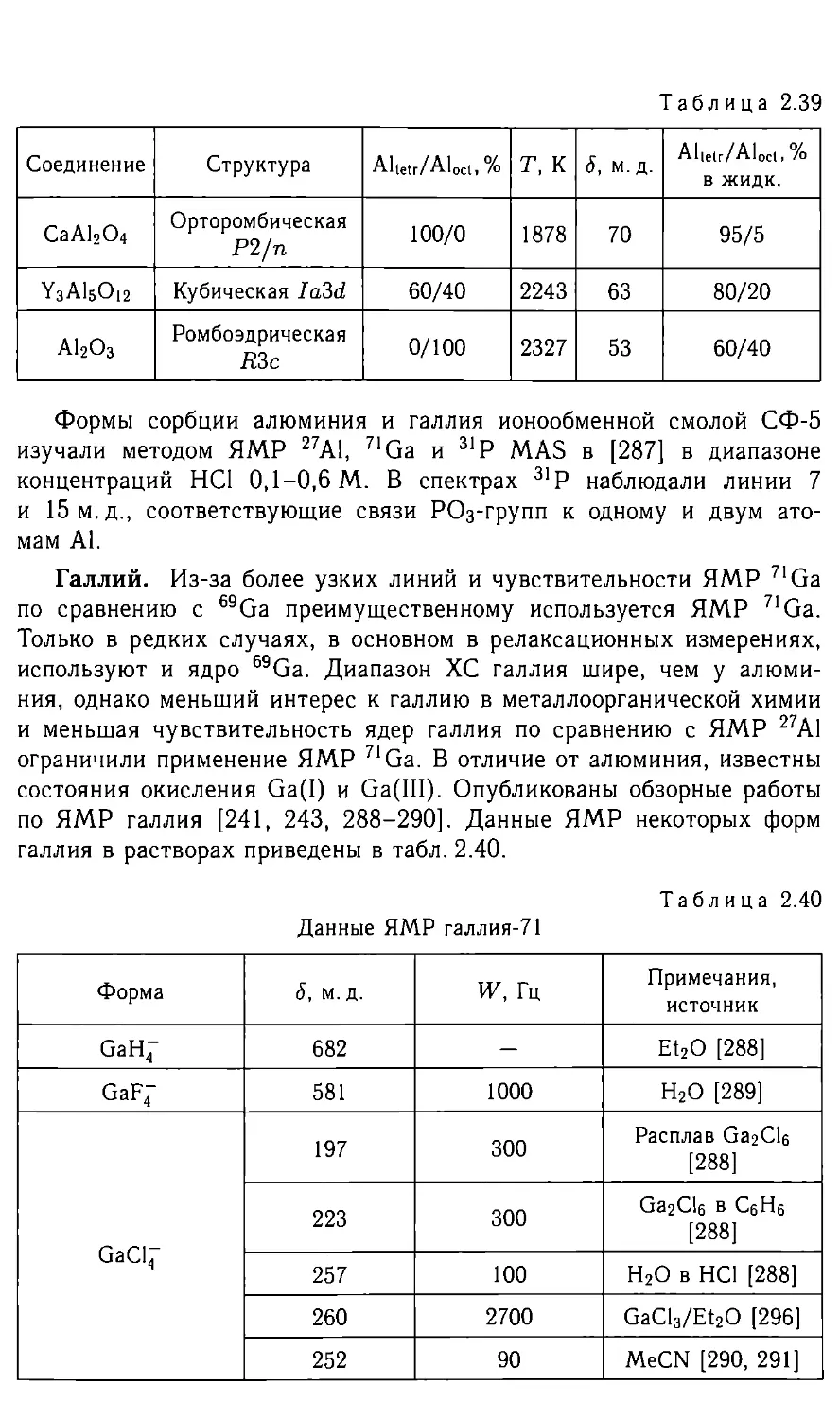
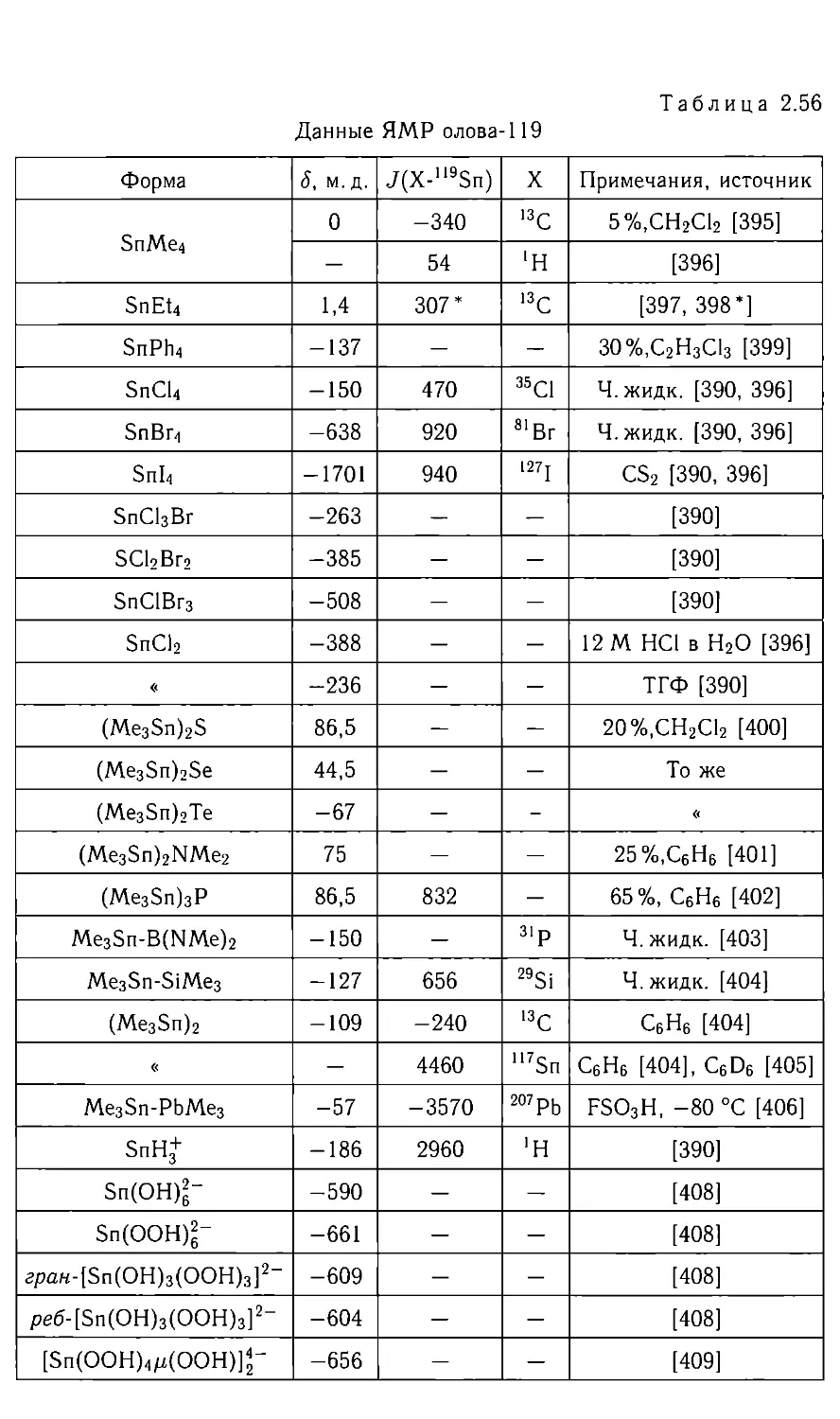
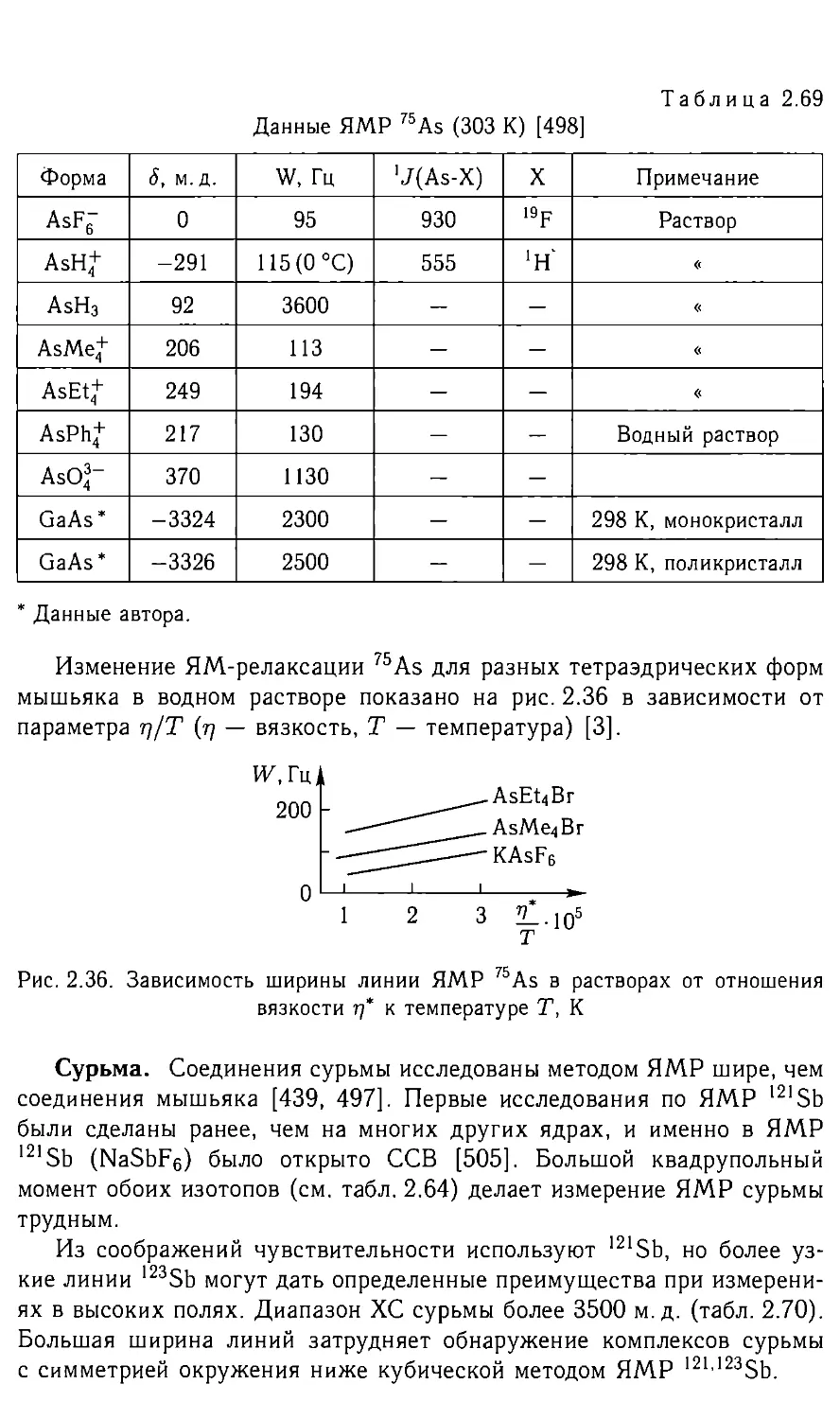
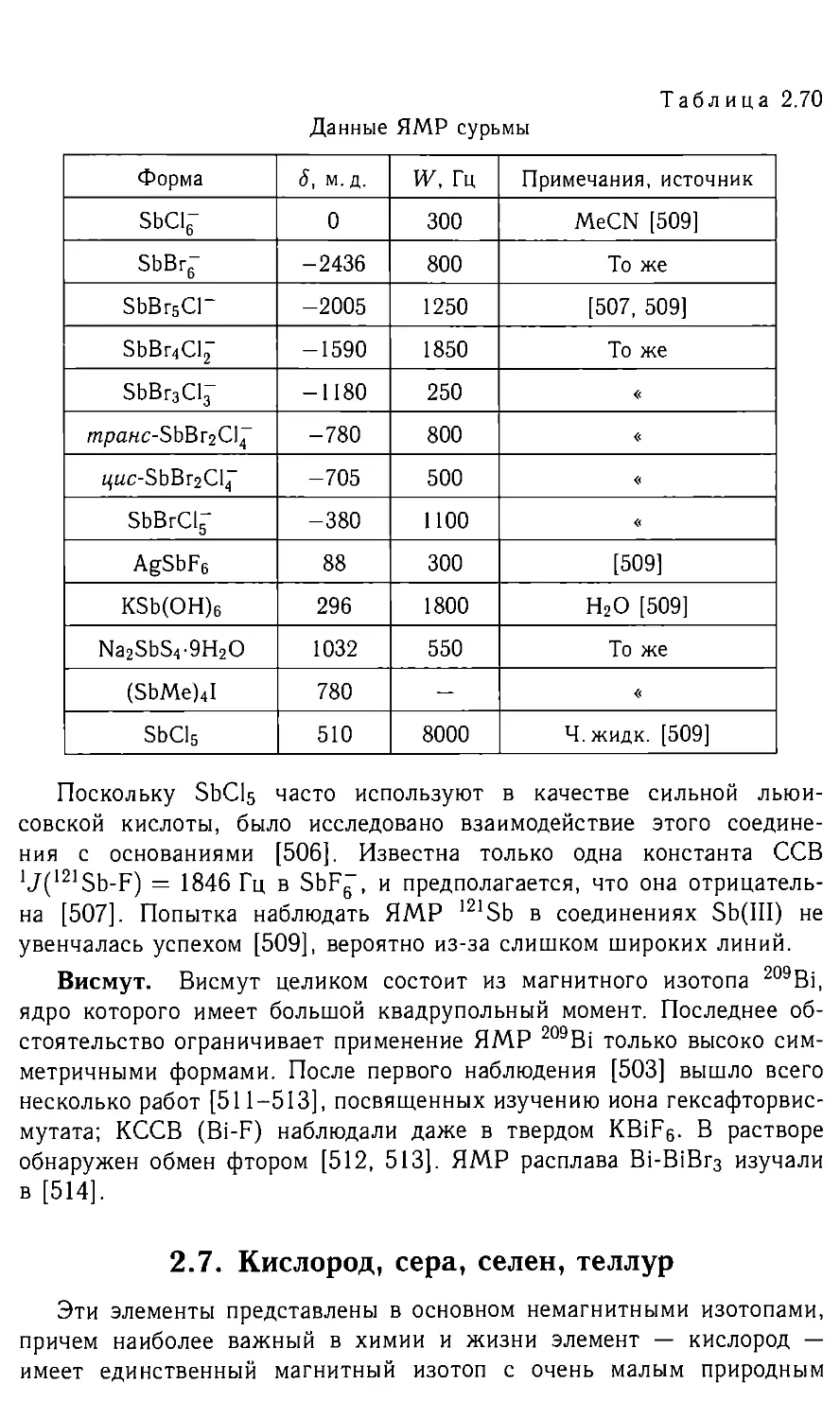
/



















































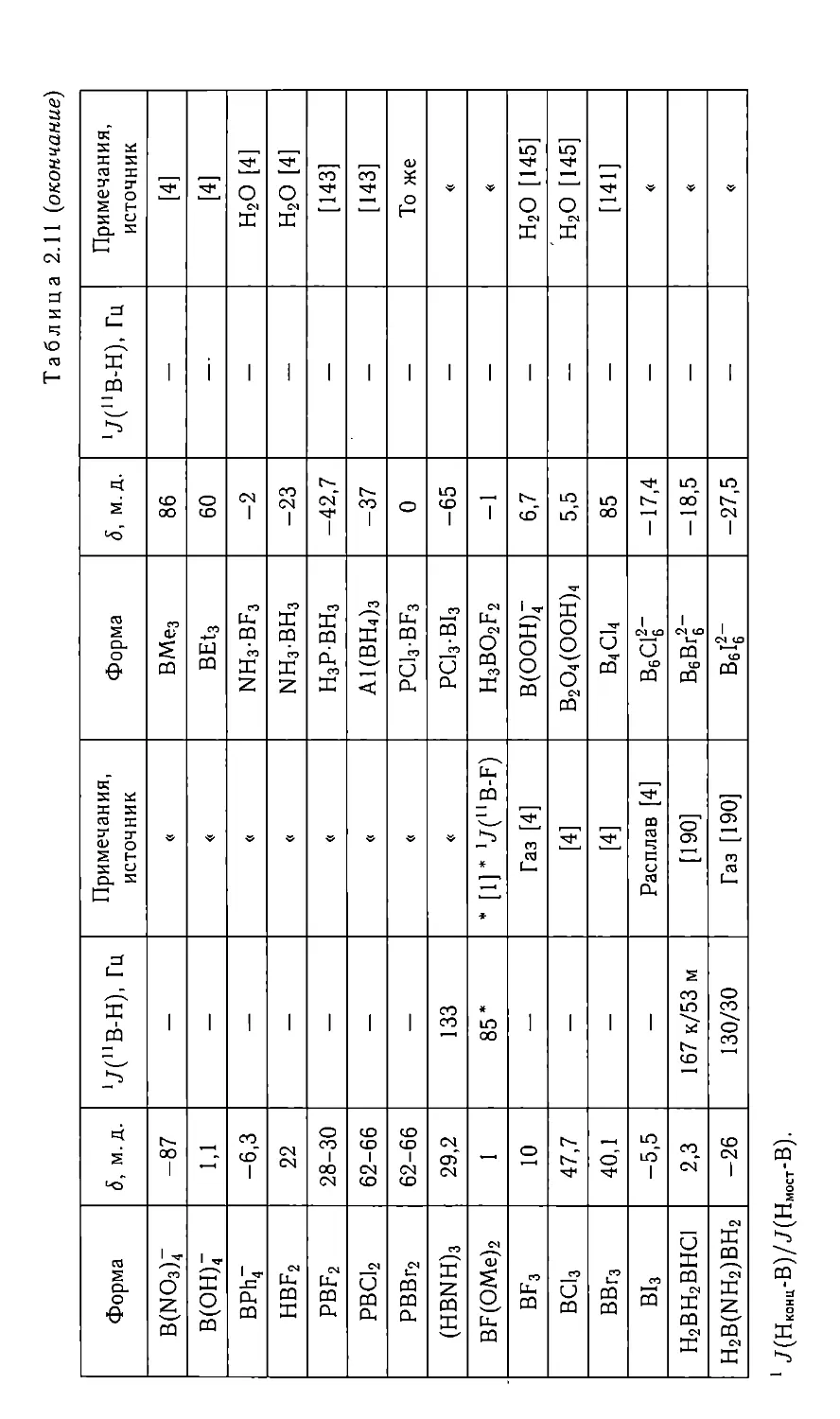



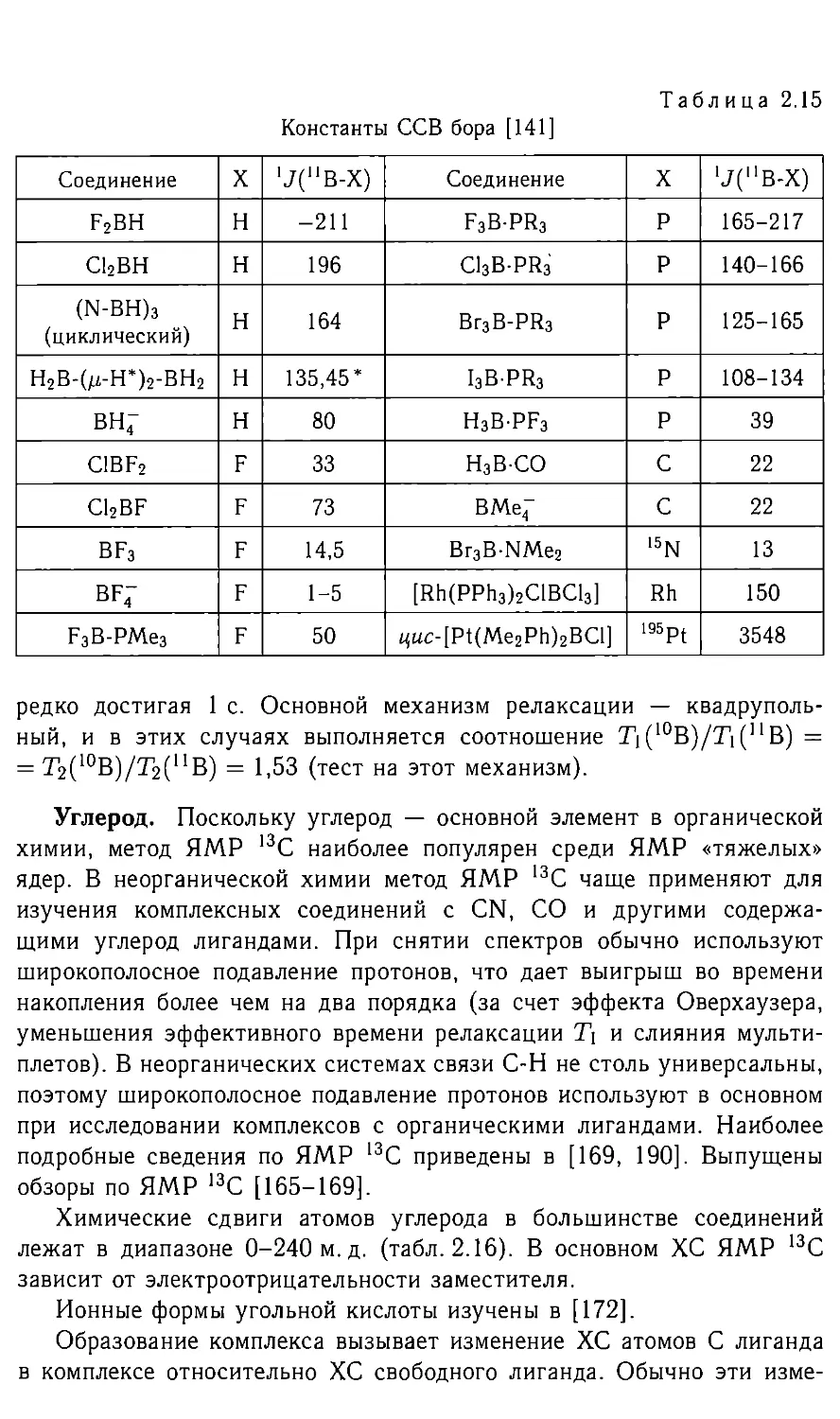














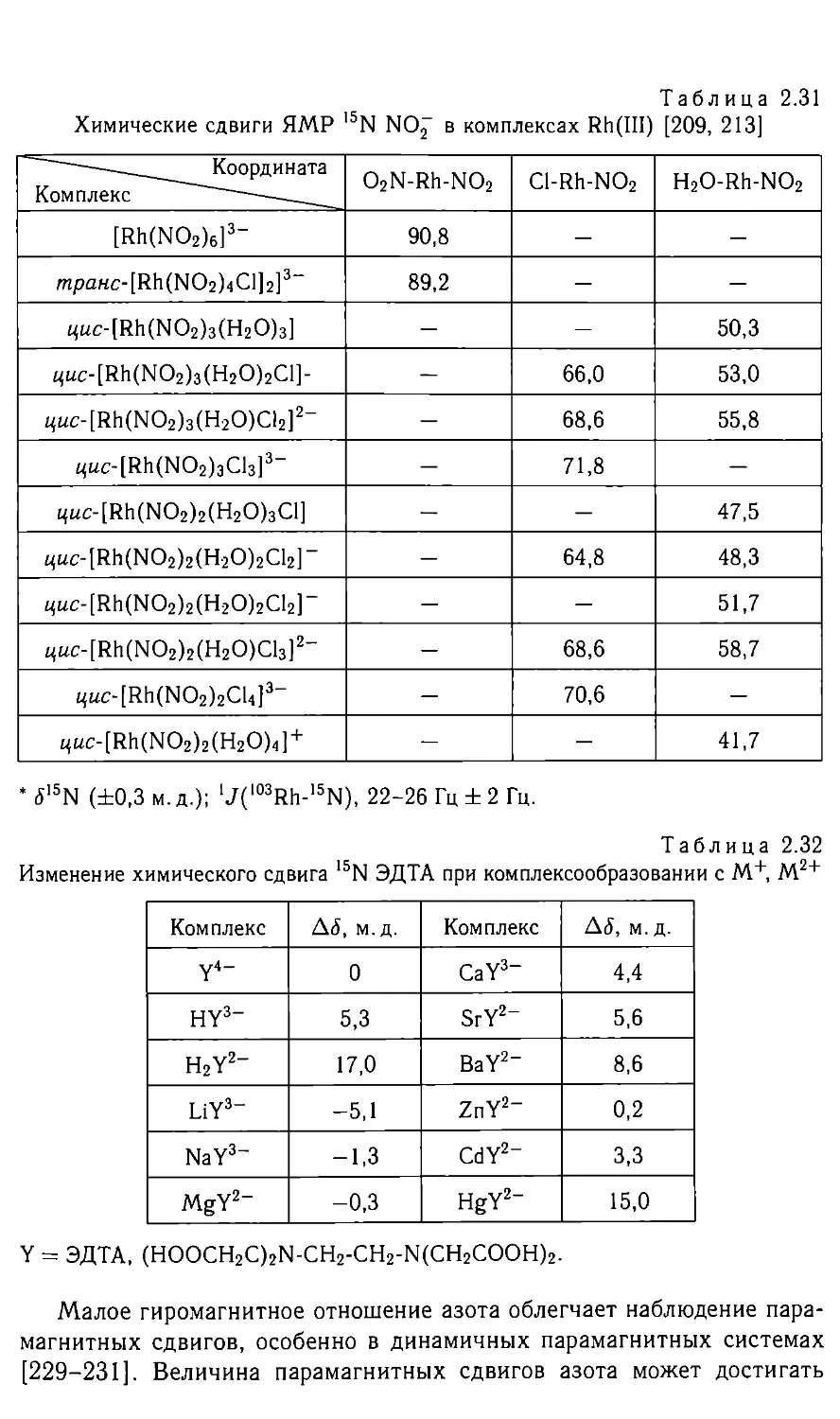

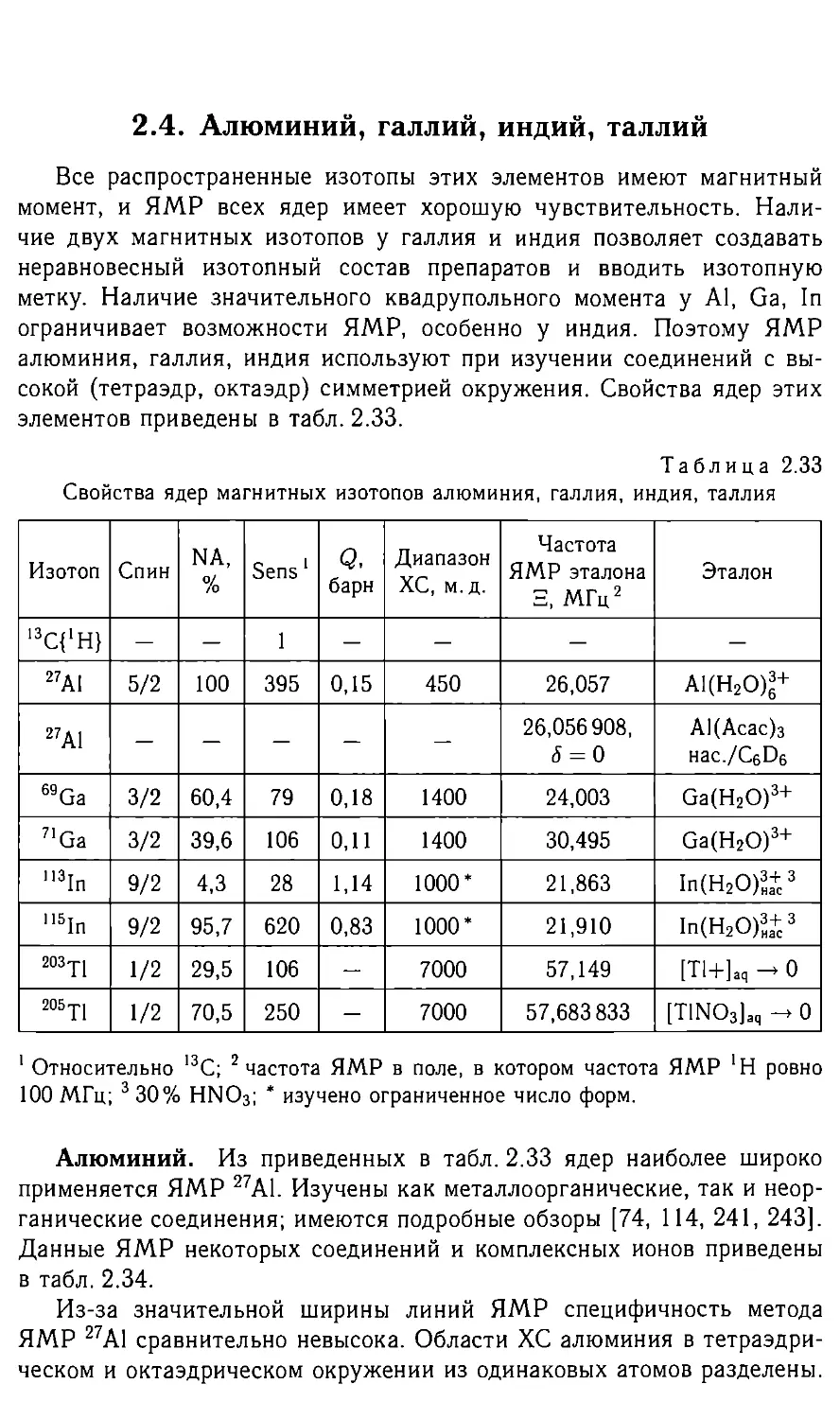












































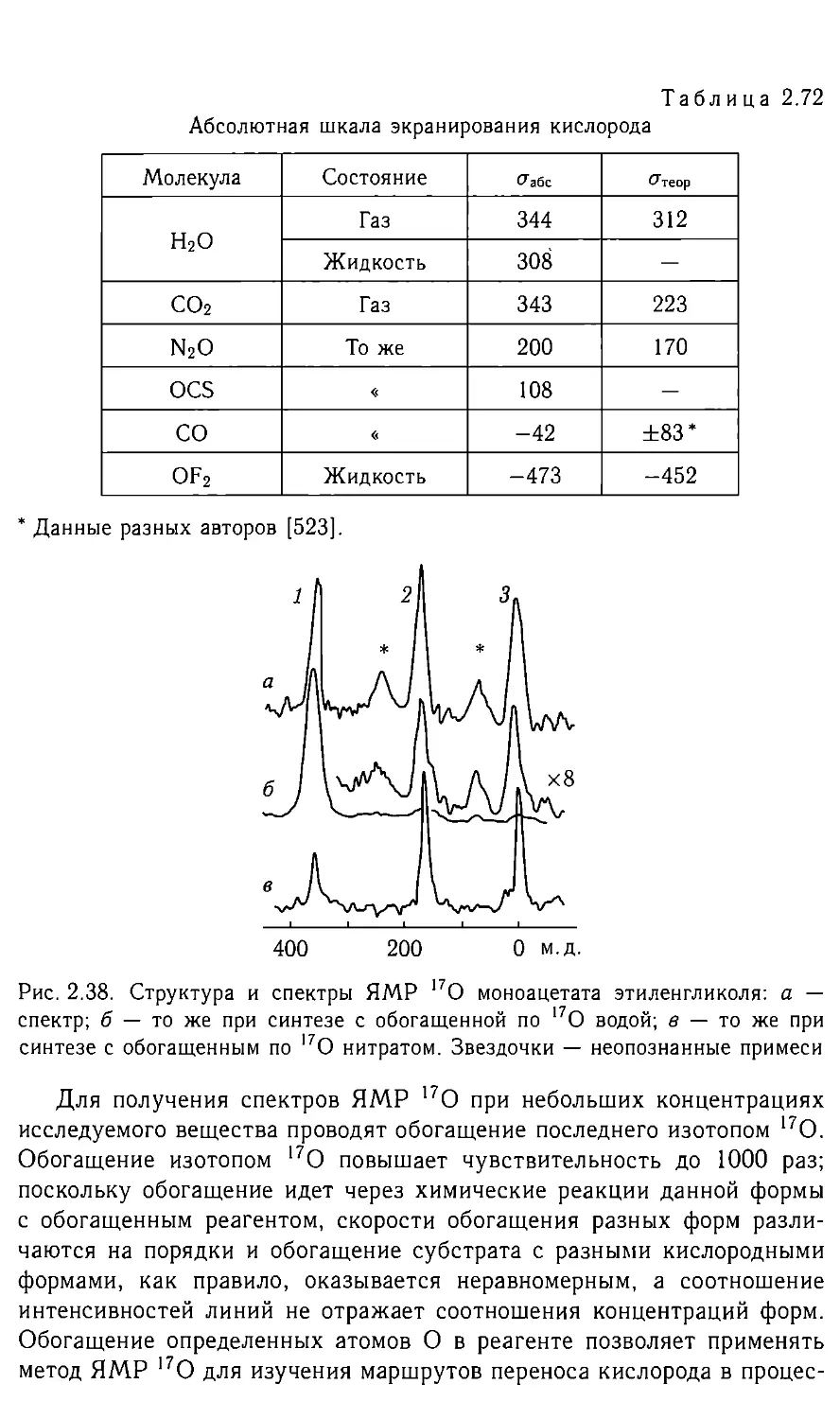


























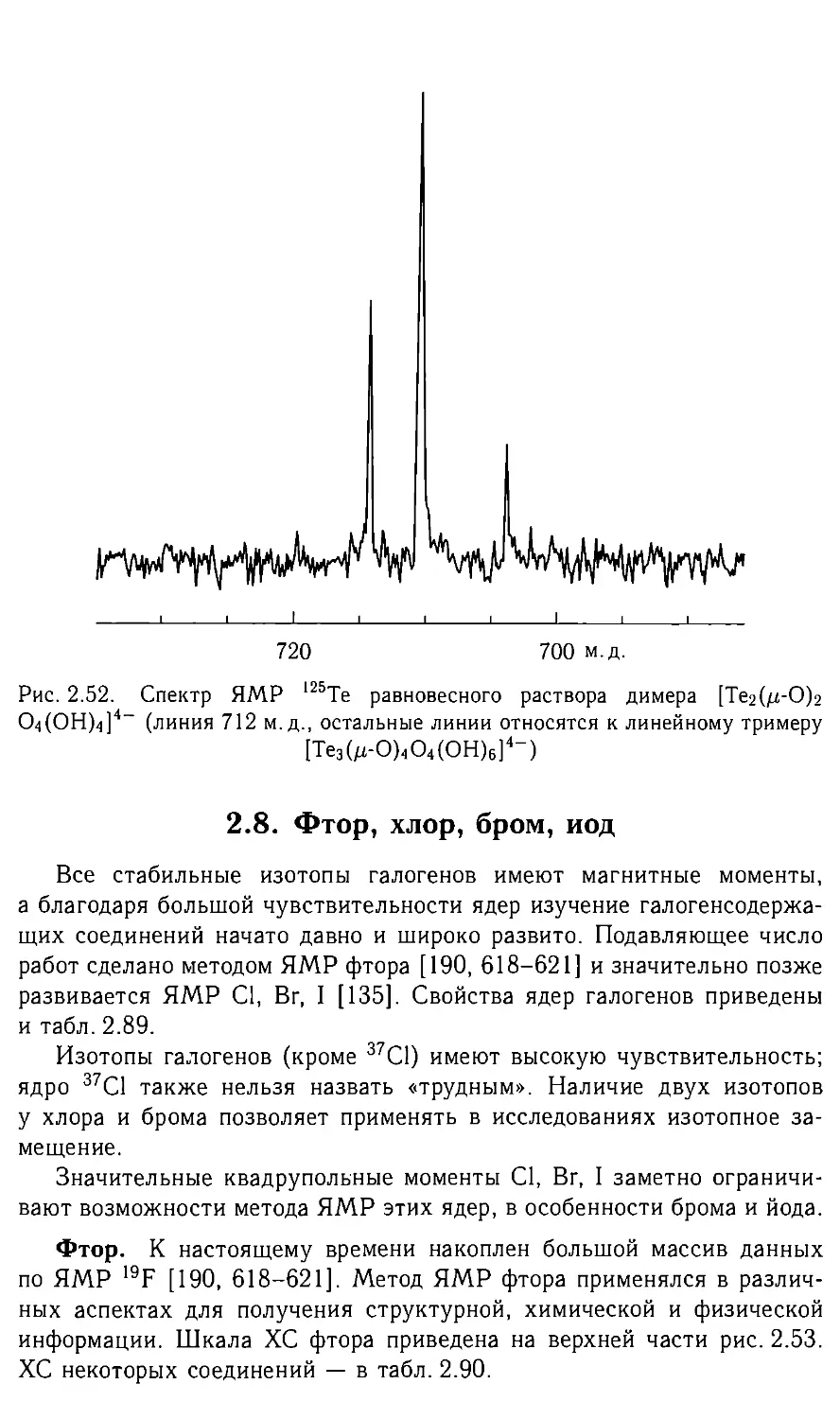

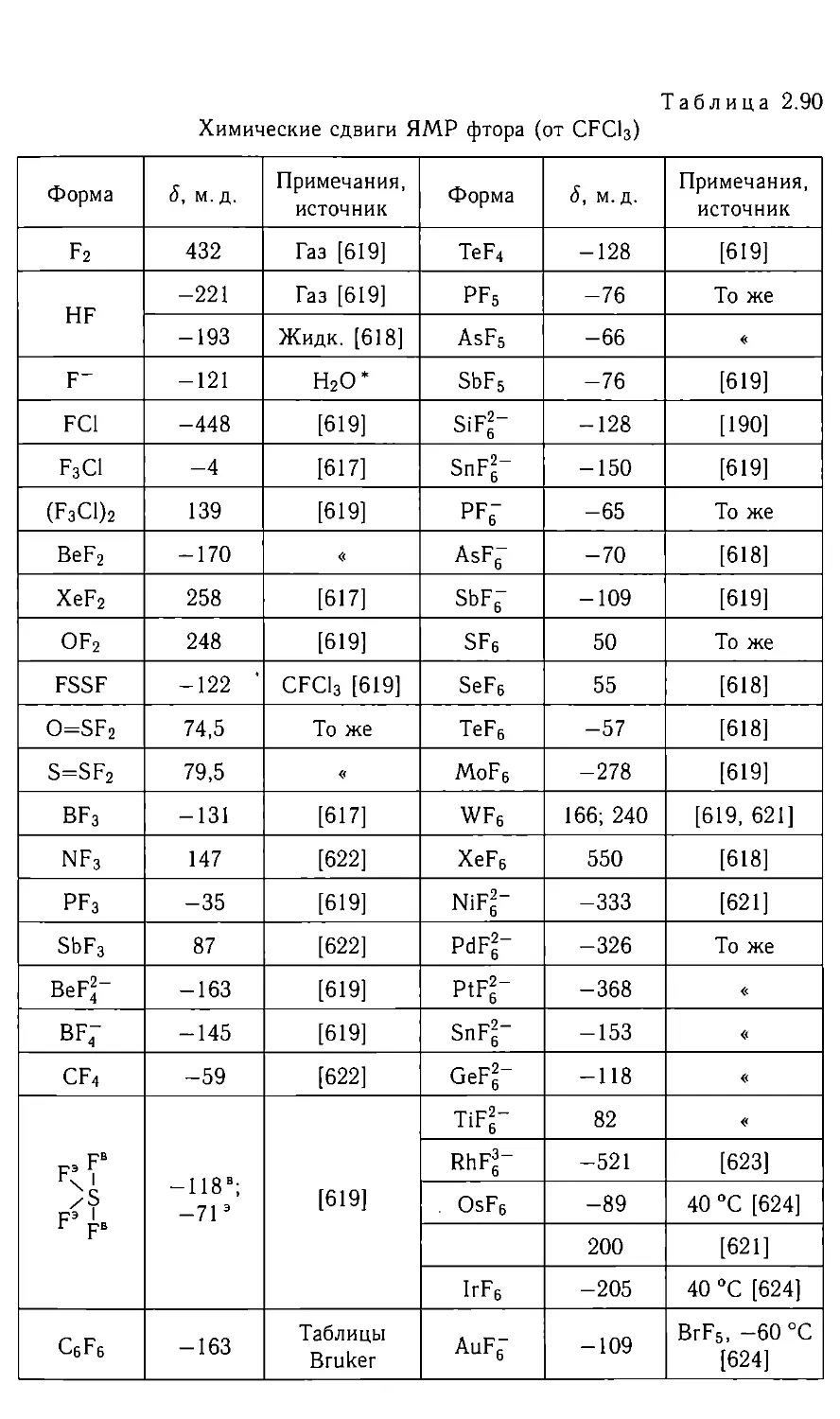






















































































































































































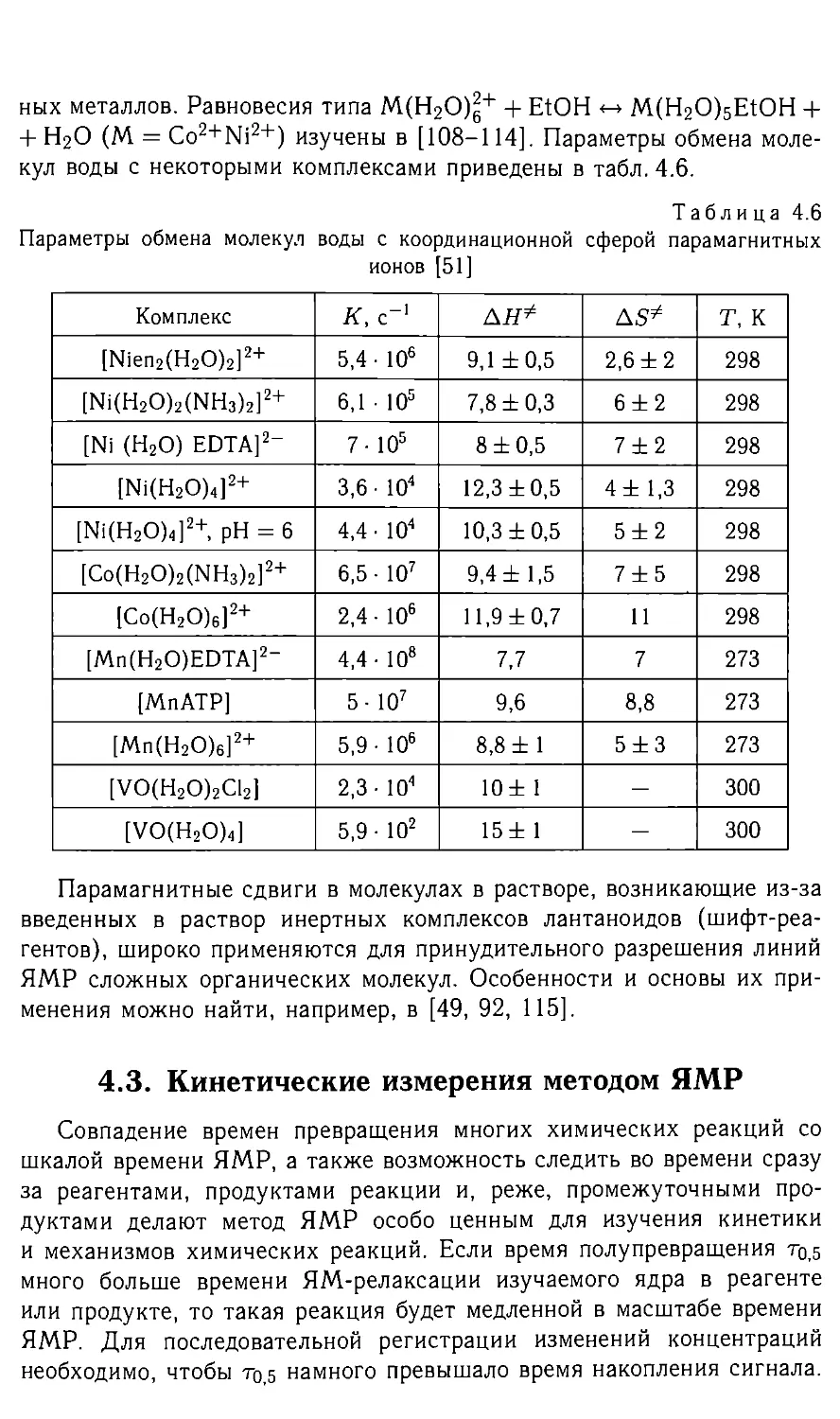





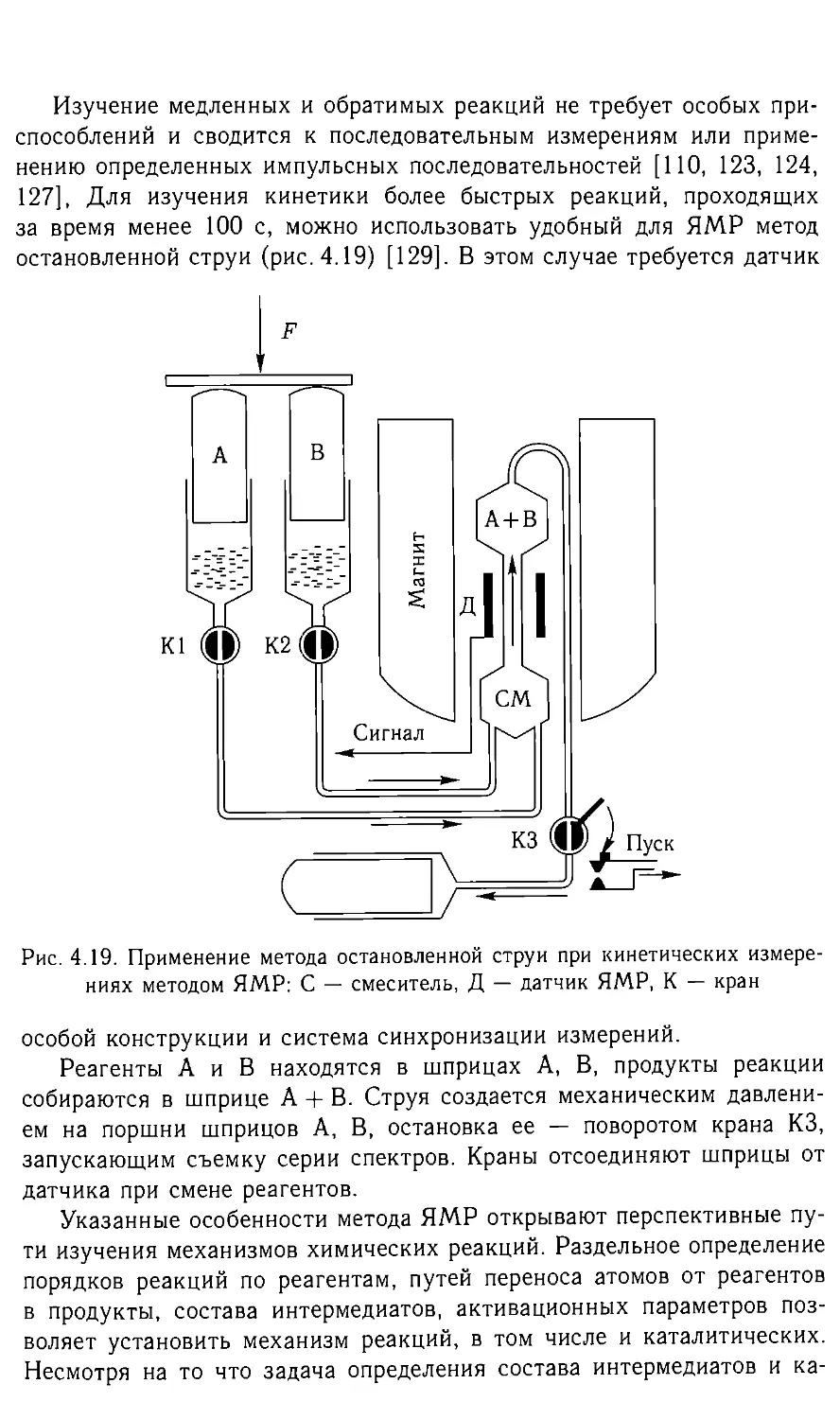



















Автор: Федотов М.А.
Теги: физика химия магнитный резонанс ядерный резонанс растворы и жидкости координационная химия
Год: 2009




Текст
М.А. ФЕДОТОВ
ЯДЕРНЫЙ
МАГНИТНЫЙ
РЕЗОНАНС
в неорганической
и координационной
химии
М. А. Федотов
ЯДЕРНЫЙ МАГНИТНЫЙ
РЕЗОНАНС
В НЕОРГАНИЧЕСКОЙ
И КООРДИНАЦИОННОЙ ХИМИИ
(растворы и жидкости)
ПРЕДИСЛОВИЕ
В 70-е гг. XX столетия произошел качественный скачок в технике
спектрометров ядерного магнитного резонанса (ЯМР); возможности
метода ЯМР многократно возросли и, как следствие, резко расшири-
лись области применения метода в химических исследованиях. К этому
привели: разработка импульсных спектрометров ЯМР с детектирова-
нием сигнала ЯМР после импульсного возбуждения спиновой системы
ядер, открытие алгоритма быстрого фурье-преобразования, развитие
электроники и появление мини-ЭВМ (что позволило разработать мно-
гоядерные и многоцелевые импульсные спектрометры ЯМР с фурье-
преобразованием), создание магнитов со сверхпроводящими соленоида-
ми (криомагнитов, что позволило получить поля свыше 20 Т, а малый
расход гелия определил их повсеместное применение). В результате
чувствительность метода возросла в тысячи раз, резко расширились
методические возможности, импульсные спектрометры с криомагнита-
ми стали рабочим инструментом химической лаборатории.
Если к началу 70-х годов ЯМР был рабочим инструментом для
органиков и химиков, работающих в химии фтора с использованием
ядер *Н, 19F, 31Р (измерения на других ядрах были экзотическими), то
с 70-х стали возможны измерения на ядрах большинства элементов,
и метод ЯМР тем самым становится рабочим инструментом и для
химиков-неоргаников. Среди других физических методов метод ЯМР
является наиболее характеристичным (избирательным) хотя бы пото-
му, что он — чисто изотопный метод и в ЯМР наблюдают только
выбранный изотоп; избирательность в наибольшей мере проявляется
при исследовании жидких образцов.
Методом ЯМР изучают вещества, которые содержат изотопы с яд-
рами, обладающими магнитным моментом (магнитные изотопы). Такие
изотопы есть почти у каждого элемента, однако свойства их ядер
(величина магнитного момента, наличие и величина квадрупольного
момента) и их взаимодействие с электронами в соединениях неодина-
ковы, поэтому возможности метода ЯМР резко различаются даже для
изотопов одного и того же элемента.
Изданные в СССР книги по ЯМР предназначены в основном для
химиков-органиков и, кроме основ метода, содержат сведения только
о ЯМР основных ядер, входящих в органические молекулы (|Н, 13С,
31Р, 15N). В постсоветское время литература по ЯМР неорганических
форм в жидкой фазе практически не издавалась. В то же время стре-
мительно расширяющаяся область применения спектроскопии ЯМР
в неорганической химии, а также постепенное обновление парка спек-
трометров ЯМР в России требует обеспечения специалистов, особенно
начинающих, соответствующей литературой, и данная книга призвана
уменьшить образовавшийся дефицит.
СПИСОК ОБОЗНАЧЕНИИ
7 — гиромагнитное отношение
<5 — химический сдвиг в шкале <5
Hq — напряженность постоянного магнитного поля
Н\ — напряженность высокочастотного магнитного поля
I — спин ядра
ио — частота ЯМР
— частота ядерного квадрупольного резонанса
сг — экранирование ядра
W — ширина линии ЯМР
Асас — ацетилацетонат
aq — акватированный
ах — аксиальный
Ви — бутил
Ср — циклопентадиенил
dppe — 1,2-бис(дифенилфосфино)этан
Cit — цитрат
еп — этилендиамин
Et — этил
NA — природное содержание изотопа
ох — оксалат
гРг — изопропил
Me — метил
Ph — фенил
Ру — пиридин
Pz — пнразил
Sens — чувствительность с учетом природного содержания
Tart — тартрат
АТФ — аденозинтрифосфат
верш., в — вершина
ГМФА — гексаметилфосфортриамид
гран — граневой изомер
ГЭП — градиент электрического поля
КССВ — константа спин-спинового взаимодействия
м. д. — миллионные доли
мост. — мостиковый
нас. - насыщенный
нас. р-р — насыщенный раствор
осн. — основание
реб — реберный изомер
ССВ — спин-спиновое взаимодействие
ССИ — спад свободной индукции
тв. — твердый
ТГФ — тетрагидрофуран
ТК — температурный коэффициент
ТМС — тетраметилсилан
ТМФ — триметилфосфат
транс — транс-изомер
ХС — химический сдвиг
центр. — центральный
цис - цис-изомер
ч.жидк. — чистая жидкость
ФВК — фосфор вольфрамовая гетерополикислота
э — экватор
ЭДТА — этилентетраацетат
ЭПР — электронный парамагнитный резонанс
ЭСП — электронные спектры поглощения
Глава 1
ЯДЕРНЫЙ МАГНИТНЫЙ РЕЗОНАНС -
МЕТОД И ЭКСПЕРИМЕНТ
1.1. ЯМР в жидкой фазе
Ядерный магнитный резонанс (ЯМР) является неразрушающим
и универсальным по отношению к фазовому состоянию методом ис-
следования свойств соединений и веществ. Однако методика исследо-
вания, равно как и получаемая информация, существенно зависят от
агрегатного состояния вещества (твердого — аморфного или кристал-
лического), жидкого, жидкокристаллического, адсорбированного или
газообразного. Отличие ЯМР в жидкостях (газах) от твердофазного
ЯМР возникает из-за быстрого хаотичного движения молекул, которое
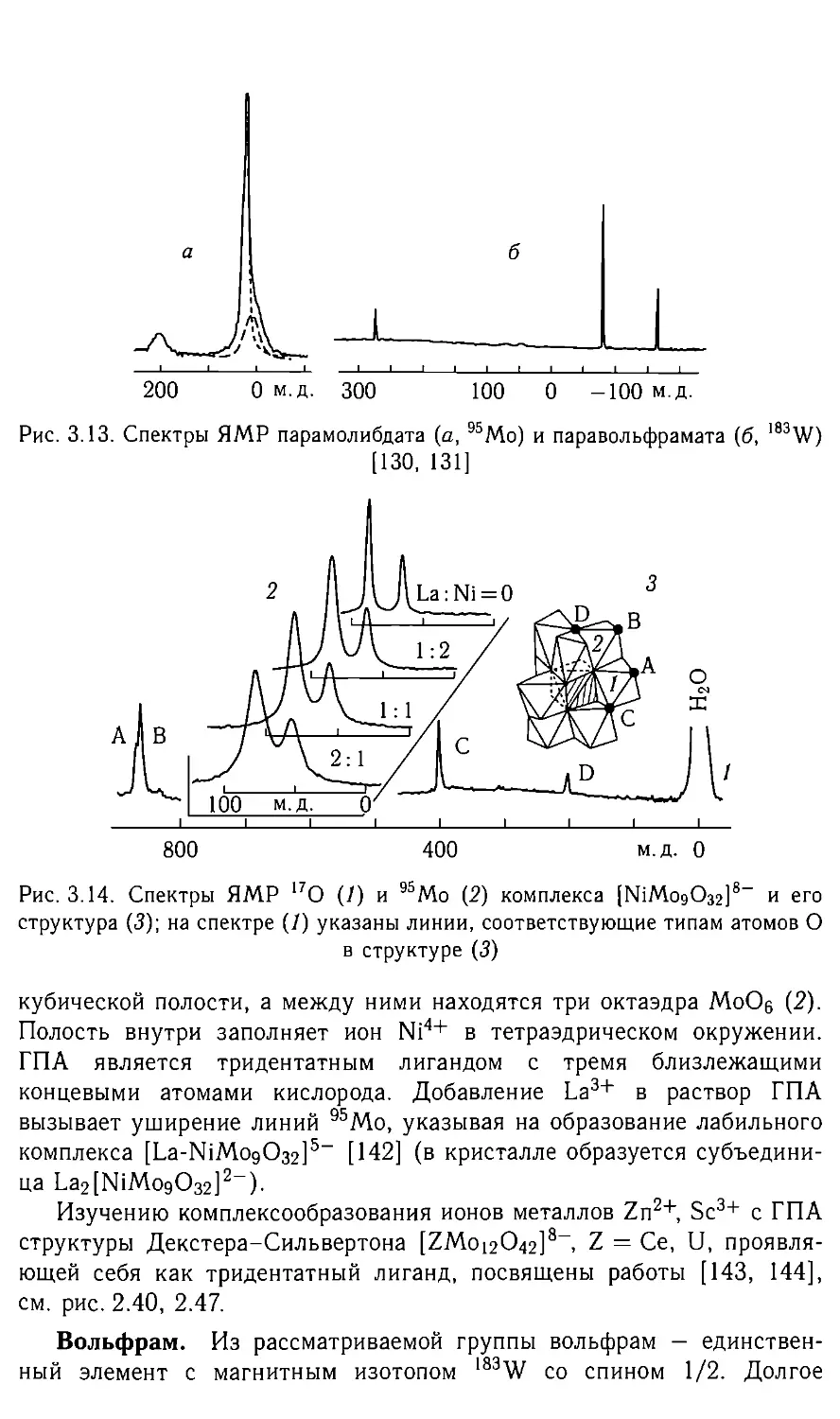
усредненяет как компоненты тензора химического сдвига, так и ди-
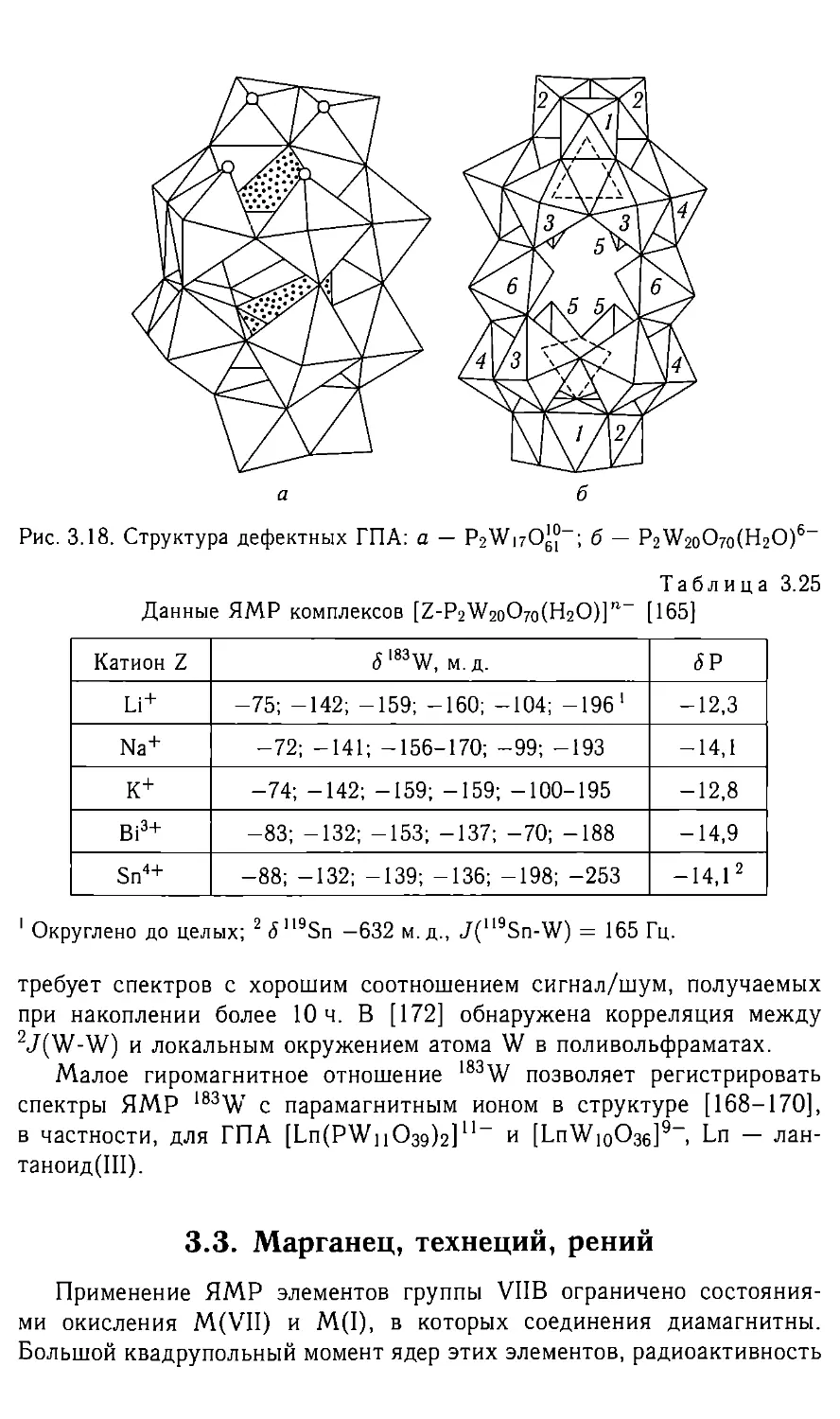
поль-дипольные и квадрупольные взаимодействия. При этом спектры
ЯМР существенно упрощаются по сравнению со спектрами твердого
тела (с соответствующей потерей определенной информации), а линии
ЯМР сужаются. Свойства магнитных ядер приведены в [1, 2].
Возможности спектрального метода определяются чувствитель-
ностью (минимальным количеством вещества, требуемого для полу-
чения информативного спектра), и специфичностью (способностью
раздельно регистрировать определенные состояния вещества, молекул,
атомов). Специфичность не тождественна разрешающей способности
метода, поскольку последняя в значительной мере зависит от уровня
техники в данной области, в то время как специфичность характеризу-
ет возможности метода, ограниченные только природными свойствами
измеряемых величин.
Для метода ЯМР показателем специфичности может служить от-
ношение диапазона химических сдвигов данного ядра при разных
окружениях к типичной для него ширине линий ЯМР. Для разных
ядер специфичность значительно различается, но и при самой низкой
специфичности, как правило, наблюдаются ядра только одного изотопа.
При очень узких линиях ЯМР специфичность ограничена разрешением
вает чувствительность и специфичность метода и позволяет улавливать
тонкие различия химического окружения и электронной структуры
атомов. Мультиплетность линий ЯМР показывает тип и количество
магнитных ядер, связанных с данным ядром. Спектр ЯМР в жидкой
фазе характеризуется следующими параметрами: положением, мульти-
плетностъю, шириной и интенсивностью линий.
Химический сдвиг (ХС) в основном связан с окружением ядра в
молекуле (комплексе); он возникает из-за экранирования постоянного
магнитного поля Hq на ядре электронами, участвующими в химической
связи. Из-за этого поле на ядре немного меньше, чем приложенное
к образцу:
Яя = Я0(1-а), (1.1)
а величина <т называется экранированием. Она безразмерна и невели-
ка и измеряется обычно в миллионных долях (м.д.). Экранирование
электронами, не участвующими в химической связи (заполненными
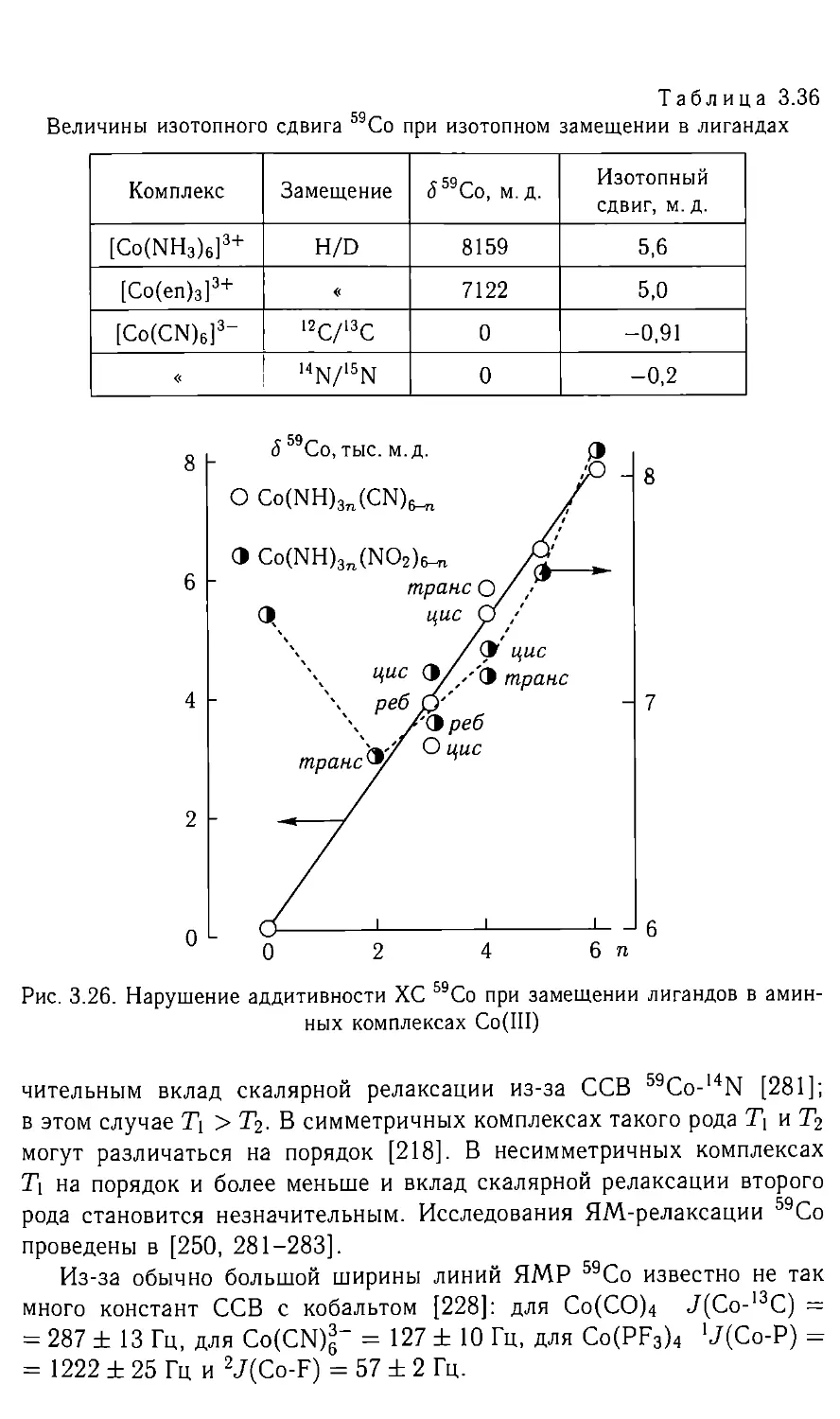
электронными оболочками) постоянно с точностью до единиц м.д.
для всех соединений данного элемента, хотя оно возрастает к концу
Периодической таблицы до 1500 м.д. [3].
Часто разделяют два вклада в ХС: внутримолекулярный (который
обычно преобладает) и межмолекулярный. Первый вклад зависит толь-
ко от окружения данного ядра в молекуле (комплексе). Межмолекуляр-
ный вклад обусловлен взаимодействием с другими молекулами (он мо-
жет стать преобладающим, если соседняя молекула парамагнитная).
В свою очередь, внутримолекулярный вклад делят на парамагнитный
и диамагнитный; и тот, и другой имеют локальный и нелокальный
вклады:
О- = a,doc + <4,n + afoc + ofon • (1.2)
Локальные вклады возникают из-за электронных токов, локали-
зованных в атоме с изучаемым ядром, нелокальные — от токов на
соседних атомах и делокализованных токов, например, кольцевые токи
в молекуле бензола. Для диамагнитного вклада у атомов в s-состоянии
предложена формула [4, 5]
2 z
<‘-3>
тС
к
где rk — расстояние к-ro электрона от ядра, z — заряд ядра, т —
масса электрона, С — скорость света, (г^1) — усредненное значение
величины. Изменение диамагнитного вклада определяет ХС протонов,
а для ядер тяжелее лития ХС определяется изменением парамагнитно-
го вклада, обусловленного нарушением симметрии электронного рас-
пределения и дающего отрицательный вклад в экранирование. Для
качественной оценки парамагнитного локального вклада используют
выражение [5, 6]
” = 2^’ (14)
где fj-o — магнитная проницаемость вакуума, //в — магнетон Бора,
(г-3) — усредненная величина куба обратного радиуса валентных
электронов, Q — член, учитывающий асимметрию валентной оболочки,
Д.Е — эффективная энергия возбуждения, являющаяся усредненной
величиной энергии возбуждения каждого электрона AEi. При этом
необходимо учитывать (г-3) и ДЕ, всех электронов, образующих
связь. Преобладающий вклад в ДЕ дают низколежащие переходы,
энергию которых можно определить из оптических спектров, и, соглас-
но (1.4), можно найти зависимость ХС от энергий низших переходов.
Такие зависимости известны для ядер 17О, 59Со, I03Rh и др.
С ростом электроотрицательности лиганда заряд координирующего
атома становится более положительным, (г“3) и дезэкранирование
(уменьшение экранирования) увеличиваются. На этом основаны кор-
реляции ХС и электроотрицательности заместителя, ХС и плотности
заряда на ядре. Члены SQ и ДЕ не являются независимыми. С ро-
стом асимметрии окружения уменьшается разделение уровней ДЕ, т. е.
члены SQ и ДЕ могут влиять на экранирование в одном направле-
нии. Удовлетворительные количественные расчеты экранирования пока
возможны для небольших молекул, и для углерода, азота, кислорода
и фтора они показывают [4], что вкладом можно пренебречь,
а вклад ст£оп может достигать нескольких процентов от и afoc.
Вклады <7|оС и близки по величине, но противоположны по знаку.
Для приведенных ядер вклад почти не зависит от окружения (для
13С в молекулах CH3CN, СО, С2Н2, HCN меняется всего от 259,1 до
260,6 м.д.) и изменение ХС определяется в основном членом af .
Спин-спиновое взаимодействие. При наличии в молекуле
нескольких ядер с ненулевым спином между последними возникает
косвенное спин-спиновое взаимодействие (ССВ), приводящее к рас-
щеплению линий ЯМР на мультиплеты. Впервые мультиплетность
была обнаружена в спектре ЯМР 121 Sb соединения NaSbFe [7].
ССВ наблюдается как между разными, так и одинаковыми, но
неэквивалентными ядрами в молекуле, причем не только через одну,
но и несколько связей. В том случае, когда спины взаимодействующих
ядер антипараллельны, знак ССВ считают положительным, а при
параллельности спинов — отрицательным. При взаимодействии ядер А
и X линия ЯМР ядра А расщепится на 2/х + 1 компонент, где
1Х — спин ядра X. Аналогично, линия ЯМР ядра X расщепится на
27а + 1 компонент, где 1А — спин ядра А; константа ССВ J(A-X)
(КССВ) в спектрах ЯМР ядер А и ядер X для данной молекулы
одинакова. В зависимости от наличия ССВ и соотношения между
разнесением линий А и X и КССВ J(A-X) спектры ЯМР делят
на спектры нулевого порядка (отсутствие ССВ); первого порядка
(АХ-спектры), когда J < Д(А1,А2), и спектры высших порядков
(АВ-спектры), когда J ~ Д(А],А2) (Д^.Аг) — расстояние между
линиями ЯМР в частотной шкале). В последнем случае определение
ХС и J требует квантовомеханического анализа. Более подробное
рассмотрение можно найти в [2, 5, 8-10]. Наличие в одной молекуле
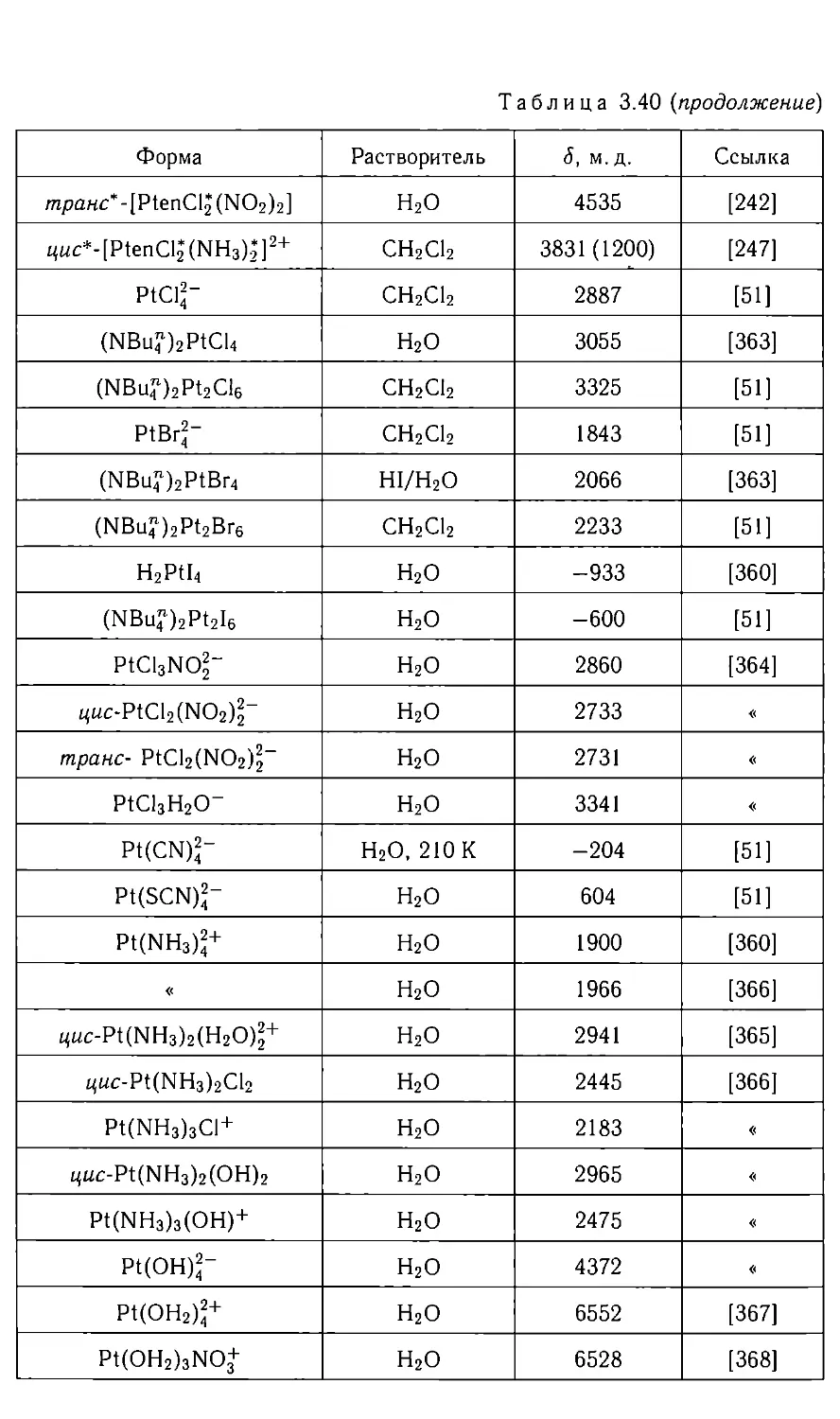
нескольких неэквивалентных ядер с большим содержанием магнитного
изотопа в неорганических соединениях встречается редко. Кроме того,
обычно ядра неорганических соединений имеют большой диапазон ХС,
что приводит к разнесению линий ЯМР, превышающему КССВ. Все
это приводит к сравнительно редкому наблюдению спектров высших
порядков в растворах неорганических веществ, особенно в высоких
магнитных полях. В неорганических растворах чаще наблюдают ССВ
между разными ядрами, чем одинаковыми. Величина J зависит от
свойств и числа связей, через которые идет ССВ, а также геометрии
молекул [2]: , , , ,
J = (1.5)
Ej — £i
где суммирование идет по всем молекулярным орбиталям; i — основ-
ные, j — возбужденные состояния; Ei и е^ — энергии молекулярных
орбиталей; 71.2 — гиромагнитные отношения ядер, В — константа.
Наиболее важными вкладами в J, как и для парамагнитного вклада
в ХС, являются вклады от низколежащих возбужденных состояний
(с небольшой энергией возбуждения). Видно, что величина J пропор-
циональна произведению гиромагнитных отношений и может сильно
различаться для сходных по характеру связей; для разных изотопов
одного и того же элемента константы пропорциональны их гиромагнит-
ным отношениям: , ,.п ,
J(X-‘°B) = 7(10В)
J(X-“B 7(ИВ)‘
В том случае, когда соседнее ядро представлено магнитными
и немагнитными изотопами элемента, спектр будет состоять из муль-
типлетов с наложенными на них одиночными линиями, с интенсивно-
стями, соответствующими природному содержанию изотопов.
Ядерная магнитная релаксация. Важную информацию можно
извлечь из времен ядерной магнитной релаксации Т\ и Т?. Время ядер-
ной магнитной релаксации Т\ (спин-решеточной или продольной релак-
сации) определяет время установления теплового равновесия ядерных
спинов с окружающей средой (решеткой) и соответствует затуханию
компоненты Mz суммарного вектора намагниченности ядерных спинов
вдоль поля Но. Время ядерной магнитной релаксации Т2 определяет
время расфазировки суммарного вектора намагниченности ядерных
спинов, которое соответствует затуханию поперечных компонент Мх
и Му указанного вектора, вызванного спин-спиновым взаимодействи-
ем. Поэтому Т2 называют еще временем поперечной, или спин-спино-
вой релаксацией. Времена Т\ и Т2 определяют условия регистрации
спектров ЯМР и успешное проведение эксперимента ЯМР. Подроб-
но механизмы релаксации изложены в [10, 11, 12], а их теория —
в [5, 12, 13].
Время поперечной релаксации связано с естественной шириной
линии ЯМР TVi/2 на уровне полувысоты соотношением
^1/2 = (1.6)
' тгТг
Релаксация спиновой системы в жидкости отличается от релак-
сации в твердом теле из-за быстрого молекулярного движения. Ка-
чественно релаксацию можно представить как результат флюктуации
локальных магнитных полей, происходящих из-за различных взаи-
модействий спинов (диполь-дипольного, косвенного спин-спинового,
квадрупольного и других). Для спин-решеточной релаксации (с вре-
менем Т\) важны компоненты спектра флюктуации на частоте резо-
нанса wq = 2тп'о, а для спин-спиновой (с временем Т2) — компоненты
с частотами шо и низкочастотные компоненты ш ~ 0. Кроме того, в Т\,
и Т2 дают вклад компоненты спектра флюктуации с частотой 2wq:
J(w) = j K{T)eiuJTdT. (1.7)
— сю
Молекула в жидкости мало меняет состояние своего движения меж-
ду столкновениями, поэтому среднее время между столкновениями
называется временем корреляции движения тс. Последнее определяет
частотный спектр флюктуации [9], который является фурье-преобразо-
ванием функции корреляции Д(т). Эта функция показывает, сколько
времени молекула «помнит» состояние своего движения после очеред-
ного соударения:
Д(т) = Дое|т|/тс. (1.8)
При описании движения в трехмерном пространстве обычно поль-
зуются сферическими координатами г, 0, р и вводят функции
Уо = т-3(1 — 3cos20), У = r-3sin0cos0exp(i99),
Уз = г-3 sin2 0 ехр(гу>).
Соответствующие функции корреляции (г = 0, 1,2):
тад = (^(Ф7(* + <)),
(1.9)
где Y* — функция, комплексно сопряженная с Y; угловые скобки
означают усреднение по ансамблю ядер.
Из (1.7)—(1.9) можно найти функции спектральной плотности,
обычно используемые в выражениях для Т\^.
J°=l^n'‘2^=c°TT^' (|10)
15r (1 + и тс) 1 + ш тс
л = ,,, =с'тт^' (111)
1 ОГ ( 1 + hJ Тс ) 1 + W Тс
т 16ТС Тс /1
Вид функций для
площадь под кривыми
15(1 + и тс) 1 + и тс
разных тс показан на рис. 1.1. Заметим, что
Ti-тз постоянна. Отсюда становится понятным,
Рис. 1.1. Функции спектральной плотности флюктуаций локального магнитно-
го поля для времен корреляции тс: л > л > тз
что как большие, так и малые времена корреляции имеют малый вклад
в спектр на частоте cjq и приводят к большим величинам Т\. В то же
время при больших тс (см. рис. 1.1, кривая л) велика низкочастотная
компонента спектра флюктуации, действующая на 7г, и в этом случае
Т1 3> 7г (заторможенное движение).
Для релаксации ядерных спинов важную роль играют следующие
взаимодействия:
а) магнитное диполь-дипольное;
б) электрическое квадрупольное;
в) взаимодействие, вызванное анизотропией тензора экраниро-
вания;
г) скалярное взаимодействие (ССВ);
д) спин-вращательное взаимодействие.
В общем случае их влияние на релаксацию можно записать в виде
т-* 1 = л, = дс2Ж),
где величина Ес характеризует силу конкретного взаимодействия. Ко-
гда в молекуле имеется несколько ядер с большим магнитным мо-
ментом (*Н, 19F), преобладающим будет диполь-дипольное взаимодей-
ствие. Показано [13], что в молекуле, содержащей два ядра с разными
спинами (I и S), для ядра I
= 7/7s 2^ + 1) (у2 ^o(wr — + J\ (w/) + ;
(1.13)
выражение для ядра S аналогично приведенному.
Для спин-спиновой релаксации
^ = 7hi^5(s+i)x
/11 3 3 3 \
х ( r — w.s) + т (cj;) + = Ji (ws) + - + w.s)) >
\О 24 4 2 о /
(1.14)
и аналогично R% для ядра S.
В случае двухспиновой системы с одинаковыми спинами I
= ^MW + O^iW + 'Ww,)), (1.15)
о7Г
R2 = -^ 7/W + 1) fl ^о(О) + Ш) + | Л(2ш;)У (1.16)
Для многоспиновой системы
Л1 = -^3h2I(I+ 1)У {[дыи + [Л(2ш7)Ь}, (1.17)
107Г
г,к
и для /?2 аналогично. Формулы (1.13)—(1.17) пригодны как для внут-
римолекулярных, так и для межмолекулярных вкладов в релаксацию.
Межмолекулярный вклад обычно невелик по сравнению с внутримо-
лекулярным, но становится значительным при взаимодействии с элек-
тронными спинами парамагнитных примесей; например, протонную
релаксацию многих органических жидкостей определяет парамагнит-
ный молекулярный кислород, растворенный в них. Для внутримоле-
кулярного вклада в релаксацию в двухспиновой системе, используя
выражения (1.13)—(1.17), получим
4 1 2
r^ = ELJL_ /(/+1)
1 1 п 1 О ' '
Ютг г
4,2 /
Сут = ( Зтс +
2 2Отг г6 \
( тс 4тс \
2 2 ' , , л 2 2 J
у 1 + ш тс 1 + 4ш тс /
5тс 2тс \
7“ 2~2 ' 1 . и 2 2 ) •
1 + и тс 1 + 4ш тс /
(1-18)
(1.19)
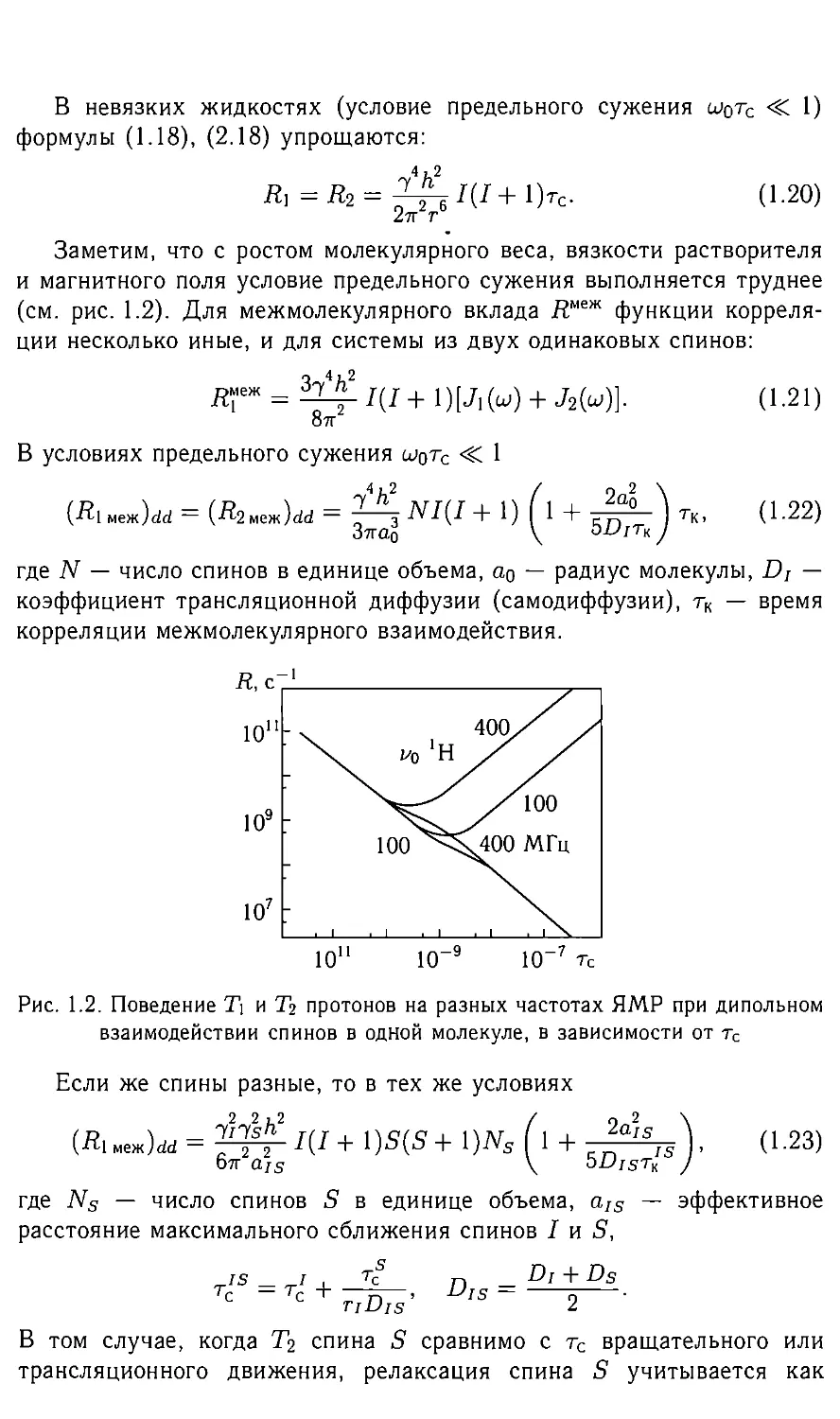
В невязких жидкостях (условие предельного сужения wo^c + 1)
формулы (1.18), (2.18) упрощаются:
^=^ = -441(1+1)^. (1.20)
2тг г
Заметим, что с ростом молекулярного веса, вязкости растворителя
и магнитного поля условие предельного сужения выполняется труднее
(см. рис. 1.2). Для межмолекулярного вклада /?ыеж функции корреля-
ции несколько иные, и для системы из двух одинаковых спинов:
дмеж= 3^/(/+1)[Ji(w) + J2H] (121)
о7Г
В условиях предельного сужения wo^c + 1
(Д1 меж)^ = (Д2меж)^ = NI(I +1)61 + Тк, (1.22)
Зтгао \ у
где N — число спинов в единице объема, o.q — радиус молекулы, D, —
коэффициент трансляционной диффузии (самодиффузии), тк — время
корреляции межмолекулярного взаимодействия.
Рис. 1.2. Поведение 7) и Тг протонов на разных частотах ЯМР при дипольном
взаимодействии спинов в одной молекуле, в зависимости от тс
Если же спины разные, то в тех же условиях
2 2 ,2 / 9 г \
(Д1меж)^ = 1)S(S+ 1)NS 1 + J JSJS , (1.23)
бтг aIS \ 5Distk )
где Ns — число спинов S в единице объема, a[S — эффективное
расстояние максимального сближения спинов I и S,
В том случае, когда Тг спина S сравнимо с тс вращательного или
трансляционного движения, релаксация спина S учитывается как
вклад во время корреляции тс-1 = tr-1 Н-т^1 для внутримолекулярного
и тс-1 = т,^1 +тд 1 для межмолекулярного вклада, где тя, т0 — времена
корреляции вращательного и трансляционного движений; rs — время
релаксации спина S. Обычно rD, тя т3, но в растворах парамаг-
нитных ионов они могут быть сравнимы. Межмолекулярный вклад
важен при малом внутримолекулярном вкладе, а также при наличии
парамагнитных примесей в растворе.
В неорганических жидкостях и растворах диполь-дипольное взаи-
модействие играет гораздо меньшую роль, чем в растворах органиче-
ских веществ, поскольку ядра с большим магнитным моментом, свя-
занным с исследуемым ядром, встречаются редко. Кроме того, большая
часть ядер элементов имеет квадрупольный момент (Г > 1/2), поэто-
му релаксацию спиновой системы чаще определяют квадрупольные
взаимодействия. Для квадрупольных ядер при условии предельного
сужения (wqtc < 1) [13]:
R}=R2= 3(2J + 3) f 1 + (^e2QQ\ )
4ОГ2(2Г — 1) \ 3 J\ h J
где 7/^1 — параметр асимметрии, e2qQ/h — константа квадрупольного
взаимодействия, q — квадрупольный момент, Q — градиент электриче-
ского поля (ГЭП) на ядре.
Разнообразие величин ГЭП и квадрупольных моментов у ядер в раз-
ных соединениях приводит к диапазону времен релаксации от долей
микросекунд до десятков секунд. Поскольку для жесткой молекулы
квадрупольное взаимодействие полностью внутримолекулярное, время
релаксации является мерой времени корреляции ее вращательного
движения и при независимом определении константы квадрупольного
взаимодействия можно определить тя. Картина существенно усложня-
ется, когда ядро входит в состав лабильного комплекса, в подвиж-
ный фрагмент молекулы, а также при нарушении условия предельного
сужения [13].
Вклад в релаксацию из-за анизотропии ХС можно оценить следую-
щим образом [11, 15]. Эффективное магнитное поле, действующее на
ядро, R = Но(1 — с)> где а — тензор экранирования. В жидкости ком-
поненты тензора <Jkk усредняются: сг0 = (охх + + <т22)/3, при этом
процесс усреднения приводит к релаксации ядерного спина. При осе-
симметричном тензоре экранирования можно получить формулы [13]:
per _ 1 2гг2/д \2 / 2тн \
R, (тйй/
Ж = (вг, + тА-Д
9и \ 1 + ЧЦТН /
(1-25)
упрощающиеся в условиях предельного сужения wztr 1. Из (1.25),
(1.26) видно, что вклад в релаксацию за счет анизотропии тензора
экранирования растет с увеличением напряженности магнитного поля
Hq и при w/Tr —> 0 отношение Ti/Tz 7/6. Этот вклад может быть
значительным в релаксации тяжелых ядер.
Скалярная релаксация возникает только" между разными спина-
ми I и S и может быть как внутри-, так и межмолекулярной. Этот
механизм релаксации возникает за счет ССВ. Роль спина S может
играть и неспаренный электрон. Энергия взаимодействия определя-
ется константой ССВ J, а в случае неспаренного электрона — кон-
стантой сверхтонкого взаимодействия А. Для скалярного механизма
важны только флюктуации локального поля, вызываемые спином S.
Они возникают или за счет переориентации спина S (со временем
релаксации т5), или за счет малого времени жизни тв в лабильном
комплексе в процессе химического обмена (в этом случае константы А
или J зависят от времени); временем корреляции в этих случаях будет
величина
Tsc =Ts' +тв'- (1-26)
Скалярная релаксация, вызванная изменением во времени кон-
стант А или J (тв < ts), называется скалярной релаксацией первого
рода, а вызванная переориентацией спина S (т3 < тв) — скалярной
релаксацией второго рода. Для реализации скалярного механизма ре-
лаксации необходимо, чтобы 2тг7тс 1, естественно, что в этом случае
расщепление на ядре S не наблюдается. Показано [13], что
flfc = |27rJS(S+l)f —Тс -з-Д (1.27)
° \ 1 + (ш/ - шз) тс /
ДГ = |27rJS(S+l)frc+ Тс Д (1.28)
о \ 1 + - шз) тс)
При (ш/ — шд)тс <§; 1 вклад скалярной релаксации существен толь-
ко в Т2, что приводит к Т\ Т%. Вклад имеет максимум при
(cj/— cj.s)tc = 1, а при увеличении или уменьшении тс Rsc умень-
шается.
За счет вращения молекулы электроны и ядра создают магнит-
ное поле, флюктуации которого приводят к спин-вращательной релак-
сации. Частота вращения молекулы и в J-м вращательном состоя-
нии и = hJ/(4Tr2IB), где 1В — момент инерции молекулы; магнитный
момент, создаваемый этим вращением, пропорционален частоте враще-
ния. В молекуле НС1 за счет ее вращения на ядре 1Н создается поле
около 10 Э. Спин-вращательный вклад в релаксацию [8]:
RsR= ^lRkTC^Tjt (1.29)
где Сдф — (2С± + Cfi), Сэф — усредненный тензор спин-вращательного
взаимодействия. В качестве времени корреляции берут время тс, кото-
рое молекула проводит в состоянии с определенным угловым моментом.
Показано [16], что т3 связано со временем корреляции вращательного
движения тс соотношением tctj = IR/(fikT). Особенностью спин-вра-
щательной релаксации является противоположная другим механизмам
зависимость от тс. Данный механизм релаксации может быть суще-
ственным для небольших молекул и для ядер, имеющих большой диа-
пазон ХС, поскольку распределение электронов, приводящее к боль-
шому диапазону ХС, способствует и сильному спин-вращательному
взаимодействию [16].
Состояние объекта, изучаемого спектральным методом, будет ка-
заться стационарным или изменяющимся во времени в зависимости от
характерной для метода шкалы времени. Состояния будут выглядеть
стационарными, если время их жизни больше временного разреше-
ния метода. Временное разрешение ограничено естественной шириной
спектральных линий — более широкие линии соответствуют корот-
коживущим состояниям, и разрешение метода во времени связано
с разрешением по частотам соотношением А/ Ат ~ 1, вытекающим
из соотношения неопределенности Гейзенберга АЕ At ~ h. Поэтому
состояния, время жизни которых At ~ 10-6 с, в оптических спектрах
выглядят стационарными, а в спектроскопии ЯМР — короткоживущи-
ми. Процессы, происходящие быстрее 1/Д/ или 1/6 f (Af — шири-
на линии ЯМР, 6f — расстояние между линиями разных состояний
в частотной шкале), будут в шкале времени ЯМР быстрыми, а при
обратном условии — медленными. В зависимости от ядер, образцов
и измеряемого параметра ЯМР шкала времени может оказаться в диа-
пазоне от 1 мкс до часа. В этом интервале лежат характерные времена
многих химических процессов, что что делает возможным изучение
их кинетики методом ЯМР. Если в каком-то процессе ядро меняет
свое окружение, то такое изменение состояний называют химическим
обменом. При этом меняются параметры ЯМР (ХС, Т\, Т%, J). При
медленном переходе из одного состояния в другое регистрируется
сначала исходное, затем наложение исходного и конечного состояний,
из которых первое уменьшается, а второе со временем увеличивается
и, в конце концов, остается единственным. При равновесии наблюдают
равновесные доли этих состояний.
Ускорение обмена состояниями приводит сначала к уширению ли-
ний ЯМР, если они не наложены друг на друга, затем их слиянию,
а при очень быстром обмене наблюдается одна узкая линия, положение
которой будет средневзвешенным от положения линий исходных со-
стояний с учетом их долей. Сравнивая спектры с разными скоростями
обмена (за счет изменения температуры), можно определить харак-
терние скорости процесса обмена. Диапазон измеряемых скоростей
10“5-1 с, в зависимости от конкретных систем; более медленные изме-
нения обнаруживаются последовательной записью спектров. Например,
если обмениваются состояния с линиями ЯМР, отстоящими в спектре
данного ядра на 10 кГц, то для их усреднения необходима скорость
обмена более 104 с-1. Если же обмениваются состояния, одно из кото-
рых характеризуется мультиплетом с константой J ~ 10 Гц, а другое —
без расщепления, то при скорости обмена ~ 10 Гц мультиплет будет
смазываться, что позволяет оценить скорость и такого медленного
обмена. Подобное применение метода ЯМР для изучения процессов
химического обмена изложено в [2, 9].
Широкое применение метода ЯМР в неорганической химии на
иных, чем 'Н и 19F, ядрах стало возможным при развитии импульс-
ных методов ЯМР с накоплением и фурье-преобразованием сигнала
ЯМР. Использование в этих методах удобного для накопления сигнала
спада свободной индукции (ССИ) позволило резко повысить чувстви-
тельность метода и перейти к измерениям на ядрах большей части
элементов при обычных для химиков концентрациях растворов. Посто-
янство частоты и поля в импульсных спектрометрах позволяет быстро
переходить от измерений ЯМР на одном ядре к измерениям на дру-
гих, применять многочасовое накопление сигнала. Новые возможности
метода появились при использовании высоких полей сверхпроводящих
магнитов. Поэтому описанные здесь особенности в первую очередь
касаются экспериментов, выполняемых на импульсных спектрометрах
с криомагнитами.
1.2. Измерение параметров ЯМР в растворах
Спектрометры ЯМР. Особенности эксперимента ЯМР обсуж-
даются в [14].
В эксперименте находят параметры ЯМР одного из ядер изучаемой
системы:
а) условие резонанса
ИЬнн = у- Я0(1 - Олин)! (1-30)
б) интенсивность линий;
в) скорость установления равновесий в спиновой системе;
г) параметры взаимодействия разных спинов (константы ССВ-
КССВ, динамика переноса поляризации спинов).
По способу нахождения условий резонанса различают стационар-
ные и импульсные спектрометры ЯМР. В стационарных спектромет-
рах резонанс находят изменением (разверткой) одного из параметров
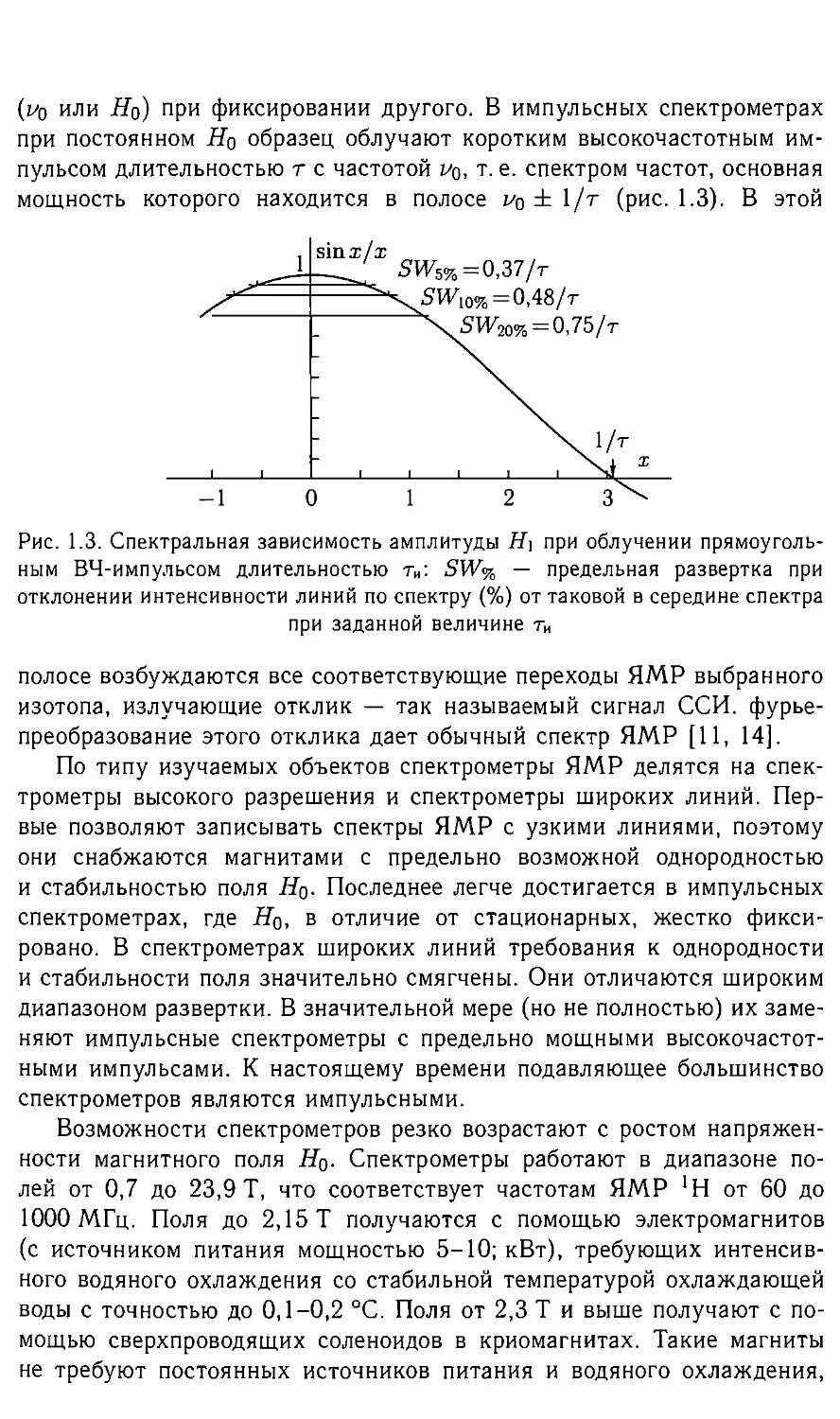
(И) или Hq) при фиксировании другого. В импульсных спектрометрах
при постоянном Hq образец облучают коротким высокочастотным им-
пульсом длительностью т с частотой ио, т. е. спектром частот, основная
мощность которого находится в полосе (рис. 1.3). В этой
Рис. 1.3. Спектральная зависимость амплитуды Н] при облучении прямоуголь-
ным ВЧ-импульсом длительностью ти: SW% — предельная развертка при
отклонении интенсивности линий по спектру (%) от таковой в середине спектра
при заданной величине ти
полосе возбуждаются все соответствующие переходы ЯМР выбранного
изотопа, излучающие отклик — так называемый сигнал ССИ. фурье-
преобразование этого отклика дает обычный спектр ЯМР [11, 14].
По типу изучаемых объектов спектрометры ЯМР делятся на спек-
трометры высокого разрешения и спектрометры широких линий. Пер-
вые позволяют записывать спектры ЯМР с узкими линиями, поэтому
они снабжаются магнитами с предельно возможной однородностью
и стабильностью поля Но- Последнее легче достигается в импульсных
спектрометрах, где Но, в отличие от стационарных, жестко фикси-
ровано. В спектрометрах широких линий требования к однородности
и стабильности поля значительно смягчены. Они отличаются широким
диапазоном развертки. В значительной мере (но не полностью) их заме-
няют импульсные спектрометры с предельно мощными высокочастот-
ными импульсами. К настоящему времени подавляющее большинство
спектрометров являются импульсными.
Возможности спектрометров резко возрастают с ростом напряжен-
ности магнитного поля Но- Спектрометры работают в диапазоне по-
лей от 0,7 до 23,9 Т, что соответствует частотам ЯМР *Н от 60 до
1000 МГц. Поля до 2,15 Т получаются с помощью электромагнитов
(с источником питания мощностью 5-10; кВт), требующих интенсив-
ного водяного охлаждения со стабильной температурой охлаждающей
воды с точностью до 0,1-0,2 °C. Поля от 2,3 Т и выше получают с по-
мощью сверхпроводящих соленоидов в криомагнитах. Такие магниты
не требуют постоянных источников питания и водяного охлаждения,
но для поддержания сверхпроводимости соленоида в них требуют-
ся хладоагенты (жидкие гелий и азот), с расходом 300-1000 (Не)
и 2000-8000 (N2) литров в год. Условие резонанса (1.30) соблюдается
в узком диапазоне частот/полей и требует их высокой стабильности.
При записи спектров ЯМР в стационарных спектрометрах последова-
тельно проходят условия резонанса для ядер с различными ХС, меняя
магнитное поле при постоянной частоте облучения/детектирования или
наоборот, меняя частоту при постоянном стабилизированном поле Но-
Большинство линий ЯМР ’Н лежит в диапазоне 10 м. д. (10-5 от Но),
т. е. при z/q = 100 МГц диапазон изменений Но всего 23 мкТ. В ЯМР
других ядер диапазон ХС много больше (для 13С — 300 м.д., для
59Со — 20000 м.д.). Основной проблемой создания резонансных усло-
вий является стабилизация магнитного поля, поскольку требования
ЯМР превышают абсолютную точность метрологических стандартов;
обычно для стабилизации поля используют сигнал ЯМР внешнего
образца или вещества, введенного в образец. Высокая стабильность
(дрейф поля ~ 100 м.д. в год) и однородность поля криомагнитов поз-
воляют во многих случаях работать без стабилизации поля и вращения
образца, что существенно упрощает эксперимент ЯМР.
Другой проблемой является детектирование резонанса, поскольку
сигнал ЯМР крайне мал. ЯМР относится к самым малочувствитель-
ным по концентрации методам исследования веществ. Для выделения
сигнала из шумов использован весь арсенал радиотехники — фильтра-
ция, техника боковых полос, перенос спектра в другие области частот,
синхронное и фазовое детектирование, накопление сигнала и др.
С разработкой на рубеже 60-70-х годов XX в. импульсных спек-
трометров ЯМР с фурье-преобразованием стационарные спектрометры
в большой мере утратили свои позиции. Хотя фурье-спектроскопия
не является полностью эквивалентной заменой стационарных методик
ЯМР, она многократно расширяет возможности метода ЯМР. Только
при записи очень широких линий стационарные методы имеют пока
определенные преимущества перед фурье-спектроскопией.
Блок-схема импульсного спектрометра приведена на рис. 1.4. В фу-
рье-спектрометре ЯМР жестко стабилизированы и частота, и поле Hq;
резонансные условия поддерживаются с точностью 10-8-10-1°. Устрой-
ство развертки отсутствует. Мощный высокочастотный импульс воз-
буждает все переходы ядра в нужном диапазоне частот.
Чем шире этот диапазон, тем короче должен быть импульс. При
этом максимальный сигнал получается после так называемого 90-гра-
дусного импульса, удовлетворяющего условию 7Я1ТНМП = тг/2 — пово-
роту вектора ядерной намагниченности на 90° во вращающиеся систе-
ме координат, где Н\ — амплитуда высокочастотного магнитного поля
в катушке датчика. После 90-градусного импульса вектор ядерной на-
Огибающая ССИ затухает со временем по экспоненте:
A(t) = Ао [1 - ехр (1.31)
L \ 12/J
если неоднородность поля HqAHq W (эрстед); при обратном соот-
ношении скорость затухания определяется величиной ДН0 (так назы-
ваемое неоднородное уширение).
Огибающая сигналов эха в последовательности CPMG (Сагг-Рег-
cell-Meibum-Gill) позволяет измерить как величину Т2, так и коэффи-
циент диффузии D молекулы с ядром, наблюдаемым при ЯМР:
A(t) = Aoexpf-^- -7W), (1.32)
\ 12 /
7 — гиромагнитное отношение ядра, G — градиент поля Hq по оси z.
При небольших градиентах вторым членом показателя можно прене-
бречь, и измеряется величина Т2.
Амплитуда сигнала после импульса 90, х с задержкой t в последо-
вательности D позволяет измерить время Т\-.
A(t) = Ао - 2А0ехр (-^-Y (1.33)
\ Il /
Проводя последовательно серию измерений с нарастающими tlt можно
построить огибающую и определить величину Т\.
Спин-спиновое взаимодействие соседних одинаковых ядер (АВ-си-
стема) или иного соседнего ядра (АХ-система) расщепляет линию на
2/ + 1 компонент (/ — суммарный спин соседних эквивалентных ядер),
усложняя тем самым спектр ЯМР и уменьшая соотношение S/N. При
этом в случае расщепления на п одинаковых спинов интенсивность
компонент меняется в соответствии с распределением треугольника
Паскаля:
Суммарный спин Число компонент Интенсивность компонентов Название сигнала
0 1 1 Синглет
1/2 2 1 1 Дублет
1 3 1 2 1 Триплет
3/2 4 13 3 1 Квартет (квадруплет)
2 5 14 6 4 1 Пентет
5/2 6 1 5 10 10 5 1 Секстет
3 7 1 6 15 20 15 6 1 Септет и т. д.
В случае расщепления на ядре X со спином, равном п, наблюдается
п + 1 компонент равной интенсивности. Например, спектр ЯМР иона
[NH4]+ на ядре 14N представляет пентет с соотношением компонент
1 : 4 : 6 : 4 : 1 (расщепление на 4 протонах, I = 2), в то время как
спектр ЯМР 'Н того же иона состоит из трех компонент равной интен-
сивности (расщепление на ядре I4N, 7=1). Естественно, константы
ССВ в обоих спектрах одинаковы.
ССВ снимается облучением образца на частоте ЯМР ядер, вызыва-
ющих расщепление, до насыщения их линий. Такое подавление спин-
спинового взаимодействия называется гетероядерным, если соседнее
ядро относится к другому изотопу, и гомоядерным, если оно представ-
ляет тот же изотоп, что и исследуемое ядро. Взаимодействие спина
одного ядра с другим нередко происходит так, что релаксация одного
спина приводит к релаксации другого. В этом случае изменения засе-
ленности спиновых состояний одного ядра будут вызывать изменение
заселенности другого. При насыщении линии резонанса (ядерного или
электронного) соседнего спина интенсивность сигнала наблюдаемого
ядра может измениться (ядерный эффект Оверхаузера, т. е. динамиче-
ская поляризация ядер). Этот эффект может быть положительным (уве-
личение сигнала) или отрицательным (уменьшение сигнала); при этом
подавляемое ядро должно иметь большее гиромагнитное отношение.
Эффект Оверхаузера широко используется в ЯМР |3С органических
соединений, где подавление протонов сопровождается троекратным
увеличением сигнала 13С и сокращением Д 13С, что позволяет быстрее
накапливать сигнал.
Обычно в случае гетероядерного подавления образец облучается
в полосе частот, охватывающей все линии ЯМР подавляемого ядра.
Для упрощения спектров при гомоядерном взаимодействии применя-
ется другой вариант двойного резонанса — тиклинг. В этом случае
вторую частоту при малой мощности облучения устанавливают на
различные линии резонанса, что позволяет выделить связанные спин-
спиновым взаимодействием ядра или некоторые линии из набора пе-
рекрывающихся линий. Для некоторых ядер гетероядерный двойной
резонанс является основным методом наблюдения. Например, наблю-
дая в ЯМР *Н расщепление от ядра 103Rh в гидридных комплексах
родия и подавляя ССВ (1H-103Rh) на частоте ЯМР l03Rh, составили
шкалу ХС l03Rh для гидридных комплексов родия [25] до прямых
измерений на ядрах l03Rh. При исследовании молекул и комплексов,
содержащих несколько магнитных ядер (’Н, 19F, 11 В, 31Р и др.),
получают спектры, усложненные взаимодействием нескольких ядер.
Для их упрощения применяют поочередное или совместное подавле-
ние ядер (двойной и тройной резонансы). Стандартные спектрометры
снабжаются системой подавления только протонов, что ограничивает
использование методов двойного и кратных резонансов для объектов
неорганической химии.
Особенности эксперимента ЯМР при изучении жидкостей. Из-
мерение химических сдвигов и констант спин-спинового взаимодей-
ствия. Из-за неоднородности магнитного поля спектрометра сигнал
ЯМР уширен. При узких линиях повышение однородности поля (т. е.
разрешения) увеличивает отношение S/N и возможность наблюдения
малых изменений ХС и малых J. Для наблюдения без искажения
линий с Д/ = 0,1 Гц при uq = 108 Гц неоднородность поля Но в образце
должна быть менее 10~9. В криомагнитах магнитное поле создается
соленоидом из тончайшего сверхпроводящего провода, находящегося
при температуре кипения гелия (4 К), в котором течет ток ~50А.
Применением набора катушек разной формы (шиммов) создают малые
дополнительные поля разной конфигурации, уменьшающие градиенты
поля, что позволяет на порядок улучшить однородность поля. В крио-
магнитах при настройке внутренних сверхпроводящих шиммов дости-
гается однородность до Ю“(6’7) в объеме 1 см3. Шиммами, встроен-
ными в «теплое» отверстие магнита, удается улучшить однородность
поля еще на порядок. Вращением образца можно усреднить градиенты
поля, направленные перпендикулярно оси вращения [26], и уменьшить
неоднородность поля в несколько раз. При этом эффективная величина
неоднородности
АЯэф = 7<(ДЯ)2)0.
где ((ДГГ)2) — среднеквадратичное отклонение напряженности поля
от средней величины, 0 — период вращения образца. Повышение
однородности поля с помощью вращения образца широко используется
в ЯМР высокого разрешения. Этим разрешение улучшается примерно
на порядок. Вращение не усредняет градиенты поля вдоль оси вра-
щения (Z-градиент в криомагнитах). Заметим, что однородность 10-7,
достаточна для работы без вращения образца для большинства ядер,
что существенно упрощает процесс измерения.
Поле внутри образца отличается от внешнего Но на величину,
зависящую от его магнитной проницаемости р и формы. Объемная
восприимчивость растворов и жидкостей может различаться более чем
на 10-6 ед. СГСМ, а в случае растворов парамагнитных частиц — до
10~5. Однородность поля в образце сохраняется, если он имеет форму
эллипсоида (частными случаями которого являются сфера и бесконеч-
ный цилиндр). Обычно образец наливают в цилиндрическую ампулу
так, что его длина L больше диаметра d, и в дальнейшем образец
рассматривают как длинный (бесконечный) цилиндр. Это приближение
неплохо работало в спектрометрах с электромагнитами и стандартными
ампулами d = 5 мм, в которых короткая приемная катушка помещена
далеко от конца ампулы.
Поле в образце с объемной магнитной восприимчивостью Xv [27],
Н = I1 + (т “ к) Ч Н°' (134)
где к, — размагничивающий фактор, равный 4тг/3 — для сферы, 2тг —
для цилиндра, ориентированного поперек поля, и нулю — для ци-
линдра, направленного по полю. Величины ХС, измеренные в разных
растворителях при несферической форме образца, будут отличаться
на величину ^Д», т.е. до 2 м.д. для набора обычно используемых
растворителей. На деле образец не может иметь форму длинного
цилиндра. В ЯМР иных ядер, кроме 'Н и 19F, обычно используются
ампулы d= 10 мм и более. В этих условиях образец является корот-
ким цилиндром. Измерительная катушка в большинстве импульсных
спектрометров расположена у конца ампулы, и даже при большом
столбе жидкости форму образца следует аппроксимировать полубес-
конечным цилиндром. Для реальной ампулы 5 мм, расположенной по
полю, экспериментально определенная величина к, находится в преде-
лах -0,5-;—0,9 [26]. Пренебрежение поправкой на восприимчивость
образца совершенно не приемлемо в спектрах ЯМР 1Н, где подавляю-
щая часть соединений имеет линии в диапазоне всего 10 м.д. В этом
случае для точного отсчета ХС в раствор вводят внутренний эталон,
как правило, тетраметилсилан (ТМС) и ХС отсчитывают от его линии,
тем самым исключая влияние восприимчивости образца.
В неорганических жидкостях и водных растворах для большинства
ядер применение внутренних эталонов невозможно, поэтому отсчет
ведут от внешних эталонов. Учитывая большие, чем у протонов, диапа-
зоны ХС других ядер (исключая 7Li и 9Ве) и то, что погрешности изме-
рения ХС ранее были довольно велики, поправок на восприимчивость
обычно не вводили. Заметим, что в измерениях ранее 1970-х годов
спектрометры имели электромагниты, ось ампулы в которых направ-
лена поперек поля Но, тогда как в криомагнитах — вдоль магнитного
поля. Поэтому ХС, измеренные спектрометром с электромагнитом,
должны корректироваться на величину —2тгДху/3 относительно ХС
сферического образца, и на величину 4тгДуу/3 — для криомагни-
та [27]. Поскольку измерения на иных, чем 1Н и 13С, ядрах делаются
относительно внешнего эталона и поправка на восприимчивость обыч-
но не вводится, различие Д^у образца и эталона на 10-6 ед. СГСМ
приводит к различию ХС, измеренных на указанных магнитах, на
~5 м.д. Погрешности такого рода уже попали в литературу [28].
Возможность в высоких полях отсчета ХС точнее 0,1 м.д., вовлече-
ние в практику ЯМР многих ядер с узкими линиями ЯМР делает учет
этих поправок актуальным.
Наиболее универсальным способом точного измерения ХС является
отсчет от линии внешнего эталона с учетом поправки на восприим-
чивость. Различие возникает даже при установке в ампулу эталона
в коаксиальном капилляре. Поправка зависит от формы образца и его
ориентации, см. формулу (1.34). Для сферического образца поправки
не требуется. Однако в этом случае для измерений нужны специальные
ампулы и резко уменьшается объем образца, т. е. чувствительность
прибора.
Другим способом учета xv является использование какой-либо ча-
стицы в растворе как внутреннего эталона при резонансе на ядре, отли-
чающемся от измеряемого. Например, водные растворы неорганических
веществ часто содержат инертный ион CIO4 , имеющий стабильный
ХС на ядре 35С1 в широком диапазоне pH, что позволяет измерить
внутреннее поле в образце. Напротив, линия ЯМР дейтерированной
воды, используемая для стабилизации поля при съемке спектров вод-
ных растворов, заметно смещается в зависимости от ионного фона
раствора и обычно не может быть использована для введения поправок
на восприимчивость.
Использование криомагнитов делает ЯМР для многих неорганиче-
ских систем рабочим физическим методом. Большее разнесение и раз-
решение линий позволяет наблюдать линии, которые сливаются в более
низких полях. Перенос спектров в область высоких частот особен-
но важен для ядер с малым гиромагнитным отношением (низкими
частотами ЯМР), для которых в обычных полях можно наблюдать
только узкие линии ЯМР из-за низкой чувствительности и сильного
акустического «звона» контура датчика, перекрывающего сигнал ССИ
широких линий в импульсных спектрометрах. Например, на ядрах
14N в поле 7 Т обычно разрешаются линии с разностью в несколько
миллионных долей и легко регистрируются линии шириной в 1-5 кГц,
что практически невозможно в поле 2 Т; это многократно увеличивает
возможности ЯМР азота. С другой стороны, в высоких полях труднее
выполняется условие предельного сужения, что в ряде случаев «сма-
зывает» расщепление ССВ в спектрах и снижает их информативность.
Например, расщепление ССВ от азота и хлора в спектрах ЯМР 17О
ионов NO^ и CIO4, видимое в поле 2,11 Т, в поле 9Т обычно не
наблюдается; аналогичная ситуация и в спектрах 19F [30].
Согласно рекомендациям ИЮПАК, принято отсчитывать ХС в шка-
ле 6, для которой сдвиги в сторону слабого поля (высокой частоты)
считают положительными:
5СИГН = Ю6 ~ . (1.35)
^эталон
Такая шкала использовалась ранее в ЯМР 1Н, для которой за ноль
был принят ХС ТМС. Для ЯМР ’Н использовалась также и т-шкала,
для которой ХС увеличивался с ростом поля, а ХС ТМС был принят
равным 10 м.д. Для других ядер ранее использовались шкалы, в кото-
рых сдвиги считались положительными от эталона в сторону высокого
поля, т. е. увеличения экранирования (шкала ст).
При диапазоне ХС ядра более 1000 м.д. возникают дополнитель-
ные погрешности. Для правильного отсчета ХС частота спектрометра
должна совпадать с частотой эталона 6 — 0; в противном случае при
отсчете ХС ~ 1000 м.д. ошибка достигает нескольких м.д. [29].
Широкое использование внешних эталонов и измерения на «сла-
бых» ядрах, для которых накопление сигнала эталона также явля-
ется длительной процедурой, привело к тому, что для определения
положения линий ЯМР часто используется абсолютная шкала ЯМР.
В этой шкале указывается величина 5 — частота ЯМР данного об-
разца в поле, в котором частота ПМР ТМС равна 100 МГц. Иногда
ХС отсчитывают от некоторой величины 5 = А. Для выражения ХС
в виде Н или относительно Н — А, следует устанавливать точную
частоту спектрометра, которая может на сотни м. д. отличаться от
частоты, указанной в названии спектрометра. Разумеется, привязка
по внешнему эталону требует введения поправки на восприимчивость
образца. В табл. 1.1 приведены величины Н для эталонных растворов
ЯМР некоторых ядер.
При большом диапазоне ХС (> 1000 м.д.) и использовании разных
эталонов пересчет ХС из одной шкалы в другую следует производить
по формуле [31] А
£ = _ ю6 Е (1.36)
где 6 — ХС в шкале эталона с ХС 2, 6А —ХС в шкале эталона с ХС
Параметры ЯМР измеряются с разной точностью: если ХС узких
линий измеряется с внутренним эталоном с точностью до 0,01 м.д.
(0,3 м.д. с внешним эталоном и учетом поправки на восприимчивость,
и 2-3 м.д. без учета поправки), a J до долей Гц, то измерение времен
релаксации, ширины линий и их интенсивностей делается гораздо гру-
бее. Вещества, рекомендуемые в качестве эталонов для разных ядер,
приведены в [29] (табл. 1.1).
