/































Текст
НАУЧНО-ИССЛЕДОВАТЕЛЬСКИЙ ИНСТИТУТ ГВФ
. Инженер-майор М. О. ХАЙКИН
АНАЛИЗ
АВИАЦИОННЫХ ТОПЛИВ, МАСЕЛ
И ОХЛАЖДАЮЩИХ ЖИДКОСТЕЙ
ЛАБОРАТОРНАЯ ТЕХНИКА
и
ПРИГОТОВЛЕНИЕ РАСТВОРОВ
X’ I&. J
|*йама*|****М^Г - - - - - ' ....
Реда н ц и о н н о -н з д а т е л ь с к и й отдел Аэрофлота
Москва 1943
НАУЧНО-ИССЛЕДОВАТЕЛЬСКИЙ ИНСТИТУТ ГВФ
mi
Инженер-майор М. О. ХАЙКИН
АВИАЦИОННЫХ ТОПЛИВ, МАСЕЛ
И ОХЛАЖДАЮЩИХ ЖИДКОСТЕЙ
ЛАБОРАТОРНАЯ ТЕХНИКА
И ПРИГОТОВЛЕНИЕ РАСТВОРОВ
Под редакцией
инженер-майора К. К. ПАПОК
6МВЛ10ТЕК1
м—зх s 7?
РЕДАКЦИОННО-ИЗДАТЕЛЬСКИЙ ОТДЕЛ АЭРОФЛОТА
Москва 1943
»4.ои<» '»«оа
ЛАБОРАТОРНАЯ ТЕХНИКА
Подготовка химической посуды к анализу
Химическая посуда должна быть абсолютно чистой. Если
посуда не загрязнена жиром и прочими нерастворимыми в, во-
де 'Beiu'ecTlBaiMH, достаточно вымыть
ее водой, предпочтительно — теплой.
Поэтому в лаборатории всегда долж-
на быть горячая вода. Посуда, тща-
тельно вымытая в теплой воде1, как
правило, ополаск1ивает1ся 2—3 раза
дестиллироиаНной 'водой.
Воли посуда должна быть идеаль-
но чИстой, например в- случае осаж-
дения, то ее пропаривают, т.е. моют
струей водяного пара. Для этого
пользуются весьма простым! присно
соблением (риС. 1). Хорошо, если в
лабораторий имеется металлический
парообразователь с водомерной труб-
кой, применяемый -обычно для пере-
гонки о .водяным Шаром. Такой паро-
образователь впо'лне можно исполь-
зовать для пропаривания посуды.
!В тех случаях, когда химическая
посуда не поддается мытью водой,
употребляют хромовую смесь. Для
этого растворяют кристаллический
двухромовокислый калий в купорос-
ном масле — 5% по весу (хромовую
смесь можно готовить м из менее
концентрированной серной кислоты).
При осторожном нагревании в фар.
форовой чашке получается однород-
ный раствор, который и применяет-
ся для мытья посуды. Хромовой
Рис. 1.
смесью осторожно обливают стенки загрязненной посуды, за-
тем выливают ее обратной сосуд, в котором смесь хранится, да-
ют посуде постоять 1—2 минуты, после чего хорошо.промывают
3
ее теплой водой и споласкивают aecnwiJimpoBaiiiiHOH водой.
Приготовленная хромовая смесь служит довольно! долго.
При отсутствии в лаборатории хромовой смеси для мытья
посуды, загрязненной 'осмолом и т. п., можно применять креп-
кую серную кислоту или .концентрированный {.дю 40%) раствор
щелочи (едкого1 натра). После использования крепкая кислота
и концентрированная щелочь должны выливаться в специаль-
ную склянку или банку, а не в раковину.
Наконец, для удаления из загрязненной посуды органиче-
ских веществ, ие растворяющихся bi воде, кислоте и щелочи,
употребляются различные органические растворители: бензин,
спирт, этиловый (серный) эфир, петролейный эфир, ацетон, че-
тыреххлори|стый углерод и др. 'Эти растворители являются бо-
лее или <менее ценными продуктами, после истользо1В1ани1я их
нужно собирать и периодически регенерировать.
При мытье посуды обычно* пользуются различного вида
щетками, ершами и пр., что значительно облегчает работу.
Химическую посуду считают чистой, когда на стенках ее
пет приставших и нестахающ1И1Х капель. Недостаточно про-
мытую посуду моют вторично, —до тех пор, пока она не ста-
нет совершенно чистой.
После того как посуда вымыта, она должна быть высуше-
на. Наиболее распространена сушка на колышках. Для этой
цели служит специальная доска. Чистая посуда насаживается
на колышки], вделанные в доску под углом, и расположенные
параллельными рядами, и оставляется на них, пока не высох-
нет.
Для ускорения сушки вымытую посуду продувают возду-
хом при .помощи меха или резиновой груши. При этом стек-
лянная трубка, соединенная с мехом или грушей, вводится в
сосуд почни до, дна, а сам сосуд медленно поворачивается над
огнем горелки или над электрической плиткой. Такая осторож-
ная сушка горячим воздухом, значительно' ускоряет работу и
дает возможность быстро получить сухую посуду. Сушить в
присутствии огня посуду, при мытые которой были применены
растворители, не разрешается.
Другой способ ускоренной сушки состоит в споласкивании
вымытой посуды этиловым спиртом, а затем этиловым эфи-
ром. Пары эфира удаляются продуванием воздуха на холоду.
Можно высушить посуду также в сушильном шкафу при
80—10О°С. После сушки посуду следует продуть грушей (в
горячем виде) для того, чтобы предупредить конденсацию
влаги на станках.
'Специальной сушкой в сушильном шкафу при 105—110°С
получают .абсолютно сухую посуду, доведенную, как говорят,
до постоянного веса.
Охлаждение посуды перед взвешиванием производят в эк-
сикаторе, обеспечивающем .атмосферу, свободную от паров во-
ды; притертые края эксикатора '(шлиф) для герметичное,та сма-
зывают 'В1а1зел1и|;тс!м.
Фильтрование
Операция отделения твердых частиц какого-либо (вещества
от жидкости, в которой это вещество находится, должна про-
изводиться наиболее целесообразно как с точки зрения мак-
симального сохранения твердой или жидкой фазы (смотря по
необходимости), так и в смысле быстроты всей операции в це-
лом. Нагретые жидкости, как правило, фильтруются легче, чем
холодные.
После того как вся жидкость с осадком вылита на фильтр,
сосуд, 1в. котором она нйходилврь, тщательно с1полсс-нут и про
мывная жидкость также вылита на- фильтр,— осадок подвер-
гается промывке!. Промы1ва1Н1ие продолжают, пока в фильтрате,
стекающем! с воронки, -не обнаруживается больше того веще-
ства, которое должно быть отмыто-. Эта операция лрсоодится
с затратой -возможно малого кодичества жидкости, что зави-
сит от характера промываемого осадка: кристаллические осад-
ки требуют значительно .меньше жидкости, и времени, для про-
мывки, чем осадки аморфного характера.
В процессе того или иного определения отфильтрованный
и промытый ссадок иногда должен быть высушен. -Для этого
при подготовке- к определению делают складчатый фильтр, по-
мещают его в бюкс .и .вместе ю последним супцтой в суш; (ль-
ном шкафу при 105—110°С доводят до постоянного веса. По
еле отделения осадка от жидкости и- промывки1 на фильт-
ре с ним! проводят точно такую же операцию, кек и при под-
готовке к определению.
Значительно удобнее в таких* случаях применять тигли
Г уча или стеклянные фильтры по* типу шоттовских; последние
должны быть более высоких номеров (.№ 3 и 4), указывающих
на сравнительно небольшой диаметр пор фильтрующей стек-
лянной пластинки. Наоборот, фильтры под № 1 -и 2 имеют
пластинки с крупными порами, через которые большая часть
жидкостей фильтруется слишком быстро, и при небольшой
величине кристаллов осадка всегда возможны его потери.
Выпаривание и кристаллизация
В практике лабораторной работы иногда. приходится совер-
шенно выпаривать жидкость и -оставлять один осадок, а чаще
всего -выпаривать ее частично с тем, чтобы только сократить
объем раствора-. Выпаривание обычно ведут ib фарфоровых или
стеклянных чашках и другой посуде различных размеров. Ра-
створ нагревают с помощью любых нагревательных прибо-
ров — в зависимости от характера, выпариваемой жидкости.
Особая осторожность должна быть ,проявлена при выпарива-
нии органических горючих растворителей; работа с ними мо-
жет вестись только в отсутствии огня — на водяной бане или
на закрытой электроплитке.
Если выпаривание производится <в большой чашке, то жид-
кость должна быть в нее налита так, чтобы до краев остава-
лось не 1ме1же 2 ом. В маленькой чанже жидкость не должна
занимать более 2/3 по высоте.
В процессе выпарки растворов солей происходит выпаде-
ние их bi осадок; такое же явление имеет место и три охлаж-
дении насыщенных растворов.
В лабораторной работе часто приходится очищать кристал-
лические вещества путем перекристаллизации. Для этого при-
готовляют насыщенный при высокой температуре раствор дан-
ного вещества, после чего быстро охлаждают его, чтобы по-
лучить выделяющееся в осадок вещество, по возможности,
мелкокристаллическим; в этом виде оно содержит наимень-
шее !коДи1чесшво маточного раствора, Й следовательно, и по-
сторонних примесей. Отделив выпавший осадок при помощи
фильтрования, упаривают маточный раствор примерно' .наполо-
вину и вторичным охлаждением выделяют новую порцию твер-
дого вещества.
Тщательная очистка продукта >в лаборатории достигается
его многократной перекристаллизацией.
Перегонка
Перегонка, или дести1Лйяция, жидкостей необходима! для
очистки данного' продукта <5т посторонних примесей, а также
для разделения жидкостей, отличающихся между собой по фи-
зико-химическим свойствам. Обычно для этой цели собирают
простой прибор, состоящий из колбы Вюрца, холодильника и
приемника (рис. 2).
При перегонке жидкость в колбе должна занимать не бо-
лее 2/з ее по объему {перед работой колба должна быть тща-
тельно вымыта и высушена). Отводная трубка должна, входить
на пробке bi форштосс холодильника приблизительно на 5 см.
Гермометр вставляют в колбу на пробке таким образом, чтобы
ртутный шарик его .находился на одном уровне с местом при-
соединения отводной трубки к шейке колбы Вюрца,
Необходимо следить з.а< тем, чтобы в процессе перегонки
жидкость слишком не бурлила —- иначе мелкие брызги ее мо-
гут попасть в отводную трубку и испортить дестилляцию. Для
6
более1 ровного кипения в колбу перед перегонкой помещают
несколько стеклянных капилляров.
Особенно важно соблюдать чистоту форштосса холодиль-
ника и приемника, так как в них попадает уже отогнанная
жидкость' ‘В качестве приемника может быть использован лю-
бой сосуд, но лучше ставить колбы — Эрленмейера или обыч-
ную плоскодонную; горлышко приемника с проходящим в не-
го концом форштосса холодильника должно быть закрыто' ва-
той для предупреждения попадания в дестиллат пыли и пр
Первую небольшую порцию дестиллата (20—30 мл) рекомен-
дуется выбрасывать.
Экстрагирование
В тех случаях, когда из смеси каких-либо жидких или твер
дых компонентов необходимо извлечь одно или несколько ве-
ществ!, пользуются зкстрагк|ро1вание!м. Эта операция часто
встречается bi лабораторной практике и в простейшем случае,
когда имеют дело со смесью жидких 'веществ-, представляет
собой обработку этой смеси в делительной воронке при! помо-
щи различных растворителей. Однако при анализе топлив и
масел приходится пользоваться также экстракцией другого
вида,— когда экстрагируемое вещество является составной
частью твердого тела.
Для экстрагирования из твердого тела обычно' применяют
аппарат Сокслета, состоящий из трех частей: колбы, экстрак-
7
тора и холодильника. Все части аппарата соединяются /пред-
почтительно при помощи шлифов; реже —- на пробках (рис. 3).
После того как аппарат тщательно вымыт, высушен и кол-
ба соединена с экстрактором, при/отопляют из фильтроваль-
ной бумаги патрон, в котором помещают продукт, подлежа-
щий экстрагированию. Особенно удобно пользоваться для этой
Рис. з.
цели гот,о1В1Ы1ми штампованными патронами йэ
специально’ обработанной целлюлозы — по .типу
патронов Шлейхера и Шюля. Патрон вклады-
вают затем в экстрактор таким образом, чтобы
верхний край патрона был немного ниже сифо-
на, т. е. чтобы растворитель мог целиком омы-
вать патрон с находящимся в нем продуктом.
Далее bi экстрактор заливают растворитель,
дают ему стечь через сифон в колбу, после че-
го наливают еще примерно половину того же
количества., соединяют экстрактор с холо-
дильником и начинают нагревание аппарата.
Экстрагирование должно вестись таким об-
разом, чтобы в течение часа происходило ,3—4
С'ифо!нирс1вани1я; более быстрое сифонирова1ние
свлзано to недостаточно спокойным и равномер-
ным кипением жидкости в колбе, а замедлен-
ное |сйфс'Еирован1и1е может привести к неполно-
му извлечению вещества.
В том случае, когда извлекаемое вещество
более 'или. менее заметно’ окрашено, конец эк-
стракции устанавливается по окраске жидкости
в экстракторе и капель, стекающих из. него в
колбу: о нН должны быть бесцветны. Обычно
после этого дают стечь жадности из экстракто-
ра еще 2—3 раза, и заканчивают операцию.
Если же экстрагируемое вещество бесцветно,
то приводится вести операцию достаточ|но
долгое время, чтобы обеспечить полноту извле-
чения. После экстракции растворитель из кол-
бы отгоняется обычным путем.
При экстрагировании нужно помнить следую-
щее: Ь) .работать только с чистым раствори-
телем, иначе можно загрязнить извлекае-
мое вещество, и 2) брать вполне опре-
деленный растворитель, выкипающий в. известных нам преде-
лах — например, петроиейный эфир с точками кипения от 40
до 6'0° С или от 50 до 70С|С.
