/


































































































































































































Текст
Experimental Methods
in Polymer Chemistry
Physical Principles and Applications
Jan F. Rabek
Department of Polymer Technology,
the Royal Institute of Technology, Stockholm,
Sweden
A Wiley-Interscience Publication
John Wiley & Sons
Chichester • New York • Brisbane • Toronto
Я.РАБЕК
ЭКСПЕРИМЕНТАЛЬНЫЕ МЕТОДЫ
В ХИМИИ
ПОЛИМЕРОВ
1
В 2-х частях
Перевод с английского
доктора хим. наук Я. С. Выгодского
под редакцией
академика В. В. Коршака
МОСКВА. МИР. 1983
ББК 24.7
Р 12
УДК 541. 64
Рабек Я.
Р 12 Экспериментальные методы в химии полимеров: в 2-х частях.
Пер. с англ.— М.: Мир, 1983.— 384 с., ил.— ч. 1.
Первое полное издание, в котором изложены основы практически всех физи-
ко-химических методов исследования полимеров. Автор, шведский ученый, останав-
ливает внимание читателя на деталях наиболее распространенных методик исследо-
вания. описания способов измерения, аппаратуре, возможностях применения каждого
метода. Полнота охвата материала в монографии делает ее также прекрасным
справочным пособием. В русском издании книга выходит в двух частях.
В ч. 1 рассмотрены общие вопросы химии полимеров, различные методы опреде-
ления молекулярного веса полимеров и ряд спектроскопических методов исследования
макромолекул.
Книга предназначена для химиков, работающих в области исследования, произ-
водства и применения полимеров, а также для студентов и аспирантов вузов.
р 1807000000-121
041 (01J-83
ББК 24.7
547
Редакция литературы по химии
© 1980 by John Wiley & Sons Ltd.
All rights reserved.
Authorised translation from the English
language edition published by John Wi-
ley & Sons Ltd.
(g) Перевод на русский язык, «Мир», 1983
ПРЕДИСЛОВИЕ
К РУССКОМУ ИЗДАНИЮ
Современная полимерная химия представляет собой область
науки, впитавшую в себя многие положения органической и не-
органической химии, физической и коллоидной химии, физики твер-
дого тела и других научных дисциплин. Это объясняется многооб-
разием химических структур высокомолекулярных соединений и
процессов их образования, спецификой свойств полимеров и приво-
дит к тому, что интерес ко многим, особенно промышленным поли-
мерам, не ослабевает уже на протяжении более 50 лет. Вместе
с тем необходимо отметить, что анализ полимеров, часто плохо
растворимых и не плавящихся до начала термического разложения,
сопряжен во многих случаях со значительными экспериментальны-
ми трудностями. Сказанное касается и изучения процессов образо-
вания высокополимеров. При этом, хотя задачу синтеза новых по-
лимеров нельзя считать более простой по сравнению с их анали-
зом, все же, вероятно, в идеале соотношение между химиками,
занимающимися исследованием полимеров, и химиками-синтетика-
ми должно быть существенно больше единицы.
Сложности, возникающие при исследовании полимеров, их хи-
мического строения, структуры и свойств, решаются с привле-
чением разнообразнейших физических и физико-химических мето-
дов, которые в ряде случаев модернизованы и модифицированы
для анализа высокомолекулярных соединений. Известен и ряд ме-
тодов, разработанных специально для изучения полимерных ве-
ществ. Представлялось бы, наверное, очень желательным, чтобы
химик-полимерщик в совершенстве владел всеми существующими
методами исследования полимеров. Однако, поскольку это вряд ли
осуществимо, минимально необходимо знание основ различных фи-
зико-химических методов, их возможностей. Только при этом, оче-
видно, можно ожидать наибольшей эффективности использования
тех или иных методов исследования полимеров и только тогда мо-
жет быть достигнута большая плодотворность творческого союза
между химиками, занимающимися синтезом полимеров, и физико-
химиками, которые изучают эти полимеры.
В книге Я. Рабека изложены основы различных физико-хими-
ческих методов, применяемых в настоящее время для изучения
процессов образования и свойств полимеров. Следует отметить, что
отдельные методы, широко используемые в различных областях
химии и физики, достаточно подробно освещены в специальных
монографиях и обзорах, где зачастую описываются различные
тонкости и последние достижения в развитии того или иного ме-
6
Предисловие к русскому изданию
тода. Поэтому научным работникам, только приступающим к ис-
следованиям в области полимеров, трудно ориентироваться в мно-
гообразии существующих методов, их основных принципах и воз-
можностях. Поэтому несомненной заслугой автора является то, что
он в одной книге представил практически все применяемые в на-
стоящее время методы анализа высокомолекулярных соединений,
включая и самые современные, и описал их в простой и доходчи-
вой форме.
В книге, состоящей из 40 глав, основное место, естественно,
уделяется описанию различных методов исследования полимеров.
Представлены все методы определения молекулярных весов поли-
меров, их молекулярновесового распределения, обсуждаются раз-
нообразные спектральные методы, применяющиеся для анализа
строения и структуры гомо- и сополимеров: УФ-, ИК-, КР-спектро-
скопия, эмиссионная спектроскопия, спектроскопия ЯМР, масс-
спектроскопия, спектроскопия ЭПР, нейтронное рассеяние, анниги-
ляция позитронов. Ряд глав посвящен хроматографическим мето-
дам, таким, как газовая и жидкостная хроматография, в том числе
и при высоких давлениях, тонкослойная хроматография, ионооб-
менная хроматография, ситовая хроматография, включая гель-про-
никающую хроматографию, хроматография с обращением фаз.
Методы анализа структуры полимеров обсуждаются при рассмот-
рении электронной микроскопии, рентгеноструктурного анализа, ди-
фракции электронов и ряда других методов. Физические свойства
полимеров оцениваются с помощью таких методов, как дилатомет-
рия, определение температур плавления и стеклования полимеров,
их электрических характеристик, анизотропии, диффузии и поверх-
ностного натяжения. Представлены также методы исследования
различных видов деструкции полимеров.
Важную роль в книге играют два небольших по объему ввод-
ных курса (гл. 1 и 26), в которых приводятся важнейшие понятия
полимерной химии, касающиеся структуры полимеров, их стерео-
химии, конформации, морфологии. Несомненным достоинством этих
глав (и всей книги в целом) являются содержащиеся в них опре-
деления разнообразных, в том числе и очень распространенных
терминов, что делает данную книгу полезным справочным посо-
бием. Определения, которые имеются в этой книге, трудно подчас
найти даже в энциклопедических изданиях. (Однако следует иметь
в виду, что в некоторых случаях приводимые автором определения
несколько отличаются от тех, которые широко используются в оте-
чественной литературе.) Многочисленные фотографии, приведен-
ные в главе, посвященной морфологии полимеров, несомненно спо-
собствуют лучшему усвоению вопросов, связанных с кристаллиза-
цией полимеров и организацией различных надмолекулярных
структур.
Еще одна заслуживающая на наш взгляд особого внимания
глава (гл. 2) отводится автором для описания взаимодействий
Предисловие к русскому изданию
7
полимеров с растворителями, играющими первостепенную роль в
различных методах анализа полимеров. При рассмотрении раство-
римости и совместимости полимеров автор приводит весьма обшир-
ные табулированные данные о растворимости полимеров в раз-
личных растворителях, смешиваемости растворителей, параметрах
растворимости различных растворителей. Кратко рассмотрены во-
просы набухания сетчатых полимеров и образования жидкокри-
сталлических растворов полимеров.
Учитывая требования об использовании в литературе Между-
народной системы единиц (СИ), в книге выделена специальная
глава (гл. 40), в которой описана эта система единиц и приводятся
таблицы, облегчающие переход от системы единиц СГС в СИ и
наоборот.
Обилие рассмотренных методов исследования и связанное с
этим огромное количество публикаций предопределили и особые
требования к форме -представления библиографических данных.
В книге имеется единый список литературы, состоящий из двух
частей: обзорной литературы [1—1482] и периодической литера-
туры [2001—7350]. Это позволило избежать неизбежного в ином
случае повторения литературных ссылок. Такая обильная библио-
графия, насчитывающая таким образом около 7000(!) наименова-
ний, позволяет желающим подробно ознакомиться с различными
особенностями рассматриваемых в книге методов и последними до-
стижениями в их развитии.
Мы надеемся, что данная монография, представляющая собой
как справочное, так и методическое пособие, будет очень полезна
студентам химико-технологических и химических вузов и факуль-
тетов университетов, аспирантам и научным работникам, присту-
пающим к исследованиям в области химии высокомолекулярных
соединений; несомненный интерес она представит и для опытных
исследователей, посвятивших себя полимерной науке.
В. Коршак
Я. Выгодский
Памяти моего отца, профессора Т. И. Рабека, ос-
нователя института технологии полимеров (1950—
1965) при Вроцлавском политехническом универ-
ситете (Польша), выдающегося ученого и педа-
гога, который ввел меня в мир полимерной хи-
мии.
знать что-то обо всем и все о чем-то ...
ПРЕДИСЛОВИЕ АВТОРА
Исследования в области макромолекулярной химии требуют
надлежащих знаний физико-химических методов, используемых в
этой отрасли науки. Для успешной трактовки экспериментальных
данных весьма важно также хорошее знакомство с применяемой
аппаратурой. Полимерщик сегодня обязан быть в курсе основ этих
методов и их практических приложений, иными словами, он дол-
жен разбираться в физических принципах, лежащих в основе того
или иного метода, и в экспериментальных методиках, включая опи-
сание способов измерения,характеристик приборов и методов при-
готовления образцов для исследований, области их практического
использования. Он должен уметь также производить необходимые
расчеты и знать, как следует представлять результаты. Как я об-
наружил, многие химики-полимерщики в своей повседневной ра-
боте нуждаются в практическом руководстве в форме ясных ре-
комендаций, не осложненных математическими и теоретическими
рассуждениями.
В этой книге предпринята попытка изложения данных в про-
стой, доступной и последовательной форме, без вхождения в де-
тали физико-химической теории. В намерения автора книги не
входили подробное описание различного оборудования и выдача
тех или иных рекомендаций по выбору соответствующих приборов
для решения специфических задач. Рассмотрены лишь те характе-
ристики, которые важны для понимания современных эксперимен-
тальных методик. Вкратце обсуждаются условия эксперимента, в
отдельных случаях для облегчения анализа указываются и неко-
торые особенности его проведения, например условия приготовле-
ния образцов для исследования или способы очистки измеритель-
ных кювет. В ряде случаев такие сведения могут оказаться чрез-
Предисловие автора
9
вычайно полезными. По этой же причине в книгу включены УФ-,
ИК- и ЯМР-спектры наиболее распространенных растворителей,
использующихся в спектральном анализе, а также таблицы для
перевода различных единиц, применяемых в спектроскопии.
В ответ на понятное всем требование единообразия Междуна-
родный союз по теоретической и прикладной химии (ИЮПАК)
рекомендует пользоваться Международной системой единиц (СИ),
представляющей собой, по существу, стандартизованные единицы
МКС. В США, однако, большинство химиков все еще предпочи-
тают пользоваться системой единиц СГС вместе с такими обыч-
ными неметрическими единицами, как калория и атмосфера. Еди-
ницы СИ все чаще появляются в учебных пособиях и особенно в
журналах, поэтому важно знать обе системы единиц — СГС и СИ.
До тех пор пока единицы СИ не будут приняты повсеместно, оста-
нется необходимость перехода от одной системы к другой. По этой
причине в книге параллельно используются обе системы. Глава 40
посвящена переходам от одной системы к другой, в ней даются
также определения наиболее распространенных констант, напри-
мер универсальной газовой постоянной, числа Авогадро, констан-
ты Больцмана.
В начале каждой главы указана литература, имеющая отноше-
ние ко всему рассматриваемому в ней вопросу. В конце всех раз-
делов приводятся ссылки на обзорную литературу [О: 1 —1482] и
периодическую литературу [П: 2001—7349], которые посвящены
отдельным аспектам той или иной проблемы. Эти ссылки не яв-
ляются исчерпывающим списком литературы по вопросам, которые
обсуждаются в этой книге, но тем не менее они могут оказать
существенную пользу в дальнейших исследованиях.
Книга рассчитана на студентов и научных работников, нуждаю-
щихся в ясном и кратком вводном курсе в экспериментальную
химию полимеров, в котором содержались бы также сведения экс-
периментального и методического плана, в том числе касающиеся
интерпретации и представления полученных данных.
Книга состоит из сравнительно небольших по объему разделов,
в которых дается краткое и систематическое описание физической
и экспериментальной химии полимеров. Как подсказывает автору
его педагогический опыт, такой порядок изложения наиболее удо-
бен для студентов, дипломников и технологов, уровень знаний и
практические навыки которых могут быть очень различными.
В частности, это относится к студентам из развивающихся
стран, где промышленность только начинает создаваться, и по-
тому к доступности изложения предъявляются повышенные тре-
бования.
Эта книга может также быть полезной при последующем изу-
чении специальных монографий и оригинальных статей.
Я благодарен редакторам и издателям журналов и книг за
разрешение использовать в книге материал из этих изданий и не
10
Предисловие автора
могу не выразить благодарность за долготерпение членам моей
семьи, которые из-за работы над книгой были лишены моего об-
щества на протяжении бесчисленных вечеров, уик-эндов и отпу-
сков.
Ян Рабек
Глава 1
КРАТКИЕ СВЕДЕНИЯ
О ПОЛИМЕРНЫХ СТРУКТУРАХ
Обзорная литература: 124, 127, 402, 424, 456, 457, 503, 637, 680, 737, 747,
804, 871, 909, 943, 1040, 1078, 1081, 1083, 1120, 1134, 1139, 1178, 1244, 1256, 1289,
1298, 1299, 1302, 1336, 1349, 1350, 1396, 1429.
1.1. ОБЩИЕ ОПРЕДЕЛЕНИЯ
Очень большая молекула называется макромолекулой, макромо-
лекулярным (высокомолекулярным) соединением или полимерной
молекулой. Если соединение содержит лишь небольшое число
структурных звеньев, его обычно называют олигомером.
Полимером называется макромолекулярное вещество, которое
обозначают термином полимер, соответствующим названием, напри-
мер поливинилхлорид, и формулой, например (—СН2—СНС1—)п,
где п — число структурных звеньев (элементарных звеньев), кото-
рые называют мерами или мономерными единицами.
Макромолекулярное, или высокомолекулярное, соединение все-
гда состоит из смеси гомологических полимерных соединений. Не-
значительные различия в составе обусловлены наличием концевых
групп, случайными ветвлениями, изменениями в ориентации меров,
нерегулярностью в последовательности различных типов меров
(в сополимерах) и в ряде случаев нерегулярностями другого типа *.
Регулярный полимер — это полимер, построенный из элемен-
тарных звеньев одного и того же типа.
Элементарное звено — это наименьшая из возможных повто-
ряющаяся единица.
Номенклатура структур регулярных линейных полимеров
имеется в публикациях, приведенных в списке обзорной литера-
туры [О: 1453, 1454].
Изомерные полимеры — это полимеры, имеющие практически
одинаковый процентный состав, но различную молекулярную
структуру.
Гомополимер — это полимер, состоящий из молекул, содержа-
щих (без учета концевых групп, мест ветвления и других незна-
чительных нерегулярностей) один, два или более типов различных
химических структур в регулярной последовательности, т. е. стати-
стически расположенных. .
* Предложено реальные полимеры, содержащие аномальные звенья, обо-
значать как разнозвенные полимеры (Коршак В. В. Разнозвенность полимеров. —
М.: Наука, 1977, с. 301). — Прим. ред.
12
Глава 1
Сополимер — это полимер, состоящий из молекул, содержащих
большое число звеньев двух или нескольких различных химиче-
ских типов в нерегулярной последовательности.
К сополимерам относят продукт сополимеризации' даже в том
случае, если полагают, что он состоит лишь из двух типов меров,
чередующихся регулярно в полимерной цепи. Регулярность в таких
чередующихся сополимерах, по всей вероятности, никогда не яв-
ляется особенно строгой.
Терполимер — это сополимер, в котором расположения двух
(трех, четырех или более) химически различающихся звеньев об-
разуют нерегулярную форму.
Линейный полимер (сополимер)—это полимер, в котором
звенья каждой молекулы связаны в цепную структуру (рис. 1.1).
линейный полимер разветвленный. полимер сшитый полимер
Рис. 1.1. Схематические изображения структур основных цепей.
Разветвленный полимер — это полимер, состоящий из молекул,
имеющих разветвления: между точками ветвления, а также вдоль
ветвлений структура цепеподобна (рис. 1.1).
Повторяющееся звено линейного полимера — это часть моле-
кулярной цепи такого состава, что соединением большого числа
данных звеньев друг с другом гипотетически может образовываться
вся макромолекула (за исключением концевых групп, которые все-
гда химически отличаются от других структурных звеньев цепи,
точек ветвления и т. п.).
Мономерное звено, или мер, линейного полимера — это такая
часть макромолекулы, которая содержит атомы такого же типа и
в том же количестве, что и реальный или гипотетический мономер.
Элементарное звено и мер могут быть как одинаковыми, так
и отличаться друг от друга. В полиэтилене, например, мер
(—СН2—СН2—) больше, чем элементарное звено (—СН2—).
В полимерах, получаемых в результате полимеризации, моле-
кулярный вес повторяющихся звеньев (например, СН2—СНС1—)
равен молекулярному весу мономера (например, СН2=СНС1).
В полимерах, получаемых поликонденсацией, молекулярный
вес элементарных звеньев [например, —NH—(СН2)/г—СО—] все-
Краткие сведения о полимерных структурах
13
гда отличается от молекулярного веса мономера [например.
NH2—(СН2)П—СООН] из-за выделения в процессе реакции низко-
молекулярных продуктов (например, воды).
Степень полимеризации можно описать как число (среднее)
повторяющихся звеньев (включая те, которые находятся в начале
и в конце макромолекулярной цепи) в молекуле.
Степень полимеризации (СП) связана с молекулярным весом
полимера следующим соотношением:
СП = М/т, (1.1)
где М — молекулярный вес полимера, m — молекулярный вес по-
вторяющегося звена.
Сшитый полимер — это полимер, построенный из макромоле-
кул, образующих трехмерную сетку (см. рис. 1.1).
При рассмотрении структуры полимера следует иметь в виду
следующие два аспекта:
а) полимерную конфигурацию, которая определяет форму цепи
полимера, образованную основными валентными связями;
б) полимерную конформацию, которая определяет форму цепи
полимера, обусловленную вращением вокруг основных валентных
связей.
1.2. СТЕРЕОХИМИЯ МАКРОМОЛЕКУЛ
В термин структурная регулярность включаются следующие два
понятия:
а) регулярность повторяемости (структурная изомерия), т. е.
регулярность, с которой повторение следует вдоль полимерной
цепи;
б) стереорегулярность (тактичность, или пространственная изо-,
мерия), т. е. регулярность пространственного порядка повторяю-
щегося звена.
1.2.1. Структурная изомерия
Винильная полимеризация таких мономеров, как СН2—CRR' (где
R =/= R', R'#=H), обычно протекает по типу присоединения «голова
к хвосту» и приводит к образованию структурно регулярных поли-
меров, содержащих последовательности типа (—СН2—CRR'—
—СН2—CRR'—). Группу CRR' называют голова, а группу СН2 —
хвост. Полимеры содержат структуры «голова к голове»
(—СН2—CRR'—CRR'—СН2—) и «хвост к хвосту» (—CRR'—СН2—
—СН2—CRR'—) (иногда называемые синсефалическими последо-
вательностями).
Такие мономеры, как
ch2=cr—сн==сн2
J 2 3 4
14
Глава 1
где R Н, могут подвергаться 1,4-, 1,2- или 3,4-полимеризации.
В результате протекания полимеризации двух последних типов об-
разуются структурно регулярные последовательности (структуры I
и 11 соответственно)
-CHa-CR-
сн
сна
I п
1.2.2. Пространственная изомерия
Различные конфигурации в регулярном полимере возникают при
наличии стереоизомерных центров. Стереоизомерными центрами в
макромолекулярных цепях являются: а) двойные связи, ответствен-
ные за цис-тране-конфигурации (цис-транс-изомерия); б) атомы
углерода, соединенные с двумя различными группами R и R' (тет-
раэдрические стереоизомерные центры).
1.2.3. цис-транс- Изомерия
^ис-Изомеры (111) или транс-изомеры (IV) образуются при 1,4-по-
лимеризации таких мономеров, как CH2=CR—CH=CH2(R=#H).
Структурно регулярные полимеры III и IV называют соответ-
ственно цис-тактическими и транс-тактическими.
V
-СНг сна— —СН2 с
/с=сС V хсн„-
R Н I
К
III IV
1.2.4. Тетраэдрическая стереоизомерия
Атактический полимер — это полимер, у которого стереоизомерные
конфигурации при всех центрах основной цепи являются совершен-
но беспорядочными.
Изотактический полимер — это тактический полимер, у кото-
рого одинаковыми являются стереоизомерные конфигурации отно-
сительно основной цепи.
Синдиотактический полимер — это тактический полимер, у ко-
торого стереоизомерные конфигурации регулярно чередуются от-
носительно основной цепи.
На рис. 1.2 представлены фишеровские проекции для тактиче-
ских полимеров общей формулы (—СН2—CHR—)п и гипотетиче-
ски вытянутые зигзагообразные цепи изотактического и синдиотак-
тического полимеров.
Краткие сведения о полимерных структурах
15
Две связи цепи, соседние с углеродным атомом, представляю-
щим стереоизомерный центр, обозначаются знаками плюс и минус
(рис. 1.3). С конфигурационных позиций идентичными являются
такие два мономерных звена, у которых соответствующие связи
характеризуются одинаковым рядом знаков плюс и минус.
В изотактических полимерах общей формулы (—СН2—CHR—)п
два мономерных звена являются конфигурационно энантиоморф-
Рис. 1. 2. Различные конформации полимерных цепей,
а —изотактический полипропилен; б—синдиотактический полипропилен.
ными, когда соответствующие связи характеризуются чередова-
нием противоположных знаков
-СН2-5±2-СН-^-СН2-^СН-^-СН2-^±-’-СН—
С") | <+) (') | ( + ) (-) |
R R R
В синдиотактических полимерах общей формулы (—СН2—
—CHR—)п связи характеризуются последовательностью одинако-
вых знаков
^СН2^СН^СН2-к±СН-Ш-СН^СН--
L 1 ।
R R R
16
Глава 1
При характеристике стерической изомерии полимеров общей
формулы (—СН2—CRR'—)п удобно рассматривать структуры ло-
кализованных сегментов цепей.
(+) R=H
Рис. 1.3. Тетраэдрические стереоизомерные центры. Обозначение связей, соседних
со стереоизомерным центром.
Последовательность двух следующих друг за другом мономер-
ных звеньев в структурно регулярной полимерной цепи, например
(—СН2—CRR'—)rt, называется тактическим расположением, или
тактической диадой.
Рис. 1.4. Различные типы тактических расположений, или тактических диад.
а —изотактическое расположение, или изотактическая диада (лезо-метиленовая группа
с гетеростерической симметрией), б—синдиотактическая диада (рацемическая метиленовая
группа с гомостерической симметрией).
Изотактическое расположение, или изотактическая диада, а
также синдиотактическое расположение, или синдиотактическая
диада, в гипотетической вытянутой (зигзаг) конформации пока-
заны на рис. 1.4.
Центральная метиленовая группа изотактического расположе-
ния обозначается как .мезо-метиленовая группа, или гп-единица.
Краткие сведения о полимерных структурах
17
Центральная метиленовая группа при синдиотактическом рас-
положении обозначается как рацемическая метиленовая группа,
или r-единица. Последовательность трех следующих друг за дру-
гом мономерных звеньев в структурно регулярных цепях полиме-
ров типа (—СН2—CRR'—-)„ на-
зывается тактической триадой.
Все три содержащиеся в
основной цепи центра простран-
ственной изомерии имеют одина-
ковую конфигурацию в изотакти-
ческой триаде. Центральная еди-
ница в изотактической триаде
называется i-единицей и эквива-
лентна последовательности mm.
Находящиеся в основной цепи
центры пространственной изоме-
рии чередуются в синдиотактиче-
ской триаде (рис. 1.5). Централь-
ная единица в синдиотактической
триаде называется s-единицей и
эквивалентна последовательно-
сти гг.
В гетеротактической триаде
(рис. 1.5) находящиеся в основ-
ной цепи два соседних центра
пространственной изомерии име-
ют одинаковую конфигурацию, а
третий центр — противоположную
конфигурацию. Центральная еди-
ница гетеротактической триады
называется h-единицей и эквива-
лентна последовательности mr
или rm.
Шесть тактических тетрадных
последовательностей мономер-
ных звеньев в полимере типа
(-—СН2—CRR'—)п обозначаются
Рис. 1.5. Различные конформации
тактических триад.
а —изотактическая триада, г —единица или
последовательность mm', б — синдиотакти-
ческая триада, s-единица или последова’
тельность гг; в — гетеротактическая триада,
^-единица или последовательность rm.
символами mmm, mmr=rmm,
rmr, mrr=rrm, mrm и rrr.
Симметрия метиленовой группы, в которой протоны различают-
ся по симметрии, называется гетеростерической, а симметрия ме-
тиленовой группы, в которой протоны эквивалентны, — гомостери-
ческой.
Тактические полимеры типа (—СН2—CRR'—)п являются моно-
тактическими, когда элементарное звено содержит только один
центр изомерии в основной цепи.
Диазот актический полимер ~ это полимер, элементарное звено
которого содержит в качестве компонентов основной цепи дв^
18
Глава 1
углеродных атома, последние имеют по два различных боковых
заместителя, пространственная ориентация которых позволяет счи-
тать молекулу изотактической относительно конфигурации вокруг
атомов основной цепи каждого типа, рассмотренной отдельно.
Существует два вида диизотактических полимеров, отличаю-
щихся по стерическим конфигурациям относительно таких двух
типов углеродных атомов (рис. 1.6).
1. эритро-Диизотактический полимер — это диизотактический
полимер, в котором конфигурации при двух находящихся в основ-
Фишеровская
проекция
н —|— R
Н------R'
Н-----R
Н — — R'
Н-----R
Н----• R'
Н —— R
Н-----R*
Рис. 1.6. Различные конформации цепей диизотактического полимера.
а — эрмтро-диизотактическая конфигурация; б—трро-диизотактическая конфигурация.
ной цепи центрах стерической изомерии в повторяющемся звене
одинаковы.
а) В гипотетически вытянутой (зигзагообразная конформация)
молекуле эритро-диизотактического полимера общей формулы
(—CHRCHR'—)л все заместители одного типа (R) располагаются
по одну сторону от плоскости атомов цепи, тогда как все другие
заместители (R') находятся по другую ее сторону.
б) В фишеровской проекции все заместители R и R' находятся
по одну сторону от линии, представляющей основную цепь.
в) В ньюменовском изображении заслоненного образования
двух последовательных атомов цепи и присоединенных к ним ато-
мов и групп, в котором последующий атом цепи накладывается на
предшествующий атом цепи, R накладывается на R' и Н на Н.
2. трео-Диизотактический полимер — это диизотактический по-
димер, в котором конфигурации при двух находящихся в основной
Краткие сведения о полимерных структурах
19
цепи центрах стерической изомерии являются противоположными
в каждом повторяющемся звене.
а) В гипотетически вытянутой (зигзагообразной конформации)
молекуле трео-диизотактического полимера общей формулы
(—CHRCHR'—)п заместители обоих типов, R и R', находятся по
одну сторону от плоскости атомов основной цепи.
б) В фишеровской проекции заместители R и R' находятся по
разные стороны от линии, представляющей основную цепь.
в) В ньюменовском изображении заслоненного образования
двух последовательных атомов цепи и присоединенных к ним ато-
мов и групп, в котором последующий атом накладывается на
предшествующий атом, R накладывается на Н и Н на R'.
Обзорная литература: 648.
В диизотактических полимерах общей формулы (—CHR—
—CHR'—)п два тетраэдрических стереоизомерных центра дают две
конфигурации, которые определяются, как это было указано выше.
Рис. 1.7. Диизотактические конфигурации: эритро(а) и трео(б). Пары (+, —)
или (—, +) определяют относительную конфигурацию эритро, тогда как пары
(—, —) или (+, +) определяют относительную конфигурацию трео.
1. эритро-Диизотактическая конфигурация (рис. 1.7), характе-
ризующаяся парами знаков (—) (+) или (+) (—).
1111
R R' R R'
го
Глава I
2. трео-Диизотактическая конфигурация (рис. 1.7), характери-
зующаяся парой знаков (—) (—) или (+) ( + ).
_CH—---—CH СИ—('-СН (~^
1111
R R' R R'
Обзорная литература: 127, 135, 175, 177, 327, 336, 385, 402, 428, 457, 637,
648, 680, 737, 966—968, 1078, 1091, 1120, 1136, 1178, 1289, 1299, 1336, 1349, 1396,
1416, 1452.
Периодическая литература: 2513, 2520, 2923, 3520, 5283, 5284, 5286, 5458,
5471, 5974.
Определения для дисиндиотактических полимеров (эритро- или
трео-конфигурация) аналогичны использующимся при обозначении
соответствующих диизотактических полимеров.
1.2.5. Статистика последовательностей
Значительный теоретический интерес, особенно при изучении сте-
реоспецифической полимеризации, представляет распределение
конфигурационных последовательностей в цепях винильных поли-
меров.
Наиболее распространено описание последовательностей с по-
мощью:
а) статистики Бернулли, согласно которой результат какого-то
события характеризуется одной-единственной вероятностью, кото-
рая не зависит от результатов всех предыдущих событий;
б) статистики Маркова, по которой распределение тактических
расположений зависит только от природы предшествующей после-
довательности тактических расположений.
Более подробные сведения об этих распределениях и механизме
их возникновения можно почерпнуть из специальных монографий
и оригинальных работ.
Обзорная литература: 175, 578, 843, 872, 1416.
Периодическая литература: 2924, 5968.
Для расчета структур сополимеров и терполимеров, в которых
используется статистика Маркова, разработаны программы для
ЭВМ [О: 579; П: 3924].
1.3. СОПОЛИМЕРЫ
Обзорная литература: 10, 27, 225, 226, 270—272, 933, 936, 978, 1046, 1244.
1.3.1. Типы сополимеров
По структуре сополимеры можно разделить на перечисленные
ниже группы (рис. 1.8).
Краткие сведения о полимерных структурах
21
1. Статистические сополимеры образуются в массе, растворе,
водной суспензии или эмульсии в присутствии свободнорадикаль-
ных инициаторов перекисного типа или окислительно-восстанови-
Рис. 1.8. Схематическое изобра-
жение сополимеров различных
типов.
а —статистический сополимер;
б—сополимер с упорядоченными
последовательностями; в-регулярно
чередующийся сополимер; г — блок-
сополимер; б —привитой сополимер;
е — сшитый сополимер.
тельных систем. Возможно также инициирование под действием
облучения.
2. Статистические сополимеры с упорядоченным распределе-
нием последовательностей образуются при регулируемом дозирова-
нии мономеров в ходе процесса сополимеризации.
3. Регулярно чередующиеся сополимеры образуются в ходе
специально разработанных методов сополимеризации. Реакцион-
ную способность полярных мономеров можно увеличить, комплек-
суя их с галогенидами металлов или алюминийорганическими га-
логенидами. Такие мономерные комплексы участвуют в реакции
одноэлектронного переноса с некомплексованным мономером или
другим электронодонорным мономером.
4. Привитые сополимеры (разд. 1.3.2).
5. Блок-сополимеры (см. разд. 1.3.3).
6. Сшитые сополимеры образуются путем создания свободно-
радикальных центров в предварительно полученном полимере в
присутствии мономера. Прививка происходит по этому центру, а
в результате реакции обрыва по механизму рекомбинации обра-
зуется сшивка.
22
Глава 1
1.3.2. Привитые сополимеры
Все привитые сополимеры построены из основной полимерной цепи
(А), к которой привит ряд последовательностей (В) (рис. 1.9).
Привитые сополимеры обычно получают радикальной, анион-
ной или катионной (аддиционной) полимеризацией или полимери-
Основная цепь
—д----д------д---1---А---А-----А—
I I
| | Привитая
В В цепь
В
В
Рис. 1.9. Схематическое изображение привитого сополимера.
зацией с раскрытием цикла мономера в присутствии предвари-
тельно полученного реакционноспособного полимера.
Реакции прививки и образующиеся в результате их привитые
сополимеры описываются следующими параметрами:
а) Полная степень превращения (%)==
__ Суммарный вес привитого сополимера
Вес загруженного мономера
б) Степень прививки (%) = в-^-спРивитых «епей . 100;
7 г ’ Вес исходного полимера ’
в) Эффективность прививки (%)==
__________Вес привитых цепей_______
Суммарный вес привитого сополимера
• 100;
• 100;
Г) Вес («%)=р---------привитых цепей------------
7 ' 7 Суммарный вес привитого сополимера ’
д) Частота прививки = Среднее число привитых последователь-
ностей в расчете на привитую цепь.
Трудно точно охарактеризовать структуру привитых сополиме-
ров. Представляет также затруднение измерение длины привитых
цепей и их полидисперсности. Дополнительные осложнения возни-
кают из-за отсутствия ясности в вопросе об определении центров
прививки в цепи основного полимера. До настоящего времени эти
проблемы относятся к числу нерешенных.
Обзорная литература: 225, 226, 270—272, 933, 1046, 1244.
Краткие сведения о полимерных структурах
23
1.3,3. Блок-сополимеры
Блок-сополимеры построены из химически разнородных сегментов,
концы которых соединяются друг с другом (рис. 1.10).
Обычно блок-сополимеры получают последовательной виниль-
ной анионной полимеризацией, полимеризацией с раскрытием цик-
ла или поликонденсацией.
Характеризовать блок-сополимеры труднее, чем гомополимеры
или однородные статистические сополимеры. Трудность представ-
Рис. 1.10. Схематическое изображение блок-сополимеров различных типов.
а—блок-сополимер типа А—В; б—блок-сополимер типа А—В—А; в—блок-сополимер типа
-(А— В— )л; а--трехлучевой блок-сополимер; б —шестилучевой блок сополимер.
ляет определение длины блоков и структуры блок-сополимеров
(например, установить различие между А—В и А—В—А). Число
сегментов в блок-сополимере иногда можно рассчитать исходя из
метода синтеза их. В блок-сополимерах типа А—В и А—В—А по
определению имеются соответственно одна и две точки соедине-
ния. В блок-сополимерах (—А—В—)„ расстояние между межсег-
ментными связями можно вычислить, зная молекулярный вес
блоков.
Обзорная литература: 10, 11, 27, 156, 225, 226, 270—272, 416, 933, 935, 936,
9?8, 1244, 1281, 1431,
24
Глава 1
1.3.4. Распределение последовательностей
и тактичность в сополимерах
Три различных типа сополимеров — статистические
АААВААВВАВВВ, регулярно чередующиеся АВАВАВАВАВАВАВ
и блок-сополимеры АААВВВАААВВВ — характеризуются различ-
ными распределениями звеньев А и В по основной полимерной
Таблица 1.1
Особенности структуры сополимеров а
с различным распределением мономерных звеньев А и В
Мономеры Диады Триады А АА ААА [ АВ ВА ] В вв ввв
ВАА ААВ ABB BBA
Тетрады ВАВ АААА ААВА АВАА АВА ABBA
ВАЛА АААВ ААВВ ВВАА BABA АВАВ | ВВВА 1 АВВВ
Пентады ВААВ ААААА BABB ВВАВ АААВА АВААА ВВВВ ABABA
ВАААА ААААВ ВВААА ВВААА ВААВА АВААВ 1ВВАВА АВАВВ
ВАААВ ВААВВ ВВААВ ВВАВВ
АВВВА ААВВА АВВАА ААВАА
ВВЙВА АВВВВ ААВВВ ВВВАА ВАВВА АВВАВ ВАВАА ААВАВ
АВВВВ ВАВВВ ВВВАВ ВАВАВ
а Harwood Н. in: Characterization of Materials and Research: Ceramics
and Polymers, J. J. Burke, V. Weiss, (eds.), Chapter 11, Syracuse, 1975, p. 316.
цепи. Имея различную структуру, такие полимеры по свойствам
могут существенно отличаться друг от друга, несмотря на то что
состав их одинаковый. Многие свойства сополимеров зависят от
относительного содержания различных диад, триад, тетрад, пентад
или других, более высших последовательностей (табл. 1.1). Важ-
ную информацию о структуре сополимеров можно получить при
изучении различных химических и физических свойств,
Краткие сведения о полимерных структурах
25
Химическая активность функциональных групп в сополимерах
зависит от пространственных эффектов, участия соседних групп и
ионного взаимодействия. Так, в частности, реакционная способ-
ность звеньев А в сополимере АВ может быть различной в зави-
симости от того, входят такие звенья в триады AAA, ААВ (ВАА)
или ВАВ. На реакционную способность звеньев А в различных
триадах могут также оказывать влияние конфигурационные эф-
фекты.
Физические свойства сополимеров часто зависят от распреде-
ления последовательностей мономеров, вместе с тем на практике
большинство определяемых свойств являются средними от вкла-
дов различных типов структурных звеньев.
Обзорная литература: И, 272, 576—578, 944.
Периодическая литература: 3129, 3704, 5743, 6210, 6496.
1.3.5. Определение композиционной неоднородности
сополимеров
Композиционная неоднородность сополимеров может быть обуслов-
лена условиями их образования или преднамеренным смешением.
Для оценки композиционной неоднородности существуют сле-
дующие методы: гель-проникающая хроматография (ГПХ) с од-
новременным определением дифференциального показателя пре-
ломления и ультрафиолетового и/или инфракрасного спектров;
тонкослойная хроматография (ТСХ); седиментация до достижения
равновесного градиента плотности.
Указанные методы дают полезную информацию о реакциях,
протекающих при синтезе сополимеров, и/или природе промыш-
ленных полимеров.
Периодическая литература: 4686, 5294, 6126, 6509.
1.3.6. Исследование распределения последовательностей
и тактичности
Для определения расположения мономерных звеньев и конфигу-
рационных структур гомополимеров и сополимеров используются
следующие методы:
1. Физические методы: определение степени кристалличности,
температуры стеклования, температуры плавления, изучение теп-
лоты полимеризации (сополимеризации), инфракрасная спектро-
скопия, спектроскопия комбинационного рассеяния, нейтронная
спектроскопия, ЯМР-спектроскопия, измерение дипольных мо-
ментов;
2. Химические методы: селективная деструкция с последующим
анализом продуктов деструкции, пиролитическая газовая хрома-
тография, пиролитическая масс-спектроскопия, циклизация внутри
и между последовательностями, изучение реакционной способно-
сти полимеров.
26
Глава 1
1.3.7. Циклизация внутри последовательностей
и между ними
Реакции циклизации имеют место в тех случаях, когда соседние
заместители в цепях гомо- и сополимеров могут реагировать с
образованием циклических структур с пяти- и шестичленными цик-
лами. Реакции циклизации весьма полезны при изучении структур
сополимеров и терполимеров.
Обзорная литература: 578.
Периодическая литература: 3811, 4330—4332, 6351, 6352.
1.4. КОНФОРМАЦИЯ МАКРОМОЛЕКУЛ
Конформация молекулы определяется как одно из отличных от
других пространственных положений, принимаемых ее атомами
или группами атомов при вращении вокруг одинарных связей, или
как изменение в относительных расположениях данного звена, со-
единенного несколькими одинарными связями.
Молекулы с неперекрываемыми конформациями называются
конформерами, ротамерами или конформационными изомерами.
Подробную информацию о существовании и стабильности кон-
формеров можно получить из энергетических расчетов.
Обзорная литература: 106, 135, 175, 402, 457, 571, 630, 637, 1083, 1134, 1349.
Периодическая литература: 2006, 2031, 2287, 2288, 3134, 3417, 3359, 3350,
4899, 5089, 5908, 6681.
1.4.1. Внутренние углы вращения в полимерах
Описать конформацию основной полимерной цепи можно с по-
мощью точного определения последовательности внутренних углов
вращения (рис. 1.11).
Внутренний угол вращения (б) —это угол, образованный двумя
плоскостями LiL2 и L2L3, который, как видно, выражает конфор-
мацию цепи вокруг связи L2: а) угол 6 положительный при вра-
щении по часовой стрелке на угол менее 180°; б) угол 6 отрица-
тельный при вращении против часовой стрелки на угол менее 180°.
Ориентация данной связи относительно двух предшествующих
связей цепи обозначается по Банну А, В, С, а по Тадокоро Т, G, G
соответственно (рис. 1.12):
А(7), транс (6= 180°),
В(6), левая го ил (б = +60°),
C(G), правая гош (б = —60°).
Указанные три основные последовательности связей в угле-
родных цепях, образованных одинарными связями (рис. 1.13), яв-
ляются единственными разрешенными в рамках зигзагообразного
связывания. В том случае, когда лейтмотив цепи или геометриче-
ское повторяющееся звено содержит более чем одну последова-
Краткие сведения о полимерных структурах
27
Рис. 1.11. Условные обозначения для
углов внутреннего вращения (д).
Рис. 1.12. Обозначение по Банну (А, В, С) и по Тадокоро (Т, G, G) для кон-
формации С—С.
Конформация: а—В (G) левая гош\ б — к(Т} транс-, в—С (О) правая гош.
28
Глава 1
тельность связей одного и того же типа, она обозначается соответ-
ствующим цифровым индексом, например Т2, (Т’С)з и т. п. Воз-
можны также смешанные случаи, например TGTG или (ТС)2(ГО)2.
Рис. 1.13. Три последовательности связей в углеродных цепях с простыми свя-
зями, которые соответствуют типу зигзагообразных связей;
Конформация: а —А (Г) транс; 6 — B(G) левая гош; в — G (G) правая гош.
G^ {TG)3 TGTG (TG)Z(TG)Z T^GT^G
Рис. 1.14. Пять конформаций цепей, основанные на ориентациях зигзагообразных
связей Т, G и G [П: 2642].
Согласно Тадокоро, различные конформации цепи и характе-
ристические периоды идентичности обозначаются так, как это
указано в табл. 1.2 и на рис. 1.14.
Для обозначения предпочтения различных конформаций поль-
зуются характеристическим отношением (Z):
Z = (1.2)
где — среднеквадратичное расстояние между концами для слу-
чая свободного вращения цепи [см. уравнение (2.30)], п — число
связей, I — длина связи,
Краткие сведения о полимерных структурах
29
Это отношение можно рассчитать с помощью статистической
механики, исходя из энергетики конформаций относительно корот-
Таблица 1.2
Характеристические периоды идентичности
в различных конформациях цепей
Период идентичности Период идентичности
тип конформации длина, А тип конформации длина, А
т2 2,5 (™)з 6,2
5,0 (™)з _ 6,2
TGTG 4,4 T3GTZG 8,8
g4 3,6 (TG)2 (TG)2 8,5
ких участков цепи или модельных соединений. Его можно также
определить экспериментально (разд. 2.11).
Обзорная литература: 16, 457, 1427.
Периодическая литература- 2002, 2004, 2005, 2125, 2151, 2286, 2422, 2423,
2444, 2503, 2525—2527, 2640—2642, 2645—2647, 2902, 2977, 3410, 3417, 3422, 3550,
3620, 3721, 3854, 3969, 4618, 4764, 4943, 4985, 4986, 5087, 5138, 5189, 5315, 5447,
5449, 5451, 5452, 5453, 5455, 6113, 6202, 6306, 6367, 6567, 6624—6626, 6628, 6634,
6668, 6713, 6743, 6811, 7102, 7270, 7277, 7278.
1.4.2. Номенклатура спиральных конформаций
Многие природные и синтетические полимеры имеют спиральную
Рис. 1.15. Номенклатура спиральной конформации.
Этот тип структуры описывают с помощью особой номенклатуры
спиральной конформации.
Известны две системы такой номенклатуры (рис. 1.15).
1. Обозначение по номенклатуре точечных спиралей [точки,
решетки, образованные пересечением спиралей (//)]:
ДФя = 2лр/Р = ЪяЦи (1.3)
kzR = p = c[u (1.4)
30
Глава 1
2. Обозначения по номенклатуре винтовых осей (S):
Дф5 = 2л/и (1.5)
\Zs — y(c/u) (1.6)
Обе системы обозначений связаны между собой выражением
Y/ = ew+l (1.7)
где ДФ — угол вращения; Дг —перемещение вдоль оси спирали Z;
Р— эквивалент перемещения на один виток спирали (шаг спи-
рали) по оси спирали Z; р — проекция расстояния между после-
дующими эквивалентными точками на спирали вдоль оси спира-
ли Z; t — число (целое) витков спирали в периоде идентичности с;
с — кристаллографический период идентичности, параллельный оси
спирали Z; и — число (целое) точек, или лейтмотивов, в спирали,
отвечающее периоду с; у — положительное целое число (>0); 6 —
положительное целое число (^0). Оси хну атомов компонентов
относительно какого-то произвольно выбранного начала спираль-
ной структуры не обозначаются.
Важно подчеркнуть, что обе системы вместо мономерного зве-
на, или мера, используют термин мотив на виток (Р/р или u/t),
представляемый одной точкой на спирали.
Обзорная литература: 16, 135, 273, 402, 457, 928, 1048, 1134, 1427.
Периодическая литература: 2051, 2502, 2503, 2902, 2903, 2917, 3677, 4171,
4613, 4775, 4943, 5183, 5189, 5285, 5287, 5290, 5315, 5407, 5448, 5454, 5456, 5459,
5460, 5467, 5954, 6362, 6611, 6669, 6689, 6696, 6764, 7018, 7032, 7217, 7268.
Кристаллографические данные по номенклатуре точечных спи-
ралей суммированы в таблицах, имеющихся в работах [О: 16, 958].
1.5. РАЗВЕТВЛЕННЫЕ ПОЛИМЕРЫ
5
Рис. 1.16. Схематическое изображение корот-
ких и длинных ветвлений.
а —короткоцепочечные ветвления; б—длинноцепочеч-
ные ветвления.
О,
Разветвленным полимером называют полимер, в котором есть бо-
ковые ответвления от основной цепи. В зависимости от условий
реакции растущая поли-
мерная цепь может под-
вергаться короткоцепо-
чечному или длинноцепо-
чечному ветвлению
(рис. 1.16), при этом ка-
ждое из ветвлений имеет
тот же состав, что и
основная полимерная
цепь. Свойства полимеров
зависят как от количе-
ства, так и от распреде-
ления по цепи ветвлений
обоих типов.
Большинство разветвленных полимеров относится к одной из
следующих трех групп (рис. 1.17): а) правильные (регулярные)
Краткие сведения о полимерных структурах
31
звездообразные, б) пра-
вильные (регулярные)
гребнеобразные и в) ста-
тистические древообраз-
ные.
Ветвление в полимер-
ных молекулах математи-
Рис. 1.17. Схематическое изображение различ-
ных типов ветвления.
а —регулярное звездообразное ветвление (3 луча);
б — регулярное звездообразное ветвление (4 луча);
в —регулярное гребнеобразное ветвление; а —стати-
стическое древообразное ветвление.
чески описывается в виде
функции числа точек вет-
влений, которые они со-
держат. Такая математи-
ческая функция назы-
вается числом точек вет-
вления и обозначается буквой g (разд. 2.5.15). Способы определе-
ния g с помощью характеристической вязкости и ГПХ рассмат-
риваются в разд. 9.1.4 и 25.15.
Обзорная литература: 532, 538, 630, 1226.
Периодическая литература: 2099, 3818—3821, 5137, 5139, 6432, 6582.
1.6. СШИТЫЕ ПОЛИМЕРЫ
Сшитыми полимерами называют полимеры, которые имеют трех-
мерную сетчатую структуру и вследствие этого являются нерас-
творимыми.
Сшивка по структуре может быть идентична основной цепи
или отличаться от нее.
Сшитый продукт, который набухает в растворителях, назы-
вается гелем: Гели очень маленького размера (300—1000 мкм) на-
зываются микрогелями.
Микрогели из-за присущей им высокой плотности ветвлений
ведут себя как плотно упакованные сферы и суспендируются в
растворителях.
Гелеобразование начинается после достижения определенной
степени превращения, называемой гелъ-точкой или точкой геля.
Сшитые полимеры можно характеризовать следующими пара-
метрами:
1. Длина цепи в сетке, определяющая число элементарных
звеньев между двумя точками ветвления.
2. Точка ветвления, являющаяся центром, из которого исходит
более двух цепей.
3. Среднечисловой молекулярный вес цепи сетки, определяе-
мый как _ __
(MH)c==Mra/fc, (1.8)
где (Мп)с — среднечисловой молекулярный вес сшивки, Мт —
средний молекулярный вес мономерного звена, — степень
32
Глава !
ветвления, которая определяется как
____________Число молей сшитых мономерных звеньев__________
Общее число молей мономерных звеньев в реакционной системе
(1.9)
4. Плотность сшивания (или степень сшивания) (Г) представ-
ляющая собой число сшитых мономерных звеньев на первичную
цепь и определяющаяся как
r=(WX (i.io)
где ($U)o — среднечисловой молекулярный вес первичной цепи
(первичная цепь — это линейная молекула перед сшиванием),
(Мп) с — среднечисловой молекулярный вес сшивки.
Рис. 1.18. Схематическое изображение дефектов сетки.
а'-непрореагировавшие функциональные группы или концы цепей; б—замкнутые петли;
в — сшитая петля; г — запутанности цепей; д — запутанности петель.
5. Молярная концентрация эффективных цепей сетки, которая
определяется как _ ___
[Afc]eff = [AfJ {1-2 («ШУ (l.H)
(моль/г в системе СГС или моль/кг в системе СИ), где [Мс]eft —
молярная концентрация эффективных цепей сетки, [Мс] — моляр-
ная концентрация всех присутствующих цепей для (Мп)о > (Мп)с,
(Мп)о и (Мп) с определены выше. При очень высокой степени сши-
вания, когда число свободных концов слишком велико, уравнение
(1.11) становится неприменимым. Число свободных концов цепей
возрастает по мере уменьшения среднечислового молекулярного
веса первичной цепи.
Обзорная литература: 286, 402, 482, 630, 1107, 1245, 1246.
Периодическая литература: 2290, 3250, 3259, 3718, 3735, 3751, 4086, 4412,
4555, 5271, 5356, 6391.
Сшитые полимеры характеризуются наличием нескольких типов
дефектов сетки (рис. 1.18): а) непрореагировавшие функциональ-
Краткие сведения о полимерных структурах 33
ные группы или концы цепей; б) замкнутые петли; в) запутанно-
сти. Такие дефекты влияют на эластические свойства полимерных
сеток.
Обзорная литература: 455, 630, 1203, 1246.
Периодическая литература: 3248, 3249, 3992, 4702, 5736, 5909, 6485, 6809,
6815.
1.7. СМЕСИ ПОЛИМЕРОВ
Обзорная литература: 153, 777, 868, 1046, 1128, 1244.
Полимерной смесью (полисмесъю) называют физическую смесь
двух или более различных полимеров и/или сополимеров, которые
не связаны ковалентными связями.
Полимерные смеси можно приготовить несколькими способами:
а) механическим смешением на вальцах или в экструдерах;
б) полимеризацией одного мономера в присутствии другого по-
лимера;
в) испарением или осаждением смеси растворов полимеров;
г) коагуляцией смеси полимерных латексов.
Совместимость в полисмесях нельзя определить однозначно.
Свое происхождение этот термин ведет от смешения двух жидко-
стей. Когда жидкости смешиваются с образованием гомогенной
и однофазной смеси, то говорят, что такие жидкости совместимы.
С термодинамических, кинетических и механических позиций го-
могенная однофазная полимерная смесь практически невероятна.
Совместимость в полимерных смесях — это характеристика
того, насколько полисмесь может сильно приблизиться к однофаз-
ной смеси и представляет собой относительную меру степени ге-
терогенности полимерной смеси.
Периодическая литература: 4051, 4343, 4481, 5334, 5730, 5799, 6295.
1.7.1. Определение совместимости полисмесей
Существует ряд способов определения совместимости полимерных
смесей.
1. Растворение в общих растворителях. При разделении фаз
компоненты несовместимы. Поскольку фазовое разделение зависит
от концентрации полимера и температуры, то такой способ яв-
ляется достаточно произвольным и дает лишь качественную кар-
тину.
2. Отливка пленок. Отливают пленку из гомогенного разбав-
ленного раствора полисмеси. Образование мутной, крошащейся
пленки указывает на несовместимость. Этот способ также весьма
груб и произволен.
3. Качество расплавов. Расплавленную полисмесь прессуют в
тонкие листы. Если такие листы прозрачные, это свидетельствует
о совместимости, тогда как мутность означает несовместимость.
И этот метод является произвольным и грубым.
2 Зак. 238
34
Глава 1
4. Температура стеклования. Если полисмесь проявляет две
различные температуры стеклования, отвечающие соответствую-
щим полимерам, то такая смесь несовместима. При наличии у по-
лисмеси только одной температуры стеклования смесь считается
совместимой (разд. 32.5.8).
5. Измерение динамических механических характеристик. Это
наиболее чувствительный метод, который широко используется на
практике. При испытании на тор-
Рис. 1.19. Электронная микрогра-
фия при сканировании скола по-
верхности полимерной смеси поли-
стирол — полиэтилен (75/25 по ве-
су) [П: 3982].
зионном маятнике получают соот-
ветствующие кривые для отдельных
полимеров и их смесей и сравни-
вают полученные результаты. Для
совместимой смеси характерно на-
личие максимума демпфирования,
лежащего между максимумами со-
ответствующих компонентов, тогда
как несовместимая смесь дает два
максимума затухания при темпера-
турах, отвечающих соответствую-
щим полимерам смеси. Динамиче-
ский механический метод может
также давать информацию о моду-
ле сдвига или модуле упругости.
Наличие на кривой «модуль — тем-
пература» нескольких переходов
указывает на несовместимость по-
лисмеси.
6. Микроскопия. Методом опти-
ческой микроскопии с контрастиро-
ванием фаз определяется гетерогенность при размерах порядка
0,2—10 мкм. Во многих случаях гетерогенность можно оценить
с помощью электронной микроскопии, позволяющей определить
гетерогенность в частицах до 0,01 мкм (рис. 1.19). Гетерогенность,
обнаруживаемая микроскопическими методами, весьма относи-
тельна.
7. Малоугловое рассеяние рентгеновских лучей (МРРЛ).
При описании совместимости полимерных смесей желательно,
чтобы приводились данные о составе полисмеси и характеризова-
лись ее компоненты, о предыстории образца, а именно о способе
его приготовления, о методе, использовавшемся для изучения сов-
местимости, и приборе, на котором проводились эти измерения, об
экспериментальных результатах и выводах из них.
Периодическая литература: 2199, 2982, 3881, 3982, 4938, 5031, 6379, 6383,
6587, 7046, 7052.
Многочисленные данные о совместимых полимерах приведены
В Polymer Handbook [О: 154].
Глава 2
ИЗУЧЕНИЕ ВЗАИМОДЕЙСТВИЯ ПОЛИМЕРОВ
С РАСТВОРИТЕЛЯМИ
Обзорная литература: 94, 127, 247, 386, 402, 456, 622, 623, 747, 943, 1067,
1178, 1289, 1302, 1307, 1336, 1429.
Взаимодействие макромолекул и различных растворителей мо-
жет вызывать разрушение отдельных межмолекулярных когезион-
ных сил, приводя к структурным перегруппировкам.
Идеальный растворитель характеризуется способностью раство-
рять какое-то количество полимера в температурной области, огра-
ниченной температурой кристаллизации раствора или низкотемпе-
ратурным расслоением и температурой, при которой давление па-
ров раствора превышает 1/760 атм. На практике такие идеальные
растворители для полимеров отсутствуют.
Нерастворитель характеризуется неспособностью растворять
какое-то количество полимера при любой температуре и атмосфер-
ном давлении. В табл. 2.1 приведен список растворителей и не-
растворителей для распространенных полимеров. Более подробные
данные суммированы в Polymer Handbook [О: 487].
Некоторые полимеры хорошо растворяются в смесях
растворителей. Смешиваемость растворителей показана на
рис. 2.1.
Многочисленные данные о физических свойствах большин-
ства известных растворителей приведены в Polymer Handbook
10: 452].
Некоторые растворители могут в определенной степени вызы-
вать сольватацию полимера (первоначальное взаимодействие с по-
лимером). Второй стадией сольватации является растворение.
Остаточное содержание растворителя в образце полимера оце-
нивают, нагревая отлитые пленки в вакууме до постоянного веса.
При этом изменение веса, как полагают, обусловлено удалением
остаточного растворителя.
Обзорная литература: 141, 160, 161, 262, 574, 766, 777, 872.
Периодическая литература: 2162—2165, 2428, 2429, 3193, 3203, 4044, 4120,
4449, 5704, 5705, 5993, 6308, 6876—6878.
2.1. ИЗМЕНЕНИЕ ОБЪЕМА ПРИ СМЕШЕНИИ
Изменение объема при смешении может иметь место, когда рас-
творитель и твердый полимер смешиваются друг с другом. Изме-
ренный объем (V) отличается от суммы объемов (Vo) обоих ком-
понентов до смешения.
2*
Таблица 2.1.
Растворители и нерастворители для полимеров [О: 487]
Растворители и нерастворители а
Полимер ацетон спирты (метанол, этанол) алифатические углеводороды (гексан, гептан) ароматические углеводороды (бензол, толуол) сероуглерод четыреххлористый углерод хлороформ циклогексан циклогексанон диметилформамид диоксан сложные эфиры простые эфиры нитробензол метиленхлорид тетрагидрофуран вода
Полиакриловая кислота, атактическая + — — + +
Полиакриламид — — — — — — — — +
Полидиены: полибутадиен полиизопрен — — + + + + 4- — — — + + —
Полиэтилен высокой плотности — — 4* > 133°С — + > 200°С — —
низкой плотности — — 4- 80°С — — —
Полиизобутилен — — + + + + 4- — — + —
Полиметилакрилат + — — + + 4- + + + — + —
Полиметилметакрилат + — — + + 4- — + + 4~ — + —
Полипропилен, атакти- ческий — — + — — — — — — — + —
Полисилоксаны, обыч- ные — — 4- + + 4- + —
Полистирол — — — + + + 4- >35°С + + — + —
Поливинилацеталь + + — + + 4- + + — + —
Поливинилацетат + — + — 4- + + + —
Поливиниловый спирт — — — — — + — — — — +
Поливинилхлорид высокомолекулярный низкомолекулярный — — — — — — —- + + + + + + + + + + —
Простые виниловые эфиры, обычные + + + + + + —
Поливинилиденхлорид — — — — — — __ — + — 4" (гор.) —
Принятые обозначения: + растворяется в, жидкости; — не растворяется в данной жидкости.
38
Глава 2
I не смешиваются
Г~1 смешиваются
Рис. 2.1. Смешиваемость различных растворителей.
Уксусная, кислота____
Ацетон---------------
Ацетонитрил__________
Бензол_______________
Бутанол______________
Четыреххлористый углерод
Хлороформ________
Циклогексан______
Циклопентан______
дихлорэт ан______
Метиленхлорид____
Димотилсрормамид-
Диметйлсульфоксид.
Диоксан__________
Этилацетат_______
Этанол..
Диэтиловый 3tpup_
Гептан___________
Гек сан__________
Метанол._________
Метилэтилкетон_
трет-Октан_______
Пентан___________
трет-Пропанол____
Дапропиловый эфир.
Тетрахлорэтан—
Тетрагидрофуран
Трихлорэтан
Вода
Ксилол.
Коэффициент термического расширения (а) определяется диф-
ференцированием аналитического выражения наблюдаемых изме-
нений объема как функции температуры (Г):
a==F Cdr)p-Const
Периодическая литература: 3407, 3408, 3280, 5613.
2.2. КОНЦЕПЦИЯ СВОБОДНОГО ОБЪЕМА
В РАСТВОРАХ
Свободный объем (Vf) в растворе при температуре Т определяется
как объем, вызываемый термическим движением молекул и нахо-
дится из формулы
Vf = VT-VQ, (2.2)
где Vt — суммарный объем раствора при температуре Т (в кельви-
нах); Vo — теоретический молярный объем наиболее плотной упа-
ковки молекул растворителя при 0 К.
Изучение взаимодействия полимеров с растворителями
39
Доля свободного объема (Vf) определяется как отношение сво-
бодного объема (Vt) к полному объему (Ут):
,,_Vf _ VT~Vo
vf~ VT
VT
(2.3)
(см. также разд. 32.2).
Обзорная литература: 585.
Периодическая литература: 3095—3097,
3211, 3477, 4055, 4886, 5601, 6186.
2.3. КРИВЫЕ ОСАЖДЕНИЯ И ТЕТА-ТЕМПЕРАТУРА
Температура осаждения — это температура, при которой становит-
ся возможным визуальное определение мутности. Температура оса-
ждения (Тр) определяется при медленном охлаждении (1—
2°С/10 мин). Эта температура в пределах 0,2°C должна согласо-
Со де ржание па лимер а, об. доля
Рис. 2.2. Фазовая диаграмма для си-
стемы полимер — растворитель.
Рис. 2.3. Кривые осаждения в виде
зависимости Тс от vM^2.
ваться с температурой, при которой мутность исчезает при нагре-
вании.
Температура осаждения данного полимера в данном раствори-
теле как функция объемной доли твердого вещества может быть
представлена в виде фазовой диаграммы (рис. 2.2).
Критическая температура осаждения (Тс) находится из фазо-
вой диаграммы как максимум на кривой зависимости температуры
осаждения от объемной доли твердого вещества. Приведенную на
рис. 2.2 кривую называют кривой точки мутности или кривой оса-
ждения.
Кривые осаждения, построенные в виде зависимости Тс от
О1/2 (где v — удельный объем полимера и М — молекулярный вес
полимера), представляют собой прямые линии (рис. 2.3).
Тета-температура (температура Флори) (0) является темпера-
турой, при которой взаимодействия между полимером и
40
Глава 2
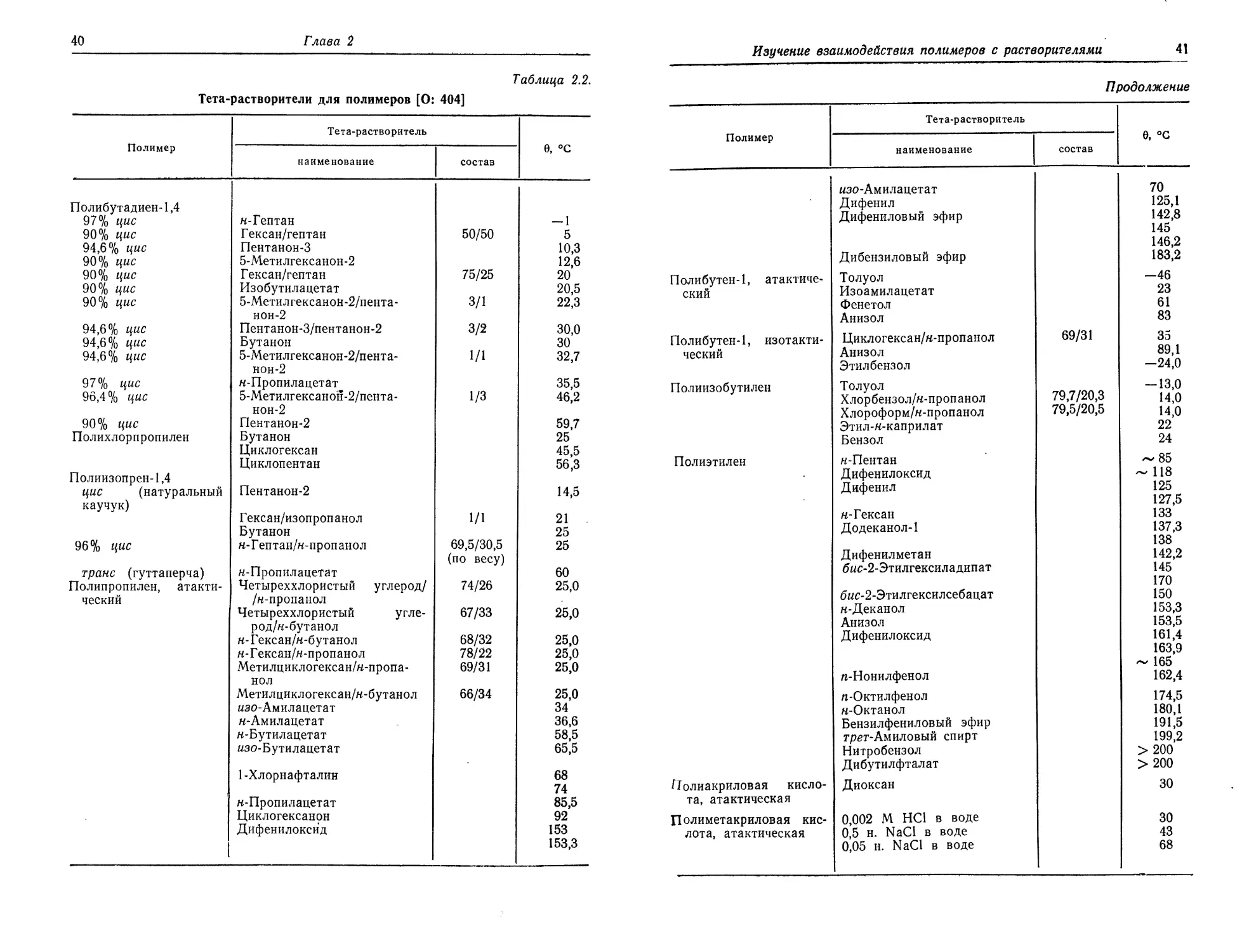
Тета-растворители для полимеров [О: 404]
Таблица 2.2.
Полимер Тета-растворитель е, °с
наименование состав
Полибутадиен-1,4
97% Цис «-Гептан -1
90% цис Гексан/гептан 50/50 5
94,6% цис Пентанон-3 10,3
90% цис 5-Метилгексанон-2 12,6
90% цис Гексан/гептан 75/25 20
90% Цис Изобутилацетат 20,5
90% Цис 5-Метилгексанон-2/пента- нон-2 3/1 22,3
94,6% цис Пентанон-З/пентанон-2 3/2 30,0
94,6% цис Бутанон 30
94,6% Цис 5-Метилгексанон-2/пента- нон-2 1/1 32,7
97 % цис «-Пропилацетат 35,5
96,4% цис 5-Метилгексанон-2/пента- нон-2 1/3 46,2
90% цис Пентанон-2 59,7
Полихлорпропилен Бутанон Циклогексан Циклопентан 25 45,5 56,3
Полиизопрен-1,4
цис (натуральный каучук) Пентанон-2 14,5
Гексан/изопропанол Бутанон 1/1 21 . 25
96% цис «-Гептан/«-пропанол 69,5/30,5 (по весу) 25
транс (гуттаперча) «-Пропилацетат 60
Полипропилен, атакти- ческий Четыреххлористый углерод/ /н-пропанол 74/26 25,0
Четыреххлористый угле- род/«-бутанол 67/33 25,0
н-Гексан/н-бутанол 68/32 25,0
н-Гексан/«-пропанол 78/22 25,0
Метилциклогексан/н-пропа- нол 69/31 25,0
Метилциклогексан/н-бутанол «зо-Амилацетат «-Амилацетат н-Бутилацетат «зо-Бутилацетат 66/34 25,0 34 36,6 58,5 65,5
1-Хлорнафталин н-Пропилацетат 68 74 85,5
1 Циклогексанон Дифенилоксид 92 153 153,3
Изучение взаимодействия полимеров с растворителями
41
Продолжение
Полимер Тета-растворитель 6, °C
наименование состав
«зо-Амилацетат Дифенил Дифениловый эфир 70 125,1 142,8 14^
Дибензиловый эфир 146,2 183,2
Полибутен-1, атактиче- ский Толуол Изоамилацетат Фенетол Анизол -46 23 61 83
Полибутен-1, изотакти- ческий Циклогексан/я-пропанол Анизол Этилбензол 69/31 35 89,1 -24,0
Полиизобутилен Толуол Хлорбензол/я-пропанол Хлороформ/я-пропанол Этил-я-каприлат Бензол 79,7/20,3 79,5/20,5 — 13,0 14,0 14,0 22 24
Полиэтилен я-Пентан Дифенилоксид Дифенил я-Гексан Додеканол-1 Дифенилметан бяс-2-Этилгексиладипат бяс-2-Этилгексилсебацат я-Деканол Анизол Дифенилоксид я-Нонилфенол -85 - 118 125 127,5 133 137,3 138 142,2 145 170 150 153,3 153,5 161,4 163,9 — 165 162,4
я-Октилфенол я-Октанол Бензилфениловый эфир трет-Амиловый спирт Нитробензол Дибутилфталат 174,5 180,1 191,5 199,2 > 200 >200
Полиакриловая кисло- та, атактическая Диоксан 30
Полиметакриловая кис- лота, атактическая 0,002 М НС1 в воде 0,5 н. NaCl в воде 0,05 н. NaCl в воде 30 43 68
42
Глава 2
П родолжение
Полимер Тета-растворитель 0, °C
наименование состав
Полиметилметакрилат, Хлороформ —273+50
атактический Дихлорэтан —233+50
Бензол —223+50
Метилметакрилат -163+50
Бутанон --98
Этилацетат --98
Толуол -65+Ю
Ацетон -55+10
Бензол/н-гексан 70/30 20
Бензол/изопропанол 38/62 20
Диоксан/н-гексан 59/41 20
З-Метилбутанон-2/н-гексан 83/17 20
Диоксан/циклогексан 53/47 20
Полистирол, атактиче- н-Бутилформиат -9
ский 1-Хлор-н-декан 6,6
Гекси л-jw-кси лол 12,5
Циклогексан/толуол 86,9/13,1 15
транс-Декалин 18,2
трннс-Декалин/^нс-Декалин 79,6/20,4 19,3
транс-Декалин 23,8
Бензол/н-гексан 39/61 20
Бензол/изопропанол 66/34 20
Диоксан/н-гексан 38/62 20
Диоксан/изопропанол 55/45 20
Бутанон/изопропанол 85,7/14,3 23
Бензол/циклогексанол 38,4/61,6 25
Бензол/н-гексан 34,7/65,3 24
Бензол/метанол 77,8/22,2 25
Бензол/изопропанол 64,2/35,8 25
Бутанон/метанол 88,7/11,3 25
Циклогексан 34
Поливинилацетат, атак- Метанол 6
тический Этанол/метанол 80/20 17
60/40 26,5
50/50 34
40/60 36
Бутанон/изопропанол 73,2/26,8 25
З-Метилбутанон/н-гептан 73,2/26,8 25
Гептанон-3 29
З-Метилбутанон/н-гептан 72,7/27,3 30
Ацетон/изопропанол 23/77 30
6-Метилгептанон-З 66
Поливиниловый спирт Вода -97
Поливинилхлорид, атак- Тетрагидрофур ан/вода 100/11,9 30
тический 100/9,5 30
Изучение взаимодействия полимеров с растворителями
43
растворителем отсутствуют (равны нулю), т. е. линейные цепные
молекулы имеют невозмущенные размеры.
При тета-температуре второй вириальной коэффициент А2 = О
(разд. 2.10).
В табл. 2.2 приведены тета-температуры для некоторых рас-
пространенных систем полимер — растворитель. Более подробные
данные можно найти в Polymer Handbook [О: 404].
Рис. 2.4. Кривые осаждения, не зави-
сящие от молекулярного веса.
В одном и том же растворителе тета-температура для раз-
ветвленных полимеров всегда ниже, чем для соответствующих ли-
нейных гомологов. Она зависит от длины и числа ветвлений, т. е.
от молекулярной структуры полимера. Если степень ветвления не-
значительна, то значения тета-температур для линейного и раз-
ветвленного полимеров сближаются.
Кривые осаждения, построенные как зависимости (Т — 0)А11/2
от vMi/2, называют кривыми осаждения, независимыми от молеку-
лярного веса, т. е. кривые, полученные для различных гомологов
одного и того же полимера, являются продолжением одна другой
(рис. 2.4).
Тета-температура для линейного полимера характеризует дан-
ную систему полимер — растворитель и не зависит от молекуляр-
ного веса полимера.
Обзорная литература: 125, 262, 367, 403, 456, 766, 1336.
Периодическая литература: 2026, 2500, 2693, 2818, 2831, 2985, 3055, 3109,
3110, 3121, 3176, 3276, 3402, 3719, 3964, 4299, 4483, 4579, 4657, 4660, 4662, 4664,
4667, 4717, 4789, 4790, 5421, 5422, 5495, 5578, 5583, 5586—5588, 4976, 6009, 6066,
6148, 6250, 6380—6382, 6474, 6421, 6475, 6477, 6478, 6510, 6595, 6639, 6921, 7147,
7151.
2.3.1. Способы определения тета-температур
Существует несколько способов определения тета-температу-
ры (6).
1. Метод фазового равновесия. По этому способу определяют
температуру фазового разделения (критическая температура оса-
44
Глава 2
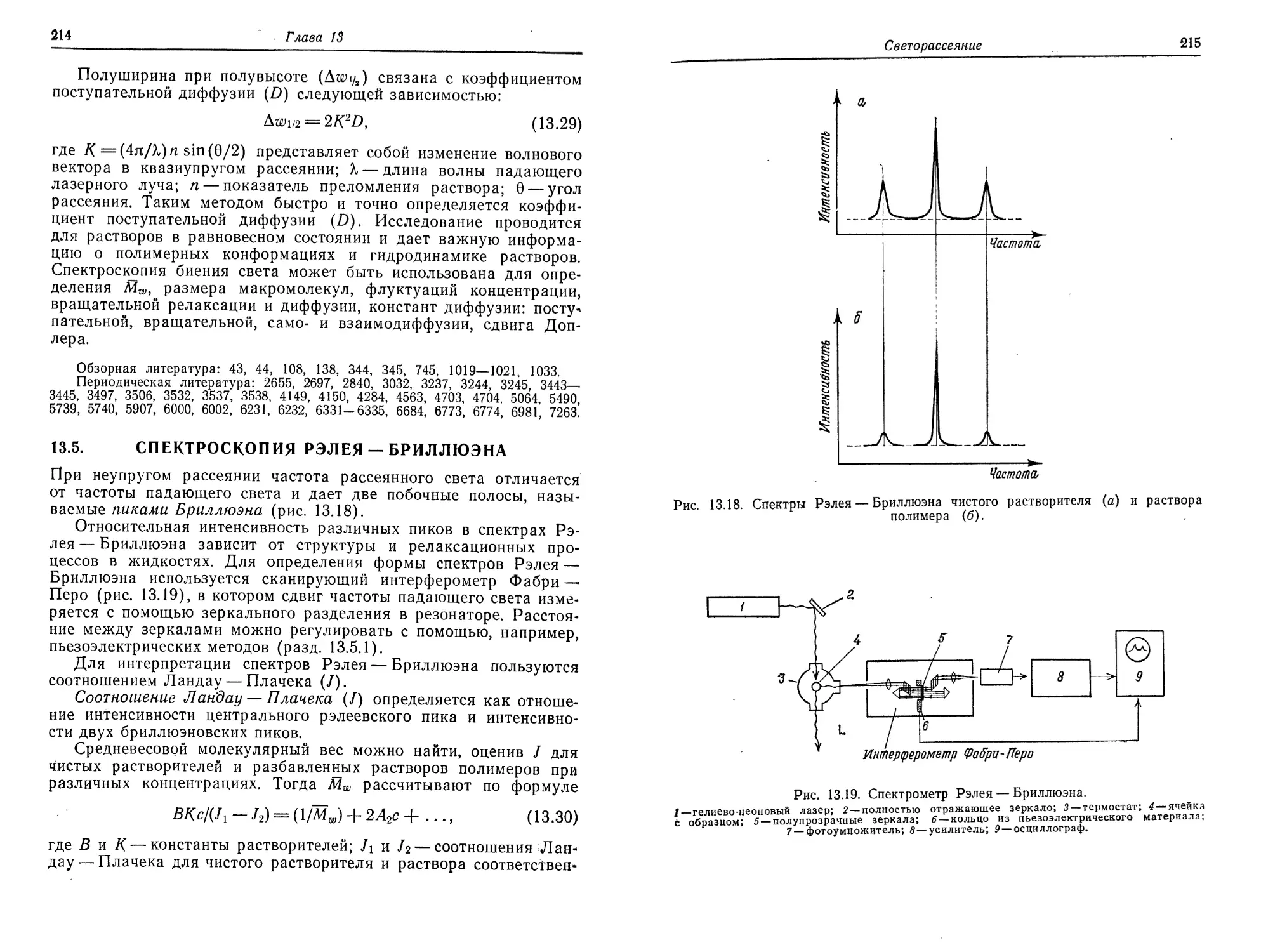
----------------а._____________2________________________________
ждения) для нескольких концентраций полимера известного сред-
нечислового молекулярного веса (Мп) и отмечают максимальную
критическую температуру осаждения (Тс). Тета-температуру мож-
но найти из кривой зависимости Тё1 от М~1/2 (рис. 2.5), используя
уравнение Флори
_L = -L + v' ' (2 4)
Те е 80'ГМ1'2 ’ 1 1
где Тс — критическая температура осаждения, 0 — тета-темпера-
тура, Vi — молярный объем растворителя, v— удельный объем по-
лимера, Т — энтропия смешения, М — молекулярный вес полимера.
Отрезок, отсекаемый на оси ординат, равен 1/0, а наклон пря-
мой дает tg а = Vi/iJOT. При определении тета-температуры для
смешанных растворителей необходима экстраполяция на бесконеч-
ное разбавление. Этот метод можно использовать только для слу-
чая разделения в двух жидких фазах.
2. Метод, основанный на определении второго вириалъного ко-
эффициента. В основу этого метода положен тот факт, что при
тета-температуре вторые вириальные коэффициенты равны нулю
(разд. 2.10). Наклон п/с (рис. 5.2) равен нулю, если растворитель
является тета-растворителем. Можно использовать все методы, с
помощью которых определяют второй вириальный коэффициент,
а именно эбулиоскопию (разд. 6.1.1), криоскопию (разд. 6.2.1),
седиментационное равновесие (разд. 8.3.1), светорассеяние
(разд. 13.1.6).
3. Метод титрования до точки мутности. Согласно этому ме-
тоду, растворы полимера различной концентрации титруются не-
растворителем до появления первых признаков помутнения
(разд. 4.2). Затем строят зависимость логарифма концентрации не-
растворителя от логарифма концентрации раствора полимера в
точке мутности и экстраполируют ее на 100%-ный полимер. Смесь
растворителя с нерастворителем в этой точке отвечает тета-смеси.
При использовании этого метода необязательно знать молекуляр-
ный вес полимера.
4. Метод, связывающий вязкость и молекулярный вес. В основу
этого метода положено то, что экспонента а в уравнении Марка —
Хоувинка — Сакурады (разд. 9.1.2) равна 0,5 для статистического
клубка в тета-растворителе.
Обзорная литература: 404, 456.
Периодическая литература: см. периодическую литературу к разд. 2.3.
2.4. ВЕРХНЯЯ И НИЖНЯЯ КРИТИЧЕСКИЕ
ТЕМПЕРАТУРЫ РАСТВОРЕНИЯ
Кривые точки помутнения, или кривые осаждения, для различных
систем полимер — растворитель имеют различный вид (рис. 2.6—
2.8). Максимумы и минимумы на этих кривых получили название
верхнего порога осаждения (ВПО) и нижнего порога осаждения
Изучение взаимодействия полимеров с растворителями
43
(НПО). Эти точки на кри-
вых указывают на верхнюю
и нижнюю критическую
температуры растворения,
соответственно ВКТР и
НКТР. (Примечание. ВКТР
и НКТР не имеют ничего
общего с действительной
температурой, при которой
наблюдается расслое-
ние).
На фазовой диаграмме
раствора полимера имеются
две области ограниченной
смешиваемости: область ни-
же ВКТР, связанная с тета-
температурой, и область вы-
ше НКТР.
Различные системы по-
лимер—.растворитель могут
иметь совершенно непохо-
жие фазовые диаграммы.
Для некоторых систем
ВКТР<НКТР (рис. 2.6), но
для других, например ха-
рактерных для ряда сильно
полярных систем (полиокси-
этилен — вода), ВКТР >
> НКТР и наблюдается
замкнутая петля раствори-
мости (рис. 2.7).
В отдельных случаях
кривые осаждения имеют
значительно более сложный
вид, например для системы
полистирол — ацетон (рис.
2.8).
Обзорная литература: 21, 125,
262, 456, 767, 1302.
Периодическая литература:
2056, 2224, 2225, 2243, 2295, 2374,
2819, 2996, 2999, 3000, 3001, 3064,
3122, 3123, 3133, 3281, 3469, 3504,
3719, 3861, 4667, 4791, 4792, 4794,
4866, 4923, 5042, 5202, 5203,5495,
5497, 5546, 5702, 5704, 5706, 5707,
6145—6149, 6309, 6380, 6414, 6429,
6686, 7149.
Рис. 2.6. Фазовая диаграмма для системы
полимер — растворитель для случая
ВКТР < НКТР (например, полистирол —
циклогексанон).
Содержание полимера., off. доля
Рис. 2.7. Фазовая диаграмма для системы
полимер — растворитель для случае
ВКТР > НКТР (например, полиоксиэти-
лен — вода или поливиниловый спирт —
вода).
Содержание полимера, off. доля
Рис. 2.8. Фазовая диаграмма для системы ,
полистирол — ацеюн.
46
Глава 2
2.5. РАСТВОРИМОСТЬ ПОЛИМЕРОВ
В СМЕШАННЫХ РАСТВОРИТЕЛЯХ
Возможны три комбинации низкомолекулярных жидкостей в сме-
шанных растворителях:
а) растворитель — нерастворитель;
б) нерастворитель — нерастворитель (явление, в результате ко-
торого сочетание двух нерастворителей образует хороший смешан-
F
Рис. 2.9. Поверхность бинодали для
тройной системы и постоянного дав-
ления, показывающая монотонное
уменьшение растворимости полимера
(Р) при изменении состава смешан-
ного растворителя от чистого Li до
чистого Ь2 [П: 7146].
ный растворитель полимера, называется со растворимостью или
синергизмом);
в) растворитель — растворитель.
На рис. 2.9 показаны кривые точки помутнения для случая мо-
нотонного изменения силы смешанного растворителя в зависимости
от состава. Каждая точка на затененных поверхностях представ-
ляет точку помутнения (т. е. мутность, которую можно обнаружить
визуально). При температурах, лежащих ниже этих поверхностей,
компоненты полностью смешиваются, тогда как растворы, обозна-
ченные точками, лежащими на обратной стороне поверхностей,
разделяются на две фазы. При узком молекулярновесовом распре-
делении полимера поверхности точки мутности идентичньк с под
верхностями бинодали, т. е. конечные точки всех связывающих
линий располагаются на поверхностях точек помутнения.
Обзорная литература: 125, 262, 764.
Периодическая литература: 2441, 2998, 3100, 3172, 4146, 4955, 5882
2.6. НЕСОВМЕСТИМОСТЬ РАСТВОРОВ ПОЛИМЕРОВ
Явление несовместимости наблюдается, когда достаточно разбав-
ленные растворы различных полимеров в одном и том же раство-
рителе не смешиваются, а разделяются на две фазы. При умень-
шении концентрации полимера или достаточном повышении тем-
пературы двухфазная система превращается в однофазную. Экспе-
риментально несовместимость полимерных систем определяют с
помощью спинодали — кривой, разделяющей нестабильную и мета-
Изучение взаимодействия полимеров с растворителями
47
стабильную области на фазовой диаграмме. Спинодаль (рис. 2.10)
можно построить по ряду экстраполированных точек, где рассея-
ние света от еще гомогенной фазы стремится к неопределенности,
Содержание полимера 2, об. доля
Рис. 2.10. Схематическая диаграмма
спинодали при постоянной темпера-
туре. Точки спинодали найдены экс-
траполяцией (разд. 13.1.1)
до нуля при постоянном содержании
одного полимера и изменяющейся
объемной доле другого [П: 6913].
Рис. 2.11. Кривая Зимма (разд.
13.1.6) для избыточного рассеяния
системы полимер 1 -р полимер 2 в
данном растворителе. Состав, отве-
чающий спинодали, находится как
точка пересечения линии 0 = 0 с го-
ризонтальной осью.
каждую такую точку получают из кривой Зимма как точку пере-
сечения экстраполированной линии 0=0 с горизонтальной осью
(рис. 2.11).
Обзор'ная литература: 262, 777.
Периодическая литература: 2497, 2747, 3133, 3177, 3719, 4656, 4665, 4713,
4794, 5202, 6065, 6250, 6307, 6913—6915, 6978.
2.7. ТЕРМОДИНАМИЧЕСКИЙ ПОДХОД
К РАСТВОРИМОСТИ
Термодинамика дает следующее описание смешения (растворения)
полимера с растворителем:
а) для аморфного полимера
Д0т==ДЯт-ГД5т
(2.5)
в калориях на моль (СГС) или джоулях на моль (СИ);
б) для кристаллического полимера
= АЯт - TASm + ДЯГ (2.6)
в калориях на моль (СГС) или джоулях на моль (СИ), где
Аб?т— изменение свободной энергии системы при смешении
[кал/моль (СГС) или джоуль/моль (СИ)], \Нт — изменение эн-
тальпии системы при смешении [кал/моль (СГС) или джоуль/моль?
48
Глава 2
(СИ)]; \Sm — изменение энтропии системы при смешении
[кал • моль-1-°C-1 (СГС) или джоуль-моль-1-К-1 (СИ)]; \Hf —
теплота плавления повторяющихся звеньев полимера [кал/моль
(СГС) или джоуль/моль (СИ)]; Т — термодинамическая темпера-
тура (К).
До настоящего времени нет теоретического решения для рас-
творимости полукристаллических полимеров. Трудной является и
проблема предсказания растворимости кристаллических полимеров
[О: 622, П: 2642].
Свободная энергия (&Gm) для системы полимер — растворитель
находится из уравнения
\Gm = RT [п! In V} + п2 In v2 + XV\V2 Оч + яш2)], (2.7)
где R— универсальная газовая постоянная (разд. 40.2); Т — тер-
модинамическая температура (К); ni и п2 — число молей раство-
рителя и полимера соответственно; m = V2/V1 — отношение мо-
лярного объема полимера и растворителя; Ui и v2 — объемные доли
растворителя и полимера соответственно (во многих публикациях
вместо и v2 встречаются символы epi и ф2)> которые находят из
следующих зависимостей:
I?! = пх1(пх + mn2), (2.8)
v2 = тп21(пх + тп2), (2.9)
% — параметр взаимодействия Флори — Хаггинса (безразмерный)
(разд. 2.9).
В уравнении (2.7) первые два члена отражают конфигурацион-
ную энтропию смешения и всегда отрицательны. Третий член, для
того чтобы изменение свободной энергии (AGm) было отрицатель-
ной величиной, должен быть или очень маленьким положительным
числом, или отрицательным числом. Процесс будет протекать, если
он сопровождается отрицательным изменением свободной энергии,
т. е. большим увеличением энтропии с последующим незначитель-
ным увеличением энтальпии или ее уменьшением.
Энтропия смешения (&Sm) для системы полимер — раствори-
тель всегда положительна и возрастает при смешении. В твердом
полимере макромолекулы перепутаны и молекулярная подвиж-
ность ограничена сегментальным броуновским движением. В рас-
творе полимера макромолекулы распутаны и цепи могут пе-
ремещаться относительно друг друга, отдаляться друг от друга и
обладать различными конфигурациями. Число степеней свободы
возрастает, резко увеличивая энтропию.
Энтальпия смешения (&Нт) (называемая также теплотой сме-
шения) для системы полимер — растворитель определяется по
уравнению Гильдебранда — Скетчарда\
= (2.10)
Изучение взаимодействия полимеров с растворителями
49
в калориях на моль (СГС) или джоулях на моль (СИ), где Vm—
суммарный объем смеси, ДЕ] и ДЕ2— энергии испарения раство-
рителя и полимера соответственно [кал/моль (СГС) или
джоуль/моль (СИ)]. Отношение ДЕ/V называется плотностью
энергии когезии и представляет собой энергию испарения на 1 моль,
т. е. является мерой количества энергии, необходимой для пре-
одоления всех межмолекулярных сил в 1 моле жидкости.
Корень квадратный из плотности энергии когезии называется
параметром растворимости и характеризуется специальным сим-
волом (б):
М-тТ (2-“)
| (кал-см-3)°'5 (СГС) или (Дж-м-у5 (СИ)].
Уравнение (2.10) для энтальпии смешения может иметь и иную
форму:
A^=Vm(d1-d2)2^2 (2.12)
в калориях на моль (СГС) или джоулях на моль (СИ), где 61
и б2—параметры растворимости растворителя и полимера соот-
ветственно. При этом соблюдаются следующие общие правила:
а) наилучшим растворителем для данного полимера является
тот, параметр растворимости которого равен или наиболее близок
параметру растворимости полимера, т. е. если б] » б2 (61 — б2 <Z
<1,7—2,0), то Д/7 ж 0 и полимер растворяется в данном рас-
творителе;
б) растворимость отсутствует при 61 — б2 > 2,0, тогда Д// 0.
Параметр растворимости растворителя (61) можно найти из
его энтальпии испарения (Д/Л):
б( = [(Д/Л - /?7>Ч1/2 (2.13)
Параметр растворимости смеси растворителей (бт) опреде-
ляется из следующего уравнения:
д __ XiVidi + X2V262 tn 1Л\
m~ XlVl + X2V2 ’ }
где Xi и х2— молярные доли компонентов 1 и 2; Vi и У2— моляр-
ные объемы компонентов 1 и 2; 61 и б2 —параметры растворимости
компонентов 1 и 2. Уравнение (2.14) справедливо до тех пор, пока
нет изменения объема при смешении.
Термодинамика дает также описание смешения (растворения)
полимера и растворителя с использованием химических потенциа-
лов (р).
Разница между химическими потенциалами растворителя в
растворе (pi) и в чистом виде (р°) находится из уравнения
Др = pt — pi =RT [in (1 — v2) + (1 — 1/х) v2 + ХЦ2], (2.15)
где х — среднее число сегментов в полимерной цепи.
50
Глава 2
Растворимость полимера в данном растворителе наблюдается,
когда Ар—отрицательная величина.
Обзорная литература: 21, 24, 94, 109, 228, 229, 262, 456, 458, 494, 622, 623,
1067, 1146.