Поскольку времена релаксации и ширина линии зависят от вели-
чины тс (которая является функцией температуры, вязкости раство-
рителя, размера изучаемых частиц), то данные их измерений должны
сопровождаться описанием условий эксперимента. Измерение времен
релаксации с точностью 10% не требует особых ухищрений; для до-
стижения погрешности 5% требования к соотношению S/N и точности
установки параметров импульсов заметно возрастают. Измерения с точ-
Таблица 1.1
Частоты ЯМР некоторых ядер в абсолютной шкале экранирования [14]
Ядро Эталонный раствор Эталон (<5, м.д.) Е, МГц
'Н (CH3)4Si-TMC Тетраметилсилан (0) 100
13с СНзСООН Уксусная кислота (170,0) 25,149280
тмс Тетраметилсилан (0) 25,145004
14n [N(CH3)4]I, 0,075М/Н20 Тетраметиламония иодид (от CH3NO2) 7,233876
i5N ch3no2 Нитрометан (0) 10,136783
|7О Н2О Вода (0) 13,556510
19f C6F6 Гексафторбензол (—163 м.д. от CFC13) 94,078500
29Si ТМС ТМС (0) 19,867184
31 р (СН3О)3Р Триметоксифосфин (141 м.д. от Н3РО4) 40,486455
l03Rh Rh(Acac)3/CHCl3 7рис-ацетилацетонат родия (8358 м.д.) 3,16
195pt Na2PtCl6/H2O Гексахлороплатинат (4521 м.д.) 21,4
ностью 2% требуют тщательной установки параметров спектрометра,
стабилизации температуры образца и S/N > 100.
Правильное измерение времен релаксации возможно в спектро-
метрах, обладающих хорошей однородностью высокочастотного поля
в объеме образца. Последнее требует, в частности, чтобы образец
находился полностью внутри катушки датчика. Подобные измерения
возможны не на всех импульсных спектрометрах. Многочисленные
методики измерений Т\ и 7г, а также времени спиновой диффузии при-
ведены в [11, 14, 18]. Преимущества того или иного метода зависят от
исследуемой системы. Для измерения коэффициента диффузии необ-
ходимо применять постоянные или импульсные градиенты магнитного
поля, что возможно только в специальных датчиках.
Измерение концентраций. Интегральная интенсивность линии
ЯМР (площадь под линией поглощения) пропорциональна числу ядер
в активной зоне катушки датчика и может быть использована для
измерения концентраций [34]. Спектр из нескольких линий сразу дает
мольное отношение концентраций неэквивалентных атомов. При этом
надо снять спектр в условиях отсутствия насыщения, т. е. для импульс-
ных спектрометров при ти 90°, периоде повторения Тп > 37). Также
должно выполняться условие ти 6f/5, где 6f — диапазон частот
спектра, занимаемый линиями образца и эталона.
Абсолютное измерение концентраций возможно введением в рас-
твор инертного соединения с данным ядром. Для водных растворов
выбор таких соединений проблематичен. Альтернативой служит съем-
ка спектра с внешним эталоном.
В этом случае при одновременном измерении спектров образца
и эталона (например, в коаксиальных ампулах) при S/N > 50 возможно
измерение относительных интенсивностей линий (т. е. концентраций
ядер) с погрешностью менее 5%. При этом линии эталона и измеряе-
мого комплекса не должны накладываться друг на друга. Кроме того,
цифровое разрешение при накоплении и в спектре ЯМР, т. е. число
адресов памяти ЭВМ, приходящееся на период сигнала ССИ и ширину
линии ЯМР, должно быть достаточным (не менее 10 на ширину линии
и образца, и эталона). Дополнительная погрешность может возникнуть
из-за скин-эффекта, если проводимость образца высока. В этом случае
поле Hi в центре образца, где находится эталон, будет слабее, чем
в образце, что приведет к систематическому завышению определяемых
концентраций (см. также гл. 4).
Следует учитывать неравномерность частотного спектра возбужда-
ющего импульса т (рис. 1.3). На рис. 1.3 указаны границы развертки
для погрешности интенсивности линий в спектре, не превышающей
5, 10 и 20%. Уменьшение длительности т при сохранении амплитуды
сигнала требует увеличения мощности импульса т. Последняя ограни-
чена как параметрами передатчика, так и электрической прочностью
датчика. С понижением частоты ЯМР пропорционально возрастает
амплитуда Н\ возбуждающего поля и опасность электрического пробоя
датчика.
Дополнительную информацию можно получить измерением спек-
тров в разных полях. При невозможности применения методов двой-
ного и множественных резонансов измерением в разных полях можно
выделить мультиплеты, так как константы ССВ не зависят от поля. Та-
кие измерения нужны для определения компонент тензоров ХС и квад-
рупольного взаимодействия и механизмов релаксации [17]. Обычно
информативность спектров повышается при переходе к высоким полям.
Во многих системах с химическим обменом переход в другое поле ока-
зывается переходом к другому типу обмена (от быстрого к промежуточ-
ному или от промежуточного к медленному), что позволяет определять
как параметры ЯМР, так и кинетические параметры обменивающихся
ядер (см. раздел 4.3).
Некоторые особенности эксперимента ЯМР высокого разрешения
и возможности увеличения его точности описаны в [2, 18, 19, 34-36].
ЛИТЕРАТУРА К ГЛАВЕ 1
1. Эмсли Дж. Элементы / Пер. с англ. Е. А. Краснушкиной. — М.: Мир,
1993. - 256 с.
2. Эмсли Дж., Финей Дж., Сатклиф Л. Спектроскопия ЯМР высокого разре-
шения. Т. 1 / Пер. с англ. Б. А. Квасова; Под ред. Э. И. Федина. — М.: Мир,
1968. - 630 с.
3. Jameson C.J., Gutowsky H.S. Calculation of chemical shifts. I. General formu-
lations and Z-dependence // J. Chem. Phys. 1964. V. 40, №6. P. 1716-1724.
4. Ebraheem K.A., Webb G.A. Some skreening constant calculation for carbon,
nitrogen and oxygen nuclei //J. Magn. Reson. 1978. V. 30, №2. P. 211-215.
5. Сликтер Ч. Основы теории магнитного резонанса / Пер. с англ. Н.Н. Кор-
ста и др.; Под ред. Г. В. Скроцкого. — М.: Мир, 1981. — 448 с.
6. Karplus М., Pople J.A. Theory of carbon NMR shifts in conjugated
molecules // J. Chem. Phys. 1963. V. 38, № 12. P. 2803-2807.
7. Dharmatti S.S., Weawer H.E. Nuclear magnetic resonance lines of antimony
in antimony hexafluoride ion // Phys. Rev. 1952. V. 87, № 4. P. 675.
8. ЖункеА. Ядерный магнитный резонанс в органической химии / Пер. с нем.
О. С. Чижова, Ю. С. Шабарова. — М.: Мир, 1974. — 176 с.
9. Керрингтон А., Мак-Лечлан Э. Магнитный резонанс и его применение
в химии / Пер. с англ. И.Н. Марова. — М.: Мир, 1970. — 448 с.
10. Gunter Н. NMR Spectroscopy. — Chichester-N. Y.: J. Wiley, 1980. Рус. пер.:
Гюнтер X. Введение в курс спектроскопии ЯМР / Пер. с нем. Ю. А. Усты-
нюка, Н.М. Сергеева. — М.: Мир, 1984. — 478 с.
11. Фаррар Т., Беккер Э. Импульсная фурье-спектроскопия ЯМР / Пер. с англ.
Б. А. Квасова; Под ред. Э. И. Федина. — М.: Мир, 1973. — 166 с.
12. Вашман А. А., Пронин И. С. Ядерная магнитная релаксация и ее примене-
ние в химической физике. — М.: Наука, 1979. — 236 с.
13. Абрагам А. Ядерный магнетизм / Пер. с англ, под ред. Г. В. Скроцкого. —
М.: ИЛ, 1963. - 551 с.
14 Martin М I Martin G J Dplnuarh J -J Practical NMR snectrosconv
16. McClung R.E.D. Rotation diffusion of spherical top molecules in liquids //
J. Chem. Phys. 1969. V. 51, №9. P. 3842-3852.
17. Mariott A.A., Farrar T.C. 35C1 and l9F NMR spin-lattice relaxation time
measurments and rotational diffusion in liquid CIOF3 // J. Chem. Phys. 1971.
V. 54, № 1. P. 64-71.
18. Дероум Э. Современные методы ЯМР для химических исследований /
Пер. с англ. Ю.М. Демина, В. А. Черткова; Под ред. Ю.А. Устынюка. —
М.: Мир, 1992. — 401 с.
19. Blumich. В. Essential NMR for Scientists and Engineers. — Berlin: Springer
Verlag, 2005. Рус. пер.: Блкмих Б. Основы ЯМР / Пер. с англ. Н.Е. Ага-
пова. — М.: Техносфера. 2007. — 160 с.
20. Braun S., Kalinowski H.-О., Berger S. 150 and More Basic NMR Experi-
ment. — Weinheim: Wiley-VCH, 1998. — 596 p.
21. Waugh J.S., Wang C.H., Huber L.M., Void R.L. Multiple-pulse NMR exper-
iments // J. Chem. Phys. 1968. V. 48, № 2. P. 662-670.
22. Ellett J.D., Waugh J. S. Chemical shifts concertina // J. Chem. Phys. 1969.
V. 51, №7. P. 2851-2858.
23. Hyde E.H., Kennedy J.D., Shaw B.L. Magnetic double resonance measur-
ments of rhodium, phosphorus, fluorine and proton magnetic resonance pa-
rameters in some rhodium(I) and rhodium(III) compounds // J.C. S. Dalton tr.
1977. № 16. P. 1571-1576.
24. Bloch F. Nuclear induction // Phys. Rev. 1946. V. 70, № 2. P. 460-472.
25. Леше А. Ядерная индукция / Пер. с нем.; Под ред. П.М. Бородина. —
М.: ИЛ, 1963. - 684 с.
26. Ионин Б.И., Ершов Б.А. ЯМР спектроскопия в органической химии. —
Л.: Химия, 1967. — 327 с.
27. Lagodzinskaya G. V, Klimenko Yu. Measurments and correction for magnetic
susceptibility with solenoidal magnet NMR spectrometer // J. Magn. Reson.
1982. V. 49, № 1. P. 1-7.
28. Heats E., Howarth O. W. Vanadium-51 and oxygen-17 nuclear magnetic reso-
nance study of vanadate (V) equilibria and kinetics // J.C.S. Dalton tr. 1981.
№5. P. 1105-1110.
29. Hoffman R.E., Levy G. C. Modern Methods of NMR Data processing and Data
Evaluation // Progr. Nucl. Magn. Reson. 1991. V. 23, №3. P. 211-258.
30. Лапташ H.M., Федотов M.A., Масленникова И. Г. Гидролиз летучего
оксофторотитаната аммония по данным ЯМР l9F, |7О, 49Ti // Ж. структ.
химии. 2004. Т. 45, № 1. С. 77-87.
31. NMR and Periodic table / Ed. В. E. Mann, R. К. Harris. — L.: Acad. Press,
1984. - P. 245.
32. Федотов М.А. Возможности метода ЯМР в аналитической химии рас-
творов неорганических веществ // Ж. аналит. химии. 1999. Т. 54, № 1.
С. 17-22.
33. Ward A.J., Morris A., Healy И. A. The use of trifluoroacetic acid in magnetic
susceptibility measurments by NMR spectroscopy // J. Magn. Reson. 1974.
V. 16, №2. P. 357-359.
34. Попл Дж., Шнейдер В., Бернстейн Г. Спектры ядерного магнитного резо-
нанса высокого разрешения / Пер. с англ. В.Ф. Быстрова и др.; Под ред.
Н. Д. Соколова. — М.: ИЛ, 1962. — 592 с.
35. Posener D. W. Precision in measuring resonance spectra // J. Magn. Reson.
1974. V. 14, №2. P. 121-128.
36. Лундин А.Г., Федин Э.И. ЯМР-спектроскопия. — М.: Наука, 1986. —
223 с.
Глава 2
МАГНИТНЫЙ РЕЗОНАНС ЯДЕР
НЕПЕРЕХОДНЫХ ЭЛЕМЕНТОВ
2.1. Водород
Неорганическая химия водорода, щелочных и щелочноземельных
металлов имеет много общего, что отражается и в ЯМР этих элемен-
тов. Важным аспектом химических свойств этих элементов являются
свойства их ионов в водной среде. ЯМР всех этих ионов имеет общие
черты. Некоторые свойства магнитных ядер указанных элементов при-
ведены в табл. 2.1.
Химия водорода проявляется в основном в трех электронных про-
цессах: образование ионов Н+ (потеря электрона), Н“ (получение
электрона), возникновение ковалентной связи (общей электронной па-
ры). ЯМР атомов Н, образующих связь, используется в неорганической
химии значительно реже, чем органической. Такое применение ЯМР
'Н находит в химии неводных и смешанных с водой растворов, где
по ЯМР 'Н растворителя можно следить за свойствами раствора,
и в химии комплексов, где с помощью ЯМР *Н лиганда можно изучать
структуру комплекса и его химические свойства.
Масштаб ХС ЯМР *Н «70 м.д., тем не менее обычно узкие линии
ЯМР 'Н обеспечивает высокую специфичность метода. Обычно изме-
нение ХС ЯМР *Н определяется диамагнитным вкладом в экраниро-
вание, но в случае гидридных комплексов металлов — парамагнитным
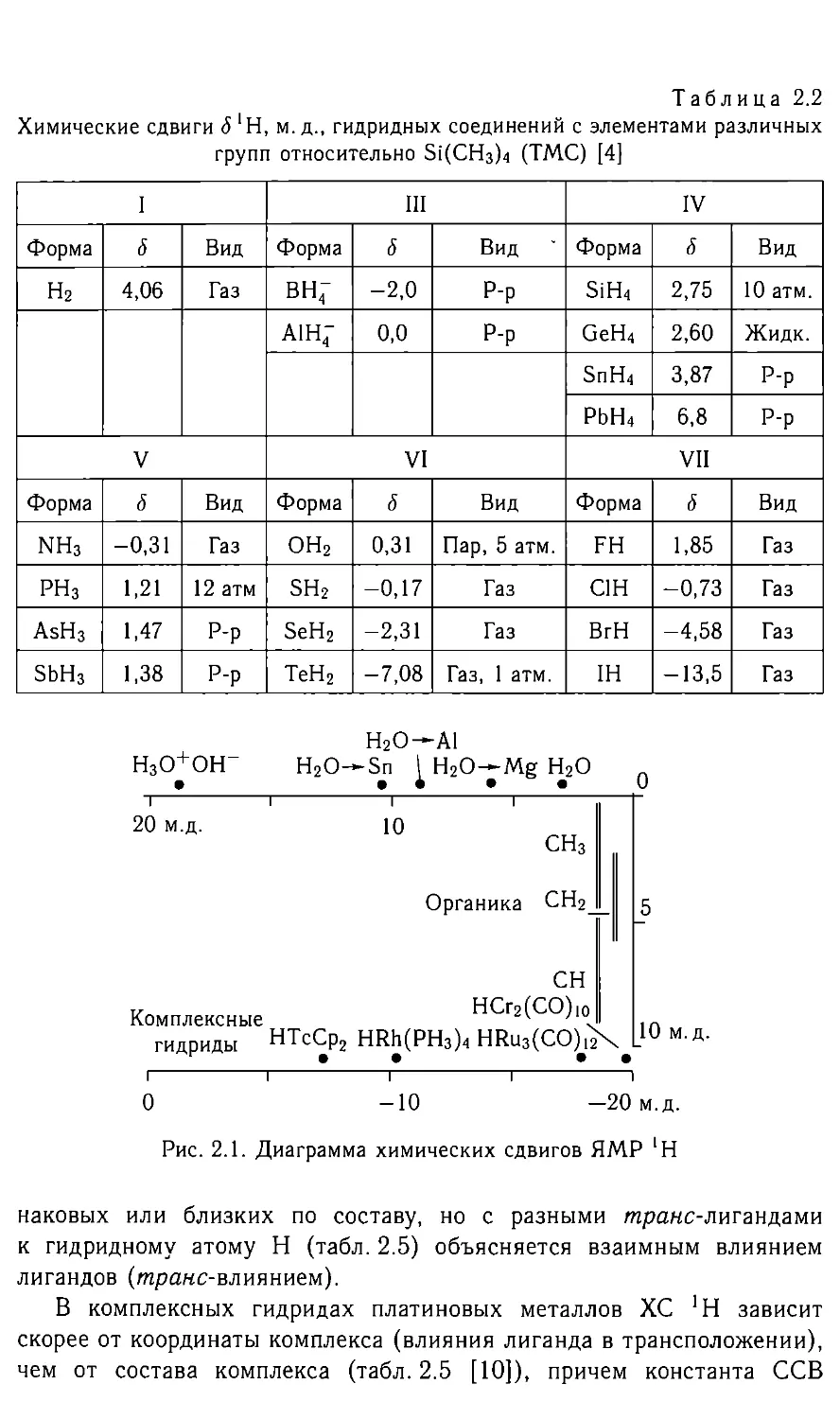
вкладом [1-3]. ХС некоторых гидридов в приведены в табл. 2.2 [4, 6].
Диаграмма химических сдвигов ЯМР *Н (рис. 2.1) показывает за-
висимость в изменении ХС от типа связи. Изотопный эффект ХС при
переходе от одного изотопа водорода к другому невелик (не более
0,5 м.д.), и обычно ХС 1Н, 2D и 3Н считают одинаковыми [1]. В слу-
чае парамагнитных сдвигов их величина зависит от гиромагнитного
отношения, и измерения на разных изотопах дают дополнительную
возможность для определения структур и свойств парамагнитных ком-
плексов.
При переходе в жидкое состояние на ХС существенное влияние
оказывает водородная связь (табл. 2.3).
Таблица 2.1
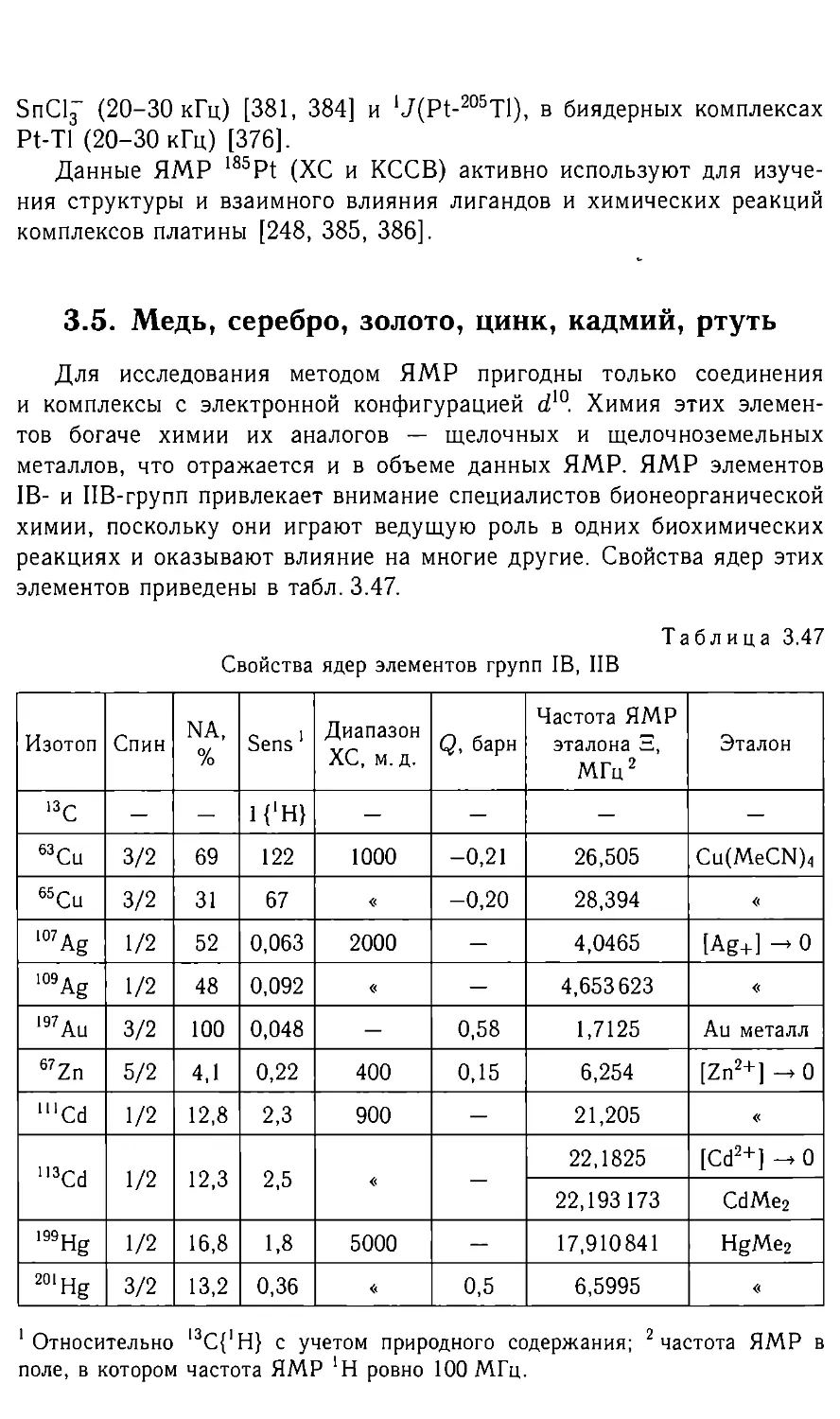
Свойства ядер магнитных изотопов групп IA и ПА
Изотоп Спин NA, % Sens 1 Диапа- зон XC, M. Д. Q, барн Частота ЯМР эталона 5, МГц2 Эталон
13С 1/2 1,1 1{‘H} — — 25,145000 ТМС
н 1/2 99,98 1900 20 — 100,000000 ТМС
2d 1 0,02 0,003 20 0,003 15,350650 d|2-TMC
Зн 1/2 — 23003 20 — 106,6635 3Н-ТМС
7 Li* 3/2 93 510 15 -0,045 38,864 Акваион при беско- нечном разбавле- НИИ
23Na 3/2 100 175 80 0,12 26,451
39К* 3/2 93 0,9 120 0,055 4,6665
85 Rb 5/2 72 14 250 0,25 9,655
87 Rb* 3/2 28 92 250 0,12 32,721
133Cs 7/2 100 89 400 — 13,117
9Be 3/2 100 26 40 0,052 14,053 Акваион при беско- нечном разбавле- нии
25Mg 5/2 10 0,6 30 0,22 6,12164
43Ca 7/2 0,15 0,02 40 -0,05 6,728
87Sr 9/2 7 0,4 — 0,36 4,3335
l37Ba* 3/2 11 1,5 800 0,28 11,113
1 Относительно |3С; 2 частота ЯМР в поле, в котором частота ЯМР
*Н ровно 100 МГц; 3 при содержании 100%; 4 5 = 6,121 636 МГц (0,18М
MgSO«-7H2O/D2O); * имеется другой магнитный изотоп с содержанием не ме-
нее 5 %.
Температурная зависимость ХС воды от 350 до 77 К и эффект
конденсации при переходе жидкой воды в лед изучены в [5].
Многие переходные металлы образуют комплексные гидриды по
большей части в составе карбонильных, фосфиновых и циклопента-
диенильных комплексов [6].
Квантовомеханические расчеты ХС гидридных атомов Н в рам-
ках теории Рэмзи [7] проведены для октаэдрических [8] и плоско-
квадратных [9] гидридных комплексов. На уровне расчетов тех лет
показано, что ХС таких атомов должен иметь отрицательный знак
(табл. 2.4), а различие в ХС гидридного протона в комплексах, оди-
Таблица 2.2
Химические сдвиги <5 *Н, м.д., гидридных соединений с элементами различных
групп относительно Si(CH3)4 (ТМС) [4]
I III IV
Форма д Вид Форма д Вид Форма 5 Вид
н2 4,06 Газ вн; -2,0 Р-Р SiH4 2,75 10 атм.
aih4- 0,0 Р-Р GeH4 2,60 Жидк.
SnH4 3,87 Р-р
PbH4 6,8 Р-р
V VI VII
Форма 6 Вид Форма 6 Вид Форма 6 Вид
NH3 -0,31 Газ ОН2 0,31 Пар, 5 атм. FH 1,85 Газ
РН3 1,21 12 атм SH2 -0,17 Газ C1H -0,73 Газ
AsH3 1,47 Р-Р SeH2 -2,31 Газ BrH -4,58 Газ
SbH3 1,38 Р-Р ТеН2 -7,08 Газ, 1 атм. IH -13,5 Газ
Н2О—Al Н3О+ОН“ Н2О—Sn H2O-j-Mg Н2О 3 м.д.
iiii 20 м.д. Ю сн3 Органика СН2_ СН rz НСгг(СО)1о Комплексные v . гидриды НТсСр2 HRh(PH3)4 HRu3(CO)*i2 • 5 1
i i i i i 0 —10 —20 м.д. Рис. 2.1. Диаграмма химических сдвигов ЯМР 'Н
наковых или близких по составу, но с разными транс-лигандами
к гидридному атому Н (табл. 2.5) объясняется взаимным влиянием
лигандов (транс-влиянием).
В комплексных гидридах платиновых металлов ХС *Н зависит
скорее от координаты комплекса (влияния лиганда в трансположении),
чем от состава комплекса (табл. 2.5 [10]), причем константа ССВ
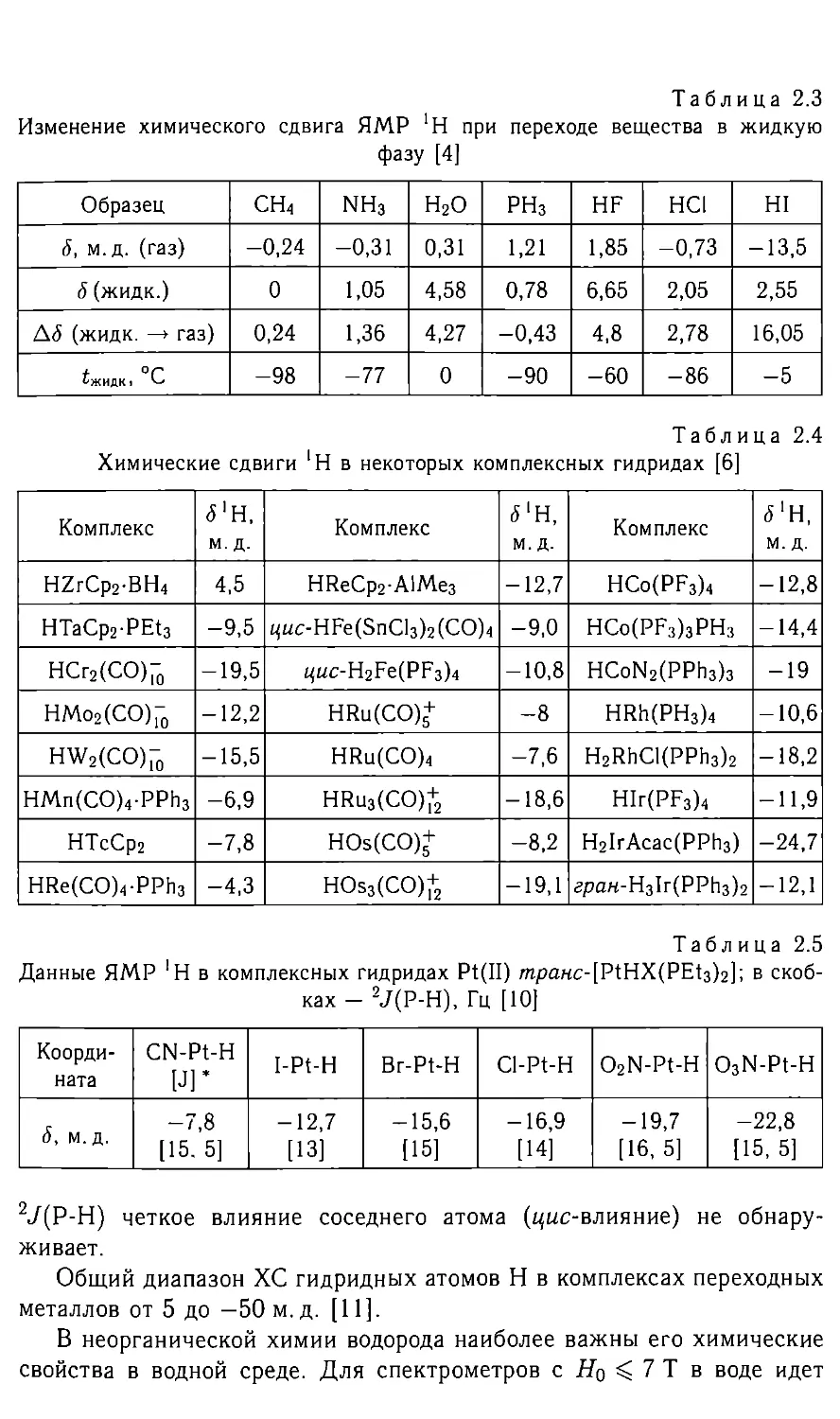
Таблица 2.3
Изменение химического сдвига ЯМР 'Н при переходе вещества в жидкую
фазу [4]
Образец сн4 NH3 Н2О РН3 HF HCI HI
6, м.д. (газ) -0,24 -0,31 0,31 1,21 1,85 -0,73 -13,5
<5 (жидк.) 0 1,05 4,58 0,78 6,65 2,05 2,55
Д<5 (жидк. —> газ) 0,24 1,36 4,27 -0,43 4,8 2,78 16,05
у. ОГ t-ЖИДК « -98 -77 0 -90 -60 -86 -5
Таблица 2.4
Химические сдвиги 'Н в некоторых комплексных гидридах [6]
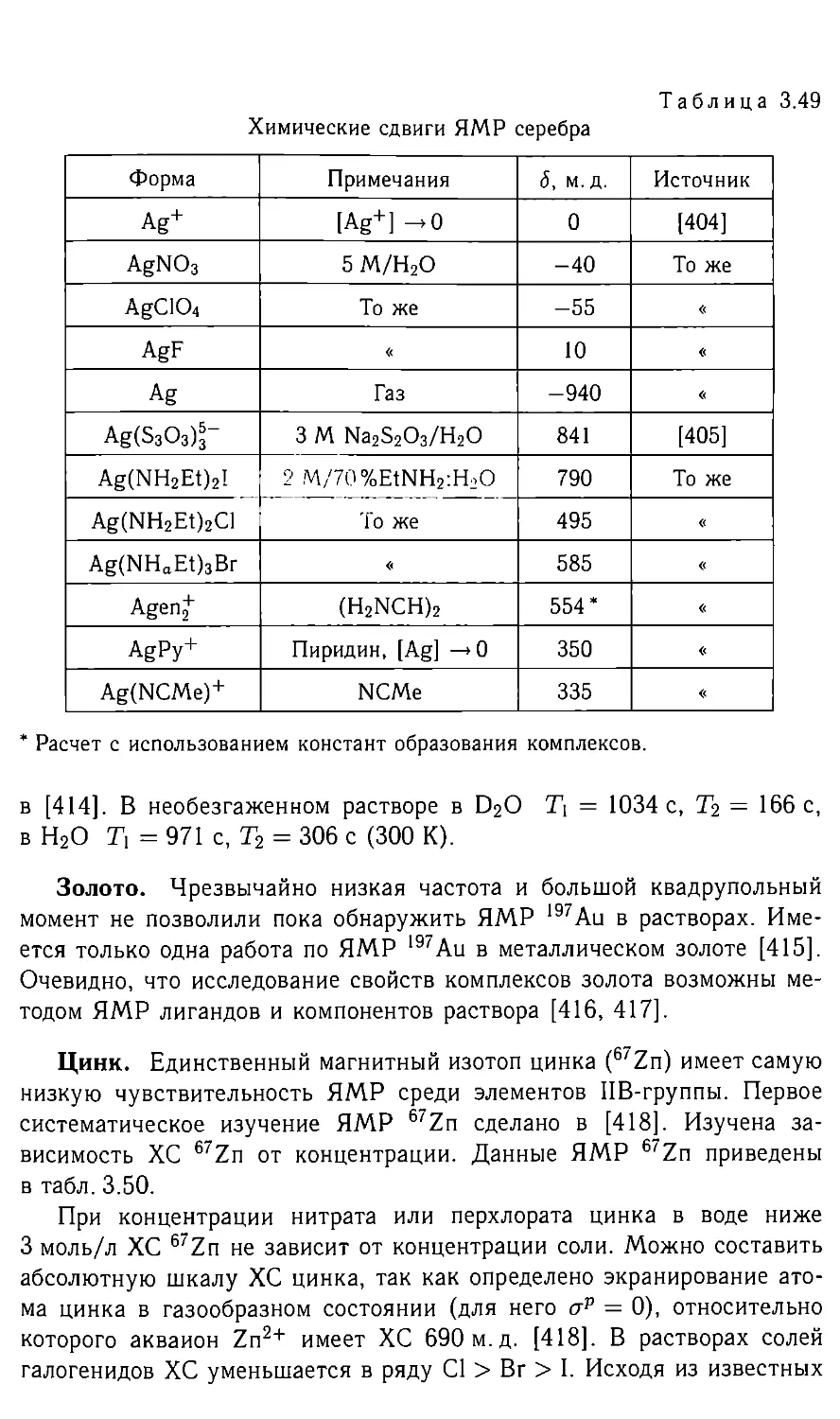
Комплекс <5'Н, м.д. Комплекс <5'H, M. Д. Комплекс 5‘H, M. Д.
HZrCp2-BH4 4,5 HReCp2-AlMe3 -12,7 HCo(PF3)4 -12,8
НТаСр2-РЕ1з -9,5 цис-НРе(5пС1з)2(СО)4 -9,0 HCo(PF3)3PH3 -14,4
НСг2(СО)[-0 -19,5 ^uc-H2Fe(PF3)4 -10,8 HCoN2(PPh3)3 -19
НМо2(СО)- -12,2 HRu(CO)+ -8 HRh(PH3)4 -10,6
HW2(CO)70 -15,5 HRu(CO)4 -7,6 H2RhCl(PPh3)2 -18,2
HMn(CO)4-PPh3 -6,9 HRu3(CO)+ -18,6 HIr(PF3)4 -11,9
НТсСр2 -7,8 HOs(CO)+ -8,2 Н21гАсас(РРИз) -24,7
HRe(CO)4-PPh3 -4,3 HOs3(CO)+ -19,1 apaH-H3Ir(PPh3)2 -12,1
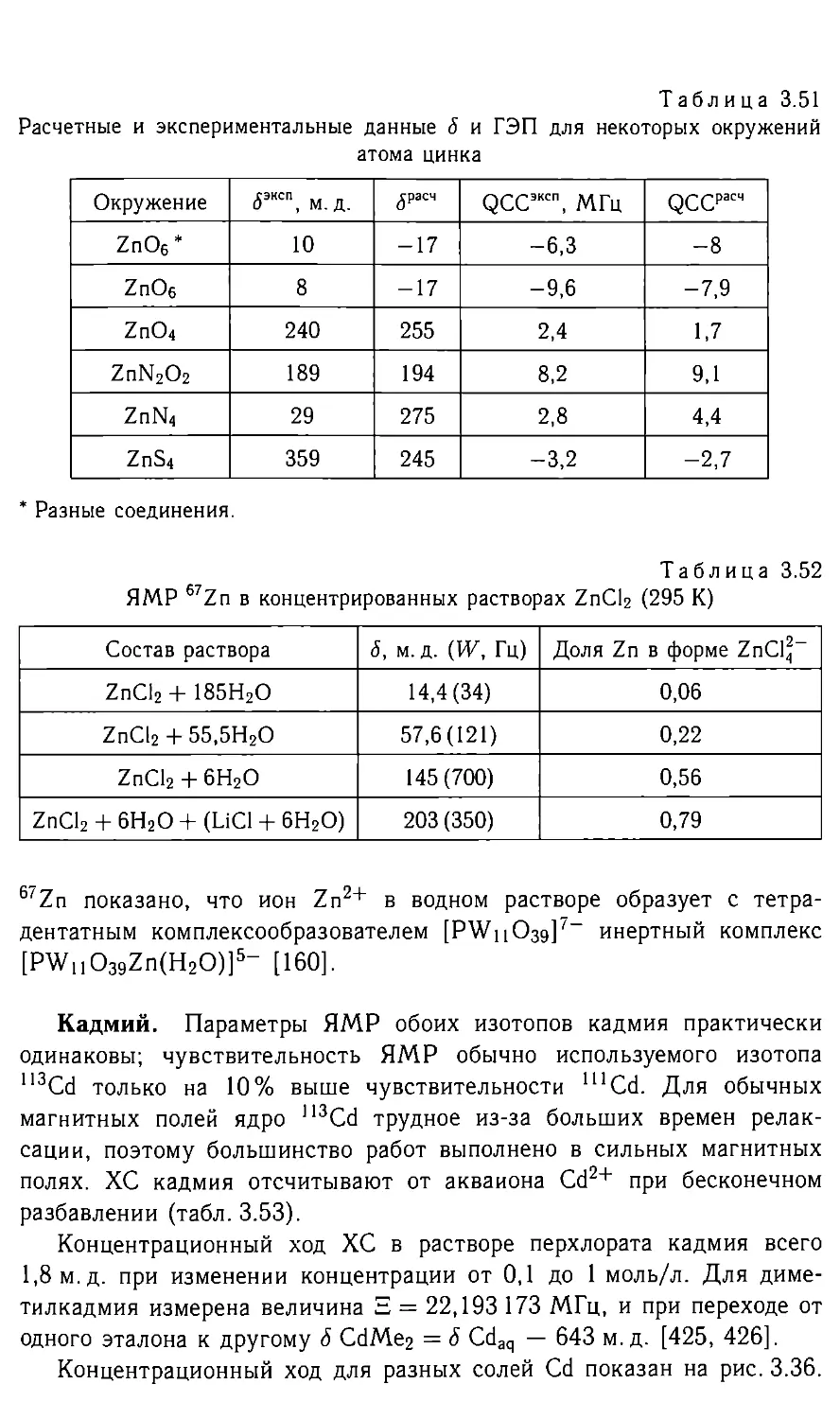
Таблица 2.5
Данные ЯМР 'Н в комплексных гидридах Pt(II) транс-[РШХ(РЕ1з)2]; в скоб-
ках - 2J(P-H), Гц [10]
Коорди- ната CN-Pt-H [J]’ I-Pt-H Br-Pt-H Cl-Pt-H O2N-Pt-H O3N-Pt-H
<5, м. д. -7,8 [15. 5] -12,7 [13] -15,6 [15] -16,9 [14] -19,7 [16, 5] -22,8 [15, 5]
2J(P-H) четкое влияние соседнего атома (цис-влияние) не обнару-
живает.
Общий диапазон ХС гидридных атомов Н в комплексах переходных
металлов от 5 до —50 м. д. [11].
В неорганической химии водорода наиболее важны его химические
свойства в водной среде. Для спектрометров с Hq 7 Т в воде идет
быстрый в шкале ЯМР 'Н обмен протонов, причем скорость обмена
минимальна при нейтральной кислотности [12]. ХС воды в большой
степени определяется межмолекулярным вкладом вследствие наличия
водородной связи. Водородная связь дезэкранирует протоны воды, при-
чем сдвиг достигает 4 м.д. (табл. 2.2). Природа ХС, возникающего
из-за водородной связи, описана в [4]. Вода, введенная в малых ко-
личествах в неполярные растворители, способна устанавливать с ними
водородную связь с образованием комплексов 1 : 1 [13].
В ХС воды водных растворах электролитов дают вклады как ка-
тионы, так и анионы; протоны координированной воды катионов непе-
реходных элементов сдвинуты в слабое поле, что вызывает в разбав-
ленных растворах сдвиг линии воды в слабое поле, пропорциональное
концентрации электролита. Сдвиг растет с увеличением заряда кати-
она. В концентрированных растворах пропорциональность нарушает-
ся. Анионы оказывают в основном разрыхляющее структуру действие
(уменьшают эффект водородной связи [4, 14]), которое увеличивается
с ростом размера аниона.
Наблюдаемый ХС 'Н воды является усредненной величиной всех
вкладов и может быть рассчитан по формуле [14]
55,5 <5цаб = ^sTTih -|- ^55,5 — mh — Fmz^ + SFmz, (2.1)
где §s — ХС координационной воды, SN = —4,48 + 0,00967 — ХС
воды с учетом температурного хода, т — моляльная концентрация
электролита, h и z — координационное число и заряд катиона, F —
фактор, учитывающий разрыв водородных связей анионами, SF — сдвиг
свободной воды (равный ХС парообразной воды). ХС ПМР воды в ко-
ординационной сфере иона (в скобках — гидратационное число КЧ)
для Ве2+ равно 8,04 (КЧ 4); для Mg2+ - 5,55 (КЧ 6); для А13+ - 8,74
(КЧ 6); для Ga3+ - 8,98 (КЧ 6); 1п3+ - 7,22 (КЧ 6) и Sn4+ - 10,1
(КЧ 6).
В растворах солей щелочных металлов ХС воды в основном опреде-
ляется разрыхляющим структуру действием (разрыв водородных свя-
зей) аниона, а не координацией воды к катиону. В этих растворах
обмен протонов воды с координационной сферой катионов обычно
быстрый в шкале ЯМР (рис. 2.2 [15]).
С повышением поля Но изменение шкалы ЯМР приводит к тому,
что обмен может выглядеть промежуточным или медленным. Заметим,
что скорость обмена воды с координационной сферой (кислородный
обмен) коррелирует с ХС координационной воды. Для катионов Са2+,
Cd2+, Ga3+, Mg2+, Sr2+, Th4+, Zn2+, ZrO2+ [4] ХС координационной
воды коррелирует с первой константой гидролиза К:
[М(Н2О)6]’,+ + Н2О Н3О+ + [МОН(Н2О)5]"_|. (2.2)
Рис. 2.2. Константа скорости К (25°, с *) обмена молекулами воды гидратных
сфер ионов металлов в водных растворах [15]
Для ионов 1Ю2+, РЬ2+ и А13+ эта корреляция не наблюдается, что слу-
жит указанием на частично ковалентный характер связи у последних
катионов. Поэтому авторы [16] полагают, что по сопоставлению рК
и ХС координационной воды можно определить степень электростати-
ческого взаимодействия в связи координационной воды. Обмен прото-
нов по реакции гидролиза (2.2) можно замедлить введением кислоты.
Поэтому в некоторых случаях координационную воду можно наблю-
дать в кислых растворах (например, в концентрированных растворах
А1С1з при /о ~ 100 МГц линии разделены уже при -11 °C) [17].
Исследование протонного окружения октаэдрического полиядерного
гексависмутиния [В1бО4(ОН)4]6+ [18] (рис. 2.3), у которых атомы О,
равно как и ОН-группы находятся на гранях поликатиона как цз-мо-
стики, в водно-ацетоновых растворах показало, что протоны воды
в данном растворе находятся в свободном и координированном виде.
Ниже 213 К наблюдали две линии — 5,8 м. д. (^-ОН-группы полика-
тиона) и 4,3 м. д. — обменную между свободной и координированной
водой. В то же время из данных ЯМР 17О [19] следует, что при 295 К
обмен протонов ОН-групп поликатиона с водой — медленный в шкале
времени ЯМР 17О, и линии атомов О (225 м.д.) и ОН-групп (95 м.д.)
поликатиона разделены (рис. 2.3).
Аналогичный цз-протон в октаэдрическом борате [BgHy]-, связан-
ный с тремя атомами В на одной грани октаэдра, при 300 К мигрирует
по всем граням, а при 190 К фиксируется на одной грани, при этом его
ХС не меняется [20]; такое положение протона разделяет атомы В на
1
500 300 100 0 м.д.
Рис. 2.3. Спектр ЯМР |7О водного раствора перхлората [В^ОДОН^]64- (2)
и структура поликатиона (/) [19]
две группы неэквивалентных атомов В по три в каждой. Остальные 6
протонов являются концевыми у каждого атома В.
ХС воды кислых водных растворов определяются влиянием 3 ча-
стиц (оксония, аниона и молекул кислоты) и будут результатом обмена
между частицами, входящими в уравнение
НА+Н2О = Н3О+ + А“.
Интересно, что ХС гидроксония и гидроксила практически одина-
ковы [21] (6 — 18,8 м.д.). В более ранних работах влияние аниона
учитывали не точно, что приводило к существенному различию ХС,
определенному в растворах разных кислот. Ряд работ [3] посвящен
определению констант диссоциации кислот в концентрированных рас-
творах. Степень диссоциации кислоты а можно получить из уравнения
=5,а + 52(1 — а),
где р = З.т(2 — х) — параметр, показывающий долю протонов в Н3О+
х — мольная доля кислоты, <51 — ХС Н3О+, 62 — наблюдаемый сдвиг
моногидрата кислоты, а термодинамическую константу диссоциации К
определяют из уравнения К13 = а/(1 — а)С, где а — активность, С —
концентрация кислоты. В апротонных растворителях, например SO2,
ХС кислотного протона коррелирует со значением р/С кислоты в во-
де [4]. Этим методом можно оценить р/С, определив значение только
одного ХС.
Сильные кислоты влияют на ХС протонов малополярных раствори-
телей. Так, в этилацетатных растворах фосфорвольфрамовой кислоты
H3[PWi204o] (ФВК) с ростом ее концентрации меняется ХС всех
протонов этилацетата (табл. 2.6 [22]): этилацетат
1 2 з
СН3-СН2-С-О-С(О)-СН3.
Обзор методов измерения кислотности с использованием ЯМР из-
ложен в [23] и цитируемой там литературе.
Таблица 2.6
Химические сдвиги 'Н этилацетата, м.д. от ГМДС [22]
[ФВК], ^''--\моль/л Атом 0 0,1 0,2 0,3 0,4
Н1 1,31 1,35 1,41 1,45 1,50
Н3 1,32 2,12 2,19 2,24 2,30
Н2 4,16 4,21 4,28 4,30 4,36
н2* — 3,89 3,95 3,98 4,04
* Наблюдали два квартета в присутствии ФВК.
ЯМР 'Н широко используют при изучении сольватации и состояния
ионов и молекул в смешанных растворителях. Большинство раство-
рителей содержит протоны в различных функциональных группах,
и по изменению ХС удается определить не только координационное
число, но и геометрию комплекса, расположение молекулы раство-
рителя в нем, а также термодинамические параметры сольватации.
Изучена сольватация Ве2+ в растворах Н2О/ДМСО и в водно-ацето-
новых растворах [24], хлорида лантана в водно-метанольных раство-
рах [25], щелочных и щелочно-земельных ионов в водно-ацетоновых
растворах [26]. Добавление неводного растворителя к воде замедляет
протонный обмен, и появляется возможность прямого наблюдения ко-
ординационной воды. Применение неводных растворителей позволяет
снижать температуру измерения и эффективно замедлять динамиче-
ские процессы. Условия изучения сольватации катионов в неводных
растворителях аналогичны тем же в водных растворах, с той раз-
ницей, что легче реализуется условие медленного обмена протонов
растворителя и сольватной оболочки; протоны последней наблюдаются
при этом раздельно от растворителя. Примерами таких исследований
могут служить: изучение метанольных растворов двух- и трехзарядных
катионов [27], солей алюминия и галлия в спиртах [28], ряда солей
в уксусной кислоте [29], солей Мп(П) в ДМСО [30], сольватации ани-
онов [31], строения 1-й и 2-й гидратных сфер ионов Еи3+ и Sm3+ [32].
ЯМР *Н применяется для изучения комплексообразования про-
тонсодержащих лигандов с диамагнитными и парамагнитными иона-
ми, в особенности лабильных комплексов. При образовании диамаг-
нитных комплексов наблюдаются небольшие сдвиги протонов лиган-
да и изменение их времен релаксации. Более значительные вклады
в релаксацию наблюдаются у комплексов с катионами, имеющими
квадрупольные ядра (А13+, Со3+ и т. д.). При медленном обмене по
линиям лиганда в комплексе можно определить сольватационное число
и геометрию комплекса. Температурные измерения позволяют во мно-
гих случаях определить энтальпии и энтропии комплексообразования.
Измерения на ядре комплексообразователя и других ядрах лиганда
позволяют получить дополнительную к данным ЯМР 'Н информацию.
Комплексообразование Со3+ с малоновой и этилмалоновой кислотами
('Н и 13С) изучено в [33]; комплексов M(IV) и M(VI) с винной
и лета-винной кислотами — в [34]; смешаннолигандных комплексов
Со(Ш) с диметилглиоксимом (DH), дифенилгуанидином (ДФГУ) со-
става СоХ(ЭН)2-ДФГУ, X = Cl, Вг, I, NCO, NCS, NCSe, NO2 - в [35]
(обнаружена корреляция ХС метильной группы диметилглиоксима
с электроотрицательностью лиганда X); пентадиенильных комплексов
Hg и Мо, содержащих связь Hg-Mo, — в [36]. Комплексообразование
Y(III) с НТФ — нитрилотриметиленфосфоновой кислотой — в [37].
В случае лабильных комплексов при быстром обмене лиганда на-
блюдается усредненный ХС закомплексованного и свободного лиган-
дов; по концентрационной зависимости ХС можно определить ХС ли-
ганда в комплексе. По температурной зависимости ХС можно оценить
скорость обмена и энергию его активации (см. гл. IV). По интенсив-
ности линий ЯМР 'Н при медленном обмене можно определить число
лигандов в комплексе.
Н-D обмен в алюмо- и галлогидридах изучены в [38], в комплексе
[А1(Н2О)6]3+ в [39].
Изучению строения оболочки Zr(IV) в водном растворе посвящены
ряд работ [40-43] и др. При растворении солей ZrOCl2, Zr(OH)2CI2
в воде стабилизируется тетраядерный циркониевый гидроксокомплекс
[Zr(OH)2(H2O)4]4'r, структурно охарактеризованный, данные ЯМР 'Н
которого пока остаются дискуссионными (рис. 2.4).
По данным [40], в растворах ZrOCl2 наблюдали одиночную линию
с ХС 7,84 м. д. В [41] изучены растворы Zr(OH)2Cl2-7H2O разных
производств; наблюдали линии с ХС 7,34; 7,77 (преобладает); 7,95
и 8,83 м.д. с небольшой вариацией ХС и интенсивностей линий образ-
цов разных производителей. В [42] изучен гидролиз Zr(IV); в процессе
гидролиза наблюдали одну линию с ХС 8 м. д. В [43] исследовали
растворы [Zr(OH)2(H2O)4]®+ методами ЯМР 'Н и 17О. В водно-ацето-
новых растворах при -70 °C наблюдали две линии протонов, в ЯМР
Н20 он2
Н20
Н20
но он
Н2О ОН2 Н2О он2
Рис. 2.4. Строение иона [Zr(OH)2(H2O)4]®+
17О — одну широкую линию, с соотношением О : Zr = 2 : 1. Авто-
ры полагают, что в комплексе координационная вода двух типов —
[/^ОНЫНдО^НгО)"]®+, причем время жизни (НгО)1 ~ 0,1 с, вто-
рой — много короче.
При наложении линий лиганда в комплексе и свободного разделить
их помогает введение в раствор сдвигающего реагента (шифт-реагента,
чаще комплекса редкоземельного иона), который сдвигает линии сво-
бодного лиганда в слабое поле тем сильнее, чем ближе геометрически
к молекуле шифт-реагента наблюдаемый протон.
В ЯМ-релаксации протонов в жидкостях преобладает внутримоле-
кулярный вклад (см. гл. 1). Для многоатомных молекул обнаружена
линейная связь между Т\ и коэффициентом самодиффузии D [44].
Анизотропное вращение молекул координационной воды НгОс акваио-
на алюминия обнаружено при изучении температурного хода Т\ НгОс
(<5 = 9,2 м.д.) [45].
В водных растворах парамагнитных ионов быстрый протонный об-
мен приводит к наблюдению одной линии ЯМР 'Н. В этом случае
парамагнитный вклад в ЯМ-релаксацию преобладает над другими; по
нему появляется возможность исследовать свойства гидратированных
парамагнитных ионов, а также находить их концентрацию. В опреде-
ленных случаях ЯМ-релаксационный метод существенно превосходит
другие методы анализа этих ионов. Применение дейтерозамещенных
лигандов облегчает наблюдение парамагнитных сдвигов (поскольку
парамагнитные вклады в ширину линии пропорциональны квадрату
гиромагнитного отношения, а парамагнитные сдвиги — первой степени,
переход от ’Н к 2D резонансу уменьшает ширину линий в 43 раза,
а сдвиги — в 6,5 раз), что позволяет выделить разные вклады в па-
рамагнитный сдвиг и релаксацию. Подробнее ЯМР и ЯМ-релаксация
в парамагнитных растворах рассмотрены в гл. 4.
Для установления структуры неорганических ионов и молекул ме-
тод ЯМР 'Н применяется тем реже, чем меньше разнообразие неор-
ганических протонсодержащих молекул по сравнению с органически-
ми. В [20] исследовано строение аниона [ВбН7]_ и миграция его
цз-протона.
Установлено существование в растворе стереохимически гибких
анионов В4Н“, В5Н", В6НП, B7Hf2> ('Н, ИВ) [32]; [В3Н8]- [В3Н7Х]~
[В3НбХ2]- [46, 47], изучено состояние иона BF^ (*Н, 11 В, 19F) [48].
Данные ЯМР *Н аниона [В3Н8]“ и его замещенных аналогов приведе-
ны в табл. 2.7. Подвижность этих анионов приводит к эквивалентности
Таблица 2.7
Данные ЯМР ’Н некоторых триборидов [46]
Анион <51Н, м. д. от ТМС 'J(H-"B), Гц н н н н н—в2—в3— н Н Х(Н)
[в3н8]- 0,18 32,5
[В3Н7С1]- 1,46 18 (В3) 25 (В1'2)
[B3H7(NCO)]- 1,22 —
[В3Н6С12]- 2,57 65 (В1)
[B3H6C1(NCS)]~ 2,3 —
как протонов, так и атомов В; спектр *Н представляет децет (мульти-
плет из 10 линий) от расщепления на трех атомах ИВ (с подспектром
от расщепления на |0В). Замещение замедляет обмен и выявляет неэк-
вивалентность атомов В с разными J(B-H).
В [49] изучены растворы NaSnH3 в жидком аммиаке (установлено
существование равновесия между SnH^“ и SnH4, построены молекуляр-
ные орбитали этих комплексов); взаимодействие Н2О и NH3 в водно-
аммиачных растворах исследованы в [50].
Данные ЯМ-релаксации щелочных металлов позволяют изучать
микроструктуру водных растворов электролитов [51]. ЯМ-релаксация
*Н широко применяется для изучения структуры концентрированных
растворов электролитов, обычно в сочетании с ЯМР других ядер (ка-
тионов и анионов). Так, например, изучены структуры 11 М водного
раствора L1C1 [52] и концентрированного раствора ZnCl2 [53].
Примером использования ЯМР 2D для изучения комплексообразо-
вания в экстрактах UO2+ является [54]. Другие работы для изучения
экстракции металлов методами ЯМР приведены в гл. IV. ЯМ-релак-
сация *Н широко используется для изучения коллоидных растворов;
систем с поверхностью раздела, гелеобразных, гетерогенных, жидко-
кристаллических систем, Ряд работ посвящен изучению молекул в ад-
сорбированном (например, [55]), интеркалированном [56] состояниях
и соединений включения (клатратах) [57], однако их обсуждение вы-
ходит за рамки этой книги. Информативность и особенности ЯМР 'Н
воды в гетерогенных системах обобщены в книге [58] и отражены
в обширной литературе к ней.
2.2. Щелочные и щелочноземельные металлы
Химические свойства щелочных и щелочноземельных металлов
определяются валентными s-электронами; их ионы обладают оболоч-
кой инертных газов, и ХС в растворах возникают из-за искажения
электронной оболочки при столкновении частиц (фактор SQ в форму-
ле (1.4)). Вследствие малой энергии ионизации легко образуются ионы
и ионные соединения, поэтому химия этих элементов преимущественно
ионная, за исключением лития, бериллия и магния, которые образуют
и ковалентные связи.
Щелочные металлы. Все щелочные металлы имеют изотопы
с магнитным моментом ядра; их свойства приведены в табл. 2.1. Литий,
калий и рубидий имеют по два магнитных изотопа, что расширяет
возможности применения ЯМР их ядер.
Ионы щелочных металлов в растворах образуют лабильные сольва-
ты или комплексы, и только в последнее время с помощью сложных
полидентатных лигандов (краун-эфиров и криптандов) были получены
инертные в масштабе времени ЯМР комплексы щелочных металлов
и даже застабилизированы их отрицательные ионы [59]. Мерой диа-
пазона ХС ядер щелочных металлов можно считать разность ХС иона
в водном растворе при бесконечном разбавлении и закомплексованного
иона (например, криптата), или свободного иона (в газовой фазе)
и акватированного [60]. Из табл. 2.8 видно, что диапазон ХС возрастает
Таблица 2.8
К оценке диапазона ХС щелочных металлов
Изотоп 7 Li 23Na 39К 87 Rb l33Cs
<7* [60], М.Д. -И ±2,8 -60,5 ± 1 -101 ±5,4 —212 ± 1,2 -344 ±5,8
М- -15 -62 — -185 -300
<7* = 1 — g-|(NM.R)/g'I(free atom), g,(NM.R) — величина ядерного магнетона,
определенная из ХС акваиона при бесконечном разбавлении; g'/free atom) —
та же величина, определенная из измерений атомных пучков металла.
от Li к Cs. Из-за лабильности комплексов при изменении соотношения
металл/лиганд наблюдают усредненные параметры ЯМР:
= j _ g,(NMR)
g/free atom) ’
g^NMR) — величина ядерного магнетона, определенная из ХС аква-
иона при бесконечном разбавлении; g^free atom) — та же величина,
определенная из измерений атомных пучков металла.
Наличие квадрупольных моментов у всех ядер щелочных металлов
приводит к ЯМ-релаксации по квадрупольному механизму. Это обу-
словило широкое применение метода магнитной релаксации этих ядер,
в особенности для Li и Na, где диапазон ХС мал. Напротив, ХС 133Cs
и в меньшей степени 39К зависят от окружения иона, и их ЯМР широко
используется при изучении свойств растворов. Поскольку измерение
ХС менее трудоемко по сравнению с релаксационными измерениями,
измерения спектров более доступны для химических исследований.
В то же время при ЯМ-релаксации по чисто квадрупольному механиз-
му Т\ = Т%, по ширине линии ЯМР определяется и время релаксации.
Последнее возможно тогда, когда другие вклады в ширину линии
несущественны.
Для большей части ионов щелочных и щелочноземельных металлов
ЯМ-релаксация является наиболее информативным методом изучения
их состояния в растворе. Для интерпретации релаксационных данных
предлагаются различные модели ближнего окружения иона в растворе,
и по соответствию зависимостей ЯМ-релаксации от различных пара-
метров (концентрации, температуры, растворителей и т. д.), рассчитан-
ных по этим моделям и экспериментальных, можно судить о справед-
ливости модели состояния иона в растворе. Квадрупольная релаксация
ядер с нечетным спином в растворах рассмотрена в [61-63]. В общем
случае ЯМ-релаксация спинов с I > 1 не экспоненциальна; она являет-
ся суммой I + 1/2 экспонент для полуцелого и I экспонент для целого
спина:
Mz - Мо = Мо\^ Лехр(--М; Мх = Мо\^ ЛехрГ-^-У (2.3)
X -*2г/ X -*2г/
г г
Для ядра с I = 3/2 предэкспоненциальные коэффициенты не за-
висят от релаксационных параметров, и релаксацию можно выразить
аналитически [62]. В этом случае продольная намагниченность затуха-
ет в виде суммы 0,2ехр(—T|t) + 0,8exp (—rtf,'), а поперечная — в виде
0,6ехр(-S1Z) + 0,4ехр(-S2i), где
И = Р /С2 2< Г^=РТТ7—2’ 5i = f fTc + Тсг 2У> (2-4)
1 + cj тс 1 + 4ш2Тс 2 у 1 4- cj тс J
р (_____
2 Ь Wrc2
Тс
1 + 4ш:
(2 \ 2
27regQ\ (2 5)
n j
При условии предельного сужения штс С 1, И = гг = si = зг,
продольная и поперечная намагниченности затухают экспоненциально
со временем релаксации Т\ = Т%-
При наличии обмена между двумя состояниями (А и В) затухание
в общем случае выражается суммой четырех экспонент. Однако в том
случае, когда для состояния А осуществляется условие (штс)А С 1,
и время пребывания ядра в состоянии А тА > тв, релаксация будет
почти экспоненциальной с ri ~ 0,2ci + 0,8сг; гг ~ 0,6di + 0,4^2. где
Ci = (n)A +
Ра
Рв(ггВ + Тв)
di = (si)A +
f (5г)в[(Дг)в + тв *]
I 1Wb+T.-']
+ (Дш)2
(2.6)
(2.7)
причем i = 1,2; рА/рв = тА/тв; рА +рв = 1; Дш — шА - шв. При шт С 1
поперечную и продольную релаксации можно считать экспоненциаль-
ными для всех нечетных спинов; при шт 3> 1 поперечная релаксация
выражается суммой экспонент с сопоставимыми вкладами. Для I = 5/2
и 7/2 соотношение этих вкладов зависит от шт; эти зависимости для
7 = 7/2 приведены на рис. 2.5.
В растворах свободные ионы обычно находятся в условиях шт 1,
т. е. наблюдается экспоненциальная релаксация. Тогда
(2\
1 + v (2-8)
О j
3(2/ + 3)
W — ширина линии ЯМР на полувысоте; £ = —*-------------П =
е QzzQ « /
= —ь — константа квадрупольного взаимодействия (в единицах
частоты); фактор асимметрии ту = qxx — qyy/qZz (0 < ту < 1).
Фактор С, показывает, что с ростом спина при прочих равных время
релаксации удлиняется:
Спин 1 3/2 5/2 3 7/2 9/2
С 0,375 0,1 0,024 0,015 0,0102 0,0056
Ось z выбирается в направлении градиента. Эффективный градиент
qzz = 9zz(l - Too) значительно превышает ГЭП (g°z), создаваемый за-
рядом. Здесь 7оо — фактор антиэкранирования Штернхаймера, возни-
кающий из-за возмущения электронов заполненных оболочек.
Неточность теоретического определения 7^ затрудняет нахождение
квадрупольных моментов ядер из данных по ЯМ-релаксации; расчет-
a
б
Рис. 2.5. Продольная (а) и поперечная (б) релаксации ядер со спином 7/2
в отсутствии обмена (I, II) и при двухместном обмене (III, IV), в зависимости
от параметров шт и х
ные значения 1 - 700 для Rb+ и Cs+ равны соответственно 51 и 99 [64].
721,2 зависит от вязкости ту* и абсолютной температуры Т, поскольку
величина тс пропорциональна ту*/Т. При постоянном ГЭП измерение
Т?1>2 позволяет определить относительные изменения тс.
В случае образования комплекса с ионом для комплекса может
выполняться условие о?отс > 1, и тогда будут обмениваться между
собой состояния с wt < 1 и шт > 1. Расчетные данные для двух-
местного обмена ядра с I = 7/2, рд > Рв, штА = 0,001 и штв = 5
в функции Рв9в./Ра9а приведены на рис. 2.5, где дА,в — ГЭП в месте А, В
(предполагается ГЭП аксиальной симметрии). Продольная релаксация
экспоненциальна и в случае двухместного обмена в широком диапа-
зоне значений параметра х — рв<1в/РА<1к, в то время как поперечная
релаксация будет экспоненциальной при х 10-3. Параметр х можно
регулировать за счет изменения рв/Рл-
Развиты два подхода к интерпретации ЯМ-релаксации квадруполь-
ных ядер в растворах солей щелочных металлов. Авторы [65-69]
рассматривают ГЭП как следствие статического искажения окружения
ядра из-за ухода одного лиганда сольватной сферы, в результате чего
симметрия окружения перестает быть кубической. К этому же приво-
дит и образование ионной пары с анионом. Напротив, Деверелл [70]
считает, что координационная сфера искажается из-за соударений с мо-
лекулами и ионами, т. е. искажения носят динамический характер.
Обзор работ по применению ЯМ-релаксации для изучения состояния
ионов в бинарных растворителях сделан в [71]. Наблюдения макси-
мумов в концентрационных зависимостях в растворах бинарных
растворителей [68] противоречат модели Деверелла. Выполнено мно-
жество работ по изучению состояния щелочных и щелочноземельных
ионов в растворах.
Эффекты экранирования ядер щелочных и щелочноземельных эле-
ментов изучены менее подробно, чем их ЯМ-релаксация. Данные по
ХС и ЯМ-релаксации в чистых жидкостях должны быть экстрапо-
лированы к бесконечному разбавлению, однако в жидкостях с ма-
лой диэлектрической проницаемостью даже при низких концентрациях
трудно исключить образование ионных пар. В общем экранирование
ионов возрастает с увеличением донорной способности координирую-
щей группы молекулы растворителя.
Данные по ХС 23Na показывают [72], что координация атомами
азота дезэкранирует, в то время как координация атомами кислорода
экранирует ядро Na+. Зависимость ХС от электронодонорной способ-
ности выражают кривыми ХС — рК [72] или ХС — донорное число
Гутмана [73, 74] (Na+), [74] (К+, Cs+). Для Li+ такой зависимости не
найдено, что указывает на иной механизм экранирования [75].
Влияние на экранирование смешанной сольватации изучали в
[72, 76].
В водных растворах галогенидов щелочных металлов обнаружена
нелинейная зависимость ХС металла [77, 78] (и галогенидов [79])
от концентрации соли (рис. 2.6, 2.7). Поскольку катионы щелочных
Рис. 2.6. Химические сдвиги щелоч-
ных металлов в водных растворах их
иодидов и нитратов
Рис. 2.7. Зависимость хими-
ческих сдвигов ЯМР l33Cs
от концентрации растворен-
ной в воде соли
металлов (и галогенид-ионы) имеют оболочку благородных газов, то
возникновение экранирования возможно только как результат дина-
мического искажения (взаимопроникновения) внешних электронных
оболочек при соударении с противоионами и молекулами растворителя.
Теоретическая оценка влияния соударений на ХС ионов М+ приведены
в [70, 80]. Измерения ХС 133Cs в области малых концентраций Cs+
проведены на рис. 2.7 [80]. Сопоставление экспериментальных данных
с теоретическими зависимостями совпадают от малых концентраций
до 5 моль/л [80]. Влияние концентрации на ХС данного металла воз-
растает в ряду ОН- < С1“ <Вг“ <1~, а при том же анионе — в ряду
металлов Li < Na < К < Rb < Cs.
Там же измерена зависимость ХС 133Cs растворов CsCl в разных
растворителях, отсчитанных от 0,1 М CsCl в воде; ХС лежат в диапа-
зоне от +58 (ДМСО) до —54 м.д. (МеОН). Растворы n-алконатов Cs
в соответствующих карбоновых кислотах (п — 0-8) изучены в [81].
Диапазон ХС Li+ не превышает величины сдвига в результате вли-
яния растворителя; только при комплексообразовании с краун-эфирами
в растворе стабилизируется ион Li- с ХС ~ —15 м.д.
Больший, чем у лития, диапазон ХС, удобная для измерений ши-
рина линии ЯМР 23Na и широкое использование растворов натриевых
солей в промышленности и науке вызывают постоянный интерес к ис-
следованиям растворов иона Na+ методом ЯМР 23Na. Обзоры таких
работ сделаны в [73, 74].
Вследствие сравнительно низкой чувствительности ЯМР 39К изме-
рения ЯМ-релаксации 39К для изучения свойств растворов или ком-
плексообразования применяется редко. Преимущественное комплексо-
образование гетерополианиона PWnO^ с Li+ при избытке К+ уста-
новлено в [82] по сужению линии ЯМР 39К при ЯМР-титровании
растворов калиевой соли этого аниона твердым хлоридом лития.
Состояние ионов К+ и Cs+ в растворах изучают преимущественно
по изменению ХС ЯМР этих ионов. При одинаковой концентрации
аниона ХС Cs+ (К+) растет с увеличением размера аниона ([77, 78],
рис. 2.8).
Рис. 2.8. Зависимость химического сдвига примесного иона цезия от концен-
трации моно- и полиядерных анионов: / — [FelllPWii04o]6-; 2 — [PdCl4]2-;
3 - СГ; 4 - ОН“; 5 - [CelvMo12O42]8-; 6 - [LaW10O36]9"; 7 - СЮ4"
Это свойство может быть использовано для индикации в растворе
полиядерных анионов и коллоидных частиц [83], например, в процессе
образования нанометровых частиц Ru(II) [84].
Состояние иона Li+ в растворах изучено довольно подробно
[85-87]. Исследована ЯМ-релаксация в водном растворе (и ее темпе-
ратурная зависимость) [85], и в переохлажденном 11 М растворе LiCl
[88]; гидратационное число (~5) определено в [89]. Взаимодействие
с парамагнитным ионом Ni2+ изучалось в [90], обмен между Li+
и Na+ молекулами основных растворителей — в [91].
Поскольку квадрупольный момент 23Na довольно велик, ширина
линии превышает 10 Гц даже в маловязких растворах. При шотс » 1
релаксация описывается суммой двух экспонент [61], однако откло-
нение от экспоненциального затухания обычно мало. При анизотроп-
ном движении в жидкокристаллических растворителях, биологических
системах, мембранах, гелях, клатратах наблюдается и неэкспоненци-
альная релаксация. В жидкокристаллических растворителях возможно
раздельное наблюдение переходов и определение константы квадру-
польного взаимодействия [92]. ЯМ-релаксация 23Na применена для
изучения микроструктуры глинистых гелей лапонита [93].
Состояние иона Na+ в водных растворах изучалось в растворах
солей галогенидов [67, 69, 70-74, 76, 78, 87, 93-102]. Установлено, что
скорость ЯМ-релаксации растет в ряду анионов С1~ < Вг“ ~ I- <F“
[66, 69, 94, 100]. Состояние Na+ в присутствии анионов NOT, С1О^“,
СЮ^, ReO^, ВГО7, IO", SCN-, BF-, PF-, HSO7, SO2-, Pof" иссле-
довали в [97], алюминатов — в [103, 104]. Состояние Na+ в расплаве
NaF-AlF3-Al2O3 изучали в [105]. Расплавы солей К методом ЯМР 39К
изучены в [106].
Изучение комплексообразования ионов М+ с криптандами привело
к открытию щелочных анионов: способность криптандов к комплексо-
образованию так велика, что они экстрагируют катион из растворов
в ТГФ, метиламине. Потеря электрона приводит к образованию М-,
наблюдаемого по отдельной линии при низкой температуре, ХС которой
не зависит от растворителя. Отсутствие влияния растворителя показы-
вает, что Зз-орбитали эффективно экранируют 2р-орбитали иона Li- от
влияния растворителя. Комплексообразование с криптандами изучали
в [107], Na+ в [108], К+ в [109], Rb+ в [109-110], Cs+ в [111-115].
Константы устойчивости щелочных ионов в растворах 18С6 и криптан-
да С222 в ацетоне и ацетонитриле определены в [107, 112]. Состояние
Rb в растворах краунэфира 18-crown-6 (18С6) в разных растворителях
при 22 °C изучено в [ПО]. Щелочные металлы ограниченно растворя-
ются органике; Cs не растворяется в краунэфирах 12С4 и 15С5, однако
в их растворе в ТГФ растворяется до 5% Cs. При 193 К в растворе
15С5/ТГФ фиксируются три линии с ХС 66, 2 и —300 м.д., отнесенные
к [Cs+Le-], [Cs+L] и Cs-, соответственно [115]. Криптанды образуют
с ионами М+ лабильные комплексы 1 : 1, а ионы М2+ — такие же
инертные комплексы; определены константы образования комплексов
и их зависимость от растворителя.
ЯМР щелочных металлов активно используется в изучении ионного
обмена в тканях и других биологических объектах, например, ускоре-
ния транспорта Cs+ через липидную мембрану [116], что относится
уже к вопросам бионеорганической химии.
Щелочноземельные металлы. Чувствительность ЯМР щелочно-
земельных элементов, за исключением 9Ве, невысока. Однако значение
метода ЯМР возрастает в связи с тем, что для Ве2+, Mg2+, Са2+ не
работают методы ЭПР, ЭСП. Все магнитные ядра элементов 2А-группы
имеют квадрупольный момент, наименьший из которых у 43Са. Барий
имеет два магнитных изотопа, причем различие в чувствительности
ЯМР у них компенсируется меньшей шириной линии у менее распро-
страненного изотопа.
Диапазоны ХС ядер трудно оценить единообразно. Для 9Ве в орган-
обериллиевых соединениях диапазон ХС около 25 м.д. [117]. В ком-
плексе с аденозинтрифосфатом определен его ХС — 260 м.д. [117],
однако авторы [119] полагают, что этот результат завышен более чем
на порядок.
В MgO линия ЯМР 25Mg сдвинута от акваиона на +25 м.д. [120].
Для 43Са сделано мало измерений из-за малой чувствительности ядра.
Комплекс Са2+ с ЭДТА дает линию +20 м.д. от акваиона, причем
обмен между акваионом и комплексом медленный [119]. Концентра-
ционная зависимость ионов 25Mg, 43Са, 35С1 в растворах перхлоратов
магния, кальция, стронция изучена в [121]. Для 135-137Ва известен ХС
между свободным ионом и акваионом 800 ±20 м.д. [122]. Обзор по
применению ЯМР 25Mg, 43Са в биологии сделан в [119].
Механизм ЯМ-релаксации ядер элементов 2А-группы в основном
квадрупольный, за исключением 9Ве, у которого в водных раство-
рах имеется и диполь-дипольный вклад, а при повышенных тем-
пературах — спин-вращательный [74]. Измерения ЯМ-релаксации
при переменной вязкости показывают, что основную роль играют не
статические, а динамические искажения окружения ядер. Для вод-
ных растворов скорость релаксации при бесконечном разбавлении
R2 = 4,5 с"1 для 25Mg [123], для 43Са - 0,75 с-1 [124, 127], для 87Sr -
205 с-1 [134] и для 135Ва — 1700 с-1 [122]. ЯМ-релаксацию в невод-
ных растворах изучали для 25Mg [126], где показано, что большая
ширина линии в растворах MgCl2, MgBr2 в ацетоне, ТГФ, ДМФ обу-
словлена сильным катион-анионным взаимодействием (вероятно, в ви-
де ионных пар). Добавление I- приводит к образованию ионов Mg^X-
и сужению линий. В ЯМ-релаксацию малых ионов (Ве2+, Mg2+, Са2+)
заметный вклад может дать внешняя координационная сфера. Рас-
чет этих вкладов и экспериментальные данные обсуждаются в [127],
рассмотрение концентрационных эффектов и влияния других ионов
сделано в [125]. ЯМ-релаксация Mg2+ в водных растворах рассмотрена
в [125, 127], в водно-метанольных растворах — в [128], сольватация
Ве2+ в ацетонитрильных растворах — в [129]. По зависимостям 7)
25Mg, 43Са, 87Sr и 137Ва от концентрации изучены гидратация М2+
и влияние анионов, которое дает вклад в ЯМ-релаксацию при боль-
ших концентрациях [130]. Константы квадрупольного взаимодействия
43Са в комплексах Са2+ с бидентатными и циклическими лигандами
оценены по величинам Д 43Са в [131]. Состояние ионов Mg2+ и Са2+
в водных растворах изучали в [124], в различных растворителях —
в [132].
В ряде случаев определены термодинамические параметры и кон-
станты образования комплексов. Концентрационная зависимость ХС
водных растворов щелочноземельных ионов мала [119]. Концентра-
ционная зависимость ХС акваиона магния ^2 м.д. при [Mg2+] —
0-4 г-ион/л [133], а для 43Са около 10 м.д. [124]. Концентрацион-
ная зависимость ХС 87Sr акваиона Sr2+ в растворах 5гС1г около
1,5 м. д./г-ион, а ХС 2М раствора 5гС1г относительно бесконечно
разбавленного +3,4 м.д. [3]. (В [134] полагают, что концентрационная
зависимость более 10 м.д.) Ширина линии ЯМР 87Sr 0,3 М раствора
SrCl2 при 40 °C в поле 7 Т около 50 Гц; большая ширина линии ЯМР
137Ва не позволила оценить концентрационный ход ХС ЯМР 137Ва
в водных растворах [122].
Полагают, что изменения ХС 25Mg, 43Са, 87Sr, 137Ва определяются
парамагнитным вкладом в экранирование, и для качественной интер-
претации [135] используется теория Кондо [136], однако для провер-
ки правильности выбранных моделей не хватает экспериментальных
данных. Экспериментальные величины разностей между ХС акваиона
и свободного иона Д° = 5 Maq — 5 Mf неточны, поскольку величина
<5М[ определяется из оптических измерений весьма грубо; для Ва
Д° = 800 м.д., для 43Са и 87Sr неизвестен даже знак Д°. Экраниро-
вание увеличивается при введении оксоанионов и уменьшается при
введении галогенид-ионов. В воде Са2+ экранирован на 17 м.д. больше,
чем в метаноле [124].
Ионы элементов 2А-группы образуют комплексы с рядом низкомо-
лекулярных лигандов, что наблюдается по изменению ХС и ЯМ-ре-
лаксации. Из концентрационных измерений можно определить ХС
и константы образования комплекса (см. гл. 4). Температурные измере-
ния помогают определить эти параметры. Использование обогащенного
изотопом 43Са препарата (61%) позволило изучить комплексообразо-
вание Са2+ с ЭДТА, которая образует комплексы, наблюдаемые по
ЯМР 43Са раздельно от акваиона Са2+ [131]. КССВ с ядрами эле-
ментов 2А-группы известны только для 9Ве, благодаря этому по ЯМР
9Ве идентифицирован ион [ВеБзНгО]- в водном растворе (NH4)BeF4
и найдена величина КССВ 'J(Be-F) = 35 Гц [137]. Металлоорганиче-
ские соединения магния изучены в [138].
2.3. Бор, углерод, азот
Определенная общность химических свойств бора, углерода, азо-
та побуждают к совместному рассмотрению свойств ЯМР их ядер
(табл. 2.9).
Бор и азот имеют по два магнитных изотопа, а углерод — один,
причем чувствительность последнего на три порядка меньше чувстви-
тельности *Н. Низкое природное содержание изотопов *°В, 13С, l5N
позволяет использовать эти ядра в качестве изотопных меток при соот-
ветствующем изотопном обогащении с последующим детектированием
методом ЯМР. Изотопы 1О'ИВ и 14N имеют квадрупольные ядра; в ряде
случаев это затрудняет получение структурной информации. ЯМР 15N
Таблица 2.9
Свойства ядер магнитных изотопов бора, углерода, азота
Изо- топ Спин NA, % Sens 1 Q, барн Диапазон ХС, м.д Частота ЯМР эталона Е, МГц2 Эталон
|°В 3 19,6 7,4 0,086 150 10,743 8563 В(ОМе3)з
"в 3/2 80,4 260 0,036 150 32,083 971 4 BF3-OEt2
13с 1/2 1,1 1{'Н) — 300 25,145 0005 Si(CH3)4
14n 1 99,6 1,9 0,016 1000 7,226 3296,7 CH3NO2
i5N 1/2 0,4 0,007 — 1000 10,136 78367 ch3no2
1 Относительно |3С; 2 частота сигнала ЯМР в поле, в котором частота ЯМР 'Н
равна 100,0 МГц; 3 CDCI3; 4 15% об. CDCI3; 5 CeDe; 6 зависит от температуры
и растворителя; 7 применяется для органических веществ, <5 = 6 м.д. от NO^",
0,1 М в CDCI3; для неорганических веществ удобнее использовать насыщен-
ный раствор NO^.
применяют для определения констант ССВ, а также для определения
ХС при большой ширине линий или недостаточном их разрешении
в ЯМР 14N из-за квадрупольного уширения. Измерения ЯМР 15N
затруднены из-за низкого природного содержания и долгих времен
релаксации; при природном содержании изотопа даже в высоких полях
криомагнитов они требуют длительного накопления.
Подавление протонов дает небольшой выигрыш при записи спек-
тров ЯМР ядер 15N, так как оно сопровождается отрицательным эф-
фектом Оверхаузера (ослаблением сигнала) из-за отрицательного гиро-
магнитного отношения этих ядер; выигрыш дает слияние мультиплетов
и уменьшение эффективного времени релаксации этих ядер. Подав-
ление протонов применяется только при наличии ССВ с протонами,
однако в этих случаях более эффективны методы переноса спиновой
поляризации (INEPT и другие). Чувствительность ЯМР к окружению
атомов В, С, N показывает табл. 2.10.
Бор. Оба изотопа бора имеют магнитные моменты, однако из-за
40-кратного преимущества в чувствительности и меньшей ширины
линий применяется изотоп 11 В. В некоторых случаях ЯМР |0В мо-
жет дать дополнительную информацию из-за того, что константы
ССВ у него втрое меньше, чем у 11 В. Подробные обзоры сделаны
в [4, 139-141].
Диапазон ХС бора около 150 м.д. Метод ЯМР бора имеет хоро-
шую специфичность, поскольку ширина линий ЯМР ИВ невелика.
Изотопный эффект ХС 1ОВ-ИВ не превышает 0,1 м.д., и обычно им
пренебрегают.
Таблица 2.10
Чувствительность ХС ядер "В, l3C, l4N к окружению [3]
Ядро: ИВ 13С 14n
Форма <5ИВ, м.д. Форма 5 13С, м.д. Форма <5 l4N, m. д.
вн~ -41 СН4 2,3 NH3 -283
ВМез 87 С(СМе3)4 — NMe3 -365
B(NMe2)3 27 CN- 167,4 N2H4 -312
В(ОМе)3 18 С(ОМе)4 — NO3~ 0
BF3 10 cf4 — NF3 —7
ВС13 47 СС14 96,7 CINO 224
B(SBu?)3 66 CS2 192,8 SN3+ -12
Измерения, как правило, делают относительно внешнего эталона
без введения поправки на восприимчивость образца. Показано, что ХС
чувствительны к электроотрицательности лиганда и характеру тг-свя-
зывания орбиталей [142], найдена корреляция между экранированием
ядра бора и плотностью р-заряда [190].
Данные ЯМР некоторых соединений бора приведены в табл. 2.11.
Обычно наблюдают только прямые константы ССВ *7(иВ-Н); для
концевых протонов они составляют 100-200 Гц, для мостиковых —
50-80 Гц; в [144] найдены J(10B-HB) от 14 до 28 Гц. Сравнение ХС
13С и “В в одинаковом окружении проведено в [144].
Бор образует большой набор полиядерных соединений, и для опре-
деления их структуры и состояния в растворах широко применяется
метод ЯМР бора. Для упрощения спектров комплексов, содержащих
протоны, широко применяется двойной резонанс. Изучены спектры
ЯМР различных боранов [139, 140], структуры некоторых из них при-
ведены на рис. 2.9. Спектр ЯМР ИВ диборана ВгН8 представляет собой
триплет триплетов и соответствует двум эквивалентным атомам В
и двум типам протонов (2 — одного и 4 — другого типа) с разными
КССВ.
В анионе [BgHy]- при 300 К наблюдают быстрый внутрикомплекс-
ный обмен протонами [140], из-за чего атомы бора эквивалентны. Ани-
он представляет собой октаэдр с группами ВН по вершинам и одним
/13-протоном (Н7) на одной грани. При 190 К /13-протон фиксируется
на грани, при этом наблюдали два типа атомов В: —6,7 и -15,2 м.д.
(гр.), 1 : 1, и 2 типа протонов 1,2 и 5,6 м.д: (Н7) с 1J(1IB-H7) = 13 Гц.
Химические сдвиги бора в борных соединениях
Таблица 2.11
Форма 5, м. д. ‘J("B-H), Гц Примечания, источник Форма 5, м. д. ‘J(HB-H), Гц Примечания, источник
BF3O(Et)2 0 — — н6в2 17,5 135/461 —
вн- -41,2 80 Н2О [190] В5н9 -52,7 176 Вершина [144]
-13,1 166 Основание
NaBH4 -38,7 82 Н2О [4] В5Н8Вг -36,5 — Вершина [3]
-12,5 — Основание
LiBH4 -38,2 75 [4] в5н81 -55 — Вершина [4]
-11,8 — Основание
в2н7- 0,1 106 Н2О [190] NaBO2-B(OH)4- 1,3 — Н2О [4]
В3Н- -30,1 33 Н2О [190] NaBO3 5,5 — То же
hbf4 0,1 — 50% в Н2О [4] МагВ^О? 9,9 — Н2О [4]
NaBF4 -2,3 — Н2О [4] К2В4 О? 7,5 — Н2О [4]
NH4BF4 -1,8 — Н2О [4] (NH^B^y 10,3 — «
bci; 6,6 — Н2О [4] в508 14 — «
ВВг4- -24,1 — [1] В(ОН)з 18,8 — «
BI4- -128 — То же В(ОМе)3 18,1 — «
Таблица 2.11 (окончание)
Форма 6, M. Д. ‘JC'B-H), Гц Примечания, источник Форма 5, м.д. ‘JC'B-H), Гц Примечания, источник
B(NO3)4- -87 — « ВМе3 86 — [4]
В(ОН)4- 1,1 — « BEt3 60 — [4]
BPh; -6,3 — « NH3-BF3 —2 — Н2О [4]
hbf2 22 — « NH3-BH3 -23 — Н2О [4]
pbf2 28-30 — « Н3Р-ВН3 -42,7 — [143]
PBC12 62-66 — « А1(ВН4)3 -37 — [143]
РВВг2 62-66 — « PCI3-BF3 0 — То же
(HBNH)3 29,2 133 « PC13-BI3 -65 — «
BF(OMe)2 1 85* * [1]* 'jfB-F) H3BO2F2 -1 — «
BF3 10 — Газ [4] В(ООН)4- 6,7 — Н2О [145]
BCI3 47,7 — [4] В2О4(ООН)4 5,5 — Н2О [145]
BBr3 40,1 — [4] В4СЦ 85 — [141]
BI3 -5,5 — Расплав [4] В6С12- -17,4 — «
H2BH2BHCI 2,3 167 к/53 m [190] В6Вг*- -18,5 — «
H2B(NH2)BH2 -26 130/30 Газ [190] в61^- -27,5 — «
1 J(H
КОНЦ
-B)/J(HM0CT-B).
Рис. 2.9. Строение боранов ВзНб (а); ВзНв (б), замещенного борана
В5НдРе(СО)з и спектры ЯМР борана BsHgFe(CO)3 на ядрах НВ (в)
Спектры В4Н10 и В5Н9 соответствуют двум неэквивалентным ато-
мам В с соотношением 1:1 и 1 : 4 в соответствующей структуре
(см. рис. 2.9) [4]. В нестабильном пентаборане-11 три неэквивалентных
атома В, причем линии атомов В3 представляют собой триплеты, рас-
щепленные на двух концевых протонах (расщепление на мостиковых
протонах из-за малой константы ССВ не проявляется), а атомы Bi и Вг
дают дублеты. По спектрам ЯМР контролировался синтез соединения
В3Н9Ее(СО)з (см. рис. 2.9) [146]. Спектры ЯМР ”В и ’Н показывают,
что атом железа включен в основание пирамиды, причем с одной
стороны расположен рядом с атомом В, а с другой — через протон.
При изучении замещенных боранов установлены места замещения. Так,
в BsHgCl замещается атом водорода в основании, а не при вершине
пирамиды [147].
Взаимодействие борогидридов щелочных металлов с галогенидами
алюминия в растворителях разной природы изучали в [148]. Отмечено
образование алюмоборогидрида А1Н(ВН4)г с ХС В 36 м.д. в растворе
ТГФ и ^(“В-Н) = 83 Гц.
Взаимодействие с непереходными и переходными металлами умень-
шает лабильность борогидридов вплоть до образования инертных
комплексов. Взаимодействие ВН^ с переходными металлами изучено
в [149] (табл. 2.11). Динамику обмена в комплексе У(ВН4)з, раство-
ренного в толуоле, в диапазоне 193-323 К изучали в [150].
Железосодержащие бораны изучены в [151], галогенобораны
XnBl0Hf-_n, (X = Cl, Вг, I) - в [152].
Ионы боратов в водных растворах изучали в [145, 153-157]. Вли-
яние концентрации и катионного состава на ХС и КССВ ионов BF4
Таблица 2.12
Данные ЯМР комплексов металлов с тетрагидроборатом [149]
Форма NaBH4 А1(ВН4)3 Sc(BH4)31 Y(BH4)3 Zr(BH4)42
<5В, м. д. -24 -37 -18,7 -23,2 -8,0
<5Н, м. д. -2,0 -1.3 0,5 1,35 1,65
J(“B-H), Гц 82 89 80 84 90
J(M-H), Гц 0 44 30 <2 28
<5М, м. д. — 71,1 113,3 1 — 40,72
1 От насыщенного раствора 5сС1з/О2О; 2 от (CsHs^ZrBrs/CeDs.
Таблица 2.13
Химические сдвиги 11В ферроборанов [153]
Форма <5B* <5 B2 <5 B3
Fe2(CO)6B3H7 4,2 12,4 4,2
[Fe2(CO)6B3H6]- 2,4 8,6 33,9
[Fe3(CO)l0HBH]- 58 — —
[Fe4(CO)12BH]2- 153 — —
[Fe4(CO)i2(AuPPh3)2B]- 192 — —
и BF3OH на ядрах ИВ и 19F изучено в [154]. Разбавленные растворы
боратных ионов Н3ВО3, NaBO2, Na2B4O7, NH4B5O8 изучены в [155];
в растворах присутствовали только В(ОН)з и В(ОН)^-, переходящие
друг в друга в зависимости от pH. Изотопный эффект 1ОВ-ИВ менее
0,03 м. д.
Равновесия полиборатных ионов в водных растворах изучены
в [157].
Исследование равновесия
В(ООН)4-^
НОО\ / О—О\ / ООН2-
В В
НОО' \ 0-0 / \оон
в водно-перекисных растворах проведено в [145], определены ХС пе-
рекисных ионов (см. табл. 2.14) и энергия активации димеризации
(11 кДж/моль).
Образование бороуглеродных перекисных комплексов изучено
в [158].
Таблица 2.14
Химические сдвиги ИВ пероксорборатных комплексов [145]
Форма <5, м.д. Форма <5, м. д.
В(ОН)4- 1,0 В2О2(ОН)|- 3,2
В(ОН)з(ООН)- 2,4 В2ОО(ОН)2- 4,8
В(ОН)2(ООН)2" 3,8 В2(ОО)2(ОН)2(ООН)2- 6,0
В(ОН)(ООН)Г 5,1 В2(ОО)2(ООН)42- 7,3
В(ООН)4- 6,4
Реакцию ВХ3 с HPO2F2 (X = CI, Вг) изучали в [159]; определены
спектральные характеристики иона [В(РОгр2)4]-: <5(ИВ) = -4,3 м.д.,
2У(ИВ-Р) = 8 Гц; 5 (31Р) = -31,5 м.д.; <5(19F) = -85 м.д., *J(P-F) =
= 986 Гц.
Биядерные комплексы бора с оксиэтилидендифосфоновой кислотой
(ОЭДФ) изучены в [160] (ОЭДФ = Ме2С{РО(ОН)г}2-
Удвоение карборана (Мез51)2СгВ4Нб в (Me3Si)2C4BgHio (рис. 2.10)
изучено в [161]; линия атомов С — синглет с 5 = 105,4 м. д., ХС атомов
бора приведены на рисунке.
Рис. 2.10. Строение карборана (МезЗО^ВвНю: <5(В1,В12) = -20,8 м.д.,
<5 (В4) = 10 м.д., <5(В5, В10) = -17,3 м.д., <5(В6, В11) = 6,2 м.д., <5 (В9) =
= 8,9 м.д.; <5(С-В2) = 105,4 м.д., <5(СН3) = 0,1 м.д.
Образование боратных эфиров в водных растворах и вине с молоч-
ной, малоновой и винной кислотами изучено в [162].
При термическом разложении аммиачного борана NH3BH3 в апро-
тонном растворителе по одной из реакций возникают олигомеры
(СНгВНг)™, п = 2-5, ХС бора в которых почти не зависит от п,
5 = -11,1 ±0, 1 м.д. [163].
Избранные КССВ бора с разными ядрами приведены в табл. 2.15.
ЯМ-релаксация бора редко используется при изучении неорганиче-
ских систем. Обычно времена релаксации лежат в пределах 10-100 мс,
Таблица 2.15
Константы ССВ бора [141]
Соединение X ’JC'B-X) Соединение X 'J("B-X)
F2BH H -211 F3B-PR3 P 165-217
С12ВН H 196 C13BPR3 P 140-166
(N-BH)3 (циклический) H 164 Br3B-PR3 P 125-165
Н2В-(ц-Н*)2-ВН2 H 135,45* I3B-PR3 P 108-134
вн4~ H 80 H3B-PF3 P 39
C1BF2 F 33 H3BCO c 22
ci2bf F 73 BMe4“ c 22
BF3 F 14,5 Br3B-NMe2 l5N 13
bf4- F 1-5 [Rh(PPh3)2ClBCl3] Rh 150
F3B-PMe3 F 50 цис-[Pt(Me2Ph)2BCl] 195Pt 3548
редко достигая 1 с. Основной механизм релаксации — квадруполь-
ный, и в этих случаях выполняется соотношение Т\ (10B)/Ti (’1 В) =
= ТгС^/ТгС'В) = 1,53 (тест на этот механизм).
Углерод. Поскольку углерод — основной элемент в органической
химии, метод ЯМР 13С наиболее популярен среди ЯМР «тяжелых»
ядер. В неорганической химии метод ЯМР 13С чаще применяют для
изучения комплексных соединений с CN, СО и другими содержа-
щими углерод лигандами. При снятии спектров обычно используют
широкополосное подавление протонов, что дает выигрыш во времени
накопления более чем на два порядка (за счет эффекта Оверхаузера,
уменьшения эффективного времени релаксации 1\ и слияния мульти-
плетов). В неорганических системах связи С-Н не столь универсальны,
поэтому широкополосное подавление протонов используют в основном
при исследовании комплексов с органическими лигандами. Наиболее
подробные сведения по ЯМР 13С приведены в [169, 190]. Выпущены
обзоры по ЯМР 13С [165-169].
Химические сдвиги атомов углерода в большинстве соединений
лежат в диапазоне 0-240 м. д. (табл. 2.16). В основном ХС ЯМР 13С
зависит от электроотрицательности заместителя.
Ионные формы угольной кислоты изучены в [172].
Образование комплекса вызывает изменение ХС атомов С лиганда
в комплексе относительно ХС свободного лиганда. Обычно эти изме-
Таблица 2.16
Химические сдвиги ЯМР |3С
Форма <5, м.д. Источник Форма <5, м. д. Источник
СН4 2,3 [190] СНС1з 77,5 [165]
То же -2,3 [165] СМ" 168,5 [165]
СНз Вг 10 [165] То же 177 [190]
СН31 -20,7 [165] СО 180 [170]
CH3NO2 75 [190] НСООН 165 [190]
СН3ОН 47 [190] (K+)NCS- 134 [165]
CH3CN 118 [165] CH3SCN 113,5 [165]
CH3NC 157,5 [165] С(ОН)3- 177 [165]
Пиридин, а, /3, 7 151,125; 137 [165] Rh6(CO)i5 191; 209; 248 Рис. 2.11 [170], отнесение в тексте
НС(ОН)2+ 184 [165] СО2 125,4 [171]
СО2- 166 Na2CO3/H2O, pH И
cs2 194 [165] НСО3“ 218 [190]
Ni(CO)4 193 [171] НСО3“ 161,2 [172]
Fe(CO)5 215 [171] Н2СО3 162,9 [172]
CH3CI 24,9 [165] Н3СО+ 164,9 [172]
нения невелики, поскольку углерод не является координирующим ато-
мом. Такого же порядка эффекты наблюдают при сольватации ионов.
В координированных молекулах этанола (табл. 2.17) знак Д<5 ме-
няется при переходе от СНг- к СНз-группе. Увеличение ХС от Mg2+
Таблица 2.17
Изменение химического сдвига ЯМР |3С растворителей при координации
к ионам непереходных металлов [173]
Катион Растворитель t, °C Д<5СНг, м. д. Д<5СНз, м.д.
Mg2+ EtOH 70 1,5 0,8
А13+ EtOH -30 -5,6 2,2
А13+ ДМСО -70 — 1,9
к А13+ соответствует в 2,5 раза большей плотности заряда Ze2/r2 [174].
Координация спиртов происходит атомом кислорода, поэтому она вы-
зывает экранирование атома кислорода, дезэкранирование ближайшего
атома С и экранирование следующего:
/О —Сх
М ) СН
^о—сх
Изменение ХС при координации ацетилацетона показано
в табл. 2.18 (растворы в дейтерохлороформе). Координация ацетил-
ацетона вызывает экранирование атомов С в цикле, образованного
связями, и дезэкранирование в СНз-группах.
Таблица 2.18
Химические сдвиги ЯМР 13С в ацетилацетонатах металлов
Форма/ссылка 6 (СО), м. д. <5(СН) 6 (СНз)
Н-Асас [174] 192,4 101 24,8
Ве(Асас)2 [174] 192,8 101,2 24,8
А1(Асас)з [174] 191,8 101,9 27,8
Со(Асас)з [174] 189 97,9 27,8
Rh(Acac)3 [3] 189 99,4 26,7
Pd(acac)2 [3] 186,9 101,3 24,8
Влияние координации и влияние растворителя близки по величине.
Изменение ХС донорного атома углерода при координации гораздо
больше, чем у других атомов лиганда; диапазоны ХС донорного атома
Ph 120-190 м.д., Me - (-30)-50 м. д., СО - 150-280 м.д., Ср -
70-125 м.д. [166]. В табл.2.19 показаны вариации ХС метила в ком-
плексах Pt(IV).
Координация приводит к сильным различиям ХС СО-групп. Так,
в кластере Rhs(C0i5) (рис. 2.11) наблюдают три типа карбонильных
групп: с 6 = 191 м.д. для СО-групп, координированных к атомам родия
в вершинах; с 5 = 200 м.д. для групп СО, координированных к трем
экваториальным атомам родия; мостиковые с 6 = 248 м.д. Отнесение
линий сделано методом двойного резонанса 13C{103Rh} [170]. Подобные
изменения типичны при образовании карбонильных комплексов пере-
ходных металлов (см. табл. 2.20).
Таблица 2.19
Химические сдвиги и КССВ 13С метильной группы в комплексах Pt(II)
/npaHc-[Pt(Me2Ph)2MeL]+ [176]
L <5, м.д. 'j(Pt-C) ± 2 Гц L <5, м. д. 'j(Pt-C) ± 2 Гц
сн2=сн2 5,6 615 MeNC -7,1 510
Пиридин -22,7 598 СО 0 509
AsPh3 0 548
Таблица 2.20
Химические сдвиги ЯМР 13С в цианидных комплексах
Форма 6 13С, м. д. Лит. Форма 6 13С, м. д. Лит.
CN" 165,1 [177] Ni(CN)42“ 135,5 [177]
HCN 112,4 То же Pd(CN)4“ 131,2 [177]
Au(CN); 104,2 « То же 131,9 [179]
То же 105,2 [179] Pt(CN)2- 125,0 [177]
Ir(CN)^- 110,9 То же То же 125,7 [179]
Pd(CN)2" 104,2 « Pt(CN)2- 84,7 [179]
Рис. 2.11. Строение кластера Rh5(CO)i5
Цианидные комплексы с донорным атомом углерода широко исполь-
зуются в химии и химических технологиях, хотя требования экологии
заставляют ограничивать их применение или замыкать оборот токсич-
ных соединений.
Константы стабильности цианокомплексов, Т1(Ш) определены в
[180]. Данные ЯМР 13С и 205Т1 приведены в табл. 2.21.
Таблица 2.21
Данные ЯМР цианокомплексов металлов
Соединение <513C, м. Д. <52O5TI, м.д. ‘J(C-2O5T1), Гц <514N (l¥, Гц) Лит.
Tl3+(aq) — 2088 — — [180]
ticn2+ 141,2 2310 14640 — «
T1(CN)+ 142 2414 13750 — «
T1(CN)° 147,4 2848 7950 — «
T1(CN); 145 3010 5440 — «
HCN 115 — ’J(C-H) = 271 «
CN“ 167,4 — — — <<
K4 [Re(CN)7]/D2O 140,3 — — -95 (850 ± 50) [181]
K4[Fe(CN)6]/D2O 177,6 — — -94 (850) «
K3[Co(CN)6]/D2O 139,8 — — -94(1450) «
[W(CN)8]4- 146,9 — — — «
[Os(CN)6]4- 142,8 — — — «
ЯМР 13С используется при изучении комплексообразования с кра-
ун-эфирами и криптандами. В комплексах с криптандом С222 (схема
строения на вставке) можно раздельно различить криптаты разных
щелочных и щелочноземельных металлов (табл. 2.22), что можно ис-
пользовать в химическом анализе.
В оксалатных комплексах Pt {цисДРДОНЦох]2-, транс-[Pt(0H)2
ох]2-, цис, mpaHC-[PtC12(OH)2Ox]2-} оксалат подсоединен бидентатно
атомами О, связанными с разными атомами С оксалата, и ХС углерода
в этих комплексах почти не меняется (167,3 ± 0,3 м.д.) [182].
Комплексы Ni(II) с ацетат-ионом в уксусной кислоте изучены в [185];
удивительно, что наибольшее парамагнитное уширение (~500 Гц) испы-
тывает линия метильного атома С, а не линия ближнего к донорному
атому О карбоксильного атома (~ 100 Гц).
Таблица 2.22
Химические сдвиги криптатов щелочных и щелочноземельных металлов [178]
Форма 6С1, м.д. 6С2, м.д. 6 С3, м.д.
С222 56,7 70,3 71,4
[Са-С222]2+ 54,5 70,0 70,4
[Sr-C222]2+ 54,4 68,4 70,7
[Ва-С222]2+ 54,9 68,4 71,2
[Li-C222] + 55,6 69,2 70,4
[Na-C222] + 55,6 68,4 69,4
[К-С222] + 54,5 68,2 71,1
С222
В [183] процесс разложения мочевины исследован методом ЯМР
|3С, 14,15N с использованием диенового комплекса Pt(II) [dienPtX].
Константы ССВ ',7(С-Н) зависят от заряда на ядре и s-характера
орбиталей атома С, участвующих в образовании связи, и лежат в преде-
лах 90-320 Гц; прямая константа 'ДС-Н) = 500а2, где а — s-характер
орбитали, образующей связь. КССВ через две и три связи на порядок
меньше.
Константы ССВ с другими ядрами редко превышают 100 Гц, за
исключением ядер металлов (табл. 2.23).
Таблица 2.23
Константы спин-спинового взаимодействия 13С с металлами [166]
Соединение X J(C-X), Гц Соединение X J(C-X), Гц
(LiMe3)4 7Li + 15 Ag(CH2PPh3)2Cl2 109 Ag 96
[Y(CH2SiMe3)4]- 89 у 28 CdMe2 113Cd 538
[V(CO)6]- 51 v 146 HgMe2 l99Hg 690
[Мо(СО)6]“ 95 Mo 68 TlMe3 205j] 1930
[W(CO)6]- 183W 126 GeMe4 73Ge 18,7
WMe6 l83W 43 SnMe4 119Sn 340
Fe(CO)s 57 Fe 23,4 SiMe4 29Si 50
[Rh(CO)4]“ l03Rh 75 TeMe2 l25Te 160
Pt(CO)2Cl2 195Pt 1576 PbMe4 207Pb 250
КССВ с платиновыми металлами отражает взаимное влияние ли-
гандов (табл. 2.24). В таблице представлены КССВ 13C-Pt в метильных
комплексах Pt(IV) [167].
Таблица 2.24
Соединение 'j(Pt-C), Гц Соединение 'j(Pt-C), Гц
гран-[Р1Ме3(Н2О)з] + 791 цис-[Р1Ме2(Н2О)4]2+ 511
гран-[Р1Ме3(МО2)3]2- 691 [PtMe2(H2O)3OH] 635
apaH-[PtMe3(CN)3]2- 499 [PtMe3Br(H2O)3] 592
Времена релаксации Т\ в органических соединениях находятся
в пределах 1-100 с. Ядра, не связанные с протонами, часто имеют
Т1 100 с, за исключением ядер, связанных с квадрупольными ядрами,
ССВ с которыми может существенно сократить Т\. В необезгаженных
образцах Т\, как правило, не превышает 20 с вследствие наличия
растворенного молекулярного кислорода. Измерение Т\ является эф-
фективным методом различения линий атомов С в спектрах, однако для
этого требуется на порядок больше, чем для получения спектра ЯМР.
Поскольку спектры ЯМР 13С обычно измеряют, растворяя вещество
в органических растворителях, ниже приводятся данные ЯМР попу-
лярных растворителей (табл. 2.25).
Азот. Два магнитных изотопа (14N, 1=1 и l5N, I = 1/2) суще-
ственно расширяют возможности ЯМР азота. В низких полях (^ 2 Т)
возможности метода ЯМР 14N ограничены плохим разрешением ли-
ний из-за большой ширины линий при низкой симметрии окружения
атома N (релаксация по квадрупольному механизму). Для ЯМР l5N
требуется обогащение образца изотопом или долгое накопление из-за
низкого природного содержания и больших времен релаксации. Подав-
ление протонов не усиливает сигнал связанных с ними ядер 15N из-за
отрицательного гиромагнитного отношения, но дает выигрыш за счет
уменьшения времени релаксации и слияния мультиплетов.
Данные ЯМР азота (табл. 2.26) отражены в ряде монографий и об-
зоров [3, 165, 186-190]. В качестве эталона для органических соеди-
нений используют нитрометан; его ХС зависит от температуры и рас-
творителя (до 7 м.д.). В необезгаженных органических растворителях
присутствует растворенный азот воздуха в концентрации ~0,1 М/л,
давая узкую линию с ХС —68 н—74 м.д. от нитрометана, в зависи-
мости от растворителя. Следует заметить, что 1) в ранних работах
использована ст-шкала ХС, 2) данные измерений ХС азота-14 на анало-
говых спектрометрах 80-100 МГц для многих соединений отличается
от современных данных до 100 м. д. Поэтому следует предпочитать
Таблица 2.25
Химические сдвиги |3С популярных растворителей
Соединение Формула 6 Соединение Формула 6
Ацетон Ме2СО 39,5; 205; 1 1 Нитрометан MeNO2 62,8
Ацетонитрил MeCN 1,3; 118,1 Пиридин C5H5N 123,5; 135,5
Бензол с6н6 128,7 149,5
Дихлорметан СН2С12 53,7 Метанол МеОН 49
Дисульфид углерода CS2 192,8 Этанол ЕЮН 12,7; 56,7
Диметил- сульфоксид Me2SO 39,5 Тетрагидро- фуран С4Н4О 25,3; 67,2
Диметилфор- мамид Me2NCHO 30,1; 35,2; 162,5 Толуол С6Н5Ме 21,3; 138,5
Формамид MeNO 165,1
Диоксан С6Н4О2 66,3 Хлороформ CHCI3 77,4
Диметило- вый эфир Ме2О 59,4 Тетрахлорид углерода СС14 96,7
Диэтиловый эфир Et2O 17,1; 67,4 Муравьиная кислота НСООН 165
Уксусная кислота МеСООН 21,1; 177,3
1 В водных растворах ХС ацетона зависит от соотношения ацетон/вода; при
10% ацетона его ХС 5,5 и 215 м.д. [184], там же изучено протонирование
ацетона в этих растворах.
данные 15N или 14N, полученные на высокополевых спектрометрах.
Для водных растворов удобнее использовать ион NO^“ как внешний
эталон, линия которого близка к линии нитрометана. Растворимость
N2 в воде гораздо ниже, чем в органике, и появление в спектре линии
растворенного азота указывает на низкую концентрацию растворенного
вещества.
В сильных полях эффективность ЯМР l4N гораздо выше. Чувстви-
тельность ЯМР l4N в поле 7 Т позволяет при ширине линий менее
1 кГц работать с концентрациями ^0,05 г-ат/л, а на ядре 15N записы-
Таблица 2.26
Химические сдвиги ЯМР некоторых соединений азота (приведено к NO^")
Форма 6, м.д. Источник Форма 5, м. д. Источник
NO3“ 0 [189] SCN“ -152 [190]
MeNO2 6 [189] CN" -112 [190]
NH3 безводн -391 [187] NH3C1“ -366 [187]
NH3 -383/-378 [189] MeCN -131 [3]
nh+ -354,5 [206] MeSCN -126 [189]
C(NO2)4 -48 [189] MeNC- -218 [189]
n2h4-h2o -346 [192] Пиридин -74 [190]
n2 -14 [189] NO2“ 235 [3]
N|* -62 4-67 [3] HNO3, 100% -52 [190]
(H2N)2CO -300 ** hno2 200 [193]
NC13 -162 [194] N2O4 чист. —26° -5 [195]
* Раствор воздуха в растворителях; “ данные автора.
вать приемлемые спектры чистых веществ в разумное время (~ 1 ч на
природном содержании изотопа с применением релаксанта).
При использовании нитрометана как внутреннего эталона следует
учитывать зависимость его ХС от растворителя (табл. 2.27) [196].
Таблица 2.27
Химические сдвиги азота 0,3 М растворов нитрометана в различных раство-
рителях
Растворитель 5, м. д. Растворитель <5, м.д. Растворитель 6, м.д.
Ч. жидк. 0 СНС13 -3,8 D2O 2,0
ecu -7,1 ДМСО 2,0
ХС азота в водных и сернокислых растворах азотной кислоты яв-
ляется линейной функцией кислотности Гаммета Н^, зависимость ХС
от концентрации показана на рис. 2.12 [197].
Нитро- и нитрозоформы, стабилизированные в кислотах при по-
ниженных температурах, исследованы в [198]. Зависимости ХС этих
форм от температуры и концентрации кислоты показаны в табл. 2.28.
Рис. 2.12. Поведение химического сдвига l4N нитрата в азотнокислых и серно-
кислых растворах
Таблица 2.28
Влияние условий на химические сдвиги нитро- и нитрозоформ
Форма Растворитель t, °C Цвет <S15N, м.д.
3 М Mg(NO3)2 H2O 22 Бесцветный 1 0
no2~ D2O 25 « 234
hno2 3% D2SO4 25 « 199
NO+ HSO3F 25 « 7
« 18% D2SO4 25 « 1
hno2 10% d2so4 -5 « 199
« 20% D2SO4 -5 « 199
« 30% D2SO4 -5 « 203
« 40% D2SO4 -5 Голубой 194
NO+ 40% D2SO4 -5 4 -15
hno2 50% D2SO4 -10 « 189
« « 0 Темно-голубой 190
« « 10 Бесцветный 189
NO+ « -10 Голубой -21
« « 0 Темно-голубой -21
« « 10 Бесцветный -22
Сложившееся мнение о низкой информативности ЯМР l4N для
спектров, снятых в высоких полях, верно лишь отчасти; на рис. 2.13
представлен спектр замещенного триазола, в котором разрешены все
атомы N молекулы[199]; при этом ширина линии используется для
отнесения атомов азота [200].
Изотопный эффект ХС (14N 15N) мал (не более 0,5 м.д.) [187].
Шкала ХС ЯМР азота представлена на рис. 2.14. Она может быть
800 600 400 200 0 -200 -400 м.д.
....11 111 11 11 I 11 11 I 11 I 11 I 11 11......11 11 I I 11............1 I I 11111111
Нитридо IIIIIIIIHHIIIIIIIIIIIIIIIIIIIIHIIIIIIIIIIIIIIII Аммины ШЖНННН
7Т г , n -I л -т. ItlllllllllllltHHIIIIIIIIIHIII IIIIIIIIIIIIII Д М U Ц kl IIIIIIIIIII1IIIUII
/ДИЗЗОТ llnlllllllllnlllllllllltlllllnIIIIIIIIIIIIII n-ivirinoi iniiiiiiiiiiiinil
Цианаты, фульминаты М—CNO НИИ ИШИМ NCO
Азиды M^-NQ-Np-N7 O'lHimiHHm ЦННШИ а
/зт
Гидразидо (—2) аШ /ЗШ tA=Na-Np-R
Арилазины НННШШ М—Ру
Нитрилы ИНННИИИ M--NCR
Имидо М—NR ННИИИИНИИНИИИ Циано М—CN
M = N М—NO
IIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIII о Нитрозил
m-(m-no)-m иниии иинини) m-no+
Нитрато щщ
Нитрито ИИИИИН НИН» Нитро О - N
Гипонитро HIIHIIIIIIIIIIIIIIIIIIIIHIIIIIIIIIIIIIIIIIIIII М<^ ||
N2 в растворе I О - N
Нитрометан |
...........................................................ин.....................................
800 600 400 200 0 -200 -400 м.д.
Рис. 2.14. Диаграмма химических сдвигов ЯМР азота, связанного с металлом;
N2 — азот растворенного воздуха
сделана абсолютной, так как по измеренной константе спин-вращатель-
ного взаимодействия в молекуле аммиака рассчитано экранирование
ядра азота 264,5 м.д. [189] и абсолютное экранирование
аабс = 246 - <r(NH3)
или
^абс = —146 — ^(MeNOj).
Рассчитанные компоненты тензора экранирования азота в некото-
рых соединениях приведены в [189]. При измерении ХС азота необхо-
димо учитывать фазовое состояние вещества и растворитель, так как
из-за водородной связи эффект среды велик и может менять знак. Так,
для NH3 Ожидк — сггаз = 20 м.д., а для HCN — 12 м.д. Азот легко
образует водородную связь с молекулами растворителя, что вызывает
низкополевой сдвиг линии ЯМР азота при образовании связи через
протон, соединенный с азотом, и высокополевой при образовании связи
непосредственно с азотом [189].
Имеется определенная общность шкал ХС азота и 13С. Показано,
что ХС азота определяет внутримолекулярный вклад. Тенденцию из-
менения ХС азота можно оценить, исходя из приближения средней
энергии для парамагнитного вклада в экранирование:
а? = -роРв „ (2-9)
27г(г3)2рДЕ
Изменение электроотрицательности заместителя меняет (г3), по-
этому наблюдается корреляция между ХС и электроотрицательностью
заместителя. Изменение ДЕ1 приводит к корреляции 6/Х: более окра-
шенные соединения азота имеют меньшую величину ДЕ и больший
сдвиг в слабое поле (А — длина волны ЭСП). В ХС азота большие
вклады дает взаимодействие неподеленной пары п —>тг*.
Координация азота к металлу вызывает значительный (20-140 м.д.)
сдвиг линии по сравнению со сдвигом свободного лиганда. Так, в амин-
ных с!6-комплексах Co(III), Ru(II), Rh(III) и ^-комплексах Pt(Il) высо-
кополевой сдвиг достигает 100 м.д. от линии жидкого NH3 [201-203].
Наблюдается транс-влияние (~30 м.д.) при переходе от СоИ1(ЫН3)б
к Coni(NH3)sX, причем это влияние уменьшается в ряду X: HSO^ «
« NO^ и Н2О > С1_ >Вг“ > NH3 > NO^ [201]. Изучение нитро-
комплексов платиновых металлов [204-211] и енаминных комплексов
Pt(IV) [212] привело к понятию координатного сдвига [191, 212]
(см. раздел 3.4). Анализ данных ЯМР донорных атомов азота показал,
что ХС N-лиганда зависит не только от координации к платиновому
металлу, но и от лиганда в транс-положении L, т. е. от координаты
L-M-N. Значение ХС атома N на данной координате является ее
характеристикой и слабо зависит от других лигандов комплекса.
Спектр ЯМР 14N комплекса транс*-[Р1епС12*МНзС1]_ показан на
рис. 2.15. Видно, что ХС линий атомов N этилендиамина четко разли-
Рис. 2.15. Спектр ЯМР 14N водного раствора транс-[Р1е«С12МНзС1] +
чаются из-за разных лигандов в транс-положении; линия NH3 расщеп-
лена на трех протонах и ядре платины — редкий случай наблюдения
СТС линии координированного аммиака [212].
На рис. 2.16 показаны координатные сдвиги координаты NH3-
Rh(III)-X в комплексах разного состава; видно, что ХС азота выбранной
Транс-лиганд
Н,0
С1
Вг й
I й
N Нз 1ЛППП1Ш и ий
-NSC й
ОН й
-SCN й
CN й
___I_____I_____1_____I_____I______I__
-400 -420 -440 м.д.
Рис. 2.16. Диаграмма координатных сдвигов азота аммиака в комплексах
родия(Ш) [305]; для несвязанного лиганда 5(МНз) = —383 м.д
координаты находится в узких пределах, а координаты с разными
противоионами четко различаются.
В табл. 2.29 приведены ХС азота на координатах Х-М NO2 и
X-M-NH3 ряда металлов.
Данные табл. 2.29 показывают, что для слабых лигандов в транс-
положении ХС наблюдаемого лиганда дальше от ХС свободного лиган-
да, чем для сильных транс-лигандов.
Таблица 2.29
Химические сдвиги ЯМР азота5 координированных лигандов в нитро- и пен-
тааминных комплексах [191]
Координата Pd(II) Pt(ll) Co(III) Rh(III) 1г(Ш)
O2N-M-NO2 87-90 44-51 — 89-91 54-56
CI-M-NO2 79-80 55-59 — 65-72 32-40
H2O-M-NO2 47 — — 42-59 13-21
H3N-M-NH3 — —412-е- -437 -432 -442
CI-M-NH3 — — -450 -440 -466
H2O-M-NH3 — — —439 4- —461 -451 -479
1 м.д., относительно [NO3 ]; 5(NO2 ) = 234 м.д., 5(МНз) = —383 м.д.
В табл. 2.30 показаны координатные сдвиги азота аммиака для
разных транс-лигандов и металлов и изменение ХС при координации.
Большие изменения ХС соответствуют слабым транс-лигандам.
Влияние растворителя на ХС аммиака изучено в [214].
В табл. 2.31 показаны ХС группы NOg в аквахлоронитрокомплексах
родия(Ш).
Если высокополевой сдвиг наблюдается и при координации аминов
к d6- и с/8-ионам металлов, то в комплексах металлов с заполненной
оболочкой наблюдают низкополевой сдвиг. Комплексы щелочных и ще-
лочноземельных металлов с ЭДТА изучены в [215] (табл. 2.32).
Сдвиг недонорных атомов N линейных амбидентатных лигандов
(NCO-, NCS-, NNN-, CNO-) меньше сдвига координирующего ато-
ма N и обычно имеет противоположный знак. NO^ обычно коор-
динируется монодентатно к платиновым металлам атомом кислорода,
и сдвиг азота «8 м.д. в сильное поле [216, 217]. Линии координиро-
ванных N02-hohob лежат в области ХС NO^ [191].
Для S-связанного роданида с Ag, Zn, Hg, Si, Ge, Sn, P, Ru, Ni,
Pt, Cd ХС приведены в [218-220], для N-связанного роданида с Rh,
Ir, Pd, Pt, Hg — в [219-220]. XC 15NCS_ приведен в [220]. Линии
координирующего атома N смещены на 8-70 м.д. в сильное поле по
сравнению со свободным SCN-, в то время как концевые атомы N
в S-координированных роданид-ионах имеют небольшой низкополе-
вой сдвиг [201, 218-222]. Биядерные комплексы [NbgCSjKNCS^]4-,
[Nb2(SSe)(NCS)s]4_ изучены в [223]. Сильная зависимость ХС иона
SCN- от pH требует осторожности к данным [219-221]. Обзор по
нитрозильным комплексам металлов представлен в [224].
Таблица 2.30
Координатные сдвиги аммиака в комплексах Co(III), Rh(III), 1г(Ш), Pd(II),
Pt(II) [191]
Координата 3, ±2 м. д. От NH3, 3NH3 = -379
Со(Ш): H2O-Co-NH3 -439 4- -461 1 -60 4--82
CI-C0-NH3 -427 4- -450 -48 4--61
H3N-C0-NH3 -415 4--437 -37 4- -48
I-C0-NH3 -404 -25
O2N-CO-NH3 -407 -28
Rh(III): H2O-Rh-NH3 -432 4- -451 -53 4- -72
Cl-Rh-NH3 -440 -61
HO-Rh-NH3 -423 -44
SCN-Rh-NH3 -421 -42
NCS-Rh-NH3 -407 -28
H3N-Rh-NH3 -409 4--421 -30 4- -42
NC-Rh-NH3 -397 -18
Ir(III): H2O-Ir-NH3 -479 -100
Cl-Ir-NH3 -466 -87
H3N-Ir-NH3 -442 -62
Pd(II): H2O-Pd-NH3 -412 -33
H3N-Pd-NH3 -430 -51
Pt(II): H2O-Pt-NH3 -437 4- 443 -58 4--64
HO-Pt-NH3 -435 -56
Cl-Pt-NH3 -419 4-437 -40 4- -58
H3N-Pt-NH3 -415 4--425 -36 4- -46
Br-Pt-NH3 -419 -40
1 Показан диапазон ХС для данной координаты разных комплексов.
Образование пероксоазотной кислоты изучено в [225], комплексы
в растворах серы в аммиаке SyNH-, SsN-, S4N~, S-N-S-NH- изу-
чены в [226, 227]. Комплексы урана с бидентатными комплексонами
иминодиацетатом и оксидиацетатом изучены в [228].
Таблица 2.31
Химические сдвиги ЯМР 15N NO^ в комплексах Rh(III) [209, 213]
—_______ Координата Комплекс ~~~ -—_______ O2N-Rh-NO2 Cl-Rh-NO2 H2O-Rh-NO2
[Rh(NO2)6]3- 90,8 — —
транс- [Rh(NC>2)4Cl] г]3- 89,2 — —
4uc-[Rh(NO2)3(H2O)3] — — 50,3
4uc-[Rh(NO2)3(H2O)2Cl]- — 66,0 53,0
i4uc-[Rh(NO2)3(H2O)C12]2“ — 68,6 55,8
4Uc-[Rh(NO2)aCl3]3- — 71,8 —
цис-[РЬ(МО2)2(Н2О)зС1] — — 47,5
4uc-[Rh(NO2)2(H2O)2C12]_ — 64,8 48,3
4uc-[Rh(NO2)2(H2O)2C12]_ — — 51,7
4uc-[Rh(NO2)2(H2O)Cl3]2- — 68,6 58,7
4uc-[Rh(NO2)2Cl4]3_ — 70,6 —
4uc-[Rh(NO2)2(H2O)4] + — — 41,7
* <515N (±0,3 м. д.); 'j(l03Rh-15N), 22-26 Гц ± 2 Гц.