8
Взвешивание
Взвешивание на аналитических (рис. 4) или техно-химиче-
ских 'весах представляет собой обычную операцию, провюди-
мую в любой лаборатории при химическом анализе. Для того,
чтобы взвешивание давало правильные результаты, необходи-
мо соблюдать определенные условия и следить за весами, как
за прибором, требующим постоянного ухода. Важнейшими ка-
чествами весов являются верность и чувствительность, а эти
Рис. 4
качества сохраняются только тогда, когда с весами обраща-
ются правильно. Накопление пыли и грязи на деталях весов,
влажная атмосфера, наличие паров кислот в воздухе — все это
способствует быстрому изнашиванию весов и отказу в работе.
Поэтому весы необходимо устанавливать в помещении с отно-
сительно сукой асмосферой, вдали от источников тепла ийи хо-
лода, причем работу с кислотами и прочими летучими реаген-
тами в таком помещении' производить не следует. Весы не
должны также подвергаться действию прямых солнечных лу-
чей. Внутри футляра аналитических весов, .как правило, долж-
но находиться какое-либо водопоглощающее вещество; для
этого стаканчик емкостью около 1'00 мл наполняют наполовину
концентрированной серной кислотой и помещают в футляр.
Периодически (ие реже одного раза1 в месяц) кислоту необхо-
димо' менять.
Осторожное обращение с весами при сборке, установке, а
также в процессе работы гарантирует долголетнюю и безот-
казную их работу. Во время взвешивания весы должны быть
максимально устойчивы и не подвергаться сотрясению; поэто-
му они помещаются на подставке, которой может служить до-
ска на консолях, прикрепленных к капитальной стене, или ка-
9
менная отшлифованная плита, также вделанная в капитальную
стену. Переставлять весы с места на место следует только в
случае действительной необходимости.
Для того, чтобы показания .весов всегда были правильны-
ми, нужно- следить за отвесом, устанавливая его при помощи
винтовых ножек — острие к осПрчю. Hie надо браться руками
за «тайлические 1Г1ике№|рювфг’ы|е, серебренные или золочен
ные 'части (весов,, та1к как это мосйет привести к иеточ1ным pte-
?ультапа)м взвешивания и ржавлению деталей. ,
Разновески к аналитическим и техно-химическим весам
обычно золопягся, серебрятся или никелируются. Они должны
хранитыоя -й с1п1е1циа|льно1м. фу'тляре и требуют не менее
аккуратного -отношения, чем сами весы. Категорически запре-
щается брать разновески руками, как бы чисты они ни были,—
для этого существует .пинцет, помещающийся -в футляре в спе-
циальном углублении1, (При надлежащем обращении разновески
служат очень долго-; их поверхность годами сохраняется неиз-
менной.
Начиная работать с весами, прежде всего устанавливают
так называемую нулевую т-о-чку, т. е. выясняют, показывает ли
стрелка весс-в нулевое делен1 'а на шкале при не-аррет ирован-
ных весах; таксе положение стрелки свидетельствует о равно-
плечности ibcc-cb. Практически ве'с-ы никогда не бывают равно-
плечими, а разноплечность весов обусловливает перевес одно-
го из плеч коромысла.
Для регулировки небольших изменений в -весе плеч коро
мысла на концах последнего вделаны 1вмнтосб|раз!я.ые: стержни,
по которым пе ремещаются небольшие гайки. Приближение
гайки в- сторону средней призмы, на 'которой покоится коро-
мысло, по закону рычага уменьшает вес данной части- коро-
мысла-, и наоборот — передвижение гайки в противоположном
направлении увеличивает вес данной части коромысла. Откло-
нения от нулевой точки (влево- или вправо) устраняются при
помощи этих гаек. Отклонение сфелки весов влево говорит
о том, что правая часть коромысла тяжелее- и нужно -подвер-
нуть правую гайку или отвернуть гайку на /левом конце коро-
мысла, чтобы CTpealKa пришла ® нуде вое положение. Наоборот,
при отклонении стрелки вправо, следует отвернуть гайку, на-
ходящуюся на правом! конце коромысла, или подвернуть ле-
вую гайку. Установление нулевой точки аналитических весов
необходимо производить перед каждым новым взвешиванием
или серией взвешиваний.
Все эти процедуры должны выполняться с большой осто-
рожностью и аккуратностью, чтобы не сбить весов. Регули
ров-ать гайки на коромысле при отклонениях стрелки ст нуле-
10
вой точки ‘Следует каждый раз на 2—3 витка, а когда откло-
нения становятся ничтожными (на 1—2 деления шкалы;, гайки
лишь слегка повертываются bi нужном направлении.
Решающее значение для весов имеет чувствительность. Она.
определяется следующим образом: устанавливают нулевую
точку весов, затем арретируют их и накладывают рейтер на
первое деление шкалы, имеющейся на коромысле, что отве-
чает нагрузке в 1 мг. Далее опускают арретир и отсчитывают,
на сколько делений откланяется стрелка от нулевой точки
при такой .нагрузке; эта величина и будет характеристикой
чувствительности весов1. Для аналитических весов средней чув-
ст1вите'лыности достаточно, если отклонение) стройки Л и на-
грузке в 1 мг составит 3 деления шкалы. Меньшая чувстви-
тельность приведет к более грубому взвешиванию, большая
чувствительность заметно удлинит период колебаний стрелки,
т. е. замедлит операцию взвешивания. Отсчет показаний стрел-
ки следует вести только1 после 4—5 колебаний.
Чувствительность весов (вернее — периоде ко тебапий ко-
ромысла) регулируется при помощи подвижной гайки на их
стрелке. Перемещение гайки вверх повышает чувствитель-
ность, но замедляет период колебаний; наоборот, орусканме
гайки приводит к ухудшению чувствительности, т. е". к уско-
рению периода колебаний. Регулировка чувствительности весов
производится при их установке.
Точность и чувствительность аналитических весов с тече-
нием времени изменяются; меняются также и действительные
веса разновесок. Поэтому весы и разновески пКриодайски
должны подвергаться специальной прочерке контролерами Па-
латы мер и весов'.
Приступая к взгеш'ватию на анал тических весах, сто-
рожно приподнимают переднюю дверцу, вносят посуд}' (бюкс,
колбу и т. д.) с продуктом и помещают ее обычно на левую
чашку весов; после этого опускают переднюю и и рывают
правую боковую дверцу, через которую наклидын. от разто-
BieteKH на правую Чашку. Самое ®звеш|т.1з1а£ти1е прс« .'". -т ри
закрытых г)верца.1х. Левой рукой работают! ю 'фретиром ( ю г|ии.
мают и опускают чашки весов), а в правой — держат пинцет,
при помощи которого накладывают рп невески. П юледаяя
операция прюизводится только при аррегчр- ванных ве« ах, т. е.
когда чашки покоятся на подставках. Отступлю те т эт о
правила .приводит к тому, что весы сбивчотся и вообш ? очень
быстро портятся, чт.С' объясняется чс’-ии щи чем ребер агат о-
вых призм, на которых качаются чащк i весов, а тп :е коро-
мысло
Разновески нужно накладывать на середину прг-з.» . чашки
11
весов, а не на края ее, с таким расчетом, чтобы чашка не пе-
рекашивалась. При отсчете качаний стрелки чашки весов не
должны колебаться; колебание приостанавливают легким под-
нятием и опусканием арретира.
Взвешивание предметов в нагретом состоянии не допу-
скается; предварительно их выдерживают в эксикаторе для
принятия температуры окружающей среды. Запрещается .также
/взвешивание твердых веществ. непосредственно на чашках ве-
сов, гак как от соприкосновения г ними последние портятся.
В таких случаях пользуются часовыми стеклами, бюксами, ам-
пулами и т. д. Взвешивание на бумаге допускается только на
технических весах,— и то лишь веществ негигроскопических
(не изменяющихся под действием водяных паров из воздуха).
При взвешивании нужно следить за тем, чтобы на посуде,
с которой приходится работать, не оставалось жирных или
каких-либо дюуги'х пятен от прикосновения рук.
После того как взвешивание закончено, подсчитывают раз-
новески, находящиеся на чашке весов. Подсчет ведут, начи-
ная с крупных разновесок. Записав вес в целых граммах и от
делив его запятой, подсчитывают затем доли грамма, которые
вписывают после запятой. Так, например, если на чашке стоят
разновески 20 г, 10 г, 2 г, 500 мт, '20 мг и рейтер находится
точно на 4-м делении шкалы коромысла, то вес взвешиваемого
предмета составит: 32,5'240 г. баписав вес, снимают разновес-
ки — также начиная с наиболее /крупных, и одновременно
проверяют записанный вес. Такая самопроверка при взвеши-
вании обязательна.
По окончании взвешивания весы 'арретируют и в. гаком виде
оставляют до -следующего взвешивания. В промежутках меж-
ду взве/ши/ваииями разновески должны на/ходиться в футляре,
а рейтер должен быть снят с коромысла.
ПРИГОТОВЛЕНИЕ ДЕСТИЛЛИРОВАННОЙ ВОДЫ
'Вода из водопровода «или из другого источника водоснаб-
жения может быть перегнана, с соблюдением необходимых
условий, для получения дест;иллирова1ной воды. Обычная, не-
перегнанная вода не годится для производства химических
испытаний, так как содержит в себе более или менее значи-
тельные количества различных солей, кислот, аммиака и т. д.
Перегонка может быть произведена весьма простым спосо-
бом— нагреванием какого-либо сосуда с водой и отводом об-
разующихся при кипении воды паров через холодильник в со-
ответствующий приемник, где и собирается дестиллированная
(перегнанная) вода. В качестве холодильника может сложить
металлическая трубка (змеевик), охлаждаемая водой. Материа-
12
лом для холодильника лучше всего брать .медь, освинцован-
ную изнутри. Нужно строго 'Следить, чтобы холодильник и
приемник были совершенно чистыми; при этом первая порция
дестиллированной воды должна выбрасываться, особенно, по-
сле некоторого перерыва, в работе установки.
Наиболее простая и удобная уота!нов1кй для дестилляцми
(.перегонки) (представлена на рис. 5.
Рис. 5.
Приемником может служить стеклянная бутыль, баллон и
пр., где дестиллмрованная вода сохраняется довольно долго,
будучи плотно закрыта пробкой.
Для получения дестиллированной воды в лаборатории в
случае необходимости' могут быть применены колба любой
формы, ;в которой нагревается вода, холодильник Либиха или
шариковый и: какая-либо склянка или банка в качестве прием-
ника.
Для постоянного употребления .в лаборатории дестиллиро-
ванная вода должна быть налита .в склянку с тубусом' (или без
тубуса) емкостью 3—6 л. Тубус закрывается пробкой с отвер-
стием, сквозь которое проходит .изогнутая стеклянная трубка с
надетой на ее наружный конец резиновой трубкой с зажимом.
Верхнее (горловое) отверстие склянки плотно закрывается
пробкой, тоже с отверстием Bi центре; последнее предназна-
чается для предохранительной ампулы, наполняемой натронной
известью. Натронная известь поглощает углекислоту из возду.
13
ха, иначе дестилли,рова1Н!ная вода, насыщаясь углекислотой, ста-
новится в>се более кислой. Если свежую д£С1ТИШлйровйН;н1ую во-
ду принято считать ие!йт1рапьиой, й. te. ймеющей pH1) около 7,
то вода, .постоявшая некоторое время открытой на воздухе,
приобретает более низкий pH, например около 6. Расширенная
часть ампулы, заполненная натронной известью, должна быть
плотно закрыта с обеих сторон ватой. Общий вид склянки (без
тубуса) с дестиллированной водой представлен на рис. 6.
ПРИГОТОВЛЕНИЕ РАСТВОРОВ (ОБЩИЕ ПОНЯТИЯ).
ТАБЛИЦЫ
С операцией растворения того или иного вещества для ана-
лиза в лабсратсрии приходится иметь дело почти ежедневно.
Различают растворы водные и неводные. К числу последних
относятся растворы в органических растворителях, как эфир,
бензол, спирт .и пр. Что касается водных растворов, то все
Oii.и — как титрованные, так и нетитрованные — готовятся ис-
ключительно с дестиллированадой водой.
') pH—концентрация ионов водорода, определяющая не общую, а
активную кислотность раствора. Кислая область лежит до pH = 7; при
pH— 7 доводится тик •называемая нейтральная точка, шли течка перехода,
а выше pH — 7 лежит щелочная область.
14
Нетитрованные растворы
Для приготовления нетитрованных 'растворов растворяе-
мые 'вещеотва юзнешиваюигя на пехнонхимических весах. Взве-
шенное количество вещества, (в данном случае имеются в виду
соли) переводится в соответствующую посуду, в. которой го
говят раствор, а затем в нее добавляется вода в необходимом
количестве. Приготовление раствора в весовом выражении
отличается от тех случаев, когда задается, объем. Так, напри-
мер, если нужно приготовить 1 кг 10 % -ного раствора какого-
нибудь вещества, то прежде всего рассчитывают, сколько на-
до 'отвесить этого вещества;
100 . . . 10
1 000..........х
сое гав ля е тся про п орци я :
10-1000 ._п
л=---juj—= 100 г.
Следовательно, вещества надо взять 100 г, а воды: 1 000-
— 100 = 900 г, или приблизительно столько1 же миллилитров.
Этот объем воды и отмеривается мерным цилиндром.
Если же надо, получить 1 л 10 %-.кого раствора данного ве-
щее гва, то сначала по справочнику устанавливают удельный
вес такого раствора и отсюда получают вес требующегося ко-
личества 10%-ного раствора. Так, если удельный вес раствора
1,128, то 1 л его .весит: 1 000 X 1,1'28 = 1 128 г. Затем расчет
ведется, Как указано (выше: ।
100..........10
1 128 . . . . х
1. 128 - 10
100
х -
=112,8
Следовательно, для приготовления 1 кг и 1 л раствора не-
обходимы различные количества вещества.
Оба расчета даны для безводного вещества, например со-
ли, не содержащей кристаллизационной воды; в случае вод-
ной соли расчет несколько видоизменяется. Так, если требует-
ся приготовить 1 кг 10 %-ного раствора. Na2SO4, то, зная, что
кристаллический сернокислый натрий содержит 10 частей
кристаллизационной воды, ведут расчет следующим образом.
