
(предполагается, что на жидкофазной границе не возникает разности
потенциалов) вольтметром (рис. 5.1).
Можно также считать, что общий результат рассматриваемого-
процесса в растворах состоит в выравнивании концентраций
путем шереноса п молей иона А+ из раствора с концентрацией
[А+] справа в раствор с концентрацией [А+] слева. Изменение
свободной энергии в этом процессе описывается уравнением [см.
уравнения (3.2) и (3.3)]
AG = Gy,^ — GcnpaBa = П (GA+)cnpaBa — П (GA)uneBa =
=Д (бА+)слева-\-nRT In (аА+)слева— п (Ga+)справа—nRT In (аА+)справа
(5.3)
AG=nRT In ([ А+]акая/[ А+]справа),
Предполагается, что растворы достаточно разбавлены, т. е.
аА+ = [А+]'; для ионов А+ в обоих растворах выбрано одинаковое
стандартное состояние. Поскольку AG — отрицательная величина,
если система не совершает работы PV (табл. 3.1), то
AG = — nFE
(5.4>
И наконец, объединяя уравнения (5.3) и (5.4), получаем уравнение
(5.5>
Е = -(RT/F) In ([А+]СЛева/[А+]справа)
Уравнение (5.5) представляет собой уравнение Нернста для
частного случая (см. общее уравнение в разд. 5.Б), и описывает
электрохимический потенциал, соответствующий данной разности
концентраций двух идентичных во всех остальных отношениях
растворов. Наконец, важно отметить, что в разобранном примере
электрохимический потенциал определяется только ионами А4*,.
эффективно переносимыми, благодаря тому что используемый
электрод (из металла А)"откликается (т. е. металл А
растворяется или осаждается из раствора) лишь на изменения
концентрации ионов А+. Если бы другие ионы также могли вступать в реак-
124 ЧАСТЬ 1
ции с этим электродом (см. ниже), то они бы также считались
эффективно переносимыми и измеряемый электрохимический
потенциал отражал бы также и их концентрации.
Пример. Мембранный потенциал в живых клетках
Живая клетка активно накапливает ионы К+ и выводит ионы Na+.
Типичное соотношение концентраций К+ внутри и снаружи клетки составлйет около
20:1
1^ 1внутрн/[^ Снаружи ^^»
Можно рассчитать соответствующий электрохимический лотенциал на мембране
из частного уравнения Нернста (5.5) при физвологической температуре 37 °С:
Р ^ (1,987 кал-моль-1 -град-*) (310 К) (4,185 Дж/кал) ((> «п«. . (()().
Д^~ (1 моль электрона) (96500 Кл/моль) {*>*"*) 'g {*")•
Полученное расчетом значение потенциала близко к экспериментально
определенному Для живой клетки в состоянии покоя. Изменения мембранного
потенциала живой клетки, сопровождающие передачу нервных импульсов или
мышечное сокращение, обусловлены возникающим при этом потоком ионов К+ (ив
клетки) и Na+ '(в клетку); это приводит к резкому падению потенциала,
регистрируемому вольтметром с помощью микроэлектродов, находящихся внутри и
снаружи живой клетки.
Пример. Ионоселективные мембранные электроды
Концентрационному элементу, изображенному на рис. 5.1, в принципе можно
сообщить чувствительность к концентрации любого иона. Однако можно создать
условия* при которых регистрируемый параметр (сила тока или напряжение
потенциала) будет селективен лишь к нескольким нли даже только к одному виду
ионов; для этого надо заменить (неселектнвный) КС1 в солевом мостике
веществом, связывающим только исследуемый ион (рис. 5.2).. При выборе
селективного промежуточного переносчика ионов существуют три возможных материала:
стекло, жидкость и твердое вешество.
Стеклянный ионоселективный электрод: измерения рН. Впервые
предложенный ионоселективный электрод представлял собой просто тонкую (0,05 мм)
стеклянную пластинку (мембрану). При замачивании ее в течение неркольких часов
в воде катионы металлов, обычно присутствующие в стекле, замещаются иа
поверхности стекла ионами Н+. После этого мембрана приобретает
электропроводность: при связывании протонов на одной стороне мембраны ионы Na+
передвигаются внутри стекла от одного силикат-иоиа к другому и в результате протоны
переходят в раствор с другой стороны стеклянной мембраны. При
соответствующем выборе типа стекла (кварц и пирекс ие -годятся) с мембраной могут
связываться только ионы Н+, н, следовательно, ток будет идти через мембрану
только при трансмембраниой разности концентраций протонов. Возникающий при
этом электрохимический потенциал описывается уравнением (5.5). Если
концентрация протонов по одну сторону мембраны известна (пусть, например,
концентрация протонов равна — 0,12 моль/л, что соответствует активности 0,10), то,
измерив электрохимический потенциал, можно определить активность иона
водорода в исследуемом (правом на рис. 5.2) растворе, и, следовательно,
рН = —lg(flH+),
£=(i?T/F) In ^^^н^рва.^ = (—^Г//) 1фН)справа — 1 ](2,303). (5.6)
125 ТЕРМОДИНАМИКА В БИОЛОГИИ
ШШ
':'■':'■'•'•:'•:
'.'•'•:'*■
■-/.]'■.:[[:
'.:':'•:'•:'•
■1***^'1 •
':'•'•'•:'■:
[■]■]■]■
ш
•:<
'■■'.:
•:•;.;
§>
|
£
&
■ 0~
t ■
вольтметр
K-J Пористая мембрана
'.''•'• '.'•'.'•:'•'.'•'.':'•'•'.'•'.'•''''<''•'.-'.'•' '••'.''•''•'.'''.'•
::::::•:•:::•:::•:•:::::•:•:::•:::•:;:::;:;:;;
ЪШХ>ЖШ&±^УЖА
::::::::::::i:::::::::::;i;::::i;::::::i;::::3
::;:■':•■::■ J:::::::::::::::::.:::::':::.::::.::i::i
'•:'-:'-:::::'-i '■'■'■:'■'•'■:'■ •;'■■■::.::::'-:::-: •:'•:'•:■•:•
• ■:■:'■•'■':'/:•'::'•:'-'■'•:'■ ■'•:'■-•'•: ■:'■:'■: ■:':'•:'■:'■:
1111Й11111
lllllillllflli
lliiiiilllliiliii
<D*
Пористая
::::•:>:::;•:•:•:•:•;•:::• .мембрана
:^-:-:-:->:::-:-:-:<^ ' "' "
?::::::.::i:.::::::::::::.:i::t-
t&V&ttWftW
t|:i|i:i:ii|:j!;!i!!l!:iH!«
мшшшш
::.\:Селектианв1щ
:'.:::aeuiecmao-\:':\
:'::=:;:':.::-::::.:::.:::.:;::.::3
$&&#&&М
§»
ектр
§
¥
I
I"I*i*!*t*!*t* ?*Г'1*^'!*г* S;«;«;!i« !*'•!« '**l*i'l •'. •Г*г,;«;«1'!'Г*^'*'!'Г*Г'!"Г'>"":*!-1
::::;i--:=;iil:;:;i=:;i;i:i;i=i--.:-;::=.:--a
Рис. 5.2. Упрощенная схема работы ионоселективного мембранного электрода.
Разность химических потенциалов растворов слева и справа от мембраны, обусловленная
разностью активностей (концентраций) иона I+, измеряется как разность потенциалов
(напряжение) между двумя идентичными электродами (см. обычную концентрационную цепь
на рис. 5.1). Однако ток в цепи появляется, если только слева от Границы раздела (между
раствором н пористой мембраной) ноны 1+ связываются с веществом-переносчиком М,
а справа от границы раздела ноны 1+ освобождаются от комплекса Ml (показано
соответствующими уравнениями внизу схемы). При условии, что вещество М, переносящее ионы,
селективно относительно связывания 1+ (т. е. М связывает I+, но не связывает другие ионы),
измеряемый электрический потенциал отражает только концентрацию 1+ в правой ячейке,
поскольку концентрация 1+ внутри ионоселективного электрода в левой ячейке известна.
Большинство стеклянных электродов позволяет измерять концентрации не
только Н+, но и других ионов, например Na+, особенно при высоких рН, когда
концентрация протонов намного меньше концентрации других ионов в растворе.
Состав некоторых стеклянных электродов позволяет увеличить чувствительность
электрода к конкретному катиону: так, теперь можно изготовлять стеклянные
мембранные электроды, селективные (до известной степени) к Na+, TI+, К+, Си+,
NHj, Rb+, Cs+, Li+ или Ag+. Однако селективность этих электродов несравнима
с высокой селективностью электродов других типов (см. ниже).
Жидкостной ионообменный электрод: коэффициенты активности. Заменив
пористое стекло (предыдущий пример) на раствор, содержащий вещество,
специфически связывающее данный ион, можно расширить область применения (т. е.
чувствительность н селективность) такого электрода для детектирования мио-
гозарядных катионов и анионов (стеклянный электрод чувствителен только к
концентрации однозарядных катионов). Например, если в качестве
специфического вещества использовать органическую фосфорную кислоту, связывающую ион
кальция намного прочнее, чем большинство других катионов, то электрод
приобретает селективность по кальцию. Наконец, поскольку в этом случае перенос
1 моля кальция соответствует переносу двух молей электронов, то уравнение
(5.4) можно переписать:
AG = — nzFE
(5.7)
И*6К~~ ЧАСТЬ I
09
5
I
I
+А0 -
+20
-20
Активность или концентрация Саг*1моль/Л
Рис. 5.3. Зависимость потенциала жидкостного ионообменного кальциевого
электрода от концентрации (/) и активности (2) растворов (чистого) хлорида
кальция.
Согласно уравнению (5.8), при десятикратном изменении активности кальция потенциал
изменяется на (RTI2F) =29,6 мВ. Поскольку концентрация кальция, соответствующая
данному потенциалу, всегда выше активности кальция, коэффициент активности кальция
всегда меньше единицы и уменьшается с увеличением концентрации кальция [см. уравнение
(3.29) и рис. 3.1]. (Данные любезно представлены фирмой Orion Research Incorporated,
Cambridge, Mass.)
где п число молей переносимых ионов (в данном случае Са2+), z— число
переноса электронов в расчете на 1 моль переносимых ионов (в данном случае
z=2). Следовательно, при использовании в качестве раствора сравнения (внутри
электрода, как и в случае комбинированного стеклянного рН-електрода*)
раствора с фиксированной концентрацией кальция э. д. с. жидкостного ионообменного
Са2+-электрода, возникающая в присутствии кальция в исследуемом растворе,
дается уравнением
Е—const -\-(RT/2F) In (aa2+),
(5-8).
где Е характеризует активность кальция в исследуемом растворе. Благодаря
тому что измеряемое напряжение пропорционально логарифму активности
кальция, чувствительность электрода сохраняется в очень широком интервале
концентраций кальция (рис. 5.3). Кроме того, поскольку 9. д. с. электрода
определяется активностью кальция, измерения в растворе с известной концентрацией
иона кальция позволяют прямо определить коэффициент активности иона при
этой концентрации. Из измерений электрохимических потенциалов были
получены многие известные значения коэффициентов активности ионов в растворе:
«Caa+=Yca2+^Caa+-
(3.96)
В литературе описаны жидкостные ионообменные мембранные электроды,
селективные к Са2+, NOi", СО|~, СЮ7, Cu2+, BF7,Mg2+, Mn2+, C1- и к
некоторым ионогенным органическим соединениям. Увеличение числа известных
селективно связывающих ионы веществ позволяет надеяться, что скоро будут
сконструированы новые подобные электроды, однако уже с повышенной
чувствительностью и селективностью (как к указанным, так и к другим ионам).
Твердофазные мембранные электроды. Одно из препятствий для широкого
использования вышеописанных (жидкостных ионообменных) электродов
обусловлено их хрупкостью, поскольку обычно для-создания надежных условий доступа
исследуемых иоиов к ионоселективному переносчику необходимо закапсулировать
раствор сравнения в электрод с очень тонкой мембраной. Этот недостаток пре-
127ТЕРМОДИНАМИКА В БИОЛОГИИ
«О
§ 80
2
ад
$
ад
JS
* О / 2 3 4
Концентрация белка, мг/м/i
Рис. 5.4. Изменение потенциала мембранного электрода на основе
кристаллического Ag2S в растворах двух разных белков сыворотки кровн [4].
одолевается, если в качестве ионоселективного компонента применить твердые
или кристаллические материалы. Например, LaF3— 'практически нерастворимое
твердое вещество, обладающее повышенной проводимостью ионов F~ по
сравнению с любыми другими ионами. Поэтому твердофазный мембранный электрод
можно сконструировать, поместив между растворами (сравнения и исследуемым)
в качестве ионоселективного (в данном случае по ионам F-) «барьера»
монокристалл LaF3. Для процесса в электроде также справедливо уравнение (5.8) при
г—1. Используя вместо LaF3 кристаллический AgjS (ионный проводник Ag+),
можно создать твердофазный электрод, чувствительный к Ag+, S2-, Cu2+, Pb2+,
Cd2+, I-, Br- и С1~, поскольку эти ионы либо замещают Ag+, либо связываются
с Ag+ на поверхности кристалла. Совсем недавно обнаружено, что электрод на
основе кристаллического Ag2S чувствителен к присутствию в исследуемом
растворе молекул белка, причем при одних и тех же концентрациях белков э. д. с.
электрода различна в растворах разных белков (рис. 5.4). Представляется
вероятным, что взаимодействие ионов Ag+ с цистеиновыми остатками в белке,
зависящее от числа дисульфидных связей в молекуле белка, лежит в основе
селективности электрода. Наконец, было обнаружено, что для данного белка э. д. с.
электрода на основе Ag2S зависит от присутствия антител, специфичных к этому
белку, так что кажется возможным сконструировать твердофазный электрод, у
которого с поверхностью мембраны был бы связан соответствующий рецептор
гормона или антитело. С помощью такого электрода можно было бы обнаружить
многие иммуноактивные соединения в крови без каких-либо специальных
приготовлений исследуемого образца. Заманчиво предположить, что с помощью таких
методов когда-нибудь окажется возможным детектировать различные
заболевания, причем основным прибором будет простой рН-метр с соответствующим ионо-
селективным электродом.
5.Б. ТОПЛИВНЫЕ ЭЛЕМЕНТЫ
Возвратимся к эксперименту, изображенному на рис. 5.1, и
допустим, что разделенные мембраной растворы содержат два
разных химических вещества (например, Си2+ и Zn2+ как показано
на рис. 5.5), а не одно и то же вещество в разных концентрациях.
Тогда, согласно рис. 5.5, перенос по такой цепи 1 моля электронов
приводит к тому, что в левом растворе 72 моля металлического Zn
Альбумин сыворотки
человека
128 ТЕРМОДИНАМИКА В БИОЛОГИИ
ь
N
€>~
Вольтметр
е^
ZnZ+UZn2*=D
Zn -*Zr?*+2e
Сиг+(ЛСи^ = 1)
?
о
Сиг+ + 2е -»Cu
Суммарный процесс: Zn + Сиг+ (аС(1г+ *1).->Си + Zn2* (а Zn2+ = 1)
Рис. 5.5. Электрохимический топливный элемент.
Прохождение по цепн 1 r-экв. электронов приводит к превращению lft г-атома
металлического цинка в ионы Zir4- и '/г г-атома иоиов СиН- в металлическую медь. В зависимости
от направления движения электронов по цепи (или направления внешнего напряжения) на
поверхности электрода осаждается цинк или медь.
окислится до Zn2+, а в правом 7г моля ионов Си2+ восстановится
до металлической Си
Zn + Cu2+ —>■ Zn2+ + Cu (5.9)
Или в более общем случае
аА + ЬВ >• cC + dD (3.4)
из уравнений (4.3) и (5.7) можно получить уравнение Нернста
Erx = Erx-(RT/zF) In—-j
°AaB
(5.10)
где Е --- Наблюдаемый электрохимический потенциал,
соответствующий реакции (3.4), концентрации компонентов в растворе
приравнены активностям, R — газовая постоянная, Т — абсолютная
температура,, Д — заряд 1 моля электронов, г — число переноса
электронов, ёбпффвождающего превращение 1 моля реагента. В ча-
129 ТЕРМОДИНАМИКА В БИОЛОГИИ
стности, когда все компоненты находятся в стандартных
состояниях (аА=ав = ас=аъ=\)
р _ р _ —__
£« - t-rx - nzF
(5.Г1)
где п — число молей реагента, превращаемых в продукт, AGxr —
изменение стандартной свободной энергии в результате реакции
(3.4). Наконец, поскольку принято определять AGrx и AG°rx на
1 моль реагента, превращаемого в реакции, уравнения (5.11) и
(5.7) часто записывают без множителя п [см. уравнение (5.12)].
5.Б.1. АНАЛИТИЧЕСКИЕ ПРИМЕНЕНИЯ: AGTXt AHrx/bSrx
И Ка ИЗ ЭЛЕКТРОХИМИЧЕСКИХ ИЗМЕРЕНИИ
Согласно вышеизложенному, измерение Егх с помощью
электрохимического элемента, в одной половине которого находятся
реагент А и продукт D, а в другой — реагент В и продукт С,
представляет собой прямое измерение AGrx, характеризующей
превращение 1 моля реагента (реагентов) в продукт (продукты) при
данных концентрациях всех присутствующих компонентов
Ь0ГХ = -zFErX
(5.12)
где г—число молей электронов, проходящих по цепи при
превращении моля реагента. Следовательно, уравнения (5.10) и (5.12)
объединяются и, измеряя Етх при нескольких различных
концентрациях компонентов, можно получить AG°X, из которой
рассчитывается константа равновесия Ка (см. пример ниже).
Поскольку часто интерес представляет лишь одна электродная
реакция из всех протекающих в электрохимическом элементе,
для измерения ее потенциала (электродного потенциала) удобно
считать, что потенциал другого электрода (сравнения) равен нулю.
(Е°гХ =0). В качестве такого электрода сравнения обычно
используется стандартный водородный электрод
V2H2 (iaTM) *=* Hjjj+^j _j_e; (5.13)
[Н+] = 1,0 моль/л в идеальном растворе
Е°=0,000 для реакции в обоих направлениях.
Следовательно, для* исследуемой электродной реакции Е —
положительна, если при сопряжении со стандартным водородным
электродом реакция, согласно уравнению (5.13), приводит к
восстановлению Н+ до Нг.
130 ЧАСТЬ 1
Пример. Восстановление цитохрома с
Цитохром с — гемсодержащий белок, широко распространенный в живых
организмах, играет важную роль в реакциях электронного переноса при
аэробном метаболизме. Поэтому механизм восстановления и окисления гемового
железа представляет общий интерес. Оказывается, что в реакции
Cytin (Fe3+)+e ч==* Cyt" (Fe2+)
(5.14)
для восстановленного Cyt11 и окисленного Cyt™ оптическая плотность при 550 им
значительно различается, так что легко определить их относительные
концентрации. На рис. 5.6 приведена зависимость экспериментальной Егх от
относительного содержания восстановленного » окисленного цитохрома с [измерения
проводились относительно стандартного водородного электрода; см. [уравнение (5.13)].
Из уравнения (5.10) легко определить стандартный потенциал этой реакции,
составляющий +0,244 В при 22 °С, так что реакция (5.15) самопроизвольна:
CytpH 7,0 + 1/2Н2 (1 атм) > CytpH 7,0 Т"Н[Н+]=10-* моль/л в идеальном растворе
Е°;х= +0,244 В. (5.15)
Кроме того, из угла зависимости на <рис. 5.6 можно сделать вывод, что
превращение Cyt111 в Cyt11 характеризуется значением z=l, так что реакция
происходит с переносом одного электрона.
Значения стандартных окислительно-восстановительных потенциалов ряда
биохимически важных реакций приведены в табл. 5.1. Обратите внимание, что
потенциалы соответствуют восстановлению указанных веществ в реакциях и что
оии измерены относительно стандартного водородного электрода [уравнение
+1,0
>
о
>
о
■3»
-2,0
-1,0-
0,35 0,30 0,25 0,20 0,15
Потенциал, 3
Рис. 5.6. Относительное содержание восстановленной и окисленной форм
цитохрома с при различных потенциалах электрода (относительно стандартного
водородного электрода) [5]. ,
Тангенс угла наклона прямой дает число переноса электронов, а потенциал прн равном
/ [Cyt"] \
содержании обеих форм цитохрома с I lg гг>Л1Ь =0) —стандартный электрод-
[Cyt"I]
ный потенциал данной реакции.
|31 ТЕРМОДИНАМИКА В БИОЛОГИИ
Таблица 5.1
Стандартные окислительно-восстановительные потенциалы некоторых
электродных реакций3 (относительно стандартного водородного электрода;
[Н+] = 10-7 моль/л в идеальном растворе)
Число электронов г,
реагент Продукт участвующих
(окислительная форма) (восстановительная форма) в переносе 1 моля
вещества
а-Кетоглутарат
Ацетат
Ферридоксин
2Н+ -(рН 0,0)
NAD+
NADP+
Липоат (окисл.)
Глутатион (окисл.)
Ацетальдегид
Пируват
Фумарат
(Цитохром Ь)3+
Дегидроаскорбат
Убихинои (окисл.)
'(Цитохром с)3+
Феррицяанид
атм) + 2НрН 7
Сукцинат+С02
Ацетальдегид
Ферридоксин
Нг (1 атм)
NADH+H+
NADPH+H+
Липоат (восст.)
Глутатион (восст.)
Этанол
Лактат
Сукцинат
(Цитохром &)2+
Аскорбат
Убихинон
Убихннон (восст.)
Ферроцианид
Н^О
2
2
1
2
2
2
2
2
2
2
2
1
2
2
1
1
2
—0,67
—0,60
—0,43
—0,41
—0,32
—0,32
—0,29
—0,23
—0,20
—0,19
+0,03
+0,07
+0,08
+0,10
+0,22
+0,36
+0,82
аУсловия: рН 7,00, активность всех остальных компонентов равна 1, температура 25 °С.
(5.13)], на котором газообразный водород окисляется до ионов Н+. Наконец, как
для данных (Дбг*) табл. 4.1, за стандартную концентрацию Н+ в перечисленных
в табл. 5.1 реакциях следует принять концентрацию Ю-7 моль/л; тогда шкала
Е" более точно отражает физиологические условия.
Пример. Определение очень больших или очень малых констант равновесия
Когда значение константы равновесия близко к единице, концентрации
реагентов и продуктов часто можно определить, непосредственно измеряя
оптическую плотность растворов. Однако для многих интересных реакций (например,
связывание фермента с ингибитором, гормона с рецептором, антитела с
антигеном, лекарственного вещества с рецептором) константа равновесия может быть
столь велика, что концентрацию свободного компонента невозможно измерить.
В таких случаях простым способом определения константы равновесия могут
оказаться измерения электрохимического потенциала, как показано в следующем
примере.
13 2 ЧАСТЬ 1
Рассмотрим диссоциацию сульфида ртути в воде:
HgS ► Hg2+ + S2-
(5.16)
Из таблицы стандартных окислительно-восстановительных потенциалов,
аналогичной табл. 5.1, легко найти подходящие электродные реакции, из который;
можно составить электрохимический элемент с соответствующим значением
э.д.с. Е\
Hg ► Hg2+ +2е Е° — —0,854 В
2е +.HgS ► Hg + S3- Е° = —0,70 В
HgS ► Hg2+ 4 S2-
E°= —1,554В
Полученное значение стандартного электрохимического потенциала реакции
(3.16) используется для расчета изменения стандартной свободной энергии
A^r=—zf£° = —2-96500(—1,554)=+300000 Дж/моль (5.11)
Из AG°X легко получить константу равновесия (произведение растворимости)
для растворения HgS в воде при 25 СС (при этом образуются ионы Hg2+ и S2-)
ag;x=— rt \пПр, (3.11)
Пр=ехр Г -зооооодж 1
н F [(1,987 кал-моль-1 град-1) (298 К) (4,185 Дж/кал)]'
Пр.=2,7.10-53=[Hg2+j [S2"],
где [Hg2+] и [S2~] — концентрации ионов Hg2+ и S2- в насыщенном растворе
HgS.
Растворимость HgS, следовательно, равна (2,7-Ю-53) '2=5,210~27 моль/л,
что соответствует диссоциации одной молекулы HgS в ~300 л раствора.
Очевидно, что столь низкую концентрацию прямо измерить невозможно] В связи с
высоким сродством белков и серосодержащих аминокислот к ртути в последнее
время проявляется усиленный интерес к ртутно-сериым комплексам; при этих
исследованиях намерение электрохимического потенциала оказывается
простейшим методом определения, какая из нескольких прочных связей ртуть — сера
является наиболее прочной и может, следовательно, определять содержание ртути в
организме.
Пример. Определение АНТХ и ASrx из температурной зависимости Етх
Поскольку соотношения, связывающие AGrx с Етх и AGrx с АНГх и ASrx
известны, ASrx и АНГХ из температурной зависимости Етх определяются следующим
образом:
&Grx = —zFE
гх
AGrx^AHrX— ГА5гхпри постоянной температуре (из табл. 1.3), так что
А5гх = -{dAGrxldT)P = zF (дЕгх/дТ)Р
(5.12)
(5.17)
и
АНТХ—AGrx+TASrx при постоянной температуре
или
АНГХ = -гЕЕгх 4 гРТ (дЕгх/дТ)Р
(5.18)
33 ТЕРМОДИНАМИКА В БИОЛОГИИ
1$ Таким образом, измерения Етх и его изменения с температурой позволяют
млределить все принципиальные термодинамические характеристики: AGr*.
Щпгх, Д5гх и К«, AGrx, AHrx и Д5Г, в уравнениях (5.12), (5.17) и (5.18)
приведены к 1 молю реагента.
кв.2. ПРЕПАРАТИВНЫЕ ПРИМЕНЕНИЯ: ЗЛЕКТР00САЖДЕНИЕ
;Й устройство аккумуляторов
В предыдущих разделах данной главы рассмотрены
электрохимические эксперименты, основной целью которых было
определение некоторых свойств (концентрации, Ка, AGrx„ AHrx, ASrx)
реакционной системы. В этом разделе кратко излагается несколько
^опытов, которые используются для получения вещества или
электрического тока (и, следовательно, энергии) с помощью укрупнен-
;яых электрохимических элементов. В табл. 5.2 приведены
стандартные окислительно-восстановительные потенциалы для ряда
широко используемых электродных реакций.
Пример. Электроосаждение как метод разделения компонентов смеси
В ч. 2 будет рассмотрена ионообменная хроматография, служащая методом
препаративного разделения смеси двух или. более ионогенных веществ. В этом
случае основным признаком, по которому осуществляется разделение, является
различие в зарядах, размере и/или в форме двух типов ионов. Электрохимические
элементы позволяют осуществить разделение по другому признаку, основанному-
на разности электрохимических потенциалов двух химически неидентичных ионов.
Например, рассмотрим раствор, содержащий ~0,1 моль/л Zn2* и Cd2+.
Из табл. 5.2 находим
Zn2+(lM)+2e —» Zn E°=—0,76В
и
Cd2+(lM)-{r2e —- Cd £°=—0,40В
Осаждение металлического Cd начнется (т. е. £V*>0) при внешнем
напряжении более отрицательном, чем
£=_0,40--°^-lg(l/0,l),
£ = —0,43 В
{относительно стандартного водородного электрода). Можно считать, что ион
Cd2+ удален количественно, когда внешнее напряжение достигнет значения, при
кото-ром {Gd2+] = 10_6 моль/л:
Е = — 0,40 ^i-lg(l/10-e)=—0,58В.
Наконец, ионы Zn2+ начнут осаждаться при лотенциале
£,= -0,76—^- lg(l/0,l) = -0,79B.
Таким образом, при напряжении на катоде от —0,58 до —0,79 В (относительно
стандартного водородного электрода) можно количественно осаждать иоиы
кадмия, не изменяя исходную концентрацию ионов цинка в растворе.
Таблица 5.2
Стандартные электродные окислительно-восстановительные потенциалы [6]
Электрод
Электродная реакция
Е°. В
Li | Li+
К|К+
Cs|Cs+
Ва | Ва2+
Са | Са2+
Na | Na+
Mg i Mg2+
Al | AP+
Zn|Zn2 +
Fe | Fe2+
Cd | Cd2+
Sn | Sn2+
Pb | Pb2+
Fe | Fe3+
Pb [ PbS04
Pt|D2|D+
Pt|H2jH+
Pt|Sn2+, Sn*
Pt | Cu+, Cu2+
Pt|S202-, S40|-
Cu | Cu2+
PtlI.II-
Pt | Fe(CN)*-, Fe(GN)|"
Pt | Fe2+, Fe«+
Ag | Ag+
Hg|Hg2+
Pt | Hg|+, Hg2*
Pt I Br21 Br-
Pt | Mn02 | Mn2*, Н+
Pt | Сг3+, Сг20?.-, Н+
Pt|Cl2|Gl-
Pt | Ce3+, Ce*+
PbS041 PbOs, 501", Н+
Pt | Co2+, Co3+
Pt | SO2-, S20i-
Pt | Ca | Ca(OH)21 OH-
Pt | H2P02, HPOf,-, OH-
Zn | ZnOfr, OH-
pt | so!-, s°4~« 0H"
Pt IH21 OH-
Ni | Ni(OH)21 OH-
Pb | PbC03 | C01"
Pt | OH", HOa
Кислые растворы
Li+ + e у—f Li
K++e <=± К
Cs+e ^=*: Cs
Ba2+ + e 4=fc Ba
Ca2+ -J- 2e -i—>• Ca
Na++e ^
Mg2* + 2e ^
Al3+ _f_ Зе ч=
Zn2+ +2«ч=
Fe2+ + 2e 3=
Cd2+ + 2e +
Sn2+ 4. 2e ^1
Pb2+ + 2e ч=
± Na
=fc Mg
=fc AI
=fc Zn
=± Fe
=fc Cd
=fe Sn
=±: Pb
Fe3+ 4.3e ~j—»• Fe
PbS04 +2e *=fc Pb +SO|-
2D+ 4.2e ^
2H+ 4. 2e ц
Sn*+ 4. 2e *
Cu2+ 4- e =?=£: Cu+
S402.- + 25=J=>: 2S201-
Cu2+ + 2e 1—>• Cu
I2 4.2e =?=±: 21-
Fe(CN)I-+e ^=fc Fe(CN)<
Fe3+ 4. e т—>- Fe2*
Ag+ + e =j=fc Ag
Hg2++2e =<==fc Hg
2Hg2+ + 2e 3F
Вг2 + 2е зр=ь
,4-
Hg|+
012 -j- ^c < <■ 2Br~
Mn02 4- 4Н+ 4. 2e -;—** Mn2+ + 2H20
Сг20|-4- 14Н+ +6e -<—>• 2Сг*++<7Н20
Cl2 + 2e -i—► 2C1-
Ce4+ + e 7—>■ Ce3+
Pb02 4- SOI" 4- 2e 4- 4Н30+ ч=Ь
T—»- PbS04 4. 6H20
Co«+ 4- e т—> Co2+
S20|" + 2e 7—>• 2S0J-
Щелочные растворы
Ca(OH)2 + 2e ^=± 20H" + Ca
HPO|- 4. 2e ч=Ь НгРОз" + 30H~
ZnOfr + 2H20 + 2e *==±. Zn 4.40H-
SOl" 4- H204- 2e ч=±: S0|- 4.20H-
2H20 + 2e *=z Ha + 20H-
Ni(OH)a + 2e -?—+ Ni + 20H"
РЬСОэ + 2e ^=fc Pb + C0|-
НОГ + H20 + 2e 4—»■ 30H-
—3,045
—2,925
—2,92S
—2,90
—2,87
—2,714
—2,37
—1,66
—0,765
—0,440
—0,403
—0,136
—0,12&
—0,036
—0,35
—0,0034
0,0000
+0,15
+0,153
+0,17
+0,337
+0.535S
+0,69
+0,771
+0,7991
+0,854
+0,92
+ 1,0652
+ 1,23
+1,33
+ 1,3595
+1,61
+1.6&
+1,82
+1,9»
—3,03
— 1,57
—1,216
—0,93
—0,828?
—0,72
—0,506
+0,88
ТЕРМОДИНАМИКА В БИОЛОГИИ
ffv Методы, аналогичные описанному, используются для разделения в растворе
1огих ионных и нейтральных соединений и их выделения. Поскольку скорость
;аждения (определяемую силой тока, проходящей по цепи), степень осаждения
зределяемую количеством электричества, проходящим по цепи) и селективность
данным соединениям (зависит от выбора внешней разности потенциалов, как
£; примере, приведенном выше) легко контролировать, выбирая подходящую силу
$&#а, длительность процесса и внешнее напряжение, электрохимическое осажде-
Йие представляет собой привлекательный метод препаративного получения
относительно чистого вещества из многокомпонентной смеси.
'." Многообещающим для практики процессом является электролиз воды, при
котором образуются газообразные водород и кислород:
t НгОжвдк ► H2(laTM)+1/202(iaTM) E°rx=— °.401 В.
I
!'... Поскольку главной проблемой потребления электроэнергии при
промышленном производстве является его неравномерность в течение суток, а наиболее
эффективный режим производства электроэнергии — постоянная мощность
производства энергии, было предложено использовать записанную выше реакцию для
«хранения» энергии [газообразный водород хранить при высоком давлении до
появления потребности в энергии, а затем в условиях, при. которых эта реакция
Самопроизвольно) идет справа налево, регенерировать затраченную для полу-
тения водорода (электро) энергию; для того чтобы не подвергаться опасности,
юаботая с Н2 при высоком давлении, наиболее вероятным реальным решением
|редставляется хранение Нг не в газообразном состоянии, а в каком-либо ином
Уиде: например, адсорбированным или в,виде твердого комплекса Нг с каким-
нибудь подходящим веществом].
пример. Аккумуляторы
if-!
f ■
*, Схема наиболее важного и одного нз самых простых аккумуляторов
показана на рис. 5.7. Потенциал цепи легко найти в табл. 5.2:
\ Катод: РЬ02 + 4H+ + 2е > РЬ2+ 4 2Н20
Катодная реакция сопровождается немедленным осаждением сульфата свинца:
РЬ*+ + SOf- *• PbS04
осадок
Анод: РЬ > РЬ2++ 2<?
Анодная реакция сопровождается немедленным осаждением сульфата свинца:
Pb2+ + SOl~ *■ PbS04
осадок
Суммарная реакция в элементе
РЬ 4 SOJ- »■ PbS04 4 2е Е° = -0,35 В
осадок
РЬ02 + 4H+ + SO|- 4 2е ► PbS04 + 2Н20 Е° = — 1,68 В
РЬ 4 РЮ2 4- 4Н+ 4 2SOJ- * 2PbS04 + 2Н20 Е° = — 2,03 В
Автомобильный аккумулятор обычно состоит из трех или шести ячеек,
соединенных вместе так, чтобы суммарное напряжение составляло ~6 или ~12 В.
Продуктом обеих реакций на катоде и на аноде является сульфат свинца PbSC»4,
который крепко пристает к электроду, так что, если аккумулятор разряжается
(весь свинец и диоксид свиица превращены в сульфат), ее можно перезарядить,
прикладывая обратное по знаку напряжение, несколько превышающее обычно
136 ЧАСТЬ 1
•g!> <Л ,/Я
Рис. 5.7. Схема свинцового аккумулятора [7].
Параллельно установленные свинцовые пластины чередуются с пластмассовыми. Свинцовые
пластины соединены последовательно через одну с помощью шины. Анодные пластины
покрыты тонкодисперсным металлическим свинцом, а катодные — диоксидом свинца такого
же качества. Вся система находится в пластмассовом кожухе, заполненном серной кислотой1
с концентрацией —3 моль/л.
создаваемое свежей батареей. При этом сульфат свинца превращается вновь в-
свинец (на аноде) или в диоксид свинца (на катоде). Если разрядка
аккумулятора продолжается долго, то сульфат свинца постепенно отделяется от
электродных пластин и накапливается на дне аккумулятора, после чего аккумулятор
больше ие может работать («перезарядка» такого аккумулятора невозможна).
Из-за постепенного электролиза воды в аккумулятор необходимо время от
времени добавлять воду, чтобы она покрывала электродные пластины.
Автомобильный аккумулятор очень тяжел, так как для производства
мощного тока, требуемого при запуске двигателя машины (см. задачи в конце
главы), необходимо большое количество свинца. Количество электричества,
запасаемого в аккумуляторе, удобно выражать в ампер-часах (см. задачи в конце
главы), а'количество энергии в ватт-часах. Наиболее серьезные препятствия для
использования свинцовых аккумуляторов в качестве источника энергии
электромобиля состоят в следующем: большая масса (причем обычные свинцовые
аккумуляторы «хранят» всего лишь ~25 Вт-ч на 1 кг массы батареи) и
неспособность выдержать несколько сотен циклов «зарядка—разрядка», которые
необходимы при эксплуатации электромобиля.
Анализ данных, приведенных в табл. 5.2, указывает на то, что наиболее
привлекательным элементом для. использован и я в качестве материала анода
для-аккумуляторов должен быггь литий, обладающий исключительно высоким
окислительно-восстановительным потенциалом и очень малой массой. Однако литий
реагирует с водой (I) и средой для аккумуляторов на основе литиевых
электродов до сих пор служили только органические растворители (при комнатных
температурах) или расплавленные соли (если рабочая температура батареи должна
достигать 400 °С). Совсем недавно в нескольких исследовательских центрах США
было обнаружено (в основном случайно), что можно создать новый тип
аккумулятора с литиевым анодом, в котором в качестве катодного реагента
используется, например, оксихлорид фосфора РОС13. Характеристики новых опытных
образцов превосходили более чем в 10 раз те же характеристики обычных
свинцовых аккумуляторов: например, на 1 кг массы аккумулятора новой серию,
приходилось ~500 Вт-ч. Однако простой расчет показал,'что если использовать
аккумуляторы на основе литиевых электродов в качестве источника энергии 20 млн.
Hf ТЕРМОДИНАМИКА В БИОЛОГИИ
Ввродских электромобилей, то не хватит всего количества лития, которым может
«аотолагать человечество даже к концу века.
|; Электрохимические элементы также предлагались в качестве средства
производства энергии взамен тепловых двигателей (устройств, превращающих тепло
§t?механическую энергию, а затем в электрическую), использующих органическое
купливо (природный газ, фракции иефти или каменноугольный газ). Схематично
йроцесс состоит в превращении исходного топлива в водород и диоксид
углерода'. Газообразный водород вводится в реакцию с атмосферным кислородом
(реакция, обратная электролизу), и выделяемая энергия используется для
производства постоянного электрического тока. Постоянный ток затем можно
преобразовать с помощью конвертора в переменный и с помощью трансформатора
увеличить напряжение до требуемого в линии лередачи. Утверждают, что с помощью
цраких элементов можно было бы увеличить к. п. д. использования топлива
приблизительно с 30% в обычных тепловых электростанциях более чем до 40%
благодаря ликвидации больших потерь энергии на трение движущихся деталей
$» обычном двигателе.
Задачи
k. Осмотическая концентрация солей в морской воде составляет ~ 1 моль/л.
■/.. Пусть осмотическую энергию, заключенную в 1 м3 морской воды, можно вы-
'i' свободить за Л с (например, добавив 1 м3 пресной воды к большому объему
'; морской).
а. Рассчитайте мощность (в ваттах), выделяемую в этом процессе.
б. Если течение всех рек в мире переносит ~ 10е м3 воды в 1 с, какая общая
мощность в принципе может быть получена при быстром добавлении соли к
речной воде («засоление», т. е. процесс, обратный опреснению морской воды)?
Сравните со средним уровнем современного мирового производства
электроэнергии, составляющим ~7-10и Вт.
в. Рассчитайте высоту столба воды, соответствующего осмотическому
давлению морской воды и затем энергию «засоления» воды сравните с высотой
водопада в устьи реки. (Устройство для извлечения энергии, соответствующей
осмотическому давлению морской воды, описано в работе \3].)
г. Д1ожно представить себе концентрационный элемент, с одной стороны
которого находится морская вода, а с другой—пресная вода, затем мы мржем
рассчитать электрохимический потенциал переноса солей из морской воды в
пресную (а не перенос воды в обратном направлении, как это делалось при
расчете осмотического давления). В цепи из нескольких элементов (рис. 5.8)
может быть получено максимально возможное напряжение
'макс
=(2NRT/F) ln((aCOJIb)MOpcK/(flc0jlb)
речи/»
где N — число пар мембран в диализной батарее. Считая, что морская вода
представляет собой 0,057 М раствор NaCl, а речная вода — 0,0259 М раствор
NaCl (предполагается, что оба раствора идеальные), рассчитайте Умакс при
20 °С для N=30.
д. Электроэнергию можно получить из этой системы, используя «нагруженный»
резистор RL, в соответствии с известным уравнением (W (Вт)—мощность,
/ (А)—сила тока, R (Ом)—сопротивление):
W^PR
или
I=V/R=VmKC/(R+R6aT)
Для #=8,8 Ом, #бат=20 Ом и Умакс из п. «г». Рассчитайте мощность,
производимую такой батареей.
В действительности мощность, выделяемая такими системами,
определяется скоростью прохождения воды (под действием осмотического давления) или
138 ЧАСТЬ 1
г
vww-
Морская
вода
Речная
вода
\
~\
Ч
Ма+- ►
:<ЬСГ
ffYf
Na+-
! <3tr=Cr
К
Ш
<*
Элюася
Рнс. 5.8. Схема диализной батареи. [8].
е — электрод, с н а — соответственно катноно- и анионообменные мембраны, R ^ —
сопротивление (нагрузка). Батарея непрерывного действия, в рабочем состоянии все ячейки
заполнены (морской и речной водой соответственно).
ионов (процесс, аналогичный протекающему в батарее) через
соответствующую полупроницаемую мембрану. Однако грубые расчеты свидетельствуют о
том, что к. п. д. таких систем таковы, что электростанция, использующая 10%
расхода воды реки Миссисипи с 25% к. п. д., могла бы производить 1000 МВт,
(сравнимо с самыми большими современными гидроэлектростанциями).
2. Покажите, что E°'=E°+(RTfzF)ln(lO-7)x, где Е° — стандартный электродный
окислительно-восстановительный потенциал относительно водородного
электрода с [Н+] = 1 моль/л в идеальном растворе; Е°' — стандартный
окислительно-восстановительный потенциал реакции при рН=7,0; z—число электронов,
переносимых при превращении 1 моля реагента,, х — число г-экв протонов,
вступающих в реакцию при превращении 1 моля реагента.
3. Рассчитайте электрохимический потенциал реакции
Zn2+(lM)-j-H2(l атм) ► Zn (металл)+2Н+(рН 5,76)
Zn2++2е —•* Zn Е°=— 0,763 В
4. Стандартный окислительно-восстановительный потенциал £° реакции с
участием красителя метилеиового синего составляет +0,53 В в реакции
ад
LH3C
V
/
голубой
s <CHl
i4
CH,J
++
+ 2H+ + 2e
н3сх
1_"з
\
/Г
-N
У
сн3
НаС A S H СНзл
бесцветный
+ +
39 ТЕРМОДИНАМИКА В БИОЛОГИИ
*.. ИЛИ
|( Dy(lM)+2H+(lM)+2e —► DyH2(lM)
V
I Если интенсивность синего окрашивания в растворе метилеяового синего сви-
!> детельствует, что доля синей формы красителя составляет 45%, электрохими-
■: ческнй потенциал раствора (относительно стандартного водородного
электрода) равен +0,04 В, рассчитайте рН раствора метиленового синего.
% Для реакции
Ox+z/2H2 —K,Red^zH+
уравнение Нернста принимает вид
E=E°+(RT/zF) ln([Ox]/[Red]).
Постройте график зависимости измеренного электрохимического потенциала Е
от доли молекул в восстановленном виде:
f _ [Red]
/Red — [Ox] + [Red]
для 2=1 и для 2=2. Покажите, как такую зависимость (кривая потенцномет-
рического титрования, примером которого служит обычное рН-титрование)
можно использовать для определения Е° и 2 реакции.
'6. а. Для запуска двигателя автомобиля свинцовый аккумулятор должен
выделить 150 А (1 А=Кл/с). Рассчитайте количество окисляемого свинца, -если
запуск двигателя длится 3 мин (обычные условия для эксплуатации
автомобиля в раннее утреннее время в холодное время года).
б..Если аккумулятор содержит 100 А-ч, каково минимальное количество
мелкодисперсного свинца (в граммах) должно быть запрессовано в поры катодной
пластины?
7. Для конкретного электрохимического элемента (для которого 2=2) £Vx =
= +0,425 В и общее изменение энтропии AS°x = + 17,2 кал-моль-1-град-1 при
30 °С. Рассчитайте общее изменение энтальпии в этой реакции и скорость
изменения э. д. с. с температурой (dErxfdJ).
Литература
1. Barrow G. M., Physical Chemistry for the Life Sciences, McGraw-Hill, New
York, 1974, Chapter 8. Хорошее изложение вопросов общей электрохимии,
в частности относящихся к равновесным процессам с переносом фосфата.
2. Skoog D. A., West D. M., Fundamentals of Analytical Chemistry, 2nd ed., Holt,
Rinehart & Winston, New York, 1969. Хорошее обсуждение общих вопросов,
посвященных ионоселективным электродам. Один из последних обзоров в
этой области — см. статью Rechnitz G. A., Chem. Eng. News, 27 Jan., 1975,
pp. 29—35.
3. Norman R. S., Science, 186, 350 (1974).
4. Alexander P. W., Rechnitz G. A., Anal. Chem., 46, 860 (1974).
5. Betso S. R„ Klapper M. H.t Anderson L. В., J. Am. Chem. Soc, 94, 8197
(1972).
6. Latimer W. M., Oxidation Potentials, 2nd ed., Prentice-Hall, Englewood Cliffs,
N. J., 1952.
7. Hutchinson £., Chemistry, W. B. Saunders Co., Philadelphia, 1959, p. 266.
8. Weinstein J. N., Leitz F. В., Science, 191, 557 (1976).
ЧАСТЬ 2
УСПЕХИ И НЕУДАЧИ
ГЛАВА 6
ЗАДАЧА О СЛУЧАЙНЫХ БЛУЖДАНИЯХ
Большинство физических и биологических проблем, если
отвлечься от специальной терминологии, удается с хорошим
приближением решить с помощью трех общих математических
подходов: дифференциального исчисления, вероятностного анализа и
линейной алгебры. Наиболее важные для биологии проблемы,
решаемые методами дифференциального анализа и линейной
алгебры, рассматриваются позже; в этой главе обсуждаются
случайные вероятностные процессы.
Знакомясь с историей математики, можно прийти к выводу,
что развитие теории вероятности получило толчок благодаря
решению задач, обусловленных широким увлечением азартными
играми при дворе короля Людовика XIV. После нескольких
проигрышей игроки быстро приходили к выводу, что в среднем
выигрыша можно добиться, заключая лари на то, что при четырех
подбрасываниях игральной кости появится по меньшей мере одна
шестерка. Интуитивно они ожидали, что при 24 бросках двух
костей можно рассчитывать на выпадение двух шестерок (т. е. 4:6 =
= 24:36). Однако во время игры обнаруживалось, что все
обстоит не так и, что в действительности более вероятно, что при 24
бросках две шестерки не появятся! Этот «игровой парадокс» был
впервые объяснен выдающимся французским математиком
Ферма; от решения этой задачи Ферма пошел гораздо дальше — к
созданию' математической теории вероятности.
Этот эпизод служит не просто забавным введением в теорию
вероятности — он показывает, что интуиция часто ведет в
ложном направлении, поэтому, как правило, большинство
интуитивных (т. е. нестрогих) выводов, которые будут рассмотрены ниже,
необходимо проверять на конкретном примере, прежде чем идти
дальше.
Перед рассмотрением задачи о случайных блужданиях
следует прежде всего понять, почему ею вообще стоит заниматься; для
этого существует несколько причин. Во-первых, из числа
немногих поразительно интересных физических задач данна,я является
особенной, поскольку имеет простое решение. Во-вторых, сам
процесс решения иллюстрирует большинство узловых моментов ста-
g-41 УСПЕХИ И НЕУДАЧИ
рристики и вероятности, которые могут затем проявиться и в
биофизических приложениях. Кроме того, решение этой задачи
демонстрирует отличный пример использования обоснованных
приближений; в противоположность общепринятым представлениям
большинство важных физических задач были решены не путем
сложных математических построений, требующих интеллектуальной
виртуозности, а с помощью разумных допущений. И наконец, решение
задачи о случайных блужданиях помогает понять физический
смысл таких различных биофизических процессов и явлений, как
"диффузия, радиоактивное излучение, электрический шум, а также
объяснить некоторые проблемы, возникающих при исследованиях
.размеров полимеров в растворе и интерпретации линий в
спектрах. Перейдем к решению задачи.
Большая часть математического анализа случайных событий
может быть сведена к двум аксиомам:
если Р(А)—вероятность* события Л,
Р(В)—вероятность события В,
А и В—независимые события (два подбрасывания одной и той
же монеты, радиоактивный распад двух нестабильных ядер и
т. д.),
то
Р(А и В)=Р(А).Р(В), (6.1)
где Р(А и В)—вероятность того, что произойдут оба события А
и В (например, распад обоих нестабильных ядер за время
наблюдения) .
Другая аксиома описывается уравнением
Р (А или В) ==Р (А)+Р (В), (6.2)
где Р(А или В) — вероятность того, что произойдет либо событие
А, либо событие В (например, произойдет распад либо первого,
либо второго нестабильного ядра). Обе эти аксиомы являются
просто формальными интуитивными утверждениями: вероятность,
что при первом подбрасывании «идеальной» монеты выпадает
«орел», а при втором подбрасывании — «решка», составляет Р
(«орел» сначала и «решка» потом) = 7г*72=74; аналогично
вероятность выпадения «орла» или «решки» при данном
подбрасывании составляет 72 + 72 = 1, т. е. можно утверждать, что в
идеальном эксперименте один из этих двух результатов будет получен.
* Под вероятностью конкретного результата понимают предположение о
наиболее вероятной доле большого числа попыток, в которых будет достигнут
конкретный результат. Например, вероятность выпадения любого конкретного
значения (например, 4) три одном броске игральной кости составляет '/в, так
как кость имеет шесть плоскостей и, следовательно, может быть получено шесть
эквивалентных результатов, из которых желательным является лишь один.
142 ЧАСТЬ 2
Наша задача будет состоять в применении этих идеальных
случаев к идеальной проблеме, а затем в объяснении и
доказательстве того, почему полученный результат применим к реальным
проблемам.
Пример. «Игровой парадокс»
Теперь можно объяснить игровой парадокс. Поскольку при одном броске
кости вероятность иевыпадения шестерки составляет 5/е, вероятность невыпадения
шестерки при 1, 2, 3. и 4-м бросках одной кости составляет произведение
вероятностей для (Независимых) отдельных бросков 5/б-5/б*5/б-5/б=0,48, т, е. <50%.
Таким образом, появление шестерки по крайней мере один раз при четырех
бросках характеризуется вероятностью (1—0,48) =0,52, т. е. >50%. Аналогично
вероятность невыпадения шестерки одновременно на двух костях при 24
независимых бросках двух костей составляет (а5/зб)24=0,51, так что вероятность
противоположного результата (т. е. что при 24 бросках выпадение хотя бы одного
«дубля» шестерок) составляет (1—0,51) =0,49, т. е, <50%.
Для извлечения максимальной (будущей) пользы из задачи
о случайных блужданиях вначале рассмотрим следующую
ситуацию.
Мешки и шары. Пусть имеется N мешков, в каждом из
которых содержится равное (большое) число белых и черных шаров
О Ф Ф
Ф О О
Мешок A/s/
>
Ф Ф О
О Ф О
Мешок №2
Ф О Ф
О ф О
Мешок /V!J
• • •
О О Ф
Ф О Ф
Мешо'кЛ'Ы
Теперь, допустим, вам завязали глаза и вы вытягиваете по
одному шару из каждого мешка (повязка на глазах гарантирует
независимость ваших действий). Найдем вероятность, что вы
вытащили т черных и (М—т) белых шаров. Для решения удобно
разбить задачу на две части.
Часть L Введем в задачу упрощение: мешки предварительно
разбиты на две группы из m и (N—т) мешков, так что надо лишь
вытащить черный шар из каждого т мешков, а из (N—т)
мешков— белый шар. Поскольку в каждом мешке число белых и
черных шаров одинаково, вероятность вытаскивания черного шара
при одной попытке составляет 7г.
Рассмотрим группу из т (отобранных заранее) мешков.
Вероятность того, что из первого мешка будет вытащен черный шар,
составляет '/г- Вероятность вытащить черный шар из второго
мешка также составляет '/г- Согласно аксиоме (6.1), вероятность,
что из первого и второго мешков будут вынуты черные шары,
составляет 1/2-1/г= (7г)2- Применяя аксиому (6.1) к третьему и всем
последующим действиям, легко показать, что вероятность того, что
•143 УСПЕХИ И НЕУДАЧИ
,>- ■ ■■
из всех т мешков будет вынуто по черному шару, составляет
(V2)m.
Аналогично вероятность вынуть по белому шару из (М—т)
мешков в другой группе составляет (V2)N~m. Наконец, поскольку
действия, совершаемые с двумя группами мешков, независимы,
аксиому (6.1) можно использовать еще раз, чтобы получить решение
Первой части исходной задачи, т. е. чтобы показать, что при
одной попытке вероятность извлечения черных шаров из каждого
из предварительно выбранных т мешков и белых шаров из
каждого из остальных (Л/"—т) мешков составляет ('/г)"** (lli)N^m-
Может показаться, что выбранный путь мучительно сложен, а
результат достаточно тривиален, но на самом деле таким образом
решена половина задачи о случайных блужданиях.
Часть 2. Осталось найти число способов, которым можно
выбрать m мешков из начального общего числа N мешков.
Во-первых, в общем случае число способов для того, чтобы отобрать один
мешок из N мешков, составляет
М1=М(М— \)(М—2).--3-2.1, (6.3)
где появление № объясняется тем, что существует М способов
отбора первого мешка, (N—1) способов — второго мешка и т. д.,
так что аксиома (6.1) прямо приводит к уравнению (6.3). И
наконец, можно прийти к уравнению (6.3) по-другому: находя число
способов составления двух групп из m и (N—т) мешков и затем
отбирая мешки по одному в каждой из групп:
N1 = (Число способов отбора мешков в группу из т
и в группу из (N—т) мешков)-т! (ЛГ—т)\
следовательно, число способов, какими можно составить из
мешков две данные группы, равно
т
т\ (N — т)\ '
Объединим обе части решенной задачи. Пусть
Рх(т)—вероятность вытаскивания т черных шаров при одной попытке, если
шары вытаскиваются по одному из каждого N мешков; тогда
Первый множитель в уравнении (6.4) указывает число
способов отбора т мешков в одну группу и (N—т) мешков в другую
группу, а второй множитель — вероятность того, что каждый шар,
вытянутый по одному из т мешков, — черный, а каждый шар из
(7V—т) мешков — белый.
Чтобы отвести любые сомнения, которые могли возникнуть в
ходе вышеописанного вывода, следует проверить правильность по-
144 ЧАСТЬ 2
лученного результата. Простой тест состоит в том, чтобы
удостовериться, что вероятность вытаскивания ни одного, или одного,
или двух, или трех и т. д., или N черных шаров составляет единицу
(один из этих результатов наверняка будет получен). С помощью
аксиомы (6.2) этот тест формально можно записать следующим
образом:
^(0)+^(1) + ^(2) + --+^(Л/)-2 Р„(т) = 1? (6.5)
Ответ на этот вопрос прямо вытекает из знакомого выражения
бинома
(а+Ь)» =а" +Na»-*b+ • • • + rtl(/iOT)l а"-»*»-|- • • • +6", (6.6)
если только обратить внимание на то, что уравнения (6.5) и (6.6)
одинаковы для случая
а=Ь = V„ (6.7)
так что
(a-f 6)" = 1" = 1, (6.8)
т. е.
S*atN = 1 (6.9)
т=0
что и следовало доказать.
Такой способ проверки часто называют нормировкой, и далее
он периодически используется.
Задача о случайных блужданиях молекулы в одномерном
пространстве заключается в том, что молекула может перемещаться
лишь вперед или назад в одном направлении с равной
вероятностью, и уравнение (6.4) характеризует вероятность того, что т
■перемещений из общего числа N перемещений направлено
вперед. Казалось бы, что задача решена, однако результат не удается
выразить в подходящей для практического применения форме —
обычное затруднение, возникающее три решении биофизических
задач. Как показано позже, переход от числа перемещений к
расстоянию несложен, более серьезную помеху представляют
содержащиеся в выражении факториалы.
Вычисление факториалов особенно усложняется при решении
биофизических задач, когда числа под. ними очень велики.
Поскольку молекула совершает миллиарды перемещений в секунду,
наблюдаемые характеристики (константа диффузии, скорость
седиментации, электрофоретическая подвижность) связаны с
величинами, усредненными по множеству молекул, и для их оценки
требуется интегрирование. Ясно, что интегрирование по т урав-
H5 УСПЕХИ И НЕУДАЧИ
нения (6.4) должно включать огромное число параметров, и для
расчета такого интеграла даже с помощью ЭВМ потребовалось
бы много времени. Однако при большом N несложный расчет
приводит к очень хорошему приближению для 1п(М) (см.
приложение, а также задачи в конце главы)
\n(N\)ttN\n(N)—Nt (6.10)
известному как приближение Стирлинга, и теперь вместо N
слагаемых в правой части уравнения (6.10) содержится всего два.
Это наглядное упрощение позволяет перейти от скучной задачи с
классическим решением [уравнение (6.4)] к рассмотрению
интересных молекулярных приложений.
Чтобы использовать приближение Стирлинга (6.10), надо
прологарифмировать обе части уравнения (6.4):
^)-ml/im)l(V2)" (6.4)
lnPM(m)=\nN\—\nml— \n{N— m)l+A/ln(V2) (6.11)
Тогда, согласно приближению Стирлинга, каждый из слагаемых
In {Ml), ln(m!) и In [(Л/"—m)\] можно представить в
соответствующей форме и подставить в уравнение (6.11):
\nPN{m)--=N\nN —mlnrn—(N— m)\n(N—m)+Л/In(V2) (6.12)
Теперь для получения искомого результата заменим N в
первом слагаемом уравнения (6.12) на [m+(N—m)]:
NlnN=mlnN+(N—m)lnN, (6.13)
так что
lnPN(m)=
=m(\nN—lnm)-f(W—m)[\nN—ln(W—m)]+ЛПп (V2) = (6.14)
-mln ^- + (^-m)ln¥^+^ln(V2). (6.15)
Во-вторых, используем свойство логарифмов (см. приложение)
\n(a/b) = — \n(b/a) (6.16)
применительно к первым двум слагаемым в уравнении (6.15):
\nPN(m) = -m)n |— (N—m)\n {N~m) -f#ln (V2). (6.17)
В третьих, предположим (доказательство будет сделано после
проведения всех преобразований), что m — среднее число черных
шаров, вынутых из N мешков, равное
m=W/2, (6.18)
146 ЧАСТЬ 2
поскольку вероятность вынуть черный inajp при одной попытке
составляет Уг- Введем новую переменную
х = т—т (6Л9)
и подставим m = #+(W/2) в уравнение (6.17):
1п^И=-(,+^),п(^±^)-
-(т--*)1п(—I— )+Ы1п^ <6-20>
Перепишем уравнение (6.20)
1пРЛ,И=-(д;+4)ь[4-( Ч~Т-)]-
-(4-)m[4(i-^)]+^m(J-) (6.2.)
