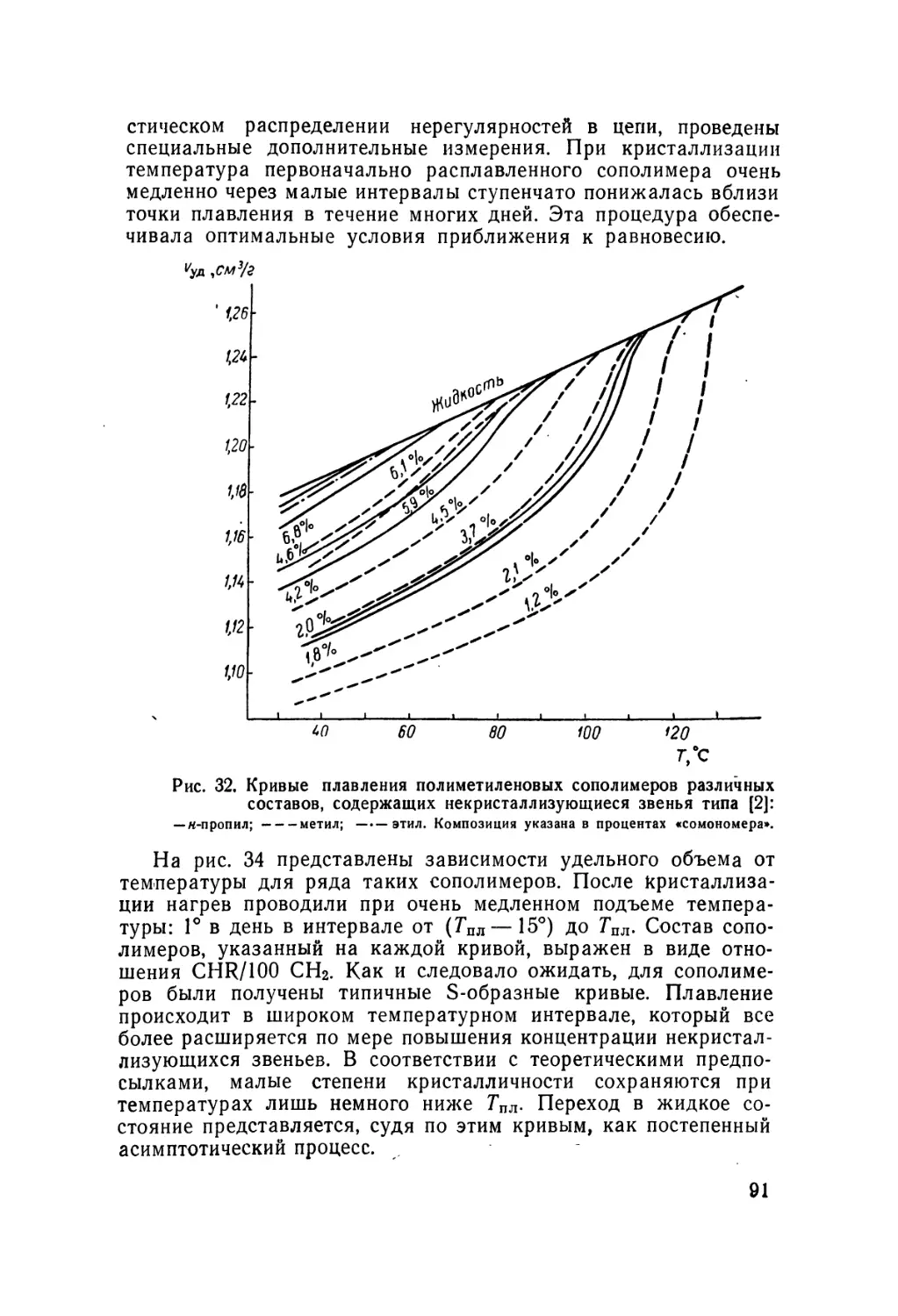
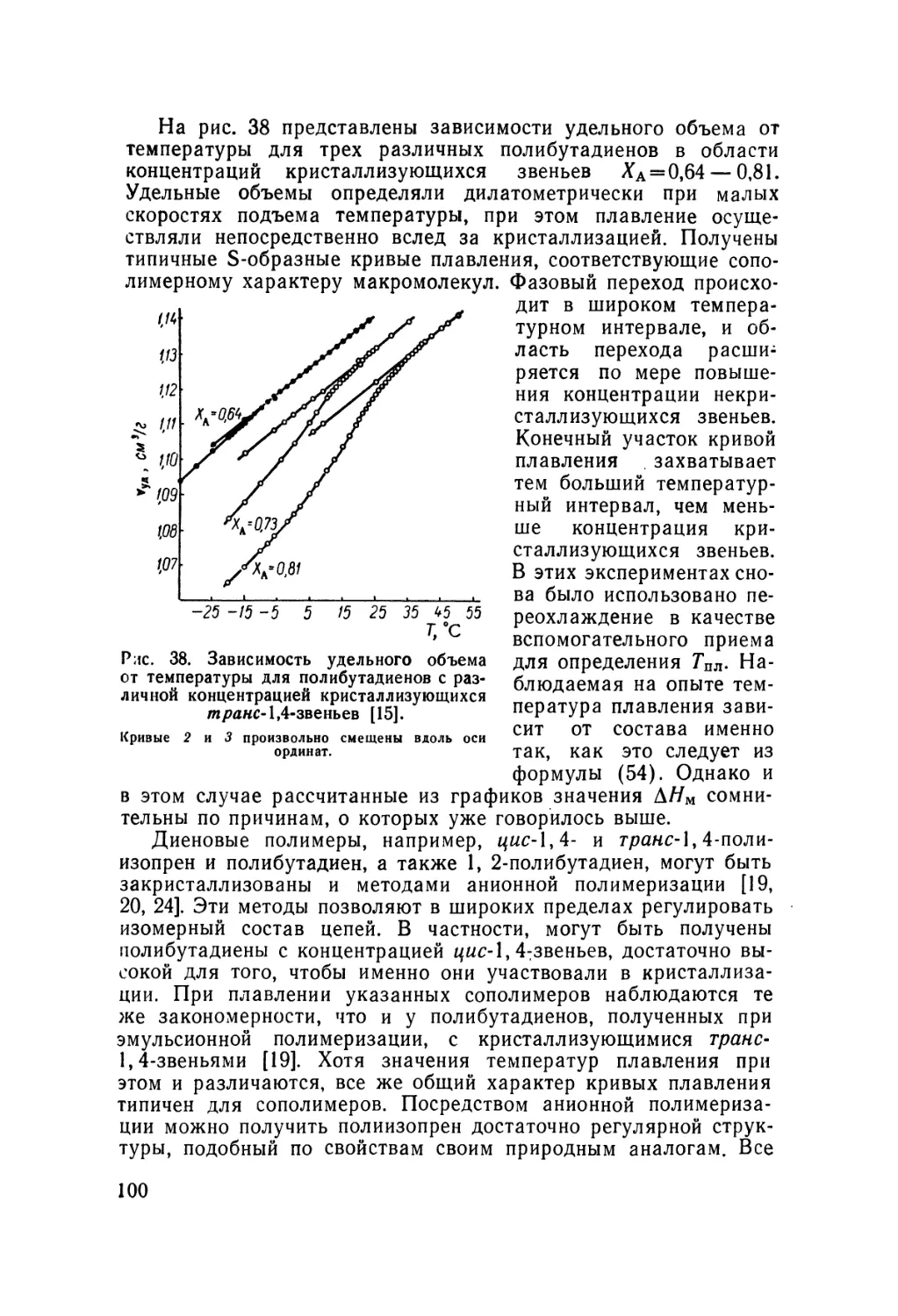
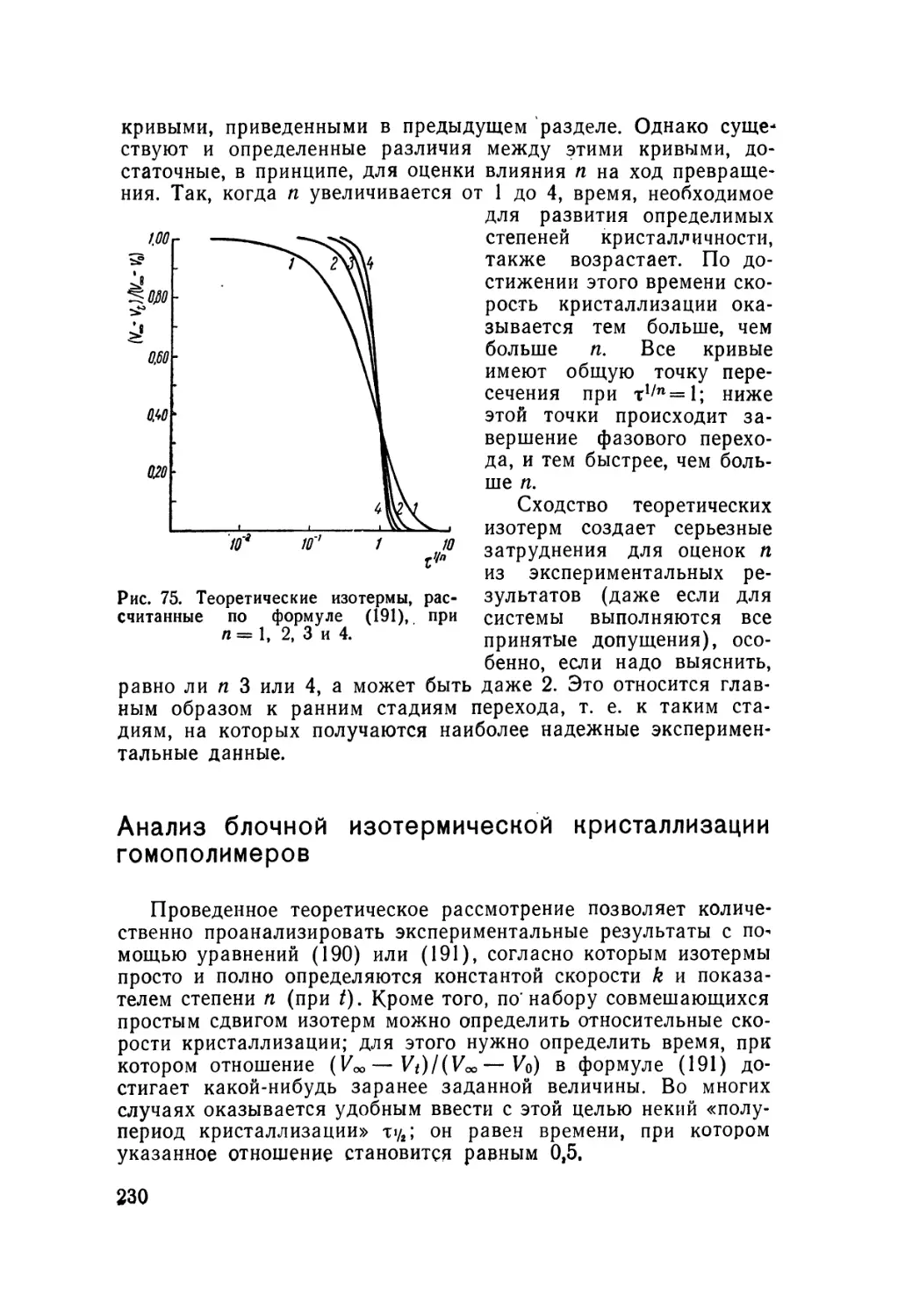
/













































































































































































































































































































































Автор: Манделькенрн Л.М.
Теги: химия термодинамика полимеры морфология кинетика переводная литература издательство химия кристаллизация
Год: 1966




Текст
CRYSTALLIZATION
OF POLIMERS
LEO MANDELKERN
Professor of Chemistry
Florida State University
MCGRAW-HILL BOOK COMPANY
NEW YORK-SAN-FRANCISKO-TORONTO-LONDON
1364
ЛЕО МАНДЕЛЬКЕРН
КРИСТАЛЛИЗАЦИЯ
ПОЛИМЕРОВ
Перевод с английского
под редакцией доктора физ.-мат. наук
С. Я. ФРЕНКЕЛЯ
ИЗДАТЕЛЬСТВО „ХИМИЯ"
МОСКВА-ЛЕНИНГРАД
1966
УДК 541.6: 542.65
В книге доступно и на высоком научном уровне
изложены термодинамические, кинетические и
морфологические представления о различных механизмах
кристаллизации.
Автор книги Л. Манделькерн, видный специалист
по физике полимеров, внес существенный вклад в эту
быстро развивающуюся область науки.
Книга представляет большой интерес как для
научных, так и для инженерно-технических работников, а
также может быть использована в качестве учебного
пособия аспирантами и студентами,
специализирующимися по физике и химии полимеров,
2-5-3
Предисловие
редактора
Механические, физические и вообще эксплуатационные
свойства полимеров в значительной мере определяются их фазовым
состоянием и надмолекулярной структурой. При решении
вопроса о способности полимерного материала или изделия
выдерживать те или иные нагрузки в определенном диапазоне
температур особое значение приобретают проблемы
устойчивости фазовых состояний, равновесий и переходов.
Безотносительно к детальной надмолекулярной организации
полимеров их теплостойкость (т. е. температурный предел, в
котором сохраняются эксплуатационные свойства) определяется
в основном температурами стеклования и кристаллизации. В тех
случаях, когда полимер работает главным образом в
ориентированном и кристаллическом состояниях (например, волокна),
доминирующей характеристикой, определяющей теплостойкость,
становится температура плавления, в то время как общий
комплекс механических свойств непосредственно определяется
надмолекулярной организацией.
Проблемы надмолекулярной организации и ее связи с
механическими свойствами получили широкое освещение в
последних работах школы В. А. Каргина, а также в исследованиях
ряда зарубежных школ — Р. Хоземанна, А. Келлера, Р. Штейна,
П. Флори. Автор предлагаемой книги, профессор Лео Мандель-
керн, являющийся представителем последней школы, много
сделал в области термодинамической теории плавления и
кристаллизации полимеров. Круг рассматриваемых в книге вопросов
ограничен именно этими проблемами и не затрагивает более
общие проблемы надмолекулярной организации полимеров.
Зато проблемы кристаллизации и плавления рассмотрены со
скрупулезными подробностями, особенно в части, касающейся
термодинамики этих фазовых превращений.
Как отмечается в предисловии автора, книга состоит из трех
частей. В первых семи главах дается анализ факторов,
определяющих температуру перехода кристалл — жидкость. В
основу автором положена аналогия между влиянием второго
компонента на температуру кристаллизации простой жидкости
и эффектами любых «примесных» факторов на температуру
плавления идеального (т. е. строго стереорегулярного) гомопо-
лимера. Такими «примесными» факторами в равной мере могут
быть низкомолекулярные жидкости (растворители или
пластификаторы) или любые нарушения регулярности цепи — гетеро-
звенья в сополимерах, точки ветвлений и сшивок, инверсия
характера присоединения смежных звеньев (изо-, синдио-, цис-л
транс-) и т. д. Автору удается на множестве примеров показать,
что во всех случаях «примесные» факторы вызывают депрессию
точки плавления, выражаемую однотипными формулами.
При общности и в некоторых случаях неожиданности такого
подхода (любые гомополимеры, в химическом смысле ведут
себя при плавлении и кристаллизации подобно сополимерам),
автор внимательно исследует специфику действия каждого
фактора, что позволяет предсказать характер фазовых превращений
при совместном действии одновременно нескольких факторов.
Изложение принципиальных проблем чередуется с очень
ценными методическими указаниями, касающимися сведения к
минимуму кинетических и релаксационных факторов, которые
могут исказить результаты измерений.
С общих позиций проведен также анализ энтропии и
энтальпии плавления: показано, в частности, что основная часть
энтальпии затрачивается при плавлении на разупорядочение
индивидуальных цепей. Это позволяет автору единообразно описать
плавление трехмерных кристаллических систем и кооперативных
переходов в «линейно-кристаллических» системах типа
полипептидных моноспиралей или полинуклеотидных мультиспиралей.
Специальный интерес представляет глава 7, где рассмотрены
фазовые переходы в ориентированных системах, подверженных
действию растягивающей силы. Эта глава имеет важное
значение для понимания поведения волокноподобных и некоторых
биологических систем. В частности, показана принципиальная
возможность приготовления из простых полимеров обратимых
сократительных систем, перерабатывающих химическую
энергию в механическую на основании тех же принципов, которые
реализуются в биологических системах.
Проблемы кинетики кристаллизации изложены в главе 8.
Стремясь к максимальной общности изложения, автор исходит
из формальной кинетики кристаллизации низкомолекулярных
веществ, анализируя в дальнейшем изотермы кристаллизации
в полимерах при постепенном наложении всех тех «примесных»
факторов, которые рассмотрены в предыдущих главах.
Изложение ведется в терминах первичной и вторичной нуклеаций и
роста кристаллической фазы. При этом впервые подвергается де-
6
тальному анализу своего рода обратная связь, возникающая в
ходе кристаллизации между кристаллической и аморфной
фазами. Эта обратная связь обусловливается тем, что по мере
развития процесса аморфная фаза непрерывно обогащается
«примесями», и соответственно непрерывно меняется скорость
нуклеации и роста. Формально это можно выразить как
непрерывное уменьшение степени переохлаждения расплава. В этой
части также содержится ряд ценных методических указаний, В
частности о способах кристаллизации плохо кристаллизующихся
полимеров.
Автор обосновывает простое общее правило:
кристаллизацию надо вести в присутствии плохого растворителя. Такой
растворитель незначительно влияет на равновесную температуру
плавления, но существенно понижает температуру стеклования,
увеличивая разность этих температур и тем самым создавая
более благоприятные кинетические условия для кристаллизации.
По аналогии с термодинамической частью в этой главе также
показаны черты сходства кинетики истинной кристаллизации
трехмерных систем с переходами в «линейно-кристаллических»
системах: в обоих случаях при увеличении степени
переохлаждения скорость кристаллизации проходит через максимум.
Глава 9, представляющая собой третью часть книги,
посвящается морфологическим проблемам. Как уже отмечалось,
собственные интересы автора связаны с кристаллизацией в
обычном значении этого слова, и поэтому он ограничивается
рассмотрением сферолитов и ламелярных единичных кристаллов,
не касаясь общих проблем надмолекулярной организации в
полимерах. Однако даже при такой ограниченной постановке
проблемы7 Л. Манделькерн по существу пренебрегает работами
других школ в области морфологии кристаллов. Автор
слишком категоричен в дискуссии с Келлером по вопросу о
регулярности складок цепей и упорядоченности больших граней,
пренебрегая' экспериментальными доводами в пользу последнего
представления. Между тем, на проходившем недавно в Праге
Международном Симпозиуме по макромолекулярной химий
C0/VIII—4/IX 1965 г.) Келлер привел весьма убедительные
данные, показывающие, что пластинчатые кристаллы с
совершенными гранями реально существуют и уж во всяком случае
не представляют исключения из правила, как пишет
Манделькерн.
Далее, Манделькерн совершенно не касаетсй
фундаментального вопроса о причинах возникновения складок, хотя на этот
счет имеется хорошо обоснованная строгая теория. Здесь
уместно подчеркнуть, что образование складок не обязательно
является термодинамически выгодным процессом, и пластинчатые
кристаллы с полностью развернутыми цепями значительно
стабильнее обычных складчатых кристаллов. Это было показано в
7
серии работ Вундерлиша с сотрудниками еще до выхода книги
Манделькерна в свет: под высокими давлениями D—5 тыс. атм)
Вундерлиш получил полиэтиленовые кристаллы толщиной до
3 мк.
Умолчание о работах, не согласующихся с взглядами автора,
в том числе и об американских работах, вызвало серьезную
критику книги в научной^ печати США.
В известной мере сказанное относится и к главе 8, где автор
чересчур увлекается аналогией с низкомолекулярными
веществами и не пытается обратить внимание на некоторые
специфические черты, качественно отличающие кинетику
кристаллизации полимеров от кинетики кристаллизации простых
веществ. Речь идет о проблеме критического ядра; на том же
Симпозиуме в Праге рядом авторов (Р. Хоземанн, Е. Хуземанн,
А. Келлер и А. Ковач) были приведены интересные данные по
кинетике кристаллизации при контролируемых линейных
размерах ядер. В случае сравнительно коротких цепей контроль
осуществляется просто выбором молекулярного веса: в отдельных
случаях он слишком мал, чтобы могла образоваться хоть одна
складка. В другом варианте (Е. Хуземанн) ядром является
макромолекула эфира (трикарбонилата) целлюлозы, образованная
жесткой цепью, несколько раз сложенной на себя; длина такого
ядра около 700 А. Наконец, третий вариант состоит в
использовании блок-сополимера полистирола и полиоксиэтилена, в
котором к кристаллизации способна лишь полиоксиэтиленовая
часть; размер этого блока можно контролировать в ходе
приготовления полимера. (Следует заметить, что это вообще первый
случай получения пластинчатых монокристаллов в гетерополи-
мере; вопреки ожиданиям, некристаллизующажся часть даже
стабилизирует пластинку с поверхностей).
Однако этих данных Л. Манделькерн мог и не знать во
время работы над книгой; они показывают лишь, что подход
автора к кинетическим проблемам в главе 8 не является
единственно возможным.
Хотя перечисленные обстоятельства и снижают несколько
познавательную ценность глав 8 и особенно 9, мы не считали
себя вправе существенно исправлять или дополнять авторский
текст, ибо стремились при переводе книги в максимальной
степени сохранить стилистические особенности и манеру
изложения автора; поэтому некоторые необходимые дополнения и
замечания сделаны в виде подстрочных примечаний редактора.
В этих же примечаниях даны ссылки на некоторые новейшие
работы, выполненные уже после выхода американского издания
в свет.
Мы надеемся, что книга принесет пользу не только
специалистам по физике и химии, работающим в области
высокомолекулярных соединений, но и технологам, в особенности работаю-
8
щим над проблемами взаимосвязи морфологии и
термомеханических свойств волокон. Кроме того, книга бесспорно может
представлять интерес для лиц, занимающихся вопросами
биологического моделирования. Много полезного и интересного
найдут в ней преподаватели, аспиранты и студенты старших курсов,
специализирующиеся в области физики и химии полимеров.
Перевод выполнен сотрудниками лаборатории физической
химии полимеров Института высокомолекулярных соединений
АН СССР — Ю. Н. Пановым (предисловие автора, главы 1—4
и 6), В. Г. Барановым (главы 5 и 8) и Т. И. Волковым (главы
7 и 9).
С. Я. Френкель
Предисловие
автора
В последние годы мы были свидетелями бурного роста
научной деятельности. Естественным следствием этого явилось
опубликование ряда работ, содержащих в большей своей части
интересную и важную информацию по самым разнообразным
вопросам. Однако лишь немногие авторы брали на себя труд
критически проанализировать и систематизировать весь
богатый, но разрозненный материал. Нигде эта проблема не
достигла такой остроты, как в области изучения свойств и поведения
высокомолекулярных соединений.
Несколько позднее развитие представлений о молекулярной
структуре полимеров привело к тому, что систематические
количественные исследования этого класса соединений стали
возможны лишь около 30 лет назад. Однако за этот короткий
период наши знания о полимерах быстро возрастали. Непрерывно
возникавшие новые проблемы привлекали внимание
исследователей во всех основных областях точных и естественных наук.
В подобной ситуации неизбежно стало подразделение науки о
полимерах на множество специальных областей. По мнению
автора, некоторые из этих областей могут быть в настоящее
время подвергнуты критическому, а в ряде случаев,
окончательному анализу. Такой анализ должен способствовать
установлению прямых связей между различными научными
дисциплинами, изучающими проблемы строения и свойств полимеров,
а также разработке более общих и фундаментальных
вопросов.
В предлагаемой вниманию читателя книге, стимулом к
появлению которой явились перечисленные проблемы,
рассматриваются явления, связанные с участием макромолекул в фазовых
переходах. Сам термин «кристаллизация» в применении к этим
явлениям оправдан тем, что по крайней мере одна из фаз имеет
высокоупорядоченную структуру. Книга состоит из трех частей.
После относительно беглого введения в природу цепных ма-
10
коомолекул рассматриваются проблемы фазового равновесия с
позиций термодинамики и статистической механики, при этом
использован обширный экспериментальный материал. Вторая
часть книги посвящается вопросам кинетики кристаллизации,
причем, рассмотрение умышленно ведется формально, что
позволяет выявить общие механизмы, лежащие в основе
кристаллизации. Фазовое равновесие и кинетические механизмы
должны, в принципе, обусловливать морфологические характеристики
кристаллического состояния, излагаемые в последнем разделе
книги. Вопросы морфологии в последние годы исследовались
особенно интенсивно. Предложено множество различных
концепций, все еще подвергающихся непрерывному пересмотру.
В связи с этим в книге обращено внимание не столько на
детальное морфологическое описание структур, сколько на
характерные общие черты морфологии, физическая сущность которых
может быть объяснена на основе положений, развитых в
предыдущих главах.
Хотя многие из рассматриваемых в этой книге вопросов
представляются сегодня достаточно изученными, некоторые
важные проблемы нуждаются в дальнейшем исследовании.
Автор надеется, что ему удалось по крайней мере четко
сформулировать такие проблемы. В конце каждой главы помещена,
хотя и неполная, но включающая все основные работы,
библиография. Поскольку при изложении материала основной акцент
был сделан на общие принципы, автор руководствовался только
этими соображениями при выборе материала для иллюстраций.
Естественно, что при подобном выборе отдается предпочтение
наиболее хорошо проверенным результатам. В настоящей книге,
по-видимому, также не удалось избежать этой
профессиональной привычки. Поскольку основные закономерности,
установленные на относительно простых макромолекулярных
соединениях, зачастую применимы и к значительно более сложным
системам биологического происхождения, автор старался всюду,
где это возможно, описывать явления кристаллизации и
плавления с единых позиций, охватывающих все типы и классы
макромолекул безотносительно к их различному происхождению и
функциям.
Автор считает своей большой удачей, что он в течение
нескольких лет имел приятную возможность сотрудничать с
профессором Флори. Автор глубоко обязан ему не только
введением в круг вопросов, которым посвящена эта книга, но
пониманием общих проблем науки и науки о полимерах, в
частности. Как будет ясно при чтении книги, изложение в
значительной мере основывается на тех идеях, которые автор
почерпнул от Флори в период работы с ним, а также из более поздних
его работ. Однако автор полностью несет ответственность за
само изложение и интерпретацию фактов.
11
Также показано, что среднеквадратичное расстояние звеньев
такой цепи от центра тяжести (т. е. среднеквадратичный радиус
инерции) и среднеквадратичное расстояние между ее концами
связаны соотношением:
/г2\'/2
Следовательно, для принятой модели линейные размеры
цепи пропорциональны корню квадратному из степени
полимеризации и во много раз меньше, чем полная длина вытянутой
макромолекулы; наиболее вероятны сильно свернутые конфор-
мации. Расчеты размеров цепей со свободно сочлененными
звеньями сделаны также и для тех случаев, когда имеется
более чем один вид связи и валентный угол [2, 4]. Таким образом,
не представляет труда сравнить действительные (измеренные)
средние размеры реальных цепей с рассчитанными для
свободно сочлененных аналогов.
В реальных цепях свобода внутреннего вращения вокруг
каждой связи ограничена потенциальными барьерами. Хотя
полная количественная оценка всех тормозящих вращение
факторов крайне затруднительна, все же из общих соображений
Следует ожидать, что взаимодействие между орбиталями
валентных электронов соседних связей сообщает потенциальной
функции угловую периодичность [5]. Однако на форму этой
функции сильнейшее влияние могут оказывать стерические
затруднения и взаимодействия соседних боковых радикалов.
Тормозящий потенциал (потенциал торможения) * для
простых связей в полимерной цепи, по-видимому, имеет ту же
форму, что и для аналогичных связей в низкомолекулярных
органических молекулах [6]. Например, вращательные состояния
(поворотные изомеры) молекулы этана описываются тормозящим
потенциалом с тройной симметрией, тогда как потенциал
центральной связи молекулы бутана уже отличается от него. Хотя
в потенциальной функции этой связи еще сохраняются три
минимума, энергии их уже не равны. Самый глубокий минимум
соответствует плоской, или ту?бшс-конформации. Другие два
минимума относятся к свернутым гош-формам, которые
получаются из транс-форм поворотом на ±120°. Энергии двух
гош-форм одинаковы и превышают энергию транс-формы
примерно на 50р—800 кал/моль.
Предполагалось, что подобная форма потенциала прило-
жима и к заторможенному вращению в линейной
макромолекуле полиэтилена. Следовательно, для этого полимера формой
с самой низкой энергией является плоская, целиком транс-кон-
формация, которая соответствует полностью вытянутой цепи.
* [Имеется в виду потенциал заторможенного внутреннего вращения
участков цепи макромолекулы. — Прим. редактора.]
18
Однако хотя 7у?а«с-состояние и выгодно энергетически, гош-со-
стояние также становится возможным при благоприятных
температурах, в результате чего полиэтилен способен принимать
крайне нерегулярные конформации.
Для макромолекул с более сложной структурой, чем
полиэтилен, характер тормозящего потенциала резко изменяется, и
в этих случаях потенциальная функция содержит минимумы,
соответствующие наиболее вероятным углам вращения. Можно
считать, что поворот вокруг каждой связи действительно
соответствует одному из разрешенных минимумов. Вращение вокруг
связи, таким образом, ограничивается узкими диапазонами
углов, которые могут рассматриваться как дискретные состояния
[7]. В пределах этого приближения удается применить
математический метод, позволяющий количественно описать
конформации цепи и учесть корреляции вращательных состояний
соседних связей. Статистическая сумма для такой цепи может
быть рассчитана с помощью одномерной модели Изинга, ранее
примененной для описания ферромагнетизма [8, 9]. В
результате можно вычислить средние размеры отдельной
изолированной реальной цепи. Установлено, что соответствующие величины
неизмеримо больше вычисленных для цепей со свободным
вращением.
Среднеквадратичные размеры реальных полимерных цепей
могут быть измерены различными физико-химическими
методами *. В табл. 1 представлены отношения этих размеров для
реальных невозмущенных цепей к среднеквадратичным
размерам, вычисленным для тождественной по структуре свободно
сочлененной цепи.
Наблюдаемые размеры макромолекул во всех случаях
больше вычисленных в предположении свободного вращения. Это
несомненно отражает сильнейшее влияние заторможенности
внутреннего вращения на конформацию макромолекулы. Самые
большие различия наблюдаются для производных целлюлозы.
Однако изменение размеров молекул этих полимеров с
повышением температуры указывает на увеличение свободы
вращения. Размеры молекул полистирола и полиакрилонитрила
относительно высоки и несомненно обусловлены влиянием боковых
заместителей. Для натурального каучука, полиизобутилена, по-
лидиметилсилоксана и полиэтилена рассматриваемое отношение
(табл. 1) оказывается ниже среднего значения. Ограниченные
данные, имеющиеся для белков и полипептидов в
неупорядоченном состоянии, также не выявляют каких-либо необычных кон-
формационных характеристик. Эти макромолекулы обладают
размерами, сравнимыми с наблюдаемыми для более простых
цепных молекул.
* Методы и принципы этих измерений подробно описаны в работе [2].
2*
19
Автор выражает искреннюю благодарность за великодушную
помощь многим друзьям и коллегам. Доктор Беккедаль
критически просмотрел большую часть рукописи и оказал тем самым
автору неоценимую услугу. Критические замечания и советы
были также получены от докторов Фокса, Хольцворса, Макро-
вича и Мак-Интайра. Доктор Райе и господин Диорио любезно
предоставили некоторые электронно-микроскопические и
рентгенографические данные, использованные автором для
иллюстраций.
За разрешение воспроизвести ранее опубликованные
материалы автор также выражает глубокую признательность
редакциям журналов и издательствам, а именно: Техническому
факультету Университета Киуши; Annals of the New York
Academy of Science; Chemical Reviews; Die Makromolekulare Che-
mie, John Wiley and Sons, Ink.; Journal of the American
Chemical Society; Journal of Applied Physics; Journal of Cellular and
Comperative Physiology; Journal of Physical Chemistry; Journal
of Polymer Science; Kolloid — Zeitschrift; Polymer, Proceedings
of the National Academy (U. S.); Proceedings of the Royal
Society; Review of Modern Physics; Rubber Chemistry and
Technology; Science и Transaction of the Faraday Society.
Лео Манделькерн
Таллахасси, Флорида
Май, 1963
Условные обозначения
основных физико-химических величин
с — относительная концентрация сшитых звеньев;
F*— свободная энергия аморфного состояния;
FK — свободная энергия кристаллического состояния;
А/^м — изменение свободной энергии на мономерное звено;
А/^п — свободная энергия фазового перехода;
Д^см — свободная энергия смешения;
&FCM— свободная энергия смешения без изменения внутренней
структуры макромолекулы;
А/^р —свободная энергия разупорядочения макромолекулы;
AF* — критическая свободная энергия;
А/7*— свободная энергия при эластической деформации аморфной сетки;
&FV— изменение свободной энергии на единицу объема;
А/м — изменение свободной энергии плавления на молекулу;
/°—равновесная сила;
Д//м — энтальпия плавления на мономерное звено;
Д//м — полная энтальпия плавления на мономерное звено;
Д#а—энтальпия аморфной фазы;
А//к—энтальпия кристаллической фазы;
АЯСМ — энтальпия смешения;
Д//р — энтальпия плавления при химической реакции;
А//р—энтальпия растворения;
ДЛ' — энтальпия плавления на моль эквивалентных статистических
элементов;
kH—константа скорости инициирования;
kf — константа скорости роста спирали;
kb — константа скорости обратного процесса;
ks — константа кристаллизации трехмерного роста;
kd — константа кристаллизации двухмерного роста;
La — длина волокна в аморфном состоянии;
LK — длина волокна в кристаллическом состоянии;
LH — длина аморфного волокна в изотропном состоянии;
L0 — длина аморфного волокна в отсутствие сшивок;
(г2) — среднеквадратичное расстояние между концами цепи;
А^м — энтропия плавления на мономерное звено;
ASy, — энтропия изменения объема при плавлении;
(ASm)^ — энтропия плавления при постоянном объеме;
AS0 — энтропия плавления несшитой системы;
AS^ — конфигурационная энтропия стандартного состояния сшитой
системы;
AS" — энтропия перехода от стандартного к реальному состоянию;
13
Тпл — температура плавления;
^пл — равновесная температура плавления;
^пл — изотропная температура плавления;
7\i — температура фазового перехода;
Тс — температура сокращения;
Т™—температура плавления смеси;
Т^л — температура плавления кристаллической фазы;
Тк — температура кристаллизации;
V2 — молярный объем полимерного звена;
V| — молярный объем растворителя;
VM — молярный объем мономерного звена или сегмента;
V0— удельный объем аморфного полимера;
Vt — удельный объем кристаллизующего полимера в момент времени /;
Кх>—удельный объем кристаллизующегося полимера при времени
tf = oo;
vv —объемная доля растворителя;
v2 — объемная доля полимера;
1 —Я — степень кристалличности;
1 — A (t) — степень кристалличности полимера в момент времени
кристаллизации /;
?—линейный размер кристаллита или ядра;
р—поперечный размер кристаллита или ядра;
?*, р* — соответствующие критические значения;
?°, Р° — равновесные размеры кристаллита;
Рк—плотность кристаллической фазы;
Рж — плотность жидкой фазы;
fAi — химический потенциал растворителя в смеси;
\х2—химический потенциал полимера в смеси;
М-2 — химический потенциал чистого растворителя;
\i2—химический потенциал чистого полимера;
(лм— химический потенциал на мономерное звено в смеси;
М^ — химический потенциал чистого полимера на мономерное звено;
|л* — химический потенциал кристаллической фазы на мономерное звено;
И* — химический потенциал жидкой фазы на мономерное звено;
Mi* — химический потенциал растворителя в жидкой фазе;
Hi — химический потенциал 1-го компонента системы;
о—поверхностная энергия;
о&— поверхностная энергия боковой грани;
ат—поверхностная энергия торца;
Oq — поверхностная энергия боковой грани кристаллита;
а? — поверхностная энергия торца кристаллита;
0% — поверхностная энергия боковой грани ядра;
а? — поверхностная энергия торца ядра;
Ту — полупериод кристаллизации;
Ф — термодинамический потенциал;
Фк — термодинамический потенциал полностью кристаллического
волокна;
Фа — термодинамический потенциал полностью аморфного волокна;
%х — термодинамический параметр взаимодействия;
ij>j — энтропийный характер взаимодействия,
Основные
структурные
представления
Введение
Одно из важнейших достижений науки за последние три
десятилетия — установление того факта, что полимерные
молекулы представляют собой реальные цепи, построенные из очень
большого числа мономерных звеньев, соединенных между собой
ковалентными связями. Это свойство присуще всем
макромолекулам независимо от их происхождения, различной химической
и стереохимической структуры. Следовательно, этот класс
соединений, включающий как относительно простые синтетические
полимеры, полученные в лаборатории, так и более сложные
природные вещества, можно изучать с единых позиций.
Характерные термодинамические, гидродинамические, физические и
механические свойства, которыми обладают полимерные вещества,
могут быть объяснены главным образом их ковалентной
структурой и обусловленными ею большими размерами
индивидуальных молекул.
Несмотря на то, что эти молекулы содержат в цепи тысячи
звеньев, макромолекулярные системы все же сохраняют
способность существовать, как и все другие вещества, в различных
фазовых состояниях. Два фазовых состояния — жидкое и
кристаллическое, присущие низкомолекулярным соединениям,
обнаружены также и у полимеров. Жидкое, или аморфное,
состояние характеризуется наличием некоторой свободы' вращения
вокруг простых связей, соединяющих атомы «хребта» цепи *.
Поэтому полимерная молекула принимает большое число
пространственных форм, или конформаций **.
* [Имеется в виду цепь главных валентностей. — Прим. редактора.]
** Л. Манделькерн в своей монографии применяет всюду термин
«конфигурация» макромолекул. Однако в настоящее время в стереохимии
полимеров под термином «конфигурация» подразумевают взаимное расположение
атомных групп в молекуле, зафиксированное при ее образовании и
изменяющееся только при разрыве или обмене химических связей. Что же
касается внешней формы, которую приобретает макромолекула в результате
теплового движения звеньев или которая фиксируется в определенных
15
У большинства молекул в аморфном состоянии звенья
различных цепей располагаются в пространстве случайно и
практически не определяют взаимное расположение своих соседей.
Однако при соответствующих условиях (температура, давление
или растягивающее напряжение, а также растворитель) может
происходить самопроизвольное упорядочение отдельных
участков цепных молекул. Это упорядочение является результатом
того, что в действительности звенья ориентированы не
произвольно относительно друг друга, а повернуты на некоторые
углы, определяемые величинами потенциальных барьеров,
препятствующих свободному вращению. Следовательно, в
противоположность аморфному или жидкому фазовому состоянию
полимера в целом отдельные макромолекулы существуют теперь
в состоянии конформационного порядка. Упорядоченные цепи
или их участки обычно образуют регулярную трехмерную
решетку с параллельной упаковкой осей цепей. В зависимости от
условий кристаллизации геометрическая форма отдельных
молекул может быть полностью вытянутой, спиральной или
складчатой. Существенно то, что у полимеров возникает состояние
трехмерной упорядоченности, которое в основных чертах
аналогично кристаллическому состоянию низкомолекулярных
веществ. Этот весьма общий вид пространственного расположения
цепных макромолекул называется кристаллическим состоянием
полимеров.
Очевидно, что отдельная полимерная молекула, обладающая
высокой степенью структурной и стерической регулярности,
должна .быть способна к кристаллизации. Действительно
кристаллизацию наблюдают для большого числа таких полимеров.
Кроме того, установлено, что даже значительное количество
нерегулярности в цепях может не нарушить процесс
кристаллизации. Однако даже для полимеров, обладающих регулярной
структурой, должны быть найдены кинетические благоприятные
условия, в которых кристаллизация проходит за конечный,
доступный для наблюдения, промежуток времени.
Например, полиизобутилен — полимер явно регулярной
структуры, может быть легко закристаллизован при
растяжении. Долго считалось, что этот полимер не кристаллизуется в
иных условиях. Позже было показано, что кристаллизация
может быть вызвана просто охлаждением, но для этого требуется
даже при оптимальной температуре длительное время
(несколько месяцев). Таким образом, кинетические факторы в этом
случае оказываются определяющими, и не удивительно, что неко-
условиях «вторичными» взаимодействиями мономерных звеньев, то ее
называют «конформацией». Поскольку Манделькерн под словом «конфигурация»
подразумевает именно конформацию цепей, термин «конфигурация» в нашем
переводе всюду заменен на «конформацию»»
16
торые полимеры, казалось бы с регулярной структурой, до сих
пор не были закристаллизованы. В качестве примера можно
указать на поли-ж-фторстирол, поли-я-хлорстирол и поли-2-ви-
нилнафталин, причем это лишь малая часть известных
случаев [1]. Хотя доказано, что эти полимеры имеют стериорегуляр-
ную структуру, подходящие условия кристаллизации для них до
сих пор не найдены.
Многие важные свойства полимерных систем обусловлены
конформацией отдельных цепей [2, 3]. В особенности это
относится к их поведению при кристаллизации. Поэтому
целесообразно перед основным изложением проблем кристаллизации
полимеров кратко рассмотреть общие принципы, используемые
для описания конформаций макромолекул.
Структура неупорядоченных цепей
Пространственная геометрия макромолекул зависит от длин
связей между атомами главной цепи, валентных углов и
возможности (хотя бы потенциальной) внутреннего вращения
мономерных звеньев вокруг простых связей. Конформация хребта
цепи (фиксированные длины связей и валентные углы)
полностью определяется углами вращения вокруг каждой из его
простых связей. Число конформаций, доступных данной
молекуле, является результатом большого набора «разрешенных»
углов вращения вокруг каждой простой связи.
Для статистического расчета размеров реальных
макромолекул в первом приближении исходят из гипотетической модели
свободно сочлененных мономерных звеньев, вращение которых
никак не заторможено. Геометрические свойства такой модели
могут быть точно вычислены, если не принимать во внимание
взаимодействия дальнего порядка, т. е. между парами
структурных элементов, далеко отстоящими друг от друга [2]. Размеры
цепи удобно характеризовать либо расстоянием между ее
концами, либо расстоянием произвольного звена от центра тяжести
макромолекулы. Поскольку число возможных конформаций цепи
чрезвычайно велико, практически вычисляется распределение
расстояний между ее концами. Соответствующая функция
распределения имеет вид гауссовой кривой, передний квадрат
расстояния между концами цепи оказывается равным
<г2)сс = л/2
где /—длина;
п — число связей в цепи.
Индексы указывают, что речь идет об изолированной цепи
со свободным вращением.
2 Л. Манделькерн
17
Таблица I
Размеры неупорядоченных полимерных цепей
Полимер
Полиэтилен
Полистирол (атактиче-
ский)
Полиизобутилен . . .
Полиметилметакрилат .
Полиакриловая кислота
Натуральный каучук
A,4-^ис-полйизопрен)
Гуттаперча A,4-т/?д«с-
полиизопрен) ....
Полиакрилонитрил . .
Полидиметилсилоксан .
Трибутират целлюлозы
Тринитрат целлюлозы
Поли-у-бензил-?-глута-
Коллаген
(неупорядоченная форма) • • • 1
t, °С
140
25
70
24
95
30
30
0-^60
30
30
20
30
90 |
130
30
25
25
[<'2>/<'\с],/2
1,85
2,44
2,35
1,93
1,84
2,2
1,96
1,70
1,45
2,6
1,6
4,9
2Л
1,8
4,5
2,0
1,15
1
! п 1
[4]
И
[Ю]
[2]
[2]
[И]
[И
12
13
14
[15]
[4]*
[4]*
* *f2>c с~вычислен0 в предположении транс-формы
пептидного звена*.
Хотя для всех исследованных полимеров
среднеквадратичные размеры превосходят размеры свободно сочлененной цепи,
они ни в одном случае не приближаются к длине полностью
вытянутой цепи. На первый взгляд это не согласуется с
выраженной тенденцией отдельных связей принимать дискретные
поворотные состояния. Поэтому экспериментальные результаты
следует рассматривать как указание на то, что даже
незначительная свобода вращения приводит к возникновению весьма
нерегулярных свернутых конформаций.
Структура упорядоченных цепей
При некоторых условиях возможны дальнейшие ограничения
внутреннего вращения. Определенная связь или
последовательность связей будет теперь ограничена углом вращения, который
соответствует самому глубокому минимуму потенциальной
кривой, описывающей заторможенность вращения. В связи с этим
20
Рис. 1. Макроскопическая
модель упорядоченной структуры
участков полиэтиленовых
цепей [16].
возникает высокоупорядоченная структура цепи, полностью ут^
ратившая изменчивость конформаций, характерную для
неупорядоченной цепи. Например, для полиэтилена транс-положение
звена представляет собой ориентацию с самой низкой энергией.
Когда последовательные связи цепи принимают эту ориентацию,
возникает конформация полностью вытянутого плоского зигзага,
характерная для многих полимеров, включая полиамиды,
полиэфиры, производные целлюлозы,
полидиены и одну из структурных
форм цепи полипептидов (рис. 1).
В виниловых полимерах с общей
формулой (—СН2—CHR—)п
разрешенное поворотное состояние
зависит от конфигураций
последовательных асимметричных атомов
углерода, несущих на себе радикалы R.
Поскольку в изотактических
полимерах этим атомам углерода свой-
свенна одинаковая тетраэдрическая
конфигурация, плоская
зигзагообразная конформация цепи
невозможна из-за стерического
перекрывания соседних R-rpynn. В
трансположении соседние радикалы должны находится на
расстоянии от 2,5 до 2,6 А друг от друга. Это расстояние настолько
мало, что должно привести к очень большому перекрыванию
соседних групп. Решение возникающей проблемы может быть
облегчено, если предположить наличие чередующихся связей
в гош-положении. При подобной геометрии цепи радикалы R
оказываются достаточно разделенными. Если подобное
чередование осуществляется регулярно, т. е. происходит
последовательная транс-гош-ориентация связей, цепь приобретает
спиральную конформацию. Так как существует два гош-положения,
то, если повороты осуществляются всегда в одном и том же
направлении, из рассматриваемой молекулы может образоваться
либо правая, либо левая спираль. В тех случаях, когда
радикалы R не слишком массивны, спираль содержит три
мономерных звена на виток, как это видно из рис. 2, а [15, 17]. В
подобной спиральной конформаций заместители достаточно
удалены друг от друга. Наименьшие расстояния между несзязан-
ными атомами углерода становятся теперь в изотактическом
полипропилене 3,2 А, а в изотактическом полистироле — 3,3 А.
Полимеры, содержащие более массивные боковые группы,
нуждаются в большем пространстве для их размещения, и в
результате образуются более развернутые спирали. На рис. 2, б,
в, г показаны несколько типичных вариантов таких спиралей.
Легко видеть, что их витки содержат больше звеньев, чем
21
простейшая Згспираль. Например, виток спирали поли-3-метил-
бутена-1 с боковым радикалом —CH(CH3h включает четыре
мономерных звена. Эта массивная боковая группа обусловливает
более сильное перекрывание, поэтому угол гош-связи изменяется
от 120 до ~100°, а четкое
транс-положение,
соответствующее 0°, изменяется
примерно на —26°.
В полимерах, у которых
разветвление происходит у
второго атома боковой цепи,
как, например, в поли-4-
метилгексене-1, спираль
содержит семь мономерных
звеньев в двух
последовательных витках (рис. 2,6).
Структура этого полимера
объясняется на тех же
общих основаниях, однако
здесь отклонение от чистых
транс- и гош-положений
меньше, чем в предыдущем
случае. Поли винил нафталин
и поли-о-метилстирол
образуют спирали (рис. 2, г) с
четырьмя звеньями в витке,
а спираль поли-ж-метилсти-
рола содержит одиннадцать
мономерных звеньев в трех
витках. Изотактический
полиметил метакрилат
образует спираль с пятью
звеньями в двух последовательных
витках.
Это краткое рассмотрение объясняет разнообразие
спиральных структур изотактических полимеров в зависимости от
природы заместителя при а-углеродном атоме. В синдиотактических
полимерах замещенные углеродные атомы обладают
чередующейся D, L-тетраэдрической конфигурацией, поэтому стериче-
ские помехи, создаваемые соседними боковыми группами, не
так велики, как в изотактических структурах, и цепь может
приобрести плоскую (или почти плоскую) и полностью
вытянутую упорядоченную структуру. В этом случае каждая связь
находится в ту?а«с-положении. Вытянутые структуры
действительно наблюдаются для поливинилхлорида и поли-1,2-бутадиена.
Геометрический период такой цепи, заключающий в себе два
мономерных звена, приблизительно в два раза превосходит со-
а 6 д г
Рис. 2. Макроскопические модели
некоторых типичных упорядоченных
спиральных структур изотактических
полимеров [16]:
а) R = -CH3, -С2Нб, -СН2-СН2-СН-(СН3J,
-СН=СН2, -О-СНз, -0-СН2-СН-(СНаJ,
-О
б) R = -CH2-CH(CH3)-C2H5, -CH2-CH-(CH3J;
22
ответствующий период незамещенной полиэтиленовой цепи.
Однако упорядоченная структура синдиотактических полимеров не
обязательно должна быть плоской. Так, например, имеются
данные, согласно которым синдиотактическии полипропилен
обладает спиральной структурой.
У полиизобутилена регулярное чередование незамещенных и
дважды замещенных углеродных атомов вызывает сильное
перекрывание боковых метильных радикалов. Эти стерические
помехи не удается устранить никакими комбинациями
поворотов вд^руг связей, сводящимися к транс- или гош-положениям.
Связи в этой молекуле, следовательно, обладают весьма
специфической потенциальной функцией, никак не похожей на
потенциал с тройной симметрией, описывающий внутреннее вращение
в полиэтилене и других цепных молекулах с углеродным
скелетом. Можно показать, что полиизобутилен приобретает в
результате спиральную структуру, которая возникает при
повороте каждой связи на 82° от т/?аяс-положения. В этой спирали
восемь мономерных звеньев заключены в пяти витках,
образующих период спирали [17, 18]. Регулярно организованная
структура возникает в тех случаях, когда направление, или знак
поворота одинаковы для каждой связи, а статистически
неупорядоченная, — когда знак вращения у разных связей различен.
Политетрафторэтилен также образует упорядоченную
структуру. Полностью упорядоченная конформация представляет
собой слабо закрученную спираль с 13 С^-группами в периоде
[19]. Каждая связь цепи повернута на 20° от точного
транс-положения. Причина этой деформации заключается в том, что
при структуре плоского зигзага несвязанные атомы фтора
относительно мало удалены друг от друга. Вращение вокруг
каждой связи цепи опять уменьшает перекрывание.
Спирально-упорядоченные цепные структуры не
ограничиваются макромолекулами с углеродным скелетом. Они также
встречаются у полипептидов, белков и нуклеиновых кислот.
Весьма важной упорядоченной структурой у полипептидов
является а-спираль, предложенная Полингом, Кори и Брэнсо-
ном [20]. В этой структуре (в противоположность вытянутой
конформации полипептидной цепи) образуется максимально
возможное число внутримолекулярных водородных связей
между карбонильным кислородом и аминным азотом. Водородные
связи образуются между «первым» и «четвертым»
аминокислотными остатками вдоль цепи. Возникает «нецелочисленная»
спираль, содержащая 3,6 остатка на виток. Пептидная группа —
плоская, как это следует из данных кристаллографического
анализа низкомолекулярных веществ с аналогичной структурой, и
каждая СО и NH группы участвуют в образовании водородной
связи. На рис. 3 показаны для сравнения а-спиральная
упорядоченная полипептидная цепь и 3,5гспираль, образованная
23
изотактическим виниловым полимером. В последнем случае
структура не стабилизируется каким-либо внутрицепным
связыванием.
Один из важных полипептидов, поли-?-пролин, является по-
лииминокислотой и поэтому не способен к образованию
внутримолекулярных водородных связей. Однако под влиянием стери-
ческих факторов в полиЪролине возникает упорядоченная кон-
формация (поли-/,-пролин II), представляющая собой спираль
с плоской имидной группой в транс-
положении [21].
Упорядоченные структуры
нуклеиновых кислот включают более,
чем одну цепную молекулу.
Структура дезоксирибонуклеиновой
кислоты (ДНК), предложенная Уотсо-
ном и Криком [22] и Уилкинсом [23],
включает две взаимнопереплетен-
ные спиральные цепочки,
напоминающие винтовую лестницу.
«Ступеньки» этой «лестницы»,
делающие структуру стабильной,
образованы водородными связями,
которые соединяют комплементарные
Рис. 3. Сравнение пептидной пуриновые и пиримидиновые осно-
а-спирали (а) и 3,5гспирали (б) вания.
изотактического полимера [16]. Хотя мы и не пытались
обсудить эту проблему детально,
совершенно ясно, что в цепных молекулах может возникать
практически бесконечное множество упорядоченных структур.
Каждая такая структура зависит от химической природы молекулы
и получается в результате фиксации специфических и вполне
определенных поворотов звеньев и ориентации связей вдоль
цепи. Процессу упорядочения могут также способствовать
специфические внутримолекулярные взаимодействия. Схематически
кристаллизацию в этом случае можно рассматривать как
процесс упаковки отдельных упорядоченных молекул в
организованную трехмерную решетку. Хотя ориентации связей
соответствуют минимуму энергии цепи в целом, может происходить
дальнейшее снижение свободной энергии, если атомы хребта
цепи и боковые радикалы разных молекул определенным
образом располагаются друг относительно друга.
Форма отдельных молекул, как показывает рентгенострук-
турный анализ, обычно соответствует конфигурациям связей
(или конфигурации последовательности связей) с минимальной
энергией [24]. Это соответствие может быть несколько модифи*
цировано межмолекулярными взаимодействиями, способными
вызвать некоторые нарушения структуры отдельных молекул.
24
Роль упаковки цепи становится определяющей, когда
существует выбор между конформациями с почти равной энергией.
По-видимому, такова ситуация при кристаллизации гидрохло-
рированного каучука и некоторых простых и сложных
полиэфиров [17].
Расположение атомов в кристаллических участках полимера
определяется обычными методами рентгеноструктурного
анализа [25]. Хотя единичные кристаллы обычно недоступны для
лиц, занимающихся кристаллографией полимеров, многие
характеристики элементарной ячейки, такие как
кристаллографическая система, размеры (ячейки), положение атомов
определены для большого ряда полимеров. При этом установлено, что,
как правило, длины связей, углы и другие элементы структуры
сохраняют свою нормальную величину. Мономерные-звенья
играют ту же роль, что «свободные» молекулы в кристаллах
низкомолекулярных органических соединений. Элементарная
ячейка обычно не содержит целую макромолекулу. Возможны
случаи, когда через элементарную ячейку проходят несколько
цепей (нередко 1—8 звеньев).
Морфологические особенности
Когда изучаются структурные особенности кристаллического
полимера, помимо геометрии элементарной ячейки, необходимо
принимать во внимание поликристаллический характер
структуры. Поликристалличность сейчас же становится очевидной
при анализе рентгенограмм. На полимерных системах можно
получить несколько характерных типов дифракции
рентгеновских лучей под большими углами. Если полимер
некристаллический, дискретные брэгговские рефлексы отсутствуют.
Наблюдается только диффузное гало, как показано на рис. 4
(натуральный каучук при 25° С).
Типичная рентгенограмма полимера, закристаллизованного
при охлаждении, приведена на рис. 5 для линейного
полиэтилена. Здесь уже наблюдаются дискретные брэгговские
рефлексы. Они имеют форму ряда концентрических окружностей.
Фотография весьма похожа на порошковую рентгенограмму для
кристаллических низкомолекулярных веществ. Эту форму
кристаллизации полимеров называют статистической, так как с
макроскопической точки зрения здесь нет в среднем
предпочтительной ориентации кристаллографических направлений.
Однако возможны и другие формы кристаллизации, когда
развиваются и наблюдаются типы предпочтительной ориентации,
например нативное состояние многих биологических
макромолекул, таких как фибриллярные белки. В других полимерах
25
аналогичные условия кристаллизации могут быть достигнуты
при механическом растяжении.
Рис. 4. Рентгенограмма аморф- Рис. 5. Рентгенограмма
линейного натурального каучука. ного полиэтилена
закристаллизованного при охлаждении.
Примеры рентгенограмм для трех одноосноориентирован-
ных кристаллических полимеров — натурального каучука, линей-.
Рис. 6. Рентгенограммы одноосноориентированных
кристаллических полимеров:
а—натуральный каучук; б— линейный полиэтилен; в — нативное коллаге-
новое волокно.
ного полиэтилена и нативного фибриллярного белка
коллагена — приведены на рис. 6. Вследствие предпочтительной
ориентации различных кристаллографических плоскостей рефлексы
сократились до дискретных пятен. Картины, полученные для
натурального каучука и полиэтилена, напоминают хорошо разви-
26
тые единичные кристаллы с круговой симметрией относитель*
но оси, перпендикулярной падающему пучку рентгеновских
лучей.
Следует отметить, что, несмотря на большое сходство этих
рентгенограмм с получаемыми для обычного монокристалла, на
них все же может быть легко обнаружено диффузное гало. При
одной и той же кристаллографической структуре, т. е. в
отсутствие полиморфизма, наблюдаемые брегговские периоды одина*
ковы независимо от того, ориентирован образец или нет.
Возможны и действительно наблюдались и другие типы ориентации.
Примером этого может служить двухосная ориентация, когда
полимерные цепи располагаются в одной плоскости.
Существуют достаточно убедительные указания на то, что
на всех доступных изучению уровнях надмолекулярной морфо*
логии развиваются вполне
определенные и хорошо различимые, хотя
и не до конца расшифрованные,
организованные структуры.
Малоугловое рассеяние рентгеновских
лучей выявляет дискретные
структуры с линейными размерами до
сотен ангстрем [26, 27]. Типичная
картина рассеяния рентгеновских
лучей под малыми углами от высо-
коориентирован'ного волокна из
линейного полиэтилена показана на
рис. 7. В этом образце
разрешаются несколько диффракционных
порядков, соответствующих большому
периоду 410±20 А. При малых
углах, кроме дискретного максимума,
появляется еще и диффузное
рассеяние. Рассеяние света от тонких
пленок кристаллических полимеров
также выявляет наличие структурных элементов с размерами
порядка тысяч ангстрем [28].
При наблюдении таких гомополимерных пленок в
поляризационном микроскопе неизменно обнаруживаются сильно дву-
лучепреломляющие сферолитные структуры. Пример такого
рода кристаллического тела, выросшего в тонкой
полиэтиленовой пленке, показан на рис. 8 [29]. Более детально эти
структуры будут обсуждены ниже. Пока же ограничимся замечанием,
что их существование является доказательством дальнейшей
надмолекулярной организации полимеров на уровне микронных
структур. Изучение в электронном микроскопе тонких пленок
или реплик с поверхностей скола кристаллических полимеров
дает микрофотографии типа рис. 9 [30]. Таким путем удается
Рис. 7. Картина рассеяния
рентгеновских лучей под малыми
углами от одноосноориентиро-
ванного кристаллического
линейного полиэтилена B6].
27
наблюдать структуры, построенные из очень тонких
закрученных пластинок (ламелей).
Рцс. 8. Поляризационная микрофотогра- Рис. 9. Электронная микро-
фия сферолитных структур, выросших фотография линейного поли-
в линейном полиэтилене [29]. этилена, закристаллизованного
из расплава [30].
Когда гомополимеры закристаллизованы из сильно
разбавленных растворов, образуются либо ромбовидные пластинки,
Рис. 10. Электронная микрофотография Рис. 11. Электронная
микролинейного полиэтилена (средневязкост- фотография линейного поли-
ный молекулярный вес ^ = 50000), этилена (средневязкостный
изотермически закристаллизованного Мл = 50000), изотермически
при 89° С из разбавленного раствора закристаллизованного при
в тетралине. 60° С из разбавленного
раствора в тетралине.
либо кристаллы, обладающие дендритной структурой.
Несколько типичных электронных микрофотографий таких кристаллов,
высаженных из разбавленного раствора, показано на рис. 10
2а
и 11. Характер роста кристаллов зависит от молекулярного веса
полимера и условий кристаллизации, в частности температуры
и растворителя. Весьма примечательной особенностью
пластинчатых монокристаллов является их толщина, обычно не
превосходящая 100—200 А. Анализ локальных электронных дифракто-
грамм от малых участков таких кристаллов показывает, что
одна и та же цепь проходит через них многократно. Таким
образом, внутри кристалла полимерная цепь должна приобретать
складчатую конформацию. Более подробно складчатые конфор-
мации цепей и природа пластинчатых кристаллов рассмотрены
в главе 9.
Выше мы пытались осветить те специфические трудности,
которые возникают при описании детальной структуры и кон-
формации отдельной изолированной полимерной цепной
молекулы. Ясно, что детальная организация таких молекул в
кристаллическую структуру порождает новые еще более сложные
проблемы. В следующих главах делается попытка развить
систематические представления о природе кристаллического
состояния полимеров.
Литература
1. F. D a n u s s о, Polymer, 3, 423 A962).
2. P. J. Flory, Principles of Polymer Chemistry, Ithaca —New York, 1953.
3. P. J. Flory, Lectures in Materials Science, New York, 1963, p. 27.
4. P. J. Flory, Protein Structure and Function, Brookhaven Symp. Biol.,
13, 89 A960).
5. E. B. Wilson Jr., in «Advances in Chemical Physics», vol. II, New
York, 1959.
6. S. Mizushima, Structure of Molecules and Internal Rotation, New
York, 1954.
7. M. В. В о л ь к е н ш т е й н, J. Polymer Sci., 29, 441 A958).
8. Н. A. Kramers, G. H. Wannier, Phys. Rev., 60, 252 A941).
9. G. F. Newell, E. W. Mont roll, Revs. Mod. Phys., 25, 353 A953).
10. T. G. Fox, Jr., P. J. Flory, J. Am. Chem. Soc, 73, 1909 A951).
11. H. L. Wagner, P. J. Flory, J. Am. Chem. Soc, 74, 195 A952).
12. W. R. К rigb a urn, J. Polymer Sci., 28, 213 A958).
13. P. J. Flory, L. Mandelkern, J. Kin singer, W. B. Schultz,
J. Am. Chem. Soc, 74, 3364 A952).
14. L. Mandelkern, P. J. Flory, J. Am. Chem. Soc, 74, 2517 A952).
15. M. L. Hunt, S. Newman, H. A. Scheraga, P. J. Flory, J. Phys.
Chem., 60, 1278 A959).
16. G. Natta, P. Corradini, Rubber Chem. Technol., 33, 703 A960).
17. С W. Bunn, D. R. Holmes, Disc. Faraday Soc, 25, 95 A958).
18. A. M. L i q u о г i, Acta Cryst., 8, 345 A955).
19. С W. В u n n, E. R. H о w e 11 s, Nature, 174, 549 A954).
20. L. Pauling, R. B. Corey, H. R. Branson, Proc Natl. Acad. Sci.
U. S., 37, 205 A951).
21. P. M, С о w a n, S. M с G a v i n, Nature, 176, 501 A955).
29
22. J. D. Watson, F. H. Crick, Nature, 171, 737, 964 A953); Proc. Roy.
Soc. (London), Ser. A, 223, 80 A954).
23. M. H. F. Wilkins, A. R. Stokes, H. R. Wilson, Nature, 171, 738
A953).
24. G. Natta, P. Corradini, P. G a n i s, J. Polymer Sci., 58, 1191
A962).
25. C. W. В u n n, Chemical Crystallography, London, 1946.
26. L. Mandelkern, С R. Worth ington, A. S. Posner, Sci., 127,
1052 A958).
27. A. S. Posner, L. Mandelkern, С R. Worth ington, A. F. D i o-
rio, J. Appl. Phys., 31, 536 A960); 32, 1509 A961).
28. R. S. Stein, R. H. Doremus, B. W. Roberts, D. Turnbull
(eds.), «Growth and Perfection of Crystals», New York, 1958, p. 549.
29. F. P. Price, J. Polymer Sci., 37, 71 A959).
30. R. Eppe, E. W. Fischer, H. A. Stuart, J. Polymer Sci., 37, 721
A959).
,ава Плавление
гомополимеров
Введение
При переходе чистого гомополимера из кристаллического
(или частично кристаллического) в аморфное состояние его
физические и механические свойства, морфологические и
структурные характеристики и термодинамические параметры
претерпевают соответствующие изменения. Так, например, в
кристаллическом состоянии гомополимер представляет собой твердое
высокопрочное вещество, в то время как в расплавленном
состоянии он может уже приобрести свойства жидкости с низкой
текучестью. Однако, если молекулярный вес образца достаточно
высок, расплав приобретает каучукоподобные свойства (высокую
эластичность). Влияние кристалличности на механические
свойства выражается в понижении модуля упругости после плавления
в 103—105раз; в частности, механическую прочность волокон
можно объяснить наличием в них ориентированных
кристаллических участков.
В образцах с хорошо развитой кристалличностью рассеяние
рентгеновских лучей под большими углами дает четкие
рефлексы от большого числа плоскостей, которые накладываются на
диффузное рассеяние. После плавления эти рефлексы исчезают,
и остается только широкое гало. Процессу перехода обычно
сопутствуют также четкие изменения в инфракрасных спектрах.
Кроме того, наблюдаются скрытые изменения энтальпии и
объема, которые обычно связывают с фазовым переходом.
Резкие различия в свойствах между двумя состояниями
полимеров аналогичны тем, которые возникают при плавлении
кристаллов низкомолекулярных соединений. Последний процесс
достаточно хорошо изучен и формально может быть описан как
фазовый переход первого рода. К такому типу переходов
применимы законы фазового равновесия. Совершенно общая
термодинамическая природа этих законов и возможность их
использования для объяснения широкого круга явлений позволяет
установить, носит ли наблюдаемое подобие между процессами
31
кристаллизации и плавления низкомолекулярных соединений и
полимеров чисто внешний характер, или же оно действительно
отражает различное проявление одного и того же процесса.
Теоретическое описание фазового перехода первого рода для
однокомпонентнои системы при постоянном давлении сводится
к тому, что температура перехода не зависит от относительного
содержания какой-либо из находящихся в равновесии фаз.
Плавление прерывно, т. е. носит бесконечно резкий характер;
температура плавления Тпл является характеристической величиной.
Она выражается известной формулой
где АЯ-и AS — энтальпия и энтропия плавления.
Абсолютно резкий переход может наблюдаться на опыте
лишь при условии, что система обладает вполне совершенным
внутренним порядком в кристаллической фазе. Более того,
необходимо также, чтобы кристаллы были больших размеров: при
этом понижается до минимума избыточная составляющая
свободной энергии, обусловленная наличием поверхностей раздела
(или перехода) между двумя фазами. Эта поверхностная
энергия повышает свободную энергию плавления, что очень важно
при плавлении реальных полимерных кристаллов. Отклонения
от определенных таким образом идеальных условий должны
неизбежно приводить к расширению температурного интервала
плавления.
Сформулированный критерий применим в равной степени к
кристаллам любых соединений. Длинные цепные молекулы,
упакованные в совершенную решетку у кристаллитов достаточно
больших размеров, находятся в типично кристаллическом
состоянии. Тот факт, что одна молекула может быть заключена во
множестве элементарных ячеек, не влияет на это заключение.
Поэтому можно ожидать, что процесс плавления подобных
систем, как и любых других кристаллических веществ, является
фазовым переходом первого рода. Разумеется это
предположение не имело бы никакой силы, если бы оно не подтвердилось
экспериментом и строгой молекулярной теорией.
Очевидно, чрезвычайная сложность кристаллического
состояния полимеров объясняется их поликристаллическим характером
и тем, что кристаллизующиеся звенья не являются
изолированными структурными элементами, а ковалентно связаны в цепные
молекулы, содержащие многие тысячи звеньев. Наблюдения
выявляют существование множества различных типов
надмолекулярного порядка в полимерах [1—3]. Однако это совсем не
означает, что кристаллическое состояние характеризуется набором
промежуточных степеней упорядоченности между жидкостью и
совершенным кристаллом. Если бы такой набор соответствовал
32
равновесному состоянию, было бы вообще бессмысленно
говорить о кристаллической фазе в полимерах или пытаться
систематически исследовать ее переход в жидкое или аморфное
состояние. Вполне реальное существование «морфологического
спектра» обусловлено чисто кинетическими причинами, в первую
очередь принципиальной невозможностью разрешения
«конфликта» между требованиями равновесия, с одной стороны, и
конечной скорости кристаллизации, с другой. В результате
этого конфликта возникают различные мезоморфные и метаста-
бильные состояния.
Чтобы упорядоченные области кристаллического полимера
можно было рассматривать как отдельную фазу, должны
удовлетворяться обычные термодинамические условия. Химический
потенциал чистой фазы одного компонента должен быть
постоянным во всей этой фазе и зависеть только от температуры
и давления. Совершенно ясно, что в слабо развитых
кристаллических системах эти условия не выполняются независимо от
того, состоит ли система из связанных полимерных сегментов или
свободных мономерных звеньев. В подобных условиях
химический потенциал зависит также от степени упорядоченности и
раз;.:ера кристаллитов. Вопрос, в какой мере можно
приблизиться к идеальному кристаллическому состоянию, должен
решаться экспериментально по узости температурного интервала
плавления и воспроизводимости температуры плавления.
Именно эти вопросы, имеющие фундаментальное значение для
понимания свойств кристаллических полимеров, мы прежде всего и
рассмотрим.
Природа процесса плавления
Опыты, предназначенные для изучения процесса плавления
и выяснения природы этого перехода, должны проводиться в
таких условиях, чтобы обеспечивалось наиболее полное
приближение к состоянию равновесия. Самое общее рассмотрение
факторов, влияющих на образование кристаллической фазы из
расплава, показывает, что это требование наилучшим образом
выполняется, если кристаллизация осуществляется очень
медленно при температуре, близкой к температуре плавления, или
если уже образовавшаяся кристаллическая фаза подвергается
длительному отжигу при повышенных температурах. В
подобных экспериментах должны измеряться такие
термодинамические параметры, которые чувствительны к очень малым
изменениям степени кристалличности.
Многие общепринятые для низкомолекулярных веществ
методы оказываются совершенно неприменимыми для определения
температуры плавления полимеров [4]. В частности, во многих
3 Л. Манделькерн
33
ед
15
с;
<3
:Ю
случаях, когда эти методы основаны на определениях изменении
физических или механических свойств полимеров, они
оказываются недостаточно чувствительными, и поэтому измеренная
температура плавления является лишь кажущимся подобием
искомой величины.
При изучении плавления любых веществ широко
используются измерения теплоемкости. В принципе этот метод может дать
ценную термодинамическую информацию. Данные Вундерлиха
и Доля [5] для образцов линейного полиэтилена (рис. 12)
типичны для иенабухшего гомополимера. В этом опыте перед
измерениями расплав полимера медленно
охлаждался. Характер кривой на
рис. 12 напоминает Я-переход,
присущий превращениям типа
порядок— беспорядок в бинарных
сплавах. Теплоемкость быстро возрастает
в интервале 120—137° С, достигает
максимума, стремительно падает
и затем принимает постоянное
значение. Для идеального фазового
перехода первого рода в одноком-
понентиой системе теплоемкость
при температуре перехода должна
обращаться в бесконечность.
Поскольку на опыте это не
наблюдается, напрашивается заключение, что
плавление полимерных систем не
может рассматриваться как
фазовый переход первого рода.
Однако, как следует из детального рассмотрения [6],
классификация перехода только по форме кривой плавления по
меньшей мере затруднительна. Всегда существует вполне
определенная возможность, что область перехода заметно сузится при
дополнительном отжиге или при осуществлении режима очень
медленного нагрева. Вполне вероятно также и то, что при
сужении температурного интервала, в котором ведутся измерения,
произойдет больший подъем теплоемкости. Поэтому описание
процесса и классификация перехода на основе поверхностного
анализа кривой плавления в равной мере затруднительны и
произвольны. Естественно, что требуется более детальное изучение
плавления, включающее исследование влияния конкретных
условий кристаллизации и последующего отжига на температуру
фазового перехода.
Важность подобной постановки вопроса выяснилась
благодаря работам Вуда и Беккедаля [7] — пионеров изучения
кристаллизации и плавления натурального каучука. Они показали,
что, если вслед за кристаллизацией проводить плавление при
80 100 120 1UQ
Т,°С
Рис. 12. Зависимость
теплоемкости от температуры для
линейного полиэтилена Мар-
лекс-50 [5].
34
Рис. 13. Зависимость температурного
интервала плавления натурального каучука
от температуры кристаллизации [7а].
больших скоростях нагрева (порядка 0,1 град!мин),
наблюдаемая температура плавления зависит от условий кристаллизации.
В частности, температура плавления оказывается функцией
температуры кристаллизации.
Кривые плавления, полученные названными авторами на
образцах, подвергнутых изотермической кристаллизации при
различных температурах, представлены на рис. 13. Найденные
температуры плавления
изменяются в интервале 0—
30° С, возрастая с
увеличением температуры
кристаллизации. Качественно
подобные же результаты
получены и для ряда
других полимеров, когда
кристаллизация и плавление
проводились
аналогичным образом [8—10].
Тот факт, что
температура кристаллизации
оказывает решающее
влияние на температуру
плавления, еще не может сам
по себе служить доказательством отсутствия равновесного
кристаллического состояния и равновесной температуры плавления
полимера. Установлено, что, если вслед за кристаллизацией
натурального каучука [11] и других полимеров [9—13]
осуществляется режим очень медленного нагрева, можно получить
вполне воспроизводимую температуру плавления, независящую от
термической предыстории образцов. В частности, температура
плавления не зависит от условий кристаллизации, в том числе
и от температуры кристаллизации, и всегда значительно выше,
чем наблюдаемая при больших скоростях нагрева.
Полученные результаты можно объяснить тем, что при
температурах кристаллизации, значительно ниже температуры
плавления, упоминавшиеся кинетические факторы обусловливают
морфологические изменения или ограничения, затрагивающие
совершенство и размеры кристаллических областей и
понижающие их термодинамическую стабильность. Эти эффекты не
могут быть устранены применением высоких скоростей подъема
температуры. Следовательно, экспериментальные данные,
полученные в таких условиях, нельзя интерпретировать с позиций
равновесного состояния. Однако, если после кристаллизации
образец нагревать очень медленно, может уже проявиться
влияние процессов отжига и перекристаллизации. Приведенные выше
экспериментальные результаты показывают, что в данном
случае удается приблизиться к условиям равновесия.
3*
35
1,04
Щ
0,960\
ОШ
от
50
юо
Для большинства полимеров простым и точным методом
изучения процесса плавления являются измерения удельного
объема в зависимости от температуры. Достоинство этого метода —
в возможности строгого контроля температуры при очень
медленном нагревании.
На рис. 14 приведены некоторые характерные результаты,
полученные описанным методом для плавления гомополиме-
ров различных типов. В этих опытах
процесс кристаллизации никак не
контролировался, но при плавлении
осуществлялся медленный подъем
температуры. Чувствительные методы
регистрации показали, что, если образец
длительное время выдерживается при
температуре ниже точки плавления, все
же происходит частичное плавление и
перекристаллизация. На рис. 14
указаны окончательные значения
удельных объемов, полученные после
выдержки образца при соответствующей
температуре около 24 ч. При
выполнении этого условия плавление
происходит очень резко, и основная часть
кристаллической фазы исчезает в узком
интервале 3—4°.
Температура, при которой исчезают
последние следы кристалличности,
хорошо определяется: наиболее
существенно при этом то, что в точке плавления на кривых
зависимости удельного объема от температуры наблюдается резкий
излом. Эта точка должна, следовательно, соответствовать
исчезновению наиболее упорядоченных областей, которые
образовались в конкретных условиях кристаллизации при всех
кинетических ограничениях; возможно, однако, что часть этих
областей образовалась или доупорядочилась во время проведения
опыта (плавления). Основываясь на подобных результатах,
можно предположить, что, если при изменениях теплоемкости с
температурой придерживаться режима очень медленного
нагревания, сходство плавления с переходом Я-типа исчезает.
Другой возможный путь приближения к равновесным
условиям заключается в проведении кристаллизации из расплава при
постоянной температуре, по возможности близкой к температуре
плавления. В подобных опытах неизбежна длительная
кристаллизация, необходимая для достижения предельно высоких
степеней кристалличности [14].
Экспериментальные результаты [15] для нефракционирован-
ного линейного полиэтилена Марлекс-50 приведены на рис. 15.
150
1°С
Рис. 14. Зависимость
удельного объема от
температуры для гомополимеров
различных типов [4]:
/— 'полиметилен; 2—полиокси-
этилен. 3—полидекметиленадипат.
36
Кривая / соответствует образцу, который перед исследованием
был закристаллизован из расплава в режиме, аналогичном
примененному при измерениях теплоемкости, показанному на рис. 12.
Полученный в этих условиях удельный объем образца
составляет при 25° С 1,041 см3/г. При очень медленном подъеме
температуры, порядка Г в день, в ходе нагрева успевает
происходить частичное плавление и перекристаллизация, как это уже
наблюдалось ранее для других гомополимеров. Процесс
плавления относительно резок,
причем последние следы
кристалличности опять
исчезают при хорошо
определяемой температуре,
равной 137,5±0,5°С. Эта
температура на несколько
градусов выше
определенной методом измерения
теплоемкости для того же
самого полимера, что
обусловлено, бесспорно,
значительно более
медленным подъемом
температуры, применяемом при
измерений удельных
объемов.
Кривая 2 на рис. 15
соответствует процессу
плавления того же
полимера, но предварительно
кристаллизовавшегося из
расплава в течение 40 дней при 130° С, т. е. на 7,5 град ниже
температуры плавления, и затем медленно (за 24 ч) охлажденного
до комнатной температуры. При соблюдении таких условий
кристаллизации удельный объем получается равным уже 1,018 см3/г
при 25° С, что соответствует более высокой степени
кристалличности и, по-видимому, развитию более близких к совершенным
кристаллитов. В этом случае при предшествующем плавлению
медленном нагреве процесс частичного плавления и
перекристаллизации сведен до минимума. Тем не менее, область
плавления существенно сужается, хотя значение самой температуры
плавления не изменяется.
Следовательно, можно показать, что в случае очень
медленной кристаллизации или очень медленного плавления или того
и другого вместе взятых существует вполне определенная
температура плавления, при которой исчезают последние следы
кристалличности. Эта температура хорошо воспроизводима и не
зависит от условий кристаллизации и термической предыстории
щ
1}25о\
- 1,20о\
1fl50\
WOO
2
-20 0 20 40 60 80 100 120 М 160 16
Т,°С
Рис. 15. Зависимость удельного объема
от температуры для нефракционированного
линейного полиэтилена Марлекс-50 [15]:
/ — перед плавлением образец очень медленно
охлаждали от расплавленного состояния до
комнатной температуры; 2—образец кристаллизовали в
течение 40 дней при 130* С и затем охлаждали до
комнатной температуры.
37
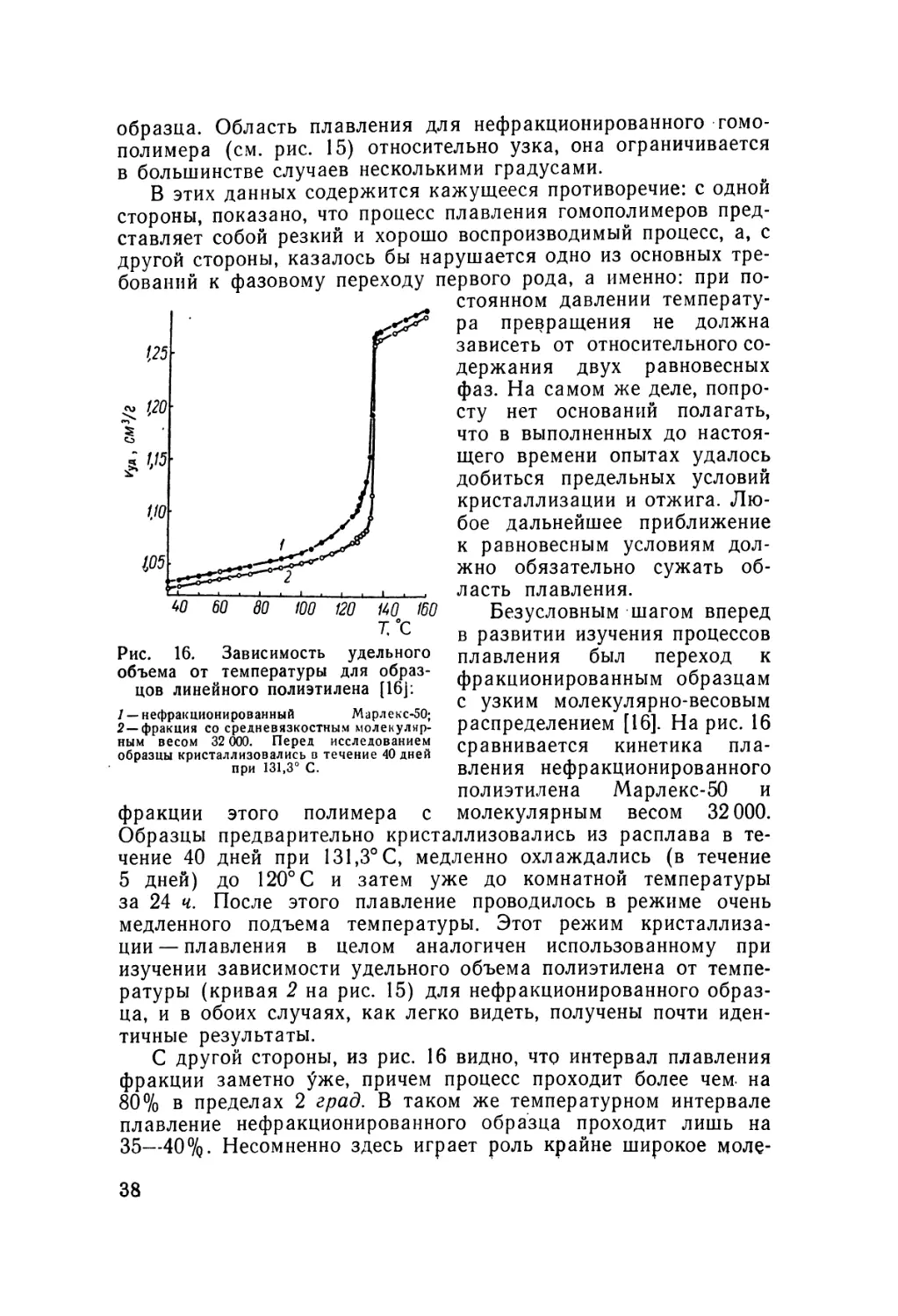
образца. Область плавления для нефракционированного гомо-
полимера (см. рис. 15) относительно узка, она ограничивается
в большинстве случаев несколькими градусами.
В этих данных содержится кажущееся противоречие: с одной
стороны, показано, что процесс плавления гомополимеров
представляет собой резкий и хорошо воспроизводимый процесс, а, с
другой стороны, казалось бы нарушается одно из основных
требований к фазовому переходу первого рода, а именно: при
постоянном давлении
температура превращения не должна
зависеть от относительного
содержания двух равновесных
фаз. На самом же деле,
попросту нет оснований полагать,
что в выполненных до
настоящего времени опытах удалось
добиться предельных условий
кристаллизации и отжига.
Любое дальнейшее приближение
к равновесным условиям
должно обязательно сужать
область плавления.
Безусловным шагом вперед
в развитии изучения процессов
плавления был переход к
фракционированным образцам
с узким молекулярно-весовым
распределением [16]. На рис. 16
сравнивается кинетика
плавления нефракционированного
полиэтилена Марлекс-50 и
молекулярным весом 32 000.
Образцы предварительно кристаллизовались из расплава в
течение 40 дней при 131,3° С, медленно охлаждались (в течение
5 дней) до 120° С и затем уже до комнатной температуры
за 24 ч. После этого плавление проводилось в режиме очень
медленного подъема температуры. Этот режим
кристаллизации— плавления в целом аналогичен использованному при
изучении зависимости удельного объема полиэтилена от
температуры (кривая 2 на рис. 15) для нефракционированного
образца, и в обоих случаях, как легко видеть, получены почти
идентичные результаты.
С другой стороны, из рис. 16 видно, что интервал плавления
фракции заметно уже, причем процесс проходит более чем- на
80% в пределах 2 град. В таком же температурном интервале
плавление нефракционированного образца проходит лишь на
35—40%. Несомненно здесь играет роль крайне широкое моле-
Щ 60 80 ШО 120 140 160
т/с
Рис. 16. Зависимость удельного
объема от температуры для
образцов линейного полиэтилена [16]:
1 — нефракционированный Марлекс-50;
2 —фракция со средневязкостным
молекулярным весом 32 000. Перед исследованием
образцы кристаллизовались в течение 40 дней
при 131,3° С.
фракции этого полимера с
38
кулярно-весовое распределение полиэтилена Марлекс-50 [17],
причем особое значение имеет высокое содержание
низкомолекулярных фракций. Это подтверждается тем фактом\ что у
закристаллизованной в аналогичных условиях
высокомолекулярной фракции (М = 490 000) процесс плавления чрезвычайно
резок. Последние следы кристалличности для этой фракции
исчезают при 138,7° С, т. е. выше температуры плавления не-
фракционированного образца A37,5°С).
Кривые плавления, характерные для образцов с узким моле-
кулярно-весовым распределением, уже вполне сравнимы с
полученными для низкомолекулярных соединений. Даже у
последних плавление может стать из прерывного более диффузным
либо при быстром охлаждении образцов из расплава до
неравновесных замороженных состояний, либо при наличии примесей.
Различие между полимерами и низкомолекулярными
соединениями, следовательно, является скорее количественным, чем
качественным. В случае полимерных систем необходима особо
тщательная постановка опытов для устранения возмущающих
кинетических и релаксационных факторов и максимально
возможного приближения к равновесным условиям.
Конечная длина любой реальной полимерной молекулы
вносит в систему некоторый элемент беспорядка, обусловленный
концами цепи. Кинетическое влияние этих концов аналогично
роли примесей в низкомолекулярном веществе. Если конечные
группы не удается упаковать в кристаллическую решетку,
температурный интервал плавления не может бесконечно сужаться
даже при идеальных условиях кристаллизации и отжига.
Однако, вопреки ожиданиям, последние следы кристалличности
всегда исчезают при строго определенной температуре, так как
плавление гомоиолимеров практически неизбежно должно
происходить в небольшом, но конечном температурном интервале.
Это следует из весьма тщательно проведенных экспериментов.
Согласно требованиям теории предельная температура
определяется как равновесная температура плавления Тпл. Она
представляет собой температуру плавления гипотетического
совершенного макроскопического кристалла *. В предельном
(теоретически) случае гомогенного полимера бесконечно высокого
молекулярного веса, образующего идеально упорядоченную
кристаллическую фазу, плавление должно быть прерывным и
происходить при строго определенной температуре. Теоретически
'даже диффузный процесс плавления может рассматриваться как
фазовый переход первого рода [18]. Следует отметить, что для
* Здесь под термином «совершенный» понимается кристалл с самой
низкой свободной энергией. Так как даже в равновесных условиях
кристаллическая решетка может содержать определенные дефекты, он не обязательно
должен представлять собой морфологически идеальный кристалл с
абсолютно совершенной внутренней структурой,
39
линейного полиэтилена эта температура хорошо согласуется с
температурой плавления гипотетического высокомолекулярного
«-парафина, полученной экстраполяцией данных для
низкомолекулярных гомологов [13, 19—21].
Чтобы между двумя макроскопическими фазами одноком-
понентной системы поддерживалось равновесие при постоянной
температуре, давление не должно зависеть от объема, т. е. от
относительного содержания той или иной фазы. Зависимость
удельного объема от давления при различных температурах для
1860
1240
а
w
If 1,2
vyA, см3/г
Рис. 17. Зависимость удельного объема от давления при
различных температурах для линейного полиэтилена Мар-
лекс-50 [22].
полиэтилена приведена на рис. 17 [22]. Постоянство давления
при изменении объема, характерное для фазового перехода
первого рода, соблюдается при температуре перехода. Поэтому,
безотносительно к природе изучаемого образца, можно
заключить, что фазы должны находиться в равновесии. В данном
случае, это, разумеется, кристаллическая и аморфная (жидкая)
фазы полиэтилена.
Поскольку экспериментальные результаты подтверждают
предположение о том, что превращение кристалл — жидкость в
полимерах обладает характеристиками фазового перехода
первого рода, любые количественные описания следует вести в
терминах такого фазового перехода. Этот важный вывод вытекает
из формального анализа экспериментальных данных и не
требует конкретизации структурных особенностей какой-либо из
40
фаз. Обоснованность данного заключения может быть
подвергнута и более строгому анализу, если распространить концепцию
фазового равновесия на кристаллические сополимеры и
многокомпонентные системы и принять во внимание влияние
деформации на кристаллизацию и плавление. В следующих главах
дается более подробный анализ этих, казалось бы, различных
проблем с единой точки зрения.
Перечисленные выводы прямо противоположны сделанным
при одностороннем морфологическом изучении процессов
плавления [2, 22]. Как уже отмечалось ранее, изучение плавления
требует термодинамического анализа. Этот анализ показывает,
что при температуре плавления между жидкой и
кристаллической фазами существует равновесие. В принципе, однако, нет
никакой причины, которая препятствовала бы развитию и
поддержанию равновесия между двумя фазами при конечных
степенях кристалличности. Поэтому можно совершенно однозначно
определить свойства кристаллической фазы, если только
принять во внимание, что должны соблюдаться условия равновесия.
Однако в случае кристаллизации в неравновесных условиях
однозначное определение свойств кристаллической фазы уже
невозможно. Структура этой фазы зависит теперь от способа
развития кристалличности. Достижение высоких степеней
кристалличности, отвечающих близким к равновесным условиям,
представляет собой трудную задачу, для решения которой нужно
согласовать условия опыта с возможным механизмом
кристаллизации. Нетрудно убедиться, что мы здесь снова встречаемся
с проблемой, общей для кристаллизации как полимеров, так и
низкомолекулярных соединений, и все различия носят чисто
количественный характер.
«Извитый» характер свободных цепей макромолекул привел
в прошлом к интуитивному предположению, что кристаллическое
состояние с наименьшей свободной энергией реализуется при
значительном содержании в системе аморфных областей. Это
заключение основано на том, что понижение энтальпии в такой
гетерогенной системе полностью компенсируется выигрышем
энтропии, обусловленным конформационной лабильностью
«аморфных» участков цепей. Из этого следует, что по мере повышения
температуры, «кристаллические» сегменты цепи будут
постепенно переходить в аморфное состояние, и аморфные области
будут расти. Процесс плавления должен был бы при этом
осуществляться непрерывно в широком интервале температур.
Хотя гипотеза и кажется правдоподобной, если исходить из свойств
единичной цепи, она полностью противоречит реальным
наблюдениям кристаллизации и плавления полимеров в блоке.
Интерпретация этого процесса требует использования
методов статистической механики. Это, в свою очередь, требует
определенной молекулярной модели для описания системы.
41
Теоретические расчеты были выполнены для двух совершенно
различных моделей [24, 25]. В первом случае рассматриваемая
молекулярная цепь могла лишь один раз проходить через
произвольный кристаллит. Во втором случае принималось, что
данная цепь входит в кристаллит многократно; разумеется, что
линейные размеры такого кристаллита много меньше длины этой
цепи.