Сначала рассчитывают на безводную соль:
100 . .
1000 .
. 10
. х
1000-10
х - 100 - =100 г-
Молекулярный вес Na2SO4—142,07, a Na2SO4-10HoO—322,23.
Нам нужно 100 г безводной соли, -а количество водной со-
ли определится из пронюрции (цифры несколько округлены).
142 . . . .322,2 322,2-100 О1СП
100.... X ^=—142~ 226,9 г.
Следовательно, надо взять для растворения 226,9 г
15
Na2SO4-10H2O. Воды в этом случае потребуется: 1000—226,9- =
= 773,1 г.
Нужно помнить, что при приготовлении раствора никогда
не сливается сразу все необходимое количество воды в сосуд
с веществом, взятым для растворения. Следует несколько раз
ополоснуть ту посуду, в которой данное вещество отвешива-
лось или отмеривалось, добавляя эти ополоски bi сосуд с ра-
створом, и лишь после этого влить остаток воды.
На каждой склянке с раствором обязательно должна быть
этикетка с тошным названием вещества, из которого приготов-
лен раствор, с указанием концентрации (крепости) последнего,
с датой (изготовления ,и фамилией лица, приготовившего раст-
вор.
Титрованные растворы
Титрованные (точные) растворы нужны для точных
аналитических работ. Необходимое количество (вещества, на-
пример, соли, для приготовления титрованного раствора взве-
шивается на аналитических весах; взвешивание производится
на часовом стекле, в бюксе, либо в стаканчике.. Затем
В' тщательно/ вымытую, сполоснутую дестиллированной водой
и высушенную посуду (например, мерную колбу) встав-
ляют чистую, сухую воронку и высыпают в нее взятую наве-
ску вещества маленькими порциями. Часовое стекло или
бюкс споласкивают над воронкой дестиллированной водой из
промывалки и небольшими количествами воды переводят ве-
щество в колбу. Воронку споласкивают несколько раз дестил-
лированной водой, причем, вынимая ее, промывают кончик —
тоже в/ .колбу. Наконец, последнюю доливают точно до метки
цестиллированнО'й водой.
В аналитической работе обычно употребляются так назы-
ваемые нормальные растворы. Нормальный — это такой раст-
вор, в 1 л которого содержится грамм-эквивалент данного ве
щества. Например, грамм-эквивалент /соляной кислоты HCI
составляет 36,46 г, а грамм-эквивалент едкого натра NaOH —
40,01 г. Следовательно', в 1 л нормального раствора этих ве-
ществ содержится соответственно 36,46 г кислоты и 40,01 г
щелочи, В большинстве случаев, однако, готовят более сла-
бые растворы: полунюрмальные, децинормальные ц другие.
В 1 л полун'ормалъного раствора данного’ вещества содержит-
ся половина его грамм-эквивалента, а в 1л децинормального
раствора.— ’/io грамм-эквивалента.
При приготовлении раствора двуосновной кислоты, напри-
мер серной H2SO4, подсчитывают ее молекулярный вес:
Н2=1,008 • 2=2,106; S=32,07; О4=16- 4=64; всего 98,086;
16
Так как кислота двуооновмая, для получения ее грамм-эквива-
лента делят молекулярный вес тополям:
—’О —= 49,043 г.
Таким образом, растворяя 49,043 г кислоты в дестиллиро-
ванной воде и доводя раствор точно до 1 л, получают 'нор-
мальный раствор серной кислоты. Аналогичным путем посту-
пают и со щелочью. Если же требуется получить нормальный
। раствор соли двуосновной кислоты, то подсчитывают моле-
кулярный вес соли и делят его на основность кислоты, из ко-
торой эта соль получена. Это и будет грамм-эквивалент нашей
соли.
Приготовленный раствор обязательно должен быть прове-
рен при помощи титрования соответствующим раствором. дру/.
ого вещества, нормальность которого точно известна. Еслпр
приготовленный раствор немного отступает от нужной нор,-
мальности, то вводят поправочный коофициент. j 5
Существуют методы приготовления раствороВ', поэволфв-
щие избежать 'провер1ки последних и сохранить в то же вр'фй
необходимую точность. Для этого применяются так называем
мые фиксаналы — запаянные ампулы того или иного вещества^
содержащие его точно в таком количестве, какое необходимо^
для получения 1 л раствора соответствующей концентрации?^
Так., если имеется ампуИа с |йадпи1аыо «НС1 N/10», то доста-
точно растворить содержимое ампулы в; дестиллированной во-
де и довести раствор точно до 1 л, чтобы получить децинор-
малыную соляную кислоту, не нуждающуюся в проверке. Про-
верку рекомендуется производить только в случае особо точ- ,
\ных работ. Титр такой кислоты, определяемый весовым содер
' жанием вещества в 1 мл раствора, составит 0,003646. При ра-
створении фиксанала необходимо обращать внимание на то, -
чтобы Bice содержимое ампулы, без малейших потерь, было пе. <
реведено в- колбу, где приготовляется раствор. Для этого при-
меняется специальное приспособление (боёк), представляющее
. собой стеклянную палочку с зубчатым венчиком в средней ее
части, этим, венчиком боёк) укрепляется в (специальной
‘воронке, вставляющейся в горлышко мерной колбы.
’< # Затем’ ампула своим нижним концом, имеющим выемку
тонкого стекла, надевается на заостренный верхний конец
бойка, им разбивается и все содержимое ампулы выливается
в колбу. (Вверху ампулы имеется вторая выемка, тоже из тон-
кого. стекла, которое осторожно' пробивается, и вся ампула
тщательно споласкивается внутри из носика ирамы валки с ,де>
тилтированной водой (рис. 7). Также тщательно* споласкивается
в колбу и боёк с воронкой, а затем и горлышко колбы, после
чего раствор доводится до метки и хорошо перемешивается.
17
При применении фиксанала надо иметь в виду его све-
жесть. Во всяком случае, употребление фиксанала, содержа-
щего какие-либо посторонние образования, .включения и пр.
или изменившего свою первоначальную
ся. Кроме того, надписи на поверх-
ности ампул делаются иногда раст-
воримой краской; поэтом)г перед
разбиванием ампулы необходимо
смыть (лучше — с мылом) такую
надпись, чтобы краска не попала
в приготовляемый
окраску, <не допускает -
рас гзор.
Рис. 9.
Рис. я.
Титрованные растворы содержатся в склянках с предохра-
нительными ампулами (в зависимости от характера раствора).
Склянка соединяется с бюреткой емкостью 25 или 50 мл или
же с 'мнкробюреткой (рис. 8 и 9). Кран бюретки нужно слег-
ка смазывать вазелином —• это обеспечивает проворачивание
крапа с достаточной легкостью.
18
Таблица 1
Атомные веса химических элементов
|№ п/п. Элемент Химиче- ский сим- вол Междуна- родный атомный вес (1935 г.) Е Е Элемент Химиче- ский сим- вол Междуна- родный атомный *•0 (1935 г.)
1 Азот . . . N 14,001 44 Натрий . . Na 22,997
2 Алюминий .... А1 26,97 45 Неодим Nd 144,27
3 Аргон А 39,94. 46 Неон . . ... Ne 20,183
4 Барий Ва 137,36 47 Никель Ni 58,69
5 Бериллий . . Be 9,02 48 Олово . . Sn 118,70
6 Бор . . В 10,82 49 Осмий . . Os 191,5
7 Бром Вг 79,916 50 Палладий . . . Pd 106,7
8 Ванадий ... V 50,95 51 Платина . . Pt 195,23
9 Висмут . ... Bi 209,06 52 Празеодим Pr 140,92
10 Водород н 1,0078 53 Радий . . . Ra 225,97
11 Вольфрам .... W 184,0 54 Радон . . . Rd 222,0
12 Гадолиний . . . Gd 157,3 55 Рений Re 186,31
13 Галлий . . Ga 69,72 56 Родий ..... Rh 102,91
14 Гафний Hf 178,6 57 Ртуть ..... Hr 200,61
15 Гелий Не 4,062 58 Рубидий Rb 85,44
16 Германий .... Ge 72,60 59 Рутений . . Ru 101,7
17 Гольмий .... Но 163,5 60 Самарий Sm 150,43
18 Диспрозий . . . Dy 162,46 ы Свинец . . Pb 207,22
19 Европий .... Eu 152,0 62 Селен Se 78,96
20 Железо ... Fe 55,84 63 Сера . . S 32,06
21 Золото . Au 197,2 64 Серебро . . Ag 107,88b
22 Индий In 114,76 65 Скандий Sc 45,10
23 Иод ....... J 126,92 66 Стронций . . Sr 87,63
24 Иридий Jr 193,1 67 Сурьма Sb 121,76
25 Иттербий .... Yb 173,04 68 Таллий . ... T1 204,39
26 Иттрий . . Y 88,92 69 Тантал Ta 181,4
27 Кадмии .... Cd 112,41 70 Теллур Те 127,61
28 Калий К 39,096 71 Тербий Tb 159,2
29 Кальций .... Ca 40,08 72 Титан . ... Ti 47,90
30 Кислород . . 0 16,000 73 Торий . . . . Th 232,12
31 Кобальт Co 58,94 74 1улий . Tu 169,4
32 Колумбий .... Св 92,91 75 Углерод . . . C 12,00
33 Кремний .... Si 28,06 76 Уран U 238,14
34 Криптон .... Kr 83,7 77 Фосфор . . . P 31,02
35 Ксенон Xe 131,3 78 Фтор F 19,00
36 Лантан .... La 138,92 79 Хлор . . Cl 35,457
37 Литий Li 6,94 80 Хром . . Cr 52,01
38 Лютеций .... Lu 175,0 81 Цезий Cs 132,91
39 Магний Mg 24,32 82 Церий . . Ce 140,13
40 Марганец . . . Mn 54,93 83 Цинк Zn 65,38
41 Медь ... < u 63,57 84 Цирконий .... Zr 91,22
42 Молибден . . . Mo 96,0 85 Эрбий Er 167,64
43 Мышьяк .... As 74,91
19
Таблица 2
Удельный вес - градусы Боме___________________ Удельный вес - весовые проценты
. I I -Я I I . . Л П _
Градусьп Боме Удель- ный вес Градусы Боме Удель- ный вес го S Q. - 1 L_ U2 Удель- ный вее Градусы Боме Удель- ный вес Г радусы Боме Удель- ный вес Уд. вес при 15°/4°С (в безвоз- А- Для минеральных кислот
Весовой процент Уд. вес npi >5°/4°С (в безвоз- Весовой процент
А. Для жидкостей тяжелее воды, при 15°С душном простран- НС1 HNOS H2SO4 душч>м прострлн- HCI HNO3 H2SO4
0 1,000 13 1,099 26 1,220 39 1,370 53 1,580 стве) 1тве)
1 1,007 14 1,107 27 1,230 40 1,384 54 1,598 1,000 0,16 0,10 0,09 1,230 36,78 31 11
1,005 1,15 1,00 0,83 1,2 ю 37 ,'53 3] *70
2 1,014 15 1,116 28 1,241 41 1,397 55 1,616 1,610 2,14 1,90 1,57 1,240 38,29 32’28
1,015 о, 12 2,80 2,30 1,245 — 39,05 32’86