Таблица 2.32
Изменение химического сдвига 15N ЭДТА при комплексообразовании с М+, М2+
Комплекс Д(5, м.д. Комплекс Д(5, м. д.
Y4" 0 CaY3- 4,4
HY3- 5,3 SrY2- 5,6
h2y2- 17,0 BaY2- 8,6
LiY3“ -5,1 ZnY2- 0,2
NaY3- -1,3 CdY2- 3,3
MgY2- -0,3 HgY2- 15,0
Y = ЭДТА, (HOOCH2C)2N-CH2-CH2-N(CH2COOH)2.
Малое гиромагнитное отношение азота облегчает наблюдение пара-
магнитных сдвигов, особенно в динамичных парамагнитных системах
[229-231]. Величина парамагнитных сдвигов азота может достигать
десятков тысяч м.д. [230]. Растворы щелочных металлов изучены
в [232]. Атомы щелочных металлов имеют один неспаренный электрон,
и наблюдаемые парамагнитные сдвиги как NH3, так и металла при
концентрации последнего 10% достигают сотен м.д. (рис. 2.17). Ин-
Рис. 2.17. Зависимость химических сдвигов 14N аммиака и 23Na натрия в ам-
миачном растворе натрия от концентрации металла
тересно, что наблюдается отрицательный температурный коэффициент
ХС азота.
Константы ССВ с ядром 14N редко наблюдают из-за большой ши-
рины линий ЯМР, и данные о них получены в основном из ЯМР 15N.
При этом J(X-14N) = -0,713J(X-15N). Не всегда КССВ через одну
связь больше КССВ через две связи. Константы ССВ с протонами
~ 100 Гц; V(15N-H) = 73 Гц для NH4+ и 134 Гц для HC=NH+ [189];
‘J(O-14N) = 35 Гц в NO3“ [208], а 1 J(Pt-15N) превышают 600 Гц [220].
Методом ЯМР 14,15N изучены процессы нитрования хлоридов пла-
тиновых металлов (Ru, Rh, Pd, Ir, Pt) в [204-210, 233-235]. Проме-
жуточные комплексы при нитровании иридия исследовали в [205, 208,
211], комплексы родия — в [206, 209], комплексы палладия и плати-
ны — в [207].
ЯМ-релаксация 14N проходит по квадрупольному механизму.
В [236] изучена ЯМ-релаксация в водных растворах солей нитратов
в диапазоне температур 3-80 °C. Обнаружено анизотропное вращение
нитрат-иона, измерены времена корреляции вращательного движения
вокруг разных осей NO^. Изучение температурной зависимости Т2
14N дало аргументы в пользу додекаэдрического строения комплекса
[Мо(СМ)в]4- в растворе.
2.4. Алюминий, галлий, индий, таллий
Все распространенные изотопы этих элементов имеют магнитный
момент, и ЯМР всех ядер имеет хорошую чувствительность. Нали-
чие двух магнитных изотопов у галлия и индия позволяет создавать
неравновесный изотопный состав препаратов и вводить изотопную
метку. Наличие значительного квадрупольного момента у Al, Ga, In
ограничивает возможности ЯМР, особенно у индия. Поэтому ЯМР
алюминия, галлия, индия используют при изучении соединений с вы-
сокой (тетраэдр, октаэдр) симметрией окружения. Свойства ядер этих
элементов приведены в табл. 2.33.
Таблица 2.33
Свойства ядер магнитных изотопов алюминия, галлия, индия, таллия
Изотоп Спин NA, % Sens 1 Q, барн Диапазон ХС, м.д. Частота ЯМР эталона S, МГц2 Эталон
13С{‘Н} — — 1 — — — —
27А1 5/2 100 395 0,15 450 26,057 А1(Н2О)3+
27А1 — — — — — 26,056908, 8 = 0 А1(Асас)з нас./С6О6
69Ga 3/2 60,4 79 0,18 1400 24,003 Ga(H2O)3+
71Ga 3/2 39,6 106 0,11 1400 30,495 Ga(H2O)3+
"3In 9/2 4,3 28 1,14 1000* 21,863 In(H2O)3+3
ll5In 9/2 95,7 620 0,83 1000* 21,910 In(H2O)3a+3
2O3T1 1/2 29,5 106 — 7000 57,149 [Tl+]aq 0
205-p| 1/2 70,5 250 — 7000 57,683 833 [TlNO3]aq 0
1 Относительно 13С; 2 частота ЯМР в поле, в котором частота ЯМР ’Н ровно
100 МГц; 3 30% HNO3; * изучено ограниченное число форм.
Алюминий. Из приведенных в табл. 2.33 ядер наиболее широко
применяется ЯМР 27А1. Изучены как металлоорганические, так и неор-
ганические соединения; имеются подробные обзоры [74, 114, 241, 243].
Данные ЯМР некоторых соединений и комплексных ионов приведены
в табл. 2.34.
Из-за значительной ширины линий ЯМР специфичность метода
ЯМР 27А1 сравнительно невысока. Области ХС алюминия в тетраэдри-
ческом и октаэдрическом окружении из одинаковых атомов разделены.
Таблица 2.34
Данные ЯМР 27А1 комплексных форм алюминия
Форма <5, м. д. W, Гц Примечания, ссылка
[А1(Н2О)6]3+ 0 3 0,1 М HNO3/H2O [242]
[А1(ОН)4]- 80 80 КОН в Н2О [242]
[А1(ОН)6]3- 0 Узкая Н2О [243]
[A1F]2+ 0,7 620 То же
[A1F2]+ 1,3 800 «
А1(С2О4)з 16 150 Н2О [243]
А1(Асас)з 0 93 С6Н6[243]
[A1SO4(H2O)5] + -3,7 5 H2SO4/H2O [245]
[А1(ОН)2(Н2О)8]4+ 4,6 450 [269]
[А11з(ОН)32]7-, октаэдры 12 Широкая Н2О [243]
[А11з(ОН)32]7_, тетраэдр 62,5 50 Н2О [249]
[aiwI2o40]5- 71,2 20 Н2О [250]
[A1WhA1O39H2O]6- 7 770 Н2О [251]
[AIWHAIO39H2O]6- 70 30 То же
[PWhA1O39H2O]4- 2,3 200 «
[А1(ОН)6Мо6О|8]3- 15,4 50 Н2О [251]
Со(А1С14)2 -225 — С6Н6 [241]
Со(А1Вг4)2 -144 — То же
ХС атомов А1 в октаэдрическом кислородном окружении лежат в преде-
лах 20ч—10 м.д., атомов алюминия в тетраэдрическом окружении —
в пределах 80-60 м.д., атомов 5-координированного алюминия — от
30 до 50 м.д. Эта тенденция соблюдается и для других окружений
алюминия.
Спектр раствора металлического алюминия в изопропиловом спирте
приведен на рис. 2.18 [252].
Относительно широкие линии ЯМР 27А1 обычно препятствуют на-
блюдению констант ССВ; некоторые КССВ приведены в табл. 2.36.
Измерения ЯМ-релаксации алюминия относительно мало.
По ЯМ-релаксации 27А1 проверяли модели взаимодействия аква-
тированного иона алюминия с другими частицами раствора [74].
Таблица 2.35
Данные ЯМР 27А1 форм в органических растворах
Форма 3, м. д. W, Гц Примечания Ссылка
[А1С13(МеОН)3] 13,2 60 МеОН [74]
[А1 (МеОН)6]3+ 3,1 200 А1С1О4/МеОН «
То же 4 36 CDC13 «
гран-[А1(МеОН)з(Н2О)3]3+ 1,8 16 МеОН «
[А1С12(ЕЮН)4] + 9,6 360 10% EtOH/CDCl3 об. «
[А1(ЕЮН)6]3+ 2,9 13 То же «
[А1С14]~ 102 17 СН2С12 [244]
[А1Вг4]“ 80 35 MeCN [245]
[ап4]- -28 58 То же То же
[А1Н4]- 100 420 Et2O [246]
А12С16 102 126 То же То же
А12Вгб 95 104 То же То же
А1216 39 90 Et2O [247]
А12Ме6 156 450 Ч. жидк. [246]
А1(Асас)з 0 93 с6н6 [245]
[Al(MeCN)6]3+ -34 73 MeCN [245]
[А1(ОМе)з]4 тетраэдрический 58 4400 Al/МеОН (283 К) [252]
[А1(ОМе)з]4 5-координатный 29 4500
гран-[А1(ОЕ1)з(Е1ОН)з] -0,4 55 Al/EtOH (297 К) То же
pe6-[Al(OEt)3(EtOH)3] 1,4 230
[Al(OiPr)3]4 тетраэдрический 65 4200 А1/гРгОН (296 К) То же
[А1(ОгРг)3]4 5-координатный 35 3500
реб-[А1(ОгРг)3(ОНгРг)3] 4,6 730
гран-[А1(ОгРг)3(ОНгРг)3] 3,2 150
Из времен релаксации оценены некоторые КССВ (табл. 2.32). При
симметричном окружении А1 обнаружен, кроме квадрупольного,
скалярный вклад в ЯМ-релаксацию. Детальное изучение релаксации
13С и 27А1 установило различие тс для разных атомов комплекса
Рис. 2.18. Спектр раствора алюминия в изопропиловом спирте на частоте
104 МГц
Таблица 2.36
Константы косвенного спин-спинового взаимодействия алюминия-27
Форма 'J(AI-X) X Растворитель Ссылка
L1A1H4 172 'H — [241]
[А1Н4]- 110 ‘H H2O To же
Li[AlD4]“ 26 2d — [253]
LiAlMe4 71,5 l3C ДМФ «
А12Ме6 91 13C Et2O «
А1С13РМе3 265 31 p CH2C12 «
A1(BH4)3NH3 9 "B C6H6 «
Al(MeCN)g+ 22 l4N* MeCN [243]
[A1C13(NCS)]- 40 14n* — «
Aici; 650 35C1* — «
А1Вг4- 750 8lBr* — «
[A1F6]3- 19 l9F H2O [254]
* Оценка по ЯМ-релаксации 27А1.
А1 > СНг > СНз [243]. Константы квадрупольного взаимодействия
QCC в ацетилацетонатах металлов определены в [255] (табл. 2.37).
ЯМР 27А1 широко используют при изучении состояния алюминия
в растворах. В кислых водных растворах ион А13+ координирован ше-
Таблица 2.37
Константы квадрупольного взаимодействия для М(Асас)з
Ядро 27А1 71 Ga 1|51п
Q, барн 0,15 0,12 0,83
Радиус (М3+), пм 51 62 81
QCC, МГц 3,1 6,9 120
стью молекулами воды (доказано методом ЯМР *Н [258] и 17О [257]),
которые обмениваются со свободной водой довольно медленно, и при
снижении температуры удается наблюдать линию координационной во-
ды отдельно от свободной. Сольватация А1(Ш) рассмотрена в [245, 248,
258]. При растворении безводных солей AlgXg (X = Cl, Br, I) в невод-
ных растворителях наблюдают две линии А1Х^“ и А1Ц+, где L —
молекула растворителя [245, 256]. В растворах алкоксидов алюминия,
образованных при растворении алюминия в безводных спиртах, обра-
зуются полиядерные алкоксисоединения с тетраэдрическим, 5-коорди-
нированным и октаэдрическим окружением алюминия [252].
Смешанную сольватацию наблюдали в растворах AHHgOV
ОР(ОМе)з]6-:г в нитрометане [259], где по ЯМР 27А1 удалось
наблюдать все возможные формы смешанных сольватов; изомерия
в комплексах одинакового состава не наблюдалась, по-видимому из-за
недостаточного разрешения. Обнаружены ацидокомплексы акваиона
А1(Ш) с SQ2- [249, 260] и РО3" [262, 203]. По ЯМР 27А1 и в том,
и в другом случае наблюдаются две линии с ХС около —3 и —7 м. д.
(см. табл. 2.31). Наличие нескольких (до пяти) линий в ЯМР 31Р
таких растворов показывает, что линия -7 м.д., а возможно, и —3 м.д.
является суперпозицией линий от нескольких комплексов [263].
Комплексообразование А1С1з, А1Вгз с этилацетатом изучено в [264].
В бензольных растворах образуются как тетраэдрические комплексы
А1На1з-ЕЮАс и [AlHah]-, так и октаэдрические [Al(EtOAc)g]3+. В аце-
тонитрильных растворах вторым лигандом выступает растворитель.
Амфотерные свойства алюминия приводят к образованию как ка-
тионных, так и анионных форм в водных растворах; в щелочных
растворах А1(Ш) существует в виде алюминат-иона А1(ОН)^" [266].
В таких растворах наблюдают одну линию с ХС около 80 м.д., ширина
которой зависит от состава раствора и условий [265].
Формально [А1(Н2О)б]3+ является кислотой, и растворы солей алю-
миния имеют кислую реакцию из-за гидролиза акваиона А1. При их
нейтрализации идут реакции гидролиза и поликонденсации акваиона
алюминия, которые многократно изучались методом ЯМР 27А1. Ак-
туальность таких исследовании вызвана широким применением как
самих реакций, так и их продуктов — гидроксида и оксида алюми-
ния в промышленности и технологиях. В результате ряда исследова-
ний [249, 267-278] установлено, что поликонденсация идет по схеме:
А13+ + Н2О^ АЮН2+ + Н+-;
А1ОН2+ + Н2О -> А1(ОН)2+ + Н+-;
2А1ОН2+ -> [А12(ОН)2]4+;
р А13+ + жН2О А1р(ОН)3р- + жН+,
и перед осаждением заканчивается образованием частиц золя разме-
ром 2 нм.
В зависимости от состава исходного раствора и условий осаждения
процесс поликонденсации идет по двум направлениям:
А13+ -+ А12(ОН)4+ А113О4(ОН)7+ золь АМОН)3*"*
—> осадок гидрогеля гидроксида А1(Ш)
и
А13+ -> А12(ОН)4+ -> А1р(ОН)3р-9А12(ОН)4+ -+
золь Alm(OH)3m-nAl2(OH)2+ —> осадок.
Комплексы А1,3О4(ОН)7+ (A1i3) и некоторые из А1р обнаруживаются
по ЯМР 27А1. Структура Al]3 предполагается в растворе такой же, как
и в кристалле (рис. 2.19, [242, 267]). При этом по ЯМР 27А1 наблюдают
обычно центральный атом в тетраэдре (62,5 м.д.) [249], а широкая
линия октаэдров этого комплекса маскируется линией акваиона А13+ и
широкими линиями других комплексов с ХС 10-12 м.д. [278], однако в
специальных условиях линию октаэдров Ali3 удалось наблюдать [243].
Переход алюминия от акваиона к AI13 по мере добавления основания
к 0,2 М раствору хлорида алюминия показан на рис. 2.19. При этом до
соотношения ОН : А1 = 2,2 не менее 95% алюминия было представлено
только двумя комплексами А13+ и AI13.
Появление в растворе частиц золя сопровождается резким увеличе-
нием ширины линии ЯМР 35С1 хлор-иона [267]. Сопоставление соста-
вов растворов, определенных методом ЯМР 27А1, с рентгенографиче-
скими данными твердых фаз, полученных из этих растворов, позволило
установить зависимость структуры гидроксида А1(Ш) от вида гидрок-
сокомплексов в растворе перед осаждением геля гидроксида [272].
Во фторидных растворах алюминия информативность ЯМР алю-
миния невысока из-за обмена фторид-иона, хотя разные по составу
системы исследовались неоднократно [254, 261, 279].
Рис. 2.19. Изменение относительной доли а алюминия в составе моноядерных
комплексов А1 (/), комплекса Ali3 (2) и ширина линии ЯМР хлора (3) при
добавлении основания к 0,2 М раствору А1С13 в воде. Вверху — структура
комплекса А1]3
Комплексы [А1(Н2О)пРб-п]"-3 лабильны в масштабе времени ЯМР
при комнатной температуре. Обмен замедляется при низких темпера-
турах (ниже —50 °C), при которых невозможно исследовать водные
растворы и линии ЯМР алюминия широкие. Такие температуры до-
стигаются в водно-органических или в водно-пероксидных растворах.
В водно-пероксидных растворах удалось снять спектры при темпера-
турах до 233 К [279], при которых отдельные формы разрешаются
в спектрах ЯМР 19F (рис. 2.20). В спектрах 27А1 разрешение хуже,
однако для AlFg- определена *J(A1-F) = 19 Гц.
Дальнейший прогресс ожидается при измерениях в сильных
(^600 МГц) полях, в которых обмен эффективно замедляется. Состав
и строение фторокомплексов алюминия в водно-перекисных растворах
и растворах минеральных кислот методом ЯМР 27AI, 19F, 'Н изучали
в [279]. Обзор на тему о комплексообразовании F- в растворах
алюминия сделан в [280].
Комплексы с лимонной, малоновой, щавелевой кислотами изучены
в [282]. В оксалатных растворах при pH 0,5 наблюдали линии 27А1 6,8
и 11,9 м.д., отнесенных к А1ох+ и А1(ох)г соответственно. В области
pH 1-9 проявлялась третья линия 16,3 м.д., отнесенная к комплексу
[А1(ох)3]3-. ХС соответствующих комплексов лимонной и малоновой
кислот близки к приведенным выше.
Ряд работ посвящен изучению соединений алюминия в расплавлен-
ном состоянии. В [283] изучены алюминаты LaA^Ozj, Y3AI5O12 и оксид
AI2O3. При 1900-2400 К наблюдали одиночные, предположительно об-
Рис. 2.20. Спектры ЯМР в водно-перекисных растворах А1(МОз)з и NH^F
при разных соотношениях F/A1: а — l9F, vo = 75,26 МГц; б — спектр 27А1
комплекса [AlFg]3-, = 20,86 МГц
менные линии с ХС 70, 63 и 53 м.д. соответственно. В [284] изучены
расплавы AI2O3 :СаО в диапазоне отношений от 100% AI2O3 до 20%.
При 2500 К ХС линейно меняется от доли AI2O3 от 60 (100% AI2O3) до
85 м.д. (30% AI2O3). Ширина линии ЯМР при этом была в пределах
100-260 Гц.
В [285] методами ЯМР 27А1, 23Na, 19F и |7О изучали расплавы,
соответствующие составу электролизной ванны для получения алюми-
ния. В расплаве NaF-AlFs-A^Os ХС 27А1 менялся линейно от 0 до 6%
AI2O3 от 20 до 48м.д., оставаясь далее постоянным до 10% AI2O3.
Интерпретация данных ЯМР всех ядер привела к выводу о наличии
в системе ряда комплексов алюминия (табл. 2.38).
Таблица 2.38
Химические сдвиги форм в расплавах системы NaF-AlF3-A12O3
Форма <527А1, м.д. Форма <527А1, м.д.
ЛЮ®" 90-55 aif; 38
ЛЮ7" 30-40 A1F2- 20
А1О|- 20 4- -20 A1F|- 1,4ч- —13
Плавление минерала меняет соотношение тетраэдрических и окта-
эдрических окружений алюминия (табл. 2.39 [283]).
Таблица 2.39
Соединение Структура Altetr/Aloct, % Т, К 5, м. д. Ahelr/Aloct, % В жидк.
СаА12О4 Орторомбическая Р2/п 100/0 1878 70 95/5
УзА15О12 Кубическая Ia3d 60/40 2243 63 80/20
А12О3 Ромбоэдрическая ЛЗс 0/100 2327 53 60/40
Формы сорбции алюминия и галлия ионообменной смолой СФ-5
изучали методом ЯМР 27А1, 71 Ga и 31Р MAS в [287] в диапазоне
концентраций НС1 0,1-0,6 М. В спектрах 31Р наблюдали линии 7
и 15м.д., соответствующие связи РОз-групп к одному и двум ато-
мам А1.
Галлий. Из-за более узких линий и чувствительности ЯМР 71 Ga
по сравнению с 69Ga преимущественному используется ЯМР 71 Ga.
Только в редких случаях, в основном в релаксационных измерениях,
используют и ядро 69Ga. Диапазон ХС галлия шире, чем у алюми-
ния, однако меньший интерес к галлию в металлоорганической химии
и меньшая чувствительность ядер галлия по сравнению с ЯМР 27А1
ограничили применение ЯМР 71 Ga. В отличие от алюминия, известны
состояния окисления Ga(I) и Ga(III). Опубликованы обзорные работы
по ЯМР галлия [241, 243, 288-290]. Данные ЯМР некоторых форм
галлия в растворах приведены в табл. 2.40.
Таблица 2.40
Данные ЯМР галлия-71
Форма 5, м.д. W, Гц Примечания, источник
GaH4- 682 — Et2O [288]
GaF“ 581 1000 Н2О [289]
GaCl4- 197 300 Расплав Са2С1б [288]
223 300 Са2С1б в СбНб [288]
257 100 Н2О в НС1 [288]
260 2700 GaCl3/Et2O [296]
252 90 MeCN [290, 291]
Таблица 2.40 (окончание)
Форма 6, M. Д. W, Гц Примечания, источник
GaChBr- 211 110 To же
GaCl2Br; 171 120 To же
GaCIBr; 118 113 To же
GaBr; 67 110 To же
69 100 H2O в НВг [288]
GaChNCS- 225 — MeCN [292]
GaCl2(NCS); 194 — То же
GaCl(NCS)7 157 — То же
Gai; -505 100 MeCN [290]
Ga(OH); 192 600 NaOH/H2O [289]
Ga(OD); 220 152 NaOH/H2O [289]
Ga(MeCN)|+ -72 — MeCN [293]
Ga(H2O)3+ 0 — H2O [290]
Са(ДМФ)3+ -25 120 ДМФ [294]
Ga(GaC14) -650, Ga(I)‘; 247, Ga(III) 13; 230 С6Нб (1 % мае.) [297] 300 К
Ga(GaBf4) -644, Ga(I) * 25 СбНб (3% мае.) [297]
62, Ga(III) 270 300 К
GaF3- -55 J(71Ga-F) = 245 Н2О2, 253 К [281]
[Ga(OH)6Mo6O18]3- 52 5000 Н2О [251]
[GaWl204o]5“ 219,2 50 То же
* Зависит от концентрации и растворителя.
Существует корреляция между ХС одинаковых соединений алюми-
ния и галлия [243].
Константы спин-спинового взаимодействия 71Ga-14N в роданидо- и
цианогалогенидных комплексах алюминия и галлия измерены в [292];
J(71Ga-14N) ~ 3000 Гц в [Ga(NCS)g]3~ [323]. КССВ галлия с протона-
ми и дейтонами измерены в [297] (табл. 2.41).
Таблица 2.41
Данные ЯМР галлия изотопомеров гидрид-иона галлия
Изотопомер GaH4- GaH3D- GaH2D2 GaHD“ GaD4
<571Ga, м.д. 0 0,12 0,23 0,35 0,52
J(69Ga-H) 516,0 515,2 512,4 510,5 —
J(69Ga-D) — 80,3 80,1 80,0 78,7
J(7lGa-H) 655,7 654,0 652,5 650,5 —
J(7lGa-D) — 101,9 101,0 100,7 100,1
ЯМ-релаксация галлия в тетраэдрических комплексах GaCI^Br^^
изучена в [295]. Изменение Т\ и Тг (табл. 2.42) показывает увеличение
ГЭП при снижении симметрии окружения ядра, а различие времен ре-
лаксации указывает на дополнительный, кроме квадрупольного, вклад
в релаксацию, возможно, из-за ССВ Ga-галоген.
Таблица 2.42
ЯМ-релаксации галлия в смешанных галогенгаллатах
Ион Симметрия Tt, 71 Ga, MC Ti, 69Ga, mc T2, 7lGa, mc T2, 69Ga, mc
GaCl; Td 15 3,5 9,5 2,4
GaChBr- Сзи 6,6 2,9 3,0 1,9
GaCl2Br^ C2v 5,5 2,7 2,9 1,6
GaClBr^" C'iv 6,1 2,8 2,8 1,6
GaBr^ Td 12 3,2 5.0 2,5
На рис. 2.21 изображены спектры алюминия-27 и галлия-71 атомов
А1 и Ga в одном и том же окружении (узкая линия в спектрах —
примесь акваионов); удивительно, что при полной идентичности окру-
жения М(1П), заряда комплекса и практически одинаковой молеку-
лярной массе (т. е. одинаковой величине тс молекулярного движения)
ширина линии ЯМР галлия на порядок больше линии алюминия, хотя
соотношение квадрупольных моментов ядер обратное.
Методом ЯМР галлия изучен ряд комплексов Ga(III) в растворах:
фторокомплексы Ga(III) в системе GaCb-HF-HgO — в [289], где по
ЯМР 19F найдены четыре комплекса: GaF^, ОаРз-НгО, ОаРг-НгО,
GaF2+-H2O, а по ЯМР 71Ga наблюдали одну широкую линию; смешан-
ные комплексы галогенидов галлия в ацетонитрильных растворах —
в [295]; цитратные комплексы — в [299]; роданидные — в [292];
взаимодействие Ga3+ с фосфатом — в [300]; галогенидные комплексы
50
0 м.д.
Рис. 2.21. Спектры ЯМР 27А1 и 7lGa в анионах [М|пМоб(ОН)бО18]3 , М = А1
(/), Ga (2), структура анионов (3)
в неводных растворах — в [301, 302]. Влияние растворителей — эфиров
на ХС ОаС1з изучали в [301]. Соединения Ga(I) изучены в [298].
Индий. Работы по ЯМР индия ограничены изучением тетраэдри-
ческих ионов, так как линия ЯМР слишком широка при более низкой
симметрии окружения ядра из-за большого квадрупольного момента.
Все измерения сделаны на ядре 1151п, кроме специальных случаев. Дан-
ные ЯМР 1151п приведены в табл. 2.43. Отмечена малая зависимость
ХС ионов 1пХ^" от растворителя [305]. Существует корреляция между
ХС одинаковых соединений индия и галлия [243].
Обмен молекул сольватной оболочки с растворителем вызывает
уширение линий, из-за чего многие 6-координированные комплексы
индия не наблюдаются в ЯМР. Дальнейший прогресс в химическом
приложении ЯМР 1151п, равно как и ЯМР галлия и алюминия, возмо-
жен при переходе к высоким полям 9 Т), где улучшение специфично-
сти, чувствительности и эффективное замедление обмена существенно
расширяет возможности метода.
Известны КССВ индия *J(115In-F) 420 Гц [302] в InF^-.
Таллий. В спектроскопии ЯМР обычно используют изотоп 205Т1,
поскольку различия свойств ядер для ЯМР несущественны. Известны
два состояния окисления таллия: Т1(1) и Т1(Ш); последнее из них
более характерно. Диапазон ХС Т1(1) около 3500 м.д., Т1(Ш) — около
4000 м.д. Общий диапазон около 7000 м.д. за счет перекрывания диа-
пазонов ХС Т1(1) и Т1(Ш) [241].
Ион Т1+ легко замещает Na+ и К+ в биологических и биохимиче-
ских объектах, что позволяет использовать его как изотопную метку
Таблица 2.43
Данные ЯМР индия-115
Форма 5, м. д. W, Гц Растворитель, источник
1п(Н2О)36+ 0 375 Н2О [303]
1п(Н2О)3+ 10 1800 Н2О:НС1О4 = 20 : 1 [304]
« -30 5000 Н1:Н2О:Ме2СО = 1 : 15 : 45 [304]
In2(SO4)3 -18 300 Н2О [303]
InCi; 440 680 Et2O [303]
« 441 370 СН2С12 [305]
1пС13Вг- 398 — То же
1пС12Вг^ 323 — «
InClBr^" 252 — «
InBr^ 186 — «
« 180 375 Et2O [303]
1пВг31— 11 — СН2С12 [305]
1пВг21^ -176 — То же
InBrl^ -372 — «
inBri; -570 — «
« -583 250 Et2O [303]
« -578 1080 Концентрированный HI [303]
« -600 190 Н1:Н2О:Ме2СО = 1:15:45
1пС13Г 250 — [304]
1пС1212“ 11 — То же
1пСП“ -266 — СН2С12 [305]
для ЯМР в биологических исследованиях; при этом надо помнить
о высокой токсичности таллия. Обзоры по ЯМР талия опубликова-
ны в [241, 243, 306]. ХС некоторых соединений таллия, приведены
в табл. 2.44.
ХС таллия зависит от концентрации, температуры (до 5 м.д./К)
[306] и растворителя. Т1(1) плохо растворяется в воде, но прекрасно
растворим в аммиаке. На рис. 2.22 приведена зависимость ХС 205Т1(1)
от его концентрации в аммиаке.
Таблица 2.44
Химические сдвиги таллия
Форма 6, м.д. Условия Ссылка
Ме3Т1 5093 н-Пентан, 24 °C [307]
4753 Et2O, 24 °C [307]
4504 10% в ацетоне, —70 °C [308]
Et3Tl 4963 н-Пентан, 24 °C [307]
Ме2Т1ОН 3636 Н2О, 24 °C То же
Ме2Т11 3594 1,2 М в ДМСО «
Ме2Т1Вг 3575 ДМСО, нас. р-р «
Me2TlF 3413 То же «
Me2TlNO3 3478 1 М в Н2О «
3514 Н2О, [Т1] -> 0 [309]
3450 ДМСО, [Т1] -> 0 [309]
Т1С1з 2581 Ч. жидк., 25 °C [ЗЮ]
2307 Н2О, [Т1] -> 0 [ЗН]
T1(NO3)3 2592 0,1 М в HNO3, [ЗИ]
Т1Вг3 1235 0,3 М в Н2О [ЗИ]
T1NO3 0 Н2О, [Т1] 0 [312]
1708 н-Бутиламин, [Т1] —> 0 [312]
783 Пиридин, [Т1] —> 0 [241]
360 ДМСО, [Т1] -> 0 [241]
145 ДМФ, [Т1] -> 0 [312]
Т1С1О4 35; 369 МеОН, [Т1] -> 0 ДМСО, [Т1] 0 [312, 313]
T1BF4 1958 н-Бутиламин, [Т1] —> 0 [312]
TlCl; 2630 — [243]
Т1Вг4- 1318 — [243]
Величина констант ССВ *<7(С-Т1) превышает 10 кГц [314, 315],
2J(H-2O5T1) = -269 Гц в метилталлии [308]. Примеры КССВ приведены
в табл. 2.45 [306].
г205т'1 .. _
О 1 1, М. Д.
Рис. 2.22. Зависимость химического сдвига таллия от концентрации TINO3
в его аммиачном растворе
Таблица 2.45
Константы спин-спинового взаимодействия 205Т1
Форма Раство- ритель 'J(2O5T1-C) 2J(2O5T1-C) ’J(2O5TI-H) J(2O5T1-X)
PhTl(OAc)2 дмсо 10718 527 — — —
d2o — — — 912 (3J) —
Ме3Т1 Ацетон 1930 — — -269 (2J) —
(T10Et)4 Чист, жидк. — — — — 2560 (2O3T1)
Т1Н4- Вода — — 6144 — —
Sn8Tl5“ еп — — — — 795 C19Sn)
ЯМ-релаксация 205Т1 и 203Т1 проходит сходно, не зависит от частоты
ЯМР, и Т\ Т%- В водных растворах ЯМ-релаксация Т1+ сильно за-
висит от концентрации растворенного кислорода: Т\ меняется от 24 мс
при давлении О2 1 ат до 1,8 с в дегазированном образце; при обычной
атмосфере Т\ ~0,1 с [316]. Для иона TI+ в дегазированном метаноль-
ном растворе Т\ = 0,91 с [317]. Основной вклад в ЯМ-релаксацию
дает спин-вращательное взаимодействие [317, 318], однако возможен
и вклад анизотропии ХС [241]. Для неводных растворов последний
вклад может превалировать [306]. ЯМ-релаксация Т13+ изучена слабо,
однако в растворе НС1 ширина линии ЯМР около 10 кГц, а в растворе
НВг — 5 кГц; в дегазированном растворе МегТ1+ Т\ = 0,56 с [332].
Водные растворы солей Т1(1) изучены в [307, 319-328], растворы
в неводных растворителях — в [312, 313, 329, 330], конкурирующая
сольватация — в [312, 313, 329]. Замена воды на тяжелую воду меняет
ХС незначительно (~ 10 м.д.), но в комплексе с этилендиамином ХС
при этом меняется на 100 м.д. [316]. Комплексообразование Т1+ с кра-
унэфирами и криптандом [С222] изучены в [306]. Ион Т1+ образует
с краунэфиром 18С6 комплекс 1:1, при этом наблюдали быстрый
обмен между сольватироваными и закомплексоваными ионами Т1+.
ХС Т1+ в некоторых растворителях и комплексах с краун-эфирами
приведены в табл. 2.46.
Таблица 2.46 ХС Т1+ в некоторых растворителях [334]
Лиганд Растворитель 5 Tls, м. д. 6 ТГГ, м. д. DB18C6 — дибензо-18- краун-6 DCH18C6 — дицикло- гекса-18-краун-6 15С5 — 15-краун-5 18С6 — 18-краун-6
15С5 ДМСО 366 215
18С6 ДМСО 366 -27
ДМФ 124 -180
МеОН 512 -70
DB18C6 ДМСО 366 199
ДМФ 124 -110
МеОН 512 -35
DCH18C6 ДМСО — ИЗ
С222 ДМСО — 62
Пиридин — 44
6 Tls — ХС в растворителе, 6 Т1СГ — ХС захваченного краун-эфиром.
Изменение сольватной сферы Т1+ меняет ХС в пределах 2500 м. д.
Растворы солей Т1(Ш) изучены слабее [307, 310, 311, 324, 331].
Концентрационная зависимость ХС в растворах Т1(МОз)з в HNO3
изучена в [325], зависимость от pH — в [311]. Изменения концентра-
ции, pH и противоиона перекрывают диапазон 2000 м.д. Изучен ряд
комплексов с органическими лигандами и органоталлиевых соедине-
ний [307, 310, 332, 333].
2.5. Кремний, германий, олово, свинец
Кроме неорганических соединений, эти элементы образуют мно-
жество элементоорганических соединений. Металлические свойства
элементов усиливаются от кремния к свинцу, и если кремний образует
много соединений, аналогичных органическим, то для свинца часть из
них несвойственна.
За исключением 73Ge, магнитные ядра этих элементов имеют спин
1/2; для них существует обширная база КССВ. Диапазон ХС возрас-
тает от кремния к свинцу; малый диапазон ХС кремния объясняется
тем, что для него нехарактерно образование соединений с кратными
связями. Частота ЯМР 73Ge лежит ниже стандартного диапазона ча-
стот многоядерных спектрометров ЯМР, однако многие стандартные
датчики высокого разрешения диапазона P-W настраиваются на эту
частоту.
Чувствительность ядер к изменению окружения показана
в табл. 2.47. Подавление протонов дает небольшой выигрыш при записи
Таблица 2.47
Свойства ядер магнитных изотопов кремния, германия, олова, свинца
Изотоп Спин NA, % Sens 1 Диапазон ХС, м.д. Q, барн Частота ЯМР эталона Е, МГц2 Эталон
29Si 1/2 4,7 0,7 250 0 19,867 184 Si(CH3)4
73Ge 9/2 7,6 0,2 1200 -0,22 3,488315 Ge(CH3)4
ll7Sn — 7,6 6,5 2000 — 35,632 295 Sn(CH3)4
ll9Sn — 8,6 8,5 2000 — 37,290665 Sn(CH3)4
2°7pb — 22,6 3,9 3500 — 20,920597 Pb(CH3)4
1 Относительно |3С{'Н}; 2 частота ЯМР в поле Но, в котором частота ЯМР 'Н
ровно 100 МГц.
спектров ЯМР ядер 29Si, 73Ge, ll9Sn, так как оно сопровождается
отрицательным эффектом Оверхаузера из-за отрицательного гиромаг-
нитного отношения этих ядер; выигрыш дает слияние мультиплетов
и уменьшение эффективного времени релаксации этих ядер.
Для неорганических систем в большинстве случаев необходимость
в подавлении протонов не возникает из-за отсутствия связи М-Н. При
исследовании растворов неорганических веществ подавление протонов
применяется только при наличии ССВ с протонами, однако в этих
случаях более эффективны методы переноса спиновой поляризации
(INEPT и другие).
Чувствительность ХС ядер к изменению окружения возрастает от
кремния к свинцу.
Кремний. У кремния один магнитный изотоп. Низкое природное
содержание и долгие времена релаксации Т\ затрудняет регистрацию
ЯМР 29Si.
Времена релаксации 29Si редко бывают менее 20 с, поэтому эффек-
тивно применение методик с вынужденным восстановлением равнове-
сия спиновой системы (DEPT и другие). В то же время Т\ анионов
в силикатных растворах на порядок меньше.
Большая часть работ по ЯМР 29Si посвящена изучению кремний-
органических соединений. Существующие обзоры [335, 337, 339-341]
показывают возрастающее применение метода ЯМР. Общий диапазон
ХС такой же, как у ядра 13С. Шкала ХС некоторых соединений крем-
ния показана на рис. 2.23 и в табл. 2.48.
I----1 SiX2“
SiX5“ I----1
I----------------------1 XnSiE4_n
I—I R3SiNR2
X2SiH2 I----1
I----------1 X3SiH
I------------------------------------1 X3SiCl
I---1 SiO4
I---1 RSiO3
I I RSiO2 R _ алкил, арил
R3SiO I I X — галоген
I---1 R4Si E — переходный
металл
i____________I___________I____________I
100 0 -100 -200 М.д.
Рис. 2.23. Шкала химических сдвигов ЯМР 29Si
Аналог метана силан образует соединения со всеми галогенами и их
произвольной комбинацией SiHal4. Химические сдвиги замещенных
силанов отображены на рис. 2.24. Существует линейная корреляция
ХС 29Si-27Al форм в одинаковом тетраэдрическом окружении [341], при
этом диапазон изменения ХС у кремния вдвое больше, чем у алюми-
ния; в то же время как корреляции 29Si-73Ge, 29Si-119Sn нелинейны.
Эффекты замещения Н-Cl в ЭНПС14_П спектрах ЯМР 29Si и 13С про-
тивоположны (рис. 2.25 [340]).
Таблица 2.48
Химические сдвиги ЯМР 29Si некоторых форм кремния
Форма 5, м. д. Примечания, источник
SiMe4 0 Ч. жидк. [336]
SiH4 -9,9 [337]
SiCl4 -16 Ч. жидк. [336]
SiBr4 -90 То же
[Me2SiO]4 -20 «
[Me2SiO]3 -10 «
Me3SiI 8,6 «
Me3SiBr 26,4 «
Me3SiF 31,1 Ч.жидк. [336], 'j(Si-F) = 275 Гц
MesSiCb 32 Ч.жидк. [336]
MeSiCl3 8 То же
(SiH3).3N -40 Ч. жидк. [4]
[SiMoi2O40]4- -81,8 0,3 М р-р Н2О [338]
[SiMo^ МоУдО40]6_ -83,6 То же
Рис. 2.24. Химические сдвиги ЯМР 29Si галогенсиланов SiH4_IHalI
Полуэмпирические расчеты экранирования 29Si сделаны в [342]
с учетом только парамагнитного вклада в приближении средней энер-
гии. Для ядра 29Si не очевидно, что изменение экранирования опре-
деляется только парамагнитным вкладом, и не ясно, можно ли прене-
Рис. 2.25. Корреляция химических сдвигов ЯМР |3С и 29Si в замещенных
метанах СНПС14_П и силанах SiHnC14-n
бречь вкладом в экранирование d-орбиталей. В [343] показано наличие
линейной корреляции между ХС и электроотрицательностью для ато-
мов кремния, связанных с лигандами, имеющими неподеленную пару
электронов.
Ряд работ [344-351] посвящен химии растворимых силикатов.
В табл.2.49 представлены обозначения типов силикатных тетраэдров
с разными типами связей с такими же тетраэдрами и их диапазоны ХС.
Таблица 2.49
Обозначение типов атомов кремния групп SiC>4 и диапазон их химических
сдвигов
Тип кремнекислородного тетраэдра Тип <5 29Si, M. Д.
Моносиликат Qo -664- -72
Концевые группы Qi -75 4- -80
Средние группы в циклических формах Q2A —79 4- -82
Средние группы в нециклических формах q2 -85 4- -89
Группы разветвления Q3 -94 4- -100
Группы разветвления циклических формах Q3A -85 4- -92
Группы, сшивающие полимеры Q4 -100 4- -120
Диапазон ХС силикатов невелик [342], и спектр раствора мета-
силиката натрия (рис. 2.26 [344]) покрывает его (отнесение линий
в табл. 2.50). Широкое использование силикатов в промышленности,
строительстве и науке вызывает продолжение исследования растворов
силикатов с разными катионами и модулями, и приведенный блок
[343-361] далеко не исчерпывает их список. В более поздних рабо-
Qo
Q,l?Qf3Q3 Qi
-80 -100 -120 -140 м.д.
Рис. 2.26. Спектр ЯМР 29Si водного раствора метасиликата натрия на частоте
79,5 МГц
тах [356, 357, 361] идентифицированы и суммированы многие поли-
ядерные формы, данные по которым сведены в табл. 2.50.
В конкретизации форм силикатов важную роль сыграла двумерная
методика ЯМР DOSY, учитывающая скорость диффузии форм [362].
В [349] были изучены растворы SiC>2 в водных КОН или NaOH,
с большими концентрациями Na (К) 8 М/л и Si 4 М/л. При этих
концентрациях максимальная величина Т\ была 3,5 с для Q2A, для
остальных форм времена Т\ были короче. По мере старения растворов
Т\ всех форм сокращались.
В [352] были изучены обогащенные по кремнию (95%) образцы,
полученные растворением SiO2 в водном КОН при 100 °C; измерены
0,6 М растворы с модулем К : Si, равным 1:1, 1,5 : 1 и 2 : 1 на
частоте ЯМР 99,36 МГц, при 273 К для исключения обменных про-
цессов. Появление групп мультиплетов позволило определить, кроме
мономера, 5 форм (2-4, 7, 8 табл. 2.50) и выделить мультиплеты
[2J(Si-Si) = 4-7 Гц], относящиеся к каждой форме. В этой работе, как
и многих других, отсчет ХС ведется от линии Q°, положение которой
зависит от состава и концентрации раствора.
Концентрированные растворы силикатов натрия с разными моду-
лями на частоте ЯМР 49,7 МГц и природным с содержанием 29Si
изучены в [354]; определено распределение форм в зависимости от
модуля и константы образования некоторых форм.
В работе [354, 355] изучены растворы в D2O силикатов с моду-
лями Na, К, Rb 1, в диапазоне температур 268-423 К. С ростом
температуры обменные процессы смазывают линии остаются только 4
широкие линии Q°, Q'+Q2^ Q2+Q3A, Q3. Измерены T\ 2 разных форм,
предложена схема взаимопревращения форм.
Использование тетраалкиламмониевых катионов при [Si] 0,3-0,6
М/л позволяет получать спектры с большим разрешением по сравне-
нию с растворами катионов металлов [345], что позволило определить
ряд новых форм.
Таблица 2.50
Химические сдвиги, м.д., 29Si в водных растворах силикатов [354]
№ Форма Структура 1 -<5 (Q°) -<5 (Q1) -<5 (Q2) -<5 (Q2a)2 -<5 (Q3) -<5 (Q3A)
1 Мономер • 71,3 — — — — —
2 Димер • • — 79,8 — — — —
3 Тример линейный * л л — 79,3 88,2 — — —
4 Тример циклический V — — — 81,4 — —
5 Тетрамер линейный - — 79,6 87,5 — — —
6 Тетрамер циклический , — — — 87,3 — —
7 Тример циклический замещенный * — 79,2 — 81,1 89,4 —
8 Тетрамер циклический связанный —< — — — 85,5 93,2 —
9 Тетрамер циклический замещенный <В/— — 79,2 — 87,1 А; 87,4 в 95,3 —
10 Пентамер бициклический в! >А — — — 81, 2а; 87,6 в — 88,4
И Гексамер призматический — — — — — 88,4
Таблица 2.50 (окончание)
№ Форма Структура 1 -<5 (Q°) -<5 (Q1) -<5 (Q2) -<5 (О2Д)2 -<5 (Q3) -<5 (Озд)
12 Гексамер трициклический ( - Ji 3 — — — 87,4 — 87,9а; 88,8В; 96,0е
13 Гексамер трицикличе- ский (ванна) 4 — — — 81,8 — 88,1
14 Гексамер трицикличе- ский (кресло) — — — 82,1 — 89,2
15 Тетрамер циклический двусвязный — — — 85,9 — 92,7
16 Гептамер - — — р — — 90,2 А; 89,2 в
17 Кубан —<< — — — — — 98,6
18 Декамер призматический — — — — — 98-98,5
1 Отмечены только атомы Si; 2 циклические формы.
Обзор форм силикатов в природных водах сделан в [357]. Самодиф-
фузия форм и обмен в силикатных растворах изучен в [358]. В резуль-
тате изучения равновесий и времен релаксации кремния обнаружена
форма кубан (табл. 2.50) [359]. Зависимость ХС отдельных форм от
состава некоторых форм обнаружена в [360]. Использование разных
типов ЯМР-спектроскопии кремния вместе с молекулярным моделиро-
ванием позволило выявить новые типы форм и изомерию некоторых
форм [361].
Расплавы некоторых силикатов, а также масс-спектры были изуче-
ны в [363]. При температурах ~ 1000 К регистрируется только одна
обменная линия [364].
Синтезированы замещенные германием призматический гексамер и
кубан [365], данные ЯМР приведены в табл. 2.51.
Таблица 2.51
Данные ЯМР 29Si монозамещенных германием полиэдров [365]
Форма Atom —5, м. д. 2J(Si-Si), Гц
Ао—-рМ A 97,9 7,5
" ‘а M 100,2 7,5
. J/м А о ОМ X 99,7 7,5
А<хоА A 89,3 4,3
M<L J)M M 90,1 4,3
X 88,3 4,3
Кислородные мостики в силикатах можно заместить на пероксомо-
стики, получив тем самым пероксосиликаты [366].
Активно исследуются алкоксисиликаты Si(0Alk)4 [367-369]. Гид-
ролиз и поликонденсацию этоксисилана изучали в [367-368], форми-
рование метоксисилана в щелочных метанольных растворах — в [369].
В алюмосиликатах также реализуются различные типы соединения
А1 с тетраэдрами SiOi; их ХС представлены в табл. 2.52.
Измерен ряд КССВ с фтором [371]. Во фторсиланах FISiH4_1.
(х — 1-4) величина ^(Si-F) равна 281, 298, 275 и 145 Гц соответ-
ственно. Увеличение электроотрицательности может и увеличивать,
и уменьшать КССВ кремний-фтор. Минимальная величина 1 J(Si-F) в
SiF6 равна НО Гц [372-373]. Константы ССВ с другими ядрами мень-
ше. В соединениях X(SiH3)3 ’J(Si-H) лежит в диапазоне 208-213 Гц
(X = N, Р, As, Sb, [371]), 'j(Si-P) = 42 Гц в P(SiH3)3, ’J(Si-N) = 6 Гц
в N(SiH3)3 [374, 375], 2J(Si-Si) = 4-8 Гц в разных формах силикатных
растворов [352, 361].
Таблица 2.52
Химические сдвиги ЯМР кремния группы SiO^ в алюмосиликатах [370]
Полоса Тип алюмокремниевого тетраэдра —8, м. д. 8 27А1, м.д.
Q0 — 0 —
А Q'+Aio; 3,9; 4,2; 4,5 80
B + Q1 Цепь 6,8-8,6 —
Q2A — 9,2-10,5 —
С Вставки в структуру алюмината в виде Q2 11,6; 11,9; 12,3 —
D q'q2q> 13,0 74; 66; 52*
Е Qi 14,3 —
Q2B — 15,2-17,5 —
Q3A — 17,5-19 —
Q3B — 21-27 —
* Неразрешенные линии в разных отношениях.
Основными механизмами релаксации ядра кремния являются ди-
поль-дипольный (при наличии в молекуле ядра !Н или 19F, и спин-
вращательный. ЯМ-релаксацию 29Si в водных растворах силикатов
металлов изучали в [376], где обнаружен заметный диполь-диполь-
ный вклад от ядер катионов. Механизмы спин-решеточной релаксации
в растворе калиевых силикатов изучены в [377], величины Т\ разных
форм приведены в табл. 2.53.
При повышенных температурах свой вклад вносит обмен, заметный
уже при 40 °C; влияние температуры (-5-200 °C) изучено в [377].
Германий. Низкое гиромагнитное отношение и малое природное
содержание единственного магнитного изотопа 73Ge (см. табл. 2.47)
ограничивает применение ЯМР 73Ge в химии. В обычных полях (^ 2 Т)
из-за акустического звона датчика регистрируются только узкие линии
ЯМР комплексов с симметричным окружением германия в невязком
растворителе. Переход к высоким полям (Но 7 Т) существенно улуч-
шает положение; становится возможной регистрация линий с шириной
более 1 кГц и концентрацией германия в растворе более 0,1 г-ат/л.
Данные ЯМР 73Ge приведены в табл. 2.54.
Таблица 2.53
Времена спин-решеточной релаксации кремния в некоторых формах [377], 25 °C
Форма Тип Ti, с
Мономер Q0 3,8
Димер Q* 2,6
Линейный тример Q1 2,8
Q2 1,4
Циклический тример Q2 4,4
Замещенный циклический тример Q1 —
Q2 3,1
Q3 2,3
Линейный тетрамер Q1 2,7
Q2 —
Циклический тетрамер Q2 2,4
Призматический гексамер Q3 59
Кубический октамер (кубан) Q3 9,8
Теоретическое исследование экранирования галогенгерманов сдела-
но в [384], где экранирование представлено суммой вкладов от каждого
лиганда и дополнительных вкладов от взаимодействия лигандов:
Sij : ^ = а + Ыг6г + CijSij. (2.10)
г ij
Хорошую аддитивность при замещении показывает спектр эквимо-
лярной смеси GeCU и GeBr4, в которой наблюдали все смешанные
формы (рис. 2.27 [379]). Экранирование в хлорном тетраэдре близ-
ко к таковому в кислородном тетраэдре как в замещенных германа-
тах с кислородсодержащими лигандами, так и в молибдогерманатах
(см. табл. 2.54). В восстановленном смешанно-валентном диамагнит-
ном молибдогерманате [GeMo^Моц}04о]6- ХС почти такой же, как
и у окисленной формы аниона [GeMoigO^]4-, в то время как ширина
линии заметно увеличена.
Данных о КССВ 73Ge известно не много. Они получены из опре-
деления Ферми-контактного вклада в Тг и носят оценочный характер
(табл. 2.54).
Таблица 2.54
Данные ЯМР 73Ge
Форма <5. м. д. Ti, mc T2, mc 'J(Ge-X), Гц X Примечания, источник
GeCU 34 590 160 ^25 35C1 Ч. жидк. [379-381, 383]
31 — 500 — — CS2
GeBr4 -312 450 200 — — [381]
— 88 81 64 79Br Толуол, 298 К [384]
GeCl3Br —47 380 — — — Смесь GeCl4 и GeBr4 [379]
ОеС^Вгг -131 220 — — — То же
GeClBr3 -219 250 — — — «
Gel4 -821 51 50 220 ,27I Толуол, 318 К [380, 391]
Ge2Ig -1103 — — — — Примесь в Gel4
Gel4 -800 — — — — CS2 [380]
Ge2l6 -1081 — — — — Примесь в Gel4
Ge(OMe)4 -36 — 30 — — Ч. жидк. [380]
GeMe4 0 — 740 — — То же
GeEt4 18 — 140 — — «
Ge(C3H7)4 -6 — 100 — — «
Ge^H^ 6 — 65 — — «
[GeMo12O40]4- -26 — 30 — — Н2О [385]
[GeMo^ Мо^О40]6“ -31 — -6 — — То же
Ge(NMe2)4 or GeMe4 49,4 — 14,5 — — C6D6, 303 К [383]
Ge(NEt2)4 47 — 2,4 — — C6D6, 303 К [383]
Ge(NPr2)4 22 — 1,6 — — То же
Ge(NCS)4 -443 — 6,4 — — Ацетон-бб [381, 383]
Ge(NCO)4 -88,9 — 18,7 — — C6D6, 303 К [382, 383]
[Ge(NCO)6]2- -444 — 6,4 — — Ацетон, 303 К [381, 383]
150 0 -150 м.д. -450
Рис. 2.27. Спектр ЯМР 73Ge смеси GeCU и GeBf4
В механизм ЯМ-релаксации 73Ge основным вкладом является
квадрупольный, однако для тетрагалогерманов значителен контактный
вклад [386, 387].
В [388] измерено абсолютное экранирование в метане, силане,
германе и станнане (табл. 2.55).
Таблица 2.55
Абсолютное экранирование и частоты в газообразных ЭН4
Форма X Ox, M. Д. он, м.д. /ix, яд. магнетон Частота МГц Примечания
СН4 l3C 196,77 30,609 0,702 370 7 — —
SiH4 29Si 482,85 27,625 -5 550520 99,4462169 300 к
SiMe4 « — — — 99,456 595 4 300 к
'H — — — 500,607466 Жидк.
GeH4 73Ge 1981,7 27,774 -0,878 241 3 17,457062 5 300 К
GeMe4 « — — — 17,462 762 5 Жидк., 300 К
SnMe4 ll9Sn 3476,01 33,508 -1,045 067 7 — —
Химические свойства Sn и РЬ во многом схожи. Магнитные изото-
пы есть у каждого из этих элементов, причем у всех изотопов, спин
равен 1/2, т. е. ЯМР не осложнен квадрупольными взаимодействиями.
Свойства ядер приведены в табл. 2.47.
Наличие нескольких стабильных изотопов у олова позволяет изу-
чать изотопный обмен их соединений методом ЯМР. Обзоры по ЯМР
Sn и РЬ сделаны в [391, 393].
Олово. Кроме указанных в табл. 2.47 изотопов, магнитный момент
имеет также изотоп 115Sn, содержание которого в природе 0,35%, из-за
чего он практически не используется в ЯМР.
Свойства изотопов ll7Sn и ll9Sn в ЯМР почти одинаковы. Обычно
измерения ведут на изотопе II9Sn из-за более высокой чувствительно-
сти. Кроме прямого детектирования [389, 390] используют и методы
двойного резонанса [391, 392]. Большое и отрицательное гиромаг-
нитное отношение ll9Sn предопределяет ограниченное использование
эффекта Оверхаузера для идентификации оловоорганических соедине-
ний. ХС некоторых соединений, отсчитанные от Бп(СНз)4, приведены
в табл. 2.56. Данные о ХС приведены также в обзорах [393, 394].
Аналогично ХС других тяжелых ядер, изменение ХС олова происхо-
дит за счет парамагнитного вклада в экранирование. Замещение лиган-
да в тетракоординированных комплексах олова приводит к изменению
ХС более чем на 300 м.д., однако аддитивности при замещении обычно
не наблюдают; в том случае, когда координирующие атомы одинаковы
или сходны по электронной структуре (S/Se), аддитивность наблюда-
ется. В парамагнитном вкладе в экранирование для олова учитываются
5d- и 5р-электроны. Вклад в экранирование от d-электронов в тетра-
координированных комплексах олова мал, и эффекты экранирования
описывают изменением факторов Р5р и (г~3)5р; фактор ДЕ считают
примерно постоянным [398]. В комплексах с переходными элементами
фактор ДЕ может значительно меняться [405]. Наблюдались [406]
корреляции между ХС ll9Sn и ст-константами.
Для аналогичных соединений олова и свинца существует линейная
корреляция ХС (рис. 2.28). Примером определения структуры комплек-
Рис. 2.28. Корреляция химических сдвигов ЯМР 119Sn и 207РЬ в аналогичных
органических соединениях олова и свинца
сов методом ЯМР олова и протонов служит работа [413], в которой
по спектрам ЯМР подтвержден синтез и существование в растворе
Таблица 2.56
Данные ЯМР олова-119
Форма <5, m. д. J(X-119Sn) X Примечания, источник
SnMei 0 -340 13c 5%,CH2C12 [395]
— 54 'H [396]
SnEt4 1,4 307* 13c [397, 398*]
SnPlu -137 — — ЗО%,С2Н3С13 [399]
SnCU -150 470 35C1 Ч. жидк. [390, 396]
SnBf4 -638 920 81Br Ч. жидк. [390, 396]
Snl« -1701 940 ,27I CS2 [390, 396]
SnChBr -263 — — [390]
SCl2Br2 -385 — — [390]
SnCIBrs -508 — — [390]
SnCi2 -388 — — 12М НС1 в Н2О [396]
« -236 — — ТГФ [390]
(Мез5п)25 86,5 — — 20%,СН2С12 [400]
(Me3Sn)2Se 44,5 — — То же
(Me3Sn)2Te -67 — - «
(МезЗп)2ММе2 75 — — 25%,С6Н6 [401]
(Ме35п)зР 86,5 832 — 65%, С6Н6 [402]
МезЗп-В(\Ме)2 -150 — 31 p Ч.жидк. [403]
Мез5п-51Мез -127 656 29Si Ч.жидк. [404]
(Me3Sn)2 -109 -240 13C С6Н6 [404]
« — 4460 117Sn С6Н6 [404], C6D6 [405]
МезЗп-РЬМез -57 -3570 207 Pb FSO3H, -80 °C [406]
SnH3+ -186 2960 'H [390]
Sn(OH);?- -590 — — [408]
Sn(OOH)g“ -661 — — [408]
гран-[5п(ОН)з(ООН)3]2- -609 — — [408]
pe6-[Sn(OH)3(OOH)3]2- -604 — — [408]
[Sn(OOH)4M(OOH)]42~ -656 — — [409]
комплекса [HRh(SnC13)s]3 (рис. 2.29). В комплексе два типа атомов
олова: один атом в /прайс-положен и и к протону, четыре — в плоскости,
Рис. 2.29. Строение комплекса [НКЩБпОз^]3 (а) и схема его спектра ЯМР
ll9Sn (б). Высота штрихов соответствует интенсивности компонент
перпендикулярной к координате H-Rh-Sn1. Взаимодействие между яд-
рами 1Н, 103Rh, 117Sn и 119Sn дает сложный спектр, из которого опреде-
лены КССВ: ^(H-Rh) = 10; ^(Rh-Sn) (см. табл. 2.59); 2J(H-Snl) = 60;
2J(H-Sn2) = 1110; 2J(Sn‘-117Sn2, цис) = 1772; 2J(119Sn1-117Snl,цис) =
= 1992; 2J(Snl-ll7Snl, транс) = 25420.
Набор данных о КССВ ll9Sn с другими ядрами весьма пред-
ставителен. Наличие двух изотопов со спином 1/2 позволяют лег-
ко определять КССВ Sn-Sn. Здесь наблюдали самые большие КССВ
^(•^Sn-'^Sn) 21248 Гц в комплексе [Ки(ЗпС1з)5С1]4- [407]; некото-
рые константы ССВ приведены в табл. 2.57. Константы ССВ 'J(C-Sn)
зависят от гибридизации а-атома углерода и меняются от -1200 Гц для
sp-, —500 Гц для sp2- до —300 Гц для зр3-гибридизации [417]. КССВ
в соединениях Sn(II) не проявляются из-за лабильности комплексов;
величина КССВ для тетрагалогенидов олова оценена из ЯМ-релакса-
ции: 'Д'^Зп-С) = 470 Гц для SnCl4, *J(119Sn-Br) = 920 Гц для SnBr4,
1J(119Sn-I) = 940 Гц для Snl4 [420]. В [418] приведены КССВ через две
связи, которые могут превышать 10 кГц для 119Sn (табл. 2.57).
В ЯМ-релаксации тетрагалогенидных соединений олова преобла-
дает скалярный вклад, в других соединениях — спин-вращатель-
ный [396].
Sn(II) образует лабильные комплексы с галогенидами. В [419, 420]
изучены растворы фторидов и хлоридов олова и их тетрагалоидных
комплексов в разных растворителях (табл. 2.58).
В [421] исследованы концентрационные зависимости ХС олова вод-
ных растворов солей олова(П). В отличие от растворов хлорида и фто-
Таблица 2.57
Максимальные КССВ между двумя ядрами X-Y
X Y 2J, цис, Гц 2J, транс, Гц
,3С 13С 3,2 35
31 р 'Н -17,5 158
« |3С 9 104
« l5N 3 61
« 19F 32 140
« 31 р -8 +610
« 119Sn 214 4188
« '"Hg 397 3879
ll9Sn 'Н — 1740
<< 117Sn 2364 12862
рида олова, ХС в растворах сульфата олова не зависит от концентрации
соли 1 моль/л.
Комплексы олова(1У) инертны в масштабе времени ЯМР, имеется
большой массив данных ХС и КССВ Sn(IV). Данные ЯМР комплексов
с лигандом SnCl^ представлены в табл. 2.59.
Растворы гексагалогенидов олова(1У) изучены в [423, 424]
(табл. 2.60).
Замещение гидроксогрупп на пероксогруппы в [Sn(OH)e]2- и поли-
конденсацию комплексов изучали в [408].
Свинец. Хотя первые данные ЯМР свинца опубликованы еще
в 1957 г. [430], данных по ЯМР свинца гораздо меньше, чем по ЯМР
олова, вследствие меньшей чувствительности и более широких линий.
ХС у 207РЬ отсчитывают от РЬ(СНз)4. Как и у 119Sn, действие концен-
трации комплекса и растворителя на ХС невелико. Данные ЯМР 207РЬ
приведены в табл. 2.61.
Линейная корреляция между ХС свинца и олова (рис. 2.28) по-
казывает втрое большую, чем у олова, чувствительность ХС свинца
к изменению окружения. Чувствительность ХС свинца к замещению
лиганда по одной координате тетраэдрического комплекса показана
в табл. 2.61. Изменения ХС у свинца, как и у олова, определяются
парамагнитным вкладом. Данные по твердым образцам показывают,
что важную роль в экранировании 207РЬ играет координационное чис-
ло. Эффект Оверхаузера может успешно использоваться при изучении
свинцовоорганических соединений.
Таблица 2.58
Данные ЯМР растворов галогенидов олова [419, 420]
Соединение Растворитель <5119Sn, м. д. * 8 i9F, м. д.
SnF2 ДМСО -630 -86
ДМФА — -89
EtOH — -106
SnCl2 ДМСО -360 —
ДМФА -310 —
EtOH -270 —
SnF2:SnCl2 2 : 1 ДМСО -540 —
SnF2:SnCl2 1 : 1 ДМСО -500 —
SnF2:SnCl2 1 : 2 « -460 —
0,15 M Snl2/1 M SnMe4 ГМФА -172 [420]
0,04 M SnI2/0,2 M SnMe4 « -185 «
0,05 M SnI2/0,8 M SnMe4 ДМСО -631 «
0,03 M SnI2/0,4 M SnMe4 << -670 «
Bu2SnF4 ДМСО -240 -156
ДМФА -195 -159
EtOH — -163
Bu2SnCl4 ДМСО -210 —
ДМФА -145 —
EtOH -75 —
* 6119Sn от SnMe4, 6 i9F от CFCI3, КССВ Sn-F не проявлялись.
Константы ССВ у свинца по величине меньше, чем у олова [405]
(табл. 2.61), ССВ с углеродом зависит от гибридизации а-атома угле-
рода, и '7(РЬ-С) меняются от 1500 для sp-, 1100 для sp2- до 700 Гц
для зр3-гибридизации [417].
В продольной ЯМ-релаксации РЬСЦ [428] доминирует спин-враща-
тельный, а в поперечной — скалярный механизм релаксации. В кон-
центрированном водном растворе перхлората свинца заметен вклад
анизотропии ХС в Т\, в разбавленном растворе преобладает спин-
вращательный механизм [431, 432]. В органосвинцовых соединениях
Т, =0,1-1 с [426].
Таблица 2.59
Данные ЯМР комплексов с лигандом SnCl3
Комплекс 6, M. Д. J(X-ll9Sn) X J(ll9Sn-117Sn) Примечания, источник
[Ru(SnCl3)5Cl]4- -168 (э) 846 "Ru 21250, транс [407]
« -149 (a) — — 2938 (осн-осн) То же
« — — — 2581 (осн-верш) «
[Os(SnCl3)5Cl]4- 531,8 (э) 1290 1870s 18600, транс [407, 410]
<< 581,8 (a) — — 2169 (осн-осн) «
« — — — 1542 (осн-верш) «
[RhCl2(SnCl3)4]3’ -208 581 103Rh 2158 [413]
[RhCl3(SnCl3)3]3- -426 742 103Rh 2804 3 М НС1 в Н2О [413, 418]
« -411 — — 2817
[RhCl5(SnCl3)]3- -992 864 IO3Rh — НС1 в Н2О [412]
« -932 860 103Rh — То же
« -914 850 l03Rh — «
[RhCl4(SnCl3)2]3“ -654 796 103Rh — «
« -637 791 103Rh — «
« -626 780 103Rh — «
[RhCl(SnCl3)5]3- -100 547 103Rh 1952 «
<< -100 — — — —
[Rh(SnCl3)6]3- -36 — — —3264, цис ЗМ НС1 в Н2О [411]
<< — — — 12862, транс «
[Rh(SnCl3)5]- 8,5 806 103Rh ' 3634 [412]
[HRh(SnCl3)5]3- -1,2‘Э 592 103Rh — [413], ') Sn1,
« 912) 524 103Rh — 2) Sn2, рис. 29
[Pt(SnCl3)5]3~ -142 — — 6230 [418]
« -133 — — 6387 То же
[PtCl2(SnCl3)2]2- -387 — — 2485 «
[Pd(SnCl3)5]3- 69 — — 7550 [422]
Таблица 2.60
Данные ЯМР комплексов [SnHale]2
Комплекс <5, m. д. 'j(ll9Sn-F) Примечания
SnCl|“ -733 — CH2CI2 [423]
i^uc-SnCUF2- -686 2263 To же
apaH-SnC^Fj- -697 2056 «
ijuc-SnC^F^- -717 1821; 2132 <<
SnF*- -811 1579 «
ijuc-SnCUBr2- -220 — «
apaH-SnCl3Br2- -1344 — «
4uc-SnCl2Br2- -1571 — «
SnBfg- -2074 — «
SnCl5OH2- -675 — H2O [424]
ijuc-SnCl^OH)2- -643 — To же
pe6-SnCl3(OH)2- -620 — «
pe6-SnFCl2(OH)2" -622 2030 «
Таблица 2.61
Данные ЯМР свинца
Форма <5, m. д. J(X-Pb), Гц X Примечания, источник
PbMe4 0 250 l3C 85%, толуол [425]
PbEt4 73 — — Ч.жидк. [425]
Pb(NO3)2 -2961 — — 1 М в Н2О [426]
Pb(C104)2 -2950 — — 1 М в Н2О [427]
PbCl4 — 705 35C1 [428]
PbO2 4450 — — Тв. [427]
PbO желтая, массикот 3750 — — То же
PbO красная, глет -5850 — — «
PbS 750 — — Тв. [430]
PbSe 1050 — — То же
PbTe 50 — — «
Таблица 2.61 (окончание)
Форма <5, м. д. J(X-Pb), Гц X Примечания, источник
РЬС12 -4750 — — Тв. [427]
PbSO4 -4550 — — Тв. [427]
Pb 11150 — — Металл [430]
МезРЬ-РЬМез -281 -286 207РЬ 40%, С6Н6 [425]
МезРЬ-БпМез -324 -57 119Sn 30%, С6Н6 [425]
МезРЬ-SeMe 380 -1170 77Se 20%, С6Н6 [425]
Me3Pb-NMePh 227 261 15n То же
Me3Pb-PPh2 41 -1335 31 р 30%, С6Н6 [425]
Me3Pb-B(NMeCH2)3 -362 1330 "в 30%, ТГФ [425]
Халькогеноплюмбаты РЬгСИд изучены в [435] (табл. 2.62).
Таблица 2.62
Данные ЯМР халькогеноплюмбатов Pb2Chj“
Анион <5 207Pb, м. д. <577Se <5125Te J(Pb-Se), Гц J(Pb-Te)
Pb2S2- 3553 — — — —
Pb2S2Se2- 3497 -17 — 98 —
Pb2SSe2” 3421 -57 — 122 —
Pb2Se2“ 3302 -99 — 153 —
Pb2S2Te2“ 3261 — -589 — 818
Pb2SSeTe2“ 3108 -147 -640 159 868
Pb2STe2- 2671 — -762 — 946
PbSe2Te2- 2894 -190 -699 188 915
PbSeTe2- 2375 -280 -817 227 994
PbTe2- 1727 — -927 — 1074
Анионные кластеры Srig^Pb^ (0-9), SngTl5-, Sng-^Ge!].- (х =
= 1-9), Sn8-xPbxTl5- (х = 0-8) и другие, полученные растворением
металлических сплавов олова, свинца, германия с щелочными метал-
лами в жидком этилендиамине, изучены в [434] (табл. 2.63).
Таблица 2.63
Химические сдвиги 119Sn, м.д. и J(ll7Sn-ll9Sn), Гц (в скобках)
Кластер Катион: Na К Cs
Sn<- -1230(256) -1210(363) -1115(293)
SnePb4- -1272(262) -1251 (275) -1152(303)
Sn7Pb^ -1319(281) -1298(293) -1194(325)
SnePbj- -1366 (317) -1344(323) -1235 (369)
Sn5Pb}- -1420 (366) -1397(378) -1281 (418)
Sn4Pb^ -1480 (427) -1456 (455) -1333 (534)
Sn3Pbg- -1538(520) —1512 (—) -1381 (610)
Sn2Pb}“ —1579 (—) -1553 (-) -1414(753)
SnPbg- —1610(—) -1587 (-) -1438 (-)
Sn8Ge4-SnGe8~ -1223 (258) -1208 (268) —
Sn8TI5- —1237(—) —1214(—) —
5п2ВЦ~ -1169(412) -1137(456) —
-1674(1638) -1575(1638) —
2.6. Фосфор, мышьяк, сурьма, висмут
Разнообразие соединений Р, As, Sb, Bi, их широкое промышленное
и научное использование дают большие возможности для применения
метода ЯМР этих ядер. В отличие от азота, эти элементы выступают не
только как доноры, но и как акцепторы электронов. Координация ато-
мов этих элементов разнообразна. Они могут образовывать 3-связные
соединения пирамидальной конфигурации (РН3, AsCla, SbPha); 4-связ-
ные в виде тетраэдра (РН^-) или тригональной бипирамиды (БЬгРу),
5-связные в виде тригональной бипирамиды (PF5, SbCls) или квад-
ратной пирамиды (SbPhs) и 6-связные октаэдрические (PFjT, AsF^,
Sb(OH)g~). Все распространенные изотопы этих элементов имеют маг-
нитный момент, их свойства даны в табл. 2.64.
Все ядра этих элементов относятся к высокочувствительным в
ЯМР, однако наличие большого квадрупольного момента у ядер мы-
шьяка, сурьмы и висмута резко ограничивает возможности метода
ЯМР этих ядер, особенно при применении импульсных спектрометров.
Фосфор. Биологическое значение фосфора привело к развитию
мощной промышленности удобрений и средств защиты растений, а так-
же широкому развитию органической, биоорганической и бионеоргани-
ческой химии фосфора.
Таблица 2.64
Свойства ядер магнитных изотопов фосфора, мышьяка, сурьмы, висмута
Изо- топ Спин NA, % Sens 1 Диапа- зон ХС, м.д. Q, барн Частота ЯМР эталона =, МГц2 Эталон
31 р 1/2 100 125 700 0 40,480737 85% Н3РО4
75 As 3/2 100 47 800 0,3 17,180 AsF"/MeCN
— — — — — 17,1229 GaAs
121Sb 5/2 57 71 3600 -0,53 — SbCl6-
123Sb 7/2 43 37 3600 -0,68 — SbCl”
209Bi 9/2 100 250 3500 -0,4 16,017649 Me4NBiF“/Me2CO
1 Относительно '3С{‘Н}; 2 частота ЯМР в поле Но, в котором частота ЯМР 'Н
ровно 100 МГц.
Высокая чувствительность и специфичность ЯМР 31Р привели к его
разнообразному применению в химии, биологии и медицине, вплоть до
изучения метаболизма фосфора in vivo на высших животных и челове-
ке. Выпущен ряд монографий и обзоров по ЯМР 31Р и аспектам его
применения [190, 435-439].
Высокая реакционная способность соединений фосфора обычно не
позволяет использовать внутренний эталон в неорганических субстра-
тах, поэтому используют внешний — 85% Н3РО4. Обычно коррекцию
на магнитную восприимчивость делают редко, часто не указывают и
температуру измерения, поэтому различие с ранними измерениями ХС
достигает ±5 м.д. Кроме того, ранние работы выполнены в шкале ст, с
противоположными знаками ХС по сравнению с современной шкалой 6.
Поскольку ампулы в спектрометрах с электромагнитами устанавлива-
ются поперек поля Hq, а в датчиках высокого разрешения в криомагни-
тах вдоль поля, возникают различия в ХС, полученных на указанных
спектрометрах без учета поправки на восприимчивость на 5-7 м.д.
[440]. Даже для Н3РС>4(85%) разброс данных более 1 м.д. [435].
В зависимости от pH в растворе преобладает одна из протонирован-
ных форм фосфата: Н4РО4+, Н3РО4, НгРО^, НРО^-, РО4 (рис. 2.30)
[3]. Первая форма имеет наиболее узкую линию ЯМР.
Диапазон ХС 31Р для Р(Ш) 250 ч—460 м.д., для P(V) 100 4-
4—50м.д., что связано с наличием неподеленной пары электронов.
Чувствительность ХС Р(Ш) к окружению показана в табл. 2.65 [190].
ХС некоторых соединений фосфора приведены в табл. 2.66.
Экранирование фосфора обычно интерпретируют согласно [435,
441-443], с учетом углов связи, электроотрицательности заместителей
fl?3Na, с-1 <531P, м.д.
48 г
О 4 8 12 pH
Рис. 2.30. Зависимость химического сдвига ЯМР 31Р иона фосфата и ЯМ-ре-
лаксации 23Na от pH в фосфатных растворах
Таблица 2.65
Химические сдвиги ЯМР 31Р трехвалентного фосфора
Окружение Н С N О F Cl S
Соединение РН3 РМе3 P(NMe2)3 Р4О6 PF3 РС13 P(SMe)3
<5, м. д. -240 -62 121,5 ИЗ 97 220 125
Таблица 2.66
Данные ЯМР фосфора
Форма <5, м. д. J(P-X), Гц X Источник
Н3РО4 85 % 0 — —
Р4 -448 — — [435]
РН3 -238 — — [190]
рн+ -101 — — [439]
PF3 97 1410 19F [444]
РС13 219 115 35С1 То же
РВг3 227 350 81Вг
Р1з 178 — — [190]
POF3 -35,5 1060 19F
РОС13 5,4 — — [435]
РОВг3 1,9 — — То же
ОРС13 29 — — «
ОРВг3 -112 — — «
Таблица 2.66 (окончание)
Форма <5, м. д. J(P-X), Гц X Источник
N-PCI2-N РС12 РС12 N-PCh-N -6 ‘ — — [435]
Р —0—Р l\ °Z| 0 X 0 I/OJ Р —0—Р 16 — — «
Q- СО — о, | СО со | СО X 00 |/\| Сс СО CL, из — — «
(OC)3Fe(PPh3)3 -9,5 — — «
(OC)3Ni(PPh3) 47 — — «
р2о<- -6 — — [190]
[О3Р-РО3]2- 9 — — То же
[О3Р-Р*О2Н]- 7; 24 (Р*) 480 31 р [435]
— 94; 444 (Р*) 'н —
[НО2Р-О-РО2Н]2~ -5 660 н То же
[О3Р-О-Р*О2Н]3- -3 620 н «
—2 (Р‘) 17 31 р —
0 [О3Р-Р*-РО3]2“ 0 36,5 340 3! р «
12,2 (Р*) — — —
0 [О3Р-О-Р*О-РО3]6- 0 —4 18,7 31 р «
-18 (Р*) — — —
0 0 [03Р—0—Р*0—Р*0—РО3]7- 0 0 -5 — — [190]
-18 (Р‘) — — —
и -тг-характера связи с заместителем. Если в ряду соединений меняется
один из перечисленных параметров, наблюдается аддитивность ХС.
Изотопные эффекты ХС 16О-18О изучены в [445-447]; для тиофос-
фатов РОз53- величина составляет около 0,03 м.д. [446],
Из спектра изогипофосфат-иона (рис. 2.31) видно, что ХС атомов
30 0 30 м.д.
Рис. 2.31. Строение (а) и спектр ЯМР 31Р изогипофосфат-аниона (б)
Р1 и Р2 различаются всего на 1 м.д. (табл. 2.66), хотя в окружении Р1
атом О замещен на протон, однако по КССВ (Н-Р) атомы однозначно
определяются.
Имеются обширные данные о константах ССВ J(P-X) [190, 438,
447]. Они зависят от состояния окисления и больше по абсолют-
ной величине у P(V): *J(P-H) = 400-1100 Гц по сравнению с 200 Гц
для Р(Ш). Константы ССВ увеличиваются с ростом электроотрица-
тельности заместителя; для связей Р-С меняют знак в зависимо-
сти от состояния окисления фосфора: '7(РП1-С) = -25-0, 'J(PV-C) =
= 454-300 Гц; *J(P-F) = 800-1500 Гц и нечувствительны к состо-
янию фосфора. В фосфатах V(P-O) 80-160 Гц; там же изучены
изотопные эффекты 17О-18О в ХС и КССВ фосфора [445]. Кон-
станты *J(P-P) = -400-500 Гц [447], а в гидридных комплексах
платины(П) менее 20 Гц, в то время как 1 J(P-Pt) = 2000-4000 Гц,
i<uc-2J(P-119Sn) = 250-300 Гц, транс-2,7(P-"9Sn) = 1700 Гц [448].
Времена релаксации Т\ 31Р лежат в пределах 1-100 с [444]; осо-
бенно долгие времена наблюдают в растворах полифосфатов со второй
координационной сферой из слабомагнитных атомов. Долгие времена
Т\ позволяют определять термодинамические и геометрические пара-
метры молекул: энергии активации скорости обмена лигандов [444,
449-450], длину цепи полифосфата [451] и т. д.
Зависимости Т) 31Р и ядерного эффекта Оверхаузера от pH, тем-
пературы, концентрации и магнитного поля изучены в [449], где пока-
зано, что в тщательно очищенных от парамагнитных ионов растворах
Н3РО4 Ti не зависит от pH и основной вклад в 7] - диполь-диполь-
ный от !Н. Вследствие протонирования ХС фосфат-иона зависит от
pH, а ЯМ-релаксация 23Na этих растворов указывает на образование
ионных пар Н^РОд Na+ (рис. 2.30) [3].
Методом ЯМР 31Р исследовано взаимодействие теллуридов Nb, Мо,
W и Re с PCI5 как хлорирующим агентом [452].
ЯМР 31Р использован при исследовании водных растворов фосфа-
тов и полифосфатов [190, 435], комплексов фосфата и пирофосфата
с алюминием [261, 263, 495], фосфорных гетерополисоединений ва-
надия, вольфрама и молибдена [385, 437-460]. Фосфат может заме-
щать аммиак в гексааминном комплексе родия(Ш) [461]; в комплексе
[Rh(NH3)sPO4] фосфат образует с родием(Ш) и полиядерные катионы,
в которых он преимущественно играет мостиковую роль [462]. С се-
леном получено соединение P4Se8, в котором обнаруживается набор
изотопомеров вследствие того, что селен представлен несколькими
изотопами с одним магнитным, 77Se, с содержанием 7,6%. Соответ-
ственно, каждый изотопомер имеет свой спектр ЯМР данного соеди-
нения; суперпозиция этих спектров при природном содержании 77Se
проанализирована в [463].
Характер изменения ХС 31Р при поликонденсации фосфата схож
с характером изменения ХС 29Si при поликонденсации ЗЮд-. Поли-
конденсация фосфата алюминия также проходит через набор сосуще-
ствующих полиядерных комплексов [261] (рис. 2.32 [263]).
Рис. 2.32. Спектры ЯМР 2,А1 (а) и 31Р (б) в алюмофосфатных растворах:
РО4 : Al = 1, СД1 = 0,3 г-ат/л; 1,3 — без добавления NaOH; 2,4 — при
добавлении NaOH в отношении NaOH : Al = 1,5
Фосфат-ион образует различные гетерополианионы (ГПА) с воль-
фрамом, молибденом, ванадием [PWgC^]9-, [PWnOsg]7- (PWn),
[РМ12О40]3_-, [Р2М|8О62]6- (М = W, Mo), [P2W17O61]10- (P2W17),
[PVi4C>42]8_ (табл. 2.67) и другие; при этом в ГПА РМ[2 вольфрам
может замещаться на молибден, ванадий, ниобий и другие металлы
Таблица 2.67
Данные ЯМР некоторых фосфорных гетерополианионов со структурой Кеггина
Анион <5P, м. д. Анион <5P, м.д.
ро3- 5,7 [PO4WMonO36]3- -6,4
H-[PO4W12O36]3~ -15 [Р04Мо^МоУ‘036]5- -6,0
[PO4Wn VO36]4- -14,9 [ThIV(P2W17O61)2]16- -8,3-13,6
[PO4WnMoO36]3- -12,9 [U,v(P2W17O61)2]16- 10,3-10,1
[PO4WnTiO36]5- -13,7 a-[P2Moi8C>62]6 -2,95
[РО4Мо12О3б]3 -3,9 (0-[P2Moi8O62]6 -2,47; -1,95
почти целиком без изменения структуры ГПА [385]. ХС этих соеди-
нений лежат в пределах от 2 до -15 м.д. (рис. 2.33); при этом при
Рис. 2.33. Зависимость ХС ЯМР 31Р гетерополианионов РМ12-1Э1О40 от со-
става полиметаллатной сферы: / — М = W, Э = V; 2 — М = W, Э = Mo
замещении W на изоэлектронный и с близким по ионному радиусу Мо
наблюдается аддитивность ХС, в то время как при замещении W на V
такой аддитивности нет.
ЯМР 31Р «чувствует» поворот фрагмента ГПА, изменение распо-
ложения атомов ванадия в PV^W^-t О40 и [Р2М18О62]6- [385, 454,
455, 464], изменение числа замещающих атомов [454], протонирование
ГПА (рис. 2.34) [385, 455]; на рисунке видно, что с ростом заряда
сила кислот ослабевает, и если 4-5-зарядные ГПА не протонируются,
Рис. 2.34. Зависимость ХС ЯМР 31Р фосфорвольфрамванадиевых гетерополи-
анионов [PVnWi2-n0.io]('3+n)_ от pH раствора: 1 — п = 0, 2 — п=\, 3 —
п = 2, 4 — п = 3
то 6- и 7-зарядные ГПА протонируются уже при pH — 5. По ЯМР
31Р изучали образование смешанновалентных ГПА [385, 457, 465],
гидролиз ГПА в щелочах М(1), М(П) [466], комплексообразование
«дефектных» ГПА PWn, P2W17 с ионами металлов [385, 467-481].
Сопоставляя данные ЯМР 31Р и других ядер ГПА, удается полу-
чить информацию о структуре ГПА в растворе, установить наличие
ГПА близкого состава, что в последнем случае невозможно сделать
другими методами [385] (рис. 2.35). В водных растворах состав ГПА
Н3РО4
О
—2 —4 м.д.
Рис. 2.35. Спектр ЯМР 31Р раствора ГПК НбРУзМод040: / — линия ГПА
[PVMon04o]4-, 2 — группа линий ГПА [PVsMoiqC^o]5-, 3 — группа линий
ГПА [PV3Mo9O40]6-, 4 - линии ГПА [PV4M08O40]7- [443]
сохраняется до концентраций ГПК 0,01 моль/л, при дальнейшем раз-
бавлении ГПА диссоциируют [482]. Состояние ГПК в уксуснокислых
растворах изучали в [385], продукты термического разложения ГПК
a-H2P2WI8O62-nH2O - в [484].
ЯМР фосфора широко используется при исследовании комплексов
переходных металлов с содержащими фосфор лигандами Со(Ш) [461],
Rh(III) [461, 485], Мп(П) [486], Au(I) [487, 488], Pt(II) [489, 490],
Pd(II) [489, 491], UO^ [492], Nb(V) [493] и непереходных металлов
Sn(II), Pb(II) [494, 495]. Пятикоординированные комплексы Со(1) и
Ni(II) с тетрадентатным Р-лигандом Р(РРЬг)з (РРз, PPh=P', ^3-Р=Р2)
изучены в [496] (табл. 2.68).
Таблица 2.68
Данные ЯМР 5-координированных комплексов
Соединение T, К <5Pi, м.д. <5Рг, м.д.
СоВг(РРЬз) 200 49,6 171,9
CoI(PPh3) 200 52,7 184,4
Co(CN)(PPh3) 200 67,2 172,6
[NiBr(PPh3)]AsF6 240 34,0 151,9
[NiI(PPh3)]AsF6 240 38,2 167,1
[Ni(CN)(PPh3)]AsF6 240 47,8 155,2
Комплексообразование с определенными фосфорными лигандами
может быть использовано для количественного анализа на катионы
металлов методом ЯМР 31Р. Так, лиганд [(Me2N)2OP]2NMe может
использоваться для анализа на А1 на фоне Ga, In [178].
Мышьяк. Остальные элементы группы VA имеют большие квад-
рупольные моменты и, несмотря на сравнимую с 31Р чувствитель-
ность, используются в практике гораздо реже [568]. Для 75As пер-
вое систематическое изучение опубликовано только в 1977 г. [498,
499]. В соединениях As(III) линии очень широки — даже для AsH3
Т\ = 95 мкс при 25 °C [499, 500], и измеренные ХС относятся к As(V)
(табл. 2.69) [438, 497, 498, 501]. Эталоном служит ион AsF^, у ко-
торого J(As-F) = 930 Гц. Из данных ЯМ-релаксации J(As-H) = 92 Гц
у АэНз [500].
Эффекты замещения у As(V) в 2-3 раза больше, чем у 3IP(V).
ЯМ-релаксация 75As изучена лучше, чем эффекты экранирования [499,
500, 502, 503]. Константы квадрупольного взаимодействия в соедине-
ниях KAsFe, AsMe4Br, AsPh4Cl лежат в пределах 1,1-1,6 МГц [501].
В ацетонитрильном растворе [504] изучен комплекс [As(OTeF5)6]_,
575As —29м.д., ширина линии 165 Гц, 'J(F-Te) = 3408, 2J(As-Te) =
= 420 Гц.
Таблица 2.69
Данные ЯМР 75As (303 К) [498]
Форма <5, м. д. W, Гц 'j(As-X) X Примечание
AsF- 0 95 930 19f Раствор
AsH+ -291 115(0 °C) 555 *H «
AsH3 92 3600 — — «
AsMe^ 206 113 — — «
AsEt+ 249 194 — — «
AsPh+ 217 130 — — Водный раствор
AsO3- 370 ИЗО — —
GaAs* -3324 2300 — — 298 К, монокристалл
GaAs * -3326 2500 — — 298 К, поликристалл
* Данные автора.
Изменение ЯМ-релаксации 75As для разных тетраэдрических форм
мышьяка в водном растворе показано на рис. 2.36 в зависимости от
параметра rj/Т (т? — вязкость, Т — температура) [3].
VP, Гц
200
0
AsEt^Br
AsMeiBr
KAsF6
j___i____i______
1 2 3 £.ю5
T
Рис. 2.36. Зависимость ширины линии ЯМР 75As в растворах от отношения
вязкости rf к температуре Т, К
Сурьма. Соединения сурьмы исследованы методом ЯМР шире, чем
соединения мышьяка [439, 497]. Первые исследования по ЯМР 121 Sb
были сделаны ранее, чем на многих других ядрах, и именно в ЯМР
121 Sb (NaSbFe) было открыто ССВ [505]. Большой квадрупольный
момент обоих изотопов (см. табл. 2.64) делает измерение ЯМР сурьмы
трудным.
Из соображений чувствительности используют l21Sb, но более уз-
кие линии l23Sb могут дать определенные преимущества при измерени-
ях в высоких полях. Диапазон ХС сурьмы более 3500 м.д. (табл. 2.70).
Большая ширина линий затрудняет обнаружение комплексов сурьмы
с симметрией окружения ниже кубической методом ЯМР 121,123Sb.
Таблица 2.70
Данные ЯМР сурьмы
Форма <5, m. д. W, Гц Примечания, источник
SbCl6“ 0 300 MeCN [509]
SbBr- -2436 800 To же
SbBr5Cr -2005 1250 [507, 509]
SbBr4Cl^ -1590 1850 To же
SbBraCl^" -1180 250 «
транс-5ЬВг2С1^ -780 800 «
^uc-SbBr2Cir -705 500 «
SbBrCl5- -380 1100 «
AgSbF6 88 300 [509]
KSb(OH)6 296 1800 H2O [509]
Na2SbS4-9H2O 1032 550 To же
(SbMe)4I 780 — «
SbCl5 510 8000 Ч.жидк. [509]
Поскольку SbCls часто используют в качестве сильной льюи-
совской кислоты, было исследовано взаимодействие этого соедине-
ния с основаниями [506]. Известна только одна константа ССВ
*J(121Sb-F) = 1846 Гц в SbF(7, и предполагается, что она отрицатель-
на [507]. Попытка наблюдать ЯМР 121Sb в соединениях Sb(III) не
увенчалась успехом [509], вероятно из-за слишком широких линий.
Висмут. Висмут целиком состоит из магнитного изотопа 209В1,
ядро которого имеет большой квадрупольный момент. Последнее об-
стоятельство ограничивает применение ЯМР 209Bi только высоко сим-
метричными формами. После первого наблюдения [503] вышло всего
несколько работ [511-513], посвященных изучению иона гексафторвис-
мутата; КССВ (Bi-F) наблюдали даже в твердом KBiFg. В растворе
обнаружен обмен фтором [512, 513]. ЯМР расплава Bi-BiBf3 изучали
в [514].
2.7. Кислород, сера, селен, теллур
Эти элементы представлены в основном немагнитными изотопами,
причем наиболее важный в химии и жизни элемент — кислород —
имеет единственный магнитный изотоп с очень малым природным
содержанием. Свойства магнитных изотопов приведены в табл. 2.71.
Ядра 17О и 33S относятся к «трудным» ядрам: 17О — из-за малого
Таблица 2.71
Свойства ядер магнитных изотопов халькогенов
Изо- топ Спин NA, % Sens 1 Диапа- зон ХС, М.д. О, барн Частота ЯМР эталона Е, МГц2 Эталон
170 5/2 0,037 0,012 1500 -0,025 13,556 5103 Н2О
— — — — — 13,564269 Ацетон, 301 К
33S 3/2 0,76 0,032 600 -0,055 7,676 602 4 М (NH4)2SO4/H2O
77Se 1/2 7,6 1,0 1900 0 19,091 523 Me2Se
,25Те 1/2 7,0 4,2 3100 0 31,549 802 Ме2Те
1 Относительно 13С{'Н}; 2 частота ЯМР в поле Но, в котором частота ЯМР ‘Н
ровно 100 МГц; 3 расчет от Е ацетона.
природного содержания, 33S — из-за малого природного содержания
и широких линий ЯМР. Отрицательные гиромагнитные отношения 17О
и |25Те приводят к тому, что подавление ССВ с протонами уменьшает
сигнал ЯМР, и определенный выигрыш достигается только в ЯМР
|25Те за счет уменьшения времен релаксации и слияния мультиплетов.
Кислород. Большая часть неорганической химии является химией
соединений кислорода, поэтому числа работ с использованием ЯМР
17О превышает число работ по ЯМР 77Se, 33S и 125Те, вместе взя-
тых. Кислород образует связи со всеми элементами, кроме Не, Ne, и,
возможно, Аг; тип связи меняется от почти чисто ионной до почти
чисто ковалентной. Небольшой квадрупольный момент 17О во многих
случаях приводит к временам релаксации, удобным для накопления
сигнала в импульсных спектрометрах, и в поле 7 Т сигнал от образца
с концентрацией определенных атомов О более 0,2 г-ат/л на природ-
ном содержании изотопа может быть накоплен за 1-4 ч. Обогащение
изотопом 17О повышает чувствительность на 3 порядка; поскольку
обогащение идет через химические реакции данной формы с обога-
щенным реагентом, скорости обогащения разных форм различаются на
порядки, и обогащение субстрата с разными кислородными формами,
как правило, оказывается неравномерным, а соотношение интенсивно-
стей линий не отражает соотношения концентраций форм. Обогащение
определенных атомов О в реагенте позволяет применять метод ЯМР
17О для изучения маршрутов переноса кислорода в процессе реакции,
кинетики изотопного обмена с целью выяснения механизмов химиче-
ских реакций.
Данные по ЯМР 17О имеются в обзорах и монографиях [3, 190,
385, 515-521], диаграмма ХС приведена на рис. 2.37.
1500
£ о юоо
о500°оЬЧ’0 м-д-
> 2 й I °
М-<
Рис. 2.37. Диаграмма химических сдвигов ЯМР |7О
Не слишком узкие линии ЯМР |7О позволяют увеличивать объем
образца без влияния неоднородного уширения, а небольшие величины
Т\ — записывать линии ЯМР чистых органических соединений и на
стационарных спектрометрах, используя высокие уровни поля Hi [522].
Измерения успешны для маловязких жидкостей с небольшим молеку-
лярным весом. Снижение вязкости за счет нагревания и небольшого
разведения невязким растворителем существенно улучшает возможно-
сти регистрации спектров.
Большая ширина линии 1 кГц) многих органических соединений
снижает информативность метода, и только переход к сильным полям
делает метод ЯМР 17О эффективным для решения проблем строения
органических молекул и их состояния в растворах. На основании
измерений константы спин-вращательного взаимодействия в [523] рас-
считано абсолютное экранирование ядра кислорода в молекуле С|7О
(7абс = —42 ± 17 м.д. от НгО, и составлена абсолютная шкала экра-
нирования ядер кислорода (табл. 2.72). Можно отметить, что данные
расчетов совпадают с данными эксперимента в пределах погрешностей.
Органические молекулы сравнительно редко содержат более двух
неэквивалентных атомов О; примером спектра молекулы с тремя неэк-
вивалентными атомами О является представленный на рис. 2.38 спектр
ЯМР 17О моноацетата этиленгликоля [524]. С ростом числа атомов О
(т. е. и молекулярного веса) вращение молекул замедляется, что при-
водит к уширению линий и наложению их друг на друга. Диапазон
ХС |7О в неорганических молекулах и ионах примерно вдвое больше,
чем в органических (табл. 2.74, 2.75), а разнообразие связей в одном
соединении приводит к многокомпонентным спектрам (рис. 2.38) [385].
Таблица 2.72
Абсолютная шкала экранирования кислорода
Молекула Состояние 0”абс СТтеор
Н2О Газ 344 312
Жидкость 308 —
СО2 Газ 343 223
N2O То же 200 170
OCS « 108 —
СО << -42 ±83*
of2 Жидкость -473 -452
* Данные разных авторов [523].
400 200 0 м.д.
Рис. 2.38. Структура и спектры ЯМР 17О моноацетата этиленгликоля: а —
спектр; б — то же при синтезе с обогащенной по 17О водой; в — то же при
синтезе с обогащенным по |7О нитратом. Звездочки — неопознанные примеси
Для получения спектров ЯМР 17О при небольших концентрациях
исследуемого вещества проводят обогащение последнего изотопом 17О.
Обогащение изотопом |7О повышает чувствительность до 1000 раз;
поскольку обогащение идет через химические реакции данной формы
с обогащенным реагентом, скорости обогащения разных форм разли-
чаются на порядки и обогащение субстрата с разными кислородными
формами, как правило, оказывается неравномерным, а соотношение
интенсивностей линий не отражает соотношения концентраций форм.
Обогащение определенных атомов О в реагенте позволяет применять
метод ЯМР 17О для изучения маршрутов переноса кислорода в процес-
се реакции, кинетики изотопного обмена с целью выяснения механиз-
мов химических реакций. Основным обогащенным препаратом являет-
ся вода с обогащением до 90% |7О; обогащение вещества проводят,
растворяя его в обогащенной по |7О воде.
Примером неравномерного обогащения является обогащение аниона
РгМощО^ в котором атомы О «колец» не обогащаются (см. рис. 2.39
Рис. 2.39. Структура (/) и спектры ЯМР |7О раствора гетерополианиона
РгМщвО^- в обогащенной изотопом |7О воде (2), в природной воде (3)
и [458]); аналогично в анионах ХМощО®^ (рис. 2.40) атомы кислорода
в мостиках Х-О-М не обмениваются с водой [525]. Поэтому исполь-
зование обычного для других ядер соответствия интенсивности линии
и концентрации ядер в данном случае ограничивается препаратами
с природным содержанием 17О или равномерно обогащенными этим
изотопом.
В неорганических системах по ЯМР |7О можно следить за состоя-
нием воды и других растворителей, в состав которых входит кислород,
и ионов, содержащих кислород. Вследствие высокой концентрации
воды как растворителя ее сигнал легко регистрируется, по нему можно
получать важную для химии водных растворов информацию, даже если
другие компоненты, содержащие кислород, не регистрируются. Воду
используют в ЯМР 17О в качестве внешнего эталона. Линию воды ча-
сто используют и как внутренний эталон в спектрах водных растворов.
Следует помнить о многих причинах, которые смещают линию
воды. Значительный вклад водородной связи в ХС воды выражается
в отличии ХС водяного пара (—36 м.д.) от ХС воды [526] и темпера-
турной зависимости ХС воды (изменение на -9 м.д. от 25 до 215 °C),
зависимости ХС воды от растворителя (табл. 2.73, [517]). Изменение
водородной связи вносит вклад и в изотопный эффект при переходе от
1000
500
0 М.д.
Рис. 2.40. Структура (/) и спектры ЯМР |7О аниона СеМо^С^ в природной
по изотопному составу воде, г/0 = 40,7 МГц (2) и обогащенной по изотопу |7О
воде, г/о = 12,2 МГц (3)
Таблица 2.73
Химические сдвиги ЯМР 17О воды в разных растворителях
Растворитель <5, м. д. Растворитель <5, м.д.
Диоксан -18,3 Метанол -13,0
Ацетон -12,1 Этанол -5,9
Ацетонитрил -н,з Пиридин -8,6
Аммиак 9,0 Хлороформ -10,3
НгО к D2O (—3,1 м.д.). ХС воды зависит от растворенных ионов из-за
вклада гидратных сфер ионов и изменения водородной связи.
Моляльные вклады в ХС воды от щелочных ионов лежат в пределах
от -0,5 (Cs+) до -1 м.д. (Li+); от ионов галогенов от 1,3 (С1_) до 2,35
(I-). Особенно велики вклады парамагнитных ионов. Парамагнитные
сдвиги в координационной сфере иона и моляльные сдвиги редкозе-
мельных ионов приведены в табл. 2.74. Заметные сдвиги (до 10 м.д.)
вызывает изменение кислотности водных растворов.
Химические сдвиги некоторых кислородных форм приведены в
табл. 2.75.
Таблица 2.74
Парамагнитные сдвиги ЯМР 170 координированной воды
Ион <5Р, м. д. W, кГц т, к Источник Ион <5Р, м.д. Источник
Ni2+ 1660 51 244 [517] ТЬ3+ -3820* [201]
Fe2+ 2200 16 234 [517] Dy3+ -3000* То же
Со2+ 2020 — 263 [518] Но3+ -2550’ «
рг3+ 450* — — [517] Ег3+ -1550* «
Nd3+ 570* — — [517] Тт3+ -1087* «
Gd3+ -3860* — — [517] УЬ3+ -295* «
* Сдвиг на 1 г-ион, растворенный в 1 кг воды.
Таблица 2.75
Химические сдвиги некоторых кислородных форм
Форма <5, м. д. Примечания Форма <5, м.д. Примечания
vo4- 571 Н2О, pH И [530] Оз 1598 Конц. [517]
СгО“ 835 То же — 1032 Мост.
МоО4“ 540 « Н2О2 174 30% в Н2О,
wo4- 420 « so3 188 [517]
МпО“ 1219 « so2 513 Жидк. [530]
ТсО; 749 « SOC12 292 [530]
ReO4“ 569 « SO2C12 298 [517]
СЮ4- 290 « so2f2 148 То же
RuO4 1119 Н2О [517] H2SO4 140 «
OsO4 796 Ч. жидк. [530] SO2- 167 «
CrO2Cl2 1460 СС14 [530] so3 235 [516]
Сг2О2- 1125 То же, конц. S2O3 228 То же
345 Мост. [517] O2N-NO2 420 «
РОС13 216 Ч. жидк. [517] C^N-NO 855 [515]
ро3- 94 90 °C [517] o2n-nq 425 То же
NaaHAsO4 98 Нас. р-р [530] Cl-NO 915 «
Таблица 2.75 (окончание)
Форма <5, m. д. Примечания Форма <5, м. д. Примечания
СО7- 192 [530] H2O -36 Газ [523]
ВгО^“ 297 [517] СО2 64,5 Газ, 7 атм [523]
СЮ” 290 To же N2O 107 Газ, 3 атм [523]
С1О4- 414 « — 115 СНС13 [523]
HNO3 298 100% [530] ocs 200 Газ, 9 атм [523]
NaNO3 412 1 M/H2O osc 205 Жидк. [523]
no2_ 670 [517] co 350 Газ, 2 атм [523]
SeO; 204 To же Ni(CO)4 362 [516]
IO3- 206 « Мп2(СО)ю 355 То же
ю4- 250 ДМСО [531] Fe(CO)5 388 «
O2F2 647 [516] of2 781 Газ, 5 атм [523]
C1OF3 331 To же — 799 Жидк., 133 К [523]
C1O3F 302 << XeOF4 313 [517]
C1O2F 384 « Te(OH)6 — [517]
сю+ 784 « Te(OOH)6 — [534]
UO2+ 1118 ±12* [532, 533] Sn(OH)6 — [409]
NpO2+ 2802 ±99* [532, 533] Sn(OOH)6 — [409]
SiO4- 44 [535] Xe(OH)6 278 [517]
* Указан диапазон ХС в зависимости от растворителя.
ЯМР |7О оксоионов (NO^“, SO4“, РО4“ и др.) более чувствителен
к взаимодействию с другими компонентами раствора, чем ЯМР других
ядер этих ионов (рис. 2.41 [536, 538]). В спектрах на ядрах 14N и 17О
наблюдали координированный к родию нитрат и коодинированную
к родию воду (рис. 2.41). Линии ЯМР 14N — 7,5 -j—8,5 м.д. относятся
Рис. 2.41. Спектры ЯМР l4N (А) и |70 (В) раствора Rh(OH)3 в азотной
кислоте; в спектре l4N 3 типа координированного к родию NO^ (<5 ~ —7 м.д.)
и свободного NO3- (<5 = —17 м.д.); в спектре 17О линии координированного
нитрата (430 и 250 м.д.) и координированной к родию воды (—140 м.д.)
к координированному нитрату, линия -19 м.д. — к HNO3 при кис-
лотности 12 М; интенсивные линии ЯМР |7О 0 м.д. и 410 м.д. отно-
сятся к свободным воде и нитрату, а линии 440 и 250 м. д. относятся
к недонорным и донорным атомам координированного к родию нитрата,
линия —140 м.д. — к координированной к родию воде на координате
НгО-КИ-ОНг. Из спектра следует координация нитрат-иона к родию
одним, а не двумя атомами кислорода [538].
Данные ЯА4Р органических форм представлены в табл. 2.76, фос-
фатных форм и чувствительность ЯМР |7О к протонированию и поли-
конденсации фосфатов показана в табл. 2.77 [537].
В спин-спиновом взаимодействии 17О с другими ядрами доминирует
контактное взаимодействие, хотя можно выделить небольшой спин-
орбитальный вклад [517]. Константы ССВ Х-17О можно определить
только из спектров ЯМР кислорода, поскольку в спектре ядер X ком-
поненты ССВ неразличимы из-за малого содержания 17О. В образцах,
обогащенных по 17О до нескольких процентов, возможно определение
J(X-O) по спектру ЯМР на ядре X. Известны константы ССВ со
многими ядрами (табл. 2.78). Наблюдение КССВ осложнено квадру-
польным уширением линий ЯМР 17О и обменом соседнего ядра (часто
встречается в ОН-группах).
Основной механизм релаксации 17О — квадрупольный, однако из-за
разнообразия связей атомов О возможны и другие вклады в релак-
сацию. Времена релаксации |7О не превышают долей секунды, для
небольших молекул ~ 10 мс. Они резко уменьшаются с ростом моле-
Таблица 2.76
Химические сдвиги некоторых органических кислородных форм [521]
Форма 6, м. д. Форма S, M. Д.
МеОН -37 MeNO2 569
ЕЮН 6 MeC(O)NH2 303
гРгОН 38 (H2N)2CO 196*
Ме2О -53 Me2SO 20
Et2O 7 МегЗОг 163
Ме2СО 569 (EtO)4Si 9
НОАс 251 SiO«- 44 [591]
* Данные автора.
Таблица 2.77
Данные ЯМР |7О фосфатов
Форма 6, M. Д. 'J(P-O), Гц
O-P- P-O-P -PO2-
H3PO4 79 — — 74
h2po4- 90 — — 90
HPO4- 100 — — 92
P<V 110 — — 90
O3P-O-PO3 104 123 — 102
Триполифосфат 105 124 97 —
кулярного веса и вязкости растворителя. Измерения времен релакса-
ции 17О применяют для изучения подвижности кислородсодержащих
молекул, определения скоростей обмена таких молекул в сольватных
оболочках парамагнитных ионов [566]. Ширина линии воды резко
меняется при переходе от истинного раствора к коллоидному. Напри-
мер, по ширине линии ЯМР |7О воды можно наблюдать образование
частиц золя при поликонденсации акваиона А1(Ш) перед осаждением
гидроксида алюминия [267]. Влияние ионов Na+ К+ и органики на
Тг 17О воды, которое связывают с изменением ее структуры, изучено
в [539, 540].
Таблица 2.78
Константы спин-спинового взаимодействия 17О [517]
Форма X J(O-X), Гц Форма X J(O-X), Гц
Н2О 'Н 90 ±2 МпО4“ 55Мп 29 ±3
МеОН •н 85 ±5 МоО2- 95 Мо 40,5 ±0,8
F2O 19f 424 XeOF4 129Хе 692 ± 10
Н3РО4 31 р 106 сю4- 35С1 85,5
OPFCI3 31 р 201 vo3- 51V 62 ±3
OPF3 31 р 184 [J(F-P) = 31] СгО2- 53Сг 10 ±2
ю4- 1271 340 [515] NO3“ 14N 35 [193]
ReO^ 187Re 50 — — —
Областью широкого применения ЯМР 17О является определение
структурных параметров и типа структуры полиоксометаллатов V,
Mo, W, Nb, Та [385, 516]. Химические свойства полиоксометаллатов
(ПОМ) определяются структурой их анионов (ПА).
ЯМР 17О для ПОМ более универсален, чем ЯМР других атомов,
так как кислород — общий элемент всех ПОМ. В ПА имеются атомы О
разного структурного типа: концевые О=М и мостиковые с координа-
ционным числом до 6. Первые спектры ЯМР |7О ПА разных структур
показали, что линии ЯМР атомов О разного типа хорошо разрешены,
и вид спектра характеристичен для определенной структуры [520], что
ставит этот метод в ряд структурных.
По следующим причинам метод ЯМР 17О следует отнести к струк-
турным методам.
1. ХС однотипных в структуре атомов О, связанных с атомами
разных металлов, различаются [520].
2. Имеется связь ХС и длины (кратности) связи О-М (корреляция
б-do-M, рис. 2.42, 2.43) [541-545].
3. Число линий соответствует числу атомов О разного типа, а ин-
тенсивности линий — числу атомов данного типа [520, 546, 547].
4. Экранирование при росте координационного числа атома О уве-
личивается.
5. Линии мостиков М-О-М* лежат между линиями мостиков
М-О-М и М*-0-М* [517, 530, 546].
6. Линии концевых атомов О спектре ЯМР 17О и линий М в спек-
тре на ядрах металла расположены зеркально [385, 455, 543, 546-548].
Рис. 2.42. Корреляция ХС ЯМР |7О и кратчайшего расстояния кислород-
молибден для полиоксомолибдатов (заряд анионов опущен)
Рис. 2.43. Корреляция ХС ЯМР 17О и кратчайшего расстояния кислород-
металл для полиоксометаллатов
7. Ширина линии зависит от градиента электрического поля (ГЭП)
на ядре О, (ширина линий ЯМР W квадрупольных ядер: Иб/г про-
порциональна квадрату ГЭП), что позволяет дифференцировать линии
одного и того же ПА по симметричности окружения атома О.
Структура некоторых полиоксоанионов и их спектры показаны на
рис. 2.44-2.46, данные ЯМР — в табл. 2.79.
В ГПА Р2М05 (рис. 2.44) кольцо из 5 октаэдров MoOg скрепле-
но сверху и снизу тремя атомами тетраэдров РО4; четвертый атом
последних свободен, но в спектре линии атомов О тетраэдров не
разрешены [542].
В структуре ИПА MgO^ (рис. 2.45) все атомы металла эквива-
лентны; анион содержит 6 концевых атомов О, 12 мостиковых и один
внутренний атом О. В этой структуре редкий пример 6-координиро-
Таблица 2.79
Химические сдвиги ЯМР |70 некоторых полиоксометаллатов [385]
Анион Тип (число) атомов 0 Анион Тип (число) атомов 0
Конц. Мост. Внутр.1 Конц. Мост. Внутр.1
AsMoi2O3^ 938(12) 572,553 Не регистр.2 w6o2- 772 413 -81
РМо12О30- 936 583,550 78 vw5o?9- 1217; 731 562 -75
SiMol20<- 928 580,555 41 — 395 —
GeMoi20^ 944 574,558 Не регистр. 762 389 —
pw12o30- 770 429,410 68 (w5o18)42- — 430 -6
SiW12O^ 761 427,405 27 655,640 416 —
h2w12o!;0- 729 414,410 75 Y(W5Oi8)8- — 345; 323 17
VW,iVO30- 1282 557,558 502 677,655 — -35
752,743 417,407 — Th(W5Oie)3- — 358; 342 17
CoW1203~ 817 929,297 Не регистр. — 875,646 — -49
CoW12O«0- 773 671,521 Не регистр. U(W5Oie)3- 834 386 —
CeMoi2O3^ 842 335 288 A1(OH)6Mo602j- 836 377 17
CeMoi20®^ 897 212 2813 Co(OH)6Mo602- 807 376 17
UMoi20®2- 898 184 276 TeMo60®4- 825 383 180
Мо602^ 834 559 -27 iMo6o3; — 387 255
1 Мостики металл-гетероатом; 2 не регистрируются из-за большой ширины линии; 3 +8Н+.
Рис. 2.44. Спектр ЯМР 170 на частоте 40,7 МГц ГПА [Р2М0 5O23]6 в вод-
ном растворе и его строение; ожидаемое соотношение интенсивностей линий
А : В : (С + О) = 10:5:8 соответствует наблюдаемому
Рис. 2.45. Производные структуры MgOig: а, б — схема строения аниона
МеО^ в виде атомов и октаэдров; в — структура WiqOj^; г — структура
LnCWsOis);!-, Ln — лантаноид
ванного кислорода, экранирование которого превышает экранирование
кислорода в молекуле воды. При замещении одного атома М иным
металлом (ИПА M*M5Oig) спектр резко изменяется: появляются 2
линии атомов О=М в соотношении 1 : 5, линия мостиков М-О-М*
(4 атома), близкие линии мостиков М-О-М, связывающих кольцо,
и таких же мостиков, связывающих кольцо с атомом М, противополож-
ным замещенному (4 + 4 атома). Спектр ЯМР 17О декавольфрамата по
количеству линий совпадает со спектром M*M5Oig, а неэквивалентных
атомов W всего два (рис. 2.46).
ГПА [ZlvMoi2O42]8- (структура Декстера-Сильвертона) имеют 24
концевых и 18 мостиковых (6 наружных и 12 внутренних) атомов
О; внутренние атомы образуют додекаэдрическую полость, координи-
A
2
С
_ В
Йе
--У /
3
Рис. 2.46. Структура декавольфрамата WioOj2 (/) и его спектры ЯМР 183W
(2) и 17О (3) [545]
рующую атом ZIV (Се, Th, U). Спектр ЯМР 17О устанавливает три
типа атомов О в анионе СеМе^О^" с соотношением интенсивностей
4:3:1 (рис. 2.47); сопоставление с корреляцией «ХС-длина связи»
Рис. 2.47. Структура аниона [UMO12O42]8 и зависимость ХС ЯМР 17О этого
аниона от добавления к раствору Sc3+
(см. рис. 2.42) показывает высокую однородность связей О-Mo; раз-
брос длин связи М=О менее 0,3 пм, М-О-М — менее 0,1 пм, в то
время как структурные данные кристалла дают разброс этих величин
соответственно 0,7 (±0,5) и 0,8 (±0,2) пм, что является эффектом
упаковки [525, 549].
Концевые атомы этих ГПА образуют тридентатный лиганд, и в рас-
творе ГПА может образовывать лабильные комплексы с редкоземель-
ными элементами; добавление в раствор СеМо^О3^ катионов Sc3+,
La3+ меняет положение линий как концевых, так и мостиковых ато-
мов О (рис. 2.47) [536]. Из-за высокого заряда эти анионы в кислых
растворах протонированы, и добавление основания к раствору так же
смещает линии атомов кислорода каркаса (рис. 2.48). В то же время ли-
Рис. 2.48. Зависимость ХС ЯМР |7О аниона [СеМо^С^]8 от добавления
к раствору основания
нии «внутренних атомов» Се-ОМо почти не меняют своего положения
при добавлении катиона или щелочи, и не обмениваются кислородом
с водой изотопом (рис. 2.48) [525].
Спектр ЯМР 17О из 4-х линий с соотношением 3 : 3 : 3 : 1 является
паспортом структуры Кеггина (рис. 2.49). Линия атомов центрального
тетраэдра ХО4 может не наблюдаться, если гетероатом имеет квад-
рупольное ядро с содержанием 100% (As, Al, Ga) и линия ЯМР
кислорода уширена из-за релаксации по скалярному механизму.
В спектрах ЯМР 17О при растворении в обогащенной изотопом
17О воде атомы ГПА обогащаются изотопом, однако скорости обмена
резко различаются [546]; в частности, практически не обмениваются
атомы центрального тетраэдра О4Х, и некоторые другие атомы каркаса.
Эти мостики наблюдаются только при природном содержании изотопа,
Рис. 2.49. Структура аниона Кеггина ХО4М|2О36
(см. спектры ГПА [РгМо^Обг]6-, производного от аниона Кеггина,
рис. 2.39).
«Зеркальность» спектров ЯМР 17О и ЯМР 51V, 183W, а также ин-
дикация неэквивалентных ядер металла по линиям ЯМР 17О концевых
атомов О показана на рис. 2.50 [385]. Заметим, что в обычных полях
линии концевых атомов О у неэквивалентных атомов металла часто
не разрешаются, и это правило проявляется в спектрах, измеренных
в полях Но 5 Т; однако и регистрация спектров ЯМР на природ-
ном содержании 17О для полиоксометаллатов, т. е. при концентрации
С 1 г-ат/л, возможна только в высоких полях.
Корреляция «ХС —длина связи» позволяет оценивать длину связи
М-0 и выявлять особенности структуры, связанные с изменением рас-
стояний меньше пикаметра. Совпадение ХС 17О для анионов РМо^О3^
и AsMo^O3^, РгМо^О^ и AsgMoigO^ [550] показывает их тожде-
ственное строение в растворах, доказывает синтез мышьяковых ана-
логов фосфорномолибденовых гетерополианионов и позволяет ожидать
у мышьяковых аналогов структурных параметров, аналогичных пара-
метрам фосфорных анионов.
Шкала ХС ЯМР 17О определяется изменением парамагнитного
вклада в экранирование, хотя и изменение диамагнитного вклада в раз-
ных соединениях достигает 50 м.д. [517]. В связи с определяющим
значением парамагнитного вклада наблюдают корреляции 6-Д“1 [530],
<5 —порядок тг-связывания [551], <5 —атомный заряд [552]. Для связи
О-М в полиоксометаллатах, в которых металл находится в (^-элек-
тронной конфигурации, порядок связи соответствует расстоянию О-М,
и сокращение связи соответствует увеличению ковалентности, которое
сопровождается уменьшением расстояния между основным и возбуж-
денным уровнями молекулы [519, 520]. При одинаковой длине связи
ХС в слабое поле (дезэкранирование) уменьшается в ряду металлов
a
Рис. 2.50. «Зеркальность» спектров ЯМР концевых атомов кислорода и сосед-
них атомов металла для анионов [РУцС^г]9- (а) и a-^WiaCfe]6- (б)
V > Mo > W (см. рис. 2.43), что соответствует изменению положения
возбужденных уровней. В сильно асимметричных мостиках М-О-М
ХС определяется расстоянием до ближайшего атома М, с которым
образуется более ковалентная связь и который, давая максимальный
вклад в разрыхляющие орбитали, определяет положение низколежа-
щих возбужденных уровней. Характеристика связи в асимметричных
мостиках с помощью среднего расстояния приводит к заниженной
оценке степени ковалентности связи [544]. Асимметричные мостики
характерны для ПОМ Mo(VI) и V(V).
Сходное изменение д-связывания в изоструктурных органических
соединениях с разными гетероатомами приводит к корреляции ХС ядер
этих атомов в рядах изоструктурных соединений, например к кор-
реляции ХС ЯМР 13С и 17О в соединениях R1-CH2-R2 и R1-O-R2
[553], корреляции между ХС азота и 17О в соединениях с NO-rpyn-
пой [195].
Из табл. 2.80 видно, что характерные ХС концевых атомов О=М в
ГПА с одним концевым атомом О для О=Мо 928-944 м.д., для O=W
729-772, для O=V 1217-1280 м. д.; для ГПА с двумя концевыми
Таблица 2.80
Изменение ХС карбонильного атома С ацетона в зависимости от растворителя
(<5 МегСО ацетона 205 м.д.)
Растворитель Д<5, м. д. Растворитель Д(5, м.д.
Ацетон 0 Хлороформ 2,3
Циклогексан -2,4 Метанол 3,7
Бензол -0,8 Уксусная кислота 6,2
Этанол —2 Муравьиная кислота 0,1
СС14 -1,3 Трифторуксусная кислота 14,1
1,4-Диоксан 0 Серная кислота 39
атомами ХС О=Мо 834-898 м.д. ХС мостиков Мо-Мо 583-553 м.д.,
W-0-W 429-410, V-O-W 558-562 м.д. В ГПА с кобальтом Со(П)
и Со(Ш) парамагнитны (в последнем случае наблюдают редкое тетра-
эдрическое состояние Со(Ш)), и в ХС 17О для мостиков и в меньшей
мере для концевых атомов наблюдаем парамагнитный вклад [554]
(рис. 2.51). В вольфрамванадиевом ГПА ванадий находится в тетраэдре
Рис. 2.51. Спектр ЯМР 17О парамагнитного ГПА [C0W12O40]6 : 1 — 293 К;
2 - 333 К
как центральный атом и в октаэдре как атом внешней металлатной обо-
лочки, что отражается в двух линиях ЯМР 51V с ХС —565 и —523 м.д.
соответственно [555].
Кроме приведенных выше структур существует и другие типы
структур [385] (VioO^-, ViO^, М7О®7 (М = Mo, W), a-MosO^,
/З-МовО^ , H2W12O42 , NiMogOj2 ); ГПА, производные от структуры
Кеггина (X2Mi8O^, М = Mo, W, PWi4O^, P2W2iO7i(Н2О)^,
HsgBsWssO^), дефектные ГПА Кеггина XWnO^, X2W|70g°-,
XWgO^J, и их комплексы с различными катионами типа MXWnO^,
/(XWnOgg)^, MX2WI7O"-, Z(X2W17O6I)|-,- M4(OH)n(XW9O34)r.
M4(OH)2(P2WisOse)^6-• Поворот фрагмента (поворотная изомерия)
приводит к новому ГПА, атомы W могут замещаться на Mo, V
почти в произвольном соотношении. Кроме того, многие ПОМ
можно восстановить на несколько электронов — образуются так
называемые «сини» молибденовых ПОМ и «коричневые» вольфрамовых
ПОМ — смешанновалентные гетерополианионы. В восстановленных
на четное количество ГПА неспаренные электроны делокализованы
или локализованы таким образом, что их магнитные моменты ском-
пенсированы и не препятствуют наблюдению спектров ЯМР, а сами
ГПА диамагнитны. По особенностям спектров ЯМР 17О и других ядер
аниона судят о локализации неспаренных электронов [456, 465, 556].
Изотопный обмен является мощным методом изучения состояния
и стабильности реагентов, кинетики и механизмов химических реак-
ций. Применение ЯМР 17О для исследования изотопного кислородного
обмена имеет те преимущества, что можно раздельно наблюдать за
кинетикой обмена неэквивалентных атомов кислорода в реагентах.
Исследование изотопного обмена кислородом в водных растворах дека-
ванадата ViqO|^ и фосфорванадийвольфрамовых и молибденовых ге-
терополикислот [547, 548] позволило определить скорости изотопного
обмена различных атомов О в структуре, выяснить влияние замещения
атомами V атомов Мо и W в анионе на его стабильность, предложить
механизм диссоциации гетерополианионов. Число существующих форм
ПОМ превышает 200, и значение ЯМР 17О для их идентификации
и изучения свойств ПОМ трудно переоценить.
ЯМР 17О используют при изучении карбонилов металлов, хотя
изменение ХС в зависимости от металла не так велико, как для 13С
(см. табл. 2.16). В кетонах ХС радикала СО зависит от растворителя
в пределах ±3 м.д., но в кислотах протонирование вызывает сдвиг на
порядок больше (табл. 2.79).