Общим для обеих моделей является предположение, что
процесс кристаллизации носит трехмерный характер. В обоих
случаях расчет приводит к заключению, что превращение
кристалл — жидкость представляет собой фазовый переход первого
рода. Теория предсказывает, что плавление происходит резко
и прерывно при строго определенной температуре. Играющая
существенную роль в упоминавшемся интуитивном подходе к
плавлению энтропийная составляющая, обусловленная
неупорядоченными сегментами цепи при температурах, ниже Гпл,
никак не проявляется. Тщательный анализ показывает, что этот
результат является прямым следствием трехмерной природы
кристаллита. Требования, чтобы конечные звенья
кристаллических последовательностей (будь то последовательности,
образованные многими цепями или одной цепью) все лежали в
плоскости, перпендикулярной оси цепей, настолько строго, что в
условиях равновесия фаз концентрация звеньев в аморфных
областях должна быть существенно ограничена. Степень
кристалличности A-Х) будет очень высока, приближаясь к единице,
тогда как равновесная длина кристаллита будет приближаться
к размерам вытянутой макромолекулы.
Эти расчеты объясняют, почему плавление полимеров
является относительно резким процессом, который можно
рассматривать как фазовый переход первого рода. Однако можно
ожидать, что большая равновесная длина кристаллита, которая
должна устанавливаться ниже температуры плавления, на
самом деле трудно достижима. Если же длина кристаллита ? не
достигает своего равновесного значения, то оптимальная
устойчивость системы понижается. При этих условиях [241:
где In D = 2o/RT (а обозначает избыток свободной энергии на
моль звеньев, образующих концы кристаллита).
Так как Т соответствует температуре плавления
кристаллитов длины ?, из формулы B) видно, что понижение температуры
плавления по сравнению с равновесной тем меньше, чем
больше величина ?. Формула B) вполне аналогична полученной при
расчетах температуры плавления кристаллитов, ограниченных
в продольном направлении длиной ?, но никак не ограниченных
42
в поперечном направлении (перпендикулярном оси цепи).
Понижение температуры плавления обусловлено существованием
избыточной свободной энергии концов кристаллита, вносящей свой
вклад в полную свободную энергию плавления. С другой
стороны, образование и развитие кристаллической фазы зависит
от кинетических факторов, могущих непосредственно влиять на
величину ?. Поэтому относительно низкие неравновесные
температуры плавления, наблюдаемые особенно в режиме быстрого
нагрева, могут отражать также чисто кинетические ограничения
величины ?.
Влияние молекулярного веса
После изложенного выше понятно, что влияние
молекулярного веса на равновесный процесс плавления непосредственно
проявляется через концентрацию концевых звеньев; при этом не
имеет значения, участвуют ли эти звенья в кристаллизации или
нет. Если концы цепи не входят в кристаллит по стерическим
ограничениям, то они могут рассматриваться как инородная
примесь, которая ведет себя подобно некристаллизующемуся
компоненту. Тогда можно вывести простое соотношение между
температурой плавления и степенью полимеризации [24]. Если
концы цепи входят в кристаллическую фазу, следует различать
несколько возможностей их расположения. Например,
концевые звенья могут быть статистически распределены в пределах
фазы, либо попарно занимать смежные ячейки. Последнее
соответствовало бы структуре молекулярных кристаллов,
образованных w-алканами, когда концевые звенья цепей образуют четкие
кристаллографические плоскости.
Для системы, обладающей первоначально «наиболее
вероятным» молекулярно-весовым распределением
Wj^xQ-pfp*-1 C)
концевые группы распределены статистически вдоль
параллельно упакованных цепей.
В формуле C):
wx — весовая доля молекул, состоящих из х звеньев;
р-— параметр, представляющий собой вероятность присо
единения нового звена к растущей цепи и. имеющий
смысл степени завершенности реакции полимеризации.
Если статистически распределенные «инородные» звенья
исключаются из кристаллитов, то прямой термодинамический
расчет [24, 26] позволяет установить, что:
J \-= -*—!- D)
^ол Т°ал Д/Ум хп
43
гДе Т'пл—температура плавления образца, имеющего средне-
численную степень полимеризации хп\
ГЦЛ — температура плавления «чистого» (концевые звенья
не учитываются) полимера с бесконечно высоким
молекулярным весом;
Д//м— теплота плавления на мономерное звено цепи.
Величина 2/хп представляет собой молярную долю некри-
сталлизующихся звеньев, а сама формула D) отражает
условия фазового равновесия для этого случая.
Согласно формуле D), по мере возрастания хп резко
уменьшается влияние молекулярного веса полимера на его Гпл.
Однако лишь тщательный
экспериментальный анализ зависимости
температуры плавления
относительно низкомолекулярных
полимеров от молекулярного веса
позволит заключить, в какой мере
верно предположение, что концы
цепей исключаются из
кристаллической фазы.
Ивэнс, Майтон и Флори [8]
изучали температуры плавления
образцов полидекаметиленадипа-
та с наиболее вероятным молеку-
лярно-весовым распределением.
Некоторые характерные
результаты этой работы представлены
на рис. 18 в форме,
соответствующей формуле D). Видно, что это
соотношение удовлетворяется вплоть до самых низких
значений хп (олигомеры). Более того, величина Д#м, вычисленная по
наклону прямой на рис. 18, хорошо согласуется с Д#м,
полученными другими методами. Хотя регистрируемая температура
плавления и связана с исчезновением кристаллитов, образованных
молекулами с относительно большим молекулярным весом, она
однозначно зависит от хп-
Данные рис. 18 относятся к полиэфирам, молекулы которых
имеют на концах гидроксильные и карбоксильные группы. Были
также приготовлены низкомолекулярные полимеры декамети-
лен-адипината с массивными концевыми группами, такими, как
бензоат, а-нафтоат и циклогексил, и для этих моделей также
была определена температура плавления. В этом случае
результаты измерений также согласуются с формулой D). В
пределах ошибок эксперимента рассчитанные значения Д#м
совпадают между собой независимо от природы и размеров концевых
групп. Так как очень мало вероятно, что массивные группы
входят в кристаллит, можно заключить, что концевые звенья, неза-
Щ
зщ
щ
W
60
70 кя
]80
'0,2 0/* 0,6 0,8
f/xn
Рис. 18. Зависимость температуры
плавления от среднечисленной
степени полимеризации хп для
полиметиленадипината с гидрок-
сильными и карбоксильными
концевыми группами, представленная
согласно формуле D).
44
висимо от их размеров, как правило, не входят в кристаллиты.
Этот вывод подтверждается характером зависимости Тил от
средней степени полимеризации.
В случае узких фракций расчет зависимости Тил от
молекулярного веса усложняется, так как теперь уже концевые звенья
не распределены абсолютно беспорядочно в системе
параллельно упакованных цепей. Для модели, в которой цепь только один
раз проходит через данный кристаллит, найдено [24], что:
_i__4-=—.i±i E)
Гпл Tl ДЯМ х
где 6 = [1-(?<>-ВД-1;
С0 —равновесная длина кристаллита.
Таким образом, формулы D) и E) показывают, что за
исключением образцов с крайне низким молекулярным весом
Гдл практически не зависит от степени полимеризации. При
обычно наблюдаемых значениях Д//м зависимость Тпл от
молекулярного веса должна практически исчезнуть при степенях
полимеризации порядка нескольких сотен. Это было показано
как на полиэфирах и полиамидах [8] с наиболее вероятным мо-
лекулярно-весовым распределением, так и на узких фракциях
полиэтиленсукцината [27] и полиэтилена [28].
Литература
1. С. S. Fuller, W. О. Baker, J. Chem. Educ, 20, 3 A943); W. С.
Baker, С. S. Fuller, N. R. Pape, J. Am. Chem. Soc, 64, 776 A942).
2. H. A. S t u a r t, Ann. N. Y. Acad. Sci., 83, 3 A959).
3. H. W. Wycof f, J. Polymer Sci., 62, 83 A962).
4. L. M a n d e 1 k e г n, Chem. Rev., 56, 903 A956).
5. B. W underlie h, M. Dole, J. Polymer Sci., 24, 201 A957).
6. J. F. Mayer, S. F. Streeter, J. Chem. Phys., 7, 1019 A939).
7a. L. A. Wood, N. Bekkedahl, J. Appl. Phys., 17, 362 A946).
7b. L. A. Wood, N. Bekkedahl, J. Res. Natl. Bur. Stand., 36, 489 A946).
8. R. D. Evans, H. R. M i g h t о n, P. J. F 1 о r y, J. Am. Chem. Soc, 72,
2018 A950).
9. P. J. F1 о г у, L. Mandelkern, H. K. Hall, J. Am. Chem. Soc. 73,
2532 A951).
10. W. E. Mochel, J. T. Maynard, J. Polymer Sci., 13, 235 A954).
11. D. E. Roberts, L. M a n del kern, J. Am. Chem. Soc, 77, 781 A955).
12. L. M a n d e 1 k e r n, F. A. Q u i n n, Jr., D. E. Roberts, J. Am. Chem.
Soc, 78, 926 A956).
13. F. A. Quinn, Jr., L. Man del kern, J. Am. Chem. Soc, 80, 3178
A958).
14. L. Mandelkern, in R. H. Do rem us, B. W. Roberts, D. Turn-
bull (eds.), «Growth and Perfection of Crystals», New York, 1958, p. 499.
15. LMandelkern, Rubber Chem. Technol., 32, 1392 A949).
16. R. Chiang, P. J. Flory, J. Am. Chem. Soc, 83, 2857 A961),
45
IV. L. Tung, J Polymer Sci., 24, 333 A957).
18. A. J. Rutgers, S. A. Wouthuysen, Physica, 4, 515 A937).
19. W. E. Garner, K. van Bibber, A. M. King, J. Chem. Soc, 1931,
1533.
20. К. Н. Meyer, A. J. van d e r W у к, Helv. chim acta, 20, 83 A937).
21. M. Broadhurst, J. Chem. Phys., 36, 2578 A962).
22. S. M a t s u о к a, J. Polymer Sci., 42, 511 A960).
23. В. А. Каргин, Г. Л Слонимский, Усп. хим., 24, 785 A955).
24. P. J. Flory, J. Chem. Phys., 17, 223 A949).
25. P. J. F 1 о г у, J. Am. Chem. Soc, 84, 2857 A9.62).
26. P. J. Flory, Principles of Polymer Chemistry, Ithaca— New York, 1953,
p. 568.
27. K. U e b e r r e i t e r, G. К a n i g, A. S. Brenner, J. Polymer Sci., 16,
53 A955).
28. L. H. Tung, S. Buckser, J. Phys. Chem., 62, 1530 A958).
Глава 3 Плавление
гомополимеров
в присутствии
низкомолекулярных
жидкостей
Введение
Действительно ли плавление полимерной системы
представляет собой фазовый переход первого рода, может быть
установлено при изучении влияния добавки низкомолекулярного
компонента на этот переход. На основании формальных
рассуждений можно совершенно строго получить соответствующие
соотношения, используя классические методы. Мы обсудим эту
проблему подробно, поскольку можно строго охарактеризовать
рассматриваемую систему и сделать количественные
предсказания о ее поведении. Для удобства сравнения целесообразно
рассматривать отдельно более концентрированные системы, т. е.
набухшие полимеры, и разбавленные растворы. В последнем
случае необходимо также различать сравнительно гибкие
цепные молекулы и молекулы с «жесткостью формы»,
характеризующиеся крайне асимметричными конформациями.
Концентрированные системы
Концентрированные полимерные системы характеризуются
статистическим распределением сегментов цепей по объему
раствора или гетерогенной смеси. Свободная энергия смешения
полимера и растворителя в аморфном состоянии в этом случае
хорошо описывается формулой Флори — Хаггинса [1, 2], которая
позволяет вычислить разность химических потенциалов
растворителя в присутствии полимера \iu и чистого растворителя \хЬ
fxj - ц° = ЯГ [in A - v2) + (l _ -L) w2 + Xl 1,2] F)
где Vo — объемная доля полимера в смеси;
х—степень полимеризации;
Xi — свободная энергия взаимодействия полимера с
растворителем [3].
47
Соответствующая разность химических потенциалов для
полимерной молекулы в смеси по сравнению с чистым жидким
(аморфным) полимером может быть выражена соотношением [3]:
ц2 - $ = RT [In A - v2) - (х -1) A - v2) + %гх A - v2f] G)
Химический потенциал в расчете на моль мономерных
звеньев или сегментов может быть получен делением обеих
частей соотношения на число молекул растворителя,
приходящихся на одну макромолекулу — xVt/VM. Таким образом [3]:
,M-,0M = wb-[i^i+(l-l)(l-t,2)+Xl(l-^] (8)
где Км и Vi — молярные объемы мономерного звена или
сегмента и растворителя, соответственно.
Для полимеров с высоким молекулярным весом соотношение
(8) упрощается до
Им ~ Им = К? -pf К1 - ^ — Xi <1 — v2y\ (9)
Эти выражения описывают термодинамические свойства
двухкомпонентного полимерного расплава.
Химические потенциалы компонентов раствора или смеси в
кристаллической фазе не могут быть выражены в столь общей
форме. Они зависят не только от числа присутствующих
компонентов, но и от характера закона их смешения. При этом
дополнительные ограничивающие условия не могут быть
выражены в терминах общих свойств полимерной цепи, а зависят от
химической и кристаллической структур соответствующего
полимера. Однако нетрудно различить случаи совместимости и
несовместимости компонентов раствора в кристаллической фазе.
В первом случае роль растворителя должна учитываться при
определении химических потенциалов обоих компонентов.
Для большинства полимерных растворов или набухших
систем проблема существенно упрощается в связи с тем, что
растворитель не способен проникать в кристаллическую решетку
из-за стерических ограничений. Это соответствует бинарной
жидкой смеси, в которой лишь один из компонентов кристаллизуется
во всей области композиций. Согласно условиям фазового
равновесия, кроме равенства температур и давлений, требуется
также и равенство химических потенциалов кристаллизующегося
компонента в обеих фазах. Поэтому при температуре
плавления набухшего полимера (в равновесных условиях) требуется,
чтобы
KJ-tf-tf-Й (Ю)
где верхние индексы относятся к кристаллической и жидкой
фазам, а за эталонное состояние принят чистый расплав.
48
Разность химических потенциалов мономерного звена в
кристаллическом и жидком состояниях может быть записана
следующим образом:
К - Им = ~ ^м = - № ~ * Д5М) A1)
Если определить отношение АЯМ/Д5М как Гпл, то выражение
(И) можно переписать в виде:
Им-и°м = АЯмА-^ A2)
При этом неявно принимается, что Д#м и А5М сами по себе
от температуры не зависят. Используя выражение (9) для
М'м —[i°M получаем
— L= RVm [(l — tf2) — Xi A — уяJ] A3)
1 пл л пл ^м'му 1
в качестве основного соотношения для определения понижения
температуры плавления в присутствии растворителя.
Соотношение A3) совершенно аналогично обычному
выражению для температуры плавления бинарной смеси
низкомолекулярных соединений, в которой кристаллизуется лишь один из
компонентов. Отличие для полимерной системы заключается в
иной форме выражения для активности кристаллизующегося
компонента в расплавленной фазе. Согласно соотношению A3),
понижение температуры плавления зависит от объемного
содержания растворителя в смеси и от значения
термодинамических параметров системы полимер — растворитель. При прочих
равных условиях, большее понижение температуры плавления
должно наблюдаться в случае хорошего (меньше величины xi),
нежели в случае плохого растворителя.
Размеры молекул растворителя также влияют на
температуру плавления; понижение Гпл должно быть большим в
растворителях с меньшим молярным объемом. Чем больше
величина АЯМ, тем меньше ожидаемое понижение температуры
плавления.
Теплота плавления на моль мономерных звеньев А#м
представляет собой важную «внутреннюю» термодинамическую
характеристику сегментов или звеньев кристаллического
полимера. Ее не следует путать с теплотой плавления, или скрытым
изменением энтальпии Д#м, получаемой при
калориметрических измерениях. Последняя величина неизбежно
обусловливается начальной степенью кристалличности системы. Д//м
всегда меньше АЯМ, за исключением предельного случая полностью
кристаллического полимера, когда эти две величины будут
совпадать.
Л- Манделькерн
49
Для тех систем, в которых растворитель не входит в
кристаллическую фазу, процесс плавления количественно
описывается выражением A3). Как было указано, следует ожидать,
что различные типы растворителей будут влиять на
температуру плавления одного и того же полимера, даже при
одинаковых концентрациях, различным образом. На рис. 19
представлены зависимости температуры
плавления линейного
полиэтилена от концентрации растворителя
в ряде систем [5, 6].
Как видно из рис. 19, в
присутствии тетралина и а-хлорнаф-
талина наблюдается
непрерывное понижение температуры
плавления по мере разбавления
смеси, как это и следовало ожидать
для случая набухания в
относительно хороших растворителях.
Кривые / и 2, которые
представляют результаты измерений в
я-бутилфталате и о-нитротолуоле,
носят совершенно отличный
характер. При добавлении малых
количеств растворителя
наблюдается небольшое понижение
температуры плавления. Однако по
достижении некоторой критической концентрации растворителя
температура плавления перестает зависеть от дальнейшего
разбавления.
Подобные результаты в присутствии плохих растворителей
были получены для политрифторхлорэтилена [7], поли-N, N'-
себацоилпиперазина [8] и разветвленного полиэтилена [9].
Постоянству температур плавления в этих смесях может быть
дано простое объяснение, если принять во внимание, что во всех
случаях в расплавленном состоянии сосуществуют две несме-
шивающиеся жидкие фазы. Именно этим подобные системы
отличаются от более общего случая, когда при плавлении
возникает одна двухкомпонентная жидкая фаза.
Таким образом, в некоторых специальных случаях при
температуре плавления должны сосуществовать три фазы. Как
следствие правила фаз, при условии постоянства давления,
температура плавления не должна зависеть от состава такой
тройной смеси. Следовательно, на основании экспериментальных
данных МОЖ1НО заключить, что при температуре плавления
кристаллические макромолекулярные системы удовлетворяют
требованиям правила фаз. В свою очередь это заключение можно
рассматривать как термодинамическое доказательство действи-
щ
135
130
125
ми
115
110
^Ч™
&2 >*
-3 ^
о k
0.2 0{> 0,6 0.5 1,0
Рис. 19. Зависимость температуры
плавления полиэтилена от
объемной доли растворителя v{ в
бинарной системе [5].
/ — «-бутилфталат; 2 — о-нитротолуол;
3 — а-хлорнафталин; 4 — тетралин.
50
тельного существования макроскопической кристаллической
фазы в смесях полимеров с растворителями.
Для тех систем, которые показывают непрерывное
понижение температуры плавления с уменьшением концентрации
полимера, можно непосредственно сравнить экспериментальные
данные с формулой A3). Согласно этой формуле начальный
наклон на графиках 1/Тил в функции от концентрации
растворителя Vi=(\— v2) должен быть обратно пропорционален Д//м-
Любые отклонения от линейности на графиках этого типа
обусловливаются конечной величиной параметра Xi- Так. как xi
тоже зависит от температуры, можно принять
где В — плотность энергии взаимодействия (когезии) пары
полимер — растворитель [10].
Тогда выражение A3) можно переписать в форме:
_J 1__
Г™ ^ t.Vnh-m.JL.) (,4)
Предположив, что xi обратно пропорционально абсолютной
температуре, мы совершенно пренебрегли энтропийной
составляющей xi- Более правильным было бы приравнять Xi к сумме
BVilRT+(xl2 — ^\), где г|>1 — некоторый энтропийный параметр,
характерный для данной пары полимер —- растворитель [10].
Если первое слагаемое умножить на v2v то получится
выражение для теплоты разбавления по Ван-Лаару; второе слагаемое
представляет собой дополнительный вклад в энтропию
разбавления сверх рассчитанного на основе простой решеточной
модели. Вторым слагаемым можно пренебречь лишь в том случае,
если исследования проводятся в очень узком интервале
температур (последнее условие обычно соблюдается). Хотя сама по
себе величина Д//м, рассчитанная по экспериментальным
данным, едва ли сильно искажается из-за введенного приближения,
значение В может уже существенно превосходить истинный
параметр теплоты смешения по Ван-Лаару.
Температура плавления набухших полимеров,
закристаллизованных и вновь расплавленных в условиях, близких к
равновесным, должна, следовательно, удовлетворять выражению
A4). Зависимость A/Г„л — l/T°aJl)/vi от vxlTnji должна быть
прямолинейной; по отрезку оси ординат, отсекаемому этой прямой
линией, можно вычислить Д#м для данного полимера, если
известно отношение VJVV
На рис. 20 и 21 представлены типичные экспериментальные
зависимости, обработанные в соответствии с выражением A4),
4*
51
для двух совершенно различных полимеров: натурального
каучука [11] и полидекаметилентерефталата [12]. Каждому из
полимеров давали набухать до различных степеней в разных
растворителях. Как видно из рис. 20 и 21, во всех случаях
экспериментальные точки хорошо укладываются на прямую линию.
Отрицательный наклон на рис. 20 указывает на положительное
значение величины В для
взаимодействия натурального
каучука с каждым из трех
0.5|
^ ОМ
Q6
0,5
CD
^Q5
^Q5
Q3
" ^^^т^—L
^^^^1
"~~~Г^-1^__
0,5 W 15
Ь,/Тяа)-Ю3
Рис. 20. Зависимость
0/7-пл-1/ОЬот^/7-пл для
натурального каучука в
присутствии различных растворителей
[111:
/ — декан; 2—тетрадекалин; 3— метил-
олеат.
0,5 1,0 1,5 2,0
Рис. 21. Зависимость
Ъ\1Тцл Для
полидекаметилентерефталата в присутствии
различных
растворителей [12]:
/ —бензонитрил; 2—бензофенон.
растворителей. С другой стороны, небольшой положительный
наклон, наблюдаемый для систем с полидекаметилентерефтала-
том, указывает на отрицательное значение этой величины.
Линейные зависимости такого же типа наблюдались для
столь большого числа систем полимер — растворитель [5], что
выражение A4) можно безусловно считать экспериментально
подтвержденным. Более того, если исключить величину
молярного объема растворителя, то Д//м, вычисленная для данного
полимера, оказывается одинаковой во всех растворителях.
Некоторые характерные значения Д#м, полученные из такого рода
экспериментов, приведены в табл. 2. Значения Д#м для
каждого из полимеров практически совпадают' друг с другом.
Таким образом, экспериментальные данные подтверждают
предположение о том, что величина Д#м представляет собой
свойство мономерного звена кристаллизующейся цепи и не за*
висит от характера применяемого растворителя. Все сказанное
выше позволяет на основании простого анализа заключить, что
при температуре плавления устанавливается равновесие между
чистой кристаллической полимерной фазой и двухкомпонент-
ной жидкой фазой.
52
Таблица 2
Значение теплот плавления для полимеров
в разных растворителях
Полимер
Растворитель
кал/моль
Полиэтилен [6]
Натуральный
чук [11]
кау-
Полидекаметилен-
терефталат [12]
Полихлортрифтор-
этилен [7]
Этилбензоат . .
о-Нитротолуол
Тетралин . . .
а-Хлорнафталин
Тетрадекан . .
Метилолеат . .
Додекан ....
Бензонитрил
Бензофенон . .
Толуол ....
Мезитилен . .
о-Х лорбензотриф то-
рид
Циклогексан . .
930
935
990
970
1040
980
1100
11600
10 400
1220
1 100
1260
-1330
Рис. 22. Зависимость (\/Тпл —
"-/^пл)/и1 от °^ъемн°й Д°ли
растворителя vx для коллагена,
набухшего в этиленгликоле [136].
Изучение понижения температуры плавления при набухании
было проведено Флори и Гарретом [136] при исследовании
термодинамических переходов кристалл — жидкость в
фибриллярных белках, в частности в
коллагене. С помощью чувствительной
дилатометрической техники
определена температура плавления
коллагена (из сухожилия хвоста
крысы и ахиллесова сухожилия
быка), набухшего до
различных степеней в безводном эти-
ленгликоле. Как видно из
рис. 22, зависимость температуры
плавления от концентрации эти-
ленгликоля подчиняется
выражению A4), если не принимать
во внимание область очень
малых концентраций полимера. Из отрезка оси ординат на этом
графике вычислено значение Д//м, равное 24 кал/л или
2250 кал/моль пептидных звеньев (Гпл принята равной
418° К).
Эти результаты показывают, что процесс плавления более
сложных кристаллических макромолекул, таких как
фибриллярные белки, может быть также рассмотрен в свете общей
проблемы фазового равновесия. Следовательно, они могут изучаться
53
и анализироваться методами, используемыми для более простых
полимерных систем.
Однако при анализе фазовых диаграмм таких систем
возникают некоторые осложнения. Трудно, например, получить
значение температуры плавления чистого ненабухшего
полимера. Существует также вероятность того, что растворитель
может войти в кристаллическую решетку и стать состазной частью
кристаллографической структуры. Например, поглощение воды
коллагеном и нуклеиновыми кислотами сопровождается
усилением экваториальных рентгеновских рефлексов [14, 15], что
указывает на проникновение растворителя в упорядоченную фазу.
При этих обстоятельствах условия равновесия, определяемые
соотношением A0), уже не достаточны, и должны быть
выполнены некоторые дополнительные требования. В частности
необходимо, чтобы удовлетворялось равенство:
Mf-Hj 05)
Более того, выражение A1) не может быть применено для
оценки (ц? — \х°му если в кристаллической фазе происходят
какие-либо взаимодействия или смешение молекул растворителя
и полимера. Простейшим примером такой системы может
служить образование твердого раствора в упорядоченном
состоянии. Следовательно, если мы имеем дело с равновесием двух
фаз в двухкомпонентной системе и оба компонента содержатся
в обеих фазах, то для вывода соотношения, связывающего
температуру плавления и концентрации компонентов, необходимо
выразить активность как функцию концентрации и структуры
каждого компонента в каждой фазе. В этих случаях анализ
экспериментальных данных более сложен, чем это следует
из выражения A4), и не может быть легко обобщен.
При анализе результатов, полученных на коллагене [13],
были введены некоторые упрощения. Так, принималось, что
определенное количество растворителя (независимо от его
общего объема) устойчиво связано с белком, тогда как остальная
часть при температуре плавления может свободно переходить
в аморфную область. В этих условиях выражение A0) снова
будет однозначно определять условия равновесия, химический
потенциал мономерного звена в кристаллической фазе будет
независимым от композиции системы в целом и равенство A4)
вновь будет выполняться. Одна-ко в этом случае Тпл соответ^
ствует уже температуре плавления не чистого ненабухшего
полимера, а комплекса полимера с растворителем и, таким
образом, зависит от природы низкомолекулярного компонента.
Это приближение должно неизбежно привести к большей
неопределенности в оценке Д#м, чем в условиях однокомпонентной
кристаллической фазы.
54
Ранее внимание было обращено главным образом на
понижение температуры плавления чистого гомополимера в
присутствии растворителя. Требования равновесия могут также быть
проанализированы при конечных условиях кристалличности
(Х<1) при температурах ниже Гпл. Если растворитель
однородно распределен по аморфной фазе, то концентрация
полимера в этой фазе изменяется со степенью кристалличности
A-Я) согласно формуле:
-1 щХ A6)
1 — v2 + v2K
При фазовом равновесии химический потенциал
мономерного звена в чистой кристаллической фазе должен быть равным
этому потенциалу в жидкой фазе при концентрации,
определяемой выражением A6). Отсюда
J !_ = _*_ Jjl
7\ Г°пл *"м Ух
K-XiWJ] 07)
где 7\ — температура, при которой поддерживается равновесие
между полимером со степенью кристалличности
A—X) и растворителем с объемной долей v'v
Следовательно, формула A7) определяет равновесную
степень кристалличности, которая существует при температуре 7\
при полной концентрации полимера v2. Если Х=1, тог^^11^
и вновь получается выражение. A3).
Как и в случаях гомополимеров, из выражения A7)
следует, что последние остатки кристалличности исчезнут при
строго определенной температуре. Однако для набухших систем
температурная область плавления должна расширяться, причем
диффузность перехода будет возрастать с увеличением
концентрации растворителя. Это предположение подтверждено
экспериментально для большого числа набухших полимеров [5, 6].
Чанг и Флори [16] исследовали зависимость удельного
объема от температуры набухшего в а-хлорнафталине
полиэтилена в области его плавления для проверки предположения
о том, что фазовое равновесие также может существовать при
конечных степенях кристалличности. При проведении опытов
тщательно контролировались условия кристаллизации, что
позволило легко определять степени кристалличности по удельным
объемам. Результаты экспериментов приведены на рис. 23.
Точки представляют собой- экспериментальные данные, тогда
как пунктирные линии построены согласно выражению A7),
причем в расчетах использованы значения АЯМ и %i» ранее
полученные из зависимостей температуры плавления от
концентрации для данного полимера. При степенях кристалличности
55
до 50% в изученном диапазоне композиций теоретически
ожидаемые и экспериментальные результаты превосходно
согласуются. В системах с меньшим
содержанием растворителя
наблюдаются незначительные
расхождения между теоретическими и
экспериментальными данными, что можно
приписать увеличению затруднений
в установлении равновесия.
Эти наблюдения
исчерпывающим образом доказывают
существование фазового равновесия при
достижении заметного уровня
кристалличности. Одна из фаз обладает
свойствами жидкой смеси, тогда
как другая — свойствами чистого
Щ
/,/5.
В Щ
ш
W0
л.
/
НО 120
на, концентрация которого
дана в весовых (до,) и
объемных (Vi) ДОЛЯХ.
130 НО
Т,°С
Рис. 23. Зависимость удель- кристаллического полимера. Таким
ного объема от температуры образом, еще раз экспериментально
для полиэтилена (М = 55000) подтверждено, что частично кри-
в присутствии а-хлорнафтали- сталлический полимер,
закристаллизованный из концентрированного
раствора или в блоке, содержит и
кристаллические и аморфные обла-
теоретическая кривая рас- СТИ. ПреДПОЛОЖеНИе [18, 191 О ТОМ,
считанная согласно формуле A7) при г L J
—„~ . „ ,л_ v что такие системы представляют
собой лишь несовершенные или
возмущенные кристаллы, лишенные
аморфного компонента, при
термодинамическом анализе
экспериментальных данных не подтверждается,
и, следовательно, морфологические основания этих предпосылок
нуждаются в пересмотре.
- экспериментальные
значения;
Гпл = 137'°° С; ЛЯм = 194° кал1М(>ль\
^3= 1,5 кал/см*, в предположении,
что существует равновесие между
кристаллической и жидкой фазами:
7-и;, = 0,313, Vi = 0,245 при 128,0° С;
2 — «/2 = 0,589, Vi =0,505 при
120,5° С [16].
Разбавленные растворы
Плавление или растворение кристаллических полимеров при
больших разбавлениях часто рассматривается как процесс,
обратный кристаллизации полимеров из разбавленных растворов.
Растворение приводит к разделению первоначально плотно
упакованных молекул и обычно сопровождается
одновременным изменением их конформации. Однако иногда
индивидуальные макромолекулы сохраняют даже при высоких степенях
разбавления типичные для кристаллического состояния
конформации. Это бывает в тех случаях, когда стерические факторы
благоприятствуют сохранению жестких избирательных ориен*
56
таций связей или когда кристаллическая структура
поддерживается внутримолекулярными взаимодействиями, приводящими
к образованию стабильных «нехимических» связей.
В последнем случае дальнейшее изменение
термодинамических свойств среды может привести *к внутримолекулярному
структурному превращению, в результате которого
макромолекулы переходят в конформацию, характерную для аморфного
состояния. Такое внутримолекулярное превращение принято
называть переходом спираль — клубок, в соответствии с теми кон-
формациями цепи, которые имеют молекулы в исходном и
конечном состояниях. Рассмотрим сначала поведение при
кристаллизации обычных макромолекул, у которых в разбавленном
растворе не сохраняется упорядоченная конформация.
В отличие от концентрированных систем гомогенный
разбавленный раствор характеризуется неоднородным
распределением сегментов по объему системы [10]. Индивидуальные
молекулы изолированы друг от друга; следовательно, имеются
области раствора, в которых полимер отсутствует. Эта отличи*
тельная черта разбавленных растворов полимеров очень важна
при рассмотрении перехода кристалл — жидкость. Ранее
использованная решеточная модель полимерных растворов теперь
становится нереальной и нуждается в пересмотре. Только в
случае достаточно плохого растворителя, когда макромолекулы
относительно свободно проникают друг в друга, .старая модель
все же может быть применена и к разбавленным системам *.
Химический потенциал растворителя в разбавленном
растворе выражается по современной теории, в вириальной
форме [10]. С помощью уравнения Гиббса — Дюгема может быть
вычислен химический потенциал мономерного звена в
расплавленной жидкой фазе. Однако при проведении таких расчетов
оказывается, что поправки слишком малы, чтобы заметно
повлиять на температуру плавления. Следовательно, в данном
случае для вычисления химического потенциала мономерного
звена все же может быть использовано выражение (8).
Если химический потенциал звена в кристаллической фазе
снова выразить соотношением (И), то при температуре
плавления должно удовлетворяться равенство:
Это выражение очень похоже на соотношение A3),
полученное для более концентрированных систем. Отклонение
от этой формы при лг-^оо будет вероятным только при
* [Манделькерн имеет в виду не столько истинный, сколько коллоидный
раствор или нерастворимый, но набухающий полимер, — Прим,
редактора.]
57
чрезвычайно высоких разбавлениях; другие отклонения, которые
могут иметь место при очень малых значениях х,
экспериментально обнаружить трудно. Следовательно, зависимость
температуры плавления от состава или отношения растворимости
выражается непрерывной функцией, которая охватывает всю
область концентраций.
Зависимость температур растворения в тетралине от
концентрации для тщательно расфракционированных образцов
полиэтилена, первоначально
закристаллизованных в блоке при высоких
температурах, приведены на рис. 24 [20].
Результаты этих измерений показаны черными
кружками, которые ложатся на одну
кривую с температурами плавления,
полученными дилатометрическим методом
для высоких концентраций полимера
(белые точки). Таким образом, в согла-
ф сии с теорией, зависимости температуры
v< плавления от состава во всей области
концентраций могут быть представлены
непрерывной функцией, описываемой
выражением A3).
Однако, если предшествующая
измерениям Гпл кристаллизация проводится
из раствора заданной концентрации, то
между температурами плавления (или
точками растворения) образцов с
разной термической, предысторией
существует отличие. Материал,
закристаллизованный из раствора, неизменно
обладает более низкой температурой плавления. Температуры
плавления в тетралине того же полиэтилена, закристаллизованного
из раствора, показаны на рис. 24 белыми квадратиками.
Различия впервые появляются приблизительно при 50%-ном
содержании полимера и становятся все более существенными по мере
дальнейшего разбавления.
Для очень разбавленных растворов наблюдается разница в
температурах плавления около 12 град. Поскольку жидкое
состояние в обоих случаях одинаково, несовпадение температур
плавления должно быть обусловлено различием структуры
кристаллических фаз. Более низкая температура плавления,
наблюдаемая после кристаллизации из раствора, указывает на то,
что в этих условиях получается метастабильная
кристаллическая форма, которая может возникать по различным причинам.
Существенно, что обычной морфологической формой,
наблюдаемой после кристаллизации из разбавленных растворов,
являются пластинчатые кристаллы, состоящие из сложенных цепей.
Рис. 24. Зависимость
температуры
растворения от объемной доли
растворителя vx для
полиэтилена в тетралине:
О —температура плавления,
определенная
дилатометрически; ф —температура
растворения образца,
закристаллизованного из расплава;
? — температура растворения
образца,
закристаллизованного из раствора |20|.
58
Мы ограничимся здесь замечанием, подчеркивающим, что
для проверки теоретических предпосылок экспериментальные
данные для температур плавления необходимо подвергнуть
весьма тщательному анализу, особенно с учетом термической
предыстории образцов (подробнее см. гл. 8 и 9). Во всем
диапазоне концентраций следует пользоваться значениями Тпл для
наиболее стабильных образцов; поэтому при определении Д#м
не рекомендуется проводить измерения в разбавленных
растворах, если только не исследуются полимеры,
закристаллизованные в блоке.
Когда рассматривается полная область концентраций от
разбавленного раствора до чистого полимера и в качестве
второго компонента используется хороший растворитель, диапазон
температур плавления может быть очень широк. При этом
сомнительно сделанное в неявной форме предположение о том,
что ASM и А//м не зависят от температуры, и следует принять
вр внимание энтропийную составляющую xi- Хотя выражение
A3) по-прежнему будет описывать равновесие, входящие в него
термодинамические величины уже могут более не быть
постоянными.
Зависимость температуры плавления от концентрации
полимера, с другой стороны, определяет температурный предел
растворимости кристаллического полимера в заданном
растворителе. При определенной концентрации температура плавления
не очень чувствительна к молекулярному весу за исключением
области очень малых молекулярных весов. Следовательно,
кристаллизация полимеров из разбавленных растворов никак не
может служить эффективным методом фракционирования
даже при достижении условий равновесия растворимости. Более
вероятно, что при охлаждении жидкой фазы процесс
кристаллизации полимера будет обусловливаться главным образом
кинетическими факторами. Проходящая по мере охлаждения
кристаллизация вызовет дополнительные трудности при
фракционировании полидисперсного образца. Чтобы осуществить
эффективное фракционирование дробным осаждением из
разбавленного раствора, необходимо предотвратить кристаллизацию и
отделять полимер в виде жидкой фазы *. Для этого необходимо
предварительно провести подробный анализ фазовых диаграмм.
Переход спираль—клубок
Хотя независимые полимерные цепи в очень разбавленных
растворах обычно имеют конформацию статического клубка,
* [Это очень существенное обстоятельство. К сожалению оно не всегда
принимается во внимание при фракционировании кристаллизующихся
полимеров, например полиамидов или полиэфиров. См. также [61], гл. 2.— Прим.
редактора.]
69
из этого правила имеются важные исключения, затрагивающие,
главным образом, макромолекулы биологического
происхождения или их модели. Физико-химическими измерениями
установлено, что многие простые синтетические полипептиды, находясь
в очень разбавленных растворах в подходящих (слабо
взаимодействующих) растворителях, способны существовать в виде
независимых а-спиралей [21—23]. Аналогично и синтетические
полинуклеотиды могут сохранять в разбавленных растворах
жесткие асимметричные конформации, представляющие собой
составные спирали (мультиспирали) из двух или трех
переплетенных цепей [24—27].
Природные нуклеиновые кислоты, а также многие
фибриллярные белки способны растворяться с сохранением
внутримолекулярной организации, присущей твердому состоянию.
Например, можно растворить фибриллярный белок коллаген,
сохранив характерную упорядоченную структуру триспиральных
протофибрилл [28, 29]. В перечисленных примерах сохранение
и стабильность упорядоченной структуры обусловлены
наличием специфических вторичных внутренних связей. У а-спи-
ральных структур возникают внутримолекулярные водородные
связи между пептидными группами основной цепи. У мульти-
спиральных структур возникают межцепные водородные связи.
Специфические типы внутримолекулярных связей, однако,
не обязательны для сохранения в разбавленном растворе
упорядоченных структур, присущих твердому кристаллическому
состоянию. Так, синтетический поли-?-пролин, имитирующий
структуру коллагена, может существовать в растворе как в
форме клубка, так и в форме спирали [30]. Поскольку этот
полимер является полииминокислотой, образование в нем
внутримолекулярных водородных связей невозможно.
Спиральная форма в этом случае несомненно сохраняется вследствие
стерических ограничений, обусловленных громоздкими пирроли-
диновыми кольцами.
Вполне вероятно существование и других макромолекул, у
которых стерические ограничения затормаживают свободное
вращение вокруг связей так сильно, что даже в разбавленных
растворах сохраняют присущую кристаллическому состоянию
конформацию. Сохранение исходной конформации может быть
обусловлено и полярными силами без участия водородных
связей.
Когда отдельные изолированные молекулы существуют в
спиральной форме, изменение окружающей среды, в
особенности температуры или характера (активности) растворителя,
может вызвать разрушение упорядоченной структуры, причем цепь
приобретает конформацию статистического клубка. Это
обратимое внутримолекулярное превращение, именуемое переходом
спираль — клубок, представляет собой наиболее элементарное
60
^ 5 6 7 8 9 10 11 12 13
рН
Рис. 25. Влияние рН на переход спираль —
клубок в поли-1-глутаминовой кислоте [23].
проявление плавления или кристаллизации полимеров. Переходы
спираль — клубок впервые были исследованы на опыте Доти * и
его сотрудниками [24—26]. Например, молекулы простого
полипептида поли-?-глутаминовой кислоты в разбавленных
нейтральных и щелочных средах существуют в форме
статистических клубков. Однако при снижении рН до ~5 стабилизируется
упорядоченная форма а-спирали. Переход спираль — клубок
проявляется в изменении различных физико-химических свойств
раствора, как это видно
из рис. 25. По мере
уменьшения заряда макромоле- эд
кулы, т. е. уменьшения i
степени ионизации, на- *Ч0
блюдается сильное увели- _эд|
чение удельного оптиче- «J
ского вращения [а].
Этому увеличению
сопутствует также повышение
характеристической
вязкости [т|]. Узкая область
рН, в которой происходят
эти изменения, указывает на кооперативный характер перехода,
напоминающий, таким образом, в сильной мере обычное
плавление. *
Аналогичный тип перехода также наблюдается у синтетичен
ских полинуклеотидов. Полиаделиновая и полиуридиловая
кислоты, например, при смешении в разбавленном растворе
образуют упорядоченную биспираль, проявляющую все свойства
индивидуальной молекулы. Поэтому вполне уместно говорить
о внутримолекулярном переходе, который происходит при
нагревании вблизи 50° С, как показано на рис. 26. Это конформа-
ционное изменение также оказывает влияние на ряд физико-
химических свойств раствора, в том числе на поглощение света
(при 250 ммк) и оптическое вращение. Все изменения
происходят в очень узком температурном интервале, что также
характерно для плавления. Разрушение упорядоченной структуры
мультицепных молекул в разбавленном растворе
сопровождается разделением отдельных цепей и переходом каждой из них в
конформацию статистического клубка. При тщательном
соблюдении условий эксперимента может быть достигнуто полное
восстановление природных мультиспиральных упорядоченных
структур даже в случае таких сложных систем, как коллаген
[31] и дезоксирибонуклеиновая кислота [32].
* [Так называемая конформация полипролина II. Она, однако, не
вполне устойчива и может переходить в неупорядоченную конформацию
полипролина I. — Прим. редактора.]
61
т
щ
0,8
0.7
0,6
50
05
Ш |
О)
1150 &
\200 I
250'
\Ж
20
40 60
80
Г/С
Внутримолекулярные переходы были подробно
проанализированы теоретически. Рассмотрим здесь в некоторых деталях
теорию перехода тех полипептидных цепей, которые существуют
в разбавленном растворе в форме а-спиралей, либо
статистического клубка. Наличие упорядоченной спиральной
структуры обусловлено
стабилизацией формы макромолекулы
внутримолекулярными
водородными связями между соседними
звеньями.
Полинг и Кори [33] показали,
что атом водорода каждой амид-
ной группы основной цепи
образует водородную связь с
карбонильным кислородом третьей
вдоль цепи предшествующей
группы, т. е. попарно связаны
каждое первое и четвертое звено.
Таким образом, ориентация
связей последующих звеньев
обусловливается положением
предшествующих звеньев, и это создает
тенденцию к поддержанию
постоянной конфигурации смежных
звеньев вдоль всей цепи.
Как подчеркнул Шеллман[34],
для обеспечения свободы
вращения мономерного звена для раз-
упорядочения одного витка
спиральной цепи, находящейся в
а-конформации, необходимо
разорвать три водородные связи
в трех последовательных пептидных звеньях. Эти жесткие
условия перевода витка спирали в состояние с
конфигурационной свободой статистического клубка и обусловливают
кооперативный характер перехода спираль — клубок. Так как на
переход цепи в состояние статистического клубка с более
высокой энтропией затрачивается определенная энергия (энтальпия),
то неупорядоченная форма цепи становится более
энергетически выгодной. Поэтому переход спираль — клубок при
изменении температуры, давления или состава растворителя
должен быть относительно резким, напоминая истинное
плавление.
Количественно эта проблема была сформулирована в очень
ясной и простой форме Шеллманом [34]. Он предположил, что
отдельные макромолекулы существуют либо только в форме
Рис. 26. Переход спираль —
клубок з синтетических полинуклео-
тидах. Изменение удельного
вращения и оптической плотности
от температуры в разбавленном
растворе комплекса,
образованного полиадениловой и полиури-
диловой кислотами, при ионной
силе раствора 0,15 и рН=7 [27].
w
спирали (И), либо клубка (С). Для равновесного процесса
Н $ С A9)
§>-"-«*(=№) B°>
где А/7,— разность свободной энергии между упорядоченной и
статистической формами в расчете на одну молекулу.
При достаточно большом молекулярном весе можно
пренебречь влиянием концов цепей.
Тогда:
где Л/^—изменение свободной энергии при переходе на
мономерное звено;
х— степень полимеризации.
Когда AFM = 0, концентрация молекул в каждой из форм
одинакова. Поскольку предполагается, что степень
полимеризации велика, весьма малые изменения AFM вблизи точки д/,м = 0
могут вызвать очень резкие изменения отношения (С)/(H).
Развитие одной конформации за счет другой при изменении
независимой переменной, такой как температура или состав
(активность) окружающей среды, может происходить
достаточно резко, чтобы походить на фазовый переход. Так как х
велико, энтальпия плавления на всю молекулу также будет
велика. Следовательно, константа равновесия К должна очень
быстро изменяться с температурой. Чтобы оценить ширину
перехода, надо определить изменение с температурой
относительной концентрации молекул, имеющих конформацию
статистического клубка. При температуре перехода Т=Ти когда Д/^О,
скорость изменения этой концентрации может быть выражена
как в работе [34]:
/л {(С)/[(С)+ (//)]} \ __ лгАЯм
Поскольку Д#м должно быть порядка нескольких
килокалорий на моль, переход относительно резок и обладает, казалось
бы, характеристиками перехода первого рода. Однако
бесконечно резким он становится только в предельном случае чистых
полипептидных цепей бесконечно большого молекулярного
веса. Если молекулярный вес не велик, область перехода
значительно расширяется.
Формулировка рассмотренной проблемы основывается на
предположении, что отдельные макромолекулы существуют
полностью в одной или в другой из двух возможных конформации.
Для молекул большого молекулярного веса это допущение
нельзя считать вполне удовлетворительной гипотезой. Хотя
63
спиральная форма представляет собой состояние с минимальной
энтальпией, а статистический клубок — с наивысшей
конфигурационной энтропией, наиболее стабильному термодинамическому
состоянию с минимумом свободной энергии могут
соответствовать промежуточные конформации цепи, образованные
чередующимися спиральными и неупорядоченными областями *.
Необходимо исследовать возможность такого
сосуществования внутримолекулярных «фаз» более строгими методами
статистической механики. За минувшее десятилетие предложено
несколько в общем сходных теоретических методов решения
сформулированной задачи [35—43]. В качестве модели во всех
случаях предложен одномерный упорядоченный набор
аминокислотных остатков. Далее для модели рассматривалось общее
число способов распределения вдоль цепи образовавшихся или
необразовавшихся водородных связей, причем концы цепей
рассматривались как специальные состояния звеньев. Это число
возможных распределений последовательностей водородных
связей ограничивается стереохимическими требованиями а-спи-
рали; с учетом таких ограничений рассчитывается
статистический вес каждого распределения.
В общих чертах сформулированная таким образом проблема
аналогична одномерной решетке Изинга, используемой как
модель для расчета в теории ферромагнетизма. Различные
теории отличаются в деталях определения статистической суммы
изолированной цепи. Однако все они приходят к одному и тому
же общему заключению, а именно: довольно резкий переход все
же возможен даже в том случае, когда в одной и той же
молекуле действительно сосуществуют спиральные и
неупорядоченные последовательности звеньев.
Для иллюстрации этого общего вывода рассмотрим, вкратце,
теорию, развитую Зиммом и Бреггом [35]. В случае простой
полипептидной цепи однородной структуры переход зависит от
двух параметров. Первый из них, обозначаемый s, может быть
приравнен к KUx в выражении B0), в то время как второй а
входит в статистическую сумму для каждой упорядоченной
последовательности звеньев, следующей за неупорядоченным
участком цепи. Величина —RTIn о означает разность изменений
свободной энергии при образовании «нуклеаций» одного
упорядоченного участка цепи из трех пептидных остатков, ранее
находящихся в неупорядоченном состоянии, и при присоединении
тех же трех звеньев к уже существующему витку. Следователь*
но, величина с, по порядку много меньшая единицы, пред-
* Сформулированная здесь проблема принципиально отличается от
обсуждавшейся в гл. 2. В данном случае анализируется одномерная система,
тогда как ранее рассматривались трехмерные кристаллиты, ограниченные
торцовыми кристаллографическими "плоскостями, в которых заканчиваются
все кристаллические участки цепей,
64
ставляет собой обратную меру кооперативности взаимодействий,
благоприятствующей образованию а-спирали *.
Для оценки расчета статистической суммы цепи
используется матричный метод. Для достаточно длинных цепей
относительное содержание звеньев в спиральной конформации
равно [35]:
е = 4^ B2)
dins v
где х — наибольший корень уравнения, равный
Х = j {1 + s ± [A -5J+ 4о8)Ц B3)
Согласно теории, критическое значение s равно единице.
Вблизи этого критического значения происходит переход из
статистической конформации в спиральную. Резкость перехода
зависит от величины а и от длины цепи. Для низких
молекулярных весов для 8 получается более сложное выражение, из
которого вытекает, что цепи представляют собой либо
статистические клубки, либо содержат лишь один ненарушенный
спиральный участок. Когда длина цепи достигает порядка
нескольких сотен амидных остатков, переход становится крайне
резким и практически перестает зависеть от молекулярного
веса. Если а=1 (полное отсутствие кооперативности),
взаимодействия состояний последовательных звеньев не происходит,
и концентрация связанных водородными связями мономерных
звеньев лишь монотонно возрастает с увеличением s.
Напротив, если а приближается к нулю, что указывает на
очень интенсивное кооперативное взаимодействие, при 5=1
наблюдается чрезвычайно острый переход.
Можно показать, что для цепей с большим молекулярным
весом ширина перехода характеризуется областью, в которой 5
изменяется между предельными значениями, определяемыми
неравенством:
-a,/2<lziL<a,/a B4)
s
В этой области относительное содержание звеньев
спиральной конформации уменьшается примерно от трех четвертей до
* [Это несколько сложное статистическое определение параметра
кооперативности 6 отражает основной физический факт: предшествование витка
повышает вероятность образования следующего витка; виток как бы играет
роль «матрицы», облегчающей присоединение следующих звеньев в
аналогичной конфигурации. С этим связан выигрыш свободной энергии — RT In a. —
Прим. редактора].
5 Л. Манделькерн
65
одной четверти. Так как — RT In 5 равно ДГМ вблизи Tt> где
s~l, то:
ds
АЯМ
dT ~~ RT*
B5)
Отсюда следует, что температурный интервал перехода
может быть выражен в хорошем приближении формулой:
2RTW*
ДГ:
АЯМ
B6)
Переход становится более резким, если Д#м увеличивается
или с уменьшается.
Для количественной проверки этой статистико-механичеекой
теории были использованы в основном результаты опытов,
проведенных на разбавленных
растворах образцов поли-у-бензил-
L-глутамата различных
молекулярных весов в смесях дихлор-
уксусной кислоты и дихлорэтана
[45]. Результаты представлены на
рис. 27. Хорошее согласие
между теоретическими и
экспериментальными данными достаточ-
и0 но очевидно; из рис. 27 следует,
что резкость перехода возрастает
с увеличением молекулярного
веса. Наилучшая сходимость
результатов получается при
использовании для расчетов
значений о = 2 • Ю-4 и Д#м =
= 900 кал/моль. В соответствии с
выражением B6) значения а и
Д#м указывают на то, что
ширина области перехода для очень
высокомолекулярных образцов
должна составлять
приблизительно 5°.
Поскольку все рассмотренные выше теоретические
соотношения выведены в предположении о равновесном распределении
спиральных и неупорядоченных участков вдоль цепи, то
определенные значения термодинамических параметров а и Д#м
имеют смысл лишь в том случае, если условия равновесия
действительно соблюдаются.
У более сложно организованных мультиспиралей нуклеа-
ция беспорядочных клубкообразных областей внутри
упорядоченной структуры связана с разрывом большого числа
межпептидных или межнуклеотидных водородных связей. Следова-
о ю 20
Т-Тпл,град
Рис. 27. Сравнение теоретических
и экспериментальных данных для
перехода спираль — клубок в
разбавленном растворе поли-у-бензил-
/.-глутамата в смеси дихлорэтана
и дихлоруксусной кислоты.
Экспериментальные точки показывают
зависимость удельного оптического
вращения от температурного параметра
*" *" где Г —температура опыта;
-температура фазового перехода
Г-Т,
пл
(плавления). Сплошные линии
представляют собой наилучшее приближение
дл! образцов с различной степенью
полимеризации л.
66
тельно, статистические параметры, соответствующие а, для
таких систем должны быть значительно меньше, чем для моно-
а-спиралей. Таким образом, при прочих равных условиях
температурная область перехода для мультицепной структуры,
по сравнению с единичной цепью, должна быть уже.
Ясно, что существует определенная аналогия между
переходом спираль — клубок и процессом одномерной
кристаллизации. Ландау и Лифшиц [46] показали в общем виде, что
термодинамическое равновесие между двумя произвольно
протяженными гомогенными «одномерными» фазами, соприкасающимися
в одной точке, невозможно. Обе фазы неизбежно должны в
какой-то степени смешиваться друг с другом. Расчеты перехода
спираль — клубок, проведенные на основе статистической
механики, согласуются с этим заключением. Именно указанная
характеристика одномерных систем обусловливает диффузный
характер перехода и допускает сосуществование в определенной
температурной области двух фаз.
Таким образом, строго говоря, переход типа спираль —
клубок не может быть квалифицирован как истинный фазовый
переход *. Однако, несмотря на диффузный характер превращения,
зависимость температуры перехода от термодинамических
переменных системы вполне соответствует требованиям теории
фазовых равновесий.
Рассматриваемый тип перехода уникален в том смысле, что
не требует одновременного участия многих молекул. Поэтому
он развивается на очень малом ограниченном участке полной
фазовой диаграммы, где не проявляются межмолекулярные
взаимодействия. При увеличении концентрации спиральных
макромолекул эти взаимодействия уже начинают проявляться,
усиливая кооперативный характер перехода. С увеличением
концентрации полимера переход из одномерного постепенно
превращается в трехмерный. Таким образом, в концентрированных
системах переход спираль — клубок даже с формальной точки
зрения становится неотличимым от обычного плавления плотно
упакованной кристаллической фазы.
Полагают, что глобулярные белки состоят из коротких
жестких спиральных областей, сочлененных менее
упорядоченными гибкими участками цепи. Детальное изучение
рентгенограмм подтверждает эту гипотезу на примере гемоглобина и
миоглобина [47, 48]. Такой тип организации элементов цепи
приводит к образованию общей молекулярной структуры с
относительно низкой асимметрией. Несмотря на это, и здесь
* [С подобным заключением не следует спешить. Многое зависит от
способа определения фазового перехода [62, 63]; кроме того, спиральные
системы не одномерны, а трехмерны, хотя различные направления в спирали и
неравноценны. — Прим. редактора].
5*
67
наблюдается довольно резкий структурный переход. Область
перехода оказывается значительно уже, чем это можно было бы
предполагать, зная ограниченную длину спиральных участков.
Шерага объясняет это кажущееся противоречие с теорией
тем, что взаимодействия между боковыми группами соседних
звеньев в пределах отдельной спирали должны сужать область
перехода [49], По-видимому, большую роль играет
невозможность независимого «плавления» отдельных спиральных
участков в глобулярной молекуле. Если спирали достаточно плотно
упакованы в белковой глобуле, макромолекула может
рассматриваться как концентрированная система, и кооперативный
характер перехода становится более выраженным *.
Переходы, не связанные с изменениями
молекулярных конформации
Одномерноупорядоченная структура полимерной
молекулы накладывает жесткие ограничения на ее форму. В
рассмотренных случаях внутримолекулярного упорядочения возникли
весьма асимметричные стержневидные (спиральные или мульти-
спиральные) молекулярные конформации с длинами, намного
превосходящими поперечник. Такие молекулы могут
существовать в разбавленном растворе совершенно независимо друг от
друга с беспорядочным распределением ориентации больших
осей. Подобный раствор вполне изотропен.
Однако по мере повышения концентрации стержневидные
макромолекулы уже не могут ориентироваться произвольно.
Действительно, достаточно плотная упаковка при абсолютно
беспорядочной ориентации длинных частиц принципиально
невозможна. Вследствие стремления системы к состоянию с
минимальным свободным объемом (и, соответственно, с
минимальной свободной энергией) должна спонтанно возникнуть
некоторая корреляция ориентации соседних макромолекул,
захватывающая тем больший объем раствора, чем выше концентрация.
В предельном случае плотная упаковка становится возможна
лишь при параллельной ориентации всех макромолекул.
Эти качественные соображения позволяют предсказать, что
при высоких концентрациях полностью дезориентированный или
изотропный раствор макромолекул асимметричной формы
вообще не может существовать. Следовательно, при повышении
концентрации полимера либо должно происходить изменение моле-
* [По-видимому, это обстоятельство доказано экспериментально в
работах [64, 65], но следует считаться и с возможностью непосредственного
фазового перехода типа глобула — клубок, который не обязательно связан с
существованием кристаллической фазы [66]. — Прим. редактора].
68
кулярной конформации, либо устанавливаться упорядоченное
расположение молекул.
Теория подобного рода превращений была развита Флори
[50]. Он использовал обычную решеточную модель,
позволяющую в данном случае рассчитать число пространственных
конфигураций, которые способны принять в объеме раствора Пг
жестких стержневидных молекул с асимметрией х (отношение
длины молекулы к ее поперечнику) в присутствии я4 молекул
растворителя, и вычислить степень их ориентации относительно
какого-либо направления. Если использовать обычное
соотношение для теплоты смешения по Ван-Лаару, то свободная
энергия смешения может быть выражена как
AF
-^р- = пх In vx + п2 In v2 — (п{ -f уп2) X
Xln[l— v2 (--")] - n2 fin (xy*) -//+ 1] + %lxnivl B7)
где vx и v2 — соответствующие объемные доли;
у — параметр, представляющий собой меру, или
индекс, дезориентации молекул.
Параметр у может изменяться от единицы (полностью
упорядоченная структура) до х (состояние полного беспорядка).
Если 0=1, формула B7) сводится к выражению для свободной
энергии смешения при образовании регулярного раствора, а
если у = х — к выражению для свободной энергии смешения
жестких полимерных цепей [51]. Таким образом, при заданной
молекулярной асимметрии х формула определяет зависимость
свободной энергии смешения от концентрации полимера и пара*
метра дезориентации у.
При неизменной концентрации и молекулярной асимметрии
величина &FCM/kT по мере увеличения параметра
дезориентации у сначала проходит через минимум, а затем достигает
максимума. Так как величина параметра дезориентации не
ограничивается никакими внешними условиями, у всегда стремится
принять значение, соответствующее минимуму AFCM„ Как
показывает анализ, при заданных v2 и х значение у,
удовлетворяющее этому условию, представляет собой меньшее из двух
решений уравнения:
— ^7[*-«*(-7)] B8)
Если v*2 — минимальная концентрация, при которой вообще
возможно решение этого уравнения, необходимым условием су*
ществования изотропной фазы (т. е. состояние полного
молекулярного беспорядка) является v2 < v*T Можно показать, что
<~И'-7) »
69
представляет собой максимальную концентрацию, при которой
возможно существование устойчивой изотропной фазы, или
минимальную концентрацию, при которой появляется стабильная
анизотропия раствора (т. е. состояние частичной равновесной
упорядоченности макромолекул).
Таким образом, максимальная концентрация, при которой
взаимные ориентации макромолекул совершенно независимы,
для больших значений х пропорциональна обратной величине
осевого отношения. Это заключение вытекает только из учета
асимметрии молекул и не связано с допущениями о наличии
каких-либо межмолекулярных сил. Легко показать, что в
отсутствии растворителя (^2=1) самоупорядочение системы
должно 'происходить уже при осевых отношениях порядка 2е.
Из выражения для свободной энергии B8) и условия
равновесия B7) можно вычислить химические потенциалы каждого
компонента в обеих фазах —в изотропной фазе, где у=х и в
фазе, где молекулы находятся в состоянии некоторого
равновесного порядка (неполностью дезориентированы). Условия
равновесия между двумя фазами при постоянной температуре
и давлении могут быть получены, если приравнять, как обычно,
химические потенциалы обоих компонентов в каждой из фаз.
Сначала мы примем, что имеет место атермическое
смешение, когда %i = 0, т. е. что между молекулами не существует
никаких прямых взаимодействий. Изменения химических
потенциалов каждого из компонентов при этом таково, что
разделение на две фазы, изотропную и упорядоченную, должно
происходить при относительно высоком разбавлении [50]. При #=100
в условиях равновесия двух фаз ^2 = 0,0806 для разбавленной
фазы и i>2 = 0,1248 для несколько более концентрированной.
Более разбавленная фаза изотропна, в ней ориентация
частиц не зависит от положения ближайших соседей. Более кон-*
центрированная фаза крайне анизотропна, поскольку
макромолекулы в ней достаточно хорошо ориентированы относительно
некоторой общей оси. Эту фазу называют «тактоидной» *.
Разделение фаз в указанном случае происходит только вследствие
асимметрии частиц без участия каких-либо специфических
межмолекулярных взаимодействий. Когда молекулярная
асимметрия возрастает, концентрация полимера в обеих фазах
уменьшается; однако в тактоидной фазе она никогда не будет
значительно больше, чем в сосуществующей с ней изотропной.
По мере возрастания величины х отношение концентраций
полимера в обеих фазах приближается к предельному
значению 1,56. Если параметр %t принимает значение несколько
большее нуля, в пределах ограниченной области %i возникает теоре-
* [Термин заимствован из коллоидной химии. «Тактоидный» означает
«образованный палочковидными частицами», — Прим, редактора].
70
тическая возможность сосуществования двух анизотропных
фаз.
Таким образом, в системе могут существовать две
гетерогенные области; в одной из них осуществляется равновесие между
двумя относительно концентрированными анизотропными
фазами, а в другой — между разбавленной изотропной и этими
двумя анизотропными фазами. При л:= 100 две гетерогенные
области сливаются, когда %i достигает значения 0,07. Для
величины /1, равной или большей
' 0,07, равновесие поддерживается
только между разбавленной
изотропной и одной
концентрированной анизотропной фазами.
Полная диаграмма фазового
равновесия в такой системе для х =
= 100 приведена на рис. 28. Во
всех фазах конформация
полимерных молекул остается
неизменной.
Из диаграммы (рис. 28)
видно, что сравнительно небольшое
увеличение /i может вызвать
резкое изменение концентрации
полимера в анизотропной фазе от
а2 ~0,15 до v2 > 0,9.
Следовательно, достаточна малая
положительная энергия
взаимодействия, чтобы исключить почти
весь растворитель из
анизотропной фазы. Концентрация полимера в изотропной фазе остается
при этом незначительной. Величина параметра взаимодействия,
при котором происходит такое разделение фаз, относительно
мала и во всяком случае намного меньше значения' 0,5, при
котором происходит разделение двух жидких фаз в растворе
статистических свернутых макромолекул бесконечно большого
молекулярного веса.
Причина указанного различия заключается в значительно
большей конфигурационной энтропии, которой обладают
статистически свернутые макромолекулы в разбавленных растворах.
Энергия смешения жестких беспорядочно ориентированных
стрежневидных макромолекул намного меньше этой
конфигурационной энтропии и приближается к величине, соответствующей
закону смешения для идеальной системы. Поэтому уже
небольшие энергии взаимодействия оказываются достаточными, чтобы
сделать изотропную фазу неустойчивой.
Специфические взаимодействия между боковыми группами
Цепи, которые не учитываются в выражении для свободной
0.2 0,4 Ofi Ofi ijo
Рис. 28. Теоретический расчет
фазового равновесия в растворах
стержневидных частиц [50].
По оси абсцисс отложены композиции
фаз v2 для лг=100, по оси ординат —
значение параметра взаимодействия Xi-
Кривая для изотропного раствора
расположена слева, а для упорядоченных фаз —
справа. Гетерогенные области
располагаются между этими двумя кривыми.
71
энергии смешения по Ван-Лаару, должны приводить к
дальнейшему упорядочению концентрированной или осажденной фазы.
В случае асимметричных однородных по структуре частиц
достаточно лишь небольшое снижение энтропии, чтобы в системе
возникла упорядоченность, характерная для кристаллического
состояния. Несмотря на неточности, присущие решеточной
модели, выведенные на ее основе соотношения удовлетворительно
объясняют спонтанное образование фибриллярных структур из
разбавленных растворов
макромолекул, в частности
образование макроскопических
фибриллярных белковых и иных макро-
молекулярных систем как in
vitro, так и в клетках. Величина
параметра %i может изменяться при
вариации состава растворителя
или температуры. В зависимости
от относительного вклада в
этот параметр энтальпии и
энтропии он может возрастать или
уменьшаться при повышении
температуры.
Переходы рассмотренного
типа встречаются главным
образом среди полимеров
биологического происхождения и их
моделей. Робинсон, Вард и Биверс
[50] наблюдали, что
разбавленные растворы поли-у-бензил-1-
глутамата в ряде органических
растворителей, где
поддерживается а-спиральная форма,
спонтанно образуют двулучепреломляющую фазу при концентрации,
зависящей от степени полимеризации. У а-спирали отношение
осей должно увеличиваться с возрастанием молекулярного веса.
Существование тактоидной фазы в совершенно различных
растворителях указывает на то, что она образуется безотносительно
к возможности избирательных взаимодействий с растворителем,
как это и следует из теории. Высокая асимметрия молекул
является главным фактором, определяющим разделение
фаз.
Характерный пример разделения фаз в диоксановых
растворах этого полимера представлен на рис. 29 [44]. Концентрации
полимера в сосуществующих при равновесии фазах выражены
в зависимости от отношения осей или степени полимеризации.
Двулучепреломляющая фаза только слегка более
концентрирована, чем разбавленная изотропная фаза. Согласно теоретиче-
* 50\
I
Отношение осей
20 W 60 80 100
. 30\
| /01
t W
/
200 W0 600 800 1000 1200
Степень полимеризации
Рис. 29. Зависимость
концентрации сосуществующих
изотропной (/) и тактоидной B) двулуче-
прелрмляющей фаз для поли-у-
бензил-/.-глутамата в диоксане при
комнатной температуре от
средней степени полимеризации или
от отношения осей макромолекул,
имеющих форму а-спирали [44].
Пунктирные кривые рассчитаны по
теории фазового равновесия в растворах
стержиевидных частиц.
72
скому расчету, пунктирные линии соответствуют составу тех же
двух фаз.
Как видно из рис. 29, отношение концентраций,
наблюдаемых на опыте фаз, достаточно хорошо согласуется с
рассчитанным. Однако реальная зависимость концентрации полимера в
каждой из фаз от осевого отношения заметно отличается от
теоретической, особенно при малых степенях полимеризации.
Все же, как показал Германе [53] на том же образце поли-у-бен-
зил-?-глутамата, максимальная объемная доля полимера г>2»
при которой растворы однородны и изотропны, правильно
предсказывается формулой B9).
Таким образом, несмотря на некоторые количественные
несоответствия, можно заключить, что образование разбавленной
двулучепреломляющеи фазы — следствие только асимметрии
частиц и происходит по указанным выше причинам.
Согласно теории при малом положительном значении
параметра взаимодействия %i концентрация полимера в
анизотропной фазе становится очень высокой. Разделение фаз будет
происходить при критический величине этого параметра, не
сопровождаясь изменениями молекулярной конформации цепей. Хотя
до сих пор и не удалось подтвердить эти теоретические
предсказания количественно, все же можно с этой точки зрения
комментировать отдельные экспериментальные результаты.
Например, поли-?-пролин может существовать в
разбавленном водном растворе в форме очень ^асимметричных спиралей
(полипролин II) с ту?а«с-конфигурацией звеньев цепи
относительно имидной связи [30]. При повышении температуры
выпадает в осадок концентрированная полимерная фаза. Можно
показать, что при выделении этой фазы не происходит никаких
изменений конформации отдельных макромолекул, хотя она и не
может в принципе меняться в результате транс-цис-перехоца
[30]. При охлаждении вновь возникает разбавленный изотропный
раствор [54]. Наблюдаемому обратимому фазовому переходу
можно дать качественное объяснение на основании развитой
теории, если предположить, что xi увеличивается при
повышении температуры, что нередко наблюдается в полярных
системах.
Согласно данным по светорассеянию растворенные молекулы
коллагена (точнее тропоколлагена) имеют асимметричную
форму с длиной 3000 и поперечником 15А [29]. Если изменяется
температура или состав среды, в разбавленных растворах этих
макромолекул возникают различные типы агрегатов или
фибрилл, характер которых зависит от величины рН, ионной силы
и других внешних факторов [55]. Триспиральная структура на-
тивных молекул не изменяется при указанных превращениях,
а надмолекулярная фибриллярная структура всегда может быть
регенерирована.
73
Эти процессы снова представляют собой примеры переходов,
происходящих без каких-либо изменений в молекулярной
конформации. Однако теперь возникающее организованное
состояние уже зависит от специфических взаимодействий между
молекулами, причем они регулируются характером растворителя.
Развитая теория может служить основой для более глубокого
количественного анализа результатов, полученных на
коллагене, а сами эти результаты могут служить отправной
точкой для моделирования биологических процессов фибрилло-
генеза.
Различные рассмотренные фазовые равновесия и переходы
в системах полимер — растворитель можно изобразить
схематически, как это показано на
рис. 30. Процесс /
представляет собой обычное плавление
или кристаллизацию
полимеров, сопровождаемую конфор-
мационными изменениями. При
этом аморфная фаза ///
может содержать или не
содержать растворитель, но
состояние / всегда будет
соответствовать чистой кристаллической
фазе. Переход этой категории
был рассмотрен на стр. 47 и
56. Образование изотропного
разбавленного раствора II, в
котором молекулы сохраняют
конформационные
характеристики кристаллического состояния /, обозначено как процесс 2.
Его можно также рассматривать как обычное растворение, но с
сохранением молекулярной конформации, в отличие от процесса /.
Обратный процесс представляет собой образование чистой
упорядоченной фазы из разбавленного раствора анизотропных
молекул. Переход спираль —клубок обозначен как процесс 3.
Разбавленная тактоидная фаза V образуется из разбавленной
изотропной фазы в результате процесса 2' при незначительном
увеличении концентрации полимера.
Эта схематическая диаграмма указывает на существование
определенного сходства между различными процессами и на
важность рассмотрения полной области концентраций для
всестороннего описания бинарных полимерных систем. Например,
переход спираль — клубок (процесс 3) можно рассматривать
как проявление процесса плавления / в разбавленном растворе.
Однако для коллагена установлена обусловленность
макроскопического (обычного) плавления внутримолекулярным переход
дом спираль — клубок,
Рис. 30. Схематическое
представление переходов и фазовых
равновесий, включающих упорядоченные
конформации полимерных цепей [44]:
/ — плотная кристаллическая фаза; /' —
тактоидная анизотропная фаза раствора; // —
изотропный разбавленный раствор спиралей;
/// — статистические клубки.
74
В некоторых полимерных системах при низкой концентрации
полимера на фазовой диаграмме сосуществуют три фазы:
чистая упорядоченная, содержащая статистические клубки и
образованная отдельными асимметричными молекулами. Эта точка
по своему характеру аналогична эвтектической точке,
отражающей сосуществование твердой, жидкой и газообразной фаз
низкомолекулярного вещества *.
Плавление, вызванное химическими реакциями**
Процессы плавления и кристаллизации могут быть
обусловлены не только изменением таких параметров, как температура,
давление и концентрация, но также и некоторыми химическими
реакциями. Чтобы сдвинуть равновесие, необходимо изменить
химический потенциал мономерного звена в одной или в обеих
фазах. Это может быть вызвано любым взаимодействием
(реакцией) между компонентами растворителя и функциональными
группами полимерной цепи. Одна из фаз будет при этом
развиваться за счет другой. Химические реакции могут протекать во
всей области концентраций, не ограничиваясь разбавленными
растворами, в которых макромолекулы изолированы друг от
друга. Характер реакций, однако, может зависеть от
концентрации полимера, которая, поэтому, должна всегда приниматься во
внимание.
В двух- или многокомпонентных растворах, включающих
полимеры, могут протекать самые разнообразные реакции.
Например, один из типов реакций может заключаться в
образовании комплексов между каким-либо компонентом
растворителя и определенными боковыми группами полимерной цепи.
* [Все же схема (рис. 30) не исчерпывает существа вопроса и
многообразия морфологических форм. Так, например, далеко не очевидно (и есть
опыты, свидетельствующие о противоположном), что одна и та же твердая
кристаллическая фаза / будет получаться в результате процессов / и 2.
В случае полипептидов, проявляющих в кристаллическом состоянии а-C-по-
лиморфизм, две различные кристаллические фазы могут получаться, в
принципе, и при кристаллизации из области //, но в условиях, когда спирали
неустойчивы.
Наконец, при фазовых переходах, в которых участвуют жесткие стерж-
невидные молекулы, кинетические факторы играют не меньшую роль, чем
при обычной кристаллизации (см. гл. 8 и 9). Так, при высоких
концентрациях поли-у-бензил-А-глутамата переход типа //—/' осуществляется не в
результате одноактного процесса 2', а в серии последовательных превращений,
включающей различные морфологические формы [67]. — Прим. редактора].
** [Под химическими реакциями Манделькерн понимает в этом
параграфе любые сильные взаимодействия с растворителем, приводящие к
образованию более или менее устойчивых молекулярных комплексов. — Прим.
редактора].
75
Вполне вероятно, что стерические затруднения, возникающие в
результате образования такого комплекса, сделают
невозможным переход макромолекул в кристаллическое состояние. Это
соответствует крайнему случаю, при котором комплексообразо-
вание неизбежно приводит к аморфизании. В других случаях
оно может лишь затруднять кристаллизацию или изменять
характер кристаллической решетки.
В первом из этих случаев комплексообразование будет
сдвигать равновесие кристалл — жидкость в сторону,
благоприятствующую плавлению, а во втором — изменит температуру
плавления в ту или иную сторону до значения, характерного для
новой кристаллической структуры. Возможен также вариант,
когда лишь часть звеньев полимерной цепи претерпит
структурные изменения (при малой концентрации комплексообразую-
щего агента). Тогда гомополимер превратится в сополимер,
кристаллизация и плавление которого подчиняются совсем иным
закономерностям (см. гл. 4).
Можно предположить, что у кристаллизующихся
полиэлектролитов равновесие между двумя состояниями будет зависеть
от электростатических эффектов *. Например, весьма
маловероятно, что заряженная группа и связанный с ней противоион
могут войти в обычную кристаллическую решетку неионизиро-
ванного полимера. Экспериментальные исследования
показывают, что у таких полимеров, действительно, плавление может
быть вызвано изменением ионного состава окружающей среды.
Так, например, в разбавленных растворах поли-/,-глутамино-
вой кислоты и поли-/,-лизина [23] переход спираль — клубок
может быть вызван изменением величины рН. Поли-1-лизин
содержит аминогруппу в боковом радикале мономерного звена,
положительно заряженную ниже рН=9,5 и нейтральную выше рН =
= 10,5. Установлено, что спиральная форма существует только в
незаряженном состоянии, так что при понижении рН происходит
изотермический переход в форму статистического клубка.
Подобно этому поли-А-глутаминовая кислота обладает стабильной
спиральной формой при рИ ниже 5, когда карбоксильные
группы боковых радикалов практически не ионизованы. Переход в
форму статистического клубка происходит при повышении рН.
Стабильность упорядоченной структуры других
полиэлектролитов аналогичным образом определяется ионной силой или рН.
Так, температура плавления ДНК зобной железы теленка
понижается с 86 до ~25° С при уменьшении ионной силы или
повышении величины рН [56]. В отсутствие постороннего
«поддерживающего» электролита Гпл снижается вообще ниже комнатной
* [В соответствии с определением автора, ионизацию или связывание
противоионов можно также трактовать как химические реакции, — Прим. ре*
доктора].
76
температуры. Аналогичное поведение характерно также и для
синтетических полирибонуклеотидов.
Мы можем заключить, таким образом, что размещение
близких заряженных боковых групп в упорядоченном состоянии
термодинамически менее благоприятно, чем в аморфном состоянии.
Сдвиг равновесия между двумя состояниями может,
следовательно, происходить при изменении величины рН среды. В
дополнение к этому Шерага также предположил [57], что помимо
чистых электростатических эффектов рН может изменить в
полипептидах и белках характер водородных связей между
боковыми радикалами. Разрыв водородных связей, если они
являются составной частью кристаллической структуры, будут
способствовать плавлению. Химические реакции, которые
вызывают образование или нарушение межцепных поперечных
связей, также должны влиять на стабильность упорядоченной
структуры. (К более подробному рассмотрению роли поперечных
связей мы вернемся в гл. 6). Это обстоятельство существенно при
анализе поведения полипептидов и белков, принимая во
внимание относительную легкость, с которой можно контролировать
химическими методами межцепные дисульфидные связи.
Помимо уже рассмотренных, можно представить себе и многие
другие типы химических реакций, способных влиять на переход
кристалл — жидкость, однако нет никакой необходимости
рассматривать здесь все возможные варианты.
Для количественного описания переходов кристалл —
Жидкость, совмещенных с химической реакцией, необходимо точно
установить, в какой фазе происходит реакция и как она
отражается на химическом потенциале мономерного звена. Саму
реакцию можно описывать обычными методами химического
равновесия; следует выяснить, как химическое равновесие
накладывается на фазовое. Возникающие при этом многочисленные
возможные варианты следует анализировать отдельно. В качестве
примера можно рассмотреть простейший тип реакции комплек-
сообразования, протекающей только в аморфной фазе [34, 58].
Рассмотрим полимерную молекулу Р, содержащую п
боковых групп, каждая из которых способна образовывать в
аморфной фазе комплекс с компонентом растворителя С согласно
схеме:
Р + гС?РСг C0)
где полная концентрация полимера Р и комплекса PC остается
постоянной. Константа равновесия реакции может быть
записана в форме:
где К — константа равновесия индивидуальной элементарной
реакции комплексообразования.
77
Предполагается, что участки молекулы, на которых
развертывается реакция, взаимосвязаны. Комбинаториальный
множитель в уравнении C1) равен числу способов, которыми комплек-
сообразующий агент может быть распределен по п различным
группам.
Степень завершенности реакции может быть выражена
следующим образом:
где ас — активность реагента С.
Изменение свободной энергии при реакции составляет:
д/?р = _ п$т In A + Кас) C3)
Изменение химического потенциала на мономерное звено
равно:
*С - Им = - na*t In A + Кас) C4)
где jx* — химический потенциал звена, принявшего участие в
комплексообразовании;
NA — молярная доля звеньев цепи, содержащих реакционно-
способную боковую группу.
Если на мономерное звено цепи приходится больше одной
реакционноспособной группы, в выражение C4) следует ввести
соответствующий численный множитель. При соблюдении
обычных условий фазового равновесия выражение, характеризующее
изменение температуры плавления, принимает форму:
1 1 R VM , оч WA
'пл Тпл ДЯм V\ Ши
В случае экспериментов, проводимых при постоянных
концентрациях полимера (обычно в разбавленных растворах),
второе слагаемое в правой части выражения C5) характеризует
понижение температуры плавления при заданном составе в
результате химической реакции. Если произведение Кас мало [37],
то:
RTlR(v2)NAKac
где Tnjl(v2) и Тпл(щ) — температуры плавления системы
соответственно до и после реакции комплексообразования.
Структурные переходы в глобулярных белках,
обусловленные воздействием мочевины [59, 60], подчиняются соотношению
C6). В этом случае предполагается, что мочевина в равной ме-
78
ре взаимодействует с карбонильным кислородом и амидным
водородом пептидной группы. Поэтому правая часть формулы
C6) должна быть умножена на 2.
В общем случае, особенно для открытых систем, следует
применять более полную формулу C5), второе слагаемое которой
характеризует влияние химической реакции на температуру
плавления. Например, если имеет место очень сильная
тенденция к комплексообразованию (К велико), то достаточно совсем
малых изменений активности (или концентрации) комплексооб-
разующего компонента, чтобы вызвать заметный сдвиг фазового
равновесия. В подобных случаях нетрудно наблюдать
процесс плавления или кристаллизации в изотермических условиях.
И наоборот, если К мало, требуются большие изменения
активности, чтобы понизить температуру плавления. Однако в
непосредственной близости точки плавления малые изменения ас
могут все же в сильной степени изменять концентрацию свободных
и связанных компонентов, вследствие фазового перехода.
Приведенные примеры иллюстрируют характерные типы
химических процессов, вызывающие изменение температуры
плавления. Все процессы, изменяющие химический потенциал
мономерного звена, должны влиять на характер фазового
равновесия. К обсуждению влияния химических реакций на плавление
мы еще вернемся при рассмотрении процессов сокращения и
механохимии молекул фибриллярных белков в гл. 7.
Литература
1. М. L. Huggins, J. Phys. Chem., 46, 151 A942); Ann. N. Y. Acad. Sci.,
41, 1 A942); J. Am. Chem. Soc, 64, 1712 A942).
2. P. J. F lory, J. Chem. Phys., 10, 51 A942).
3. P. J. Flory, Principles of Polymer Chemistry, Ithaca-—New York, 1953,
p. 513.
4. P. J. Flory, R. R. Garrett, S. Newman, L. M a n d e 1 k e r n, J.
Polymer Sci., 12, 97 A954); Ref. 3, p. 568ff.
5. L. M a n d e 1 k e r n, Rubber Chem. Technol., 32, 1392 A959).
6. F. A. Quinn, Jr., L. Man del kern, J. Am. Chem. Soc, 80, 3178
A958); 81, 6533 A959).
7. A. M. В u e с h e, J. Am. Chem. Soc, 74, 65 A952).
8. P. J. Flory, L. M a n d e 1 k e r n, H. K. Hall, J. Am. Chem. Soc, 73,
2532 A951).
9. R. B. Richards, Trans. Faraday Soc, 42, 10 A946).
10. P. J. Flory, W. Rf Krigbaum, Ann. Rev. Phys. Chem., 2, 383 A951);
Ref. 3, p. 508ff.
11. D. E. Roberts, L. Mandelkern, J. Am. Chem. Soc, 77, 781 A955).
12. P. J. Flory, H. D. Be don, E. H. Keefer, J. Polymer Sci., 28, 151
A958).
13a. R. R. Garrett, P. J. Flory, Nature, 177, 176 A956).
13b. P. J. Flory, R. R. Garrett, J. Am. Chem. Soc, 80, 4836 A958).
14. A. 1. Z a i 4 e s, Kp\L Z.? 16f 265 A954)..
79
15. M. Feughelman, R. Langridge, W. E. Seeds, A. R. Stokes,
H. R. Wilson, С W. Hopper, M. H. F. W i 1 к i n s, R. K. Barclay,
L. D. H a m i 11 о n, Nature, 175, 834 A955).
16. R. Chiang, P. J. Flory, J. Am. Chem. Soc, 83, 2857 A961).
17. В. А. Картин, Г. Л. Слонимский, Усп. хим., 24, 785 A955).
18. Н. A. S t u a r t, Ann. N. Y. Acad. Sci., 83, 3 A959).
19. P. G e i 1, J. Polymer Sci., 47, 65 A960).
20. J. B. Jackson, P. J. Flory, R. Chiang, Trans. Faraday Soc, 59,
1906 A963).
21. P. Doty, A. M. Holtzer, J. H. Bradbury, E. R. В lout, J. Am.
Chem. Soc, 76, 4493 A954); P. Doty, J. H. Bradbury, A. M.
Holtzer, J. Am. Chem. Soc, 78, 947 A956); J. T. Yang, P. Doty, J. Am.
Chem. Soc, 78, 498 A956); 79, 761 A957).
22. P. Doty, K. Iamhori, E. Klemperer, Proc. Natl. Acad. Sci. V. S.,
44, 474 A958); P. Doty, A. W a d a, J. T. Y a n g, E. R. В lout, J.
Polymer Sci., 23,851 A957).
23. P. Doty, Revs. Mod. Phys., 31, 107 A959).
24. R. С Warner, J. Biol. Chem., 229, 711 A957).
25. A. Rich, D. R. Da vies, J. Am. Chem. Soc, 78, 3548 A956); G. F el-
sen f eld, A. Rich, Biochim. et Biophys. acta, 26, 457 A957); A. Rich,
Nature, 181, 521 A958); A. Rich, Biochim. et Biophys. acta, 29, 502
A958).
26. J. R. Fresco, P. Doty, J. Апъ Chem. Soc, 79, 3928 A957).
27. P. Doty, H. В о e d t k e r, J. R. Fresco, B. D. Hall, R. H a s e 1 k о г n,
Ann. N. Y. Acad. Sci., 81, 693 A959).
28. С Cohen, Nature, 175, 129 A955); J. Biophys. Biochem. Cytol., 1, 203
A955).
29. H. Boedtker, P. Doty, J. Am. Chem. Soc, 77, 248 A955); 78, 4267
A956).
30. W. F. Harrington, M. Sela, Biochem. et Biophys. acta, 27, 24A958).
31. R. V. Rice, Proc. Natl Acad. Sci. U. S., 46, 1186 A960).
32. J. Marmur, D. Lane, Proc Natl Acad. Sci. U. S., 46, 453 A960);
P. Doty, J. Marmur, J. Eigne г, С Schildkraut, Proc. Natl. Acad.
Sci. U. S., 46, 461 A960).
33. L. Pauling, R. B. Corey, H. R. В ransom, Proc. Natl. Acad. Sci.
U. S., 37,205 A951).
34. J. H. S с h e 11 m a n, Compt. Rend. Trar. Lab. Carlsberg, Ser. Chim., 29,
230 A955).
35. F. H. Zimm, J. K. Bragg, J. Chem. Phys., 28, 1246 A958); 31, 526
A959).
36. J. H. Gib bs, E. A. DiMarzio, J. Chem. Phys., 28, 1247 A958); 30,
271 A959).
37. L. Peller, J. Phys. Chem., 63, 1194, 1199 A959).
38. S. A. R i с e, A. W a d a, J. Chem. Phys., 29, 233 A958)
39. T. L. H i 11, J. Chem. Phys., 30, 383 A959).
40. J. H. S с h e 11 m a n, J. Phys. Chem., 62, 1485 A958).
41. В. Н. Zimm, J. Chem. Phys., 33, 1349 (I960).
42. K. Nagai, J. Phys. Soc Japan, 15, 407 A960); J. Chem. Phys., 34, 887
A961).
43. S. L i f s о n, A. R о i g, J. Chem. Phys., 34, 1963 A961).
44. P. J. Flory, J. Polymer Sci., 49, 105 A961).
45. B. H. Zimm, P. Doty, K. I so, Proc. Natl. Acad. Sci. U. S., 45, 1601
A959).
46. Л. Д. Ландау, Е. М. Лифшиц, Статистическая физика, Гостехиздат,
1951.
47. J. С. К е n d r e w, R. E. D i с k e r s о п, В. Е. Strandberg, R. G. Hart,
D. R. Da vies, D. С Phillips, V. С S h о re, Nature, 185, 422 A960).
48. M. F. P e r u t z, M. G. R о s s m a n n, A. F. С u 11 i s, H. M u i n h e a d,
G. W i 11, А. С, Т. N о г t h, Nature, 185, 416 A960),
ao
49. H. A. S с h e r a g a, J. Phys. Chem., 65, 699 A961).
50. P. J. Flory, Proc. Roy. Soc. (London), Ser. A, 234, 73 A956).
51. P. J. Flory, Proc. Roy. Soc. (London), Ser. A, 243, 60 A956).
52. C. Robinson, Trans. Faraday Soc, 52, 571 A956); Disc. Faraday Soc,
25, 29 A958).
53. J. Hermans, J. Col. Sci., 17, 638 A962).
54. F. С о r n i с к, L. M a n d e 1 к е г п, неопубликованные данные.
55. J. H. Highberger, J. Gross, F. O. Schmitt, J. Am. Chem. Soc,
72, 3321 A950); Proc Natl Acad. Sci. U. S., 37, 286 A951).
56. P. Doty, J. M a r m u r, N. S u e о k a, Brookhaven Symp. Biol., 12, 1
A959).
57. H. A. Scheraga, J. Phys. Chem. 64, 1917 A960).
58. P. J. Flory, J Cellular Сотр. Physiol., 49, (Suppl. 1) 175 A957).
59. J. G. Foss, J. A. Schellman, J. Phys. Chem., 63, 2007 A959). *
60. A. Nakajima, H. A. Scheraga, J. Am. Chem. Soc, 83, 1575 A961).
61. С. Я. Френкель, Введение в статистическую теорию полимеризации,
Изд. «Наука», 1965.
62. L. Pel le r, J. Phys. Chem., 63, 1194 A959).
63. М. В a u e r, L. N о s a n о w, J. Chem. Phys., 38, 578 A963).
64. J. Hermans, H. Scheraga, J. Am. Chem. Soc, 83, 3283 A961).
65. С. Я. Френкель, В. П. Кушнер, Биохимия, 28, 535 A963).
66. А. К. Крон, О. Б. Птицын, Тезисы докладов XII научной
конференции ИВС АН СССР, 1965, стр. 17; О. Б. Птицын, Ю. Е. Эйзнер,
Высокомол. соед., 10, 3 A965).
67. В. Г. Баранов, Т. И. Волков. С. Я. Френкель, ДАН СССР, 162,
836 A965).
@ Л. Манделькерн
Глава 4
Плавление
сополимеров
Введение
Введение в полимерную цепь звеньев, отличающихся от
основной цепи по химическому составу, стереохимическим
параметрам или по структуре, накладывает определенные
ограничения на процессы плавления и кристаллизации. Вполне вероятно,
что в таких макромолекулах не все звенья цепи могут
принимать участие в кристаллизации. По характеру поведения при
кристаллизации и по логическим соображениям к классу
сополимеров удобно отнести любые полимерные цепи, мономерные
звенья которых любым способом отличаются друг от друга.
Поэтому мы будем причислять к этой категории не только
истинные (химические) сополимеры, но также и макромолекулы,
состоящие из изомерных звеньев. Полимеры с другими типами
нерегулярности цепи, например точками ветвления, также
удобно включить в эту группу. Однако по причинам, которые будут
далее изложены, системы, обладающие межцепными
поперечными связями, необходимо рассматривать отдельно.
Проблемы, связанные с кристаллизацией и плавлением
сополимеров, не могут быть сформулированы единым образом.
Это обусловлено тем, что при кристаллизации сополимеров не
всегда требуется a priori, чтобы все типы звеньев принимали
участие в превращении. Введение различных звеньев
обусловливает многообразие процесса кристаллизации. Оно
определяется условиями кристаллизации, концентрацией звеньев
различных типов, характером их распределения по длине цепи и сте-
реохимическими взаимосвязями, которые существуют между
ними.
Таким образом, при теоретических расчетах неминуемо
приходится пользоваться теми или иными моделями, но на опыте
необходимо строго учитывать конкретные особенности
изучаемой реальной системы. Наиболее простой из возможных
случаев, достаточно часто встречающийся у обычных синтетических
сополимеров и некоторых более простых цепнь?х молекул, зз-
88
ключается в том, что кристаллизоваться способен лишь один
сорт звеньев, преобладающий в цепи. Однако возможны и такие
случаи, когда в одну и ту же кристаллическую решетку входят
звенья различных сортов.