и используем свойство
\n(a.b)=\na-\-\nb. (6.22)
Уравнение (6.21) можно упростить:
ш/>яи=-(*+4Н1+тг)-(т--*)1п(1—тг) (6-23)
Если подбросить монету, например, 1000 раз, число случаев
появления «орла», вероятно, будет близко к 500. Для
разбираемого примера с шарами и мешками аналогично при большом числе
вытаскиваний т близко к т, т. е.
-^=4«к <6-24>
![Используя приближения, не достаточно указать, что величина
мала, — надо определить, что она мала по сравнению с какой-то
другой, как это сделано в уравнении (6.24).]
Теперь вернемся к рассмотрению обоих логарифмических
членов в правой части уравнения (6.23). Для a<Cl ряд Тейлора (см.
приложение), на который можно разложить функцию 1п(1±а),
можно аппроксимировать двумя членами:
1п(1 ±а)«±а—£, (6.25)
Используя приближение (6.25) для преобразования уравнения
(6.23), легко показать, что
lnP„(m)=~^- (6.26)
147 УСПЕХИ И НЕУДАЧИ
ИЛИ
PN(m)=e~^iN\ (6.27)
Трудности, которые встретились при решении уравнения (6.4),
содержащего факториалы, удалось преодолеть с помощью двух
приближений (Стирлинга и Тейлора); однако необходимо вновь
проверить нормировку PN(m), которая могла пострадать в
результате использования этих приближений. Поскольку дискретные
переменные под факториалами заменены непрерывной переменной
х [уравнение (6.27)], сумму в уравнении (6.5) следует заменить
интегралом для проверки условия нормировки (см. рис. 6.1 и
относящееся к нему обсуждение). Нам надо найти постоянную А,
чтобы
ЛГ/2
A f PN(x)dx=lt (6.28)
—ЛГ/2
где х может изменяться от —N{2 до + /V/2, что соответствует
интервалу OssCmssCAf. Наконец, поскольку при больших х функция
ехр[—2х2/М] близка к нулю, область под этой функцией [т. е.
интеграл в уравнении (6.28)] будет перенебрежимо мала при
больших х и пределы интегрирования можно расширить до ±оо.
Используя формулу известного определенного интеграла (см.
приложение)
-{-оо
§<r-**dx=Y-T> <6-29)
—оо
легко оценить постоянную А:
-{-ос
A jV<2*W) dx== А ]/i^.= 1 (6.30)
или
-V
2
nN
(6.31)
и получить конечный результат
Р» М = УШ- е-2*"" - Уж '-' ^тт
(6.32)
6.А. РАВНЫЕ ШАНСЫ: ПЕРЕМЕЩЕНИЕ НА РАССТОЯНИЕ
Пусть молекула начинает двигаться из некоторого начального
положения и совершает N перемещений, причем каждое
перемещение с равной вероятностью совершается вперед или назад и
148 ЧАСТЬ 2
расстояние, на которое перемещается молекула за одно
перемещение, равно единице
■ 1 1 I 1 1 1 1 1 I I I I I 1 1! I ■
о
у (расстояние)
Рассмотрим ситуацию, когда молекуле разрешается
передвигаться лишь в одномерном (Пространстве. Задача состоит в том,
насколько далеко от исходного положения сместится молекула
через N (перемещений? Формально эта задача идентична только
что решенной, т. е. задаче о черных шарах, вынимаемых то
одному из N мешков, в каждом из которых находится равное
количество черных и белых шаров. Решение дается уравнением (6.32), и
для получения окончательного решения надо лишь заменить
переменные (число перемещений на пройденное расстояние). Пусть
даны:
'/ — длина одного перемещения,
v — число перемещений в секунду,
^продолжительность всех перемещений (в секундах).
Следовательно, общее число перемещений N составляет
N=vt (6.33)
Пусть т — число перемещений влеред из общего_ числа
перемещений N, (N-^m) — число перемещений назад, т — среднее число
перемещений вперед для множества перемещений N, равное
«в1Г' (6-34)
у — (суммарное) расстояние, лройденное за время t (т. е. в
результате N перемещений), тогда
y=mlf{N—m)(—l) (6.35)
Согласно уравнению (6.34), N=2m, так что уравнение (6.35)
можно переписать:
у=21(т—~т)
или
(6.36)
На этом этапе необходимо понять принципиальное свойство
вероятности, позволяющее из известных свойств лишь одной частицы
рассчитывать средние значения физических характеристик для
аолуляции из множества частиц. Какова физическая основа для
J49 УСПЕХИ И НЕУДАЧИ
а
PN(m)
PN(m)
(т;- I) mi(.ml + /)
J
/
f 1
^f
—-*"^
dm~^
«e-
m
/77;
"t
Рис. 6.1. Вероятность наблюдения дискретного (о) и непрерывного (б)
результата т* (см. текст).
перехода от суммы (дискретных величин) к интегралу
(непрерывной величины)? Ответ можно сразу же получить, если обратиться:
к рис. 6.1.
При регистрации дискретного результата (числа,
выпадающего на игральной кости, «орла» или «решки» на монете) значение
Ря(т) характеризует вероятность обнаружения этого
конкретного результата. Таким образом, если число вариантов — конечная
величина, то вероятность выбора любого из них строго
определена. Однако если регистрируемый результат — непрерывно
изменяющаяся величина (функция), например расстояние, то вероятность
нахождения частицы на каком-либо одном фиксированном
расстоянии равна нулю (поскольку число возможных локализаций"
бесконечно), и вместо этого надо искать вероятность локализации
частицы в конкретной области. На рис. 6.1, а площадь
заштрихованной области равна
Площадь=Р^ (mt) » [{тг -\~l)—mi\=PN (mt). (6.37)
Физический смысл этой величины (площади) — вероятность
наблюдения результата в области т/^т<С,тгЧ-1). При
исследовании дискретного результата такое определение неинформативно»
но по аналогии теперь можно рассматривать (бесконечно малую)
площадь, выделенную на рис. 6.1, б, которая характеризует
вероятность обнаружения результата в интервале т^тКтг+йт.
Поэтому для непрерывно изменяемых экспериментальных
параметров
Pjj (m) dm = Вероятность наблюдения результата в
интервале от т до (т + dm)
Наконец, так же как среднее любой дискретной величины
выражается суммой
т=тй• PN (m0)-f mx• PN К)+"Л(щ)-\ \-mN• PN(mN) (6.38)
150 ЧАСТЬ 2
среднее непрерывной переменной выражается интегралом
(6.39)
N
(т) = \ mPN (m) dm (среднее значение т, если т—непре-
J рывный параметр)
рывный параметр)
Пример. Среднее значение дискретного наблюдаемого параметра
Пусть подбрасывают две игральные кости, и необходимо предсказать
средний результат для большого числа таких опытов. Существует 6-6=36 возможных
результатов (шесть значений на одной кости умножить на шесть на другой
игральной кости). Вероятность появления двух единиц составляет Узе, вероятность
получения на двух костях в сумме тройки — 2/3б (2 на первой и 1 на второй
кости или наоборот) и т. д. для всех остальных вариантов. Рзб(2) = 1/зб),
^Рзб(3)=2/зб и т. д., и для получения среднего значения надо найти сумму
произведения каждого параметра на вероятность:
т=2( !/„) + 3(*/зв) + 4(3/36) + 5(V36) + 6(5/3в) + 7(в/зв) +
+8(6/зв) +9(*/м) + Ю(э/36) +11 (2/зв) +12(V*) =252/36 - 7.
Пример. Среднее значение непрерывного параметра:
среднее число перемещениий вперед при случайных блужданиях
Если перемещения осуществляются путем случайных блужданий и число
таких перемещений достаточно велико, то число перемещений вперед т можно
рассматривать как непрерывно меняющийся параметр. Тогда, согласно уравнению
(6.32),
PN(m)dm=^Y-^re"2<m~<N/2)yN) dm =
=вероятность того, что после N
перемещений молекула
совершила от т до (m-f-dm)
перемещений вперед I (6.40)
Среднее, число перемещений вперед тогда определяется из уравнения (6.39):
ЛГ
(т) = Уж Jme_2 (m~(M/2))2'Ndm. (6.41)
m=0
Поскольку из известных формул для определенных интегралов самой
близкой к уравнению (6.4!) является формула
со
1
xe-x2dx,
то представляется разумным заменить переменные
x=m—(N/2), (6.19)
dx=dm.
151 УСПЕХИ И НЕУДАЧИ
Тогда
+ЛГ/2
—N/2 — JV/I
+ЛГ/2
e-wtdx].
(6.42)
Наконец, поскольку N велико и при больших т площадь под функцией
ехр{—2>[т—(W/2)12/jV} пренебрежимо мала, допустимо расширить пределы
интегрирования в уравнении (6.42) до ±<х>. Первый интеграл в уравнении (6.42)
исчезает (подумайте почему), а решение второго интеграла дает уравнение (6.30),
т. е., как и ожидалось,
<m> = N/2
(6.43)
Средний разброс числа перемещений вперед выводится так же просто.
Поскольку молекула с равной вероятностью совершает больше, или меньше
перемещений вперед по сравнению с Af/2.
< (m—<m» > =<m>— <m>=0.
Однако в действительности необходимо знать абсолютный разброс числа
перемещений вперед (т. е. число перемещений вперед, отличающееся от (т),
больше или меньше (т)), описываемый уравнением (см.. задачи в конце главы)
(6.44)
где
Y4-
среднеквадратичное отклонение от числа перемещений вперед.
Возвратимся к задаче о случайных блужданиях. Если
Pn(m)dm— вероятность того, что молекула совершила от т до
(m+dm) перемещений, a Pt(y) — вероятность того, что та же
молекула за время t оказалась в положении между у и (y-\-dy), то
PN(m)dm=Pt(y)dy, (6.45)
поскольку для передвижения на расстояние у совершается т
перемещений. Используя уравнения (6.36), (6.40) и
dm = {l/2l)dy (6.46)
и заменяя переменные, уравнения (6.32), (6.36), (6.45) и (6.46)
объединяют:
^^/w^^*
(6.47)
Из уравнения (6.47) легко показать (см. задачи в конце
главы) , что
<0> = О, (6.48)
352 ЧАСТЬ 2
Рис. 6.2. Трехмерное случайное блуждание.
ИЛИ
V<yz>=iVu'.
(6.49)
Хотя преобразования, приведшие к этой важной формуле
(6.49) > основаны на рассмотрении «блуждания» в одномерном
пространстве, можно показать, что подобное же уравнение
Описывает «блуждание» молекулы в трехмерном пространстве в
направлениях х или у или z, если длина одного перемещения /
У (г*) = I YN
(6.50)
где результат конкретного «передвижения» показан на рис. 6.2.
Далее уравнение (6.50) применяется для предварительной
оценки размера растворенных молекул полимеров.':
Размер растворенных макромолекул
Для описания формы моЛекул синтетических или природных
биологических полимеров в растворе простыми и наиболее
полезными моделями являются сфера, эллипсоид, жесткая палочка и
статистический клубок (см, также обсуждение седиментации и
вязкости, помещенное ниже в этой части).
Для того чтобы получить представление о форме
статистического клубка, надо зафиксировать один конец 'полимера в
исходной точке и затем локализовать каждую последующую связь в
цепи путем случайного трехмерного перемещения от исходной
точки; следовательно, уравнение (6.50) описывает среднее расстояние
между концами цепи при условии, что / — длина одной связи, а
N— число связей в полимерной цепи. Существует ряд методов,
153 УСПЕХИ И НЕУДАЧИ
например светорассеяние, позволяющих выбрать
наиболее.вероятные среди возможных вариантов конфигурации данной
макромолекулы. К числу молекул, точно описываемых с помощью
перечисленных выше моделей, относятся: инсулин (сфера), гаммаглобу-
лин (эллипсоид), вирус табачной мозаики (жесткая палочка) и
натуральный каучук (статистический клубок). Проявление
особого внимания к форме молекул обусловлено тем, что биологические
функции большинства макромолекул тесно связаны с трехмерным
расположением длинных цепей, из которых построен биополимер.
Развертывание двойной спирали ДНК можно, например, описать
переходом жесткой палочки в статистический клубок, и этот
переход характеризуется параметром, подобным параметрам
обычных фазовых переходов, например температуре плавления льда_
Причиной инактивации ферментов йод воздействием
экстремальных рН, температуры или концентрации соли часто являются
нарушение при этом высокоорганизованной структуры
макромолекулы и переход ее в более статистическую структуру. Модель
статистического клубка может использоваться при исследованиях:
структуры полимеров в растворе. Если известна молекулярная
масса (и, следовательно, число связей в цепи) полимера, то
можно рассчитать размер соответствующего статистического клубка;
затем полученный результат сопоставить с экспериментально
определенным и на основании этого делать выводы о силах,
удерживающих вместе сегменты полимерной цепи.
Полиизопрен (натуральный каучук) состоит из длинных це*
пей, содержащих от 500 до 5000 изопреновых звеньев
СН3 "I
—сн2-с=сн-сн2—\N
и ведет себя в растворе как идеальный статистический клубок. Наг
первый взгляд такие свойства каучука трудно объяснить,
поскольку для любого реального полимера при случайном «блуждании»
существуют определенные нереализуемые локализации
сегментов, потому что две связи в данной цепи не могут занимать одну
и ту же область пространства. Для более простого двухмерного*
случайного «блуждания» такая ситуация показана на рис. 6.3.
Ясно, что не все конфигурации, возникающие при «случайном
блуждании», могут реально существовать; при отборе /вероятных
структур остаются такие, которые характеризуются средним
расстоянием между концами цепи, существенно превышающим
величину, характерную для истинного статистического клубка (при-
строгом расчете оказывается, что оно завышено иа ~20%).
Аномальное поведение полиизопрена впервые было объяснено Флори,
предположившим, что силы притяжения между сегментами цепи-
превышают силы отталкивания; в натуральном каучуке эти силы
достаточно сильны для того, чтобы действительное расстояние
154 часть 2
\ ^ >
~1_П
-lH
^
^ и3
Рис. 6.3. Иллюстрация «исключенного объема» в двухмерном полимере со
статистически возникшей конформацией [31.
Поскольку два атома не могут занимать в пространстве одно и то же положение,
некоторые конфигурации при идеальном статистическом перемещении невозможны. Остальные
конфигурации характеризуются средним расстоянием между концами цепи, которое
больше, чем если бы рассматривались все конфигурации (см. текст).
между концами цепи соответствовало величине, характерной для
идеального статистического клубка.
Натуральный латекс представляет собой аморфное и
пластичное вещество, но после обработки его однрхлористой серой S2CI2
между цепями образуются поперечные сшивки, расположенные
далеко друг от друга:
сн3
—сн2—с—сн—сн2—
н
ci к
—СН— С— СН—СН2—
сн.
i
V.
у
и изопреновые связи больше уже не могут далеко
«проскальзывать» друг относительно друга; полимер приобретает способность
155 УСПЕХИ И НЕУДАЧИ
к обратимому растяжению — свойство, необходимое для его
применения в качестве материала автомобильных шин. Впервые
поперечное сшивание было осуществлено путем обработки серой при
нагревании (поэтому такой процесс называется вулканизацией).
Аналогичный принцип лежит в основе укладки волос «перманент»:
повышенная температура способствует каталитическому
образованию дисульфидных связей, благодаря чему повышается
жесткость волос, состоящих из белковых молекул.
В частности, для синтетических полимеров расчеты на модели
статистического клубка позволяют исследовать влияние
растворителя на полимеры. В «хорошем» растворителе, связывающимся с
полимером на большом числе сегментов, полимер набухает (т. е.
принимает форму, описываемую большим средним расстоянием
между концами цепи), поскольку полимерная цепь уже не можег
свернуться так же, как в отсутствие растворителя. Аналогична
«плохой» растворитель приводит к сжатию цепи, обусловленному
силами отталкивания между растворителем и поверхностью
макромолекулы.
Аналогичные эффекты характерны и для более типичных
биологических полимеров, в цепи которых находится множество
заряженных групп. Действие зарядов, как правило, приводит к
стабилизации (обычно жесткой) структуры ферментов и нуклеиновых:
кислот путем образования электростатических связей между
разными сегментами молекулы. Введение высокополярных или
заряженных соединений, таких, как мочевина, гуанидин, соль или
детергент, вызывает разрушение этих электростатических связей и
создает возможность для разворачивания биомакромолекулы.
Например, в высококонцентрированном солевом растворе размер-
натриевой соли карбоксиметилцеллюлозы (заряженная
макромолекула) совпадает с размером целлюлозы (незаряженная
макромолекула) . В последнее время обнаружено, что для получения
компонентов биологических мембран при выделении мембранносвя-
занных ферментов и рецепторов большое практическое значение
имеют детергенты. Использование детергентов основано также на
Их способности связываться и разворачивать большинство молекул
белков. Белки принимают при этом форму жесткой палочки,
длина которой коррелирует с молекулярной массой полипептида.
Большое затруднение при определении молекулярной массы
белка вызывает то обстоятельство, что почти все способы
определения зависят от формы белковой молекулы; введение в раствор
белка детергента или гуанидингидрохлорида приводит к
разворачиванию большинства полипептидов и переходу в общую для них
форму (жесткой палочки или статистического клубка); затем
полипептиды можно сравнивать только на основании данных о размере
{молекулярной массе).
156 ЧАСТЬ 2
6.Б. ТРАНСЛЯЦИОННАЯ ДИФФУЗИЯ
Трансляционная диффузия вещества через границу раздела
затрудняет, а иногда, напротив, является полезной во многих
биофизических измерениях. Для понимания принципиальных
закономерностей, на которых основано разделение компонентов
смеси в ультрацентрифуге, при хроматографии и электрофорезе,
обратимся к процессу диффузии. При иммунологическом анализе
распространение антигена по агаровой пластине осуществляется
путем диффузии. Скорость диффузии можно использовать для
расчета молекулярной массы крупных молекул, что представляет
одну из основных (и наиболее сложных) задач в биохимии.
Наконец, прежде чем рассматривать активный перенос через
мембранные границы раздела, необходимо познакомиться с процессами
пассивной диффузии.
Вначале выполним не очень строгий вывод уравнений
диффузии для того, чтобы можно было сравнить эти уравнения со
строго полученным решением задачи о случайных блужданиях.
Особенно полезны эти уравнения три последующем обсуждении
электрофореза и седиментации, при которых помимо диффузии
через границу раздела имеет место перемещение самой границы
под действием электрического или центробежного шля.
Вывод уравнения диффузии
Физические законы .представляют собой интуитивные
заключения, которые нельзя вывести из более простых утверждений и
-следствия из которых не (противоречат эксперименту. К числу
таких заключений относятся законы механики и термодинамики;
таков же и закон потока Фика. Фик. предположил, что поток,
вещества из области с одной концентрацией в область с другой
концентрацией пропорционален разности этих концентраций по
аналогии с потоком тепла,, пропорционального разности; температур
Хталец быстрее.замерзает в жидком азоте,, чем. в ледяной воде!).
Поток = — D (-£- ] • (Площадь поперечного сечения, (6.51)
^ у ' через которую проходит поток),,
•где Z) — коэффициент пропорциональности; перед выражением
стоит знак минус, поскольку поток направлен в направлении
уменьшения концентрации; символ производной указывает, что закон
распространяется на бесконечно малое изменение в концентрации
При переходе границы раздела. Уравнение (6.51) записано в
гораздо более общем виде, чем требуется для рассматриваемых
•здесь применений: подобное же уравнение описывает
пропорциональность потока тепла градиенту температуры,
пропорциональность потока жидкости градиенту давления или
пропорциональность потока зарядов градиенту напряжения (электрического
«давления») .
157 УСПЕХИ И НЕУДАЧИ
* s
*~ Начальное состояние}
Поток наружу
Рис. 6.4. Эксперимент для исследования диффузии.
В нулевой момент времени все вещество находится в тонком слое прн у=0. Уравнение
(6.52) описывает скорость изменения концентрации вещества в интервале у—(y+dy) к
моменту времени t. Принимается, что исследуемый слой занимает единичную площадь.
Для проверки интуитивного утверждения, описываемого
уравнением (6.51), достаточно рассмотреть ситуацию при диффузии,
которая шрямо сравнима с задачей о случайных блужданиях.
Допустим, что в нулевой момент времени бесконечно тонкий слой
красителя окружен с обеих сторон растворителем; через
некоторое время краситель распределится в растворителе и его
концентрацию в любом тонком слое можно определить (например)
путем измерений оптической плотности растворов. Уравнение (6.51)
выражено терминами потока, а не общего количества вещества, но
можно рассмотреть скорость изменения концентрации в любом
Выбранном тонком слое (рис. 6.4). i
Обратимся к рис. 6.4 и рассмотрим область в интервале от у
до {y-\~dy). Для удобства предположим, что площадь
поперечного сечения данного тонкого слоя равна единице, так что объем
составляет (с1уЛЛ)=йу. Скорость изменения концентрации в слое
158 ЧАСТЬ 2
со временем выражается разностью потока красителя в
рассматриваемую область и из нее:
дс Поток внутрь (моль/с)—Поток наружу (моль/с) >■ , _, _1ч
Ж Объем зоны (см*) моль-(см «с ),
дс_ __ \ ду )у~т~ \ ду jy+dy /g ^2)
dt ду \ • /
Здесь вновь возникает та же трудность, что и раньше при
решении задачи о случайных блужданиях: результат описан точно,
но выражен неудобно. Выбор бесконечно малой области можно
сделать полезным следующим образом. Если концентрация в
момент t является функцией расстояния, то градиент концентра-
дс
ции -5— также зависит от расстояния, допустим как f(y). Тогда в
пределах любого малого интервала функцию можно
охарактеризовать ее значением и значением ее производных в близких
точках, используя ряд Тейлора (см. приложение):
/ (У)УЧ (Уо) +Г (У)* (У-Уо) +{^]н-(У-УоГ+ ■ • • ■ (6-53)
дс
Уравнение (6.53) можно применить для оценки -%- в
интервале y+dy
Замещая в уравнении (6.52) правую часть уравнения (6.54)
на h^L^. получаем
дУ1
V+dy
_дс _ д%_
dt'~D dif
уравнение диффузии (6.55)
Уравнение (6.55)—это известное уравнение'диффузии: оно
относится к числу наиболее важных уравнений физики,
большинство из которых приводится в этой книге. Для решения уравнения
(6.55) требуется использовать специальные методы
(разработанные Фурье); описание которых здесь опущено; получаемый
результат имеет простой вид:
°(У) _ * е-мш
с (0) 2 VnDt
(6.56)
где с(у)—концентрация вещества на расстоянии у в момент tr
а с(0)—исходная концентрация вещества в точке начала
отсчета в нулевой момент времени.
159 УСПЕХИ И НЕУДАЧИ
Насколько хорошо уравнение (6.56) согласуется с данными,
получаемыми при экспериментальном исследовании диффузии,
показывает рис. 6.5. Легко показать (см. задачи в конце главы), что
^расстояние между точками изгиба, согласно теоретическому
уравнению (6.56), составляет 2\2Dt. Кроме того, поскольку левая
часть уравнения (6.56) соответствует вероятности обнаружения
молекулы данного красителя в точке у, абсолютное значение
среднего расстояния от исходной точки в момент времени t можно
найти из уравнения
+00
<У2> =
^а*)*-"""*'
(6.57)
(или просто
V(y2> = Vwt
(6.58)
Но ситуация, описываемая уравнением (6.56) и показанная на
рис. 6.5, идентична той, которая рассматривалась при решении
задачи о случайных блужданиях:
У(у*у = /м2 - Уш*
(6.59)
где N — общее число перемещений, / — длина одного
перемещения, v — число перемещений в единицу времени, t — длительность
'Ш перемещений.
\ Из уравнений (6.58) и (6.59) следует
D = -k~vP (в одномерном пространстве)
(6.60)
? Распространение диффузионной задачи на двух- или
трехмерное пространство сопровождается довольно громоздкими
алгебраическими преобразованиями, хотя в основе лежит подход,
совершенно аналогичный одномерной задаче (см. задачи в конце
&лавы) и приводит к формулам
D = (-т- I v/2 (диффузия в двухмерном пространстве)
D = I -г-1 v/2 (диффузия в трехмерном пространстве)
(6.60а)
(6.606)
|i. Уравнения (6.60) — (6.606) описывают связь между
интуитивной (эмпирической, физической) моделью, основанной на непре-
160 ЧАСТЬ 2
Расстояние
Рис. 6.5. Диффузия СО-гемоглобина в Н26
из тонкого слоя [5].
Сплошная кривая построена теоретически по
уравнению (6.56).
рывном (потоке, и
дедуктивной (микроскопической,
статистической) моделью,
основанной: на множестве
малых случайных
(перемещений. Причина острого
интереса физиков к
'Статистическому .анализу обусловлена
тем, что точти !все
физические модели 'строятся с
учетом поведения одной или
двух частиц в данный
момент. Для 'применения
расчетов, сделанных на таких
(моделях, к
макроскопическому эксперименту с
участием огромного числа
частиц 'всегда требуется
провести какое-либо урреднение
[уравнения (6.49) и (6.57)].
Проблема диффузии
'Относится к числу тех немногих проблем, для которых можно
установить строгое соответствие между (микроскопической) моделью и
(макроскопической) реальностью.
На данном этапе разумно задать вопрос, почему молекулы
должны вести себя так, как если бы они очень быстро и
хаотично перемещались. Чтобы этот вопрос был менее гипотетическим,
можно рассмотреть движение частиц пыльцы, легко наблюдаемое
с помощью обычного микроскопа (броуновское движение).
Детальные эксперименты показали; что статистическое движение
таких частиц нельзя объяснить неравномерным испарением
растворителя, капиллярными силами, действующими в краевых
точках, кавитацией (образованием мелких пузырьков),
неравномерным нагреванием, силами притяжения между частицами или
каким-либо внешним влиянием. Объяснение этого явления состоит в
том, что любая данная частица со всех сторон бомбардируется
молекулами растворителя и, хотя в среднем результирующая сила
равна нулю, т. е. частица остается на своем исходном месте, в
любой данный момент времени суммарная сила, действующая на
частицу, может быть не равной нулю и, таким 'образом, заставляет
ее передвигаться. Статистическое движение молекул (частиц)
надо принимать во внимание при рассмотрении любых химических
реакций (молекулы должны столкнуться прежде, чем они Смогут
прореагировать). Обсуждая действие ферментных катализаторов,
ускоряющих реакции, диффузия на короткие расстояния будет
детально анализироваться, чтобы определить, как часто
сталкиваются реагирующие молекулы друг с другом.
[61 УСПЕХИ И НЕУДАЧИ
Применения. К
.принципиальным применениям
уравнения диффузии (6.55) отно'сят-
гя: 1) диффузия из точки, 2)
диффузия из одного слоя в
другой и 3) диффузия через
мембрацу. Диффузия из
одного слоя в другой происходит
1ри хроматографии,
электрофорезе и седиментации и
рассматривается в соответствую-
ецих(разделах книги и
несколько позже в этом раздел е.
Диффузия через мембрану
обсуждалась (п,ри анализе равнове-
аия Доннана (гл. 2), а здесь
ниже приведены \два примера
диффузии из точки,
постоянно встречаемые в
иммунологии.
Пример. Иммунодиффузия
/ Иммунная система человеческого организма основана на существовании
ряда больших белков (антител), циркулирующих в крови и проявляющих
тенденцию локализоваться в определенных органах. Функция этих антител — опознать
йаобые чужеродные (и предположительно вредные) токсины или антигены и
вывести их из циркуляции путем образования прочного комплекса антиген —
антитело. Например, бактерии опознаются по характерным олигосахаридным
группам, находящимся на их поверхности; антигены» белковой природы — по
характерным олигопептидным фрагментам в данном белке и т. д. Большинство имму-
^охимических исследований направлено на изучение процессов образования
комплексов антиген — антитело, часто сопровождающихся осаждением
комплекса, как, например, при стандартных тестах групповой специфичности крови.
■Л" Чтобы понять суть метода иммунодиффузии, надо рассмотреть влияние н»
растворимость комплекса антиген — антитело соотношения концентраций этих
Двух веществ. Как видно на рис. б.б, если антитело или антиген находится в
избытке, осаждение происходит в очень незначительной степени.
Одно из простых объяснений этой ■зависимости (рис. 6.6) иллюстрируется
фсемой, приведенной на рис. 6.7. При малых соотношениях антитела к антигену
[образуются растворимые комплексы антиген — антитело, для которых характер-
;йо низкое содержание в комплексе и антигена, и антитела (рис. 6.7, г, дне).
#1ри больших значениях отношения антитело : антиген полифункциональная
природа антитела позволяют образоваться крупным агрегатам (преципитатам),
примем наиболее компактные Соответствуют наибольшим значениям отношений
антитело : антиген.
Важно подчеркнуть, что, когда раствор антитела приводится в контакт с
„Раствором антигена, подоса преципитации наблюдается только при соотношениях
(ёнтитело :антиген, близких к положению максимума на кривой рис. 6.6.
;\ Радиальная иммунодиффузия. В клинической иммунологии обычное
диагностическое измерение состоит в определении количества индивидуальных гамма-
даобулинов (иммуноглобулинов) в сыворотке крови. При стандартном анализе в
.Маленькую чашку Петри наливают растворы агар-агара (отрицательно заряжен-
Увеличение количества
добавленного антигена при
постоянном количестве антисыворотки
.....
} Антиген
) Антисыворотка
Количество добавленного антигена
Рис. 6.6. Типичная кривая преципитации»
при добавлении антигеиа в раствор с
фиксированным количеством антител-
162 ЧАСТЬ 2
а
9 Антиген (Ао)
^ Антитело (АЬ)
б
/JpuucmumambtNa: Ag = 4,3
АЬ:Ад = 3,0
Растворимые АЫАд = 0 75
комплексы (Аь3Ад4')
АЬ.'Ад = 0,67
(АЬ2Ад3)
АЬ.'Ад = 1,1
^ t
Ab:Ag = 0,5
(АЬАд2)
Рис. 6.7. Схематическое изображение особенностей пространственной структуры
■комплексов антиген—антитело (Ag — АЬ) [7].
«а — антитела в избытке; б — максимальное количество преципитата; в—е — антиген в
избытке.
ного полисахарида) и антисывороткн (антитела), специфической к исследуемому
иммуноглобулину (антигену); при стоянии 1%-ная смесь антитела с агаром
превращается в относительно жесткий гель (диффузия в такой среде происходит с
небольшой контролируемой скоростью). Затем в пластине геля вырезают
небольшую лунку и в нее вводят сыворотку, содержащую иммуноглобулин. По
мере радиальной диффузии из лунки специфического к данному антителу антигена
в геле становится заметным «кольцо», положение которого соответствует
оптимальному для преципитации отношению концентраций антитела к антигену
(рис. 6.8). Поскольку диаметр кольца растет с увеличением концентрации
антигена (см. задач» в конце главы), этот тест позволяет быстро, точно и
экономично измерить количество гаммаглобулина в исследуемой сыворотке. С помощью
разных антител можно определить содержание соответствующих антигенов. Если
Рис. 6.8. Определение иммуноглобулина методом радиальной иммунодиффузни
(фото) Г81.
Диаметр кольца преципитации характеризует концентрацию иммуноглобулина (см. текст).
163 УСПЕХИ И НЕУДАЧИ
Антигене! Антиген №
Г\
Антитела к антигенам //? и А/2
а
А
6
Рис. 6.9. Типичные результаты, получаемые при иммунохимическом анализе
двойной диффузией в геле.
Две верхние луики заполнены растворами разных антигенов, нижняя — раствором
иммуноглобулинов (антител).
а — серологическая идентичность: иммуноглобулины взаимодействуют с обоими антигенами
одинаково; б — частичная серологическая идентичность: один антиген взаимодействует с
Ъбоими антителами, а другой антиген — лишь с одним антителом; в — серологическая пе-
идентичность: взаимодействие каждого антигена с антителом независимо. (См. текст.)
ч:месь антител вводят в агар еще до начала гелеобразования, то одновременно
появляется несколько колец; однако для оценки гетерогенности фракции антител
более удачным является метод Ухтерлони.
Двухмерная диффузия (метод Ухтерлони). Одной из стандартных задач,
Стоящих перед иммунологом, является определение иммунного подобия между
антигенами из двух различных источников. При создании вакцил или
антисывороток важно установить аналогичность взаимодействия антигена с антителом для
■обеих сывороток, чтобы удостовериться в том, что в них нет нежелательных
комбинаций компонентов. Для стандартного метода, используемого для этой цели»
вновь необходим тонкий агаровый слой на стеклянной пластинке, однако без
предварительного введения антитела. В геле вырезают три маленькие лункйу
располагая их в вершинах равностороннего треугольника (рис. 6.9); в нижнюю
лунку вводят антитела, а в верхние лунки помещают соответствующие
исследуемые антигены.
Большинство наблюдений соответствует трем возможным ситуациям:
A. Серологическая идентичность. Если антитело не может отличить один
антиген от другого, образуется непрерывная дуга преципитации (рис. 6.9, с).
Б. Серологическая неидентичность. Если два антигена реагируют с антитела*
ми совершенно различно (т. е. реакции не имеют ничего общего), то
появляются две дуги преципитации (рис. 6.9, в).
B. Частичная серологическая идентичность. Присутствуют два типа антител:,
оба антитела образуют преципитат с антигеном слева и лишь одно —
преципитат с антигеном справа (рис. 6.9,6). В эксперименте правый антиген,
преципитирует со своим антителом, а другое антитело распространяется
через область преципитации до контакта с левым антигеном.
Понятно, что дуга преципитации выпукла по направлению к лунке,
содержащей молекулу (антитела) с самой низкой скоростью диффузии. Следовательно,.
^Прямая полоса преципитации появляется, если антиген и антитело имеют прибли-
364 часть 2
зительно одинаковую молекулярную массу (позже будет рассмотрена
количественная взаимосвязь скорости диффузии и молекулярной массы), н это полезно
для оценки молекулярной массы антигена, поскольку большинство антител
имеют приблизительно одинак'овую молекулярную массу (~ 150 000).
Методы иммунодиффузии исключительно чувствительны: с помощью
радиальной иммунодиффузии можно обнаружить ^ 3 мкг/мл антигена в сыворотке.
Эти методы позволяют определить число антигенов в смеси и идентифицировать
антигены в разных препаратах. Они служат также для проверки чистоты
вакцин и других биологически активных антигенов, в том числе ферментов и
гормонов. Образование преципитатов в геле используется для диагностического
анализа сыворотки крови при таких инфекционных заболеваниях, как гистоплазмоз,
бластомикоз и коцидомикоз. Недавно было показано, что кроличью сыворотку,
содержащую антитела к миоглобину, можно использовать для обнаружения
миоглобина в моче, выделяемого при поражениях сердца; с помощью методов
иммунодиффузии можно обнаружить миоглобин через несколько часов после
начала сердечного приступа с целью его ранней диагностики.
Пример. Диффузия на границе раздела
Диффузия иа границе раздела представляет интерес, поскольку она лежит в
основе метода определения константы диффузии макромолекул, позволяющего
■оценивать молекулярную массу, а также в основе многих препаративных и
аналитических методов разделения (очистки) смесей макромолекул, предшествующих
.любому исследованию функции на молекулярном уровне.
Измерения константы диффузии. Рассмотрим систему, которая вначале имеет
резкую границу раздела, отделяющую слой растворителя от слоя раствора
макромолекулы с концентрацией [С] о (рис. 6.10). Если толщина слоя раствора по
обе стороны границы раздела достаточно велика, то можно предположить, что
концентрация макромолекул при удалении от границы раздела совпадает с
исходной fCJ0 или равна нулю; при этих условиях решение уравнения (6.55)
приводит к зависимости, связывающей концентрацию макромолекул и расстояние от
границы раздела
u=hf/2)Y"5i
[С1, = -£М1--^. \ e-*du\. (6.61)
и=0
Зависимости концентрации вблизи границы раздела от времени,
рассчитанные по уравнению (6.61), 'Представлены иа рнс 6.10. i[Определенный интеграл
Со
^
4°°
Се
3
5Г
Ъ
S.
1
*
*»
4 4С°
V
чл
V
ио
ч
\\
\\
V
Расстояние g
31*
5)
5*
§
S
$
а-^.
%<v
*?>
4, 1
э
«ft
Гра
■ ■ "
t очень мало
.'
•
V
*^^\
"^%\
V4 i
г^У )\ Ч^-
fboemomrmy
Рис. 6.10. Результаты изучения диффузии с начальной резкой границей раздела
в момент времени /=0 [3, с. 354].
УСПЕХИ И НЕУДАЧИ
Тангенс
угла наклона*4л1)
50000 г оо ооо
Время, с
150 000
(Рис 6.11. Определение коэффициента диффузии овальбумина измерениями
площади и высоты зависимости концентрационного градиента от расстояния в
конкретные моменты времени [5].
Поскольку в момент 7=0 граница раздела не идеальна, прямая на графике не проходит
|} через нуль.
% уравнении (6.61) представляет собой известную функцию ошибок Гаусса; его
«вначения приведены во многих математических таблицах.]
Как расчет, так и эксперимент упрощаются при анализе не самой
концентрации, а концентрационного градиента д[С]/ду
L дУ' \y.t ' 2/nDt
№ е-
yi/4Dt
(6.62)
Вид зависимости градиента концентрации от расстояния в заданные
моменты времени также показан на рис. 6.10.
Константу диффузии можно найти, определяя либо концентрацию
(например, измеряя оптическую плотность растворов при подходящей длине волны),
либо ее градиент (измеряя показатель 'преломления; см. разд. 14.А).
а. Константа диффузии из измерений концентрации. Из уравнения (6.61)
можно показать (см. задачи в конце главы), что квадрат расстояния между
точками, в которых [С]=;[С]0/4 и [C]=3i[C]o/4, пропорционален времени,
проведшему от исходного момента существования границы раздела
(^от[С]"о/4 до ЗССЫ2^ 3,64^. (6.63)
Следовательно, D можно определить из угла наклона экспериментальной
зависимости (Д«/)2 от времени.
б. Определение константы диффузии из градиента концентрации
Из уравнения (6.62) легко показать, что площадь под кривой для
зависимости градиента концентрации от расстояния равна
Площадь = —[С]0 (6.64)
в то время как высота пика при «/=0 составляет
[С1о
Следовательно,
Высота пика д [С] /ду — у — — ■ /~ут.
у — О *
Площадь _ y~jrt
Высота ~ *
(6.65)
(6.66)
и зависимость (Площадь/Высота)2 от t — прямая с тангенсом угла наклона
AnD (рис. 6.11).
166 ЧАСТЬ 2
Рибонуклеаза
Химотрипсиноген
Овальбумин
Гемоглобин
Тропомнозин
Фибриноген
Коллаген
ДНК
Вирус табачной мозаики
13 683
23 200
45 000
68 000
93 000
330 000
345 000
6 000 000
40 000 000
Таблица 6.1
Коэффициенты диффузии макромолекул в водном раствореа [3, с. 358, 361]
Макромолекула Молекулярная масса ^20 °С НгО **°7 см2/с
11,9
9,5
7,76
6,9
2,24
2,02
0,69
0,13
0,53
коэффициенты диффузии приведены к значениям, которые должны были ,бы
наблюдаться при 20 *С в чистой воде, если размер и формы макромолекул постоянны. ~
Очевидно, что большие молекулы движутся медленней, и, как будет
показано, коэффициенты диффузии обратно пропорциональны молекулярной массе.
Примеры приведены в табл. (6.1). Корреляция между молекулярной массой.и
константой диффузии не описывается каким-либо законом, поскольку еще не
объяснено влияние формы макромолекулы на скорость ее диффузии. Этот вопрос
рассматривается после обсуждения вязкости.
Задачи
1. Приближение Стирлинга lnN!=MnAf — N. является фундаментальным при
решении многих статистических задач, и в том числе (нормального)
распределения Гаусса, лежащего в основе явлений, связанных с диффузией. Выведите
это приближение (выполняется для больших N) из Nl=N(M—\)(N—2)...
...3-2-1. (Примечание. Рассмотрите зависимость IniV от N.)
2. Покажите, что лри случайных блужданиях в одномерном пространстве
[уравнение (6.44)] среднеквадратичное число перемещений из некоторого
начального положения после N перемещений составляет
V((m- (m)f)
3. Покажите, что при случайных блужданиях после N перемещений среднее
расстояние от начального положения равно
<«/> = 0
Покажите также, что при случайных блужданиях в одномерном .пространстве
после N перемещений среднеквадратичное расстояние от начального положения
составляет [уравнение (6.49)]
где I — длина одного перемещения.
£7 УСПЕХИ И НЕУДАЧИ
^ В данной главе показано, что при случайных перемещениях из начальной
точки вероятность достижения молекулой области у—(y+dy) составляет
Pt (У) dy = [llV^tDt] exp [—yVADt] dy.
С помощью преобразований, практически аналогичных используемым при
решении задач 1 и 2, можно показать, что среднеквадратичная ошибка этого
распределения составляет .[уравнение (6.58)]
V<F) = V^Dt
и это является наиболее часто упоминаемым свойством случайного блуждания
в одномерном пространстве. Для того чтобы лучше понять физический смысл
этого результата, рассчитайте ширину зависимости Pt(y) от у> принимая за
меру этой ширины расстояние между точками изгиба по обе стороны от
исходной точки. Сравните полученный результат с уравнением (6.58).
Изобразите (приблизительно) зависимость Pt(y) от у и покажите локализацию точек
изгиба и среднеквадратичную ширину.
i С помощью измерений светорассеяния (см. разд. 15.А) полиметилена (т. е.
насыщенного линейного углеводорода) определено среднеквадратичное
расстояние между концами полимерной цепи, составляющее 85 А. Предполагая,
что макромолекула не имеет разветвлений и что свобода перемещений ее
связей не ограничена, а) рассчитайте молекулярную массу полимера (не забудьте
учесть атомы водорода), приняв среднюю длину связи С—С, равной 1,54 А;
б) покажите, что, поскольку в реальной молекуле связи не свободны (тетра-
С
эдрический угол / ч составляет ~ 109,5°), как введение этого условия
С С
качественно повлияет на выше полученный результат.
I. Константа диффузии сахарозы в воде составляет 4-10-6 см2/с
а. Рассчитайте среднее (среднеквадратичное) расстояние (в см), которое
типичная молекула сахарозы пройдет за 1 ч.
б. За сколько времени молекула сахарозы продиффунднрует от центра к
внешней стенке кровеносного сосуда (диаметр сосуда 8-lfr"6 м). Полученный
результат помогает понять, почему, чтобы обеспеч'ивать эффективный обмен
питательных веществ и> продуктов метаболизма между кровью и окружающи-
. ми тканями, сосуды должны быть такими узкими.
■. Для случая двухмерной или трехмерной диффузии при условии, что в
нулевой момент времени все диффундируемое вещество находится в некотором
начальном положении, вероятность обнаружить молекулу в двухмерной
области с координатами х—\x-\-dx) и у—iy+dy) в момент времени t составляет
: Pt (X, у) dxdy = {-^ ) <-W«*> fW dxdy,
а вероятность обнаружить молекулу в трехмерной области х—(x-\-dx),
■ У—(У+dy) и z—(z-f-dz) в момент времени t составляет
Pt (х, у, z) dxdydz == [-Щ^'2е-{хг1Ш) e~W4Dt> e~^Dt> dxdydz.
Рассчитайте среднеквадратичное расстояние от исходного положения до
положения молекулы через время t а) для случая двухмерной диффузии; б) для
случая трехмерной диффузии, в) Используйте полученный результат для
подтверждения уравнений (6.60).
|* Уравнение латеральной диффузии в одномерном пространстве соответствует
^ уравнению (6.55) для диффузии через единичную площадь поперечного сече-
168 ЧАСТЬ 2
дс , д2с
ния -дт =^дГ2 • Уравнение латеральной диффузии в трехмерном пространстве
выражается соответствующим образом:
дс
-6f-=Dv*c,
где
уг _ ^2^2 л. дуду2 + d2/dz2.
а. Подтвердите, что решением уравнения трехмерной латеральной диффузии
является формула
с = со{ ~^ЬТ )'2'ехр f_^2 +у2 + z2>/4D'] *
б. Найдите точки перегиба на зависимости с от г (изменение концентрации
от расстояния в данный момент времени t).
9. Предположим, что при радиальной иммунодиффузии все исследуем'ое
вещество в нулевой момент времени сконцентрировано в начальной точке
(концентрация Су). Концентрация в любой другой точке плоскости (кроме начальной)
через время t описывается уравнением двухмерной диффузии
(r^)=c1(^DT)^p[-rV4Dtl
Допустим, что наблюдаемая преципитация антитела с антигеном
происходит только при одной конкретной критической концентрации сКрит, которая
достигается в момент времени t\ на расстоянии г=\ см от начальной точки.
Наконец, допустим, что эксперимент повторяется с другой начальной
концентрацией с2 в той же начальной точке. Рассчитайте радиус кольца
преципитации в зависимости от начальной концентрации вещества в начальной точке при
условии, что длительность каждого опыта t\. (Ср. полученный результат с
данными, приведенными на рис. 6.8.)
10. Диффузия на границе раздела фаз лежит в основе двух способов
определения коэффициента латеральной диффузии из измерений концентрации (или
концентрационного градиента) в зависимости от расстояния от начальной
границы раздела. Выведите уравнения (6.63) — (6.65), лежащие в основе этих
методов, из уравнений (6.61) н (6.62),^
Литература
1. Davidson N., Statistical Mechanics, McGraw-Hill, New York, 1962, pp. 280—
285. Хорошее, рассмотрение проблемы случайных блужданий.
2. Тэнфорд Ч. Физическая химия полимеров. М.: Химия, 1965. Хорошее
рассмотрение статистического клубка и трансляционной диффузии.
3. Мороветц Г. Макромолекулы в растворе. М.: Мир, 1969.
4. Lamm О., Poison A., Biochem. J., 30, 528 {1936).
5. Sehon A. #., in: Sensitivity Chest Diseases, Harris M. C, Shure N.t Davis,
Philadelphia, 1964.
6. Davis B. D., Dulbecco R., Eisen H. N.t Ginsberg H., Wood W. В., Microbiology,
Harper & Row, New York, 1967.
7. Tahey J. Т., Makelvey E. M., J. I. Immun., 94, 34 (1965).
[ABA 7
1ЫНУЖДЕНН0Е ПЕРЕДВИЖЕНИЕ
?A. ЭЛЕКТРОФОРЕЗ
щ Большинство биомолекул — амфолиты; это означает, что в
молекуле присутствует большое число подвижных протонов,
характеризующихся различными рКа- Из индивидуальных рКа можно
[см. гл. 3) рассчитать изоэлектрическую точку pi, т. е. значение
>Н, при котором в макромолекуле содержится равное число
отрицательных и положительных зарядов, и, следовательно,
поведение макромолекулы соответствует поведению незаряженной
^нейтральной) частицы. При рН<р1 макромолекула обладает
доложительным суммарным зарядом, при рН>р1 — отрицатель-
1ым суммарным зарядом. В данном случае важно, что при рН
Ьаствора, отличном от значения pi, макромолекула несет
электрический заряд и должна двигаться под действием электрического
юля. Для объяснения закономерностей такого движения прежде
:его необходимо разобраться, почему заряженная молекула в
растворе движется с постоянной скоростью,, пропорциональной
напряженности наложенного электрического поля. Действие на
молекулу электрического поля аналогично действию постоянной си-
|ы, так что интуитивно следует ожидать, что молекула должна
Ускоряться по направлению поля. Далее для объяснения влияния
юрмы молекулы на явления переноса (при диффузии,
седиментации, электрофорезе) необходимо установить важное
соотношение между подвижностью иона и коэффициентом диффузии. И
наконец, для предсказания результата электрофоретического
эксперимента следует использовать эмпирическую модель непрерывно-
потока, описывающую движение иона.
1очему ион под действием постоянной силы движется
постоянной скоростью?
При наложении электрического поля Е (градиент напряжения,
|радиент электрического «давления») частица с зарядом Q
испытывает действие постоянной суммарной силы
ma—QE, (7.1)
f-^e m — масса, а — ускорение.
170 ЧАСТЬ 2
Изолированная частица (например, в вакууме) 'под действием
такой силы стала бы двигаться с 'постоянным ускорением. Однако
при движении иона в водном растворе он должен предолевать
силу трения, пропорциональную его скорости движения:
(7.2)
где / — коэффициент трения. При наложении электрического
поля частица начинает ускоряться до достижения максимальной
скорости (подобно предельной скорости парашюта), при которой сила
трения уравновешена (движущей) силой электрического поля.
В результате равновесия этих сил скорость движения частицы
становится постоянной:
dt~
или
Максимальная скорость = —п~ *== —г—
(7.3)
Микроскопическая природа трения, связь между электрофорезом
и трансляционной диффузией
Прежде всего надо рассмотреть микроскопическую модель,
согласно которой предполагается, что молекула претерпевает
множество столкновений в секунду (модель случайных блужданий);
тогда можно рассчитать перемещение молекул между
столкновениями. Чтобы определить среднее время между столкновениями
т, используем следствие из закона идеальных газов (известное
уже на самых ранних шорах обучения химии в высшем учебном
заведении).
Средняя поступательная кинетическая энергия =
I 2 3 , гп
(7.4)
где k — постоянная Больцмана, т — масса частицы, Т —
абсолютная температура и ^Тепл — средняя скорость молекул газа. Хотя
остальные рассуждения, таким образом, основываются на
свойствах газов, получаемый результат, как вскоре будет очевидно,
носит общий характер. '
Введем следующие обозначения:
х—среднее время между столкновениями (в секундах на
столкновение); .
v — частота столкновений (в столкновениях в секунду);
171 УСПЕХИ И НЕУДАЧИ
/ — средняя длина одного перемещения между столкновениями
(в единицах длины на столкновение);
vтепл — скорость теплового движения (в единицах длины на
столкновение) причем
T=-f. (7.5)
Рассмотрим конкретную молекулу сразу же после
столкновения. Хотя молекула обладает очень большой средней
подвижностью, ее средняя скорость равна нулю, так как скорость
является векторной величиной, а молекула с равной вероятностью
может передвигаться в любом направлении. Под действием
электрического поля (заряженная) "молекула будет непрерывно
ускоряться вплоть до следующего столкновения:
ma=QE (7.1)
или
а=И. (7.6)
При этом молекула сместится на конечное расстояние (ат2/2)
в направлении у; если х — средняя продолжительность
передвижения, то суммарная скорость молекулы между столкновениями
при движении вперед составляет
Средняя суммарная скорость между столкновениями
при движении впеРеД=:|^г=:|^-. (7-7)
На данном этапе удобно ввести ионную подвижность \i
(отношение средней скорости перемещения молекулы вперед к
напряженности электрического поля):
V-Ш—ьГ- (7'8)
Тепловая подвижность (случайно направленная) немного
превышает подвижность, наведенную электрическим полем, поэтому
■направление движения после любого столкновения (в среднем)
является полностью случайным, и после любого столкновения
средняя скорость молекулы вновь равна нулю. Умножая правую
часть уравнения (7.8) на
vr=l (7.9)
и подставляя вместо х
т^-!—, (7.10).
итепл
получаем
^^г,*— ■ <7Л1>
*"штепл
172 часть 2
Наконец, подставляя вместо иТепл ее выражение из уравнения
(7.4) и используя уравнение (6.606), описывающее трехмерное
""б" Г получаем
(7.12)
Уравнение (7.12), связывающее подвижность иона в
электрическом поле с диффузией в отсутствие электрического поля,
можно (проверить экспериментально, поскольку можно исследовать
зависимость \л от температуры, а затем сравнить рассчитанные
значения D с результатами прямых измерений, допустим, при
изучении диффузии через границу раздела. Оказывается, что
уравнение (7.12) имеет общий характер, хотя его вывод сделан
для частного случая. Так часто бывает в физике, и это служит
дополнительным аргументом для использования простых моделей.
Сопоставим модель непрерывного потока, описываемую
уравнением (7.3), с микроскопической моделью, описываемой
уравнением (7.12), при этом получаем еще'один общий результат
(7.13)
Уравнение (7.13) выполняется для молекул любой формы, и,
поскольку зависимость D от формы молекулы хорошо известна
(см. обсуждение вязкости), поведение нейтральных или
заряженных молекул любой формы при диффузии или электрофорезе
легко можно объяснить.
Что такое электрофоретический эксперимент?
До сих пор макроскопическое и микроскопическое
рассмотрения совпадали в том отношении, что заряженная молекула в
растворе (или в геле) движется с постоянной скоростью,
определяемой, ее подвижностью (отношением скорости иона к
напряженности электрического поля). Кроме того, выше выведено
соотношение, связывающее ионную подвижность с коэффициентами
диффузии и трения. Впервые электрофоретический опыт проводили,
осторожно наслаивая разбавленный раствор соли на раствор, в
котором находились исследуемые заряженные макромолекулы, и
наблюдая за перемещением границы раздела под действием
электрического поля (в аппарате Тизелиуса), как показано на рис. 7.1.
Рисунок изображает электрофорез макромолекул с
отрицательным зарядом, но с разной ионной подвижностью.
УСПЕХИ И.НЕУДАЧИ
t-t
Разбавленный
солевой
раствор
Только А
Общая концентраций
макромолекул
Общая концентрация
Макромолекул
Рис. 7.1. Разделение двух макромолекулярных соединений А и В в электрофоре-
тическом аппарате раиией конструкции.
Оба типа молекул заряжены отрицательно, и А под влиянием электрического Поля движется
быстрее, чем В. На графиках рядом со «схемой» устройства изображена зависимость общей
концентрации макромолекул от^расстояния по трубкам устройства.
При использовании аппарата, изображенного на рис. 7.1,
наиболее трудно сформировать и сохранить резкую исходную
границу раздела, что необходимо для хорошего разрешения.
Поэтому современные методы основаны на использовании таких сред,.
jiKaK бумага или гель, уменьшающих конвективное смешивание и:
почти не препятствующих движению ионов под действием
электрического поля. Раствор высокомолекулярных веществ инъектируют
!в виде тонкой полосы, например, в гель, после чего «включают»
электрическое поле. Для получения результата измеряют
содержание вещества в элюате тонкого фрагмента геля, как показано
для положительно заряженных молекул на рис. 7.2.
Из предыдущего анализа диффузии из тонкого слоя ясно, что
^через любую единичную площадь поперечного сечения будет
двигаться поток, равный
д[С)
Диффузионный поток =—D—j-
(6.51)
174 ЧАСТЬ 2
X
*
* J
!
_
/Нулевой момент времени
At
S
Момент времени t f
<*r
—
f
Движение
иона
Рис. 7.2. Диаграмма для анализа экспериментов по гель-электрофорезу.
Вещество в начале в момент t=Q находится в виде узкой полосы в положении #=0;
в следующие моменты времени t=t анализируется поток вещества внутрь и наружу из
области с единичной плошадью поперечного сечення с координатами {у, y+dy). Если
принять, что макронон заряжен положительно, то при включении электрического поля он
будет двигаться вправо.
Кроме того, действие электрического поля должно вносить в этот
поток дополнительный вклад:
Электрофоретический поток=(|*£)[С), (7.14)
где (цЕ) — средняя скорость движения иона в электрическом
поле. Суммарная скорость изменения концентрации в интервале
У—(У+riy) вновь получается из разности потоков внутрь
интервала и из него:
d [С] _ -D (д [C\ldy)y + |*Е [С\у +Р{д [C\ldy)y+dy - цЕ [C\y+dy
dt1
\-\-dii
(7.15)
д[С]
Поскольку и [С] и —ду
иараметра можно использовать ряд Тейлора
зависят от у, для описания каждого
_djc] г а [см , г_а_ Jjcn .
№y+dy ~ [ {ду) \^[ ду ду I ау'
(6.54)
(7.16)
Подставляя уравнения (6.54) и (7.16) в уравнение (7.15),
получаем
д[С) __п д2[С] nF*[C\
dt
(7.17)
Для объяснения смысла уравнения (7.17) достаточно заменить
переменные на осях координат, перемещающихся с той же скоро-
Щ5 УСПЕХИ И НЕУДАЧИ
Электрическое
поле; диффузии
нет
1 = 0 о
1 = 1 '->
f = 2г о
т=.3/ о
Диффузия;
электрического
поля нет
Электрофорез;
электрическое попе
и диффузия
-У
Рис. 7.3. Зависимости концентрации ионов от расстояния в разных условиях.
Эффект одного электрического поля описывается уравнением (7.3); влияние одной диффу-
зин описывается уравнениями (6.55) и (6.56); влияние диффузии совместно с электрическим
полем (при электрофорезе) описывается уравнениями (7.17) и (7.19).
стью, с какой ионы движутся под действием>электрического поля
(см. задачи в конце главы):
y'=y-liEt. (7.18)
В новых координатах получаем простое уравнение
(7.19)
д[С] _п *[С\
at ~и д(У'у •
Уравнение (7.19), таким образом, показывает, что в системе
координат, движущейся вместе с ионами в процессе электрофореза»
диффузия происходит так, как если бы ионы были неподвижны.
Другими словами, диффузия и движение ионов — процессы
независимые (рис. 7.3).
Методы электрофореза и их применения
Гель-электрофорез. Электрофоретические методы относятся к
числу лучших современных способов разделения и анализа смесей
макромолекул (в частности, белков) и выделения этих
макромолекул. Эти методы применяются и в медицинской диагностике, и
при определении чистоты биохимических препаратов (как
показано ниже). В качестве носителей чаще всего используют гели из
агара, крахмала, ацетата целлюлозы или полиакриламида.
Методически электрофорез осуществляют следующим образом.
Исследуемый раствор образца вводят в колонку или тонкий слой геля. За-
176 ЧАСТЬ 2
/
Введение
образца.
7
к.
rt a2 at Альбумин
Рис. 7.4. Электрофоретическое поведение белков нормальной сыворотки крови
^человека при электрофорезе в агаровом геле (анод находится справа, катод —
слева) [4, с. 12].
тем гель погружают в подходящий буфер и накладывают
электрическое поле, подсоединяя электроды к обоим концам геля. Разные
макроионы в образце начинают мигрировать в геле с разными
(постоянными) скоростями и их локализацию в геле в конце опыта
можно определить по окрашиванию и/или по поглощению света в
разных местах (по длине) колонки или слоя геля. Результаты,
получаемые при электрофорезе сыворотки человека в агаровом геле,
представлены на рис. 7.4.