Периодическая литература: 2008, 2062, 2063, 2187, 2188, 2359, 2418, 2440,
2812, 3003, 3400, 3413, 3872, 3990, 4122, 4140, 4162, 4165—4168, 4359—4365, 4447,
4659, 4664, 5072, 5093, 5392, 5423, 5703, 5704, 5893, 6025, 6180, 6276, 6278, 6286—
6289, 6398, 6399, 6476, 6690, 6692, 6706, 7144.
Примечание. В Polymer Handbook суммированы подробные
данные по изменению теплот, энтропии и объема для систем по-
лимер — жидкость [О: 164], приведены теплоты растворения ряда
распространенных полимеров [О: 323], а также многочисленные
данные по взаимодействию полимера с растворителем и параметры
растворимости [О: 230, 1413].
Теплоты смешения (АЯт) разбавленных растворов полимеров
определяют путем микрокалориметрических исследований.
Обзорная литература: 243.
Периодическая литература: 2096, 3137, 3570, 4152, 4355, 4363, 4901, 5880,
6588, 6806.
2.8. ТРЕХМЕРНЫЙ АНАЛИЗ РАСТВОРИМОСТИ
Трехмерный параметр растворимости (б) можно вычислить из
уравнения
в = V№+ «* + «?). (2-16)
где 6d —дисперсионная сила, dh — способность к образованию во-
дородных связей (которая пропорциональна прочности водород-
Рис. 2.12. Схематическое изображение
типичного объема взаимодействия
для концепции Хансена о трехмерном
параметре растворимости.
ной связи), бр — полярное и индукционное взаимодействие. Пара-
метры растворимости для различных растворителей приведены в
табл. 2.3,
Изучение взаимодействия полимеров с растворителями
51
Таблица 2.3
Параметры растворимости для некоторых растворителей [О: 93] 4
Растворитель «р
Метанол 7,42 6,3 10,9
Этанол 7,73 4,3 9,5
Пропанол-1 7,75 3,3 О л 8,5 8,0 7,7 7,8 6,8
Пропанол-2 Бутанол-1 7,70 7,81 3,0 2,8 2,8 2,2
Изобутанол Пентанол-1 7,4 7,81
2-Этилбутанол 7,70 2,1 1 <2 6,6 5,8
2-Этилгексанол 7,78 1,0
Метилизобутилкарбинол 7,47 1,6 л л 6,0 6,6
Циклогексанол 8,50 2,0
Этиленгликоль 8,25 5,4 12,7
Пропиленгликоль Бутандиол-1,3 Глицерин льКрезол Диэтиленгликоль 8,24 8,10 8,46 8,82 7,86 4,6 4,9 5,9 2,5 7,2 П,4 10,5 14,3 6,3 10,0
Дипропиленгликоль а 7,77 9,9 9,0
Этиллактат 7,80 3,7 6,1
н-Бутиллактат 7,65 3,2 5,0
Диацетоновый спирт 7,65 4,0 5,3
Уксусная кислота а 7,1 3,9 6,6
Муравьиная кислота 3 7,0 5,8 8,1
Масляная кислота а 7,3 2,0 5,2
Уксусный ангидрид Вода а 7,5 6,0 5,4 15,3 4,7 16,7
Вода — мочевина (насыщ) * 8,8 13,0 15,2
2-Метоксиэтанол 7,9 4,5 8,0
2-Этоксиэтанол 7,85 4,5 7,0
2-Бутоксиэтанол 7,76 3,1 5,9
2-Этоксиэтилацетат 7,78 2,3 5,2
2- (2-Метоксиэтокси) этанол z 7,90 3,8 6,2
2- (2-Бутоксиэтокси)этанол 7,80 3,4 5,2
Диметилдиэтиленгликоль 7,70 3,0 4,5
Этилацетат 7,44 2;6 4,5
н-Бутилацетат 7,67 1,8 3,1
Изобутилацетат 7,35 1,8 3,7
Изоамилацетат 7,45 1,5 3,4
Изобутилизобутират 7,38 1,4 2,9
у-Бутиролактон 9,26 8,1 3,6
Пропиленкарбонат 9,83 8,8 2,0
Этиленкарбонат 9,50 10,6 2,5
Диэтиловый эфир 7,05 1,4 2,5
Фуран 8,70 0,9 2,6
Тетрагидрофуран 8,22 2,8 3,9
52
Глава 2
Изучение взаимодействия полимеров с растворителями
53
Продолжение
Растворитель «р
Диоксан а 9,30 0,9 3,6
Метилальа 7,35 0,9 4,2
Анизол а 8,7 2,0 з,з
Четыреххлористый углерод 8,65 0,0 1 с? 0,0
Хлороформ 8,65 1,5 2,8
Метиленхлорид 8,91 3,1 3,0
Этиленхлорид 9,20 2,6 2,0
1,1,1-Трихлорэтан 8,25 2,1 1,0
1-Хлорбутан 7.95 2,7 1,0
Трихлорэтилен 8 78 1,5 2,6
2,2-Дихлордиэтиловый эфир а 9,20 4,4 1,5
Хлорбензол 9,28 2,1 1,0
о-Дихлорбензол 9,35 3,1 1,6
Циклогексилхлорид 8,50 2,7 1,0
Хлорпропанол 8,58 2,8 7,2
Эпихлоргидрин 9,30 5,0 1,8
1-Бромнафталин 9,94 1,5 2,0
Ацетон 7,58 5,1 3?4
Метилэтилкетон 7,77 4,4 2,5
Метилизобутилкетон 7,49 3,0 2,0
Метилизоамилкетон 7,80 2,8 2,0
Диизобутилкетон 7,77 1,8 2,0
Изофорон 8,10 4,0 3,6
Ацетофенон 8,55 4,2 1,8
Циклогексанон 8,65 4,1 2,5
Мезитилоксид 7,97 3,5 3,0
Бензальдегид 9,15 4,2 2,6
Ацетонитрил 7,50 8,8 3,0
Бутиронитрил 7,50 6,1 2,5
Нитрометан 7,70 9,2 2,5
Нитроэтан 7,80 7,6 2,2
2-Нитропропан 7,90 5,9 2,0
Нитробензол 8,60 6,0 2,0
Формамид 8,40 12,8 9,3
Диметилформамид 8,52 6,7 5,5
Анилин 9,53 2,5 5,0
Этаноламин 8,35 7,6 10,4
Дипропиламин 7,50 0,7 2,0
Диэтиламин 7,30 1,1 3,0
Циклогексиламин 8,45 1,5 3,2
Пиридин 9,25 4,3 2,9
Морфолин 9,20 2,4 4,5
Диэтилентриамин 8,15 6,5 7,0
Бензонитрил 8,50 6,5 2,5
1Ч-Метилпирролидон-2 8,75 6,0 3,5
Пирролидон-2 9,5 8,5 5,5
Ди мети л ацетамид 8,2 5,6 5,0
Продолжение
Растворитель 6Р
Тетраметилмочевина 8,2 4,0 5,4
Этиленциангидрин 8,4 9,2 8,6
Г ексаметилфосфорамид 9,0 4,2 5,5
Триметилфосфат 8,2 7,8 5,0
Триэтилфосфат 8,2 5,6 4,5
Диэтилсульфид 8,25 1,5 1,0
Сероуглерод 9,97 0,0 0,0
Диметилсульфоксид 9,00 8,0 5,0
Диметилсульфон 9,3 9,5 6,0
Бензол 8,95 0,5 1,0
Толуол 8,82 0,7 1,0
Ксилол 8,65 0,5 1,5
Этилбензол 8,70 0,3 0,7
Стирол 9,07 0,5 2,0
Тетралин 9,35 1,0 1,4
Гексан 7,24 0,0 0,0
Циклогексан 8,18 0,0 0,0
а Величины неточные.
Кривая растворимости для данного полимера может быть пред-
ставлена как сфера, построенная вокруг точки (рис. 2.12). Эта
сфера определяет зону растворимости данного полимера.
Общее правило состоит в том, что а) любая жидкость, нахо-
дящаяся внутри сферы, служит истинным растворителем поли-
мера; б) жидкость, находящаяся вне сферы, является нераство-
рителем; в) жидкость, находящаяся вблизи границы сферы, может
частично смешиваться с полимером.
Трехмерный анализ растворимости оказывается полезным при
решении ряда технических проблем, особенно в лакокрасочной
промышленности.
Обзорная литература: 93.
Периодическая литература: 2484, 3024, 3025, 3523, 3888, 3889, 3891, 4379,
4616, 6761.
2.9. ПАРАМЕТР ВЗАИМОДЕЙСТВИЯ
ФЛОРИ —ХАГГИНСА
Параметр взаимодействия Флори (часто называемый параметром
взаимодействия Флори — Хаггинса) (%) характеризует меру взаи-
модействия между каким-то определенным растворителем и
54
Глава 2
данным полимером. Он является параметром свободной энергии и
содержит как энтропийный, так и энтальпийный член.
1. Для систем неполярный или малополярный полимер — рас*
творитель % находят из выражения
Х = Хз + Хн (2.17)
(безразмерная величина), где %$— энтропийный член, равный об-
ратной величине координационного числа решетки (г), т. е. равен
Таблица 2.4
Методы определения параметра взаимодействия
Флори — Хаггинса (х)
Метод Периодическая литература
1. Метод определения равновесного 2493, 2494, 2535, 2559, 2578, 3223, 3424,
набухания 3403, 4313, 5066, 5383, 5763, 6310, 6691
2. Мембранная осмометрия 4727, 5579, 6610
3. Парофазная осмометрия 2057, 2300, 2302, 2303, 2493, 2494, 2848,
3608, 5424, 5468, 6690
4. Седиментация — ультрацентри- фугирование до равновесия 2841
5. Вискозиметрический 2535, 2577, 3003, 3416, 3762, 5066, 6310,
6433, 6614
6. Оптический 2107, 2968, 3002, 3421
7. Обращенная газовая хроматогра- 2581, 3136, 3138, 3165, 3168, 3578, 3580,
фия 3871, 4891—4894, 5470, 5585, 5708, 6260,
6606, 6607, 6622, 6690, 6766
обратной величине числа ближайших соседей молекулы или сег-
мента в растворе; %s— характеристика, зависящая от темпера-
туры; — энтальпийный член:
Хн = (1/Ж)(62-61)* 2, (2.18)
Vi — молярный объем растворителя; R — универсальная газовая
постоянная (разд. 40.2); Т — термодинамическая температура (К);
61 и 62— параметры растворимости растворителя и полимера со-
ответственно.
2. Для систем полярный полимер — растворитель параметр рас-
творимости полимера (62) в уравнении (2.18) нужно заменить на
трехмерный параметр растворимости (разд. 2.8).
Параметр взаимодействия Флори (%) зависит от температуры
и от концентрации при сильном варьировании концентраций поли-
мера. Этот параметр необходимо определять для каждой пары
полимер — растворитель.
Параметр взаимодействия Флори — Хаггинса (%) можно найти
при определении второго вириального коэффициента (Д2)
(разд. 2.10), а также различными другими методами (табл. 2.4).
Изучение взаимодействия полимеров с растворителями
55
Обзорная литература: 141, 254, 262, 456, 872, 943, 1302.
Периодическая литература: 2440, 2663, 2693, 3124, 3281, 3407, 3414, 3419,
3423, 3425, 3591, 4075, 4077, 4162, 4472, 5612, 5703, 5705, 6162, 6163, 6700.
2.10. ВТОРОЙ ВИРИАЛЬНЫЙ КОЭФФИЦИЕНТ
Второй вириальный коэффициент (А2) характеризует взаимодей-
ствие между растворителем и полимером и выражается согласно
теории Флори — Хаггинса — Кригбаума как
= (4 -0 «) ДX) = V (1 - 4) (£) J(X), (2.19)
где %—параметр взаимодействия Флори (разд. 2.9); и— удель-
ный объем полимера; Vi — молярный объем растворителя; ф—
энтропия смешения; 0 — тета-температура; Т — температура, при
которой имеет место взаимодействие между полимером и раство-
рителем; J(X)—параметр, зависящий от молекулярного веса, при-
роды растворителя и температуры и равный единице при тета-тем-
пературе, где % = 0,5.
Второй вириальный коэффициент можно определить различи
ными методами, причем в зависимости от способа определения он
выражается по-разному:
а) осмометрией (разд. 5.2):
^=^дг(4-х); (2.20)
б) эбулиометрией (разд. 6.1.1):
в) криоскопией (разд. 6.2.1):
IW)
г) светорассеянием (разд. 13.1.6):
Л2 = -27г(т-0 (2-23)
Pp^S 42 7
Расшифровка символов имеется в соответствующих разделах.
Размерность Д2 зависит от способа его оценки.
Общее правило состоит в том, что
а) для «хороших» растворителей А2 — большая величина;
б) для «плохих» растворителей А2— маленькая величина;
в) при А2 = 0 полимер выпадает из раствора.
Второй вириальный коэффициент (А2) зависит от ряда фак-
торов:
56
Глава 2
а) температуры (с повышением температуры А2 уменьшается);
б) системы полимер — растворитель;
в) молекулярного веса и молекулярновесового распределения
(МВР) полимера;
г) тактичности полимера;
д) молекулярной формы макроионов.
Обзорная литература: 1429.
Периодическая литература: 2337, 2740, 2741, 2750—2752, 3003, 3053, 3206,
3302, 3420, 3571, 3822, 4085, 4152, 4295, 4463, 4746, 4751, 4761, 4770, 4850, 5514,
5608, 5640, 5690, 5832, 5879, 6094, 6281, 6284, 6287, 6417, 6578, 6595, 6923, 7143,
7145, 7331.
Обширные данные о втором вириальном коэффициенте полиме-
ров в растворах имеются в Polymer Handbook [О: 410].
2.11. НЕВОЗМУЩЕННЫЕ РАЗМЕРЫ ЛИНЕЙНЫХ
ЦЕПНЫХ МАКРОМОЛЕКУЛ
Размер макромолекул в растворе нельзя строго определить, по-
скольку молекулярный клубок меняется в форме со временем,
причем эти изменения конформации обусловлены броуновским дви-
жением. Молекулярный размер меняется при переходе от одной
Рис. 2.13. Схематическое изображе-
ние изолированного молекулярного
клубка. Центр тяжести молекулы на-
ходится в точке 0; г — расстояние ме-
жду концами цепи, s — радиус инер-
ции относительно центра тяжести
макромолекулы.
молекулы к другой идентичной массы и структуры, и поэтому этот
размер можно описать лишь исходя из усредненных свойств
(рис. 2.13):
1. Среднеквадратичное расстояние между концами (г2) линей-
ной цепной молекулы, которое равняется
г2 = г2а2 (2.24)
в квадратных сантиметрах (СГС) или квадратных метрах (СИ),
где — среднеквадратичное расстояние между концами невоз-
мущенной цепи [см2 (СГС) или м2 (СИ)], а — параметр набу-
хания,
Изучение взаимодействия полимеров с растворителями
57
2. Среднеквадратичный радиус инерции (s2), который выра-
жается формулой
s = s2a2 (2.25)
в квадратных сантиметрах (СГС) или квадратных метрах (СИ),
где s2 — среднеквадратичный радиус инерции невозмущенной цепи,
a — параметр набухания.
Для линейных полимеров
r2 = 6s2 (2.26)
Возмущенные размеры — это размеры цепи в данном раство-
рителе, где проявляются межмолекулярные взаимодействия между
макромолекулами и растворителем.
Невозмущенные размеры — это размеры цепи в данном рас-
творителе, определяемые только длинами связей и углами между
ними.
Тета-температура (температура Флори) — это такая темпера-
тура данного растворителя Т — 0, при которой линейные цепные
молекулы имеют невозмущенные размеры.
Обширные данные по тета-растворителям приведены в Polymer
Handbook [О: 404].
Невозмущенные размеры линейной полимерной цепи (г2 или
s2) зависят от молекулярного веса (Мп). Отношения типа
гЦмп или sllMn зависят лишь от структуры цепи, но не от рас-
творителя или температуры.
Размеры полимерных цепей в разбавленных растворах опреде-
ляются следующими показателями:
1. Взаимодействиями ближнего порядка, например ограниче-
ниями в углах связей и стерическими ограничениями внутреннему
вращению. Эти взаимодействия определяются среднеквадратичным
расстоянием (г2) между концами невозмущенной цепи или средне-
квадратичным радиусом инерции (Sg) невозмущенной цепи. Доля
эффекта взаимодействия такого рода определяется величиной Сж
(2.27)
П->оо / i
где ni — число f-го типа связи длиной Ц.
2. Взаимодействиями дальнего порядка, которые зависят от
взаимодействия полимера с растворителем и определяются пара-
метром набухания (а) и параметром исключенного объема (г),
связанными между собой выражением
lim а* = const • z, (2.28)
Z~»oo
где х— константа, не зависящая от г. Разными теориями были
предложены значения х, колеблющиеся от I до 6,67.
58
Глава 2
Параметр набухания (а) —это отношение размера цепи в дан-
ном растворителе (возмущенные размеры) —г2 или s2 — ив тета-
растворителе (невозмущенные размеры)—г2 или s2 — при опре-
деленной температуре:
(^_(^
(г*)1'2 (О"2 ' ’
(безразмерная величина).
В том случае, когда полимер находится в хорошем раствори-
теле, параметр набухания (а) больше единицы, это означает, что
действительные возмущенные размеры (г2 или $2) превышают зна-
чения невозмущенных размеров (г2 или s2). Параметр набуха-
ни_я (а) увеличивается также с увеличением молекулярного веса
(Л1я), поскольку Т > 0 число некомпенсированных взаимодействий
между сегментами цепи возрастает с ростом их числа. Параметр
набухания зависит также от силы растворителя.
Исключенный объем — это объем, из которого данная поли-
мерная молекула эффективно исключает все другие молекулы.
Исключенный объем является результатом фактического оттал-
кивания между полимерными молекулами, обусловленного про-
странственными соображениями. Каждая молекула в очень раз-
бавленном растворе в хорошем растворителе будет стремиться
исключить из объема, который она занимает, все другие молекулы.
Состояние свободного вращения — это гипотетическое состояние
цепи, в котором ограничения в углах связей остаются, но стери-
ческие препятствия внутреннему вращению ослаблены.
Среднеквадратичное расстояние между концами цепи при сво-
бодном вращении (г2^) можно найти для данной структуры цепи
по следующей формуле:
= (2.30)
где п — число связей, I — длина связи, 0 — валентный угол связи.
Влияние пространственных затруднений на средний размер
цепи (а), называемый также параметром жесткости полимерной
цепи, выражается как
го
°—
Величина о безразмерная и не зависит от числа связей (п).
Невозмущенные размеры цепей линейных полимеров можно
определить перечисленными ниже методами:
1. Измерение характеристической вязкости (разд. 2.13).
2. Светорассеяние, например метод асимметрии в тета-раство-
рителе (разд. 13.1.5), а также метод Зимма в тета-растворителе
^разд. 13.1.6.)
3. Малоугловое рассеяние рентгеновских лучей.
Изучение взаимодействия полимеров с растворителями
59
4. Из зависимости напряжения от температуры неразбавленных
или набухших образцов при разном г0.
Обзорная литература: 402, 456, 457, 943, 1307, 1429, 1430.
Периодическая литература: 2031, 2040, 2052—2054, 2147, 2148, 2217, 2339,
2358, 2419, 2444, 2499, 2533, 2542, 2579, 2651, 2666, 2825, 2831, 2864, 3114, 3135,
3147, 3148, 3179, 3187, 3188, 3197, 3198, 3204, 3402, 3416, 3459—3461, 3499, 3547,
3805, 3840, 3841, 4483, 4695, 4724, 4767, 4771, 4792, 4805, 4844, 4851, 4877, 4878,
4940, 5086, 5088, 5118, 5141, 5142, 5152, 5182, 5347, 5387, 5405, 5524, 5547, 5699,
5836, 5852, 5881, 6094, 6105, 6114, 6154, 6191, 6338, 6341, 6451, 6723, 6836, 6867,
6899, 6922, 6293, 6994, 7042, 7145, 7326.
Невозмущенные размеры линейных цепных молекул приведены
в [О: 786].
2.12. НЕВОЗМУЩЕННЫЕ РАЗМЕРЫ
РАЗВЕТВЛЕННЫХ МАКРОМОЛЕКУЛ
Разветвленные макромолекулы имеют большую среднюю плот-
ность сегмента, чем линейные цепные неразветвленные макромо-
лекулы того же молекулярного веса, и характеризуются меньшим
объемом клубка.
Разветвленные макромолекулы в растворе характеризуют сле-
дующие параметры:
1. Число точек ветвления (g), которое представляет собой от-
ношение размеров цепей разветвленных (зОв) и линейных
макромолекул одинакового молекулярного веса в тета-раствори-
теле:
-2
Я = (2.32)
S0L
(безразмерная величина), где s2— радиус инерции. Число точек
ветвления является характеристикой только статистически раз-
ветвленного полимера, имеющего три- и тетрафункциональные
точки ветвления и образующего как моно-, так и полидисперсные
системы.
2. Параметр набухания (ав), представляющий собой отноше-
ние размера цепи одного и того же разветвленного полимера в хо-
рошем растворителе (гв или s2^ и в тета-растворителе (ГрВ или
$ов)»
“В-(4Г
(2.33)
(безразмерная величина).
Параметры g и ав можно определить для любого разветвлен-
ного полимера, но для этого нужно знать их характеристики в
тета-растворителе.
60
Глава 2
Обзорная литература: 456.
Периодическая литература: 2693, 2744, 3843, 4087, 4746, 4877, 5140, 5153,
5513, 6969, 6979, 7338.
2.13. ИЗМЕРЕНИЕ НЕВОЗМУЩЕННЫХ РАЗМЕРОВ
ЦЕПЕЙ ПО ВЯЗКОСТИ РАЗБАВЛЕННЫХ
РАСТВОРОВ ПОЛИМЕРОВ
Расширение клубка в разбавленном растворе определяется из
уравнения Флори — Кригбаума:
а5 - а3 = 2CMW (1 - Q/T) Mif2 (2.34)
где 'F—энтропия смешения, 0 — тета-температура, Т — темпера-
тура, при которой имеет место расширение клубка, М — молеку-
лярная масса полимера, См— величина, которая находится из вы-
ражения
,2Ж)
В этом выражении v — удельный объем полимера, Na — число
Авогадро (разд. 40.3), Vi — молярный объем растворителя, rf} —
среднеквадратичное расстояние между концами цепи.
Расширение клубка в разбавленном растворе зависит от мо-
лекулярного веса и термодинамического взаимодействия с раство-
рителем. При повышении температуры выше тета-температуры
расширение клубка возрастает. При понижении температуры рас-
ширение клубка уменьшается, приближаясь к нулю при тета-тем-
пературе. При этой температуре клубок имеет присущие ему не-
возмущенные размеры.
Невозмущенные размеры цепи линейных полимеров можно
определить с помощью следующих зависимостей:
\ 1. Уравнение Флори — Фокса:
[г)]ЛГ=ФГл = Ф(г2)3/2, (2.36)
где [tj]—характеристическая вязкость (разд. 9.1), ЯТ — средний
молекулярный вес полимера, Ф —функция, связанная с гидроди-
намическим поведением макромолекул в растворе —для узкой
фракции и тета-условий, ф = Фо = 2,8-1021 моль-1 (Фо называется
также константой Флори), Vh — гидродинамический объем (т. е.
объем, занимаемый макромолекулой [см3 (СГС) или м3 (СИ)],
г2 — среднеквадратичное расстояние между концами цепи.
При замене г2 в уравнении (2.36) на г2а и выделении члена
foAl"1 получаем уравнение, которое позволяет вычислить размеры
невозмущенных цепей (г2):
fo] = Фо (гШ"‘)зрлТ1/2а3 = ' (2.37)
Изучение взаимодействия полимеров с растворителями
61
где а — параметр набухания, равный
а3 = [т)]/[г)]в,
(2.38)
h]e — характеристическая вязкость при тета-температуре, Kq —
константа для данного полимера, не зависящая от растворителя
Рис. 2.14. Типичная кривая зависимо-
сти Кураты — Штокмайера — Фикс-
мана для линейного полимера в дан-
ном растворителе при постоянной
температуре.
Рис. 2 15. Типичная кривая зависимо-
сти Фокса — Флори — Шефгена для
линейного полимера в данном рас-
творителе при постоянной темпера-
туре.
температуры, молекулярного веса (исключая область низких зна-
чений молекулярного веса)
Кв = Ф0(^М’')3/2 (2.39)
Ле можно найти из данных о характеристической вязкости при
тета-температуре: _
[П]е = *№ (2.40)
2. Уравнение Кураты — Штокмайера — Фиксмана (рис. 2.14)
[П: 4771, 6583]: _ _
[П] М~т = Ка + О,51ВФоМ1/2, (2.41)
где
В = 2гЯ(4-%)ДлГ1 (2.42)
v — удельный объем полимера, Vi —- молярный объем раствори-
теля, % — параметр взаимодействия Флори (разд. 2.9), Na — число
Авогадро (разд. 40.3).
Зависимость [т]] от Л41/2 представляет собой прямую линию,
отсекающую на ординате отрезок, равный Ле, с тангенсом наклона
а = О,51ФоВ (рис. 2.14).
62
Г лава 2
Рис. 2.16. Типичная кривая зависимости Берри для линейного полимера в дан-
ном растворителе при постоянной температуре.
Рис. 2.17. Типичная кривая зависимости Инагаки — Судзуки — Кураты для ли-
нейного полимера в данном растворителе при постоянной температуре.
Рис. 2.18. Типичная кривая зависимости Камиде — Мура для линейного полимера
в данном растворителе при постоянной температуре.
Изучение взаимодействия полимеров с растворителями
63
3. Уравнение Флори — Фокса — Шефгена (рис. 2.15) [П: 3416]:
[л]2/3 = ЛГ1/3 = №/3 + Кв3см [Г]]-1, (2.43)
где С — константа для данной системы полимер — растворитель.
4. Уравнение Берри (рис. 2.16) [П: 2376]:
[п] 1/2М"1/4 = Ке2 + О,42ЛТфо/Ш МГ* (2.44)
д) Уравнение Инагаки — Судзуки — Кураты (рис. 2.17) [П:
4220]:
[П]4/5М-2/s = 0,786Ке/3 + 0,454Ке'3Ф3/3В2;3Л41/3 (2.45)
е) Уравнение Камиде— Мура (рис. 2.18) [П: 4387]:
—In К + In {2 [(а - l/2)_1 - 2]’1 + 1} = (а - l/2) In Мо - In Кд, (2.46)
где К и а — константы уравнения Марка — Хоувинка — Сакурады,
Мо—среднегеометрическое значение молекулярных весов иссле-
дуемых образцов.
Обзорная литература: 255, 456, 457, 943, 1429.
Периодическая литература: 2231, 2237, 2298, 2393, 2450, 2452, 2650, 2994,
3179, 3194—3198, 3202, 3205, 3206, 3402, 3408, 3415, 3416, 3452, 3805, 3843, 4445,
4454, 4463, 4479, 4483, 4572, 4575, 4771—4773, 5085, 5189, 5388, 5491, 5516, 5770,
5771, 5925, 5961, 6104, 6154, 6303, 6506, 6583, 6638, 6769, 6985, 7145, 7218, 7221,
7252, 7253.
2.14. ОБРАЗОВАНИЕ ЖИДКОКРИСТАЛЛИЧЕСКИХ
РАСТВОРОВ
В растворе линейные гибкоцепные полимеры дают беспорядочные
клубки (рис. 2.19, а). Если же цепи построены из жестких фраг-
ментов и связаны таким образом, что цепь может вытягиваться
а 5 в г
Рис. 2.19. Схематическое изображение состояний полимера в растворе.
а —статистические клубки; б —статистические стержни; в —стержни в жидкокристаллических
образованиях; г—нематический порядок в одном образовании.
в одном направлении, тогда они представляют стержнеподобные
образования, и в идеализированном изображении их можно пред-
ставить в виде прямых линий, расположенных хаотически
(рис. 2.19,6). На жесткость и объем, занимаемый каждой поли-
мерной молекулой, оказывает влияние ассоциация с раствори-
телем.
При увеличении концентрации стержнеподобных молекул часть
полимера может растворяться с образованием областей, в которых
64
Глава 2
Рис. 2.20. Диаграмма, отражающая
связь между параметром взаимодей-
ствия (%) растворителя и полимера и
объемной долей (v2) стержнеобраз-
ных полимерных молекул при обра-
зовании тактоидальной фазы рас-
твора.
расположение полимерных цепей с ассоциированным растворите-
лем приближается к параллельной упаковке стержней (рис. 2.19, в).
Такие упорядоченные области называются мезоморфным, или
жидкокристаллическим, состоянием, причем они образуют фазу, не
совместимую с изотропной фазой.
Если стержнеподобные цепи находятся в примерно параллель-
ном порядке, но не организованы вдоль всей цепи, то такая упо-
рядоченная фаза называется не-
матической или нематикой
(рис. 2.19,а).
В расплавах низкомолекуляр-
ных жидкокристаллических со-
единений, молекулы которых оди-
наковой длины и имеют на кон-
цах группы, обусловливающие
полярную ассоциацию, слоевые
состояния называются смектиче-
скими или смёктикой.
Упорядоченная фаза органи-
зуется в микроскопические доме-
ны различного размера и направ-
ления ориентации. Жидкокри-
сталлические растворы являются
оптически анизотропными, т. е.
деполяризуют плоскополяризо-
ванный свет. Растворы хаотиче-
ски расположенных полимерных
молекул оптически изотропны.
Поведение жестких стержнеподобных полимеров в растворе ил-
люстрируется диаграммой, на которой приведена зависимость
параметра взаимодействия полимера с растворителем (параметр
Флори—Хаггинса, %) от объемной доли (ц2) стержнеподобных
полимеров (рис. 2.20). При постоянном % с ростом объемной доли
полимера вначале существует одна-единственная фаза с хаотиче-
ским расположением полимера, затем узкая гетерогенная область
хаотической фазы, находящейся в смеси со второй упорядоченной,
или тактоидальной, фазой, и, наконец, вся система становится так-
тоидальной.
На образование жидкокристаллических растворов влияет мно-
жество факторов, в их числе следующие:
а) структура полимера (вытянутые цепи);
б) молекулярный вес (должен превышать какую-то минималь-
ную величину);
в) растворимость (должна быть достаточно большой, чтобы
быть выше критической концентрации);
г) температура (от нее зависят растворимость и интервал жид-
кокристаллического состояния).
Изучение взаимодействия полимеров с растворителями
65
Содержание полимера, вес.0/-
Рис. 2.21. Влияние содер-
жания полимера и образо-
вания жидкокристалличе-
ской фазы на вязкость массы.
Существует ряд признаков, характерных для жидкокристалли-
ческих растворов.
1. Опалесценция. Жидкокристаллические растворы являются
мутными, хотя они и не содержат нерастворенного вещества. Эта
мутность обусловлена дифракцией света, проходящего через жид-
кокристаллические домены, имеющие различные размеры и на-
правления. При перемешивании (даже слабом) такой системы
легко можно заметить опалесценцию (или жемчужный блеск), ко-
торая быстро пропадает при прекращении перемешивания.
2. Деполяризации плоскополяризованного света. При помеще-
нии под микроскоп тонкого слоя раствора жидкокристаллического
полимера в случае скрещенных поляри-
заторов наблюдаются окрашенные доме-
ны (голубые, зеленые, красные, желтые),
вид которых зависит от типа системы и
предыстории образца.
3. Точка критической концентрации.
Растворы полимеров с вытянутыми це-
пями характеризуются внезапным или
очень резким изменением вязкости мас-
сы при увеличении концентрации поли-
мера в точке, где начинается жидкокри-
сталлический порядок (рис. 2.21). В ле-
вой части рис. 2.21 вязкость возрастает
обычным образом, характерным для изо-
тропных растворов, однако затем в
точке критической концентрации система
будет «принимать» дополнительное количество полимера лишь в
том случае, если может образоваться жидкокристаллическая фаза.
Когда это происходит, то вязкость уменьшается, содержание упо-
рядоченной фазы увеличивается. Если такая смесь фаз не является
особенно вязкой, фазы при стоянии или в центрифуге могут быть
отделены друг от друга. Упорядоченная, анизотропная фаза, бо-
лее обогащенная полимером, чем изотропная фаза, находится вни-
зу, когда плотность растворителя меньше плотности полимера.
Периодическая литература: 3404, 3405, 5351.
2.15. НАБУХАНИЕ СШИТЫХ ПОЛИМЕРОВ
В РАСТВОРИТЕЛЯХ
В жидкости, растворяющей несшитый полимер, соответствующий
сшитый полимер набухает. Степень набухания определяется взаи-
модействием между полимером и растворителем, которое зависит
от потенциала набухания и потенциала упругости, зависящего от
плотности сшивки.
Равновесие достигается, когда эти два потенциала, которые
действуют в противоположных направлениях, становятся равными
3 Зак. 238
66
Глава 2
друг другу (увеличение упругой энергии цепей, образующих сетку,
уравновешивает уменьшение свободной энергии при смешении по-
лимерных сегментов с молекулами растворителя). Это приводит
к хорошо известному уравнению набухания Флори:
1 = 2 _ (р/У1)[1п(1 + +
m m R3--Ы ' ’
где (Мп)с — среднечисловой молекулярный вес цепи сетки, (A4n)0—
среднечисловой молекулярный вес первичной цепи (первичной
Продолжительность набухания
называется линейная молекула перед
сшиванием), v — удельный объем по-
лимера, У] — молярный объем раство-
рителя, vs — конечная объемная доля
полимера при равновесном набуха-
нии, % — параметр взаимодействия
Флори полимера с растворителем
(разд. 2.9).
Величина равновесного набухания
связана с природой системы поли-
мер— растворитель и дает информа-
цию о природе сшивки и армирова-
Рис. 2.22. Типичные кривые НИЯ.
скорости набухания. Для определения конечной объем-
ной доли полимера при равновесном
набухании (в процентах) необходимо поместить образец извест-
ной плотности в выбранный растворитель и выдерживать его в
нем до тех пор, пока взвешиванием не будет установлено на-
сыщение полимера растворителем. Полагая, что в полимере отсут-
ствуют экстрагируемые фракции и что весь поглощенный раство-
ритель вызывает набухание, конечную объемную долю полимера
при равновесном набухании находят из следующей зависимости:
(^1-Го)РР
Vs~ li^ops 100 ’
(2.48)
где IFo — вес образца до набухания, IFi— вес образца после на-
бухания, рр — плотность полимера, ps — плотность растворителя.
Для того чтобы получить кривою скорости набухания (рис. 2.22),
необходимо проделать ряд гравиметрических определений и найти
увеличение объема или веса (в процентах) в зависимости от вре-
мени набухания. Точное определение зависимости набухания от
времени требует большого числа экспериментальных данных, осо-
бенно на начальных стадиях набухания. Весовой метод обычно
связан с обрывом процесса набухания, что с экспериментальной
точки зрения представляет неудобство. Точность данных, получае-
мых этим методом, весьма низка.
Изучение взаимодействия полимеров с растворителями
67
Для проведения золь-гель-анализа образцы полимера выдер-
живают в данном растворителе при определенной температуре
(например, 20 °C) в течение какого-то промежутка времени (от
минут до сотен часов). Затем высушиванием остатка образца в
вакууме при комнатной температуре оценивают количество геля.
Количество золя рассчитывают как разность между весом об-
разца до и после набухания.
Содержание золь-фракции (в процентах) определяется как от-
ношение
Золь-фракция = 3оль3+ЬГе— • ЮО (2.49)
Обзорная литература: 456.
Периодическая литература: 2589, 2617, 2618, 2826, 3433, 3434, 3686, 3845,
4043, 4227, 4376, 4379, 4818, 5008, 5009, 5198, 5237, 5479, 5764, 6057, 6063, 6319,
6320, 6486, 6799, 6829, 7296, 7297.
Глава 3
СРЕДНИЕ МОЛЕКУЛЯРНЫЕ ВЕСА
Обзорная литература: 23, 125, 127, 159, 402, 424, 456, 681, 871, 1022, 1082,
1120, 1336, 1350, 1396.
При использовании системы СИ в расчетах определенные труд-
ности могут возникать при оперировании с безразмерными зна-
чениями молекулярных весов. В связи с этим рекомендуется ино-
гда вместо молекулярного веса пользоваться величиной молеку-
лярной массы, которую также можно определять как молярную
массу, иначе говоря, как массу 1 моля вещества в килограммах
на моль (СИ).
В системе СИ моль является основной единицей, определяемой
как масса числа частиц, равного количеству атомов, находящихся
в 0,012 кг углерода-12. Это число, равное 6,023-1023 моль-1, назы-
вается числом Авогадро (разд. 40.3).
В настоящее время применяется следующая терминология для
определения молекулярного веса (и молекулярной массы), причем
необходимо отдавать себе отчет в том, что при использовании
разных методов оценки молекулярного веса (молекулярной мас-
сы) измеряют совершенно независимые друг от друга свойства
полимера, поэтому различия в получаемых данных могут быть
довольно значительными.
Среднечисловой молекулярный вес {среднечисловая молекуляр-
ная масса), Мп — это суммарный вес (масса) растворенных частиц,
деленный на суммарное число молей в них:
i i
(3.1)
где Ni — число молей частиц i в веществе; Mi — молекулярный вес
частиц /; NiMi — действительный вес частиц i. Среднечисловой мо«
лекулярный вес (Мп) очень чувствителен к наличию даже неболь-
шой примеси макромолекул низкого молекулярного веса.
Значение Мп можно экспериментально определить методами
мембранной осмометрии, эбулиометрии, криоскопии и анализа со-
держания концевых групп; все эти методы зависят от числа на-*
ходящихся в системе молекул.
Средневесовой молекулярный вес {среднемассовая молекуляр-
ная масса) определяется как
(3.2)
Средние молекулярные веса
69
Средневесовой молекулярный вес (Mw) очень чувствителен к на-
личию небольших количеств по весу высокомолекулярных макро-
молекул.
Значения Mw можно экспериментально определить методами
светорассеяния и/или седиментационного равновесия, которые за-
висят от веса присутствующих частиц.
Z- и (Z + 1)-Средние молекулярные веса [Z- и (Z + 1)-средние
молекулярные массы], Mz и Mz+i равны
(3.3)
Я+1 = Е JV/A1//E N,M3i (3.4)
Могут быть рассчитаны и более высокие степени усреднения мо-
лекулярных масс, получаемые при увеличении показателя степени
при Mi.
Средневязкостный молекулярный вес (средневязкостная моле-
кулярная масса), Mv, определяется как
Mv = [Е NiM^/X NiMi]'" (3.5)
Если а — 1, то_Ж = Mw.
Значение Mv можно найти лишь с помощью вискозиметриче-
ских определений (разд. 9.1.2).
Между различными средними молекулярными весами суще-
ствует следующая взаимосвязь (см. рис. 3.2):
Мп <MV ^MW<MZ < M2+i (3.6)
Примечание. В этой книге во всех уравнениях используются
значения средних молекулярных весов, а не средних молекуляр-
ных масс.
Обзорная литература: 23, 116, 124, 125, 127, 159, 248, 250, 309, 630, 943, 1289,
1366.
Периодическая литература: 2222, 4023, 5023, 5310.
3.1. ВЫРАЖЕНИЕ МОЛЕКУЛЯРНОВЕСОВОГО
РАСПРЕДЕЛЕНИЯ: ГРАФИЧЕСКИЕ МЕТОДЫ
Применяют три типа графического изображения молекулярновесо-
вого (молекулярномассового) распределения (МБР).
1. Кривая интегрального молекулярновесового распределения —
это зависимость кумулятивной весовой доли от молекулярного
веса (рис. 3.1). Кумулятивная весовая доля, соответствующая мо-
лекулярному весу, например Mi, представляет собой весовую долю
всех частиц полимера с молекулярным весом, меньшим или рав-
ным
70
Глава 3
Функция, отражающая соотношение между кумулятивной весо-
вой фракцией и молекулярным весом, называется функцией ин-
тегрального распределения, ЦМ).
2. Кривая дифференциального молекулярновесового распреде-
ления — это зависимость весовой
Рис. 3.1. Типичная кривая интегрального
молекулярновесового распределения.
Рис. 3.2. Типичная кривая дифференциаль-
ного молекулярновесового распределения.
доли от молекулярного веса
(рис. 3.2). Площадь, огра-
ниченная кривой дифферен-
циального молекулярнове-
сового распределения и аб-
сциссой, равна единице.
Функция, представляю-
щая соотношение между ве-
совой долей и молекуляр-
ным весом, называется
функцией дифференциаль-
ного весового распределе-
ния, W(M).
Кривую дифференциаль-
ного молекулярновесового
распределения можно полу-
чить графическим диффе-
ренцированием кривой ин-
тегрального молекулярно-
весового распределения.
3. Кривая числового рас-
пределения— это зависи-
мость мольной доли фрак-
ции от молекулярного веса.
Функция, представляю-
щая соотношение между
мольной долей и молекуляр-
ным весом, называется
функцией числового распре-
деления, N(M).
Соотношения между раз-
личными функциями, о ко-
торых шла речь выше, описываются следующими уравне-
ниями:
dI(M)/dM==W(M)
м
I(M)=^W(M) dM,
о
(3.7)
(3.8)
(3.9)
' ' Al ' J M
о
Средние молекулярные веса
71
$ W (М) dM = 1
/(со) =1
J N(M)dM= 1
О
(3.10)
(3.11)
(3.12)
Обзорная литература: 35, 125, 309, 630, 689, 1022, 1309.
Периодическая литература: 2306, 2491, 3076, 3927, 4666, 5398, 6861.
3.2. ФУНКЦИИ РАСПРЕДЕЛЕНИЯ
Иногда молекулярновесовое распределение полимеров может быть
описано с помощью аналитических функций с одним или двумя
параметрами. Предложены многочисленные функции молекуляр-
новесового распределения. Наиболее распространенными из них
являются: 1) распределение Шульца, 2) распределение Танга,
3) распределение Гаусса, нормальное в логарифмической системе
координат.
Подробные данные об этих распределениях можно найти в
оригинальных публикациях.
Обзорная литература: 72, 527, 630, 1314.
Периодическая литература: 2822, 3519, 3714, 4137, 4817, 6272, 6860, 7053.
3.3. ПОКАЗАТЕЛИ МОЛЕКУЛЯРНОВЕСОВОГО
РАСПРЕДЕЛЕНИЯ
Для описания молекулярновесового распределения полимера кроме
кривой пользуются также численным показателем. Для характе-
ристики молекулярновесового распределения предложено несколь-
ко показателей:
а) показатель полидисперсности, определяемый как Mw/Mn\
б) фактор неоднородности (U)
(3.13)
в) корень квадратный из фактора неоднородности (А)
А = U42 = - I]1'2; (3.14)
г) показатель полидисперсности для концов более высокомо-
лекулярных цепей (g)
g = [(M2/Mj-l]1/2 (3.15)
Наиболее часто используется показатель полидисперсности, ко-
торым называется отношение средневесового молекулярного веса
к среднечисловому молекулярному весу, Mw/Mn. Это отношение
Глава 3
равно единице для полимера однородного молекулярного веса и
становится больше единицы для широкого распределения. Он за-
висит от условий образования полимера (табл. 3.1).
Таблица 3.1
Наиболее характерные значения MwjMn [О: 127, 309]
Полимер MwlMn
Гипотетический монодисперсный полимер Реальные «монодисперсные» живущие полимеры Полимер, полученный методом полимеризации (обрыв в результате рекомбинации) Полимер, полученный методом полимеризации (обрыв в результате диспропорционирования) или полимер поликонденсационного типа Винильные полимеры, полученные с высокой сте- пенью превращения Полимеры, полученные с автоускорением Координационные полимеры Разветвленные полимеры 1,000 1,01-1,05 1,5 2,0 2-5 5-10 8-30 20-50
Обзорная литература: 1219, 1309.
Периодическая литература: 4132, 4978, 6275, 7332.
Показатели полидисперсности можно найти в Polymer Hand-
book [О: 72].
Глава 4
ФРАКЦИОНИРОВАНИЕ ПОЛИМЕРОВ
Обзорная литература: 23, 124, 125, 127, 247, 309, 402, 456, 681, 1082, 1112,
1314, 1350.
Под фракционированием полимера понимается разделение
этого вещества на различные по молекулярному весу частицы для
получения однородных фракций.
Между собой макромолекулы могут различаться по следую-
щим трем основным признакам: по молекулярному весу, по хими-
ческому составу, по молекулярной конфигурации и структуре.
Таблица 4.1
Типичные данные фракционирования
Фракция Весовая доля Средний молекулярный вес
1 101 мх
2 £02 м2
3 £03 м3
• • •
• .
Z Wi Ml
Общепринятое название фракционирование полимеров подра-
зумевает фракционирование по молекулярному весу.
Методы фракционирования делятся на следующие группы:
а) фракционное растворение;
б) фракционирование с помощью хроматографии;
в) фракционирование седиментацией;
г) фракционирование диффузией;
д) фракционирование путем ультрафильтрации через пористые
мембраны;
е) фракционирование зонной плавкой.
Данные фракционирования обычно представляют в таком виде,
как это сделано в табл. 4.1. Фракции располагают в порядке по-
вышения молекулярного веса. Весовые фракции wi w2, ..., wi—•
это веса каждой фракции, деленные на суммарный вес образца.
Средние молекулярные веса фракций обычно определяют вискози-
метрически или каким-то иным методом.
Фракции полимера не однородны по молекулярновесовому
распределению (рис. 4.1). Кривые распределения фракций
74
Глава 4
перекрывают друг друга. Степень перекрывания зависит от эффек-
тивности фракционирования, но ни один из экспериментальных ме-
тодов разделения не позволяет полностью исключить перекры-
вание.
Рис. 4.1. Различное распределение фракций.
Обзорная литература: 98, 247, 275, 309, 319, 331, 370, 486, 560, 649, 689, 707,
765, 766, 937, 1137, 1163, 1164, 1186, 1231, 1302, 1311, 1352.
Периодическая литература: 3015, 3016, 3832.
4.1. ФРАКЦИОНИРОВАНИЕ ПО РАСТВОРИМОСТИ
4.1.1. Фракционное осаждение
Фракционное осаждение — это последовательное осаждение поли-
мерных фракций из раствора путем а) добавления нерастворителя,
или осадителя, б) удаления растворителя испарением; в) пониже-
ния температуры; г) изменения давления при нижней критической
температуре растворения.
При использовании первых трех способов сначала выпадают
наиболее высокомолекулярные фракции, тогда как последними
осаждают в первую очередь более низкомолекулярные фракции.
Обзорная литература: 319, 707, 765, 770, 937, 1112, 1311.
Периодическая литература: 4384, 4663.
4.1.2. Метод добавления нерастворителя
В методе добавления нерастворителя постепенное осаждение по-
лимерных частиц из раствора происходит путем введения смеши-
вающегося нерастворителя.
Образец полимера растворяют в соответствующем раствори-
теле (0,1—1,0 вес.°/о) в трехгорлой колбе для фракционирования
(рис. 4.2, а). При энергичном перемешивании и постоянной темпе-
ратуре в атмосфере инертного газа постепенно добавляют осади-
тель. После введения определенного количества добавление еще
Фракционирование полимеров
75
одной капли нерастворителя вызывает появление мутности, не ис-
чезающей при перемешивании. Такой раствор оставляют для вы-
зревания на несколько часов. После этого добавляют дополни-
тельное количество нерастворителя, пока раствор не станет «мо-
лочным». Затем нагревают его до повышения температуры на не-
сколько градусов. Когда раствор вновь становится прозрачным, его
постепенно при энергичном перемешивании охлаждают до перво-
начальной температуры. Через несколько часов осажденную фрак-
Рис. 4.2. Сосуды для фракционирования.
а —фракционирование по методу добавления осадителя; б — фракционирование по методу
испарения растворителя.
цию полимера можно отделить через кран у дна сосуда или путем
декантации, сифонирования или отсасывания с помощью шприца.
Оставшийся раствор обрабатывают дополнительным количе-
ством нерастворителя, используя ту же самую методику.
Фракции полимера отделяют и высушивают. Последнюю фрак-
цию получают испарением жидкости.
Основным недостатком этого варианта метода фракционного
осаждения является то, что каждая фракция содержит в заметном
количестве компоненты более низкого молекулярного веса; этот
эффект получил название эффекта хвоста, или эффекта шлейфа.
С целью уменьшения этого эффекта каждую фракцию необхо-
димо подвергнуть рефракционированию. Для того чтобы избежать
увеличения объема жидкости, рекомендуется сочетать фракцион-
ное осаждение с процессом испарения. Недостаток этого метода
заключается в том, что фракционное осаждение с 4—6 рефрак-
ционированиями требует примерно месяца.
76
Глава 4
Обзорная литература: 560, 770, 1256.
Периодическая литература: 2492, 2529, 2701, 3401, 3902, 4621, 4622, 5075,
5227, 5240, 5246, 5303, 5391, 5457, 5551, 5575, 6296, 6783, 6879, 6882.
Системы растворитель — нерастворитель для ряда полимеров
приведены в табл. 2.1. Более подробно этот вопрос освещен в
Polymer Handbook [О: 487].
4.1.3. Метод испарения растворителя
В методе испарения растворителя последовательное осаждение
полимерных частиц из раствора полимера в смеси растворителя
с нерастворителем осуществляют путем регулируемого испарения
более летучего растворителя.
Образец полимера, растворенный в соответствующем раство-
рителе (0,1—1,0 вес. %), помещают в сосуд для фракционирования
(рис. 4.2,6). При тщательном перемешивании и постоянной тем-
пературе в атмосфере инертного газа добавляют постепенно не-
растворитель до достижения точки мутности. Испарение смеси
растворителя с нерастворителем осуществляется с помощью тока
сухого нагретого газа, который пропускают через сосуд, находя-
щийся под разрежением, которое создается водоструйным насо-
сом. Степень осаждения можно контролировать с помощью тур-
бидиметра. Когда раствор достигает определенной степени мут-
ности, ток воздуха сначала замедляют и затем совсем прекращают
подачу воздуха. После этого раствор нагревают на несколько
градусов до тех пор, пока он не станет прозрачным, и при энер-
гичном перемешивании охлаждают его постепенно до первоначаль-
ной температуры. Через несколько часов выпавшую полимерную
фракцию можно отделить через кран на дне сосуда, а также де-
кантацией, сифонированием или отсасыванием с помощью шприца.
Преимуществами этого метода являются уменьшение объема си-
стемы по мере протекания фракционирования, непрерывность из-
менения состава смеси растворитель — нерастворитель, исключение
локальной, более высокой концентрации нерастворителя, а также
возможность увеличения в 2 раза загрузки полимера на том же
самом оборудовании.
Обзорная литература: 560, 770.
4.1.4. Метод понижения температуры
В методе понижения температуры последовательное осаждение
полимерных частиц из раствора осуществляют путем регулируе-
мого охлаждения.
Образец полимера растворяют в соответствующем нагретом
растворителе. Необходимо обратить внимание на то, чтобы при
высоких температурах не протекала деструкция полимера. Охла-
Фракционирование полимеров
77
ждение проводят при энергичном перемешивании с такой ско-
ростью, чтобы медленно приближаться к квазиравновесному со-
стоянию; обычно это занимает несколько часов. Размер фракции
зависит от разницы температур. Улучшению процесса фракциони-
рования может способствовать добавление небольших количеств
нерастворителя. Этот метод является весьма простым и не требует
специального оборудования, можно использовать обычный сосуд
для фракционирования (см. рис. 4.2).
Обзорная литература: 560, 770.
Периодическая литература: 2313, 2828—2830, 3421, 4257, 4471, 4687, 5443.
4.1.5. Метод изменения давления
В методе изменения давления последовательное осаждение прово-
дят при нижней критической температуре растворения в изотер-
мических условиях путем изменения давления системы.
Вначале выпадает самая низкомолекулярная фракция. В об-
ласти низких молекулярных масс эффективность метода плохая.
4.2. ТУРБИДИМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
Турбидиметрическое композиционное титрование — аналитический
метод оценки молекулярновесового распределения путем непрерыв-
ного осаждения полимерных молекул из очень разбавленного рас-
твора постепенным добавлением нерастворителя. Вначале выпа-
дают наиболее высокомолекулярные молекулы, мутность при этом
возрастает, т. е. наблюдается увеличение оптической плотности
раствора (т) (разд. 13.1.2). При добавлении большего количества
нерастворителя осаждаются более низкомолекулярные молекулы
и мутность непрерывно возрастает.
С помощью калибровки фракциями известного молекулярного
веса можно найти соотношение между количеством нерастворителя
и молекулярным весом. Величина мутности является характери-
стикой количества полимера.
Турбидиметрическое температурное титрование — аналитиче-
ский метод оценки молекулярновесового распределения путем не-
прерывного осаждения полимерных молекул из очень разбавлен-
ного раствора плавным изменением температуры.
В прибор для турбидиметрического титрования (рис. 4.3) вхо-
дят монохроматический источник света; турбидиметрическая ячей-
ка объемом 25—100 мл, помещенная в термостат, в котором можно
изменять температуру от 20 до 200 °C с колебаниями +0,01 °C;
система для перемешивания (скорость перемешивания в турбиди-
метрической ячейке должна быть как можно более низкой, чтобы
избежать образования нестабильных растворов, но раствор при
этом должен оставаться гомогенным); устройство для введения
78
Глава 4
^2
1 з
3
Рис. 4.3. Прибор для тур-
бидиметрического титро-
вания.
/ — источник света; 2 — бю-
ретка с осадителем; 3 — изме-
рительный фотоэлемент:
4— термостат; 5—турбиди-
метрическая кювета; 6 — проз-
рачные окна.
нерастворителя, которое характеризуется постоянной воспроизво-
димой скоростью потока, — последнюю необходимо регистрировать
(скорость добавления нерастворителя лежит между 0,05 и
2,0 мл/мин); фотоэлемент с умножителем и регистрирующим
устройством (интенсивность рассеянного света измеряется под
углом 90°).
В зависимости от среднего молекулярного веса и типа системы
растворитель — нерастворитель начальная концентрация образца
полимера колеблется в интервале от 0,5 до 50 мг на 100 мл. При
более высоких концентрациях могут на-
блюдаться коагуляция и выпадение хлопьев
полимера.
Турбидиметрическое титрование, кото-
рое позволяет определить самое первое по-
явление мутности (титрование до точки
мутности), следует проводить при различ-
ных концентрациях полимера с последую-
щей экстраполяцией полученных результа-
тов до 100 вес. %-ной концентрации поли-
мера или при одной концентрации, но с
использованием ряда образцов с различ-
ным молекулярным весом.
При турбидиметрическом титровании,
которое измеряет увеличение мутности,
обычно ограничиваются лишь одной кон-
центрацией одного полимерного образца.
При турбидиметрическом титровании
возможно много ошибок, поэтому необхо-
димо введение поправок. К важнейшим из
них относятся:
1. Поправки на изменение объема жидкости. Концентрация не-
растворителя в жидкости не изменяется линейно с объемом нерас-
творителя, введенного в турбидиметрическую ячейку.
2. Поправки на изменение концентрации полимера в жидкости.
Концентрация полимера не изменяется линейно с объемом нерас-
творителя, введенного в турбидиметрическую ячейку.
3. Поправки на концентрацию осажденного полимера.
4. Поправки на изменение относительного показателя прелом-
ления осажденных частиц и жидкости. Для гомополимеров пока-
затель преломления не меняется. В случае сополимеров есть раз-
личие в показателе преломления между сополимерами.
5. Поправки на размер частиц (см. теорию светорассеяния в
разд. 13.1.3).
Турбидиметрический метод удобен при изучении
а) молекулярновесового распределения полимера;
б) процессов образования полимеров (периодический и непре-
рывный процессы, получение полимеров в массе, растворе, суслен-
Фракционирование полимеров
79
зии или эмульсии, влияние температуры, катализатора или скоро-
сти введения мономеров в зону реакции);
в) процессов деструкции в массе или растворе;
г) процессов сополимеризации (процессов получения привитых
и блок-сополимеров); позволяет получить ценную информацию о
степени образования сополимеров и количестве гомополимеров.
Обзорная литература: 275, 309, 325, 403, 512, 560, 607, 689, 774, 775, 1017,
1111, 1164, 1171, 1311, 1325, 1366.
Периодическая литература: 2023, 2083, 2084, 2310, 2420, 2509, 2685, 2863,
2912, 2973, 3013, 3014, 3301, 3303, 3304, 3584, 3650, 3705, 3715, 3785, 3796, 3798,
3842, 3913, 3920, 3941, 4084, 4090, 4138, 4211, 4697, 4747, 4748, 4793, 5110, 5226,
5328, 5329, 5350, 5435, 5624, 5684, 5697, 5762, 5733, 5734, 5940, 6135, 6245, 6303,
6350, 6504, 6525, 6563, 6615, 6720, 6760, 6872, 6896.
4.3. КУМУЛЯТИВНОЕ ФРАКЦИОНИРОВАНИЕ
Кумулятивное фракционирование — это метод, который заклю-
чается в растворении полимера в растворителе и последующем до-
бавлении к нему относительно большого объема (одной трети)
нерастворителя. Смесь центрифугируют и выпавший полимер от-
деляют от жидкости. Эту процедуру повторяют с использованием
более сильного осадителя для удаления из раствора большего ко-
личества полимера. Характеризуют количество и молекулярный
вес полимера из каждого такого раствора, что позволяет построить
кривые молекулярновесового распределения.
Обзорная литература: 84, 689.
Периодическая литература: 2306, 2406, 2529, 2582—2584, 2967, 3699, 5084,
5966, 6495.
4.4. ФРАКЦИОННОЕ РАСТВОРЕНИЕ
Фракционным растворением называется метод, который заклю-
чается в том, что готовят полимер в соответствующем физическом
состоянии, после чего из него экстрагируют фракции все более и
более высокого молекулярного веса, используя для этого ряд элю-
ентов с повышающейся от фракции к фракции растворяющей
способностью. При таком методе вначале экстрагируется наиболее
низкомолекулярная фракция, тогда как наиболее высокомолеку-
лярная фрация выделяется последней.
Существует ряд методов фракционного растворения:
1. Прямая экстракция — это прямое и последовательное элюи-
рование образца полимера жидкостями с повышающейся раство-
ряющейся способностью в экстракторе (аппарат Сокслетта) при
определенной температуре.
Обзорная литература: 331, 370, 406, 560.
Периодическая литература: 3142, 4100, 4517, 4729, 4995, 5420, 6928, 7214.
80
Глава 4
под давлением
2
Рис. 4. 4. Прибор для фракционного
растворения в колонке.
/ — емкость со смесью растворителя и оса-
дителя; 2— нагреватель; 3 — колонка с об-
разцом; 4 — инертный носитель, покрытый
Полимером; 5 —инертный носитель; 6 — кол-
лектор для сбора фракций.
2. Экстракция пленки. Такая операция может быть осуществ-
лена двумя способами:
а) Периодический процесс. На алюминиевой фольге погруже-
нием ее в раствор полимера отливают полимерную пленку тол-
щиной 5—10 мкм. Затем пленку сушат и разрезают на кусочки
размером 1 X 3 см. Площадь по-
верхности для 500—1000 мг по-
лимера должна составлять 600—
1200 см2. После этого кусочки по-
крытой фольги помещают в кол-
бу Эрленмейера и экстрагируют
пленку 100 мл соответствующей
смеси растворителя с осадителем
при осторожном взбалтывании.
После достижения равновесия
элюент удаляют и вносят вторую
порцию растворителя с более
сильной растворяющей способ-
ностью. Эту процедуру продол-
жают до тех пор, пока фракцио-
нирование не завершится.
б) Непрерывный процесс. Рас-
твор полимера наносят в виде
тонкого покрытия на обе сторо-
ны медленно движущейся ленты,
при прохождении через зону суш-
ки растворитель испаряется. Тон-
кая пленка подвергается экстрак-
ции в ряде трубок, содержащих смеси растворитель — нераство-
ритель с повышающейся растворяющейся способностью, которые
находятся при постоянной температуре.
Обзорная литература: 406, 560, 707.
Периодическая литература: 2357, 3534—3536, 3833.
3. Экстракция в колонке — в этом методе полимер наносят на
инертную подложку (например, песок или стеклянные шарики),
которой заполнена колонка, и проводят последовательное элюи-
рование жидкостями с повышающейся растворяющей способ-
ностью. Градиент жидкостей колеблется от 100%-ного нераство-
рителя до 100%-ного растворителя с изменением состава раство-
рителя во времени в логарифмической шкале. Наиболее обычный
способ создания такого градиента заключается в использовании
сосуда для смешения, снабженного хорошей мешалкой, в который
вначале вносят нерастворитель, а затем часть его, удаленную из
смесительной камеры в колонку, заменяют растворителем, добав-
ляя последний с той же скоростью.
Фракционирование полимеров
81
По колонке движутся полимерные молекулы, извлекаемые
жидкой смесью, которая пропускается через колонку под давле-
нием азота (рис. 4.4). Колонку можно подогревать, можно про-
водить экстракцию кипящей смесью растворителя с нераствори-
телем. Если колонка имеет температурный градиент, то экстракция
в колонке становится осадительной хроматографией (разд. 4.7.2).
Для получения молекулярновесового распределения из собран-
ных фракций элюента выделяют полимер, определяют его коли-
чество и молекулярный вес для каждой фракции и строят на
основании таких данных кривые интегрального и дифференциаль-
ного распределения.
Обзорная литература: 80, 109, 406, 707, 1057, 1186.
Периодическая литература: 2193, 2248, 2392, 2505, 2702, 2709, 3050, 3052,
3342, 3446, 3474, 3590, 3625, 3812, 3911, 3912, 4022, 4066, 4134, 4531, 4728, 4816,
4976, 4977, 5380, 5551, 5553, 5589, 5686, 5858, 5876, 6243, 6279, 6314, 6386,
6742, 6781, 6826, 7212, 7213.
4. Экстракция коацервата. Согласно этому методу, полимер по-
следовательно экстрагируют из коацервата. Коацерватом назы-
вается обогащенная полимером жидкая фаза, которая отделяется
при добавлении к раствору полимера нерастворителя.
В этом методе нерастворитель добавляют к раствору до тех
пор, пока практически весь полимер не перейдет в коацерват. Уда-
ляют разбавленный раствор полимера, содержащий самые низко-
молекулярные фракции, и выделяют из него полимер. Затем коа-
церват экстрагируют смесью с несколько большей растворяющей
способностью. Эту процедуру повторяют до завершения фракцио-
нирования.
Обзорная литература: 406, 707, 1111.
Периодическая литература: 2391, 2657, 3076, 3081, 3254, 3763, 3905, 4383,
4385, 4386, 4388, 4390—4392, 4395—4399, 4976, 4977, 5443, 5576, 5595, 5996, 6805,
6807, 6860.
4.5. ФРАКЦИОНИРОВАНИЕ РАСПРЕДЕЛЕНИЕМ
МЕЖДУ НЕСМЕШИВАЮЩИМИСЯ
РАСТВОРИТЕЛЯМИ
Распределение между несмешивающимися растворителями пред-
ставляет собой метод, при котором полимерные молекулы распре-
деляются по их молекулярному размеру между двумя несмеши-
вающимися жидкостями различной растворяющей способности.
Разделение проводится путем обычной противоточной экстракции.
Периодическая литература: 2587, 2754, 2755, 3254, 4448, 6879, 6752.
В том случае, когда растворители смешиваются при высоких
температурах, а при пониженной температуре происходит фазовое
разделение, становится возможным количественное разделение
82
Глава 4
отличающихся между собой по химическому строению полимеров
из растворов в соответствующих гомогенных смесях растворителей.
Периодическая литература: 4761.
4.6. ФРАКЦИОННАЯ КРИСТАЛЛИЗАЦИЯ
Фракционная кристаллизация — это метод, предполагающий по-
следовательное выделение макромолекул из раствора полимера
путем кристаллизации при различных температурах. Кристаллиза-
цию можно вызвать путем быстрого перемешивания. Вначале кри-
сталлизуются наиболее высокомолекулярные макромолекулы, оса-
ждаясь на мешалке в виде тонких длинных фибриллярных кри-
сталлов. Этот метод относится к числу плохо воспроизводимых.
Периодическая литература: 2058—2060, 2395, 2495, 2496, 2498, 4378, 4473,
4477, 4522, 4661, 5750, 5752, 5643, 5884, 5903, 7227, 7228, 7229.
4.7. ФРАКЦИОНИРОВАНИЕ С ПОМОЩЬЮ
ХРОМАТОГРАФИИ
4.7.1. Адсорбционная хроматография
Адсорбционная хроматография (хроматография на активном носи-
теле) представляет собой метод, основанный на том, что адсорб-
ция полимерных частиц на активном носителе зависит от молеку-
лярного веса.
Существует два общих метода адсорбционной хроматографии:
1. Фронтальный анализ. По этому способу активным носителем
заполняют колонку. Раствор полимера пропускают через колонку
сверху вниз и собирают его на выходе из колонки (рис. 4.5).. Кон-
центрация в элюате меняется в соответствии с собранным объе-
мом и представляет собой последовательные фронты, что обуслов-
лено различной адсорбцией молекулярных частиц на активном но-
сителе (см. также гл. 23).
2. Анализ, основанный на элюировании, можно осуществлять
в следующих двух вариантах:
а) Элюирование в колонке. По этому методу небольшим ко-
личеством полимера пропитывают верхнюю часть носителя, на-
полняющего колонку. Элюирование полимерных частиц проводят
подходящим растворителем. Различные полимерные частицы дви-
жутся вниз по колонке с разной скоростью и собираются в конце
концов в элюате на выходе из нее. В методе элюирования с гра-
диентной колонкой элюент представляет собой жидкость с повы-
шающейся растворяющей способностью (разд. 23.13).
б) Тонкослойная хроматография (ТСХ). По этому методу не-
большое количество полимера наносят на стеклянную пластинку,
покрытую силикагелем (разд. 23.15). Затем такую стеклянную пла-
Фракционирование полимеров
83
стинку помещают вертикально в сосуд, содер-
жащий соответствующий растворитель. По
мере продвижения растворителя вверх с ним
захватывается и каждый компонент полимер-
ного вещества, но с разной скоростью. Этим
методом можно разделить полимерные части-
цы по химическому составу, молекулярному
весу, стереорегулярности. Решающим факто-
ром в определении молекулярновесового рас-
пределения является точный анализ концен-
трации полимера как функции расстояния
вдоль пути его элюирования. Наиболее точ-
ные результаты дает прямое денситометриче-
ское сканирование хроматограмм с помощью
спектроденситометра.
Периодическая литература: 4215, 4403, 4404, 5311,
5625—5627, 5630 (см. также литературу к разд. 23.18).
4.7.2. Осадительная хроматография
Осадительная хроматография (называемая
также методом Бейкера — Уилльямса)—это
метод, согласно которому небольшое количе-
ство полимера наносят на инертный носитель,
например, стеклянные шарики диаметром
40—70 мкм, заполняют таким обработанным
Рис. 4.5. Прибор для
хроматографического
фракционирования
по методу фронталь-
ного анализа.
1 — емкость с раствором
полимера; 2 —колонка;
3 — активный носитель;
4—коллектор для сбора
фракций.
носителем колонку,
после чего проводят последовательное элюирование полимера с
использованием жидкости с возрастающей растворяющей способ-
ностью. Градиент жидкости колеблется от 100%-ного нераствори-
теля до 100%-ного растворителя с изменением состава раствори-
теля во времени в логарифмической шкале.
Вдоль колонки устанавливается температурный градиент. Верх-
нюю часть колонки поддерживают при более высокой температуре
(100—200°C), чем ее дно, где температура комнатная (рис. 4.6).
Частицы полимера движутся по колонке сверху вниз, причем
между осажденной фазой и насыщенным раствором происходит
непрерывный обмен. Распределение зависит от размера молекул.
Определить молекулярновесовое распределение можно, собирая
элюированные фракции, выделяя из них полимер, для которого
оценивают количество и молекулярный вес, и, наконец, строя на
основании полученных данных кривые интегрального и дифферен-
циального распределения.
Осадительная хроматография может также проводиться и в
отсутствие температурного градиента, в этом случае она превра-
щается в непрерывное фракционирование фракционным раство-
рением [экстракция в колонке (разд. 4.4)].
84
Глава 4
Рис. 4.6. Прибор для
хроматографического
фракционирования
по методу Бейкера —
Уилльямса.
/—емкость с растворите-
лем; 2 —колонка; 3—инер-
тный носитель, покры-
тый полимером; 4 — инер-
тный носитель; 5—нагре-
ватель; б —коллектор для
сбора фракций.
внутри пор геля и
Обзорная литература: 80, 152, 689, 853, 1056, 1057,
1163, 1172, 1186
Периодическая литература: 2206, 2470, 2637, 2702,
2703, 2705, 2713, 2797, 2964, 2965, 3052, 3091, 3428,
3625, 3672, 3784, 3812, 3813, 3839, 3890, 4005, 4177,4325,
4349, 4375, 4531, 4532, 4600, 4601, 4620, 4642, 4728,
5231, 5257, 5327, 5341, 5554, 5589, 5679, 5766, 5858,
5865, 6058, 6239, 6240, 6241, 6242, 6279, 6285, 6455,
6457, 6487, 6885, 7026, 7215.
4.7.3. Гель-проникающая хроматография
Гелъ-проникающая хроматография (ГПХ) —
это метод, в котором разделение полимерных
молекул основано на различных объемах вну-
три пористых частиц геля, которые доступны
молекулам растворенного вещества разного
размера.
В колонку помещают маленькие частички
геля с разным размером пор. Растворитель
заполняет промежуточное пространство ме-
жду частичками и все поры геля. Образец
растворяют, вводят в колонку и извлекают
весь полимер пропусканием того же самого
растворителя. В результате ограничений, на-
кладываемых размером пор на большие моле-
кулы, существует значительный объем пор,
доступный полимерным молекулам меньшего
размера, которые свободно диффундируют
между частичками его. В итоге маленькие мо-
лекулы характеризуются большей длиной пробега и вследствие
этого более сильно удерживаются при элюировании. Разделение
молекул происходит только по их длине и, по-видимому, является
нечувствительным к структуре (разд. 25.3).
Обзорная литература: 30, 32, 689 (см. также литературу к разд. 25.3).
4.7.4. Фазораспределительная хроматография
Фазораспределительная хроматография (ФРХ)—метод разделе-
ния полимеров путем распределения образца между растворите-
лем и полимерной фазой очень высокого молекулярного веса,
которой в виде тонкого слоя покрывают неактивный носитель,
например стеклянные шарики диаметром 0,1 мм. Разделение ста-
новится более эффективным при понижении температуры раство-
рителя, который необходимо поддерживать при температуре, ле-
жащей ниже тета-температуры образца (рис. 4.7).
Разделение проводят в приборе, показанном на рис. 4.8.
Периодическая литература: 2758.
Фракционирование полимеров
85
Удерживаемый объем, мл
Рис. 4.7. Эффективность разделения в
фазораспределительной хроматогра-
фии двух образцов А и В в зависи-
мости от температуры (Т).
Рис. 4.8. Прибор для фазораспреде-
лительной хроматографии.
1 — емкость с растворителем; 2 — колонка
с образцом; 3 — эталонная колонка; 4 —сте-
клянные шарики, покрытые полимером;
5 —стеклянные шарики; 6— нагреватели;
7 —дифференциальный рефрактометр;
8 — коллектор для сбора фракций.
4.8. ФРАКЦИОНИРОВАНИЕ СЕДИМЕНТАЦИЕЙ
Ультрацентрифугирование представляет собой классический метод
определения молекулярного веса высокомолекулярных полимеров и
кривых их молекулярновесового распределения. В седиментацион-
ном анализе используют следующие три общих метода:
а) метод скоростной седиментации (разд. 8.1);
б) метод седиментационного равновесия (разд. 8.2);
в) метод градиента плотности (разд. 8.4).
Обзорная литература: 689, 890 (см. также литературу в гл. 8).
4.9. ФРАКЦИОНИРОВАНИЕ ДИФФУЗИЕЙ
Фракционирование полимеров по их диффузии — это другой ана-
литический метод. Известны два общих метода:
а) Изотермическая диффузия (при постоянной температуре).
В этом случае полимерные молекулы диффундируют с различной
скоростью из раствора в граничный слой в зависимости от их
86
Глава 4
молекулярного веса. Константы диффузии можно определить опти-
ческими методами и выразить через них полидисперсность образца
(разд. 8.1.5).
Обзорная литература: 224, 689.
Периодическая литература: 2696, 2697, 3061, 3062, 3508, 3658, 4273, 4274,
5112, 5279, 5321, 5322, 6929.
б) Термическая диффузия. Согласно этому методу, вследствие
температурного градиента возникает тепловое циркулирование мо-
лекул в полимерном растворе и их разделение. Раствор полимера
впрыскивают в каскад колонок. Каждый каскад представляет со-
бой резервуар с температурным градиентом между верхней и ниж-
ней поверхностями. При таком температурном градиенте молекулы
движутся по направлению к нижней поверхности. То, что нахо-
дится на дне резервуара колонки 1 (каскад 1), становится исход-
ной загрузкой длй колонки 2 (каскад 2), содержимое на дне ко-
лонки 2 становится загрузкой для колонки 3 и т. д. на протяжении
8 каскадов, в которых пропускается только продукт со дна со-
судов.
Обзорная литература: 408, 689.
Периодическая литература: 3636—3639, 3641, 3642, 3648, 3834, 4083, 4136,
4673, 4676, 4677, 4719, 4774. 4812—4815, 5400, 6758, 6775, 6776.
4.10. ФРАКЦИОНИРОВАНИЕ ПУТЕМ
УЛЬТРАФИЛЬТРАЦИИ ЧЕРЕЗ ПОРИСТЫЕ
МЕМБРАНЫ
Ультрафильтрация через пористые мембраны представляет собой
метод диффузии молекул через ряд мембран различной пористо-
сти. Скорость диффузии зависит от молекулярного размера и сте-
пени проницаемости мембран. Высокопористые мембраны готовят
из чистых биологически инертных нитрата целлюлозы, ацетата
целлюлозы, регенерированной целлюлозы и других полимеров. Эти
мембраны называются поверхностными фильтрами. В противопо-
ложность глубинным фильтрам, полученным из волокнистых ма-
териалов, они отличаются исключительно высокой эффективностью
удерживания, что обусловлено их весьма однородной пористой
структурой и одинаковым размером пор. Большая часть вещества
при фильтрации раствора задерживается на поверхности (эффект
сита).
Фракционирование на глубинных фильтрах осуществляется ме-
ханически в результате адсорбции через толщу материала фильт-
ра. Они состоят из хаотически расположенных волокон или очень
толстых листов спрессованного гранулированного материала. Раз-
личия в природе волокон или размерах зерен и сопутствующие им
различия в упаковке и толщине приводят к тому, что размер кана-
лов глубинного фильтра может в определенной степени колебаться.
Фракционирование полимеров
87
Обычно такие фильтры используют в качестве эффективных пред-
варительных фильтров. На фильтрацию не влияют колебания дав-
ления внутри фильтрующей системы или pH раствора, подвергаю-
щегося фильтрации, в некоторой степени фильтрацию ухудшают
вещества со значительным поверхностным натяжением. К умень-
шению эффекта фильтрации и скорости пропускания может приве-
сти засорение поверхности фильтра.
Слоистые мембраны получают путем прикрепления однородной
тонкой пористой пленки из нитрата целлюлозы к картонной под-
ложке, изготовленной из высокоочищенной целлюлозы. Такая
пленка имеет губкоподобную структуру, характерную для поверх-
ностных фильтров с хорошо определенными размерами пор. До-
вольно высокая прочность подложки во влажном состоянии обес-
печивает создание слоистых мембран с прочностью, достаточной
для того, чтобы выдержать усилия, развивающиеся при фильтра-
ции под давлением. Слоистые мембраны сохраняют работоспособ-
ность даже при использовании для фильтрации жидкостей с пуль-
сирующей подачей. Пленочный слой определяет производитель-
ность, химическую стабильность и эффективность удерживания
фильтра. Он работает как микротонкое сито.
При наличии достаточно прочной подложки пористые мембран-
ные фильтры могут выдерживать давления вплоть до 800 кГ/см2.
Максимальное давление определяется конструкцией фильтрующей
аппаратуры. На практике рабочее давление в фильтрующих устрой-
ствах обычно не превышает 5—6 атм. Большинство промышленных
мембран пригодно для использования при температурах до 80—
125 °C.
Обзорная литература: 249.
Периодическая литература: 2208, 2571, 3713, 3763, 3999, 4000, 4088, 5258,
6095, 6523, 6949, 7088.
4.11. ФРАКЦИОНИРОВАНИЕ ЗОННОЙ ПЛАВКОЙ
Фракционирование методом зонной плавки основано на том, что
при равновесии в твердой и жидкой фазе раствора концентрации
твердого растворенного вещества различаются между собой. Ко-
лонку заполняют плохим твердым растворителем и на него сверху
помещают небольшое количество полимера, который затем рас-
творяют путем нагревания узкой зоны (рис. 4.9).
После затвердевания нагревают более низкую зону, плавят ее
и снова дают ей затвердеть. Таким путем колонку обрабатывают
постепенно, шаг за шагом. Частички расплавленного полимера
продвигаются вниз по колонке с разной скоростью в зависимости
от размера молекулы. В конце концов полимер распределяется по
всей длине колонки и извлекается путем разрезания в твердом со-
стоянии на несколько порций с последующим удалением раство-
рителя.
88
Глава 4
Рис. 4.9. Прибор для зонной плавки.
Обзорная литература: 249.
Периодическая литература: 3288, 4959, 4976, 4977, 5734, 6130.
4.12. ФРАКЦИОНИРОВАНИЕ ТЕЧЕНИЕМ
ПОД ВЛИЯНИЕМ ПОЛЯ
Фракционирование течением под влиянием поля (ФТП)—это та-
кой метод элюирования, при котором прикладывается внешнее си-
Рис. 4.10. Схематическое изображение фракционирования по методу течения под
влиянием поля.
а — канал течения; б—параболический профиль течения в канале.
ловое поле или градиент под углом 90° к потоку через всю поверх-
ность канала. Поле взаимодействует с растворенным веществом и
продвигает его к дальней стенке, формируя таким образом тонкий
равновесный слой, находящийся против стенки.
Толщина слоя различна для каждого полимера и зависит от
прочности сцепления между полем и макромолекулами и прису-
щего им коэффициента диффузии. Слой, который течет вдоль ка-
нала (рис. 4.10), имеет параболический профиль, причем скорость
жидкости наибольшая вблизи центра канала. Макромолекулы
Фракционирование полимеров
89
большего размера сдвигаются к стенке сильнее, чем меньшие мо-
лекулы. При таком методе вначале элюируются маленькие части-
цы, а большие частицы — в конце.
Внешнее силовое поле создается центробежными или грави-
тационными силами, различиями в электрических потенциалах или
температурах или другими полями.
Не считая настоящего фракционирования на колонке, все дру-
гие аспекты этого метода, в том числе введение образца, разделе-
ние, детектирование, обработка данных и возможность сбора об-
разца, такие же, как и при обычном хроматографическом фракцио-
нировании. ФТП можно с успехом использовать для разделения
макромолекул с широким набором молекулярных весов (от 103
до 1012, что соответствует размеру частиц от 10~3 до 1 мкм).
Обзорная литература: 510, 511.
Периодическая литература: 3636, 3644, 3646, 3647, 6775, 7230, 7231.
Глава 5
МЕМБРАННАЯ ОСМОМЕТРИЯ
Обзорная литература: 23, 114, 125, 127, 309, 681, 1040, 1082, 1301, 1307, 1350.
Если чистый растворитель отделен полупроницаемой мембра-
ной от того же растворителя, находящегося в растворе, их химиче-
Капилляры
Рис. 5.1. Принцип
измерения осмоти-
ческого давления.
ские потенциалы не равны друг другу. Это обу-
словливает перенос растворителя через мембра-
ну к раствору. Разность давлений по обе сторо-
ны от мембраны в состоянии равновесия назы-
вается осмотическим давлением и оценивается
разностью высот в капиллярах раствора и рас-
творителя (Л/г) (рис. 5.1).
Интервал молекулярных весов, которые мож-
но определить с помощью мембранной осмомет-
рии, составляет 5-Ю3—5-Ю5 и даже 106. Ниж-
ний предел задается проницаемостью мембраны
(диффузией частиц низкого молекулярного ве-
са), тогда как верхний предел определяется тем
наименьшим осмотическим давлением, которое
поддается измерению.
Обзорная литература: 77, 306,635,779,1003,1323,1365.
Периодическая литература: 2869, 4085, 4804, 6092,
6397, 7142.
5.1. ОПРЕДЕЛЕНИЕ ОСМОМЕТРИЧЕСКИМ МЕТОДОМ
СРЕДНЕЧИСЛОВОГО МОЛЕКУЛЯРНОГО ВЕСА
Для полидисперсного линейного полимера среднечисловой молеку-
лярный вес (Мп) можно рассчитать по данным осмометрии, ис-
пользуя следующие выражения:
4-Вс + Сс2+.... (5.1)
С Мп
= Д2с + Д3с2+...Y (5.2)
- = +Г2с + ГзС2+ ...), (5.3)
С мп
размерность которых дин-л на грамм-сантиметр (СГС) или джоуль
на килограмм (СИ). В этих выражениях л — осмотическое дав-
Мембранная осмометрия
91
ление, определяемое как
л — р^Л/г
(5.4)
р — плотность растворителя; g— ускорение силы тяжести
(0,981 м-с~2); Л/г— давление, выражаемое через высоту столба
растворителя (см); с — концентрация раствора; (л/с)—приведен-
ное осмотическое давление-, R— универсальная газовая постоянная
(разд. 40.2); Т — термодинамиче-
ская температура (К); В, А, Г2 и
С, Аз, Гз_—вириальные коэффи-
циенты; Мп — среднечисловой мо-
лекулярный вес. _
Для того чтобы получить Мп
в надлежащих единицах, необхо-
димо, чтобы выражение л/с име-
ло размерность дин-л-г-1 и R =
= 0,08207 л • атм • моль*"1 • К-1
(СГС) или размерность л/с была
Дж-кг-1 и R = 8,314 Дж-К-1
моль-"1 (СИ).
Поскольку третий член
(А3с2) пренебрежимо мал, то
уравнение (5.2) принимает вид
я/с = ЯГ(1/М„ + А2с) (5.5)
Рис. 5.2. Зависимость приведенного
осмотического давления (л/с) от
концентрации.
Тогда кривая зависимости л/с от с представляет собой_прямую ли-
нию, отсекающую на ординате отрезок, равный RT/Mn, с накло-
ном А2 (рис. 5.2). Если измерения проводятся в 0-условиях, то л/с
не зависит от с и равно
nlc = RTlMn (5.6)
На практике осмотическое давление измеряют как разность
высот жидкости в капиллярах с раствором и растворителем (Л/г)
в сантиметрах. Для экстраполяции на бесконечное разбавление
необходимо знать осмотическое давление при нескольких концен-
трациях (например, 2,0, 4,0, 6,0 и 8,0 г/л). Среднечисловой моле-
кулярный вес можно найти из кривой на рис. 5.2 или из выра-
жения
Мп
RT
(л/с)с~й
(5.7)
При обработке осмотических данных очень важно выражать экс-
периментальные данные в соответствующих единицах.
92
Глава 5
5.2. ОПРЕДЕЛЕНИЕ ОСМОТИЧЕСКИМ МЕТОДОМ
ВТОРОГО ВИРИАЛЬНОГО КОЭФФИЦИЕНТА
В осмометрии второй вириальный коэффициент (Л2) выражается
как
л = (5.8)
Ррмs к 2 7
где ps — плотность растворителя, рр — плотность полимера, Ж —
молекулярный вес растворителя, % — параметр взаимодействия
Флори (разд. 2.9).
Единицы, в которых выражается второй вириальный коэффи-
циент, зависят от вида используемого уравнения (табл. 5.1).
Таблица 5.1
Различные способы выражения приведенного осмотического давления (л/с)
и второго вириального коэффициента
^Уравнение Второй вириаЛьный коэффициент (В, ЛТг)
в единицах СГС в единицах СИ
— = + Вс + Сс2 С мп ДИН «л • Г-2 Дж • м3 • кг-2
-=RT(^- + Aic + A3c2'} С \Мп J Л • МОЛЬ • Г-2 м3 • моль • кг~2
— = ^-[(1 + Г2с + Г3с2)] С мп Л • г-1 М3 • КГ-1
Второй вириальный коэффициент (А2) экспериментально мож-
но вычислить из значений я/с для двух концентраций Ci и с2, ис-
пользуя выражение - . „ . _
л _ (я/с)а Crc/c)i ' /н q\
Л2“ /?Т(С2_С1)
1л«молЬ‘Г~2 (СГС) или м3-моль-кг~2 (СИ)].
А2 можно найти и из наклона кривой на рис. 5.2: tga=/42.
5.3. ОСМОТИЧЕСКОЕ ДАВЛЕНИЕ
ВЫСОКОКОНЦЕНТРИРОВАННЫХ ПОЛИМЕРНЫХ
РАСТВОРОВ
Приведенное осмотическое давление (л/с) для сильно концентри-
рованных растворов полимеров выражается как
А = АТ(1+Г2С+ГзС2+...), (5.10)
Мембранная осмометрия
93
Рис. 5.3. Зависимость
Концентрация (с)
где член (1 + Г2с + Г3с2 + ...)
дает относительное отклонение от
идеального раствора. Вириаль-
ные коэффициенты Г2 и Гз яв-
ляются функциями системы рас-
творитель— растворенное веще-
ство, температуры, молекулярно-
го веса растворенного вещества
и молекулярновесового распреде-
ления.
Преобразование уравнения
(5.10) дает
(•V- 1)| = Г2 + Г3с, (5.11)
где Д] = 4Д. Зависимость (-^— 1)— от с представляет собой
мп \ * с
прямую линию, отсекающую на ординате отрезок, равный Г2
(рис. 5.3). Гз находят как тангенс угла наклона прямой: tg а = Г3.
Обзорная литература: 456.
Периодическая литература: 2367, 6947, 6948.
5.4. МЕМБРАННЫЕ ОСМОМЕТРЫ
Мембранные осмометры можно разделить на две большие группы:
статические и динамические осмометры.
Статические осмометры конструируются следующих двух ти-
пов: 1) вертикальные осмометры (например, осмометр Хельфрица)
(рис. 5.4) и 2) горизонтальные осмометры (например, осмометр
Шульца) (рис. 5.5).
Статический осмометр состоит из двух ячеек: ячейки для рас-
твора, показанной на рис. 5.4 и 5.5, и ячейки для растворителя,
представляющей собой обычный стеклянный сосуд, куда помещают
ячейку с раствором. Эти ячейки разделяют одна или две полупро-
ницаемые мембраны. После заполнения одной ячейки чистым рас-
творителем, а другой — раствором известной концентрации осмо-
метр помещают в термостат (+0,01 °C). После установления изо-
хронного давления измеряют разность высот жидкости в капилля-
рах, соединенных с каждой ячейкой. Изохронное давление — это
давление, при котором суммарный объемный поток равен нулю.
Основной недостаток статического метода состоит в большой про-
должительности (от нескольких часов до нескольких дней) дости-
жения равновесного осмотического давления. Это время опреде-
ляется главным образом тем временем, которое требуется раство-
рителю для перехода в ячейку с раствором через полупроницаемую
мембрану.
94
Глава 5
Обзорная литература: 635, 1040, 1323.
Периодическая литература: 2609, 2869, 3316, 3317, 3505, 3553, 3651, 3998,
5107, 5247, 6182, 6271, 6273, 6507, 7335.
Динамические осмометры конструируются в виде автоматиче-
ских приборов, измеряющих скорость течения через полупрони-
цаемую мембрану, с регулированием давления таким образом, что-
бы полезный поток был равен нулю. Важнейшим достоинством ди-
намического метода является возможность измерения изохронного
Уровень
растворителя.
Осмометр в разрезе
(вид едоку)
Рис. 5.4. Узел кюветы в вертикальном ос-
мометре Хельфрица.
1 — капилляры; 2—ячейка с раствором; 3—вну-
тренний фланец; 4— мембрана; 5 —внешний фла-
нец.
Уровень
растворителя
Осмометр в разрезе
(вид сбоку)
Рис. 5.5. Узел кюветы в гори-
зонтальном осмометре Шульца.
/ — капилляры; 2—ячейка с раство-
ром; 3— внутренний фланеп; 4-мем-
брана; 5 —внешний фланец.
давления через небольшой промежуток времени (5—10 мин) и
меньший перенос растворенного вещества.
Наиболее распространены следующие динамические осмометры:
1. Осмометр Mechrolab (рис. 5.6). В этом осмометре горизон-
тальная мембрана разделяет две ячейки: одну верхнюю для рас-
твора и другую нижнюю для растворителя. К ячейке, содержащей
растворитель, присоединен стеклянный капилляр. Один его конец
соединен гибкой трубкой с емкостью, содержащей растворитель.
В капилляр вводится пузырек воздуха, который прерывает свето-
вой луч, освещающий фотоячейку. Этот эффект используется для
выравнивания осмотического тока с помощью сервосистемы. Ха-
рактеристикой осмотического давления служит разность высот
жидкости в капилляре, соединенном с ячейкой раствора, и в ре-,
зервуаре с растворителем.
Мембранная осмометрия
95
Рис. 5.6. Схема осмометра. Mechrolab.
/ — капилляр; 2—ячейка с раствором; .3 —мембрана; 4 — ячейка с растворителем; 5 —усили-
тель; 6— фотоэлемент; 7 —пузырек воздуха; 8 — источник света; 9 — емкость с растворителем;
10 — подъемник; // — сервомотор.
Рис. 5.7. Схема осмометра Shell.
/ — ячейка с растворителем; 2 — мембрана; 3—ячейка с раствором; 4—металлическая диа
фрагма; 5—емкостный датчик; 6 — емкостно-чувствительный осциллятор; 7 —усилитель;
8 — самописец; 9 — манометрическая трубка с растворителем; 10—сервомотор.
96
Глава 5
2. Осмометр Shell (рис. 5.7). В этом осмометре также горизон-
тальная мембрана разделяет две ячейки: верхнюю для раствори-
теля и нижнюю для раствора. Одной из стенок в ячейке с раство-
ром, выполняющей роль пластины конденсатора, является гибкая
металлическая диафрагма. Конденсатор представляет собой часть
сервомеханизма, с помощью которого регулируется давление в
ячейке с растворителем. При проникании растворителя через мем-
брану диафрагма отклоняется, возникающее при этом изменение
частоты тока генерирует сигнал, приводящий в действие сервоме-
ханизм. Равновесие регистрируется на самописце, функционирую-
Рис. 5.8. Схема осмометра Melabs.
/ — ячейка с раствором; 2—мембрана; 5 —ячейка с растворителем; 4 — диафрагма из нержа-
веющей стали; 5—подвод энергии; 6 — тензодатчик; 7 — усилитель; 8 — самописец.
щем от этого сервомеханизма, причем наклон выписываемой кри-
вой дает возможность следить за прохождением растворенного ве-
щества через мембрану. Объем образца составляет 8—10 мл. Рав-
новесное осмотическое давление можно оценить уже через 15—
30 мин с точностью ±0,01 см. Область определяемых молекуляр-
ных весов составляет 10 000—30 000, измерения можно проводить
при температуре 20—60° (и даже до 130° в определенных моди-
фикациях прибора).