3 1,021 16 1,125 29 1,251 42 1,411 56 1,634 1,020 4,13 3,70 3,03 1,250 39,82 33,43 34,00 34,57 35 14
» 1,025 5,15 4,60 3,76 1,255 40',58
4 1,029 17 1,133 30 1,262 43 1,421 57 1,653 1,030 6,15 5,50 4,49 1,260 41,34
1,035 7,15 6,38 5,23 1,265 42 10
5 1,036 18 1,142 31 1,274 44 1,439 58 1,672 1,040 8,16 7,26 5,96 1,270 42,87 За'71
6 7 1,043 1,051 19 20 1,152 1,161 32 33 1,285 1,296 45 46 1,453 1,468 59 60 1,692 1,712 1,043 1,050 1,05а 1,160 9,16 10,17 11,18 12,19 8,13 8,99 9,84 10,63 6,67 7,37 8,07 8,77 1,275 1,280 1,285 1,290 — 43,64 44,4] 45,18 45,95 36,29 36,87 37,45 38,03 38,61 39 19
47 1,483 61 1,732 1,065 13,19 11,51 9,47 1,295 46,72
8 1,059 21 1,170 34 1,308 1Д70 14,1/ 10,19 1,300 47,49
48 1,498 62 1,753 1,0/5 1а, 16 13,15 10,9и 1,305 • 48,26 39 >7 40,35 4Э.93 41,50 42,08 42,66 43,20 43,74 44,28 44,82 45,35 4 >,88 46,41 46,94 47,47 48,00 48,53 49,06 49,59 50,11 50,63 51,15 51,66 52,00 52,63 53,11 53,59 54,07 54,55 55,03 55,50
9 10 11 1,065 1,074 1,082 22 23 24 1,180 1,190 1,200 35 36 37 1,320 1,332 1,345 49 50 51 1,514 1,530 1,547 63 64 65 1,775 1,797 1,820 1,080 1,085 1,>90 1,095 1,100 1,105 16,15 17,13 18,11 19,06 20,01 20,97 13,95 14,/4 15,53 16,32 17,21 17,89 11,60 12,30 12,99 13,с7 14,35 15,03 1,310 1,315 1,320 1,325 1,330 1,33а — 49,07 49,89 50,71 51,53 52,37 53,22
12 10 1,091 Б. Д 1,0000 25 ляд 20 1,210 < И Д К О с 0,9359 38 е й 30 1,357 легче 0,8795 52 о Д I 40 1,563 1, при] 0,8295 66 2,5°С 50 1,842 0,7849 к 1,110 1,115 1,1.0 1,125 1,130 21,92 22,86 23,82 24,/8 25,75 18,67 19,4э 20,23 21,00 2, ,77 15,71 16,35 17,01 17,66 18,31 1,340 1,345 1,3,0 1,355 1,. 60 — 54,07 54,93 55,79 56,66 57,57 58,48 59,39 60,30 61,27 62,24 63,23 64,25 65,30 66,40 67,50 68,63 69,80 70,98 72,17 73,39 74,68 75,98 77,28 78,60
11 12 13 14 15 16 17 18 19 20 0,9932 0,9865 0,9799 0,9733 0,9669 0,9605 0,9512 0,9480 0,9420 | 21 22 23 24 25 26 27 28 29 0,9300 0,9241 0,9183 0,9125 0,9068 0,9012 0,8957 0,8902 0,8808 31 32 33 34 За 36 37 38 39 0,8742 0,8690 0,8639 0,8588 0,8538 0,8488 0,8439 0,8391 0,8343 41 42 43 44 45 46 47 48 49 0,8248 0,8202 0,8156 0,8111 0,8066 0,8022 0,7978 0,7936 0,7892 51 52 53 54 55 56 57 58 59 60 0,7807 0,7766 0,7725 0,7684 0,7643 0,7604 0,7565 0,7526 0,7 lt>7 0,7449 1 1 1,135 1,140 1,145 1,150 1,155 .,160 1,16а 1,170 1,175 1,180 1,185 1,190 1,195- 1,200 1,205 1,210 1,21 > 1,220 1,225 26,70 27, 6 28,61 29,57 30,55 31,52 32,49 33, чб 34,42 35,39 36,31 37,23 38.16 39,11 22,54 23,31 24,08 24,84 25.60 2б,3б 27.17 27,8ч 23, ЬЗ 29,38 30,11 30,88 31,62 32,36 33,0м 3 >,8'2 34, ла 3 >,28 36, иЗ 18,96 19,61 20,26 20,91 21,55 22,19 22,83 23, 17 24,12 24, /6 25,40 26,04 26,Ь8 27,..2 2/, 95 8,08 29,21 9,84 30,48 1,365 1,3~0 1,375 1,380 1,385 1,390 1,395 1,400 1,405 1,410 1,415 1,423 1,425 1,430 1,435 1,440 1,445 1,4о0 1,455 1 II1II1II1 1 1 II 1 1 II
Уд. вес при 15°/4°С (в безвоз- душном простран- стве) Весовой процент Уд. вес при 15°/4°С (в безвоз- душном простран- стве) Весовой процент
НО HNO, H2SO.j НС1 HNO3 H2SO4
1,460 79,98 55,97 1,670 74,51
1,465 — 81,82 56,43 1,675 — — 74,97
1,470 — 82,90 56,90 1,680 — — 75,42
1,475 — 84,45 57,37 1,685 — — 75,86
1,480 — 86,05 57,83 1,690 — — 76,30
1,485 — 87,70 58,28 1,695 — — 76,73
1,490 — 89,60 58,74 1,700 — 77,17
1,495 — 91,60 59,22 1,705 — 77,60
1,500 — 94,09 59,70 1,710 —- 78,04
1,505 — 94,39 60,18 1,715 — — 78,48
1,510 — 98,10 60,65 1,720 — — 78,92
1,515 — 99,07 61,12 1,725 — — 79,36
1,520 — 99,67 61,59 1,730 — — 79,80
1,525 — - — 62,06 1,735 — — 80,24
1,530 — — 62,53 1,740 — 80,68
1,535 — — 63,00 1,745 — — 81,12
1,540 —. — 63,43 1,750 — — 81,56
1,545 • — 63,85 1,755 — — 82,00
1,550 — — 64,21 1,760 — •82,44
1,555 — 64,67 1,765 — — 82,88
1,560 — - 65,08 1,770 — — 83,32
1,565 — — 65,49 1,775 — — 83,90
1,570 — 65,90 1,780 — — 84,50
1,575 — — 66,30 1,785 — — 85,10
1,580 — — 66,71 1,790 — .— 85,70
1,585 — 67,13 1,795 — — 86,30
1,590 — 67,59 1,800 '— — 86,90
1,595 — 68,05 1,805 — — 87,60
1,600 .— — 68,51 1,810 — — 88,30
1,605 — 68,97 1,815 — — 89,05
1,610 .— 69,43 1,820 — — 90,05
1,615 — 69,89 1,825 — — 91,00
1,620 — — 70,32 1,830 — 92,10
1,625 — 70,74 1,835 —- S3,43
1,630 — — 71,16 1,840 — — 95,60
1,635 — — 71,57 1,8405 — — 95,95
1,640 — — 71,99 1,8410 — — 97,00
1,645 72,40 1,8405 — — 98,70
1,650 — —- 72,82 1,8400 — — 99,20
1,655 — — 73,23 1,8395 — — 99,45
1,660 — — 73,64 1,8390 — 99,70
1,665 — 74,07 1,8385 — — 99,95
22
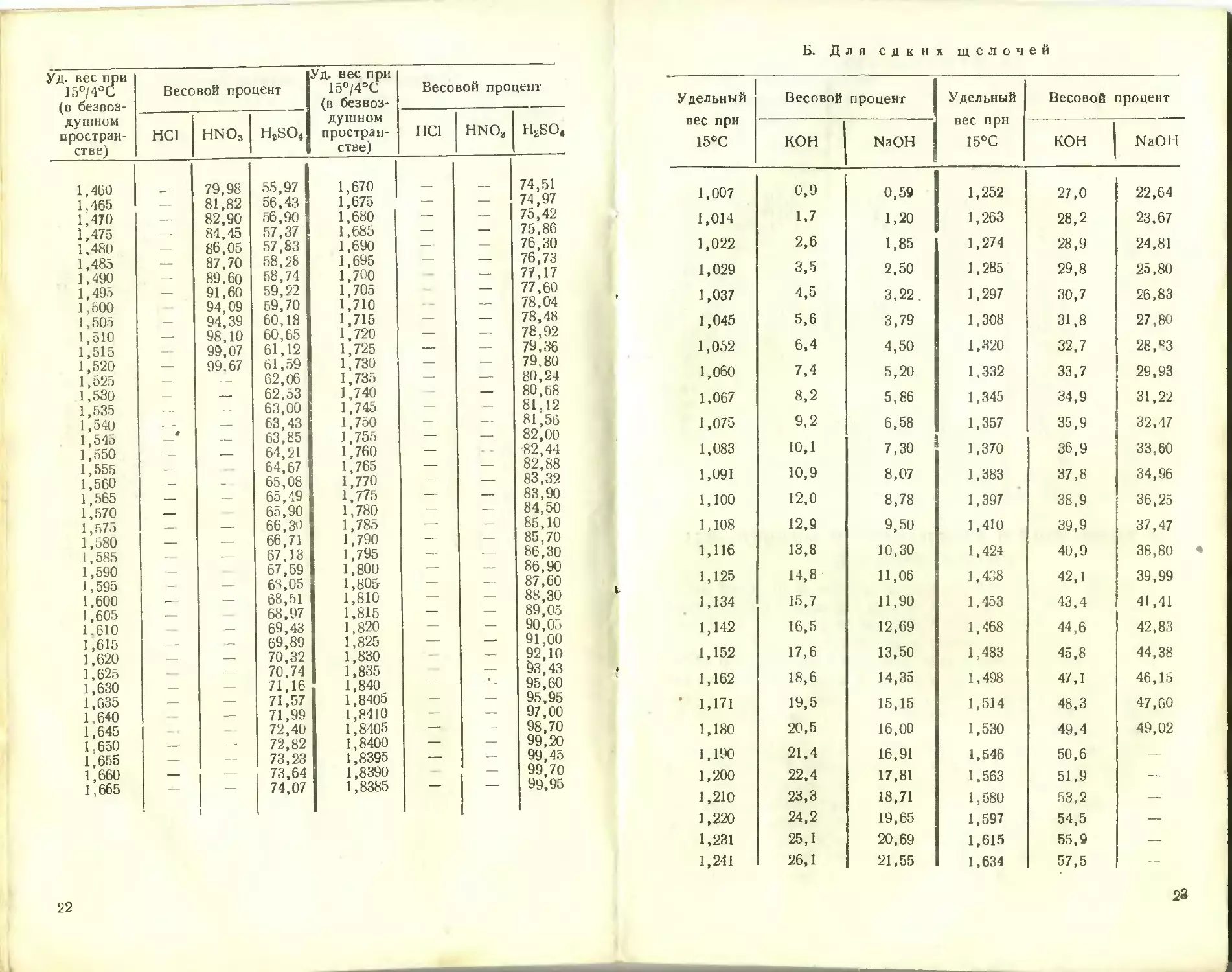
Б. Для едких щелочей
Удельный вес при 15°С Весовой процент Удельный вес при 15°С Весовой процент
КОН NaOH КОН NaOH
1,007 0,9 0,59 1,252 27,0 22,64
1,014 1,7 1,20 1,263 28,2 23,67
1,022 2,6 1,85 1,274 28,9 24,81
1,029 3,5 2,50 1,285 29,8 25,80
1,037 4,5 3,22. 1,297 30,7 26,83
1,045 5,6 3,79 1,308 31,8 27,80
1,052 6,4 4,50 1,320 32,7 28, ”3
1,060 7,4 5,20 1,332 33,7 29,93
1,067 8,2 5,86 1,345 34,9 31,22
1,075 9,2 6,58 1,357 35,9 32,47
1,083 10,1 7,30 1,370 36,9 33,60
1,091 10,9 8,07 1,383 37,8 34,96
1,100 12,0 8,78 1,397 38,9 36,25
1,108 12,9 9,50 1,410 39,9 37,47
1,116 13,8 10,30 1,424 40,9 38,80
1,125 14,8 11,06 1,438 42,1 39,99
1,134 15,7 11,90 1,453 43,4 41,41
1,142 16,5 12,69 1,468 44,6 42,83
1,152 17,6 13,50 1,483 45,8 44,38
1,162 18,6 14,35 1,498 47,1 46,15
’ 1,171 19,5 15,15 1,514 48,3 47,60
1,180 20,5 16,00 1,530 49,4 49,02
1,190 21,4 16,91 1,546 50,6 —
1,200 22,4 17,81 1.563 51,9 —
1,210 23,3 18,71 1,580 53,2 —
1,220 24,2 19,65 1,597 54,5 —
1,231 25,1 20,69 1,615 55,9 —
1,241 26,1 21,55 1,634 57,5 —
28
В. Для аммиака
Удельный вес при 15°С Весовой процент nh8 Удельный вес при 15°С Весовой процент NHS Удельный вес при 15°С Весовой процент NHa
4,000 0,00 0,958 10,47 0,916 23,03
0,998 0,45 0,956 11,03 0,914 23,68
0,996 0,91 0,954 11,60 0,912 24.33
0 994 1,37 0.952 12,17 0,910 24,99
0,992 1,84 0,950 12,74 0,908 25,65
0,990 2,31 0,948 13,31 0,906 26,31
0,988 • 2,80 0,946 13,88 0,904 26,98
0,986 3,30 0,944 14,46 0,902 27,65
0,984 3,80 0,942 15,04 0,900 28,33
0,982 4,30 0,940 15,64 0,898 29,01
0,980 4,80 0,938 16,22 0,896 29,69
0,978 5,30 0,936 16,82 0,894 30,37
0,976 5,80 0,934 17,42 0,892 31,05
0,974 6,30 0,932 18,03 0,890 31,7S
0,972 6,80 0,930 18,64 0,888 32,50
0,970 7,31 0,9‘. 8 19,25 0 886 33,2i
0.968 7.82 0,926 19,87 0,884 34,10
0,966 8,33 0,924 20,49 0,882 34,95
0,964 8,84 0,922 21,12 — —
0,962 9,35 0,920 21,75 — —
0,960 9,91 0,918 22,39 — —
АЦИДИМЕТРИЯ И АЛКАЛИМЕТРИЯ. ИНДИКАТОРЫ
(ОБЩИЕ ПОНЯТИЯ)
При количественном определении кислот и щелочей поль-
зуются методами, объединяемыми под «даванием ацидиметрии
и алкалиметрии. Эти методы основаны на реакциях нейтрали-
зации одних веществ другими bi водных растворах.
Методы ацидаметрии и алкалиметрии позволяют по объе-
мам растворов! реагирующих веществ в момент окончания ре-
акции определить содержание анализируемого вещества «а ос-
нове эквивалентности количеств веществ!, содержащихся в
данных объемах.
Основные положения ацидиметрии и алкалиметрии находят
свое объяснение в теорий, электролитической диссоциации
Аррениуса.. Согласно этой теории все кислоты, основания и
соли в водных растворах находятся в диссоциированном со-
стоянии (состояние распада, .молекулы соединения на две ча-
сти). Степень диссоциации вещества, электролита изменяется с
разбавлением раствора; математически это выражается зако-
ном разведения Оствальда, основанным, в свою очередь, на
24
законе действующих масс в применении его к растворам элек-
тролитов, т. е. веществ’, проводящих электрический ток и раз-
лагающихся при этом на свои составные части. По этому за-
кону степень диссоциации увеличивается с разбавлением.
В зависимости от степени диссоциации вещества различают
сильные и слабые электролиты. К сильным относятся такие
электролиты, которые в растворах средней концентрации дис-
соц1иирсва1ны на 50 % и бодее. .
Для определения конца реакции при титровании в методах
ацидиметрий и а|л1ка,лим1е1тр1ии пользуются (и|нд|И1катора1м1И. По-
следние довольно резко различаются между собой и по своему
химическому строению принадлежат к различным группам ор-
ганических соединений. Способность реагировать на водород-
ный, или гидроксильный, ион, внешне выражающаяся в изме-
нении окраски., позволила объединить все подобные вещества
в одну группу «индикаторов», т. е. указателей конца реакции.
Все индикаторы являются или слабыми кислотами,, или слабы-
ми основаниями, и даже небольшого’ количества водородных
или гидроксильных ионов достаточно для того, чтобы изме-
нить их окраску при титровании.
Например, из индикаторов, наиболее употребительных при
определений киюлот и основа|н'ий, метилоранж представляет
собой слабое основание, не диссоциированное (не распавшееся
на ионы) в щелочном растворе и окрашенное в желтый цвет.
В кислой среде, находясь в диссоциированном виде, метил-
оранж окрашивает раствор в различные оттенки красного цве-
та. Другой индикатор—‘фенолфталеин — является слабой ки-
слотой; будучи диссоциированной в щелочном растворе, она
придает ему розовую окраску.