Зависимость ХС атомов ацетона от его доли в водных растворах
приведена в табл. 2.81.
Обмен между изомерами бисульфит-иона, SO3H", 6= 195 м.д. и
HSO^“, 6 = 175 м.д. и водой, а также обмен с водой дисульфит-иона
S20g— изучен в [557].
В [558, 559] изучены замещенные серой молибдаты и вольфраматы
(табл. 2.82).
Таблица 2.81
Данные ЯМР 13С, |70 ацетона в водных растворах в зависимости от концен-
трации
[H2O] [Me2CO] <5|3С Me2CO, м.д. <517O Me2CO
0 13,64 206 527,7
0,54 13,50 206,5 570,7
1,13 13,36 206,7 569,6
2,24 13,09 207,2 567,3
3,89 12,68 207,9 563,6
7,86 11,71 209,5 555,3
13,02 10,44 210,8 548,2
22,15 8,20 213,1 539,2
33,33 5,45 215,1 534,0
Таблица 2.82
Данные ЯМР [MOnS4-n]2-, М = Mo, W
Анион <5|7О, м.д. <533S (I¥, Гц) <595 Mo <5'83W
WS4 — 159(13) — 3769
WS3O 579 57(140) — 2760
WS2O2 532 -56(220) — 1787
WSO3 482 -186(300) — 841
WO4 422 — — 0
MoS4 — 345(38) 2258
M0S3O 699 240(170) 1654
M0S2O2 633 123 (250) 1066
M0SO3 585 -25 (200) 497
Mo04 525 — 0
H2O 0 —
5 M Cs2SO4/H2O — 0
Из данных табл. 2.82 следуют линейные корреляции <533S— 1/Д,
<533S(Wn)-<533S(Mon), <517O(Wn)-<517O(Mon), <5Wn-<5Mon, <533S(Wn)-
<517O(Wn), <533S(Mon)-<517O(Mon).
Расчеты ab initio ХС 170 гидроксилов к кремнию и алюминию
сделаны в [560]; из данных расчетов (<5Si(OH)4 12-16 м.д., <5-S(OH)3
10-17, <5=Si-ОН 14-27, <5Al2O(OH)g“ — Зм.д.) следует, что наблю-
дение гидроксилов в спектрах ЯМР 17О водных растворов на фоне
мощной линии Н2О затруднительно.
Синтезом в обогащенной Н217О воде и перёосаждением в обычной
удалось получить раствор обогащенного по 17О катиона [Ali3(ОН)32]7+
[561], и получить спектр ЯМР 17О, состоящий из двух линий 55 м.д.
(А1О4) и 32 м.д. (октаэдры А1О6); структура катиона — на рис. 2.19.
В инертных диамагнитных комплексах с переходными металлами,
так же, как и для N-лигандов, наблюдают значительный сдвиг линий
донорных атомов кислорода лиганда в сильное поле, и ХС донорного
атома О определяется координатой, к которой принадлежит лиганд.
Наиболее отчетливо это выражено для металлов VIII группы, и больше
всего данных известно для лиганда Н2О. Так, координированная вода
в комплексе Co(NH3)sH2O имеет ХС —130 м.д. [515]; в комплексах
Rh(IIl) ХС Н2О на координате H2O-Rh-NO2 6 — -62 4- -72 м. д., Н2О-
Rh-CI 6 — -99, H2O-Rh-H2O 6 — -141 [193]. Z/uc-влияние наиболее
выражено в комплексах с нитрит-ионом в транс-положении. Линия
ЯМР 17О координированного NO^“ (<5) сдвинута от свободного нитрата
(412 м.д.) на 165 м. д. (рис. 2.41 [538]).
Координированный к Мо пероксид не обменивается кисло-
родом с концевым атомом О, а так же с водой: в растворе
[0=Mo(CN)4(02)]{N(PPh3)2] в СН2С12 при 273 К наблюдали две
линии с 6 705 (W = 1800 Гц, О=Мо) и 6 487 м.д. (W = 80 Гц,
О2-Мо) [562].
Изучение водного обмена координационной воды (Н2ОС) трехъ-
ядерного комплекса родия [Rh3(p3-O)(p-O2CCH3)6(OH2)3]+ [563]; ХС
кислорода в комплексе: р3-0 — 6 = 9, в карбоксильной группе <5=131,
Н2О — 6 — -58 м.д. В тетраядерном комплексе [Zr4(OH)a(H2O)i6]8+
наблюдали одну широкую линию 6 = 180 м.д. с интенсивностью
[О] : [Zr] = 2 [43]; авторы полагают, что эта линия относится к одному
из типов Н2ОС (с медленным обменом), однако положение линии про-
тиворечит такому отнесению. Механизмы обмена Н2ОС, 6 — -132 м.д.,
в комплексе [Pd(OH2)4]2+ изучены в [525, 536]; измерены скорости
и энергетические параметры обмена.
В растворе комплекса [TcO2F4]~ (раствор NMe4 соли в MeCN)
обнаружена линия 17О с 6 = 1204 м.д. на координате O-Tc-F с нераз-
решенной СТС и *J("Tc-O) ~26 Гц и линия примесного комплекса
[TcO2F3] с д = 1236 м.д. и такой же константой ССВ [565].
Сера. Единственный магнитный изотоп серы 33S имеет природное
содержание на порядок больше, чем содержание 17О, однако из-за низ-
Таблица 2.83
Данные ЯМР серы-33
Форма <5, m. д. W, Гц Источник Примечание
RbgSO4 0 — [570] 4 M в Н2О
cs2 -332 170 [567] —
H2S -503 1700 [567] Н2О
MeSH 131 — [568] —
Me2S 86 — [568] —
Me2SO -404 — [567] Ч. жидк.
M.62SO2 193 3 [569] Ч. жидк.
C12SO2 -45 50 [569] Ч. жидк.
f2so2 -327 — [568] —
osc -594 — To же —
Сульфолан 38 2600 [568] —
Et2S 330 — [569] —
s8 -208 — [569] —
SF6 -177 — [568] —
SO2 375 — To же —
SOC12 224 — « —
HFSO3 516 — « —
K2MoS4 33 60 « —
(NH4)2S2O3 34,7 90 * Н2О, 295 К
Na2S -583 2700 [575] 1 М NaOH/H2O
SO2(NH2)2 -15 260 [574] D2O
S4N4O2 -35 1400 [574] ТГФ
H2SO4 —9; 108 500; 2300 [567, 575] 5 М в Н2О конц.
o2sf2 -41 — [575] -50 °C
ZnS -563 ^65 [575] Тв., сфалерит
* Данные автора.
кого гиромагнитного отношения чувствительность ЯМР 33S всего в 1,5
раза выше, чем ЯМР 17О. Большой квадрупольный момент приводит
к широким линиям ЯМР 33S, из-за чего специфичность метода ЯМР
серы невысока. Низкая частота ограничивает использование ЯМР серы
в обычных полях. Возможности метода существенно возрастают в по-
лях 7 Т.
Измеренные ХС (табл. 2.83) относятся к малым и симметричным
молекулам. Довольно симметричное окружение серы позволило изме-
рить ХС ряда сульфонов [569, 572]. В ионе тиосульфата два неэкви-
валентных атома серы, зарегистрирована более узкая линия 35 м.д.
центрального атома S. Съемки в высоких полях и при повышенных
температурах уточнят данные, приведенные в табл. 2.82.
Используя данные измерений константы спин-вращательного взаи-
модействия для молекулы OCS (которая для линейных молекул связана
с парамагнитным вкладом в экранирование), рассчитанный для сво-
бодного атома серы диамагнитный вклад и данные ЯМР на частоте
27,7 МГц, авторы [568] построили абсолютную шкалу экранирования
серы. Величины 5 некоторых соединений приведены в табл. 2.84.
Таблица 2.84
Величины частот S некоторых соединений серы
Соединение 5, МГц Условия
(NH4)2SO4 7,676602 4 M, Н2О
CS2 7,673457 —
SO2 7,688 850 —
SF6 7,674646 —
H2S 7,672 141 —
Известна только одна константа ССВ ^(S-F) = 251 Гц для SF6
[573].
Из-за ограничений по ширине линии и чувствительности метод
ЯМР 33S вряд ли можно широко использовать в химии неорганических
соединений серы. Определенную информацию можно получить из ЯМР
33S молекул со сравнительно узкими линиями (SO^, MoS^,
SF6). Однако другие ядра этих форм обычно имеют более узкие линии
при такой же или большей чувствительности ЯМР (17О, 95Мо, 19F),
что делает применение ЯМР последних ядер предпочтительным.
Обнаружены линейные корреляции <533S(W„)-<517O(W„),
<533S(Mon)-<5170(Моп) при замещении кислорода на серу в МО4
(М = Mo, W, табл. 2.81 [558, 559]).
Влияние противоиона и pH на ХС 33S сульфата мало, но комплек-
сообразование с Mg2+, А13+ вызывает заметное уширение линий [576].
Полиядерные азотносерные соединения S4N4, S4N4O2, (5МОС1)з и
другие изучены в [574].
Селен. Среди элементов группы кислорода селен имеет благо-
приятный с точки зрения ЯМР магнитный изотоп — 77Se. Первые
измерения ЯМР селена относятся к 50-м г. г. [577]. В связи с развитием
химии селеноорганических соединений большинство работ по ЯМР
селена лежит в этой области. Многие данные ЯМР селена получены
методом двойного резонанса, однако для неорганических соединений
преобладают прямые измерения спектров.
В качестве внешнего эталона использовали водный раствор селени-
стой кислоты Н2ВеОз [577], SeOCl2 [578], диметилселен [579, 580], ко-
торый наиболее употребителен. Заметим, что диалкилселенам присущ
температурный коэффициент ХС (~0,4 м. д./К); другие соединения
вряд ли составляют исключение. Наиболее представительный набор
данных по ЯМР селена приведен в [515], ХС некоторых соединений
и ионов приведен в табл. 2.85.
Полагают, что изменение ХС 77Se определяется парамагнитным
вкладом в экранирование. Наибольшая величина этого вклада (самое
низкое экранирование) оказывается в соединениях с неподеленной па-
рой электронов и электроотрицательными атомами [515].
Примеров изучения растворов неорганических соединений селена
немного; в [581] изучена зависимость ХС селена от pH в растворах
селенистой кислоты; с ростом pH от —1 до 13 наблюдали посте-
пенное превращение форм НзЗеО^- (-1257 м.д.) —> H2SeO3 (-1282) —»
—> HSeO^ (-1303) —> SeOj“ (-1256 м.д.). Такой ход отличается от
монотонного хода ХС 31Р при депротонировании фосфорной кис-
лоты: Н4РО+ (0) Н3РО4 (0) Н2РО4" (0,5) НРО2“ (3) РО3"
(5,5 м.д.) [435]. Поэтому при приготовлении эталонных растворов на
основе Н25еОз надо выбирать значения pH ~5, при котором ХС от pH
меняется мало и в растворе преобладает анион HSeO^ [581]. Там же
установлена корреляция между силовой константой ст-связи O-Se и ХС
77Se. В органических соединениях существует линейная корреляция
между ХС 77Se и 125Те.
Почти все КССВ 77Se получены при регистрации ЯМР ядра X или
двойного резонанса X{Se}; в последнем случае устанавливается знак
константы. Поскольку гиромагнитное отношение 77Se положительно,
ПК и n J имеют одинаковый знак. Наибольшее число данных относится
к КССВ Se-P; знак J(P-Se) всегда отрицателен, причем формаль-
но одинарная связь соответствует величине 'J(P-Se) = -200-500 Гц,
а формально двойная связь 'J(P-Se) = 500-1000 Гц. Это позволя-
Таблица 2.85
Химические сдвиги ЯМР селена-77
Форма <5, м. д. Источник Примечания
H2Se -226 [578] Ч. жидк.
Me2Se 0 [579] ’ Ч. жидк.
Et2Se 233 [579] Ч. жидк.
Ph2Se 402 [579] Ч. жидк.
(CF3)2Se 694 [578] Ч. жидк.
(PF3)2Se 701 [578] Ч. жидк.
Me2SeO 812 [579] Н2О
Me2SeCl2 448 [579] СН2С12
MeSeCl3 890 To же СН2С12
^uc-(Me2Se)2PtCl2 120 « CH2C12
mpaHC-(Me2Se)2PtCI2 135 « СН2С12
Sn(SeMe)4 -127 [582] Ч. жидк.
(SiH3)2Se -666 [583] CH2C12
(GeH3)2Se -612 [583] СН2С12
H2SeO3 1282 [579] Н2О
Na2SeO3 1253 [577] Н2О, нас. р-р
Кз2еО4 1024 [577] Н2О, нас. р-р
SeO3 944 [577] Н2О, нас. р-р
HSeO3F 1001 [578] SO2
HSeO3Cl 1003 To же SO2
SeOF2 1378 « Ч. жидк.
SeOFCl 1478 « Ч. жидк.
SeOCl2 1479 « Ч. жидк.
SeOBr2 1559 « Ч. жидк.
SeF4 1092 « Ч. жидк.
SeCU 1154 « ДМФ, нас. р-р
H2SeCl6 1451 « Н2О
SeF6 610 « Ч. жидк.
ет делать некоторые структурные отнесения [584, 585]. Константы
ССВ с оловом равны 1000-1500 Гц и положительны [582], в то
время как *J(Pb-Se) = -1170 Гц в MePbSeMe [405]. В MeSeTeMe
!J(Te-Se) = —169 Гц (7(125Те) < 0) [586]. Константа ССВ с 195Pt за-
висит от координации платины: в PtX3SeMe2 'J(Pt-Se) = 670; 507
и 234 Гц, а в PtX5SeMe2 'J(Pt-Se) = 259; 101 и -68 Гц (X = С1,
Вг и I, соответственно) [587]. Величины ’J(Se-H) лежат в пределах
40-70 Гц, ^(C-Se) — в пределах -10 ч—70 Гц [515], 'J(F-Se) —
1000-1500 Гц [588, 589]. В [590] наблюдали КССВ S-Se = 47-60 Гц.
Селен образует циклические соединения различной ядерности, в ко-
торых селен может быть замещен на серу или теллур. В [591] изу-
чены катионы S^-Se2^.- В зависимости от числа и расположения за-
местителей, ХС 77Se был у S3Se2+ 1933 м.д., цис-525е2+ — 2002,
mpaHc-S2Se2 — 1869 м.д.
Иодидные катионы Se^^4" (SeA (<5 = 979 м.д.) : SeB (6 —
= 1554 м.д.) = 1:1), J(AB) = 235 Гц, J(AB') = 54 Гц и Se6I2+
(SeC (5 = 1312 м.д.) : SeD (5 = 484 м.д.) = 2:1), J(C-D) = 275 Гц,
J(C-D') = НО Гц обнаружены в растворах SO2 при —80 °C [592].
Циклические соединения Se6 и Se? изучены в [593]. Циклическое
соединение Se3S2, в котором атомы серы связаны между собой, изу-
чено в [594]; соединение образует конфигурации «кресло» и «ванна»,
каждая из которых имеет по 4 изомера, различающиеся положением
атомов серы в цикле, с ХС в области 978-987 м.д. Циклические селе-
новые соединения SnSea_n изучены в [595-597]. Соединения SnSe8-n
(п = 1-7) в растворе CS2 и расплаве при 430 °C изучены в [595]; таких
соединений с изомерами 28, с 6 в диапазоне 580-700 м.д. ХС между
одинаковыми формами в растворе и расплаве различаются на величину
~ 10 м. д., что определяет величину температурного коэффициента ХС
^0,025 м.д./К. В [596] изучены SnSee-n. Приводятся данные ЯМР
77Se изотопомеров S3Se3 (всего 5 изомеров); из-за неэквивалентности
атомов Se наблюдали J(Se-Se) 30-50 Гц. Катионы Se3 и Вещ в раство-
рах SO2 изучали в [597], в отличие от гибкого See, Seto жесткий из-за
звена с параллельными цепочками.
Сера, селен и теллур образуют с Мо и W соединения с кубическим
остовом M4E4Li2 (М = Mo, W; Е = S, Se, Те), причем все ребра
куба состоят из М-Е [598]. Данные ЯМР [W4E4(CN)I2]6~ приведены
в табл. 2.86.
Подобные соединения синтезированы из таллия и селена —
[(Tl4Se4)Se]4- и [(Tl4Se4)Se2]4- [599], и получены данные ЯМР 77Se,
203,205-р|
Дигалогениды селена SeSX2 изучены в [594] (табл. 2.87).
Таблица 2.86
Данные ЯМР комплексов [W4(/i3-E)4(CN)i2]6
Форма <5183W, м.д.1 <5 77Se/l25Te2 J(W-E), Гц
[W4(№-S)4(CN)12]6- -1659 — —
[W4(№-Se)4(CN)12]6- -1026 1496 55
[W4(M3-Te)4(CN)l2]6- -94 2061 156
1 От WO2-; 2 от Ме2Е.
Таблица 2.87
Данные ЯМР 77Se сероселеновых дигалогенидов в смеси Se2Cl2 и S2CI2
Форма d77Se, м.д. Примечание Форма <5 77Se, м. д.
BrSe-SeBr 1105 ClSe-SCl 1277
1172 Ч. жидк. BrSe-SCl 1184
ClSe-SeCl 1264 CISe-SBr 1283
1274 Ч. жидк. CISe-SBr 1269
BrSe-SeCl 1184 SeCl4 1439
BrSe-SBr 1201 SeOCl2 1479
Селеноплюмбаты и теллуроплюмбаты РЬгСИ^- изучены в [433]
(табл. 2.62).
В ряде работ [600, 601] изучены кластеры на основе октаэдра
Reg, скрепленного ^3-Е-лигандами (Se, Те). В [600] изучены растворы
соединений К4[Ре6Те8-ж5е;г (CN)g]; в зависимости от замещения ли-
нии ЯМР селена лежат в диапазоне — 325 4—608 м.д., линии ЯМР
125Те в диапазоне -764 ч—1302 м.д. Se (Те) может быть замещен
бромом [601]; в комплексе [(RegSeyBrJBrg] число неэквивалентных
атомов Se три, в то время как наблюдали только две линии, —223
и —282 м.д. с соотношением 4 : 3. Интерпретация данных ЯМР 77Se
получена в представлении ХС Se как суммы вкладов от нахождения
атома на ребре, грани и диагонали, общей с атомом Вг; для комплекса
[(Яе65е6Вг2)Вгб] существует 3 изомера по положению атомов брома,
и в каждом разное количество неэквивалентных атомов. При равном
количестве изотопомеров по принятой схеме получили всего три линии
ЯМР -117, -192 и -200 м.д. в соотношении 6:1:2, что близко
к реальному спектру.
Димерные ниобиевые селенаты Nb2(^SeSe^)2L4, L = ох, асас, ди-
тиокарбамат, изучены методом ЯМР 77Se в [602].
Теллур. Хотя наблюдение ЯМР |25Те проще, чем 77Se (и по
чувствительности, и из-за меньших величин 7\ у |25Те), данных по
ЯМР 125Те значительно меньше. В подавляющем большинстве они
получены методом двойного резонанса [604, 607]. Неорганические си-
стемы изучают в основном прямым методом ЯМР 125Те [608]. Везде
в качестве внешнего эталона применяют ТеМег, температурный коэф-
фициент которого 0,14м.д./К [603]. Поведение экранирования ядра
125Те в зависимости от окружения аналогично тому же у 77Se, о чем
говорит линейная корреляция между ХС 77Se и 12эТе в изоструктур-
ных соединениях [607]. Некоторые из данных [515, 608] приведены
в табл. 2.88.
Таблица 2.88
Химические сдвиги ЯМР теллура-125
Форма <5, м. д. Примечания Источник
Ме2Те 0 Ч. жидк [515]
Ph2Te 688 СН2С12 [515]
(C6F5)2Te -5,4 С2Н4С12 *
ТеС14 1138; 1962 Ме2СО ТГФ [515]
ТеВг4 1442 ДМФ То же
TeF4 1271; 1313 MeCN SO2, 200 К [607, 610]
TeF- 1142 Ме2СО, 313 К [610]
Ме2ТеС12 749 СН2С12 [515]
Me2TeF2 1231 C2H4C12 •
(CF3)2TeCl2 1114 — [606]
(CF3)2TeF2 1187 — То же
Н2ТеС16 1402 2 М НС1/Н2О [515]
H2TeBr6 1356 1 М НВг/Н2О То же
H2TeI6 857 1 М Н1/Н2О «
К2ТеО3 1732 2 M/D2O «
Н2ТеО6 712; 707 1 M/D2O 1 М/Н2О 4 *
TeIV(OTevlF5)4 1341, 557(a) 554(e) O2C1F, — 110 °C [607]
Таблица 2.88 (окончание)
Форма <5, м. д. Примечания Источник
Telv(OTeVIF5)4 1289, 536(e) S0C1F, -130 °C [610]
Xe(OTeF5)4 48 CFC13, 24 °C [607]
XeOTeF+ 576 SbF5, -83 °C [607]
As(OTeF5)5 541 S0C1F, 22 °C [610]
[Те2(^-О)2О4(ОН)4]4- 710 Н2О [607]
[Те2(^-ОО)2О4(ОН)4]4- 778 Н2О [607]
[Те3(^-О)4О4(ОН)6]4- 703, 714 Н2О, средний атом *
* Данные автора.
Уменьшение ширины линии при снижении температуры указывает
на спин-вращательный механизм релаксации в приведенных в [608]
соединениях (измерения сделаны в поле 9,4 Т; Т\ около 10 с). В рас-
творах Na2TeOs и К3ТеО3 в воде — 2,5 с [609]; спин-вращательный
механизм релаксации отмечен и для ТеМег [603].
Константы ССВ 125Те-Х больше, чем аналогичные 77Se-X; пер-
вые константы ССВ с протонами измерены в [611], ^(Н-Те) = 59 Гц
в НгТе, 58 в H3Si-TeH и 52 Гц в H3GeTeH. Положительный знак
'J(F-Te) в Et2N-TeF5 установлен в [612], где для экваториальных
атомов 'J(F-Te) — 3970, а для атома F в транс-положении к N
'J(F-Te) — 3060 Гц. Величины прямых констант ССВ с фосфо-
ром около 2000 Гц [613], 'J(Se-Te) = —169 Гц в MeTeSeMe [586],
‘J(Te-ll9Sn) = -1385 Гц в (Me3Sn)2Te [400], ‘J(Te-Pt) = -1553 Гц
в PtCl3TeMe2 [587]. Из-за отрицательного гиромагнитного отношения
125Те знаки nJ и ПК противоположны.
Процесс замещения оксо- и гидроксолигандов в димерах теллурата
Te2(OH)4Og- (рис. 2.52) пероксидом изучен в [534]. 7- и 8-коорди-
нированные фториды теллура описаны в [614]. Для [TeFy] 6 125Те =
= 327 м.д., 'J(Te-F) = 2876 Гц.
В [615] линии 683 и 658 в соотношении 2 : 1 также отнесены
к тримеру, причем спектры 125Те при обогащении изотопом на 93%
относятся к системе АВ2. Эти линии появляются при щелочном гидро-
лизе Те(ОН) 6 и. возможно, относятся к тримеру, связанному гидрок-
сомостиками.
Данные элементоорганических соединений S, Se, Те приведены
в [616].
Рис. 2.52. Спектр ЯМР |25Те равновесного раствора димера [Тег(^-О)г
О4(ОН)4]4- (линия 712 м.д., остальные линии относятся к линейному тримеру
[Те3(^-О)4О4(ОН)6]4-)
2.8. Фтор, хлор, бром, иод
Все стабильные изотопы галогенов имеют магнитные моменты,
а благодаря большой чувствительности ядер изучение галогенсодержа-
щих соединений начато давно и широко развито. Подавляющее число
работ сделано методом ЯМР фтора [190, 618-621] и значительно позже
развивается ЯМР Cl, Вг, I [135]. Свойства ядер галогенов приведены
и табл. 2.89.
Изотопы галогенов (кроме 37С1) имеют высокую чувствительность;
ядро 37С1 также нельзя назвать «трудным». Наличие двух изотопов
у хлора и брома позволяет применять в исследованиях изотопное за-
мещение.
Значительные квадрупольные моменты С1, Вг, I заметно ограничи-
вают возможности метода ЯМР этих ядер, в особенности брома и йода.
Фтор. К настоящему времени накоплен большой массив данных
по ЯМР 19F [190, 618-621]. Метод ЯМР фтора применялся в различ-
ных аспектах для получения структурной, химической и физической
информации. Шкала ХС фтора приведена на верхней части рис. 2.53.
ХС некоторых соединений — в табл. 2.90.
Таблица 2,89
Свойства ядер магнитных изотопов галогенов
Изо- топ Спин NA, % Sens 1 Диапазон ХС, м. д. <2, барн Частота ЯМР эталона Е, МГц2 Эталон
13с — — 1{‘Н} — — — —
,9f 1/2 100 1570 1100 0 94,093 795 CC13F
35С1 3/2 75 6,7 1200 -0,08 9,799041 ± 10 [Cl-]aq ^0
37С1 3/2 25 1,3 1200 -0,063 — [Cl-]aq ^0
79Вг 3/2 51 75 2500 0,33 — [Br-]aq -» 0
81 Вг 3/2 49 92 2500 0,28 27,009 765 ±30 [Br“ ]aq —► 0
1271 5/2 100 177 4100 -0,79 20,007 328 ±20 [Г]аЧ-»0
1 Относительно 13С{'Н}; 2 частота ЯМР в поле Hq, в котором частота ЯМР *Н
ровно 100 МГц.
Рис. 2.53. Шкала ХС ЯМР l9F (а) и спектр ЯМР 19F смеси ZrF2- и HfF2" (б)
В экранировании 18F важную роль играет парамагнитный вклад.
Для двухатомных молекул экранирование коррелирует с ионностью
связи: ковалентный характер связи увеличивает ар и уменьшает ad,
Таблица 2.90
Химические сдвиги ЯМР фтора (от CFCI3)
Форма <5, м. д. Примечания, источник Форма <5, м. д. Примечания, источник
f2 432 Газ [619] TeF4 -128 [619]
HF -221 Газ [619] PF5 -76 To же
-193 Жидк. [618] AsF5 -66 «
F“ -121 Н20* SbF5 -76 [619]
FC1 -448 [619] SiF*- -128 [190]
F3C1 —4 [617] SnF*- -150 [619]
(F3C1)2 139 [619] PF- -65 To же
BeF2 -170 « AsF- -70 [618]
XeF2 258 [617] SbF- -109 [619]
of2 248 [619] SF6 50 To же
FSSF -122 ' CFCI3 [619] SeF6 55 [618]
o=sf2 74,5 То же TeF6 -57 [618]
s=sf2 79,5 « MoF3 -278 [619]
BF3 -131 [617] WF6 166; 240 [619, 621]
NF3 147 [622] XeF6 550 [618]
PF3 -35 [619] NiF*- -333 [621]
SbF3 87 [622] PdF’- -326 To же
BeF*- -163 [619] PtF’- -368 «
BF4- -145 [619] SnF*- -153 «
cf4 -59 [622] GeF’- -118 «
ai «a U- —C/D—U- Л / \ff> 1 — 118B; -713 [619] TiFg- 82
RhF^- -521 [623]
OsF6 -89 40 °C [624]
200 [621]
IrF6 -205 40 °C [624]
C6F6 -163 Таблицы Bruker AuF- -109 BrF5, -60 °C [624]
Таблица 2.90 (окончание)
Форма 8, м. д. Примечания, источник Форма <5, м. д. Примечания, источник
SiF4 -174 [619] (XeF6)4 109 CF3C1O2FC1, 133 К [624]
GeF4 -172 То же 238UF6 764 (28)" CFC13, 28 °C [624]
SeF4 64 « 235uf6 764 (550) CFCI3, 28 °C [624]
CF3COOH -78 « IF7 170 [618]
XeF4 438 [617] ReF7 345 [618]
C1F5 247; 412 [617] PuF6 660 [621]
* Зависит от ионного фона; ’* в скобках указана ширина линии; в — вершина,
э — экватор.
что в общем уменьшает экранирование. Расчеты [625] дали значение
(<т)абс 410 м.д. для HF, что позволило построить абсолютную шкалу
экранирования для 19F [619]. В табл. 2.91 приведены ХС некоторых
соединений в газообразном состоянии.
Таблица 2.91
Химические сдвиги некоторых фторсодержащих газов
Форма <5, м. д. Форма <5, м. д. Форма 8, м. д.
f2 432 BF3 -116 C1F -448
SF6 50 SiF4 -174 CCI3F, газ —7
(C1F3)2 139 HF -221 CCI3F, жидк. 0
C1F3 -14
Изменение crd сравнительно мало, и основное изменение ХС опре-
деляется вкладом ар, поэтому в бинарных соединениях [626] экрани-
рование ядра фтора коррелирует с электроотрицательностью.
Экранирование в октаэдрических фторокомплексах обсуждено в
[621]. Эффекты растворителя в ЯМР 19F доходят до 10 м.д.; такой
же порядок величины дает и водородная связь — различие между ХС
HF в жидкой и газовой фазе около 20 м.д. [625]. ХС некоторых газов
приведены в табл. 2.92. Константы ССВ с другими ядрами достигают
нескольких килогерц (табл. 2.92).
Таблица 2.92
Константы спин-спинового взаимодействия с ядром фтора
Форма J(F-X), Гц X Источник Форма J(F-X), Гц X Источник
NF3 160 ,4N [374] SbF- 1843 12lSb [372]
PF3 1441 31 p To же TeF6 3688 l25Te To же
BF3 10 "B « MoFe 44 95Mo «
SiF4 178 29Si « TiF2- 33 49Ti [627]
pf5 916 31 p « GeF2" 98 73Ge To же
PF6“ 710 31 p « PtF2- 1072 195p| «
SF6 252 33s [573] RhF3- 244 103Rh [623]
AsF- 930 75 As [372] (XeF6)4 325 l29Xe [624]
ЯМ-релаксация фтора редко превышает 10 с. Во многих соедине-
ниях большое значение имеет кросс-релаксация F-H. В обычных усло-
виях лиганд F- фторидных комплексов часто подвижен и фиксируется
в комплексе при низких температурах, что дает в наблюдаемую ширину
линии большой обменный вклад. Кроме того, бывает, что одни лиган-
ды лабильны при других инертных фторид-лигандах комплекса [279].
ЯМ-релаксация гектафторидов Sc, Ti, Ge изучена в [628].
Фтор образует соединения или ионы 3Fg со многими элемента-
ми, что создает благоприятные условия наблюдения КССВ с квад-
рупольными ядрами, а также ЯМР на центральном ядре. Пример
спектра ЯМР 19F ионов ZrFg- и HfFg- в водном растворе показан на
рис. 2.53.
Проведены расчеты экранирования ядер фтора в различных соеди-
нениях: F2 [629], GeF4, SiF4, CF4, SFe, SeFg, TeFg, UFe [630, 631].
Корреляции XC 19F и параметров, характеризующих донорные свой-
ства партнеров (частота валентных колебаний, ст-константы Гаммета,
электроотрицательность), обсуждаются в [632]. Корреляции ХС 19F —
энергия во фторкислородных соединениях найдены в [639]. Данные
по фторидным комплексам алюминия обобщены в [279, 643, 644].
Комплексы в расплаве, моделирующим таковой в электролизере плавки
алюминия, изучены в [645]. Анионы IFgO-, TeFgO^, TeF^, IF^ and
TeFg- изучены в [614]. Ступенчатое комплексообразование в системах
F--PO4- и TeF6-NNN~, TeF6-NMe2 изучено [640].
Данные о ХС HF, HOF, F2, OF в жидком и газообразном состояни-
ях и в водных растворах приведены в [190, 625, 636-638]; в [636] до-
казано существование гипофторной кислоты HOF. Сольватация ионов
в безводном пероксиде водорода изучена в [641], SF2 в газовой фа-
зе — в [642]. Образование полифторсерных кислот изучено в [646].
Фторидные комплексы бора изучены в [647, 648] и комплексы олова
в [649, 650], комплексы, процессы гидролиза гексафторантимоната —
в [651], спектры и взаимодействие с органическими растворителями
[TeFs]- — в [610, 652]. Данные ЯМР l9F гидроксоантимонатов приве-
дены в табл. 2.93.
Таблица 2.93
Данные ЯМР 19F гидроксофторидоантимонатов [651]
Форма 8 19F, м.д. Форма <5 i9F, м. д. J(F-F)
[SbF6]- 121, J('21Sb-F) = = 1934 Гц apaH-[SbF3OH)3]“ 122 —
[SbF5OH]- 109 pe6-[SbF3OH)3]- 127 86
i4uc-[SbF4OH)2]- 117 4uc-[SbF2OH)4r 126 96
mpaHC-[SbF4OH)2]- 118 транс- [5ЬРгОН)4]_ 130 —
Фторборатные комплексы изучены в [648]; большую роль метод
ЯМР l9F сыграл в химии кремнефтористых соединений [653-655].
ЯМР 19F широко используется для изучения состояния и реакций
замещения лигандов в растворах фторидных комплексов переходных
металлов и их соединений. Влияние донорных свойств лигандов на
ХС 19F атома F в транс-положении к лиганду в комплексах TiFsL,
TaFgL, SI3F5L, M0F5L, WF5L изучены в [633, 634]; изменение ХС
между крайними случаями достигает 400 м. д. Изменение ХС 19F в сме-
шанных галогенидных комплексах W, Мо в зависимости от струк-
турных данных рассмотрено в [635]. Растворы VOF3, NbFs, TaF5,
MOO2F4, WO2F4 изучены в [633, 656-658]; фторокомплексы оксопе-
роксовольфраматов — в [658, 659]; реакции замещения в растворах
фторгаллатов — в [289]. Превращения гексафторидных комплексов
Rh(III), Ru(IV), Os(IV) в водных растворах — в [623, 660]; этилен-
диаминовый и пентааминовый фторидные комплексы Со(Ш) изучены
в [661], смешанные фторокомплексы родия — в [662], оксофторидные
комплексы осмия — в [663, 664]. Оксофторидные комплексы рения
изучены в [665], фторидные комплексы вольфрама с /?-дикетонатами —
в [666]. Образование пероксофторотитанатов исследовано в [667], са-
моассоциации дифторида уранила — в [668], влияние противоиона на
образование мицеллярных структур — в [669]. Гидролиз оксофторти-
таната изучен в [670]. Строение и кислотные свойства оксофторидных
комплексов технеция изучены в [565]. Большую роль метод ЯМР 19F
сыграл в химии ксенона [624].
Хлор, бром, иод. Ядра этих галогенидов имеют квадрупольный
момент, возрастающий от хлора к йоду (см. табл. 2.88), что ограничива-
ет возможности измерения ХС соединений и комплексов с концевыми
атомами галогенидов, линии которых часто слишком широки (напри-
мер, ширина линии ЯМР 35С1 в CCI4 при 300 К около 15 кГц [671]).
Наиболее представительные данные по ХС 35С1.
Данные ЯМР С1, Вг, I приводятся в [135, 672]. В неорганических
растворах преобладают ионные формы галогенидов, в виде ионов в
водных растворах или ионных пар в неполярных растворителях. Линии
галогенид-ионов существенно уже, масштаб дает ширина линий 0,5 М
водных растворов NaX, X — галогенид [679]:
X 35С1 37С1 79 Вг 8|Вг 1271
W, Гц 8 5 480 330 1680
Сравнение экранирования в пергалогенатах показано в табл. 2.94.
Таблица 2.94
Данные ЯМР ионов ХО^ [684]
Соединение X <5(X), м.д. J(X-O), Гц 8 C'O), м.д. Условия
Et4NCIO4 35C1 1009 83 293 0,1 M/MeCN
Et4NBrO4 81Br 2489 440 361 To же
Et4NIO4 127I 4121 489 243 To же
HBrO4 8lBr 2478 433 359 2,3 M/H2O
NaIO4 l27I 4089 — 238 0,9 M/H2O
Зависимость ХС галогенид-ионов от концентрации в водных рас-
творах изучена в [79].
Хлор. Хотя ширина линий ЯМР 37С1 меньше, чем 35С1, использо-
вание ядра 37С1 не дает существенного преимущества в определении
ХС, поскольку изменение частоты у ядра 37С1 также меньше (про-
порционально гиромагнитному отношению), вследствие чего выигрыш
в разрешении линий всего в 1,34 раза. В то же время из-за умень-
шения соотношения сигнал/шум (меньшее естественное содержание
35С1) погрешности определения ХС возрастают. Данные по ХС хлора
приведены в табл. 2.95.
Таблица 2.95
Химические сдвиги ЯМР хлора
Форма T, к 8, м. д. (W, кГц) Примечания, источник
СГ 293 0 H2O, [СГ] 0
НС1 298 15±0,3(0,1) Водный раствор, конц.
С12 298 370 ± 150 Ч. жидк. [673]
300 430 ± 75 0,5 М H2SC>4 в Н2О, насыщенный раствор при 8 атм
С13“ 300 -150± 150 [674]
СН3С1 298 40 [675]
298 50 ± 22 (22) Et2O [671]
СН2С12 298 226 ±6 (7,8) Et2O [671]
СНС13 — 410 [750, 754]
298 401 ±8(7,7) Чист. ж. [671]
ссц — 560 [675]
299 540 ± 50 [135]
298 599 ±8 (13) Et2O [671]
CFC13 299 470 ± 50 [135]
SiCU 298 195 ±3(3,6) Чист. ж. [671]
РС13 299 370 ± 90 [135]
POCI3 « 430 ± 56 То же
PSCI3 « 530 ± 90 «
SOC12 « 660 ± 40 «
S2 Cl 2 « 480 ± 40 «
SnCU 298 120 ±60 «
« 138 ± 15(15,5) Чист. ж. [671]
« 135 ± 8 (12,6) С6Н6 [671]
TiCl4 303 873 ±2 [136]
298 865 ± 2 (0,8) Чист. ж. [677]
[TiOCl4]2- 293 24 ±4 TiCl4/KOHu. НС1 [677]
Таблица 2.95 (окончание)
Форма т, к 5, м. д. (W, кГц) Примечания, источник
GeCU 299 170 ±60 [135]
298 247 ±7 (7,7) Чист. ж. [671]
А1С1з « 150 ±60 [676]
СгОгОг « 603 ± 13 То же
VOCI3 285 790 ± 13 «
VCI4 — 990 ± 50 [678]
СЮ3- 295 1050 [675]
299 1130± 10 Нас. р-р КС1О3 *
СЮ4- 301 946 ±6 10 М НС1О4 [676]
1003 NaClC>4 в НгО, нас. р-р [679]
* Данные автора.
Диапазон ХС ЯМР хлора около 1200 м.д. Диамагнитный вклад в
экранирование возрастает от F к I (для свободного атома F <rd — 464,
для С1 — 1150, для Вг — 3080 и I — 5450 м.д. [680]), однако он не
определяет ХС, поскольку мало меняется от соединения к соединению
и примерно такой же, как у свободного атома [681, 682].
В парамагнитном вкладе в экранирование (4.1) влиянием факто-
ра SQ обычно пренебрегают, а наиболее чувствительным фактором
при образовании связи считают (г-3). Поскольку от этого фактора
зависит и константа квадрупольного взаимодействия, можно ожидать
корреляции между ХС и указанной константой в ряду соединений,
в которых ДД можно считать постоянным [135]:
d eqQlh
Сабе = с + const- ;
Д(г 3)пр
в свободных ионах галогенидов парамагнитный вклад должен быть
равен нулю. В ионных кристаллах [136] и в водных растворах взаи-
модействие с окружением приводит к появлению парамагнитного вкла-
да [683, 684]. Концентрационная зависимость ХС хлора и йода для
водных растворов солей щелочных металлов приведена на рис. 2.54,
2.55. Как и для катионов [78, 80], зависимость резко увеличивается
с увеличением атомного веса катиона и аниона.
<535С1, водные растворы
Рис. 2.54. Зависимости химиче-
ского сдвига ЯМР 35С1 от концен-
трации соли
Рис. 2.55. Зависимости химиче-
ского сдвига ЯМР 1271 от кон-
центрации соли
Многие теоретические оценки изменения ХС трудно проверить из-
за грубости экспериментальных определений.
Для измерения физических и химических характеристик состоя-
ния галогенсодержащих молекул и ионов широко применяется метод
ЯМ-релаксации, данные которого (табл. 2.96) показывают возможно-
сти ЯМР 35С1 для разных соединений.
Все константы ССВ получены из ЯМ-релаксации ядер, связанных
с атомами хлора, кроме прямо измеренных 1J(F-35C1) для иона C1F^“
[136] и V(I7O-35C1) для СЮ^ [577] (табл. 2.97).
Образование лабильных комплексов с парамагнитными ионами
приводит к появлению парамагнитных вкладов в ХС и ЯМ-релак-
сации галогенид-ионов. Вклад в 1\,2 галогенид-ионов пропорциона-
лен 72 [135], поэтому отношение времен релаксации R(35C1)/R(37C1) =
= [-у(35С1)/-7(37С1)]2 = 1,44; чисто квадрупольные вклады в релаксацию
относятся как Q2(35C1)/Q2(37C1) — 1,61. Очевидно, различить эти вкла-
ды в реальных системах затруднительно.
Парамагнитные сдвиги ЯМР 35С1 были обнаружены в кислых рас-
творах Fe3+ [685]; распределение ацидокомплексов в них в зависи-
мости от концентрации НО изучено по ЯМР |7О и 35С1 в [686].
Из этих данных парамагнитный сдвиг в ионе FeCl^" составляет 3,6%
и A/h — 7 • 107 рад/с. По ЯМР 35С1 изучено взаимодействие иона
Cl" с Со2+ [687-689]; VO2+ [690]; Сг3+, Fe3+, Со2+, Ni2+, Мп2+
и Fe(CN)g“ [694]; иона С1~ с ионами лантаноидов [695]; ионов С1О^
и Мп2+ [696]. Взаимодействие Lu3+ с С1О^“ в водно-ацетоновых
Таблица 2.96
Данные ЯМ-релаксации хлора-35 и частоты ЯКР [135]
Форма T, К Ti, мкс T2, мкс i/q, МГц
СС14 298 — 22 81,3
СНС1з 298 — 25 76,6
СН2С12 298 — 50 72,5
СН3С1 Комн. — 100 68
CFCI3 298 38 — 79,6
C1CN 287 — 30 83,4
SiCl4 299 — 79 40,8
GeCh 299 — 41 51,4
SnCU 299 — 22 48,2
РЬСЦ 298 — 5 45,4
TiCl4 298 — 400 12,2
PC13 299 — 60 52,2
AsCl3 299 — 23 50,2
BC13 298 — 122 43,2
HC1 395 765 — 67,5
Cl2 299 — 41 109
VOC13 298 — 153 23,1
CrO2Cl2 299 — 107 31,4
CIO3F 298 1300 — 19,2
POCI3 299 — 27 57,9
SbCl5 299 — 16 57,2
SOC12 299 — 38 64,0
SO2C12 299 — 30 75,4
CIO.]’ (aq) 295 53* 16 59,8
CIO; (aq) 301 27 • 104 27- 104 —
GaCl]“ (aq) 298 — 8-15 32,2
Данные автора.
Таблица 2.97
Константы спин-спинового взаимодействия с ядром хлора-35
Форма ‘J(35C1-X) X Форма ‘J(35C1-X) X
НС1 41 н РЬС14 705 2°7pb
РС13 120 31 р C1F3 ' 260 l9F
СНС13 23 13с C1F5 192 (а), 20 (э)‘ «
SnCU 375 ,19Sn C1OF3 195 «
SnCl3I 378 ,19Sn C1F+ 337 «
С1О4- 85,5 17О
а — вершина, э — основание пирамиды комплекса.
растворах изучено в [697], определено гидратационное число Lu3+,
равное 7. Процесс щелочного гидролиза [PdC^]2- изучали в [692]
с определением свободного С1“ методом ЯМР 35С1 и взаимодействия
Cs+ с анионами и коллоидными частицами по ЯМР l33Cs; перед оса-
ждением Pd обнаружены две зоны золя, в которых линии ЯМР 133Cs
различаются на 70 м. д. Взаимодействие [Аи(еп)2]3+ с С1“ изучено
в [663]. Обмен С1“ и С1г изучен в [698].
Вследствие наличия большого квадрупольного момента ХС брома
определяется грубо; измерения обычно проводят на изотопе 81Вг, у
которого линии ЯМР более узкие. В [699] измерены ХС ВгО^“, равный
+2484 м.д. от Вг-, и ’7(Вг-О) — 425 Гц. Данные ЯМР 8|Вг приведены
в табл. 2.98 [135].
Таблица 2.98
Данные ЯМР и ЯКР брома-81
Форма T, К Ti, мкс T2, мкс j(8lBr-X), Гц X i7q, МГц
НВг 295 17 — 62 *H 448
РВг3 278 0,33 0,32 360 31 p —
SnBr4 294 — 0,75 960 119Sn 330
CdBr+ (aq) 298 — 0,1 — — —
CdBr4“ (aq) 298 — 0,17 — — —
HgBr4“ (aq) 300 — 0,68 — — 332
ZnBr4~ (aq) 298 — 0,25 — — —
BrF+ — — — 1697 ,9f —
В ЯМР |271 измерен ХС иона 10^ (<5 1410 м.д. от I-), который и
определяет диапазон ХС 1271. Из-за квадрупольного уширения данные
ЯМР 1271 сводятся, как и у брома, к временам релаксации и КССВ
(табл. 2.99) [135].
Таблица 2.99
Данные ЯМР йода
Форма Г, к Ti, мкс T2, мкс V(I27I-X), Гц X i/q, МГц
Snh 427 0,15 — 940 ll9Sn 1390
SnCl3I 298 — 0,16 1638 « 1355
SnClI3 400 — 0,12 1097 « 1355
ю4- 298 — 200 — — —
Hgl4- (aq) 300 — 0,21 — — 840
IF" (HF) 307 1000 — 2730 ,9F —
IF? — 57 — 2100 « —
CH3I — — — 60 l3C —
Изучение растворов [XF6] [ВЬгР]i] (X = Cl, Вг, I) [700] позволило
определить ХС и КССВ для всех ядер галогенов (табл. 2.100).
Таблица 2.100
Данные ЯМР катионов [XFg]+ [700]
Катион [XFg] + <5(X), м.д. J(F-X), Гц T|, c W, Гц
35cif+ 814,3 339 1,3 3,6
37cif+ 814,7 283 2,6 3,2
79BrF+ 2079,9 1575 0,025 25
8lBrF+ 2079,3 1699 0,035 17
'27if+ 3380,6 2744 0,0065 56
ЯМР С1“, Вг-, I- применяется для изучения обменных реакций
в растворах с этими ионами, линии ЯМР которых довольно узки.
Взаимодействие с катионом или другой частицей приводит к уширению
линии ЯМР, которое при быстром обмене со свободным ионом уда-
ется наблюдать. Из него можно получить информацию о параметрах
обмена; такие исследования выполнены для реакции X- + Хг Хз
(для X = С1 [673, 674] и для X = Вг в [699], для X = I в [701,
702]). Для реакций М2+ + X MX, MX + X <-> МХг изучено вза-
имодействие ионов Cd2+, Zn2+, Hg2+ с Вг [703—705]; Cd2+, Zn2+
с I- [703]; Hg2+ c Cl- [706]. Изучено взаимодействие трехвалентных
катионов с СГ: Ga3+ [707, 708], In3+ и Т13+ [709]. По ЯМР 35С1
обнаружено внешнесферное взаимодействие с инертными в масштабе
времени ЯМР хлороаквакомплексами Rh(IIl) [710]. При исследовании
процесса формирования нанометровых частиц Ru(III) [84] с параллель-
ным измерением концентрации свободных СГ по сигналу ЯМР 35С1
определен их состав [Ри(ОН)С12(Н2О)г]п- Определение концентрации
СГ методом ЯМР 35С1 используется при изучении гидролиза ком-
плексных хлоридных соединений металлов [692, 712]. Методом ЯМР
ядер галогенов изучаются взаимодействия между катионами щелочных
металлов и галогенид-анионами для установления структуры растворов
электролитов [79]. Гидролиз фторидов тантала изучен в [711]; данные
некоторых комплексов в процессе гидролиза раствора Та в 38% HF
приведены в табл.2.101.
Таблица 2.101
Данные ЯМР 19F фторидных комплексов тантала
Комплекс <5i, м.д. <$2, М. Д. J(F!-F2), Гц
TaF5(OH)- 30,5 —4 35,8
TaF5O2- -14 -95 37,7
цис-ТаГ (ОН)^ 25 -10 31
z^uc-TaFiOj- -21,5 -121,5 —
транс-Тар4ОН(Н2О) 20 — —
Ta2(M-O)F2- 33 9 33
2.9. Инертные газы
Элементы группы VIIIA, кроме аргона, имеют магнитные изото-
пы (табл. 2.102). Первые измерения ЯМР благородных газов сделаны
в 50-х годах [503, 713, 714]; имеются обзоры [715, 716]. ЯМР этих
элементов нашел применение в молекулярной физике; отсутствие хими-
ческих соединений (кроме Кг и Хе) делает термин «химический сдвиг»
для Не, Ne некорректным.
Успешно развивающаяся химия ксенона и благоприятные свойства
ядра |29Хе позволили составить значительный массив данных по ЯМР
соединений ксенона [589, 717, 718] и подробно изучить особенности
магнитного экранирования его ядра. В обычных условиях ксенон —
атомарный газ, и в этом случае экранирование его ядра чисто диа-
Таблица 2.102
Свойства ядер магнитных изотопов инертных газов
Изо- топ Спин NA, % Sens Диапазон ХС, м. д. <2, барн Частота ЯМР эталона =, МГц Эталон
13с — — 1{'Н) — — — —
3Не 1/2 10~6 2300 1 — 0 76,1812 3Не, газ
21 Ne 3/2 0,29 0,012 — 0,09 7,898 Ne, газ
83Кг 9/2 11,6 0,41 — 0,15 3,862 Кг, газ
,29Хе 1/2 26,2 10,4 7400 0 27,856 XeOF4/MeCN
131Хе 3/2 21,2 1,1 « -0,12 8,257 4
1 При содержании изотопа 100%; 2 рассчитано из величины ядерного момента.
магнитное (~5800 м.д.). Экранирование Хе в жидком и газообразном
состоянии пропорционально плотности [720]:
ст (м. д.) = сто — 0,55р (амага)
(1 амага — плотность газа при нормальных условиях). Добавление
других газов вызывает значительный вклад в экранирование:
а[Т, р(Хе), р(А)] = ст0 + ^ер(Хе) + ст^(А) + <т2Хер2(Хе) + ст2Ар2(А).
Данные ЯМР 129Хе приведены в табл. 2.103.
Таблица 2.103
Данные ЯМР 129Хе
Форма <5, м. д. J(Xe-F), Гц Примечания, источник
XeOF2 0 1123 Ч.жидк. 25 °C [717]
Хе -5351 — n-C6F14, 25 °C [717]
Хе(П): Xe(OSeFf,)2 -2200 37 CFCl3 [589]
Xe(OTeF6)2 -2379 31 То же
F5SeO- Хе- OTeFs -2289 — «
FXeOSeF5 -1952 37 «
FXeOTeF5 -2067 30 «
XeOTeF+ -1608 — [716]
Таблица 2.103 (окончание)
Форма <5, m. д. J(Xe-F), Гц Примечания, источник
XeF2 -2023 5550 CFC13 [589]
-1905 5630 SO2CIF, 25 °C, нас. р-р [715]
-1750 5616 2,7 M в BrF5 [715]
-1708 5583 То же
Xe(OSO2F)2 -1613 — HSO3F, -84 °C [715]
-1572 — HSO3F, -90 °C [715]
FXeOSO2F -1616 — [716]
XeOSO2F+ -1306 — [716]
FXeF-MoOF4 -1383 5117 BrF5, -80 °C [715]
6139 То же
FXeFWOF4 -1331 5051 BrF5, -66 °C [715]
6196 То же
XeF+ -514 7210 BrF5, 25 °C [715]
FXeN(SO2F*)2 -1997 5586; 19* BrF5, -58 °C [719]
Xe(IV): XeF5“ 13 159; 1400 HF, 25 °C [715]
XeF4 253 3823 BrFs, 25 °C, нас. р-р [715]
XeF2F*+ 595 2384; 2609* [716]
Xe(OTeF5)4 -436 3506 [716]
Xe(VI): XeO3 217 — Н2О [717]
(XeF6)4 -35 330 (F5S)2O, -118 °C [717]
XeF6 -45 330 [716]
XeO2F2 73 1217 «
171 1213 25 °C [735]
XeF4F*+ 132 1400; 159* [716]
OXeF4 -30 1131 25 °C [735]
OXeF+ 237 1032 [736]
Xe(VIIl): XeOg- 2077 — 0,5 М в Н2О [715]
Вклады высших порядков много меньше вкладов первого порядка <7\
(табл. 2.104).
Таблица 2.104
Величина вкладов ст^, м.д. • см3/моль, при 25 °C [721]
Газ А а? Газ А _А Vi Газ А <4
Аг -3120 СН« -6200 Хе -12280
СО2 -3820 Кг -6070 no2 -17800
CF< -4330 НС1 -7680 о2 -21000
Фториды и оксофториды Хе изучали методом ЯМР 19F [722-725].
ЯМР 129Хе дает прямую информацию о координационной сфере Хе;
ранние работы сделаны методом двойного резонанса 19F{l29Xe} [718];
в настоящее время преобладают прямые измерения на ядре 129Хе [589,
712, 726, 727].
Важную роль в ХС Хе играют [414] координационное число
Хе, гибридизация связывающих орбиталей, ионность связи; фак-
торы (г-3)бр, (r-3)5d и ДЕ в парамагнитном вкладе в экрани-
рование примерно постоянны. Экранирование Хе меняется в ряду
Xe(VIII) < Xe(VI) < Xe(IV) < Xe(II) [726]. Величина V(Xe-F) лежит
в пределах 95-7200 Гц и коррелирует с расстоянием Xe-F (для Хе(П)
на рис. 2.56 [715]). Известны также *J(129Xe-O) = 692 Гц (XeOF4
Рис. 2.56. Корреляция КССВ F-Xe и расстояния ксенон — фтор для фторксе-
ноновых соединений
[728]), 2J(l29Xe-Se) = 130 Гц (FXeOSeF5, [729], 2J(129Xe-Te) = 540 Гц
(FXeOTeF5, [729]), 1 J(129Xe-15N) = 307 Гц (FXe(SO2F)2 [719]). Време-
на релаксации 129Хе в газообразном ксеноне около 1 ч, в жидком —
около 1 мин, в изученных оксофторидах — 0,1-1 с [713].
ЯМР благородных газов широко применяется для изучения моле-
кулярного движения, так как движение их атомов не осложнено вра-
щательными и вибрационными степенями свободы и взаимодействие
с другими молекулами непосредственно передается на ядро. Наиболее
подходящими для этого изотопами являются 129Хе и 3 Не. 3 Не отлича-
ется очень долгими временами релаксации (до 2400 с [714, 715, 728]).
Ядра других изотопов в сжиженных газах имеют величину Т\ = 24 с
(21Ne [817]); 0,6 с (83Кг [731]); 0,04 с (131Хе [732, 733]).
В [716] приведены константы квадрупольного взаимодействия 83Кг
и 131Хе для некоторых соединений криптона и ксенона (табл. 2.105).
Таблица 2.105
Константы кварупольного взаимодействия CQQ |e2gQ//i|
Форма CQQ 83Кг, МГц hv, см-1 Форма CQQ l31Xe, МГц hv, cm’1
KrF2 978 1,07 XeF2 2450 1,456
KrF+.AsF" 1070 1,17 (FXe)2F-AsF~ 2642 1,514
KrF+-SbF“ 1090 1,19 Xe(OTeF5)2 2450 1,402
Кг+ 4р5 * 915 1,00 ХеС12 1800 1,032
KrCIF 13,9 0,15 XeF< 2620 3,0
ХеСЦ 1640 1,98
OXeF4 1156 —
XeO2F+-SbF“ 868 —
XeFg 492 —
* Атомные пучки.
Гидролиз XeFg изучен в [734]. Оксофторосоединения Хе изучены
в [735] (табл. 2.106).
Таблица 2.106
Данные ЯМР соединений ксенона с группой OTeF^ [735]
Форма <5Xe 3 l9F 'j(Xe-F) J(Xe-X) X Раствори- тель
Xe(OTeF5)4 -646 — — CFC13, 25°
FXe(OTeF5)3 -436 16,4 3506 1032 (э) Те —
— — — 1292 (в) — «
mpa«c-F2Xe(OTeF5)2 -216 11 3503 1166 Те «
4uc-F2Xe(OTeFs)2 -243 -8,6 3714 1059 Те «
F*F2Xe(OTeF5) -26 5,9; -12* 3552; 3433* 1192 Те «
Таблица 2.106 (окончание)
Форма <5Xe 6 19f 'j(Xe-F) J(Xe-X) X Раствори- тель
OXe(OTeF5)4 -204 — — 1351 — To же
OXeF(OTeF5)3 -157 111,3 1206 — — «
транс-ОХер2(ОТер5)2 -106 108,2 984 1535 Те «
цис-ОХер2(ОТер5)г -118 112,6 1074 1536 Те «
OXeF*F2(OTeF5) -66 106,8*; 103 1148*; 931 1364 Те «
— — — 83 FFTe SO2C1F
O2Xe(OTeF5)2 131 — — 1684 Те -74°
O2XeF(OTeF5) 154 — 1046 1856 |7О CFC13,
XeOF4 24 316 (<517O) 1146 704 ,7O 25°
XeO2F2 171 302 (<517O) 1213 521 — —
в — вершинные, э — экваториальные атомы F.
Свойства ксенона, адсорбированного на цеолитах и пористых алю-
мофосфатах изучали в [727, 737], в том числе и на катализаторах
[738-740]. Зависимость ХС ксенона от давления (рис. 2.57) является
отражением формулы (2.9).
Рис. 2.57. Зависимость ХС 129Хе, адсорбированного на разных типах пористых
фосфатов, от относительной плотности ксенона
Хе в клатратах и твердых телах изучен методом ЯМР 129Хе в [741].
ЛИТЕРАТУРА К ГЛАВЕ 2
1. NMR and Periodic table / Ed. В. E. Mann, R. К. Harris. — L.: Acad. Press,
1984. P. 49-86.
2. Akitt J. W. Hydrogen and Its Isotopes. Multinuclear NMR / Ed. J. Mason. —
N.Y.-L.: Plenum Press, 1987. P. 171-187.
3. Федотов M.A. ЯМР в растворах неорганических веществ. — Новоси-
бирск: Наука, 1986. — 196 с.
4. Эмсли Дж., Финей Дж., Сатклиф Л. Спектроскопия ЯМР высокого раз-
решения. Т. 1 // Пер. с англ. Б. А. Квасова; Под ред. Э.И. Федина. —
М.: Мир, 1968. - 630 с.
5. Pfrommer B.G., Mauri F., Louie S.G. NMR Chemical Shifts of Ice and
Liquid Water: the effect of Condensation // J. Amer. Chem. Soc. 2000. V. 122,
№ 1. P. 123-129.
6. Kaecz H.D., Saillant R.B. Hydride comlexes of the transition metals //
Chem. Revs. 1972. V. 72, №2. P. 231-281.
7. Karplus M., Pople J.A. Theory of carbon NMR shifts in conjugated
molecules // J. Chem. Phys. 1963. V. 38, № 12. P. 2803-2807.
8. Buckingham A.D. Stephens P J. Proton Chemical Shifts in the NMR Spectra
of Transition-metal Hydrides. Octahedral Complexes // J. Chem. Soc. 1969.
№ 8. P. 2747-2759.
9. Buckingham A.D., Stephens P.J. Proton Chemical Shifts in the NMR Spec-
tra of Transition-metal Hydrides. Square-Planar Platinum(II) Complexes //
J. Chem. Soc. 1964. № 11. P. 4583-4587.
10. Chatt L, Shaw B.L. Hydrido-complexes of Platinum(II) // J. Chem. Soc.
1962. № 12. P. 5075-5084.
11. Geoffroy G.L., Lehnman J.R. Hydride complexes of Ruthenium, Rhodium
and Iridium // Adv. Inorg. Chem. Radiochem. 1977. V. 20. P. 189-290.
12. Meiboom S. Nuclear magnetic resonance study of the proton transfer in
water // J. Chem. Phys. 1961. V. 34, №2. P. 375-388.
13. Coetzee J.F., Hussam A. NMR study of speciation of water at low concen-
tration in hydrogen bond acceptor solvents // J. Solut. Chem. 1982. V. 11,
№ 6. P. 395-407.
14. Akitt J. W. Proton chemical shifts of water in cationic hydration complexes
and their contribution to water shifts in electrolyte solutions // J. C. S. Dalton
tr. 1973. № 1. P. 42-49.
15. Коттон Ф., Уилкинсон Дж. Основы неорганической химии / Пер. с англ.
М. Н. Варгафтика — М.: Мир, 1979. — 678 с.
16. Axtmann R.C. Proton NMR spectra of hydrated cations // J. Chem. Phys.
1959. V. 30, № 1. P. 340-341.
17. Schuster R.E., Fratiello A. Proton magnetic resonance solvation study of
aqueous solution of AICI3 // J. Chem. Phys. 1967. V. 47, № 5. P. 1554-1555.
18. Grethe I., Toth I. *H NMR Studies of Bi3+-HO-System: Stoichiometric
Composition of the Hexanuclear Complex and Rate of Proton Exchange of
coordinated H2O and HO- in Mixed Acetone I Water Solution // Inorg.
Chem. 1985. V. 24, № 15. P. 2405-2407.
19. Федотов M.A., Юхин Ю.М., Шубин. А. А., Удалова T.A. Исследова-
ние строения ди-2-этилгексилфосфата тетрагидроксогексависмутата(Ш) //
Ж. неорг. химии. 1998. Т. 43, № 2. С. 307-310.
20. Привалов В.И., Тарасов В.П., Меладзе М. А. и др. Миграция и скалярная
связь протона в анионе [ВвНуХ]- по данным ЯМР “В, ‘Н // Ж. неорг.
химии. 1986. Т. 31, №5. С. 1113-1119.
21. Akitt J. W. Proton chemical shift and hydration of hydronium ion in aqueous
acids // J.C.S. Dalton tr. 1973. № 1. P. 49-52.
22. Чуваев В.Ф., Ониани Э.С. Исследование методом ЯМР ’Н гидратно-
сольватного взаимодействия H3PW12O40 с диэтиловым эфиром и этилаце-
татом // Ж. неорг. химии. 1999. Т. 44, №8. С. 1397-1401.
23. Федотов М.А., Максимов Г.М., Игнашин С.В. Возможности метода
ЯМР разных ядер в определении кислотности растворов гетерополикислот
и других минеральных кислот // Ж. неорг. химии. 2002. Т. 47, №12.
С. 2031-2037.
24. Frahm J. A proton NMR study on the kinetics of protolysis and water
exchange in mixed solvent systems. Berillium(II) nitrate in water-acetone (de)
and water-dimethylsulfoxide (de) mixtures // Ber. Bunsenges. phys. Chem.
1980. Bd. 84, №8. S. 754-758.
25. McCain D. C. NMR study of LaCh solutions in water-methanol mixed sol-
vents Ц J. Inorg. Nucl. Chem. 1980. V. 42, №8. P. 1185-1187.
26. Fukui H., Miura K., Ugai T, Abe M. Proton magnetic resonance study of
ion hydration in acetone // J. Phys. Chem. 1977. V. 81, № 12. P. 1205-1209.
27. Butler R.N., Symons M.C. NMR study of electrolyte solutions: cation and
anion shifts in hydroxylic proton resonance of methanol // Tr. Faraday Soc.
1969. V. 65, № 556. P. 945-949.
28. Richardson D., Alger T.D. PMR studies of aluminium(III) and gallium(III)
in methanol and ethanol. Determination of solution number and exchange
rate // J. Phys. Chem. 1975. V. 79, № 15. P. 1733-1739.
29. Akisanya A., Ayanabadejo F.A. PMR solvent shifts in acetic acid solutions
of some inorganic salts // Spectrochim. Acta. 1976. V. 32a. № 2. P. 223-226.
30. Von-Goldammer E., Kreich W. Proton NMR relaxation studies on solution
of manganese(II) perchlorate in dimethylsulfoxide // Ber. Bunsenges. phys.
Chem. 1978. Bd. 82, № 5. S. 463-468.
31. Symons M.C., Thomas U.K. Solvation of anions by protic solutions NMR
and JR correlations // J.C.S. Faraday tr. I. 1981. №8. P. 1891-1897.
32. Чижик В.И., Кабаль К., Родникова М.А., Гусман А. Микроструктура
гидратных оболочек редкоземельных ионов по данным ЯМР релаксации //
Коорд. химия. 1988. Т. 14, №3. С. 349-352.
33. Amirhaeri S., Farago M.E., Ghick A.P., Smith M.A.R. *H and l3C NMR
studies on malonic and ethylmalonic acids and their cobalt(III) complexes //
Inorg. Chim. Acta. 1979. V. 33, № 1. P. 57-61.
34. Целинский Ю.К., Пестряков Б.В., Галат В.Ф. Исследование комплек-
сообразования винной и метавинной кислот с металлами IV-VI групп
методом ЯМР низкого разрешения // Коорд. химия. 1979. Т. 5, №5.
С. 661-605.
35. Шафранскнй В.Н., Стратулат А. А., Дранка П.В. Спектры ПМР коор-
динационных соединений кобальта(Ш) с диметилглиоксимом и дифенил-
гуанидином//Ж. неорг. химии. 1981. Т. 26, №7. С. 1807-1811.
36. Kubicki М.М., Kergoat R., Guerchais J.E. et al. Biscyclopentadyenyl com-
plexes with transition metal-mercury bonds // J. Organomet. Chem. 1981.
V. 219, №3. P. 329-343.
37. Ларченко B.E., Григорьев А.И., Литвинова Л.А., Попов К.И. Изучение
комплексообразования иттрия с нитрилотриметилфосфоновой кислотой
методом ЯМР // Ж. неорг. химии. 1987. Т. 32, №3. С. 596-599.
38. Тарасов В.П., Бакум С. И., Привалов В. И. и др. Дейтон-протонный обмен
в алюмо- и галлогидридах // Коорд. химия. 1983. Т. 9, №7. С. 882-890.
39. Akitt J. VT., Elders J.M., Howarth О. W. Longitudinal relaxation of protons in
partially deuterated [A1(H2O)6]3+ // J.C.S. Faraday Trans. I. 1989. V. 85,
№ 8. P. 2035-2046.
40. Костромина H.A., Шелест В.П., Шевченко Ю.Б., Терновая Т.В. Иссле-
дование состояния ионов циркония и гафния в водных растворах методом
ЯМР Ц Укр. хим. ж. 1984. Т. 50, №9. С. 922-924.
41. Арсенин К.И., Малинко Л.А., Шека И.А. и др. Спектры ПМР водных
гидроксохлоридов циркония и гафния // Ж. неорг. химии. 1991. Т. 36,
№ 10. С. 2665-2671.
42. Наппапе S., Bertin Е, Bouix J. Etude de 1’hydrolyse de Zr(lV) par spectro-
metric Raman et RMN du proton // Bull. Sci. Chim. France. 1990. V. 124,
№ 1. P. 43-49.
43. Aberg M., Glaser J. 17O and 'H NMR study of the tetranuclear hydroxozir-
conium complexes in aqueous solution // Inorg. Chim. Acta. 1993. V. 206,
№ 1. P. 53-61.
44. Сазонов А.М. Связь самодиффузии и магнитной релаксации молекул в
жидкости // Ж. физ. химии. 1987. Т. 61, №4. С. 1053-1058.
45. Miura К., Hashimoto К., Fukui Н. et al. NMR Study of Hydration.
Anisotropic Rotation Motion of Water Molecule Bound to the Aluminium
Ion // J. Phys. Chem. 1985. V. 89, № 23. P. 5098-5101.
46. Arihchaiy M., Morris J.H. High Field (360 MHz) proton NMR spectra of
the octahydroborate (1-) ion, [ВзН8]_ and its monosubstituted ([ВзНуХ]-)
and disubstituted [ВзН8Х2]- derivatives // Inorg. Chim. Acta. 1985. V. 103,
№ 1. P. 31-35.
47. Remmel B.J., Johnson H.D., Javorivski I.S., Shore S.G. Preparation and
NMR studies of the stereochemically nonrigid anions B4H^, BsH^, B8H[j
and ByH“2 // J. Amer. Chem. Soc. 1975. V. 97, № 19. P. 5395-5403.
48. Stungis G.E., Rugheimer J.H. NMR of HBF4 // J. Chem. Phys. 1971. V. 55,
№ 1. P. 263-267.
49. Birchall T„ Pereira A. The NMR spectrum of the stannyl ion //
J.C.S. Chem. comm. 1972. №20. P. 1150-1151.
50. Alei M., Florin A.E. The nature of water species in water-ammonia solution
as inferred from proton and oxygen-17 NMR observations // J. Phys. Chem.
1969. V. 73, №4. P. 863-867.
51. Chizhik V.I. NMR relaxation and microstructure of aqueous electrolyte
solution // Mol. Phys. 1997. V. 90, №4. P. 653-659.
52. Harmon J.F., Suiter E.J. Molecular motion in supercooled liquides. Nuclear
magnetic relaxation of deuterons and protons in HM-aqueous lithium chlo-
ride // J. Chem. Phys. 1978. V. 82, № 17. P. 1938-1942.
53. Nakamuva K, Shimokawa S., Futamata K., Shimoji M. NMR relaxation of
water in concentrated zinc chloride solution // J. Chem. Phys. 1982. V. 77,
№ 6. P. 3258-3262.
54. Пронин И.С., Вашман А. А. Изучение комплексообразования в трибутил-
фосфатных экстрактах нитрата уранила с помощью ЯМР спектроскопии
2Н, l4N, 31Р // Ж. неорг. химии. 1987. Т. 32, №3. С. 728-731.
55. Rakiewicz E.F., Benesi A.J., Grutzeck M.W., Kwen S. Determination of
the State of Water in Hydrated Cement Phases Using Deuterium NMR
Spectroscopy // J. Amer. Chem. Soc. 1998. V. 120, №25. P. 6415-6416.
56. Kanzaki Y., Watanabe N., Maeda K., Matsumoto O. NMR Spectra of Wa-
ter Molecule Intercalated in Layered Transition-Metal Dichalcogenides //
J. Phys. Chem. 1987. V. 91, №11. P. 2727-2729.
57. Рипмейстер Дж. А., Ратклиф К. Вклад спектроскопии ЯМР в исследо-
вании клатратов // Ж. структ. химии. 1999. Т. 40, №5. С. 809-821.
58. Манк В.В., Лебовка Н.И. Спектроскопия ядерного магнитного резонанса
воды в гетерогенных системах. — Киев: Наукова думка, 1988. — 203 с.
59. Ceraso J.M., Dye J.L. 23Na NMR spectrum of the sodium anion // J. Chem.
Phys. 1974. V. 61, №4. P. 1585-1587.
60. Beckman A., Boklen R.D., Elke D. Precision Measurments of the Nuclear
Magnetic Dipole Moments of 6Li, 7Li, 23Na, 39K, 41К // Z. Phys. 1978.
Bd. 270. H.3. S. 173-186.
61. Hubburd P.S. Nonexponential nuclear magnetic relaxation by quadrupol
interaction // J. Chem. Phys. 1970. V. 53, №3. P. 985-987.
62. Bull T.E. Nuclear magnetic relaxation of spin 3/2 nuclei in chemical ex-
change // J. Magn. Reson. V. 8, № 3. P. 342-353.
63. Bull T.E., Forsen S., Tunner D.L. Nuclear magnetic relaxation of spin 5/2
and spin 7/2 including the effect of chemical exchange // J. Chem. Phys.
1979. V. 70, №6. P. 3106-3111.
64. Сликтер Ч. Основы теории магнитного резонанса / Пер. с англ. Н.Н. Кор-
ста и др.; Под ред. Г. В. Скроцкого. — М.: Мир, 1981. — 334 с.
65. Валиев К. А. К теории квадрупольнои релаксации ядерных спинов //
Ж. эксперим. теор. физики. 1960. Т. 38, №4. С. 1222-1232.
66. Валиев К.А., Хабибуллин В.М. Ядерный магнитный резонанс и структура
водных растворов // Ж. физ. химии. 1961. Т. 35, № 10. С. 2265-2274.
67. Валиев К.А. Исследование растворов электролитов в дипольной жид-
кости методом магнитного резонанса. Теория магнитной релаксации //
Ж. структ. химии. 1962. Т. 3, №6. С. 653-661.
68. Hertz H.G. Magnetic relaxation by quadrupole interaction of ionic nuclei in
electrolyte for infinite dilution // Ber. Bunsenges. phys. Chem. 1973. Bd. 77,
№7. S. 531-540.
69. Hertz H.G., Holz M., Keller G. et al. Nuclear magnetic relaxation of alkali
metal ions in aqueous solutions // Ber. Bunsenges. phys. Chem. 1974. Bd. 78,
№ 5. S. 493-509.
70. Deverell C. Nuclear magnetic shielding of hydrated alcali and halide ions in
aqueous solutions // Mol. Phys. 1969. V. 16, №4. P. 491-500.
71. Holz M., Weingarther H., Hertz H.G. Nuclear magnetic relaxation of al-
kali halide nuclei and preferential solvation in methanol-water mixtures //
J.C.S. Faraday tr. I. 1977. V. 73, № 1. P. 71-83.
72. Delville A., Detellier A., Gerstmans A., Laszlo P. The theoretical interpreta-
tion of sodium-23 NMR chemical shifts and quadrupolar coupling constants,
as supported by new experimented evidence // J. Magn. Reson. 1981. V. 42,
№ 1. P. 14-27.
73. Laszlo P. Sodium-23 nuclear magnetic spectroscopy // Angew. Chem. Int.
ed. Engl. 1978. V. 17, №4. P. 254-260.
74. Annual reports on NMR spectroscopy. V. 9/ Ed. G.A. Webb. — L.: Acad.
Press, 1979. P. 126-216.
75. Cohen Y.M., Dye J.L., Popov A. I. Lithium-7 NMR study of lithium cryptates
in various solvents // J. Phys. Chem. 1975. V. 79, № 13. P. 1489-1491.
76. Bloor EG., Kidd R.G. Solvation of sodium ions studied by 23Na NMR //
Can. J. Chem. 1968. V. 46, №22. P. 3425-3430.
77. Lutz O. Kernresonanzuntersuchungen an Elektrolytlosungen. I. Der Einfluss
von Anionen und paramagnetischen Kationen auf die Cs133 Kernresonan-
zlinie // Z. Naturforsch. 1967. Bd. 22A, №2. S. 286-288.
78. Deverell C., Richards R.E. Nuclear magnetic resonance studies of alkali
metal halide solutions. I. Cation resonanse // Mol. Phys. 1966. V. 10, №6.
P. 551-564.
79. Deverell C., Richards R.E. Nuclear magnetic resonance studies of alkali
metal halide solutions. II. Anion resonance // Mol. Phys. 1969. V. 16, №5.
P. 421-439.
80. Halliday J.D., Richards R.E., Sharp R.R. Chemichai shifts in nuclear reso-
nances of caesium ions in solutions // Proc. Roy. Soc. Lond. 1969. V. A313.
P. 45-69.
81. Мирная T.A., Яремчук Г. Г., Трачевский В. В. Ядерный магнитный ре-
зонанс 133Cs н-алканатов Cs // Ж. неорг. химии. 1991. Т. 36, №5.
С. 1269-1272.
82. Максимовская Р.И., Федотов М.А., Максимов Г.М. Взаимодействие ге-
терополианиона PWnO^, с одновалентными катионами по данным ЯМР
разных ядер // Ж. неорг. химии. 1985. Т. 30, №4. С. 918-924.
83. Fedotov М.А. NMR tools for detection of colloids and polynuclear species //
Abstracts of NATO ARW. — St. Peterburg, 2001. P. 88.
84. Троицкий С.Ю., Федотов M.A., Новгородов Б.Н. и др. Исследование
процесса формирования нанометровых частиц Ru(IlI) // Ж. структ. химии.
2007. Т. 48, № 1. С. 143-147.
85. Мазитов Р.К., Самойлов О.Я., Брюшкова Н.В. и др. Температурная
зависимость времени спин-решеточной релаксации ядер 7Li в водном
растворе хлорида лития // Ж. структ. химии. 1975. Т. 16, № 4. С. 546-567.
86. Maciel G.E., Hancock J. К., Lafferty L.F. et al. Lithium-7 chemical shifts of
lithium perchlorate and bromide in various solvents // Inorg. Chem. 1966.
V. 5, № 4. P. 554-557.
87. Popov A. I. Multinuclcar NMR studies of alkali ions in nonaqueous solvents //
Pure AppL Chem. 1979. V. 51, № 1. P. 101-110.
88. Sutter E. J., Harmon J. F. Molecular motion in supercooled liquids. Pulsed
NMR of 7Li in ИМ aqueous lithium chloride // J. Phys. Chem. 1975. V. 79,
№8. P. 1978-1961.
89. Sutter E.J., Updegrove D.M. Hydration number of Li ion by pulsed magnetic
resonance // Chem. Phys. Lett. 1975. V. 36, № 1. P. 49-50.
90. Hirata F., Friedman H.L, Holz M., Hertz H.G. NMR relaxation study of
lithium ion-nikel(2+) ion interaction in aqueous solutions // J. Chem. Phys.
1980. V. 73, № 12. P. 6031-6038.
91. Мишустин А. И., Подковырин Л. И., Кесслер Ю.М. Механизм обмена
молекул основных растворителей у катионов лития и натрия по данным
спин-решеточной релаксации ядер ионов // Коорд. химия. 1980. Т. 6, №2.
С. 236-242.
92. Chen D.M., Reeves L. U7 Sodium magnetic resonance in lyotropic nematic
phases and implications for observation in living systems // J. Amer. Chem.
Soc. 1972. V. 94, № 12. P. 4384-4386.
93. Porion P., Faugere M.P., Gherardi B., Delvielle A. 23Na Nuclear Quadrupo-
lar Relaxation as a Probe of the Microstructure and Dynamics of Aqueous
Clay Dispersion: An Application to Laponite Gels // J. Phys Chem. 1998.
V. B102, № 18. P. 3477-85.
94. Eisenstadt M., Friedman H.L. Nuclear magnetic relaxation in ionic solution.
Relaxation of 23Na in aqueous solutions of vaious diamagnetic salts //
J. Chem. Phys. 1967. V. 46, №6. P. 2182-2193.
95. Eisenstadt M., Friedman H.L. Nuclear magnetic relaxation in ionic solution.
Relaxation of 23Na in aqueous solutions of NaCl and NaClO^ // J. Chem.
Phys. 1966. V. 44, №4. P. 1407-1415.
96. O’Reilly D.E., Peterson E.M. Relaxation rates of alkali metals and chloride
nuclei in aqueous solution. Quadrupole moment of 133Cs // J. Chem. Phys.
1969. V. 51, № 11. P. 4906-4908.
97. Hall C., Richards R.E., Schulz G.N., Sharp R.R. Magnetic relaxation of
23Na and 81 Br in aqueous solutions of the alkali halides // Mol. Phys. 1969.
V. 16, №6. P. 529-554.
98. Ионов В. И., Мазитов Р.К., Самойлов О.Я. Зависимость ближней гидра-
тации высаливаемого катиона от гидратации катионов, высаливателя по
данным ЯМР // Ж. структ. химии. 1969. Т. 10, № 3. С. 407-410.
99. Чижик. В. И., Ермаков Ю.А. Квадрупольная релаксация ядер ионов лития
и натрия в водных растворах электролитов // В кн.: Ядерный магнитный
резонанс. Вып. 4. — Л.: ЛГУ, 1971. С. 60-65.
100. Speight Р. A., Armstrong R.L. Spin-relaxation in solutions of electrolytes.
Relaxation of 7Li and 23Na in aqueous solutions of alkali halides // Can.
J. Chem. 1967. V. 45, №8. P. 2493-2503.
101. Wertz J. E., Jardetzki O. Nuclear spin resonance in aqueous sodium ion //
J. Chem. Phys. 1956. V. 25, № 2. P. 357-358.
102. Templeman G.L, Van Geet A.L. Sodium magnetic resonance of aqueous salt
solutions // J. Amer. Chem. Soc. 1972. V. 94, № 16. P. 5578-5582.
103. Мальцев Г.З., Малинин Г.В., Машовец В.П. О структуре алюминатных
растворов // Ж. структ. химии. 1965. Т. 6, №3. С. 378-383.
104. Moollenar R.J., Evans J.C., McKeever C.D. The structure of the aluminate
ion in solution at the high pH // J. Phys. Chem. 1970. №20. P. 3629-3636.
105. Lacassagne V, Bessada C., Florian P. et al. Structure of High-Temperature
NaF-AlFa-A^Os Melts: a Multinuclear NMR Study // J. Phys. Chem. 2002.
V. 106B, №8. P. 1862-1869.
106. Schmidt E., Popov A. I. Exploratory Studies of Some Molten Potassim Salts
by Potassium-39 NMR // Spectr. Lett. 1981. V. 14, № 11-12. P. 787-794.
107. Boss R.D., Popov A.L Competitive NMR study of the complexation of
several cations with 18-crown-6, l,10-dibenzo-18-crown-6, and cryptand-222
in nonaqueous solutions // Inorg. Chem. 1986. V. 25, № 11. P. 1747-1750.
108. Kintzinger J.P., Lehn J.M. 23Na quadrupole coupling constants in different
coordination shells from 23Na and 13C Fourier transform NMR measurments
in sodium cryptates // J. Amer. Chem. Soc. 1974. V. 96, № 10. P. 3313-3314.
109. Shporer M., Luz Z. Kinetic of complexation of potassium ion with
dibenzo-18-crown-6 in methanol by 39K nuclear magnetic resonance //
J. Amer. Chem. Soc. 1975. V. 97, №3. P. 665-666.
110. Khazareli S., Dye J.L., Popov A.L Rubidium-87 NMR Studies of Rubidium
Salts and Complexes with 18-Crown-6 and Cryptand-222 in Solutions //
Spectrochim. Acta. 1983. V. 39A, № 1. P. 19-21.
111. Mei E., Popov A.L, Dye J.L. Cesium-133 NMR study of complexation by
cryptand C222 in various solvents: evidence for exclusive and inclusive
complexes // J. Amer. Chem. Soc. 1977. V. 99, №20. P. 6532-6536.
112. Mei E., Lin L., Popov A.L Determination of stability constants of cesium
2-cryptand complexes in nonaqueous solvents by cesium-133 NMR // J. So-
lut. Chem. 1977. V. 6, № 11. P. 771-778.
113. Rounaghi G., Popov A.I. l33Cs NMR study of the cryptand 222-Cs+ complex
in binary solvent mixtures // Polyhedr. 1986. V. 5, № 12. P. 1935-1939. Ibid.
1986. V. 5, №8. P. 1323-1333.
114. Wehrly F. W. Nuclear Magnetic Resonance of the Less Common Quadrupolar
Nuclei // Ann. Reports on NMR Spectroscopy. 1978. V. 9. — L.: Acad. Press,
1979. P. 126-216.
115. Ellaboudy A.S., Pyper N.C., Edvards P.P. First Simultaneous Observation
of the 133Cs NMR Cs+, Cs- and Cs+e in a Metal Solution // J. Amer. Chem.
Soc. 1988. V. 110, №5. P. 1618-1620.
116. Riddell F.G., Arumugam S., Patel A. Nigericin-mediated transport of cesium
ion through phospholipid bilayers studied by a 133Cs magnetization transfer
NMR technique // Inorg. Chem. 1990. V. 29, № 13. P. 2397-2398.
117. Kovar R.A., Morgan G.L. Berilliuim-9 and hydrogen-1 magnetic resonance
studies of berillium compounds in solution // J. Amer. Chem. Soc. 1970.
V. 92, № 17. P. 5067-5072.
118. Bryant R.G. Magnesium-25 NMR in aqueous phosphate solutions //
J. Magn. Reson. 1972. V. 6, №2. P. 159-166.
119. Forsen S., Lindman B. Calcium and Magnesium NMR in Chemistry and
Biology. Annual reports on NMR spectroscopy. V. Ila / Ed. G.A. Webb. —
L.: Acad. Press, 1981. P. 183-226.
120. Fedotov M.A., Gerasimova G.F. 25Mg and 17O in MgO and Ni^Mgi-^O
solid solutions // React. Kinet. Catal. Lett. 1983. V. 22, № 1-2. P. 113-117.
121. James D. W., Cutler P.G. Ion-Ion Interaction in Solution. Group 2. Perchlo-
rates in Water // Aust. J. Chem. 1986. V. 39, № 1. P. 137-147.
122. Lutz O., Oehler H. Barium-135 and -137 Fourier transform NMR and NQR
studies // Z. Phys. 1978. Bd. 288a, № 1. S. 11-15.
123. Holz M., Guenter S., Lutz O. et al. Magnesium-25 (2+) ion nuclear mag-
netic relaxation in aqueous solution // Z. Naturforsch. 1979. Bd. 34a, №8.
S. 944-949.
124. Lutz O., Schwenk H., Ohl H. Fourier transform magnetic resonance studies
of25Mg and 43Ca // Z. Naturforsch. 1975. Bd. 30a, №9. S. 1123-1127.
125. Simeral L., Maciel G.E. Fourier transform 25Mg NMR study of aqueous
magnesium (II) electrolytes // J. Phys. Chem. 1976. V. 80, №5. P. 552-557.
126. Heubel P.H., Popov A. I. Magnesium-25 NMR studies of magnesium salts
and complexes in nonaqneous solvents // J. Soln. Chem. 1979. V. 8, №4.
P. 283-291.
127. Lindman B., Forsen S., Lilja H. Quadrupole relaxation ot aqueous alkaline
earth ions // Chem. Scripta. 1977. V. 11, №2. P. 91-92.
128. Bouhoutsos-Brown E., Bryant R.G. Magnesium-25 NMR in melhanol-water
solutions // J. Inorg. NucL Chem. 1982. V. 43, № 12. P. 3213-3215.
129. Wehrli F.W., Wehrli S.L. Solvation of berilliurn chloride in acetonitrile
studied by high-field berillium-9 NMR // J. Magn. Reson. 1982. V. 47, № 1.
P. 151-156.
130. Helm L., Hertz H.G. The hydration of the alkaline earth metal ions Mg2+,
Ca2+, Sr2+ and Ba2+, a NMR study, involving the quadrupole moment of the
ionic nuclei // Z. phys. Chem. (BRD). 1981. Bd. 127, № 1. S. 23-44.
131. Drakenberg T. Calcium-43 NMR relaxation times and quadrupole coupling
constants for some small calcium complexes // Acta Chem. Scand. 1982.
V. 36a. № 1. P. 79-82.
132. Bouhoutsos-Brown E., Murk-Rose D., Bryant R.G. High resolution alkaline
earth NMR // J. Inorg. NucL Chem. 1981. V. 43, № 10. P. 2247-2278.
133. Eilenberg M., Villemin M. Resonance magnetique nucleare le deplacement
chimique de la raie du magnesium 25 // Compt. Rend. Acad. Sci. 1968.
V. 266b. №23. P. 1430-1433.
134. Banc K.J., Schwenk A. 87-strontium NMR studies // Z. Phys. 1973. Bd. 265,
№2. S. 165-171.
135. Lindman B., Forsen S. Chlorine, bromine and iodine NMR. NMR Basic
Principles and Progress. V. 12. — Berlin: Springer Verlag, 1976. — 365 p.
136. Kondo J., Yamashita J. Nuclear quadrupolar relaxation in ionic crystals //
J. Phys. Chem. Solids. 1959. V. 10, №4. P. 245-253.
137. Kotz ]. C., Schaffer R., Clouse A. High resolution NMR spectroscopy of some
berillium-containing compouds. Berillium-9 and fluorine-19 spectra // Inorg.
Chem. 1967. V. 6, №3. P. 620-622.
138. Benn R., Rufinska A. High-Resolution Metal-NMR Spectroscopy of
Organometallic Compounds // Angew. Chem. Int. Ed. Engl. 1986. V. 25,
№ 10. P. 861-881.
Бор
139. Noth H., Wracmeyer B. Nuclear magnetic resonance spectroscopy of boron
compounds. NMR Basic Prine, and Progr. V. 14. — Berlin: Springer Verlag,
1978. - 481 p.
140. Siedle A.R. Ann. reports on NMR spectroscopy. V. 12 / Ed. G. A. Webb. —
L.: Acad. Press, 1982. P. 177-182.
141. Kennedy J. W. Boron. Multinuclear NMR / Ed. J. Mason. — N.Y.-L.:
Plenum Press, 1987. P. 221-58.
142. Thompson R.G., Davis J.C. Electronegativity effects on 11В chemical shifts
in tetrahedral BX^ ions // Inorg. Chem. 1975. V. 4, № 10. P. 1464-1467.
143. Kroner J., Wracmeyer B. HB-chemical shifts of diboranes, polyboranes,
carboranes and coupling constants ^("B-'H), и ^(^B-1^) //J.C. S. Fara-
day tr. II. 1976. V. 72, № 12. P. 2283-2290.
144. Spielfogel B., Nutt W.R., Izidore R.A. Correlation between carbon-13 and
boron-11 chemical shifts. Carbenium ions and their trigonal boron analogs //
J. Amer. Chem. Soc. 1975. V. 97, №6. P. 1609-1610.
145. Чернышов Б.Н., Васева Ю.А., Бровкина О.В. Исследование пероксобо-
ратных комплексов в водных и водно-ацетоновых растворах пероксобората
методом ЯМР НВ // Ж. неорг. химии. 1991. Т. 36, № 2. С. 460-463.
146. Shore S.G., Ragani J.D., Smith R.L. et al. Preparation and NMR spectra of
B5HgFe(CO)3, an analog hexaboranes (10) // Inorg. Chem. 1979. V. 18, №3.
P. 670-673.
147. Shaeffer R., Shoolery J.N., Jones S. Structures of halogen substituted bo-
ranes // J. Amer. Chem. Soc. 1958. V. 80, № 11. P. 2670-2673.
148. Бакум С.И., Болотина И.В., Кузнецова С.Ф. Изучение взаимодействия
борогидридов щелочных металлов с галогенидами алюминия в среде орга-
нического растворителя методом ЯМР "В // Ж. неорг. химии. 1994. Т. 39,
№7. С. 1173-1183.
149. Mancini М., Boudcard Р., Burns Q.C. et al. Bounding in Transition-Metal
Tetrabotates: A Multinuclear Magnetic Resonance Study of (CsHsJgScfB^)
and Sc(BH4)3 and Some Comments on Isolobality of BH^“, Halide and
7?5-C5H5 Groups // Inorg. Chem. 1984. V. 23, №8. P. 1072-1078.
150. Бойко Г.Н., Кравченко C.E., Семененко K.H. ЯМР-исследование химиче-
ского обмена и квадрупольной релаксации в Y(BH4)3-2C4H3O // Изв. АН
СССР. Сер. хим. 1981. №6. С. 1199-1207.
151. Rath N.P., Fehlner Т.Р. Interaction Iron with Boron in Metal-rich Metallob-
oranes Resulting in Large Deshilding and Rapid Relaxation Processes of the
Boron-11 Nucleus // J. Amer. Chem. Soc. 1988. V. 110, № 16. P. 5345-5349.
152. Srebny H.-G., Preetz W., Marsmann H.C. Darstellung HB NMR- und
Schwingung-spektren isomerenreiner halogenhydrododecaborate ХпВцН|^_п;
X = Cl, No = 1-3; X = Br, No = 1, 2; X = I, No = 1 // Z. Naturforsch.
1984. Bd. 39B, № 1. S. 6-11. Ibid. №2. S. 189-196.
153. Epperlein B. W., Lutz O., Schwenk A. Fourier-kernresonanzuntersuhungen
an 10B und "B in wassrigen losung // Z. Naturforsch. 1975. Bd. 30a, №8.
S. 955-958.
154. Akitt J. W. Multinuclear NMR studies of aqueous solutions of tetrafluorobo-
rate salts // J.C.S. Faraday tr. I. 1975. V. 71, №7. P. 1557-1572.
155. Janda R., Heller G. Boron-11 NMR spectroscopic studies on aqueous
polyborate solutions // Z. Naturforsch. 1979. Bd. 34b, №7. S. 1078-1083.
156. Baltz R., Brdndle U., Kammerer E. et al. nB and 10B Investigation in
Aqueous Solutions // Z. Naturforsch. 1986. Bd.41A, №5. S. 737-742.
157. Selentane C.G. High Field “B NMR of Alkali Borates. Aqueous Polyborane
Equilibria // Inorg. Chem. 1983. V. 22, № 26. P. 3920-3924.
158. Бровкина О.В., Чернышов Б.Н. Образование перекисных комплексов бор-
углеродного состава по данным ЯМР пВ, l3C, l9F // Ж. неорг. химии.
1989. Т. 34, №2. С. 299-303.
159. Dove M.F., Hibbert R.C., Norman L. Reactions of Boron Compounds
with Difluorophosphoric Acid; Characterization of the Tetrakis(difluorophos-
phato)borate Anion by Multinuclear ("B, l9F, 31P) NMR Spectroscopy //
J.C.S. Dalton tr. 1984. №12. P. 2719-2721.
160. Костромина H.A., Крятова О.П., Третьякова H.H., Трачевский В. В.
Биядерные комплексы бора с оксиэтилидендифосфоновой кислотой //
Ж. неорг. химии. 1997. Т. 42, №3. С. 420-425.
161. Hosmane N.S., Deghan M., Davies S. Significance of the Trimethylsi-
lyl Moiety in Sinthetic Carborane Transformation: Conversion of
nido-[(CH3)3Si]2C2B4H6 to п16о-[(СНз)з51]2С2ВюНю by Thermal Elimina-
tion of Trimethylsilane, an Important Advance in Carborane Chemistry //
J. Amer. Chem. Soc. 1984. V. 106, №21. P. 6435-6436.
162. Lutz 0., Veil E., Brandie U. et al. HB NMR Investigation of Borate Esters
in Aqueous Solutions and Wine // Z. Naturforsch. 1986. V. 41A, №8.
S. 1041-1044.
163. Wang J.S., Geanangel R.A. HB NMR Studies of the Thermal Decomposition
of Ammonia-Borane in Solution // Inorg. Chim. Acta. 1988. V. 148, №2.
P. 185-190.
Углерод
164. Леви Г., Нельсон Г. Руководство по ядерному магнитному резонансу уг-
лерода-13/ Пер. с англ. Н.М. Сергеева. — М.: Мир, 1975. — 296 с.
165. Mann В.Е. The Common Nuclei. NMR and Periodic table Ed. В. E. Mann,
R. K. Harris. — L.: Acad. Press, 1978. P. 87-105.
166. Mann B.E. Carbon. Multinuclear NMR / Ed. J. Mason. — N. Y.-L.: Plenum
Press, 1987. P. 293-303.
167. Mann B.E., Taylor B.F. 13C NMR data for organometallic compounds. —
L.: Acad. Press, 1981. — 326 p.
168. Spectroscopic properties of inorganic and organometallic compounds.
V. 11. - L.: Acad. Press, 1979. P. 1-157.
169. Mann B.E., Taylor B.F. 13C NMR data for organometallic compounds //
J. Magn. Reson. 1982. V. 47, №3. P. 539-543.
170. Heaton В. T, Strona L. Carbon-13 NMR studies of rhodium carbonyl clusters
on ressurization with CO/H2 // J.C.S. Dalton tr. 1982. №6. P. 1159-1164.
171. Bramley R., Figgis B.N., Nyholm R.S. I3C and 17O NMR spectra of
metal carbonyl compounds // Tr. Faraday Soc. 1962. V. 58, №478(10).
P. 1893-1896.
172. Rasul D., Prakash R. V, Zdunek L.Z. et al. Carbonic Acid and Its Mono- and
Diprotonation: NMR, ab initio, and IGLO Investigation // J. Amer. Chem.
Soc. 1993. V. 115, №6. P. 2236-2238.
173. Stocton G. W., Martin J.S. A carbon-13 NMR study of cation solvation in
alcohols // Can. J. Chem. 1974. V. 52, № 5. P. 744-748.
174. Hammel J.C., Smith J.A.S. Electric field effects in NMR spectroscopy. Sub-
stituted /3-diketone complexes//J. Chem. Soc. A. 1970. № 11. P. 1855-1859.
175. Беляев А.В., Венедиктов А.Б., Федотов M.A., Храненко С.П. Иссле-
дование взаимодействия хлоридов родия(Ш) с ацетилацетоном // Коорд.
химия. 1985. Т. 11, №6. С. 794-804.
176. Pregosin P.S., Kunz R. W. 31P and 13C NMR of Transition Metal Phosphine
Complexes. NMR Basic Principles and Progress. V. 16 / Ed. P. Diehl,
E. Fluck, R. Kosfeld. — Berlin: Springer Verlag, P. 90-144.
177. Molien F., Helm L., Abou-Hamdan A., Merbach A.E. Mechanistic Di-
versity Covering 15 Orders of Magnitude in Rates: Cyanide Exchange
on [M(CN)4]2“ (M = Ni, Pd, Pt) // Inorg. Chem. 2002. V.41, №7.
P. 1717-1727.
178. Федоров Л.А., Родехузер Л., Дельпеш Дж.Дж. и др. Раздельное наблю-
дение комплексов двух- и трехзарядных катионов непереходных металлов
в их смесях в растворе // Ж. неорг. химии. 1993. Т. 34, № 1. С. 140-143.
179. Pesek J.J., Mason W.R. Carbon-13 Magnetic Resonance Spectra of Diamag-
netic Cyano Complexes // Inorg. Chem. 1979. V. 18, №4. P. 924-928.
180. Blixt J., Gyori B., Glaser J. Determination of Stability Constants for Thal-
lium(lll) cyanide complexes in aqueous Solution by Means of l3C and 2O5T1
NMR//J. Amer. Chem. Soc. 1989. V. Ill, №20. P. 7784-7791.
181. Taure T. I3C and HN Nuclear Magnetic Resonance of K4[Re(CN)7]-2H2O and
Some Related Cyano Metal Complexes // Inorg. Chim. Acta. 1989. V. 163,
№2. P. 131-132.
182. Dunham S.O., Larsen P.L., Abbott E.H. Nuclear Magnetic Resonance In-
vestigation of the Hydrogen Peroxide Oxidation of Platinum(II) Complexes //
Inorg. Chem. 1993. V. 32, № 10. P. 2049-2055.
183. Agnew N.H., Appleton T. G., Hall J.R. Multinuclear NMR Study of Some Di-
and Trimethylplatinum(IV) Complexes // Aust. J. Chem. 1982. V. 35, №5.
P. 881-894.
184. Shen L.-F., Du Yu.-R., Shao Q.-F., Mao Sh.-Z. I3C, 17O and *H NMR Study
of the Protonation of Acetone in Water // Magn. Reson. Chem. 1987. V. 25,
№ 7. P. 575-578.
185. Матвеев В. В. Комплексы никеля(П) с ацетат-ионом в уксусной кислоте
по данным ЯМР // Коорд. химия. 1994. Т. 20, № 7. С. 516-517.
Азот
186. Annual Reports on NMR Spectroscopy. V. 1 IB / Ed. G. A. Webb. — L.: Acad.
Press, 1981. — 502 p.
187. Mason J. Nitrogen NMR in Inorganic, Organometallic and Bioinorganic
Chemistry // Chem. Revs. 1981. V. 81, № 3. P. 205-227.
188. Mason J. Nitrogen. Multinuclear NMR / Ed. J. Mason. — N.Y.-L.: Plenum
Press, 1987. P. 335-367.
189. Гордон А., Форд P. Спутник химика / Пер. с англ. Е.Л. Розенберга,
С. И. Коппель. — М.: Мир, 1976. — 541 с.
190. Эмсли Дж., Финей Дж., Сатклиф Л. Спектроскопия ЯМР высокого
разрешения. Т. 2 / Пер. с англ. Ю. С. Константинова и др.; Под ред.
В. Ф. Быстрова, Ю. Н. Шейнкера. — М.: Мир, 1968. — 468 с.
191. Федотов М.А. Ядерный магнитный резонанс донорных атомов как ин-
струмент для определения строения комплексов платиновых металлов
в растворах // Изв. РАН. Сер. хим. 2003. № 4. С. 743-755.
192. Buchanan G. W. Application of 15N Spectroscopy to the Study of Molecular
Binding Phenomena // Tetrahedron. 1989. V. 45, №3. P. 581-604.
193. Беляев А.В., Федотов M.А., Венедиктов А.Б., Храненко С.П. Сб. Бла-
городные металлы: химия и технология. Ч. 2 / / Под ред. В. Г. Торгова,
Ф. А. Кузнецова. — Новосибирск: Ин-т неорган. химии, 1989. С. 5-54.
194. Wrackmreyer В. 14N NMR Spectroscopy of N-chloroamines // Spectrochim-
ica Acta. 1987. V. 43A, №9. P. 1187-1188.
195. Anderson L-О., Mason J. Oxigen-17 Nuclear Magnetic Resonance.
Oxygen-Nitrogen Groupings // J.C.S. Dalton tr. 1974. №2. P. 202-205.
196. Mason J., Larkworthy L.F., Moore E.A. Nitrogen NMR Spectroscopy of
Metal Nitrosil and Related Compounds // Chem. Revs. 2002. V. 102, №4.
P. 913-934.
197. Лагутенков В.В., Скороваров Д.И., Ульянов В. С. и др. Связь между
параметрами специфической сольватации и химическим сдвигом ЯМР 14N
ионов NO^“ в водных растворах электролитов // Ж. физ. химии. 1986.
Т. 60, №5. С. 1299-1301.
198. Prakash G.K. S., Heider L., Olah G.A. I5N NMR Spectroscopic Investigation
of Nitrous and Nitric Acids in Sulfuric Acid Solution of Varying Activities //
Inorg Chem. 1990. V. 29, № 24. P. 4965-4968.
199. Федотов M. А. Многоядерный ЯМР нитро-1,2,4-триазолов // Abstr VIII
Int. Workshop on Magn. Reson (Spectroscopy, Tomography and Ecology).
Rostov, Don. Sept. 11-16. 2006. P. 196.
200. Witanowski M., Stefaniak L., Webb G.A. Nitrogen-14 Line Width as an Aid
in Nitrogen Chemical Shifts Assignments // J. Magn. Reson. 1979. V. 36,
№2. P. 227-237.
201. Bromley R., Figgis B.N., Nyholm R.S. Nuclear magnetic resonance in tran-
sition metal complexes // J. Chem. Soc. A. 1967. №6. P. 861-863.
202. Lehman J. W., Fung B.M. Nitrogen-15 chemical shifts in cobalt(III) penthane
complexes // Inorg. Chem. 1972. V. 11, № 1. P. 214-215.
203. Alei M.J., Vergamini P.J., Wageman W.E. Nitrogen-15 NMR of
cis-diamino-platinum(II) complexes in aqueous solutions // J. Amer. Chem.
Soc. 1979. V. 101, № 18. P. 5415-5417.
204. Беляев А. В., Венедиктов А.Б., Федотов M. А., Храненко С.П. Комплекс-
ные нитриты родия(Ш) в водн. растворах // Коорд. химия. 1986. Т. 12,
№ 5. С. 690-699.
205. Венедиктов А.Б., Федотов М.А., Коренев С.В., Беляев А.В. Исследо-
вание процессов нитрования гексахлорокомплексов иридия методом ЯМР
I5N // Коорд. химия. 1989. Т. 15, №4. С. 556-560.
206. Храненко СП., Федотов М.А., Беляев А.В. Изучение превращений ком-
плексов [Rh(NO2)IClyH2O6-I-y]3^I-w методом ЯМР // Коорд. химия.
1990. Т. 16, №7. С. 991-996.
207. Федотов М.А., Коренев С.В., Беляев А.В. Изучение взаимопревращений
[PtCln(NO2)4-n]2- методом ЯМР 195Pt и 15N // Коорд. химия. 1990. Т. 16,
№9. С. 1272-1276.
208. Венедиктов А. Б., Коренев С.В., Федотов М.А., Беляев А.В. ЯМР спек-
троскопическое исследование промежуточных продуктов нитрования гек-
сахлоро комплексов иридия (IV) и (III) // Коорд. химия. 1990. Т. 16, № 10.
С. 1400-1405.
209. Храненко С.П., Федотов М.А., Беляев А. В. Синтез и исследование
TpaHC-K3[Rh(NO2)4C12] // Коорд. химия. 1991. Т. 17, №6. С. 860-863.
210. Емельянов В.А., Федотов М.А., Беляев А.В. Исследование обратимых
нитро-нитрозо превращений в комплексах рутения(П) методом ЯМР 15N,
|7О, 99Ru // Ж. неорг. химии. 1993. Т. 38, № 11. С. 1842-1848.
211. Коренев С. В., Венедиктов А. Б., Малкова В. И., Федотов М. А. Синтез
и свойства тринитротриаква комплекса иридия(Ш) и его производных //
Коорд. химия. 1999. Т. 25, №9. С. 696-698.
212. Федотов М.А., Федотова Т.Н., Голованева И.Ф. Координатные сдвиги
,4N аминолигандов в комплексах Pt(IV) и их использование для опреде-
ления состава и геометрического строения комплексов // Ж. неорг. химии.
1997. Т. 42, №6. С. 1003-1008.
213. Федотов М.А. Ядерный магнитный резонанс донорных атомов как ин-
струмент для определения строения комплексов платиновых металлов
в растворах // Изв. АН. Сер. хим. 2003. № 4. С. 743-755.
214. Litchman W.M., Alei М., Florin А.Е. A Study of Nitrogen-15 NMR shifts
in Liquid 15ИНз — Solvent Mixtures // J. Amer. Chem. Soc. 1969. V. 91,
№ 24. P. 6574-6579.
215. Hagen R., Warren J. P., Hunter D.H., Roberts J. D. 15N and 13C Spectra
of Complexes of Ethylenediaminetetraacetic Acid (EDTA) with Closed-Shell
Metal Ions // J. Amer. Chem. Soc. 1973. V. 95, № 17. P. 5712-5716.
216. Федотов M.A., Беляев А.В. Прямое наблюдение инертных нитратоком-
плексов платиновых металлов методом ЯМР 14,I5N в водном растворе //
Коорд. химия. 1994. Т. 20, №8. С. 613-615.
217. Chew K.F., Healy М.А., Khalil M.I., Logan L. Nitrogen-14 NMR study of
some diamagnetic covalent metal nitrates // J.C.S. Dalton tr. 1975. № 13.
P. 1315-1318.
218. Howarth O.W., Richards R.E., Venanzi L.M. The l4N chemical shifts in
metal-thiocyanate complexes // J. Chem. Soc. 1964. №9. P. 3335-3337.
219. Bohland H., Miihle E. 14N-NMR-Untersuchungen an einigen diamagnetis-
chen Metall- und Triphenylzinnthiocyanatverbindungen sowie Kaliumseleno-
cyanat // Z. anorg. allg. Chem. 1970. Bd. 379, №2. S. 273-278.
220. Pregosin P.S., Strett H, Venanzi L.M. 15N, 31P and 195Pt studies of thio-
cyanate complexes // Inorg. Chim. Acta. 1980. V. 8, №2. P. 237-242.
221. Schprorer M., Ron G., Loevenstein A., Navon G. Study of some cyanometal
complexes by NMR. Chemical shifts and line width of l4N and 13C reso-
nances // Inorg. Chem. 1965. V. 4, № 3. P. 558-361.
222. Chew K.F., Derbishire W, Logan N. I4N chemical shifts of isocyanides and
cyanates // J.C.S. Chem. Comm. 1970. №24. P. 1708.
223. Мищенко А.В., Федоров В.E., Колесов Б.А., Федотов М.А. Синтез и
свойства биядерных халькогенидных комплексов ниобия // Коорд. химия.
1989. Т. 15, №2. С. 200-204.
224. Mason L, Larkworthy L.F., Moore Е.А. Nitrogen NMR Spectroscopy of
Metal Nytrosil and related compounds // Chem. Revs. 2002. V. 102, №4.
P. 913-934.
225. Lehnig M., Kirsch M., Korth H.-G. I5N CIDNP Study of Formation and
Decay of Peroxonitric Acid: Evidence for Formation of Hydroxyl Radicals //
Inorg Chem. 2003. V. 42, № 14. P. 1275-1287.
226. Chivers T., McIntyre D.P., Schmidt K.J., Vogel H.J. I4N and 15N NMR
Characterization and the Identification in Sulphur-Ammonia Solution of the
S7N- Ion // J.C.S. Chem Comm. 1990. № 19. P. 1341-1342.
227. Chivers T., Smith K. Characterization of the ЗгИгН- Ion by 14N and 15N
NMR Spectroscopy // J.C.S. Chem Comm. 1990. № 19. P. 1342-1344.
228. Jiang J., Renshaw J. C., Sarsfield M.J. et al. Solution Chemistry of Uranyl Ion
with Iminodiacetate and oxydiacetate: A Combined NMR/EXAFS and Poten-
tiometry/Calorimetry Study // Inorg. Chem. 2003. V. 42, № 4. P. 1233-1240.
229. McGarvey B.R., Kurland R.J. NMR of paramagnetic molecules / Ed.
G. N. La Mar, W. Horrock, R. N. Holm. — N. Y.: Acad. Press, 1973. — 678 p.
230. Schwarzhans K.E. NMR-Spectroskopie an paramagnetische Kom-
plexverbindungen // Angew. Chem. 1970. Bd. 82, №24. S. 975-982.
231. Молин Ю.Н., Счастнев П.В., Чувылкин Н.Д. Распределение спиновой
плотности в комплексах Ni(II) с этилендиамином и 1,3-диаминопропа-
ном // Ж. структ. химии. 1971. Т. 12, №3. С. 403-407.
232. Niibe М., Nakamura Y. Nuclear Magnetic Resonance Studies of 14N and
*H in Metal-Ammonia Solutions // J. Phys. Chem. 1984. V. 88, №23.
P. 5608-5614.
233. Емельянов В.А., Федотов M.A., Беляев А.В. Кинетика нитрования нит-
розо пентахлорорутений(П) иона // Ж. неорг. химии // 1992. Т. 37, № 12.
С. 2717-2721.
234. Венедиктов А. Б., Федотов М. А., Коренев С. В. Превращения гексанитро-
родиат(Ш) иона в хлорнокислых и солянокислых средах // Коорд. химия.
1999. Т. 25, №4. С. 285-289.
235. Беляев А.В., Федотов М.А., Шагабутдинова С.Н. Исследование процес-
са нитрования сульфатов Rh(III) методом ЯМР 103Rh, 17О, 14,15N // Тез.
докл. Межд. Чугаевск. Межд. конф, по коорд. химии. — Одесса, 2007.
С. А15-А17.
236. Nicolas А.М.Р., Wasilishen R.E. A nuclear magnetic resonance study of
aqueous solution of several nitrate salts // Canad. J. Chem. 1987. V. 65, № 5.
P. 951-956.
237. Brownlee R.T., Shehan B.P., Wedd A.G. 95 Mo and 14N Relaxation Time
Measurements Conforming that [Mo(CN)e]6“ is Dodecahedral in Aqueous
Solution // Inorg. Chem. 1987. V. 26, № 13. P. 2022-2024.
238. Bonner F.T, Donald C.E., Huges M.N. Stoicheiometric and Nitrogen-15
Labeling Sytudies on the Hiponitrous Acid — Nitrous Acid Reactions //
J.C.S. Dalton tr. 1989. №3. P. 127-133.
239. Jaszunski M., Szymanski S., Christiansen O. et al. NMR Properties of N^“ //
Chem. Phys. Lett. 1995. V. 243, №2. P. 144-150.
240. Au-Yeung S.C.F., Kwong К. W., Buist R.J. Multinuclear NMR Study of the
Crystal Field Strength of the Nitro Ligand and the Empirical Estimation of
the 59Co NMR Chemical Shifts of Cobalt Nitro Complexes // J. Amer. Chem.
Soc. 1990. V. 112, №22. P. 7482-7488.
Алюминий, галлий, индий, таллий
241. Hinton J. F., Briggs R.W. Group III — Aluminium. Gallium. Indium, and
Thallium // NMR and Periodic table / Ed. В. E. Mann. R. K. Harris. —
L.: Acad. Press, 1978. P. 279-308.
242. Johansson G., Lundgren G., Sillen L.G., Soderquist R. On the crystal struc-
ture of basic aluminium sulphate and the corresponding selenate // Acta
Chim. Scand. 1960. V. 14, №3. P. 769-771.
243. Akitt J. W. Aluminum. Gallium. Indium and Thallium // Multinuclear NMR /
Ed. J. Mason. — N. Y.: Plenum Press, 1987. P. 259-291.
244. Kidd R.-G., Truax. R.D. The occupance of mixed tetrahaloaluminate iones
shown by aluminiun-27 NMR spectroscopy // J. Amer. Chem. Soc. 1968.
V. 90, № 24. P. 6867-6869.
245. Haraguchi H., Fujiwara S. Aluminium complexes in solution as studied by
aluminum-27 NMR // J. Phys. Chem. 1969. V. 73, № 10. P. 3467-3473.
246. Lauterbur P.C., Hopkins R.C., King R.W. et al The NMR spectra of
aluminium borohydride trimethylamine // Inorg. Chem. 1968. V. 7, №5.
P. 1025-1027.
247. O'Reily D.E. NMR chemical shifts of aluminium: experimental data and
variational calculations // J. Chem. Phys. 1960. V. 32, №4. P. 1007-1012.
248. Movius W.G., Matwiyoff N.A. Proton and aluminium-27 NMR studies of
aluminium (III) perchlorate in N,N-dimethy!formamide // Inorg. Chem. 1967.
V. 6, №4. P. 847-849.
249. Akitt J. W., Greenwood N.N., Khandelwall B.L., Lester G.D. 27A1 NMR
studies of the hydrolysis and polymerisation of the hexaaquo-aluminium
(III) // J. C. S. Dalton tr. 1972. № 5. P. 604-610.
250. Akitt J. W., Farthing A. Aluminium-27 NMR studies of heteropolyanions
containing aluminium as heteroatom // J.C.S. Dalton tr. 1981. №7.
P. 1615-1616.
251. Федотов M.A., Казанский Л.П. Изучение состояния гетерополианионов
Mo(VI) и W(VI) с алюминием(Ш) и галлием(Ш) в водных растворах
методом ЯМР 27А1, 7lGa и других ядер // Изв. АН СССР. Сер. хим. 1988.
№9. С. 2000-2003.
252. Федотов М.А., Молчанов В. В., Зотов Р.Н., Тузиков Ф.В. О состоянии
А1(Ш) в спиртовых растворах алкоксидов алюминия по данным ЯМР
(27А1, 13С) и малоуглового рентгеновского рассеяния // Ж. неорг. химии.
2008. Т. 53, № 10. С. 1735-1742.
253. Тарасов В.П., Привалов В.И., Горбик А. А., Бакум С.И. Состояние ион-
ных пар в растворах алюмогидридов лития и тетрабутиламмония по дан-
ным ЯМР 27А1, *Н, 2D // Коорд. химия. 1985. Т. 11, № 12. С. 1626-1634.
254. Коньшин В.В., Чернышов Б.Н., Ипполитов Е.Г. Прямая ЯМР-идентифи-
кация гексафторалюминат-ионов в водно-перекисных растворах // ДАН
СССР. 1984. Т. 278, №2. С. 370-372.
255. Dechter J.J., Hendriksson U., Kovalevski J., Nilsson A.-C. Metal nucleus
quadrupole coupling constant in aluminium, gallium and indium acetylaceto-
nates // J. Magn. Reson. 1982. V. 48, № 3. P. 503-511.
256. Delpuech J.J., Khaddar M.R., Peguy A.A., Rubini P.R. Octahedral and
tetrahetlral solvates of the aluminium cation. A study of the exchange of
free and bound organophosphorus ligands by NMR spectroscopy // J. Amer.
Chem. Soc. 1975. V. 97, № 12. P. 3373-3379.
257. Fiat D., Connick R.E. Oxygen-17 Magnetic Resonance Studies of Ion Sol-
vation. The Hydration of Aluminium (III) and Gallium (III) Ions // J. Amer.
Chem. Soc. 1968. V. 90, №3. P. 608-615.
258. Shuster R.E., Fratiello A. Proton Magnetic Resonance Solvation Study
of Aqueous Solutions of AICI3 // J. Chem. Phys. 1967. V. 47, №4.
P. 1554-1555.
259. Canet D., Delpuelch J. J., Khaddar M.R., Rubini P. Direct observation of sol-
vated species by 27A1 NMR // J. Magn. Reson. 1973. V. 9, №2. P. 329-330.
260. Akitt J. W., Farnsworth J.A., Letellier P. NMR and Molar-Volume Stud-
ies of the Complex Formed between Aluminium(III) and Sulfate Anion //
J.C.S. Faraday trans. I. 1985. V. 81, № 1. P. 193-205.
261. Akitt J.W., Greenwood N.N., Khandelwall B.L., Lester G.D. NMR and
Raman studies of the Aluminium Salts. Containing Phosphoric Acids and
Fluoride Ions // J. Chem. Soc. A. 1971. № 15. P. 2450-2457.
262. Karlik S.J., Elgavish G.A., Pillai R.P., Eichborn G.L. Aluminium-27 NMR
studies of Al(III)-phosphate complexes in aqueous solutions // Inorg Chem.
1983. V. 22, №3. P. 525-529.
263. Федотов M.A., Мудраковский И. Л., Мастихин В.М. и др. Зависимость
химических сдвигов ЯМР 27А1 и 31Р в алюмофосфатах от состава вто-
рой координационной сферы // Изв. АН СССР. Сер. хим. 1987. № 10.
С.2340-2342.
264. Тарасов В.П., Киракосян Г. А., Рандаревич С.Б., Буслаев Ю.А. Комплек-
сообразование тригалогенидов алюминия с этилацетатом по данным ЯМР
27А1 // Коорд. хим. 1989. Т. 15, №7. С. 888-893.
265. Akitt J. IV., Gessner W., Weinberger M. High Field Aluminium-27 NMR
Investigation of Sodium Aluminate Solution // Magn. Reson. Chem. 1988.
V. 26, № 12. P. 1047-1050.
266. Еремин Н.И., Волохов Ю.А., Миронов B.E. Некоторые вопросы структу-
ры и поведения алюминатных растворов // Успехи химии. 1974. Т. 43. В.
2. С. 224-251.
267. Федотов М.А., Криворучко О.П., Буянов Р.А. Взаимодействие анионов
исходных солей с продуктами гидролитической полимеризации акваионов
алюминия (III) // Изв. АН СССР. Сер. хим. 1977. № 12. С. 2647-2651.
268. Федотов М.А., Криворучко О.П., Буянов Р.А. Исследование гидроли-
тической поликонденсации акваионов алюминия(Ш) как промежуточного
этапа формирования гидрогелей алюминия методом ЯМР на различных
ядрах // Ж. неорг. химии. 1978. Т. 23, №4. С. 2326-2331.
269. Криворучко О.П., Федотов М.А., Буянов Р.А. О влиянии способа до-
бавления к растворам основания на состав продуктов поликонденсации
акваионов А1111 // Ж. неорг. химии. 1978. Т. 23, №8. С. 2242-2244.
270. Федотов М.А., Криворучко О.П., Буянов Р.А. Зависимость состава про-
дуктов полимеризации акваионов А1И| от концентрации исходных раство-
ров // Изв. АН СССР. Сер. хим. 1977. № 10. С. 2183-2186.
271. Федотов М.А., Криворучко О.П., Головин А.В., Буянов Р.А. Особенно-
сти гидролитической полимеризации акваионов металлов в смешанных
водных растворах солей FeUI-Alln // Изв. АН СССР. Сер. хим. 1977. № 2.
С.473-475.
272. Криворучко О.П., Буянов Р.А., Федотов М.А., Плясова Л.М. О меха-
низме формирования байерита и псевдобемита // Ж. неорг. химии. 1978.
Т. 23, №7. С. 1798-1803.
273. Akitt J. W., Farthing A. New 27А1 NMR studies of the hydrolysis of the
aluminium (III) cation // J. Magn. Reson. 1978. V. 32, № 2. P. 345-352.
274. Akitt J. U7., Farthing A. Aluminium - 27 NMR studies of the hydrolisis of
aluminium(III). Gel-permetation chromatography // J.C.S. Dalton tr. 1981.
№7. P. 1601-1608.
275. Akitt J. W., Farthing A., Howarth O. W. Aluminium-27 NMR studies of
hydrolisis of aluminium(III). Stopped-Flow kinetic studies // J.C.S. Dalton
tr. 1981. №7. P. 1609-1614.
276. Akitt ]. W., Farthing A. Aluminium-27 studies of hydrolisis of aluminium
(III). Hydrolisis using sodium carbonate // J.C.S. Dalton tr. 1981. №7.
P. 1617-1623.
277. Akitt J. W., Farthing A. Aluminium-27 NMR studies of hydrolisis of alu-
minium(III). Slow hydrolisis using aluminium metal // J.C.S. Dalton tr.
1981. №7. P. 1624-1628.
278. Bottero Y., Cases J.M., Fissinger F., Poirier J.E. Studies of hydrolized alu-
minium chloride solutions. Nature of aluminium species and composition of
aqueous solutions // J. Phys. Chem. 1980. V. 84, №22. P. 2933-2939.
279. Коньшин В.В., Чернышов Б.Н., Ипполитов Е. Т. Состав и строение фто-
рокомплексов алюминия в водных растворах перекиси водорода и неорга-
нических кислот // Изв. АН СССР. Сер. хим. 1987. №8. С. 1707-1711.
280. Петросянц С.П., Буслаев Ю.А. Комплексообразование А1(Ш) в раство-
рах // Ж. неорг. химии. 1999. Т. 44, № 11. С. 1766-1776.
281. Киракосян Г.Ф., Ложков С.А., Галузина Т.В. и др. Сольватация ионов
в безводном пероксиде водорода // Ж. неорг. химии. 1990. Т. 35, №9.
С. 2306-2311.
282. Greenaway F. Т. Aluminium-27 NMR Study of Aluminium(III) interaction
with Hydroxy Catboxylic Acids // Inorg. Chim. Acta. 1986. V. 116, №1.