3. Осмометр Melabs (рис. 5.8). В осмометре этого типа серво-
механизм отсутствует. Осмотическое давление измеряют непосред-
ственно с помощью тензодатчика, прикрепленного к гибкой диа-
фрагме в ячейке с растворителем.
Обзорная литература: 45, 125, 309, 400, 723, 1323.
Периодическая литература: 2610, 2932, 4095, 5659, 6017, 6088.
Данные, полученные с помощью автоматических осмометров,
могут быть введены в ЭВМ. Эти данные включают сведения о рас-
творителе, весовых загрузках, разбавлении, подпоре растворителя.
Мембранная осмометрия 97
Выходными данными являются значения п/с, (л/с) о, второго
вириального коэффициента и стандартных отклонений.
Обзорная литература: 311.
5.4.1. Мембраны
Мембраны должны быть изготовлены из материала, который не
растворяет используемый растворитель. Наиболее распространены
гель-целлофановые мембраны, выдержанные в воде. Их получают
осаждением ксантогената целлюлозы и после формования никогда
не высушивают. Доступны также мембраны из гидратцеллюлозы,
ацетата и нитрата целлюлозы, полиуретанов, полихлортрифторэти-
лена (типа Кель-F) и даже из стекла типа викор с размером
ячеек 40 А.
Перед тем как использовать мембраны в органическом раство-
рителе, их вымачивают в воде и доводят до кондиции, что вклю-
чает замену воды на растворитель. Если растворитель не смеши-
вается с водой, мембрану обрабатывают сначала этанолом или
ацетоном и лишь затем растворителем, который будет использо-
ваться в эксперименте. К мембранам, выпускающимся промыш-
ленностью, прилагаются инструкции по подобной обработке.
Обзорная литература: 45, 282, 307, 398, 635.
Периодическая литература: 2093, 2301, 2738, 2911, 2931, 3300, 3401, 3553,
3651, 4114, 4213, 4766, 5106, 5167, 5179, 5248—5250, 5696, 5816, 6060, 6416, 6524,
6382, 6932, 6933, 6946, 7166, 7233.
5*4.2. Экспериментальные проблемы в мембранной
осмометрии
В мембранной осмометрии возможны следующие эксперименталь-
ные осложнения:
1. Разрушение мембраны, которое трудно заметить в статиче-
ских осмометрах; при использовании динамических автоматиче-
ских осмометров просачивание легко обнаружить по нестабильно-
сти базовой линии.
2. Асимметрия мембраны, выражающаяся в том, что при за-
полнении обеих ячеек осмометра идентичными растворителями
все-таки возникает разница в давлении между ячейками. Этот эф-
фект обусловлен просачиванием, сдавливанием мембраны, влия-
нием растворенного вещества и температурными градиентами.
3. Всплывание мембраны, которое вызывается разницей в дав-
лении и может быть обнаружено по изменению давления при до-
бавлении растворителя в ячейку, предназначенную для него, или
при удалении растворителя из этой ячейки.
4. Преждевременное разрушение мембраны.
5. Наличие растворенного воздуха. Перед тем как заполнить
ячейки, растворитель и раствор следует дегазировать.
Обзорная литература: 1323.
Периодическая литература 3305, 4762, 5815, 6290, 6521, 6522, 6524, 6944,
6945, 6997.
4 Зак. 238
Глава 6
МЕТОДЫ ИССЛЕДОВАНИЯ, СВЯЗАННЫЕ
С ОЦЕНКОЙ КОЛЛИГАТИВНЫХ СВОЙСТВ
Обзорная литература: 23, 125, 127, 128, 309, 402, 681, 1082, 1350.
К коллигативным свойствам раствора относятся свойства, ко-
торые определяются числом молекул, находящихся в системе.
Методы, основанные на этих свойствах, включают: а) эбулио-
метрию, б) криоскопию, в) изотермическую дистилляцию (изопие-
стический метод), г) парофазную осмометрию.
Таблица 6.1
Коллигативные свойства полимеров в растворе
Эффект Эффект изменения свойства для полимеров различного молекулярного веса
Мп = 1000 Мп = 5000 Мп=20 000
Повышение температу- ры кипения, °C оо ю о ел о о 1 1 Ю К1 5•10“3 1,5 - 10^3
Понижение температу- ры замерзания, °C 5 - 10”2 1 • 10“2 2,5- 10~3 —.3
Понижение давления паров, мм рт. ст. 8- 10“2 1,5« 10 2 4- 10 3
Оцениваемые эффекты пропорциональны молярной концентра-
ции растворенного вещества. Среднечисловой молекулярный вес
(Мп) легко рассчитать, если известна весовая концентрация. Для
каждого метода необходимо проводить измерения при нескольких
концентрациях и экстраполировать на бесконечное разбавление.
В табл. 6.1 представлены значения различных эффектов для пере-
численных методов.
6.1. ЭБУЛИОМЕТРИЯ
Эбулиометрия базируется на различии между температурами ки-
пения раствора и чистого растворителя.
Используя эбулиометрические данные, можно рассчитать Мп
полидисперсных линейных полимеров по- следующим формулам:
— = ^- + Яс + СС2+ .... (6.1)
С Мп
^ = к/Х + Л2с+Л3с2+...Y (6.2)
с \ Мп J
-^ = ^(1+Г2С+ГзС2+...), (6.3)
с Мп
Методы, связанные с оценкой коллигативных свойств
99
Рис. 6.1. Зависимость приве-
денного повышения температу-
ры кипения (ДТ/с) от койцен-
размерность которых кельвин-метр на грамм (СГС) или кель-
вин-кубический метр на килограмм (СИ), где АГ — повышение
температуры кипения (К); с — концентрация полимера; КГ/с—
приведенное повышение температуры кипения\ К— эбулиоскопи-
ческая константа (величина, характеризующая повышение темпе-
ратуры кипения, вызываемое 1 молем растворенного вещества)
= (6-4)
где R— универсальная газовая постоянная (разд. 40.2); Ts — тем-
пература кипения растворителя (К); Ms— молекулярный вес рас-
творителя; ps — плотность растворите-
ля; &HS — теплота испарения раство-
рителя; В, А2, Г2 и С, А3, Гз — вири-
альные коэффициенты. Размерность
Ке зависит от того, в каких единицах
выражены все другие параметры: R,
Тs, MS, Ps, MiS.
В табл. 6.2 приведены эбулиоско-
пические константы для ряда раство-
рителей. Поправки на давление
dKe/dp, которые также указаны в
этой таблице, следует прибавлять к К
для каждого миллиметра давления
выше 760 мм рт. ст. (1 мм рт. ст. =
== 1/760 атм = 133,322 Н-м-2).
Мп можно рассчитать из кривой,
представленной на рис. 6.1.
Эбулиометрия применима для определения молекулярных весов
вплоть до 50 000 и даже 100 000—170 000.
Обзорная литература: 124, 423, 424, 618, 810, 1133, 1446.
Периодическая литература: 3675.
6.1.1. Определение с помощью эбулиометрических
измерений второго вириального коэффициента
В эбулиометрии второй вириальный коэффициент (А2) определяет-
ся по формуле
где R— универсальная газовая постоянная (разд. 40.2); Т — тер-
модинамическая температура (К); р? — плотность полимера; &HS —
теплота испарения растворителя; % — параметр взаимодействия
Флори (разд. 2.9).
А2 можно найти по тангенсу угла наклона (tga=A2) кривой
зависимости КГ/с от концентрации (рис, 6,1).
4*
100
Глава 6
Таблица 6.2
Эбулиоскопические константы для различных растворителей
Растворитель Точка кипения. °C °С-моль“1 (мм рт. ст.)—1
Уксусная кислота 118,0 3,07 0,8
Ацетон 56,2 1,71 0,4
Бензол 80,1 2,54 0,7
Бромбензол 156,1 6,12 1,6
Сероуглерод 40,3 2,34 0,6
Четыреххлористый углерод 76,7 5,03 1,3
Хлороформ 61,2 3,64 0,9
Циклогексан 80,4 2,79 0,7
Этанол 78,3 1,19 0,3
Этилацетат 77,1 2.77 0,7
Этиловый эфир 34,6 2,10 0,5
н- Гексан 68,7 2,80 0,7
Иодбензол 188,4 8,87 2,1
Метанол 64,7 0,84 0,2
Метилацетат 57,1 2,15 0,5
Метилэтилкетон 79,6 2,28 0,4
Нитробензол 210,8 5,24 1.3
«-Октан 125,6 4,25 1,0
Фенол 181,8 3,56 0,9
Толуол 110,7 3,33 0,8
Вода 100,0 0,51 0,1
6.1.2. Эбулиометр
Основной частью эбулиометра (рис. 6.2) является маленький ки-
пятильник с платиновым нагревателем. Непосредственно над на-
гревательным элементом находится раструб насоса Коттрела, в
котором пузырьки пара продвигают кипящий раствор к выходу
из насоса и над гнездом термопары. На выходе из гнезда термо-
пары находится измерительный спай. Эталонный спай окружен
двойной паровой рубашкой. После конденсации пары возвращают-
ся в кипятильник через каплеотбойник. Эбулиометр помещают в
сосуд Дьюара.
Лимитирующим фактором в применении эбулиометрии для
определения молекулярных весов полимеров является чувстви-
тельность измерения температуры. К наиболее распространенным
датчикам относятся термисторы и термоэлементы (ЫО-5—
2-10~6 °C).
Обзорная литература: 517, 518, 1133.
Периодическая литература: 2143, 2432—2435, 3051, 3163, 3338, 3432, 3673,
3674, 3676, 3907, 4057, 4585, 4871, 5983, 6083, 6253, 6443, 7317.
Одной из новейших разработок является ротационный эбулио-
метр. Прибор включает вращающиеся сосуды, служащие для
Методы, связанные с оценкой коллигативных свойств
101
установления полного равновесия между жидкостью и паром
(рис. 6.3). Температуру кипения можно задавать уменьшением
Рис. 6.2. Эбулиометр.
У—платиновый нагреватель; 2 — кипятиль-
ник; 3 — насос Коттрела; 4 — эталонный
спай термопары; 5—холодильник; 6~капле-
отбойник; 7—измерительный спай термо-
пары.
1
4
Рис. 6.3. Ротационный эбулиометр.
/ — регулятор давления; 2—мотор; 3 —эта-
лонный термистор; 4— нагреватель; 5—воз-
душный вентилятор; 6— измерительный
терМИстор.
давления с помощью регулятора давления в пределах 100—
760 мм рт. ст. Повышение температуры кипения регистрируется
двумя термисторами, включенными в мостик Уитстона;
Периодическая литература; 6483.
6.2. КРИОСКОПИЯ
Криоскопия базируется на различии между температурами замер-
зания раствора и чистого растворителя.
Используя криоскопические данные, можно рассчитать Мп по-
лидисперсного линейного полимера с помощью следующих
102
Глава 6
уравнений:
— = -S-4-Bc + Cc24-.... (6.6)
с мп
^L = Kc(4- + A2c + A3c2+...\ (6.7)
с \Мп )
^ = ^-(1 + Г2с + ГзС2+ ...), (6.8)
с Мп
размерность которых кельвин-метр на грамм (СГС) или кель-
вин-кубический метр на килограмм (СИ), где АТ — понижение
температуры плавления (К); с — концентрация полимера; &Т/с —
приведенное понижение температуры плавления; Кс — криоскопи-
ческая константа (величина, характеризующая понижение темпе-
ратуры плавления, вызываемое 1 молем растворенного вещества^,
определяемая как
Кс~ — R.TsMslps&Hs, (6.9)
где К— универсальная газовая постоянная (разд. 40.2); Ts — тем-
пература замерзания растворителя (К); Ms — молекулярный вес
Таблица 6.3
Криоскопические константы для различных растворителей
(в единицах СГС)
Растворитель Точка плавления, °C 10-3.хс, °C-моль 1
Уксусная кислота 16,6 3,9
Бензол 5,5 5,1
Камфора 178,0 40,0
Диметилсульфоксид 18,4 4,8
Дифенил 70,0 8,0
Дифениламин 53,0 86,0
Дибромэтилен 10,0 П,8
Нафталин 80,2 6,8
Нитробензол 5,7 7,0
Фенол 41,0 7,4
Сукцинонитрил 58,8 20,3
Трифенилметан 92,5 12,4
Уретан 50,0 5,1
Вода 0,0 .1,8
растворителя; ps— плотность растворителя; \HS — теплота плавле-
ния растворителя; В, Аг, Г2 и С, Л3, Гз — вириальные коэффициен-
ты. Размерность Кс зависит от того, в каких единицах выражены
все другие параметры: /?, 7\, Ms, ps и AHS.
В табл. 6.3 приведены криоскопические константы для ряда рас-
творителей.
Мп можно рассчитать из кривой, представленной на рис. 6.4.
Методы, связанные с оценкой коллигативных свойств
103
Криоскопия применима для определения молекулярных весов
вплоть до 50 000.
Рис. 6.4. Зависимость приведенного
понижения температуры плавления
(ДГ/с) от концентрации.
Обзорная литература: 125, 515, 518, 1133.
Периодическая литература: 2155, 4787.
6.2.1. Определение с помощью криоскопических
измерений второго вириального коэффициента
В криоскопии второй вириальный коэффициент (Л2) выражается
как
л2=тй-(4-х)- (6-10)
где 7? — универсальная газовая постоянная (разд. 40.2); Т — тер-
модинамическая температура (К); р?— плотность полимера;
&HS — теплота плавления растворителя; %— параметр взаимодей-
ствия (разд. 2.9).
А2 можно найти по тангенсу угла наклона (tga=X2) кривой
зависимости \Т/с от концентрации (рис. 6.4).
6.2.2. Криоскопические ячейки
Основной частью криоскопической ячейки (рис. 6.5) является труб-
ка из стекла пирекс с воздушной рубашкой. Нижнюю часть крио-
скопической ячейки помещают в термостат, поддерживаемый при
температуре на 1,5 К ниже температуры плавления растворителя.
Жидкость в криоскопической ячейке перемешивают с помощью
магнитной мешалки. В качестве датчика используют термистор
(1-10“4°С). Образец вводят в ампуле. Приблизительно 5 г очи-
щенного растворителя помещают во взвешенную криоскопическую
ячейку. Ячейку затем устанавливают в термостате и находят кри-
вую замерзания чистого растворителя (рис. 6.6). После этого
ячейку нагревают, вводят известное количество образца и записы-
вают вторую кривую замерзания.
104
Глава 6
Рис. 6.5. Криоскопическая ячейка.
/ — воздушная рубашка; 2—магнитная ме.
шалка; 3—термистор.
Обзорная литература: 515.
Периодическая литература: 2155, 2378, 3432, 3668, 3669, 4485, 5466, 6389,
6569, 6884, 6958, 7138.
6.3. ИЗОТЕРМИЧЕСКАЯ ПЕРЕГОНКА
(ИЗОПИЕСТИЧЕСКИЙ МЕТОД)
Изопиестический метод основан на изотермическом переносе рас-
творителя, обусловленном градиентом концентраций. Дистилляция
будет протекать из одного раствора в другой до выравнивания дав-
ления паров (изопиестические условия).
Мп можно рассчитать по формуле
J^==J-lim-^, (6.11)
мп Ms с->0 Ср
где Ms — молекулярный вес стандартного вещества; cs — концен-
трация стандартного раствора; ср — концентрация раствора поли-
мера.
Изопиестическим методом можно измерять молекулярные веса
вплоть до 20 000 с точностью ±5%.
Обзорная литература: 125, 1133, 1446
6.3.1. Прибор для изопиестической перегонки
Для изопиестической перегонки применяется прибор в виде Н-труб-
ки («салазки»), показанный на рис. 6.7. В двух составляющих ее
частях — трубке А и трубке В находятся стандартный раствор
известной весовой и молярной концентрации и раствор полимера
известной весовой и неизвестной молярной концентрации (по 5 мл
в каждой трубке), которые запаиваются под вакуумом. По дости-
жении равновесия такие «салазки» помещают в термостат
Методы, связанные с оценкой коллигативных свойств
105
(±0,001 °C) и постоянно покачивают несколько раз в минуту так,
чтобы растворы переливались из одного конца трубки в другой, на
протяжении нескольких дней или даже недель. Большая продол-
жительность требуется для более разбавленных растворов. В ка-
честве растворителей обычно используют ацетон при 25 °C и бен-
зол при 45 °C. При применении жидкостей с высоким давлением
паров равновесие достигается
быстрее. В качестве эталонного
вещества часто используются
азобензол (мол. вес 182,2) и ме-
тилстеарат (мол. вес 298,5).
Стандартные вещества должны
иметь высокую степень чистоты
и низкую летучесть. Для удале-
ния следов влаги добавляют не-
большие количества сульфата
кальция. Концентрации изопие-
Рис. 6.7. Прибор («салазки») для оп-
ределения молекулярного веса изо-
пиестическим методом.
стических растворов (по дости-
жении равновесия) обычно опре-
деляют путем измерения конеч-
ных объемов жидкостей в двух
частях Н-трубки. Предельное от-
1 — осушители; 2—эталонный раствор;
3 — раствор полимера.
ношение концентрации при бесконечном разбавле-
нии (с = 0) можно найти экстраполяцией равновесных данных,
полученных по крайней мере для четырех различных Н-трубок,
заполненных растворами различной концентрации.
Обзорная литература: 125, 1133, 1446.
Периодическая литература: 2862, 3609, 6129, 6396.
6.4. ПАРОФАЗНАЯ ОСМОМЕТРИЯ
Парофазная осмометрия основана на законе Рауля, согласно ко-
торому в идеальном растворе парциальное давление пара каждого
компонента пропорционально его мольной доле. Понижение дав-
ления пара (Др) можно выразить как
Ар = ₽1 — Pz = Pin2 = Pi~, (6.12)
где Др—величина понижения давления пара; pi—давление пара
чистого растворителя; р2— давление пара раствора; п2 —мольная
доля раствора; Vi =Mi/pi — молярный объем растворителя; Mi —
молекулярный вес растворителя; pi — плотность растворителя;
М2 — молекулярный вес растворенного вещества.
Предельное понижение давления паров (Др/с)с=0 составляет
(6.13)
106
Глава 6
Если растворенным веществом является полимерный образец, то
М2 можно заменить на среднечисловой молекулярный вес (Мп):
(^) =P1V,^- (6.14)
\ с Jc=O мп
Парофазная осмометрия применима для определения молеку-
лярных весов растворимых полимеров вплоть до 20 000.
Обзорная литература: 125, 309, 866.
Периодическая литература: 2382, 3181, 4393, 4394, 4397, 6983.
6.4.1. Парофазные осмометры
Парофазные осмометры состоят из двух частей: термоизолирован-
ной измерительной камеры (рис. 6.8) и электронного записываю-
щего устройства. Два термисторных шарика, образующие два пле-
Рис. 6.8. Парофазный осмометр.
/ — изолированные блоки; 2—термистор;
3—гнездо для шприца; 4 — шприц; 5—чашка
с растворителем.
Рис. 6.9. Зависимость 1g Мп от
lg(W)c=0.
ча моста сопротивлений, помещают в термостат, насыщенный па-
рами растворителя. С помощью направленных шприцев каплю
растворителя наносят на один термистор (стандартный шарик),
а каплю раствора — на другой (измерительный шарик). Вслед-
ствие более низкого давления паров раствора на измерительном
шарике конденсируется растворитель, повышая температуру ша-
рика, что обусловливает разность температур между шариками и
разбаланс моста.
В состоянии равновесия разность температур (&T)S составляет
(AT)s = Ks (c/Mn + А2с2 + А3с3 +...), (6.15)
где Ks — калибровочная константа в состоянии равновесия; с —
концентрация раствора; Мп—среднечисловой молекулярный вес
Методы, связанные с оценкой коллигативных свойств
107
полимера; А2 и А3 — соответственно второй и третий вириальный
коэффициенты полимера. На практике прибор калибруют для дан-
ного растворителя, температуры, термистора с помощью вещества
известного молекулярного веса (например, нафталина) при не-
скольких концентрациях. Затем проводят расчет по формуле
<6J6)
где k — константа прибора для данных растворителя, температуры
и термисторов; AV — разбаланс моста для концентрации с.
Кривая зависимости 1g от lg(AV/e)c=0 представляет собой
прямую линию (рис. 6.9).
На результаты парофазной осмометрии оказывают также влия-
ние такие факторы, как размер капли, летучесть растворенного
вещества и процессы, лимитируемые диффузией.
Обзорная литература: 127, 518, 925, 1333.
Периодическая литература: 2022, 2382, 2383, 2614, 2704, 3181, 4394, 5216,
5364, 5381, 5474, 5693, 6409, 6410, 6759, 6803, 6911, 6983, 7029.
Глава 7
АНАЛИЗ КОНЦЕВЫХ ГРУПП
Обзорная литература: 23, 125, 127, 357, 362, 456, 681, 1082.
Анализ концевых групп применим главным образом к линей-
ным полимерам и позволяет оценить их среднечисловой молеку-
Анализ концевых групп
Таблица 7.1
Полимер Тип концевых групп Литература
Алифатические поли- амиды Аминные Карбоксильные О: 495, 1065; П: 2588, 2915, 3594, 5352, 5672, 6366, 6999, 7103, 7307 О: 495, 1065, П: 2284, 3166, 3594, 5147, 5721, 6244, 6999, 7153
Ароматические поли- амиды Аминные Карбоксильные О: 495; П: 3175, 4721, 5897 О: 495; П: 3175, 4721, 5897
Сложные полиэфиры Гидроксильные Карбоксильные О: 1212; П: 2291, 2664, 2767, 2770, 2944, 3770, 4538, 4641, 4674, 5036, 5376, 5709, 5854 О: 357, 495; П: 3374, 4537, 4674, 5036, 5148, 5854, 5979, 6926, 7004, 7006
Полинитрилы Нитрильные П: 4411
Полиоксйалкиленгли- коли и полиуретаны Гидроксильные Цианатные О: 357; П: 2439, 2620, 2662, 2664, 3026, 3885, 4011, 5049, 5658, 5997, 6572, 7096 О: 357; П: 3058, 3768, 4756, 5395
Винильные полимеры Ненасыщенные Фрагменты инициа- тора или агентов пе- редачи цеп и т. п. П: 2833, 7322 О: 59; П: 2085, 2137, 2233, 2336, 2386, 2683, 2827, 2963, 3394, 3406, 3631, 3632, 4405, 4406, 4536, 4569, 5191, 5379; 5461, 5666-5671, 5765, 6098, 6804, 6916, 7211
лярный вес (АЦ). При этом надо учитывать, что а) суммарное ко-
личество концевых групп вдвое больше числа полимерных моле-
кул; б) если полимер содержит одну концевую функциональную
Анализ концевых групп
109
группу, то число концевых групп этого типа совпадает с числом
молекул полимера.
Среднечисловой молекулярный вес можно найти по формуле
Ma = n-106/m, (7.1)
где п — 1—2 — число групп в макромолекуле, которые могут быть
определены; т — концентрация концевых групп в микроэквивален-
тах на грамм (СГС) или эквивалентах на килограмм (СИ).
Реакционная способность функциональных групп не зависит от
молекулярного веса полимера, если молекулярный вес превышает
несколько сотен грамм на моль. Практически верхний предел мо-
лекулярных весов, определяемых с помощью анализа концевых
групп, составляет 50 000.
Для обычного контрольного анализа должна быть известна
природа концевых групп (табл. 7.1).
Наиболее распространенным способом анализа концевых групп
является неводное потенциометрическое титрование, в меньшей
степени применяются ИК-, ЯМР- и масс-спектроскопия. Концевые
группы можно также определить с помощью специфических реак-
ций с радиоактивными реагентами, хромофорными веществами
(красителями) или реагентами, содержащими такие элементы, ко-
торые отсутствуют в анализируемом полимере.
В отдельных случаях анализ концевых групп позволяет оце-
нить Мп разветвленных полимеров, получаемых из полифункцио-
нальных мономеров.
В этом случае Мп можно рассчитать по формуле
(7.2)
где х — суммарная концентрация концевых групп в микроэквива-
лентах на грамм (СГС) или эквивалентах на килограмм (СИ);
у — число точек ветвления в микроэквивалентах на грамм (СГС)
или эквивалентах на килограмм (СИ).
Обзорная литература 456, 495, 596, 1006, 1065.
Периодическая литература: 2019, 6346.
Глава 8
УЛЬТРАЦЕНТРИФУГИРОВАНИВ
Обзорная литература: 6, 68, 125, 140, 181, 281, 335, 399, 489, 491, 888, 1148,
1167, 1344, 1355, 1397—1400.
Метод ультрацентрифугирования (седиментационный анализ)
основан на определении состава растворенных частиц в растворе,
помещенном в быстровращающийся ротор.
Используются следующие разновидности седиментационного
анализа:
а) скоростная седиментация (разд. 8.1),
б) анализ седиментационно-диффузного равновесия (разд. 8.2),
в) анализ седиментационного «приближения к равновесию»
(разд. 8.3),
г) седиментационное равновесие в градиенте плотности
(разд. 8.4).
8.1. СКОРОСТНАЯ СЕДИМЕНТАЦИЯ
Скоростной седиментацией называют метод, в котором оценивается
скорость перемещения частицы в растворе полимера или коллоид-
ной суспензии под влиянием сильных центробежных сил.
В процессе ультрацентрифугирования полимерного раствора
все растворенные молекулы движутся в направлении дна кюветы,
в которой находится такой раствор. Этот процесс называется се-
диментационным переносом. Спустя какой-то промежуток времени
(/) образуется слой чистого растворителя у мениска, тогда как
макромолекулы собираются в узком слое вблизи дна кюветы
(рис. 8.1).
Скорость, с которой макромолекулы передвигаются в направ-
лении дна кюветы, называется скоростью седиментации.
На скорость седиментации влияют следующие факторы:
а) центробежная сила, б) подъемная сила, в) молекулярный вес,
г) трение макромолекул.
Пограничный слой между растворителем и раствором осаждаю-
щегося полимера продвигается в направлении дна кюветы
(рис. 8.1) со скоростью, определяющейся скоростью седиментации.
Эта граница весьма размыта и расширяется со временем вслед-
ствие того, что скорость седиментации макромолекул полидис-
персного полимера различна. На вид границы влияет также об-
ратная диффузия, которая начинает оказывать влияние при воз-
никновении разности концентраций.
Ультрацентрифугирование
111
Седиментационный перенос представляют двумя графическими
изображениями:
а) концентрацию (с) на всем протяжении ячейки при седимен-
тационном переносе выражают как функцию расстояния от центра
вращения г, с — f (г) (рис. 8.1, а);
б) концентрационный градиент (dc/dr} получают путем диф-
ференцирования концентрационной кривой (рис. 8.1,6)—скорость
Рис. 8.1. Седиментационный перенос в кювете для ультрацентрифугирования в
процессе вращения, г — расстояние от центра вращения; с—концентрация.
а —изменение концентрации по высоте столбика в кювете; б —градиент концентрации, обра-
зующийся в кювете.
передвижения концентрационной кривой является мерой скорости
седиментации.
Изменение концентрации частиц в полимерном растворе {dc/dt}
при равновесии между седиментацией и диффузией характери-
зуется duффepeнцuaлbным уравнением ультрацентрифугирования
Ламма\
(%?} ==---^-(s«>2r2c-Dr ^-Y (8.1)
где r — расстояние от центра вращения до рассматриваемой точки
(объема) полимерного раствора [см (СГС) или м (СИ)]; с — кон-
центрация раствора до центрифугирования [г/см3 (СГС) или
кг/м3 (СИ)]; со — угловая скорость (скорость ротора) [град/с
(СГС) или радиан/с (СИ)]; S— коэффициент седиментации (с);
Р — коэффициент диффузии [см2/с (СГС) или м2/с (СИ)].
112
Глава 8
Коэффициент седиментации (S) —это скорость седиментации
(vs) на единицу поля (со2г), он называется также центробежным
ускорением
S = vs/(d2r = (1 — up) M/NAf (8.2)
Скорость седиментации выражается как
vs = (д2г (1 — др) MINAf, (8.3)
где v — удельный объем растворенного вещества [см3/г (СГС)
или м3/кг (СИ)]; р — плотность раствора [г/см3 (СГС) или
кг/м3 (СИ)]; (1—vp)— фактор всплытия — для разбавленных
растворов, равный
= (8.4)
М — молекулярный вес полимера; NA— число Авогадро
(разд. 40.3); f — коэффициент трения.
Коэффициент седиментации измеряется в сведбергах (S) i
1S = 1 . Ю“13с
Коэффициент диффузии (D) определяется по формуле
P = ^(1+4W + MAC2+...), (8.5)
где R — универсальная газовая постоянная (разд. 40.2); Т — тер-
модинамическая температура (К); А2 и А3 — вириальные коэффи-
циенты.
Коэффициент трения (f) пропорционален среднему линейному
размеру макромолекул и вязкости растворителя
/ = Р(Г-2)1«П> (8.6)
где Р == 6л/д, fA — фактор асимметрии — отношение между радиу-
сом инерции и радиусом, наиболее часто использующимся при
описании молекулы; г2— среднеквадратичное расстояние между
концами макромолекул; т] — коэффициент вязкости растворителя.
Обстоятельные данные по коэффициентам седиментации, коэф-
фициентам диффузии и коэффициентам трения приведены в Poly-
mer Handbook [О: 410].
Скорость седиментации сильно зависит от а) концентрации
раствора, поскольку вязкость влияет на коэффициент трения мак-
ромолекул и через последний — на коэффициент седиментации;
б) гидростатического давления, которое при вращении достигает
нескольких сотен атмосфер и влияет на парциальный удельный
объем растворенного вещества, плотность раствора, коэффициент
трения макромолекул.
При измерении скорости седиментации получают информацию
о форме полимерных частиц, молекулярном весе полимера1 назц-
Ультрацентрифугирование
113
ваемом седиментационным среднедиффузным молекулярным ве-
сом; молекулярновесовом распределении (МБР), степени ветвле-
ния разветвленных полимеров (вместе с другими методами); па-
раметрах равновесия при реакциях между полимерами.
Обзорная литература: 1167, 1278.
Периодическая литература: 2138, 2210, 2309, 2399, 2436, 2876, 3541, 3542,
3691, 3726, 3843, 4103, 4315, 4571, 4584, 4658, 4685, 4811, 5076, 5513, 5515, 5537,
5727, 6094, 6632, 6641, 6991, 6992, 7036, 7107—7110.
8.1.1. Определение формы частиц методом
скоростной седиментации
Комбинируя уравнение (8.6) с уравнением Флори — Фокса (2.36)
(разд. 2.13), получим выражение для фактора формы (0):
р = ф1/3р-1 = п[п]1/3Л11/7/. (8.7)
где Ф — функция, связанная с гидродинамическим поведением
макромолекул; Р = 6nfA; fA — фактор асимметрии — отношение
между радиусом инерции и радиусом, наиболее часто использую-
щимся при описании молекулы; г] — коэффициент вязкости рас-
творителя; [г|] —характеристическая вязкость; М — молекулярный
вес полимера; f — коэффициент трения. Для гибких макромолекул
0= 2,5-106. Для других частиц (например, сфер, тонких дисков,
тонких стержней, цилиндров, статистических клубков, полидисперс-
ных статистических клубков и жесткосвернутых клубков) величи-
на f отличается от только что упомянутой.
Периодическая литература: 5057, 5060.
8.1.2. Определение молекулярных весов полимеров
методом скоростной седиментации
Определяемый с помощью скоростной седиментации молекулярный
вес полимера (М), как правило, не равен средневесовому молеку-
лярному весу (AU). Его называют седиментационным среднедиф-
фузным молекулярным весом.
Молекулярный вес полимера (Af) можно найти из уравнений
(8.2) и (8.5)
5 М+(1~рр) , .
D ^Т(1 + Л4Д2с +...) ’ V * 7
Эффективный молекулярный вес полимера (Марр)] определяется с
помощью уравнения Сведберга
^аРР — __ у) 7Г» (8.9)
где S — коэффициент седиментации; D — коэффициент диффузии;
V — удельный парциальный объем растворенного вещества; р —
114
Глава 8
Концентрация (с)
Рис. 8.2. Зависимость \/Марр от
концентрации (с).
плотность раствора; /? — универсальная газовая постоянная
(разд. 40.2); Т — термодинамическая температура (К); Л2— вто-
рой вириальный коэффициент; с —
концентрация раствора.
Для того чтобы была возможна
экстраполяция на бесконечное разбав-
ление, необходимы измерения Марр
при нескольких концентрациях, напри-
мер 2,0; 4,0; 6,0 и 8,0 г/л. После этого
молекулярный вес полимера можно
найти из кривой, представленной на
рис. 8.2, используя выражение
^ = 4- + Л2С+... (8.10)
Отрезок, отсекаемый на оси ординат,
равен 1/М, а наклон прямой дает ’зна-
чение второго вириального коэффи-
циента tg а = А2.
Другой способ заключается в экстраполяции S и D до нулевой
концентрации, в этом случае молекулярный вес полимера вычис-
ляют непосредственно из выражения
где Sc=0 — коэффициент седиментации, полученный экстраполя-
цией до нулевой концентрации; £)с=:0 — коэффициент диффузий,
полученный экстраполяцией до нулевой концентрации.
8.1.3. Определение молекулярновесового
распределения (МВР)
методом скоростной седиментации
При ультрацентрифугировании макромолекулы высокого молеку-
лярного веса движутся в направлении дна кюветы с удельной ско-
ростью, более высокой, чем макромолекулы низкого молекулярного
веса.
Распределение скоростей седиментации зависит от концентра-
ционного градиента, который устанавливается в граничной обла-
сти, и его изменения во времени.
Периодическая литература: 6864, 6866.
8.1.4. Определение коэффициента седиментации
Коэффициент седиментации (S) находится из выражения
(8.12)
Ультрацентрифугирование
115
в сведбергах (S), где vs — скорость седиментации; о2г — центро-
бежное ускорение; со — угловая скорость (скорость ротора); г —
расстояние от центра вращения до рассматриваемой точки (объе-
ма) раствора полимера.
Разбавленный раствор полимера (например, 2,0 г/л) в квар-
цевой кювете помещают в ротор центрифуги. При вращении цен-
трифуги макромолекулы направляются с постоянной скоростью на
дно кюветы под влиянием цен-
тробежной силы (1—2 мм/ч).
Граница между раствором и
растворителем (более или ме-
нее резкая) также движется в
направлении дна кюветы. Ско-
рость продвижения этой гра-
ницы называется скоростью
седиментации (уз).
Скорость седиментации
можно определить а) методом
поглощения света, б) с по-
мощью оптической системы
с интерферометром Рэлея,
в) оптическим шлирен-мето-
Рис. 8.3. Зависимость 1/S от концентра-
ции (с).
ДОМ.
Скорость, с которой расстояние (г) между осью ротора и мак-
симумом концентрационного градиента возрастает во времени (/),
также является скоростью седиментации (t>s).
В этом случае коэффициент седиментации (S) определяется по
видоизмененному уравнению (8.12):
vs (Г2~ГЛ________
2r \t2 — ti J (О2 (Г1 + г2)
где Г1 и г2— расстояния границы между раствором и растворите-
лем (максимум концентрационного градиента) от центра враще-
ния при времени t\ и /2 соответственно.
Скорость седиментации следует измерять при нескольких кон-
центрациях (например, 2,0; 4,0; 6,0; 8,0 г/л). Тогда коэффициент
седиментации можно найти из кривой, представленной на
рис. 8.3, используя выражение
Y = yL- + ^ (8-13)
Отрезок, отсекаемый на оси ординат этой прямой, составляет
1/SC=O, a tg а == ks. Для растворов многих полимеров в хороших
растворителях ks & 1,6 [ц], где [ц] —характеристическая вязкость
(или предельное число вязкости).
116
Глава 8
Рис. 8.4. Схема кюветы для определения коэффициента диффузии.
Рис. 8.5. Диаграмма диффузии.
Рис. 8.6. Изменение градиента концентрации (dc[dx) во времени (/).
Ультрацентрифугирование
117
Константа ks не идентична со вторым вириальным коэффи-
циентом (А2), при этом А2 = ks + ko. (О константе ko можно про-
читать в разд. 8.1.5.)
8.1.5. Определение коэффициента диффузии
Коэффициент диффузии (D) определяется как
П=^-(1+Л1Л2с + ЛМ3с2+...) (8.14)
в сантиметрах квадратных в секунду (СГС) или метрах квадрат-
ных в секунду (СИ), где R — универсальная газовая постоянная
(разд. 40.2); Т — термодинамическая температура (К); Na— число
Рис. 8.7. Кривая для определения ко-
эффициента диффузии (Da).
А — площадь под диффузионной кривой;
Я—максимальная высота кривой (см.
рис. 8.5).
Рис. 8.8. Зависимость 1/D от концен'
трации (с).
Авогадро (разд. 40.3); f — коэффициент трения; М — молекуляр-
ный вес полимера; А2 и А3 — вириальные коэффициенты; с — кон-
центрация раствора.
Коэффициент диффузии можно определить путем измерения
концентрационного градиента при различных временах (/), т. е. пу-
тем измерения скорости передвижения в начале резкой границы
между растворителем и раствором. На рис. 8.4 приведена схема
кюветы для получения экспериментальных данных. В нижнюю
часть диффузной кюветы помещают полимерный раствор и отде-
ляют его перегородкой от верхней части кюветы, где находится
чистый растворитель. После удаления перегородки макромолекулы
начинают диффундировать из нижнего слоя раствора в чистый
растворитель, вследствие этого концентрация выше перегородки
возрастает в той же степени, в какой ниже перегородки умень-
шается. Градиент концентрации максимален вблизи границы; по-
нижение его при увеличении расстояния вверх и вниз от границы
118
Глава 8
Закрепленный капилляр
Рис. 8.9. Образование границы при сифонировании через закрепленный капил-
ляр. Диффузная кювета открыта сверху и снизу для симметричного течения рас-
творителя и раствора [П: 2875].
Подвижный капилляр
Рис. 8.10. Образование границы при сифонировании через капилляр, движущий-
ся сверху вниз по кювете вместе с границей. Кювета закрыта снизу. Симметрич-
ное течение у границы достигается, когда скорость перекачивания вдвое превы-
шает скорость движения капилляра (<?) [П: 2875].
Рис. 8.11. Измерительное устройство для определения градиентов концентрации
в диффузной кювете.
/ — гелиево-неоновый лазер; 2—диффузная кювета; <3—термостат; 4 — интерферометр; 5 — про-
тивоударный массивный блок.
Ультрацентрифугирование
119
имеет вид гауссовой кривой (рис. 8.5). С увеличением продолжи-
тельности измерения кривые градиента концентрации становятся
все более и более пологими (рис. 8.6). В большинстве случаев
кривые диффузии более или менее сильно отличаются от гауссовых
кривых, поэтому для определения коэффициента диффузии (D)
необходимо пользоваться площадями под кривыми диффузии. Зна-
чения констант диффузии, определяемые таким способом, полу-
чили название коэффициентов диффузии высота — площадь (Da).
Среднее значение DA можно найти из кривой на рис. 8.7 как tg а =
= Da.
Изменение градиента концентрации (dc/dx) следует измерять
при нескольких концентрациях (например, 2,0; 4,0; 6,0 и 8,0 г/л).
Тогда коэффициенты диффузии (£)с==о) можно рассчитать из кри-
вой, представленной на рис. 8.8, по формуле
Отрезок, отсекаемый на оси ординат, равен 1/Z>C==O> тогда как
tg а — kD. Константа kD не идентична второму вириальному коэф-
фициенту (А2), так как Д2 = ko + ks. (О константе ks см. в
разд. 8.1.4.)
Для достижения резкой границы между раствором и раство-
рителем предложено несколько методов: а) разрезание или ото-
двигание перегородки, разделяющей раствор и растворитель
(см. рис. 8.4); б) сифонирование через закрепленный капилляр
(рис. 8.9); в) сифонирование через движущийся капилляр
(рис. 8.10); г) сифонирование через щель в стенке кюветы.
Для измерения градиента концентрации обычно пользуются
интерферометром Рэлея с гелиево-неоновым лазером в качестве
источника света (рис. 8.11).
Обзорная литература: 529, 1276.
Периодическая литература: 2613, 2861, 2867, 2875, 2878, 2988, 3018, 4340,
4367, 5868, 6629, 6646.
8.2. АНАЛИЗ СЕДИМЕНТАЦИОННО-ДИФФУЗНОГО
РАВНОВЕСИЯ
Анализ седиментационно-диффузного равновесия позволяет опре-
делить изменение концентрации частиц в полимерном растворе или
коллоидной суспензии при достижении равновесия, устанавливае-
мого между седиментацией и диффузией под влиянием слабого
центробежного поля.
При изучении седиментационного равновесия получают инфор-
мацию о а) средневесовом молекулярном весе (7Ида) и Z-среднем
молекулярном весе (Mz) (в том случае, когда инкременты плотно-
сти и показателя преломления равны для всех полимерньц
120
Глава 8
компонентов); б) молекулярновесовом распределении полимера;
в) структуре разветвленных полимеров.
Обзорная литература; 1167.
Периодическая литература: 2842, 3543, 3544, 3546, 5390, 5623, 6055, 6093,
(6096, 6097, 6246, 6990, 7076.
«8.2.1. Определение средневесового молекулярного веса
методом седиментационно-диффузного равновесия
При достижении седиментационно-диффузного равновесия концен-
трация в каждой точке столбика раствора во времени инвариантна,
т. е. (dc/ti/)r = O, следовательно, суммарный перенос в каждой
точке столбика раствора равен нулю и уравнение Ламма (8.1)
принимает вид
4 = -sV(»- (8.16)
При подстановке S/D в уравнение Сведберга (8.8) получается сле-
дующее выражение для весового молекулярного веса (Mw):
(8Л7>
где R — универсальная газовая постоянная (разд. 40.2); Т — тер-
модинамическая температура (К); (о— угловая скорость (скорость
ротора); 5 —удельный парциальный объем растворенного веще-
ства; р — плотность раствора; с — концентрация раствора до цен-
трифугирования; г — расстояние между центром вращения и рас-
сматриваемой точкой полимерного раствора.
Интегрирование уравнения (8.16) дает
2RT / с2 — Ci х
(8.18)
где индекс 1 относится к мениску, а индекс 2 — к донной части
кюветы.
Концентрацию (с) в кювете для ультрацентрифугирования
можно найти следующими способами (см. также разд. 8.5):
1. Измерением поглощения в ряде точек столбика раствора
в кювете.
2. По интерферометрическим данным, которые позволяют полу-
чить абсолютные концентрации по всей высоте раствора в кювете.
3. Измерением с использованием шлирен-метода. В этом слу-
чае концентрация определяется интегрированием de/dr. [Приме-
чание. Измерение градиента концентрации (dc/dr) предполагает
знание удельного инкремента показателя преломления dn/dc.]
(.8.19)
dr dr / da 4 *
Ультрацентрифугирование
121
Уравнение (8.18) можно переписать в виде
дд __ 2/?Т d (In с) /q пл\
-2(1 —Spj- dp)~' (8-20)
при этом становится возможным определение величины d (In с) )d (г2)
из кривой зависимости Inc от г2 (рис. 8.12). Прямая линия на,
рис. 8.12 получается лишь для монодисперсных и однородных по-
лимерных образцов. Образец полидисперсного или неоднородного!
полимера дает кривую линию.
При изучении седиментации у полидисперсного полимера об-
наруживаются различия не только в концентрации, но и в распре-
Рис. 8.12. Зависимость Inc от г2 при
данной скорости ротора.
Рис. 8.13. Изменение по высоте
столбика раствора в кювете.
делении полимерного компонента (большие макромолекулы седи-
ментируют с большей скоростью, чем меньшие макромолекулы).
Молекулы самого высокого молекулярного веса располагаются в.
точке наивысшей концентрации. Весовой молекулярный вес (Mw)
зависит от того, какая точка в столбике раствора анализируется;
(рис. 8.13). Для определения средневесового молекулярного веса
(Л4да) полидисперсных полимеров необходимо проводить соответ*
ствующее интегрирование.
Продолжительность установления равновесия в 2—3-миллимет-
ровом слое полимерного раствора колеблется от нескольких часов
до многих дней.
8. 2.2. Определение Z-среднего молекулярного веса
методом седиментационно-диффузного равновесия
Z-Средний молекулярный вес (Д?) можно рассчитать по формуле
Ьт(£).-i(£)/(».-4 <8-2')
122
Глава 8
где R — универсальная газовая постоянная (разд. 40.2); Т — тер-
модинамическая температура (К); со — угловая скорость (скорость
ротора); v — удельный парциальный объем растворенного вещест-
ва (из литературных данных); р — плотность раствора; с — концен-
трация раствора при равновесии; г — расстояние от центра вра-
щения до анализируемой точки полимерного раствора; индекс 1
относится к мениску, индекс 2 — к донной части кюветы.
8. 2.3. Определение молекулярновесового распределения
методом седиментационно-диффузного равновесия
Молекулярновесовое распределение (МБР) определяется из изме-
нения концентрации по столбику раствора. При малой высоте
столбика практически невозможно определить все молекулярно-
весовое распределение. Лучшие результаты даст проведение ана-
лиза при различных скоростях ротора.
Обзорная литература: 61.
Периодическая литература: 3210, 5912, 6630, 7074, 7075.'
Данные, получаемые при ультрацентрифугировании, можно об-
считать с помощью ЭВМ, и при наличии результатов для ряда
равновесий можно вычислить молекулярные веса (Mw, Mz, Mz+i) и
молекулярновесовое распределение.
Периодическая литература: 4685, 6247, 6248, 6999.
8.3. АНАЛИЗ ПРИ ПРИБЛИЖЕНИИ
К СЕДИМЕНТАЦИОННОМУ РАВНОВЕСИЮ
Анализ при приближении к седиментационному равновесию (метод
Арчибальда) измеряет изменение локальной концентрации в поли-
мерном растворе или коллоидной суспензии в течение квазиравно-
весия (неустановившегося равновесия) между седиментацией и
диффузией под влиянием слабого центробежного поля.
При измерении седиментационного приближения к равновесию
получают данные о средневесовом молекулярном весе (Mw).
Обзорная литература: 1167.
Периодическая литература: 2139, 6640, 6827, 7293.
8.3.1. Определение средневесового молекулярного веса
методом Арчибальда
В седиментационно-диффузном квазиравновесии (приближении к
равновесию) концентрации на дне и в мениске инвариантны во
времени, de/dt = 0.
Ультрацентрифугирование
123
Рис. 8.14. Зависимость ]/(Mw)app от
концентрации (с).
Эффективный молекулярный вес (Mw)app определяется как
(^w)app = Ю2 _ рр) — , (8.22)
где —универсальная газовая постоянная (разд. 40.2); Т — тем-
пература (К); со — угловая скорость (скорость ротора); v— удель-
ный парциальный объем растворенного вещества; р — плотность
раствора; с — концентрация раствора до центрифугирования; г —
расстояние от центра вращения
до анализируемой точки поли-
мерного раствора.
Концентрацию в точках мени-
ска или дна кюветы (измеряется
шлирен-методом) (см. разд. 8.5)
находят интегрированием de/dr.
с = соехр(—2Sco2/), (8.23)
где S — коэффициент седимента-
ции; t — продолжительность цен-
трифугирования. Для экстрапо-
ляции на бесконечное разбавле-
ние проводят измерения (Mw)app
при нескольких концентрациях
(например, 2,0; 4,0; 6,0 и 8,0 г/л).
Тогда средневесовой молекулярный вес (Mw) можно рассчитать
из кривой, представленной на рис. 8.14, используя выражение
—Ъ- = 4-+Д2с+... (8.24)
{^w)app
Отрезок, отсекаемый прямой на оси ординат, составляет \/Mw,
тогда как наклон этой прямой дает tga=^A2 (второй вириальный
коэффициент).
Для полидисперсных полимеров метод Арчибальда является
менее точным, чем анализ седиментационно-диффузного равно-
весия.
8.4. СЕДИМЕНТАЦИОННОЕ РАВНОВЕСИЕ
В ГРАДИЕНТЕ ПЛОТНОСТИ
Седиментационное равновесие в градиенте плотности позволяет
установить изменение градиента плотности в растворе полимера,
который достигается в столбике жидкости, представляющей собой
смесь легкого и тяжелого растворителей, при равновесии между
седиментацией и диффузией под влиянием слабого центробежного
поля.
‘Измерения седиментационного равновесия в градиенте плотно-
сти дают информацию о средневесовом молекулярном весе
и химической неоднородности макромолекул.
124
Глава 8
Обзорная литература: 613, 1167.
Периодическая литература: 3098, 3313—3315, 3967, 4027, 4028, 4197, 4884,
4992, 5241, 5913, 5950, 6509.
8.4.1. Определение средневесового молекулярного веса
анализом в градиенте плотности
Градиента плотности достигают использованием смеси легкого и
тяжелого растворителей, например СбН6 и СВг4, при вращении
в ультрацентрифуге с определенной скоростью. Макромолекулы
Рис. 8.15. Установление седиментационного равновесия в анализе с градиентом
платности, г — расстояние от центра вращения; с — концентрация.
а—изменение концентрации по высоте столбика в кювете; б—градиент концентрации, обра-
зующийся в кювете.
осаждаются в направлении от мениска к дну кюветы и всплывают
со дна к мениску (рис. 8.15). В состоянии равновесия макромоле-
кулы собираются в точке, в которой плотность смеси раствори-
телей точно соответствует плотности макромолекулы в растворе.
Эта точка находится на расстоянии г0 от центра вращения.
Весовой молекулярный вес (Л4Ш) находят по формуле
мш = — (rfpfdr) J [(г _ Го)2] ) • (8.25)
где R — универсальная газовая постоянная (разд. 40.2); Т — тер-
модинамическая температура (К); со— угловая скорость (скорость
Ультрацентрифугирование
125
ротора); Го—расстояние от центра вращения до точки, в которой
плотность макромолекулы в растворе точно соответствует плотно-
сти смеси растворителей; г — расстояние от центра вращения до
анализируемой точки в столбике раствора полимера; р — плот-
ность смеси растворителей в анализируемой точке столбика рас-
твора полимера; vs — удельный парциальный объем растворенного
полимера; fs— фактор избирательной сольватации; с — концентра-
ция раствора.
Интегрирование уравнения (8.25) позволяет выразить концен-
трацию (г) как функцию (г—г0):
с = соехр[—(г —г0)2/2а2], (8.26)
где Со — концентрация в центре полосы;
2 КГ 1
* == —-----------------------------
со2го (dp/dr) vsfs Mw
Уравнение (8.26) показывает, что при равновесии макромолекулы
собираются в полосу с концентрацией, характеризующейся гауссо-
вым распределением относительно расстояния г0 со стандартным
отклонением о. _
Средневесовой молекулярный вес (Alw) можно определить ин-
тегрированием Mw по всей ширине полосы. Измерения необхо-
димо проводить при нескольких концентрациях и полученные ре-
зультаты экстраполировать на нулевую концентрацию (бесконеч-
ное разбавление).
8.4.2. Определение химической неоднородности макромолекул
анализом в градиенте плотности
Макромолекулы, имеющие различную плотность (химическая не-
однородность), дают отдельные полосы при анализе в градиенте
плотности. Анализ результатов является очень сложным и тре-
бует дополнительных данных из других измерений, в частности
молекулярновесового распределения и гетерогенности по плотно-
сти как функции молекулярного веса.
8.5. АНАЛИТИЧЕСКИЕ УЛЬТРАЦЕНТРИФУГИ
Аналитические ультрацентрифуги состоят из следующих частей
(рис. 8.16):
1. Механическая система, включающая ротор, который пред-
ставляет собой эллипсоидальный дюралюминиевый или титановый
блок с 2, 4 или 6 дырками, в которые можно вставлять стандарт-
ные кюветы. Ротор приводится во вращение электромотором через
зубчатую систему и гибкий вал. Угловую скорость электромо-
тора можно варьировать от 800 до 80 000 об/мин. Скорость ро-
тора периодически сравнивается с эталонной скоростью
126
Глава 8
синхронизированного моторчика с дифференциальной зубчатой пе-
редачей. Это позволяет поддерживать скорость ротора на постоян-
ном уровне. В новейших ультрацентрифугах ротор подвешивается
в магнитном поле и не имеет прямого контакта с приводом после
достижения рабочей скорости. Для защиты от возможных повре-
ждений ротор заключается в стальную камеру. Для того чтобы
свести к минимуму фрикционный нагрев, в стальной камере
Рис. 8.16. Аналитическая ультрацентрифуга.
/ — кювета с образцом; 2—оптическое окно; 3 —ротор; 4 — привод; 5—противовес; 6 — сталь-
ная камера; 7—система оптического контроля; 3 —фотографическая пластинка; 9 — фото-
умножитель.
создают вакуум до 10-6 мм рт. ст. с помощью ротационного мас-
лодиффузного вакуумного насоса. Используя систему нагрев —
охлаждение, регулируют температуру ротора от —10 до +40оС±
±0,1 °C. Работа при высоких температурах (выше 100°С) сопря-
жена с рядом трудностей практического плана: например, линзы
оптической системы весьма подвержены помутнению вследствие
конденсации на них паров масла.
Обзорная литература: 281, 1167. 1400.
Периодическая литература: 230о, 4685, 6054.
2. Оптическая система. Изменения, происходящие в кюветах,
можно наблюдать с помощью оптической системы. Свет от источ-
ника проходит через кювету и отражается зеркальцем в направ-
лении регистрирующей системы, в которую входит фазовая пла-
стинка и качающееся зеркальце, и фиксируется прямо на фото-
пленку.
Ультрацентрифугирование
127
Для измерения концентра-
ции или градиента концентраций
как функции высоты столбика
жидкости существуют три
оптические системы наблюде-
ния:
а) Шлирен-оптическая систе-
ма. В этой системе изменение
концентрации (с) с расстоянием
от центра вращения (г) диффе-
ренцируется с помощью специ-
ального оптического приспособ-
ления, так что градиент концен-
трации (dc/dr) становится функ-
цией расстояния (г). При этом
можно наблюдать границу как
максимум градиента концентра-
ции непосредственно на фото-
пленке. Получающаяся фотогра-
фия представляет собой двумер-
ную кривую, отражающую зави-
симость инкремента показателя
преломления раствора (dn/dc)
от расстояния в столбике жидко-
сти (рис. 8.17, а). Градиент кон-
центрации (dc/dr) пропорциона-
лен инкременту показателя пре-
ломления раствора (dn/dc) (см.
разд. 8.2.1).
б) Интерференционная опти-
ческая система. В этой системе
измеряют число интерференцион-
ных полос (рис. 8.17,6). Число
таких полос пропорционально
разности показателей преломле-
ния и как следствие этого — раз-
ности концентраций. Интерферо-
метрический метод не позволяет
оценить градиент концентрации,
однако последний можно найти
дифференцированием интерферо-
метрических данных.
в) Оптическая система, осно-
ванная на поглощении света
(рис. 8.17, в). В этой системе из-
меряют поглощение ультрафио-
летового или видимого света
границы между двумя слоями разной
концентрации при использовании трех
различных оптических систем.
а — шлирен-метод; б — интерференционный
метод; в — метод поглощения света.
Навинчивающееся кольца '
Прокладка
навинчивающегося кольца
Оправка верхнего окна
Прокладка окна
Втулка окна
Окно
Прокладка сердцевины
Алюминиевая сердцевина
Прокладка сердцевины
Окно
Втулка- окна
Прокладка окна
Оправка нижнего окна
Корпус кюветы
Прокладка отверстия
ч в корпусе
Отверстие в корпусе
Рис. 8.18. Разборная кювета для уль-
трацентрифуги [О: 1400].
128
Глава 8
с помощью фоточувствительного электрического устройства, на-
пример фотоумножителя.
Обзорная литература: 529, 1167, 1276, 1400.
Периодическая литература: 2821, 3427, 4222, 4963, 4964, 5010, 6035, 6629,
6631, 6647.
3. Кюветы. Промышленностью освоены многочисленные типы
кювет. При выборе кювет необходимо иметь в виду условия экспе-
римента:
а) тип седиментационного анализа (скоростная седиментация
или исследование седиментационного равновесия);
Рис. 8.19. Различные варианты препаративного ультрацентрифугирования,
а —обычное ультрацентрифугирование; б—ультрацентрифугирование в полосе; в —ультра*
центрифугирование в градиенте плотности.
б) тип используемой оптической системы (шлирен-метод, ин-
терференционный метод или метод поглощения света);
в) температура, растворитель и концентрация.
На рис. 8.18 показано устройство одной из стандартных кювет.
.Два оптических плоских окна из кварца или сапфира помещают
на вкладыш и закрепляют внешней оправкой из алюминиевого
сплава, имеющей с одного конца резьбу для стопорного кольца.
Раствор вводят через маленькое отверстие в секторную полость
вкладыша. Стенками канала ограничивается сектор цилиндра, об-
разующегося осью вращения, поэтому молекулы растворенного ве-
щества всегда будут осаждаться (или диффундировать) в ради-
альном направлении, чтобы избежать конвекционных токов. Ме-
жду окошками и вкладышем, а также у отверстия, через которое
Ультрацентрифугирование
129
вводится раствор, с помощью прокладок создается плотное герме-
тичное уплотнение. Наиболее употребительна не односекторная, а
двухсекторная кювета с двумя рядом расположенными секторными
полостями. В ,таких кюветах легче одновременное наблюдение рас-
твора и его растворителя (что необходимо при использовании ин-
терференционной оптической системы) и исключается необходи-
мость в проведении специальных экспериментов, учитывающих пе-
рераспределение компонентов или влияние гидростатического дав-
ления. Наиболее употребительная длина оптического пути: 1,5, 3,
6, 12, 18 и 30 мм. Для достижения стабильной работы ротора
необходимо, чтобы разность весов двух заполненных кювет (или
заполненной кюветы и ее противовеса) не превышала 0,5 г.
Обзорная литература: 1167, 1400.
Периодическая литература: 7294, 7295.
8.6. ПРЕПАРАТИВНОЕ УЛЬТРАЦЕНТРИФУГИ-
РОВАНИЕ
Препаративное ультрацентрифугирование используется для разде-
ления веществ, различающихся по молекулярному весу, и особен-
но эффективно для разделения макромолекул с плотной упаковкой,
например биополимеров. Существуют три разновидности препара-
тивного ультрацентрифугирования:
1. Обычное ультрацентрифугирование. Частицы седиментируют
в чистом растворителе или достаточно разбавленных солевых рас-
творах. Плотность растворителя или солевого раствора практиче-
ски одинакова во всей кювете (рис. 8.19, а). Наиболее быстро дви-
жущиеся частицы собираются на дне, однако они всегда загряз-
нены примесями более медленно осаждающихся частиц. Молекулы,
которые движутся медленнее, количественно отделить не удается.
2. Ультрацентрифугирование в полосе (или зонное ультрацен-
трифугирование). Частицы седиментируют в смеси растворителей,
которая не имеет значительного градиента плотности. На первой
стадии градиент плотности создается ультрацентрифугированием
чистой смеси растворителей. На второй стадии к верхней части
слоя растворителя добавляют очень тонкую пленку полимерного
раствора. При дальнейшем ультрацентрифугировании более быст-
ро движущиеся молекулы в этой пленке отделяются от более
медленно движущихся частиц и образуют одну или более полос
(рис. 8.19,6). Таким способом удается выделить макромолекулы
высокой степени чистоты.
3. Ультрацентрифугирование в градиенте плотности. Частицы
седиментируют в смеси легкого и тяжелого растворителей, напри-
мер С6Н6 и СВг4. В равновесных условиях под влиянием центро-
бежной силы частицы собираются в точке, в которой плотность
смеси растворителей точно соответствует плотности макромолекул
в растворе (рис. 8.19, в) (см. разд. 8.4).
5 Зак. 238
Глава 9
ВИСКОЗИМЕТРИЧЕСКИЕ МЕТОДЫ
Обзорная литература: 23, 125, 127, 301, 309, 621, 681, 917, 922, 943, 1082,
1120, 1350, 1396.
Деформация полимерного образца происходит в том случае,
когда приложенная сила изменяет форму и размер образца. Эти
деформации могут быть двух видов.
1. Обратимые (упругие) деформации. При приложении силы
к образцу идеального упругого полимера имеет место какая-то
определенная деформация. После удаления силы упругий образец
принимает первоначальные размер и форму. Энергия, применяе-
мая для упругой деформации, возвращается полностью.
2. Необратимые деформации. При приложении силы к идеаль-
ному вязкому образцу деформация меняется со временем и обра-
зец течет. Энергия, затраченная на необратимую деформацию, не
регенерируется.
Многие полимеры проявляют упругую деформацию в течение
коротких промежутков времени и необратимую деформацию при
продолжительном приложении силы.
Напряжение сдвига (т) в обратимой деформации определяется
как
%-=FIA (9.1)
в динах на квадратный сантиметр (СГС) или ньютонах на квад-
ратный метр (СИ), где F— приложенная сила (рис. 9.1) [дин
(СГС) или ньютон (СИ)]; А — площадь поверхности [см2 (СГС)
или м2 (СИ) ].
Деформация сдвига (у) —это мера деформации, определяемая
как
y^bxlby (9.2)
Для идеального упругого материала деформация сдвига пропор-
циональна напряжению сдвига.
Закон Гука для обратимой деформации дает соотношение ме-
жду напряжением (т) и деформацией (у) сдвига:
т = = (9.3)
где О — модуль сдвига.
Текучий полимерный образец, заключенный между двумя пла-
стинами, можно рассматривать как пакет параллельных слоев
(рис. 9.2). Верхняя пластина движется со скоростью v относитель-
но нижней пластины под влиянием силы сдвига F на площадь Л.
Вискозиметрические методы
131
Отдельные параллельные слои образца движутся со скоростями,
пропорциональными расстоянию текучего слоя от нижней пласти-
ны (ламинарное течение).
Для идеальных вязких жидкостей напряжение сдвига прямо
пропорционально производной скорости по расстоянию (dv/dy).
Рис. 9.1. Обратимая деформация. Д—
площадь; F — приложенная сила.
Закон Гука для необратимой деформации дает следующую за-
висимость между напряжением (т) и деформацией (у) сдвига:
F ( dv \ /л
T=T=4dJ <9-4)
Поскольку
dv __________________ d (dx/dt) _ d (dx/dy) ,Q
dy ~ dy ~ dt '
уравнение (9.4) можно переписать в виде
т=т)у, (9.6)
где т) — вязкость, у — скорость сдвига (градиент скорости) (с-1).
Для данного напряжения сдвига (т) и данного градиента ско-
рости [d (dx/dy) /dt] вязкость (г)) определяется выражением
(9.7)
[дин-с-см-2 (СГС) или Н-с-м~2 (СИ)]. Единица дин-с-см-2 по-
лучила название пуаз (П).
Сантипуаз равен 10-2 пуаз. Вязкость чистых растворителей состав-
ляет примерно 10-2 П, тогда как у неразбавленных (и расплавлен-
ных) полимеров вязкость достигает 1012 П и выше.
Если вязкость не зависит от градиента скорости, то жидкость
называется ньютоновской жидкостью.
5*
132
Глава 9
Если вязкость зависит от градиента скорости, то жидкость на-
зывается неньютоновской жидкостью.
Вязкость можно также определить как
п = ^. (9.8)
где В — работа, затраченная на преодоление вязкостного сопро-
тивления течению в единице объема раствора на единицу времени.
Измерение вязкости полимерных систем позволяет получить
данные о а) средневязкостном молекулярном весе (Mv)
(разд. 9.1.2); б) молекулярновесовом распределении; в) размерах
Таблица 9.1
Различные вязкостные характеристики
Правильное наименование Тривиальное наименование Величина
Коэффициент вязкости Вязкость л
Вязкостное отношение Относительная вяз- кость Удельная вязкость Лотн = Л/Ло Луд =* Лотн — 1
Число вязкости Приведенная вязкость Лпр = (Лотн 1)/^
Логарифмическое число вязкости Логарифмическая вяз- кость Ллог = In Лотн/^
Предельное число вяз- Характеристическая [nJ = lim (ЛудЛО
кости вязкость с->0 [Л] = lim (In Лотн/с) С->0
невозмущенных цепей (разд. 2.13); г) вязкостных характеристи-
ках для течения концентрированных растворов и в особенности
расплавленных полимерных образцов, имеющих значение при вы-
боре параметров для переработки полимеров такими способами,
как экструзия, каландрирование и прессование.
Предельное число вязкости (характеристическая вязкость)
([т]]) определяется как частичное увеличение вязкости одной еди-
ницы растворителя, обусловленное добавлением 1 г (СГС) или
1 кг (СИ) невзаимодействующих полимерных молекул.
Для того чтобы исключить влияние межмолекулярных взаимо-
действий, характеристическую вязкость всегда находят экстрапо-
ляцией числа вязкости (приведенной вязкости) или логарифмиче-
ского числа вязкости (логарифмической вязкости) до нулевой кон-
центрации.
Все указанные выше характеристики суммированы в табл. 9.1.
Обзорная литература: 255, 787, 992, 1466.
Периодическая литература: 2012, 2025, 2400, 3191, 5268, 5388, 5832, 4424,
6708.
Вискозиметрические методы
133
9.1. ИЗМЕРЕНИЯ ВЯЗКОСТИ ДЛЯ РАЗБАВЛЕННЫХ
РАСТВОРОВ ПОЛИМЕРОВ
Следующие вязкостные характеристики используются для описа-
ния разбавленных растворов:
1. Вязкостное отношение (относительная вязкость) (Лотн), ко-
торое определяется отношением времени истечения раствора (/)
к времени истечения чистого растворителя (/0):
Лотн ~ (9.9)
(безразмерная величина).
2. Удельная вязкость (Луд), которая представляет собой отно-
шение относительного инкремента вязкости раствора к вязкости
растворителя:
Луд = (Л ~ Ло)/Ло == Лотн — 1 (9.10)
(безразмерная величина).
3. Число вязкости (приведенная вязкость) (лпр) — это удельная
вязкость на единицу концентрации (с):
Л пр5=3 Луд/^ =z= (Лотн 1)/^ (9.11)
[дл/г (СГС) или м3/кг (СИ)], где с — концентрация полимера
[г/дл (СГС) или кг/м3 (СИ)].
4. Логарифмическое число вязкости (логарифмическая вяз-
кость) (Ллог) определяется как
Ллог In Лотн/с (9.12)
[дл/г (СГС) или м3/кг (СИ)].
5. Предельное число вязкости (характеристическая вязкость)
([л])—число вязкости (приведенная вязкость) или логарифми-
ческое число вязкости (логарифмическая вязкость), экстраполиро-
ванное к с — 0:
[т)] = Нт(^Р-) = Нт(1пт)отн/с) (9.13)
Для экстраполяции на бесконечное разбавление необходимо изме-
рять вязкость при нескольких концентрациях (например, 0,05;
0,10; 0,15; 0,20 г/100 мл). Концентрация образца не должна быть
очень высокой, поскольку при этом возможны побочные эффекты,
обусловленные межмолекулярными взаимодействиями и перепу-
тыванием цепей (в случае очень высоких молекулярных весов).
Существует несколько эмпирических формул для расчета ха-
рактеристической вязкости:
1) уравнение Хаггинса [П: 4163]
Пуд/с = In] + k' [Г)]2с, (9.14)
2) уравнение Крэмера [П: 4701]
In Потн/с = W — k" h]2 с, (9.15)
134
Глава 9
3) уравнение Шульца — Блашке [П: 6280]
Луд/^ — [л] “Ь 1лЗ Луд»
(9.16)
где k', k" и k'"— константы для данного полимера при данной
температуре в данном растворителе. Величины k' и k" связаны
соотношением
k' + k"&±, (9.17)
k' обычно составляет 0,3—0,4 и возрастает с уменьшением силы
растворителя. Для данной системы полимер — растворитель k',
Концентрация (с)
Рис. 9.3. Зависимость Пуд/с или
In Т] отн /с от концентрации.
как правило, не очень зависит от
молекулярного веса. Константы
k', k" и k"' можно найти путем
соответствующих измерений при
различных концентрациях данно-
го полимера в данном раствори-
теле при данной температуре.
Все три уравнения (9.14—
9.16) дают прямые линии, отсе-
кающие на оси ординат отрезок,
равный [т]] при с = 0 (рис. 9.3).
Обзорная литература: 125, 621, 787,
942.
Периодическая литература: 2400,
2760, 3007, 3500, 3793, 4194, 4219 4731,
4949, 5399, 5607, 5744, 5802, 6121, 6159,
6160, 7216.
Обширные данные по константам Хаггинса и Шульца — Блашке
имеются в Polymer Handbook [О: 1277].
9.1.1. Метод определения по одной точке
Этот, метод предполагает измерение вязкости при определенной
концентрации с последующим расчетом характеристической вяз-
кости по одному из приведенных выше уравнений (9.14—9.16),
если известны k', k" или k'ff.
Комбинированием уравнений (9.14), (9.15) и (9.17) можно ис-
ключить k' и k" и получить уравнение Соломона — Сьюта
[п] = 2(ПуД-1п11огн),/7с (9.18)
Метод определения характеристической вязкости по одной точ-
ке является достаточно точным только в том случае, если имеет
место линейная зависимость между с и г)уд/с.
1 Йериодическая литература: 4788, 5403, 6376, 6480.
Вискозиметрические методы
135
9.1.2. Средневязкостный молекулярный вес
Для полидисперсных линейных полимеров средневязкостный мо-
лекулярный вес (Му) определяется из уравнения Марка — Хоувин-
ка — Сакурады: __
[т)] = ДЖ (9.19)
В литературе имеются многочисленные данные о константах /С и а
[О: 786, П: 2025].
Средневязкостный молекулярный вес (Му) определяется как
М„ = [ Е ЛГ,м!+7Е J1,а, (9.20)
где К и а — константы для данного полимера при данной темпе^
ратуре в данном растворителе.
Рис. 9.4. Зависимость Марка — Хоу-
винка — Сакурады для данного поли-
мера в растворе.
Mv можно определить лишь измерением вязкости и никакими
другими методами.
На рис. 9.4 приведена типичная кривая зависимости 1g [i]]
от Му для данного полимера в данном растворителе. На данной
кривой видно отклонение от линейности при низких молекулярных
весах. Такого отклонения можно избежать путем представления
результатов в виде зависимости 1/[л] от \/Му'Ь (рис. 9.5). У по-
следней кривой линейность соблюдается даже при таких низких
молекулярных весах, как 5000.
Средневязкостный молекулярный вес (Л4И) лежит между сред-
нечисловым (Af„) и средневесовым (AU) молекулярными весами
(см. рис. 3.2);
Мп <MV <MW 1 -
При этом Mv ближе к MW) чем к Мп.
136
Глава 9
Константа а колеблется обычно в интервале 0,5 <С а <С 0,8
Более высокие значения а иногда получаются для жестких и/или
коротких молекул.
Константа а связана с силой растворителя и параметром на-
бухания а. Она зависит от термодинамических взаимодействий
между полимерными сегментами и молекулами растворителя
(разд. 2.11).
Константы К и а находят при построении зависимости 1g [т|] от
1g Мп или lgMw (рис. 9.6):
lg[n] = lgK + algM (9.21)
Для определения значений Киа необходимо использовать абсо-
лютные методы определения молекулярного веса (Мп или Л4да),
Рис._ 9.6. Зависимость 1g [4] от
1g Мп или lg Mw для данного поли-
мера.
например осмометрию, светорас-
сеяние или седиментационное
равновесие.
При этом соблюдаются сле-
дующие общие правила:
а) если полимерный образец,
использовавшийся для опреде-
ления констант Киа, имеет ши-
рокое молекулярновесовое рас-
пределение, то найденные значе-
ния К и а могут быть весьма не-
точными;
б) если полимерный образец
имеет одинаковое отношение
Mw/Mn, то правильной является
лишь константа а, тогда как кон-
станта К будет ^ошибочной;
в) при построении зависимости 1g [т]] от 1g Мп значение кон-
станты К получается завышенным; _
г) при построении зависимости lg [rj] от lg Mw значение кон-
станты К будет занижено. Монодисперсные полимерные образцы,
как правило, редко доступны, поэтому для определения Киа
обычно пользуются тщательно выбранными фракциями данного
полимера.
Применение вискозиметрических измерений для определения
средневязкостного молекулярного веса (Mw) осложнено еще рядом
факторов:
а) влиянием молекулярновесового распределения;
б) наличием ветвлений;
в) композиционной неоднородностью и статистикой последо-
вательностей в случае стереоспецифических полимеров и сополи-
меров;
г) существованием агломератов;
д) сольватацией макромолекул;
Вискозиметрические методы
137
е) перепутыванием цепей;
ж) эффектом торможения (drag effect);
е) такими физическими факторами, как адсорбция полимерных
молекул на стенках капилляров, разрыв цепей в результате среза,
локальные перегревы, обусловленные рассеянием энергии вязкого
течения.
Периодическая литература: 2025, 2091, 2102, 2170, 2304, 2379, 2504, 2649,
2694, 2860, 3179, 3180, 3192, 3200, 3201, 3306, 3318, 3519, 4040, 4069, 4187, 4382,
4551, 4599, 4771, 4802, 4917, 4949, 4990, 5076, 5083, 5118, 5217, 5251, 5293, 5389,
5442, 5524, 5770, 5812, 6336, 6488, 6635, 6787—6789, 6893, 6985, 7043, 7219, 7288,
7306.
9.1.3. Измерение вязкости для разбавленных
растворов сополимеров
Не существует одного-единственного уравнения, связывающего ха-
рактеристическую вязкость со средневязкостным молекулярным
весом линейных гетерогенных сополимеров. Так, например, три
типа сополимеров
статистический: А ААВААВВАВВВ
регулярно-чередующийся: АВАВАВАВАВАВ
блок-сополимер: АААВВВАААВВВ
имеют различные распределения звеньев А и В вдоль основной
цепи. Все эти сополимеры могут иметь одинаковые молекулярные
веса, но отличаться по характеристической вязкости. Для линей-
ных гетерогенных сополимеров уравнение Марка — Хоувинка—
Сакурады [уравнение (9.19)] теряет свою справедливость.
Обзорная литература: 100, 255, 1214.
Периодическая литература: 3120, 3199, 4210, 4344, 4345 5761, 5948, 5962,
6585, 6903.
9.1.4. Измерение вязкости разбавленных растворов
разветвленных полимеров
Уравнение Марка — Хоувинка — Сакурады [уравнение (9.19)] не
справедливо также для разветвленных полимеров. Средневязкост-
ный молекулярный вес, вычисленный с использованием этого
уравнения, для разветвленных полимеров будет ниже, чем для со-
ответствующих линейных полимеров (особенно в области более
высоких молекулярных весов). Для того чтобы расчет был пра-
вильным, необходимо знать функцию молекулярновесового рас-
пределения, функцию распределения точек ветвления, структуру
ветвлений.
Обзорная литература: 255, 534.
Периодическая литература: 2377, 2468. 2757, 3204, 3741, 3843, 4087, 4696,
4747, 4769, 5048, 5345, 5430, 5464, 5513, 5616 5632, 5677, 6902, 6903, 6339, 7337,
18ё
Глава 9
9.1.5. Капиллярные вискозиметры для измерения вязкости
разбавленных растворов
Капиллярные вискозиметры, использующиеся для измерения вяз-
кости разбавленных растворов, делают из стекла. Принцип их дей-
ствия заключается в следующем: капилляр заполняют необходи-
мым объемом исследуемой жидкости, причем столбик жидкости
должен подняться выше верхней метки над шариком, после чего
измеряют время, необходимое для опускания мениска жидкости
от верхней метки до нижней. Время истечения зависит от вязкости
жидкости и определяется величиной давления, которое входит в
уравнение Пуазейля:
П = n/?4P/8/Q = л/?4Р//8/7, (9.22)
где R— радиус капилляра, Р — давление, заставляющее жидкость
двигаться по капилляру, I — длина капилляра, Q—V/t — объем-
ная скорость истечения, V — объем жидкости, t — время исте-
чения.
Как правило, движущая сила (давление) обусловлена раз-
ностью высот уровней жидкости в вискозиметре (А). Более высо-
кое давление можно создать с помощью подключенного моностата.
Скорость сдвига (у) в капилляре
*=-£=<• (9-23)
где г — расстояние от центра капилляра, v — скорость жидкости.
При ламинарном течении скорость на расстоянии г от центра ка-
пилляра определяется как
у(г) = Р(^2-г2)/4т]/ (9.24)
Скорость изменяется параболически от нуля у стенки капилляра
(г = ^) до максимальной величины:
vmax = W/4t)/ (9.25)
Напряжение сдвига (т) в капилляре равно
т = F/A = PR/21, (9.26)
где F = пг2Р — сила на радиальном расстоянии г от центра ка-
пилляра (fmax = R, тогда F = tcR2P) , А = 2л/?/ — площадь поверх-
ности капилляра.
В капиллярной вискозиметрии существует много источников
ошибок, связанных с величиной измеряемой вязкости, природой
жидкости и геометрической конструкцией вискозиметра. Поправки
можно разделить на две группы:
1. Поправки на скорость сдвига, а именно поправки на откло-
нение жидкости от ньютоновского поведения (поправка Рабино-
вича), сжимаемость жидкости, изменения размеров капилляра при
различных температурах.
Ёискозиметрическив методы
139
2. Поправки на напряжение сдвига, а именно: поправка на
кинетическую энергию (поправка Хагенбаха), поправка на вход,
необходимая потому, что скорость жидкости не параллельна оси
капиллярной трубки, поправка на упругую энергию.
Эти поправки важны при исследовании разбавленных растворов
полимеров, при изучении самих полимеров (расплавов) некоторые
из них можно не принимать во внимание.
Уравнение Пуазейля (9.22) после внесения поправки на кине-
тическую энергию и поправки на вход принимает вид
п = Ар/ — Bp/Z = Apt (1 — В/At2), (9.27)
где г) — вязкость жидкости, р — плотность жидкости, А и В — кон-
станты для данного вискозиметра.
А и В можно найти графически из зависимости (ц/р)// от I//2.
Отсекаемый на оси ординат отрезок равен А, тогда как наклон
определяет величину (—В/А).
9.1.6. Измерение относительной вязкости
По определению относительная вязкость равняется
__________________________Вязкость раствора __ г|
Лотн Вязкость растворителя д0 ’
и тогда
л р*(1-В/Л/*)
1отн Ло р0/0(1-В/Л/2) ’
где индекс нуль относится к чистому растворителю.
В разбавленных растворах отношение р/р0 обычно
единице, так что
(9.28)
(9.29)
близко к
(9.30)
_ /(i—в/Л/2)
По™ /0(1-В/Л^)
Если время истечения чистого растворителя для данного вискози-
метра превышает 100 с, то поправка на кинетическую энергию
В/At2 по сравнению с единицей пренебрежимо мала и тогда
Лотн = ^Ао (9.31)
На рис. 9.7 приведены основные типы капиллярных вискозиметров.
а) Вискозиметр Оствальда. Движущая сила в таком вискози-
метре пропорциональна разности высот двух уровней U-образной
трубки (А). Чтобы результаты были воспроизводимы, необходимо
в каждом измерении использовать один и тот же объем жидкости.
б) Вискозиметр Кеннона — Фенске. Конструкция этого виско-
зиметра предусматривает отклонение обоих колен U-образной
трубки от вертикали, в результате этого центры поверхности жид-
кости в обоих коленах располагаются вдоль оси, которая сохраняет
140
Глава 9
Рис. 9.7. Различные типы вискозимет-
ров.
а—вискозиметр Оствальда; б —виско-
зиметр Кеннона — Фенске; в—вискозиметр
Уббелоде; г—вискозиметр с регулируемым
напряжением сдвига.
Рис 9.8. Детектор истечения в авто-
матическом вискозиметре [П: 3742].
1 — призма; 2—фотодиод; 3—вискозиметр;
4—ламцы.
вертикальность даже при нали-
чии небольшой ошибки в верти-
кальном расположении вискози-
метра.
в) Вискозиметр Уббелоде.
Движущей силой в вискозиметре
этого типа является расстояние
(h) от уровня жидкости в шари-
ке до уровня, определяемого
нижним концом капилляра.
В этом вискозиметре нет необхо-
димости проводить все измерения
при одинаковом объеме жидко-
сти. Некоторые конструкции ви-
скозиметра предполагают нали-
чие большого резервуара, из ко-
торого можно добавлять раство-
ритель к то су же раствору, пони-
жая таким образом концентра-
цию раствора.
г) Вискозиметр с регулируе-
мым напряжением сдвига. Напря-
жение сдвига (т) в капиллярном
вискозиметре пропорционально
разности высот уровней жидко-
сти (Л). В вискозиметре Уббело-
де напряжение сдвига понижает-
ся с уменьшением высоты. Суще-
ствует тип вискозиметра, назы-
ваемый вискозиметром с регули-
руемым напряжением сдвига, в
котором т различно для каждого
из шариков.
Все рассмотренные выше ти-
пы вискозиметров выпускаются в
двух вариантах: с объемом 10
или 1 мл (полумикро) и различ-
ными диаметрами капилляров.
Устанавливать вискозиметры
следует строго вертикально, ина-
че вводится дополнительная
ошибка. Во избежание невоспро-
изводимости или разброса дан-
ных желательно перед внесением
в вискозиметр отфильтровать
раствор от пыли. Для измерения
характеристической вязкости с
Васкозиметрические методы
141
ошибкой, не превышающей 1%, необходимо термостатирование
вискозиметра с отклонением температуры не более +0,01 °C.
Обзорная литература: 917, 992.
Периодическая литература: 2220, 2873, 3700, 3915, 4468, 4490, 4726, 5190,
6298, 6874.
В новейших конструкциях вискозиметры Оствальда или Уббе-
лоде снабжены стеклянной термостатированной рубашкой, в кото-
рую подают жидкость из отдельного термостата. Течение жидкости
в вискозиметре регистрируется с помощью фотоэлемента А или В,
Рис. 9.9. Логическая часть вычислительной машины для автоматического виско-
зиметра [0:7,23].
который освещается одной лампочкой, направленной на него не-
посредственно, а другой — через призму (рис. 9.8). Фотоэлемент
подает сигнал на
а) электронный таймер с точностью +0,01 с, что достигается
использованием кварцевого кристаллического осциллятора и циф-
ровой записи (индикации) времен истечения;
б) воздушный насос для автоматического контроля течения;
в) автоматическую бюретку для автоматического ввода об-
разца или заполнения вискозиметра.
На рис. 9.9 показана типичная блок-диаграмма вычислительной
схемы такого вискозиметра. Система может быть запрограммиро-
вана таким образом, чтобы вначале измерялось время истечения
полимерного раствора концентрации с, а затем после автомати-
ческого разбавления, например, концентрации 2с/3, с/2, с/3, с/6.
Расчет характеристической вязкости из обычной кривой (/— t$)/t
проводится с помощью настольного компьютера.
В оригинальных публикациях можно встретить самые разно-
образные конструкции автоматических вискозиметров.
Обзорная литература: 723.
Периодическая литература: 2873, 2946, 3132, 3686, 3742, 4170, 4552, 4820,
4839, 4887, 5253, 5254, 6411, 6446, 6526.
142
Глава 9
Данные, получаемые с помощью автоматических вискозимет-
ров, обсчитывают с помощью компьютеров. В данные, вводимые
в ЭВМ, входят времена истечения и концентрация растворов
(включая время истечения чистого растворителя, т. е. с = 0).
В результате получают характеристическую вязкость, константу
Хаггинса, кривую зависимости т]уд/с от наклон этой кривой и
стандартные отклонения.
Обзорная литература: 312.
Периодическая литература: 2681, 2946.
9.2. ИЗМЕРЕНИЕ ВЯЗКОСТИ НЕРАЗБАВЛЕННЫХ
ПОЛИМЕРОВ
Для измерения вязкости неразбавленных полимеров или их рас-
плавов применяются следующие методы:
1. Капиллярная, или пуансонно-щелевая вискозиметрия, в
основу которой положено течение жидкости под давлением.
2. Ротационная вискозиметрия, использующая вискозиметры с
сочетанием двух измерительных поверхностей: концентрических
цилиндров и плоскостей с конусом.
3. Методы, основанные на ползучести и релаксации напряже-
ний, применимые в области очень высоких значений вязкости
(в этой книге не рассматриваются).
Обзорная литература: 110, 217, 255, 259, 301 353, 395, 436, 533, 922, 1177,
1185, 1283, 1339, 1345. 1429.
Периодическая литература: 2380, 2807, 3113, 3231, 3657, 3738—3740, 3880,
4255, 4256, 5221, 5759, 5760, 5780, 6119, 6143, 6158, 6299, 6658, 6886, 6904, 7000.
9.2.1. Капиллярная вискозиметрия для измерения вязкости
расплавов полимеров
Капиллярные реометры действуют на двух принципах:
а) экструзионное давление постоянное (регулируется регуля-
тором давления), тогда как объемная скорость течения (скорость
сдвига) регистрируется как переменная величина;
б) объемная скорость течения (скорость сдвига) постоянная
(постоянный расход), тогда как экструзионное давление регистри-
руется как переменная величина.
Капиллярные вискозиметры классифицируются также по спо-
собу получения зависимости между скоростью и напряжением
сдвига;
а) скорость сдвига является независимой переменной величи-
ной, тогда как напряжение сдвига представляет собой зависимую
функцию;
б) напряжение сдвига представляет собой независимую пере-
менную величину, тогда как скорость сдвига является зависимой
функцией.
Вискозиметрические методы
143
Для многих вязких полимерных жидкостей используются сталь-
ные капиллярные вискозиметры (капиллярные реометры). Типич-
ный капиллярный вискозиметр экструзионного типа состоит из ре-
зервуара (втулки) с капиллярной трубкой, соединенной с дном
(рис. 9.10). Под высоким _
давлением полимерная жид-
кость перемещается из ре-
зервуара в капиллярную
трубку. Через капилляр
жидкость выталкивается
под действием штока, кото-
рый движется с заданной
скоростью. Объемная ско-
рость истечения определяет-
ся скоростью штока и раз-
мерами резервуара.
Вязкость полимерной
жидкости (т]) равняется
П = т/у, (9.7)
где т — напряжение сдвига:
x = FIA = PRI2l, (9.26)
F = nr2P — сила на ради-
альном расстоянии (г) от
центра капилляра (гтах =
= R, тогда F = nR2P); А =
= 2nRl — площадь поверх-
ности капилляра; Р — дав-
ление, под влиянием которо-
го жидкость движется по
Рис. 9.10. Экструзионный вискозиметр по-
стоянного давления.
1— капилляр; 2—датчик давления; 3 —самописец;
4 — образец; 5 —поршень; 6 — манометр; 7—гидра-
влический насос; 8 —ресивер; 9 — регулятор давле-
ния.
капилляру, у — скорость
сдвига.
Для ньютоновских жид-
костей
y = 4Q/W (9.32)
В общем случае, однако,
• — 4Q Г3 4- 1 (43/лЯ3)]-|
Y — nR3 L4 + 4 ’ d [In (PR/21)] J’
(9.33)
где Q = V/t— объемная скорость течения; V — объем жидкости;
t — время истечения; R — радиус капилляра; / — длина капилляра.
Давление (Р) измеряется датчиком давления, записывается на
диаграммной ленте и применяется для расчета напряжения сдвига
с использованием уравнения (9.32). В настоящее время с помощью
капиллярных вискозиметров можно фиксировать скорости сдвига
S нижним пределом 1—0,1 с-1.
144
Глава 9
Выдача и обработка данных во многом облегчаются благодаря
вычислительной технике.
Обзорная литература: 255, 533, 754, 1339, 1357.
Периодическая литература: 2190, 2218, 3737, 3876, 4482, 5244, 6118, 6740.
9.2.2. Пуансонно-щелевой вискозиметр
Пуансонно-щелевой вискозиметр представляет собой реометр с
прямоугольным поперечным сечением, образующимся системой
Рис. 9.11. Экструзионная щелевая
матрица.
Рис. 9.12. Ротационный вискозиметр
с концентрическими цилиндрами.
двух параллельных пластин (рис. 9.11). Наряду с капиллярным
реометром такой вискозиметр позволяет получить информацию о
вязкоупругих свойствах полимерных расплавов.
Периодическая литература: 3874, 3875, 3877, 3878, 3882, 4847.
9.2.3. Ротационный вискозиметр с концентрическими
цилиндрами
Цилиндрический ротационный вискозиметр применяется для изме-
рения вязкости концентрированных растворов полимеров. На
рис. 9.12 представлена схема устройства такого вискозиметра.
В вискозиметре такого типа образец раствора полимера поме-
щается между внутренним и внешним цилиндрами. Образец под-
вергается напряжению сдвига путем вращения внутреннего или
внешнего цилиндра с постоянной угловой скоростью (Q) [в граду-
сах в секунду (СГС) или радианах в секунду (СИ)].
Вискозиметрические методы
145
Напряжение сдвига составляет
т = M/2nr2ht
(9.34)
где М — крутящий момент [дин-см (СГС) или Н-м (СИ)]; г —
радиальное расстояние от оси вращения (/?i г и R2—
радиусы внутреннего и внешнего цилиндров); h — глубина погру-
жения внутреннего цилиндра в исследуе-
мую жидкость.
Скорость сдвига равняется
v= — г ,
' dr
(9.35)
где ® — угловая скорость.
Вязкость полимерного образца (т])
составляет
^ = 7 =-ж (4) (9-36>
После интегрирования уравнения (9.36)
между г = /?1, где со = 0, и г — R2i полу-
чаем уравнение Маргулиса для вязкости
полимера:
Рис. 9.13. Ротационный вис-
козиметр с конусом и плос-
костью.
Оно справедливо только для ньютоновских жидкостей, когда дви-
жение является ламинарным, нет проскальзывания у стенок при-
бора и в изотермических условиях.
Важно отметить, что напряжение сдвига и скорость сдвига
изменяются обратно пропорционально квадрату расстояния (г) от
оси вращения. Обычно их рассчитывают у поверхности внутрен-
него (r = R\) или. внешнего (г = R2) цилиндра. Напряжение
сдвига (т) и скорость сдвига (у) следует рассчитывать для одного
и того же положения вискозиметра.
Ротационные вискозиметры применимы для непрерывных изме-
рений.
Обзорная литература: 1357.
Периодическая литература: 4504, 4931, 7070, 7333.
9.2.4. Вискозиметр с плоскостью и конусом
в качестве измерительных поверхностей
Вискозиметр, у которого измерительные поверхности представляют
сочетание плоскости и конуса, применяется для измерения вязко-
сти более высоковязких жидких сред, например расплавов поли-
меров. На рис. 9.13 приведена схема геометрии такого ротацион-
ного вискозиметра. Расплав полимера помещается в зазор между
конусом и плоской круглой пластиной. Угол конуса (а) опреде-
ляется как угол между поверхностью конуса и поверхностью
146
Глава 9
горизонтальной плиты. Этот угол обычно очень мал (1—5°). Конус
вращается с постоянной угловой скоростью (Q).