Механизм действия индикаторов, в основном, был объяс-
нен ионной теорией индикаторов, предложенной Оствальдом.
По этой теории действие индикаторов основано на принципе
различной окраски диссоциированной и неджсоцицроваинюй
их ф’ОрМ.
Пределы восприятия глазом изменения окраски того или
иного раствора в. присутствии индикатора различны в зависи-
мости от характера, последнего1. Область изменения окраски,
находящаяся между предельными ее изменениями, восприни-
маемыми нашим зрением, носит название интервала превраще-
ния индикатора. В табл. 4 указаны интервалы превращения
наиболее употребительных индикаторов.
Интервал превращения индикатора имеет, по существу,
практическое значение и отвечает, как правило, изменению зна-
чения pH (концентрации водородных ионов) в пределах двух
единиц.
25
\
Таблица 4
Индикатор Интервал превраще- ния в pH Окраска раствора Раствор инди- катора Количество индикатора на 10 мл ти- труемого раствора
кислого щелочно- го
Метилоранж 3,1-4,4 Красная Желтая 0,1%-ный водный раствор 1 капля
Метилрот 4,4-6,2 ?? W 0,2% в 90%-ном спирте 1 »*
Нейтралрот 6,8-8,0 м 0» 1°/0 в 70%-ном спирте 1 п
Лакмус 5,0—8,0 » Синяя 0,5%-ный водный раствор 5 капель
Фенолфталеин 8,0-10,0 Бесцвет- ная Розовая 0,1°/0 в 90%-ном спирте 1—о ка- пель
Тимолфталеин 9,4 -10,6 Синяя 0,10/о в 90%-ном 1 капля
Ализариновая желтая 10.0 12,0 Желтая Фиолето- вая О,1°/о-НЫЙ водный раствор 1 N
В методах ацидиметрии и алкалиметрии очень важен; -вы-
бор индикатора., подходящего для титрования данного раство-
ра. Необходимо подбирать индикатор так, чтобы эквивалент-
ная точка анализируемого вещества лежала в интервале пре-
вращения индикатора. Индикатор должен .менять окраску в-
пределах изменения pH титру'еаюго раствора вблизи эквива-
лентной точки содержащегося в нем вещества. Ниже мы при-
водим в краткой форме некоторые практические указания по
выбору индикаторов при титровании.
Если титруется децинормальный раствор НС1 едким нат-
ром, то. можно применить все индикаторы, указанные в
табл. 4, имеющие интервал от pH = 4 до pH =10, т. е. начи
идя с метилоранжа до фенолфталеина включительно*.
При определении слабых кислот титрованием их сильным
основанием следует употреблять фенолфталеин, характеризую-
щийся интервало’м превращения в пределах рН=8 до рН = Ю.
Если же титруют слабую кислоту слабым основанием, то- наи-
лучшими индикаторами будут лакмус (азолитмин) и частично
нейтралрст. Резкого изменения цвета! .при титровании здесь не
происходит, и возможная ошибка составляет 0,5—1,0%.
Наконец, при. титровании, слабого основания сильной ки-
слотой в качестве индикатора могут быть применены метил-
рот, метилоранж и некоторые другие индикаторы, характери-
зующиеся интервалом в пределах pH, отвечающих кислой
среде.
Приводим способ приготовления растворов' индикаторов
метилоранжа и фенолфталеина, с которыми всегда приходится
26
иметь дело в лабораторной практике, а также растворов алка-
либлау и таинина, употребляющихся лишь в отдельных опре-
делениях.
Метилоранж. 0,02 г метилоранжа (в порошке) раство-
ряют в 100 мл горячей дестиллированной воды, дают раство-
ру остыть, после чего фильтруют его') и наполняют капель-
ницу.
Фенолфталеин. 1 г фенолфталеина (в порошке) ра-
створяют в 100 мл этилового, сп1К(рта-1ректиф®1кй;та!. Приготов-
ленный раствор наливают в капельницу. Для титровайия к
раствору анализируемого 'вещества 41р)и1ба)в|л1яю'т небольшое ко-
ийчество HHfllHiKalTopa' : 2—3 вдп!ли. I
Алкалиблау. 2 г алкалиблау марки СВ растворяют в.
100 мл этилового- спирта-ректификата. Припото1вленньгм раство-
ром наполняют капельницу.
Тан нин. 0,1 г таннива растворяют в 20 мл дестиллирэ-
ванной воды. Раствор должен быть совершенно, бесцветным.
Для каждого определения приготовляется овежий раствор ган-
нина.
ПРИГОТОВЛЕНИЕ СМЕСИ КАТВИНКЕЛЯ
ДЛЯ СУЛЬФИРОВАНИЯ
Смесь Кацвинкеля готовят на 95—97 %-ной (по весу) сер-
ной кислоте удельного веса 1,84. Обычно продажная концент-
рированная серная кислота имеет крепость в пределах 95—
96%, т. е. вполне удовлетворяет предъявляемым к ней требо-
ваниям. Однако в ряде случаев концентрация продажной ки-
слоты не превышает 92—93%, и тогда приходится смешивать
ее с олеумом, представляющим собой раствор серного ангид-
рида ЭОз в мюнюпидрате H2SO4 (иначе говоря, разбавлять
олеум крепкой серной кислотой).
Можно было бы рекомендовать моногидрат, т. е. 100%-ную
серную кислоту для получения из недостаточно концентриро-
ванной кислоты продукта необходимом крепости. Практически
этот .путь не реален, так как моногидрат — весьма дефицит-
ный продукт, а количество его, необходимое для смешения,
значительно превысило бы количество олеума.
При определении количества! серной кислоты, требующейся
для разбавления олеума,, надо знать содержание в нем сво-
бодного SO3. В свою очередь, для установления процента
свободного SO3 в олеуме должна быть известна его общая
кислотность (содержание H2SO4).
’) При растворении метилоранжа, представляющего собой или сво-
бодную диметиламиноазобензолсульфокислоту, либо ее натриевую или
аммонийную соль (Лунге), выделяется метасульфокислота.
27
Определение общей кислотности олеума
Навеску олеума около! О1,5 г берут в ампулу с тонким
оттянутым концом, который запаивают на огне перед ее изве
шиванием. После этого навеску растворяют в дистиллирован-
ной воде следующим 'образом: сначала у ампулы осторожно
обламывают 1пинц1йтоы1 тонкий конец и вместе с ним поме-
щают ампулу bi колбу Эрленмейера емкостью 250—300 мл
с притертой пробкой, куда предварительно' наливают
'около 100 мл дестиллмрованной воды. Колбу герме-
тически закрывают и энергичным встряхиванием разбива-
ют ампулу на кусочки. Кислота растворяется в воде, а
для того, чтобы весь SO3 мог поглотиться растворам, остав-
ляют колбу еще на полчаса после примерно' 15—20-минутного
встряхивания. Затем пробка и стенки колбы тщательно про-
мываются в колбу же дестиллированной водой, и весь раствор
титруется NaOiH N/1 в присутствии метилоранжа.
Приводим пример расчета общей кислотности. На навеску
олеума 0,52 г пошло при титровании 11,2 мл NaOiH N/1;
следовательно, общая кислотность:
„ 11,2-0,049038-100
------cw--------=1OV7o.
где 0,049038 — титр H2SO4 N/1.
Определение содержания свободного SO3 в олеуме
При 'определении содержания свободного SO3 в олеуме
пользуются формулой, представляющей состав олеума (д не-
сколько преобразованном виде):
х-}-*/= 100, (1)
где х— процент H2SO4;
у — процент свободного SO3.
Второе уравнение, в которое входит значение кислотности,
непосредственно определяемой опытным путем, имеет следую
щий вид:
х + ту = К, (2)
где т— отношение H2SO4 к SO3;
Д’ — общая кислотность;
х и у имеют те1 же значения, что и !bi формуле (1).
Решая систему из двух уравнений, находим значение
для у.
ав
Если вместо т подставить его численное выражение, пред-
ставляющее собой отношение молекулярных весов1 H2SO4 и
SO3:
981076 - 1 225
80,06 11 2 ’
то получим:
А-100
У “ 0,225 '
В данном примере общая кислотность оказалась равной
105,7%. Подставляя это значение в формулу, выведенную
для у, ойред|е|ляе1м процент свободного SO3 в onteyiMe: ,
105,7-100
У 0,225 '°'
/О'
Определение количества серной кислоты, необходимой для
разбавления олеума
Расчет количества се|рной кислоты, необходимой для раз-
бавления олеума; состоит и1з Двух этапов: 1) (получения 100%-
ной серной кислоты, без свободного SO3, и 2)i получения 96%.
/ной терной кислотьк. i
При расчете по первому этапу пользуются следующей фор-
мулой:
л 100--V
44ч—(4,44- а)’
где А — количеству серозой кйслоты, необходимой для
разбавления, на 100 г олеума, bi граммах:
у — количество свободного SO3 в олеуме, в %; i
а — содержание H2SO4 в кислоте, 'взятой для ра1збакв1лети1я,
в %.
Пользуясь олеумом, взятым нами для примера, и имея, на-
пример, 92%-ную серную кислоту для разбавления, получаем:
. 100 -25
А 444—(4,44-92) -7°’4 Г‘
Крепость серной кислоты, взятой для разбавления, предва
рительио определяется тем же методом, что и общая кислот-
ность олеума.
Следовательно, для получения 100%-ной серной кислоты
надо к 100 г олеума! .прибавить 70,4 г 92 %-ной H2SO4. В ре-
зультате получится 170,4 г 100%-ной серной кислоты.
Второй этап расчета решается пропорцией:
4,0..........170,4 17ПИ
. „ х=170,4 г
4,0.........х
29
составленной на основании схемы:
Иначе говоря, к 170,4 г 100%-ной серной кислоты необхо-
димо добавить еще 170,4 г 92 %-«ой H2SO4, чтобы получить
96"очную серную кислоту.
Таким образом, для того чтобы в нашем, примере из олеу-
ма шойучить 96%-ную серную кислоту, пользуясь 92 % -ной ки-
слотой для разбавления, необходимо на каждые 100 г олеума
брать 240,8 г 92%-ной серной кислоты.
Полученная серная кислота для сульфирования должна
быть проверена таким же методом, как олеум и кислота, иду-
щая на разбавление.
Получение смеси Катвинкеля
Реактив Катвинкеля представляет собой смесь серной ки-
слоты с фосфорным ангидридом, причем на каждые 100 мл
серной кислоты крепостью 96% (95—97%) ,по весу берется
30 г фосфорного 1а|нпидрида1. Отвешенное, количество' фос-
форного ангидрида быстро' обливается серной кислотой,
заготовленной в необходимом объеме. При смешении раст-
вор разогревается. Сосуд со смесью охлаждают ® экси-
каторе или под пришлифованным на гладкой поверхности стек-
лянным колпаком, чтобы предупредить поглощение смесью
влаги из воздуха.
ПРИГОТОВЛЕНИЕ РАСТВОРОВ БРОМА И МОЛИБДЕНО-
ВОКИСЛОГО АММОНИЯ ДЛЯ ОПРЕДЕЛЕНИЯ ТЕТРА-
ЭТИЛСВИНЦА
10%-ный раствор брома в четыреххлористом углероде, хлоро-
форме или дихлорэтане
Зная удельный вес брома (3,14) и четыреххлористого' угле-
рода (1,58), можно произвести расчет объемов, обоих компо-
нентов, которые необходимо смешать для получения раствора
заданной концентрации,—- не прибегая к взвешиванию этих
ядовитых веществ.
30
Берут 10'0 мл СС14 и переводят это количество в весовые
единицы: 100 • 1,58 = 158 г.
Для получения 10 % -ного раствора брома в СС14 нужно
следующее количество Вг2:
90 г СС14.......10 г Вг2 10-158 к
158 г.............. Л- ад—-17,5 г.
Пере1ведя это количество в объемные единицы, получим:
17,5 : 3,14 = 5,58 мл.
Следовательно, смешав 100 мл четыреххлористого углеро-
да с 5,58 мл (округляем: 5,6 мл) брома, получим 10%-ный
раствор.
Аналогично приготовляются и 10%-ные растворы брома в
хлороформе и дихлорэтане—с учетом удельных весов этих
растворителей: соответственно 1,49 и 1,26.
Децинормальный раствор молибденовокислого аммония
Молибденовокислый аммоний в количестве 9,8—10 г раст-
воряют в 1 л дестиллированной воды и полученный раствор
фильтруют через плотный бумажный фильтр. Затем титр
раствора устанавливают по перекристаллизованному и высу-
шенному при 105о,С до постоянного веса азотнокислому свин-
цу (или по хлористому свинцу). Для этого берут навеску соли
около 0,2 г, растворяют при нагревании в небольшом количе-
стве 2'0 % иного раствора уксуснокислого аммония и тигруки
горячий раствор уксуснокислого свинца приготовленным мо-
либдатом аммония.
Конец реакции устанавливается по капельной пробе со
свежеприготовленным 0,5 % -ным раствором таныина (.момент
появления желтого окрашивания). Раствор таннина должен
быть совершенно бесцветным. Необходимо помнить, что на
титрование поступают только горячие растворы.
При расчете пользуются следующей формулой:
А-196
П-331,2 '
где А—навеска Pb (NO3)2 ® граммах;
V — количество мм1ллилитро1в l(NH4)2 МоО4, 1йзрас1ходонан-
ное на тйтро1в1г|ние; >
Т — титр раствора (NH4) 2lMoO4.
Для удобства 1 мл молибдата аммония выражают в весо-
вом значении тетраэтилсвинца; с этой целью полученный титр
31
делят та молекулярный вес (NH4)2 Мо04 ,и умножают на мо-
лекулярный вес РЬ(С2Н5)4, т. е. 1
Г-323,4
196 •
Объединив весь расчет, получим:
/•323,4
Н-331,2 ~Х’
где х — весовое количество тетраэтилсвинца в граммах, отве-
чающее 1 .мл раствора (NH4)2 Мо04.
При применении хлористого свинца вместо азотнокислого
в фор!му|л®х везде взамен молекулярного веса Pb(NO3)2 —
331,2 подставляется молекулярный .вес РЬС12:278.