P. L21-L23.
283. Contures J.P., Massiot D., Bessada C. et al. Etude par RMN 27 d’aluminates
liquides dans domain 1600-2100 °C // C. R. Acad. Sci. Ser. C. 1990. V. 310,
№8. P. 1041-1045.
284. Рое B.T., McMillan P.F. Structure and Dynamics in Calcium Aluminate
Liquids: High Temperature 27A1 and Raman Spectroscopy // J. Am. Ceram.
Soc. 1994. V. 77, №7. P. 1832-1838.
285. Lacassagne V., Bessada C., Florian P. et al. Structure of High-Temperature
NaF-AlFj-AbOa Melts: A Multinuclear NMR Study // J. Phys. Chem. 2002.
V. 106B, №8. P. 1862-1869.
286. Coutures J.-P., Massiot D., Bessada C. et al. Etude par RMN 27 d’aluminates
liquides dans le domain 1600-2100 ° С // C. R. Acad. Sci. 1990. V. 310.
Ser. II, №8. P. 1041-1046.
287. Рандаревич С.Б., Коровина В.Ю., Жукова Н.Г. и др. Механизм сорбции
алюминия и галлия ионообменной смолой СФ-5 по данным ЯМР 31Р, 27А1,
7lGa // Ж. неорг. химии. 1992. Т. 37, № 1. С. 142-146.
Галлий
288. Akitt J. W., Greenwood N.N., Storr A. NMR studies with the gallium-71
isotope // J. Chem. Soc. 1965. №8. P. 4410-4416.
289. Буслаев Ю.А., Петросянц С.П., Тарасов В.П. ЯМР водных растворов
фторокомплексов. Фторгаллаты // Ж. структ. химии. 1974. Т. 15, №2.
С. 200-204.
290. Буслаев Ю.А., Тарасов В.П., Петросянц С.П., Мельников Н.Н. Исследо-
вание комплексообразования методом ЯМР на ядрах центральных ионов.
Неводные растворы трихлорида, трибромида и трииодида галлия(Ш) //
Ж. структ. химии. 1974. Т. 15, №4. С. 617-623.
291. Тарасов В.П., Привалов В.И., Буслаев Ю.А., Кузьмин И.А. Скаляр-
ная релаксация квадрупольных ядер // ДАН СССР. 1977. Т. 234, №3.
С. 636-639.
292. Тарасов В.П., Петросянц С.П., Киракосян Г.А., Буслаев Ю.А. Химиче-
ские сдвиги ЯМР 27А1, 69-71 Ga и константы спин-спинового взаимодей-
ствия 27A1-14N, 71Ga-14N в роданидо- и цианогалогенидных комплексах
алюминия и галлия // ДАН СССР. 1978. Т. 242, № 1. С. 156-159.
293. Lincoln S.F. Solvation of gallium(III) chloride in acetonilrile, *H and
7lGa magnetic resonance study // Aust. J. Chem. 1972. V. 25, №12.
P. 2705-2709.
294. Movius U7 G., Matviyoff N.A. Direct characterisation of the mixed complexes
gallium(III) with N,N-dimethylformamide and 2,4-pentanedione. PMR study
of first coordination sphere stoichiometry and kinetics of ligand exchange //
Inorg. Chem. 1969. V. 8, №4. P. 925-931.
295. Буслаев Ю.А., Тарасов В.П., Мельников H.H., Петросянц С.П. Перерас-
пределение лигандов в тетрагалоидных ионах галлия(Ш) // ДАН СССР.
1974. Т. 216, №4. С. 796-799.
296. Bock S., Noth. Н., Wietelman A. Solutions of Gallium Trichloride in Ethers:
A 71 Ga NMR Study and The X-Ray Srtucture of GaCU-Monoglyme //
Z. Naturforsch. 1990. Bd. 45B, №7. S. 979-984.
297. Тарасов В.П., Привалов В. И., Буслаев Ю.А. Изотопные эффекты в
69-71 Ga ЯМР // Тез. докл. 2-й Всесоюз. конф. «Спектроскопия ЯМР тя-
желых ядер элементоорганических соединений». — Иркутск, 1983. С. 88.
298. Smidbaur R., Zafiropolus Т, Bublak W. et al. High Resolution and Solid
State NMR Investigations of Subvalent Gallium Compounds // Z. Natur-
forsch. 1986. Bd. 41A, № 1-2. S. 315-318.
299. Glickson J.D., Pither T.P., Webb J.E., Ganis R.A. Hydrogen-1 and gal-
lium-71 NMR study of gallium citrate in aqueous solutions // J. Amer. Chem.
Soc. 1975. V. 97, №7. P. 1679-1683.
300. Lenkinski R.E., Chang C.H., Glickson J.D. Gallium-71 and phosphorus31
NMR studies of the interaction of gallium with phosphoric acid in aqueous
solution // J. Amer. Chem. Soc. 1978. V. 100, № 17. P. 5383-5386.
301. McGarvey B.R., Taylor M. J., Tuck D.G. Gallium-71 NMR studies of anionic
gallium halide species in nonaqueous solutions // Inorg. Chem. 1981. V. 20,
№7. P. 2010-2013.
302. Colton R., Dakternieks D., Hauenstein J. Gallium-69, Gallium-71 and In-
dium-115 NMR Studies of Group 3 Tetrahalometallale(III) Anions // Aust.
J. Chem. 1981. V. 34, №5. P. 949-955.
Индий
303. Cannon Т.Н., Richards R.E. Magnetic resonance studies of solutions, con-
taining 115In // Tr. Faraday Soc. 1966. V. 62, №552(6). P. 1378-1387.
304. Fratiello A., Davis D., Peak S., Schuster R.E. Hydrogen-1 and Indium-115
NMR Hydration and Complex Formation Study of Indium Halide Solution in
Water-acetone Mixtures // Inorg. Chem. 1971. V. 10, №8. P. 1627-1632.
305. McGarvey B.R., Trudell C O., Tuck D.G., Victoriano L. Coordination Com-
pounds of Indium. 115In NMR Studies of Anionic Indium Species in Non-
aqueous Solutions // Inorg. Chem. 1980. V. 19, № 11. P. 3432-3436.
Таллий
306. Hinton J.F. Thallium NMR Spectroscopy // Magn. Reson. Chem. 1987. V. 25,
№ 8. P. 659-669.
307. Koppel H., Dallorso J., Hoffmann G., Walther B. 2O5T1 NMR-spektroskopisc-
ne Uutersuchungen an Thallium(I) und Organothallium(III) verbindungen //
Z. anorg. allg. Chem. 1976. H. 427, № 1. S. 24-36.
308. Hildenbrandt K., Dreeskamp H. Indirekte Kernkopplung einiger Methyl-.
Vinyl-. Phenylverbindungen der Elemente Cd. Sn. Hg. TI und Pb // Z.phys.
Chem. (NF). 1970. Bd. 69, №3-4. S. 171-182.
309. Hinton J.F., Brisgs R. W. Infinite-dilution chemical shifts of Thallium-205 in
(CH3)2T1(III) ion // J. Magn. Reson. 1977. V. 25, №3. P. 555-557.
310. Schneider W.G., Buckingham A.D. Mercury, thallium and lead resonance //
Disc. Faraday Soc. 1962. №34. P. 147-159.
311. Figgis B.N. NMR chemical shifts in solutions containing 2O5T1 // Tr. Faraday
Soc. 1959. V. 55, №7. P. 1075-1085.
312. Dechter J. J., Zink J.I. Preferentional Solvation of the Thallous Ion and its
Relationship to Thallium-205 NMR chemical shifts // Inorg. Chem. 1976.
V. 15, №7. P. 1690-1694.
313. Dechter J. J., Zink J.I. Solvent Dependence of 205TI(I) NMR Chemical Shifts //
J. Amer. Chem. Soc. 1975. V. 97, № 11. P. 2937-2942.
314. Kitching W., Praeger D., Moore C.J. Arylthallium Trifluoroacetates 13C
and 19P NMR: the substituent effect of the bis(trifluoroacetato)thallium(III)
group // J. Organomet. Chem. 1974. V. 70, № 3. P. 339-342.
315. Kitching W., Moore C.J., Doddrell D., Adcock W. 13C NMR spectra
arylthaliumtrifluoro-acetates: a clarifications // J. Organomet. Chem. 1979.
V. 94, № 3. P. 469-472.
316. Bacon M., Reeves L. W. Nuclear magnetic relaxation rates of thallium(I)-205
in aqueous solution with respect to complexing of molecular oxygen //
J. Amer. Chem. Soc. 1973. V. 95, № 1. P. 272-273.
317. Chan S.O., Reeves L. W. NMR studies of thallium(I)-205 in aqueous solu-
tion // J. Amer. Chem. Soc. 1974. V. 96, № 2. P. 404-410.
318. Kitching W., Praeger D., Moore C.J. Arylthallium Trifluoroacetates 13C and
i9F NMR: the subslituent effect of the bis(trifluoroacetato)thallium (III)
group // J. Organomet. Chem. 1974. V. 70, № 3. P. 339-342.
319. Gasser R.P., Richards R.E. Ion-pair formation in thallous solution // Mol.
Phys. 1959. V. 2, №3. P. 357-361.
320. Freeman R., Gasser R.P., Richards R.E., Wheeler D.H. Chemical shifts of
thallium resonance spectra in solutions of thallous salt // Mol. Phys. 1959.
V. 2, № 1. P. 75-84.
321. Sheriff R. E., Williams D. Nuclear gyromagnctic rations Tl(III) // Phys. Rev.
1951. V. 82. P. 651-655.
322. Poss H.L. The magnetic moment of 2O5T1 // Phys. Rev. 1947. V. 72, №7.
P. 637.
323. Poss H.L. On the magnetic moments of 13C, 19F, 2O3T1, and 2O5T1 // Phys.
Rev. 1949. V. 75, №4. P. 600-606.
324. Proctor W.G. The magnetic moment 2O3T1 // Phys. Rev. 1949. V. 75, № 3.
P. 522-523.
325. Gutowsky H.S., McGarvey B.R. NMR in thallium compounds // Phys. Rev.
1953. V. 91, № 1. P. 81-86.
326. Herbinson-Evans D., Phipps P.B., Williams R.J. Charge transfer phenomena
in some inorganic complexes. The physical properties of thallium(I) hexa-
cyanoferrate(III) // J. Chem. Soc. 1965. № И. P. 6170-6173.
327. Freeman R., Gasser H.P., Richards R.E. Thallium NMR spectra // Mol.
Phys. 1959. V. 2, №3. P. 301-308.
328. Vaughan R. W., Anderson D.H. Thallium NMR in thallous iodide // J. Chem.
Phys. 1970. V. 52, № 10. P. 5287-5290.
329. Hinton J.P., Briggs R. W. Thallium-205 NMH spectroscopy. 2O5T1 chemical
shifts a sensitive probe for preferential solvation anil solution structure //
J. Magn. Reson. 1975. V. 19, №3. P. 393-397.
330. Dechter J. J., Zink L.I. A 2O5T1 NMR method of determining solvation effects
on alkali ion selectivity // J. Amer. Chem. Soc. 1976. V. 98, № 3. P. 845-846.
331. Haffner S., Nachtrieb N.H. NMR in molten salts. Chemical shifts in thallium
halide-alkali halide mixtures // J. Chem. Phys. 1965. V. 42, №2. P. 631-636.
332. Burke P.J., Matthews R. W. The influence of anion, solvent and concentration
on thallium-205. carbon-13 and proton NMR parameters of the dimelhylthal-
liuin(III) ion // J. Organomet. Chem. 1976. V. 118, №2. P. 129-134.
333. Sheldrick G.M., Yesinowski J.P. Proton and thallium-203 and 205 NMR and
INDOR study of dimeric dimethylthallium derivatives // J.C.S. Dalton tr.
1975. № 10. P. 870-875.
334. Srivanavit C., Zink J.I., Dechter J. J. A Thallium NMR Determination of
Polyether Cation Selectivity Sequence and Their Solvent Dependence //
J. Amer. Chem. Soc. 1977. V. 99, № 18. P. 5876-5881.
Кремний, германий, олово, свинец
335. Kitzinger I.-P., Marsman H. Oxygen-17 and Silicon-29 // NMR Basic Prin-
ciples and Progres / Ed. P. Diehl, E. Fluck, R. Kosfeld. — Berlin: Springer
Verlag, 1981. V. 17. - 235 p.
336. Frankiss S.G. NMR spectra of some substituted methanes // J. Phys. Chem.
1963. V. 67, №4. P. 752-755.
337. Kennedy J. D., McFarlane W. Silicon. Germanium. Tin and Lead. Multinu-
clear NMR / Ed. J. Mason. — N. Y.: Plenum Press, 1987. P. 305-333.
338. Федотов M.A., Казанский Л.П. Спектры ЯМР 29Si в гетерополианионах
кремния // Коорд. химия. 1984. Т. 10, №3. С. 423-425.
339. Group IV — Silicon, Germanium, Tin and Lead // NMR and Periodic table /
Ed. В. E. Mann, R. K. Harris. — L.: Acad. Press, 1978. P. 309-377.
340. Annual reports on NMR spectroscopy. V. 9 / Ed. by G.A. Webb. — L.: Acad.
Press, 1979. P. 221-318.
341. Marsman H. 29Si-NMR spectroscopic results // NMR Basic Prine, and Progr.
V. 17. — Berlin: Springer Verlag, 1981. P. 66-235.
342. Engelhardt G., Radeglia R., Janke H. et al. Zur Interpretation 29Si
NMR chemischer Verschiebung // Org. Magn. Reson. 1975. V. 5, №12.
P. 561-566.
343. Janes N., Oldfield E. Prediction of Silicon-29 Nuclear Magnetic Resonance
Chemical Shifts Using a Group Electronegativity Approach: Applications
to Silicate and Alumosilicate // J. Amer. Chem. Soc. 1985. V. 107, №24.
P. 6769-6775.
344. Engelhardt G., Zeigan D., Janke H. et al. 29Si-NMR-spektroskopie an Sili-
catlosungen // Z. anorg. allg. Chem. 1975. H.418, № 1. S. 17-28.
345. Marsman H. 29Si NMR studies on silicate solutions in water // Z. Natur-
forsch. 1975. Bd. 29b, №7-8. S. 495-499.
346. Gould O.R., Lowe B.M., McGilp N.A. Investigation of aqueous sodium
metasilicate solutions by 29Si NMR spectroscopy // J.C.S. Chem. comm.
1974. № 17. P. 720-721.
347. Engelhardt G., Altenburg W., Hoebbel D., Wicker W. 29Si-NMR-spec-
troskopie an Silikatlosimgen. Untersuchungen zur Kondensation der Monok-
iesersauere // Z. anorg. allg. Chem. 1977. H. 428, № 1. S. 43-50.
348. Engelhardt G., Altenburg W., Hoebbel D., Wieker W. The silicon-29 NMR
spectra of low-molecular weight silic acid // Z. anorg. allg. Chem. 1977.
H. 437, №2. S. 249-252.
349. Harris R.K., Newman R.H. 29Si NMR studies ot aqueous silicate solutions //
J.C.S. Faraday tr. II. 1977. V. 73, №9. P. 1204-1205.
350. Engelhardt G., Janke H., Hoebbel D., Wieker W. Strukturuntersuchungen
an Silikatanionen in wassriger Losungen mit Hilfe der 29Si NMR Spek-
troskopie // Z. Chem. 1974. Bd. 14, №3. S. 109-110.
351. Harris R.K., Newman R.H. 29Si NMR Studies of Aqueous Silicate Solu-
tions //J.C.S. Faraday tr.2. 1977. V. 73, №8. P. 1204-1215.
352. Harris R.K., Knight С. T. G. Silicon-29 Nuclear Magnetic Resonance Studies
of Aqueous Silicate Solutions // J.C.S. Faraday tr.2. 1983. V. 79, №10.
P. 1525-1538.
353. Sjoberg S., Olman L.-O., Ingri N. Equilibrium and Structural Studies of
Silicon(IV) and Aluminium(III) in Aqueous Solution. Polysilicate Formation
in Alkali Aqueous Solution. A Combined Potentiometric and Silicon-29 NMR
Study // Acta Chem. Scand. 1985. V. A39, № 2. P. 93-107.
354. Kinrade S.D., Swaddle T.W. Silicon-29 Studies of Aqueous Silicate Solu-
tions // Inorg. Chem. 1988. V. 27, №23. P. 4253-4259.
355. Kinrade S.D., Swaddle T.W. Silicon-29 Studies of Aqueous Silicate Solu-
tions. 2. Transverse 29Si Relaxation and the Kinetics and Mechanism of
Silicate Polymerisation // Inorg. Chem. 1988. V. 27, №23. P. 4259-4264.
356. Hendricks W.M., Bell A.T., Radke C.J. Effect of Organic and Alkali Metal
Cations on Distribution of Silicate Anions in Aqueous Solutions // J. Phys.
Chem. 1991. V. 95, №23. P. 9513-9518.
357. Sjoberg S. Silica in aqueous environments // J. Non-Crystal. Solids. 1996.
V. 196, № 1. P. 51-57.
358. Bahlman E.K.E., Harris R.K. Metcalfe K. et al. Silicon-29 NMR
self-diffusion and chemical-exchenge studies of concentrated sodium silicate
solutions // J.C.S. Faraday tr. 1997. V. 93, № 1. P. 93-98.
359. Kinrade S.D., Knight С. T. G., Pole D.L., Syvitski R. T. Silicon-29 NMR of
Tetraalkylammonium Silicate Solutions. 1. Equilibria. 29Si Chemical Shifts,
and 29Si Relaxation // Inorg. Chem. 1998. V. 37, № 17. P. 4272-4277.
360. Couti R., Fernandez L. Etude du passage de 1’etat colloidal a 1’etat ion-
ique de solutions de silicates sodiques par spectrometries RMN 29Si et
infrarouge // J. Chim. Phys. 1998. V. 95, № 2. P. 384-387.
361. Cho H, Felmy A.R., Craciun P. et al. Solution State Structure Determination
of Silicate Oligomers by 29Si NMR Spectroscopy and Molecular Modeling //
J. Amer. Chem. Soc. 2006. V. 128, № 7. P. 2324-2335.
362. Harris R.K., Kinnear K.A., Morris G.A. et al. Silicon-29 diffusion ordered
NMR spectroscopy (DOSY) as a tool for studying aqueous silicates //
J.C.S. Chem. Comm. 2001. №51. P. 2422-2423.
363. Harris R.K., Howard J. A. К., Samadi-May body A. et al. A New Silicate
Clatrate Hydrate: An X-Ray Diffraction and Magnetic Resonance Study of
a System with Octametric Silicate Anions and Trivalente Cations // J. Solid
St. Chem. 1995. V. 120, №2. P. 231-237.
364. Fernan /., Stebbius J.F. High-Temperature 29Si NMR Investigation of Solid
and Molten Silicates // J. Amer. Chem. Soc.1990. V. 112, №2. P. 32-39.
365. Knight C.T.G., Kirkpatrick R.G., Oldfield E. Silicon-29 NMR Structural
Characterization of Two Novel Germanosilicate Solution // J. Amer. Chem.
Soc. 1986. V. 108, № 1. P. 30-33.
366. Kinrade S.D., Holah D.G., Hill G.S. et al. The Peroxysilicate Question.
29Si Evidence for the Role of Silicates in Alkaline Peroxide Brightening of
Mechanical PulP // J. Wood Chem. Technol. 1995. V. 15, №2. P. 203-222.
367. Буевич. А.А., Жебровская Е.Б., Шалимов Б.З., Шевелевич P.C. Исследо-
вание гидролитической поликонденсации тетраэтоксисилана и триэтокси-
бора методом ЯМР 29Si и ИВ // Ж. прикл. химии. 1990. Т. 63, №3.
С. 660-668.
368. Sugahara Yo., Inoue Т., Kuroda К. 29Si NMR study on co-hydrolis processes
in Si(OEt)4-Rsi(OEt)3-EtOH-water-HCl systems (R = Me, Ph): effect of R
groups // J. Mater. Chem. 1997. V. 7, № 1. P. 53-59.
369. Samadi-Maybodi A., Harris R.K., Azizi S.N., Kenwright A.M. Silicon-29
NMR Study of the Formation of Monomethoxysilic Acid in Methanilic Alka-
line Silicate Solutions // Magn. Reson. Chem. 2001. V. 39, № 8. P. 443-446.
370. Kinrade S.D., Swaddle T. W. Direct Delection of AlumoSilicate Species in
Aqueous Solution by Silicon-29 and Aluminium-27 NMR Spectroscopy //
Inorg. Chem. 1989. V. 28, № 10. P. 1952-1954.
371. Ebsworth E.A. V., Sheldrik G.M. NMR spectra of silicon derivatives // Tr.
Faraday Soc. 1966. V. 62, № 528(12). P. 3282-3286.
372. Annual reports on NMR spectroscopy. V. 11b / Ed. G. A. Webb. — L.: Acad.
Press, 1981. — 502 p.
373. Mason J. Nitrogen NMR in inorganic, organometallic and bioinorganic chem-
istry // Chem. Revs. 1981. V. 81, №3. P. 205-227.
374. Mutterties E.L., Phillips W.D. Structure and exchange processes in some
inorganic fluorides by NMR // J. Amer. Chem. Soc. 1959. V. 81, №5.
P. 1084-1088.
375. Bramley R., Figgis B.N., Nyholm R.S. Nuclear Oxigen Magnetic Resonance
in Transition Metal Complexes // J. Chem. Soc. A. 1967. №6. P. 861-863.
376. Kinrade S.D., Swaddle T. U7 Mechanisms of Longitudinal 29Si Nuclear Mag-
netic Relaxation in Aqueous Alkali-Metal Silicate Solutions // J. Amer. Chem.
Soc. 1986. V. 108, №23. P. 7159-7162.
377. Knight S.T.G., Harris R.K. Silicon-29 NMR Studies Of Aqueous Silicate
Solutions. 8. Spin-Lattice Relaxation Times and Mechanisms // Magn. Reson.
Chem. 1986. V. 24, № 10. P. 872-874.
378. Kinrade S.D., Swaddle T.W. Aqueous Silicate Exchange Dynamics and
29Si Nuclear Magnetic Relaxation: Importance of Protonation Equilibria //
J.C.S. Chem. Comm. 1986. №2. P. 120-121.
Германий
379. Тарасов В. П., Привалов В. И., Дробышев С. Г., Буслаев Ю.А. Статический
и вибрационный вклады в квадрупольную релаксацию ядер германия-73
в [GeClnBr4-n] //ДАН СССР. 1983. Т. 272, №5. С. 1176-1179.
380. Kaufmann 7., Sahm W., Schwenk К. A. 73Ge NMR studies // Z. Naturforsch.
1971. Bd. 26a, № 9. S. 1384-1389.
381. Kupce E., Upena E., Trusule M., Lukevics E.J. Use of l4N and 73Ge NMR
Spectroscopyfor Elucidation of Product of the Reaction between GeCl4 and
KSCN in Acetone // Polyhedr. 1989. V. 8, №22. P. 2641-2644.
382. Kupce E., Lukevics E.J. The 73Ge Chemical Shift of Hexacoordinated Ger-
manium in the Ge(NCS)g“ Anion // J. Magn. Reson. 1988. V. 79, №2.
P. 325-327.
383. Купце Э.Л., У пена Э.А., Трушуле М.А., Лукевиц Э. Спектры ЯМР
73Ge аминогерманов // Изв. АН Латвийск. ССР. Сер. хим. 1988. № 3.
С. 359-60.
384. Kidd R. G., Spinney Н. G. Germanium-73 NMR Spectra of Germanium Tetra-
halides // J. Amer. Chem. Soc. 1973. V. 95, № 1. P. 88-90.
385. Федотов KI.А., Максимовская Р.И. Структурные аспекты в химии по-
лиоксометаллатов V, Mo, W // Ж. структ. химии. 2006. Т. 45, № 6.
С. 961-984.
386. Harazono Т., Tanaka К., Takeuchi К, Fukutomi И. Relaxation Mechanism
of Germanium-73 Nuclei in Tetrahalogermanes // Inorg. Chem. 1987. V. 26,
№23. P. 3851-3854.
387. Harazono T., Tanaka K., Takeuchi Y. Relaxation Mechanism of Germa-
nium-73 in Tetrabromogermanes and Tetraiodogermanes at High Tempera-
ture // Inorg. Chem. 1989. V. 28, № 10. P. 1838-1815.
388. Makulski W., Jakowski K., Antisek A., Jastrunski M. Gas-Phase NMR Mea-
surment. Absolute Shielding Scales, and Magnetic Dipol Moments of 29Si
and 73Ge Nuclei // J. Phys. Chem. 2006. V. 110, №40. P. 11462-11466.
Олово
389. Hunter B.K., Reeves L. W. Chemical shifts for compounds of the group IV
elements silicon and tin // Canad. J. Chem. 1968. V. 46, №8. P. 1399-1414.
390. Burke J.J., Lauterbur P.C. 119Sn NMR spectra // J. Amer. Chem. Soc. 1961.
V. 83, №2. P. 326-331.
391. Annual reports on NMR spectroscopy. V. 5A / Ed. E. F. Mooney. — L.: Acad.
Press, 1972. P. 353-393.
392. Harrison P.G., Ulrich S.E., Zuckerman J.J. l,9Sn chemical shifts by the
double resonance of organotin compounds // J. Amer. Chem. Soc. 1971. V. 93,
№21. P. 5398-5402.
393. Progress of nuclear magnetic resonance spectroscopy. V. 11 / Ed. J. W. Em-
sley. J. Feeney. L. H. Sutcliffe. — Oxford: Pergamon, 1977. P. 115-118.
394. Hani R., Geanangel G.A. Tin-119 NMR in coordination chemistry // Coord.
Chem. Rev. 1982. V. 44, № 2. P. 229-246.
395. Davies A. G., Harisson P. G., Kennedy J.D. et al. Magnetic double resonance
studies of 119Sn chemical shifts in organic compounds // J. Chem. Soc.(C).
1969. №8. P. 1136-1141.
396. Lassigne C.R., Wells E.J. High resolution 119Sn NMR studies by pulse
Fourier transform // Can. J. Chem. 1977. V. 55, №6. P. 927-931.
397. McFarlane W., Maire J.C., Delmas M. Heleronuclear magnetic double reso-
nance studies of ll9Sn chemical shifts in ethyltin derivatives // J. C. S. Dalton
tr. 1972. № 17. P. 1862-1865.
398. Tupciauskas A.P., Sergeev N.M., Ustynyk Yu.A. Shielding constants of
tin-119 nuclei // Org. magn. reson. 1971. V. 3, №6. P. 655-659.
399. Kennedy J.D., McFarlane W., Pyne G.S. Magnetic double resonance studies
of tin-119 chemical shifts in compounds with tin-sulphur bonds and related
species // J.C.S. Perkin tr.II. 1975. № 11. P. 1234-1239.
400. Kennedy J.D., McFarlane W. Magnetic double resonance studies of trimethyl
chalcogenides // J. Organomet. Chem. 1975. V. 94, № 1. P. 7-14.
401. Van den Berghe E. V., Van der Kelen G.P. A study of the *H and 119Sn NMR
spectra of (CH3)4_„Sn(NR2)n compounds (R — CH3. C2H5) // J. Organomet.
Chem. 1973. V. 61, №4. P. 197-205.
402. McFarlane W., Rycroff D.S. Magnetic double resonance studies of
trimethylstannyl-phosphines and their complexes // J.C.S. Dalton tr. 1974.
№ 18. P. 1977-1979.
403. Kennedy J.D., McFarlaue W., Pyne G.S., Wracmeyer B. Nuclear spin-spin
coupling between tin and other directly bound elements // J.C.S. Dalton tr.
1975. №5. P. 386-390.
404. Mitchell T.N. NMR 'j(Sn-Sn) coupling constants in hexaorganotins //
J. Organomet. Chem. 1974. V. 70, № 1. P. C1-C2.
405. Kennedy J.D., McFarlane W., Wrackmeyer B. Indirect nuclear spin-spin
coupling of lead-207 to other magnetic nuclei // Inorg. Chem. 1976. V. 15,
№6. P. 1299-1302.
406. Webster J. R., Jolly W.L. The stannonium ion in acid solution // Inorg. Chem.
1971. V. 10, №5. P. 877-879.
407. Lassigne C.R., Farrigia L.J., James B.R. Heteronuclear secondorder coupling
effect in high-resolution tin NMR spectra // J. Magn. Reson. 1981. V. 43,
№ 3. P. 488-490.
408. Приходченко П.В., Привалов В.И., Трипольская Т.А., Ипполитов Е.Г.
Комплексообразование олова(1У) в водно-пероксидных растворах по
данным ЯМР-спектроскопии // Ж. неорг. химии. 2001. Т. 46, №12.
С. 2060-2065.
409. Приходченко П.В., Устинова Е.А., Федотов М.А., Ипполитов Е.Г. Оли-
гомерные формы олова(1У) в системе Rb2Sn(OH)e-H2O-Rb2Sn(OOH)6 по
данным спектроскопии ЯМР (119Sn, |7О) // Ж. неорг. химии. 2004. Т. 49,
№ 10. С. 1685-1691.
410. McFarlane W., Maire J. С., Delmas М. Heleronuclear magnetic double reso-
nance studies of 119Sn chemical shifts in ethyltin derivatives // J.C.S. Dalton
tr. 1972. № 17. P. 1862-1865.
411. Tupciauskas A.P., Sergeev N.M., Ustynyk Yu.A. Shielding constants of
tin-119 nuclei // Org. Magn. Reson. 1971. V. 3, № 6. P. 655-659.
412. Kennedy J.D., McFarlane U7, Pyne G.S. Magnetic double resonance studies
of tin-119 chemical shifts in compounds with tin-sulphur bonds and related
species//J.C.S. Perkin tr.II. 1975. №11. P. 1234-1239.
413. Крутько Д.П.. Пермин А. Б., Петросянц В. С., Реутов О. А. Синтез, спек-
тры ЯМР и строение гидридных комплексов родия со связями Rh-Sn //
Изв. АН СССР. Сер. хим. 1984. № 12. С. 2787-2790.
414. Jameson C.J., Gutowsky H.S. Calculation of chemical shifts. I. General
formulations and Z-dependence // J. Chem. Phys. 1964. V. 40, №6.
P. 1716-1724.
415. Harris D.H., Lappert M.F., Poland J.S. Binuclcar organometallic com-
pounds. Nuclear magnetic doubly resonance studies of tin-119 chemical shifts
in compounds with transition metal-to-tin bounds // J.C.S. Dalton tr. 1975.
№4. P.311-316.
416. Kroth H.J., Schumann H., Kuivila H.G. et al. Tin-119 chemical shifts of
ortho, meta, para, 2,6- and polysubstituted arilotrimethyltin derivatives and
related organotin compounds // J. Amer. Chem. Soc. 1975. V. 97, №7.
P. 1754-1760.
417. Федоров Л. А. Спектроскопия ЯМР металлоорганических соединений. —
М.: Наука, 1984. — 248 с.
418. Moriyama Н., Aoki Т., Shinoda S., Saito Y. Photo enhanced catalytic de-
hydrogenation of propano-2-ol with homogenious rhodium-tin complexes //
J.C.S. Perkin tr. 1982. №3. P. 369-374.
419. Кокунов Ю.В., Ершова M.M., Разгоняева Г. А., Буслаев Ю.А. Исследова-
ние взаимодействия дигалогенидов олова(П) и диалкилолова(П) в невод-
ных растворах методом ЯМР // Ж. неорг. химии. 1999. Т. 44, № 3.
С. 457-463.
420. Yen Н.-М.М., Geanangle R.A. ll9Sn NMR Spectra of Tin(II) Halides //
Inorg. Chim. Acta. 1981. V. 52, № 1. P. 113-118.
421. Кокунов Ю.В., Раков И.Э. Исследование соединений Sn(II) в водных
растворах методом ЯМР ll9Sn // Коорд. химия. 1990. Т. 16, № 12.
С. 1616-1620.
422. Карымова Р.Х., Альт Л. Я., Агапов И. А., Дуплякин В. К. Изучение про-
цессов в растворах Р4(П)-оловохлоридных комплексов в нитрометане,
методами ЯМР 119Sn и ЯГР 119Sn спектроскопии // Ж. неорг. химии. 1991.
Т. 36, № 2. С. 464-468.
423. Dillon К.В., Marshall A. Tin-119 Nuclear Magnetic Resonanse Studies of
Some Mixed Halogeno-Complexes of Tin(IV) // J. C. S. Dalton tr. 1984. № 5.
P. 1245-1247.
424. Кокунов Ю.В., Раков И.Э. Смешанные галоидные комплексы олова(П)
и олова(1У) в водных растворах // Ж. неорг. химии. 1998. Т. 43, №6.
С. 1021-1024.
Свинец
425. Kennedy J.D., McFarlane W. Lead-207 shielding in organometal com-
pounds // J. C. S. Dalton tr. 1977. № 23. P. 2332-2337.
426. Maciel G.E., Dallas J.L. 207Pb pulse Fourier transform NMR. A promising
new tool for studies in lead chemistry // J. Amer. Chem. Soc. 1973. V. 95,
№ 9. P. 3039-3040.
427. Piette L.H., Weaver H.E. NMR chemical shifts of 207Pb in a few com-
pounds // J. Chem. Phys. 1958. V. 28, №4. P. 735-736.
428. Hawk R.M., Sharp K.R. Field-dependent relaxation and absolute nuclear
shielding of 207Pb in liquid PbCL // J. Chem. Phys. 1974. V. 60, № 3.
P. 1009-1017.
429. Weinberg I. Chemical shifts of the 207Pb NMR in some polar semiconduc-
tors // J. Chem. Phys. 1962. V. 36, № 4. P. 1112-1113.
430. Rocard J.M., Bloom M., Robinson L.B. NMR of lead-containing com-
pounds // Can. J. Chem. 1957. V. 37, №4. P. 522-525.
431. Hawk R.M., Sharp R.R. Nuclear magnetic relaxation of lead-207 in a aque-
ous medium // J. Magn. Reson. 1973. V. 10, №3. P. 385-387.
432. Hawk R.M., Sharp R.R. Field dependence of nuclear magnetic relaxation
of 207Pb in aqueous Pb(ClC>4)2 // J. Chem. Phys. 1974. V. 60, №4.
P. 1522-1527.
433. Bjorgvinsson M., Mercier J.F., Kehheth M.M. et al. Multinuclear mag-
netic resonance study of the dilead trichalcogenide anions Pb2SnnCh3_n
(Ch = Se. Те), and МгЗе^- (M = Sn. Pb) and the Crystal Structure
of (2,2,2-crypt-K+)2Sn2Tej“ and (2,2,2-crypt-K+)2Sn2Sej“ // Inorg. Chem.
1993. V. 32, № 26. P. 6046-6055.
434. Wilson W.L., Rudolph R. W., Lohr L.L. et al. Multinuclear NMR Character-
ization of Anionic Cluster of the Main-Group Elements Ge. Sn. Tl. Pb, and
Bi in Nonaqueous Solution // Inorg. Chem. 1986. V. 25, № 10. P. 1535-1541.
Фосфор, мышьяк, сурьма, висмут
435. Crutchfield М., Dungan С.Н., Van Vazer J.P. P31 nuclear magnetic reso-
nance. — N.Y.: Interscience. 1967. — 452 p.
436. Annual reports on NMR spectroscopy. V. 1 / Ed. E. F. Mooney. — L.: Acad.
Press, 1968. P. 91-134.
437. Annual reports on NMR spectroscopy. V. 5b / Ed. by E.F. Mooney. — L.:
Acad, press, 1973. — 441 p.
438. Mann B.E. The Common Nuclei // NMR and Periodic table. Ed. В. E. Mann.
R. K. Harris. — L.: Acad. Press, 1978. P. 100-105.
439. Dixon K.R. Phosphorus to Bismuth // Multinuclear NMR / Ed. J. Mason. —
N.Y.: Plenum Press, 1987. P. 369-403.
440. Lagodzinskaya G. V, Klimenko Yu. A. Measurment and correction for mag-
netic susceptibility with solenoidal magnet NMR spectrometer // J. Magn.
Reson. 1982. V. 49, № 1. P. 1-7.
441. Purdela D. Theory of 37P NMR chemical shifts. Bond-angle dependence //
J. Magn. Reson. 1971. V. 5, № 1. P. 23-26.
442. Тарасевич А. С., Егоров Ю.П. К теории химического сдвига на ядрах
тетракоординированного фосфора // Теор. и экспер. химия. 1975. Т. 2,
№2. С. 193-199.
443. Valkovich Р.В., Conger J. L., Castiello F.A. et al. Dependence of 3/P chemi-
cal shifts on oxygen-phosphorus bond angles in phosphate elhers // J. Amer.
Chem. Soc. 1975. V. 97, № 4. P. 898-900.
444. Leipert T.K., Freeman W.J., Haggle J. H. Nuclear magnetic relaxation on 3/P
in phosphorus halides // J. Chem. Phys. 1975. V. 63, № 10. P. 4177-4186.
445. Sammons R.D., Frey P.A., Bruzik K., Tsai M.-D. Effect of l7O and l8O on
31P NMR: Further Investigation and Application // J. Amer. Chem. Soc.
1983. V. 105, № 16. P. 5455-5461.
446. Arnold J.R. P., Love G. The Canonical Structure and 31P NMR Properties Of
Inorganic Thiophosphate // J.C.S. Chem. Comm. 1986. № 19. P. 1487-1488.
447. Progress NMR spectroscopy. V. 6 / Ed. by J. Emsley, J. Feeney, L. H. Sut-
cliff. — Oxford: Pergamon, 1970. P. 61-118.
448. Богдашкина В. И., Пермин А. Б., Петросян В. С. и др. Изучение взаимо-
действия гидридных комплексов платины с галогенидами двухвалентного
олова методом ЯМР 31Р и *Н // Изв. АН СССР. Сер. хим. 1982. №5.
С. 1033-1037.
449. McCain D. С., Markley J. С. Phosphorus-31 spin-lattice relaxation in aque-
ous orthophosphate solutions // J. Amer. Chem. Soc. 1980. V. 102, № 17.
P. 5559-5565.
450. Вашман А. А., Пронин И.С. Ядерная магнитная релаксация и ее приме-
нение в химической физике — М.: Наука, 1979. — 224 с.
451. Glonek Т, WangP.L, Van VazerJ.R. Phosphorus-31 spin-lattice relaxation.
Inorganic ring and chain phosphates // J. Amer. Chem. Soc. 1976. V. 98,
№ 25. P. 7968-7973.
452. Шеер M., Федин В.П., Федоров B.E. и др. О взаимодействии теллури-
дов ниобия, молибдена, вольфрама и рения с пентахлоридом фосфора //
Ж. неорг. химии. 1987. Т. 32, №7. С. 1769-1771.
453. Martinez E.J., Girardet J.-L., Maerschalk C., Morat C. Determination of New
Stable Aluminium Complexes with Pyrophosphate and Fluoride in Aqueous
Solution // Inorg. Chem. 1999. V. 38, № 21. P. 4765-4770.
454. Massart H., Contant R., Fruchart J.M. et al. 31P NMR studies of molybdic
and tungstic heteropolyanions. Correlation between structure and chemical
shifts // Inorg. Chem. 1977. V. 16, № 11. P. 2916-2921.
455. Максимовская Р.И., Федотов M.A., Мастихин B.M. и др. Исследование
состояния фосфорномолибдованадиевых гетерополикислот в водных рас-
творах методом ЯМР // ДАН СССР. 1978. Т. 240, № 1. С. 117-120.
456. Казанский Л.П., Федотов М.А., Потапова И. В., Спицын В. И. ЯМР
|7О и 31Р в восстановленных смешанно-валентных гетерополианионах
с делокализованной электронной парой // ДАН СССР. 1979. Т. 244, №2.
С. 372-376.
457. Лебедева Л.И., Ванникова Е.В. Изучение растворов фосформолибденовой
(ФМК) и фосфорвольфрамовой (ФВК) гетерополикислот методом ЯМР
31Р // Ж. неорг. химии. 1974. Т. 19, № 12. С. 3285-3287.
458. Максимовская Р.И., Федотов М.А., Максимов Г.М. О структуре гетеро-
полианионов PsMieO^ (М — Mo, W) в водных растворах и кислородном
обмене по ЯМР 17О // Изв. АН СССР. Сер. хим. 1983. №2. С. 247-252.
459. Сергиенко В.С., Порай-Кошиц М.А., Федотов М.А. и др. Рентгено-
структурное и 17О-ЯМР исследования фосфорномолибденованадиевых ге-
терополикислот // Ж. структ. химии. 1977. Т. 18, № 5. С. 976-977.
460. Клевцова Р.Ф., Юрченко Э.Н., Глинская Л. А. и др. Структура
и свойства фосформолибдованадиевых гетерополисоединений состава
Эд[РМобУб04о]-тН20 (Э — Н, Na). Кристаллическая структура
НазН6[РМо6Уб04о] I6H2O // Ж. структ. химии. 1981. Т. 22, № 6. С. 49-61.
461. Seel F., Bohrstedt G. 31P-kernresonanzspektrometrische Untersuchungen an
phosphato-cobalt(III) — und rhodium(III)-Komplexen // Z. anorg. allg. Chem.
1977. H. 435, №8. S. 257-267.
462. Беляев А. В., Федотов М.А. Первое наблюдение методом ЯМР (3|Р, |7О,
103Rh) комплексообразования родия(Ш) с фосфорной кислотой // Ж. неорг.
химии. 2008. Т. 53, № 5. 826-829.
463. Tatershall В. W., Blachnik R. Phosphorus-31 and Selenium-77 Nuclear Mag-
netic Resonance Spectra of Tetraphosphorus Triselenide // I.C. S. Dalton tr.
1988. №. P. 2055-2057.
464. Казанский Л.П., Федотов M.A., Птушкина M.H., Спицын В. И. Геомет-
рическая изомерия в гетерополианионах Ц ДАН СССР. 1975. Т. 224, №4.
С. 866-868.
465. Kazansky L.P., Fedotov М.А. Phosphorus-31 and oxygen-17 NMR evidence
of trapped electrons in reduced 18-molybdo diphosphate(V). РгМо^О^ //
J.C.S. Chem. Comm. 1980. № 14. P. 644-645.
466. Максимовская P. И. Исследование гидролиза ГПК H3PW12O40 методом
ЯМР 31Р // Ж. неорг. химии. 1998. Т. 43, № 12. С. 1960-1972.
467. Максимовская Р.И., Федотов М.А., Максимов Г.М. Взаимодействие
анионов PWnOf^ с катионами по данным ЯМР // Ж. неорг. химии. 1985.
Т.ЗО, №4. С. 918-924.
468. Федотов М.А., Максимовская Р.И., Максимов Г.М., Матвеев К.И. Вза-
имодействие гетерополианиона PWnO^ с катионами Zn2+, Cd2+. Hg2+.
Pb2+ no данным ЯМР l83W и других ядер // Ж. неорг. химии. 1987. Т. 32,
№3. С. 647-521.
469. Федотов М.А., Максимов Г.М., Матвеев К.И. Спектры ЯМР 17О, 183W
и состояние в растворах комплексов Y(III). La(lII). Ce(IV). Th(IV).
Lu(III) с гетерополианионом PWnO^ // Коорд. химия. 1990. T. 16, №2.
С. 203-206.
470. Fedotov М.А., Pertsikov B.S., Danovich D.K. I7O, 3IP and 183 NMR
spectra of paramagnetic complexes with the heteropoly tungstate anion
[Ln(PWnO39)2]11_ and their constitutionin aqueous solytion (Ln — rare
earth element) // Polyhedron. 1990. V. 9, № 10. P. 1249-1256.
471. Федотов М.А., Персиков Б.С., Данович Д.К. Спектры ЯМР |70, 3|Р,
183W и состояние в растворах парамагнитных комплексов с гетерополи-
вольфрамат-ионом [Ln(PW i ] Ogg)2]11 — // Коорд. химия. 1991. Т. 17, №2.
С. 234-242.
472. Федотов М.А., Детушева Л.Г., Кузнецова Л.И., Лихолобов В.А. Ком-
плексы железа(Ш) с гетерополианионом PWiiO^ в водных растворах по
спектроскопическим (ЯМР 31Р, 17О, 183W, ИК, ЭСП) и магнетохимиче-
ским данным // Ж. неорг. химии. 1993. Т. 38, №3. С. 515-525.
473. Кравченко Р.Л., Федотов М.А., Максимовская Р.И., Кузнецова Л. И.
Синтез и состояние в концентрирированных растворах комплексов Сг(Ш)
с лакунарными гетерополианионами [PWn Ogg]7- и [P2W2o07o]10- // Ж.
неорг. химии. 1994. Т. 39, №4. С. 629-634.
474. Fedotov М.А., Samokhvalova Е. Р., Kazansky L.P. 17О and l83W NMR
diamagnetic and paramagnetic shifts in heteropoly tungstates XWioO"6-
(X = Ln, Th, U) // Polyhedron. 1996. V. 15, № 19. P. 3341-3351.
475. Detusheva L. G., Fedotov M.A., Kuznetsova L.I. et al. Titanium (IV) polynu-
clear hydroxocomplexes stabilized in solutin by [PWnOgg]7- heteropoly
anion // React Kinet Catal Lett. 1996. V. 59, №2. P. 367-374.
476. Детушева Л.Г., Федотов M.A., Кузнецова Л. И. и др. Синтез и спек-
тральные свойства полиядерных гидроксокомплексов титана(1У), стабили-
зированные в растворе гетерополианионом [PWnOgg]7- // Изв. АН. Сер.
хим. 1997. №5. С. 914-920.
477. Kholdeeva О.A., Maksimov G.M., Maksimovskaya R.I. et al. A dimeric
titanium containing polyoxometallate. Synthesis, characterization, and catal-
ysis of HoOg-Based thioether oxydation // Inorg. Chem. 2000. V. 39, № 17.
P. 3828-3837.
478. Детушева Л.Г., Кузнецова Л.И., Федотов М.А. и др. Комплексообр-е
палладия(П). меди(П). железа(Ш). ванадия(У) с гетерополианионом
PWgO^- по данн. ЯМР 31Р, 183W, 51V и ИК-спектроскопии // Коорд.
химия. 2001. Т. 27, № 12. С. 890-897.
479. Максимов Г.М., Максимовская Р.И., Кожевников И.В. Гетерополикисло-
ты. производные от комплексов аниона PWnO^ с катионами металлов //
Ж. неорг. химии. 1992. Т. 37, № 10. С. 2279-2286.
480. Zhang С., Howell R.C., Scotland К. В. et al. Aqueous Speciation Studies
of Europium(III) Phosphotungstate // Inorg. Chem. 2004. V. 43, №24.
P. 7691-7701.
481. Contant R., Abessi M., Thouvenot R., Herve G. Dowson Type Het-
eropolyanions. Synthesis and 31P, 51V, and 183 W NMR Structural
Investigation of Octadeca(molybdo-tungsto-vanado) Diphosphates related
to the [H2P2W12O48]12“ anion // Inorg. Chem. 2004. V. 43, №12.
P. 3597-3604.
482. Максимов Г.М. Состояние гетерополикислот в разбавленных растворах
по данным ЯМР 31Р // Ж. неорг. химии. 1998. Т. 43, №9. С. 1568-1570.
483. Холдеева О.А., Максимовская Р.И. Исследование состояния P-Mo(W)-re-
терополикислот в среде АсОН-НдО методом ЯМР 31Р, 51V // Ж. неорг.
химии. 1992. Т. 37, №6. С. 1349-1354.
484. Максимовская Р.И., Максимов Г.М. Термические превращения гетеропо-
ликислоты a-H2P2WieO62-nH2O по данным ЯМР 31Р // Ж. неорг. химии.
1995. Т. 40, №8. С. 1363-1368.
485. Slack D.A., Greveling I., Baird M.C. Phosphorus-31 NMR studies of cat-
alytic systems, containing rhodium complexes of chelating chiral and achiral
diphosphines // Inorg. Chem. 1979. V. 18, № 11. P. 3125-3132.
486. Pisaniello D., Lincoln S.F., Williams E.H. The exchange of trimethylphos-
phate on manganese(II). A phosphorus-31 NMR study // Inorg. Chim. Acta.
1978. V. 31, №2. P. 237-240.
487. Mays M.J., Vernano P.A. Structure and bonding in gold(I) compounds.
A phosphorus-31 NMR study of structure of some gold(I) phosphine corm-
plexes in solution // J. C. S. Dalton tr. 1979. № 6. P. 1112-1115.
488. Vollano J.N., Picker D.H., Statler J. A. Synthesis and 3IP NMR Spectroscopy
of a Series of Gold(I) Amine Phosphine Cations // Inorg. Chem. Acta. 1989.
V. 115, № 1. P. 31-34.
489. Альт Л.Я., Фоменко Л.В., Юрченко Э.Н., Троицкая А.Д. Исследование
галогенид-, роданид- и 5пС1з-содержащих триалкилфосфитных комплек-
сов Pd(II) и Pt(Il) методом ЯМР 31Р спектроскопия // Коорд. химия. 1980.
Т. 6, № 4. С. 594-598.
490. Goel А.В., Goel S. Preparation and phosphrus-31 NMR studies of cationic
methylplatinum(II) complexes containing mixed ligands // Inorg. Chim. Acta.
1982. V. 59, №2. P. 237-240.
491. Favez R., Roulet R. The system PdX2(PMe3)2 and trimethylphosphine in
dichloromethane solution as studied by pnospliorus-31 NMR // Inorg. Chem.
1981. V. 20, №5. P. 1598-1601.
492. Bakas A.E., Hounslow A.M., Lincoln S.F., Maeji N.J. A preparative and
phospliorus-31 NMR study of the coordination of three phosphate ligands to
dioxouranium (VI) // Aust. J. Chem. 1982. V. 35, №6. P. 1489-1492.
493. Rehder D., Bechthold H.-С., Paulsen K. A multinuclear NMR study of
[V(PF3)6]- and [Nb(PF3)6]- // J. Magn. Reson. 1980. V. 40, №2.
P. 305-310.
494. Dean P.A., Phillips D.D., Polesek L. A phosphorus-31 NMR spectroscopic
study of complexation of tin(II) and lead(II) by some phosphines, phosphine
oxides and related ligands // Can. J. Chem. 1981. V. 59, № 1. P. 50-61.
495. Mathieu L., Thivole P., Delman M., Berger M. Phosphorus-31 NMR spec-
troscopy of mixed aqueous sodium pyrophosphate and stannous chloride
solution. Evidence for pyrophosphate-tin complex formation // J. Magn.
Reson. 1982. V. 46, №2. P. 332-337.
496. Hohman W.H., Kountz D.J., Meek D. W. Phosphorus-31 NMR Studies and
X-ray Structures of Five-Coordinate Cobalt(I) and Nickel(II) Complexes of
Tris(2-(diphenylphosphino)ethyl)phosphine // Inorg. Chem. 1986. V. 25, №5.
P. 616-623.
Мышьяк
497. Harris R.K. Group V — Arsenic. Antimony, and Bismuth // NMR and
Periodic table / Ed. B.E. Mann. R.K. Harris. — L.: Acad. Press, 1978.
P. 379-382.
498. Baldman G., Pregosin P.S. Arsenic-75 Nuclear Magnetic Resonance. A
Study of Some Arsenic Salts // J. Magn. Reson. 1977. V. 26, №2.
P. 283-289.
499. Jones B.D., Uehling E. A. NMR of Arsenic and Antimony in Liquid AsFs and
SbCl5 // J. Chem. Phys. 1971. V. 36, №6. P. 1091.
500. Burnett L.J., Zeltman A.H. Pulse NMR Relaxation Study of Liquid Arsine //
J. Chem. Phys. 1972. V. 56, №9. P. 4695-4701.
501. Larsen D. W. Proton Magnetic Resonance Study of Ion-pairing Effects on
Arsenic-75 Quadrupole Relaxation in Tetraalkylarsoniuin Ions // J. Phys.
Chem. 1971. V. 75, №9. P. 3880-3885.
502. Arnold M. S. J., Packer K. J. Nuclear spin relaxation in electrolyte solution.
Some comments on validity of the relationship ot correlation times with
shear viscosity in multicomponent system // Mol. Phys. 1968. V. 14, № 3.
P. 241-248.
503. Proctor W.G., Yu F.C. On the Nuclear Magnetic Moments of Several Stable
Isotopes // Phys. Rev. 1951. V. 81, № 1. P. 20-30.
504. Collins M.J., Schrobilgen G.J. Study of the OteF5 Donor Properties
of Te(OTeF5)4 by 75As and 125Te NMR Spectroscopy: Preparation and
Characterization of the [TeFI(OTeF5)3_I]+ Cations. [TeFI(OTeF5)4_I].
As(OTeF5)5. As(OTeF5)^ // Inorg. Chem. 1985. V. 24, № 17. P. 2608-2614.
Сурьма
505. Dharmatti S.S., Weaver H.E. Nuclear magnetic resonance of antimony in
antimony hexafluoride ion // Phys. Rev. 1952. V. 87, № 4. P. 675.
506. Stilbs P., Olofsson G. A *H, and 12/Sb NMR study of ionization reactions
in antimony pentachloride donor-acceptor adduct systems in solution // Acta
Chim. Scand. 1974. V. 28a. №6. P. 647-655.
507. Kidd R.G., Spinney H.G. Antimony-121 NMR study of hexahaloantimonate
anions // Can. J. Chem. 1981. V. 59, № 20. P. 2940-2944.
508. McFarlane W., Wood R.J. The sign of the Sb-F spin-spin coupling constant //
J.C.S. Chem. comm. 1969. №6. P. 262.
509. Kidd R.G., Matthews R.W. Antimony-121 NMR in some antimony (V)
compounds // J. Inorg. Nucl. Chem. 1975. V. 37, №3. P. 661-663.
510. Goetz-Grandmont G.J., Leroy J.F. Chloroantimonates (V) - new synthesis
and l2lSb NMR study // Z. anorg. allg. Chem. 1983. Bd. 496, № 1. S. 40-46.
Висмут
511. Fukushima E. NMR of KBiFe powder: the sign of Bi-F spin-spin coupling and
the Bi quadrupole coupling // J. Chem. Phys. 1971. V. 55, № 5. P. 2463-2466.
512. Morgan К., Yayer B.G., Schrdbilgen G.J. Bismuth NMR spectroscopy: 209Bi
and 19F high resolution NMR spectra of hexafluorobismutate(V) ion //
J. Magn. Reson. 1983. V. 52, № 1. P. 139-142.
513. Ильин Е.Г., Суховерхое В. Ф., Шарабарин А.В., Буслаев Ю.А. Константа
спин-спинового взаимодействия J(Bi-F) в BiF^" // ДАН СССР. 1983.
Т. 271, №3. С. 652-653.
514. Dupree R., Gardner J. A. Bismuth-209 NMR in molten bismuth-bismuth (III)
bromide mixtures // J. Phys. Colloq. (Orsay. Fr.) 1980. № C. 8. P. 20-23.
Кислород, сера, селен, теллур
515. Rodger С., Sheppard N., McFarlane C., McFarlane M. Oxygen, Sulphur,
Selenium and Tellurum. NMR and Periodic table / Ed. by H.K. Harris,
B.E. Mann. — L.: Acad. Press, 1978. P. 383-419.
516. Klemperer W.G. I7O NMR spectroscopy as structural probe // Angew. Chem.
Int. ed. 1978. V. 17, №4. P. 246-253.
517. Kintzinger J.P. Oxygen NMR - characteristic parameters and applications //
NMR Basic Prine, and Progr. V. 17. — Berlin: Springer Verlag, 1981.
P. 1-64.
518. Furin G.G., Rezvukhin A.I., Fedotov M.A., Yakobson G.G. Nuclear mag-
netic resonance spectra of polyfluoroaromatic compounds // J. Fluor. Chem.
1983. V.22, №2. P. 231-252.
519. Казанский Л.П. Химические сдвиги ЯМР 17О и энергия первого переноса
заряда в изо- и гетерополианионах // Коорд. химия. 1977. Т. 3, №3.
С. 327-331.
520. Filowitz М., Но R.K., Klemperer W.G., Shum W. ''О nuclear magnetic
resonance of polyoxometallates. Sensitivity and resolution // Inorg. Chem.
1979. V. 18, № f. P. 93-103.
521. McFarlane W., McFarlane H. С. E. Oxygen. Multinuclear NMR / Ed. J. Ma-
son. — N.Y.: Plenum Press, 1987.
522. Christ H.A., Diehl P., Schneider Hr., Dahn H. Chemische verbindnngen in
der Kernmagnetische Resonanz von 17O Verbindungen // Helv. Chim. Acta.
1961. V. 44, № 1. P. 865-880.
523. Wasillshen R.E., Mooibrock S., McDonald J.B. A more reliable oxygen-17
absolute chemical shielding scale // J. Chem. Phys. 1984. V. 81, № 3.
P. 1057-1059.
524. Кузнецова Н.И., Лихолобов В.А., Федотов M.A., Ермаков Ю.И. Меха-
низм образования моноацетата этиленгликоля из этилена в уксуснокислом
растворе, содержащем нитрит лития и ацетат палладия // Изв. АН СССР.
Сер. хим. 1982. № 12. С. 2799-2801.
525. Котванова М.К., Казанский Л.П., Федотов М.А., Торченкова Е.А. Изу-
чение протонирования гетерополианионов ZMo^O^" в водных раство-
рах с использованием ЯМР 17О // Коорд. химия. 1984. Т. 10, №8.
С. 1062-1067.
526. Florin А.Е., Alei М. 17О NMR shifts in НаО liquid and vapor // J. Chem.
Phys. 1967. V. 47, № 10. P. 4268-4269.
527. Chmelnick A.M., Fiat D. Oxygen-17 magnetic resonance study of hydration
of the ferrous and nikelous ions // J. Amer. Chem. Soc. 1971. V. 93, № 12.
P. 2875-2877.
528. Fiat D., Luz Z., Silver L. Solvation of Co (II) in methanol and water, enriched
in oxygen-17 // J. Chem. Phys. 1968. V. 49, № 3. P. 376-379.
529. Reuben J., Fiat D. Oxygen-17 and proton shifts in aqueous solutions and the
nature of aqua- and mixed complexes // J. Chem. Phys. 1969. V. 51, № 11.
P. 4409-4417.
530. Figgis B.N., Kidd R.G., Nyholm. R.S. Oxygen-17 NMR of inorganic com-
pounds // Proc, of the Roy. Soc. 1962. V. 262a. № 1339. P. 469-480.
531. Kuznetsova N.I., Fedotov M.A., Likholobov V.A., Yermakov Yu.I. Mech-
anism of catalytic oxidation olefins by periodic acid in acetic solution of
palladium acetate // J. Molec. Catal. 1986. V. 38, № 2. P. 263-271.
532. Щербаков В.А., Гусев Ю.К., Маширов Л.Г. и др. ЯМР-исследование
растворов перброматов уранила и нептунила // Радиохимия. 1984. Т. 24,
№ 5. С. 700-703.
533. Щербаков В. А., Маширов Л. Г. Спектроскопия |7ОМР солей нептуни-
ла и уранила в различных растворах // Радиохимия. 1984. Т. 24, №5.
С. 703-708.
534. Устинова Е.А., Приходченко П.В., Федотов М.А. Равновесие в воднопе-
роксидных растворах теллурата цезия по данным ЯМР |25Те спектроско-
пии // Ж. неорг. химии. 2006. Т. 51, №4. 662-666.
535. Kinrade S.D. Oxigen-17 NMR Study Of Aqueous Potassium Silicates //
J. Phys. Chem. 1996. V. 100, № 12. P. 4760-4764.
536. Самохвалова E.A., Федотов M.A., Татьянина И.В., Торченкова Е.А.
Изучение взаимодействия скандия(Ш) с уранмолибденовой кислотой ме-
тодами спектроскопии // Ж. неорг. химии. 1989. Т. 34, №4. С. 660-663.
537. Gerothonassis I.R., Sheppard N. Natural Abundance l7O NMR Spectra of
Some Inorganic and Biologically Important Phosphates // J. Magn. Reson.
1982. V. 46, №3. P. 423-439.
538. Федотов M.A., Беляев А. В. Прямое наблюдение инертных нитратоком-
плексов платинов металлов методом ЯМР H,15N в водном растворе //
Коорд. химия. 1994. Т. 20, № 8. С. 613-615.
539. Окроян Г.Р., Кушнарев Д.Ф., Калабин Г.А., Пройдаков А.Г. Спин-спи-
новая релаксация ядер 17О в спектрах водных кластеров Na+ и К+ //
Ж. структ. химии. 2002. Т. 43, № 2. С. 263-266.
540. Калабин Г.А., Каницкая Л.В., Кушнарев Д.Ф. Количественная спектро-
скопия ЯМР природного органического сырья и продуктов его переработ-
ки. — М.: Химия, 2000. — 408 с.
541. Miller K.F., Wentworth R.A. Oxygen-17 NMR spectra of certain of
oxomolybdenum(VI) complexes and influence of multiplicity of the
molybdenum-oxygen boud // Inorg. Chem. 1979. V. 18, №4. P. 984-988.
542. Федотов M.A., Максимовская Р.И., Молчанова О. А., Ильясова А. К.
Спектры ЯМР 17О дифосфатполиоксомолибдатов // Изв. АН СССР. Сер.
хим. 1980. №3. С. 709-713.
543. Fedotov M.A., Maksimovskaya R.L, Kazanskii L.P. Structure of vanadium
phosphate anion in solution from 17O, 51V and 31P NMR data // React. Kinet.
Catal. Lett. 1981. V. 16, №2-3. P. 185-189.
544. Максимовская Р.И., Федотов М.А. Химические сдвиги ЯМР 17О полиок-
сометаллатных анионов // Ж. структ. химии. 1981. Т. 22, № 1. С. 160-162.
545. Федотов М.А., Казанский Л.П., Спицын В.И. Химические сдвиги ЯМР
|7О и l83W в полиоксовольфраматах и длины связей вольфрам-кислород //
ДАН СССР. 1983. Т. 272, №5. С. 1179-1186.
546. Федотов М.А., Максимовская Р.И. Скорость обмена кислородом вана-
дофосформолибденовых гетерополианионов с водой по ЯМР |7О // ДАН
СССР. 1978. Т. 240, № 1. С. 128-131.
547. Федотов М.А., Максимовская Р.И., Бегалиева Д.У., Ильясова А.К.
Спектры ЯМР 17О и изотопный обмен кислорода в водных растворах фос-
форванадийвольфрамовых гетерополикислот // Изв АН. Сер. хим. 1980.
№7. С. 1477-1480.
548. Максимовская Р.И., Федотов М.А. Спектры ЯМР 31Р, 51V и |7О фос-
форвольфрамванадиевых гетерополикислот // Ж. неорг. химии. 1985.
Т. 30, № 1. С. 103-108.
549. Татьянина И. В., Молчанов В.П., Ионов В.М. и др. Кристаллическая
структура церимолибденовой и уранмолибденовой гетерополикислот //
Изв. АН СССР. Сер. хим. 1982. №4. С. 863-867.
550. Воловая И.Я., Максимовская Р.И... Федотов М.А., Моросанова С.А.
Синтез и спектры ЯМР 17О молибденомышьяковых кислот состава 1:12
и 2 : 18 // Ж. неорг. химии. 1984. Т. 29, №6. С. 1468-1472.
551. Kidd R. G. Relationship between oxygen-17 NMR chemical shifts and тг-bond-
ing to oxygen in the dichromate ion // Can. J. Chem. 1967. V. 45, №6.
P. 605-607.
552. Май Л. А. Корреляция между химическим сдвигом ЯМР ,7О и атом-
ным зарядом кислорода // Изв. АН Латв. ССР. Сер. хим. 1981. №4.
С. 402-404.
553. Delseth С., Kintzinger J.-P. Resonance magnetique nucleare de l3C et l7O
d’etheres aliphatiques. Effets 7 entre les atomes d’oxygene et carbone // Helv.
Chim. Acta. 1978. V. 61, №4. P. 1327-1334.
554. Казанский Л.П., Федотов М.А. Спектры ЯМР 17О и 183W в парамагнит-
ных гетарополианионах // Коорд. химия. 1988. Т. 14, №7. С. 939-942.
555. Деркач Л.В., Маркес Риос А., Максимовская Р.И. Кристаллические ге-
терополисоединения в системе Wvl-Vv-H2SC>4 // Ж. неорг. химии. 1989.
Т. 34, № 10. С. 2531-2537.
556. Казанский Л.П., Федотов М.А., Спицын В. И. ЯМР кислорода-17 в диа-
магнитных и парамагнитных гетерополианионах молибдена и вольфрама //
ДАН СССР. 1977. Т. 223, № 1. С. 152-154.
557. Horner D.A., Connick R.E. Kinetic of Oxygen Exchange between the Two
Isomers of of Bisulfite Ion. Disulfite Ion (ЗгОд-), and Water As Studied by
Oxygen-17 Nuclear Magnetic Resonance Spectroscopy // Inorg Chem. 2003.
V. 42, №6. P. 1884-1894.
558. Belton P.S., Cox I.]., Harris R.K., O'Connor M.J. Sulfur-33 and Oxygen-17
NMR Studies of Thiomolybdates and Thiotungstates // Aust. J. Chem. 1986.
V. 39, № 7. P. 943-952.
559. Gheller S.F., Hambley T.W., Rodgers J.R. et al. Synthesis and Charac-
terization of Complex of Thiomolybdates and Tungstates with Copper(I)
and Silver(I) Cyanides. Including 95Mo and 183W NMR Properties and
the Crystal and Molecular Structures of (Pr41N)2[(CN)CuS2MoS2CuCN].
(PrJ1N)2[(CN)AgS2WS2] and (Ph4As)2[(CN)CuS2MoS2CuCN]-H2O // In-
org. Chem. 1984. V. 23, № 16. P. 2519-2528.
560. Xue X., Kanzaki M. Ab initio Calculation of the 17O and 'H NMR Parame-
ters for Various OH Groups: Implications to the Speciation and Dynamics ol
Dissolved Water in Silicate Glasses // J. Phys. Chem. 2001. V. 101B, № 17.
P. 3422-3434.
561. Thompson A.R., Kunwar A.C., Gutowsky H.S., Oldfield E. Oxygen-17 and
Aluminium-27 Nuclear Magnetic Resonance Spectroscopic Investigation of
Aluminium Hydrolysis Products // J.C.S. Dalton tr. 1987. №7. 2317-2322.
562. Postel M., Brevard C., Arzoumanian H, Riess J.G. 17O NMR as a Tool for
Stadiing Oxygenated Transition Metal Derivatives; First Direct l7O Observa-
tion of Transition-Metal-Bonded Peroxidic Oxygen Atoms. Evidence for the
Absence of Oxo-Peroxo Oxygen Exchange in Molybdenum(VI) Compounds //
J. Amer. Chem. Soc. 1983. V. 105, № 15. P. 4922-4926.
563. Houston J.R., Ping Y., Casey W.H Water Exchange from the Oxo-Centred
Rhodium(III) Trimer [РЬз(/хз-О)(/х-О2ССН3)б(ОН2)з] + : A High-Pressure
,7O NMR Study // Inorg. Chem. 2005. V. 44, № 14. P. 5176-5182.
564. Helm L., Elding L.I., Merbach A.E. Water-Exchange Mechanism of
Tetraaquapalladium(II). A Variable-Pressure and Vaiable-Temperature
Oxygen-17 NMR Study // Helv. Chim. Acta. 1984. V. 67, №9.
P. 1453-1460.
565. Casteel W.J., Dixon D.A., LeBond N. et al. Lewis-Acid Properties of
Technetium(VII) Dioxide Trifluoride. TcO2F3: Characterization by 19F, l7O,
and 99Tc NMR Spectroscopy and Raman Spectroscopy. Density Functional
Theory Calculation of TcO2F3. M+TcO2F^ [M = Li, Cs, N(CH3)4], and
TCO2F3 CH3CN, and X-Ray Crystal Structure of Li+TcO2F^ // Inorg. Chem.
1998. V. 37, №2. P. 340-353.
566. Swift T.J., Connick R.E. NMR- relaxation mechnisms of 17O in aqueous
solutions of paramagnetic cations and lifetimes of water molecules in the first
coordination sphere // J. Chem. Phys. 1962. V. 37, №2. P. 307-320; Errata.
Ibid. 1964. V.41, №8. P. 2553-2554.
Сера
567. Retcofsky H.L., Friedel R. A. Sulfur-33 Magnetic Resonance of Selected
Compounds // J. Amer. Chem. Soc. 1972. V. 94, № 19. P. 6579-6584.
568. Wasylishen R.E., Connor C., Friedrich J. O. An approximate absolute 33S nu-
clear magnetic shielding scale // Can. J. Chem. 1984. V. 62, № 5. P. 981-985.
569. Harris D.L., Evans S.A Sulfur-33 NMR spectroscopy of simple sulfones.
Alkyl-substituent induced chemical shift (SCS) effects // J. Org. Chem. 1982.
V. 47, № 17. P. 3355-3348.
570. Lutz 0., Nepple A., Schwenk A. NMR investigation and nuclear magnetic
moment 33S // Z. Naturforsch. 1973. Bd. 28a, №8. S. 1370-1372.
571. Lutz 0., Nepple A., Nolle A. I7O and 33S fourier transform NMR studies in
thiosulphate and thiomolybdate solutions // Z. Naturforsch. 1976. Bd. 31a,
№ 8. S. 978-980.
572. Faure R., Vincent E.J., Ruiz J.M., Lena L. Sulfur-33 NMR studies of sulfur
compounds with sharp resonance lines, 33S NMR spectra of some sulfones
and sulfonic acid // Org. magn. reson. 1981. V. 15, №4. P. 401-403.
573. Gillespie R.L, Quail J. W. Sulfur and silicon isotope effect in fluorine NMR
spectroscopy // J. Chem. Phys. 1963. V. 39, № 10. P. 2555-2557.
574. Belton P.S., Woolins J.D. 33S and l4N Nuclear Magnetic Resonance of Some
Sulfur-Nitrogen Compounds // Magn. Reson. Chem. 1986. V. 24, №12.
P. 1080-1082.
575. Belton P.S., Cox I.J., Harris R.K. Line Width, Sensitivity and Artefacts in
33S Spectra: The Case of Aqueous Sulfides // Magn. Reson. Chem. 1986.
V. 24, № 11. P. 1004-1007.
576. Belton P.S., Cox I.J., Harris R.K. Medium Effects on 33S NMR of Inorganic
Sulphate // Magn. Reson. Chem. 1986. V. 24, № 2. P. 171-174.
Селен
577. Walchli H.E. Nuclear magnetic resonance. Measurments of selenum // Phys.
Rev. 1953. V.90, №2. P. 331-332.
578. Birchall T., Gillespie R. J., Verkris S.L. NMR of some selenium compounds //
Can. J. Chem. 1965. V. 43, №4. P. 1672-1679.
579. McFarlane W., Wood R.J. Nuclear magnetic double resonance studies
organoselenium compounds // J.C.S. Dalton tr. 1972. № 13. P. 1397-1407.
580. Anderson S.J., Goggin P.L., Goodfellow R.J. Study of the coordination be-
haviour of the thiocyanate and cyanate-ions in some platinum(II) complexes
by heteronuclear resonance speetroscopy // J.C.S. Dalton tr. 1976. №19.
P. 1959-1964.
581. Kolshorn H., Meier H. Selenum-77 magnetic resonance. pH dependence of
chemical shifts of 77Se in aqueous selenous acid // J. Chem. Res.(S). 1977.
№ 3. P. 338-339.
582. Kennedy J.D., McFarlane W. Nuclear magnetic double resonance studies of
organotin selenides // J.C.S. Dalton tr. 1973. №20. P. 2134-2139.
583. Arnold D.E., Dryburg J. S., Ebsworth E.A., Rankin D. W. Exchange reactions
of bromodifluorophosphine with silyl- and germyl-dorivatives of the group VI
elements // J.C.S. Dalton tr. 1972. №22. P. 2518-2522.
584. McFarlane W., Rycroft D.S. Determination of chemical shifts by nuclear
magnetic triple resonance: the nature of the phosphorus selenium bond in
organophosphorus selenides // J. C. S. Chem. comm. 1972. № 15. P. 902-903.
585. Pinnell R.P., Megerle C.A., Manatt S.L., Kroon P.A. NMR of phosphorus
compounds. Evidence for steric effects on the 31P-77Se coupling and 31P
chemical shifts // J. Amer. Chem. Soc. 1973. V. 95, №3. P. 977-978.
586. Pfisterer G., Dreeskamp H. Indirecte kernspinkopplung in den Methyl-. Silyl-
und Hydridverbihdungen der VI-Hauptgruppe // Ber. Bunsenges.phys. Chem.
1969. Bd. 73, №7. S. 654-661.
587. Goggin P.L., Goodfellow R.L, Haddock S.R. Platinum-donor atom spin-spin
coupling constants in complexes with dimethylselenidc ami dimethyltel-
luride // J.C.S. Chem. comm. 1975. №5. P. 176-177.
588. Seppelt K. Die Struktur des Selentetrafluorid in Losungcn // Z. anorg.allg.
Chem. 1975. H.410, № 1. S. 12-18.
589. Seppelt K, Rupp H.H. l29Xe Kernresonanzuntersuchungen von Xenon-
verbindungen. Xenon(II)-derivative // Z. anorg. allg. Chem. 1974. H. 409,
№ 3. S. 338-342.
590. Pekenen P., Hiltunen Y., Lailinen R.S., Pakkanen T. 77Se NMR Spectro-
scopic Study of Fluxional Selenium Sulfide l,2,3,4,5-Se5S2 // Inorg. Chem.
1990. V. 29, № 15. P. 2770-2772.
591. Collins M.L, Gillespie R.J., Sawyer J.F., Schrobilgen G.L Selenium-77
NMR and Crystal Structure of (Se3S)2(Sb4Fi7)(SbFe)3 Containing a Dis-
ordered Mixture of SajSe^^ Cations // Inorg. Chem. 1986. V. 25, №12.
P. 2053-2057.
592. Carnell M.M., Grein F., Murhie M. et al. The Identification in Solution
by 77Se NMR Spectroscopy of the Novel Se4I2+ cation and the Study of
its Equilibrium with Seelj-1- and Sel^- // J.C.S. Chem Comm. 1986. № 3.
P. 225-227.
593. Laitinen R.S., Pakkanen T.A. 77Se NMR Study of the Sulfur-Selenium
Binary System // J.C.S. Chem. Comm. 1986. № 17. P. 1381-1382.
594. Milne J. Selenium Sulfur Dihalides SeSX2 (X = Br, Cl): A Raman and
77Se NMR Spectroscopic Study // J.C.S. Chem. Comm. 1991. №15.
P. 1048-1049.
595. Chivers T., Laitinen R.-S., Schmidt K.L 77Se NMR spectroscopic study of
the molecular composition sulfur-selenium melts // Can. J. Chem. 1992.
V. 70, №3. P. 719-725.
596. Laitinen R.S., Pakkanen T.A. 77Se NMR Spectroscopic Characterization of
Selenium Sulfide Ring Molecules SenSe-n // Inorg Chem. 1987. V. 26, № 16.
P. 2598-2603.
597. Burns R.C., Collins M.L, Gillespie R.L, Schrobilgen G.S. 77 Se NMR Study
of Se|+ and Se2 J: Examples of Rigid and Fluxional Species. Disproportion of
Se2J at Low Temperatures // Inorg. Chem. 1986. V. 25, № 25. P. 4465-4469.
598. Fedin V.P., Kalinina /. V., Samsonenko D.G. et al. Synthesis. Structure, and
Properties of Molybdenum and Tungsten Cyano Complexes with Cuboidal
М4(мз-Е)4 (M = Mo, W; E = S, Se, Те) Cores // Inorg. Chem. 1999. V. 38,
№9. P. 1956-1965.
599. Campbell J., Mercier H.P.A., Santry D P. et al. First Example of Tallium
Chalcogenide Cages Sintheses, 77Se, 2O3T1 and 2O5T1 NMR Study Tl4Sej~ and
TLiSeg Anions. X-ray Crystal Structure of (2,2,2-crypt-K+)3Tl5Se3 . and
Theoretical Study // Inorg. Chem. 2001. V. 40, №2. P. 233-254.
600. Федоров B.E., Ткачев С. В., Наумов Н.Г. и др. Ступенчатое замещение
g3-Te лигандов в октаэдрическом кластерном ядре [Re6Te8]2+: ЯМР-спек-
троскопическое доказательство равновесия между химическими форма-
ми // Ж. неорг. химии. 1998. Т. 43, № 10. С. 1683-1693.
601. Яровой С. С., Солодовников С.Ф., Ткачев С.В. и др. Синтез, структура
и исследование методом ЯМР 77Se комплекса (PPh4)[{Re6Se?Br}Br6] //
Изв. АН. Сер. хим. 2003. № 1. С. 65-69.
602. Sokolov M.N., Gushchin A.L., Tkachev S. V. et al. New Synthesis of
Nb2(g-Q2)4+ (Q = S, Se) Complexes with Bidentate Ligands. Their Iso-
merism and Electronic Structure // Inorg. Chim. Acta. 2005. V. 368, № 7.
P. 2371-2383.
Теллур
603. Granger P., Chapelle S. Relaxation Mecanism of l25Te and 123Te and Tem-
perature Effect of Chemical Shifts // J. Magn. Reson. 1980. V. 39, №2.
P. 329-334.
604. McFarlane W., Berry FJ., Smith B.C. 'H{l25Te} HeteronuclearMagnetic
Double Resonance in Some Organotellurum Compounds // J. Organomet.
Chem. 1976. V. 113, №2. P. 139-141.
605. Birchall T., Myers R.D., De-Waard H., Schrdhilgen G.J. Multinuclear NMR
and Mossbauer study of pentafluirooxotellurate (OTeF8) derivatives of tellu-
rum. iodine and xenon: spectroscopic study of the relative electronegativities
of fluoride and pentafluorooxotellurate // Inorg. Chem. 1982. V. 21, № 3.
P. 1068-1073.
606. Gombler W. NMR spektroskopische Untersuchungen an Chalkogen-
verbindungen // Z. Naturforsch. 1981. Bd. 36b, №5. S. 535-543.
607. McFarlane H.C., McFarlane W. Studies of tellurum shielding by heteronu-
clear magnetic double resonance in representative series of compounds //
J.C.S. Dalton tr. 1973. №22. P. 2416-2418.
608. Chadha R.K., Miller J. M. 125Te Fourier transform NMR studies of tellurium
compounds // Can. J. Chem. 1982. V. 60, № 17. P. 2256-2258.
609. Buckler K. U., Kronenbitter J., Lutz O., Nolle A. l23Te and l25Te Fourier
transform NMR investigation // Z. Naturforsch. 1977. Bd. 32a, №11.
S. 1263-1265.
610. Кокунов Ю.В., Афанасьев B.M., Густякова М.П., Буслаев Ю.В. Иссле-
дование взаимодействия аниона TeFs с некоторыми органическими рас-
творителями методом ЯМР l9F и |25Те // Коорд. химия. 1989. Т. 15, №3.
С. 348-353.
611. Glidewell С., Rankin D. W., Sheldrick G.M. NMR studies of mixed group 4 /
group 6 hydrides // Tr. Faraday Soc. 1969. V. 65, № 558(6). P. 1409-1412.
612. Traser G. 117., Peadcock R.D., McFarlane U7. A magnetic double resonance
study of EtaNTeFj: sign of the F-Te spin-spin coupling constants // Mol.
Phys. 1969. V. 17, №3. P. 291-296.
613. Нуретдинов И. А., Логинова Э.И. Спин-спиновое взаимодействие ядер 31Р
и |25Те в теллурофосфорорганических соединениях // Изв. АН СССР. Сер.
хим. 1973. № 12. С. 2827.
614. Christe К. О., Sanders J.C.P., Schrobilgen G.J., Wilson К. О.
High-Coordination Number Fluoro- and Oxofluoro-Anions IFgO-, TeFgO^,
TeF7“, IF" and TeF2- // J.C.S. Chem Comm. 1991. № 13. P. 837-840.
615. Inanio M. l25Te NMR Evidence for the Exitence of Trinuclear Tellurate Ion
in Aqueous Solution // Chemistry Letter. 1996. № 1. P. 17-18.
616. Lutz 0. Group VI Elements other than Oxygen. The Multinuclear Approach
to NMR Spectroscopy / Ed. J.B. Lambert, F. G. Riddel. — Doddrecht:
D. Reidel Publ. Co., 1983. P. 389-403.
Галогениды
617. Lindman B., Forsen S. Chlorine, Bromine and Iodine. Multinuclear NMR /
Ed. J. Mason. — N.Y.: Plenum Press, 1987. P.421-438.
618. Jameson C. Fluorine. Multinuclear NMR / Ed. J. Mason. — N.Y.: Plenum
Press, 1987. P. 437-446.
619. Emsley J.W., Phillips L. Fluorine chemical shifts // Progress nucl. magn.
reson. spectroscopy. V. 7 / Ed. J. W. Emsley. J. Feeney. L. H. Sutcliffe. —
Oxford: Pergamon, 1971. — 529 p.
620. Габуда С.П., Гагаринский Ю.В., Полищук С. А. ЯМР в неорганических
фторидах. — М.: Атомиздат, 1978. — 205 с.
621. Габуда С.П., Земсков С. В. Ядерный магнитный резонанс в комплексных
соединениях. — Новосибирск: Наука, 1976. — 86 с.
622. Драго Р. Физические методы в химии. Т. 1 / Пер. с англ. А. А, Соловья-
нинова; Под ред. О. А. Реутова — М.: Мир, 1981. — 422 с.
623. Шипачев В.А., Земсков С.В., Ткачев С.В. Исследование поведения
гексафторродиат-иона в растворах // Коорд. химия. 1980. Т. 6, №8.
С. 1237-1240.
624. Seppelt К., Bartlett N. NMR studies of hexafluorides // Z. anorg. allg. Chem.
1977. Bd. 436, №9. S. 122-126.
625. Hindermann D.K., Cornwell C. D. Fluorine and proton NMR study of gaseous
hydrogen fluoride // J. Chem. Phys. 1968. V. 48, № 5. P. 2017-2025.
626. Урсу И. Магнитный резонанс в соединениях урана. — М.: Энергоатомиз-
дат, 1983. — 216 с.
627, Dean Р.А., Evans D.P. Spectropcopic studies of inorganic-fluorocomplexes.
The 19F NMR and vibrational spectra of hexafluorometallates of group IVA
and IVB // J. Chem. Soc. A. 1967. №4. P. 698-701.
628. Tarasov V.P., Buslaev Yu. A. 39K and 45Sc nuclear magnetic relaxation for
the hexafluoroscandate, hexafluorotitanate and hexafluorogermanate ions in
aqueous solutions // J. Magn. Reson. 1977. V. 25, № 1. P. 197-203.
629. Hinderman D.K., Williams L.L. Nuclear magnetic shielding in Fj //J. Chem.
Phys. I960. V. 50, № 7. P. 2839-2841.
630. Mohanti S. Paramagnetic contribution of magnetic shielding constant in
some tetrahedral and octahedral molecules // J. Chem. Phys. 1973. V. 59,
№8. P. 4415-4417.
631. Mason J. Nuclear magnetic shielding in the fluorides of elements //
J.C.S. Dalton tr. 1975. № 14. P. 1426-1431.
632. Игнатов M.E., Ильин Е.Г., Швец А. А., Буслаев Ю.А. Химические сдвиги
ЯМР i9F комплексов 3°-переходных металлов и донорные способности
лигандов // Коорд. химия. 1978. Т. 4, № 12. С. 1818-1827.
633. Ильин Е.Г., Буслаев Ю.А. Применение метода ЯМР 19F к исследованию
фторокомплексов 3°-переходных металлов // Коорд. химия. 1978. Т. 4,
№8. С. 1178-1194.
634. Игнатов М.Е., Ильин Е.Г., Швец А.А., Буслаев Ю.А. Химические сдвиги
ЯМР 19F фторокомплексов и донорные свойства лигандов // ДАН СССР.
1978. Т. 240, №4. С. 865-868.
635. Буслаев Ю.А., Кокунов Ю.В. Фториды хрома, молибдена, вольфрама //
Коорд. химия. 1983. Т. 9, №6. С. 723-751.
636. Hindman J.C., Svirmikas A., Appelman Е.Н. Proton and fluorine NMR
observations on hypofluorous acid. HOF // J. Phys. Chem. 1972. V. 57, № 11.
P. 4542-4543.
637. Alzewel K.A. Relaxation time measurments on fluoride ions in aqueous
solutions // Z. phys. Chem. (Leipz.). 1974. Bd. 255, № 1. S. 193-198.
638. Martin J.S., Fujiwara F. Y. High resolution NMR spectra of hydrogen fluo-
ride in solution and in bihalide ions. Nuclear spin coupling in strong hydrogen
bonds // J. Amer. Chem. Soc. 1974. V. 96, №25. P. 7632-7637.
639. Ghibaudi E., Colussi A. J., Christe K.O. Extended Correlation between O-F
Energies and l9F NMR Chemical Shifts in Fluoroxy Compounds // Inorg.
Chem. 1985. V. 24, № 18. P. 2869.
640. Шамахова H.H., Плахотник В.И., Ильин Е.Г. Ступенчатое комплексо-
образование в водных фторфосфорных системах // Коорд. химия. 1989.
Т. 15, № И. С. 1504-1509.
641. Киракосян Г.А., Логинов С.В., Галузина Т.В. и др. Сольватация ионов
в безводном пероксиде водорода по данным ЯМР // Ж. неорг. химии.
1990. Т. 35, №9. С. 2306-2311.
642. Gombler U7, Schaebs J., Willner H. I9F NMR Chemical Shifts of SF3 in the
Gas Phase // Inorg. Chem. 1990. V. 29, № 14. P. 2697-2698.
643. Коньшин В. В., Чернышов Б.Н., Ипполитов Е.Г. Состав и строение фто-
рокомплексов алюминия в водно-спиртовых растворах // Коорд. химия.
1986. Т. 12, № 10. С. 1345-1350.
644. Петросянц С.П., Малярик М.А., Буслаев Ю.А. Фтороалюминокарбок-
силатные комплексы алюминия // Ж. неорг. химии. 1996. Т. 41, №11.
С. 1838-1844.
645. Bassada A. C.,Anghei Е.М. 11 В, 23Na, 27А1 and l9F Study of Solid and Molten
Na3AlF6-Na2BnO7 // Inorg Chem. 2003. V. 42, № 12. P. 3884-3890.
646. Dean Р.А. W., Gillespie R.J. Formation and fluorine-19 NMR spectra of the
polysulfuric acids. Н(50з)„Р. Existance of the polyfluorosulluric acids in
SbF5-HSO3F // J. Amer. Chem. Soc. 1970. V. 92, № 8. P. 2362-2364.
647. Щетинина Г.П., Бровкина О.В., Чернышов Б.И. ЯМР ИВ, l9F растворов
гидроксофторборатов в уксусной и пероксоуксусной кислоте // Ж. неорг.
химии. 1985. Т. 30, №8. С. 2161-2162.
648. Чернышев Б. И., Щетинина Г.П., Колзунов В. А., Ипполитов Е. Т. Иссле-
дование системы борная кислота — бифторид аммония методом ЯМР //
Ж. неорг. химии. 1980. Т. 25, №6. С. 1468-1474.
649. Чернышев Б.Н., Диденко И. А., Ипполитов Е. Т. Исследование образова-
ния пероксофторстаннатов в растворах перекиси водорода методом ЯМР //
ДАН СССР. 1982. Т. 262, № 3. С. 626-629.
650. Кокунов Ю.В., Гуля А.П., Афанасьев В.М. и др. Фторидные комплексы
олова (II) в растворе // Коорд. химия. 1988. Т. 14, №5. С. 648-654.
651. Чернышов Б.Н., Василюк И. С. Изучения процессов гидролиза гексафто-
рантимоната щелочных металлов методом ЯМР 19F // Коорд. химия. 1986.
Т. 12, № 10. С. 1354-1357.
652. Афанасьев В.М., Сахаров С.Г., Густякова М.П. и др. Спектр ЯМР
19F комплексного аниона [l26TeF6]_ // Коорд. химия. 1987. Т. 13, №2.
С. 279-280.
653. Margrave J.L., Sharp K.G., Wilson Р. W. The reaction of silicon difluoride
with iodine and properties of some iodofluorosilanes // J. Inorg. Nucl. Chem.
1970. V. 32, №6. P. 1813-1816.
654. Margrave J.L., Sharp K.G., Wilson P.W. The reaction of silicon difluo-
ride with iodotrifluoromethane // J. Inorg. Nucl. Chem. 1970. V. 32, №6.
P. 1817-1826.
655. Johansson R.B., Farrar T.C., Brinckman F.E., Coule T.D. NMR studies
of inorganic fluorides: high resolution 19F spectra of Si2?6 and (SiFa^O //
J. Chem. Phys. 1960. V. 44, № 3. P. 962-964.
656. Буслаев JO.А., Щербаков В.А. Химические сдвиги и спектры ЯМР 19F
в растворах фторидов элементов V и VI групп // Ж. структ. химии. 1966.
Т. 7, № 3. С. 344-350.
657. Буслаев Ю.А., Кокунов JO B., Бочкарева В.А. и др. Реакции замещения
и взаимное влияние лигандов в оксогалогенидных комплексах молибдена
и вольфрама // Ж. неорг. химии. 1974. Т. 19, №5. С. 1196-1202.
658. Буслаев Ю.А., Петросянц С.И., Тарасов В.П. ЯМР водных растворов
фторокомплексов оксопероксовольфраматов // Ж. структ. химии. 1970.
Т. 11, №6. С. 1023-1027.
659. Postel М., Riess J. G., Calves J. Y., Guerchais J. Structural and stereodynamic
studies on molecular early transition metal derivatives. NMR study of iso-
lated tungsten dioxofluoride adduct of type WO2F3 // Inorg. Chim. Acta.
1979. V.32, №2. P. 175-180.
660. Шипачев В.А., Земсков С.В. Изучение превращений гексафторокомплек-
сов Pt(IV), Os(IV) в водных растворах // Коорд. химия. 1982. Т. 8, №7.
С. 990-993.
661. Dean P.A. W. Synthesis of cis-[Coen2(MF6)2]MF6 (M = As, Sb): Cobalt(IlI)
Complexes Containing ry’-MF^ Characterizable by NMR // Inorg Chim.
Acta. 1986. V. 114, № 1. P. L1-L3.
662. Киракосян Г. А., Тарасов В. П. ЯМР 19F смешанных фторокомплексов
родия(Ш) в растворах // Коорд. химия. 1992. Т. 18, № 1. С. 86-92.
663. Gerken М., Dixon D.A., Schrobilgen G.J. The OsO^F-, OSO4F2-, and
OsO3F^. Anions. Their Study by Vibrational and NMR Spectroscopy
and Functional Theory Calculations, and the X-ray Crystal Structures of
[N(CH3)4][OsO4F] and [N(CH3)4][OsO3F3] // Inorg. Chem. 2000. V. 39,
№ 19. P. 4244-4255.
664. Christe K.O., Dixon D.A., Mack H.G. et al. Osmium Tetrafluoride Dioxide,
cis-OsO2F4 // J. Amer. Chem. Soc. 1993. V. 115, №24. P. 11279-11284.
665. Christe K.O., Dixon D.A., LeBlond N. et al. Lewis-Acid Behavior
of ReO2F3: Synthesis of (Re02F3)oo. Re2O2F7- Re2O4F7, Re3O6F^
and ReO2F3(CH3CN) and Study by NMR and Raman Spectroscopy,
and Dencity Functional Theory Calculations; and X-ray Structures of
[Li][Re2O2F4], [K] [Re2O4F7], [K][Re2O4F7-] [2ReO2F3], [Cs][Re3O6Fl0],
and ReO2F(CH3CN)2 CH3CN // Inorg. Chem. 1999. V. 38, №10.
P. 2340-2358.
666. Кокунов Ю.В., Бочкарева В.А., Сахаров С.Г. и др. Оксофторидные ком-
плексы вольфрама с /3-дикетонатами и /3-трикарбонильными соединения-
ми // Коорд. химия. 1988. Т. 14, № 12. С. 1652-1657.
667. Чернышов Б.Н., Диденко Н.А. Образование и стереохимия пероксофто-
ротитанатов по данным ЯМР l9F // Ж. неорг. химии. 1984. Т. 34, №8.
С. 2002-2011.
668. Кварацхели Ю.К., Петров В.М., Браверман И. Б. и др. Исследование
самоассоциации дифторида уранила в водной среде методом ЯМР 19F //
Ж. неорг. химии. Т. 33, №6. С. 1368-1371.
669. Bossev D.P., Matsumoto М., Nakahara М. *Н and 19F NMR Study of
the Counterion Effect on the Micellar Structures Formed by Tetraethy-
lammonoum and Lithium Perfluorooctylsulfonates // J. Phys. Chem. 1999.
V. 103B, №39. P. 251-8258.
670. JIanmaui H.M., Федотов M.A., Масленникова И.Г. Гидролиз летучего
оксофторотитаната аммония по данным ЯМР 19F, |7О, 49Ti // Ж. структ.
химии. 2004. Т. 45, № 1. С. 77-87.
Хлор, бром, иод
671. Fedotov М.А., Malkina O.L., Malkin V.G. 35 /37C1 NMR Chemical Shifts
and Nuclear Quadrupole Couplings for Some Small Chlorine Compounds //
Chem. Phys Lett. 1996. V. 258, №2. P. 330-335.
672. Lindman B., Forsen S. The Halogens — Chlorine, Bromine and Iodine //
Multinuclear NMR / Ed. J. Mason. — N. Y.: Plenum Press, 1987.
P. 421-438.
673. Hall C., Kydon D.W., Richards R.E., Sharp R.R. The chlorine-35 NMR
spectrum of Cl2 // Mol. Phys. 1970. V. 18, №5. P. 711-712.
674. Hall C., Richards R.E., Sharp R.R. Multisite chemical exchange effects in
the NMR spectra of quadrupolar nuclei: the aqueous Cl-CI2 system as an
example // Proc. Roy. Soc. (Lond.). 1970. V. A 318. № 1532. P. 119-141.
675. Sayto Y. 35C1 and 37Cl magnetic resonance of simple chlorine compounds //
Can. J. Chem. 1965. V. 43, №9. P. 2530-2534.
676. Johnson K.J., Hunt J.P., Dodgen H. W. Chlorine-35 NMR study of the shifts
and line shapes of some liquids inorganic chlorides // J. Chem. Phys. 1969.
V. 51, № 10. P. 4493-4496.
677. Федотов M.A., Зенковец Г.А., Гаврилов В.Ю. Исследование состояния
Ti(IV) в концентрированных солянокислых растворах методом ЯМР раз-
ных ядер // Ж. неорг. химии. 1999. Т. 44, №6. С. 973-976.
678. Pratt D. W., Myers R.J. NMR spectrum of liquid VCI4 // J. Chem. Phys.
1969. V. 50, №1. P. 555-556.
679. Blaser J., Lutz O., Stelnkilberg W. NMR studies of 35C1, 37C1, 79Br and 81Br
in aqueous solutions // Z. Naturforsch. 1972. Bd. 27a, № 1. S. 72-76.
680. Dickinson W.C. Hartree computation of the internal diamagnetic field for
atom // Phys. Rev. 1950. V. 80, № 4. P. 563-566.
681. McGurk J., Norris C.L., Tigelaar H.L., Flygare W.H. Molecular magnetic
properties of FC1 // J. Chem. Phys. 1973. V. 58, №7. P. 3118-3120.
682. Gierke T.D., Flygare W.H. An empirical evaluation of the individual elements
of the nuclear diamagnelic shielding tensor by the atom dipole methods //
J. Amer. Chem. Soc. 1972. V. 94, №21. P. 7277-7283.
683. fkenberry D., Das T.P. Nuclear magnetic shielding in alkali halide crystals //
Phys. Re V. 1965. V. 138a. №3. P. 822-828.
684. Dove M.F.A., Sandlers J.C.P., Appelman E.H. Comparative Multinuclear
(35C1, 79,81 Br, l27I and 17O) Magnetic Resonsnce Study of Perhalogate Anions
XO^ (X = Cl, Br, or I) in Acetonitrile Solution // Magn. Res. Chem. 1995.
V. 33, № 1. P. 44-58.
685. Shulman R.G. NMR and hyperfine infractions in paramagnetic solutions //
J. Chem. Phys. 1958. V. 29, №4. P. 945-947.
686. Zellman A.H., Morgan L.O. NMR of oxygen-17 and chlorine-35 in aqueous
hydrochloric solutions of iron(III) // J. Phys. Chem. 1966. V. 70, №9.
P. 2807-2813.
687. Zeltman А. П., Morgan L.O. NMR of oxygen-17 and chlorine-35 in aqueous
hydrochloric acid solutions of cobalt(II). Relaxation and chemical exchange //
J. Phys. Chem. 1969. V. 73, №8. P. 2689-2696.
688. Zeltman A.H., Matvijoff N.A., Morgan L.O. NMR of oxygen-17 and chlo-
rine-35 in aqueous hydrochloric acid solution of cobalt(II). Line shifts and
relative abundance of solution species // J. Phys. Chem. 1968. V. 72, № 1.
P. 121-127.
689. Chesnut D.B. 35C1 NMR contact shifts in aqueous cobaltous chloride solu-
tions // J. Chem. Phys. 1960. V. 33, №4. P. 1234-1237.
690. Zeltman A.H., Morgan L.O. Ligand substitution processes of complex ox-
ovanadium(IV) species in aqueous hydrochloric acid solutions. NMR of
oxygen-17 and chlorine-35 // Inorg. Chem. 1971. V. 10, № 12. P. 2739-2745.
691. Wertz J.E. Hydration effects in chlorine-35 nuclear spin resonance //
J. Chem. Phys. 1956. V. 24, №2. P. 484.
692. Троицкий С.Ю., Федотов M.A., Лихолобов В.А. Изучение продуктов
гидролиза солей палладия(П) // Изв. АН СССР. Сер. хим. 1993. №4.
С. 675-679.
693. Дон Г.М., Федотов М.А., Голованева И.Ф., Гладкая A.LU. Комплексо-
образование бисэтиленди-аминового комплекса золота(Ш) с хлорид-ионом
по данным ЯМР 35С1 // Ж. неорг. химии. 1994. Т. 39, № 1. С. 135-137.
694. Weingaertner Н., Mueller С., Hertz H.G. Composition of the first coordina-
tion sphere of nickel(2+) in concentrated aqueous nickel chloride and nickel
bromide solutions // J.C.S. Faraday tr. II. 1979. V. 75, № 11. P. 2712-2734.
695. Barbalat-Rey F. RMN de 35C1 en solution avec des ions paramagnetiques //
Helv. Phys. Acta. 1969. V. 42, №3. P. 515-538.
696. Reimarson P. NMR studies of the interactions between manganese (2+) and
perchlorate ions in aqueous solution // J. Magn. Reson. 1980. V. 38, №2.
P. 245-252.
697. Brucher E., Glaser J., Grenthe I., Puigdomenech I. On the Hydration of the
Lutetium(III) Ion in Water Acetone Mixtures // Inorg. Chim. Acta. 1985.
V. 109, №2. P. 111-116.
698. Doddgen H. W., Jordan A.D., Jordan R.B. NMR investigation of exchange
chlorine and chlorine ion in aqueous solution // J. Phys. Chem. 1973. V. 77,
№ 17. P. 2149-2153.
699. Щербаков В.А., Гусев Ю.К., Маширов Л.Г. и др. ЯМР-исследование
растворов перброматов уранила и нептунила // Радиохимия. 1989. Т. 24,
№ 5. С. 700-703.
700. Lehman J.F., Schrobilgen G.J., Christe К. О. et al. X-ray Crystal Structures
of [XF6][Sb2Fu] (X = Cl, Br, I), 3537C1, 79’81Br, and l27I NMR Studies and
Electronic Structure Calculations of the XF^ Cations // Inorg. Chem. 2004.
V.93, №22. P. 6905-6921.
701. Myers O.E. Kinetics of the trifluoriodide equilibrium // J. Chem. Phys. 1958.
V. 28, №6. P. 1027-1029.
702. Genser E. E., Connlck R.E. Exchange of iodide ion with triiodidc ion studied
by NMR // J. Chem. Phys. 1973. V. 58, № 3. P. 990-996.
703. Hertz H.G. Austauschgeschwindigkeiten brom- und jodhaltiger lonen in
wassriger Losung aus der Linienbreite der magnetischen Kernresonanz //
Z. Electrochem. 1961. Bd. 65, № 1. S. 36-50.
704. Hertz H. G. Ermitthing der Austaschgeschwindigkeit aus der Breite magnetis-
cher Kernresonanzlinien von Atomkernen mit Quadrupolmoment // Z. Elec-
trochem. 1960. Bd. 64, № 1. S. 53-54.
705. Garnett M. W., Halstead T.K. Bromine NMR study of the reorientation of
bromomercurio-N-acetyl-l-phenylalanine. bromomercurio-cinnamit acid and
bromomercuriomethane in water // J.C.S. Faraday tr. II. 1974. V. 70, № 12.
P. 1921-1927.
706. O'Reilly D.E.. Schacher G.E., Schug K. Rate exchange of halide nuclei
with mercury(II) in aqueous solutions // J. Chem. Phys. 1963. V. 39, №7.
P. 1756-1768.
707. Lincoln S.F., Saudercock A.C., Stranks D.R. Chloride exchange processes
on gallium(III) in concentrated aqueous dichloride solutions: a chlorine-35
and gallium-71 magnetic resonance study // J.C.S. Dalton tr. 1975. №8.
P. 669-673.
708. Lincoln S.F., Sandercock A. C., Stranks D.R. A 35Cl nuclear relaxation study
of the exchange of chloride ion on tetrachlorogallate(III) ion // J.C.S. Chem.
comm. 1972. № 19. P. 1069-1070.
709. Lincoln S.F., Sandercock A. C., Stranks D.R. A 35C1 NMR study of chloride
exchange on indium(III) and thallium(lll) in aqueous hydrochloric acid solu-
tion // Aust. J. Chem. 1975. V. 28, №9. P. 1901-1905.
710. Беляев А. В., Федотов M.А. О состоянии Rh(III) в концентрированных
водных растворах по данным ЯМР l03Rh и 35С1 // Коорд. химия. 1983.
Т. 9, №9. С. 1252-1257.
711. Ильин Е.Г., Зозулин А.Н., Буслаев Ю.А. Основной гидролиз фторидов
тантала по данным ЯМР 19F // Докл. РАН. 1994. Т. 335, № 5. С. 597-601.
712. Беляев А.В., Федотов М.А., Корсунский В.И. и др. О строении поли-
ядерных хлоридов родия(Ш) // Коорд. химия. 1984. Т. 10, № 7. С. 911-918.
Инертные газы
713. Brun Е.О., Oeser J., Staub H.H., Telschow G.G. The nuclear magnetic
moments of Xe129 and Xe131 // Phys. Rev. 1954. V. 93, №4. P. 904.
714. Anderson H.L. Precise measurment of the gyromagnetic ratio of He3 // Phys.
Rev. 1949. V. 76, № 10. P. 1460-1470.
715. Schrobilgen G.J. The Noble Gases // NMR and Periodic table / Ed.
H.K. Harris. В. E. Mann. — L.: Acad. Press, 1978. P. 459-464.
716. Jameson C.J. The Noble Gases // Multinuclear NMR / Ed. J. Mason. —
N.Y.: Plenum Press, 1987. P. 463-478.
717. Seppelt K., Rupp H.H. l29Xe-Kernresonanzuntersuchungen von Xe non-
verbindungen. Einfache Xenonderivate // Z. anorg. allg. Chem. 1974. H. 409,
№3. S. 331-337.
718. Brown Т.Н., Whipple E.B., Verdier P.H. High resolution l9F and 129Xe
magnetic double resonance spectrum of XeOF^ // J. Chem. Phys. 1963. V. 38,
№ 12. P. 3029-3030.
719. Sawyer J. F., Schrobilgen G.J., Sutherland S.J. X-Ray Crystal and Multinu-
clear NMR Study of FXeN(SC>2F)2. The First Example of a Xenon-Nitrogen
Bond // J.C.S. Chem. Comm. 1982. №4. P. 210-211.
720. Cowgill D.F., Norberg R.E. Local-magnetic-field Shift in Natural Xenon //
Phys. Rev. 1972. V. 6b, №5. P. 1636-1638.
721. Jameson C. J., Jameson A. К., Gutowsky H.S. Density dependence of l29Xe
chemical shifts in mixtures of xenon and other gases // J. Chem. Phys. 1970.
V. 53, №6. P. 2310-2321.
722. Giellespie R.J., Schrobilgen G.J. Fluorine-19 NMR study of the XeF^ cation
and F5XeSO3F // Inorg. Chem. 1974. V. 13, № 6. P. 765-770.
723. Giellespie R.J., Netzer A., Schrobilgen G.J. Fluorine-19 NMR studies of
some xenon(II) compounds // Inorg. Chem. 1974. V. 13, №6. P. 1455-1459.
724. Giellespie R.J., Schrobilgen G.J. Fluorine-19 NMR and Raman spectro-
scopic study of the (FXe)2SO3F+ cation. Preparation and characterization of
(FXe)2SO3F+AsF6' // Inorg. Chem. 1974. V. 13, №7. P. 1694-1698.
725. Giellespie R.J., Schrobilgen G.J. The XeF^, XeOF^, and XeO2F+ cations.
Preparation and characterization by fluorine-19 NMR spectroscopy // Inorg.
Chem. 1974. V. 13, № 10. P. 2370-2374.
726. Schrobilgen G.J., Holloway J.H., Granger P., Brevard C. Xenon-129 Pulse
Fourier-transform NMR Spectroscopy // Inorg. Chem. 1978. V. 17, №4.
P. 980-987.
727. Jto T., FrassardJ. Xenon-129 NMR study of xenon, adsorbed on Y-zeolytes //
J. Chem. Phys. 1982. V. 76, № 11. P. 5225-5229.
728. Gamblin R.L., Garner T.R. Polarization and relaxation processes in 3He
gas // Phys. Re V. 1965. V. A138, № 4. P. 946-960.
729. Seppelt K., Rupp H.H. 129Xe-Kernrezonanzuntersuchungen von Xenon-
verbindungen. Einfache Xenondervate // Z. anorg. ailg. Chem. 1974. H. 409,
№3. S. 331-337.
730. Henry R., Norberg R.E. Pulsed NMR in 2iNe in solid and liquid neon //
Phys. Rev. 1972. V. 6b. №5. P. 1645-1653.
731. Cowgill D.F., Norberg R.E. Spin-lattice relaxation and chemical shifts of 83Kr
in solid and liquid krypton // Phys. Rev. 1973. V. 8b. № 11. P. 4966-4974.
732. Adrian F.J. Quadrupolar relaxation of l3lXe in xenon gas // Phys. Rev. 1965.
V. A138, №2. P. 403-410.
733. Warren W. W., Norherg R.E. Nuclear Quadrupole Relaxation and Chemical
Shifts of 131 Xe in Liquid and Solid Xenon // Phys. Rev. 1966. V. A148, № 1.
P. 408-412.
734. Reuben J., Samuel D., Selig H, Shamir J. Hydrolysis of XeFe // Proc. Chem.
Soc. 1963. №9. P. 270.
735. Schumacher G.A., Schrobilgen G.J. Preparation of O2XeF2_I(OTeF5)I.
OXeF4-!/(OTeF5)!/. and XeF4_y(OTeF5)y (x = 0-2, у = 0-4) and Study by
l29Xe and 19F NMR and Raman Spectroscopy: The Oxygen Primary Isotopic
Effect in the l29Xe NMR Spectra of XeO2F2 and XeOF4 // Inorg. Chem.
1984. V. 23, № 19. P. 2923-29.
736. Mercier H.P.A., Sanders J.C.P., Schrobilgen G.J., Tsai S.S. The
Oxofluoroxenon(VI) Cation: X-ray Crystal Structure of XeOFT-SbF~ and a
Solution l7O 129Xe Nuclear Magnetic Resonance Study of the '',18O-Enriched
XeOF+ Cation // Inorg. Chem. 1993. V. 32, №4. P. 386-393.
737. Chen Q.J., Springel-Huet M.A., Frassard J. l29Xe NMR of Xenon Adsorbed
on the Molecular Sieves AIPO4-5, SAPO-5, MAPO-5, and SAPO-37 // Chem.
Phys. Lett. 1989. V. 129, № 1. P. 117-121.
738. Terskich V. V., Mudrakovskii I.L., Mastikhin V. M. !29Xe Nuclear Magnetic
Resonance Studies of the Porous Structure of Silica Gels // Faraday Tr. 1993.
V. 89, № 10. P. 4239-4243.
739. Terskich V. V., Mastikhin V.M., Okkel’ L.G. et al. An Application of l29Xe
NMR of Adsorbed Xenon to the Study of Silica-based Catalyst / Ed. J. Frais-
sard. — Netherlands: Kluwer. 1997. — 165 p.
740. Romanenko К. V., Ру X., D’Espinose de la Caillerie J.B. et al. 129Xe NMR
Study of Pitch-based Activated Carbon Modified by Air Oxidation / Pyrolysis
Cycles: A New Approach to Probe the Micropore Size // J. Phys. Chem. B.
2006. V. 110, № 6. P. 3055-3060.
741. Ripmeester J.A., Ratcliffe C.I., Tse J.S. The Nuclear Magnetic Resonance
of 129Xe trapped in Clatrates and some other Solids // J.C.S. Faraday Tr. 1.
1988. V. 4, № 11. P. 3731-3745.
Глава 3
МАГНИТНЫЙ РЕЗОНАНС ЯДЕР
ПЕРЕХОДНЫХ ЭЛЕМЕНТОВ
В главе рассмотрены элементы с частично заполненной d- или
/-электронной оболочкой и Zn, Cd, Hg, хотя в соединениях они яв-
ляются с£10-элементами. Во многих состояниях окисления переходные
элементы парамагнитные, и в них регистрация ЯМР обычно невоз-
можна; однако в состояниях dP, f°, d10 и /14, а также в ряде случаев
в состояниях с четным числом валентных электронов соединения диа-
магнитны и доступны для исследования методом ЯМР. Соединения
лантаноидов, кроме La(III) и Lu(III), парамагнитны.
В переходных металлах основную роль в химической связи играют
d- и /-электроны. Участие их дает основной (парамагнитный) вклад
в ХС, который наблюдают как у ядер самих переходных элементов, так
и у ядер, связанных с ними. Большие диапазоны ХС (рекордный у 59Со,
18000 м.д. = 1,8%) отражают величину этого вклада. Для оценки
последнего обычно пользуются приближением средней энергии [1].
При отсутствии детальных представлений о возбужденных состояниях
волновых функций, экранирование представляют в виде
В качестве меры диапазона ХС переходных элементов предложено [2]
изменение экранирования в тетрагалогенидных комплексах от МСЦ до
МЦ. Направление этого смещения принимается нормальным у 13С —
экранирование возрастает в ряду
С1 < Вг < I;
положение фтора в этом ряду неопределенное [3]. Большинство пере-
ходных металлов имеет нормальный галогенидный ряд экранирования,
а у Ti, V, Nb, Си зависимость противоположна.
Богатая химия переходных элементов предлагает широкое поле
деятельности для метода ЯМР. Обзор ЯМР переходных металлов
с квадрупольными ядрами сделан в [4, 5].
3.1. Скандий, иттрий, лантан, титан,
цирконий, гафний
Все эти элементы имеют магнитные изотопы со значительным при-
родным содержанием; в соединениях они обычно в (^-состоянии и,
следовательно, могут быть исследованы методом ЯМР. Свойства их
ядер приведены в табл. 3.1.
Таблица 3.1
Свойства ядер магнитных изотопов элементов групп IIIB и IVB
Изо- топ Спин NA, % Sens 1 Диапа- зон XC, M. Д. Q, барн Частота ЯМР эталона S, МГц2 Эталон
13с 1/2 — 1{‘H} — — 1{‘Н} —
45Sc 7/2 100% 570 — -0,22 24,2943 [Sc3+(aq)] —+ 0
89у 1/2 100 0,22 — 2,7 4,897 775 [Y3+(aq)] —+ 0
138 La 5 0,09 0,18 300 0,21 13.1933 [La3+(aq)] -» 0
,39La 7/2 99,9 112 300 5,68 14.1263 «
l7lYb 1/2 14,3 4,4 800 — 17,499 306 Yb(77-C5Me5)2/ ТГФ
175Lu 7/2 97,4 58 — 5,7 11,4073 —
47Ti 5/2 7,3 0,3 1700 0,29 5,642 757 ± ± 10 TiCl4, d-MePh, 80% мае.
49Ti 7/2 5,5 0,4 1700 0,24 5,644 259 ± ± 10 TiCl4, d-MePh, 80% мае.
5,639051 TiF4/NH4F/ H2O
91 Zr 5/2 11,2 2,0 1000 -0,21 9,297 303 Ср27гВгг/ТГФ
,77Hf 7/2 18,5 0,22 — 4,5 3,1203 —
179Hf 9/2 13,8 0,06 — 5,1 1.8693 —
235g 7/2 0,72 1,6- 10“4 — — 1,79223 UF6
’Относительно 1ЭС{’Н} с учетом природного содержания; 2 частота ЯМР в
поле Но, в котором частота ЯМР ’Н ровно 100 МГц; 3 рассчитано из величины
ядерного момента.
Из всех приведенных магнитных изотопов только 89Y не имеет
квадрупольного момента. Слишком большой квадрупольный момент за-
трудняет наблюдение ЯМР ядер 138La, 175’176Lu, 179Hf. Однако наиболее
распространенный изотоп лантана 139La имеет умеренный квадруполь-
ный момент и широко используется в ЯМР. Химическое поведение Sc,
Y, La более схоже с щелочноземельными металлами, чем с переход-
ными. Они в основном образуют лабильные комплексы, а образование
тетрагалоидных анионов для них нехарактерно. Преобладает степень
окисления М(Ш). Информацию об окружении их в растворе получают
скорее из данных ЯМ-релаксации, чем ЯМР.
Скандий. Высокая чувствительность и умеренный квадрупольный
момент ядра позволяют применять ЯМР 45Sc для изучения растворов
с концентрациями до 10-4 г-ат/л. Отсчет ХС ЯМР 45Sc ведут от
акваиона Sc3+ при бесконечном разбавлении.
Концентрационная и анионная зависимости ХС измерены в [6]
(рис. 3.1), изотопный эффект ХС акваиона при замещении ЩО на D2O
Рис. 3.1. Зависимость хим. сдвига 8 ЯМР 45Sc (А) в водных растворах
Зс(СЮ4)з (Л и ScCl3 (2) и ширины линии W тех же растворов, от их
концентрации
около 6 м.д. Линейность концентрационной зависимости при С < 1
г-ион/л указывает на взаимодействие с анионами; для водных раство-
ров 5с(С1О4)з наклон концентрационной зависимости —3,6 м.д./г-ион,
для ScBr3 он -2,5 и для ScCl3 +0,5 м.д./г-ион. Экстраполяция к ну-
левой концентрации для растворов нитрата, сульфата и иодида скандия
не совпадает с общей для других солей точкой; это указывает на
образование ацидокомплексов или ионных пар; на это же указывает
и ширина линий ЯМР в разбавленных растворах [7]. При низкой
температуре разрешается СТС в комплексе Sc с триметилфосфатом
(рис. 3.2).
Исследование ЯМ-релаксации в водных растворах проведено в
[8-11], влияние образования ацидокомплексов с галогенид-ионами —
-10 -20 -30 М.д.
Рис. 3.2. Спектр ЯМР 45Sc раствора [5с(ТМФ)е](СЮ4)з в MeCN, 238 К
в [12, 13], изучение иона ScF|“ — в [14, 15]; там же рассчита-
на константа квадрупольного взаимодействия (9,5 МГц), при том что
'J(Sc-F) = 180 Гц; исследование неводных растворов — в [16, 17].
Данные ЯМР 45Sc некоторых комплексов приведены в табл. 3.2. В сме-
шаннолигандных комплексах проявляется изомерия (табл. 3.3).
Иттрий. Низкое гиромагнитное отношение 89Y и спин 1/2 приво-
дит к долгим временам релаксации и малой чувствительности, и это
определяет трудности детектирования ЯМР. С момента первого из-
мерения [18] вышло несколько работ по ЯМР 89У [19-21]. Изотоп-
ный эффект растворителя для акваиона близок к таковому у Sc3+
(4,3 м.д.). Концентрационные зависимости для растворов перхлората,
хлорида и нитрата иттрия сходятся и одной точке с наклонами —1,1
и 7,5 м. д./г-ион. Данные ЯМР 89Y приведены в табл. 3.4.
Концентрационные зависимости ХС [У(ЭДТА)]-, У(СНзСОг)з,
YCI3, Y(NO3)3, У(С1О4)з изучены в [22]; при изменении концентрации
соли от 0 до 1 моль/л ХС меняется от 0 до «10 м.д. Измерения при
низких температурах в водно-ацетоновых и в водно-ацетон-фреоновых
смесях позволили обнаружить ацетоновый сольват иттрия отдельно от
акваиона, а также нитратокомплекс иттрия (рис. 3.3 [23]).
Ширина линии ЯМР 89Y в растворах постоянна в диапазоне pH
0-6 [18]; ЯМ-релаксацию 89Y и растворах нитрата иттрия изучали
в [21]. Механизм релаксации в основном диполь-дипольный и спин-
вращательный; в 1 М растворе 7) = 180 с, а в 3 М растворе — 85 с.
При таких временах релаксации значительный выигрыш во времени
регистрации могут дать методы с переносом поляризации с протонов
(для металлоорганических соединений), а также особые стационарные
методы [24].
Лантаноиды. Лантан-139 — магнитный изотоп, на котором сде-
лано преобладающее количество измерений на ядрах лантаноидов.
Преобладающее состояние окисления лантаноидов — М(Ш), в котором
они, кроме La и Lu, парамагнитны, а заключающий ряд лантаноидов
Таблица 3.2
Данные ЯМР 45Sc комплексов скандия
Комплекс Раство- ритель С, моль/л <5, м. д. (W, Гц) J(Sc-X), Гц Примечание
Sc(H2O)3+ н2о 0,1 0(142) — [17]
ScF3- н2о — 2,3 (40) J(Sc-F) = 172 To же
ScClg- MeCN <0,02 249 (340) — «
ScBrg- MeCN — 288 (275) — «
Sc(NCS)g“ MeCN 0,5 27 (50) — «
5с(ТМФ)3+ MeCN ~0,l -20,7 (35) 2J(Sc-P) = 37 «
5с(ДМФ)3+ ДМФ ~0,l 33 (320) — «
Sc(TMK)g+ MeCN 0,05 -56 (5) — ScCl3, HC1 10-12 M [4]
ScC14(H2O)2- H2O 1 124-144 — ScCl3, HC1 6-8 M [4]
ScC12(H2O)+ H2O 1 88-73 — ScCl3, HC1 1-3 M [4]
ScCI(H2O)j+ H2O 1 0,5-19 — [4]
Sc(NCS)g“ MeCN — 27 — To же
Sc(OH)7 NaOH 0,005 -116 — «
ScAcaCj- C6H6 — 86 — «
ScCp3 C6H6 — 14 — —
Sc(Acac)3 CHC13 — 89(1800) — *
ТМФ — триметилфосфат, ТМК — тетраметилкарбамид; * данные автора.
лютеций имеет столь большой квадрупольный момент, что обнаруже-
ние его ЯМР проблематично.
Из-за преобладающего природного содержания и меньшего квад-
рупольного момента используют 139La, хотя известны измерения и на
ядре 138La [26, 27]. Чувствительность ХС акваиона лантана к замеще-
нию на тяжелую воду 1,6 м.д. [27]; таким образом, чувствительность
ХС акваионов к замещению уменьшается в ряду 45Sc > 89Y > 139La.
Некоторые данные ЯМР 139La приведены в табл. 3.5 [4].
Таблица 3.3
Данные ЯМР 45Sc смешаннолигандных комплексов скандия [ScClnU-„] [17]
Комплекс 6, м.д. (W, Гц), L = ТМФ <5, м.д. (W, Гц), L = ДМСО 8, м.д. (ГУ, Гц), L = ДМФ
ScClg- 249 (340) 249 249
ScCl5L2- — — —
цис-ЗсСЬЦ" 179 (55) — 189
mpaHc-ScCU Ц" — 187 —
pe6-ScCl3L3 139(21) 164(55) 163 (42)
гран-5сС1зЬз 123 (63) 155(330) 160(168)
4uc-ScC12L+ 88 (30) 126(200) 187(138)
mpaHC-ScC^L^ 72(180) — 73
ScC1L2+ 33(150) — —
ScL3+ -20,7(35) 44(32) 33
Таблица 3.4
Данные ЯМР 89У в водных растворах
Соединение С, моль/л <5, м. д.1 Ссылка
К[У(ЭДТА)] 1 126 [4]
У(СН3СО2)з 1 37 То же
УС13 2,8 9 «
УВгз 2 7 «
Y(NO3)3 0,75 -1,9 «
« 3 -19 «
Y(W5O18)9- 0,4 -19 Н2О [25]
Y(PW11O39)21“ 0,3 -6 «
‘От 1,5 М раствора У(С1О4)з/0,2 D2O + 0,8Н2О.
В водных растворах наблюдают быстрый обмен координационной
воды La3+ со свободной. Влияние анионов на ХС La3+ изучено в [11,
27]. Зависимость ХС l39La от количества введенных НС1, HNO3, НСЮ4
при постоянной концентрации La3+ (0,3 г-ион/л) измерена в [11]; вве-
дение кислоты (до 10 моль/л) вызывает сдвиг на -70 м.д. для HCI
Рис. 3.3. Спектр ЯМР 89Y раствора У(1\Юз)з с добавлением HCIO4 в смеси
7% НгО+67% ацетон +26% фреон, 168 К, при соотношении СЮ^ : NO^: а —
СЮ4" 100%; б — 5:1; в — 2:1; г— 1:1; 6— 1:2
и +70 м. д. для HNO3; в 8 М растворе НСЮ4 в воде <5 (La3+) — 20 м. д.,
Т2 139La в водном растворе около 2 мс [28, 29]. Поведение ХС La3+
для разных противоионов в смеси ДМФ/НгО в зависимости от доли
последней показано на рис. 3.4 [30].