Напряжение сдвига равняется
т = ЗЛ4/2л7?3, (9.38)
где М — крутящий момент [дин-см (СГС) или Н-м (СИ)]; У? —
радиус конуса.
Скорость сдвига равняется
у = Q/a, (9.39)
где Q — постоянная угловая скорость [град/с (СГС) или радиан/с
(СИ)]; a — угол конуса [град (СГС) или радиан (СИ)].
Вязкость расплава полимера
т] = т/у = ЗаЛ1/2лУ?3О = kM!Q>, (9.40)
где k = За/2лУ?3— константа, определяемая геометрией прибора.
С помощью таких вискозиметров можно измерять скорости
сдвига с нижним пределом 10~3 с-1.
Обзорная литература: 255, 533, 1339, 1357, 2381, 3739, 5760.
Глава 10
ОПТИЧЕСКИЕ МЕТОДЫ
Обзорная литература: 58, 87, 114, 139, 166, 167, 188, 207, 240, 241, 468, 647,
744, 782, 813, 849, 873, 874, 981, 990, 991, 1086, 1206, 1220, 1230, 1243, 1257, 1415.
10.1. СВОЙСТВА ЭЛЕКТРОМАГНИТНОГО ИЗЛУЧЕНИЯ
Электромагнитная энергия — это форма энергии, распространяю-
щейся в пространстве без переноса массы. Поведение электро-
магнитного излучения может быть связано с его волновым или
корпускулярным характером. На рис. 10.1 показана плоскополя-
ризованная волна одной частоты, называемая монохроматическим
лучом. Плоскополяризованное электромагнитное поле характери-
зуется тем, что электрический вектор Е колеблется в одной пло-
скости, тогда как вектор магнитного поля Н колеблется в другой
плоскости, перпендикулярной электрическому полю. Реально в
большинстве случаев электромагнитное излучение является непо-
ляризованным, т. е. Имеет электрический и магнитный вектор во
всех ориентациях, перпендикулярных направлению распростра-
нения.
Различные виды электромагнитного излучения обычно харак-
теризуются длиной волны (А,) или частотой (v).
Длиной волны (%) называется длина цикла или расстояние
между двумя последующими максимумами или минимумами
(рис. 10.1).
Обычно пользуются следующими единицами длин волн (СГС
и СИ):
1мкм = 1СГ6 м = 10“4 см (мкм — микрометр)
1нм = 10-9 м = 10"7 см (нм — нанометр)
1А = Ю-10 м = 10"8 см (А — ангстрем)
1 ангстрем — единица длины волны, равная 1/6438,4696 длины
волны красной линии Cd. (Примечание. Не пользуйтесь единицами
старой системы, например микроном или миллимикроном.)
Большинство работников в области конструирования оптиче-
ских приборов, фотохимии и оптики выражают длины волн в на-
нометрах, тогда как спектроскописты обычно длину волны обо-
значают в ангстремах.
Частотой (v) называется число циклов в единицу времени (с-1
или Гц). (Примечание. 1 герц = 1 цикл в секунду.)
148
Глава 10
Длина волны и частота связаны выражением
X = u/v = c/v, (10.1)
где'у — скорость распространения.
В вакууме все виды электромагнитного излучения имеют оди-
наковую скорость распространения (с), равную 2,9979-1010 см-с-1
(СГС) или 2,9979-108 м-с-1 (СИ).
Частота представляет собой единственную истинно характери-
стическую величину данного вида излучения, поскольку как ско-
Рис. 10.1. Плоскополяризованная электромагнитная волна одной частоты. Е —
электрический вектор; Н — магнитный вектор.
рость (а), так и длина волны (%) зависят от среды, в которой
распространяются электромагнитные волны.
Волновое число (v) — это число волн на единицу длины:
v=l/Z, = v/c (10.2)
[см-1 (СГС) или м-1 (СИ)].
Единицы волнового числа и длины волны связаны между собой
следующими выражениями:
см’* = ^Т Ю4(СГС); Юв(СИ) (10.3)
В приложении Б даются таблицы для перевода волнового числа
(см-1) в длину волны (мкм).
Электромагнитный спектр приведен на рис. 10.2.
Согласно корпускулярной теории электромагнитного излучения,
формой энергии являются маленькие пучки или частицы, называе-
мые фотонами.
Фотоны — это квантованные электромагнитные волны.
Фононы — это квантованные колебательные волны.
Оптические методы
149
Вид излучения Частота, Гц Длина волныtM
Космическое
Гамма,
Рентгеновское
Ультрафиолетовое
Видимое <
Инфракрасное
ю24-
ю23-
- 10"14
ю^'-10-13
10 - 4Л-12
ю20-
ю1*?
1018-
Микроволновое (радиолокация) Г
Ультракоротковолновое гигагерцы
(радио и ТВ)
Коротковолновое (радио) -{мегагерцы
-10’12
-1О’10
-10‘9
-Ю‘8
-ю~7
10'6
-10’5
-10’4
-10-3
г2
ю17-
ю16-
1015-’
ю14-
ю13-'
ю12-'
ю11 - .
ю9 < 0
108 ~~
Длинноволновое (радио)
Радиошумы
Электрические шумы
килогерцы
герцы
10
10е -
105 -
10* -
ю3
ю2
Ю -*
10
ю2
-ю3
^-ю4
'-ю5
-106
ю7
нанометры
микрометры
сантиметры
метры
километры
Рис. 10.2. Электромагнитный спектр.
В каждом случае энергия молекулы находится по уравнению
Планка:
E = hv, (10.4)
где h — постоянная Планка, равная 6,626-10-27 эрг-с (СГС) или
6,626-10~34 Дж-с (СИ); v — частота света в случае фотонов или
частота колебания в случае фононов (с-1).
10.2. СПЕКТРОСКОПИЧЕСКИЕ МЕТОДЫ
В ИССЛЕДОВАНИИ ПОЛИМЕРОВ
Обзорная литература: 53, 63, 65, 71, 81, 102, 190, 192, 207, 212, 240, 274, 337,
364, 389, 390, 405, 476, 612, 693, 730, 795, 878, 961, 962, 991, 1012, 1037, 1105, 1158,
1179, 1192, 1213, 1292, 1305, 1379, 1393.
Спектроскопические методы основаны на измерении интенсив-
ности и длины волны энергии излучения и спектрах, обусловлен-
ных переходами между характеристическими энергетическими со-
стояниями.
На рис. 10.3 представлены различные области электромагнит-
ного спектра вместе с названиями для ряда спектроскопических
150
Глава 10
100 МэВ МэВЮОШ 1кэВ rfwr1 25000 см'1 ЗЗиг1 1см^ _I I I I 5 1 1 1 f0iS- IO10 fO8 I I I I § £ s- J- 1- 1 1- •5- Радио Спектроскопия ЯМР Вращение молекул:спин электронов, спин ядер
§ Инфракрасная Микроволновая Микроволновая и ЭПР- спектроскопия
ближняя\ Средняя\Дальняя Г ИК 1 ИК 1 И к | ИК-спектрометрия Молекулярные колебания
Перенос внешних электронов
I 1 Ваку у мн. ЯР Бя^,и^> У Ф-спектро- метрия
Рентгеновские лучи Рентгеновская спектроскопия Перенос внутренних электронов
o“ Гамма-лучи Гамма- спектроскопия Ядерные реакции
Энергия, единицы Частота, Гц Плана волны, единицы Спектральная область Оптический метод изучения Переходы
Видимая спектрофотометрия
Рис. 10.3. Электромагнитный спектр и оптические методы для его изучения.
Оптические методы
151
Таблица 10.1
Классификация спектроскопических методов по типам спектров
Спектры поглощения Спектры излучения Спектры КР
Ультрафиолетовая и видимая спектрофотометрия Инфракрасная спектрофото- метрия Микроволновая спектроско- пия Спектрометрия кругового ди- хроизма Спектроскопия поглощения рентгеновских лучей Спектроскопия ядерного маг- нитного резонанса (ЯМР) Спектроскопия электронного парамагнитного (спинового) резонанса (ЭПР) Эмиссионная спектро- скопия Спектрофлуорометрия Спектрофосфоромет- рия Спектроскопия КР
методов. Все спектры можно разделить на три основных типа
(табл. 10.1):
1) спектры поглощения (гл. 14 и 15);
2) спектры излучения (гл. 16);
3) спектры комбинационного рассеяния (гл. 17).
10.2.1. Абсорбционная спектроскопия
Суммарная энергия молекулы складывается из энергии электро-
нов (Еэл), колебательной энергии (ЕКОл), вращательной энергии
(Евр) и трансляционной энергии (Етр).
Величина АЕ представляет собой энергию молекулы при по-
глощении электромагнитного излучения и составляет
АЕ = АЕЭЛ + АЕКОЛ + АЕвр + АЕтр, (10.5)
где АЕЭЛ— расстояние между разрешенными электронными энер-
гетическими уровнями, АЕкол расстояние между разрешенными
колебательными энергетическими уровнями (обычно АЕ кол при-
мерно в 10 раз меньше АЕЭЛ); АЕвр— расстояние между разрешен-
ными вращательными энергетическими уровнями (АЕвр в 10—
100 раз меньше АЕКОЛ); АЕтр— расстояние между разрешенными
трансляционными энергетическими уровнями (величина АЕтр чрез-
вычайно мала и в абсорбционной спектроскопии не играет суще-
ственной роли).
При пропускании пучка монохроматического излучения через
полимерный образец оно может в зависимости от своей длины вол-
ны (Л) частично поглотиться специфическими группами молекулы
152
Глава 10
полимерного образца. На экспериментальной кривой зависимости
интенсивности пропускания от длины волны излучения при длинах
волн, отвечающих максимальному поглощению, будут наблюдаться
минимумы. Такую кривую называют спектром поглощения об-
разца.
На рис. 10.4 приведено схематическое изображение спектра
поглощения при различных длинах волн, соответствующих уль-
Злектронный спектр поглощения Колебательный спектр поглощения
Волновое число, см”1
Рис. 10.4. Спектр поглощения.
трафиолетовой (УФ) области (А, = 0,1—0,4 мкм), видимой обла-
сти (% = 0,4—0,8 мкм), инфракрасной (НК) области (% = 1 —
50 мкм).
Пик поглощения (полоса) характеризуется положением и ин-
тенсивностью пика (полосы) поглощения.
Положения пиков поглощения в ультрафиолетовой и видимой
областях выражаются в тех же самых единицах, что и их длины
волн (X), т. е. в ангстремах, микрометрах, нанометрах.
Положения полос поглощения в ИК-спектрах можно обозна-
чать в тех же единицах, что и их длины волн (ангстремах, микро-
метрах или нанометрах), частоты (с-1 или Гц) или волновые чис-
ла (см-1).
10.3. НЕСПЕКТРОСКОПИЧЕСКИЕ ОПТИЧЕСКИЕ
МЕТОДЫ ИССЛЕДОВАНИЯ ПОЛИМЕРОВ
С помощью неспектроскопических оптических методов не снимают
спектры, не изучают переходы между характеристическими энер-
гетическими состояниями, но исследуют взаимодействия между
электромагнитным излучением и веществом, которые приводят к
изменению направления или физических свойств электромагнит-
Оптические методы
153
ного излучения. При неспектроскопических методах возможны сле-
дующие специфические взаимодействия.
1. Рефракция, которая представляет собой искривление излу-
чения при прохождении из одного вещества в другое и может быть
связана с различием в скоростях распространения в двух средах
(гл. 11).
2. Отражение, наблюдающееся при попадании излучения на
границу между двумя веществами, где существует изменение по-
казателя преломления.
3. Рассеяние, представляющее собой взаимодействие электро-
магнитного излучения с частицами на его пути с индуцированием
осцилляций электрических зарядов вещества. Индуцированные ди-
поли излучают во всех направлениях вторичные волны (гл. 13).
4. Интерференция, которую можно определить как изменение
интенсивности волнового излучения в результате комбинирования
или наложения двух или более волн:
а) деструктивная интерференция происходит при уменьшении
интенсивности;
б) при росте интенсивности наблюдается конструктивная ин-
терференция', это явление используется в оптических интерферен-
ционных фильтрах, которые можно применять в ближней ультра-
фиолетовой, видимой и ближней инфракрасной областях
(разд. 10.4).
5. Дифракция, которая возникает, когда волны электромагнит-
ного излучения проходят через щель или распространяются за
край непрозрачного препятствия; они всегда в некоторой степени
заходят в область, не подвергающуюся прямому облучению.
6. Дисперсия, которую можно определить как разделение смеси
волн различной длины на составляющие компоненты.
7. Поляризация, которая имеет место, когда амплитуды век-
торов становятся несимметричными относительно направления рас-
пространения; устранение всех плоскостей, за исключением одной,
приводит к плоской поляризации (разд. 10.3.1).
8. Дихроизм, который представляет собой селективную абсорб-
цию одной колебательной плоскости излучения (разд. 35.5).
10.3.1. Поляризованное излучение
Большинство источников дает электромагнитное излучение, в ко-
тором колебания электрического и магнитного векторов происходят
с одинаковой амплитудой во всех направлениях перпендикулярно
направлению распространения (рис. 10.5, а). Такое излучение на-
зывается неполяризованным излучением.
Если под влиянием какого-то вещества амплитуда неполяри-
зованного излучения меняется не однозначно в разных направле-
ниях, то электромагнитное излучение будет поляризоваться. Когда
134
Глава 10
колебание в каком-то направлении полностью исключается, излу-
чение называется линейно-поляризованным или плоскополяризо-
ванным (рис. 10.5,6).
Электрическое колебание может постоянно вращаться как по
часовой, так и против часовой стрелки, соответственно пучок будет
Рис. 10.5. Контур, который оставляет электрический амплитудный вектор на раз-
ных стадиях поляризации.
а — неполяризованное излучение; б—линейная поляризация; в — круговая поляризация.
г — эллиптическая поляризация.
право- и леьо-циркулярно-поляризованным (рис. 10.5, в). Ампли-
туда такой волны вращается по часовой или против часовой стрел-
ки, оставляя в пространстве спиральный след (рис. 10.6), и при
наблюдении спирали сзади выгля-
Рис. 10.6. Амплитуда волны с круго-
вой поляризацией вращается по часо-
вой стрелке, оставляя в простран-
стве спиральный след. Показана
только одна длина волны.
дит как эллипс (рис. 10.5,а). Пу-
чок в такой проекции называется
эллиптически-поляризованным.
Относительную интенсивность
двух ортогональных компонент
колебаний в луче можно опреде-
лить с использованием пары —
поляризатора и анализатора. Они
разлагают свет на ортогональные
колебания и селективно выклю-
чают один из видов таких коле-
баний. При облучении линейно-
поляризованным светом интен-
сивность, пропущенная анализа-
тором, составляет:
а) 100%, если направление
колебания падающего облучения
параллельно характеристическому направлению колебаний анали-
затора (рис. 10.5, а);
б) любое значение между 100 и 0% в зависимости от того,
возникает при расщеплении благоприятная или неблагоприятная
компонента;
в) 0%, если направление колебаний падающего света перпен-
дикулярно благоприятному направлению анализатора, — в этом
Оптические методы
155
случае падающее облучение не создает компоненты, пропускаемой
в направлении колебаний (рис. 10.5, б).
Степень поляризации (Р), или поляризуемость, равняется
Р = (10.6)
где /' и /" — максимальная и минимальная интенсивности, наблю-
даемые через анализатор, вращающийся на 360°.
Эффективность поляризатора зависит от его отдачи, исследуе-
мой области спектра и угловой апертуры.
Поляризаторы можно разделить на следующие категории:
1. Дихроичные поляризаторы селективно поглощают одну из
ортогональных колебательных компонент (рис. 10.7). В дихроич-
ных поляризаторах применяются материалы различного рода.
Рис. 10.7. Определение линейно-поляризованного излучения при помощи поля-
роидных дисков.
а — нескрещенные поляризаторы; б—скрещенные поляризаторы.
а) Поляроидные пластинки типа Н и К, которые часто ис-
пользуются в видимой области и ближней ПК-области. Их изго-
тавливают из пленок поливинилового спирта. Листы Н-типа вна-
чале подвергают вытяжке, с тем чтобы сориентировать молекулы
в направлении растяжения, после чего пропитывают иодом. Мо-
лекулы 12 образуют комплексы с полимером, благодаря чему они
также выстраиваются надлежащим образом. Компонента падаю-
щего света, имеющая колебания, параллельные оси связи моле-
кул 12, поглощается, тогда как соответствующая компонента под
углом 90° пропускается. В пленке типа К селективное поглощение
обеспечивается в результате каталитической дегидратации поли-
винилового спирта с последующим параллельным выстраиванием
двойных связей путем вытяжки.
156
Глава 10
б) Пленки пиролитического графита, используемые для поля-
ризации в инфракрасной области от 2500 см-1 до микроволнового
диапазона.
2. Поляризаторы отражения частично поляризуют свет за счет
отражения от диэлектрика (рис. 10.8). Отражательная способ-
деполяризованный
свеп
Линейно -
поляризованный
сеет
Рис. 10.8. Поляризация света с помощью поляризатора отражения с пластин-
ками, расположенными под углом поляризации.
ность двух ортогонально-поляризованных компонент находится из
уравнений Френеля:
П 1 зт2(Ф-Ф') ,1П7х
2 мп2(Ф + Ф')’ (10‘7'
1 tg2 (Ф - Ф')
2 tg2 (Ф + Ф') ’ (10.8)
где Ф — угол падения (и отражения); ф' — угол преломления.
Степень поляризации можно увеличить включением в систему
дополнительных пластинок, каждая из которых будет исключать
Рис. 10.9. Поляризация света с помощью поляризатора двойного преломления,
изготовленного из кристалла кальцита.
еще какую-то определенную часть вертикальной компоненты. По-
ляризаторы отражения широко используются в ИК-области (при-
меняют от 4 до 6 пластин из хлорида серебра, селена, германия).
3. Поляризаторы двойного преломления собираются из секций,
представляющих собой кристаллы кальцита (СаСО3). Кристалл
расщепляет падающий свет на пучки, которые пропускаются при
различных углах вследствие различия в показателях преломления
(рис. 10.9). Поляризаторы двойного преломления дают два полных
Оптические методы
157
спектра линейно-поляризованного света. Поляризованный в ре-
зультате двойного преломления свет, образующийся при исполь-
Рис. 10.10. Поляризация света с помощью призмы Николя.
зовании СаСОз, применяется для поляризации в области 220—
1800 нм (2200А—1,8 мкм), тогда как поляризованный свет, по-
лучающийся при использовании кристаллов первичного кислого
фосфата кальция, применяется в области до ~185 нм.
z
Рис. 10.11. Поляризация света с помощью призмы Рошона — Волластона.
Призмы Николя (или поляризаторы Николя) изготавливают из
кристаллов кальцита с минимальным чередованием поверхностей
в природном кристалле (рис. 10.10). Падающий луч расщепляется
на обыкновенный и необыкновенный лучи. Путем раскалывания
158
Глава 10
кристалла под соответствующим углом и склеивания половинок
канадским бальзамом получают изотропное вещество с nD = 1,55,
в котором на границе раздела обыкновенный луч полностью от-
ражается. Поэтому лишь необыкновенный луч распространяется
в исходном направлении. Он появляется как луч, параллельный
падающему пучку, но пространственно отделенный от него. Приз-
мы Николя можно использовать только для поляризации в види-
мой области спектра, поскольку канадский бальзам не пропускает
УФ-облучение.
Призмы Глена—Томпсона изготавливают из кристаллов каль-
цита, которые устанавливаются таким образом, что оптическая
ось перпендикулярна фронтальной и тыльной поверхностям. По-
ляризаторы с воздушным зазором между призмами пригодны для
поляризации света в УФ-области, тогда как призмы, склеенные
канадским бальзамом, можно применять только для поляризации,
в видимой области.
Призмы Рошона — Волластона (рис. 10.11) изготавливают из
кристаллов кальцита или кусочков кварца, которые сначала раз-
резают, а затем скрепляют вместе глицерином или касторовым
маслом (прозрачны до 230 нм).
Обзорная литература: 468, 981, 1205, 1206, 1243, 1257, 1275, 1353, 1415.
10.4 . ОПТИЧЕСКИЕ МАТЕРИАЛЫ
В фотометрической аппаратуре решающим фактором является
спектральная прозрачность веществ, использующихся для изме-
рительных кювет, линз, призм и окон.
Характеристики пропускания многих материалов в УФ-области
представлены в табл. 10.2. Некоторые кристаллические вещества
имеют несколько полос поглощения. В табл. 10.3 приводятся пере-
чень кристаллов и их предельные полосы поглощения в коротко-
волновой области. Характеристики пропускания тех или иных ма-
териалов особенно важно знать при выборе материалов для окон,
предназначенных для использования в ИК- и УФ-спектроскопии.
Оптические волокнистые пучки представляют собой гибкие про-
зрачные волокна из кварца (для УФ-области) и синтетических
полимеров (для видимой области) и предназначены для «транс-
портирования света» в отдаленные части приборов или экспери-
ментальных приспособлений. Пропускание зависит от полного
внутреннего отражения от стенок (рис. 10.12).
Фильтры являются приспособлениями, которые поглощают, от-
ражают или отклоняют все частоты, кроме одной частоты или об-
ласти частот, от оптического пути. Существуют различные виды
фильтров.
1. Абсорбционные стеклянные фильтры селективно отсекают
часть спектра источника света. На рис. 10.13 приведены кривые
пропускания для ряда фильтров такого типа.
Оптические
159
Таблица 10.2
Ориентировочные пределы длин волн, пропускаемых различными оптическими
материалами и водой при температуре, близкой к комнатной [О: 242]
Ориентировочная А для
указанного процентного
Материал Толщина, мм пропускания, А
50% зо% ю%
Оконное стекло (обычное) 1 3160 3120 3020
3 3300 3230 3140
10 3520 3420 3300
Оптическое стекло (белый 1,8 3270 3200 3090
крон) Пирекс (корнинг 774) 1 3060 2970 2800
2 3170 3090 2970
4 3300 3190 3100
Корекс D [пирекс, корнинг 1 2780 2670 2500
9-53 (9700)] 2 2880 2800 2670
4 3040 2920 2810
Корекс А 2,9 2480 2430 2400
Викор 790 2 > 2540
Викор 791 1 2150 2130 2120
2 2230 2170 2130
4 2360 2250 2170
Кварц, кристаллический 5 1850
Ю 1930 1920 1860
Кварц, прозрачный плавленый 10 1940 1810 1720
Супрасил 10 1700 1680 1660
Сапфир 0,5
Флуорит (CaF2), природный 5 1350
10 1570 1450 1380
CaF2, синтетический 3 1220
Фторид лития (синтетиче- 5 1070
ский) 10 1420 1270 1150
Плексиглас (полиметилмет- 2,5 3220 3100 2970
акрилат) 5,0 3380 3250 3110
10,0 3500 3420 3260
Вода (дистиллированная) 20 1880 1860 1850
40 1920 1880 1860
80 2020 1940 1880
2. Интерференционные фильтры — это оптические фильтры,
основанные на интерференции между многократно отраженными
лучами (рис. 10.14). Их изготавливают из чрезвычайно тонких
слоев диэлектрика, расположенных сэндвичеобразно между полу-
отражающими металлическими (обычно серебряными) пленками.
Интерференционные фильтры можно получить для области от
~2000 см-1 (5 мкм) до ближнего ультрафиолета включительно.
Характеристики полос пропускания интерференционных фильтров
показаны на рис. 10.15.
3. Жидкие трансмиссионные фильтры готовят растворением
неорганических солей (NaNOa, К2Сг2О7) или органических
160
Глава 10
Рис. 10.12. Внутреннее отражение в пучке оптических волокон.
Рис. 10.13. Кривые пропускания некоторых абсорбционных фильтров.
Рис. 10.14. Поперечный разрез интерфе-
ренционного фильтра.
Рис. 10.15. Характеристики полос
пропускания интерференционных
фильтров. Ашах — длина волны пика;
Т — пропускание; HW — ширина на
полувысоте пика.
красителей, использующихся в пищевых продуктах, в воде; они
пригодны для использования в области 4000—7200 А.
4. Жидкие фильтры с узкой полосой пропускания предназначе-
ны для отделения узких спектральных областей проходящего света.
Оптические методы
161
Таблица 10.3
Предельные полосы поглощения некоторых кристаллов
в коротковолновой области
Материал Предельная полоса, нм Материал Предельная полоса, нм
Кварц (SiO2) 120 TiO2 414
Сапфир (А!20з) 140 Agl 444
Кальцит (СаСО3) 200 ZnSc 480
NaCl 210 CdS 510
Алмаз 232 GaP 550
Zn S 345 Cu2O 590
ZnO 393 GaAs 885
AgCl 390 PbS 3550
Таблица 10.4
Состав и характеристики фильтров с узкой полосой пропускания [П: 7140]
№ Пропускание, А । | Состав фильтра
1 2600—3900 200 г СоС12-6Н2О 4- 100 г NiCl2-6H2O на 1 л смеси’ 65% этанола и 35% воды, содержащей 1 моль/л НС1
2 2900-3900 200 г СоС12-6Н2О + 100 г NiCl2-6H2O на 1 л смеси 55% ди- метилформамида и 45% воды, содержащей 1 моль/л НС1
3 3100-3900 200 г СоС12-6Н2О 4- 100 г NiCl2-6H2O на 1 л смеси 35% эта- нола, 25% ацетона и 40% воды, содержащей 1 моль/л НС1 200 г СоС12-6Н2О на 1 л смеси 70% этанола и 30% воды, содержащей 1 моль/л НС1
4 2600—4600
5 2900-4500 200 г СоС12-6Н2О на 1 л смеси 65% диметилформамида и 35% воды, содержащей 1 моль/л НС1
6 3100-4600 200 г СоС12-6Н20 на 1 л смеси 75% ацетона и 25% воды, содержащей 1 моль/л НС1
а Смешанный растворитель получали следующим образом: в мерную колбу объемом
100 мл вносят 65 мл 99,5%-ого этанола, добавляют 20 мл 5 М НС1 и доводят объем до метки
водой. Ту же самую процедуру использовали при приготовлении других смесей раствори-
телей.
Таблица 10.5
Состав растворов, которые используются для фильтров,
предназначенных для выделения света с длиной волны 2537 А
ртутной лампы среднего давления [О: 242]
Толщина слоя раствора, см Компоненты
5 5 1 5 27,6 г NiSO4-6H2O на 100 мл водного раствора 8,4 г CoSO4-7H2O на 100 мл водного раствора 0,108 г 12 4- 0,155 г KI на 1000 мл воды С12 (газ) при 1 атм и 25 °C
§ Зак 238
162
Г лава 10
В табл. 10.4 приведены данные о составе таких фильтров и их
характеристики. Для выделения света с длиной волны 2537 А ис-
пользуется особый состав фильтра, он указан в табл. 10.5.
В ряде публикаций можно найти сведения о различных кон-
струкциях и приложениях волновой и геометрической оптики.
Обзорная литература 242, 647, 709, 744, 782, 813, 873, 874, 981, 1086, 1230
Периодическая литература 4223, 7140
10.5. ИСТОЧНИКИ СВЕТА
Для спектроскопических измерений желательно иметь постоянный
источник сравнительно невысокой интенсивности Интенсивность
источника (число фотонов, выделяющихся в секунду) должна быть
стабильна во времени.
При поставке многих приборов в них отсутствуют спектры из-
лучения ламп, которые в них использованы Для определения
Испытываемая
Лампа,
-> Монохроматор - > Детей, тор [-> Фотометр
Рис 10 16 Блок-схема прибора для измерения спектров излучения источников
света
спектров излучения применяют фотометры (радиометры). Суще-
ствует два типа фотометров:
а) фотодиодные радиометры (фотометры), показывающие сиг-
нальный ток от фотодиода, сигнальный ток пропорционален под-
водимому излучению;
б) фотоэлектронные умножительные радиометры, которые по-
казывают сигнальный ток от фотоумножителя, сигнальный ток
пропорционален подводимому излучению.
Эти приборы можно использовать для измерения относитель-
ного излучения в широких пределах.
Спектр лампы можно создать, используя монохроматор и ра-
диометр (рис. 10.16). Лампу закрепляют у входной щели моно-
хроматора. Интенсивность света, падающего на образец, измеряют
с интервалами от 2 до 50 нм в области длин волн 250—800 нм.
Затем строят зависимость интенсивности, которую показывает при-
бор, от длины волны, предварительно вводя поправку на измене-
ние дисперсии призмы в исследуемой области спектра. Данные о
дисперсии призмы можно получить из инструкции по монохрома-
тору, которая прикладывается к прибору.
Для фотохимических исследований желательно иметь источник
света, испускающий несколько интенсивных частот.
Обзорная литература: 451, 1227.
Оптические методы
163
10.5.1. Дейтериевые источники света
Дейтериевая дуга концентрируется в источнике диаметром 1 мм,
который дает континуум, не содержащий пиков поглощения в об-
ласти от <180 до 400 нм (рис. 10.17). Суммарный выход в види-
Рис 10 17. Спектральный выход дейтериевого излучателя.
мой и ИК-областях очень мал, в результате чего уровни сигнала
света также низки. Дейтериевые источники света используются во
многих стандартных УФ-спектрометрах.
10.5.2. Лампы на инертном газе
Лампы на инертных газах среднего давления являются хорошим
источником близко расположенных частот в ультрафиолетовой и
коротковолновой видимой частях спектра (табл. 10.6).
Таблица 10.6
Спектральная область, которую дают лампы
среднего давления е инертным газом
Инертный газ
Длина волны, нм
Аргон Гелий Криптон Неон Ксенон 390-440 380-450 430-450 340-360 460-480
Дуговые ксеноновые лампы высокого давления имеют непре-
рывный спектр от 190 до 750 нм, являясь прекрасными источни-
ками света для использования в ультрафиолетовой и видимой об-
ластях спектра.
10.5.3. Ртутные дуговые лампы
В основу классификации дуговых ртутных ламп положено давле-
ние внутри баллона дуги: низкое, среднее и высокое. Каждый тип
6*
164
Глава JO
характеризуется специфическим спектром и интенсивностью и при-
годен для использования в тех или иных областях спектроскопии
и фотохимии.
Ртутные лампы низкого давления, в которых давление паров
ртути составляет 10~3 мм рт. ст., дают излучение преимущественно
при двух так называемых резонансных частотах, 1849 и 2537 А.
Таблица 10.7
Распределение энергии в ртутных лампах низкого
и среднего давления [О: 242]
Относительная энергия
Длина волны, А ртутная лампа низкого давления ртутная лампа среднего давления
13 673 15,3
11287 — 12,6
10 140 — 40,6
5770—5790 10,14 76.5
5461 0,88 93,0
4358 1,00 77,5
4045—4078 0,39 42,2
3650—3663 0,54 100,0
3341 0,03 9,3
3126-3132 0,60 49,9
3022—3028 0,06 23,9
2967 0,20 16,6
2894 0,04 6,0
2804 0,02 9,3
2753 0,03 2,7
2700 — 4,0
2652—2655 0,05 15,3
2571 6,0
2537 100,0 16,6
2482 0,01 8,6
2400 —— 7,3
2380 8,6
2360 6,0
2320 — 8,0
2224 — 14,0
Линия 2537 А является наиболее интенсивной, тогда как интен-
сивность линии 1849 А зависит от условий возбуждения. Одна из
особенностей ламп низкого давления заключается в большой про-
должительности ее работы (2000—10000 ч и более) и надежности.
В ртутных лампах среднего давления давление ртутных паров
составляет от 10-3 мм рт. ст. до 1 атм. Интенсивность света лампы
возрастает с повышением давления, при этом повышается также
число различных испускаемых спектральных полос.
В табл. 10.7 сопоставлены спектры ртутных ламп низкого и
среднего давления.
Оптические методы
165
Ртутная
лампа
высокого
давления
__Источник
энергии
Воздух Воздух
Рис. 10.18. Вид корпуса ртутной лампы высо-
кого давления с воздушным охлаждением.
Кварцевая линза
Ртутные лампы высокого давления работают при внутренних
давлениях порядка нескольких сотен атмосфер. Спектры излуче-
ния характеризуются на-
личием широких полос в
отличие от дискретных
линий в спектрах ртут-
ных ламп низкого и сред-
него давления. Ртутные
лампы высокого давле-
ния применяются глав-
ным образом в фотохи-
мических исследованиях.
Средняя продолжитель-
ность работы такой лам-
пы составляет от 10 до
80 ч в зависимости от
эффективности охлажде-
ния и числа включений.
Эти лампы следует охлаж-
дать воздушным венти-
лятором или водой, что-
бы предотвратить разрыв
баллона. Необходимо,
чтобы вода циркулирова-
ла вокруг лампы с очень
большой скоростью, пред-
отвращающей образова-
ние пузырьков пара, и
была бы очень чистой,
чтобы обеспечить макси-
мальное пропускание и
минимальную электро-
проводность.
На рис. 10.18 и 10.19
показаны типичные кор-
пусы для ртутных ламп
высокого давления с воздушным и водяным охлаждением соответ-
ственно.
вентилятор
Ртутная
капля
Кварцевая
линза
Охлаждающая
вода —«
Источник
энергии
Ртутная лампа
высокого давления
Рис. 10.19. Вид корпуса ртутной лампы высо-
кого давления с водяным охлаждением.
Обзорная литература: 242, 451, 1086.
10.6. ЛАЗЕРЫ
Название «лазер» образовано первыми буквами английского вы-
ражения Light Amplication by Stimulated Emission of Radiation —
усиление света за счет вынужденного излучения.
Основные процессы, происходящие в лазерах, схематически по-
казаны на рис. 10.20.
166
Глава 10
1. Поглощение происходит при абсорбции атомом фотона.
Энергия последнего превращается во внутреннюю энергию атома,
т. е. электрон переходит на более высокий энергетический уровень.
Скорость поглощения = IBN0, (10.9)
где I — интенсивность излучения, В — коэффициент Эйнштейна для
индуцированного поглощения, No — число атомов в основном со-
стоянии.
Верхний Возбужденное
уровень 1 ————. состояние
Нижний Основное
уровень 0--------------Nq состояние
1 —I--
б—L-----
Помещение
Самопроизвольное Индуцированное
испускание испускание
Рис. 10.20. Различные абсорбционные и эмиссионные процессы.
2. Самопроизвольное испускание имеет место при возвращении
возбужденного атома в основное состояние. Обычно время жизни
возбужденного состояния составляет 10-9—10-4 с.
Скорость самопроизвольного испускания = ANlt (10.10)
где А — коэффициент Эйнштейна для самопроизвольного испуска-
ния, N\ — число атомов в возбужденном состоянии.
3. Индуцированное испускание наблюдается, когда дополни-
тельный фотон с энергией, точно равной энергии поглощенного
фотона, сталкивается с атомом, находящимся в возбужденном со-
стоянии. Этот фотон соединяется со вторым фотоном от атома в
возбужденном состоянии, что обусловливает усиление потока фо-
тонов. Волна второго фотона по фазе совпадает с волной, кото-
рая вызвала его выделение, что приводит к полностью когерент-
ному по фазе излучению.
Скорость индуцированного испускания = ICNh (10.11)
где С — коэффициент Эйнштейна для индуцированного испуска-
ния.
Оптические методы
187
При анализе этих трех процессов можно увидеть, что скорость
увеличения заселенности уровня 1 составляет
Скорость0>1 = IBN0, (10.12)
скорость снижения заселенности уровня 2 равна
Скорость! >о = A + 1СМ{, (10.13)
тогда при равновесии
IBNq^ANi + ICN! (10.14)
Необходимым условием для действия лазера является наличие
инверсной заселенности энергетических уровней, т. е.
ICN[ > IBN0, или N[ > Af0,
в противном случае поглощение будет преобладать над испуска-
нием. Для того чтобы создать условия для реализации инверсной
заселенности, необходимо достичь соответствующих параметров
Частично.
Рис. 10.21. Принцип действия лазера.
для действия лазера. Увеличение заселенности атомов в возбу-
жденном состоянии иногда называют оптической накачкой (опти-
ческим методом возбуждения).
Для усиления индуцированного испускания лазерное устройство
помещают в оптический резонатор — длинную трубку, закрытую
двумя зеркалами, одно из которых полупрозрачно (рис. 10.21).
Расстояние между зеркалами представляет собой целое кратное
требуемой длины волны. В оптическом резонаторе прирост энергии
наблюдается при заданной длине волны (резонансная частота), а
168
Глава 10
потеря энергии происходит при других длинах волн. Фотоны, дви-
жущиеся вдоль оси трубки, отражаются торцевыми зеркалами, то-
гда как фотоны, распространяющиеся в других направлениях, гиб-
нут. При движении фотонов по трубке индуцированное испускание
вызывает быстрый рост их интенсивности, усиливаемый при по-
вторных отражениях. Часть движущегося пучка может покидать
трубку через полупрозрачное зеркало.
Ниже приведены наиболее важные свойства лазеров.
1. Лазерное излучение является высокомонохроматическим
(узкая ширина линии).
2. Лазерные лучи исключительно интенсивны. Вся энергия ла-
зера концентрируется в очень узком параллельном пучке, имею-
щем относительно постоянную площадь сечения на больших рас-
стояниях.
3. Лазерные лучи, будучи сильно сфокусированными, позво^
ляют исследовать или облучать образцы очень малых размеров.
4. Лазерные лучи, будучи очень хорошо коллимированными,
позволяют упростить оптическую геометрию, использующуюся в
приборах для спектроскопии.
5. Лазерные лучи характеризуются четко выраженной поляри-
зацией, поэтому они особенно полезны для точных измерений де-
поляризации.
Внимание. На лазерный луч нельзя смотреть даже в темных
очках. Необходимо принимать меры предосторожности, чтобы при
установке зеркал лазерный луч не отражался в глаза.
Обзорная литература: 209, 279, 365, 393, 485, 497, 523, 695, 742, 811, 939,
1038, 1064, 1152, 1156, 1210, 1319, 1358, 1376.
10.6.1. Гелиево-неоновый лазер
Гелиево-неоновый лазер имеет оранжево-красное излучение при
длине волны 6329 А с выходной мощностью порядка нескольких
милливатт. Пропускание лазерного излучения имеет место между
энергетическими уровнями неона, гелий же используется для опти-
ческой накачки неона и создания инверсной заселенности. При
пропускании через гелий электрического тока его атомы переходят
в возбужденные состояния в результате столкновения со свобод-
ными электронами и затем ступенчато спускаются на соответ-
ствующие энергетические уровни. Те атомы, которые попадают на
уровни 23s и 2Js, остаются там в течение длительного времени.
Постепенно атомы собираются на тех уровнях, заселенность кото-
рых достаточно высока. При столкновении возбужденного атома
гелия с невозбужденным атомом неона возбуждение переносится
на последний. Две другие линии наблюдаются при 3,39 и 1,15 мкм
(рис. 10.22).
Типичный гелиево-неоновый лазер показан на рис. 10.23. В него
входит трубка из стекла пирекс (длиной 35 см и диаметром 2 мм)
Оптические методы
169
с электродами на боковой поверхности и окнами из плавленого
SiO2, установленными под углом Брюстера на концах трубки. В та-
ком виде выходящий лазерный луч является поляризованным. Ла-
зерную трубку вначале вакуумируют, затем заполняют гелием при
давлении ~2,5 мм рт. ст и неоном при 0,5 мм рт. ст. Электроды
Стадия I:
возбуждение
атомов Не
8 результате
столкновений
с электронами
Стадия 1Г-
перенос
энергии
при столкно-
вении Не и №
Стадия Z-
возбуждение
атомов Ne
в результате
столкновений
с электронами
Стадия III:
лазерное,
индуцированное
испускание
света,
Рис. 10.22. Схема энергетических уровней, играющих роль в гелиево-неоновом
лазере.
Окно с у г,
Брюстера
Полностью
отражающее
зеркало
HejNe
Трубка
с газовой
плазмой
Частично
пропускающее
зеркало
_ Лазерное
чх ,г ^индуцированное
-AS**- ^испускание
света
Высокочастотный
генератор
Рис. 10.23. Схема газового гелиево-неонового лазера.
подсоединены к источнику высокого напряжения постоянного тока
(4000 В). Зеркала устанавливаются вне трубки перпендикулярно
оси с точностью до нескольких секунд дуги. Одно зеркало обычно
имеет максимальную отражательную способность (99,99%), дру-
гое, находящееся на выходе, — 99,0%. Зеркала должны быть из
готовлены из многослойных диэлектрических пленок, осажденных
на оптически полированном материале. Металлические зеркала
170
Глава 10
нельзя использовать вследствие высокого поглощения (4%). Рань-
ше в лазерах устанавливались плоские зеркала, теперь большин-
ство газовых лазеров имеют сферические зеркала, которые легче
настраивать.
10.6.2. Аргоновый ионный лазер
Аргоновый ионный лазер имеет сине-зеленое излучение при 4880,
4965 и 5145 А с выходной мощностью порядка 500 мВт— 1 Вт или
более. К сожалению, срок службы аргонового ионного лазера не-
велик, что делает его в настоящее время слишком дорогим для
обычных работ.
Возбуждение в аргоновом ионном лазере происходит в две
стадии:
1. В результате неупругого столкновения с электронами атомы
аргона (с конфигурацией внешних электронов 3s23p6) переходят
в состояние Аг+ (с конфигурацией 3s23p5).
2. Следующее столкновение с электроном обусловливает пере-
ход внешних электронов иона аргона (Аг+) в одно из нескольких
4р-состояний, в результате последующего перехода в 4$-состояние
возникает линия излучения. Таким образом, переходы между раз-
личными уровнями в 4р- и 4$-состояниях сопровождаются излуче-
нием при 4880, 4965 и 5145 А.
Наиболее интенсивна линия при 4880 А, которая является един-
ственной, если количество аргона мало.
10.6.3. Рубиновый лазер
Рубиновый лазер относится к категории твердотельных лазеров.
Они обычно характеризуются более высокой выходной мощностью,
чем газовые лазеры. Инверсная заселенность в рубиновых лазерах
достигается путем оптической накачки.
Рубин представляет собой монокристалл оксида алюминия, в
котором небольшая часть алюминиевых ионов заменена на ионы
Сг3+. Глубина красного цвета зависит от концентрации ионов Сг3+:
а) светло-розовый рубин содержит Сг3+ в концентрации 0,05%
или менее;
б) темно-красный рубин содержит Сг3+ в концентрации до 1%.
При попадании белого света на кристалл рубина голубой и зеле-
ный цвета поглощаются, а красный отражается. Кроме этих полос
поглощения, объясняющих цвет рубина, имеются две узкие линии
флуоресценции при 6943 А (уровень 7?i) и 6928 А (уровень /?2).
Лазерный переход при 6943 А происходит обычно между уровнем
7?i и основным состоянием. При использовании специальных зеркал
с высокой отражательной способностью при 6928 А и низкой при
6943 А лазер можно перевести в режим работы с линией /?2
(рис. 10.24). При оптической накачке рубинового лазера исполь-
Оптические методы
171
зуют импульсные лампы, заполненные ксеноном. Поглощая свет,
ионы Сг3+ переходят с нижнего уровня на верхние, затем быстро
спускаются на /^-уровни с образованием фотонов. Уровни R мета-
. Синяя полоса
Зеленая полоса
5 Безызлуча- тельный переход Безызлучательный переход
1 Возбуждение, светом разрядной трибы. Лазерное индуцированное испускание света л 2.^6943 X Лазерное индуцированное испускание света о Л= мгвА
Рис. 10.24. Схематическая диаграмма энергетических уровней в рубиновом лазере.
Рубиновый
стержень
Цилиндрический эллиптический
резонатор
Ксеноновая
импульсная лампа
Рис. 10.25. Схематическое изображе-
ние оптического резонатора, содер-
жащего рубиновый стержень и ксено-
новую разрядную лампу.
стабильны, при этом их время жизни Rx составляет 3,2-10-2 с.
Если накачка продолжается, то ионы накапливаются на /?-уров-
нях, причем достигается сильная инверсная заселенность, приво-
дящая к возникновению лазерного эффекта.
Типичный рубиновый лазер показан на рис. 10.25. Ксеноновую
импульсную лампу устанавливают вдоль одной оси цилиндриче-
ского эллиптического резонато-
ра. Вдоль другой оси размещают
рубиновый стержень диаметром
3—20 мм и длиной 20—250 мм.
Эллиптический резонатор направ-
ляет почти весь свет от импульс-
ной лампы на рубиновый стер-
жень. При высоких температу-
рах, необходимых для оптической
накачки рубина, большинство
ксеноновых ламп не могут рабо-
тать в непрерывном режиме и
поэтому являются импульсными.
Один конец рубинового стержня
крыши» и полируется. Полуугол
этой «верхушки крыши» больше критического угла. Свет, распро-
страняющийся вдоль оси стержня и падающий на указанный его
конец, отражается на 180° и возвращается на ось. Другой конец
стержня отрезается под углом Брюстера, поэтому в какой-то сте-
пени поляризованный свет никак не отражается на этой поверх-
ности. Снаружи устанавливается выходное зеркало.
вырезается в форме «верхушки
172
Глава 10
Эффективность подобного рубинового лазера очень мала и
обычно не превышает величины 10~3. Так, например, если газо-
разрядные импульсы имеют мощность 1000 Вт-с, то выходная
мощность рубинового лазера менее 1 Вт-с. Выходную мощность
можно существенно увеличить путем использования специальных
модифицированных рубиновых лазеров, называемых гигантскими
импульсными лазерами (лазеры, генерирующие гигантский им-
пульс) с помощью метода модулированной добротности (Q-kom-
мутация).
Q-Коммутация получила свое название от коэффициента до-
бротности (сохранение энергии/потеря энергии в секунду), кото-
рый для резонатора коммутируется от низкого до высокого зна-
чения. Больших выходных мощностей можно достичь при пол-
ностью закрытом выходном зеркале и продолжении накачки.
В этом случае инверсная заселенность превысит пороговую вели-
чину, соответствующую условиям для открытого зеркала. Если те-
перь осуществить правильную ориентацию зеркала, то избыточная
инверсия дает очень высокий начальный поток фотонов и возни-
кает большой импульс при уменьшении инверсии до состояния
равновесия.
Существуют два пути импульсного уменьшения потерь:
1. Закрепление зеркала на вращающейся оси. В каждый мо-
мент времени, когда зеркало перпендикулярно оси стержня, на вы-
ходе возникает гигантский импульс. Таким способом можно соз-
дать импульс шириной порядка 100 нс и мощностью пика порядка
100 МВт.
2. Помещение в резонатор электрооптического затвора. Перво-
начально коэффициент поглощения высок. Свет от рубинового
стержня затем отбеливает затвор, например фталоцианин ванадия,
делая его прозрачным. Затем свет отражается зеркалом с высо-
кой отражательной способностью, и его инверсия превышает по-
роговую величину. Избыточная инверсия быстро переходит в им-
пульс с высокой мощностью пика, продолжительность импульса,
менее 20 нс.
10.6.4. Лазеры на органических красителях
Лазер на красителях относится к категории твердотельных лазе-
ров. Действие лазера основано на флуоресцентных переходах боль-
ших органических молекул в растворе, например в воде или
спирте. Выходная мощность их достигает 107—108 Вт, продолжи-
тельность импульса 10-9 с. Для получения излучения лазера испу-
скания в интервале длин волн от 3400 А до 1,2 мкм пригодны
сотни промышленных красителей (рис. 10.26). Красители, которые
могут применяться в качестве активных сред лазера, должны со-
держать хромофорные структуры (отмечены жирными линиями),
Оптические методы
173
Длина волны флуоресценции обычно зависит от размера хромо-
фора, причем с увеличением размера хромофора длина волны
возрастает, т. е. возникает более
красное излучение. Свойства кон-
кретного лазера на красителе
определяются спектральными и
молекулярными характеристика-
ми используемого красителя.
Действие лазера на красите-
ле зависит от электронных пере-
ходов в возбужденные синглет-
ное, а затем триплетное состоя-
ния. Лазерный процесс начинает-
ся с поглощения света от источ-
ника возбуждения (обычно ги-
гантского импульсного лазера
или специальных импульсных
ламп), в результате чего молеку-
лы красителя переходят из основ-
ного состояния (So) в вышележа-
щие возбужденные синглетные
состояния OS, 2S) (рис. 10.27).
Лазерное испускание излучения
Длина волны, нм
тЗОО
происходит из возбужденного
синглетного состояния (*S) в
основное состояние (So). Часть
молекул красителя будет участво-
вать в процессе интеркомбина-
-400
—500
-600
1000
Рис. 10.26. Схематическая диаграмма,
отражающая область длин волн, ха-
рактерных для различных красителей
(а) и лазеров (б) накачки.
ционной конверсии до триплетного состояния (JT). Молекулы, ко-
торые достигнут триплетного состояния, сохраняют его и исклю-
чаются из лазерного процесса, поэтому важно избежать интер-
комбинационной конверсии молекул в триплетное состояние. Этого
174
Глава 10
Синглетное состояние
О'§
gh
5 у
Синглетное состояние
^(нтеркомбинационная конверсия
f v (следует избегать)
ТЛ......... Триплетное состояние
Основное
состояние
Рис. 10.27. Схематическая диаграмма энергетических уровней в лазерах на кра-
сителях.
Полностью
отражающее
зеркало
Гигантский рубиновый
лазер, работающий
У в импульсном режиме
Полупрозрачное
зеркало
Выход лазера
на красителе
Кювета с раствором красителя
к-------------
Лазер на красителе
Гигантский рубиновый
лазер, работающий Полупрозрачное
в импульсном режиме зеркало
б
-> Выход лазера
-> на красителе
Кювета с раствором красителя
i^_-------------
Лаверна красителе
Рис. 10.28. Схема лазера на красителе,
а —поперечное возбуждение; б—продольное возбуждение.
Оптические методы
175
можно добиться путем использования источника возбуждения, ко-
торый достигает порога лазера до начала индуцированного испу-
скания. Для этого необходимы источники света, которые дают
мощность несколько сотен киловатт за долю микросекунды (это
гигантские импульсные лазеры или специальные импульсные лам-
пы). В последние годы разработана методика контроля концентра-
ции триплетного состояния с помощью химических гасителей и
механического воздействия.
Лазеры на красителях можно разделить на следующие три
группы:
а) с накачкой лазером; б) с возбуждением импульсными лам-
пами; в) непрерывного излучения.
Лазер на красителях с накачкой лазером показан на рис. 10.28.
Для его установки возможны: а) геометрия поперечного возбу-
ждения и б) геометрия продольного возбуждения.
При использовании гигантского рубинового или Nd-стеклян-
ного лазера можно создать излучение лазера на красителе в ин-
тервале длин волн от 3400 А до 1,2 мкм (рис. 10.26).
Обзорная литература: 545, 1152, 1235.
10.7. ОПТИЧЕСКИЕ ДЕТЕКТОРЫ
Оптические детекторы используются во всех фотометрических ме-
тодах и бывают следующих типов: а) фотоэмиссионные, б) фото-
проводящие, в) фотоэлектрические, г) тепловые, д) фотографиче-
ские. Спектральные характеристики ряда оптических детекторов
представлены в табл. 10.8.
10.7.1. Фотоэмиссионные детекторы
Фотоэмиссионные детекторы основаны на фотоэлектрическом эф-
фекте, который представляет собой испускание электронов щелоч-
ными металлами (цезий, натрий и калий), на которые попадает
световая энергия. Такой фотоэлектрический детектор называется
фотоэлементом (рис. 10.29). Катод покрывается одним из указан-
ных выше щелочных металлов. Фотон с энергией Е = hv ударяет
катод и вызывает испускание электрона. Если электростатический
потенциал между катодом и анодом положителен, то электроны
направляются к аноду и ток регистрируется амперметром.
Характеристикой фотоэлемента является кривая спектральной
чувствительности (рис. 10.30), в которой квантовая эффективность
фотоэлемента (Q) определяется как число электронов, испускае-
мых на один поглощаемый квант. Подробные данные по оптиче-
ским и электрическим характеристикам можно найти в описаниях
к детекторам.
Фотоумножители представляют собой устройства, которые уси-
ливают один фотоэлектрон на пять и более порядков величины,
176
Глава 10
Таблица 10.8
Спектральные характеристики некоторых детекторов излучения
Тип Чувствительный элемент Наилучший интервал длин волн, мкм
Фотонные детекторы
Трубка фотоумножителя Катод: оксиды металлов групп I и V 0,16-0,7
Вакуумный фотоэлемент Оксиды щелочных металлов 0,2-1
Фотоэлектрическая ячейка Полупроводник на металле 0,4-0,8
Фотопленка или фотопластин- Зерна AgX на эмульсии 0,2-1,2
ка
Ячейка с фотосопротивлением I. PbS, PbSe, InSb II. Ge, Си, Au или Zn акти- вированный (для работы при ~5 К) 0,7-4,5 2—15 или 2—100
Тепловые детекторы
Термопара Спай различных металлов с черненой полоской в каче- стве абсорбера 0,8-40
Болометр (мостиковый детек- тор) Проволочное сопротивление или стружка термистора с черненой полоской 0,9-40
Пневматическая ячейка (эле- мент Голея) Черненая мембрана или га- зовая камера 0,8-1000
Пироэлектрический детектор Сегнетоэлектрический кри- сталл с постоянной элек- трической поляризацией 0,8-1000
Рис. 10.29. Фотоэлемент, в котором
напряжение, дающее нулевой ток,
пропорционально частоте падающего
света.
Рис. 10.30. Спектральная чувствитель-
ность некоторых стандартных фотокатод-»
цых поверхностей.
Оптические методы
177
Кроме фотоэмиссионного катода, они имеют несколько динодов,
которые поддерживаются при последовательно возрастающих по-
тенциалах (рис. 10.31). Обычно при переходе от одного динода
к следующему разность потенциалов возрастает примерно на
100 В. Разность потенциалов обусловливает ускорение электронов
настолько, что удар каждого из них о поверхность динода вызы-
вает испускание трех-четырех вторичных электронов. Эти вторич-
ные электроны затем ускоряются при движении к следующему
Эмитированный.
Рис. 10.31. Схема измерительной системы с использованием фотоумножителя.
диноду, и этот процесс еще раз повторяется. Таким образом, один
электрон с катода может быть усилен до одного миллиона элек-
тронов на аноде после десяти таких стадий. Последний динод, ко-
торый поддерживается при высоком положительном напряжении
относительно катода, заземляется, вследствие чего напряжение на
выходе становится низким.
Из-за большого внутреннего усиления фотоумножительные
трубки используют лишь при низких интенсивностях света. При
повышении мощности экспозиция трубки освещением даже уме-
ренного уровня может приводить к необратимым изменениям по-
верхности катода. Поэтому фотоумножители всегда заключают в
светонепроницаемые корпуса с регулируемой величиной окошка.
Для работы фотоумножителя необходим источник высокого на-
пряжения (порядка 1200 В). Хотя многие фотоумножители рабо-
тают в интервале напряжений 900—1000 В, источник высокого
напряжения должен быть отрегулирован на интервал 500—1200 В.
Характеристика усиления фотоумножителя в большой степени за-
висит от высокого напряжения. Поэтому источник высокого на-
пряжения должен работать очень стабильно.
Рядом с фотоумножителем в непосредственной близости от него
устанавливают предварительный усилитель. Его функция заклю-
чается в превращении слабого электрического импульса (высокий
импеданс), подаваемого фотоумножителем, в сильные импульсу
178
Глава 10
(низкий импеданс), которые по проводам передаются на линейный
усилитель.
Для измерения очень низкой интенсивности света используется
аппаратура, называемая счетчиком фотонов. При низких интенсив-
ностях фотоусилительная трубка дает индивидуальные (отдель-
ные) сигналы тока, которые можно считать. Такие индуцируемые
фотонами импульсы нужно отличать от импульсов темнового тока,
возникающих внутри трубки по иным причинам. Амплитуда их су-
щественно ниже. Для пропускания импульсов с интенсивностью
выше минимальной можно использовать амплитудный дискрими-
натор импульса, который является эффективным усилителем с
дифференциальным вводом. Число проходящих импульсов реги-
стрируется соответствующим устройством прибора.
Обзорная литература: 87, 139, 470, 702, 837, 1275.
10.7.2. Фотопроводящие и фотоэлектрические детекторы
Фотопроводящие детекторы — это твердые детекторы, известные
как полупроводники, электрическое сопротивление которых при об-
лучении (освещении) существенно уменьшается.
Фотоэлектрические детекторы — это особый вид полупроводни-
ков, которые генерируют электрический потенциал при оптическом
возбуждении.
Обзорная литература: 9, 25, 980, 1334, 1414.
10.7.3. Тепловые детекторы
Тепловые детекторы — это твердотельные детекторы, которые при
поглощении света повышают свою температуру. Их применяют в
ближней, средней и дальней ИК-спектроскопии.
Термопара состоит из спая различных металлов. Термопары,
используемые как детекторы излучения, покрывают ламповой са-
жей, и они поглощают почти весь падающий свет.
Болометры — это детекторы, в которых сопротивление металла
или полупроводника зависит от окружающей температуры.
Термоэлемент — это большое число (пучок) термопар.
Элемент Голея (индикатор ИК-излучения)—это детектор, в
основу действия которого положено расширение нагретого газа
(этот тип детекторов одинаково чувствителен при всех длинах
волн).
Обзорная литература: 647, 782, 874, 980, 1215, 1229, 1391.
10.7.4. Фотопленка
Чувствительным детектором излучения является фотографическая
эмулъсия. Эмульсия представляет собой кристаллы галогенида се-
Оптические методы
179
Рис. 10.32. Зависимость плот-
ности изображения от логариф-
ма экспозиции для фотогра-
фической эмульсии.
ребра, диспергированные в прозрачной среде, например в жела-
тине, которая сильно набухает в воде. Эмульсию наносят на пленку
или тонкую стеклянную пластинку. При экспозиции кристаллы
галогенида серебра, подвергшиеся облучению, дают скрытое изо-
бражение. В результате последующей химической обработки
(проявления) получается черный налет серебра в месте скрытого
изображения. Поскольку кристаллы галогенида серебра очень ма-
ленькие, эмульсии обеспечивают высокое пространственное разре-
шение. Обычный рабочий интервал — это ультрафиолетовая и
видимая области, однако вместе со специальными добавками, по-
вышающими светочувствительность, этот интервал можно расши-
рить до ближней ПК-области (230—880 нм). В зависимости от
светочувствительности (низкая, сред-
няя и высокая) и контрастности (сред-
няя, высокая и очень высокая) произ-
водят различные эмульсии. Светочув-
ствительность определяется в услов-
ных единицах ASA, например 400 ASA
характеризует очень быструю эмуль-
сию, а 0,004 ASA — очень медленную
эмульсию. Контрастность выражается
величиной гамма (1—5). Характери-
стика эмульсии представлена кривой
на рис. 10.32. Плотность (D) налета
связана с логарифмом экспозиции, ко-
торая представляет собой произведе-
ние интенсивности излучения на вре-
мя. Контрастность эмульсии пропор-
циональна наклону кривой, а чувстви-
тельность связана с интервалом, где кривая является прямой ли-
нией. Быстрая низкоконтрастная эмульсия дает кривую с низким
наклоном, линейная часть которой соответствует очень малому
времени выдержки.
Если необходимо записать сигнал очень низкой интенсивности,
а продолжительная экспозиция неосуществима, то адекватную ха-
рактеристику нельзя получить даже при использовании быстрой
эмульсии той же контрастности. В этом случае можно использовать
технику превуалирования путем мгновенного облучения открытой
пластинки и пленки несколько раз на определенном расстоянии.
Затем обработанные таким способом фотоматериалы используют
для записи необходимого сигнала низкой интенсивности. Если пре-
вуалирование сделано правильно, то даже под действием радиации
очень низкой интенсивности суммарная характеристика будет рас-
полагаться в линейной области.
Для записи сигнала очень высокой интенсивности на пути луча
можно установить фильтры средней плотности или ступенчатый
вращающийся сектор, пропускание которого зависит от его
180
Глава 10
радиуса. Такой способ позволяет весьма точно ослаблять излу-
чение.
Плотность налета обычно определяется фотоэлектрическим ска-
нированием, для чего используется спектроденситометр. Перед тем
как рассчитать отношение плотности к внутреннему стандарту, фо-
новая плотность вычитается.
Обзорная литература: 772, 839, 1293, 1300.
Глава 11
ПОКАЗАТЕЛЬ ПРЕЛОМЛЕНИЯ
Обзорная литература: 125, 167, 263, 757, 912, 991.
Показатель преломления (показатель рефракции} (п) веще-
ства представляет собой отношение скорости света в вакууме к
его скорости в данном веществе и находится из закона Снелла
(рис. 11.1):
где а — угол падения, р — угол преломления.
Величина показателя преломления зависит не только от типа
вещества, но и от длины волны света (X) (рис. 11.2).
Показатель преломления вещества зависит от его молекулярной
структуры, эта зависимость известна под названием закона Ло-
ренца — Лоренца'.
ALr_L ~ А яр = (112)
п2 + 2 3 М ’ U1’2'
где Р — поляризуемость молекул,
Р = Ж (11.3)
W— число молекул в единице объема; а — поляризуемость одной
молекулы, или мера подвижности электронов в молекуле, а воз-
растает с увеличением числа электронов; р — плотность вещества;
Rx— молярная рефракция (или молекулярная рефракция) при
постоянной длине волны; М — молекулярный вес вещества.
При переходе от одного углеводородного полимера к другому
поляризуемость существенно не меняется. Из этого факта с учетом
уравнения (11.3) следует, что нет существенного различия и в по-
казателях преломления таких полимеров (обычно у углеводород-
ных полимеров п 1,5). Замена атомов водорода на галогены
приводит к изменению поляризуемости полимеров и соответственно
к изменению их показателя преломления (табл. 11.1).
Разность показателя преломления (dn) полимера в растворе
равна
dn = n — пъ (Н.4)
+ = (11.5)
где п — показатель преломления раствора; п\— показатель пре-
ломления растворителя; /?2— показатель преломления полимера..
182
Глава 11
Показатель преломления полимера в растворе зависит от тем-
пературы, длины волны света, природы растворителя, весовой
концентрации, молекулярного веса растворенного вещества. Зави-
Рис. 11.1. Геометрия рефракции
Рис. 11.2. Зависимость показателя
преломления (п) от длины волны
света (X).
света.
симость от молекулярного веса растворенного вещества наиболее
ярко выражена для низкомолекулярных полимеров (олигомеров).
При увеличении степени полимеризации выше 10 показатель пре-
Таблица 11.1
Показатели преломления ряда полимеров [О: 757]
Полимер Показатель преломления
Политетрафторэтилен 1,35—1.38
Полихлортрифторэтилен 1,39—1,43
Ацетат целлюлозы 1,46-1,50
Поливинилацетат 1,47—1,49
Полиметилметакрилат 1,485—1,4 )
Полипропилен 1,49
Поливиниловый спирт 1,49-1 ,53
Фенолформальдегидный полимер 1,5—1,7
Полиизобутилен 1,505-1,51
Полиэтилен 1,512-1,519 (-25 °C)
Полиизопрен (натуральный каучук) 1,519 (25 °C)
Полибутадиен 1,52
Полиизопрен (синтетический каучук) 1,5219 (20° С)
Полиамид 1,54
Поливинилхлорид 1,54-1,56
Полистирол 1,59 — 1,60
Поливинилиденхлорид 1,60-1,63
ломления отражает весовое количество полимера в единице объема
и практически не зависит от молекулярного веса исследуемого по-
лимера.
Показатель преломления
183
Удельный инкремент показателя преломления раствора (dn/dc)
определяется как
= (11.6)
de с х
где с — концентрация растворенного вещества.
Этот инкремент при с^О можно найти из уравнения Лорен-
ца — Лоренца:
dn («1+2)2 / р2/?2 Р1-^1
de 6П1Р2 \ М2 Mi
(П-7)
где pi и р2 — плотности растворителя и полимера соответственно;
ni — показатель преломления растворителя; и R2— молекуляр-
ные рефракции растворителя и повторяющихся звеньев полимера
соответственно; Mi и М2— молекулярные веса растворителя и по-
вторяющихся звеньев полимера соответственно.
Если измерения проводят при высоких концентрациях и раз-
личных температурах, то удобно выражать концентрацию как ве-
совую долю (w).
Удельный инкремент показателя преломления раствора (dn/dc)
можно также выразить в виде dn/dw.
тНЧЖ <и-8)
где с — pw— концентрация растворенного вещества; р — плот-
ность раствора; w — весовая доля растворенного вещества. При
очень низких концентрациях членом w можно пренебречь, и
уравнение (11.8) принимает вид
dn dn .
———р—_ (11.9)
dw r de v '
Показатель преломления . (/г), разность показателя преломле-
ния (dn) и удельный инкремент показателя преломления (dn/dc)
используются в основном в ультрацентрифугировании (гл. 8), све-
торассеянии (гл. 13), хроматографии (гл. 25) и двойном лучепре-
ломлении (разд. 35.3).
Обзорная литература: 125, 167, 753, 912.
Периодическая литература: 2034, 2114, 2131, 2257, 2328, 2329, 2691, 2692,
3476, 3477, 3767, 3997, 4041, 4042, 4173, 4174, 4224, 4440, 4819, 4835, 4967, 4968,
5082, 5493, 5998, 6020, 6031, 6091, 6249, 6277, 6873, 6986, 7316.
В литературе имеются подробные табличные данные об удель-
ных инкрементах показателя преломления для многих систем по-
лимер— растворитель [О: 155, 283, 401, 651],
184
Глава 11
11.1. ОПРЕДЕЛЕНИЕ ИНКРЕМЕНТА ПОКАЗАТЕЛЯ
ПРЕЛОМЛЕНИЯ
Различия в показателях преломления измеряются непосредствен-
но с использованием специальных дифференциальных рефракто-
метров как отклонение монохроматического пучка света, проходя-
щего через границу показателя преломления в секционной кювете
(рис. 11.3). Раствор помещают в полую призму 60° и окружают
чистым растворителем, находящимся в прямоугольной кювете.
Луч падающего
ч 'ста.
Рис. 11.3. Дифференциальный рефрактометр с секционной кюветой.
/ — линза; 2— призма; 3 —прямоугольная кювета; 4— окуляр микрометра; 5— перекрестие^
С помощью оптической системы изображение тонкой щели фоку-
сируется на перекрестии окуляра микрометра. Разность показа-
теля преломления (dri) можно найти из выражения
dn = n — 2/tg(a/2) ’ (11.10)
где п — показатель преломления раствора, — показатель пре-
ломления растворителя; d — отклонение, f — фиксированное фокус-
ное расстояние линзы; a — краевой угол кюветы.
Инкремент показателя преломления (dn/dc) можно получить
из наклона кривой зависимости п от с.
Дифференциальная кювета заполняется и опорожняется с по-
мощью шприца. Кювета должна быть плотно закрыта, чтобы не
было испарения жидкости, вызывающего температурный градиент
в кювете и изменение вследствие этого показателя преломления.
Кювету необходимо термостатировать с отклонением температуры
не более ±0,01 °C. Кювета имеет два фиксированных положения,
отличающихся на 180° друг от друга и ориентированных таким
образом, что переднее и заднее окна кюветы перпендикулярны
потоку света. Благодаря такому расположению возможно опреде-
ление разности отклонения, возникающего при прохождении лучом
сначала растворителя, а затем раствора (положение 1) или вна-
чале раствора и лишь затем растворителя (положение 2).
Вначале оба отделения кюветы заполняют растворителем. От-
клонение для системы растворитель — растворитель находят как
dl — d^ (в положении 2) —(в положении 1) (11.11)
Показатель преломления
185
После этого из одного отделения удаляют растворитель и вместо
него вводят раствор. Отклонение для системы раствор — раствори-
тель находят из выражения
d2 — d2 (в положении 2) — d2 (в положении 1) (11.12)
Отклонение для нулевого количества растворителя (d) составляет
d = d2-dx (11.13)
Разность показателя преломления (dn) можно найти из соотно-
шения
dn = kd, (11.14)
где k — константа кюветы.
Обзорная литература: 263.
Периодическая литература: 2038, 2574, 2712, 6282.
11.2. АВТОМАТИЧЕСКИЙ ДИФФЕРЕНЦИАЛЬНЫЙ
РЕФРАКТОМЕТР
В некоторых методах, например ГПХ, используют автоматический
дифференциальный рефрактометр (рис. 11.4). Свет от источника
проходит через щель и линзу, которые коллимируют пучок света.
Рис. 11.4. Автоматический диффе-
ренциальный рефрактометр.
/ — источник света; 2 — линза; 3 — поток
через кювету; 4—образец; 5 —эталон;
6 — зеркало; 7 —оптическая установка нуля;
8— фотоумножитель; 9 — усилитель; 10—
перьевой самописец.
Параллельный пучок света проходит через кювету, содержащую
образец и эталонную жидкость, и попадает на зеркало. Зеркало
отражает пучок снова через кювету с образцом и эталоном на
линзу, которая фокусирует его на детектор. Расположение сфоку-
сированного луча (а не его интенсивность) определяется углом
отклонения, образующимся вследствие различия в показателях
преломления в двух частях кюветы. При попадании луча на де-
тектор генерируется выходной сигнал. Этот сигнал усиливается и
записывается на самописце. С помощью ^специального оптического
стекла луч отклоняется в ту или другую сторону для установки
на нуль выходного сигнала. Дифференциальные рефрактометры
очень чувствительны к изменениям температуры. С увеличением
температуры увеличивается уровень шумов.
Периодическая литература: 2257, 2404, 4322, 5336, 5523.
186
Глава 11
11.3. ОПРЕДЕЛЕНИЕ УДЕЛЬНОГО ОБЪЕМА
РАСТВОРЕННОГО ВЕЩЕСТВА ИЗ ПОКАЗАТЕЛЯ
ПРЕЛОМЛЕНИЯ РАСТВОРА
Соотношение между удельным объемом растворенного вещества
(v) и инкрементом плотности (dp/dw) выражается как
и1-15)
где v — удельный объем растворенного вещества, р — плотность
раствора, w — весовая доля растворенного вещества.
Инкремент плотности определяется как dp/dw и связан с
dp/dc:
или
й-М-й!1-<>)] ом?)
Из уравнения Лоренца — Лоренца можно найти общее выра-
жение для зависимости между удельным инкрементом показателя
преломления и инкрементом плотности
7 = 001.18)
где п — показатель преломления, — удельная рефракция чистого
растворителя, равная
/?2 — удельная рефракция чистого растворенного вещества (поли-
мера), равная
индекс 1 относится к чистому растворителю, индекс 2 — к поли-
меру. Удельная рефракция (/?) — постоянная величина для дан-
ного вещества при определенной длине волны.
Уравнение (11.18) можно записать в виде
_ rt2 - 1 ( Г \
Р п^ + 2 k/?i + №-₽i)® )
(11.19)
Дифференцирование уравнения (11.19) по w дает зависимость ме-
жду инкрементом плотности и удельным инкрементом показателя
Показатель преломления 187
преломления (dn/dw):
dp _________________________1_______ / 6/2 \ dn
dw [/?t + (/?2 — /?i) зу ] \ (п2 + 2)2 7 dw
--------TL-?1-------(_(и 20)
[Ri + (R2-Ri)w]2 \n2 + 2j Ui.zuj
Расчет можно существенно упростить, используя графическую
экстраполяцию.
Периодическая литература: 2257, 2508, 3996, 4172, 4173, 5523, 5595,
5640, 6053, 6249, 6251.
Глава 12
ОПТИЧЕСКАЯ АКТИВНОСТЬ
В МАКРОМОЛЕКУЛЯРНЫХ СИСТЕМАХ
Обзорная литература: 3, 115, 175, 232, 241, 274, 289, 329, 330, 374, 467,
682, 693, 844, 849, 945, 991, 1159, 1295, 1343, 1418, 1439.
Оптической активностью характеризуются полимеры, содержа-
щие асимметрический атом углерода в основной или боковой цепи,
например поли-а-аминокислоты —(—NH—CHR—СО—)п— или
поли-3-метилпентен-1 соответственно.
Оптическая активность заключается во вращении плоскости ли-
нейно-поляризованного света вокруг оси распространения луча.
Это явление наблюдается при прохождении света через некото-
рые полимеры в твердом и жидком состоянии, объяснить его можно
взаимодействием между веществом и право(D)- или лево (L) вра-
щаемым поляризованным светом.
Для проявления в полимерах оптической активности необходи-
мо, во-первых, чтобы макромолекула не обладала зеркальной сим-
метрией и, во-вторых, чтобы число структурных элементов, имею-
щих асимметрическую структуру определенного D- или L-типа, не
было одинаковым; в противном случае, даже если отдельный эле-
мент структуры оптически активен, суммарный эффект будет равен
нулю, так как вращение, обусловленное D-элементами, компенси-
руется равным по величине и противоположным по знаку враще-
нием, вызываемым L-элементами.
Присутствие асимметрических центров может быть обнаружено
и оценено с помощью методов дисперсии оптического вращения
(кругового двойного лучепреломления), когда показатели прелом-
ления неодинаковы (пъ Ф пС), и молекулярного кругового дихроиз-
ма при различии в коэффициентах поглощения (eD ¥= sL).
Подробные данные об оптически активных полимерах можно
найти в Polymer Handbook [О: 526].
12.1, ДИСПЕРСИЯ ОПТИЧЕСКОГО ВРАЩЕНИЯ
Дисперсией оптического вращения (ДОВ) называется зависимость
угла вращения плоскости плоскополяризованного света оптически
активным полимером от длины волны падающего света.
Угол вращения плоскости поляризации (а) равняется
a = ^-(nL-nD), (12.1)
где а выражается в градусах (СГС) или радианах (СИ); d — тол-
щина слоя оптически активного образца, через который проходит
Оптическая активность в макромолекулярных системах
189
свет; Ml и nD — показатели преломления лево- и правовращаемого
поляризованного света.
Удельное вращение, или оптическая активность, ([a]f) опреде-
ляется как
= (12.2)
где а — угол [в градусах (СГС) или радианах (СИ)], измеряемый
в поляриметре при температуре Т при использовании монохрома-
тического света с длиной волны X; с — концентрация раствора
оптически активного полимера [г/см3 (СГС) или кг/м3 (СИ)]; / —
толщина слоя раствора полимера [дм (СГС) или м (СИ)].
Молярное вращение (или молекулярное вращение) ([<₽]() опре-
деляется как
[<р]1 = 10а/с,/ = М [а]{/1000 (12.3)
[град• см2• дмоль-1 (СГС) или радиан-м2-моль-1 (СИ)], где М —
молекулярный вес исследуемых оптически активных соединений
(в случае полимеров М — молекулярный вес элементарного звена
макромолекулы); а — концентрация раствора оптически активного
вещества [дмоль/л (СГС) или моль/м3 (СИ)]; / — толщина об-
разца [дм (СГС) или м (СИ)].
В современных спектрополяриметрах можно измерять углы а
даже порядка миллиградусов (СГС) или миллирадиан (СИ) при
толщине образца порядка 1 мм, тогда как в оптических поляри-
метрах толщина образца около дециметра.
Для того чтобы иметь возможность сравнивать данные, полу-
ченные в разных растворителях, необходимо вводить поправоч-
ный коэффициент на показатель преломления растворителя. Мо-
лярное вращение, полученное с учетом такой поправки, называется
приведенным молярным вращением [(Фпр)[] и составляет
/ 3 М[а](
(ФпР)х~ (/г2+ 2) 100- (12.4)
[град-см2-дмоль-1 (СГС) или радиан• м2-моль-1 (СИ)], где п —
показатель преломления растворителя.
12.2. КРУГОВОЙ ДИХРОИЗМ
Круговой дихроизм (КД) представляет собой неодинаковое в за-
висимости от длины волны поглощение плоскополяризованного
света оптически активным полимером.
Для оптически активного полимера молекулярный круговой ди-
хроизм можно найти по уравнению
lM = ®L-eD = i(lg^-lg^) = 2£^- (12.5)
190
Глава 12
[град-см2-дмоль“1 (СГС) или радиан • м2-моль-1 (СИ)], где eL
и 8d — различные молярные коэффициенты поглощения для лево-
и правовращаемого поляризованного света при длине волны X;
/о — интенсивность падающего света с длиной волны X; Л и Id —
различные интенсивности лево- и правовращаемого поляризован-
ного света с длиной волны X; / — толщина образца [см (СГС)
или м (СИ)]; с — концентрация раствора оптически активного об-
разца [моль/л (СГС) или моль/м3 (СИ)].
Иногда используется термин молекулярная эллиптичность
а<):
[е]1 = 3300 [Ае]1 (12.6)
12.3. ЭФФЕКТ КОТТОНА
Зависимость дисперсии оптического вращения и кругового ди-
хроизма от длины волны обозначается как эффект Коттона и мо-
жет быть представлена в форме трех различных кривых дис-
персии:
1. Аномальная кривая дисперсии вращения (рис. 12.1). Важ-
нейшей характеристикой дисперсной полосы является то, что ин-
Длима волны,
Рис. 12.1. Типы аномальных кривых дисперсии.
1 — дисперсия оптического вращения; 2—круговой дихроизм.
тенсивность оптической активности в центре полосы равна нулю,
простираясь по обе стороны от него с измеряемой интенсивностью.
Кривые называются положительными и отрицательными в зависи-
мости от того, что наблюдается при большей длине волны: пик
или впадина, иногда называемые положительным или отрицатель-
ным экстремумом (соответственно).
2. Плавная кривая дисперсии (рис. 12.2). Такая кривая харак-
теризуется увеличением вращения при уменьшении длины волны
и называется положительной или отрицательной в зависимости от
Оптическая активность в макромолекулярных системах
191
того, поднимается она или спускается при уменьшении длины
волны.
3. Кривая множественного эффекта Коттона. На кривых этого
типа имеется несколько пиков и впадин. Они характерны для длин
Рис. 12.2. Типы плавных кривых дисперсии.
1 — положительная кривая; 2—отрицательная кривая.
волн, связанных с множественными полосами поглощения. Разли-
чают также внутренний (характеристический) эффект Коттона,
обусловленный асимметрическими хромофорами, уже присутствую-
щими в макромолекуле, и внешний эффект Коттона, обусловлен-
ный индуцированными асимметрическими хромофорами.
12.4. СПЕКТРОПОЛЯРИМЕТРЫ
Для записи дисперсии оптического вращения и кругового дихроиз
ма используются выпускаемые промышленностью спектрополяри
метры, имеющие приставки для исследования кругового дихроизма.
На рис. 12.3 показаны основные части спектрополяриметра
Свет от монохроматического источника проходит через поляриза-
тор, становясь плоскополяризованным, т. е. колебания его проис-
ходят только в одной плоскости, ориентация которой определяется
ориентацией поляризатора. Затем свет проходит через исследуе-
мый образец, после чего попадает на анализатор, представляющий
собой призму, аналогичную поляризатору. В простых поляримет-
рах детектором является человеческий глаз или электровакуумный
фотоэлемент.
Интенсивность света, достигающего детектор, меняется при
вращении анализатора, являясь минимальной, когда плоскость
пропускания анализатора находится под углом 90° к плоскости
поляризации падающего на него света. Вообще говоря, нуль поля-
риметра устанавливается вначале в отсутствие оптически актив-
ного образца, при заполнении кюветы только одним растворителем.
Затем заполняют кювету раствором образца, после этого опреде-
ляют угол, на который нужно повернуть анализатор, чтобы вернуть
192
Г лава /2
Интенсивность зависит 9
1
Рис. 12.3. Оптическая система спектрополяриметра. ----------_
/ — источник света; 2 — конденсорная кварцевая линза; 3 — монохроматор; 4— коллиматор;
5—поляризатор; 6 — трубка с образцом; 7—анализатор; 8 — фотоумножитель; 9— усилитель;
10 — самописец.
Рис. 12.4. Оптическая система спектрополяриметра с механической установкой
нуля.
1 — источник света; 2—коиденсорная кварцевая линза; 3—монохроматор; 4— коллиматор;
'—поляризатор; 6 — модулирующий мотор; 7—трубка с образцом; 8 — анализатор; 9—серво-
мотор; 19 — фотоумножитель; // — усилитель; 12— самописец.
193
—-te.
Оптическая активность в макромолекулярных системах
свет в нулевое положение; этот угол и будет характеристикой
оптического вращения раствора.
В автоматически записывающих спектрополяриметрах ис-
пользуется одна из следующих двух систем нулевого от-
счета:
1. Устройство с механической балансировкой нуля, в котором
анализатор или поляризатор повертывается с помощью сервоме-
ханизма (рис. 12.4).
2. Устройство, в котором для установки нуля используется элек-
трический фарадеевский эффект; положения анализатора и поля-
Рис. 12.5. Оптическая система спектрополяриметра с электрической установкой
нуля, основанной на эффекте Фарадея.
1 — источник света; 2 — конденсорная кварцевая линза; 3—монохроматор; 4 — коллиматор;
5—поляризатор; 6 — ячейка модулятора переменного тока; 7—трубка с образцом; 8 — ком-
пенсаторная ячейка постоянного тока; 9 — анализатор; 10— фотоумножитель; // — усилитель;
12 — самописец.
ризатора являются фиксированными, а вращение, вызываемое
образцом, компенсируется электрически с использованием эф-
фекта Фарадея (в магнитном поле оптическая активность ис-
чезает).
Кювета Фарадея представляет собой цилиндрическую ячейку,
обычно заполненную водой, которую помещают внутри катушки,
подсоединенной к постоянному току (рис. 12.5). Плоскость поля-
ризации поляризованного света, проходящего через кювету, вра-
щается. Если катушку подсоединить к источнику переменного тока,
7 Зак. 238
104
Глава 12
то направление плоскости поляризации будет осциллировать с той
же частотой, что и переменный ток.
Измерение кругового дихроизма оптически активного поли-
мера предусматривает оценку разности между коэффициентами
поглощения образца в право- и левовращаемом поляризованном
свете. Для получения циркулярно-поляризованного света световой
поток должен сначала быть плоскополяризованным, после чего его
надлежит пропустить через приспособление, которое расщепляет
его на право- и левовращаемые поляризованные компоненты. Это
достигается путем торможения одной компоненты относительно
другой точно на одну четверть длины волны. Известны два типа
устройств для расщепления плоскополяризованного света на цир-
кулярно-поляризованные компоненты:
1. Призменные модуляторы, например ромб Френеля, которые
помещают на пути луча сравнения. Помещая образец в оба луча
спектрофотометра, автоматически измеряют разность в поглоще-
нии, обусловленном право- и левовращаемым поляризованным
светом.
2. Электрооптические модуляторы, например кювета Покелса,
которая представляет собой вырезанную по оси z пластинку пер-
вичного кислого фосфата аммония, тетрагональный кристалл кото-
рого в отсутствие электрического поля имеет только одну оптиче-
скую ось (ось г, вдоль которой проходит световой луч), а в элек-
трическом поле — две оптические оси, т. е. проявляется эффект
Покелса. Приложением переменного тока высокого напряжения
(порядка киловольт) перпендикулярно оси z индуцируемые элек-
трооптически оси будут попеременно располагаться под углом +45
и —45° к первоначальной плоскости поляризации. В результате
плоскополяризованный свет сначала будет превращаться в право-
вращаемый, а затем в левовращаемый поляризованный свет в
течение одного цикла переменного тока. Если образец является
циркулярно-дихроичным, т. е. по-разному поглощает право- и ле-
вовращаемый поляризованный свет, то световой луч, контролируе-
мый детектором, будет эллиптически поляризованным. Таким об-
разом, электрооптические модуляторы обеспечивают прямое изме-
рение кругового дихроизма.
Для измерения- дисперсии оптического вращения и кругового
дихроизма используются кюветы следующих двух видов: целиком
закрытые кюветы и кюветы с завинчивающейся крышкой. Важно,
чтобы кювета не деформировалась. Деформации можно обнару-
жить по отклонению от линейности базовой линии, когда кювета
заполнена водой. Кюветы необходимо вставлять в прибор все-
гда только одной и той же стороной по отношению к световому
пучку.
Калибровку прибора следует проводить с использованием рас-
твора камфорсульфокислоты (1 мг/мл) в воде в кювете толщиной
Оптическая активность в макромолекулярных системах
195
1 см, которая имеет пик кругового дихроизма при 290 нм с ам-
плитудой +0,009 единиц поглощения. В спектре дисперсии опти-
ческого вращения такая кювета имеет амплитуду 0,4° с пиком
при 305—270 нм.
12.5. ИСПОЛЬЗОВАНИЕ МЕТОДОВ ДИСПЕРСИИ
ОПТИЧЕСКОГО ВРАЩЕНИЯ И КРУГОВОГО
ДИХРОИЗМА В ИССЛЕДОВАНИИ ПОЛИМЕРОВ
Исследования ДОВ и КД имеют особенно большое значение при
изучении оптически активных полимеров, например винильных
полимеров с оптической активностью в основной цепи, простых и
сложных полиэфиров, полиальдегидов и биополимеров, например
полипептидов и белков в а-спиральной конфигурации.
Обзорная литература: 175, 427, 682, 693, 931, 1041, 1173, 1418, 1439.
Периодическая литература: 2003, 2110, 2111, 2543, 2648, 2716, 2809, 2857,
2942, 2943, 2980, 3125, 3130, 3146, 3708—3713, 4175, 4654, 5076, 5119, 5535, 5644,
5645, 5838—5840, 6059, 6292, 6293, 6816, 6839, 6892, 7222, 7291, 7299.
Глава 13
СВЕТОРАССЕЯНИЕ
Обзорная литература: 99, 125—127, 166, 236, 263, 264, 309, 320, 381, 382, 425,
500, 650, 733—735, 776, 797, 846, 899, 943, 994, 995, 1016, 1250, 1289, 1308, 1311.
В идеальной гомогенной среде свет не рассеивается, если же
свет проходит через неоднородную среду, он рассеивается во всех
направлениях. Растворы макромолекул всегда можно рассматри-
вать как негомогенные среды. Из-за беспорядочного диффузного
движения макромолекул образуются области различной концен-
трации. Такие различающиеся по концентрации участки раствора
имеют разные диэлектрические константы и как следствие различ-
ные показатели преломления относительно всей массы жидкости,
в результате они выступают в качестве центров рассеяния.
Различают два вида светорассеяния.
1. Упругое светорассеяние (рэлеевское рассеяние), характери-
зующееся тем, что падающий и рассеянный свет имеют одну и ту
же частоту (рэлеевская линия). Интенсивность рэлеевского света
(интенсивность рэлеевской линии) оценивается обычно широко-
угловой (разд. 13.1) и малоугловой спектроскопией (разд. 13.3).
При движении центры рассеяния испытывают низкочастотный
доплеровский сдвиг по отношению к частоте падающего света,
в результате чего рассеянный свет перестает быть чисто монохро-
матическим (квазиупругое рассеяние). Вместо одной рэлеевской
линии наблюдается рэлеевский пик. Форму пика можно оценить
с помощью спектроскопии биения света, называемой также спек-
троскопией рэлеевских широких полос (разд. 13.4).
2. Неупругое рассеяние (рэлеевско-бриллюэновское рассеяние),
характеризующееся тем, что частота рассеянного света отличается
от частоты падающего света. Подобное светорассеяние оценивается
с помощью спектроскопии Рэлея — Бриллюэна (разд. 13.5).
Свет, который рассеивается частицами большого размера, мож-
но увидеть невооруженным глазом; это явление называется эффек-
том Тиндаля.
Интенсивность рассеянного света в разбавленных растворах
полимеров составляет всего 10-4 от интенсивности падающего све-
та, измерить такую слабую интенсивность можно только при ис-
пользовании фотоумножителя. При взаимодействии видимого света
с частицами индуцируется осциллирующий диполь, испускающий
рассеянный свет.
В случае вертикально поляризованного пучка света диполи
осциллируют в г-направлении (рис. 13.1, а).
Светорассеяние
197
В случае горизонтально поляризованного света диполи осцил-
лируют в направлении оси у (рис. 13.1,6).
Существует следующая зависимость между углами 0О и 0^:
sin2 0У + sin2 0Л = 1 + cos2 0, (13.1)
где 0 — угол между лучом рассеянного света и падающим лучом.
Если рассеивающая частица изотропна, то вертикально поля-
ризованный падающий свет дает лишь вертикально поляризован-
z рассеяния света.
Интенсивность
рассеяния света
Рис. 13.1. Рассеяние света маленькими частичками с вертикально (а) и гори-
зонтально (б) поляризованным светом.
ный рассеянный свет, тогда как горизонтально поляризованный па-
дающий свет дает только горизонтально поляризованный рассеян-
ный свет.
Если рассеивающая частица анизотропна, то вертикально (или
горизонтально) поляризованный падающий свет дает как верти-
198
Глава 13
кально, так и горизонтально поляризованный рассеянный свет.
Этот эффект называется деполяризацией рассеянного света. Рас-
сеяние таким образом является большим, чем теоретически
ожидаемое. Деполяризацию можно учесть путем введения попра-
вочного коэффициента, так называемого фактора Кабанна. Для
макромолекулярных растворов этот коэффициент обычно очень
близок единице.
Обзорная литература: 236.
Периодическая литература: 2351, 3221, 4130.
13.1. ШИРОКОУГЛОВОЕ РАССЕЯНИЕ СВЕТА
13.1.1. Рассеяние маленькими частицами в растворе
Если величина рассеивающих частиц мала по сравнению с длиной
света (менее Х/20), то интенсивность рассеянного света (7е) оди-
накова во всех направлениях (сферическая симметрия) (рис. 13.2).
Рис. 13.2. Диаграмма интенсивности рассеяния света 70 для частиц размером
менее Х/20.
Интенсивность рассеянного света (/о) пропорциональна поля-
ризуемости (а) и молекулярному весу (Л1) рассеивающих частиц
и составляет
/0 «= 8л4а270# (1 + cos2 0)/Z2A4 V, (13.2)
где /о — интенсивность падающего света, X — длина волны падаю-
щего света, / — расстояние от источника рассеяния, при котором
измеряется I®.
N/V = cNa/M, (13.3)
где N—число рассеивающих частиц; V — объем, занимаемый рас-
сеивающими частицами; с — концентрация рассеивающих частиц
в растворе; Na — число Авогадро (разд. 40.3); М — молекулярный
вес рассеивающих частиц; 0 — угол между рассеянным и падаю-
щим светом; а — поляризуемость рассеивающих частиц:
niM dn
а ~ 2nNA de ’
(13.4)
Светорассеяние
199
п\ — показатель преломления растворителя; dn/dc— удельный ин-
кремент показателя преломления (гл. 11).
Комбинируя уравнения (13.2) и (13.4), можно написать
^е = /е/2//0 = /С(1 +соз20)Ж (13.5)
где известно как рэлеевское отношение; К—константа, равная
/ dn \2
(13.6)
13.1.2. Определение молекулярного веса маленьких частиц
методом светорассеяния
Для небольших изотропных частиц (диаметром менее ?t/20) мо-
лекулярный вес можно найти из уравнения Дебая
Rc/R^X/M, (13.7)
где К— константа, определяемая уравнением (13.6); с — концен-
трация рассеивающих частиц в растворе; Re— рэлеевское отно-
шение, которое можно найти из уравнения (13.5); М — молекуляр-
ный вес рассеивающих частиц.