ПРИГОТОВЛЕНИЕ РАСТВОРОВ СОЛЯНОЙ ИЛИ
СЕРНОЙ кислоты
Для приготовления деци.иормальногю раствора соляной ки-
слоты обычно пользуются концентрированной химически чи-
стой кислотой удельного веса 1,19. Такая кислота содержит
37,23 весовых процента HCL; это знаиит, что в> 100 г кислоты
содержится 37,23 г чистой ЙО. Для приготовления 1 л N/10
раствора НО, содержащего 3,645 г 1Н.С1, необходимо следую-
щее количество кислоты:
или
9 79
fir-8-2 -•
Отмерив 8,2 .мл концентрированной кислоты bi мерную кол-
бу емкостью 1 л, доливают ее до метки дестиллированной во-
дой, тщательно перемешивают и устанавливают титр получен-
ного раствора.
Титр раствора кислоты устанавливается по химически чи-
стому карбонату натрия (углекислому натрию, соде), .который
для этой цели готовится из чистого бикарбоната натрия. 50 г
продажной соли растворяют в 500 мл воды, фильтруют и вы-
паривают раствор при температуре не выше 40°С до момента
выделения кристаллов. Затем раствору дают остыть и, предо-
хранив от пыли, оставляют, пока не выделится околю 3/4 взя-
того количества соли. Осторожно сливают маточный раствор,
осадок промывают один раз холодной водой, после чего отжи-
мают его между листами фильтровальной бумаги и сушат при
120°С в течение 1—1% часов.
Несколько- граммов! сухого бикарбоната натрия очень осто
рож)но прокашливают1 в платиновом или никелевом тигле; пос-
32
ледн1и1й (при этом погружается ипесок (песча|з|ой ба1нИ настолько,
чтобы он доходил снаружи до уровня соды в тигле. Прокали-
вание ведется в интервале температур 250—300°С и продол-
жается (при этой температуре) полчаса при частом перемеши
ваним содержимого тигля, чтобы не получалось сплавления
соли. После охлаждения ® эксикаторе и взвешивания тигель
снова нагревают и опять взвешивают, чтобы убедиться в пол
ноте прокаливания. Повышение температуры прокалив-шик
сверх 300°С недопустимо, так как ведет к разложению соды. По-
лученную чистую соду хранят в бюксе с (притертом крышкой.
Три навески Na^CO3, около 0,2 г каждая, растворяют в
100 мл дестиллированной воды и прибавляют две капли ме-
тилоранжа. Затем начинают титрование; приготовленным paci-
вором кислоты, установив его уровень в бюретке на нулевом
делении. Титрование ведется медленно при непрерывном поме-
шивании до того' момента, пока желтая окраска раствора не
перейдет .в оранжевую.
Этот оттенок следует сопоставить с оттенком, получаю-
щимся при добавлении к дестиллированной воде с индикато
ром такого количества кислоты, которое вызывает оранжевую
окраску. Объем воды и количество- индикатора, в этом слепом
опыте должны быть примерно такими1 же, как и в основном
определении. Если в слепом опыте не удается получить оран-
жевую окраску, а проба дестиллированной воды от одной
капли раствора кислоты становится уже розовой,— то титро-
вание соды ведут до момента, пока оттенок раствора не сде-
лается средним между окраской пробы воды с метилоранжем
и окраской воды с метицюрайжем и одной каплей раствора ки-
слоты (иначе говоря, наблюдают первое изменение окраски
титруемого раствора). Расчет ведется по следующей формуле:
V- U,005303 '
где А—навеска углекислого натрия в граммах;
V — количество раствора кислоты, израсходованного на
титрование, в миллилитрах;
К — нормальность .кислоты.
Титр (фактический) приготовленного раствора кислоты по-
лучают, умножая нормальность раствора К на титр точно де-
цинормалыного раствора кислоты. В данном случае для соля
ной кислоты НС1 N/10 фактический титр составит:
Т = /С-0,003646.
Если К меньше единицы, го приготовленный раствор ока-
жется слабее децинормального, и наоборот,— если К больше
единицы, то раствор будет крепче децинормального. Напрн-мер:
33
Т = 0,9880.0,003646 = 0,003602 (раствор слабее децинор.
мяльного);
Т = 1,0298-0,003646 = 0,003722 (раствор крепче децинор-
мального).
На склянку с титрованным раствором наклеивают этикет-
ку, на которой записывают титр кислоты до шестого знака
после запятой, нормальность ее до четвертого знака после за-
пятой, а также да!ту установим титра. !
Всякое титрование1 должно производиться не менее двух
раз. Бюретки, мерные колбы и прочая посуда, употребляю-
щаяся при установке титра, должна быть тщательно вымыта,
высушена и несколько раз сполоснута испытуемым раствором
(отдельными порциями).
Если нужно получить более слабый раствор кислоты, на-
пример N/2'О, то берут соответственно меньшие количества ее;
в остальном эти растворы готовятся так же, как и более
крепкие.
Растворам соляной кислоты разной крепости соответствует
различное весовое содержание вещества в 1 л раствора:
НС1 N/1 — 36,465 г, НС1 N/2 — 18,232 г, HCI N/5 — 7,293 г,
HCI Ы/10— 3,646 г, HCI N/20— 1,823 г, HCI N/100 — 0,3646 г.
Приготовление децинормального раствора серной кислоты
ничем не отличается от приготовления децинормального раст-
вора соляной кислоты,— только берут другие количества ве-
щества, соответственно молекулярному весу.
ПРИГОТОВЛЕНИЕ РАСТВОРА СЕРНОВАТИСТОКИСЛОГО
НАТРИЯ (ГИПОСУЛЬФИТА) ДЛЯ ОПРЕДЕЛЕНИЯ ИОД-
НОГО ЧИСЛА
Основой иодометрии является следующая реакция:
2Na2S2O3 -|— J2 = 2NaJ NajS^jOg.
Если к раствору, содержащему мод, приливать из бюрет-
ки раствор гипосульфита натрия (серноват,истокислого натрия,
иначе —• тиосульфата натрия) в присутствии кракмала, играю-
щего роль индикатора и синеющего’ при самом незначитель-
ном избытке иода,— то синяя окраска раствора исчезает, как
только весь, иод, согла1с1но приведенному .уравнению, перейдет
в йодид натрия NaJ. Эта реакция является одной из наиболее
чувствительных bi аналитической химии. В большинстве слу-
чаев принято! пользоваться для ана1ливй1 децийормальным раст-
вором гипосульфита натрия.
Эквивалентный вес иода и его. атомный вес в, граммак от-
вечают одному молю гипосульфита. 'Следовательно., для полу-
чения 1 л децинормального. раствора его необходимо было бы
34
брать ггочйо 0,1 моля чистой крИсТалличеюкой 'соли Na2S2O4X
X 5Н2О. /
Однако раствор гипосульфита при стоянии, как правило,
выделяет серу; этот процесс разложения обусловливается за-
грязнениями, содержащимися в- растворе (стерилизованные ра-
створы гипосульфита, не содержащие углекислоты, могут со-
храняться долгое -время). Поэтому децинормальный раствор
NasSa'Og готовится из следующего расчета. Молекулярный вес
кристаллической соли равен 248,32. Для приготовления деци-
нормального раствора нужно взять 24,83 г соли, или округ-
ленно 2’5 г. Обычно приготовляют 5 л раствора, для чего от-
вешивают 125 г гипосульфита.
Растворение соли производится bi свеже;1ро,кипяченно|й и
быстро остужен'ной дестиллирован'ной воде. Через 8—14 дней
устанавливают титр раствора, двумя методами: 1) посредством
чистого иода и- 2) при помощи бикроШта ке|жя ’).
Установка титра гипосульфита натрия посредством чистого
иода
Чистый иод, при помощи которого устанавливается титр
Na2S2O3, цриготозляется путем- возгонки. Для этого бе-
р г тщательно вымытый и высушенный стаканчик емкостью
100—200 мл, на дно которого насыпают 2—3 г иода. Реко-
мендуется перед вС'Згенкой растереть иод -в ступке с несколь-
кими кристалликами йодистого калия для того, чтобы освобо-
дить код от возможных примесей хлора и брома. Сверху ста-
канчика помещают небольшой кристаллизатор с холодней во-
дой. Дно кристаллизатора должно быть совершенно чистым и
сухим. Далее- стаканчик устанавливается >на асбестовой сетке
п осторожно1 подогревается; при этом иод возгоняется на дно
кристаллизатора в виде мелких и крупных табличатых кри-
сталлов, легко отстающих о г стекла при небольшом сотрясе-
нии.
Вместо кристаллизатора может быть использована колба
Для разгонки по Энглеру, через которую -медленно пропускает-
ся водя2) (рис. 10).
Собрав возогнаиные кристаллы иода на чистое часовое
стекло-, их помещают в эксикатор, где выдерживают (высуши-
вают) над едким калием- в течение суток; шлиф эксикатора не
1) Рекомендуется устанавливать титр раствора гипосульфита натрия
но двум методам для большей точности.
2) Автор пользуется случаем принести благодарность инж. Л. И. Са-
ранчук и другим своим коллегам, сделавшим замечания по изложенным
здесь отдельным вопоосам, связанным с упрощением техники лаборатор-
ной работы.
35
должен быть при этом смазан жирам. |Возопнанный мод хра-
нится в бюксе с хорошо притертой крышкой, г очищаемом в
эксикатор Самая усте|нов1ка титра гипосульфита произво-
дится, как описано ниже.
Рис. 10.
Две-три 'навески чистого, не содержащего иода, йодистого
калия по 2—2,5 г берутся в небольшие стакагччики для взве-
шивания с хорошо притертыми крышками; сюда же приливают
по 2—3 мл дестиллированной воды. Закрыв стаканчики, точно
взвешивают их на аналитических .весах. Затем в каждый ста-
канчик прибавляют по 0,4—0,5 г чистого- иода, быстро закры
вают их крышками и снова взвешивают Разность в весе рав-
няется количеству иода.
Растворение йодистого калия в воде связано со значитель-
ным понижением температуры, вследствие чего стаканчики
покрываются снаружи влагой и не могут быть немедленно
взвешены. Поэтому влага должна быть удалена фильтроваль-
ной бумагой; стаканчики выдерживаются 15 минут в весовом
шкафу, вытираются замшей или полотном и через 5 минут
взвешиваются.
Растворение иода в концентрированном растворе иодисто-
г-о калия идет чрезвычайно быстро. Далее стаканчик вводят
в горлышко наклоненной колбы Эрленмейера, содержащей
200 мл дестиллированной воды и около- 1 г йодистого калия,
36
( .
быстро снимают крышку и опускают стаканчик на дно кол-
бы; крышку затем также опускают в колбу. Все эти предо-
сторожности 'Необходимы для того, чтобы избежать каких-
либо потерь иода при приготовлении его раствора.
На практике удавалось получить достаточно точные ре-
зультаты и при более простом методе приготовления раствора
иода. В стаканчик с притертой крышкой берут необходимую
навеску чистого иода, быстро пересыпают ее в колбу Эр-лен-
мейера с притертой пробкой, на дне которой находится кон-
центрированный раствор йодистого калия: 2—2,5 г KJ в
3—5 мл дестиллиро1ванн'ОЙ воды. Стаканчик закрывают крыш-
кой и взвешивают. По разности определяют величину
навески чистого иода. Содержимое колбы с концентри-
рованным раствором KJ <и растворенным в нем иодом разбав-
тяют затем 200 мл дестиллированной воды.
Полученный раствор иода с точно известным его содержа-
нием титруют из бюретки при 'непрерывном взбалтывании ра-
створом гипосульфита натрия до слабо желтой окраски; после
этого- прибавляют 2—3 мл свежеприготовленного раствора
крахмала -(появляется 1си'Г!е-ф'И1олетовая окраска!) и продолжают
титровать до обесцвечивания раствора.
В .результате двух-трех определений, получив количество
ми!лли.л1ит1ров раствора1 гипосульфита, израиходоваДного на
титрование раствора иода, вычисляют среднюю величину тит-
ра раствора гипосульфита натрия — подсчитывают, какое ко-
личество иода отвечает 1 мл раствора гипосульфита.
Например, на- навеску чистого иода в 0,4591 г пошло при
титровании 36,4 мл раствора гипосульфита!. В действительно-
сти навеска отвечает 0,4591 : 0,0127 = 36,15 мл децинормаль-
пюго раствора. Отсюда:
= 0,9931.
Выражая титр гипосульфита по иоду, имеем:
TNagS,Os = 0,01270 • 0,9931 = 0,012612.
Титр раствора гипосульфита может быть вычислен и дру
г им методом:
-г / ч навеска иода
Na2S2Og (п0 иоду) количество мл Na2S2O3’
а к- Т
0,01270’
37
Установка титра гипосульфита натрия при помощи двухромо-
вокислого калия
Двухромовокислый калий относится к числу исходных ве-
ществ, которыми часто пользуются для установки титра ги-
посульфита «атрия. Ввиду того что нетюсредственн'ое титро-
вание К2СГ2О7 гипосульфитом не удается, прибегают к методу
замещения, при помощи которого устанавливают связь между
названными веществами, исходя из следующего уравнения:
KaCrgOy -|- 6KJ ф- 14НС1 = 8КС1 + 2СгС13 -1- /Н2О - J2.
Следовательно, если к раствору йодистого, калия в соляной
кислоте прибавить двухромовокислый калий, то хромовая ки-
слота восстанавливается «а холоду до соли трех1валентиого
хрома (зеленого цвета), освобождая эквивалентное количество
свободного иода.
Приготовляют 1 л децинормального раствора К2Сг2О7, для
чего отвешивают на аналитических весах 4,903 г чистой сухой
соли (дважды перекристаллизс1В1а1Мной из продажного продук-
та и высуЖ1н!:той при 125—130°С до псстойдного веса), раот
воряют соль в дестиллированной вю.де и доводят до метки в
литровой колбе при 20 С. В колбу Эрленмейера емкостью око
ли 600 мл, содержащую 10 мл концентрированной соляной ки
слоты и 3 г йодистого калия, растворенные в 50 мш дестил-
лированной воды, приливают 25—40 мл приготовленного де-
ц анормального раствора бихромата калия. В течение 5 минут
в темноте .заканчивается реакция, после чего разбавляют ра
ствср до 300 мл и титруют раствором гипосульфита натрия
в присутствии крахмала в качестве индикатора. Бурая окраска,
характерная для раствора в начале титрования, переходит1 за-
тем в фиолетово-синюю и далее — в зеленую (цвет раствора
с эли окиси хрома), каковую и считают концом титрования.