Аналогично тому как использование мелкозернистой
фотопленки привело к улучшению качества фотографий (увеличилось число
передаваемых деталей и полутонов), внедрение электрофореза в
биохимические исследования и в медицинскую диагностику было
весьма (полезным, поскольку позволило анализировать смеси
биологических жидкостей, разделять их на компоненты и определять
их индивидуальные характеристики. Наибольший интерес среди
биологических жидкостей представляет кровь; она поэтому
наиболее полно исследована. После удаления из крови клеток остается
жидкость, которую называют плазмой, а если кровь свернулась,
то выделившаяся при этом жидкость называется сывороткой (сы-
Таблица 7.1
Среднее содержание фракций белков
в нормальной плазме крови человека
Белок2
Альбумин
арГлобулины
а2-Глобулины
(З-Глобулины
Фибриноген
у-Глобулины
Содержание, %
50
4
12
13
8
13
аНазвания белков плазмы крови основаны иа
их положении при электрофорезе; функции
компонентов плазмы крови описаны в любом
учебнике биохимии.
УСПЕХИ И НЕУДАЧИ
К».
кротка отличается от плазмы только отсутствием в ней фибрино-
5на, см. табл. 7.1).
удаление
Кровь »- Плазма
свертывание
клеток
удаляется фибриноген
Сыворотка
|рнмер. Гель-электрофорез при диагностике заболевания
На рис. 7.5 показана типичная картина, наблюдаемая при электрофорезе
блков сыворотки крови и характерные изменения в этой картине,
сопровождающие три патологических состояния. Благодаря тому что эти эксперименты"
прошлись в прозрачном геле ацетата целлюлозы, локализацию белков можно об-
Ьружить, измеряя поглощение света (см. рисунок). Условия эксперимента ука-
|$ны в подписи к рисунку. В последнее время предпринято также несколько по-
itok установить взаимосвязь между уровнем гликопротеинов в сыворотке кро-
с-возникновением атеросклероза; в связи с этим интересно отметить, что Среди
|йстралийских аборигенов заболевание атеросклерозом встречается гораздо реже,
|£м среди городского населения, а уровень гликопротеинов в сыворотке крови у
Альбумин <*\
Альбумин «1 «2
Рис. 7.5. Электрофореграммы при электрофорезе в геле ацетата целлюлозы
нормальной сыворотки крови (а), а также сыворотки крови при янфекциониом гепа-
■ц тите (б), при липидном нефрозе {в) и при гамма-миеломе (г) [7].
Названия белковых фракций указаны в табл. 7.1. Концентрации белков определяются по
?т оптическому поглощению (см. ч. 4).
178 ЧАСТЬ 2
\
2Q0OOO
г
&
«j
i
Qt
*
S»
I
^1
100000
80000
60000
50000
40000
30000
20000
10000
О 0,2 04 0,6 Цв 1,0
Электрофоретическая подвижно&ть
Рис. 7.6. Взаимосвязь между молекулярной массой (в логарифмической шкале) и
электрофоретической подвижностью белков, предварительно развернутых путем
восстановления дисульфидных связей и обработки додецилсульфатом натрия [8].
них также ниже. Существенного улучшения селективности разделения удалось
достичь с помощью недавно разработанного метода диск-электрофореза (см.
ниже), что должно повысить значение электрофореза для диагностики. /
Диагностическая ценность электрофореза проявляется также при анализе
белков двух других биологических жидкостей: спинномозговой жидкости и мочи.
Единственная трудность при этом состоит в том, что, хотя белковые
компоненты обеих этих жидкостей аналогичны компонентам сыворотки крови, их
относительные концентрации ниже, чем в сыворотке, в 100 и в 1000 раз соответственно.
Наиболее характерные изменения относительного содержания белков в
спинномозговой жидкости обнаруживаются при обширном склерозе, менингите и
энцефалите и частичные — при дегенеративных заболеваниях или при инфильтрации
новообразованиями (раковыми). Появление белков в моче является
чувствительным показателем нарушения функций почек: обычно почки при образовании
мочи отфильтровывают почти все сывороточные белки, ио при почечных
заболеваниях белки могут попадать в мочу. Поскольку электрофоретическая
подвижность — функция молекулярной массы, степень расстройства органа можно
частично оценить по размеру белков, проникающих через почки в мочу.
Определение относительно небольших «белков Бене — Джонса», соответствующих «легким»
цепям иммуноглобулинов (гаммаглобулинов), является диагностическим
показателем миеломы и лимфатической лейкемии и в меньшей степени первичной
макроглобулинемии. При проведении всех этих тестов экспериментальные
условия идентичны- используемым при электрофорезе сыворотки; отличие состоит
лишь в том, что концентрацию белков перед электрофорезом следует повысить
в 100 или в 1000 раз с помощью диализа под давлением (см. разд. 2.В).
Скорость, с которой белки могут двигаться в растворе (при седиментации,
диффузии) или в геле (при хроматографии, электрофорезе), существенно
зависит от формы макромолекул. Определение молекулярной массы неизвестного
— >> Миозин
ъ-Пзлактоаидаза
^Фоарори/нзза А
Трансферрин
Каша/юза
Овальбумин
Пепсин^
сс-Химотрипсипоген
УСПЕХИ И НЕУДАЧИ
Изаэлектрическое фокусирование
б градиенте рН
Катод (—)
РН/Р1 , * .
Л С л
|р*1
^Р*2
Шр13
9
-<±>-
—<+)—•&--©—
6
■ вается
W////M
Белок
1
f
о.
Анод&\
Ряс. 7.7. Поведение трех белков с соответствующими изоэлектрическимн. точками
I pli, рЬ и р1з в схематичном опыте по электрофокусированию [2].
Нри рН>р1 молекула белка заряжена отрицательно (и, следовательно, перемещается но
колонке вниз), а при рН<р1 — положительно (и, следовательно, перемещается по колонке
jpJepx). Поэтому молекулы каждого белка перемещаются в электрическом поле до тех пор,
; пока достигнут уровня, на котором рН геля совпадает с pi этого белка.
глка (с неизвестной формой) .представляет собой, следовательно, сложную
задачу. В настоящее время существует два общих метода для того, чтобы вызвать
практически у любого белка изменение конформаци» до одной и той же формы:
\) 6 М. гуанидингидрохлорид разрушает третичную структуру белков так, что
они принимают форму, с хорошим приближением соответствующую
статистическому клубку, и 2) додецилсульфат натрия (широко применяемый детергент) в
Концентрации Г^5'1СН моль/л связывается с белками по всей длине полипеп-
агидиой цепи, и это приводит к тому, что они ведут себя как жесткие палочки
Июстоянного диаметра, длина которых пропорциональна молекулярной массе по-
^ипептидной цепи. [В обоих случаях —S—S-связи, соединяющие разные
пептидные цепи, должны быть восстановлены (разрушены) до обработки гуанндином
или детергентом.] Установлено, что после обработки детергентом электрофоре-
Тическая подвижность хорошо коррелирует с молекулярной массой
денатурированного белка (рис. 7.6), в то время как коэффициент диффузии (или
подвижности) коррелирует с молекулярной массой интактных белков (табл. 6.1). При
изучении неизвестного белка надо лишь внести образец, содержащий этот белок и
несколько стандартных белков с известными молекулярными массами, в поли-
акриламидный гель (после предварительной обработки детергентом) и после
Электрофореза путем экстраполяции найти молекулярную массу исследуемого
белка; это наиболее часто применяемый способ для приблизительной оценки
молекулярной массы* белков.
180 ЧАСТЬ 2
Рис. 7;8. Разделение компонентов гемоглобина изоэлектрическим
фокусированием [9].
1 — изменение рН п0 длине колонки; 2 — поведение различных белков (измеряется
оптическое поглощение).
Изоэлектрическое фокусирование в градиенте рН. Принцип
изоэлектрического фокусирования иллюстрирует рис. 7.7. Этот
метод основан на том, что любой амфолит (молекула с двумя или
более диссоциирующими протонами) положительно заряжен при рН
ниже его изоэлектрическои точки и отрицательно заряжен при рН
выше его изоэлектрическои точки; при изоэлектрическом значении
рН амфолит нейтрален и, следовательно, не движется в
электрическом поле (детальное обсуждение см. в разд. З.В). Следовательно,
в геле (гель вновь используется для уменьшения диффузии) с
градиентом рН по длине колонки данный белок при наложении
электрического поля будет двигаться до достижения слоя геля с рН,
отвечающим изоэлектрическои точке данного белка (рис. 7.7 и
7.8).
Пример. Разделение компонентов гемоглобина изоэлектрическим фокусированием
Один из - наиболее наглядных примеров использования изоэлектрического
фокусирования, приведен на рис, 7.8. В этом-случае доказана возможность
разделения; двух белков, изоэлектрическне точки которых отличаются всего лишь на
0,02 единицы рН!
Иммуноэлектрофорез. В этом методе электрофорез проводят
как обычно в слое агарового (отрицательно заряженного) или ага-
розного (нейтрального) геля; затем по обеим сторонам слоя
параллельно оси миграции прорезают «траншеи», которые заполняют
раствором антител. При этом выбираются антитела, специфические
к определенным белкам (антигенам), разделяемым при
электрофорезе; в результате каждая реакция антигена с антителом
сопровождается образованием полосы преципитации. Как видно на
рис. 7.9, группу белков с одной и той же электрофоретической
подвижностью удается разделить на компоненты благодаря реакции
с антителами, поскольку в этой группе различны концентрации
компонентов и, следовательно, скорости радиальной диффузии
этих компонентов.
УСПЕХИ И НЕУДАЧИ
аг -Макроглобулин
Церц/юллазлшн
Гаптогловин
ссг- Гликопротеин
О
У
fi
Альбумин
тс. 7.9. Иммуноэлектрофоретические эксперименты [4].
|Тосле проведения обычного электрофореза в небольшие «траншеи» по краям полосы геля
даомещают специфические антитела. При диффузии антител к центру полосы там, где анти-
?6ло реагирует со специфическим к нему антигеном, образуется кольцо преципитации, а —
^Неизвестное антитело (в «траншеях») оказывается специфическим к v-глобулииу, ссггло-
,булину и альбумину; б — с помощью полифуикциональной аитисыворотхи (содержащей мно-
|й> антител) группа ссгглобулннов разделяется на несколько компонентов белковой природы
й(благодаря тому что эти компоненты присутствуют в разных концентрациях, они
диффундируют в наружном направлении с разной скоростью и образуют различающиеся полосы
j препипитации).
В настоящее время уже известно, что под названием «гамма-
•глобулины» объединяется большое число белков —
иммуноглобулины IgG, IgA и IgM (см. любой современный учебник
иммунологии). Иммуноэлектрофорез применяется для обнаружения
изменения концентрации этих иммуноглобулинов; считается, что
каждый иммуноглобулин образуется группой клеток (клоном),
образовавшихся из единичной клетки-предшественницы, и оценка
индивидуальных иммуноглобулинов имеет огромное значение,
например, для дифференциальной диагностики вида опухоли костного
^мозга, а также для решения вопроса о доброкачественности
(уровень иммуноглобулинов относительно постоянен во времени, в
течение нескольких месяцев) или злокачественности (уровень
иммуноглобулинов увеличивается со временем) таких опухолей.
182 ЧАСТЬ 2
Начало опыта
Концентрирование
Глициновый
4у/рер(вНв,Ъ) [;
Гель для введения
образца (рн 6,3)
Концентрирующий
гель (рН8,Э)
&азаеляющий гель
(рИ9,5)
Г/гициновый
■*— ДрН
Разделение
ведущий Ведомый
ион оон
белок
'
т
к
УУУУУУ.
•■■■•;<>•.-.-
'-'А '-Л- "
. .4 .V. .
•:•:•:
Ml*:
{СП (Глицин)
Рис. 7.10. Схема проведения диск-электрофореза [10].
Цель применения разных гелей и различающихся по рН буферов объясняется в тексте.
Результат состоит в том, что различные белковые компоненты в образце «тащатся» чуть
сзади фронта ионов С1- при перемещении С1- вниз, так что как раз перед попаданием в
гель, в котором происходит обычное электрофоретическое разделение, белки образуют очень
тонкий слой. Отдельные белковые полосы после разделения, следовательно, очень узки, и это
позволяет различить белки, подвижность и молекулярная масса которых различаются очень
незначительно.
Диск-электрофорез. При любом обычном электрофоретическом
опыте разрешение, достигаемое при разделении нескольких
компонентов, зависит от двух факторов: ширины зоны образца в начале
эксперимента и обусловленного диффузией размывания ее в
процессе электрофореза. Теория метода диск-электрофореза
разработана Орнстейном, а его методическая сторона — Дэвисом. Этот
метод позволил уменьшить ширину зоны исходного образца до
нескольких микрон вместо нескольких сантиметров при менее
совершенных методиках электрофореза. Заметно улучшилась
разрешающая способность метода электрофореза. Схема прибора показана
на рис. 7.10.
Сущность метода состоит в использовании буферов с разными
рН на концах колонки геля. Гель состоит из трех областей
(сверху вниз): стартовой области, в которую вводится исходная смесь
макромолекул, концентрирующей области, отделяющей стартовую
область от третьей разделяющей области, в которой происходит
саморазделение компонентов. Стартовый и концентрирующий гели
имеют сравнительно крупные поры, уменьшающие дуффузию, но не
снижающие скорость движения макроионов под действием
электрического поля. Разделяющий гель более мелкопорист, что замедляет
движение макромолекул соответственно их молекулярной массе
УСПЕХИ И НЕУДАЧИ
щ- улучшает разделение, обусловленное различной электрофорети-
рзской подвижностью этих молекул. Для концентрирования мак-
йойонов используются гели с разными рН: необходимо, чтобы при
Цереходе в разделяющий гель ионы двигались в виде узкой
полосы. В стартовом и в концентрирующем геле содержатся ионы С1~;
Керхний резервуар и разделяющий гель содержат глицин. Глицин
Обладает двумя диссоциирующими протонами с p/Ci =2,7 и р/С2 =
|1=9,8. Следовательно, при рН 9,5 глицин в разделяющем геле
состоит из приблизительно равных количеств +H3N—СН2—С02 и
fcN—СН2—COJ (следовательно, средний заряд молекул глицина
Отрицательный) и обладает соответственно большой ионной
подвижностью в электрическом поле (см. разд. З.В):
М'СГ ~ Иг лицин (рН 9,Б) »^елок. (7.20)
; При рН 8,3 в стартовом и в концентрирующем гелях глицин
|гочти нейтрален и его ионная подвижность даже меньше, чем у
макроионов:
М'СГ > И-белок ■> И-глицин (рН 8.3)'
(7.21)
В начале опыта под действием электрического поля ионы С1~
в стартовом геле быстро движутся. Казалось бы, что они
должны обогнать другие отрицательно заряженные ионы, но при
небольшом размышлении ясно, что это бы привело к созданию за
ионами С1~ зоны с суммарным положительным зарядом — так как
разделение зарядов приводит к возникновению градиента
напряжения (электрического поля), движущиеся сзади отрицательно
заряженные ионы белка «тянутся» за ионами С1~ (подобно тому,
как косяк рыб ловится движущейся сетью) для уменьшения
возникающего за зоной ионов С1~ положительного заряда. Теперь
ясно, зачем надо изменить рН в разделяющем геле; как только
ионы С1~ (и следующие тут же за ними белки и глицин) входят
в разделяющий гель, рН увеличивается и ионы глицина с
соответствующим отрицательным зарядом обгоняют белки и движутся
непосредственно за ведущими ионами С1~. Белки в этой области
геля, следовательно, движутся со скоростями, определяемыми их
ионной подвижностью и способностью проникать через мелкие
поры этого геля. Конечный результат этого процесса состоит в том,
что все макроионы перед входом в разделяющий гель
концентрируются и образуют очень тонкую зону. (Одна из функций
концентрирующего геля состоит в том, чтобы предохранить от
размывания резкую границу раздела рН при формовании стартового
геля непосредственно перед началом опыта.)
Пример. Улучшенное разделение компонентов смеси
На рис. 7.11 показаны (наверху, справа) результаты обычного
гель-электрофореза нормальной сыворотки крови человека. Если разрезать гель на кусочки
(соответствующие заштрихованным зонам) и подвергнуть белок нз каждого
такого кусочка диск-электрофорезу (рис. 7.10), то каждая группа белков разделит-
184 ЧАСТЬ 2
\
Ос,-Гло6улан
' 23
Ю 15 20 25 30 35 АО
Номер фракции
1
®j& -Глобулин
Рис. 7.11. Разделение и идентификация с помощью диск-электрофореза
фракций белков сыворотки крови, полученных обычным тель-электрофорезом из
нормальной сыворотки крови человека [5].
<;я на компоненты, как показано на соответствующих диаграммах (слева). Хотя
труппу гаммаглобулииов ие удалось хорошо разделить, метод иммуноэлектрофо-
реза можно использовать для идентификации компонентов этой группы.
Пример. Определение числа субъединиц в феркенте
Роль субъединиц в ферментах 'более подробно описана в ч. 3. Хотя у многих
ферментов молекулярная масса выше 100 000, накапливается все больше
доказала)*
UJ
>СО + + -
coco*.+СО-
-со-
(5
1
I
Рис. 7.12. Схематическая диаграмма обнаружения субъединиц фермента путем
смешивания сукцинилированного и нативного ферментов с последующим
анализом дополнительных полос при электрофорезе смеси (пример из практики) '[11].
УСПЕХИ И НЕУДАЧИ
ьств о том, что большинство основных субъединиц ферментов имеют молеку-
:pHb»e массы от 15 000 до 50 000. Поскольку взаимодействие между субъедиии-
;ми часто можно связать с механизмами, контролирующими эффективность дей-
'вия ферментов (и соответственно с относительной важностью разных путей
угаболизма), выяснение числа субъединиц в ферментах представляет огромный
ferepec. Однако, хотя субъединицы могут быстро разъединяться » соединяться,
йи большой константе ассоциации прямое наблюдение субъединиц может быть
бтрудиено (так как большинство субъеднниц агрегировано). Хорошим подхо-
ри для решения этой задачи может быть использование химической модифика-
|ни белка с последующим использованием электрофореза.
';■• Химическая модификация состоит в превращении (положительно заряжен-
ifx) остатков лизина в белке в (отрицательно заряженные) карбоксильные
jjfynubj
о=<>о ^ *
;' Допустим для простоты, что исследуемый фермент — димер и что в реакции
укцинилирования затрагивается лишь один остаток лизина на субъединицу,
ферментный препарат разделяют на две части., и в одной нз них проводят сукци-
^лирование. Затем нативнььй н сукцинилированный фермент смешивают, и если
&ть субъединицы, а димер может диссоциировать, то равновесная смесь
содержит частицы с дополнительными (двумя или одним) отрицательными зарядами
Ь сравнению с нативным ферментом. Следовательно, если в, электрофореграмме
jiiecH нативного и сукцинилированного ферментов обнаруживается три, а не две
Олосы, это служит прямым доказательством димерной структуры фермента.
Ьтя этот Метод имеет некоторые ограничения (химически модифицированная
^бъединица может вести себя не так!^ как нативная), он был успешно использо-
ан для изучения четвертичной структуры альдолазы и- для изучения состава
аталитической субъединицы аспартаттранскарбамилазы, являющейся тримером
рис. 7.12).
Изотахофорез. Объединение принципов электрофореза с движу-
цейся границей раздела н непрерывного электрофореза лежит в
>снове нового метода изотахофореза, многообещающего для
аналитического разделения небольших катионов или анионов,
подобных метаболитам .и электролитам человеческого организма (в
отличие от методов разделения больших белковых ионов,
рассматривавшихся до сих пор).
is В типичном опыте лри электрофорезе со свободной границей
|рис. 7.1) ионы с большей подвижностью движутся в растворе
*еред более медленными ионами, что приводит к разделению сме-
m на зоны, свободные от ионов и содержащие один или оба иона.
Однако при обсуждении диск-электрофореза (рис. 7.10) было
Сказано, что ионы с большей скоростью движения могут обогнать
Медленные ионы только на ограниченное расстояние, прежде чем
[градиент напряжения, образующийся за быстрыми ионами,
возрастет настолько, что начнет «подтягивать» медленные ионы и
заставит их двигаться с той же скоростью, что и быстрые ионы
|isotacho — равная скорость). Следовательно, если прюводить опыт
§ свободной границей раздела достаточно долго, то все ионы в
186 ЧАСТЬ 2
Электропро-
водность с'
Расстояние вдоль
вертикальной трубки —*•
Рис. 7.13. Изотахофореграмма некоторых нуклеотидов [6, с. 44].
Электропроводность и ее производная отложены в зависимости от расстояния вдоль
трубки (прибор изображен на рис. 7.1). Как указано в тексте, отличительная особенность
этой диаграммы состоит в том, что ордината характеризует природу иуклеотида, а
абсцисса (расстояние) — количество каждого аниона в образце.
разных зонах (рис. 7.1) будут двигаться практически с одинаковой
скоростью.
Изотахофореграмма очень похожа на изображенную на рис. 7.1
(внизу), но интерпретировать ее надо совершенно иначе. При
обычных опытах электрофореза со свободной границей раздела
абцисса («ступеньки», на рис. 7.1) дает качественную информацию
(т. е. указывает на тип присутствующей в смеси макромолекулы),
а ордината (общая концентрация макромолекул) —
количественную (указывает содержание каждого компонента в смеси). При
стационарном движении границы раздела (при изотахофорезе)
ситуация противоположна и значение по ординате дает качественную
информацию, а значение по абциссе — количественную. Такое
различие двух методов просто объяснить: для нового метода значения
ординаты (обычно электропроводности, которая характеризует об-
17 УСПЕХИ И НЕУДАЧИ
|цую концентрацию ионов) различны для химически неидентичных
^онов (например, Na+ и К+), в то время как степень отставания
ведомого иона от ведущего иона (расстояние между «ступеньками»
jta рис. 7.1) определяется разностью их концентраций. Поскольку
i настоящее время метод изотахофореза еще очень редко
используется в биологии, здесь ему уделено немного внимания; однако
1ринципы этого метода подробно обсуждаются в обычных
учебниках физической химии при рассмотрении чисел переноса. На рис.
ЦЗ приведена типичная экспериментальная изотахофореграмма,
Юлученная при анализе смеси нескольких биологически интерес-
шх анионов. Аналогичный способ количественного анализа,
основанный на'разности ионных подвижностей компонентов смеси
электролитов, использовался для определения калия, натрия, кальция,
Магния и лития в сыворотке крови.
lb. СЕДИМЕНТАЦИЯ
Крупные частицы, суспендированные в воде, осаждаются под
Действием силы тяжести, если их плотность выше плотности
раствора. Суспензия биологических макромолекул самопроизвольно
ie разрушается, так как скорость (случайно ориентированного)
Теплового движения молекул намного выше (ориентированной в
)?дном направлении) скорости движения под действием силы тя-
кести. Например, потенциальная (гравитационная) энергия
макромолекулы с молекулярной массой 100 000 между двумя
точками, отстоящими друг от друга на 1,0 см, составляет
' Ег?ав=т§кж (100000/6.10"2в) кг-(10 м/с2)-(10-2 м) =
& =1,7.10-2ЭДж (7.23)
[для гравитационного ускорения принято приближенное значение
[О м/с2). Порядок величины тепловой энергии той же самой
молекулы (см. ч. 5) оценивается значением
? £Г=(1,38-10-23 Дж.К-1)(2,93.102К)=4.10-21 Дж, (7.24)
*
I е. лочти в 200 раз больше. Ультрацентрифуга — устройство для
^усиления» гравитационной составляющей скорости движения мо-
|екул (для осаждения макромолекул потенциальная энергия
Должна быть выше энергии теплового движения).
' Различия в скорости седиментации (осаждения) обеспечивают
возможность разделять макромолекулы в смеси по их размеру и
Плотности. Равновесная седиментация позволяет определять
абсолютные величины молекулярной массы и служит для оценки
гетерогенности смеси. Объединение обоих методов позволяет точно
рмерить молекулярную массу и получить информацию о разме-
|ах и степени гидратации (количестве связанной воды)
макромолекулы. Хотя новейшие хроматографические и электрофоретиче-
188 ЧАСТЬ 2
ские методы существенно вытеснили использование
ультрацентрифуги в качестве инструмента для препаративного разделения
белков в растворе, большинство значений (причем их число очень
велико) молекулярных масс белков, приведенных в литературе,
найдено седиментационными методами. Для характеристики
размеров и формы нуклеиновых кислот чаще всего используют
методы седиментации; эти же методы широко применяются для
изучения равновесий между субъединицами и мультимерными
структурами, характерными для белков, нуклеиновых кислот и других
полимеров, а также для разделения компонентов биологических
мембран.
7.Б.1. СКОРОСТЬ СЕДИМЕНТАЦИИ
Интерпретация электрофоретических экспериментов
относительно проста, благодаря тому что действующая сила
(электрическое поле) при проведении эксперимента не меняется во времени.
В противоположность этому при седиментационных исследованиях
действующая на макромолекулу сила (центробежная сила)
меняется с расстоянием от оси вращения: Гц -возрастает в процессе
осаждения молекулы на дно кюветы; это затрудняет точное
описание движения молекул (т. е.. получение решения с конечным
числом параметров). Два упрощения, однако, помогают получить
математическое решение этой задачи: если силы, действующие на
молекулу, компенсируют друг друга или если суммарный поток
молекул через данную область раствора равен нулю, решение
возможно. Все седиментационные методы основаны на
использовании одной или другой ситуации; вначале рассмотрим случай
взаимной компенсации сил, действующих на макромолекулу.
Схема процесса седиментации в опыте показана на рис. 7.14
для (обычного случая) макромолекулы с плотностью,
превышающей плотность окружающего ее раствора.
Суммарная центробежная сила, действующая на 1 моль
макромолекул с молекулярной массой М, описывается произведением
эффективной массы макромолекул на ускорение о)2г (где <d —
угловая скорость вращения)
Fn=[Масса 1 моля макромолекул—
—Масса раствора, вытесняемого 1 молем макромолекул] «©V, (7.25)
т. е. макромолекула осаждается, если ее плотность превышает
плотность раствора. Масса 1 моля макромолекул равна
молекулярной массе М и
Масса раствора, вытесняемого 1 молем макромолекул =
_ г (раствора) г (вещества) см3 (раствора, вытесняемого __
см3 (раствора) ' моль (вещества) ' 1 г вещества)
) I _}
= Рраст„ор М VBW (7.26)
УСПЕХИ И НЕУДАЧИ
/о о о о о о о о\
(о о о о о о о о|
оооооооо
/OOOOOOOOOl
fo ooooooooj
ооооооооо
/о оооооооо о\
fo ооооооооо!
оооооооооо
/О ООООООООО OI
(о оооооооооо\
ооооооооооо
1о оооооооооо ol
(о оооооооооо о\
d
!
!
!
Мениск
I
)
(оооооооо'
'оооооооо
оооооооо
/ооооооооо
ООООООООО
ооооооооо
/ОООООООООО^
'оооооооооо
оооооооооо
/ооооооооооо
'ооооо оооооо
хил пял гдч.1,1,11,»:отгс1Хюо\
t -t\
/ = ft
оооооооо
оооооооо
оооооооо
оооо ооооо
ооооооооо
ооооооооо
оооооооооо
оооооооооо
оооооооооо
t = tz
Дно \
к/озегы.
?Рис. 7.14. Развитие седимеитационного эксперимента при очень высокой центро-
[ бежной силе [1, с. 366J.
я —схема распределения макромолекул иа трех разных стадиях эксперимента; б н в —
изменение концентрации и градиента концентрации в зависимости от расстояния от центра
,вращения г (включая эффект диффузии иа границе раздела) в соответствующие моменты
времени.
где рраствор — плотность раствора, а увеш (парциальный удельный
Побьем растворенного вещества) — увеличение объема при
добавлении 1 г растворяемого вещества к очень большому объему
раствора (большой объем раствора нужен для условия
независимости концентрации макромолекул от добавления небольшого
дополнительного количества вещества; см. гл. 2)*. Тогда суммарная
центробежная сила выражается следующим образом:
ы Fu=£dVIM—Мувещ.рраствор]. (7.27)
* В гл. 2 уже встречался аналогичный параметр: парциальный моляльный
Ьбъем V, определенный как объем раствора, вытесняемый 1 молем растворенного
вещества V=Mt>.
190 ЧАСТЬ 2
Как и при электрофорезе, движению под действием
центробежной силы противодействует сила трения (направленная
противоположно), пропорциональная скорости движения макромолекул:
dr
гтр— Nf .
(7.28)
[число Авогадро N введено в уравнение (7.28), поскольку оно
относится не к одной частице, а к 1 молю частиц]!. Следовательно,
макромолекула в ультрацентрифуге ускоряется под действием
центробежной силы, пока эта сила не уравновесится силой,
трения; после этого скорость седиментации становится постоянной
(определяемой условием /гтр+77ц=0):
dr
Nf~dl' =Мй>гГ [1—УвещРраствор].
(7.29)
Введем коэффициент седиментации s (отношение скорости
седиментации к центробежной силе) и после соответствующих
преобразований получаем
М=-
Nfs
(1 — увещРраствор)
где
(7.30)
kT
Наконец, вспомнив, что f = -p- [уравнение (7.13)] и Nk=R,
получаем искомый результат
М =
RTs~
L* (1 — ^вещРраствор)
(7.31)
Уравнение (7.31) лежит в основе одного из прямых методов
определения молекулярной массы. Экспериментальное
определение D обсуждалось в разд.^б.Б (см. также ч. 6), а измерения
плотности и температуры не вызывают каких-либо особых
затруднений. Коэффициент седиментации определяют, пользуясь
преобразованным уравнением (7.30):
dr/r dinr
~~dT~
dt
=^©2s.
(7.32)
Таким образом, зависимость логарифма расстояния от центра
вращения до центра границы раздела от времени описывается
прямой, угол наклона которой пропорционален s (рис. 7.15); по-
fl УСПЕХИ И НЕУДАЧИ
I
100
Вргмя, мин
150
^ис. 7.15. Определение коэффициента седиментации. [Данные Гриффиса и Мар-
шелла (Griffith P. R.t Marchall A. G.).]
'i— расстояние от центра вращения до центра границы раздела; изучалась седиментация
" врмеита целлобиозилгидролазы из разъедающего древесину грибка Trichoderma viride.
Скорость ротора 48 400 об./мин.
сольку обычно s имеет порядок Ю-13 с, принято выражать эти
»еличины в сведбергах:
15= Ю-13 с. (7.33)
Принято приводить s и D для разбавленного водного раствора
1ри 20 °С. Соответствующие поправки в значении плотности
раствора, парциального удельного объема и вязкости т| учтены урав-
1ениями (7.34) и (7.35), причем предполагается, что jp пропорцио-
[ально т| (разд. 7.В). [При использовании этих уравнений без-
)говорочио подразумевается, что молекулярные свойства раство-
>енного вещества (например, конформация) не меняются с
температурой или концентрацией, что часто не выполняется.]
i
$20 °С. Н20 —St °С, раствор
D
ЛГ'С. раствор | (1 — РР)20°С, НаО
П20 °С, Н20 J (1 — ~VQ)T оС> раствор'
293 К Л/Лг-с. раствор\
n i ^зо i\ ^ i 47--L,, раствор 1
'20 «С. Н20 =DT -С, раствор ^ -^ 1 I ^^ )
(7.34)
(7.35)
\ Как показано в следующем разделе, вязкость просто
характеризует силу трения, замедляющую осаждение макромолекул, так
;что поправки на вязкость в выражениях для s и D интуитивно
^оправданы. Другая .поправка для величины s обусловлена
разностью плавучести объекта при измерениях в воде при 20 °С и
измерениях при температуре Т в растворе макромолекул. Наконец,
температурная поправка для величины D связана с тем, что D
пропорциональна абсолютной температуре [см. уравнение (7.13)].
^ Коэффициент седиментации s дает информацию о форме, раз-
192 ЧАСТЬ 2
мерах и степени гидратации макромолекулы; для более
подробного анализа необходимо^ предварительно кратко рассмотреть
вязкость в растворах несферических молекул; этот вопрос
обсуждается в следующем разделе.
7.Б.2. СЕДИМЕНТАЦИОННОЕ РАВНОВЕСИЕ
Достаточно простая для анализа результатов ситуация
возникает при седиментации, когда через данное сечение вращающего-,
ся раствора суммарный поток макромолекул равен нулю. Поток
макромолекул через площадь поперечного сечения А
складывается из потока наружу, вызванного седиментацией, и потока внутрь,
вызванного диффузией. При равновесии концентрация на любом
данном уровне раствора в кювете 'постоянна, и через эту область
суммарный поток равен нулю. Эту ситуацию описывает
аналитическое выражение, которое можно вывести из уравнения (6.51)
Поток внутрь^— DA-^-, (6.51)
ПОТОК Наружу = ° ~ v^Ppacr^P) MiohA [С] ^ (?щ
Уравнение (7.36) Вытекает из факта, что
I(* —^вещРраствор) Щ №*г] = (Суммарная масса) ■ (Ускорение) =
=Центробежная сила (наружу) на
1 моль растворенного вещества
в то время как 1/f ^-(направленная наружу) скорость движения на
единицу (направленной наружу) силы, действующей на молекулу:
читатель может легко убедиться, что размерность потока в
уравнении (7.36) выражена в единицах массы, обнесенной к единице
времени и к данной площади. Поскольку при равновесии в
данной области раствора не происходит суммарного накапливания
или снижения количества вещества во времени, то
(Поток внутрь) + (Поток наружу]).=О (7.37)
или ...^
D ~ЬГ = (! - WpacTBop) AfaV [C]/Nf. (7.38)
ьт
Подставим (вновь) f=-=- и R = Nk и сделаем преобразования:
-^gL = rf In [C1 = ^г(1-^веЩРраСТвор) ф (? щ
Чтобы, выразить уравнение (7.39) в удобном для использования
при интерпретации экспериментальных данных виде, следует его
УСПЕХИ И НЕУДАЧИ
Ось
вращений
О
^Уравновешиваю- Кювета]
тая кювета сраст-
вором
!
I
t=0
t=t,
t
Мениск
t *
I Мене
*-*,
Дно
Мениск
f f
I /*Л?/у
Дно
t-*oo
(равновесие) j
Мениск
>0
Мениск
Рис. 7.16. Развитие опытов по седиментациоияому равновесию.
— схема опыта; б — образец после достижения равновесия; в — разные стадии прибли-
:ения к равновесию, г — расстояние от центра вращения, т — молярная концентрация иа
расстоянии г.
фоинтегрировать в интервале от Т\ до г2 (соответствующая
концентрация макромолекул равна [C]i и [С] г)
i„ хгл 1« хгл М<°2 ^ — РрастворРраствор) , « о.
In [С]2 — In [С]г = 2#т V* ~~ ^
(7.40)
Опытным путем надо измерить зависимость концентрации
макромолекул в растворе от расстояния от оси вращения; измерения
концентрации удобно проводить в оптически прозрачной ультра-
центрифужной кювете с помощью соответствующего оптического
Устройства (например, спектрофотометра или рефрактометра). За-
|>ем строят зависимость ln[C]i от г2: тангенс угла наклона^прямой
гропорционален молекулярной массе макромолекулы, причем
коэффициенты пропорциональности легко найти из
экспериментальных данных. Необходимость определения коэффициента диффузии
отпадает, что является важной особенностью этого метода опре-
[еления молекулярной массы.
Приближение к седиментационному равновесию (и его дости-
[ение) легко определить графически (рис. 7.16).
(,■ Потрясающая точность определения молекулярных масс
методом седиментационного равновесия иллюстрируется данными в
Табл. 7.2, приведенными для веществ, молекулярная масса
которых вычислена также из их химических формул. Существенный
недостаток метода состоит в том, что для достижения равновесия
194 часть 2
342,3
13 683
14 305
25 767
341,5
13 740
14 500
25 670
Таблица 7.2
Сравнение молекулярных масс, полученных методом седиментационного
равновесия с использованием уравнения (7.40) (зависимость концентрации
от расстояния от оси вращения), с величинами, рассчитанными
на основании химической формулы для тех же соединений [14]
Молекулярная масса, Молекулярная масса
Вещество рассчитанная из химической по данным седиментационного
формулы равновесия
Сахароза
Рибонуклеаза
Лизоцим
Химотрипсииоген А
обычно требуется не менее суток. Однако можно не относить
временные затраты к большим недостаткам метода, поскольку
точность определения молекулярной массы больших молекул в
растворе— основной критерий его полезности.
Термодинамическое описание седиментационного равновесия.
Ранее равновесие уже определено как состояние, при котором
отсутствует суммарный поток растворенного вещества. Однако
также показано (табл. 2.1), что такому состоянию точно отвечает
условие dG=0. Кроме того, с помощью уравнения (З.Зб) можно
рассчитать разность химических потенциалов для растворов
макромолекул ярного вещества в точках Т\ и гг (рис. 7.16), в которых
молярная концентрация вещества равна соответственно Ш\ и m%:
Овещ ta) -6вещ (rx) =RT In щ-RT In щ =RT In {Щ1т,). (7.41)
Нетрудно найти работу, произведенную при переносе 1 моля
молекул с молекулярной массой М против центробежной силы
(ускорение которой составляет а)2£, где а)— угловая скорость
центрифуги) из точки Т\ в точку Гг:
Сила=т<о2г,
Га
Работа = I Fdr,
п
Работа=V2M (1 -^вещрраствор) со2 {г\-г% (7.42)
Множитель (1—УвещРраствор) вносит поправку на плавучесть,
обусловленную тем, что при переносе вещества молекулы
растворителя должны сдвинуться со своего места. При равновесии
разность свободных энергий, обусловленная концентрационным
градиентом, возникающим в центрифужной кювете [уравнение 7.41)],
равна разности свободных энергий, вызванной разностью
центробежных энергий в тех же двух точках [уравнение (7.42)]. При-
195 УСПЕХИ И НЕУДАЧИ
равнивание уравнений (7.41) и (7.42), таким образом, приводит к
искомому выражению [ср. с уравнением (7.40)] для
экспериментально определенной молекулярной массы М в условиях седимен-»
тационного равновесия:
м
(1
2ИТ\п(тг/т^)
— ^вещРраствор)
-Л)
Значение уравнения (7.43) [идентичного (7.40)] состоит в
том, что оно получено исключительно путем термодинамического
анализа безотносительно какой-либо конкретной модели,
описывающей движение макромолекул в центрифуге. Следовательно,,
нет ничего удивительного в том, что экспериментальные данные в-
табл. 7.2 столь хорошо согласуются с уравнением (7.40),
выведенным с помощью простой модели диффузии. Аналогично можно
показать, что однооараметровое выражение осмотического
давления (уравнение 2.40) и результаты, получаемые при
светорассеянии (см. ч. 4), соответствуют лишь приближениям первого порядка
к более общему (термодинамическому) результату. Хотя
выбранная модель (диффузия, случайные блуждания, груз на пружине)
интуитивно считается удовлетворительной и является
информативной, термодинамический результат более общий и точный,
поскольку не зависит от какого-либо конкретного механизма исследуемого
процесса.
Метод Арчибальда. Еще один широко используемый метод се-
диментационного анализа основан на допущении, что в любой
момент времени поток растворенного вещества через поверхность
раствора (мениск) и через дно центрифужной пробирки (кюветы)
равен нулю (т. е. переноса вещества нет). Следовательно, для
поверхности раствора и дна кюветы справедливо уравнение (7.40) г
которое можно использовать для определения молекулярной
массы согласно уравнению (7.43), причем надо лишь найти
экспериментально концентрации вещества в этих двух областях.
Практически, однако, концентрации в этих областях не измеряют, а
находят путем экстраполяции зависимости концентрации от расстояния
(рис. 7.16) либо к поверхности раствора, либо к дну кюветы.
Получаемое этим способом значение молекулярной массы не столь
точно, как определяемое измерениями при истинном равновесии в
кювете (поскольку при таких измерениях число прямо определяемых
данных гораздо больше), но, благодаря тому что не требуется
достижения полного равновесия, этот метод более быстрый.
7.Б.З. СЕДИМЕНТАЦИЯ В ГРАДИЕНТЕ ПЛОТНОСТИ
Рассмотренные выше седиментационные методы позволяют
определять молекулярную массу очищенного макромолекулярного
вещества, однако они плохо подходят для измерения молекуляр-
196 ЧАСТЬ 2
Обвем раствора, взятого со дна центрифужной хюввты, мл
Рис. 7.17. Кривая седиментации в- градиенте сахарозы смеси трех известных
ферментов (£-галактозидазы, каталазы и алкогольдегидрогеназы) и образца
очищенной нативной ацетилхолинзстеразы [15]. v
/ — активность разных проб /к субстратам указанных трех известных ферментов; 2— аце-
тшгхолинэстеразная активность (служит индикатором относительного количества двух
основных форм фермента).
бых масс в смеси нескольких разных макромолекулярных веществ.
Смеси макромолекул с разной молекулярной массой вначале
необходимо разделить, например, с помощью центрифугирования в
градиенте плотности, т. е. в среде, плотность в которой плавно
уменьшается ш высоте кюветы (от дна центрифужной кюветы до
поверхности раствора),
При проведении скоростной седиментации в градиенте
плотности сахарозы (рис. 7.17) макромолекулы осаждаются со
скоростью, приблизительно пропорциональной их коэффициентам
седиментации. Следовательно, если в исследуемый образец ввести
макромолекулы с известным коэффициентом седиментации, то после
центрифугирования и анализа (аликвотных частей) раствора на
присутствие каждого из макромолекулярных веществ, можно опре-
197 УСПЕХИ И НЕУДАЧИ
делить коэффициенты седиментации всех исследуемых
макромолекул в растворе (рис. 7.17). Если градиент сахарозы создан так,
что макромолекулы с разной молекулярной массой осаждаются
с (разной) постоянной скоростью, то метод называется изокинети-
ческим центрифугированием в градиенте плотности.
При седиментационном равновесии градиент шлотности можно
образовывать в ходе эксперимента, используя, например,
раствор соли CsCl (рис. 7.19). Данная макромолекула в таком
растворе либо всплывает, либо погружается, шока не достигнет изо-
пикнической точки в кювете, в которой плотность раствора равна
плотности данной макромолекулы (т. е. Рраствор = (^;—1, Не-
\ °вещ /
сложный расчет (см. задачи в конце главы) показывает, что если
плотность раствора линейно изменяется на небольшом
расстоянии, то макромолекулы с данной молекулярной массой
распределятся по закону Гаусса с центром в изопикнической точке.
Большое достоинство обоих методов центрифугирования в градиенте
плотности состоит в том, что после разделения комшонентов смеси
три центрифугировании пробы раствора можно отбирать на
разной высоте центрифужной кюветы и определять содержание в них
исследуемых компонентов химически (и, следовательно, очень
селективно) .
Пример. Центрифугирование смеси ферментов
в изокннетическом градиенте сахарозы
Среду с градиентом плотности надо 'приготовить до центрифугирования,
например, путем осторожного последовательного наслаивания растворов сахарозы»
с уменьшающейся плотностью в центрифужную кювету. Тонкий слой (с
меньшей плотностью) раствора макромолекулярного вещества затем наслаивают иа
поверхность раствора сахарозы я осуществляют центрифугирование, при
котором различные ферменты осаждаются с неодинаковой (постоянной) скоростью.
Затем из центрифужной кюветы на разной высоте отбирают пробы раствора и
подвергают их анализу иа присутствие различных ферментов (рис. 7.17). В этой
работе оказалось возможным, используя три фермента с известными
коэффициентами- седиментации, определить относительные количества двух форм аце-
тилхолинэстераз,ы.
Пример. Репликация ДНК
Наиболее известным примером использования центрифугирования в
градиенте плотности служит исследование спаривания молекул ДНК, с помощью
которого получено прямое доказательство двухцепочечной структуры ДНК в
растворе. Вскоре после того, как Уотсон и Крик выдвинули гипотезу о механизме
репликации ДНК (рис. 7.18), быт выполнен следующий эксперимент (рис. 7.19),
Бактерии, выращенные в среде, меченной тяжелым азотом 'SN, были внезапно
перенесены в среду, содержащую только 14N. Согласно гипотезе репликации
Крика — Уотсона, к концу первого поколения должны быть обнаружены (двух-
цешочечные) молекулы ДНК с молекулярной массой, точно равной среднему
значению для молекулярной массы ДНК* (содержащей только ,SN) и молекулярной
массы ДНК (содержащей только ,4N). После двух поколений ДНК бактерий
должны представлять собой равную смесь 19NuN-flHK и 1*iNuN^HK Эти
J 98 ЧАСТЬ 2
Рис. 7.18. Механизм репликации ДНК, предложенный Криком и Уотсоном [16],
После одной репликации каждая дочерняя молекула состоит из одной родительской цепи
(темная), спаренной с вновь синтезированной цепью (светлая). Поскольку родительская
(единичная) цепь остается интактной, во втором и следующих поколениях две молекулы
будут содержать по одной родительской цепн. '
предсказания полностью подтвердились в эксперименте .(рис. 7.19). Великолепное
разрешение, достигаемое с помощью метода центрифугирования в градиенте
плотности, очевидно: массы 16N и |4N отличаются всего на '/is,, а в нуклеотиде
содержится 8% азота. Данные, приведенные на рис. 7.19, свидетельствуют о том,
что удается разделить молекулы, разность молекулярных масс которых
составляет всего ~0,5—1%.
7.В. ВЯЗКОСТЬ
Вязкость характеризует движение молекул под действием
силы механического сдвига, так же как ионная подвижность и
коэффициенты седиментации характеризуют отклик системы на дей-
99 УСПЕХИ И НЕУДАЧИ
а
Поколения
Рис. 7.19. Применение центрифугирования в градиенте плотности [17].
л —фотографин УФ-спектров поглощения с полосами ДНК, появляющимися при
центрифугировании лизатов бактерий, анализируемых в разные моменты времени после
добавления к растущей культуре, меченной l5N, избытка субстратов, содержащих HN. Данные
получены через 20 ч центрифугирования при 44 770 об./мин (условия описаны в тексте).
Плотность раствора CsCl увеличивается слева направо. Области одинаковой плотности на
каждом спектре занимают одинаковое положение по горизонтали. Время анализа
измеряется с момента добавления I4N в единицах поколений. Время смены поколений оценивали
измерениями роста бактерий.- б — мнкроденситометрический анализ полос ДНК, показанных
иа соответствующих фотографиях. Смещение пера микроденситометра от нулевой линии
прямо пропорционально концентрации ДНК- Степень захвата радиоактивной метки в
образец ДНК соответствует относительному положению его полосы между полосами
полностью меченной и не содержащей метки ДНК (нижний рисунок, служащий уровнем отсчета
плотности). Проверку вывода о том, что ДНК а полосе промежуточной плотности мечена
наполовину, проводилась сравнением с интервалом при смешивании поколений 0 и 1,9. При
учете относительных количеств ДНК во всех трех пиках оказывается, что пик с
промежуточной плотностью находится в центре на расстоянии 50±2% между положениями пиков
"N н lbN.
ствие электрической или центробежной силы. Определение
вязкости позволяет описать поведение потока жидкости в узком
капилляре, и изучение такого потока служит наиболее простым
способом измерения вязкости. Наконец, вязкость можно связать с
200 ЧАСТЬ 2
коэффициентами трения для молекул разной формы, и эта связь
дает ряд способов определения формы молекул из данных
исследования седиментации, диффузии или вязкости.
При описании сил притяжения, действующих между 1023
молекулами, с помощью единственного параметра (вязкости) ясно, что
любой предполагаемый механизм должен быть вероятностным. Пусть
две параллельные плоскости, имеющие площадь А, находятся
друг от друга на расстоянии d (в приближении это очень тонкие
слои жидкости). Какую силу надо приложить, чтобы верхняя
-^v
т
1Z
плоскость двигалась относительно нижней со скоростью у?
Представляется вероятным, что при увеличении площади плоскостей,
сближении плоскостей и увеличении их относительной скорости
потребуется большая сила. Наконец, понятно, что сила,
необходимая для смещения одной плоскости относительно другой, зависит
от конкретной жидкости, в которой происходит этот процесс; все
эти интуитивные предположения можно записать в виде общего
уравнения
л»
(7.44)
где г\ — вязкость среды, F — сила, необходимая для создания
разности скоростей величиной в единицу, если площадь плоскостей
принята равной единице и оии находятся друг от, друга на
расстоянии, равном единице. Уравнение (7.44) характеризует один из
очень важных аспектов при анализе любого эксперимента, а
именно отделение геометрии эксперимента от исследуемого истинного
(физического) свойства системы.
7.В.1. ПОТОК ЖИДКОСТИ В КАПИЛЛЯРЕ
При рассмотрении потока через капилляр никаких новых
предположений, помимо уравнения (7.44), не требуется, но геометрию
эксперимента необходимо изменить. Рассмотрим теперь цилиндр
радиусом Ro и длиной L. Какую силу надо приложить для
проталкивания меньшего цилиндра жидкости внутри наружного
цилиндра со скоростью v? Замечая, что расстояние между цилиндрами
I'j УСПЕХИ И НЕУДАЧИ
*-v
равно разности (Ro—R) и что площадь поверхности движущегося
дилиндра равна 2kRL, из уравнения (7.44) получаем
F(Rp-R)
Поскольку
то в данном случае
V' 2nRLi\ '
Давление=Сила/Площадь,
F
(7.45)
Р=-
яЯа
Подставим выражение для силы F в уравнение (7.46):
, _ nR*P(R0 — R) _ PR(R0-R)
2nRLr\ 2Li\
V-
(7.46)
(7.47)
Наконец, считая, что R бесконечно близко к Ro, так что (R0—
—#)-»—dR и v-^dv, и интегрируя от поверхности капилляра до Ri
{вспомните, что в уравнении (7.44) у->0 при d->-0]j, получаем
скорость жидкости в любой точке внутри капилляра:
°§1 р Rl
о*1=1цг<*з-*&
(7.48)
Зависимость скорости жидкости от положения внутри
капилляра описывается, таким образом, простой параболой (рис. 7.20).
Скорость
течения.
Рис. 7.20, Распределение скоростей жидкости в капилляре с радиусом Ro
согласно уравнению (7.48) для ламинарного потока.
202 ЧАСТЬ 2
Такой поток служит примером
ньютоновского или ламинарного (слоистого)
потока, в котором все линии потока прямые в~
противоположность неньютоновскому
потоку, в котором в линиях потока
наблюдаются завихрения. Турбулентное течение
считают важным условием инициирования
процесса свертывания крови,
обусловленного значительными силами сдвига вблизи-
краев (неровного) пореза, что способствует
агглютинации тромбоцитов на неровной
поверхности. Образование тромба в
нормальной сосудистой системе предотвращается,,
так как внутренние поверхности
кровеносных сосудов имеют гладкие стенки,
препятствующие адгезии тромбоцитов и, таким
образом, препятствующие выходу из них
прокоагулянтов. Аналогично поток кровиг
из пореза, совершенного острым предметом,
ближе к ламинарному, чем поток из рваной
раны; поэтому при порезах бритвой
медленно образуется сгусток крови. Сгустку
трудно образоваться также в сосуде с
высокой скоростью потока крови (например,
в артерии), так как кровь столь быстро
уносит прокоагулянты, что их концентрация не
успевает достичь уровня, достаточного для
тромбообразования.
Простым устройством для измерения
вязкости является вискозиметр Оствальда-
(рис. 7.21), где измеряется время истечения определенного объема
жидкости, ограниченного двумя рисками а к Ь. Полученный
результат представляется как скорость жидкости [уравнение (7.48)], для
чего рассчитывается расход жидкости (объем/время); необходимо
учесть, что объем жидкости, протекающей через (бесконечно
малое) поперечное сечение (от R до R-\-dR) за единицу времени,
равен произведению скорости движения жидкости на площадь
бесконечно малого тонкого кольца v(2jtRdR). Суммарный поток
жидкости через всю площадь поперечного сечения капилляра,
следовательно, получается интегрированием:
Рис. 7.21. Вискозиметр
Оствальда [19].
Вязкость определяют по
времени, необходимому для
того, чтобы мениск жидкости
спустился от риски а до
риски Ъ (см. текст).
«=До
Поток (объем/время) =■
пР
2Lt\
JW-
R2] RdR,
V
t
nPRl
8Lr|
(7.Щ
203 УСПЕХИ И НЕУДАЧИ
[Интересно напомнить, что впервые уравнение (7.49) было
выведено анатомом Пуазейлем, изучавшим поток крови по
кровеносным сосудам.] Поскольку давление столба жидкости в опыте
пропорционально плотности раствора р, вязкость можно
определить из уравнения
-5- = const-/. (7.50)
Однако кинетическая энергия жидкости, вытекающей из
капилляра, выше, чем для жидкости в резервуаре наверху. Если принять
это во внимание, появляется дополнительный фактор, которым
можно пренебречь, если диаметр капилляра достаточно мал и
время течения t достаточно большое (несколько минут),
-^- =canst.*+const'.(l#). (7.51)
На практике вязкость получают, определяя отношение времен
протекания исследуемой жидкости и жидкости с известной
вязкостью в вискозиметре Оствальда:
Робразец'образец
^образец — ^стандарт п *
^стандарт* стандарт
(7.52)
Вискозиметр со стеклянным капилляром наиболее прост в
изготовлении и эксплуатации, но в нем жидкость находится под
действием постоянной (большой) силы сдвига, определяемой
высотой столба жидкости. Такие удлиненные молекулы, как ДНК,
обладают тенденцией при высокой скорости сдвига ориентироваться
длинной осью параллельно линиям потока, и для измерения их
вязкости из-за необходимости низких скоростей сдвига
требуются сложные вискозиметры, основанные на принципе измерения
механического или магнитного момента, требуемого для
вращения двух коаксиальных цилиндров друг относительно друга с
известной скоростью. Как описывается в ч. 4, ориентация несфериче-
•ских молекул при течении под действием силы сдвига позволяет
Определять форму молекулы.
7.В.2. ВЯЗКОСТЬ, КОЭФФИЦИЕНТ ТРЕНИЯ
И ФОРМА МАКРОМОЛЕКУЛ В РАСТВОРЕ
Если переписать уравнение (7.44) в виде
F=fo, (7.53)
то коэффициент трения / при скольжении одной плоскости
раствора относительно другой определяется простым соотношением
f=FditfA, (7.54)
>где у] —вязкость растворителя.
204 ЧАСТЬ 2
Вращение
вокруг оси а
Вращение
вокруг оси Ь
вытянутый эллипсоид
Сплющенный эллипсоид
Рис. 7.22. Вытянутый и сплющенный эллипсоиды вращения [1].
Дальнейшее уточнение геометрии задачи со сдвигом для
определения силы трения, создаваемого сферической
макромолекулой или макромолекулой более сложной формы, не представляет
трудностей, Однако утомительно. По существу, все это
включается в уточненное определение вязкрсти [уравнение (7.44)] для
жидкости между двумя плоскостями бесконечно малой площади
с бесконечно малым расстоянием между ними,' двигающейся с
бесконечно малой разностью скоростей; затем с помощью
обычных способов дифференцирования, (усложненных лишь для
задачи в трехмерном пространстве) приходим к уточненному виду
уравнения (7.44) и, интегрируя его по всем элементам поверхности
изучаемого объекта, получаем выражение вида Сила=/• Скорость.
Выражения для коэффициента трения при некоторых простых
формах объектов приведены в табл. 7.3, а вид вытянутого и
сплющенного эллипсоидов показан на рис. 7.22.
Влияние формы молекулы на коэффициент трения (табл. 7.3)
более наглядно при сравнении с коэффициентом трения, который
следовало бы ожидать, если бы молекула была сферой того же
объема (рис. 7.23). Поскольку сила трения прямо связана с
площадью поверхности исследуемого объекта и поскольку сфериче-
УСПЕХИ И НЕУДАЧИ
Таблица 7.3
[Коэффициенты трения, соответствующие некоторым простым геометрическим
формам движущихся тел
>„• форма
Коэффициент треиия
Обозначения
А' ■
(Сфера
Сплющенный
>■ эллипсоид / =
R (l-ftVtf)1'»
Щ ° (Ь/а)2/Чп{[1 + (1 -b2/a2)1/2J/*/a}
Вытянутый
' эллипсоид /
_ (fl2/62 —1)1/а
~6ЯТ1/?0 (a/&)2/3tg-i(aV62-l)^
Вытянутая f _ fi r,
(а/й)
2/з
палочка
(3/2)1/з{21п[2(а/6)]-0>11}
Я — радиус сферы
а —, большая ось, Ь —
малая ось, Яо—
радиус сферы равного
объема,
Ro={ab*)lh
а — большая ось, Ъ —
малая ось,
R0—радиус сферы равного
объема,
/?0=.(а^)1/з
а —г половина длины,
Ъ — радиус, /?0 —
радиус сферы равного
объема,
До=.(ЗЬ2а/2)1/з
ский объект из всех объектов равного объема имеет
наименьшую поверхность (см. задачи в конце главы), можно ожидать,
что в общем случае коэффициенты трения для сферических
объектов выше соответствующих величин для «эквивалентной» сферы
того же объема (рис. 7.23).
Гидродинамическую форму макромолекулы (т. е. ее форму в
растворе) экспериментально можно определить с помощью
любых методов, применяемых при определении коэффициента
трения, а также независимо, измеряя эффективный радиус молекулы;
тогда /о можно рассчитать из уравнения
/0 = 6m\R
Закон Сгокса
(7.55)
где R — радиус макромолекулы, принимаемой за сферу.
Радиус молекулы, как -правило, определяют, измеряя
светорассеяние или малоугловое рентгеновское рассеяние (см. ч. 4) или
рассчитывая парциальный удельный объем молекулы и ее
молекулярную массу. Если бы на поверхности молекулы не было гид-
2,5
2,0
Si:
1,Ь
f П
-
Ж^
&r
I 1 1
JO 20
Отношение осей, а/6
30
Рис. 7.23. Зависимость коэффициента трения от асимметрии молекулы для разных
эллипсоидов вращения [1].
fo — коэффициент трения сферы, имеющей объем, равный объему рассматриваемого
эллипсоида.
ратной оболочки, ее парциальный удельный объем описывался бы
уравнением
— (Объем молекулы) (Число молекул/моль)
веп* Число молей
=Объем 1 г вещества в растворе,
4nR*N
'■'вещ
ЗМ
п г ш - у/»
* [4nN Wbc«J
(7.56)
Любая реальная макромолекула несет определенное
количество 6 (граммы растворителя на 1 г сухого вещества) связанного с
ней растворителя (с парциальным удельным объемом ^растворитель),
и поэтому ее эффективный размер возрастает:
— AnN ^вец1 т~ "^растворитель!