Рассеяние света молекулами определяется следующей зависи-
мостью:
= (13.8)
где /о—интенсивность падающего света; I — интенсивность света,
прошедшего через образец; / — толщина образца, т—мутность об-
разца.
Мутность (т) можно выразить как интегрированное рэлеев-
ское отношение:
л
т = 2л Rq sin 8 dQ, (13.9)
о
где Re— рэлеевское отношение, определяемое уравнением (13.5),
0 — угол рассеяния. Для небольших изотропных частиц диаметром
менее Х/20 выражение для мутности равно рэлеевскому отноше-
нию, измеренному при угле рассеяния 0—90° и экстраполирован-
ному к 0 = 0ч:
т = -^/?9о.=-^-/?о« (13.10)
Молекулярный вес можно найти из следующего уравнения
Дебая-.
Яс/т = (1/М) + Л2с+ (13.11)
где /7=16лК/3 [К—константа, определяемая из уравнения
(13.6)]; с—концентрация рассеянных частиц в растворе; Д2 —
второй вириальный коэффициент (разд. 2.10). Зависимость Нс/ъ
200
Глава 13
от с представляет собой прямую линию, отсекающую hj оси орди-
нат отрезок \/М (рис. 13.3). Наклон tg а — А2 дает второй вири-
альный коэффициент.
Обзорная литература: 320, 1250.
Периодическая литература: 3106.
Рис. 13.3. Зависимость Hcfx от кон-
центрации (с).
13.1.3. Рассеяние растворами макромолекул
Для макромолекул, имеющих диаметр клубка больше Х/20, напри-
мер виниловых полимеров со степенью полимеризации выше 500,
Рис. 13.4. Диаграмма интенсивно-
сти рассеяния света /одля частиц
е размером более %/20.
Рис. 13.5. Рассеяние света от различных
точек длинной макромолекулы.
интенсивность рассеянного света (/е) зависит от угла, под кото-
рым проводится наблюдение (рис. 13.4), т. е. интенсивность сфе-
рически несимметрична.
Светорассеяние
201
При рассеянии света от двух различных точек (z и /) длинной
макромолекулы (рис. 13.5) интерференция при нулевом угле рас-
сеяния отсутствует, но наблюдается при малых углах рассеяния и
усиливается при более широких углах рассеяния.
При рассмотрении интерференции рассеяния от различных эле-
ментов i и / для рассеивающей макромолекулы разной формы не-
обходимо в расчеты вводить поправочный фактор рассеяния (Ре).
Фактор рассеяния (Ре) определяется как отношение интенсив-
ности рассеяния под углом 0(Ре) к этой интенсивности, экстрапо-
лированной к 0 =О°(Ро°):
Ре = Ре/Ро° (13.12/
Для макромолекул любой формы Ре = 1 при 0 = 0°. С увеличе-
нием 0 величина Ре уменьшается. Фактор рассеяния связан с ра-
диусом инерции ($2) и зависит от формы частиц (сферы, тонкие
диски, тонкие палочки, цилиндры, статистические клубки, поли-
диСперсные статистические клубки и жесткие клубки). Из-за слож-
ности математическое выражение фактора рассеяния для любой
заданной формы рассеивающей частицы здесь не приводится.
Обзорная литература: 166, 381, 382, 846, 1016.
Периодическая литература: 2020, 2021, 2293, 2311, 2312, 2563, 2653, 2654,
2757, 3103, 3104, 3106, 3222, 3346, 3361, 3489, 4101, 4151, 4371, 4377, 4533-4535,
4701, 5154, 5215, 5620, 5711, 5738, 5980, 6115, 6274, 6520, 6736, 6817, 6897, 6940,
6954, 7220, 7292.
13.1.4. Определение средневесового молекулярного веса
методом светорассеяния
Для частиц большого размера (макромолекул) диаметром более
Х/20 средневесовой молекулярный вес Mw можно найти из урав-
нения
Rc/RQ=\/MwPQ + 2A2c + ..., (13.13)
где К—константа, определяемая из уравнения (13.6); Rq — рэ-
леевское отношение, которое можно найти из выражения (13 5):
Ре—фактор рассеяния, определяемый из уравнения (13.12), М» —
средневесовой молекулярный вес; с — концентрация рассеивающих
частиц в растворе; А2— второй вириальный коэффициент
(разд. 2.10), Mw можно также рассчитать, исходя из мутности,
вычисленной при измерении избыточного рассеяния при 0 = 90°.
ЗНс/8л/?9э° = (1 !MW Pw) + 2 42с + ..., (13.14)
где Н = 16л/(/3 [Л’— константа. определяемая из уравнения
(13.6)];/?9о°—рэлеевское отношение при угле измерения 0=90°;.
Рэо°—фактор рассеяния для 0=90°. Для определения Mw мето-
дом светорассеяния существуют два различных метода обра-
ботки экспериментальных результатов: 1) метод асимметрии
(разд. 13.1.5) и 2) метод Зимма (разд. 13.1.6).
202
Глава 13
13.1.5. Метод асимметрии
Для того чтобы найти фактор рассеяния (Ре), необходимо знать
коэффициент асимметрии.
Коэффициент асимметрии (z) —отношение интенсивностей рас-
сеяния под углами, симметричными относительно 90°:
z = Ze/Z<iso—е> (13.15)
По мере увеличения размера молекул z возрастает для любой
выбранной величины 0.
Коэффициент асимметрии связан также с фактором рассеяния
(Ре):
z == Ре/Р(18о-е) (13.16)
В результате измерения при двух различных углах, например 45
и 135°, находят значение z:
Z = /45°/^135° == Р45°/Р135°, (13.17)
а 7эо° является прямой характеристикой рэлеевского отношения
Рэо°.
Рэлеевское отношение при нулевом угле (Ро°) равняется
Ро° = Рэоо/Рэо0 = /э0°/Р90° (13.18)
Отношение I/P900 обычно называют коэффициентом корректировки
молекулярного веса.
Коэффициент асимметрии зависит от концентрации частиц, по-
этому экспериментальные данные нужно экстраполировать к нуле-
вой концентрации. График зависимости (1/z)— 1 от с имеет вид
прямой (с — концентрация раствора).
Величина гс=0 используется при расчете коэффициента коррек-
тировки молекулярного веса для перехода от (Кс/Т?9о°)с«о к
(Кс/7?о°)с=о [см. Уравнение (13.13), из которого можно рассчитать
.
Значение гс=0 дает величину характеристического размера рас-
сеивающих частиц, если принять, что форма молекулы соответ-
ствует одной из теоретических моделей (сферы, палочки, стати-
стические клубки и т. п.). Обычно принимают, что молекула по-
лимера имеет конфигурацию статистического клубка.
Обзорная литература: 1016, 1250.
Периодическая литература: 2401, 3033, 3222.
13.1.6. Метод Зимма
Фактор рассеяния (Ре) для частицы с конфигурацией статисти-
ческого клубка определяется по уравнению Зиммах
1/PQ = 1 + (8n2/9X2)^2 sin2(0/2), (13Д9)
Светорассеяние
203
где К — длина волны падающего света; г2— среднеквадратичное
расстояние между концами полимерной цепи, имеющей форму ста-
тистического клубка (разд. 2.11); 0 — угол между рассеянным и
падающим светом.
Подставляя выражение (13.19) в уравнение (13.13), получим
7£ = ¥_[(1+U)^sin2Tl + w + --- (13'20)
или
^ = i + ^sin2(0/2) + 2AC+ ... (13.21)
Л0
где К — константа, определяемая из уравнения (13.6); с — кон-
центрация раствора; — рэлеевское отношение, определяемое из
уравнения (13.5); Mw — средневесовой молекулярный вес; Л2—
второй вириальный коэффициент (разд. 2.10),
<13'221
1. График зависимости Rc/Rq от с (при 0 = const) представ-
ляет собой прямую линию (рис. 13.6).
2. График зависимости Kc/Rq от sin2(0/2) (при с = const)
также представляет собой прямую линию (рис. 13.7).
В случаях 1 и 2 измерения при различных концентрациях и
возможно больших углах 0 (например, 45, 60, 75, 90, 105° ...)
дают ряд прямых линий, которые можно экстраполировать до
ординаты, обеспечивая возможность определения (Кс/7?0)С==О и
(Kc//?0)sin, (0/2)=о.
3. График зависимости (Лс/7?0)С=О от sin2 (0/2) представляет
собой прямую линию (рис. 13.8) .
4. График зависимости (Кс//?0)зШ2(0/2)=о от с также представляет
собой прямую линию (рис. 13.9).
Анализ уравнения (13.21) показывает следующее:
5. Для с = 0 {KcIR^^^IM^ + K' sin2(0/2).
6. Для sin2(0/2) = 0 (W/?0)s.n2(0/2)=o = (l/Ma,) + 2^2c+ ...
Прямые линии_ на рис. 13.8 и 13.9 отсекают на оси ординат от-
резок, равный 1/Мда.
На рис. 13.8 tg а = R', на основании чего можно рассчитать
среднеквадратичный радиус (г2) для полимерной цепи, имеющей
конфигурацию статистического клубка.
На рис. 13.9 tga=2X2, что позволяет найти второй вириаль-
ный коэффициент.
Как правило, экстраполяцию, показанную на рис. 13.8 и 13.9,
проводят не раздельно, а преимущественно с использованием од-
ной диаграммы (называемой диаграммой Зимма), представляющей
собой зависимость Kc/Rq от 100 с + sin2(0/2) (рис. 13.10).
Рис. 13.7. Зависимость Kc/Rq от
sin2 (0/2).
Концентрация (с)
Рис. 13.6. Зависимость Kc)Rq от
концентрации (с).
л
i/a7w
YJ____________________
Концентрация (с)
Рис. 13.9. Зависимость Kc/Rq от
концентрации для sin2 (0/2) = 0.
Рис. 13.10. Диаграмма Зимма.
Светорассеяние
205
1. Линии с небольшим наклоном (тонкие линии) отражают за-
висимость Kc/Rq от 100с + sin2 (0/2) для определенной постоян-
ной концентрации с. Путем экстраполяции этих линий до пересе-
чения с линией, абсцисса которой равна 100 с при 0 = 0, получают
ряд точек, отражающих зависимость Rc/Rq = f(c) при sin2(0/2) =
= 0.
2. Линии с большим наклоном (жирные линии) характеризуют
зависимость Rc/Rq от 100с + sin2 (0/2) для постоянных углов рас-
сеяния. Экстраполяцией этих линий до пересечения с соответствую-
щей величиной абсциссы sin2(0/2) при с = 0 получают ряд точек,
отражающих зависимость Rc/Rq = f (sin2(0/2)) при с = 0.
3. При данном методе экстраполяции обе линии могут пере-
секать ординату в одной и той же точке, что является критерием
справедливости уравнения (13.21).
Обзорная литература: 320, 776, 1016, 1350.
Периодическая литература: 2033, 2102, 2145, 2344, 2350, 2656, 2732, 2806,
2974, 3206, 3412, 3695, 3759, 3844, 4085, 4205, 4301, 4372, 4483, 4575, 4628, 4751,
4849, 5256, 5345, 5640, 5914, 5950, 6069, 6318, 6384, 6561, 6562, 6596, 6699, 6724,
6900, 7145, 7332, 7336.
13.1.7. Определение второго вириального коэффициента
методом светорассеяния
Второй вириальный коэффициент (Л2) можно выразить как
(13.23)
где — плотность растворителя, рр — плотность полимера, Ms —
молекулярный вес растворителя, % — параметр взаимодействия
Флори (разд. 2.9).
Экспериментально А2 находят, измеряя отношение c/Rq при
двух концентрациях, С\ и с2:
I к [(«,, - (С//ад
(13.24)
[см2-моль«г-2 (СГС) или м2-моль-кг-2 (СИ)], где —константа,
определяемая из уравнения (13.6); — рэлеевское отношение,
определяемое из уравнения (13.5); с — концентрация раствора.
Второй вириальный коэффициент также равен тангенсу угла
наклона кривой, представленной на рис. 13.9.
Обычно значение Л2, полученное из измерений рассеяния света,
хорошо соответствует его величине, найденной при осмометриче-
ских исследованиях (разд. 5.2).
Периодическая литература: 4463, 4850, 5186, 5514, 6291, 6901.
206
Глава 13
13.1.8. Определение среднеквадратичного расстояния между
концами полимерной цепи методом светорассеяния
Среднеквадратичное расстояние между концами полимерной цепи
(г2) можно рассчитать по методу Зимма (разд. 13.1.6). Измерения
рассеяния света дают размеры Z-средней величины (г2).
При использовании вискозиметрических данных (разд. 2.13)
нужно применять среднечисловую величину (г2). Равенство
г2 = г2=г2„ (13.25)
справедливо только для хорошо фракционированных полимеров.
Для линейных полимеров
г2 = 6s2, (13.26)
где s2 — среднеквадратичный радиус инерции (разд. 2.11).
Обзорная литература: 263.
Периодическая литература: 2145, 3721, 4174, 4301, 4302, 4463, 4696, 6105,
6338, 6897, 7332.
13.1.9. Приборы для измерения светорассеяния
Приборы для измерения светорассеяния имеют следующие части
(рис. 13.11 и 13.12):
1. Источник света. В качестве источника света в фотометрах
старой конструкции для измерения света применяли ртутную лам-
пу высокого давления в комбинации с соответствующими фильт-
рами с целью получения монохроматического пучка (зеленая линия
ртути при 546 нм и синяя линия при 436 нм). В последнее время
стали использовать лазерные лучи (рис. 13.12). Лазеры — идеаль-
ные источники света для фотометров, предназначенных для
измерения рассеяния света. Лазерные лучи являются монохро-
матическими, в высшей степени коллимированными, интенсивными
лучами, которые можно полностью поляризовать, при этом их
поперечное сечение может быть очень мало (точечный источник)
(разд. 10.6).
Обзорная литература: 846.
Периодическая литература: 3448, 4488, 4989, 4991, 7298.
2. Кювета, помещенная в термостат. Для измерения светорас-
сеяния используют кюветы различных типов. Применение конусо-
образных кювет позволяет свести к минимуму отражения от
границы раздела стекло — жидкость (рис. 13.13). В обычных
фотометрах используют кюветы объемом 8—30 мл, в лазерных
фотометрах можно пользоваться кюветами объемом в 103—104 раз
меньше. Кювету помещают в термостат, заполненный жидкостью,
показатель преломления которой соответствует показателю прелом-
Светорассеяние
207
Рис. 13.11. Оптическая система фотометра, предназначенного для измерения
светорассеяния под углами 45, 90 и 135°.
/ — источник света; 2—конденсорная линза; 3 —монохроматический фильтр; 4—коллиматор;
5—поляризатор; 6 — кювета для светорассеяния; 7 — анализатор; 8 — фотоэлемент.
Рис. 13.12. Лазерный фотометр для измерения светорассеяния.
1— лазер; 2—поляризатор; 3 — частично пропускающее зеркало; 4 — полностью отражающее
Зеркало; 5 — кювета для светорассеяния; 6—термостат; 7 —анализатор; 8 — фотоумножитель;
9 — усилитель; 10 — коррелятор; //—усилитель разбаланса; 12 — самописец.
208
Глава 13
электрических приборов,
i
а б
Рис. 13.13. Кюветы для изме-
рения светорассеяния.
а — кювета для измерения под угла-
ми 45, 90 и 135°; б—кювета для из-
мерения под любыми углами (в кю-
ветах такого типа можно проводить
ультрацентрифугирование).
ления содержимого кюветы. Специальный сосуд Дьюара позво-
ляет проводить измерения рассеяния света в интервале температур
от —40 до 200 °C.
Обзорная литература: 263.
Периодическая литература: 2375, 2573, 2749, 3049, 3238, 3362, 3447, 3724,
3908, 4312, 4488, 4626, 5633, 6832, 6898, 7332. .
3. Детекторы для измерения рассеяния света. Интенсивность
рассеянного света измеряется с помощью фоточувствительных
например фотоумножителей. Интенсив-
ность рассеянного света нужно срав-
нивать с интенсивностью первичного
пучка света. Эти две величины отли-
чаются между собой примерно на
шесть порядков, поэтому неудобно из-
мерять оба пучка при полной чувстви-
' тельности фотоумножителя. Для опре-
деления констант данного прибора не-
обходимо проводить калибровку, ко-
торая осуществляется в два этапа:
а) Для калибровки постоянного
рабочего стандарта используют пер-
вичный стандартный раствор (кремне-
вольфрамовая кислота, H4SiWi204o,
Af = 2879 в 1 Af NaCl). Постоянный
рабочий стандарт (мутный блочный
полимер или опаловое стекло), приме-
няемый вместе с нейтральным фильт-
ром для уменьшения интенсивности света, вмонтирован в прибор
или ставится на место кюветы для рассеяния.
б) В ходе исследования светорассеяния полимерных растворов
время от времени для проверки правильности калибровки прово-
дят измерения на постоянном рабочем стандарте.
В современных лазерных фотометрах сравнивается интенсив-
ность рассеянного и падающего света путем измерения выходных
характеристик от двух фоточувствительных электрических прибо-
ров (фотоумножителей) (рис. 13.12).
4. Приспособление для записи и обработки экспериментальных
данных. Выдача и обработка данных в значительной степени об-
легчаются благодаря использованию автоматической записи и
компьютерной техники.
Обзорная литература: 412, 846.
Периодическая литература: 3032, 3909, 4989, 6802.
В литературе можно найти подробное описание различных кон-
струкций фотометров для измерения рассеяния света.
Светорассеяние
209
Обзорная литература: 774.
Периодическая литература: 2179, 2297, 2375, 2507, 2573, 2838, 2877, 2989,
3105, 3362, 3447, 3724, 3759, 3908, 3909, 3989, 4030, 4312, 4502, 4714, 4720, 4778,
4874, 4991, 5196, 5618, 5633, 5642, 5844, 6283, 6388, 6825, 6898, 7136, 7298, 7332.
Анализ с помощью ЭВМ позволяет получить более достовер-
ные и точные данные. Опубликован ряд программ ЭВМ для об-
работки результатов измерения светорассеяния.
Обзорная литература: 733, 884.
Периодическая литература: 2576, 2612, 2953, 3333, 3807, 3909, 4533, 4823,
4989, 5096, 5267, 5289, ббб9, 6109, 6237, 6387, 6802, 6996, 7024, 7205.
Подробные сведения о коэффициентах асимметрии и рассеяния
частицами имеются в Polymer Handbook [О: 284].
Опубликованы многочисленные таблицы, в которых суммиро-
ваны функции рассеяния [О: 366, 1200; П: 4989].
13.1.10 . Приготовление образцов для измерения
рассеяния света
Растворы, предназначенные для измерения рассеяния света, со-
вершенно не должны содержать пыли. Пыль удаляют путем филь-
трации через мелкопористые фильтры и/или высокоскоростной се-
диментацией в центрифуге. Фильтра- Подача
цию лучше всего проводить с исполь-
зованием целлюлозных мембранных
фильтров, диаметр пор которых 0,2—
0,5 мкм. Мембраны такого типа мож-
но использовать с большинством орга-
нических и неорганических раствори-
телей. Для таких агрессивных сред,
как сильные кислоты и основания, в
некоторых случаях применяют пори-
стые стеклянные или фарфоровые
фильтры. На рис. 13.14 показано ти-
пичное приспособление для фильтра-
ции полимерных растворов. Чтобы из-
бежать флуоресценции от примесей,
которые могут искажать получаемые
результаты при рассеянии света, рас-
творители, предназначенные для изме-
рения светорассеяния, должны быть
спектрально чистыми.
Наличие примесей (пыли) обыч-
но обнаруживается по замет-
ил створа
----®под
давлением
LZZ3
Мембранный
фильтр
t—I
Кювета для измерения
——' светорассеяния
Рис. 13.14. Устройство для
фильтрации полимерных раст-
воров через мембранный
фильтр.
ным отклонениям в кривой Зимма
ших 45°.
при углах, мень-
Обзорная литература: 263, 1016, 1250.
Периодическая литература: 5091, 5732, 6226, 6283, 6782.
210
Глава 13
Интенсивность рассеяния удовлетворительной величины можно
измерить, когда drt/dc >> 0,05 см3/г. В полимерных растворах, где
с = 0,01 г/см3, разность показателей преломления раствора и рас-
творителя составляет примерно 0,002 единицы. Для того чтобы
определить Mw с точностью ±2%, необходимо знать инкременты
показателя преломления с точностью ±1% и разность показате-
лей преломления с точностью ±2-10-5.
Обзорная литература: 1016.
Периодическая литература: 2405, 2572, 2665, 3105, 3455, 3996, 4172. 6984.
13.1.11 . Применение светорассеяния в исследовании полимеров
Измерения интенсивности рассеяния света разбавленным раство-
ром полимера дают информацию о Mw (от 100 до 107), форме
полимерных молекул, взаимодействии между полимером и раство-
рителем, частицах в суспензии, оптических характеристиках от-
дельных точек рассеяния (однородные или слоистые сферы и ци-
линдры).
Обзорная литература: 99, 125, 126, 263, 264, 320, 425, 500, 650, 734, 735, 776,
797, 846, 899, 943, 994, 995, 1016, 1250, 1289, 1308, 1331.
Периодическая литература: 2573, 3063, 3179, 3191, 3206, 3694, 3869, 3909,
4085, 4205, 4301, 4302, 4370, 4424, 4848, 4989, 5860, 6336, 6452, 6540, 6963.
13.2. ИМПУЛЬСНО-ИНДУЦИРУЕМОЕ КРИТИЧЕСКОЕ
РАССЕЯНИЕ
Импулъсно-индуцируемое критическое рассеяние — это метод изме-
рения рассеяния света растворами полимеров в изотермических
условиях (рис. 13.15).
Рис. 13.15. Спектрометр для измерения импульсно-индуцируемого критического
светорассеяния.
1 — гелиево-неоновый лазер; 2 —ячейка для измерения светорассеяния; 3—импульсный термо-
нагреватель; 4—световоды; 5—фотоумножители; 6—термистор; 7 — усилитель; 8 — насос;
3—нагревательный элемент; 10—охлаждающий элемент; // — самописец.
Тонкостенный капилляр диаметром 1 мм заполняют несколь-
кими микролитрами раствора полимера. Вдоль оси такой ячейки
Светорассеяние
211
пропускают луч гелиево-неонового лазера. Свет, рассеянный рас-
твором полимера после быстрого регулирования температуры, за-
писывают под углами 30 и 90°. По световодам свет поступает в
фотоумножители. Для измерения температуры используются тер-
мисторы.
Ячейку помещают в проточную водяную систему, снабженную
небольшим нагревательным элементом, расположенным вверх по
течению от ячейки для поддержания ее при температуре выше
точки мутности. Температуру регулируют путем выключения на-
гревателя. Тогда содержимое ячейки быстро охлаждается до тем-
пературы проточной воды.
Этот метод можно использовать для изучения явления фазо-
вого разделения в несмешивающихся системах жидкость—жид-
кость, для изучения фазовых диаграмм и определения спинодаль-
ных точек, для исследования совместимости полимерных смесей и
процессов кристаллизации.
Обзорная литература: 367, 731.
Периодическая литература: 3720, 4631.
13.3. МАЛОУГЛОВОЕ ЛАЗЕРНОЕ СВЕТОРАССЕЯНИЕ
В малоугловом лазерном рассеянии света измерения проводят в
интервале углов 2—10°, поэтому, если Mw не превышает 106, из-
мерения следует осуществлять только при одном угле. Используе-
мые в широкоугловом светорассеянии сложные экстраполяции
(диаграммы Зимма) в этом случае оказываются ненужными. По-
скольку объем рассеяния геометрически определен, значения Mw,
получаемые при малоугловом рассеянии света, представляют со-
бой абсолютные величины. К минимуму сводятся и проблемы
осветления образцов для исследования, последнее обстоятельство
связано с очень маленьким (0,1 мкл) объемом рассеивания.
Mw можно рассчитать из следующего уравнения:
Kc/RQ = (l/Mw) + 2A2c (13.27)
или
Ма = (Кс/7?е) - 2А2с ’ (13-
где К — константа, определяемая по уравнению (13.6); с — кон-
центрация рассеивающих частиц в растворе; Re—рэлеевское от-
ношение, определяемое по уравнению (13.5); А2— второй вири-
альный коэффициент (разд. 2.10). Для определения величину
/?е измеряют для нескольких концентраций растворов. Зависимость
Rc/Re от с представляет собой прямую линию, отсекающую на оси
ординат отрезок, равный \/Mw. Тангенс угла наклона этой прямой
равен 2Л2 (tga = 2А2).
Если А2 известен, то, используя уравнение (13.27), можно рас-
считать определив лишь одно значение Это существенно
212
Глава 13
облегчает непрерывный контроль молекулярного веса полимера,
необходимый при исследованиях методом ГПХ.
Метод малоуглового лазерного рассеяния света обладает сле-
дующими преимуществами:
а) метод не сопряжен с деструкцией полимеров;
б) обеспечивает возможность абсолютной калибровки при опре-
делении молекулярного веса;
в) очень маленький объем рассеяния сводит к минимуму
проблемы осветления образцов;
г) из-за высокой чувствительности можно исследовать неболь-
шие и разбавленные образцы;
д) конструкция измерительной кюветы позволяет исключить
фоновое рассеяние;
е) благодаря малой инерционности возможно проведение ки-
нетических исследований;
ж) длинноволновый лазерный излучатель позволяет исключить
флуоресценцию;
з) благодаря удобной и быстрой настройке определение моле-
кулярного веса занимает считанные минуты.
Малоугловое лазерное рассеяние света может найти примене-
ние для самых различных целей, в том числе и указанных ниже:
а) определение молекулярного веса;
б) изучение молекулярновесового распределения вместе с ГПХ,
при этом можно в значительной степени избежать необходимости
введения поправочных коэффициентов на расширение полосы
(разд. 25.8);
в) исследование макромолекулярной кинетики;
г) определение флуоресценции и нефелометрии от одной ча-
стицы;
д) исследование молекулярных взаимодействий (вириальных
коэффициентов);
е) определение молекулярных конформаций (радиуса инер-
ции);
ж) изучение молекулярной анизотропии (деполяризации);
з) рэлеевская спектроскопия (коэффициенты диффузии);
и) бриллюэновская спектроскопия (звуковая модуляция);
к) рефрактометрия.
Обзорная литература: 889.
Периодическая литература: 4486, 4488, 5636, 5637.
13.4. СПЕКТРОСКОПИЯ БИЕНИЯ СВЕТА
Спектроскопия биения света (называемая также спектроскопией,
основанной на доплеровском сдвиге, или рэлеевской спектроско-
пией ширины полосы) используется для оценки формы (ширины
линии) пика Рэлея.
Светорассеяние
213
Этот метод основан на следующем принципе. Вследствие того
что спектр падающего света подвержен влиянию эффекта Доп-
лера, частота молекул, движущихся в растворе, несколько меняет-
Рис. 13.16. Лазерный оптический спектрометр, основанный на самобиении света.
1 — гелиево-неоновый лазер; 2— частично пропускающее зеркало; 3—полностью отражающее
зеркало; 4 — вращающаяся ячейка с образцом; 5—термостат; 6 — фотоумножитель; 7 — уси-
литель; 8 — самописец.
ся. Сдвиг Доплера измеряют путем оптического «смешения» рас-
сеянного света с эталонным лазерным лучом (рис. 13.16). Резуль-
тирующий гетеродин (или частота биения) равен разности частот
Рис. 13.17. Спектры падающего лазерного луча (а), света, рассеянного вра-
щающимся образцом (б), тока фотоумножителя (в).
эталонного и рассеянного пучков. При знании этого частотного
сдвига и геометрии возможен прямой расчет скорости движения
молекул в растворе.
Процесс оптического самобиения позволяет осуществлять сдвиг
первоначального спектра, концентрированного при частоте падаю-
щего света, к частотам порядка начальной ширины линии, как это
схематически показано на рис. 13.17.
214 Глава 13
Полуширина при полувысоте (Дгеи/Д связана с коэффициентом
поступательной диффузии (D) следующей зависимостью:
Дшш = 2К2£>, (13.29)
где К = (4л/%)ц sin (0/2) представляет собой изменение волнового
вектора в квазиупругом рассеянии; X — длина волны падающего
лазерного луча; п — показатель преломления раствора; 0 — угол
рассеяния. Таким методом быстро и точно определяется коэффи-
циент поступательной диффузии (Z)). Исследование проводится
для растворов в равновесном состоянии и дает важную информа-
цию о полимерных конформациях и гидродинамике растворов.
Спектроскопия биения света может быть использована для опре-
деления MWt размера макромолекул, флуктуаций концентрации,
вращательной релаксации и диффузии, констант диффузии: посту-
пательной, вращательной, само- и взаимодиффузии, сдвига Доп-
лера.
Обзорная литература: 43, 44, 108, 138, 344, 345, 745, 1019—1021. 1033.
Периодическая литература: 2655, 2697, 2840, 3032, 3237, 3244, 3245, 3443—
3445, 3497, 3506, 3532, 3537, 3538, 4149, 4150, 4284, 4563, 4703, 4704, 5064, 5490,
5739, 5740, 5907, 6000, 6002, 6231, 6232, 6331-6335, 6684, 6773, 6774, 6981, 7263.
13.5. СПЕКТРОСКОПИЯ РЭЛЕЯ — БРИЛЛЮЭНА
При неупругом рассеянии частота рассеянного света отличается
от частоты падающего света и дает две побочные полосы, назы-
ваемые пиками Бриллюэна (рис. 13.18).
Относительная интенсивность различных пиков в спектрах Рэ-
лея — Бриллюэна зависит от структуры и релаксационных про-
цессов в жидкостях. Для определения формы спектров Рэлея —
Бриллюэна используется сканирующий интерферометр Фабри —
Перо (рис. 13.19), в котором сдвиг частоты падающего света изме-
ряется с помощью зеркального разделения в резонаторе. Расстоя-
ние между зеркалами можно регулировать с помощью, например,
пьезоэлектрических методов (разд. 13.5.1).
Для интерпретации спектров Рэлея — Бриллюэна пользуются
соотношением Ландау — Плачека (/).
Соотношение Ландау — Плачека (/) определяется как отноше-
ние интенсивности центрального рэлеевского пика и интенсивно-
сти двух бриллюэновских пиков.
Средневесовой молекулярный вес можно найти, оценив J для
чистых растворителей и разбавленных растворов полимеров при
различных концентрациях. Тогда Mw рассчитывают по формуле
W/(/1-/2) = (l/A4J + 2X2c+(13.30)
где В и К — константы растворителей; и /2 — соотношения Лан*
дау — Плачека для чистого растворителя и раствора соответствен-
Свегорассеяние
215
Рис. 13.18. Спектры Рэлея — Бриллюэна чистого растворителя (а) и раствора
полимера (б).
Рис. 13.19. Спектрометр Рэлея — Бриллюэна.
I—гелиево-неоновый лазер; 2—полностью отражающее зеркало; 3 —термостат; 4—ячейка
6 образцом; 5 —полупрозрачные зеркала; 6 — кольцо из пьезоэлектрического материала;
7 —фотоумножитель; 8—усилитель; 9 — осциллограф.
216
Глава 13
но; с — концентрация рассеивающих частиц; Д2 — второй вириаль-
ный коэффициент.
Обзорная литература: 287, 745, 967.
Периодическая литература: 2906, 4981, 5272, 5273, 5737, 6684.
Спектроскопия Рэлея — Бриллюэна может быть использована
при исследовании размера макромолекул в растворе (в том числе
молекул разветвленных полимеров), диффузионных процессов в
растворе, полимерных сеток, сверхзвуковой скорости и затухания
звука в растворах.
Обзорная литература: 43, 287, 288, 343, 745, 1033, 1045, 1074, 1199, 1230.
Периодическая литература: 2017, 2320, 2449, 2585, 2616, 2624, 2652, 2717,
2718, 3509—3511, 4017, 4018, 4268, 4287, 4550, 4668, 4669, 4375, 4935, 5163, 5255,
5275, 5304, 5373, 5374, 5384—5386, 5542, 5712—5720, 5907, 5945, 5804, 6001, 6090,
6439, 6529, 6722, 6773, 6774, 7181, 7238.
13.5.1. Интерферометры Фабри—Перо
Показанный на рис. 13.20 интерферометр Фабри — Перо состоит
из двух стеклянных или кварцевых пластинок. Две внутренние по-
верхности плоско отполированы в пределах до 0,05—0,005 от длины
Рис. 13.20. Интерферометр Фа-
бри — Перо.
/ — стеклянные пластинки; 2—полу-
прозрачные зеркала; 3 —регулиро-
вочные винты.
волны рабочего диапазона и покры-
ваются металлическими пленками с
высокой отражательной способностью
(серебро, золото или алюминий) или
чередующимися слоями материалов с
высокой и низкой диэлектрической про-
ницаемостью. Для того чтобы умень-
шить отражение, стеклянные пластин-
ки имеют небольшой краевой угол
(0,1°) между главными поверхностями.
Между двумя сильно отражающи-
ми пленками, разделенными воздуш-
ной прослойкой (Z), происходят мно-
гократные отражения (рис. 13.21).
Обычно полезный интервал в ин-
терферометре Фабри — Перо ограни-
чен первыми десятью полосами.
Применяются сканирующие интерферометры Фабри — Перо
двух типов:
1. Интерферометр старой конструкции, в котором оптическое
расстояние между пластинками можно регулировать варьирова-
нием показателя преломления (ц) среды между пластинками
(рис. 13.22). В вакууме показатель преломления п0 = 1,000000,
тогда как для воздуха показатель преломления п = 1,000290. Ин-
тервал между п и nQ достаточно велик, в результате чего интер-
Светорассеяние
217
Падающая волна
с амплитудой £0
Многократные
отражения
Отраженная
волна
Пропущенные
волны с ампли-
О2 ту дама ^1,£г,Г3....Ея
Рис. 13.21. Многократные отражения, наблюдающиеся между двумя сильно от-
ражающими пленками, которые находятся на расстоянии I друг от друга.
Рис. 13.22. Сканирующий интерферометр Фабри — Перо старой конструкции, в
котором варьированием давления осуществляют изменения показателя прелом-
ления и оптического расстояния между пластинками.
1 — интерферометр Фабри — Перо; 2—вакуумная камера; 3 —оптическое окно; 4 — фотоумно-
житель; 5—усилитель; 6 —самописец.
Интерферометр
Рис. 13.23. Сканирующий интерферометр Фабри — Перо новой конструкции, в
котором сделанное из пьезоэлектрического материала кольцо под влиянием
электрического поля обеспечивает изменение расстояния между пластинками.
/ — кольцо из пьезоэлектрического материала; 2—фотоумножитель; 3— усилитель; 4—осцил-
лограф.
218
Глава 13
ферометр захватывает несколько свободных спектральных обла-
стей.
2. Интерферометр новой конструкции, который работает на
принципе варьирования расстояния между пластинками (/). Одна
пластинка закрепляется на кольце из пьезоэлектрического мате-
риала (рис. 13.23). При действии на такой материал элек-
трического поля он расширяется или сжимается, что и вызывает
изменение расстояния /. Колебание частоты интерферометра син-
хронизируется с частотой сканирования осциллографа. При под-
соединении выхода фотоумножителя к входу осциллографа можно
непосредственно наблюдать получаемый спектр.
Обзорная литература: 1220, 1257.
Глава 14
УЛЬТРАФИОЛЕТОВАЯ И ВИДИМАЯ
СПЕКТРОСКОПИЯ
Обзорная литература: 71, 89, 192, 513, 672, 730, 1093, 1182, 1213, 1262, 1393.
При поглощении полимерами ультрафиолетового (УФ) и ви-
димого (ВИ) света происходят переходы электронов на более вы-
сокие энергетические уровни, в результате чего наблюдается ти-
пичный электронный спектр поглощения (см. рис. 10.4).
Ультрафиолетовая и видимая спектроскопия дает информацию
о кратных связях и ароматическом сопряжении в макромолекулах.
В систему сопряжения в полимерах с участием кратных связей
могут также вовлекаться неподеленные пары электронов кисло-
рода, азота и серы.
Человеческому глазу полимеры, поглощающие свет в интервале
длин волн между 400 и 800 нм (видимый свет), представляются
окрашенными. Смещение поглощения от 400 нм в область больших
длин волн приводит к постепенному потемнению окраски от жел-
той, оранжевой, красной к зеленой, синей, фиолетовой и, наконец,
черной.
Поглощение света макромолекулами происходит лишь в том
случае, когда разность между двумя энергетическими уровнями в
точности отвечает энергии кванта:
kE = hv = E2-Eu (14.1)
где h — постоянная Планка [6,626 -10-27 эрг-с (СГС) или
6,626-10-34 Дж-с (СИ)]; v — частота (с-1); Е2 и Е\ — энергия од-
ной макромолекулы в конечном (высокий уровень) и начальном
(низкий уровень) состоянии соответственно.
Хромофоры — это функциональные группы, которые поглощают
электромагнитное излучение независимо от того, возникает при
этом окраска или нет. Так, карбонильная группа С=О является
хромофором, поглощающим в области 280 нм, в то же время ке-
тоны— бесцветные вещества. В табл. 14.1 приведены примеры ор-
ганических хромофоров, которые встречаются в полимерных со-
единениях. Использование ультрафиолетовой и видимой спектро-
скопии для исследования полимеров в значительной степени
обусловлено наличием в молекулах полимеров некоторых из этих
хромофорных групп.
Ауксохромы — это функциональные группы, например —ОН,
—OR, —NH2 и др., которые, вступая в сопряжение с хромофором
за счет своих неподеленных электронов> становятся частью новену
более протяженного хромофора,
220
Глава 14
При поглощении макромолекулой ультрафиолетового или ви-
димого света определенной энергии только один электрон перехо-
дит на более высокий энергетический уровень, все остальные элек-
троны в первом приближении не затрагиваются. Образующееся
в таком варианте возбужденное состояние характеризуется корот-
ким временем жизни порядка 10-6—10~9 с, а затем наиболее ве-
роятен перенос одного электрона с верхней занятой молекулярной
Таблица 14.1
Типичные хромосомы и их характеристики
Хромофор Длина волны (Чпах)’ нм Коэффициент молярной экстинкции (етах)
с=с 175 14 000
185 8 000
с=с 175 10 000
195 2 000
223 150
с=о 160 18 000
185 5 000
280 15
с=с- с=с 217 20 000
( 184 60 000
{ 200 4 400
(.255 204
орбитали на нижнюю возможную незаполненную орбиталь. В ряде
случаев можно наблюдать несколько переходов, дающих ряд полос
поглощения в спектре полимера. Разрешены не все переходы от
заполненных к вакантным орбиталям. Когда такой переход «за-
прещен», интенсивность соответствующей полосы поглощения так-
же является низкой (гл. 16).
На рис. 14.1 представлены относительные энергии молекуляр-
ных орбиталей, имеющие значение в электронной спектроскопии
полимеров. На относительные энергии молекулярных орбиталей в
полимерах оказывают влияние многие факторы, которые необхо-
димо знать для понимания электронной спектроскопии макромо-
лекул.
При поглощении макромолекулой ультрафиолетового или ви-
димого света возникает полоса поглощения, так как на колеба-
тельные и вращательные эффекты накладываются электронные
переходы.
В некоторых случаях на интенсивность и положение полосы
поглощения влияет природа растворителя, причем в результате
полоса может сдвигаться как в длинноволновую, так и в коротко-
волновую область. Согласно старой номенклатуре (которая по-
степенно забывается):
Ультрафиолетовая и видимая спектроскопия
221
с=о
тт-^тт1
п-*-тт'
п-*-сг'
Рис.
молекулярных орбиталей и различ-
ные типы электронных переходов,
встречающиеся в электронной спек-
троскопии.
14.1. Относительные энергии
С-С С=С
ст->- er*
а) батохромный сдвиг (или
красный сдвиг) — это сдвиг
полосы в сторону длинных
волн;
б) гипсохромный сдвиг (или
синий сдвиг) —это сдвиг по-
лосы в сторону коротких
волн;
в) гиперхромный эффект —
повышение интенсивности погло-
щения;
г) гипохромный эффект — по-
нижение интенсивности поглоще-
ния.
Для нахождения ультрафио-
летовых спектров органических
соединений низкого молекуляр-
ного веса рекомендуются следую-
щие пособия: [О: 290, 478, 626,
792, 1037, 1472, 1476].
Применение ультрафиолето-
вой спектроскопии в исследова-
нии полимеров ограничено соеди-
нениями, содержащими хромо-
форные группы, это, в частности,
полиены, ароматические полимеры, полимеры с карбонильными
группами.
Периодическая литература: 2295, 3143, 4407, 4803, 6167, 7134.
14.1. КОЛИЧЕСТВЕННЫЙ АНАЛИЗ
В УЛЬТРАФИОЛЕТОВОЙ И ВИДИМОЙ
спектроскопии
Количественный анализ в спектрофотометрической практике осно-
ван на использовании закона Ламберта — Бера:
A = lg(IQ/I) = scl (14.2)
(безразмерная величина), где /о — интенсивность падающего света
(квант-с-1); / — интенсивность света, прошедшего через раствор
или пленку образца (квант-с-1); lg(/0/7) называется поглоще-
нием (А) раствора, а также оптической плотностью (D) или экс-
тинкцией (£); е — коэффициент молярной экстинкции:
ъ = А/с1 (14.3)
[[л-моль”1-см-1 (СГС) или мЗ-кг^-м"1 (СИ)].
Коэффициент е является постоянной величиной для данного
соединения при данной длине волны. При больших значениях е
222
Глава 14
Рис. 14.2. Перекрывающиеся спектры
двух компонентов, I и II, и спектр
смеси двух компонентов, I -f- II.
Пропускание, %
100 90 80 70 60 50 40 30 20 10 О
°»1 0,2 0,3 0,4 0,5 0,60,70,8 1,0 сэ
Поглощение
Рис. 14.3. Связь между пропусканием и поглощением.
Рис. 14.4. Схематическое изображе-
ние спектров поглощения в виде за-
висимости поглощения от длины
волны (а) и в виде зависимости
пропускания от длины волны (б).
Положительное
отклонение /
Рис. 14.5. Проверка выполнимости
закона Ламберта — Бера, или ка-
либровочные кривые для количе-
ственного анализа.
Ультрафиолетовая и видимая спектроскопия
223
удобно пользоваться его логарифмом (1g е). В случае смеси двух
или более компонентов поглощение (экстинкция) является куму-
лятивной характеристикой:
А = Z (е^! + 82с2 + • • • + (14.4)
Концентрацию двух компонентов можно определить, если известны
четыре значения коэффициента молярной экстинкции и измерения
проведены при двух длинах волн:
А = I (Sjtj + е2с2) для А/, (14.5)
А" = I Для (14.6)
Длины волн V и X" выбирают из такого расчета, чтобы один ком-
понент сильно поглощал при этих длинах волн, а поглощение
другого компонента было значительно меньше (рис. 14.2).
Рис. 14.6. Проверка выполнимости закона Ламберта — Бера для спектра погло-
щения при различных длинах волн %! и Х2.
а — спектр поглощения; б—калибровочные кривые.
Между поглощением (А) и пропусканием (Т) существует сле-
дующая зависимость (рис. 14.3):
Л = 1g (/оД) = - 1g Т = 1g (100/7) (14.7)
В приложении В имеется таблица для перехода от пропускания
к поглощению и обратно.
Современные двухлучевые спектрофотометры позволяют непо*
средственно записывать поглощение или пропускание. Спектры
поглощения строятся . таким образом, что на ординате отклады-
вается поглощение (А) (или е, или 1g е) (рис. 14.4, а) или про-
пускание (Т) (рис. 14.4,б), а на абсциссе — длина волны (X).
В спектрофотометрии также используются такие показатели,
как процентное пропускание 1ОО///о, процентное поглощение
100 (/о — /) //о, дробное поглощение (/0 — I) /h-
Если известны коэффициент молярной экстинкций (е), толщи-
на кюветы. (Z) и поглощение (А), то концентрацию хромофорного
224
Глава 14
вещества можно найти количественно, используя закон Ламбер-
та — Бера (14.2).
Для точного количественного анализа вначале нужно подтвер-
дить справедливость закона Ламберта — Бера для исследуемого
случая. С этой целью измеряют поглощение (Л) при нескольких
концентрациях (с) при определенной длине волны и фиксирован-
ной толщине кюветы (рис. 14.5). Если закон Ламберта — Бера вы-
полняется в исследованном интервале концентраций, то должна
получиться прямая линия, выходящая из начала координат. От-
клонения от данного закона обозначаются как положительные или
как отрицательные. Иногда закон Ламберта — Бера выполняется
при одной длине волны (М) и не выполняется при другой длине
волны (%2) (рис. 14.6).
При проведении количественных измерений поглощения могут
быть различные ошибки: химические, инструментальные или лич-
ные, индивидуальные.
14.2. ПРИБОРЫ ДЛЯ ЭЛЕКТРОННОЙ
спектроскопии
14.2.1. Двухлучевой спектрометр
Двухлучевой УФ-ВИ-спектрометр (рис. 14.7) имеет следующие
основные части.
1. Источник света, который состоит из вольфрамовой лампы
накаливания для длин волн более 375 нм и разрядной дейтерие-
вой лампы для длин волн менее 379 нм (разд. 10.5).
Первоначальный пучок света после прохождения, через моно-
хроматор (дифракционная решетка или кварцевая призма) делит-
ся с помощью вращающегося зеркального сектора на два одина-
ковых пучка. Он попеременно пропускает пучок в один канал (от-
крытый сектор) и отражает его во второй (зеркальный сектор).
2. Зона образца.
3. Фотометрическая оптическая система нуля. Для измерения
поглощения образца должны быть сопоставлены интенсивности
пучков, прошедших через образец и сравнительную кювету. Два
пучка после прерывателя попеременно подаются на детектор (фо-
тоумножитель) и усиливаются. Если интенсивности одинаковы,
то выходной сигнал после усилителя отсутствует. При любом раз-
личии в интенсивностях появляется выходной сигнал, имеющий ча-
стоту прерывателя. Этот сигнал затем усиливается и приводит в
действие аттенюатор, который вводится в сравнительный луч или
выводится из него. Аттенюатор представляет собой тонкую пло-
скую гребенку, расстояние между зубцами которой линейно уве-
личивается с расстоянием. Доля открытого пространства в гребенке
определяет степень пропускания луча, которую можно линейно из-
менять в очень узких пределах. В зависимости от фазы сигнала
Ультрафиолетовая и видимая спектроскопия
225
разбаланса движение аттенюатора регулируется таким образом,
чтобы увеличить или ослабить интенсивность сравнительного луча,
пока она не сравняется с интенсивностью луча, прошедшего через
образец. Когда интенсивности пучков сравняются и никакого сиг-
нала не будет, движение аттенюатора прекращается. Положение
Рис. 14.7. Схема автоматического двухлучевого УФ-ВИ-спектрофотометра с
оптической системой нуля.
/ — источник излучения; 2— монохроматор; 3 — аттенюатор; 4—кювета сравнения; 5—преры-
ватель; 6—зона образца; 7—кювета с образцом; 8—зеркало; 9 — детектор; 10 — усилитель;
// — сервомотор; 12 — самописец.
аттенюатора является характеристикой относительного поглощения
образца: передача его положения на цифровое устройство дает
показание поглощения.
Для измерения поглощения с большей точностью, особенно при
больших поглощениях (Л >> 1), лучше пользоваться электриче-
ской схемой измерения отношения интенсивностей.
4. Система записи результатов. Значение отношения интенсив-
ностей сравнительного и рабочего пучков (/0Д) подается на перье-
вой самописец, который рисует зависимость поглощения (Л) от
длины волны (X) в равномерной шкале (интервал 0—2 единицы
поглощения).
Имеются разнообразные спектрометры, работающие в областях
от вакуумной ультрафиолетовой до дальней инфракрасной (от
~50 нм до 1000 мкм).
Обзорная литература: 838, 991, 1213, 1275.
Периодическая литература: 5046, 5489, 5683, 7067.
14.2.2. Кюветы для образцов и кюветы сравнения
Обычно применяются различные типы прямоугольных кварцевых
кювет толщиной 0,1; 1,0; 10,0 см (рис. 14.8). Такие кюветы доро-
гие и хрупкие. После каждого использования их нужно тщательно
8 Зак. 238
226
Глава 14
очищать от раствора полимера. Это можно осуществлять, исполь-
зуя весьма простой прибор, показанный на рис. 14.9. Иногда кю-
Рис. 14.9. Про-
стое приспо-
собление для
мытья спектро-
скопической кю-
веты.
Рис. 14.8. Различные типы кювет для УФ-ВИ-спектроскопии.
а — стандартная прямоугольная открытая кювета; б — стандартная от-
крытая МЙКрокювета; в —стандартная прямоугольная кювета с крышкой;
г —стандартная микрокювета с крышкой; д — стандартная цилиндриче-
ская мйщюкювета с крышкой; е — проточная кювета; эм; —проточная
кювета (Автоматическое или непрерывное течение); з — проточная кю-
вета с тремя выводами для подсоединения вакуума и введения образ-
ца; и —стандартная прямоугольная термостатируемая кювета; к — стан-
дартная цилиндрическая термостатируемая кювета; л —кюветы со спе-
циально уплотнёнными для работы в вакууме окнами и градуированным
затвором [0:1473].
веты очищают мягкой бумагой, смоченной растворителем. Хра-
нить кюветы следует в специальных коробках и никогда не тро-
гать оптические поверхности руками.
14.2.8. Кюветы высокого давления для оптических исследований
Для спектроскопических и других исследований растворов под дав-
лением 1000—10 000 атм необходимы кюветы специальной кон-
струкции. Основная трудность при создании таких кювет заклю-
чается в том, что под влиянием давления в оптических окнах
возникает значительное и переменное двулучепреломление. Опти-
ческие окна, предназначенные для работы под высоким давлением,
изготавливают из стекла пирекс или кварца. Во избежание утечки
раствора используют сложную уплотнительную систему. Кюветы,
предназначенные для работы под высоким давлением, можно ис-
пользовать в спектрометрах для снятия ультрафиолетовых, види-
мых и инфракрасных спектров, изучения флуоресценции, фосфо-
ресценции, светорассеяния и т. п.
Периодическая литература: 2868, 2874.
14.2 й воды 400 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 S 1 1 1
<3 гг 350 1 о о 1 1 1 66 1 1 1 1 1 1 1 1 1 1 1 1
<3 о SC SC Я м 320 1 ю о 1 1 1 о 1 1 1 1 1 1 1 1 1 1 ВО ю оо о> 1 1
о сх
S ч ч S О о со о о ь- ь- о ст> о 1 ОФЮ 5>оост> : 1 1 1 о 1 1 1 1 ю ю ь- о 1 1
S
о й г, л гР 290 1 о — ю 00 О О 1 ю сч'ш 00 00 1 ! 88 ! 1 1 1 10 93 1 1
•носител 1Н (нм), 280 оо ю оо сч сч г- ст> 1 сч со со'ь- Г- GO,оо 1 1 ОО 1 1 1 1 1 о о 1 1
471] 1 см от шах boj 270 1 1 ю ш 1 -Ф 00 1 ю о °о СЧ Ю LO о 1 1 СО 1 1 1 1 1 о 00 1 1 •
>пии [О: 1 ювете толщиной i различных дле 260 со о 1 1 ° 1 СО 1 10 при 261 70 1 1 о 1 1 1 1 1 10 98
о Ьй О О. Ьй Ф Е 250 1 1 I О Ь' 1 сч о 1 1 ООО ю о о 1 ю о о о о о о о 1 1 О CG
х сх Й Я 240 93 1 1 1 06 1 1 во ю 00 00 сч о о о со 00 о 1 1 СО во О СУ>
¥ е И м 8Й о 230 о 1 1 1 ю 1 1 во со со со ь- ь- 1 СО 00 оо О ВО со со оо 1 1 ВО ю О> 00
ДЛЯ н о сх к 220 80 1 1 1 о ю 1 1 о сч сч сч ю ю 1 о со ь- о о сч со 1 1 94 60
Е Ф о я д 210 1 1 1 1 ю 1 1 1 о о со со 1 о ю — о ю о — со 1 1 сч о о со
В СХ О ва Я 3 S к 200 1 1 1 1 1 1 1 1 1 1 1 1 со 1 о 1 1 1 1 90 10
Си S £ 195 1 1 1 1 1 1 1 1 1 1 1 06 1 1 1 1 70
SSp. х И в дел а я д в ч а Ч о я К й сч V оо сч ю ш СО -Ф сч сч о о сч 00 — со со сч сч ю ю ю —« о о сч сч сч о 8 210 < 190 во ю о ООО сч сч сч 260 190 200
Растворитель ю <4 S к « о Ф Е < ч о СО Е ф ю Четыреххлористый углерод Хлороформ Циклогексан 6 Диметилформамид Димстилсульфоксид в 1,4-Диоксан 6 Этанол 6 (95%-ный) Этанол (абсолютный) 1 Этилацетат Гексан6 (нефтяная фракция) 1,1,1,3,3,3- Гексафторпропа- нол-2б Метанол 6 Пропанол-2 6 Тетрахлорэтилен Трифторуксусная кислота 2,2,2-Трифторэтанол 6 2,2,4-Триметилпентан 6
а Приблизительная длина волны, при которой относительное пропускание через кювету толщиной 1 см снижается до 10%.
6 Для измерения при коротких длинах волн растворитель непосредственно перед использованием следует продуть азотом,
в Цифры в СКобках указывают пропускание после продувки азотом в течение 15 мин.
8*
228
Глава 14
14.3. РАСТВОРИТЕЛИ ДЛЯ УЛЬТРАФИОЛЕТОВОЙ
СПЕКТРОСКОПИИ
Обычно ультрафиолетовые и видимые спектры снимают в очень
разбавленных растворах (10~4—10-6 моль/л). В исследуемом диа-
пазоне длин волн растворитель должен быть прозрачным.
Длина волн ы, мкм
Длина волны.' м/см
Частота, см~*
Рис. 14.10. Спектры пропускания различных растворителей (толщина кюветы
10 мм, относительно воды) [О: 1472].
/ — гептан; 2—метанол; 3—диметилформамид; 4—диметилсульфоксид; 5—гексан; 6—этанол;
7—диоксан; 3 —бензол; 9—апетон; 10— изооктан; 11 — изопропанол; 12—диэтиловый эфир;
13—тетрагидрофуран; 14— пиридин; 15— ацетонитрил; 16— циклогексан; 17—метиленхлорид;
18—хлороформ; 19—четыреххлористый углерод.
В табл. 14.2 указаны нижние пределы длин волн для растворите-
лей, обычно использующихся в спектроскопии. При длинах волн
ниже указанных пределов растворитель характеризуется избыточ-
ным поглощением, что не позволяет записывать поглощение образ-
ца в линейной шкале. Полные УФ-спектры некоторых обычных
растворителей, использующихся в спектроскопии, показаны на
рис. 14.10.
Глава 15>
ИНФРАКРАСНАЯ СПЕКТРОСКОПИЯ
Обзорная литература: 56, 71, 89, 127, 191, 274, 313, 316, 339, 359, 389, 431,
437, 554, 669, 730, 913, 965, 991, 1060, 1120, 1158, 1213, 1267, 1282, 1332, 1393.
При прохождении инфракрасного света через образец полимера
некоторые из частот поглощаются, тогда как другие пропускаются.
Наблюдаемые при поглощении инфракрасного света переходы свя-
заны с колебательными изменениями в молекуле. Разные связи,
содержащиеся в полимерах (С—С, . С=С, С—О, С=О, О—Н,
N—Н и т. д.), имеют различные частоты колебаний. Присутствие
этих связей в полимерах можно определить путем идентификации
характеристических частот как полос поглощения в инфракрасном
спектре.
Инфракрасная спектроскопия полимеров касается интервала
частот 650—4000 см-1 (15,4—2,5 мкм). Область частот 650—10 см-1
называется дальней инфракрасной, а область 4000—12 500 см-1 —
ближней инфракрасной областью.
Таблица перехода от длин волн в микрометрах к волновым
числам в обратных сантиметрах приводится в приложении Г.
Следующие пособия полезны для отыскания инфракрасных,
спектров низкомолекулярных органических веществ: [О: 96, 97,
101, 378, 453, 580, 845, 911, 1061, 1283—1285, 1430, 1468, 1471,
1477—1479].
Для идентификации инфракрасных спектров полимеров, пласт-
масс, каучуков и различных добавок, применяющихся в процессах
образования и переработки полимеров, рекомендуется пользо-
ваться следующими источниками: [О: 46, 124, 211, 239, 360, 405,
580, 582, 608, 652—657, 693, 701, 730, 982, 1213, 1268, 1324, 1441,
1445] .
Известны два основных типа молекулярных колебаний
(рис. 15.1):
1. Валентные колебания — это ритмическое движение вдоль
оси связи, при этом межатомное расстояние то увеличивается, то
уменьшается.
2. Деформационные колебания заключаются в а) изменении
углов между связями с общим атомом или б) движении группы
атомов относительно остатка молекулы, не затрагивающем атомы
в самой группе.
Молекула, содержащая п атомов, имеет Зп колебательных сте-
пеней свободы, три степени свободы описывают вращения и три —
трансляции, оставшиеся 3/г — 6 степеней свободы представляют
230
Глава 15
собой колебательные степени свободы основных колебаний. Пра-
вило Зп — 6 не применимо к СН2-группе, так как последняя пред-
ставляет собой только часть молекулы.
Редко когда наблюдается
колебаний, так как обертоны
симметричные
валентные
(^СНг)
асимметричные
валентные
(«'ggCHZ )
а
Ф Ф
, плоскостные
деформационные
или ножничньк
(85СН2)
внеплоскостные
деформационные,
или веерные
(и> СН2)
Фл zP
внеплоскостные
дец> ормац ионные,
или крутильные
(тСН2)
5
плоскоотные
деформационные,
или маятниковые
</>СН2)
точно теоретическое число основных
(кратные значения данной частоты)
и комбинационные тоны (сумма
двух других колебаний) приво-
дят к увеличению числа полос, а
вследствие других явлений число
их уменьшается. К числу послед-
них явлений относятся следую-
щие: 1) попадание основных ча-
стот вне пределов области 650—
4000 см-1 (15,4—2,5 мкм);
2) слишком слабые для наблю-
дения основные частоты; 3) рас-
положение основных частот очень
близко друг к другу, так что они
сливаются; 4) наличие вырожден-
ной полосы от нескольких погло-
щений одной и той же частоты в
молекулах с высокой симметрией;
5) неспособность проявления ря-
да основных колебаний в инфра-
красной области вследствие от-
сутствия требуемого изменения
дипольного характера молекулы.
15.1.
Колебания СНг-группы.
и минус характеризуют
перпендикулярные пло-
ПРИБОРЫ ДЛЯ
ИНФРАКРАСНОЙ
СПЕКТРОСКОПИИ
15.1.1.
Рис. 15.1.
Знаки плюс
движения,
скости страницы в направлении к
читателю и от читателя [О: 1213].
а—валентные колебания! б—деформацион-
ные колебания.
Двухлучевой
спектрометр
Двухлучевой инфракрасный спек-
трометр (рис. 15.2) имеет сле-
дующие части:
1. Источник излучения, который образуется при нагревании
электрическим током до 1000—1800 °C штифта — оксидов цирко-
ния, тория и церия (7100—1000 см-1) или карбида кремния
(5500—600 см-1). Инфракрасное излучение затем делится на
два пучка и с помощью системы зеркал направляется на
образцы.
2. Зона образца, в которой можно разместить кюветы самых
различных конструкций. Сравнительный и рабочий пучки проходят
через кювету сравнения и кювету с образцом,
Инфракрасная спектроскопия
231
3. Фотометр — оптическая система нуля. Для измерения погло-
щения образца нужно сопоставить интенсивности сравнительного
пучка и луча, прошедшего через образец (рабочего пучка). Пучок
сравнения и рабочий пучок проходят через аттенюатор и гребенку
соответственно и отражаются системой зеркал на вращающееся
секторное зеркало, которое попеременно отражает или пропускает
пучки на монохроматическую щель. После прерывателя пучки по-
падают на детектор (термопара) и усиливаются. При одинаковой
Рис. 15.2. Схема устройства автоматического двухлучевого инфракрасного
спектрофотометра.
1 — источник излучения; 2—зеркала; 3 —кювета с образцом; 4 — зона образца; 5 — кювета
сравнения; 6—аттенюаторная гребенка; 7—гребенка; 8 — сервомотор; 9 — прерыватель;
10—монохроматор; 11 — детектор (термопара); 12 — усилитель; 13 — самописец.
интенсивности обоих пучков выходной сигнал на усилителе отсут-
ствует. При наличии разности интенсивностей образуется выходной
сигнал, имеющий частоту прерывателя. Этот сигнал усиливается
и приводит в действие аттенюатор, который вводится в сравни-
тельный пучок или выводится из него как отклик на сигнал, по-
даваемый детектором под влиянием рабочего пучка. Положение
аттенюатора характеризует относительное поглощение образца, пе-
редача этого положения на цифровое устройство дает количе-
ственное выражение поглощения.
4. Система записи результатов. Отношение интенсивностей
сравнительного и рабочего пучков (/0//) поступает на перьевой
самописец, который в линейной шкале рисует зависимость пропу-
скания (Т) от волнового числа (v).
Инфракрасные спектрометры выпускаются для областей от
4000 до 600 см-1 (и даже вплоть до 200 см*1).
* В настоящее время стала обычной автоматизация записи ин-
фракрасных спектров путем цифрового преобразования сигна-
ла спектрометра. На бумажной ленте приводятся положение,
№
Глава 15
интенсивность и полуширина полос поглощения. С помощью элек-
тронного программирующего устройства регулируются циклы вво-
да образца, сканирование по длинам волн и цифровая запись. На-
ряду с цифровыми данными, автоматически печатающимися на
машинке, приборы также выдают обычные спектры. Число спек-
тров, имеющихся в различных атласах, каталогах или других
публикациях, превышает 100 000. В настоящее время стало возмож-
ным использование цифровых данных и программ, имеющихся в
каталогах спектров [О: 1468, 1477—1479]..
Обзорная литература: 257, 340, 926, 1060, 1120, 1438.
Периодическая литература: 2116 3235, 3257, 3747, 4320, 4342, 4353, 4699»
5143, 5674.
15.1.2 . Оптические материалы для ИК-спектроскопии
Оптические материалы, предназначенные для использования в
ИК-спектроскопии, должны быть прозрачными (пропускать ИК-из-
Рис. 15.3. Области прозрачности различных инфракрасных оптических материя*
лов [О: 1471].
лучение) в диапазоне 10 000—10 см-1 (1—1000 мкм). Наиболее
часто используют перечисленные ниже материалы.
1. Хлористый натрий (NaCl), пригодный для применения в об-
ласти 5000—625 см-1 (2—16 мкм). Пластинки из NaCl можно
использовать при повышенных температурах вплоть до 250°C, но
они непригодны при низких температурах. Их нужно беречь от
термоудара и резких механических нагрузок. Пластинки из NaCl
легко нарезать металлической пилой. Помещая такие пластинки на
металлические или стеклянные плиты, их обрабатывают суспен-
зией карборунда в воде или тонкой карборундовой бумагой, после
чего полируют на притире полирующим составом, который смешан
с небольшим количеством воды. Окончательную полировку про-
водят в сухой атмосфере. Полированные пластинки хранят в экси-
каторе.
2. Бромистый калий (КВг), пригодный для применения в об-
ласти 5000—400 см-1 (2—25 мкм). КВг мягче и пластичнее NaCl,
его используют в качестве материала окон в жидкостных кюветах,
Инфракрасная спектроскопия
233
а также как основу при изготовлении прессованных дисков или
таблеток.
3. Хлористое серебро (AgCl), пригодное для применения в об-
ласти 5000—435 см-1 (2—23 мкм). AgCl не растворяется в воде
и органических растворителях, это очень мягкий, неколющийся
материал. Полировать его чрезвычайно трудно. AgCl можно рас-
плющить практически до любой толщины, его легко прессовать
и плавить. Под действием света с длиной волны 500 нм AgCl
быстро темнеет, что приводит к уменьшению пропускания. AgCl
используют в качестве материала в низкотемпературных кюветах.
ИК-спектры ряда стандартных оптических материалов, исполь-
зующихся в ИК-спектроскопии, приведены на рис. 15.3 и в при-
ложении Д.
Обзорная литература: 946.
Периодическая литература: 5178.
15.1.3 . Обращение с оптическими материалами,
использующимися в ИК-спектроскопии,
и их хранение
Оптические материалы, использующиеся в ИК-спектроскопии, под-
вержены действию воды и ее паров. При сорбции воды поверх-
ность солевой пластинки местами мутнеет и рассеивает попадаю-
щее на нее инфракрасное излучение. Пластинки из NaCl и КВг
можно использовать при относительной влажности 30—40% в те-
чение нескольких часов. Хранить их следует в эксикаторе. Ни в
коем случае нельзя их трогать руками, брать пластинки можно
лишь в хирургических перчатках. После использования солевые
пластинки необходимо тщательно промыть растворителем, высу-
шить под лампой (за исключением пластинок из AgCl) в сухой
атмосфере.
Обзорная литература: 946, 1066.
15.2. МЕТОДИКА ПРИГОТОВЛЕНИЯ ОБРАЗЦОВ
15.2.1. Твердые полимеры
Для записи спектров твердых веществ используются следующие
приемы:
1. Прессование таблеток (дисков) с КВг. Мелко измельченный
полимер тщательно размешивают с бромистым калием. На 2 мг
полимера берут 100—200 мг, прессуют прозрачную таблетку. КВг
необходимо обезводить сушкой при 105 °C. Если можно, то перед
измельчением образец полимера и КВг следует нагреть до 40°C
под инфракрасной лампой, для того чтобы избежать конденсации
атмосферной влаги. Последняя дает широкую полосу поглощения
234
Глава 15
в интервале 3500—3400 см~1. Измельчение обычно проводят в ага-
товой ступке в течение 10—15 мин или вибрационной мельнице
(1—2 мин). Важно, чтобы дисперсия была мелкой и однородной,
так как плохо измельченные смеси в таблетке больше рассеивают
свет, чем его пропускают. Размер частиц должен быть меньше
2 мкм. Для получения качественной таблетки прессование прово-
дят при высоких давлениях (15 мин при давлении 2 тонны и еще
15 мин при давлении 8—10 тонн) в вакуумируемых пресс-формах
(рис. 15.4). Эти формы конструируют таким образом, чтобы можно
Рис. 15.4. Вакуум-форма для приго-
товления таблеток (дисков) КВг
[О: 1470].
/ — основание формы; 2—герметизирующая
прокладка; 3 —таблетки; 4 — цилиндр
пресс-формы; 5—пуансон; 6—герметизи-
рующая прокладка к пуансону; 7 —образец;
8 — трубка для подсоединения к вакууму.
Рис. 15.5. Принцип действия уик-
стика для приготовления образца.
1 — образец с сорбентом; 2—уик-стик;
3—крышка; 4~элюент.
было проводить удаление воздуха из таблетки, находящейся под
давлением, с помощью подсоединенного вакуум-насоса. Получен-
ные таким способом таблетки обычно имеют толщину 0,3 мм и
диаметр 13 мм. Края таблеток следует тщательно обработать. Хо-
рошо сделанная таблетка прозрачна. Для изготовления качествен-
ных таблеток необходимы определенные навыки. Следующие со-
веты могут оказаться полезными:
а) следует использовать порошкообразный мелкозернистый
КВг, при наличии крупных зерен в таблетке могут быть белые
пятна;
б) из-за влаги поверхность таблетки может быть грубой, ше-
роховатой;
в) недостаточная прозрачность таблетки является часто след-
ствием неудовлетворительного размалывания полимера в смеси с
порошкообразным КВг, а также слишком низкого давления или
плохого удаления воздуха;
г) при слишком продолжительном размалывании таблетка мо-
жет стать мелоподобной;
Инфракрасная спектроскопия
235
д) такие эластомеры, как, например, каучук, нужно измельчать
в вибрационной мельнице при низкой температуре (—40°C).
2. Приготовление таблеток с КВг из микрообразцов. При про-
ведении анализов методом тонкослойной хроматографии
(разд. 21.15) получают микрообразцы, которые затем должны быть
подвергнуты ИК-спектроскопии.
После выделения образца, содержащего анализируемое соеди-
нение, из поля хроматограммы удаляют сорбент, вещество извле-
кают соответствующим растворителем, фильтруют для удаления
сорбента и, наконец, смешивают с КВг.
Помещая пористый треугольник прессованного КВг, фирмен-
ное название которого «уик-стик» (Wick-Stick), в маленький стек-
лянный пузырек, закрытый таким образом,
что испарение происходит только в сере-
дине пузырька, можно осуществить филь-
трацию сорбента и нанесение образца на
КВг в один прием. Сорбент, содержащий
требуемый образец, соскребают с пластин-
ки для тонкослойной хроматографии и пе-
реносят в стеклянный пузырек, содержа-
щий уик-стик, с помощью воронки с тонким
носиком, для того чтобы сорбент не садил- -
ся на верхнюю половину прессованного
треугольника. Добавляют соответствующий
элюент и закрывают пузырек крышкой с
отверстием.
Использование уик-стика показано на рис. 15.5. Прессованный
треугольник из КВг имеет высоту 2,5 см, ширину у основания
0,8 см и толщину 0,2 см. Растворитель поднимается по этому тре-
угольнику под влиянием капиллярных сил, испарение происходит
преимущественно на вершине треугольника, где и осаждается ис-
следуемый образец. Для ускорения испарения растворителя тем-
пературу можно поддерживать на 10—20 °C ниже температуры ки-
пения растворителя и продувать пузырек током воздуха, направ-
ляемым через крышку.
Периодическая литература: 3596.
3. Лиофильный способ. По этому методу измельченный поли-
мер (около 200 мг) помещают в маленькую емкость, содержащую
~2 г КВг в 5 мл воды. Эту емкость соединяют со специальным
вымораживающим устройством (рис. 15.6). К такому устройству
можно одновременно подсоединить несколько колбочек с различ-
ными образцами. Затем растворы в колбочках охлаждают до тем-
пературы замерзания и дают вакуум. Сублимирующийся лед со-
бирается в холодильнике, заполненном сухим льдом. Спустя 6 ч
дегидратация полностью завершается, при этом полимер
Сухой лев
(СО»)
Рис. 15.6. Прибор для
лиофильной сушки.
236
Глава IB
полностью покрывается КВг. Затем образец измельчают в агато-
вой ступке и готовят таблетку обычным способом.
Обзорная литература: 210.
Периодическая литература: 2089, 4102, 7290.
4. Суспензионный способ. Полимер в виде мелких частиц дис-
пергируют в капле суспензионной жидкости, например жидком
Волновое число, cM~f
5000 3000 20001500 1000900800 700
Нуйол
Гексамар&утодиен
Келъ-F
2 3 4 5 6 7 8 9 10 11 12 13 14 15
Длина волны, мкм
Рис. 15.7. Области прозрачности различных жидкостей для приготовления сус-
пензий [О: 1471].
парафине (нуйол), гексахлорбутадиене или хлорфторуглеводоро-
дах (Кель-К). Затем суспензию помещают между двумя пластин-
ками из NaCl, причем толщина слоя при этом составляет 0,1 —
0,01 мкм.
Такие суспензионные жидкости прозрачны в широком диапа-
зоне частот (рис. 15.7 и приложение Ж).
15.2.2. Изотропные пленки
Существует несколько способов приготовления изотропных пленок.
1. Сухой метод. Испаряют растворитель из полимерного рас-
твора, нанесенного на горизонтальную поверхность между двумя
проволочками (рис. 15.8). Стеклянную пластинку тщательно моют
и протирают мягкой тряпочкой, смоченной раствором дихлорди-
метилсилана в четыреххлористом углероде. После испарения рас-
творителя пластинку выдерживают в воде. После такой обработки
пластинка имеет водоотталкивающую поверхность, от которой
большинство полимеров можно легко отделить. Наиболее равно-
мерные по толщине пленки получают при медленной сушке, для
чего пластинку с нанесенным раствором полимера покрывают пе-
ревернутым маленьким лабораторным стаканом. Для удаления
следов растворителя пленку прогревают в вакууме.
Для приготовления пленок одинаковой толщины очень удобна
их отливка на целлофановой подложке. Для этого конец стеклян-
ной трубки диаметром 2—3 см накрывают смоченным в воде цел-
лофаном, который при высыхании натягивается, давая совершенно
гладкую поверхность (рис. 15.9). Такую трубку или, точнее, коль-
цо помещают на столик и наливают на целлофан отмеренное ко-
личество раствора полимера (1—10 вес. %). Скорость испарения
Инфракрасная спектроскопия
237
растворителя можно уменьшить, закрывая целлофан часовым стек-
лом; таким способом получают ровную, гладкую пленку. После
Рис. 15.8. Отливка полимерной
пленки на стеклянной пластинке.
1 — столик; 2—проволочки; 3— раствор по-
лимера; 4 — стеклянная пластинка.
Рис. 15.9. Отливка полимерной плен-
ки на поверхности целлофана.
/ — раствор полимера; 2—целлофановая
пленка; 3 — стеклянное кольцо или стеклян-
ная трубка.
испарения растворителя полимерную пленку отделяют от целло-
фана, для облегчения снятия пленки целлофан весьма сильно
увлажняют. Перед снятием спек-
тра полимерную пленку необхо-
димо высушить в вакууме.
Можно полимерную пленку
наносить непосредственно на пла-
стинку из NaCl, для чего испа-
ряют растворитель из полимерно-
го раствора, налитого на поверх-
ность пластинки.
2. Мокрый метод. Полимер-
ную пленку получают осажде-
нием из раствора на стеклянной
пластинке, которую погружают в
осадитель полимера, смешиваю-
щийся с растворителем, исполь-
зующимся для приготовления
раствора полимера.
3. Расплавный способ. Этот
способ заключается в плавлении
полимера, заключенного между
двумя плоскими полированными
металлическими пластинками,
Рис. 15.10. Получение пленки для
ИК-исследований из расплава поли-
мера между двумя плоскими поли-
рованными металлическими пластин-
ками.
1 — рамка, с помощью которой задается
толщина образца; 2— полированные метал-
лические пластинки; 3—образец полимера.
зазор между которыми устанавливается с помощью специальной
рамки. Плавят полимер под высоким давлением при необходимой
238
Глава 15
температуре (рис. 15.10). Быстрое охлаждение способствует боль-
шей амортизации образца.
Толщина пленок для ИК-спектроскопии должна составлять
5—200 мкм. Толщину пленки определяют с помощью микрометра.
15.2.3. Ориентированные пленки
Для получения ориентированных пленок для ИК-спектроскопии
используют вытяжку или вальцевание полимерных пленок.
1. Вытяжка. Подвергающиеся вытяжке пленки должны быть
равномерны по толщине и ширине и иметь края, отрезанные ост-
рым ножом, бритвой или скальпелем. Если на краях пленки
имеется какой-то надрез, то он при вытяжке еще увеличится. Одни
полимеры легко вытягиваются при комнатной температуре, дру-
гие, которые при этом рвутся, следует вытягивать при повышенных
температурах или в присутствии пластификатора. В ряде случаев
в качестве пластификатора может выступать небольшое количество
растворителя для данного полимера. Эффективен также и водяной
пар.
2. Вальцевание. Механическое вальцевание проводят между
цилиндрическими валками маленькой мельницы. Для тонких пле-
нок, использующихся в ИК-спектроскопии, такую операцию про-
водить сложно. Вальцевание проходит легче, если пленка отлита
на деформируемом материале, например свинце, кадмии или хло-
риде серебра, который при вальцевании становится тоньше вместе
с полимерной пленкой.
Наиболее часто используются листы из хлорида серебра, ко-
торый по механическим свойствам близок к свинцу и прозрачен
в ИК-области спектра. Благодаря этому необязательно удалять
AgCl из полимерной пленки. Если это все же необходимо, то сво-
бодную полимерную пленку можно получить путем удаления хло-
рида серебра растворением его в водном растворе тиосульфата
натрия.
3. Многие полимерные растворы образуют пленки с некоторой
ориентацией молекул, если в процессе сушки их постоянно под-
вергать сдвигу, например перемещая раствор в заданном направ-
лении с помощью плоской ленты. Высокомолекулярные полимеры,
как правило, ориентируются легче, чем их низкомолекулярные
аналоги.
15.2.4. Вырезание полимерных образцов
Некоторые полимерные образцы являются слишком толстыми для
того, чтобы с них можно было снимать ИК-спектры, причем их
толщину нужно уменьшить, не меняя физического состояния пу-
тем плавления или растворения. Существуют два способа умень-
шения толщины пленки.
Инфракрасная спектроскопия
239
1. Вырезание микротомом. Образец вставляют в маленький
металлической блок (рис. 15.11) и затем отрезают стеклянным
ножом, в качестве которого выступает
край только что разбитого куска стек-
лянной пластинки. Нельзя определить
угол направления резки такого ножа;
этот угол очень сильно зависит от
природы полимерного образца. Режу-
щую поверхность смазывают разбав-
ленным водным детергентом или та-
кими органическими растворителями,
как спирты, ацетон или лигроин.
К блоку образцы прикрепляют с по-
мощью эпоксидной смолы, однако
есть опасность, что такие смолы могут
реагировать с исследуемым образцом.
После вырезания образцы закрепляют
Рис. 15.11. Вырезание полимер-
ного образца с помощью мик-
ротома.
1 — микротом; 2 — образец полимера;
3 — эпоксидная смола; 4—металли-
ческий блок.
в соответствующих рамках или на солевых пластинках.
2. Шлифование хоном тонкодисперсного оксида алюминия.
Обзорная литература: 70, 618.
Периодическая литература: 2106, 2969—2971, 3899, 3923, 4262, 6484.
15.2.5. Удаление из спектров пленочных образцов
интерференционных полос
В спектрах свободных пленок могут наблюдаться интерференцион-
ные полосы. Их можно исключить путем отливки полимерной
пленки непосредственно на солевой пластин-
ке; покрытием пленки иммерсионной жидко-
стью и
легким
тонкой
4
2.
Рис. 15.12. Полимер-
ный образец, поме-
щенный в иммерсион-
ную жидкость для
уменьшения рассея-
ния от поверхности.
1 — клей; 2—пластинки из
NaCl; 3—иммерсионная
жидкость; 4 — образец по-
лимера.
запрессовкой ее на солевой пластинке;
матированием пленки с помощью очень
шлифовальной шкурки.
15.2. .
Уменьшение рассеяния от
поверхности образцов
Некоторые полимеры, будучи мелко измель-
ченными, вытянутыми, вальцованными или
вырезанными, могут иметь такую поверхность,
которая в значительной степени способна к
рассеянию инфракрасного излучения. Для
снижения этого эффекта образцы погружают
в соответствующую иммерсионную жидкость,
например жидкий парафин (нуйол), гексахлорбутадиен или хлор-
фторуглеводород (Кель-F). Такие иммерсионные жидкости про-
зрачны в широком интервале длин волн (см. рис. 15.7 и приложе-
ние Ж) и не оказывают влияния на большинство полимеров.
240
Глава 15
Полимерный образец закрепляют между двумя солевыми пластин-
ками так, как это показано на рис. 15.12. Для герметизации реко-
мендуется пользоваться жидким белковым клеем (секотин). Клеев,
содержащих фенол (обнаруживается по запаху), следует избегать.
Обзорная литература: 405, 618.
15.2.7. Приготовление образцов полимерных гелей
для спектроскопических исследований
Для исследования нерастворимых полимерных гелей их зажимают
между солевыми пластинками или запрессовывают между пла-
стинками из хлорида серебра. Таким способом редко удается по-
лучить образец с равномерной толщиной, но снять с него удовле-
творительные спектры все же можно. Советуют также подвергать
гель набуханию в том или ином растворителе и уже потом зажи-
мать между пластинками. Полосы растворителя можно компенси-
ровать путем регулирования толщины кюветы сравнения в инфра-
красном спектрометре.
Обзорная литература: 618.
Периодическая литература: 5306, 6183.
15.2.8. Приготовление волокон для спектроскопических
исследований
Спектроскопические исследования натуральных и синтетических
волокон предполагают сохранение или разрушение формы волокна.
В методиках, при которых волокно сохраняется неизменным,
исследуют отдельные волокна или пучки волокон (микроспектро-
скопия и ослабленное полное внутреннее отражение).
В методиках, связанных с разрушением волокна, исследова-
ниям подвергают прессованные с КВг диски, пленки, отлитые из
раствора, пряжу, пленки, полученные горячим прессованием, ди-
ски, сформированные холодным прессованием, продукты пиролиза
и гидролиза.
Обзорная литература: 76, 405, 608, 926, 1249, 1390, 1441, 1445, 1462.
15.2.9. Микроспектроскопия отдельных волокон
Для исследования отдельных волокон используют приставку с
отражающим микроскопом. Диаметр волокна при этом обычно
больше требуемого, толщину уменьшают с помощью микротома,
шлифования (не влияющего на ориентацию волокна) или вальце-
вания (влияющего на ориентацию).
Микротомирование осуществляют путем закрепления волокна
В подходящем воскообразном материале, например парафине, кол-
Инфракрасная спектроскопия
241
лодии или полиметилметакрилате, после чего вырезают с помощью
стеклянного ножа под углом 90°.
Обзорная литература: 70, 1249.
Периодическая литература: 2106, 2451, 3310, 3496, 3899, 4262, 6637.
15.2.10. Спектроскопия пучков волокон
Для получения параллельного пучка волокон используют U-образ-
ные металлические зажимы (рис. 15.13, а) или волокна закреп-
ляют между двумя солевыми пластинками (рис. 15.13,б). Удов-
Рис. 15.13. Устройства для зажима волокон для ИК-исследований.
а—U-образные металлические зажимы; б—две пластинки из NaCl.
Пучок параллельна
с лалее иных волокон
летворительные спектры получаются и для сухих волокон, но по-
верхность их рассеивает ИК-излучение. С целью уменьшения рас-
сеяния волокна смачивают иммерсионной жидкостью.
Обзорная литература: 1249.
Периодическая литература: 3031, 3456, 5282, 5509, 6128, 6469, 6470.
15.2.11. Жидкие полимеры
Жидкие полимеры, например силиконовые масла, исследуют в
виде жидких пленок. Каплю исследуемой жидкости помещают
между двумя солевыми пластинками, которые затем закрепляют
в металлической рамке. В луч сравнения ставят солевую пла-
стинку двойной толщины. При исследовании жидкостей с низкой
вязкостью применяют кюветы для жидких сред, использующиеся
также при изучении растворов.
15.2.12. Растворы
Вследствие того что все растворители имеют специфическое погло-
щение, выбор растворителя для инфракрасной спектроскопии со-
пряжен с известными трудностями. На рис. 15.14 и 15.15, а также
в приложении Ж приведены спектры большинства распространен-
ных растворителей, применяющихся в ИК-спектроскопии?
242
Глава 15
Толщина
слоя
4000
0,2мм Е
0,05мм Е
0,2мм Е
Ацетонитрил
Бензол
Сероуглерод
Четыреххлористый углерод 0,2мм
Хлороформ
Дейтерохлороформ
Дихлорэтан
Диметилрормамид
н-Гептан
Гексахлор бу та -
.диен-1,3
Гель-Г №3
Метиленхлорид
Жидкий парарин
Пиридин
Тетрахлорэтилен
0,2 мм
0,2 мм
0,2 мм
о,О5мм
0,2 ММ
*
*
0,2 мм
*
0,2 мм
о,2мм
Полиэтилен (порошок,) 0,2мм
Волновое число, см~1
3000 2000 1000 /00
rWW
i ui i
1Щ
JLL^
Рис. 15.14. Интервалы прозрачности различных растворителей [О: 1471]. Белые
окна на диаграмме отвечают областям, в которых данный растворитель пропу-
скает более 25% падающего света. Звездочками в колонке, где указана тол-
щина слоя, помечены растворители, которые анализировали в виде жидкой
пленки между пластинками из CsI. Данные для н-гептана и метиленхлорида
добавлены автором книги.
Волновое число, см4
5000 3000 2000 1500 1000 900 800 700
Сероуглерод
Метиленхлорид
Хлороформ
Четыреххлористый углерод
Тетрахлорэтилен
Метиленбромид
Бромоформ
2 3 4 5 6 7 8 9 /0 11 12 13 14 15
Длина волны, мкм
Рис. 15.15. Интервалы прозрачности для сероуглерода и галогенсодержащих рас-
творителей [О: 1213]. В интервалах между линиями пропускание при толщине
J мм превышает 25%.
Инфракрасная спектроскопия
243
С помощью двухлучевых инфракрасных спектрофотометров
автоматически обеспечивается исключение поглощения, обуслов-
ленного растворителем. Для этого используют кювету, заполнен-
ную чистым растворителем, что и позволяет компенсировать спе-
цифическое поглощение растворителя. В случае очень сильного
поглощения полная компенсация не достигается. Очень важным
условием является необходимость использования сухих раствори-
Рис. 15.16. Разъемная кювета для жидких сред [О: 1470].
1 — основание и задняя крышка; 2 — прокладка; 3 — заднее окно; 4 — прокладка между окнами,
определяющая толщину кюветы; 5 — переднее окно; 6 — отверстия для заполнения кюветы
раствором; 7—прокладка; 8 — передняя крышка; 9 — колпачковая гайка; 10 — входные отвер-
стия и заглушки.
телей, поскольку вода сильно поглощает при 3710 см-1 (2,7 мкм)
и 1630 см-1 (6,15 мкм).
Концентрация полимера в растворе составляет обычно 3—10%,
но иногда можно использовать даже 20%-ные растворы.
Наиболее распространены кюветы следующих трех типов:
а) герметично закрытые кюветы (для полимерных растворов ис-
пользуются редко) (рис. 15.16); в) кюветы с регулируемой тол-
щиной (рис. 15.17).
Кюветы для жидких сред состоят из двух окон, представляю-
щих собой солевые пластинки, между которыми помещается пере-
городка соответствующей толщины (0,1—0,01 мм), которая также
определяет объем исследуемого образца. Кюветы самых различ-
ных конструкций доступны в настоящее время. Раствор в кювету
вводят капиллярной капельницей или шприцем.
В кювете с регулируемой толщиной прокладка отсутствует.
Одно окно закрепляют в корпусе кюветы, тогда как другое пере-
мещается в поршневом уплотнении с помощью тщательно отгра-
дуированного микрометрического винта. Регулирование толщины
кюветы очень важно для облегчения компенсации поглощения
растворителя и достижения требуемой толщины исследуемого
образца раствора. Особое внимание следует обращать на про-
мывку после работы кювет такого типа и избегать их поврежде-
ния из-за чрезмерного зажимания.
Если с кюветами для ИК-спектроскопии не обращаться надле-
жащим образом, они быстро выходят из строя (разд. 15.1.3).
244
Глава 15
Толщину кюветы (/) определяют путем измерения толщины
прокладки микрометром с круговой шкалой или из интерференцион-
ных полос (рис. 15.18), используя выражение
/ = n/2Av, (15.1)
в сантиметрах, где п — число полных интерференционных полос
в определенном интервале волновых чисел Av (см-1). Величина
Av постоянна для данной толщины ячейки. На рис. 15.19 показана
Рис. 15.18. Образование
интерференционных по-
лос между близко рас-
положенными окнами
кюветы.
Рис. 15.17. Кювета с
регулируемой толщиной
[О: 1470].
/ — задняя крышка; 2—вход-
ные отверстия и заглушки;
3—профилированный бара-
бан микрометра; 4—отбор-
тованное окно.
Длина волны, мкм
7 8 9 Ю и
----!---]------г-
WWWVW
1 I__I_ I_
1400--------------1200_1000
Волновав число, см~*
Рис. 15.19. ИК-спектр
пустой кюветы.
интерференционная картина для пустой кюветы. Для получения
отдельных интерференционных полос окна кюветы должны быть
очень ровными, измерения следует проводить сразу же после по-
лировки окон. Поскольку со временем толщина кюветы меняется
из-за постепенного сжатия прокладки или эрозии окон, ее надле-
жит периодически проверять.
Для определения толщины кюветы используют также интерфе-
рометрический метод.
Обзорная литература: 1066.
Периодическая литература: 4281, 6633.
15.2.13. Многопроходные кюветы
Многопроходные кюветы используют для ИК-анализа фракций,
собираемых при газовой хроматографии. С помощью трех зеркал
в кювете (рис. 15.20) обеспечивается до 16 проходов (расстояние
в 1 м) через образец, полученный испарением. Для максимальной
отражательной способности и минимального износа во времени
зеркала покрывают золотом. Кювету нагревают с помощью элек-
трического тока до 250°C для испарения образца. Кюветы такого
Инфракрасная спектроскопия
245
Рис. 15.20. Многопроходная кювета.
/ — зеркала; 2—многопроходная кювета.
типа позволяют исследовать образец объемом 10 мл, содержащий
1% полимера. Такую кювету можно присоединить непосредственно
к газовому хроматографу и затем к инфракрасному спектрометру.
Периодическая литература: 3023, 7066.
15.3. КАЧЕСТВЕННЫЕ ИССЛЕДОВАНИЯ
ПОЛИМЕРОВ С ПОМОЩЬЮ ИНФРАКРАСНОЙ
СПЕКТРОСКОПИИ
Применение инфракрасной спектроскопии для характеристики по-
лимеров включает следующие основные исследования:
1. Определение структуры. Наибольшую ценность в инфракрас-
ной области для определения структуры имеет область частот ха-
рактеристических групп. Есть несколько способов представления
данных «группа — частота». Рис. 15.21 представляет обширную
картину длин волн, составленную в соответствии с классами сое-
динений. После снятия инфракрасного спектра выбираются наибо-
лее интенсивные полосы и по карте пытаются найти группы, соот-
ветствующие этим полосам. При этом могут возникать осложне-
ния, связанные с тем, что характеристические частоты часто пе-
рекрываются. В связи с этим нужно привлекать дополнительные
данные, в частности относительные интенсивности различных по-
лос, другие спектроскопические данные или какие-либо имею-
щиеся в распоряжении физические или химические характери-
стики. После отнесения наиболее сильных полос следует сделать
отнесение полос средней и даже слабой интенсивности. При этом
также может возникнуть ряд проблем, обусловленных наличием
обертонов и комбинационных тонов. Несомненную пользу оказы-
вает определение спектров модельных соединений. Наконец, в
соответствии с правилами отбора следует предположить ряд
4000
3500
3000
2500
2000
1800
1600
1400
1200
1000
800
Алканы
Алкены
Алкины
Ароматические
углеводороды j
Простые эфиры
Спирты |
/саоЛ) "У"
.(Из.) __|__
J I __г£-
Кислоты |
Г'11ЛМГ1Л1.1в п
— О'—;—неси ММ. I J__!_
---—симм. J_______I ।
лЗнаФталины
Г^Г“|~П
f J -4——' (винил I -сн=с(
—1—' ' Т-СН- I н
400см-1
ьэ
от
Сложные эфиры
Альдегиды
Кетоны ।
Ангидриды
AwudtJ I
4000
3500
2,50
Амины
вац-тризамещенныи I - । 1 ।
неси м м. | J д_ - 1 i । j
^Диафталины । | -j- } j
алифатические! r
ароматические J г
— j-1— -------[первичные^ спирты ‘
— ——j—1— вторичные i _!__________I
— |—J—j—!—j третичные | J________1
"j :—q—j—j—j—J--------|-ароматические J__L
-—1—J. карбоновые кислоты -co-ОН - I
ованныи ХЗПППКГПП /глии л ~-(1 ’ /
~ ngpvvnvbjmc AUVJlUlilDl -bU“UH
ио»изовднньш карбоксил (соли,цвиттер-ионы)1
[формиаты1 1—!L L h-co-o-rI I °
ацетаты J—<____l i-c.w.-cn-a-n I
ацетаты J—< L-CH.-C0-O-R Д_ _£• । I
пропионаты -j, I ,а , Т~ IT I ~i Г
бутираты и т.д. | _!_1 I i_ТТ | i 1
акрилаты . --!| = CH-C0-O-R ' ib_l ' Г
Фумараты J i . п ) i’T’i ~—г
малеаты | „ I i I 1 .1 , . Т| -j—p
бензоаты-фталаты । Oco-o-r i i.'TTi i_L
-* —г-алифатические{альТ>е-“СН2-СНО I L I I I
ч —ароматические/гиды |O-CH0 J___I , с I ~f
алифатические^ ' I CH3-C0-CHz 1 I. Г I I
I автоматические J№™H“ <yco-c —1_' , , I,
-< <“*> I—{-а*«Лм IJ—I—I_____Lco-nh, / 1 Cf I. I
“I I H=s^spss7 |-«s£" ффррТ
3OOO 2500
2000 1800
1600 1400
(m.)