Рис. 3.4. Зависимость химического сдвига l39La (а) и приведенной к одинако-
вой вязкости ширины линии (6) от отношения ДМФ/НоО для разных солей
лантана
Таблица 3.5
Данные ЯМР 139 La (от [LaCl3] —+ 0)
Форма 8, м.д. (W, Гц) Растворитель
La-ЭДТА 570 H2O
La(NO2)3- 342 (600) —
LaCl3 -60(260) MeOH
LaN(CH2CO2)3 106(500) «
La3+ 100 НОАс
La3+ 36(1000) 5 M/NH4SCN
La(OSMe2)3+ 82(1800) —
La3+ 100(1300) 10М/НС1
LaCl3 7 3 М/Н2О
La(NO3)3 -8,5 2,7 М/Н2О
La(NO3)3 -13 2,7 M/D2O
LaCl3 3 (250) 0,4 М/Н2О
LaCl3 10 3 М/Н2О
La(C104)3 0 (250) 0,36 М/Н2О
La(C104)3 -25 (2000) 0,3 М/Н2О-7,5 М/НС1О4
La(NCS)47“ * 196 (4400) —
La(NO3)3- * -60(260) —
La(NO3)3 0 (700) 0,29 М
La(NO3)3 -75(1700) 0,29 М, 10M/HNO3
* Есть данные с меньшим числом лигандов.
ЯМ-релаксация 139La показывает, что нитрат-ион образует внутри-
сферный комплекс с La3+ [31], а СЮ^“ — внешнесферный. Ширина
линий в водно-органических растворах нитрата, хлорида и перхлората
лантана показана на рис. 3.4, б. В метанольных растворах La3+ обра-
зует биядерные комплексы (6 139La = 293 м.д.) [32]. Параметры ЯМР
лантана в ацетонитрильных растворах показаны в табл. 3.6 [32].
Влияние на ЯМ-релаксацию 139La взаимодействий с ЭДТА, ацетат-
и роданид-ионами изучено в [34], с макромолекулами — в [35]. Ком-
плексы лантана с дитиокарбаматами исследованы в [36].
Таблица 3.6
Данные ЯМР l39La в MeCN (294 ± 2 К)
Комплекс Концентрация, моль/л S, м. д. (ГК, Гц) Примечания
[La(H2O)6]3+ 0,16 0(140) СЮ4-СОЛБ
[La(MeCN)6]3+ 0,2 -129(380) 004-СОЛЬ
[La(NO3)6]3+ 0,06 -60 (260) NBuJ-соль
[Еа(ДМФ)8]3+ 0,21 -39(1700) NBu^-соль
[La{P(NMe2)3O}6]3+ 0,2 66 (930) При избытке P(NMe2)3O
[Та(ДМСО)8]3+ 0,21 82(1800) 004-СОЛЬ
[La(NO2)6]3- 0,07 342 (600) NBu4nNO2 в избытке
[LaCl6]3- 0,05 851 (200) NBuJCl в избытке
[LaBr6]3- 0,11 1090 (500) NBu"Br в избытке
[La(en)]“ 0,2 40 (3800) 004-СОЛЬ
[La(en)4]n“ 0,2 400 (3800) 004-СОЛЬ
[La(dien)3]3+ 0,18 450 (2400) С104-СОЛЬ
Из других лантаноидов имеются данные ЯМР 171 Yb в циклопента-
диенильных соединениях Yb(II) (табл. 3.7 [37]).
Таблица 3.7
Данные ЯМР 171 Yb
Соединение Растворитель Т, К 6, м. д. (W, Гц) ТК, м.д./К
Yb(77-C5Me5)2(OEt2)2 Et2O 308 36 (90) +0,25
Yb(77-C5Me5)2(NC5H5)2 NC5H5 338 249 (70) + 1,46
Yb(NR2)2(OEt2)2 Et2O 193 614(70) -0,18
Yb(NR2)(/i-NR2)2 Толуол 263 796 (90) +0,35
УЬ(77-С5Ме5)2(ТГФ)2 ТГФ 173 87(24) +0,69
Титан. Редкой особенностью ЯМР титана является возможность
наблюдения ЯМР сразу на обоих магнитных изотопах, линии которых
разнесены на 271 м.д. [38] (в поле 7,05 Т на 4600 Гц). Малое гиро-
магнитное отношение в сочетании со значительным квадрупольным
моментом ядра позволяет изучать только симметричные структуры. Ис-
следованы тетрагалогениды титана и ион TiFg-, что дает возможность
оценить диапазон ХС ЯМР титана. Галогенидный ряд титана обращен
по сравнению с 73Ge: наблюдается уменьшение экранирования в ряду
CI > Вг > I в соответствии с энергиями низших переходов 34 000 см -1
(T1CI4), 28 680 (TiBr4), 9400 см -1 (ТЩ) [38]. Данные ЯМР соединений
титана приведены в табл. 3.8.
Таблица 3.8
Данные ЯМР титана
Соединение Растворитель &Ti, m. д. (W, Гц) Ссылка
TiF4 H2O 0 [4]
« 48% HF/H2O -5 [38]
TiCl4 4. жидк. 1162 [4]
TiBr4 C6H6 1675 [4]
Til4 C6H6 2440 [4]
CpTiX2, X = (K222)2Ti(CO)6 MeCN -227 [39], J(49Ti-C) = 23
TiFg- 40% HF 0, 'J(Ti-F) = 59 TiO0i2F4i6, 273 К [42]
[Ti2(/z-O)F10]4- « 22(1400) «
В смеси TiCl4 и TiBr4 наблюдается одна линия (для каждого изото-
па титана), сдвиг которой меняется соответственно долям компонентов;
это указывает на быстрый обмен лигандов в растворах тетрагалоге-
нидов титана при «300 К. ХС T1F4 в 48%-ном водном растворе HF
не зависит от концентрации титана; в водных растворах линия T1F4
уширена [38]. Константа ССВ в TiFg- J(F-Ti) = 33 Гц (определена по
ширине линии ЯМР 19F) [40]. Возможности ЯМР титана обсуждены
в [4, 41].
При растворении оксифторида титана состава TiOo,2F4i6 в 40% HF
наблюдали две линии 49Ti [41] — узкую (Ом.д., TiFg-) и широкую
(22м.д.). Наблюдаемая при этом линия кислорода 52 м.д. отнесена
к ^-ОН в димере [FsTi-^-OH-TiFs]3-.
Растворы Т1С14 в концентрированной водной НС1 изучены в [43];
надежно зарегистрировать ЯМР на ядрах титана не удалось, одна-
ко по ЯМР 17О наблюдали два типа концевых атомов О=Т1 812
и 791 м. д., что означало наличие в растворе двух типов оксохлоридных
комплексов титана. Доля последних возрастала по мере содержания
воды в растворе вместе с увеличением доли полиядерных комплек-
сов, не наблюдаемых в спектрах ЯМР 17О. Предполагается, что ли-
ния 791 м.д. относится к [ОТ1С14]2-, а линия 812 м.д. — к димеру
[(Т1ОС13)2]2-.
Широкое применение металла титана как конструкционного матери-
ала и его соединений (пигмента и наполнителя в производстве красок,
пластмасс, волокон, резиновой и бумажной промышленности), а также
в гетерогенных и гомогенных [44] катализаторах имеет следствием
большое количество работ по ЯМР титана. Использование ЯМР в
сверхвысоких полях (600 МГц для 'Н) позволят провести исследования
тех форм, которые не доступны из-за большой ширины линии и слабого
сигнала.
Цирконий. Чувствительность ЯМР 91 Zr выше, чем у титана. Хотя
определение гиромагнитного отношения сделано еще в 1957 г. [45],
первые работы по определению ХС появились только в 1981 г. Сравне-
ние ХС 91 Zr в ионах ZrXg- (X = Cl, F) показало, что ядро 91 Zr более
экранировано в хлориде, чем во фториде [46]. Измерены [46] эффекты
замещения в тетраэдрических комплексах циркония (табл. 3.9).
Таблица 3.9
Данные ЯМР циркония-91
Соединение Растворитель 8 Zr, м. д. W, Гц 8 |3С, м. д.
Cp2ZrCl2 ТГФ -122 276 116,2
Cp2ZrClBr ТГФ -66 237 —
Cp2ZrBr2 ТГФ 0 19 116,2
Cp2ZrI2 ТГФ 126 134 115,1
Cp4Zr ТГФ — — 113,5
(NH4)2ZrF6 D2O -191 50 -48’
H2ZrCl6 НС1 601 245 —
* 19р
Константа ССВ Zr-H в Zr(BH4)4 использована в [47] как индикатор
межмолекулярного взаимодействия (2J(Zr-H) = —28 Гц, ’J^’B-Zr) —
= 18 Гц). Довольно большая ширина линии ЯМР 91 Zr в ZrF|“ в вод-
ных растворах (500 Гц при 25 °C в поле 9,4 Т) указывает на обмен
фторида и то, что применение метода ЯМР 91 Zr ограничено изучением
высокосимметричных форм.
Гафний. Низкая чувствительность, низкая частота ЯМР и боль-
шой квадрупольный момент наиболее благоприятного для измерений
изотопа 177Hf не позволили пока обнаружить сигнал ЯМР гафния
в жидкой фазе.
3.2. Ванадий, ниобий, тантал, хром,
молибден, вольфрам
Богатая неорганическая и металлоорганическая химия этих элемен-
тов стимулирует применение метода ЯМР их ядер; банк данных ЯМР
этих элементов значителен. Каждый из этих элементов существует
в нескольких состояниях окисления; состояния с нечетным числом
электронов парамагнитны и могут наблюдаться в ЯМР в полиядер-
ных формах в спин-спаренном состоянии. Парамагнитные формы этих
элементов наблюдают методом ЭПР, и для них существует большой
банк данных [48-49 и др.]. Свойства «магнитных» ядер этих элементов
приведены в табл. 3.10.
Ванадий. ЯМР 51V применяется в неорганической химии с 1965 г.
[50], и данные ЯМР ванадия включены в ряд обзоров [4, 51-53].
Сигнал ЯМР ванадия удобен для накопления из-за умеренной величи-
ны Т[ и не слишком короткого Т? (ширина линии обычно 20-200 Гц).
Это позволяет широко использовать ЯМР ванадия в неорганической
химии до концентраций 10-4 г-ат/л и дает возможность адекватно
сопоставлять данные ЯМР и электронной спектроскопии. Линии ЯМР
50V в одних и тех же соединениях в 16 раз шире, чем ЯМР 51V;
учитывая более низкую частоту ЯМР 50V и низкую чувствительность,
применение его малоперспективно. Изотоп 50V можно использовать
как метку для изучения кинетики изотопного обмена методом ЯМР
ванадия, для изучения механизмов реакций или для синтеза меченых
изотопом препаратов для ИК-спектроскопии.
Применение высоких полей существенно расширяет возможности
метода, и для разрешения сигналов в спектрах требуются поля 9 Т.
Диапазон ХС V(V) простирается от 2570 ([VSe^3-) до —895 м.д.
(VF5). Изменение ХС ванадия определяется парамагнитным вкладом
в экранирование [52]. Расчет диамагнитного вклада (для VO3- —
1708 м.д., CpV(CO)4 — 1712 м.д., V(CO)g — 1718 м.д.) показывает,
что он почти не меняется. Парамагнитный вклад в экранирование
Таблица 3.10
Свойства ядер магнитных изотопов групп VB-VIB
Изо- топ Спин NA, % Sens 1 Диапазон ХС, м.д. Q, барн Частота ЯМР эталона S, МГц2 Эталон
13с — — Н'н) — — 1{‘Н) —
50 V 6 0,25% 0,21 2500 0,2 9,9700 VOC13
51у 7/2 99,8 720 « -0,05 26,228 «
93Nb 9/2 100 910 2500 -0,2 24,4425 NbOCl3/ MeCN
181 Та 7/2 100 69 8000 3 11,9974 Et4NTaCl6/ MeCN
53Сг 3/2 9,6 16 2000 0,03 5,652 2 M Na2CrO4/H2O
95Мо 5/2 15,7 9,7 4000 0,12 6,5145 2 M Na2MoO4/H2O
97 Мо 5/2 9,5 6,1 « 1,1 6,652 «
ie3W 1/2 14,4 0,02 7000 — 4,157064(10) 2 M Na2WO4/H2O
4,161 733(20) WF6
'Относительно 13С{'Н} с учетом природного содержания; 2 частота ЯМР в
поле Но, в котором частота ЯМР 'Н ровно 100 МГц.
в приближении средней энергии для ядра ванадия можно выразить как
аР = const 'У*1, (3.1)
А.Е
где АЕ, (r~3}3d и Du связаны с общей силой лигандов, нефелоксетиче-
ским эффектом и ковалентностью связи соответственно. В зависимости
от состояния окисления ХС ванадия меняется от 440 до -790 м. д. для
V(V), от -870 до -1540 м.д. для V(I) и от -1679 до -1950 м.д. для
V(—I). Данные ЯМР 51V приведены в табл. 3.11, 3.12.
Диаграмма состояния ванадия в водных растворах, полученная хи-
мическими методами (см. рис. 3.5), в целом согласуется [65] с данными
ЯМР, хотя последний обнаруживает большее число форм.
Состояние V(V) в водных растворах методом ЯМР 51V изучали
в ряде работ, начиная с [50]. В щелочной области существует равно-
Таблица 3.11
Данные ЯМР 51V моноядерных форм ванадия
Форма Раствори- тель <5, м. д. J(X-V), (ИС Гц) X Источник
VOF3 ТГФ -757 — . — [54]
VOCI3 Ч. жидк 0 — — То же
VOBr3 То же 432 — — «
СС14 417 — — «
VO(NEt2)3 MeCN -365 — — «
VPCl2OMe Ч. жидк. -290 38 31 р «
v(co)6- — -1592 166 13с [52, 55, 56]
V(CO)5L, L = NH3 — -1670 — — То же
PH3 — -1850 170 31 р [57]
PF3 — -1961 490 31 р То же
P(OR)3 — -1915 355-370 31 р R-алкил [52, 58]
AsPh3 — -1800 — — [57]
SbPh3 — -1881 — [54]
BiEt3 — -1743 — — То же
[V(CO)2CN]2- — -1850 — [59]
транс- [V(CO)2(PF3)4]_ MeCN -1948 506 31 р То же
V(PF3)6- — -1946 510 31 р [61]
[VO2(C2O4)212- Н2О -533 — — [62]
vo2ci2- MeCN -301 — — То же
vo2f2- ДМСО -64 — — «
VOCl(Acac)3 ТГФ -344 — — «
vo+ HNO3/H2O, pH = 0 -545 850 — [50, 63]
pH = 2 -541 580 — [66]
Таблица 3.12
Данные ЯМР 51V ванадия в анионных формах в кислородном окружении в
водных растворах
Форма 6, M. Д. W, Гц Примечания, источник
vof -534 — pH = И
[(VO3)4]4- -573 — pH = 8 (A1)
[VW5O19]3- -522 25 pH = 2,8 (D) [69]
-522 60 pH = 1,7 [70]
[V2W4O19]4- -507 45 pH = 4-8,5 (D) [69]
-513 110 pH = 2 [64]
[V3W5O19]5- -499 — pH = 7 (D) [69]
-484 75 pH = 8 [69]
[V10O2e]6- -510; -492; -418 — pH = 7 (B) [70]
[HV10O28]5- -519; -502; -422 — pH = 3 (B) [70]
[PVWnO40]4- -551 40 pH = 1 (C) [52]
[PVMoHO40]4- -530 50 pH = 0-3 (C) [71]
[SiVWnO40]5- -548 40 pH = 5,5 (C) [36]
[H2VWnO40]7- -539 97 pH = 4 (C) [62, 64]
[VaV3W9O40]7- -552* — A — центральный атом (С) [68]
-533; -530; -529 — Периферийные атомы в разных изомерах
-528,5; -527; -522 — —
-508; -502; -480 — —
[PV2‘V12O42]9- -522*; -572; -587 — pH = 4
-528*; -581; -594 — pH = 1,5 (Е) [73]
1 Индекс указывает тип структуры рис. 3.6; * атомы вне каркаса Кеггина.
весне между ионами VO^-, УгО4-, (УОз)"-, их протонированными
формами и неидентифицированными ионами. Ход ХС в зависимости от
pH приведен на рис. 3.5.
Рис. 3.5. Диаграмма состояния ванадия в водных растворах (а) и зависимости
ХС ЯМР 51V разных форм ванадия от pH. Точки соответствуют измерени-
ям при C(V) 0,0001 г-ат/л (б): А — VO3- и его протонированные формы;
В — УгО?- и его протонированные формы; С — неопознанные формы; D —
[(VO3)4]4-; Е - [(VO3)5]5-; F - [V10H2e]6-; G - VO2+
<551V, м.д.
Протонирование вызывает значительные изменения ХС в области
pH от 10 до 7 и соответствует переходам
V2O^“ -> HV2O3“ -> H2V2O2“ H3V2O7(B),
аналогично изменению ХС 31Р от pH фосфат-иона. Линии —573 (D)
и -580 м.д. (Е) относятся к циклическим формам (УОз)4“ и (УОз)^-
(отнесение подтверждается спектрами ЯМР 17О, из которых п не
определяется). Более детальное отнесение линий в [66, 67] надо по-
лагать предположительным. Данные ЯМР полиядерных форм ванадия
обсуждаются в [68] и приведены в табл. 3.12, схема структур — на
рис. 3.6.
Спектр ЯМР 5IV декаванадата [VioO^]6- приведен на рис. 3.7.
В комплексах и молекулах с высокой симметрией окружения ва-
надия наблюдаются константы ССВ, некоторые из них приведены
в табл. 3.13.
В комплексах Ср(СО)3Р7з величина 'J(V-P) возрастает в ряду
РН3 < PR3 < P(NR2)3 < P(OR)Ph2 < P(OR)2Ph < P(OR)3 < PF3 (R -
алкил) и коррелирует с электроотрицательностью лиганда.
Ширина линий ЯМР 51V определяется квадрупольным механизмом
релаксации [79], т. е. величинами ГЭП и тс вращательного движения
Рис. 3.6. Строение анионов: а — [(VO3)4]4 ; б — [УщОгв]6 ; в — ванадофосфат
[PV14O42]9-
-420 -460 -500 -540
Рис. 3.7. Спектр ЯМР 51V декаванадата
Таблица 3.13
Константы спин-спинового взаимодействия ванадия-51
Форма 'J(V-X), Гц X Источник
CpVH(CO)3 20,3 *H [52]
V(CO)6 166 13c [55-56]
(Me3SiO)3VNBul 95 14 N [75]
vo; 62 17O [67]
vof; 116-140* ,9F [77, 78]
VF- 88 ,9f [78]
[vw5o19]3- 11 le3W [62]
CpV(CO)2PZ3 80-430 31 p [52]
Зависит от температуры.
комплекса (иона). Во многих системах наблюдаемую ширину линии
определяет химический обмен. Диапазон ширины линий в состояниях
без химического обмена простирается от 1,5 Гц [V(CO)g], симметрия
Од, до нескольких килогерц в симметрии Cs.
При подкислении раствора ванадата образуется полиоксоанион де-
каванадат (рис. 3.5, б) и его протонированные формы. В этом анионе
Таблица 3.14
Данные ЯМР 51V и 170 пероксованадатов
Форма <55iV(i7O) Форма <551V(17O) Форма <551V(I7O)
vo3- -541(-) VO3(OH)2- -539(-) VO2(OH)2- -560(-)
VO3(OO)3- — VO2OH(OO)2 769 (580) V(OH)2(OO)2(H2O)2- —
VO2(OO)3- — VOOH(OO)2- -771 (1030) V(OO)3“ -700(1070)
VO(OO)3" 769 (600) VOH(OO)2- -737 (-) -738 (-)
vo+ -830(980) VO(OO)+ 628(1050)
-546(-)
Форма <551V(i7O) Форма <551V(17O)
[{VO2(OO)}2O]4- -636(-) [HO2(OO)VOV*O(OO)2]3- -624; -743*
HO3VOV*O(OO)3- -556; -743 * [{VO(OO)2}2OH]3- -766(1060)
H4(oo)2ov*ovo2(oo)] -671 (930); -674* (1210)
три типа атомов ванадия с соотношением 2:4:4 дают три линии ЯМР
с соответствующим соотношением интенсивностей (см. табл. 3.12). При
кислотности выше изоэлектрической точки (pH 1-3), в зависимости
от концентрации ванадия в растворе, образуется ион [VO(H2O)x]2+
с быстрым обменом молекул координационной воды.
Атомы ванадия изоморфно замещают атомы Мо и W в изо- и ге-
терополимолибдатах и -вольфраматах; и ХС, и ширины линий за-
висят от структуры и состава аниона (табл. 3.13) [62, 64, 68, 69,
71]. Ванадий также образует гетерополианион ванадофосфат PV14O42
[73, рис. 11.3, (Е)]. Изучению оксо- и пероксокомплексов ванадия по-
священа работа [63], где были идентифицированы 11 пероксованадатов
с ХС от —746 до -623 м. д. Отмечено, что при связи фосфора и ванадия
(непосредственно или через атом кислорода) экранирование ядер 51V
и 31Р меняется антибатно [52, 71]. Изучение образования пероксоком-
плексов ванадия в сильных полях [80] уточнило формы, обнаруженные
в [63] (табл. 3.14). Заметим, что ХС ЯМР 17О 1000-1200 м.д. относит-
ся к концевому к ванадию атому О.
ХС 51V имеют заметный температурный ход вследствие увеличения
парамагнитного вклада из-за уменьшения ДЕ1 (роста заселенности
возбужденных уровней) с температурой (табл. 3.15).
Таблица 3.15
Химические сдвиги 51V и их температурные коэффициенты ТК(<5) комплексов
ванадия [81]
Форма 6, м. д., 300 К ТК(<5), м.д./К
[V(CO)(PF3)5] -1963 -0,32
Et4N[V(CO)6] -1955 -0,30
[CpV(CO)4] -1533 -0,61
[CpV(CO)3P(p-C6H4F)3] -1322 -0,70
[CpV(CO)3py] -519 -1,17
[СрУ(СО)3ТГФ] -107 -1,5
[У(МО)2(ТГФ)4] 282 -1,23
В полиоксоанионах ХС 51V коррелирует со средним ^-фактором
восстановленных форм этих анионов [64]. ХС замещенных серой вана-
датов и их зависимость от pH изучены в [82] (табл. 3.16).
ХС ванадия в замещенных гексавольфраматах, ГПА структур Кег-
гина и Доусона измерены в [83]; там же приведен спектр ЯМР
всех пяти изомеров аниона [PV2Wio04o]5-. Оксофторидные и оксо-
хлоридные комплексы V(V) изучены в [84]. Лабильные комплексы
Таблица 3.16
Данные ЯМР 51V оксотиованадатов [82]
Форма 55IV Форма 551V Форма 55IV
VO3- -541 VO3OH2- -539 V2O|" -561
VO3S3" -250 VO2SOH2- -121 v2-s*- 1457
VO2S3- 184 VOS2OH2- >230
VOS3- 740 VOS3H2- 748
VS3" 1395 VS4H2- 1392
ванадата с пирофосфатом и арсенатом изучены в [85]. Диванадаты
типа [У2О3(Н2О)8-хи]4“ж (L = Cl-, NO^, HSO^) изучены в [86].
В [87] изучено комплексообразование VOCI3 в растворах лигандов
(табл. 3.17).
Таблица 3.17
Взаимодействие VOCI3 с лигандами в растворах при 303 К по данным ЯМР 51V
Лиганд Раствор <5, м. д. Лиганд Раствор <5, м. д.
NEt3 Ч. жидк. -3,4 MeCN CHCI3 -20
Пиридин СС14 -28; -59; -412 Ме2СО Ч. жидк. 39; -388
MeCN СС14 -114,-391 ДМФ Ч. жидк. -366
Ниобий. Метод ЯМР ниобия применяется гораздо реже, чем
ЯМР ванадия, хотя чувствительность последнего ниже. Химическое
приложение ЯМР ниобия начато в 1965 г. [77], однако известно не
так много работ [51, 88-96]. Эталон NbOCla/MeCN имеет ширину
линии ~700 Гц [89], что позволяет измерять ХС в поле спектрометров
400 МГц с точностью ~1 м.д. Диаграмма ХС ниобия приведена на
рис. 3.8.
Методом ЯМР ниобия изучено замещение хлора на бром
в NbBr^Clg-^ и найдены все 10 возможных изомеров в этой
системе [89, 91, 93].
Экранирование в комплексах NbCIsX [98] увеличивается в ряду
I- < < Вг- < С1- < МеСН^" < SCN- < NCS- < F-, т. е. прояв-
ляется обратная галогенидная зависимость ХС. Экранирование 93Nb
в разнолигандных комплексах ниобила NbOXY (X, Y = С1—, Вг-, F-,
NCS-) изучено в [94]; из-за короткой связи Nb=O ХС определяется
лигандами в плоскости комплексообразователя (сам комплекс ниобила
по геометрии близок к квадратной пирамиде), а транс-лиганд к кис-
лороду практически не влияет на ХС.
2000
1000
0
-1000
-2000 м.д.
Рис. 3.8. Диаграмма химических сдвигов 93Nb
Комплексы со связями Nb-S и Nb-Se изучены в [95], фосфи-
новые производные СрЫЬ(СО)д — в [96]. Проявляется корреляция
ХС-(ДЕ)-1, однако в [91] считают, что изменение парамагнитного
вклада определяет только 2/3 изменения ХС в системе ЫЬС1жВгб-ж.
Известны КССВ 'J(Nb-F), лежащие в пределах от 342 Гц (NbF^“) [88]
до 410 Гц (NbOF^) [94]. Децет спектра 19F иона NbF^ показывает
расщепление на ниобии (I = 9/2), рис. 3.9 [97]. В [98] изучены карбо-
Рис. 3.9. Спектр 19F иона NbF6
нилы металлов группы V, в том числе и ниобия. В растворах CH2CI2
солей наблюдали линии ЯМР 93Nb с параметрами: для Na3[Nb(CO)s]
<5 = -2138 м.д. (РК = 1100 Гц), для Na[Nb(CO)6 6 = -2136 м.д.,
1 J(Nb-C) = 236 Гц, для Na[Nb(CO)5NH3] 6 = -1860 м.д. (W =
= 2000 Гц). Температурный коэффициент ХС для Е14Ы[ЫЬ(СО)б]/ТГФ
-0,15 м.д./К [99]. В политиометаллате [NbgSiy]4- [100] два типа
ниобия в отношении 1 : 5; в растворе MeCN при 335 К наблюдали
две линии с соответствующими параметрами 6 — 1'109 м.д. (2600 Гц)
и 8 = 1208 м.д. (2400 Гц).
Для отнесения линий ЯМР металла в разнолигандных комплексах
в тех случаях, когда не наблюдается КССВ металл-лиганд, применя-
ются эмпирические подходы, основанные на аддитивности характерных
инкрементов ХС. Для отнесения линий в спектрах комплексов с двумя
лигандами предложено два подхода.
В парной модели [89] инкремент соответствует числу определенных
ребер октаэдра в комплексе (всего 12 инкрементов). В транс-модели
[91] инкрементом является диагональ (координата) комплекса — всего
6 инкрементов (рис. 3.10).
транс-
цис-
Рис. 3.10. Набор всех возможных октаэдрических комплексов с двумя разными
лигандами
Примеры расчетов приведены в табл. 3.18 для [NbClxВг^-.,.] .
Тантал. Большой квадрупольный момент 181 Та делает ЯМР 181 Та
малоинформативным. В растворе тантала в HF : HNO3 =1:1 наблю-
дали линию шириной 36 кГц при комнатной температуре и 10 кГц
при 80 °C, предположительно относя ее к TaF^" [101]. Определено
отношение 7(l8lTa): 7(2D) = 0,7798 ± 0,0002. Изучение растворов ком-
плексов тантала на частоте ЯМР 8 МГц позволило оценить диапазон
ХС тантала-181 >8000 м.д. ([102], табл. 3.19).
Таблица 3.18
Пример расчета ХС металла в октаэдрическом комплексе по схемам [89] и [91]
№ Комплекс Ребро [89] Cl-Cl Cl-Br Br-Br 0"расч1 <^наб бТрасчН Диагональ [91] С1-М-С1 Cl-M-Br Вг-М-Вг
1 NbCl6- 12 0 0 0 0 (±0,5) 0 3 0 0
2 NbClsBr" 8 4 0 143,2 -126 145,3 2 1 0
3 цис-МЬгСиВг- 5 6 1 -246 290,6 1 2 0
4 mpawc-NbCUBr^ 4 8 0 286,4 -253 211,67 2 0 1
5 apaH-NbChBr^" 3 6 3 -371 436 0 3 0
6 реб-МЬС1зВг7 2 8 2 -378 357 1 1 1
7 цис-МЬС12Вг7 1 6 5 -492 502,3 0 2 1
8 транс-МЬС12ВС“ 0 8 4 -497 423,3 1 0 2
9 NbClBr" 0 4 8 -616 568,6 0 1 2
10 NbBr" 0 0 12 635 -635 635 0 0 3
rj(Cl-Cl) = 0, rj(Br-Br) = 52,92, т/(С1-Вг) * = 35,8; * среднее комплексов rj(Cl-Cl) = 0, Tj(Cl-Br)* = 145,3, r/(Br-Br) = 211,67; * среднее комплексов
Таблица 3.19
Данные ЯМР 181 Та соединений тантала
Соединение Концентрация, растворитель <5, м.д. (IV, Гц)
Et4N[TaCl6] 0,6 M/Me2CO;MeCN 1:1 0 (4300)
K2TaF7 Нас./48 % HF -2295(28700)
Et4N[Ta(CO)6] 0,6 М/ТГФ:МеСИ 5:1 -3450 (6700)
Хром. Характерное для соединений хрома состояние окисления
Сг(Ш), в котором хром парамагнитен с долгим временем электронной
релаксации, исключает эти соединения из области применения ЯМР
хрома; ЯМР наблюдают в соединениях Cr(VI). Из-за квадрупольно-
го уширения ЯМР можно наблюдать только для атомов Сг в сим-
метричном окружении. Линия карбонила хрома отстоит от СгО^ на
-1795 м.д. [103], что позволяет оценить диапазон ХС ЯМР 53Сг.
Концентрационная зависимость ХС СгО^- около -5 м.д./г-ион; ка-
тионы увеличивают экранирование в ряду Na+ < К+ < NH^“ < Cs+.
Из ЯМР 31Р карбонилфосфиновых комплексов известна КССВ
'J(Cr-P) = 153 Гц [4], для СгО3” J(Cr-O) = 10 Гц.
ЯМ-релаксация 53Сг в СгО3- изучена в [83], причем Т\ = 55 мс
(1 г-ион/л) и мало зависит от концентрации хромат-иона. Для этого
иона определена константа квадрупольного взаимодействия QCC =
= 0,75 МГц и КССВ ‘J(53Cr-17O) = 10 Гц [76]; йз ЯМР 31Р кар-
бонилфосфиновых комплексов известна КССВ *J(Cr-P) = 153 Гц [4].
Влияние заместителей на ХС 53Сг в комплексах Cr(CO)sXY изучено
в [104] (табл. 3.20).
Таблица 3.20
Параметры ЯМР комплексах Cr(CO)sXY в растворах ТГФ при 293 К
X Y <5, м.д. (W, Гц) от внешнего Сг(СО)в
н nh2 85 (2000)
СНз nh2 103(1400)
« NMe2 165 (400)
« OMe 187 (900)
« CN 21 (50)
Ph nh2 159(1150)
NMe2 199 (280)
CN 21 (150)
МеО MeO 109 (350)
ЕЮ NEt2 145(140)
Молибден. Молибден имеет два магнитных изотопа 95Мо и 97Мо,
из которых последний практически не используют в ЯМР из-за боль-
шего квадрупольного момента (и соответствующего уширения линий
ЯМР) и меньшей чувствительности. С момента наблюдения ЯМР
95Мо и определения его магнитного момента [105] опубликованы де-
сятки работ по химическому применению ЯМР 95Мо, половина из
которых относится к изучению металлоорганических соединений Мо.
ХС молибдена отсчитывают от очень узкой линии иона МоО^- при
экстраполяции к бесконечному разбавлению; влияние катионов и изо-
топного замещения H2O/D2O (~1м.д.) изучено в [106]. Наиболее
сильно на ХС влияет ион Li+ (—2,4 м. д. • л/г-ион), менее всего Na+
(0,7 м. д. • л/г-ион). Раствор молибдата натрия в D2O при концентрации
1,7 моль/л имеет ХС = 0. ХС некоторых ионов и комплексов показаны
в табл. 3.21.
Таблица 3.21
Данные ЯМР молибдена-95
Форма Раствори- тель <5, м. д. (W, Гц) Ссылка
МоО2- [MoO2-] -^0 0(0,5) [107]
Mo03S2“ H2O 487(10) To же
Mo02S2~ To же 1067(3) «
MoOS2- << 1654 (0,7) «
M0S2- « 2259(0,3) «
MeOH 2230 [108]
MeCN 2207 To же
ДМСО 2176 <<
MoSe2- MeCN 3339 «
CuCNMoS4 MeCN 998 (38) «
МоО2(Асас)2 ДМФ -45(225) [109]
[Mo03Edta]4“ H2O 63 (290) [Hl]
Mo(CN)4- ДМФ -1309 (75) [Hl]
[Mo70^ H2O, 293 К 210(760); 36(310); 15(900) [68]
[A1Mo6(OH)6018]3- H2O, 320 К -21 (400) [H3]
[GaMo6(OH)6018]3- To же -3 (350) [H3]
[CoMo6(OH)6018]3- H2O, 323 69 (470) [H4]
[InMo6(OH)8018]3- H2O, 320 16(600) [H3]
[TeMo6024]6- H2O, 330 10(200) [H5]
[Mo2 O4(H2O)6]2+ 1 — 535-565 [4]
[{Mov(;z-0)(C204)(H20)}2]2- H2O, 323 520 (430) [116]
[Mo2(^-0)4Edta]2- D2O 612 [117]
[Mo!,v(M-0)4cit2]4- H2O, 333 к 36(360); -54 (380) *
Таблица 3.21 (окончание)
Форма Раствори- тель <5, м. д. (Ш, Гц) Ссылка
[Mo'v04(H20)9]4+ 2 — 920-1162 [4]
[Мо!,у(ц-0)2(МеСОО)6(Н20)з]2+ d2o 1061 [И8]
[Moiv(M-0)4*tart3(H20)3]2+ Н2О, 323 921 [И8]
[Р2Мо5023]6- Н2О, 320 -11 (550) [Н9]
Н2[Р2Мо502з]4" — -18(550) [И9]
[Со2Мо,о(ОН)4Оз4]6 Н2О, 323 153(190); 47 (330) [И9]
[NiMOgOifj]3- Н2О, 323 87(100); 63(100) [Н9]
[SiMoi204o]4 Н2О, 320 К 19(450) [ИЗ]
[GeMo12O40]4- « 34(250) То же
[РМо|2О40]3“ « 16(1300) «
[AsMo,204o]3 « -9 (2600) «
[СеМо12 042]8- Н2О, 295 К 0(50) [143]
[UMo12042]8 * * * *- Н2О, 295 К -206 (400)3 [144]
Мо Металл 5848 (500) [Ю6]
AgMoCU Тв. 82 (80) То же
1 Димеры Mo(V) на основе остова МО2О4; 2 тримеры Mo(IV) на основе остова
Мо^СЛ; tart — тартрат, cit — цитрат; 3 сдвиг и уширение линии ЯМР 95Мо за
счет парамагнетизма U(IV); * данные автора.
В ионах МоО^- и MoS^- обнаружено влияние изотопного замеще-
ния на ХС (16О/18О — 0,25 м.д. на атом, 32S/34S — 0,09 м.д. на атом)
[123]. Линейная корреляция 6-Д.Е обнаружена в [124] для комплексов
MoO^S2!^, означающая, что ХС молибдена в этом ряду определяется
энергией возбуждения комплекса.
Известен ряд КССВ 95Мо: V(95Mo-17O) = 40,3 Гц (МоО2“)[76,
125], J(85Mo-14 *N) [126], ‘J(95Mo-P) = 219 Гц, Mo(PF3)6 [127]; обычно
наблюдению КССВ препятствует большая ширина линий ЯМР.
Ширина линии ЯМР значительно превышает 10 Гц, и только в сим-
метричных структурах уменьшается до 1 Гц и менее. Значительного
сужения линии можно достичь за счет уменьшения вязкости раствора
и повышения температуры измерения. Специфичность ЯМР 95Мо зна-
чительно повышается с переходом в сильные магнитные поля [113].
ЯМ-релаксация 95Мо в монозамещенных карбонильных комплексах
изучена в [128]; изучение ЯМ-релаксации комплекса [Mo(CN)g]4-
[129] указывает на 12-координированное состояние комплекса в вод-
ных растворах.
При подкислении раствора ЫагМоС^ при pH 6 линия ЯМР стано-
вится шире и исчезает при дальнейшем подкислении [106], что связано
с образованием полиядерных оксомолибдатных ионов и обменом атома-
ми кислорода последних с водой. Равновесия и формы в этом процессе
изучены в [130]. Диаграмма состояний приведена на рис. 3.11.
Рис. 3.11. Распределение форм в процессе кислотного гидролиза аниона
МоО24~ : 1 - Мо70^; 2 - а-Мо80426-; 3 - ^-MogO4^; 4 - Мо36О^2
Ряд работ посвящен изучению замещенных карбонильных комплек-
сов Мо, данные которых сведены в табл. 3.22.
Основные формы в процессе (без учета протонирования) МоуО9^- —>
-> а-МодО4^ -»/3-Мо8О4^ -> Мо360®72.
Парамолибдат МоуО®^- изучен в [130, 131]. При 293 К в нем наблю-
дается обмен фрагментами структуры, полностью разрешенный спектр
ЯМР 17О на частоте 40,7 МГц наблюдали при 273 К. Полиядерные
формы в растворах показаны на рис. 3.12.
Спектр ЯМР 95Мо парамолибдата МоуО9^ по положению линий
ЯМР аналогичен спектру l83W изоструктурного пара-вольфрамата
W7O®4“ [131] (рис. 3.13).
В полиоксомодибдатах молибден находится в искаженных октаэд-
рах, и линии ЯМР 95Мо имеют значительную ширину [113].
В изоструктурных полимолибдатах ХМ012О40 ширина линии ЯМР
95Мо зависит от центрального атома и растет в ряду Ge < Si < Р < As.
В последнее время ЯМР 95Мо все шире применяется для определения
Таблица 3.22
Данные ЯМР 95Мо карбонильных комплексов молибдена
Форма Растворитель <5, м.д. (IV, Гц) Ссылка
Мо(СО)в ТГФ -1856(0,7) [HI]
Mo(CO)5L, L = PPh3 PEt3 -1857 (15) [112]
L = PPh3 CHC13 -1743 (35) То же
L = AsPh3 To же -1751 (153) «
L = SbPh3 « -1865(186) «
L = PF3 — -1860 [121]
L = PC13 — -1523 [121]
4uc-Mo(CO)4 (PEt 3 )г CH2C12 -1756(30) [122]
транс-Мо(СО)4 (PEt3)2 CH2C12 -1810(110) «
4UC-Mo(CO)4(AsPh3)2 CH2CI2 -1577(190) «
ffipaHC-Mo(CO)4(AsPh3)2 Толуол -1757(5) <<
цис-Мо(СО)4(РРЬ3)2 CH2C12 -1556 (50) <<
транс-Mo(CO)4 (NEt3)2 CH2C12 -1327 (30) «
цис-Мо(СО)4(МеСМ)2 MeCN -1307(50) «
i{uc-Mo(CO)4(NEt3)2 CH2CI2 -1152 (20) «
цис-Мо(СО)4(ру)г CH2CI2 -1046(80) «
4uc-Mo(CO)4[(OPh)3]2 CH2CI2 -1754 (40) «
Рис. 3.12. Строение форм в процессе гидролиза МоО| : 1 — Мо?024 ; 2 —
а-МовО^-; 3 — Д-МовО^; 4 — Мо360|“2
структуры и поведения комплексов и металлоорганических соединений
[108-110, 132-140] и оказывается иногда более информативным, чем
ЯМР 31Р фосфорных лигандов [141].
На рис. 3.14 изображены спектры ГПА [NiMogOag]8- и его
структура, состоящая из двух триплетов МО3О13 (/) сверху и снизу
200 0 м.д. 300 100 0 -100 м.д.
Рис. 3.13. Спектры ЯМР парамолибдата (а, 95Мо) и паравольфрамата (б, 183W)
[130, 131]
Рис. 3.14. Спектры ЯМР 17О (/) и 95Мо (2) комплекса [ММодОзг]8- и его
структура (3); на спектре (/) указаны линии, соответствующие типам атомов О
в структуре (3)
кубической полости, а между ними находятся три октаэдра MoOg (2).
Полость внутри заполняет ион Ni4+ в тетраэдрическом окружении.
ГПА является тридентатным лигандом с тремя близлежащими
концевыми атомами кислорода. Добавление La3+ в раствор ГПА
вызывает уширение линий 95Мо, указывая на образование лабильного
комплекса [La-NiMogC^]5- [142] (в кристалле образуется субъедини-
ца La2[NiMo9O32]2-).
Изучению комплексообразования ионов металлов Zn2+, Sc3+ с ГПА
структуры Декстера-Сильвертона [ZMo^C^]8-, Z = Се, U, проявля-
ющей себя как тридентатный лиганд, посвящены работы [143, 144],
см. рис. 2.40, 2.47.
Вольфрам. Из рассматриваемой группы вольфрам - единствен-
ный элемент с магнитным изотопом l83W со спином 1/2. Долгое
время низкая чувствительность ЯМР 183W не позволяла использовать
его в химических исследованиях, хотя первое из них было сделано
в 1962 г. [145]. Развитие техники ЯМР и переход к высоким полям
позволяют получать приемлемые спектры с обычными концентрациями
(~0,2 г-ат/л) в разумное время (4-10 ч в поле 7 Т), что ставит ЯМР
183W в разряд рабочих методов исследования. Подробные обзоры дан-
ных ЯМР 183W содержатся в [2, 4, 51]. Чувствительность ХС ЯМР
вольфрама к изменению окружения вдвое выше, чем у 95Мо [146].
Такая чувствительность и очень узкие линии ЯМР 183W позволяют
исследовать тонкие эффекты химической связи.
В качестве эталона в ЯМР используют водный раствор Na2WO4
или WFg. Оба эти эталоны неудобны: в первый трудно ввести релак-
сационный агент, поскольку ион вольфрамата стабилен только в силь-
нощелочной среде, в которой парамагнитные ионы металлов выпада-
ют в осадок, а без релаксационного агента даже в поле 7 Т время
накопления сигнала насыщенного раствора Na2WO4 более 1 ч. Темпе-
ратура кипения WFg 17 °C; кроме того, сигнал без подавления ССВ
с фтором представляет собой септет. Более удобным вторичным этало-
ном является водный раствор кремневольфрамовой гетерополикислоты
H4SiW1204o, в котором достигается концентрация по вольфраму до
8 г-моль/л и в который легко вводится релаксационный агент — ион
Сг3+. В этом случае сигнал эталона проявляется при накоплении 1-3
ССИ. ХС и константы ССВ некоторых соединений и ионов вольфрама
приведены в табл. 3.23.
Таблица 3.23
Данные ЯМР вольфрама-183
Форма Растворитель <5, м. д. J(W-X), Гц X Источник
WO*" H2O, pH = 11 0 10 ,7O [147]
WF6 4. жидк. 1122 41 19f —
WC16 CS2 -2180 — — [148]
WOF“ — 1727 71 58“ I9f —
WF5OMe — 1163 43э; 33“ 19f [148]
W(CO)6 — -3500 124 ,3C [4, 150]
W(CO)5PBu3 CH2C12 — 231 31 p [146]
WO3 Тв. 2000 — — [145]
W Металл 11700 — — «
в — вершина; э — экватор.
Методом ЯМР 183W интенсивно изучаются структуры изо- и гете-
рополивольфраматов и их превращения в водных и неводных средах
(обзоры [68, 151], табл. 3.24).
Таблица 3.24
Данные ЯМР полиоксовольфраматов
Полианион <5i83W, м.д. <5X <5M (IV, Гц) Примечания
w6o?9- 59 — — Me2NCHO [152]
vw5o39- 76,4; 75,9' (4: I)2 — -522(25, V) [153]
V2w4of9- 74; 65 (1 : 1) — -507(45, V) [69, 154]
a-SiW12O4- -92 — — Me2NCHO [152]
-104 -74,6 — H2O [155, 156]
a-SiW12O50- (2e) -43 — — [68]
w7o&4- 269; -92; -179 (1:4:2) — — H2O, pH = 7 [131]
Mo6W0264- -47; 264 3 — — [157]
/3-SiW12O^ -103,5; -104; -120 (1:2:1) — — Me2NCHO [161]
« -110; -115; -130 (1:2:1) — — H2O [157]
a-PW12O30- -99 -15 — H2O [68, 157]
Na-a-PWiiO^- -104; -110; -134; -99; -154; — 1184 -10,7 — H2O [164]
a-PVWnO^- -106; -72; -101; -99; -109; -104 -14,9 -557 (V) H2O [68, 158]
PTiWnO®- -71; -105; -118; -119; -128 -13,7 — H2O [68, 159]
PWi1O3gZnH2O5- -74; -142; -132; -159; -108; -131 -11,7 1 (250, Zn) [68, 160]
Таблица 3.24 (окончание)
Полианион <5i83W, м.д. <5X <5 M (TV, Гц) Примечания
PWnO39CdH2O5- PWnO39CdCI6- -105; -150; -134; -165; -110; -124 — -45(260, Cd) [68, 160]
PWnOagHgHsO5- -97; -147; -135; -165; -104; -123 — 102(130, Cd) «
PWHO39Pb5“ -79; -150; -130; -86; -106-114 -11,3 -2180(250, Hg) «
a-P2W18O®2- -97; -148; -135; -162; -106; -129 -11,7 4169(200, Pb) «
/?-p2w18o«2- -128; -174(1:2) -12,8 — H2O, pH = 1,6 [162]
(SiWnO39Al)2OВ * * * 12- -82; -110; -96; -111 -12,1 — H2O, pH = 1,4 [163]
a-BW,< -102; -104; -127; -131; -151; -196 — 13(2100) [155]
a-GeWi2O40- -131 — — H2O [155]
-82 — — «
a-ZnWi2O®(7 -56 — — «
a-H2W12O®0- -113 — — «
1 Значения ХС округлены до целых, кроме случаев, когда расстояние меж-
ду линиями м.д.; 2 соотношение интенсивностей линий; 3 ХС относятся
к разным изомерам MogWO®^; 4 порядок ХС соответствует нумерации атомов
рис. 3.18.
В гетерополианионах со структурой Кеггина наблюдается корреля-
ция XC-(A£?)-1 при замещении центрального атома (рис. 3.15) [155].
В однотипных гетерополианионах наблюдают корреляцию между ХС
и средним расстоянием вольфрам-кислород 5-r0_w [154].
По ХС 183W четко различаются а- и /3-изомеры структуры Кег-
гина, отличающиеся поворотом одной тройки октаэдров WO3 на 60°
вокруг оси, проходящей через центральный атом и атом О, общий для
Рис. 3.15. Корреляция ХС ЯМР l83W и энергии низших электронных переходов
в гетерополивольфраматах XW12O40
атомов этой тройки. В /3-изомере атомы становятся неэквивалентными
и разбиваются на группы из трех, шести и трех атомов, что дает
спектр ЯМР вольфрама из трех линий с отношением интенсивностей
1:2:1 [152, 157]. Подобный поворот в анионе PgWigO^ приводит
к спектру ЯМР 183W из четырех линий, соответствующему четырем
типам атомов в структуре с количеством в группах 3; 6; 6 и 3
(рис. 3.16) [162].
Рис. 3.16. Структура а-изомера структуры Доусона P2WieOg^ и спектр ЯМР
l83W /3-изомера ГПА P2Wi8O|^"; внешние линии —107,7 и —186,6 м.д. от-
носятся к половине ГПА с повернутым триплетом, внутренние линии 127,4
и 167,3 м.д. — к невозмущенной половине
Отсутствие или замещение одного атома в а-изомере структуры
Кеггина делает анион зеркально-симметричным с шестью типами ато-
мов W, из которых пять попарно эквивалентны и один атом (противо-
положный замещенному) не имеет партнера (рис. 3.17), в соответствии
с чем получается спектр из шести линий с соотношением интенсивно-
стей 1 : 2 : 2 : 2 : 2 : 2 [152].
-90 -НО -130 -150м.д.
Рис. 3.17. Нумерация атомов W в дефектном анионе Кеггина XWnOj9 (а)
и влияние катиона на положение линий в спектре ЯМР l83W (б)
Интенсивно изучают «дефектные» анионы, у которых нет одного
или нескольких октаэдров в структуре, например XWjjOgg, кото-
рый является своеобразным неорганическим тетрадентатным лигандом.
Их взаимодействие в водных растворах с однозарядными катионами
изучено в [164], двухзарядными — в [160].
Спектры растворов солей PWnO^ (PWn) с однозарядными ка-
тионами показаны на рис. 3.17 [164]. Наблюдаемые спектры усред-
нены быстрым обменом между свободным состоянием аниона PWn
и комплексом M_-PWn- В растворе вакансия PWn всегда за-
полнена катионом. В случае двухзарядного катиона обмен замед-
ляется и время жизни катиона М2+ в комплексе увеличивается
с уменьшением радиуса катиона [160]. Оценка скорости обмена да-
ет ряд, в котором время жизни иона металла в комплексе растет:
К+ < Na+ < Li+ < Pb2+ < Hg2+ < Cd2+ < Mg2+ < Zn2+; цинк образу-
ет стабильный в шкале времени ЯМР 183W комплекс ZnPWnO8^.
Кроме PWn, в водном растворе существуют как таковые дефектные
ГПА - P2Wi7O^-, P2W2007o(H20)6- и др. (рис. 3.18).
Спектр ЯМР 183W ГПА P2Wi70g°~ (P2W]7) состоит из 11 линий,
по аналогии с PWn, спектр ГПА P2W2qO7q(H2O)6~ — из 6, согласно
обозначениям эквивалентных атомов рис. 3.18, б. Последний «захваты-
вает» одно- и двухзарядные катионы в вакансию в центре ГПА, образуя
лабильный комплекс, или прочно присоединяет к октаэдрам 5, как,
например, Sn(IV) [165]. Данные комплексов приведены в табл. 3.25.
Циклопентадиенильные комплексы CpW(CO)3Me, CpWOMeL]L2,
CpWSMeL]L2, изученные в [171], перекрывают диапазон ХС -1300 4-
4- 4000 м. д.
Из-за малого гиромагнитного отношения 183W наблюдаемые
константы ССВ невелики. В металлоорганических соединениях
!J(P-W) достигают 300 Гц; другие константы значительно меньше.
Для установления структуры гетерополианионов активно используют
2J(W-O-W) (5-30 Гц) [157, 166, 167]; выявление этих констант
Рис. 3.18. Структура дефектных ГПА: а — Рг^ЛуО^® ; б — P2W2oO?o(H20)6
Таблица 3.25
Данные ЯМР комплексов [Z-P2W2o07o(H20)]n- [165]
Катион Z 6 183W, м.д. <5 P
Li+ -75; -142; -159; -160; -104; -196 1 -12,3
Na+ -72; -141; -156-170; -99; -193 -14,1
к+ -74; -142; -159; -159; -100-195 -12,8
Bi3+ -83; -132; -153; -137; -70; -188 -14,9
Sn4+ -88; -132; -139; -136; -198; -253 -14,12
1 Округлено до целых; 2 6 ll9Sn —632 м. д., J(119Sn-W) = 165 Гц.
требует спектров с хорошим соотношением сигнал/шум, получаемых
при накоплении более 10 ч. В [172] обнаружена корреляция между
2J(W-W) и локальным окружением атома W в поливольфраматах.
Малое гиромагнитное отношение 183W позволяет регистрировать
спектры ЯМР 183W с парамагнитным ионом в структуре [168-170],
в частности, для ГПА [Ln(PWnO3g)2]11- и [LnWioOae]9-, Ln — лан-
таноид(Ш).
3.3. Марганец, технеций, рений
Применение ЯМР элементов группы VIIB ограничено состояния-
ми окисления M(VII) и М(1), в которых соединения диамагнитны.
Большой квадрупольный момент ядер этих элементов, радиоактивность
изотопа искусственного "Тс налагают дополнительные ограничения
при использовании метода ЯМР этих элементов. Свойства их магнит-
ных ядер показаны в табл. 3.26. ЯМР всех элементов группы VIIB
Таблица 3.26
Свойства ядер магнитных изотопов группы VIIB
Изо- топ Спин NA, % Sens 1 Диапазон ХС, м. д. Q, барн Частота ЯМР эталона Е, МГц2 Эталон
13с — — 1{'Н} — — — —
55Мп 5/2 100% 340 3500 0,56 24,665 МпО4-
"Тс 9/2 0 740 4600 -0,1 22,508 304 ТсО4-
185Re 5/2 37 94 7000 2,8 22,514 ReO4“
187 Re 5/2 63 164 — 2,6 22,744 «
'Относительно |3С{'Н} с учетом природного содержания; 2 частота ЯМР в
поле Но, в котором частота ЯМР ’Н ровно 100 МГц.
имеет высокую чувствительность. Наличие двух изотопов у рения
позволяет изучать вопросы химии рения методом изотопного заме-
щения с применением ЯМР, однако большая ширина линий делает
метод ЯМР в большинстве случаев неэффективным. Данные ЯМР
этих элементов приведены в [2, 4, 51].
Марганец. Набор данных по ЯМР 55Мп состоит из параметров
ЯМР МпО^ и карбонилов марганца. Характерное для марганца состо-
яние Мп(П) недоступно для ЯМР из-за парамагнетизма комплексов.
Диаграмма ХС 55Мп приведена на рис. 3.19 [4, 173-176].
Концентрационная и катионная зависимость ХС МпО^“ изучена
в [177]; чувствительность ХС к наличию катиона возрастает в ря-
ду Li+ < Mg2+ < К+ < Ва2+ и для Ва2+ достигает —9,5 м.д./г-ион.
Температурная зависимость ХС +0,3 м. д./К. Зависимость ХС МпО^“
от растворителя изучена в [178], наибольшее отклонение достигает
18 м.д. Эффекты изотопного замещения кислорода в MnO^j" рассмот-
рены в [179], где найден сдвиг 0,6 м.д. на замещение 16О-18О с кон-
стантой времени замещения около 7 дней. В комплексах [(СО^МпХЦ,
где X — галоген, L — P(As)Ra, ХС перекрывают диапазон —900 +
+ -1700 м.д.[180].
ЯМ-релаксация изучалась в [181, 182]. По характеру температур-
ной зависимости Т\ показано, что квадрупольная релаксация МпО^
+ 1000
-1000 -2000 -3000 м.д.
Мп(СО)5-
Mn(co);i
Mn2(CO)10_nPZ3n
HMn(CO)5_n(PF3)n
Mn(CO)5R
Mn(CO)5C(O)R
Mn(CO)5ElvR3
Mn(CO)5X
Mn(CO)5CO2Et
Mn(CO)5Co(CO)4
Mn(CO)4LL“
Mn(CO)6_n(PZ3)n
о
о
+ 1000
I------1
I------1
-1000 -2000
775-c5R5Mn(CO)3_nLn
Mn(NO)3L
MnO4“
—3000 m. д.
Рис. 3.19. Диаграмма химических сдвигов 55Mn
определяется не только вращательным, но и колебательным движением
иона; для него же найдена КССВ *J(Mn-17O) = 29 Гц [80]. КССВ
'ДМп-С) измерена в растворе [Mn(CNEt)g]BF4 в CD3CN [183].
Технеций. Технеций — искусственный элемент, однако он изучен,
в отличие от многих других искусственных изотопов, и методом ЯМР.
Он образуется в процессе радиоактивного распада урана и и извлекает-
ся из отработанного ядерного топлива. Эталоном в ЯМР "Тс служит
пертехнетат-ион [184]. Его изучению посвящена большая часть работ
по ЯМР "Тс [182, 184, 185]. При взаимодействии ТсО^ с KrF2 и XeFs
получены оксифториды технеция, охарактеризованные методом ЯМР
"Тс, 17О и 19F [186, 187]. В щелочной среде получен анион ТсНд .
Данные ЯМР "Тс приведены в табл. 3.27 [186], диаграмма ХС — на
рис. 3.20.
Изотопный эффект замещения 16О-18О изучен в [185]; спектр ЯМР
|7О и "Тс изотопомеров ТсО^" приведен на рис. 3.21. Изотопный сдвиг
"Тс на одно замещение 0,45 м.д. В водном растворе ТсО^- ("Тс) Т\ =
= 0,13 с [108], а Т2 — 0,1 с; температурная зависимость Т\ показывает
вклад колебательных движений иона в релаксацию [106].
Таблица 3.27
Данные ЯМР технеция-99
Форма <5 "Tc (W) Ссылка Ti, mc 'j(Tc-X), Гц X
Тео; 0(3) [168] 130 131,4 l7O
ТсНд- -3622 (22) « 23 24 'H
TcO3F 43,7(23) « 18 140 170
ТсО+ 161 (670) « 1 — —
[{TcO2F2]2(M-O)] 246(135) « 1 87; 259 17o, l9F
TcO2F3 396 (375) « 3 — —
ifuc-TcO2F^“ 247 [189], HF — 260 19f
транс- [TcO2(CN)4]3- 806 (640) [186] — — —
[Tc(CO)5Br] -1630(186) [188] 0,0028 — —
[Tc2(CO)10] -2477(1,5) [188] 0,42 — —
F3P(CO)4Tc-Tc*(CO)5 -2632* [191], CHC13, 337 К — 1750(P), 19 i9F
[Tc(CNBu‘)6-PF3 -2450 [190] — — —
[Тс(ТМФ)6]-ВРЬ4 -1907(73) [190] — 910 31 p
[Тс(ДМФЭ)3]С1 -1657(23) [190] — 910 31 p
[Tc{P(OMe)3}6]-BPh4 -1837 [192], CDC13 — — —
-1658 — — — —
CNBu1 — mpem-бутилизонитрил, ТМФ — триметилфосфит, ДМФЭ —
1,2-бис(диметил-фосфино)этан.
Tc(V)
Tc(III)
Tc(I)
Tc(O)
Tc(VII) TcD2F3 I | TcO4 TcHg2- |
TcOCl4"
-------------------1 TcO2(CN)3-
Tc(CO)[(PMe3)2Ph]3Cl3|----1 Tc(CN)'~
TcBr(CO)3(PPh3)2 |-----1 Tc(CO)dppe(MeCN)
Tc2(CO)10
5000 м.д.
0
-4000
Рис. 3.20. Диаграмма химических сдвигов "Тс
Рис. 3.21. Спектр ЯМР |70 (а) и влияние изотопного состава кислорода на ХС
ЯМР "Тс пертехнетат-иона (б)
Рений. Измерения ЯМ-релаксации 185Re, 187Re в растворе перре-
нат-иона [195, 196] показали большую ширину (5-15 кГц) линии ЯМР,
что даже в поле 7 Т составляет 70-200 м. д. Такая большая ширина для
высокосимметричного иона показывает, что применение ЯМР рения
в химических исследованиях ограничено. Известные данные приведены
в табл. 3.28.
Таблица 3.28
Данные ЯМР рения на частотах 20,26 МГц (185Re) и 20,47 МГц (l87Re) [197]
Соединение Растворитель <5,87/,85Re TV187 Re, TV 185 Re, кГц
NaReO4 0,8 M, Н2О 0 13,5; 14,2
2 М, Н2О 0 20; 14,2
(Ph4P)ReO4 Нас., ДМФ/толуол 4 : 1 0 3,3; 3,6
(Et4N)ReS4 0,2 М MeCN/CH2Cl2 3435/3200 1,96; 1,96
[Re(CO)6]Cl-HCl 0,3 М MeCN -3400 2,3; 2,6
Данные таблицы определяют диапазон ХС рения не менее 7000 м. д.
3.4. Элементы группы VIII
Магнитные изотопы имеют все элементы группы железа. Как и для
других переходных металлов, парамагнетизм многих соединений этой
группы препятствует наблюдению и использованию ЯМР на ядрах этих
элементов. Свойства их ядер приведены в табл. 3.29.
Таблица 3.29
Свойства ядер магнитных изотопов подгруппы железа
Изо- топ Спин NA, % Sens 1 Диапазон ХС, м. д. Q, барн Частота ЯМР эталона =, МГц2 Эталон
13с — — 1{'H} — — — —
57 Fe 1/2 2,2% 0,0014 8200 — 3,237798 Чист. Fe(CO)s
59Со 7/2 100 532 18000 0,4 23,727449 K3[Co(CN)6]/ D2O
61N1 3/2 1,2 0,081 1000 0,16 8,936050 Ni(CO)4/ d-MePh
99Ru 3/2 12,7% 0,13 9000 0,076 4,605 127 K4[Ru(CN)6]/ d2o
101 Ru 5/2 17,1 0,51 « 0,44 5,162 344 20 °C
103Rh 1/2 100 0,06 13000 — — = = 3,16 МГц
« — — — — — 3,186 447 Rh(Acac)3
« — — — — — 3,155 671, <5= 1370 Rh металл
l05Pd 5/2 22,2 0,47 — 0,6 4,58572 [PdCl6]2-/ HC1:HNO3
1870s 1/2 1,6 0,004 — — 2,282 343 OsO4/CDC13
1890s 3/2 16,1 0,71 — 0,8 — «
l91Ir 3/2 37,3 0,02 — 1,5 — ЯМР не набл.
193Ir 3/2 62,7 0,04 — 1,4 — —
195 Pt 1/2 33,8 6,4 14000 — — -= 21,4 МГц
« — — — — — 21,496 770 Na2PtCl6/D2O (<5 = 4522)
'Относительно 13С{'Н} с учетом природного содержания; 2 частота ЯМР в
поле Нд, в котором частота ЯМР 'Н ровно 100 МГц.
ЯМР этих элементов отличает большой диапазон ХС и его высокий
температурный коэффициент. Данные ЯМР обсуждены в ряде обзоров
и монографий [3, 51, 198-202].
Природное содержание 57Fe и 61 Ni позволяет проводить исследова-
ния с изотопным замещением, используя ЯМР как детектор изотопной
метки.
Железо. Чувствительность ЯМР 57Fe очень мала и превышает
только чувствительность ЯМР 1870s. В жидкости ЯМР 57Fe наблю-
дают только в диамагнитных соединениях железа: карбонилы Fe°,
ферроцены Fe(II), К2[Ре(С1Ч)б].
Первое прямое наблюдение ЯМР 57Fe в Fe(CO)s потребовало 20 ч
накопления сигнала [203]. Точное определение гиромагнитного отно-
шения сделано в [203, 204]: i/57Fe(CO)e/^(2D2O) = 0,210921 4 [204].
Данные ЯМР 57Fe приведены в табл. 3.30.
Таблица 3.30
Данные ЯМР железа
Соединение <557Fe Ссылка
Fe(II): [FeCp2]-C6H6 1532 [205]
[РеСр2]-замещенные бензолы 1530-2070 [205, 206]
[FeCp2]H 460 [207]
[РеСр(РМез)2]Н 1309, J(Fe-P) = 59 310 К [217]
« 1294 300 К [217]
[FeCp2](77-C6H6) 1593 [208]
K4[Fe(CN)6] 2497 [205]
Fe(0): [Fe(CO)s] neat 0 [205]
Карбонилы 582-1562 [205]
[Fe(CO)2(PMe3)2CS2] 1220 209
8220 [216]
ХС 1,5 М раствора ферроцена в бензоле 1553 м.д. относительно
Fe(CO)5; ХС замещенных ферроценов лежат в пределах нескольких со-
тен миллионных долей от Fe(C5H5)2, что дает общий диапазон ХС 57Fe
около 2000 м. д. Изучению ферроценов посвящены работы [205-207,
210-214]. Обнаружено, что ХС 57Fe отражает наклон плоскостей цик-
лопентадиенильных колец друг относительно друга [214].
Константы ССВ V^Fe-^C) = 23,4 Гц в Fe(CO)5 и 3-7 Гц в фер-
роценах [207, 208, 259], 'J(57Fe-P) = 26 ± 1 Гц в замещенных фосфи-
нами карбонилах железа [215]. Времена релаксации 57Fe в Fe(CO)5
Т112 > 1 с [203].
Кобальт. Кобальт занимает особое место в спектроскопии ЯМР.
Обнаруженный в 1951 г. [105] ЯМР 59Со использовался в дальнейшем
для тщательного изучения различных эффектов в ЯМР, поскольку
чувствительность ХС ЯМР 59Со к окружению исключительно велика
и имеется большой набор соединений с разным типом связей, изучен-
ных методом оптической спектроскопии [51]. Методом ЯМР можно
изучать соединения с состояниями окисления Со(Ш), Со(1) и Со(—I);
эталоном служит комплексный ион Co(CN)g-. Большая чувствитель-
ность позволяет регистрировать и широкие линии ЯМР, а чувстви-
тельность ХС — наблюдать на них эффекты экранирования, обычно
видимые только на узких линиях.
Диаграмма ХС 59Со представлена на рис. 3.22.
СоОб
CoO4XY
k>01N4A I
C0N9X — CoSe
CoSeg I ---- Co(NO)2XPZs
-----Co(CN)5X
Корбонилы Со0 -- _|
Со
10000 5000 0 м.д.
Рис. 3.22. Диаграмма химических сдвигов ЯМР 59Со
Данные ЯМР некоторых соединений приведены в табл. 3.31.
Таблица 3.31
Данные ЯМР кобальта
Форма Растворитель <5, м.д. (W, Гц) Ссылка
Co(CN)9- Н2О 0(5) [218]
Co(CN)5L, l = ОН- 0,4 М/Н2О 1840(5300) [219]
Ns" То же 1820(6100) То же
NO“ « 1400(18000) «
Вг“ « 1220(11400) «
Г « 780(10000) [220]
Таблица 3.31 (продолжение)
Форма Растворитель <5, м.д. (ТЕ, Гц) Ссылка
. Co(NH3)|+ H2O 8175(170) [218]
Co(NH3)5L, L = NO2- To же 7600(175) [219]
N3“ « 8820 (400) [218]
H2O « 9150(4000) [218]
OH- « 9100(1900) [220]
NCS“ « 8330(1320) [218]
SCN” « 8600(1540) [218]
c2o2- « 9130 [221]
CO2- « 9060(1950) [218]
F" « 9520(1750) То же
cr « 8890(1460) «
Br“ « 8820(1750) [219]
r « 8760(2100) [219]
SSO2- <1 8233 (480) [309]
OS2O2- « 7002(1200) [309]
Co(NO2)|- « 7440(175) [220]
Coerij- — 7122 (205) [220]
Co(Acac)3 — 12500(175) [220]
Co(CO3)3- — 14070 (510) [220]
Со(С2О4)з — 13000(440) [218]
Co[(EtO)2PS2]3 — 8880 [220]
Co(Et2NCS2)3 — 6450 (520) [220]
Co(Me2NCSe2)3 — 6760 [223]
NaCo(CO)4 Пентан -3100 [222]
Cl3SnCo(CO)4 — -2950 [222, 224, 225]
I3SnCo(CO)4 — -2570 [224, 225]
Ph3SnCo(CO)4 — -3170 4
Ph3PbCo(CO)4 — -2950 4
Таблица 3.31 (окончание)
Форма Растворитель <5, м.д. (ГТ, Гц) Ссылка
Ph3GeCo(CO)4 — -3050 [224, 225]
Ph3SiCo(CO)4 — -3270 «
Co4(CO)i2 3 * : 1, гексан -1520*; -250 [226]
Co2(CO)e Пентан -2100 [222]
Гексан -1860 [227]
K2Co(PF3)4 — -4220 [228]
Co(N3)g“ — 12530(210) [218]
Co(NCS)g- — 8187(160) [218]
[CoOH(NH3)4]2 — 9180(170) [229]
[Co{(OH)2Co(NH3)4}3]6+ 1 * : 3 14850(1710)*; 10060 (6200) [226]
[Co(NH3)6]C13 — 8100 [233]
[Co(NH3)5CO3]NO30,5H2O — 900 «
[Co(NH3)3CO3C1]-H2O — 1165 «
K[Co(NH3)2(CO3)2]H2O — 1174 «
К3[Со(СОз)3]-ЗН2О — 1395 «
[Co(OH)6Mo6Oie]3- Н2О 14070(85) [И4]
[Co2(OH)4Moi0O34]6 — 12275(1400) [И4]
Растворитель и температура сильно влияют на ХС 59Со иона
Co(CN)g~; кислотность и концентрация противоиона влияют меньше
(эффекты менее 10 м.д.). В неводных растворителях добавление кисло-
ты приводит к изменениям ХС до 350 м.д. [230]. В некоторых работах
эталоном служат другие формы кобальта, и ХС можно пересчитать
по формулам [277]: 5[Co(CN)|"] = <5[Соеп3] + 7120 = <5[Co(NH3)9+] +
+ 8150 = <5[Со(Асас)3] + 12500 м.д. Вследствие большого диапазона
ХС 59Со пересчет из одной шкалы (6’) в другую (5) следует делать по
формуле [51]
6 = 5' J + 106 (3.2)
однако поправка S’/Н имеет значение только для узких линий и при
измерениях в высоких полях. Для ядер с диапазоном ХС 1000 м.д.
Е'/Е=1.
Температурные коэффициенты ХС d(j/dT приведены в табл. 3.32
[51]. Большой набор диамагнитных комплексов Со(Ш) представляет
широкое поле для исследования химической связи и влияния разных
факторов на ХС 59Со.
Таблица 3.32
Температурные коэффициенты ХС ЯМР
Соединение Растворитель ТК, м.д./К
K3Co(CN)6 Н2О -1,38
Co(NH3)6Cl3 То же -1,42
Со(Асас)3 Толуол -2,29
СНС13 -2,96
Na3Co(NO2)6 Н2О -2,85
В основном изменении экранирования 59Со
а = ad + ар = А — const
LAD
преобладает парамагнитный вклад. Экранирование возрастает в ряду
лигандов F < О < N < С согласно спектрохимическому ряду [278],
который соответствует способности лиганда вызывать расщепление
d-орбиталей кобальта. У комплексов с С-, N-, О- и F-лигандами на-
блюдается корреляция ст(Д.Е_1) [232-236]. Это связывают с опреде-
ляющим влиянием фактора Д.Е-1 в парамагнитном вкладе, что поз-
воляет считать другие факторы примерно постоянными. Однако эта
корреляция не выполняется для As-, S-, Se-лигандов [223, 232, 236].
Спектрохимический ряд
I<Br<Cl<F<O<N<C
не совпадает с рядом по возрастанию экранирования
F < С1 < Вг < С < I,
которое образует нормальный галогенидный ряд, противоположный
ожидаемому из изменений Д.Е-1; аналогичная картина наблюдается
и в экранировании 103Rh комплексов Rh(III), RhCl^F^OJg-z [237],
поскольку Со(Ш) и Rh(III) изоэлектронны. Из этого следует вы-
вод, что для лигандов со связывающими атомами элементов тре-
тьего и следующих периодов фактор Д.Е-1 не остается постоян-
ным при замещении лигандов, а уменьшается в зависимости от
способности лиганда увеличивать {г)а- Мерой этой способности яв-
ляется нефелоксетический ряд [148], основанный на уменьшении
разнесения термов, наблюдаемых в электронных спектрах в ряду
F < НгО < С < NH3 < NCS < Cl ~ CN < Вг < I; значение нефелоксе-
тического эффекта обсуждалось в [238]. Изменения факторов {r~3}d
и орбитального Du трудно разделить, поскольку они часто меняются
симбатно. При увеличении ковалентности связи оба фактора уменьша-
ются по сравнению с их величиной для свободного иона; действие кова-
лентности связи на ХС обсуждалось в [239]. Интерпретации ХС окта-
эдрических <76-комплексов переходных металлов посвящен обзор [198].
При интерпретации эффектов экранирования наиболее часто ис-
пользуют зависимость экранирования от ДЕ1-1. Корреляция <5-ДЕ-1
наблюдается для многих переходных металлов. Обычно она выражена
четко для комплексов с низколежащими возбужденными электронными
состояниями, для которых переходы наблюдают в видимом свете или
ближнем УФ-диапазоне.
Такие корреляции построены для комплексов с N-лигандами в
[234], S- в [240], S-, О-лигандами — в [51]; данные для комплексов
CoNg и CoOg ложатся на одну, а для CoSg и CoPg — на другую прямую.
Наклон этих прямых является мерой факторов (r-3)d-Du, и для коор-
динирующих атомов третьего и следующих периодов он меньше, чем
для атомов второго периода. Вклад в экранирование в первую очередь
зависит от координирующих атомов лиганда, и если считать вклады от
лигандов независимыми, можно рассчитать инкременты каждого ли-
ганда. Такой подход не учитывает взаимного влияния лигандов, из-за
которого ХС изомеров комплексов одинакового состава различаются
до 500 м.д. Обычно цис-изомеры более экранированы, чем транс-
изомеры, а реберные (осевые) — более, чем граневые [228, 241], хотя
имеются и исключения. Для выделения эффектов взаимного влияния
лигандов могут использоваться данные ЯКР, которые коррелируют
с данными ЯМР [242].
Координатный сдвиг. В первых наблюдениях ЯМР донорных
атомов кислорода в системе Rh(III)-HCl-H2O [237] обнаружено,
что ХС ЯМР 17О координированной воды (НгО0) в комплек-
сах [RhCU^O^]3”1 имеет всего два значения (-99 ± 1
и -141 ±1 м.д.) в восьми разных аквахлорокомплексах, которые
проявились в спектрах ЯМР 103Rh (х от 1 до 5). Наблюдение
только двух значений ХС логично отнести к наблюдению НгОс
на двух координатах комплекса: НгО-Rh-Cl и H2O-RI1-OH2 этих
комплексов; различие ХС возникало из-за транс-влияния, типичного
для комплексов металлов группы платины и кобальта.
Впервые сдвиг ХС донорных атомов при координации (координа-
ционный сдвиг) был обнаружен для гидридных протонов комплексов
[243-244]; он направлен в сильное поле по отношению к ХС сво-
бодного лиганда. Для наблюдаемого донорного атома ХС меняется
при замещении транс-лиганда к нему, что объяснили для гидридных
протонов транс-влиянием [244]. Таким образом, для наблюдаемого
донорного атома в лиганде ХС определяется транс-лигандом X, ме-
таллом М и самим лигандом L, т. е. координатой комплекса X-M-L
(рис. 3.23), и, в меньшей мере, составом комплекса. Сдвиг донорного
Координаты X-M-L
Z-M-Y X-M-L
Q-M-P Z-M-Y
Рис. 3.23. Координаты октаэдрического (X-M-L; Y-M-Z; Q-M-P) и плоскоквад-
ратного (X-M-L; Y-M-Z) комплексов
атома на определенной координате X-M-L определен как координат-
ный сдвиг [246, 247].
В табл. 3.33 показано смещение ХС при координации для некоторых
лигандов [246], а на рис. 3.24 — координатные сдвиги НгОс.
Транс- Rh лиганд Pd, Pt □
Н2О □ 1
он 1
сг □ 1
NO2“
NH3
р-ОН-
p-NO3-
P-SO2- 1
<5, м.д. _J L—
О -50 -100 -150
Рис. 3.24. Координатные сдвиги |7О координированной воды в комплексах
Rh(III), Pd(II), Pt(II)
Таблица 3.33
Смещение линий донорных атомов лигандов при их координации
Лиганд Донорный атом Металл ХС свободного лиганда 1 Смещение при координации от свободного лиганда
NH3 N Со(Ш) —383 м.д. '-114- -78
« Rh(III) — —14 Ч- —68
« Pt(II) — -22 4- -60
NO2“ « Ru(ll) 234 — 136 4- -166
« Rh(III) — -142 4- -192
<< Pd(II) — -144 4- -186
<< Pt(II) — -175 4- -190
« Pt(IV) — -225 4--231
« Co(III) — — 135 4- —162
Еп « Rh(III) -357 -24 4- -53
« Pt(II) — 20 4- -18
NO3“ О Rh(III) 412 <517О = — 138ь, 33е.
Н2О О — 0 -70 4- -144
СНзСОО" О Rh(III) 270 -71
СО с Rh(III) 180 -184- -21
CN" с Pt(II) 169 -11 4- -43
с Pt(II) — -35 4- -83
Р(СН3)з р Pt(IV) -61 +49 + +59
Р(ОС2Н5)з р Pd(II) 139 -70
1 м.д., относительно [NO3 ] (азот), воды (17О), тетраметилсилана (13С), 85%
Из РО« (3|Р); 2 донорный атом О нитрата; 3 два других атома нитрата. В * * * *
В том случае, когда состав раствора (металл и возможные лиганды)
известен, ЯМР на ядрах металла и лиганда позволяет частично или
полностью определить строение комплекса без выделения его в твер-
дую фазу (и дальнейших структурных исследований), если известны
координатные сдвиги какого-то лиганда [246-248].
Комплексы с N-лигандами изучены в [249-259]; в табл. 3.34 при-
ведены данные некоторых дву- и трехзамещенных аминных комплек-
сов Со(Ш).
Таблица 3.34
Данные ЯМР 59Со замещенных аминных комплексов Co(III), N=NHs
Соединение <5, м.д. (W, Гц) Ссылка
z4uc-[CoN4(ONO)2] + 6875 [259]
z4uc-[CoN4(OH)2] + 6192 То же
цис- [CoN4(NO2)2] + 7286 «
цис- [CoN4(H2O)2] + 6370 «
транс- [CoN4(SSC>3)2]_ 8356 (540) [261]
транс- [CoN4 (SO3) (OSO2) ] “ 10363 [262]
транс- [Соеп2(55Оз)2] + 9312(480) [261]
цис- [Coen2(SSO3)(OS2O2)]_ 8508 (720) [261]
гран-[СоДз(ОМО)з] 6645 [259]
apaH-[CoN3(NO2)3] 7215 То же
гран-[Со1Чз(Н2О)з] 5560 «
pe6-[Codien(SeS03)3]2- 9154(760) [261]
реб-[СоДеп(О5еО2)з]3- 5878(910) [261]
Комплексы с О-лигандами изучены в [233, 242-268], Р-лиганда-
ми — в [221, 261, 267, 268], S-лигандами — в [253, 261], разноли-
гандные комплексы и эффекты изомерии — в [219, 233, 241, 254, 259,
262, 273]. Влияние водородной связи, второй координационной сферы
и противоионов рассмотрено в [230, 249, 265, 269, 275, 276], изотопные
эффекты — в [277-279].
ХС карбонато-аминных комплексов в виде их твердых солей и их
ЭСП изучены в [233] (табл. 3.35).
Изотопное замещение одного из атомов лиганда вызывает заметное
смещение линии ЯМР 59Со [277, 279]. H-D-замещение в комплек-
сах [Со(МН3)б]3+, [Соеп3]3+ приводит к многокомпонентным спектрам
59Со, соответствующим набору изотопомеров; на рис. 3.25 показан
спектр из линий всех возможных изомеров первого комплекса, полу-
ченных при растворении соли в HDO. По подобным спектрам мож-
но рассчитать содержание изотопов в воде и определить содержание
дейтерия в воде. Изменение ХС 59Со при изотопном замещении в ли-
Таблица 3.35
ЯМР и электронные свойства карбонатоаминных комплексов
Соединение (N=NHs) <5, м.д. А, см 1
[CoN6]3+ 8100 —
[CoN5C03]-N03-5H20 9000 2Q440; 27170
цис-[СоМиСОз]-НОз-0,5Н2О 9620 19080; 27700
[CoN3C03C1]H20 11650 18150; 28050
K[CoN2(CO3)2]-H2O 11740 17270; 23770
K2[CoNCO3(NO2)3]-H2O 7400 21740; 28940
Кз[Со(С03)з]-ЗН20 13950 15870; 26450
оо
оо
СО
Рис. 3.25. Наблюдение всех изотопомеров [CoNeH^Dis-x]34’ в спектре ЯМР
59Со при растворении Кз[Со(МНз)б] в HDO
гандах и температурные коэффициенты ХС 59Со-комплексов показаны
в табл. 3.36 [278].
Полиядерные комплексы изучали в [229, 280]. Обычно используе-
мое в ЯМР 59Со правило аддитивности выполняется не всегда; таким
примером является ход ХС в комплексах Со(МНз)п(МО2)б-п (рис. 3.26)
[273]). ЯМР 59Со использован для различения энантиомеров в жидко-
кристаллических системах при добавлении к ним [Со(еп)3]3+ [265].
В ЯМ-релаксации 59Со определяющим является квадрупольный
вклад. В том случае, когда координирующим атомом является атом
с квадрупольным ядром (например, у N-лигандов), может быть зна-
Таблица 3.36
Величины изотопного сдвига 59Со при изотопном замещении в лигандах
Комплекс Замещение <5 59 Со, м. д. Изотопный сдвиг, м. д.
[Co(NH3)6]3+ H/D 8159 5,6
[Со(еп)3]3+ « 7122 5,0
[Co(CN)6]3- 12с/13с 0 -0,91
« 14n/15n 0 -0,2
Рис. 3.26. Нарушение аддитивности ХС 59Со при замещении лигандов в амин-
ных комплексах Со(Ш)
чительным вклад скалярной релаксации из-за ССВ 59Co-14N [281];
в этом случае Т\ > Т%. В симметричных комплексах такого рода Т\ и
могут различаться на порядок [218]. В несимметричных комплексах
Т\ на порядок и более меньше и вклад скалярной релаксации второго
рода становится незначительным. Исследования ЯМ-релаксации 59Со
проведены в [250, 281-283].
Из-за обычно большой ширины линий ЯМР 59Со известно не так
много констант ССВ с кобальтом [228]: для Со(СО)4 J(Co-13C) =
= 287 ± 13 Гц, для Co(CN)3' = 127 ± 10 Гц, для Co(PF3)4 'J(Co-P) =
= 1222 ± 25 Гц и 2J(Co-F) = 57 ± 2 Гц.
Никель. В первой работе по точному измерению частоты ЯМР
61 Ni [284] определено отношение частот ЯМР 61 Ni и 17О в Ni(CO)4:
p(61Ni)/ р(17О) = 0,658 944(5), что с учетом <517О = 362 м.д. и р(17О)/
p(2D) = 1,132 352 в Н2О позволяет определить для Ni(CO)4 2 =
= 8,932 985 МГц (±90 Гц).
Известные работы по ЯМР 61 Ni содержат данные по замещенным
карбонилам никеля. В [285] приведена диаграмма ХС 61Ni (рис. 3.27);
т-1-—।------ч—----——т-
1000 500 0 -500 м.д.
Рис. 3.27. Диаграмма химических сдвигов 61 Ni
комплексы NiL4 (L — фосфин, фосфит) изучены в [286]. Данные ди-
и трикарбонильных комплексов приведены в табл. 3.37.
Таблица 3.37
Данные ЯМР 61 Ni ди- и трикарбонильных комплексов в растворе ТГФ (298 К)
Соединение <5, м. д. (IV, Гц), z/q = 26,8 МГц J(Ni-P)
(CO)2Ni(PMe3)2 104(240) —
(CO)2Ni(dppe) -160(1200) —
(CO)2Ni(PPh2H)2 -3(630) —
(CO)2Ni(PPh2Cl)2 91 (3000) —
(CO)2Ni(PPh3)2 91 (750) —
(CO)2Ni(OPPh3)2 -294 (2700) —
(CO)3Ni(PMe3) 26(140) 230
(CO)3Ni(dppe) -2(-) 210
(CO)3Ni(PPh2H) 5(190) 234
(CO)3Ni(PPh2Cl) 48(-) 236
(CO)3Ni(PPh3) 24(16) 236
(CO)3Ni(OPPh3) -130(—) 351
Общий диапазон ХС 61Ni изученных соединений более 1000 м.д.;
известны КССВ J(Ni-P) = 200 Гц в комплексах Ni(CO)3L и 400-500 Гц
в комплексах NiL4.
Платиновые металлы. Широкое использование платиновых ме-
таллов и их соединений в промышленности, новейшей технике и тех-
нологии вызывает неослабевающий интерес к химии этих элементов,
многие проблемы которой решаются с помощью метода ЯМР. Каждый
из этих элементов имеет магнитные изотопы, свойства ядер которых
приведены в табл. 3.29. ЯМР всех платиновых металлов, кроме 195Pt,
имеет крайне низкую чувствительность, поэтому ядерный магнитный
резонанс иридия в растворах пока не обнаружен, а прямые измерения
ЯМР на 99,101 Ru, 103Rh, 1870s, 105Pd сделаны сравнительно недав-
но [204, 288-290]. Измерения ЯМР на этих ядрах осложнены боль-
шими диапазонами ХС, большими ширинами линий квадрупольных
ядер и долгими временами релаксации ядер со спином 1/2. Обзоры [4,
51, 291, 293] содержат наиболее полные данные по ЯМР платиновых
металлов.
Рутений. В последнее время ЯМР рутения активно развивает-
ся [295-306]. В большинстве случаев более выгодно использовать
для ЯМР изотоп 99Ru из-за меньшего квадрупольного момента ядра,
несмотря на меньшую чувствительность. Чувствительность ЯМР "Ru
вшестеро выше, чем у 17О, поэтому удивительно, что рутений столь
долго находился вне поля зрения ЯМР. Данные ЯМР "Ru приведены
в табл. 3.38.
Измерены константы ССВ J(89RuC) = 44,8 в Ru(CN)g~;
J("Ru-l7O) = 23,4 Гц в RuO4; J("Ru-119Sn) - 846 Гц в Ru(SnCl3)45'
[294].
ЯМ-релаксация "Ru имеет квадрупольный характер. Из-за низ-
кой чувствительности ЯМР рутения широкие линии несимметричных
форм могут быть «утеряны» на фоне узких линий, поэтому про-
явление в спектре всего рутения в растворе часто проблематично.
ЯМ-релаксация "Ru использована для изучения подвижности класте-
ра Et4N[RuCo3(CO)12] [300].
В [297] методами ЯМР "Ru и 15N изучена кинетика нитрования
комплекса рутения [RuNOCls]2-. В спектре ЯМР рутения (рис. 3.28)
наблюдали исходный комплекс [RuNOCls]2- (<5 = 4192 м.д.) и ко-
нечный в процессе нитрования [RuNOOH(NO2)4]2-, 5 = 3482 м.д.,
и примесный ион [RuCle]4-. В спектре 15N виден нитрующий агент
NO./, 6 = 231 м.д. и атомы N на координате O2N-Ru-NO2 комплекса
[RuNOOH(NO2)4]2-, 6 = 94 м. д.
Механизмы взаимопревращения [RuNOOH(NO2)4]2- <-> i{uc-[RuNO
(OH)3(NO2)(H2O)]“ и промежуточные комплексы исследованы в [298].
Таблица 3.38
Химические сдвиги ЯМР рутения-99
Форма Растворитель Г, К <5, м. д. (ИС Гц) Ссылка
КС11 H2O Комн. 11361 [294]
[Ru(CN)6]4- D2O 293 0 [294]
[Ru(NH3)6]3+ Нас, D2O 7821 [294]
[RuCl6]4- H2O 295 3967 (49) [297]
[Ru(NO2)6]4~ H2O 295 5892(150) [298]
RuO4 1 M/CCI4 2021 [294]
[RuNOCls]2- H2O 295 4192(440) [297]
[RuNO(NO2)3Cl2]2- H2O 295 3555 (785) [301]
[RuNOOH(NO2)4]2~ Ацетон 323 3488(133) [296]
« H2O 303 3500 (68) «
« H2O 343 3520 (74) «
[RuNOOH(NO2)3(H2O)]- H2O 303 3635(140) «
« H2O 343 3688 (98) «
pe6*-[Ru(NO2)3NO(OH)2]2- H2O 295 3704(170) [298]
транс* - [Ru(NOOH)*(NO2)2*H2OOH]- H2O 295 3614(300) «
pe6-[Ru(OH)3NO NO2H2O|- H2O 295 4102 (450) «
Ru(Cp)2 Нас., CH2C12 346 -3291 [294]
Ru2(CO)i2 Нас. СбН6 297 -3299 [294]
Ru2(CO)6C14 0,3 M в ацетоне 297 -817 То же
0,3 М пиридин 297 -1215 «
Cs[Ru(CO)3C13] Ацетон 311 -807 «
EtOH 313 -1020 «
D2O/HC1 296 -927 «
d2o 296 -749 «
(Et4N)4[Ru(SnCl3)5Cl] Нас., MeCN 303 -467 «
[Ru(bpy)3]Cl2 Нас., D2O 297 2497 <<
[Ru(bpy)3](PF3)2 Нас., MeCN 297 2588 «
1 В шкале ХС "Ru.
Рис. 3.28. Спектры ЯМР "Ru (а) и l5N (б) раствора комплекса [RuNOCls]2
в процессе его нитрования
Внешнесферный перенос электронов в гексааквакомплексах Ru(II/III)
методами ЯМР "Ru, 17О и ЭПР, а также влияние парамагнитных
ионов Си2+ и Ni2+ на процесс нитрования комплексов рутения(Ш)
изучен в [301].
Активно изучают методом ЯМР "Ru металлоорганические ком-
плексы и комплексы с органическими лигандами, такие как комплекс
[Ru(bpy)s]а+ (известный катализатор фоторазложения воды), родствен-
ные комплексы [295] и другие [303-304].
Ряд работ посвящен расчету ХС рутения ([305] и цитируемые там).
Родий. Родий — единственный из платиновых металлов элемент
с содержанием магнитного изотопа 100%. В ЯМР ядер, связанных
с родием, наблюдают характерные дуплеты из-за ССВ, и сравнительно
давно для измерения ХС 105Rh применяют методы двойного резонан-
са [51, 307]. Однако для многих комплексов, в которых нет непо-
средственной связи родия с «хорошим» ядром, т. е. нет разрешенных
дублетов, применение методов двойного резонанса невозможно и ХС
103Rh определяется прямым измерением ЯМР, первое из которых было
сделано только в 1979 г. [289]. В высоких полях Hq 7 Т измерения
на ядре 103Rh возможны в разумное время, и при концентрации родия
1 г-ат/л в диапазоне 500 м.д. приемлемый сигнал может быть накоплен
за 2 ч.
Накоплен большой объем данных ЯМР 103Rh [51, 202, 308], однако
большая часть их относится к металлоорганическим соединениям и по-
лучена методами двойного резонанса. Данных комплексов с неоргани-
ческими лигандами, полученных прямыми измерениями ЯМР 103Rh,
существенно меньше.
Среди соединений родия нет таких, которые могли бы служить
эталоном и регистрировались за 15-30 мин, что возможно при концен-
трации 5-10 г-ат/л. Поэтому ХС 103Rh отсчитывают от 5 = 3,16 МГц,
для чего измеряют частоту ЯМР сигнала и магнитное поле; пересчи-
тав частоту ЯМР к полю, в котором частота ПМР тетраметилсилана
108 Гц, определяют тем самым величину S для сигнала. В этой шкале
ХС ЙЬ(Асас)з равен 8358 м.д. [307], и его также используют как
эталон. Приведение к той или иной шкале делается по ЯМР других
ядер или по сигналу ЯМР-стабилизатора поля спектрометра. При этом
для точных измерений надо учитывать поправку на восприимчивость
образца (при внешнем эталоне для стабилизации) и влияние среды на
ХС эталона при внутреннем эталоне. Большой температурный коэф-
фициент ХС, зависимость ХС от растворителя и других компонентов
раствора требуют специальных мер при измерениях ХС с точностью
~ 1 м.д. Масштаб изменения ХС от температуры и растворителя пока-
зан на рис. 3.29.
-550 -500 -450 -400 -200 -100 м.д.
Рис. 3.29. Температурная зависимость ХС ЯМР 103Rh комплекса [RhX(PPh)a]-,
X = CN-, NCO-, С1- в дихлорметане (сплошная линия) и хлороформе
(пунктир)
Вследствие большого диапазона ХС родия пересчет из одной шкалы
в другую следует делать по формуле (3.2). Запись спектров ЯМР 103Rh
приходится проводить в узком интервале ХС ~1000 м.д., поскольку
измерение во всем диапазоне ХС родия требует неоправданно боль-
ших объемов памяти данных и времени накопления. Шкала ХС 103Rh
приведена на рис. 3.30, ХС некоторых соединений и комплексов —
в табл. 3.39.
Измерения ХС ЯМР 130Rh комплексов Rh(III), как и изоэлектрон-
ного ему Со(Ш), определяются парамагнитным вкладом в экранирова-
ние; наблюдаются корреляции типа 5-(ДЕ)_| длинноволновых пере-
Таблица 3.39
Химические сдвиги ЯМР родия
Форма <5, м. д. от - = 3,16 МГц Условия, источник
Rh(I): [RhCN(PPh3)3] -449 СНС13, 300 К [309]/J(Rh-P) = 144
[RhNCO(PPh3)3] -168 СНС13, 300 к [309]/J(Rh-P) = 173
[RhNCS(PPh3)3] -156 СНС13, 300 к [309]/J(Rh-P) = 175
[RhN3(PPh3)3] -70 СНС13, 300 К [309]/J(Rh-P) = 180
[RhBr(PPh3)3] -142 DCM, 280 К [310]/J(Rh-P) = 192
[RhI(PPh3)3] -267 СНС13, 260 К [310]/J(Rh-P) = 194
Rh(III): Na3[Rh(NO2)6] 5580 Н2О, 295 К [311]
[Rh(NO2)3(H2O)3] 6570 Н2О, 295 К [311]
Na3[RhCl6] 8302 Н2О, 298 К [237]
7985 НС1 водн., 313 К [313]
Na3[RhBr6] 7077 НВг водн., 313 К [312]
[Rh(H2O)6]3+ 9936 НС1О4, 298 К [314]
« 9866 НС1О4, 276 К [315]
[Rh(H2O)6]3+ 9910-9922* HNO3 водн., 300 К [316, 317]
[Rh(H2O)6]3+ 9989* H2SO4 водн, 300 К [318]
[RhAcac3] 8358 СНС13, [307]
[Rh2(/z-OH)2(H2O)8]4+ 9997 НС1О4 водн, 298 К [319, 320]
[Rh2(M-OH)(/z-NO3)(H2O)8]3+ 9936-9947 HNO3 водн., 300 К [317]
[Rh2(M-SO4)2(H2O)8]3+ 10060 H2SO4 водн, 300 К [318]
[Rh(SCN)6]3- 2726 Н2О, 298 К [321]
Rh(-I): N(PPh)2[Rh(CO)4] -644 MeCN, 298 К [308]
N(PPh)2[Rh3S2(CO)4] " -413 ТГФ, 177 К [322]
[Rh6(CO)16] -426 СНС13, 333 К [323]
Rh (металл) -1356 [324]
Зависит от концентрации HNO3; ** треугольная бипирамида; *** октаэдр.
£
ю
m
II
[i]
тыс. м.д.
10 8 6 4 2 О
|------------------------------------------|Rh(PPh3)3XClH I
Н20 СГ ВГ N02“ SCN“
RhX4(SMe2-)2 Cl |-----------------1 I
|--1 Полиядерные pe6-RhX3(PMe3)3 |----------1
сульфаты Rh(III)
|---------------------------------------1 pe6-RhX3(SMe2)3
apa«-RhX3(PMe3)3
apa«-RhX3(PMe3)3Cl3 I |—|
mpa«c-RhX2(PMe3)+ |----
I—I mpaHC-RhX2(SMe2)^
Рис. 3.30. Диаграмма химических сдвигов родия
ходов (например, d-d-переходов [325]). Этим же вкладом объясняется
большой температурный коэффициент ХС. Однако может наблюдаться
и обратный ход зависимости ХС-ДЕ, например в хлороаквакомплексах
Rh(III), который может быть вызван влиянием радиального фактора
(г-3), поскольку при замещении лиганда более тяжелым величина
фактора (г-3) уменьшается, что ведет к экранированию центрального
ядра [325]. Экранирование родия в водных растворах сильно зависит от
ионного фона. Изменение pH, увеличение концентрации ионов может
менять ХС комплекса до 50 м.д. [326].
Известно большое количество КССВ с родием; в гидридах
Rh(III) *J(Rh-H) = 15-30 Гц и значительно меньше в гидридах Rh(I)
[327]. В комплексах [RhFx(H2O)6_z]3-3: 'j(Rh-F) = 50-250 Гц [308,
328]; >J(Rh-15N) = 10-24 [309]; ‘J(Rh-Si) = 20-90 [308];
'J(Rh-P) = 70-250 Гц; ‘J(Rh-Se) = 30-140; ‘J(Rh-Te) = 70-140 Гц;
j(Rh-Sn) = 520-740 Гц [308, 329].
Комплексные хлориды родия изучали в [237, 313, 326, 330], реак-
ции комплексных хлоридов родия в [331, 332], комплексные нитриты
родия изучали в [311, 333, 334], реакции нитрования родия изучали
в [248, 335, 336]. Состояние родия в азотнокислых растворах изучали
в [316, 317, 337-339], сернокислые растворы родия — в [314, 318,
340-342]. Для последних характерно разнообразие форм в растворе
(рис. 3.31).
Комплексообразование Rh(III) с фосфорной кислотой изучали в
[343], каталитический окислительный гидролиз Н2РО^ с помощью
Rh3+ — в [344]. Комплексы родия(Ш) с парамагнитным енаминокето-
ном изучали в [345].
10600 10400 10200 10000 м.д. 9800
Рис. 3.31. Спектр ЯМР l03Rh сернокислого водного раствора Rh(lII) на частоте
12,77 МГц
Палладий. Палладий относится к трудным ядрам из-за низкой
чувствительности и очень широких линий. ЯМР 105Pd в металле на-
блюдали в [346]; была измерена температурная зависимость сдвига
Найта. Для распространенных соединений Pd(II) характерна плоско-
квадратная координация, которой сопутствуют большие ГЭП и корот-
кие времена релаксации квадрупольных ядер. Высокосимметричные
комплексы характерны для Pd(IV), но он не стабилен в растворах:
Pd(IV) восстанавливается до Pd(II), окисляя даже воду. В присутствии
мощного окислителя — в растворе Н2Р6СЦ в НО : HNO3 = 3:1 ион
PdClg- существует несколько часов, и при концентрации 1,8 г-ион/л
и температуре 298 К в таком растворе удалось наблюдать линию
ЯМР l05Pd с шириной 1200 Гц [289]. Этот сигнал сЕ = 4,58572 МГц
(±25 Гц) смещен от линии металла в слабое поле на 32000 м.д. Темпе-
ратурный коэффициент его (2,3 м.д./К) близок к таковому для линии
ЯМР l03Rh иона [RhCU]3- [326]. При 323 К ширина линии возрастает
до 2500 Гц, указывая на обмен лигандов. В растворе H2PdC14 в насы-
щенном растворе С12/НС1 линия 105Pd имеет ХС ±28 м.д. от первой,
показывая тем самым влияние ионного фона.
Возможно, ЯМР 105Pd комплекса [PdCl^]2- в водном растворе
с концентрацией 0,2М наблюдали в [347], с шириной линии 8 кГц. На-
блюдение не было проверено накоплением сигнала 105Pd при смещении
магнитного поля Но на величину, превышающую ширину линии, хотя
положение этой линии относительно PdClg- не противоречит таковому
для подобных комплексов платины
Основным способом изучения комплексов палладия методом ЯМР
является ЯМР лигандов и среды [348-351]. Для изучения реакций
хлорирования и нитрования палладия использовали ЯМР 14>,5n и 35С1
[348]; при этом лиганд С1- в комплексах не наблюдается методом ЯМР
35С1, однако концентрация свободного хлорида в растворе легко опре-
деляется и может быть использована для определения материального
баланса. Для определения строения комплексов и их концентрации
использовали координатные сдвиги азота и кислорода (см. рис.3.25).
Для изучения строения комплексов палладия использовали ЯМР 17О
[349], для изучения процессов гидролиза и поликонденсации ЯМР
133Cs [350, 352].
Осмий. Прямое наблюдение ЯМР осмия осложнено как на ядрах
1870s (низкая чувствительность и долгие времена релаксации [204]),
так и на ядрах 1890s (широкие линии ЯМР [353], ситуация близ-
ка к ЯМР 105Pd), Оценка времен релаксации 1870s в OsO4, [204]
(1 < Т1 < 26 с; 0,7 < Т2 < 12 с) и 1890s в расплаве OsO4 [353]
(Т2 = 230 мкс) является примером прямого наблюдения ЯМР осмия.
По-видимому, шкала ХС ЯМР осмия будет составлена методом двой-
ного резонанса, поскольку в кластерных соединениях осмия наблюдали
константы *J(H-1870s) — 14-38 Гц [354]. Для этого требуются «силь-
ные» ядра (1Н, 19F, 31Р), поскольку для наблюдения сателлитов от
1870s (природное содержание 1,6%) необходимо хорошее соотношение
S/N. Активно изучают оксофторидные комплексы осмия [355, 356].
Иридий. Оба изотопа иридия имеют магнитный момент, одна-
ко малая чувствительность и очень большой квадрупольный момент
каждого ядра делают проблематичными как прямое наблюдение ЯМР
иридия, так и методами двойного резонанса. Однако комплексы 1г(Ш)
могут характеризоваться по ЯМР лигандов, как в [248, 357-359].
Платина. Довольно высокая чувствительность ядра 195Pt, боль-
шой диапазон ХС, богатая химия платины и важность платины и ее
соединений для химической промышленности и новейших техноло-
гий привели к тому, что ЯМР платиновых металлов — это в боль-
шой мере ЯМР 195Pt. ХС соединений платины лежат в диапазоне
4000 4—2000 м.д., хотя общий диапазон ХС 14000 м.д. за счет дале-
ко отстоящей линии ЯМР PtFg-. Диаграмма ХС 195Pt приведена на
рис. 3.32, ХС некоторых соединений — в табл. 3.40-3.42. Большому
диапазону ХС соответствует большая чувствительность ХС соедине-
ний платины к концентрации, температуре, растворителю. Для PtClg-
изменение ХС от растворителя достигает 400 м.д., для PtCl2- —
200 м.д., причем сравнение 6- и 4-координированных комплексов по-
казывает, что влияние молекул растворителя по оси z в послед-
них несущественно. Большой температурный коэффициент ХС PtClg-
(^1м.д./К) осложняет использование PtClg- как эталона. В ней-
тральных комплексах платины температурный коэффициент ХС лежит
в пределах 0,13-0,82 м.д./К [51, 360, 361].
На рис. 3.33 показан спектр ЯМР 195Pt раствора при лигандном
замещении Cl- —> NO^-, в котором одновременно присутствуют линии
всех комплексов [PtCln(NC>2)4-n]2- [364].
-Ц-------1-------1-------1----------------1-------1------L
Cl Br CN I
Pt(II)X2— I--------1----------1----1
NH3
Pt(II)N4 N02 i i i En/2
Pt(II)Cl3N |--1 H Pt(II)Cl3P
O, Sb, Те
Pt(II)Cl3L S, Se |—| As
Карбонильные кластеры |--------1
Pt(0)(PR3)4 |-----
Рис. 3.32. Диаграмма химических сдвигов l95Pt
Таблица 3.40
Химические сдвиги ЯМР платины
Форма Растворитель <5, м. д. Ссылка
Na2PtCI6 D2O, 293 К 4522 [51]
гран- PtCl3F2- H2O 7605 «
транс- PtC^OH)2- « 5778 [369]
PtCl5Br2- « 4236 [51]
гран-РЮзВг2- << 3635 «
PtClBr2“ << 2990 «
PtBr2- << 2651 «
(NBu?)2PtCI6 « 4783 [360]
H2PtI2- « -1528 [360]
Na2Pt(CN)6 H2O, 323 К 672 [51]
Pt(en)3(C104)2 H2O, 323 К 3579 [360]
транс*-[PtenCl2ClNH3] + ТГФ, 295 К 3864 (650) [247]
транс* -[PtenC12Py2]2+ H2O, 297 К 3911 (820) «
Таблица 3.40 (продолжение)
Форма Растворитель 8, m. д. Ссылка
mpaHC*-[PtenCI2(NO2)2] H2O 4535 [242]
4Mc*-[PtenCl2*(NH3);]2+ CH2C12 3831 (1200) [247]
ptci2- CH2C12 2887 [51]
(NBu?)2PtCl4 H2O 3055 [363]
(NBu4n)2Pt2Cl6 CH2C12 3325 [51]
PtBr2- CH2C12 1843 [51]
(NBu4n)2PtBr4 HI/H2O 2066 [363]
(NBuJ)2Pt2Br6 CH2C12 2233 [51]
H2PtI4 H2O -933 [360]
(NBu4n)2Pt2I6 H2O -600 [51]
PtCl3NO2~ H2O 2860 [364]
4izc-PtCl2(NO2)2- H2O 2733 «
транс- PtCl2(NO2)2~ H2O 2731 «
ptci3H2cr H2O 3341 «
Pt(CN)2- H2O, 210 К -204 [51]
Pt(SCN)2- H2O 604 [51]
Pt(NH3)2+ H2O 1900 [360]
« H2O 1966 [366]
4Mc-Pt(NH3)2(H2O)2+ H2O 2941 [365]
4izc-Pt(NH3)2Cl2 H2O 2445 [366]
Pt(NH3)3Cl+ H2O 2183 «
4Uc-Pt(NH3)2(OH)2 H2O 2965 «
Pt(NH3)3(OH)+ H2O 2475 «
Pt(OH)42- H2O 4372 «
Pt(OH2)2+ H2O 6552 [367]
Pt(OH2)3NO+ H2O 6528 [368]
Таблица 3.40 (окончание)
Форма Растворитель 5, m. д. Ссылка
Pt(OH2)3SO4 H2O 6626 [368]
Pt(OH2)3PO4H+ H2O 6697 «
Pt(NO2)2“ CH2C12 2353 [364]
Pt(en)2“ CH2C12 1493 [362]
Pt(SCN)2- CH2C12 565 [291]
Pt(SCN)(NCS)j“ (CD3)2CO 1085 «
mpaHC-Pt(SCN)2(NCS)2- « 1723 <<
mpaHC-PtCl(SnCl3)(PEt3)2 « -260 «
mpaHC-Pt(SnCl3)2(PEt3)2 — -632 «
Pt2Cl2(SnCl3)2(PEt3)2 — 254 «
От S = 21,4 МГц.
Таблица 3.41
Данные ЯМР l95Pt полиядерных оксонитрокомплексов Pt(II,IV)
Форма 5, м.д. (W, Гц) pH
4Mc-[Pt(NO2)2(H2O)2] 3151 <2,5
[Pt2(№-OH)2(NO2)4]2- 3444 —
[Pt3(№-O)2(NO2)6]4- 3558 (500) ^8,6
[Pt4(№-O)4(NO2)12]4- 7461 (450) ^2
[Pt7(^3-O)6(NO2)12]8- 3783 —
Широко исследованы карбонильные комплексы Pt(II), Pt(I) и Pt(O).
Соединения Pt(I) в принципе парамагнитны, однако димеры диамагнит-
ны. И в них можно наблюдать ЯМР на всех ядрах комплекса. Изучение
процессов замещения галогенид-ионов в комплексах [Pt2X4(CO)2]2-,
(X = Cl, Вг, I) показало возможность произвольного замещения лиган-
дов X друг на друга; предложены механизмы реакций замещения [370].
Диапазон ХС 195Pt в комплексах —200-640 м.д., ХС 13С около 160 м.д.
и ’J(Pt-C) —2000 Гц и почти не меняются при замещении галоге-
нид-ионов, a ’j(Pt-Pt) «5000 Гц указывает на прямую связь между
Таблица 3.42
Данные ЯМР биядерных комплексов Pt-Tl
Форма 'j(Pt-205Tl), Гц 5Pt 5T1
[Pt(CN)6]2- — 655 —
[(NC)5Pt-Tl(H2O)I]2+ 71060 474 786
[(NC)5Pt-TlCN(H2O)4]- 57020 383 1371
[(NC)5Pt-Tl(CN)2]2- 47260 184 1975
[(NC)5Pt-Tl(CN)3]4- 38760 68 2224
T1(CN)4]- — — 3010
[Pt(CN)4]2- — 213 —
о
LO
00
сч
2
сч о
ео ео оо
lo ео
сч сч сч сч
ц1Ц lHj .ill,
Рис. 3.33. Спектр ЯМР 195Pt раствора Na2PtCl4 после добавления к нему
Nal5NO2 в соотношении Pt : NO^" = 1:2
атомами металла. Обратимые превращения карбонильных комплексов
[Pt3(CO)6]2- изучали в [371].
В тетраэдрическом комплексе Pt4(CO)5L4 из-за содержания магнит-
ного изотопа 195Pt 34% возможны 4 магнитно неэквивалентных изото-
помера (0-4 атома 195Pt в тетраэдре) с различными типами спектров
l95Pt и 31Р при L = PEt3 [372, 373]. Наблюдаемый спектр является
суперпозицией частных спектров с их вероятностным весом. Спектр
ЯМР 195Pt и 31Р комплекса приведен на рис. 3.34 [372].
Полиядерные оксонитрокомплексы Pt(II,IV) изучены в [374, 375],
табл. 3.41, биядерные гетерометаллические цианидные комплексы Pt-Tl
с расстоянием М-М « 260 пм изучены в [376] (табл. 3.42).
Сульфоксидные биядерные комплексы Pt(II) с двойной ОН-связью
изучены в [362].
Влияние замещения на ХС 195Pt показано в табл. 3.43-3.45.
a
Рис. 3.34. Спектр ЯМР l95Pt раствора кластера Pt4(CO)5(PEt)4 в толуоле: а —
спектр; б — схема спектра при размещении трех изотопов 195Pt в кластере;
в — то же при двух изотопах 195Pt
Таблица 3.43
Эффекты замещения в ЯМР l95Pt в комплексах PtX3L [51]
L X L X
Cl Вг I Cl Br I
С = О 1264 563 -953 PF3 907 183 -1370
СН2=СН2 1748 1060 — AsMe3 1360 664 -913
С = NMe 1487 806 -730 SbMe3 1390 805 -1109
N = СМе 2511 1827 — OH2 3330 — —
NHMe 2670 2114 529 SMe2 1766 1118 -440
NMe2 2818 2282 — SeMe2 1764 1957 -596
РМе3 1033 414 -973 TeMe2 1474 707 -995
Р(ОМе)3 1037 371 -1060 SOMe2 1535 892 —
Комплекс Pt(CN)g- имеет температурный коэффициент (0,5 м.д./К),
мало зависящий от растворителя. Большая чувствительность ХС 195Pt
позволяет наблюдать изомеры (табл. 3.40, 3.44-3.46) и изотопомеры
комплексов [372, 373]. Различие между изомерами в хлоробромных
комплексах как Pt(II), так и Pt(IV) невелико; подобная картина
Таблица 3.44
Эффекты замещения лигандов в ЯМР l95Pt комплексов платины(П) [51, 364]
L Комплекс
PtCl3L цас-Р1С12Ь2 mpaHC-PtCl2L2 PtClL3 PtL«
CNMe 1487* 665 — 203 -318
NMe2 2818 — 2647 — —
РМе3 1033 125 583 -146 -358
AsMe3 1360 242 753 -211 —
SbMe3 1390 -79 — -746 —
SMe2 1776 982 1102 523 —
Br_ 2661 2408 2418 2138 1843
NO2“ 2959 2734 2732 2584 2346
ХС 195Pt относительно S = 21,4 МГц.
Таблица 3.45
Эффекты последовательного замещения лигандов в ЯМР 195Pt комплексов пла-
тины(1У) [51]
L Комплекс
PtCl5L ^ac-PtChl^ mpaHC-PtCUL2 pe6-PtCl3L3
CNMe 3323 2447 — —
PMe3 3060 1931 2367 1348
AsMe3 1359 2162 2502 1449
SMe2 3656 2788 2850 —
и в комплексах [RhCl^Bre-x]3- [312]. Заметим, что отнесение линий
ЯМР этих комплексов платины сделано в предположении статистиче-
ского распределения форм; наблюдаемое в комплексах Rh(III) примерно
одинаковое различие ХС изомеров одной формы и ХС соседних по со-
ставу форм [313] ставит вопрос о дополнительных аргументах в поль-
зу отнесения линий хлоробромных комплексов платины. В комплек-
сах PtC12(NO2)2- различие между цис- и транс-изомерами невелико
и разрешается только на высокополевых спектрометрах (рис. 3.33).
В ряде комплексов Pt(II) наблюдается корреляция <5-(ДЕ)-1 [51],
подтверждающая преобладающее влияние парамагнитного вклада и
фактора ДЕ1 в этом вкладе; экстраполяция к ДЕ оо (ар —> 0) дает
диамагнитное экранирование Pt(II), равное 5900 м.д. [51]. Однако при
замещении галогенами наблюдается противоположный ход зависимо-
сти ХС — ДД (как и в комплексах Rh(III) [237]), что показывает преоб-
ладание влияния других факторов в парамагнитном вкладе. В ЯМ-ре-
лаксации l95Pt большое значение имеют спин-вращательный [362]
и скалярный механизмы релаксации.
Имеется много данных о КССВ с платиной; в гидридах ’J(Pt-H) =
= 700-1300 Гц, причем величина констант согласуется с транс-
влиянием лигандов [51, 364-366]; в комплексах со связанным уг-
леродом *J(Pt-В * * * * 13C) 2000 Гц [380]. Для N-связанных NC-, NCS-
'J(Pt-14N) = 250-500 Гц [381]. Величина КССВ 195Pt с азотом и фос-
фором зависит от состояния окисления платины и транс-влияния
лигандов [51, 361, 382]. По константам можно различать изоме-
ры комплексов Pt(II) — цис-изомеры имеют большие константы,
чем транс-изомеры (для 15 *N J цис/J транс = 1,2 [382], для 3|Р
Jцис/Jтранс = 1,4 [383]). Необычное взаимное влияние лигандов,
приводящее к изменению знака констант ССВ, обнаружено в комплек-
сах с SeMe2, ТеМег.
Гидролиз [PtCU]2- изучен в [387] при концентрации Pt 0,05 М/л
и добавлении 1 М NaOH. Данные ЯМР 195Pt [387], приведенные к S =
= 21,6 МГц вместе с данными 17О, представлены в табл. 3.46.
Таблица 3.46
Данные ЯМР комплексов, обнаруженных в процессе гидролиза PtCl2-
Комплекс 6 l95Pt, L = ОН 5l95Pt, L = OH2 <517OH2OC
ptci?- 2877 — —
PtCl3L 3328 3374 -86 (Cl-Pt-OH2)
циС-Р1С\2^2 3648 3748 —
транс-Р1С\2^2 3884 3915 —
PtClL3 4022 4208 -102 (H2O-Pt-OH2)
PtL4 4339 4590 -72 (H2O-Pt-OH)
В биядерных комплексах платины наблюдали необычно большие
величины J(Pt-Pt) до 9 кГц [51], хотя обычно они гораздо меньше
[376].
Обогащение изотопом 15N позволило обнаружить изомерию в ком-
плексе Pt(CNS)4~ (N-S-изомеризация); кроме наблюдаемого в твердом
состоянии иона Pt (SCN)^-, после старения раствора наблюдали ионы
Pt(NCS)4~, Pt(NCS)(SCN)3- и Pt(NCS)2(SCN)2-, которые однозначно
отнесены по мультиплетности сигнала ЯМР l95Pt [381]. Необычно
большие величины *J(Pt-ll9Sn) получены в комплексах с лигандом
SnCl3 (20-30 кГц) [381, 384] и *J(Pt-205Tl), в биядерных комплексах
Pt-Tl (20-30 кГц) [376].
Данные ЯМР 185Pt (ХС и КССВ) активно используют для изуче-
ния структуры и взаимного влияния лигандов и химических реакций
комплексов платины [248, 385, 386].
3.5. Медь, серебро, золото, цинк, кадмий, ртуть
Для исследования методом ЯМР пригодны только соединения
и комплексы с электронной конфигурацией d10. Химия этих элемен-
тов богаче химии их аналогов — щелочных и щелочноземельных
металлов, что отражается и в объеме данных ЯМР. ЯМР элементов
IB- и ПВ-групп привлекает внимание специалистов бионеорганической
химии, поскольку они играют ведущую роль в одних биохимических
реакциях и оказывают влияние на многие другие. Свойства ядер этих
элементов приведены в табл. 3.47.
Таблица 3.47
Свойства ядер элементов групп IB, ПВ
Изотоп Спин NA, % Sens 1 Диапазон XC, m. д. Q, барн Частота ЯМР эталона S, МГц2 Эталон
13С — — 1{‘H} — — — —
63Си 3/2 69 122 1000 -0,21 26,505 Cu(MeCN)4
65Си 3/2 31 67 « -0,20 28,394 «
107 Ag 1/2 52 0,063 2000 — 4,0465 [Ag+] -> 0
109 Ag 1/2 48 0,092 « — 4,653623 «
197 Au 3/2 100 0,048 — 0,58 1,7125 Au металл
67Zn 5/2 4,1 0,22 400 0,15 6,254 [Zn2+] -> 0
"‘Cd 1/2 12,8 2,3 900 — 21,205 «
ll3Cd 1/2 12,3 2,5 « — 22,1825 [Cd2+] -> 0
22,193 173 CdMe2
‘"Hg 1/2 16,8 1,8 5000 — 17,910841 HgMe2
201 Hg 3/2 13,2 0,36 << 0,5 6,5995 «
‘Относительно |3С{‘Н) с учетом природного содержания; 2 частота ЯМР в
поле, в котором частота ЯМР 'Н ровно 100 МГц.
Каждый элемент группы IB и ПВ имеет магнитный изотоп, однако
метод ЯМР стал широко применяться только в последнее время вслед-
ствие низкой чувствительности ядер этих элементов (кроме меди). Все
они, кроме 197Au, позволяют проводить изотопные замещения и ис-
пользовать метод ЯМР как детектор изотопной метки. Данные ЯМР
этих элементов имеются в [2, 5, 51, 388].
Медь. Применение ЯМР ограничено химией Cu(I) и значитель-
ной шириной линий, что снижает специфичность метода ЯМР 63Си.
Первое измерение ЯМР 63Си сделано в [389]. Состояние Cu(I) ста-
билизируется тг-акцепторными лигандами; комплексы Cu(I) лабильны,
и измерений поэтому мало. Данные о параметрах ЯМР меди приведены
в табл. 3.48.
Таблица 3.48
Данные ЯМР меди-63
Форма Примечания 6, м.д. (ГЕ, Гц) 'J(63Cu-P), Гц Ссылка
Cu(CN)3- — 501 — [390]
Си(Пиридин)^ 0,05 М/Пиридин 111 (880) — [391]
Си(РМе3)4 CH2CI2/CD2CI2 286(150) 790 [397]
Си[Р(ОМе)3] + 0,04 M/P(OMe)3 83 1214 [391, 392]
Cu[P(OEt)3] + 0,1 M/P(OEt)3 92 1224 [392]
Cu(MeCN)4BF4 0,1 M/MeCN 0(540) — [390]
Cu2HgI4 Тв. -270 — [2]
CuCl To же -319(2500) — [393]
CuBr <r -321 (2600) — То же
Cui <r -381 (3400) — «
Cu(AsMe3)^ CH2C12/CD2C12 -42 (6000) — [397]
Cu(SbMe3)+ <r -6(180) — «
Cu(SbPh3)+ « -249 — «
Cu Газ. -1820 — [394]
Подробные исследования комплексов Cu(I) с пиридином и ацето-
нитрилом сделаны в [395, 396]. Зависимость параметров ЯМР 65Си от
состава растворителя показана на рис. 3.35.
Комплексы с фосфорными лигандами изучены в [395, 396, 398],
N-тридентатными лигандами — в [399]; растворы и суспензии галоге-
Мольная доля пиридина
Рис. 3.35. Химический сдвиг и ширина линии 65Си в зависимости от состава
растворителя MeCN/CsHsN
нидов Cu(I) изучены в [400]. Температурная зависимость ХС в галоге-
нидах меди до точки плавления изучена в [401].
ЯМ-релаксация в тетрафосфитных комплексах меди изучена в
[402], зависимость ширины линии ЯМР 65Cu Cu(MeCN)4 в ацетонит-
риле от температуры изучена в [403].
Серебро. Обычно для ЯМР используется изотоп 109Ag, чувстви-
тельность ЯМР которого в 1,5 раза выше, чем у 107Ag; в остальном
свойства ядер примерно одинаковы. Отсчет ХС ведут от акваиона при
бесконечном разбавлении, для которого Е = 4,653 623 МГц [204]. Рабо-
чим эталоном служит водный раствор AgNOa (9 моль/л) с релаксантом
Ге(МОз)з (0,25 моль/л), 6 — —М м.д. [404]. ХС некоторых соединений
и ионов приведены в табл. 3.49.
Зависимости ХС 109Ag от концентрации и анионов близки к за-
висимости для щелочных металлов; концентрационная зависимость
почти линейна [404, 406]; оксоанионы смещают линию ЯМР серебра
в сильное, а галогенид-ионы — в слабое поле. S-координированные
комплексы изучены в [405], N-координированные комплексы — в [405,
407]. Общий диапазон ХС серебра около 1000 м.д. Более подробно
ХС в галогенидах серебра изучены в [408]; ХС комплексов в невод-
ных растворах — в [409, 410]; смешанная сольватация Ag+ в водно-
аминовых растворах — в [411]; взаимодействие с нитроксильными
радикалами — в [412]. Для усиления сигнала ЯМР серебра используют
перенос спиновой поляризации [413].