С подобными проблемами приходится сталкиваться и при
исследованиях белков, у которых полипептидные цепи состоят
примерно из 20 сортов строго определенным образом
чередующихся вдоль цепи аминокислотных остатков. При наличии сте-
реохимической идентичности этих звеньев можно предположить,
что все они способны войти в одну и ту же решетку. Однако в
отсутствие такой идентичности подобное предположение уже не
будет справедливым. Способность лишь определенных звеньев
входить в кристаллическую решетку не только влияет на
характер плавления этого важного класса макромолекул, но и
определяет характер их рентгенограмм. Они не могут быть
правильно расшифрованы без знания того, какие из 20 сортов
звеньев способны кристаллизоваться. Это, в особенности,
проявляется при образовании так называемой сложно-складчатой
структуры в фибриллярных белках. Полипептидные цепи
находятся при этом в р-конформации. В такой системе стереохими-
чески идентичны лишь пептидные группы, тогда как различные
боковые радикалы зачастую не могут встраиваться в общую
кристаллическую решетку. Поэтому и возникает своеобразная
слоистая структура, в- которой слои представляют собой
правильные двухмерные кристаллические решетки [78, 79].
Предположение, что все звенья входят в решетку, может
привести к серьезным ошибкам при интерпретации микроструктуры
цепей.
При изучении общей проблемы плавления сополимеров
необходимо не только выяснить влияние композиционных и
структурных изменений на температуру плавления, но также выявить
различия между возможными формами кристаллизации.
Теория
Теория равновесной кристаллизации сополимеров
предложена Флори [1]. Следуя ей, мы рассмотрим модель сополимера,
который состоит только из одного типа способных
кристаллизоваться звеньев, обозначаемых как А-звенья. В расплавленном
(аморфном) состоянии эти звенья совершенно беспорядочно
распределены по объему полимера, но в каждой отдельно взятой
цепи имеется характерное распределение длин непрерывных
последовательностей А-звеньев, обусловленное конкретным
механизмом сополимеризации. Эти способные кристаллизоваться
последовательности сочленены некристаллизующимися участками
6*
83
цепи, образованными иекристаллизующимися В-звеньями или
смесью А- и В-звеньев. Таким образом, кристаллическое
состояние должно включать кристаллиты различной длины.
Длину кристаллической последовательности удобно
обозначить через ?, что означает число А-звеньев данной цепи,
которые пересекают кристаллит от одного конца до другого.
Продольному развитию кристаллита препятствует существование в
полимерной цепи некристаллизующихся В-звеньев. Поперечное
развитие кристаллитов обусловливается доступностью, или
концентрацией, А-последовательностей нужной длины в еще неза-
кристаллизовавшейся массе и уменьшением свободной энергии,
которое происходит при переходе сплошной последовательности
А-звеньев из аморфной в кристаллическую фазу.
Для количественного описания кристаллизации в качестве
одной из характеристик вводится вероятность Р^ того, что
произвольно выбранное А-звено в аморфном полимере
локализовано внутри сплошной последовательности по крайней мере из?
таких звеньев. Пусть, далее, Wj представляет собой вероятность
того, что звено, произвольно выбранное в аморфных областях
сополимера, является А-звеном, которое, к тому же входит в
последовательность из / А-звеньев, ограниченную с обоих концов
В-звеньями. Наконец, пусть Р^;- представляет вероятность того,
что за произвольно выбранным А-звеном следует в
определенном направлении еще по крайней мере ? — 1 звеньев этого же
типа. Тогда [1]:
(J-Z + Dw, |
Pi.j-—j i>i\ C7)
и
Pc.?,>t„_f; "-'+¦'"
Решение для последовательных значений ? приводит к
уравнению:
wz = Z(Pz-2Pz+l+Pl+2) C8)
Уравнения C7) и C8) общие и не зависят от присутствия
или отсутствия кристаллитов. Величины Р$ и w^ могут быть
отнесены как к исходному расплавленному полимеру, так и к
частично кристаллическому. На основе этих уравнений нетрудно
установить условия равновесия между двумя фазами.
В полностью расплавленном сополимере до развития какой-
либо кристалличности
•S--fe5- C9)
84
где NA — общее число А-звеньев в сополимере;
ХА — соответствующая молярная доля;
v°—число последовательностей из ? А-звеньев,
первоначально присутствующих в расплаве.
Предположим, что вероятность следования за некоторой
А-группой другой А-группы не зависит от числа
предшествующих им А-групп в последовательности. Эта вероятность
обозначается через р. Тогда уравнение C9) можно переписать так:
о С*аО-/»У
wt = D0)
С учетом уравнения C7) для первоначально расплавленного
сополимера получаем:
Р\ = ХкР1-х D1)
Если кристаллизация протекает в условиях
термодинамического равновесия, то вероятность Р\ того, что в
некристаллической области данное А-звено локализовано в
последовательности по крайней мере из ? таких звеньев, выражается в виде
Р?=ехр(-^) D2)
где Д/^— стандартная свободная энергия плавления
последовательности ? А-звеньев, принадлежащей
кристаллиту длины ?.
Д/7^ может быть выражена следующим образом
Л^ = гл/?м —2а D3)
где Д/\,— свободная энергия плавления на моль звеньев;
о—поверхностная свободная энергия на звено,
расположенное на конце кристаллита.
Принимается, что кристаллиты достаточно велики в
направлениях, поперечных осям цепей, так что вкладом избыточной
поверхностной энергии боковых граней в Д/^ можно пренебречь.
Тогда соотношение D2) можно записать в виде
Я|?=.1ехр(-Че) D4)
где
D5)
Ми I 1 1 \
R [т Tlj
И
D = exp(--^) D6)
Если в равновесии с расплавом находятся кристаллиты длин
?, ?+1 и ? + 2, то, комбинируя D4) и C8), получим выражение
85
для остаточной концентрации последовательностей А-звеньев
длиной ? в расплаве:
w\ = ID-1 [1 - exp (- 9)]2 exp <- ?9) D7)
Таким образом, необходимое и достаточное условие
кристаллизации можно представить неравенством
Р\>Р\ D8)
для одного или более значений ?. Подобно этому, условие
wl ^ wl для °ДН0Г0 или более значений ? также необходимо,
но уже недостаточно для кристаллизации.
Выражения D0) и D7), описывающие начальное и
равновесное распределение последовательностей в расплаве, являются
функциями параметра ?. Следовательно, имеется критическое
значение ?=?*, при котором обе функции распределения равны.
Аналитически это выражается как:
-{1п{РХА/р)+21п[A-р)/A-е-«)]}
** - д + \пр {™}
При ?<?**?>? больше, чем w°^ тогда как для ?>?*,
происходит обратное. Таким образом, ?* представляет собой
предельный размер, при котором кристаллиты могут существовать в
равновесии; при меньших размерах последовательностей
А-звеньев равновесное состояние кристаллитов уже поддержать нельзя.
С учетом D1) и D4) условие D8) можно переписать в
форме:
у/>^ е'% E0)
За исключением сополимеров, обладающих аномально
высокой тенденцией к правильному чередованию звеньев разных
типов, 1/D больше, чем XJp. Поэтому неравенство E0) можно
упростить до
е > — in/? Ei)
откуда следует:
1 1 Я
7* ДЛМ {пР
E2)
пл
Таким образом, должна существовать предельная
температура, выше которой условие E2) не может быть выполнено.
При более высоких температурах кристаллизация протекать не
может. Ясно, что эта температура представляет собой
равновесную температуру плавления сополимера Tnjl. Следовательно,
1 l R In/? E3)
Тип Кл А"м
86
Таким образом, можно ожидать, что для сополимеров только
с одним типом способных к кристаллизации звеньев понижение
температуры плавления (по сравнению с гомополимером типа А)
будет зависеть от теплоты плавления на моль
кристаллизующихся звеньев и от вероятности р образования сплошных
последовательностей этих звеньев, но не от среднего состава.
Параметр Д связанный с поверхностной свободной энергией концов
кристаллитов, не появляется в явной форме в выражении для
понижения температуры плавления.
Формула E3) выведена для системы, находящейся в
состоянии термодинамического равновесия. При этом подразумевается,
что продольный рост кристаллитов нарушается лишь за счет
некристаллизуемых звеньев, тогда как их развитие в поперечном
направлении целиком контролируется термодинамическими
факторами. При любых отклонениях от равновесия предлагаемый
анализ экспериментальных данных уже неприменим; и должны
быть приняты во внимание кинетические и релаксационные
факторы.
Если указанные условия удовлетворяются, то зависимость
температуры плавления от концентрации кристаллизующихся
звеньев определяется, в конечном счете, соотношением
вероятности р генерирования сплошных последовательностей А-звеньев
и общим составом. Для статистических сополимеров р может
быть приравнено к ХАу и уравнение E3) преобразуется:
1 1 D
— s-« — 1п*А E4)
Тпл К, ДЯМ А
Для упорядоченных или блок-сополимеров, характеризуемых
очень большими последовательностями кристаллизующихся
звеньев, p^XA> и понижение температуры плавления не столь
велико, как у статистических сополимеров. Для сополимеров
с тенденцией к правильному чередованию звеньев, напротив,
р<ХА, и наблюдается значительно большее понижение
температуры плавления *.
Соотношение E4) позволяет ожидать у статистических
сополимеров относительно большое понижение температуры
плавления. Например, для статистического сополимера
углеводородного типа с Д#м=1000 кал/моль и 7^ = 400°К при ХА=0,8,
вычисленное понижение температуры плавления составляет 61°.
* Если тенденция к правильному чередованию звеньев в сополимере
очень велика, то отношение Ха/р может превзойти 1/D. Следовательно,
неравенство E1) больше не выполняется, и условие E2) становится
неприменимым. Все же требования неравенства E0) могут удовлетворяться и
для очень малых значений ?. В этом специальном случае кристалличность
развивается посредством преимущественного образования кристаллитов
меньшей длинц,
87
При XA = Ofi снижение температуры плавления должно
достигать 115°.
Теория позволяет также оценить концентрацию А-звеньев,
которые находятся в кристаллическом состоянии при
температурах ниже Гпл. Эта оценка получается при усреднении по всем
последовательностям А-звеньев, включенным в кристаллиты.
Такой метод расчета приводит к некоторой переоценке степени
кристалличности, так как последовательности с длиной,
превышающей ?, также могут входить в кристаллиты длиной ?.
Если wjf—концентрация последовательностей из ? звеньев,
включенных в кристаллит, то
w\ = w\ — w\ E5)
a wK — доля А-звеньев в кристаллическом состоянии равна:
оо со
^к= 2>к= 2 («?-«$ E6)
Используя выражения D0) и D7) для w\ и w\,
соответственно, получаем
«v = ^(t-P)Vvf/><i-/>r2-'-e(i-*-T2 +
+ t,[(l-p)-,-(l-»-T1]) E7)
где ?*— критическое значение ?, определяемое формулой D9).
С помощью выражения E7) можно построить теоретические
кривые, описывающие изменение степени кристалличности с
температурой; разумеется, при этом приходится задаваться
определенными численными значениями входящих в выражение
E7) параметров. Соответствующие кривые представлены на
рис. 31 для статистических сополимеров с различными
концентрациями кристаллизующихся звеньев. Теоретические кривые
заметно отличаются от ожидаемых и наблюдаемых для гомо-
полимеров.
При сравнимых температурах уровень кристалличности
заметно понижается с повышением концентрации некристаллизую-
щихся звеньев. Наиболее характерной чертой кривых на рис. 31
является крайняя размытость кривых плавления.
Кристалличность исчезает в очень широкой температурной области, причем,
ширина диапазона плавления возрастает с повышением
концентрации некристаллизующихся звеньев. Так, даже для полимера,
содержащего только 5 мол.% В-звеньев, остаточная 50%-ная
степень кристалличности исчезает в температурном интервале
25 град. Таким образом, даже введение в цепь незначительного
числа нерегулярностей заметно изменяет картину плавления,
88
Другой отличительной чертой кривых на рис. 31 является
сохранение хотя и малой, но конечной величины
кристалличности в сравнительно большом температурном интервале перед
ее полным исчезновением. Ширина этого интервала существенно
увеличивается при повышении концентрации некристаллизую-
щихся звеньев. Для сополимера с 60% кристаллизующихся
звеньев она составляет приблизительно 30 град. Хотя полная
степень кристалличности,
сохраняющаяся в этих условиях, и
очень мала, она все же
существенна. Исчезновение этой
остаточной кристалличности
определяет равновесную температуру
плавления.
Рассмотренные характерные
черты плавления сополимеров
обусловлены, главным образом,
широким распределением длин
кристаллизующихся
последовательностей в статистических
сополимерах с вытекающим
отсюда распределением возможных
продольных размеров
кристаллитов. При определенной
температуре лишь те
последовательности могут принимать участие в
процессе кристаллизации, длины
которых превосходят ?*, причем
сам параметр ?* зависит от температуры. При изменении
температуры изменяется как состав, так и распределение
последовательностей в аморфной фазе.
В свою очередь, эти факторы определяют концентрацию
А-звеньев, способных к дальнейшей кристаллизации. Таким
образом, диффузные кривые плавления получают естественное
объяснение. Форма кривых плавления, хотя и зависит от
величины параметра Z), но не очень чувствительна к нему. По мере
приближения к температуре плавления ?* достигает очень
большой величины, тогда как концентрация последовательностей,
которые превышают эту длину, становится очень малой. Этим
объясняется вероятность длительного сохранения малых
степеней кристалличности при температурах чуть ниже Гпл (рис. 31).
Из-за их диффузного характера кривые плавления на рис.31
не представляются типичными для фазового перехода первого
рода. Однако кривые плавления такого типа являются
естественным следствием химического строения статистических
сополимеров. Более того, при Гпл исчезает wK и все ее
производные, так что наблюдается истинная прерывность, и
"*пл может
к
0,8
0,6
V*
0,2
. —^^^
¦ ^^"^^^^N**=0>95
;\. \&-деЧ
-^ NA-«ff\ \
чл-де \ \
>^ ¦ . X^j \j Vj
200 250 300 350 Ш
т,°к
Рис. 31. Теоретические кривые,
описывающие, согласно
формуле E7), зависимость
содержания А-звеньев wK от температуры
для статистических сополимеров
различного состава.
Вертикальные стрелки обозначают
температуры плавления сополимеров. Для
приведенного случая Т® = 400 К, Д/Л. =
пл м
= 1000 кал/моль и lnD = — 1.
89
быть теоретически идентифицирована как равновесная
температура плавления сополимера. Эта температура представляет
собой истинную термодинамическую температуру плавления, при
которой исчезают все кристаллиты, образованные из очень
длинных последовательностей А-звеньев. Именно ее следует
подставлять в формулу E4). Ниже Гпл будет сохраняться конечная
степень кристалличности, тогда как выше Гпл она исчезает. Тот
факт, что существование этой температуры предсказывается
теоретически, очень важен, так как ее определение на опыте по
полному исчезновению очень малых степеней кристалличности
представляет собой чрезвычайно трудную задачу.
Существует множество наблюдений, показывающих, что
плавление сополимеров на самом деле представляет собой
диффузный процесс, причем кристалличность исчезает в очень широкой
температурной области. В дополнение к этому также известно,
что достичь высокого уровня кристалличности в сополимерах
значительно труднее, чем в гомополимерах. Таким образом,
главные теоретические представления подтверждаются
экспериментально. Но так как нерегулярности могут вводиться в
полимер и без изменения химической природы мономерного звена,
диффузный характер плавления многих «гомополимеров» (если
судить по их составу, а не структуре) не всегда связывается с
принципиально сополимерным характером таких систем.
Следовательно, легко сделать ошибочный вывод, о том, что плавление
гомополимеров также носит диффузный характер. С другой
стороны, теория ясно демонстрирует, что детальный анализ
процесса плавления в условиях, близких к равновесным, дает очень
чувствительный метод выявления нерегулярностей цепи в
кристаллизующемся полимере.
Общие экспериментальные результаты
Наблюдаемый процесс плавления для некоторых типичных
сополимеров может теперь быть проанализирован с развитых
выше теоретических позиций. В качестве типичных примеров
рассмотрим поведение при плавлении ряда сополимеров,
состоящих главным образом из метиленовых звеньев —СН2— с
небольшим содержанием включенных в цепь звеньев типа—CHR—
[2], где R может быть я-С3Н7, СН3 или С2Н5. Образцы были
приготовлены сополимеризацией диазометана с соответствующими
количествами высших диазоалканов. Чтобы убедиться в стати-
* [Согласно развиваемой Л. Манделькерном концепции, гомополимером
можно назвать лишь абсолютно стереорегулярный полимер, — Прим.
редактора].
90
стическом распределении нерегулярностей в цепи, проведены
специальные дополнительные измерения. При кристаллизации
температура первоначально расплавленного сополимера очень
медленно через малые интервалы ступенчато понижалась вблизи
точки плавления в течение многих дней. Эта процедура
обеспечивала оптимальные условия приближения к равновесию.
vyA ,см3/г
I 1 I 1 1 1 ' ' ! I I Л
М 60 80 ЮО 120
Рис. 32. Кривые плавления полиметиленовых сополимеров различных
составов, содержащих некристаллизующиеся звенья типа [2]:
— «-пропил; метил; —•—этил. Композиция указана в процентах «сомономера».
На рис. 34 представлены зависимости удельного объема от
температуры для ряда таких сополимеров. После
кристаллизации нагрев проводили при очень медленном подъеме
температуры: Г в день в интервале от (Гпл— 15°) до ГПл- Состав
сополимеров, указанный на каждой кривой, выражен в виде
отношения CHR/100 СН2. Как и следовало ожидать, для
сополимеров были получены типичные S-образные кривые. Плавление
происходит в широком температурном интервале, который все
более расширяется по мере повышения концентрации некристал-
лизующихся звеньев. В соответствии с теоретическими
предпосылками, малые степени кристалличности сохраняются при
температурах лишь немного ниже Гпл. Переход в жидкое
состояние представляется, судя по этим кривым, как постепенный
асимптотический процесс.
91
Тщательный анализ данных вблизи температуры плавления
не обнаруживает никаких признаков прерывности. Это также
согласуется с теорией, которая, хотя и предсказывает наличие
прерывности как таковой на кривых плавления, но в то же
время, исходя из оценки размеров этой прерывности, объясняет
практическую невозможность ее
обнаружения на опыте. Таким образом,
теоретически ожидаемую ГПл
прямым путем измерить невозможно.
Для косвенного
экспериментального определения Тпл приходится
использовать переохлажденное
состояние. Температуру, при которой
исчезают заметные отклонения от
переохлаждения и принимают за
экспериментальное значение Тпл.
Именно так определенную
температуру плавления лучше всего
использовать для проверки теоретических
соотношений.
На рис. 33 пунктирная линия
показывает зависимость температуры
плавления от состава, вычисленную
по формуле E4) при подстановке
Д#м = 970 кал/моль СН2 и ТПЛ =
= 411,7° К [2]. Экспериментальные
точки располагаются заметно ниже
расчетной кривой.
Дифференциальный термический анализ метил- и
этилзамещенных полиметиленов дает по существу те же самые
результаты [3]. Следует указать, что такие пониженные
значения экспериментальных температур плавления, по сравнению с
теоретическими, вообще довольно часто встречаются у
сополимеров [4]. Частично эти расхождения могут быть объяснены
уже упомянутой невозможностью точного прямого
экспериментального определения Гпл. (Напомним, что при температурах,
лишь немного меньших истинной температуры плавления,
степень кристалличности падает до столь низкого значения, что
уже не может быть обнаружена обычными экспериментальными
методами.)
Следует отметить, что экспериментальные точки для
температур плавления этиловых и «-пропиловых сополимеров
укладываются на рис. 33 на одну и ту же линию. Однако для соло-
лимеров, содержащих боковые метильные группы, аналогичная
линия проходит несколько выше. Тот факт, что в этом случае
экспериментальные точки не экстраполируются к Т°ПЛ при
понижении концентрации «сомоцомера, означает, что малые кон*
2 4 6 8 Ю
Число апкипьных боковых групп
на 100 звеньев СН2
Рис. 33. Зависимость
температуры плавления полиметиле-
новых сополимеров от
состава [2]:
«Сомономеры»: о—«-пропил;
Д — этил, ф—метил;
теоретическая кривая по формуле E4).
92
центрации боковых метильных групп оказывают весьма слабое
влияние на Тпл. Это можно объяснить образованием твердого
раствора, т. е. кристаллической фазы, в которую при
равновесии с равным успехом входят звенья —СН2— и —СН(СН3)—.
С другой стороны, монотонное понижение Тпл в этиловом и н-
пропиловом ряду указывает на отделение чистой полиметиле-
новой кристаллической фазы.
На первый взгляд, кажется, что данные противоречат
результатам, полученным рентгенографическим путем для
аналогичных систем [5, 6]. В этих
исследованиях
зарегистрировано увеличение размеров
оси а элементарной
ячейки полиэтилена по мере
введения в цепь метиловых,
этиловых, н-пропиловых и
«-бутиловых боковых групп.
Однако, рентгенограммы
получены при температурах,
весьма далеких от ГПл. При
таких температурах
кристаллиты с некоторым
содержанием «сомономерных»
звеньев могут сохранить
устойчивость, которая
исчезает при более высоких
температурах.
Другим важным
фактором является уменьшение
размеров кристаллитов при
повышении концентрации
«сомономера». Такое
изменение размеров
кристаллитов резко понижает точность определения истинных
рентгенографических периодов. Поэтому несоответствие фазовых
диаграмм и рентгенографических данных носит скорее кажущийся,
нежели реальный характер.
Степень кристалличности может быть вычислена по
удельному объему, как это показано на рис. 34, для н-пропиловых и
этиловых сополимеров. Пунктирные линии на рис. 34 вычислены
согласно формулам D9) и E7) при использовании одинаковых
параметров Тпл, Д//м и InD для всех сополимеров. Параметр/?
приравнен к молярной концентрации СН2-звеньев в каждом
сополимере. Так как Тпл и А//м определяются независимо,
остается лишь один произвольный параметр In D, который
приходится подбирать для анализа всего экспериментального
w?
100 120
Т/С
Рис. 34. Зависимость степени
кристалличности (вычисленной по удельному
объему) от температуры для
сополимеров метилена с и-пропиловым (—) и
этиловым (—-•—) заместителями [2].
Теоретические кривые ( ) вычислены по
формулам D9) и E7).
93
семейства кривых. При правильном подборе этого параметра
наилучшее согласие между теорией и экспериментом получается
в температурной области, где степень кристалличности
изменяется наиболее быстро. По абсолютной величине наблюдаемая
степень кристалличности отличается от теоретической в области
как малых, так и больших значений.
Отклонения в области низких степеней кристалличности
обусловлены, с одной стороны, уже упоминавшейся
ограниченной чувствительностью существующих методов измерения, а, с
другой, — кинетическими затруднениями, возникающими при
необходимости сведения воедино крайне редких длинных
последовательностей кристаллизующихся А-звеньев (что требуется для
образования стабильных кристаллитов при высоких
температурах).
Отклонения при высоких степенях кристалличности могут
быть отнесены за счет пространственных ограничений,
накладываемых уже образовавшимися при более высоких
температурах кристаллическими областями, на дальнейшую
кристаллизацию в ходе охлаждения. Возникшая на начальных стадиях
кристаллизации надмолекулярная организация может
препятствовать дальнейшей кристаллизации оставшихся
кристаллизующихся последовательностей.
Для получения наилучшего согласия с опытом величина
InD была приравнена —11,5, что соответствует удивительно
большой свободной энергии поверхности раздела, оо= 170эрг/см2
или 4600 кал/моль цепей, выходящих из грани @01). Это
значение гораздо больше соответствующей величины, обычно
приписываемой неполимерным кристаллам, однако делать прямые
сравнения нельзя. У кристаллов, образованных из полимерных
цепей, существует составляющая свободной энергии, связанная
с диссипацией кристаллического порядка при прохождении
цепей через поверхностный слой (см. гл. 6 и 9). Большая
величина свободной энергии поверхности раздела для @01) граней
кристаллитов приобретает особое значение при обсуждении
вопроса о возникновении зародышей кристаллизации в
полимерных системах.
Несмотря на отсутствие полного согласия между
теоретическими и экспериментальными данными (см. рис. 32 и 34),
обусловленного в значительной мере несовершенством
экспериментальных методик, кривые, представленные на этих рисунках,
обладают всеми основными характеристиками вычисленных
теоретически кривых плавления. Следовательно, они в общем
типичны для плавления статистических сополимеров,
безотносительно к характеру введенных некристаллизующихся
звеньев.
94
Сополиэфиры и сополиамиды
Сополимеры, полученные поликонденсацией, обычно
характеризуются параметром вероятности генерирования
последовательностей р, который не зависит ни от состава сополимера, ни
от степени конверсии. Более того, в таких системах величина
р может быть приравнена к молярной доле кристаллизующихся
звеньев, которую легко определить аналитически. Плавление со-
полиэфиров и сополиамидов происходит в широком температур*
ном интервале, и кривые плавления, снова имеют S-образную
форму [7, 8]. Поэтому точно определить температуру плавления
очень трудно.
Зависимости температуры плавления от состава для
некоторых образцов сополиэфиров и сополиамидов приведены на
рис. 35. Сополимеры этого типа, звенья которых кристаллизуются
независимо друг от друга, проявляют определенные
характерные свойства. Например, температура плавления зависит только
от состава и не зависит от химической природы второго
компонента, что иллюстрируется данными для сополимеров полиэти-
лентерефталата и полигексаметиленадипамида. Полученные
результаты подтверждаются данными рентгеноструктурного
анализа, который показывает, что лишь один тип звеньев
принимает участие в кристаллизации. Все это хорошо согласуется со
статистическим распределением кристаллизующихся
последовательностей в сополимере. Характерно, что когда
концентрация второго компонента становится достаточно велика, он может
уже сам начать кристаллизоваться за счет первого компонента.
Зависимость температуры плавления от состава для этого
компонента характеризуется самостоятельной кривой, так что
пересечение двух независимых кривых дает типичный
эвтектический минимум. Такое поведение характерно для бинарных
статистических сополимеров с кристаллизующимися звеньями
обеих типов во всем диапазоне составов.
При тщательно проведенных, согласно требованиям теории,
экспериментах зависимости температуры плавления от состава
на каждой из ветвей кривых рис. 35 должны были бы
описываться формулой E4). В соответствии с этим на рис. 36
представлены зависимости l/TnjI от —\пХА для некоторых типичных
полиэфиров и полиамидов. Как видно из рис. 36, данные для
температуры плавления, в согласии с теорией, хорошо
укладываются на прямую линию. Во всех случаях зависимости линейно
экстраполируются к температуре плавления чистого гомополи-
мера. Аналогичные результаты получены и для других
полиэфиров и полиамидов.
Наклон прямых линий на этом типе графиков определяется
величиной А#м. Так, например, из данных по зависимости
95
температуры плавления от состава для полиэфиров дикаметилеи-
адипата — декаметиленизофталата получено значение Д#м =
= 3800 кал/моль повторяющихся звеньев*. Эта величина суще*
ственно ниже, чем 10200 кал/моль, которая получена для того
же полимера при изучении влияния низкомолекулярных
жидкостей на его температуру
плавления.
Таким образом, хотя теория
правильно предсказывает харак-
0,6 0,8 W
Рис. 35. Зависимость
температуры плавления от состава для
различных сополиэфиров и сополи-
амидов [9—11]:
ф-гполи(этилентерефталат-адипинат);0~
поли(этилентерефталат-себацамид); и —
поли(гексаметиленадипамид-себацамид);
Q — поли(гексаметиленадипамид-капро-
амид).
2,3
?1
3,0
?fl
2,2
2,0
2,2
2Р
¦г^%~~-%*~~~*~^'
1 1— ,.. 1
/
"~
-^^4
0,1
0.2 0,3 Oh 0,5
Рис. 36. Зависимость \/Тпл от
— 1пА"А длясополиамидов и
сополиэфиров:
/ — сополимер N, ЛГ'-себацоилпиперазина
с изофталоилпиперазином DJ; 2 —
сополимер декаметиленадипата с декаметилен-
изофталатом |8|; 3 — сополимер капро-
амида с гексаметиленадипамидом |10|;
4 —сополимер этилентерефталата с эти-
ленадипатом |9].
тер зависимости температуры плавления статистических
сополимеров от состава, измеренная величина Д#м значительно ниже
определенной другим методом. Отклонение носит общий
характер, что подтверждается результатами измерений на нескольких
различных системах.
В табл. 3 приведены результаты для систем, у которых Д//м
было определено по зависимости температуры плавления гомо-
полимера от содержания в системе низкомолекулярной
жидкости, а также из зависимости температуры плавления от
содержания некристаллизующегося компонента. Метод II дает
несколько заниженное значение Д#м по сравнению с первым. Это
кажущееся различие происходит либо от неточностей
экспериментальной методики, либо от последующего анализа.
* [В случае полиэфиров и полиамидов этот термин, обычно, более
уместен, чем «мономерное звено», — Прим» редактора].
96
Таблица 3
Сравнительные данные экспериментальных
значений ЛЯМ, кал/г
Полимер
Полидекаметиленадипинат
Полидекаметиленсебацинат
Поли-N, N'-себацоилпипе-
Полидекаметиленсебаца-
мид
Метод измерения Д#м 1
I
13,4 [8]
13,8 [8]
19,8 [7]
23 [8]
п
36 [12]
36 [8J
24,5 [7]
24,5 [13]
Примечание. I — Д#м определяли из зависимести 1/7*пл
от In Яд; II —Д#м определяли из зависимости Т гомополимера
от содержания в системе растворителя.
Возникшая проблема подобна ранее рассмотренной в связи
с систематическим занижением экспериментальной температуры
плавления по сравнению с теоретической у полиметиленовых
сополимеров. Занижение Д//м обусловлено, главным образом,
малочисленностью кристаллитов, образованных из длинных
последовательностей А-звеньев даже при полном равновесии. И в этом
идеальном случае наблюдалась бы лишь фиктивная
температура плавления из-за ограниченной чувствительности
экспериментальных методов.
Согласно теории, разность между регистрируемой
температурой при малых, но конечных степенях кристалличности и
истинной температурой плавления возрастает с понижением
концентрации А-звеньев. Зависимость между составом сополимера и
экспериментально определяемой температурой плавления в
действительности может быть приблизительно выражена
соотношением, аналогичным по форме E4). Температура плавления,
регистрируемая при малом, но конечном проценте
кристалличности, может быть вычислена по формуле E7). Результаты
такого расчета приведены на рис. 37, где использованы те же
параметры, что и при построении кривых рис. 31. При расчете
рассматривался (гипотетический) статистический сополимер,
содержащий до 40% некристаллизующихся звеньев.
Кривые рис. 37 представляют собой зависимости
температуры плавления от состава для разных уровней
чувствительности. Кривая 1 представляет собой идеальный случай, когда
можно совершенно точно зафиксировать момент исчезновения
длинных кристаллических последовательностей. Остальные кривые
представляют случаи, когда на самом деле сохраняется 1, 2 и
5% А-звеньев в кристаллическом состоянии, хотя измерения
7 Л. Манделькерн
97
указывают на полное исчезновение кристалличности. В
пределах ошибки отсчета температуры во всех случаях получаются
прямолинейные зависимости, внешне согласующиеся с
теоретическими по формуле E4), причем наклон кривых непрерывно
возрастает с уменьшением чувствительности метода.
Таким образом, если применить к реальным
экспериментальным данным формулу E4), неизменно будут получаться
заниженные значения А#м. Например, если для рассматриваемого
гипотетического полимера
Ю'/ъ'к Д#м=1000 кал/моль, то
при сохранении 1%
кристалличности, не
регистрируемого
экспериментальной техникой, эта
величина снизится до
780 кал/моль. Поэтому
экспериментальное
значение АЯМ сильно зависит
от чувствителньости
метода определения
кристаллического порядка. Это
приводит к
возникновению принципиальных
трудностей при интерпретации
зависимостей
температуры плавления от состава
статистических
сополимеров. Хотя и наблюдается
кажущееся согласие
экспериментальных данных
с формулой E4),
получаемое на ее основе
экспериментальное значение
ДЯМ, как правило, будет ошибочным. Следовательно, более
правильно определять значение Д#м из зависимости температуры
плавления гомополимера от содержания в системе
низкомолекулярной жидкости.
Рис. 37. Зависимости вычисленных
значений \/Тпд от — In XA для статистических
сополимеров в предположении различного
уровня чуствительности
экспериментальных методик:
1—регистрируется полное исчезновение
кристалличности; 2—4 — не регистрируется 1, 2 и Ъ%
остаточной кристалличности, соответственно.
При вычислениях по формуле E7) принимали
rj? =400°K, Mfx =1000 кал/моль, lnD = -l.
Геометрический изомеризм
Химически идентичные, но изомерные звенья цепи могут
придавать кристаллизационному поведению макромолекулы
выраженный сополимерный характер. Например, полимеры,
полученные из 1,3-диенов, могут содержать различные типы нерегу-
лярностей цепи. У полибутадиена могут существовать следую-
98
щие три типа структуры цепей:
("хИхн)
A,4-транс)
(Х_Х\
V н н /
A,4-цис)
[ Н )
I
сн2—с—
II
( сн2 J
A,2-виниловая)
Таким образом, полиизопрен, полихлоропрен, полибутадиен и
другие полимеры этого класса обладают звеньями, которые
могут существовать в транс-1,4- и цис-l, 4-конфигурациях, а также
содержать виниловые боковые радикалы в D- или
/.-конфигурациях. В природных диеновых полимерах подавляющее число
звеньев находится либо в цис- (каучук гевеи), либо в транс-
конфигурации (гуттаперча). Эти полимеры легко
кристаллизуются, и их поведение при плавлении типично для гомополиме-
ров. Однако состав или микроструктура цепей синтетических
диеновых полимеров, определяющие их кристаллизационное
поведение, зависят от методов и механизма полимеризации. Как
и следовало ожидать, сосуществование цис- и граяс-конфигура-
ций в одной цепи вызывает заметные отклонения от присущего
гомополимерам поведения при плавлении и кристаллизации.
Полихлоропрен [14] и полибутадиен [15], полученные методом
радикальной эмульсионной полимеризации, содержат ряд
пропорций различных звеньев, причем изомерный состав
сополимеров определяется температурой полимеризации [14, 16—18].
Количественный анализ микроструктуры этих полимеров проведен
с помощью ИК-спектроскопии. В области композиций, где
поведение при кристаллизации изучалось в деталях, способны
кристаллизоваться только 77?аяс-1,4-звенья. В благоприятных
условиях такие полимеры могут кристаллизоваться даже при
наличии 40—45% некристаллизующихся звеньев [15].
7*
99
На рис. 38 представлены зависимости удельного объема от
температуры для трех различных полибутадиенов в области
концентраций кристаллизующихся звеньев ЯА=0,64— 0,81.
Удельные объемы определяли дилатометрически при малых
скоростях подъема температуры, при этом плавление
осуществляли непосредственно вслед за кристаллизацией. Получены
типичные S-образные кривые плавления, соответствующие сопо-
лимерному характеру макромолекул. Фазовый переход
происходит в широком
температурном интервале, и
область перехода
расширяется по мере
повышения концентрации
некристалл изующихся звеньев.
Конечный участок кривой
плавления захватывает
тем больший
температурный интервал, чем
меньше концентрация
кристаллизующихся звеньев.
В этих экспериментах
снова было использовано
переохлаждение в качестве
вспомогательного приема
для определения Тпл.
Наблюдаемая на опыте
температура плавления
зависит от состава именно
так, как это следует из
формулы E4). Однако и
в этом случае рассчитанные из графиков значения Д//м
сомнительны по причинам, о которых уже говорилось выше.
Диеновые полимеры, например, цис-\,4~ и транс-], 4-поли-
изопрен и полибутадиен, а также 1, 2-полибутадиен, могут быть
закристаллизованы и методами анионной полимеризации [19,
20, 24]. Эти методы позволяют в широких пределах регулировать
изомерный состав цепей. В частности, могут быть получены
полибутадиены с концентрацией цис-1,4-звенъеву достаточно
высокой для того, чтобы именно они участвовали в
кристаллизации. При плавлении указанных сополимеров наблюдаются те
же закономерности, что и у полибутадиенов, полученных при
эмульсионной полимеризации, с кристаллизующимися транс-
1,4-звеньями [19]. Хотя значения температур плавления при
этом и различаются, все же общий характер кривых плавления
типичен для сополимеров. Посредством анионной
полимеризации можно получить полиизопрен достаточно регулярной
структуры, подобный по свойствам своим природным аналогам. Все
-25 -15 -5 5 15 25 35 45 55
ГС
Рас. 38. Зависимость удельного объема
от температуры для полибутадиенов с
различной концентрацией кристаллизующихся
/я/?аяс-1,4-звеньев [15].
Кривые 2 и 3 произвольно смешены вдоль оси
ординат.
100
же цепи этого синтетического полиизопрена содержат
небольшое количество изомерных транс-\\ 4-звеньев. Как ни мало это
количество, оно оказывается достаточным, чтобы затруднить
кристаллизацию по сравнению с природным гомополимером.
По-видимому, эти затруднения обусловлены некоторым
понижением температуры плавления из-за наличия некристаллизую-
щихся звеньев [4].
В диеновых полимерах, содержащих в первоначальном
состоянии только транс- или только ^ыс-конфигурацию звеньев,
можно химическими методами изменить конфигурацию части
звеньев цепи, не нарушая при этом скелетных связей. Реакции
этого типа могут быть проведены с помощью тиоловых кислот
[25], двуокиси серы [26] и излучений высокой энергии при
использовании подходящих сенсибилизаторов [27]. Изомеризация
должна происходить по тем же механизмам, что и в случае
низкомолекулярных олефинов. Такие механизмы включают
взаимодействие со свободным радикалом, в результате которого
происходит образование промежуточного аддукта с временным
превращением двойной связи в простую.-При распаде аддукта
происходит регенерация двойной связи; получится ли при этом
та же самая или новая конфигурация звена, зависит от
концентрации взаимодействующих веществ и равновесия реакции,
определяемого условиями ее проведения. Таким путем можно
вызвать далеко идущую изомеризацию цепей при сравнительно
небольшой концентрации введенных в систему агентов.
Спектроскопический анализ подтверждает осуществление в
указанных условиях [28] ^ыс-транс-изомерного перехода у диено-
зых полимеров. Кунин, Хиггинс и Уотсон [28] показали, что
ИК-спектры натурального каучука и гуттаперчи, обработанных
двуокисью серы, выявляют структурные изменения,
усиливающиеся с течением времени, пока в результате реакции не
устанавливается равновесие между изомерными формами.
Например, после обработки двуокисью серы первоначально чистых
qwc-1,4- или трансА, 4-полиизопрена в течение 24 ч при 140° С
возникают, судя по ИК-спектрам, совершенно идентичные
структуры, т. е. для каждого из полимеров образуется
равновесная смесь звеньев в цис- и г/ишс-конфигурациях. Установлено,
что в равновесии сосуществуют 57% транс- и 43% ^wc-двойных
связей. Это отношение цис- и т/?аяс-конфигураций полностью
соответствует равновесному составу смеси цис- и транс-изоме-
ров З-метилпентена-2 при той же самой температуре.
С точки зрения кристаллизационного поведения полимеров,
при этих методах обработки превращают гомополимер в
сополимер. Если реакция протекает беспорядочно, то должен
образоваться статистический сополимер, содержащий значительно
большую долю инверсий структуры, чем это можно было бы
ожидать по концентрации реагента. Поэтому следует ожидать
101
существенных изменений температуры плавления, процесса
плавления, кинетики кристаллизации и других связанных с
кристаллизацией свойств полимера. Эти предположения действительно
оправдываются.
Температура плавления гуттаперчи, например, может
настолько понизиться, что полимер при комнатной температуре
оказывается аморфным веществом, обладающим
высокоэластическими свойствами, которые присущи некристаллическим
полимерам при температурах выше температуры стеклования.
Подобно этому, достигается и заметное понижение температуры
плавления цис-l, 4-полибутадиена при изомеризации части
звеньев из цис- в граяс-конфигурацию [27]. Как будет показано
в гл. 8, сополимерный характер таких полимеров неизбежно
должен приводить к сильному запаздыванию кристаллизации. Это
согласуется с экспериментальными наблюдениями [27]. Следует
особо подчеркнуть, что принципы, определяющие характер
кристаллизации или плавления статистических полимеров
совершенно не зависят от природы нерегулярностей цепи или от того,
каким способом эти нерегулярности введены в макромолекулы.
Химическая формула участка полипептидной цепи может
быть записана следующим образом (в скобках указано
мономерное звено):
О R'"
II I
I н
н
н
1 у
н о
I Н ||
\c/N\d/C\
I
R"
Полипептид, содержащий одинаковые радикалы R, является го-
мополимером, и его кристаллизационное поведение зависит
только от характера аминокислотных остатков, из которых он
состоит. С другой стороны, если в цепи содержатся различные
радикалы R, выявляется сополимерный характер
макромолекул *.
В дополнение к химическим различиям в повторяющихся
звеньях здесь также возможно существование определенного
типа геометрического изомеризма идентичных звеньев. Согласно
Полингу и Кори [29], связь между углеродным атомом,
содержащим карбонильный кислород, и атомом азота
приблизительно на 40% двойная, ввиду резонанса между структурами:
Н н
Cv I Cv I
\С—N4 и >C=N +
|! \С ~СК \С
О
См. сноску на стр. 90.
102
В результате такого, «частично двойного», характера связен
амидные группы должны быть почти плоскими. Следовательно,
как это уже указывалось, и здесь должен существовать выбор
между цис- и грдяс-конфигурацией.
н о
н
Л/
L R
N-
н
^/
I
R J
Н
R I
Н
//ис-конфигурация амидной
группы в полипептидной цепи
Г^аяс-конфигурация амидной
группы в полипептидной цепи
Согласно Полингу и Кори [29] и Мацудзиме [30], транс-кон-
фигурация амидных групп полипептидных цепей, как правило,
более стабильна. Если все звенья макромолекулы полимера
обладают одинаковой конфигурацией, предполагается гомопо-
лимерный характер его поведения при кристаллизации и
плавлении. С другой стороны, если в полимере сосуществуют две
различные конфигурации хотя бы идентичных звеньев, можно
ожидать типичное уже для сополимеров поведение при
кристаллизации (за исключением того случая, когда звенья в обеих
конфигурациях способны входить в одну и ту же
кристаллическую решетку).
Следовательно, по аналогии с результатами для диеновых
полимеров, превращение гомополипептида в сополимер может быть,
в принципе, осуществлено изомеризацией звеньев из одной
конфигурации в другую. Эта изомеризация должна включать
поворот вокруг связи между карбонильным углеродом и азотом и
может быть осуществлена с помощью некоторых химических
реакций. В частности, такое превращение вполне возможно, если
реакция включает образование переходной структуры, в которой
«частично двойной» характер пептидных связей утрачивается
лишь временно. В других случаях опредленные реакции*, такие
как специфическое связывание некоторых ионов-, могут
способствовать стабилизации одной из двух резонансных структур.
Отдельные пептидные связи могут при этом терять свой
«частично двойной» характер. В этом случае восстановление
двойного характера связей потребует осуществления обратной
реакции. Развивается ли геометрический изомеризм или пептидные
связи приобретают характер простых, термодинамическая
стабильность кристаллического состояния, по сравнению с
жидким состоянием, существенно понижается.
Рассмотрим различные механизмы плавления полипептидных
гомополимеров. Плавление может происходить при сохранении
* [Здесь понятию «реакция» придается тот же смысл, что и на
стр. 75. — Прим. редактора].
103
цис- или гране-конфигурации в расплавленном состоянии, по
аналогии с плавлением чистых цис- или граяс-полиизопренов.
Возможно также и возникновение цис-транс-пзомерии, которое
должно понизить стабильность системы и привести к ее
плавлению. Вероятно развитие структуры цепей с большей свободой
вращения при условии, что некоторые или все пептидные связи
утратят свой «частично двойной» характер. Такой структурный
процесс тоже должен закончиться плавлением.
Микроструктура цепей в жидком состоянии, т. е.
конфигурация их звеньев, во всех этих случаях будет различной, хотя
макромолекулярная конформация статистического клубка будет
одна и та же. Именно поэтому следует очень тщательно
различать причины, вызывающие плавление, и структурные
характеристики цепей в получающемся расплаве. В целом, однако,
легкость и множество химических способов, которыми в
полипептидных системах может быть осуществлен переход кристалл —
жидкость, в том числе изотермический, не должны вызывать
удивление.
Стереоизомеризм
У определенных классов полимеров возможно
существование стереоизомерных химических идентичных звеньев.
Концентрация и последовательность распределения стереоизомеров
вдоль цепи оказывает существенное влияние на
кристаллизацию и плавление таких полимеров. Важным классом
полимеров с асимметричными или псевдоасимметричными атомами
углерода является ряд с общей формулой:
f Н X )
1
L_c_c-
I I
I H Y \п
где X и Y — два различных боковых радикала.
Полимеры этого типа могут быть получены из а-олефинов и
соответствующим образом замещенных виниловых мономеров.
Более сложный тип стереоизомеров образуется в том случае,
если каждый углеродный атом содержит различные заместители,
как в случае полимеров, приготовленных из 1,2-замещенных
этиленовых мономеров. В рассмотренном более простом случае,
если произвольно представить цепь в виде плоского зигзага,
радикалы X или Y могут быть расположены по одну или по обе
стороны плоскости зигзага, и такое же расположение может
повторяться или не повторяться в соседних мономерных звеньях.
104
Если псевдоасимметричные атомы углерода обладают
идентичными конфигурациями, т. е. расположение радикалов X и Y
относительно плоскости зигзага одинаково вдоль всей цепи либо
эти конфигурации чередуются или изменяются правильным
образом вдоль цепи, то можно ожидать, что такие
макромолекулы будут кристаллизоваться, как гомополимеры. Вполне
возможно также, что конфигурации звеньев цепи чередуются
совершенно нерегулярным образом. Цепи приобретают сополимер-
ный характер, и уже не удивительно, что в течение длительного
времени полимеры рассматриваемого типа не могли быть
закристаллизованы из-за недостаточной стереорегулярности
микроструктуры цепи.
В серии выдающихся исследований Натта с сотрудниками в
совершенстве разработали специальные методы анионной
полимеризации [31], позволяющие получать кристаллизующиеся
полимеры из виниловых мономеров, содержащих различные
заместители, и из а-олефинов. Метод осуществления стерического
контроля в этих полимеризационных процессах до сих пор не
вполне ясен. Вероятнее всего при полимеризации проявляется
направляющее действие каталитического комплекса. Позднее
на основе термодинамических принципов, ранее
сформулированных Хаггинсом [32], стереорегулярные полимеры из виниловых
мономеров удалось получить и радикальной полимеризацией
[33—36]. Предполагается, что в этом случае стереоконтроль
обусловливается направляющим влиянием растущего конца
цепи. Вариация стереорегулярности достигается с помощью
изменения температуры полимеризации; при этом используется
небольшая разница энергий активации присоединения звеньев
в двух возможных конфигурациях [33, 37].
Могут быть рассмотрены два предельных варианта
микроструктуры цепи. В первом случае все звенья цепи обладают
идентичными конфигурациями — это изотактический полимер.
Во втором случае осуществляется правильное чередование двух
возможных конфигураций; такой полимер называют синдиотак-
тическим [31].
Полимерная молекула вовсе не обязательно должна быть
полностью изотактической или синдиотактической. В принципе
может существовать бесконечно большое число промежуточных
микроструктур, начиная с полностью статистического
распределения последовательностей в той или иной конфигурации, когда
в кристаллизации может участвовать лишь один тип звеньев,
до предельных структур упорядоченных (стереоблочных)
сополимеров, где синдиотактические и изотактические
последовательности (стереоблоки) могут кристаллизоваться независимо,
в пределах одной молекулы. Звенья, изотактические друг к
другу, могут быть правращены в синдиотактические только в
результате разрыва и восстановления ковалентных связей.
105
Сополимерный характер стереонерегулярных полимеров
открывает возможность систематического анализа и исследования
их свойств на основе изложенной выше теории Флори [1].
Практически здесь следует попросту приложить общую теорию к
рассмотрению влияния различных типов нарушения стереорегу-
лярности на кристаллизацию и плавление. Кольман [38]
рассмотрел эту проблему в предположении, что образование
стереопоследовательностей определяется чисто случайными
процессами. Основной задачей является вывод соотношений,
связывающих параметр вероятности генерирования
последовательностей р и вероятность возникновения либо синдиотакти-
ческой, либо изотактической последовательности в растущей
цепи.
Для примера рассмотрим схему радикальной полимеризации
винилового мономера с нормальным присоединением звеньев
голова к хвосту [38]:
Г Н
I
-С-
Н Y
4
Г Н Y л
-С-С-
Н X
3
Н Y
-С— С-
Н X
2
Н X
-С—С-
Н Y
1
Звенья 3 и 2 присоединены друг к другу в изотактической
конфигурации, а 3 и 4 — в синдиотактической. Можно говорить,
соответственно, об изо- или синдиотактическом присоединении.
Константы скорости роста цепи при изотактическом или
синдиотактическом присоединении звеньев соответственно
обозначаются ka и ^. В первом приближении считают, что ka и k^
не зависят от предшествующих присоединений звеньев, включая
и последнее. На основании этого предположения вероятность а
изотактического присоединения равна:
kn E8)
а =
ka + b$
а вероятность синдиотактического присоединения равна:
Р =
*3
K + fy
Eу)
Вероятность того или иного типа присоединения,
следовательно, постоянна. Однако эти две вероятности не равны друг
другу, за исключением особого случая, когда ka = k„, из чего
следует, что а = р = 0,5. Полимеры с такой микроструктурой
называются атактическими. Но следует помнить, что ka вовсе
необязательно равно &р. Разность в свободных энергиях
активации изо- и синдиотактического присоединения приводит к раз-
106
личиям в величинах ka и k^. В частности, даже при малой раз-
ности энтальпии активации один тип присоединения начинает
преобладать над другим по мере понижения температуры
{33, 37].
Таким образом, в рассматриваемом случае концентрация
кристаллизующихся звеньев и распределение их сплошных
последовательностей однозначно определяется относительными
скоростями роста цепи при изо- и синдиотактическом
присоединении.
В условиях, когда в кристаллизации могут принимать
участие только изотактические последовательности, р и ХА
формул C7) и E7) совпадают и могут быть приравнены к а-доле
звеньев, расположенных изотактически по отношению к их
ближайшим соседям в заданном направлении. Аналогичный
результат получается, если возможна кристаллизация только синдио-
тактических звеньев. Тпл может быть определена как
равновесная температура плавления либо полностью изотактического,
либо полностью синдиотактического полимера.
Следовательно, кристаллизация и плавление полученных сте-
реонерегулярных полимеров (см. рис. 30) должны подчиняться
тем же закономерностям, которые были уже установлены для
обычных статистических сополимеров, описанных выше.
Решение проблемы сводится к количественному определению
концентрации звеньев каждого типа.
Может существовать и другой механизм присоединения
звеньев при радикальной или ионной полимеризации, при ко
тором стереорегулярные последовательности не возникают по
такому простому статистическому закону. Вероятность
определенного присоединения может зависеть от характера
предыдущих присоединений, в частности от конфигурации последнего
звена растущей цепи. В такой ситуации приобретают значения
различные условные вероятности. При учете влияния
конфигурации только последнего звена возможна следующая схема
расчета [38]. Если два последних звена цепи находятся в изотакти-
ческой конфигурации, то аи представляет условную вероятность
того, что следующее добавляемое звено также присоединится
изотактически, тогда как (Зи — условную вероятность
синдиотактического присоединения. Аналогично, ас — условная
вероятность того, что за синдиотактическим присоединением последует
изотактическое, а рс — условная вероятность следования
синдиотактического присоединения за синдиотактическим. Тогда
«с+Рс=1 J
Безусловная вероятность (а) того, что два произвольно
выбранных последовательных звена расположены в изотактической
107
конфигурации, получается суммированием по всем возможным
вариантам предшествующего расположения. Таким образом:
Р = РРс + «Ри J ( '
Если стереопоследовательности генерируются в результате
такого процесса, то ХА можно приравнять к a, a p — к аи, при
условии, что кристаллизуются только изотактические
последовательности звеньев. Принимая во внимание небоходимые и
достаточные условия кристаллизации сополимеров, неравенство E0)
можно переписать в форме:
т1~1 >-&*'* F2)
При образовании стереопоследовательностей в благоприятных
условиях а>аи предельная температура, при которой возможна
кристаллизация (температура плавления), выражается таким
образом:
п~ = 1паи F3)
Таким образом, для установления соотношения между
температурой плавления и стереосоставом недостаточно определение
одной лишь композиционной переменной а. Необходимо также
знать и условную вероятность аи. Однако, если всякая
корреляция присоединений отсутствует (а = аи) или имеет место
неблагоприятная корреляция (а<аи), то выражение для
температуры плавления может быть получено из формулы типа E2), где
р заменено на а. Аналогичные результаты, разумеется,
получаются и в том случае, когда только синдиотактические
последовательности звеньев способны кристаллизоваться.
Здесь снова следует строго разграничивать полностью
регулярные полимеры, т. е. содержащие либо только изотактические,
либо только синдиотактические последовательности, когда
ожидается характерное для гомополимеров кристаллизационное
поведение, и неполностью регулярные полимеры, даже если
кристаллизуются звенья только одного типа. В последнем случае
ожидается поведение при кристаллизации и плавлении,
характерное для сополимеров.
Таким образом, хотя полученные различным образом стерео-
регулярные полимеры и можно называть изотактическими или
синдиотактическими, следует помнить, что на самом деле
обычно всегда имеются некоторые нарушения стереорегулярности
цепи. Хотя предложенная статистическая схема образования сте-
реорегулярных последовательностей и не обязательна, все же
ясно, что в конечном счете характер кристаллизации полимеров
108
определяется полной концентрацией кристаллизующихся звеньев
и распределением их последовательностей вдоль цепи.
Экспериментальные исследования указывают, что
неполностью стереорегулярные полимеры действительно ведут себя, в
этом смысле, как сополимеры. Хотя количественное
сопоставление провести трудно, качественные подтверждения этого вывода
имеются. Так, используя метод
анионной полимеризации и
изменяя условия реакции, в
частности тип катализаторов, Нат-
та [39] приготовил из
пропилена ряд кристаллических
образцов, температура плавления
которых изменялась в
пределах от 176° С (предполагается,
что эта температура
характеризует полностью изотактиче-
ский полимер) до 106° С;
достижимая степень
кристалличности убывала, соответственно
от 85 до 20%. Эти
результаты подтверждают сополимер-
ный характер кристаллизации
таких полимеров и тот факт,
что кристаллизующиеся звенья
образуют беспорядочно
распределенные по цепи
последовательности.
Как можно было ожидать,
экспериментальная
зависимость удельного объема от
температуры у этих полимеров
в общих чертах воспроизводит
теоретическую — для
плавления сополимеров [40]. Ньюмен
подробно изучил зависимость
удельного объема от температуры для различных растворимых
фракций полипропилена [41]. Результаты его исследований
приведены на рис. 39. Уменьшение стереорегулярности
сопровождается понижением, температуры плавления и увеличением
диффузности плавления. Соответствующие кривые плавления
аналогичны таковым для статистических сополимеров, содержащих
химически различающиеся мономерные звенья. Это обусловлено,
разумеется, тем, что кристаллизация и плавление в обоих
случаях подчиняются одним и тем же принципам.
При гомогенной радикальной полимеризации таких
виниловых мономеров, как метилметакрилат [33], винилацетат [35],
Рис. 39. Зависимость удельного
объема от температуры для нефрак-
ционироваиного образца
полипропилена и его четырех растворимых
экстрактов [41]:
1 — эфирный экстракт, закаленный; 2—пен-
тановыи экстракт, отожженный; 5—гексано-
вый экстракт, отожженный; 4 — триметилпен-
тановый экстракт, отожженный; 5 — исходный
полимер, отожженный; 6 — чистый
кристаллический полимер.
109
винилхлорид [36] и изопропил-или циклогексилакрилат [34],
контроль стереорегулярности может быть осуществлен посредством
изменения температуры реакции. По мере понижения
температуры полимеризации кристаллизация получаемых полимеров
заметно облегчается. Это может быть отнесено за счет разности
величин ka и ?р и предпочтительности присоединения звеньев в
определенной конфигурации при понижении температуры
реакции. В детально изученных системах по мере понижения
температуры полимеризации происходит в основном развитие синдио-
тактических последовательностей [35, 36, 42].
Одна из главных проблем для полимеров вообще и для
неполностью стереорегулярных в частности заключается в их
потенциальной возможности кристаллизации, которая может
существовать, даже если они получены в аморфной или не в явно
кристаллической форме. С этой проблемой часто приходится
сталкиваться при исследовании сополимеров различного типа,
и для ее правильного решения необходимо принимать во
внимание все детали механизма и кинетики кристаллизации,
рассмотренные в гл. 8. Установлено, например, что некоторые не
полностью стереорегулярные полимеры, как, например, полистирол,
полученный на олефиновых катализаторах [43], или полиметил-
метакрилат, приготовленный методами радикальной или ионной
полимеризации [33, 44, 45], не получаются сразу в
кристаллической форме. Однако при воздействии специфическими
растворителями или жидкостями, в которых образцы способны
набухать при повышенных температурах, полимеры могут перейти
в кристаллическую форму [33, 43—45].
Затруднения при кристаллизации могут быть связаны,
прежде всего, с большим понижением температуры плавления из-за
наличия даже небольших количеств нерегулярности цепи.
Вероятно, что в рассмотренных случаях из-за нерегулярной
структуры цепей происходит столь сильное понижение температуры
плавления по сравнению с полностью стереорегулярным гомопо-
лимером, что она слишком приближается к температуре
стеклования. Процесс кристаллизации при этом чрезвычайно
затрудняется по кинетическим причинам, хотя и остается
термодинамически выгодным.
Кристаллизация полимера может быть облегчена обработкой
растворителем или добавлением к нему пластифицирующих
жидкостей, понижающих температуру стеклования в большей
степени, чем температуру плавления. Так, например, добавление
растворителя с большим молярным объемом, но малой
растворяющей способностью (положительные xi) согласно формуле A3)
приводит лишь к минимальному понижению температуры
плавления полимера при данной концентрации. Понижение же
температуры стеклования неразбавленного полимера зависит главным
образом от концентрации, а не от характера растворителя.
ПО
Следовательно, используя плохие растворители, можно
существенно увеличить интервал между температурами
стеклования и плавления по сравнению с неразбавленной системой. С
кинетической точки зрения, при этом создаются более
благоприятные условия для кристаллизации. Растворители упомянутого
типа, действительно, были с успехом применены для
осуществления кристаллизации.
Таким образом, хотя полимерные цепи сами по себе могут
обладать достаточной для кристаллизации регулярностью
структуры, кристалличность не будет развиваться до тех пор, пока
для нее не будут созданы кинетически благоприятные условия.
Нельзя определенно и категорически утверждать, что полимер и
в особенности сополимер не способен кристаллизоваться, если
не достигнуты оптимальные условия кристаллизации *.
Возможность более простой регистрации, когда не полностью
стереорегулярный полимер получается в кристаллической
форме непосредственно в ходе его приготовления, определяется
конкретными условиями полимеризации. Еще задолго до детальных
исследований процессов полимеризации, приводящих к
образованию стереорегулярных полимеров, было известно, что
некоторые полимеры (например, поливинилхлорид, полиакрилонитрил,
политрифторхлорэтилен и поливиниловый спирт) получаются
обычно сразу в кристаллической форме, несмотря на большую
вероятность стереохимических нерегулярностей. Нередко в
подобных случаях рентгеноструктурныи анализ не подтверждает
с полной определенностью наличие развитой кристалличности.
Однако особенно для поливинилхлорида [46, 47] и полиакрило-
нитрила [48], анализ свойств этих полимеров в растворе и
механических свойств дал явные подтверждения их кристалличности.
Последующее получение указанных полимеров новыми
методами, обеспечивающими повышенную регулярность цепей,
также подтвердило эти наблюдения [36, 49].
Разветвленность
Другим типом нерегулярности цепей, влияющим на процессы
кристаллизации и плавления, является разветвленность,
поскольку точки ветвления отличаются в структурном отношении
от остальных звеньев цепи. Пример введения коротких
ветвлений одинаковой длины уже был приведен для сополимеров
полиэтилена. Экспериментальная оценка эффекта разветвленности
* [Все же следует помнить, что возможны случаи, когда абсолютно сте-
реорегулярные полимеры принципиально не могут кристаллизоваться из-за
стерических помех, вносимых массивными боковыми группами. — Прим.
редактора].
Ill;
может быть произведена при сравнении плавления линейного и
разветвленного полимера с одинаковыми мономерными
звеньями. С этой точки зрения очень удобен полиэтилен, поскольку он
может быть получен в любой из структурных форм.
Измерения теплоемкости [50, 51] и зависимости удельного
объема от температуры [52, 53] указывают, что плавление
разветвленного полиэтилена происходит в
очень широком температурном
интервале: приблизительно 50%
кристалличности исчезает в интервале более 40°. Это
резко отличается от поведения при
плавлении ранее рассмотренного линейного
аналога (см. гл. 2).
Сравнение двух типов полимеров
дано на рис. 40. Широкая область
плавления разветвленного полиэтилена (кривая
2) характерна., для плавления*полетмеров,
нерегулярности
то
от[
5 0Щ
^ ож\
от
ж 70 90 но W 150 содержащих
7, °С ~
в - цепи.
С другой стороны, плавление линейного
полиэтилена характерно для гомополи-
меров. Оно происходит очень резко,
причем приблизительно 70%
кристалличности исчезает в узком интервале
температур порядка 3—4 град. Как уже
указывалось, при более строгих условиях
кристаллизации процесс плавления может происходить в еще более
узком температурном интервале. Можно предположить, что
другие разветвленные кристаллические полимеры будут подобным
же образом изменять характер плавления по сравнению со
своими линейными аналогами.
Рис. 40. Зависимость
удельного объема от
температуры [53]:
/ — линейный полиэтилен
(полиметилен):
2—разветвленный полиэтилен.
Упорядоченные сополимеры*
До сих пор мы рассматривали плавление и кристаллизацию
сополимеров, кристаллизующиеся звенья которых были
совершенно беспорядочно или почти беспорядочно распределены
вдоль цепи. Однако могут быть получены сополимеры с
совершенно иным типом распределения последовательностей. Особый
интерес представляют два предельных случая распределения.
Одним из них является упорядоченный, или блок-сополимер, у
которого, в противоположность статистическому сополимеру,
кристаллизующиеся звенья располагаются в очень длинные
непрерывные последовательности. В другом случае наблюдается
[Имеются в виду блок-сополимеры. — Прим. редактора].
112
сильная тенденция к строгому чередованию кристаллизующихся
и некристаллизующихся звеньев. Так как соотношение между
параметром вероятности генерирования последовательностей р
и составом в этих двух крайних случаях различно, следует
ожидать, что поведение этих сополимеров при плавлении или
кристаллизации также будет совершенно различным.
При строгом чередовании двух структурно различающихся
звеньев вполне вероятно образование новой кристаллической
решетки. Например, мономерные звенья сополимера этилена и
окиси углерода в молярном отношении 1 : 1 имеют вид (СН2—
—СН2—СО) п. Такой полимер был действительно получен [54].
Он обладает кристаллической структурой, отличной от
.наблюдаемой у полиэтилена, особенно в направлении оси с.
Температура плавления полимера 180° С, т. е. значительно выше, чем у
полиэтилена. Эта структура сохраняется, даже если отношение
этилена к окиси углерода возрастает до 1,1 : 1. Дальнейшее
увеличение относительного содержания этилена приводит к
возникновению характерной для
полиэтилена кристаллической ячейки.
Параметр вероятности
генерирования последовательностей
упорядоченного или
блок-сополимера значительно превосходит
молярное содержание
кристаллизующихся звеньев Ад.
Следовательно, температура плавления
не понижается в такой сильной
мере, как у статистического
сополимера того же состава, и,
согласно формуле E7), интервал
плавления становится заметно
более узким.
Эти предположения
подтверждаются наблюдениями Коффи
и Мейрика [55], которые изучали зависимости температуры
плавления от состава в ряду упорядоченных сополиэфиров полиэти-
ленадипината и полиэтиленсебацината. В противоположность
результатам, полученным для статистических сополимеров того
же самого состава (см. рис. 35), температура плавления
упорядоченных сополимеров до определенного момента не зависит от
второго компонента. Соответствующие результаты приведены на
рис. 41.
В таких сополимерах происходит плавление только
основного компонента, и поэтому температура плавления в широком,
хотя и ограниченном, диапазоне композиций не зависит от
изменения состава. Однако, если рассматривать полный
диапазон, оказывается, что зависимость температуры плавления от
т °с
во
70
60
50
[ " * * ' т^\
1_
¦" • •
0.2 0,4 0,6 0,6 1,0
Содержание этиленадипината, мол доли
Рис. 41. Зависимость
температуры плавления упорядоченных
сополимеров этиленадипината и
этиленсебацината от состава [55].
8 Л. Манделькерн
из
состава описывается ступенчатой кривой. Это отражает тот
факт, что при достаточно большой концентрации «главным»
становится второй компонент.
Введение в цепь различных мономерных звеньев в виде
сплошных блоков позволяет изменять химический состав
полимерных молекул без изменения физических и механических
свойств, связанных с высокими температурами плавления и
уровнями кристалличности. Сохранение указанных свойств
показано на примере блок-сополимеров этилентерефталата и окси-
этиленгликоля [56]. Введение в виде блока 30% второго
компонента приводит лишь к небольшому снижению температуры
плавления. Это существенно отличается от поведения
статистических сополимеров того же состава.
Возможность получения упорядоченных сополимеров не
ограничивается сополиэфирами или сополиамидами. Наряду со сте-
реоблочными полимерами полипропилена [39] и
блок-сополимерами этилена и пропилена '[57], известны также блок-сопол«меры
изотактического и синдиотактического полиметилметакрилата
[44] *.
Весьма вероятно, что упорядоченные сополимеры более
сложного строения, чем рассмотренные, могут существовать среди
природных фибриллярных белков и, возможно, нуклеиновых
кислот. Хотя фибриллярные белки состоят из большого числа
химически различающихся аминокислотных остатков, тем не менее,
многие из этих остатков могут входить в одну и ту же
кристаллическую решетку. Если бы все мономерные звенья принимали
участие в процессе кристаллизации, следовало бы ожидать
типичное для гомополимеров поведение при плавлении, несмотря
на химически неоднородный состав цепи. Однако, если
определенные звенья исключаются из кристаллизации вследствие сте-
рических затруднений, то процесс плавления изменит свой
характер на типичный для сополимеров.
Следовательно, a priori не требуется поддержание стехиоме-
трической идентичности между общим составом белка или
нуклеиновой кислоты и составом звеньев цепи, участвующих в
кристаллизации. Это обстоятельство весьма важно при
интерпретации физико-химических процессов в таких системах, связанных
с переходом кристалл — жидкость. Кроме того, при
интерпретации рентгенограмм от фибриллярных белков необходимо знать,
какие именно звенья способны к кристаллизации, так как
только они вносят вклад в дискретное рассеяние.
Таким образом, для правильного понимания свойств
фибриллярных белков и, в частности, явлений, связанных с их кристал-
* [Существует также множество других блок-сополимеров с атактиче-
скими блоками, не упоминаемых автором из-за того, что они принципиально
неспособны к кристаллизации. — Прим. редактора].
114
лизацией, необходимо не только определить концентрацию
мономерных звеньев, действительно способных кристаллизоваться,
но и выяснить характер их распределения вдоль полипептидной
цепи.
Для фибриллярных белков возможен случай, когда способные
к кристаллизации аминокислотные остатки (соответствующие
А-звеньям) объединены в один блок, а неспособные к
кристаллизации — в другой блок. Два эти стереохимически
различных блока могут чередоваться вдоль цепи. Подобное
расположение блоков по схеме эквивалентно упорядоченным полиэфирам.
С другой стороны, возможно и чисто статистическое
распределение кристаллизующихся и некристаллизующихся звеньев в
цепи. Выяснить, какой тип распределения имеет место в
действительности, можно лишь при комбинации структурных и
термодинамических исследований, сопровождаемых анализом
физических и механических свойств.
Хотя фиброину шелка может быть приписана
рентгенографическая структура, в которую встраиваются все химически
различающиеся звенья его цепи [58], на самом деле он обладает, по-
видимому, характерным для упорядоченных сополимеров
распределением аминокислотных остатков вдоль макромолекулы.
Анализ [59] небольших пептидных фрагментов, содержащихся в
неполных гидролизатах фиброина шелка, не согласуется в
предположением о существовании какой-либо регулярной
химической периодичности в цепи. Скорее можно предположить
структуру, в которой определенные типы звеньев концентрируются в
достаточно протяженных блоках.
С помощью фермента химотрипсина можно осуществить
специфическое расщепление полипептидной цепи в местах
расположения тирозиновых остатков, что позволяет выделить две
основные части макромолекулы [60]. Одна часть цепи, составляющая
60%, содержит только глициновые, аланиновые и сериновые
остатки и дает характерную рентгенограмму порошка,
подобную наблюдаемой на нативном фиброине. Другая часть
содержит все массивные аминокислотные остатки, а также небольшие
количества глицина, аланина и серина. Эти аналитические
результаты согласуются с предположением К. Мейера [61] о том,
что глициновые, сериновые и аланиновые остатки образуют
кристаллические области полимера, тогда как прочие остатки
связаны с аморфными или некристаллическими областями.
Концепция частичной кристаллической структуры фиброина
шелка дополнительно подтверждается при исследовании
механических свойств. Лукас с соавторами [62] изучали эластические
свойства волокон, полученных от различных видов шелкопрядов
и соответственно содержащих разное относительное количество
аминокислотных остатков с массивными или длинными
боковыми группами.
8*
115
Как и следовало ожидать, волокна, содержащие 90%
глицина и аланина, относительно мало растяжимы, поскольку
степень их кристалличности очень велика. С другой стороны, по
мере увеличения концентрации аминокислотных остатков с
большими боковыми радикалами волокна становятся более-.растя-
жимыми, что хорошо согласуется с заметным увеличением
аморфной фазы. Механические свойства, дополненные
изложенными выше сведениями о распределении последовательностей
аминокислотных остатков, определенно свидетельствуют в
пользу того, что фиброин шелка может рассматриваться как блок-
сополимер. Другие фибриллярные белки, в частности
относящиеся к классу кератина или коллагена, также могут быть
отнесены к этой категории сополимеров.
Сокристаллизация и изоморфное замещение
Фибриллярны и глобулярные белки, как и нуклеиновые
кислоты, обладают кристаллической структурой, в которой сходные
по общей структуре, но все же различающиеся химически
мономерные звенья входят в одну и ту же кристаллическую решетку.
Кристаллографический анализ указывает на стереохимическую
идентичность многих аминокислотных остатков и нуклеотидов *.
При благоприятных обстоятельствах те же особенности
наблюдаются и у простых синтетических сополимеров. В подобных
случаях говорят, что звенья неспособны сокристаллизоваться.
Рентгеноструктурный анализ выявляет при этом лишь одну
кристаллическую структуру, присущую главному компоненту.
Зависимость температуры плавления от состава в таких системах
отлична от наблюдаемой у статистических или упорядоченных
сополимеров.
В литературе опубликовано несколько примеров изоморфного
замещения у сополиамидов [63—67]. Однако до сих пор не
сформулированы определенные принципы структурной совместимости
сомономеров, благоприятствующей сокристаллизации. Эдгар и
Хилл [63], например, основываясь на нормальных значениях длин
связей и валентных углов, провели расчеты, из которых следует,
что расстояние между карбоксильными группами в терефтале-
вой и адипиновой кислотах почти идентичны. Это может быть
достаточным основанием для частичного взаимного замещения
двух дикарбоновых кислот в кристаллической решетке. Такое
* [Следует еще раз подчеркнуть, что это сходство отражает
идентичность тех участков мономерных звеньев, которые непосредственно образуют
цепь главных валентностей; различия же сопряжены с боковыми группами. —
Прим. редактора].
116
so о
замещение действительно наблюдается для сополимеров гекса-
метиленадипамида и терефтальамида. Температура плавления
указанных сополимеров возрастает монотонно с увеличением
концентрации терефталевой кислоты. На соответствующей
кривой нет минимума, который всегда наблюдается, если
кристаллизуется только один из сомономеров. Таким образом, можно
заключить, что здесь произошла сокристаллизация двух типов
звеньев.
Известны и другие подобные системы [64, 66, 67], но в целом
необходимо установление более определенных и общих правил
совместимости. Действительно, одинаковая длина сомономерных
звеньев еще недостаточна для сокристаллизации. Это следует,,
например, из того, что сополиэфиры, соответствующие
упомянутым сополиамидам, уже не сокристал-
лизуются. ттх. ,
Сокристаллизация наблюдается 2т у'
также среди виниловых сополимеров
[68]. Статистическое введение изоморф- 2щ
ных звеньев вызывает непрерывное
изменение температуры плавления, 2щ
которая принимает значения, проме- L"
жуточные по отношению к чистым го- [
мополимерам. Так сополимеры о-фтор- 0.2 Qj* 0,6 ofl W
стирола и стирола обладают одинако- Содертаниеп-фторстиропа,
1 г мол. доли
выми кристаллическими структурами
и практически одинаковыми постоян- РиСф 42. Зависимость
темными решетки. Изоморфизм наблю- пературы плавления сопо-
дается во всей области составов. Об- лимеров стирола и л-фтор-
разование смешанных кристаллов в стирола от состава [68].
этой системе может быть обусловлено
одинаковым периодом идентичности каждого гомополимера:
и очень малой разницей размеров атомов фтора и
водорода.
Однако сокристаллизация наблюдается и тогда, когда
соответствующие гомополимеры обладают различными
кристаллическими структурами. Например, стабильные кристаллические
модификации полистирола и поли-я-фторстирола обладают
различной симметрией, причем первый образует спираль третьего,,
а второй — четвертого порядка. Тем не менее, сополимеры
кристаллизуются во всей области композиций [68], и температура
плавления оказывается строго линейной функцией состава, как:
это видно из рис. 42. Это уже само по себе свидетельствует о
сокристаллизации.
Возможность образования твердых растворов в сополимерах
полиметилена, содержащих метильные боковые радикалы, уже
упоминалась.
117
Набухшие сополимеры
По аналогии с гомополимерами кристаллизация
сополимеров в присутствии низкомолекулярного растворителя происходит
во всей области концентраций. Зависимость температуры
плавления от концентрации полимера описывается формулой,
аналогичной A3) [62], с заменой Тпл на температуру плавления нена-
бухшего сополимера. Экспериментально эта зависимость
подтверждена для большого числа не полностью стереорегулярных
полимеров, таких как политрифторхлорэтилен [70], полиакрило-
нитрил [71] и полипропилен [72].
Кристаллизация гомополимера из разбавленного раствора
приводит к появлению двух отдельных фаз, легко разделяемых
механическими средствами. Рассматриваемое явление
обсуждалось выше, а наблюдаемая необычная морфология была
представлена на рис. 10 и 11. С другой стороны, при кристаллизации
статистических сополимеров из разбавленных растворов
произвести механическое разделение фаз очень часто оказывается уже
невозможным. Впрочем, этот вопрос нуждается в более
тщательном и систематическом исследовании, которое помогло бы
установить количественные соотношения между концентрацией
полимера, термодинамическими свойствами растворителя и
композицией сополимера, при которой механическое разделение фаз
становится невозможным. В подобных условиях полимерные
молекулы заполняют весь объем системы, и разбавленный раствор
с очень высокой текучестью превращается в систему с высокой
вязкостью. Этот процесс, обычно называемый гелеобразованием
или желатинизацией, является специфическим проявлением
кристаллизации.
Такого рода резкие изменения механических свойств при
кристаллизации из разбавленных растворов наблюдались в
различных системах, например, поливинилхлорид в диоктилфталате
[47], полиакрилонитрил в диметилформамиде [48],
нитроцеллюлоза в этиловом спирте [73] и метилцеллюлоза в воде [74].
Вследствие нарушений стереорегулярности поливинилхлорид и
полиакрилонитрил проявляют при кристаллизации и плавлении
свойства сополимеров, тогда как у неполностью замещенных
производных целлюлозы, по-видимому, сокристаллизация
мономерных звеньев также оказывается неполной. Кристаллизация
этих систем в разбавленных растворах может происходить либо
при повышении, либо при понижении температуры, так как
растворимость определяется величиной и знаком свободной
энергии разбавления по сравнению со свободной энергией
плавления. Две указанные целлюлозные системы служат примерами
кристаллизации, сопровождающейся гелеобразованием и
происходящей при повышении температуры [73, 74].
i 18
По-видимому, все полимерные системы, которым
свойственен такой тип гелеобразования, кристаллизуются, как
сополимеры. Следует, однако, добавить, что все упомянутые полимеры
обладают или должны обладать очень протяженными макро-
молекулярными конформациями из-за малой
термодинамической (равновесной) гибкости цепей.
Сополимерный характер молекул приводит к образованию»
геля потому, что даже при полном соблюдении условий
равновесия далеко не все звенья могут принимать участие в
кристаллизации, так что лишь небольшая их часть переводится в
кристаллическую фазу. В результате большое число незакристалли-
зованных звеньев оказывается связанным в единую сетчатун>
систему кристаллическими областями; эта система занимает весь
объем раствора, что и придает ему высокую вязкость и
характерную для геля жесткость.
У сополимеров, особенно у кристаллизующихся из
разбавленного раствора, прирост числа цепей в поперечном
направлении кристаллитов ограничен, так как нерегулярная структура
цепей не позволяет им вновь возвратиться в кристаллит, из
которого они вышли. Без этого возвращения поперечный рост
неможет быть очень интенсивным (подробнее см. гл. 9).
Рассматриваемый эффект, сопровождающийся также запаздыванием
развития кристаллитов в продольном направлении, неизбежно
ограничивает число звеньев, участвующих в кристаллизации.
Специфический процесс желатинизации не следует путать
с другими типами гелеобразования в полимерных системах.
В частности, надо различать образование геля в результате
кристаллизации и возникновения сетки без фазового перехода.
Последний тип гелей, или трехмерных сеток, может быть
образован введением достаточного числа межцепных поперечных связей*
в набор полимерных цепей или при полимеризации мульти-
функциональных сомономеров [75]. Именно потому, что
образование гелей может происходить по различным механизмам, и
возникает путаница. Аморфный сшитый гель при набухании в
растворителе обладает такими же механическими свойствами,,
как и гели, образовавшиеся при кристаллизации сополимеров.
Однако гели, возникшие в результате кристаллизации, могут быть
расплавлены до состояния золя или гомогенного раствора. Как
раз это обстоятельство и может служить основой для
идентификации гелей различных типов.
Флори и Гарретт [76] показали, что классическая гелевая
система желатина—вода возникает в результате перехода
жидкость—кристалл; поэтому процесс гелеобразования или
превращения в золь может быть отнесен к фазовым переходам
первого рода.
Согласно работам Марсланда [77], с подобным типом
перехода золь — гель связан целый ряд биологических процессов,
119'
таких как амебоподобное движение или развитие складок,
непосредственно предшествующее делению клеток в митотическом
цикле. По-видимому, в подобных процессах важную роль играют
кристаллизуемые белковые молекулы. Исследования влияния
давления или температуры на прекращение амебоподобного
движения или на торможение образования складок показывают, что
в таких процессах проявляются общие закономерности,
аналогичные описываемым уравнением Клаузиуса — Клапейрона. Это
означает, что превращения золь — гель, соответствующие
указанным процессам, связаны с переходом между двумя
различными фазами, скорее всего кристаллической и жидкой.
Литература
1. P. J. F 1 о г у, Trans. Faraday Soc, 51, 848 A955)
!2. М. J. Richardson, P. J. Flory, J. B. Jackson, Polymer, 4, 221
A963).
3. B. Ke, J. Polymer Sci., 61, 47 A962).
4. L. M a n d e 1 k e r n, Rubber Chem. Technol., 32, 1392 A959).
5. P. R. Eichorn, J. Polymer Sci., 31, 197 A958).
•6. P. R. S w a n, J. Polymer Sci., 56, 409 A962).
7. P. J. Flory, L. Man del kern, H. K. Hall, J. Am. Chem. Soc, 73,
2532 A951).
8. R. D. Evans, H. R. M i g h t о n, P. J. Flory, J. Am. Chem. Soc, 72,
2018 A951).
9. O. B. E d g a r, E. E 11 e г у, J. Chem. Soc, 1952, 2633.
10. S. Sonnerskog, Acta chem. scand., 10, 113 A956).
11. E. F. I z а г d, J. Polymer Sci., 8, 503 A952).
12. L. Mandelkern, R. R. Garrett, P. J. Flory, J. Am. Chem. Soc,
74, 3939 A952).
13. P. J. Flory, H. D. Bed on, E. H. Keefer, J. Polymer Sci., 28, 151 A958).
14. J. T. Maynard, W. E. Мое he 1, J. Polymer Sci., 13, 235 A954).
15. L. Mandelkern. M. Try on, F. A. Quinn, Jr., J. Polymer Sci., 19,
77 A956).
16. E. J. Hart, A. W. Meyer, J. Am. Chem. Soc, 71, 1980 A949).
17. F. E. Condon, J. Polymer Sci., 11, 139 A953).
18. J. L. Binder, Ind. Eng. Chem., 46, 1727 A954).
19. G. Lanzavecchih, Ind. chim. beige, Suppl. 2, 347 A959).
20. G. N a 11 a, P. С о г г a d i n i, Angew Chem., 68, 615 A956).
21. F. W. Stavely и др., Ind. Eng. Chem., 48, 778 A956).
22. S. E. Home, Jr., J. P. К i e h 1, J. J. S h i p m a n, V. L. F о 11,
С F. Gibbs, E. A. Wilson, E. B. Newton, M. A. Reihart, Ind.
Eng. Chem., 48, 784 A956).
•23. H. E. Adams, R. S. Stearns, W. A. Smith, J. L. Binder, Ind.
Eng. Chem., 50, 1508 A958).
24. G. Natta, P. С о г г a d i n i, Atti Accad. Naz. Lincei, 19, 230 A953).
25. J. I. Cuneen, F. W. Shipley, J. Polymer Sci., 36, 77 A959).
26. J. I. Cuneen, W. F. Watson, J. Polymer Sci., 38, 521 A959).
27. M. Golub, J. Polymer Sci., 25, 373 A957).
28. J. I. Cuneen, G. M. С H i g g i n s, W F. Watson, J. Polymer Sci.,
40, 1 A959).
29. L. Pauling, R. B. Corey, H. R. Branson, Proc Natl Acad. Sci.
U. S., 37, 205 A951).
120
30. S. Mizushima, Advances in Protein Chem., 9, 299 A954).
31. G. Natta, J. Polymer Sci., 16, 143 A955); G. N a t t a, P. Pino,
P. С о г г a d i n i, F. D a n u s s o, E. M a n t i с a, G. M a z z a n t i, G. Mo-
raglio, J. Am. Chem. Soc, 77, 1708 A955).
32. M. L. H u g g i n s, J. Am. Chem. Soc, 66, 1991 A944).
33. T. G. Fox, W. E. G о о d e S. G r a t с h, С M. H u g g e 11, J. F. К i n-
caid, A. Spell, J. D. Stroupe, J. Polymer Sci., 31, 173 A958).
34. B. S. Garrett, W. E. G о о d e, S. G га ten, J. F. К i n с a i d, С L. Le-
v e s q u e, A. Spell, J. D. Stroupe, W. H. W a t n a b e, J. Am. Chem.
Soc, 81, 1007 A959).
35. J. W. L. F о r d h a m. G. H. McCain. L. E. Alexander, J. Polymer
Sci., 39, 335 A959).
36. J. W. L. F о r d h a m, P. H. Burleigh, С L. Sturm, J. Polymer Sci.,
41, 73 A959).
37. J. W. L. F о r d h a m, J. Polymer Sci., 39, 321 A959).
38. B. D. Coleman, J. Polymer Sci., 31, 155 A958).
39. G. N a 11 a, J. Polymer Sci., 34, 531 A959).
40. F. D a n u s s o, G. M о r a g 1 i o, W. G h i g 1 i a, L. M о 11 a, G. T a 1 a-
mini. Chim. Ind. (Milan), 41, 748 A959).
41. S. Newman, J. Poly me r Sci., 47, 111 A960).
42. J. D. Stroupe, R. E. Hughes, J. Am. Chem. Soc, 80, 2341 A958).
43. J. L. R. Williams, J. Van Den В e r g h e, W. J. Dulmage,
K. P. D u n h a m, J. Am. Chem. Soc, 78, 1260 A956); J. L. R. W i 11 i a m sv
J. Van Den Berghe, K. R. Dunham, W. J. Dulmage, J. Am.
Chem. Soc, 79, 1716 A957).
44. T. G. Fox, B. S. Garrett, W. E. Goode, S. G г a t с h, J. F. К i n-
caid, A. Spell, J. D. Stroupe, J. Am. Chem. Soc, 80, 1768 A958).
45. А. А. Короткое, С. П. М и ц е н г е н д л е р, У. Н. К р а с у л и н а,.
Л. А. Волкова, Высокомол. соед., 1, 1319 A959).
46. P. M. Doty, H. L. Wagner, S. F. Singer, J. Phys. Col. Chem., 55„
32 A947).
47. A. T. W a 11 e r, J. Polymer Sci., 13, 207 A954).
48. J. Bisschops, J. Polymer Sci., 12, 583 A954); 17, 89 A955).
49. V. S. E 11 i s, K. S. M i n s k e y, E. E. R у 1 о v, D. N. В о г t, High
Molecular Weight Compounds, 1, 1403 A959).
50. H. С Raine, R. B. Richards, H. Ryder, Trans. Faraday Sot., 41„
56 A945).
51. M. Dole, W. P. He 11 in gen, N. R. Larson, J. A. Wethington„
J. Chem. Phys., 20, 781 A952).
52. E. Hanter, W. G. Oakes, Trans. Faraday Soc, 41, 49 A945).
53. L. Mandelkern, M. H e 11 m a n, D. W. Brown, D. E. Roberts,.
F. A. Q u i n n, Jr., J. Am. Chem. Soc, 75, 4093 A953).
54. Y. С h a t a n i, T. T a k i z a w a, S. M u r a h a s h i, Y. S a k a t a, Y. N i s h i-
mura, J. Polymer Sci., 55, 811 A961).
55. D. Coffey, T. J. M e у r i с k, Proc Rubber Technol. Conf., 2nd, 1954.
p. 170.
56. D. Coleman, J. Polymer Sci., 14, 15 A954).
57. E. G. К о n t о s, E. K- E a s t e г b г о о k, R. D. Gilbert, J. Polymer Sci.,.
61, 69 A962).
58. R. E. Marsh, R. B. Corey, L. Pauling, Biochim. et Biophys. acta,
16, 1 A955).
59. F. Lucas, J. Т. В. Shaw, S. G. Smith, Adv. Protein Chem., 13, 107'
A958).
60. F. Lucas, J. T. B. Shaw, S. G. Smith, Nature, 178, 861 A956); Bio-
chem. J., 66, 468 A957).
61. К. Н. М е у e r, H. M a r k, Chem. Ber., 61, 1932 A928).
62. F. Lucas, J. T. B. Shaw, S. G. Smith, J. Textile Inst., 46, T-140
A955).
63. O. B. E d g a r, R. H i 11, J. Polymer Sci, 8, 1 A952).
12L
64. AJTu, R.D.Evan s, J. Polymer Sci., 20, 5361 A959).
65. F. В Carmer, R. G. В e a m a n, J. Polymer Sci., 21, 237 A956).
66. P. R. Thomas, Т. С T r a u t e г, G. J. Tyler, Paper presented before
International Union of Pure and Applied Chemistry, Symposium on Macro-
molecules, Wiesbaden, 1959.
67. M. Levine, S. С Temi n, J. Polymer Sci., 49, 241 A961).
68. G. N a 11 a, Makromol. Chem., 35, 94 A960).
69. P. J. F lory, J. Chem. Phys., 17, 223 A949).
70. A. M. Beuc he, J. Am. Chem. Soc, 74, 65 A952).
71. W. R. Krigbaum, N. Tokita, J. Polymer Sci., 43, 467 A960).
72. F. D a n u s s o, Atti Accad. Naz. Lincei, 25, 520 A958).
73. S. Newman, W. R. Krigbaum, D. K. Carpenter, J. Phys. Chem.,
60, 648 A956).
74. E. Hey man, Trans. Faraday Soc, 31, 846 A935).
75. P. J. Flory, Principles of Polymer Chemistry, Ithaca — New York, 1953,
pp. 347ff., 454ff.
76. R. R. Garrett, P. J. Flory, Nature, 177, 176 A956); P. J. Flory,
R. R. Ga rrett, J. Am. Chem. Soc, 80, 4836 A958).
77. D. A. Mar si and, D. E. S. Brown, J. Cellular Сотр. Physiol., 20, 295
A942); J. V. Land an, A. M. Zimmerman, D. A. M a r s 1 a n d,
J. Cellular Сотр. Physiol., 44, 211 A954); D. A. M а г s 1 a n d, J. V. L a n-
d a n, J. Exptl Zool., 125, 507 A954).
78. E. H. В am ford, A. Elliott, W. E. H a n 1 y, Synthetic polypeptides,
New York, 1954.
79. С. Я. Френкель, Полимеры, Физический энциклопедический словарь,
т. 4, Изд. «Советская энциклопедия», 1965.
Термодинамические
параметры
Введение
В связи с тем, что плавление полимерных систем может быть
истолковано как фазовый переход первого рода, равновесная
температура плавления полимера является вполне
определенным параметром. Поэтому представляет интерес выяснить,
каким образом температура плавления гомополимеров зависит от
химических и структурных свойств мономерного звена. Так как
температура плавления определяется отношением теплоты к
энтропии плавления (в расчете на мономерное звено), именно им
должно быть уделено особое внимание. Поскольку эти
величины определены экспериментально для многих полимеров,
следует сосредоточить внимание непосредственно на молекулярной
интерпретации температур плавления макромолекул. Однако
было бы преждевременно полагать, что уже в настоящее время
могут быть сформулированы какие-либо количественные
закономерности. Это обусловлено главным образом недостатком
общих принципов, на основе которых удавалось бы установить
взаимосвязь температур плавления с химическим составом даже
для самых простых мономерных веществ. Тем не менее обзор
литературных данных, относящихся к различным полимерам,
представляется все же полезным, ибо он может помочь
выявлению наиболее характерных особенностей рассматриваемой
проблемы.
При определении температур плавления полимеров
измерения следует обязательно проводить в условиях
термодинамического равновесия. Поэтому термодинамические свойства
необходимо изучать на хорошо отожженных образцах,
закристаллизованных вблизи температуры плавления.
Изменения механических свойств или структурных
характеристик, которые неизбежно происходят при плавлении, могут
быть использованы для нахождения области плавления; однако
наиболее удобны измерения подобного типа для
определения больших различий в температурах плавления. Истинно
123
равновесная температура плавления должна соответствовать
исчезновению абсолютно совершенных монокристаллов. Поэтому
•следует стремиться к оптимальным режимам
кристаллизации, в наибольшей степени приближающимся к условиям
равновесия.
Опыт показывает, что соблюдение указанных
экспериментальных требований и особенно повышение эффективности
отжига неизменно приводят к получению более высоких значений
температуры плавления для самых различных полимеров.
Повышение экспериментальной температуры плавления
достигается также при очень низких скоростях нагревания,
осуществляемого сразу же после кристаллизации, особенно, если
кристаллизация проводилась вблизи температуры плавления.
Температура, теплота и энтропия плавления
В табл. 4 представлены основные термодинамические
параметры, характеризующие плавление гомополимеров. Они
найдены посредством метода измерений понижения точки
плавления полимеров при набухании в низкомолекулярных
жидкостях (стр. 51). Температуры плавления чистых и набухших
полимеров определены при очень малых скоростях
нагревания.
Для удобства сравнения А#м различных полимеров эта
величина во всех случаях поделена на молекулярный вес
мономерного звена М0. Полученная при этом удельная теплота,
отнесенная к 1 г кристаллического полимера приведена в табл. 4.