ср.
1200 1000
800
600
2.75 3>ОО 3,25 3,50 3,75 4,00 4,5 5,0
5.5 6,0
6.5 7,0 7,5 8,0 9,0 10,0 Ji 12 13 14 15
400см'(
20 25мкм
; ! ; । । > I I 1
J- ‘—Iвторичные амины -
хлоргийрат-1— 1
Имины I 1_______ _
Нитрилы'
Смешанные
соединена;
третичные амины |—
J—<—1 c-NHjcr
имины I
1| замещенные ими ни
нитрил -СзЫ—!—!—L—
1зоцианиЙ -н’=с‘—!—[--
'«Ill'll
Х=С»Х (изоцианаты, производные
| | |«риено,|ИпЛ)|
гТ I П
пцие
Неорганические
и их производные
хлор-
йром-
conul
ср.
ср.
Отнесения
соединения ----------
| серв-кислород |
фосфор-кислород
—< SH
____ PH
CH?O-(Si,P, ORS)
5;н
сульфат-ион
СО СВЯЗЬЮ:
угольной кислоты 0=С(0-Я), ,1.
иминокарбонат hn-с(о-Н)г l£_!(
снммпн. кол.
хлорформиат
хлорангидрид С=0
с»о вал кол
—C-Ц вал кол
С-С вал. кол.
— —j--.ЫНд«р. кол.
111 р-
-I —С«Хвал. кол.
_а , ------ 1(50Аг‘
сульфонат-ион i R-
_сильфокислота R-S0.H !—|----------:—!
эфир серной кислоты! R-O-SO^-O-RI_, 1
эфир сульфокислоты Я-О-5О3-Н4_____L .
сульфонамид . I R-S03-NH2
сульфон ч R-SO*-R
сульфоксид R-S0-R . —1
фосфат-ион _1 I I
эфир фосфорной KucnoiM(R-O),P-> о|—.
углероЪ-кислород | карйонат-ион I (со.)
| | | | I зфир угольнойкислотыо=с(о-«)г
азот-iw слорой
эфир азотной кислоты R-0-N0,
нитросоединения R-NO3_____
I ”1 J I | I I I
эфир азотистой кислотыR- О-но __
нитрозосоеоинения r-no 1 i 1
. эммоний (NH4)* _L_ III I । । ।
.он
I 1 I I I
, мн вал. кол.
-< сн вал. кол.
:C-F (ненас.1
F -I — Z« f f
CBrt и
Инфракрасная спектроскопия
С-О вал. кол.
С—N вал кол.
_!___। с—Свал кол.
4000 3500 3000 2500 2000 1800 1600 1400 1200 1000 800 600 400см-’
Л--------1------1----1----й——1----1------1----1_______|______( (____1___I I I__________1_ J___I__l_J_J________I___I
2,50 2,75 3,00 - 3,25 3,50 3,75 4.00 4,5 5,0 5,5 6,0 6.5 7.0 7,5 8,0 9,0 10,0 И 12 13 14 15 20 25мкм
Фис. 15.21. Характеристические инфракрасные частоты поглощения для различных групп, расположенных в соответствии
с классами соединений (П: 2940].
Интенсивность полосы: с—сильная, ср. —средняя, сл. —слабая. Символом 2v обозначены обертоны.
248
Глава 15
возможных моделей и сравнить их с экспериментальными резуль-
татами. Полный колебательный анализ представляет значительные
трудности. В табл. 15.1 указаны работы, посвященные колебатель-
Таблица 15.1
Колебательный анализ различных полимеров с помощью
инфракрасной спектроскопии
Полимер Литература
Полиакрилонитрил
Полибутадиен
Полибутен-1
Полихлоропропен
Полиэтилен
Полиэтилен-2,6-нафта-
лат
Полиэтилентерефталат
Полиизопропилен
Полиметилакрилат
Полиметилметакрилат
Полипентен-1 и поли-4'
метилпентен-1
Полипропилен
Полистирол
Политетрафторэтилен
Полиуретаны
Поливинилацетат
Поливиниловый спирт
Поливинилхлорид
Поливинилиденхлорид
П: 2150, 4908, 4915, 6297
О: 565; П: 2407, 2409, 3370, 3702, 4678, 6400
П: 2882, 4996, 5445, 6891, 7247
П: 3364, 5156, 5477
О:-780; П: 2204, 2227, 2228, 2605, 4007, 4054, 4742,
4932, 5209, 5281, 5316, 5485, 5574, 6353, 6461, 6553,
6745, 6792, 6793, 7305
П: 5639
О: 780; П: 2191, 2462, 3776, 4909, 5281, 5309
П: 2408, 2410, 3036, 3077, 3701, 6044
П: 3850
П: 2299, 5855
П: 3566
О: 780; П: 3438, 3743, 3746, 4212, 4910—4916, 4995,
5188. 5318, 5319, 5767, 6468, 6671, 6673, 6793
О: 780;'П: 4907, 5560, 5673, 6673, 6675, 6676, 7157
П: 2598, 4906, 5102, 5378, 7320
П: 2524, 6344, 6725
П: 3851, 3852
О: 780; П: 4741, 4913, 5408, 6665, 6666, 6674, 6678,
6679, 6681
О: 780; П: 2205, 3319, 3777, 4733, 4736—4740, 5307,
5604, 5344, 6297, 6364, 6373, 6746, 6748
О; 780; П: 4740, 5439, 5440
ному анализу различных полимеров; эта литература может быть
полезной при определении конфигурации и конформации поли-
меров.
2. Определение структуры сополимеров. В ряде случаев с по-
мощью ИК-спектроскопии можно получить сведения о распреде-
лении последовательностей и тактичности в сополимерах. Так как
полосы поглощения находятся очень близко друг от друга и часто
перекрываются, то интерпретация таких спектров весьма затруд-
нена. Иногда также трудно бывает решить, обусловлены ли наблю-
даемые различия в данной области спектра нескольких сополи-
меров только изменениями в составе сополимеров или также влия-
нием неодинаковой микрогетерогенности.
Обзорная литература: 578.
Периодическая литература: 2615, 3235, 3318, 3624,
Инфракрасная спектроскопия
№
15.4. КОЛИЧЕСТВЕННЫЙ АНАЛИЗ
В ик-спектроскопии
Количественный анализ в инфракрасной спектроскопии основы-
вается на использовании закона Ламберта — Бера:
A = \g(I0/I) = acl, (15.2)
где А — поглощение (безразмерная величина); /0 — интенсивность
падающего инфракрасного излучения (или интенсивность облу-
чения, прошедшего через кювету сравнения); / — интенсивность
Рис. 15.22. Два способа построения базовой линии.
инфракрасного излучения, прошедшего через образец; а — коэф-
фициент поглощения (л*г-1 -см-1); с — концентрация раствора
(г-л-1); I — толщина кюветы с образцом (см).
Для того чтобы можно было сделать поправки на частичное
рассеяние инфракрасного излучения и перекрывание соседних пи-
ков поглощения, нужно, как это показано на рис.. 15.22, построить
базовую линию и оценить /0 как интенсивность пропускания отно-
сительно базовой линии.
250
Глава 15
Поглощение (Л) при определенной частоте (см-1) равняется
A = \g(I0/I) = lg(AC/AB) (15.3)
Для количественного анализа следует выбирать полосу в обла-
сти, в которой пропускание образца постоянно и составляет не
менее 25%.
На ИК-спектр влияет ряд факторов:
а) перекрывание полос;
б) различие в аморфно-кристаллическом состоянии образца;
Рис. 15.23. Схематическое изображение ИК-спектров аморфной (/) и кристал-
лической (2) областей полукристаллического полимера.
в) различие в конфигурации мономерных звеньев в полимерной
цепи, например различные стереохимические формы (синдитакти-
ческая и изотактическая);
г) образование водородных связей;
д) межмолекулярное взаимодействие в смесях полимеров или
привитых сополимерах.
На эти факторы следует обратить внимание перед тем, как на-
чать калибровку. Стандартные и модельные системы, которые
используют для калибровки, должны быть похожи на исследуемые
образцы.
Для сравнительных количественных измерений нужно использо-
вать пленки одинаковой толщины, однако это условие трудновы-
полнимо. Для того чтобы исключить влияние толщины, часто
применяют метод внутреннего стандарта. Для этого к порошко-
образному полимеру или его раствору добавляют известное коли-
чество вещества, имеющего легко определяемую полосу поглоще-
ния, например КЮ4, Pb(CNS)2- Путем сравнения поглощения
исследуемого полимера при определенной длине волны с погло-
щением внутреннего стандарта можно проводить количественное
определение, не заботясь о толщине слоя.
Обзорная литература: 1324, 1447.
Инфракрасная спектроскопия
251
15.5. ОПРЕДЕЛЕНИЕ СТЕПЕНИ КРИСТАЛЛИЧНОСТИ
ПОЛИМЕРОВ МЕТОДОМ ИНФРАКРАСНОЙ
СПЕКТРОСКОПИИ
Степень кристалличности (%с), определяемая методом ИК-спектро-
скопии, выражается как
= (15.4)
где Аа и Ас — поглощения при определенной частоте (см-1)
аморфного и кристаллического пиков соответственно (рис. 15.23).
Таблица 15.2
Частоты поглощения, используемые в инфракрасной спектроскопии
для определения кристалличности полимеров [О: 1427]
Полимер Поглощение, см 1
кристаллическая область аморфная область
Полиэтилен Политетрафторэтилен Полипропилен, изотактиче- ский Полипропилен, синдиотакти- ческий Поливиниловый спирт Поливинилхлорид Полихлортрифторэтилен Поли-2-хлорбутадиен Поли-г{мс-2-метилбутадиен Поли-тра«с-2-метилбутадиен, а Поли-тра«с-2-метилбутадиен, Р Полиэтиленсебацинат Полиэтилентерефталат Найлон-6 Найлон-6,6 Найлон-6,10 Целлюлоза Поли-3,3-бис (хлорметил) ок- сациклобутан, а Поли-3,3-бис (хлорметил) ок- сациклобутан, р 71, 730, 1050, 1178, 1889 809, 842, 894, 997 866, 977 1146 955, 1226, 1254, 1373, 1428 440, 490, 1290 952 ИЗО 800, 870, 890 750, 800, 890 806, 970 972 848 835, 936, 964, 970, 1029, 1201 936 852, 940 1372 525, 1318 525, 1310 1300, 1352, 1268 770 790, 1158 1131, 1199, 1230 2920 754 840 840 840 720, 847 790, 898, 1042 980, 990, 1076, 1118, 1170 1139 1126 2900
В табл. 15.2 приведены соответствующие частоты поглощения
для ряда полимеров.
Обзорная литература: 16, 713, 1427.
Периодическая литература: 2536, 3047, 3351, 4020, 5187, 5281, 5574, 5615,
6663, 6796, 6797, 7305, 7318.
252
Глава 15
15.6. ИНФРАКРАСНАЯ СПЕКТРОСКОПИЯ
ПОВЕРХНОСТИ ПОЛИМЕРНЫХ КРИСТАЛЛОВ
Иногда ИК-спектроскопией можно воспользоваться для изучения
природы складывания цепей на поверхности полимерных кристал-
лов, полученных из разных растворов. Отношение инфракрасного
поглощения для аморфной и кристаллической полос зависит от
природы использующегося растворителя и термической предысто-
рии полимера.
В полимерах с двойными связями среднее число мономерных
звеньев на складку можно рассчитать по ИК-спектрам продуктов
эпоксидирования.
Периодическая литература: 2226, 4016, 4633, 4639, 4735, 6255, 6564, 6565.
15.7. ИЗМЕРЕНИЕ МИКРОЗЕРКАЛЬНОГО ОТРАЖЕНИЯ
Для качественного контроля пленок на отражательных поверх-
ностях, например покрытиях пластиков, лаковых покрытиях, ла-
ках на консервных банках или эпоксидных покрытиях, обычно
Рис. 15.24. Приспособление для
микрозеркального отражения.
/ — образец; 2—зажим для образца; 3—зад-
няя стенка; 4—зеркала; 5—блок для креп-
ления зеркал.-.
применяют метод микрозеркаль-
ного отражения. Приспособление
(рис. 15.24) закрепляют непо-
средственно в кассете для обыч-
ных кювет, которая привинчи-
вается к основанию отделения
для образца спектрофотометра.
Достигаемое при этом пропуска-
ние обычно достаточно для изме-
рений на образцах под углом 20°
с помощью призмы, составленной
из двух плоских зеркал с алю-
миниевым покрытием. Образец
своей отражательной поверх-
ностью покоится на апертурной
пластинке, защищенной политет-
рафторэтиленовым покрытием.
Образцы удерживаются за счет
силы тяжести или пружинным
зажимом, который прикреплен к
пластинке для образца. В зависимости от диаметра апертуры и
зеркала с алюминиевым отражающим слоем пропускание обычно
колеблется в следующих пределах:
Диаметр апертуры, мм
2
Пропускание, %
28—-30
68-70
80-81
Инфракрасная спектроскопия
253
15.8. СПЕКТРОСКОПИЯ ВНУТРЕННЕГО ОТРАЖЕНИЯ
Спектроскопия внутреннего отражения (называемая также спектро-
скопией ослабленного полного отражения) представляет собой ме-
тод, основанный на внутреннем отражении. Падающий свет вна-
чале направляется на покрывающее образец вещество с высоким
показателем преломления (п\) под углом (0), большим критиче-
ского угла (0Сг), и после некоторого проникания вглубь образца
отражается от него, причем образец имеет более низкий показа-
тель преломления (п2) (рис. 15.25).
Когда полимерный образец, селективно поглощающий излуче-
ние, находится в контакте с отражающей поверхностью, некото-
рые частоты падающего пучка поглощаются, тогда как остальные
Таблица 153
Характеристики материалов для спектроскопии внутреннего отражения,
использующихся в видимой и УФ-областях
Материал Область пропускания, мкм Показатель пре- ломления («1) при 400 мкм
Вольфрамат кальция 300—3500 1,91
Титанат бария 400-6500 2,40
Титанат стронция 400—4000 2,65
Кристаллический кварц 200—2500 1,56
Оптическое стекло (флинт- 200-1500 1,45
глас)
Сапфир 200-6500 1,78
частоты пропускаются и отражаются. Такое ослабленное излуче-
ние измеряют и выражают как зависимость от длины волны, при
этом получается спектр поглощения образца, называемый спек-
тром внутреннего отражения.
Путем увеличения числа отражений можно усилить поглоще-
ние (рис. 15.26). При использовании внутреннего рефлектора раз-
мером 50X50X2 мм с углом падения 45° достигается примерно
25 отражений.
Спектроскопия внутреннего отражения применима к ультра-
фиолетовой, видимой и инфракрасной областям. В табл. 15.3 и
15.4 указаны материалы, пригодные для использования в качестве
пластинок для внутреннего отражения в различных областях
спектра.
Большинство инфракрасных спектрофотометров можно исполь-
зовать с приставкой для многократного внутреннего отражения
(МВО), которая устанавливается в отделении для образца. Су-
ществует много различных оптических систем, применяющихся в
МВО:
254
Глава 15
Рис. 15.25. Однократное внутреннее отражение. Условие для однократного пол^
ного внутреннего отражения: п\ > п2 и 0 > Всг.
<3
•§
Падающий
луч полимера
X. ।.1 Л .
Кристалл Образец
Образец Отраженный
Рис. 15.26. Многократные внутренние отражения.
полимера
Рис. 15.27. Оптические системы для многократных внутренних отражений с
двумя (я) и четырьмя (б) зеркалами [О: 1470].
1 — задняя стенка; 2 — зеркало; 3—основание; 4—кристалл.
1. Двухзеркальная оптическая система (рис. 15.27, а): девять
отражений со стандартным кристаллом, угол падения 45°. Неза-
висимо от толщины образца положение кристалла не меняется
относительно оптического пути. Благодаря простоте конструкции
такая система наиболее пригодна в обычной лабораторной прак-
тике.
2. Четырехзеркальная оптическая система (рис. 15.27,6):
25 отражений со стандартным кристаллом, углы падения 30, 45 и
60°. С помощью системы такого типа легко регулировать глубину
проникания света, при этом значительно увеличивается интенсив-
ность в спектрах веществ с низким поглощением.
Обзорная литература; 76, 571, 572, 768, 1369, 1374, 1389.
Отрицательные характеристики Хрупкий, рассыпается под да- влением На воздухе окисляется Высоко токсичен. Нельзя ис- пользовать с водными рас- творами Подвержен коррозии, вслед- ствие чего нельзя оставлять в контакте с металлически- ми поверхностями Очень дорог Очень дорог
Положительные характеристики Самый высокий среди извест- ных материалов показа- тель преломления и абсо- лютная нерастворимость Высокий (второй по поряд- ку) показатель преломле- ния, очень твердый и инертный кристалл Устойчив (не рассыпается) под высоким давлением Отражательные пластинки легко вырезаются из ру- лонного листа с помощью обычной бритвы; мягкий, легко деформирующийся Нерастворимость (идеаль- ный материал для всех жидких образцов)
Критический Угол (есг), град СМ О о о СМ СМ СО ’Ф
Показатель преломления (щ) при 10 мкм О СМ 00 о ’Ф СО О -Ф Q0 ГТ со" см" U см" см"
Область пропускания, мкм 00 «о CM LO СО 00 СД _Ч СО СО СМ г- см Till II 1 Ю ю 'Ф со со СМ о" o' o' —" см"
Материал 5 С4 03 s 'Т s'? 04 sc О Я 2 - *- S ’н 1 -г’- OJ S <У « и s w SU SCO Р-Н s I gstf £ N ф cu ф'-" ф^ & X X UH
256
Глава 15
Спектроскопия внутреннего отражения используется для ка-
чественной идентификации разнообразных полимерных образцов,
например пленок, клеев, бумаги и бумажных покрытий, порошков,
красок, волокон и пеноматериалов; изучения мономолекулярных
слоев; изучения молекулярной ориентации (спектроскопия поля-
ризованного внутреннего отражения) в полимерных пленках и
вытянутых волокнах; для определения оптических констант; изуче-
ния загрязнения поверхностей при машинной переработке, руками
человека или в контейнерах; для исследования процессов окисле-
ния и/или разложения полимерных поверхностей; изучения диффу-
зии в полимерные материалы и выпотевания различных компонен-
тов на поверхности; количественного анализа полимерных мате-
риалов.
Обзорная литература: 18, 76, 334, 572, 1389.
Периодическая литература: 2684, 2780, 3112, 4564, 6062, 6687, 6689, 7069.
15.9. ИНФРАКРАСНАЯ ОТРАЖАТЕЛЬНО-
АБСОРБЦИОННАЯ СПЕКТРОСКОПИЯ
Отражательно-абсорбционная спектроскопия основана на инфра-
красном излучении, поляризованном параллельно плоскости па-
дения. При угле падения (0) (рис. 15.28), лишь на несколько гра-
Рис. 15.28. Расположение образца для отражательно-адсорбционной спектро-
скопии с использованием многократных отражений под углом 0 от металличе-
ческих зеркал.
дусов меньшем 90°, падающий и отраженный лучи суммируются,
давая стоячую волну с заметной амплитудой у поверхности ме-
таллического зеркала; подобная волна может взаимодействовать
с молекулами сорбированной пленки.
Из-за высокой интенсивности поглощения для параллельно
поляризованного света и больших углов падения, не обусловлен-»
ных геометрической длиной пути инфракрасного излучения в орга-
нической пленке, инфракрасная спектроскопия этого типа резко
отличается от обычной ИК-спектроскопии.
Изменение отражательной способности (АТ?), обусловленное
наличием на отражающем металлическом зеркале полимерной
Инфракрасная спектроскопия
257
пленки, для 0° 0 80° составляет
др___ zed / 9,212 sin2 0
где е — коэффициент молярной экстинкции для полимера; с — кон-
центрация поглощающего полимера; d — толщина пленки; п — по-
казатель преломления поглощающего полимера; 0 — угол падения.
Область образца
о спектрофотометре
(15.5)
Падающий
луч
Металлическое
зеркало
- Л монохроматору
Зеркало
Полимерная
мта- заднее
зеркало
Рис. 15 29. Расположение образца для
скопии с использованием многократных
пленка
отражательно-абсорбционной спектро-
отражений от металлических зеркал.
На рис. 15.29 показано, как располагается образец для отра-
жательно-абсорбционной инфракрасной спектроскопии. Отражаю-
щее устройство может быть вмонтировано в обычный ИК-спектро-
метр. Инфракрасный свет, поляризованный параллельно плоскости
падения, можно получить с помощью поляризатора из AgBr (ди-
фракционная решетка), который ставится перед входной щелью
монохроматора.
Периодическая литература: 2465, 3475, 3764—3766.
15.10. ИНФРАКРАСНАЯ ФУРЬЕ-СПЕКТРОСКОПИЯ
Инфракрасная фурье-спектроскопия представляет собой метод, в
котором вместо монохроматора применяется интерферометр, на-
пример интерферометр Михельсона (рис. 15.30). Интерферометр
состоит из двух плоских зеркал, расположенных под углом 90°
друг к другу, и расщепителя луча под углом 45° к зеркалам. Одно
зеркало крепится стационарно, другое может перемещаться с по-
стоянной скоростью в направлении, перпендикулярном его фрон-
тальной поверхности.
9 Зак 238
258
Глава 15
Таблица 15.5
Расщепители света в средней и близкой
инфракрасной областях
Покрытие Подложка Интервал длин волн, мкм
Fe2Q3 Кварц 0,65—2,5
Fe2O3 CaF2 1,0-5,0
Ge КВг 2,7-25,0
Ge CsI 10,0-50,0
Расщепитель пучка делит падающий свет — 50% пропускается,
остальные 50% отражаются. Он представляет собой тонкое пле-
Падающий луч
Подвижное
зеркало
Закрепленное
зеркало
Расщепитель лучка
Компенсатор
— К детектору
Рис. 15.30. Схема интерферометра
Михельсона.
-* О
Рис. 15.31. Выходной сигнал для
интерферометра Михельсона как
функция расположения зеркала (х)
для монохроматического (а) и ши-
рокополосного (б) источника света.
Нуль относится к положению зер-
кала, при котором обеспечивается
одинаковый оптический путь для
двух плеч интерферометра.
му всех косинусных осцилляций,
ночное покрытие, нанесенное на
оптически ровную подложку
(табл. 15.5). Такой же толщины
подложка (называется компен-
сатором) помещается в одно пле-
чо интерферометра для уравни-
вания длин оптических путей в
обоих направлениях.
Если падающий свет является
монохроматическим, то сигнал де-
тектора (или интерферограмма)
проходит ряд максимумов (оба
световых - луча будут в одной фазе,
когда они возвращаются в расще-
питель пучка) и минимумов (при
возвращении в расщепитель пуч-
ка оба световых луча находятся
не в фазе). При постоянном дви-
жении зеркала сигнал осциллиру-
ет от максимума к минимуму (для
движения зеркала на каждую
четверть волны) (рис. 15.31, а).
Если же падающий свет явля-
ется полихроматическим, то сиг-
нал детектора (или интерферо-
грамма) представляет собой ре-
зультирующий сигнал для каж-
дой частоты падающего света.
Каждая входящая частота может
быть обработана независимо, по-
этому выходной сигнал (рис.
15.31,6) представляет собой сум-
которые вызываются всеми опти-
ческими частотами, содержащимися в падающем полихроматиче-
ском излучении.
Инфракрасная спектроскопия
259
Интерферограмма также содержит информацию об интенсив-
ности каждой частоты в спектре.
Рис. 15.32. Блок-диаграмма основных частей инфракрасного фурье-спектрометра.
/ — интерферометр Михельсона; 2—источник ИК-и'злучения; 3 —отделение для образца;
4 — кювета сравнения и кювета с образцом; 5 —двухлучевая оптика; 6 — детектор; 7 — анали-
тический цифровой преобразователь; 3 —контроллер; 9 — основной компьютер; 10— цифровая
печать; // — дисковая память.
С помощью компьютера выходная информация дается в циф-
ровом выражении и преобразуется в частотный домен: каждая
индивидуальная частота выделяется из сложной интерферограммы
Таблица 15.6
Колебательный анализ различных полимеров
с помощью инфракрасной фурье-спектроскопии
Полимер Литература Полимер Литература
Полиакрилонитрил Полибутадиен Полихлоропрен Полиэтилен О: 302 П: 2927, 3916, 4417, 5741 О: 302 О: 302; П: 5660, 5662 Полиэтилентерефта - лат Полипропилен Полистирол Поливинилхлорид П: 3141 О: 302 О: 302, 668' П: 5661 О: 302
(фурье-преобразование). Затем эти сигналы преобразуются в при-
вычный инфракрасный спектр. На рис. 15.32 приведена блок-диа-
грамма основных компонентов инфракрасного фурье-спектро-
метра.
Запись, обсчет и преобразование всего спектра занимают всего
лишь несколько секунд. (В обычных спектрометрах для записи
спектра требуется несколько минут.)
9*
260
Глава 15
Инфракрасная фурье-спектроскопия используется для опре-
деления слабых сигналов, изучения очень разбавленных раство-
ров образцов (0,5%-ная концентрация, 20 мкг образца), изучения
сорбированных монослоев (например, следов чернил на бумаге),
изучения ИК-спектров на отдельных кристаллах (например, кри-
сталле бензола диаметром 300 мкм), исследования в водных рас-
творах в области 950—1550 см-1, для колебательного анализа
(табл. 15.6), исследования конформационно-чувствительных ин-
фракрасных полос.
Обзорная литература: 73, 95, 302, 542—544, 639, 659, 668, 834, 840, 841, 883,
1330.
Периодическая литература: 2925—2928, 4129, 4632, 5661, 5663, 6663.
15.11. БЛИЖНЯЯ ИНФРАКРАСНАЯ СПЕКТРОСКОПИЯ
К ближней инфракрасной области относится интервал частот от
12 500 до 4000 см-1. Полосы поглощения в ближней инфракрасной
области — это преимущественно обертоны инфракрасных низко-
частотных полос (рис. 15.33). Из-за сложного характера перекры-
вающихся полос в ближней инфракрасной области отнесение полос
достаточно затруднено.
Для снятия ИК-спектров в ближней инфракрасной области
можно модифицировать обычные инфракрасные спектрофотометры,
заменив в них призму NaCl/KBr на призму из плавленного оксида
кремния, кварца, фтористого лития или кальция и добавив более
чувствительный детектор. Многие промышленные УФ-ВИ-спектро-
фотометры сконструированы таким образом, что позволяют иссле-
довать и ближнюю инфракрасную область.
Кюветы для образцов изготавливают из специальных сортов
стекла или кварца, они имеют толщину от 0,1 до 10 см.
Трудности представляет выбор подходящего растворителя для
ближней ИК-спектроскопии; это обусловлено тем, что в этом слу-
чае не пригодны растворители, в которых имеются группы О—Н,
N—Н и даже С—Н. На рис. 15.34 представлены характеристики
пропускания некоторых растворителей, пригодных для использо-
вания в ближней ИК-спектроскопии. Наиболее часто применяются
четыреххлористый углерод, являющийся идеальным растворите-
лем для ближней ИК-спектроскопии, сероуглерод, метиленхлорид.
Полимерные пленки для анализа должны быть достаточно тол-
стыми.
Применение ближней инфракрасной спектроскопии для харак-
теристики полимеров позволяет проводить исследования в следую-
щих основных направлениях:
1. Количественный анализ функциональных групп органиче-
ских соединений. К наиболее важным функциональным группам,
которые изучали этим методом, относятся С—Н-группы: концевые
метиленовые и метиновые группы дают особенно хорошо разре-
Инфракрасная спектроскопия
261
Илина, волны, мкм
Поглощение С-Н -0.02 -о> 4 0.3 0,2 1 <0.2-6 1
Концевая н 1 7,2 4,2
Концевая -СН^^;СНг W и *ч
Концевая = сн м м/0 ^0
г^с-СН=СН м »0 15
х'С''СНг"О (оксетан) м и н м И нч
—СПз H-f 0,02 ->o,t -* 03
>снг •40,02 . и *—*0.1 Н025
>с-н
— СН(ароматич) —4 н«0 ,1 0,1 м
—СН/альдегид) И 0,5
—СН (Формиат) <4(П
Поглощение N-H — I Моматич. тиамин [алищатич. ч “•0 ж 4 0,2 * HI- * 7 30 н 4 30 1-54 м2
чми Сароматцч. амин аяицзатпич. ж '^5 <20 *н1
— NHjaJwud 0,?> ал. ЗМ* 05 (00 4100
амид 0,5 '0,4 50-100
>NH имид м
—мнг гидразин ж 0,5 *-<0,5 н
Поглощение О-Н —ОН спирт иг 4 50
-ОН гидроперекись[а^^ 1>41 н2 н/ НО в 30 Изо <80
несвязан. -^^{свяган водород свя 1 нЗ (неп ост UPh (НО) 200
—ОН карбоновая кислота. н 1 - ю-юо
-ОЦ гликоль [ f, | г1 50 50 *4 50 \ 1-50 н м 2( -80 4 1 н 1 3-100 ^5-40
ОН вода гор Г.2 ЗОН »7,
.. =NQH окси» _ м *200
Н с невозможно, гидрат.) н
Поглощение других функци- ональных групп -SH н 0,01
>РН о,2
>с=о Ч 43
. -C3N <0/
Рис. 15.33. Поглощение различных функциональных групп в ближней ПК-обла-
сти [О: 519]. Числа, расположенные рядом с полосами, указывают коэффициент
молярной экстинкции.
длина волны, мкм
Четыреххлористый углерод
Сероуглерод
Хлоророрм
(стабилизованный этанолом)
Метиленхлорид
Диоксан
Ди-х-бутиловый эфир
Диметиловый эфир
триэтиленгликоля
Гептан
Бензол
Ацетонитрил
Диметилформамид
Диметилсульфоксий
Рис. 15.34. Характеристики пропускания некоторых растворителей в ближней
ИК-области [О: 519]. Сплошными линиями обозначены области, в которых
возможно использование данного растворителя; числа указывают максимально
желаемую толщину слоя в сантиметрах.
262
Глава 15
шаемые инфракрасные спектры; N—Н-группы: основное валентное
колебание при 2,8—3,0 мкм, первый обертон — 1,5 мкм и второй
обертон—1,0 мкм; О—Н-группы: основное валентное колебание
при 2,7—3,0 мкм; С=О-группы: первый обертон карбонильных
групп при 2,8—3,0 мкм; S—Н, Р—Н, нитрильные и другие группы.
2. Исследование водородных связей.
3. Изучение взаимодействия растворителя с растворенным ве-
ществом.
Обзорная литература: 519, 716, 717, 1324.
Периодическая литература: 3247, 3457, 3472, 3473, 5276, 7115.
15.12. ДАЛЬНЯЯ ИНФРАКРАСНАЯ СПЕКТРОСКОПИЯ
Дальняя инфракрасная область четко ограничена частотами от
200 до 10 см-1. Многие спектроскописты относят к дальней инфра-
красной области частоты ниже 650 см-1, но их лучше определять
как часть средней инфракрасной области (4000—200 см-1).
Область 650—200 см-1 можно исследовать с помощью стан-
дартных спектрометров. Ниже 200 см-1 требуются спектрометры
специальной конструкции с дифракционными решетками и транс-
миссионными фильтрами.
Окна в кюветах изготавливают из йодистого цезия (исполь-
зуются в области до ~50 мкм), кристаллического кварца (исполь-
зуются в области выше 50 мкм, от 4,5 до 45мкм непрозрачны);
полиэтилена различных марок, который в большинстве случаев
прозрачен в области выше 25 мкм, но слабо поглощает при 50 и
150 мкм; алмаза, который прозрачен во всей дальней инфракрас-
ной области.
Необходимо полностью удалять пары воды из образцов и кю-
веты путем продувки сухим воздухом или азотом или вакуумиро-
ванием, в области 50 мкм имеется широкая полоса поглощения,
обусловленная вращательными движениями молекул воды.
Дальняя инфракрасная спектроскопия, охватывающая область
650—200 см-1, имеет большое значение при изучении ароматиче-
ских, галогенсодержащих, фосфорсодержащих и металлоорганиче-
ских полимеров. Поглощение в этой области весьма чувствительно
к общим изменениям молекулярной структуры, например измене-
нию тактичности, конформации и молекулярной подвижности.
Обзорная литература: 101, 654, 693, 938.
Периодическая литература: 2108, 2226, 2385, 2791—2793, 3073, 3099, 3393,
3485—3488, 3697, 3698, 3793, 4178, 5320, 6107, 6592, 6744, 6747, 6673, 7114.
Глава 16
ЭМИССИОННАЯ СПЕКТРОСКОПИЯ
Обзорная литература: 75, 92, 133, 179, 180, 212, 242, 274, 328, 451, 463, 550,
610, 705, 791, 897, 952, 991, 1009, 1086, 1105, 1362, 1367, 1380, 1384.
Энергия, испускаемая как люминесценция (флуоресценция
и/или фосфоресценция), образуется за счет поглощенной энергии
падающего света.
Большинство молекул в невозбужденном виде находится в ос-
новном синглетном состоянии (So) (рис. 16.1). При поглощении
света единственными возбужденными состояниями, которые могут
достигаться непосредственно, являются возбужденные синглетные
состояния (Si, S2, S3, ..., Sn). Состояние Si отличается от So по
многим параметрам, и их следует рассматривать как химически
различные. В возбужденных синглетных состояниях спины элек-
тронов антипараллельны. Время жизни возбужденного синглет-
ного состояния (Si) составляет 10~8—10~10 с.
При инт ер комбинационной конверсии (безызлучательная инвер-
сия спина) молекула, находящаяся в возбужденном синглетном
состоянии (Si), может достигнуть первого триплетного состояния
(71). Время жизни триплетного состояния (7J колеблется от не-
скольких минут до 10-3 с.
Более высокие триплетные состояния (Т2, Тз, ..., Тп) обра-
зуются только при поглощении нового фотона молекулой, находя*
щейся в самом низком триплетном состоянии Т\. Такой процесс
получил название триплет-триплетного поглощения.
Электронно-возбужденная молекула может дезактивироваться
путем испускания излучения, называемого люминесценцией. Су-
ществует несколько видов испускания:
1. Флуоресценция — излучательный переход из самого нижнего
синглетного возбужденного состояния Si в синглетное основное
состояние (So).
2. Фосфоресценция — излучательный переход из самого ниж-
него триплетного возбужденного состояния (7\) в основное син-
глетное состояние (So).
3. Эксимерная флуоресценция — излучательный переход, свя-
занный с распадом эксимерного комплекса, образованного самым
нижним возбужденным синглетным состоянием (Si) и основным
синглетным состоянием (So) (разд. 16.1).
4. Замедленная флуоресценция — излучательный переход, обу-
словленный разложением эксимерного комплекса, образованного
возбужденными триплетными состояниями (7\) (разд. 16.1).
264
Глава 16
Как связаны между собой спектры поглощения и испускания,
хорошо видно из рис. 16.2.
Электронно-возбужденная молекула может также дезактиви-
роваться в результате следующих безызлучательных процессов:
1. Внутренняя конверсия — безызлучательный переход между
двумя различными электронными состояниями одинаковой муль-
типлетное™, т. е. сингл ет-синглетный переход или три-
плет-триплетный переход (7\->Ti). В процессе такого типа элек-
тронно-возбужденная молекула может выделять энергию в виде
теплоты.
2. Безызлучательный перенос энергии — перенос энергии от мо-
лекулы в синглетном (или триплетном) возбужденном состоянии
(донор) к молекуле, находящейся в основном состоянии (акцеп-
тор).
3. Диссоциация — это процесс, в котором энергия возбуждения
расходуется на инциирование разложения молекулы на свободные
радикалы.
Мономолекулярное время жизни (т) возбужденного состоя-
ния— время (с), необходимое для того, чтобы концентрация мо-
лекул в возбужденном состоянии уменьшилась в е раз по сравне-
нию с первоначальной величиной.
Собственное радиационное время жизни (то) возбужденного со-
стояния— время (с), необходимое для того, чтобы молекула
дезактивировалась путем испускания при условии, что безызлуча-
тельные процессы дезактивации отсутствуют. Между мономолеку-
Эмиссионная спектроскопия
265
лярным и радиационным временем жизни выполняются следующие
соотношения:
а) для флуоресценции
= (16.1)
б) для фосфоресценции
где <Df и Фр — квантовые выходы флуоресценции и фосфоресцен*
ции соответственно.
Длина, волны, mkja
Рис. 16.2. Соотношение между спектрами поглощения и испускания.
Квантовый выход люминесценции определяется как число кван-
тов, выделяющихся на один поглощенный квант возбуждения.
Квантовый выход флуоресценции (Of) составляет
ф ___ Число испускаемых квантов флуоресценции (16 3)
f Число квантов, поглощенных системой до ' * '
синглетного возбужденного состояния
Квантовый выход фосфоресценции (Фр) составляет
ф ___ Число испускаемых квантов фосфоресценции (16 4)
н Число квантов, поглощенных системой до \ • /
триплетного возбужденного состояния
Измерение квантового выхода связано с большими трудностями.
16.1. ЭКСИМЕРЫ И ЭКСИПЛЕКСЫ
Эксимер представляет собой молекулярный димерный ассоциат,
образующийся между возбужденной молекулой в самом нижнем
возбужденном синглетном состоянии (Si) и молекулой в синглетном
266
Глава 16
основном состоянии (So). В основном состоянии эксимеры не ста-
бильны, но устойчивы в электронно-возбужденном состоянии:
S1 + S0->(S1...S0) (16.5)
При разложении эксимерного комплекса можно наблюдать экси-
мерную флуоресценцию, которая отличается от «обычной» флуо-
ресценции:
(Sj ... S0)->Sj + So + Эксимерная флуоресценция (16.6)
Для эксимерной флуоресценции, которая зависит от температуры,
характерен широкий, размытый спектр (рис. 16.2).
Эксимеры могут также возникать при реакции двух возбуж-
денных триплетных состояний (бимолекулярное триплет-триплет-
ное взаимодействие):
Г1 + Г1^(Г1..,Г1) (16.7)
При разложении такого эксимера может наблюдаться замедлен-
ная флуоресценция, которая также отличается от обычной флуо-
ресценции:
(7\ ... 7\)—>S0 +So + Замедленная флуоресценция (16.8)
Замедленная флуоресценция представляет собой испускание,
имеющее спектральные характеристики обычной флуоресценции,
но со значительно большими временами нарастания и затухания.
Часто длительность затухания того же порядка, что и у фосфо-
ресценции. Замедленная флуоресценция зависит от квадрата ин-
тенсивности падающего света, это объясняется тем, что триплет-
ное состояние (Т1) возникает лишь от первоначального возбужде-
ния через возбужденное синглетное состояние (Si), и для возник-
новения флуоресценции такого типа необходимо протекание двух
этих процессов. Замедленная флуоресценция чувствительна к дей-
ствию кислорода, который эффективно тушит триплетное со-
стояние.
Эксимеры образуются в концентрированных полимерных рас-
творах или в твердом состоянии. Для того чтобы возбужденная
молекула образовала эксимер с другой молекулой, последняя во
время возбуждения должна приблизиться к возбужденной моле-
куле на расстояние 3—4 А. Возможность образования эксимера за-
висит также от пространственного расположения пары, молекул и
параллельности выстраивания жестких плоскостных ароматиче-
ских колец в полимерах.
Эксиплексом называется комплекс, существующий только в
электронно-возбужденных состояниях. Эксиплексы образуются
Эмиссионная спектроскопия
267
между возбужденными молекулами донора (D*) и молекулами
акцептора (А) или возбужденными молекулами акцептора (А*)
и молекулами донора (D):
D* + A-*(D“A+)**-*(D + A~)*->D* A+->DA*; (16.9)
A*+D->(A-D+)*«-*(A+D“)*->A* D«-»AD*
Комплекс с переносом
заряда.
(16.10)
Флуоресцентное испускание от эксиплекса зависит от полярности
растворителя. С увеличением полярности растворителя наблю-
дается уменьшение квантового выхода флуоресценции.
В отдельных случаях триплетные эксиплексы можно обнару-
жить при исследовании фосфоресценции и триплет-триплетного
поглощения. /
Обзорная литература: 138, 464, 528, 974, 1242.
Периодическая литература: 2041, 2414, 3019, 3035, 3066—3069, 3451, 3465,
3466, 3479, 3630, 3910, 4065, 4142, 4244—4246, 4611, 4612, 5095, 5195, 5507, 5835,
6015, 6016, 6482, 6753—6756, 6905, 7002, 7240, 7271.
17.2. ФЛУОРЕСЦЕНЦИЯ
нижнего
основное
Флуоресценция — это излучательный переход из самого
синглетного возбужденного состояния
состояние (So) (рис. 16.1).
Для построения спектра флу-
оресценции (рис. 16.3) на орди-
нате откладывают интенсивность
флуоресценции If (в условных
единицах), а на абсциссе — дли-
ну волны % (в нанометрах или
ангстремах).
Интенсивность флуоресценции
(If) раствора можно рассчитать,
используя закон Ламберта — Бе-
ра:
= /0(1 — 10-ес')Фл, (16.11)
где If — интенсивность флуорес-
ценции (квант/с); /0— интенсив-
ность падающего света; е — молярный
с — концентрация раствора (моль/л); / — толщина образца (см);
— квантовый выход флуоресценции. Поскольку кванты флуорес-
ценции не монохроматичны, абсолютный квантовый выход флуо-
ресценции определить очень сложно; кванты неравномерно рас-
пределены по изучаемому направлению и могут самопоглощаться
(Si) в синглетное
Длина, волны, нм
16.3. Спектр- флуоресценции
для данной молекулы.
а; я
си у
^5=
Рис.
коэффициент экстинкции;
268
Глава 16
раствором. Дополнительные трудности обусловлены поляризацией
и рефракцией.
Более просто определяется относительный выход флуоресцен-
ции, особенно если есть возможность сравнить флуоресценцию
исследуемого вещества с флуоресценцией стандартного соедине-
ния, выход флуоресценции (<DfS) которого известен (табл. 16.1).
Таблица 16.1
Стандартные флуоресцирующие вещества [О: 1086]
Соединение Растворитель фь
Флуоресцеин Водный карбонат-бикарбо- натный буфер, pH 9,6 0,85
Родамин В Этанол при низких концен- трациях раствора 0,69
Бисульфат хинина 0,1 н. H2SO4, низкая концен- трация раствора 0,55 (0,46)
Нафтол-2 Водный раствор, 10-3 М в 0,05 М боратном буфере, pH 10 0,21
Профлавин Водный раствор, 10~4 М в 0,05 М ацетатном буфере, pH 4 0,27
Широко использующимися стандартными соединениями являются
растворы сульфата и бисульфата хинина в 1 н. водном растворе
H2SO4.
Квантовый выход флуоресценции (Ф^) неизвестного соедине-
ния можно найти, используя следующее уравнение:
/ f в
ф/ = -^Гфм-
где If и IfS — интенсивности флуоресценции неизвестного соедине-
ния и стандартного соединения соответственно; 8 и е5 — молярные
коэффициенты экстинкции неизвестного и стандартного соедине-
ния соответственно; с и cs — концентрации неизвестного и стан-
дартного соединения в растворе.
Относительную интенсивность флуоресценции следует измерять
в той же самой кювете с одним и тем же пучком монохромати-
ческого света. Одинаковыми должны быть геометрия излучаемой
области, положение щелей спектрографов и оптический диапазон
света флуоресценции, попадающего на детектирующую систему.
Спектры флуоресценции, получаемые в виде зависимости сиг-
нала фотоумножителя от частоты, называются кажущимися спек-
трами флуоресценции. Необходимо вводить поправку к наблюдае-
мому сигналу фотоумножителя для получения величин, которые
пропорциональны числу квантов в секунду. (Это является главной
Эмиссионная спектроскопия
269
проблемой при определении относительного квантового выхода
флуоресценции.)
Истинные спектры флуоресценции (пропорциональные числу
квантов на единичный интервал частоты) получают из кажущихся
спектров, деля ординату, соответствующую каждому значению ча-
стоты, на коэффициент чувствительности детектирующей системы
для этой частоты.
Площади под кривыми истинных спектров флуоресценции двух
растворов (неизвестного и стандартного соединений) пропорцио-
нальны их общим интенсивностям флуоресценции (If и IfS).
Квантовый выход флуоресценции неизвестного соединения
можно найти по уравнению
Площадь под кривой истинного спектра
1г флуоресценции неизвестного соединения
фс = Ofs — = -р;------------------------------/-------- • Фк-
г Ifs Площадь под кривой истинного спектра 1
флуоресценции стандартного/Соединения
(16.13)
Многие типы современных спектрофотометров позволяют реги
стрировать истинные спектры флуоресценции.
С флуоресценцией могут кон-
курировать перечисленные ниже
молекулярные процессы.
1. Тушение путем внутренней
конверсии (Si->S0).
2. Интеркомбинационная кон-
версия (Si -> Ti).
3. Безызлучательный перенос
энергии.
4. Тушение примесями. Здесь
следует отметить, что при изме-
рении флуоресценции необходи-
мо использовать очень чистые
растворители и изучаемые ве-
щества.
5. Тушение кислородом.
Вследствие этого обстоятельства
Рис. 16.4. Зависимость In If от вре-
мени t.
интенсивность флуоресценции данного раствора необходимо изме-
рять дважды: в инертном газе (например, азоте или аргоне) и в
среде, насыщенной кислородом.
6. Концентрационное тушение и образование эксимеров.
После удаления источника возбуждения интенсивность флуо-
ресценции уменьшается экспоненциально, подчиняясь закону реак-
ции первого порядка:
h = Ладе-'4,
(16.14)
где Zf(O) — начальная интенсивность флуоресценции (t = 0); If —
интенсивность флуоресценции в момент времени t после импульса
270
Глава 16
света; t — время; т — среднее (мономолекулярное) время жизни
возбужденного синглетного состояния.
После интегрирования уравнения (16.14) получаем
lnZf = ln/f(o)-(//T) (16.15)
Из кривой зависимости 1п7/ от времени / находят время жизни
возбужденного синглетного состояния (рис. 16.4).
Обзорная литература: 92, 179, 435, 549, 796, 923, 997, 1086.
16.3. ОБОРУДОВАНИЕ ДЛЯ ФЛУОРЕСЦЕНТНОЙ
СПЕКТРОСКОПИИ
16.3.1. Флуоресцентные спектрофотометры
Флуоресцентный спектрометр, предназначенный для определения
истинных спектров флуоресценции (рис. 16.5), состоит из следую-
щих частей:
1. Источник света, которым являются дейтериевая газоразряд-
ная лампа для длин волн ниже 375 нм и ксеноновая лампа для
Рис. 16.5. Схема флуоресцентного спектрофотометра.
1 — источник возбуждения; 2—монохроматор; 3 — кварцевая пластинка; ^-контрольная кювета;
5— фотоумножитель Рг; 6— усилитель; 7—кювета с образцом; 3 —фотоумножитель Pi;
9 — разностный самописец.
области 250—1200 нм. Вместо ксеноновой лампы в некоторых
спектрофотометрах используется ртутная лампа среднего или вы-
сокого давления.
2. Монохроматоры. Монохроматоры, применяющиеся во флуо-
ресцентных спектрофотометрах, должны иметь высокие яркость и
разрешение.
3. Система оптического контроля.
Эмиссионная спектроскопия
271
а) Небольшая часть пучка монохроматического света отра-
жается прозрачной кварцевой пластинкой на контрольную кювету,
в которой находится флуоресцирующий раствор, например раствор
родамина В. Флуоресцирующие растворы, выбранные для кон-
трольной кюветы, должны в идеале иметь постоянный выход флуо-
ресценции и поглощать практически весь поток падающего света
при толщине слоя 1 мм или менее во всем рассматриваемом спек-
тральном интервале. Испускаемая флуоресценция регистрируется
фотоумножителем Р2. После усиления сигнал от Р2 подается на
разностный самописец.
б) Основная часть начального пучка света проходит через про-
зрачную кварцевую пластинку и через кювету с раствором образ-
ца, расположенные под прямым углом к оптической оси прибора,
который регистрирует флуоресценцию.
Измерительное устройство, предназначенное для определения
интенсивности флуоресценции от кюветы с образцом, состоит из
анализирующего монохроматора такого же тип^, что и монохро-
матор возбуждения, снабженного чувствительным фотоумножите-
лем Pi. После усиления сигнал от фотоумножителя Pt поступает
в разностный самописец.
4. Система записи результатов. Сигнал от разностного само-
писца поступает на перо, которое рисует зависимость исправлен-
ной интенсивности флуоресценции If от длины волны, давая истин-
ные спектры флуоресценции.
Обзорная литература: 242, 836, 949, 1008, 1086, 1380.
Периодическая литература: 3017, 3286, 4810, 4965, 6087, 7137, 7289.
16.3.2. Методы измерения длительности флуоресценции
Измерения длительности флуоресценции (затухания флуоресцен-
ции), составляющей порядка нескольких микро- или миллисекунд,
не вызывает особых затруднений. Большинство же флуоресцирую-
щих веществ имеет длительность флуоресценции порядка 10~8—
10-10 с, измерение затухания флуоресценции в таком интервале
времен представляет значительные трудности.
Известны два метода определения времени’затухания флуорес-
ценции порядка наносекунд: а) импульсные методы и б) методы,
связанные со сдвигом фаз.
Обзорная литература: 949, 1361.
16.3.3. Метод счета отдельных фотонов
Монофотонный или однофотонный корреляционный метод оцени-
вает время испускания отдельных фотонов флуоресценции. За ну-
левое время принимается начало вспышки импульсной лампы или
272
Г лава 16
электрического импульса, во времени связанного с разрядом им-
пульсной лампы. Скорость счета фотонов составляет порядка
100—200 С”1. Время поступления каждого фотона, отнесенное к
указанному нулевому времени, регистрируется электронным
устройством (преобразователь время — амплитуда), данные об
амплитуде импульса — в многоканальном анализаторе амплитуды
импульса. Данные, хранящиеся в памяти такого анализатора,
Рис. 16.6. Схема прибора для измерения длительности флуоресценции с реги-
страцией флуоресценции, возбуждаемой отдельной вспышкой.
/ — генератор импульсов; 2 — тиратрон и фотолитическая импульсная лампа; 3—монохрома-
тор; 4 — кювета с образцом; 5—фотоумножитель; 6 — усилитель; 7 —преобразователь время -
амплитуда; 3 — многоканальный анализатор амплитуды импульса; 9— запись на пишущей
машинке и ленте; 10 — осциллограф; // — самописец.
подаются в перфоратор, печатающее цифровое устройство или на
диаграммную бумагу самописца. Блок-диаграмма такого прибора
приведена на рис. 16.6. Аналогичное оборудование используется
в ядерной спектроскопии.
Обзорная литература: 484, 1361, 1435.
Периодическая литература: 2475, 4834, 5017.
16.3.4. Осциллографический импульсный метод
С помощью осциллографического импульсного метода измеряют
длительность флуоресценции, возбуждаемой отдельной вспышкой.
Для этого используют наносекундную импульсную лампу, чувстви-
тельный фотоумножитель и осциллограф (рис. 16.7). С помощью
такого метода измеряют амплитуды при наборе времен, отсчитан-
ных от фиксированного нулевого времени. Каждый раз измеряе-
мый переход запускает систему, в результате чего амплитуда
определяется при времени, большем на Д/? чем при предыдущее
Эмиссионная спектроскопия
273
измерении. Результат в виде серии точек на экране осцилло; рафа
появляется со значительным запаздыванием.
Рис. 16.7. Схема установки импульсного фотолиза с осциллографом для изме-
рения длительности флуоресценции.
1 — источник высокого напряжения; 2— наносекундная импульсная лампа; 3 — монохроматор;
4—фотоумножитель; 5—кювета с образцом; 6 — дискриминатор; 7 — замедлитель; 8—осцилло-
граф; 9— многоканальный анализатор амплитуды импульса; 10 — самописец.
Обзорная литература: 1361.
Периодическая литература: 4184, 6556.
16.3.5. Метод, связанный со сдвигом фаз
Метод определения длительности флуоресценции, связанный со
сдвигом фаз, основан на том, что молекула, возбужденная сину-
соидально модулированным световым сигналом, дает флуоресцен-
цию той же самой частоты, но сдвинутой по фазе относительно
возбуждающего света.
Длительность флуоресценции (т) определяется измерением
разности фаз возбуждающего и испускаемого света при известной
угловой частоте возбуждающего света:
t^e = <oT, (16.16)
274
Глава 16
где 0 — фазовый угол, со — частота модулированного возбуждаю-
щего света.
Нужно использовать модулированный источник света и фазо-
чувствительный детектор. Блок-диаграмма типичного прибора для
измерения длительности флуоресценции этим методом приведена
на рис. 16.8.
Модулированный источник света создается модуляцией прило-
женного к газоразрядной лампе напряжения или пропусканием
Рис. 16.8. Схема прибора, основанного на сдвиге фаз, для измерения длитель-
ности флуоресценции.
/ — источник модулированного света; 2— монохроматор; 3—кювета с образцом; 4—фото-
умножитель; 5—фиксированный замедлитель; 6 — переменный замедлитель; 7—суммирующий
усилитель; 3 —детектор, чувствительный к сдвигу фаз.
равномерного светового пучка через устройство с переменной
оптической плотностью. Для модуляции света используются сле-
дующие средства: а) ячейки Керра, б) электрооптические (твер-
дые) модуляторы света, в) сверхзвуковые модуляторы, г) ампли-
тудно-модулированный высокочастотный газовый разряд, д) высо-
кочастотные разряды, работающие при заданной частоте.
Для определения сдвига фаз сопоставляют фазы при частоте
модуляции или после преобразования частот.
Метод, основанный на сдвиге фаз, позволяет измерить длитель-
ность флуоресценции порядка 10~9—10~10 с,
Эмиссионная спектроскопия
275
Обзорная литература: 132, 179, 1160, 1361.
Периодическая литература: 2194, 2415, 2416, 2562, 2570, 2811, 4400, 4988,
5243, 6227—6229, 6494, 6591, 7011.
16.4. ПОЛЯРИЗОВАННАЯ ФЛУОРЕСЦЕНЦИЯ
Поляризацию флуоресценции можно характеризовать системой
оптических координат (х, у, г) (рис. 16.9):
а) линейно-поляризованный свет возбуждения проходит вдоль
оси у и имеет электрический вектор (Pi) в направлении оси г;
Рис. 16.9. Система оптических координат для измерения степени поляризации
флуоресценции (Р/).
б) поляризованная компонента флуоресценции измеряется че-
рез анализатор в направлении оси г/, имея электрический вектор
Р2, лежащий в плоскости г—х.
Степень поляризации флуоресценции (Pf) равняется
> б) 7fll ~ 7fJL
f Zfll +
(16.17)
где If\\ измеряется, когда поляризатор Pi и анализатор Р2 уста-
новлены параллельно друг другу (Р1||Р2); измеряется, когда
поляризатор Pi и анализатор Р2 установлены перпендикулярно
друг другу (Pi_LP2).
Для измерения поляризованной флуоресценции можно восполь-
зоваться оптической системой (рис. 16.10), состоящей из следую-
щих частей:
а) Источник света — ртутно-ксеноновая лампа. Начальный пу-
чок света после прохождения через кварцевые конденсоры моно-
хроматируется с помощью монохроматора с дифракционной ре-
шеткой или кварцевой призмы и поляризуется ультрафиолетовым
поляризатором Рь
б) Кювета для раствора или расплава полимера.
в) Испускаемая образцом флуоресценция проходит через ана-
лизатор Р2 и узкополосный фильтр и затем измеряется фотоумно-
Глава 16
жителем. После усиления сигнал от фотоумножителя попадает в
самописец.
г) Оси поляризации поляризатора Pi и анализатора Р2 уста-
навливаются параллельно или перпендикулярно друг другу для
измерения и соответственно.
Метод поляризованной флуоресценции используется для изуче-
ния молекулярной ориентации в твердых полимерах (разд. 35.11)
Рис. 16.10. Оптическая система для измерения степени поляризации флуорес-
ценции раствора или расплава полимера.
/ — источник излучения; 2— монохроматор; 3—коллиматор; 4—поляризатор (Pi); 5 —кювета
с образцом; 6— анализатор (Рг); 7 —узкополосный фильтр; 8—фотоумножитель; 9— усили-
тель; 10 — самописец.
и времен релаксации, связанных с подвижностью полимерных мо-
лекул (разд. 16.5).
Обзорная литература: 796, 974.
Периодическая литература: 2029, 5498, 5503, 5512.
16.5. ИЗУЧЕНИЕ МОЛЕКУЛЯРНОЙ ПОДВИЖНОСТИ
ФЛУОРЕСЦЕНТНЫМ МЕТОДОМ
Время вращательной релаксации для микроброуновского движе-
ния в растворах и расплавах полимеров составляет 10-12—10~5 с.
Длительность возбуждения для флуоресцентной молекулы
обычно 10-10—10-7 с.
Длительность возбуждения можно использовать в качестве
эталонной временной шкалы для измерения времен релаксации
молекулярной подвижности в полимерных системах. Флуоресцент-
ные группы химически вводят в полимерные молекулы или просто
диспергируют флуоресцирующие соединения в полимерной систе-
ме» все зависит от целей исследования.
Эмиссионная спектроскопия
m
Если время релаксации (р) молекулярной подвижности в си-
стеме становится сравнимым с временем жизни возбужденного со-
стояния (т), то направления молекулярных осей молекул вслед-
ствие броуновского движения в период возбуждения будут хаоти-
ческими, в результате наблюдается частичная деполяризация
флуоресценции.
Для системы, в которой оптически анизотропные флуоресци-
рующие молекулы распределены статистически, степень такой
вращательной деполяризации флуоресценции можно связать с
вращательными движениями молекулы следующей зависимостью:
Pfo/Pf == 1 + А (т/р), (16.18)
где Pf — степень поляризации, определяемая уравнением (16.17);
Pfo — предельно достижимая степень поляризации, когда враща-
тельное движение молекулы полностью прекращается (р->оо);
А — константа, зависящая от используемой для измерения опти-
ческой системы; т — длительность флуоресценции (с); р — время
релаксации молекулярной подвижности (с).
Выход флуоресценции (Ф^) молекул, способных к внутреннему
вращению, в значительной степени зависит от вязкости и темпе-
ратуры среды. Это явление можно охарактеризовать как внутрен-
нее тушение флуоресценции, обусловленное вращением части мо-
лекулы относительно ее остатка (отклоняющейся от плоской кон-
формации в течение возбуждения). Для таких молекул изменение
выхода флуоресценции можно выразить через время релаксации
внутреннего вращения р':
Ofo/<£f==l+B(r/p')> (16.19)
где Ф/о— предельный выход флуоресценции при прекращении вну-
треннего вращения (р'->оо); В — коэффициент пропорционально-
сти; т — длительность флуоресценции (с); р' — время релаксации
внутреннего вращения (с).
Измерение степени поляризации описано в разд. 16.4, а изме-
рение выхода флуоресценции — в разд. 16.2.
Обзорная литература: 974.
Периодическая литература: 5499—5501, 5619.
16.6. ФОСФОРЕСЦЕНЦИЯ
Фосфоресценция — излучательный переход из самого нижнего
триплетного возбужденного состояния (Л) в синглетное основное
состояние (So) (рис. 16.1).
Фосфоресценция обычно наблюдается только в вязких средах
или низкоплавких стеклах. В жидких средах фосфоресценция
очень слаба из-за протекания следующих двух реакций:
278
Глава 16
1. Триплетное тушение — безызлучательная реакция между
возбужденным триплетным состоянием (7\) и молекулой тушителя
(Q), например кислородом или примесями:
Л + Q—(или продукты) (16.20)
2. Триплет-триплетная аннигиляция — это реакция между
двумя возбужденными триплетными состояниями (71) (бимолеку-
лярное триплет-триплетное взаимо-
действие), в результате которого об-
разуется триплетный эксимер:
П + Л-ЧЛ...Л) (16.21)
При разложении такого эксимера
наблюдается замедленная флуорес-
ценция
(?!... ТJ+ So + Замедленная
флуоресценция, (16.22)
Длина, волны, нм которая приводит к уменьшению кван-
Рис. 16.11. Спектр флуоресцеп- тового выхода фосфоресценции.
ции для данной молекулы. При использовании чрезвычайно
чистых растворителей и тщательном
удалении кислорода из системы можно измерить затухание фос-
форесценции и в жидких средах.
Для построения спектра фосфоресценции на ординате отклады-
вают интенсивность фосфоресценции (1Р) в условных единицах,
а на абсциссе — длину волны (X) в нанометрах или ангстремах
(рис. 16.11).
После удаления источника возбуждения фосфоресценция осла-
бевает, подчиняясь закону реакций первого и второго порядка:
= + (16.23)
где ki и k2 — различные константы скорости для различных про-
цессов.
Обзорная литература: 92, 242, 1086.
Периодическая литература: 4024, 4339, 6505.
16.7. ОБОРУДОВАНИЕ ДЛЯ ФОСФОРЕСЦЕНТНОЙ
спектроскопии
16.7.1. Фосфоресцентные спектрофотометры
Для разделения фосфоресценции и флуоресценции необходимо ис-
пользовать спектрофосфориметр — вращающийся цилиндр со ще-
лью (рис. 16.12) или прерыватели с дисковым приводом
(рис. 16.13), в которых пучок падающего света и пучок испускае-
Рис. 16.12. Схема фосфоресцентного спектрофотометра с применением спектро-
фосфориметра.
/ — источник возбуждения; 2—монохроматор; 3— спектрофосфориметр; 4—вращающийся
цилиндр со щелью; 5 —сосуд Дьюара, в котором находится образец; б—монохроматор;
7—фотоумножитель; 8—усилитель; 9 — самописец.
Рис. 16.13. Прерыватель в виде диска с прорезями (вращающееся секторное
колесо).
/ — монохроматор; 2—прерыватель; 3—кювета с образцом.
Рис. 16.14. Кювета с образцом} помещенная в сосуд Дьюара с жидким азотом.
/ — сосуд Дьюара; 2—жидкий азот; 3 — кювета с образцом; 4—кварцевые окна.
280
Глава 16
мого света периодически прерываются. В спектрофосфориметре
используется вращающийся цилиндр со щелью, который окружает
сосуд Дьюара, содержащий образец (рис. 16.14). Излучение об-
разца регистрируют только в течение темнового периода, когда
короткоживущая флуоресценция полностью исчезает.
Кварцевые сосуды Дьюара, содержащие образец, имеют не-
посеребренные полоски, через которые проходит возбуждающий
свет и фосфоресценция.
Остальные части блок-схемы спектрофосфориметра (рис. 16.12)
не отличаются от частей прибора, использующегося для регистра-
ции спектров флуоресценции (разд. 16.3.1).
Обзорная литература: 1008, 1086, 1440.
Периодическая литература: 3950, 5749.
16.7.2. Измерение длительности фосфоресценции
Измерение длительности фосфоресценции (затухания фосфорес-
ценции) во временном интервале порядка 1 —10-4 с не сопряжено
с какими-то серьезными экспериментальными трудностями,
Рис. 16.15. Схема установки
с импульсной лампой, исполь-
зующейся для измерения дли-
тельности фосфоресценции.
1 — батарея конденсаторов; 2—им-
пульсная лампа; 3— узкополосный
фильтр; 4—кювета с образцом;
5 — прерыватель; 6 — монохроматор;
7—фотоумножитель; 8 — усилитель;
9 — самописец.
Затухание фосфоресценции обычно измеряют методом импульс-
ного фотолиза. Блок-схема прибора для измерения длительности
фосфоресценции с помощью импульсного фотолиза показана на
рис. 16.15.
Промышленностью выпускаются импульсные лампы с време-
нами затухания менее 10~4 с и мощностью несколько джоулей.
Интенсивная вспышка света освещает реакционный сосуд и вызы-
вает фотовозбуждение молекул. Свет фосфоресценции проходит
через монохроматор и измеряется с помощью фотоумножителя.
Эмиссионная спекЪроскопия.
281
После усиления выходной сигнал от фотоумножителя поступает
на самописец.
Обзорная литература: 179, 242, 791, 1086.
16.8. ИМПУЛЬСНАЯ КИНЕТИЧЕСКАЯ
СПЕКТРОСКОПИЯ
Метод импульсной кинетической спектроскопии применяется для
изучения триплетных состояний и импульсного фотолиза моле-
кул. При поглощении молекулами исследуемого вещества интен-
сивной вспышки света в относительно большой концентрации
образуются свободные радикалы или другие неустойчивые ча-
стицы.
Импульсную кинетическую спектроскопию можно также ис-
пользовать для изучения важнейших процессов в фотохимических
системах, например триплет-триплетного поглощения, гибели три-
плетного возбужденного состояния (7\) и т. л.
На рис. 16.16 показана схема аппаратуры для импульсного
фотолиза вместе с измеряющим спектрофотометром. Новые ча-
стицы, образующиеся в реакционном сосуде под действием им-
пульса света, можно изучить, регистрируя их спектры поглоще-
ния с помощью фотографической пластинки или в виде сигнала
на экране осциллографа. Свет, необходимый для анализа системы
после облучения, фотолизирующим импульсом света, получают с
помощью дополнительной спектроскопической анализирующей им-
пульсной лампы. Фотолитическая и аналитическая импульсные
лампы связаны таким образом, что можно контролировать интер-
вал времени между возбуждением от фотолитического импульса
и появлением анализирующегося светового пучка.
Длительность вспышки (т) представляет собой интервал вре-
мени от 1/е максимальной интенсивности на восходящей части
кривой до соответствующей точки нисходящей части кривой для
рассеянного света (рис. 16.17). Рассмотрим простой контур RCL
(рис. 16.18), где R— сопротивление, обусловленное лампой после
начала разряда (зависит от длины лампы, обычно R составляет
примерно 5 Ом или менее), С — емкость, которая является пере-
менной величиной, но обычно изменяется от 1 до 20 мкФ, L — ин-
дуктивность, обусловленная конденсатором: при очень коротких
т индуктивность должна быть очень мала, чтобы можно было
поддерживать большие значения емкости. Из контура RCL видно,
что длительность вспышки составляет
x^Rc (16.24)
При сопротивлении 5 Ом и заряде конденсатора до 2 мкФ дли-
тельность вспышки около 10 мкс. Поэтому наиболее короткожи-
вущие частицы, которые можно изучать в таких условиях, должны
иметь длительность вспышки более 10 мкс.
282
Глава 16
Рис. 16.16. Схема прибора для импульсного фотолиза.
/ — источник высокого напряжения; 2—спектрографическая лампа; 3—узкополосный жидкий
фильтр; 4— импульсная лампа; 5 —батарея конденсаторов; 6 — кювета с образцом; 7 —пара-
болический отражающий экран; 8 — монохроматор; 9 — фотоумножитель; 10 — усилитель;
11 — осциллограф; 12—самописец.
Вспышка
Рис. 16.17. Зависимость интенсивности вспышки от времени.
Рис. 16.18. Контур RCL.
Эмиссионная спектроскопия
283
Энергия, получаемая от импульса, равна
е=±С72
£
(16.25)
в джоулях, где С — емкость, V — напряжение, приложенное к кон-
денсатору. Для конденсатора емкостью 2 мкФ, заряженного до
10 кВ, Е = 100 Дж. Для того же самого конденсатора, заряжен-
ного до 20 кВ, Е — 400 Дж. Обычно в установках для импульсного
фотолиза используемые энергии составляют от 100 до 500 Дж.
Анализ данных импульсной кинетической спектроскопии стро-
Рис. 16.19. Изменение интенсивности света (отклонение луча осциллографа, х)
при образовании и гибели новых возбужденных частиц, измеренное при одной
длине волны (2i).
ится на предположении, что ответ фотоумножителя линейно зави-
сит от интенсивности возбуждающего света.
Спектры поглощения вновь образовавшихся частиц фотографи-
руются через различные интервалы времени после фотолитической
вспышки на фотографической пластинке, установленной на спек-
трографе, либо регистрируются фотографированием изображения
на экране осциллографа. Спектры поглощения можно также хра-
нить, используя магнитную память.
На рис. 16.19 представлен типичный спектр поглощения, сня-
тый с изображения на экране осциллографа в ходе образования и
гибели новых частиц после вспышки, измеренный только при одной
длине волны. Отклонение луча осциллографа (х) пропорционально
интенсивности света (/):
lQ = kxQ и I — kx, (16.26)
где k—константа, которая зависит от чувствительности детек-
тора. Величину k определять не нужно, так как
А = 1g Цо//) = 1g (х0/х) = гкс1, (16.27)
284
Глава 16
где —поглощение при длине волны X; 8х— молярный коэффи-
циент экстинкции при той же длине волны; с—концентрация об-
разовавшихся частиц; / — толщина образца.
С помощью этого уравнения можно определить зависимость
относительной или абсолютной концентрации изучаемого соеди-
нения от времени и использовать полученные данные для кинети-
ческих расчетов.
Обзорная литература: 1026, 1054, 1086.
Периодическая литература: 2342, 2564, 2586, 2804, 2866, 2870—2872, 2879,
3390, 4679, 5045, 5871, 7059.
16.9. НАНОСЕКУНДНАЯ ИМПУЛЬСНАЯ
СПЕКТРОСКОПИЯ
При использовании обычной аппаратуры для импульсной кинети-
ческой спектроскопии минимальная длительность вспышки, ко-
торую можно измерить, составляет примерно 1 мкс. Для изучения
Рис. 16.20. Схема установки для наносекундной импульсной спектрографии.
/ — рубиновый лазер; 2—кристалл первичного кислого фосфата аммония; 5—расщепитель
пучка; 4—элемент замедления; 5—полностью отражающее зеркало; 6—источник контроль-
ной флуоресценции; 7—кювета с образцом; 8—монохроматор; 9 — фотоумножитель; 10— уси-
литель; // — осциллограф; 12—самописец.
процессов, затрагивающих синглетные состояния, а также связан-
ных с образованием других очень активных частиц, следует ис-
пользовать наносекундную импульсную спектроскопию, в которой
применяется рубиновый лазер (разд. 10.6.3). Схема какого при-
бора показана на рис. 16.20. Световой импульс от рубинового ла-
зера с модулированной добротностью (ванадилцианин) проходит
через кристалл определенного типа, например кристалл первич-
ного кислого фосфата аммония, в результате чего частота удваи-
вается. Около 20% излучения при 694 нм, входящего в удвоитель
частоты, выходит при длине волны 347 нм. Затем свет проходит
Эмиссионная спектроскопия
285
через ряд фильтров и линзу и попадает на расщепитель пучка
света. Часть пучка направляется в кювету с образцом, остальная
часть импульса попадает на зеркало и от него возвращается на-
зад. В результате этот луч запаздывает относительно того пучка,
который был направлен на образец, причем время запаздывания
соответствует двойному расстоянию от зеркала до расщепителя.
После возвращения в расщепитель свет отражается на раствор
сильно флуоресцирующего соединения, например 1,Г,4,4'-тетрафе-
нилбутадиена-1,3, непрерывно испускающего при большей длине
волны (400—600 нм), чем у возбуждающего импульса. Этот им-
пульс флуоресценции затем возвращается в расщепитель, после
чего попадает в кювету с образцом и, наконец, в фотоумножитель.
Запаздывание контрольного импульса относительно возбуждаю-
щего импульса может составлять примерно ЮСГ нс.
Периодическая литература: 3356, 3357, 3449, 3630, 4971, 5872, 5873.
16.10. ХЕМИЛЮМИНЕСЦЕНЦИЯ И ТЕРМОЛЮМИНЕС-
ЦЕНЦИЯ
Хемилюминесценция — это испускание света в результате опреде-
ленных химических реакций, протекающих без возбуждения све-
том. Глубокое понимание механизма возбуждения ограничено не-
Рис. 16.21. Схема прибора для
измерения хемилюминесценции
и/или термолюминесценции.
1 — нагревательный блок; 2—регуля-
тор температуры; 3— образец поли-
мера; 4 — фильтр; 5—фотоумножи-
тель; 6—усилитель; 7 — самописец-
болыпим числом систем, связанных с газовыми реакциями и реак-
циями в растворах. Во многих случаях хемилюминесценцию мож-
но рассматривать как испускание света скорее от колебательно-
возбужденных, нежели от электронно-возбужденных состояний.
Хемилюминесценцию можно наблюдать при окислительной де-
струкции полимеров как результат реакции обрыва путем реком-
бинации свободных радикалов.
Термолюминесценция — это испускание света, наблюдаемое при
нагревании образца и обусловленное некоторыми определенными
реакциями, например термоокислением.
На рис. 16.21 показан прибор для изучения хемилюминесцен-
ции и термолюминесценции.
286
Глава 16
Образец полимера помещают на поверхность нагревательного
блока с контролируемой температурой, который находится внутри
светонепроницаемого корпуса. После этого образец нагревают в
атмосфере какого-либо газа, например кислорода. Свет, испускае-
мый полимерным образцом, проходит через стеклянный фильтр и
попадает в фотоумножитель. После усиления сигнал от фотоумно-
жителя подается в самописец.
Обзорная литература: 1086, 1103, 1104, 1201, 1362, 1372.
Периодическая литература: 2154, 2244, 3733, 3834, 4937, 5233, 5346, 5817,
5946, 5947, 6037, 6215, 7135.
16.11. ПРИМЕНЕНИЕ ЭМИССИОННОЙ
СПЕКТРОСКОПИИ ДЛЯ ИССЛЕДОВАНИЯ
ПОЛИМЕРОВ
Области применения эмиссионной спектроскопии для характери-
стики полимеров включают изучение молекулярной подвижности
макромолекул в растворах, изучение естественной флуоресценции
полимеров и биополимеров, изучение взаимодействия полимеров
с красителями, изучение примесей в промышленных полимерах,
исследование фотодеструкции и фотостабилизации полимеров,
изучение процессов сенсибилизации (сенсибилизированной фото-
полимеризации, фотодеструкции, фотоотверждения).
Обзорная литература: 22, 451, 469, 796, 977, 996, 1086, 1228.
Периодическая литература: 2067—2082, 2317, 2395, 2659, 2810, 3048, 3118,
3119, 3458, 3901, 4141, 4238, 4592, 4593, 5094, 5195, 5462, 5562, 5688, 6481, 6889,
6890.
Глава 17
СПЕКТРОСКОПИЯ КОМБИНАЦИОННОГО
РАССЕЯНИЯ
Обзорная литература: 36, 71, 150, 191, 193, 212, 274, 313, 377, 431, 477, 514,
602, 604—606, 669, 693, 694, 761—763, 832, 835, 1045, 1154, 1286, 1296, 1297.
Спектроскопия комбинационного рассеяния (спектроскопия
КР, раман-спектроскопия) связана с изменением частоты света,
рассеиваемого молекулами того или иного вещества. Эффект ком-
бинационного рассеяния обусловлен энергетическим обменом
между рассеивающей молекулой и фестоном падающего света.
В процессе такого типа происходит переход молекулы из одного
энергетического состояния в другое, компенсирующийся соответ-
ствующим изменением энергии. Основное уравнение, описывающее
этот процесс, представлено ниже:
Av0 + Е{ -= hvr + Е2, (17.1)
где h — постоянная Планка; Vo— частота падающего, или возбуж-
дающего, света (фотон); vr — частота рассеянного света (фотон);
Ei и Е2— начальная и конечная энергии молекулы соответственно.
Сдвиг частот уг — Vo = Av может быть по знаку положительным
или отрицательным. Абсолютная величина этого различия полу-
чила название частоты КР (раман-частоты). Набор таких частот
рассеивающих частиц дает спектр КР.
Линия КР с частотой vr < v0 называется раман-стоксовой ли-
нией.
Линия КР с частотой vr > v0 называется раман-антистоксовой
линией (рис. 17.1).
Стоксовы линии отвечают переходам молекулы из более низ-
кого в более высокое энергетическое состояние, при этом энергия
фотона уменьшается. Антистоксовы линии соответствуют перехо-
дам молекулы из возбужденного состояния в более низкое энерге-
тическое состояние, при этом энергия фотона возрастает.
Сдвиг частоты в спектроскопии КР выражается в волновых
числах:
= (17.2)
где с — скорость света.
Столкновения фотонов возбуждающего света, обладающих
энергией hvQ, с молекулами могут быть упругими, т. е. без изме-
нения энергии, что соответствует рэлеевскому рассеянию, и не*
упругими. В последнем случае возможны следующие два варианта:
а) в результате неупругого столкновения молекула испытывает
квантовый переход на более высокий энергетический уровень, при
288
Глава 17
этом энергия фотона уменьшается и он рассеивается при понижен-
ной частоте (Av — отрицательная величина); б) когда молекула
находится не на самом низком энергетическом уровне, при столк-
новении с фотоном она может перейти на более низкий энергети-
ческий уровень, и в этом случае рассеяние фотона происходит при
повышенной частоте (Av — положительная величина).
Сдвиги КР (частоты КР) не зависят от частоты возбуждаю-
щего света vo и эквивалентны энергетическим изменениям, наблю-
дающимся при переходах рассеивающих частиц и, следовательно,
Возбуждающий Рассеянный
свет свет
Стоксово КР
hvr< hv0
Перенос энергии
отрицателен
Антистоксово кр
hvr > hv0
Перенос энергии
положителен
Рис. 17.1. Схематическое изображение наиболее важных процессов при рас-
сеянии молекулами света.
являются характеристическими для данного перехода. Линии, ле-
жащей в низкочастотной области по отношению к линии возбуж-
дающего света (Av — отрицательная величина), отвечает идентич-
ная линия, располагающаяся в высокочастотной области (Av —
положительная величина). Величина отрицательного отклонения
больше, чем соответствующее положительное отклонение. При
температурном равновесии заселенность более высокого уровня
становится ниже заселенности более низкого уровня, причем умень-
шение экспоненциально зависит от энергии.
Сдвиги КР, обусловленные рассеянием, соответствуют колеба-
тельным или вращательным переходам молекул и могут наблю-
даться в видимой области.
В спектроскопии КР фотон как таковой никогда иё наблюда-
ется, но он проявляется в том, что, возбуждая молекулы, вызы-
вает в них колебательный или вращательный переход.
Не следует путать начальный энергетический уровень (Еп)
молекулы с основным состоянием молекулы (Ео) (рис. 17.1).
Спектроскопия комбинационного рассеяния
289
При Еп = Ет и, следовательно, vntn = 0 рассеивающая моле-
кула остается в неизменном состоянии и наблюдается рэлеевское
рассеяние.
При Еп =# Ет наблюдается эффект КР. Необходимым условием
для комбинационного рассеяния является то,, что энергия /rvo воз-
буждающего фотона должна превосходить разность энергий
Ет—Еп между конечным и начальным состояниями происшедшего
перехода.
17.1. ОБОРУДОВАНИЕ ДЛЯ СПЕКТРОСКОПИИ КР
17.1.1. Спектрометры КР
Суммарная интенсивность молекулярного рассеяния, включая рэ-
леевское, для жидких образцов и видимого света составляет при-
мерно 10~5 от интенсивности возбуждающего света, причем лишь
1 % этой величины приходится на спектр КР.
Для газов, молекулярная плотность которых ниже, соответ-
ственно ниже и интенсивность, в связи с чем, как правило, иссле-
дования нужно проводить на образцах большего объема. Именно
в связи со всем этим необходимы интенсивные источники монохро-
матического света и спектрографы, характеризующиеся высокой
яркостью.
Спектрометр КР (рис. 17.2) состоит из следующих основных
частей:
1. Источник света. Нельзя использовать источники, дающие
полихроматический свет, поскольку каждая линия возбуждает
свой собственный спектр КР и такие спектры могут перекрывать
Друг Друга. Из-за недостаточной избирательности большинства
фильтров применение их также ограничено.
Источник света, использующийся в приборах для спектроско-
пии КР, должен давать сильное монохроматическое излучение.
В связи с этим применяют дуговые ртутные лампы специальной
конструкции, испускающие интенсивную синюю линию при 4357 А
и сильную зеленую линию при 5461 А. Выбранный возбуждающий
свет не должен поглощаться исследуемым образцом и давать
флуоресценцию, которая бы маскировала спектр КР или вызы-
вала фотодеструкцию данного образца. Иногда поэтому при ис-
следовании светочувствительных веществ предпочитают приме-
нять гелиевые газоразрядные лампы, которые дают интенсивные
линии при 5876 и 6678 А.
В некоторых промышленных спектрометрах КР в качестве
источника света используются лазеры (лазерная спектроско-
пия КР).
Большинство лазеров генерирует одну или лишь несколько воз-
буждающих линий высокомонохроматического света, испускае-
мого в четко фиксированном направлении (разд. 10.6).
10 Зак. 238
290
Глава 17
Наиболее мощными лазерными источниками для спектроско-
пии КР являются гелиево-неоновый ионный лазер с сильной ли-
нией испускания при 6328 А; аргоновый ионный лазер с сильными
возбуждающими линиями при 4880 и 5145 А и более слабыми
линиями при 4579, 4765, 4965 и 5017 А; криптоновый ионный ла-
зер с возбуждающими линиями при 6471, 5682, 5308 и 5208 А. Бла-
Рис. 17.2. Схема лазерного спектрометра КР.
1 — лазер; 2— прерыватель; 3 — полностью отражающее зеркало; 4 — полупрозрачное зеркало;
5—кювета с образцом; 6 — полусферические линзы; 7 —призма с углом Брюстера; 8 — линзы;
9 — поляризатор; 10— монохроматор; 11 — фотоумножитель; 12 — усилитель; 13 — коррелятор;
14 — усилитель разбаланса; 15—самописец.
годаря использованию лазеров аппаратура для спектроскопии КР
существенно упростилась.
2. Система освещения образца. Для получения очень высокой
освещенности образца и эффективной фиксации излучения КР,
испускаемого маленьким объемом, применяют традиционную
схему «90°» (луч испускаемого излучения проходит перпендику-
лярно к направлению наблюдения) или «180°» (рис. 17.4 и 17.6).
Испускаемое образцом рассеяние направляется во входную щель
монохроматора.
3. Монохроматоры. В большинстве промышленных спектромет-
ров КР используются один или два монохроматора с дифракцион-
ной решеткой (системы Черни — Тэрнера), которые позволяют
разделить надлежащим образом слабые линии КР и возбуждаю-
щее излучение.
4. Система записи результатов. Лучше всего фиксировать ча-
стоты линий на фотографических пластинках, однако для этого тре-
буется облучать образец в течение многих часов. Для определения
интенсивностей линий используется фотоумножитель в комбинации
Спектроскопия комбинационного рассеяния 291
с усилителем и самописцем. При измерении интенсивности линий
с колеблющейся интенсивностью возбуждающего света необхо-
димо записывать отношение сигнала КР и луча сравнения. В боль-
шинстве промышленных спектрометров КР используют фотогра-
фические или фотоэлектрические системы записи. Спектры запи-
сывают в линейной шкале волновых чисел.
Обзорная литература: 514, 603, 673, 832, 1297, 1420.
Периодическая литература: 4561.
17.1.2. Устройства для образцов
В зависимости от физического состояния образца для записи спек-
тров КР используются различные приспособления.
1. Газы и пары. Газовые кюветы конструируются как кюветы
многократного отражение (рис. 17.3), в которых световой пучок
проходит туда и обратно много раз, например 100 раз при длине
кюветы 25 мм. Два плоских зеркала устанавливаются под не-
большим углом (ф ж 5°) друг к другу. Падающий пучок образует
угол 9 с нормалью к дну рефлектора, а первый отражающийся
луч образует угол 0 — ф с нормалью к верху рефлектора. При сле-
дующем отражении от верхней пластинки угол становится 0 — Зф
и т. д. Поэтому каждый последующий ход пучка все меньше отли-
чается от его пути при предыдущем ходе и приближается к нор-
мали к одной из отражающих пластинок. Когда наконец пучок
становится перпендикулярным верхней или нижней пластинке, он
отражается сам на себя, повторяет в обратной последовательности
свой путь по кювете и выходит из нее в точке, находящейся совсем
рядом с точкой входа пучка. Внутри кюветы луч света совершает
примерно 100 ходов, что соответствует длине пути порядка 1,5 м.
2. Жидкости. Большие количества жидких образцов можно
исследовать непосредственно в ампулах, используя схему освеще-
ния «90°» (рис. 17,4, а) или «180°» (рис. 17.4, б). Для исследова-
ния небольших жидких образцов в тех же схемах «90°» и «180°»
(рис. 17.5) используют стеклянные или кварцевые капилляры.
3. Твердые вещества. Исследования твердых образцов можно
проводить при освещении по схеме как «90°», так и' «180°»
(рис. 17.6). Хорошие спектры КР с твердых образцов получить
трудно. Наряду с комбинационным рассеянием твердое вещество
рассеивает возбуждающий свет. Внесено много усовершенствова-
ний для того, чтобы справиться с этими трудностями. Нужно очень
тщательно подходить к выбору типа кюветы для образца, размера
кристалла, толщины образца, его положения и условий приготов-
ления. Монокристалл рассеивает лучше, чем мелкий порошок.
В случае полимеров целесообразно использовать твердые стержни..
При применении порошков оказывается пригодной методика прес-
сования таблеток с КВг. Исследованию могут быть подвергнуты
образцы волокон, пластиков и пленок. Можно изучать анизотропное
10*
292
Глава 17
Рис. 17.3. Кювета многократного от-
ражения.
Рис. 17.4. Исследование жидкости в
ампулах при освещении по схеме
«90% (а) и «180% (б).
Рис. 17.5. Исследование жидкости в капиллярах при освещении по схеме
«90°» (а) и «180% (б).
Рис. 17.6. Исследование твердых образцов при освещении по схеме «90%
и «180%.
а—порошок в пробирке; б—полимерный блок; в—отдельное волокно; е—волокно, намотан-
ное на стеклянную палочку; д—пучок волокна.
Спектроскопия комбинационного рассеяния
293
рассеяние света от ориентированных образцов, например вытяну-
тых волокон или пленок.
Обзорная литература: 514, 602—604.
Периодическая литература: 4010.
17.2. ПОЛЯРИЗАЦИЯ КОМБИНАЦИОННОГО
РАССЕЯНИЯ СВЕТА
Рассеянный свет можно разделить на две компоненты: компоненту
1У(1±), поляризованную перпендикулярно плоскости х—г, и ком-
Рис. 17.7. Оптическая система координат для измерения степени поляризации
рассеянного света.
поненту /г(/ц), которая поляризована параллельно этой плоскости
(рис. 17.7).
Степень поляризации (р) рассеянного света определяется от-
ношением Iy/Iz или 71//ц. Величины р всегда характеризуют рас-
сеяние под углом 90° к направлению возбуждающего света.
Когда рассеивающая молекула изотропна, например четырех-
хлористый углерод, величина р равна нулю, т. е. линия Рэлея пол-
ностью поляризована.
Суммарная интенсивность рассеянного света (7) равна
/ = /, + /, (17.3)
Для изучения поляризации комбинационного рассеяния света
можно использовать как неполяризованный (Ez о и £х=#0),
так и плоскополяризованный возбуждающий свет (Ех = 0). В этом
случае степень деполяризации обозначают соответственно как рп
и рр.
17.3. ПРИМЕНЕНИЕ СПЕКТРОСКОПИИ КР
ПРИ ИССЛЕДОВАНИИ ПОЛИМЕРОВ
Каждая рассеивающая полимерная частица дает свой собствен-
ный характеристический колебательный спектр КР, который ис-
пользуют для качественных измерений. На спектроскопию КР не
294
Глава 17
влияет наличие воды. По появлению новых линий в спектре КР
можно обнаружить новые молекулярные образования, возникаю-
щие при химическом взаимодействии. Интенсивность характери-
стической линии КР пропорциональна объемной концентрации
частиц и позволяет проводить количественный анализ. Для интер-
претации конкретного спектра с учетом молекулярных свойств
Таблица 17.1
Колебательный анализ различных полимеров с помощью
спектроскопии КР
Полимер Литература
Полибутадиен Полиэтилен Полиэтиленоксид Полиэтилентерефталат Полиизопрен Полиоксиметилен Полипропилен Поливинилхлорид Поливинилиденфторид Политетрафторэтилен Сополимер этилена с пропи- леном П: 2927, 2975, 3332 П: 2463, 2597, 3344, 3490, 3491, 3582, 4006, 4007, 5486, 6463, 6600 П: 3267, 3917, 5161 П: 2462, 5928 П: 2976 П: 5943 П: 5878, 6216, 6793, 6930 П: 2265, 4638, 4925, 6073-6075 П: 4624 П: 2464, 3886, 4635, 5731, 5944 П: 3492
данного соединения необходимо более подробно рассмотреть кван-
товомеханическую теорию эффекта КР.
Широкому использованию спектроскопии КР для исследования
полимеров частично препятствуют следующие факторы:
а) низкая интенсивность комбинационного рассеяния света
для многих полимеров, составляющая примерно 10~8 от интенсив-
ности возбуждающего света;
б) наличие во многих случаях сильного фонового излучения,
отличающегося от рассеяния Рамана, Тиндалла и Рэлея;
в) наличие флуоресцирующих Примесей.
Области практического использования спектроскопии КР для
характеристики полимеров включают изучение конфигурации и
конформации цепей гомо- и сополимеров, образования спиралей;
полимерных кристаллов и межламеллярных взаимодействий в
них; кристаллической и аморфной ориентации в полимерах; тек-
стуры, особенно при использовании низкочастотной спектроско-
пии КР; молекулярной подвижности в растворе; полимерных рас-
плавов сетчатых полимеров и гелей; влияния напряжения на по-
лимеры; процессов деструкции.
В табл. 17.1 приведены примеры колебательного анализа спек-
троскопии КР для разнообразных полимеров.