(7.57)
Существует два простых способа нахождения коэффициента
трения / из экспериментальных данных:
1. Сольватация отсутствует, асимметрия максимальна. В этом
случае 6 в уравнении (7.57) считают равным нулю и, используя
рис. 7.23, определяют соответствующее отношение осей, которое
связано с величиной fffo [f0 рассчитан из уравнения (7.55), a f
!07 УСПЕХИ И НЕУДАЧИ
определен экспериментально, например с помощью измерений ко-
зффициента диффузии D= -j—].
2. Сольватация максимальна, молекулы имеют сферическую
[форму. В этом предельном случае предполагается, что
макромолекула имеет сферическую форму и 6 в уравнении (7.57)
увеличивают до тех пор, пока /, рассчитанный по уравнению (7.55), не
!совпадет с экспериментальным значением /.
\ Коэффициент диффузии D=— был использован для
расчета размеров молекул ряда белков; полученные данные
приведены в табл. 7.4. Поскольку молекула любого белка не обладает
правильной сферической формой и содержит связанную воду,
наиболее вероятное объяснение полученных результатов состоит в том,
что степень гидратации белка составляет 6 = 0,2 г/г (относительное
содержание гидратной воды).
' Существует еще один способ исследования формы
макромолекул из гидродинамических характеристик, основанный на
измерении вязкости раствора. До сих пор рассматривался коэффициент
[Трения (табл. 7.3), характеризующий замедление молекулы при
>ее движении в растворе под действием силы сдвига, причем
замедление зависит от формы молекулы и от вязкости растворителя.
;Родствеиная (и более сложная) задача состоит в определении
влияния суспензии больших молекул на характер потока,
поскольку, например, присутствие суспендированных сфер приводит к
искажению потока растворителя вблизи этих (больших) сфер, что
[влияет на измеряемую вязкость раствора. Эйнштейн (1906 г.)
показал, что в суспензии жестких сферических частиц (когда эти
!частицы намного крупнее молекул растворителя, так что
растворитель можно считать непрерывной средой) измеряемая вязкость
[раствора у] связана с вязкостью растворителя ^0 уравнением
I
Ч = 11оО +v«J>)
(7.58)
|где Ф —доля объема, занимаемого частицами в суспензии; для
(сферических частиц v = 2,5. Симха (1940 г.) вывел уравнения, ана-
i логичные приведенным в табл. 7.3, для эллипсоидов с
произвольным соотношением осей; результаты такого расчета
приведены на рис. 7.24.
I Пользуясь определением удельной вязкости
■"уд
и приведенной вязкости
ЛуА = Л=^' (7-59)
[т)] = 1ип(луд/с) (7.60>
с->0 ,УА'
Таблица 7.4
Размеры* белковых молекул, рассчитанные из их коэффициентов диффузии в предположении, что молекула представляет
собой либо идеальную сферу, либо она полностью лишена гидратноЙ воды [см. текст, для расчета
использовались уравнения (7.55) и (7.57)] [1]
Белок
Рибонуклеаза
0-Лактоглобулин
Сывороточный альбумин
Гемоглобин
Каталаза
Тропомиознн
Фибриноген
Коллаген
Мнозии
Молекулярная
масса,
дальток
13 683
35 000
65 000
68 000
250 000 \
93 000
330 000
345 000
493 000
"белок, «^
0,728
0,751
0,734'
0,749
0,730
0,710
0,710
0,695
0,728
Константа
диффузии6
D'107, см2/с
11,9
7,82
5,94
6,9
4,1
2,24
2,02
0,69
1,16
Максимальная
сольватация (f/f0=l)
0
0,35
0,72
1,07
0,36
0,70
23,0
8,4
218
49
о
R. А
18,0
27,4
36,1
31,0
52,2
96
106
310
215
Максимальная асимметрия
(о=0)
f/fo
1,05
1,16
1,25
1,05
1,15
а/Ь для
вытянутого
эллипсоида
3,4
4,9
6,5
3,4
4,9
62
31
300
100
Компромисс
(6=0.2);
а/Ь для
сплющенного
эллипсоида
2,1
3,7
4,9
2,1
3,6
а о —степень сольватации белка (граммы воды на I г сухого белка). R находят, подставляя экспериментально определяемое значение
коэффициента трення f=kT/D в уравнение (7.55) (предполагается, что макромолекула имеет форму сферы; вязкость для воды приняла равной 0,01 г/см).
Значение R подставляют в уравнение (7.57) и находят 6. f рассчитывают как R из экспериментально измеренного значения коэффициента
трения f=kT/D. Считают 6 равным нулю и рассчитывают R из уравнения (7.57). Затем рассчитывают /о по уравнению (7.66) и отношение flft, с
помощью которого и рис. 7.23 находят величину отношений осей а/Ь сплющенного эллипсоида.
6 Значения" (20 "С, вода) рассчитаны по уравнению (7.35).
УСПЕХИ И НЕУДАЧИ
3 /О
Отношение осей а/6
IS
'Рис. 7.24. Коэффициент вязкости v в зависимости от отношений осей вращения
эллипсоидов [уравнение (7.61)] [1].
i
(с— концентрация растворенного вещества) и уравнением (7.58),
можно получить следующее соотношение (см. задачи в конце гла-
Ъы):
М = V (Увещ +^растворитель)- (7*61)
Уравнение (7.60) исключает концентрационную зависимость
Иуд, поскольку г)уд должно быть пропорционально концентрации
вещества в растворе, по крайней мере при очень низких
значениях последней.
Далее уравнение (7.61) можно использовать практически так
же, как уравнения, описывающие связь коэффициента трения с
формой молекулы. Измерив приведенную вязкость раствора,
можно предположить, что либо v=2,5 (сфера), и тогда найти степень
гидратации 6 по уравнению (7.61), либо 6=0 (максимальная
асимметрия), и тогда рассчитать v по уравнению (7.61), а затем с
помощью рис. 7.24 найти соотношение осей, характеризующее
форму макромолекулы в растворе. Полученные с помощью
такого метода результаты для ряда белков (тех же, что и в табл. 7.4)
приведены в табл. 7.5.
Гидродинамические формы разных белковых молекул
изображены на рис. 7.25 с соблюдением единого масштаба.
Другие применения измерений вязкости основаны на
(эмпирической) зависимости вязкости раствора от молекулярной
массы растворенного вещества
[ч! = КМа
(7.62)
где а=0 для сферы, а = 0,5 для идеального статистического
клубка 'И а=1,8 для длинной жесткой палочки. Для полимера, значе-
Таблица 7.5
Размеры белковых молекул, найденные из значений их истинной вязкости [уравнение (7.61 )]8 [1]
Белок
Молекулярная — „, г , „,
масса «2. смэ/r [tj], смэ/г
Максимальная
сольватация (v=2,5)
6 (г/т)
о
Ro, A
Максимальная
асснметрия (6=0)
а/Ь для
V вытянутого
эллипсоида
Компромисс (в=0,2)
для v
а/Ь для
сплющенного
эллипсоида
Рибонуклеаза
Р-Лактоглобулин
Сывороточный альбумин
Гемоглобин
Каталаза
Тропомиозин
Фибриноген
Коллаген
Миозин
13 683
35 000
65 000
68 000
250 000
93 000
330 000
345 000-
493 000
0,728
0,751
0,734
0,749
0,73
0,74
0,710
0,695
0,728
3,30
3,4
3,7
3,6
3,9
^52
27
1150
217
0,59
0,61
0,75
0,69
20
10,1
460
86
19,3
26.6
33,7
34
91
112
400
257
4,5
4,5
5,0
4.8
70
38
1660
298
3,9
3,9
4,4
4,1
29
20
175
68
3.6
3,6
4,0
3,8
2,9
2,9
3,3
3,1
а Обозначения см. табл. 7.4.
fj УСПЕХИ И НЕУДАЧИ
Относительные размеры разных белков
Масштаб
rl
100 A Na+cr Глюкоза
Яичный альбумин Инсулин fi-Лактоглобулин
42000 36000 АОООО
Альбумин
69000
Гемоглобин
68000
fli-ГлобулиН
90000
CCf-Липопротеин
200000
J5j -/lunonpomeuH
1300000
/-Глобулин
156000
Фибриноген
АООООО
Эдестин
310 000
Зеин
50000
Желатин (недеградированный)
550000
£ис. 7.25. Размеры эллипсоидов вращения, которые наилучшим образом
объясняют гидродинамические свойства (вязкость и коэффициент трения) различных
белковых молекул [20].
'йия К и а у которого определены экспериментально, уравнение
■(7.62) позволяет оценить молекулярную массу; это уравнение
особенно полезно для характеристики синтетических полимеров.
С другой стороны, если молекулярная масса, например, белка
известна, то уравнение (7.62) позволяет охарактеризовать поведение
етолипептидной цепи в растворе (рис. 7.26). На рис. 7.26
приведена зависимость приведенной вязкости от молекулярной массы; по
тангенсу угла наклона прямой находят значение а, которое
свидетельствует о том, что при денатурации в 6 М гуанидингидро-
хлориде белки ведут себя как статистические клубки (0=0,66),
а ДНК — в большей степени как длинн-ая палочка (о=1,13).
212 ЧАСТЬ 2
4 5 6 7
IgM
Рис. 7.26. Взаимосвязь коэффициента вязкости от молекулярной массы.
/ — разные белки, денатурированные 6 М гуанидннгидрохлоридом [21]; 2— нативные ДНК
из разных источников :[21а]. Угол наклона аависимостн характеризует форму молекулы;
|[см. уравнения (7.62) и след.].
Определение приведенной вязкости широко используется при
исследованиях разворачивания (денатурации) белков: нагревание
глобулярного белка и#и обработка его кислотой или основанием,,
гуанидннгидрохлоридом или мочевиной, вызывает переход белка
в статистический клубок и приведенная вязкость в его растворе
возрастает [см. уравнение (7.62)], а при денатурации такого
белка, как коллаген или миозин, с характерной формой длинной
палочки из-за уменьшения степени асимметрии молекулы
приведенная вязкость падает. /
Вязкость является показателем протекания ферментативных
реакций, в которых происходит распад макромолекулярных
субстратов, например, при действии рибонуклеазы, деполимеризующей
РНК, или при действии гиалуронидазы на гиалуроновую кислоту;
роль обладающего высокой вязкостью сополимера N-ацетилглюкоз-
амина и глюкуроновой кислоты состоит в ограничении
распространения инфекционных агентов благодаря образованию «цемента»
между клетками. Наконец, измерения вязкости позволяют оценить,
степень сшивки в полимерах [при сшивке статистического клубка
значение а в уравнении (7.62) уменьшается]; исследования
вязкостных характеристик свидетельствуют о том, что полисахарид дек-
стран— разветвленный полимер глюкозы,— образуемый при
ферментации сахарозы определенными бактериями, приобретает
большую степень разветвленности при увеличении его молекулярной
массы.
7.Г. ВЫНУЖДЕННОЕ ПЕРЕДВИЖЕНИЕ. ПОЛЯРОГРАФИЯ
Большинство химических реакций, проведенных в обычных
условиях (допустим, смешиванием реагентов), необратимы, т. е.
(ГСПЕХИ И НЕУДАЧИ
к'йё происходят в обратном направлении. Большая ценность
Етрохимических методов состоит в том, что с их помощью мож-
)беспечить обратимость реакции и, следовательно, можно опре-
1ть константу равновесия данной реакции (гл. 5). Однако в слу-
Цразбавленных растворов (аВещ«/явещ) наблюдаемый окисли-
шо-восстановительный потенциал зависит от отношения кон-
граций, а не от абсолютной концентрации окисленной или вос-
*овленной формы
Е=Е°+^\п^=Е°+-^Ы-Шг (при25°С) (7.63)
[все обозначения имеют обычный смысл (см. гл. 5).
[Полярография — электрохимический метод измерения абсолют-
концентраций восстанавливающихся соединений, который поз-
^яет проводить определение даже в присутствии других
способах к восстановлению соединений; этот метод применяется очень
фоко, например в качестве недорогого метода обнаружения
Вгдов металлов в окружающей среде или для диагностики рако-
|к заболеваний. Экспериментально исследуется зависимость тока
Цотока «электрической жидкости») от наложенного напряжения
электрического давления»); поэтому следует начать изложение
рассмотрения протекания зарядов в электрической цепи.
[Ir Для обычной электрической цепи сила тока пропорциональна!
Ьшнему напряжению i=V7R, где R — электрическое сопротивле-
йе. Однако если в. цепь включен раствор электролитов, в который
^гружены два электрода, то
^отекание тока по цепи воз-
шно, если внешнее напря-
шие превзойдет минималь-
Je напряжение, необходимое
[я начала химической реак-
ги (кривые 1 и 2 на рис. 7.27).
|£ак следует из уравнения
рГ.63), минимальное внешнее §
^пряжение возрастает на *°
|),059/z) В при каждом деся-
^кратном уменьшении
концентрации восстанавливаемых
|еществ [Ох]. Интересная
ситуация возникает, когда
концентрация [Ох] настолько ма-
}а, что на кривой появляется
1лато (кривые 3 и 4).
Говорят, что электрод
«поляризован» всякий раз, когда его
|пектрический потенциал из-
"* Г££££.1~Ч Н^
-ЭОС-6не(инее г О
Рис. 7.27. Зависимость силы
стационарного тока от приложенного постоянного
напряжения (схема изображена на
рис. 7.28).
Концентрация (моль/л) восстанавливаемого
соединения [Ох]: / — 0,1; 2 — 0,01; 3 — 0,001;
4 — 0,0005. Все растворы содержат 1 М КС1
для увеличения проводимости при
минимальном приложенном напряжении.
214 часть 2
JC источнику
напряжения
К источник*/
напряжения
Катод
/Гч ©©©©
©0© 0©
Иача/гьное
состояние
■ у = 0
Катод
©
0
0
© ©
-jr»0
0^0 едги
© © ©0®
Стационарное
состояние
Рис. 7.28. Концентрация молекул восстанавливаемого соединения вблизи
поверхности плоского электрода в перемешиваемом растворе (см. текст).
меняется при прохождении по нему тока, как в случаях,
показанных на рис. 7.27. Поляризационное сопротивление (начальная
область на кривых рис. 7.27) появляется в результате падения
потенциала t'R, вызванного сопротивлением цепи. Концентрационная
поляризация (плато на кривых рис. 7.27) связана с диффузией (см.
рис. 7.28; именно это явление является главным предметом
дальнейшего обсуждения). /
На рис. 7.28 показано, что происходит с первоначально
гомогенным раствором, содержащим вещества, способные к
химическому восстановлению в катодной области яри наложении
напряжения, достаточного для восстановления этих веществ.
Предполагается, что восстанавливаемые соединения приближаются к
поверхности катода путем диффузии (какое-либо кулоновское
притяжение игнорируется) и очень быстро (по сравнению со
скоростью диффузии к электроду) восстанавливаются на его
поверхности. Следовательно, в непосредственной близости от катода
возникнет недостаток восстанавливаемых молекул, и это приводит к
созданию концентрационного градиента, изображенного справа на
рис. 7.28, и стационарный ток, который может протекать по цепи,
будет ограничен скоростью, с которой восстанавливаемые
молекулы могут диффундировать к катоду.
Силу ограниченного диффузией тока легко рассчитать. Поток
ионов через границу раздела на катоде (в молях на единицу
площади в единицу времени) определяется согласно первому закону
диффузии Фика
Поток = ■ -п- {*№
=-ч^ь
(6.51)
УСПЕХИ И НЕУДАЧИ
Гальванометр
г-©-
Резервуар с
ртутью
Азот, освобожденный
От кпп/мрпдп.. у-
Ртутный
анод
\(:
IV
I WWWWWv '
п
щ
I
Продувание
Юд может
. - Исследуемый
раствор с избыт
ком инертной
CO/1U
Рис. 7.29. Схематическое изображение полярографа.
азотом используют для удаления растворенного кислорода, поскольку кясло-
сам восстанавливаться и влиять на диффузионный ток, измеряемый в
эксперименте.
(■де £>ох — константа диффузии восстанавливаемых молекул. Из
&исла электронов z, переносимых в реакции, и из заряда F 1 г-экв.
Электронов сразу же получаем силу диффузионного тока
/ д [Ох]
т
i=—zFAD
Ох!
ду
У=0
(7.64)
Поскольку начальное условие [Ох] = [Ох];0 для всех у (т. е.
раствор гомогенен в нулевой момент времени), можно решить
/равнение (6.55) — второй закон диффузии Фика:
( д [Ох] \ _ [Ох]0
ду
(7.65)
(:iDOx01/3 '
Подстановка уравнения (7.65) в (7.64) приводит к искомому
результату
1дифф = zFA [Ox]0 (-^§Н
(7.66)
где А — площадь поверхности катода, находящаяся в контакте
с раствором.
На этом этапе могло бы казаться, что аналогичный путь
анализа надо применить для описания диффузии восстановленных
молекул к аноду, так что возникающий ток должен описываться
сложной комбинацией катодного и анодного процессов.
Нововведение, приведшее к созданию метода полярографии и принесшее
216 ЧАСТЬ 2
Рис. 7.30. Зависимость диффузионного тока от времени при использовании
капельного ртутного электрода [уравнение (7.69)].
автору, Ярославу Гейровскому, Нобелевскую премию, состояло в
создании аппарата с очень маленьким катодом и очень большим
анодом (рис. 7.29); в этих условиях текущий по цепи ток
ограничен исключительно диффузией восстанавливаемых молекул к
катоду. Кроме того, учитывая, что, поскольку загрязнения в
результате .адсорбции и других нежелательных необратимых реакций
особенно существенны для электрода малого размера,
поверхность катода необходимо непрерывно освежать, что легко
достигается при использовании капельного ртутного электрода
(рис. 7.29).
К новой геометрии катода (сферическому катоду) применить
предыдущий путь анализа проще, чем к плоской поверхности
катода. Объем данной капли ртути непосредственно перед
падением равен
4/3лг3=(т/р)/>
(7.67)
где г — радус капли, р — плотность ртути, т — масса ртути,
вытекающей в единицу времени, и t—интервал времени между
падениями двух капель. Решая уравнение (7.67) относительно г,
можно выразить площадь капли ртути:
4яг»=А=&иа/з **/з, (7.68)
где k — постоянная (включающая я и плотность ртути). Затем,
подставляя выражение площади [уравнение (7.68)] в уравнение
(7.66), получаем
*дифф
= /Cz/tt2/3/VeD$[Ox]0 (уравнение Ильковича) (7.69)
или просто
Уш
*дИфф==С0ПЙ 'ZD°* [°XJo-
(7.70)
Согласно уравнению (7.69), при падении данной капли ток
прерывается, а изменение его во времени описывается функцией
7б-порядка (рис. 7.30). Переменный ток (зубцы на рис. 7.30)
можно сгладить и превратить в постоянный ток (в среднем) с
помощью детектора (гальванометра) с достаточно низкой постоян-
"УСПЕХИ И НЕУДАЧИ
JV
%$Сатод
[Ох]
[Red]
[Ох].
Раствор
[Reo*]0 = 0
fc. 7.31. Схема концентрирования компонентов на поверхности катода (слева)
¥ ив объеме раствора (справа).
времени (см. гл. 8); можно показать, что средний ток равен
максимального тока.
| Уравнения (7.65) — (7.70) основаны на предположении о том»
iro ток лимитирует диффузия и, более строго, что концентрация
Постанавливаемых молекул на поверхности электрода практиче-
Йй равна нулю. В общем случае, однако, электрического «давле-
|я» (внешнего напряжения) может не хватать для создания это-
Ь, предельного условия. Для того чтобы преобразовать уравнение
t.70) и написать его в форме, соответствующей общему виду,
^дходящему для сравнения с экспериментом, рассмотрим поверх-
рсть катода, изображенную на рис. 7.31. (Если в начальный мо-
рят восстановленные молекулы отсутствуют, то эффективную
рнцентрацию [Red] о в объеме раствора можно принять равной
|гдю.) Уравнение Ильковича (7.70) теперь можно выразить в
рлее общем виде:
f t=const.zD$([Oxl0—[Oxl), (7Jla)
i=canst.zDj&i [Red], (7.716)
если диффузия — лимитирующая стадия процесса, при котором
рх]-^0, то
i ^^onst.zD$[Ox]0, (7.70)
е. получается уравнение, аналогичное (7.716).
Решая уравнения (7.70) и (7.71а) относительно [Ох] и урав-
:ение (7.716) относительно [Red]; получаем
[Ox^JwMnj (7.72а)
const-«D^» 1 '
[Red] = -
const -zD](ld
(7.726)
218 ЧАСТЬ 2
X
ej
/77
»3
S
*
ui
о
^
i f J.
\j
| /
и
J
/'
/ 1
/ i
И .
J 1 1
.
К
'
Еанешм (относительно нас.к.э)
Рис. 7.32. Полярограмма катодного восстановления катиона.
Потенциал линейно изменяется во времени.
Подстановка уравнений (7.72а) и (7.726) в уравнение Нернста
<7.63) приводит к
0,059 Л„( DRed \V2 , 0,059
Е^Е°
| 0>059 lgfDRed\V' I °'Q59 ig /" 'Д"ФФ —' \ (7.73)
Вид исходной кривой на рис. 7.27 теперь понятен из уравнения
(7.73): диффузионный ток (ордината плато для кривых 3 к 4)
непосредственно характеризует концентрацию восстанавливаемых
молекул в объеме раствора согласно уравнению (7.70); при этом
напряжение три полувысоте полярографической волны (силе тока)
составляет
где
л пел / п_ . \1/«
(7.75)
(7.74)
F F0 I. °'059 Ь ( DR*d Yh
-V*:
(Часто хорошее приближение получается, если принять £i/2=
— Е°у поскольку DRed и £>ох обычно имеют один порядок.)
График зависимости, описываемой уравнением (7..73), приведен на
рис. 7.32.
Практические приложения полярографии основаны или на
определении г (числа электронов, переносимых при превращении
молекулы реагента в продукт) либо [Ох]0 (концентрации данного
восстанавливаемого вещества в растворе), согласно уравнению
(7.70), или на определении £уа либо Е° (см. гл. 5: стандартный
•электродный потенциал Е°) по уравнению (7.74).
Пример. Обратима ли данная химическая реакция?
£" — одна из лучших характеристик для точного определения констант
равновесия (гл. 5). Однако найденные расчетом значения констант равновесия
надежны только в том случае, когда электрохимический процесс, к которому они
относятся, обратим. Простым (и общепринятым) критерием для решения
вопроса об обратимости реакции является определение тангенса угла наклона зависи-
Щ- УСПЕХИ И НЕУДАЧИ
-Ю,2 0 -0,2 -0,4- -0,6 -0,8 -1,0 -1,2 -1,4
Логпенциа/т ртутного электрода (относительно лас.к.э.')
~у>
7.33. Полярограмма водного раствора, содержащего Ю-4 моль/л Cu2+, T.1+
f и Zn2+ и в качестве вспомогательного электролита 0,1 моль/л KN03 [23].
Потенциал линейно изменяется во времени.
Е от lg
р'дифф—П.
: если найденное значение совладает с 0,059/г [где z —
|(ожительное целое число, согласно уравнению (7.73)], то реакция обратима.
1анном случае при обратимости реакций на электродах они происходят го-
Ы,о быстрее, чем диффузия любого из веществ к электроду.
I
рюмер. Одновременное селективное определение нескольких
| восстанавливаемых компонентов
| На рис. 7.33 показана полярограмма, полученная для раствора, содержаще-
нравные концентрации трех восстанавливаемых соединений, стандартные окис-
^ельно-восстановительные потенциалы которых достаточно различаются, так
ft возможно хорошее разрешение при полярографическом определении. Поля-
Графия — одни из простейших н наиболее дешевых методов определения сле-
Фых количеств ионов и особенно полезна для определения таких ионов, как
г+, для которых прямой химический анализ вызывает затруднения. .[Диффу-
(жный ток 1дифф прямо пропорционален концентрации восстанавливаемых
селений; см. уравнение (7.70).]
220 ЧАСТЬ 2
Н202*2НгО (
-Е&,(относительно нас. к. э.)
Рис. 7.34. Полярограмма восстановления молекулярного кислорода.
Пример. Число переноса электронов в данной реакции
Благодаря тому что диффузионный ток £дИфф пропорционален числу переноса
электронов при исследуемой реакции восстановления, это число легко определить
сравнением 1ДИфф в исследуемой реакции с 1дИфф для той же концентрации
раствора (например, ионов Zn2+) в реакции с известным числом переноса
электронов.
Например, как показано иа рис. 7.34, восстановление молекулярного
кислорода происходит в две стадии. Однако в присутствии фермента каталазы высота
первой волны возрастает, свидетельствуя, что каталаза катализирует диспропор-
ционирование перекиси водорода на воду и кислород: НгОг^'/гОа+НгО.
Следовательно, каталаза обеспечивает возможность превращения кислорода в воду
в условиях, не требующих использования более высокого напряжения
(электрического «давления»), которое потребовалось бы для появления второй волны в
отсутствие катализатора.
Можно привести и другой интересный пример: восстановление кобальта(3+)
в витамине В^— двухэлектронный процесс, в результате которого образуется
стабильный кобальт(1+) (необычная степень окисления кобальта). Становится
все более очевидным, что биологические функции соединений, содержащих иоиы
металлов, обусловлены искаженной геометрией окружающих лигандов, и пример
с витамином В^ один из первых продемонстрировал особенности бионеоргаии-
ческих комплексов.
Пример. Стехиометрия и устойчивость комплексов металлов
Для равновесия типа
Аравн
М*+ + рХ =*=* МХР (7.76)
можно показать, что зависимость —Еуя от —1&[Х] описывается прямой с
(отрицательным) тангенсом угла наклона 0,059 р[п. Такая зависимость приведена
иа рис. 7.35 для комплекса меди (И) с тиоаспирииом. Из данных, представленных
на рисунке, оказывается, что угол наклона этой зависимости равен 0,181, так
что р=0,181 :0,059=3,07. Можно также найти, что в данном случае константа
связывания равна 2,2-10". Детальное рассмотрение таких методов можно найти
в книге [22]. >
Пример. Полярографическое определение лекарственных веществ,
витаминов и гормонов
Полярографически можно определять широкий круг лекарственных веществ,
включая стрептомицин (структура показана ниже), хлорамфеиикол и сульфаиил-
УСПЕХИ И НЕУДАЧИ
3,60
|
1 3,40
й
3
£ 3.20
I
3,00
1 ) 1 1 . 1 I 1 I I I 1
0,08 0,10 0,12 0,14 0.16 0,18 0,20
~ '/2 (комплекс)
^ис. 7.35. Графический способ определения стехиометрии комплекса меди(И) с
тиоаспирином (см. текст) [11, с. 186].
Ймидные производные. В стрептомицине восстанавливается, вероятно, альдегидная
Группа
Р
1}
Стрептомицин.
У
\ Точность полярографического анализа выше точности микробиологического
*еста (см. обсуждение иммунодиффузии) при определении количеств
^200 мкг/см3.
I. Полярография позволяет определять несколько витаминов серии В и
витамин С (аскорбиновую кислоту). В случае витамина С—сильного
восстановителя — наблюдается двухэлектронное окисление на аноде
Витамин С
нонхнонсн
он он
С==С — С=С— ?± — С—С— + 2Н+ +
Л U он он И
V
2е
222 ЧАСТЬ 2
~Евнешнее (.относительно нас.к.э.)
Рис. 7.36. Полярограмма сыворотки крови человека в электролитной среде,
содержащей 0,001 моль/л хлорида кобальта, 0,1 моль/л аммиака и 0,1 моль/л
хлорида аммония [11, с. 163].
К числу стероидов, которые можно определять полярографически, относятся
кортикостерон, дезоксикортикостерон, прогестерон и тестостерон.
Пример. Использование полярографии при диагностике рака
В 1933 г. было обнаружено, что в присутствии иона кобальта на поляро-
грамме сыворотки крови человека появляется двойная волна со специфическим
максимумом (показан стрелкой на рис. 7.36) на второй волне. Позднее было
показано, что причиной такого вида зависимости является катализируемое белком
восстановление иона водорода на поверхности электрода (в отсутствие белка
восстановление иона водорода происходило бы при более отрицательных
напряжениях).
В любом случае интересная особенность заключается в том, что высота
двойной волны отличается лри анализе сыворотки здорового человека и больного
раком. Для увеличения этого различия используется эмпирический прием,
состоящий в измерении отношения высот полярографических волн при анализе
сыворотки, гидролизованной КОН, и сыворотки, обработартой сульфосалициловой
кислотой и профильтрованной; это отношение называют протеиновым, индексом.
В нормальной сыворотке протеиновый индекс равен 2,4±0,17. У больных раком
индекс заметно выше, как и у больных некоторыми другими заболеваниями,
такими, как артрит, подагра, шизофрения и язва желудка. Важность правильного
Диагноза переоценить невозможно — по последним оценкам, две трети больных,
погибающих от рака, могли бы быть спасены с помощью современных способов
лечения, если бы заболевание было у них обнаружено на несколько месяцев
раньше.
Задачи
1. Уравнения (6.55) и (7.19) демонстрируют независимость диффузии и элект-
рофоретической подвижности. Выведите уравнение (7.19) из уравнений (7.17)
и (7.18).
Замечание. Используйте правило последовательного дифференцирования.
в
УСПЕХИ И НЕУДАЧИ
Что произойдет при изоэлектрическом фокусировании, схематично
изображенном на рис 7.7, если анод соединить с основным концом ячейки, а катод —
& с кислым?
|i Используя данные рис. 7.15 подтвердите, что для рассматриваемого белка
{ 5=4,37 S.
|, Рассмотрим седиментационное равновесие в градиенте плотности. В области,
примыкающей к изопикнической точке г0, (стр. 195) для данрюго высокомо-
\i лекулярного вещества в растворе предположите, что градиент плотности
приблизительно линеен:
dp dp
р = (1 /увещ) 4- (г — г о) -д~, ~df да const.
Подставьте это выражение в уравнение (7.39), описывающее условие
обычного седиментационного равновесия, и решите его, выразив концентрацию
макромолекул как функцию расстояния от оси вращения г. Как будет
зависеть ширина данного распределения от массы макромолекулы?
3. На рис. 7.23 показано, что молекулы несферической формы при движении в
■'\ растворе испытывают большее сопротивление трения, чем сферические
молекулы того же объема. В основном этот эффект вызван тем, что
несферические молекулы обладают большей площадью поверхности. Убедитесь в этом,
сделав следующие расчеты:
а. Рассчитайте площадь поверхности сферической макромолекулы с
радиусом 10 А.
б. Рассчитайте площадь поверхности куба того же объема и сравните с
результатом, полученным в пункте а.
в. Рассчитайте площадь поверхности правильного цилиндра того же объема,
если длина цилвндра равна радиусу цилиндра или в 10 раз больше радиуса
цилиндра.
в. Какова размерность (в единицах системы МКС) ионной подвижности р,
коэффициента трения i/, коэффициента седиментации s, коэффициента
латеральной диффузии D при одно-, двух- и трехмерной диффузии и вязкости ц?
7. Для данного белка известно, что £>говс, н2о=3,7-10~7 см2/с; i>=0,72 см3/г и
молекулярная масса 300 000.
Принимая, что вязкость воды 0,01 г/см, рассчитайте
а. размер (диаметр) сферической молекулы белка;
б. степень гидратации б такой молекулы;
в. отношение осей для молекулы белка, имеющей форму вытянутого
эллипсоида и не содержащей гидратную воду;
г. отношение осей для молекулы белка, имеющей форму вытянутого
эллипсоида и содержащей гидратную воду (6=0,2);
Замечание. Сформулированные задачи (а — г) решаются (относительно
размеров, формы и степени гидратации макромолекулы) из данных диффузии.
8. Размеры, форму и степень гидратации макромолекул можно найти прямо из
данных вязкости с помощью уравнения (7.61) (см. следующую задачу).
Выведите уравнение (7.61) из уравнения (7.58) с помощью определений
удельной и приведенной .вязкости (стр. 207).
9. С помощью уравнения (7.61) и рис. 7.24 по данным, приведенным в трех
первых колонках табл. 7.5, рассчитайте значения, приведенные в шести
последних колонках этой таблицы. Эта задача служит примером определения
размера и/или степени гидратации макромолекул из измерений вязкости.
1в. Покажите, что из уравнения (7.74) следует вид графической зависимости
тока от напряжения, показанный иа рис. 7.32.
224 ЧАСТЬ 2
Литература
Трение
1. Тэнфорд Ч. Физическая химия полимеров. М.: Химия, 1965.
Электрофорез
2. Haglund H., Isoelectric Focusing in pH Gradients, Methods of Biochemical
Analysis, 19, 1 (1970).
3. Wieme R. /., Agar Gel Electrophoresis, Elsevier, 1961.
4. Cawley L. P., Electrophoresis and Immunoelectrophoresis, Little Brown & Co.,
Boston, 1969.
5. May pep t. P. Диск-электрофорез: теория и практике электрофореза в поли-
акриламидном геле. М.: Мир, 1971.
6. Everaerts F. M., Mulder А. /., Verheggen Th. P. E. M„ Isotachophoresis:
Analytical Tool in Electrophoresis, American Laboratory (Dec, 1973), pp. 37—
45.
7. Clinica Chimica Acta, 672 (1960).
8. Trayer H. R. et al., J. Biol. Chem., 246, 4486 (1971).
9. Haglund H., Science Tools, 14, 17 (1967).
10. Davis B. /., Ornstein L., Ann. N. Y. Acad. Sci., 121, 321, 404 (1964).
11. Meighen E. M. et al., Proc Natl. Acad. Sci. U. S. A., 65, 234 (1970).
Седиментация
12. Coates /. H., Ultracentrifugal Analysis, in: Physical Principles and Techniques
of Protein Chemistry, Part B, S. J. Leach (Ed.), Academic Press, New York,
1970.
13. Chervenka С. Н., A Manual of Methods for the Ultracentrifuge, Spinco
Division, Beckman Instruments Inc., Stanford Industrial Park, Palo Alto,
California, 1969.
14. van Holde К.. Е., Physical Biochemistry, Prentice-Hall, Englewood Cliffs, New
York, 1971. .
15. Morrod P. /., Marshall A. G., Clark D. G., Biochem. Biophys. Res. Commun.,
63, 335 (1975).
16. Crick F. H. C, Watson /. D., Proc. Roy. Soc. London, Ser. A, 223, 80
(1954).
17. Meselson M. S., Stahl F. W., Vinograd J., Proc. Natl. Acad. Sci. U. S. A.,
44, 671 (1958).
Вязкость
18. Bradbury /. H., Viscosity, in: Physical Principles and Techniques of Protein
Chemistry, Part B, S. J. Leach (Ed.), Academic Press, New York, 1970.
19. Shoemaker D. P., Garland C. W., Experiments in Physical- Chemistry, McGraw-
Hill, 1967.
20. Moore W. L, Physical Chemistry, Prentice-Hall, Englewood Cliffs, New York,
1972.
21. Reisner A. H., Rome /., Nature, 222, 558 (1969).
2\a.Eigner J., Doty P., J. Mol. Biol., 12, 549 (1965).
Полярография
2$. Purdy W. C, Electroanalytical Methods in Biochemistry, McGraw-Hill, New
York, 1965.
23. Schaap W. В., in: Moore W. J., Physical Chemistry, Prentice-Hall, Englewood
Cliffs, New York, 3rd ed., p. 410, 1963.
8
tKAH ВЕРОЯТНОСТЬ:
1ПРЕДЕЛЕНИЕ ПУАССОНА
ш
ИЗМЕРЕНИЕ РАДИОАКТИВНОСТИ
Шрирода радиоактивности и взаимодействие излучения с ве-
>м являются предметом исследования современной кванто-
JH механики; в этой главе будет рассмотрено использование ра-
Чгактивных изотопов для изучения локализации и концентрации
^логически активных химических соединений. Использование
|го метода основано на том, что вероятность того, что любое
щое ядро испустит излучение в течение периода наблюдения
£сть, за 1 с) намного меньше единицы и что в образце с Сюль-
И* количеством радиоактивных ядер среднее число
регистрируете импульсов пропорционально времени наблюдения.
Таким образом, условия данной задачи очень похожи на усло-
задачи с мешками и шарами, рассмотренной ранее; отличие
Цтоит только в том, что вероятность исследуемого события (на-
' >дения радиоактивного распада) a<til, а не а=7г, как
считать при выводе уравнения (6.4). Образно говоря, наблюдение
|диоактивности подобно поиску черного шара в мешке, почти все
рры в котором белые: вероятность обнаружения черного шара
щюактивного распада) в данном мешке (ядре) намного мень-
единицы, и надо узнать, сколько черных шаров (распадов)
>жно обнаружить при обследовании большого числа мешков
|дер). Путем интуитивного рассуждения, в точности аналогично-
приведшему к уравнению (6.4), можно получить уравнение
PN(m)
т
m\{N — m)\
am}j{N-m)
4ji . '
(8Л)
*е Ря(т) —вероятность вытаскивания т черных,шаров при ноис-
jfe шаров в N мешках, если для извлечения шара из мешка
девается одна попытка, а — вероятность того, что при любой одной
Попытке будет вынут черный шар, Ь — вероятность того, что при
|юбой одной попытке будет вынут белый шар. При применении
равнения (8.1) к решению задачи определения радиоактивности
щ других задач этой главы) предполагается, что вероятность вы-
226 ЧАСТЬ 2
нуть черный шар при одной попытке (или вероятность распада
любого из нестабильных ядер за время наблюдения) мала:
а«1. (8.2)
но общее число попыток (ядер) очень велико:
#»1, (8.3)
так что среднее число черных шаров, обнаруженных в данном
опыте, составляет
aN=ni, (8.4)
где т — любое число от нуля до ^ 1000.
Из уравнения (8.4) найдем a=m/N и подставим его в
уравнение (8.1)
^и=^br(l-)",(■-i),Л'-n,• м
где ясно, что
д+6 = 1. (8.6)
Уравнение (8.5) теперь можно привести к более удобному виду
D i ^ .VI (m\m(i m \N-m
Если N-*-oot все три параметра в уравнении (8.7) приводятся
к виду
ШпЛ=_^ (8.8а)
N-*oo ml
\imB=e-^ (по определению е*) (8.86)
N-*oa
limC=l (см. задачи в конце главы) (8.8в)
Л/-»оо
Уравнение (8.1) при ограничениях, описываемых уравнениями
(8.2) — (8.4), следовательно, приводится к виду
тт
Рм(т)=-тТе-т (8>9)
представляющему собой знаменитое распределение Пуассона.
УСПЕХИ И НЕУДАЧИ
3,25
JL
-Гм м I Пч
0 1 23456789 10
7»
$>ис. 8.1. Два биномиальных распределения, приближающихся к пределам Гаусса
f и Пуассона, указанным в табл. 8.1,
id— (0,5+0,5)10; б — (0,95+0,05)30. Обратите внимание на асимметрию кривой на правом
\\ графике.
ь
Чтобы проверить этот вывод, оценим среднее число событий
<т>, используя определение среднего значения дискретного
переменного [уравнение (6.38)]:
N _
<m>=lim 5]m
m'
т
N^-jo
m=Q
m!
e~m _
—e~m lim
==me~m lim
—me~mem
m - 2m2 . 3m? ■
l! ' 2| ' 3J 1
mil ' m
m 1 2! 1
<m> = m = Na
1 W
Mw
слг
V!
]-
]-
(8.10)
что согласуется с интуитивно полученным уравнением (8.4). В
качестве упражнения (см. задачи в конце главы) читатель может
самостоятельно доказать важное и полезное свойство
распределения Пуассона _
(8.11)
< (т — (т))2) =m=Na
Таким образом, средний результат большого числа отдельных
измерений радиоактивности составляет m = Na с
среднеквадратичным отклонением, равным у m = zh']/Na.
Смысл уравнений (8.9) и (8.11) легче всего понять, если
обратиться к рис. 8.1. В табл. 8.1 приведены основные
математические уравнения, используемые в этой главе.
228 часть г
Таблица 8.1
Иллюстрация общего происхождения распределения Пуассона
и нормального распределения (Гаусса) как особых случаев биномиального
распределения3
N\ кг
PN(m) = ти»-ш amb m
Биномиальное распределение
N\
m\(N — m)l
fl=r6 = (V2); n-»°° fl<i; л^-voo
Нормальное распределение (Гаусса) Распределение Пуассона
PN{m) = у*_ е-т-<г«»ыы Ры{т) в а^е-т
т\
<т> = ЛГа = 4 <■> — »«
и V<(m —<m»a> =
V<{m - <т»2> = /<т> =/М*
* Обратите внимание, что, хотя алгебраический вид распределения Пуассона отличается
от распределения Гаусса, принципиальное свойство (стандартное отклонение от среднего
результата) одинаково в обоих случаях.
При изучении радиоактивности: наиболее важным в
распределении Пуассона является тот факт, чтоГесли т — среднее число
импульсов, наблюдаемых за данное время, то за любой
конкретный промежуток времени будет зарегистрировано т импульсов;
m отклоняется от т не более чем на ±ут. Поскольку число ям-
пульсов растет с длительностью наблюдения (moct), а ошибка
измерения увеличивается пропорционально]/т, то, чтобы умень*
шить относительную ошибку —=1/_L в два раза (рис. 8.2), дли-
т V т
тельность измерения надо увеличить в четыре раза.
Пример
Сколько радиоактивных импульсов необходимо измерить, чтобы
относительная ошибка результата составила ±2%?
Решение. (1/Vm)=0,02,
m=2500 (±50) имп.
Прежде чем перейти к рассмотрению некоторых .приложений,
необходимо обсудить две практические трудности, связанные с
УСПЕХИ И НЕУДАЧИ
К.)
т
|^8.2. Ошибка при определении радиоактивности (числа импульсов) в
зависимости от среднего количества импульсов.
|цессом подсчета импульсов: фоновую радиацию (обусловлен-
космическими лучами, природной радиоактивностью окруже-
|) и время разрешения самого счетчика.
рСазалось бы, радиоактивность образца (наблюдаемое в еди-.
времени число импульсов)
(8.12)
л— t
>еделяется простой разностью суммарной радиоактивности (об-
|ец+фон) Лобщ и активности фона (в отсутствие образца),
л л л
■** —■Г1общ "фон*
|Однако ошибка измерения радиоактивности образца
<т—
(8.13)
(8.14)
(8.15)
(8.16)
(8.17/
Если активность фона измеряется за время /фош а активность-
бразца измеряют за время ?0бщ, то можно показать, что
отношение времени измерения должно составлять
{
[адывается из ошибок измерения А0бЩ и Лф0Н:
а = у о^бщ+Сфон=
У *1
~Я
общ
+
Щон
ф°н
-VI
А&
общ | j
общ 'фон
'фо
Щ *|/ ^общ
)Н V Лфои
(8.18)
Следовательно, если радиоактивность образца, например, п
;евять раз превышает радиоактивность фона, то измерения радио-
(ктивности образца целесообразно проводить в у!Г=3 раза доль*
Зе, чем измерения фона.
230 часть 2
ё |— " ■ ■ —- - — ■■ -■ ' — ■ ■ ■ ■■ ■ ■,.,— ■ ■
Пример
Радиоактивность образца (вместе с фоном) составляет 900 имп./мии, а
радиоактивность фона — 225 имп./мии. Сколько времени надо измерять активность
образца и активность фона, чтобы суммарная активность образца была
определена с ошибкой 2%?
Решение. Активность образца равна 900—225=675 имп./мин. Если ошибка
измерения равна 2%, то отклонение от этой величины составляет 0,02-675=
«=14 имп./мин. Следует найти отношение времен измерения
'фон ~~ У 225 ~^
или
*общ—^фон*
Подставим в уравнение (8.17)
«,« 900 . 225-2
14я =у- \~
*общ 'общ
И получим
'общ =6,9 МИН
и
'фон=3»5 МИН»
Известно, что любой счетчик радиоактивности после
регистрации данного импульса в течение некоторого короткого времени не
способен зарегистрировать следующий импульс (для счетчиков
Гейгера это время равно 100 мкс, для сцинтилляционных
счетчиков оно <10 мкс). Если период отсутствия чувствительности, или
мертвое время, обозначим т, а наблюдаемую радиоактивность —
Лнабл нмп./с, то в течение 1 с время неработоспособности
счетчика составит всего Лнабл*т;. За это время будет пропущено Л«
•Лнабл-т имп. (где А— истинное испускаемое образцом число
импульсов в 1 с). Следовательно,
А ^Наблюдаемые импульсы-f-Ненаблюдаемые импульсы
или
^==4набл+^^иабл^ (8Л9)
и
^набл
* — ^иаблт
А=,_7°- (8-20)
Для определения мертвого времени t счетчика необходимо
измерить число импульсов, регистрируемое в единицу времени для
1) образца № 1; 2) образца № 2 и 3) образцов № 1 и № 2.
Исправленные (истинные) активности (число импульсов в единицу
времени) образца № 1 и образца № 2 должны затем в сумме дать
исправленную активность образцов № 1 и № 2:
УСПЕХИ И НЕУДАЧИ
|скольку ?<1 и т2<Ст, уравнение (8.21) можно упростить:
г ^1 ~\~ А2— ^i+з /о 99\
|мер
Л Счетчик Гейгера зарегистрировал 1242 имп./с для образца № 1, 1371 имп./с
образца № 2 и 2209 имп./с для образцов № 1+№ 2 (вместе). Рассчитайте
^твое время счетчика и ошибку измерения, если соответствующая поправка
;введеиа. Фон составляет 3 имп./с, и им можно пренебречь.
Решение
т = —
1242 + 1371—2209
2-1242-1371
= 1,19-10-* с
гинная активность (число импульсов в секунду) для образца № 1 составляет
л1=пг
набл
1242
1-Л„аблТ ~ 1-1242 (1,19- Ю-4) •
i4i = 1460 имп./с, так что невведение поправки на мертвое время приводит к
гибке измерения ~ 15%.
(зотопное разбавление и метод радиоактивных индикаторов
Современная медицинская диагностика часто в какой-то сте-
(ени основана на определении количества данного метаболита в
конкретной жидкости или ткани организма. Однако, хотя в
принципе нужное вещество можно выделить с высокой степенью
чистоты, получить его с высоким выходом обычно невозможно. Метод
[зотопного разбавления позволяет обойти эту трудность, и на нем
сновано широкое использование радиоизотопов в медицине (см.
римеры).
При изотопном разбавлении радиоактивность-служит в
качестве характеристики концентрации радиоактивного материала,
рели По граммов меченого соединения с удельной активностью Ао
рмешать с ппсСл граммами такого же соединения, не содержащего
радиоактивный индикатор, то удельная активность смеси
уменьшится:
Л=Л0[—^ 1
(8.23)
{так же как концентрация обычного реактива падает при его
разведении растворителем). Сделав необходимые преобразования
уравнения (8.23), можно определить количество неактивного
соединения:
An —A
Яиссл — Я0
(8.24)
232 часть я
Пример. Определение общего объема крови в организме человека
а. Введение радиоактивной метки в плазму крови. Сывороточный альбумин
человека (белок с молекулярной массой 68 000, в норме присутствует в крови
человека) иодируют ш1 (т. е. ковалентно соединяют с иодом) по некоторым
остаткам тирозина в ojwo-положении к гидроксильной группе. 10 см8 такого
раствора альбумина с активностью 1 мкКи/см3 (1 Ки=3,7-10'° имп./с) вводят в
локтевую вену; после внъекции равновесие в кровеносной системе наступает
через ~10 мнн« Затем у подопытного (человека) берут образец крови объемом
3 см3 и измеряют в нем радиоактивность.
Если радиоактивность 3 см3 образца крови составляет 3287 имп./мнн,
активность фона 3 см3 образца крови (взятой у того же человека до введения ему
меченого альбумина) —175 имп./мни, а активность меченого альбумина (3 см3),
использованного для инъекции при разведении 1 :200 (приблизительно в таком
соотношении находятся объем введенного раствора меченого альбумина и объем
крови) — 5702 нмп./мнн, то объем плазмы составляет
Объем плазмы^200'5702 ^'7f-'^7-175)-10 см°=3550 см>=3,5 л
Наконец, относительный объем клеток крови можно определить,
центрифугируя образец крови и измеряя объем осажденных клеток. В данном примере
объем клеток крови, по-виднмому, равен 2,8 л, тогда общий объем крови
(плазма плюс клетки) равен 6,3 л.
б.Введение радиоактивной метки в эритроциты. 51Ст инкубируют с образцом
крови пациента до тех пор, пока большая часть 51СгО!~ не связывается с
эритроцитами. Затем добавляют восстановитель |(пусть, аскорбат натрия) для
восстановления непрореагировавшего CrgO|~ до Сг3+, Сг34" почти исключительно
связывается с белками плазмы. После инъекции, достижения равновесия и отбора
крови, как ранее,,можно определить радиоактивность образца меченой крови до
инъекции, радиоактивность цельной крови послб"инъекцнн н радиоактивность
плазмы (получаемой центрифугированием) после инъекции и вычислить объем
эритроцитов.
Измерение объема крови особенно нужно при решении вопроса о необходн-
мости переливания-крови или плазмы при острых кроволотерях, ожогах или
хирургических щоках; таким образом было спасено много человеческих жизней..
Объем кровн в организме худых людей приблизительно прямо
пропорционален массе тела и составляет у мужчин ~80 см'/кг. У толстых людей, однако,
Отношение объема кровн к массе тела меньше из-за того, что в жировой ткани
мало кровеносных сосудов. Объем крови у женщин ~65 см3/кг, в среднем на
20% меньше, чем у мужчин, поскольку у женщин выше содержание жировых
тканей.
Хотя общий объем крови .может до известной степени изменяться при
заболеваниях, более чувствительным диагностическим показателем является
отношение объема эритроцитов к общему объему кровн, так называемый гематокрит.
При острой анемии гематокрнт из-за нехватки эритроцитов может уменьшаться
(норма для мужчин 40%, для женщин 36%) до 15%. В противоположность
этому избыток эритроцитов (гематокрит до 70%) появляется у людей,
длительное иремя находящихся на больших высотах, или у больных с опухолью крове-
творящих органов. Наконец, поскольку в основном возврат объема крови к
норме после резких изменений (например, после потребления большого объема воды)
регулируется почками, кинетические измерения объема кровн при
преднамеренном изменении ее объема служит средством контроля за функциями почек. Во
Всех этих случаях использование радионзотояных методов позволяет быстро
получать точные результаты.
'УСПЕХИ И НЕУДАЧИ
|4ф. Функция щитовидной железы
гя физические свойства (стабильного) 1271 и (радиоактивного) ш1
разлился, их химические свойства одинаковы, так что 1311 является естественным
>активным индикатором для исследования метаболизма иода и его соеди-
Jty связанного с функцией щитовидной железы. Щитовидная железа обла-
?<уннкальной способностью избирательно концентрировать н удерживать иод
^Последующего превращения о иодотирозин н затем в тироиин.
|ча. Поглощение иода. Хотя общее количество иода в организме человека
прицельно постоянно н на него сравнительно мало влияет,-нарушение, функций
ШДной железы, скорость поглощения иода является хорошим диагиостиче-
; показателем активности щитовидной железы; в норме щитовидная железа
1ение 1 ч поглощает 0,5—6,8% общего количества иода, циркулирующего в
шзме; эта величина увеличивается до 2,5—6,8% при гиперфункции щитовид-
, железы и 7меиьшае,1ся ДО 0—1,3% при микседеме. Поглощение ш1 легко
грять, определяя радиоактивность в некоторой точке иа Шее больного, рас-
|юженной прямо над щитовидной железой; для калибровки используют
неразменный образец ,3,1, помещенный на таком же расстоянии от счетчика:.
£, б. Иод, связанный с белком. Определить-поглощение иода непросто, посколь-
гиперактивная щитовидная железа быстро концентрирует иод, однако она
»ускает его (также быстрее, чем в норме) в ток крови в связанном с белком
не. Радиоактивный 1511 вводят пациенту, как описано выше, но на этот раз
>сле уравновешивания) берут пробу крови, выделяют из образца крови бел-
*ую фракцию (допустим, осаждением) и измеряют радиоактивность в этой
^акцин и в остальной части образца: отношение количества иода, связанного
Мелком, к общему циркулирующему 13Ч служит хорошей характеристикой ак-
jhocth щитовидной железы.
пример. Локализация опухолей мозга
Определенные изотопы, например 74As или 64Си, излучают позитроны. Сразу
fee после эмиссии позитрон распадается на два гамма-луча, направленных в нря-
\6 противоположных направлениях. Мозговые опухоли проявляют тенденцию к
!}Нцеитрированию таких ионов; прикладывая счетчики к противоположным
стоим головы пациента и фиксируя только те события, которые регистрируются
5оими счетчиками одновременно, можно с довольно высокой точностью лока-
иовать положение опухоли,
1рнмер. Пернициозиая анемия
При этом заболевании содержание витамина Ви в крови уменьшается и со-
гавляет только небольшую часть относительно его содержания в норме. Бнохнь
(ическая причина заболевания, по-видимому, связана с отсутствием определен-
юго мукопротеииа в кишечио-желудочном тракте; в норме этот белок соеди*
1яется с витамином В^, способствуя повышению растворимости витамина и era
Поглощению кровью. Поскольку витамин Bia содержит кобальт, простейший те<~р
^ia это заболевание заключается в следующем. Пациенту дают некоторое
количество витамина Bi2, меченого 58Со, а затем через ^8 ч определяют уровень
;<8Со в крови.
|1ример. Контроль циркуляции крови
Препятствия (сгустки, тромбоз) или сужения различных кровеносных
сосудов также легко детектировать. Для этого перед предполагаемым пораженным
участком вводят 24NaCl н регистрируют радиоактивность в разных точках
исследуемого сосуда. Аналогичный тест используют при контроле работы сердца:
гводят эритроциты, меченные "Сг, в вену и измеряют радиоактивность в аорте.
234 ЧАСТЬ 2
Пример. Длительность жизни эритроцитов
При многих анемиях срок жизни эритроцитов отличен от нормы,
составляющей —120 сут. В образец крови пациента добавляют клетки, меченные 5,Сг, и
вводят одну часть образца пациенту и другую часть здоровому человеку; при
этом в обоих случаях можно получить неблагоприятные результаты. Если
обнаружено, что срок жизни эритроцитов слишком мал в обоих случаях, то
заболевание связано с аномалией самих клеток. Если же срок жизни эритроцитов мал
у больного н нормалей у здорового человека, то это свидетельствует о том, что
в крови больного существует какой-то агент, разрушающий эритроциты.
Вышеприведенные примеры иллюстрируют наиболее типичные
применения радиоактивных методов, в том числе и метода
изотопного разбавления, в медицинской диагностике. Метод
радиоактивных индикаторов широко применяется и в биохимии: в метаболи-
зируемый предшественник вводят метку и определяют степень
обогащения меткой в последующих продуктах метаболизма;
таким образом удается выяснить химические механизмы, с помощью
которых молекулы трансмутируются из одной формы в другую.
Примеры такого использования метода можно найти в учебниках
биохимии.
8.Б. ЭЛЕКТРИЧЕСКИЙ ШУМ
При биофизических исследованиях регистрация данных, как
правило, производится с помощью какого-нибудь электрического
устройства, например рН-метра и других подобных приборов или
самописцев, регистрирующих функции организма. В
противоположность тому, что можно было бы ожидать, работоспособность
таких устройств иногда в большей степени определяется
внутренним шумом устройства, чем слабостью" регистрируемого
сигнала,— определяющим фактором является отношение сигнал/шум.
Поскольку биологические концентрации большинства веществ
низки по сравнению с оптимальными для экспериментального
измерения, ясно, что для прямого изучения интересных веществ, как
правило, требуется регистрация слабых сигналов. Чтобы успешно
бороться с шумами, поэтому необходимо иметь' представление о
природе шума, наблюдаемого в данном электрическом устройстве.
К счастью, природу некоторых видов электрического шума можно
достаточно просто объяснить с помощью модели случайных
блужданий.
8.Б.1. ИМПУЛЬСНЫЙ ШУМ
Любая электрическая цепь, в которую включены вакуумные
трубки или транзисторы, содержит один или более разрывов: в
вакуумной трубке катод отстоит от анода или, например,
разрывы образуются в точках контактов в транзисторе. Измерение
электрического тока заключается в подсчете электронов, прохо-
УСПЕХИ И НЕУДАЧИ
IX через такие контакты. Однако, как и при измерении радио-
живности, вероятность того, что любой данный электрон прой-
через контакт, намного меньше единицы, в то время как через
|ку контакта пройдет потенциально большое число электронов.
|ая ситуация анализировалась выше: если число электронов,
|ходящих через контакт в единицу времени, составляет 1/е (где
\gjt-ток, а е — число единиц заряда на электроне), то общее
ю электронов, проходящих по цепи за некоторое время т,
шо
— /т
т=—
е
(8.25)
>шибка измерения Am вычисляется так же, как ошибка изме-
шя радиоактивности образца с большим количеством ядер:
Дт
-V4--V*-
(8.26)
\ Отношение сигнал/шум, следовательно, составляет
(8.27)
Уравнение (8.27) показывает, что для увеличения отношения
1тиала^к шуму в два раза, необходимо увеличить сигнал (входной)
% четыре раза (а не в два, как можно было бы ожидать). Чтобы
>нять смысл фактора т в уравнении (8.27), надо выяснить, для
вакой цели используется данная электрическая цепь. Обычно
требуется, чтобы данная цепь реагировала на конкретное изменение
\гк можно быстрее: самописец спектрофотометра должен
регистрировать сигнал за время, в течение которого монохроматор ска-
[рует исследуемую область длин волн; рН-метр должен регист-
|ировать значение рН за время, за которое титрующий раствор
«водится в ячейку, и т. д. Постоянная т характеризует кратчайшее
|время, за которое цепь должна оказаться способной
зарегистрировать отклик системы. В электротехнике более типичной
характеристикой цепи является ширина полосы пропускания частот
Ширина полосы = —.
(8.28)
Ширина полосы, следовательно, соответствует наибольшей
частоте, при которой устройство может реагировать на изменение
сигнала. Импульсный шум, следовательно, можно уменьшить,
«отфильтровывая» более высокие частоты, т. е. уменьшая ширину
полосы или (что то же самое) увеличивая постоянную т данной
электрической цепи.
236 ЧАСТЬ 2
Рис. 8.3. Осциллограмма дробового шума в электрической цепи фотоэлемента
(детектора в спектрофотометре) [6],
I — постоянная времени депи в десять раз превышает постоянную времени цепи, которой
соответствует диаграмма 2. Согласно уравнению (8.27), амплитуда шума в кривой 1
уменьшена по сравнению с нижней кривой приблизительна в К)1/» раз.
Результаты, полученные в предыдущем разделе,
количественно описываемые уравнением (8.27), в действительности всего
лишь отражают эксперимент. Ясно, что отношение сигнала к шуму
можно улучшить, увеличивая интенсивность сигнала, например
увеличивая щель в спектрофотометре. Однако разрешение при
этом падает, т. е. уменьшается способность установить различие в
поглощении света при незначительно различающихся частотах.