Энтропия плавления ASM на мономерное звено находится из
отношения АЯм/Гпл (Т'пл выражается в этом случае в градусах
Кельвина). Для сравнения на какой-то рациональной основе
энтропии плавления полимеров, различающихся по величине
мономерных звеньев, ASM было разделено на число единичных
связей в главной цепи мономерного звена. Полученный таким
образом параметр приведен в последней колонке табл. 4.
Калориметрически можно определить некоторую условную теплоту
плавления А#м различных полимеров, однако, зная степень
кристалличности системы до плавления, можно затем по А//м
оценить энтальпию фазового перехода А#м. Результаты для
полимеров, в отношении которых отсутствуют данные, полученные
более точными методами, приведены также в табл. 4.
Необходимо отметить, что соответствующие величины Гпл и
А#м носят приблизительный характер, ибо калориметрические
измерения, как правило, проводились на плохо отожженных
образцах и при больших скоростях нагревания.
124
Таблица 4
Термодинамические параметры, характеризующие плавление
гомополимеров
Полимер
7° .
'пл'
°С
кал/моль
кал/г
кал/(моль'град)\
АИМ определено по понижению температуры плавления
при набухании
Полиэтилен [1] .
Полипропилен [2]
Полиизопрен, 1,4-цш;
(натуральный
каучук) [3]
Полиизопрен,
1,4-транс
(гуттаперча) [4] ** . .
Полистирол [5] .
Полихлоропрен
\Л-транс [6]3*
Полихлортрифтор-
этилен [7] . . .
Поливинилфто-
рид [8] ....
Полидекаметилен-
адипинат [9] . .
Полидекаметилен-
себацинат [10] .
Полидекаметилен-
ацелат [11] . .
Полинонметилен-
ацелат [11] . .
Полидекаметилен-
терефталат [11]
Полигексаметилен-
терефталат [И]
Политетраметилен-
терефталат [12]
Политетраметилен-
изофталат [12] .
Поли-N, N'-себа-
циолпипера-
зин [13] ....
Полидекаметилен-
себацианамид [11]
Полидекаметилен-
ацеламид [11] .
Трибутират
целлюлозы [14] ...
Тринитратцеллюло-
зы [15] ....
Нитратцеллюлоза
B,44) [16] . . .
Трикаприлат
целлюлозы [17] ...
| 137,5*
176
28
74
239
80
210
197
79,5.
80
69
65
138
160,5
230
152,5
180
216
214
207
>700
617
116
I 960 |
2600
1050
3040
2000
2000
1200
1800
10 200
12000
10000
10300
11000
8500
7 600
10100
6200
8300
8 800
3000
900—1 500
1350
3100
68,5
62,0
15,3
45,1
19,2
22,6
10,3
39
36
35
31
33
36
34
33
45
24,5
24,5
27
8,1
3,0—5,0
5,2
5,7
2,34
5,78
3,46
8,75
3,9
5,68
2,49
3,87
29
34
29
30,5
27
19,5
15,1
23,7
13,7
17
18
6,2
1.5
1,51
8,0
125
Продолжение
Полимер
°С
кал/моль
кал! г
кал/(моль-град)\
AS/связь,
кал/град
Полиоксиэти-
лен [18] . . .
Полиоксимети-
лен [19] . . .
Полиакоилони-
трил '[20] . .
Коллаген [21] .
66
180
317
1454*
1980
1590
1200
2 250
45
53
23
24
5,35
3,5
2,0
5,85
ДЯМ определено по колориметрическим измерениям
на чистом полимере
Полиэтилентере-
фталат [23] . . .
Полиэтиленсебаци-
нат [24]
Полигексаметилен-
аднпамид [25] . .
Полигексаметилен-
себацианамид [25]
Поликапроамид [26]
Полиоксиметилен
[27, 28]
267
76
267
226
225
180
5500
8000
10300
12000
5100
1780
28,1
30,5
45
43
45
59,2
10,2
23
19,1
24
10,3
3,94
Д//м определено по зависимости температуры
плавления от гидростатического давления
Политетрафторэтилен [29] ....
327
A атм)
335
F9 атм)
1460
14,6
2,9
2,68
3,5
1,0
1,95
U
1,65
1,36
1,32
1,46
3,94
1,45,
* Температура плавления наиболее высокомолекулярной фракции равна 138,5° С B2].
** Для наиболее высокоплавкого полиморфа.
3* Экстраполировано к 100% содержания 1,4-т/?а«г-изомера.
<* Экстраполировано по данным, относящимся к плавлению в этиленгликоле.
Зависимость температуры плавления от гидростатического
давления р описывается уравнением Клаузиуса — Клапейрона:
dTl
dp
ш 7U
F4)
где АУМ — изменение объема на мономерное звено,
происходящее при плавлении.
Если известны AVM и степень кристалличности, величина
Д#м может быть легко найдена с помощью соответствующих
опытов.
126
Установленные таким образом термодинамические
параметры полимера, для которого перечисленные выше методы не
применимы, приведены в табл. 4.
Данные, приведенные в табл. 4, свидетельствуют об
отсутствии простой или очевидной корреляции между температурой
и теплотой плавления. Теплоты плавления для всех
рассмотренных полимеров можно разделить на две большие категории, к
одной из которых относятся величины порядка нескольких
тысяч калорий на моль мономерных звеньев и ко второй —
теплоты плавления, равные ~ 10000 кал/моль. При этом многие из
высокоплавких полимеров имеют низкую теплоту плавления и
наоборот, полимеры, имеющие низкую температуру
плавления, характеризуются относительно большой теплотой
плавления.
При обсуждении физического смысла величины Тпл следует
помнить, что Д#м и ASM представляют собой соответственно
разность энтальпий и энтропии в жидком и кристаллическом
состояниях. Поэтому должны быть охарактеризованы свойства
полимеров в этих двух состояниях и их изменения при
соответствующих фазовых переходах.
Соответствующие термодинамические данные для
углеводородных полимеров свидетельствуют о том, что параметры Т°пл и
ДЯМ, определенные экспериментально для линейного
полиэтилена, хорошо согласуются с величинами, полученными
экстраполяцией Т°пл и АЯМ низкомолекулярных нормальных парафинов
на большие молекулярные веса.
Температура плавления нефракционированного образца
полиэтилена равна 137,5° С (см. табл. 4). Для
высокомолекулярной фракции эта величина повышается уже до 138,5° С, так как
ее кристаллическое состояние более совершенно. Для
полипропилена температура плавления 176° С также несколько
занижена, ввиду его неполной изотактичности. Молярная теплота
плавления полипропилена выше, а удельная ниже
соответствующих теплот плавления полиэтилена. В работе [37] показано, что
температура плавления синдиотактического полипропилена
неопределенной степени стереорегулярности несколько выше, чем
изотактического. Однако надежные данные о различии в
энтропии и теплоте плавления этих двух стереоизомерных форм
отсутствуют.
Примером высокоплавкого полимера, обладающего
относительно низкой теплотой плавления является изотактический
полистирол с 7,Пл = 239°С, а удельная теплота плавления
сравнима с теплотой плавления натурального каучука, температура
плавления которого значительно ниже. Ясно, что для полимеров
такого типа значение Тпл в основном определяется энтропией
плавления.
127
Плавление полимеров диенового ряда также соответствует
закономерностям, наблюдаемым при плавлении их мономерных
аналогов. Хорошо известно, например, что цис-изомеры
производных этилена более легкоплавки, чем соответствующие транс-
изомеры; поэтому неудивительно, что 1,4-7у?а«с-полиизопрен
плавится при более высокой температуре нежели 1,4-цис-нзо-
мер. Аналогичным образом, 1, 4-чис-полибутадиен имеет
температуру плавления + ГС [37], в то время как 1, 4-г/?аяс-полибу-
тадиен 148° С.
У полиэфиров и полиуретанов, имеющих группу СН = СН в
главной цепи температура плавления ^ыс-изомеров также ниже,
чем у граяс-изомеров [38]. Например, температура плавления
цис-изомера полиуретана, полученного поликонденсацией 1,4-
циклогександиола и метилен бисD-фенилизоцианата),
значительно ниже температуры плавления соответствующего транс-
изомера. Однако, когда в качестве двухосновного спирта
выбирается 1,4-циклогександиметанол, температуры плавления
обоих стереоизомеров близки по величине [39].
Изотактический 1,2-полибутадиен имеет температуру
плавления 120° С [40], в то время как для синдиотактического
Гпл=154°С [41]. Более высокая температура плавления
синдиотактического полимера, по сравнению с его изотактическим сте-
реоизомером, наблюдается также для полиметилметакрилата
[42] и полипропилена. Причина этого, однако, остается неясной
ввиду отсутствия надежных, пригодных для сравнения, величин
Д#м и Д5М.
Многие полимеры, полученные на основе нелинейных а-оле-
финов, таких как 4, 4-диметилгексен-1 и З-фенилбутен-1, имеют
температуру плавления порядка 350° С [43], температура
плавления поли-я-ксилена равна 412° С [44]. Поскольку этим
полимерам могут быть приписаны значения Д//м того же порядка,
что и обычно предсказываемые и наблюдаемые для
углеводородов, следует предположить, что крайне высокая их Т^л
обусловлена очень низкой энтропией плавления.
Полимеры, относимые обычно к категории эластомеров,
должны иметь очень низкую температуру стеклования и либо не
кристаллизоваться вообще, либо в закристаллизованном
состоянии иметь низкую температуру плавления. Три полимера,
относящиеся к этой категории, а именно: натуральный каучук, поли-
изобутилен и полидиметилсилоксан имеют соответственно
следующие температуры плавления: 28 [3], 5 [45] и около —40° С
[46]. Следует полагать, что теплота плавления полиизобутилена
не выходит за обычные пределы, характерные для
углеводородов, и нет никаких оснований полагать, что для полидиметилси-
локсана этот параметр почему либо должен быть ниже.
Следовательно, столь низкие температуры плавления этих полимеров
128
и их принадлежность к категории эластомеров определяются
относительно большим значением ASM при плавлении.
Галогенсодержащие полимеры принадлежат к классу,
характеризуемому высокими температурами плавления при
относительно низких теплотах плавления, как это и следует из
сопоставления термодинамических параметров для политрифтор-
хлорэтилена и поливинилфторида, приведенных в табл. 4.
Температура плавления политетрафторэтилена находится
вблизи 330° С. Теплота плавления этого полимера, определенная
при изучении зависимости температуры плавления от
гидростатического давления, оказывается меньше, чем для полиэтилена.
Причину повышенных ГпЛ, наблюдаемых для этого класса
полимеров, надо по-прежнему искать в той значительной роли,
которую играет энтропия в процессах плавления.
Алифатические полиэфиры плавятся при более низких
температурах, чем полиэтилен, как и аналогичные мономерные
эфиры. Обнаружена [47] общая тенденция к уменьшению
температуры плавления при возрастании числа эфирных групп в цепи.
Такой результат противоречит тому, что следовало бы ожидать,
если бы в кристаллическом состоянии возрастала
интенсивность межмолекулярных взаимодействий, обусловленная
полярными эфирными группами. Однако два полиэфира,
содержащие в цепи наибольшее относительное число эфирных групп в
этом отношении аномальны. Действительно, полиэтиленсукцинат
плавится при 108° С, а полиэтиленмалонат даже при комнатной
температуре — жидкость [48].
Полиэфиры, содержащие четное число групп СН2, плавятся
при более низких температурах, по сравнению с полиэфирами
с нечетным числом этих групп; в свою очередь, Д#м для
последних несколько выше, чем для первых. По-видимому, это
обстоятельство и объясняет различия в температурах плавления у
этих двух типов полиэфиров.
Введение кольцевых структур в полимерную цепь приводит
к значительному возрастанию температуры плавления,, по
сравнению с соответствующими алифатическими полимерами, что
показано на примерах полиэфиров [49] и полиангидридов [50]. Так,
полисебациановыи ангидрид плавится при 83° С, в то время как
полимеры, полученные из двухосновных ароматических кислот,
имеют температуры плавления в области 150—300° С.
Табл. 4 показывает далее, что ароматические полиэфиры
плавятся при более высоких температурах, чем их алифатические
аналоги, но оба типа полиэфиров имеют сравнимые величины.
Значительное повышение температуры плавления
ароматических полимеров должно быть обусловлено понижением энтропии
плавления. Однако величины энтропии плавления, рассчитанные
на единичную связь, для ароматических полиэфиров даже
несколько выше, чем для линейных.
9 Л, Манделькерн
129
Следовательно, предложенный выше метод сравнения
энтропии плавления различных полимеров становится
неколичественным, если полимерная цепь содержит кольцевые структуры. Из
ароматических полиэфиров политетраметилентерефталат
плавится при значительно более высокой температуре, чем его
изомер политетраметиленизофталат. Это различие снова может
быть отнесено за счет повышенной энтропии плавления поли-
тетраметиленизофталата. Опять можно отметить сходство
закономерностей плавления мономерных и полимерных веществ, так
как известно, что я-дипроизводные бензола имеют более высокие
температуры плавления, по сравнению с другими изомерами.
Алифатические полиамиды плавятся при более высоких
температурах, чем полиэтилен; в противоположность полиэфирам, с
ростом относительного числа полярных групп в цепи, здесь
наблюдается возрастание Тпл. Это может быть объяснено
способностью амидных групп образовывать водородные связи. В
согласии с хорошо известной общей закономерностью [47, 49]
алифатические полиамиды, содержащие нечетное число групп в
мономерном звене, плавятся при более высокой температуре,
чем их аналоги с четным числом групп в повторяющейся
единице. Полимеры, состоящие из звеньев различной четности,
обладают некоторой промежуточной температурой плавления. Это
различие между полимерами с четным и нечетным числом
групп в повторяющейся единице цепи отражает изменение
температур плавления соответствующих мономерных аналогов.
Такое изменение Г„л наблюдается, например, для
низкомолекулярных нормальных парафинов, исчезая, однако, с ростом длины
цепи. В случае полиэфиров, для которых температуры
плавления могут быть точно измерены различие Г„л полидекаметилен-
себацината и полидекаметиленацелата достигают 11 град.
Разница в температурах "плавления соответствующих полиамидов
всего 2 град. Отметим, что во многих случаях [47—49]
температуры плавления, значениями которых мы пользовались для этих
обобщений, были измерены при больших скоростях нагревания.
Сравнение полученных таким путем температур плавления,
может привести к значительным ошибкам, достигающим в
некоторых случаях 20 град.
Замена атома водорода в NH группе на алкильную группу
сильно снижает температуру плавления. Так, например,
полимер N-метилундекановой кислоты [N—CH3—(CH2)ioCO]n
плавится при 60° С, в то время как незамещенный полимер —
при 182° С [51]. Снижение Тпл объясняется в данном случае
уменьшением способности полимера к образованию
водородных связей при замещении амидного водорода на алкильную
группу.
130
Сравнение данных табл. 4 для алифатических полиамидов и
соответствующих полиэфиров показывает, что полиамиды
плавятся при более высоких температурах, нежели полиэфиры. В то
же время теплоты плавления полиамидов значительно ниже,
хотя, казалось бы, что они обладают потенциально большей
способностью к образованию межцепных водородных связей.
Следовательно, в данном случае снова определяющую роль играет
энтропия плавления, и можно утверждать, что алифатические
полиамиды более высокоплавки, потому что их энтропия
плавления ниже.
Два из трех полиамидов (см. табл. 4), для которых
определены точные термодинамические характеристики, способны
образовывать межмолекулярные водородные связи, третий, поли-N, N'-
себацоилпиперазин этой способности не имеет. Несмотря на
различие в температурах плавления удельные теплоты плавления
этих трех полимеров сравнимы по величине.
Оценивая роль, которую играет образование водородных
связей в повышении температуры плавления, нельзя упускать из
виду их влияние и на энтропию плавления. Найденные
калориметрически для трех полиамидов значения Д#м сходны по
величине ив то же время значительно выше, чем определенные по
снижению температуры плавления в набухшем состоянии для
двух аналогичных полимеров. Хотя главная причина этого
несоответствия не совсем ясна, можно считать, что при
калориметрических исследованиях значительные погрешности
получаются при определении степени кристалличности.
Полиуретаны содержат функциональную группу
— (О—СО—NH) и сходны с полиамидами в том, что способны
образовывать водородные связи между полярными группами;
в то же время, они содержат и типичные для полиэфиров связи
О—СН2. Поэтому не удивительно, чтр температура плавления
полиуретанов занимает промежуточное положение между Т1Л
полиэфиров и полиамидов [47]. Для них по-прежнему
выполняется правило, согласно которому полимеры, содержащие
четное число групп СН2 в мономерном звене, плавятся при более
высокой температуре, чем полимеры с нечетным числом таких
групп.
Целлюлоза и ее производные являются высокополярными
полимерами с очень высокой температурой плавления. Однако
теплоты плавления этого класса полимеров крайне низки.
Например, тринитроцеллюлоза, имеющая самую высокую из всех
известных для полимеров температуру плавления, в то же время
обладает наименьшей теплотой плавления.
Таким образом, факторы полярности и структуры цепи,
влияющие на ГПл, должны отражаться на энтропии плавления.
Низкие теплоты плавления, приведенные в табл. 4, соответствуют
9*
131
полностью кристаллическому полимеру; в обычных условиях,
когда полимер лишь частично кристалличен, непосредственно
измеряемая энтальпия плавления должна быть значительно
ниже. Поэтому понятно, что на кривых охлаждения трибутирата
целлюлозы [52] нет плато или «остановки» вблизи температуры
плавления; неудивительно и то, что независимо от состояния
полимера теплоты растворения целлюлозы и ее производных
практически совпадают [53—55]. Эти факты, однако, не следует
объяснять отсутствием кристалличности в таких полимерах или
отсутствием фазового перехода между кристаллическим и
жидким состоянием, как это иногда предполагается.
Пониженная температура плавления трикаприлата
целлюлозы, по сравнению с трибутиратом, обусловлена увеличением
энтропии плавления. Последнее может быть приписано
дополнительному выигрышу в конфигурационной энтропии за счет
боковых эфирных групп. При увеличении длины боковых групп три-
замещенных производных целлюлозы Тпл вначале уменьшается,
достигая некоторого минимума, и затем снова начинает расти.
Это явление, типичное для линейных полимеров с длинными
боковыми алькильными группами, наблюдается также у поли-
а-олефинов.
Температура плавления полиоксиэтилеиа намного меньше,
чем у полиэтилена. Большую энтропию плавления
полиоксиэтилеиа можно объяснить его повышенной гибкостью в жидком
состоянии из-за периодического повторения в цепи эфирных звеньев.
Полиоксищропилен обладает почти одинаковой с полиокси-
этиленом температурой плавления. Это означает, что либо
введение метильных групп не влияет на ASM и Д//м, либо, если
изменения все-таки происходят, отношение этих параметров
остается прежним. Казалось бы, определяющую роль здесь
играет отношение числа метиленовых групп к числу атомов
кислорода. Однако Тш полиоксиизобутилена равна 156° С [58].
Температуры плавления полиоксиметилена более чем на
100 град выше, чем у полиоксиэтилеиа. Удельная теплота
плавления, как и энтропия плавления в расчете на единичную связь
у этого полимера несколько больше, чем у полиоксиэтилеиа.
Влияние относительного числа эфирных групп в
повторяющемся звене на температуру плавления, по-видимому, может
быть проиллюстрировано на примере политиоэфиров [47]. Так,
температура плавления равная 145° С для полимера,
содержащего в мономерном з'вене одну группу (ClbbS падает до 65° С
с увеличением числа групп СН2 в повторяющемся звене.
Полимеры, имеющие в цепи дисульфидные связи, плавятся при более
низких температурах, чем соответствующие политиоэфиры,
откуда следует, что полярность атомов серы не влияет
существенным образом на температуру плавления.
132
Полиакрилонитрил является еще одним примером полярного
высокоплавкого полимера с малой теплотой плавления.
Соответствующая низкая энтропия плавления этого полимера
показывает, что выигрыш в конфигурационной энтропии при
переходе из кристаллического состояния в расплавленное
незначителен. Все кристаллизующиеся полимеры эфиров акриловой и ме-
такриловой кислот имеют сравнительно высокие температуры
плавления. Ничего большего, однако, сказать о полимерах этого
класса нельзя, пока для них не будут определены необходимые
для оценки Тплтермодинамические параметры.
Плавление фибриллярного белка коллагена изучено очень
подробно и его термодинамические параметры известны.
Исследования Флори и Гарретта [21] показали, что Д#м и ASM,
найденные для этого природного полимера, очень близки к
соответствующим характеристикам для более простых синтетических
полимеров. И теплота, и энтропия плавления имеют нормальные
значения; так, например, удельные теплотышлавления коллагена
и синтетических полиамидов практически совпадают. Какое бы
ни было повышение стабильности кристаллического состояния,
которое можно приписать образованию водородных связей, оно
незаметно, если не принимать во внимание, что вклад их в
устойчивость структуры намного меньше ожидаемого.
Основную часть мономерных звеньев в молекуле коллагена
составляют глицин, пролин и оксипролин. Хотя содержание
амино- и иминокислотных остатков меняется от образца к
образцу, в коллагене позвоночных и беспозвоночных содержание
глицина остается постоянным и составляет приблизительно треть
всех звеньев. Несмотря на различия в составе коллагенов,
существует взаимосвязь между температурой плавления (опре*
деленной при фиксированной полной концентрации белка) и
содержанием иминокислот.
Густавсон [59] показал, что температура плавления возра-*
стает с увеличением количества оксипролина. Увеличение
стабильности отнесено за счет вовлечения гидроксильных групп
оксипролина в образование водородных связей; тем самым
предполагалось, что возрастание Тпл обусловлено в основном
увеличением Д#м, и возможные изменения ASM считались
незначительными. Позже установлено [60—62], что существует
зависимость между температурой плавления и общим содержанием
иминокислотных остатков. Гарретт предположил [54], что
причиной увеличения температуры плавления является уменьшение
ASM, обусловленное ростом общего содержания пролина и
оксипролина. Возрастание концентрации пирролидиновых колец в
главной цепи должно приводить к подавлению
конфигурационной свободы молекулы в расплавленном состоянии. Поэтому,
если концентрация иминокислотных остатков в цепи не влияет
133
на характер кристаллической фазы, увеличение этой
концентрации неминуемо приводит к понижению энтропии плавления, и,
следовательно, к повышению температуры плавления. В
принципе, повышенная стабильность может быть достигнута в
фибриллярных белках при соответствующем воздействии на
конфигурационные свойства расплава. Развитие более
термодинамически стабильных структур не обязательно требует или
предполагает увеличения концентрации водородных связей в
кристаллическом состоянии.
В работах [63—65] были измерены температуры плавления
других биологически важных макромолекул, синтетических поли-
нуклеотидов и природных нуклеиновых кислот. В
упорядоченном состоянии молекула дезоксирибонуклеиновой кислоты
состоит из двух спирально переплетенных цепей.
Кристаллографическая структура, определенная Криком и Уотсоном [66],
допускает только один способ образования пар гетероциклическими
основаниями, входящими в состав каждой из этих цепей.
Анализ состава нуклеиновых кислот показывает, что концентрация
пуриновых оснований равна концентрации пиримидиновых
оснований; поэтому образование пар через водородную связь, по
статистическим соображениям, возможно только между адени-
ном (А) и тимином (Т), и между гуанином (Г) и цитозином (Ц).
При плавлении цепи разделяются и переходят в
беспорядочно свернутое состояние.
Были определены температуры плавления в разбавленных
растворах ряда дезоксирибонуклеиновых кислот различного
состава [63—65]. Состав оснований, выраженный процентным
содержанием пар Г — Ц, изменялся от 0 до 65 и температура
плавления возрастала с ростом концентрации этих пар. Для
синтетических нуклеиновых кислот, содержащих только пары А—Т,
Тш равна 65° С; для природных дезоксирибонуклеиновых
кислот, содержащих около 65% пар Г—Ц она увеличивается до
95° С. Этот факт можно объяснить тем, что обе пары способны
к образованию различного числа водородных связей [67]; так
пара Г—Ц содержит три, а пара А—Т — только две
водородных связи, следовательно различные пары вносят не одинаковый
вклад в стабильность спирали [63, 64].
Поэтому различие в температурах плавления
дезоксирибонуклеиновых кислот следует объяснять интенсивностью
водородных связей между цепями, составляющими кристаллическую
молекулу. Это сразу же позволяет на формальных основаниях
приписать Д#м определяющую роль в плавлении. Влиянием
энтропии плавления в данном случае можно пренебречь,
несмотря на то, что она вносит существенный вклад в процессы
плавления более простых полимеров. Вопрос о том, существуют
ли значительные различия конформаций нуклеиновых кислот
различного состава, требует еще уточнения.
Ш
Синтетические полирибонуклеотиды также способны
образовывать упорядоченные мультиспирали, состоящие из одинаковых
цепных молекул [68, 69]. В этом случае возможны другие типы
пар оснований (и сопутствующие им водородные связи). Для
четырех таких комплексов можно сравнить температуры
плавления, так как они были определены при сходных условиях [64].
Две биспиральные системы, состоящие из комплементарных пу-
риновых и пиримидиновых оснований, а именно: полиаденило-
вая — полиуридиловая кислота и полицитозиновая — полиино-
зиновая кислота, имеют температуры плавления соответственно
75 и 90° С. Тройные спирали, имеющие в своем составе только
пуриновые основания, такие как поли(И + И + И) * и
поли(А + И + И) имеют температуры плавления 40 и 60° С,
соответственно. Различия в температурах плавления,вероятно,
следует объяснять несовпадением интенсивностей водородных
связей.
Энтропия плавления
Попытки найти взаимосвязь между температурами
плавления полимеров и межмолекулярными взаимодействиями,
используя в качестве меры этих взаимодействий плотность энергии
когезии в расчете на мономерное звено, практически всегда были
безуспешны [47, 48, 70, 71], так как при этом во внимание
принимается только Д#м. В связи с отсутствием простой
зависимости между Тпл и Д//м не удивительно, что многие полимеры с
низкими плотностями энергии когезии плавятся при высокой
температуре, и наоборот, многие низкоплавкие полимеры
обладают высокой плотностью энергии когезии.
Из сопоставления температур плавления и характеризующих
плавление термодинамических параметров большого числа
полимеров становится очевидной роль энтропии плавления [70] в
определении величины Гпл. Это обстоятельство особенно
очевидно в случае высокоплавких полимеров, для которых неизменно
наблюдаются относительно малые значения ASM. Так как
величина ASM определяет различие энтропии мономерного звена в
кристаллическом и жидком состояниях, всегда следует
учитывать оба эти состояния.
Нарушение порядка в кристаллах может вызвать такие
отклонения от регулярности, присущей кристаллическому
состоянию, что они, в свою очередь, вызовут изменение энтропии этого
состояния. Поскольку уровень беспорядка возрастает с
увеличением температуры, энтропия плавления, вообще говоря, должна
убывать с возрастанием температуры плавления.
* [И — инозин, — Прим. редактора],
135
Было показано [25, 72, 73], например, что некоторые поли-*
амиды при повышенных температурах претерпевают
полиморфный переход от триклинной к гексагональной форме.
Гексагональная упаковка допускает большую вращательную свободу
хребта цепи и тем самым приводит к возрастанию энтропии
кристаллического состояния. Принимая во внимание более высокие
температуры плавления полиамидов, по сравнению с
соответствующими полиэфирами [11], можно в связи с этим
предположить, что полиэфиры не могут существовать в гексагональной
форме.
В целом, однако, обобщение, утверждающее, что повышение
конфигурационного беспорядка приводит к увеличению
температуры плавления, требует дополнительных обоснований. Они
могут быть получены при структурных исследованиях различных
полимеров вблизи температуры плавления.
Заметно различающиеся структурные характеристики
молекулы полимера в кристаллическом и жидком состояниях должны
проявляться в увеличении конфигурационной энтропии при
плавлении. В жидком состоянии молекулы полимера могут
приобретать самые различные конформации, от беспорядочно
свернутого клубка до удлиненной стержневидной частицы, в
зависимости от типа полимера. Приписываемая конкретным цепям
конформация зависит от природы мономерных звеньев и
характера их взаимодействия. Тормозящие потенциалы,
препятствующие свободному вращению мономерных звеньев относительно
друг друга, определяются стерическими отталкиваниями и
притяжениями между соседними радикалами.
Конфигурационная энтропия жидкого состояния зависит,
следовательно, от конформации и относительных размеров
отдельных полимерных молекул. Поэтому энтропия плавления
должна зависеть от конформационных свойств цепи в
расплавленном состоянии. Таким образом, известные различия в
конформации дают основание ожидать больших изменений
энтропии плавления при переходе от одного класса полимеров к
другому.
Информация об усредненных конформациях отдельных
полимерных молекул может быть получена при изучении некоторых
гидродинамических и оптических свойств их разбавленных
растворов. Подобными измерениями установлено, например, что
натуральный каучук [74], полиизобутилен [75] и полидиметилсил-
оксан [76] существуют в форме сильно свернутого клубка, как
это следует из относительно малых отношений ((го)Дг2)/оI/2
(см. табл. 1, стр. 20).
Термодинамический анализ показывает, что относительно
низкие температуры плавления этих полимеров и объясняются,
в первую очередь, энтропийными эффектами. Сильно свернутая
форма цепи обеспечивает возможность осуществления весьма
136
большого числа конформаций в расплаве и приводит,
соответственно, к высокой энтропии плавления. Другой класс
полимеров, таких как производные целлюлозы и полиакрилонитрил,
характеризуется весьма протяженной конформацией цепей. Га-
логенсодержащие полимеры также могут быть отнесены к этой
категории. Полимеры, молекулы которых развернуты в жидком
состоянии при плавлении испытывают не столь большое
возрастание конфигурационной энтропии. Следовательно, в этом
случае можно предсказать уменьшение энтропии плавления.
При всех прочих равных условиях указанные полимеры
имеют более высокую температуру плавления только из-за кон-
формации их цепи, откуда следует, что высокие температуры
плавления вовсе не обязательно связаны с большими теплотами
плавления. Другие полимеры имеют конформаций,
промежуточные между этими двумя, и соответственно характеризуются
промежуточными значениями энтропии плавления.
Чтобы вычислить точный выигрыш в конфигурационной
энтропии при плавлении, из экспериментально определяемой
величины ASM следует вычесть вклад, вносимый эффектом
изменения объема. Формально можно записать
(?),-(*),-*
где а — изобарный температурный коэффициент объемного
расширения;
р—изотермический коэффициент объемного расширения
при изменении давления.
Тогда при Гпл равновесной температуре плавления при
давлении в 1 атм — изменение энтропии, вызываемое изменением
объема, выразится следующим образом:
Если предположить, что
ASM = ASK + (ASM)K F7)
легко получить (ASM)V, изменение энтропии при постоянном
объеме и плавлении при Г„л.
Для ряда полимеров имеются данные, необходимые для
вычисления (ASM)v. Оценки (ASM)y, полученные на основе этих
данных с помощью соотношения F7), приведены в табл. 5.
Из табл. 5 ясно, что изменение объема, происходящее при
плавлении, обычно вносит ощутимый вклад в экспериментально
наблюдаемую энтропию плавления. Для натурального каучука,
например, приблизительно половина изменения энтропии, может
быть отнесена исключительно за счет изменения объема. Таким
137
Таблица 5
Значения энтропии плавления при постоянном
объеме для различных полимеров калЦмоль • град)
Полимер
Полиэтилен . .
Натуральный
каучук ....
Гуттаперча * . .
Полиоксимети-
Полиоксиэтилен
Полистирол . .
Политетрафторэтилен ....
AS
м
2,29
3,46
8,75
3,5
5,35
3,9
1,Н
&Sy
0,52
1,8
3,7
2,1
1,13
1,3
0,38
(*Sm)V
1,77
1,7
5,1
1.4
4,22
2,6
0,76
Литература
[1]
[3]
14]
[27]
[831
[5]
[27]
Для наиболее высокоплавкого полиморфа.
образом, при любом количественном анализе влияния
молекулярной конформации на плавление необходимо предварительно
рассчитать энтропию плавления при постоянном объеме.
Конформация цепи и кристаллизация
Приведенный в предыдущем разделе анализ выявляет
определяющую роль, которую конформация цепи в расплавленном
состоянии играет при плавлении. Метод количественной оценки
стабильности упорядоченной полимерной фазы с учетом
конформации цепи был развит Флори [84, 85]. В его расчетах для
описания смеси полимера и разбавителя используется известная
модель квазирешетки. Статистическая сумма, вычисляемая для
отдельной цепи, учитывает поворотно-изомерный характер
полимерной молекулы.
Методы, развитые Волькенштейном с сотрудниками [86—88]
и Лифсоном [89], основываются на предположении, что
состояние каждой связи в цепи соответствует минимуму потенциала
заторможенного вращения. Каждая из подобных
предпочтительных ориентации связи рассматривается как дискретное
состояние, и конформация цепи определяется последовательным
набором таких состояний. Однако состояние произвольно выбранной
связи зависит от состояний ее непосредственных соседей.
Поэтому рассматривая проблема в сущности идентична модели
Изинга (для описания ферромагнетизма). Принципы расчета
для такой модели разработаны достаточно хорошо [90, 91], и,
138
поэтому, зависимость ориентации данной связи от ориентации
ее соседей может быть учтена в явной форме.
Для вычисления статистической суммы цепи определяют
некоторую матрицу U=||ttij||. Элемент этой матрицы иц
представляет собой вероятность перехода или статистический вес
того, что поворотное состояние / следует за состоянием /. Все
возможные последовательности состояний, соответствующим
образом взвешенные, включены в эту матрицу. Используя ранее
развитые методы, статистическую сумму для длинноцепной
молекулы, охватывающую все возможные конформации
изолированной полимерной молекулы, можно записать следующим
образом
Z^spur(U*-2)«G>*-2 F8)
где со — наибольшее собственное значение матрицы U.
Это собственное значение можно интерпретировать как
параметр, определяющий эффективное число доступных для связи
состояний, следовательно, оно является мерой относительного
разнообразия конформации цепи. В этом смысле
рассматриваемый параметр можно использовать для описания «гибкости»
цепи.
Статистическая сумма системы, содержащей /г2 цепей,
каждая из которых характеризуется статистической суммой F8),
и tii молекул растворителя, при отношении молярных объемов
полимера и растворителя, равному х, может быть определена
как [84, 85]:
X^V^exp^^L F9)
В уравнении F9) <7i и Яг представляют собой внутренние
статические суммы молекул растворителя и полимера,
соответственно; z — координационное число решетки, соответствующее
числу доступных направлений первой связи цепи, а степень
симметрии полимерной молекулы выбрана равной 2*.
Используя формулу Стирлинга, свободную энергию
смешения чистого растворителя и упорядоченного полимера можно
записать в виде
AFCM = RT [щ In о, + л2 In v2 + ixxn2vx] — n2RT [in^ + (х — 2) In ¦-] G0)
где rti и п2 означают число молей соответствующего компонента.
* Соотношение F9) совпадает с первоначально определенной в работе
[84] статистической суммой, если учесть, что <о-1«1 — Л В исходном
расчете f определено, как доля связей, имеющих энергетически невыгодные
ориентации.
139
Уравнение G0) можно переписать так:
^см = ^с°м + ^р G1)
Таким образом свободную энергию смешения можно разбить
на два члена, один из которых зависит от состава и не зависит
от со. Второй — зависит только от со и, следовательно, от конфор-
мационных свойств цепи. AFCm представляет собой энергию
смешения цепных молекул без изменения их внутренней структуры,
т. е. имеющих одно и то же со, в начальном и конечном
состоянии.
Нетрудно убедиться, что Д/\?м — привычное выражение для
свободной энергии смешения, получаемое в теории Флори —
Хаггинса [92, 93]. AFp соответствует изменению свободной
энергии при переходе идеально упорядоченной полимерной молекулы
(все связи имеют преимущественную ориентацию) в абсолютно
неупорядоченное состояние, при котором утрачивается
корреляция в расположении соседних цепей и статистическая сумма
отдельной молекулы равна со*. Разделение свободной энергии
смешения на две составляющих, одна из которых зависит только
от состава, а другая только от внутримолекулярной конформа-
ции, имеет очень важные следствия.
Для чистого полимера v2=l, так что &FCM = AFV.
Неупорядоченное состояние будет стабильным, при А/7Р<0, или, как
следует из уравнения G0) при выполнении следующего
неравенства;
Для больших значений х это неравенство сводится к
о>*>2,72 G3)
Таким образом, если приведенное выше условие не
выполняется, то более стабильным будет упорядоченное состояние, в
котором полимерные молекулы уложены параллельно. В случае
цепей с ограниченной свободой беспорядочная упаковка
оказывается затрудненной из-за недостатка свободного объема, т. е.
отсутствия доступного пространства для расположения соседних
молекул. Поэтому предельная концентрация полимера в данном
объеме может быть достигнута только для упорядоченной
системы.
Указанное обстоятельство является прямым следствием
ограничения, накладываемого на внутримолекулярную структуру
для таких систем и выражаемого неравенством G3). Переход
от неупорядоченного состояния к параллельной упаковке
является кооперативным процессом, так как выигрыш в свободной
энергии, достигаемый при упорядочении, реализуется лишь при
140
условии, что все цепи в заданном объеме одновременно
становятся параллельными.
Критерий стабильности фазы определяется величиной
функции распределения для индивидуальной цепи. Обычно функция
распределения монотонно убывает с температурой, имея
наибольшее значение при высоких температурах; она асимптотически
стремится к единице по мере приближения температуры к
абсолютному нулю. Поэтому существует определенная вероятность,
что при некоторой температуре будет выполняться условие со = е.
При этой температуре должен присходить спонтанный
переход от упорядоченного состояния, стабильного при низких
температурах, к беспорядку. Возможность указанного перехода
возникает только как следствие внутримолекулярных
взаимодействий. Учет влияния ил'и роли межмолекулярных сил для
предсказания подобного перехода пока не требовался.
Существование перехода такого типа для реальных цепей зависит от
величины о и соответствующего ей температурного коэффициента.
С этой точки зрения температура плавления полимера и его кон-
формационные свойства взаимообусловлены величиной оэ.
Кристаллизацию, или развитие трехмерно упорядоченной
структуры, можно рассматривать как процесс, протекающий в
два этапа. Первый этап заключается в кооперативном
упорядочении системы в результате внутримолекулярного процесса,
описанного выше. Для завершения кристаллизации необходимо уже
развитие продольного порядка. В системе гомополимеров или
полимеров, обладающих достаточной регулярностью
структурных единиц цепи, оно осуществляется небольшим (порядка
периода вдоль цепи) смещением параллельно ориентированных
молекул относительно друг друга. В результате смежные
радикалы занимают взаимные положения, соответствующие структуре
кристаллической решетки. В этом и состоит второй этап
кристаллизации, управляемый уже межмолекулярными
взаимодействиями и сопровождающийся соответствующим уменьшением
свободной энергии.
Следовательно, промежуточное состояние с параллельной
упаковкой цепей менее стабильно, чем трехмерно упорядоченное,
и переход к последнему, если не принимать во внимание
возможные кинетические помехи, должен происходить спонтанно *.
Следует ожидать, что в свободную энергию плавления вносит вклад
* Доказательством этого заключения является тот факт, что некоторые
полимеры, такие как полиакрилонитрил и поливинилтрифторацетат, имеют в
кристаллическом состоянии только поперечный порядок [94]. Отсутствие
трехмерного порядка здесь можно объяснить либо кинетическими
трудностями, либо стереоизомерной природой мономерных звеньев. Однако, ввиду
относительно протяженных конформаций цепей у этих полимеров, некоторая
регулярность во взаимном расположении звеньев все же необходима, если
141
как внутримолекулярные, так и межмолекулярные
взаимодействия.
Изложенная выше теория дает основания предполагать, что
вклад внутримолекулярных взаимодействий в процесс
плавления весьма значителен. Однако в противоположность переходам
в разбавленных растворах, при кристаллизации и плавлении в
блоке важную роль играют также и межмолекулярные эффекты.
Поэтому переход оказывается значительно более резким, чем
в случае чисто одномерного упорядочения.
При добавлении в систему растворителя конкуренция за
свободный объем между отдельными элементами цепи ослабевает,
и сохранение неупорядоченной аморфной фазы оказывается
более вероятным, по сравнению с чистой полимерной системой.
Можно показать, что для атермической (%i=0) смеси
высокомолекулярного полимера и растворителя условие стабильности
неупорядоченной фазы сведется к неравенству:
ю > ещ G4)
Отсюда следует, что при понижении v2 значение со,
необходимое для устойчивости аморфной фазы, может сильно
уменьшиться. Поэтому, в принципе, плавление может быть
осуществлено простым разбавлением.
Влияние ограничений конфигурации цепи на процессы
кристаллизации и плавления зависит от статистической суммы (при
условии, разумеется, что ее критическое значение, определенное
выше, находится в доступной температурной области); для
реальных полимеров требуется вычисление со с подстановкой
потенциалов заторможенного вращения. Для этой цели принято
использовать потенциальную функцию «-бутана, который может
существовать в трех поворотноизомерных формах *.
Транс-положение энергетически более выгодно, по сравнению с двумя
«свернутыми» гош-состояниями, соответствующими углам
вращения + 120° и —120°. Если транс-состояние принять за
исходное, и различие в свободной энергии между транс- и
гош-состояниями обозначить е, то
<о = 2? + 1 G5)
гяе g=exp(—e/kT).
В соответствии с общепринятыми обозначениями, неравенство
е>0 показывает, что грая^-состояние энергетически более
выгодно, если же е<0, то предпочтительнее любые из двух гош-
состояний. Однако для полиэтиленовой цепи последовательность
двух гош-состояний различного знака ( + 120°, —120°)
запрещена из-за сильных стерических взаимодействий углеродных
атомов, так называемого «пентанового запрета». Поэтому вероят-
* ]См. Т. М. Б и р ш т е й н, О. Б, П т и ц ы н, Конформация
макромолекул, Изд. «Наука», 1964. — Прим. редактора].
142
ность такой последовательности будет иметь нулевой
взвешивающий фактор [27, 85]. При этом ограничении [75]:
»=^Ь/1+йУ <76>
Соотношение G6) совместно с F8) дает функцию
распределения для изолированной полиэтиленовой цепи в расплавленном
состоянии. В качестве примера можно также рассмотреть
потенциальную функцию заторможенного вращения,
допускающую лишь два энергетически выгодных положения. Для такого
случая:
© = * + 1 G7)
Графически зависимость о от температуры для этих трех
случаев представлена на рис. 43. Пунктирная горизонтальная
линия соответствует у сло-
вию о) = е. При наиболее
вероятной транс-конфор-
мации (е>0)со никогда
не достигает е ни для
потенциальной функции с
двумя минимумами, ни
для потенциальной
функции с тремя минимумами
и «пентановым запретом»,
независимо от различия
между температурами или
энергиями поворотных
изомеров. Для
потенциальной функции с
тремя минимумами без
ограничений в
последовательности поворотных
состояний со превосходит е при
очень малых значениях
переменной e/kT.
Если соотношение действительно справедливо для
полиэтилена и разность энергий транс- и гош-изомера составляет
выигрыш в 500 кал/мольу как это принято в работе [95], фазовый
переход из кристаллического в жидкое состояние невозможно
представить без участия межмолекулярных взаимодействий.
Если же для некоторой цепи энергетически выгоднее гош-поло-
жение, что постулируется для полиоксиэтилена [27, 96], то со
может легко превзойти критическое значение.
Приведенные примеры, которые, возможно, отражают
некоторые свойства реальных цепей, показывают, что необходимые
для фазового перехода критические величины со вполне дости-
Рис. 43. Зависимость (о от г/kT для
различных энергетически выгодных
состояний поворотных изомеров:
/ — два энергетически выгодных состояния с равной
энергией; 2 —три энергетически выходных
состояния с «пентановым запретом», два из которых имеют
одинаковую энергию; 3 — то же, что и кривая 2,
но без «пентанового запрета». Горизонтальная
пунктирная линия соответствует условию w=*e:
из
жимы. Однако, как показано на примере полиэтилена, это еще
не означает, что кристаллизация в блоке, даже при
благоприятных условиях, происходит только в результате
внутримолекулярных взаимодействий.
Согласно определению AFP и соотношению G0), отнесенная
к единичной связи часть свободной энергии плавления,
обусловленная внутримолекулярными конформационными изменениями
при упорядочении полимерных цепей для полимера с большим
молекулярным весом, равна:
д/?м,/ = -*Ппт <78>
Соответственно:
"'»,-«"(у)+'"'тг <*»
Выражения G9) и (80) определяют скрытые энтропию и
энтальпию плавления, обусловленные лишь одним
внутримолекулярным упорядочением. Указанные выражения получены на
основе решеточной модели при обязательном предположении о
сохранении постоянного объема системы, и все изучаемые
термодинамические параметры должны соответствовать этому
требованию.
Следовательно, температуру плавления чистой полимерной
системы Гпл, соответствующую данной модели, следует
определить, как температуру плавления при постоянном объеме. Т'пл
не совпадает с Тил, температурой плавления при 1 атм\ обычно
последняя ниже Т'пл.
При Тил критические условия выполняются когда ы = е или,
что то же, AFm, j = 0. Тогда:
/ас ч I RTd\n®\ /01ч
(ASm,/V = —ОТ—) , <81>
Если все взвешивающие множители в матрице U равны нулю,
единице или ?=ехр(е/&Г), то
/до \ _ рт(din® d\ng\ _ (din® tdt \
( M'''Tnn~ Win*' dT Jr=7/ -\d\ng'TJT)T=T-
(82)
ПЛ
ПЛ
Так как е = ел — Tes, если е^ и es выбираются независимыми
от температуры, скрытое изменение энтропии при Т'цл, обусло-
144
вченное только внутримолекулярными эффектами, равно:
тогда как
(^„,/)г;л = (еА)(-шу)г_г-
" пл
В связи с тем, что первый множитель в правой части
равенства (84) соответствует разности энтропии для двух ориентации
связи, параметр (d In со/я? In g) > должен определять долю свя-
~у пл
зей, обладающих энергетически выгодной конформацией в
процессе упорядочения. Следовательно, если внутримолекулярные
процессы вносят определяющий вклад в процесс плавления,
энтропия плавления обратно пропорциональна температуре
плавления. Действительно, при G) = ng+1, что соответствует
большинству потенциальных функций заторможенного вращения при
отсутствии ограничений в последовательности поворотных
состояний, соотношения (84) и (85) сводятся к следующему:
(^„,/V =е41-7) I
пл ч 'I
I (86)
пл ' пл \ * I }
Эти соотношения были ранее получены Флори [84]. Если,
однако, существует какая-то взаимосвязь между
последовательными поворотными состояниями, то параметр (d In co/d In g) уже
зависит от детальных характеристик тормозящего потенциала.
При независимо определенном Т'ил, величины (Д5М> 7) » и
пл
(Д//м> j) ' могут быть и в этом случае рассчитаны теорети-
пл
чески. Сравнение их с экспериментальными результатами,
пересчитанными на постоянный объем, позволит оценить вклады
внутримолекулярных эффектов в процесс плавления.
Конформационные свойства реальных цепей и трудности,
возникающие при заполнении произвольно выбранного объема
вытянутыми, беспорядочно расположенными друг относительно
друга полимерными цепями, составляют основу молекулярной
интерпретации кристаллизации и плавления. Что касается
внутримолекулярного вклада, то энтропия плавления должна иметь
определяющее влияние на температуру плавления. Таким
образом может быть дано рациональное объяснение тому факту, что
многие высокоплавкие полимеры с низкими энтропиями
плавления сравнительно сильно (как это следует из измерений в
разбавленных растворах) развернуты в расплавленном состоянии.
10 Л. Манделькерн
(84)
(85)
Полиморфизм
Полимеры могут кристаллизоваться в различных
структурных модификациях и, следовательно, им присущ
кристаллический полиморфизм, по аналогии с низкомолекулярными
веществами. Например, углеводородные 7у?а«с-полимеры, гуттаперча
и полибутадиен, обладают различными кристаллографическими
структурами, соответствующими полиморфизму мономерных
тралс-углеводородов. На основании стереохимических исследо-
дований Банн [97] предсказал возможности существования
четырех различных кристаллических модификаций гуттаперчи
(т/яшс-1,4-полиизопрена). Три таких полиморфных формы
наблюдались и описаны [97, 98]. Две из них образуются при
простом охлаждении до соответствующих температур, тогда как
третья возникает только при растяжении. Полиморфизм
свойственен не только простым полимерам, но белкам [99] и
синтетическим полипептидам [100].
Полиморфизм, наблюдаемый в .полимерах, может быть
разделен на две больших категории. К первой относятся полимеры,
цепи которых способны в кристаллическом состоянии
приобретать существенно отличные упорядоченные конформации. В
результате различаются периодичности цепей и соответственно
размеры элементарных ячеек. К этой категории полимеров
относятся граяс-полиуглеводороды, а также а- и р-структуры,
наблюдаемые у полипептидов и фибриллярных белков.
При другом типе полиморфизма конформация и
периодичность вдоль цепи остаются неизменными, но способ, которым
она упаковывается в элементарной ячейке, изменяется. Этот тип
полиморфизма характерен для полиамидов.
Например, у полигексаметиленадипамида [72] и полигексаме-
тиленсебацамида [73] асимметричная упаковка в базальной
плоскости триклинной ячейки переходит к гексагональной при
возрастании температуры без изменения структуры плоского
зигзага цепи. Для изотактических полимеров, у которых цепи
имеют спиральную конформацию, должны быть рассмотрены
возможности появления правой и левой спиралей при одном и
том же типе решетки, что также может привести к
модификациям кристаллической структуры. Предполагается, что
подобные модификации возникают в полипропилене [37]. Однако
возможности полиморфизма сохраняются и в том случае, когда
кристаллы состоят только из правых или только из левых
спиралей.
Наблюдаемые полиморфные модификации определяются
условиями кристаллизации и не зависят от многих
кинетических факторов. Превращение одной формы в другую может быть
146
обусловлено изменением температуры, давления, напряжений
или состава полимера. Этот переход может происходить либо
непосредственно, либо при плавлении одной модификации и
последующей перекристаллизации. Однако с помощью прямых
наблюдений далеко не всегда удается различить эти два типа
перехода. Лишь определение свободной энергии плавления
каждой из полиморфных форм делает возможными заключения об
относительной стабильности модификаций и оценку других
термодинамических параметров перехода.
На рис. 44 приведены схематические диаграммы двух
возможных способов превращения одной полиморфной формы в
F
Рис. 44. Схематическая диаграмма зависимости свободной
энергии (при постоянном объеме) от температуры для
двух кристаллических модификаций (/ и //) и жидкого
состояния (///) одного и того же полимера.
другую. Так как в качестве переменной выбрана одна лишь
температура, свободная энергия каждой фазы может быть
представлена кривыми на плоской диаграмме.
Как видно из рис. 44а форма // более высокоплавка, по
сравнению с формой / (что показано местами пересечений
соответствующих трех кривых), и имеет более низкую свободную
энергию при всех температурах ниже ее температуры плавления.
Следовательно, эта форма более термодинамически стабильна
при всех температурах, тогда как форма / является метаста-
бильной модификацией полимера, с температурой плавления
ниже, чем у формы //.
На рис. 446 показан другой случай. Хотя форма / является
теперь более высокоплавкой модификацией, картины обратной
(рис. 44а) не наблюдается, так как форма // по-прежнему
стабильнее при низких температурах (обладая при этом более
низкой температурой плавления). Когда температура возрастает,
кривая свободной энергии формы // пересекает кривую
формы /, и происходит переход кристалл—кристалл. При
некоторых промежуточных температурах форма / стабильнее формы //,
вплоть до температуры плавления.
10*
147
В случае систем с другими внешними переменными (кроме
температуры), например при учете давления или приложенных
напряжений, подобным же образом следует анализировать
трехмерные фазовые диаграммы, где линии заменены поверхностями
свободных энергий индивидуальных фаз. Добавление
растворителя к жидкой фазе приводит к уменьшению свободной энергии
при всех температурах и соответствующему изменению условий
стабильности каждой из кристаллических фаз.
Следовательно, каждое полиморфное состояние
количественно описывается его свободной энергией на мономерное звено.
Наиболее стабильной формой является та, которая имеет более
низкую, по сравнению с некоторым стандартным состоянием при
заданной температуре, свободную энергию. Если в качестве
стандартного состояния выбрать полностью жидкий полимер, то
наиболее стабильной будет форма с наивысшей свободной
энергией плавления. Так термодинамические критерии стабильности
могут быть сформулированы без привлечения кинетических
наблюдений. Более того, так как подобные наблюдения зависят от
легкости, с которой образуется та или иная полиморфная
модификация, они вообще могут привести к ошибочным
заключениям. Поскольку свободная энергия плавления всегда может
быть определена по понижению температуры плавления при
набухании, использование подобных экспериментальных методов
для формального описания полиморфизма в полимерных
системах не представляет труда.
В настоящее время, однако, известна лишь одна такая
полимерная система. Это —гуттаперча, природный г/?алс-1,4-поли-
изопрен [4]. Для нее были определены теплоты плавления двух
полиморфных состояний при отсутствии внешних напряжений.
Было показано, что более низкоплавкая форма метастабильна
во всем температурном диапазоне своего существования.
Предложенный выше метод анализа, по-видимому, также
может быть полезным при изучении перехода а—>р в
синтетических полипептидах и фибриллярных белках. Для таких систем
кристаллическая модификация, легче остальных возникающая
в определенном растворителе и при заданной деформации, еще
не обязательно является самой устойчивой [101, 102].
Деформация, предшествующая кристаллизации, повышает свободную
энергию жидкого состояния. Поэтому одна из модификаций
может теперь стать стабильнее другой, или кристаллизация мета-
стабильной модификации может быть облегчена
деформационным процессом. Проблемы, связанные с этими эффектами
деформации, могут быть, как и в предыдущих случаях, разрешены
при определении свободной энергии плавления каждой из форм.
Чрезвычайно интересно, в частности, выяснить, находятся ли
встречающиеся в природе кристаллические молекулы в наиболее
термодинамически выгодной модификации.
148
Литература
1 F A. Quinn, Jr., L. Mandelkern, J. Am. Chem. Soc, 80, 3178
' A958).
2 F Danusso, G. Moraglio, E. Flores, Atti Accad. Naz. Lincei,
' 25,520 A958).
3. D. E. Roberts, L. Mandelkern, J. Am. Chem. Soc, 77, 781 A955).
4 L Man de Ike г n, F. A. Quinn, Jr., D. E. Roberts, J. Am. Chem.
' Soc, 78, 926 A956).
5 R Dedeurwaerder, J. F. M. Oth, J. Chim. phys., 56, 940 A959).
6 W. E. Mochel, J. T. Maynard, J. Polymer Sci., 13, 235 A954).
7 A. M. В u e с h e, J. Am. Chem. Soc, 74, 65 A952).
8. D. I. S a p p e r, J. Polymer Sci., 43, 383 A960).
9 L. Mandelkern, R. R. Garrett, P. J. F1 о r y, J. Am. Chem. Soc,
74, 3939 A952).
10 R. D. E v a n s, H. R. M i g h t о n, P. J. F 1 о г y, J. Am. Chem. Soc, 72,
2018 A950).
11 P. J. Flory, H. D. Be don, E. H. Keefer, J. Polymer Sci., 28, 151
A958).
12. A. Co nix, R. Van Kerpel, J. Polymer Sci., 40, 521 A959).
13. P. J. Flory, L. Mandelkern, H. K. Hall, J. Am. Chem. Soc, 73,
2532 A951).
14. L. Mandelkern, P. J. Flory, J. Am. Chem. Soc, 73, 3026 A951).
15. P. J. Flory, R. R. Garrett, S. Newman, L. Mandelkern,
J. Polymer Sci., 12, 97 A954).
16. S. Newman, J. Polymer Sci., 13, 179 A954).
17. P. G о о d m a n, J. Polymer Sci., 24, 307 A957).
18. L. M a n d e 1 к e r n, J. Appl. Phys., 26, 443 A955).
19. M. Inouc, J. Polymer Sci., 51, 518 A961).
20. W. R. Krigbaum, N. Так it a, J. Polymer Sci., 43, 467 A960).
21. P. J. Flory, R. R. Garrett, J. Am. Chem. Soc, 80, 4836 A958).
22. R. F. Chiang, P. J. Flory, J. Am. Chem. Soc, 83, 2857 A961),
23. С W. S m i t h, M. D о 1 e, J. Polymer Sci., 20, 37 A956).
24. B. Wunderlich, M. Dole, J. Polymer Sci., 24, 201 A957).
25. R. С Wilhoit, M. Dole, J. Phys. Chem., 57, 14 A953).
26. M. Dole, B. Wunderlich, Makromol. Chem., 34, 29 A959).
27. H. W. Starkweather, Jr., R. H. Boyd, J. Phvs. Chem., 64, 410
A960).
28. W. H. Linton, H. H. Goodman, J. Appl. Polymer Sci., 1, 179 A959);
С F. Hammer, T. A. Koch, J. F. Whitney, J. Appl. Polymer Sci.,
1, 169 A959).
29. P. L. McGear, H. С Duus, J. Chem. Phys., 20, 1813 A952).
30. A. H. Etessani, M. F. Saurger, J. Inst. Petrol., 25, 253 A939).
31. K. van Nes, H. A. Van West en, Aspects of the Constitution of
Mineral Oils, New York, 1951, p. 105.
32. W. E. Garner, K. van Bibber, A. M. King, J. Chem. Soc, 1931,
1533.
33. К. Н. Meyer, A. J. A. van der Wyk, Helv. chim. acta, 20, 83
A937). J
34. M. Dole, W. P. Hettinger, Jr., N. R. Larson, J. A. Wething-
o* t°Sv Jr'; J- Chem- physM 21» 731 A952).
U' Г; Y' BiHmeyer, J. Appl. Phys., 28, 1114 A957).
*• M. В г о a d h u r s t, J. Chem. Phys., 36, 2578 A962).
ъ г о aAt a> Makromol. Chem., 35, 94 A960).
?q fC ?. Marvel, С H. Young, J. Am. Chem. Soc, 73, 1066 A951).
oy- u- J. L у m a n, J. Polymer Sci, 55, 507 A961).
149
40. Q. Natta, L. Porri, P. Corradini, D. Morero, Atti Acad. Naz,
Lincei, Mem., Classe Sci., 20, 560 A956).
41. G. Natta, P. Corradini, Atti Acad. Naz. Lincei, Mem., Classe Sci.,
19, 229 A955).
42. T. G. Fox, B. S. Garrett, W. E. G о о d e, S. Grate h, J. F. К i n-
caid, A. Spell, J. D. S troupe, J. Am. Chem. Soc, 80, 1768 A958);
Т. С Fox, W. E. Goode, S. Cratch, С. М. Huggett, J. F. Kin-
caid, A. Spell, J. D. S troupe, J. Polymer Sci., 31, 173 A958).
43. T. W. Campbell, A. C. Haven, Jr., J. Appl. Polymer Sci., 1, 73
A959).
44. M. H. Kaufman, H. F. Mark, R. B. M e s г о b i a n, J. Polymer Sci.,
13, 3 A954). i
45. R. M. Kell, B. Bennett, P. B. Stickney, Rubber Chem. Technol.,
31, 499 A958).
46. С E. W e i r, W. H. L e s e r, L. A. W о о d, J. Res. Natl Bur. Stand., 44,
367 A950).
47. С W. В u n n, J. Polymer Sci., 16, 323 A955).
48. С W. Bunn, in R. E. Hill (ed.), «Fibers from Synthetic Polymers»,
Amsterdam, 1953, p. 310.
49. R. H i 11, E. E. W a 1 к e r, J. Polymer Sci., 3, 609 A948).
50. A. Co nix, J. Polymer Sci., 29, 343 A958).
51. R. Ae lion, Ann. chim. (Paris), 3, 5 A948).
52. W. O. Baker, С S. Fuller, Ind. Eng. Chem., 38, 272 A946).
53. В. А. Карги н, Г. Л. Слонимский, Усп. хим., 24, 785 A955).
54. В. А. К а р г и н, J. Polymer Sci., 30, 247 A958).
55. В. А. К а р г и н, Высокомол. соед., 2, 466 A960).
56. С. J. М а 1 m, J. W. М е п с h, D. С. К е n d а 11, G. D. H i a 11, Ind. Eng.
Chem., 43, 684, 688 A951).
57. F. P. Re d i n g, J. Polymer Sci., 21, 547 A956).
58. S. Kambara, A. Takahadi, Makromol. Chem., 58, 226 A962).
59. K. H. G u s t a v s о n, The Chemistry and Reactivity of Collagen, ch. 9,
New York, 1956.
60. K. A Piez, J. Am. Chem. Soc, 82, 247 A960).
61. R. E. В urge, R. D. Hynes, J. Mol. Biol., 1, 155 A959).
62. R. R. Garrett, P. J. S e e, Flory in Discussion, Brookhaven Symp.
Biol., № 13, «Protein Structure and Function», 1960, p. 229.
63. J. M a r m u r, P. D о t y, Nature, 183, 1427 A959).
64. P. Doty, H. В о e d t k e r, J. R. Fresco, R. H a s e 1 k о r n, M. L i 11,
Proc Natl Acad. Sci. U. S., 45, 482 A959).
65. J. M а г m u r, P. D о t y, J. Mol. Biol., 5, 109 A962).
66. J. D. W a t s о n, F. H. С. С г i с k, Nature, 171, 737 A953).
67. L. Pauling, R. B. Corey, Arch. Biochem. Biophys., 65, 164 A956).
68. R. С Warner, J. Biol. Chem., 229, 711 A957).
69. D. R. Da v ies, A. Rich, J. Am. Chem. Soc, 78, 3548 A956); A. Rich,
Nature, 181, 521 A958); A. Rich, Biochim. et Biophys. acta, 29, 502
A958); D. R. Da vies, A. Rich, J. Am. Chem. Soc, 80, 1003
A958).
70. L. M a n d e i k e r n, Chem. Rev., 56, 903 A956).
71. M. D о 1 e, Adv. Polymer Sci., 2, 221 A960).
72. R. В г i 11, J. Prakt. Chem., 161, 49 A942).
73. W. P. S 1 i с h t e r, J. Polymer Sci., 35, 82 A959).
74. H. L. Wagner, P. J. Flory, J. Am. Chem. Soc, 74, 195 A952).
75. T. G. Fox, Jr., P. J. Flory, J. Am. Chem. Soc 73, 1909 A951).
76. P. J. Flory, L. M a n d e 1 k e r n, J. К i n S i n g e r, W. B. S h u 11 z,
J. Am. Chem. Soc, 74, 3364 A952).
77. L. Mandelkern, P. J. Flory, J. Am. Chem. Soc, 74, 2517 A952).
78. M. L. Hunt, S. Newman, H. A. S с h e r a g a, P. J. Flory, J. Phys.
Chem., 60, 1278 A956).
79. A. M. Holzer, H. В e n о i t, P. Doty, J. Phys. Chem., 58, 624 A954),
150
ЯП Р J Flo г у, О. К. Spur г, D. К. Carpenter, J. Polymer Sci., 27,
231 A958).
81 R. А. О r i a n i, J. Chem. Phys., 19, 93 A951).
Я9 J С S 1 a t e r, Introduction to Chemical Physics, New York, 1939.
ЯЗ G N Malcolm, С L. D. Ritchie, J. Phys. Chem., 66, 852 A962).
g4 P J. F lory, Proc. Roy. Soc. (London), Ser. A, 234, 60 A956).
85 P J. F lory, J. Polymer Sci., 49, 105 A961).
86 M. В. Вол ькен штейн, О. Б. Птицын, ДАН СССР, 78, 657 A951).
87 М. В. Волькенштейн, J. Polymer Sci., 29, 441 A958).
88 О. Б. Птицын, И. А. Шаронов, ЖТФ, 27, 2744, 2762 A957).
89 S L i f s о n, J. Chem. Phys., 29, 80 A958); 30, 964 A959).
90 H. A. Kramers, G. H. Wannier, Phys. Rev., 60, 252 A941).
9l' G. F. Newell, E. W. M о n t г о 11, Rev. Mod. Phys., 25, 353 A953).
92 M. L. Hug gins, J. Phys. Chem., 46, 151 A942); Ann. N. Y. Acad. Sci.,
43, 1 A942).
93 P. J. Fl о ry, J. Chem. Phys., 10, 51 A942).
94. С R. В о h n, J. R. S с h a e f g e n, W. O. S t a 11 о n, J. Polymer Sci.,
55,531 A961).
95. P. J. F 1 о г у, С. А. Н о e v e, А. С i f e r r i, J. Polymer Sci., 34, 337
A959).
96. T. Uchida, Y. Kurita, M. Kubo, J. Polymer Sci., 19, 365 A956).
97. С W. Bunn, Proc. Roy. Soc. (London), Ser. A, 180, 40 A942).
98. D. Fisher, Proc. Phys. Soc. (London), Ser. B, 46, 7 A953).
99. W. T. A s t b u г у, Н. J. Woods, Phil. Trans. Roy. Soc. London, Ser. A,
232 333 A934).
100. С. Н. В a m f о r d, W. E. H a n b y, F. H a p p e y, Proc. Roy. Soc.
(London), Ser. A, 205, 30 A951).
101. E. R. В 1 о u t, С d e L о z e, S. M. Bloom, G. D. F a s m a n, J. Am.
Chem. Soc, 82, 3787 A960).
102 A. F. D i о r i o, L. M a n d e 1 k e r n, E. R. L i p p i n с о 11, J. Phys. Chem.,
66, 2096 A962).
Плавление
сшитых полимеров
Введение
Особый интерес представляет определенная форма
нерегулярности в цепях, связанная с образованием между звеньями
различных линейных макромолекул поперечных связей
(сшивок). По мере увеличения числа таких сшивок в системе
постепенно развивается трехмерная сетчатая структура, достигающая
макроскопических размеров. Такие структуры будем называть
бесконечными сетками *.
Согласно теории [1, 2], начальное образование сетки
происходит в том случае, если концентрация сшивок с превосходит
критическую величину:
с* = -=±— «J- (87)
X в 1 Xq
где хв — средневесовая степень полимеризации исходного
несшитого полимера.
При этой концентрации в нерастворимую сетку связаны еще
далеко не все полимерные цепи. Дальнейший рост сетки, или
гель-фракции, зависит от молекулярно-весового распределения
исходного Линейного полимера [3]. Однако обычно полное
образование сетки завершается уже тогда, когда концентрация
межцепных поперечных связей достигает нескольких процентов.
Если бы роль сшивок сводилась только к понижению
степени упорядоченности системы, то их влияние на
кристаллизацию можно было описать по аналогии с влиянием сомономера
в сополимерных системах в терминах «золь-эффекта»**. Но
дальнейшее теоретическое рассмотрение и экспериментальные
наблюдения опровергают это предположение.
* [В русской литературе их называют «сетками» или «трехмерными
полимерами». — Прим. редактора].
** [Понижение температуры плавления одинаковым образом
увеличивается с ростом концентрации растворителя или химически связанного
источника нерегулярности. — Прим. редактора].
152
В противоположность другим типам нерегулярностей
поперечные связи оказывают специфическое влияние на плавление,
поскольку они в действительности соединяют различные цепи.
Помимо того, вследствие пространственных ограничений,
включение поперечно связанных звеньев в кристаллическую ячейку
затрудняется, что, безусловно, может привести к изменению
процесса кристаллизации, по сравнению с аналогичной
системой, но с несвязанными звеньями.
При теоретическом рассмотрении образования сетки обычно
предполагают, что поперечные связи беспорядочно распределены
по объему образца. Однако нет никакой необходимости вводить
какие-либо ограничения, касающиеся взаимных конформаций
полимерных цепей в момент возникновения сетки. Хотя обычные
сетки образуются из статистически свернутых полимерных
клубков, этот вариант представляет собой лишь частный случай.
Например, сетки могут также возникать из деформированных
систем или из систем, в которых в момент образования сетки
цепи частично или полностью упорядочены. В этом случае
можно считать, что сами сшивки статистически распределены в
пространстве. На основании теоретических данных можно
предположить [4] наличие зависимости результирующих свойств
такой системы от характера расположения цепей во время
образования сетки.
Следовательно, при обсуждении свойств систем, состоящих
из сшитых макромолекул, в частности их кристаллизационного
поведения, необходимо обращать серьезное внимание на
условия, предшествующие образованию поперечных связей в системе.
Сшивка звеньев различных полимерных цепей может быть
вызвана химической реакцией, например, при вулканизации
натурального каучука [5], или в некоторых благоприятных случаях,
под действием ионизирующего облучения высокой энергии [6].
Многие природные высокомолекулярные системы в ходе их
синтеза образуют достаточное число сшивок, так что в
расплавленном состоянии они обладают большинством характеристик
бесконечной сетки. В дальнейшем для упрощения мы ограничимся
рассмотрением идеализированной совершенной сетки, которая
не содержит растворимых макромолекул и в равной мере
свободных концов цепей.
Для дальнейшего количественного описания сетки [4, 7, 8] в
аморфном или жидком состоянии за единичные цепочки
примем линейные участки макромолекул между двумя соседними
сшивками. Удобно характеризовать такую цепочку вектором г,
равным среднему расстоянию между ее конечными звеньями,
т. е. между сшитыми звеньями. Число цепей v должно быть
равно числу таких звеньев, сшитых межцепными поперечным»
связями,
153
Пусть Л^о —общее число звеньев цепи в сетке, тогда c = v/N0.
Таким образом, сетку можно характеризовать числом единичных
цепей и функцией распределения из векторов в пространстве.
При деформации сетки эта функция распределения изменяется
так же, как и макроскопические размеры сетки, и происходит
аффинное преобразование координат сшивок. Кроме того,
предполагается, что конформация индивидуальных цепей
подчиняется гауссовской статистике.
Обычно за стандартное состояние сшитой системы
принимается изотропная сетка с компонентами среднеквадратичных
расстояний между концами цепей х2 = y2^=z\ = /"o/3. В
стандартном состоянии определенное таким образом
среднеквадратичное расстояние между концами единичных цепей совпадает
с невозмущенной длиной эквивалентной свободной цепи. При
любом заданном состоянии сетки, в котором х2, у2 и z2 —
декартовы компоненты среднеквадратичных расстояний между
концами единичных цепей, энтропия сетки включает составляющую
т[-т|-<*«+*+*«>]
характеризующую внутренние конформации цепей [4, 7]. Вклад
в общую энтропию статистического распределения поперечных
связей по объему V образца составляет [4, 9]:
-^- In V -|- const
для тетрафункциональных связей (сшивки между двумя
молекулами).
Следовательно, разность энтропии в заданном и стандартном
состояниях может быть выражена следующим образом:
&+* + Ъ+1+Ща)] (88)
2
где {a) — (x2y2z2/x2y2z2y*. Параметр (а) представляет собой,
среднегеометрическое значение линейного расширения сетки в
заданном состоянии по сравнению со стандартным. При этом
объемы стандартного и рассматриваемого состояний могут не
совпадать друг с другом.
Для сетки, образованной из изотропных статистически
свернутых полимерных цепей, (а) = 1 при температуре и объеме
сетки, которые были в момент сшивки. С другой стороны, сетка,
возникшая при сшивании нестатистически распределенных
цепей, характеризуется величиной (а), которая зависит от
размеров молекул и их расположения; при сохранении начального
объема и температуры у такой сетки значение (а) может быть
как больше, так и меньше единицы.
154
Этого беглого анализа способов образования и описания
сеток достаточно для того, чтобы перейти к рассмотрению
плавления кристаллизующихся сшитых систем.
Теория плавления изотропных сеток
Температура плавления ненапряженной изотропной сетки
Гпл может быть выражена в самом общем виде как отношение
энтальпии к энтропии плавления. В свою очередь, энтропия
плавления может быть представлена как сумма трех аддитивных
составляющих: AS0 — энтропия плавления в отсутствие
ограничений, накладываемых на конформацию цепи при сшивке; AS^—
изменения конфигурационной энтропии в стандартном
состоянии ((а) = 1), обусловленные появлением поперечных связей;
А5э — изменения энтропии при переходе от стандартного
состояния к реальному. В последнем случае (а) принимает
значение, характерное для данной структуры сетки.
Температура плавления изотропной сетки, следовательно,
может быть выражена соотношением:
1 A5°+A5J + as;
Она зависит от строения сетки и от характера ее
происхождения.
Если структура сетки не затрудняет кристаллизацию сшитых
звеньев, то можно предположить, что АН и AS0 не зависят от
концентрации этих звеньев. Тогда AS0 совпадает с энтропией
плавления чистого несшитого полимера, а отношение AS0/АН —
с равновесной температурой плавления Тпл чистого полимера.
Если же стерические факторы таковы, что сшитые звенья
исключаются из кристаллизующихся областей, то указанные
величины изменяются соответствующим образом. Присутствие сшитых
звеньев в расплавленной фазе и их отсутствие в
кристаллической приводит к увеличению AS0 (по сравнению с несшитым
полимером) на величину Re из расчета на моль звеньев цепи.
По той же причине температура плавления должна
соответственно уменьшается, если АН при сшивке не меняется.
Для сеток, образованных из статистически свернутых цепей,
AS°X должно обязательно равняться нулю, так как сшивки между
звеньями распределяются статистически. Подобный процесс
поперечного связывания не оказывает влияния на
характеристики конфигурационной энтропии статистических несшитых
цепей. Для этого случая AS? также должно равняться нулю.
155
Однако при сшивке полимерных цепей, расположенных в
исходном состоянии параллельно, используемые для расчетов
параметры должны быть изменены. Если (а) известно, А5э может
быть рассчитано теоретически [4]. Относительная величина Л5э
в выражении (89) должна быть малой. Однако вследствие
характера расположения цепей во время образования сшитой
системы определенная доля начальной высокой степени
упорядоченности сохраняется и в структуре сетки. Эти элементы
упорядоченности будут сохраняться независимо от любых
последующих превращений, которым сетка может подвергаться, если
только они не включают разрыв поперечных связей.
Когда образование сетки происходит при условии одноосной
упорядоченности, требуется, чтобы звено одной цепи, которая
должна быть сшита, присоединялось к некоторому строго
предопределенному звену соседней цепи.
Таким образом, несмотря на то, что сшивки распределяются
в пространстве беспорядочно, сшиваемые звенья не могут быть
выбраны статистически. Так как сетчатая структура сохраняется
даже в жидком состоянии, в результате такой предопределенной
сшивки происходит уменьшение конфигурационной энтропии
системы. Последнее обстоятельство отличает рассматриваемую
систему от сетки, образованной при беспорядочной сшивке
статистических цепей. Уменьшение конфигурационной энтропии
проявляется в уменьшении общей энтропии плавления, которое
включается в составляющую AS*.
Чтобы рассчитать Д5° сетки, образованной из идеально
ориентированных цепей, необходимо вычислить вероятность
того, что звенья, включенные в сшивку, будут расположены в
некоторых определенных положениях относительно друг друга.
Результаты такого вычисления [4] могут быть представлены
формулой:
«..„(J^-' + 'uu) т
где С —безразмерный коэффициент порядка единицы;
Nc — число статистических элементов сетки*. Подобное
выражение было выведено Шелманом для учета влияния
межцепных сшивок на стабилизацию упорядоченных полипептидных
цепей.
На основе этого можно вывести количественные соотношения
для равновесной температуры плавления различных типов
кристаллизующихся сеток.
* Статистический элемент сетки находится в том же самом отношении
к звену единичной цепи, как и статистический элемент «эквивалентной
статистической цепи» к повторяющемуся звену реальной цепи. См, [7], стр, 410.
156
Для сеток, образованных из статистических цепей, при
условии, что сшитые звенья могут кристаллизоваться без
ограничений (сшитые и несшитые звенья стерически неразличимы), не
должно наблюдаться никаких изменений в температуре
плавления, по сравнению с несшитым полимером. С другой стороны,
если сшитые звенья не могут входить в кристаллическую
решетку, то:
и должно происходить понижение температуры плавления.
Формула (91) может рассматриваться как предельная
форма выражения E4), которое описывает температуру плавления
статистических сополимеров, содержащих с некристаллизую-
щихся звеньев.
Когда сетка образована из идеально ориентированных цепей
и сшитые звенья включаются в кристаллизацию на равных
правах со «свободными» звеньями, с достаточным приближением
должно выполняться соотношение:
1 1 RC /9 З...Л (92)
м U 4 /
С Пл Щ
Здесь k — число мономерных звеньев, соответствующее
одному статистическому элементу.
Согласно соотношению (92), равновесная температура
плавления такой сетки должна увеличиваться по сравнению с
температурой плавления исходной несшитой системы. Если участие
сшитых звеньев в кристаллизации ограничено, то этот эффект
будет частично скомпенсирован появлением в формуле (92)
составляющей, эквивалентной правой части формулы (91).
Мы можем теперь на основании изложенных теоретических
соображений перейти к рассмотрению экспериментальных
данных, касающихся температур плавления сеток, образованных в
различных условиях.
Температура плавления сетки,
образованной статистическими цепями
Статистическая сшивка неупорядоченно расположенных
цепей происходит при обычной (химической) вулканизации
каучука, радиационной сшивке каучука при комнатной
температуре или радиационной сшивке полиэтилена при температуре
выше точки его плавления (когда он находится в каучукоподобном
167
состоянии). Такие сетки могут быть закристаллизованы из
расплава простым охлаждением с последующим определением
равновесной температуры плавления. Некоторые результаты для
типичных сеток этого вида представлены на рис. 45. Сетки были
получены при облучении полиэтилена при температуре выше
его точки плавления [11], а также вулканизацией натурального
каучука серой, либо перекисью ди-трег-бутила [12]. Температура
плавления во всех случаях была определена при малых
скоростях нагрева, осуществляемого
сразу после кристаллизации.
Для двух различным
образом завулканизированных сеток
натурального каучука снижение
температуры плавления зависит
только от концентрации сшитых
звеньев и не зависит от
характера химической реакции,
посредством которой была
осуществлена вулканизация. Наиболее
важным результатом является тот
факт, что введение уже весьма
малого количества сшивок
вызывает заметное снижение
температуры плавления. У
натурального каучука снижение
температуры плавления на 20 град
происходит при сшивке лишь 1%
звеньев. У сшитого полиэтилена
наблюдается снижение
температуры плавления на 30 град при сшивке 0,5% звеньев цепи.
Сходные результаты получены и для температур плавления
вулканизированного ^ис-полибутадиена [14]. Сильное снижение
температуры плавления сеток рассмотренного типа представляет
собой общее явление.
Если снижение температуры плавления обусловлено лишь
тем, что сшитые звенья исключаются из кристаллической фазы,
то формула (91) может быть использована для анализа данных
рис. 45. Согласно формуле (91), при использовании в расчетах
величины Д#м для натурального каучука или полиэтилена
следовало бы ожидать снижения температуры плавления всего на
2—3 град. Очевидно, что теоретические расчеты в данном случае
не подтверждаются экспериментальными, поэтому возникает
вопрос о применимости формулы (90).
При выводе уравнения (90) принималось, что система
находится в условиях равновесия. Это означает, что развитию
кристаллизации в направлении цепей могут препятствовать только
статистически распределенные некристаллизующиеся звенья, в
Рис. 45. Зависимость понижения
температуры плавления от
концентрации сшивки с в
полимерной сетке, образованной из
статистических цепей [11, 12]:
Д —расплавленный полиэтилен, сшитый
под действием ионизирующего
облучения; О — натуральный каучук,
вулканизованный серой; 0 —натуральный каучук,
вулканизованный перекисью трет-д.ибу-
тила.
158
то время как кристаллизация в поперечном направлении
неограниченна. Если эти условия не выполняются и возникает менее
совершенная кристаллическая система, то выражение (91) уже
не может быть применено. В этом случае будет наблюдаться
более сильное снижение температуры плавления. Имеется
достаточно экспериментальных данных, свидетельствующих о
неприменимости формулы (91) при кристаллизации сеток
рассматриваемого типа.
Изучение кристаллических сеток полиэтилена и натурального
каучука методом рассеяния рентгеновских лучей под большими
углами [11, 12] указывает на то, что увеличение плотности
сшивки влечет за собой прогрессирующее расширение рефлексов от
различных кристаллических плоскостей. Это может быть
связано с уменьшением размеров кристаллитов, дальнейшим
нарушением кристаллического порядка или с возникновением
внутренних напряжений. Независимо от того, какой из этих
эффектов вызывает расширение полос рентгеновской дифракции,
каждый из них может понижать температуру плавления.
Следовательно, главной причиной такого большого снижения
температуры плавления является сильное ограничение возможности
установления совершенного кристаллического порядка в
системе даже после тщательного отжига. Совершенно очевидно, что
это ограничение вызвано наличием сшивок. Постоянные сшивки
препятствуют установлению поперечной упорядоченности при
упаковке полимерных цепей, необходимой для образования
достаточно больших кристаллитов. Участие в кристаллизации
звеньев, смежных со сшитыми, также может быть
затруднено или невозможно. Поэтому и развитие продольной
кристаллической упорядоченности ограничивается в большей
степени, чем это следует из простого учета концентрации
сшивок.
Кун и Майер [14] показали, что в набухших полимерных
сетках температура замерзания низкомолекулярного растворителя
значительно снижается, по сравнению с температурой
замерзания чистой жидкости. Для сеток натурального каучука,
набухших в бензоле, или для смешанных сеток полиакриловой
кислоты и поливинилового спирта, набухших в воде, понижение
температуры замерзания растворителя обусловлено с
концентрацией сшивок: она непрерывно возрастает с увеличением
плотности сшивки. При этом можно достичь снижения температуры
замерзания на 21 град, что связано с ограничением размеров
образующихся кристаллов за счет структуры сетки и
присутствия сшитых звеньев.
Таким образом," наличие сшивок в малых концентрациях не
только влияет на кристаллизацию сетки, но и кристаллизация
присутствующего в геле жидкого компонента также сильно
ограничивается.
159
Температура плавления сеток,
образованных из одноосноориентированных
цепей
Частично упорядоченные сетки могут быть получены из
полимерных цепей, находящихся в одноосноориентированном
состоянии (частным случаем этого состояния являются волокна)
в противоположность статистическому типу сеток, которые
рассмотрены в предыдущем разделе. Такие предварительно
ориентированные сетки образуются из растянутого натурального
каучука под действием ионизирующего облучения [12, 15]. В
подобных случаях перед плавлением и определением равновесной
температуры плавления образца последнему дают возможность
сократиться или отрелаксировать и только после этого
кристаллизуют в свободном состоянии простым охлаждением. Лишь при
таких условиях
измеренную температуру
плавления можно сравнивать с
температурой плавления
сетки, образованной из
того же самого полимера
под действием
одинаковых агентов, но при
начально статистически
свернутых полимерных цепях.
Зависимость
температуры плавления от
концентрации сшитых звеньев
для указанных двух
предельных типов сеток
натурального каучука приведена на рис. 46. Как видно из рисунка,
сетке, образованной из статистических цепей (кривая /),
соответствует понижение температуры плавления того же порядка,
что и в ранее рассмотренном случае для вулканизированного
натурального каучука. Для сеток, образованных из
предварительно одноосноориентированных цепей (кривая 2),
наблюдается лишь очень малое понижение температуры плавления с
увеличением концентрации сшивок в изучаемой области
концентраций.
Следовательно, независимо от степени сшивки ГЦЛ для сеток
ориентированных цепей выше, чем для сеток, образованных
из неупорядоченных цепей. Более того, различие Тпл этих двух
типов сеток возрастает при увеличении степени сшивки. Таким
образом, мы имеем убедительный пример повышения
стабильности, которую система приобретает при сшивке
ориентированных цепей. Главной причиной этого является пониженная кон-
I i . ^ i
5 Ю /5 20
с-Ю\ мол.%
Рис. 46. Зависимость равновесной
температуры плавления изотропных сеток
натурального каучука, образованных при
облучении, от концентрации сшивки [12]:
7 —статистические цепи; 2—цепи, одноосноориен-
тированные в момент образования сетки.
160
Таблица 6
Сравнение равновесных температур плавления
для сеток натурального каучука
р-108
5,0
7,5
10,0
12,5
15,0
(г /г" -1/7-" Лю3
V ' пл. с пл. о/
наблюдаемое
0,049
0,080
0,110
0,163
0,180
теоретическое
*«1
0,062
0,084
0,109
0,133
0,155
*»з
0,052
0,073
0,093
0,113
0,131
Примечание. ГЦЛ с и Т*л —равновесные
температуры плавления сеток, образованных из статистических и
ориентированных цепей*
фигурационная энтропия жидкого состояния, возникающего при
образовании сетки такого типа.
Согласно формуле (92), если сшитые звенья участвуют в
кристаллизации, должно происходить непрерывное увеличение
равновесной температуры плавления. Однако поскольку реально
этого не наблюдается, следует заключить, что в данном случае
сшивка препятствует процессу кристаллизации. Если же
предположить, что неконфигурациониые эффекты сшивок одинаковы
для обоих типов сеток, то сравнение их температурного
поведения может быть осуществлено на основе выражения (92). С
учетом этого ожидаемая разность температур плавления может
быть вычислена для различных с.
Результаты расчета сеток, образованных из
неупорядоченных и предварительно ориентированных цепей, приведены в
табл. 6. При расчете была использована величина Д#м=
= 1050 кал/моль, а параметру k приписывали значения 1 и 3.
Если сравнение делается при одинаковых значениях с, то
наблюдаемые различия в температурах плавления хорошо
согласуются с теоретически ожидаемыми для всей области
концентрации сшивок, в которой могут быть получены
кристаллизующиеся сетки. Очень хорошее согласие получается для
концентрации сшивок меньше 1%. Небольшие различия между
расчетными и измеренными величинами ГпЛ при высоких с могут
быть отнесены за счет очень большого понижения температуры
плавления, наблюдаемого в этой области концентраций для
сеток, образованных из статистических цепей.
И Л. Манделькерн
161
Следовательно, удается количественно подтвердить
предположение, согласно которому при образовании сеток из одноос-
ноориентированных цепей происходит заметное уменьшение
конфигурационной энтропии жидкого состояния. Можно сказать,
что в данном случае образуется частично упорядоченная
жидкость.
Температура плавления сеток,
образованных из кристаллических
неориентированных цепей
Так как вследствие сшивания одноосноориентированных
полимерных цепей образуется сетка с очень необычными
свойствами, возникает вопрос, какой вид или какая степень
межмолекулярной упорядоченности требуется, чтобы эти свойства
возникли. Например, частично кристаллические недеформиро-
ванные полимеры обладают высоким уровнем
межмолекулярной упорядоченности, так как отдельные участки макромолекул
вынуждены располагаться в трехмерной сетке параллельно
друг другу. Эта упорядоченность ближнего характера,
поскольку отдельные кристаллические области располагаются в
объеме полимера беспорядочно. Вопрос о том, влияет ли
наличие такой упорядоченности на свойства возникающей
изотропной сетки, должен быть решен на основе анализа
экспериментальных данных.
Исследования показали, что равновесная температура
плавления сеток, образованных из кристаллического, но
неориентированного линейного полиэтилена, существенно отличается от
температуры плавления сеток, полученных из того же самого
полимера, только в условиях, когда цепи находились в
расплавленном состоянии. Для последнего типа сеток по мере
сшивания наблюдается непрерывное понижение ТпЛ> как это
показано на рис. 45.
С другой стороны, у сеток, образованных из
кристаллических полимеров, температура плавления уменьшается только
на 6,5 град и становится независимой от концентрации
поперечных связей вплоть до очень высоких степеней сшивки [И].
Температуры плавления сшитых систем определялись после
предварительного плавления и перекристаллизации. Результаты
наблюдений приведены в табл. 7 (а) и 8 (а). Образцы
охарактеризованы количеством связываемого при равновесном
набухании ксилола Vi при 130° С.
Уменьшение величины v\ указывает на прогрессирующее
увеличение значения с. Для наименее набухшей сетки, обр'азо-
162
Таблица 7
Свойства полиэтиленов, радиационно сшитых
при 17° С
а
Vi
0,97
0,95
0,83
0,74
0,61
0,39
0,28
пл
131
131
131
131
131
131
132
б
т
пл
102
104
105,3
106.2
ПО
114
^(пР»Гпл)
0,88
0,83
0,77
0,69
0,58
0,36
Таблица 8
Свойства полиэтиленов, радиационно
сшитых при 175° С
а
Vi
0,93
0,91
0,87
0,76
0,70
0,49
т
пл
134
132,5
130
128
115,5
107.5
101
б
т
пл
101
100,2
100
100,6
94
"i("p"V)
0,84
0,85
0,80
0,71
0,68
ванной при 17°С (в высококристаллическом состоянии),
содержание сшитых звеньев составляет 2,5% [11]. Несмотря на
большое изменение величины с, Тпл после небольшого уменьшения
в начале не изменяется по сравнению с этой величиной для
чистого полимера.
Большая стабильность кристаллического состояния сеток,
образованных из неориентированных, но кристаллических
цепей, по сравнению с сетками, образованными из аморфных
полимеров, может быть объяснена (качественно) так же, как
для сеток из растянутого натурального каучука. Хотя до
образования сетки кристаллиты были статистически распределены
относительно друг друга, участки цепей внутри кристаллитов
ориентированы параллельно. Поэтому при сшивке
преимущественно кристаллического полимера уже не может возникнуть
совершенно беспорядочная сетка. Характер сшивки предопреде-
11»
163
лен локальной ориентацией цепей в кристаллитах.
Следовательно, у таких сеток следует ожидать увеличение равновесной
температуры плавления.
Можно заключить, что для развития частичной
упорядоченности в жидком состоянии макроскопическая ориентация не
обязательна. Ограничению упорядоченности будет
сопутствовать уменьшение энтропии плавления.
Проявление молекулярной упорядоченности в жидком
состоянии подтверждается далее прямым микроскопическим
наблюдением расплавов полимеров [11, 16].
Рис. 47. Микроструктура линейного полиэтилена
(марлекс-50), облученного дозой 120 Мрад при
25° С [11].
Фотография сделана после выдержки образца в течение
1/2 ч при 150° С.
Как показано на рис. 47, для таких систем интенсивное
двойное лучепреломление, обусловленное наличием сферолитов,
сохраняется при температурах, выше температуры плавления.
В противоположность этому у полиэтилена, сшитого в
расплавленном состоянии, при температурах выше Т^л двойное
лучепреломление уже не наблюдается. Наличие двойного
лучепреломления в первом случае может быть непосредственно
связано с образованием необычайной (упорядоченной) жидкой
структуры.
Интенсивность электронного пучка, используемого в
электронно-микроскопических исследованиях тонких
кристаллических полимерных пленок, обычно достаточна для образования
сшивок. Поэтому неудивительно, что после предварительного
исследования (на электронном микроскопе) кристаллических
полиамидных и полиэтиленовых пленок они сохраняют упоря-.
доченную структуру и при последующих исследованиях уже в
расплавленном состоянии.
164
Зти результаты не могут служить доказательством того, что
жидкое состояние в полимерах вообще является
упорядоченным [17]. По указанным причинам частичная ориентация
жидкости представляет собой очень интересный, но исключительный
случай, обусловленный характером расположения цепей во
время образования сетки.
Плавление набухших сеток
Кристаллизация полимерной сетки может также
происходить в условиях ее контакта с низкомолекулярной жидкостью,
или растворителем. Наиболее простым для расчета является
случай контакта сетки с избытком однокомпонентной жидкой
фазы, что соответствует термодинамически открытой системе.
При плавлении сетка в аморфном состоянии впитывает в себя
большое количество жидкости; степень происходящего при этом
набухания зависит от структуры сетки, температуры и
термодинамического параметра взаимодействия для данной системы
полимер — растворитель. И наоборот, при кристаллизации
растворитель полностью удаляется из сетки, если он не может
входить в кристаллическую решетку.
При температуре плавления кристаллическая полимерная
фаза находится в равновесии со смешанной фазой, состоящей
из аморфного полимера и впитанного растворителя. В свою
очередь, последняя фаза должна находиться в равновесии с
фазой чистого растворителя. Три различные фазы должны
сосуществовать при температуре плавления.
Следовательно, условия равновесия для компонентов каждой
фазы имеют вид:
Hm-Hm = ^m-Vm (93)
иГ-ц? = 0 (94)
где [ij; и \icM — химические потенциалы полимерных'звеньев в
кристаллической и смешанных фазах;
[i°M — химический потенциал полимерного звена чистого
расплава.