Времена релаксации Ag+ в ЩО (Д = 962 с, Тг = 157 с) и D2O
(1115 и 146 с соответственно) показывают трудности детектирования
ЯМР серебра [404]. Влияние дегазации и выдержки в течение 300
часов 1 М раствора нитрата серебра на ЯМ-релаксацию 109Ag изучена
Таблица 3.49
Химические сдвиги ЯМР серебра
Форма Примечания 8, м.д. Источник
Ag+ [Ag+] ^0 0 [404]
AgNO3 5 M/H2O -40 To же
AgC104 To же -55 «
AgF « 10 «
Ag Газ -940 <f
Ag(S3O3)|~ 3 M Na2S2O3/H2O 841 [405]
Ag(NH2Et)2I 2 M/70%EtNH2:H2O 790 To же
Ag(NH2Et)2Cl To же 495 «
Ag(NHaEt)3Br « 585 «
Agen+ (H2NCH)2 554* <<
AgPy+ Пиридин, [Ag] —»0 350 «
Ag(NCMe)+ NCMe 335 «
* Расчет с использованием констант образования комплексов.
в [414]. В необезгаженном растворе в D2O Т\ — 1034 с, Т? — 166 с,
в Н2О Г, = 971 с, Т2 = 306 с (300 К).
Золото. Чрезвычайно низкая частота и большой квадрупольный
момент не позволили пока обнаружить ЯМР 197Au в растворах. Име-
ется только одна работа по ЯМР 197Au в металлическом золоте [415].
Очевидно, что исследование свойств комплексов золота возможны ме-
тодом ЯМР лигандов и компонентов раствора [416, 417].
Цинк. Единственный магнитный изотоп цинка (67Zn) имеет самую
низкую чувствительность ЯМР среди элементов ПВ-группы. Первое
систематическое изучение ЯМР 67Zn сделано в [418]. Изучена за-
висимость ХС 67Zn от концентрации. Данные ЯМР 67Zn приведены
в табл. 3.50.
При концентрации нитрата или перхлората цинка в воде ниже
3 моль/л ХС 67Zn не зависит от концентрации соли. Можно составить
абсолютную шкалу ХС цинка, так как определено экранирование ато-
ма цинка в газообразном состоянии (для него <тр = 0), относительно
которого акваион Zn2+ имеет ХС 690 м.д. [418]. В растворах солей
галогенидов ХС уменьшается в ряду С1 > Вг > I. Исходя из известных
Таблица 3.50
Химические сдвиги ЯМР цинка-67
Форма Примечания <5, м.д. (W, Гц) Источник
Zn2+ H2O, [Zn] -> 0 0 [419]
То же MeOH -19 • To же
« ДМФ -27 «
ZnCl+ H2O 30 «
ZnCI2 To же 295 «
ZnCl^ « 119 «
ZnCl2- « 253 «
ZnBr2- « -36 «
Zn(NH3)2+ 1 M Zn(NO3)2/ll M NH3:H2O 288(65) [420]
Zn(CN)2- Zn(CN)2/3N NaCN:H2O 284 (40) [420]
Zn(OH)2- 0,6 M, ZnO/16 M KOH 220 (230) [421]
Zn(NO3)2 1 M/H2O 0(47) «
« 2 M/H2O -1,9(89) «
ZnBr2 0,25 M/HBr 171 (42) «
ZnCl2 2 M/H2O 93 (200) «
Zn(C104)2 2 M/H2O -1,4(37) <<
Znl2 1 M/H2O -36(70) «
[PWnO39ZnOH2]5- 0,3 M/H2O 1 (250) [160]
констант и предполагая быстрый обмен [418], рассчитаны ХС отдель-
ных форм.
Квантовомеханический расчет ХС и констант квадрупольного взаи-
модействия (QCC) сделан в [422] (табл. 3.51).
В растворе ZnC12 по мере насыщения раствора увеличивается доля
тетраэдрического иона ZnCl^- [423] (табл. 3.52).
ЯМР 67Zn растворов солей цинка в неводных и смешанных рас-
творах изучены в [425]; в органических растворителях линии ЯМР
67Zn уширены. При введении в растворы ЭДТА линия ЯМР 67Zn
исчезает, Важная роль цинка в биохимических реакциях стимулиро-
вала изучение комплексообразования акваиона Zn2+ с биологически
активными молекулами и аминокислотами [423, 424]. Методом ЯМР
Таблица 3.51
Расчетные и экспериментальные данные <5 и ГЭП для некоторых окружений
атома цинка
Окружение <5ЭКСП, м.д. £расч QCC3Kcn, МГц QCCpac4
ZnO6* 10 -17 -6,3 -8
ZnOg 8 -17 -9,6 -7,9
ZnO4 240 255 2,4 1,7
ZnN2O2 189 194 8,2 9,1
ZnN4 29 275 2,8 4,4
ZnS4 359 245 -3,2 -2,7
* Разные соединения.
Таблица 3.52
ЯМР 67Zn в концентрированных растворах ZnC12 (295 К)
Состав раствора <5, м.д. (ГК, Гц) Доля Zn в форме ZnCl2
ZnCl2 + 185Н2О 14,4(34) 0,06
ZnCl2 + 55,5Н2О 57,6(121) 0,22
ZnCl2 + 6Н2О 145 (700) 0,56
ZnCl2 + 6Н2О + (LiCl + 6Н2О) 203 (350) 0,79
67Zn показано, что ион Zn2+ в водном растворе образует с тетра-
дентатным комплексообразователем [PWnOsg]7- инертный комплекс
[PW11O39Zn(H2O)]5- [160].
Кадмий. Параметры ЯМР обоих изотопов кадмия практически
одинаковы; чувствительность ЯМР обычно используемого изотопа
113Cd только на 10% выше чувствительности lllCd. Для обычных
магнитных полей ядро ll3Cd трудное из-за больших времен релак-
сации, поэтому большинство работ выполнено в сильных магнитных
полях. ХС кадмия отсчитывают от акваиона Cd2+ при бесконечном
разбавлении (табл. 3.53).
Концентрационный ход ХС в растворе перхлората кадмия всего
1,8 м.д. при изменении концентрации от 0,1 до 1 моль/л. Для диме-
тилкадмия измерена величина S = 22,193 173 МГц, и при переходе от
одного эталона к другому <5 CdMe2 = <5 Cdaq — 643 м.д. [425, 426].
Концентрационный ход для разных солей Cd показан на рис.3.36.
Таблица 3.53
Химические сдвиги ЯМР кадмия
Форма Примечания <5, м. д. Источник
Cd(NO3)2 H2O, [Cd] -> 0 0
CdCl£- Tb. 162 • [427]
CdCl2" Tb. 474 [428]
То же ДМСО 442 То же
CdBr2“ 0,1 M в H2O 399 <<
То же Tb. 365 [429]
Cd(SCN)2" 0,1 M в H2O 174 [428]
cdi2- 0,1 M в H2O 71 То же
Tb. 70 [427]
CdS Tb. 703 [429]
/S CN\2- Cd C=C \s cn/2 0,5 M в ДМСО 813 [426]
Cd(NH3)*+ 1 M в NH3:D2O = 1 288 [430]
Cd34TA*+ Н2О 85 [426, 431]
Cd(Gly)2+ * Н2О, -45 °C -77 [430]
Cd(Gly)2+ То же 55 То же
Cd(Gly)2+ То же 153 «
Cd(Gly)2+ То же 252 «
Cd(en)3 Н2О 346 [432]
[PW, 1039CdCl]6- Н2О 102 [160]
[PWnO39CdH2O]6- Н2О -45 То же
* Gly — глицин.
Комплексообразование Cd(II) с галоген-ионами изучалось в [427,
428, 433]; с Р-лигандами — в [434, 435]; с S- и Se-лигандами — в [426,
434-438]; с аминокислотами — в [430, 439, 440]. Комплексы Cd(II) при
комнатной температуре обычно лабильны, и ХС отдельных форм опре-
Рис. 3.36. Химические сдвиги ЯМР кадмия в водных растворах его солей: / —
Cd(NO3)2; 2 - Cd(C104)2; 3 - CdSO4; 4 - Cdl2; 5 - CdBr2; 6 - CdCl2
деляют расчетом, используя известные константы равновесия и сдвиги,
наблюдаемые при разных соотношениях кадмий/лиганд. При низких
температурах линии комплексов с разным лигандным окружением раз-
решаются, как, например, в системе Cd-глицин [440], Cd—I- [441].
Константы ССВ с 113Cd обычно не наблюдаются из-за лабильности
комплексов.
ЯМ-релаксацию 113Cd в растворах перхлората кадмия изучали
в [428]; в растворе Cd(C104)2 Т\ — 25 с, много меньше. Добавление
хлорид-иона вызывает резкое уширение линии ЯМР кадмия до 450 Гц
при Cd : Cl = 1; дальнейшее увеличение доли С1“ сужает линию,
которая при избытке С1~ имеет ширину 30 Гц. ЯМ-релаксацию 113Cd
в растворе с ЭДТА изучали в [431] в диапазоне pH 3-13. В области
стабильности комплекса с ЭДТА изменение Т\ от поля (20 с при 2,35 Т
и 8 с при 9,4 Т) показывает, что анизотропия ХС не может полностью
объяснить ЯМ-релаксацию 113Cd.
Ртуть. Первые работы по ЯМР 199Hg относятся к концу 50-х
годов [442], и вскоре же было установлено, что ртуть имеет большой
диапазон ХС [443]. Большая часть работ по ЯМР ртути относит-
ся к изучению ртутьорганических соединений. Обзоры данных ЯМР
199Hg содержатся в [444]. Изотоп 201 Hg почти не используется в ЯМР
из-за более низкой чувствительности и широких линий. ХС 199Hg
отсчитывают от диметилртути, для которой Е = 17,910841 МГц [2],
хотя в некоторых работах используют и другие эталоны. Диаграмма ХС
ртути приведена на рис. 3.37 [51, 202], а данные ЯМР — в табл. 3.54.
Линейная координация большинства соединений ртути приводит к
большой анизотропии ХС [445], которая дает значительный вклад в
ЯМ-релаксацию 199Hg.
м.д. 20000 10000 0
300кИ'1,2к ' Hgl' l,2kl NaHg2 ' HgMeJ ' ' ^Hg(aTOM)
Сверхпроводник
. , 0 , g ,
(Bu3P)2HgX2 Cl НЧ1C1H-----11 K2HgX4
CN I---------1 I HgX2
CF3HgX Mel------II
C6F5 I—I PhHgX
I--------iRHgX
alkyl2Hg Mel---1 Bu‘
Li2Hg(SiR3)4 I I Hg2+(aq)l lHg+(aq)
LiHgSiMe3
(R3MIV)2Hg SiEt3l-----1 Sn(C6F5)3
Cl I Brl II HgX^“
м w1-------------l(Bu3P)Hg,X4
[Mv,(CO)3Cp]2Hg II I
Cr
-1-----------1---1-----1--1---1-----1--
2000 0 -2000 -4000 м.д.
Рис. 3.37. Абсолютная шкала ядерного экранирования (а) и диаграмма ХС
ЯМР ртути (б)
Изменения ХС 199Hg, определяемые парамагнитным вкладом, обыч-
но выражаются в приближении средней энергии [388] в виде ар =
— const(r-3)6PPu/A.E, где Ри — фактор асимметрии, зависящий от
заселенностей бр-орбиталей и играющий основную роль в изменении
<тр. Уменьшение заселенности, участвующей в sp-гибридизации 6р2-ор-
битали, приводит к уменьшению Ри, т. е. к уменьшению ар и сдвигу
линии ЯМР в сильное поле.
Особенностью ХС 199Hg является противоположный знак измене-
ний ХС под влиянием одних и тех же заместителей по сравнению с 13С
и ‘Н. Наблюдается большой эффект растворителя (свыше 300 м.д.
для HgCl2, см. табл. 3.55); для HgMe2 найдена линейная корреляция
между ХС и потенциалом ионизации молекул растворителя в ряду
C6H6_3;F3; (х = 0-6) [444].
Еще более сильная зависимость ХС у галогенидов ртути, в осо-
бенности у Hgl2 [452]. В ряде работ изучены фосфиновые ком-
плексы Hg(II) и димеры типа P2Hg2 Х4 (Р — фосфин, X — гало-
Таблица 3.54
Химические сдвиги ЯМР ртути
Форма Примечания <5, м. д. Источник
HgMe2 Ч. жидк. 0 [51]
Газ -2,4 [445]
Hg(C104)2 H2O, [Hg] 0 -2253 [446]
Hg2+ Н2О -1453 [446]
HgCl*- Н2О -808 [447]
HgCl3- 0,5 М в СН2С12 -1183 [2]
HgBr3- То же 01935 То же
Hgl3" « -2822 «
HgCl2 Н2О, [Hg] 0 -1153 [446]
0,5 М в Н2О -1203 [447]
0,5 М в EtOH -1497 [448]
HgBr2 0,5 М в EtOH -2152 [2]
Hgi2 1 М в ДМСО -3106 [2]
Hg(CN)2 2 М в Пиридин -1070 [449]
Hg(SiMe3)2 — 481 [450]
1 М в С3Нб 499 [451]
Ме2О 411 То же
Hg(SiH3)2 С6Н6 196 «
Hg(SiCl3)2 CgDe -1001 «
Ме2О -1177 <<
Hg(GeH3)2 -147 [450]
Hg(PMe3)2 0,1 М в D2O -1159 [2]
K2HgI4 Н2О, нас. раствор -3451 [451]
[PWHO39Hg-H2O]5- Н2О -2180 [160]
ген), в которых наблюдали КССВ 'J(P-H) = 4-7 кГц [153, 278-283];
’J(Hg-Si) = 1-4 кГц в соединениях Hg(SiRs)2 [451].
Константы ССВ ‘J(199Hg-C) зависят от гибридизации а-атома уг-
лерода и меняются от 2500 Гц для sp-гибридизации до 700-500 Гц для
$р3-гибридизации. КССВ (Hg-Se) ~2200 Гц, J(Hg-Te) ~ 6500 Гц [463].
Использование констант ССВ для изучения структуры и состояния
форм ртути в растворах подробно освещено в [459].
Цианидные комплексы Hg(ll) изучались в [460, 461]; смешанные
тетрагалогенидные комплексы — в [463]; комплексы со связью Hg-
Мо - в [449].
Комплексы с Р-лигандами изучены в [462, 463], с S-лигандами —
в [464], с S-краун-эфирами Hg(9S3)2, 6 — -275 м.д., Hg(12S4)2, 6 =
= -1120 м.д. — в [465], комплексы с Se- и Те-лигандами — в [466],
комплексы Hg с цистеином — в [467]. Иллюстрацией лабильности в
растворе комплекса ацетата ртути является изменение ХС 199Hg по
мере добавления этиламина (рис. 3.38) [468].
Рис. 3.38. Изменение ХС I99Hg раствора Hg(CH3COO)2 в ДМСО по мере
добавления Е1гМН
Анизотропия ХС для молекул HgMe2 и MeHgX (X = Cl, Вг, I)
изучена в [445]. Для линейных молекул определяющим ЯМ-релакса-
цию является вклад анизотропии ХС [469], что приводит к неожи-
данно коротким (и благоприятным для регистрации ЯМР) временам
релаксации порядка секунд. Этот вклад возрастает с ростом поля, что
дополнительно увеличивает преимущества спектрометров с высокими
магнитными полями для ЯМР ртути. В тетраэдрических комплексах
[Hg(CN)4]2- вклад механизма анизотропии ХС в релаксацию незна-
чителен [469]. ЯМ-релаксация 2o;Hg изучена в [470]. Для триады
Zn-Cd-Hg доступна абсолютная шкала экранирования [446], посколь-
ку известна разность между ХС акваиона и ХС свободного атома:
-690 м.д. для 67Zn, -1106 м.д. для 1I3Cd и —2432 м.д. для 199Hg.
В этой шкале с учетом ХС металлической ртути общий диапазон ХС
ртути превышает 20000 м. д.
ЛИТЕРАТУРА К ГЛАВЕ 3
Скандий, иттрий, лантан
1. Webb A. Background Theory of NMR Parameters. NMR and Periodic table
Ed. by R.K. Harris, В.E. Mann. L. Acad. Press, 1978. P 49-86.
2. Annual reports on NMR spectroscopy. V 10a / Ed. by G.A. Webb. L..
Acad. Press, 1980. P 2-74.
3. Mason J. Patterns of Nuclear Magnetic Shielding of Transition-Metal Nu-
clei // Chem. Revs. 1987. V 87, №6. P. 1299-1312.
4. Rehder D. Early Transition Metals. Lanthanides and Actinides. Multinuclear
NMR / J. MaSon. ed. — N. Y.: Plenum Press, 1987. P 479-519.
5. Annual reports on NMR spectroscdpy. V 9A / Ed. by G.A. Webb. L..
Acad. Press, 1979. P. 126-216.
6. Lutz 0. The magnetic moment of 45Sc Phys. Lett. 1969. V 29a, №2.
P 58-59.
7 Fratiello H. Lee R. E. Schuster R. E. A proton magnetic resonance hydra-
tion study of scandium, yttrium and thorium perchlorates in waier-acetone
mixtures // Inorg Chem. 1970. V 9, №2. P. 391-392.
8. Буслаев Ю.А. Петросянц С.П., Тарасов В.П., Чагин В.И. ЯМР 4aSc
водных растворов солей скандия // Ж. неорг. химии. 1974. Т 19, №7.
С. 1790-1792.
9. Буслаев Ю.А. Тарасов В.П., Буслаева М.Н., Петросянц С.П. Исследо-
вание водных растворов электролитов методом ЯМР на ядрах катионов //
ДАН СССР. 1973. Т. 209, № 4. С. 882-884.
10. Melson G.A. Olsanski D.J.. Roach E. T High resolution 45Sc NMR — new
tool for studies of scandium compounds in solution // J.C.S. Chem.comm.
1974. №6. P. 229-230.
И. Тарасов В.П. Киракосян Г. А. Троц С. В. Буслаев Ю.А. Исследование
водных растворов солей лантана(Ш) и скандия(Ш) методом ЯМР 139La
и 45Sc // Коорд. химия. 1983. Т 9, №2. С. 205-210.
12. Melson G.A., Olsanski D.J. Rahimi A. К. Coordination chemistry, of scan-
dium. Detection of complex formation in solution by 4°Sc NMR spec-
troscopy // Spectrochim. Acta. 1977 V. 33a, №3. P. 301-304.
13. Киракосян Г.А., Тарасов В.П. Хлороаквакомплексы скандияДП) в водных
растворах // Коорд. химия. 1982. Т. 8, №2. С. 261-262.
14. Pfadeahauer Е.Н., McCain D.C. NMR of fluoroscandate anion. ScFf- in
aqueous solutions // J. Phys. Chem. 1970. V. 74, № 17. P. 3291-3293.
15. Tarasov V P., Buslaev Yu.A. 19F and 45Sc nuclear spin relaxation for the hex-
afluorotitanate, hexafluoroscandate, and hexafluorogermanate ions in aqueous
solution//J. Magn. Reson. 1977. V. 25, №1 P 197-203.
16. Буслаев Ю.А., Киракосян Г.А., Тарасов В.П. Исследование состояния
Sc(III) в неводных растворах методом ЯМР 45Sc высокого разрешения //
Коорд. химия. 1980. Т. 6, №3. С. 361-371.
17. Kirakosyan G.A., Tarasov V.P., Buslaev Yu.A. 45Sc NMR Study of Scan-
dium Complex Formation in Non-Aqueous Solutions // Magn. Reson. Chem.
1989. V. 27, №2. P. 103-111.
18. Crawford M.F., Olson N. Nuclear moment of 89Y // Phys. Rev. 1949. V. 76,
№ 10. P. 1528.
19. Hassler C., Kronenbitter J., Schwenk A. 89Y nuclear magnetic resonance
studies // Z. Phys. 1977. Bd. 280a, №2. S. 117-123.
20. Adam R.-M., Fazakerley G. V., Reid D.G. 89Y spin-spin relaxation times are
not pH dependent // J. Magn. Reson. 1979. V. 33, № 3. P. 655-658.
21. Levy G.C., Hinaldy P.L., Bailey J.T. Yttrium-89 NMR. A possible
spin-relaxation probe for studying metal ion interaction with organic
ligands // J. Magn. Reson. 1980. V. 40, № 1. P. 167-173.
22. Holloway C.E., Mastracci A., Walker J. M. Yttrium-89 Magnetic Resonance
Study of Simple Coordination Complexes // Inorg. Chim. Acta. 1986. V. 113,
№2. P. 187-91.
23. Fratiello A., Kubo-Anderson V., Bolinger T. et al. A Hydrogen-1. Chlo-
rime-35, and Yttrium-89 NMR Complex Formation Study of Aqueous
Y(C1O4)3 and Y(NOa)3 Solutions // J. Magn. Reson. 1989. V. 83, №2.
P. 358-370.
24. Grueninger K.D., Schwenk A., Mann B.E. Direct observation of rhodium-103
NMR and relaxation investigation by steady-state techniques // J. Magn.
Reson. 1980. V. 41, №2. P. 354-357.
25. Федотов M.A., Самохвалова Е.П., Максимов Г.М., Торченкова Е.А.
ЯМР 89Y в иттриевых гетерополивольфраматах // Ж. структ. химии. 1989.
Т. 30, №5. С. 174.
26. Lutz О., Oehler Н. Lanthanum-138 and lanthanum-139 NMR studies //
J. Magn. Reson. 1980. V. 37, №2. P. 261-267.
27. Kruger H., Lutz O., Oehler H. Nuclear magnetic moments and ratios of
quadrupole moments of 135Ba, 137Ba, 138La, 139La by NMR spectroscopy //
Phys. Lett. 1977. V. A62, №2. P. 131-132.
28. Reuben J. Lanthanum-139 as NMR probe of macromolecular dynamics //
J. Amer. Chem. Soc. 1975. V. 97, № 13. P. 3823-3824.
29. Nakamura K., Kawamura K. A nuclear magnetic relaxation of 139La in ionic
aqueous solution // Bull. Chem. Soc. Jap. 1971. V. 44, №2. P. 330-334.
30. Chen Z., Deteller C. A I39La and 17O nuclear magnetic Resonance study of
nature of La(III) first coordination sphere in acetonitrile solution of hydrated
lantanum nitrate // Can. J. Chem. 1994. V. 72, №8. P. 1792-1829.
31. Тарасов В.П., Киракосян Г. А., Буслаев Ю.А. и др. Исследование в водно-
диметилформамидных растворов лантана(Ш) методом ЯМР 139La // Коорд.
химия. 1985. Т. 11, №7. С. 913-917.
32. Reuben L Longitudinal relaxation rates of lanthanum-139 in aqueous salt
solution // J. Phys. Chem. 1975. V. 79, №20. P. 2154-2157.
33. Smith L.S., McCain D.C., Wertz D.C. A preliminary report on the coordina-
tion of lanthanum(III) in lanthanum chloride-methanol solutions // J. Amer.
Chem. Soc. 1976. V. 98. № 17. P. 2125-2128.
34. Evans D.F., Missen. P.H. Lanthanum-139 Nuclear Magnetic Resonanse Spec-
tra of Lanthanum Complexes // J.C.S. Dalton tr.1982. №6. P. 1929-1932.
35. Rinalti P.L., Khan S.A., Choppin G.B., Levy G.C. Lanthanum-139 NMR
chemical shifts // J. Amer. Chem. Soc. 1979. V. 101, №5. P. 1350-1351.
36. Smith L.S., Wertz D.L. Solute structuring in aqueous lanthanum(III) chloride
solutions // J. Amer. Chem. Soc. 1975. V. 97, №9. P. 2365-2368.
37. Avent A.G., Edelman M.A., Lappert M.F., Lawless G.A. The First High
Resolution Direct NMR Observation of f-Block Elements // J. Amer. Chem.
Soc. 1989. V. 111, № 9. P. 3423-3424.
Титан, цирконий, гафний
38. Kidd R. G., Mathews R. V., Spinney H. G. Titanium-47 and titanium-49 NMR
in titanium(!V)-halogen compounds // J. Amer. Chem. Soc. 1972. V. 94,
№ 19. P. 6686-6689.
39. Dean P.A. Evans d. F. Spectroscopic studies of inorganic fluorocomplexes.
The 19F NMR and vibrational spectra of hexafluorometallates ol group IVA
and IVB // J. Chem. Soc. A. 1967. №4. P. 698-701.
40. Chi K.M., Frerichs S.R., Philson S.B., Ellis J.E. Highly Redused
Organometallic Sinthesis. Isolation, and Characterization of Hexacarbonylti-
tanates // J. Amer. Chem. Soc. 1988. V. 110, № 1. P. 303-304.
41. Hao N., Sayser B.G., Denes G. et al. Titanium-47 and -49 NMR spec-
troscopy: chemical application // J. Magn. Reson. 1982. V. 50, № 1. P. 50-63.
42. Лапташ H.M., Федотов M.A., Масленикова И. Г. Гидролиз летучего
оксофторотитаната аммония по данным ЯМР 19F, |7О, 49Ti // Ж. структ.
химии. 2004. Т. 45, № 1. С. 77-87.
43. Федотов М.А., Зенковец Г.А., Гаврилов В.Ю. Исследование состояния
Ti(IV) в концентрированных солянокислых растворах методом ЯМР раз-
ных ядер // Ж. неорг. химии. 1999. Т. 44, №6. С. 973-976.
44. Максимов Г.М., Максимовская Р.И., Холдеева О.А. и др. Структура и
свойства гетерополикислоты Нз(Р\УцТЮзЭ)2О // Ж. структ. химии. 2009.
Т. 50, № 4. С. 648-649.
45. Brun Е., Oeser L, Staub Н.Н. Magnetic moment and spin of 4oZr91 // Phys.
Rev. 1957. V. 105, №6. P. 1929-1930.
46. Sayer B.G., Hao N., Denes G. et al. Zirconium-91 NMR spectroscopy: the
first chemical study // Inorg. Chim. Acta. 1981. V. 48, № 1. P. 53-55.
47. Sayer B.G., Thompson J.I., Hao N. et al. 9lZr NMR spectrum of Zr(BHi)4:
the Zr-H coupling constants as probe for fast intramolecular process // Inorg.
Chem. 1981. V. 20, № 11. P. 3748-3750.
Ванадий, ниобий, тантал
48. Альтшулер С. А., Козырев Б.М. Электронный парамагнитный резонанс
соединений элементов промежуточных групп. — М.: Наука, 1972. — 672 с.
49. Власова М.В., Каказей Н. Т., Калиниченко А.М., Литовченко А. С. Ра-
диоспектроскопические свойства неорганических материалов. — Киев: На-
укова думка, 1987. — 719 с.
50. Howarth О. W., Richards R.E. NMR study of poly vanadate equilibria by use
of vanadium-51 // J. Chem. Soc. 1965. №2. P. 864-870.
51. Kidd R.G., Goodfellow R.J. The Transition Metals. NMR and the Periodic
Table / Ed. R. K. Harris, В. E. Mann. - L.: Acad. Press, 1978. P. 195-278.
52. Rehder D. A survey of 5IV-NMR spectroscopy // Bull. Magn. Reson. 1982.
V. 4, № 1-2. P. 33-83.
53. Rehder D., Polenova T, Biihl M. Vanadium-51 NMR // Ann.reports NMR
spectr.2007. V. 62. P. 49-114.
54. Rehder D. A study of vanadium-51 shielding in VO3+ compounds // Z. Natur-
forsch. 1977. Bd. 32b, №7. S. 771-775.
55. Яржемский В. Г., Тарасов В.П., Нефедов В. И. Константы ядерного спин-
спинового взаимодействия в оксоанионах // Коорд. химия. 1983. Т. 9,
№ 10. С. 1329-1335.
56. Bodner G.M., Todd L.J. Fourier transform carbon-13 NMR study of
transition metal carbonyl complexes // Inorg. Chem. 1974. V. 13, №6.
P. 1335-1338.
57. Rehder D. Fotochemische Substitutionreactionen am Hexacarbonyl-
vanadat(-l) //J. Organomet. Chem. 1972. V. 37, №2. P. 303-312.
58. Rehder D. 51V chemical shifts and 5IV-3IP coupling in carbonyl complexes
of vanadium // J. Magn. Reson. 1977. V. 25, № 1. P. 177-182.
59. Rehder D., Schmidt J. 5IV-NMR und IR Untersuhungen an V(CO)5EZ3-com-
plexes // J. Inorg. Nucl. Chem. 1974. V. 36, №2. P. 333-337.
60. Kececi A., Rehder J. IR, 31P, 55Mn und l85'187 Re NMR spectroskopische
Untersuchungen an einigen Carbonylmangan- und -rhenium complexen //
Z. Naturforsch. 1981. Bd. 36b, № 1. S. 20-26.
61. Rehder D., Berthold H.-С., Paulsen K. A multinuclear NMR study of
[V(PF3)6]' and [Nb(PF3)6]" // J. Magn. Reson. 1980. V. 40, №2.
P. 305-310.
62. O’Donnell S.E., Pope M.T. Applecations of vanadium-51 and phospho-
rus-31 NMR spectroscopy to the study of iso- and heteropoly-vanadates //
J.C.S. Dalton tr. 1976. №21. P. 2290-2297.
63. Howarth O. W., Hunt J.R. Peroxocomplexes of vanadium(V): a vanadium-51
NMR study // J.C.S. Dalton tr. 1979. №9. P. 1388-1391.
64. Казанский Л.П., Спицын В.И. ЯМР ванадия-51 в гетерополи- и изополи-
анионах // ДАН СССР. 1975. Т. 223, № 2. С. 381-384.
65. Санников Ю.И., Золотавин В.Л., Безруков И.Я. Исследование гидролиза
соединений пятивалентного ванадия // Ж. неорг. химии. 1963. Т. 8, №4.
С. 923-933.
66. Heath Е., Howart О. 117. Vanadium-51 and oxygen-17 NMR study of
vanadate(V) equilibrium and kinetics // J.C.S. Dalton tr. 1981. №5.
P. 1105-1110.
67. Habaeb M.A., Hileman D.E. Vanadium-51 FT-NMR investigation of meta-
vanadale ions in aqueous solutions // Can. J. Chem. 1980. V. 58, №21.
P. 2255-2261.
68. Федотов M.A., Максимовская. Р.И. Структурные аспекты ЯМР в химии
полиоксометаллатов V, Mo, W // Ж. структ. химии. 2006. Т. 47, № 5.
С. 961-984.
69. Максимовская Р.И., Ильясова А.К., Бегалиева Д.У. и др. Идентифи-
кация смешанных ванадийвольфрамовых полиоксокомплексов в водных
растворах по ЯМР |7О и 51V // Изв. АН СССР. Сер. хим. 1984. № 10.
С. 2169-2174.
70. Howarth O.W., Jarrold М. Protonation of the decavanadate(6-) ion:a vana-
dium-51 NMR study // J.C.S. Dalton tr. 1978. №5. P. 503-506.
71. Максимовская Р.И., Федотов M.A., Мастихин B.M. и др. Исследование
состояния фосфорномолибдованадиевых гетерополикислот в водных рас-
творах методом ЯМР // ДАН СССР. 1978. Т. 240, № 1. С. 117-120.
72. Максимовская Р.И., Федотов М.А. Спектры ЯМР 31p.51v и 17О фос-
форвольфрамванадиевых геторополикислот // Ж. неорг. химии. 1985.
Т. 30, № 1. С. 103-108.
73. Fedotov М.А., Maksimovskaya R.I., Kazansky L.P. Structure of vanadium
phosphate anion in solutions from 17O, 51V and 31P NMR data // React.
Kinet. Catal. Lett. 1981. V. 16, №2-3. P. 185-189.
74. Lauterbur P. C., King R.B. Carbon-13 NMR spectra of transition metal
cyclopentadienyl and carbonyl derivatives // J. Amer. Chem. Soc. 1965. V. 87,
№ 11. P. 3266-3207.
75. Nugent W.A., Harlow R.L. Stable first-row transition metal alkylimido-
complexes: X-ray crystal and molecular structure of N-(l-adamantylimi-
do)tris(trimethylsiloxy)vanadium(V) // J.C.S. Chem. comm. 1979. №7.
P. 342-343.
76. Lutz O., Nepple W., Nolle A. Indirect spin-spin coupling between ‘'O and
other quadrupolar nuclei in oxoanions // Z. Naturforsch. 1976. Bd. 31a, №9.
S. 1046-1050.
77. Hatton J. V., Saito Y., Schneider W.G. NMR investigation of some Group V
metal fluorides and oxyions // Can. J. Chem. 1965. V. 43, № 1. P. 47-50.
78. Howell J.AS., Moss K.C. NMR studies on fluorine-containing compounds.
Tetrafluorooxovanadate(V) ions // J. Chem. Soc. A. 1971. №2. P. 270-272.
79. Haid E., Kohlein D., Kossler G. e. a. Spin-lattice relaxation times of 33S,
51V, S3Cr and 55Mn in tetrahedral oxoanions in aqueous solutions // J. Magn.
Reson. 1983. V. 55, № 1. P. 145-150.
80. Harrison A. T., Howarth O.W. High-Field Vanadium-51 and 17O Nuclear
Magnetic Resonance Study of Peroxovanadates // J.C.S. Dalton tr. 1985.
№6. P. 1173-1177.
81. Jameson C., Rehder D., Hoch M. Isotope and Temperature Dependence of
Transition-Metal Shielding in Complexes M(XY)e // J. Amer. Chem. Soc.
1987. V. 109, № 9. P. 2589-2594.
82. Harrison A. T, Howarth O.W. Vanadium-51 Nuclear Magnetic Resonance
Study of Sulfido- and Oxosulfidovanadate(V) Species // J.C.S. Dalton tr.
1986. №7. P. 1405-1409.
83. Laparulo-Loftus M.A., Pope M.T. Vanadium-51 NMR Spectroscopy of
Tungstovanadate Polyanions. Chemical Shifts and Line-Width Pattern
for Identification of Stereoisomers // Inorg. Chem. 1987. V. 26, №13.
P. 2112-2120.
84. Hibbert R.C. Vanadium(V) Fluoride Complexes in Organic Solvents. A Vana-
dium-51 and Fluorine-19 Nuclear Magnetic Resonance Study // J.C.S. Dal-
ton tr. 1986. №47. P. 751-753.
85. Guesser M.J., Tracey A.S., Parkinson K.M. The Interaction of Vanadate
with Pyrophosphate. Phosphate and Arsenate // J. Amer. Chem. Soc. 1986.
V. 108, № 20. P. 6929-6934.
86. Курбатова Л.Д., Медведева Н.И., Курбатов Д.И., Максимовская Р.И.
Хлоридные комплексы оксованадия(У) // Ж. неорг. химии. 2002. Т. 47,
№ 12. С. 2023-2026.
87. Wiedemann С., Rehder D. The Solution Structures of VOCI3 Complexes of
Neutral Nitrogen and Oxygen Donors: A Vanadium-51 NMR Spectroscopic
Study // Inorg. Chim. Acta. 1986. V. 120, № 1. P. 15-20.
88. Buslaev Yu. A., Kopanev V.D., Tarasov V.P. 93Nb NMR in solutions of the
halides of niobium(V) // J.C.S. Chem. comm. 1971. № 19. P. 1175-1177.
89. Kidd R.G., Spinney H.G. Niobium-93 NMR study of hexahaloniobates and
pentahalohiobium-acetonitrile adducts // Inorg. Chem. 1973. V. 12, №9.
P. 1967-1971.
90. Буслаев Ю.А., Копанев В.Д., Синицына C.M., Хлебодаров В. Г. Исследо-
вание пента- и оксогалогенидов ниобия в растворах ацетонитрила методом
ЯМР 93Nb и 19F // Ж. неорг. химии. 1973. Т. 18, №9. С. 2567-2569.
91. Tarasov V.P., Buslaev Yu.A., Privalov V.I. Spin relaxation and magnetic
shielding of 93Nb niclei in [NbClnBr6_n]- // J. Mol. Phys. 1978. V. 35, № 4.
P. 1047-1055.
92. Reuben J. Lanthanum-139 as NMR probe of macromolecular dynamics //
J. Amer. Chem. Soc. 1975. V. 97, № 13. P. 3823-3824.
93. Ильин Е.Г., Тарасов В.П., Ершова M.M. и др. Исследование комплексов
ниобия(У) со смешанными лигандами во внутренней сфере методом ЯМР
93Nb Ц Коорд. химия. 1978. Т. 4, №8. С. 1370-1379.
94. Буслаев Ю.А., Тарасов В.П., Синицына С.М. и др. Изучение методом
ЯМР 93Nb строения разнолигандных комплексов ниобила // Коорд. химия.
1979. Т. 5, №2. С. 189-196.
95. Тарасов В.П., Синицына С.М., Копанев В.Д. и др. Изучение мето-
дом ЯМР 93Nb разнолигандных комплексов ниобия(У) со связью Nb=S
и Nb=Se // Коорд. химия. 1980. Т. 6, № 10. С. 1568-1580.
96. Berthold Н. С., Render D. Preparation and IR, phosphorus-31, and nio-
bium-93 NMR spectroscopic investigations of phosphine derivatives of
7?5-C5H5(CO)4 // J. Organomet. Chem. 1981. №3. P. 305-315.
97. Aksenes D.W., Hutchison S.M., Packer К. J. Nuclear spin relaxation and
chemical exchange effects in the 19F nuclear magnetic resonance spectrum
of the hexafluoroniobate ion // Mol. Phys. 1968. V. 14, №4. P. 301-309.
98. Barybin M.V., Ellis J. E., Pomije M.K. et al. Synthesis and Properties
of Homoleptic Carbonyl and Trifluorophosphane Niobates: [ЫЬ(СО)б]~,
[Nb(PF3)6]", and [Nb(CO)5]3~ // Inorg. Chem. 1998. V. 37, №25.
P. 6518-6527.
99. Jameson C.J., Rehder D., Hoch M. Effect of Rovibrational Averaging on the
93Nb Chemical Shift in the [\Ь(СО)б]~ Ion Based on NMR and Vibrational
Spectra // Inorg. Chem. 1988. V. 27, № 20. P. 3490-3495.
100. Sola J., Do Y., Berg J.M., Holm R.H. Soluble Sulfides of Niobium(V) and
Tantalum(V): Synthesis. Structure, and Properties of the Fivefold Symmetric
Gages [M6Si7]4- И Inorg. Chem. 1985. V. 24, № 11. P. 1706-1713.
101. Erich L.C., Gossard A.C., Hartless R.L. Magnetic moment of 18lTa //
J. Chem. Phys. 1973. V. 59, №8. P. 3911-3912.
102. Rehder D., Basler W. Tantalum-181 Solution NMR Spectroscopy // J. Magn.
Reson. 1986. V. 68, № 1. P. 157-160.
Хром, молибден, вольфрам
103. Epperlein В. U7, Kruger H., Lutz O. et al. S3Cr Fourier transform NMR
studies // Z. Naturforsch. 1975. Bd. 30a, № 10. S. 1237-1240.
104. Hafner A., Hegelus L.S., de Week G. et al. Chromium-53 NMR Studies of
Pentacarbonylchromium-Carbene Complexes // J. Amer. Chem. Soc. 1988.
V. 110, №25. P. 8413-8421.
105. Proctor W. G., Yu F.C. On nuclear moments of several stable isotopes //
Phys. Rev. 1951. V. 81, № 1. P. 20-30.
106. Kautt W.D., Kruger H., Lutz O. et al. Fourier transform NMR studies of
95Mo and 97Mo // Z. Naturforsch. 1976. Bd. 31a, №3/4. S. 351-356.
107. Kroneck P. NMR investigation of 17O.33S, 95Mo and 97Mo in thiomolyb-
dates // Z. Naturforsch. 1980. Bd. 35a, № 2. S. 226-229.
108. Gheller S.F., Gazzana P.A., Masters A.F. et al. Molyhdenum-95 NMR of
molybdenum-sulfur and -selenium species // Inorg. Chim. Acta. 1981. V. 54,
№4. P. L131-L132.
109. Christensen K.A., Miller P.T., Minelli M. et al. Molybdenum-95 NMR
spectra of dioxomolybdenum(VI) complexes // Inorg. Chim. Acta. 1981. V. 56,
№ 2. P. L27-L28.
110. Freeman M.A., Schultz F.A., Reinlley C.N. Carbon-13, Oxygen-17 and
Molybdenum-95 NMR Studies of Oxomolybdenum(VI) Complexes // Inorg.
Chem. 1982. V. 21, №2. P. 567-576.
111. Lutz O., Nolle A., Kroneck P. Use of 95Mo NMR spectroscopy as new
approach of structural analysis of diamagnetic molybdenum complexes //
Z. Naturforsch. 1976. Bd.31a, №5. S. 454-456.
112. Masters A.F., Bossard G.E., Adrian G.T. et al. Application of 95Mo NMR
Molybdenum(O) Carbonyl Derivatives of Phosphines, Phosphites and Related
ligands // Inorg. Chem. 1983. V. 22, №6. P. 903-911.
113. Федотов М.А. Особенности ЯМР 95Мо в полиоксомолибдатах // Изв. АН
СССР. Сер. хим. 1984. №5. С. 1166-1168.
114. Федотов М.А., Гаврилова Л.О., Татьянина И.В., Торченкова Е.А. Спек-
тры ЯМР 170 , 59Со,95Мо и состояние полиоксомолибдатов с гетероатомом
в октаэдрическом окружении в водных растворах // Ж. неорг. химии.
1991. Т. 36, № 1. С. 194-198.
115. Gheller S.F., Sidney М., Masters A.F. et al. Application of Molybdenum-95
Spectroscopy. Polyoxomolybdates // Austral. J. Chem. 1984. V. 37, №9.
P. 1825-1832.
116. Старцев A.H., Шкуропат С. А., Климов О.В. и др. Синтез и строение ок-
салата молибдена(У) (NgHs^IMoy(^-0)2^204)2(^0)2 // Коорд. химия.
1991. Т. 17, №2. С. 229-233.
117. Nagasawa A., Sasaki Yo., Ito Т. et al. I83W NMR Spectra of Some Di- and
Trinuclear Clusters of Tungsten(III). (IV) and (V) // Chem. Lett. 1987. № 7.
P. 1271-1274.
118. Климов О.В., Федотов М.А., Кочубей Д.И. и др. Нанесенные на AI2O3
катализаторы, приготовленные с использованием би- и трехъядерных со-
единений молибдена // Кинет, катализ. 1995. № 3. С. 330-336.
119. Федотов М.А., Максимовская Р.И., Молчанова О.А., Ильясова А.К.
Спектры ЯМР 17О дифосфатполиоксомолибдатов // Изв. АН СССР. Сер.
хим. 1980. №3. С. 709-713.
120. Samotus A., Kanas A. Dudek М et al. 1:1 Molybdenum(VI) citric acid
complexes // Transition Met. Chem. 1991. V. 16, № 3. P. 495-499.
121. Ruiz-Morales Y., Ziegler T.A. Theoretical Study of 3IP and 95Mo NMR
Chemical Shifts in Mo(CO)sPR3 (M = Cr, Mo; R = H, CH3, CgHs, F and Cl)
Based on Density Functional Theory and Gauge Including Atomic Orbitals //
J. Phys. Chem. 1998. V. 102A, №23. P. 3970-3976.
122. Alyea E.C., Somogyvari A. Molybdenum-95 NMR Studies on Distributed
Molybdenum(O) Carbonyls // Can. J. Chem. 1988. V. 66, № 3. P. 397-400.
123. Luiz O., Nolle A. Sulfur isotope effect on 95Mo nuclear magnetic shielding
in MoS9- // Z. Phys. Bd. 282a, №2. S. 157-158.
124. Nikatsu/i H., Sugimoto M., Saito S. Electronic Origin of 95Mo NMR Chem-
ical Shifts in Molybdenum Complexes. Relationships between Exitation En-
ergy end Chemical Shifts // Inorg. Chem. 1990. V. 28, № 17. P. 3095-3097.
125. Void R.R., Void R.L. Nuclear relaxation and spin-spin coupling in aqueous
solutions of molybdate // J. Chem. Phys. 1974. V. 61, № 10. P. 4360-4361.
126. Minelli M., Hubbard J.L., Cristensen K.A., Enemark J.H. Observation
of 9SMo-14N spin-spin coupling in multiniclear NMR studies on
(t?s-C5H5)Mo(CO)2NO and related compounds // Inorg. Chem. 1983. V. 22,
№ 18. P. 2652-2653.
127. Jaither P., Wohlgenaunt W. Molybdenum-95 NMR of series of phosphine
and phosphite substituted molybdenum carbonyls Mo(CO)8-nL. n = 1-5 //
Monatsch. Chem. 1982. V. 113, №6-7. P. 699-703.
128. Brownlee R.T.C., Shenan B.P., Wedd A.G. Application of 95Mo NMR
Spectroscopy. XVIII. Relaxation Times. Quadrupole Coupling Constants and
Partial Field Graduents in [Mo(CO)5L] Complexes // Aust. J. Chem. 1988.
V. 41, №9. P. 1457-1464.
129. Brownlee R. T. C., Shenan B.P., Wedd A. G. Application of 95Mo NMR Spec-
troscopy. 17. 9SMo and l4N Relaxation Time Measurments Confirming that
[Mo(CN)8]4- Is Dodecahedral in Aqueous Solution // Inorg. Chem. 1987.
V. 87, № 13. P. 2022-2024.
130. Maksimovskaya R.L, Maksimov G.M. 90Mo and l7O Studies of Aqueous
Molybdate Solutions // Inorg. Chem. 2007. V. 46, № 9. P. 3688-3695.
131. Maksimovskaya R.L, Burtseva K.G. 17О and 183W Studies of the
Paratungstate Anion in Aqueous Solutions // Polyhedron. 1985. V. 4, №9.
P. 1559-1562.
132. Masters A.F., Bossard G.E., Adrian G.T. et al. Application of 95Mo NMR
Molybdenum(O) Carbonyl Derivatives of Phosphines. Phosphites and Related
Ligands // Inorg. Chem. 1983. V. 22, №6. P. 903-911.
133. Dysart S., Georgii J., Mann B.E. Molybdenum-95 NMR spectra of some
molybdenum carbonyl and related compounds // J. Organomet. Chem. 1981.
V. 213, №2. P. C10-C12.
134. Bailey LT., Clark H.L, Levy G.C. Molybdenum NMR studies of trifluo-
rophospine complexes // Inorg. Chem. 1982. V. 21, №5. P. 2085-2087.
135. Jaither P., Wohlgenaunt W. Molybdenum-95 NMR of series of phosphine
and phosphite substituted molybdenum carbonyls Mo(CO)e_nL. n = 1-5 //
Monatsch. Chem. 1982. V. 113, №6-7. P. 699-703.
136. Le-Gall J. Y., Cublcki M.M., Pettillon S. Y. Attlication of95Mo NMR in study
of organo-molybdenum carbonyl complexes // J. Organomet. Chem. 1981.
V. 221, №3. P. 287-290.
137. Kubicki M.M., Kergoat R., Le-Gall J. Y. et al. The molybdenum-mercury
bond. A multinuclear ('H, l3C, 31P, 95Mo and l99Hg) NMR study of
(775-C5H5)(CO)3LMoHgX; L=CO. P(OMe)3. PPh3; X = Cl, Br, I, S2COEt.
S2P(OEt)2 and Mo(CO)3(t?5-C5H5) complexes and the crystal and molecular
structure of (7?5-C5H5)(CO)3MoHgS2CNEt2 // Aust. J. Chem. 1982. V. 35,
№8. P. 1543-1554.
138. Alyea E.C., Lenkinski R.E., Somogyvari A. Molybdenum-95 NMR of stud-
ies of molybdenum-phosphorus compounds // Polyhedron. 1982. V. 1, № 1.
P. 130-132.
139. Alyea E. C., Topich J. Molybdenum-95 NMR spectra of dioxomolybdenum(VI)
Shiff base complexes // Inorg. Chim. Acta. 1982. V. 65, № 3. P. L95-L96.
140. Masters A. F., Brownlee R.T., O’Connor M.L., Wedd A.G. Application ol
molybdenum 9SNMR spectroscopy. Arenomolybdenum tricarbonyl deriva-
tives // Inorg. Chem. 1981. V. 20, № 12. P. 4183-4186.
141. Masters A.F., Brownlee R.T., O’Connor M. J. et al. Molybdenum-95 NMR
Applications to Substituted Carbonyls // J. Organomet. Chem. 1980. V. 195,
№2. P. C17-C20.
142. Гаврилова Л.О., Федотов M.A., Татьянина И.В., Торченкова Е.А. Ис-
следование аниона [NiMog032]6~ и его взаимодействие с ионами d-
и f-элементов в водных растворах методом ЯМР 170,95Мо и других ядер //
Ж. неорг. химии. 1990. Т. 35, №8. С. 2100-2103.
143. Петрухина М. А., Федотов М. А., Татьянина И.В., Торченкова Е.А. Изу-
чение комплексообразования церимолибденовой кислоты с ионами Zn2+
методом ЯМР |7О, 9SMo, 67Zn // Ж. неорг. химии. 1989. Т. 34, №4.
С. 849-852.
144. Самохвалова Е.П., Федотов М.А., Татьянина И.В., Торченкова Е.А.
Изучение взаимодействия скандия(Ш) с уранмолибденовой кислотой ме-
тодами спектроскопии // Ж. неорг. химии. 1989. Т. 34, №3. С. 660-663.
145. Narath A., Wallace D.C. NMR in cubic sodium tungsten bronzes // Phys.
Rev. 1961. V. 127, №3. P. 724-729.
146. Andrews G.-T., Colquhoun I.J., McFarlane W. Transition metal chemical
shifts in complexes of molybdenum(O) and tungsten(O) // J.C.S. Dalton tr.
1982. № 12. P. 2353-2358.
147. Banck J., Schwenk A. I83W tungsten NMR studies // Z. Phys. 1975. Bd. 20b,
№ 1. S. 75-80.
148. McFarlane W., Noble A. M., Winfield J. M. Magnetic double-resonance study
of some derivatives of tungsten hexafluoride and tungsten oxotetra-fluoride //
J. Chem. Soc. A. 1971. №8. P. 948-953.
149. Kozik M., Hammer C.F., Baker L. C. W. Direct Determination by 183W NMR
of the Locations of Added Electrons in ESR Silent Heteropoly Blues. Chemical
Shifts and Relaxation Times in Polysite Mixed-Valence Transition-Metal
Species Ц J. Amer. Chem. Soc. 1986. V. 108, № 10. P. 2748-2749.
150. Kohler F.H, Kolder H.J., Fischer E.O. l3C-l83W-kopplungen als Sonde fur
Metall-carbin-, carben-, und -alkyl-Bindungen // J. Organomet. Chem. 1975.
V. 95, №2. P. C19-C20.
151. Chen Ya.-G., Gong Ji., Qu L.-Yu. Tungsten-183 nuclear magnetic resonance
spectroscopy in the study of polyoxometallates // Coord. Chem. Rev. 2004.
№ 1. P. 245-260.
152. Gansow O.A., Ho R.K., Klemperer W.G. Hypotesis of bond alternation mech-
anism for charge delocalization which determine tungsten-183 chemical shifts
in (7?5-C5-H5) Ti(PWnO39)4“ // J. Organomet. Chem. 1980. V. 187, №3.
P. C27-C39.
153. Maksimovskaya R.I., Chumachenko N.N. 51V and l7O NMR Studies of the
Mixed Metal Polyanions in Aqueous V-Mo Solutions // Polyhedron. 1987.
V.6, № 10. P. 1813-1821.
154. Федотов M.A., Казанский Л.П., Спицын В. И. Химические сдвиги ЯМР
17О и 183W в полиоксовольфраматах и длины связей вольфрам-кислород //
ДАН СССР. 1983. Т. 272, №5. С. 1179-1185.
155. Acerete R., Hammer С. F, Baker L.C. Direct Tungsten-183 NMR a Powerful
Tool for Heteropoly and Isopolytungstate Chemistry // J. Amer. Chem. Soc.
1979. V. 101. P. 267-259.
156. Федотов M.A., Казанский Л.П. Спектры ЯМР 29Si в гетерополианионах
кремния // Коорд. химия. 1984. Т. 10, №3. С. 423.
157. Acerete R., Hammer C.F., Baker L.C. Tungsten-183 NMR of Heteropoly-and
Isopoly-Tungstates. Theoretical Considerations // J. Amer. Chem. Soc. 1982.
V. 104, №20. P. 5384-5390.
158. Doma.il P.L 1- and 2-Dimentional 183W and 5IV NMR Characterization of
Isopolymetallates and Heteropoly Metallates // J. Amer. Chem. Soc. 1984.
V. 106, № 25. P. 7677-7687.
159. Максимов Г.М., Максимовская Р.И., Кожевников И. В. Гетерополикисло-
ты, производные от комплексов аниона PWnO^ с катионами металлов //
Ж. неорг. химии. 1992. Т. 37, № 10. С. 2279-2286.
160. Федотов М.А., Максимовская Р.И., Максимов Г.М., Матвеев К. И. Вза-
имодействие гетерополианиона PWnO^ с катионами Zn2+. Cd2+. Hg2+.
Pb2+ no данным ЯМР l83W и других ядер // Ж. неорг. химии. 1987. Т. 32,
№3. С. 647-651.
161. Lefebvre /., Chauveau F., Doppelt P., Brevard C. Tungsten-183 NMR Spec-
troscopy: 2J(W-W) Coupling. Structural Application to 1-12 Heteropoly-
Tungstates // J. Amer. Chem. Soc. 1981. V. 103, № 15. P. 4589-4591.
162. Максимовская Р.И., Федотов M.A., Максимов Г.М. О структуре гете-
рополианиона PsMieOg^- (М = Мо, W) в водных растворах и кислородом
обмене по ЯМР 17О // Изв. АН СССР. Сер. хим. 1983. №2. С. 247-252.
163. Максимов Г.М., Федотов М.А. О синтезе кремнеалюмовольфрамовой
гетерополикислоты // Ж. неорг. химии. 2001. Т. 46, №3. С. 384-386.
164. Максимовская Р.И., Федотов М.А., Максимов Г.М. Взаимодействие ге-
терополианиона PWnO^" с одновалентными катионами по данным ЯМР
разных ядер // Ж. неорг. химии. 1985. Т. 30, №4. С. 918-924.
165. Maksimovskaya R.I., Maksimov G.M. NMR Studies of Heteropolyanion
[P2W2oC>7o(H20)2]l0- Complexes with Metal Cations // Inorg. Chem. 2001.
V. 40, №6. P. 1284-1290.
166. Brevard C., Schimpf R., Tourne G., Tourne C.M. Tungsten-183 NMR:
a Complete and Unequivocal Assignement of Tungsten-Tungsten Connectiv-
ities in Heteropolytungstates vie Two-dimentional l83W NMR Techniques //
J. Amer. Chem. Soc. 1983. V. 105, №24. P. 7059-7063.
167. Knoth W.H., Domallle P.J., Roe D.C. Halometal Derivaties of W^PO3^- and
Related l83W Studies // Inorg. Chem. 1983. V. 22, №2. P. 198-201.
168. Kazansky L.P., Fedotov M.A. Paramagnetic l83W NMR chemical shifts in
heteropolytungstates // J.C.S. Chem. comm. 1983. №8. P. 417-419.
169. Федотов M.A., Перциков Б.С., Данович Д.К. Спектры ЯМР |7О, 31Р,
le3W и состояние в растворах парамагнитных комплексов с гетерополи-
вольфрамат-ионом [Ln(PWnO3g)2]11- // Коорд. химия. 1991. Т. 17, №2.
С. 234-242.
170. Федотов М.А., Самохвалова Е.П., Казанский Л.П. Химические сдвиги
ЯМР 17О, l83W и состояние гетерополианионов [ZWioOge]”- в водных
растворах. Z — редкоземельный элемент // Коорд. химия. 1995. Т. 21,
№9. С. 709-715.
171. Ma Y., Demou Р., Faller J. W. A l83W NMR Study of Mononuclear Tung-
sten(IV) Methyl Complexes Containing Terminal Oxo-. Sulfido-, and Imido
Ligands // Inorg. Chem. 1991. V. 30, № 1. P. 62-64.
172. Kazansky L.P. 183W NMR of polyoxotungstates. Correlation of chemical
shifts and 2Jjv-iv coupling with local geometry // Chem. Phys. Lett. 1994.
V. 223, № 2. P. 289-296.
Марганец, технеций, рений
173. Calderazzo F, Lucken E.A.C., Williams D.F. The s5Mn NMR spectra of
alkyl- and acylmanganese pentacarbonyls and related compounds // J. Chem.
Soc A. 1967. № 1. P. 154-158.
174. Onaka S., Sasaki Y. ll9Sn-Mossbauer and *H- and ssMn-NMR spectroscopic
studies of a series of compounds. Ra_xXx; Sn-Mn(CO)e // Bull. Chem. Soc.
Jap. 1971. V.44, №3. P. 626-730.
175. Bankroft G.M., Clark H.C., Kidd R.G. et al. Manganese-55 NMR Studies
of Compounds Containing Mn-M (М-Si; Ge. Sn. Pb) Bonds and the Sign
of e2qQ in Manganese(I) Compounds // Inorg. Chem. 1973. V. 12, №4.
P. 728-731.
176. Miles W.J., Garnett B.B., Clark R.J. Manganese NMR of the Phosphorus
Tritluoride Derivatives of Manganese Pentacarbonyl Hydride // Inorg. Chem.
1969. V. 8, № 12. P. 2817-2818.
177. Lutz O., Steinkilberg W. 55Mn NMR studies in aqueous permanganate
solution // Z. Naturforsch. 1974. Bd. 29a, № 10. S. 1467-1470.
178. Popov A. I. Alkali metal NMR and vibrational studies on solvates in non-
aqueous solvents // Pure Appl. Chem. 1971. V. 41, № 3. P. 275-290.
179. Haase A. R., Lutz O., Muller M., Nolle A. On 55Mn nuclear magnetic shield-
ing in permanganate // Z. Naturforsch. 1976. Bd.31a, № 11. S. 1427-1428.
180. Grobe J., Kopp W. 55Mn Shielding in Carbonylmanganese Halids Containing
Phosphido- and Arsenido Bridges // Inorg. Chim. Acta. 1986. V. 115, № 1.
P. L7-L8.
181. Ireland P.S., Deckert C.A., Brown T.L. Manganese-55 NMR linewidth and
quadrupole coupling constants in solution: partial field gradient parameters
in Mn(I) complexes // J. Magn. Reson. 1976. V. 23, №3. P. 485-491.
182. Тарасов В.П., Привалов В. И., Буслаев Ю.А. Магнитная релаксация квад-
рупольных ядер в тетраоксокомплексах // ДАН СССР. 1983. Т. 269, № 3.
С. 640-644.
183. Nielson R.M., Wherland S. First Observation of Resolvalble Coupling be-
tween Mn-55 and C-13 // Inorg. Chem. 1984. V. 23, №20. P. 3265-3266.
184. Buckingham M.L, Hawkes G.E. The 99Tc and l7O NMR spectra of TcO^ —
the first detailed reports of a 99Tc resonance // Inorg. Chim. Acta. 1981.
V. 56, №2. P. L41-L42.
185. Тарасов В.П., Привалов В.И., Киракосян Р.А. и др. Изотопные эффекты
в ЯМР "Тс и спин-спиновое взаимодействие |7О-99Тс в анионах ТсО^“ //
ДАН СССР. 1982. Т. 263, №6. С. 1416-1418.
186. Franklin K.L, Lock С.J., Sayer В. С., Schrobilgen G.L. Chemical Applica-
tions of Technetium-99 NMR Spectroscopy: Novel Technetium (VII) Species
and their Characterization by Multinuclear NMR // J. Amer. Chem. Soc.
1982. V. 104, № 20. P. 5303-5306.
187. Franklin К. J., Lock C.J., Sayer B.G., Schrobilgen G.L. The preparation of
technetium oxyfluorides and their characterization by 99Tc, 17O and 19F NMR
spectroscopy // J. Fluor. Chem. 1982. V. 21, № 1. P. 1-68.
188. Findeisen M., Kaden L., Lorenz B. et al. "Тс-NMR on Tc Carbonyl Com-
pounds // Inorg. Chim. Acta. 1987. V. 128, № 1/2. P. L15-L16.
189. Casteel W.J., Dixon D.A., LeBlond N. Et al. Lewis-Acid Properties of
Technetium(VII) Dioxide Trifluoride. TcO2F3: Characterization by 19F, 17O,
and 99Tc NMR Spectroscopy and Raman Spectroscopy. Density Functional
Theory Calculation of TcO2F3, M+TcOgF^ [M = Li, Cs, N(CH3)4], and
TcO2F3-CH3CN, and X-ray Crystal Structure of M+TcO2F^ // Inorg. Chem.
1998. V. 37, №2. P. 340-353.
190. Findeisen M., Lorenz B., Abram U. et al. "Тс-NMR an Kationen
Technetium(I)-Komplexen // Z. Chem. 1989. V. 29, № 1. S. 29-30.
191. Grim C.C., Clark R.J., Rosanske R. The 19F and 99Tc Spectrum of
ax-Te2(CO)g(PF3). Inorg Chim. Acta 1989. V. 159, №2. P. 137-139.
192. Davidson A., Kronaugl J.F., Jones A.G. et al. 99Tc NMR Spectroscopy of
Technetium(I) Phosphino- and Phosphite Complexes // Inorg. Chem. 1988.
V. 27, № 18. P. 3245-3246.
193. Спицын В.И., Тарасов В.FL, Герман К.Э. и др. Симметрия 4°-комплек-
сов технеция по данным ЯМР "Тс // ДАН СССР. 1986. Т. 290, №6.
С. 1411-1414.
194. O’Conell L.A., Pearlstein R.M. Davidson A et al. Technetium-99 NMR
Spectroscopy: Chemical Shift Trends and Long Range Coupling Effects //
Inorg. Chim. Acta. 1989. V. 161, № 1. P. 39-43.
195. Dwek R.A., Luz Z., Shporer M. NMR of sodium perrenate // J. Phys. Chem.
1970. V. 74, № 10. P. 2232-2233.
196. Kececi A., Behder D. IR, 31P, 55Mn und 185,187Re-NMR spektroskopische
Lfntersuchungen an einigen Carbonylmangan-und -rhenium -Komplexen //
Z. Naturforsch. 1981. Bd. 36b, № 1. S. 20-26.
197. Milller A., Krickelmeyer E., Bogge H. [ReS^-, [ReSg]-, [ReOSe]-: Simple
Synthesis with S7~ Solutions. Structural Data, and l85/,e7Re NMR Studies //
Chimia. 1986. V. 40. 4635.
198. Juranic N. Ligand Field Interpretation of Metal NMR Chemical Shifts in
Oktahedral d6 Transitional Metal Complexes // Coord. Chem. Revs. 1989.
V. 96, № 2. P. 253-290.
199. Киракосян Г. А. Мультиядерный магнитный резонанс в исследовании
неорганических координационных соединений благородных металлов //
Коорд. химия. 1993. Т. 19, № 7. С. 507-525.
Железо, кобальт, никель
200. Goodfellow R.J. Group VIII Transition Metals. Multinuclear NMR / Ed.
J. Mason. — N. Y.: Plenum Press, 1987. P. 521-561.
201. Pregosin P.S. Platinum-195 Nuclear Magnetic Resonance // Coord. Chem.
Rev. 1982. V. 44, №2. P. 247-291.
202. Федотов М.А. Ядерный магнитный резонанс в растворах неорганических
веществ. — Новосибирск: Наука, 1986. С. 135-149.
203. Schwenk A. 57Fe NMR studies in the diamagnetic Fe(CO)s // Phys. Lett.
1970. V. 31a. №9. P. 513-514.
204. Sahm W., Schwenk A. Precision measurments of magnetic moments of
nuclei with weak NMR signal // Z. Naturforsch. 1974. Bd. 29a, № 12.
S. 1763-1766.
205. Haslinger E., Robien W., Schldgl K., Wassensteiner W. 57Fe-Kernreso-
nanzspektroscopie von Ferrocenderivative // J. Organomet. Chem. 1981.
V. 218, № 1. P. C11-C14.
206. Коридзе А. А., Астахова H.M., Петровский П.В., Луценко А.И. Магнит-
ное экранирование железа-57 в железоорганических комплексах и участие
атома металла в стабилизации а-ферроценил-карбениевых ионов // ДАН
СССР. 1978. Т. 242, № 1. С. 117-120.
207. Коридзе А.А., Петровский П.В., Губин С.П. и др. Константы спин-спи-
нового взаимодействия *J(57Fe-13C) и химические сдвиги 57Fe в ферроце-
нах // Изв. АН СССР. Сер. хим. 1975. №7. С. 1675.
208. Коридзе А.А., Петровский П.В., Астахова Н.М. и др. Циклогексаениль-
ные комплексы переходных металлов: ЯМР 13С-{*Н} и 13С-{‘Н, 57Fe}
спектроскопическое исследование производных железа-57 // ДАН СССР.
1980. Т. 255, № 1. С. 117-120.
209. Brevard С., Schimpf R. Phosphorus-Irradiation INEPT Experiments on Spin
1 /2 Metal Nuclides. Application to 103Rh, l83W, and 57Fe // J. Magn. Reson.
1982. V. 47, №3. P. 528-534.
210. Koridze A.A., Petrowskii P.V., Gubin S.P., Fedin E.I. *J(57Fe-13C) spin
coupling constants and 57Fe chemical shifts in ferrocenes. The structure
of metallocenyl carbenium ions // J. Organomet. Chem. 1975. V. 93, № 2.
P. C26-C30.
211. Haslinger E., Robien W., Schldgl K., Weissensteiner W. Ferrocene deriva-
tives. Iron-57 NMR spectroscopy of ferrocene derivatives // J. Organomet.
Chem. 1981. V. 218, № 1. P. C11-C14.
212. Haslinger E., Koch K, Robien W., Schldgl K. Ferrocene derivatives. 57Fe
NMR spectroscopy of ferrocenes // Monat. Chem. 1983. V. 114, №4.
P. 495-499.
213. Koridze A.A., Astakhova N.M., Petrovskii P. V. Iron-57-carbon-13 coupling
constants in a ferrocenyl carbocations. Direct metal participation in the
stabilization of metallocenyl carbocations // J. Organomet. Chem. 1983.
V. 254, № 3. P. 345-360.
214. Коридзе А.А., Астахова H.M., Петровский П.В. и др. Химический сдвиг
ЯМР 57Fe — чувствительный индикатор отклонения циклопентадиениль-
ных колец ферроцена от параллельности // Изв. АН СССР. Сер. хим.
1983. №8. С. 1928-1929.
215. Mann В.Е. Measurments of the Coupling Constants *J(57Fe-13C) and
*J(57Fe-3lP) in Phosphine and Carhonyl Complexes of Iron // J.C.S. Chem.
Comm. 1971. № 19. P. 1173-1174.
216. Baltzer L., Landergren M. 57Fe NMR Spectroscopy of Carbonyl Iron Por-
phyrins. A New Probe for Heme-Ligand Interaction // J.C.S. Chem. Comm.
1987. № 1. P. 32-34.
217. Benn R., Brenneke H., Frings A. et al. Indirect Two-Dimentional Heteronu-
clear NMR Spectroscopy. (31P, 57Fe) Spectra of Organoiron Complexes //
J. Amer. Chem. Soc. 1988. V. 110, № 17. P. 5661-5668.
218. Au-Yeung S. C., Eaton D.K. A model for estimation °9Co NMR chemical
shifts and line width and it applications to cobalt dioxygen complexes //
Canad. J. Chem. 1983. V.61, № 10. P. 2431-2441.
219. Matwiyoff N.A., Wageman W.E. Cobalt-59 NMR spectra of unsymmetrical
cobalt(III) complexes // Inorg. Chim. Acta. 1970. V. 4, №3. P. 460-462.
220. Yamasaki A., Hoyama T., Fujiwara S., Nakamura K. NMR stud-
ies of cobalt complexes. Cobalt-59 NMR spectrum of the hex-
akis(trimethylphosphite)cobalt(III) complex // Bull. Chem. Soc. Jap. 1978.
V. 51, №2. P. 643-644.
221. Biradar N.S., Pujak M.A. NMR and electronic spectral study of oxalatoam-
mine cobalt(III) complexes // Z. anorg. allg. Chem. 1978. Bd.379, №1.
S. 88-94.
222. Kanekar C.R., Dhingra M.M., Marathe V.R., Nagarian R. Ligand field
strength of xantane and thioxantane // J. Chem. Phys. 1967. V. 46, №5.
P. 2009-2010.
223. Martin R. L., White A. H. Cobalt-59 NMR of sulfur, selenium and arsenic
chelates of cobalt(III) // Nature. 1969. V. 223, №5204. P. 394-396.
224. Speiss H. W., Sheline R.K. Anisotropic chemical shifts in trigonal cobalt
carbonyl containing metal - metal bonds // J. Chem. Phys. 1970. V. 53, №8.
P. 3036-3041.
225. Kohen M.A., Kidd D.R., Brown T.L. The structures of Co^CO)^ and
Со4(СО)цР(ОСНз)з in Solution // J. Amer. Chem. Soc. 1975. V. 97, №15.
P. 4408-4409.
226. Mooberry E. S., Pupp M., Slater J. L., Sheline R.K. Quadrupole coupling
and anisotropic chemical shifts in dicobalt octacarbonyl at liquid nitrogene
temperature // J. Chem. Phys. 1971. V. 55, №8. P. 3655-3660.
227. Haas H., Sheline R.K. On the structure of Co<(CO)i2 in solution // J. Inorg.
Nucl. Chem. 1967. V. 29, №3. P. 693-698.
228. Lucken E.A., Noack K., Williams D.F. 59Co NMR study of organocobalt
compounds // J. Chem. Soc. A. 1967. № 1. P. 148-154.
229. Hackbusch W, Rupp H.H., Wieghardt K. Cobalt-59 NMR study of
somepolynuclear cobalt(III) complexes // J.C.S. Dalton tr. 1975. №11.
P. 1015-1018.
230. Eaton D.R., Rogerson С. V., Sandercock A.C. Hydrogen bonding involving
the hexa-cyanocobaltate(lll) anion. Cobalt-59 NMR studies // J. Phys. Chem.
1982. V. 86, №8. P. 1365-1371.
231. Yajima F., Koike Y., Yamasaki A., Fujiwara S. Cobalt-59 NMR study of
cobalt(III) complexes. Empirical rules for the cobalt 59 chemical shifts and
linewidth of CoII,(en)I(NH3)6-iLy-type complexes // Bull. Chem. Soc. Jap.
1974. V. 47, №6. P. 1442-1446.
232. Dharmatti S.S., Kanekar C.H. Chemical shifts in the NMR of 59Co and the
ligand field theory // J. Chem. Phys. 1960. V. 31, №5. P. 1436-1437.
233. Biradar N.S., Pujak M.A. NMR and Electronic spectral studies of carbonato-
ammine cobalt(III) complexes // Z. anorg. allg. Chem. 1972. H.391. №1.
S. 54-59.
234. Freeman R., Murray G.R., Richards R.E. Cobalt nuclear resonance spectra //
Proc. Roy. Soc. 1957. V. 242A, № 1231. P. 455.
235. Biradar N.S., Pujak M.A. Chemical shifts of 59Co in some cyanatocobalt(III)
complexes // Inorg. Nucl. Chem. Lett. 1971. V. 7, №3. P. 269-273.
236. Weiss R., Verkadi J.G. Cobalt-59 NMR studies on hexakis(phosphite)-
cobalt(III) complexes // Inorg. Chem. 1979. V. 18, №2. P. 529-530.
237. Федотов M.A., Беляев А.В. ЯМР 17O и 103Rh хлороаквакомплексов po-
дия(Ш) // Коорд. химия. 1984. Т. 10, №9. С. 1236-1242.
238. Juranic N. Significance of nephelauxetic effects in interpretation of cobalt-59
NMR frequensies of cobalt(III) complex compound // Inorg. Chem. 1980.
V. 19, №4. P. 1093-1095.
239. Juranic N. 59Co NMR study of the metal-ligand covalency in cobalt (III)
complexes // Inorg. Chem. 1983. V. 22, №3. P. 521-524.
240. Jenkinson J.J., Levonson W, Perry R., Spicer M.D. Cobalt(III) Complexes
of Bi- and Tetra-Thioethers: Sinthesis. Properties, and Co-59 NMR Data //
Dalton tr. 1989. №3. P. 453-455.
241. Juranic N., Celap M.B., Vucelic D. et al. The carlbon-13 and cobalt-59 NMR
study of mixed cobalt(III) complexes containing the glycinato ligand // Inorg.
Chim. Acta. 1977. V. 25, №3. P. 229-232.
242. La Rossa R.A., Brown T.L. Cobalt-59 nuclear quadrupole and NMR spectra
of cobaloximes // J. Amer. Chem. Soc. 1974. V. 96, № 7. P. 2072-2081.
243. Buckingham A.D., Stephens P.J. Proton Chemical Shifts in the NMR Spectra
of Transition-metal Hydrides. Octahedral Complexes // J. Chem. Soc. 1969.
№ 8. P. 2747-2759.
244. Buckingham A.D., Stephens P.J. Proton Chemical Shifts in the NMR Spec-
tra of Transition-metal Hydrides. Square-Planar Platinum(II) Complexes //
J. Chem. Soc. 1964. № 11. P. 4583-4587.
245. Pidcock A., Richard R.E., Venanzi L.M. 195Pt-3lP nuclear spin coupling and
the nature of the trans-effect in platinum complexes // J. Chem. Soc. A.
1966. № 10. P. 1707-1710.
246. Федотов M.A. Ядерный магнитный резонанс донорных атомов как ин-
струмент для определения строения комплексов платиновых металлов
в растворах // Изв. АН. Сер. хим. 2003. №4. С. 743-755.
247. Федотов М.А., Федотова Т.Н., Голованева И.Ф. Координатные сдвиги
l4N аминолигандов в комплексах Pt(IV) и их использование для опреде-
ления состава и геометрического строения комплексов // Ж. неорг. химии.
1997. Т. 42, №6. С. 1003-1008.
248. Беляев А.В., Федотов М.А., Венедиктов А. Б., Храненко С.П. Изуче-
ние нитрокомплексов платиновых металлов методом ЯМР. Благородные
металлы: химия и технология. Ч. 2 / Под ред. В.Г. Торгова, Ф.А. Кузне-
цова. — Новосибирск: ИНХ, 1989. С. 5-54.
249. Wi.li.nski J., Kurland R.J. Cobalt-59 NMR shifts and the acidity of amine
protons in tris(ethylendiamine)cobalt(III) // Inorg. Chem. 1973. V. 12, №9.
P. 2202-2203.
250. Rose K.D., Bryant R.G. Cobalt-59 and nitrogen-14 NMR relaxation in hex-
anitrocobaltate(III) ion // Inorg. Chem. 1979. V. 18, №8. P. 2130-2131.
251. Doddrell D.M., Pegg D. T., Bendall M.R. Field dependence of chemical shifts
in cobalt(III) NMR spectroscopy. Theoretical predictions // Aust. J. Chem.
1979. V. 32, № 1. P. 1-10.
252. Juranic N., Celap M.B., Vucelic D. et al. A cobalt-59 NMR study of
cobalt(III) complexes containing amino carboxylato ligands // J. Coord.
Chem. 1979. V. 9, №2. P. 117-123.
253. Тарасов В.П., Капанадзе Т.Ш., Цинцадзе Г.В., Буслаев Ю.А. ЯМР 59Со
комплексов Со(Ш) с азот- и серосодержащими лигандами Ц Коорд. химия.
1983. Т. 9, №5. С. 647-657.
254. Yajima F., Koiko К, Sakai Т., Fujiwara S. Cobalt-59 NMR study of iso-
merization reactions of mer- and fac-Coen(NHa)30H2+ions and trans-, cis-,
trans- and cis, с!з-Соеп(ННз)з(ОН)^ // Inorg. Chem. 1972. V. 11, №9.
P. 2054-2057.
255. Гуля А.П., Старым М.П., Батыр Д.Т. Спектры ЯМР 'Н и 59Со транс-
диоксиминов кобальта(Ш) с ацетонитрилом и циклогексилизонитрилом Ц
Коорд. химия. 1981. Т. 7, № 1. С. 108-111.
256. Pujak М.А. A spectroscopic study of azidoammine cobalt(III) complex // Ind.
J. Chem. 1980. V. A19, №4. P. 384-385.
257. Pujak M.A., Biradar H.S. NMR and magnetic susceptibility studies of
nitroammine cobalt(III) complexes // J. Ind. Chem. Soc. 1980. V. 57, №8.
P. 783-780.
258. Аблов А.В., Попа Э.В., Мазус М.Д. и др. Исследование катиона
[Со(ННз)зО1еп]2+ методами рентгеноструктурным и ЯМР °9Со Ц Коорд.
химия. 1979. Т. 5, К° 2. С. 287-293.
259. Капанадзе Т.Ш., Тарасов В.П., Цинцадзе Г.В., Буслаев Ю.А. Геометри-
ческая и связевая нитрито-нитро- изомеризация октаэдрических аминных
комплексов Со(Ш) // Коорд. химия. 1987. Т. 13, №9. С. 1261-1271.
260. Hagen K.I., Schwab С.М., Edvards J. О., Sweigart D.A. Application of S9Co
NMR to Co(lII) Porphyrins. Linear Relationship between 59Co and °‘Fe
Chemical Shifts // Inorg. Chem. 1986. V. 25, № 7. P. 978-983.
261. Капанадзе Т.Ш., Тарасов В.П., Цинцадзе Г.В., Буслаев Ю.А. Амииоком-
плексы с О-, S-, Se- и теллуросодержащими конкурирующими аминоли-
гандами // Коорд. химия. 1988. Т. 14, №5. С. 666-675.
262. Капанадзе Т.Ш., Элердашвили М.А., Тарасов В.П. и др. Сульфатоком-
плексы Со(Ш) со смешанной координационной сферой // Коорд. химия.
1989. Т. 15, №2. С. 248-55.
263. Капанадзе Т.Ш., Кокунов Ю.В., Цинцадзе Г. В., Буслаев Ю.А. Дисте-
реоизомерия комплексов Со(1П) с этилендиамином, 1,3-пропилендиамином
и 1,2-бутилендиамином // Коорд. химия. 1990. Т. 16, №5. С. 666-72.
264. Капанадзе Т.Ш., Элердашвили М.А., Гуля А.П., Буслаев Ю.А. Реакции
mer-fac изомеризации трис-(2-аминоэтанола) кобальта(Ш) // Ж. неорг.
химии. 1991. Т. 36, №5. С. 1212-1216.
265. lida М., Mizuno Yu., Koine N. 59Со NMR Spectroscopic Chiral Discrimina-
tion of [Со(еп)з]3+ Enantiomers in lonjc Interacting Systems // Bull. Chem.
Soc. Jap. 1995. V. 68, №5. P. 1337-1344.
266. Navon G. A search for the thermally populated high-spin excited state of
hexaaquocobalt (3+) by cobalt-59 NMR // J. Phys. Chem. 1981. V. 85, № 24.
P. 3547-3549.
267. Au- Yeung S. C., Eaton D.R. Characterization of cobalt dioxygen complexes
by means of high field 59Co NMR // Inorg. Chim. Acta. 1983. V. 76, №2.
P. 141-144.
268. Cran H., Edlund U., Holak T.A. A 59Co NMR Chemical Shift and Relaxation
Study of Preferential Solvation Toward Tris(acetylacetonato)cobalt(III) //
Magn. Reson. Chem. 1987. V. 25, №5. P. 497-501.
269. Schuman H., Meissner M., Kroth H.-J. NMR Untersuchungen an
Ogranoelement(IVB)-phosphinen, 59Co NMR, IR und UV-spektroskopische
Untersuchungen an Organo-element(IVB)-phosphin-substituirten Komplexen
des Tricarhonylnitrosylcobalt // Z. Naturforsch. 1978. Bd. 38b, №12.
S. 1489-1490.
270. Juranic H., Celap M.B., Vucelic D. et al. Chemical shifts in cobalt-59
NMR spectra of low-symmetry ligand field cobalt(III) complex compounds //
J. Magn. Reson. 1979. V. 35, №3. P. 319-327.
271. Dodvad S.S., Datar M.G. NMR study of cobalt-59 in some cobalt(III)
complexes // Ind. J. Chem. 1979. V. 17A, № 1. P. 100-102.
272. Yajima F., Yamasaki A., Fujiwara S. Cobalt-59 NMR study of acid dissotia-
tion equilibria in aquapentaamminecobalt(III) perchlorate and cis- and trans-
aquaamminebis(ethylene-diamine) cobalt(III) bromide // Inorg. Chem. 1971.
V. 10, №10. P. 2350-2352.
273. Juranic N., Celap M. V., Vucelic D. et al. Non-additivity of ligand effects on
59Co NMR chemical shifts in Co(III) complexes // Spectrochim. Acta. 1979.
V. 35a. №8. P. 997-1002.
274. Dhake P.M., Haidar B.C. Structural investigations of Co(III) and
Rh(III)complexes of a-benzilmonoxime // J. Ind. Chem. Soc. 1978. V. 55,
№ 1. P. 18-23.
275. Rose K., Bryant R.G. The insignificance of second coordination sphere inter-
action in coball-59 NMR relaxation: a more precise assignement // J. Magn.
Reson. 1979. V. 35, №2. P. 223-226.
276. Laszlo P., Stokis A. Accurate and sensitive determination by new 59Co
NMR method of electron acceptance and hydrogen bond donation by protic
solvents // J. Amer. Chem. Soc. 1980. V. 102, №20. P. 7818-7820.
277. Bendal R.M., Doddrell D.M. Proton-deuterium isotope shifts in 59Co NMR //
Aust. J. Chem. 1978. V. 31, №5. P. 1141-1143.
278. Rehder D. Application of transition Metal NMR Spectroscopy in Coordination
Chemisry // Chimia. 1986. V. 40, №6. P. 186-199.
279. Fedotov M.A., Muzykantov V. S., Slyudkin O.P. Atomo-molecular mecha-
nism of cobalamines H-D exchange in aqueous solutions: NMR 59Co studies
of isotopic kinetics. Тез. межд. Конф. «Физические методы исследования
катализа на молекулярном уровне». Институт катализа СО РАН: Новоси-
бирск, 1999. Р. 153.
280. Hidai М., Matsuraka Н., Koyasu Yu., Uchida Ya. Metallo-selective Substitu-
tion Reactions by Amines or Phosphines in НРиСоз(СО)12 // J.C.S. Chem.
Comm. 1986. № 18. P. 1451-1452.
281. Au-Yeung S.C., Buist H.J., Eaton D.R. Spin-Lattice Relaxation in Co Com-
plexes of Low Symmetry // J. Magn. Reson. 1983. V. 55, № 1. P. 24-38.
282. Doddrell D.M., Bendall R.M., Healy P.C. et al. o9Co and 13C nuclear spin
relaxation studies in solutions of symmetric, bidentante cobalt(III) com-
plexes. Crystal structure determination of tris(tropolonate) cobalt(III) // Aust.
J. Chem. 1979. V. 32, №6. P. 1219-1230.
283. Shirley W.M., Bryant R.G., Hugus Z.Z. Cobalt-59 NQR Spin-Lattice Relax-
ation Times in 1гаи5-[СоС12{еп)г] [ЩС^СЬ and trans-[CoC12(en)2]N03 //
J. Magn. Reson. 1981. V.42, №3. P. 493-497.
284. Drain L.E. The magnetic moment of 61Ni // Phys. Lett. 1964. V. 11, №2.
P. 111-115.
285. Benn R., Rufinska A. High-Resolution Metal-NMR Spectroscopy of Organo-
metallic // Z. Naturforsch. 1980. Bd. 35b, №5. S. 639-641.
286. Hao N., McGlinchey M.J., Sayer B.C., Schrobilgen G.S. A Nickel NMR
Study of Some d10 Nickel Complexes // J. Magn. Reson. 1982. V. 46, № 1.
P. 158-162.
287. Beringer K.D., Bliimel J. 61 Ni NMR Spectroscopy of Di- and Tricarbonyl-
nickel Complexes // Magn. Reson. Chem. 1995. V. 33, №8. P. 729-733.
Платиновые металлы
288. Brevard G., Grander P. The First Direct Detection of Ruthenium-99 and
Ruthenium-101 NMR // J. Chem. Phys. 1981. V. 75, №9. P. 4174-4177.
289. Gill D.S., Gansow О.Л., Bennis E.J., Ott K.C. The Direct Observation of
Rhodium-103 NMR // J. Magn. Reson. 1979. V. 35, № 3. P. 459-461.
290. Федотов M.A., Лихолобов В.А. Первое прямое наблюдение ЯМР 105Pd
в растворах // Изв. АН СССР. Сер. хим. 1984. №8. С. 1917-1918.
291. Pregosin P.S. The multinuclear NMR Approach. Contribution to the Chem-
istry of Platinum // Chimia. 1981. V. 35, №2. P. 43-49.
292. Burgstaller A., Voitlanger J., Ebert H. The nuclear magnetic resonance of
"Ru and 101 Ru in hexagonal close-packed ruthenium metal // J. Phys.:
Condens. Matter. 1994. V. 6, №40. P. 8335-8340.
293. Dykstra R.W., Harisson A.M. Ruthenium-99 and -101 NMR // J. Magn.
Reson. 1982. V. 40, № 2. P. 338-342.
294. Brevard C., Granger P. Ruthenium NMR spectroscopy: a promising struc-
tural and analytical tool // Inorg. Chem. 1983. V. 22, №3. P. 532-535.
295. Steel R.J., Lahousse F., Larner D., Marzin C. New rutheniuni(II) complexes
with pyridylpyrazole ligands. Photosubstitution and 1H, l3C and "Ru NMR
structural sludies // Inorg. Chem. 1983. V. 22, № 10. P. 1488-1493.
296. Федотов M. А., Беляев А. В. Исследование нитрозонитрокомплексов руте-
ния методом ЯМР разных ядер // Коорд. химия. 1991. № 1. С. 103-111.
297. Емельянов В.А., Федотов М.А., Беляев А.В. Кинетика нитрования нит-
розо пентахлорорутений(И) иона // Ж. неорг. химии. 1992. Т. 37, №12.
С. 2717-2721.
298. Емельянов В.А., Федотов М.А., Беляев А.В. Исследование обратимых
нитро-нитрозо превращений в комплексах рутения(П) методом ЯМР 15N,
|7О, "Ru//Ж. неорг. химии. 1993. Т. 38, №11. С. 1842-1848.
299. Беляев А.В., Емельянов В.А., Храненко С.П., Федотов М.А. Синтез
нитрозохлоридных комплексов рутения и механизм процессов, лежащих
в его основе // Коорд. химия. 1996. Т. 22, № 5. С. 380-382.
300. Granger Р., Richert Т., Elbayed К. et al. Microdynamic Motion of Tetrahe-
dral Clusters Studied by 59Co and "Ru NMR Relaxation // Mol. Phys. 1997.
V. 92, № 5. P. 895-902.
301. Емельянов В.А., Федотов M.A., Беляев А.В. Образование гетерометал-
лических комплексов рутения с цветными металлами // Ж. неорг. химии.
2000 Т. 45, №5. С. 813-818.
302. Bernhard Р., Helm L., Ludi A., Merbach А.Е. Direct Measurment of a
Prominent Outer-Sphere Electron Self-Exchange: Kinetic Parameters for
the Hexaaquaruthenium(II) I (III) Couple Determination by Oxygen-17 and
Ruthenium-99 NMR // J. Amer. Chem. Soc. 1985. V. 107, №2. P. 312-317.
303. Orellana G., de Mesmaeker K.A., Turro N.J. Ru NMR Spectroscopy of
Ruthenium(II) polypyridyl complexes // Inorg. Chem. 1990. V. 29, №4.
P. 882-885.
304. Gaemers S., Van Slageren J. O’Connor et al. Ru NMR Spectroscopy of
Organometallic and Coopdination Complexes of Ruthenium(II) // Organome-
tallics. 1999. V. 18, №25. P. 5238-5244.
305. Autschbach J., Zheng S. Density Functional Computations of "Ru Chemical
Shifts: Relativistic Effects. Influence of the Density Functional, and Study of
Solvent Effects on /ас-[Ри(СО)з!з] // Magn. Res. Chem. 2006. V. 44, № 11.
P. 987-1007.
306. Емельянов В.А., Федотов М.А. Состояние рутения в нитритно-нитрат-
ных азотнокислых растворах по данным ЯМР // Ж. неорг. химии. 2006.
Т. 51, № 11. С. 1923-1930.
307. Gruender К.D., Schwenk A., Mann В.Е. Direct observation of rhodium-103
NMR and relaxation investigation by steady-slate techniques // J. Magn.
Reson. 1980. V. 41, №2. P. 354-357.
308. Carlton L. Rhodium-103 // Annual Reports on NMR Spectroscopy. 2008.
V. 63. P. 49-178.
309. Ernsting J.M., Gaerners S., Elseviers C.J. 103Rh NMR spectroscopy and its
application to rhodium chemistry // Magn. Reson. Chem. 2004. V. 42, №7.
P. 721-736.
310. Brown C., Green P.J. The Phosphorus-31 and Rhodium -103 Nuclear Mag-
netic Resonance of Some Rhodium(I) and Rhodium(III) Phosphine Com-
plexes // J. Amer. Chem. Soc. 1970. V. 92, № 8. P. 2359-2362.
311. Беляев А.В., Венедиктов Б. А., Федотов M.A., Храненко С.П. Комплекс-
ные нитриты родия(Ш) в водных растворах // Коорд. химия. 1986. Т. 12,
№5. С. 690-699.
312. Mann В.Е., Spencer С.Н. l03Rh chemical shifts of [RhC15-nBrnJ3- // Inorg.
Chim. Acta. 1983. V. 76, № 1. P. 65-66.
313. Mann B.E., Spencer C.H. The identification of all ten Rh(OH2)6-nCln iso-
mers by rhodium-103 NMR spectroscopy // Inorg. Chim. Acta. 1982. V. 65,
№ 2. P. L57-L58.
314. Федотов M.A., Шипачев В.А., Левченко Л.М. Изучение аква- и аква-
сульфатных комплексов родия(Ш) методом ЯМР |7О и l03Rh в водных
и сульфатных средах // Коорд. химия. 1999. Т. 25, № 1. С. 35-39.
315. Carr С., Glaser J., Sandstrom М. IO3Rh NMR chemical shifts of all ten
[RhCl„(OH2)6-n]3-n complexes in aqueous solution // Inorg. Chim. Acta.
1987. V. 131, № 1. P. 153-156.
316. Беляев А.В., Федотов M.A., Храненко С.П., Емельянов В.А. Состояние
родия(Ш) в азотнокислых растворах // Коорд. химия. 2001. Т. 27, № 12.
С. 907-916.
317. Федотов М.А. О состоянии Rh(III) в разбавленных азотнокислых раство-
рах по данным ЯМР 14N, 17О, 103Rh // Коорд. химия. 2002. Т. 28, №8.
С. 610-617.
318. Беляев А. В., Федотов М.А., Шагабутдинова С.Н. Состояние родия(Ш)
в сернокислых растворах // Коорд. химия. 2007. Т. 33, №2. 141-145.
319. Read М.С., Glaser J., Sandstrom M., Toth I. Hydrolytic Oligomers of
Rhodium(III). A Multinuclear NMR Study of the Doubly Hydroxo-Bridged
Dimer and Trimer in Aqueous Solution // Inorg. Chem. 1992. V. 31, №20.
P. 4155-4609.
320. Spiccia L., Aramini J.M., Crimp S.J. et al. Hydrolytic Polymerization of
Rhodium(III). Characterization of Various Forms of Trinuclear Aqua Ions //
J.C.S. Dalton tr. 1997. №23. P. 4603-4609.
321. Read M. C., Glaser J., Persson I., Sandstrom M. Rhodium(III) complexes
with cyanide and Sulfur-Donor Ligands: Rhodium-103 Nuclear Magnetic
Resonance Chemical Shifts correlation // J.C.S. Dalton tr. 1994. №21.
P. 3243-3248.
322. Galli D., Garlaschelli L., Ciani G. et al. Synthesis, and Chemical
Characterization of the New Anions [ИЬз(р.з-Е)2(СО)б]1- (x = 10, 11;
E = S, Se). Crystal and Molecular Structures of [PPh3][Rh3(/A3-S)2(CO)6]
and [NMe4][Rh3(/i3-Se)2(CO)6] // J.C.S. Dalton tr. 1984. № 1. P. 55-61.
323. Ceriotti A., Londoni G., Pergola R.D. et al. Bimetallic Iron-Rhodium
Anionic Carbinyl Ciasters: [Fe2Rh(CO)^ (x = 10, 11), [FeRh4(CO)i5]2 .
[Fe2Rh5(CO)i6]- //J.C.S. Dalton tr. 1983. №7. P. 1433-1440.
324. Sogo P.B., Jeffries C.D. Nuclear magnetic moments of 35CI, l03Rh, l83W //
Phys. Rev. 1955. V. 98, №5. P. 1316-1317.
325. Mason J. Correlations in nuclear magnetic shielding. Adv. in inorganic chem-
istry and radiochemistry / Ed. by H.J. Emeleus, A.G. Sharpe. — L.: Acad.
Press, 1979. V. 22. P. 199-240.
326. Беляев А. В., Федотов M.A. О состоянии Rh(III) в концентрированных
водных растворах по данным ЯМР 103Rh и 35С1 // Коорд. химия. 1983. Т. 9,
№8. С. 1252-1257.
327. Hyde Е.М., Kennedy J.D., Shaw B.L., McFarlane W. Magnetic double res-
onance measurments of rhodium, phosphorus, fluorine and proton NMR pa-
rameters in some rhodium (I) and rhodium(III) compounds // J.C.S. Dalton
tr. 1977. № 10. P. 1571-1575.
328. Шипачев В. А., Земсков С. В., Ткачев С. В. Исследование поведения
гексафторродиат-иона в растворах // Коорд. химия. 1980. Т. 6, №8.
С. 1237-1240.
329. Moriyama Н., Aoki Т., Shinoda S., Saito V. Photoenhanced catalytic de-
hydrogenation of propan-2-ol with homogenious rhodium-tin complexes //
J.C.S. Dalton tr. 1982. №3. P. 369-374.
330. Беляев А. В., Федотов M.A., Корсунский В. И. и др. О строении поли-
ядерных хлоридов родия(Ш) // Коорд. химия. 1984. Т. 10, №7. С. 911-918.
331. Беляев А. В., Венедиктов А. Б., Федотов М. А., Храненко С. П. Иссле-
дование взаимодействия хлоридов родия(Ш) с а цетила цетоном // Коорд.
химия. 1985. Т. 11, №6. С. 794-804.
332. Казакова А. А., Федотов М.А., Венедиктов А. Б., Беляев А. В. Иссле-
дование взаимодействия хлоридов родия(Ш) с оксалат-ионами // Коорд.
химия. 1989. Т. 15, №6. С. 822-824.
333. Храненко С.П., Федотов М.А., Беляев А.В. Синтез и исследование
TpaHC-K3[Rh(NO2)«C12] // Коорд. химия. 1991. Т. 17, №6. С. 860-863.
334. Венедиктов А. Б., Федотов М. А., Коренев С. В. Превращения гексанитро-
родиат(Ш) иона в хлорнокислых и солянокислых средах // Коорд. химия.
1999. Т. 25, №4. С. 285-289.
335. Храненко С.П., Федотов М.А., Беляев А. В. Изучение превращений ком-
плексов [Rh(NO2)IClyH2O6_I_!;]3_I_y методом ЯМР // Коорд. химия.
1990. Т. 16, №7. С. 991-996.
336. Беляев А.В., Федотов М.А., Шагабутдинова С.Н. Исследование про-
цесса нитрования сульфатов Rh(III) методом ЯМР 103Rh, I7O, 14,I5N //
Ж. неорг. химии. 2009.
337. Федотов М.А., Беляев А.В. Прямое наблюдение инертных нитратоком-
плексов платиновых металлов методом ЯМР l4,l5N в водном растворе //
Коорд. химия. 1994. Т. 20, №8. С. 613-615.
338. Беляев А. В., Ренард Э.В., Храненко С.П. и др. О состоянии радиородия
в жидких высокоактивных отходах от регенерирования отработанного
топлива АЭС // Радиохимия. 2002. Т. 44, №6. С. 493-505.
339. Беляев А. В., Емельянов В. А., Храненко С.П., Федотов М.А. Исследова-
ние взаимодействия нитратных комплексов Pd, Ru и Rh с сульфаминовой
кислотой методом ЯМР // Коорд. химия. 2001. Т. 27, № 3. С. 203-213.
340. Шагабутдинова С.Н., Федотов М.А., Беляев А. В. Исследование про-
цессов индуцированной акватации сульфатных комплексов родия(Ш) //
Коорд. химия. 2007. Т. 33, №2. 146-150.
341. Беляев А.В., Ильяшевич. В.Д., Павлова Е.И. и др. Комплексообразование
Rh(III) в разбавленных растворах серной кислоты // Коорд. химия. 2007.
Т. 33, № 6. С. 458-462.
342. Беляев А. В., Федотов М.А., Шагабутдинова С.Н., Павлова Е.И.
Комплексообразование родия(П1) в сернокислых растворах // Вестник
МИТХТ. 2007. Т. 2, № 3. С. 29-35.
343. Федотов М.А., Беляев А. В. Первое наблюдение методом ЯМР (31Р, 17О,
l03Rh) комплексообразования Rh(III) с фосфорной кислотой // Ж. неорг.
химии. 2008. Т. 53, № 5. С. 826-229.
344. Yurchenko Е.П., Yuryavichus A. Yu., Fedotov М.А., Valsyunene Ya.I. Cat-
alytic oxidative hydrolysis ol H2PO^ anions in the presence of RhCh //
React. Kinet. Catal. Lett. 1989. V. 31, № 1. P. 55-60.
345. Коренев C.B., Храненко С.П., Федотов М.А. и др. Комплексы родия(Ш)
с парамагнитным енаминокетоном // Коорд. химия. 1987. Т. 13, №11.
С. 1499-1506.
346. Seitchick J.A., Gossard A C., Jaccarino V. Knight shifts and susceptibil-
ities of transition merals: palladium // Phys. Rev. 1964. V. 136A, №4.
P. 1119-1125.
347. Brevard C., Granger P. Handbook of High Resolution Multinuclear NMR. —
N.Y.: Wiley Interscience, 1981. — 202 p.
348. Беляев А.В., Федотов M.A., Коренев С.В. Изучение взаимопревращений
комплексов [PdCln(NO2)4_„]2— методом ЯМР 15N и других ядер // Коорд.
химия. 1989. Т. 15, № 11. С. 1551-1555.
349. Троицкий С.Ю., Федотов М.А., Лихолобов В.А. Изучение комплексов
Pd(II) с кислородсодержащими лигандами методом ЯМР 17О // Коорд.
химия. 1990. Т. 16, № 12. С. 1675-1677.
350. Троицкий С.Ю., Федотов М.А., Лихолобов В.А. Изучение продуктов
гидролиза солей палладия(П) // Изв. АН СССР. Сер. хим. 1993. №4.
С. 675-679.
351. Крылова Л.Ф., Диканская Л.Д., Федотов М.А. Моноциклические ком-
плексы платины(П) и палладия(П) с аминокислотами ряда глицина //
Коорд. химия. 1994. Т. 20, № 1. С. 57-59.
352. Troitsky S. Yu., Serebryakova М.А., Fedotov М.А. et al. Synthesis and study
of palladium colloids and related catalysts // J. Molec. Catal. A. 2000. V. 158.
P. 461-465.
353. Schwenk A., Zimmerman G. The ratio of the Larmor frequency of 1890s
relative to *H // Phys. Lett. 1968. V. 26A, № 6. P. 258-259.
354. Constable F.C., Johansson B.F., Geoff N.B., Taylor M.J. ie7Os-'H coupling
in hydrido-osmium ciasters as aid to assignement of molecular structures //
J.C.S. Chem. Comm. 1982. №13. P. 754-756.
355. Christe K.O., Dixon D.A., Mack H.G. et al. Osmium Tetrafluoride Dioxide.
cis-OsO2F2 // J. Amer. Chem. Soc. 1993. V. 115, №24. P. 11279-11284.
356. Gerken. M., Dixon D.A., Schrobilgen GJ. The OsC^F-, OsC^Fi;-, and
OsO3F^ Anions. Their Study by Vibrational and NMR Spectroscopy and
Density Functional Theory Calculatioms, and the X-ray Crystal Structures
of [N(CH3)4][OsO4F] and [N(CH3)4] [OsO3F3] // Inorg. Chem. 2000. V. 39,
№ 19. P. 4244-4255.
357. Венедиктов А.Б., Федотов M.A., Коренев С.В., Беляев А. В. Исследо-
вание процессов нитрования гексахлорокомплексов иридия методом ЯМР
i5N // Коорд. химия. 1989. Т. 15, №4. С. 556-560.
358. Венедиктов А.Б., Коренев С. В., Федотов М.А., Беляев А.В. ЯМР спек-
троскопии. исследование промежуточных продуктов нитрования гексахло-
ро комплексов иридия (IV) и (III) // Коорд. химия. 1990. Т. 16, № 10.
С. 1400-1405.
359. Коренев С.В., Венедиктов А.Б., Малкова В.И., Федотов М.А. Синтез
и свойства тринитротриаква комплекса иридия(Ш) и его производных //
Коорд. химия. 1999. Т. 25, №9. С. 696-698.
360. Pesek J.J., Mason W.R. Platinum magnetic resonance spectra of some plat-
inum(II) and platinum(IV) complexes // J. Magn. Reson. 1977. V. 25, № 3.
P. 519-529.
361. Freeman W., Pregosin P.S., Sze S.N., Venanzi L.M. Platinum-195 NMR
using Fourier transform techniques. The PtCl2--ion // J. Magn. Reson. 1976.
V. 22, № 3. P. 473-478.
362. Martin J.R., Abbout E.H. A platinum-195 nuclear magnetic resonance in-
vestigation of some dihydroxo bridged binuclear platinum(II) sulfoxide com-
plexes // Inorg. Chim. Acta. 1996. V. 247, №2. P. 267-271.
363. Freeman W., Pregosin P.S., Sze S.N., Venanzi L.M. Platinum-195 NMR
using Fourier transform techniques. The PtCl2_-ion // J. Magn. Reson. 1976.
V. 22, №3. P. 473-478.
364. Федотов M. A., Коренев C.B., Беляев А. В. Изучение взаимопревращений
комплексов [PtClntNOjh-n]2- методом ЯМР l95Pt и l5N // Коорд. химия.
1990. Т. 16, №9. С. 1272-6.
365. Appleton T.G., Hall J.R., Hambley T.W., Premier P.D. Reactions of the
cis-Diamminediaquaplatinum(II) Cation with Glicinamide. N-Glycylglycine,
and N-(N-Glycylglycyl)glycine. Crystal Srtucture of a Complex with Two
DiamminepIatinum(II) Cation Bound to Glycylglycenate // Inorg. Chem.
1990. V. 29, № 18. P. 3562-3569.
366. Schwenderski B.E., Lee H.D., Margerum D. W. l95Pt NMR Spectroscopy of
(15N) peptide Complexes. Inorg. Chem. 1990. V. 29, № 18. P. 3569-3578.
367. Groning 0., Drakenberg T., Elding L.I. Water Exchange and Solvolysis in
Dimethyl Sulfoxide of Square-Planar Tetraaquaplatinum(II). A Platinum-195
NMR Study // Inorg. Chem. 1982. V.21, №5. P. 1820-1824.
368. Appleton T.G., Hall J.R., Ralph S.F., Thompson C.S.M. Reactions of
Platinum(II) Aqua Complexes, 195Pt NMR Study of Reactions between
the Tetraaquaplatinum(II) Cation and Chloride. Hydroxide. Perchlorate. Ni-
trate. Sulfate. Phosphate, and Acetate // Inorg. Chem. 1984. V. 23, №22.
P. 3521-3525.
369. Dunham S.O., Larsen R.L., Abbott E.H. Nuciear Magnetic Resonance In-
vestigation of the Hydrogen Peroxide Oxidation of Platinum(II) Complexes //
Inorg. Chem. 1993. V. 32, № 10. P. 2049-2055.
370. Boag N.M., Goggin P.L., Goodfellow R.J., Herbert I. R. Preraration and NNR
Study of Salts of the Platinum(I) Anions [PtaX^CO^]2- (X = Cl, Br, or I) //
J.C.S. Dalton tr. 1983. №6. P. 1001-1007.
371. Шитова Н.Б., Альт Л.Я., Перелевская И.Г. Обратимые превращения
карбонильных комплексов платины под действием кислот и оснований
в вводно-ацетоновых растворах и на поверхности оксида алюминия //
Ж. неорг. химии. 1998. Т. 43, №5. 800-804.
372. Юданов В.Ф., Еременко Н.К., Медников Е.Г., Губин С.П. Структура
кластера Pt4(CO)s(PEt3)4 в растворе // Ж. структ. химии. 1984. Т. 25,
№ 1. 49-52.
373. Moor A., Pregosin PS., Venanzi L.M. 31Р and 195Pt NMR Studies on the
Clusters [Pt4(M2-CO)5L4], L = PEtj, РМегРЬ, PMePhj, PEtjBu*. The Molec-
ular Structure of a Monoclinic Modification of [Pt4(/X2-CO)s(PMe2Ph)4] //
Inorg. Chim. Acta. 1984. V.85, № 1. P. 103-110.
374. Привалов В.И., Лапкин В.В., Шубочкина У.Ф., Тарасов В.П. ЯМР 195Pt
водных растворов оксонитрокомплексов Pt(II) и Pt(IV) // Коорд. химия.
1988. Т. 14, №3. С. 390-393.
375. Privalov V.I., Lapkin V.V., Tarasov V.P., Buslaev Yu.A. Structure and
Hydroliysis Heterovalent Polynuclear Oxonitrocomplexes of PtI1,IV in Aqueous
Solutions by 195Pt and 15N NMR Spectroscopy // Mend. Comm. 1991. №2.
P. 59-61.
376. Jalilchvand F., Maliarik M. Sandstrom M et al. New Class of Oligonuclear
Platinum-Thallium Compounds with Direct Metal-Metal Bond. Structure
Determination of Heterometallic Cyano Complexes in Aqueous Solution by
EXAFs and Vibrational Spectroscopy // Inorg. Chem. 2001. V. 40, №16.
P. 3889-3899.
377. Pregosin P.S., Kunz R. W. 31P and 13C NMR of Transition Metal Complexes.
NMR Basic Principles and Progress. V. 16/ Ed. P. Diehl, E. Fluck, R. Kos-
feld. — Berlin: Springer Verlag, V. 19. P. 129-133.
378. Appleton T. G., Clark H. C., Manzer L.E. The trans-influence: its measurinent
and significance // Coord. Chem. Rev. 1973. V. 10, №3/4. P. 335-422.
379. MrFarlane W. The sign of the l95Pt — *H coupling constant in square planar
complexes of platmum(II) // J.C.S. Chem. Comm. 1967. № 15. P. 772-773.
380. Advance in organomet. Chemistry. V. 12/Ed. by F.G. Stone, R. West. — N. Y.:
Acad. Press, 1974. P. 135-213.
381. Anderson S.J., Goggin P.L., Goodfellow R.J. Study of the co-ordination
behaviour of the thiocyanate and cyanate ions in some platinum(II) complexes
by heteronuclear resonance spectroscopy // J.C.S. Dalton tr. 1976. №19.
P. 1959-1964.
382. Pregosin P.S., Omura H., Venanzi L.M. Use of 'j(195Pt-l5N) as probe into
the electronic structure of some platinum(II) amine complexes // J. Amer.
Chem. Soc. 1973. V. 95, №6. P. 2047-2048.
383. McFarlane W. The signs of spin-spin coupling constants in square planar
complexes of. platinum (II) // J. Chem. Soc. A. 1967. № 12. P. 1922-1923.
384. Перепалова В.И., Белый А. С., Альт Л.Я., Дуплякин В.К. Взаимодей-
ствие хлоридов платины с двуххлористым оловом в изопропиловом спир-
те // Коорд. химия. 1983. Т. 9, №2. С. 280-281.
385. Желиговская Н.Н. О количественной характеристике* трансвлияния //
В кн.: Трансвлияние в химии координационных соединений. — М.: Наука,
1979. С. 36-63.
386. Нефедов В. И. Взаимное влияние лигандов в соединениях переходных и
непереходных элементов. — М.: Наука, 1979. С. 73-92.
387. Li Wu, Schwederski В.Е., Margenum D.W. Stepwise Hydrilysis of Tetra-
chloroplatinate(II) in Base // Inorg. Chem. 1990. V. 29, № 22. P. 3578-3584.
Медь, серебро, золото
388. Goodfellow R. J. Post-Transition Metals. Copper to Mercury. J. Mason, ed. —
N. Y.: Plenum Press, 1987. P. 563-589.
389. McConnell H.M., Weaver H.E. Rate of electron exchange between cuprous
and cupric ions in hydrochloric, acid solutions by NMR // J. Chem. Phys.
1956. V. 25, №2. P. 307-311.
390. Yamamoto T., Haraguchi H., Fujiwara S. Copper NMR study
of-cyanocuprate(I) ions in solution. Formation of polynucluar species and of
mixed complexes // J. Phys. Chem. 1970. V. 74, №25. P. 4369-4373.
391. Lutz O., Oehler H., Kroneck P. Chemical shifts and coupling constants
in copper(I) compounds by copper-63 and copper-65 FT-NMR studies //
Z. Naturiorsch. 1978. Bd. 33a, №9. S. 1021-1024.
392. McFarlane W., Rycroft D.S. The presentation of nuclear magnetic triple
resonance spectra: a study of the [(СНз}зР]«Си+ cation by MINDOR and
TINDOR techniques // J. Magn. Reson. 1976. V. 24, № 1. P. 95-101.
393. Lutz O., Oehler H., Kroneck P. Copper-63 and -65 Fourier transform NMR
studies // Z. Phys. 1978. Bd. 288a, № 1. S. 17-21.
394. Ochsenbein U., Schlaepfer C. W. Copper-65 NMR studies of copper(I)
tetrapyridine and copper(I) tetraacetonitrile solvates // Helv. Chim. Ac-
ta. 1980. V. 63, №7. P. 1926-1931.
395. Gill D.S., Rodehdser L., Delpuech J.-J. Solvation of Copper(I) Perchlorate in
Mixed Solvent Systems Containing Acetonitrile. A 63Cu, 65Cu and 31P NMR
Study // J.C.S. Faraday Tr. 1990. V. 16, № 16. P. 2847-2852.
396. Gill D.S., Rodehiiser L., Rubini P., Delpuech J.-J. 63Cu and 109Ag NMR
Studies of Copper(I) and Silver(I) Salts in Mixed Solvents Containing Ni-
triles // J.C.S. Faraday tr. 1995. V.91, № 15. P. 2307-2309.
397. Hetherington A., Lewason W., Spicar M. D. Sunthesis and Copper-65 Studies
Copper(I) Stibine Complexes // Polyhedr. 1990. V. 9, № 13. P. 1609-1612.
398. Kroneck P., Lutz O., Nolle A., Oehler H. Copper-63 FT-NMR studies of
tetrahedral copper(I)-phosphorus complexes // Z. Naturforsch. 1980. Bd. 35a,
№2. S. 221-225.
399. Kujime M., Kurahashi T., Tomura M., Fudjii H. 63Cu NMR Spectroscopy
of Copper(I) with Various Tridentate Ligands: CO as Useful 63Cu Probe for
Sharping 63Cu NMR Signals and Analyzing the Electronic Donor Effect of
Ligands // Inorg. Chem. 2007. V. 46, №2. P. 541-551.
400. Endo K., Yamamoto K., Deguchi K., Matsuchita K. Synthesis and Chemical
Characterization of the New Anions [РЬз(/хз-Е)2(СО)е] (E = S, Se).
Crystal and Molecular Structures of [PPh4][Rh3(/x3-S)2(CO)6] and
[NMe4][Rh3(/i3-Se)2(CO)6] // Bull. Chem. Soc. Jap. 1987. V. 60, №8.
P. 2803-2807.
401. Becker K.D. Temperature dependence of NMR chemical shifts in copper
halides // J. Chem. Phys. 1978. V. 68, № 8. P. 3785-3792.
402. Marker A., Counter M.J. Quadrupole relaxation in tetrahedral
copper(I)-phosphite complexes: copper-63 and phosphorus-31 NMR studies //
J. Magn. Reson. 1982. V. 47, № 1. P. 118-132.
403. Irangu J.K., Jordan R.B. Copper-63 NMR Line Width Study of the Copper(I)-
Acetonitrile System // Inorg. Chem. 2003. V. 42, № 12. P. 3934-3942.
404. Burges C.-W., Koschmieler R., Sahm W., Schwenk A. 107Ag and 109Ag
NMRstudies of Ag+ ions in aqueous solutions // Z. Naturforsch. 1973.
Bd. 28a, № 11. S. 1753-1758.
405. Juker K., Sahm W., Schwenk A. 109Ag NMR studies of organic and inorganic
silver complexes // Z. Naturforsch. 1976. Bd.31a, № 12. S. 1532-1538.
406. Deverell C., Richards R.E. NMR studies of alkali metal halide solutions.
Cation resonances // Mol. Phys. 1966. V. 10, №6. P. 551-564.
407. Hennrichs P.M., Ackerman J.J., Maciel G.E. Complexation of silver(I) with
thiourea and tetramethyl thiourea in dimethylsulfoxide solution as studied by
l3C and 101 Ag NMR spectroscopy // J. Amer. Chem. Soc. 1977. V. 99, №8.
P. 2544-2548.
408. Becker K.D., Von-Goldammer E. NMR chemical shifts in silver halides //
Chem. Phys. 1980. V. 48, №2. P. 193-201.
409. Rahimi A.K., Popov A.I. Silver-109 NMR studies of some silver complexes
in nonaqueous solvents // J. Magn. Reson. 1979. V. 36, № 3. P. 351-358.
410. Henrichs P.M., Shead S., Ackerman J.J., Maciel G.E. Structural studies of
organic silver complexes in dimethylsulfoxide by carbon-13 and silver-109
NMR // J. Amer. Chem. Soc. 1979. V. 101, № 12. P. 3222-3228.
411. Kronenbitter A., Schweitzer U., Schwenk A. Silver-109 NMR studies of the
selective solvalion of the silver(l+) ion in water-ethyl amine mixtures and of
the coordination of halide anions to the silver-ethylamine solvate complexes //
Z. Naturforsch. 1980. Bd. 35a, №3. S. 319-328.
412. Endo K., Matsushita K., Deguchi K. et al. Silver-109 NMR spectra of
aqueous silver ions coordinated with nitroxide radical. Silver-tanol complex
and tanol as doping agent // Chem. Lett. A. 1982. №9. P. 1497-1500.
413. Brevard C., Van-Stein G. C., Van-Koten G. Silver-109 and rhodium-103 NMR
spectroscopy with proton polarization transfer // J. Amer. Chem. Soc. 1981.
V. 103, №22. P. 6746-6748.
414. Pfister H, Schwenk A., Zeller D. Longitudinal and Transverse 109Ag Relax-
ation of the Silver Ion in Aqueous Solution // J. Magn. Reson. 1986. V. 68,
№ 1. P. 138-145.
415. Tokita M., Haga E. NMR of gold-197 in gold metal // J. Molec. Struct.
1982. V.83, №2. P. 143-153.
416. Голованева И.Ф., Дон Г.М., Федотов М.А. и др. Стереохимическая
направленность реакции N-нитрозоамидирования координированного
1,2-пропилендиамина в комплексе золота(Ш) бис-диаминового типа //
Ж. неорг. химии. 1997. Т. 42, №3. С. 386-390.
417. Дон Г.М., Федотов М.А., Голованева И.Ф. Комплексообразование бис-
этилендиаминового комплекса золота(Ш) с хлорид-ионом по данным ЯМР
35С1 // Ж. неорг. химии. 1994. Т. 39, № 1. С. 135-137.
Цинк,кадмий, ртуть
418. Epperlein В. IF., Kruger Н., Lutz О., Schwenk A. Fourier transform NMR
studies of 67Zn // Z. Naturforsh. 1974. Bd. 29a, № 11. S. 1553-1557.
419. Maciel G.E., Simeral L., Ackerman J. J. Effect of complexation of zinc(II) on
zinc (II) chemical shifts // J. Phys. Chem. 1977. V. 81, №3. P. 263-267.
420. Li Zhi-fen, Popov A. I. Zinc-67 NMR study of zinc ions in water and in
some nonaqueous and mixed solvents // J. Solut. Chem. 1982. V. 11, № 1.
P. 17-26.
421. Cain K.J., Melendres C.A., Maroni V.A. Raman and 67Zn NMR Studies
of the Structure of Zn(II) Solution in Concentrated Aqueous Potassium
Hydroxide // J. Electrocem. Soc. 1987. V. 134, №3. P. 519-524.
422. Zhang Y., Mukherjee S., Oldfield E. 67Zn NMR Chemical Shifts and Electric
Field Gradient in Zink Complexes. A Quantum Chemical Investigation //
J. Amer. Chem. Soc. 2005. V. 127, №8. P. 2370-2371.
423. Shimizu T., Kodaka M., Hatano M. Zinc-67 and proton NMR studies of
zinc(2+)-imidazole and carboxilato complexes // Biochem. Biophys. Res.
Comm. 1982. V. 106, №3. P. 988-993.
424. Shimizu T., Hatano M. Zinc-67 NMR spectral studies of aqueous zinc(2+)
and zinc(2+)-insulin complexes // Inorg. Chim. Acta. 1983. V. 76, № 3.
P. L177-L178.
425. Kennedy J.D., McFarlane IF. Auto-assotiation in organometallic compounds:
a NMR study of methylcadmium alkoxides // J.C.S. Perkin tr.II.1977. №9.
P. 1187-1191.
426. Habercorn R.A., Que L., Gillium W.O. et al. Cadmium-113 Fourier trans-
form NMR and Raman spectroscopic studies of cadmium(II)-sulfur com-
plexes including [Cdio(SCH2CH20H)i6]4+ И Inorg. Chem. 1976. V. 15.№ 10.
P. 2405-2414.
427. Ackerman J.J., Orr T. V., Bartuska V.J., Maciel G.E. Effect of halide com-
plexation of cadmium(II) on cadmium-113 chemical shifts // J. Amer. Chem.
Soc. 1979. V. 101, №2. P. 341-347.
428. Drackenberg T., Bjork H.-О., Portanova R. Cadmium-113 NMR study of
cadmium(II) halide complexes in water and dimethylsulfoxide // J. Phys.
Chem. 1978. V. 82, №22. P. 2423-2426.
429. Lock D.C. NMR shemical shifts in CdS. CdSe and CdTe // Phys. Stat. Solid.
1972. V. 50b. № 2. P. K97-K100.
430. Cardin A.D., Ellis P.D., Odom J. D., Howard J. W. Cadmium-113 Fourier
transform NMR spectroscopy // J. Amer. Chem. Soc. 1975. V. 97, № 7.
P. 1672-1679.
431. Jensen C.F., Deshmukh S., Jakobsen H.J. et al. Cadmium-113 NMR studies
of cadmium-ethylenediamintetraacetic acid complexes // J. Amer. Chem. Soc.
1981. V. 103, № 13. P. 3659-3666.
432. Minakata M., Kitawaga S., Yagi F. Cadmium-113 NMR of Cadmium(II)
Complexes with Ligand Containing N-donor Atoms. Dependence of Chemical
Shift upon the Ligand, Baseing Chelate Ring Size Counterion and Cadmium
Concentration // Inorg. Chem. 1986. V. 25, № 7. P. 964-970.
433. Colton R., Dakternieks D. Cadmium-Ill, cadmium-113 and mercury-199
NMR studies of tetrahalometallate complexes // Aust. J. Chem. 1980. V. 33,
№ 11. P. 2405-2407.
434. Dean P.A. A ll3Cd NMR study of some complexes of cadmium with phos-
phine oxides, sulfides, and selenides // Can. J. Chem. 1981. V. 59, №23.
P. 3221-3225.
435. Carson G.K., Dean P.A. A selenium-77 and cadmium-113 NMR spectro-
scopic study of phenylselenolate complexes of cadmium // Inorg. Chim. Acta.
1982. V. 66, № 1. P. 37-40.
436. Banda R.M.H., Dance LG. Bailey et al. Cadmium Polysulfide Complexes.
[Cd(Sx)(Sy)]2-: Synthesis. Crystal and Molecular Structures, and 113Cd NMR
Studies // Inorg. Chem. 1989. V. 28, № 10. P. 1862-1871.
437. Земскова C.M., Глинская Л. А., Клевцова Р.Ф. и др. Строение и свойства
моно- и гетерометаллических комплексов кадмия, цинка, никеля, содержа-
щих диэтилдитиокарбамат-ионы и молекулы этилендиамина // Ж. структ.
химии. 1999. Т. 40, №2. С. 340-349.
438. Abrahams B.F., Winter G., Dakternieks D. NMR Studies of Anionic Cad-
mium and Mercury 1,1-Ditiolate Complexes // Inorg. Chim. Acta. 1989.
V. 162, №2. P. 211-216.
439. Kostelnik R.J., Bother-By A.A. Cadmium-113 NMR Studies of Cad-
mium(II)-ligand Binding in Aqueous Solutions // J. Magn. Reson. 1974.
V. 14, №2. P. 141-151.
440. Jakobsen H.J., Ellis P.D. Solution structure of cadmium-glycine complexes
probed by cadmium-113 NMR of supercooled aqueous solutions // J. Phys.
Chem. 1981. V. 85, № 12. P. 3367-3369.
441. Ackerman M.J. B., Ackerman J.J.H. ll3Cd Chemical shift of Cadmium-Iodide
Complexes in Supercooled Aqueous Solution // J. Amer. Chem. Soc. 1985.
V. 107, №22. P. 6413-6414.
442. Dessy R.E., Flautt T.J., Jaffe H.H., Reynolds G.F. NMR spectra of some di-
alkyl mercury compounds // J. Chem. Phys. 1959. V. 30, № 6. P. 1422-1425.
443. Schneider W.G., Buckingham A. D. Mercury, thallium and lead resonances //
Disc. Faraday Soc. 1962. №34. P. 147-185.
444. Стреленко Ю.А. Спектроскопия ЯМР 199Hg высокого разрешения
ртутьорганических соединений: Автореф. канд. дис. — М.: МГУ, 1981;
Стреленко Ю. А. ДАН СССР. 1980. Т. 252, № 4. С. 924-927.