Этот пример типичен для экспериментальных ситуаций, в которых
используют электронные измерительные приборы, —для
получения максимума информации (наивысшего разрешения в данном
случае) необходимо «пожертвовать» отношением сигнал/шум.
При решении проблемы ширины полосы приходится идти на
аналогичный компромисс; рис. 8.3 показывает, что снизить шум пера
самописца достаточно просто: для этого следует уменьшать
постоянную т (тогда перо не успеет отметить шумовые флюктуации).
Однако, чтобы перо реагировало на исследуемый сигнал
(допустим, регистрировало пик поглощения при сканировании длин
волн оптического спектра), необходимо замедлить скорость
развертки в соответствующее число раз, т. е. увеличить время
эксперимента. Это пример еще одного компромисса, на который
приходится идти при использовании электронных измерительных
приборов: если надо уменьшить шум, время эксперимента должно-
быть увеличено.
8.Б.2. ШУМ СОПРОТИВЛЕНИЯ (ШУМ ДЖОНСОНА, ШУМ НАЙКВИСТА^
ТЕПЛОВОЙ ШУМ)
Гипотеза о том, что тепловой электрический шум прямо
связан с броуновским движением (т. е. с обязательным случайным
УСПЕХИ И НЕУДАЧИ
Иристическим движением) электронов в сопротивлениях» была
И&казана Найквистом вскоре после открытия самого явления
Яконсоном в 1928 г. В любом обычном проводнике электроны мо-
№ свободно перемещаться соответственно их тепловой энергии
Ни. ч. 5). Поскольку это движение является случайным, в любой
■рный момент времени на одном конце сопротивления может ока-
Кься больше электронов, чем на другом конце сопротивления, и
Н& приводит к возникновению разности потенциалов (напряже-
■ш) между двумя концами сопротивления. В следующий момент
Иагодаря перераспределению электронов напряжение, вероятно,
Ю*ет другим. Эту ситуацию можно рассмотреть с помощью моде-
щ, согласно которой электроны могут перемещаться по сопротив-
Ьию вперед или назад, используя решение задачи с мешками и
«рами, рассмотренной в начале данной главы, практически так
ке, как оно применялось при анализе случайных блужданий и ста-
Встического клубка (разд. 6.А). Колебания напряжения можно
■ргразить формулой
lie k — постоянная Больцмана, Т—абсолютная температура,
б — омическое сопротивление резистора, В — ширина
рассматриваемой полосы частот.
г Наиболее характерная особенность импульсного шума и теп-
нового шума — их независимость от ширины полосы. По аналогии
I «белым» светом, для которого характерны равные интенсивности
|а всех частотах, этот вид шума называют «белым» шумом. Вид
шума, рассматриваемый ниже, этим свойством не обладает.
|:б.з. (1//)-шум (фликжер-шум)
!' В твердофазных электронных устройствах серьезным
источником шума на^изких частотах является так называемый фликкер*
шум, обратно зависящий от частоты (рис. 8.4). Дрейф при
измерениях тока — медленное смещение измерителя или самописца —
доставляет особенно много хлопот. Происхождение этого вида
щука неясно, но оно может быть связано со степенью дисперсности
материалов проводников. В любом случае фликкер-шум можно
сделать пренебрежимо малым, проводя все измерения на
частотах >1000 Гц; поэтому во многих устройствах, регистрирующих
Достоянный ток или медленно изменяющийся сигнал, встроены
•механические прерыватели (например, в спектрофотометре такой
прерыватель прерывает световой луч много раз в секунду) или
быстродействующие электронные переключатели для превращения
сигнала постоянного тока в сигнал переменного тока перед
регистрацией.
(8.29)
238 ЧАСТЬ 2
10
-10 —
■". ю-"
I
« 10
\
.-/2
/О
-/4
ldc = 25MKA
Уровень шума
_ Никвиста
10
100 Ю00 10 000
Частота, Гц
Рис. 8.4. Экспериментальный шум композитного сопротивления в 22 МОм под
напряжением [4].
Обратите внимание, что спектр обратно пропорционален частоте в присутствии тока /jc
и в отсутствие тока имеет место лишь «белый» тепловой шум.
8.Б.4. УСТРАНЕНИЕ ШУМА: УСРЕДНЕНИЕ СИГНАЛА
Основными средствами устранения шума являются:
фильтрование, модуляция, фазовочувствительное детектирование или
усреднение сигнала. Первый из этих способов упоминался выше;
второй основан на введении в схему прерывателя, как описано в
предыдущем разделе. Усреднение сигнала представляет собой
пример решения проблемы случайных блужданий; В качестве
простого примера можно рассмотреть измерение спектра поглощения
молекулы.
Типичный спектроскопический эксперимент (ч. 5) состоит в
измерении поглощения излучения веществом в зависимости от
изменяющейся частоты излучения; регистрация осуществляется с
помощью пера самописца, двигающегося по специально
разлинованной бумаге. При любом одном таком опыте шум нулевой
(базовой) линии может исказить измерения. Однако, если проведено
много сканирований, проявляется интересная характерная
особенность; если выбрать конкретную частоту, то положение пера
самописца при данном сканировании от шума определяется так, как
если бы перо могло двигаться только вверх и вниз на одну и ту же
величину от базовой линии. Следовательно (разд. 6.А), согласно
модели случайных блужданий, среднее расстояние, на которое
передвинется перо самописца при множестве перемещений
(множестве сканирований в данном случае), увеличивается
пропорционально корню квадратному от числа перемещений (сканирований).
Этот вывод иллюстрирует рис. 8.5, где сравниваются спектры,
полученные при одном сканировании и при множестве сканирований.
УСПЕХИ И НЕУДАЧИ
t
V
Прогестерон
Сумма 500
Сканирований
500 fit
Ыс. 8.5. Спектр протонного магнитного резонанса женского полового гормона
рогестерона, для которого видно увеличение отношения сигнал/шум при суммн-
Ьваиин большого количества индивидуальных сканирований (вертикальная шка-
а верхнего спектра сжата, чтобы облегчить сравнение с нижним спектром) [7].
Рример
|. При изучении разбавленного раствора фермента лизоцима, лнзнрующего
^леточные стенки, методом протонного магнитного резонанса обнаружено, что в
результате сложения 30 спектров (30 сканирований) одного и того же вещества
|олучается одни спектр с отношением сигнала к шуму 2:1. Предполагая, что
бгум является «белым» (т. е. случайным по частотным характеристикам),
рассчитайте необходимое число сканирований, которое в сумме позволило бы полу-
Нить спектр с отношением сигнала к шуму 7:1.
| Решение. Сигнал возрастает пропорционально числу сканирований, а шум
растет пропорционально квадратному корню из этого числа. Следовательно,
i S/JV=const-y7^
»де п — число сканирований, так что
I- 2=const."j/30.
ожно записать, что
7 =: COnSt *У~П~,
р иайтл отношение этих двух уравнений:
7/2=V^/30,
л=С/2)2.30=368 сканирований.
f В этом конкретном примере одно сканирование занимает 500 с; для того
чтобы S/W=7, исследователь должен затратить 51 ч только на запись, т. е. бо-
•лее двух суток непрерывного сканирования! Методы преобразования Фурье
'(гл. 6) сокращают это время в —1000 раз, поэтому теперь такие спектры
биологически интересных молекул стали доступны.
240 ЧАСТЬ 2
Рис. 8.6. График вероятностей Пуассона: вероятность Р того, что событие
произойдет не менее с раз при обычном (ожидаемом) числе событий m [1, с. 105].
8.В. ПОМОГЛО ЛИ ЛЕЧЕНИЕ ПАЦИЕНТАМ?
Важной проблемой в медицинской практике является
правильная оценка состояния пациента (нормальное ли оно или есть
отклонения), а также определение эффективности данного способа
лечения больного (приводит ли оно к улучшению состояния
больного). Хотя ни один показатель не является однозначным,
количественная оценка диагностического теста необходима. При срар-
нении, например, формулы крови пациента со средней формулой
крови нормальных здоровых гсациентрв или коэффициента
смертности при новой методике лечения с коэффициентом, характерным
для уже существующей, методики, оказывается полезным
использование распределения Пуассона (рис. 8.6), тем более что во
многих медицинских примерах число экспериментальных точек мало.
На рис. 8.6 охарактеризована вероятность того, что событие
произойдет_по меньшей мере С раз, если ожидаемое число
событий равно т. Например, если ожидаемое число событий равно 2,
вероятность того, что при данном наблюдении будет
зафиксировано 5 или более событий, составляет всего 20%.
Пример. Успех нового способа лечения
Из опыта лечения определенного заболевания .известно, что коэффициент
смертности при отсутствии лечения составляет 10%. При испытании нового
способа лечения иа группе из 40 больных умерло 7 человек. Согласно
распределению Пуассона, если ожидаемое число событий (в данном случае смертей)
составляет 0,10*40=4, то вероятность того, что в отсутствие лечения случится 7
или более смертей составляет всего 10%. Следбвательио, новый способ лечения
с вероятностью 90% оказывается хуже, чем старый способ.
Применимость распределения Пуассона носит исключительно
общий характер. Первое успешное совпадение эксперимента с те-
УСПЕХИ И НЕУДАЧИ
шей получено для данных, собранных Борткевичем (см. зада-
в конце главы) о вероятности гибели кавалериста в течение
)да при падении с лошади (данные соответствуют зависимости,
приведенной на рис. 8.1,6). К числу других примеров относятся:
^следование числа звонков по общественным телефонам, числа
|шибок при наборе телефонных номеров, числа эритроцитов, бак-
|ерий или вирусов при подсчете, числа смертей от рака в семье,
|кисла вакансий в Верховном суде, числа забастовок в неделю,
!аисла пуль, попадающих в цель, и т. д. (см. задачи в конце гла-
Ьы).
л'
кг. хроматография
Естественным образом распределение Пуассона используется
при разработке теоретических основ определенных хроматогра-»
фических методов (основанных на распределении жидкость —
жидкость, газ — жидкость, а также таких, как
гель-хроматография, ионообменная хроматография и аффинная хроматография).
Как обычно, вывод основывается на интуитивно предложенной
модели: предполагается, что можно рассматривать хроматографиче-
скую колонку как набор теоретических.дисков или тарелок,
каждая из которых содержит объем растворителя V (рис. 8.7).
Очевидно, что после добавления N раз бесконечно малых доз
растворителя при условии, что система приходит в равновесие после
1О0
dV
iVJL
т
dV^l
dyj^
V-dV -е-
L^L-
tcjo
V-dV-
V-dV *
«44*)
M.(,-ffi>
'^.(fX'-f)
•«•ft?
a
в
Рис. 8.7. Схема хроматографического процесса прн прерывистом потоке раствб*
рителя. '6,ч'.
В начале опыта (а) колонка рассматривается в виде набора большого ядаяач
Гипотетических «тарелок»; образен, находится в верхней тарелке. Затем (б) добйий1^р небольшой
.объем dV растворителя, « соответствующий объем dV растворителя Лдокает из колонки
<снизу). Тогда первая тарелка содержит объем dV с нулевой концеятрДцней растворенного
вещества н объем (V—dV) с концентрацией вещества i[C]0. При уравновешивании (в)
предполагается, что первая тарелка представляет собой гомогенную cpefty с концентрацией ве«
ицества [C]0[(H-yfV)/V]-"[C]0[l—(dvlv)], а концентрация вещества: во второй тарелке а*
«тавляет \C]e(dV/V). При повторном введении растворителя (объемом dV) в результате
уравновешивания происходит новое распределение вещества, среди разных гипотетических
тарелок колонки (в). Таком образом можно логически вывести биномиальное распределение*
242 ЧАСТЬ 2
каждого последующего добавления, конечная концентрация на
данной тарелке (пусть m-й, если считать сверху вниз) описывается
биномиальным распределением
|СИНС,^(^( 1-fp*. (8.30)
где [С(т)]—концентрация растворенного вещества на т-й та-
( dV \
релке. Поскольку 1~ГГ/ ^1 н ^^1 (типичный газовый
хроматограф соответствует нескольким сотням тысяч тарелок), можно
применить предельное условие распределения Пуассона к
уравнению (8.30) [см. вывод уравнения (8.9)]. Введем обозначения
а=Ц- « 1 (8.31)
Na=m=N(^}=^. (8.32)
где v — объем растворителя, прошедшего через колонку. Тогда
получим уравнение
Доля исходного материала, iC(m)] \~V) e °^
оставшегося в т-й гипоте- — pN (т) — '. ^ " _. \_ 1 .
тической тарелке I^Jo ml
(8.33)
— — 71
В уравнении (8.33) т. —-у соответствует числу прошедших
через колонку объемов растворителя на каждой тарелке.
Графическая иллюстрация уравнения (8.33) ^представлена на рис. 8.8,
на котором каждая кривая характеризует зависимость
концентрации растворенного вещества на m-й тарелке от числа объемов
тарелок, прошедших через колонку. Следует отметить, что
концентрационный профиль (т. е. зависимость концентрации от
номера тарелки для фиксированного количества элюента,
прошедшего через, колонку) вначале имеет асимметричную форму
(рис. 8.1,6) и постепенно становится все более симметричным, по
мере того как вещество в процессе движения концентрационного
пика распространяется вниз по колонке. Однако в обычной
колоночной хроматографии концентрацию растворенного вещества
регистрируют при выходе раствора с колонки; следовательно, если
при появлении первых порций вещества иа выходе с последней
тарелки колонки профиль вещества на колонке описывается узким
«пиком», то концентрационный профиль значительно расширится
к тому времени, когда более поздние доли того же пика выйдут с
колонки. Другими словами, ожидаемые концентрации вещества
во фракциях, собираемых по выходе из колонки, будут
распределяться неодинаково и при выходе с колонки последних фракций,
УСПЕХИ И НЕУДАЧИ
0,20
0,16
0,12
0,06
0,04
п
■
т = 0
= 4
т = 9
т= 15
20
28
Рис. 8.8. Зависимость относительной концентрации вещества [С(т)]/[С]о на
т-й гипотетической тарелке хроматографической колонки (рис. 8.7) от числа
объемов растворителя иа одной тарелке (v/V), прошедших через колонку [9].
Рели т — последняя тарелка колонки, то соответствующая кривая описывает концентрацию
вещества в последовательных фракциях, выходящих с колонки, как это происходит в
типичном опыте (см. рис. 8.9).
как видно из рис. 8.8, будет наблюдаться «хвост» вещества.
Практический пример показан на рис. 8.9, где представлена газо-жид-
костная хроматограмма различных аминокислот, каждая из
которых была переведена в соответствующее производное для
облегчения испарения.
Колонка для газо-жидкостной хроматографии (ГЖХ)
представляет собой стеклянную капиллярную трубку (пусть, с
внутренним диаметром 0,02 см и длиной сотни метров), покрытую
растворителем, в то время как образец (в виде пара) переносится
по колонке инертным газом-носителем, например азотом. Колонка
может быть предварительно заполнена нелетучим веществом, на
которое наносится жидкое покрытие: Разделение основано на
рапном сродстве веществ к жидкости на колонке; элюирование
прочно связанных веществ облегчается увеличением температуры
(рис. 8.9).
Газо-жидкостная хроматография стала одним из самых
универсальных средств разделения смесей низкомолекулярных
соединений (с молекулярной массой < 1000) прежде всего потому, что
поведение вещества в колонке соответствует содержание в ней
огромного числа теоретических тарелок и прохождение вещества
по колонке можно рассматривать как множество равновесных
актов (по той же причине фракционная перегонка обладает
преимуществами перед одностадийной перегонкой). Серьезный
недостаток метода состоит в необходимости испарять исследуемые
вещества; поскольку многие представляющие интерес химические со-
/.$. = Внутренний стандарт
(tiymu/tcmeapam)
(CYS-h
242'С
_L
-L
_L
j_
Л.
Z4Z°C
242'C
Изотермически
-*4«*-
225'C ZOO'C
5°Pe 1muh'
175"C 15S°C 150X
+
-эН-е-
125'C
--~2.'Св1мин
-V_L
Минуть/
75"c GS°c Температура
—s»-J Режим
Рис. 8.9. Разделение производных аминокислот на неапентилгликолевом эфире себациновой кислоты [10]
Каждый пнк соответствует б мкг аминокислоты. Колонка (1,5 мХЗ,7 мм) заполнена 0.5%-иым иеопентилглнкольсукнииатом ' »я
хромосорбе G (80/100 меш). промытом кислотой. Хроматограф Микротек (Microtek) модель 220 с пламенно-иоииааиионным^
детекторами; регистрирующий потенциометр на 1 мВ. Скорость потока N (газа-носителя) 64 мл/мин 4«ww»i«m
3245 успехи и неудачи
единения недостаточно летучи, их необходимо модифицировать
так, чтобы ввести функциональную группу, благодаря чему моди-
SHUHpoeaHHoe соединение имеет более высокое давление пара,
днако такой способ нельзя использовать при исследовании
биологических макромолекул, хроматографическое разделение
которых обсуждается ниже.
Гел ь-хром атогр аф ия
Предыдущее рассмотрение ширины распределения
концентрации вещества на хроматографической колонке проведено с
помощью искусственной модели, описывающей прерывистый поток
растворителя. Однако результат носит общий характер, и для его
вывода совсем не обязательна частная схема, показанная на
рис. 8.7. В этом разделе предлагается другая простая модель,
позволяющая объяснить положение максимума распределения
вещества (т. е. механизм, с помощью которого хроматография
позволяет разделять смесь молекул по размеру). При газо-жидкост*
ной хроматографии разделение смеси веществ основано на разном
сродстве газообразных веществ к жидкой стационарной фазе.
При гель-хроматографии вещества разделяются в основном по
размеру, а не по их химическим свойствам или растворимости)
этот метод лег в основу многих распространенных методик
выделения, очистки и идентификации биологических макромолекул.
На рис. 8.10 приведены фотографии широко используемого геля
сефадекса на основе декстрана (полимера глюкозы),
содержащего поперечные сшивки эпихлоргидрином. Хроматографическая
колонка, плотно заполненная гранулами такого геля, имеет
принципиальную структурную особенность: достаточно небольшие
молекулы растворенного вещества могут проникать в поры или ще-
Рис. 8.10. Фотографии гранул декстрана (сефадекса) прн малом увеличении
(слева) и при большом увеличении (справа) [12].
Обратите внимание на пористую поверхность гранул геля, создающих возможность
проникновения внутрь гранулы молекул достаточно малого размера.
246 часть 2
2R
ли в гранулах геля, т. е. таким
молекулам доступен гораздо больший
объем колонки по сравнению с
крупными молекулами, размер
которых не позволяет им проникать
в поры гранул геля. Таким
образом, если смесь крупных (по
сравнению с диаметром пор в геле) и
небольших молекул в растворе
нанести на гель-хроматографическую
колонку, наиболее крупные
молекулы опустятся ниже всех по
колонке, так как для больших
молекул эффективный объем колонки
меньше: они не могут проникнуть
внутрь гранул геля.
Предположим, что гранула
геля имеет внутри много крошечных
конусообразных полостей, в
которые молекула с эффективным
радиусом г может проникать на
глубину h (рис. 8.11), причем радиус основания конической полости
R и высота Я. Объем конусообразной полости равен
Объем полости =лЯ/?2/3. (8.34)
Объем, доступный молекуле, составляет для узкой длинной
области (#>/?).
Объем, доступный для молекулы « nh(R—г)9/3. (8.35)
'Объем внутри гранулы геля, доступный для молекулы, тогда
составляет
Рис. 8.11. Схематическое
изображение конической полости
внутри гранулы геля.
Молекула с эффективным радиусом
г проникает иа глубину Л; высота
полости равна Н и радиус
основания R (в данном примере Я»/?).
[Данные фирмы Pharmacia Fine
Chemicals.]
;Но поскольку
Доля доступного объема внутри гранулы=
_h(R — r)* h_/ . r_y
~~ HR* e H \ L RJ '
_h_
Н
R — r
(8.36)
(8.37)
то
Доля доступного объема внутри гранулы» 1—^-1, г<#
или (8.38)
Доля доступного объема внутри гранулы «О, г> R,
Согласно уравнению (8.38), молекулы растворенного
вещества, эффективный диаметр которых превышает диаметр пор,
должны при перемещении по колонке двигаться между гранулами (в
|47 УСПЕХИ И НЕУДАЧИ
Б)
Первыми выходят
самые крупные молекулы
\
1
2 3
80 100
Последними выходят
самый малые молекуль/
t
7
120 140
Объем м(оата}мл
160
180
Рис. 8.12. Разделение смесн белков на сефадексе G-200 (сверхтонком).
Колонка фирмы Pharmacia К 16/100, высота геля 90,6 см. Объем образца 1 мл, скорость*
потока 1,5 см/ч. Образец содержит тироглобулин (/), IgG (2), сывороточный альбумин {3),
овальбумнн (4), хнмотрнпенноген (5), цитохром с (6) н витамин В13 (7).
«наружном» объеме) со скоростью, практически не зависящей от
размера молекул. Точно так же молекулы, диаметр которых
намного меньше диаметра пор (r<^.R), должны иметь небольшую-
скорость (поскольку для этих молекул доступен практически весь
объем и внутри и снаружи гранул), также не зависящую от
размера молекул. Наконец, молекулы, эффективный диаметр которых
имеет тот же порядок, что и диаметр пор, но меньше диаметра
пор, должны перемещаться со скоростью, изменяющейся
(нелинейно) с молекулярной массой (т. е. размером). Это иллюстрируег
и рис. 8.12.
аш
%
0,05
\
ч \
0 X
2 "эЧ
&! ^ \
S &
11
•з*
S0
1Й»
<>
■с
I
'?
1
1$
**
Stf
^?
5
S?
1
1?"
1
1»
4
§
1
%»
1
*
1
<j
1
1
1
1
-—• i
/04 10s
Молекулярная масса
Юь
Рис. 8.13. Зависимость миграции (обратной величины расстояния, I/D) от
молекулярной массы белков при гель-фильтрации в тонком слое на сефадексе G-150»
(сверхтонком).
■[Данные Pharmacia Fine Chemicals;]
Размер пластины 2ОХ30 см. Толщина слоя 0,5 мм. Время 7 ч 40 мии. Угол наклона пластн-
■ы 15°. Из слоя геля белкн переносят на бумагу ватман ЗММ, обработанную 0,1%-ным рас»-
твором бромфенолового синего Гв смесн (9:1) метанола и ледяной уксусной кислоты].
Бумажную реплику промывают 6%-иой уксусной кислотой и определяют миграцию, измеряя
расстояния.
248 часть 2
Лоуи/оларуе, muni
I
i
100 150
Объем э/г/оата,л(/г
Ряс 8J 4. Разделение меченного 32Р аденовируса и лолиовируса на сефарозе 2 В,
^Данные Pharmacia Fine Chemicals.]
Колонка 2,1X56 см. Элюат: 0,002 М Na-фосфатный буфер (рН 7,2) и 0,15 М хлорид натрия.
Скорость потока 2 мл-см-1 я-1. Объем образца 1 мл.
Экспериментальную зависимость, которая хотя и нелинейна, но
поддается аналитическому описанию, для большего приближения
кривой к прямолинейному виду обычно строят в
полулогарифмическом масштабе, чтобы облегчить интерполяцию между двумя
известными точками, необходимую для оценки неизвестного
значения. Именно так и обработаны данные, приведенные иа рис. 8.13
и показывающие скорость движения молекул ряда белков,
имеющих очень близкую между собой и приблизительно сферическую
форму. Рис. 8.13 иллюстрирует применение одного из наиболее
широко используемых методов оценки^молекулярных масс
неизвестных белков; измеряя объемы элюирования для каждого
компонента смеси" неизвестных и нескольких известных белков, легко
определить молекулярную массу неизвестного белка с помощью
интерполяции. Такой способ может быть полезным и при
исследовании неглобулярных (несферических) белков: для этого все
белки в смеси приводят к общей форме (статистического клубка)
благодаря присутствию денатурирующего агента, например гуани-
дингидрохлорида. Метод можно использовать для изучения
макромолекул с гораздо большими молекулярными массами, таких,
как нуклеиновые кислоты и вирусы; с этой целью используются
несшитые гели (и, следовательно, имеющие поры гораздо
большего размера), такие, как агароза, линейный полисахарид,
состоящий из чередующихся звеньев D-галактозы и 3,6-ангидро-ь-галак-
*озы (рис. 8.14).
Кроме определения молекулярных масс и разделения смесей
макромолекул в соответствии с их размером, основное применение
гель-хроматография находит при обессоливаиии или удалении
иизкомолекулярных соединений из растворов макромолекул. Как
УСПЕХИ И НЕУДАЧИ
0,0 0,1 0,2 0,3 0,4 0,5 0,6 0,7 0,8 0,9 1,0
'( Относительный odse/и а/икшрования
V
^ис. 8.15. Зависимость молекулярной массы субъединиц белков от относительного-
Йъема элюнрования комплексов белков с додецилсульфатом натрия при хрома-
юграфнровании на пористом стекле с узким распределением пор по размеру [13].,
&елкн: / — гемоциаиин; 2 — тироглобулин; 3 — р-галактозидаза; 4 — фосфорилаза; 5 —ка-
ялаза; 6 — глутаматдегядрогеназа; 7 — овальбумин; в—пепсин; 9 — карбоангидраза; 10 —
1-химотрипсииогвн; // — трипсин; 12 — р-лактоглобулин; 13 — миоглобин; 14 — лизоцим;
' 15 — цнтохром с.
рассматривалось в разд. З.Б.1, один из простейших способов вы-
деления данного белка из смеси белков состоит в избирательном
Осаждении такой солью, как сульфат аммония; когда данный бе-
йок затем вновь растворяют, прежде чем характеризовать белок,
его необходимо избавить от соли. Так как молекулярные массы
соли и белка значительно различаются, гель-хроматография
служит простым способом, для удаления соли.
Аргументы, основанные на рассмотрении внутренней структуры-
гранулы геля, состоящей из конусообразных полостей, и приведи
щие к уравнению (8.38), не являются единственным возможным
объяснением приблизительно линейной зависимости между
объемом злюата иа.выходе и логарифмом молекулярной массы.
Можно описать структуру геля с помощью однородных
цилиндрических каналов; при входе в такой канал молекула встречает
возрастающее сопротивление трения, проявляющееся как уменьшение
кажущегося коэффициента диффузии и, следовательно, как замед-"
ление движения через гранулу по сравнению с движением по
объему раствора (снаружи гранулы). Наконец, подходящая теория"
гель-хроматографии должна учитывать влияние неодинаковой
адсорбции разных веществ на самой поверхности геля; недавно
однаружено, что кажущиеся молекулярные массы гликопротеи-
нов (белков, ковалентно связанных с углеводными остатками),
определеииые с помощью гель-хроматографии, занижены, что-
обусловлено повышенным сродством углеводного фрагмента белка
к полисахаридной матрице геля, т. е. более прочным удерживани-
,250 ЧАСТЬ 2
ем в колонке по сравнению с наблюдаемым для обычного белка
с той же молекулярной массой.
В заключение можно отметить, что необязательно использовать
гель на основе природного полимера; полистирол, хлорбутиловый
каучук и полиакриламид — синтетические полимеры, которые
также являются подходящими для гель-хроматографии; на рис. 8.15
показано, что можно применять и шарики из пористого стекла:
зависимости логарифма молекулярной массы от относительного
объема элюата линейны в области молекулярных масс от 17 000 до
385000 при использовании стекла с порами размером ~500 А.
Для разворачивания белков использовали додецилсульфат натрия,
сорбирующийся на белках так, что разные белки принимают одну
и ту же форму (палочки).
Ионообменная хроматография
При гель-хроматографии макромолекулы разделяют по их
размеру; если гель содержит заряженные группировки, то можно
разделять молекулы по заряду. Этот метод известен как ионообменная
хроматография. Наиболее широко применяемые ионообменные
смолы (гели) получают введением —S03H и —СООН (для
получения смолы с отрицательно заряженными группами) или —NR2
и —NHR (для получения смолы с положительно заряженными
группами) в гель на основе декстрана (сефадекс}, или в гель на
основе сополимера стирола и дивинил бензола (дауэкс, амберлит)
СН =СН2 СН =СН2 _с_с-С-С-С-
6-6 '999
СН =СН, —С—С—С-С—С—С—С—
Стирол Дивиниябензоп (r\\ (Y\\
Как подробно рассматривалось в разд. З.В, суммарный заряд
белка зависит от рН. Согласно рис. 8.16, можно предположить,
что если белок приготовлен при рН, при котором он, например,
заряжен отрицательно, и этот раствор затем добавлен к анионо-
обменной смоле, то белок свяжется с гелем, и для снятия белка
с колонки (в данном случае) необходимо понизить рН до
значения, при котором белок уже недостаточно отрицательно заряжен,
чтобы удерживаться на анионообмеином геле. Проводя элюиро-
вание раствором с изменяющимся рН (в рН-градиенте), можно
разделять разные макромолекулы (рис. 8.17 и 8.18), если их изо-
электрические точки различаются (рис. 8.16).
1 УСПЕХИ И НЕУДАЧИ
I
I
I
t
Изоэлектрическая
/почка'
h взаимодействие
с аниониталгц
1 U
ю
Взаимодействие с
катионита
/пи
Рис. 8.16. Зависимость суммарного заряда белка от рН.
©тмечены области рН, в которых белок связывается с анионитами и катионитамк.
1
Начало
опыта
■ Д •
О
°Рт
V V
<N—'о
2
Лдсорбция
образца
© © ©
▲
IP)
V У
**^Sa
о о о
о о о
Начало
десорбции
Конец
десорбции
Регенерация
9
О ©
о Ионы буферного раствора
" Разделяемое вещество
J /foya/ элюента (градиент концентрации)
Рис. 8.17. Схема разделения смеси двух заряженных белков с помощью
ионообменной хроматографической колонки Г14а1.
Для избирательного удаления с ионообменной колонки элюирование производилось в
градиенте рН нли концентрации соли.
Пример. Аминокислотный анализ
Колонка содержит катионообменную смолу на основе сульфированного
полистирола. рН смесн аминокислот понижают до ~3, т. е. значения, когда все
аминокислоты положительно заряжены; смесь вводят на колонку; аминокислоты А+
связываются согласно уравнению
А+ + RSOgNa-»" ?=-> RSOaA+ + Na+
Поскольку более основные аминокислоты, как правило, имеют более
положительный заряд, оии связываются в колонке более прочно. После этого рН
252 ЧАСТЬ 2
I
л
/
I"
\
А-нормальнаа тРНК ~
В-меченая
б-днпарурациЛ'ТРНК
400
\00
W0
I
I
I
I
!
Рис. 8.18. Разделение нормальной тРНК н меченой тРНК (содержащей
радиоактивный 5-фторурацил вместо нормального урацила) методом ионообменной
хроматографии.
{Данные Смита и Маршелла (SmCth J. L., Mashall A. C.).]
Здтоирование в градиенте концентрации соли. По оптической плотности при 260 нм
определяется общее содержание тРНК, а по радиоактивности — содержание меченой тРНК. Обе
РНК снимали с аиионообменной смолы (DEAE-целлтолоэы) элюнрованнем раствором с
увеличивающейся концентрацией NaCl (от 0.3 до 0,5 моль/л). Поскольку рК 5-фторурацнла
■i&,l) меньше рК урацила (9,5), РНК. содержащая 5-фторурацил, несет больший
отрицательный заряд и сильнее связывается со смолой, и поэтому для снятия ее со смолы
требуется более высокая концентрация соли. (5-Фторурацил— противоопухолевый агент,
вводимый в тРНК бактерий при выращнваини их на среде, содержащей этот препарат.)
(и концентрацию соли) на колонке постепенно увеличивают (рис. 8.19), кислые
аминокислоты нейтрализуются первыми (и, следовательно, уходят с колонки),
далее следуют нейтральные и наконец основные аминокислоты. В отличие от
тазо-жндкостной хроматографии при использовании этого метода для разделения
смесн аминокислот не надо модифицировать и превращать аминокислоты в их
тфонзводиые; однако при разделении на ионообменной колонке требуются
намного большие количества материала.
Пример. Анализ следовых количеств компонентов
Благодаря высокому сродству ионов к ионообменной смоле можно
концентрировать нужные ионы, пропуская разбавленный раствор через ионообменную
•смолу (чтобы задержать ионы) с последующим промыванием смолы малым
объемом концентрированной кислоты (или основания, или еоли) для их снятия со
смолы
Адсорбция ионов: RS03H + M+ rr» R$03M + Н+
Десорбция ионов: RSOaM + Na+ ^=fc RS08Na + М+
Регенерация смолы: RS08Na-f Н+ ч-■ Ъ RSO-H-f Na+
Этот метод особенно полезен для оценки содержания иона фтора в питьевой
«оде, содержания иоиов таких металлов, как Pb, Fe и Си в вине и других
продуктах и дли определения (особенно радиоактивных) металлов в моче. Такие
гсоны, как Cu2+, Ni2+, Mg2+, Ca2+, Na+ и К+, удерживаются колонкой
практически и а 100%, и таким образом их можно сконцентрировать из очень разбав-
УСПЕХИ И НЕУДАЧИ
1.0
I 0.5
|0.4
^ 0.3
0.1
'«юент.мл
Циетеинммг су/гьфон Треонин
кислота ^ ^ ^Серин
Аспарагинавая\
кислота
Ме/пшнин
0 а//г»фоксид
Кутаминоваа 'ЯиЦ™
ПролиН
A/IONUH
Цастуя
40 80 120 140 160
е J50-CM/тюнхагрН5,25;0,1н. цитрат Ыа •
200
240
320
1,0 И-
«и 0,5
\ | 0,4
1 0,2
* 0,
Ва/гин
Ме/пионаН
Шолейциц
/
Лизин
LL
I
\Аейцин
/Ьстидин
Тирозии
Феншимонан
J L
JtpWHUIt
эзо
370 410 450
~-уЩ25ю,2н. цитрат На—
490
50 90 130
15см колонка, pH5,28i 0,35н. цитрат ffe
1С, 8.19. Хроматограмма синтетической смеси аминокислот на смоле яз
сульфированного полистирола [15].
Ценных растворов, а также проводить анализ с помощью различных обычных
Способов. '
ч
Пример. Лекарственные препараты с замедленным действием
\ ■
X Ионообменные смолы можно использовать для приготовления таблеток на
Цсвове комплексов с различными лекарственными веществами. Такие таблетки
Наряду с достаточной механической прочностью отличаются тем, что
лекарственное вещество медленно диссоциирует и освобождается от смолы после того, как
ромплекс попадает в кишечный тракт. Для приготовления пенициллина в виде
лекарственной формы для перорального употребления использовали
анионообменную смолу; для получения пероральных лекарственных форм морфина в
качестве анестетика (обезболивающего средства) или гистамина в качестве
средства против кашля использовали катионообменную смолу. В аналогичных
случаях анионообменную смолу использовали для удаления меченого трииодтироиина
цз сыворотки крови человека, введенного для проверки функции щитовидной
железы (см. также разд. 8.А),
Пример. Разделение олигонуклеотидов
На рис. 8.20 показано, что олигонуклеотиды удается разделять иа анионо-
обмеииой смоле (в данном случае диэтйламиноэтилдекстране); разделение
основано на различий зарядов, которое обусловлено размером и составом
оснований.
a
!1
I
Мономер
LLLLL
Тример
Тетрамер
200 400 600 800 1000 1200 140016001800 2000 2200
i-мер ft-мер—
1Л1 JV.l
0,4 §
0,2 S
!
I
i
0,4
0,2
2200 2400 26002800 3000 3200 3400 3600 3800 40004200
Элюент ,мл
100 125
Э/чоетл, мл
v
| 30
I
V
^ 20
1
10 —
.—»
2. *
I
2
о ~=>
^5 *•
< <
LISJ
3
<
d
О
о
СЧ
<
*
о.
э
<
о
(Z>
50 100 150
Номер фракции
Hi
о
200
Рнс. 8.20. Примеры разделения олигонуклеотидов анионообменной хроматографи
ей иа DEAE-сефадексе.
а я б- [16]; а-Ц7].
55 УСПЕХИ И НЕУДАЧИ
Воздух
1
е
л
0,2h.H2SO4-
©
Ж
3L.
-0,2н. NaOtt
Катионообменная \^ ^]
Мембрана }ч^ _^
АнионообменнаЯ
мембрана
Рис. 8.21. Аппарат для обессоливания с помощью электроднализа с
ионообменными мембранами [13].
Вода, содержащая соль, вводится в центральную ячейку. В процессе электролиза катионы
движутся от центра влево, а анионы—от центра вправо в соответствующие ячейки; любая
диффузия катионов направо или анионов влево предотвращается односторонней ионной
проводимостью ионообменных мембран. Когда нз воды удалено достаточное количество солн.
деионизованную воду отводят из центральной ячейки и процесс повторяют.
Пример. «Обессоливание» воды
Как, возможно, читателю хорошо известно, воду можно «обессоливать»,
пропуская ее через (смешанные или последовательно соединенные) аиионо* и ка-
тионообменные смолы; именно такой принцип положен в основу использования
ряда коммерческих смол для получения деиоиизованной воды из водопроводной
воды. Однако для крупномасштабного «обессоливания» этот способ относительно
дорог из-за необходимости прерывать процесс для периодической регенерации
смолы. Ионообмеиные смолы можно изготовить в форме гибкого листа. Такие
листы можно использовать в качестве мембран при удалении минеральный ионов
из центральной ячейки и переносе их в катодную илн анодную ячейки в
аппарате, схема которого изображена на рис. 8.21. Диффузия положительных ионов
из центральной ячейки в правую ячейку, например, предотвращается
(положительно заряженной) ан ионообменной мембраной. Методика основана на том, что
ионообменная мембрана практически непроницаема для ионов одного с ней
заряда.
Кстати, можно отметить, что процесс «обессоливания» в некоторых
устройствах медицинского назначения основан на другом принципе: удаление анионов
и катионов происходит одновременно в результате реакции
RS08Ag -f- Na+ + ОТ »- RS08Na + AgCIJ
Аффинная хроматография
Аффинная хроматография— сравнительно недавно
разработанный метод*, позволяющий исключительно селективно выделить и
См. Туркова Я. Аффинная хроматография. — М.: Мир, 1980. — Прим. ред.
2S6 часть 2
Высокая удельная активность-, низкое
содержание общего белка:
соответствует 55 - кратной очистке при одной
рроводке через колонку саран/нным сорбентом
ДекфИ&поний, ммоль/я
10
НаС\,мо/1б//1 ~
0.5
-4
i
!
I
I
I
1
I
1
36 44 52 бО
Номер фракции
Рис. 8.22. Элюирование ацетилхолинэстеразы из Torpedo на сефарозе,
содержащей аффинный лигаид (Сефароза 2В) —N—(СН2) 5—С—N^ ^—N(CH3)3. Де-
[ НОН
|каметоняй— сильный ингибитор фермента; его используют для снятия ацетил-
|- холинэстеразы с аффинного носителя.
[Данные Моррода, Кларка и Маршелла (Morrod P. J., Clark D. О., Marshall A. 0.).]
очистить нужную макромолекулу из обычной для первых стадий
выделения смеси макромолекул. Метод основан на использовании
высокой специфичности связывания, характерной для
биологических макромолекул: например, ингибитор, специфический для
данного фермента, ковалентно присоединяют к гелю, поры которого
достаточно велики для фермента; фермент наносят на колонку с
таким гелем, и он связывается с иммобилизованным на колонке
ингибитором, в то время как другие белки проходят по колонке,
не вступая ни в какие взаимодействия с гелем; для снятия
иммобилизованного на колонке фермента после этого достаточно элюи-
ровать его раствором активного ингибитора этого фермента.
С помвщью этого метода конкретную биомакромолекулу
можно селективно выделить и сконцентрировать из исходного
препарата с помощью всего одной хроматографической колонки. На
практике обнаружено, что присоединение аффинного лиганда
(низкомолекулярного соединения, обладающего способностью к специ-
258 часть 2
фическому связыванию с нужной макромолекулой) прямо к
матрице обеспечивает худшие связывающие свойства, чем соединение
лиганда с матрицей геля через «ножку». Из-за необходимости
иметь большой размер пор обычно в качестве геля используют
один из природных линейных полимеров агарозы. Подробности,
касающиеся химической иммобилизации, используемой при
изготовлении аффинных носителей, можно найти в работе [9], а также
во многих последних учебниках биоорганической химии.
Наглядный пример возможности одностадийной очистки с помощью
этого метода приведен на рис. 8.22. Очевидно, что на колонке с ага-
розой, к которой ковалентно присоединен лиганд (структура
которого приведена в подписи к рисунку), большая часть
выделяемого фермента ацетилхолинэстеразы задерживалась на
колонке, а белки без ферментативной активности выходили с
колонки (приблизительно фракции 0—15). Затем в градиенте
ингибитора ацетилхолинэстеразы происходило элюирование фермента;
получали белковый пик с очень высокой удельной активностью.
В заключение для удаления каких-либо оставшихся на колонке
белков ее промывали в градиенте соли, и, как видно, при этом
получено очень мало активного фермента. В данном конкретном
примере для получения фермента с сопоставимой удельной
активностью потребовалось бы использовать не менее четырех разных
хроматографических и других стадий очистки.
Аффинные методы используются и для выделения
специфических нуклеиновых кислот, ряда ферментов, рецепторов
лекарственных веществ, гормонов и др. Если предпринимаемые в последнее
время попытки соединить антитела с агарозными гелями будут
успешными, окажется возможным избирательно удалять клетки—
переносчики заболевания, вирусы или антигены из крови
человека.
В табл. 8.2 перечислены методы, с помощью которых можно
выявить различия между разными макромолекулами в растворе,
и признаки, по которым эти различия выявляются. В таблице
указано большинство методов, рассмотренных в гл, 1—8, а также
дана ссылка, в какой главе следует искать примеры применения этих
методов.
Задачи
1. Закончите вывод распределения Пуассона, доказав уравнения (8.9).
Замечание. Для факториалов, входящих в параметр С, следует применить
более точный вид приближения Стирлинга
а затем преобразовать конечное уравнение, чтобы получить
1 1
/ т\*г' ( м \т '
С=е~
т
I1"*
<т-1/2П
J>59 УСПЕХИ И НЕУДАЧИ
Для каждого из четырех множителей вышеприведенного уравнения
необходимо найти предел прн N—*-сс.
2. Получите фундаментально полезное свойство распределения Пуассона
[уравнение (8.11)]
Y<{m-{m)f> = V~m.
3. Борткевич собрал следующие данные о вероятности гибели кавалериста в
течение года от падения лошади. Данные были получены из архивов десяти
армейских корпусов за двадцать лет (200 значений):
Число смертных Число корпус-лет, в которые произошло
случаев в год данное число смертных случаев
0 109
1 65
2 22
3 3
4 1
5 или более О
а. Покажите, что среднее число смертных случаев в год в корпусе составляет
/71 = 0,61.
б. Сравните наблюдаемое число корпус-лет, для которого характерно данное
число произошедших смертей, с предсказуемым из распределения Пуассона
при /й=0,61.
в. В качестве дополнительной проверки рассчитайте вероятность того, что в
наблюдаемом числе корпус-лет произойдет не менее одной смерти; не менее
двух; не менее трех н т. Д., и нанесите каждое значение на соответствующую
кривую (С=*1, С=2 и т. д.) на распределении. Пуассона (рис. 8.6) и
проверьте, лежат ли точки на прямой, параллельной ординате.
г. Если эта задача кажется слишком отвлеченной, представьте себе, что
приведенные выше данные получены при разном числе наблюдений под
микроскопом квадрата, в котором находится 0, 1, 2 и т. д. нормальных
эритроцита в разбавленном образце крови.
4. Если, измерения радиоактивности проводились в течение времени t,
насколько дольше надо проводить измерения, чтобы на 35-% повысить точность
определения?
5. С помощью счетчика радиоактивности найдена радиоактивность образца А
1500 имп./с, радиоактивность образца В 735 нмп./с и радиоактивность
образцов А + В (вместе) 2082 имп./с.
а. Рассчитайте мертвое время счетчика.
б. Определите «истинные» значения радиоактивности образцов А и В.
6. Пусть 2,0 г исследуемого образца X с известной радиоактивностью
1000 имп./мин добавлено в смесь веществ, содержащей некоторое количество
нерадиоактивного X. Далее предположим, что после перемешивания взято
некоторое количество X, проведена его очистка от примесей и активность
этого очищенного X равна 150 имп.-мин~1-г~1. Сколько нерадиоактивного X
присутствовало в исходной смеси?
7. Коэффициент усиления фотоумножителя составляет 106, т. е. каждый
фотон, попадающий иа экран трубки, генерирует Ю6 электронов. Если число
фотонов, попадающих на экран фотоумножителя за 1 с, равно 4 и постоянная
времени соответствующей электрической цепи составляет 0,1 мс, рассчитайте
.260 ЧАСТЬ 2
отношение сигнал/шум. (Можно (предположить, что шум определяется
дробовым эффектом фотоумножителя.)
8. Группа из 20 пациентов подверглась новому способу лечения заболеваний.
При традиционном способе лечения этого же заболевания 20% больных
умирают. Если в подопытной группе умрет 7 лациентов, насколько серьезно
доказательство того, что новый способ лечения оказался лучше или хуже
традиционного способа?
9. Покажите, что хроматографическое разрешение (отношение расстояния, на
которое продвинулся пик вещества по колонке, к ширине этого пика)
пропорционально корню квадратному из высоты столба геля в колонке.
1.0. Каково значение отношения сигнал/шум в цепи, по которой проходит ток в
5 мкА с шириной полосьв пропускания 20 кГц?
Литература
Распределение Пуассона
1. Могопеу М. /., Facts from Figures, Penguin Books, Baltimore, 1956.
Измерение радиоактивности
2. Silver S., Radiactive Isotopes in Medicine and Biology: Medicine, 2nd. ed.,
Lea and Febiger, 1962.
3. Phelan E. W., Radioisotopes in Medicine, U. S. Atomic Energy Commission,
Division of Technical Information (1966). Obtain free from USAEC, P. O.
Box 62, Oak Ridge, Tennessee.
Шум в электрических схемах
4. Brophy J., Basic Electronics for Scientists, 2nd ed., McGraw-Hill, New York,
1972.
5. Coor Т., J. Chem. Educ, 45, A533, A583 (1968).
6. Davidson N., Statistical Mechanics, McGraw-Hill, New York, 1962, p. 300.
7. Ernst R. R., Anderson W. A, Rev. Sci. Instrum., 37, 93 (1966).
Гель-хроматог рафия
8. Pescok R. E., Saunders D., in: Separation Techniques in Chemistry and
Biochemistry, R. A. Keller (Ed.), Marcel Dekker, New York, 1967.
9. Berg E. W., Physical and Chemical Methods of Separation, McGraw-Hill, New
York, 1963, p. 110.
10. Gehrke C. W., Stalling D. L., in: Separation Techniques in Chemistry and
Biochemistry, R. A. Keller (Ed.), Marcel Dekker, New York, 1967, p. 44.
11. Sephadex: Gel Filtration in Theory and Practice, Beaded Sepharose 2B-4B-6B:
Agarose Ge*, Filtration, both from Pharmacia Fine Chemicals AB, Box 175,
S-751, 04 Uppsala 1, Sweden.
12. Collins R. C, Haller W., Anal. Biochem., 54, 47 (1973).
Ионообменная хроматография
13. Ion-Exchange Resins, BDH Chemilcals Ltd., Poole, England.
14. Sephadex Ion Exchangers, a guide to ion exchange chromatography,
Pharmacia Fine Chemicals AB, Box 175, S-751, 04 Upsala, 1, Sweden.
15. Spackman D. H., Stein W. H., Moore S., Anal. Chem., 30, 1190 (1958).
16. Lloyd D. A., Mandeles S., Biochemistry, 9, 932 (1970).
17. Matthews H. R., Eur. J. Biochem., 7, 96 (1968).
Аффинная хроматография
18. Cuatrecasas P., J. Biol. Chem., 245. 3059 (1970).
клетьз
jpOCT И РАСПАД
1лАВА 9
КИНЕТИЧЕСКИЕ ПРОЦЕССЫ
РЕРВОГО ПОРЯДКА
I.
V;
i Процессы бактериального роста, радиоактивного распада, фар-
макокинетические процессы и все кинетические схемы химических
реакций основаны на общем.соотношении
d[ \/dt=±k[ ], (9.1)
Где [ ]—количество или концентрация материала, оставшиеся
к моменту времени t; знак перед k указывает на увеличение
(плюс) или уменьшение (минус) количества вещества, k
характеризует скорость процесса. Из уравнения (9.1) следует (см.
ниже) экспоненциальный характер роста или распада, наблюдаемый
в вышеперечисленных процессах. Вид уравнения (9.1) можно
объяснить с помощью статистических методов, описанных в ч. 2.
Зависимость макроскопических изменений количества или
концентрации вещества от времени экспоненциальна просто по тому,
что поведение любой одной бактерии (атома, пары
взаимодействующих молекул) носит вероятностный характер. Существует
строго определенная вероятность того, что любая одна бактерия
(атом, пара взаимодействующих молекул) претерпит изменение
в течение данного периода наблюдения, и эта вероятность не
зависит от предыстории данной системы. (Говоря проще, ни один
неустойчивый атом не знает, претерпели ли распад любые другие
нестабильные атомы в этом образце.) Чтобы понять, каким
образом из этого условия следует экспоненциальный характер
распада, рассмотрим следующий пример. Пусть в комнате находится
100 человек, каждый из которых раз в минуту подбрасывает
монету, причем любой, у кого выпадет «орел», должен выходить из
комнаты. После первой попытки в комнате останется ~50
человек; после двух попыток ~25 человек, и далее число людей,
остающихся в комнате, будет изменяться во времени приблизительно
экспоненциально. При увеличении числа людей (бактерий,
нестабильных атомов, пар химически взаимодействующих молекул),
подбрасывающих монету, как было показано в гл. 8,
относительная точность измерений значительно повышается. В небольшом
образце (рис. 9.1, а) гладкая экспоненциальная кривая сравни-
262 часть з
а
20
1
5
0
| 10
§
*
(
—— у!%
|
) 10
г
20
Время
30 40
,сут
50
6
2000000 000
1
б
£ 1 000 000 000
S
с
[ !>-
) 10 20 30
Врем я, сут
40
Рис. 9.1. Исчезновение атомов mI во времени,
в —исходное число атомов "Ч равно 20; б —исходное число атомов равно г'ООООООООО.
тельно плохо описывает постепенное снижение исследуемого
параметра; однако для большого образца (рис. 9.1,6) такое
снижение очень точно описывается экспоненциальной зависимостью.
Бактериальный рост. Радиоактивные и химические методы
определения геологического возраста
Из предыдущего примера можно ожидать, что число людей
(бактерий, нестабильных атомов, пар взаимодействующих
молекул), подбрасывающих монеты, изменяется во времени согласно
уравнению
П = [ J,.2± W"
рост
распад
(9.2>
где Т — время удвоения параметра (при его увеличении) или
уменьшение параметра в 2 раза (при его уменьшении); это
означает, что если t=T, то
*- 21 Jo
U
(9.3)
распад
т. е. данная популяция удваивается (или уменьшается в два
раза) через каждые Т (секунд).
Теперь можно показать, что при другой исходной интуитивной
позиции уравнение (9.1) приводит к тому же самому результату.
Уравнение (9.1) просто указывает, что число наблюдаемых
бактериальных делений (распадов атомов, химических взаимодейст-
£63 РОСТ И РАСПАД
Г
Ьий) должно быть пропорционально числу бактерий
(нестабильных атомов, rfap взаимодействующих молекул), присутствующих
b системе. Преобразуя уравнение (9.1)
; I 1
=dln[ ] = ±kdt
(9.4)
'проинтегрируем его от начального нулевого момента времени до
^конечного момента времени /, за которое количество вещества
изменяется от начального значения [ ]0 до конечного [ ]
i ln[ ]—ln[ ]0 = ±kt (9.5)
^или
[ ]-[ 1ов±
kt
(9.6)
'Определим период полупревращения т для роста или распада
объекта:
\/k
(9.7)
По аналогии с уравнением (9.3), если t = x, популяция вырастет
(распадется) до величины, в е раз превосходящей начальную (или
в е раз меньше начальной)
[ ] = [ Jo^1-
рост
(9.8)
распад
В кинетике химических реакций обычным параметром является
константа скорости реакции k\ при исследовании бактериального
роста и радиоактивного распада используют время
полупревращения популяции Г; при изучении кинетики быстрых реакций и
релаксационных процессов (ч. 4) используют время релаксации
системы т. Эти параметры связаны друг с другом следующим
образом. Объединим уравнения (9.2) и (9.6):
I J =e± c/t)=2± viT)=e±kt
[ U
(9.9)
Прологарифмируем уравнение (9.9):
±(*/т)=±(//Г)1п2 =
±k
или
b__L~ lnl ~ °'693
«— т — -р ^ т
(9.10)
Теперь применим результат, полученный в этом разделе, к
исследованиям некоторых процессов роста или распада.
264 часть з
Логарифмический
■ <раза" >
\ \ \ \ \ \ \ \ \
'0 123456789
Время, v
Рис. 9.2. Кривая роста популяции в культуре клеток Saccharotnyces cerevisiae
(дрожжей).
[Данные Луома. Смита н Маршелла (Luoma G., Smith J. L., Marshall A. G.).] После
начального «лаг»-нсрнода клетки входят в «логарифмическую фазу», в которой деление кле«
ток происходит с максимальной скоростью, так что число клеток экспоненциально
увеличивается во времени. С истощением питательных веществ в исходной смеси и накоплением
отходов процессов метаболкэа (справа наверху) число клеток в конце концов
выравнивается (и в пределе уменьшается).
Пример. Рост бактерий
Данные, приведенные на рис. 9.2 и описывающие поведение культуры
дрожжей, иллюстрируют экспоненциальную природу роста микроорганизмов. В
настоящее время широко распространена точка зрения, что одноклеточные
микроорганизмы потенциально бессмертны в том смысле, что жизнеспособность н
деление в культурах многих микроорганизмов поддерживаются годами. В
действительности такое же свойство присуще определенным культурам тканей
растительного и животного происхождения и культурам тканей человека. Выделение
отдельных клеток из культуры ткани человека позволяет,вырастить клоны
дочерних клеток в виде колоний, аналогичных колониям микроорганизмов.
Одна из причин интереса к росту микроорганизмов состоит в том, что рост
раковых опухолей описывается экспоненциальной зависимостью очень близкого
типа (рис. 9.3). Все современные методы лечения рака основаны на
использовании средств, подавляющих уже начавшийся рост опухоли [ионизирующая
радиация и введение солей радиоактивных элементов (ч. 5) и различных
ингибиторов синтеза метаболитов, необходимых для непрерывного роста опухоли
(гл. И)], Исследование рака заключается в основном в изучении свойств
нормальных клеток (нерастущих и неделящихся), обусловливающих процессы
роста и деления клеток.
Пример. Определение геологического возраста при помощи
радиоактивного углерода 14С
Природный углерод содержит ~99% ,2С и ~1% ibC. Кроме того, при
воздействии космических лучей на атмосферный азот образуется радиоактивный
углерод 14С; этот МС окисляется до мСОа и в конце концов усваивается растения-
I
$ РОСТ И РАСПАД
!
I
1000
100
•in
I ■ j I МШ{
— 1
\
I
1
I
1
\
1
^_ щ -
1
1 1
1 1
—*
—
1 ! m in) .. .
О 2 4 6 8 10
Время {после инокуляции ), сут
12
14
Рис. 9.3. Кривая роста популяции клеток опухоли асцита Эрлиха [7].
Опухоль состоит из отдельных клеток, размножающихся в биологической (асцнтной)
жидкости. День инъекции, содержащей 22" 10е клеток, пряняг за начало отсчета («нуль» на осн
абсцисс). Обратите внимание, что в течение первых четырех суток увеличение числа
клеток описывается логарифмической зависимостью, затем рост популяции происходит не так
стремительно, а далее число клеток уменьшается из-за их гибели. В целом кривая
соответствует приведенной на рис. 9.2. Среднее время выживания мышей, которым ннокулиро-
вана эта опухоль, составляет 14 сут. Каждая точка — среднее из шести наблюдений.
мн, а затем тканями животных. Поскольку период полураспада МС составляет
5668 лет, между образованием и распадом ,4С существует стационарное
равновесие, так что относительная концентрация 14С в живых организмах равна
~1(Н2 г "С на 1 г 12С. В современных организмах удельная радиоактивность
(иа I г углерода ,4С) Ао, следовательно, составляет
Л0=15,3±0,1 распадов-мин-1-г1
Действительно, измерения радиоактивности являются наиболее точным
способом детектирования 14С. При условии, что скорость образования 14С в
атмосфере осталась относительно постоянной « что обмен углерода между объектом
и атмосферой прекращается, когда объект умирает, сравнивая оставшуюся
14С-радиоактивиость с предполагаемой исходной активностью Ао, можно
определить, пользуясь уравнениями <(9.6) и> (9.10), возраст объекта:
А/А0=е~«1п*/т)
или
(9Л1)
На рис. 9.4 приведены некоторые данные по определению возраста с
помощью радиоактивного углерода. Сравнивая возраст объекта, определенный с
помощью радиоактивного углерода, с возрастом такого же объекта (остистой сое-
266 ЧАСТЬ 3
Возраст • /О лет
Рис. 9.4. Скорость р-распада в зависимости от возраста некоторых объектов
(возраст определялся по активности 14С) [81.
/ — ранняя полинезийская культура; 2— свитки из Мертвого моря; 3 — Птоломей; 4 —
секвойя; 5 — Стоунхеидж; 6 — гробинцы (Средний Восток); 7 — деревья, вынутые из кратер-
ного озера (Орегон); в —индейские сандалнн (Форт-рок, Орегон); 9 — ископаемые остатки
человека (Фулсом); 10 — гигантский ленивец (гипсовая пещера, Невада).
иы), найденным по числу колец на срезе старого дерева (рис. 9.5), оказывается
возможным исследовать изменение процесса образования атмосферного 14С со
временем, что особенно важно для старых образцов. Ошибки при определении
возраста измерением 14С-радиоактивности вызываются также интенсивным
сжиганием в последнее время топлив (угля, нефти), в которых почти нет 14С и
которые разбавляют современный атмосферный ИС, и особенно увеличением
образования 14С в результате ядерных взрывов (см. пример о выпадении осадков).
Наконец, следует указать, что (уже низкая^ радиоактивность ,4С в образцах
углерода снижается приблизительно до 0,1 % от исходной величины за время,
превышающее 10 периодов полураспада, так что метод определения возраста по
14С строго ограничен образцами с возрастом от 2000 до 20 000 лет.