Формула (94) устанавливает равенство химических
потенциалов растворителя в смеси и жидкой фазе *.
Изменение свободной энергии AF при образовании
смешанной фазы из ее чистых компонентов, т. е. чистого растворителя
* Рассмотрение, конечно, может быть обобщено и на случай,
включающий мультикомпонентную изотропную жидкую фазу и на распределение
компонентов между ней и смешанной фазой. Случай проникновения
растворителя в кристаллическую фазу также может быть рассмотрен.
165
и чистой изотропной аморфной сетки, складывается из двух
частей: свободной энергии смешения AFCM и эластической
свободной энергии AFQy связанной с расширением сетки при набухании
[4 и 7, стр. 578].
Согласно теории Флори — Хаггинса [18, 19]:
bFm = kT(n{\nvx + ixn{v2) (95)
где П\ — число молекул растворителя.
Используя идеализированную, но удобную теорию каучуко-
подобной эластичности, в которой предполагается, что
деформационные процессы, сопутствующие набуханию, не связаны с
изменениями внутренней энергии, обусловленной межцепными
взаимодействиями, можно записать [4]:
ЬРЬ = *?- [C (аJ а2, - In а3, - 3 - 3 In (а))] (96)
где as — коэффициент линейного расширения для набухания
сетки.
В результате суммирования формул (95) и (96) получим:
^L = ^YjL(Vl-bVl)+^[l-v2 + 2(a)(v^-v^)\ (97)
поскольку a3 = i;!f1. Как и в отсутствие сетки, м? — цЯ может
быть записано в форме:
^-^ — ДЯм^-^) (98)
где Гпл — соответствует равновесной температуре плавления
заданной сетки.
При равновесии Т=ТПЛ, так что
Первые два члена правой части формулы (99) идентичны
полученным для смеси несшитого полимера с растворителем
того же самого состава. Остальные учитывают влияние
эластической свободной энергии смешанной фазы. Для открытой
системы состав смешанной фазы v^ определяется из уравнения
(94), которое описывает равновесное набухание [7, стр. 578].
Следовательно, v2 есть равновесная величина и должна
соответствующим образом обозначаться. Она может быть
приравнена обратному значению равновесной степени набухания при
Т=ТПЛ. Для замкнутой системы, у которой состав смешанной
фазы фиксирован и изотропная жидкая фаза отсутствует,
формулы (92) и (98) удовлетворительно определяют отношения
точек плавления.
166
Из сравнения выражений (99) и A3) следует, что надо
ожидать большего понижения температуры плавления
рассматриваемой сетки, по сравнению с набором полимерных цепей
одинаковых по строению и концентрации, вследствие вклада
эластической свободной энергии. Однако в связи с тем, что обычно
встречающиеся значения с имеют порядок от 0,01 до 0,02 или
меньше, этот эффект очень слаб. Он проявляется только в том
случае, когда у2 смешанной фазы становится меньше 0,5.
Была измерена температура плавления полиэтиленовых
сеток, погруженных в избыточное количество ксилола [11]. Анализ
результатов можно провести на основе теории равновесия. Для
сеток, образованных как из статистических цепей, так и из
неориентированных кристаллических цепей, естественно наблкн
дается падение равновесной температуры плавления, по
сравнению с Тпл неразбавленных систем. Однако, как это следует из
анализа результатов, приведенных в табл. 7F) и 8F), Т*пл
упомянутых сеток зависит от плотности сшивки совершенно
различным образом. Системам ма-
кромолекулярных цепей, сшитых
в кристаллическом состоянии и
погруженных в ксилол, присуще
непрерывное и заметное
увеличение температуры плавления с
ростом концентрации сшивок. В то
же время температура плавления
сеток, образованных из
статистических цепей, слегка
уменьшается с ростом плотности сшивки.
Независимо от расположения
цепей в момент образования сетки
в последнем случае неизменно
наблюдается уменьшение Гпл с
увеличением концентрации сшивок.
Температура плавления
набухшей сетки зависит от
характера исходной сетки и от количества жидкости, впитываемой
сеткой после плавления. Для сеток, образованных при 17° С,
Гпл при отсутствии растворителя не зависит от концентрации
сшивок. Однако равновесное набухание при Tnjl(vi при Тпя), как
это следует из табл. 7 и 8, непрерывно уменьшается. Для сеток,
образованных из статистических цепей, скорость уменьшения
7^пЛ не компенсируется концентрационными изменениями в
смешанной фазе, в результате чего происходит уменьшение Тпл.
Сравнение экспериментальных результатов с теоретическим
дано на рис. 48. Сплошная линия на этом рисунке представляет
теоретическую зависимость, полученную из уравнения (99) для
02 0{* 06 08 Ю
Рис. 48. Зависимость 1/Тпл — 1/7^
от объемной доли ксилола vb
поглощенного при плавлении [11]:
ф —сетка, состоящая из кристаллических
цепей; А —сетка, образованная из
расплавленных цепей; теоретическая
зависимость согласно выражению (99)
в предположении Xi^O и с = 0.
167
%i = 0 и в пренебрежении эластической составляющей AF9.
Детальный анализ данных затруднен тем, что для этих систем
неизвестно %i и нельзя точно определить параметры сетки.
Однако для малых значений viy при которых ни
термодинамическое взаимодействие, ни эластическая составляющая AF9 не
вносят заметного вклада в понижение температуры плавления,
экспериментальные данные согласуются с простыми
теоретическими предпосылками. При уменьшении концентрации полимера
в смешанной фазе уже можно ожидать отклонения от простой
теоретической зависимости в связи с возрастанием роли
упомянутых взаимодействий. Более вероятно, что %i>0. Это
означает, что экспериментально наблюдаемые значения температур
плавления будут выше теоретических, полученных для xi = 0-
Несмотря на ограниченные возможности точного
количественного сравнения, все же заслуживает внимания тот факт,
что существующая теория явно предсказывает различия в
поведении разных типов набухших сеток при изменении
концентрации сшивок. Особое внимание следует обратить на важность
количественной оценки концентрации смешанной фазы в
открытой системе.
Фибриллярные белки
У некоторых фибриллярных белков, таких как коллаген [20]
и а-кератин, из различных слоев эпидермы [21] наблюдается
увеличение температуры плавления с ростом плотности сшивки.
У коллагена увеличение температуры плавления достигает
35 град после сшивки с помощью специальных дубящих
агентов [20]. Для различных слоев эпидермиса губы быка
температура плавления быстро уменьшается при переходе от внешних
слоев к внутренним; при этом одновременно понижается
содержание цистина (что, вероятно, может быть связано с числом
межцепных сшивок) *.
Природные фибриллярные белки представляют собой одно-
осноориентированные системы, в которых на упорядоченную
структуру наложены поперечные связи. Следовательно, в
соответствии с теоретическими предпосылками [4], с ростом
концентрации сшивок, если последние не препятствуют
кристаллизации, следует ожидать непрерывного повышения температуры
плавления. Однако плавление фибриллярных белков
практически однозначно предопределяется их погружением в
соответствующую жидкую среду. Следовательно, при равновесии кон
* [Цистин сшивает цепи посредством дисульфидных связей» — Прим. ре*
дактора].
168
центрация полимера в смешанной фазе также должна
возрастать с увеличением степени сшивки. В этой связи, по аналогии
с рассмотренными выше набухшими полиэтиленовыми сетками,
следует ожидать увеличения температуры плавления.
Таким образом, введение сшивок в систему фибриллярного
белка вызывает как изменение энтропии плавления, так и
изменение состава смешанной фазы. Оба эти эффекта приводят
к повышению температуры плавления и благоприятствуют
стабильности кристаллической фазы. Как и следует ожидать, в
опытах, проведенных в присутствии большого количества
жидкости, наблюдается строгая корреляция между плавлением
коллагена и набухающей способностью смешанной фазы [22].
Литература
1. P. J. Flory, J. Am. Chem. Soc, 63, 3097 A941).
2. W. H. S t о с k m а у e r, J. Chem. Phys., 12, 125 A944).
3. P. J. F 1 о r y, J. Am. Chem. Soc, 69, 30 A947).
4. P. J. Flory, J. Am. Chem. Soc, 78, 5222 A956).
5. D. Craig, Rubber Chem. Technol., 30, 1291 A957).
6. A. Charlesby, Atomic Radiation and Polymers, New York, 1960.
7. P. J. Flory, Principles of Polymer Chemistry, Ithaca — New York, 1953,
p. 464.
8. R. G. Treloar, The Physics of Rubber Elasticity, New York, 1947.
9. P. J. F lory, J. Chem. Phys., 18, 108 A950).
10. J. G. Schellman, Compt. Rend. Trav. Lab. Carlsberg, Ser. Chim., 29,
223 A955).
11. L. Mandelkern, D. E. Roberts, J. С. Н a 1 p i n, F. P. Price,
J. Am. Chem. Soc, 82, 46 A960).
12. D. E Roberts, L. Mandelkern, J. Am. Chem. Soc, 82, 1091 A960).
13. G. S. Trick, J. Polymer Sci., 41, 213 A959).
14. W. Kuhn, E. Peterli, H. M a j e r, Z. Electrochem., 62, 296 A958);
W. Kuhn, H. Majer, Z. phys. Chem. (Frankfurt), 3, 330 A955);
W. Kuhn, H. Majer, Angew Chem., 68, 345 A956); W. Kuhn,
H. M a j e r, Ric Sci., Suppl., A, 3 A955).
15. D. E. Roberts, L. Mandelkern, J. Am. Chem. Soc, 80, 1289 A958);
D. E. Roberts, L. Mandelkern, P. J. Flory, J. Am. Chem. Soc,
79, 1515 A957).
16. С F. Hammer, W. W. Brandt, W. L. Peti colas, J. Polymer Sci.,
24, 291 A957).
17. V. A. Kargin, J. Polymer Sci., 30, 247 A958); В. А. Карги н,
Г. Л. Слонимский, Усп. хим., 24, 785 A955).
18. М. L. Huggins, J. Phys. Chem., 46, 151 A942); Ann. N. Y. Acad. Sci.,
41, 1 A942).
19. P. J. Flory, J. Chem. Phys, 10, 51 A942).
20. К. Н. Gustavson, The Chemistry and Reactivity of Collagens, New
York, 1956, p. 227.
21. K. M. Rudall, Symposium on Fibrous Proteins, J. Soc. Dyers Colourists,
15 A946); Adv. Protein Chem , 7, 253 A952).
22. F. R. The is, Trans. Faraday Soc, 42B, 244 A946).
Ориентационная
кристаллизация
и сократимость
Введение
Характерным свойством аморфных полимеров является их
способность выдерживать большие напряжения или
деформации. В сшитой системе, образующей трехмерную сетку,
напряжение обычно релаксирует, а процесс деформации обратим.
Тем не менее, при деформации такой системы сильно возрастает
тенденция к кристаллизации, так как участки цепей между
сшивками в большей или меньшей степени распрямляются,
утрачивая свою наиболее вероятную конформацию. Это вызывает
уменьшение конфигурационной энтропии. Следовательно, при
постоянно действующем напряжении переход в кристаллическое
состояние связан с меньшей затратой энтропии. Уменьшение
общей энтропии плавления повышает температуру
кристаллизации, по сравнению с тем же веществом в отсутствие
деформации. Возрастающая тенденция к кристаллизации хорошо
демонстрируется в известных опытах с натуральным каучуком и
полиизобутиленом, которые чрезвычайно медленно
кристаллизуются в отсутствие внешнего напряжения, но с удивительной
быстротой и легкостью при растяжении.
Как правило, цепи в кристаллитах, возникших при
растяжении, ориентированы преимущественно вдоль оси растяжения.
Такая ориентация особенно типична для процессов
кристаллизации при очень больших деформациях *. Морфология,
возникающая при ориентационной кристаллизации, этим и отличается
от обычной кристаллической текстуры, которая получается при
постепенном охлаждении и характеризуется беспорядочным
распределением ориентации кристаллитов. При включении части
деформированной цепи в кристаллит среднее напряжение,
которое она испытывает на концах, уменьшается. Это заключение
* Можно, однако, указать на некоторые исключения из этого правила,
встречающиеся обычно при неизотермической кристаллизации и
недостаточных деформациях. В этом случае цепи преимущественно ориентированы
нормально к направлению растяжения [1].
170
следует как из принципа Ле Шателье, так и из более
детального молекулярного анализа [2, 3].
В соответствии с молекулярной теорией каучукоподобной
эластичности, сила, действующая на фиксированные концы цепи,
обратно пропорциональна числу статистических элементов цепи
и расстоянию между концами цепи [2, 4]*. Так как после
включения цепи в кристаллит на результирующую силу влияют
только оставшиеся аморфные участки, эта сила должна
уменьшаться при ориентационной кристаллизации. Более того,
линейные размеры аморфных участков существенно уменьшаются
из-за непропорционально большей протяженности
выпрямленных участков, «захваченных» кристаллитами. Следовательно,
при ориентационной кристаллизации происходит общее
уменьшение сокращающей силы, возникающей в системе в ответ на
растяжение.
Можно сделать вывод, что ориентация при растяжении
способствует кристаллизации, а последняя в предварительно
ориентированных полимерах уменьшает внутреннее напряжение.
Каким бы ни было напряжение, испытываемое аморфными цепями,
проекция длины беспорядочно свернутой молекулы на ось
ориентации значительно меньше ее длины в кристаллическом
состоянии. Естественно, поэтому, что плавление ориентированной
структуры вызывает сокращение, а кристаллизация —
удлинение **. Изменения размеров и напряжения могут быть, таким
образом, отнесены за счет фазового перехода кристалл —
жидкость.
Подобное поведение, которое отражает одно из наиболее
уникальных свойств полимерных цепей, обусловлено их конфор-
мационной изменчивостью. Оно не ограничивается простейшими
типами цепных молекул, но в равной мере характерно для
фибриллярных белков и других макромолекул биологического
происхождения. Многие полимеры этой категории обладают в на-
тивном состоянии упорядоченной кристаллической структурой,
поэтому при анализе некоторых свойств биологических систем,
таких как термоэластичность, а также при исследовании
механизма процессов, связанных с изменением длины (например,
мышечной деятельности), необходимо помнить о существовании
в этих системах развитой кристаллической структуры.
* Подробное изложение теории каучукоподобной эластичности см. в
монографии [5].
** [Это очевидное на первый взгляд утверждение не вполне точно.
Возможны системы (с кристаллическим полиморфизмом или образованные муль-
тиспиральными макромолекулами), где плавление связано с удлинением.
См. Т. М. Б и р ш т е й н, О. Б. Птицын, Конформации макромолекул, Изд.
«Наука», 1964, особенно гл. 10; С. Я. Френкель, Л. В. К у х а р е в а,
Б. М. Гинзбург, В. И. Воробьев, ДАН СССР, т. 165, стр. 149—152,
1965, — Прим. редактора].
171
При анализе процесса плавления ориентированной системы
необходимо установить, действительно ли процесс обратим, т. е.
образуются ли при рекристаллизации ориентированные кристалл
литы. Предварительный анализ необходим, так как не
исключена возможность, что исходное ориентированное
кристаллическое состояние не будет восстанавливаться. Возможные
неравновесные особенности процесса плавления ориентированных
полимеров обсуждались в связи с плавлением «замороженного»
каучука [6].
Известно, что при хранении (в комнатных условиях)
натуральный каучук часто становится твердым и неэластичным;
обусловлено это заметным ростом кристалличности. При первом
нагревании такого каучука точка плавления оказывается
значительно выше, нежели при равновесном плавлении (в отсутствие
каких-либо внешних сил). Это кажущееся противоречие легко
разрешается, если учесть, что у «замороженного» каучука
кристаллические области имеют преимущественную ориентацию,
несмотря на отсутствие ориентирующей внешней силы, что и
обусловливает повышение температуры плавления. После первого
плавления и последующей рекристаллизации Гпл приобретает
обычное для натурального каучука значение, так как ориента-
ционная кристаллизация вновь не развивается.
Многие полимеры можно сделать фибриллярными («волокно-
подобными»), т. е. обладающими высокой одноосной
ориентацией кристаллитов, с помощью простых механических способов.
Во многих случаях это состояние достигается ниже температуры
плавления и без приложения растягивающей нагрузки. При
плавлении, кроме обычных изменений свойств, наблюдается
также одноосное сокращение. Температура этого перехода
называется температурой сокращения *. Однако только при
некоторых специальных условиях (см. стр. 198) эта температура
может быть идентифицирована с равновесной температурой
плавления.
Как правило, в отсутствие равновесной растягивающей силы
начальное кристаллическое состояние не восстанавливается при
простом обращении процесса плавления. Даже если к системе
и приложена растягивающая сила, все равно необходимо
различать равновесную температуру плавления Тпл и температуру
сокращения Т. Для последней требуется сосуществование
аморфной и кристаллической фаз по длине волокна, причем
следует учитывать, что для инициирования плавления и
наблюдения сокращения сильно ориентированного
высококристаллического волокна может понадобиться значительный перегрев.
Поэтому, если не учитывать разницу между Т°пл и Гс, можно
* [В случае биологических полимеров часто говорят о температуре
сверхсокращения! — Прим. редактора].
172
допустить серьезные ошибки, что и произошло при
исследовании сверхсокращения коллагена [7, 8]. Таким образом,
температура сокращения не является подходящей характеристикой для
термодинамического анализа.
Хотя ориентированные кристаллические полимеры обычно
претерпевают необратимое плавление, возможностью
обратимого перехода в условиях, приближающихся к равновесию, все
же нельзя пренебрегать. Действительно, рассматривая эту про-
блему с точки зрения фазового равновесия, можно получить
важные соотношения между кристаллизацией, деформацией и
изменениями размеров [3, 4].
Однокомпонентные системы,
подверженные действию растягивающей силы
Рассмотрим сшитую фибриллярную систему, состоящую из
высокоориентированных кристаллических областей,
сосуществующих с аморфными участками, полностью лишенных
каких-либо следов кристаллического порядка. Пусть однородная
растягивающая сила / приложена к волокну в направлении его
оси. Не рассматривая возможные различия в поперечном
сечении, возникающие вследствие распределения кристаллических и
аморфных областей по длине волокна, будем считать образцы
вполне однородными по химическому составу, структуре и
поперечному сечению.
В соответствии с первым законом термодинамики, изменение
внутренней энергии Е любой системы может быть записано как
dE = dQ — dW A00)
где dQ— теплота, поглощаемая системой;
dW — внешняя работа, совершаемая системой над
окружающей ее средой.
Пусть х{ представляет экстенсивные переменные,
характеризующие систему, и Уг — соответствующие им интенсивные
переменные, тогда:
dW^^^iyidxl A01)
dE^dQ — ^y.dXt A02)
В случае однокомпонентной системы нас будут интересовать
пары — /?, V и /, L,
где р — давление (интенсивная переменная как и /), а
V wL — объем и длина волокна (экстенсивные переменные).
Для обратимого процесса dQ = TdS (где S — энтропия) и
dE=TdS — pdV + jdL A03)
173
Вводя термодинамический потенциал Гиббса
Ф=*Е + рУ — TS = H— TS A04)
где Я— энтальпия, получим из равенств A03) и A04)
dO = — SdT + Vdp + fdL A05)
Для наших целей в качестве независимых переменных
удобнее выбрать р, Т и f и записать эквивалентное соотношение:
d@ — fdL) = — SdT-{-Vdp — Ldf A06)
Чтобы система находилась в состоянии равновесия при
постоянных р, Т и /, функция Ф—fL должна быть минимальна по
отношению к любым отклонениям от этого состояния, в
частности и при изменениях степени кристалличности волокна
A-Я).
Следовательно, если существует равновесие между двумя
фазами
Р^Ч,.,-
Полный термодинамический потенциал волокнистой системы
может быть выражен следующим образом:
Ф =ЛФа+A — Я)ФК A08)
где Фк и Фа— термодинамические потенциалы полностью
кристаллического и полностью аморфного волокна
при условиях, определяемых р, Т и f.
Аналогично можно выразить и другие экстенсивные
характеристики.
Таким образом, условие равновесия запишется в форме:
Фа — /?а = Фк— fLK A09)
или
d (фа — fL*) = d (Фк — f LK) A10)
Из равенства A06) следует, что при постоянном давлении
\dT)Pt?*B*ss'dL A11)
где AS и AL — изменения энтропии и длины при плавлении
всего волокна при постоянных Г, р и /.
Для рассматриваемого обратимого процесса поглощаемое
при плавлении тепло равно:
Q = TbS = /iE + HW^/iE + p\V — fM A12)
поэтому
д5=г^Ш. A13)
174
Комбинируя A13) и A11), получим:
или в более компактной форме, предложенной Джи [9] и Фло-
ри [4]:
Г а (у/г) -I _дя
Уравнения A14) и A15) являются очевидными вариантами
уравнения Клапейрона для одномерной системы с одноосно-
ориентированными кристаллической и аморфной фазами.
Температура Т может рассматриваться как температура плавления
ТИЛ в присутствии силы / и под давлением р.
Аналогия между этим специфическим фазовым равновесием
и равновесием в системах пар — жидкость или кристалл —
жидкость в низкомолекулярных системах становится более явной,
если учесть, что —/ и L в уравнениях A14) и A15)
соответствуют давлению и объему при обычной (трехмерной)
формулировке уравнения Клапейрона. В равновесной однокомпонентной
системе пар — жидкость давление не зависит от ее объема, т. е.
не зависит от относительного содержания каждой фазы.
Подобным же образом при «одномерной» формулировке
уравнения Клапейрона подразумевается, что в однородной
однокомпонентной фибриллярной системе равновесная сила f не
зависит от относительных длин, приходящихся на аморфную и
кристаллическую фазу, при постоянных Тир. Следует ожидать,
что обычно AL<0, если Д#>0. Таким образом, из равенства
A15) следует, что f/T будет возрастать с увеличением Г, т. е.
при постоянном давлении температура плавления растет с
увеличением приложенной растягивающей силы *.
Интегрирование уравнения A15) в определенных пределах
приводит к зависимости между равновесной силой /° и
температурой плавления. Эта операция аналогична интегрированию
уравнения Клаузиуса — Клапейрона для равновесия между
жидкостью и паром. Зависимость давления от температуры
может быть получена в том случае, если известно уравнение
состояния, связывающее давление с объемом жидкости. В нашем
случае необходимо располагать уравнением состояния,
связывающим силу, которая действует на аморфную сетку, с ее
длиной. Такое уравнение может быть получено на основе теории
каучукоподобной эластичности.
При действии постоянной растягивающей силы на одноком-
понентную аморфную сетку, составленную из цепей с гауссовым
* [См., однако, сноску на стр. 171. — Прим. редактора.]
175
распределением расстояний между концами, зависимость между
силой и длиной цепи задается соотношением:
'-*".['-(?Л
где Iй — длина неориентированной аморфной сетки в
изотропном состоянии, т. е. длина ее в отсутствие внешней
силы;
Za — длина этой же сетки в ориентированном аморфном
состоянии при действии равновесной растягивающей
силы *.
Соотношение A16) может в равной мере применяться как
к сеткам, образованным макромолекулами в беспорядочной кон-
формации, так и к сеткам из сильно ориентированных цепей.
Макроскопическую равновесную длину образца можно
связать с числом цепей v и среднеквадратичным расстоянием
между концами цепи соотношением:
?ш-7(тГ<в> A18)
где о'— число векторов цепи, пересекающих плоскость перпен*
дикулярно продольной оси образца.
Для сеток из высокоупорядоченных цепей, что соответствует
рассматриваемой проблеме, L" пропорционально v'/*. В такой
системе а равно числу цепей, проходящих через поперечное
сечение образца, и, следовательно, не зависит от v. Однако
величина г2, пропорциональная числу звеньев цепи, обратно
пропорциональна v; следовательно, длина L" пропорциональна v!/»(a).
Как следует из равенства A17), для образованных таким
путем сеток значение В не зависит от v. В дальнейшем удобно
ввести величину Ьмакс, представляющую собой длину
аморфного волокна при максимальном растяжении. Тогда [4]:
B-*faL aw)
^макс
Если La значительно больше Iй, сокращающая сила может
быть выражена таким путем:
f « ZkTvn' 72 A20)
* [Равенство A16) получено для гауссовой сетки из соотношения
/=(дД/Уд?)рг<а>; выражение для ASa хорошо известно [4]].
176
A16)
A17)
Подставляя f из A16) и A15), получим, учитывая, что
AL = La — LK:
(i._iK)d[L._ig^] = ^.rf^ (i2i)
Интегрируя это уравнение в пределах V\ La и Т°пл, Т1Л1
получим
2(LaLK)[ia-^]-[(LaJ+^-3(H=^(?i:~4')A22)
где ТпЛ — равновесная температура плавления в отсутствие
внешней силы;
Тпл — равновесная температура плавления при действии
силы /, растягивающей аморфный образец до длины La.
В неявном виде соотношение между Тпл и приложенной
силой можно получить, используя уравнения A16) и A17),
исключив из первого /Я
Для сеток, образованных высоко упорядоченными цепями,
где В задается формулой A21)
20»' /1 М A23)
зя \тал К,
здесь АЛ' — теплота плавления на моль статистических
элементов цепи.
При больших деформациях, когда (LH/LaK<l и может
быть использовано равенство A20), выражение A23)
упрощается до
(IaJ — 2L*LK 2ДЛ'
^макс 3#
A24)
Интегрирование можно также провести в пределах LK и La,
что дает
IV L*y\\ I 2(^L-2^/ I !_\ A25)
(L L)[l + L'{L'f\- В [Тал rj A25)
здесь Т*л — температура плавления при L*=LK.
Используя предыдущее выражение для В, получим:
((Vt-Щ U , 2(L"K I 2ДА1 / J Ц A26)
I W Jl + ^aJJ ЗЯ [Тпя ТЬ)
12 Л. Манделькерн 177
Для больших деформаций это выражение упрощается до
(LK — LaJ _ 2Мг' / 1
LxtCiVt. За
I— -—)
A27)
При TUJl<TL уравнения A25) и A26) имеют два решения
относительно La, больше и меньше LK. При Тпл >
Т*лвещественного решения не существует, т. е. Тпл играет роль критической
температуры, выше которой кристаллическая фаза уже не
может существовать ни при каких условиях.
При значительных деформациях, позволяющих использовать
A25) и A27) можно с помощью уравнения A20) исключить
La из этих двух уравнений:
/J_\ _ 3k\n' Г LK + ГШ^_ /_1 1\ 1
^ Т ' равн ^макс L ^макс V 3/? \ 7'пл Tln /J
/_М ~ Зкш' Г L* f LK , 2А/г' /1 1__\
* ^ 'равн ^макс L ^макс V ^макс °R \ / пл Гпл у
A28)
A29)
D
1 •
У
У
'С
В
Приближенные уравнения A28) и A29) позволяют
представить плавление ориентированных систем графически (рис. 49).
Если процесс деформации начинается
при некоторой температуре, при
которой сетка находится еще в
аморфном состоянии, и если уравнение
имеет форму A20), отношение f/T будет
линейно расти с увеличением L до
момента начала кристаллизации
(точка Л). Эта точка характеризует
температуру плавления сетки при
заданной силе и удлинении. С возрастанием
степени кристалличности длина
образца растет (участок АВ). Для од-
нокомпонентной системы сила
должна оставаться неизменной вплоть до
полного завершения перехода
(точка В). Меньший из двух корней
уравнений A28) и A29) определяет это равновесие.
Предполагается, что далее напряжение будет возрастать очень резко
(почти по вертикальной прямой) в результате перехода системы
в точке В в жесткое кристаллическое состояние.
Если можно достичь состояния, при котором La>LKy на
участке DE вновь возникает аморфная фаза. В этом случае
равновесная сила соответствует большему из двух корней. При
возрастании температуры точки А и Е смещаются к С и при
некотором значении Т равновесные участки АВ и DE могут исчез*
нуть. Это соответствует Г?л— критической температуре, выше
О L
Рис. 49. Зависимость f/T
от длины L полимерных
сеток при переходе
кристалл— жидкость в
соответствии с уравнениями A28)
и A29).
178
которой кристаллизация невозможна. Восстановление аморфной
фазы на участке DE вряд ли имеет физический смысл, так как
сомнительно, что полимерная цепь способна выдержать
огромные деформации, необходимые для перехода от La к LK *.
Поэтому для реальных систем основное внимание необходимо
уделять участку OABD. Использование более общего уравнения
состояния A16) не может изменить основные характеристики
процесса, отраженные на рис. 49.
Зависимость напряжения от удлинения для аморфной сетки,
выражаемую прямой, проходящей через начало координат,
можно заменить кривой, начинающейся при L = LH, что
соответствует f/T=0 и имеющей асимптому, также проходящую через
начало координат. Соотношения силы, температуры и длины,
выраженные выше в аналитической и графической формах,
имеют совершенно общее значение. Они не зависят ни от каких
деталей и могут быть дополнительно уточнены в рамках стати-
стико — механической теории каучукоподобной эластичности.
Эксперименты Ота и Флори подтверждают основные выводы
теории, в общих чертах намеченной выше. Изучение ими
зависимости вида сила — длина— температура для фибриллярного
натурального каучука, завулканизованного в ориентированном
состоянии, убедительно подтверждают, что эти зависимости
можно трактовать как классическую проблему фазового равновесия.
Первые опыты включали определение силы /° при
температуре выше равновесной температуры плавления в отсутствие
нагрузки Гпл- В этих сложных экспериментах плавление
инициировалось выше температуры сокращения. Однако
завершение перехода предотвращалось увеличением силы или
понижением температуры или одновременным воздействием обоих этих
факторов. Таким образом, в опытах к равновесию подходили
различными путями, что гарантировало надежное определение /°.
В соответствии с теорией было установлено, что коль скоро
сосуществуют две фазы, /° не зависит от длины образца и
возрастает с увеличением температуры. Зависимость равновесной
силы от температуры плавления для полиизопреновой сетки
изображена на рис. 50. Даже в относительно небольшом темпе*
ратурном интервале, где устанавливалось равновесие,
требовалось приложение значительных сил.;
Необходимые для поддержания равновесия напряжения были
порядка 3—4 кгс/см2. Если бы равновесие устанавливалось при
более высокой температуре, напряжения были бы еще выше.
Экстраполяция кривой на рис. 50 к значению /°=0 дает
величины равновесной температуры плавления ГпЛ. В этой
конкретной системе Тал на 6° ниже температуры сокращения, наблю-
* [Автор не учитывает возможного полиморфизма. См, сноску на
стр. 171. — Прим. редактора].
12*
179
даемой в отсутствие внешней силы; это различие демонстрирует
неравновесный характер температуры сокращения, о чем
говорилось выше.
По аналогии с мономерными веществами, где анализ
зависимости температуры перехода от давления позволяет оценить
скрытую теплоту плавления или испарения, зависимость
температуры плавления от растягивающей силы в нашем случае
дает возможность определить скрытую теплоту плавления при
переходе. Для этого нужно провести
графическое интегрирование
уравнений A14) и A15), используя
экспериментальную зависимость AL от
температуры и предполагая, что Д5 и
АН постоянны внутри небольшого
температурного интервала, в котором
ведутся измерения.
Скрытые изменения AS и ДЯ,
рассчитанные этим методом, могут быть
отнесены исключительно на счет
плавления кристаллической фракции
полимера. Следовательно, AS и ДЯ
для плавления гипотетического
полностью кристаллического волокна
можно получить простым'делением
найденных значений на степени
кристалличности.
Сравнение температуры, энтальпии
и энтропии плавления,
определенных этим методом и по понижению температуры плавления
натурального каучука в присутствии низкомолекулярных
жидкостей, дано в табл. 9. Результаты, полученные двумя методами,
совпадают друг с другом, и' это убедительно показывает, что
плавление растянутых полимеров нужно трактовать с позиций
фазового равновесия.
Зависимость равновесной силы от длины образца дается
на рис. 51 при различных температурах выше равновесной
температуры плавления ГпЛ. Для конкретного образца,
представленного на рис. 51, 7'пл = 302оК. Горизонтальные сплошные
линии представляют напряжения, необходимые для поддержания
двух фаз в равновесии. Точками указана длина образца после
завершения плавления при заданных силе и температуре.
Пунктирные линии изображают рассчитанную на основе теории кау-
чукоподобной эластичности зависимость длины от приложенной
силы для аморфного состояния при любой температуре.
Зависимость силы от длины для кристаллического состояния при
303,2° К представлена восходящей вертикальной линией. Такой
30
м20
? ю
'пл
35
bS
т/с
40 45
300 305 310 315
г/к
Рис. 50. Зависимость силы,
необходимой для
поддержания фазового равновесия,
от температуры для завул-
канизованного в
ориентированном состоянии
(фибриллярного) натурального
каучука [10]:
г = 1 • 56 • 10~2 и Г" =*302°K.
180
Таблица 9
Термодинамические величины, характеризующие
плавление натурального каучука
1 Термодинамические
величины
С °к
Д//м, кал/моль . . .
ASM, кал/(град • моль)
Метод определения
из уравнения
A15) [10|
302
1280±150
4,2 ±0,4
по понижению
температуры
плавления |11]
301
1040±60
3,5 ±0,2
же ход этой зависимости можно ожидать и для других
температур, коль скоро сохраняется кристаллическое состояние.
Изображенный на рис. 51 набор изотерм, характеризующих
ориентированное кристаллическое и жидкое состояния,
соответствует обычным изотермам, р — 1/, описывающим фазовый
переход пар — жидкость для мономерных веществ. Необходимо
подчеркнуть, что в двухфазной системе сила не зависит от длины.
Возрастание температуры плавления с увеличением
приложенной растягивающей силы демонстрируется положениями
точек на различных горизонталях рис. 51. Зависимости между
силой, длиной и температурой при такой вариации внешних
условий, когда волокно проходит через область сосуществования
двух фаз, также могут быть проанализированы.
Рассмотрим, например, сетку при условиях, определяемых
точкой А на рис. 51, что соответствует фазовому равновесию в
образце длиной 8 см при 303,2° К и напряжении немного меньше
0,4 кгс/см2. Предположим, что мы хотим исследовать процесс,
в котором длина образца должна поддерживаться постоянной
при возрастающей температуре. Такой процесс будет
описываться прямой вертикальной линией, восходящей из точки А.
Разумеется, для поддержания постоянной длины необходимо
приложить растягивающую силу, которая бы компенсировала
возникающую в (плавящейся) кристаллической сетке
сокращающую силу. Эта добавочная нагрузка необходима для
предотвращения плавления при подъеме температуры; в противном
случае первоначальная длина не может быть сохранена. При
повышении температуры в ходе этого процесса до 318,2° К
развивается напряжение до 4 кгс/см2. Это напряжение того же
порядка, что и при тетаническом сокращении фибриллярной
системы мышц.
Простым повышением температуры в фибриллярном
натуральном каучуке можно развить еще большее напряжение. Оно
181
будет непрерывно увеличиваться пока сохраняется двухфазная
система, т. е. до достижения критической температуры Г„л.
Развитие таких больших напряжений при повышении температуры
обусловлено разделением горизонтальных участков изотерм
(рис. 51). В свою очередь, это разделение отражает уравнение
состояния, характеризующее аморфную фазу.
Основной вклад в сокращающую силу для аморфной фазы
обусловлен, как известно, изменениями энтропии, которые и
40
30
%20
У Щ
3/в,2°К
В;
315,7°
313,2°
310,7°
h
308,2°
t
305,7°
303,2°
2 4 6 8
L,Cm
Рис. 51. Зависимости напряжения
от длины при различных
температурах для фибриллярного
натурального каучука [10].
определяют разделение изотерм
в двухфазной области. Мы
можем, следовательно, сделать
вывод, что развивающиеся
большие напряжения является
следствием конформационной
изменчивости полимерных цепей.
Необходимо указать, однако, что
напряжение, увеличивающееся с
повышением температуры, но при
постоянной длине в двухфазной
системе, во много раз выше,
нежели в полностью аморфной
сетке с фиксированной длиной.
Этот анализ демонстрирует
фундаментальный механизм
развития больших напряжений - в
одноосноориентированных
кристаллических макромолекулярных системах. Такой механизм
является неотъемлемым внутренним свойством полимеров,
обладающих ориентированной структурой, и должен
обнаруживаться независимо от кристаллографической и химической
природы полимерных цепей и способа, которым возбуждается
плавление *.
Можно теперь рассмотреть другой процесс, взяв за
отправное состояние полностью аморфную сетку, которой соответствует
точка В на рис. 51. Если вновь задается условие постоянства
длины, то при понижении температуры, процесс должен
описываться вертикальной линией, опущенной из точки В. Так как
при этом пересекается область сосуществования двух фаз, в си^
стеме развивается ориентационная кристаллизация, и
равновесная сила соответственно убывает. К 303,2° К напряжение дол-*
жно уменьшиться примерно в десять раз. Это дает
теоретическое обоснование экспериментальным результатам Смита и Сей-
лора [12], Тобольского и Брауна [13], Джента [14], которые
наблюдали релаксацию напряжений в ходе ориентационной кри-
* См. сноску на стр. 171,
182
сталлизации сеток натурального каучука при фиксированной
длине. Молекулярная причина этого понижения уже была
обсуждена *.
Возможны также процессы, где не длина, а напряжение
поддерживается постоянным. Рассмотрим систему (в
кристаллическом состоянии), снова определяемую точкой А (рис. 51). Если
теперь напряжение поддерживается постоянным, а температура
повышается, процесс должен описываться горизонтальным
участком, который ограничен соответствующей пунктирной кривой,
представляющей границу полностью аморфного состояния.
В данном случае при переходе будет наблюдаться
четырехкратное изменение длины. Этот процесс обратим, так как
поддерживается равновесное напряжение. При возвращении к начальной
температуре переход от аморфного к кристаллическому
состоянию будет сопровождаться спонтанным удлинением.
Спонтанное увеличение длины при кристаллизации деформированных
сеток натурального каучука, выдерживаемых при постоянной
нагрузке, действительно наблюдалось [12].
Анизотропные изменения размеров наблюдаются также при
полиморфизме во время перехода из одной ориентированной
кристаллической формы в другую, например, в полибутадиене
[15] и а-кератине [16]. В этих случаях изменения размеров
отражают различную осевую периодичность двух форм. Поэтому
можно ожидать, что изменения размеров будут значительно
меньше наблюдаемых при плавлении **. Более того, так как
уравнения упругого состояния обеих форм должны быть
аналогичны, нельзя ожидать большого разделения изотерм в области
сосуществования двух фаз (в данном случае, двух
кристаллических фаз), а следовательно, и развития больших сокращающих
сил.
Если цикл плавление — кристаллизация ориентированных
сеток осуществляется в равновесныхусловиях,то при поддержании
постоянной силы система претерпевает обратимое сокращение.
* Следующее из теории и подтвержденное экспериментально уменьшение
напряжения при развитии кристалличности в одноосноориентированной
системе противоречит, на первый взгляд, обычно наблюдаемым зависимостям
напряжения от растяжения для сеток натурального каучука. Разрешение
этого кажущегося парадокса заключено в различной природе этих двух
процессов. Кристаллиты, образованные при изотермическом растяжении в
эксперименте последнего типа могут действовать как дополнительные
сшивки. При дальнейшем растяжении сегменты в аморфных областях
ориентируются гораздо сильнее, чем обычно. Это вызывает пропорционально
большее уменьшение энтропии, что, в свою очередь, сказывается в увеличении
сокращающей силы. Так как при дальнейшем удлинении кристаллизация
продолжается, этот эффект будет возрастать и соответственно ускоряться
рост напряжения.
** [Обрисованная таким образом картина несколько упрощена. Мы снова
отсылаем читателя к работам, процитированным в сноске на стр. 171.—
Прим. редактора].
183
Аналогичным образом, большие и тоже обратимые изменения
напряжения наблюдаются при поддержании постоянной длины.
Подобные процессы являются характерным внутренним
свойством макромолекулярных систем.
Предшествующий анализ и описание ограничивались чистым
однокомпонентным и однородным по сечению гомополимером.
Этот анализ, однако, нетрудно распространить на
неоднородные волокна, сополимеры и набухшие полимеры [4].
Изменения химической структуры по длине волокна и
поперечного сечения сказываются в расширении области перехода
от кристаллического к аморфному состоянию. Первичный
эффект изменения химической структуры, например в случае
сополимеров, проявляется в изменении температуры плавления при
заданной силе. Поскольку изменения в поперечном сечении
влияют на напряжение, которое определяет равновесие, то в
разных сечениях установятся различные критические
напряжения. Следовательно, для гомогенной системы возможен переход
при постоянных давлении и температуре, но в конечном
диапазоне растягивающей силы. В гомогенном по сечению волокне
этот диапазон переходит в точку, соответствующую равновесной
силе.
Более детальный анализ показывает, что зависимости,
подобные формулам A22) и A29) сохраняются и для
негомогенных волокон при условии, что они используются для
интерпретации поведения отдельных элементов волокна при фазовом
равновесии. Соответственно, линии АВ и BD на рис. 49
заменяются S-образными кривыми.
Благодаря наличию двух фазовых состояний полимерной
сетки и возможности их сосуществования в макроскопическом
образце, у ориентированных полимеров можно наблюдать
уникальные термоэластические коэффициенты. Особенно интересны
коэффициенты, характеризующие зависимость силы от
температуры и силы от длины, связанные между собой тождеством:
Так как частная производная (df/dL)p.T всегда
положительна, знак (df/dT)Pt l противоположен знаку (dL/dT)pf и оба
коэффициента обращаются в нуль одновременно при
определенной длине или силе. Зависимость длины от температуры при
постоянной силе схематически представлена на рис. 52, а для
идеализированного гомогенного волокна. При больших L характерен
незначительный положительный коэффициент теплового
удлинения, типичный для твердой кристаллической фазы. В точке
плавления свойства системы меняются скачкообразно, и далее
для расплавленного волокна характерен уже некоторый
небольшой отрицательный коэффициент теплового изменения, ожидав
184
мый для каучукоподобного вещества. Температура плавления
возрастает с увеличением силы (пунктирная линия).
Для волокон, неоднородных по химической структуре или в
поперечном сечении, область плавления расширяется.
Скачкообразное уменьшение длины теперь сглаживается в
непрерывную кривую (рис. 52,6). Термический коэффициент удлинения
Рис. 52. Схематическое изображение зависимостей
длины от температуры для идеализированного
гомогенного волокна /' > / (а) и для не
гомогенного волокна (б) [3|
все еще слабо отрицателен для сократившихся волокон и
положителен при больших растяжениях в высококристаллическом
состоянии. При промежуточных степенях кристалличности
коэффициент сильно отрицателен. Он достигает максимальной
отрицательной величины при возрастании кристалличности и затем
постепенно приобретает нормальное положительное значение.
Термические коэффициенты силы описываются аналогичным
образом при использовании равенства A30).
В реальных системах всегда следует ожидать очень сильной
зависимости термоэластических коэффициентов от растяжения,
которая является непосредственным следствием происходящих
фазовых переходов и диффузного характера плавления
неоднородных волокон. Термоэластическое поведение описанного типа
действительно наблюдалось для многих фибриллярных белков
[17-19].
Многокомпонентные системы,
подверженные действию растягивающей силы
Плавление и кристаллизация ориентированных систем
наблюдается также и для случая, когда такие системы находятся
в контакте с чистым растворителем или раствором, содержащим
низкомолекулярные компоненты. Введение новых компонентов
приводит к некоторому усложнению теоретического и
экспериментального анализов, не изменяя характера основных физических
185
процессов. Хотя с этими осложнениями и надо считаться, они
не должны затруднять понимание молекулярного механизма
фазовых переходов под нагрузкой. Это тем более существенно
в случае фибриллярных белков, необходимые опыты с которыми
могут быть проведены только в присутствии растворителя или
подходящих химических агентов.
Равенство, соответствующее A06) для многокомпонентной
системы, запишется следующим образом:
d@-fL) = --SdT + Vdp--Ldf + ]?1vLidnl A31)
Для равновесия между кристаллической и аморфной
фазами необходимо, чтобы функция Ф — fL была минимальной,
когда drii молей компонента i переходит из одной фазы в
другую при постоянных ру Т, f. Следовательно
6 (Ф — fL) = 6/i^J — бя^ =0 A32)
или
И?=^У A33)
для каждого компонента, присутствующего в обеих полимерных
фазах.
Если допустить, что кристаллическая фаза является чистой,
уравнение A33) приобретает вид:
Й-Й 034)
Аналогично, когда присутствует жидкая фаза (помеченная
индексом s), состоящая из мономерных комплексов, то при
равновесии аморфной части волокна с этой фазой должно
удовлетворяться равенство химических потенциалов:
И? = и? A35)
для каждого мономерного компонента в аморфной или
смешенной фазах.
В двух полимерных фазах химический потенциал звена
является функцией Г, /?, f и состава. Так как кристаллическая
фаза предполагается чистой получаем:
d< = -SKudt-KdP-LKudf A36)
тогда как для аморфной фазы, содержащей г компонентов,
должно удовлетворяться соотношение:
d»* — г;л + vidp-iidf+%(|?)r ^ йх% A37)
где 5ш VI и Ы — соответствующие молярные характеристики
на одно звено в этой фазе;
Xi — молярная доля /-го компонента.
186
Для двух фаз, находящихся в равновесии при постоянном р
Фм = «К A38)
A39)
(Ч ~ Ф « — (*« - si) ат + J (^ p *,
При постоянном составе аморфной фазы
Ы "Т^ 040)
где инденкс п указывает на постоянную концентрацию всех
компонентов. Постоянство состава, определяемое уравнением A40),
включает не только фиксированную концентрацию полимера, но
и постоянство концентрации низкомолекулярных компонентов,
присутствующих в этой фазе. Изменение энтропии на звено в
результате плавления при постоянных /?, Т и п задается
соотношением:
*а _ sk = ГО-*?)-№-*?) (Ш)
так что
d(f/T)
д(МТ)
Н1-Н
к
р, п м
м м (И2)
Удобно умножить числитель и знаменатель правой части
этого соотношения на пм — общее число мономерных звеньев
в волокне. Тогда уравнение A42) примет вид:
\УШУ\ =*? A43)
где Л77 = Яа — Як; AL = La — LK;
Нк и LK — энтальпия и длина полностью кристаллического
волокна при /?, Т и /;
Яа и L* — частные производные полной энтропии и длины
аморфной фазы по относительной концентрации
полимера в этой фазе X (напоминаем, что степень
кристалличности равна 1 —Я).
Таким образом, АН складывается из теплоты плавления и
дифференциальной теплоты разбавления; аналогичным образом
определяется и AL.
Необходимо различать два основных случая. В первом —
фиксировано полное содержание неполимерных компонентов; во
втором — аморфная часть волокна находится в равновесии с
жидкой фазой, содержащей значительный избыток мономерных
187
компонентов. В первом случае волокно и содержащиеся в нем
неполимерные компоненты представляют собой замкнутую
систему. Если кристаллизация и плавление происходят в одно-
компонентном растворителе, система бивариантна при
постоянном давлении. По мере плавления длина волокна уменьшается.
Состав аморфной фазы изменяется, так как концентрация
полимера в ней возрастает при фиксированном содержании
растворителя.
Условие постоянства состава для дифференциального
коэффициента уравнения A43) совпадает с условием постоянства Я,
т. е.
[д(\/Т)\р,п [д(\/Т)\р,х М
__В отличие от чистой однокомпонентной системы AS, AL и
АН зависят от состава и, соответствено, производная силы по
температуре зависит от X. Таким образом, сила в данном
случае не определяется одной только температурой, и полное
плавление при постоянной силе не происходит.
В присутствии окружающей жидкой фазы волокно с
наполняющими его низкомолекулярными компонентами представляет
собой открытую систему, так как в этом случае возможен обмен
веществом между окружающей жидкой и аморфной фазами.
Если окружающая жидкая фаза состоит из одного компонента,
система унивариантна при постоянном давлении, т. е.
равновесная сила однозначно и полностью определяется температурой.
Полное плавления происходит при постоянной силе, не
зависящей от длины образца, как и в случае чистой однокомпонентной
системы. Благодаря наличию избытка растворителя в
окружающей жидкой фазе равновесное набухание в аморфной фазе
может устанавливаться при заданных / и Т для любых значений L.
Поэтому при плавлении состав смешанной фазы остается
постоянным, откуда:
УдГ/п, п = \дт)Р, х ^ \дТ1р. l = ~~ лГ °45)
(ШЩ -Щг A46)
Величины, помеченные двумя горизонтальными штрихами,
представляют собой сумму скрытых изменений S, L и Н (хотя
в случае L этот термин не очень удачен), происходящих при
плавлении полимерного компонента и интегральных изменений
этих же характеристик при смешении таких количеств каждого
компонента, которые необходимы для достижения равновесного
состава аморфной фазы.
188
Если окружающая фаза многокомпонентна, система
перестает быть унивариантной. Хотя условия, предполагаемые
равенством A35), все еще должны выполняться, оно уже не
гарантирует постоянство состава аморфной фазы при изменении X.
При постоянном давлении равновесная сила уже не должна
зависеть только от температуры. Следовательно, по аналогии
с замкнутой системой, полное плавление и здесь не должно
происходить при постоянной силе.
Поскольку равновесие кристалл — жидкость может
регулироваться также химическими процессами *, то действие
последних может оказывать влияние на соотношения силы, длины и
температуры в одноосноориентированных системах. Формальный
анализ этой проблемы аналогичен проведенному для
нереакционно способной системы с
Ш-уЬI*г\р./.ш A47)
(±) -.
\дТ)р,п
\*(К-<)№\Р,т,п
Здесь необходимо сосрдоточить внимание на изменении
химического потенциала мономерного звена, вызванное
специфической химической реакцией, и на свойствах фазы или фаз, в
которых протекает эта реакция. Определив таким образом
условия равновесия, можно оценить дифференциальный
коэффициент (dfldT)p, п.
Для иллюстрации и облегчения анализа предположим, что
1) химическая реакция затрагивает только аморфную фазу, а
кристаллическая остается неизменной; 2) состав аморфной фазы
не зависит от А,, даже если окружающая жидкая фаза много-
компонента. Это равнозначно условию однокомпонентности
окружающей фазы. При таких условиях:
(±) ¦
—Sm-Sm -as
; Га Т.* -^ (И8>
Ы/т))ля д| A49)
Помеченные тремя штрихами величины определяются тремя
факторами: плавлением чистого полимера, интегральным
смешением компонентов до состава, определяемого п, и
изменением химического потенциала мономерного звена при
химической реакции. Таким образом, например, в предположении
независимости состава аморфной фазы от L, Д# можно записать:
Л77=Д// + Д//СМ + ДЯР A50)
* См, сноску на стр. 171,
189
где А//— теплота плавления;
А//См—интегральная теплота смешения;
Л//р—энтальпия плавления на звено в результате
химической реакции при общем составе системы,
определяемом величиной п.
При образовании простых компонентов, типа обсужденных в
гл. 3, изменение химического потенциала можно выразить как в
работе [20]:
\iM-V0M=-RT\n(l + Ka) A51)
по аналогии с уравнением адсорбции. Следовательно,
ь»9=«тгт^{§-)р,ГттТШАН° A52)
где Д//°— изменение энтальпии для стандартного состояния
при комплексообразовании.
Отлична ли соответствующая характеристика AL от нуля,
зависит от того, является ли константа равновесия реакции
функцией от приложенного напряжения. Аналогичным образом
анализируется эффект и других возможных химических реакций
при условии, что поддается оценке изменение химического
потенциала мономерного звена.
Интегрирование уравнений A45) и A46) приводит к
получению соотношений, аналогичных полученным для однокомпонент-
ной системы. Параметр А/г' включает теперь дополнительные
вклады теплот разбавления и реакции. Интегрирование должно,
разумеется, проводиться при постоянстве состава аморфной
фазы, так что используемое уравнение состояния должно
учитывать концентрацию полимера в этой фазе. Константы
интегрирования ZH и ГЦл или LK и Гпл относятся к этому
фиксированному составу. Таким образом, изменение состава влияет не
только на энтальпию и длину, но в равной мере и на L" и ГпЛ.
Флори и Опуром было определено напряжение, необходимое
для поддержания в равновесии кристаллической и аморфной
фазы частично сшитого коллагена [8]. Измерения проводились в
широкой области температур, причем волокно в этих опытах
помещалось либо в чистую воду, либо в водный раствор
роданистого калия. Величина равновесной силы при заданной
температуре почти не зависит от общей длины образца и,
следовательно, от степени завершенности перехода. Если окружающая
фаза состоит только из чистой воды, то система унивариантна и
можно ожидать результатов, предсказываемых изложенной
выше теорией. Однако данные, полученные при использовании
двухкомпонентного растворителя показывают, что приближение,
принятое для однокомпонентной системы, справедливо и в этом
случае.
190
Некоторые характерные результаты, иллюстрирующие
зависимость равновесного напряжения от температуры для этого
волокна, представлены на рис. 53. Ясно видно значительное
возрастание этого напряжения с увеличением температуры. В таких
процессах могут развиваться чрезвычайно высокие напряжения,
как уже указывалось при рассмотрении плавления
фибриллярного натурального каучука.
Ход кривых на рис. 53 плохо соответствует характеру
изменения напряжения с температурой, наблюдаемого при
деформации полностью аморфного
коллагенового волокна [8].
В этом случае при
поддержании постоянной длины
наблюдается лишь относительно ела- ^^401
40
20
3,10
• i »
ST..X .4
юг/тплХ
3,00 2,90 260
1 » 1 УР \ I
У г
у/..\
50
60 70
80
90
Рис. 53. Зависимость отношения
равновесного напряжения т* к Тпл
от температуры для коллагеновых
волокон в чистой воде (/) и Ш
растворе роданистого калия B) [8].
бое увеличение напряжения с
ростом температуры. В связи
с этим уместно отметить, что в
коллагеновом волокне,
помещенном в 2М раствор смеси
йодистой ртути и йодистого
калия (среда, способствующая
плавлению фибриллярных
белков) при поддержании
постоянной длины развивается
напряжение порядка 100 кгс/см2 [22].
Помимо роста напряжения
при повышении температуры на
рис. 53 также показаны изменения для постоянной температуры
в случае различного состава окружающей фазы. В чистой воде
при 70° С для поддержания равновесия необходимо приложить
напряжение 4,4 кгс/см2, тогда как в Ш растворе роданистого
калия напряжение при этой же температуре возрастает до
11,5 кгс/см2. Таким образом, изменение состава окружающей
жидкой фазы приводит к существенным изменениям
равновесного напряжения. Так как наклоны двух кривых на рис. 53
приблизительно одинаковы при всех температурах, основной
причиной увеличения равновесного напряжения должно быть
снижение ТпЛ с 60° С в чистой воде до 43° С в Ш растворе KSCN.
Это снижение равновесной температуры плавления
непосредственно обусловлено химическими процессами, происходящими
при взаимодействии коллагена с роданидом.
В соответствии с формулами A48) и A52), значение ДЯ/д!
можно определить по наклону кривых на рис. 53. Полагая LK
постоянной величиной, можно определить значение АН и
соответственно теплоту плавления полимера АЯМ. При этом
необходимо определить теплоту растворения АНР и оценить вклад
191
Таблица 10
Термодинамические характеристики для плавления
коллагена
Окружающая жидкая
1 фаза
1
Ш раствор KSCN .
ЗМ раствор KSCN .
60
43
14
О
., s5
*5 §
< *
1,2
0,87
0,43
ь*5
5 §
< *
—0,15
-0,10
—0,03
«4
О
*1
s^
1С ?
< *
1,35
0,97
0,46
<ъ
&
«V» 1
Л
^ 5
< §
4,1
3,1
1,6
в ЛЯ от химической реакции Д//р. Результаты расчета Д//м в
пренебрежении Д#р приведены в табл. 10 [8].
Наблюдаемое уменьшение энтальпии плавления при
увеличении концентрации KSCN может быть и не столь сильным, так
как не учитывалась величина Д#р. Изменения энтальпии,
приведенные в табл. 10, скорее относятся к изменениям на моль
пептидных звеньев в нативном волокне, нежели на моль
пептидных звеньев в кристаллическом состоянии.
Используя метод понижения температуры плавления в
присутствии низкомолекулярного растворителя, Флори и Гаррет [7]
нашли, что для системы коллаген — этиленгликоль АНМ —
= 2,25 ккал на моль звеньев в кристаллическом состоянии.
Меньшее значение Д#м, полученное в водной среде для этого
количества и по такой же методике, можно приписать значительному
содержанию аморфной фазы в нативном коллагеновом волокне.
Естественно, однако, предложить и другую альтернативу [8],
согласно которой часть воды, внедряясь в кристаллическую
решетку, образует с полимером гидратное соединение, и поэтому
характер плавления даже идентичных образцов в воде и этилен-
гликоле должен различаться.
Как бы то ни было, результаты, полученные на системах
коллаген — вода и коллаген — вода — роданистый калий,
подтверждают, что плавление под нагрузкой и в этом случае можно
интерпретировать с позиций фазового равновесия. Отсюда понятна
также возможность развития очень больших напряжений в
результате химических процессов.
Анализ рассмотренных систем значительно упрощается
благодаря их унивариаитному поведению. Наблюдаемая
независимость равновесной силы от степени завершенности перехода
означает постоянство состава в аморфной фазе. Для открытой
системы с многокомпонентной окружающей жидкой фазой это-
192
го, как правило, уже нельзя ожидать. В процессе перехода при
постоянной температуре состав аморфной фазы может
изменяться в результате, например, неравномерного распределения
низкомолекулярных компонентов между двумя фазами.
Равновесная сила должна соответствовать определенному составу
смешанной фазы, который, в свою очередь, должен зависеть от
общей длины образца. Для системы, число степеней свободы
которой больше единицы, двухфазную область при постоянном
давлении уже невозможно будет представить горизонтальной
прямой линией, как на рис. 51, такая область должна
изображаться кривой с положительным наклоном и кривизной.
Зависимость силы от длины (в области сосуществования
двух фаз) отражает эти изменения состава и, следовательно, на
нее должны оказывать влияние соответствующие изменения ГЦЛ
и отношения A///AL. Так как при постоянных давлении и
температуре /=/(L,n), то:
«-Ш,„./'+2Ш,,,л
где в сумму включены все компоненты.
При равновесии между фазами
(JL\ =(ЯЛ + У{-*Ц (*«L\ A54)
\ dL ]Tt р, рав„ \dL)TyPttl^ JU\ dtii )т% p,Ln\ dL )Tt p, paBH
Идеализированное поведение чистой однокомпонентной
системы реализуется теперь лишь в том случае, если сумма, по
той или иной причине, обращается в нуль.
Ориентационная кристаллизация и
сокращаемость при отсутствии растяжения
Установлено, что одноосноориентированные кристаллические
системы, образованные из простых полимеров, а также из
фибриллярных белков, при плавлении претерпевают сокращение.
Характерная зависимость длины от температуры при
отсутствии растягивающей силы для фибриллярного натурального
каучука и коллагена волокна, помещенного в воду, показана на
рис. 54 и 55. В обоих случаях наблюдается сильное одноосное
сокращение в узком температурном интервале. Сокращение
сопровождается исчезновением характерных черт
кристаллического состояния — дискретных рефлексов рентгеновского
рассеяния и оптического двулучепреломления. Можно, таким
образом, считать, что происходит процесс плавления. Однако при
13 Л. Манделькерв
193
охлаждении в отсутствии внешнего напряжения начальное или
нативное состояние, характеризуемое одноосноориентированной
кристаллической структурой, в данном случае полностью не
восстанавливается. Удается достичь лишь кристаллического
состояния с беспорядочной ориентацией кристаллитов.
Основная проблема, следовательно, сводится к установлению
условий, при которых переход между ориентированным
кристаллическим и жидким состояниями может быть обратимым. Как
показывает детальный анализ, данный в предыдущем разделе,
обратимость проще всего обеспечить приложением силы,
достаточной для поддержания жидкой и кристаллической фазы в
L,CM
Г/К
310 320
Рис. 54. Зависимость длины от
температуры для фибриллярного
натурального каучука при
отсутствии растягивающей внешней
силы [10].
Рис. 55. Зависимость
Lr/Z.40 от температуры
для коллагена из
сухожилия хвоста крысы при
отсутствии
растягивающей внешней силы [20].
равновесии. Однако обратимость может быть достигнута и при
отсутствии растягивающей силы, если использовать выигрыш в
изотропной длине LH, получающейся при вулканизации одноос-
ноориентированного полимера. Изотропную длину можно
выразить формулой A18).
Эта характеристика для сеток, образованных из сильно
ориентированных макромолекул существенно отличается от
аналогичной величины для сеток, образованных из беспорядочных
цепей. В последнем случае сетка неминуемо изотропна и,
следовательно, Iй может быть отождествлена с длиной образца и не
зависит от числа цепей v, образующих сетку.
В случае же цепей, которые были достаточно хорошо
ориентированы перед вулканизацией, получается сетка с совершенно
иными свойствами. Как уже указывалось ранее, длина LH
должна возрастать при этом пропорционально vl/a(a).
Предсказываемое увеличение изотропной длины
подтверждается исследованиями сеток из сшитого фибриллярного кол-
194
лагена [8, 23, 24], фибриллярного натурального каучука [25] и
сильно ориентированного линейного полиэтилена [26].
Результаты приведены на рис. 56 и 57. Поперечные связи в этих
случаях образовывались под действием мощного ионизирующего
излучения. Ординаты на соответствующих графиках
представляют относительное увеличение длины в аморфном состоянии
после вулканизации ориентированных цепей. Измерения
проводились при 25° С для натурального каучука и 140° С для
полиэтилена, чтобы по возможности удовлетворить требованиям изо-
L"/LQ
L*/La
у
-Ф
25\
2о\
15{
Щ
5 10 15 20
УТ-Ю2
Рис. 56. Зависимость L"/L0 при 25° С
от относительной концентрации с
сшитых звеньев для фибриллярного
натурального каучука [25].
к
5 Ю 15 20 25 30 35
W
Рис. 57. Зависимость LH/L0 при
140° С от дозы облучения R для
сильно ориентированных
полиэтиленовых волокон [20]:
А—У"излУчение Со60; Д —электроны
высокой энергии.
тропности. В случае полиэтилана, при дозе 1000 Мрад
сшивается около 4% звеньев [26].
Хотя ход реальных кривых и не согласуется в точности с
предсказываемым соотношением A18), в обоих случаях
отчетливо видно значительное увеличение изотропной длины.
Особенно сильно этот эффект проявляется для полиэтилена, где
происходит двадцатикратное увеличение длины без приложения
какой-либо растягивающей силы. Подобных удлинений не удается
достичь даже при механической деформации обычных аморфных
сеток. В этом случае свободному развитию деформации либо
мешает кристаллизация, либо происходит разрыв отдельных
цепей. Во всяком случае, механическая деформация не приводит
к удлинениям, сравнимым с показанными на рис. 57.
Рассмотренные полиэтиленовые сетки легко
кристаллизуются при понижении температуры. На рис. 58 приведены
некоторые типичные рентгенограммы для рекристаллизованных
волокон. Образование поперечных связей в сильно ориентированных
волоиках под действием ионизирующего облучения,
незначительно влияет на характер дифракции. Однако после сшивания,
13*
195
плавления и последующей рекристаллизации характер
получающихся рентгенограмм уже сильно зависит от относительного
числа введенных в систему сшивок. Это становится очевидным при
Рис. 58. Рентгенограммы (при комнатной температуре)
сшитых при различных дозах облучения и рекристал-
лизованных полиэтиленовых волокон [26]:
д-Я = 0, Lli/L0 = Vt6-R=l79Mpad, ?и/?0=13,7; в - Л = 353 Мрад,
1и/10 = 16,8; г-# = 660 Мрад, 1И//.0=18,3.
сравнении рентгенограмм от четырех образцов, облученных
различными дозами (рис. 58).
Для несшитого образца рентгенограммы после плавления и
рекристаллизации состоят из серии концентрических колец, что
свидетельствует о совершенно беспорядочной ориентации кри-
196
сталлитов. Другие же рентгенограммы явно указывают на
развитие преимущественной ориентации кристаллитов по мере
увеличения числа сшивок. Например, рентгенограмма,
изображенная на рис. 58, г, соответствующая волокну с относительным
содержанием сшитых звеньев порядка 2,65хЮ и LH/L0,
равному 18,3 показывает, что ось с кристаллитов снова
преимущественно ориентированна вдоль макроскопической оси волокна.
Таким образом, надмолекулярная организация одноосно-
ориентированных кристаллитов развивается в процессе
кристаллизации без всякого приложения внешней силы. Эта особенность
кристаллизации непосредственно связана с чрезвычайно
большими значениями LH/L0, достигаемыми в аморфном состоянии
при сшивке. Развивающиеся значительные коэффициенты
удлинения приводят к установлению преимущественной оси
перехода. Следовательно, ядра кристаллизации, образование
которых неминуемо предшествует фазовому переходу, также имеют
преимущественную ориентацию, что и приводит к
ориентированной кристаллизации. Имеются данные, свидетельствующие о
преимущественной одноосной
рекристаллизации сеток,
образованных из
фибриллярного натурального каучука
[10]. Преимущественная
ориентация кристаллитов в
описанных волокнистых
системах является их
неотъемлемой характерной чертой и
должна наблюдаться после
любого количества циклов
плавление —
рекристаллизация при условии
сохранения сшивок.
Таким образом,
заметные изменения размеров
должны сопровождать
плавление и кристаллизацию таких сеток. В частности, вследствие
одноосной ориентации должно происходить сокращение при
плавлении и спонтанное удлинение три кристаллизации из расплава.
Как видно из рис. 59, такие изменения размеров при фазовых
превращениях действительно происходят [26]. В начале
изображенного цикла волокно было в кристаллическом состоянии
(достигнутом после плавления сшитого волокна и
рекристаллизации). Этому состоянию соответствует рентгенограмма на
рис. 58, г. В начале, 'при нагревании до ~95° С, наблюдается
слабый положительный коэффициент удлинения, характерный
для кристаллического состояния. Плавление происходит в узком
температурном интервале и сопровождается сокращением
50
75
Ю0 125
тХ
ратуры при обратимом сокращении
полиэтиленового волокна:
ф — при нагревании; Q — при охлаждении.
Рис* 59. Зависимость L jL от темне-
197
волокна на 25%. Наблюдаемое сокращение. естественно для
одноосноориентированного кристаллического образца.
Выше температуры плавления, коэффициент теплового
удлинения снова слабо положителен, что типично для аморфного
состояния. При охлаждении - происходит кристаллизация, в
результате которой волокно восстанавливает свои первоначальные
размеры. Затем процесс нагревания можно повторить, и
плавление произойдет в том же самом температурном интервале.
Если вести охлаждение после плавления очень медленно, как
показано на рис. 59, можно перевести волокно в
переохлажденное состояние, что характерно для всех кристаллизующихся
полимерных систем. Этот эффект, проявляющийся в данном
случае в запаздывании удлинения, может быть сведен к минимуму
быстрым охлаждением до низких температур.
Таким образом, получена обратимая сокращающаяся
система, включающая фазовый переход кристалл — жидкость,
полностью циклическая и не требующая для генерирования
приложения внешней растягивающей силы. Температура сокращения
в этом случае может быть отождествлена с равновесной
температурой плавления.
Резкость наблюдаемого сокращения представляет собой
естественное следствие плавления гомополимера в полном
согласии с трактовкой этого процесса как фазового перехода
первого рода. Для статистического сополимера с точно таким же
расположением цепей и сшивок область плавления и
сокращения расширяется. Наложение на указанные системы
напряжения повышает температуру плавления, но сокращение остается
обратимым. Следовательно, волокна, подобные описанным,
могут служить в качестве рабочего вещества двигателя,
непосредственно преобразующего тепловую энергию в механическую
работу.
Как уже отмечалось выше, равновесие кристалл — жидкость
в общем виде может управляться также химическими
процессами. Таким образом, плавление и сопутствующее сокращение
могут происходить в изотермических условиях. Коль скоро был
установлен общий принцип, согласно которому в основе
процессов сокращения лежит плавление, необходимо делать
различие между собственно механизмом сокращения и тем
процессом или химической реакцией, которые вызывают или
регулируют фазовый переход.
Исходя из этой формулировки проблемы, можно рассмотреть
многие известные сокращающиеся системы с обшей точки
зрения. Необходимость такого общего подхода становится особенно
понятной, если сосредоточить внимание на поведении
различных полимеров биологического происхождения, среди которых
весьма распространена способность к сокращению под дей-
198
ствием самых различных химических агентов. Принципы
обратимости, установленные при анализе полиэтиленовых волокон,
могут служить удобной моделью при исследовании этих более
сложных систем, так как первопричина обратимых
сократительных механизмов никак не связана с детальной
кристаллографической структурой волокна.
Сокращаемость фибриллярных белков
В серии основополагающих работ Астбери с сотрудниками
[16, 27, 29] было установлено, что многие белковые системы
существуют в природе в кристаллическом состоянии. Кроме
кристалличности, эти системы также характеризуются высокой
степенью одноосной ориентации и называются, поэтому,
фибриллярными. К фибриллярным белкам относятся а- и E-кератин,
коллаген, эластоидин и мышечные волокна.
Таким образом, фибриллярные белки обладают той
специфической структурой, при которой плавление должно
сопровождаться сокращением. В некоторых из указанных систем, в
частности в кератинах, существуют межмолекулярные ковалент-
ные связи. Предполагается, что они возникают в процессе
биосинтеза уже после образования волокна и, следовательно,
накладываются на предварительно ориентированную структуру.
Поэтому здесь следует ожидать обратимой сокращаемости при
фазовом переходе кристалл — жидкость. Для фибриллярных
белков, не имеющих межмолекулярных связей или не
сохраняющих их при плавлении, возможно лишь необратимое изменение
размеров.
Тот факт, что во множестве самых различных фибриллярных
белков одноосное сокращение может вызываться большим
разнообразием химических агентов и при различных условиях,
никак не противоречит принципу, что в основе этих многообразных
конкретных процессов лежит общий механизм. Анализ
подобных систем, следовательно, должен свестись к установлению
того, действительно ли фазовый переход сопровождает изменение
размеров и обусловлена ли обратимость присутствием
благоприятно расположенных поперечных связей.
На ряде опытов продемонстрировано, что гидротермическое
сокращение коллагена (примерно в пять раз по сравнению с на-
тивным состоянием) происходит непосредственно в результате
плавления [7, 8, 30]. Как уже указывалось, однако, ни
ориентированное кристаллическое состояние, ни начальные размеры не
восстанавливаются при одном лишь охлаждении образца ниже
температуры плавления. Природа обнаруженных в коллагене
аминокислотных остатков не свидетельствует о наличии каких-
199
либо межцепных поперечных связей, способных сохраниться
в процессе плавления. Следовательно, в соответствии с
выводами, сделанными при изучении полиэтиленовых волокон, здесь
нельзя ожидать восстановления ориентированного
кристаллического состояния при отсутствии внешней растягивающей
силы.
Однако, как было показано Эвалдом [31], если коллаген в
нативном состоянии сшит (задублен) формальдегидом, то
гидротермический процесс плавление —
кристаллизация сопровождается
обратимым анизотропным
изменением размеров. Одноосноориенти-
рованная кристаллическая
структура, развивающаяся из
расплавленного состояния, подтверждается
картиной рассеяния рентгеновских
лучей под большими и малыми уг»
лами [32, 33]. Рекристаллизованное
волокно снова сокращается при
последующем нагревании, так что
процесс может проводиться
циклически [8, 34]. Пример такого рода
наблюдений приведен на рис. 60.
Как видно из рис. 60
первоначальное плавление нативного
сшитого волокна необычайно резкое;
при охлаждении происходит
спонтанное удлинение примерно до
половины исходной длины. При после-
уменьшается при нагревании более
плавно. Завершение процесса плавления фиксируется легко,
и разница между двумя температурами плавления составляет
всего несколько градусов. Диффузный характер перехода и
несколько пониженная температура плавления, возможно,
обусловлены гидролизом при относительно высоких температурах и
кинетическими факторами, которые могут затруднять
развитие кристалличности, типичной для нативного состояния. Тем
не менее, можно утверждать, что фазовый переход при
отсутствии растягивающей силы сопровождают обратимые
анизотропные изменения размеров.
Аналогичный пример сокращаемости демонстрирует
фибриллярный белок — эластоидин. В нативном состоянии его
кристаллическая структура идентична коллагеновой. Кроме того эти
белки имеют во многом сходный аминокислотный состав.
Однако эластоидин содержит 1—2% цистина, боковые группы
которого могут образовывать устойчивые межмолекулярные кова-
лентные (дисульфидны'е) связи. Не удивительно, поэтому, что
L,CM
W
!•»««••••*
ТС=69°С
I
20 W
60 80
т,°0
Рис. 60. Обратимое
сокращение сшитого (задубленного)
коллагенового волокна [34]:
7 — первоначальное плавление и
сокращение; 2—плавление после
рекристаллизации.
дующем плавлении длина
200
эластоидин обнаруживает обратимую сокращаемость и
релаксацию при плавлении и кристаллизации [35, 36].
При нагревании нативного волокна эластоидина,
помещенного в воду, в области 65°С происходит значительное
одноосное сокращение; при последующем охлаждении (при отсутствии
растягивающей силы) волокно удлиняется примерно до
половины начальной длины. После первоначального сокращения
процесс можно повторять циклически с чередованием сокращений
несколько выше 65° С и удлинений при охлаждении.
Рентгенограмма сократившегося волокна содержит только аморфное
гало, тогда как для нативного или отрелаксированного волокна
получаются характерные для ориентированной структуры
коллагена рентгенограммы.
При проведении описанных опытов волокна помещали в
жидкую среду, которая способствует понижению температуры
плавления, так что процесс
протекает без заметной де- а
струкции. Плавление может 50г
также вызываться взаимо- ^
действием некоторых групп
полимерной цепи с различ- к
ными ионами,
присутствующими в окружающей фазе. ?
Укажем, к примеру, на
зависимость температуры
плавления и сокращения
эластоидина от рН среды [37].
Результаты,
приведенные на рис. 61, а
демонстрируют зависимость
температуры плавления для
обратимых процессов от рН среды.
Аналогичные зависимости
получены и для
необратимого сокращения нативного коллагена [38]. В широкой области
рН, симметричной относительной нейтральной точки,
температура плавления постоянна, а ниже и выше этой области резко
понижается. Плавление сопровождается сокращением, а при
охлаждении обратимо восстанавливаются как размеры, так и
кристаллическое состояние.
Безотносительно к деталям действующего химического
механизма (рис. 61,6) демонстрирует важность состава смешанной
фазы (фаза набухшего аморфного полимера) на установление
температуры плавления. На рис. 61,6 показана зависимость
равновесного коэффициента набухания (при температуре
плавления) от рН окружающей среды. Примечательное соответствие
существует между степенью набухания и температурой плавления.
ч ОМ
02
2 4 6 6 10 12
рН
Рис. 61. Зависимости температуры
плавления (а) и равновесной степени
набухания (б) эластоидиновых волокон от рН
окружающей среды [37].
201
Если концентрация полимера в расплавленной фазе постоянна
при изменяющемся рН, то температура плавления также
постоянна. С уменьшением концентрации полимера температура
плавления понижается. Понятно, следовательно, что в
значительной мере влияние рН на равновесную температуру
плавления осуществляется через изменение свойств набухшего
аморфного белка.
Хорошо известно, что добавление определенных низкомоле-
куляоных реагентов в окружающую среду также влияет на
температуру плавления
(сокращения) белковых волокон.
На рис. 62 приведены
характерные изменения температуры
плавления эластоидиновых
волокон в различных средах [39].
В каждом случае одноосное
сокращение сопровождает
плавление, причем рентгенограммы
свидетельствуют о полном
исчезновении упорядоченной структуры.
Таким образом, на самом
деле плавление и соответственно
сокращение вызываются
множеством различных агентов.
Нейтральные соли примерно в
равной мере понижают температуру
плавления. Растворы мочевины
при сравнимых концентрациях в
этом смысле гораздо менее
эффектны. При охлаждении
сократившегося волокна в растворе
мочевины или нейтральных солей получаются совершенно
различные результаты. В первом случае развивается ориентированная
кристаллизация. В растворах нейтральных солей этот процесс
не идет, и волокно при охлаждении остается в аморфном
состоянии. Тем не менее, при помещении такого волокна в чистую
воду происходит почти мгновенное восстановление
упорядоченной кристаллической структуры.
Таким образом, в отличие от поведения в растворе
мочевины и в чистой воде рекристаллизация после плавления в
растворе нейтральной соли требует не только охлаждения, но и
сильного разбавления. Это указывает на то, что помимо
обычного разупорядочения структуры цепей при плавлении под
действием электролитов происходят дополнительные структурные
изменения цепей в расплавленном состоянии, затрудняющие
рекристаллизацию. Удаление электролита немедленно инициирует
кристаллизацию и удлинение.
Концентрация растдора, моль/л
Рис. 62. Зависимость температуры
плавления эластоидиновых
волокон от концентрации
низкомолекулярного реагента в
окружающей фазе [39]:
/ — мочевина; 2— LiBr; 3— CaCi2, KI;
4-KSCN.
202
Хорошо известно, что а- и р-кератины существуют в
ориентированном кристаллическом состоянии, имеют высокую
концентрацию цистиновых остатков и могут сокращаться под
действием различ-ных реагентов [40]. Александер и Гудзон
обнаружили два существенно различающихся типа процессов
сокращения в а-кератиновых волокнах.
В одном случае взаимодействие с некоторыми
специфическими реагентами сопровождается разрывом дисульфидных
мостиков, поэтому наблюдаемое сокращение, естественно,
необратимо. Здесь опять уместно провести аналогию с результатами
для сшитых полиэтиленовых волокон (см. рис. 57). В другом
случае, обратимое анизотропное изменение размеров
происходит без разрыва межцепных связей. Как мы уже неоднократно
убеждались, этот тип сокращаемости является прямым
следствием процессов плавления и рекристаллизации.
Пример превращений последнего типа, как для а-, так и
р-кератина, был впервые получен Уивеллом и Вудсом [41]. При
помещении волокна в медно-аммиачный раствор достаточной
концентрации наблюдается 20%-ное уменьшение длины при
комнатной температуре. Сокращение сопровождается характерным
изменением рентгенограмм, показывающим, что действительно
происходит плавление. Вероятно, в этом случае изотермическое
плавление вызвано реакцией комплексообразования между
определенными аминокислотными остатками белкового волокна
и медно-аммиачным раствором. При помещении сократившегося
волокна в разбавленный кислый раствор происходит
восстановление его начальной длины, а рентгенограммы приобретают
вид, характерный для ориентированного кристаллического
волокна. Таким образом, разрушение комплекса приводит к
обращению плавления, т. е. кристаллизации.
Плавление и сопутствующее сокращение кератиновых
волокон можно также проиллюстрировать на примере их
взаимодействия с водными растворами LiBr. Последний реагент действует
как универсальный преобразователь упорядоченных
полипептидных и белковых структур [42, 43]. При помещении а-кератино-
вого волокна (шерсть овцы породы Линкольн) в большой объем
раствора LiBr определенной концентрации в узком
температурном интервале проиходит резкое сокращение волокна (рис.63).
Картины дифракции рентгеновских лучей под большими
углами подтверждают, что происходит плавление. Как и для эла-
стоидиновых волокон в присутствии LiBr рекристаллизация
при охлаждении не происходит. Опять необходимо разбавление
как это изображено схематически пунктирными линиями на
рис. 63. Подобное поведение обнаруживает и р-кератин —
кератин пера [43].
На рис. 64 приведены рентгенограммы ряда образцов
кератина пера. Они соответствуют нативной одноосноориентирован-
203
М
I2Y
1
¦
U
ной кристаллической структуре (рис. 64,а), аморфному
состоянию, характерному для расплавленного сократившегося
волокна (рис. 64,6), и рекристаллизованному состоянию, в котором
полностью восстановлена
нативная структура (рис.
64, в). Все брэгговские
рефлексы нативного волокна
наблюдаются и для
регенерированных образцов. Таким
образом, здесь обратимость
осуществляется не только
в термодинамическом
смысле, но и восстанавливаются
все тонкие структурные
детали.
Плавление
ориентированной кристаллической
структуры кератина
происходит при строго
определенной температуре, зависящей
от концентрации раствора
LiBr и природы волокна.
Зависимости температуры
плавления от состава
окружающей жидкой фазы для
двух образцов а-кератино-
вых волокон приведены на
рис. 65. Первоначальное добавление LiBr в окружающую
фазу вызывает понижение Гпл. Минимум температуры плавления
50 60 70
30 ЬО 50
ГС
Рис. 63. Зависимость длины а-керати-
новых волокон от температуры в
водных растворах LiBr [42]:
а— 6,5 М LiBr, шерсть овцы; б — 4,8 Л1 LiBr,
шерсть овцы; в —10,65 М LIBr, конский волос.
Непрерывные линии —изменение длины при
нагревании, пунктирная вертикальная линия демон-
стрирует восстановление начальной длины при
погружении волокон в чистую воду.
Рис. 64. Рентгенограммы кератина пера [43]:
а — нативное волокно; б — после сокращения в 5,8 М
растворе LIBr; в —после сокращения в 7,7 М растворе LiBr и
последующего погружения в чистую воду.
достигается примерно в 1М растворе LiBr. При дальнейшем
увеличении концентрации соли температура плавления снова
возрастает. Кривые (рис. 65) очень напоминают по форме кривые,
описывающие зависимость температуры плавления от состава
для статистических сополимеров. Это может свидетельствовать
о дополнительных изменениях структуры полипептидных цепей
сверх обычного разупорядочения при плавлении.
204
В соответствии с данными, представленными на рис. 65
можно осуществить изотермическое плавление волокон изменением
состава окружающей фазы. Изотермическое плавление системы
а-кератин — LiBr — вода показано на рис. 66. Этот график
следует рассматривать справа налево. Волокно вначале
помещалось в наиболее концентрированный раствор LiBr, а затем
система постепенно разбавлялась
водой при постоянной
температуре 24° С. По достижении неко-
т *с
юо\
80\
бо\
20\
&
^ 2\
см с
/ч.
2 и 6 в Ю 12
Концентрациярастбора LiBr, моль/л
Рис. 65. Зависимость температуры
плавления (температуры
сокращения) для а-кератиновых
волокон от концентрации раствора
LiBr [42]:
7 —шерсть овцы породы Линкольн,
2— конский волос.
О 2 4 6 8 Ю 12
Концентрация растбора Li Br, моль/л
Рис. 66. Изотерма плавления и
рекристаллизации (сокращение —
удлинение) а-кератиновых всло-
кон (шерсть овцы Линкольн) в еод-
ных растворах LiBr при 24° С [42].
Измерения проводились посредством
постепенного разбавления чистой водой
концентрированных растворов LIBr (т. е.
«справа налево»).
торой концентрации LiBr, определяемой рис. 65, происходит
резкое плавление, сопровождающееся сокращением волокна.
Это состояние сохраняется в некотором интервале концентраций:
длина волокна не меняется при дальнейшем разбавлении.
Однако по достижении некоторой новой концентрации LiBr
происходит рекристаллизация и соответственное удлинение образца.
То обстоятельство, что фазовый переход может происходить
изотермически, за счет изменения концентрации соли в
окружающей фазе, открывает принципиальную возможность
использования фибриллярных макромолекул в качестве рабочего
вещества в двигателе, изотермически преобразующем
химическую энергию в механическую работу [3, 25].
Мышечные фибриллы представляют собой белковые
волокна, также существующие в природе в ориентированном
кристаллическом состоянии. Ограничивая обсуждение опытами in vitro
над изолированными волокнами, можно указать, что
значительные сокращения достигаются воздействием KI [44] и LiBr
[45], которые известны как агенты, вызывающие плавление и
205
0.9
Ofi
0,7\
¦ОД
oA
сокращение и других фибриллярных белков. Можно a priori
предположить, что в основе сокращения мышечных волокон
лежит тот же механизм.
Сокращение глицеринизированных мышечных волокон
исследовалось при погружении образцов в водные растворы,
содержащие аденозинтрифосфат (АТФ) — ре-
L f°Q. агент, участвующий в мышечной деятель-
ности в физиологических условиях [46].
Изменение длины такого волокна в результате
изменения концентрации АТФ в
окружающей фазе при комнатной температуре
показано на рис. 67.
Как видно из рис. 67, изотермический
кооперативный фазовый переход
вызывается очень небольшим увеличением
концентрации АТФ. Этот переход представляет
собой плавление, что подтверждается и
структурными изменениями. Действительно,
рентгенограммы показывают, что натив-
0002 0004 ная ориентированная кристаллическая стру-
Концентрация'растбора ктура типа а-кератина полностью исче-
АТФ, моль/л зает после завершения сокращения [42]. Это
Рис. 67. Зависимость вполне согласуется с наблюдениями, про-
сокращения (относи- веденными на других фибриллярных бел-
тельного изменения ках. Рекристаллизация и восстановление
длины) глицеринизи- длины волокна не происходят в этой систе-
^локоГоГ!ГонцеЫн" ме ни ПРИ охлаждении, ни при разбавле-
трации АТФ в окру- нюи, что указывает на отсутствие
сохраняющей фазе [47]. няющихся (ковалентных) межцепных
связей в волокне. Сокращение,
сопровождающее плавление, наблюдается также для глицеринизированных
мышечных волокон в системах АТФ — глицерин — вода и
АТФ — этиленгликоль — вода. Большие и резкие изменения
длины происходят при относительно небольших изменениях
состава растворителя или температуры [48].
Приведенные результаты показывают, что в опытах in vitro
со всеми фибриллярными белками можно осуществить
анизотропные изменения размеров, являющиеся следствием фазового
перехода между ориентированным кристаллическим и аморфным
состояниями. Эти результаты вполне аналогичны полученным на
фибриллярном полиэтилене и других полимерах с более простой
структурой *.
* Эти выводы могут быть распространены на переходы в разбавленных
растворах, в которых участвует более чем одна молекулярная цепь.
Обратимость переходов спираль — клубок в разбавленных растворах ДНК
существенно облегчается введением ковалентных связей в упорядоченном
состоянии, которые стабилизируют двойную спираль [49, 50],
206
Несмотря на многообразие реагентов, способных вызывать
сокращение фибриллярных белков, лежащий в основе их
действия механизм один и тот же. Многие важные природные
процессы, включая биологические, связаны с сокращаемостью на
молекулярном уровне. Во всех этих процессах макромолекулы
обычно находятся в ориентированном кристаллическом
состоянии на некоторой стадии сократительного цикла.
Обоснованность применения изложенных общих принципов к таким
реальным биологическим процессам как механохимия мышечной
деятельности, митоз, движение хромосом и подвижность клетки
требует дальнейшего анализа на системах, функционирующих
in vivo. Косвенные данные, полученные в опытах
нефизиологического характера in vitro, показывают плодотворность
исследований в этом направлении.
Следует ожидать, что в естественных действующих системах
процессы регулирования чрезвычайно сложны и включают серии
химических реакций. Это дополнительно затрудняет проведение
прямых опытов. В принципе, однако, если происходит фазовый
переход, его характерные черты должны проявляться
достаточно четко даже на фоне сопутствующих процессов. Особенно
интересна ситуация, при которой волокно или часть волокна
обладает ферментативной активностью. В этом случае волокно, в
одном или другом фазовом состоянии, может влиять на
концентрацию реагентов в окружающей фазе, которые, в свою очередь,
могут благоприятствовать либо кристаллизации, либо
плавлению. Соответственно, должны происходить легко
регистрируемые изменения размеров или, если приложено растягивающее
усилие» напряжения.
Механохимия
Выше мы показали, что характерная для полимеров
способность к сильным обратимым деформациям позволяет, в
принципе, использовать длиноцепные молекулы для преобразования
тепловой или химической энергии в механическую работу.
Естественно предположить, что деформация полимерных тел всегда
связана с конформационными изменениями отдельных молекул.
Нет никаких априорных причин и для исключения
биологических процессов, сопряженных с макроскопическими
изменениями размеров, из этой общей категории, хотя в отдельных
специальных случаях механизмы деформации могут быть иной
природы. С другой стороны, было бы удивительно, если бы природа
не «использовала» конформационную изменчивость
макромолекул [51].
Необходимо различать деформации и связанные с ними кон-
формационные изменения макромолекул, ограничивающиеся
207
аморфной фазой, и деформационные процессы, включающие
фазовый переход кристалл — жидкость. Высокоэластическая
деформация некристаллических сеток при температуре выше
температуры стеклования относится к первой категории. При такой
каучукоподобной деформации на молекулярном уровне
происходит увеличение длины аморфных цепей соответственно
приложенному макроскопическому напряжению.
С этим явлением тесно связаны деформации при набухании
полимерных сеток, погруженных в большой объем растворителя.
Набухание или сжатие сетки может быть вызвано изменениями
интенсивности термодинамических взаимодействий полимер —
растворитель или различными химическими реакциями.
Например, в случае полиэлектролитных сеток степень набухания
зависит от величины рН или ионной силы окружающей среды.
Деформации, связанные с аморфной фазой, обычно изотропны,
если только не приложены слишком большие напряжения или
a fi 6 л
\ju_\0_
d1——Т \ ъ
* -^. I т-
L L
Рис. 68. Диаграмма цикла Карно для машины с чистым
полимером в качестве рабочего вещества:
а —полимер все время в аморфном состоянии; б —полимер при
наличии изотермического фазового перехода.
нет механических препятствий для изотропного набухания [52].
Что же касается второй категории, т. е. анизотропной
деформации и развития релаксационных напряжений при переходах
кристалл — жидкость, то ей была посвящена вся эта глава.
Для оценки возможности использования преобразования
тепловой энергии в механическую работу, рассмотрим цикл Карно;
на природу рабочего вещества при этом не накладывается
никаких ограничений. Необходимо, однако, чтобы все процессы
могли развиваться обратимо, и чтобы обмен получаемого и
выделяемого рабочим телом тепла происходил при постоянной
температуре. Соответственно, процессы, при которых температура
рабочего вещества меняется, должны быть обратимо
адиабатическими.
Вспоминая аналогию между /?, V и —/, L-диаграммами,
можно построить диаграмму обратимого цикла Карно тепловой
машины с аморфным полимером в роли рабочего вещества,
изображенную на рис. 68, а [22]. AD и СВ — изотермы; полимер на-
208
ходится в контакте с тепловым резервуаром большой емкости
при температурах соответственно 7\ и Т2. АВ и CD — обратимые
адиабаты для системы, изолированной от окружающей среды.
Обратимая тепловая машина может быть построена из любого,
способного деформироваться вещества, для которого
напряжение при постоянной длине растет с увеличением температуры, а
адиабаты напряжение— длина имеют больший наклон, чем
соответствующие изотермы. Эти критерии согласуются с ранее
обсужденными термоэластическими свойствами аморфных
полимеров. Машина такого типа реализована в
саморазогревающемся маятнике, описанном Вигандом, где роль рабочего
вещества выполняет каучук*. Термодинамическая эффективность
машины, показанной на рис. 68, а, рассчитывается, как хорошо
известно, непосредственно из диаграммы цикла Карно и зависит
только от двух рабочих температур. Площадь ABCD
представляет работу, выполняемую за один цикл.
Если происходит не каучукоподобная деформация, а
фазовый переход, то изотермические процессы представятся
горизонтальными линиями (сила в этом случае не зависит от длины),
как показано на рис. 68, б. При одинаковых адиабатах в обоих
циклах совершаемая работа больше для цикла с фазовым
переходом, который аналогичен использованию конденсации пара
в обычном цикле Карно. Термодинамическая эффективность
остается неизменной, так как она определяется только двумя
рабочими температурами.
Циклы с использованием фазового перехода имеют более
высокий коэффициент полезного действия, независимо от
молекулярной природы рабочего вещества, и поэтому более
предпочтительны. В случае фибриллярных макромолекул ориентированная
кристаллическая структура дает возможность осуществить
фазовый переход. Именно благодаря переходам такого типа в
природных системах возможны предельно резкие процессы
сокращения; рабочее вещество при фазовом переходе является более
чувствительным и эффективным преобразователем тепловой
энергии в механическую работу.