Спектроскопия комбинационного рассеяния
295
Обзорная литература: 42, 74, 149, 151, 211, 492, 762, 898.
Периодическая литература: 2198, 2463, 2466, 2523, 2739, 2766, 2772, 2972,
3173, 3267, 3335, 3493, 3494, 3626, 3727, 3918, 3919, 3949, 4009, 4627, 5113, 5160,
5158, 5159, 5162, 5225, 5592, 5746, 5878, 5927, 5929, 6217, 6463—6465, 6489, 6490,
6599, 6612, 6777, 6778, 6930, 7286, 7319.
17.4. СРАВНЕНИЕ ИК-СПЕКТРОСКОПИИ
И СПЕКТРОСКОПИИ КР
Как инфракрасная спектроскопия, так и спектроскопия КР пред-
ставляют собой методы исследования колебательных спектров по-
лимеров. Однако между этими двумя спектральными методами
существуют глубокие различия, касающиеся физических основ
процессов. В ряде случаев можно использовать лишь тот или иной
метод исследования, тогда как в других случаях эти методы пре-
красно дополняют друг друга. Поэтому любая научная лабора-
тория, в которой проводится исследование полимеров, должна
быть оснащена спектрометрами обоих видов, только при этом
можно получить максимум информации о структуре полимеров на
основании колебательных спектров.
Между указанными двумя спектроскопическими методами су-
ществует несколько различий, которые необходимо принимать во
внимание (табл. 17.2).
1. Наиболее интенсивные инфракрасные полосы обусловлены
полярными связями, например О—Н, N—Н или С = О, тогда как
наиболее интенсивные полосы в спектрах КР обусловлены свя-
зями с близким к симметричному распределением заряда, напри-
мер С—С, С = С и S—S. В результате этого ИК-спектроскопию
обычно используют для изучения заместителей в полимерах, тогда
как спектроскопию КР предпочитают применять для определения
конформации полимерных цепей.
2. Вода весьма интенсивно поглощает инфракрасное излуче-
ние, тогда как комбинационное рассеяние воды является очень
слабым, поэтому спектроскопия КР особенно полезна для изуче-
ния водных растворов, например при исследовании конформа-
ционных изменений биополимеров в водных растворах в зависи-
мости от pH, ионной силы и температуры.
3. Подготовка образцов в спектроскопии КР значительно про-
ще, чем в ИК-спектроскопии.
ИК-спектроскопия и спектроскопия КР — это методы, которые
дополняют друг друга.
Обзорная литература: 74, 211, 1224.
Периодическая литература: 2462, 2927, 3030, 3492, 4624, 6492, 7007.
296
Глава 17
<3 S' а to Q Группы, цающие интенсивные линии в спектрах ИК и КР интервал частот, см 2800—3000 2200—2300 2100—2300 500-1400
P [O: 211] колебания Валентные алифа- тические С—Н Валентные C = N Валентные Si—Н Валентные С—X (X — галоген)
грах ИК и KI 1 я я ч V интервал частот, см"”1 § со 1 1 1600-1700 2100—2250 400-500 600-700 950-1050 1500-1700 1575-1630 5 450-550
ностях полос поглощения в спек* Группы, дающие интенсивнь в спектрах КР колебания Валентные ароматические С—Н Валентные С = С Валентные С = С Валентные S=S (Se—Se и т. п.) Валентные С—S (С—Se и т. и.) Скелетные ароматического кольца Плоскостные колебания аро- матических С = С Валентные N = N Симметричные валентные (Si—О—Si)
s и X <u № tt К 1 линии интервал частот, см"” 1600-1800 900-1300 3000-3400 650-850 3100-3300 1000-1100
Различи Группы, дающие интенсивные в ИК-спектрах колебания Валентные С = О Валентные С—0 Валентные О—Н (связанные водородными связями) Деформационные внепло- скостные С—Н Валентные N—Н (связанные водородными связями) Асимметричные валентные Si—О—Si
Спектроскопия комбинационного рассеяния
297
17.5. ВНУТРИЦЕПНЫЕ КОЛЕБАНИЯ В ПОЛИМЕРАХ
Любая спиральная молекула полимера имеет бесконечное число
колебаний, которые можно разделить на группы с конечным чис-
лом колебаний, причем каждая группа будет характеризоваться
разностью фаз (6) между колебательным размещением соответ-
ствующих атомов в соседних звеньях.
Правила отбора для инфракрасного поглощения и комбина-
ционного рассеяния зависят от угла вращения (0) вокруг оси
цепи на одно повторяющееся звено.
1. Инфракрасные полосы поглощения обусловлены невырож-
денными колебаниями при разности фаз 6 = 0 или вырожденными
колебаниями, когда разность фаз 6 = 0.
2. Полосы комбинационного рассеяния обусловлены невырож-
денными колебаниями при разности фаз 6 = 0, вырожденными
Рис. 17.8. Частотно-дисперсионные кривые изолированной цепи полиэтилена
[П: 6749].
колебаниями при разности фаз 6 = 0 и колебаниями с разностью
фаз 6 = 20.
Если полимерная цепь имеет двойную ось, пересекающую спи-
раль под углом 90°, то такая цепь относится к диэдрической фак-
торной группе; тогда невырожденные колебания с разностью фаз
6 = 0, называемые A-колебаниями, разделяются на Арколебания,
неактивные под инфракрасным излучением, но активные при ком-
бинационном рассеянии, и А2-колебания, инфракрасно активные,
но неактивные при комбинационном рассеянии. Ар и А2-колебания
симметричны и антисимметричны соответственно относительно
двойной оси.
При нейтронном рассеянии (разд. 18.3) не существует правила
отбора, связанного со спиральной симметрией.
Например, в полиэтилене, имеющем зигзагообразные цепи, по-
вторяющееся звено состоит из трех атомов СН2 и существует
298
Глава 17
девять колебательных степеней свободы. Пять из них (vi—V5) яв-
ляются симметричными и четыре (v6—v9) антисимметричны.
Частота
V1
V2
V3
V4
Vs
V6
V7
V8
V9
Отнесение
Симметричные валентные колебания СН2
Ножничные колебания СН2
Веерные колебания СН2
Валентные колебания С—С
Деформационные колебания С—С—С
Антисимметричные валентные колебания СН2
Крутильные и маятниковые колебания СН2
Маятниковые и крутильные колебания СН2
Внутреннее вращение С—С
На рис. 17.8 показана зависимость между колебательными ча-
стотами и разностью фаз. Различные типы колебаний в полиэти-
леновых цепях приведены на рис. 15.1.
Например, при изменении разности фаз (6) от 0 до л маятни-
ковое колебание v7 СН2-группы (6 = 0) превращается в крутиль-
ное колебание v7 (б = л), тогда как крутильное колебание СН2-
группы v8 (6 = 0) переходит в маятниковое колебание vs (б = л).
Полиэтиленовая цепь имеет двойную винтовую ось, и угол вра-
щения (0) вокруг оси цепи на повторяющееся звено СН2 равен л.
Инфракрасные полосы и линии КР возникают от колебаний в цепи
при разности фаз б = 0 и б = л.
Обзорная литература: 74, 752, 1287, 1290, 1443.
Периодическая литература: 2385, 3099, 3824, 4053, 4624, 4734, 4742, 4906,
4932, 5122, 5123, 5316, 5318, 5320, 5484—5486, 5841, 6112, 6462, 6463, 6466, 6613,
6661, 6667, 6670, 6671, 6682, 6744, 6747, 6749, 6750, 7320.
Глава 18
АНАЛИЗ НЕЙТРОННОГО РАССЕЯНИЯ
Обзорная литература; 62, 64, 168, 392, 752, 1135, 1442.
18.1. СВОЙСТВА НЕЙТРОНОВ
Нейтрон представляет собой неустойчивую частицу с массой,
равной
тп~ 1,67482- 10"24 г (СГС) = 1,008665 ед. = 1,67482- 10“27 кг (СИ),
и временем полураспада примерно 12 мин. При распаде нейтрона
образуются протон, электрон и нейтрино. Так как у нейтрона нет
никакого заряда, на него не влияют магнитное и электростатиче-
ское поля. Он может отклоняться лишь в результате столкновения
с другими частицами. Отсутствие заряда в значительной степени
объясняет большую проникающую способность нейтрона. Остано-
вить его может лишь очень толстый барьер.
В зависимости от скорости нейтроны делятся на следующие
типы:
1. Тепловые нейтроны, которые своим названием обязаны тому,
что их средняя энергия равна средней кинетической энергии моле-
кул газа при комнатной температуре; средняя скорость таких ней-
тронов составляет 2200 м/с, а энергия их равна 0,025 эВ. Они
образуются в результате замедления быстрых нейтронов при мно-
гочисленных столкновениях последних с ядрами.
2. Промежуточные нейтроны имеют энергию порядка 0,5 эВ—
10 кэВ.
3. Быстрые нейтроны имеют энергию порядка 10—20 кэВ.
4. Релятивистские нейтроны имеют энергию > 20 кэВ.
Так как нейтрон — электрически нейтральная частица, то нет
такого барьера, который не позволял бы даже медленным нейтро-
нам достигать атомных ядер. Нейтроны могут взаимодействовать
с ядрами по одному из следующих путей:
1. Упругое рассеяние (упругое столкновение) ответственно в
первую очередь за замедление нейтронов. При упругом столкнове-
нии суммарная кинетическая энергия и суммарный момент ней-
трона и ядра, с которым он сталкивается, не изменяются, т. е. не
происходит потерь энергии в виде электромагнитного излучения.
Наиболее часто замедлителями выступают такие элементы, как
углерод и водород.
Когерентное рассеяние представляет собой особый вид упру-
гого рассеяния (дифракции) нейтронов на кристаллах. Для
300
Глава 18
объяснения когерентного рассеяния нейтроны надо рассматривать
как волны.
2. Неупругое рассеяние (неупругое столкновение) предпола-
гает потери суммарной энергии сталкивающихся частиц.
3. Реакция захвата — это поглощение нейтрона ядрами, сопро-
вождающееся повышением энергии и образованием высокоэнер-
гетического состояния ядра. Избыточная поглощенная энергия
повторно испускается в виде какой-то частицы или фотона.
Таблица 18.1
Сечения ядра для нейтронного рассеяния
Рассеяние Сечение, барн
Н D С N О F С1
Некогерентное 79,7 2,2 0,03 0,4 0,04 0,2 2,9
Когерентное 1,8 5,4 5,5 11,0 4,2 3,8 12,2
Как правило, уравнения, описывающие нейтронное рассеяние,
весьма сложные и поэтому здесь не приводятся.
Сечением ядра (о) называется вероятность протекания реак-
ции. Суммарное сечение равно сумме сечения поглощения (<та),
называемого также сечением реакции (о>), и сечения рассеяния
(о»):
О( = аа + а, (18.1)
Сечение выражается в барнах: 1 барн = 10-24 см2 (СГС) =
= 10~28 м2 (СИ).
Сечение рассеяния для водорода приблизительно на один по-
рядок больше, чем для большинства других элементов (табл. 18.1).
18.2. ПРИБОРЫ ДЛЯ АНАЛИЗА
НЕЙТРОННОГО РАССЕЯНИЯ
18.2.1. Источники нейтронов
Для получения нейтронов используются два процесса: а) бомбар-
дировка ядер в ускорителях и б) расщепление в реакторе.
Экспериментальная точность или разрешение спектров ней-
тронного рассеяния зависит от интенсивности потока нейтронного
пучка. Чем выше интенсивность потока, тем лучше разрешение.
18.2.2. Спектрометры рассеяния нейтронов
Существует два класса спектрометров нейтронного рассеяния:
1. Время-пролетные спектрометры (рис. 18.1). Пучок нейтронов
из реактора проходит через бериллиево-висмутовый фильтр, на-
Анализ нейтронного рассеяния
301
холящийся в жидком азоте. Такой фильтр пропускает нейтронный
луч с энергией 5,3 МэВ, который падает на исследуемый образец
и рассеивается во всех направлениях. Поток рассеянных нейтро-
нов прерывается вращающимся коллиматором, распределение
энергий нейтронов измеряется счетчиком пропорциональности на
Рис. 18.1. Схема устройства время-пролетного нейтронного спектрометра.
/ — активная зона реактора; 2— бериллиево-висмутовый фильтр; 3 —защита реактора;
4—образец; 5—вращающийся коллиматор; 6—детекторы на BF3; 7 —подвижный экран.
BF3. Время, затраченное нейтронами на прохождение пути от пре-
рывателя до детекторов, записывается или регистрируется в элек-
тронном анализаторе времени пролета.
2. Спектрометры на кристаллах с тройной осью (рис. 18.2).
Пучок нейтронов из реактора проходит через коллиматор и па-
дает на монохроматический кристалл. Этот кристалл отбирает
монохроматические нейтроны с длиной волны М согласно уравне-
нию Брэгга
— 2dM sin 0jW, (18.2)
где п — целое число 0, 1, 2, 3, ..., называемое порядком; М —
длина волны монохроматических нейтронов, рассеянных монохро-
матическими кристаллами; йм — расстояние между соседними
плоскостями в монохроматическом кристалле; 0м — половина угла
отклонения рассеянных нейтронов от направления падающих ней-
тронов.
Рассеянные нейтроны падают на образец, рассеиваются в пре-
делах угла ф и анализируются путем сканирования, согласно урав-
нению Брэгга
п%2 — %dA sin Од, (18.3)
где п — любое целое число 0, 1, 2, 3, ..., называемое порядком;
Х2— длина волны нейтронов, рассеянных анализирующим
Глава 18
302
Л.
кристаллом; с1а — расстояние между соседними плоскостями в ана-
лизирующем кристалле; 0л— половина угла отклонения рассеян-
Рис. 18.2. Схема устройства нейтронного спектрометра на кристаллах с трой-
ной осью.
1 — активная зона реактора; 2— защита реактора; 3 — кристаллический монохроматор; 4 — об-
разец; 5 —детектор на BFg; 6~подвижный экран.
ных нейтронов от направления нейтронного пучка, рассеянного
образцом.
Спектрометры на кристаллах с тройной осью характеризуются
высоким разрешением и обычно используются для изучения моле-
кулярных колебаний в кристаллах и направления поляризации.
Обзорная литература: 62, 64, 208, 214, 300, 392, 670, 752, 854, 1135.
Периодическая литература: 2363, 3665, 4193, 6230.
18.2.3. Детекторы нейтронов
Детекторы нейтронов основаны на образовании ионов под дейст-
вием излучения. В счетчике пропорциональности для тепловых
нейтронов с этой целью используется бор и особенно изотоп 10В:
10В + —> 7Li + 4Не (а-частица) (18.4)
На каждый захваченный нейтрон выделяется одна а-частица.
а-Частицы представляют собой мощные ионизаторы (одна а-ча-
стица с энергией 2,5 МэВ дает примерно 80 000 ионных пар), спо-
собные дать очень существенный импульс.
Типичный детектор нейтронов состоит из длинного полого ме-
таллического цилиндра, заполненного трифторидом бора — газом,
обогащенным изотопом 10В. Цилиндр выполняет роль катода, а
проволочка, расположенная коаксиально, является анодом. Ней-
Анализ нейтронного рассеяния
303
трон, проходящий через газообразный BF3, захватывается в ре-
зультате реакции с 10В, при этом выделяется а-частица.
18.2.4. Приготовление образцов
К виду, форме и размеру образца не предъявляется никаких тре-
бований. Это может быть жидкость или твердое вещество, послед-
нее в виде пленки, порошка, волокна и т. д. Образцы закрепляют
между кусочками алюминиевой фольги в держателе образца, за-
щищенном кадмием. Для уменьшения рассеяния воздухом держа-
тель образца помещают в вакуумную ячейку. Высокотемпера-
турный спектр получают при использовании электрических на-
гревательных элементов, вделанных в держателе образца. Для
измерений при низких температурах образец охлаждают с по-
мощью рубашки с жидким воздухом. —
18.3. ПРИМЕНЕНИЕ АНАЛИЗА НЕЙТРОННОГО
РАССЕЯНИЯ ДЛЯ ИЗУЧЕНИЯ
СТРУКТУРЫ ПОЛИМЕРОВ
Анализ нейтронного рассеяния позволяет получить ценную инфор-
мацию о нормальных и межцепных колебаниях в полимерах. Ней-
троны с низкой энергией могут рассеиваться полимерным образ-
цом и терять ча'сть своей энергии, которая эквивалентна харак-
теристическим молекулярным колебательным частотам образца.
Возбуждающие нейтроны должны иметь узкое распределение по
энергиям и среднюю энергию, близкую к энергии низкочастотных
движений молекул рассеивающего вещества. При этих энергиях
длины волн нейтронов сравнимы с атомными расстояниями. Рас-
сматриваемый метод анализа позволяет оценить также сечения
нейтронного рассеяния полимеров, конформации полимеров в стек-
лах, каучуках и растворах (особенно при малоугловом рассеянии
нейтронов), структуру полимерных сеток.
Есть два существенных различия между анализом нейтрон-
ного рассеяния и абсорбционной спектроскопией (или спектроско-
пией КР):
а) на спектры нейтронного рассеяния практически не оказы-
вают влияния квантовые правила, поэтому можно наблюдать все
колебательные частоты молекулы;
б) анализ нейтронного рассеяния наиболее чувствителен к
движениям, затрагивающим атомы водорода, так как сечение рас-
сеяния для водорода примерно на порядок больше, чем для боль-
шинства других элементов (разд. 18.1).
Обзорная литература: 752, 854, 1135, 1382.
Периодическая литература: 2028, 2055, 2064, 2066, 2095, 2180, 2214—2216,
2345, 2346, 2363, 2790, 2984, 2985, 3054, 3055, 3252, 3384, 3665, 3879, 4021, 4052,
4573, 4574, 4586—4589, 4658, 4751, 4927, 5001, 5124, 5401, 5402, 5538, 5796, 5826,
6038, 6141, 6142, 6150, 6151, 6220—6223, 6477, 6492, 6620, 6621, 6833—6835, 6894,
7077, 7078, 7273—7275.
Глава 19
АНАЛИЗ АННИГИЛЯЦИИ ПОЗИТРОНОВ
Обзорная литература: 520, 562, 583, 916, 1266, 1377.
19.1. СВОЙСТВА ПОЗИТРОНОВ
Различают электроны двух типов: отрицательный электрон (нега-
трон) (е~) и положительный электрон (позитрон) (е+). Эти две
частички идентичны и отличаются по заряду: —1 у негатрона и
+ 1 у позитрона. Одна из них является античастицей другой.
Масса электрона
те — 9,1091 • 10"28 г (СГС) = 9,1091 • 10"31 кг (СИ)
Заряд электрона
е = 4,802 • 10’10 эл.-ст. ед. (СГС) = 1,60210 • 10~19 А • с (СИ)
При взаимодействии позитрона с исследуемым веществом его
скорость в течение очень короткого промежутка времени
(<С 10-11 с) уменьшается до тепловых скоростей. Средняя скорость
таких позитронов получила название тепловой, так как равна
средней скорости молекул газа при комнатной температуре.
Спустя период, называемый временем жизни позитрона, по-
следний аннигилирует с электроном, при этом наблюдается выде-
ление двух у-квантов, называемых аннигиляционными фотонами'.
е+ + е--*2у (19.1)
Среднее время жизни позитронов в конденсированных средах со-
ставляет 0,1—5 нс (1 нс = 10~9 с).
Путем исследования аннигиляционных фотонов получают ин-
формацию об электронно-позитронном состоянии в момент анни-
гиляции, зависящем от химической и физической структуры об-
разца.
19.2. ЭКСПЕРИМЕНТАЛЬНЫЕ МЕТОДЫ
Существует три общих метода анализа аннигиляции позитронов:
1. Метод угловой корреляции, основанный на том, что суммар-
ный момент пары электрон — позитрон передается двум анниги-
ляционным фотонам:
а) если этот момент равен нулю, то два фотона испускаются
р строго противоположных направлениях*
Анализ аннигиляции позитронов
305
б) если этот момент отличен от нуля, то угол (0) между на-
правлениями движения фотонов будет меньше 180°, причем этот
угол зависит от момента аннигилирующей пары.
2. Метод доплеровского уширения, основанный на измерении
распределения энергии аннигиляционных фотонов с применением
эффекта Доплера. Вследствие эффекта Доплера с увеличением
скорости аннигилирующей пары распределение энергий аннигиля-
ционных фотонов становится шире. С помощью этого метода оце-
нивается также распределение момента аннигилирующей пары.
3. Метод определения времени жизни, основанный на измере-
нии длительности существования позитрона. В этом методе источ-
ником позитронов служит изотоп 22Na в форме 22NaCl. Одновре-
менно с позитроном источник испускает также у-квант. Аннигиля-
ционные фотоны регистрируются двумя разными Детекторами.
Разделение во времени сигналов от двух детекторов определяет
время жизни позитрона.
Обзорная литература: 4, 520, 521, 562, 916, 1266.
Периодическая литература: 5012, 5013.
19.3. ОБОРУДОВАНИЕ ДЛЯ АНАЛИЗА
АННИГИЛЯЦИИ ПОЗИТРОНОВ
19.3.1. Источники позитронов
Позитроны образуются при распаде двух радиоактивных изото-
пов: 22Na и 65Zn.
Источник позитронов можно легко получить испарением воды
из капли 22NaCl, помещая остаток между двумя тонкими листоч-
ками из металла или полимера. Если такие листочки достаточно
тонкие, то в них аннигилируют не более 20% позитронов. Радио-
активность такого источника не превышает 50 мКи, и нет необхо-
димости в специальной защите.
1 кюри (1 Ки) — единица радиоактивности, определяемая как
3,70-1010 распадов в секунду.
19.3.2. Гамма-сцинтилляционные счетчики
При взаимодействии гамма-излучения с определенными вещест-
вами, называемыми флуорами (иногда называемыми фосфорами),
наблюдается слабая вспышка (сцинтилляция) видимого света.
В гамма-сцинтилляционных счетчиках используются кристаллы
иодида натрия, содержащие следы иодида таллия в качестве акти-
ватора (кристаллы Nal/TlI).
Для определения таких небольших сцинтилляций используется
фотоэлемент, который обладает очень высокой чувствительностью
306
Глава 19
и плотно прикрепляется к кристаллу, чтобы испускаемый свет
эффективно передавался фоточувствительному катоду. Для этой
цели используется фотоумножитель (разд. 10.7.1).
Обзорная литература: 781.
19.3.3. Система измерения времени жизни позитрона
В системах этого типа сигналы от детекторов преобразуются в
электрический сигнал, амплитуда которого пропорциональна раз-
ности времен их регистрации. Этот сигнал поступает в многока-
нальный анализатор, который регистрирует амплитуду сигнала,
т. е. время жизни позитрона, и накапливает эту информацию. При
регистрации аннигиляций многочисленных позитронов результаты,
накапливаемые в многоканальном анализаторе, отражают распре-
деление позитронов по времени жизни, т. е. число позитронов, ко-
торые имеют определенное время жизни как функцию этого вре-
мени. Это распределение обычно называют спектром времен жизни.
19.3.4. Приготовление образцов
Специальных требований к виду, форме и размеру образцов для
исследования не существует. Это может быть жидкость или твер-
дое вещество, последнее в виде пленки, порошка или волокон.
Важно, чтобы все позитроны, испускаемые источником, задержи-
вались и аннигилировали в образце. Поэтому образец должен
быть достаточно толстым и иметь поверхностную плотность более
0,1 г/см2.
19.4. ПРИМЕНЕНИЕ АНАЛИЗА АННИГИЛЯЦИИ
ПОЗИТРОНОВ ДЛЯ ИССЛЕДОВАНИЯ
СТРУКТУРЫ ПОЛИМЕРОВ
Аннигиляция позитронов наблюдается для всех полимеров, для
которых имеются опубликованные данные. Перед аннигиляцией
позитрон с электроном дает граничное состояние, называемое по-
зитронием (и имеющее собственный символ Ps). Есть два различ-
ных типа таких состояний:
а) ортопозитроний (орто-Ps), у которого спины позитрона и
электрона параллельны (триплетное состояние) (75% всех
форм), — время его жизни примерно 140 нс, при его распаде воз-
никают три фотона;
б) парапозитроний (napa-Ps), у которого спины позитрона и
электрона антипараллельны (синглетное состояние) (25% всех
форм), — время его жизни примерно 0,125 нс, при его распаде
испускаются два фотона.
Анализ аннигиляции позитронов
307
Процесс аннигиляции позитронов в значительной степени за-
висит от структуры полимера и может применяться как экспери-
ментальный метод изучения свойств полимеров.
Аннигиляцию позитронов использовали при изучении радиа-
ционной твердофазной полимеризации. В ходе полимеризации
спектр времен жизни изменяется.
Обзорная литература: 19, 113, 136, 194, 195, 1407
Периодическая литература: 2850—2852, 2781, 3376, 3791, 4253, 4254, 4283,
4542—4544, 4966, 5549, 5550, 6573-6576, 6660, 6741, 6779, 6780.
Глава 20
СПЕКТРОСКОПИЯ ЯДЕРНОГО МАГНИТНОГО
РЕЗОНАНСА
Обзорная литература: 1, 2, 39, 52, 54, 55, 71, 91 120—122, 124, 174, 176, 177,
212, 213, 256, 274, 277, 321, 324, 373, 389, 391, 409, 413, 474, 506, 548, 561, 669, 671,
693, 730, 755, 789, 819, 824, 847, 878, 879, 903, 915, 960, 991, 1039, 1051, 1052, 1114,
1115, 1158, 1175, 1204, 1213, 1221, 1225, 1383, 1393.
Спектроскопия ядерного магнитного резонанса (ЯМР) представ-
ляет собой метод, фиксирующий переходы между энергетическими
уровнями магнитных ядер во внешнем магнитном поле. Спектро-
скопия ЯМР связана с поглощением образцом, помещенным во
внешнее магнитное поле, энергии электромагнитного излучения
в области радиочастот. Поглощение является функцией магнитных
свойств некоторых атомных ядер, содержащихся в молекуле. Кри-
вая зависимости поглощения энергии радиочастот от внешнего
магнитного поля дает спектр ЯМР.
Атомные ядра можно классифицировать по их ядерным спи-
нам. Лишь те ядра поглощают электромагнитное излучение, у ко-
торых спиновое квантовое число (ЛЬ) больше нуля.
Спиновое квантовое число (ЛЬ) зависит от массового числа и
атомного номера ядра следующим образом:
Массовое число Атомное число Спиновое квантовое число (Му)
Нечетное Нечетное или четное V2, 72, 72,...
Четное Четное 0
Четное Нечетное 1, 2, 3, ...
Магнитные яч^а
У ряда ядер Mi = 0, например это ядра 12С, 16О и 32S (угловой
момент у них отсутствует), у других ядер Mi Ф 0, так
М;= Г; 1Н (протон), 3Н, 13С, l5N, 19F, 31Р |
ЛЬ== 1:2Н (дейтерий), l4N
Л17> 1: 10В, 11В, 17О, 35С1
В табл. 20.1 представлены свойства магнитных ядер.
Все ядра, у которых спиновое квантовое число Mi =# 0, обла-
дают магнитным дипольным моментом, или магнитным моментом
(цлг), который возникает вследствие движения (вращения) заря-
женной частицы. Ядра, обладающие электрическими квадрупо-
лями, могут взаимодействовать как с магнитным, так и с электри-
ческим градиентом поля.
Спектроскопия ядерного магнитного резонанса
309
Таблица 20.1
Характеристики некоторых магнитных ядер [О: 1213]
Изотоп Распростра- ненность, % ^пиковое число (Z) Магнитный момент (Ц) Частота ЯМР в поле 10 кГс, МГц Относительная чувствительность
99,9844 1/2 2,79268 42,576 1,000
2Н 1,56- 10-2 1 0,85739 6,5357 9,64 • 10—2
Зн — 1/2 2,9788 45,414 1,21
10В 18,83 3 1,8005 4,575 1,99- 10-2
ив 81,17 3/2 2,6880 13,660 0,165
12С 98,9 0 — —к —
13с 1,108 1/2 0,70220 10,705 1,59- 10-2
I4N 99,635 1 0,40358 3,076 1,01 • 10~3
15N 0,365 1/2 0,28304 4,315 1,04- 10“3
16О 99,76 0 — — —
17О 3,7-10-2 5/2 1,8930 5,772 2,91 • 10“2
19р 100 1/2 2,6273 40,005 0,834
28Si 92,28 0 — —
29Si 4,70 1/2 0,55548 8,458 7,85 • 10-2
30Si 3,02 0 — — ——
31р 100 1/2 1,1305 17,236 6,64 - IO”2
32 S 95,06 0 — — —
33S 0,74 3/2 0,64274 3,266 2,26“ 10-3
34S 4,2 0 —— —- —
35C1 75,4 3/2 0,82091 4,172 4,71 - 10“3
37C1 24,6 3/2 0,68330 3,472 2,72 - 10“3
79Br 50,57 3/2 2,0991 10,667 7,86- 10-2
81Br 49,43 3/2 2,2626 11,499 9,84 • IO-2
127J 100 5/2 2,7937 8,519 9,35- IO-2
Ниже перечислены характеристики протона (!Н).
1. Масса
тр == 1,6730 • 10"24 г (СГС) = 1,007277 ед. = 1,6730 - 10~27 кг (СИ),
где ед. — универсальная единица массы (ед. = 1,661-10-27 кг).
2. Заряд.
3. Характеристический угловой момент, или спин. Это вектор,
обозначаемый символом 1. Компонента спинового вектора I в раз-
личных направлениях имеет только значение ±1/2Й. Символ h —
= h/2n читается как «h бар» и
h == 1,054 • КГ27 эрг • с (СГС) = 1,054 - 10"34 Дж • с (СИ),
где h — постоянная Планка:
h = 6,626 • 10"27 эрг • с (СГС) = 6,626 • 10“34 Дж • с (СИ)
4. Магнитный момент. Это вектор, обозначаемый’ символом gw-
Он связан с характеристическим угловым моментом I следующей
зависимостью:
Wv= + ^I, (20.1)
310
Глава 20
где gN называется ядерным g-фактором, безразмерная величина
(gN = 5,5855 для протона); — ядерный магнетон, равный
5,0509-10-24 эрг-Гс-1 (СГС) = 5,0509-10-27 А-м2 (СИ).
При наложении внешнего статического магнитного поля (Яо)
такие магнитные ядра, как протон }Н, претерпевают прецессию,
частота которой (<оо или Vo) определяется уравнением Лармора:
®о = ~?#о, (20.2)
v0 = Шо/2л, (20.3)
где Hq — напряженность приложенного внешнего статического маг-
нитного поля [гаусс (СГС) или тесла (СИ), 1 тесла (Т)= 104
Таблица 20.2
Частоты прецессии (v0) при различной напряженности магнитного поля (Но)
Ядро Vo при различных Но (Гс), МГц
14000 21000 23000 51000 58000 71000
’Н 60 90 100 220 250 300
2Н 9,2 13,8 15,3 33,7 38,4 46,0
13с 15,1 22,6 25,2 55,0 62,9 75,5
14N 4,3 6,5 7,2 15,8 17,9 21,5
19р 56,5 84,7 93,0 206,5 203,4 282,0
31р (Свободный элек- трон) 24,3 3,9ХЮ4 36,4 40,5 89,2 101,5 121,5
гаусс (Гс) ]; у — гиромагнитное отношение (различное для разных
ядер):
Y = 2лдоЛГ/ЛМ z; (20.4)
— магнитный момент протона; h — постоянная Планка; Mi —
спиновое квантовое число. Типичные приблизительные значения
частот прецессии v0 для различных ядер приведены в табл. 20.2.
Прецессия протона, помещенного во внешнее статическое маг-
нитное поле, например Н0 = 14 000 Гс (СГС) = 1,4 Т (СИ), про-
исходит с частотой Vo = 60 МГц (60 млн. раз в секунду). При на-
ложении магнитного поля //0 = 71 000 Гс (СГС) =7,1 Т (СИ),
vo = 300 Мгц (см. табл. 20.2).
Под влиянием внешнего статического магнитного поля (Но)
магнитные ядра способны принимать различные ориентации отно-
сительно этого поля. Число возможных ориентаций составляет
2М/ + 1, где Mi — спиновое квантовое число.
Протон (’Н), спиновое квантовое число которого Mi — 1/2, в
статическом магнитном поле (//0) может иметь лишь две ориента-
ции (рис. 20.1):
а) совпадающую с направлением приложенного поля (парал-
лельная ориентация, нижний энергетический уровень);
Спектроскопия ядерного магнитного резонанса
311
б) противоположную направлению приложенного поля (анти-
параллельная ориентация, верхний энергетический уровень).
При прецессии протон (*Н) в параллельной ориентации может
поглощать энергию (ДЕ) от высокочастотного генератора и пере-
ходить в антипараллельную ориентацию (это явление называется
зеемановским расщеплением для протона) при одном важном
4VQ
MrWZ
Ларморова
Mr
Вращающееся
ядро^>
Е^-МПд^Н^
-1/Я 1
•%
^Вращающееся
прецессия Антипараллельная
ориентация
На Ларморова,
л прецессия
Ее
Параллельная
ориентация
Напряженность приложенного
магнитного поля (Но)
Рис. 20.1. Уровни энергии (а), характеризующие зеемановское расщепление,
для протонной системы в скрещенных магнитном (Яо) и радиочастотном (7Л)
полях и кривая ЯМР-поглощения (б).
условии: частота прецессии должна совпадать с частотой высоко-
частотного генератора (это явление называется ядерным магнит-
ным резонансом). Поглощенная энергия регистрируется в виде
спектра ЯМР.
Поглощенная энергия (ДЕ = hv) очень мала [порядка
10-4 кДж-моль-1 (СИ) при наложении магнитного поля Но =
= 1,4 Т (СИ)], но может излучаться обратно в виде радиоволн
С частотой 60 МГц, которая фиксируется детектором высоких ча-
312
Глава 20
стот, указывая на то, что резонансное состояние действительно
достигнуто.
Если заселенность двух уровней (нижний и верхний энергети-
ческие уровни) выравнивается, поглощение энергии прекращается,
приводя к уменьшению наблюдаемого резонансного сигнала. Это
явление называется насыщением сигнала ЯМР.
При обычном условии измерения заселенность двух уровней
не является одинаковой, поскольку ядра с более высокой энергией
непрерывно возвращаются на нижний энергетический уровень.
Важную роль в потере (релаксации) энергии ядер с более высо-
кой энергией играют два безызлучательных процесса:
1. В процессе спин-решеточной релаксации разность энергии
ДЕ передается на соседние атомы, находящиеся в той же самой
молекуле или в молекулах растворителя.
2. В процессе спин-спиновой релаксации разность энергии ДЕ
переносится на соседние ядра.
Скорости релаксации возбужденных состояний, обусловленной
указанными двумя процессами, имеют большое значение, их опре-
деляют следующие параметры:
1. Время полужизни процесса спин-решеточной релаксации
(Т1), обычно называемое временем спин-решеточной релаксации
(в секундах).
2. Время полужизни процесса спин-спиновой релаксации (Г2),
обычно называемое временем спин-спиновой релаксации (в се-
кундах).
Время релаксации — это время, за которое величина разности
заселенностей спиновых уровней уменьшается в е раз.
Существует следующая зависимость между временем релакса-
ции (Д/) и уширением линии (Av) — так называемый принцип не-
определенности-. Д£. д/ А/2я const; (20.5)
Дv • Д/1/2л const (20.6)
Общее правило заключается в том, что при малом Д/ (в твер-
дых полимерах) Av велика (быстрая релаксация) и это ведет к
появлению в спектре ЯМР широких линий поглощения; если же
Д/— большая величина (растворы полимеров в невязких раство-
рителях), то Av мала (медленная релаксация) и в спектре ЯМР
имеются узкие линии поглощения.
Другими словами, если 1\ и Т2 малы (в твердых полимерах),
то время жизни возбужденных ядер невелико (примерно 10~5 с)
и это дает очень широкие линии поглощения в спектре ЯМР;
когда же Т\ и Т2 велики (растворы полимеров в невязких раство-
рителях), то время жизни возбужденных ядер большое (примерно
1 с), что приводит к появлению в спектре ЯМР очень резких ли-
ний поглощения.
Процесс спин-решеточной релаксации очень чувствителен к
температуре, так как при повышении температуры частоту кол§-
Спектроскопия ящерного магнитного резонанса
313
баний решетки возрастает. У полимеров зависимость 7\ от темпе-
ратуры представляет собой кривую с минимумом, соответствую-
щим началу характеристических типов молекулярных переходов
(разд. 20.9).
20.1. ИНТЕРПРЕТАЦИЯ СПЕКТРОВ ЯМР
При интерпретации спектров ЯМР важную роль играют такие
четыре характеристики, как положение линии (разд. 20.1.1), ин-
тенсивность линии (разд. 20.1.2), расщепление линии (разд. 20.1.4)
и ширина линии (разд. 20.10) в спектре ЯМР.
20.1.1. Положение линии в спектре ЯМР
Частота прецессии всех протонов, содержащихся в полимерном
образце (например, атомы водорода в СНз-, СН2- и СН-группах),
при наложении внешнего статического магнитного поля (Но) не
одинакова, причем точная величина для определенного протона
зависит от его химического окружения, т. е. степень экранирова-
ния данного протона при атоме углерода зависит от индуктивного
эффекта других групп протонов, присоединенных к углероду. По-
этому сдвиг частоты получил название химического сдвига.
Химический сдвиг — это разность в положении поглощения
данного протона и протона стандарта.
Для двух различных групп протонов положения химического
сдвига в спектре различны. Химический сдвиг зависит от напря-
женности постоянного магнитного поля (Но) и от частоты пере-
менного поля.
Измерение частоты прецессии (положение поглощения) груп-
пы ядер в абсолютной шкале частот сопряжено с большими труд-
ностями. Обычно измеряют разность частот относительно выбран-
ного стандарта.
В спектроскопии ЯМР используют внутренние стандарты, ко-
торые растворяют в исследуемом растворе, и внешние стандарты,
которые в запаянных капиллярах погружают в исследуемый рас-
твор.
Наиболее распространенным стандартным веществом является
тетраметилсилан (ТМС):
СНз
I
Н3С— Si—СНз
I
СН3
ТМС — химически инертное, магнитно-изотропное, легколетучее
(т. кип. 27 °C) вещество, благодаря высокой летучести его можно
легко удалить из образца. Он растворяется в большинстве орга-
нических растворителей, но не растворяется в воде и D2O. Его
можно вводить как внутренний стандарт в раствор исследуемого
314
Глава 20
вещества (1 вес.%). Даже при низкой концентрации ТМС дает
один интенсивный резкий сигнал, находящийся в области более
сильного поля, чем сигналы большинства других протонов. При
использовании в качестве растворителей воды или D2O ТМС мож-
но использовать в виде внешнего стандарта.
/
60 МГц
н0
600 540 480 420 360 300 240
10 “ ~ * - -
О
180 120 60
3 *
7
0\ 8, млн’1 или м. д.
9 8 7
1 2 3
6 5 4
4 5 6
2
8
9
\
V
1100
11
100 МГц
Но
1000 900 800 700 600
10
О
9 8 7 6
12 3 4
500
5
5
400
4
6
300
3
7
200
2
8
100
J
О Гц
О 8, млн'*
10 г
или м.д.
9
Рис. 20.2. ЯМР-Шкала при 60 МГц и 100 МГц
[О: 1213].
Для растворов в Н20 и D20 в качестве стандартного вещества
обычно используют натриевую соль З-(триметилсилил) пропан-
сульфокислоты:
СНз
I
СН3— Si—CH2CH2CH2SO3"Na+
I
СНз
Для выражения химических сдвигов можно пользоваться следую-
щими шкалами (рис. 20.2):
1. Частотная шкала, причем частота дается в герцах (Гц).
При выражении химических сдвигов в герцах нужно указывать
напряженность приложенного магнитного поля. Результаты сле-
дует представлять в этой шкале в тех случаях, когда спектры до-
статочно сложны или необходимо привести константы спин-спи-
нового взаимодействия (/).
2. б-Шкала (безразмерная). При выражении в этой шкале ве-
личина химического сдвига не зависит от значения Но или v0 и
получается путем деления частоты (v), при которой наблюдается
сигнал, на частоту переменного ВЧ-поля (v0) и умножения на
1066. Химические сдвиги в 6-шкале выражаются в миллионных до-
лях (млн-1 или м. д.).
3. т-Шкала. В этом случае
т—10 —6 (20.7)
Следует отметить, что 6 — положительное число.
Спектроскопия ядерного магнитного резонанса
315
Резонанс протонов ТМС наблюдается точно при 60 МГц, когда
приложенное внешнее статическое магнитное поле (Hq) имеет на-
пряженность 14092 Гс (в этой точке), 6 = 0 и т = 10. Положения
сигналов от других протонов оценивают относительно ТМС (млн-1
ИЛИ М. Д.).
На химические сдвиги оказывают влияние такие факторы, как
а) эффекты экранирования и разэкранирования, связанные с
Алифатические алициклические
/3-Замещенные алифатические
Ацетиленовые
а -Монозамещенные алира ти чес кие
(х-Дизамещенные алифатические
Олефиновые
Ароматические и гетероциклические
Альдегиды
I_I__I___I__I_[___I__I_1___I_1
10 987654321 06
0 1 234 5 6 7 8 9 Ю т
Рис. 20.3. Ориентировочные значения химических сдвигов протонов в некото-
рых органических соединениях [О: 1213].
меняющейся электроотрицательностью молекулы; б) вандервааль-
сов эффект разэкранирования, обусловленный наличием групп,
которые находятся в стерически затрудненных положениях, и
вследствие этого электронное облако такой группы стремится к
электростатическому отталкиванию электронного облака, окру-
жающего протон; в) эффекты анизотропии, обусловленные нали-
чием л-электронов, например, в алкенах, карбонильных группах,
ароматических ядрах и др.
Обзорная литература: 91, 409, 671, 697, 730, 934, 1085, 1213, 1270.
Интерпретация спектра ЯМР органического соединения начи-
нается с установления химических сдвигов различных атомов во-
дорода в молекуле с помощью корреляционных таблиц химиче-
ских сдвигов и каталогов спектров ЯМР.
На рис. 20.3 для примера показаны приблизительные значения
химических сдвигов для протонов в некоторых органических соеди-
нениях.
Следующие публикации могут быть полезными при отыскании
химических сдвигов и спектров ЯМР:
Обзорная литература: 2, 171, 176, 213, 615, 643, 1204, 1213, 1383, 1480, 1481.
Периодическая литература: 6428,
316
Глава 20
20.1.2. Интенсивность линий в спектрах ЯМР
Интенсивность означает общую силу сигнала. Это суммарная энер-
гия, поглощенная образцом при резонансе.
(Примечание. В оптической спектроскопии слово «интенсив-
ность» означает величину ординаты при записи сигнала пером
самописца. Здесь интенсивность — это
. высота линии.)
Ь---- Интенсивность линии в ЯМР-спек-
тре -это площадь под кривой ЯМР-спек-
тра поглощения. Площадь под каждым
сигналом ЯМР в спектре пропорциональ-
J на числу соответствующих атомов водо-
рода в этой группе. Измерение площа-
дей пика в ЯМР-спектрометре прово-
дится автоматически путем интегрирова-
ния каждого сигнала, причем интегриро-
Рис. 20.4. ЯМР-Спектр по-
глощения (а) и интеграл
спектра поглощения (б).
ванная величина представляется на
спектре в виде непрерывной линии, ка-
ждая ступенька которой отвечает появ-
лению нового сигнала (рис. 20.4). Вы-
сота ступеньки пропорциональна площади пика. Для широких пи-
ков точность интегрирования ниже, чем для узких пиков.
Интегральная кривая протонного ЯМР-спектра смеси позво-
ляет оценить относительное содержание компонентов. Проведение
количественного анализа этим методом особенно целесообразно,
когда разделение смеси на отдельные компоненты трудно или не-
возможно.
20.1.3. Определение молекулярного веса с помощью
ЯМР-спектроскопии
Интегральная интенсивность пика поглощения в протонном ЯМР-
спектре зависит только от того, соответствует ли молярная кон-
центрация вещества числу ядер в молекуле, ответственных за дан-
ный пик.
Интегральная интенсивность в расчете на ядро или на 1 моль
одинакова для всех веществ в образце.
При добавлении определенного количества вещества известного
молекулярного веса (стандарт ws/Ms в молях) к навеске иссле-
дуемого вещества неизвестного молекулярного веса (неизвестное
вещество w/M) справедливо следующее уравнение:
is/ns __ 1/п
ws/Ms w/M ’
(20.8)
где Is и I — интегральные интенсивности пика стандартного и ис-
следуемого вещества соответственно; ns и п — число ядер, ответ-
ственных за пики стандарта и исследуемого вещества соответ-
Спектроскопия ядерного магнитного резонанса
317
ственно; ws— навеска стандарта; w — навеска неизвестного веще-
ства; Ms и М — молекулярные веса стандарта и исследуемого
вещества соответственно.
Используя уравнение (20.8), можно легко рассчитать молеку-
лярный вес (М) исследуемого вещества.
Этот метод применим тогда, когда индивидуальные резонанс-
ные пики хорошо отделяются от остальной части спектра иссле-
дуемого вещества и стандарта. В качестве стандартных веществ
можно использовать йодоформ (СШз) или 1,3,5-тринитробензол.
Обзорная литература: 55, 769, 819.
20.1.4. Расщепление линий в спектре ЯМР
Расщепление энергетических уровней магнитных ядер пред-
ставляет собой явление, приводящее к увеличению числа энерге-
тических уровней в результате наложения магнитного поля на
систему, содержащую магнитные ядра.
Расщепление ЯМР-спектральных линий вызывается спин-спи-
новым взаимодействием между соседними протонами и зависит от
числа ориентаций спинов, которые могут иметь эти соседи. Это
явление получило название спин-спинового расщепления или спин-
спинового. взаимодействия.
Число пиков (мультиплетность), наблюдаемое в сигнале ЯМР
для группы протонов, не связано с числом протонов этой группы,
но зависит от числа протонов соседних групп. Существует пра-
вило (правило п-\- 1), согласно которому мультиплетность сигнала
от группы протонов равна (/г-j-l), где п — число соседей. (Из-
вестно несколько исключений из этого правила.)
В табл. 20.3 приведены различные варианты спин-спинового
расщепления в ЯМР. Наиболее простыми случаями спин-спино-
вого расщепления являются следующие:
1. Спиновая система АХ, которая дает спектр типа АХ
(рис. 20.5). Эта система представляет два вицинальных протона,
которые имеют различное химическое и магнитное окружение и
которые резонируют при различных положениях в ЯМР-спектре.
Они дают не одиночные пики (синглеты), а дублеты. Расстояние
между линиями каждого дублета одинаково и называется кон-
стантой спин-спинового взаимодействия J (измеряется в герцах).
Сигнал от НА появляется в виде дублета из-за двух ориента-
ций спина протона Нх — параллельной (низкая энергия) и анти-
параллельной (высокая энергия). Точно так же сигнал от протона
Нх появляется в виде дублета из-за двух ориентаций спина про-
тона НА — параллельной (низкая энергия) и антипараллельной
(высокая энергия).
2. Спиновая система АХ2, которая дает спектр типа АХ2
(рис. 20.6). Для этой системы существуют три различные возмож-
ные комбинации спина:
318
Глава 20
а) спин ядра двух протонов X имеет параллельную ориента-
цию относительно протона A(ff);
б) спин ядра двух протонов X имеет антипараллельную ориен-
тацию относительно протона А(||);
Таблица 20.3
Возможные варианты спин-спинового взаимодействия в спектрах ЯМР [О: 55]
Спиновая система Тип резонанса
А А-резонанс
А2 Синглет
А3 Синглет
А/п Синглет
А-резонанс Х-резонанс
АХ 1:1 дублет 1 : 1 дублет
ах2 1:2:1 триплет 1 : 1 дублет
АХ3 1 :3 : 3:1 квартет 1 :1 дублет
АХП (п 4- 1)-членный мультиплет 1 :1 дублет
А2Х2 1:2:1 триплет 1:2:1 триплет
А2Хз 1 :3: 3 : 1 кварт ет 1:2:1 триплет
А2Хд (п + 1 )-членный мультиплет 1:2:1 триплет
AXrt (п + 1)-членный мультиплет (т 4-1)-членный мультиплет
А-резонане М-резонанс Х-резонанс
АМХ Пара дублетов Пара дублетов Пара дублетов
амх2 Пара триплетов Пара триплетов Пара дублетов
АМХз Пара квартетов Пара квартетов Пара дублетов
A/nMpXrt (Р + О от (п-f 1) (т 4- 1) от (п + 1) (т 4- 1) от (р + 1)
в) один протон X имеет спин с параллельной ориентацией, а
другой протон X — спин с антипараллельной ориентацией (|f
или f|).
Знак f означает параллельную ориентацию спинов. Знак |
означает антипараллельную ориентацию спинов. Эти три спино-
Спектроскопия ядерного магнитного резонанса
&19
вые ориентации позволяют предположить, что сигнал от протона
А должен быть в виде триплета. Вероятность двух спиновых
ориентаций (а) и (б) одинакова, но вероятность третьей спино-
вой ориентации (в) в два раза больше, вследствие этого интен-
сивность сигнала, связанного с таким состоянием, должна вдвое
Спиновая система А (или X)
Гап
резонанса:
А- резонанс
резонанс
Спиномм 'стш, АХ2
^х(1) Нхи)
?—?—
синглет
Тип резонанса:
Спиновая система
А-резонанс 1 2:1 триплет
^резонанс 1 1 дублет
Спиновое взаимодействие между протоном Нд
и двумя соседними протонами Нх«) и НХ(^
А-резонанс I : { дублет
X -резонанс f : f дублет
t
Расщепление сигналов для двух
вицинальных протонов НА и Нх
Возможная
ориентация ft N If ||
спинов обе Параллельная Обе
Х(1)цХ(2) парал- иантипарал- антипарал-
лельные лельная лельные
Рис. 20.5. Картина спин-спинового
расщепления для спиновых систем
А (или X) и АХ.
Рис. 20.6. Картина спин-спинового
расщепления для спиновой системы
АХ2.
превышать интенсивность линий, обусловленных первыми двумя
состояниями, поэтому соотношение интенсивностей в триплете
составляет 1:2:1 (рис. 20.6).
3. Соотношение теоретических интенсивностей линий для три-
плетов, квартетов, квинтетов и т. д. можно предсказать, используя
треугольник Паскаля для биномиального разложения (рис. 20.7).
Внешние линии в мультиплетах могут иметь настолько низкую
интенсивность, что их не удается обнаружить, если не переснять
эту часть спектра в более растянутой шкале.
На константу спин-спинового взаимодействия (J) влияет ряд
факторов:
520
Глава 20
1. Геминальное взаимодействие, относящееся к протонам,
деленным только двумя связями (/ « 10—18 Гц).
раз-
п
Относителъная интенсивность
О
Синглет
Ду5лет
Триплет
Квартет
Квинтет
-1 Секстет
V>
8 1
2
3
4
5
6
7
8
r’rW'l
Y4y6Y4Y1'
Y5y1°y10y5y
'i y6 у15у^°у 15ув'
Y7xr2l4r35Y35Y2lY
8 28 56 70 56 28
Рис. 20.7. Треугольник Паскаля.
2. Вицинальное взаимодействие, относящееся к протонам,
деленным тремя связями
раз-
Гц).
S 5—
Спектры спиновой
Рис. 20.8.
системы АХ для различных
соотношений Д6/Л
(/ 0—12 Гц, обычно порядка 8
3. Взаимодействия цис(Т ;
14 Гц) и транс(1 » 11 — 19 Гц).
4. Ароматические взаимодействия
орто 7 10 Гц, Jмета 2—3 Гц,
Jпара ~ 0—1 Гц).
5. Аллильное взаимодействие (Jж
« 0-2 Гц).
6. Дейтериевое взаимодействие,
относящееся к взаимодействию прото-
на !Н с дейтероном 2Н, проявляюще-
муся в виде триплета с относительной
интенсивностью линий 1:1:1.
7. Гетероядерное взаимодействие,
относящееся к взаимодействию прото-
на (!Н) с некоторыми другими маг-
нитными ядрами, например 13С, 15N,
19F, 31Р, и определяющее возникнове-
ние сложных спектров.
Спектры первого порядка обра-
зуются, если разделение между муль-
типлетами (разность химических сдви-
гов между сигналами) намного пре-
вышает константу спин-спинового взаимодействия (Л6>-6/).
В этом случае будет наблюдаться невозмущенный спектр
(рис. 20.8).
Спектроскопия ядерного магнитного резонанса 321
Спектры второго порядка образуются, если разделение между
мультиплетами (разность химических сдвигов между сигналами)
мало (А6 </) (рис. 20.8). В этом случае спектры сильно иска-
жены, сигналы сближаются, внутренние пики становятся больше
за счет внешних пиков, меняется положение линий. Такие спектры
обычно называют АВ-спектрами (или спектрами сильного взаи-
модействия), что характеризует значительно большую близость
химических сдвигов друг к другу, чем в случае системы АХ.
Если два протона имеют одинаковый химический сдвиг
(А/ — 0), например геминальные протоны в стерически незатруд-
ненных СН2-группах, то взаимодействия между спинами в спек-
тре ЯМР не обнаруживаются и наблюдается только один пик
(рис. 20.8).
Многие спектры второго порядка были анализированы расчет-
ным путем и приводятся в литературе.
Если получается спектр второго порядка, то можно применить
ряд методик для их упрощения (разд. 20.5 и 20.6).
Обзорная литература: 1213, 1383.
20.2. АППАРАТУРА ДЛЯ ЯМР-СПЕКТРОСКОПИИ
20.2.1. ЯМР-Спектрометры
ЯМР-Спектрометры состоят из следующих основных частей
(рис. 20.9).
Рис. 20.9. Принципиальная схема ЯМР-спектрометра.
1 — радиочастотный генератор; 2 — источник магнитного поля; 3 — магнит; 4 — ампула с образ-
цом; 5 —генератор развертки; 6 — приемник; 7—записывающее устройство; 8 — компьютер.
1. Магнит. В ЯМР-спектрометрах используются три типа маг-
нитов:
а) Постоянный магнит с напряженностью магнитного поля
14 000 Гс. Приборы с такой напряженностью магнита обычно
11 Зак 238
322
Глава 20
мость достигается
называют приборами на 60 МГц. Такие спектрометры используют
только для определения спектров протонов !H.
б) Электромагнит с напряженностью магнитного поля 21 000
(23 000) Гс. Приборы с магнитом 23 000 Гс обычно называют спек-
трометрами на 100 МГц.
в) Магниты со сверхпроводящими соленоидами с напряжен-
ностью магнитного поля 51 000 и 71 000 Гс. Приборы с магнитом
51 000 Гс обычно называют спектрометрами на 220 МГц, а с маг-
нитом 71 000 Гс — спектрометрами на 300 МГц. Сверхпроводи-
огружением электромагнитов в жидкий гелий
при 4 К, при этом электрическое сопротивле-
ние практически снижается до нуля и металл
становится сверхпроводником.
Важнейшим условием при создании ЯМР-
спектрометров является обеспечение вокруг
образца однородного поля. Для того чтобы
скомпенсировать малейшую неоднородность
поля основного магнита, необходимо исполь-
зовать дополнительные катушки. Настраивая
надлежащим образом эти катушки, создают
контурное магнитное поле.
Наконечники магнита должны иметь боль-
ший диаметр, чем ширина воздушного зазо-
ра. Температуру воздуха для постоянного
магнита и температуру охлаждающей воды
для электромагнита следует тщательно кон-
Приемная
катушка
| |
Катушка
возбуждения ,
Катушка развертки
Рис. 20.10. Расположе-
ние образца и катушек
в магнитном зазоре.
тролировать.
2. Радиочастотный генератор. Это кристаллический осцилля-
тор, отрегулированный на определенную частоту, например 60 МГц
для магнита 14 000 Гс; 100 МГц для магнита 23 000 Гс; 220 МГц
для 51 000 Гс и 300 МГц для 71 000 Гс. Вырабатываемое высоко-
частотным генератором поле накладывается на образец с по-
мощью катушки возбуждения.
'3. Приемник радиочастот, с помощью которого детектируется,
усиливается и фильтруется ЯМР-сигнал.
4. Генератор развертки, который позволяет менять напряжен-
ность магнитного поля Но. Выходной сигнал от генератора раз-
вертки синхронизируется с разверткой осциллографа или само-
писца по оси х. Регулируемые электромагнитные катушки назы-
ваются катушками развертки.
5. Устройство для закрепления образца, вставляемое в зазор
магнита и представляющее собой две катушки. Приемная катушка
своей осью ориентирована перпендикулярно направлению глав-
ного магнитного поля и оси катушки возбуждения (рис. 20.10).
Ампула с образцом закрепляется в легкой турбинке, причем воз-
душная струя подбирается таким образом, чтобы обеспечивалась
устойчивая скорость вращения порядка 30 Гц.
Спектроскопия ядерного магнитного резонанса
323
6. Система записи результатов. Осциллограф и самописец син-
хронизируются с выходом генератора развертки. Присоединенный
к ЯМР-спектрометру небольшой компьютер может осуществлять
операцию, называемую накоплением.
20.2.2. Накопитель
При низкой концентрации полимера (менее 0,5 вес.%) отношение
сигнал/шум в спектре ЯМР из-за необходимости работы при боль-
ших усилениях понижается. При этом трудно отличать истинные
пики ЯМР от базовой линии с шумами. Если провести многократ-
ное сканирование, то сумма всех шумовых сигналов будет равна
нулю. Сигналы, поступающие из ЯМР-спектрометра, хранятся в
дисковой памяти компьютера. После усреднения до нуля всех шу-
мовых сигналов истинные ЯМР-сигналы от образца будут появ-
ляться в том же самом месте спектра и процесс накопления при-
ведет к увеличению отношения сигнал/шум. Вместо накопителя
такого типа (CAT computer) можно использовать цифровое на-
копление сигнала (DSA).
20.2.3. Ампулы для образцов, применяемые
в ЯМР-спектроскопии
На результаты ЯМР-измерений может влиять тип ампулы для
образца. При отклонениях от цилиндрической симметрии образца
и/или ампулы для образца возникает неоднородность магнитного
Рис. 20.11. Стандартная микрокюве-
та и ампула с образцом, применяю-
щиеся в ЯМР-спектроскопии.
/ — тефлоновый стержень; 2—держатель
микрокюветы; 3 — стандартная микрокюве-
та; 4 — стандартная ампула с образцом.
поля. Подобные отклонения от симметрии приводят к значитель-
ным колебаниям в значениях химического сдвига и ширины
линии.
Используется несколько типов микрокювет (рис. 20.11). Микро-
кювета состоит из стандартной ЯМР-ампулы, фторопластового
держателя и стандартной микрокюветы емкостью 30—100 мкл,
в которую вводят исследуемый образец. Микрокювета с образцом
11*
324
Глава 20
и фторопластовый держатель помещают в ЯМР-ампулу с по-
мощью тефлонового стержня, который затем удаляют. Простран-
ство вокруг шарика микрокюветы ниже фторопластового держа-
теля заполняют четыреххлористым углеродом, таким образом сво-
дится к минимуму возможность раскачивания микрокюветы с об-
разцом при вращении ампулы.
Ампулы для ЯМР-исследований дороги, по возможности их
следует промывать и использовать повторно. Самым лучшим спо-
собом очистки ампулы является ее промывка растворителем сразу
же после использования. Нельзя пользоваться для очистки раство-
ром хромпика, так как при этом могут остаться следы парамаг-
нитных ионов хрома, которые при последующих съемках вызы-
вают уширение пиков.
Периодическая литература: 2539, 4993.
20.3. РАСТВОРИТЕЛИ, ИСПОЛЬЗУЕМЫЕ
В ямр-спектроскопии
Наиболее резкие ЯМР-спектры получают только в невязких рас-
творах. В ряде случаев выбор растворителя для полимеров пред-
ставляет определенные трудности. Растворители, имеющие низкую
Таблица 20.4
Растворители для ЯМР-спектроскопии
Растворитель Формула Ориентиро- вочная величина б для примеси Ш Т. кип., °C Т. пл., °C
Сероуглерод cs2 46 -108,5
Четыреххлористый углерод СС14 —— 77 -23
Дейтероуксусная кислота CD3COOD 13 и 2 118 16,6
Дейтероацетон CD3COCD3 2 56 -95
Дейтероацетонитрил CD3CN 2 82 -44
Дейтеробензол Тяжелая вода (оксид дейте- СбЭв 7,3 80 5,5
рия) d2o 5 101,5 3,8
Дейтерохлороформ CDCls 7,3 61 -63
Дейтерометанол CH3OD 3,4 65 —98
Дейтеропиридин C5D5N 7,5 115 -42
Дейтеротолуол Дейтеротрифторуксусная кис- C6D5CD3 7,3 и 2,4 НО -95
лота CF3COOD 13 72 — 15
Дейтеродиметилсульфоксид (CD3)2SO 2 189 18
Гексахлорацетон cciscoccu — 203 -2
вязкость, отличаются высокой летучестью, а для того чтобы сни-
зить диполярное уширение, необходимо снимать ЯМР-спектры при
Спектроскопия ядерного магнитного резонанса
325
повышенных температурах (100—150°C). Типичными раствори-
телями для полимеров являются обычные органические раствори-
тели, в которых водород замещен на дейтерий (табл. 20.4,
рис. 10.12). Изотопная чистота дейтерированных растворителей
должна по возможности приближаться к идеальной. Хорошие
ЯМР-спектры полимеров получают при концентрациях 1—2%.
Сероуглерод
Четыреххлористый углерод
Тяжелая вода, (оксид дейтерия)
Дейтероуксусная кислота,
Дейтероацетон
Дейтероацетонитрил
Дейтеробензол
Дейтерохлоророрм
Дейтерохлороформ с i% тетраметилсилана
Т^йтеродиметилсулыроксид
Дейтерометанол
Дейтеропиридин
Дейтеротрисрторуксусная кислота.
Тетраметил силан
Рис. 20.12. Растворители, использующиеся в ЯМР-спектроскопии. Обозначены
области, в которых вероятно нахождение слабых полос, обусловленных при-
месями, в частности неполностью дейтерированных аналогов. Точное положение
этих полос зависит от условий эксперимента, особенно температуры и концен-
трации [О: 1471].
ЯМР-Спектр вещества, растворенного в одном растворителе,
может несколько отличаться от спектра, полученного при исполь-
зовании более полярного растворителя, поэтому важно во всех
работах, связанных с ЯМР-спектрами, указывать растворитель.
При замене одного растворителя на другой ЯМР-сигналы для
протонов, присоединенных к углероду, как правило, сдвигаются
очень незначительно. Иная ситуация наблюдается для протонов
групп ОН, NH и SH, сигналы от которых смещаются более за-
метно при использовании растворителей с различной полярностью
(внутри- и межмолекулярные водородные связи). При высоких
концентрациях, когда роль водородных связей усиливается, про-
тоны групп ОН, NH и SH появляются при более высоких б, чем
в сильно разбавленных растворах. Резонансное положение для
этих протонов чувствительно к температуре: с повышением темпе-
ратуры величины б снижаются, это обусловлено ослаблением во-
дородных связей при более высоких температурах.
Полимерные растворы, используемые для ЯМР-спектроскопии,
совершенно не должны содержать нерастворимых частиц (или
326
Глава 20
геля). Их удаляют путем фильтрации через микропористые филь-
тры и/или высокоскоростной седиментации в центрифуге (разд.
13.1.10).
20.4. ДЕЙТЕРИРОВАНИЕ В ЯМР-СПЕКТРОСКОПИИ
В полимерах оксид дейтерия (D2O) вступает в обменную реак-
цию с подвижными протонами, например протонами групп ОН,
NH или SH:
ROH + D—О—D —> ROD + Н—О—D, (20.9)
RCOOH + D—О-D RCOOD + H—O-D (20.10)
Пики, которые ранее наблюдались для подвижных протонов JH,
исчезают или уменьшаются, а при примерно 56 появляется новый
пик, соответствующий Н—О—D. Можно снимать ЯМР-спектр не
в D2O, а в другом растворителе, а затем добавить несколько ка-
пель D2O в исследуемый раствор и после встряхивания снять
спектр еще раз.
20.5. РЕАГЕНТЫ, ВЫЗЫВАЮЩИЕ
КОНТАКТНЫЙ СДВИГ
С помощью реагентов, вызывающих контактный сдвиг, удается
растянуть картину ЯМР-поглощения без повышения напряжен-
ности приложенного магнитного поля (рис. 20.13). К таким реа-
гентам относятся комплексы редкоземельных элементов (ланта-
ноидов) с органическими лигандами:
а) трис-(дипивалоилметанато) европий [Eu(DPM)3];
б) трис-1,1,1,2,2,3,3-гептафтор-7,7-деметил-3,5-октандионатоевро-
пий [Eu(FOD)3];
в) т/шс-2,2,6,6-тетраметил-3,5-гептан-4,6-дионатоевропий
[Eu(FHD)3];
Eu(DPM)3:
Eu(FOD)3:
Eu(FHD)3:
R, = R2 == Me3
R, == CMe3, R2 = C3FT
R, = R2 = C2F5
При использовании реактивов, вызывающих контактный сдвиг,
в качестве растворителей наиболее целесообразно применять че-
тыреххлористый углерод, дейтеробензол и дейтерохлороформ.
При проведении анализов в присутствии таких реактивов нуж-
но указывать молярное отношение сдвигающего реактива к иссле-
дуемому веществу, концентрацию, тип растворителя и т. п.
Спектроскопия ядерного магнитного резонанса
327
Молярное
отношение
реагента,
вызывающего
контактные
сдвиг,
к веществу
0:0
Н0
НЬ
8
Рис. 20.13. Влияние на ЯМР-спектр реагентов, вызывающих контактный сдвиг.
Обзорная литература: 1211, 1213.
Периодическая литература: 2686, 4070, 4229, 5577.
20.6. МЕТОД СПИНОВОЙ РАЗВЯЗКИ
20.6.1. Двойной резонанс
Двойной резонанс (двойное облучение) представляет метод, в ко-
тором в ЯМР-спектрометре находятся два источника радиоволн.
Этот метод используется для упрощения ЯМР-спектров. В случае
двух соседних протонов НА и Нх возникает мультиплетность сиг-
налов (рис. 20.14). Протон НА появляется как дублет из-за двух
спиновых ориентаций — параллельной (низкая энергия) и анти-
параллельной (высокая энергия)—протона Нх. Протон Нх также
появляется в виде дублета из-за двух спиновых ориентаций — па-
раллельной (низкая энергия) и антипараллельной (высокая энер-
гия)— протона НА. При прикладывании к протону Нх второго
сильного ВЧ-поля на частоте его резонанса переходы между двумя
спиновыми состояниями (параллельная и антипараллельная ори-
ентации) протона Нх становятся очень быстрыми, в результате
чего время жизни протона Нх в любом из его спиновых состояний
является слишком коротким, чтобы было разрешено его взаимо-
действие с протоном НА. Поэтому резонанс протона На можно
328
Глава 20
наблюдать только один раз,
коллапсировать в синглет (j
На нх
-с----с—
I I
синглет
Рис. 20.14. Двойной резонанс
(спиновая развязка) системы
АХ.
а ранее проявлявшийся дублет будет
ис. 20.14). То же самое случится при
облучении вместо протона Нх ядра
НА. Этот процесс получил название
спиновой развязки.
Для осуществления такой опе-
рации необходимо снадбить ЯМР-
спектрометр двумя ВЧ-генератора-
ми. Один протон облучается силь-
ным сигналом ВЧ-поля на его ре-
зонансной частоте, тогда как дру-
гие ядра подвергаются сканирова-
нию для определения того, на ка-
кие из них влияет подавление от
облученного протона.
Кроме полной спиновой развяз-
ки (полное подавление мультипле-
тов) и селективной спиновой раз-
вязки (частичное подавление муль-
типлетов) существуют еще три
вида частичной спин-развязки:
а) спин-тиклинг (расщепление
линий на мультиплеты);
б) ядерный эффект Оверхаузера
(увеличение сигналов поглоще-
ния) ;
в) межъядерный двойной резо-
нанс (МДР*) (изменение интенсив-
ности сигнала более чувствительного ядра при облучении менее
чувствительного ядра).
Обзорная литература: 67, 91, 177, 631, 730, 1115, 1149, 1213.
20.6.2. Спин-тиклинг
Спин-тиклинг представляет собой метод, предполагающий значи-
тельно менее интенсивное облучение ядра, чем это необходимо
для полной или селективной спиновой развязки. Эффект состоит
в увеличении числа линий при спин-спиновом взаимодействии. Эта
методика редко используется в ЯМР-спектроскопии полимеров.
Обзорная литература: 91.
20.6.3. Ядерный эффект Оверхаузера
Ядерный эффект Оверхаузера возникает при близком простран-
ственном расположении двух протонов Нл и Нв (или двух углерод-
* Иногда в качестве названия этого метода используется сокращение
ИНДОР от английского Internuclear Double Resonance. — Прим, персе.
Спектроскопия ядерного магнитного резонанса
329
них атомов 13Са и 13Св), когда становится возможным взаимо-
действие их флуктуирующих магнитных векторов (каждый оказы-
вает влияние на процесс спин-решеточной релаксации другого).
Двойное облучение при сигнале НА приводит к поглощению
и испусканию для Ид. В результате этого через флуктуирующие
магнитные векторы возбуждается релаксационный механизм про-
тона Нв. Спин-решеточная релаксация протона Нв ускоряется,
способствуя увеличению интенсивности ЯМР-сигнала Нв на
10—50%.
Эксперименты с использованием этого эффекта следует прово-
дить в атмосфере, не содержащей кислорода, так как молекуляр-
ный кислород парамагнитен и может вносить вклад в процессы
спиновой релаксации (разд. 20.9).
Обзорная литература: 91, 144, 975, 1001, 1002, 1241.
Периодическая литература: 3971, 3974, 4334, 4335, 4760, 4830, 5168, 6202,
6205, 6209, 6214, 6225, 6479.
(Примечание. Несколько статей, в которых обсуждается использо-
вание методики ядерного эффекта Оверхаузера при ЯМР-спектро-
скопии полимеров, приведено в числе ссылок в разд. 20.7 и 20.9.)
20.6.4. Межъядерный двойной резонанс
Методика межъядерного двойного резонанса (МДР) используется
для изучения взаимодействий между двумя ядрами с резко разли-
чающейся относительной чувствительностью, например !Н и 13С,
и 15N.
Обычно если такие ядра взаимодействуют, то становится не-
возможным получение спектра менее чувствительного ядра. При
применении МДР-методики такой спектр можно регистрировать
косвенно, оценивая высоту пика в спектре высокочувствительного
ядра при облучении ядра с низкой чувствительностью с помощью
узкополосной частоты. Облучение пика с низкой интенсивностью
приводит к пропорциональному уменьшению интенсивности пика
ядра с высокой чувствительностью. Кривая зависимости указан-
ных изменений от частоты развертки (в герцах) дает картину
спектра низкой интенсивности. МДР-спектры записываются прямо
на обычный ЯМР-спектр, при этом МДР-пики накладываются на
первоначальные переходы, на которые они влияют. МДР-методика
является весьма трудной и требует больших затрат времени.
Обзорная литература: 91, 730, 1213.
20.7. 13С-ЯМР-СПЕКТРОСКОПИЯ
Спин ядра 13С равен 1 = и может наблюдаться методом ЯМР
при следующей частоте прецессии (v0) (см. табл. 20.2): Vo ~
«15,1 МГц при 77о = 14ООО Гс (прибор на 60 МГц) и
Ур « 25,2 МГц при #о = 23000 Гс (прибор на 100 МГц).
330
Глава 20
Относительное содержание 13С составляет только 1,1% содержа-
ния 12С. Резонанс 13С имеет всего лишь 1,6% чувствительности
протонного резонанса, время релаксации для 13С больше, чем для
протона 1Н, и составляет несколько минут. Протонный ЯМР-сиг-
нал от связи SSC—41 расщепляется на дублет (симметрично рас-
положенный по обе стороны от основного пика !Н). Эти пики
известны под названием сателлитные ^С-пики.
Константы спин-спинового взаимодействия J для 13С—!Н и
13С—12С—!Н составляют ~100 и ~25 МГц соответственно, т. е.
они большие, и поэтому интерпретация 13С-ЯМР-спектров может
быть затруднена из-за наложения ^С-ДГмультиплетов. Для
упрощения 13С-спектра проводят полную спиновую развязку ядер
13С от всех протонов путем использования методики двойного ре-
зонанса (разд. 20.6.4). При этом получается простой спектр, со-
стоящий из серии синглетов, соответствующих каждому типу ато-
мов углерода, содержащихся в исследуемом образце.
Поскольку спиновая развязка может идти вразрез с временами
релаксации, то усилению сигнала 13С-пиков способствует приме-
нение ядерного эффекта Оверхаузера (разд. 20.6.3).
Одним из способов преодоления трудностей, обусловленных
низким естественным содержанием и малым магнитным момен-
том 13С, является использование методики накопления (разд. 20.2.2)
с многократным последовательным сканированием. Для десяти-
кратного усиления сигнал/шум требуется 100 сканирований, что
занимает довольно много времени, так как одно сканирование тре-
бует примерно 3 мин.
Обзорная литература: 61, 200, 201, 299, 690, 814, 815, 947, 1091, 1271.
20.7.1. Применение 13С-ЯМР-спектроскопии
в исследовании полимеров
Применение 13С-ЯМР-спектроскопии в химии полимеров включает
исследование стереохимии макромолекул (табл. 20.5), в том числе
структурной изомерии, пространственной изомерии, конформации
макромолекул и конформации спирали, коротко- и длинноцепного
ветвления, структуры сшитых гелей, механизма полимеризации,
механизма окисления и деструкции полимеров.
Периодическая литература-: 3029, 3216, 3629, 3810, 3971, 3974, 3988, 4258,
4545, 4608, 4722, 4760, 4956, 5125, 5126, 5131, 5151, 5362, 5556, 5968—5971, 6061,
6181, 6205, 6208, 6211, 6214, 6502, 6558, 6729, 7266, 7321.
20.8. ЯМР-СПЕКТРОСКОПИЯ
С ФУРЬЕ-ПРЕОБРАЗОВАНИЕМ (ФП)
Я^Р-фурье-спектроскопия {импульсная ЯMP-спектроскопия} пред-
ставляет собой метод, основанный на использовании ряда корот-
ких радиочастотных импульсов (~30 мкс) вместо непрерывного
сигнала, применяемого в обычной ЯМР-спектроскопии.
Спектроскопия ядерного магнитного резонанса
331
Таблица 20.5
13 С-ЯМР-спектроскопия полимеров
Полимер
Литература
Оксиэтилцеллюлоза
Поли-Ы-акрилиминоалканы
Полиакрилонитрил
Полиалкенсульфиды
Полибутадиен-1,2
Полибутадиен-1,4
Полибутен-1
Полихлоропрен
Полициклические бутадиены
Полиэтилен
П олиэтиленоксид
Полиизопрен
Полиметакрилонитрил
Поли-З-метилбутен-1
Поли-4-метилгексен-1
Полиметилметакрилат
Поли-а-метилстирол
Поли-М-формилпропиленимины
Полипентадиен
Полипентамер
Полипропилен
Полипропиленимин
Полипропиленоксид
Полистирол
Полистиролпероксид
Поливиниловый спирт
Поливинилацетат
Поливинилкарбазол
Поливинилхлорид
Поливинилметиловый эфир
Поливинилпиридин
Сополимер бутадиена с изопреном
Сополимер бутадиена с метакрило-
нитрилом
Сополимер бутадиена со стиролом
Сополимер бутадиена с пропиленом
Сополимер этиленоксида с пропилен-
оксидом
Сополимер этилена с пропиленом
Сополимер а-метилстирола с мет-
акрилонитрилом
Сополимер пропилена с бутадиеном
Сополимер пропилена с этиленом
Сополимер стирола с акрилонитри-
лом
П: 3127
П: 4258
П: 2213
П: 3358
П: 3298, 3290, 3561, 5223
П: 2448, 2880, 2881, 2949, 2950, 3128,
3239, 3289—3292, 3298, 3561, 3562,
3563, 5323, 5960, 5971, 5974, 6617,
6727, 6731, 6732, 6771
П: 5151
П: 3262
П: 5933
П: 6498
П: 5125
П: 3186, 3239, 3789, 5348, 6728, 6729,
6733, 6734
П: 4230, 4231
П: 6730
П: 2948
П: 4326
П: 3299
П: 3868
П: 3293, 3599
П: 2726
П: 2152, 3008, 3182, 4232, 4234, 4326,
5968, 5972, 5973, 7156, 7309, 7310,
7313
П: 3867
П: 6204
П: 3263,4233,4326, 4831,5132, 5969, 5973
П: 2670
П: 5534, 7178, 7180
П: 7179
П: 4480, 6843
П: 2001, 2124, 2721, 2724, 2725, 4228,
5813, 6205
П: 4326
П: 2575, 3629, 4987, 5128
П: 4956
П: 3297
П: 6328
П: 2369
П: 7061
П: 7308
П: 3296
П’ 2722 5975
П: 2517,’ 2723, 2727, 3008, 3294, 4326,
5982, 6181, 6726, 6814, 7090, 7311
П: 6205, 6557
332
Глава 20
При протонной ЯМР-фурье-спектроскопии образец облучают в
фиксированном магнитном поле сильным ВЧ-импульсом, содер-
ЯМР-
Фурье -
спектрометр
t
Временной домен
а —
Компьютер
Частотный домен
TVVVXAAAAAAAA/
+
Одычный спектр ЯМР
Рис. 20.15. Схема
ЯМР-спектроскопии с
фурье-преобразова-
нием.
жащим все частоты протонного диапазона.
Протоны каждого типа поглощают соответ-
ствующую им частоту из радиоимпульса и,
взаимодействуя друг с другом, образуют все
энергетические подуровни взаимодействия.
После снятия импульса все протоны под-
вергаются релаксационным процессам и испу-
скают поглощенную энергию и энергию взаи-
модействия одновременно.
Эти испускаемые энергии образуют вре-
менной домен (рис. 20.15, а)—сложную кар-
тину взаимодействий, — который быстро зату-
хает. Этот выходной сигнал обсчитывается
компьютером и преобразуется в частотный до-
мен (рис. 20.15,6)—каждая индивидуальная
частота отфильтровывается от сложной кар-
тины взаимодействия (фурье-преобразова-
ние). Затем сигналы приводятся к привычно-
му ЯМР-спектру (рис. 20.15, в).
Для записи, обсчета и преобразования все-
го спектра требуется лишь несколько секунд,
тогда как снятие обычного ЯМР-спектра за-
нимает несколько минут.
ЯМР-фурье-спектроскопия используется
при определении слабых сигналов, изучении
образцов при очень низких концентрациях
(10—50 мкг), исследовании образцов (ядер)
с низким естественным содержанием и малым
магнитным моментом, например 13С, опреде-
лении времен спин-решеточной релаксации
(разд. 20.9).
Обзорная литература: 187, 414, 415, 429, 430, 475, 624, 947.
Периодическая литература: 2353, 3944, 4235, 5710, 6205, 6206.
20.9. ЯДЕРНЫЕ МАГНИТНЫЕ ДИПОЛЬНЫЕ
ВЗАИМОДЕЙСТВИЯ
Каждое магнитное ядро в молекуле дает мгновенный магнитный
диполь, величина которого пропорциональна магнитному моменту
ядра.
Такое флуктуирующее магнитное поле диполя зависит от ве-
личины ядерных моментов, расстояния между ядрами в твердом
веществе, концентрации обладающих магнитными ядрами моле-
кул в растворе, частотного распределения молекулярного дви-
жения.
Спектроскопия ядерного магнитного резонанса
333
Время корреляции (тс) отражает время, в течение которого
два ядра сохраняют определенную относительную ориентацию:
а) для вращательного процесса, связанного с внутримолеку-
лярной релаксацией, тс характеризует время, необходимое для
вращения молекулы на угол, рав-
ный одному радиану;
б) для поступательного процес-
са, обусловленного межмолекуляр*
ной релаксацией, тс характеризует
время, которое необходимо молеку-
ле для продвижения на расстояние,
равное одному диаметру молеку-
лы.
Как внутримолекулярная, так и
межмолекулярная релаксация за-
висят от вязкости образца, размера
и формы макромолекул.
На рис. 20.16 показана зависи-
мость от тс:
а) для быстрого молекулярного
движения
Рис. 20.16. Зависимость Т\ и Т2
от времени корреляции (тс).
1/tc^2jiv0 и
- тс;
(20.11)
б) для очень медленного движения
l/rc^2ftv0 и l/r^l/Tc (20.12)
Время корреляции, равное
= 1/2jiv0, (20.13)
где vo — частота ларморовой прецессии [см. уравнение (20.3)],
соответствует наиболее эффективной спин-решеточной релакса-
ции, тогда как при более коротких или длинных тс величина Т\
возрастает (рис. 20.16).
Рис. 20.16 отражает также зависимость Т2 от тс:
а) при коротких тс (быстрое молекулярное движение) Т2 сов-
падает с Л;
б) при больших тс (медленное молекулярное движение) Т2
отличается от 7\: при больших временах корреляции диполь-ди-
польные взаимодействия становятся более эффективными, что при-
водит к уширению линий.
Диполь-дипольное взаимодействие для пары идентичных ядер,
имеющих спин I ~ у (например, протонов), дает статическое ло-
кальное магнитное поле (Hi)
7/z = Зк/r3 (cos20 — 1),
(20.14)
где ц— магнитный момент протона, г — расстояние между двумя
протонами (рис. 20.17), 0 — угол между линией, соединяющей два
384
Глава 20
протона, и направлением приложенного магнитного поля (7¥0) •
Как правило, наблюдаются следующие случаи влияния локаль-
ного поля на ширину линий.
1. В твердых полимерах при фиксированном угле 0 статиче-
ское локальное поле (Hi) достаточно велико (Hi ж 14 Гс для
протона при расстоянии 1А), что обусловливает большую ширину
линий в ЯМР-спектре.
2. При движении макромолекул в твердых полимерах угол 0
начинает изменяться, что приводит к усреднению локального
поля, вызывающему частичное сужение линий в ЯМР-спектре.
Рис. 20.17. Пара протонов !Н в магнитном поле Но.
3. Если движение макромолекул беспорядочное и быстрое
(малые Тс), величина cos20 усредняется по всем возможным про-
странственным ориентациям. При достижении cos20 = y локаль-
ное поле равно нулю (Я/ = 0), что приводит к резкому сужению
резонансных линий. Поэтому ЯМР-спектры растворов полимеров
надо снимать при повышенных температурах, когда тс мало.
Т{ и Т2 связаны с шириной, формой и интенсивностью полос
поглощения в спектре ЯМР. Эти характеристики можно получить
следующим образом:
1. Т\ определяют непосредственно при быстрых сканированиях
и наблюдении сигнала сразу же после помещения образца в маг-
нитное поле.
2. Т2 вычисляют из ширины линий, если оно больше 1 с,
Avi/272~ 1/л, (20.15)
где Vi/2 — ширина линии на полувысоте пика. Для жидкостей
Г2 » Гь и поэтому этот метод применять нельзя.
3. Когда Т\ <2—3 с или Г2 > ~1 с, то обычно пользуются
импульсной ЯМР-спектроскопией (разд. 20.8).
4. Истинную величину можно определить методом спинового
эха (в этой книге не рассматривается).
Обзорная литература: 91, 146, 174, 177, 318, 831, 885, 1150, 1223.
Периодическая литература: 2087, 2119—2121, 2168, 2186, 2195, 2506, 2518,
2522, 2600, 2661, 2670—2672, 2769, 2920, 2981, 3041, 3079, 3219, 3220, 3224, 3225,
3256, 3435, 3436, 3524—3530, 3616, 3628, 3629, 3745, 3786—3788, 3790, 3942, 3943,
3970, 3971, 3976, 4025, 4074, 4128, 4229, 4235, 4251, 4333—4335, 4440, 4556,
4650—4652, 4821, 4822, 4830, 4831, 4950, 4951, 4953, 4998—5000, 5070, 5169—5171,
Спектроскопия ядерного магнитного резонанса
335
5173, 5367, 5426, 5494, 5497, 5541, 5972, 6107, 6108, 6110, 6209, 6225, 6402, 6426,
6440, 6441, 6500, 6501, 6514, 6559, 6584, 7010, 7101, 7346 (см. также литературу
к разд. 20.11).
20.10. ВТОРОЙ МОМЕНТ ЛИНИИ СПЕКТРА ЯМР
Второй момент линии спектра ЯМР (А//2) является функцией фор-
мы и ширины сигнала ЯМР и равняется
Д//2= jj (Н$ f(H)dH, (20.16)
— оо — оо
где /(//)—форма линии, т. е. зависимость амплитуды резонанс-
ного сигнала от напряженности (Н) магнитного поля, (Н— Но) —
характеристика (в гауссах) отклонения каждой точки спектра от
центрального положения резонанса Но.
Сигнал ЯМР может быть представлен в виде спектра погло-
щения или спектра первой производной (рис. 20.18). Ширина на
полувысоте (Avi/2) (рис. 20.18, а) и ширина между пиками (Avpp),
т. е. расстояние между экстремумами (рис. 20.18, б), равны 1/лТ2,
где Т2 — время спин-спиновой релаксации.
С увеличением температуры ширина сигнала ЯМР уменьшается
(рис. 20.19). ___
Второй момент ЯМР (А/f2) связан с ориентацией молекуляр-
ных цепей в жестких твердых полимерах следующей зависи-
мостью:
A^ = -^-^r;-.6(3co^9-1)2 (20.17)
а
в квадратных гауссах (СГС) или квадратных теслах (СИ), где
0 = 4/(/+1)§2л,Р2,) (20.18)
I — спиновое квантовое число исследуемого ядра; gn— ядерный
g-фактор; Pjv — ядерный магнетон, N — число ядер, по которым
проводится суммирование; гц— расстояние между исследуемыми
ядрами i и j-, 9 — угол между линией, соединяющей ядра i и /, и
направлением приложенного магнитного поля.
Эффект молекулярного движения может проявляться в умень-
шении второго момента А//2, когда частота, обусловленная дви-
жением, превышает ширину резонансного спектра (в единицах ча-
стоты) .
На практике уменьшение второго момента наблюдают при по-
вышении температуры образца и увеличении частот, связанных с
молекулярным движением (рис. 20.20).
Уменьшение АН2 при повышении температуры обусловлено
тепловым движением атомов, приводящим к уменьшению членов,
336
Глава 20
£ Сигнал , Сигнал
Рис. 20.18. Сигналы ЯМР в виде спектра
поглощения (а) и первой производной (б)
этого спектра.
20.19. Уменьшение ширины
сигналов ЯМР при повышении температуры.
Рис. 20.20. Зависимость второго момента ЯМР
(Д№) от температуры для различных типов
полимеров,
Спектроскопия ядерного магнитного резонанса
337
входящих в уравнение (20.17), — среднего значения (3cos20 — 1)
и /~6.
Обзорная литература: 145, 146, 885, 1347.
Периодическая литература: 2013, 3038, 3260, 3830, 3836, 4015, 4442, 4939 (см.
также литературу к разд. 20.9).
20.11. ПРИМЕНЕНИЕ ЯМР-СПЕКТРОСКОПИИ
ШИРОКИХ ЛИНИЙ
Измерения релаксации протонов при прямом наблюдении ЯМР-
спектров широких линий используются в основном при изучении
морфологии полимеров и молеку-
лярной подвижности.
ЯМР-сигнал жестких молекул
в кристаллической или аморфной
фазе полимерного образца ниже
температуры стеклования пред-
ставляет собой относительно ши-
рокую линию. Ширина на полу-
высоте такой линии составляет
примерно 10—20 Гс. Уширение
линии обусловлено локальными
магнитными полями протонов об-
разца. При увеличении подвиж-
ности молекул ширина уменьша-
ется, что объясняется уменьше-
Рис. 20.21. Сигнал ЯМР широких ли-
ний для частично кристаллического
полимера.
нием среднего времени, при ко-
тором два ядра остаются в данной относительной ориентации.
Поэтому, например, сигнал молекул в образце аморфного поли-
мера имеет ширину на полувысоте только 0,1 Гс или даже менее.
В результате влияния подвижности на ширину линии ЯМР-
сигнал частично кристаллического полимера выше температуры
стеклования состоит из двух компонент (рис. 20.21):
а) узкая компонента, обусловленная сегментальной подвиж-
ностью цепей, находящихся в аморфной области.
б) широкая компонента, обусловленная движением жестких
цепей в кристаллических областях и жесткоцепной фракцией,
иногда содержащейся в аморфных областях.
Изучение широких линий ЯМР-спектров позволяет также полу-
чить информацию о структуре расплава, например, образование
пучков параллельных цепей вызывает отклонения от лоренцевой
формы сигнала ЯМР.
Обзорная литература: 20, 174, 461, 581, 885, 1063, 1145, 1222.
Периодическая литература: 2196, 2221, 2368—2370, 3065, 3171, 3277, 3279,
3382, 3703, 4127, 4441, 4557, 4583, 4675, 4745, 4840, 4958, 5175, 5361, 5543, 5591,
5814, 5885, 6211, 6235, 6261, 6326, 6422, 6424, 6444, 6445, 6968, 7126, 7302—7304
(см. также литературу к разд. 20.9).
338
Глава 20
Таблица 20.6
'Н-ЯМР-спектроскопия полимеров
Полимер
Литература
Полиацет альдегид
Полиакриламид
Полиакрилаты
Полиакриловая кислота
Полиакрилонитрил
Полибутадиен
Полихлоропрен
Полициклические бутадиены
Сложные полиэфиры
Простые полиэфиры
Полиэтилен
Полиэтиленоксид
Полиизопрен
Полиметакриловая кислота
Полиметилакрилат
Полиметилакрилонитрил
Полиметилметакрилат
Поли-а-метилстирол
Полифторбутадиен
Полипропилен
Полипропиленоксид
Полипропйленсульфид
Полистирол
Полиуретаны
Поли-Ы-винилацетамид
Поливинилацетат
Поливиниловый спирт
Поливиниламин
Поливинилкарбазол
Поливинилхлорид
Поливинилхлоридсульфон
Поливинилфторид
Поливинилкетон
Поливинилпиридин
Поливинилиденфторид
Сополимер метилметакрилата с мет-
акриловой кислотой
Сополимер метилметакрилата с хло-
ропреном
П: 3709
П: 7265
П: 2905, 5133, 7284
П: 5127
П: 5394
П: 3946, 3947, 4348, 6184, 6185, 6604,
6731, 6735, 7350
П: 3363, 3365, 3369, 3597, 5573
П: 5933
П: 5768
П: 2602, 3189, 4952, 5446, 5617, 5959
О: 753; П: 3321, 3368, 3728
П: 6738
П: 3186, 6207, 6264, 6735
П: 4607
П: 5134
П: 4243, 5130, 6327, 6645, 7207
О: 173; П: 2520, 3366, 3367, 3369, 3521
3945, 5957, 6497, 6499, 6500
П: 4883, 5404, 5492
П: 6824
П: 3366, 3368, 3409, 3418, 3520, 3975,
3977, 4464, 4961, 5567, 5568, 6198,
6326, 6530, 6531, 6787, 7167, 7168,
7312, 7314, 7315
П: 2945, 5557, 5558
П: 4259
П: 2516, 2601, 3549, 3972, 6329, 6619,
7276
П: 6618
П: 5393
П: 5958
П: 3126, 5360, 7178
П: 5393
П: 3994, 6843, 7103, 7283
П: 2240, 2461, 2514, 2515, 2521, 2761,
2854, 3319, 3940, 3973, 4318, 4319,
5803, 5956, 6197, 6365, 6785, 6786,
7089, 7285
П: 2673
П: 7128
П: 5129, 5236
П: 4987, 5128, 7031
П: 7125
П: 4317
П: 3261
Спектроскопия ядерного магнитного резонанса
339
20.12. ПРИМЕНЕНИЕ ЯМР-СПЕКТРОСКОПИИ
ВЫСОКОГО РАЗРЕШЕНИЯ
К областям применения ЯМР-спектроскопии высокого разреше-
ния для характеристики полимеров относятся: изучение конфи-
гурации полимерных цепей (форма цепей полимера, образованная
основными валентными связями); исследование конформации по-
лимерных цепей (форма цепей полимера, обусловленная враще-
нием вокруг основных валентных связей); анализ распределения
последовательностей и тактичности в полимерах и сополимерах;
установление разницы между полимерными смесями, блок-сополи-
мерами, чередующимися сополимерами и статистическими сополи-
мерами; исследование переходов спираль — клубок; изучение мо-
лекулярных взаимодействий в полимерных растворах, диффузии
в полимерных пленках, совместимости полимеров и полимерных
смесей; исследование процессов сшивания; изучение механизма
роста цепи при винильной полимеризации.