445. Kennedy J.D., MeFarlane W. Studies of 199Hg nuclear shielding anisotropyes
and their relation to isotropic chemical shifts // J.C.S. Faraday tr. II. 1976.
V. 72, №9. P. 1653-1659.
446. Kruger H., Lutz 0., Nolle A., Schwenk A. Fourier transform NMR studies
of 199Hg // Z. Phys. A. 1975. V. 273, № 4. P. 325-330.
447. Goldfrey P.D., Heffernan M.L., Kerr D.F. 199Hg wide line NMR spectra
of chlorine complexes of mercury // Aust. J. Chem. 1964. V. 17, K° 6.
P. 701-704.
448. Goodfellow R.J., Stobard S.R. Heteronuclear magnetic double resonance
spectra of some tin and mercury cyclopentadienyl derivative: characteristics
of fast exchange // J. Magn. Reson. 1977. V. 27, № 1. P. 143-146.
449. Maciel G.E., Borzo M. Pulse Fourier Transform 199Hg // J. Magn. Re-
son. 1973. V. 10, № 3. P. 388-390.
450. Cradok S., Ebsworth E.A., Hosmane N.S., Mackay K.M. Disilylmercury and
Digermylmercury // Angew. Chem. Int. ed. 1975. V. 14, №2. P. 167-168.
451. Albright M.J., Schaaff T.F., Hovland A. K., Oliver J. P. Metal-silicon bonded
compounds. A 199Hg FT NMR study of some silylmercury derivatives and
selected organomercury compounds // J. Organomet. Chem. 1983. V. 259,
№ 1. P. 37-50.
452. Peringer P. l99Hg NMR studies of the mercury (II) halides // Inorg. Chim.
Acta. 1980. V. 39, № 1. P. 67-70.
453. Colton R., Dakternieks D. Phosphorus-31 and mercury-199 NMR studies on
mercury (II) halide-tributyl phosphine complexes // Aust. J. Chem. 1980.
V. 33, № 5. P. 955-963.
454. Colton R., Daklernieks D. Phosphorus-31. cadmium-113 and mercury-199
NMR studies of cadmium halide and mixed cadmium(II) - mercury(II)
halide complexes with tributylphosphine // Aust. J. Chem. 1980. V. 33, №8.
P. 1677-1684.
455. Bond A.M., Colton R., Dakternieks D., Hank K. W. Comparison of electro-
chemical data for induction of mercury(II) dihalide diphosphine complexes in
dichloromethane and mercury-199 and phosphorus-31 NMR data // Inorg.
Chem. 1982. V. 21. P. 117-122.
456. Colton R., Dakternieks D. Phosphorus-31 and mercury-199 NMR studies of
mercury(II) complexes with some phosphines // Aust. J. Chem. 1981. V. 34,
№ 2. P. 323-333.
457. Goggin P.L., Goodfellow R.J., McEvan D.M., Kessler K. NMR studies (31P
and l99Hg) of complexes Hg2X^ (PBuJ^. (X - Cl. Br. I) and their reactions
with NBuJX // Inorg. Chim. Acta. 1980. V. 44, №3. P. L111-LU2.
458. Colton R., Daklernieks D. Phosphorus-31. mercury-199 and selenium-77
NMR studies of ligand exchange reactions in mercury(II) halide complexes //
Aust. J. Chem. 1980. V. 33, №7. P. 1463-1470.
459. Федоров Л.А. Спектроскопия ЯМР металлоорганических соединений. —
М.: Наука, 1984. - 248 с.
460. Bain R.L. Multinuclear NMR studies of the system: mercuric
cyanide-mercuric ion-heavy water in perchlorate media // J. Inorg. Nucl.
Chem. 1981. V. 43, № 10. P. 2481-2483.
461. Peringer P. Mercury-199 NMR studies of mixed cyanomercury compounds //
Inorg. Nucl. Chem. Lett. 1980. V. 16, №4. P. 206-209.
462. Dakternieks D. 31P and 199Hg Studies of some Mercury(II) Complexes Con-
taining Tricyclohexylphosphine and Tributhylphosphine // Inorg. Chim. Acta.
1984. V. 89, №3. P. 209-211.
463. Bond A.M., Colton R.t Elner J. Multinuclear Magnetic Resonance (31P, '7Se,
l99Hg) and Electrochemical Studies of Nonlabile Mercuty(II) Complexes with
Group 15/Group 16 Donor Ligands // Inorg. Chem. 1988. V. 27, №10.
P. 1697-1702.
464. Земскова C.M., Селезнева И.М., Федотов M.A. и др. Синтез, строение
и термические свойства разнолигандных диэтитдитиокарбаматогалогенид-
ных комплексов цинка(П), кадмия(П), ртути(П) // Сиб. хим. журнал. 1991.
Вып. 1. С. 115-121.
465. Helm M.L., Helton G.P., Van Derveer D.G., Grant G.J. Mercury-199 NMR
Studies of Thiacrown and Related Complexes: The Crystal Structures of
[Hg(18S6](PF6)2 and [Hg(9S3)2]C104 // Inorg. Chem. 2005. V. 44, №16.
P. 5196-5705.
466. Burns R.C., Devereux L.A., Granger P., Schrobilgen G.J. Prepapation and
Multinuclear Magnetic Resonance Study of the Zintl Anion HgCh22- (Ch =
Se and / or Те). Chemical Shift and Coupling Constant correlatrion // Inorg.
Chem. 1985. V. 24, № 17. P. 2615-2624.
467. Логинова O.H., Пещевицкий Б.И., Федотов M.A. Комплексообразование
двухвалентной ртути с цистином // Изв. СО АН. Сер. хим. 1987. № 1,
вып. 1. С. 61-64.
468. Fraginals R., Barba-Behrens N., Contreras R. Study of Interaction of Lewis
Bases with Мегсигу(П) Chloride and Mercury(II) Acetate by Mercury-195
NMR // Spectrochimica Acta. 1989. V. 45A, №5. P. 581-584.
469. Wasylishen R.E., Lenkinski R.E., Roger C. Mercury-199 NMR relaxation
in some mercury(II) compounds // Canad. J. Chem. 1982. V. 60, № 16.
P. 2113-2117.
470. Heinmann P.A., Greenwood I.A., Simpson J.H. Quadrupole perturbation
effects upon the 199Hg magnetic resonance. Relaxation due to an anisotropic
perturbation // Phys. Rev. A. 1981. V. 23, №3. P. 1209-1214.
Глава 4
НЕКОТОРЫЕ ПРИЛОЖЕНИЙ
МЕТОДА ЯМР
4.1. ЯМР в аналитической химии
Определение концентраций элементов и соединений. С
ача метода ЯМР — определение форм в растворе (жидкое-
эения, при этом определяется соотношение интенсивносте
гментов молекулы, например групп СН3 : СН2 : СН : ОН в
ртов, что входит уже в сферу аналитической химии. Дл
метод ЯМР особенно нагляден, поскольку в спектре ЯМ)
гветствуют различным окружениям выбранных атомов (г
ого изотопа), причем интегральная интенсивность линий
/ет числу атомов в каждом окружении, т. е. наблюдаемые
цстают в их мольном отношении. Пропорциональность
центрации ядер выбранного элемента, исключительная изб
ть (виден только выбранный изотоп, а линия соответствует
ному окружению), слабое влияние состава раствора, конце!
грогенных примесей, возможность измерений в широком Д1
овий делают привлекательным использование ЯМР в ан;
й химии. Хотя основной задачей жидкофазного ЯМР i
яется определение строения и некоторых свойств форм в
1], попутно метод использовали как аналитический, опред
целевого продукта и/или количество примесей или относи
эсолютные концентрации форм.
ЯМР как аналитический метод применяется более 50 лет
для решения задач анализа органических веществ (ЯМР
имии фтора (19F), затем, по мере увеличения чувствите
ктрометров, и других элементов на ядрах их изотопов.
Обзор литературы по ЯМР до 1982 г., касающейся анализе
еских растворов, сделан в [3]; имеются соответствующие 1
1 [4, 5], более поздняя оценка возможностей ЯМР для элег
Для оценки концентраций можно использовать сигнал ССИ, ам-
плитуда которого в его начале пропорциональна сумме интенсивностей
всех линий спектра, т. е. содержанию изотопа в образце, сигнал ко-
торого наблюдают. Амплитуда сигнала для датчика с соленоидальной
катушкой пропорциональна факторам [9]
+7W+1Wo/2(?0 /лп
Vs = const —i' ° (4.1)
Kl
(7 — гиромагнитное отношение ядра, Hq — магнитное поле спектро-
метра), откуда видно, что она пропорциональна числу ядер в катушке
датчика N, фактору заполнения 0 и добротности катушки датчика Q.
Величина 7 входит в параметр «чувствительность ядра»; очевидно, что
с ростом Но измерение амплитуды сигнала легче.
Фактор 0 будет тем же при одинаковых ампулах образца и эталона,
однако добротность контура Q зависит от свойств образца, в первую
очередь от его проводимости. Если спектрометр снабжен генератором
качающейся частоты для настройки датчика (воблером), то по кривой
поглощения воблера можно оценить изменение добротности контура с
образцом и скомпенсировать изменение величины Q соответствующим
коэффициентом, хотя точность определения Q таким способом вряд
ли лучше 20%. Кроме того, поскольку измерения ведутся на высокой
частоте, возбуждающее поле Н[ может ослабляться внутри образца по
сравнению с его внешним слоем, и из-за того что высокочастотное поле
вытесняется в проводящем образце к его периферии (скин-эффект).
Последний фактор преодолевается подбором эталона с проводимостью
такой же, как у образца. В случае коаксиального размещения эталона
требуется калибровка эталона образцом с известной концентрацией и
той же проводимостью, что и у измеряемых образцов.
Погрешности возрастают при оценке концентраций линий широ-
ких линий с коротким спадом ССИ; поскольку регистрация сигнала
начинается с некоторой задержкой после возбуждающего импульса.
В результате наблюдаемая амплитуда может значительно отличаться от
истинной. Поэтому при большом различии свойств образца и эталона
ошибка измерения может быть 50% и более. Источники ошибок при
количественных измерениях рассмотрены в [12].
Как видно из формулы (4.1), амплитуда сигнала ССИ пропорцио-
нальна количеству спинов в катушке датчика, т. е. их концентрации
в единице объема. Поэтому концентрацию элемента можно определить:
а) прямым измерением амплитуды сигнала ССИ изотопа данного
элемента и сравнением ее с сигналом эталона,
б) измерением образца вместе с эталоном (внутренним или внеш-
ним) и сравнением интегральных интенсивностей линий в спектре,
в) связыванием определяемого элемента в инертный в шкале ЯМР
комплекс и определением концентрации по спектру ядра лиганда.
Как показывает практика, точность определения концентраций ме-
тодом ЯМР сравнительно невелика (5-50% по ССИ, 2-30% по соот-
ношению линий в спектре при отсутствии насыщения и неравномерно-
сти возбуждения спектра), однако во многих случаях "такая точность
достаточна; в то же время сама процедура измерений и подготовки
растворов значительно упрощается по сравнению с подготовкой пробы
и последующим химическим анализом. Задачи вскрытия пробы такие
же, как и для химического анализа, поэтому они здесь не обсуждают-
ся. По скорости анализа метод можно отнести к экспрессным.
Наиболее просто измерить концентрацию по амплитуде ССИ.
В этом случае последовательно измеряют амплитуды ССИ образца
и раствора с определенной концентрацией (в идентичных ампулах,
с одинаковым растворителем). Водные растворы электролитов сильнее
поглощают ВЧ-поле, что уменьшает фактор Q (формула (4.1)), и для
уменьшения погрешности измерения надо подбирать концентрацию
электролитов эталона близкой к концентрации образца. Поскольку
точность измерения с учетом указанных факторов обычно не лучше
5%, измерения амплитуд на экране монитора не ухудшает общей по-
грешности. В то же время при достаточно долгом времени релаксации
и подборе эталона измерения на экране дают точность лучше 5%.
Концентрацию можно определить и по соотношению интегральных
интенсивностей линии вещества и эталона. При измерении интен-
сивностей с внутренним эталоном последний растворяют в образце
в сравнимых с определяемой формой количествах; эталон должен
быть инертным по отношению к компонентам раствора. В спектре
линии эталона не должны совпадать с линией определяемой формы.
В этом случае изменение добротности и неточность настройки датчика
одинаково сказываются на сигналах эталона и образца и тем самым
исключаются.
Как правило, в неорганических растворах найти инертный раство-
римый эталон для ядер, иных, чем *Н и 13С, трудно, поэтому обычно
используют внешний эталон, наливая его в 5-миллиметровую ампулу,
которая коаксиально вставляется в 10-миллиметровую ампулу с образ-
цом (рис. 4.1). В спектре сравниваются интенсивности линий образца
и эталона, при этом точность измерения можно повысить в 2-3 раза.
Для некоторых ядер трудно подобрать эталон с близкой к образцу
и удобной для накопления линией, например, в спектрах 7Li. В этом
случае можно использовать для сравнения более концентрированный
раствор того же образца в коаксиальной ампуле, добавив в него па-
рамагнитный ион (Fe3+, Сг3+, Мп2+) до концентрации ~ 0,1 г-ион/л,
а б
Рис. 4.1. Сравнение концентраций ядер образца и эталона с неразрешенными
линиями: а — расположение ампулы в датчике (К — катушка датчика, О —
образец); б — использование коаксиальных ампул при налагающихся линиях
образца и эталона (Э — эталон); спектр ЯМР (/) и его интеграл (2) при
парамагнитной добавке в эталоне
тогда линия образца раздвоится (см. рис.4.1) и интенсивности линий
образца и эталона легко определить.
Практически все элементы имеют стабильные магнитные изотопы,
однако удобные для измерения концентраций около 20. Часть элемен-
тов находятся в растворе в состоянии, невозможном для детектиро-
вания. В этом случае используют подходящий для данного элемента
(соединения) лиганд и определяют концентрацию закомплексованного
лиганда [4].
Ниже приведены изотопы, удобные для измерений концентраций
(полужирный шрифт), редко применяемые для этого, и изотопы, полу-
чить сигнал ЯМР которых достаточно трудно (курсив).
'Н, 7Li, 9Ве, "В, 19F, 23Na, 27Al, 3|Р, 35С1, 45Sc, 5IV, 55Mn, 59Co, 8lBr,
93Nb, "Tc, "3Cd, ll5In, l33Cs, 139La, 2O5T1
13C, '“N, 39K, 53Cr, 65Cu, 67Zn, 87Rb, ll9Sn, 207Pb
2D, 15 N, 17 O, 25Mg, 29Si, 33S, Ca, *9Ti, 57Fe, 61 Ni, 89Sr, 89 У, 91Zr, 95Mo, 99 Ru,
103Rh, * 103 * 105Pd, 137Ba, mHf, ™Ta, 183 W, 185 Re, 187Os, 193/r, 197 Au, >99Hg, 209Bi
Переходные элементы в определенной степени окисления парамаг-
нитны, у других мала чувствительность (например, Y, Rh), и их кон-
центрации определяют по ядрам лиганда после образования с ними
комплекса. Ряд лигандов для этого испытан в [4]. При комплексообра-
зовании наибольшие сдвиги относительно свободного состояния имеют
донорные атомы (так называемый координационный сдвиг). Лиганды
с О- и N-донорными атомами малопригодны для этого, поскольку
ширина линий ЯМР 17О и 14N при комплексообразовании резко уве-
личивается и их интенсивность измеряется с большой ошибкой. Более
удобны фосфорные лиганды, а также регистрация ЯМР 13С ближайше-
го к донорному атома С. Для ионов лантаноидов и актиноидов удобны
как N-лиганды (NTA с регистрацией 'Н), так и полиядерные тетра-
дентатные лиганды — дефектные гетерополианионы (ГПА) [WsOis]6-,
[PWi 1 Озэ]7- и [Р2W17O61 ]11 -, которые образуют с первыми комплексы
1 : 2 (рис. 4.2) [13-15].
Рис. 4.2. Схема строения комплекса ионов РЗЭ с ГПА [Z^W^Oei)г]17
В лиганде [P2W17O61)] |0“ (который существует как таковой в вод-
ном растворе) один атом Р расположен близко к иону РЗЭ и имеет
большой парамагнитный сдвиг, а другой — дальше от него, мало
меняющий ХС и служащий репером. При избытке P2W17 все ионы РЗЭ
находятся в закомплексованном виде и их относительное содержание
пропорционально интенсивностям линий (рис.4.3 [4]). Определение
Рис. 4.3. Определение концентраций РЗЭ по спектру ЯМР 31Р
концентраций лантаноидов в их смеси с помощью комлексообразования
с NTA показано на рис. 4.4.
Рис. 4.4. Определение концентраций РЗЭ по спектру ЯМР 'Н комплексоната
с нитрилотриацетатом (NTA)
Данные комплексов с ГПА PWnO^g приведены в табл. 4.1.
Таблица 4.1
Химические сдвиги ЯМР 31Р комплексов [Ln(PWi ] Оз9)2]1, Ln — лантаноид
[78]
Ln La Се Рг Nd Sm Eu Gd
8, м. д., 293 К -12,0 -19,3 -14,6 -24,6 -15,4 -1,2 —
323 К — -17,6 -15,2 -22,1 -14,8 -2,4 -3(4000) *
Ln Tb Dy Но Ег Tm Yb Lu
5, м. д., 293 К -223 -108 -109 61,4 217 32 -13,2
323 К -169 -98 -90 48,8 164 26,1 —
* Указана ширина линии, Гц.
Метод ЯМР принципиально изотопный, и его используют в этом
качестве в лабораториях и промышленности для определения кон-
центрации и соотношения изотопов в соединениях (например, 1H/2D,
6Li/7Li и др.). При определении примеси одного изотопа в соединении
с преобладанием другого изотопа, как например 2D в НгО или 'Н
в D2O (и то, и другое имеет практическое значение, в том числе
и для самого метода ЯМР), по ССИ точность составит 5-10% для
2D в НгО при содержании ~ ICC4 моль/л, а для ’Н в D2O — при
содержании 'Н ~ 10-6. Для определения отношения Н/D в воде ис-
пользован изотопный эффект в ЯМР 59Со комплекса [Со(МНз)б]3+ [12]
(см. рис. 3.26). В другом методе определения Н/D используется гидра-
тация ионов и различие ХС ионов в Н2О и D2O, и ХС иона зависит от
Н/D. Наибольшую чувствительность имеет ЯМР l9F в растворах HF
и KF [17].
Для определения концентрации парамагнитных ионов развит маг-
нито-релаксационный метод [5]. Из-за резкого различия времен релак-
сации молекул растворителя в сольватной сфере и свободном состоя-
нии и быстром обмене молекул между этими состояниями измеряемая
скорость релаксации R пропорциональна концентрации парамагнитных
ионов в растворе в широком интервале концентраций:
Л1.2 = <х [М₽]. (4.2)
-1 1,2
Систематическое изложение методик измерения концентраций, изу-
чения конкурирующей сольватации приведено в [5]. Чувствительность
метода ЯМ-релаксации сравнима с чувствительностью других методов
(табл. 4.2), а в некоторых условиях превышает ее. В полярных рас-
Таблица 4.2
Чувствительность разных методов определения концентрации ионов [5]
Метод Чувствительность, г-ион/л
Потенциометрический IO-4
Фотометрический КГ6
Поляриметрический 1(Г5
ЯМ-релаксация 3- 10“7
Спектрофотометрия 5- 10“8
творителях она может на 2-4 порядка превышать чувствительность
метода ЭПР и магнитной восприимчивости. Метод не требует особой
подготовки образца (кроме случаев низких концентраций, когда необ-
ходимо удалять из образца растворенный кислород); мало чувствите-
лен к гетерогенности и солевому фону образца; может встраиваться
в технологическую линию; в некоторых случаях это может иметь
решающее преимущество. Недостатком метода является то, что он
пригоден для растворов определенного парамагнитного иона или их
смеси постоянного состава. Он не пригоден для определения какого-то
иона из смеси парамагнитных ионов, если неизвестен состав этой
смеси.
Другим приложением метод ЯМ-релаксации является определение
доли одного из компонентов в сложных продуктах без выделения этого
компонента. При наличии двух фаз с резко отличающимися времена-
ми Т<2, долю этих фаз в образце можно определить по ССИ.
Изменение логарифма амплитуды спада свободной индукции в от-
сутствии насыщения показано на рис. 4.5. Отрезок А соответствует
Рис. 4.5. Определение соотношения твердой (А) и жидкой (В) фазы в образце
маргарина по ЯМ-релаксации 'Н
доле фракции с быстрым затуханием («твердая» фаза). Время получе-
ния данных соответствует экспресс-анализу [II].
Подобные измерения имеют перспективу в пищевой промышленно-
сти, а также используются в ЯМ-томографии для увеличения контра-
ста отдельных тканей.
Применение метода ЯМР для измерения кислотности. Для
сравнения силы кислот в растворах используют в основном два метода:
1) определение констант протонной диссоциации и 2) индикаторный
метод, основанный на разной способности кислот протонировать осно-
вание:
В + Н+^ВН+. (4.3)
По измеренному методом оптической спектроскопии соотношению
[ВН+]/[В] определяется функция кислотности Гаммета Hq как крите-
рий кислотности раствора.
Первый метод применяется для разбавленных растворов, второй
испытывает трудности, например, при определении силы окрашенных
кислот или при оптическом поглощении компонентами раствора. Осо-
бые сложности возникают при определении кислотности ГПК — по-
лиоксосоединений молибдена, вольфрама, ванадия и некоторых других
элементов, которые составляют обширный класс минеральных кис-
лот [18].
Давно предпринимались попытки использовать метод ЯМР, а имен-
но ЯМР 1Н, для определения концентрации протонов; его применение
основано на том, что ХС протонов гидроксония НзО+ (18,8 м.д.) [19]
отличается от ХС воды (4,8 м.д.), а обмен протонами — быстрый
в масштабе времени ПМР. В результате положение наблюдаемой линии
является средневзвешенным [9]:
<5наб=р(а<5| + |(1 -а)52) +2(1 -p)W (4.4)
где р — За;, х — мольная доля кислоты, 5; — ХС гидроксония, <52 —
ХС моногидрата кислоты, а — степень диссоциации кислоты. Здесь
предполагается гидратированная форма протона в виде Н3О+. Фор-
мула (4.3) учитывает и влияние аниона кислоты. Такие работы по
оценке кислотности активно велись в 1960-1970 гг. и цитированы
в [19, 20]. Формула (4.4) предполагает гидратированный протон только
в виде гидроксония, в то время как найдены ди- и более гидратиро-
ванные формы гидратированного протона с другими, чем гидроксоний,
ХС. Применение ПМР для определения кислотности осложняется при
наличии в растворе других катионов и анионов, поскольку каждый
компонент дает свой вклад в ХС протонов раствора [21].
Для определения кислотности можно использовать данные ЯМР
индикаторов или молекул субстрата, если они являются слабыми ос-
нованиями. Оценка изменения параметров ЯМР 14N и 17О некоторых
оснований при изменении pH от 1 до 12 сделана в [22], оценка ос-
новности спиртов методом ЯМР 13С сделана в [23], гексаметилбензола
в [24], оценка основности ряда кетонов методом ЯМР 'Н — в [25].
Замечено, что в водных растворах H2SO4 ХС примесного нитрата
линейно связан с функцией кислотности Гаммета Hq [26]:
H0 = pKBH+-\gC^, (4.5)
С'В
где и Св и Свн-ь — концентрации основания и его протонированной
формы.
В [27] для определения кислотности методом ЯМР 13С предложен
индикатор мезитилоксид, у которого кето-енольное равновесие меняет-
ся в зависимости от степени протонирования по уравнению:
Ме2С2=С‘Н—С—Me + АН Ме®С2-С‘Н=С-Ме + А"
О ОН
Ме2С2=С‘ Н-С-Ме . (4.6)
ОН
Разность ХС 13С атомов С1 — С2 меняется на ~50 м.д. при переходе
от нейтрального раствора к 100% H2SO4.
В [28] предложены и сделаны измерения кислотности ряда кислот
(НСЮ4, H2SO4, Н3РО4) с помощью индикаторов алкенонов R-C(O)-
CH=CRIR2, кето-енольные равновесия которых зависят от степени про-
тонирования.
Возможности ЯМР тяжелых ядер для определение кислотности
ГПК по параметрам ядер индикатора Гаммета ортонитроанилина
(ОНА) и молекул субстрата изучены в [29]. Там определены параметры
ЯМР 13С, l4N ОНА. Метод часто применялся для определения кислот-
ности минеральных кислот и ГПК, а также растворителей (10% воды
в ацетонитриле и 10% воды в уксусной кислоте), часто используемых
для проведения каталитических реакций. На рис. 4.6 приведена зави-
Рис. 4.6. Зависимость ХС ЯМР 13С атомов opmo-анилина от кислотности
в растворах CF3SO3H в СНзСООН/Ю% HjO (номера кривых соответствуют
номерам атомов в молекуле орто-нитроанилина)
симость ХС 13С от концентрации «сверхкислоты» CF3SO3H в растворе
90% уксусной кислоты. Для измерения кислотности логично исполь-
зовать разность ХС атомов С2 и С1, компенсируя тем самым влияние
растворителя и другие факторы.
На рис. 4.7 приведена зависимость ХС азота ортонитроанилина от
концентрации трифторсульфоновой кислоты. Из-за значительной ши-
рины линии точность определения кислотности по 14N ниже, чем по
13С, однако время измерения спектров азота в 2-3 раза меньше. Вид
nh2
Рис. 4.7. Зависимость ХС ЯМР l4N нитрогруппы (/, 3) и аминогруппы (2, 4)
в растворах CF3SO3H в СНзСООН/Ю% Н2О (/, 2) и воде (3, 4)
спектра ЯМР 14N ОНА (0,1 М) в ацетонитрильных растворах CF3SO3H
приведен на рис. 4.8. Видно, что протонируется группа NH2: линия
NH2 с ростом кислотности сдвигается и сужается из-за повышения
симметрии окружения азота вследствие протонирования.
Рис. 4.8. Спектр ЯМР 14N 0,1 М раствора ор/гео-нитроанилина в ацетонитриле
при 50 °C; а - 0,1 М CF3SO3H; б - 1 М CF3SO3H
С той же целью изучен и другой индикатор — 2,6-диметил-7-пиррол
(ДМП) [30]. В растворах ГПК и ОНА, и ДМП обнаружили дополни-
тельный по сравнению с минеральными кислотами вклад в ХС атомов
индикаторов из-за взаимодействия индикатора с ГПА и, строго говоря,
непригодны для определения кислотности ГПК.
4.2. Изучение комплексообразования
и структуры комплексов
Диамагнитные комплексы. Методом ЯМР в принципе можно
определить состав, строение комплекса, константу образования ком-
плекса, энтальпию и энтропию активации комплексообразования. Ре-
альные подходы и определяемые параметры зависят, во-первых, от
времени жизни комплекса и, во-вторых, от параметров ЯМР ядер
конкретной системы. Для установления состава стабильных в шкале
времени ЯМР комплексов используют те параметры (5, J, Т\, Т2},
которые зависят от состава комплекса. Эта задача, в принципе, не
отличается от задачи установления строения органической молекулы,
но чаще используют иные, чем 'Н и В * * * * 13С, ядра. Если удается наблю-
дать ССВ на ядре комплексообразователя от ядер донорных атомов
лигандов, то однозначно устанавливается число этих лигандов. Это
возможно с Н-, F-, 15N-, Р-лигандами. В случае лабильного комплекса
добиться наблюдения КССВ можно снижением температуры. Диапа-
зоны времен жизни форм т, измеряемые различными экспериментами
ЯМР, представлены на рис. 4.9.
105 1 03 10 1 0,1 Ю-3 10“5
__I___________I___________I_____I_____I__________I___________1_
|Анализ формы линии 13С,14 Nj
(Анализ формы линии 'Н
. (Двойное облучение 13С,’ Н|
(Flow эксперимент Н ।
|Flow-stop эксперимент 1Н,14 N|
(Установление равновесия
||3С (Установление равновесия
|17О,15 N (Установление изотопного равновесия
Рис. 4.9. Диапазоны времени жизни, измеряемые различными методами ЯМР.
Для других ядер, чем указанные, возможно измерение и более коротких времен
жизни
В том случае, когда природное содержание магнитного изотопа
лиганда менее 15%, в составе комплекса в среднем оказывается не
более одного лиганда с данным ядром; тогда число лигандов мож-
но определить по интенсивности боковых компонент, как, например,
в случае лиганда SnCl^.
В том случае, когда металл или лиганд наблюдаются раздельно
в комплексе и свободном виде, можно определить константу образо-
вания комплекса. Для этого по интенсивностям линий ЯМР опре-
деляют равновесные концентрации форм. Применение комьпьютерных
программ для определения констант стабильности из ХС и параметров
ЯМ-релаксации обсуждается в [31].
Во многих случаях комплекс остается лабильным при разных усло-
виях эксперимента, и в этом случае параметры ЯМР будут усреднен-
ными от их значений в комплексе и свободном состоянии, а константы
ССВ часто не наблюдаются вообще.
При образовании лабильного комплекса 1 : 1 по реакции
M + L-^ML (4.7)
в условиях быстрого обмена по зависимости ХС от соотношения M/L
можно определить ХС ядра в комплексе и константу образования
комплекса к [33]. Теоретическая зависимость наблюдаемого ХС от
IgL-lgM Ig/cMo
Рис. 4.10. Зависимость наблюдаемого ХС <5на6 от соотношения М : L при быст-
ром обмене в условиях равновесия М + L <-> ML (а) и параметра Л от кМ0 (б)
Равновесные концентрации металла, свободного лиганда и комплек-
са (М, L и ML соответственно) при общей концентрации металла Mq
и лиганда Lq связаны соотношениями
Мо = М + L, Lq = L + ML, ML = к М L, (4.8)
и наблюдаемый ХС ядер металла:
гнаб ___ г ।
х
X {1 + k(L0 + Мо) - ^[1 +fc(Mo + Lo)]2-4fc2MoLo }, (4.9)
где 6К и <5ml — ХС металла в отсутствие лиганда и в составе комплекса.
Выражения для любого ядра лиганда можно получить заменой в (4.8)
8п на 6L.
Наклон кривых <5наб при Lo —> О
<ггб\ -k5^~S'-'
dLo у £_>() 1 + кМд
(4.Ю)
Величина <5ML не определяется непосредственно в эксперименте.
Ее можно получить из (4.9), сняв концентрационные зависимости при
существенно различных Mq. Низкая чувствительность метода ЯМР
ограничивает диапазон изменений Мо- Анализ кривизны линий на
рис. 4.10 по двум точкам А (при Lo = Мо) и В (при Lo = 2М0)
позволяет исключить <5ML и оценить к:
А = 6"аб -6п = (1 + 2кМ0 - V1 + 4Ш0), (4.11)
£K1v1q '
В = йвнаб = (1 + - 71 + 6Шо + ™о) (4-12)
ГО J Кх Q \ V J
Л _ А - 0,5В _ j В _ j 1 + 3fcM0 - У1 + 6fcM0 + k2Ml >
А 2А ~ 2(1 +2fcM0- -/1 +4Ш0
(4.13)
Из графиков рис. 4.10 видно, что к можно определить, когда Л
находится в пределах значений 0,1-0,4, т. е. в диапазоне кМо от 0,1
до 100. Погрешность определения ЛДЛ/Л ~ (ДА/А + ДВ/В)/Л. Вне
диапазона 0,05 Л 0,45 можно указать только границу величины
кМо. Более точный результат можно получить подгонкой расчетной
кривой с варьируемыми к и ML к экспериментальной, для чего создан
ряд программ. С использованием численных методов можно в благо-
приятных случаях определить наиболее вероятный тип образующегося
комплекса. Естественно, при определении параметров комплексообра-
зования необходимо точно выдерживать концентрации реагентов и тем-
пературу измерения.
Исследуя температурную зависимость параметров ЯМР, можно
определить параметры активации обмена лигандов ДВМ и ДВ^. Для
систем, у которых концентрационная зависимость ХС мала, более ин-
формативными параметрами являются времена релаксации. При иссле-
довании состояния ионов и молекул в водных растворах ЯМР на ядрах
ионов, растворенных молекул и самой воды !Н, 2D и 17О дают вза-
имодополняющую информацию. Высокая концентрация молекул воды
позволяет проводить измерения на ядрах всех указанных изотопов при
их природном содержании, а доступность D2O — менять соотношение
Н/D произвольно.
Замена *Н на 2D позволяет разбавить протоны и снять межмолеку-
лярный вклад в параметры ЯМР. При химическом обмене вклады за
счет различия частот ЯМР обменивающихся состояний в 7h/?d ~ 6,56
раза меньше у 2D, чем у 1Н, что позволяет определить эти вкла-
ды. Сравнение параметров ЯМР 17О и 'Н (2D) позволяет различить
обмен молекул воды и протонный обмен. Набор данных всех ядер
дает возможность критически проверить различные модели ближнего
окружения молекул и ионов (рис. 4.11). В приближении бесконечного
Рис. 4.11. Модель ближнего окружения ионов А- и К+ при их взаимодействии
в полярном растворителе. Стрелками показано направление дипольного момен-
та молекул воды
разбавления параметры ЯМР будут зависеть только от взаимодействия
с молекулами воды, при этом макросвойства воды не отличаются от
свойств чистой воды. Для большей части ядер щелочных и щелочно-
земельных металлов наиболее чувствительными параметрами ЯМР яв-
ляются времена релаксации. По ЯМ-релаксации можно определить ха-
рактер окружения иона. Большинство ядер ионов имеет квадрупольный
момент, и механизм их релаксации в основном квадрупольный. В слу-
чае чисто квадрупольного механизма релаксация зависит от градиента
электрического поля на ядре (ГЭП) и времени корреляции вращения
иона тс. Но ионы щелочных и щелочноземельных элементов имеют
конфигурацию инертных газов со статическим ГЭП = 0, и в растворах
ГЭП имеет динамический характер.
Существуют разные модели возникновения ГЭП: в [34] предложили
модель возникновения за счет искажения комплекса из-за соударений
с молекулами растворителя (динамическое искажение). В этой модели
трудно оценить время межмолекулярных столкновений; ей также про-
тиворечит наблюдение максимумов в концентрационных зависимостях
в смешанных растворителях [34]. В [35-39] предложена модель воз-
никновения ГЭП из-за искажения окружения в момент отрыва лиганда
от иона, т. е. образования комплекса с координационным числом на 1
меньше, чем обычно, и других статистических искажений (обзор работ
в [40, 41]). В рамках этой модели можно представить разные случаи:
а) полностью случайное распределение молекул около иона (СР),
б) неориентированная сольватация (НС),
в) полностью ориентированное расположение молекул около
иона (ОР).
При бесконечном разбавлении и полной диссоциации соли ион-
ионными взаимодействиями можно пренебречь и ЯМ-релаксация опре-
деляется только взаимодействием с растворителем:
/?! = ! = 67Г(2/ + 3) [ре^1 +7oo)N (4 14)
' 5/г2/2(2/+1)
где р= (2е + 3)/(5е) — фактор поляризации, е — диэлектрическая
проницаемость, Q — квадрупольный момент, 7ТО — фактор антиэкра-
нирования Штернхеймера, d определяется в зависимости от модели
сольватации.
Для модели СР 2
d = dl = —Cs50|T501, (4.15)
Го
где т — дипольный момент молекул растворителя, Csoi и tS0| — кон-
центрация и время корреляции вращательного движения молекул рас-
творителя, го — расстояние минимального сближения иона и молекулы
растворителя. В модели FRD ожидается линейный ход зависимости от
состава растворителя, что на деле не выполняется.
Для модели НС
di = d2 — -Д m2nS0|TS0| + гд. (4.16)
4тгг0
где di — член типа вышеприведенного, но с величиной Tq, соот-
ветствующей второй координационной сфере, nso| — сольватационное
число.
Для случая ОР предполагается, что диполь ориентирован по ра-
диусу комплекса:
d = d3 = —Д m2nS0|TS0i(l - е-6А) + diTg. (4.17)
4тгг
Здесь А — ширина распределения, описывающего отклонение комплек-
са от кубической симметрии (nsoi = 4,6,8).
Рассчитаны [37] времена релаксации квадрупольных ядер ионов
в дипольном растворителе в предположении свободной диффузии ди-
полей молекул растворителя около иона и при том условии, что ион не
образует стабильных комплексов с растворителем. По мнению авторов,
такая модель описывает ЯМ-релаксацию галоген-ионов в водных рас-
творах. При этом
^г = СИ1е0та(1г + 7“)|,№1:. (4.18)
a2h2VkT
где т — дипольный момент молекулы растворителя, 2а — расстояние
ядро-диполь, N/V — число молекул растворителя в единице объема,
т?* - вязкость, С = 2,31 (1,03; 0,246; 0,105) для I = 1 (3/2, 5/2, 7/2)
соответственно. Для раствора 1~ расчет величины Ri дал завышен-
ную в 23 раза величину относительно экспериментальной. Причину
расхождения авторы видели в большой погрешности величины 1 +
и отличии микро- от макровязкости, величину которой использовали
в (4.19). Несколько другая модель обсуждается в [42], где считается,
что галогенид-ион окружен молекулами воды и ГЭП создается за их
счет. С ростом концентрации соли растет вклад катионов, которые
отделены от аниона сольватной оболочкой (см. рис.4.11). В случае
больших катионов возможен вклад прямых взаимодействий анион-ка-
тион. Для объяснения ЯМ-релаксации предложена электростатическая
модель [39], пригодная как для анионов, у которых молекулы воды
около иона почти свободны, так и для сильно гидратированных ионов
(Li+, Na+), у которых окружение считали жестким. Авторы полагали,
что ГЭП возникает из-за искажений электрического поля, создавае-
мого электронами самого иона в области, очерченной ионным радиу-
сом окружающими диполями воды. Дополнительный вклад происходит
из-за отличия молекул НгО от точечных диполей. В случае слабо соль-
ватированных ионов I-, Вг-, С1“ возмущения создают молекулы первой
координационной сферы, в случае прочно сольватированных ионов за
дистанцию взаимодействия принимают радиус первой координацион-
ной сферы, а возмущения создают молекулы второй координационной
сферы. Если вклад от статических искажений отсутствует (ион сфе-
рической симметрии или первая координационная сфера кубической
симметрии), то
д _ 24тг2(2Г + l)[eQm(l + 7оо)]2С1гТ11) ^д,
5Г2(2Г — l)/i2rg ’
где т — дипольный момент Н2О, го — расстояние от ядра иона до
молекул воды, вызывающих возмущение (tq = гион + гв, Св — концен-
трация молекул воды). При наличии координационной сферы вместо
гион используют радиус координационной сферы. Время корреляции
1/тш — \/тТ + 1/т£, где тг — время корреляции реориентации дипо-
ля воды относительно вектора ион-вода, а т* — время корреляции
вращения этого вектора. Расчеты, проведенные по формуле (4.19),
показывают, что модель удовлетворительно описывает ЯМ-релаксацию
в водных растворах при бесконечном разбавлении (табл. 4.3) [39]. При
повышении концентрации соли все больший вклад дает взаимодействие
с противоионами и влияние ионов на структуру воды. Заметим, что
влияние ионов зависит от типа растворителя: с уменьшением полярно-
сти растворителя влияние противоиона увеличивается и может сказы-
ваться при концентрации 0,1 моль/л и ниже. Описание ЯМ-релаксации
катионов и анионов в неразбавленных растворах приведено в [35, 42].
Таблица 4.3
Сравнение расчетных и экспериментальных параметров ЯМ-релаксации в вод-
ных растворах
Ядро, ион Q, барн * 1 + 7оо тв, пм Г*, пм Тв, ПС Л?асч /?ГСП
7Li+ -0,042 0,74 208 — 5 0,037 0,027
23Na+ 0,11 5,1 240 290 2,5 24,2 16,2
39 к+ 0,09 18,3 273 — 2,5 13,3 24
85Rb+ 0,31 48,2 288 — 2,5 435 420
133 Cs+ -0,04 111 307 — 2,5 0,054 0,075
27А13+ 0,15 3,3 — 200 7,5 4,0 7,5
69Ga3+ 0,19 10,5 — 210 7,5 216 350
35СГ -0,079 57,6 321 — 2,5 40 42
в1Вг- 0,28 100 336 — 2,5 1350 1050
127,- -0,75 180 356 — 2,5 5650 5270
* В настоящее время величины квадрупольных моментов уточнены.
Структура первой координационной сферы в водных растворах при
высокой концентрации ионов по ЯМ-релаксации ’Н и 2D изучена в
[43, 44].
Обмен молекул воды между первой, второй координационными
сферами иона и растворителем — быстрый для 'Н и 2D, и Т\ воды
усредняется для этих состояний. В [44] установлено наличие второй
координационной сферы с удвоенным числом молекул по сравнению
с первой. При этом подвижность молекул в первой координационной
сфере ниже, а во второй — выше, чем у свободной воды. Из анализа
температурных зависимостей сделан вывод об увеличении координа-
ционного числа некоторых ионов (Li+, С1_) с 4 до 6 с ростом тем-
пературы. Сравнение координационных чисел, найденных из данных
ЯМ-релаксации протонов, большую точность их определения по срав-
нению с найденными другими методами.
Надежно гидратационное число определяется методом ЯМР в слу-
чае раздельного наблюдения линий воды гидратной оболочки и сво-
бодной воды. Такие наблюдения сделаны методом ПМР для А13+,
Mg2+ и некоторых других ионов. Поскольку обмен молекулами воды
(атомами кислорода) гораздо медленнее, чем протонами, по ЯМР 17О
раздельное наблюдение линий воды происходит намного легче, чем
в ПМР; переход к более высоким полям также облегчает раздельное
наблюдение.
Метод ЯМ-релаксации катионов применен для определения свобод-
ной энергии Гиббса AG°p их пересольватации [45-47]. Для реакции
пересольватации
YAn + nB YBn + nA, (4.20)
где А, В — растворители, Y — катион, AG?p = —RTink. Для ла-
бильных сольватов ЯМ-релаксация катиона будет усредненной от всех
возможных сольватов YAmBn_m (п — координационное число) с их
равновесной долей для данного соотношения растворителей. Измене-
ние R\ при замещении растворителя А растворителем В будет иметь
максимум, положение которого определяется величиной к (рис. 4.12):
к^п = Х™зх/(1 — Х™ах). Максимум R\ сдвинут в сторону более соль-
Рис. 4.12. Возможные варианты хода ЯМ-релаксации иона при замещении
растворителя в растворе; Хв — доля растворителя В. Пример см. на рис. 3.35
ватирующего растворителя. Полученные величины AG?p сольватов
YAmBn_m (п — координационное число) коррелируют с их равновес-
ной долей для данного соотношения растворителей.
Полученные величины AG°p коррелируют с донорным числом Гут-
мана [48]. Многие работы посвящены определению параметров обмена
сольватных сфер парамагнитных ионов.
Парамагнитные комплексы. Электронно-ядерное взаимодействие
на три порядка сильнее ядерно-ядерного. Оно приводит к большим
сдвигам линий ЯМР (парамагнитным сдвигам) и большим парамагнит-
ным вкладам в ЯМ-релаксацию, вследствие чего ЯМР на ядре самого
парамагнитного иона обычно не наблюдается. Для наблюдения пара-
магнитных сдвигов должно удовлетворяться условие \/те gJ3e/haN,
где те — время корреляции электронно-ядерного взаимодействия;
1/те = 1/Т\е + 1/т, где Tie — время релаксации электронного спина,
т — время жизни ядра в комплексе. Это значит, что наблюдение
ЯМР на ядрах лиганда в парамагнитном комплексе возможно или при
малом времени нахождения лиганда в комплексе, или при коротком
времени электронной релаксации иона. Общее правило таково, что хо-
рошо разрешенные спектры парамагнитного комплекса наблюдают или
в ЯМР (тогда спектры ЭПР плохие), или в ЭПР (тогда плохие спектры
ЯМР). Из параметров ЯМР парамагнитных растворов можно извлечь
информацию о строении парамагнитного комплекса, его концентрации,
кинетике обмена со свободным лигандом, внутри- или внешнесферном
взаимодействии с другими частицами раствора. Анализ параметров
ЯМР парамагнитных комплексов дан в работах [49-54].
Электронно-ядерное взаимодействие в растворенном комплексе при-
водит к изотропному парамагнитному сдвигу, в котором различают два
вклада: контактный (Ферми) и дипольный (псевдоконтактный) [55-58];
разделение этих вкладов сложно [49], однако в ряде случаев один
из них преобладает. При условиях: Т, е т, {/т (Emax - Emin)//i,
отсутствии расщепления в нулевом поле для S < 3/2 контактный вклад
в парамагнитный сдвиг выражается [53] как
/ ДЯ\С = -/3S'(S'+l)(g||A + g±B)
\ Н J &пыкТ ’ { ’
где А = 0,5aNgs^; g,, g± — компоненты g-тензора парамагнитного
иона, В = 0,5aNgs±-, aN — константа электронно-ядерного взаимодей-
ствия, gs — компоненты соответствующего мультиплета спектра ЭПР.
Дипольный вклад
(ддур = -^2g,(s' + O(gH -gj)(1 - 3coS2e) ,, 99.
k H ) 9kTR3 ' ( j
В случае изотропного g-тензора при чисто кубическом окружении иона
gn = Д±> ДяП = Ssi. = 2, и дипольный вклад исчезает. Величины Дтах
и E'min — соответствующие значения зеемановской энергии иона при
различных ориентациях относительно магнитного поля. Чувствитель-
ность разных ядер к взаимодействию различна, и константа взаимо-
действия при нахождении неспаренного электрона на s-орбитали при-
ведена в табл. 4.4 [51]. Соотношение между парамагнитным сдвигом
Таблица 4.4
Константы электронно-ядерного взаимодействия
Ядро a,N Ядро O.N
Гс Отн. ед. Гс Отн. ед.
’н 508 1 |7О -1659 24,1
2D 78 1 l9F 17160 35,9
14n 557 15,2 23Na 224 1,67
15n -781 15,2 31 р 2676 17,9
,3С 1119 8,76 35С1 1672 33,6
и вкладом в ширину линии ЯМР, от которого зависит разрешение
линий, также зависит от ядра. В самом деле, в простейшем случае [51]
= _.^S(S+I)
Н &1%знг
и парамагнитный вклад в ширину линии ЯМР (при условии tcws 1,
где ws — частота ЭПР)
= (4.24)
12 Oib
откуда видно, что с уменьшением гиромагнитного отношения ядра
условия наблюдения парамагнитных сдвигов становятся благоприятнее.
Теории релаксации в парамагнитных растворах посвящено много
работ [53, 59-64], в сжатом виде она представлена в [50]. С учетом
ws Wj уравнения для релаксации ядра в координационной сфере
парамагнитного иона могут быть представлены как
1 = h4nsS(S + 1} ( Зтс 7тс \ 2S(S+ 1)Л2те , .
Т? ЗОтА6 U+<4tc2 1+^т2/ 3(1+4те2) ’ ' ;
1 = h2y2Ny2sS(S + 1) Л Зтс 13тс \
Тк бОтгМ VC 1 + ^т2 1 + 4т2)
+ g(_g+‘M2.(Te + ^£__), (4.26)
з 1 + WsTe
где А — ^saNг — расстояние от ядра до неспаренного электрона.
При условии ШдТс 1 вклад сверхтонкого взаимодействия можно вы-
делить:
J_____1_ = S(S+ 1)А2 ( те \
T,fc Tfc 3 ( ^”е + 1 । , ,2 _2 ) ’
*2 Л ° \ 1+ч?5Те/
(4.27)
В растворах лабильных комплексов лиганд часть времени проводит
в свободном состоянии и параметры ЯМР лиганда легко измерить. На-
блюдаемые параметры будут зависеть от соотношения лиганда в ком-
плексе и свободного, скорости их обмена и параметров ЯМР лиганда
в комплексе. Варьируя концентрацию лиганда и температуру, можно
изменять соотношения между разными частями парамагнитного вклада
и получать данные о кинетических и термодинамических параметрах
комплекса. Формулы (4.25)-(4.27) очерчивают круг задач, для решения
которых используют ЯМР в парамагнитных растворах.
При образовании с парамагнитными ионами инертных комплексов
линии ЯМР *Н лиганда наблюдаются только в комплексах с ионами,
имеющими короткое время электронной релаксации (Со2+, Ni2+, лан-
таноиды), причем обычно видны только удаленные от иона протоны
лиганда. В лабильных комплексах можно наблюдать все протоны, при
этом парамагнитный сдвиг и ширина линий убывают по мере удаления
протонов в лиганде от координирующего атома, как бы раздвигая
линии спектра лиганда. На этом явлении основано применение сдви-
гающих реагентов (шифт-реагентов).
Изучение парамагнитных сдвигов позволяет определить структуру
и геометрию комплекса, состояние окисления иона, взаимное влияние
лигандов и т. д. [49-62].
Примером таких работ служит изучение структуры комплексов
Мп(П) с этилендиаминтетрауксусной кислотой (ЭДТА) [67], хелат-
ных комплексов никеля [68], комплексообразования в растворах РЗЭ
[69-72], комплексов Со(П) [73].
Парамагнитные сдвиги в ГПА, возникающие внутри инертных ком-
плексов на ядрах 13С, 14N, 17О, 31Р, 183W изучали в [76-81]. Вели-
чина гиромагнитного отношения позволяет наблюдать спектры ЯМР
и парамагнитные сдвиги и в том случае, когда парамагнитный ион
находится в структуре комплекса, а парамагнитный ион имеет короткое
время электронной парамагнитной релаксации. Такие сдвиги наблю-
дали в «гантельных» анионах [L^WsOis)»]9- и [Ln(PWnO39)2]11-,
в которых Ln3+ — парамагнитный ион лантаноида [13, 78-81].
На рис. 4.13 показаны спектры ЯМР 17О анионов [LaCWsOig)»]9- и
[Pr(W50ia)2]9- и спектр ЯМР l83W последнего; ХС атомов вольфрама
[Ln(W50ia)2]9_ показаны на рис. 4.14 [80, 81].
Парамагнитные сдвиги атомов Р и W в ГПА [Ln(PWnO39)2]11-
показаны на рис. 4.15.
Спектры ЯМР 17О парамагнитных комплексов [CoW1204o]n“,
(Со(П), п — 6, Со(Ш), п - 5) показаны на рис. 2.51 [77]. В комплексе
Со(Ш) парамагнитен из-за тетраэдрического окружения, не характер-
ного для Со(Ш). Данные ЯМР этих ГПА приведены в табл. 4.5. При
экстраполяции ХС парамагнитных комплексов к Т —> оо парамагнитные
сдвиги минимизируются, и ХС близки к таковым у диамагнитного
ГПА [H2Wi204o]6-
Механизм возникновения сдвигов ядер молекул, не входящих во
внутреннюю сферу парамагнитных ионов, обсуждался в [75, 76].
Изучение распределения электронной плотности и связи в ком-
плексах. Наибольшее число работ такого плана выполнено для ком-
плексов Со(П) и Ni(II), время электронной релаксации которых доста-
точно коротко из-за модуляции расщепления в нулевом поле. Плос-
коквадратные комплексы Ni(II) диамагнитны, и парамагнитные сдвиги
возможны только в тетраэдрических комплексах (основное состоя-
ние 3Ti). Обычно у них преобладает контактный вклад в парамагнит-
ный сдвиг; у комплексов Со(П) может быть значительным и диполь-
______I_____I_____I_____I I_______I_____I_____I I______I____
800_________400_____________________0 м.д.
Рис. 4.13. Спектры ЯМР 17O анионов [LaCWsOieh]9- (/) и [PrCWsOieh]9- (2),
спектр ЯМР 183W [Pr(W5O18)2]9- (3) и структура [Ln(W50i8)2]9- (4)
ный вклад. Теория парамагнитных сдвигов этих комплексов изложена
в [82]. По парамагнитным сдвигам изучены переходы комплексов Ni(II)
из диамагнитной плоскоквадратной в парамагнитную тетраэдрическую
координацию в комплексах типа NiL2X2, L = PR3 (X — галоген) [84],
определены и скорости межструктурных переходов [83-86].
Комплексы Cu(II) обычно не дают хороших спектров ЯМР из-за
большого Т]е, однако для удаленных ядер 1Н, 2D и 13С линии ЯМР
могут быть достаточно узкими, чтобы их наблюдать.
Активно изучаются нитрозильные комплексы Fe(III) [87]. Широко
изучают /3-дикетонатные комплексы переходных металлов М(Асас)з,
M(Acac)2L2 [88]. Имеются данные о парамагнитных сдвигах октаэд-
рических комплексов Fe(III) и Fe(II) [89-90], Со(П) и Со(Ш) в одина-
Рис. 4.14. Парамагнитные сдвиги в анионах [Ln(W50i8)2]9 : / — атомы WA,
2 — атомы WB
Рис. 4.15. Парамагнитные сдвиги 31Р (/) и 183W, (2) и 183Wi (3), м.д., в ком-
плексах [Ln(PWi 1039)2]11 - (для Tb-Yb парамагнитные сдвиги 183W различа-
ются); нумерация атомов [PWuOas]7- на рис. 3.18
ковом тетраэдрическом окружении [CoW^O^]”- [77], Мп(П) с изо-
цианатными лигандами [91]. В комплексах с лантаноидами часто пре-
обладает дипольный вклад в изотропный сдвиг. ЯМР-спектроскопиче-
ские методы исследования лантаноидных комплексов изложены в [92].
Предложенная в [93] для дипольного вклада температурная зависи-
Таблица 4.5
Данные ЯМР |70, 183W парамагнитных ГПА в сравнении с диамагнитным
[H2W12O40]6-
Анион 5I83W, м.д. (И/, Гц) 5 17О, м. д.
293 К 333 К Т —» оо 293/303 К •333 К Т —+ оо
[Co(II)W12O40]6- -915(180) — — 759 (293 К) 770 850 (А)
694 652 347 (С)
526 512 407 (В)
[Co,nW12O40]5- -2000(40) -1780 -182 929 (303 К) 876 346 (С)
— — 817 813 773 (А)
297 313 473 (В)
[H2W i2C>4o]6 -НО 722
414
410
мость 1/Т2 подтверждена экспериментально в [94], где парамагнитный
сдвиг выражен в виде
ДЯИЭ0 = ДЯс + ДЯй|р = £ + 4. (4.28)
Т т2
В диеновых комплексах лантаноидов от Се до Но преобладает контакт-
ный вклад, для остальных — дипольный [95].
Многочисленны работы по изучению комплексов U(IV). Заметные
дипольные взаимодействия должны были бы возникать из-за вырож-
денного основного состояния 5/2 конфигурации U(IV). Фактически
же изотропные сдвиги от U(IV) не определяются дипольным вкладом.
Оба вклада оказались важными в комплексах U(IV) с глицином [97],
различными аминокислотами [98], U(NCS)8(NEt4)4 [99-100] и /3-дике-
тонатами [101].
Внешнесферные комплексы. Многие комплексы координируют
лиганд к первой координационной сфере, по реакции типа Сг(Асас)з +
+ L <-> Cr(Acac)3L, и изучению таких внешнесферных комплексов по-
священо много работ [54, 102-107]. Измерения позволяют установить
структуру комплекса и термодинамические параметры комплексообра-
зования.
Определение кинетических и термодинамических параметров
сольватации. Методика определения кинетических и термодинамиче-
ских параметров аналогична таковой для диамагнитных комплексов и
систематически изложена в [95, 97, 98] на примере акваионов переход-
ных металлов. Равновесия типа M(H2O)g+ ± EtOH M(H2O)sEtOH +
+ Н2О (М = Co2+Ni2+) изучены в [108-114]. Параметры обмена моле-
кул воды с некоторыми комплексами приведены в табл. 4.6.
Таблица 4.6
Параметры обмена молекул воды с координационной сферой парамагнитных
ионов [51]
Комплекс K, c“‘ ДЯ* Д5* т, к
[Nien2(H2O)2]2+ 5,4- 106 9,1 ±0,5 2,6 ±2 298
[Ni(H2O)2(NH3)2]2+ 6,1 • 105 7,8 ±0,3 6±2 298
[Ni (Н2О) EDTA]2- 7 105 8 ±0,5 7±2 298
[Ni(H2O)4]2+ 3,6 104 12,3 ± 0,5 4± 1,3 298
[Ni(H2O)4]2+, pH = 6 4,4- 104 10,3 ±0,5 5±2 298
[Co(H2O)2(NH3)2]2+ 6,5- 107 9,4 ± 1,5 7±5 298
[Co(H2O)6]2+ 2,4- 106 11,9 ±0,7 11 298
[Mn(H2O)EDTA]2- 4,4 108 7,7 7 273
[MnATP] 5- 107 9,6 8,8 273
[Mn(H2O)6]2+ 5,9 • 106 8,8 ± 1 5±3 273
[VO(H2O)2C12] 2,3- 104 10± 1 — 300
[VO(H2O)4] 5,9- 102 15± 1 — 300
Парамагнитные сдвиги в молекулах в растворе, возникающие из-за
введенных в раствор инертных комплексов лантаноидов (шифт-реа-
гентов), широко применяются для принудительного разрешения линий
ЯМР сложных органических молекул. Особенности и основы их при-
менения можно найти, например, в [49, 92, 115].
4.3. Кинетические измерения методом ЯМР
Совпадение времен превращения многих химических реакций со
шкалой времени ЯМР, а также возможность следить во времени сразу
за реагентами, продуктами реакции и, реже, промежуточными про-
дуктами делают метод ЯМР особо ценным для изучения кинетики
и механизмов химических реакций. Если время полупревращения год
много больше времени ЯМ-релаксации изучаемого ядра в реагенте
или продукте, то такая реакция будет медленной в масштабе времени
ЯМР. Для последовательной регистрации изменений концентраций
необходимо, чтобы год намного превышало время накопления сигнала.
В этих условиях метод ЯМР отличается от других методов изучения
кинетики реакций [116] только своей уникальной специфичностью,
В зависимости от скорости реакции и целей исследования выбирается
ядро с подходящими параметрами ЯМР. Применяя высокочувствитель-
ные ядра, можно изучать реакции, протекающие за секунды. Другим
приложением метода ЯМР является изучение кинетики обратимых
реакций в диапазоне т0>5 = 0,1 —10—6 с. Например, по ЯМР 'Н показано
существование интермедиата Co(NH3)3+ в реакции замещения лиган-
дов в пентааминных комплексах кобальта (4.29)—(4.31) и определено
его время жизни [117].
(NH3)5CoX (NH3)5CoY + X, (4.29)
(NH3)5CoX (NH3)5H2O + X, (4.30)
н2о
(NH3)5CoY ^MNH3)5H2O + Y. (4.31)
н2о
Интересно, что кинетика замещения протона на дейтерий в ком-
плексе [(NH3)5Co(H2O)]3+ определяется по ЯМР 59Со (рис. 4.16).
Рис. 4.16. Спектр ЯМР 59Со Кз[Со(1ЧНз)б] при растворении в 98% D2O через
4 ч (а); содержание дейтерия в изотопомерах (б): 1 — сразу после растворения
К3[Со(ХОз)б], 2 — через 4 ч, 3 — через 23 ч; точки — экспериментальные
интенсивности линий ЯМР, линии — расчетные данные
Возможность одновременного измерения концентраций реагентов
и продуктов обеспечивает взаимный контроль измерений. Дополни-
тельную информацию можно получить, изучая изотопный обмен после
введения изотопной метки. Обогащая или обедняя какой-либо реагент
изотопом, на ядрах которого ведутся измерения, можно изучать кине-
тику изотопного обмена и следить за маршрутами переноса определен-
ных атомов из реагента в продукты. Скорости установления изотопного
равновесия меньше скоростей химических реакций, приводящих к это-
му равновесию; кроме того, кинетика изотопного обмена всегда опи-
сывается уравнением первого порядка, независимо от типа химических
реакций [118]. Поскольку в этом случае каждая точка кинетической
кривой означает накопление спектра, измерениям доступны процессы
с временем полупревращения от 10 с до суток в зависимости от ядер.
Метод ЯМР использует стабильные изотопы, безопасные по сравнению
с радиоактивными изотопными метками.
Высокая специфичность метода ЯМР определяет его преимущество
перед другими методами изучения изотопного обмена и позволяет
следить за изменением изотопного содержания отдельных фрагментов
и атомов молекулы. Так, структурно различные атомы в полиоксо-
соединениях [PMoi2-IV3;04o](3+Ib, [PVIW12_I04o](3+Ib, [Vi0O28]6-,
[Р2Мо18О62]6- в водных растворах обмениваются с водой с разной
скоростью [119-121]. Кинетику изотопного обмена с водой нитратных
форм Rh(III) в азотнокислых растворах нитрата родия изучали в [122].
Изотопные метки возможны для изотопов с природным содержанием
менее 100%. Возможности ЯМР как изотопного метода определяет
номенклатура доступных соединений со стабильными изотопами. Осо-
бенно велики возможности изучения H-D-обмена.
Метод ЯМР активно применяют при изучении кинетики обратимых
реакций (химического обмена). Если в процессе обратимой реакции
ядро переходит из одного состояния в другое, отличающееся парамет-
рами ЯМР (ХС, Ti, Тг, J), то в зависимости от соотношения между
скоростью обмена этими состояниями и параметрами ЯМР отдельных
состояний наблюдаемые параметры отличаются от стационарных [123,
124]. Если состояния различаются положением линий ЯМР, то при
медленном обмене наблюдаются оба состояния; по мере убыстрения
обмена сначала происходит уширение линий (рис. 4.17), затем они
сливаются в одну с шириной, близкой к разносу линии (из-за чего она
может и не наблюдаться), затем эта линия сужается (обменное суже-
ние). В аналитическом виде общее решение громоздко и в наиболее
компактном виде приведено в [124]
При обозначениях Д(1/Т2) = 1/Т2 - 1/Т2а, £ = Т^ъ/тъ, и = ТгвД^дв
величины Т\ и Т2 могут быть выражены:
1[
Т|лТ1в Та.'
1 . , G±x/G2 + H2
— =а2±^-----з----,
ш* + । I —G ± х/бг + Я2
-б ----------
(4.32)
(4.33)
(4.34)
(4.35)
2
где
a
Рис. 4.17. Изменение положения линий ЯМР и их ширины при медленном
двухместном обмене (а) и зависимость приведенных параметров ЯМР от
скорости обмена (б): А = Т^/рТ™6 — Т^/рТ^', £ = Т^/т; v = ТгцДш
(4.36)
1
Т1А
1
т1А
1
2tia
1
1
2tib
1 _
Л в
^2 = 2т- +
_ _ 1-
В 'Ть
/д =
-р;
/в =
1
ТаЬ
ТЬ
1
2т2в ’
1
= р;
ть
1 -р.
р
_1_.
ть’
(4.37)
= 1
1
4
/а + /в = 1-
Здесь Tiab — времена релаксации в состоянии А, В; та>ь — времена
жизни ядра в этих состояниях; /А1 /в — доли ядер в состояниях А, В.
Выражения упрощаются в тех случаях, когда р = 0,5 и р <£. 1. Случай
р = 0,5 реализуется при конформационных переходах в молекуле, когда
оба состояния энергетически эквивалентны. Подробный анализ разных
случаев сделан в [50].
В зависимости от соотношения TiAB, Тглв- ДшАВ возмож-
ны те или иные упрощения. При изучении химических равновесий,
сольватации ионов часто выполняется условие р <£. 1. Тогда выраже-
ния (4.32)-(4.36) упрощаются, в особенности для предельных случаев.
В случае быстрого обмена при р <§; 1
1
2^наб
ТЬ I 1 1а -Ив I
= — + А
Т1Л Tib
1
Т|Наб
2
ДшАВ;
тъ
= ~ РТаЬ^-Шкъ',
lit.
(4.38)
шнаб = шА + рДшАВ.
(4.39)
Для Т\ условие быстрого обмена выполняется легче, чем для Т2,
а при большом ДшАВ условие быстрого для других параметров ЯМР
обмена не приводит к обменному сужению. В этом случае при р 1
ДшАВ « - »
ть
тЬл Т2в
Индекс «наб» относится к величинам, наблюдаемым в эксперименте.
В этом случае медленного обмена при р 1 обычно наблюдают только
линию А, для которой
1 = 1 , Р_.
ТГ6 Г1Л ть’
1 = 1 , Р_
Т2наб Т2а + ть •
(4.40)
(4.41)
Смещение линии А мало, и его можно оценить из рис. 4.18 [124],
где изменение параметров выражено в безразмерных величинах. Пара-
Рис. 4.18. Температурный ход ЯМ-релаксации при малом вкладе ДшАВ (а) и
при его преобладании (6)
метр ть можно менять в широких пределах температурой. Меняя ее,
можно переходить из области быстрого в область медленного обме-
на, и по зависимостям параметров ЯМР от температуры определить
кинетические и термодинамические (ДТТ^ и Д5^) параметры обмена,
Подобным образом изучают сольватацию парамагнитных ионов. Резкое
различие параметров ЯМР молекул в сольватной сфере этих ионов
и молекул растворителя позволяет упростить равенства (4.25, 4.26)
[108, 109]. Линия ЯМР в таких растворах наблюдается при р 1;
кроме того, в них Ti,2b Следуя [ПО], при условии отсутствия
зависимости ширины линии от частоты (что приводит к
форме линий), парамагнитный вклад в Т2
1 = J______L = Р^2 + Т2^ь' + ^ав)
Т? ТГ Т2а ть[^-'+Т2в'Г + ^в
лоренцевои
(4.42)
(4.43)
при р — 0.
чем р <g; 1,
'гь [(^2п ' + ТЬ ') + Aw,\B
где Ашд — измеренный сдвиг линии А от ее положения
Равенства (4.42), (4.43) справедливы при более сильном,
условии р <§; Т2наб/Т2Р = Т2а/(^2а + Т//). Чем больше обменный вклад
АшЛ, тем меньшее значение р надо выбирать при эксперименте.
Возможны условия, при которых (4.42) упрощается. Условие
1 /?2в Ашав. При этом 1 /Тр = р/(Т2ъ + ть), а при медленном обмене
1 /Т2Р = р/ть, и из температурной зависимости в полулогарифмическом
масштабе определяется энтальпия активации обмена
h (АН* AS\ , , АН* .. ...
ТЬ = ЙГ^\~ВТ-^:)' -ln^P = const+ —. (4.44)
При быстром обмене (ть Т2в) i/IJ = Р/^ъ, в этом случае
1/^2в = Ст, где С — константа, не зависящая от температуры и ча-
стоты, а 1/т = 1/Те + 1/тв. При 1/Те 1/тв 1/Т2р = СрТе, а при
Те 2> т 1 /Т2Р = Срт. В сумме температурная зависимость представлена
на рис. 4.18, а.
Условие <§; 1/Т22в при медленном обмене (Дш2в <§; 1/т^)
приведет к 1/Т2р = р/ть, а при быстром (1/т^ » Дш2в < 1/ть72в)
к 1/Т2Р =рАш2вТ(,. При еще более быстром обмене (1/ть2 2> \/тьТ2ъ 2>
Аш2в) 1/Т2р =р/Т2в, что приводит к температурной зависимости на
рис. 4.18, б.
Условие 1/Т22в ~ Аш2в. Этот случай наиболее сложен для анализа
и приводит при медленном обмене (АшЛВ, 1 /Т2ъ 1/ть2) к 1/^Т = р1тъ,
а при быстром (Ашлв, 1/72в < l/r^) к 1/^? = Р/^2в + ртт,Дш2в. Во всех
рассмотренных случаях на низкотемпературной ветви 1/Тр — р/ть, а на
высокотемпературной 1/Тр = Срть- Предполагается, что электронно-
ядерное взаимодействие проходит по контактному механизму. Экспе-
риментально трудно провести измерения в столь широком диапазоне
температур, чтобы пройти обе ветви температурной зависимости.
При многоместном обмене анализ существенно усложняется [128,
130], и определенные выводы возможны в частных случаях, при кон-
кретном соотношении фаз, или при р2,... ,pk 1 •
Изучение медленных и обратимых реакций не требует особых при-
способлений и сводится к последовательным измерениям или приме-
нению определенных импульсных последовательностей [ПО, 123, 124,
127], Для изучения кинетики более быстрых реакций, проходящих
за время менее 100 с, можно использовать удобный для ЯМР метод
остановленной струи (рис. 4.19) [129]. В этом случае требуется датчик
Рис. 4.19. Применение метода остановленной струи при кинетических измере-
ниях методом ЯМР: С — смеситель, Д — датчик ЯМР, К — кран
особой конструкции и система синхронизации измерений.
Реагенты А и В находятся в шприцах А, В, продукты реакции
собираются в шприце А + В. Струя создается механическим давлени-
ем на поршни шприцов А, В, остановка ее — поворотом крана КЗ,
запускающим съемку серии спектров. Краны отсоединяют шприцы от
датчика при смене реагентов.
Указанные особенности метода ЯМР открывают перспективные пу-
ти изучения механизмов химических реакций. Раздельное определение
порядков реакций по реагентам, путей переноса атомов от реагентов
в продукты, состава интермедиатов, активационных параметров поз-
воляет установить механизм реакций, в том числе и каталитических.
Несмотря на то что задача определения состава интермедиатов и ка-
талитически активных комплексов в процессе реакции трудна из-за их
малых времени жизни и концентрации, она успешно решается [124];
трудности растут при увеличении скорости реакций и активности ка-
тализаторов, которые могут быть уменьшены снижением температуры
эксперимента.
4.4. Технологические приложения метода ЯМР
Высокая специфичность и оперативность метода ЯМР, отсутствие
химического воздействия на образец, возможность непрерывного из-
мерения параметров открывают многообразные пути его применения
в промышленности. Внедрению метода ЯМР мешает сложность аппа-
ратуры и ее эксплуатации, высокая стоимость спектрометров, исследо-
вательский характер самого метода. Развитие методов ЯМР, с одной
стороны, и развитие химических и иных технологий, вовлечение в тех-
нологию все более тонких и точных приемов, требующих оперативных
химических анализов, с другой стороны, показывают необходимость
расширения областей применения метода ЯМР в технологии. Укруп-
нение масштабов производства, переход к комплексным, экологиче-
ски замкнутым технологиям переработки сырья делают экономически
обоснованным применение и дорогих спектрометров ЯМР, если другие
методы не обеспечивают необходимый технологический контроль.
Наиболее разнообразное приложение метод ЯМР может найти
в практике заводских лабораторий химических и металлургических
производств для определения качества сырья, промежуточных и конеч-
ных продуктов, технологических растворов и т. д. Обычно химический
анализ на производстве осложнен примесями; более того, в сырье по-
лезный компонент часто содержится на уровне нескольких процентов,
и высокая специфичность метода ЯМР, «неприхотливость» к парамет-
рам образца и быстрота анализа определяют области применения ме-
тода и преимущества перед другими методами анализа. Использование
ЯМР в заводских лабораториях отличается от обычной исследователь-
ской практики только спецификой образцов, и возможности метода
описаны в предыдущих главах.
Основным ограничением метода была (и в определенной мере оста-
ется) низкая чувствительность, однако в технологических процессах
стремятся к повышению концентраций, часто встречаются растворы,
близкие к насыщению, при анализе которых для многих методов воз-
никают определенные проблемы; для метода ЯМР таких проблем не
возникает. Для технологических целей перспективно использование
ядер с высокой чувствительностью: 1Н, 7Li, 19F, 23Na, 27А1, 31Р, 45Sc,
51V, 59Со, 93Nb, 139La.
Многие пути применения метода ЯМР в промышленности опреде-
лились уже давно [132-140].
Изучение процессов экстракции. Экстракция как процесс кон-
центрирования и очистки элемента (вещества) активно используется
в химических исследованиях и производственных процессах. Изучение
экстракции входит в процесс создания химической технологии извле-
чения многих элементов.
Ряд работ посвящен изучению экстракции, поскольку ряд попу-
лярных экстрагентов (трибутилфосфат (ТБФ), ди-2-этилгексилфосфат
(Д2ЭГФ) и другие) позволяют обнаруживать их по ЯМР 31Р. В [141]
изучено комплексообразование Zr(IV) с Д2ЭГФК. Экстракция UO2
ТБФ изучена в [142, 153]. Комплексообразование висмута с Д2ЭГФ
изучено в [143]. Взаимодействие WO3- и МоО3- с 1-оксиэтилиден-
дифосфоновой кислотой изучено в [144]. Аддукты La и Се с Д2ЭГФ
циркония изучены в [145]; экстракцию В1(Ш) Д2ЭГФ изучали в [143,
147]. Экстракцию скандия изучали в [148, 149], ванадия — в [150],
теллура в [151, 154], влияние фона цветных металлов на экстракцию
нитрозоформ рутения — в [152].
Контроль состава и компонентов сырья и продуктов. Методом
ЯМ-релаксации протонов можно определить концентрации парамаг-
нитных ионов в широких пределах [5], что может быть использовано
для контроля состава растворов гальванических ванн [155], анализа
состава металлов [134, 135].
Определению влажности различных продуктов посвящены рабо-
ты [156, 159-161]. Измерение влаги высушиванием часто портит обра-
зец и затруднено для огне- и взрывоопасных веществ: методом ЯМР
подобные измерения можно делать в потоке без порчи образцов. Точное
определение влажности сырья и продуктов необходимо для технологи-
ческих целей, безопасного хранения, определения качества продукта.
Методом ЯМР определяют и оптимальные условия хранения продуктов
при низких температурах [158], влажность угольной шихты [159],
апатитового концентрата (содержание влаги 1-1,5%, допуск 0,1%)
[160], хлопка [161].
ЯМР l9F может быть использован для контроля состава содержа-
щих фтор растворов, содежания фтора в фармацевтических препара-
тах [162], для количественного анализа Si-Ge-сплавов [163] (исполь-
зовано различие в ХС ионов SiFg- и GeF|“ после растворения образца
в HF, причем в этом случае отношение Si/Ge определяется с точностью
2%), доли гафния в соединениях Мг/гРб (использовано различие ХС
19F ZrF9” и HfF?-).
ЯМР 31Р может быть использован для определения количества
и типа фосфорных соединений и ионов в растворах, начиная с концен-
траций фосфора 10-3 г-ат/л [164], содержания фосфора в апатитовом
концентрате [165]. ЯМР натрия и алюминия применяли для контроля
состава технологических растворов алюминиевого производства [166].
Метод ЯМР может широко применяться для технологического контро-
ля растворов гидрометаллургидеского производства.
Особые требования предъявляются к соединениям стабильных изо-
топов, и метод ЯМР, будучи в принципе изотопным методом, может
активно использоваться при анализе технологических полупродуктов
и конечных продуктов производства соединений стабильных изотопов.
Высокая стоимость последних окупает применение дорогих спектро-
метров ЯМР. Так, метод ЯМР использован при контроле разделе-
ния изотопов урана [167], приготовлении меченых дейтерием соедине-
ний [168].
Применению метода ЯМР в производстве химических реактивов
и особо чистых веществ посвящен обзор [169].
Ключевая роль катализа в решении многих проблем химической
технологии, биотехнологии и экологии вызывает соответствующий ин-
терес к изучению катализа, катализаторов, процессов приготовления
последних, адсорбции и других сопутствующих явлений, что отраже-
но в ряде обзоров [170-172]. Приготовление подложек катализато-
ров (AI2O3, SiC>2, цеолиты, алюмофосфаты и другие пористые носи-
тели) происходит в жидкой фазе. Процессы приготовления и работу
гомогенных катализаторов, поиск ключевых интермедиатов изучает
ЯМР в жидкой фазе [18, 27, 131]. Например, состояние компонентов
в растворе катализатора [РУзМодО^]6- показывает спектр ЯМР 31Р
(рис. 2.35) [173].
Определенный интерес технологов может привлечь возможность
измерения методом ЯМР скорости потоков [174, 175], определения
соотношения фаз в гетерогенных потоках (пульпе) [175], определения
концентраций парамагнитных частиц (парамагнитных ионов, свобод-
ных радикалов) [176]. ЯМР в земном поле применяется в геофизиче-
ских исследованиях [179], в том числе для карротажа скважин и по-
иска водоносных слоев грунта, археологии [177-179, 193]. Изучению
природного сырья и продуктов ее переработки посвящена книга [180].
Все шире используется ЯМР в медицинской технике. Развивитие
ЯМР-томографии [181], позволяющей с разрешением 1 мм делать «сре-
зы» в любой плоскости пациента и создавать объемное изображение
фрагмента организма привело к повсеместному ее применению. Ин-
формативность ЯМР-томографии много выше информативности рент-
геновского снимка, поскольку ткани организма резко различаются со-
держанием протонов; дополнительное увеличение контраста объектов
делается по различию времен релаксации, при этом в интересующую
область может быть введен дополнительный релаксант. Для томогра-
фии могут быть использованы также ядра 31Р, 23Na. Метод ЯМР
применяют для измерения потока крови [182], диагностики злокаче-
ственных опухолей, однако эти применения ЯМР выходят за рамки
данной работы.
Наиболее широко ЯМР применяется в пищевой и фармацевтиче-
ской промышленности и для определения влажности различных ве-
ществ. Методом ЯМР определяют содержание жиров в семенах мас-
личных культур [139, 140, 183-185]; соотношение твердой и жидкой
фаз в пищевых жирах [185, 186]; содержание воды в крахмальных
и других гелях, являющихся основой многих пищевых, фармацевти-
ческих и парфюмерных продуктов, от которого зависят свойства этих
продуктов [187-192]; определение содержания воды или другой жидко-
сти может быть сделано в течение секунд, что позволяет осуществлять
постоянный технологический контроль.
Для химика возможность определения происхождения абсолютно
чистого соединения выглядит как ересь, однако для этанола это можно
определить по изотопному распределению 2D в фрагментах EtOH по
спектрам ЯМР дейтерия (рис. 4.20.). Соотношение линий в спектре
Рис. 4.20. Спектры ЯМР 2D на частоте 61,4 МГц этанола гидролизного (а)
и ректификата (б), 298 К
ЯМР *Н этанола групп СН3, СН2 и ОН 3:2:1 в спектрах на ядрах
2D совершенно иное. Этанол получают из органического сырья вслед-
ствие многостадийных реакций, катализируемых ферментами. Извест-
но, что реакции с дейтерированными реагентами проходят значительно
медленнее, чем с их протонными аналогами, а скорость изотопного
переноса гораздо меньше скоростей сопутствующих реакций [118].
В процессе получения сами реакции зависят от исходного сырья и ка-
тализируются разными ферментами, при этом изотопное равновесие не
достигается. Это и позволяет определить сырье, из которого получен
этанол, по спектрам ЯМР дейтерия.
С использованием ЯМР 2D проводят паспортизацию и контроль
винных продуктов [194].
Тенденции в развитии техники ЯМР свидетельствует о благопри-
ятных перспективах применения метода в неорганической химии, как
в научных исследованиях, так и в химических и иных технологиях.
Особенно перспективно применение метода ЯМР при изучении концен-
трированных растворов и расплавов, где многие методы исследования
перестают работать, и технологических процессов с участием жидкой
фазы для непрерывного контроля состава технологических растворов
и качества продуктов.
ЛИТЕРАТУРА К ГЛАВЕ 4
1. Золотов Ю.А., Верещагин В. И. История и методология аналитической
химии. — М.: Академия, 2007. — 462 с.
2. Johnson L.F., Shoolery J.N. Determination of Unsaturation and Average
Molecular Weight of Nature Fats by Nuclear Magnetic Resonance // Anal
Chem. 1962. V. 34, №9. P. 1136-1139.
3. Федоров Л. А. Литература по спектроскопии ЯМР высокого разрешения
и проблемы аналитической химии // Ж. аналит. химии. 1983. Т. 38, № 1.
С. 154-159.
4. Федоров Л.А., Ермаков А.Н. Спектроскопия ЯМР в неорганическом ана-
лизе. — М.: Наука, 1989. — 245 с.
5. Попель А.А. Применение ядерной магнитной релаксации в анализе неор-
ганических веществ. — Казань: Изд-во Казан, ун-та, 1975. — 173 с.
6. Федотов М.А. Возможности метода ЯМР в аналитической химии рас-
творов неорганических веществ // Ж. аналит. химии. 1999. Т. 54, № 1.
С. 17-22.
7. Ando /., Webb G.A. Some Aspects of High-Pressure NMR // Magn. Reson.
Chem. 1986. V. 24, № 7. P. 557-567.
8. Holz M. Electroforetic NMR // Chem. Soc. Revs. 1994. V. 23, № 3.
P. 165-174.
9. Эмсли Дж., Финей Дж., Сатклиф Л. Спектроскопия ЯМР высокого
разрешения. Т. 2 / Пер. с англ. Ю. С. Константинова и др.; Под ред.
В. Ф. Быстрова, Ю. Н. Шейнкера. — М.: Мир, 1968. — 630 с.
10. NMR and Periodic Table I Ed. B.E. Mann, R. K. Harris. — L.: Acad. Press,
1978. - 459 p.
11. Федотов M.A. ЯМР в растворах неорганических веществ. — Новоси-
бирск: Наука, 1986. — 196 с.
12. Shoolery J.N. Quantitative measurements // Encyclopedia NMR. V. 5 / Ed.
D.M. Grant, R.K Harris. -N.Y.: Wiley, 1996. P. 3907-3916.
13. Kazansky L.P., Fedotov M.A. Paramagnetic 183W chemical shifts in het-
eropoly tungstates // J.C.S. Chem. Comm. 1983. №8. P. 417-419.
14. Федотов M.A., Молчанов В.И., Казанский Л.П. и др. Химические сдвиги
ЯМР фосфора-31 гетерополикомплексов Z^WiyOgi)"-, Z = Се, Th, UIV
и их строение в растворах // ДАН СССР. 1979. Т. 245, №2. С. 376-380.
15. Федотов М.А., Максимов Г.М., Матвеев К. И. Спектры ЯМР 17О, 183W
и состояние в водных растворах комплексов Y(III), La(III), Ce(IV),
Lu(III) с гетерополианионом PWnO^“ // Коорд. химия. 1990. Т. 16, №2.
С. 203-206.
16. Russell J.G., Bryant R.G. A Co-59 Nuclear Magnttic Resonance Reagent for
The Determination of Hydrogen / Deuterium Ratios // Inorg. Chim. Acta.
1983. V. 151, №2. P. 227-229.
17. Jarett R.M., Saunders M. The Use of Various Nuclei as Probes in a New
NMR Method for Obtaining Proton I Deuteron Fractionatuon Data //
J. Amer. Chem. Soc. 1986. V. 108, №24. P. 7549-7553.
18. Максимов Г.M. Достижения в области синтеза полиоксометаллатов и изу-
чения гетерополикислот // Усп. химии. 1995. Т. 64, №3. С. 480-496.
19. Akitt J. IV. Proton chemical scift and hydration of hydronium ion in aqueous
acids // J.C.S. Dalton tr. 1973. № 1. P. 49-52.
20. Arnett E.M., Scorrano G.R. Protonation and solvation in strong aqueous
acids // Adv. Phys. Org. Chem. 1976. V. 13. P. 83-151.
21. Akitt J. IV. Proton chemical scift of water in cationic hydration complexes and
their contribution to water shifts in electrolyte solutions // J.C.S. Dalton tr.
1973. № 1. P. 42-49.
22. Bagno A.. Comuzzi C., Scorrano G. A novel Method for Determination of
Ionization Sites in Polyfunctiona! Acids and Bases by NMR Relaxation Rate
Measurements // J.C.S. Perkin tr.II. 1993. №2. P. 283-284.
23. Chandler Xv.D.. Lee D.G. Evaluation of KBH+ constants for weak organic
bases // Canad. J. Chem. 1990. V. 68, № 10. P. 1757-1761.
24. Farcasiu D., Marinoi G., Miller G., Kastrup R.V. Acidity measurments
from protonation of aromatics. Protonation of hexamethylbenzene by trifluo-
romethanesulfonic acid and its use to relate acidities of strong superacid to
acidities of acid included in the traditional Hammett scale // J. Amer. Chem.
Soc. 1985. V. Ill, № 18. P. 7210-7213.
25. Levy G.C., Cargoli J.D., Racela W. NMR determination of ketone basidity
and the use of ketones as indicator for evaluation of medium acidity //
J. Amer. Chem. Soc. 1970. V. 92, №21. P. 6238-6246.
26. Лагутенков В. А., Скороваров Д.Н., Ульянов В. С. и др. Связь между
параметрами специфической сольватации и и хим. сдвигом 14N ионов
NO^ в водных растворах электролитов // Ж. физ. химии. 1986. Т. 60,
№5. С. 1299-1301.
27. Farcasiu D., Ghenciu A., Miller G. Evaluation of Acidity of Strong Cata-
lysts // J. of Catalysis. 1992. V. 134, № 1. P. 118-125.
28. Farcasiu D., Ghenciu A. Acidity function from 13C NMR // J. Amer. Chem.
Soc. 1993. V. 115, № 23. P. 10901-10908.
29. Федотов M.A., Максимов Г.М., Игнашин С.В. Возможности ЯМР раз-
ных ядер в определении кислотности растворов гетерополикислот //
Ж. неорг. химии. 2002. Т. 47, № 12. С. 2031-2037.
30. Максимов Г.М., Головин А. В. Применение ЯМР 13С индикатора 2,6-ди-
метил-7-пиррола для оценки кислотности растворов гетерополикислот //
Ж. неорг. химии. 2004. Т. 49, №8. С. 1362-5.
31. Huskens J., Lammerds Н., van Bekkum Н., Peters J.A. Determination of
Stability Constants of Metal Complexes from NMR Chemical Shifts and
Relaxation Rates Using Spreadsheet Computer Program // Magn. Reson.
Chem. 1994. V. 32, № 9. P. 691-698.
32. Fujiwara H., Sakai F., Sasaki Y. Analysis of the concentration dependence
of NMR parameters by means of chemical equilibria // J. Phys. Chem. 1979.
V. 83, № 18. P. 2400-2404.
33. Федотов M.A., Максимовская P. И., Максимов Г. М., Матвеев К. И. Вза-
имодействие гетерополианиона с катионами по данным ЯМР и других
ядер // Ж. неорг. химии. 1987. Т. 32, №3. С. 647-651.
34. Deverell С. Nuclear, magnetic shielding of hydratetl alkali and halide ions in
aqueous solutions // Mol. Phys. 1969. V. 16, № 5. P. 491-500.
35. Hertz H., Holz M., Keller G. et al. Nuclear magnetic relaxation of alkal metal
ions in aqueous solutions // Ber. Bunsenges. Phys. Chem. 1974. V. 78, № 5.
P. 493-509.
36. Валиев К.А. К теории квадрупольной релаксации ядерных спинов //
Ж. эксп. теор. физ. 1960. Т. 38, №4. С. 1222-1232.
37. Валиев К. А., Хабибуллин Б.М. Ядерный магнитный резонанс и структура
водных растворов // Ж. физ. химии. 1961. Т. 35, № 10. С. 2265-2274.
38. Валиев К. А. Исследование растворен электролитов в дипольной жидкости
методом магнитного резонанса. Теория магнитной релаксации // Ж. струк-
тур. химии. 1962. Т. 3, № 6. С. 653-661.
39. Hertz Н. G. Magnetic relaxation by quadrupolar interaction of ionic nuclei in
electrolyte [or infinite dilution // Ber. Bunsenges. phys. Chem. 1973. V. 77,
№7. P. 531-540.
40. Annual reports on NMR spectroscopy. V. 9 / Ed. G.A. Webb. — L.: Acad,
press, 1979. P. 126-216.
41. Laszlo P. Sodium-23 nuclear magnetic spectroscopy // Angew. Chem. Int.
ed. 1978. V. 17, №4. P. 254-266.
42. Hertz H.G., Holz M., Kliite R. et al. 35C1, 8lBr and l2'I NMR in aqueous
solutions of alkali halides // Ber. Bunsenges. phys. Chem. 1974. V. 78, № 1.
P. 24-35.
43. Langer H., Hertz H.G. The structure of the first hydration sphere of ions in
electrolyte solutions. A nuclear magnetic relaxation study // Ber. Bunsenges.
phys. Chem. 1977. V. 81, № 5. P. 478-490.
44. Chizhik V.I. NMR relaxation and microstructure of aqueous electrolyte
solution // Mol. Phys. 1997. V. 90, №4. P. 653-659.
45. Мишустин А.И., Подковырин А.И., Кесслер Ю.М. Прямой метод опре-
деления энергии Гиббса пересольватации катионов Ц ДАН СССР. 1979.
Т. 245, №6. С. 1420-1423.
46. Мишустин А.И., Подковырин А.И., Кесслер Ю.М. Определение свобод-
ных энтальпий переноса катионов между растворителями методом квад-
рупольной релаксации их ядер. I. Теория метода // Ж. физ. химии. 1981.
Т. 55, №1. С. 56-60.
47. Мишустин А. И. Определение свободных энтальпий переноса катионов
между растворителями истодом квадрупольной релаксации их ядер. П.
Катионы лития в диполярных апротонных растворителях // Ж. физ. хи-
мии. 1981. Т. 55, № 1. С. 61-64.
48. Гутман В. Химия координационных соединений в неводных растворите-
лях. — М.: Мир, 1971. — 230 с.
49. Драго Р. Физические методы в химии. Т. 2 / Пер. с англ.^А. А. Соловья-
нинова; Под ред. О. А. Реутова. — М.: Мир, 1981. — 456 с.
50. Ватман А.А., Пронин И.С. Ядерная магнитная релаксация и ее приме-
нение в химической физике. — М.: Наука, 1979. — 224 с.
51. Annual reports on NMR spectroscopy. V. 6A / Ed. E.F. Mooney. — N.Y.:
Acad. Press, 1975. P. 2-145.
52. Annual reports on NMR spectroscopy. V. 9 / Ed. G.A. Webb. — L.: Acad,
press, 1979. P. 2-143.
53. NMR of paramagnetic molecules / Ed. G.N. La Mar, W. D. Horrocks,
R.H. Holm. - N.Y.: Acad. Press, 1973. P. 2-52.
54. Некипелов B.M., Замараев К. И. Измерение методой ЯМР геометри-
ческих параметров парамагнитных металлокомплексов в растворах //
Ж. физ. химии. 1978. Т. 59, № 9. С. 2155-2175.
55. McConnell Н.М., Holm С.Н. Proton resonance shifts in nickelocene //
J. Chem. Phys. 1957. V. 27, № 1. P. 314-315.
56. Bloembergen N. Comments on «Proton relaxation times in paramagnetic
solutions» Ц J. Chem. Phys. 1957. V. 27, №2. P. 595-596.
57. McConnell H.M., Chesnut D.B. Theory of isotropic hyperfine interactions in
7r-electron radicals // J. Chem. Phys. 1958. V. 28, № 1. P. 107-117.
58. McConnell H.M., Robertson R.E. Isotropic nuclear resonance shifts //
J. Chem. Phys. 1958. V. 29, №6. P. 1361-1365.
59. Козырев Б.М., Ривкинд А.И. Об исследовании комплексообразования в
растворах методом протонного резонанса // ДАН СССР. 1955. Т. 98, № 1.
С. 97-98.
60. Ривкинд А.И. Исследование комплексообразования в растворах методом
протонного резонанса // ДАН СССР. 1955. Т. 100. С. 933-936.
61. Ривкинд А.И. Протонная релаксация и ближний порядок в парамагнит-
ных растворах // ДАН СССР. 1955. Т. 102, №6. С. 1107-1110.
62. Ривкинд А. И. Протонная релаксация в смесях H2O-D2O, содержащих
парамагнитные ионы // ДАН СССР. 1957. Т. 112, №2. С. 239-240.
63. Bloembergen N. Proton relaxation limes in paramagnetic solutions //
J. Chem. Phys. 1957. V. 27, №2. P. 572-573.
64. Solomon I. Relaxation processes in system of two spins // Phys, Rev. 1955.
V. 99, № 2. P. 559-565.
65. Bloembergen N.H., Purcell E.M., Pound R. V. Relaxation effects in nuclear
magnetic resonance absorption // Phys. Rev. 1948. V. 73, №7. P. 679-712.
66. Bernheim R.A., Brown Т.Н., Gutowsky H.S. Temperature dependence of
proton relaxation times in aqueous solutions of paramagnetic ions // J. Chem.
Phys. 1959. V. 30, №4. P. 950-956.
67. Oakes J., Smith E.G. Structure ol manganese(III) EDTA complex in aqueous
solution by relaxation nuclear magnetic resonance // J.C.S. Faraday tr. II.
1981. №2. P. 299-308.
68. Пилипенко A. T., Мельникова H.B., Страшно В. В. Исследование методом
ПМР кинетики обмена лигандов в системах, содержащих хелаты никеля и
гетероциклические амины // Коорд. химия. 1979. Т. 5, № 9. С. 1372-1375.
69. Сальников К). И., Девятов Ф. В., Давлетбаева И.М. Изучение комплексо-
образования гадолиния(Ш) с винной кислотой методом ядерной магнитной
релаксации // Ж. неорг. химии. 1979. Т. 24, № 7. С. 1838-1842.
70. Чернышев Б.Н., Кавун В. Я. Исследование комплексообразования и гид-
ратации в растворах солей // Коорд. химия. 1979. Т. 5, № 9. С. 1309-1314.
71. Кавун В. Я., Чернышев Б.П., Костин В. И., Сергиенко В.Н. Комплексооб-
разование иттербия(Ш) и тулия(Ш) с нитрат-ионами в водно-ацетоновых
растворах по данным ПМР и ИК-спектроскопии // Ж. неорган, химии.
1981. Т. 26, №9. С. 2375-2383.
72. Сухно И. В., Панюшкин В. Т., Бузько В.Ю., Арутюнян М.Н. Межчастич-
ные взаимодействия в системе РЗЭ — уксусная кислота по данным ЯМР
(’Н) спектроскопии // Ж. неорг. химии. 2003. Т. 48, №5. С. 857-861.
73. Everhardt D.S., Evilia B.F. NMR observation of mixed complexes formation
and stereochemical non-rigity in cobalt(II) complexes // Inorg. Chim. Acta.
1979. V. 32, № 1. P. 81-86.
74. Szczepaniak L., Bryant R.G. Proton nuclear magnetic relaxation in aqueous
copper(II) amine chelate solution // Inorg. Chim. Acta. 1991. V. 184, № 1.
P. 7-11.
75. Матвеев В. В. О возможном механизме внешнесферных смещений сигна-
лов ПМР в водных растворах парамагнитных ионов // Теор. эксперим.
химия. 1982. Т. 18, №6. С. 742-744.
76. Кессених Ф.В., Атаев А., Плахутин Б.Н., Федотов М.А. Изотропный
парамагнитный сдвиг ЯМР ядер азота и углерода в комплексах никеля
и кобальта // Хим. физика. 1993. Т. 12, № 12. С. 1687-1696.
77. Казанский Л.П., Федотов М.А. Спектры ЯМР |7О и l83W в парамагнит-
ных гетерополианионах // Коорд. химия. 1988. Т. 14, №7. С. 939-942.
78. Fedotov М.А., Pertsikov B.S., Danovich D.K. 17O, 3IP and 183 NMR
spectra of paramagnetic complexes with the heteropoly tungstate anion
[Ln(PWiiO39)2]11_ and their constitutionin aqueous solytion (Ln — rare
earth element) // Polyhedron. 1990. V. 9, № 10. P. 1249-1256.
79. Федотов M.A., Перциков Б. С., Данович Д. К. Спектры ЯМР |7О, 3|Р,
l83W и состояние в растворах парамагнитных комплексов с гетерополи-
вольфрамат-ионом [LnfPWnOagh]"- // Коорд. химия. 1991. Т. 17, №2.
С. 234-242.
80. Федотов М.А., Самохвалова Е.П., Казанский Л.П. Химические сдвиги
ЯМР |7О, l83W и состояние гетерополианионов [ZWioOag]”- в водных
растворах, Z — редкоземельный элемент // Коорд. химия. 1995. Т. 21,
№9. С. 709-715.
81. Fedotov M.A., Samokhvalova E.P., Kazansky L.P. I7O and 183W NMR
diamagnetic and paramagnetic shifts in heteropoly tungstates XWioO^-
(X = Ln, Th, U) // Polyhedron. 1996. V. 15, № 19. P. 3341-3351.
82. McGarvey B. Theory of the NMR Paramagnetic Shifts of Pseudotetrahe-
dral Complexes of Ni(II) and Co(II) // Inorg. Chem. 1995. V. 34, №24.
P. 6000-6007.
83. Que L., Pignolet L.H. Dynamic stereochemistry of tris-chelate complexes.
Tris(Dithio-carbamato) complexes of manganese(III), vanadium(III),
chromium(III), gallium(III) and indium(III) // Inorg. Chem. 1974. V. 13,
№2. P. 351-356.
84. Stolzer O., Unger E. Alkyl- and aryl-fluorophosphmes as ligand in
transition-metal complexes with metals in positive oxydation slates.
Nickel(II) and cobalt(II) halide complexes of di(t-butyl) fluorophosphines //
J.C.S. Dalton tr. 1973. №17. P. 1783-1788.
85. Grim S.O., Satek L.C. On the 31P spectrum of [(п-С4Н9)зР]з№С12 //
Z. Naturforsch. 1973. Bd. 28b, №9/10. S. 683-684.
86. Ниворожкин Л.Е., Константиновский Л.Е., Воронов M.A. и др. Спек-
тры ЯМР и конфигурационное равновесие в растворах 1,1-замещенных
бис-тиосалицилальдиминатов никеля // Ж. неорг. химии. 1973. Т. 18, №9.
С.2456-2459.
87. Basosi R., Gaggelli Е., Tiezzi Е., Valensin G. Nitrosyliron complexes with
mercapto-purines and -pirimidines studied by nuclear magnetic and electron
spin resonance spectroscopy // J.C.S. Perkin tr. II. 1975. №5. P. 423-428.
88. Обыночный А.А., Сагдеев P.3., Молин Ю.Н., Резвухин А.И. Стереоспе-
цифические закономерности сверхтонких взаимодействий в комплексах
Ni(AA)2 с циклическими и линейными аминами // Коорд. химия. 1975.
Т. 1, №6. С. 817-823.
89. Strouse J. I3C NMR studies of ferrous citrates in acidic and alkaline solutions.
Implications concerning the activ site of aconitase // J. Amer. Chem. Soc.
1977. V. 99, №2. P. 527-530.
90. Schneider J. UZ, Luoma J.R., Cusick J.P. A spectroscopic study of bonding
and structure in dihalotetrakis(toluolisocianide)iron(II) complexes // Inorg.
Chim. Acta. 1974. V. 10, №3. P. 203-209.
91. Reiff W.M., De Simone R.E. Electronic ground slates of di-
cyanobis(diimine)iron(III) compounds // Inorg. Chem. 1973. V. 12, №8.
P. 1793-1796.
92. Бабайлов С.П., Кригер Ю. Г. ЯМР-спектроскопические методы исследо-
вания молекулярной структуры парамагнитных комплексных соединений
лантаноидов в растворах и их использование для комплексов с краунэфи-
рами // Ж. структ. химии. 1998. Т. 39, №4. С. 714-730.
93. Bleany В. NMR shifts in solution due to lanthanide ions // J. Magn. Reson.
1972. V. 8, № 1. P. 91-100.
94. Hill H.A., Williams D., Zarb-Adaml N. Origin of isotropic shifts in lan-
thanide complexes: a study of the temperature dependence of the *H NMR
spectra of the tetrakis-N,N-dimet-hylthiocarbamatolantanate(III) anions //
J.C.S. Faraday tr. II. 1976. V. 72, №9. P. 1494-1502.
95. Birnbaum E.R., Stratton S. Studies of ethylenediamine complexes of the
lanthanide(III) perchlorates., rising NMR spectroscopy // Inorg. Chem. 1973.
V. 12, №2. P. 379-383. ''
96. Костромина H.A., Черногоренко В.Б., Тасыбаева Ш.Б. Исследование
комплексообразования меди(П) с фосфористой и фосфорной кислотой //
Укр. ж. химии. 1993. Т. 59, № 2. С. 115-119.
97. Miyake С., Imoto S. NMR of uranium glycinate // Chem. Phys. Lett. 1974.
V. 24, № 4. P. 606-607.
98. Neven C., Folcher G., Laurent A.M. Etude de complexes uranium(IV) acides
amines par electrochemie, spectroscopic d’absorption et resonance magne-
tique nucleare // J. Inorg. Nucl. Chem. 1976. V. 38, №6. P. 1223-1226.
99. Folcher G., Marqueti-Ellis H., Rigny P. et al. Etude spectroscopique d’un
complexe d’uranium(IV) a haute simmetrie U(NCS)e[N(CaH6)i]4 // J. Inorg.
Nucl. Chem. 1976. V. 38, №4. P. 747-753.
100. Miyake C., Sakurai H., Imoto S. PMR and magnetic susceptibility of
tetrakis-(dibenzoyl-methanato) uranium(IV) in solutions // Chem. Phys. Lett.
1975. V. 30, №2. P. 273-275.
101. Folcher C., Keller N., Klener C., Paris J. Kinetics of ligand exchange between
a uranium(IV) /З-diketonate and free /3-diketonate // Can. J. Chem. 1977.
V. 55, №20. P. 3559-3561.
102. Некипелов B.M., Шупик А. И., Замараев К. И. Изучение механизма рас-
творимости комплексоз переходных металлов методом ядерного магнитно-
го резонанса // Ж. физ. химии. 1975. Т. 49, №4. С. 1061-1063.
103. Шупик А.И., Лезина В.П., Некипелов В.М., Замараев К.И. Изучение
внешнесферных аддуктов Сг(АА)з методом ядерного магнитного резонан-
са // Ж. физ. химии. 1975. Т. 49, №4. С. 1063-1064.
104. Некипелов В.М., Щупик А.Н., Замараев К. И. Изучение методом протон-
ного магнитного резонанса внешнесферных аддуктов Со(П) // Ж. физ.
химии. 1975. Т. 49, №4. С. 1028-1029.
105. Свитым Р.В., Бучаченко А.Л., Яблонский О.П. и др. Комплексообра-
зование гидроперекисей с ацетилацетонатами переходных металлов //
Кинетика и катализ. 1976. Т. 17, № 1. С. 73-78.
106. Chan S.O. Eaton D R. The structure of the second coordonation sphere of
transition metal complexes: NMR Tj measurments // Can. J. Chem. 1976.
V. 54, №8. P. 1332-1340.
107. Китайгородский A. H., Некипелов В. M., Замараев К. И. Определение вре-
мени жизни хлороформа в сольватной оболочке трис-ацетилацетонатов
Сг(Ш) и Fe(III) // ДАН СССР. 1977. Т. 235, № 3. С. 622-625.
108. Swift T.J., Connick R.E. NMR-relaxation mechanism of 17O in aqueous
solutions of paramagnetic cations and lifetimes of water molecules in the first
coordination sphere // J. Chem. Phys. 1962. V. 37, №2. P. 307-320.
109. Swift T.J., Connick R.E. Errata: NMR-relaxation mechanism // J. Chem.
Phys. 1964. V. 41, №8. P. 2553-2554.
110. Granot /., Fiat D. Effect of chemical exchange on the transverse relaxation
rate of nuclei in solutions containing paramagnetic ions // J. Magn. Reson.
1974. V. 15, №3.' P. 540-548.
111. Матвеев В. В. Комплексы Со(П) и Ni(II) с этиловым спиртом в вод-
ных растворах по данным ПМР // Ж. физ. химии. 1976. Т. 50, № 1.
С. 220-221.
112. Счастнев П.В., Сагдеев Р.З., Душкин А. В. и др. Исследование комплек-
сов Ni(II) с водой и насыщенными спиртами методом ЯМР 'Н и 13С //
Ж. структ. химии. 1976. Т. 17, №4. С. 620-628.
113. Попель А. А., Захаров А.В. Исследование реакций обмена лигандов в эти-
лен-диаминовых комплексах меди (II) // Ж. неорг. химии. 1974. Т. 19,
№5. С. 1345-1348.
114. Шапник М.С., Гильманов А.Н., Ермакова В.Е., Музеев И.Х. Установле-
ние состава и расчет констант образования комплексных ионов меди (II)
с сульфосалицилат-анионом и этилендиамином по временам ПМ-релакса-
ции // Ж. неорг. химии. 1975. Т. 20, № 11. С. 3117-3119.
115. Панюшкин В. Т. Спектрохимия координационных соединений редкозе-
мельных элементов. — Ростов-на-Дону: Изд-во Ростов, ун-та. 1983. —
150 с.
116. Экспериментальные методы химической кинетики / Под ред. Н.Н. Эмма-
нуэля, Г. Б. Сергеева. — М.: Высш, школа, 1980. — 374 с.
117. Jackson W.G., McGregor В. С., Jurisson S.S. On the existence and lifetime
of the intermediate in substitution reactions of (1МНз)5СоХл // Inorg. Chem.
1987. V. 26, №8. P. 1286-91.
118. Бродский А.И. Химия изотопов. — M.: Изд. АН СССР, 1957. — 595 с.
119. Федотов М.А., Максимовская Р.И. Скорости обмена кислородом фос-
форванадиевых гетерополианионов с водой по ЯМР 17О // ДАН СССР.
1978. Т. 240, № 1. С. 128-131.
120. Федотов М.А., Максимовская Р.И., Бегалиева Д.У., Ильясова А. К.
Спектры ЯМР |7О и изотопный обмен в водных растворах фосфорвана-
дийволъфрамовых гетерополикислот // Изв. АН СССР. Сер. хим. 1980.
№7. С. 1477-1480.
121. Максимовская Р.И., Федотов М.А. О структуре гетерополианиона
РгМ^О^ (М = Мо, W) и кислородном обмене по ЯМР |7О // Изв. АН
СССР. Сер. хим. 1983. №2. С. 247-252.
122. Федотов М.А. О состоянии Rh(III) в разбавленных азотнокислых раство-
рах по данным ЯМР l4N, 17О, I03Rh // Коорд. химия. 2002. Т. 28, № 8.
С. 610-617.
123. Драго Р. Физические методы в химии. Т. 1 / Пер. с англ. А. А. Соловья-
нинова; Под ред. О. А. Реутова. — М.: Мир, 1981. С. 334-336.
124. McLaughlin A. A., Leigh J.S. Relaxation times in system with chemical
exchange: approximate solutions for nondilute case // J. Magn. Reson. 1973.
V. 9, № 2. P. 296-304.
125. Заводовский А. Г. Спин-решеточная релаксация и молекулярная динамика
переохлажденной воды // Ж. физ. химии. 1989. Т. 58, № 8. С. 2132-2135.
126. Kimura К. A New Formalism of Chemical Exchange near the Region of
Intermediate Rate // . J. Magn. Reson. 1983. V. 52, № 1. P. 13-22.
127. Lindman B., Forsen S. Chlorine, bromine and iodine NMR. NMR basic
Prine, and Progr. V. 12. — Berlin: Springer Verlag, 1976. — 365 p.
128. Reeves L. W., Shaw K.N. NMR studies of multi-site chemical exchange. II.
Hindered rotation in N,N-dimelhylcarbonyl fluoride // Can. J. Chem. 1971.
V. 49, №22. P. 3671-3682.
129. Kuhne R.O., Schaffhauser T., Wokaun A., Ernst R.R. Study of transient
chemical Reactions by NMR. Fast stopped-flow Fourier transform experi-
ments // J. Magn. Reson. 1979. V. 35, № 1. P. 36-37.
130. Reeves L. W., Shaw K.N. NMR studies of multi-site chemical exchange.
I. Matrix formulation of Bloch equations // Can. J. Chem. 1970. V. 48, № 23.
P. 3641-3653. Ibid. 1971. V.49, №22. P. 3671-3682.
131. Babushkin D.E., Talsi E.P. Multinuclear NMR spectroscopic charakteriza-
tion of Co(III) species: Key intermediates of cobalt catalyzed autooxidation //
J. Molec. Catal. A. 1998. V. 130, № 1. P. 131-137.
132. Померанцев H.M. Применение ядерного магнитного резонанса для анали-
тических целей // Завод, лаб. 1960. Т. 26, № 8. С. 950-956.
133. Шумиловский Н.Н., Скрипко А. Л., Король В. С., Ковалев Г. В. Методы
ядерного магнитного резонанса. — М.: Энергия, 1966. — 140 с.
134. Бородин П.М., Свентицкий Е.Н., Чижик В.Н. Применение метода ядер-
ного магнитного резонанса для исследования жидких сред // Завод, лаб.
1966. Т. 32, №5. С. 531-542.
135. Финкельштейн А.Д. Ядерный магнитный резонанс и его применение к
исследованию металлов // Завод, лаб. 1961. Т. 27, № 1. С. 58-61.
136. Хачатуров А. С. Изучение свойств высокомолекулярных соединений ме-
тодом ядерного магнитного резонанса // Завод, лаб. 1965. Т. 31, №8.
С. 948-956.
137. Бородин П.М., Мельников А.В., Морозов А.А., Чернышов Ю., Ядер-
ный С. магнитный резонанс в земном поле. — Л.: Изд. ЛГУ, 1967. —
232 с.
138. Декабрун Л.Л. Физические основы измерительных методик, использу-
ющих явление ядерного магнитного резонанса // Измерения, контроль,
автоматизация. 1974. №3. С. 3-10.
139. Щербаков В.Г., Купченко Е.И., Аспиотис Е.Х. Определение содержания
масла в единичном семени методом спинового эха // Изв. вузов. Пищ.
технология. 1973. №2. С. 122-124.
140. Conway T.F., Earie F.R. NMR for determining oil content of seeds //
J. Amer. Oil Chem. Soc. 1963. V. 40, №5. P. 265-268.
141. Вашман А. А., Мухин И.В., Пронин И.С. и др. Изучение комплексов
ди-2-этилгексилфосфата циркония методом ЯМР 31Р // Ж. неорг. химии.
1987. Т. 32, №5. С. 1050-1054.
142. Пронин И. С., Вашман А. А. Изучение комплексообразования в трибутил-
фосфатных экстрактах нитрата уранила с помощью ЯМР спектроскопии
2Н, HN, 3,Р // Ж. неорг. химии. 1987. Т. 32, № 3. С. 728-731.
143. Федотов М.А., Юхин Ю.М., Шубин А.А., Удалова Т.А. Исследова-
ние строения ди-2-этилгексилфосфата тетрагидроксогексависмутата(Ш) //
Ж. неорг. химии. 1998. Т. 43, №2. С. 307-310.
144. Толкачева Е. О., Сергиенко В. С., Илюхин А. В., Мешков С. В. Взаимодей-
ствие анионов WO4 и МоО4 C.l-оксиэтилидендифосфоновой кислотой по
данным спектроскопии 31Р и рентгеноструктурного анализа. Кристалличе-
ская структура Na8[W6Oi7(L*)2]-26H2O и NagWyC^- 14Н2О // Ж. неорг.
химии. 1997. Т. 42, № 5. С. 752-764.
145. Пронин И.С., Вашман А.А., Шорохов И.А. и др. ЯМР спектроско-
пия комплексов лантана и церия ди-2-этилгексилфосфатом циркония //
Ж. неорг. химии. 1988. Т. 33, № 11. С. 2775-2778.
146. Grethe Toth. I. 'Н NMR Studies of Bi3+-HO-System: Stoichiometric
Composition of the Hexanuclear Complex and Rate of Proton Exchange of
coordinated H2O and HO- in Mixed Acetone/Water Solution // Inorg. Chem.
1985. V. 24, № 15. P. 2405-2407.
147. Федотов M.A., Юхин Ю.М., Удалова T.A. Исследование строения
ди-2-этилгексилфосфатов висмута методом ЯМР на 31Р, 17О и других
ядрах // Ж. неорг. химии. 1997. Т. 42, №9. С. 1540-1546.
148. Коровин В.Ю., Рандаревич С.Б., Бодарацкий С.В., Трачевский В.В. Экс-
тракция скандия из сернокислых растворов ТБФ и ТВЭКС-ТБФ по дан-
ным ЯМР 31Р и 45Sc // Ж. неорг. химии. 1990. Т. 35, №9. С. 2404-2408.
149. Коровин В.Ю., Рандаревич С. Б., Бодарацкий С. В. и др. Механизм экс-
тракции скандия(Ш) Д2ЭГФК и ТВЭКС — Д2ЭГФК по данным спектро-
скопии ЯМР 31Р, 45Sc // Коорд. химия. 1991. Т. 17, №4. С. 561-567.
150. Курбатова Л.Д., Кодесс М.И., Максимовская Р.И. Изучение экстракции
ванадия(У) трибутилфосфатом методом ЯМР 51V и 31Р // Ж. неорг.
химии. 1996. Т. 41, № 12. С. 2111-2112.
151. Иванова С.И., Ткачев С.В., Иванов И.М. Химия экстракции теллура(У1)
растворами триалкилбензиламмоний хлорида в толуоле с добавками ка-
приловой кислоты из солянокислых растворов. Ж. неорг. химии. 2001.
Т. 46, № 4. С. 694-699.
152. Торгов В. Г., Шульман Р.С., Ус Т.В. и др. Влияние цветных металлов
на экстракцию нитронитрозоформ рутения(П) трибутилфосфатом из ней-
тральных растворов // Ж. неорг. химии. 2003. Т. 49, №7. С. 1221-1227.
153. Cemire А.Е., Jansen A.F., Marat К. А 21Р and l5N NMR Study of the
Extraction on Uranyl Nitrate by Di-2-Ethylhexyl Phosphoric Acid. Inorg //
Chim. Acta. 1985. V. 110, №3. P. 237-241.
154. Baker L.-J., Taylor J. Raman and 125Te NMR Spectroscopic Evidence on the
Solvent Extraction of Tellurum(IV) Species from HCI Solutions // J. Raman
Spectr. 1994. V. 25, № 3. P. 845-847.
155. Попель А. А., Сапрыкова З.А. Применение метода ядерной магнитной
релаксации для анализа сплавов и электролитов гальванических ванн //
Завод, лаб. 1965. Т. 31, №8. С. 957-959.
156. Rollwitz W.L. Moisture measurments in various hydroscopic materials using
NMR // Humidity and moisture. 1965. V. 2, №4. P. 137-147.
157. Steffa R.K., Blanck C.A., Wierbicki E., Cooper G.E. Application of NMR in
meat research // Food Res. 1969. V. 24, №5. P. 210-217.
158. Чернышев B.M., Бабкин А.Ф., Головкина Т.Н. и др. Применение ядер-
ной протонной релаксации для характеристики процессов замораживания
пищевых продуктов // Холодил. Техника. 1974. №4. С. 30-35.
159. Белых Л.Г., Куролени О.А., Скрипка А.Л. Измерение влажности уголь-
ной шихты методом ядерного магнитного резонанса // Завод, лаб. 1963.
Т. 29, №2. С. 168-172.
160. Слуцкий М.А. Применение ЯМР для определения влажности апатитового
концентрата // Завод, лаб. 1975. Т. 41, № 1. С. 54.
161. Child T.F., Jones D. UZ Broad-line NMR measurment of water assessibility
in cotton and wood-pulp celluloses // Cellulose Chem. Technol. 1973. V. 7,
№ 6. P. 525-534.
162. Warren R.J., Bender A. D., Stainger D.B., Zarembo J.E. Determination of
fluorine in organic and inorganic pharmaceutical compounds by high reso-
lution NMR spectrometry interfaced with computer system // Anal. Chem.
1978. V. 50, №3. P. 426-428.
163. Кучеряев А.Г., Лыдыкина B.M., Лебедев В.А. и др. Количественный
анализ сплава кремния — германия методом ядерного магнитного резо-
нанса // Завод, лаб. 1970. Т. 36, № 12. С. 465-471.
164. Шкаравский Ю. Ф., Лыпчак К.А., Черногоренко В. Б. Методы опреде-
ления фосфора в неорганических материалах. Современное состояние //
Завод, лаб. 1968. Т. 34, № 1. С. 3-14.
165. Кричевский Е.С., Слуцкий М. А. Применение метода ЯМР для экспресс-
ного определения и контроля содержания фосфора в апатитовом концен-
трате // Завод, лаб. 1968. Т. 34, №4. С. 435-436.
166. Чижик В. И., Чернышев Ю. С., Фролов В. Ф. и др. Прибор для определения
концентрации натрия и алюминия в технологических растворах алюмини-
евого производства // Завод, лаб. 1980. Т. 46, №3. С. 250-253.
167. Ursu I., Vilcu N. Uranium enrichment effect in uranium solution by pulsed
NMR methods // Rev. Roum. Phys. 1979. V. 24, №6. P. 561-565.
168. Рознятовский В.А., Чертков В.А., Гришин Ю.К. Изотопный анализ
дейтерированных органических соединений с помощью тройного ЯМР
13С{'Н, 2Н} // Завод, лаб. 1980. Т. 46, №9. С. 805-809.
169. Кессених А. В. ЯМР в промышленном производстве химических реактивов
и особо чистых веществ. — М.: НИИТЭХИМ, 1990. — 67 с.
170. Zamaraev K.I., Mastikhin V.M. New possibilities of NMR spectroscopy in
studies of adsorption and catalysis // Coll. Surf. 1984. V. 12, №. P. 401-427.
171. Mastikhin V.M., Zamaraev K.I. NMR Studies of Heterogeneous Catalysis //
Z. Phys. Chem. Neue Folge. 1987. Bd. 152. S. 59-80.
172. Иванова И. И. Спектроскопия ЯМР in situ в гетерогенном катализе: до-
стижения и перспективы // Росс. хим. журн. 1998. Т. 42, № 1-2. С. 67-85.
173. Федотов М.А., Максимовская Р.И. Структурные аспекты ЯМР в химии
полиоксометаллатов V, Мо, W // Ж. структ. химии. 2006. Т. 45, №6.
С. 961-84.
174. Матвеева Н.П., Вечерухин Н.М. Исследование гидродинамических эф-
фектов методом ЯМР в земном поле // Ядерный магнитный резонанс.
Вып. 7. - Л.: Изд. ЛГУ, 1988. С. 130-136.
175. Кенндел А. Метод ЯМР определения in situ относительных объемов и ин-
дивидуальных скоростей потоков в двухкомпонентных смесях // Приборы
научн. иссл. 1979. № 12. С. 47-52.
176. Жерновой А. И. Устройство для измерения времен релаксации в текущей
жидкости методом ЯМР // Завод, лаб. 1975. Т. 41, № 12. С. 1466-1467.
177. Эйткин М. Дж. Физика и археология / Пер. с англ. И. М. Беккермана,
В. С. Березинского; Под ред. Г. Н. Петровой. — М.: Изд. иностр, литера-
туры, 1963. — 258 с.
178. Смекалова Т.Н., Мельников А.В., Сухаржевский С.М., Горид А.Л. При-
менение радиоспектроскопических методов для исследования археологиче-
ских и геологических объектов // Ядерный магнитный резонанс. Вып. 7. —
Л.: Изд. ЛГУ, 1988. С. 3-7.
179. Lambert J.B., Frye J.S., Poinar G.O. Amber from the Dominican republic
analysis by nuclear magnetic resonance spectroscopy // Archeometry. 1985.
V. 27, № 1. P. 43-51.
180. Калабин Г.А., Каницкая Л.В., Кушнарев Д.Ф. Количественная спекиро-
скопия ЯМР природного органического сырья и продуктов его переработ-
ки. — М.: Химия, 2000. — 407 с.
181. Damadian R. Field focusing NMR (FONAR) and the formation of chemical
images in man // Philos. Trans. Roy. Soc. Lond. B. 1980. V. 289, № 1037.
P. 489-500.
182. Wyrwich A.M. Nuclear magnetic resonance of circulating blood // J. Magn.
Reson. 1982. V. 46, № 1. P. 176-179.
183. Van Putte K., Van den Enden J. Pulse NMR as quick method-for the
determination of the solid fat content in partially crystallized fats // Phys.
Rev. E: Sci. Instr. 1973. V. 6, №7. P. 910-912.
184. Van Putte K., Van den Enden J. Fully automated determination of solid
fat content by pulsed NMR // J. Amer. Oil Chem. Soc. 1974. V. 51, №7.
P. 316-320.
185. Макаренко В.Л., Грищенко А.Д., Аввакуумов А. А., Бабкин А., Исследо-
вание Ф. твердой и жидкой фазы в молочном жире импульсным методом
ЯМР // Изв. Вузов. Пищ. технология. 1975. № 1. С. 72-74.
186. Templeman G.J. Evaluation of several pulsed NMR techniques for solid-in-fat
determination of commersial fats // J. Food. Sci. 1977. V. 42, № 3.
P. 432-435.
187. Abezedarskaya L.A., Miftalhutdinova F.G., Fedotov V.D. Proton relaxation
in some protein solutions and gels // Mol. Biol. 1967. V. 1, №4. P. 384-390.
188. Mousseri J., Steinberg M. P., Nelson A.I. Bound water capacity of corn starch
and its derivative by NMR // J. Food. Sci. 1974. V. 39, № 1. P. 114-116.
189. Chield T.F., Pryce N.G. Steady-state and pulsed NMR studies of gelation in
aqueous agarose // Biopolymers. 1972. V. 11, №2. P. 409 429.
190. Woessner D.E., Snowden B.S. Pulsed NMR study of water in agar gels //
I. Coll. Interf. Sci. 1970. V. 34, № 2. P. 290-299.
191. Conway T.F., Coh.ee R.F., Smith R.J. NMR moisture analyser shows big
potential // Food Engn. 1957. V. 29, № 1. P. 80-83.
192. Miller B. S., Kaslow H.D. Determination of moisture by NMR and oven
method in wheat, flour, doughs and dried fruits // Food. TechnoL 1963. V. 16,
№5. P. 142-145.
193. Burnett L.J., Jackson J.A. Remout (Inside-out) NMR. II. Sensitivity of
NMR detection for external samples // J. Magn. Reson. 1980. V. 41, № 3.
P. 406-410.
194. Айххофф У. Фирма Bruker — от университетской лаборатории до меж-
дународной группы приборостроения // Росс. хим. ж. 1996. Т. 40, № 1.
С. 26-39.
ФЕДОТОВ Мартин Александрович
ЯДЕРНЫЙ МАГНИТНЫЙ РЕЗОНАНС В НЕОРГАНИЧЕСКОЙ
И КООРДИНАЦИОННОЙ ХИМИИ
РАСТВОРЫ И ЖИДКОСТИ