Можно оценить работу, совершаемую на одном
изотермическом участке цикла, сопровождающегося полным плавлением
ориентированного волокна. При температуре плавления Гпл и
растягивающей силе должно удовлетворяться условие:
Фа_Фк = /д? = _№ A55)
При этом предполагается, что Фк не изменяется в процессе
деформации, тогда как фа может быть выражено:
Ф* (Г™ I) - Фа <Г„л. 0) + АФ^-(Тпл, () A56)
* [Принцип «саморазогревания» основан на том, что при растяжении
каучука выделяется тепло, а при сокращении поглощается. — Прим.
редактора].
14 Л. Манделькерн
209
где ДФ*— изменение термодинамического потенциала в
аморфном состоянии при Гпл, обусловленное возрастанием
высокоэластической деформации при изменении силы
от нуля до /.
Следовательно
ДФ/ (Ты. 0) + АФэа (Гм> /) = f M шп - W A57)
где
АФ/ (Гпл. 0) = Фа (Ты, 0) - Фк (Гпл, 0) A58)
Разлагая ДФ/(ГПЛ, 0) в ряд вблизи равновесной температуры
плавления Гпл» получим:
ДФ, (Гпл, 0) = ДФ, (Т°ш, 0) + [-^jAj о (Гпл - Т°пл) A59)
'пл
АФ/(^Ы.0)*-(ГПЛ-Г2Л)А^ A60)
Для системы, где ДФЭ=—TASQ
**fru-r°,)AS° + rnjlAS. A61)
W » - Т°пл bS°f + Тпл (ASa + ^S0f) A62)
Произведенная волокном при плавлении работа зависит от
двух слагаемых. Одно из них не зависит от силы, а другое
зависит от нее через Гпл и Д5Э. В изотропном варианте, когда
приложенная сила равна нулю, совершаемая работа с
неизбежностью также равна нулю, кроме того при Т=ТпЛ, Д? = 0 так
что W снова обращается в нуль.
Таким образом, совершаемая за один цикл работа проходит
через максимум при увеличении температуры и силы; это
неминуемо следует из существования критической температуры
Гпл. Две последние величины Пл и AZ, связаны уравнением
A28). При малых силах, когда Гпл лишь незначительно
превышает Тпл и Д5Э мало, совершаемая работа должна быть
порядка R кал/моль. Это сопоставимо с величинами, наблюдаемыми
для биологических систем.
Более общий интерес, особенно в связи с биологическими
системами, представляют химические машины, которые работают
изотермически. Например, природные мышечные волокна можно
рассматривать как преобразователи химической энергии в
механическую работу, действующие под влиянием обратимых
напряжений, генерируемых в рабочем веществе — фибриллярных
белках мышц [22]. Можно представить себе изотермическую
химическую машину, действующую по аналогии с тепловой [22, 54].
210
Вместо двух резервуаров тепла, волокно поддерживается в
контакте с двумя большими ваннами, одна из которых содержит,
при постоянном химическом потенциале, реагент,
осуществляющий плавление, а другая — «поглотитель» этого реагента
(«нейтрализатор»). Процессы, протекающие при постоянном
химическом потенциале, изображаются линиями, которые по аналогии
с изотермами тепловой машины называют изопотенциалами [54].
Перевод волокна из состояния с одним химическим потенциалом
в другой происходит как в изолированной системе, общий
состав которой остается неизменным. Эти процессы изображаются
линиями, которые называются изофорами и соответствуют
адиабатам теплового цикла. В последнем случае, при поддержании
постоянной энтропии, происходит изменение термического
потенциала или температуры. В случае фибриллярного рабочего
вещества химическая машина будет функционировать, если
развивающееся при постоянной длине напряжение возрастает с
увеличением концентрации реагента, а наклон изопотенциалы
(df/dL)ill меньше наклона изофоры (df/dL)n .
Следствие кристаллизации рабочего вещества в химической
машине оказывается таким же, как и в тепловой машине. При
работе между одинаковыми уровнями химического потенциала
с общими изофорами, эффективная работа, произведенная за
один цикл, больше при наличии фазового перехода на изопотен-
циальном участке. Изопотенциальный фазовый переход
обеспечивает выполнение необходимых соотношений между изофор-
ными и изопотенциальными зависимостями сила — длина.
Известно, по крайней мере, несколько фибриллярных белковых
систем, которые могут служить в качестве рабочего вещества и
реагента химической машины с изопотенциальным фазовым
переходом.
Для простой машины, состоящей только из одного
компонента (кроме рабочего вещества), помещенного в два
резервуара с химическими потенциалами \х1 и \iu эффективная
работа, произведенная за один цикл [54], равна
U7 = (|х! — цп)Дл A63)
где An — количество низкомолекулярного реагента,
перешедшего из одного резервуара в другой.
Непосредственным источником работы, выполненной при
сокращении, является, главным образом, увеличение энтропии при
плавлении. Однако, для полного цикла, когда рабочее вещество
возвращается в свое начальное состояние, все изменения
должны отражаться и на окружающей среде. Основной источник
совершаемой работы — изменение свободной энергии при
перемещении образцов из одного резервуара в другой.
14*
211
Более сложные типы химических машин, включающих
многокомпонентные системы, подробно обсуждены Качальским с
сотрудниками [54] на основе таких же принципов. Приведенный
анализ идеализированных циклов отнюдь не подразумевает, что
реальные системы должны точно соответствовать им. Он дает
лишь основу для понимания механохимических процессов,
протекающих с участием макромолекул.
Литература
1. Т. Т. Li, R. J. Volungis, R. S. Stein, J. Polymer Sci., 20, 194
A956); J. T. Judge, R. S. Stein, J. Appl. Phys., 32, 2357 A961).
2. P. J. Flory, J. Chem. Phys., 15, 397 A947).
3. P. J. Flory, Sci., 124, 53 A956).
4. P. J. F 1 ory, J. Am. Chem. Soc, 78, 5222 A956).
5. Л. Т р е л о а р, Физика упругости каучука, ИЛ, 1953.
6. D. Е. Roberts, L. M a n d e 1 k e г п, J. Res. Natl Bur. Stand., 54, 167
A955).
7. P. J. Flory, R. R. Garrett, J. Am. Chem. Soc, 80, 4836 A958).
8. P. J. Flory, O. K. Spurr, Jr., J. Am. Chem. Soc, 83, 1308 A961).
9. G. Gee, Quart. Rev. (London), 1, 265 A947).
10. J. F. M. Oth, P. J. Flory, J. Am. Chem. Soc, 80, 1297 A958).
11. D. E. Roberts, L. Man del kern, J. Am. Chem. Soc, 77, 781 A955).
12. W. H. Smith, С P. Say lor, J. Res. Natl Bur. Stand., 21, 257 A938).
13. A. V. Tobolsky, G. M. Brown, J. Polymer Sci., 17, 547 A955).
14. A. N. Gen t, Trans. Faraday Soc, 50, 521 A954).
15. G. Natta, P Corradini, Rubber Chem. Technol., 33, 703 A960).
16. W. T. Astbury, in A. Wasserman (ed.), «Size and Shape Changes
of Contractile Polymers», New York, 1960, p. 78.
17. H. В. В u 11, J. Am. Chem. Soc, 67, 533 A945).
18. A. Weber, H. Weber, Biochim. et Biophys. acta, 7, 214, 339 A951).
19. M. M о r a 1 e s, J. В о 11 s, Disc Faraday Soc, 13, 125 A953).
20. P. J. Flory, J. Cellular Сотр. Physiol., 49, (Suppl. 1), 175 A957).
21. H. A. Scheraga, J. Phys. Chem., 64, 1917 A960).
22. M. G. M. Pry or, in J. A. V. Butler, J. T. Randa 1 (eds.), «Progress
in Biophysics», vol. I, Butterworth — Springer, 1950, p. 216.
23. J. F. M. Oth, Roll. Z., 162, 124 A959).
24. O. G e r n g г о s s, L. R. К a t z, Koll. Beih., 23, 368 A926).
25a. D. E. Roberts, L. M a n d e 1 k e r n, P. J. Flory, J. Am. Chem. Soc,
79, 1515 A957).
25b. D. E. Roberts, L. Man del kern, J. Am. Chem. Soc, 80, 1289A959).
26. L. M a n d e 1 k e r n, D. E. Roberts, A. F. D i о r i o, A. S. Posner,
J. Am. Chem. Soc, 81, 4148 A959).
27. K. Bailey, W. T. Astbury, К. М. Rirdall, Nature, 151, 716 A943).
28. W. T. Astbury, Proc Roy. Soc (London), Ser. A, 134, 303 A947).
29. W. T. A s t b u r y, Trans. Faraday Soc, 34, 378 A948).
30. B. A. Wright, N. M. Wiederhorn, J. Polymer Sci., 7, 105 A951).
31. A. Ewald, Z. physiol. Chem., 105, 135 A919).
32. R. S. Be a r, Adv. Protein Chem., 7, 69 A952).
33. R. V. R ice, Proc Natl Acad. Sci. U. S., 46, 1186 A960).
34. J. F. M. Oth, Koll. Z., 162, 124 A959).
35. E. Faure-Freme t, J. Chim. phys., 34, 126 A937).
36. G. Champetier, E. F a u r e - F re me t, J. Chim. phys., 34, 197 A937).
37. L. Mandelkern, W. T. Meyer, J. Polymer Sci., 49, 125 A961).
212
38. F. G. Lennox, Biochim. et Biophys. acta, 3, 170 A949).
39. L. M a n d e 1 к e r n, W. T. Meyer, A. F. D i о r i o, J. Phys. Chern., 66,
375 A962).
40. P. Alexander, R. F. Hudson, Wool, Its Chemistry and Physics, New
York, 1954, p. 55.
41. C. S. Whewell, H. J. Woods, Symposium on Fibrous Proteins,
Society of Dyers and Colourists, 1946, p. 50.
42. L. M a n d e 1 к e r n, J. C. H a 1 p i n, A. F. D i о r i o, A. S. P о s n e r,
J. Am. Chem. Soc, 84, 1383 A962).
43. L. M a n d e 1 к е г n, J. C. H a 1 p i n, A. F. D i о r i o, J. Polymer Sci., 60,
531 A962).
44. W. J. Bo wen, K. L а к i, Am. J. Physiol., 185, 92 A956).
45. L. M a n d e 1 к e r n, J. С. Н a 1 p i n, неопубликованные данные.
46. W. J. В о wen, J. Cellular Сотр. Physiol., 49, (Suppl. 1), 267 A957).
47. L. M a n d e 1 k e r n, A. S. P о s n e r, A. F. D i о г i о, К. L a k i, Proc. Natl
Acad. Sci. U. S., 45, 814 A959).
48. С A. J. H о e v e, Y. A. W i 11 i s, D. J. Martin, Biochemistry, 2, 279
A963).
49. E. P. Geiduschek, Proc. Natl Acad. Sci. U. S., 47, 950 A961).
50. J. M а г m u r, L. Grossman, Proc. Natl Acad. Sci. U. S., 47, 778
A961).
51. P. J. Flory, Brookhaven Symp. Biol., N° 13, Protein Structure and
Function, 1960, p. 89.
52. P. J. Flory, Conference on Contractility, Mellon Institute, Pittsburgh,
January 27—30, 1960.
53. W. B. Wiegand, Trans. Inst. Rubber Ind., 1, 141 A925).
54. А. К a t с h a 1 s k y, S. L i f s о n, I. M i с h a e 1 i s, H. Z w i с k, A. W a sser-
mann (ed.), «Size and Shape*Changes of Contractile Polymers». New
York, 1960, p. 1.
Кинетика и механизмы
кристаллизации
Введение
До сих пор при обсуждении проблемы кристаллического
состояния полимеров мы в основном имели дело с равновесными
условиями и процессами. Хотя тот факт, что плавление
полимеров связано с проблемой фазового равновесия можно считать
твердо установленным, его строгое экспериментальное
подтверждение весьма затруднительно.
.. Из теории фазового равновесия следует, что при
кристаллизации полимеров высокого молекулярного веса и регулярности
можно в принципе достичь очень высоких уровней
кристалличности. Однако, как мы уже могли убедиться, в реальных
полимерных системах такие высокие степени кристалличности
обычно не наблюдаются. Одна из основных причин такого
расхождения теоретических и экспериментальных данных объясняется
тем, что кристаллическая фаза развивается с конечными
скоростями при температурах, достаточно низких, по сравнению с
температурой плавления; поэтому состояние, наблюдаемое в
большинстве реальных систем, соответствует условиям, весьма
далеким от равновесия.
Строение и свойства достигнутого морфологического
состояния являются результатом конкуренции между кинетическими
и релаксационными факторами, с одной стороны, и
требованиями термодинамического равновесия, с другой. Наличие
подобной конкуренции свойственно не только полимерам, но и
большинству низкомолекулярных веществ, претерпевающих переход
из жидкого в кристаллическое состояние.
Для иллюстрации этих общих соображений рассмотрим,
каким образом какое-либо простое свойство, например, удельный
объем или плотность, зависит от условий кристаллизации [1].
На рис. 69 представлена зависимость удельного объема при
25° С для нефракционированного линейного полиэтилена (мар-
лекс-50) от температуры кристаллизации. Кристаллизация
проводилась в изотермических условиях и время ее выбиралось
достаточно большим для того, чтобы при измерении удельного
214
объема не происходило заметной дополнительной
кристаллизации. При относительно низких температурах кристаллизации
удельные объемы изменялись в пределах от 1,06 до 1,04, что
соответствует обычно определяемым для этого полимера величинам.
Однако при повышении температуры кристаллизации
наблюдается относительно резкое падение удельного объема,
который, начиная примерно со 100° С, становится очень
чувствительным к малейшим изменениям температуры. Более низкие
удельные объемы соответствуют более
высоким степеням кристалличности,
которые, как известно, достигаются
при проведении кристаллизации
вблизи точки плавления.
Пунктирная линия на рис. 69,
представляющая экстраполяцию
удельного объема на более высокие,
нежели наблюдаемые обычно
температуры кристаллизации,
показывает определенную возможность
того, что при дальнейшем повышении
температуры кристаллизации могут
быть достигнуты еще большие
плотности. Из экстраполяции, в
частности, следует, что в случае
кристаллизации при температуре
плавления удельный объем должен стать
равным ~1,00. Эта величина
соответствует значению плотности
полностью кристаллического полимера,
найденному Банном [2] при
определении кристаллической структурь
образование почти идеального макроскопического
монокристалла. Однако экспериментальное подтверждение
экстраполированного участка кривой потребовало бы столь длительных
измерений, что его можно считать практически неосуществимым.
Таким образом, можно продемонстрировать чрезвычайно
широкую вариацию плотности при 25° С для одного и того же
полимера.
В цитированных опытах плотность непосредственно зависела
от температуры кристаллизации. Однако не только удельный
объем или плотность, но и другие физические, а также
термодинамические и механические свойства очень чувствительны к
условиям перехода жидкость — кристалл. Можно ожидать, что
морфология полимера или его кристаллическая текстура также
в значительной степени определяются условиями кристаллизации.
Таким образом, для полного понимания свойств и
поведения полимерных кристаллических систем необходимо помимо
?
?
?
W6[
Щ
щ
1М
щ
w\
'" 20 W 60 80 100 120 \U0
- т,°с
Рис. 69. Зависимость
удельного объема при 25° С от
температуры кристаллизации для
нефракционированного
линейного полиэтилена (мар-
лекс-50) [1].
Время кристаллизации выбирается
таким образом, чтобы процесс
протекал в основном при выбранной
температуре.
полиэтилена, и означает
215
знания равновесных характеристик иметь определенные
сведения о механизме перехода в кристаллическое состояние. Такую
информацию, в принципе, можно получить, изучая кинетику
перехода жидкость — кристалл.
Все экспериментальные исследования при этом можно
разделить на две большие категории. К первой относятся опыты, в
которых определяется валовая скорость развития
кристаллической фазы из переохлажденной жидкости в изотермических
условиях. В опытах такого типа необходимо регистрировать
изменение каких-либо свойств полимера, весьма чувствительных к
изменениям степени кристалличности. Плотность полимера как раз
и является таким чувствительным и к тому же удобным для
измерений свойством. В некоторых благоприятных случаях
возможно также применение и других методов, таких как
инфракрасное поглощение или деполяризация проходящего света.
Вторая широко распространенная категория исследований
состоит в прямом микроскопическом наблюдении скорости
образования и последующего роста отдельных сферолитов в
изотермических условиях. Именно на основании получаемых таким
образом двух типов информации следует делать заключение о
механизмах кристаллизации. При этом, однако, следует считаться
с одним серьезным ограничением, заключающемся в отсутствии
прямых данных об образовании и росте в блочных полимерах
отдельных кристаллитов.
Экспериментальные исследования
кристаллизации чистых гомополимеров
Впервые валовая скорость кристаллизации чистого гомопо-
лимера в изотермических условиях была измерена Беккедалем
[3] при исследованиях натурального каучука. Некоторые
типичные изотермы кристаллизации из расплава линейного
полиэтилена [4] и натурального каучука [5] приведены на рис. 70 и 71.
В обоих случаях за ходом кристаллизации следили по
изменениям удельного объема.
Для полиэтилена приведена зависимость степени
завершенности перехода от времени, а для натурального каучука —
уменьшение относительного объема. В опытах такого рода,
когда расплавленный образец быстро охлаждается до температуры,
при которой ведется наблюдение, существует вполне
определенный промежуток времени, в течение которого кристаллизация не
наблюдается. Совершенно очевидно, что этот промежуток
времени определяется только чувствительностью применяемого
прибора (детектора кристалличности). Начиная с момента, когда
кристалличность уже поддается оценке, процесс протекает со
216
все возрастающей скоростью и носит автокаталитический
характер. По истечении какого-то времени достигается
псевдоравновесный уровень кристалличности. Однако процесс
кристаллизации на этом не заканчивается и продолжается еще очень долго,
хотя и с весьма малой скоростью.
Весь процесс протекает плавно, без каких-либо резких
изменений или прерывностей на изотермах; S-образные кривые,
приведенные на рис. 70 и 71, Л1/в/о
типичны для всех изучен- ' °0
ных гомополимеров.
Если плавление,
предшествующее
кристаллизации, проводится до конца
и в условиях,
исключающих деструкцию
полимера, то изотермы кристал-
200 400 000
Время, мин
Рис. 70. Зависимость
(К»— Vt)l(Yоо— vo) 0T времени
кристаллизации линейного
полиэтилена из расплава [4].
На кривых показана температура
кристаллизации.
в 12
Время, ч
Рис. 71. Изменение удельного объема
taV в процессе кристаллизации
натурального каучука при различных
температурах [5].
На кривых показана температура
кристаллизации.
лизации превосходно воспроизводятся и не зависят от
температуры отправного расплава [6]. Этот вывод справедлив и тогда,
когда температура расплава лишь немного выше температуры
плавления; важно лишь, чтобы плавление было полным.
В настоящее время трудно судить, в какой мере эта
воспроизводимость изотерм сохраняется при начальных температурах
расплава, намного превосходящих температуру плавления, хотя
уже сейчас имеются некоторые данные о плохой
воспроизводимости в этом случае. Скорее всего, подобные результаты
означают, что при выбранной температуре расплава перечисленные
выше условия не могут быть соблюдены, или что скорость
охлаждения выбрана неправильно и кристаллизацию уже нельзя
рассматривать, как истинно изотермическую.
217
Другой не менее важной характеристикой процесса
кристаллизации является очень сильная зависимость его скорости от
температуры кристаллизации. В качестве примера на рис. 72
приведена зависимость времени, требуемого для достижения
половинного уровня кристалличности, от температуры
кристаллизации в очень широком интервале температур для
натурального каучука [5]. Аналогичные зависимости наблюдались для
самых различных гомополи-
меров, например, для поли-
этилентерефталата [7], поли-
этиленадипината [8] и поли-
этиленсукцината [9].
Как видно из рис. 72,
вблизи Тпл скорость
кристаллизации очень мала, и
появление кристалличности
не наблюдается в течение
весьма длительного
времени. По мере понижения
температуры кристаллизации
скорость ее растет,
проходит через максимум и затем
снова падает практически до
максимума совпадает с
областью перехода полимера в стеклообразное состояние. Однако
для многих полимеров при температурах ниже Т°т
кристаллизация протекает настолько быстро, что оказывается крайне
трудным изучить изотермическую кристаллизацию во всей области
температур и определить температуру, при которой скорость
кристаллизации максимальна.
Высокую чувствительность скорости кристаллизации к
температуре вблизи Гпл хорошо иллюстрируют изотермы,
приведенные на рис. 70. При 129° С возникновение кристалличности
невозможно определить по крайней мере в течение 400 мин.
Однако понижение температуры кристаллизации всего на 1 град
приводит к тому, что при той же чувствительности метода
регистрации кристалличность определима уже через 6 мин, т. е.
скорость кристаллизации возрастает более чем в 60 раз. Это
показывает, что температурный коэффициент скорости
кристаллизации действительно очень велик.
На рис. 73 по данным рис. 70 построены зависимости
отношения (Voo— Vt)l(Vco — V0) (т. е. степени завершенности
перехода) от логарифма времени при фиксированных температурах
кристаллизации. Эти кривые по-прежнему сохраняют все
признаки изотерм. Легко видеть, что каждая из новых изотерм
может быть совмещена с соседней простым сдвигом вдоль гори-
-50 -40 -30 -20 -10 0 +10 +20
ГС
Рис. 72. Зависимость скорости
кристаллизации натурального каучука от
температуры [5].
Скорость определяется обратной величиной
полупериода кристаллизации, т. е. времени,
требуемого для достижения половинного уровня
кристалличности.
нуля. Интервал температур ниже
218-
зонтальной оси [4, 6]. Этим характерным свойством обладают
изотермы всех до сих пор изученных гомополимеров (в точном
значении этого термина, см. гл. 4) и не только вблизи Гпл, но
практически во всей области температур кристаллизации [3,
10]*. Из этих наблюдений следует, что температурный коэффи*
циент скорости
кристаллизации остается неизменным -*ДО«
в течение большей части
процесса.
Таким образом, с чисто
экспериментальной точки
зрения оказывается
возможным, используя
приведенные переменные,
включающие время и
температуру, построить одну
обобщенную изотерму,
исчерпывающим образом описывающую
температурно-временную
кинетику кристаллизации
данного гомополимера.
Что касается
микроскопических исследований, то
тщательно проведенные
эксперименты показывают, что
в тонких полимерных
пленках при определенных
условиях может происходить
спорадический во времени и
пространстве рост сфероли-
тов [8, 11, 12].
Специальные опыты показывают, что при полном плавлении
последующей повторной кристаллизации образца сферолиты
не обязательно возникают на старых местах. Однако Шарплс
[13] установил, что в полидекаметилентерефталате
сферолиты хотя и образуются во времени случайно, но появляются
всегда в одних и тех же местах. Можно заключить, что в этой
системе рост сферолитов всякий раз начинается на одних и
тех же фиксированных в пространстве неоднородностях. Во
многих других работах тоже отмечается тенденция сферолитов
появляться при повторных циклах кристаллизации и
плавления в одной и той же точке в поле зрения микроскопа
[14-18].
Время, мин
Рис. 73. Зависимость (VM— V^HV^— V0)
от логарифма времени кристаллизации
линейного полиэтилена [4].
На кривых показана температура кристаллизации.
* Полная суперпозиция изотерм реализуется при степенях
завершенности кристаллизации по крайней мере 80%. Имеется лишь некоторая
неопределенность в отношении «хвоста» изотермы.
219
Ричарде и Хоукинс [12] показали, что в случае разветвленного
полиэтилена такой результат объясняется исключительно
неполным плавлением полимера. Появление сферолитов в одном и том
же месте при повторных циклах кристаллизации и плавления
у других полимеров может быть объяснено либо этой, же при-
50 100
бремя, мин
Рис. 74. Зависимость скорости роста сферолитов
от времени для полиэтиленадипината М = 9900 [8а].
На изотермах показана температура кристаллизации.
чиной, либо тем, что в расплаве имеется конечное число
дискретно расположенных центров нуклеации.
Вблизи температуры плавления скорость, с которой
возникают сферолиты, очень сильно зависит от температуры
кристаллизации и быстро увеличивается с ее понижением. Например,
у полидек&метиленадипината скорооть возникновения центров
сферолитизации уменьшается на пять порядков при повышении
температуры кристаллизации от 67 до 72° С [II].
220
В результате большого числа экспериментальных работ
установлено, что в самых различных полимерных системах при
фиксированной температуре радиус растущего сферолита
линейно увеличивается во времени и широком диапазоне
температур кристаллизации. Это хорошо согласуется с
автокаталитическим характером изотерм, приведенных на рис. 70 и 71.
Скорость линейного роста сферолитов также весьма чувствительна
к изменениям температуры [8, 11, 18]. Например, в работе
Такаянаги [8а] показано, что по мере понижения температуры
кристаллизации скорость роста сферолитов в полиэтиленади-
пинате вначале растет, достигая максимума при некоторой
температуре, а затем начинает падать. Типичные для этой системы
результаты приведены на рис. 74.
Ясно видно, что скорость роста постоянна при
фиксированных температурах кристаллизации, а наклон прямых зависит
от температуры. Имеется явное качественное сходство между
зависимостью валовой скорости кристаллизации и скорости
роста линейных размеров сферолитов от температуры
кристаллизации. Температуры, при которых скорости роста в обоих
типах экспериментов максимальны, в случае полиэтиленадипината
почти совпадают. Однако при значительном увеличении общего
уровня кристалличности сферолиты начинают заполнять
большую часть объема системы, и их перекрывание, разрушение
(коллапс) и взаимное подавление роста чрезвычайно
затрудняют дальнейшее измерение их размеров. Поэтому возможности
микроскопической техники как количественного метода
ограничены областью умеренных степеней кристалличности.
Математическое описание кинетики
фазовых превращений
Возникновение новой фазы в исходной, например кристалла
в жидкости, включает в себя ее зарождение и последующее
развитие. Первая часть процесса называется нуклеацией, а
вторая — ростом. В большинстве случаев, представляющих интерес,
изотермическая кристаллизация может быть описана с
помощью уравнений, содержащих в качестве основных параметров
частоту нуклеации N и скорости роста G{ каждой из
кристаллографических плоскостей. Эти уравнения должны определять
валовую скорость кристаллизации, т. е. переход вещества (в
молях) из жидкой в кристаллическую фазу, как функцию
времени.
Конкретные условия кристаллизации и возможные
ограничения процесса во времени или пространстве учитываются при
составлении этих уравнений,
221
Сначала рассмотрим процесс кристаллизации
низкомолекулярных веществ. Стационарную частоту нуклеации на единицу
массы, еще не перешедшей в другую фазу, обозначим через
N', тогда число зародышей, образующихся за время dx, равно:
dn = N(x)[l—X(x)]dx A64)
где Х(х).— доля вещества, перешедшего в новую фазу за
время т.
Если w(t, т)—масса к моменту / некоторого растущего
центра, который возник в момент т (т</) и далее рос без
ограничений, то:
X (t) = J w (tt x)dn = J w (t, x) N' (t) [1 — X (т)] dx A65)
о о
Соотношение A65) можно переписать следующим образом:
х (t) = JEsl f v (t, т) n (т) [i - x (t)] dx A66)
Рж J
0
•
где N(x)— частота нуклеации на единицу еще не перешедшего
в другую фазу объема;
v(t, х) — соответствующий объем растущего центра;
Рк и Рж — плотности кристаллической и жидкой фазы.
Соотношение A65), выведенное фон Голером и Заксом,
будем именовать приближением свободного роста. При этом
приближении принимается, что развитие отдельного растущего
центра полностью независимо как от массы, уже перешедшей в
другую фазу, так и от роста других центров. В частности,
эффектами подавления одного растущего центра другим в этом
приближении пренебрегают.
Чтобы решить уравнение A66) относительно X(t),
необходимо определить v(t, т) и N(x). Примем упрощающее
предположение, что параметр N(x) не зависит от количества
вещества, уже претерпевшего фазовый переход, т. е. является
постоянной величиной. Такое. допущение достаточно оправдано,
ибо во многих случаях, в частности для однокомпонентных
систем, рост новой фазы действительно происходит по линейному
закону. Если теперь обозначить векторы линейного роста в
направлениях х, у, z через (/#, Оу, Gz, то, пренебрегая
подавлением, можно записать:
v(t,x) = fGxGyQ,(t-x)* A67)
Предполагается, что векторы линейного роста неизменны
в процессе роста; фактор / определяет геометрию растущего
центра.
222
Сделанное ранее предположение о линейности роста
зародыша новой фазы (ядра кристаллизации) означает, что
скорость изменения объема растущего центра пропорциональна
площади его поверхности. Таким образом, в этом случае
скорость роста контролируется процессом, происходящим на
поверхности раздела кристалл — жидкость. Если же, с другой
стороны, предположить, что скорость роста новой фазы
определяется скоростью диффузии кристаллизующихся звеньев или
сегментов к растущей поверхности, то линейный размер
зародыша должен увеличиваться пропорционально корню
квадратному из времени. В этом случае v(t, т) зависит уже от
Для случая линейного роста в двух направлениях
v(t, x) = fGxGyy(t-xy A68)
где у определяет фиксированный размер в третьем
направлении.
Для одномерного роста находим аналогичную формулу:
v(t, T) = lGxyty2{t-i) A69)
В качестве примера рассмотрим случай сферически
симметричного и линейного роста зародыша новой фазы. При этом
^ = 4л/3 и Gx=Gy=Gz=G. Уравнение A66) можно теперь
переписать в виде:
X = 4? GW-^- f (* — тK A — X) dx A70)
0
Для начальной стадии перехода это соотношение можно ап-
роксимировать следующим:
X» 4— &йР 071)
3 рж
Формулу A71) также следует определить, как приближение
свободного роста. Принимая во внимание отправные
предположения, нетрудно убедиться, что полное решение уравнения A66)
приводит к нереалистическим результатам.
При более точном описании кинетики фазовых переходов
необходимо принимать во внимание взаимное подавление роста
соседних растущих центров. Эта проблема рассмотрена в работах
Джонсона и Мела [20], Аврами [21] и Ивенса [22] с
использованием весьма сходных методов [23]. Во всех этих работах было
принято, что при подавлении рост центров не замедляется,
а прекращается вообще. Математический анализ основан на
замене реальной системы некоторым ее гипотетическим
аналогом, в котором нуклеация может происходить во всем
объеме системы, Это подразумевает существование фиктивных
223
зародышей, которые могут возникать в участках системы, уже
претерпевших фазовый переход.
Общее число зародышей, как фиктивных, так и реальных,
образовавшихся за время dx, определяется соотношением:
dri = N' A -X) dx + N'X dx = N' dx A72)
•
где N'Xdx — число зародышей, которые возникли бы в части
массы X, если бы она перешла в другую фазу.
Удобнее вести расчет массы переходящего в другую фазу
вещества, не учитывая подавление роста, а рассматривая вместо
этого образование и рост фиктивных зародышей. Тогда
объемная доля вещества, претерпевшая фазовый переход, может быть
представлена в форме:
X' = f w (/, т) dnf = [ w (*, т) N' (т) dx A73)
о о
а объемная доля вещества, в действительности претерпевшая
переход, связана с X' соотношением:
-gr-1-X A74)
Строгий математический вывод формулы был дан Аврами
[21]. Физический смысл этого соотношения может быть понят
следующим образом. Рост произвольно выбранного реального
центра замедляется в результате подавления при
соприкосновении с областями, в которых фазовый переход уже произошел.
Поэтому действительная доля массы, перешедшей в другую
фазу за время dt, будет меньше dX\ В связи с тем, что
зародыши, как реальные, так и гипотетические, возникают
беспорядочно по всему объему, то и вкрапления новой фазы будут
также беспорядочно распределены в пространстве.
Таким образом, та доля массы в окрестности растущих
центров, которая способна перейти в новую фазу, равна общей доле
массы, переходящей в кристаллическое состояние. В этом
случае ожидаемое реальное приращение новой фазы определится
уравнением A74).
Интегрируя это уравнение, получаем:
A^l-expf-A") A75)
Подставляя X' из уравнения A73) в A75), имеем:
t 1
f w (/, т) N' (т) dx A76)
о J
X = 1 — ехр
или
X = 1 — ехр
-g- JV(*. т)Ж(т)</т
Рж
О
A77)
224
Таблица 11
Значения показателей степени п для различных
типов нуклеации и роста
Рост
Трехмерный . .
Двумерный . .
Одномерный . .
Гомогенная
нуклеация
линейный
рост
4
3
2
рост,
контролируемый
дуффузией
5/2
2
3/2
Гетерогенная
нуклеация *
линейный
рост
3<л<4
2<л<3
1<л<2
* Определяемая соотношением A82).
Эти выражения определяют закономерности, которым
подчиняются фазовые переходы в однокомпонентных мономерных
системах.
Интегралы A76) и A77) могут быть вычислены, если
известны законы образования и роста зародышей новой фазы.
Это даст возможность определить скорость образования новой
фазы. Если к моменту /=0 устанавливается стационарная
скорость нуклеации, не меняющаяся по мере роста новой фазы,
то jV(t) можно рассматривать как константу.
При таком упрощающем предположении можно найти
аналитические решения уравнения A76) и A77) для различных
процессов роста. Так, для трехмерного линейного роста имеем:
1 я рк
In
1-*- 3 р„
¦ NG4* === kst*
A78)
Показатель степени при / в этой формуле, который в
дальнейшем будет обозначаться п, равен четырем, указывая на то,
что мы имеем дело с гомогенной нуклеацией и трехмерным
ростом.
В случае диска фиксированной толщины /к, растущего ра-
диально по линейному закону:
Рк
In
1^ = 4^^^^
— Л О рж
A79)
Аналогично для одномерно растущей системы находим я = 2.
Величины п при гомогенной нуклеации в системах, где скорость
образования зародышей не зависит от времени и где их рост
линеен, приведены в табл. 11.
Когда рост развивающегося центра контролируется
диффузионными процессами, протекающими в участках системы, не
претерпевших переход, линейные его размеры увеличиваются
пропорционально /,/г. Если учесть эти ограничения, то можно
15 Л. Манделькерн
225
получить соотношения, очень сходные с выведенными выше.
Однако, как это показано в табл. 11, величины показателя
степени при / будут различны при одной и той же геометрии
роста".
Отказ от произвольных ограничений, требующих
однородности и постоянства скорости нуклеации, открывает множество
других возможностей, как и при отказе от постоянства констант
скорости роста. Рассмотрим теперь, к чему приведет отказ от
некоторых ограничивающих условий, наложенных на скорость
роста.
Если, например, предположить, что зародыши образуются в
результате реакции первого порядка с константой скорости
роста v, или что в системе имеется фиксированное число исходных
потенциальных зародышей или неоднородностей с
вероятностью развития v, то число их, остающееся без развития
через время t=v/, задается следующим соотношением [21]:
W = jVexp(— т) A80)
Отсюда скорость нуклеации
N' = Wexp(— т) A81)
Соотношение A77) для случая трехмерного роста может
быть тогда записано в виде:
. 1 8nGW рк Г , „ л . . (v*J . / v/3 \1 /1СОч
Когда параметр vt велик, что означает высокую вероятность
роста всех потенциальных ядер в системе, то формула A82)
сводится к следующему:
IiiT-i-в- = ^ -?*- GW*» A83)
1-Х 3 рж
Это соотношение совпадает с формулой, полученной для
системы с однородной нуклеацией и линейным ростом в двух
направлениях. В другом крайнем случае, когда vt мало,
соотношение A82) примет вид:
In т-Цг = 4- — GMvt* A84)
1 — Л О рж
Оно по форме соответствует соотношению A78). Последнее
приближение означает, что не все зародыши начали свое
развитие в момент / = 0. Вместо этого предполагается, что зародыши
активируются с постоянной скоростью в течение всего процесса
кристаллизации.
В более реалистическом режиме, занимающем
промежуточное положение между двумя рассмотренными крайними
случаями, ядра исчерпываются при некоторой конечной степени за-
226
вершенности перехода. В этом случае показатель степени п
имеет среднее значение между 3 и 4. Легко удостовериться,
что если скорость нуклеации подчиняется формуле A81), в
двух крайних случаях для различных геометрических типов
роста может быть получено соотношение вида:
tojZTjT-W A85)
Как указано в последней колонке табл. 11 при двумерном
росте 2 ^ я ^С 3, а при одномерном 1 ¦< п < 2. Очевидно, что
даже при таких относительно мягких ограничениях, наложенных
на процессы нуклеации и роста зародышей, детали геометрии
роста и способа нуклеации не могут быть определены из
изотерм кристаллизации на основании одной лишь оценки
величины экспонента п. В случае изотерм, подчиняющихся
соотношению A85), все члены зависящие от температуры, заключены
в параметр k, который не зависит от времени /. Поэтому можно
построить координатную систему, абсциссой которой будет
некоторое приведенное время, т. е. параметр, включающий
температуру и истинное время. В этой системе координат кинетика
фазового перехода описывается точно так же, как в • обычной
системе, где по оси абсцисс отложено время.
Следовательно, если построить графики экспериментальных
величин 1 —X в функции от / при различных температурах,
получающиеся изотермы должны обладать свойством
суперпозиции. Однако, если процесс описывается более полным
уравнением A82), изотермы могут быть совмещены простым сдвигом
лишь в том случае, когда отношение G/v не зависит от
температуры.
На основании этих общих положений может быть развита
формальная теория кинетики кристаллизации полимерных
систем. Наблюдаемая на опыте возможность совмещения
изотерм простым сдвигом и вытекающее из нее постоянство
температурного коэффициента скорости кристаллизации дает
возможность полагать, что процессы нуклеации и роста зародышей
происходят параллельно. В свою очередь, обычно наблюдаемое
линейное увеличение радиуса растущего сферолита позволяет
заключить, что рост контролируется диффузионными
процессами, протекающими на границе раздела сферолит — расплав.
Подобные же результаты, при введении этих допущений,
вытекают также и из анализов Джонсона, Мела [20] и Аврами [21].
Однако в теории для мономерных веществ содержится
необходимое предположение о том, что фазовое превращение
происходит до конца. В полимерах абсолютная кристалличность если и
достижима, то очень редко. Поэтому необходимо при
теоретическом рассмотрении учитывать факторы, мешающие
возникновению и развитию кристалличности. Это обстоятельство во многих
15»
227
отношениях усложняет анализ эффекта подавления роста, так
как теперь уже нельзя считать, что не перешедшая за время /
в кристаллическую фазу масса полимера способна
кристаллизоваться.
Для полимерных систем соотношение, аналогичное A74),
может быть записано в виде [6, 24]:
dXT = \-U{t) A86)
dX
где U(t) -—«эффективная доля» массы, претерпевшей фазовый
переход за время t.
Она определяется как та часть общей массы, в которой
дальнейший рост кристаллической фазы невозможен. В эту
величину входят как действительно закристаллизовавшаяся
масса, так и аморфные сегменты цепей, по каким-либо причинам
не способные к кристаллизации в момент t. Предполагается [25],
что перешедшая в другую фазу «эффективная доля»
пропорциональна действительно перешедшей в кристаллическую фазу
массе, причем коэффициент пропорциональности равен 1/1—Л(оо).
Поэтому весовая доля кристаллического полимера в конце
процесса равна 1—Х(оо). Произвольно принимается, что
коэффициент пропорциональности не зависит от времени. При малых
степенях завершенности перехода dXg^dX', и рассматриваемое
дополнительное затруднение устраняется. Если принять
"<'> = T=W <187>
то интегрирование уравнения A86) дает:
tol~V-Mco)]"T^Wy(<) A88)
Так как здесь величина X'(t) та же, что и для мономерных
веществ, можно воспользоваться уравнением A73) и получить:
, , t
1 1 Ркр г •
-1 vTTi 1/ 41=1 ГТ^ГТ^ VQ> *)N(x)dT A89)
1 — Х/[\— Я(оо)] 1 —Л(оо) рж J
о
Чтобы найти зависимость X от времени, снова необходимо
определить детали нуклеации и процессов роста. Если провести
интегрирование A89) при тех же допущениях, что и в случае
низкомолекулярных веществ, можно получить следующее
общее уравнение кинетики:
,п Т=Х/11-Х<ро)] = 1-1 (со) Ы" A90>
Интерпретация показателя степени п здесь та же, что и
ранее, т. е. для его оценки по-прежнему можно пользоваться
табл. 11.
1п-
228
Результаты, вытекающие из соотношений A78) и A90),
могут иметь лишь ограниченное применение, так как при
выводах коэффициент скорости роста G принимался постоянным и
рассматривалась только одна из множества возможных зависи-г
мостей скорости нуклеации от времени. Эти ограничения
следует иметь в виду при сравнении экспериментальных
результатов с теоретическими.
Теоретические изотермы непосредственно дают массу,
перешедшую в кристаллическую фазу, однако этот параметр редко
можно определить из опыта. Для того, чтобы иметь
возможность сравнивать теорию с экспериментом, полученные
соотношения необходимо выразить через непосредственно измеряемые
параметры. Например, если проводится измерение удельного
объема, то соотношение A90) может быть переписано в виде:,
ьт^—вд** <191>
где V0 — начальный объем (объем расплава в момент /=0);
Voo — объем в конце процесса;
Vt — объем в момент t.
Нетрудно вывести аналогичные соотношения и для других
относительно легко измеримых величин, меняющихся в
процессе фазового превращения. Так, если измерить относительное
уменьшение объема в процессе перехода a(t) = (Vo—Vt)/Vo, то
соотношение A91) можно переписать в форме:
Если же степень кристалличности 1—X(t) определить как
(VK-Vt)l{V*-VK)*9 то:
1 —Я@ —[1 —Я (со)] { 1 — ехр [- г_1{оо) Ып] } A93>
Для малых степеней кристалличности соотношение A93)
сводится к 1—%(t)^ktn, что соответствует свободному росту.
На рис. 75 приведены теоретические изотермы, соответствую*
щие уравнению A91) при я=1, 2, 3 и 4. В соответствии;
со сказанным выше, температурно-временной параметр;
1/[1 — %{oo)]ktn обозначен как т; тогда величина т!/",
отложенная по оси абсцисс и пропорциональная времени, и есть то!
«приведенное время», о котором говорилось на стр. 227.
Рассмотрим некоторые характерные черты кривых рис. 75.
Общий вид этих изотерм очень сходен с экспериментальными;
* Это определение 1 — К основывается на предположении, что в
частично кристаллическом полимере удельные объемы кристаллической и
аморфной фазы аддитивны,
229:
кривыми, приведенными в предыдущем разделе. Однако
существуют и определенные различия между этими кривыми,
достаточные, в принципе, для оценки влияния п на ход
превращения. Так, когда п увеличивается от 1 до 4, время, необходимое
для развития определимых
степеней кристалличности,
также возрастает. По
достижении этого времени
скорость кристаллизации
оказывается тем больше, чем
больше я. Все кривые
имеют общую точку
пересечения при т1/п=1; ниже
этой точки происходит
завершение фазового
перехода, и тем быстрее, чем
больше п.
Сходство теоретических
изотерм создает серьезные
затруднения для оценок п
из экспериментальных
результатов (даже если для
системы выполняются все
принятые допущения),
особенно, если надо выяснить,
равно ли п 3 или 4, а может быть даже 2. Это относится
главным образом к ранним стадиям перехода, т. е. к таким
стадиям, на которых получаются наиболее надежные
экспериментальные данные.
Рис. 75. Теоретические изотермы,
рассчитанные по формуле A91), при
п = 1, 2, 3 и 4.
Анализ блочной изотермической кристаллизации
гомополимеров
Проведенное теоретическое рассмотрение позволяет
количественно проанализировать экспериментальные результаты с по-*
мощью уравнений A90) или A91), согласно которым изотермы
просто и полно определяются константой скорости k и
показателем степени п (при /). Кроме того, по'набору совмешающихся
простым сдвигом изотерм можно определить относительные
скорости кристаллизации; для этого нужно определить время, при
котором отношение (!/«> — Vt)/(Voo— Vo) в формуле A91)
достигает какой-нибудь заранее заданной величины. Во многих
случаях оказывается удобным ввести с этой целью некий
«полупериод кристаллизации» ту2; он равен времени, при котором
указанное отношение становится разным 0,5.
230
Для количественного анализа экспериментальных
данных может также использоваться графическое построение
In ln{(l/oo— Vt)l(Voo—Vq)] в функции от In/. Если процесс
описывается той упрощенной теорией, которая предложена в
предыдущем разделе, то из прямолинейного графика,
полученного при таком построении, могут быть определены параметры
п и k. Однако двойное
понижает
чувствительность этого метода к
тонким деталям процесса; в
частности, из анализа
исключается очень важный
интервал времени,
соответствующий развитию
весьма малых степеней
кристалличности.
Более прямым и,
возможно, более детальным
методом сравнения
теоретических и
экспериментальных данных является
построение графиков
(Voo-Vt)l(Voo-Vo) в
функции от lg/,/n при
различных значениях
параметра п.
Экспериментальные данные также
строятся на этом графике с
lg/, откладываемым по
абсциссе. Сдвигая затем
экспериментальные
кривые вдоль горизонтальной
оси до наилучшего
совпадения с одной из
теоретических кривых, можно оценить степень соответствия
теоретических и экспериментальных данных и определить наиболее
вероятные значения величин пик.
На рис. 76 приведены изотермы кинетики кристаллизации
полиэтиленадипината, по данным Такаянаги [8а], с
использованием метода двойного логарифмирования. Каждой температуре
кристаллизации соответствует своя прямая линия. Наклон
линий во всех случаях равен 4. Все это говорит о том, что процесс
в данном случае хорошо описывается уравнением A91).
«Буквальная» интерпретация найденной величины п = 4 означает,
согласно табл. 11, что в этой системе происходит с постоянной
скоростью спорадическая нуклеация с параллельным трехмер*
ным линейным ростом.
логарифмирование неминуемо резко
i^t.MUH
Рис. 76. Зависимость log [log A/д)] от
времени для фракционированного
полиэтиленадипината с М = 9900 [8а].
На изотермах показана температура кристаллизации
231
Изотермы для натурального каучука имеют сходную форму
независимо от давления или температуры кристаллизации [25].
zoo т доо woo 3200 бш
2000 то 8000 16000 32000
Время, мин
Рис. 77. Зависимость (V^— Vt)l(V^ — vt) 0T времени для
натурального каучука при —20° С (а) и 0° С (б) и давлениях
(кгс/см2), указанных на кривых [25].
Некоторые типичные результаты приведены на рис. 77.
Суперпозиция этих изотерм продемонстрирована на рис. 78.
Экспериментальные точки соответствуют изотер-
^ мам, полученным при определенных
температурах и давлениях. Произвольная
временная шкала выбрана так, чтобы
совмещение точек на одной кривой было
наилучшим. Линия, проведенная
соответственно формуле A91), показывает, что
большая часть перехода подчиняется
рассмотренной теории. В этом случае я = 3.
Отклонения от теории начинаются в
конце перехода, когда скорость
кристаллизации понижается, по сравнению
с предсказываемой. Это несоответствие
начинает проявляться при степени
кристалличности порядка 25%.
Анализ кинетических исследований,
выполненных на целом ряде других
полимеров [4, 6—10, 14, 15, 26—30], также
выявляет хорошее согласие с уравнением
A91). Таким образом, приведенные выше
результаты можно рассматривать как типичные для
кристаллизации гомополимеров.
Рис. 78. Пример
совместимости изотерм
натурального каучука [25].
д-0° С, 0 кгс/см*\ П-0° С,
ВО0кгс/см2; О-25° С
400 кгс/см2; сплошная линия
проведена согласно
соотношению A91) при л = 3.
232
Более тщательное сравнение теоретических и
экспериментальных данных можно осуществить при сопоставлении
исходных значений для двух фракций линейного полиэтилена
различного молекулярного веса.
Изотермы каждой из фракций по-прежнему совместимы
(хотя изотермы различных фракций уже не совместимы). На
рис. 79 приведены типичные изотермы для М = 24 000 и 360000
совместно с кривыми, рассчитанными по формуле A91) при
% о,ьо
0,60
0/,0
020
Рис. 79. Зависимость (V^—V^KV^—Vq) от log/ узких
фракций линейного полиэтилзна [31].
а-/И = 24 00>, <5-М = ЗСО0ОО.
Пунктирные линии, соответствующие уравнению A91), сдвинуты по оси
log / до наилучшего совпадения с экспериментальными.
/2 = 2 и /2 = 3 [31] и дающими наилучшее совпадение с
экспериментальными кривыми. Для фракции с М = 24 000 кривая /2 = 3
лучше описывает в. приведенных («нормализованных»)
координатах переход до степени завершенности порядка 7з, что
соответствует степени кристалличности около 25%. Дальше процесс
развивается со скоростью, заметно меньшей, чем
предсказывается теорией. Что касается фракции с М = 360 000, то здесь
практически невозможно сделать выбор между /2 = 2 и /2 = 3.
Отклонения от теории того же типа, что у первой фракции также
начинаются по достижении степени кристалличности
приблизительно 20—25%. Очень похожие результаты для фракции
линейного полиэтилена в той же области молекулярных весов
опубликованы Гордоном с сотрудниками [31а].
Очевидно, что форма изотерм для фракций с различными
молекулярными весами должна быть различна. Это
продемонстрировано на рис. 80 для двух уже рассмотренных фракций
полиэтилена ц для нцзкомолекулярной фракции с М = 5300.
1 1 1 1 1—
» \
/?¦-<? 11
,. 1
б
¦—,-,. -1,-.
V
V
At
\1
1
Ф
\v
1 \
233
- 1.0
0.6 \
OA
Изотермы для каждой фракции сдвинуты вдоль горизонтальной
оси так, чтобы добиться их максимального совпадения. Полимеры
с очень короткими цепями обладают рядом уникальных
особенностей, которые рассмотрены в следующем разделе. Здесь мы
сосредоточим внимание на двух высокомолекулярных фракциях.
Хотя различия в форме изотерм и незначительны, они все же
вполне отчетливы и поэтому должны приниматься во внимание
при полном анализе. Вполне
очевидно, что кристаллизация более
высокомолекулярной фракции протекает
медленнее [32].
Во многих исследованиях
показано, что высокомолекулярным
фракциям соответствует меньшая степень
кристалличности C3—35]. (Заметим,
что изотерма очень
низкомолекулярной фракции имеет совершенно
другую форму и соответствует
максимальному — из трех сравниваемых
процессов — запаздыванию
кристаллизации). Чтобы эти данные укладывались
в рамки предлагаемой теории,
влияние молекулярного веса на кинетику
кристаллизации должно
непосредственно проявляться на стадиях ну-
клеации или роста, или на обеих
сразу. Хотя при сравнении теоретических
и экспериментальных данных мы
фиксировали внимание на различного
рода несоответствиях, все же нельзя
игнорировать обычно наблюдаемое хорошее согласие между
опытом и теорией на разных стадиях фазового перехода. Это
согласие приобретает особое значение, если учесть те
жесткие упрощающие предположения, которые были приняты при
выводе соотношения A91).
Проведенное теоретическое рассмотрение позволяет
адекватно истолковать особенности формы изотерм и вполне
удовлетворительно объясняет их повсеместно наблюдаемую
способность к суперпозиции. Поэтому имеются все основания для
вывода, что, как и в случае низкомолекулярных веществ,
основными механизмами, определяющими развитие кристалличности
в полимерах, являются нуклеация и процессы дальнейшего
роста. Кинетика кристаллизации полимеров подчиняется самым
общим математическим законам фазовых переходов [20, 21].
Следовательно, кинетический анализ кристаллизации
согласуется и дополнительно подкрепляет выводы, сделанные при
непосредственном изучении процесса плавления, а именно —
let
Рис. 80. Сравнение формы
изотерм кристаллизации для
фракций линейного
полиэтилена различного
молекулярного веса:
/-М-5300; 2-Ж = 364 000;
3-Ж = 2400.
234
плавление и кристаллизация полимеров являются типичными
фазовыми превращениями. То обстоятельство, что переход от
жидкой фазы к кристаллической в полимерах, как правило, не
достигает полного завершения, не мешает рассматривать этот
процесс с позиции фазового равновесия [36].
Отмеченные выше различия между теорией и экспериментом
требуют дальнейшего исследования. Можно сделать общий
вывод: наблюдаемая скорость кристаллизации меньше
предсказываемой. В тех системах, которые были исследованы детально,
заметное запаздывание кристаллизации начинается по
достижении степени кристалличности порядка 25—30%. Более того,
форма изотерм зависит от молекулярного веса (для фракций);
чем выше молекулярный вес, тем более длителен процесс
кристаллизации.
Следовательно, необходимо пересмотреть упрощающие
допущения, сделанные при выводе соотношения A91). В связи
с тем, что все наблюдаемые отклонения от теории явно зависят
от степени кристалличности, достигнутой к определенному
критическому моменту, необходимо сосредоточить внимание на
возможности, что нуклеация и скорость роста могут зависеть от
степени завершенности перехода.
Существенной особенностью математического рассмотрения
было молчаливое допущение, что N и G не зависят от
продолжительности фазового перехода. Это допущение было сделано,
в основном, для упрощения математического анализа. Кроме
того, оно основывалось на том, что для гомополимеров высокого
молекулярного веса структура расплава остается неизменной по
мере развития фазового перехода. Однако именно это
предположение далеко не всегда справедливо и структура расплава
может меняться.
Действительно, в процессе кристаллизации аморфные
участки цепей конечного размера оказываются отделенными друг
от друга кристаллическими последовательностями звеньев.
Некоторые из этих изолированных участков из-за малости
размеров уже не могут принимать участия в актах нуклеации (см.
стр. 238). В результате эффективная концентрация мономерных
звеньев, способных кристаллизоваться, по мере развития
процесса постепенно уменьшается. Следовательно, коэффициент
пропорциональности, введенный в уравнение A87), не
обязательно является константой. Целесообразно рассматривать его
как функцию степени завершенности кристаллизации, причем
U{t) растет со временем быстрее, чем X, Введение этого
обобщения в теорию должно привести к лучшему согласию с
опытом.
Количественный анализ проблемы изоляции аморфных
участков (при кристаллизации) единичной полимерной цепи, хотя
и не применим непосредственно к трехмерной кристаллизации,
235
все же показывает, что ~25% мономерных звеньев «выпадают»
из процесса, если они расположены в аморфных
последовательностях ограниченного размера [37].
Кроме перечисленных факторов, проявляющихся
исключительно при кристаллизации цепных молекул, необходимо учесть
и некоторые другие. Предположение о линейной скорости роста
хотя и следует из прямых наблюдений за ростом сферолитов
при малых степенях кристалличности, но требует пересмотра,
когда степень кристалличности становится значительной. По
мере уменьшения концентрации аморфного материала
диффузия полимерных сегментов к поверхности раздела кристалл —
жидкость может стать основным процессом, определяющим
скорость кристаллизации. В этом случае радиус развивающейся
области начнет увеличиваться пропорционально квадратному
корню из времени, и процесс кристаллизации соответственно
замедлится. Поскольку коэффициент диффузии макромолекул
в расплаве зависит от молекулярного веса, ясно, что он должен
влиять как на скорость кристаллизации, так и на форму
изотермы.
Кроме того, если нуклеация катализируется конечным
числом неоднородностей, которые спорадически активируются с
течением времени, давая начало росту кристаллических областей,
то расходование этих неоднородностей до окончания процесса
должно привести к теоретическим изотермам, соответствующим
формуле A82). При правильном выборе скорости гетерогенной
нуклеации можно рассчитать изотермы, которые совпадают с
наблюдаемыми на опыте.
Таким образом, существует целый ряд причин, связанных
с тонкими физическими деталями реального процесса
кристаллизации и вызывающих отклонения от предложенных выше
простых зависимостей. Возникновение серьезных расхождений
можно ожидать по достижении достаточно высоких кристаллич-
ностей. Следовательно, именно эта стадия фазового перехода
нуждается в значительно более тщательном теоретическом
анализе.
Если предположить, что одно первичное ядро дает начало
одному сферолиту, то при прямом измерении скорости
нуклеации и скорости роста сферолита, по соотношению A78) может
быть определена константа скорости кристаллизации ks. Этот
параметр можно сравнить с соответствующей величиной, полу*
чаемой из измерений валовой скорости кристаллизации. При
этом для ряда систем наблюдалось очень хорошее совпадение
величин [11, 38]. В табл. 12 приведены результаты, полученные
для полидекаметиленсебацината. На основании этих данных
можно сделать вывод о том, что процессы образования и роста
сферолитов в гомополимерах тесно связаны с общим развитием
кристалличности.
236
Таблица 12
Константы скорости кристаллизации ks
для полидекаметиленсебацината |11а]
т, °с
72,6
71,6
70,7
69,7
68,6
67,7
66,7
ks (определена
дилатометрически)
5,51
4,30
4,32
1,00
2,38
1,28
1,50
КГ15
106
10"!п
10"°
ю-8
10~5
.кг4
г, °с
72,0
71,1
70,1
69,1
68,1
67,1
k$ (определена
микроскопически
по росту
сферолитов)
2,оз- ю-!!
6,84-10
1.56-10"*
5,00-10"'°
7,85-10 8
2,02-10 5
Температурный коэффициент
кристаллизации гомополимеров
В связи с тем, что изотермическая кристаллизация
полностью описывается при сделанных предположениях о механизмах
нуклеации и роста, необычайно высокий температурный
коэффициент скорости кристаллизации должен, естественно,
определяться температурными коэффициентами скорости этих двух
процессов. Поэтому займемся каждым из температурных
коэффициентов в отдельности.
Если две фазы однокомпонентной системы находятся в
равновесии при Тпл, и фаза В имеет более низкую свободную
энергию при температурах ниже Гпл, то понижение температуры
вовсе необязательно вызовет спонтанный рост этой фазы. Перед
образованием макроскопической новой фазы процесс должен
пройти через стадию, когда система содержит относительно
малые частицы растущей фазы. При этом имеется возможность
сосуществования в равновесии старой фазы А и вкрапленных в
нее мельчайших областей фазы В при температурах ниже ГПл,
в связи с тем, что понижение термодинамического потенциала,
которое характеризует нормальное развитие макроскопической
фазы в объеме, может быть скомпенсировано вкладом
поверхностной энергии малых частиц. Следовательно, условия
стабильности определяются относительными вкладами энергий
поверхности и объема частицы новой фазы.
Таким образом, нуклеациго можно определить как
зарождение новой фазы внутри исходной, а зародыши — как малые
структурные элементы растущей фазы. Зародыши могут
возникать однородно (гомогенно) из-за статистических флуктуации
237
в исходной фазе; однако этот процесс может катализироваться
присутствием в системе неодиородностей *.
До обсуждения проблемы нуклеации в полимерных системах
сначала рассмотрим формальную теорию нуклеации для
низкомолекулярных веществ. Свободная энергия образования в
гомогенных условиях сферического зародыша радиуса г из расплава
низкомолекулярного вещества может быть выражена
следующим образом:
Д/7 = — -i лг3 &Fy + 4яг2у A94)
где &FV — изменение объемной свободной энергии на единицу
объема;
у — свободная энергия на единицу площади.
Графическое изображение этой функции дано на рис. 81. По
мере увеличения радиуса зародыша А/7 возрастает, достигая
максимума при г=г*. Затем свободная энергия резко падает и
становится отрицательной. В максимуме
г = /** =
2а
А/\,
bF = bF*--
V
16 л; а3
A95)
A96)
j (A/VJ
Используя обычное приближение
AFV = ДЯУ (Гпл—Т)/ТПЛу эти
отношения можно переписать в виде:
2оТпл
Рис. 81. Схематическая
диаграмма изменения
свободной энергии образования
сферического зародыша.
/•• =
АНуМ
AF* = -
16я
3 (АЯ^J (Д7У
A97)
A98)
Здесь Гпл — Т отождествлено с ДГ и характеризует степень
переохлаждения.
Необходимые условия термодинамической стабильности
достигаются при Д/г=0, так что
За
А/\,
A99)
* [В сущности, зародыши представляют собой типичные гетерофазные
флуктуации; однако, из-за цепного характера полимерных молекул и
высокой вязкости среды (расплава), время их жизни может на несколько
порядков превосходить время жизни гетерофазных флуктуации в
низкомолекулярных системах. Поэтому их легко наблюдать. По-видимому, даже при
развитии жидкокристаллической фазы в растворах некоторых спиральных
макромолекул образованию трехмерного порядка предшествует появление
«жидких сферолитов», которые можно трактовать как гигантские
гетерофазные флуктуации. — Прим. редактора].
238
Зародыши, меньшие, чем г*, нестабильны и исчезают, так как
А/7 возрастает с увеличением г. Однако как только радиус
зародыша превысит критическую величину, размер его может
увеличиваться беспредельно, так как теперь AF уменьшается с
увеличением г. Таким образом, А/7* представляет собой
энергетический барьер, после преодоления которого развивается устойчивая
новая фаза.
Сущность классической теории нуклеации, как это следует
из соотношения A94), сводится к определению свободной
энергии А/7 образования зародыша в гомогенных условиях. При этом
рассчитывается вклад структурно совершенной фазы конечного
размера в А/7 и к этому вкладу добавляется избыточная
поверхностная свободная энергия, обусловленная появлением границы
раздела между фазами *. Эта классическая концепция,
принадлежащая Гиббсу [39], подразумевает, что размер зародыша
достаточно велик, так что он однороден внутри, а его внешняя
поверхность имеет вполне четкие границы. При очень больших
переохлаждениях, когда AT велико, радиус зародыша достигает
атомарных размеров. При этом определенные выше условия уже
не выполняются, и возникает необходимость в более сложных и
неочевидных теориях [40, 41]. Однако, несмотря на эти
осложнения, классическая теория нуклеации имеет широкое применение.
Простота приведенного анализа в значительной мере
является следствием того, что при рассмотрении нуклеации мы
исходили из сферической симметрии зародышей. Полимерные же
молекулы скорее всего образуют зародыши асимметричной формы.
Естественно, поэтому, обсудить проблему образования
зародышей цилиндрической формы в низкомолекулярных веществах.
Можно также рассмотреть и другие геометрические формы
зародышей, однако результаты при этом будут отличаться только
некоторыми постоянными коэффициентами.
Свободную энергию образования цилиндрического зародыша,
содержащего р молекул в поперечном сечении и ? молекул по
длине цилиндра, можно записать в виде:
^F = 2? Ущ аб + 2рат - ?р /±fu B00),
где А/м — свободная энергия плавления на молекулу;
аб—поверхностная свободная энергия боковой
поверхности цилиндра на молекулу;
* С точки зрения развиваемой здесь теории, совершенная фаза обладает
самой низкой свободной энергией (или самым низким термодинамическим
потенциалом), возможной при тех ограничениях, которые были наложены на
систему. Следовательно, наличие дефектов равновесного типа, например
вакантных мест в решетке, учитывается автоматически. Возможность
существования в макроскопическом кристалле или кристаллите дефектов
неравновесного типа, способных, в определенных условиях, развиваться, не имеет
отношения к существу рассматриваемой проблемы.
239
ат—поверхностная свободная энергия торца цилиндра на
молекулу.
Свободная энергия деформации и свободная энергия краев
цилиндра считаются пренебрежимо малыми [42]. Свободная
энергия, представленная соотношением B00), имеет
экстремальную точку (седловину). Координаты этой точки могут быть
определены из.равенства:
(дД/7<Э?)р = 0 и (дДА7др)с«0
Из этого условия могут быть определены критические
размеры кристаллического зародыша:
B01)
B02)
В экстремальной точке
Snal a_
Для удобства дальнейшего рассмотрения введем ряд новых
безразмерных переменных:
р~=4-
р*
р* =
?* =
= AF*
4ла26
А/2м
4ат
8 лаб ат
д/7 =
АЛ
B04)
Соотношение B00) при этом можно переписать в виде:
bF = 2Zpb + p—2lp B05)
На рис. 82 приведено графическое_изображение этого
соотношения при постоянных значениях AF. Энергетический барьер,
который необходимо преодолеть, определяется из условия Д^=1
и находится в экстремальной точке ? = р=1. Стабильное
состояние зародышей достигается при пересечении контурной линии
А^=0.
Таким образом, для достижения стабильности зародыша
требуется увеличение его размеров, по сравнению с критическими.
В экстремальной точке начинается большое число различных
путей перехода к устойчивому состоянию. Однако, следование по
некоторым из этих путей бесплодно.
Из рис. 82 видно, что независимо от величины ?р должно
быть больше единицы, чтобы AF могло Достигнуть отрицатель-
24Q
ных значений. С другой стороны, ? может быть фиксированным
и рарным единице, но стабильность будет все же достижима
при р, большем 4. Следовательно, для стабильности достижение
сверхкритического размера в направлении ?, не обязательно.
Обязательное условие образования стабильности зародыша,
следующее из соотношения B05), сводится к тому, чтобы ? превы-
Рис. 82. Контурные диаграммы, построенные в соответствии с
уравнением [205], для различных постоянных значений &F = AF/&F*.
шало V2, даже если в поперечном направлении возможен
неограниченный рост. Хотя невозможно предсказать a priori,
единственный путь превращения асимметричного зародыша
критического размера в термодинамически устойчивый кристаллит,
всегда имеется возможность определить запрещенные варианты
этого пути.
До сих пор мы ограничивались рассмотрением однородной
нуклеации, при которой единственной причиной возникновения
зародышей являются статистические флуктуации в расплаве.
Однако хорошо известно, что инородные тела, границы
включений и пустоты могут катализировать процесс нуклеации [43—45].
JQ Д. Манделькерн
241
В работе [46] было показано, что в пустотах потенциальные
центры нуклеации могут сохраняться даже при температурах
выше точки плавления. При наличии в системе различных не-
однородностей частота нуклеации зависит от термической
предыстории образца, если только при рассматриваемой
температуре не происходит полного разрушения всех
потенциальных зародышей. При охлаждении образца ниже температуры
плавления зародыши все еще могут возникать как в пустотах,
так и на поверхности инородных включений присутствующих в
системе. В отсутствие потенциальных зародышей выражение
для А/7,,, свободной энергии неоднородного образования
зародышей, качественно сходно с выражением для свободной
энергии однородной нуклеации. Для рассматриваемых случаев
показано, что А/^н¦= /(в) А/7 , где функция /(8) принимает
значения между нулем и единицей. Зависимость свободной энергии
от температуры при обоих способах нуклеации одинакова, хотя
при неоднородной нуклеации расходуется меньшая свободная
энергия.
Другой подлежащий рассмотрению тип зародышей
возникает уже на развившейся кристаллической грани;
следовательно, этот тип нуклеации связан с образованием своего рода
монослоя при одновременной адсорбции на этой грани достаточно
большого числа молекул. Свободная энергия образования
такого зародыша шириной в р мономерных звеньев и длиной в ?
звеньев равна:
А/7 = 2атр + 2?аб — р? bfM B06)
Критические условия для этого двухмерного зародыша
определяются соотношениями:
B07)
B08)
B09)
Отметим, что ?г соответствует минимальному размеру, при
котором развивается термодинамически устойчивый кристалл,
даже если поперечные размеры растут неограниченно.
Следовательно, в любом реальном процессе кристаллизации
при поверхностной нуклеации такого типа для образования
устойчивого кристаллита продольные размеры (?) должны
превышать критическую величину &. Этим двумерная нуклеация
отличается от трехмерной, где устойчивость может быть
достигнута без превышения ?*¦
242
р2 =
Ь2 =
AF2 =
2аб
А/м
2ат
AfM
AfM
Используя теорию переходного состояния, Торнбалл и
Фишер [47] получили выражение для скорости нуклеации в
конденсированных системах. Равновесная скорость нуклеации на
единичный объем с достаточной степенью точности задается
соотношением:
N = N0 ехр ( ^ ] B10)
где ED — свободная энергия активации переноса через
поверхность раздела жидкость —зародыш;
N0 — rtikT/h;
пх — число молекул в единице объема жидкостей.
Это выражение для скорости имеет ряд уникальных свойств,
отличающих его от обычных соотношений для активационных
химических процессов. Напомним, что Д77* обратно
пропорционально \/(Тпл~ТJ, поэтому из соотношения B10) следует, что
скорость нуклеации равна нулю при абсолютном нуле и Гпл и
имеет максимальное значение .при некоторой промежуточной
температуре. При температурах немного ниже Гпл скорость
нуклеации имеет очень большой отрицательный температурный
коэффициент в основном из-за составляющей ехр (—AF*/RT).
Дальнейшее понижение температуры вызывает медленный рост
скорости нуклеации, затем она достигает максимума и начинает
падать. При температурах ниже максимума, поскольку в
экспоненте начинает доминировать энергия активации переноса,
температурный коэффициент становится положительным.
Температурная зависимость процесса нуклеации, вызываемого
присутствием инородных включений или поверхностных неоднород-
ностей, имеет тот же характер, что при однородной нуклеации,
отличаясь лишь численным коэффициентом.
Высокая чувствительность скорости нуклеации к степени
переохлаждения или пересыщения в многокомпонентных
системах хорошо установлена на мономерных веществах. Например,
Торнбалл [48] показал, что образец ртути может быть выдержан
в течение 1 ч при переохлаждении в 43 град без каких-либо
фазовых изменений, однако дальнейшее охлаждение всего на
3 град приводит к отвердеванию образца в течение 1 мин.
Подобная же зависимость скорости кристаллизации от
температуры наблюдается, как уже отмечалось, и для полимерных систем.
Поэтому вполне уместно предположить, что теория нуклеации
может быть использована для объяснения наблюдаемых
температурных коэффициентов скорости кристаллизации полимеров.
При разработке теории нуклеации, применимой к цепным
молекулам, используются те же формальные приемы, что и при
выводе основных соотношений для мономерных веществ.
Избыточная поверхностная свободная энергия, соответствующая
выбираемой геометрии роста, складывается с объемной свободной
16*
243
энергией, необходимой для образования равновесной
кристаллической фазы конечных размеров. Однако в случае полимеров
конечного молекулярного веса необходимо знать не только
общую геометрию роста, но и способ, которым полимерная цепь
уложена внутри кристаллита. Поэтому модель зародыша
должна быть специализирована, по сравнению с
низкомолекулярными системами.
Выше было показано, что модель кристаллита, в которой
разные участки одной и той же полимерной цепи принадлежат
различным кристаллитам, исключая тем самым возможность
многократного вхождения цепи в один кристаллит, непригодна
в большинстве реальных случаев [49]. Это вытекает из стериче-
ских требований, налагаемых на кристаллиты больших
поперечных размеров *. Однако указанные ограничения не
распространяются на кристаллические участки, состоящие из небольшого
числа цепей; следовательно, такая модель может быть
использована при рассмотрении процесса нуклеации.
Свободная энергия образования зародыша из пачки,
содержащей N полимерных цепей, каждая из которых состоит из
х мономерных звеньев, задается соотношением [24, 50]:
где ?— число мономерных звеньев вдоль образующей
зародыша цилиндрической формы;
р—число моиомерных звеньев в поперечном его сечении;
стт — избыточная свободная энергия, приходящаяся на одно
звено, выходящее из грани кристаллита,
перпендикулярной направлению полимерной цепи.
Соотношение B11) сложно и мало пригодно для анализа.
Однако в случае больших молекулярных весов, для которых
х —юо и ?<С*, это соотношение можно записать в более простой
форме:
^F = 2?аб (ярI'* + 2рст - ?р А/м B12)
Видно, что в такой форме это выражение полностью
совпадает с соотношением B00). Таким образом, для выбранной
модели свободная энергия образования зародыша из полимеров
бесконечно большого молекулярного веса равна свободной
энергии образования зародыша из эквивалентного количества
«свободных» мономерных звеньев при одном и том же способе их
расположения. Поэтому свободные энергии в обоих случаях
одинаковы, и р*, ?* и Д/7* для рассматриваемого случая задаются
* Это важное заключение выведено и детально проанализировано в гл. 9.
244
соотношеними B01), B02) и B03). В этом приближении цепной
характер молекулы не играет никакой роли, так как свободной
энергией взаимодействия концов молекулы можно пренебречь.
Может быть рассмотрена и другая модель зародышей, в
основе которой лежит наблюдаемый при кристаллизации из
разбавленных растворов пластинчатый монокристалл или ламел-
лярная структура блочных кристаллических гомополимеров.
Принимается, что зародыш состоит из абсолютно регулярно
сложенных полимерных цепей [51]. Никаких ограничений или
условий относительно расположения концов молекулы не вводится.
Поэтому из предшествующего анализа сразу следует, что эта
модель совпадает с асимметрично организованным набором
свободных мономерных звеньев, а цепной характер макромолекулы
снова никак не проявляется. Неудивительно, что, как и выше,
свободная энергия образования зародыша задается
соотношением B00), а критические параметры системы соотношениями
B01), B02) и B03). Период складывания (в ядре) может быть
отождествлен с ?, в то время как р соответствует числу
кристаллических последовательностей, проходящих через поперечное
сечение.
Таким образом, предложенные до настоящего времени
теории первичного акта нуклеации, строго говоря, применимы
только для полимеров бесконечно большого молекулярного веса.
Все теории формально совпадают как друг с другом, так и с
классической теорией нуклеации низкомолекулярных веществ.
Единственные различия могут быть связаны со значениями
поверхностной свободной энергии различных моделей на границе
раздела фаз. Эти величины не поддаются оценке при любом
теоретическом расчете. В связи с тем, что для полимеров
поверхность свободной энергии, изображенная на рис. 82, по-прежнему
определяет условия кристаллизации, ограничения,
накладываемые на переход от зародыша критического размера к
устойчивому кристаллиту, остаются в силе и для полимеров.
Характерная особенность всех теорий нуклеации — сильная
зависимость критических размеров зародыша от степени
переохлаждения. Соотношения B02) и B01) можно переписать
в виде:
С—ОТГ&- B13)
9'=-щ^ B14)
Графическое изображение этих соотношений при различных
параметрах от/АНм и аб/АЯм приведено на рис. 83 и 84. Хотя
?* и р* зависят от соответствующих отношений свободной
энергии поверхности раздела к теплоте плавления, качественный
245
характер зависимости этих параметров от температуры
малочувствителен к их величине. При Гцл критические размеры
бесконечны и резко, хотя и непрерывным образом, уменьшаются при
понижении температуры. Большим степеням переохлаждения
(АГ порядка 40—50 град для рассматриваемого случая)
соответствуют очень малые критические размеры, которые почти
нечувствительны к изменениям температуры. Однако не вполне
Ю 30 50 70 90 W 130
т;с
Рис. 83. Зависимости ?* от Т, построенные по
уравнению B13).
Указаны значения отношения о /Д#м; Т'пд8*13^0 С.
ясно, в какой мере справедливы выводы классической теории
нуклеации при столь малых размерах полимерных зародышей,
возникающих при очень больших переохлаждениях. Из
соотношений A98) следует, что Д/7* также очень сильно зависит от
температуры, оставаясь при этом непрерывной функцией.
Дальнейшее обсуждение должно подчеркнуть важность
процесса нуклеации для развития кристаллической фазы в
полимерных системах, хотя оно не решит и не .объяснит всех
поставленных проблем. В частности, необходимо понимать, что нет
никаких априорных причин, по которым структура критического
зародыша, будь то полимерного или низкомолекулярного, в
какой-либо степени должна отражать свойства возникающего
в конечном счете кристаллита.
246
Основные выводы теории нуклеации мономерных веществ
были экспериментально подтверждены при исследовании
кристаллизации металлов [52, 53], простых молекулярных
жидкостей [54, 55] и низкомолекулярных я-алканов [56]. Найденные
зависимости скорости нуклеации от степени переохлаждения
находятся в очень хорошем соответствии с соотношением B10) и
доказывают применимость классической теории нуклеации к
переходу жидкость — кристалл.
Применимость классической
теории нуклеации к процессам
(TJWIMHO2
9
го w бо до юо по
гХ
Рис. 84. Зависимости р*
от 7\ построенные по
уравнению B14).
Указаны значения отношения
°б/д"м;гпл=,37'5°с-
2 3 * S.
(Ш№)
Рис. 85. Зависимость log N
от (Т2пл/Т) A/A7f (/) и
(Тпл/Т) A/М) B) для поли-
декаметиленсебацииата
[Па].
кристаллизации полимерных систем в первую очередь
доказывается исследованием температурных коэффициентов скорости
образования и роста сферолитов и общей скорости
кристаллизации. Можно воспользоваться соотношением B10), понимая под
tii концентрацию полимерных сегментов в единице объема.
Тогда из соотношений A98) и B10) можно найти стационарную
скорость однородной нуклеации:
N==N0 exp
Li»
I. RT
RT A//* (MJ
Для удобства перепишем это соотношение в виде:
N = N0 exp
kfi
RT Т(ЬТ)*ш
B15)
B16)
При этом сегменты играют ту же роль, что молекулы, из
которых образуются молекулярные кристаллы мономерных веществ.
Для экспериментальной проверки соотношения B15) можно
воспользоваться результатами Флори и Мак Интайра по
247
определению скорости образования центров сферолитов в
расплавленном полидекаметиленадипинате [Па]. При малых значениях
ДГ определяющую роль в соотношении B15) играет второй член.
Следовательно, в этом температурном интервале зависимость
\gN от (Т1Л/Т)A/&ТJ должна иметь вид прямой линии.
Поэтому, если измеряемая на опыте скорость тождественна
величине N, получается прямая линия, изображенная на рис. 85.
В этих опытах скорость нуклеации изменяется на четыре
порядка в области переохлаждений 11 — 16 град. Следовательно,
большой отрицательный температурный коэффициент, наблюдаемый
вблизи Гпл, вполне может быть объяснен в терминах
классической теории нуклеации. #
На рис. 85 по тем же данным построена зависимость \ogN
от (Тил/Т) A/АГ); снова получается прямая линия. И та и
другая зависимости согласуются с экспериментальными данными
при достижимой точности определения равновесной температуры
плавления. Это справедливо для большинства, если не для всех,
опытных данных по определению температурного коэффициента
процесса кристаллизации полимеров. Модель простого
трехмерного зародыша, описываемая соотношениями B04), приводит
к линейной зависимости log iV от (T^JT) A/ДГJ. Линейная
зависимость log jV от (TnJT) A/Д7) также теоретически допустима
и может быть объяснена либо образованием монослойных
зародышей [см. соотношение B09)], либо тем, что трехмерный
зародыш имеет крайне большую «линейную» свободную энергию
ребер или краев [42, 51].
Развитие сферолитов-при монослойной нуклеации возможно
только в очень редких случаях при наличии весьма специального
типа неоднородностей. Поэтому, очевидно, в большинстве
случаев первичный акт нуклеации заключается в образовании
трехмерного зародыша. Более того, если только различные
избыточные свободные энергии не имеют аномальных значений,
логарифм скорости нуклеации прямо пропорционален A/А71J.
Выше было показано, что линейная скорость роста
сферолитов для полимеров самых различных типов вблизи Гпл
характеризуется отрицательным температурным коэффициентом. Для
всех полимеров, исследованных в широкой области температур,
максимальная скорость роста сферолитов наблюдается
приблизительно при тех же температурах, что и максимальная валовая
скорость кристаллизации. Из приведенных выше данных
следует, что рост сферолитов контролируется процессами вторичной
нуклеации. Поэтому необходимо различать кристаллиты,
возникающие в результате первичной нуклеации, и кристаллиты,
приводящие к увеличению размеров сферолитов.
248
В принципе информацию относительно актов вторичной
нуклеации, и тем самым о деталях механизма роста, можно
получить, исследуя температурный коэффициент скорости роста
сферолитов. Для гомополимеров скорости линейного роста
сферолитов вблизи Тпл подчиняются соотношению вида:
\g G = const -
«
ЯТ(ТПЛ-ТГ
B17)
(Ur)(l/M)!0*
10
4 5
где т=1 или 2, а С{ определяет вклад свободной энергии
поверхности раздела и теплоты плавления.
К сожалению, точность опытных данных не позволяет
сделать выбор между двумя значениями /п. Типичный пример
приведен на рис. 86 для полидекамети-
ленсебацината. Видно, что оба типа
зависимости от температуры
выражаются прямыми линиями. Точно
такие же результаты получены для всех
исследованных систем [4]. Для
решения этого важного вопроса
необходимо более точное определение скоростей
роста и значения Тпл.
В связи с тем, что рост сферолита
включает образование новых
кристаллитов вблизи уже существующих,
можно предположить, что вновь
возникающие кристаллиты зарождаются на
поверхности старых [18, 57]. Это
предположение приводит к
экспоненциальной зависимости скорости роста от
(Тпл/Т) A/АГ), поскольку нуклеация
в этом случае должна включать образование
мономолекулярных слоев полимерных сегментов. Однако при таком процессе
нуклеации необходимо, чтобы была обеспечена возможность
последующего роста в направлении оси цепей. В противном
случае, как уже указывалось, размер образующегося кристаллита
не сможет обеспечить его устойчивости.
Но кроме рассмотренной существует также возможность
трехмерной вторичной нуклеации. Присутствие ранее
возникшего кристаллита может определенным образом влиять на
ориентацию и организацию соседних аморфных областей и тем
самым способствовать нуклеации на одном из смежных
участков. При этом критическое значение свободной энергии,
необходимое для вторичной нуклеации, должно быть меньше, чем при
первичной однородной нуклеации. Этот механизм согласуется
с экспериментальными данными, приведенными на рис. 85 и 86.
Наклон линии, представляющей процесс трехмерной нуклеации
Рис. 86. Зависимость \g G
от (Т2ПЛ/Т) A/ДГJ (/) и
(Тпл/Т) A/Д7-) B) для поли-
декаметиленсебацината
[Па].
249
на рис. 86, составляет приблизительно половину
соответствующего наклона линии на рис. 85. Рассмотрение этих типичных для
полимеров результатов ясно показывает, что процесс нуклеации
контролирует скорость роста сферолитов. Однако такие
кинетические исследования не дают никакой информации о детальном
строении самих зародышей.
Чтобы найти выражение температурного коэффициента
валовой скорости блочной кристаллизации, необходимо определить
температурные зависимости процессов первичной и вторичной
нуклеации. Первичная нуклеация может быть представлена
соотношением типа B16), тогда как способ вторичной нуклеации
(происходящей при росте сферолитов), как мы убедились, не
может быть определен однозначно. Следовательно, при выборе
выражения, описывающего весь процесс, всегда будет
существовать некоторая неопределенность.
В качестве примера рассмотрим трехмерную вторичную
нуклеацию, которая не связана с монослойным осаждением
полимерной цепи на грани уже существующего кристаллита.
Покажем, что эти допущения находятся в формальном согласии с
экспериментальными результатами. Будем считать, что
критическая свободная энергия, необходимая для образования
вторичного зародыша, в а раз меньше, чем при первичной
нуклеации [11]. Отсюда: _
nED [1 + (п— 1)а]кТ2пл
In^ = In/.0 ^- >(д7у B18)
Если константа скорости роста выражена через полупериод
кристаллизации ту2 (см. стр. 230), вместо выражения B18)
можно также пользоваться следующим *:
1 1 ED [l + {n—l)a]kT*
In = — — - — — — B19*
т1/§ лAпЛ0 + 1п1п2) RT n ^iy'
Определяющим при малых переохлаждениях является
последний член соотношений B18) и B19). Следовательно, в этой
области температур зависимость In ks или In (l/ti/2) от ТПл/Т(кТJ
должна быть линейной.
Соответствующие данные приведены на рис. 87. При
Т^л = 80 С наблюдается явное искривление. Однако при
выборе Г™ —83 С получается прямая линия. В другом месте было
показано, что последняя оценка равновесной температуры
плавления максимально приближается к истине. Таким образом, в
пределах точности определения равновесной температуры
плавления процесс действительно описывается уравнением B18).
* Если вторичная нуклеация носит двумерный характер, получаются
соотношения такого же типа, но с более сложным температурным
коэффициентом [51].
250
Следует отдавать себе ясный отчет в том, что, хотя абсолютная
неопределенность значения Гпл мала, она может играть важную
роль, так как при исследованиях рассматриваемого типа
определяющую роль играет степень переохлаждения, а не абсолютная
температура.
Аналогичный анализ валовой скорости кристаллизации
может быть проведен и для нефракционированного линейного
полиэтилена (марлекс-50) [4]. При использовании непосредственно
•2
Dp
6
-ю
•К
•Гб
\\
\\
°\ \_
Л
\ /nfl"<W°C \
} V
^
\b-«fc
°\.
12 3*567
(Tn'nIW/ATf
Рис. 87. Зависимость \gks
oi(TL/T) 0/ДГJ Д^ по-
лидекаметиленсебацината
[11а].
ф —дилатометрические данные;
? — микроскопические
наблюдения.
Рис. 88. Зависимости lg(l/t,/j0 от
(Г^/Т4)A/Л74J для
нефракционированного линейного
полиэтилена (марлекс-50) при различ-
ных Т°пл [4].
измеряемой температуры плавления этого полимера, равной
137,5° С, на графике для lg— (рис. 88) получается прямая ли-
Чг
ния. Две другие прямые линии на этом же графике показывают,
что линейность сохраняется, даже если равновесная.температура
плавления на несколько градусов выше.
С другой стороны, недостаточная чувствительность методов
при необходимости выбора между т=1 и 2 показана на рис. 89.
Хорошо видно, что те же самые данные могут быть
представлены в виде линейной функции от ГПЛ/ГАГ. Хотя при низких
температурах и наблюдается некоторое отклонение от линейности,
полупериоды кристаллизации в этой области чересчур малы.
Поэтому, принимая во внимание неизбежные экспериментальные
ошибки, этим отклонениям нельзя придавать значение.
На рис. 90 приведена зависимость т./2 от температуры
кристаллизации для трех фракций различного молекулярного веса
и нефракционированного образца линейного полиэтилена [4, 31].
Кривые для двух низкомолекулярных фракций сходны по форме
251
и сдвинуты по температурной оси на 5 град. Частично это
различие может быть приписано более низкой температуре плавления
фракции с меньшим молекулярным весом. Объясняется это тем,
что при одной и той же абсолютной температуре, более
низкомолекулярная фракция кристаллизуется в условиях меньшего
переохлаждения. Однако форма кривой для
высокомолекулярной фракции совсем иная. При высоких температурах
высокомолекулярная фракция кристаллизуется несколько быстрее, чем
фракция с М-24000, а при
понижении температуры наблюдается
обратный эффект и уже низкомо- ь1г
U /4
Рис. 89. Зависимости lg A/т'/2) от
(Тпл/Т) A/Л74) для нефракциони-
рованного линейного полиэтилена
(марлекс-50) при различных
значениях Т^л [4].
Рис. 90. Зависимости т„ от
72
температуры кристаллизации
для фракционированных и не-
фракционированных образцов
полиэтилена [31]:
;_М = 5300; 2- Ж = 24000;
3 — Ж = 364000; 4 — нефракциониро-
ванный марлекс-50.
лекулярная фракция кристаллизуется быстрее. Кривая,
соответствующая исходному нефракционированному образцу (марлекс-
50, Мп-10000, Mv-130000) также отлична от уже
рассмотренных.
Аналогичные результаты были получены Баксером и Тунгом
[26]. Все опыты подобного рода ясно показывают, что влияние
молекулярного веса и молекулярно-весового распределения на
скорость кристаллизации даже вблизи Гпл значительно сложнее,
чем следовало бы ожидать, если бы весь эффект сводился к пре-
одолеванию энергетического барьера, учитываемому в простой
термодинамической теории нуклеации.
В связи с тем, что А#м может быть определено независимо
от каких бы то ни было кинетических опытов, в принципе, по
данным, приведенным на рис. 85—90, можно найти величину
252
параметра о26от .Выше были обсуждены различные упрощающие
предположения, на основании которых выведены соотношения
B18) и B19). Установлено также, что эти соотношения
справедливы не только для специально выбранных комбинаций при
выводе модели, но и для других. Поэтому не приходится
ожидать, что при анализе температурного коэффициента блочной
кристаллизации можно определить абсолютную величину
произведения свободных энергий поверхности раздела. Найти эту
величину можно было бы только после однозначного
определения температурного коэффициента процесса вторичной нуклеа-
ции.
Несмотря на это, полезно ввести в рассмотрение условный
параметр а = (абатK» который все же может дать некоторую
информацию о процессе кристаллизации. Этот параметр
представляет собой среднюю свободную энергию поверхности раздела,
которая находится из соотношений B18) или B19) в
предположении, что имеет место только первичная нуклеация, т. е. а=1.
Определенная таким образом величина а равна или меньше
истинного среднего значения свободной энергии поверхности
раздела, ибо не учитывается неоднородная нуклеация и
возможность снижения скорости вторичной нуклеации.
Типичные значения а для некоторых полимеров приведены
в табл. 13; величина их меняется от 3 до 6 эрг/см2. Хотя эти
значения несколько меньше, чем для металлов или
низкомолекулярных жидкостей, однако, сами по себе, на первый взгляд,
вполне правдоподобны.
Более внимательный анализ, однако, заставляет усомниться
в этом. Действительно, расчет параметра о основан на
допущении изотропности свободных энергий поверхности раздела и не
учитывает того, что для длинной цепной молекулы величина ат
может быть значительно больше, чем gq. Первая величина
определяет свободную энергию выхода цепи на поверхность
кристаллита. С другой стороны, «обычные» грани - кристаллита,
параллельные осям цепей, должны обладать поверхностной
энергией, близкой по величине к поверхностным энергиям для
мономерных веществ. Мы уже могли убедиться, что величина ат,
определенная при исследовании плавления сополимеров [58],
равна ~170 эрг/см2. Поэтому значение <Уб, получаемое при
использовании этой величины и а, лишено физического смысла
из-за своей малости.
Параметры, определяющие нуклеацию для
низкомолекулярных веществ, могут быть найдены при исследованиях
кристаллизации образца, диспергированного на небольшие изолированные
капельки [45]. Вероятность присутствия случайных неодно-
родностей в жидкой системе уменьшается пропорционально
253
Таблица 13
Характерные величины (Г, найденные по соотношению B18) и B19),
при й = 3на=1
Полимер
Метод
определения *
Наклон **
—0,6
—1,2
-2,0
—0,86
-0,69
—0,50
—0,69
—12
—4,4
—3,40
—9,35
—6,16
О Т'
эр г3/см6
58
99
195
84
68
49
68
27
76
33
90
24
а
3 9
4,7
5,8
4,4
4.1
3,7
4,1
3,0
4,3
3,2
4,5
3,9
Лите- |
ратура
[41
[31
[31
[31
[31
[25
[6]
[6]
[И]
[6]
Полиэтилен:
нефракционирован-
ный
фракционированный
М-24000
М-364000 ....
Натуральный каучук .
Полиоксиэтилен . . .
Полидекаметиленади-
пинат
Полидекаметиленсеба-
цинат
Поли-N, N'-себациолпи-
перазин
ks
ks
ks
137,5
140
143
138.5
137,5
138,5
139,5
30
66
79,5
83
183
* Данные получены или при непосредственном определении k или по полупериодам
кристаллизации ту .
** Определен по графикам зависимости \g (VTy2) или !g k$ от Тпл/Т (ATJ.
уменьшению ее объема или поверхности. Потому имеется
принципиальная возможность разделить жидкость на такие маленькие
капельки, что большую их часть можно уже считать свободными
от неоднородностей. Все неоднородности будут сосредоточены
в сравнительно малом числе капелек и практически не будут
влиять на процессы инициирования нуклеации в системе.
Следовательно, в системах, где нуклеация на случайных не-
однородностях играет существенную роль, понижение размеров
образца должно приводить к увеличению степени
переохлаждения, необходимой для начала кристаллизации.
Экспериментально показано, что небольшие капельки
вещества могут быть переохлаждены несравненно сильнее, чем
большие объемы жидкости [45, 48, 52, 56].
Например, такие металлы, как ртуть или олово в виде
капель могут быть переохлаждены на 40—120 град. Однако мелко-
диспергированные я-алканы, такие как Ci7H36 и С24Н50
выдерживают без кристаллизации переохлаждения всего лишь порядка
12—14 град [56]. Эр однозначно указывает на однородную ну-
клеацию; значения а, рассчитанные по этим данным для я-алка-
нов, равны приблизительно 10 эрг/см\
254
Кормиа, Прайс и Торнбалл [59], диспергируя линейный
полиэтилен в различных жидких средах получали капельки
полимера диаметром 3—9 мк. При температурах 122—123° С
кристаллизация не происходит, и только 5% капелек отвердевает
при 100° С. (Линейный полиэтилен в блоке кристаллизуется
почти мгновенно при температурах ниже 120°С). В области
85—87° С кристаллизуется уже почти 50% капелек, и, наконец,
все капельки отвердевают при температурах ниже 84° С. Эти
температуры соответствуют переохлаждениям порядка 55 град,
что намного больше, чем для полимера в блоке или для
маленьких капелек я-алканов *.