Сделав1 отсчет п.о бюретке, прибавляют еще несколько капель
гипосульфита, т. е. пе.ретитровы.вают раствор. Полученная зе-
леная жидкость служит свидетелем (контролем) при после
дующих титрованиях.
Если на титрование 25 мл децинормального раствора би-
хромата калия затрачено 25,2 мл раствора гипосульфита, то
Wa—Ну 0,9920.
Отсюда титр гипосульфита по иоду будет:
= 0,01270 0,9920 = 0,012598.
Можно вести титрование хромпика беря его. в виде отдель-
ных навесок, как и при титрсва1нии иода. Для этого отвеши-
вают на аналитических весах 2—3 навески около 0,1 г чистой
38
. . X
127-0,102
“49,03
0,2644 г.
сухой соли (что соответствует 20—25 мл детданюрмального
раствора КаСггО?) и переводят каждую из них в колбу
Э|рленм1ей'ера', содержащую 10 мл концентрированной соляной
кислоты, 3 г йодистого калия и 50 мл дестиллироВ1аин'ой во-
ды. Далее поступают, как описано выше, т. е. дают смеси
прореапфсвать, разбавляют ее водой и титруют гипосульфи-
том в присутствии краж-мала.
Допустим, что на навеску в 0,102 г бмхромата пошло при
титровании 20,8 мл раствора гипосульфита; тогда расчет сво-
дится к следующему:
К3Сг2О7
49,03 г
0,102 г
О гсюда
rN:loSi0s = 0,2644 : 20,8 = 0,01271.
ПРИГОТОВЛЕНИЕ РАСТВОРОВ ЕДКОГО КАЛИЯ ИЛИ
ЕДКОГО НАТРА
Для практики лабораторного исследования топлив1 и масел
особенно важны дец1инормаль:ные растворы щелочи. Последние
устанавливаются по титрованному раствору соляной кислоты,
приготовляемому, как указано выше. (см. стр. 32).
Около 6 г едкого калия1) растворяют в 1 л дестиллиро-
ванной воды. Приготовленный раствор выдерживают примерно
в течение часа возле раствора соляной кислоты, которым он
должен быть оттитрован (это необходимо для выравнивания
температуры). Затем 25 мл раствора щелочи, отмеренного по
бюретке, титруют децинермальнюй соляной кислотой в присут-
ствии двух-трех капель раствора метилоранжа. На о,. ювании
нескольких титрований берут среднее значение и рассч гтывают
нормальность раствора едкого калия:
Нормальность Н )рмалыюсгь Кодич. мл раствора НС1.
раствора КОН ’ раствора НС1 Колич. мл раствора КОН
Титр приготовле1нйого раствора едкого калия получают,
умножая .нормальность раствора К на титр точ> деципормаль-
ног’О раствора КОН, т. е.
т = К- 0,0056104.
При Л' меньше единицы .приготовленный раствор будет сла-
бее пец;г,.,нормального, а при 7< больше ед- мцы -крепче де-
ци нормального. Например:
Т -- 0,9922 • 0,0056104 0 005567 (раствор слабее децинор-
мального).
]) Приготовление ртстворов едкого натра принципиально не отли-
чается от помещаемого здесь описания; берутся только другие количесп)а
вещества, соответственно молекулярному весу.
39
Т= 1,0290 -0,0056104 = 0,005773 (раствор крепче децинор-
мального).
При титровании едких щелочей децинормальным раство-
ром соляной кислоты в .присутствии метилоранжа четкого пе-
рехода от желтого цвета к красному не наблюдается; это
имеет место только при титровании более крепкими раство-
рами. Здесь же сначала появляется оранжевая окраска, кото-
рая после прибавления 1—2 капель раствора кислоты перехо-
дит bi красную. За конечную точку титрования следует при-
нимать момент перехода от желтого к оранжевому цвету.
Установка титра раствора гцело-чи при помощи кислоты мо-
жет быть произведена также в присутствии фенолфталеина, но
при этом необходимо учесть наличие углекислоты в титруе-
мом растворе. -Дело в том, что сухие щелочи и их растворы
обладают свойством жадно притягивать углекислоту из воз-
духа, что сопровождается обра1зовани10м К2СО3 (или NagCO3 в
случае NaOH). Внешне это выражается в появлении на кусоч-
ках твердой щелочи, которая обычно имеет желтоватый цвет,
налета свеяйно-бедого цвета. Ом тем больше, чем дольше ще-
лочь подвергалась действию углекислоты воздуха.
При малом количестве углекислых щелочей вредное дей-
ствие, оказываемое углекислотой на результаты титрования,
незначительно, и им можно пренебречь. В противном случае
приходится прибегать к несколько измененному методу тит
рования. Для этого к отмеренному из бюретки коийчеспву ра-
створа едкой щелочи "прибавляют каплю фенолфталеина и
титруют раствором соляной кислоты, примерно той же концен-
трации до момента обесцвечивания розового раствора. После
этого нагревают раствор до кипения, в результате чего Снова
появляется розовая окраска; затем охлаждают его- и титруют
по капле раствором соляной кислоты до исчезновения окра-
ски. Эту операцию — последовательное кипячение, охлажде-
ние и титрование — повторяют до тех пор, пока кипячение не
вызывает больше появления окраски. Такой способ титрова-
ния кропотлив', но дает весьма точные результаты.
Титроваете едких щелочей, содержащих более или менее
значительное количество углекислых солей, может быть вы-
полнено также bi присутствии метилоранжа. Однако и ® атом
случае титрование ведут на холоду только до первого изме-
нения окраски, после чего удаляют углекислоту кипячением.
В результате раствор снова становится желтым и после охлаж-
дения дотитровывается соляной кислотой до прежнего оран-
жевого оттенка.
При титровании как с фенолфталеином, так и с метилоран-
40
жем в расчет нормальности едкой щелочи принимаются конеч-
ные результаты титрования.
Если нужно приготовить не децинормальный раствор, а бо-
лее крепкий, например полунормальный, то берут соответ-
ственно большее количество щелочи для растворения; в
остальном такйе .растворы готовитая, как чи бблее слабые ро-
ст® еры.
'Растворам едкого калия 'разной крепости соответствует
различное весовое содержание вещества в 1 л раствора:
КОН N/1 — 5'6,104 г, КОН N/2 —28,052 г, КОН N/5 —
11,221 г, КОН N/10 —5,610 г, КОН N/20—2,805 г,
КОН N/100 — 0,561 г.
Если готовят спиртовой раствор едкой щелочи, в; большин-
стве случаев' — КОН, для чего применяется, как 'правило, аб
солкл|Я'ЫЙ спирт '(ib крайнем случае, пользуются 96%-ным Спир
том-ректификатсм), то последний подвергают предварительной
обработке для предупреждения 'пожелтения'. С этой целью к 1 л
спирта прибавляют 5 мл 50 %-кого раствора NaOH или КОН
и 5 г цинковой пыли (лучше — несколько капель пергидроля),
кипятят полчаса с обратным холодильником, после чего спирт
отгоняют с дефлегматором'. При добавлении пергидроля раст-
вор перед кипячением .надо сильно .встряхнуть. В зависимости
от качества спирта однократная обработка его иногда оказы-
вается недостаточной; тогда спирт обрабатывают повторно
таким же образом.
На склянку с титрованным раствором наклеивают этикетку,
где указывается титр щелочи до шестого знака после запя-
той, нормальность ее до четвертого знака после запятой, а
также дата установки титра. Склянка обязательно' снабжается
предохранительной трубкой с натронной известью; такая же
трубка должна быть у бюретки с раствором, соединенной со
склянкой.
ПРИГОТОВЛЕНИЕ РАСТВОРА КАРБОНАТА НАТРИЯ И
СМЕСИ ВАРТА—ПФЕЙФЕРА ДЛЯ ОПРЕДЕЛЕНИЯ ЖЕСТ
КОСТИ ВОДЫ
Децинормальный раствор карбоната натрия
1 л децинормального раствора карбоната натрия содержит
5,2997 г Na2_CO3. Отвешивают 5,3 г химически чистого карбо-
ната и растворяют при нагревании в небольшом количестве де
стиллированной воды. Полученный раствор переливают в- мер-
ную колбу, доливают до метки дестмллирО|ванной водой и пе-
ремешивают. Нормальность .раствора устанавливают так же,
как это делается при приготовлении едкого калия (ом. выше,
41
I
стр. 39). В качестве индикагпара также применяется метил-
оранж.
Смесь Варта—Пфейфера
1 л децинормального раствора NaOH смешивают с 1 л де-
цинормального раствора NagCO... Смесь .проверяют по соляной
кислоте с метилоранжем в качестве индикатора.
ПРИГОТОВЛЕНИЕ РАСТВОРА АЗОТНОКИСЛОГО СЕРЕБ-
РА ПО ВИНКЛЕРУ ДЛЯ ОПРЕДЕЛЕНИЯ ХЛОРА В ВОДЕ
Такой раствор готовится растворением 4,791 г азотнокис-
лого серебра в 1 л дестил-лированной воды. В этом случае
1 мл раствора соответствует 1 мг хлор-иона.
Навеска азотнокислого серебра определяется из реакции:
AgNO3 + NaCl = NaNO3 + AgCl,
откуда навеска
AgNO3 . . Cl1
169,89 . . . 35,5
x . . . 1
x = =4,791 г AgNO,
3o,5
Проверка титра производится по перекристаллизованной со-
ли — хлористому натрию.
Хлористый натрий реагирует с AgNO3 по уравнению:
AgNO3 Ц- NaCl = AgCl -f- NaNO3.
Обычная продажная соль NaCl, даже химически чистая,
недостаточно пригодна для установки титра. Поэтому имею-
щийся в лаборатории препарат очищается следующим образом-
приготовляется на холоду насыщенный раствор соли, из кото-
рого хлористый натрий осаждается газообразным хлороводо-
родом. Последний получается путем прибавлен ня в специаль-
ную склянку по каплям из делительной ворс и репкой НС1
удельного веса 1,19 .и концентрированной серной ю слоты. Вы-
деляющийся при этом газообразный хлороводород по трубке
(через пустую промежуточную склянку) отводится в сосуд с
насыщенным раствором NaCl; конец трубки должен быть-рас-
ширен, так как выпадающая соль забивает отверстие. Осаж-
денная соль отфильтровывается, быстро промывается холод-
ной дестиллированной водой, сушится между листами фильт-
42
ренальной бумаги' в сушильном шкафу и, наконец, прокали-
вается в муфеле до темнокрасного каления (500—600°С).
Ввиду того, что хлористый натрий очень гигроскопичен,
нсрбходамо 1ка1ждый раз перед употреблением (слегка прока-
лить его в платиновом тигле в (адуфеще до прекращения потре-
скивания (10—15 мин). ।
Титр азотнокислого серебра устанавливают по хлористому
натрию посредством отдельных навесок. Для этого берут две-
три навески, растворяют в небольшом количестве воды.
(50 мл) и титруют растворам азотнокислого серебра bi присут-
ствии пяти капель 10 %-ного раствора хромата калия К.2СгО<
до изменения окраски.
Пример расчета титра
1-я 2-я Зя навеска-0,0214 г, пошло на титрование 12,90 мл AgNO. „ -0,0216 „ „ 12,90 ,, „ „ -0,0177,, „ „ „ 10,55,,
1. Отсюда титр Т _3 равен: AgNO, 169,89 . . . )12,9'0 NaCl 58,4540 0,0214
т 169,89-0,0214 1 58,454 • 12,90 = 0,004844 -
2. 169,89 . . . 12,90 . . . . 58,4540 0,0216
т 169,89-0,0216 2 58,454 • 1'2,90 = 0,004366.
3. 169,89 . . 10,55 58,4540 0,0177
т 169,89 -{0,0177 = 0,004876.
3 58,454 -10,55
Следовательно, средний титр азотнокислого серебра
7^=0,004862.
Из среднего значения титра азотнокислого серебра нахо-
дим, что 1 мл AgNO3 соответствует следующему количеству
хлора в миллмграмах:
AgNO3.............Cl
169,89 . . . . 35,5
0,004862 . ... х
43
О.,004862 • 35,5 . П1С Г11
•'=-----16ЗД9-----= ’•°'6 Cl‘-
t. e. 1 мл AgNO3 соответствует 1,016 мг Cl'.
Таким образом получаем, что титрованный раствор азотно-
кислого серебра имеет:
Т- 0,004862
х — 1,016 мт С11,
где Т — татр раствора AgNO3;
х — 1 мл раствора AgNO3.
ОТБОР И ПОДГОТОВКА ПРОБ ДЛЯ АНАЛИЗА
Анализ авиационного топлива, масла или другого продукта
только тогда дает действительную характеристику исследуе-
мого вещества, когда проба для анализа отобрана правильно.
Этот момент является решающим в оценке результатов иссле-
дования в целом.
•Отбор пробы нефтепродукта для лабораторного исследова-
ния должен быть произведен с соблюдением всех необходи-
мых мер предосторожности, предупреждающих загрязнение
отбираемого продукта, какими-либо посторонними, веществами.
Посуда, приготовленная для отбора пробы, должна быть со-
вершенно' сухой и предварительно тщательно вымытой. В це-
лом, ряде случаев, мы встречаемся о нарушением именно' этих
элементарных правил отбора пробы: образец топлива' или. ма-
сла представляется во влажной посуде, и, кроме того, еще
загрязненной различными .механическими примесями, что со-
вершенно недопустимо. Так ж.е внимательно, как к процессу
отбора, пробы, следует относиться и к отправке ее. на анализ,
если последний не производится на месте1.