Пример. Измерение геологического возраста по отношению содержания
радиоактивных изотопов: определение возраста по отношению
содержания изотопов калия н аргона
Несмотря на короткий период полураспада (относительно возраста земли),
14С удается сегодня определять благодаря его непрерывному образованию в
верхних слоях атмосферы; для большинства других изотопов, имеющих
палеонтологический интерес, 'положение иное. Так, например, в настоящее время
радиоактивный 40К представляет собой «остатки» того 40К, который присутствовал при
образовании земли. Изотопное содержание 40К во всех современных рудах
приблизительно одно и то же (0,0119 атом. %). При изоляции образца,
содержащего 40К, например, при затвердевании руды, прерывается обмен дочернего
изотопа с окружающей средой в большей степени, чем для родительского изотопа,
как в случае 14С. Поэтому методы определения возраста, основанные на
измерении радиоактивности не '4С, а другях изотопов, несколько отличаются от уже
описанного выше.
Поскольку большинство полезных руд и минералов содержат 0,1—10%
калия, общее содержание калия сравнительно легко определить гравиметрически
267 РОСТ И РАСПАД
It
Г
1
>
I
а
в
if
•в
!
о
<i
I
«О
§u
I
$
1
J.
7000 6000 SOOO 400O 3000
Условный еозрас/п по иС, годы
2000
1000
Рис. 9.5. Корреляция данных о возрасте дерева, определяемом по кольцевому
риеунку ствола и по содержанию радиоактивного углерода [6].
Настоящее время — 1950 г. н. э. Для периода с 5300 г. до н. э. по настоящее время
определялась радиоактивность образцов Pinus aristata н Sequoia gigantea (гигантская секвойя).
Если бы за этот период уровень атмосферного содержания ,4С был постоянен, то данные
бы попали на прямую с углом наклона 45°; отклонения, следовательно, отражают измене-
нения в содержании в атмосфере МС.
или пламенной фотометрией; затем можно найти содержание в образце 40К,
поскольку его доля от всего калия постоянна. Дочерний изотоп 40Аг определяется
после плавления образца в вакууме масс-опехтрометрически. Точность метода
определяется точностью (необходимой) поправки на присутствие современного
атмосферного *°Аг, Расчет возраста основывается на уравнениях (9.6) и (9.10)
(где Nd, No и N теперь означают соответственно количество дочернего изотопа в
268 часть з
настоящее время, количество родительского изотопа, находящегося в образце во
время затвердевания руды, и количество родительского .изотопа в образце в
настоящее время):
ND=N0-N (9.12)
или
=Ме+Пп2'т — N=N{enn2'T — 1) (9.13)
N
Тогда получаем
*-тегН1+%) <914>
Использование описанной методики сопровождается еще одним
затруднением: распад радиоактивного 40К происходит по двум направлениям:
^*°Са Гв= 1,47-10е лет
\4оАг ть = 1,19> Ю10 лет
Если принять это во внимание, то уравнение (9.14) принимает вид (см.
задачи в конце главы)
*= 1
In 2
1
+
L 4 о
4;]"№+')т]
(9.15)
На рис. 9.6 приведены данные, иллюстрирующие одни из самых известных
примеров определения возраста по отношению калий/аргои, показавшего, что
возраст людей как биологического вида составляет ие менее 1,75 млн. лет.
(Различные числа с индексом «КА» на рисунке шифруют разные образцы: КА1047 —
образец, найденный на 30,5 см выше уровня 'локализации представителей
Zinjanthropus; KA850—г образец, обнаруженный на 2,54 см выше уровня пола;
н КА1180 — образец, найденный на 2 м 6 см ниже уровня пола.) Измерение
отношения калий/аргон применяется для, определения геологического возраста
самых старых известных руд (— 3,5-109 лет) и руд не старше 50 000 лет. Другие
примеры применения метода можно найти в литературе, указанной в конце
главы.
Пример. Химический анализ при определении возраста
Совсем недавно разработанный и весьма многообещающий метод
определения возраста объектов от 5000 до 150000 лет основывается на двух фактах:
1) в живых организмах обычно находят только L-аминокислоты и 2) за
десятки тысяч лет происходит существенная рацемизация (взаимное превращение L-
и D-амииокислот):
k k
L ^=± D; /СРавн = ^-=1, (9.16)
где k — константа скорости (первого порядка) взаимного превращения L- и
D-изомеров. Согласно уравнению (9.1), скорость изменения содержания D-изоме-
ра должна определяться его скоростью образования (из L-изомера) и скоростью
его исчезновения (превращения в L-изомер):
-$-=k[L]-k[D]. (9.17)
.Запад
Синантроп
Восток
Второй рамам
Третий разлом
l,66'10*/iem (*А№7)
178-Юв/гепт(КАВ5о)
J,76'tO'/rem (КА1051)
(f, 74 • 706лет)(КАЮ39)
ies-W6/rem {ка ива)
(1,60- Ю6лет)(КА927Е)
1,92 Ю'/iem (НА II00)
^Шттттт mm
.'.'..] Галечный конгломерат
|£г;-;-';л.*£ /Toто к лавы
}^^-bj Pi una, аргиллит' \' "S а., [ Валунный конгломера/ft
'.■.- -\Тиф I л лл * | Туфобрекчия ила грубозернистые
.•,--ь--1 *r I.. ■ * .. I отложения грязевык потоков
, Наплавленное
отложение:
0,6040 6лет ) (KA92UA )
(/,оФ'70влет) (KA966)
1,76-Ю6лет (КАЮ4Л)
1,76-Ю6лет 1КАЮ53)
1.75 • 10 6лет (КА105 7 )
1.76 • Ю6лет(КАЮбв)
\ 1,70-Ю*лет(КАШ9)
-Лава пахоэхоэ
■Аа-угавы
-(1,76 40 6лет)(КА933)
t.65-IO6/iem(KAIOS0)
J,91 • Ю*/гет (КАIOS8)
t
J.
-=*0
,00 |
\50 |
1 \ » 1
Горизонтальная шкала, мили
Рис. 9.6. Стратиграфический срез пласта периода I в районе Олдувай Гордж (Танзания), на котором указаны
расположения окаменелых человеческих останков синантропа и образцов, возраст которых определяется
радиометрическими методами [5J.
Последовательность отложений реконструирована так, как если бы они произошла в концепериода I. По мнению Еверндена и
Куртнса значения, приведенные в скобках, являются ненадежными.
270 ЧАСТЬ 3
Уравнение (9.17) можно привести к виду, позволяющему определять
возраст органических объектов, если известно отношеине содержания изомеров
![D]/i[L], характерное для исследуемого объекта в настоящее время. Определим
(переменное х:
'где [L]o и [D]o—исходные концентрации L- и D-изомерав в момент смерти
организма (т. е. в момент начала рецемизации), и предположим, что [Ljo^tDjo,
яюсхольку в живых организмах преобладают L-изомерьп:
d[D]/dt =k({L]0-x)-k([D]0+x). (9.19)
Прежде всего из уравнения (9.18), можно заметить, что
d[D]/di = dx/dt,
так что уравнение (9.19) можно переписать:
dx/dt = k ([L]0 — ID]0) — 2kx.
Далее введем новое переменное и
(L!o-[D]„
« =
— *,
(9.20)
(9.21)
(9.22)
гак что
—dxldt = du-ldt = — 2ku. (9.23)
Теперь уравнение (9.23) ло виду аналогично уравнению (9.1) и его можно
In и — lnuo = —Ш (9.24)
проинтегрировать
или
,„ (E&jIS) _ ,„ (ibbiPI» _ ,) , 2«. (9.25)
Вновь используя уравнения (9.18), можно упростить уравнение (9.25):
-№ВД—
(9.26)
Уравнение (9.26) использовано для построения графиков на рис. 9.7. Видно,
что разность концентраций (i[L]o—[D]o) падает экспоненциально с постоянной
(——^k). Наконец, если, мертвый организм изолирован из окружающей среды
(например, путем захоронения), то общее количество аминокислот должно
остаться постоянным во времени:
[LJ+ [Dj = [L]0-HDJo (9.27)
и уравнение (9.26) можно переписать:
ULJe-JDJo) QL] + [DP ?,.
|П (Й-(D|) '([LJq + ID]0)-^
или, наконец,
(9.28)
27|1 РОСТ И РАСПАД
\
\ (U10 + [D]0)
Ul0
(fUo+fDjg)
2
Wo
0/23456
t
Единицы времени = (1я2)/2к г
Рис. 9.7. Зависимость концентрации L-изомера (верхняя кривая) и D-изомера
(нижняя кривая) от времени после начала рацемизации смеси.
{Llo и [DJo—исходные концентрации изомеров в пулевой момент времени. Кривые
описываются уравнением (9.26). Обратите внимание на то, что асимптота (пунктирная линия)
соответствует составу смеси изомеров (1 : 1).
Согласно уравнению (9.28), возраст органического объекта можно
определить путем измерений отношения [D]/I[L], характерного для объекта в
настоящее время, и отношения [D]/![L], характерного для современного объекта
органической природы и принимаемого за эталон, т. е. отношения r[D]o/[L]o. На
практике вначале выделяют исследуемую аминокислоту, например аспарагиновую,
в виде D.L-смеси с помощью ионообменной хроматографии, как описано в
раз. 8.Г, а затем ее связывают с L-лейцином для образования смеси двух диасте-
реомерных дипептидов, которые можно легко разделить за одну стадию
ионообменной хроматографии. Некоторые типичные значения отношения [D]/[L]
приведены на рис. 9.8 (окраска в реакции с ниигидрииом для L-D-дипептида слабее,
чем для L-L-дипептида, что и приводит к кажущемуся неравенству количеств L-D-
и iL-L-дипеитидов на хроматограмме рацемической смеси дипептида; это легко
исправить, введя соотвтествующий поправочный коэффициент). Образец № 1965,
исследовавшийся в Калифорнийском Университете, по данным измерения
радиоактивности углерода характеризовался возрастом 17 550±1000 лет (рис. 9.8), и,
следовательно, его можно было использовать для определения константы скорости
раЦеМИЗацИИ Каспар:
Ааспар = 1.48-10-6 лет-1 при 24 °С.
Скорость рацемизации аспартата (на основании этих данных) затем можно
использовать при определении возраста других объектов аналогичной географн-
ческой локализации (верхняя часть Олдуваи Горж, Танзания). Таким образом
найдеи возраст Эйази I Гоминида, составляющий 34 000 лет.
Точность определения возраста измерениями радиоактивности углерода
зависит от постоянства образования 14С в атмосфере; точность определения
возраста с помощью химического анализа зависит от сохранения постоянства
температуры объекта, .поскольку скорости химических реакций заметно изменяются
с температурой. Однако для химического анализа необходимо всего лишь
несколько граммов образца, а для надёжных измерений радиоактивности углерода
требуются сотни граммов образца; кроме того, исследуя аминокислоту
(например, аланин), которая рацемизуется медленнее, чем аспарапшовая кислота,
можно определять возраст костей вплоть до 100 000—150 000 лет:
Каспар ^ 7«изолейцин; «аспар Я? 3 «аланин.
1
[11
[D]
1 1 1
1 l
272 часть з
Рис. 9.8. Часть хроматограммы
регистрируемой при. аминокислотном
анализе разных образцов костей,
указывающая на присутствие диастерео-
мериых днпептидов Ь-лейцил-О-аспа-
рагиновой кислоты и Ь-лейцил-О-ас-
•парагииовой кислоты [3].
Колонка (56X0,9 см) заполнена смолой
бекман-спинко AA-I5. Колонку элюировалн
буферами с рН 3,24 (40 мии), а затем с
рН 4,25. Смена буферов и появление пиков
LLeu-D-Asp и L-Leu-L-Asp происходили на
72, 79 н 83-й мин с указанной
последовательностью элюирования. Небольшой
пик, элюнруемый между пиком изменения
буфера и пиком L-Leu-D-Asp,
соответствует глицину, который иеполн9стью
отделяется от аспарагиновой кислоты на колонке
со смолой дауэкс 50 (Н+).
Пример. Радиоактивные осадки и прочие радиоактивные загрязнения
В связи с тем что в последнее время добыча топлива органического
происхождения стала совершенно явно сокращаться, причем с каждым годом все
больше, экономическая потребность в расширении сети атомных электростанций
постоянно увеличивается. Поэтому актуальным является обсуждение опасностей,
связанных с увеличением радиоактивных отходов >при эксплуатации таких
электростанций, и влияние этого фактора на здоровье иаселения. Ниже коротко
перечислены различные источники радиации н приведены некоторые
количественные характеристики их относительной значимости.
Самый значительный на сегодня (и, вероятно, в будущем) источник
радиации — естественная радиоактивность космических лучей и их продуктов, а также
радиоактивных изотопов природного происхождения в коре земли (и,
следовательно, в строительных материалах и в пище)... Следующим по создаваемой
радиоактивности источником (около половины средней общей дозы естественной
радиации) является диагностическая радиология (рентгеновские лучи,
применение радиоактивных индикаторов для локализации заболевания и исследований
метаболизма). Влияние первого из этих двух источников избежать нельзя, а
польза от второго значительно перевешивает вред, сопровождающий его
применения. Наиболее крупным источником радиоактивности, который в принципе
можно ликвидировать, являются испытания ядерного оружия в атмосфере
(табл. 9.1). Единица дозы — бэр—определяется как количество любой
ионизирующей, радиации, обладающей эффективностью биологического воздействия,
проявляемому 1 рад рентгеновского излучения, а 1 рад соответствует 100 эрг,
поглощенным 1 г материала. Для оценки биологической эффективности величин,
приведенных в табл. 9.1, можно упомянуть, что дозы, составляющие всего
фрагмент кости быка
■)/
B/L » 0.07
jJ
Исследуемый
образец
D/L = 0,72
DL- аспараги новая
кислота
UCLA 1695
J7/L ,0,32 1 L-L;u"
■ d-Asp
Таблица 9.1
Радиоактивность на душу населения и суммарные дозы радиоактивности в США в 1960, 1970
и 2000 гг. [4]
Источник радиоактивности
1960 г.
(население США 183-10' чел.)
дозз на душу суммарная
населения, доаа, млн.
мбэр/год чел,- бэр/год
1970 г.
(население США 205-108 чел.)
доза на душу суммарная
населения, доза, млн.
мбэр/год чел.- бэр/год
2000 Г.
(население США 321-10* чел.)
доза на душу суммарная
населения, доза, млн.
мбэр/год чел. бар/год
Природные изотопы
Радиоактивные изотопы в промышленности и
научных исследованиях
Ядерная энергетика
Переработка топлива
Источники, выявленные Комиссией по Атомной
энергии (исключая испытания оружия в
атмосфере)
Испытания оружия в атмосфере
Телевизоры, товары широкого потребления,
путешествия воздушным транспортом
Диагностическая радиология
130
0,75
0,0001
—
0,01
13,0*
1,6
72
217
24
14-Ю-2
18,10-»
—
18-10-*
2,4
29-Ю-3
13,3
40
130
0,8
0,002
0,0008
0,01
4,0
2,6
72
209
27
16-Ю-2
41 Ю-5
17-10-5
20 Ю-4
0,82
40-Ю-3
14,8
43
130
0,9
0,2
0,2
0,01
4,9
1,1
72
208
42
27-iO-2
64-10-*
64- Ю-*
32-10-*
i,C
6 10~а
23,1
66
т 1963 г.
274 часть з
80 мрад, приводят к 40%-ному увеличению риска заболеть раком, если радиация
воздействовала на плод в период первьюс трех месяцев беременности (эта оценка
основана на исследовании 700 000 детей доктором Брианом Мак-Махоном в
Гарвардском медицинском институте в 1962 г.). Следует при этом отметить, что
прн увеличении на 40% очень малой величины она остается все-таки
незначительной.
Радиоактивные изотопы появляются в атмосфере в результате ядерных
испытаний как промежуточные продукты радиоактивного расщепления ядра
(табл. 9.2), как продукты взаимодействия радиоактивного излучения с воздухом
(табл. 9.3) и почвой (табл. 9.4). (1 Ки соответствует такому количеству
радиоактивного материала, в котором происходит 3,7-1010 распадов/с.) Здесь уместно
напомнить, что с 1945 г. суммарная мощность ядерных устройств, испытанных в
атмосфере, уже превысила 400 Мт. Следующим по интенсивности радиации после
испытаний ядерного оружия, как видно из табл. 9.1, идет телевидение (в
основном рентгеновское излучение цветных телевизоров), товары широкого
потребления (в основном ручные часы, циферблаты которых нанесены радиоактивной
краской) и путешествия воздушным транспортом (облучение человека в
самолете увеличивается из-за того, что самолет совершает полет выше уровня, при
котором атмосфера представляет собой естественный защитный экран от косми-
Таблица 9.2
Некоторые (приблизительные)
характеристики основных радиоактивных
изотопов, образующихся при ядерном
взрыве мощностью 1 Мт [4, с. 321]
Изотоп
•»Sr
9°Sr
«Zr
103Ru
1MRu
1311
137Cs
131 Ce
i^Ce
Период
полураспада
53 сут
28 лет
65 сут
40 сут
1 год
8 сут
30 лет
1 год
290 сут
Радиоактивность,
МКи
20,0
0,1
25,0
18,5
0,29
125,0
0,16
39,0
3,7
Таблица 9.3
Основные радиоактивные изотопы, образующиеся
в воздухе [4, с. 325]
Изотоп
ЗН
14С]
39Аг
Период
полураспада (лет)
12,3
5600
~260
Радиоактивность, Кн/Мт
<1
3,4-10*
59
75 РОСТ И РАСПАД
2,5 г-
VyW
1111
1954 1956
1958
1960
1962
1964 1966 1968 1970 1972
Рис. 9.9. Содержание 90Sr в водопроводной воде в Нью-Йорке за 1954—1971 гг,
[4, с. 371).
ческой радиации). Наряду с людьми, которые непосредственно работают с
радиоизотопами (табл. 9.1), и для остальной части населения быстро возрастает
опасность радиоактивного облучения, связанная со строительством атомных
электростанций, хотя пока эта опасность еще очень мала. Лишь недавно
атомные (гражданские) электростанции США "превысили по уровню радиоактивных
отходов военное производство плутония; к 2000 г. ожидается, что будет
накоплено около 227• I О6 л жидких радиоактивных отходов, не пригодных к
дальнейшей переработке. С 1969 г. Комиссия по атомной энергии рекомендовала
переводить высокорадиоактивные жидкие отходы в твердые для окончательного
захоронения. Удачным местом захоронения могут оказаться соляные озера,
поскольку соль обладает защищающими от радиации свойствами, аналогичными бетону
и природной пластичностью для эффективного замыкания контейнера с отхода-
Таблица 9.4
Основные радиоактивные изотопы, образующиеся
в почве [4, с. 325]
Изотоп
Период полураспада
Радиоактивность» Ки/Мт
2*Na
32р
«к
4БСа
ь«Мо
Бьре
59Fe
15 ч
14 сут
12 ч
152 сут
2,6 ч
2,9 года
46 сут
2,8.10й
1,92.10е
3-J010
4,7- Ю7
3,4.1011
1,7-Ю7
2,2.10е
276 часть з
7,0
6,0
* 5,0
£ 4,0
|з,о
I
4 2,0
1,0
"1954 1956 1958 1960 1962 1964 1966 J968
Рис. 9.10. Относительное содержание 9oSr (относительно Са) в образцах костей
человека в северной зоне с умеренным климатом (возрастной интервал 0—4
года) [4, с. 378].
/ — Канада; 2 —Дания; 3 — ФРГ; 4 — Польша; 5 —СССР (Москва): 6 — Англия
(исследовались в Западном Лондоне взрослые, в других местностях — население любого возраста).
ми, а также высочайшей способностью к рассеянию тепла по сравнению с
любыми видами горных пород. (Из стоков реактора уже удается удалять около
99,9% радиоактивных благородных газов.)
Поскольку 90Sr характеризуется очень, большим периодом полураспада
(28 лет), а также способен замещать кальций (в костях), предложено
рассматривать содержание 90Sr как лимитирующий фактор степени глобального
загрязнения продуктами ядерного расщепления, которую может выдерживать
человеческий организм. Данные, приведенные на рис. 9.9 и 9Л0, Показывают тесную
взаимосвязь между содержанием 90Sr в окружающей среде и его содержанием в
Человеческих костях; колебания в течение календарного года обусловлены
ядерными испытаниями в атмосфере; этим же можно объяснить максимум,
приходящийся на 1963 г.
Следом за мощными ядерными испытаниями в атмосфере в 1961 и- 1962 гг.
тропосферное содержание 14С увеличилось до уровня» более чем на 500%
превосходящего нормальный уровень в северной гемисфере, а содержание его в
человеческих тканях уже в 1964 г. возросло на 20—30% (относительно уровня
1950 г.). Помимо прямой опасности радиации раопад UC, входящего в состав
нуклеиновых кислот, создает опасность мутаций нз-за повреждения самой
структуры гена. В заключение следует сказать несколько слов о 13,1: из-за
способности щитовидной железы человека концентрировать этот элемент (и из-за его
интенсивного образования <при ядерном расщеплении) дозьв радиации, которым
подвергается щитовидная железа, могут оказаться относительно высокими даже
при сравнительно низкой концентрации 13,1. Так, по оценкам медиков после
испытательных взрывов 19 мая 1953 г. дозы для населения в Сант-Джордже (штат
Юта) составляли около 84 рад.
Возрастной интервал
0-4 года
- О / + 4
а 2 ж 5
7 3 • б
• а
ГЧ
277 РОСТ И РАСПАД
Задачи
1. Внутривенное введение собаке 30 мг фенобарбитала на 1 кг массы тела
обычно вызывает анестезию, необходимую при хирургическом вмешательстве.
Удаление лекарства нз организма в результате процессов метаболизма
соответствует процессу первого порядка ([ ] = [ Ьехр(—kT)) с периодом
полупревращения 4,5 ч. Если анестезирована собака массой 14 кг, сколько
-фенобарбитала (мг) надо ввести через два часа (когда анестезирующий эффект
начинает уменьшаться), чтобы полностью сохранилась исходная степень
анестезии?
2. Допустим истинный возраст биологического образца составляет 57 000 лет, и
пусть образец загрязнен 1 масс. % современным углеродом. Рассчитайте
(кажущийся) возраст, который будет установлен на основании измерений
радиоактивности углерода, если экспериментатор не учитывает наличия примесн.
3. Выведите выражение для определения возраста объекта [уравнение (9.15)] с
учетом схемы двойственного распада радиоактивного Л0К и покажите, что
общий период полураспада составляет
Г=(1п2)/(£а+Ад = 1,ЗЫ09 лет.
4. Определение возраста с помощью химического анализа основано на данных о
рацемизации L-аспарагиновой кислоты:
L Ь^ D
^аспар "*^— ^аспар
*acn.p=(M8 ± 0,09)- Ю-5 лет"1 при 24 °С
Рассчитайте возраст образца биологического происхождения, у которого
измеренное соотношение l[D]/[L] составляет 0,72, если у современного образца
соотношение l[D]/i[L] —0,07.
Литература
1. Hodgson F., Dating by Radioisotopes, Wlliam Clowes & Sons, Ltd., London,
1970. Хорошее рассмотрение вопросов определения геологического возраста
по измерениям радиоактивности.
2. Dalrymple G. В., Lanphere M. A., Potassium-Argon Dating, W. H. Freeman, San
Francisco 1969
3. Bada J. L., Proisch R., Proc. Natl. Acad. Sci. U. S. A., 70, 1331 (1973).
Принципы определения геологического возраста по данным химического анализа:
новейшие сведения о применении метода для определения возраста
биологических объектов см. Nature, 262, 279 (1976).
4. Eisenbud M., Environmental Radioactivity, 2nd ed., Academic Press, New York,
1973. Факты о радиоактивных загрязнениях, обусловленных выпадением
осадков.
5. Gramme С. S., Hay R. L., Earth Planet. Sci. Letters, 2, III—115 (1967).
6. Radiocarbon Variations and Absolute Chronology, I. U. Olsson (Ed.), Wiley
Interscience, 12th Nobel Symposium (1970).
7. Giese A. C, Cell Physiology, W. B. Saunders Co., Philadelphia, 1962, p. 521.
&. Mahan B. H., University Chemistry, 2nd ed., Addison Wesley, Reading, Massa-
chuets, 1969, p. 800.
ГЛАВА 10
КАТАЛИЗАТОРЫ
10.А. КИНЕТИКА (СТАЦИОНАРНАЯ)
ЛШХАЭЛИСА — МЕНТЕН
I0.A.1. НЕОБХОДИМОСТЬ ГИПОТЕЗЫ О СТАЦИОНАРНОСТИ
В противоположность общей точке зрения основные трудности
при исследовании многих биофизических проблем сопряжены не
с их экспериментальным изучением (что часто имеет место), а с
аналитическим выражением получаемых результатов. Иногда
аналитическое выражение имеет настолько сложный вид, что это не
позволяет объяснить исследуемый процесс. Упрощенные схемы
ферментативной кинетики служат одним из лучших примеров
того, как с помощью обоснованных приближений можно свести
сложную задачу к понятному и решаемому варианту. Такой подход
широко применяется при анализе ферментов и лекарственных
веществ, при изучении механизмов ферментативных реакций и
реакций биологических систем на воздействие лекарств.
Идея о существовании фермент-субстратного комплекса
впервые была высказана (косвенно!) Брауном в 1902 г. при
исследовании зависимости скорости инверсии сахарозы ферментом ин-
вертазой от концентрации реагентов. До этого в течение 20 лет
было собрано много данных, подтверждающих, что кинетика
химических реакций с участием ферментов, по-видимому, принципи*
•ально отличается от обычных химических реакций. В то время
как скорость большинства химических реакций пропорциональна
концентрации реагентов в системе [уравнение (9.1.), известное как
закон действующих масс], скорости ферментативных реакций
часто не зависят от концентрации реагента. Простое объяснение
этого факта состоит в том, что для успешного протекания реакции
требуется связывание реагента (субстрата) с катализатором
(ферментом) ; тогда понятно, что при достаточно высокой
концентрации субстрата фермент будет «насыщен», так что практически
весь фермент в любой данный момент времени связан с
субстратом и, следовательно» скорость суммарного химического процесса
достигнет предельного значения, пропорционального
концентрации фермента в системе, но независящего от концентрации
(избыточной) субстрата. Сформулируем задачу математически, чтобы
получить обоснование вышеизложенному логическому
объяснению.
279 РОСТ И РАСПАД
Если связывание субстратов с ферментом является важной
особенностью ферментативной реакции, следовало бы ожидать, что
простейшей общей моделью, с которой надо начать формальный
анализ, должна служить следующая:
Е
z± E*V + B
Г^ЕР+Q
11 Г И
А+В + Е EAB ■*—*• EPQ E + P+Q (ЮЛ)
I'-»- ЕВ 4- А ^-\ \\->- ЕО+Р ^-\
где А и В—два реагента, Р и Q — продукты, Е — свободный
катализатор и ЕА, ЕВ, EAB, EPQ, ЕР и EQ — разные возможные
комплексы фермента с субстратом (субстратами) и продуктом
(продуктами). Математические выражения, описывающие эту
модель, не пригодны ни для каких практических целей. Первое
упрощение состоит в игнорировании второго субстрата, что будет
обосновано позже. Остается схема
Е + А у-> ЕА -г—* ЕР ч—* Е+Р (10.2)
где А и Р — субстрат и продукт, как общепринято в литературе.
Далее необходимо игнорировать присутствие всех, кроме одного,
промежуточных соединений:
Е + А?р± ЕА?=± Е+Р (10.3)
К—1 К—2
Далее целесообразно ограничить период наблюдения только
начальным этапом реакции до тех пор, пока не накопилось
значительное количество продукта; обратная реакция между Е и Р,
приводящая к образованию ЕА, тогда может быть опущена:
Е + А *=* ЕА v Е+Р
(10.4)
Последнее упрощающее предположение состоит в том, что
концентрация фермента мала по сравнению с (начальной)
концентрацией субстрата и по сравнению с (равновесной) концентрацией
продукта. Предлагаемые упрощения интуитивно достаточно
обоснованы; необходимо еще последнее условие
I [А]0»[Е]0 1 (10.5)
поскольку концентрация комплекса ЕА должна быть постоянной.
Обратите внимание, что ограничения относительно одного
субстрата, одного промежуточного соединения и необратимого
образования продукта в дальнейшем можно легко снять, не изменяя ос-
280 ЧАСТЬ 3
новной вид результата, но допущение о стационарном состоянии
является абсолютно необходимым для описания ферментативной
кинетики в сжатом виде. (Механизмы реакций, предполагаемые
стационарной кинетикой, можно проверить методами кинетики
переходных состояний, рассматриваемыми в ч. 4,) Поскольку услот
вие стационарности, описываемое уравнением (10.5), является
практически единственным принципиальным допущением, ниже
рассматривается его необходимость и дается обоснование.
После всех упрощений для описания ферментативной реакции
(10.1) наиболее простым и полезным является уравнение (10.4);
теперь надо вернуться к элементарной схеме для создания основы,
необходимой для решения уравнения (10.4). Из следующих пяти
иллюстраций (рис. 10.1—10.5) читатель может проследить за
развитием кинетической модели, которое привело к введению
допущения о стационарности концентрации промежуточного
соединения. Алгебраические выражения, соответствующие кривым на этих
рисунках, не должны отвлекать внимания от простоты получаемых
результатов.
Ю.А.2. ОДНОСТАДИЙНАЯ ОД ПОКОМПОНЕНТНАЯ ПРЯМАЯ РЕАКЦИЯ
(10.6)
Реакция (10.6) характеризуется скоростью исчезновения
реагента А:
d[A]/dt=— k^A]
(10.7)
что аналогично распаду «первого порядка», уже рассмотренному
в гл. 9.' Это уравнение уже было решено [см. уравнения (9.1) —
(9.6)], и полученный результат графически изображен на рис. 10.1.
Время, t
Рис. 10.1.Зависимость концентрации реагента [А] и концентрации продукта [В]
от времени реакции, протекающей по уравнению (10.6).
В начальный момент продукт отсутствует [В]о«0.
$1 РОСТ И РАСПАД
б.А.3. ПРЯМАЯ И ОБРАТНАЯ РЕАКЦИИ
(10.8)
Уравнение (10.6) описывает простейшую химическую реакцию;
для процесса (10.4) необходимо допустить возможность
протекания реакции в обратном направлении:
d[A]/dt=—fci[Al-Hfe-i[B]=—d[B]/dt. (10.9)
т. е. А исчезает со скоростью, пропорциональной количеству А, и
увеличивается со скоростью, пропорциональной содержанию В в
системе. Несколько более простой случай уже был решен при
kx=k-\ при аналдзе химических методов установления
геологического возраста [(разд. 9.А, уравнения (9.16) — (9.28)]. Поскольку
"данная задача совершенно аналогична, читатель сам может
показать, что
[А]=[А]0- ^АД (l-expl-fo+fe.,)*]) (10.10)
W= vr^~7 O-^l-fo+M'J)
(10.11)
где предполагается, что в начале реакции продукта в системе нет,
т. е. [В]о=0.
Уравнения (10.10) и (10.11) проиллюстрированы графически на
рис. 10.-2. [А] и [В] экспоненциально приближаются к предельным
значениям, но из-за обратной реакции [А] не падает до нуля, и
СА],
|
8. [Ala*-/**/**-/)
I
ffpe/и я
Рис. 10.2. Зависимость концентраций компонентов А и В от времени реакции,
протекающей по уравнению (10.8).
Предполагается, что исходная концентрация В равна нулю. Особый случай, при котором
*i"•&-!, уже был проиллюстрирован ранее (см. рис. 9.7).
282 часть з
конечные предельные концентрации [А] и [В] достигаются
быстрее, чем в предыдущем примере. Из рис. 10.1 и 10.2 следует, что
реакция практически заканчивается прежде, чем концентрация
какого-либо из реагентов станет постоянной.
10.А.4. ПОСЛЕДОВАТЕЛЬНЫЕ РЕАКЦИИ
(10.12)
Теперь становится ясно, что для решения даже очень простых
кинетических моделей могут требоваться существенные
алгебраические построения! Последняя реакционная схема, которая будет
описана полностью, характеризуется уравнением (10.12):
d[A]/dt=— MA], (ЮЛЗ)
d[B]/dt=k1[A]—k2[B], (10.14)
d[P]/dt=k2[B]. (10.15)
Как было показано выше, уравнение (10.13) имеет следующее
решение:
[A] = [A]0exp(-V)
(10.16)
Подставим выражение для [А] в уравнение (10.14):
d[B]/#=MA]0exp(—ад— ^[В]. (10.17)
Решением последнего уравнения, как легко проверить, является
kt [Aj0
W = xbj[ IexP (-V) ~ exP (-V)l
(10.18)
Подставляя выражение для [В] в уравнение (10.15), находим
(10.19)
d [P)fdt = -g^ [exp(-^)-exp(-V)).
к2 — кх
Читатель легко может решить уравнение (10.19):
К [А]0 . k„ [AL
lp] = k Lk texP Н^) - Ч ~ k -k lexP (-V) - i]
2 1 Z X
(10.20)
На рис. 10.3 приведены рассчитанные концентрации [А], [В]
и [Р], если &i = 2&2, согласно чему можно предположить, что
существует момент (в процессе реакции), в который концентрация
промежуточного соединения [В] начинает уменьшаться.
[g3 РОСТ И РАСПАД
is* 100
!Рис. 10.3. Концентрационные изменения
в последовательных реакциях первого
порядка [4].
40 60
Время
80 100 120
аО.А.5. НЕКАТАЛИЗИРУЕМАЯ РЕАКЦИЯ
;С ОДНИМ ПРОМЕЖУТОЧНЫМ СОЕДИНЕНИЕМ
(10.21)
Схема реакции (10.21) напоминает схему ферментативной
реакции (10.4) с тем отличием, что при завершении реакции
реагент В не регенерируется. Реакции (10.21) соответствуют
математические уравнения
d[A]/dt=—k1[A] [B]+MX]=d[B]/#, (10.22)
d[Xj/#=MA][B]—fc-i[XJ — k2[X] (уравнения скоростей), (10.23)
d[P]/dt=k2[X\
(10.24)
и
(AJ0—[A]=[B]0— [В] (уравнение материального баланса). (10.25)
Уравнения (10.22) — (10.24) описывают скорости изменения
концентраций всех компонентов реакции; например, по уравнению
(10.22) компонент А исчезает со скоростью, пропорциональной
количеству всех присутствующих реагентов, и образуется со
скоростью, пропорциойальной количеству присутствующего X.
Уравнение (1Q.25) просто отражает тот факт, что в реакцию вступает од-г
на молекула А и одна молекула В.
Для системы четырех независимых уравнений с четырьмя
неизвестными [А], [В], [X] и [Р] решение существует; однако, к
сожалению, это решение невозможно выразить точно для общего
случая, и конечные концентрации должны приближенно (с любой
нужной точностью) определяться интегрированием с помощью
цифровой вычислительной машины. Типичное решение приведено-
на рис. 10.4 для случая k\zzk-xmk2 и [А]0^ [В]0. Качественно
поведение системы (рис. 10.4) аналогично более простому случаю
последовательных реакций (рис. 10.3).
> 84 часть з
Время
Рис. 10.4. Развитие реакции, протекающей по уравнению
А + В=е=*Х
fe3
при условии £i«£_i«/i2 и [А]0« [В]о![1. с. 224].
10.А.6. КАТАЛИТИЧЕСКАЯ РЕАКЦИЯ С ОДНИМ
ПРОМЕЖУТОЧНЫМ СОЕДИНЕНИЕМ
(10.26)
Уравнение (10.26) описывает кинетическую схему, которая
обычно служит отправной точкой в кинетике ферментативных
реакций, и отличается от предыдущего примера [уравнение (10.21)]
только тем, что в конце реакции катализатор Е регенерируется.
Соответствующие уравнения скоростей:
d [A]ldt=-kx [А] [Е]+&_г [ЕА],
d [EA]/dt=kx [A] [E] - &_! [ЕА] - &2 [ЕА-],'
d[P]/^=^[EA],
dfEj/^^-MAHEj+^tEAj+MEA]
(10.27)
(10.28)
(10.29)
(10.30)
и
[Е]0=[Е]+[ЕА] (уравнение материального баланса). (10.31)
Хотя общего решения системы уравнений (10.27) —(10.31) не
существует, результат в каждом конкретном случае вновь можно
получить интегрированием, применяя ЭВМ (рис. 10.5). Новой и
характерной особенностью кривых на рис. 10.5 является
постоянство концентрации промежуточного соединения [ЕА] и скорости
образования продукта после начального индукционного периода
(слева от вертикальной пунктирной линии) (угол наклона
зависимости [Р] от времени приблизительно постоянен). Эта область
РОСТ И РАСПАД
*>
ис. 10.5. Развитие реакции, протекающей по уравнению
( А + Е*=*ЕА »-Р + Е
? «-1
при условии £(«/г_,да£2 и [А]0>[£]о [1, с. 226].
(реакции называется стационарной областью; чем больше
начальный избыток реагента по сравнению с ферментом [А]0> [Е]0, тем
короче индукционный период, в котором не выполняется условие
стационарности ([ЕА] =const).
Условие стационарности, т.е. [EA]=const, a d'[EA]/^/«0,
позволяет значительно упростить выражения скоростей реакции
(так что вообще не нужно больше решать дифференциальные
уравнения!). Рассмотрим уравнение (10.28):
d[EA]fdt=k1[k] [E]—(^.1+^)[ЕА]=0
или
(А-1+*0#1 = [Е] [А]/[ЕА] (10.32)
Поскольку в правую часть уравнения (10.32) входят только
постоянные величины и ее вид соответствует выражению для
константы равновесия, ее обычно обозначают как константу Михаэли-
са Ка
Ka = (*-i+*i)/*i
(10.33)
/Сд можно считать константой равновесия для диссоциации
комплекса ЕА на реагенты Е и А или на продукты Е и Р.
Следовательно 1//Са характеризует прочность связывания субстрата с
ферментом: меньшее значение Ка соответствует более прочному
связыванию. Перепишем уравнение (10.32):
[ЕА1=[ЕПА]/КА. (10.34)
.386-- шьсть з
Концентрация сс/бс/прата [А]
Рис. 10.6. Зависимость скорости стационарной ферментативной реакции v от
концентрации субстрата [А] при постоянной концентрации фермента.
При высокой концентрации субстрата [А] скорость достигает предельного значения Vwe„„
макс*
Лд равна концентрации субстрата при скорости, равной половине максимальной.
В этом случае скорость образования продукта (т. е. искомая
наблюдаемая величина) пропорциональна концентрации [ЕА],
которую, однако, желательно выразить более практически полезным
уравнением, чем уравнение (10.34). Поэтому, заметив, что фер^
мент в системе должен находиться в начальный момент в виде
одной из двух форм [Е] или [ЕА], запишем
[Е]общ = [Е] +[ЕА]
(10.35)
Уравнение (10.34) можно переписать:
[ЕА]=([Е]общ-[ЕА0[А]/^А
или
[ЕА]=[Е]общ[А]/(/СА+[А]).
(10.36)
И наконец, обозначив скорость появления продукта d[P]/dt = v,
можно получить выражение для скорости суммарной реакции из
уравнений (10.36) и (10.29):
й[Р1/Л=о=йа[Е1общ[А]/(/СА+[А]). (10.37)
Зависимость скорости реакции от концентрации субстрата
показана на рис. 10.6: v не зависит от концентрации А в присутствии
избытка А и пропорциональна ей при низких концентрациях А.
Простая кинетическая модель, описываемая уравнением (10.4),
следовательно, может объяснить загадочное отсутствие
концентрационной зависимости, иногда наблюдаемое в ферментативных
реакциях, о чем упоминалось в начале этого раздела. Поскольку
истинная концентрация фермента [Е]0бщ часто неизвестна (так
как большинство препаратов ферментов гетерогенны), полезно
отметить, что при очень высоких концентрациях субстрата [А]^>
РОСТ И РАСПАД
а
Рис. 10.7. Графические способы определения V„aKc и К а ферментативной
реакции из кинетических данных.
а — зависимость v от рА [уравнение (10.40)) (зависимость имеет вид кривой титрования, для
которой «конечная» точка соответствует Умакс, а точка перегиба находится при [А]=Кл)',
б — зависимость Лайнуивера — Берка (\/v от 1/{А]). Отрезок, отсекаемый на оси ординат,
соответствует l/VMaKC> а отрезок, отсекаемый на оси абсцисс, равен — 1//Сд (нет
необходимости определять скорости реакции при очень высоких концентрациях субстрата для точной
оценки VMaKC).
^>Ка v достигает максимального значения Умакс [рис. 10.6 и
уравнение (10.37)]:
v = Vu№c[A]/{KA + [A])
(10.38)
Согласно рис. 10.6, можно дать другую простую интерпретацию Ка>
согласно которой величина /Са равна концентрации субстрата, при
которой скорость (стационарной) реакции v достигает половины
своего максимального значения:
^=^макс/2=Умак? [А]/(*А-НА]).
Решая это уравнение относительно [А], находим, что
[А]=/сА,
когда
v=Vmj2. (10.39)
Уравнение (10.38) описывает гиперболу, и нахождение
значений Умакс и /Са из графика этого уравнения (рис. 10.6) зависит от
набора данных по скоростям реакции при концентрациях
субстрата, достаточно больших для приближения к предельной скорости
Умако Поэтому нужны способы определения значений УМакс и /Са,
с помощью которых можно бы получать надежные результаты при
произвольном выборе области концентрации субстрата в
эксперименте. По аналогии с хорошо известной ситуацией при титровании
283 часть з
pH = p/CA+lg([A~]/[HA]) можно определить рН = — lg[A] и
рассмотреть уравнение
РА = РКа + lg [ (Умакс ~ V)IV]
(10.40)
которое можно вывести из уравнения (10.38) (см. задачи в конце
главы). Кинетические данные, описывающие ферментативную
реакцию, таким образом, можно проанализировать так же, как
обычно анализируются результаты рН-титрования (рис. 10.7,д):
«конечная» точка определяет Умакс, а точка перегиба р/Сд=—lg^A- Как
видно из рис. 10.7, а, для определения Умакс вновь необходимы
данные при высоких концентрациях [А], чтобы достичь
«конечной» точки.
Трудности, возникающие при использовании графиков,
изображенных на рис. 10.6 и 10.7, а, можно легко обойти, если основное
уравнение гиперболы (10.38) преобразовать в уравнение прямой.
Среди нескольких возможных уравнений прямой наиболее
популярным в ферментативной кинетике является уравнение
Лайнуивера— Берка*. Возьмем обратные величины от обеих частей
уравнения (10.38):
1Д/=(/СА+[А])/Кмакс[А]
или
I/O = (AWW) (1/[A] + (1/Кмакс)
(уравнение Лайнуивера—Берка) (10.41)
График уравнения (10.41) приведен на рис. 10.7,6. Одна из
причин широкого использования уравнения Лайнуивера — Берка
для определения Умакс и К\ по кинетическим данным
ферментативной реакции состоит в удобстве нахождения искомых величин.
Для этого строится прямая по данным, полученным при
произвольном выборе концентраций субстрата '[А], затем прямая
продолжается до пересечения с осями абсцисс и ординат.
Основными параметрами, значения которых можно определить
из зависимости Лайнуивера — Берка, являются Умакс, Да и k2.
1. Умакс ПОСКОЛЬКУ Умакс = &2 [Е]0бщ, ТО Умакс МОЖНО ИСПОЛЬЗО-
вать в качестве характеристики количества фермента,
присутствующего в неизвестном растворе (для «оценки» активности
фермента). Обычно Умакс выражают в единицах ферментативной
активности, например 1 ед. активности соответствует образованию
1 мкмоля продукта в минуту при условии, что фермент насыщен
* Внимательный читатель должен заметить поразительное сходство
уравнений (10.38), (10.40) и (10.41) с соответствующими уравнениями (3.43), (3.48) и
(3.51), применяемыми для определения констант равновесного связывания.
Алгебраическое соответствие очень близко, и мы еще вернемся к этому факту,
после того как закончим обсуждение диагностической ценности зависимости
Лайнуивера — Берка.
РОСТ И РАСПАД
SGOT
»
\
ШЬНармаОО-АОед)
V
^^^■У/Л)ЛЛ,-.У|-ЛУ.У..рЛЛ|лЛ.У^
4 6
Время t сут
8
10
Рис. 10.8. Концентрация сывороточной аслартатамииотрансферазы (SGOT),
рассчитанная из значений Кмакс, ПРИ остром нифаркте миокарда [5],
субстратом. Измерения ферментативной активности все шире
начинают применяться в медицинской диагностике. Для иллюстрации
ниже приведено несколько примеров.
Пример. Диагностика заболевания сердца
Концентрация аспартатаминотрансферазы (GOT) особенно высока в
сердечной мышце (миокарде). Инфаркт миокарда (локальное повреждение ткани
сердца из-за нарушения циркуляции крови) сопровождается резким повышением
активности GOT в сыворотке крови (рис. 10.8). Острота приступа прямо
коррелирует с увеличением уровня GOT в сыворотке крови. Детектируется активность
GOT в сыворотке крови, и получаемые значения используются как
диагностический показатель: таким образом обнаруживается до 96% инфарктов миокарда,
в то время как с помощью других методов, например электрокардиограмм
(ЭКГ), не удается поставить правильный диагноз в 25—30% случаев этого
заболевания, поскольку повышение концентрации фермента в сыворотке крови
часто наблюдается при полном отсутствии каких-либо электрокардиографических
изменений. Ферментная диагностика, следовательно, позволяет гораздо раньше
выявить внезапное поражение ткани сердца.
Пример. Дифференциальная диагностика заболеваний печени и желчных путей
Дополнительную полезную информацию можно получить, если в одном
образце сыворотки крови определять содержание нескольких различных ферментов.
Хотя концентрация любого из этих ферментов не является диагностическим
показателем патологии, число возможных вариантов заболевания можно уменьшить,
сопоставляя концентрации нескольких ферментов. Например, только при
первичном билиарном циррозе (рис. 10.9, внизу справа) высокая концентрация щелочной
290 часть з
Итр&сцадяяый Твксачеаша ч Токсический л
MOt/oSykmot паренхиматозный жо/кстатическиЦ
IllJL
Qxupems
лечат
Постнекроптческш
Лаэннека цырроз t Cmeamwetcpoi
(Мтурацаотшц
• желтуха^
{опухоль)
ill-
Обтдрацаонная
же/гтуха
- {камни}
Ji
Первичный или „ Первичный^
Метааталтческщ далиарныц
рак, - царраЬ
ILiltLi
APGOTGPTLDH
Рис. L0.9. Относительные
APGOTGPTLDH
APGOTGPTLDH
APGOTGPTLDH,
концентрации ферментов при различных указанных
патологических состояниях [6].
Ферменты указаны в тексте.
фосфатазы (К.Ф. 3.1.3.1) наблюдается одновременно с низкими концентрациям»
аепартатаминотрансферазы (GOT, К.Ф. 2.6Л.1), алайинамннотрансферазы
(К.Ф. 2.6.1.2) и лактатдегидрогеиазы (К.Ф. 1.1.1.27).
2. /Са- Параметр /Са в первую очередь используется для
выявления специфичности действия фермента, поскольку /Сд
характеризует тенденцию фермент-субстратного комплекса к диссоциации
на продукт и фермент или на фермент и субстрат. Сравнивая
значения /Са для ряда различных субстратов, можно определить,
какой тип молекул наиболее прочно связывается с ферментом;
как правило, к числу наиболее прочно связывающихся субстратов
(т. е. /Са малы) относятся «природные» субстраты (субстраты, на
которые фермент действует in vivo). Проводя такие исследования,
можно, например, классифицировать различные протеолитические
ферменты (гидролизующие белки) согласно предпочтительным
участкам их действия по полипептидной цепи (рис. 10.10). Химо-
трипсин (КФ. 3.4.4.5), как было показано, расщепляет цепь,
атакуя карбоксильный конец связи между тирозином и соседним
аминокислотным остатком; эта специфичность, однако, не является
РОСТ И РАСПАД
Щ0Щ1В/1(Пидаза ч Химотрипсин -i
Пепсин \ Трипсин ^-Амино'пояа*
пептидазъ
Ш2
аргинин
^ис. 10.10. Диаграмма, иллюстрирующая специфичность действия разных про-
теолитических ферментов [7].
Цифрами пронумерованы аминокислотные остатки, названия двух из которых (тирозин в
иргинин) приведены внизу. Полипептидазы проявляют специфичность относительно свобод-
ioro С-концевого остатка (слева) или относительно N-концевого остатка (справа) белка или
пептида. Пепсин специфичен относительно N-концевого остатка тироэииа (или фенилалани-
la) внутри белковой молекулы; химотрипсин специфичен относительно тех же остатков по
карбоксильному концу аргинина нлн лизина.
Абсолютной, и химотрипсин может связывать также остатки фени-
^аланина, триптофана и метионина. Проявление подобной
специфичности часто используется для расщепления белка по
известным участкам перед аминокислотным анализом (см. разд. 8.Г):
проводя расщепление в нескольких различных (известных)
участках и анализируя получающиеся при этом продукты, можно
установить аминокислотную последовательность в белке (рис. 10.11),
3. &2. Если концентрация фермента известна, например
при.работе с очищенными ферментными препаратами, из Умакс можно
рассчитать к*
'макс — *а [Е]общ
(10.42)
Параметр &2 характеризует эффективность ферментативного
катализа и используется для получения информации о его
механизмах*. Например, деацилирование (стадия k-i), катализируемое хи-
* £2 — каталитическая константа — может рассматриваться как число молей
продукта, образующихся в единицу времени одним молем чистого фермента,
насыщенного субстратом. Введем число превращения
Число превращения=Ы(Число активных центров в молекуле фермента). (10.43)
Следовательно, число превращения показывает количество молей продукта,
образуемых в единицу времени на одном активном центре фермента, насыщенном
субстратом.
292 часть з
Рис. 10.11. Последовательность аминокислотных остатков в ферменте лиаоциме
[8, 9].
мотрипсином, в D2O протекает в 2,5 раза медленнее, чем в Н2О;
это свидетельствует о том, что на этой лимитирующей стадии
происходит перенос протона, как следовало бы ожидать при
механизме общего основного катализа. Значение kz, определенное по
данным рН-титрования, показывает, что предполагаемая основная
группа в активном центре фермента характеризуется р/(а 6,8 (см.
о влиянии рН ниже в этой главе). Как показано в табл. 10.1,
типичные значения &2 лежат в интервале 10'—106.
Таблица 10.i
Максимальные числа превращения для нескольких
ферментов
Фермент
Максимальное число
превращения (максимальное число
молекул продукта,
образующееся в 1 с на активном
центре фермента)
Химотрипсин
Рибонуклеаза
Карбоксипептидаза
Кииаза
Каталаза
Пероксидаза
Карбоаигидраза
1000
100
100
1000
5 000 000
10
36 000 000
РОСТ И РАСПАД
|& Фермент каталаза, катализирующий диспропорционирование
;рекиси водорода Н2О2, сопровождающееся распадом на воду и
[слород, обеспечивает одну из наиболее высоких скоростей
превращения, известных для хорошо изученных ферментов. Интересно
Отметить, что каталазная ферментативная система лежит в основе
гникального защитного механизма жука-бомбардира. На кончике
}рюшка этого жука находится «реакторная железа», содержащая
25 (масс/об.) % Н2Ог. Если жук потревожен или ожидает
нападения, каталазная реакция происходит столь быстро, что за
1 с вода в железе нагревается от комнатной температуры до
}00°С и под действием высокого давления, развивающегося
образующимся кислородом, в нападающего выстреливается струя
(кипящей!) воды.
|10.Б. СНЯТИЕ ОГРАНИЧЕНИЙ МИХАЗЛИСА — МЕНТЕН
^ОТНОСИТЕЛЬНО ФЕРМЕНТАТИВНЫХ РЕАКЦИЙ
%■
Г Необходимость условия стационарности (10.5) достаточно
подробно обоснована в разд. 10.А.1—I0.A.6. В этом разделе
снимаются три других ограничения (отсутствие обратной реакции, одно
'"промежуточное соединение, один субстрат), и становится ясно, что
[проблема принципиально не меняется и остается почти столь же
шростой.
%
«10.Б.1. СУЩЕСТВОВАНИЕ ОБРАТНОЙ РЕАКЦИИ
J Если обратная реакция между ферментом и продуктом возмож?
на, то в общем виде уравнение Михазлиса — Ментен принимает
Следующий вид:
kl ka
(10.3)
Соответствующее уравнение описывает скорость изменения
концентрации промежуточного соединения ЕА:
d[Ekydt^ki [E] [А]—(*_!+*,) [EA]+fc_2[E] [P] (10.44)
и при стационарных условиях [Е]0< [А]0 и/или [Е]0<'[Р]о можно
записать d[EA]!dt~Q и решить уравнение относительно [ЕА]:
[ЕА] =ЬМ±^р: [Е]ш (10.45)
:У*
Решая уравнение материального баланса
[EJ0=[E]+[EA] (10.46)
относительно [Е], подставим решение в уравнение (10.45):
ГЕА1 — fe[A]+*-,[P])[B], n А7.
294 часть з
Наконец, подставляя выражения [Е] и [ЕА] в уравнение
скорости
d[P]/dt=v=k2[EA]~ Al2[E][P] (10.48)
получаем общее уравнение стационарной скорости
(10.49)
(*Л ГА]-*_!*_, [Р])[Е]0
v~ ft-i -f- A» -h Aj [A]" + A_2. IPJ
В отсутствие продукта ([Р]=0) уравнение (10.49) принимает
вид обычного уравнения Михаэлиса—Ментен
"макс 1 * l\C\Kf\\
^прямая ~ ^A_f_ [A] IV U,0U/
и аналогично, если концентрация субстрата падает до нуля
([А]=0), вновь получается уравнение Михаэлиса — Ментен, но на
этот раз относительно продукта реакции
.. ''макс \У\ /|Л с 1\
^обрати— др _f_ [p] (1U.01J
где Умакс и /Са имеют свой обычный смысл. Эти предельные случаи
очень важны. Во-первых, если ферментативная реакция
действительно обратима, то обратная реакция также является
каталитической, и если в начале реакции в системе находится фермент и
продукт, для обратной реакции Умакс и /Ср определяются так же
просто, как если бы измерялась скорость прямой каталитической
реакции. Во-вторых, осложнения, обусловливаемые наличием
обратной реакции, как правило, можно обойти, просто ограничивая
измерения экспериментами, в которых {Р]«0.
Анализ уравнения (10.49) свидетельствует, ч?о скорость
обратимой ферментативной реакции уменьшается по двум причинам.
Во-первых, некоторое количество продукта всегда удаляется в
результате обратной реакции [вычитаемое в числителе уравнения
(10.49)]. Во-вторых, увеличивается доля фермента, связанного в
комплексе ЕР, следовательно, этот связанный фермент не может
теперь взаимодействовать с субстратом [слагаемое &-2[Р] в
знаменателе уравнения (10.49)]. Обратимая ферментативная реакция,
описываемая уравнением (10.49), следовательно, служит
примером простой биологической регуляции на молекулярном уровне:
продукт образуется быстро, только если его мало, и он образуется
медленнее, если [Р] велико. В следующей главе будут
рассмотрены другие (более сложные) механизмы регуляции скорости
ферментативной реакции.
195 рост и распад
Щ).Б.2. РЕАКЦИИ С ЧИСЛОМ ПРОМЕЖУТОЧНЫХ СОЕДИНЕНИЙ
ПОЛЬШЕ ОДНОГО
| В обычной схеме Михазлиса—Ментен (10.4) предполагалось,
нто при связывании фермента с субстратом или с продуктом
образуется один и тот же комплекс; более разумным
представляется предположение о том, что природа этого комплекса изменяется
te ходе протекания реакции
(10.52)
Запишем соответствующие уравнения скорости
d[EA]/df=ME] [A]+£_3[EP] — (£_i+£3) [ЕА] =
=0 (при стационарных условиях)
d[EP]/dt=k3[EA} — (&_з+&2) [ЕР] =
=0 (при стационарных условиях)
d[P]A#=t>=&2[EP]
и уравнение материального баланса
[Е]0=[Е1 = [ЕА]-НЕР].
(10.53)
(10.54)
(10.55)
(10.56)
Чтобы решить эту систему уравнений, надо сделать следующие
преобразования. Уравнение (10.56) решают относительно [Е] и
подставляют вместо [Е] в уравнение (10.53); затем решают
уравнение (10.53) относительно [ЕА], подставляют полученное
выражение вместо [ЕА] в уравнение (10.54) и решают его
относительно [ЕР]; и наконец, подставляют полученное выражение вместо
[ЕР] в уравнение (10.55) и получают выражение скорости
стационарной реакции v (см. задачи в конце главы)
и =
Wi [А] [Е]0
к.гк.9 + к.гкг + кэк2 + [А] кг (кэ + fc_3 + k2)
(10.57)
Если не принимать во внимание обозначения и смысл констант,
уравнение (10.57), следовательно, имеет точно такой же вид, как
уравнение (10.38), описывающее ситуацию с одним
промежуточным соединением. (Легко показать, что допущение возможности
обратной реакции в схеме (10.52) приводит к уравнению такого
же вида, как уравнение (10.49), описывающее аналогичный
случай, но с одним промежуточным соединением.)
Таким образом, введение второго промежуточного соединения
в основную кинетическую схему Михазлиса — Ментен приводит к
результату, экспериментально неотличимому (по крайней мере в
кинетике стационарных реакций) от упрощенного случая с одним
296 часть з
промежуточным соединением. Хотя смысл кажущихся /Са и Умако
в терминах констант скоростей элементарных кинетических актов
совершенно изменен, экспериментальная зависимость (l/v) от
(I / Г А]) по-прежнему описывается прямой с углом наклона
Ка/Унькс, пересекающей ось ординаты при значении у= (1/Умакс).
Это не просто пример еще одного алгебраического упражнения;
описанный вывод указывает на одну из самых серьезных
слабостей стационарной кинетики ферментативных реакций, а именно
на неспособность детектировать несколько промежуточных
соединений в реакции. В ч. 4 будет показано, что для решения этой
важной задачи, недоступной для стационарных методов, можно
использовать совершенно иные экспериментальные условия,
приводящие к релаксации системы при предварительном
неожиданном импульсе \а не к стационарной реакции при непрерывном
химическом изменении).
10.Б.З. ДВА СУБСТРАТА: ПОЧЕМУ ОДНОСУБСТРАТНАЯ СХЕМА
ВООБЩЕ УДОВЛЕТВОРЯЕТ ЭКСПЕРИМЕНТАЛЬНЫМ ДАННЫМ?
Расчеты, приведенные в предыдущем разделе, иллюстрируют
типичную проблему при постановке научных экспериментов, за-'
ключающуюся в том, что усложненная и детализированная теория
бесполезна, если экспериментальные данные не позволяют ее
проверить! В этом разделе рассматриваются скорости реакции для
трех простейших случаев с участием двух субстратов:
статистического и последовательного (упорядоченного) механизмов и
механизма типа «пинг-понг». Доказана возможность отличить
механизм типа «пинг-понг» от двух других механизмов с помощью
обычной зависимости Лайнуивера — Берка (l/v от 1/[А]). Для
отличия статистического механизма от упорядоченного требуются
более сложные эксперименты. Схемы всех трех механизмов
приведены ниже.