В табл. 20.6 указаны примеры исследования микроструктуры
полимеров методом ЯМР-спектроскопии.
Обзорная литература: 20, 170, 172—175, 177, 178, 280, 341, 683, 778, 831,
887, 1084, 1113, 1165, 1190, 1402, 1416.
Периодическая литература: 3295, 3417, 5357, 5377, 5577, 7055, 7226.
20.13. ОЦЕНКА СТЕПЕНИ КРИСТАЛЛИЧНОСТИ
ПОЛИМЕРОВ МЕТОДОМ ЯМР-СПЕКТРОСКОПИИ
На основании данных ЯМР-спектроскопии можно определить сте-
пень кристалличности полимера (%с) по формуле (см. рис. 20.21):
Хс ___ Площадь широкой компоненты . qx
1 — Хс Площадь узкой компоненты ' * *
Обзорная литература: 555, 713, 885, 1222, 1225, 1427.
Периодическая литература: 2369, 2938, 3278, 3350, 5842, 5889, 5890, 6425,
6427, 6966, 7126, 7127.
Глава 21
СПЕКТРОСКОПИЯ ЭЛЕКТРОННОГО
ПАРАМАГНИТНОГО РЕЗОНАНСА
Обзорная литература: 33, 50, 51, 60, 71, 111, 147, 212, 274, 473, 505, 506, 573,
669, 693, 794, 799. 842, 904, 959, 988, 1005, 1050, 1071, 1088, 1095, 1157, 1166, 1175,
1193, 1248, 1279, 1326, 1375.
Спектроскопия электронного парамагнитного резонанса (ЭПР),
известная также под названием спектроскопии электронного
спинового резонанса (ЭСР), представляет собой метод, регистри-
рующий переходы между спиновыми уровнями неспаренных элек-
тронов молекулы во внешнем магнитном поле. ЭПР (ЭСР)-спектро-
скопия имеет дело с поглощением микроволновой энергии электро-
магнитного поля образцом, помещенным в такое поле. Поглощение
представляет собой функцию неспаренных электронов, содержа-
щихся в молекуле. Спектр ЭПР (ЭСР) — это зависимость погло-
щения микроволновой энергии от внешнего магнитного поля.
В атомах и молекулах электроны образуют пары. Каждому
электрону, находящемуся на определенной орбитали со спиновым
квантовым числом Ms = — у, соответствует другой электрон на
той же самой орбитали со спиновым квантовым числом Ms =
= + у. Спаренные электроны не дают спектр ЭПР. У неспарен-
ного электрона на той же самой орбитали нет соответствующего
электрона-партнера, поэтому такой электрон дает спектр ЭПР.
Основными свойствами электрона являются:
1. Масса
те = 9,112- 10“28 г(СГС) = 5,486 • 104 ед. = 9,112- 10-31 кг(СИ),
где ед. — универсальная единица массы
(1 ед. = 1,661 • 10~27 кг)
2. Заряд
е = 4,803 • 10~'° = ст. ед. (СГС) = 1,60210 • 10~19 А • с (СИ)
3. Характеристический угловой момент, называемый спином.
Это вектор, обозначаемый символом S. Компонента спинового век-
тора (S) вдоль любого направления может принимать значения
, 1 *
только ± jп.
Символ h = h/2n читается как «А бар», причем
Я = 1,054- 10-27 эрг • с (СГС)= 1,054- 10“34 Дж • с (СИ),
Спектроскопия электронного парамагнитного резонанса
341
где h — постоянная Планка,
/г = 6,626-10’27 эрг-с (СГС) = 6,626 10-34 Дж-с (СИ)
4. Магнитный момент. Это вектор, обозначаемый символом
Его связывает с характеристическим угловым моментом S сле-
дующее уравнение:
Me=-4-S, (21.1)
где g — g-фактор, называемый также лянде-^-фактором, — безраз-
мерная величина, равная 2,002319 для неспаренных электронов;
Р — магнетон Бора\
0 = 9,2740-10"21 эрг-Гс"1 (СГС) = 9,2740 • 10’24 А • м2 (СИ)
(обозначается также символом g0)-
Знак минус в уравнении (21.1) указывает на то, что вектор
магнитного момента электрона противоположен по направле-
нию угловому моменту S.
Кроме спина у электрона существует дополнительный угловой
момент (называемый орбитальным угловым моментом), обуслов-
ленный движением электрона не только вокруг своей оси, но так-
же и по орбите.
Помещенный во внешнее магнитное поле (Но) электрон со спи-
новым квантовым числом Ms — 1/2 может иметь только две ориен-
тации (рис. 21.1, а): совпадающую с направлением приложенного
поля (параллельная ориентация, нижний энергетический уровень)
и противоположную направлению приложенного поля (антипарал-
лельная ориентация, верхний энергетический уровень).
При прецессии электрон в параллельной ориентации способен
поглощать энергию (АЕ) от микроволнового источника и перехо-
дить в антипараллельную ориентацию (это явление называется
зеемановским расщеплением для электрона) лишь при соблюде-
нии следующего условия: частота прецессии должна совпадать
с частотой микроволнового источника (это явление называется
электронным спиновым резонансом). Поглощенная энергия реги-
стрируется в виде ЭСР{ЭПР)-спектра (рис. 21.1, б или в).
При сближении заселенностей двух уровней (верхнего и ниж-
него энергетического уровней) поглощение энергии прекращается
и наблюдаемый резонансный сигнал исчезает. Это явление полу-
чило название насыщения ЭСР (ЭПР)-сигнала.
В обычных условиях заселенность двух уровней не одинакова,
поскольку электроны с повышенной энергией непрерывно возвра-
щаются на нижний энергетический уровень. Важную роль в потере
(релаксации) энергии электронами с высокой энергией играют
следующие два безизлучательных процесса: процесс спин-решеточ-
ной релаксации, при котором разность энергий (АЕ) передается
на соседние атомы в той же самой или другой молекуле, и
h a
§
Mp+l/2.
A^fc£,=/n>=g/3//0
0 HQ] Параллельная
Ms=-\/Z ориентация
6
#0=0
Магнитное поле (#0)
Магнитное поле (//0)
Рис. 21.1. Зеемановское расщепление (а) спиновых уровней электрона в скре-
щенных магнитом (Яо) и микроволновом полях. ЭПР (или ЭСР)щпектр
поглощения, полученный при развертке магнитного поля (б). Первая произ-
водная сигнала ЭПР (в). «Палочный» спектр (г)—спектр первой производной
при нулевой ширине.
Спектроскопия электронного парамагнитного резонанса
343
процесс спин-спиновой релаксации, при котором разность энергий
(АВ) передается соседним электронам.
Большое значение имеют скорости релаксации, обусловленной
указанными процессами, которые определяются временем полу-
жизни процесса спин-решеточной релаксации (7\), обычно назы-
ваемым временем спин-решеточной релаксации (в секундах), а
также временем полужизни процесса спин-спиновой релаксации
(Т2), обычно называемым временем спин-спиновой релаксации
(в секундах).
Время релаксации характеризует время, за которое величина
разности заселенностей энергетических уровней уменьшается в
е раз.
7\ можно определить, наблюдая возвращение сигнала к его
равновесной интенсивности после быстрого уменьшения мощности
микроволнового возбуждения.
Т2 легко найти из ширины линии по следующему уравнению:
в обратных секундах (с-1), где g— g-фактор, р — магнетон Бора,
АЯ1/2 — ширина линии (в гауссах) на полувысоте максимальной
интенсивности (не на спектре первой производной) (рис. 21.2 или
21.3), h — константа, й —
Между временем релаксации А/ и уширением линии Av наблю-
дается следующая зависимость (принцип неопределенности):
А£ А/ ~ Л/2л ж const, (21.3)
или
Av А/ « А/2л ~ const, (21.4)
или
gP АЯ1/2 • А/ ~ Л/2л const (21.5)
Общим правилом является то, что при малом А/ величина Av воз-
растает (быстрая релаксация), что приводит к появлению в спек-
тре ЭСР (или ЭПР) широких линий поглощения; при большом
А/ величина Av мала (медленная релаксация), что обусловливает
появление в ЭСР (или ЭПР)-спектре узких линий.
Определение времен релаксации 7\ и Т2 необходимо для изуче-
ния вероятности переходов между двумя спиновыми уровнями и
позволяет оценить молекулярную подвижность в кристаллических
областях полукристаллических полимеров.
21.1. ИНТЕРПРЕТАЦИЯ ЭПР(ИЛИ ЭСР)-СПЕКТРОВ
Для интерпретации спектров ЭПР (или ЭСР) важны следующие
четыре характеристики: форма линии (разд. 21.1.1), ее интенсив-
ность (разд. 21.1.2), положение (разд. 21.1.3) и расщепление
(разд. 21.1.4).
344
Глава 21
21.1.1. Форма линии ЭПР
Спектр ЭПР (или ЭСР) можно регистрировать (рис. 21.2 и 21.3)
в виде кривой поглощения, а также в виде первой или второй про-
изводной этой кривой поглощения. (Примечание. В отличие от
ЯМР-спектрометров, которые обычно дают кривую поглощения,
Таблица 21.1
Важнейшие параметры, характеризующие лоренцеву
и гауссову линии
Параметр Линия Лоренца Линия Гаусса
Ширина на полувысоте максимальной интен- ля1/2 1/2
сивности
Расстояние между пи- ками Д#рр = -^- РР д/3 t^Hpp- ( 2 У2 ^1/2 \ In 2 7 2
Высота пика , 2 А= ( 1п2У/2 2
я 1/2 л 7 А/7
Расстояние по вертика- ли между пиками д зут 1 В = Г 2 \1/2 2 1п2
" (Д"1/2)2 ле) (Д#1/2)2
Амплитуда положитель- II и С= А / 16е3/2 In 2 \
ного пика \ 1/2 7 \ (л^1/а)2 )
Амплитуда отрицатель- Z" QO | 1 II Q D = —
ного пика \ Д/Ч/2 7 \ Д#1/2 7
ЭПР-спектрометры сразу же выдают первую производную кривой
поглощения энергии.)
Для описания формы линий спектра ЭСР обычно проводят
сопоставление с формой гауссовой и лоренцевой кривых, используя
следующие аналитические выражения:
а) кривая Лоренца (рис. 21.2)
(/ = «/(!+ М; (21.6)
б) кривая Гаусса (рис. 21.3)
у = а~Ьх> (21.7)
В табл. 21.1 приведены измеряемые экспериментальные пара-
метры, характеризующие оба типа спектральных кривых.
В ряде случаев форма кривой поглощения представляет собой
сочетание гауссовой и лоренцевой форм. Математический обсчет
точной формы кривой ЭПР (или ЭСР) весьма сложен. Для упро-
щения такой обработки и установления различия двух типов кри-
вых существуют два способа:
Спектроскопия электронного парамагнитного резонанса
345
Рис. 21.2. Лоренцева форма линий в ЭПР-спектре.
Plic. 21.3. Гауссова форма линии в ЭПР-спектре.
346
Глава 21
1. Метод определения наклонов производной линии поглоще-
ния, иллюстрируемый рис. 21.4. Для гауссовой кривой отношение
наклонов т: п = 2,2, а для лоренцевой кривой т : п = 4.
Рис. 21.4. Идентификация лоренцевой и гауссовой форм первой производной
ЭПР-спектра методом определения наклонов.
2. Метод нормирования производной линии поглощения, иллю-
стрируемый на рис. 21.5. Базовую линию разделяют на ряд отрез-
Рис. 21.5. Идентификации лоренцевой (/) и гауссовой (2) форм первой про-
изводной ЭПР-спектра методом нормирования.
ков а, где а — точка на абсциссе, отвечающая максимуму на кри-
вой, и проводят нормирование ординат к единице.
Экспериментальные способы определения ширины линии опи-
саны в разделе 21.2.4.
Обзорная литература: 646, 1088, 1375.
Периодическая литература: 3501.
21.1.2. Интенсивность линий в спектре ЭПР
Интенсивность характеризует суммарную силу сигнала и пред-
ставляет собой энергию, поглощенную образцом при резонансе.
(Примечание. В оптической спектроскопии слово «интенсивность»
характеризует величину ординаты спектра на самописце, т. е. ин-
тенсивность равна высоте линии.)
Интенсивность линии ЭПР (или ЭСР)-спектра — это площадь
под кривой ЭПР.
Спектроскопия электронного парамагнитного резонанса
347
Площадь под каждым сигналом ЭПР пропорциональна числу
неспаренных спинов в образце (на грамм, миллилитр или милли-
метр длины образца). Для того чтобы определить число свободных
радикалов в различных образцах по их ЭПР-спектрам, необходимо
вычислить интеграл сигналов поглощения, так как формы линий
могут сильно отличаться друг от друга. К сожалению, такой ра-
счет нельзя провести с высокой степенью точности (не более
30-50%).
В разд. 21.3 описано измерение абсолютного числа неспарен-
ных спинов.
21.1.3. Положение линий в спектре ЭПР
За положение линии в ЭПР (или ЭСР)-спектре обычно прини-
мается точка, в которой первая производная спектра пересекает
нулевой уровень. Для асимметричных спектральных кривых по-
рошков часто также определяют точки максимума и минимума на
первой производной спектра поглощения. Точность измерений за-
висит от ширины линии и уровня шумов.
Положение линии в спектре указывают в гауссах (или теслах).
Так как поле зависит от частоты спектрометра, положение обычно
характеризуют также с помощью g-фактора.
g-Фактор определяют как коэффициент пропорциональности
между частотой и полем, при котором наблюдается резонанс, и
находят из уравнения
ДЕ =/tv = g|3770, (21.8)
где ДЕ — энергия, поглощенная от радиочастотного источника, ко-
торый вызывает переходы между двумя состояниями электрона;
h — постоянная Планка; v — частота радиоволн (МГц); g— g-фак-
тор (g = 2,002319 для несвязанных электронов); |3 — магнетон
Бора; Hq — напряженность приложенного внешнего магнитного
поля.
g-Фактор пропорционален магнитному моменту изучаемой мо-
лекулы. Обычно g-фактор связанных неспаренных электронов в
молекуле не равен g-фактору свободных (несвязанных) элек-
тронов.
Из-за магнитных взаимодействий, обусловленных орбитальным
угловым моментом неспаренного электрона, g-фактор отклоняется
от значения 2,002319. Поскольку орбитальный угловой момент не-
спаренного электрона зависит от его химического окружения в
атоме, молекуле или кристалле, то g-фактор, вычисленный на
основании уравнения (21.8), также должен изменяться в зависи-
мости от химического окружения. Следствием этого являются зна-
чительные отклонения g-фактора от величины 2,002319, подобные
химическим сдвигам ЯМР (разд. 20.1.1).
348
Глава 21
В монокристаллах g-фактор зависит от направления внешнего
магнитного поля по отношению к кристаллографическим осям —
х, у и z. Следует использовать величины gx, gy и gz для того, что-
бы показать три возможных значения g-фактора, получаемые при
проведении экспериментов на кристалле, последовательно ориен-
тированном в трех взаимно перпендикулярных направлениях от-
Рис. 21.6. Аксиальная асимметрия,
£ц=^ g±-
а—спектр поглощения; б —первая производ-
ная спектра.
Рис. 21.7. Анизотропия ^-фактора,
gi < < ёз-
а —спектр поглощения; б —первая произ-
водная спектра.
носительно внешнего вектора поля (это явление получило назва-
ние анизотропии g-фактора).
Для кристаллов с аксиальной симметрией используются сле-
дующие выражения для g-фактора:
a) gi — при измерении g-фактора, когда ось кристалла пер-
пендикулярна направлению внешнего магнитного поля (Яо)‘>
б) gu — при измерении g-фактора, когда ось данного кристалла
параллельна направлению внешнего магнитного поля (Яо)«
Анизотропия g-фактора обусловливает ассимметрию спектраль-
ной кривой (рис. 21.6 и 21.7). В растворах при быстрых опроки-
дываниях можно наблюдать только среднюю величину g-фактора.
g-Фактор для органических радикалов близок его значению
для свободного электрона, т. е. 2,002319. По отклонению g-фак-
тора радикалов от указанного значения можно судить о структуре
свободного радикала.
Перечень g-факторов приведен в О: 447. В разд. 21.4 описы-
вается способ определения g-фактора.
Обзорная литература: 111, 1088, 1375.
Периодическая литература: 4123, 4124, 4615.
21.1.4. Расщепление линий в спектре ЭПР
Расщепление энергетических уровней электрона представляет со-
бой увеличение числа энергетических уровней в результате воз-
действия на систему с неспаренными электронами магнитного поля.
При наложении на неспаренные электроны магнитного поля
число энергетических уровней возрастает от одного (Е = Eq) до
Спектроскопия электронного парамагнитного резонанса
349
двух (см. рис. 21.1, а):
£1 = £о+^§рЯо.
Е2 — Е0 — у£|Ж
(21.9)
(21.10)
В тех случаях, когда система содержит ядра с магнитным момен-
том, например протон СН), вблизи неспаренного электрона, на
магнитный момент электрона влияет ориентация магнитного
Рис. 21.8. Диаграмма энергетических
уровней (а), отражающая сверхтон-
кое расщепление сигнала ЭПР в
результате взаимодействия неспарен-
ного электрона с ядром, спин кото-
рого М/== уС’Н). Спектр поглоще-
ния (б). Первая производная спек-
тра (в). «Палочный» спектр (г) —
относительные интенсивности линий
в спектре первой производной ха-
рактеризуются высотами линий на
диаграмме. А// — константа сверх-
тонкого расщепления.
Рис. 21.9. Диаграмма энергетических
уровней (а), отражающая сверх-
тонкое расщепление сигнала ЭПР в
результате взаимодействия неспа-
ренного электрона с двумя ядрами
со спином = — (1Н). Спектр
поглощения (б). Первая производ-
ная спектра (в). «Палочный» спектр
(г) — интенсивности линий пропор-
циональны коэффициенту биноми-
ального расширения (1 Д- х)п. &Н —
константа сверхтонкого расщепления.
350
Глава 21
момента ядра. В результате такого взаимодействия каждый
магнитный энергетический уровень электрона расщепляется на ряд
подуровней (рис. 21.8 и 21.9). Это взаимодействие электрона и
магнитного ядра называется сверхтонким взаимодействием, а рас-
щепление энергетических уровней — сверхтонким расщеплением.
Подуровней, обусловленных сверхтонким расщеплением, возникает
Относительная интенсивность
о
Синглет
2
,2
•6^,4
3
4
5
6
7
8
Дублет
Триплет
Квартет
Квинтет
^6^16 id^s^i^Секстет.
К 15^ 20 J5 6 1
4,21^35^35^21^,7^
28 56 70 56 28 8
Рис. 21.10. Треугольник Па-
скаля: биномиальное расшире-
ние, характеризующее коэф-
фициенты расширения (1-|-х)п.
(Сумма вдоль любого ряда
равняется 2П.)
столько, сколько возможно ориентаций ядерного магнитного мо-
мента.
Константа сверхтонкого расщепления (АЯ), измеряемая в
гауссах, представляет собой расстояние между двумя сверхтон-
кими линиями в спектре (рис. 21.8 и 21.9).
В результате взаимодействия неспаренного электрона и п экви-
валентных протонов возникает п + 1 линия, относительная интен-
сивность которых пропорциональна коэффициенту биномиального
разложения (1+%)п (рис. 21.10). Сводные данные о константах
сверхтонкого расщепления (А//) приведены в 0:447. В разд. 21.2.4
приводятся экспериментальные условия измерения АЯ.
Обзорная литература: 111, 1088, 1375.
21.2. ОБОРУДОВАНИЕ
ДЛЯ ЭПР(ЭСР)-СПЕКТРОСКОПИИ
21.2.1. ЭПР(ЭСР)-Спектрометры
Спектрометры электронного парамагнитного (спинового) резо-
нанса имеют следующие основные узлы (рис. 21.11):
1. Магнит. В ЭПР-спектрометрах используются три типа маг-
нитов:
а) соленоиды с воздушным сердечником, редко применяющиеся
в низкочастотных спектрометрах (100—200 Гс);
б) магниты с железным сердечником с напряженностью поля
3000—18 000 Гс;
в) сверхпроводящие магниты с напряженностью 60 000 Гс;
электромагнит погружают в жидкий гелий при 4 К, при этом элек-
Спектроскопия Электронного парамагнитного резонанса
351
тросопротивление исчезает и металл приобретает сверхпроводи-
мость.
Важнейшим условием для хорошей работы ЭПР-спектрометра
является однородность магнитного поля вокруг образца. Регуля-
рно. 21.11. Блок-схема спектрометра электронного спинового (парамагнитного)
резонанса.
/ — магнит; 2—микроволновый генератор; 3 —система автоматической подстройки
(контроля) частоты; 4 — плечо сравнения; 5—аттенюатор; 6—микроволновод; 7—мост;
8 — детектор; 9 — резонатор; 10 — катушки развертки; 11 — генератор развертки; 12 — усилитель;
13 — фазочувствительный детектор; 14 — регистрирующее устройство.
рование стабильности магнитного поля часто осуществляют с по-
мощью полупроводникового кристалла, помещаемого в магнитный
зазор, в котором возникает напряжение, пропорциональное полю,
в результате чего любые изменения в напряженности могут быть
сведены на нет с помощью тока обратной связи.
Полюсные наконечники магнита имеют большой диаметр по
сравнению с шириной воздушного зазора. Следует тщательно сле-
дить за температурой воздуха для постоянного магнита и темпе-
ратурой охлаждающей воды для электромагнита.
2. Микроволновый мост, состоящий из перечисленных ниже
частей.
а) Микроволновый генератор, который служит источником
микроволнового излучения. Наиболее распространенным генера-
тором является клистрон, который можно отрегулировать на узкий
частотный интервал. Микроволновое излучение передается по вол-
новодам. Микроволновые генераторы выпускаются со следующими
рабочими диапазонами:
Полоса Центр диапазона частот, ГГц К, мм Н апряженность магнитного поля, Гс
S 3 90 1 100
X 9 30 3 300
К 24 12 8 500
Q 35 8 12 500
Е 70 4 25 000
35i
Глава 21
Большинство спектрометров ЭПР предназначено для частоты
порядка 9,5 ГГц (Х-полоса).
Частота клистрона настраивается на резонансную частоту
микроволнового резонатора, в котором находится образец. Мощ-
ность клистронов, используемых в спектрометрах ЭПР, обычно
составляет несколько сотен милливатт. Отвод тепла, выделяемого
клистроном, осуществляется циркулирующей водой.
б) Аттенюатор (параметрический усилитель), предназначен-
ный для ослабления микроволновой мощности, поступающей в ре-
зонатор.
в) Мост (микроволновод), в котором происходит распростра-
нение микроволн. В высокочувствительных спектрометрах исполь-
Рис. 21.12. Схема двойного Т-моста.
зуется резонатор отражения и
двойной Т-образный мост
(рис. 21.12). Мощность клистро-
на делится на два плеча в фор-
ме буквы Т, если смотреть от
клистрона. Когда оба эти плеча
совершенно одинаковы, полови-
на мощности поступает в резона-
тор, а другая ее половина погло-
щается в согласователе. При ре-
зонансном поглощении равнове-
сие нарушается и сигнал отра-
жается от резонатора. Мощность
его раздваивается на два плеча
в форме буквы Т, если смотреть от резонатора. Половина этого
сигнала поглощается ферритовым вентилем, помещаемым в плече
клистрона, тогда как другая половина направляется в детектор.
г) Как правило, применяется микроволновый резонатор отра-
жательного типа, сигнал подается и выходит через одно и то же
отверстие, называемое «ирисовой диафрагмой».
Резонаторы могут быть различных размеров и форм. Наиболее
употребительны: ТЕю2— прямоугольный резонатор, применяю-
щийся при исследовании больших и особенно жидких образцов
(рис. 21.13), ТЕои — цилиндрический резонатор, пригодный для
исследования газообразных систем и жидкостей в капиллярах
(рис. 21.14). Резонаторы классифицируются по конфигурации рас-
пределения СВЧ-поля или по электрической ТЕ/, т, „-моде. Индек-
сы /, т, п представляют собой целые числа, характеризующие кон-
фигурацию поля по числу периодичности в Е (электрическом) и
II (магнитном) полях по осям координат х, у и z.
д) Детектор, предназначенный для измерения микроволновой
мощности. Это кристаллический диод или туннельный диод, ра-
ботающий как микроволновый выпрямитель. После детектирова-
ния сигнал усиливается и поступает в фазочувствительный детек-
тор, а затем после фильтрации регистрируется как сигнал ЭПР.
Спектроскопия электронного парамагнитного резонанса
353
в) Плечо сравнения. Это волновод, который направляет часть
микроволновой мощности от клистрона непосредственно в де-
тектор.
ж) Система автоматической подстройки (контроля) частоты
(АПЧ), которая используется для точной коррекции резонансной
частоты резонатора.
3. Генератор развертки, с помощью которого регулируется на-
пряженность магнитного поля (Но). Выходной сигнал от него
л*
--------------
8
Рис. 21.13. Прямоугольный резонатор (а), действующий в ТЕюг-моде. Контур
электрического поля (б). Контур магнитного поля (в).
синхронизируется со скоростью развертки луча по оси X осцил-
лографа или со скоростью движения каретки самописца. Исполь-
зующиеся электромагнитные катушки называются катушками раз-
вертки и устанавливаются на каждой стороне резонатора.
4. Запись результатов. Осциллограф и самописец синхронизи-
руются с выходным сигналом генератора развертки. С помощью
Рис. 21.14. Цилиндрический резонатор (а), действующий в ТЕон-моде. Контур
электрического поля (б). Контур магнитного поля (в).
небольшого компьютера можно осуществлять хранение данных.
По чувствительности ЭПР-спектрометры намного превосходят
ЯМР-спектрометры; это объясняется тем, что магнитный момент
электрона значительно больше, чем у протона. Из уравнения рас-
пределения Больцмана
n./ni==ie^EikTt (21л1)
где rii и П/ — заселенности верхнего и нижнего спинового уровня
соответственно, ХЕ— энергия, необходимая для расщепления уров-
ней, & —постоянная Больцмана (разд. 40.4), Г —температура
12 Зак. 238
354
Глава
(К), можно видеть, что при использовании метода ЯМР различие
в заселенностях составляет только 7 м. д., тогда как при приме-
нении метода ЭПР-спектроскопии эта разность достигает 1600 м. д.,
в связи с чем чувствительность ЭПР-спектрометров в 230 раз
выше. Энергия, необходимая для расщепления уровней ДЕ, прямо
пропорциональна напряженности магнитного поля, и поэтому уве-
личение чувствительности можно достичь повышением напряжен-
ности поля и резонансных частот. Увеличению чувствительности
также способствует понижение температуры, но поскольку послед-
няя влияет также на молекулярную подвижность, то это вызывает
известные ограничения.
Обзорная литература: 17, 1050, 1088.
21.2.2. Приготовление образцов
С помощью метода ЭПР (ЭСР)-спектроскопии проводят исследо-
вания как газов, так и жидкостей и твердых веществ.
1. Газы. ЭПР-методом изучают газы следующих трех типов:
а) газы с парамагнитными молекулами: О2, NO, NO2, С1О2
и т. п.;
б) реакционноспособные атомы: Н, N, О;
в) свободные радикалы в газовой фазе.
Основной экспериментальной трудностью является создание до-
статочной концентрации свободных радикалов в газе для полу-
чения хорошего отношения сиг-
нал/шум. При измерениях при
давлении 10—100 мм рт. ст.
(1017—1018 молекул/см3) иногда
наблюдается уширение линий.
Спектры с лучшим разрешением
получаются при давлениях 0,1 —
1 мм рт. ст. (1015—1016 моле-
кул/см3). Поэтому необходимо
иметь систему регулирования
газа, с помощью которой можно
подбирать давление, обеспечи-
вающее оптимизацию интенсив-
ности сигналов и разрешения.
С этой целью обычно исполь-
зуются следующие два метода:
создание перепада давлений для удаления избытка молекул и
разбавление инертными диамагнитными газами, например гелием
или аргоном.
2. Жидкости. В жидких средах исследуют следующие ради-
кальные частицы.
а) Радикалы, образующиеся при радиолизе. Такие радикалы
образуются в источнике радиации и затем их пропускают через
Рис. 21.15. Снятие ЭПР-спектров при
облучении образца в резонаторе
ультрафиолетовым светом.
/ — источник излучения; 2— резонатор ЭПР;
<3—кварцевая ампула с образцом; 4 — сосуд
Дьюара из кварца с жидким азотом.
Спектроскопия электронного парамагнитного резонанса
355
резонатор или же радикалы получают непосредственно в резо-
наторе под действием электронного пучка с высокой энергией.
Использование обоих методов получения радикалов сопряжено со
значительными экспериментальными трудностями.
б) Радикалы, образующиеся при фотолизе. Такие радикалы
получаются при прямом облучении образца ультрафиолетовым
Рис. 21.16. Проточная система, поз-
воляющая получать свободные ра-
дикалы в больших концентрациях.
/ — емкость для восстановителя; 2 —емкость
для окислителя; 3 —редукторы и ротаметры;
4— плоская кювета; 5— резонатор ЭПР;
6 — зона смешения; 7 —сборник.
Резонатор ЭПР
смешения
Поток i
Поток 2
Рис. 21.17. Кювета смешения типа
Диксона — Нормана для исследова-
ния ЭПР свободных радикалов, об-
разующихся в протонной системе.
светом в резонаторе, одна стенка которого имеет окно из кварца
(рис. 21.15).
в) Отрицательные и положительные ион-радикалы, образую-
щиеся в растворах.
г) Промежуточные частицы, образующиеся при химических
реакциях. Такие свободные радикалы наблюдаются преимущест-
венно в проточных системах (рис. 21.16), например при изучении
окислительно-восстановительных реакций в процессе радикальной
полимеризации. Для этого используются кюветы специальной кон-
струкции, например кюветы Диксона — Нормана (рис. 21.17). Та-
кая кювета состоит из двух трубочек с плоской камерой сверху.
Внутренняя трубочка имеет шприцевидную головку, через которую
один раствор, например восстановитель, вводится во второй рас-
твор, например окислитель. После их смешения в резонаторе сразу
же образуются свободные радикалы. Скорость пропускания обоих
растворов следует подбирать экспериментальным путем; обычно
12*
356
Глава 21
работают при скоростях порядка 1—10 см3/с. Из-за высоких ди-
электрических констант растворы исследуемых соединений в воде
или серной кислоте дают очень плохие сигналы, в связи с чем
измерения следует проводить в тонких капиллярах.
3. Твердые вещества. Свободные радикалы в твердых вещест-
вах можно получать непосредственно в резонаторе путем радио-
лиза, фотолиза, пиролиза, механического воздействия на образец
и т. п.
21.2.3. Стабилизация свободных радикалов
Из-за очень малого времени жизни многих органических и поли.
мерных свободных радикалов при комнатной температуре их
трудно определять методом ЭПР-спектроскопии. Поэтому важной
„ задачей является стабилизация
Рис. 21.18. Проточная система для
газов, предназначенная для регули-
рования температуры образца.
свободных радикалов и ион-ра-
дикалов. Эта проблема решается
несколькими путями.
1. Охлаждение до низких тем-
ператур, например до температу*
ры жидкого азота (—196feC==
= 77 К) или жидкого гелия
(—268,8 °C = 4,2 К).
Для измерений при темпера-
туре жидкого азота используют
обычный сосуд Дьюара, в кото-
ром после заполнения жидким
азотом помещается капилляр с
образцом. При помещении образ-
ца непосредственно в жидкий
азот часто возникают трудности,
обусловленные тем, что пузырь-
ки газообразного азота дают
сильный шум при записи спект-
1— резувщатор ЭПР; 2—кварцевая ампула
с образцом; 3—трубка с нагревателем;
4—термопара; 5—сосуд Дьюара с жидким
азотом; б—электрическая спираль тепло-
обменника.
ра. Этот эффект можно подавить
пропусканием в жидкий азот га-
зообразного гелия.
Для снятия спектров ЭПР
при различных температурах
(77—300 К) температуру регулируют с помощью специального
устройства, представляющего собой теплообменник (рис. 21.18).
Через медный змеевик, погруженный в жидкий азот, пропускают
сухой азот, который затем проходит через трубчатый нагреватель
и поступает в резонатор. Регулирование температуры осуществ-
ляют варьированием тока нагревателя и скорости пропускания
охлаждающего газа.
Для измерений при температуре жидкого гелия (1,2—4,2 К)
необходим специальный низкотемпературный проточный криостат.
357
—Mint
Спектроскопия электронного парамагнитного резонанса
Работа с жидким гелием создает большие трудности при проведе-
нии таких экспериментов.
2. Стабилизация свободных радикалов в кристаллических орга-
нических матрицах, например спиртах.
3. Захват свободных радикалов с помощью спиновой ловушки.
Эта методика используется для детектирования и идентификации
короткоживущих свободных радикалов путем определения ЭПР-
спектров нитроксильных продуктов присоединения (спиновых ад-
дуктов) свободных радикалов (R-) к нитрозосоединениям (напри-
мер, 2-нитро-2-метилпропану, трет-бутилнитроксиду, нитрозобен-
золу) или нитроносоединениям ( например, фенил-М-трет-бутил-
нитрону), называемым спиновыми ловушками
СН3 0“
। к
----> СН.-С----Nt
CH, R
(2.1.12)
СН3
R* + CH3-C-N=O
СН3
R” +
<21.13)
R’ +
Н О'
Nt (2.1.14)
'—V R CH
H,cx CH,
Обзорная литература: 1088.
Периодически литература: 6192—6195.
21.2.4. Калибровка развертки поля
Для измерения ширины линий и сверхтонких расщеплений необ-
ходима точная калибровка развертки поля. Это легко осуществить,
используя стандартные образцы, для которых положения линий
точно известны. Стандартный образец помещают вместе с неиз-
вестным веществом в двойной резонатор и записывают их спектры
с помощью двухканального самописца.
В качестве стандартных веществ используют ионы Мп2+ в по-
рошке SrO, которые дают спектр, состоящий из шести линий
(рис. 21.19), с шириной 1,6 Гс при 420 Гс с константой сверхтон-
кого расщепления (АЯ) 85 ±0,2 Гс, а также тетрацианэтиленовый
анион-радикал в растворе, который дает девять линий в спектре
с шириной 100 мГс при 13 Гс с константой сверхтонкого расщеп-
ления (АЯ) 1,574 Гс.
Периодическая литература: 5978, 6099, 6322.
358
Глава 21
Рис 21.19. ЭПР-спектр Мп2+ в MgO
при комнатной температуре (у «
« 9,3 ГГц) [О: 1375].
"о.
21.3. ОПРЕДЕЛЕНИЕ АБСОЛЮТНОГО КОЛИЧЕСТВА
НЕСПАРЕННЫХ СПИНОВ
Измерения концентрации спинов проводят путем сравнения инте-
гральных интенсивностей сигнала анализируемого соединения и
стандартного образца с известным числом неспаренных спинов
электронов.
К стандартному образцу предъявляются следующие требова-
ния: а) ширина и форма линии должны быть сходными с шириной
и формой линии неизвестного
вещества; б) число спинов близ-
ко числу спинов неизвестного об-
разца; в) физическая форма и
диэлектрические потери анало-
гичны соответствующим харак-
теристикам неизвестного образ-
ца; г) число спинов, ширина ли-
ний и g-фактор должны сохра-
няться неизменными (стабиль-
ность во времени и по темпера-
туре); д) время спин-решеточной
релаксации (Л) должно быть
коротким, чтобы избежать легко-
го насыщения сигнала.
Поскольку ширина линий и
концентрация спинов меняются
на несколько порядков величины, необходимо готовить ряд стан-
дартных образцов.
Используются различные вторичные стандартные образцы:
а) синтетический кристалл рубина, который можно разместить
постоянно в резонаторе, чем обеспечивается возможность одно-
временного наблюдения стандартного и неизвестного образцов;
б) карбонизованная декстроза, не меняющая своих магнитных
свойств в интервале температур 4—500 К;
в) угольный порошок (пек), содержащий КС1, товарный про-
дукт;
г) ионы Мп2+ в MgO;
д) свежеперекристаллизованный 1,1-дифенил-2-пикрилгидра-
зин (ДФПГ).
Использование двойного резонатора позволяет применять
образцы с близкими характеристиками по заполнению и положе-
нию в радиочастотном поле. Замена одного образца на другой до-
статочно проста. Взаимная перестановка образцов исключает раз-
личия, обусловленные неэквивалентностью двух измерительных
каналов.
Точность измерений концентрации спинов путем относитель-
ного сравнения со вторичным стандартом составляет 5—10%.
Спектроскопия электронного парамагнитного резонанса
359
Обзорная литература: 17, 558, 127Q.
Периодическая литература: 2759, 4133, 6412, 7204, 7242.
21.4.
при идентификации мно-
Рис. 21 20.
тров двух
дартного
Разделение цен-
спектров — стан-
и исследуемого
образцов
1—линия стандартного вещества,
2 — линия исследуемого вещества.
2
ТОЧНОЕ ОПРЕДЕЛЕНИЕ g-ФАКТОРА
Эффективные значения g-фактора обычно можно определить с
помощью полупроводникового кристалла, применяемого для регу-
лирования однородности поля, точность оценки при этом не пре-
вышает сотых, что, однако, недостаточно
гих органических и полимерных свобод-
ных радикалов.
Для того чтобы точность была поряд-
ка десятитысячных или стотысячных для
узких линий, необходимо раздельное из-
мерение частот с помощью счетчика ча-
стот или волнометра. Одновременно про-
водят измерение поля с помощью ЯМР-
измерителя магнитной индукции или по-
лупроводникового кристалла — регуля-
тора поля, откалиброванного указанным
измерителем. Затем рассчитывают g-фак-
тор из уравнения (21.8).
g-Фактор можно также определить
сравнением неизвестного образца со
стандартом. Оба образца помещают в
двойной резонатор и записывают их спектры на двухканальный
самописец. Разделение центров двух спектров дает разность на-
пряженностей магнитных полей (А//) при двух положениях об-
разца (рис. 21.20).
g-Фактор неизвестного образца рассчитывают из следующего
выражения:
ДЯ
g gs gs>>
(21.15)
где gs — g-фактор стандартного образца; АН — расстояние между
центрами двух спектров в гауссах (см. рис. 21.20); HQ — напря-
женность внешнего магнитного поля в гауссах.
В качестве стандартных образцов наиболее часто используют
положительный тетраценовый ион, g — 2,002604 ± 0,000007, а так-
же положительный периленовый ион, g — 2,002583 ± 0,000006.
Периодическая литература: 2086, 4594, 6219, 6321.
21.5. РЕАКЦИИ СВОБОДНЫХ РАДИКАЛОВ
Свободным радикалом называют атом, группу атомов или моле-
кулу, которые имеют один неспаренный электрон, занимающий
верхнюю орбиталь. К Числу свободных радикалов относятся,
360 Глава 21
например, атомарный водород (Н-), гидроксильный радикал
(ОН-), метильный радикал (Н3С-)-
Бирадикал — это частица, содержащая на верхних орбиталях
два неспаренных электрона, например молекулярный кислород
:О2 в основном или возбужденном состоянии.
Ион-радикал — это свободный радикал, имеющий положитель-
ный или отрицательный заряд, например протонированный амин-
ный радикал H3Nt или нафталиновый анион-радикал СюН8~.
Вследствие сильного стремления неспаренных электронов к
взаимодействию с другими электронами с образованием электрон-
ных пар (химических связей) свободные радикалы обычно очень
активны.
Для свободных радикалов типичны цепные реакции, протекаю-
щие в три стадии:
1. Стадия инициирования, представляющая собой процесс обра-
зования радикалов, например, радикальная пара получается при
гомолитическом разрыве двухэлектронной связи.
2. Стадия роста, которая является реакцией переноса свобод-
ных радикалов и связана с изменением места радикального цен-
тра. Существует четыре типа реакций роста:
а) реакции переноса атома, например отрыв водорода свобод-
ным радикалом с образованием нового свободного радикала:
A- + RH —> AH + R- (21.16)
б) реакции присоединения, в процессе которых свободный ра-
дикал присоединяется по двойной связи:
А* + С=С —> А—С—С* (21.17)
в) реакции расщепления, типичным примером которых явля-
ется p-р зацепление, при котором неспаренный электрон молекулы
расщепляет связь в p-положении, при этом образуется новый сво-
бодный радикал и молекула с двойной связью:
RC—С - —> R • + С=С (21.18)
г) перегруппировки, когда меняется положение свободного ра-
дикала в молекуле и образуется свободный радикал нового типа:
R
• I
R—С—СН2—R —> R—С—СН2 (21.19)
R
3. Реакции обрыва, наблюдающиеся во всех системах, где есть
свободные радикалы. Существует два типа реакций обрыва:
а) рекомбинация двух радикалов:
R- + R- —> R—R (21.20)
Спектроскопия электронного парамагнитного резонанса
361
б) диспропорционирование, связанное с переносом водорода:
R* + RCH2—CHR —> R—H + RCH=CHR (21.21)
Все перечисленные типы реакций наблюдаются при свободно-
радикальной полимеризации и деструкции, инициированной сво-
бодными радикалами.
21.6. ПРИМЕНЕНИЕ ЭПР-СПЕКТРОСКОПИИ
В ИССЛЕДОВАНИЯХ ПОЛИМЕРОВ
В основном ЭПР (ЭСР)-спектроскопия в полимерной химии исполь-
зуется для изучения свободных радикалов, образующихся в про-
цессах: полимеризации (радиационная полимеризация и фотопо-
лимеризация, свободнорадикальное инициирование, полимериза-
ция, инициированная окислительно-восстановительными система-
ми, ионная полимеризация, сополимеризация и т. д.); деструкции
полимеров (радиационная и фотодеструкция, табл. 21.2); окисле-
Таблица 21.2
ЭПР-спектроскопия полимеров
Полимер Литература
Ацетат целлюлозы Полибутадиен Полиэтилен П: 4105-4109 П: 2737, 4918, 5212, 7344 О: 987, 1088, 1306: П: 2009, 2177, 2599, 2682, 2801, 2803, 3343, 3539, 3540, 3858, 4321, 4437— 4439,4782, 4843,4918,5437, 5563, 5564, 5568-5570, 5967, 6169, 6330, 6358, 6704, 6848-6852, 6854-6858, 7021
Полиизобутилен Полиизопрен Полипропилен П: 4980 П: 2599, 2736, 4290, 4466, 5564, 7344 П: 2183, 2599, 3387, 3450, 3860, 4918, 5436, 5603, 5963, 5967, 6853, 6858, 7281
Полистирол П: 2009, 3070, 3388, 3399, 4221, 6236, 6657, 6942, 7118
Политетрафторэтилен Поливиниловый спирт Поливинилхлорид П: 2808, 3980, 4888, 6152, 6393, 6819 П: 7163 П: 2856, 3952, 4318, 4842, 4979, 4926, 5265, 5564-5566, 5938, 5965, 6656, 6818
Поливинилиденхлорид П: 5566
ния полимеров; расщепления полимерных молекул при механоде-
струкции. Этот метод применяется также для изучения стабильных
полимерных радикалов и подвижности макромолекул (с помощью
метода спиновой метки).
Обзорная литература: 107, 203, 204, 233, 234, 245, 440—446, 448, 711, 743,
987, 1088, ИЗО, 1306.
Периодическая литература: 4123, 4124, 4265, 5409—5412, 5488, 5834, 5934,
6157, 6203, 6357, 6361, 7287.
362
Глава 21
21.7. МЕТОДЫ СПИНОВОЙ МЕТКИ
И СПИНОВОГО ЗОНДА
В обоих этих методах предполагается использование стабильных
нитроксильных радикалов, например 4-Р-2,2,6,6-тетраметилпипери-
дина
Метод спиновой метки заключается в образовании ковалент-
ной связи между различными нитроксидными радикалами и диа-
магнитными полимерами, которые не имеют неспаренных электро-
нов и не дают спектров ЭПР. Измеряя ширину линий в ЭПР-спек-
трах полимеров, содержащих стабильные нитроксидные радикалы,
можно оценить молекулярные переходы.
Обзорная литература: 665, 666, 821, 1088, ИЗО.
Периодическая литература: 2184, 2628—2636, 3781, 4104, 4799, 4809, 5955,
6007, 6008, 6820, 6821, 6935, 6936, 7009.
При использовании метода спинового зонда различные ни-
троксидные радикалы смешивают с диамагнитным полимером, не
связывая их химически, и изучают поведение смесей в процессах,
связанных с релаксацией и переходами в полимерах Концентрация
нитроксидных радикалов в полимерной матрице при использова-
нии метода спинового зонда составляет 10—100 м. д.
Обзорная литература: 186, 219, 1008, ИЗО.
Периодическая литература: 2545, 2628, 2635, 3792, 4698, 4765, 4769—4781,
4783, 5413, 5942, 6007, 6203, 6820, 6937.
21.8. СПЕКТРОСКОПИЯ ЭЛЕКТРОН-СПИНОВОГО
ДВОЙНОГО РЕЗОНАНСА
21.8.1. Спектроскопия электрон-ядерного двойного резонанса
Спектроскопия электрон-ядерного двойного резонанса представ-
ляет собой регистрацию спектра при насыщении сигнала ЭПР
вследствие индуцирования ядерного магнитного резонанса ядер,
связанных с парамагнитным центром.
В основе электрон-ядерного двойного резонанса лежат следую-
щие процессы:
1. Частичное насыщение ряда определенных электронных пе-
реходов под воздействием микроволнового поля дает ЭПР-сигнал
данной амплитуды.
Спектроскопия электронного парамагнитного резонанса
363
2 При медленном изменении второго ВЧ-поля с ЯМР-часто-
тами происходит изменение в заселенности спиновых электронных
уровней.
3. Под влиянием указанного изменения меняется амплитуда
начального сигнала ЭСР и регистрируется зависимость величины
этого так называемого «различия амплитуд» от ЯМР ВЧ-поля.
Этот эффект обычно называется электрон-ядерным двойным резо-
нансом.
Зависимость амплитуды сигнала ЭСР от ЯМР ВЧ-поля назы-
вается спектром электрон-ядерного двойного резонанса.
Этот метод спектроскопии особенно полезен при недостаточном
разрешении сверхтонких линий в спектре ЭПР.
Обзорная литература: 739, 137£.
Периодическая литература: 4189.
21.8.2. Спектроскопия электрон-электронного двойного
резонанса
Спектроскопия электрон-электронного двойного резонанса пред-
ставляет собой метод, обеспечивающий высокое разрешение сверх-
тонкой структуры и позволяющий определить уменьшение интен-
сивности одного сверхтонкого перехода при одновременном насы-
щении второго сверхтонкого перехода.
Одновременный ЭСР в одном магнитном поле для двух раз-
личных переходов обусловливает необходимость облучения образ-
ца двумя микроволновыми частотами.
Данный метод спектроскопии применяется для изучения про-
цессов спин-спиновой релаксации и полезен также при исследова-
нии молекулярных движений с временем корреляции от 1О~10 до
10-3 с.
Обзорная литература: 660, 739, 1375.
Периодическая литература: 2608, 3214, 3215, 3502, 4190, 4191, 5372, 7232.
Глава 22
МАСС-СПЕКТРОМЕТРИЯ
Обзорная литература: 12, 103, 117—119, 123, 158, 212, 220. 221, 227, 2/4, 424,
4$0, 5б4, 658, 692, 750, 894, 900, 902, 908, 930, 984, 1612, 1055, 1096, 1097, 1119,
1188, 1202, 1213, 1247, 1316, 1354, 1381, 1393, 1395.
При бомбардировке органического соединения электронами
возможно протекание следующих реакций:
1. Образование положительных молекулярных ионов (катион-
радикалов):
М + е“ —> М+- + 2е" (22.1)
Если время жизни молекулярных ионов достаточно для того, чтобы
они смогли достичь детектора (около 10-6 с), то при этом наблю-
дается молекулярный пик. Положение этого пика характеризует
молекулярный вес вещества, которым является ближайшее целое
число.
2. Образование многозарядных ионов:
М + пе- —> Мп+- + (п+1)е- (22.2)
3. Образование положительных осколочных ионов и радика-
лов, возникающих при диссоциации молекулярного иона, которая
начинается через 10“10—10~3 с после бомбардировки:
М+- —> Mt + Ri-
М+- —> М2+ + R2-
(22.3)
* • •
М+. —> MJ + Rn-
4. Образование метастабильных ионов. Такие ионы образуются
в результате распада положительных осколочных ионов в области
между электростатическим и магнитным анализаторами:
—> м; + м°, (22.4)
где М + — метастабильный ион, а М° — нейтральный осколок.
Если ион М+ с массой гп\ распадается с образованием иона с
массой т2 после ускорения до входа в магнитное поле, то он реги-
стрируется в виде диффузного малоинтенсивного пика с кажу-
щейся массой т*, равной
т*«(т2)21т{
(22.5)
Масс-спектрометрия
365
Пик, обусловленный ионным током, который соответствует массе
т*, называется метастабильным пиком.
Оценка массы, отвечающей метастабильному пику, позволяет
установить, что масса m2 получается непосредственно из массы
ni\ при отщеплении нейтрального осколка М°.
5. Образование изотопных ионов (в случае органических сое-
динений, содержащих естественно встречающиеся изотопы, кото-
рые приведены в табл. 22.1). Изотопы обусловливают образование
Таблица 22.1
Изотопный состав некоторых элементов [О: 1213]
Элемент Распространенность
Углерод 12С 100 13С 1,08
Водород ‘Н 100 2Н 0,016
Азот 14N 100 15М 0,38
Кислород 160 100 17О 0,04 180 0,20
Фтор 19р 100
Кремний 28Si 100 29Si 5,10 30Si 3,35
Фосфор 31р 100
Сера 32S 100 33 s 0,78 84S 4,40
Хлор 35Q 100 37С1 32,5
Бром 79Вг 100 81Вг 98,0
Иод 127J 100
ионов с различным отношением m/е, появляющихся как пики
М + 1 и М + 2 (квазимолекулярные ионы).
В специальных таблицах можно найти отношения распростра-
ненности различных изотопов для ряда комбинаций углерода, во-
дорода, азота и кислорода.
6. Образование перегруппировочных ионов, Эти ионы возни-
кают в результате изменения расположения связей между атомами
перед диссоциацией иона.
7. Образование отрицательных ионов по одной из следующих
реакций:
а) диссоциативный резонансный захват
АВ + е“ —> А + В“ (22.6)
б) резонансный захват
АВ + е“ —> АВ- (22.7)
в) образование ионных пар
АВ + е’ —> А+ + В‘ + е~ (22.8)
В обычных условиях работы масс-спектрометров (50—100 эВ)
эффективность образования положительных ионов во много раз
больше, чем отрицательных. Фрагменты положительных ионов
дают масс-спектр.
366
Глава 22
Масс-спектр образца представляет собой кривую эксперимен-
тально определяемых интенсивностей для ряда ионов с различным
отношением массы (т) к заряду (е), т. е. m/е (рис. 22.1).
За интенсивность пика иона принимается расстояние по вер-
тикали от вершины пика до горизонтальной оси (в миллиметрах).
Высота пика пропорциональна числу ионов определенной массы.
Отношение массы к заряду (т/е)— это расстояние от'нулевой
точки, или точки начала сканирования, по горизонтальной оси.
Рис. 22.1. Фрагмент масс-спектра.
Результаты масс-спектрометрии обычно представляются в од-
ной из следующих форм:
1. Ионная интенсивность по отношению к максимальной интен-
сивности в процентах (ИИтах). Для того чтобы найти относитель-
ную интенсивность пика Z-ro иона в спектре, выбирают пик с наи-
большей интенсивностью. Интенсивность этого пика называется
ионной интенсивностью базового пика (рь). Теперь для вычисле-
ния °/оИИтах достаточно разделить высоту пика Z-ro иона (pt) на
рь и умножить на 100
ИИтах = (рг/^)- 100% (22.9)
Полученные данные представляют в виде графика, состоящего из
отдельных линий (рис. 22.2). При таком способе изображения
практически невозможно показать ионы с низкой интенсивностью,
например метастабильные ионы.
Стандартным методом представления полученных данных яв-
ляется табулирование относительных ионных интенсивностей в по-
рядке увеличения т/е.
В масс-спектрометрических каталогах обычно не бывает ука-
занного выше графического представления результатов.
2. Ионная интенсивность по отношению к суммарной интенсив-
ности в процентах (% ИИ2). Для вычисления % ИИ2 высоты всех
пиков складывают, получая суммарную ионную интенсивность
(%Pi). Затем интенсивность пика Z-ro иона делят на эту величину
Масс-спектрометрия
367
и умножают на 100
ИИе = (р;/Ер,) • 100% (22..0)
Полученные данные представляют в виде графика, состоящего
из отдельных линий (рис. 22.2).
Для каждого масс-спектра следует обязательно указывать чув-
ствительность. Обычно чувствительность приводится для наиболее
интенсивного иона или для базового пика и выражается в микро-
амперах ионного тока на микрограмм образца. Чувствительность
сильно зависит от типа исследуемого соединения.
Перед снятием спектра исследуемого образца необходимо оце-
нить фоновые пики, которые могут быть обусловлены следами ве-
*1
£Ю0г-
80-
60-
40-
20-
ol_i IIJiLlkJ Li_i______i___11 i -1j_k—i—i—i—
20 30 40 50 60 70 80 90 100 110 120 130 140 150 160 170 180
L________k.
Af
/ЛУ + 1
д^+г
m/e
Рис. 22.2. Относительная ионная интенсивность.
щества, не удаленного полностью из прибора после предыдущего
эксперимента. Пики, которые наблюдаются в подобном так назы-
ваемом «фоновом» спектре, необходимо вычесть при анализе пиков
вновь исследуемого вещества.
Следующие публикации могут оказаться полезными при интер-
претации масс-спектров органических соединений: [О: 117, 326,
564, 802, 803, 901, 902, 1213, 1261, 1316, 1461, 1463], а также
[П: 4530].
22.1. ОБОРУДОВАНИЕ ДЛЯ МАСС-СПЕКТРОМЕТРИИ
По методу разделения заряженных частиц (фокусировка) масс-
спектрометры можно классифицировать следующим образом:
1. Масс-спектрометры с отклонением под действием магнитного
поля, которые можно подразделить еще на два типа:
а) масс-спектрометры с простой фокусировкой одним только
магнитным полем, разрешение которых колеблется от низкого до
среднего (разд. 22.1.1);
б) масс-спектрометры с двойной фокусировкой сначала элек-
тростатическим, а затем магнитными полями, характеризующиеся
высоким разрешением (разд. 22.1.3).
2. Времяпролетные масс-спектрометры (разд. 22.1.4),
368
Глава 22
3. Квадрупольные масс-спектрометры (разд. 22.1.5).
4. Масс-спектрометры циклотронного резонанса.
5. Двухлучевые масс-спектрометры.
Можно также классифицировать масс-спектрометры по их раз-
решающей способности:
1. Приборы низкого разрешения (200/1 или менее), к которым
относятся масс-спектрометры устаревших конструкций.
2. Приборы среднего разрешения (500/1—5000/1), к которым
относятся современные приборы с простой фокусировкой.
3. Приборы высокого разрешения (1000/1—100 000/1 или даже
выше): к ним относятся большинство масс-спектрометров с двой-
ной фокусировкой конструкций Маттауха — Герцога или Нира —
Джонсона и некоторые другие специальные приборы.
Разрешение прибора (Л) определяется отношением
Я = М/ДМ, (22.11)
где М — более высокое массовое число, отвечающее одному из
двух выбранных пиков; ДМ — разность между двумя массовыми
числами.
Обзорная литература: 117, 231, 564, 658, 750, 809.
22.1.1 . Масс-спектрометры с простой фокусировкой
Масс-спектрометр подобного типа (рис. 22.3) имеет следующие
основные узлы:
1. Система напуска. Она включает:
а) приспособление для введения образца (разд. 22.1.2);
б) микроманометр для определения количества введенного
образца;
в) натекатель для введения образца в ионизационную камеру;
г) вакуумную систему, обеспечивающую разрешение 10-1—
10~3 мм рт. ст.
2. Ионизационная камера и камеры ускорения. Из натекателя
газовый поток поступает в ионизационную камеру, в которой дав-
ление поддерживается на уровне 10-6—10~5 мм рт. ст. и подвер-
гается в ней бомбардировке под углом 90° электронным пучком,
испускаемым горячим катодом. Положительные ионы, образую-
щиеся при взаимодействии с электронным пучком, пропускаются
через первый ускоряющий электрод с помощью слабого электро-
статического поля между выталкивающим и ускоряющим элек-
тродами. Сильным электростатическим полем между первым и
вторым ускоряющими электродами ионы разгоняются до их конеч-
ных скоростей. При прохождении пучка ионов между ускоряющи-
ми электродами достигается его дополнительная фокусировка. Для
получения спектра к трубе анализатора прикладывается магнит-
ное роле или же варьируется разность потенциалов между первым
Масс-спектрометрия
369
и вторым электродами. В результате ионы последовательно фоку-
сируются у коллектора в соответствии с отношением масса/заряд.
В современных приборах сканирование от массы 12 до массы 500
можно проводить за 1—4 мин и даже быстрее.
3. Трубка анализатора и магнит. Трубка анализатора имеет
кривизну 180°, ее изготовляют из металла и откачивают до оста-
точного давления 10~7—10~8 мм рт. ст. Через такую трубку ионный
Рис. 22.3. Масс-спектрометр с простой фокусировкой.
/ — система напуска; 2—ионный источник; 3— выталкивающий электрод; 4 — катод; анод;
3—ускоряющие электроды; 7—фокусирующие электроды; 8— ионы; 9— магнит; 10— нере-
гистрируемые ионы; 11 — анализационная трубка; /2—регистрируемые ионы; /3—коллектор
ионов; 14—щель коллектора; /5—усилитель; /6—цифровой преобразователь; /7—телетайпная
запись и перфоратор; 18—самописец.
пучок проходит на пути от источника ионов до коллектора. Важ-
нейшим требованием для правильной работы масс-спектрометров
является однородность магнитного поля вокруг анализатора.
4. Коллектор ионов и усилитель. Обычно такой коллектор со-
стоит из одной или более ограничивающих щелей и так называе-
мого «фарадеевского цилиндра» (узкий закрытый с одного конца
длинный цилиндр). Ионный пучок, подлежащий измерению, входит
в коллектор вдоль его оси, сигнал усиливается с помощью электро-
метрического усилителя или электронного умножителя.
5. Регистрация. Широко распространенной регистрирующей си-
стемой является шлейфный осциллограф, состоящий из пяти
отдельных гальванометров, которые дают одновременную запись
на фотобумаге. Чувствительность каждого гальванометра умень-
шается сверху вниз в пропорции 1:3:10:30:100. Высоты пиков
от базовой линии рассчитываются относительно следа наиболее
чувствительного гальванометра и умножаются на соответствующий
показатель чувствительности,
370
Глава 22
Современные масс-спектрометры снабжены цифровыми устрой-
ствами, позволяющими переводить сигналы интенсивности пиков
ионов и т/е в цифровую форму, после чего они могут быть вы-
ведены на телетайп, перфоленту (перфокарту) и/или на магнит-
ную ленту.
Обзорная литература: 117, 564, 658, 750, 902, 1213.
22.1.2 . Методы введения образцов, применяемые
в масс-спектрометрии
Для исследования методом масс-спектрометрии газов, жидкостей
и твердых веществ могут быть использованы обычные приемы.
Выбор той или иной методики зависит от типа системы напуска,
используемой в данном масс-спектрометре, механизма введения
образцов и физико-химических характеристик разделения. Обычно
в каждой лаборатории существуют свои приемы для введения
образцов и соответствующие приспособления.
1. Введение газов. Промышленность выпускает газовые шпри-
цы различных типов.
2. Введение жидких образцов. Благодаря большому разнообра-
зию микролитровых (~2 мкл) шприцев для жидкостей сущест-
вует много различных способов введения таких образцов. Образец
засасывают в иглу, вводят через пробку из самозатягивающейся
резины в систему ввода, после чего шприц удаляют. С помощью
шприцев вводят постоянные объемы или их калибруют для введе-
ния известных аликвотных количеств образца.
, 3. Введение твердых веществ. Образцы весом примерно 10 мкг
вводят с помощью капилляров, стеклянных чашечек или металли-
ческих трубочек. Количество вещества, подвергающегося исследо-
ванию, в значительной степени зависит от его летучести.
Обзорная литература: 117, 123, 564, 750.
Периодическая литература: 3075, 4048, 5259, 6403, 7241.
22.1.3 . Масс-спектрометры высокого разрешения
В современных масс-спектрометрах электростатическое поле пред-
шествует электромагнитному. На положительный ион, находя-
щийся в электростатическом поле, действует сила в направлении
данного поля. Траектория иона в поле при этом искривляется.
В радиальном электростатическом поле, всегда перпендикулярном
направлению пролета ионов, радиус кривизны траектории ионов
зависит от энергии иона и напряженности электростатического
поля. Электростатическое поле представляет собой анализатор
энергии вместо анализатора массы и способствует ограничению
рассеяния ионного пучка, перед тем как последний войдет в маг-
нитное поле. Рассеяние энергии является одним из основных фак-
Масс-спектрометрия
371
торов, которые обусловливают уменьшение разрешения в масс-
спектрометре с простой фокусировкой.
Существует два типа масс-спектрометров с двойной фокуси-
ровкой:
1. Масс-спектрометр с двойной фокусировкой Маттауха — Гер-
цога (рис. 22.4). В этом приборе угол электростатического поля
Рис. 22.4. Масс-спектрометр с двойной
фокусировкой Маттауха — Герцога.
/ — система напуска; 2—ионный источник;
3 —электростатическое поле; 4 — электроды
развертки; 5 —детектор ионов; 6— фотографи-
ческая пластинка; 7 — магнитное поле анали-
затора.
Рис. 22.5. Масс-спектрометр с двой-
ной фокусировкой Нира — Джонсона.
/ — система напуска; 2 —ионный источник;
3— электростатическое поле; 4 — магнитное
поле; 5 —детектор ионов.
равен 31° 51', а угол отклонения в магнитном поле 90°. После элек-
тростатического поля энергия многомассового ионного пучка ста-
новится однородной, в магнитном же поле создается заданное
распределение по массам, при этом ионный пучок с определенной
массой фокусируется в отдельной точке.
2. Масс-спектрометр с двойной фокусировкой Нира — Джон-
сона (рис. 22.5). В приборе этого типа углы отклонения как элек-
тростатического, так и магнитного полей составляют 90° и все
ионы фокусируются в одной и той же точке детектора.
Обзорная литература: 407, 564, 685, 750, 880, 881.
Периодическая литература: 2387—2389, 5135.
22.1.4 . Времяпролетные масс-спектрометры
В масс-спектрометре высокого разрешения, известном под назва-
нием времяпролетного масс-спектрометра, магнитное поле отсут-
ствует, основным элементом является трубка, в которой происхо-
дит дрейф ионов (рис. 22.6). После ускоряющего поля ионы имеют
одинаковую кинетическую энергию, но разные скорости из-за раз-
личия в массах. Они движутся по линейной траектории в трубке,
372
Глава 22
в которой отсутствует поле. Время, за которое различные ионы
проходят определенное расстояние, неодинаково. Измерение вре-
мени пролета в микросекундах положено в основу определения
значений т/е для различных ионов. На пути ионного пучка ста-
вится регулирующая сетка, которая делит поток ионов на импуль-
сы длительностью 0,25 мкс с частотой 10 000 с-1. В случае если бы
ионы попадали в трубку непрерывно, не было бы возможности
Вакуум
Рис. 22.6. Времяпролетный масс-спектрометр.
[—система напуска; 2—ионный источник; 3—выталкивающий электрод; 4—катод; 5—анод?
F—фокусирующие электроды; 7—ускоряющие электроды; 8—ионы; 9—трубка дрейфа иОнов;
10—коллектор ионов; // — усилитель; 12—осциллограф.
измерить время пролета какого-то определенного иона. В последо-
вательном импульсном режиме должны также работать ускоряю-
щая сетка и детектор. Для регистрации спектра обычно исполь-
зуется осциллограф.
Периодическая литература: 5996.
22.1.5 . Квадрупольные масс-спектрометры
В масс-спектрометре высокого разрешения используются четыре
электростатических поля (квадруполь) (рис. 22.7), тогда как маг-
Рис. 22.7. Квадрупольная электродная структура (а), дающая эквипотенциаль-
ные линии (б).
ни гное поле отсутствует. Такие масс-спектрометры известны под
названием квадрупольных масс-спектрометров (рис. 22.8). Ионы,
Масс-спектрометрия
373
поступающие из ионного источника, движутся с постоянной ско-
ростью в направлении, параллельном полям (z-направление),
осциллируя в направлениях хну. Это достигается приложением
к полюсам напряжения постоянного тока и радиочастотного поля.
Устойчиво осциллируя, ион проходит от одного конца квадруполя
Рис. 22.8. Квадрупольный масс-спектрометр.
/ — система напуска; 2—ионный источник; 3 —выталкивающий электрод; 4 — катод; 5 —анод;
6 — фокусирующие электроды; 7 — ускоряющие электроды; 8 — волновод квадрупольного пучка;
9— генератор постоянного тока и радиочастотного поля; 10— анализирующий квадруполь;
// — апертура; 12 — электронный умнаЖИХОЛь; /3 —усилитель; /4 —осциллограф; 15— много-
канальный самописец.
к другому, не сталкиваясь с полюсами. Такая осцилляция зависит
от m/е иона. Вследствие этого лишь ионы с определенным отноше-
нием т/е преодолевают всю длину анализатора, тогда как осталь-
ные ионы с неустойчивыми колебаниями сталкиваются с полюсами.
Сканирование по массам проводится путем варьирования напря-
жения постоянного тока и радиочастотного поля при постоянном
их отношении. Разрешающая способность недавно разработанных
приборов достигает 8000.
Обзорная литература: 361, 564, 750, 1213.
Периодическая литература: 2603, 3392, 5728, 6437, 6967.
22.2. МАСС-СПЕКТРОМЕТРИЯ, СОВМЕЩЕННАЯ
С ГАЗОВОЙ ХРОМАТОГРАФИЕЙ
Ряд современных приборов включает газовый хроматограф, совме-
щенный с масс-спектрометром через промежуточную зону, в кото-
рой повышается концентрация образца в газе-носителе (рис. 22.9).
Времена сканирования вполне обеспечивают получение несколь-
ких масс-спектров при выходе одного пика в газовом хромато-
графе.
374
Г лава 22
Рис. 22.9. Газовый хроматограф, совмещенный с масс-спектрометром.
1 — газовый хроматограф; 2 —промежуточная зона; 5 —масс-спектрометр.
Обзорная литература: 547, 564, 895, 1202.
Периодическая литература: 3780, 4602, 5239, 5333, 6705.
22.3. МАСС-СПЕКТРОСКОПИЯ
С ЭЛЕКТРОГИДРОДИНАМИЧЕСКОЙ
ИОНИЗАЦИЕЙ
Масс-спектроскопия с электрогидродинамической ионизацией *
представляет собой метод, в основу которого положено распыле-
ние разбавленных растворов полимеров с помощью шприца в элек-
трическом поле при напряжении ~ 10 кВ в атмосфере азота при
атмосферном давлении. При прохождении газовой смеси через
систему сопло — сепаратор образуется молекулярный пучок смеси
ионов и нейтральных молекул, имеющий сверхзвуковую скорость,
энергия измеряется с помощью коллектора ионов, включающего
выталкивающий электрод и кювету Фарадея. Оценка скорости
пучка (~500—1000 м/с) позволяет произвести расчет отношения
M/z для макроионов (М — масса иона и z— число элементарных
зарядов).
Обзорная литература: 424.
Периодическая литература: 3185, 5011, 6762.
22.4. ПРИМЕНЕНИЕ МАСС-СПЕКТРОМЕТРИИ
ДЛЯ ХАРАКТЕРИСТИКИ И АНАЛИЗА
ПОЛИМЕРОВ
Масс-спектрометрия в полимерной химии используется для изуче-
ния стереорегулярности полимеров путем термического разложе-
ния макромолекул и анализа выделяющихся газов; исследования
термической деструкции полимеров (пиролиза) путем анализа вы-
деляющихся газов (разд. 34.14); определения констант газопро-
ницаемости.
Обзорная литература: 658, 1356.
Периодическая литература: 2604, 2607, 2668, 3251, 3633, 3873, 3898, 4076,
4179—4181, 4267, 4275, 4276, 4723, 4853, 4882, 4982, 4983, 5099, 5333, 5518, 5924,
5996, 6707, 6136, 6263, 7085, 7086.
* Этот метод называется также масс-спектрометрией полевого испарения ио-
нов из раствора. — Прим, перев.
СОДЕРЖАНИЕ
Предисловие к русскому изданию........................................5
Предисловие автора . .................................................. 8
Глава 1. Краткие сведения о полимерных структурах.......................8
1.1. Общие определения............................................11
1.2. Стереохимия макромолекул.....................................13
1.2.1. Структурная изомерия...................................13
1.2.2. Пространственная изомерия..............................14
1.2.3. цис-транс-Изомерия ....................................14
1.2.4. Тетраэдрическая стереоизомерия.........................14
1.2.5. Статистика последовательностей.........................20
1.3. Сополимеры ...................................................20
1.3.1. Типы сополимеров.......................................20
1.3.2. Привитые сополимеры ...................................22
1.3.3. Блок-сополимеры .......................................23
1.3.4. Распределение последовательностей и тактичность в сополи-
мерах ..................................................... 24
1.3.5. Определение композиционной неоднородности сополимеров 25
1.3.6. Исследование распределения последовательностей и тактич-
ности ................................................... .... 25
1.3.7. Циклизация внутри последовательностей и между ними . . 26
1.4. Конформация макромолекул ....................................26
1.4.1. Внутренние углы вращения в полимерах...................26
1.4.2. Номенклатура спиральных конформаций....................29
1.5. Разветвленные полимеры.......................................30
1.6. Сшитые полимеры..............................................31
1.7. Смеси полимеров............................................. 33
1.7.1. Определение совместимости полисмесей...................33
Глава 2. Изучение взаимодействия полимеров с растворителями ..... 35
2.1. Изменение объема при смешении................................35
2.2. Концепция свободного объема в растворах......................38
2.3. Кривые осаждения и тета-температура..........................39
2.3.1. Способы определения тета-температур ...................43
2.4. Верхняя и нижняя критические температуры растворения ... 44
2.5. Растворимость полимеров в смешанных растворителях............46
2.6. Несовместимость растворов полимеров.........................46
2.7. Термодинамический подход к растворимости.....................47
2.8. Трехмерный анализ растворимости..............................50
2.9. Параметр взаимодействия Флори — Хаггинса ........ 52
2.10. Второй вириальный коэффициент................................55
2.11. Невозмущенные размеры линейных цепных макромолекул ... 56
2.12. Невозмущенные размеры разветвленных макромолекул .... 59
2.13. Измерение невозмущенных размеров цепей по вязкости разбавлен-
ных растворов полимеров.................................. . . , . 60
376
Содержание
2.14. Образование жидкокристаллических растворов................... 63
2.15. Набухание сшитых полимеров в растворителях....................65
Глава 3. Средние молекулярные веса .....................................68
3.1. Выражение молекулярновесового распределения: графические ме-
тоды ..............................................................69
3.2. Функции распределения ........................................71
3.3. Показатели молекулярновесового распределения..................71
Глава 4. Фракционирование полимеров...................................73
4.1. Фракционирование по растворимости...........................74
4.1.1. Фракционное осаждение................................74
4.1.2. Метод добавления нерастворителя.......................74
4.1.3. Метод испарения растворителя..........................76
4.1.4. Метод понижения температуры...........................76
4.1.5. Метод изменения давления..............................77
4.2. Турбидиметрическое титрование...............................77
4.3. Кумулятивное фракционирование...............................79
4.4. Фракционное растворение.....................................79
4.5. Фракционирование распределением между несмешивающимися
растворителями .................................................81
4.6. Фракционная кристаллизация..................................82
4.7. Фракционирование с помощью хроматографии....................82
4.7.1. Адсорбционная хроматография...........................82
4.7.2. Осадительная хроматография............................83
4.7.3. Гель-проникающая хроматография........................84
4.7.4. Фазораспределительная хроматография ........ 84
4.8. Фракционирование седиментацйей..............................85
4.9. Фракционирование диффузией..................................85
4.10. Фракционирование путем ультрафильтрации через пористые мем-
браны ...........................................................86
4.11. Фракционирование зонной плавкой........................... 87
4.12. Фракционирование течением под влиянием поля.................88
Глава 5. Мембранная осмометрия..........................................90
5.1. Определение осмометрическим методом среднечислового молекуляр-
ного веса.......................................................90
5.2. Определение осмотическим методом второго вириального коэффи-
циента .........................................................92
5.3. Осмотическое давление высококонцентрированных полимерных
растворов ......................................................92
5.4. Мембранные осмометры......................................93
5.4.1. Мембраны............................................97
5.4.2. Экспериментальные проблемы в мембранной осмометрии 97
Глава 6. Методы исследования, связанные с оценкой коллигативных свойств 98
6.1. Эбулиометрия...................................................98
6.1.1. Определение с помощью эбулиометрических измерений вто-
рого вириального коэффициента . . ................99
6.1.2. Эбулиометр ....... ..... ..... 100
Содержаний
$77
6.2. Криоскопия .................................................101
6.2.1. Определение с помощью криоскопических измерений второго
вириального коэффициента......................................ЮЗ
6.2.2. Криоскопические ячейки..................................ЮЗ
6.3. Изотермическая перегонка (изопиестический метод)............104
6.3.1. Прибор для изопиестической перегонки...................104
6.4. Парофазная осмометрия.......................................105
6.4.1. Парофазные осмометры...................................106
Глава 7. Анализ концевых групп .... *.................................108
Глава 8. Ультрацентрифугирование . . . .............................ПО1
8.1. Скоростная седиментация.......................................ПО
8.1.1. Определение формы частиц методом скоростной седиментации 113
8.1.2. Определение молекулярных весов полимеров методом ско-
ростной седиментации...........................................ИЗ
8.1.3. Определение молекулярновесового распределения (МБР) ме-
тодом скоростной седиментации.................................114
8.1.4. Определение коэффициента седиментации.................114
8.1.5. Определение коэффициента диффузии......................117
8.2. Анализ седиментационно-диффузного равновесия.................119
8.2.1. Определение средневесового молекулярного веса методом се-
диментационно-диффузного равновесия...........................120
8.2.2. Определение /-среднего молекулярного веса методом седи-
ментационно-диффузного равновесия.............................121
8.2.3. Определение молекулярновесового распределения методом
седиментационно-диффузного равновесия .......................122
8.3. Анализ при приближении к седиментационному равновесию . . . 122
8.3.1. Определение средневесового молекулярного веса методом
Арчибальда....................................................122
8.4. Седиментационное равновесие в градиенте плотности............123
8.4.1. Определение средневесового молекулярного веса анализом в
градиенте плотности.....................................124
8.4.2. Определение химической неоднородности макромолекул ана-
лизом в градиенте плотности............................125
8.5. Аналитические ультрацентрифуги...............................125
8.6. Препаративное ультрацентрифугирование.......................129
Глава 9. Вискозиметрические методы.....................................130
9.1. Измерения вязкости для разбавленных растворов полимеров . . 133
9.1.1. Метод определения по одной точке.......................134
9.1.2. Средневязкостный молекулярный вес......................135
9.1.3. Измерение вязкости для разбавленных растворов сополимеров 137
9.1.4. Измерение вязкости разбавленных растворов разветвленных
полимеров.....................................................137
9.1.5. Капиллярные вискозиметры для измерения вязкости разбав-
ленных растворов............................................ 138
9.1.6. Измерение относительной вязкости........................139
9.2. Измерение вязкости неразбавленных полимеров..................142
9.2.1. Капиллярная вискозиметрия для измерения вязкости рас-
плавов полимеров..............................................142
9.2.2. Пуансонно-щелевой вискозиметр..........................144
9.2.3. Ротационный вискозиметр с концентрическими цилиндрами 144
9.2.4. Вискозиметр с плоскостью и конусом в качестве измеритель-
ных поверхностей..............................................145
378
Содержание
Глава 10. Оптические методы .......................................... 147
10.1. Свойства электромагнитного излучения........................147
10.2. Спектроскопические методы в исследовании полимеров .... 149
10.2.1. Абсорбционная спектроскопия...................... ... 151
10.3. Неспектроскопическиё оптические методы исследования полимеров 152
10.3.1. Поляризованное излучение ............................153
10.4. Оптические материалы........................................158
10.5. Источники света.............................................162
10.5.1. Дейтериевые источники света .........................163
10.5 2. Лампы на инертном газе............................ 163
10.5.3. Ртутные дуговые лампы................................163
10.6. Лазеры .....................................................165
10.6.1. Гелиево-неоновый лазер...............................168
10.6.2. Аргоновый ионный лазер...............................170
10.6.3. Рубиновый лазер......................................170
10.6.4. Лазеры на органических красителях....................172
10.7. Оптические детекторы .......................................175
10.7.1. Фотоэмиссионные детекторы............................175
10.7.2. Фотопроводящие и фотоэлектрические детекторы .... 178
10.7.3. Тепловые детекторы...................................178
10.7.4. Фотопленка ..........................................178
Глава 11. Показатель преломления ....................................181
11.1. Определение инкремента показателя преломления.............184
11.2. Автоматический дифференциальный рефрактометр ...... 185
11.3. Определение удельного объема растворенного вещества из по-
казателя преломления раствора...................................186
Глава 12. Оптическая активность в макромолекулярных системах .... 188
12.1. Дисперсия оптического вращения .....................188
12.2. Круговой дихроизм.........................................189
12.3. Эффект Коттона............................................190
12.4. Спектрополяриметры .......................................191
12.5. Использование методов дисперсии оптического вращения и кру-
гового дихроизма в исследовании полимеров.................195
Глава 13. Светорассеяние................................................198
13.1. Широкоугловое рассеяние света ...............................198
13.1.1. Рассеяние маленькими частицами в растворе.............198
13.1.2. Определение молекулярного веса маленьких частиц мето-
дом светорассеяния .......................................... 199
13.1.3. Рассеяние растворами макромолекул.....................200
13.1.4. Определение средневесового молекулярного веса методом
светорассеяния ...............................................201
13.1.5. Метод асимметрии......................................202
13.1.6. Метод Зимма..........................................202
13.1.7. Определение второго вириального коэффициента методом
светорассеяния ..................................... . , . . 205
Содержание
379
13.1.8. Определение среднеквадратичного расстояния между кон-
цами полимерной цепи методом светорассеяния .... 206
13.1.9. Приборы для измерения светорассеяния.................206
13.1.10. Приготовление образцов для измерения рассеяния света 209
13.1.11. Применение светорассеяния в исследовании полимеров 210
13.2. Импульсно-индуцируемое критическое рассеяние . .............210
13.3. Малоугловое лазерное светорассеяние.........................211
13.4. Спектроскопия биения света..................................212
13.5. Спектроскопия Рэлея — Бриллюэна.............................214
13.5.1. Интерферометры Фабри —Перо . .........................216
Глава 14. Ультрафиолетовая и видимая спектроскопия....................219
14.1. Количественный анализ в ультрафиолетовой и видимой спектро-
скопии ...........................................................221
14.2. Приборы для электронной спектроскопии......................224
14.2.1. Двухлучевой спектрометр..............................224
14.2.2. Кюветы для образцов и кюветы сравнения..............225
14.2.3. Кюветы высокого давления для оптических исследований 226
14.3. Растворители для ультрафиолетовой спектроскопии............228
Глава 15. Инфракрасная спектроскопия ................................ 229
15.1. Приборы для инфракрасной спектроскопии......................230
15.1.1. Двухлучевой спектрометр...............................230
15.1.2. Оптические материалы для ИК-спектроскопии.............232
15.1.3. Обращение с оптическими материалами, использующимися
в ИК-спектроскопии, и их хранение............................233
15.2. Методика приготовления образцов.............................233
15.2.1. Твердые полимеры......................................233
15.2.2. Изотропные пленки.....................................236
15.2.3. Ориентированные пленки................................238
15.2.4. Вырезание полимерных образцов....................... 238
15.2.5. Удаление из спектров пленочных образцов интерференцион-
ных полос . .................................................239
15.2.6. Уменьшение рассеяния от поверхности образцов .... 239
15.2.7. Приготовление образцов полимерных гелей для спектро-
скопических исследований ....................................240
15.2.8. Приготовление волокон для спектроскопических исследо-
ваний ...................................................... 240
15.2.9. Микроспектроскопия отдельных волокон.................240
15.2.10. Спектроскопия пучков волокон..........................241
15.2.11. Жидкие полимеры.......................................241
15.2.12. Растворы .............................................241
15.2.13. Многопроходные кюветы.................................244
15.3. Качественные исследования полимеров с помощью инфракрасной
спектроскопии .....................................................245
15.4. Количественный анализ в ИК-спектроскопии....................249
15.5. Определение степени кристалличности полимеров методом инфра-
красной спектроскопии ............................................ 261
15.6. Инфракрасная спектроскопия поверхности полимерных кристаллов 252
15.7. Измерение микрозеркального отражения........................252
15.8. Спектроскопия внутреннего отражения.........................253
15.9. Инфракрасная отражательно-абсорбционная спектроскопия . . 256
15.10. Инфракрасная фурье-спектроскопия............................257
15.11. Ближняя инфракрасная спектроскопия..........................260
15.12. Дальняя инфракрасная спектроскопия..........................262
380
Содержание
Глава 16. Эмиссионная спектроскопия..................................263
16.1. Эксимеры и эксиплексы.....................................265
16.2. Флуоресценция.............................................267
16.3. Оборудование для флуоресцентной спектроскопии.............270
16.3.1. Флуоресцентные спектрофотометры.....................270
16.3.2. Методы измерения длительности флуоресценции .... 271
16.3.3. Метод счета отдельных фотонов.......................271
16.3.4. Осциллографический импульсный метод.................272
16.3.5. Метод, связанный со сдвигом фаз.....................273
16.4. Поляризованная флуоресценция..............................275
16.5. Изучение молекулярной подвижности флуоресцентным методом 276
16.6. Фосфоресценция ...........................................277
16.7. Оборудование для фосфоресцентной спектроскопии............278
16.7.1. Фосфоресцентные спектрофотометры ...................278
16.7.2. Измерение длительности фосфоресценции...............280
16.8. Импульсная кинетическая спектроскопия.....................281
16.9. Наносекундная импульсная спектроскопия....................284
16.10. Хемилюминесценция и термолюминесценция....................285
16.11. Применение эмиссионной спектроскопии для исследования по-
лимеров .........................................................286
Глава 17. Спектроскопия комбинационного рассеяния....................287
17.1. Оборудование для спектроскопии КР.........................289
17.1.1. Спектрометры КР.....................................289
17.1.2. Устройства для образцов.............................291
17.2. Поляризация комбинационного рассеяния света...............293
17.3. Применение спектроскопии КР при исследовании полимеров . . 293
17.4. Сравнение ИК-спектроскопии и спектроскопии КР.............295
17.5. Внутрицепные колебания в полимерах........................297
Глава 18. Анализ нейтронного рассеяния................................299
18.1. Свойства нейтронов.........................................299
18.2. Приборы для анализа нейтронного рассеяния..................300
18.2.1. Источники нейтронов..................................300
18.2.2. Спектрометры рассеяния нейтронов.....................300
18.2.3. Детекторы нейтронов..................................302
18.2.4. Приготовление образцов ..............................302
18.3. Применение анализа нейтронного рассеяния для изучения струк-
туры полимеров...................................................303
Глава 19. Анализ аннигиляции позитронов..............................304
19.1. Свойства позитронов.......................................304
19.2. Экспериментальные методы .................................304
19.3. Оборудование для анализа аннигиляции позитронов...........305
19.3.1. Источники позитронов................................305
19.3.2. Гамма-сцинтилляционные счетчики................. 305
19.3.3. Система измерения времени жизни позитрона . . . 306
19.3.4. ПригбтовЛёниё образцов ........................ 306
Содержание
381
19.4, Применение анализа аннигиляции позитронов для исследования
структуры полимеров................................................306
Глава 20. Спектроскопия ядерного магнитного резонанса ........ 308
20.1. Интерпретация спектров ЯМР............................ . . . 313
20.1.1. Положение линии в спектре ЯМР......................313
20.1.2. Интенсивность линий в спектрах ЯМР.................316
20.1.3. Определение молекулярного веса с помощью ЯМР-спектро-
скопии ....................................................316
20.1.4. Расщепление линий в спектре ЯМР....................317
20.2. Аппаратура для ЯМР-спектроскопии..........................321
20.2.1. ЯМР-Спектрометры ..................................321
20.2.2. Накопитель ........................................323
20.2.3. Ампулы для образцов, применяемые в ЯМР-спектроскопии 323
20.3. Растворители, используемые в ЯМР-спектроскопии............324
20.4. Дейтерирование в ЯМР-спектроскопии........................326
20.5. Реагенты, вызывающие контактный сдвиг.....................326
20.6. Метод спиновой развязки...................................327
20.6.1. Двойной резонанс...................................327
20.6.2. Спин-тиклинг..................................... 328
20.6.3. Ядерный эффект Оверхаузера.........................328
20.6.4. Межъядерный двойной резонанс.......................329
20.7. 13С-ЯМР-Спектроскопия ....................................329
20.7.1. Применение 13С-ЯМР-спектроскопии в исследовании поли-
меров ................................................. ... 330
20.8. ЯМР-Спектроскопия с фурье-преобразованием (ФП)............330
20.9. Ядерные магнитные дипольные взаимодействия................332
20.10. Второй момент линии спектра ЯМР..........................335
20.11. Применение ЯМР-спектроскопии широких линий...............337
20.12. Применение ЯМР-спектроскопии высокого разрешения .... 339
20.13. Оценка степени кристалличности полимеров методом ЯМР-спек-
троскопии .................................................... 339
Глава 21. Спектроскопия электронного парамагнитного резонанса .... 340
21.1. Интерпретация ЭПР (или ЭСР)-спектров........................343
21.1.1. Форма линии ЭПР.......................................344
21.1.2. Интенсивность линий в спектре ^ПР.....................346
21.1.3. Положение линий в спектре ЭПР.........................347
21.1.4. Расщепление линий в спектре ЭПР.......................348
21.2. Оборудование для ЭПР (ЭСР)-спектроскопии................... . 350
21.2.1. ЭПР (ЭСР)-Спектрометры ...............................350
21.2.2. Приготовление образцов . . » . .......................354
21.2.3. Стабилизация свободных радикалов......................356
21.2.4. Калибровка развертки поля.............................357
21.3. Определение абсолютного количества неспаренных спинов . . . 358
21.4. Точное определение g-фактора................................359
21.5. Реакции свободных радикалов.................................359
21.6. Применение ЭПР-спектроскопии в исследованиях полимеров 361
21.7. Методы спиновой метки и спинового зонда.....................362
21.8. Спектроскопия электрон-спинового двойного резонанса .... 362
21.8.1. Спектроскопия электрон-ядерного двойного резонанса . . 362
21.8.2. Спектроскопия электрон-электронного двойного резонанса 363
382 Содержание
Глава 22. Масс-спектрометрия .......................................... 365
22.1. Оборудование для масс-спектрометрии..........................367
22.1.1. Масс-спектрометры с простой фокусировкой..............368
22.1.2. Методы введения образцов, применяемые в масс-спектро-
метрии .......................................................370
22.1.3. Масс-спектрометры высокого разрешения.................370
22.1.4. Времяпролетные масс-спектрометры......................371
22.1.5. Квадрупольные масс-спектрометры ......................372
22.2. Масс-спектрометрия, совмещенная с газовой хроматографией . . 373
22.3. Масс-спектроскопия с электрогидродинамической ионизацией 374
22.4. Применение масс-спектрометрии для характеристики и анализа
полимеров..........................................................374
Уважаемый читатель!
Ваши замечания о содержании книги, ее оформ-
лении, качестве перевода и другие просим присылать
по адресу: 129820, Москва, И-110, ГСП, 1-й Риж-
ский пер., д. 2.
я. Рабек
ЭКСПЕРИМЕНТАЛЬНЫЕ МЕТОДЫ
В ХИМИИ ПОЛИМЕРОВ.
Ч. I
Научный редактор И. Н. Лаврова
Мл. научный редактор Н. Н. Устякова
Художник В. Г. Сорокин
Художественный редактор М. Н. Кузьмина
Технический редактор Г. Б. Алюлина
Корректор В. И. Киселева
ИБ № 3167
Сдано в набор 08.06.82. Подписано к печати 10.01.83.
Формат 60Х90’/1в. Бумага типографская № 2.
Гарнитура латинская. Печать высокая. Объем
12 бум. л. Усл. печ. л. 24. Усл. кр.-отт. 24. Уч.-изд.
л. 22,42. Изд. № 3/2034. Тираж 4200 экз. Зак. 238.
Цена 3 р. 60 к.
ИЗДАТЕЛЬСТВО «МИР»
129820, Москва. И-110, ГСП
I-й Рижский пер., 2.
Ленинградская типография № 2 головное пред-
приятие ордена Трудового Красного Знамени
Ленинградского объединения «Техническая книга»
им. Евгении Соколовой Союзполиграфпрома при
Государственном комитете СССР по делам
издательств, полиграфии и книжной торговли.
198052, г. Ленинград, Л-52, Измайловский про-
спект, 29.