Отбор проб топлива и масла
При взятии пробы горючего из тары небольшой емкости —
бочки или бидона— их перекатывают несколько, раз в разные
стороны, после чего берут пробу из любого места!. В этом слу-
чае отбирают пробу из каждой бочки или бидона, а при нали-
чии значительной партии продукта — ив каждой третьей или
пятой емкости!, в зависимости от величины партии.. Если содер-
жимое какой-либо бочки в представленном партии по внешним
признакам окажется подозрительным,, то из этой бочки берут
пробу независимо от количества! взятых из партии' проб.
44
Когда продукт при,ходит >в цистерне, то обычно берут верх-
нюю, среднюю и нижнюю пробы. Бее .три пробы смешиваются
затем в равных соотношениях В1 одну общую пробу.
Наконец, при отборе образца топлива или масла из бака
самолета предварительно, спускают воду, скопившуюся в от-
стойнике, после чего уже берут пробу для анализа.
Особенно, тщательно следует перемешивать при отборе про-
бы ©одержимое бочек или другой посуды с авиационным ма-
слом в зимнее время. На дне такой посуды при пониженной
температуре может выделяться некоторое количество пара-
фина — церезина, содержащегося bi .масле; главная масса про
дукта (жидкая составная часть) приобретает тогда физико-хи-
мические свойства, отличные от свойств .первоначального про-
дукта. Поэтому бочки с авиационным маслом перед отбором
пробы нужно медленно прокатывать взад и (вперед несколько
(12—15) раз и, кроме тю|го, перемешивать их; содержимое ib
вертикальном направлении, т. е. в направлении оси бочки.
Проба продукта отбирается -в количестве 1 л; при этом по-
суда должна, быть заполнена примерно на 9/ы так, чтобы сво-
бодное пространство составляло не менее 7io ее объема. Это
нужно и для обеспечения целости пробы при расширении про-
дукта с повышением температуры, а главным образом для того,
чтобы можно было тщательно .перемешать пробу перед ана-
лизом.
Отбор пробы свинцового бензина
Не .меньшая тщательность должна быть проявлена при пе-
ремешивании только, что приготовленного свинцового бензина.
Этот вопрос нуждается в специальных указаниях, так как,
несмотря на кажущуюся простоту операции, смешения бензина
со свинцовой жидкостью, на практике юна далеко не всегда
проходит как следует. Сплошь и радом получаются более или
менее .заметные расхождения между результатами анализа и
количеством фактически заданной свинцовой жидкости только
потому, что не была правильно, отобрана, проба! из приготов-
ленной партии' свинцового бензина. Это объясняется большой
разницей в удельном весе между свинцовой жидкостью и бен-
зином той или иной марки: удельный вес свинцовой жидкости
равен 1,5, а удельные веса бензинов колеблются в пределах
0,700—0,750. При заливе свинцовой жидкости в посуду с бен-
зином последний растворяет лишь небольшую часть ее, основ-
ная же масса свинцовой жидкости, не успевая .раствориться,
быстро сседйет /н!а дно. В результате проба снизу даст завы-
цйенные цифры содержащий тетраэтилсвинца, а проба, сверху—
за'н1иже..ц1ные, что и наблюдается на практике.
4э
Поэтому свежеприготовленный бензин в посуде небольшой
емкости перед отбором пробы должен быть очень хорошо пе-
ремешан веслом в течение примерно 15 мин. то касается ци-
стерн, даже небольших, то .нередки случаи, когда та сое пере-
мешивание оказывается недостаточно эффективным. 'Несколь-
ко лучшие результаты получаются здесь при прокатке ци-
стерн (авто) 'в течение 2Е—30 минут, но самым лучшим и
наиболее правильным методом является тщапеитьное и полное
перемешивание их содержимого насосом.
Перемешивание насосом, создающее циркуляцию бензина,
заключается в том, что продукт забирается одним шлангом
со дна цистерны и выбрасывается другим шлангам у Bepxnei о
уровне бензина. Необходимую продолжительность работы на-
соса находят, как частное от деления общего объема свинцо-
вого продукта на производительность насоса в единицу вре-
мени.
Снятие нагара
Для снятия нагара, образовавшегося на деталях авиадви-
гателя в процессе его работы и преднавнаменного для анали-
за, мы рекомендуем пока механический способ, используемый
нами на практике bi течение достаточно продолжительного
времени (методы химического удаления нагара с целью от-
бора пробы для анализа нами почти не прорабатывались).
Нагар снимается с детали при помощи шпателя, сделанного
по возможности из мягкого (Mefflatoa, чтобы не повредить по-
верхность детали и не засорить пробу гас сторс нним веществом.
С этой точки зрения особенно важна осте рожность при соскре
быванпи пленки нагара; в крайнем случае лучше пойти на не-
достаточное удаление нагара с детали, чем повредить послед-
нюю и загрязнить пробу. Всю операцию по снятию нагара
производят на развернутом листе т альки, из которой и дела-
ют пакетик с отобранным образцом нагара (лучше пробу на-
гара помещать непосредственно в бюкс или чашку с притер-
той крышкой). Если -нагар соскоблен неосторожно и деталь
авиадвигателя повреждена, то в массе пробы при тщательном
рассмотрении можно обнаружить блестки — стружки металла
Способы отбора проб
Наиболее вцлстой способ отбора (Пробы топлива или ма
ела — это отбор при помощи бутылки емкостью 0,5—1 л с
достаточно широким горлышком. Обычно этот способ приме-
няется при отборе проб из цистерны или нефтехранилища с
разных глубин. Предварительно бутылка плотно1 закрывается
пробкой, привязанной на веревке, конец которой находится у
чб
лида, отбирающего пробу. Затем бутылку опускают на (верев-
ке же 'в цистерну до уровня того слоя, из которого, жела
тельно отобрать пробу; при этом, если имеют дело с ма1слом,
то бутылку перед спуском необходимо чем-нибудь утяжелить
(наиболее целесообразно надеть на 'нее металлическое кольцо).
Спустив бутылку до нужной глубины, выдергивают пробку и
дают посуде нале лниться до краев, что узнают по прекраще-
нию выделения пузырьков воздуха. (Вытянув! наполненную бу-
тылку, опоражнивают ее ® чистую сухую посуду.
Для отбора проб из цистерн употребляется также желонка
(рис 11). Она опускается через верхнее отверстие (лаз) ци-
стерны и медленно догружается до дна,. В процессе погруже-
ния желонка автоматически наполняется.
Рис 11.
Пробоотборник Кутушева ((На'учно-иссл’едювательский ин-
ститут Гражданского воздушного флота) может быть приме-
нен при отборе проб как из бочек, так и из цистерн. Он пред-
ставляет собой стеклянный резервуар а (рис. 12) емкостью
около 150 ми, 1в который йпаяна (Стеклянная Трубка б, нижним
концом выходящая наружу; этот отросток в может быть уд-
линен при помощи каучука и стеклянной трубки. Верхний ко-
нец стеклянной трубки загнут в виде пе'тли внутри резервуара.
Для отбора пробы верхний конец стеклянной трубки ре-
зервуара г, который, в свою очередь, может быть наращен
47
при помощи стеклянной же трубки с каучуком, зажимают
пальцем и вводят прибор в бочку (или в другую посуду) с
продуктом. Открыв верхний конец трубки, дают резервуару
заполниться продуктом, подлежащим испытанию; затем при-
поднимают пробоотборник над бочкой и дают стечь излишку
продукта. После этого спускают отобранную пробу через
трубку д (в нижней части резервуара), на которую надет кау-
чук с зажимом:, iBi заранее приготовленную посуду.
При отборе проб и!з цистерн целесообразно пользоваться
пробоотборником о резервуаров большей емкости — примерно
на 500 мл. Кроме того, верхняя трубка прибора должна быть
значительно удлинена при помощи каучука.
Подготовка проб для анализа
Отобранную пробу плотно закупоривают, что особенно
важно для горючего, и наклеивают этикетку с указанием на-
звания продукта, его сорта, даты и места, отбора пробы и т. д.
.(случается, что некоторые данные при сдаче пробы Epi анайив
неизвестны). Нужно помнить, что подробные сведения на эти-
кетке могут быть Только полезны и, наоюбсрот,— недостаток
данных часто наносит вред.
Вместо наклеивания этикетки можно привешивать к посу-
де с пробои фанерную дощечку (бирку), на которой делаются
соответствующие записи.
При отправке образца топлива: на анализ ,в далеко отстоя-
щую лабораторию необходимо пробку, закрывающую посуду
с пробой, заливать парафином или сургучом для предупрежде-
ния улетучивания легких фракции продукта.
Пробы свинцового бензина с момента, отбора не должны
подвергаться действию света, так как на свету выпадает бе-
лый осадок свинцовых соединений, который затем не удается
растворить простым встряхиванием пробы. Поэтому образцы
свинцового бензина держат до момента анализа в шкафу, в
темноте или обертывают плотной черной бумагой.
Также не рекомендуется подвергать действию света пробы
горючего, содержащего риформинг- или крекинг-фракции.
В таких случаях, кроме того, следует максимально наполнять
бутылку, сста1вляя в ней лишь минимальное количество воз-
духа.
Все образцы топлив и масел перед началом анализа тща-
тельно: перемешиваются для получения совершенно однород-
ного продукта. Это условие с точки зрения качества анализа
не менее важно,, чем правильный отбор пробы. Образцы топ-
лив, вследствие' малой вязкости перемешиваются очень легко
от руки; для этого, достаточно: 10—15 секунд. Что касается
масел, то пробы свежего: продукта также могут быть переме-
4S
шаны от руки; при этом надо не взбалтывать бутылку, а
только переворачивать ее примерно 15—20 раз в минуту, в за-
pJU'CHMO'CTMi от вязкости масла. Продолжительность перемеши-
вания— около 2 'минут.
Другое дело — перемешивание пробы отработанного' масла.
В этом случае приходится учитывать не только' высокую вяз-
кость продукта, но и явления коагуляции (осаждения) механи-
ческих (примесей, углеродистых частиц и т. п. на дне и cmeiH-
ках посуды с пробой; особенно’ это имеет место1 при большом
разрыве во времени между отбором пробы и ее анализом. По-
этому для перемешивания проб отработанного масла мы реко
мендуем механическую (или ручную) болталку, представляю-
щую собой железную раму с гнездами для бутылок, вращаю-
щуюся вокруг своей оси(ри|с. 13). Пробы жестко закрепляются
Рис. 13.
в гнездах, после чего рама приводится в движение. Скорость
вращения регулируют таким образом, чтобы один оборот
длился примерно 4’ сек., чтс составляет 15 об/мин. Продол-
жительность вращения — 20—25 минут. При отсутствии бол-
танки можно перемешивать пробы и от руки, соблюдая уста-
новленные скорость перевертывай^» бутылки; и общую про-
должительность пере;ме;щи!вй(:(ия.
(Подготовка пробы нагара, снятого' с деталей авиадвигате-
ля, состоит в том, чтобы хорошо перемешать ее, т. е. гомо-
генизировать. Для-этого отобранную пробу тщательно расти-
рают в ступке до исчезновения комочков и получения совер-
шенно однородной, тонко-порошкообразной массы. Если тре-
4J
буется объединить нагар, снятый, .например, с различных
поршней, -в одну пробу, то одновременно с растиранием! произ-
водят и перемешив1ание' пробы. При большом количестве на-
гара допускается измельчение (растирание) не всей пробы, а
некоторой ее части, но не менее одной четверти всего образ-
ца; в этом случае необходимое смешивание нескольких проб
в Одну производится до измельчения нагара.
Измельченная проба нагара помещается в. бюкс с притер-
той крышкой или в небольшую колбу с пробкой. Сделав! эти-
кетку с надписью, подобной надписям -на пробах топлив и ма-
сел, считают подготовку пробы нагара законченной. В этом
виде образец поступает на анализ и может сохраняться дли
тельное время.
УЗ о?
ОГЛАВЛЕНИЕ
Стр
Лабораторная техника...................... .....................3
Подготовка лимичоской посуды к анализу.....................3
Фильтрование............................................... .5
Выпаривание и кристаллизация.................................5
Перегонка....................................................Ь
Экстрагирование............................................7
Взвешивание..................................................9
Приготовление дестиллированной воды .......................... , 12
Приготовление растворов (общие понятия). Таблицы.................14
Нетирроваиные. растворы . . . . ... 15
Титрованные растворы , , ... , . 18
Атомные веса химических элементов 19
Удельный вес — градусы Боме . . .20
Удельный вес — вековые проценты.................., .21
Ацидиметрия и алкалиметрия. Индикаторы (общие понятия) , 24
Приготовление смеси Кацвинкеля для сульфирования . 27
Определение общей кислотности олеума . . , ... 28
Определение содержания свободного SOg в олеуме ... 28
Определение количества серной кислоты, необходимой для раз-
бавления олеума 29
Получение смеси Катвиикеля . ......................30
Приготовление растворов брома и молибденовокислого аммония для
определения тетраэтилсвинца ....................................: 30
Ю’/о-ный раствор брома в четыреххлористом углероде, хлороформе
или дихлорэтане - . . . .... .30
Дец'инормзльный раствор жмибденовокиелого аммония . 31
Приготовление растворов соляной или серной кислоты .... 32
Приготовление раствора серноватистокислого натрия (гипосульфита)
для определения иодного числа 4 ........................34
Установка титра гипосульфита натрия посредством чистого иода 35
Установка титра гипосульфита натрия при помощи двухромово-
/кислого калии ........................._................38
Приготовление растворов едкого калия или- едкого натра .... 39
Приготовление раствора карбонада натрия и смеси Варта—Пфейфера
для определения жесткости воды и 41
Децинормальный раствор карбоната натрия . . . 41
Смесь Варта—Пфейфера! ..................42
Приготовление .раствора азотнокислого серебра по Винклеру для опре-
деления хлора .в -воде....................................... 42
Отбор и подготовка проб для анализа ...... 44
Отбор проб топлива и масла ... • • 44
Отбор пробы свинцового бензина . ..............45
Снятие нагара............................................ лк
Способы отбора проб • • ' ля
Подготовка проб для анализа .... • ... 8
Редактор К. К. Папок.
Подписано к печати 18.XII.43 i
Печ. л. 3,25. Уч.-изд. я. 3,5.
Л52750. Тип. РИО Аэрофлота. Москва Старопавскяй, 5. Зак. 1242/214к