Статистический механизм
Е + Ат-rfe Е\
ЕР з=г* Е + Р
Е + В + ЕВ
ЕАВ т=*= EPQ (10.58)
t t
EQ ,=fc E+Q
Упорядоченный (последовательный) механизм
E +A ,=fc ЁА *=± EAB 4=* EPQ ^=fc EQ ^=fc E+ Q (10.59)
Механизм типа «пинг-понг»
E -f. A 7—»- EA ч—►• EP 7—*• E' + P
E' + B ^=fc EB -j—E- EQ 7—>: E + Q,
При реакции, происходящей по статистическому и
упорядоченному механизмам, оба субстрата должны связаться с ферментом
,}
(10.60)
РОСТ И РАСПАД
того, как реакция произойдет; при механизме типа «пинг-понг»
фмент сначала связывается и осуществляет превращение перво-
субетрата, а затем происходит взаимодействие со вторым
субгратом. Кинетика реакций (10.58) — (10.60) описывается
уравнениями
vsss. тш
П
Cq+Ci[A]+C8[B] + [A][B]
(статистический или упорядоченный механизм) (10.61)
^[А][В] * /1лсоч
С \А] 4-С FBI 4- ГА] ГВ1 » механизм типа «пинг-понг», (10.62)
£де С0, С\ и Сг — комбинации разных констант скоростей в
соответствующих механизмах.
г Анализ уравнений (10.61) и (10.62) приводит к полезным и
интересным выводам. Прежде всего, если один из реагентов (пусть,
$5) присутствует в большом избытке относительно другого
реагента ([В]о>[А]о), то уравнения (10.61) и (10.62) упрощаются до
одинакового вида
«"-■гг&г- (1(Ш)
>
•Уравнение (10.63) точно совпадает с выражением, ранее
полученным при рассмотрении схемы Михазлиса — Ментен для реакции
с одним субстратом [ср. с уравнением (10.38)]. Эта ситуация
типична для любой ферментативной реакции (например, гидролиз
эфиров), при которой одним из реагентов является вода,
например для многих ферментативных процессов в желудочно-кишечном
тракте (с ферментами: трипсин, химотрипсин, пепсин и т. д.).
Кроме того, важной особенностью этих уравнений скоростей двухсуб-
стратных реакций является то, что при постоянной концентрации
второго субстрата (пусть, В) и изменении концентрации первого
субстрата А зависимость Ifv от 1/[А] описывается прямой. Однако
если из данных, полученных при другой (постоянной)
концентрации В, построить другую зависимость l/v от 1/[А], то зависимости
Лайнуивера — Берка будут выглядеть, как на рис. 10.12.
Уравнения (10.61) и (10.62) и соответствующие им зависимости на
рис. 10.12 показывают, что механизм типа «пинг-понг»
(зависимости Лайнуивера — Берка при нескольких фиксированных
значениях [В] образуют семейство параллельных прямых) можно
отличить от статистического или упорядоченного механизма
(зависимости Лайниувера — Берка при нескольких фиксированных
значениях [В] образуют семейство прямых, пересекающихся при
отрицательном значении (1/[А] и значении (l/v), которое может
быть положительным, отрицательным или равным нулю). К числу
298 часть з
1/о
Ф
(риксиров
Упорядоченный или
статистический
механизм
(риксиров
Механизм типа
пинг-понг"
//ГА]
Рис. 10.12. Зависимость (обратная) скорости ферментативной реакции от
концентрации первого субстрата [А] при нескольких различных (фиксированных)
концентрациях второго субстрата [В].
При механизме типа «пинг-понг» наблюдается семейство параллельных прямых, а при
упорядоченном и при статистическом механизмах семейство прямых всегда пересекается в
точке ниже, выше или на оси абсцисс слева от оси ординат.
примеров упорядоченного механизма относятся реакции,
катализируемые NAD- и NADP-зависимыми дегидрогеназами; примером
типичного статистического механизма может служить действие
креатинкиназы (фосфотрансферазы); механизм типа «пинг-понг»
характерен для реакций пиридоксаль-зависимой трансаминазы.
Для установления различий между статистическим и
упорядоченным механизмами требуются дальнейшие исследования (ингиби-
рования или изотопного обмена).
В этом разделе показано, что основную кинетическую схему
Михаэлиса — Ментен (10.4) можно обобщить, допуская
возможность обратной реакции фермента с продуктом, присутствие
многих промежуточных соединений и присутствие двух субстратов,
способных реагировать с ферментом раздельно («пинг-понг») или
одновременно (статистически или упорядоченно). Алгебраические
сложности при этом легко обойти, используя зависимость Лайнуи-
вера — Берка {\jv от 1/[А]), которая соответствует прямой: 1) ин-
гибирующее влияние продукта сводится к минимуму ограничением
измерений начальных скоростей реакций (так, чтобы эффективная
концентрация продукта [Р] равна нулю); 2) поскольку вид
уравнения скорости одинаков для ситуаций с одним и с большим
числом промежуточных соединений, введение в схему дополнительных
промежуточных ферментсубстратных комплексов или комплексов
фермента с продуктом бессмысленно; 3) влияние второго субстрата
подавляется проведением экспериментов при условиях, при
которых концентрация второго субстрата постоянна и/или избыточна.
Удивительно, что простая схема Михаэлиса — Ментен не только
определяет простейший механизм, которым можно объяснить
зависимость скоростей ферментативных реакций от концентрации
субстрата и фермента, но при попытке обобщить эту схему и
РОСТ И РАСПАД
шменить ее к более реальным (и, следовательно, более слож-
ш) ситуациям дает сравнительно мало новой информации. Важ-
гое исключение из вышесказанного составляет информация
(получаемая экспериментальными исследованиями стационарной
ферментативной кинетики) относительно механизмов, которыми
^лекарственные вещества, яды, гормоны и природные метаболиты
регулируют скорости конкретных ферментативных реакций.
Однако даже при анализе действия ингибиторов достаточно
рассматривать фермент, связывающийся всего лишь с одним
субстратом с образованием всего лишь одного промежуточного
соединения, игнорируя обратную реакцию фермента с продуктом.
Значение вопросов, изложенных в разд. 10.А и 10.Б, следовательно,
состоит в установлении степени приближения (хотя и очень грубого),
■приводящего к получению полезной информации при анализе
реальных ферментативных реакций.
В заключение следует кратко остановиться на до сих пор не
упоминавшемся математически важном методе, основанном на
использовании меченых реагентов. Например, механизм типа «пинг-
понг» легко идентифицировать при использовании одного меченого
субстрата, поскольку меченым окажется лишь один продукт и
метка в продукте появится даже в отсутствие второго субстрата.
Легко представить (но сложно описать) возможные эксперименты
с применением меченых субстратов, которые можно использовать
для выявления последовательности стадий реакции в относительно
сложных кинетических схемах,— большое количество примеров
читатель сможет найти в работе [3].
Задачи
1. Для простой равновесной кинетической схемы
А-1
В
выведите выражения для ([А] и [В] в зависимости от времени при начальном
условии: в нулевой момент времени [В]=0. [Ответ. Уравнения (10 10) и
(10.11).]
2. Для случая двух последовательных реакций первого порядка '
А ► В > Р
выведите выражения для [А], [В] и [Р] в зависимости от времени при
начальном условии: в нулевой момент времени [В] = [Р]=0. [Ответ. Уравнения
(10.16), (10.18) и (10.20).]
3. Для обычной схемы ферментативной реакции Михаэлиса — Ментен
Е -f A 4=fc ЕА ► Е + Р
покажите, что
pA=p/fA+lg[VMaKr°] (10.40)
300 ЧАСТЬ 3
где
рА = — 1£[А]; К а = (fe_i + k2)/kv VMaKC = ^[Е]^
и
v = d [P]/dt.
4. Выведите уравнение стационарной скорости для схемы Михаэлиса — Ментен
с допущением возможности обратной' реакции [уравнение (10.49), разд.
10.Б.1].
5. Выведите уравнение стационарной скорости для схемы Михаэлиса — Ментен
с двумя промежуточными соединениями [уравнение (10.57), разд. 10.Б.2].
6. Выведите уравнение стационарной скорости для схемы Михаэлиса—Ментен
по механизмам типа «пинг-понг» с участием двух субстратов [уравнение
(10.62), разд. 10.Б.З].
7. Выведите уравнение стационарной скорости для схемы Михаэлиса — Ментен
с участием двух субстратов по последовательному механизму [уравнение
(10.61), разд. 10.Б.З].
Е 4- А < > ЕА ' > ЕАВ —^> Е 4-Продукты,
fe-i «-a
8. Возьмите обратные величины от уравнений (10.61) и (10.62) для скоростей
двухсубстратиых реакций и покажите, что зависимости Лайнуивера — Берка
(1/и от l/[A]), действительно имеют вид, показанный на рис. 10.12.
Литература
Почти все опубликованные материалы, в которых рассматриваются вопросы
скоростей некаталитических (или каталитических) химических реакций, очень
похожи, поскольку почти все они основаны на данных классической работы
Фроста и Пирсона (или Клелаида). В перечисленных ниже ссылках
результаты этих работ описаны детально и в наиболее легкодоступном виде.
1. Mahler H. R., Cordes Е. #., Biological Chemistry, Harper & Row, New York,
1966. Хорошее рассмотрение с привлечением примеров из совремеииой
экспериментальной практики.
2. Dixon M., Enzymes, Academic Press, New York, 1964. Старая, но все* еще
хорошая книга.
3. Cleland W. W., in: The Enzymes, Vol. 2, P. D. Boyer (Ed.), Academic Press,
New York, 1970. Самый формальный, самый общий и подробный, однако и
самый трудный для усвоения материала подход.
4. Moore W. /., Physical Chemistry, 3rd. ed.. Prentice-Hall, Englewood Cliffs,
New York, 1962, p. 267.
5. Coodley E. L., in: Diagnostic Enzymology, E. L. Coodley (Ed.), Lea and Febi-
ger, Philadelphia, 1970, p. 41.
6. Zimmerman H. J., Seeff L. В., in: Diagnostic Enzymology, E. L. Coodley (Ed.),
Lea and Febiger, Philadelphia, 1970, p. 29.
7. Giese A. C, Cell Physiology, W. B. Saunders Co., Philadelphia, 1962, p. 307.
8. lolles P., Proc. Roy. Soc, 167B, 350 (1967).
9. Canfield R. E., J. Biol. Chem., 238, 2698 (1963).
ABA 11
Регуляция скоростей
ФЕРМЕНТАТИВНЫХ РЕАКЦИЙ
ь
I»
'.'" В разд. I0.A показано, что скорость ферментативной реакции
||рямо пропорциональна количеству субстрата. Следовательно;
'если под действием фермента субстрат исчезает из реакционной
^меси, система Михаэлиса—Ментен саморегулируется; чем
большие субстрата в смеси, тем выше скорость его удаления. Однако
«если под действием фермента происходит образование продукта,
to схема Михаэлиса — Ментен не обеспечивает регуляции,
поскольку скорость образования продукта не зависит от его
концентрации. В разд. 10.Б.Г показано, что в присутствии обратной
реакции схема Михаэлиса — Ментен регулирует количество продукта,
поскольку скорость его образования падает с ростом его
концентрации. Однако оба этих типа регуляции естественны для действия
фермента, которое вызывает превращение субстрата в продукт.
В этой главе рассматривается несколько типов регуляции,
основанных на присутствии химических соединений, самих по себе не
являющихся субстратами или продуктами, однако способных связы-
заться с ферментом на участках, которые могут быть удалены от
каталитического центра фермента. Действие таких агентов
обеспечивает тонкий (высокочувствительный) контроль за скоростями
ферментативных реакций и служит средством биохимической
компенсации внешних воздействий окружающей среды (например, из^
менения рН) путем изменения скоростей соответствующих фермен*
тативных реакций.
П.А. ЛЕКАРСТВЕННЫЕ ВЕЩЕСТВА, ЯДЫ И ГОРМОНЬЬ
ТИПЫ ИНГИБИРОВАНИЯ И АКТИВАЦИИ ФЕРМЕНТОВ
Некоторые лекарственные вещества неспецифичны в
структурном отношении; это означает, что их активность коррелирует с
некоторыми физико-химическими свойствами, такими, как
поверхностное натяжение в их растворах, растворимость или степень
ионизации, в большей степени, чем с присутствием в их структуре
каких-либо специфических функциональных химических группировок.
К числу таких лекарственных веществ относятся анестетики
общего действия (хлороформ, диэтиловый эфир), физиологическая ак-
302 часть з
тивность которых коррелирует с размером их молекул и с
растворимостью в органических неполярных растворителях; к другой
аналогичной группе веществ относятся некоторые наркотические
и возбуждающие агенты (пикротоксин, димфлин), действие
которых основано на неспецифическом связывании с внешним слоем
мембран и изменении ионной проницаемости синаптических
мембран (нервных синапсов). Однако обнаружено много
лекарственных веществ и ядов, действие которых основано на изменении ими
эффективности каталитического действия специфических
ферментов. Благодаря тому что скорости обычных химических реакций
намного ниже скоростей каталитических реакций,
избирательность действия таких специфических агентов обеспечивает
эффект «химического усиления», благодаря которому всего лишь
несколько молекул лекарственного вещества могут оказывать
колоссальное физиологическое действие (например, высокоактивный
препарат оубаин, действующий на сердечную мышцу,
максимально эффективен в количествах всего десятка молекул лекарства на
одну клетку). Исследования ингибиторов ферментов преследуют
двоякую цель. Во-первых, влияние структуры химических
соединений на их ингибирующую активность позволяет выяснить, какие
особенности структуры ингибитора принципиально важны для его
действия, и рационально разрабатывать на этой основе лекарства
и противоядия. Во-вторых, эту же самую информацию часто
удается использовать для выяснения природы естественной функции
фермента, подавляемой ингибиторами.
Как и в случае ферментативной кинетики, начнем с
рассмотрения простейшей общей модели, которая могла бы объяснить
действие ингибиторов. Затем сделаем ряд упрощающих
предположений для облегчения анализа этой модели и сравнения с
экспериментом, а затем используем полученные выводы для рассмотрения
некоторых примеров действия ингибиторов различного типа.
Обычная классификация ингибиторов основана-на их влиянии
на зависимость Лайнуивера — Берка (Ijv от 1/[А]), т. е.
изменяет ли присутствие ингибитора угол наклона зависимости и
величину отрезка, отсекаемого на оси ординат, или только одну из этих
величин. Для того чтобы принцип этой классификации был
понятен, вначале рассмотрим общий механизм
Elf ХЕА->Е+Р
*\ А (11Л)
где К\— константа диссоциации комплекса EI (EI^E+I), /Са—'
константа диссоциации комплекса ЕА(ЕА^=Е-Т-А), и т. д. Напри-
РОСТ И РАСПАД
jjUep, на стадии
прямая
Е -Ь I "( > EI
ь
обратная
Al =='гобратная/'гпрямая>
При рассмотрении аналогичной сложной кинетической схемы
в предыдущей главе [уравнение (10.1)] оказалось целесообразным
проанализировать некоторые специальные предельные случаи.
Для схемы, описанной уравнением (11.1), какпоказано в табл. 11.1,
существует три простые предельные ситуации, две из которых
принципиально важны в эксперименте. Во-первых, если
связывание с ингибитором I полностью предотвращает связывание
фермента с субстратом A[ia=oo в уравнении (11.1)], I называют
конкурентным ингибитором (а процесс — конкурентным ингибировани-
ем). В противоположном случае (а=1), при котором связывание
с I не влияет на связывание с А, I называют неконкурентным
ингибитором (процесс неконкурентного ингибирования). Еще
одна предельная ситуация, называемая бесконкурентным ин-
гибированием, возникает, когда ингибитор может связываться с
ферментом только в виде его комплекса с субстратом A[/G = oo
в уравнении (11.1)]. В общем случае (а принимает произвольные
значения) ингибирование называют смешанным. Предельные
ситуации перечислены в табл. 11.1 (и изображены на рис. 11.4). Да-
Таблица 11.1
Предельные случаи общего процесса ингибирования ферментов
[уравнение (ИЛ)] при которых графические данные [зависимость
Лайнуивера — Берка (1/и от 1/[А])] достаточно просто интерпретируются
Предельные
условия
[уравнение
Тип ингибирования
Механизм ингибирования Практическое значение
a
->- оо
a = i
*i =
оо
Ъф 1, оо
К\Ф оо
Конкурентное
ингибирование
Неконкурентное
ннгибироваиие
Бесконкурентн ое
ингибнроваиие
Смешанное ииги-
бнрование
Связывание с
ингибитором предотвращает
связывание с
субстратом
Связывание с
ингибитором не влияет на
связывание с субстратом
Ингибитор может
связываться только после
связывания субстрата
Общий случай
[уравнение (11.1)]
Многочисленные
примеры (см.
текст)
Многочисленные
примеры (см.
текст)
Редкие примеры
Обычный случай
при
экспериментальных
исследованиях
04 ЧАСТЬ 3
лее будут рассмотрены конкурентное и неконкурентное ингибиро*
вания — предельные случаи, представляющие принципиальный
интерес при экспериментальных исследованиях.
И.АЛ. КОНКУРЕНТНОЕ ИНГИБИРОВАНИЕ
Кинетическая схема конкурентного ингибирования получается,
если изъять из общей схемы, описываемой уравнением (ИЛ),
соединение EIA:
Е+А^ЕА-^Е-И^ к Е к
Iei ea-^e+p
Е+1^Е[ Г (11-2)
EI+A-»
Даже если образование комплекса EI является кинетическим
«тупиком» (из него не образуются продукты), ингибитор
эффективно выводит из реакции некоторое количество свободного
фермента Е, которое иначе могло бы связываться с субстратом А.
Поэтому концентрация комплекса ЕА в присутствии ингибитора
понижена и скорость реакции (пропорциональная [ЕА]) падает.
Уравнения скоростей реакции в стационарном состоянии ([Е]о"С
<[А]о, [1]о) запишутся следующим образом:
d .[ЕА]/Л=—(*-i +kz) [ЕА] +&! [Е] [А] =
=0 (в стационарном состоянии), (П.З)
d [El]/dt=~k_3 IEI] -f k3 [E] [1] =
=0 (в стационарном состоянии). (11.4)
Уравнения (11.3) и (11.4) удобно перегруппировать для
определения К а и /G:
[Ej [A] _ k.x + fea _„ (константа Михаэлиса— /до 33)
[ЕА] kx А Ментен для субстрата А) I ■ /
и
*-з _у (константа диссоциации /и ч\
[EI] A3 1 комплекса EI).
Обозначив общую начальную концентрацию фермента [Е]0,
получаем уравнение материального баланса
[Е]в=[Е]+[ЕАЖИ]. (11.6)
И наконец, поскольку целью является определение скорости
ферментативной реакции,
dlP]/dJ=v=k2[EA]. (11.7)
|05 РОСТ, И РАСПАД
Вначале надо решить уравнение (11.б)- относительно [Е] и
юдставить полученное выражение вместо [Е] в уравнения (10.33)
1 (11.5):
гг _ ([Е]0-1ЕА]_]Е1]ИА]
АА —
[ЕА]
к _ (1Е]0-[ЕА]-1Е1])П]
Д1 — fEl]
Затем, решая уравнение (11.9) относительно [EI]
Гр>Т1 ([ЕЗо — fEA]) [II
(11.81
(11.9^
(11.10),
подставляя это выражение в уравнение (11.8) и решая его
относительно [ЕА], получаем
LEA] =-
Ki [E]0 [A]
\ЧКкЛ-Кх\к\ + КхКк
(ПЛ\у
Последнее уравнение при подстановке в искомое уравнение
скорости (11,7) приводит к выражению
,_ ^[E]0^i[A]
V-
Ki [A] + К a [I] + KiKa
(11.12)
которое показывает, что при достаточно больших [А] скорость
реакции v приближается к предельной максимальной скорости
VMaKC = ME]0:
v==-
макс
Ki [A]
^iIAI + KaIU+^i^a
(11.13)
Беря от обеих частей уравнения (11.13) обратные величины,
получаем
Конкурентное ингибирование (l/v) =я
(ИЛ*)
Уравнение (11.14) надо сравнить с результатом Михаэлиса
Ментен в отсутствие конкурентного ингибитора
Ингибитора нет (-f) = (^Д-) + ^ (-^ )
(10.41)
Сравнение уравнений (11.14) и (10.41) позволяет сделать
вывод, что в присутствии постоянной концентрации^ конкурентного
306 ЧАСТЬ 3
0,6 мкмоль/л
[I] ш0,4 МКМО/76/4
[II =0,2мкмо/1ь/л
Ш 'О
0,05
У\А\ (л/мкшль)
Рис. 11.1. Конкурентное ингибироваиие аце-
тилхолннэстеразы фнзостнгмииом
(зависимости l/v от 1/[А1) прн разных
(фиксированных) концентрациях физостигмина.
[Неопубликованные данные Бенбазата и Мар-
шелла).
Субстрат А — ацетилтиохолнн. Из этих данных
было определено, что /^=4.6'10~7 моль/л. Поскольку
^Сд=7,610-5 моль/л, ясно, что физостигмии
является очень сильным конкурентным ингибитором.
ингибитора изменение
зависимости Лайнуивера — Бер-
ка (1/и от 1/[А]) состоит в
увеличении угла наклона
при неизменной величине
отрезка, отсекаемого на оси
ординат (рис. 11.1).
Качественные следствия
из этих результатов легко
объяснимы. Поскольку
ингибитор I и субстрат А
конкурируют друг с другом за
один и тот же участок
связывания на молекуле
фермента и поскольку в обоих
случаях связывание
является обратимым, ясно, что при
достаточно большом
избытке [А] относительно [I]
практически весь фермент
будет находиться в
комплексе ЕА и скорость
реакции будет равна той же
максимальной предельной
скорости ферментативной
реакции УМакс. При
пониженных концентрациях А,
однако, ингибитор I
выводит некоторое количество
фермента из сферы реакции
в виде комплекса EI, так
что концентрация ЕА (и,
следовательно, скорость
реакции и) понижается.
Пример. Ингнбирование ацетилхолинэстеразы физостнгмнном
При передаче нервного возбуждения импульс обязательно попадает на
нервные или нервио-мышечиые синалтические окончания. При достижении импульсом
предсинаптического окончания нервного волокна из сотен микроскопических
мешочков (везикул) выделяется химическое вещество — медиатор. Как только
молекула медиатора диффундирует через сииаптическую щель к рецепторному
белку иа постсинаптическом нервном окончании или мышечном волокне
генерируется новый импульс. Чтобы постсинаптияеекий нерв или мышечное волокно
могли подготовиться к восприятию следующих импульсов, молекулы медиатора
Должны удаляться из щели сразу после связывания с рецептором; это
обеспечивает фермент, локализованный на внешних краях щели. Если химическим
медиатором служит ацетилхолин, как в случае парасимпатической нервной системы
(контролирующей замедление ритма сердца, сужение коронарных артерий и
сужение зрачков глаз), ферментом, разрушающим медиатор, служит ацетилхолии*
встераза. Если медиатором служит норадреналин, как в случае симпатической
307 РОСТ Й РАСПАД
Участок
связывания
Каталитический
участок
CHa-N СН2 ГГН Л
с=£д*~
сн,
н
с
СНз-NH-CO-O—СУ С-
сня
-с-
сн,
у vw
СНа -бНа
Рис. 11.2. Активный центр фермента ацетилхолннэстеразы.
нервной системы (контролирующей ускорение ритма сердца, расширение
коронарных артерий и зрачков глаз), ферментом, разрушающим медиатор, является
катехол-о-метилтрансфераза.
Считается, что в ацетилхолинзстеразе существуют различные каталитический
и связывающий центры (рис. 11.2). В молекуле фнзостигмина имеется
положительно заряженный атом четвертичного азота, благодаря чему физостигмии
эффективно конкурирует с нейтральным субстратом ацетилхолином за один и тот
же участок связыванния на молекуле фермента и, следовательно, тормозит
катализ, препятствуя связыванию субстрата. Физиологический эффект действия
фнзостигмина состоит в том, что ацетилхолии больше не разрушается после
стимулирующего воздействия на данное нервное или мышечное волокно и поэтому
накапливается в синаптической щели и продолжает стимулировать следующий
нейрон или мышцу. Этот эффект называют парасимпатомиметическим, поскольку
он аналогичен нормальному возбуждению парасимпатических иервов.
Разобранный пример показывает, каким образом физиологическое возбуждение может
возникать в результате ингибирования химической реакции. Конкурентное инги-
бирующее действие фнзостигмина очевидно из зависимостей Лайнуивера — Берка
в присутствии различных (фиксированных) количеств физостигмииа (рис. 11.1).
Пример. Лекарственные сульфамидные препараты
Еще более 100 лет назад было известно, что различные красители обладают
выраженным сродством к бактериям определенных типов. Предполагая, что
некоторые красители могут каким-то образом убивать эти бактерии, фирма
I. G. Farbenindustrie (Германия) проводила испытания действия новых
красителей на бактериальные культуры, и таким образом в 1935 г. был открыт первый
сульфонамидный препарат (сульфамид), способный препятствовать дальнейшему
размножению гемолитических стрептококков у мышей. В результате
исследований буквально тысяч синтезированных с тех пор соединений стало ясно, что
сульфонамидная группировка является необходимым функциональным
компонентом (см. примеры ниже) таких препаратов.
308 ЧАСТЬ 3
N
Сулмратиазол
(при острых инфекциях)
гт\
n-H,NCeH4S02NHCONHz
Сульфвцстамид (для
местного применения)
Сульфадиаэич
(обладает низкой
токсичностбю )
л H2NCeH<S02NHC(NH2)=NH
Сулырагуамидин
(при дизентерии)
7 Механизм действия сульфонамидов состоит в конкурировании с л-амииобен-
зойной кислотой при биосинтезе коэнзиматически активной формы фолевой
кислоты в метаболизме бактерий
С №
ОН || '--..
NH—СН
\
соон
сн,
4NH—R
/
сн2
соон
Сульфамидные препараты являются бактериостатиками (т. е.
предотвращающими размножение бактерий), ио не бактерицидами (т. е. они не убивают
бактерии) к пневмококкам, стрептококкам, стафилококкам, кишечным бактериям
Е. cdli н к возбудителям гангрены и чумы. Сегодня многие сульфамидные
препараты заменены продуцируемыми микроорганизмами антибиотиками с гораздо
более сложной химической структурой (пенициллин, террамиции, ауреомицин).
При изучении путей метаболизма у разных организмов одной из наиболее
интересных проблем является установление различий между путями метаболизма в
втих организмах и у человека для создания терапевтических лекарственный
препаратов, которые могли бы обладать направленным иигибирующнм действием на
ферментные системы" возбудителя заболевания, которые отсутствуют или менее
важны в организме человека (см. обсуждение химиотерапии рака в конце
разд. 11. А).
I1.A.2. НЕКОНКУРЕНТНОЕ ИНГИБИРОВАНИЕ
При неконкурентном ингибировании ингибитор не влияет на
связывание субстрата с ферментом, но препятствует превращению
субстрата в продукты своим присутствием в составе
фермент-субстратного комплекса
е+а4^еа-^е+р^
К-1
к в к
EI
# *
ЕА
ЕА + I +g=* EIA -» EI + PJ
Е + Р
(11-15)
i09 рост й распад
Читатель сам может показать (см. задачи в конце главы), что
:истема реакций по уравнению (11.15) описывается уравнениями
:коростей и материального баланса (11.16а) — (11.16д):
d[EA]/#=~(^i+fc2) [ЕА]+МЕ] [А]—
-^3[ЕА][1]-[-А:_з[Е1А], (11.16а)
d[EI]/*=^[E][I] + ^i[EIA]-^3[EIl-^[EI][A], (11.166)
d[ElA\/dt=k1[El][A]+k3[EA)[\)~{k_1-\-k_3)[ElA]t (11.16в)
d[P]A#=fc2[EAl, (11.16г)
[Е]0=[Е1 + 1ЕА]+[ЕЦ = [Е1А], (11.16д)
И приводит к уравнению скорости, которое при преобразовании
выглядит следующим образом ([Е]о<С[А]0, [1]о):
Ка 1\ (неконкурентное /ц j7\
■f'
V #1 Д^макс ' ^макс [Л] ) ИНГйбирОВаНИе),
110=Ы^) + {7^)Щ (ингиби^ювания нет) (10.41)
Сравнение уравнений (П.17) и (10.41) показывает, что в
присутствии постоянной концентрации неконкурентного ингибитора
Таблица 11.2
Предельные случаи кнгибированйя ферментов и их влияние
на зависимость Лайнуивера — Берка
Тип ингибирова-
ния
Механистическое
представление
Свойства зависимости Лайнуивера—Берка
(I/O от 1/[А]>
отрезок на оси
У
угол наклона
отрезок на оси
х
Ингибитора нет Схема Михаэлн-
са — Меитеи
Конкурентное
ингибирова-
ние
Неконкурентное
ингибирова-
вие
Связывание
ингибитора с
ферментом
предотвращает
связывание с
субстратом, и наоборот
Связывание
ингибитора с
ферментом не
влияет на связывание
субстрата, ио
комплекс EIA ие
превращается в
EI и продукт
Ка
макс
макс
макс
(
1 +
'макс
\ ''макс / \ 'макс /
1
X —
Ка
1
('+«
Ка 1 +
Ка
310 ЧАСТЬ 3
изменение зависимости Лай-
нуивера — Берка (1/и от
1/[А]) состоит в увеличении
угла наклона и увеличении
величины отрезка,
отсекаемого на оси ординат, при
неизменной величине
отрезка на оси абсцисс (рис. 11.3;
приведен экспериментальный
пример). Хотя
неконкурентное ингибирование в чистом
виде встречается редко,
аналогичный механизм можно
использовать для
объяснения активированного
связывания и кооперативного
связывания субстратов
ферментами (см. аллостерия далее
в этой же главе).
В табл. 11.2
перечислены основные характеристики конкурентного и неконкурентного ин-
гибирования.
-'/«а
1/Ш
Рис. 11.3. Неконкурентное торможение
протонами катализируемой а-химотрипсииом
реакции гидролиза ацетил-Ь-трилтофаиами-
да [8].
ПАЗ. СМЕШАННОЕ ИНГИБИРОВАНИЕ
На рис. 11.4 схематично представлены два предельных случая
ингибирования ферментов, при которых связывание ингибитора
либо препятствует связыванию субстрата (конкурентное
ингибирование), либо не влияет на связывание субстрата (неконкурентное
ингибирование). Можно ожидать (и это наблюдается в
действительности), что большинство обратимых ингибиторов оказывают
действие, промежуточное между этими двумя крайними случаями,
и такое действие называют смешанным ингибирова'нием. Хотя
алгебраическое выражение уравнений скорости в этом случае
становится громоздким, графический анализ остается простым и
приводит к зависимостям, которые лежат между предельными
случаями конкурентного и неконкурентного ингибирования, как показано
на рис. 11.5.
Особенно полезным при анализе данных о скорости
ферментативной реакции в присутствии ингибиторов оказывается способ,
основанный на использовании перегруппированных уравнений
(11.14) и (11.17):
1
(,+w)+(TR&n-)w
Кмакс \ * ' [А] Г\ *l!W[A]
(конкурентное ингибирование),
(11.18)
311 РОСТ И РАСПАД
CS5
Фермент Ингибиторы Субстрат
Конкурентное
ингибироеание
фермент £ и А £ и I
Неконкурентное
шеибироеание
СЕБЕ
Фврнент Ей А Е и I EuAul
Рис. 11.4. Схематические, диаграммы конкурентного и неконкурентного ингибиро-
ваиия ферментов.
При конкурентном ннгнбнрованнн субстрат А не может уже связываться с ферментом, как
только между ферментом и ингибитором образовался комплекс. Прн неконкурентном ин-
гибнрованин связывание субстрата с ферментом не зависит от присутствия ингибитора,
однако до тех пор, пока ингибитор связан с ферментом, ферментативная реакция происходить
не может.
И
(неконкурентное ингибирование) (11.19)
Сопоставление уравнений и способов графического анализа, используемых для определения константы
равновесия диссоциации Л\ (или константы, Мнхаэлиса — Ментен Ка) из измерений доли участков связывания,
занятых лигаидом (или из измерений стационарных скоростей ферментативной реакции), при равновесном связывании
низкомолекулярного лиганда с макромолекулой (или при ферментативном катализе превращения субстрата)8
Равновесное связывание
Стационарная ферментативная кинетика
E+nl *===* EI„; Kx =
[Е] Ц}
1ЕЦ
для каждого из п мест
Е + А
-1
ЕА >- Е + Р; д-А==
Уиакс-ЫЕ1]
[Е] [I] *_х + h
[EI]
*i
Прямая зависимость
v =
*; строить V от [\]
п
п = lim v; Kj = [I] при v = -y
[1]->оо
Прямая завися- Умакс f A] rA1
мость v = лТ+ТО СТР0ИТЬ U ^ [A1
°иакс
Умакс = Hm v; tfA = [А] прн v - —%
[А]->~
Зависимость Бьерру-
ма (кривая
титрования)
п — v
pi = pATi + lg ——; строить v от 1g [I]
п
n = limv; p/<"i = pi при v = ~2"
iff П]-*»
Зависимость
«lg Доза»
pA = рКл + Igj-^ ); строить v от Jg[AJ
УИакс = Нто;
lg [A]->oo
р/Сд = рА при v =* —^
макс
Обратная зависимость
(зависимость Беие-
зи«— Хильдебран-
да)
■^-■г+^Хтп) строить 1/v
от 1/[1]
п г= (Отрезок, отсекаемый иа оси у)'1;
К\ = — (Отрезок иа оси х)~х
Зависимость Скэтчар-
да
у
1П
^-к7 — -к7 строить
n s= Отрезок, отсекаемый на оси х;
Отрезок на оси у \~г
«-(•
v — число занятых участков на одну (среднюю)
макромолекулу
Зависимость
Лайнуиве-
ра — Берка
К а (
1
" "VW ([А])+ Умакс'
строить l/i» от 1/[AJ
^макс*"* (Отрезок, отсекаемый на оси у)-1
К\ = — (Отрезок, отсекаемый на оси х)~г
Зависимость
Эди
V
[А]
макс
Ка
1ГГ" -лТ"); строить
Умакс = (Отрезок на оси х)
К а = — (Тангенс угла наклона)-1
[AJ
ОТ V,
макс
[ЕА]
[Е]общ
(средняя доля
участков
связывания, занятых
субстратом)
314 ЧАСТЬ 3
Неконкурентное
Конкурентное
Смешанное
IN цитирование „ г) Шгибирование v -а иншбирооание v
Кип/ее 'маке . "Лнииг
Прямой.
#emo<f
Кривые
титрования
'маке
Р*д - ig [A]
Зависимости
/1айнуивера-6ерт
Неконкурентное
ингибирование
Зависимости
Диксона
P*A-iflM
рЛд-lg W
Конкурентное
t_ ингибирование * £А J2
Рис. 11.5. Графические способы анализа кинетических данных для
ферментативных реакций в присутствии конкурентного или неконкурентного ингибитора
или ингибитора смешанного типа.
В девяти верхних рисунках скорость ферментативной реакции в зависимости от
концентрация субстрата [А] определяется в присутствии или в отсутствие фиксированной
концентрации ингибитора (I]. На двух зависимостях Диксона скорости определяются прн двух
разных фиксированных концентрациях субстрата в зависимости от концентрации ингибитора.
Использование разных способов анализа обсуждается в тексте.
Уравнения (11.18) и (11.19) служат основой для построения
зависимости Диксона (l/v) от [1] при постоянной [А]. Такие
зависимости (рис. 11.5 и обсуждение этого рисунка) удобны для
экспериментального определения Ki-
Как и при изучении связывания макромолекул с
низкомолекулярными соединениями (разд. З.В), существует много способов
графического анализа экспериментальных данных. В табл. 11.3
сопоставлены обычные методы анализа равновесного связывания
315 РОСТ И РАСПАД
и кинетики ферментативных реакций в отсутствие ингибиторов.
Как и при равновесном связывании, недостаток прямых
зависимостей и кривых титрования для определения величин VMaKC и /Са
при описании кинетики ферментативной реакции состоит в том, что
измерения необходимо проводить в широкой области
концентраций субстрата. Среди зависимостей, представленных прямыми
(Лайнуивера — Берка и Эди), зависимость Эди (аналогичная
зависимости Скэтчарда при обычном равновесном связывании)
обладает теми же самыми преимуществами при графическом
анализе данных ферментативной кинетики (оптимальный учет
среднестатистического веса данных, полученных при разных
концентрациях); более простой вид зависимости для описания
реакций с многоточечным фермент-субстратным взаимодействием.
Однако в литературе почти всегда используется зависимость
Лайнуивера — Берка (аналогичная графической зависимости обратных
величин при равновесном связывании). Причины такого
предпочтения отчасти обусловлены исторически, а в некоторых случаях
объясняются тем, что зависимость Лайнуивера — Берка позволяет
легко установить тип ингибирования (если в системе
присутствуют ингибиторы). Наконец, следует отметить, что с помощью
любого из вышеназванных способов можно определить константу
ингибирования Ки если известна концентрация субстрата [А].
Однако, если субстрат имеет полимерную природу (полинуклеотид,
полипептид, полисахарид), фермент может действовать на него на
разных участках полимерной цепи и тогда концентрацию
потенциально расщепляемых ферментом связей в субстрате оценить
оказывается не просто. В таких случаях простейшим графическим
способом определения Ki является зависимость Диксона, которую
удается использовать даже при отсутствии информации об абсолютном
значении концентрации субстрата.
В разд. 11.А описан ряд кинетических схем, уравнений и
графиков, используемых для исследования и количественной оценки
эффективности и механизма торможения ферментативных реакций
различными ингибиторами. Одним из наиболее важных примеров
этих методов, нашедших применение в последнее время, служит
идентификация и установление механизма действия активных
ингибиторов биосинтеза нуклеиновых кислот, которые часто
проявляют противоопухолевое действие (см. пример ниже).
Пример. Химиотерапия рака
Лечение рака осуществляется главным образом с помощью хирургического
вмешательства и облучения. Хирургия позволяет достичь хороших результатов,
если опухоль локализована и относительно доступна: в основном именно
благодаря хирургическим методам лечения продолжительность жизни увеличилась до
5 лет и более у 85% больных раком кожи, у 60% женщин с раком груди и у
40% больных со злокачественной опухолью толстой кишки. Если из-за своей
локализации опухоль неоперабельна, особенно полезно бывает облучение, с по-
316 ЧАСТЬ 3
ч
Оксимочевина
Арабиноэил-
цитозин
Ингибирует
ДНК-
полимеразу
№
блокирует
восстановление цаГивалооой
кислоты до дезоксици-
тидияоаой кислоты
flop*
tfUQ
and"'
5-Фторураци/]
блокирует тими-
дилатеинтетазу
подимое для ее
превращения вгиыи-
диловую кислоту
Ингибирует
взаимные
превращения
пуринов
6-Меркапто пурин
6-Тиогуанин
блокирует
биосинтез
пуриновых
оснований
Метотрексот
Ингибирует редцктрзу
дегадрофолаевои
кислоты, задерживая ее
переход в mempatpo-
лиевдю
Алш/шрующие
агенты
Поперечные
сшивки
нитрозомочешны
Прокарбазин
Деполимеризует
Адриал-шцин
Дактиномициш
Даунорубицин
Митрамицин
связываются с
ДНК, блокируя
образование
РНК
\.-Аспарагиназа
Гидролизиет i-acna-
рагин
РНК
(транспортная,рабасоАЮлбная,информационная)
Белковый синтез
(ферменты)
Рис. 11.6. Механизм действия нескольких противоопухолевых агентов,
нарушающих репликацию клеток [9].
мощью которого удается продлить срок жизни на Б и более лет у 80% детей,
больных ретинобластомой (рак глаз), у 75% больных болезнью Ходжкинса
{опухоль лимфатической системы») и у 50% больных раком носоглотки. Однако
наибольшую трудность представляет лечение опухолей, рост которых
сопровождается образованием метастаз (т. е. частичным отсоединением злокачественных
клеток от первичного места локализации и перемещением с циркулирующей
кровью или лимфатической жидкостью в другие органы» тела больного).
Поскольку опухоли методами клинического анализа не обнаруживаются до того,
как их масса составляет ^109 клеток, после хирургического ил»
радиобиологического удаления первичной опухоли множество метастаз могут оставаться
необнаруженными. Именно в этих случаях широкбе применение начинает находить
химиотерапия.
317 РОСТ И РАСПАД
Характерной особенностью опухолевых клеток является их быстрый рост и
репликация; поэтому основным направлением химиотерапии является
торможение клеточного роста. К ингибиторам клеточного роста относятся
антиметаболиты, участвующие в биосинтезе нуклеиновых кислот в качестве конкурентных
ингибиторов специфических ферментов этого процесса (рис. 11.6); таким
ингибитором, например, является метатрексат, блокирующий редуктазу фолевой
кислоты в самом начале биосинтеза пуринов. Другие примеры показаны на рис. 11.6.
На рис. 11.6 показано также, что действие прокарбазина (производное метил-
гидразина) состоит в деградации клеточной ДНК; действие антибиотиков
(например, актиномицина D) заключается в неспецифическом связывании путем.
интеркаляци» с двойной спиралью ДНК, мешающем процессу транскрипции;
L-аспарагиназа гидролизует аоларагин в крови и этим способствует голоданию
определенных видов опухолевых клеток, которым требуется внешний источник
аспарагина. Одним из примеров последних достижений химиотерапии рака
служит постоперационное лечение остеогенной саркомы (детской опухоли костей);
5-летнее выживание после первичной терапии, проводимой вслед за ампутацией
пораженной конечности, составляет < 20%; лучших результатов не удается
получить в основном из-за метастазов, появляющихся в легких больного через год
после хирургического вмешательства. Использование массивных доз метатрекса-
та с последующим введением цитр офор ум-фактор а (аналог фолевой кислоты,
служащий заместителем природного продукта редуктазы фолевой кислоты и,
следовательно, не взаимодействующий с ингибированиым ферментом) привело в
огромном большинстве случаев к отсутствию метастазирования даже через
26 месяцев после операции. Схемы лечения, основанные на использовании
комбинаций из нескольких агентов, указанных на рис. 11.6, повысили вероятность
длительного выживания при остром лейкозе с 0 до более 50%.
В 1954 г. можно было ожидать, что из каждых четырех больных раком один
проживает 5 лет и более. К 1975 г. уже один из трех больных проживал ^ 5 лет.
Наиболее существенный вклад в такое 30%-ное увеличение коэффициента
вылечивания внесли успехи химиотерапии (только в США благодаря применению
химиотерапии сохраняется ~ 55 000 чел. жизней в год).
Необходимость детальной информации о механизме ингибирования
ферментов для клинического применения хорошо иллюстрируется более подробным
рассмотрением действия двух основных противоопухолевых препаратов: метатрекса-
та и 5-фторурацила. 5-Фторурацил является конкурентным ингибитором тимиди-
латсинтетазы — фермента, катализирующего синтез тимидшювой кислоты из
дезоксиуридиловой кислоты; как отмечалось ранее, метатрексат ингибирует
редуктазу фолевой кислоты. Можно было бы ожидать, что при совместном
использовании этих двух агентов препарат должен обладать повышенной
противоопухолевой активностью, и в действительности во многих химиотераяевтиче-
ских программах лечения рака используются смеси препаратов. Однако
оказывается, что продуктом ингибируемой метатрексатом реакции является кофактор
(см. разд. П.Б.4), необходимый для связывания 5-фторурацила с тимидилат-
синтетазой. Следовательно, действие 5-фторурацила при добавлении метатрекса-
та должно ослабевать и эффект совместного действия обоих препаратов должен
быть меньше суммарного эффекта при использовании, каждого из них
поодиночке! Эта гипотеза была проверена на культуре опухолевых клеток мыши, и она
может оказаться верной при терапии человека. Понимая это, можно надеяться
ликвидировать антагонизм между этими двумя препаратами с помощью третьего
соединения, которое могло бы заместить 5,10-метилентетрагидрофолевую кислоту
(продукт реакции, ингибируемой метатрексатом) в роли кофактора при
связывании 5-фторурацила с тимидилатсинтетазой; перспективным кандидатом на эту
роль, по-видимому, является витамин природного происхождения, фолиновая
кислота (5-формилтетрагидрофолевая нислота). В данном случае выяснение деталей
механизма действия ингибиторов ферментов приводит к получению данных,
имеющих прямое влияние на их перспективное клиническое использован»* (щ.
работу i[24]).
318 ЧАСТЬ 3
П.Б. ДРУГИЕ ТИПЫ РЕГУЛЯЦИИ ФЕРМЕНТОВ
В разд. 10.Б.1 было показано, что накапливание продукта
тормозит скорости ферментативных реакций и, следовательно,
«автоматически» способствует установлению стационарной
концентрации продукта (скорость образования продукта наибольшая, когда
его мало, и уменьшается с увеличением количества продукта).
Однако, поскольку основной функцией ферментативных реакций
является образование продукта, а не ускорение достижения
равновесия, на практике большинство таких реакций сдвинуты в одну
сторону, так что связывание продукта с ферментом обязательно
намного слабее, чем связывание субстрата с ферментом.
Следовательно, для торможения большинства ферментативных реакций
потребовалась бы очень высокая концентрация продукта. В разд.
11.А показано, что гораздо более тонкая регуляция обеспечивается
благодаря присутствию внешних агентов (ингибиторов), которые
при связывании с ферментом не претерпевают химических
изменений, но связывание которых с ферментом может препятствовать
связыванию или превращению субстрата (или и тому и другому).
Еще более тонкая регуляция скоростей ферментативных
реакций достигается, если (как будет показано ниже) скорость
ферментативной реакции ускоряется (фермент активируется) в
присутствии субстрата или внешних агентов. Это, по-видимому,
характерно для ферментов, состоящих из нескольких субъединиц, и
субъединичная структура может быть простейшим способом
общего биологического решения проблемы тонкой регуляции скоростей
реакций метаболизма. Наконец, зависимость скорости
ферментативной реакции от рН служит средством, с помощью которого
отдельные стадии процесса метаболизма могут компенсировать
изменения во внешнем окружении (рН); в следующем разделе
рассматривается простой механизм рН-регуляции ферментативных
реакций.
11.Б.1. рН-РЕГУЛЯЦИЯ СКОРОСТЕЙ ФЕРМЕНТАТИВНЫХ РЕАКЦИИ
Типичная зависимость ферментативной активности от рН, как
показано на рис. 11.7 для фермента гистидазы, описывается коло-
колообразной кривой. Поскольку разные ферменты проявляют
оптимальную активность при разных рН, очевидно, что пути
метаболизма могут изменяться при изменении рН, осуществляемом самим
организмом, или в результате внешнего воздействия. Влияние рН
объясняется с помощью предположения (в простейшем случае),
что фермент может существовать в трех ионных формах, ЕН|+,
ЕН+ и Е, из которых только в одной форме, например ЕН+,
фермент способен к связыванию субстрата и к каталитическому акту.
Поэтому прежде всего следует определить относительные концент-
319 РОСТ И РАСПАД
Рис. 11.7. Активность фермента
гистидазы в зависимости от рН
при действии на гистидин
(субстрат) [10].
Кружки — экспериментальные
данные; сплошная линия построена по
уравнению (11.23).
рации этих форм фермента в зависимости от рН:
где
ЕН1+ 4=fc EH+ у—»• Е
к __ [ЕН+] [НП
Л1— [ЕН2+] '
(11.20)
Кш=-
[Е] [Н+]
(ЕН+)
(11.21)
Из уравнения (11.21) можно показать (см. задачи в конце
главы), что относительная доля каждой из форм фермента
составляет
[ЕН22+]/[Е]общ -
[ЕН+]/[Е]общ =
[Е]/[Е]общ =
[H+J2
1Н+р + к, [н+] + ед
Кг [Н+]
IH+P + ZCilH+l+Zd^
К} К 2
[H+J2-f KiIH+1 + ZdK,
(11.22а)
(11.226)
(11.22в)
Поскольку предполагается, что связываться с субстратом и
катализировать его превращение может только ферма ЕН+, и
поскольку Умакс пропорциональна концентрации этой формы
фермента, колоколообразная форма зависимости активности фермента от
рН объясняется уравнением (11.23) [которое следует из
уравнения (11.226)], как видно на кривых на рис. 11.8:
VWc/Cx [H+J (11<23)
VI
макс
[Н+]2 + Кг [Н+1 + Кг*г '
где Умакс — максимальная скорость ферментативной реакции,
которая бы наблюдалась, если бы весь фермент находился в
активной форме ЕН+, а оставшийся множитель характеризует долю об-
320 ЧАСТЬ 3
а>
I I I I I I I J
рК, EH*
н
S^ 0,5
II
II
о;
1 1
EH|*
-
—
-
1 U'
1 1 1 '
. P*1 PA"?
1
Ns^.
I 1
E
—
—
—
7 8 9 10 11 12
PH
Рис, 11.8. Относительное содержание различных форм фермента ЕН§+, ЕН+ и Е
в зависимости от рН [2, с. НО].
Для двух ионизируемых групп $Ка составляют: а — 5 н Ю; 6 — 7 н 8. Совпадение таких
теоретических кривых с экспериментальными данными проиллюстрировано на рис. 11.7.
щего количества фермента, находящуюся в форме ЕН+.
Уравнение (11.23) хорошо согласуется с экспериментальными данными
(рис. 11.7).
Легко показать (см. задачи в конце главы), что максимум ко-
локолообразной кривой на рис. 11.8. находится при рН, которое
равно половине разности между p/Ci и р/С2, т. е.
fH+J[EH+]M =
/*ЗС = [Н+]
оптим
(11.24)
Область рН, при которых активность фермента составляет не
менее половины максимальной, можно рассчитать из [Н+], при
которой доля ЕН+ равна половине своего максимального
'Значения (см. задачи в конце главы), т. е. надо найти корни
уравнения
[Н+]« - (К, + 4[Н+]0ПГНМ) [н+] + [Н+]20ПГНМ - О
(11.25)
321 РОСТ И РАСПАД
Выведенное уравнение объясняет изменение предельной
скорости (Умакс) при большом избытке субстрата. Для объяснения
изменения скорости при пониженных (т. е. обычных) концентрациях
субстрата необходимо рассмотреть несколько более сложную
модель, учитывающую, что константа равновесия протонирования
свободного фермента может отличаться от константы,
характеризующей фермент-субстратный комплекс. Будем по-прежнему
предполагать, что только форма ЕН+ способна связывать субстрат и
катализировать реакцию
Е ЕА
+н+
*•« +н+
еаг
А _j_ ЕН з=* ЕНА —^*. EH + Продукты (11.2вХ
+н
Кп +Н+
Keai
ЕН ЕН2А
Можно показать, что механизм реакции (11.26) приводит к
обычному результату типа зависимости Михаэлиса — Ментен:
4- = (1^макс)+(^аКсЖА) (l/fAj), (11.27)
где (кажущиеся) Умакс и К'а зависят от рН:
у VuaKa (\ 1 28^
%9г — к l + tH+i^i+*wih+] п 1 «ох
Аа-Аа I + IH+I/^oi+KWIH+J (U'^
(VMaKc и /Са соответственно максимальная скорость и константа
Михаэлиса — Ментен, которые бы были определены, если бы весь
фермент находился в форме ЕН+). Как и в предыдущих примерах
с участием двух субстратов, многих промежуточных соединений,
ингибиторов и т. д., это усложнение (рН-зависимость связывания
и катализа в. ферментативной реакции) приводит к результату
того же вида, что и простая схема Михаэлиса — Ментен.
11.Б.2. ПОЧЕМУ У ФЕРМЕНТОВ ЕСТЬ СУБЪЕДИНИЦЫ?
Частично биологический контроль на молекулярном уровне
основан на' специфичности ферментов, благодаря которой при
изменении концентрации субстрата или ингибитора изменяются
скорости всего лишь одной или нескольких химических реакций. Если
представить себе биологическую машину в виде устройства с
большим количеством контрольных «кнопок», то, чем выше специфич-
322 часть з
I 0,6
I
Lo.4
* 0,2
■о
^0 10 20 30 40 50
[Аспартат], ммоль/л
Рис. 11.9. Зависимость скорости появления продукта от концентрации субстрата
(аспартата) [11].
Ферментативная активность выражена в единицах на мнкрограмм каталитически активного
полнпептнда. / — одиночная каталитическая субъединнца фермента; 2 — ассоциированный
фермент с многомерной структурой, состоящей из двух каталитических и грех «регулягор-
ных» полипептидных субъединнц.
ность ферментов, тем больше имеется кнопок, что обеспечивает
очень сложную систему регуляции. Однако, для каждой кнопки,
по-видимому, следует учитывать и ее «пороговую»
чувствительность, т. е. насколько должно быть велико возмущение, требуемое
для отклика системы. Субъединичная структура многих ферментов
способствует увеличению чувствительности системы, так что малое
изменение концентрации в значительной степени влияет на
скорость ферментативной реакции. Например, на рис. 11.9 показана
зависимость скорости ферментативной реакции от концентрации
субстрата в реакции, происходящей под действием каталитической
субъединицы фермента аспартаттранскарбамилазы (кривая /),
и под действием фермента, в котором каталитическая субъединица
объединена с регуляторной (кривая 2). Видно, что при катализе
ассоциированным ферментом зависимость скорости реакции от
концентрации субстрата имеет гораздо более характерную форму,
чем при действии одной каталитической субъединицы.. Поскольку
эта реакция служит первой стадией биосинтеза пиримидиновых
нуклеотидов, по-видимому, необходим тонкий контроль скорости
этой реакции; это осуществляется благодаря сигмоиднои форме
зависимости скорости реакции от концентрации субстрата,
которая объясняется ниже с механических позиций.
Сигмоидные кинетические зависимости, подобные приведенной
на рис. 11.9, наблюдаются в многих ферментативных реакциях;
Известно не менее полдюжины механизмов для их объяснения.
Существование большого числа механизмов, по-видимому, связано
с тем, что на молекуле фермента имеется несколько участков
связывания с субстратом [часто дополнительный (дополнительные)
участок (участки) локализован (локализованы) на разных субъ-
323 РОСТ И РАСПАД
•единицах], так что скорость реакции увеличивается с ростом
числа молекул субстрата, связавшихся с данной молекулой фермента.
Например, сигмоидная кинетическая зависимость появляется, если
фермент может образовывать комплекс ЕАг, характеризующийся
большей скоростью превращения, чем исходный комплекс ЕА.
В простейшем случае, когда ЕА полностью неактивен,
стационарное образование продукта описывается уравнением
h k2[A] *з
Е + A 4=fc ЕА >■ ЕЛ2 > Е + Л + Р
*—1
&Л&3 [Е1о №
V~~ Ъ (А, + k2) + kjb [A] + Vi I M2 '
Следовательно, при очень большой концентрации субстрата
[А] скорость достигает значения Умакс, которое определяется
количеством фермента в системе [Е]0; при очень малой [A] v
увеличивается пропорционально квадрату [А], и в результате
зависимость v от [А] приобретает сигмоидную форму. Ключевой
особенностью этого механизма является приобретение
фермент-субстратным комплексом большей реакционной способности при
увеличении числа молекул субстрата, связанных с молекулой
фермента; это означает, что связывание двух молекул субстрата на
ферменте на двух участках не является независимым. (Можно
показать, что если связывание одной молекулы субстрата не зависит
от связывания второй молекулы субстрата с той же молекулой
фермента, то зависимость v от [А] имеет обычную
гиперболическую форму.)
Согласно современной биохимической терминологии, аллосте-
рия означает увеличение (или уменьшение) скорости
ферментативной реакции, обусловленное связыванием эффектора
(субстрата или ингибитора) на участке молекулы фермента, удаленном от
активного центра. Предыдущий пример [уравнение (11.30)]
иллюстрирует аллостерическую активацию фермента субстратом.
Неконкурентное ингибирование (разд. 11.А. 2) можно рассматривать
как аллостерическое торможение. Ниже обсуждаются две
наиболее популярные в последнее время модели, объясняющие
аллостерическую активацию субстратом с помощью ряда простых
гипотетических свойств многомерного фермента; аллостерическое
торможение можно объяснить таким же образом.
В 1965 г. Моно, Уайман и Шанже опубликовали статью,
которая стала очень часто цитироваться (гораздо чаще, чем другие
теоретические статьи по биохимии) благодаря тому, что в ней
приведено объяснение сигмоидальных кривых связывания кислорода
с тетрамерным белком (гемоглобином). Такая форма кривых
связывания часто наблюдается при связывании субстратов или
ингибиторов с различными многомерными ферментами, реакции
которых локализованы в точках разветвления путей метаболизма.
(11.30)
(11.31)
*Ш*
'л
ГЕ*][А] _КЯ
fc*AJ 4
^
Е»А
[ЕдА]]А]_ 2К&
Л
(£гА1[а; _ 2*
KrAaJ 3
ЕЛА2
А. А
'Л
ILrA3) 2
Л
***
АД
~1А
^
'№-<*
Рис 11.10. Схематическое описание модели связывания субстрата с ферментом,
обладающим- четвертичной структурой [12].
В качестве примера рассмотрен фермент тетрамерной структуры. Фермент может
существовать только в двух состояниях (изображены в виде четырех кружочков нли четырех
квадратиков). Предполагается, что каждая субъединица связывает одну молекулу
субстрата; процесс характеризуется одной и той же константой диссоциаций. Статистические
факторы исключаются, например: диссоциация комплекса En Ад на ЕрА и А может происходить
двумя способами, во связывание А с Е»А может происходить тремя способами, в резуль»
тате чего в выражение [En A],[A]/(E^A2j вводится статистический фактор, равный */з.
Степень неоперативности (S-обраэная кривая связывания) при связывании субстрата
ферментом определяется относительными значениями констант равновесия (см. уравнение (11.34)
и следующие].
Предложенная модель основана на том, что фермент состоит из
идентичных субъединиц; данная субъединица может существовать
в. одной из двух конформаций; но если одна субъединица данной
молекулы фермента обладает конкретной конформациеи, то все
другие субъединицы в этой молекуле должны также обладать
этой же конформациеи (рис. 11.10). Таким образом, фермент,
обладающий субъединичной структурой, может существовать только
в двух конформационных состояниях: ER и Ет. Чтобы объяснить
сигмоидальную кинетическую зависимость, достаточно предполо-
325 РОСТ И РАСПАД
жить, что свободный фермент в основном существует в одной кон-
формации, пусть в виде Ег; тогда константа равновесия L
определяется выражением
L=w »' <п-32>
и субстрат обладает повышенным сродством к молекуле в конфор-
мации Ел: _^ ..£
К _ [Ег] [А] [ER] [А] _к .