Снова используя классическую теорию скорости нуклеации,
можно по соотношению B03) для свободной энергии
образования зародыша определить величину сг=56. Если теперь принять,
что свободная энергия поверхности раздела я-алканов
совпадает с величиной 06 для полиэтилена, можно найти ат« 170 эрг/см2.
Это значение хорошо согласуется с полученным ранее при
исследованиях плавления сополимеров.
Большие переохлаждения, наблюдаемые для дисперсий
полимеров, показывают, что кажущиеся низкие значения а,
получаемые для блочных образцов, объясняются наличием
неоднородной нуклеации. Однако во многих случаях форма изотерм и
способ образования сферолитов указывают на то, что
неоднородная нуклеация протекает спорадически во времени и
пространстве. Интерпретация малоуглового рентгеновского рассеяния и
исследований по плавлению сополимеров, даже независимо от
опытов с диспергированными полимерами, приводит к очень
большим значениям ат (см. гл. 9).
Наиболее важным выводом из этих исследований является
то, что у самых различных полимеров величины ат необычно
велики, по сравнению со свободными энергиями поверхностей
раздела в низкомолекуляриых веществах. Это означает, что в
полимере присутствуют какие-то особые поверхности раздела,
которые, очевидно, оказывают большое влияние на морфологию и
текстуру возникающей в итоге кристаллической системы.
До сих пор основное внимание было направлено на
процессы кристаллизации вблизи температуры плавления, когда
большие значения температурного коэффициента скорости
* В этих опытах [59] подразумевается, что капельки целиком состоят из
полиэтилена. Однако, к сожалению, некоторые диспергирующие среды были
таковы, что в исследуемой области температур внутри капелек могло
происходить разделение на две жидкие фазы, с участием полимера в обеих.
Таким образом, если до кристаллизации условия в какой-то мере
приближались к равновесным, некоторое неопределенное, но конечное количество
диспергирующей жидкости могло присутствовать в капельках.
Следовательно, наблюдаемые температуры могли не соответствовать кристаллизации
чистого полимера,
255
благоприятствуют теоретическому анализу. При значительных
переохлаждениях второй член соотношений B18) и B19)
перестает играть определяющую роль, и возникает
необходимость рассматривать все соотношение в целом.
При упрощающих допущениях, что k0, ED и К не зависят от
температуры, скорость кристаллизации максимальна при
температуре Ти которая определяется соотношением:
B20)
Максимальная скорость определяется соотношением:
In fcs = In k0 — [1 -f (n — 1) a] k ¦
T V{Tn
37-, - Tn
1
7\У * (Тпл-Т)\
B21)
Реальный процесс кристаллизации натурального каучука в
широкой области температур полностью описывается этим
соотношением. Свидетель-
Ьп-W
ством тому служат
приведенные на рис. 91 данные
для кристаллизации
натурального каучука при
атмосферном давлении и
давлении 400 кгс/см2. То
обстоятельство, что реальный
процесс подчиняется такой
сравнительно простой
закономерности, особенно
удивительно, если принять во
внимание явления релаксации и
переноса в блочных
аморфных полимерных системах,
особенно вблизи
температуры стеклования. Область
температур кристаллизации
на 40—70 град выше
температуры стеклования; в этой области ожидается и
экспериментально наблюдается в полностью аморфных системах сильное
увеличение ED с ростом температуры [60]. Возможно, что в
области температур, в которой проводятся опыты по кристаллизации,
имеется компенсирующий вклад других членов, ответственный
за кажущееся постоянство ED.
По-видимому, большее значение имеет тот весьма интересный
экспериментальный факт, что отношение 7УГПЛ, равное 0,83
при 1 атм лишь незначительно убывает с повышением давления.
Табл. 14 показывает, что это отношение практически одинаково
и для других самых различных полимеров. Если считать
соотношение B20), справедливым, то из данных табл. 14 следует, что
т,°с
Рис. 91. Зависимости \gks от
температуры для натурального каучука при
атмосферном давлении (/) и 400 кгс/см2
B) [221].
Кривые проведены в соответствии с
уравнением B21).
256
Таблица 14
Взаимосвязь между температурой максимальной скорости
кристаллизации 71! и температурой плавления различных полимеров;
полупериоды кристаллизации при Тх
Полимер
I
Тх, °К
ri/rn
т1/2 при Г,
Литература
Натуральный
каучук
Полиэтилентере-
фталат
Полиэтиленадипи-
нат
Полиэтиленсукци-
нат
Полипропилен . . .
Полигексаметилена-
дипамид
303
537
332
379,5
— 453
1 537
249
453
271
303
348
— 423
0,83
0,84
0,815
0,78
— 0,77
0,79
2,5 ч
40 сек
3 мин
4 мин
5 сек
[5, 10, 25)
[7]
[8]
[9]
[29]
[29]
отношение ED/k также одинаково по величине для всех
полимеров.
Хотя отношения 7УГПЛ сходны для различных полимеров,
абсолютные значения констант скорости и соответственно
временные характеристики кристаллизации в максимуме могут
различаться на несколько порядков. Для иллюстрации этого в
табл. 14 приведены значения полупериодов кристаллизации при
максимальных скоростях ряда гомополимеров.
Как видно из табл. 14, наблюдается
определенная-зависимость времени кристаллизации от температуры плавления. Чем
выше температура плавления, тем больше скорость
кристаллизации при сравнимых температурах. Вспоминая сделанное ранее
(гл. 5) заключение о связи, которая существует между
конформацией отдельной цепи и температурой плавления, можно
распространить его и на время кристаллизации. Цепи с более
развернутой конформацией, хотя и кристаллизуются по тому же
механизму, что и при любой конформации, претерпевают более
быстрый фазовый переход. Большие скорости кристаллизации
высокоплавких полимеров могут быть объяснены, поэтому,
сходством конформации цепей в обоих фазовых состояниях.
Кристаллизация полимеров,
содержащих структурные нерегулярности
В предыдущих главах мы убедились в существовании
серьезных различий между процессами плавления чистых
гомополимеров и полимеров с различными структурными нерегулярностями,
17 Л. Манделькерн
257
такими как сомономерные звенья, точки ветвления, сшивки и
концевые группы. Эти нарушения играют роль, аналогичную
роли чужеродных примесей или некристаллизующихся вторых
компонентов при кристаллизации низкомолекулярных веществ.
Они приводят к понижению точки плавления и расширяют
температурную область плавления.
Хорошо известно, что введение второго компонента в простые
жидкости может значительно изменить скорость нуклеации и
кристаллизации из расплава
[61, 62]. Фазовые диаграммы
таких двухкомпонентных
систем определяют в
значительной мере характер
процесса кристаллизации, так
как последний чувствителен
к различиям композиции
двух фаз. Добавление к
полимеру истинного второго
компонента оказывает
подобное же действие. Однако
эти некристаллизующиеся
компоненты могут быть
введены непосредственно в
полимерную цепь. Таким
образом, можно ожидать, что
не только термодинамика,
но и кинетика
кристаллизации сополимеров имеет
свою специфику, по
сравнению с чистыми гомополиме-
рами.
Прямые кинетические
исследования подтверждают
этот вывод. На рис. 92 приведены примеры кинетических
изотерм для полибутадиена, полученного эмульсионной
полимеризацией и содержащего 80% кристаллизующихся транс-1,4-
звеньев. Его температура плавления равна 37±ГС. Кривые
явно отличаются от изотерм чистого гомополимера; они не
совмещаются при сдвиге, их форма зависит от температуры
кристаллизации. Однако в случае кристаллизации при больших
переохлаждениях форма кривых уже начинает напоминать
изотермы для блочных полимеров. Это становится ясно при
сравнении экспериментальных изотерм с пунктирной кривой на
рис. 92, которая проведена в соответствии с уравнением A91)
для /г = 3. При повышении температуры кристаллизации
переход замедляется, и изотермы веерообразно распределяются
относительно оси времени,
258
соответственно валовую скорость
бремя, пин
Рис. 92. Зависимости (V^— V/)/(Kuo— V0)
от времени кристаллизации
полибутадиена, содержащего 80% транс-1,4-
звеньев.
На каждой изотерме указана температура
кристаллизации. Пунктирная кривая проведена
в соответствии с уравнением A91).
Аналогичные изотермы наблюдались при кинетических
исследованиях разветвленного полиэтилена [63, 64], сшитых
полимеров [65] и не полностью стереорегулярного полипропилена [66].
По-видимому, такая кинетика характерна для кристаллизации
полимерных цепей, содержащих структурные нерегулярности.
Результаты, полученные для разветвленного полиэтилена, при-
о
0,2
1,0
5 10 50 100 500 1000 500010000
дремя, пин
Рис. 93. Зависимости (V^—Vt)l(V^—V0) от
времени кристаллизации разветвленного полиэтилена.
Для каждой изотермы указана температура кристаллизации.
ведены на рис. 93; можно отметить подобие этих данных с
изображенными на рис. 92.
Для количественного объяснения этих результатов будем
считать, что элементы нерегулярности структуры («гетеро-
звенья») в процессе превращения исключаются из
кристаллической фазы. Это полностью подтверждается анализом кривых
плавления многих сополимеров *. Следовательно, при развитии
кристаллизации расплавленная фаза обогащается некристалли-
зующимся компонентом, и температура плавления непрерывно
понижается. Поскольку при изотермическом переходе
температура постоянна, фактически уменьшается степень
переохлаждения, причем тем сильнее, чем больше степень завершенности
кристаллизации. Таким образом, можно ожидать уменьшения
скоростей первичной и вторичной нуклеации при развитии
процесса. Уравнение A91), которое описывает кинетику
кристаллизации гомополимеров в блоке, выведено в предположении
постоянства этих скоростей при развитии процесса. Необходимы,
следовательно, дальнейшие уточнения, включающие учет изменений
состава и структуры расплава, а также растущей степени
кристалличности образца в целом.
* Как уже указывалось, иногда возможна ситуация, при которой
вторичные компоненты способны внедряться в кристаллическую решетку
основного компонента или даже образовывать с ним новую кристаллическую
структуру. Мы не будем здесь останавливаться на этих особых случаях.
17*
259
В качестве первого шага к обобщениям кинетических
уравнений рассмотрим кристаллизацию сополимера, образованного
звеньями типа (ср. гл. 4) А и В. Пусть эти звенья распределены
в полимерной цепи хаотически, и только звенья А могут входить
в кристаллическую решетку [67]. Удобно характеризовать
степень завершенности хода при времени t параметром 0
(степенью конверсии):
е^ [1 —Я@1
[1-Я (со)]
где Я(оо)— доля аморфного вещества при полном завершении
перехода;
k(t)— соответствующее количество в момент t.
Скорость нуклеации при степени конверсии 8 выразится
уравнением: т
N @) = N0XBA exp (- |f - ^fj B22)
где Ха — молярная доля звеньев А в расплаве;
Д/^е — свободная энергия образования ядра критических
размеров на стадии процесса, определяемой
значением 0.
•
Пользуясь для выражения N(Q) характеристиками состава
полностью расплавленной фазы, можно записать:
N @) = N (°) -ф ехР ( ^г~/ B23)
где N@) — стационарная скорость нуклеации при ^=0,
которая соответствует составу Х\.
Для ядер цилиндрической формы
* $ло1оТ
д^=-дпг <224>
где AF(Q)~AHM[To-T)/TQl a
TQ — равновесная температура плавления системы при
степени конверсии 6.
Она находится из выражения, описывающего понижение
температуры плавления для статистических сополимеров:
11 R Х*А
= In—?- B25)*
* Использование уравнения B25) включает предположение, что Ге
зависит только от ^а/^а»т- е- от химического состава расплава. Однако, как
следует из теоретических, и экспериментальных данных (гл. 4), для
сополимеров при конечных уровнях кристалличности возможно дальнейшее
уменьшение TQ, обусловленное характером распределения длин
кристаллизующихся последовательностей. В нашем случае, следовательно, допускается
переоценка TQ для статистических сополимеров.
269
Так как отношение ХУх°х может быть выражено в виде:
Х°А l+(WB/WA)(MA/MB){l-Qll-b(co))/WA}
уменьшение скорости первичной нуклеации запишется так:
Уменьшение скорости нуклеации может быть легко
рассчитано, если известны k и Д#м для отправного чистого гомополи-
мера.
Результаты такого модельного расчета для статистических
сополимеров этилена при МА/МВ = 0,44 и 1— Х(оо)=0,75
приведены на рис. 94. При введении
незначительного количества сомоно-
мера должно происходить
небольшое, но регистрируемое уменьшение
скорости нуклеации при больших
переохлаждениях, особенно на
последних стадиях перехода. Однако
уменьшение переохлаждения или
незначительное повышение
концентрации звеньев типа В приводит ужек;
значительному уменьшению
скорости нуклеации, причем это
изменение должно быть ощутимо даже
на ранних стадиях
кристаллизации.
Подобные эффекты обусловлены
непрерывным понижением
фактического переохлаждения (Те — Т) по
мере развития перехода. При
больших номинальных степенях
переохлаждения относительное
изменение фактического переохлаждения
в течение перехода мало, так что скорость нуклеации
изменяется незначительно. Однако при малых номинальных
переохлаждениях изменение 7е с конверсией таково, что должно
происходить заметное уменьшение скорости нуклеации. Этот эффект
становится более ощутимым при увеличении исходной
концентрации звеньев В,
Теперь необходимо вывести общее уравнение, описывающее
кинетику фазовых превращений и учитывающее исключение не-
кристаллизующихся звеньев из решетки. По сути дела, этот
вывод включает учет изменений скорости первичной нуклеации и
N@)
ЖГ\т
02 0.4 0.6 0.6 1.0
Рис. 94. Теоретические
зависимости — In [N (в)/ЛГ @)] от
степени конверсии 9 для двух
статистических сополимеров
указанного на графике
состава [67].
На кривых показана температура
начального (номинального)
переохлаждения.
261
скорости роста при вычислении интеграла в уравнении A77).
С этой целью выразим скорость первичной нуклеации в виде:
!LLxl = h(x) B28)
N@)
Скорость линейного роста G(z) можно записать в форме
G@)g(z)v где G@)—начальная скорость при t=0.
Тогда:
v (t, т') = KG (О)"-1 [Ф (/, т')]" B29)
где
t
Ф it, 1) = J" g (z) dz B30)
т'
и более общее выражение для валовой скорости фазовых
превращений приобретает вид:
где А = A//г)(ркр/рж)^@)О@)Л.
Так как скорость роста предположительно определяется
процессом нуклеации, /г(т) и g(x), вообще говоря, являются
функциями l—k(t). При g(x) и Л(т), равных единице, уравнение
B31) совпадает с A77).
Уравнение B30) не имеет простого аналитического решения
и может быть решено лишь численными методами. Для расчета
принималось, что рост происходит в результате трехмерной
нуклеации, а уменьшение скорости нуклеации оценивалось, как
это было сделано выше. Результаты приведены на рис. 95, где
представлена зависимость 1—8 от безразмерной переменной
(</t*J, где т*/а— гипотетический полупериод кристаллизации,
который наблюдался бы, если бы уравнение A91) было
справедливо.
Как следует из уравнения A90), некристаллизующиеся
звенья цепи, по-видимому, должны вызвать значительные
изменения формы изотермы в сравнении с чистыми гомополимерами.
Должна также утрачиваться способность к суперпозиции и тем
в большей степени, чем больше концентрация некристаллизую-
щегося сомономера и меньше переохлаждение. Однако при
достаточно высоких переохлаждениях формы изотерм для
сополимера и гомополимера во многом сходны. Пунктирные линии на
рис. 95 представляют изотермы, рассчитанные по уравнению
A91), при постоянных скоростях нуклеации и роста; я = 4.
Сравнение экспериментальных изотерм с этим простым
случаем свидетельствует о запаздывании кристаллизации под влия-
262
нием нер^гулярностей. Наблюдается характерный «веер»
изотерм, хотя в области низких степеней кристалличности все
изотермы проявляют тенденцию к совмещению. Изменение формы
изотерм происходит несмотря на то, что тип нуклеации и
характер кристаллического роста у «сополимеров» такой же, как и у
чистых гомополимеров. Различия обусловлены исключительно
изменением состава расплава вследствие исключения из
кристаллической фазы нерегулярных участков цепи при развитии
превращения.
Необычайная чувствительность изотерм кристаллизации даже
к незначительным нарушениям структуры хорошо
демонстрируется на рис. 95 для случая 1^в=0,01. Хотя отклонений от
О 0,5 1,0 0 0,5 1,0 0 0,5 1,0
Рис. 95. Теоретические зависимости 1—9 от \g(t/xu) для
трех различных сополимеров [67].
На кривых показана температура переохлаждения. Пунктирные кривые
проведены в соответствии с уравнением A91) для п = 4.
уравнения A91) не происходит до 1 —6 = 0,6, при более высоких
уровнях кристалличности наблюдается небольшое, но
бесспорное отклонение от теоретического для гомополимеров.
Изучение кинетики кристаллизации, таким образом, дает метод
регистрации небольших количеств некристаллизующихся звеньев.
Теоретические изотермы на рис. 95 сходны с
экспериментальными для полибутадиена и разветвленного полиэтилена (см.
рис. 92 и 93). Типичные «размазанные» изотермы названных
систем можно, следовательно, объяснить их сополимерным
характером (в том смысле, который придается этому термину в гл. 4)
и вытекающими отсюда изменениями состава и структуры
расплавленной фазы в процессе кристаллизации. Это объяснение
согласуется с характером плавления таких систем и может
рассматриваться как доказательство сосуществования
кристаллических и аморфных областей в сополимерах. Изотермы могут
соответствовать упрощенному варианту уравнения кинетики A91)
лишь в том случае, если экспонент п изменяется со степенью
263
завершенности кристаллизации; подтверждается это
.отдельными экспериментальными исследованиями.
Такой подход, однако, приводит к ошибочной интерпретации
механизма, так как соответствие экспериментальных изотерм
уравнению A91) теперь означало бы, что характер процессов
нуклеации и роста изменяется при развитии кристаллизации.
Как уже указывалось, на самом деле эти данные имеют вполне
естественное объяснение без допущения изменений характера
процессов нуклеации и роста, если учесть, что состав расплава
непрерывно меняется.
Другие факторы, вызывающие изменение AF* в ходе
кристаллизации, можно, в принципе, проанализировать аналогично.
Нетрудно убедиться, что даже в случае чистых гомополимеров
структура расплава будет меняться при возрастании степени
кристалличности. Поэтому параллельно с результирующим
изменением будет меняться и форма изотерм. Если бы результаты
интерпретировались в терминах постоянной скорости нуклеации
(характеризующей начальные стадии перехода), это неизбежно
привело бы к заключению об изменении способа нуклеации или
роста в процессе кристаллизации, что, разумеется, вовсе
необязательно.
Тот факт, что статистические сополимеры и сшитые полимеры
кристаллизуются, причем, довольно легко, показывает, что для
инициирования кристаллизации цепных молекул вовсе не
требуется образования ядра с регулярно сложенными цепями.
Подобная упаковка звеньев невозможна в сополимерной или
сшитой системе. Добавим к этому, что А/7* представляет собой
критическую свободную энергию, необходимую для образования
ядра из звеньев типа А в расплаве заданного состава. Если бы
звенья типа В могли одинаковым образом входить в
кристаллическую решетку в виде неравновесных дефектов, как было
принято [68, 69], то А/7* не изменялось бы значительно с изменением
величины 8. Поэтому не было бы причин ожидать появления
размазанных изотерм, подобных наблюдаемым на опыте.
Температура плавления сравнительно низкомолекулярных
полимеров зависит от длины цепи из-за значительной
концентрации в подобной системе свободных концов цепей. В
соответствующей области молекулярных весов при изотермической
кристаллизации концентрация свободных концов цепей в расплаве
возрастает, что приводит к понижению кажущейся температуры
плавления. Процесс кристаллизации низкомолекулярных
полимеров сильно отличается, однако, от процесса, в котором
низкомолекулярные цепные молекулы (типа я-алканов) образуют
молекулярные кристаллы. В последнем случае концы цепей
различных молекул лежат в одной из главных кристаллографических
плоскостей. Поэтому в процессе кристаллизации не происходит
никакого изменения в составе расплава, и изотермы в этом слу-
264
чае должны соответствовать рассчитанным для постоянных
скоростей нуклеации и роста. Изотермы же для низкомолекулярных
полимеров должны быть совсем иными.
Это более или менее априорное заключение подтверждается
исследованием низкомолекулярных образцов полиоксиэтилена
[70] и фракции линейного полиэтилена с молекулярным весом
5300 [31]. Изотермы характерным образом отличаются от кривых
для фракций более высокого молекулярного веса, приведенных
на рис. 80. В рассматриваемом случае изотермы менее крутые,
и характерное для кристаллизации ускорение не наблюдается.
Хотя тенденция к веерообразному распределению относительно
оси времени и имеется, она не столь выражена, как у
статистических сополимеров, и изотермы сохраняют способность к
суперпозиции вплоть до относительно высоких конверсии.
Кристаллизация полимеров
в присутствии низкомолекулярных жидкостей
Количественные исследования кинетики кристаллизации
полимеров в присутствии низкомолекулярных жидкостей
проводили главным образом методом наблюдения за валовой
скоростью кристаллизации вблизи температуры плавления. Кроме
того, проводились и непосредственные микроскопические
наблюдения роста кристаллов из очень разбавленных растворов
полиэтилена [71]. Разбавление расплава должно оказывать
влияние как на свободную энергию нуклеации, так и на
процессы переноса, играющие определенную роль при нуклеации и
росте кристаллов.
Поэтому кинетика кристаллизации из концентрированных и
умеренно концентрированных растворов (т. е. содержащих до
70% низкомолекулярного компонента) имеет свою специфику.
Процесс кристаллизации вновь характеризуется высокими
отрицательными температурными коэффициентами. Для заданного
состава изотермы, полученные при различных температурах,
весьма подобны по форме и с достаточной точностью могут быть
совмещены. Однако при понижении концентрации полимера
форма и характер изотерм изменяются. Процесс кристаллизации при
разбавлении замедляется, изотермы становятся более пологими.
Характерные примеры приведены на рис. 96 для ряда смесей
полиоксиэтилена и дифенилового эфира. Сплошные кривые
получены в результате приблизительного совмещения реальных
изотерм. Каждая кривая соответствует определенному составу.
Для сравнения на рис. 96 приведены также изотермы
неразбавленного полимера (пунктирные кривые), вычисленные по
формуле A91) при п = 4.
265
При малом содержании растворителя экспериментальные и
теоретические кривые, соответствующие неразбавленному гомо-
полимеру, хорошо согласуются. Однако по мере понижения
концентрации полимера расхождения между кривыми возрастают,
становясь существенными не только на конечной, но и на
начальной стадии процесса. Таким образом, исчезает даже внешнее
подобие между расчетной изотермой для гомополимера и
экспериментальными кривыми для разбавленных систем. Для
формального объяснения этих результатов можно было бы приписать
параметру п произвольно уменьшающееся значение, зависящее
Рис. 96. Изотермы кристаллизации полиоксиэтилена
в присутствии дифенилового эфира.
Сплошные линии представляют составляющую (см. текст) изотерму
для каждой системы. На кривых показано значение vx — объемная
доля растворителя. Пунктирные линии соответствуют
теоретическим изотермам при ««=4, рассчитанным по уравнению A91).
Положение каждой изотермы на абсциссе произвольно. Короткие
вертикальные штрихи на оси абсцисс соответствуют десятичным
порядкам [24).
от степени завершенности перехода и состава. Однако, если
принять этот метод, то следовало бы допустить, что характер
процессов нуклеации и роста изменяется. Мы видели, что для кри«
сталлизации сополимеров это оказалось ошибочным
допущением, которое может привести к ложным выводам. При
кристаллизации полимеров в присутствии низкомолекулярной жидкости
по мере протекания процесса также происходит изменение
состава расплава. В рассматриваемом случае это уменьшение
термодинамической движущей силы благоприятствует
кристаллизации. Кроме того, диффузия сегментов цепей к поверхности
раздела кристаллит — жидкость должна приобретать все
возрастающее значение для процесса роста по мере разбавления
системы. По этой причине следует также ожидать замедления
роста.
Изотермы для смесей линейного полиэтилена и а-хлорнафта-
лина в сравниваемом интервале концентраций [4, 31] сходны с
изотермами, приведенными на рис. 96. При дальнейшем же пд-
266
нижейии концентраций полимера наблюдается значительное
изменение формы изотерм. Они вновь приобретают сходство с
изотермами, характерными для чистых гомополимеров. Результаты,
полученные для очень разбавленных растворов, приведены на
рис. 97 и 98; сходство с кинетикой для неразбавленного гомопо-
Рис. 97. Зависимость
(V^-VdKV^-Vo) от
времени кристаллизации
линейного полиэтилена
из разбавленного
раствора в а-хлорнафталине.
Содержание полимера в
исходной смеси 0,25 вес. %. На
кривых показана температура
кристаллизации.
лимера очевидно. Именно в этом диапазоне низких
концентраций происходит выпадение из раствора пластинчатых или
дендритных единичных кристаллов (см. гл. 9).
Подводя итоги, можно сказать, что форма изотерм и,
следовательно, обусловливающие их кинетические процессы в
значительной степени зависят от концентрации. После
первоначального разбавления характерная для гомополимеров изотерма
искажается и размазывается, но вновь появляется в очень
разбавленных системах. Однако было бы преждевременно сделать
из этого вывод, что механизмы кристаллизации одинаковы в
области очень высоких и очень низких концентраций, а в
промежуточной области действуют иные механизмы. Для интерпретации
Время, мин
Рис. 98. Зависимость
(^-^)/(^оо-^0)ОТ
времени кристаллизации
линейного полиэтилена
из разбавленного рас:
твора в а-хлорнафталине.
Содержание полимера в
исходной смеси 0,05 вес. %. На
кривых показана температура
кристаллизации.
267
механизмов, действующих во всем диапазоне концентраций,
требуется более детальный анализ экспериментальных данных.
Предыдущий анализ кристаллизации сополимеров
показывает, что всегда следует принимать во внимание влияние
изменений состава расплава в ходе кристаллизации на величину
свободной энергии нуклеации. Свободная энергия образования
ядра из расплава в присутствии смешивающейся с ним низкомо-
лекулярной жидкости может быть выражена уравнением [24]:
A/7 (v2) = ?р Д/м - 2рат - 2?аб (я/рI* + RT In vg B32)
Последний член в уравнении B32) представляет собой
энтропийный вклад в свободную энергию, обусловленный
вероятностью выбора р кристаллических последовательностей звеньев
из двухкомпонентной смеси. Другие члены уравнения сходны с
членами уравнения для нуклеации в неразбавленном гомополи-
мере, а А/м представляет собой объемную свободную энергию
плавления при заданном составе. Для концентрированных и
умеренно концентрированных растворов, где концентрация
сегментов однородна во всем растворе, v2 может быть отождествлено
с действительной объемной долей полимера, присутствующего в
системе. Однако мы уже видели, что в диапазоне низких
концентраций можно представить раствор как состоящий из двух
различимых типов областей: «среды», в которой сегменты
полимерных цепей отсутствуют, и занятых отдельными клубками
координационных сфер, где концентрация сегментов, хотя и мала,
но конечна. Это и есть та концентрация, которую необходимо
использовать в уравнении B32); разумеется, ее ни при каких
обстоятельствах нельзя отождествлять со средней концентрацией
раствора в целом.
Анализ приведенных выше (см. рис.. 82) поверхностей
свободной энергии дает для координат точки минимума:
p=4oT-2RTlnv2
Апо\
Следовательно, свободная энергия, необходимая для
образования ядра с минимальной устойчивостью, выражается
соотношением:
8яа!<тт — AnRTai In v<,
где А/м — сумма свободных энергий плавления и разбавления в
расчете на одно звено.
268
Ее можно приближенно выразить в форме [24]:
где Гпл — температура плавления системы смеси заданного
состава. Следовательно
Snoia. — AnRToi In v2 ~2
Д/" <«*> - (ДЯмА7-J Т™ <236>
и стационарную скорость нуклеации можно выразить
уравнением:
N Ы = N0v2 exp [- D{ 2)rt L2ZJ B37)
Вследствие того, что понижение концентрации полимера
оказывает влияние на предэкспоненциальный множитель, энергию
активации переноса и А/7*, стационарная скорость нуклеации
должна существенно изменяться при разбавлении. Особый
интерес представляют изменения Д/7*.
При эффективной концентрации полимера порядка
нескольких процентов, что типично для номинально разбавленного
раствора, Д/7* приобретает чрезмерно большое значение из-за
наличия в уравнении B35) члена с In v2. Поэтому нуклеация не
происходит с измеримой скоростью при переохлаждении, сравнимом
с переохлаждением в системах, где более высокая концентрация
полимера. Таким образом, необходимо значительно увеличить
переохлаждение, чтобы понизить число последовательностей,
принимающих участие в акте нуклеации. Это необходимо даже
в том случае, если в процессе нуклеации участвует только одна
молекула, так как сегменты цепи должны быть выбраны в
пределах координационной сферы с эффективной средней
концентрацией v2 и перенесены в чистую кристаллическую фазу.
Разбавление расплава происходит либо при
непосредственном добавлении второго (мономерного) компонента, либо'в ходе
кристаллизации полимера и выпадения его из бинарной
системы. Для систем с высокой концентрацией по мере протекания
кристаллизации температура равновесия между
кристаллической и жидкой фазами понижается в соответствии с выведенным
соотношением A4). Переохлаждение уменьшается, и происходит
понижение скорости нуклеации, что вызывает замедление
валовой скорости кристаллизации. Эти выводы находятся в
качественном соответствии с изотермами, приведенными на рис. 96.
Однако с целью детального количественного сравнения
необходимо было бы провести расчет теоретических изотерм для
систем полимер — низкомолекулярная жидкость при
соответствующих изменениях скорости нуклеации.
269
При более высоких разбавлениях, когда v2 имеет порядок
0,10 или менее, изменение степени переохлаждения в процессе
изотермической кристаллизации обычно очень незначительно.
Действительно, так как в этом диапазоне v2 температура
плавления мало чувствительна к концентрации полимера, то ее
понижение при дальнейшем разбавлении оказывается всего
порядка 1 град или менее. Результирующее практическое
постоянство переохлаждения вполне согласуется с формой изотерм,
наблюдаемой в этом концентрационном диапазоне. Форма
изотерм приближается теперь к таковым, рассчитанным для
системы с постоянными скоростями нуклеации и роста. При этом
принимается, что логарифмический член (с In v2) в уравнении B35)
не испытывает значительных изменений в зависимости от
концентрации. Если бы в формулу подставлялись номинальные
(средние) концентрации, то, несомненно, это не было бы
справедливо для разбавленных растворов. Таким образом,
приводимые результаты подчеркивают необходимость использования
истинной, эффективной, концентрации полимера в разбавленных
растворах.
Необходимо теперь детальнее рассмотреть причины сходства
между изотермами для неразбавленного блочного гомополимера
и для очень разбавленного раствора (у2^0,01). Теоретическое
объяснение формы изотерм первого типа основано на том
допущении, что взаимные помехи при развитии смежных центров
вызывают прекращение роста. Это приводит к наблюдаемым 5-об-
разным изотермам. Очевидно, в очень разбавленных растворах
взаимодействия растущих центров не могут иметь серьезного
значения. Таким образом, ясно, что изотермы сходной формы
могут быть получены при двух совершенно различных исходных
условиях. В разбавленных растворах можно допустить, что рост
происходит на изолированных центрах, расположенных на
значительных расстояниях, и не мешающих друг другу. Поэтому
в данном случае прекращение роста является следствием
понижения содержания кристаллизующегося компонента в жидкой
фазе. Эти предложения аналогичны тем, которые были
использованы для истолкования кинетики осаждения мономерных
веществ из пересыщенных разбавленных растворов [45].
Для разбавленной системы рост обусловлен либо теми
процессами, которые протекают на поверхности раздела между
частицей и раствором, либо диффузией растворенного компонента в
жидкой фазе. Диффузный поток тем больше, чем меньше
размеры частиц. При этих обстоятельствах скорость роста должна
контролироваться процессами, протекающими на поверхности
раздела. При увеличении размеров частиц сопутствующее
уменьшение потока вещества к поверхности стремится ослабить
влияние диффузных процессов. Детальный обзор методов
математического анализа кинетики осаждения приведен в работе Торн-
270
балла [45]. Хотя конкурирующие процессы диффузионного
контроля и контроля поверхностью раздела могут быть описаны
совместно, для наших целей достаточно рассматривать оба
процесса как протекающие независимо друг от друга.
Помимо механизма роста необходимо также уточнить
природу акта инициирования или нуклеации. Новые частицы могут
либо возникать спорадически во времени, либо появляться
одновременно в самом начале процесса осаждения. При
допущении, что ядра образуются спорадически во времени,
получающееся интегральное уравнение не может быть решено
аналитически. Однако если допустить, что определенное число п
пренебрежимо малых частиц инициируется в момент /=0, и это число
остается неизменным в ходе процесса *, то легко показать, что
для диффузионно-контролируемого роста [45]:
где X—доля, подвергшаяся превращению;
D—коэффициент диффузии;
kd—константа.
Для контроля роста поверхностью раздела [45] имеем:
I 1П[ (X^-lf J + VS tg 2+T^ = kin Ut = *'' (
где kt — другая константа;
и — константа скорости реакции для процесса,
протекающего на поверхности раздела.
Зависимость доли, не подвергшейся превращению A—Л'),
от приведенного времени kt приведена на рис. 99 в соответствии
с уравнениями B38) и B39). Изотермы для процессов,
контролируемых диффузией и поверхностью раздела, совершенно
аналогичны друг другу и заметна их S-образная форма. Они
похожи на изотермы для чистого блочного полимера. На рис. 99
это показано пунктирной линией, которая рассчитана по
уравнению A91) при я=3. Точечная линия представляет собой
типичную экспериментальную изотерму для кристаллизации из
очень разбавленного раствора. Если не считать самых
начальных и конечных участков изотерм, для роста, контролируемого
поверхностью раздела, получается очень хорошее
соответствие между экспериментальной кривой и кривой,
рассчитанной по уравнению B39). Расхождения могут быть приписаны
* Это допущение, противопоставляющееся спорадическому образованию
новых центров во времени, сделано исключительно для упрощения
вычислений. Аналогично проблему при кристаллизации неразбавленных полимеров
мы решили раньше, рассматривая фиктивные зародыши и поэтому не
учитывая подавление роста в явном виде.
271
*?щ
значительным упрощениям, введенным при анализе. По-видимому,
на самом деле при / = 0 не образуется фиксированное число частиц,
Более того, приближаясь к концу процесса, когда размер частиц
увеличивается, процессы диффузии приобретают все большее
значение. Это проявляется в запаздывании кристаллизации,
которое наблюдалось
экспериментально.
Таким образом, можно
сделать вывод (не рассматривая
морфологию или внешний вид
образующихся кристаллов), что
кристаллизация полимеров из
разбавленных растворов протекает
в соответствии с теми же
кинетическими законами, что и
кристаллизация низкомолекулярных
веществ при сравнимых условиях.
Для малых значений X
уравнения B38) и B39) сведутся к
виду:
1 — х = ехр A - Ып) B40)
Рис. 99. Зависимость доли
вещества, еще не подвергшейся
превращению A — X), от условного
времени для кристаллизации из
разбавленного раствора:
1 и 2 — теоретические изотермы при
диффузионном контроле и поверхностью
раздела, рассчитанные по уравнениям B38)
и B39), соответственно; 3 —
теоретическая изотерма для кристаллизации чистого
гомополимера, рассчитанная по
уравнению A91), при л = 3; 4 —типичная
экспериментальная изотерма для
кристаллизации из разбавленного раствора.
с n = z/2 при диффузионном
контроле и /г = 3 при контроле
поверхностью раздела. Это
уравнение имеет тот же вид, что и
уравнение A79), выведенное для
кинетики кристаллизации
неразбавленных блочных гомополимеров.
Интересно проследить за
изменениями константы скорости
кристаллизации и ее
температурного коэффициента при
изменении концентрации полимера. Для удобства можно избрать в
качестве меры скорости время т0,э, при котором степень
завершенности перехода достигает 10%. Таким путем можно провести
рациональное сравнение систем с различной концентрацией
полимера, избежав осложнений, связанных с изменениями
переохлаждения при изотермической кристаллизации. Независимо
от номинальной концентрации, начальные участки изотерм
соответствуют упрощенной математической формулировке
кинетики фазовых превращений, выражаемой уравнением A91).
Поэтому при анализе экспериментальных данных мы исходим из
обычного выражения для константы скорости k. Таким образом:
ks (v2) = k0 (v2) exp
[-
nEr
(v2) 1 + (я-1)д
RT
RT
AF* <»,)
B41)
272
откуда
log
т0,9
log kQ (v2) + log log
-1-
0,9 J
Ed(v2) [IrK/i-DaJA^fra)
RT 2,3 RT
B42)
Здесь AF*(v2) определено уравнением B36). Следует
напомнить, что v2 не обязательно представляет номинальную
концентрацию, а скорее соответствует истинной концентрации
полимера в координационной сфере набухшего клубка. В
концентрированных системах обе
величины совпадают. s
Уравнение B42) показь: *~'^
вает, что при незначитель- ~-
ном переохлаждении
зависимость log(l/r0,9) от
температурной переменной
Т2ш/Т (&Т)° должна быть
линейной при подстановке
равновесной температуры
плавления. Для
разбавленных систем это означает, что
температура плавления или
растворимости (при
соответствующей концентрации)
должна определяться для
образца,
закристаллизованного в блоке и хорошо
закаленного [72]. Рис. 100. Зависимость lg A/т0,9) от тем
На рис. 100 приведена пературной переменной (т1л/т)A /ДТ)
охватывающая широкий ди- для нефракционированного линейного
апазон концентраций серия полиэтилена в а-хлорнафталине.
кинетических кривых,
построенных согласно
уравнению B36) с учетом
сказанного о Гпл. В соответствии с теоретическими данными, эта
серия представлена семейством прямых линий, наклон которых
постепенно увеличивается при понижении концентрации
полимера, вплоть до 1%. При дальнейшем уменьшении концентрации
полимера наклон остается постоянным, даже если она
понижается еще в 100 раз. На основании этих данных можно
сделать вывод, что процесс кристаллизации продолжает
контролироваться нуклеацией даже при очень высоких разбавлениях.
Этот вывод соответствует данным, полученным при
микроскопическом наблюдении роста кристаллов в этом же диапазоне
концентраций [70].
OLIWW
На прямых показана исходная объемная доля
полимера в расплаве [31].
18 Л. Манделькерн
273
В соответствии с уравнением B42), наклон прямых линий
может быть выражен соотношением:
1 + (п — 1) а
8яа|ат
4лоб Т log v2
2,3#Д#2
д//:
B43)
Для полиэтилена в интересующем нас интервале температур
отношение наклона при концентрации v2 к наклону для чистого
полимера (v2=l) выражается формулой:
Наклон (v2) _ v __ л 2,3RT log v2 ^ 1 0,9\ogv2
Наклон (v2 = 1)
= /< = !
2Д//М (ат/Д//м)
«1-
ат/ДЯм
B44)
На рис. 101 показаны зависимости наклона S и отношения
наклонов /С, полученных по прямым линиям рис. 100, от — log v2.
Обе зависимости сохраняют
линейность до высоких разбавлений,
характеризуемых объемной долей
полимера у2=0,03. Однако, при
дальнейшем разбавлении линейность
утрачивается, и для ее сохранения
следовало бы перейти к
эффективным концентрациям полимера в
координационной сфере клубка,
которые значительно выше средних
значений v2. Если бы эти
эффективные концентрации имели порядок
у2 = 0,02—0,03 формула B41) все
еще была бы справедлива.
Указанный диапазон v2 представляет собой
вполне реальную оценку истинной
концентрации сегментов в пределах
координационной сферы
изолированного набухшего полимерного клубка. Этот вывод
подтверждается постоянством наклона при переходе концентрации от
1% к 0,01%.
По линейным участкам кривых рис. 101 можно определить
величины Об1АНм и ат/АЯм. В зависимости от значений,
придаваемых п и а, аб/А//м колеблется в пределах от 1 до 2 • 10~2, а
ат/АЯм=0,75. Будучи взяты сами по себе, эти значения энергии
поверхности раздела очень малы. Однако низкие значения ат и
аб, вычисленные на основании одного лишь анализа эффекта
разбавления, не отличаются от величин, полученных при
анализе температурного коэффициента кристаллизации
неразбавленного блочного полимера. Фактически, произведение ото2б,
вычисленное по данным рис. 101, находится в хорошем количе*
ственном соответствии с произведением, полученным из наклона
кривой 1 на рис. 99. Поэтому применительно к исследованиям
Рис. 101. Зависимость
наклона 5 и отношения наклонов К
(по данным рис. 100) от — \g v2.
274
эффекта разбавления, когда вопрос об абсолютных значениях
энергий поверхностей раздела не ставится, предпосылки,
лежащие в основе уравнения B41), вполне приемлемы.
Можно считать, что в пределах от чистого полимера до
раствора концентрации порядка 3% изменения температурного
коэффициента (наклоны прямых на рис. 100) могут быть целиком
приписаны члену, содержащему In v2 в выражении для А/7*, где
v2 отождествляются с номинальной (средней) концентрацией.
При более высоких разведениях необходимо заменить среднюю
концентрацию полимера эффективной, полагая ее равной ~3%.
Влияние общего состава системы на шкалу времени при
кристаллизации также можно определить по рис. 100. При
одинаковом значении температурной переменной (Тил/Т)(\/кТJ
константы скорости понижаются при разбавлении. В более
разбавленных системах необходимо значительно большее
переохлаждение, чтобы процесс кристаллизации мог протекать с
измеримой скоростью. Действительно, полученные данные
свидетельствуют о том, что при незначительном переохлаждении
кристаллизация разбавленных систем практически невозможна. Для
достижения постоянной скорости необходимо непрерывно
повышать переохлаждение при разбавлении даже в случае очень
разбавленных растворов. Эти наблюдения противоречат
существующему мнению о том, что в разбавленном растворе нуклеа-
ция протекает внутримолекулярно, в пределах отдельных,
изолированных молекул. Однако для достаточно разбавленного
раствора неизбежно должна быть достигнута точка, в которой
нуклеация становится внутримолекулярной.
Хотя в принципе можно провести последовательный анализ
влияния растворителя на кинетику кристаллизации при
переохлаждениях, мы рассматривали здесь только роль нуклеации
и не фиксировали внимания на процессах переноса и
релаксации. При более детальном анализе полностью игнорировать
возможные изменения этих механизмов в результате разбавления
уже нельзя.
Другой кинетический процесс, протекающий в очень
разбавленных растворах полипептидов и нуклеиновых кислот, связан
с регенерацией упорядоченной спиральной структуры из
статистических клубков. Можно допустить, что превращение клубка
в спираль (или обратный процесс) протекает через
последовательность ступеней, каждая из которых включает одно звено на
границе между спиральными и неупорядоченными участками
цепей. Ввиду принципиальной обратимости этих процессов
элементарные акты прямого и обратного переходов конкурируют
между собой. Поэтому валовой процесс аналогичен одномерной
диффузии или одномерной последовательности случайных
блужданий, причем направление элементарного шага («выбор»)
зависит от относительной вероятности каждой из конкурирующих
18*
275
ступеней. До начала роста спиральной формы, описанного
выше, необходимо инициирование спиральной последовательности.
Оно может произойти на конце цепи, которая в исходном
состоянии представляет собой абсолютно неупорядоченный
статистический клубок, или же (что связано с большими трудностями)
может начинаться в некоторой точке развивающейся цепи, еще не
затронутой переходом.
Превращение отдельной изолированной цепи из клубка в
спираль можно схематически представить в форме [73]:
cn <~^ cn_lnl ^=±сЛ_2я2 <=^ ... с1нп_1 <=^ нп
Hb Rb Rb b
где Cn^iHt — макромолекула, состоящая из последовательности
п — / неупорядоченных звеньев, смежной с
«линейно-кристаллической» спиральной
последовательностью /-звеньев;
?и — константа скорости инициирования;
kf—константа скорости роста спирали;
kb — константа скорости обратного процесса.
Скорость этого обратного процесса можно считать равной
скорости инициирования, помноженной на математическое
ожидание того, что возникающий спиральный блок в конечном итоге
охватит всю молекулу.
Используя методы стационарной кинетики [74] или
математической теории игр [73], можно получить следующее выражение
для скорости образования спирали:
чг = кАх-т}) B45)
где Сн и Сс — концентрации «спиральных» и неупорядоченных
звеньев.
Отношение falfa можно приравнять /С_п,
где К — константа равновесия реакции спираль^статистиче-
ский клубок. Вблизи температуры плавления, или
перехода
^=*иСс-^(-ДГ) B46)
где А//м — изменение энтальпии на одно мономерное звено при
переходе от спирали к клубку;
AT— переохлаждение, при котором протекает процесс.
Если допустить, что статистический клубок представляет
собой устойчивую форму при высоких температурах, то при
понижении температуры ниже ГПл величина (АНм/RT2) (—AT)
должна увеличиться. Если же принять, что константа fa аналогична
константе скорости химической реакции, то она должна умень-
276
шаться с понижением температуры. При небольших
переохлаждениях в уравнении B46) доминирует величина
(AHM/RT2) (—ДГ), так что вблизи температуры перехода
температурный коэффициент скорости спирализации должен быть
отрицательным. При больших переохлаждениях, напротив,
должна доминировать экспоненциальная зависимость kn от
температуры, и температурный коэффициент скорости спирализации
должен стать положительным.
Поэтому при понижении температуры
скорость должна проходить через
максимум. Здесь сразу же
бросается в глаза сходство между
температурной зависимостью для
одномерного роста спирали и для
трехмерной кристаллизации полимеров.
Однако, как показывает сравнение
соответствующих уравнений, это
сходство лишь качественное.
Теория для простой (однотяже-
вой) спирали может быть
распространена на спирали из двух или
более цепных молекул. Кинетический
порядок процесса изменится, так
как следует ожидать, что скорость
инициирования должна зависеть от
числа цепей, участвующих в
образовании мультиспирали.
Флори и Уивер [75] исследовали
скорость, с которой протекает рес-
пирализация обратимо
денатурированного коллагена. Они наблюдали
заметный отрицательный температурный коэффициент, однако
он зависел от температуры сильнее, чем это следовало бы из
уравнения B46). Кроме того, процесс соответствовал кинетике
первого порядка, что противоречит представлениям, связанным
с триспиральной структурой коллагена. Чтобы обойти эту
трудность, необходимо было допустить, что определяющая скорость
ступень превращения связана с образованием неустойчивой
промежуточной формы, которая может представлять собой од-
нотяжевую спираль в конформации полипролина II.
Росс и Стюртевант [76] исследовали скорость взаимодействия
полирибоадениловой кислоты (поли А) и полирибоуридиловой
кислоты (поли У), приводящего к образованию упорядоченного
комплекса поли (А+У) A:1). Рентгенографический анализ
показал, что волокна этого комплекса имеют биспиральную
структуру, подобную структуре натуральной дезоксирибонуклеиновой
кислоты (ДНК). Наблюдавшаяся кинетика этого процесса была
й^/VNaCl
0t25Mti&C\
20
40
60 1С
Рис. 102. Зависимость
кажущейся константы скорости
второго порядка k от
температуры при образовании
комплекса поли (А + И) [76].
Вертикальные линии обозначают
температуры перехода для двух
указанных на кривых концентраций соли.
Концентрация полимерного фосфата
3,6 • 10Ж.
277
сложной. Процесс бйачале подчиняется кинетике второго
порядка, которая к концу перехода отклоняется в сторону
первого порядка. Это подтверждает ту точку зрения, что
инициирование комплекса поли (А + У) сопряжено с образованием
нескольких пар оснований, скрепленных водородными связями, за
которым следует рост упорядоченных спиральных областей. При
слиянии.равных объемов эквимолекулярных растворов
индивидуальных полирибонуклеотидов начальная скорость спирализа-
ции пропорциональна квадрату концентрации однотяжевых
макромолекул.
Скорость образования комплекса поли (А + У),
определенная по кажущейся константе скорости второго порядка,
показана на рис. 102 в зависимости от температуры, при которой
происходило перемешивание компонентов. При понижении
температуры ниже температуры перехода скорость повышается,
проходит через отчетливый максимум и затем вновь уменьшается
при дальнейшем снижении температуры. Температура
максимальной скорости зависит от температуры перехода и
повышается вместе с нею. Наличие максимума зависимости скорости от
температуры подтверждает теорию, изложенную выше.
Литература
1. L. Mandelkern, A. S. Posner, A. F. Diori, D. E. Roberts, J.
Appl. Phys., 32, 1509 A961).
2. С. W. В u n n, Trans. Faraday Soc, 35, 483 A939).
3. N. Bekkedahl, J. Res. Natl Bur. Stand., 13, 411 A934).
4. L. Mandelkern, in R. H. D о r e m u s, B. W. Roberts, D. Turn-
bull (eds.), «Growth and Perfection of Crystals», New York, 1958.
5. L. A. W о о d, N. В e k k e d a h 1, J. Appl. Phys., 17, 362 A946).
6. L. Mandelkern, F. A. Q u i n n, Jr., P. J. F1 о г y, J. Appl. Phys., 25,
830 A954).
7. W. H. Cobbs, Jr., R. L. Burton, J. Polymer Sci., 10, 275 A953).
8a. M. Takayanagi, Mem. Fac. Eng., Kyushu Univ., 16C), 111 A957).
8b. M. Takayanagi, T. Yamashito, J. Polymer Sci., 22, 552 A956).
9. K. Ueberreiter, G. Kanig, A. S. Brenner, J. Polymer Sci., 16,
53 A955).
10. A. N. Gent, J. Polymer Sci., 18, 321 A955).
11a. P. J. Flory, A. D. Mclntyre, J. Polymer Sci., 18, 592 A955).
lib. A. D. Mclntyre, Ph. D. Thesis, Cornell University, Ithaca—New
York, 1956.
12. R. B. Richards, S. W. Hawkins, J. Polymer Sci., 4, 515 A949).
13. A. S h a r p 1 e s, Polymer, 3, 250 A962).
14. P. W. Al len, Trans. Faraday Soc, 48, 1 A952).
15. L. B. Morgan, Phil. Trans. Roy Soc. London, Ser. A, 247, 13, 23 A954).
16. L. B. Morgan, F. D. Hartley, F. W, Lord, Ric. Sci., Suppl. A, 25,
577 A955).
17. F. P. Pri ce, J. Am. Chem. Soc, 74, 311 A952).
18. B. B. Burnet, W. F. M с D e v i t, J. Appl. Phys., 28, 1101 A957).
19. F. V о n G о 1 e rt G. S а с h s, Z. Phys., 77, 281 A932).
278
20. W. A. Johnson, R. F. Me hi, Trans. Am. Inst. Mining Met. Engrs, 135,
416 A939).
21. M. Avrami, J. Chem. Phys, 7, 1103 A939); 8, 212, A940); 9, 177
A941).
22. R. U. Evans, Trans. Faraday Soc, 41, 365 A945).
23. J. E. Burke, D. Turnbull, Progress in Metal Physics, vol. 3, New
York, 1952, p. 220.
24. L. M a n d e 1 к e r n, J. Appl. Phys., 26, 443 A955).
25. G. M. Martin, L. Mandelkern, J. Appl. Phys., 34, 2312 A963).
26. S. В u с к s e r, L. H. T u n g, J. Phys. Chem., 63, 763 A958).
27. J. H. Griffith, B. G. R a n b y, J. Polymer Sci., 38, 107 A959).
28. В. К a hie, Z. Elektrochem., 61, 1318 A957).
29. J. H. M a gi 11, Polymer, 2, 221 A961); 3, 35 A962).
30. J. D. Hoffman, J. J. Weeks, W. M. Murphey, J. Res. Natl Bur.
Stand., A, 63, 67 A959).
31. L. Mandelkern, F. A. Quinn, Jr., Abstracts, 134th Meeting of the
American Chemical Society, Chicago, III, September, 1958.
31a. W. Banks, M. Gordon, R. J. Roe, A. Sharpies, Polymer, 4, 61
A963).
32. S. Matsuoka, C. J. Aloisio, Bull. Am. Phys. Soc, Ser. II, 8C),
A963).
33. R. Chiang, P. J. Flory, J. Am. Chem. Soc, 83, 2857 A961).
34. L. H. T u n g, S. В u с к s e r, J. Phys. Chem., 62, 1530 A958).
35. H. Kojima, K. Yamaguchi, Chem. High Polymers (Tokyo), 19, 715
A962).
36. P. J. Flory, Nature, 124, 53 A956).
37. F. Go r nick, J. L. Jackson, J. Chem. Phys., 38, 1150 A963).
38. B. V. Fa Ik a i, H. A. Stuart, Koll. Z., 162, 138 A959).
39. J. W. Gibbs, Collected Works, vol. 1, New York, 1931, pp. 55—372.
40. R. С Tolman, J. Chem. Phys., 16, 758 A948); 17, 118, 333 A949).
41. F. P. Buff, J. G. Kir к wood, J. Chem. Phys., 18, 991 A950).
42. R. S. Brad ley, Quart. Rev. (London), 5, 315 A951).
43. D. Turnbull, J. Chem. Phys., 18, 198 A950).
44. J. H. Hollomon, Thermodynamics in Metallurgy, Cleveland, 1950,
pp. 161—177.
45. D. T u r n b u 11, Solid State Physics, vol. 3, New York, 1956.
46. D. T u r n b u 11, J. Chem. Phys., 20, 411 A952).
47. D. T u г n b u 11, J. С F i s h e r, J. Chem. Phys., 17, 71 A949).
48. D. Turnbull, Principles of Solidification, in «Thermodynamics in
Physical Metallurgy», Cleveland, 1949.
49. P. J. Flory, J. Am. Chem. Soc, 84, 2857 A962).
50. P. J. Flory, J. Chem. Phys., 17, 223 A949).
51. J. D. Hoffman, J. I. Lauritzen, J. Res. Natl Bur. Stand., A, 64,
73 A960); 65,297 A961).
52. D. Turnbull, J. Appl. Phys., 20, 817 A949); 21, 1022 A950).
53. D. Turnbull, R. E. Сech, J. Appl. Phys., 21, 804 A950).
54. D. G. Thormas, L. A. K. S t a v e 1 y, J. Chem. Soc, 1952, 4569.
55. H. S. D e N о г d w a 11, L. A. K. S t a v e 1 y, J. Chem. Soc, 1954, 224.
56. D. Turnbull, R. L. Cor mi a, J. Chem. Phys., 34, 820 A961).
57. F. P. Price, Ann. N. Y. Acad. Sci., 83, 20 A959).
58. M. J. Richardson, P. J. Flory, J. B. Jackson, Polymer, 4, 221
A963).
59. R. L Cormia, F. P. Price, D. Turnbull, J. Chem. Phys., 37, 1333
A962).
60. M. L. Williams, R. F. L a n d e 1, J. D. Ferry, J. Am. Chem. Soc, 77,
3701 A955).
61. J. H. Но И oman, D. Turnbull, Progress in Metal Physics, vol. 4.
London, 1953, p. 333.
62- B. Chalmers, Physical Metallurgy, New York, 1959, p. 249.
279
63. А. Ко vacs, Ric. Sci., Suppl. A, 25, 668 A955).
64. R. Buchdahl, R. L. Miller, S. Newman, J. Polymer Sci., 36, 215
A959).
65. J. С H a 1 p i n, L. M a n d e 1 к е г п, неопубликованные данные.
66. R. S. S t e i n, S. N e w m a n, частное сообщение.
67. F. Gornick, L. M a n d e 1 k e г n, J. Appl. Phys., 33, 907 A962).
68. В. Wunderlich, Abstract of Paper, Division of Polymer Chemistry,
American Chemical Society Meeting, Atlantic City, N. J., September, 1962,
p. 63.
69. R. К. Е b y, Abstract of Paper, Division of Polymer Chemistry, American
Chemical Society Meeting, Atlantic City, N. J., September, 1962, p. 74.
70. W. B. Barnes, W. G. Luetzel, F. P. Price, J. Phys. Chem., 65, 1742
A961).
71. V. Holland, P. Lindenmeyer, J. Polymer Sci., 57, 589 A962).
72. J. B. Jackson, P. J. F 1 о r y, R. Chiang, Trans. Faraday Soc, 59, 1906
A963).
73. P. J. F lory, J. Polymer Sci., 49, 105 A961).
74. M. Saunders, P. D. Ross, Biochem. Biophys. Res, Commun., 3, 314
A960).
75. P. J. Flory, E. S. Weaver, J. Am. Chem. Soc, 82, 4518 A960).
76. P. D. Ross, J. M. Sturtevant, J. Am. Chem. Soc, 84, 4503 A962).
Морфология
Кристаллизация полимеров из расплава
Как уже указывалось в гл. 1, на всех доступных
наблюдению уровнях надмолекулярной ориентации кристаллических
полимеров мы встречаемся с особыми характерными чертами их
морфологии. Для определения кристаллической текстуры
используются методы электронной микроскопии, рассеяния света
или рентгеновских лучей и прямого наблюдения в световом
микроскопе, позволяющие характеризовать структурные элементы
с размерами больше элементарной ячейки. Большинство
полимеров диффузно рассеивает рентгеновские лучи, но для многих
систем наблюдаются и относительно резкие дифракционные
максимумы.
Как правило, дифракционные максимумы многих порядков
обнаружены для природных фибриллярных белков, таких как
кератины [1], коллаген [2], мышечные волокна [3, 4].
Преимущественно меридианальные рефлексы найдены для некоторых од-
ноосноориентированных синтетических волокон [6—11] и
единичных полимерных кристаллов, выращенных из разбавленного
раствора [12, 13]. Многие неориентированные полимеры,
закристаллизованные из расплава, обнаруживают также дискретные
малоугловые дифракционные максимумы [14—16]. В. последнем
случае максимум на рентгенограмме не ограничен
меридиональной областью, а имеет вид круга.
Молекулярная интерпретация этих результатов важна для
понимания природы как самых кристаллитов, так и их
взаимной организации. Необходимо установить соответствие между
термодинамической природой кристаллического состояния (по
данным исследования фазовых равновесий), основными
кинетическими механизмами кристаллизации и морфологическими
наблюдениями. Именно с этой точки зрения мы рассмотрим
проблему больших периодов в малоугловом рентгеновском
рассеянии и влияние условий кристаллизации на характер
дифракционных максимумов.
281
Абсолютная интенсивность малоуглового рассеяния для
различных ориентированных и неориентированных кристаллических
полимеров изучалась Германсом и Вейдингером [17].
Тщательному исследованию были подвергнуты такие полимеры, как
целлюлоза, полиэтилен, полиэтилентерефталат, поликапроамид и
полиоксиметилен. За исключением целлюлозных волокон,
рассеяние от которых связано в основном с наличием
микрополостей [18], абсолютная интенсивность рассеяния определяется
числом и размерами флуктуации электронной плотности,
обусловленных сосуществованием кристаллической и аморфной фаз.
Это заключение совпадает с более ранними
предположениями Гесса и Киссига [19] о причине дифракционного максимума
в ориентированных волокнах. Названные авторы связывали
появление малоугловых меридианальных максимумов в
полимерных системах с прохождением развернутых ориентированных
цепей через кристаллические и аморфные участки структуры.
Периодичность обусловлена при этом достаточно правильным
чередованием упорядоченных кристаллических и разупорядочен-
ных аморфных областей, и периодическое изменение
электронной плотности отражает действительное различие плотностей
двух фазовых состояний. Сосуществование кристаллических и
аморфных областей подтверждается изучением фазового
равновесия и анализом кинетики кристаллизации. Таким образом,
подобная причина возникновения флуктуации электронной
плотности, определяющих малоугловое рассеяние, подтверждается
результатами других наблюдений.
Если, однако, не признавать существования аморфных
областей и считать, что во всех случаях мы имеем дело с сильно
дефектными кристаллами, то малоугловое рассеяние должно
вызываться только нарушениями дальнего порядка в
кристаллической решетке, и дифракционные максимумы в этом случае
должны указывать на периодичность таких нарушений [19].
Следовательно, предполагается, что правильно сложенные
кристаллиты представляют основные структурные элементы
материала, закристаллизованного в блоке, и именно они
ответственны за дифракционные максимумы. Это подтверждается
электронномикроскопическими наблюдениями поверхностей
скола, а также тем, что все дифракционные максимумы
соответствуют ограниченному диапазону размеров 50—200 А, соответ^
ствующему толщинам пластинчатых кристаллов.
Исследование кинетики выявляет сильную зависимость
морфологии от условий кристаллизации, особенно от температуры
кристаллизации. Следовательно, можно предвидеть зависимость
положения дифракционных максимумов от температуры
кристаллизации, что и подтверждается экспериментами [14, 21, 22].
В частности, тщательное изучение дифракционных максимумов
малоуглового рассеяния в широкой области температур кри-*
282
сталлизации было проведено на кристаллическом, но неориен*
тированном линейном полиэтилене. Результаты одной из серии
наблюдений для образца нефракционированного линейного
полиэтилена толщиной 1,0 мм представлены на рис. 103 [14].
Для образца, закристаллизованного в области 115—120° С,
хорошо разрешаются два максимума, численно эквивалентные
рефлексам первого и второго порядка. Незаштрихованные
90 ПО №
КС
Рис. 103. Зависимость
малоугловых периодов
рассеяния d от
температуры кристаллизации для
образца линейного
полиэтилена (марлекс-50)
толщиной 1 мм [14]:
ф — экспериментально
наблюдаемые периоды; О
—теоретически рассчитанные.
ом ом
2В,2рад
Рис. 104. Интенсивность
малоуглового рассеяния /
для образцов
полиэтилена [15]:
1 — узкая фракция с М=360 000,
закристаллизованная при 131,3° С;
2— тот же образец, но при
использовании монохроматического
пучка; 3—быстро
закристаллизованный нефракционированный
образец, при использовании
монохроматического излучения.
кружки соответствуют рефлексам первого порядка,
рассчитанным в предположении, что при более высоких температурах
кристаллизации разрешаются только рефлексы второго порядка.
Прямые наблюдения очень больших периодов, предполагаемых
в этом случае, были выполнены Полаком, Робинсоном, Чангом
и Флори [15] на фракциях линейного полиэтилена (М= 126000
и 360000), закристаллизованных при 131,3°С. Полученные
этими авторами результаты представлены на рис. 104, где
хорошо виден дискретный максимум порядка 1100 А.
Область основных периодов для нефракционированного
образца простирается от 200 до 900 А. Как и предполагалось,
величина периода сильно зависит от температуры. При
температурах кристаллизации ниже 100° С дифракционный максимум
соответствует ~250 А и мало чувствителен к температуре кри-
283
сталлизации, в то время как выше этой температуры
наблюдается резкое увеличение периода. Следовательно,
предполагать, что дифракционные максимумы в блочной системе
ограничены областью 100—200 А, нет оснований. Различия
надмолекулярной структуры высокомолекулярной фракции и нефрак-
ционированного образца выявляют влияние молекулярного веса
и молекулярно-весового распределения на морфологические
особенности кристаллического\состояния. Выше, при
обсуждении различий в процессах плавления нефракционированных
образцов и узких фракций, мы пришли к такому же заключению.
Важность этих наблюдений совершенно очевидна, как и
необходимость весьма тщательного проведения и анализа опытов
подобного рода. В частности, необычно высокое содержание
низкомолекулярных фракций в нефракционированном образце
линейного полиэтилена марлекс-50 делает возможной
избирательную кристаллизацию этих «парафиноподобных» фракций.
Приведенные результаты подчеркивают значение
изотермической кристаллизации. Для такого сравнительно быстро
кристаллизующегося полимера, как полиэтилен, возникают, однако,
определенные технические трудности во время проведения
изотермической кристаллизации при охлаждении расплава до
достаточно низких температур. При этом, как видно из табл. 15,
возможно сильное проявление влияния толщины образца на
положение дифракционного максимума.
Из табл. 15 следует, что при любой определенной
температуре кристаллизации большие периоды тонких образцов меньше,
нежели у более толстых. Различие особенно велико при низких
температурах, но несколько уменьшается при возрастании
температуры кристаллизации. Эффект толщины образца может
быть объяснен трудностями проведения изотермической
кристаллизации полимера с относительно высокой скоростью
кристаллизации при больших переохлаждениях. Меньшие значения d
для тонких образцов можно приписать различиям в скорости
охлаждения расплава. Для тонких образцов скорость
охлаждения от расплава до температуры кристаллизации выше и,
следовательно, теплота кристаллизации легче диссипирует. Таким
образом, при низких температурах условия изотермической
кристаллизации реализуются с лучшим приближением для более
тонких образцов, и значения dy полученные для них, в большей
степени приближаются к равновесным.
Но наиболее важным результатом является зависимость d от
температуры кристаллизации, особенно в области Тпл. Это
подтверждает, что d действительно определяется некоторым
характеристическим размером, связанным с размером критического
ядра [14, 22]. Если ядра по крайней мере в одном направлении
не перерастают существенно сверх критических размеров, то
должна развиваться четкая периодичность размеров кристаллитов.
284
Таблица 15
Сравнение рефлексов первого порядка для
образцов нефракционированного полиэтилена различной
толщины
Температура
кристаллизации,
°С
о
105
ПО
115
118
• 120
123
125
Период рассеяния d
1,0 мм
230
308
330
345, 380
375
380, 400
400
445
0,5 мм
277
0,1 мм
150
238
256
257
277
270
320
360—400
В соответствии с изложенной выше теорией нуклеации
размеры критического ядра р* и ?* сильно зависят от степени
переохлаждения [см. уравнения B13) и B14)].
Для подтверждения этой гипотезы на рис. 105 отложена
зависимость периода рассеяния d (первого порядка) от
безразмерной температурной характеристики ТПЛ/АТ для нефракцио-
нированных образцов линейного полиэтилена. Горизонтальная
ось на этом рисунке — нелинейная шкала температур, сильно
растянутая в области высоких температур кристаллизации и
сжатая в области больших переохлаждений. Данные для
высоких температур кристаллизации согласуются с ожидаемой
линейной зависимостью, которая экстраполируется к очень малым
периодам d при больших переохлаждениях. Однако при
кристаллизации ниже ~115° С наблюдается отклонение от
линейности, причем в достаточно широком интервале температур
изменение величины d относительно мало. Не исключена
возможность, что в этом интервале температура окружающей среды
и температура, при которой в основном проходила
кристаллизация, не идентичны. Последняя температура, конечно, выше.
Поэтому вполне возможно, что если бы удалось осуществить
изотермическую кристаллизацию при более низких
температурах, то температурная зависимость d представилась бы прямой
линией во всей области кристаллизации.
При значительных переохлаждениях безразмерная функция
Тпл/АТ практически не зависит от температуры плавления и,
возможно, одинакова для различных полимеров. Таким
образом, при кристаллизации со значительными переохлаждениями
можно всегда ожидать возникновения периода порядка 100 А
285
в согласии с опытными данными. Так, например, Эппе, Фишер
и Стюарт [16], исследовавшие кристаллизацию образца
полиуретана в стеклообразном состоянии при температурах, весьма
удаленных от температуры плавления, наблюдали
дифракционные максимумы в области 50—150 А. По-видимому, из опытов,
проведенных при значительных переохлаждениях, следует, что
дифракционные максимумы закристаллизованного в блоке
полимера в самом деле лежат в этих пределах. Тем не менее для
25
0/5075 105110 mm 120 123 125 121 128 129
nil
130
тХ
131
Рис. 105. Зависимость малоугловых периодов рассеяния d от ТПЛ/АТ для
нефракционированного линейного полиэтилена [14]:
ф —образец толщиной 1 мм; Д —толщиной 0,1мм. Верхняя шкала —нелинейная шкала
температур.
широкой области температур значения периодов не
ограничиваются указанными пределами.
Линейная зависимость на рис. 105 подтверждает
предположение, что рост критических ядер ограничен по крайней мере
в одном направлении. Для дальнейшего обсуждения следует
уточнить обозначение размеров в различных
кристаллографических направлениях. Естественно предположить, что рост
задерживается в направлении цепи, или оси с, в то время как
развитие граней кристалла в перпендикулярных к оси цепи
направлениях ничем не ограничивается. В таком случае, если принять
Гпл равной 137,5° С, из уравнения B13) и наклона прямой
линии на рис. 105 значение ат/АЯм равно 2,6. Для полиэтилена
286
это соответствует 2500 кал/моль мономерных звеньев или,
поскольку размеры элементарной ячейки известны, 95 эрг/см2.
Определяемое из опыта отношение ат/Д#м весьма чувствительно
к принятому значению равновесной температуры плавления.
Например, не чрезмерно завышенное значение 143° С для Гпл уже
дает для энергии поверхности раздела 260 эрг/см2.
Значения для ат/Д#м кажутся, на первый взгляд,
исключительно высокими, однако совпадают с полученными другими
методами. Так, анализ плавления сополимеров [23] дает
значение ат = 170 эрг/см2 [2, 24], а Корми, Прайс и Торнбал [25] из
опытов с переохлажденными каплями получили 168,5 эрг/см2.
Необходимо заметить, что анализ данных малоугловой
дифракции основан на теории гомогенной нуклеации. Влияние
гетерогенности на направлении цепи при образовании ядер может
привести к более высоким значениям ат/АЯм. Опыты с
переохлажденными каплями показывают, что, изолируя
гетерогенности, можно достичь гораздо больших переохлаждений,
нежели при обычной! блочной кристаллизации. Тем не менее,
анализ закристаллизованных в блоке образцов также дает высокие
значения от/АНм.
Таким образом, существует определенная возможность, что
при образовании асимметричного ядра каталитический эффект
гетерогенности отражается только на одном из размеров. Эта
точка зрения позволяет объяснить, почему в блочных образцах
удается достичь лишь ограниченных переохлаждений, несмотря
на то, что в обоих типах экспериментов полученные значения
От СХОДНЫ.
Необычно высокое значение ат должно быть обусловлено
молекулярной природой соответствующей поверхности раздела.
Эта поверхность, на которой осуществляется переход
связанных последовательностей звеньев из кристалла в расплав, не
имеет аналогии при кристаллизации мономерных веществ. Если
принять, что ядро с регулярными резкими складками
образовано единичной молекулой, то ат может быть
идентифицировано со свободной энергией, необходимой для образования
такой складки в цепи. С другой стороны, если в нуклеации
участвует набор молекул, а образование резких складок не
обязательно, то ат представляет сумму избыточных свободных
энергий для участков одной цепи, проходящей из полностью
упорядоченной кристаллической области в полностью разупорядо-
чениую аморфную фазу. Как уже указывалось, это изменение
упорядоченности не может произойти на коротком участке в
пределах одного звена, так как в этом случае значительная
энтальпия и низкая энтропия привели бы к чрезмерно высокому
значению сгт.
Подобный анализ малоугловой диффракции на полимерах,
с учетом заторможенности роста кристаллов в одном направ-
28?
лении, приводит к заключению, что первичной кристаллической
единицей является ламеллярный или пластинчатый кристаллит.
К такому выводу можно прийти и без электронномикроскопиче-
ского наблюдения.
Поверхностные реплики тонких пленок,
закристаллизованных из расплава или поверхности скола толстых образцов,
обнаруживают ламеллярные структуры (см. рис. 9). Хотя подобные
типы структур наблюдались для множества полимеров [16, 22,
28—30], явно недостаточно принимались во внимание условия
кристаллизации и возможные различия молекулярных весов
образцов, без чего нельзя отождествлять эти наблюдения и
искать для них корреляции с данными малоуглового
рентгеновского рассеяния. Обычно ламелли имеют толщину порядка
100—150 А*, причем оси цепей ориентированы нормально к
широкой грани, что возможно только при многократном
прохождении одной и той же цепи через кристаллит. Эти размеры в
основном и обусловливают большие периоды, наблюдаемые при
малоугловом рассеянии рентгеновских лучей на образцах,
закристаллизованных при очень сильном переохлаждении.
Результаты исследования идентичных образцов обоими методами
совпадают, если периоды и размеры лежат в области 100—200 А.
Однако, по данным Шелла [22], при возрастании
дифракционного максимума до 300—400 А размеры ламеллей,
наблюдаемых на поверхности блочного полимера, заключены в
диапазоне всего 80—140 А. Неизвестно, действительно ли такая же
ситуация сохраняется и при кристаллизации, приводящей к
дифракционным максимумам порядка 1000 А.
Если непосредственно наблюдаемые в электронном
микроскопе ламелли на самом деле ограничены по толщине в
результате кристаллизации при малых переохлаждениях, то
необходимо изменить интерпретацию дифракционных результатов; в
противном же случае следует предположить, что локальные
деформационные процессы, происходящие при сколе, оказывают
некоторое влияние на морфологию и кристаллическую текстуру
поверхности. Необходимо заметить, что поверхности скола
обычно получают при температуре жидкого азота, что гораздо ниже
температуры стеклования большинства полимеров,
представляющих интерес.
Относительно недавно выдвинутая концепция образования и
существования ламеллярных кристаллитов в гомополимерах,
закристаллизованных в блоке, по-видимому в достаточной мере
подтверждается приведенными фактами. Однако, она
дополняется теперь важным выводом о том, что полимерные цепи
внутри отдельного кристаллита регулярно и последовательно
* Недавно при исследовании поверхностей скола низкомолекулярных
фракций полиэтилена, изотермически закристаллизованных при высокой
температуре, были обнаружены ламелли толщиной до 700 А,
288
сложены, а периодичность, наблюдаемая под малыми углами,
определяется периодом складки. В то же время довольно
затруднительно отождествление больших периодов (рис. 103
и 104) с регулярной складчатой структурой, принимая во
внимание молекулярный вес и молекулярно-весовое распределение
образцов. Это затруднение можно пытаться обойти,
предположив присутствие в решетке концов цепей и других структурных
нерегулярностей. Однако такое предположение противоречит
данным для кристаллов, выращенных при малых
переохлаждениях и характеризуемых высокой температурой плавления и
резким переходом; оно противоречит также экспериментам,
демонстрирующим удаление хвостов цепей из решетки.
Разумеется нет необходимости отождествлять ламеллярные
структуры с регулярно и плотно сложенными цепями, так как
обычные цепи вполне могут образовывать петли или частично
складываться. Различные кристаллические последовательности
одной и той же молекулы могут и не проходить через смежные
участки кристаллита. Таким образом, возможно существование
некристаллических или аморфных областей, связанных с
кристаллитом. Более того, некоторые цепи могут проходить через
кристаллит только один раз и после прохождения аморфной
области войти в другие кристаллиты. Другая же часть цепей
после выхода из кристаллита может снова вернуться в него.
В принципе, следовательно, нет несоответствия между
наличием ламеллярных кристаллитов, толщина которых значительно
меньше длины вытянутой молекулы, и существованием
аморфных или жидкоподобных областей. Однако противоречия
неминуемо возникают, если принять, что кристаллит состоит только
из регулярно и плотно сложенных цепей. Подобная точка
зрения не следует ни из фундаментальной теории, ни из
тщательного анализа экспериментальных данных.
Причину образования кристаллитов ламеллярного типа
можно установить при анализе конформаций и способов упаковки
полимерных цепей вблизи поверхности раздела и в зоне полной
изотропности [32]. Вследствие непрерывности полимерной цепи
порядок, существующий в кристаллите, не может исчезать
внезапно на границе раздела, нормальной к направлению оси с.
Это обстоятельство подчеркивалось при обсуждении высокого
значения свободной энергии поверхности раздела стт.
Рассмотрим ламеллярный кристалл с неограниченными
размерами больших (перпендикулярных оси с) граней, показанный
на рис. 106 [32]. Выходящие из кристалла цепи пересекают под
прямым углом плоскость раздела ВВ и входят в соседние
области. Часть цепей далее пересекает плоскость СС,
находящуюся в полностью изотропной аморфной области. Необходимо
установить, можно ли уместить в аморфной области все цепи,
выходящие из кристалла. Если Ак — площадь поперечного
19 Л. Манделькерн
289
сечения одной цепи в кристаллической фазе, то число цепей па
единицу площади, выходящих из грани кристалла
B47>
Для подсчета аналогичного числа цепей Wa, пересекающих
плоскость СС, примем в качестве модели аморфного полимера
цепь из свободно сочлененных сегментов. Ориентации соседних
сегментов взаимонезависимы, так что конформация цепи
представляется непрерывно изменяющей направление линией. Длину
площади поперечного сечения и объем сегмента обозначим U
Аа и Va, соответственно. Чтобы сегмент, ориентированный под
углом 9 к нормали плоскости СС, мог ее пересечь, его начало
должно находиться на расстоянии не более / cos 0 от данной
плоскости.
Число сегментов (или связей), пересекающих единицу
площади плоскости СС под углами от 9 до 9 + d9, равно:
лг0</е =
VT1/ cose sinG dQ
л/2
Jsin
e^e
= v^1/cose sin e^
B48>
д С
Рис. 106. Схематическое
изображение сечения
зоны поверхности
раздела по оси с [32].
где V^1 — концентрация сегментов в
отсутствие растворителя.
Интегрирование по всем
направлениям 9 дает:
я/2
B49)
Сравнивая с уравнением B47) получим для отношения
плотностей потоков цепей в аморфной и кристаллической областях:
NK
2 Ая
B50)
Если, например, Ак/Аа порядка единицы, то не более
половины всех цепей, выходящих из грани кристалла, попадает в
полностью изотропную область. Оставшаяся часть цепей
должна каким-то образом рассеиваться. Так как они никуда не
могут исчезнуть, уже ясно, что они должны возвращаться в
кристалл.
Для более подробного анализа необходимо установить
относительное смещение плоскостей ВВ и СС. При увеличении
расстояния между ВВ и СС можно ожидать внедрения аморфных
цепей в зону между двумя плоскостями из других источников,
290
не связанных с рассматриваемым кристаллитом. Поэтому поток
цепей, выходящих из кристаллита и достигающих СС, может
оказаться меньше того предельного значения, которое следует
из уравнения B50). Этот эффект усугубляется присутствием
соседних кристаллитов, которые могут захватывать часть цепей
из аморфных областей.
Равенство B50) дает, таким образом, верхний предел той
доли цепей, которые, выходя из неограниченного в поперечных
направлениях ламеллярного кристалла, не могут вернуться в него
вновь. Это заключение получено исключительно в результате
сравнения упаковки и конформации полимерных цепей в
аморфных областях и в упорядоченной кристаллической решетке.
Относительное число цепей, которые могут расположиться в
аморфных областях, определяется отношением Аи/Аа. Если цепи
в кристаллическом состоянии развернуты и имеют конформа-
цию плоского зигзага, например в полиэтилене, и ориентации
связей в аморфном состоянии не скоррелированы, то отношение
площадей поперечных сечений должно совпадать с отношением
соответствующих удельных объемов. Это значение для
большинства полимеров было бы порядка единицы.
Следовательно, при Лк/Ла=1 и значительной поверхности
пластинок по крайней мере половина выходящих из кристалла
цепей должна вернуться в него. При этом вовсе не обязательно,
чтобы кристаллиты состояли из регулярно сложенных или пло*
ских цепей. Значительная часть цепей может попадать в
изотропную область и часть из них войдет в другие кристаллиты.
Те же цепи, которые возвращаются в кристаллит, могут до
возвращения довольно долго «путешествовать» в области раздела.
Действительно, a priori не требуется вовсе, чтобы цепь
возвращалась в кристалл в позиции, смежной с местом ее выхода из
него. Таким образом, вполне совершенные ламеллярные
кристаллиты могут возникать при кристаллизации из расплава
полимера, и это никак не противоречит экспериментальным
наблюдениям.
Нет также необходимости приписывать молекуле
обязательную «склонность» к регулярному (плотному) складыванию.
Следовательно, принципиального запрета на сосуществование
ламеллярных кристаллитов и аморфных областей не существует.
Важно лишь допустить, что складывание цепей нерегулярно.
Тогда требования и эксперимента и теории легко
удовлетворяются в предположении, что кристаллические
последовательности одной и той же молекулы проходят через кристаллит
чисто статистически, не в смежных позициях. Разумеется, в
зависимости от кристаллографических деталей и конформации
аморфных цепей можно ожидать складывание их в кристалле.
Однако не существует никаких оснований для образования
регулярной слоисто-складчатой структуры.
19*
291
Возможны и такие случаи, когда отношение AJAa
превышает единицу. Тогда NJNa окажется порядка единицы, т. е. все
выходящие из кристаллита цепи расположатся в аморфных
областях. Пример этому — а-спиральная кристаллическая форма
полипептидов. Проекция мономерного звена на ось спирали
равна 1,5 А, тогда как длина транс-пептлного звена 3,8 А. Так
как ЛкМа примерно равно обратному отношению этих длин, то
#а/#к=1,25. В этом случае было бы совсем нетрудно упаковать
неупорядоченные цепи в доступном для них пространстве.
Сделанные выводы основаны на предположении, что
поперечные размеры кристалла значительно больше его толщины.
Такое условие, однако, не применимо к ядру критического
размера. Оценка свободной поверхностной энергии боковых
граней Об показывает [см. уравнение A69)], что для полиэтилена,
закристаллизованного при обычных переохлаждениях, ядра
состоят из 20—100 звеньев. Для такого малого числа цепей можно
легко обойти пространственные затруднения, характерные для
кристаллитов. Действительно, зародышевые грани в этом
случае весьма малы, и выходя из них цепи могут расходиться в
направлениях, перпендикулярных оси первичной пачки.
Следовательно, для образования ядра не требуется какого-либо
складывания цепей.
С геометрической точки зрения критическое ядро,
образованное последовательностями различных молекул, является
наиболее вероятным зародышем структуры. При возникновении
такого ядра должны удовлетворяться лишь минимальные
требования, касающиеся кооперативное™ взаимодействий
неупорядоченных цепей. Такая пачечная модель ядра наилучшим
образом согласуется с кинетическим анализом и поэтому отражает
наиболее вероятный механизм нуклеации. Кроме того, нет
никаких оснований думать, что расположение цепей в кристаллите
и ядре должно быть одинаковым. Но это отдельная проблема.
Свою специфику имеет также и кристаллизация набухших
полимеров. При этом проявляются дополнительные специфические
факторы, влияющие как на конформацию и упаковку цепей,
так и на нуклеацию.
Некоторые интересные проблемы возникают при анализе
кристаллизации статистических сополимеров. Малое число
длинных последовательностей кристаллизующихся звеньев
затрудняет многократное прохождение одной и той же цепи через
кристаллит. Неограниченный рост кристаллита в поперечном
направлении, для которого необходимо участие звеньев из
других цепей, может быть сильно заторможен, вследствие только
что обсужденных трудностей расположения цепей в переходной
и аморфной зонах. Введение некристаллизующихся хаотически
распределенных вдоль цепи звеньев может подавить развитие
пластинчатой морфологии, характерной для отправных гомопо-
292
лимеров. Снова необходимо напомнить, что высокое значение
<7к, сравнимое со значением для чистых отправных гомополиме-
ров, было получено для статистических сополимеров этилена.
Ясно, что оно не может быть идентифицировано со свободной
энергией при образовании складчатой структуры.
Хотя мы и показали, что нет никаких препятствий к
сосуществованию больших ламеллярных кристаллитов и аморфных
областей в гомополимерах, любые экспериментальные
доказательства существования таких некристаллических областей
требуют тщательного анализа. Наиболее убедительно в этом смысле
исследование стеклования. Сравнение температур стеклования
расплавленных полимеров, а также полимеров с различными
(контролируемыми) степенями кристалличности показывает, что
для одного и того же полимера в разных фазовых состояниях
эта температура одинакова [33—35].
Именно этот факт можно считать действительно
убедительным доказательством существования аморфных областей в кри^
сталлических полимерах.
Если, с другой стороны, существовала бы только
кристаллическая, хотя и дефектная фаза, то температуру стеклования
необходимо было бы связывать со специфическими свойствами
соответствующих дефектов. Полимерный расплав при этом
следовало бы рассматривать как набор дефектных кристаллов.
А это было бы, конечно, несовместимо с тем, что при
плавлении происходит фазовый переход первого рода. Более того,,
большинство, если не все физические, механические и
реологические свойства расплава остались бы необъяснимыми. Любое
их объяснение возможно лишь на основе представлений о
высокой конфигурационной энтропии большого набора
молекулярных цепей в конформации статистического клубка.
Рентгенограммы кристаллических полимеров неизменно
содержат широкое гало, соответствующее брэгговскому периоду
в области 3—5 А. Наличие гало может, в принципе,
объясняться рассеянием как от аморфных областей, так и от
нарушений порядка в кристалле. Поэтому, используя только
рентгенографические данные и отбрасывая обширную и ценную
информацию, полученную другими методами, довольно трудно
установить молекулярную природу этого диффузного рассеяния.
Однако очень тщательный анализ Руланда [36] как диффузной,
так и дискретной дифракции на полипропилене показывает, что
гало обусловлено именно наличием аморфных областей, в
которых отсутствует какой-либо структурный порядок.
Для оценки уровня кристалличности можно использовать
различные методы. Наряду с рентгенографией, инфракрасной
спектроскопией и ядерным магнитным резонансом высокого
разрешения для этих целей используется анализ таких
термодинамических характеристик, как плотность, теплоемкость
293
и тепловое расширение. При этом делается неявное
предположение, что сосуществуют две фазы, вклады которых в измеряемые
характеристики аддитивны и пропорциональны их
относительному содержанию. Неизбежно имея дело с неравновесным
состоянием полимеров, когда кинетические факторы играют
особую роль, нужно помнить, что сравнение различных методов
исследования данного полимера имеет смысл только для
образцов, закристаллизованных в идентичных условиях и
имеющих одинаковую термическую предысторию.
Ниже представлены полученные различными методами
значения степени кристалличности A-Х) натурального каучука,
закристаллизованного при 0°С:
Рентгенографический метод [37] 0,30—0,35
Определение теплоемкости [33, 38] 0,26
» плотности [38] 0,31
» теплового расширения [38] . . . .0,31
Натуральный каучук относится к слабо кристаллизующимся
полимерам. Данные, представленные выше, показывают, что
при различных методах оценки получаются практически
совпадающие величины. Небольшие расхождения, возможно,
обусловлены чувствительностью примененных методик к различным
дефектам структуры. Тем не менее можно сделать основной
вывод, что при относительно низких степенях кристалличности
сосуществуют две фазы.
В другом предельном случае, при очень высокой степени
кристалличности, следует считаться с рядом осложнений.
Однако для отожженного образца линейного полиэтилена (полимера
с наиболее высоким известным уровнем кристалличности) Вун-
дерлих и Доль [39] получили, по калориметрическим данным,
1 — К порядка 0,90. Этот результат хорошо согласуется со
степенью кристалличности, определенной по плотности [14, 40] и
рентгенографическим путем [41] для образцов,
закристаллизованных в аналогичных условиях. При дальнейшем повышении
степени кристалличности вклад от аморфных областей
уменьшается, и возрастает роль дефектов кристаллической решетки.
Применение законов аддитивности теперь становится довольно
спорным и нельзя ожидать, что различные методы будут в
равной степени регистрировать эти дефекты.
Нарушение порядка в решетках, образованных цепными
молекулами, можно отнести или за счет внутримолекулярных
дефектов, или связать их с трудностями упаковки таких
молекул. Термодинамические методы автоматически учитывают
дефекты равновесного типа; неравновесные нарушения не
фиксируются одинаковым образом различными методами. Такие
методы как рентгенография и инфракрасная спектроскопия
различно реагируют на любые эффекты. Так, если инфракрасная
294
спектроскопия характеризует в основном межмолекулярные
взаимодействия, то рентгенография определяет трехмерный
порядок. Следовательно, если нарушения затрагивают конформа-
ции отдельных цепей, а не их упаковку, указанными двумя
методами можно получить различные значения степени
кристалличности. Кроме того, следует иметь в виду уникальную
природу областей раздела фаз. Эти области нельзя отнести ни
к аморфной, ни к кристаллической фазе, и поэтому от них
нельзя ожидать характерных для этих фаз рентгеновского
рассеяния или термодинамических свойств.
Кристаллизация полимеров
из разбавленных растворов
Кристаллы из разбавленных полимерных растворов
обладают в высшей степени интересной и важной для понимания
кристаллического состояния полимеров морфологией. Именно
их открытие стимулировало изучение морфологии и текстуры
кристаллических полимеров [12, 13, 16, 42—46]. В электронном
микроскопе видно, что эти
кристаллы состоят из тонких слоев, или ла-
мелей с постоянной толщиной
порядка 100 А и линейными
размерами, достигающими нескольких
микрон. Утолщение кристаллов при
наслаивании дополнительных ламел-
лей приводит к образованию
своего рода спиральных террас, что
соответствует механизму роста в
результате винтовых дислокаций
[47]. Макроскопические черты этой
морфологии совершенно подобны
обнаруженным для
низкомолекулярных «-алканов,
закристаллизованных из разбавленных растворов Рис. 107. Электронограмма от
[13, 48]. ромбовидного кристалла поли-
Ориентация цепей молекул в этилена,
пластинках определялась по
локальным электронограммам от избранных областей. На рис. 107
приведена типичная картина для ромбовидного кристалла
полиэтилена. Из анализа этой исключительно четкой картины сле^
дует, что направление цепей или ось с перпендикулярна
широкой грани кристалла. Более того, направления
кристаллографических осей а и Ь постоянны во всей пластинке, так что
к таким структурам вполне можно применить термин
295
«монокристалл». Так как пластинки имеют толщину всего
порядка 100—200 А, что составляет лишь небольшую долю длины
обычной полимерной молекулы, очевидно, одна и та же молекула
должна многократно пересечь данный кристалл. Следовательно,
пластинки должны состоять из сложенных полимерных цепей.
Это заключение, вполне согласующееся с выводами
структурного анализа, отнюдь не требует, чтобы молекулы были
обязательно регулярно сложены, т. е. внешняя плоскость складок
@01) не обязательно должна быть совершенно регулярной.
Хотя к настоящему времени наилучшим образом
исследованы кристаллы линейного полиэтилена, пластинчатые
кристаллы наблюдаются также при кристаллизации из
разбавленных растворов множества других полимеров. Сюда относятся
производные целлюлозы [49, 50], полиамиды {51], полиэфиры [52],
полиолефины, такие как полипропилен [13] и поли-4-метилпен-
тен-1 [53], полиакрилонитрил [54]. Ламеллярные кристаллы
образуются также при кристаллизации из разбавленных
растворов разветвленного полиэтилена [16, 55]. В последнем случае
ламелли овальны, толщина их меньше 90 А и они гораздо менее
совершенны, чем у полимеров регулярной структуры. Хорошо
ограненные ламеллярные структуры также наблюдаются при
кристаллизации из разбавленных растворов политрифторхлор-
этилена в мезитилене [56]. Для этих систем, как показывают
электронограммы, оси цепей также ориентированы
перпендикулярно к широкой грани пластинки.
Для некоторых из этих систем большое влияние на
морфологическую форму оказывает природа растворителя. Например,
триацетат целлюлозы, закристаллизованный из раствора в ни-
трометане, дает хорошо ограненные пластинки, попытки же
вырастить подобные кристаллы из других растворителей пока
еще остаются безуспешными [49]. Хотя возникают пластинки из
разбавленных растворов полиакриланитрила в пропиленкарбо-
нате, в других растворителях, таких как диметилформамид и
диметилацетамид, образуются лишь гели, не представляющие
интереса в морфологическом плане [57]. Морфологические
формы полиамидов также зависят от природы растворителя [55, 58].
Таким образом, термодинамическая природа растворителя и,
возможно, его влияние на конформацию цепи определяет в не*
которой мере характер образующихся кристаллов *.