Дг= [Ej-AJ »"Те^АГ==Д^ {П'66'
При постепенном увеличении концентрации субстрата вначале
он связывается с доступными молекулами Е*, но, поскольку
существует равновесие Ет^Ец, из молекул Ег будет образовываться
все больше молекул Ед. Следовательно, концентрация комплекса
|[Е*А] в действительности будет увеличиваться быстрее, чем если
бы она была пропорциональна концентрации [А] (т. е. в начале
рост [ЕдА] приблизительно пропорционален [А], а далее
зависимость становится более крутой, что соответствует концентрации
субстрата в степени >1). Это приводит к сигмоидальному виду
зависимости скорости ферментативной реакции от [А], поскольку
скорость реакции пропорциональна ;[ЕЛА]. Математическое
описание этой модели поразительно коротко, и им можно заняться в
качестве упражнения (см. задачи в конце главы).
Если определить
и
c=KRlKT (11.34)
то доля участков фермента Уд, занятых субстратом, описывается
уравнением, (см. задачи в конце главы):
у ^_ Lac (1 + ск)"-1 + «(1 + а)""1
А L{\^ca)" + {l+a)n
(11.35)
где п — число субъединиц в молекуле фермента.
На рис. 11.11 показаны типичные зависимости Уд_ (скорость
реакции пропорциональна доле участков фермента Уд, занятых
субстратом) от и (согласно уравнению (11.34), а пропорциональна.
i[A]) для нескольких значений с и L (константа равновесия в
реакции Ед^Ег). Теоретические кривые на рис. 11.11 согласуются с
экспериментальными данными, приведенными на рис. 11.9. Следует
отметить, что описанная модель удовлетворяет обычному
гиперболическому виду зависимости v от [А] в предельных случаях: или
при с=0 и L=l, или с —конечная величина и L — очень большая
величина.
326 ЧАСТЬ 3
Рис. 11.11. Теоретические зависимости, доли F участков фермента, занятой
субстратом, от параметра а, который пропорционален концентрации субстрата [А]
для фермента, состоящего из четырех идентичных субъединиц (я=4) [12].
Параметры L и с определены уравнениями (11.32) я (11.34). Обратите внимание на то, что
модель Моно— У аймак а—Шанже может объяснить сигмоидальный характер зависимостей
ферментативных реакций.
Выше указывалось, что ключевым моментом при объяснении
сигмоидального вида ферментативных кинетических зависимостей
для ферментов с четвертичной структурой является гипотеза об
изменении конформаций фермента при связывании с субстратом,
способствующим связыванию последующих молекул субстрата с
той же молекулой фермента. Модель, изображенная на рис. 11.10,
описывает экстремальную ситуацию, при которой связывание
субстрата сопровождается одновременным изменением конформаций,
всех субъединиц в молекуле фермента. Согласно,
модифицированной модели, связывание субстрата вызывает изменение
конформаций той субъединицы, с которой произошло связывание, а это
изменение влияет на конфоршационные состояния соседних
субъединиц (рис. 11.12); новая особенность этой модели состоит в том, что
число возможных конформаций не ограничено двумя, а зависит
от числа субъединиц, существующих в данной конформаций.
Модель, изображенная на рис. 11.12, также объясняет сигмо-
идальные кинетические зависимости и здесь обсуждаться не будет,
. А Л Ж А А
Рис, 11.12. Альтернативная ш>дель кооперативного связывания субстрата А
ферментом со структурой тетрамера [13]. ^
Предполагается, что сродство молекул субстрата к субъеднницам фермента, изображенным
в виде квадратика, выше, чем сродство к субъединицам, изображенным в виде кружочка,
и что изменение конформаций одной субъединицы благоприятствует изменению
конформаций смежных с ней субъединиц.
327 РОСТ И РАСПАД
у*
1,0
0,5
О 10 20 "«
Рис. 11.13. Теоретические зависимости доли участков, фермента, занятых
субстратом, в присутствии фиксированной концентрации активатора, ингибитора или
без добавления агентов" (норма) от а, согласно расширенной модели Моио — Уай-
мана — Шанже, для аллостерической активации или ингибирования реакции,
катализируемой ферментом с четвертичной структурой (л=4) [12].
а пропорциональна концентрации субстрата [А].
Принципиально модели, приведенные на рис. 11.10 и 11.12,
различимы (см. методы релаксационной кинетики в ч. 4), но пока таких
данных немного.
В заключение следует отметить, что рассмотренные модели,
описывающие изменения конформаций субъединиц при
связывании субстрата, легко использовать более широко для описания
конформационных изменений субъединиц при связывании молекул
эффектора, действие которых увеличивает (активатор) или
снижает (ингибитор) скорость реакции с участием многомерного
фермента. На рис. 11.13 приведены теоретически рассчитанные
зависимости, описывающие влияние фиксированной концентрации
активатора (верхняя кривая) и ингибитора (нижняя кривая) на
зависимость Уд от и в системе, в которой с=0 (т. е. субстрат
обладает сродством только к одной из двух возможных форм
фермента согласно модели Моно — Уаймана — Шанже).
Пример. Аллостерическое ингибйрование L-треоииндезаминазы
На рис. 11.14 показано сигмоидальное снижение скорости ферментативной
реакции, вызванное добавлением аллостерического ингибитора L-изолейцина к
ферменту L-треониндезаминазе. Как и в случае сигмоидального увеличения
скорости ферментативной реакции в присутствии субстрата, наблюдаемое
уменьшение скорости под действием ингибитора можно объяснить с помощью модели
взаимодействующих субъединиц.
Сигмоидальная зависимость, характеризующая связывание субстрата и
торможение изолейцииом в случае тетрамерного фермента Ь-треоннндеэаминаэы,
можно описать, если привлечь модель аллостерического ингибировання-
{рис. 11.13), схематично изображенную иа рис. 11.15. Предполагается, что су-
0 12 3 4 5
~" Относительная концентрация L-цзалейцина
Рис. 11.14. Влияние концентрации L-изолейцииа на каталитическую активность
L-треоншдезаминазы [Н].
а Е-
Е-
о о>
*~ Продукт
О О
Ингибитор (\.-<лзолейцин)
Субстрат (t-треонан)
Рис. 11.15. Схематическое представление двух конформационных состояний
L-треониндезаминаэы [15].
Е-р— связывание с ингибитором; Ей — связывание с субстратом и каталитическое
превращение субстрата.
Г329 РОСТ И РАСПАД
О 5 10 15 20
[Аспартат], м моль/а
Рис. 11.16. Зависимость скорости ферментативной реакции от концентрации
субстрата для реакции аспартата с карбамилфосфатом (контрольный опыт) в
присутствии активатора АТР или ингибитора СТР [16].
Эти зависимости следует сравнить с теоретически предсказанными согласно модели Моно —
Уаймана — Шанже (рис. 11.15). Видно, что модель может хорошо объяснить
экспериментально наблюдаемые эффекты.
ществует две конформации фермента Еи и Ег. Если субстрат связывается
только с формой Ед, то при связывании с ферментом субстрат сдвигает равновесие
Егч*Ед вправо, н начальное связывание субстрата—процесс более высокого
порядка, чем первый (т. е. зависимость описывается сигмоидальной кривой).
Аналогично, если предположить, что ингибитор связывается только с Ег, добавление
ингибитора будет сдвигать равновесие Егч=*Ея влево, оставляя меньше
активного фермента в сфере реакции и приводя к .аллостерическому торможению
реакции.
Пример. Активация и иигибирование аспартаткарбамоилтрансферазы
На рис. 11.16 показаны некоторые экспериментальные зависимости,
характеризующие активацию фермента аспартаткарбамоилтрансферазы с помощью
АТР и иигибирование этого фермента с помощью СТР.
Эти агенты, как известно, связываются с субъедикицей, отличной от той,
с которой связывается субстрат, и, следовательно, служат классическими
примерами аллостерических эффекторов.
В заключение следует указать, что тонкий контроль за
скоростями ферментативных реакций можно обеспечить, если
механизмы этих реакций приводят к сигмоидальным, а не гиперболическим,
зависимостям скорости реакции от концентрации субстрата. Оф,
ним из средств обеспечения такого контроля служит использед^
ние ферментов с четвертичной структурой, свойства которых--'dpjt*
сывают модели, изображенные на рис. 11.10 или 11.12. В обоих;
случаях важной особенностью является то, что связывание су&
страта приводит к изменению конформации части или всего
фермента.
330 ЧАСТЬ 3
11.Б.З САМОТОРМОЖЕНИЕ СУБСТРАТОМ
Относительно прямым средством регуляции скоростей
ферментативных реакций является торможение реакции при высокой
концентрации субстрата, как показано для уреазы на рис. 11.17.
Согласно сравнительно простой модели этого процесса,
связывание со второй молекулой субстрата данной молекулы фермента
эффективно препятствует осуществлению реакции
*1 *2
Е + А *==* ЕА ► Е + Р
Л-1
Аз
ЕА + А +==* ЕА,
Й-З
->■ Реакции нет
(11.36)
Читатель в качестве упражнения (см. задачи в конце главы)
может показать, что конечное выражение стационарной скорости
имеет вид
jrrn/л ^макс^д [А]
d P]A#—v=— ^— г. (11.37)
[АР+ЯА[А]+*А*А V '
При перегруппировке уравнения (11.37) очевидно, что
и—
макс
максДл
1+Яа/(А] + [А]/Яа *а + (А]
(для больших [А]) (11.38)
Следовательно, К'а легко найти из зависимости l/v от [А] при
высоких [А] (рис. 11.18), в то время как Ка и УМанс определяют
из обычной зависимости Лайнуивера — Берка (\/v от [А])^ при
очень малых [А].
о о, t о,2 о,з
Концентрация мочевины , моль/л
Рис 11 17. Самоторможение субстратом реакции разложения мочевины уреазо*
[17]-
/ — ожидаемая гиперболическая кривая в отсутствие ингибнрования; 2 — экспериментальные
данные, свидетельствующие о усилении ингибнрования при повышенных концентрациях суо-
страта.
331 РОСТ И РАСПАД
_/
умакс
V„„~r 10бУмакс=Ю 6моль-л*'-с'1
1.0
40
2,0 3.0
[А] • 102мо/1ь//1
Рис. 11.18. Типичная зависимость 1/о от [А] для ферментативной реакции,
ингибируемои высокими концентрациями субстрата [18].
Кд=10-4 моль/л. КА—10—а моль/л и VMaKC=IO-e моль-л-'-с-'.
Читатель может обратить внимание на близкое подобие
уравнения (11.37) для самоторможения реакции субстратом и
уравнения (11.23) для зависимости скорости ферментативной реакции
от рН. Действительно, оба механизма приводят к колоколообраз-
6,0 —
5,0
4,0
Г
2,0
1,0
-1
—2
-3
Lg LAI
-4
Рис. 11.19. Теоретическая зависимость скорости от логарифма концентрации
субстрата для ферментативной реакции, ингибируемои высокими концентрациями
субстрата [il8].
Кд=ю-4 моль/л, Кд^Ю-3 моль/л и ^макс=1^-в моль/л- Оптимальная скорость реакции
6.2" Ю-7 моль/л, составляющая <*/» УМакс' Д°стигается ПРИ концентрации субстрата
3,15'Ю-* моль/л.
332 часть з
ной форме зависимости ферментативной активности от логарифма
концентрации субстрата или иона водорода и оба механизма
предсказывают достижение максимальной- скорости реакции при
концентрации субстрата (или иона водорода), равной (см. задачи в
конце главы) среднему между Ка. и /Са для связывания субстрата
(или иона водорода) с ферментом (рис. 11.19). Согласно
рассматриваемому механизму самоторможения, скорость реакции
увеличивается при увеличении концентрации субстрата, но в присутствии
избытка субстрата скорость падает, обеспечивая простое средство
прямой регуляции скорости образования продукта.
П.Б.4. АКТИВАЦИЯ ИОНАМИ МЕТАЛЛОВ
Из почти 1000 известных на сегодня ферментов животного
происхождения около четверти либо содержат металлы, встроенные
в^нативную структуру фермента, либо для проявления активности
нуждаются в ионах металлов, либо активируются этими ионами,
выше уровня, проявляемого в отсутствие этих ионов. Поскольку
ферменты, содержащие прочно связанные ионы металлов,
кинетически неотличимы от ферментов без металла, в этом разделе
рассматриваются ферменты, с которыми ион металла связан
относительно слабо (т. е. может очень быстро образовать с ионами
металла ассоциаты, которые также быстро диссоциируют).
Простейший общий кинетический механизм аналогичен обычной схеме ин-
гибирования фермента [ср. с уравнением (11.1)]
ЕА^
•ы/ \+мг
Б ЕМА-»Е+Р + М'
Как обычно, общее решение для этих процессов настолько
сложно, что не имеет большой практической или теоретической
ценности. Однако поведение многих ферментов можно описать
несколькими простыми предельными случаями. Вначале
предположим, что реакция не происходит, если фермент не образовал
тройной комплекс ЕМА, и тогда возможны два предельных случая:
1. Комплекс ЕА не образуется в отсутствие М — реакция
происходит по нижнему пути [схема (11.39)]:
Rtn RA k
Е+ М *=*= ЕМ ^==fc ЕМА ► Е + Р + М
-т -А
(11-39)
(11.40)
333 РОСТ И РАСПАД
Уравнение стационарной скорости имеет вид
VW [А] [М]
1А][М]+Ка1МЛ-КаКм'
U=s
(11.41)
где /См=-7~—константа диссоциации комплекса ЕМ, /Са=&а-Ь
т
-|-«/«а — константа связывания субстрата Михаэлиса — Ментен.
2. В отсутствие А комплекс ЕМ не образуется — реакция
происходит по верхнему пути [схема (11.39)]:
ftA km k
Е + А zt==z EA 5==fc EAM ► Е + Р + М
k-A k-m
(11.42)
Уравнение стационарной скорости имеет вид
v — Умакс [(*м + [М] + [А]) - У{Км + [М] + [А])» - 4 [М] [А] ({ j 43
Наиболее часто металл участвует в ферментативных реакциях с
переносом фосфатной группы
Mg++
/ V
о- о о-
Ad—О—Р—О—Р Р—О-
О 0 0 0
АТР
о- о-
Ad—О—Р—О—Р—О-
II II
О О
ADP
(аденозиндифоарат )
о-
+ Р +Mg+*
'4
в которых в роли иона металла выступает обычно Mg2+ и иногда
Мп2+. Эти механизмы [уравнения (11.40) и (11.42)] легко
различить из зависимостей скорости ферментативной реакции от [М]
Механизм типа 6
[А]3
[AJA
[А],
\ Концентрация иона металла
Концентрация иона металла
Рис. 11.20. Графический способ анализа различий между двумя механизмами
активации металлом {уравнения (11.40) и (11.42)] путем построения зависимости
скорости стационарной ферментативной реакции о от концентрации металла [М]
при трех (фиксированных) концентрациих субстрата '[А] [19],
В обоих случаях [A]i<[Ah<:[A]t.
334 ЧАСТЬ 3
при разных фиксированных [А] (рис. 11.20). При [А]>[М] и
постоянной [М] (т. е. на прямой линии, параллельной оси ординат,
проведенной в левой области зависимостей на рис. 11.20) скорость
реакции обратно пропорциональна [А] (а), и постоянна или
прямо пропорциональна [А] (б). С помощью экспериментов такого
типа показано, что катализ с помощью дрожжевой гексокиназы
Глюкоза-f ATP-f-Mg2+ *=±: Глкжоза-б-фосфат+ADP+Mg2* (11.45)
описывается зависимостями типа а, а катализ с помощью пиру-
ваткиназы
Пируват-|-АТР-(-Мп2+ *=* Фосфоенолпируват+АОР-|-Мп2+ (11.46)
зависимостями типа б. Зависимости типа б характерны для
значительно большего числа-ферментов, чем зависимости типа а.
11.В. БИОЛОГИЧЕСКИЕ ЧАСЫ —ПРИЧУДА ПРИРОДЫ
ИЛИ ЯВЛЕНИЕ, ОБУСЛОВЛЕННОЕ ХИМИЧЕСКИМИ
ПРОЦЕССАМИ?
Одним из самых неожиданных следствий активации и ингиби-
рования химических реакций путем обратной связи является тот
факт, что на этой основе существует группа химических систем,
в реакциях которых концентрация данного промежуточного
соединения многократно возрастает и падает до достижения
равновесия. Процессы, происходящие в таких системах, в настоящее
время еще плохо поняты, это подтверждается, в частности, тем,
что все такие системы были Открыты совершенно случайно. Еще
никто не смог создать химическую колебательную систему
направленным подбором соответствующих реагентов. Для
объяснения процессов в таких системах, как правило, привлекается
простая модель, впервые предложенная Лотка в 1920 г., которую
проще всего понять на примере взаимоотношений хищников и их
жертв в популяциях животных:
T-f-K —*- 2К Трава-j-Кролики —> Увеличение количества
кроликов
К+Л —*• 2Л Кролики-f-Лисы —*• Увеличение коли- (11.47)
чества лис
Л->0 Лисы умирают
Интуитивно ясно, что при постоянном «притоке» Т (травы) К
и Л (популяции кроликов и лис) в конце концов достигнут
стационарного уровня. Однако если в климате произойдет внезапное
изменение и приток травы изменится, то популяции животных
начнут колебаться на пути к новому стационарному состоянию:
кролики будут быстро размножаться до тех пор, пока увеличение
числа лис не уменьшит число кроликов, и лисы в конце концов
335 РОСТ И РАСПАД
_ Равновесие
Время
Рис. 11.21. Изменения в концентрации промежуточного химического соединения
во времени при установлении химического равновесия в системе, обладающей
химическими колебаниями [5].
размножатся до такого большого количества, что им вновь начнет
не хватать кроликов, и весь цикл повторится снова (рис. 11.21).
Далее можно думать над тем, как выявить критическую
характерную особенность механизма (11.47), приводящую к
колебательному поведению системы. Сравнивая множество схем, в
основе которых положена оригинальная идея, выдвинутая Лотка,
можно прийти к выводу, что количество общих механизмов
довольно мало:
/1ктивпцир
*1—г>- а2 —э- • • • вп_1 -->- йг
Ингибированае
F "'
п ^„^„-^ „„„^„„-^ (iMg)
Активация
I _ L
III -*- я1^_ я2 ^_ я„-1 _^_ я„ —>-
Ингибирование
I ^ \_
iv ^-я'^я2^; ап-\ ^L an -9-
Ключевой особенностью, общей для всех этих механизмов,
является активация или торможение предшествующих стадий в
схеме с помощью обратной связи. Нет ни одной гипотетической
реакционной схемы, описывающей колебательные процессы, которая
не включала хотя бы один из четырех вышеуказанных типов
регуляции обратной связью. Схему Лотка (11.47) можно
рассматривать как вариант I (11.48), в котором существует только два
промежуточных соединения (кролики и живые лисы) и каждая
реакция имеет по крайней мере второй или более высокий порядок.
Хотя схемы химических реакций с участием трех и более соеди-
336 часть з
Рис. 11.22. Изменения
концентрации Ai во времени по
механизму, описываемому
уравнением (11.49). Предполагается,
что реагенты Ai и Аг
подводятся в систему извне с
постоянной скоростью [20].
1000 2000
Время, с
3000
йений напоминают хорошо известную физическую задачу о трех
небесных телах, которай не имеет строгого решения, в принципе
можно исследовать решение для описания колебательных
химических систем с помощью итерационных расчетов на ЭВМ.
Результат для относительно шростого конкретного механизма
модельной ферментной системы (11.49) графически изображен на
рис. 11.22, где ясно видны колебания:
:А1+Хт±А1Х-^Х + Р1^Р1
P, + Y^P,Y
Р2 + Х^Р2Х
А 1 (извне)
А а (ж**)
:A, + Y^A2Y^Y + P;
(11-49)
_|
(Зснову системы (1.1.49) составляют две ферментативные
реакции» в которых продукт одной реакции тормозит другую реакцию.
Пример. Колебательные реакции при гликолизе
Как показано на рис. 11.23, гликолиз изобилует обратными связями.
Исходя из критериев, необходимых для химических колебаний [схема
(11.49)], эту систему следует считать наиболее подходящей для
экспериментального доказательства колебательного характера процесса. В 1964 г. Чане и др.
получили такие доказательства; некоторые из данных этих исследователей
приведены на рис. 11.24. Вначале аэробная суспензия дрожжевых клеток
инкубировалась с глюкозой; затем после короткого периода приток воздуха отключался
и система переводилась к анаэробному метаболизму. Относительные
концентрации пиридинового нуклеотида NADH легко регистрировались по характерной
флуоресценции этого соединения. Кроме того, в указанные промежутки времени
в отбираемых пробах определяли содержание ATP, ADP и AMP (11.24,6*).
Интересно отметить, что изменения во времени количеств ADP, AMP и NADH
очень близки («в одной фазе»), в то время как колебания АТР и ADP отстают
друг от друга по фазе почти на 180°. Хотя эти жолебания легко объяснить с
Помощью любой обратной связи (наиболее детальное описание основывается на
машинном анализе одновременно 57 дифференциальных, уравиеиий, представляю-
Дсюс 101 химическую реакцию!), более полезный подход состоит в попытке опре*
доиггь лимитирующую для системы обратную связь. Наиболее вероятно, что о*-
u Гликоген
ATP
ъ
GLU-1P
Глюкоза.-
1
GLU* P Г
WtMWMV^VMM
Этанол
ATP
FRU-6P
ADP
FRU-U-i»l»|
DHAP
t
@
Actemi
'Л«*-
NAO*
6A3 P '
дегид
NADH
ADP
T
co2
/I
ATP
PYR
PEP
u:
ATP
ADP
ATP
~r\ lurtj-rr—1 - i ifcri——*i f 1
ADP
-Л
ADP
Рис. 11.23. Реакции метаболизма при гликолизе.
АТР — аденозин-5'-трифосфат; ADP ~- аденозин-5'-дифосфат; GLU-1-P — глюкоза-Ьфосфата
Я|— неорганический фосфат; GLU-6-P — глкжоза-6-фосфат; FRU-6-P — фруктоз а-6-фосфатг
FRU-I,6-AH-P — фруктоза-1,6-дифосфат; DHAP — диокснацетонфосфат; GA-3-P — глнцеральде-
гид-3-фосфат; NAD+ — окисленная форма никотинамндаденвндннуклеотида; NADH —
восстановленная форма инкотинамидадениндинуклеотида;- I.3PGA — 1,3-днфосфоглнцернновая
кислота; PEP — фосфоеиолпируват: PYR — пируват; Ei — фосфорнлаза; В* — фосфоглюкомут»
аза; Ез — гексокиназа; Ед — фосфофруктокнназа; Е5 — альдолаза; Е» — глицеральдегяд-$-фое*
фатдегндрогеназа; Ez — фосфоглицератквнаэа; Е» — пвруваткиваза; Е»— пнруватдекарбо*
ксилаза; Ел — алкогольдегидрогеназа; Ец — аденознв-5'-трифосфатаза.
ГС
ч
Z
«и
3
1
3
\
"20мМ
- Глюкоза
■ ■'
нив л
духа
1,3
х„ 1,1
If 0,9
Д; о,7
X
1
0,5
0,3
0,1
.
£
/"
Г
/
/
f-
(-
\
\
\
\
Vu
Л
/
Г
U
V
л
\
'
/
/
•
V
\J
/\
'Л
■< »-
Лмин Н
1
- У"
f
\
\>
-А.---
г-
-\ А/
=\/V
-У.
— АТР
<
«
AD
Р
Д
WIF
||
Рис. 11.24, Концентрационные колебания компонентов метаболизма при
гликолизе суспендированных дрожжевых клеток 1[21].
Компонеитыз NADH — восстановленная форма викотинамидадениндинуклеотвда, АТР — ад*»
нозиитрнфосфат, ADP — аденоэиндифосфат и AMP — аденозинмонофосфат.
338 часть з
ветственньш за это является фермент фосфофруктокииаза, ингибируемая адено-
аинтрифосфатом (АТР служит субстратом данного фегрмента) и активируемая
продуктами реакции ADP и фруктоза-1,6-дифосфатом.
Необходимо соблюдать большую осторожность при попытках приписать
колебаниям в процессе гликолиза какое-либо прямое значение в биологических
часах. Поскольку такое 'поведение можно ожидать от любой системы с большим
количеством обратных связей и существование таких связей может иметь своей
единственной функцией регуляцию системы (разд. 11.А и 11.Б), то
колебательные процессы, являющиеся следствием обратных связей, могут быть просто
случайными. (Затухание колебаний иа рис. 11.24 может быть заметно уменьшено
стимулированием гомеостатических биологических условий постоянной подачей
глюкозы в отличие от внезапной одноразовой порции глюкозы, использованной
для запуска ритма, показанного на рисунке, и тогда можно заставить гликоли-
тическую систему колебаться в течение нескольких часов.) Однако любопытное
спекулятивное заключение можно сделать, исходя из аналогии этого процесса с
падением камня в резервуар со стоячей водой. В том месте, в котором падает
камень, вода поднимается и опускается точно так же, как увеличивается и
уменьшается концентрация АТР при гликолизе в месте химической пертурбации,
вызываемой добавлением глюкозы; представляется поэтому логичным
предположить, что флуктуации АТР распространяются в пространстве так же, как волна
распространяется по воде из точки ее образования. Следовательно, такой процесс
может служить средством синхронизации поведения пространственно разделенных
клеток (например, в ткани сердца) согласно возникающему где-то триггерному
сигналу (синусовый узел сердца?). Колеблющиеся химические «волны»,
распространяющиеся в пространстве, в действительности наблюдались в ряде
модельных экспериментальных систем, проявляющих химические колебания.
Одно из серьезных затруднений, которое возникает при попытке объяснить
существование биологических ритмов с помощью колебательных химических
реакций, состоит в том, что биологические ритмы, как правило, характеризуются
большой продолжительностью (от нескольких секунд до нескольких суток), в то
время как химические реакции происходят гораздо быстрее: период химического
колебания (~1000 с на рис. 11.22) может быть намного дольше периода
соответствующих химических реакций (1/£=5 с в том же примере). Далее, хорошо
известно свойство осцилляторов, состоящее в том, что при «смешивании» или
«соединении» двух быстрых осцилляторов легко получить колебание с
«модулированной» частотой, которая может быть намного меньше, чем частота колебаний
любого из компонентов системы.
Более простое объяснение замедленных биологических часов с «циркадным»
или суточным ритмом может основываться на колебаниях, сопровождающих
молекулярный процесс, собственная скорость которого намного ниже скоростей
большинства метаболических реакций. Недавно предложенная модель осиоваиа
на относительно медленной (см. разд. 14.Г) латеральной диффузии белков в
мембранах. Поскольку «циркадные» ритмы сохраняются даже во многих эиуклеи-
рованиых (безъядерных) клетках и даже в присутствии ингибиторов синтеза
различных нуклеиновых кислот и белков, мембранная модель не зависит от
точных концентраций фермента и, следовательно, объясняет предыдущие
наблюдения.
Пример. Мембранная модель циркадных часов
На рис. 11.25 показано, как жидкостно-мозаичная мембрана (см. разд. 2.А.2
и 14.Г или работу 1[8]) может обеспечить колебания трансмембранного
градиента ионов, служащие основой циркадного ритма. Предполагается (для простоты),
что мембрана существует только в двух состояниях, отличающихся тем, что
мембранные белки л>ибо агрегированы" (и тогда могут активно «перекачивать» ионы
К+ через мембрану), либо распределены по мембране (« тогда не могут обес?
печивать активный транспорт). Действие света может «открывать ворота» иоиу
К+, возможно индуцируя агрегацию белков (см. в ч. 4 пример о родопсине). При
ОЬгпорецептор
Транспорту бедка
<=з к
Концентрации
Неравны
Меньше К+ *, ,•
Активная конфигурация CT1S Активна* конфигурация СТЗ
4
D
Дольше
К
Е>
Концентрации
К*'равны
Меньше К +
Пассивная конфигурация СТ 21
К*
Пассивная конфигурация СГЗ
Рис. 11.25. Схематическое изображение способа обеспечения временных
процессов жидкостно-мозаичной мембраной |[22].
Концентрации нонов и ионные потоки (транспорт) действуют в качестве системы обратной
связи для создания самогенернруемых цнркадных колебаний (см. рнс. 11.26). Свет
действует на мембрану либо прямо, либо посредством связывания гормонов, приводя к открытию
ионных «ворот» (или вызывая перенос мембранных белков по мембране в неактивное поло»
женне, предотвращая этим активный транспорт нонов) н уменьшая трансмембранный
градиент ионов для перевода .«часов» вперед или назад (см. текст и рнс. 3.47).
6 12 1Q
Циркадное время
Рис. 11.26. Гипотетические колебания в упрощенной системе (один ион и только
два состояния мембраны) [23].
Световой импульс, наложенный прямо перед СТ18, сдвигает «часы» назад к состоянию, »ж*
вивалентиому СТ13 (которое имело, место рзньше). Анзлогнчно световой импульс, наложен»
ный сразу после CTI8, сдвигает «часы» вперед.
340 часть з
последовательном чередовании в дневном цикле света и темноты поток ионов и,
4 следовательно, градиент ионов будет изменяться соответствующим образом.
Поскольку многие циркадные ритмы сохраняются и в темноте (в отсутствие света),
может существовать ие связанный с действием света специфический механизм,
изменяющий состояние мембраны. Однако иитересио отметить, что модель,
изображенная на рис. 11.25, объясняет также влияние световых импульсов на
фазовые изменения («перевод» часов) в реальных системах. Например, наложение
светового импульса как раз перед моментом, когда циркадное время (СТ) равно
18 на рис. 11.26 «откроет ворота» иону К+ и сдвинет концентрацию иоиов к
уровню, прн котором она находилась в момент СТ-13 (перевод «стрелок» часов
назад), в то время как световой импульс, (Приложенный к системе сразу после
СТ-18, сдвинул бы концентрации иона вперед по времени.
Задачи
1. Для случая неконкурентного ингибирования выведите выражение скорости
стационарной ферментативной реакции и покажите, что зависимости Лайиуи-
вера — Берка при разных (фиксированных) концентрациях ингибитора
пересекаются на оси (1 [А]). Другими словами, получите уравнение (11.17) иэ
уравнений скоростей и баланса (11.16) для механизма реакции (11.15).
2. Покажите, что в предельных случаях чистого конкурентного или
неконкурентного ингибирования [уравнения (11.14) или (11.17)] зависимости Диксона
(1/о) от [I] при нескольких (фиксированных) значениях [А] будут
пересекаться при [I] =—К\.
3. Для модели рН-контроля ферментативных реакций
ЕН|+ ч==* Ен+ =e=r* E
где только форма ЕН+ способна к связыванию субстрата и к катализу данной
реакции и
Кг = [EH+J [Н+]/[ЕН5+1 и К2= [Е] [Н+]/[ЕН+К
а. Определите долю фермента в каждой, из соответствующих форм ЕН|+,
ЕН+ и Е [уравнения (11.22)].
б. Покажите, что максимальная скорость ферментативной реакции
достигается при
[H+JonxiiM = У КгК2.
в. Покажите, что интервал рН, в котором фермент проявляет не менее
половины своей максимальной активности, определяется корнями уравнения
•[№■]» - (К, + 4 [Н+]оптам) [№•] + [Н+Ропгам = 0.
4. Одна из простейших кинетических схем, объясняющих сигмюидальный вид эа«
висимости скорости ферментативной реакции от концентрации субстрата,
основана на двухстадийном связывании*, изображенном ниже:
kl fta *3 .
Е + А *==± ЕА > ЕА, ► Е + А + Р
Найдите общее выражение стационарной скорости образования продукта по
этой схеме и для предельных случаев проанализируйте ожидаемый вид
зависимостей v от [А] н \/v от 1/[А].
в*. Сделайте полный вывод модели Моно — Уаймана — Шанже, описывающей
Ц кооперативное связывание субстрата с многомерным ферментом. На основании
341 РОСТ И РАСПАД
модели, изображенной на рис. 11.10, и пользуясь определениями
L=[ET]/[ER]; a.= [A)fKR и c = KR/KT.
где
Кт = [Ег] [А]/[ЕГА] и KR = [Ы [А]/[ЕЛА],
покажите, что для фермента, состоящего из п субъединиц, доля участков 7л»
занятых субстратом, описывается уравнением
- aLc (1 + g»)"-1 + «О + а)"'1
Уа_ L(l-f сауЧ-О+а)"
Замечание. Вначале решите случай с п=4, показанный на рис. 11.10 н
получите конечный результат индуктивным способом. Начните с доказательства,
что
[ЕдА] + 2 [ЕЯЛ2] + 3 [ЕЛА3] + 4 [ЕЛА4] +
v + [ЕГА] + 2 \ЕТА2] + 3 |Е7А3] + 4 [Е7А4] ^
ГА ~ 4 ЦЕд] if- |ЕЛА] + [ЕЛА2] + 1Е*А3] + 1ЕЛА4] + [Ег] +
+ [ЕГА1 + [ЕГА2] + [ЕГА3] + [ЕГА4]}
[EpA](l+a)3 + fE7A](l+ca)3
~ 4{[ER\{l + a)*-j-[ET)(l+ca)*}
Затем покажите, что
[ErAJ/[E^A] = cL
так что предыдущее уравнение упрощается:
- acL(l + ca)3+a (1+a)3
Уа_ L(l+ca)4+(l+a)4
6. Для самоторможения субстратом по механизму (11.36).
а. Выведите уравнение стационарной скорости [уравнение (11.37)].
4 б. Покажите, что максимальная скорость ферментативной реакции достигается
при
где Ка~—£—и KA=k-Jk3 для схемы (11.36).
в. Предскажите форму зависимости 1/о от 1/[А] в данном случае.
Литература
1. Mahler H. R„ Cordes Е. H., Biological Chemistry, Harper & Row, New York,
1966. Хорошее изложение основного материала.
2. Dixon M., Enzymes, Academic Press, New York, 1964. Хороший критический
сравнительный анализ разных графических способов описания кинетических
данных при изучении ферментативных реакций.
3. Laidler К., Bunting P. S., The Chemical Kinetics of Enzyme Action,
Clarendon Press, Oxford, 1973, Chapter П. Хорошее обсуждение явления аллосте-
рии.
4. Mildvan A. S., in: ЗЪе Enzymes, P. D. Boyer (Ed.), vol. 2, Academic Press,
New York, 1970. Хороший общий обзор механизмов ферментативного
катализа с участием ионов металлов.
342 ЧАСТЬ 3
5. Degn #., J. Chem. Educ, 49, 302 (1972). Обсуждение неферментативных
химических колебаний.
6. Walter С, Enzyme Kinetics: Open and Closed Systems, Ronald Press Co.,
New York, 1966, Chapter 8; Hess В., Boiteux A., Ann. Rev. Biochemistry, 40,
237 П971). Примеры химических колебаний в биохимических системах.
7. Segel I. И., Enzyme Kinetics, John Wiley & Sons, New York, 1975.
Энциклопедический каталог различных специальных предельных случаев выражений
скоростей стационарных ферментативных реакций.
8. Bernhardt S., Structure and Function of Enzymes, W. A. Benjamin, Inc., New
York, 1968, p. 88.
9. Maugh T. H., Science, 184, 972 (1974).
10. Walker A. C, Schmidt C. L. A., Arch. Biochem. Biophys., 5, 445 (1944).
11. ЛГог* /. S., Chan W. W.-C, J. Biol. Chem., 250, 653 (1975).
12. Monod J., Wyman J., Changeux J.-P., J. Mol. Biol., 12, 88 (1965).
13. Cornlsh-Bowden A. /., Koshland D. E., Biochemistry, 9, 3337 (1970).
14. Changeux J. P., The Control of Biochemical Reactions, Scientific American,
Inc., April 1965.
15. Bender M. L., Brubacher L. J., Catalysis and Enzyme Action, McGraw-Hill,
New York, 1973, p. 196.
16. Gerhart J. C, Curr. Top. Cell. Regul., 2, 275 (1970).
17. Bull H. В., Introduction to Physical Biochemistry, F. A. Davis, Philadelphia,
1971, p. 415; Laidler K. J., Hoare.J. P., J. Amer. Chem. Soc, 71, 2699 (1949).
18. Walter C, Steady-State Applications in Enzyme Kinetics, The Ronald Press
Co., New York, 1965, p. 80.
19. Gutfreund H., Enzymes: Physical Principles, Wiley-Interscience, London, 1972,
p. 152.
20. Spangler R. A., Snell F. M., Nature, 191, 457 (1961).
21. Betz A., Chance В., Arch. Biochem. Biophys., 109, 585 (1964).
22. Njus D., Sutzman F. M., Hastings J. W., Nature, 248, 116 (1974).
23. Диксон М., Уэбб Э. Ферменты. М.: Мир, 1966, с. 110.
24. Singer S. J., Nicolson G. L.t Science, 175, 720 (1972).
ГЛАВА 12
ФАРМАКОКИНЕТИКА:
КИНЕТИКА ХИМИЧЕСКИХ РЕАКЦИЙ
С ПЕРЕИМЕНОВАННЫМИ ПЕРЕМЕННЫМИ
12.А. ПРОДОЛЖИТЕЛЬНОСТЬ ДЕЙСТВИЯ
ЛЕКАРСТВЕННЫХ ВЕЩЕСТВ:
ВВЕДЕНИЕ И ВЫВОД ЛЕКАРСТВЕННЫХ ПРЕПАРАТОВ
Для описания механизма и кинетики действия лекарственных
препаратов используются математические уравнения, которые
рассматривались выше при анализе вероятностных процессов и
кинетики некаталитических и ферментативных химических реакций.
Вначале целесообразно обсудить скорости, с которыми
лекарственный препарат достигает участок своего действия, анализируя
процессы введения и удаления его из организма (разд. 12.А), а
затем уже молекулярные механизмы действия препарата,
определенным образом локализованного в организме (разд. 12.Б).
Благодаря близкой аналогии с вышерассмотренными схемами
кинетики химических реакций достаточно проиллюстрировать кинетику
действия лекарственного препарата одним примером из общей
практики.
Пример. Отрезвление — действие этанола на человеческий организм
Вслед за введением единичной дозы лекарственного препарата его количество
на разных стадиях можно описать общей схемой
A -J-+ В > С (12.1)
где А — вводимый препарат (допустим, в желудок или кишечно-желудочный
тракт при пероральном введении препарата), В — препарат в крови и С —
препарат выводимый в результате процессов обмена (путем секреции или
выделений). Ранее уже была решена кинетическая схема, описываемая уравнением
(12.1) (см. разд. 10.А.4, последовательные реакции первого порядка), и типичная
зависимость концентрации от времени имеет такой же вид, как приведенная на
рис. 10.3. Единственное умозрительное отличие здесь состоит в том, что объем
«реакционной ячейки» меняется в ходе реакции, поскольку при попадании, в
кровь или в другие органы препарат может и разбавляться и кои центрировать*
ся — это изменение в основном приводит к тому, что в общем случае масштабы
по оси ординат для [А], .[В] и [С] на рис. 10.3 будут различаться. Далее
рассмотрим некоторые специальные случаи схемы (12.1) н затем попользуем
полученные результаты для анализа концентрации этанола в крови человека.
Мгновенное введение и мгновенное всасывание. Если введение и всасывание,
как часто бывает, происходят гораздо быстрее, чем выведение препарата из ор^
гаиизма, то удобно предположить, что снижение концентрации препарата
происходит экспоненциально:
[В] =[В]0ехр[-М] (12,2)
344 часть з
72
время, ч
Рис. 12.1. Накопление лекарственного препарата при регулярном (через посто:
иые промежутки времени) многократном введении препарата прн условии, '
препарат всасывается мгновенно [4].
Константа скорости вывода препарата £=0,0289 ч-' (время вывода половины препарата 24
и интервал введения препарата 7"=4 ч. / — Доза=1 г; 2 — Доза=2 г для первых шести д
а далее Доза«-1 г; 3 и 4 (ломаные) — накапливание препарата при непрерывном введе>
(введение прекращается через 72 ч); 5 и 6— рассчитаны по уравнениям (12.3) и (12.4). К
вая вывода препарата, как видно, идентична кривой накопления, но обращена.
где [В]о — концентрация (мгновенно введенного и всасавшегося) препаратг
крови, возникающая сразу же после введении первой дозы. Если теперь чц
постоянные интервалы времени Т (в секундах) вводить вторую и последуют
дозы в тех же количествах, то легко показать (см. задачи в конце главы), ч
концентрация препарата в крови будет экспоненциально увеличиваться до i
стоянного уровня, достигаемого после лриема большого числа доз:
/ = Доля от постоянного конечного уровня, достигаемого после л доз =
= 1 — ехр [~hnT\ О2-
Зависимость фактического уровня препарата в крови от времени показа
на рис. 12.1. Если k2 известна, то встает самый обычный вопрос: сколько д
данного размера необходимо для достижения доли f от постоянного уровня, f
стигаемого прн приеме бесконечного числа таких доз? Согласно уравнению (12,
(<2М. задачи в конце главы), ответ описывается уравнением
rt=ln(l— /}/(—кгТ). (12.
Последний общий вопрос заключается в том, насколько большую дозу нес
ходимо ввести при данном временном интервале (допустим, 7"=4 ч) для подде
ж.ания достигнутого уровня препарата в крови. Ясно, что непосредственно пер
медеиием следующей дозы из организма ушло количество препарата, равн
EBJqO—ехр[—kit]), и это количество надо возместить.
Всасывание — процесс первого порядка; выведение — процесс нулевого я
рядка. В случае этанола и многих других препаратов организм способен вывес
Определенное максимальное количество препарата за единицу времени иезавис
ко фт того, сколько препарата находится в организме. Выведение этанола пр
Деходит путем ферментативного окисления до ацетальдегида (и в конце конц
0> уксусной кислоты) в печени. Поскольку скорость ферментативной реакции
Данном случае определяется концентрацией фермента (при большом избытке эт
*рЛа), выведение этанола является процессом нулевого* порядка относителы
545 рост и распад
1,5 2,0
Время, v
Рис. 12.2. Изменение концентрации этанола в организме со временем в
предположении, что поглощение и всасывание происходят мгновенно (см. табл. 12.1)
[1].
его концентрации. Кинетика процесса (12.1), следовательно, описывается
уравнением
d[B]/^ = Jfe1[A]~^2, (12.5)
при интегрировании которого (начальное условие [В]о=0) получаем
* [В] = [А]0 (1-ехр [VI) - V- (12.6)
Для предсказания степени интоксикации этанолом представляют интерес две
особенности полученного уравнения (12.6), Во-первых, максимальный уровень
этанола, достигаемый после введения, рассчитывается при подстановке
d[B]/dt=0 из уравнения (12.6) и составляет
.. [Этанол]^ = [А]0 - (*2/*i) - (*2*макс)* (12.7)
где /макс — время, требуемое для достижения максимального уровня этанола,
Однако, поскольку, как вскоре увидим, k2<^ki (табл. 12.1) и /макс'С (1/£г),
уравнение (12.7) свидетельствует, что максимальный уровень этанола в крови очень
близок к всему введенному количеству (рис. 12.2).
Вторая интересная особенность уравнения (12.6) состоит в том, что можно
рассчитать время,' необходимое для того, чтобы концентрация этанола в крови
снизилась ниже допустимой (например, юридически допустимой) после того, кап
этот уровень был превзойден. В качестве упражнения читатель может сам
показать, что время «отрезвления> /отреа можно выразить в знакомых единицах
(табл. 12.1):
'отрез (в часах) = 1,22 [(Количество поглощенного СаН6ОН)—4,3], (12.8]
"(где количество этанола выражено в унциях), а время полного выведения «^
алкоголя, (похмелення) описывается уравнением
'пот (в часах) = 1,22 (Количество поглощенного саН5<Щ|
В частности* для достижения допустимого (юридически)
нола в крови (т. е. до признания человека трезвым) тфи "
напитка 80-градусиой крепости (31,7% спирта) сверх^
а для выведения всего этанола из организма, после^ -
дин уже достигала юридически установленного
г
,25 ч
интоксика-
5 ч.
346 ЧАСТЬ 3
Таблица 12.1
Факторы конверсии и постоянные, используемые при определении
содержания этанола в человеческом организме как функция времени, [1]
Этанол, г
Концентрация
этанола в
жидкости
организма,
г/л
Эквивалентное
количество алкогольного
напитка 80-градусиой
крепости [40% (об./об.)
этанола]
объем, мл масса, г
Качественные физиологические и
психические реакции организма
20
40
60
80
100
0,5
1.0
1.5
2,0
2.5
63
127
190
254
317
65,17
133,74
199,06
267,5
332,8
Чувство эйфории
Расстройство (от слабого до
среднего) координации моторной системы
(юридически возможная
интоксикация)
Полное отсутствие координация и
невнятность речи
Амнезия
Индуцированный сон или потеря
сознания
*, = Ю ч-1; ftj=0,192 г-л-1-'
40 л.
соответствует 10 мл/ч. Объем жидкости тела принят равным
Введение — процесс нулевого порядка; всасывание — процесс первого
порядка; выведение — процесс нулевого порядка. Вечеринка с коктейлями. Очевидно,
что обычно никто не принимает этанол в виде единичной большой дозы. Более-
разумной моделью может служить постоянная скорость введения за данный
период времени, после которого введение прекращается. В качестве упражнения
заинтересованному читателю предлагается показать, что уровень этанола в
крови в этом случае будет изменяться, как показано на рис. 12.3.
2,5
* 2,0
I
1.5
Юридически установленный
уровень
интоксикации
%
а
!
1,5 2,0
Время, ч
Рис. 12.3. Изменение концентрации этанола в организме во времени в
предположении, что скорость поглощения постоянна в течение 1 ч, а затем поглощение
прекращается :[1].
347 РОСТ И РАСПАД
12.Б. ТЕОРИИ ДЕЙСТВИЯ ЛЕКАРСТВЕННЫХ ПРЕПАРАТОВ
12.Б.1. ДИФФЕРЕНЦИРОВАННЫЙ ОТКЛИК: ТЕОРИЯ ЗАНЯТОСТИ
И ТЕОРИЯ СКОРОСТИ
Хотя действие некоторых препаратов, таких, как анестетики
общего действия, является неспецифичным (разд. 11.А), общие
теории действия лекарственных веществ основываются на
представлениях о связывании веществ со специфическим рецептором
(часто мембранным белком, как при действии инсулина),
вызывающим биохимический отклик. В результате происходит, например,
ускорение или замедление определенной реакции обмена или
изменение проницаемости мембран по отношению к специфическим
ионам или молекулам, что в свою очередь приводит к такому
физиологическому эффекту, как, например, изменение дыхания или
ритма сердца. Согласно одной из простейших теорий, считается,
что данный организм обладает фиксированным числом,
по-видимому, независимых друг от друга эквивалентных рецепторов (как
схематично показано на рис. 12.4), с которыми может связываться
молекула лекарственного вещества.
Модели, изображенной на рис. 12.4, соответствует следующий
кинетическая схема:
D + R з-^-> DR —> Эффект (отклик) (12.10)1
-'■"■?
где D, R и DR — соответственно препарат, рецептор и комплёк.$|
препарата с рецептором и /Cd=&-i/&i — равновесная констант^
диссоциации комплекса DR. Если предположить, что (дифферент!
цированный) отклик пропорционален доле рецепторных участкчм^
Рис. 12.4. Схема расстановки в организме возможных рецепторов лекйщШщ^
препарата. ; ', -,...ку
Рецепторные участки, занятые связанными молекулами лекарственного веществ*, затемнены.
Предполагается, что действие лекарственных веществ, согласно данной теории «едйтости»,
выбывает фииолог^чХй отклик, "пропорциональный, дозе рецепторных участков, заняты*'
молекулами лекарственного вещества. Считается, что участки не эавнсят Друг ОТ друге,
так что лекарствК! молекуи одинаково хорошо может связываться е любым участком
независимо от того, заняты ли соседние рецепторнче участки.
348 часть з
занятых молекулами препарата, то максимальный эффект
достигается при [D]^>[R], т. е. когда практически все рецепторные
участки заняты. Легко заметить отчетливо выраженное подобие
уравнения (12.10) (и следствий из него) и кинетической схемы
МихаэЛиса — Ментен (10.4). Алгебраическое описание выглядит
следующим образом:
Эффект=const «(Доля рецепторных участков, занятых
молекулами препарата)
Эффект- const-™ -const- |РЯ] П2 1П
Эффект- [RJ + [DR, -const №бщ vz.u)
Если вспомнить, что
к _ *-i _ [D][R] _((Р]общ-[РЯ])([Н1общ-[РЯ]) /19 \9\
А° kT [DR] [Ш] {МЛ*)
и ограничиться случаем, когда препарат находится в большом
избытке по сравнению с рецепторами
Р]общ »[RU,, (12.13)
та уравнение (12.12) упрощается:
ЛЬ» PWWg.-[WD (I2.i4)
Решим уравнение (12.14) относительно [DR] и подставим
полученное выражение в уравнение (12.11):
Эффект=шш1.^й_ (12.,5)
Наконец, поскольку
ЭФФ^Тмакс=1,1П (Эффект) = const • 1, (12.16)
и уравнение (12.15) можно переписать:
^ . Эффектмакс [D]
общ
^W*"- /CD + lDJo6l4 "
(12.17)
Последнее уравнение удобно для сопоставления с аналогичным
кинетическим уравнением Михаэлиса— Ментен:
°=штт- (1аз8>
В табл. 12.2 приведен удобный для сопоставления перечень
„параметров, предположений и формул для описания
взаимодействий лекарственного препарата с рецептором и фермента с суб-
|)тратом. Из табл. 12.2 видно, что принципиальное различие (по-
349 РОСТ И РАСПАД
Таблица 12.2
Аиалогичные параметры, символы, предположения, уравнения и способы
графического анализа при исследованиях взаимодействий фермент — субстрат
и лекарственное вещество — рецептор
[ Взаимодействия фермент—субстрат3
Взаимодействия лекарственное
вещество—рецептор3
Обозначения
А — субстрат
Е — фермент
ЕА — фермент-субстратный
комплекс
\Р — продукты
v — начальная -стационарная
скорость реакции
I — ингибитор
D — лекарственное веществ»
(агонист)
R — рецептор
DR — комплекс лекарственного»
вещества с рецептором
Эффект—реакция, отклик
Ant — антагонист
Схема
взаимодействия
Е+A 5F
ЕА
«2
Кт
*■ Р + Е D + R т—^»- DR
Эффект
-1
Предположения Одна молекула субстрата иа
каталитическом участке
фермента
Только начальная скорость
(нет обратной реакции)
Графическое
представление
результатов
Формулы
Одна молекула лекарственного»
вещества на рецепторноь*
участке
Аналогичного предположени»
иет (ио см. «теорию
скоростей» далее в этой главе)
Связывание вещества с данны»
рецептором не зависит от
того, заняты ли уже соседние
рецепторы
ОбЩ
Эффект пропорционален [DR]
Эффект—>-Эффектмакс при»
i[DR]—-^[R]°6m
Эффект от [D] при постоянной
,''..'■/Л!''л <[R]
* 1/и от 1/[А], щщ|йретряниой (1/Эффект> от 1/[D] при по-
Мономерный фермент (нет
кооперативного влияния
связывания)
общ.
v пропорциональна [ЕА]
V »-7иакс При [ЕАрЛЙ(Е]общ
v от 1[А] при noct^ikaoH .[E]
,[Е]
о от lg[A] при
оянной [Е]
макс
[А]
Ка
стояниой [R]
•Эффект от lg[D] при
постоянной [R]
_.. Эффектмакс [D]
Эффект = ■
mm _ k.i+k2
*>-(У4ш*12\ при1ГА1 = АГА
KD =
[R1 PI *-i
PR] ~ kx '
dp»
Эффект=ЭффекТм»ке/2
Уравнения, описывающие конкурентное и неконкурентное инги-
бирование ферментов абсолютно аналогичны уравнениям»
описывающим конкурентный и неконкурентный антагонизм
при действии агонистических лекарственных препаратов
а Звездочкой *
тальных данных.
отмечены предпочтительные способы графического
■*А&.
,350 ЧАСТЬ 3
Молярная концентрация
{логарифмическая шкала)
:Рис. 12.5. Кривые, описывающие взаимосвязь логарифма дозы и отклика биоси-
хтемы при действии адреналина и норадреналина иа изолированную селезёнку
кошки '[5].
Адреналин характеризуется меньшей дозой EDit и, следовательно, является
(приблизительно в 2 раза) более активным лекарственным средством, чем норадреналин.
мимо иных наименований переменных) между кинетикой действия
•ферментов и взаимодействиями препарат—рецептор состоит в том,
каким графическим способом более выгодно представлять
полученные данные. Поскольку наибольший интерес для фармаколога
•представляют эффекты-при дозах, близких к Еййо^Кп—Дрза., при
которой эффект равен половине максимально возможного, обычно
используют зависимость эффекта от lg [D]. На рис. 12.5
показаны данные по действию адреналина и норадреналина,
вызывающих сокращение (в миллиметрах) изолированной полосы ткани
селезенки кошки. Кривые подобны кривым титрования;
сравнивая точки перегиба на этих кривых видно, что адреналин является
•более активным и его сродство при связывании почти в два раза'
^превышает сродство норадреналина. Поскольку в обоих случаях
.вид зависимостей аналогичен, можно предположить, что оба
препарата действуют по общему механизму. (Одна из причин того,
что среди лиц, использующих психотропный препарат LSD, так
часто бывают передозировки, состоит в том, что этот препарат
характеризуется исключительно малой EDso, составляющей
~ 1 мкг на 1 кг массы тела.)
Аналогию между взаимодействиями препарат^ рецептор и
^фермент — субстрат можно расширить, включив случаи
конкурентного и неконкурентного ингибирования и большинство
других ситуаций (гл. 10 и 11). В качестве типичного примера на
рис. 12.6 показан случай конкурентного ингибирования
(антагонизма) антигистамином — димедролом эффекта гистамина, действие
^которых оценивалось с помощью зависимостей типа Лайнуивера —
"Берка (в координатах [(1/Эффект) — l/[-D]]; измерялось падение
давления крови, вызываемое гистамином. Приведенные
зависимости, полученные при разных концентрациях димедрола,
пересекаются в точке, лежащей на оси ординат, что свидетельствует о
конкурентном связывании.
Согласно предыдущей модели, молекула препарата
непрерывно оказывает свое действие в течение всего времени, которое она
351 РОСТ И РАСПАД
Зрдккк.
О 0,25 0,50 0,75
1/[Гистаминдифосфат] (мкг /кг)
Рис. 12.6. Зависимость Лайнуивера — Берка, описывающая взаимосвязь между
обратной величиной отклика организма » обратной величиной концентрации ан<-
тагониста при падении давления крови, вызываемом у собак введением гистами-
на '[6].
Измерения реакции организма проводились в присутствии только гнстамииа, а также в-
присутствии разных (фиксированных) концентраций препарата антагониста димедрола.
Дозы препарата указаны на I кг массы тела испытуемого животного. Данные свидетельству-,
ют о конкурентном антагонизме (см. текст).
находится на рецепторном участке. Это одна точка зрения.
Другая точка зрения заключается в том, что воздействие препарата
происходит только в момент его связывания с рецептором
(например, как в случае появления импульсного потенциала действия,,
сопровождающего перенос нейромедиаторов на их рецепторы^ в*
постсинаптической мембране); эта модель служит основой reopj^
скоростей взаимодействия лекарственных препаратов с ре.цщщ^
рами: . '?№&?
Эффект^ (1 — f) [D] -const, 'ЩЩ-
так что эффект пропорционален скорости образования коьщщ4шш..
DR и описывается той же основной кинетической схеы<Ш$ЩШ
в которой (1—/)—доля рецепторов, не связанных с\;'пщ'*
том D. Поскольку скорости ассоциации и диссоциация, к0л|
DR при равновесии равны, то Q>#;.;£*0?f% -''■
Эффект=const • ft.J. /'>' ;>( ■•' ' (12. Щ
Кроме того; при [D]»[R]o6m И{0]/(Ло^Й^)*^ак-'что
352 часть э
Уравнение теории скоростей (12.20) описывает ту же самую
функциональную связь между эффектом и [D], как и теория
занятости [уравнение (12.17).]. Таким образом, модель
взаимодействия препарат — рецептор приводит к соотношению между [D]
и эффектом, абсолютно аналогичному соотношению между [А] и v
в ферментативной кинетике независимо от того, обусловлен ли
эффект препарата просто долей занятых рецепторных участков
-или скоростью связывания молекул препарата с рецептором.
Биологическое значение модели взаимодействия препарат —
рецептор зависит от доказательства реального существования
рецепторов, так же как модель фермент-субстратного
взаимодействия основана на доказательстве существования ферментов. В
последнее время получены доказательства существования
рецепторов, гормонов, число которых продолжает расти; некоторые из этих
рецепторов выделены: ацетилхолиновый рецептор, рецепторы
различных'половых гормонов и рецептор холерного токсина. Во
многих случаях сам рецептор представляет собой фермент, и
действие препарата состоит просто в ингибировании этого фермента с
соответствующими физиологическими последствиями. Наиболее
наглядно эффекты обоих типов демонстрируются данными,
полученными при изучении механизма действия различных
лекарственных препаратов, влияющих, на настроение и психическое состояние.
Пример. Психотропные фармакологические средства и физиологическое
действие катехоламинов
Огромное значение психотропных фармакологических средств становится
очевидным при рассмотрении данных, приведенных на рис. 12.7. Согласно этим
данным, существенное снижение числа пациентов в психиатрических больницах
(несмотря, на общий рост численности населения и. улучшение способов диагно*
сгикн психических расстройств) совпадает по времени с внедрением в практику
Я(ервого производного фенотиазина антиисихотического действия (хлорпромази-
ма). Действие этого препарата и огромного числа различных аитипсихотических
лекарств других типов легче всего понять, зная основные пути физиологического
действия симпатических нервных окончаний (рис. 12.8).
ХЛорпромазин и другие производные феиотиазина оказались очень
полезными для облегчения таких психических расстройств, как шизофрения и
различные типы маниакального состояния, а также хронических галлюцинаций и пута-
цости сознания. Транквилизаторы этого класса, по-видимому, действуют, блокируя
«ам рецептор (в данном случае медиатором служит адреналин, а не норадрена-
дин). К числу используемых антипсихотических транквилизаторов относится также
резерпин, основное действие которого состоит в том, что он истощает норадрена-
Лии, вызывая его выделение из пресннаптических везикул, в которых он хранится,
с дальнейшим быстрым разрушением его моиоаминооксидазой. К другой группе
:<&нтидепрессантов относится ряд трициклических соединений, как, например, ими-
^нн, блокирующий вторичное связывание медиатора норадреналина и,
следовательно, увеличивающий стимуляцию постсинаптнческих нервных волокон и
приводящий к накоплению медиатора в синапсе. (Фенамин также действует в роли
стимулятора, блокируя связывание норадреналина). И наконец, еще к одному классу
^нтидепрессантных препаратов относится группа ингибиторов моноаминооксида-
■Mt (МАО), таких, как, например, нпразид (больше не используется) и феиа-