* Значительно большие единичные кристаллы, видимые в оптическом
микроскопе, вырастают из разбавленных растворов гуттаперчи. Кроме того,
облучение больших единичных кристаллов мономерного триоксана приводит
к полимеризации и образованию в процессе облучения макроскопических
кристаллов полиоксиметилена. Рентгенограммы свидетельствуют, что это
Также единичные кристаллы [59, 60]. Образцы подобного типа оказываются
идеальными объектами для изучения физических и механических свойств
кристаллических полимеров,
296
Другое важное свойство пластинок — их необычайная
однородность по толщине (в пределах экспериментальной ошибки —
15 А для электронномикроскопических наблюдений). Это
также доказывается наличием максимумов четырех порядков при
рассеянии рентгеновских лучей под малыми углами. Первый
период соответствует размеру, наблюдаемому в электронном
микроскопе. Форма кристаллов может быть очень различна —
от ромбовидных Пластинок (см. рис. 10) до типичных
дендритных форм (см. рис. 11) и сильно зависит от температуры
кристаллизации и молекулярного веса полимера. Дендритные
образования легче развиваются при быстрой кристаллизации и
больших переохлаждениях [28, 61, 62], а также при очень
медленной кристаллизации и малом переохлаждении [62]. Электро-
нограммы образцов, полученных при малых и очень больших
переохлаждениях, весьма схожи, несмотря на то, что здесь
возможно действие различных механизмов. Тенденция к
образованию дендритов усиливается при повышении концентрации
раствора примерно более 1%. В
промежуточном диапазоне
переохлаждений образуются уже хорошо
нам известные пластинчатые
единичные кристаллы.
Исследование растворов узких
фракций полиэтилена в ксилоле
показывает, что температура, ниже
которой кристаллизация приводит
уже не к пластинчатым
монокристаллам, а к более диффузным
дендритоподобным структурам,
возрастает с увеличением
молекулярного веса. Верхний температурный
предел, при котором снова
образуются дендриты, а не пластинки,
также зависит от молекулярного
веса. В диапазоне молекулярных
весов 10 000—120 000 этот температурный предел возрастает
от 84 до 92° С. Поскольку равновесная температура плавления
довольно слабо зависит от молекулярного веса в этой области,
следует заключить, что морфологические характеристики
определяются не только степенью переохлаждения, при котором
ведется кристаллизация, но и 'Непосредственно молекулярным
весом. Однако в температурной области роста правильно
ограненных структур толщина кристаллов не зависит от
молекулярного веса [61, 63, 64].
Весьма примечательна зависимость толщины ламеллей от
температуры кристаллизации. Это показано на рис. 108 для
системы полиэтилен — ксилол. По мере возрастания температуры.
Рис. 108. Зависимость
толщины пластинки от температуры
кристаллизации для
полиэтилена, закристаллизованного иа
разбавленного раствора в
ксилоле [61, 63, 64].
Данные получены методами
рассеяния рентгеновских лучей под малыми
углами и электронной микроскопии.
29Г
кристаллизации период увеличивается от 90 А, при
наименьших температурах, до 150 А. Выше температуры
кристаллизации (90° С) уже не наблюдается хорошо ограненных структур,
а при очень низких температурах трудно достичь
изотермической кристаллизации. Однако, несмотря на эту ограниченность
температурного диапазона кристаллизации, изменение толщины
пластинок с температурой кристаллизации очевидно. Тем не
менее эти периоды значительно меньше наблюдаемых в блочных
полимерах.
Таким образом, возникновение пластинчатых или
дендритных форм, в которых полимерная цепь многократно проходит
через один кристалл, является широко распространенным
способом кристаллизации полимеров из разбавленных растворов.
Конкретная форма кристаллизации зависит от температуры
кристаллизации и степени переохлаждения, а также
молекулярного веса, взаимодействия с растворителем и конформации
цепи.
Несмотря на большое число и резкость рефлексов под
малыми углами, а также кажущееся совершенство пластинок,
наблюдаемых в электронном микроскопе, как электронограммы,
так и рентгенограммы таких образцов содержат гало.
Рентгенограммы от единичных кристаллов полиэтилена идентичны
полученным от высококристалличного блочного полимера с
содержанием аморфной фазы 15—20% (по плотности [65]). Для
линейного полиэтилена, включая высокомолекулярные фракции,
плотность выращенных из разбавленного раствора образований
на 2—3% меньше плотности идеального монокристалла [62, 66]
и соответствует степени кристалличности порядка 80%.
Несовпадение плотности реального единичного кристалла и
>вычисленной по размерам элементарной ячейки монокристалла
не может быть обусловлено небольшим числом звеньев,
выходящих из решетки и образующих резкие изгибы в складчатой
структуре. Внутренними нарушениями регулярности это также
нельзя объяснить, ибо потребовалась бы такая концентрация
пустот и дефектов в кристаллах, что встал бы вопрос о самом
их существовании. Кроме того, энтальпия плавления
единичных кристаллов полиэтилена, выращенных из раствора, на 7%
меньше, чем для отожженного образца, закристаллизованного
в блоке [66]. Эти наблюдения можно удовлетворительно
объяснить только присутствием заметного количества аморфного
полимера в единичном кристалле.
В связи с регулярностью структуры пластинок,
наблюдаемых в электронном микроскопе, следует предположить, что
аморфные области сосредоточены на поверхности пластинок [32].
Действительно, в электронном микроскопе ровный аморфный
слой, образующийся в процессе сушки, невозможно отличить от
кристалла, на ко юром он находится.
.298
Температура плавления кристаллов, выращенных из
разбавленных растворов, значительно ниже, чем у выросших в
блочном полимере, и зависит от температуры кристаллизации
полимера и термодинамической природы растворителя [63, 67].
Температуры плавления удобно сравнивать по точкам
растворимости, т. е. температурам растворения полимера при очень
больших разбавлениях. Например, для полиэтилена,
закристаллизованного из расплава в течение двух недель при 131° С,
точка растворимости в тетралине соответствует 110° С, тогда
как для закристаллизованного из разбавленного раствора
96° С [63]*.
Если различие в температурах плавления отнести только за
счет ограниченной толщины образцов, закристаллизованных из
раствора, то вычисленная свободная энергия поверхности
раздела окажется 1800 кал/моль «поверхностных» звеньев, или
68 эрг/см2 [62]. Это значительно меньше соответствующей
свободной поверхностной энергии, вычисленной для образцов,
закристаллизованных в блоке. Однако различие в температурах
плавления образцов, выращенных из раствора и из расплава,
существенно уменьшается в случае плохих растворителей [67],
что снова указывает на возможность влияния
термодинамического взаимодействия полимер — растворитель на процесс
кристаллизации.
Необходима какая-то теоретическая основа, объясняющая
образование ламеллярных кристаллов из разбавленного
раствора и сопутствующую ему складчатую конформацию цепей.
Все предлагаемые при этом механизмы и процессы должны,
естественно, соответствовать описанным выше физическим,
термодинамическим и морфологическим свойствам кристаллов.
Один из важнейших вопросов, способствующий выяснению
принципов построения теории, связан с выявлением
соответствий и различий в структуре и свойствах ламеллей,
полученных из разбавленного раствора и в блоке.
Ранние теории, в которых не различались кристаллизации в
растворе и блоке, основывались либо на кинетических
представлениях, связанных с нуклеацией [26, 63, 68, 69], либо на
представлениях о равновесной кристаллизации [70]. В последнем
случае делается попытка распространить на все три
кристаллографических направления некоторые факторы,
противодействующие образованию слишком протяженной решетки. Эти
«внутренние» нарушения трехмерного порядка и должны приводить
к складыванию цепи. Соответственно, предполагается, что как
первичные, так и растущие ядра состоят из регулярно и плотно
сложенных цепей. Следовательно, рост может происходить
* Эта температура незначительно изменяется в зависимости от
температуры кристаллизации,
299
только в направлениях, перпендикулярных к оси цепи, в
результате чего должна возникать регулярная складчатая структура.
Кинетический контроль размеров ядер и их зависимость от
температуры кристаллизации объясняются на основе теории
нуклеации для низкомолекулярных веществ, обсуждавшейся
выше (см. стр. 238). Поскольку, однако, в случае полимеров
имеет место мономолекулярная, но двухмерная нуклеация,
предсказываемая этой теорией термодинамическая
устойчивость кристаллитов оказывается значительно меньше
наблюдаемой экспериментально. Делались попытки обойти эти трудности
[27, 64, 70], но они включали много сомнительных
предположений, касающихся способа упаковки последовательности звеньев
на уже закристаллизовавшейся «подложке».
Мы уже видели, что соображения, связанные с конформа-
цией и упаковкой «аморфных» цепей, дают естественное
объяснение образованию ламеллярных кристаллитов в блочном
полимере. В случае разбавленных растворов это становится еще
очевиднее. Действительно, в растворе поток цепей, выходящих из
грани, диссипирует в изотропной области несравненно сильнее,
чем в блочной системе. Это обусловлено тем, что полимеру
«необходимо» резко снизить свою концентрацию от v2=l в
кристалле до средней концентрации окружающего раствора.
Равенство B50) следует поэтому записать в виде [32]:
. &-*? <251>
Частота возвращений цепи к грани кристалла, таким
образом, должна возрастать с разбавлением.
В случае бесконечно длинной цепи в предельно
разбавленном растворе вообще образовался бы лишь один кристалл, так
как все цепи, выходящие из него, в конечном счете должны в
него же вернуться. В реальных разбавленных растворах, где
цепи имеют конечную длину, расстояние между кристаллитами
достаточно велико, чтобы воспрепятствовать прохождению
одной молекулы через несколько кристаллов. Этот элементарный
анализ, в сущности, уже предсказывает выделение из
разбавленных растворов изолированных кристаллов.
Следовательно, основная проблема касается способа и
эффективности объединения звеньев, участвующих в
кристаллизации молекул, в упорядоченную решетку. Вначале должны быть
сформулированы условия равновесия, которые далее следует
проанализировать с. позиций возможных механизмов
кристаллизации.
Рассмотрим два предельных случая, подтверждающихся
микроскопическими и дифрактометрическими исследованиями.
Один из них, схематически представленный на рис. 109, а, пред-
300
п
^ ^
^щш?>
г
ставляет регулярную складчатую упаковку. Каждая молекула
закристаллизована до максимально возможной степени.
Кристаллизация при этом — фактически полная, в кристалл не
входит лишь небольшое количество образующих регулярные
складки звеньев. В другом случае, изображенном на рис. 109,6,
кристаллические последовательности соединены более или менее
беспорядочно, петлями различной длины. Возможны,
разумеется, и промежуточные структуры, когда, например,
кристаллические участки молекулы расположены в одном слое, но
сочленены нерегулярными
петлями различной длины. а
За исключением регу- - ^ ^ п- б
лярной складчатой
структуры, остальные модели имеют
область поверхности
раздела, состоящую из
неупорядоченных участков цепей,
перемешанных с
растворителем. Совершенно
очевидно, что диффузная область
раздела повышает энтропию
и соответственно
термодинамическую стабильность
кристалла.
С другой стороны, для
образования кристалла из
достаточного количества цепей, сосредоточенных в одной
небольшой области, необходимы затраты свободной энергии
согласно уравнению B32). К тому же, концентрация полимерных
цепей в аморфной зоне раздела выше, чем концентрация
раствора в целом. Требуется определенная работа против
осмотических сил, чтобы обеспечить такое сосредоточение звеньев в
переходном слое. Следовательно, при образовании
нерегулярной складчатой решетки повышение энтропии должно
компенсировать затраты свободной энергии на осмотическую работу при
образовании диффузного поверхностного слоя.
Для оценки осмотической работы примем модель
кристаллита, схематически изображенную на рис. 109,6 [32].
Предположим, что имеется v кристаллических последовательностей,
каждая длиной ?. Кристаллические последовательности связаны
аморфными петлями средней длины у. Таким образом, в
образовании кристалла участвуют (?+#)v звеньев и (?+y)v/x
молекул, где х — число звеньев в одной молекуле. В случае
регулярной складчатой структуры у имеет минимальное значение,
которое для упрощения расчетов примем равным нулю.
Образование диффузного слоя требует перемещения yv/x молекул
полимера из разбавленного раствора объемной концентрации v%
Рис. 109. Схема ламеллярного
кристаллита [32]:
л —с регулярной складчатой структурой; <5 —с
нерегулярной складчатой структурой (длина петель
различна).
301
в область поверхности раздела, где концентрация равна
В соответствии с теорией растворов полимеров затраченная
осмотическая работа равна:
A/^a^vJ^ B52)
где Xi=Vi/VM.
Значение AF0CM можно оценить по изменению
растворимости или температуры плавления.
Пусть Гдл — равновесная температура плавления в растворе
кристалла размером ? в отсутствие осмотических эффектов.
При некоторой температуре 7,/=Гпл — АГ изменение свободной
энергии при образовании кристалла, содержащего диффузную
область, равно:
AF = AF0CM-^A7- B53)
* П
' пл
Будем считать, что влияние конечного размера кристалла
одинаково в обоих случаях. Приравнивая AF к нулю и
подставляя AFocm из соотношения B52), найдем понижение
температуры плавления за счет осмотической работы при образовании
зоны раздела:
При больших х множитель xjx, который представляет собой
первый вириальный коэффициент, связанный с распределением
и взаимодействием молекулярных центров, пренебрежимо мал.
Следовательно
AT
тт&Ы?-1)*1-*-**] <2к>
Среднюю концентрацию звеньев в переходном слое и его
размеры точно определить невозможно. Однако если
произвольно выбрать толщину слоя такой, что i>2 = 0,5, то для хорошего
растворителя с xi — 0
У RTL
АГ-адг*йг B56>
Используя известные параметры для полиэтилена в раство*
рителе с молярным объемом порядка 100 см3 (#i = 7), получим:
д7, = ц# B57>
Если эффективная концентрация выше установленной, то
302
численная константа в соотношении B57) должна возрасти,
тогда как ухудшение растворителя приводит к ее уменьшению.
Понижение температуры плавления не превышает нескольких
градусов; точная величина AT зависит от упомянутых факторов
и отношения #/?. Хотя термодинамическая стабильность
изменяется при этом незначительно, не следует делать вывод, что
осмотический эффект при образовании диффузной области
пренебрежимо мал. Величина свободной энергии может
существенно влиять на процесс роста кристалла. Если бы аморфные
петли между кристаллическими последовательностями возникали
вследствие иных факторов, можно было бы ожидать
незначительного уменьшения устойчивости. Кроме того, если бы
процессы нуклеации включали образование петель, то скорости
кристаллизации (при одинаковом переохлаждении) зависели бы
от природы растворителя, особенно от его молярного объема и
термодинамического взаимодействия с полимером.
Важная статистическая проблема связана с распределением
звеньев между кристаллическими последовательностями и
переходным слоем [32]. Одна и та же цепь может входить в
пластинчатый кристаллит фиксированной толщины ? и иметь некоторое
количество кристаллических участков v<x/?,. С учетом длины
промежуточных аморфных участков, число возможных способов
упаковки равно:
C^V)= (*_v?)!v! <258>
Для расчета статистической суммы v—1 некристаллическим
участкам необходимо приписать некоторую допустимую кон-
формацию.
В случае правильно ограненного кристаллита все
граничные звенья лежат в плоскости @01). Следовательно, аморфные
петли должны начинаться и заканчиваться в этой плоскости.
Таким образом, на пространственное распределение аморфных
цепей, которое в соотношении B58) принималось совершенно
беспорядочным, накладываются серьезные ограничения.. Так как
концы петель ограничены плоскостью, проекция вектора
расстояния между концами петли на нормаль к плоскости должна
быть равна нулю.
Если растущая грань имеет тенденцию наращивать сразу
целый слой, участки выбранной молекулы будут, в основном,
входить в тот же самый слой (НО). Поэтому другая компонента
вектора расстояния между концами петли также имеет
значение, близкое к нулю. Если каждая петля сочленяет смежные
концы кристаллических последовательностей, лежащие в одной
плоскости (ПО), то и величина самого вектора расстояния
между концами петли близка к нулю. Это условие напоминает
критерий регулярного складывания за тем исключением, что
303
петли могут различаться по конформации, а их длины
превышать минимальные возможные размеры.
Как следует из теории статистических клубков, вероятность
обращения в нуль одной из проекций вектора, связывающего
концы петли, в декартовых координатах равна C/2пу)ч\ Если
d — число возможных ненапряженных размеров для каждой
петли, то число возможных состояний цепи с v
кристаллическими участками равно С(?, v) | C/2лу) \ (v— l)d/2. Среднее число
звеньев на петлю у равно (л:—v?)/(v+l) ~ (x—v?)/v и
статистическая сумма может быть выражена таким образом:
Q = С (С, v) JL ^(V2~1} exp Ш - 2vsK) B59)
где 4>=&FMIRT и sK = oK/RT.
Из соотношения B59) можно найти оптимальное число
участков, входящих в кристалл определенной толщины ?. Для
случая d=l, т. е. когда петли соединяют пары точек, беспорядочно
расположенных в плоскости @01) и при фиксированном ?,
Q максимально при наибольшем v. Области Q соответствует
весьма ограниченный диапазон Ф (т. е. Г). По одну сторону
максимума при Т<ТПЛ в равновесных условиях должна
закристаллизоваться практически вся цепь; разумеется, петли при
этом будут очень короткими. По другую сторону, при Т>ТПЛ
может образоваться лишь очень небольшое число
кристаллических участков.
При d>2, когда петли расположены вдоль слоя в
плоскости (НО), кристаллизация в зависимости от значения ср либо
проходит полностью, либо не идет совсем. В условиях
равновесия можно при этом ожидать резкую прерывность и
образование регулярной складчатой структуры, которая будет наиболее
благоприятной, ибо при ней достигается максимальная
кристаллизация молекулы. Этот вывод получает дополнительное
подтверждение, если принять во внимание осмотический эффект,
который стремится снизить концентрацию аморфного материала
на поверхности кристаллита.
Заключение о том, что складчатая структура,
благоприятствующая максимальной кристаллизации, является наиболее
устойчивым состоянием для кристаллитов фиксированной
длины, основывается исключительно на статистических
соображениях. Следовательно, для предсказания такой структуры не
нужны допущения, заимствованные из классической теории ну-
клеации низкомолекулярных веществ, или предположения, что
молекулы полимера обладают специфической «внутренней»
склонностью к складыванию [26, 63, 68, 69]. Разупорядочивание
на границе раздела и сопуствующее увеличение энтропии,
связанное с распределением цепей между кристаллическим и
аморфными участками в рассматриваемом случае не имеет места.
304
Это обусловлено тем, что аморфные петли достаточно жестко
локализованы на гранях кристалла, положение которых, в свою
очередь, фиксировано размерами кристаллических
последовательностей. Соответственно, создаются наиболее благоприятные
условия для развития кристаллизации до предельно возможной
степени. К аналогичному выводу можно прийти даже игнорируя
возвращение цепей в кристаллит [71]. Для цепи, входящей в
единичный кристаллит, размер которого меньше контурной
длины молекулы, регулярная складчатая структура удовлетворяет
условиям равновесия.
Остается еще установить, однако, действительно ли наиболее
устойчивое состояние достигается при реальных условиях
кристаллизации. Для этого нужно принять во внимание, что рост
кристалла происходит с конечной скоростью. Хорошо известно,
что при всех переходах жидкость — кристалл действуют
кинетические факторы, так что достигнутое конечное состояние
представляет собой некоторый компромисс между равновесием
и необходимостью развития процесса с конечной скоростью. Это
верно как для мономерных, так и полимерных веществ. Далее,
в разбавленном растворе полимера время релаксации больших
сегментов слишком велико по сравнению с высокой скоростью
наслаивания цепей на растущей грани. При этом молекулярный
вес должен существенно влиять на скорость роста [61].
Аномально низкие энтальпии и плотности пластинок, полученных
из разбавленных растворов, отчетливо показывают, что в
реальных условиях кристаллизации не достигается наиболее
устойчивое состояние.
Морфология и текстура пластинок определяется первичным
актом нуклеации и процессом роста. Процесс первичной нуклеа-
ции сводится к созданию своего рода фундамента, на котором
происходит дальнейший рост кристалла. В разбавленной
системе тенденция к уменьшению осмотической работы,
затрачиваемой на перемещение молекул из раствора в кристаллическую
фазу, проявляется в сокращении до минимума числа молекул,
участвующих в образовании ядра. Следовательно, в
образовании ядра максимально участвуют лишь несколько молекул.
О размерах критического ядра уже говорилось на стр. 240.
Рост кристалла в направлении, перпендикулярном к
направлению цепи, связан с «наслаиванием» сегментов полимера на
уже существующий субстрат. Большинство объяснений роста
ламеллярных кристаллов [27, 63, 69] включали предположение,
что наслаивание пол-ной последовательности «кристаллических»
звеньев осуществляется в одноактном процессе. Длина
последовательности ? определяется в момент наслаивания и полностью
соответствует критическим размерам ядер гиббсовского типа в
поверхностных монослоях. Далее делается допущение о
возможном конкурентном удалении осевшей последовательности до
20 Л. Манделькерн
305
того, как успеет присоединиться следующая. Принимается во
внимание также возможность колебаний длины заключительной
последовательности в незавершенном внешнем слое. Что
касается предыдущих последовательностей, то их длины уже не
могут изменяться в этом процессе. Очевидно, при этих
предположениях, процесс должен привести к упорядоченной структуре
со строго определенными размерами. При этом, однако, не
принимается во внимание возможность колебаний длины других
последовательностей во внешнем слое, возникающих при
удалении концевых участков каждой из них, как и возможность
продольного смещения (дислокаций) полных
последовательностей. Иными словами, основное допущение в рассматриваемой
концепции сводится к тому, что длина последовательности
однозначно определяется в момент ее наслаивания на субстрат.
Но на самом деле нельзя игнорировать возможность
флуктуации по длинам всех последовательностей во внешних слоях
[32]. В высшей степени маловероятно, что длина
последовательности полностью определяется в момент ее высаживания.
Для исследования этого вопроса предположим, что в некоторый
момент внешний слой является точной репликой предыдущего,
которому, для упрощения анализа, мы припишем постоянную
толщину"?. Зато мы не накладываем никаких ограничений на
характер складывания, полагая, что оно может быть как
регулярным, так и нерегулярным, а длина сочленяющих петель не
обязательно минимальна. Возможность флуктуации длины
последовательностей ? зависит от свободной энергии, необходимой
для удаления концевых звеньев из последовательности во
внешнем слое*. Эта энергия складывается из свободной энергии
плавления AFM на одно звено и увеличения свободной энергии
поверхности раздела 2ам в результате «обнажения» смежных
звеньев соседних последовательностей.
Для полиэтилена, например, AFM~ 2/?Д7\ Как следует из
работы Торнбалла и Корми [72] значение ам лежит в области от
50 до 100 кал/моль. За исключением чрезвычайно сильных
пересыщений изменение свободной энергии, связанное с удалением
концевых звеньев, очень мало по сравнению с RT.
Следовательно, тепловые флуктуации не требуют
значительной затраты энергии, и удаление концевых звеньев из
кристаллического слоя вполне возможно. Частичное отделение одной
цепи должно облегчить подобный процесс для соседних
последовательностей, так как переходная область при этом не
увеличивается (за счет удаления звеньев, смежных*с ранее заня-
* [Обращаем внимание на полную аналогию этой концепции механизму
роста граней реальных низкомолекулярных кристаллов. См. Я. И.
Френкель, Кинетическая теория жидкостей, избр. соч., т. 3, Изд. АН СССР,
1959, гл. VII. — Прим. редактора].
306
тыми участками). Поэтому можно ожидать, что ребра внешнего
слоя обладают рыхлой структурой, и цепи ни при каких
обстоятельствах не будут иметь регулярную складчатую конформа-
цию. Однако полное удаление последовательности из внешнего
слоя должно происходить крайне редко из-за высокого значения
свободной энергии поверхности раздела 2?ам, необходимой для
такого удаления. По этой же причине маловероятны большие
колебания длины различных кристаллических
последовательностей. Вполне возмож'ны продольные дислокации, особенно, если
регулярность последовательности нарушается петлей,
выходящей из грани кристалла.
Таким образом, внешний слой растущего кристаллита
находится в переходном флуктуирующем состоянии и все время
подвержен изменениям.
Естественно, что в конечном счете из всех таких перестроек
отбирается наиболее устойчивая, которая и закрепляется после
наращивания последующих слоев. Маловероятно, что вся
последовательность высаживается одновременно; скорее это
должен быть многоактный процесс.
Строение кристаллита вблизи растущей грани зависит от
соотношения скоростей развития одной кристаллической
последовательности и инициирования целого нового слоя. После
инициирования скорость развития кристаллической
последовательности зависит от отношения скорости встраивания к
скорости удаления звеньев. Это отношение равно ехр(—AFJRT),
откуда следует, что результирующая скорость будет увеличиваться
с возрастанием AT. При умеренных пересыщениях
температурный коэффициент роста отрицателен. При допущении же
одноактного процесса наслаивания температурный коэффициент —
положительный. В обоих случаях размер должен превышать
?* = 2ат/Д/гм, иначе удаление последовательностей будет
происходить быстрее их осаждения.
Температурный коэффициент полной скорости роста грани
кристалла зависит также от температурного коэффициента
процесса наслаивания последовательностей. При нуклеационном
контроле осаждения температурный коэффициент скорости
роста грани также отрицателен. Однако отрицательный
температурный коэффициент роста еще не обязательно свидетельствует
о нуклеационно контролируемом механизме.
Холланд и Линденмейер [61] осуществили измерения полной
скорости роста граней, нормальных к оси с, при
кристаллизации полиэтилена из разбавленного раствора. В исследуемой
области температур 80—92° С (соответствующей времени
кристаллизации от нескольких секунд до дней) обнаружены
отрицательные температурные коэффициенты роста. Эти опыты
подтверждают нуклеационный характер процессов при развитии
боковых граней кристаллов.
20*
307
Значительное число сложных факторов, влияющих на рост
кристалла, исключает возможность простой количественной
формулировки этой проблемы; к тому же нужно учесть, что мы
допустили сверхупрощение, полагая, что основная ступень
роста включает образование целого слоя. На самом деле вполне
возможно одновременное развитие нескольких слоев на
растущей грани. Хотя инициирование новой кристаллической
последовательности вероятнее всего происходит на краю складки,
можно также ожидать инициирования с конечной скоростью
и на поверхности грани.
Несмотря на эти затруднения, все же возможно объяснить
постоянство толщины ламелли, не прибегая к несостоятельному
требованию об одноактном наслаивании полной
кристаллической последовательности. Новый слой может возникнуть еще до
того, как прошло достаточное время для того, чтобы
предыдущий слой успел достичь размера и степени упорядоченности
своих предшественников. Следовательно, ? уменьшается при
росте кристалла, и полная скорость развития последовательности
понижается. Но это абсолютное уменьшение скорости роста
приводит к выигрышу времени для упорядочения внешних слоев
и увеличения ?. При увеличении ? скорость снова возрастает, и
весь цикл повторяется.
Таким образом, существует механизм саморегулирования
толщины ламеллей, обусловленный самим характером
обобщений кинетики роста. Можно ожидать, и это подтверждается
экспериментальными наблюдениями, что характеристическая
толщина кристаллита ? будет возрастать с температурой
кристаллизации. Значение ? должно лежать где-то между
размером первичного ядра и минимальным размером, необходимым
для устойчивости. Можно ожидать, поэтому, зависимости
скорости роста от молекулярного веса, что и наблюдается на опыте.
Следовательно, нельзя считать, что наиболее устойчивое
кристаллическое состояние может быть достигнуто при
конечных скоростях кристаллизации, ибо последнее условие требует
значений ?, сравнимых с контурной длиной молекулярной цепи.
По аналогичным причинам невозможно и образование
регулярной складчатой структуры. В действительности должен
возникать внешний разупорядоченный слой в полном соответствии
с измерениями плотности и энтальпии.
Зависимость температуры плавления
от условий кристаллизации
В связи с тем, что кристаллизация должна проводиться при
конечных скоростях, возникают отклонения от равновесия, ко-
308
торые проявляются в уменьшении термодинамической
стабильности и понижении температуры плавления образовавшихся
кристаллитов. Для поликристаллических веществ факторы,
способствующие понижению температуры плавления, обусловлены
конечными размерами кристаллитов, степенью их дефектности
и наличием поверхностей раздела и связующих областей. Что
касается полимеров в блоке и в растворе, то мы могли
убедиться, что их кристаллиты обладают ламеллярной структурой.
Поэтому было бы интересно исследовать влияние ограниченных
размеров кристаллитов на температуру плавления. При анализе
необходимо различать аспекты проблемы, относящиеся только
к полимерным системам или касающиеся малых кристаллитов
любых веществ. Неотъемлемой чертой этого анализа является
предположение, что температура плавления, характеризующая
определенную морфологию, может быть экспериментально
достигнута. Это значит, что нагревание, необходимое для
приближения к точке плавления, не нарушает природу кристаллитов..
Свободная энергия образования цилиндрического
кристаллита конечных размеров при температуре Т может быть
выражена следующим образом:
bF = &AfM-2po«-2V~WOK6 . B60)
где о* и а{5 — соответствующие поверхностные энергии
завершенного кристаллита *.
Влияние внутренних напряжений и вклад от поверхностной
энергии ребра в уравнении B60) незначительны, поэтому не
принимаются во внимание. Также не учитывается возможность
неравновесных внутренних дефектов в кристаллической
решетке; принимается во внимание лишь конечный размер. В ка«
жущейся точке плавления такого кристаллита Т=Т1Л,
&р(Т*пл) = 0, откуда:
*@~^ + ^<* B61)
Удобно выразить размеры кристаллита через соответствую*
щие размеры первичного ядра, т. е. ?=я?* и р|^=тр*1/2.
Напомним, что минимальные условия устойчивости при температуре
кристаллизации соответствуют л = 1 и т=2. После подстановки
уравнение B61) преобразуется:
, . ч / 1 < ,. 1 окб\
* Различие между поверхностными энергиями раздела завершенного
кристаллита и ядра введено не только в целях обобщения, но и для того,
чтобы можно было избежать предположения об идентичности конформаций
цепей в кристаллите и устойчивом ядре.
309
где Ткр—температура кристаллизации;
oj и og — свободные энергии поверхностей раздела для ядра.
Используя приближение &fM(T) =Д#МДГ, получим:
С-С=(^+^г)(^л-^) B63)
ИЛИ
^пл-^пл-^пл-^к) B64)
где а = о*/а« и р = а"/а*.
Параметры пит представляют отношения действительных
размеров кристаллита и критического ядра. Последнее может
возникнуть как в гомогенных, так и гетерогенных условиях *.
Соотношения B63) и B64) характеризуют понижение
температуры плавления кристаллита, обусловленное конечностью
его размеров. Это характерно не только для полимерных
систем.
Из приведенных равенств следует, что ТПЛ должна быть
линейной функцией Тк, причем Ф лежит в области от 0 до 1.
Условие Ф = 0 характеризует максимальную устойчивость, так как
Тпл= Гплпри любой температуре кристаллизации. При Ф= 1 Г„л
всегда равна Тк и образующиеся кристаллиты внутренне
неустойчивы.
Таким образом, для кристаллитов, полученных при
различных температурах и отличающихся только размерами, значение
7пл может быть найдено путем линейной экстраполяции
кажущейся температуры плавления. Однако оценка Ф с помощью
такого анализа еще не определяет однозначно структурные
параметры, влияющие на плавление. Действительно, в
указанном диапазоне любое значение Ф может быть получено
разнообразными комбинациями а, р, п и т.
Для полимеров, закристаллизованных в блоке, когда
размеры ламеллей очень велики в направлении, перпендикулярном
оси с, т -> оо, так что:
^л - С = •? (Кл ~ТК) = Ф' (Г°пл - Тк) B65)
Для особого случая, когда п=1 и сс=1 [73, 74]
K*-K» = j(Tl-rK) B66)
* Обоснованность этого утверждения можно видеть в следующем: если
?0 представляет размер ядра, возникшего в гомогенных условиях, а ?
—действительный размер ядра, возникшего в гетерогенных условиях, то
отношение ClC^k меньше или равно единице. Если п = ? /?JJ, ?/?* = /*'/& = я.
Аналогичные соображения приложимы и к прочим размерам.
310
или
Т° 4- Т
~* ' пл Т ' к
1 пл ~~ 2
Но даже если установлено на опыте, что Ф'=1/2, еще не
следует, что п н а должны быть равны единице. Этому условию
могут также удовлетворять и другие комбинации. Однако
простота описания плавления кристаллов конечных размеров весьма
заманчива как метод определения равновесной температуры
плавления полимеров,
особенно учитывая трудности
ее прямого определения.
Напомним ранние опыты
Вуда и Беккедаля [75],
посвященные изучению
плавления натурального каучука
(см. стр. 35). Эти
результаты в высшей степени
удобны для анализа в
соответствии с только что
изложенными принципами.
Кристаллизация образцов
проводилась изотермически, а
температура последующего
плавления определялась при
очень быстром нагревании.
Натуральный каучук
кристаллизуется настолько
медленно, что возможность
рекристаллизации при нагревании минимальна. Результаты
представлены на рис. ПО в виде зависимости Гпл от Тк. Как
видно из рис. 110, полученные данные хорошо описываются со-»
отношением B66), за исключением низких температур кри-»
сталлизации.
Аналогичные результаты получены Гофманом и Виксом [74]
для политрифторхлорэтилена. Однако анализ их более сложен,
так как неизвестно влияние присутствующих в большом
количестве нестереорегулярных цепей. Несмотря на эти затруднения,
обнаружена четкая зависимость температуры плавления от тем*
пературы кристаллизации. Кроме того, заметен некоторый рост
кажущейся температуры плавления с увеличением степени
кристалличности при фиксированной температуре кристаллизации.
Эти результаты представлены на рис. 111 в виде зависимости
ДГпл=Гпл~ Гпл от AjTk = 7"пл — Тк. В данном случае подби*
рались значения Гпл> при которых получаются прямые линии,
идущие в начало координат; эти значения в соответствии
Рис. 110. Зависимость температуры
плавления Тпл от температуры
кристаллизации Тк для натурального
каучука [73].
Линии проведены в соответствии с
соотношением B66) для Т® равной 301 и 303° К.
311
с формулой B65) представляют собой равновесную температуру
плавления. Из рис. 110—111 следует, что Гпл лежит в области
224—229° С. Это значительно выше непосредственно
наблюдаемого значения [74].
Слабое повышение стабильности с возрастанием степени
кристалличности может быть приписано изменению состава и
строения «расплава, благодаря чему кристаллизация происходит
при значительно пониженных переохлаждениях. С другой
стороны, это может
отражать упорядочение
структуры и увеличение
размеров самих кристаллитов.
Значение Ф' зависит от
Т°пл и лежит в области
0,25—0,4. Это
значительно меньше значения 0,5,
ожидаемого для случая,
когда толщину и
поверхностную свободную
энергию кристаллита и
первичного ядра можно
считать идентичными. Таким
образом, возможно
образование более устойчивых
структур, по сравнению с
предсказываемыми при
этих простых
предположениях.
Несмотря на
трудности интерпретации Ф
использование теории плавления кристаллитов конечного
размера дает возможность оценить равновесную температуру
плавления гомополимеров и сополимеров. Температура плавления,
полученная линейной экстраполяцией, по-видимому,
действительно является нижним пределом, так как не делалось никаких
допущений о влиянии внутренних дефектов на кажущуюся
температуру плавления. Необходимо отметить, что если даже
соотношение B66) выполняется независимо от значения Ф', это
еще не означает, что цепи в кристаллите сложены регулярно, а
указывает лишь на конечную толщину кристаллитов.
Как было уже показано в гл. 2, значительные
усовершенствования методики кристаллизации и отжига привели к
прогрессирующему возрастанию измеренной температуры
плавления полимеров. Например, температура плавления узкой
фракции полиэтилена, по данным Чанга и Флори [40], равна 138,5° С.
При этом измерении образец нагревался очень медленно после
Рис. 111. Зависимость ДГПЛ от &ТК для
политрифторхлорэтилена.
Верхние прямые проведены в соответствии с
формулой B66) дляФ' = 1 и 1/2- -Д —экспериментальные
значения для 1 — Л = 0,07 при ГдЛ = 2290 С; В —при
1-А. = 0,07 и г?л = 224°С; С-при 1-Я. = 0,4для
Г° =224° С 174].
312
кристаллизации при 13ГС. Линейная экстраполяция
кажущихся температур плавления в функции от температур
кристаллизации нефракционированного линейного образца (марлекс-50)
дает 71 =143 ±2° С [74]*.
Вычисленное значение Тпл для полиэтилена бесконечно
большого молекулярного веса согласуется со значением,
предсказываемым многими исследователями [76—80] на основе анализа
термодинамики плавления молекулярных кристаллов,
образованных мономерными я-алканами. Однако в случае молекулы
бесконечной длины молекулярные кристаллы не могут
образоваться, так как нет концов цепи, которые могут быть помещены
в смежные узлы решетки. Следовательно, для корректного
проведения такой экстраполяции необходимо сделать
соответствующую поправку при расчете энтропии плавления
низкомолекулярных гомологов [81]. В результате теоретически
экстраполированная температура плавления оказывается равной 145,5° С.
Таким образом, существует реальный метод, позволяющий
оценить значение / пл» избегая трудностей, связанных с прямыми
измерениями. Величины Тпл, полученные для гомополимеров [73,
74] и сополимеров [82], удовлетворительно согласуются с
результатами других опытов и теоретических расчетов. Однако, к
сожалению, предлагаемая линейная экстраполяция не имеет
твердой теоретической основы. Следовательно, из измерений
такого рода, помимо факта конечности размеров, нельзя
получить никакую иную детальную информацию о внутренней и
поверхностной структуре кристаллитов.
Сферолиты
Существование в полимерах упорядоченных
надмолекулярных структур, значительно превосходящих размерами отдельные
кристаллиты, следует из хорошо известной непрозрачности
кристаллических полимеров и подтверждается непосредственными
наблюдениями в поляризационном микроскопе. На рис. 8
(стр. 28) был приведен типичный пример такой структуры.
Наблюдая образцы при скрещенных поляроидах, замечаем
большие двулучепреломляющие области. В большинстве случаев они
состоят из концентрических двулучепреломляющих колец,
пересекаемых темным «мальтийским крестом», «перекладины»
которого параллельны и перпендикулярны направлению
поляризации. Разность хода изменяется непрерывно вдоль радиуса.
Это типичная сферолитная агрегация кристаллов или кристал-
* Важный вопрос при определении температуры плавления — удается ли
избежать рекристаллизации, несмотря на высокую скорость нагревания.
313
литов. Сферолит состоит из большого числа кристаллов,
расходящихся из одной точки, причем каждый кристалл
ориентирован вдоль радиуса сферы. Таким образом, сферолит
представляет собой определенную форму взаимной организации и
ориентации. Сферолиты обладают хорошо очерченными границами
и охватывают область, которая может достигать 0,1 м в
диаметре. Очевидное первичное требование для образования сферо-
лита — поликристалличность вещества. Однако не все
поликристаллические материалы обязательно или в действительности
образуют сферолиты. Подобные структуры наблюдаются в
таком разнообразии гомополимеров, закристаллизованных в
блоке, что напрашивается вывод о сферолитизации, как наиболее
общей форме кристаллизации
полимеров.
Образование сферолитов
характерно не только для полимеров.
Впервые этот термин использован
при описании поликристаллических
структур, обнаруженных в
изверженных породах. Сферолитные
образования наблюдаются в
различных неорганических и органических
кристаллических соединениях [83,
84]. Глобулярные белки, такие как,
например, фермент карбоксипепти-
даза, также кристаллизуются из
разбавленного раствора в сферо-
Рис. 112. Микрофотография литной форме [85]. Как было пока-
в скрещенных поляроидах Рпбингпнпм ГЯЮ ппрпр пячттр-
сферолита поли-у-бензил-*.- зано ™оинсоном i«oj, после разде-
глутамата, выращенного из раз- ления фаз в разбавленном растворе
бавленного раствора [86]. поли-у-бензил-?-глутамата в спира-
лизующих растворителях
образуются большие, хорошо очерченные сферолиты (рис. 112) *. При
наблюдении между скрещенными поляроидами оптическая
природа этих сферолитов оказывается той же, что у сферолитов
ленточного типа, образующихся при кристаллизации линейных
молекул из расплава. Следовательно, вязкость среды не влияет
решающим образом на возможность образования сферолитов.
Характерная черта сферолитов поли-у-бензил-?-глутамата —
появление полос гашения, расположенных по радиусу и хорошо
видимых в обычном свете.
Хотя сферолитная форма и является преобладающим типом
морфологии закристаллизованных в блоке гомополимеров, сфе-
* [В концентрированных растворах поли-у-бензил-А-глутамата, напротив,
образование сферолитов предшествует возникновению дальнего
жидко-кристаллического порядка (см. В. Г. Баранов, Т. И. Волков, С. Я.
Френкель, ДАН СССР, 162, № 4, 836 A965). — Прим. редактора].
314
роЛиТы возникают и бо множестве Других случаев. Можно
Предположить, что существуют определенные общие свойства, не
относящиеся непосредственно к молекулярной структуре,
определяющие образование сферолитов в различных веществах.
Почему же предпочтительнее возникают сферолитные
объединения, а не неорганизованные и несвязанные объединения
отдельных кристаллитов или кристаллических областей? Для
ответа на этот вопрос полезно исследовать оптическую природу
сферолитных картин, обращая особое внимание на то, как они
зависят от условий кристаллизации и деталей структуры цепи.
Оптические картины полимерных сферолитов зависят от
определенных черт структуры цепи и условий кристаллизации,
особенно от температуры кристаллизации, молекулярного веса
Рис. 113. Микрофотография сферолитов полиэтиленади-
пината, закристаллизованного при различных
температурах [87]:
я-0° С; 6-29° С; в-45,5° С
и структурной регулярности отдельных цепей. На рис. 113
приведены микрофотографии сферолитов полиэтиленадипината,
закристаллизованного при 0,29 и 45,5° С [87], расположенных
соответственно ниже, вблизи и выше температуры максимальной
скорости кристаллизации. При 29° С возникают обычного типа
кольцевые сферолиты с серией темных и светлых полос и
чередующимся знаком двулучепреломления вдоль радиуса. При
низких температурах кристаллизации сферолиты подобного
типа не возникают, а двулучепреломление по радиусу
отрицательно. При более высоких температурах сферолиты более
фибриллярны, не так хорошо очерчены, а двулучепреломление вдоль
радиуса положительно. Границы сферолитов, образованных
около температуры плавления, в основном хуже очерчены,
нежели при более низких температурах. Аналогичные оптические
картины получены также для гуттаперчи, закристаллизованной
в широкой области температур [88],
315
В случае высокомолекулярного полиэтилена сферолиты
кольцевого типа возникают при температурах выше предельной
для изотермической кристаллизации. Однако в
низкомолекулярных образцах (М«20 000 или меньше) растут лишь сильно
фибриллярные, а не кольцевые сферолиты [89]. Аналогичные
наблюдения проведены на низкомолекулярных образцах поли-
оксиэтилена, закристаллизованного при различных
температурах [90].
Полимеры с нерегулярной структурой и сополимеры также
образуют сферолитные структуры, но в основном некольцевого
типа [91, 92]. Однако в разветвленном полиэтилене возникают
кольцевые сферолиты, причем период (ширина кольца) зависит
от степени разветвленности [93]. Возрастание температуры
кристаллизации приводит к увеличению кольцевого периода,
который во всей возможной области температур кристаллизации
оказывается обратно пропорциональным степени
переохлаждения. При увеличении доли некристаллизующегося компонента в
цепи сферолитная структура исчезает, хотя кристаллизация все
еще происходит. Например, в сополимерах этилена не
наблюдаются сферолиты (в пределах разрешения светового
микроскопа) в образцах, содержащих более 2% боковых групп Л1-С3Н7
или 5,9% СНз [92].
По оптическим свойствам кристалла, величине и знаку дву-
лучепреломления вдоль главных направлений сферолита,
можно оценить ориентацию цепей в сферолите. Например, в случае
полиэтилена показатель преломления света, поляризованного в
направлении радиуса сферолита, выше, чем у поляризованного
перпендикулярно. Из измерений показателей преломления
сильно ориентированных фибрилл очевидно, что показатель
преломления имеет наибольшее значение в направлении длины цепей.
Отсюда можно заключить, что в сферолите оси с кристаллитов
(оси цепей) ориентированы нормально к радиусу сферолита
[94]. Рациональное объяснение этого удивительного факта дано
Банном [94]. Он показал, что радиусы сферолитов являются
направлениями роста кристаллов; при этом в полиэтилене
развивается морфология,- весьма сходная с кристаллической
структурой короткоцепных углеводородов. Кристаллы последних
растут в виде тонких пластинок, в которых цепи ориентированы
перпендикулярно широким граням. Гораздо быстрее кристаллы
растут в направлениях, перпендикулярных осям молекул,
нежели в параллельном им направлении.
Таким образом, эти наблюдения хорошо коррелируют с
установленной ниже ламеллярной природой большинства
полимерных кристаллитов. Величина двулучепреломления в сферолитах
очень мала в сравнении с вытянутыми волокнами, так что
перпендикулярная ориентация цепей далека от совершенства. Для
полимеров с сильно поляризуемыми боковыми группами, таких
316
как полиамиды и полиэфиры, показатели преломления вдоль и
поперек оси цепи сравнимы. Поэтому даже при
перпендикулярной к радиусу ориентации осей молекул, сферолит может быть
положительно двулучепреломляющим. В полиэтиленадипате
образуются, в зависимости от температуры кристаллизации,
положительные или отрицательные сферолиты [87].
Для полиамидов известны также примеры сосуществования
в одном образце положительных и отрицательных сферолитов.
В обоих случаях молекулы расположены тангенциально
относительно радиуса сферолита, но различные кристаллографические
направления при этом параллельны. Эти выводы
подтверждаются рентгенографическими исследованиями отдельных
участков сферолита с помощью острофокусной техники [95,
96]. Для полиэтилена ось Ь соответствует оси, параллельной
радиусу. Возникающие в растворах поли-у-бензил-?-глу-
тамата сферолиты обладают таким же типом ориентации.
Длинные оси молекул ориентированы нормально к радиусу
сферолита.
Развитие радиальной симметрии завершенного сферолита
проходит через начальную стадию образования и роста других
геометрических форм. Это наблюдалось при изучении
сферолитов из мономерных веществ [83, 84] и неудивительно, что
аналогичные механизмы обнаружены и для полимеров. Вначале
возникают фибриллярные или иглоподобные кристаллы,
которые затем удлиняются, разветвляясь или расходясь в виде
«веера» по аналогии с дендритным ростом. Сферическая форма
развивается постепенно; образованию ее предшествует
появление «листков», которые можно наблюдать в электронном
микроскопе. Предположение о регулярности ответвлений приводит
к заключению, что должны возникать хорошо очерченные
сферические образования, завершающие достаточно большую
последовательность ступеней роста [97]. Кроме того, если взаимные
направления ответвления для всех кристаллитов вполне
определенны, то в сферолите развивается также вполне определенный
кристаллографический порядок. Само ответвление носит
некристаллографический характер и должно зависеть от свойств
поверхностей раздела кристаллитов.
Был проведен тщательный анализ сферолитов кольцевого
или ленточного типа. В настоящее время принято считать, что
чередующиеся темные и светлые полосы возникают вследствие
периодического изменения ориентации двулучепреломляющих
единиц вдоль радиуса сферолита [28, 98]. При некоторой
ориентации двулучепреломляющих единиц происходит гашение;
такая ориентация повторяется периодически. Реплики
поверхности скола полиэтилена, закристаллизованного из расплава и
содержащего сферолиты с периодическими кольцами гашения,
обнаруживают тонкие ламелли, периодически изменяющие свою
317
ориентацию вдоль радиуса сферолита. Период идентичен на*
'блЮд^емому для колец гашения в оптическом микроскопе [45].
На рис. 114 схематически представлено изменение
ориентации кристаллитов вдоль радиуса сферолита в виде проекций
спирально расположенных двуосных эллипсоидов показателя
преломления, характеризующих оптические свойства
кристаллита [99, 100, 101]. Плоскость оптических осей перпендикулярна
радиусу сферолита OR и каждый эллипсоид повернут в одном
и том же направлении на угол, определяемый его расстоянием
Рис. 114. Схема спирального расположения
двуосных эллипсоидов показателя преломления вдоль
радиуса сферолита, объясняющая характер двулуче-
преломления в сферолитах.
от центра. Следовательно, каждое угловое положение
регулярно повторяется. Детальный анализ этой модели объясняет
периодическое гашение, появление мальтийского креста,
зигзагообразные линии и другие тонкие детали оптической структуры
кольцевых сферолитов. Для объяснения круговой симметрии
картин, наблюдаемых в поляризационном микроскопе, т. е.
правильных колец, необходимо предположить, что повороты
эллипсоидов на «смежных» радиусах совпадают по фазе. Таким
образом, вблизи любого произвольно выбранного круга, видимого
в микроскопе двумерного сферолита, ориентация оптической
индикатриссы относительно любого радиуса одинакова. Этот
вывод вытекает из экспериментальных наблюдений и не
следует непосредственно из предложенной модели.
Подтверждение такой модели получено при
рентгенографическом исследовании кольцевых сферолитов полиэтилена с
помощью острофокусной техники. Как уже указывалось, этими
опытами доказана тангенциальная ориентация цепей. Кроме
того, анализ дифракционных картин выявляет спиральную
ориентацию элементарных ячеек относительно радиально на*
правленной оси Ь. Полученное из рентгенографических измерен
ний расстояние, соответствующее половине шага спирали, сов*
318
падает с периодом гашения, непосредственно наблюдаемым в
оптическом микроскопе [96]. В связи с этим необходимо* дЗба^
вить, что в более фибриллярных сферолитах
низкомолекулярного полиоксиэтилена рентгенограммы (от различных участков
вдоль произвольно выбранного радиуса) не свидетельствуют
о периодической ориентации элементарной ячейки [90].
Следовательно, существуют большие различия в ориентации
кристаллитов в различных типах сферолитов. Это подтверждается и на
других системах. Мы уже указывали на то, что оптический
характер сферолитов зависит от факторов, связанных со структур
рой цепи, и условиями кристаллизации.
Подобное описание оптических свойств кольцевых
сферолитов еще не характеризует их исчерпывающим образом;
необходимо связать эти свойства с молекулярными характеристиками
и распространить уточненное описание на другие типы
сферолитов. В конечном счете необходимо установить всю
последовательность этапов образования и роста сферолитов. Так как
оптическое описание уже по своему характеру является
существенно феноменологическим, оно не позволяет однозначным
образом определить специфические формы структурных
элементов. Так как рост сферолитов представляет собой нуклеационно
контролируемый процесс, напрашивается предположение, что
регулярное вращение оптических индикатрисе и элементарных
ячеек вдоль радиуса сферолита обусловлены некогерентной
нуклеацией на поверхности кристаллографической плоскости,
параллельной оси цепи [69, 102].
Предполагается далее, что соответствующие
кристаллографические плоскости кристалла и ядра ориентированы
неодинаково, а повернуты относительно друг друга. Качественно такая
картина согласуется с оптическими свойствами кольцевых
сферолитов. Однако эта гипотеза объясняет свойства лишь одного
луча или радиуса. Не дается никакого объяснения совпадению
по фазе поворотов на соседних радиусах. Из кинетических
исследований также следует, что радиус сферолита растет
линейно со временем. Для сферически симметричного тела это
означает, что при постоянной температуре масса, переносимая на
единицу площади сферолита, постоянна. Площадь в данном
случае представляет собой непрерывную границу между сферо-
литом и расплавом.
При постоянстве плотности и ориентации кристаллитов и по-*
стоянной скорости нуклеации на единичной площади можно
сделать вывод, что и размер кристаллитов (в направлении роста)
постоянен независимо от времени его существования [102]. Если
эта гипотеза верна, то, учитывая предыдущие выводы, рост в
двух главных направлениях, параллельно и перпендикулярно
направлению цепи, оказывается заторможенным. Так как
рассматривается только одна ветвь сферолита, необходимо предпо-
319
ложить непрерывность границы сферолит—жидкость для объ*
яснения постоянства скорости линейного роста. В противном
случае, скорости нуклеации на участках, лишенных
кристаллитов, были бы отличны от скоростей нуклеации на поверхности
или вблизи кристаллитов, так что переносимая масса не была
бы независимой от времени. При таком рассмотрении трехмер*
ный характер сферолитов молчаливо игнорируется, так же как
их объемные характеристики.
Влияние одной растущей ветви на соседнюю представляет
серьезную проблему при описании процесса нуклеации, а также
механизма разветвлений. Хотя ясно, что рост сферолитов связан
с нуклеационным механизмом, классическая теория нуклеации
еще не может объяснить некоторые характерные черты
развития сферолитов.
Несмотря на то, что механизм образования сферолитов
полностью еще не раскрыт, уже сейчас можно утверждать, что у
всех веществ, образующих сферолиты, имеются общие харак*
терные черты. Большинство, если не все, образующие
сферолиты поликристаллические агрегаты состоят из кристаллитов,
которые анизотропны или асимметричны по форме. Ламеллярные
кристаллиты гомополимеров явно попадают под эту
классификацию. Жидкие кристаллы холестерического типа также
асимметричны и образуют сферолиты, как, например, структурно
асимметричные а-спирали поли-у-бензил-?-глутамата. В
последнем случае сферолиты наблюдаются даже при
кристаллизации из разбавленных растворов. Углерод (в модификации
графита) имеет листоподобную или слоистую структуру; в этой
системе также наблюдаются сферолиты. В расплаве
низкомолекулярных органических веществ сферолиты возникают при
добавлении небольшого количества примесей. Эти примеси
концентрируются на определенных гранях кристалла, замедляя их
рост и вызывая развитие кристалла асимметричной формы.
Сферолиты низкомолекулярных неорганических солей
образуются в результате метатеза при возможности диффузии
разбавленного раствора одного компонента в разбавленный
раствор другого компонента, взвешенного в виде вязкого геля.
Конвекция в этом геле исключена и рост определенной грани
замедляется из-за ограниченного поступления необходимых
ионов на ее поверхность.
Таким образом, если по каким-либо причинам скорости
роста кристалла в двух различных направлениях заметно
различны, это приводит к образованию асимметричных по форме
кристаллитов.
Отсутствие сферолитов в сополимерах, содержащих
структурно различные элементы, можно объяснить подавлением
роста ламеллярных кристаллитов по причинам, о которых уже
говорилось [92].
320
Близкое сходство между этими, казалось бы различными,
случаями легче понять, если учесть, что несколько первых кри*
сталлитов, возникающих в процессе первичной нуклеации в
расплавленном полимере, распределены хаотически в неизме*
ненной матрице. Следовательно, возникает, по сути дела,
система «разбавленных» анизотропных кристаллов, напоминающая
обычным образом разбавленную «молекулярную» систему. Если
принять такое обобщение, то возникает вопрос, предопределена
ли поликристалличность первичной нуклеацией в далеко
отстоящих друг от друга элементах объема. Если это так, то почему
образование асимметричных кристаллитов приводит скорее к
возникновению сферолитов, а не к беспорядочному
объединению кристаллитов? В особенности необходимо разрешить
проблему, происходит ли образование сферолитов благодаря их
повышенной устойчивости в сравнении с другими
организациями кристаллитов или вследствие кинетических факторов,
начинающих доминировать после первичной нуклеации.
Для ряда частных случаев Фулман [103] показал, что
условие повышенной стабильности выполняется. Для кристаллитов,
образованных линейными молекулами, существует высокая
анизотропия поверхностных свободных энергий. Свободная энергия
поверхности, перпендикулярной к направлению цепей, гораздо
выше, чем поверхности, ориентированной вдоль направления
цепей. Так как известно, что размеры кристаллита гораздо
меньше в направлении цепей, теорема Вульфа совершенно не
выполняется. Характерные тонкие ламеллярные кристаллиты из
линейных молекул не могут, следовательно, быть равновесными-
Организация этих неустойчивых кристаллитов в сферолит
представляет собой возможный механизм, разрешающий это
противоречие.
Литература
1. R. S. Bear, H. S. Rugo, Ann. N. Y. Acad. Sci., 53, 627 A951).
2. R. S. Be a r, J. Am. Chem. Soc„ 66, 1297 A944).
3. R. S. В e a r, Adv. Protein Chem., 7, 69 A952).
4. R. S. Bear, J. Cellular Сотр. Physiol., 49, (Suppl. 1), 303 A957).
5. H. E. Huxley, Proc. Roy. Soc. (London), Sen B, 141, 59 A953).
6. H. К i e s s i g, Koll. Z., 152, 62 A957).
7. L. M. A r n e 11, E. P. H. M e i b о h m, A. F. Smith, J. Polymer Sci., 5,
737 A950); 7, 57 A951).
8. L. G. W a 11 n e r, Monatsh. Chem., 79, 279 A942).
9. W. O. Statton, J. Polymer Sci., 22, 385 A956); W. O. Statton,
G. M. God ard, J. Appl. Phys., 28, 1111 A957).
10. B. Belbeoch, A. Guiner, Makromol. Chem., 31, 1 A959).
11. L. Man del kern, С R. Worth in gton, A. F. Diorio, Sci., 127,
1052 A958); J Appl. Phys., 31, 536 A960).
21 Л. Манделькерн
321
12. A. Keller, Phil. Mag., 2, 1171 A957); Disc. Faraday Soc, 25, 114
A958).
13. B. G. Ran by, F. F. Morehead, N. M. Walter, J. Polymer Sci., 34,
349 A960).
14. L. Mandelkern, A. S. Posner, A. F. D i о r i o, D. E. Roberts,
J. Appl. Phys., 32, 1509 A961).
15. S. S. Pollock, W. A. Robinson, R. Chiang, P. J. Flory,
J. Appl. Phys., 33, 237 A962).
16. R. Eppe, E. W. Fischer, H. A. Stuart, J. Polymer Sci., 34, 721
A959).
H. A. Stuart, Ann. N. Y. Acad. Sci., 83, 3 A959); Roll. Z., 165, 3
A959).
17. P. H. Hermanns, A. Weidinger, Makromol. Chem., 39, 67 A960).
18. W. 0. S t a 11 о n, J. Polymer Sci., 22, 385 A956).
19. K. Hess, H. Kiessig, Z. Physik. Chem. (Leipzig), 193, 196 A944);
K. Hess, H. Kiessig, Naturwiss., 31, 171 A955); K. Hess, Ric. Sci.,
Suppl. A, 594 A954).
20. R. Hoseman n, Z. Phys., 128, 1 A950); R. Bonart, R. Hosemann,
Makromol. Chem., 39, 105 A960); R. Hosemann, Polymer, 3, 349
A962).
21. H. Arnold, Roll. Z., 157, 111 A958).
22. С Sella, J. J. Trill at, Compt. rend., 246, 3246 A958); 248, 410, 1819,
2348 A959).
23. P. J. Flory, A. D. Mc In tyre, J. Polymer Sci., 18, 592 A955).
24. M. Richardson, P. J. Flory, J. B. Jackson, Polymer, 4, 221
A963).
25. R. L. Cormia, F. P. Price, D. Turnbull, J. Chem. Phys., 37, 1333
A962).
26. J. I. Lauritzen, J. D. Hoffman, J. Res. Natl. Bur. Stand., A, 64,
73 A960).
27. F. С Frank, M. To si, Proc. Roy. Soc. (London), Ser. A, 263, 323
A961).
28. A. Keller, Makromol. Chem., 34, 1 A959).
29. P. H. Geil, J. Polymer Sci., 44, 449 A960); 47, 65 A960); 51, S10
A961).
30. G. К a m p f, Roll. Z., 172, 507 A960).
31. F. Anderson, Bull. Am. Phys. Soc, Ser. 11, 8 C), A963).
32. P. J. F 1 о r y, J. Am. Chem. Soc, 84, 2857 A962).
33. N. Bckkedahl, H. Ma the son, J. Res. Natl. Bur. Stand., 15, 503
A935).
34. N. McC rum, J. Polymer Sci., 34, 355 A959).
35. M. Takayanagi, M. Yoshino, S. M i n a m i, J. Polymer Sci., 61,
57 A962).
36. W. Ruland, Acta cryst., 14, 1180 A961).
37. J. M. Go p pel, J. J. Arlman, Appl. Sci. Res., Sect. A, 1, 462 A949).
38. D. E. Roberts, L. Mandelkern, J. Am. Chem. Soc, 77, 781 A955).
39. B. W under lich, M. Dole, J. Polymer Sci., 24, 201 A957).
40. R. F. Chiang, P. J. Flory, J. Am. Chem. Soc, 83, 2857 A961).
41. H. G. Kili an, Roll. Z., 183, 1 A962).
42. R. H. S torks, J. Am. Chem. Soc, 60, 1753 A958).
43. R.J a ceo dine, Nature, 176, 301 A955).
44. P. H. Till, J. Polymer Sci., 24, 301 A957).
45. E. W. Fischer, Z. Naturforsch., 12, 753 A957).
46. W. Schlesinger, H. M. Leeper, J. Polymer Sci., 11, 203 A953).
47. F. С Frank, Disc. Faraday Soc, 5, 48 A949).
48. I. M. Dawson, V. V a n d, N. G. Anderson, Proc Roy. Soc
(London), Ser. A, 206, 555 A951); 214, 72 A952); 218, 255 A953)
49. R. St. John Man ley, J. Polymer Sci., 47, 149 A960); Nature, 189, 390
A961).
822
50. В. G. R a n b у, R. W. N о е, J. Polymer Sci., 51, 337 A961).
51. P. H. Ge i 1, J. Polymer Sci, 44, 449 A960).
52. P. H. Geil, N. K. J. Symons, R. G. Scott, J. Appl. Phys, 30, 1516
A959).
53. A. Keller, Makromol. Chem, 34, 1 A959).
54. V. F. Holland, S. B. Mitchell, W. L. Hunter, P. H. Linden-
meyer, J. Polymer Sci., 62, 145 A962).
55. P. Geil, J. Polymer Sci., 51, S10 A961).
56. В. А. К а р г и н, Н. Ф. Б а к е е в, Л. Ли HI e н, Высокомол. соед., 3,
1102 A961).
57. J. В i s s с h о р s, J. Polymer Sci., 17, 89 A955).
58. A. Kel ler, J. Polymer Sci., 36, 361 A959).
59. S. Okamura, К. Н а у a s h i, Y. К i t a n i s h i, Makromol. Chem., 47,
230, 237 A961); J. Polymer Sci, 58, 925 A962); 60, 526 A962).
60. J. L a n d o, N. M о г о s о f f, H. M о r a w e t z, B. Post, J. Polymer Sci,
60, 169 A962).
61. V. F. Holland, P. Lindenmeyer, J. Polymer Sci, 57, 589 A962).
62. J. B. Jackson, P. J. Flory, R. Chiang, Trans. Faraday Soc, 59,
1906 A963).
63. F. P. P r i ce, J. Chem. Phys, 35, 1884 A961).
64. A. Keller, A. O'Connor, Polymer, 1, 163 A960).
65. W. O. Stat ton, P. Geil, J. Appl. Polymer Sci, 3, 357 A960).
66. B. Wunderlich, M. К ash dan, J. Polymer Sci, 50, 71 A961).
67. R. F. Chiang, P. J. Flory, Abstract of Paper presented before
Division of Polymer Chemistry, American Chemical Society, New York,
September, 1960.
68. F. P. Price, J. Polymer Sci, 38, 139 A960).
69. J. D. Hoffman, J. I. L a u r i t z e n, J. Res. Natl. Bur. Stand, A, 65,
297 A961).
70. A. Peterlin, E. W. Fischer, Z. Phys, 159, 272 A960); A. Peter-
lin, E. W. Fischer, С Reinhold, J. Chem. Phys, 37, 1403 A962).
71. P. J. Flory, J. Chem. Phys, 17, 223 A949).
72. D. Turn bull, R. L. Cormia, J. Chem. Phys, 34, 820 A961).
73. L. M a n d e 1 k e r n, J. Polymer Sci, 47, 494 A960).
74. J. D. Hoffman, J. J. Weeks, J. Res. Natl. Bur. Stand, A. 66, 13
A962).
75. L. A. Wood, N. Bekkedahl, J. Appl. Phys, 17, 362 A946).
76. W. E. G a r n e r, K. v a n Bibber, A.M. King, J. Chem. Soc, 1931,
1533.
77. K. H. Mever, A. J. A. van der Wyk, Helv. chim. acta, 80, 83
A937).
78. F. W. Billmeyer, J. Appl. Phys, 28, 1114 A957).
79. K. V a n N e s, H. A. V a n Westen, Aspects of the Constitution of
Mineral Oils, New York, 1951, p. 105.
80. M. В г о a d h u г s t, J. Chem. Phys, 36, 2578 A962).
81. P. J. Flory, A. Vrij, J. Am. Chem. Soc, 85, 3548 A963).
82. L. M a n d e 1 k e r n, F. A. Q u i n n, Jr., неопубликованные данные, 1963.
83. F. В e r n a u e r, Gedrille Kristalle, Berlin, 1929.
84. H. W. Morse, J. D. H. Donnay, Am. Mineralogist, 21, 392 A936);
H. W. Morse, С H. Warren, J. D. Donnay, Am. J. Sci, 23, 421
A932).
85. J. E. Coleman, B. J. Allan, B. L. Vallee, Sci, 131, 350 A960).
86. С Robinson, Trans. Faraday Soc, 52, 571 A956); Disc. Faraday
Soc, 25,29 A958).
87. M. Takayanagi, Mem. Fac. Eng, Kyushu Univ., 16C), 111 A957).
88. G. Schuur, J. Polymer Sci, 11, 385 A953).
89. W. Banks, J. N. Hay, A. Sc harpies, G. Thomson, Nature, 194,
542 A962).
90. F. P; P r i с e, R. W. К i 1 b, J. Polymer Sci., 57, 395 A962),
21»
323
91. V. F. Н о 11 a n d, J. Polymer Sci., 43, 572 (I960).
92. J. B. J а с к s о n, P. J. F1 о г у, Polymer, 4 A963).
93. T. Naono, J. Sci. Hiroshima Univ., Ser. A, 24, 653 A960).
94. С W. Bunn, Т. С A1 cock, Trans. Faraday Soc, 41, 317 A945).
95. A. Kel 1 er, J. Polymer Sci., 17, 351 A955).
96. Y. Fujiwara, J. Appl. Polymer Sci., 4, 10 A960).
97. A. Keller, J. R. S. Waring, J. Polymer Sci., 17, 447 A955).
98. R. J. Clark, R. L. Miller, R. S. Stein, P. R. Wilson, J. Polymer
Sci., 42, 275 A960).
99. A. Keller, J. Polymer Sci., 39, 160 A959)
100. H. D. Keith, F. J. Padden, Jr., J. Polymer Sci., 39, 101, 123 A959).
101. F. P. Price, J. Polymer Sci., 39, 139 A959).
102. F. P. P rice, Ann. N. Y. Acad. Sci., 83, 20 A959).
103. R. L. Fu 11 m a n, Acta Met., 5, 638 A957).
104. G. Wulf, Z. Krist., 34, 449 A901).
Предметный указатель
Белки
водородные связи 77
кристаллизация 115
полиморфизм 146
структура 23, 60, 67, 83, 116
структурные переходы 78
фибриллярные 168 и ел.
дифракционные максимумы 281
сокращение 193, 199 и ел.
Виниловые полимеры, конформация 21
Водородные связи
в спирали ДНК 24, 134
в а-спирали полипептидов 23, 60,
62, 65, 66, 77
Волокна
меридианальные рефлексы 281
одноосная ориентация 172
плавление 184, 185, 188, 197
степень кристалличности 174
Гелеобразование 118, 119
Гемоглобин, рентгенограмма 67
Гуттаперча
конфигурация 99
размеры цепей 20
сферолиты 315
термодинамические параметры
плавления 125, 138
Дезоксирибонуклеиновая кислота
плавление
температура 76
термодинамические параметры
134
структура 24, 61, 134
Золь-эффект 152
Изомеризация 101, 103
Изомеры
геометрические 98, 128
поворотные 18, 143, 145
Изоморфное замещение 116 и ел.
принцип структурной
совместимости сомономеров 116
Каучук гидрохлорированный,
кристаллизация 25
Каучук натуральный
конформация 136
кристаллизация 172, 216, 232,
256
связь с температурой
плавления 257
скорость 218
плавление 34, 35, 52
сеток 161
теплота 53
термодинамические параметры
125, 128, 137, 138, 180
размеры цепей 19, 20
рентгенограмма 26
сокращаемость 193
степень кристалличности 294
Кератин
дифракционные максимумы 281
плавление 203, 204
сокращение 203
Коллаген
набухание 54
размеры цепей 20
рентгенограмма 26, 281
сверхсокращение 173
сокращение 199, 201
структура 60, 61, 73
термодинамические параметры
плавления 126, 133, 192
Комплексообразование 75—79
Конформация 18, 19, 73, 136
ассимметричная 47, 60
влияние
боковых групп 21, 22
растворителя 296
определение 15
325
Конформация
спиральная 21, 22, 60, 65, 68,
146
энергия 18, 19, 142, 143
Коэффициент
кристаллизации, температурный
237 и ел.
скорости
кристаллизации 219, 229
роста сферолитов 149, 247
спирализации 277
теплового удлинения 198
термоэластический 184
Кристаллизация 16, 24, 25, 33
блок-сополимеров 112
блочная 253
влияние
деформации 41, 170, 191
диффузии 270
молекулярного веса 252
разветвленности 111
способа полимеризации 99,
100
в неравновесных условиях 41
гомополимеров 28, 216 и ел., 230
и ел.
зависимость формы от
температуры кристаллизации 298
затруднения НО, 114
изоляция отдельных участков
235
изотермы 217—219, 227,230,232—
234, 259
из расплава 281 и ел.
из раствора 265 и ел., 295 и ел.
влияние растворителя 296
в реальных условиях 305
заданной концентрации 58
изотермы 271
кинетика 272
механизм 295
складчатые структуры 304
теория 299
форма кристаллов 297
кинетика 214 и ел.
интерпретация 287
кристаллическое ядро 292
механизмы 214 и ел.
набухших полимеров 292
ориентационная 170 и ел., 185,
202
полиморфизм 146 и ел.
полиэлектролитов 76
полупериод 230, 250, 251
связь времени с температурой
плавления 257
скорость 251
сополимеров 82, 84, 117
изотермы 263
Кристаллизация
кинетика 258
набухших 118 и ел.
статистических 292
теория 83, 106, 260
стереорегулярных полимеров 16,
107, 108, 111
структурно-нерегулярных
полимеров 257
сшитых полимеров 161, 165
теория 222, 228
кинетика осаждения 270, 271
отклонения от 235
трехмерная 42
Кристаллиты 32, 42
концепция образования в гомопо-
лимерах 288
ламеллярные 289, 301
ориентация цепей 163, 164, 170
проходные цепи 45, 119, 291
размеры 159
зависимость от температуры
кристаллизации 297
расчет 304, 309
складывание цепей 291, 296
Кристалличность
методы оценки 293
псевдоравновесный уровень 217
Кристаллы
пластинчатые 296
присутствие аморфного полимера
298
рост 305
температура плавления 299
форма 297, 298
Методы
ИК-спектроскопия 99, 101, 293,
294
калориметрия 294
кристаллографический анализ 116
рентгенография 25, 67, 111, 196,
197, 277, 293, 294
рассеяние под малыми углами
см. Рассеяние рентгеновских
лучей под малыми углами
электронная микроскопия,
корреляция с малоугловым
рассеянием рентгеновских лучей 288
ядерный магнитный резонанс 293
Механохимия 207
химические машины 210, 211
цикл Карно 208, 209
Миоглобин, рентгенограмма 67
Модели
зародыша кристаллизации 244,
245
Изинга 19, 64, 138
квазирешетки 138
Модели
кристаллита 144, 301
процесса плавления 41, 42
решеточные 144
свободносочлененных
мономерных звеньев 17
ядра кристаллизации, пачечная
292
Мультнспирали 60, 61, 66, 68, 135, 277
Нуклеация 66, 221, 225—231
внутримолекулярная 275
в растворах 273
монослойная 248
определение 237
параметры 253
первичная 305
свободная энергия образования
зародыша 238, 239, 244, 245,
250, 268
скорость 243, 255, 261, 269
спорадический характер 255
теория 239, 243, 246, 247
трехмерная 249
Нуклеиновые кислоты
набухание 54
переход клубок—спираль 275
структура 23, 60, 116, 135
Отжиг 33—35, 159
Параметры
вероятности генерирования
последовательности в сополимерах
95, 106, 108, 113
гибкости цепи 139
дезориентации 69
линейного расширения сегки 154
Переход
золь—гель 119
клубок—спираль 64, 66, 275
кинетика 276
полиморфный 136
спираль—клубок 57, 59 и ел., 74,
76
аналогия с процессом
одномерной кристаллизации 67 '
диффузный характер 67
кооперативный характер 62, 65,
68
фазовый см. Фазовый переход
^ис-г/?аяс-изомерный 101
Плавление
влияние
внутримолекулярных
взаимодействий 142
деформации 41, 181
когезии 135
конформации 138
Плавление
молекулярного веса 43 и сл«
химических реакций 75 и сл<
электролитов 202
волокна 184, 185, 188, 197
диффузный характер 39, 90,
185
набухших сеток 165
обратимость 173, 194
ориентированных систем 178, 183,
185
полимеров
в растворе 47 и ел., 56 и ел.
гомополимеров 31 и ел.
стереорегулярных 107, 108
сшитых 152 и ел.
теория 155 и ел.
природа процесса 33 и ел., 53
свободная энергия 32, 43, 141,
144
сополимеров 82 и ел.
блок-сополимеров 112
температура см. Температура
плавления
температурный интервал 32, 33
теплота на моль мономерных
звеньев 49, 52, 53, 96, 124 и ел,
термодинамика 41, 123 и ел.
параметры 125, 126, 137, 138
фибриллярных белков 168
энтальпия 144
энтропия 124 и ел., 135 и ел.,
144, 145, 168
Полиакриловая кислота, размеры
цепей 20
Полиакрилонитрил
гелеобразование 118
конформация 137
кристаллизация 296
кристалличность 111
плавление
температура 118
термодинамические параметры
126, 133
размеры молекул 19, 20
Полиамиды
кристаллизация 296
плавление
температура 45
термодинамические параметры
130
полиморфизм 146
Поли-у-бензил-?-глутамат
зависимость концентрации фаз от
степени полимеризации 72
переход спираль—клубок 66
размеры цепей 20
а-спираль 72
сферолиты 314, 317
327
Полибутадиен
геометрические изомеры 98, 99
зависимость удельного объема от
температуры 100
конформация 22
кристаллизация 100
термодинамические параметры
плавления 128
Поли-2-вицилнафталин
конформация 22
кристаллизация 17
Поливиниловый спирт,
кристаллизация 111
Поливинилфторид,
термодинамические параметры плавления 125,
129
Поливинилхлорид
гелеобразование 118
конформация 22
кристалличность 111
Полигексаметиленадипамид
кристаллизация 257
плавление
сополимеров 95
термодинамические параметры
126
полиморфизм 146
Поли(гексаметиленадипамид-капро-
амид), плавление 96
Поли(гексаметиленадипамид-себац-
амид), плавление 96
Полигексаметиленсебацамид,
полиморфизм 146
Полигексаметилентерефталат,
термодинамические параметры плавления
125
Поли-?-глутаминовая кислота,
переход спираль—клубок 61, 76
Полидекаметиленадипинат
зависимость удельного объема от
температуры 36
плавление
температура 44
теплота на моль мономерных
звеньев 97
термодинамические параметры
125
Полидекаметиленацилат,
термодинамические параметры плавления
125
Полидекаметиленсебацамид
плавление
теплота на моль мономерных
звеньев 97
термодинамические параметры
125
Полидекаметиленсебацинат
константа скорости
кристаллизации 237
Полидекаметиленсебацинат
плавление
теплота на моль мономерных
звеньев 97
термодинамические параметры
125, 130
рост сферолитов 249
Полидекаметилентерефталат
плавление 52
теплота 53
термодинамические параметры
125
Полидиметилсилоксан
конформация 136
размеры цепей 19, 20
термодинамические параметры
плавления 128
Полиизобутилен
конформация 136
кристаллизация 16
размеры цепей 19, 20
термодинамические параметры
плавления 128
Полиизопрен
геометрические изомеры 99
кристаллизация 100
Поликапрамид, термодинамические
параметры плавления 126
Поликристалличность 25, 32
Поли-я-ксилен, термодинамические
параметры плавления 128
Поли-/,-лизин, переход
спираль—клубок 76
Полимеризация
анионная 100, 107
методы Натта 105, 109
радикальная 99, 105—107
Полимеры
аморфные 170, 209
атактические 106
изотактические 22, 105, 128,
146
кристаллические
молекулярная картина 289
плотность 215
рентгенограмма 293
микроструктура 105
молекулы
геометрическая форма 16, 17,
68
размеры 17—20
распределение концевых групп
43
среднеквадратичный радиус
инерции 18
средний квадрат расстояния
между концами цепи 17, 18
статистическая сумма 65
строение 15
328
Полимеры
синдиотактические 22, 23, 105
стереорегулярные 106
структура
с неупорядоченными цепями 17
и ел.
с упорядоченными цепями 20
и ел.
сшитые 153, 154
из неориентированных цепей
164
из одноосноориентированных
цепей 160 и ел.
изотропные сетки 155 и ел.
из статистических цепей 157
и ел.
термодинамика расплавов 48
эластомеры 128
Поли-о-метилстирол, конформация 22
Поли-4-метилгексен-1, конформация
22
Полиметилен, зависимость удельного
объема от температуры 36
Полиметиленовые сополимеры
плавление 91, 92
степень кристалличности 93
Полиметилметакрилат
изотактический, конформация 22
кристаллизация ПО
размеры цепей 20
Поли-ж-метилстирол, конформация 22
Полиморфизм 146 и ел.
анизотропные изменения 183
стабильность форм 147
Полинонметиленацелат,
термодинамические параметры плавления 125
Полинуклеотиды
конформация 60
переход спираль—клубок 66
спиральные комплексы 135, 277,
278
строение 134, 135
Полиоксиизобутилен,
термодинамические параметры плавления 132
Полиоксиметилен, термодинамические
параметры плавления 126, 138
Полиоксипропилен,
термодинамические параметры плавления 132
Полиоксиэтилен
зависимость удельного объема от
температуры 36
изотермы кристаллизации 266
термодинамические параметры
плавления 126, 132, 138
Полипептиды
водородные связи 77
геометрические изомеры 103
переход спираль—клубок 64, 275
плавление 103
Полипептиды
полиморфизм 146
а-спирали 60, 292
структура 23
Поли-1-пролин
конформация 24
разбавленные растворы 73
структура 60
Полипропилен
зависимость удельного объема от
температуры 109
конформация 23
кристаллизация 257, 296
изотермы 259
плавление
температура 118
термодинамические параметры
125, 127
рентгенограмма 293
Поли-И-Ы'-себацоилпиперазин
плавление 50
теплота на моль мономерных
звеньев 97
термодинамические параметры
125, 131
Полистирол
кристаллизация ПО
размеры молекул 19, 20
термодинамические параметры
плавления 125, 127, 138
Политетраметиленизофталат,
термодинамические параметры плавления
125
Политетраметилентерефталат,
термодинамические параметры плавления
125
Политетрафторэтилен
структура 23
термодинамические параметры
плавления 125, 127, 138
Политрифторхлорэтилен
кристалличность 111
плавление
температура 50, 118, 311
теплота 53
термодинамические параметры
125, 129
Полиуретаны
малоугловое рассеяние
рентгеновских лучей 286
термодинамические параметры
плавления 131
Поли-ж-фторстирол, кристаллизация
17
Полихлоропрен
геометрические изомеры 99
термодинамические параметры
плавления 125
329
Поли-я-хлорстирол, кристаллизация
17
Полиэтилен
зависимость удельного объема
от давления 40
зависимость удельного объема
от температуры 37, 38, 55, 56,
112
конформация цепи 18, 19, 21
кристаллизация 217, 219
изотермы 233, 234, 259, 266
размер ядра 292
микроструктура 164
плавление
температура 45, 50
теплоемкость 34, 112
теплота 53
термодинамические параметры
125, 127, 138
поляризационная
микрофотография 28
размеры цепей 19, 20
рассеяние рентгеновских лучей
под малыми углами 283
рентгенограмма 25—27, 298
сферолиты 316
сшитый 163, 195
температура растворения 58
электронная микрофотография 28
Полиэтиленадипинат
кристаллизация 257
сферолиты 220, 221, 315, 317
Полиэтиленсебацинат,
термодинамические параметры плавления 126
Полиэтиленсукцинат
кристаллизация 257
температура плавления 45
Полиэтилентерефталат
кристаллизация 257
плавление
сополимеров 95
термодинамические параметры
126
Поли (этилентерефталат-адипинат),
плавление 96
Поли (этилентерефталат-себацамид),
плавление 96
Полиэфиры
кристаллизация 25, 296
плавление
температура 44, 45
термодинамические параметры
129, 130
Потенциал
термодинамический
волокнистой системы 174
торможения 18, 19, 136, 138, 142
Потенциальная функция
«-бутана 142
Потенциальная функция
заторможенного вращения 143
полиизобутилена 23
угловая периодичность 18
Правило фаз 50
Принцип Ле Шателье 171
Протофибриллы 60
Рассеяние рентгеновых лучей под
малыми углами 27
абсолютная интенсивность 282
влияние
температуры кристаллизации
283-286
толщины образца 285
Рекристаллизация 203, 205
Респирализация 277
Сокращаемость 193 и ел.
вызванная химическими
реагентами 207
фазовый переход 206
Сокристаллизация 116 и ел.
Сополиамиды
зависимость температуры
плавления от состава 96
изоморфное замещение 116
плавление 95 и ел.
Сополиэфиры
плавление 95 и ел.
температура, зависимость от
состава 96
Состояние
аморфное 15, 16, 31, 41, 57, 83
жидкое см. аморфное
кристаллическое 16, 31, 41, 72,
135
мезоморфное 33
метастабильное 33, 58, 147, 148
переохлажденное 197
полиморфное 148
а-Спираль 23, 60, 62, 292
Степень
кристалличности 293
влияние температуры 88, 89, 93
определение 55, 93
ориентированных полимеров
174, 178
равновесная 54
полимеризации 18
Стереоизомеризм 104 и ел.
Сферолиты 27, 28, 164, 313 и ел.
двулучепреломление 317, 318
кольцевые 316, 319
морфология роста 317
оптическая картина 315
ориентация цепей 316, 318
радиус 221, 227, 316
330
Сферолиты
размеры 314, 319
свободная энергия образования
309
скорость роста 220, 236, 247—
249, 320
спорадический характер роста
219
фибриллярные 316
Температура
кристаллизации 35, 251, 308
критическая 178
плавления см. Температура
плавления
растворения, зависимость от
концентрации 58
сокращения 172, 179
стеклования 128, 293
Температура плавления 32, 37, 42, 124
и ел., 144
блок-сополимеров 113
влияние
концентрации сшивки 168
изомеризации 102
изоморфного замещения 117
растворителя 49—55
состава раствора 58, 59
степени полимеризации 44, 45,
59, 264
химического строения 130—133
набухшей сетки 168
набухших полимеров 51, 52, 54
определение 33, 34, 92, 100
равновесная 39, 89, 90, 124, 156,
162, 166, 312
связь с временем кристаллизации
257
сополимеров 86, 87, 90, 95, 97,
102, 113, 118, 312
стереорегулярных полимеров 107,
108
сшитых полимеров 155 и ел.
Теория
Зимма и Брегга 64
каучукоподобной эластичности
175, 179
Флори
кристаллизации сополимеров
83 и ел., 106
фазовых переходов в
растворах стержневидных частиц 69
Флори — Хаггинса 140, 166
Удельный объем, зависимость от
давления 40
Удельный объем, зависимость от
температуры 36, 37, 55, 100, 109.
112, 115, 117
Уравнение
Гиббса — Дюгема 57
Клапейрона 175
Клаузиуса — Клапейрона 120,126,
175
Фазовое равновесие
при кристаллизации 235
теория 67
при плавлении 53
расчет 71
условия 55, 79, 165, 186, 190
Фазовые состояния 15
Фазовый переход 31, 100
взаимное подавление роста
соседних центров 223
изопотенциальный 211
кинетика 221, 261
кристалл — жидкость 171
первого рода 32, 34, 39, 42, 89
при сокращении 206
спираль — клубок 67, 119
Фазы
рост 221, 225, 226
сосуществование 293, 294
тактоидиая 70
Фиброин шелка
кристалличность 116
структура 115
Формула
Аврами 224, 227
Стерлинга 139
Флори — Хаггинса 47
Фракционирование 59
Целлюлоза, эфиры
гелеобразование 118
конформация 137
кристаллизация 296
размеры цепей 20
термодинамические параметры
плавления 125, 131, 132
Эластичность, молекулярная теория
171
Эластоидин
сокращение 200, 201
температура плавления 202
Элементарная ячейка 32
геометрия 25
размеры 25
Оглавление
Предисловие редактора 5
Предисловие автора 10
Глава 1. Основные структурные представления 15
Введение 15
Структура неупорядоченных цепей 17
Структура упорядоченных цепей 20
Морфологические особенности . 25
Литература 29
Глава 2. Плавление гомополимеров 31
Введение 31
Природа процесса плавления 33
Влияние молекулярного веса 43
Литература 45
Глава 3. Плавление гомополимеров в присутствии низкомолекулярных
жидкостей 47
Введение 47
Концентрированные системы 47
Разбавленные растворы 56
Переход спираль — клубок 59
Переходы, не связанные с изменениями молекулярных конфор-
маций 68
Плавление, вызванное химическими реакциями 75
Литература 79
Глава 4. Плавление сополимеров 82
Введение 82
Теория 83
Общие экспериментальные результаты 90
Сополиэфиры и сополиамиды 95
Геометрический изомеризм 98
Стереоизомеризм 104
Разветвленность 111
Упорядоченные сополимеры 112
Сокристаллизация и изоморфное замещение 116
Набухшие сополимеры 118
Литература 120
332
Глава 5. Термодинамические параметры 123
Введение 123
Температура, теплота и энтропия плавления 124
Энтропия плавления 135
Конформация цепи и кристаллизация 138
Полиморфизм 146
Литература 149
Глава 6. Плавление сшитых полимеров 152
Введение 152
Теория плавления изотропных сеток 155
Температура плавления сетки, образованной статистическими
цепями 157
Температура плавления сеток, образованных из одноосноориенти-
рованных цепей 160
Температура плавления сеток, образованных из кристаллических
неориентированных цепей 162
Плавление набухших сеток 165
Фибриллярные белки 168
Литература 169
Глава 7. Ориентационная кристаллизация и сократимость 170
Введение 170
Однокомпонентные системы, подверженные действию
растягивающей силы 173
Многокомпонентные системы, подверженные действию
растягивающей силы . . . 185
Ориентационная кристаллизация и сокращаемость при отсутствии
растяжения 193
Сокращаемость фибриллярных белков 199
Механохимия 207
Литература 212
Глава 8. Кинетика и механизмы кристаллизации 214
Введение 214
Экспериментальные исследования кристаллизации чистых
гомополимеров 216
Математическое описание кинетики фазовых превращений .... 221
Анализ блочной изотермической кристаллизации гомополимеров . 230
Температурный коэффициент кристаллизации гомополимеров . . . 237
Кристаллизация полимеров, содержащих структурные
нерегулярности 257
Кристаллизация полимеров в присутствии низкомолекулярных
жидкостей 265
Литература 278
Глава 9. Морфология 281
Кристаллизация полимеров из расплава 281
Кристаллизация полимеров из разбавленных растворов 295
Зависимость температуры плавления от условий кристаллизации . 308
Сферолиты 313
Литература 321
Лео Манделькерн
КРИСТАЛЛИЗАЦИЯ ПОЛИМЕРОВ
с. 336 УДК 541.6:542.65
Темплан 1966 г. п. 14
Издательство „Химия", Ленинградское отделение.
Невский пр., дом 28
Редактор В. Д. Пиастро
Техн. редактор Ф. Т. Черкасская
Корректоры Н. Я. Шубина, Л. А. Любович
Сдано в набор 26/11. Подписано к печати 20ДУ 1966 г.
Формат 60x90Vi6>. Бумага № 2. Печ. л. 21,0. Уч.-изд. л. 21,83.
Тираж 8000 экз. Цена 1р 67к. Заказ № 86.
Ленинградская типография № 2
имени Евгении Соколовой Главполиграфпрома
Комитета по печати при Совете Министров СССР.
Измайловский проспект, 29.
ИЗДАТЕЛЬСТВО
„ХИМИЯ"
ГОТОВЯТСЯ К ПЕЧАТИ
КАРГИН В. А., СЛОНИМСКИЙ Г. Л. Краткие очерки по
физико-химии полимеров. Изд. 2-е, доп. и пер. 15 л., 25 000 экз.,
цена 1 р. 19 к. в пер.
Книга является расширенным и переработанным
вторым изданием «Кратких очерков по физико-химии
полимеров», вышедших в 1960 г.
При переработке основное внимание было уделено
рассмотрению новейших данных по надмолекулярным
структурам и их роли в формировании свойств
полимерных тел во всех состояниях.
Книга предназначена для научных работников,
инженеров, аспирантов и студентов старших курсов,
интересующихся физико-химией полимеров.
МАРЕК О., ТОМКА М. Акриловые полимеры (Прага, 1964).
Перевод с чешского. 18 л., 6000 экз., цена 1 р. 54 к. в пер.
Акриловые полимеры, главным представителем
которых является плексиглас, превосходят остальные
пластики некоторыми свойствами, а именно: стойкостью к
атмосферным влияниям и к старению, хорошо
пропускают свет, совершенно прозрачны и окрашиваются з
любой цвет. Поэтому в мировой промышленности они
вырабатываются в большом количестве и по своему
значению занимают важное место среди
термопластических масс.
Данная книга знакомит широкий круг работников
промышленности и потребителей пластиков со
свойствами акриловых полимеров и их применением.
В книге описываются способы получения и
технология производства мономеров, их полимеризация,
свойства и методы анализа полимеров и сополимеров,
подробно рассматриваются способы переработки
полимеров и сополимеров.
Книга предназначена для научных и инженерно-
технических работников научно-исследовательских
институтов и химических предприятий, а также для
студентов средних и высших химических учебных
заведений.
335
ГОТОВЯТСЯ К ПЕЧАТИ
Вспомогательные вещества для полимерных материалов
(Справочник). Коллектив авторов, под ред. канд. хим. наук
Пиотровского К. Б. и канд. хим. наук Салнис К- Ю. 15 л.,
10 000 экз., цена 90 коп.
В справочник включены сведения о стабилизаторах,
органических ускорителях и агентах вулканизации,
замедлителях подвулканизации, ускорителях
пластификации, модификаторах полимерных материалов,
выпускаемых отечественной промышленностью, а также
намеченных к промышленному производству и находящихся
в стадии широких исследований.
По каждому продукту приводятся основные
свойства, техническая характеристика отечественных и
иностранных образцов, принципиальная схема получения,
сведения об эффективности применения и некоторые
другие данные. Для удобства пользования наряду с
химическими названиями приводятся наименования
химикатов отечественного ассортимента. В приложение
включены указатель торговых наименований продуктов
и указатель иностранных фирм, выпускающих подобные
химикаты.
Справочник предназначен для инженеров и
научных работников всех отраслей промышленности
полимерных материалов: каучука и резины, химических
волокон, пластических масс, полимерных пленочных
материалов, лаков, красок и клеев.
Справочник по пластическим массам. Полимерные материалы.
Коллектив авторов под ред. Гарбара М. И., Егорова Н. М.,
Акутина М. С. 40 л., 25 000 экз., цена 2 р. 36 к. в пер.
В справочнике собраны важнейшие сведения о
пластических массах, выпускаемых в широких масштабах
в Советском Союзе. В книге рассмотрены химические
и физико-механические свойства важнейших полимеров,
технические требования к вырабатываемым на их
основе пластмассам, приведены сведения об областях их
применения и способах переработки в изделия.
Справочник предназначен для
инженерно-технического персонала предприятий, перерабатывающих
пластические массы, а также для работников проектных
и научно-исследовательских организаций тех отраслей
народного хозяйства и техники, где используются
пластмассы и изделия из них.
Заказы направлять
книжным магазинам Союзкниги