/


























































































































































































































Автор: Афонин В.П. Гуничева Т.Н. Пискунова Л.Ф.
Теги: металлы эвм цветные металлы издательство новосибирск
Год: 1984




Текст
АКАДЕМИЯ НАУК СССР
СИБИРСКОЕ ОТДЕЛЕНИЕ
ИНСТИТУТ ГЕОХИМИИ им. АКАДЕМИКА А. П. ВИНОГРАДОВА
В. П. АФОНИН, Т. Н. ГУНИЧЕВА,
Л. Ф. ПИСКУНОВА
Рентгенофлуоресцентный
силикатный анализ
Ответственный редактор
д-р физ.-мат. наук Н. Ф. Лосев
ИЗДАТЕЛЬСТВО «НАУКА»
СИБИРСКОЕ ОТДЕЛЕНИЕ
Новосибирск -1984
УДК
Афонин В. П., Гуничева Т. Н., Пис-
кунова Л. Ф. Рентгенофлуоресцентный силикат-
ный анализ.— Новосибирск: Наука, 1984.
В монографии рассматриваются теоретические основы и
практическое применение рентгенофлуоресцентного метода
для определения натрия, магния, алюминия, кремния, фос-
фора, калия, кальция, титана, марганца и железа в горных
породах различного типа и некоторых промышленных про-
дуктах. Основное внимание уделено способам многокомпо-
нентного анализа на современной автоматической аппарату-
ре, управляемой мини-ЭВМ.
Книга рассчитана на научных сотрудников и инженерно-
технических работников аналитических лабораторий геоло-
гической службы, цементной промышленности и цветной ме-
таллургии.
Ил. 55. Табл. 49. Библиогр. 328.
Рецензенты:
А. Г. РЕВЕНКО, С. В. ЛОНЦИХ
Валерий Петрович Афонин,
Татьяна Николаевна Гуничева,
Людмила Филипповна Пискунова
РЕНТГЕНОФЛУОРЕСЦЕНТНЫЙ
СИЛИКАТНЫЙ АНАЛИЗ
___Утверждено к печати Институтом геохимии
им. академика А. П. Виноградова СО АН СССР
Редактор издательства Л. П. Голышева
Художественный редактор Г. Ф. Каманина
Художник В. В. Каширин
Технический редактор С. А. Смородинова
Корректоры Л. Л. Михайлова, Н. В. Лисина
ИБ М 23495
Сдано в набор 20.09.83. Подписано к печати 16.07.84. МН-01550. Формат 60x90 */1в.
Бумага типографская Л? 1. Обыкновенная гарнитура. Высокая печать. Усл. печ. л. 14.
Усл. кр.-отт. 14,2. Уч.-изд. л. 19. Заказ X". 789. Тираж 1500 экз. Цена 3 р. 20 к.
Издательство «Наука», Сибирское отделение. 630099, Новосибирск, 99, Советская, 18.
4-я типография издательства «Наука». 630077, Новосибирск, 77, Станиславского, 25.
1704060000—840
042(02)—84 154—84—Ш
© Издательство «Наука», 1984 г.
ПРЕДИСЛОВИЕ
В аналитической химии одним из самых трудных и в то же время
важных изучаемых объектов является минеральное сырье. Поиски
полезных ископаемых, разведка обнаруженных месторождений, под-
счет запасов и другие геологические исследования базируются на
результатах анализа минерального сырья. Знание химического со-
става лежит в основе определения и классификации всего многообра-
зия минералов, горных пород и руд. К числу наиболее трудоемких
относится силикатный анализ минерального сырья. Он выполняется
в основном химическими методами. Занимая по количеству 12 % об-
щего числа анализов в геологической службе, по трудоемкости он
составляет 40 % всего объема определений.
В последние годы благодаря научно-техническому прогрессу
в аналитической химии все шире применяются физические методы.
Внедрение их в практику связано прежде всего со все возрастающей
потребностью народного хозяйства в большом количестве анализов
различных материалов. Высокопроизводительные физические методы
позволяют автоматизировать процесс силикатного анализа. В этом
отношении наиболее перспективно применение рентгенофлуорес-
центного метода анализа (РФА). Он сравнительно легко поддается
автоматизации и по точности определения макрокомпонентов сопо-
ставим с химическими методами.
Обобщению исследований по этой теме, выполненных в лабора-
тории рентгеноспектрального анализа Института геохимии им. ака-
демика А. П. Виноградова СО АН СССР, посвящена настоящая мо-
нография. Авторы широко использовали также опыт по рентгено-
спектральному анализу (РСА) силикатов, накопленный в нашей
стране и за рубежом. Некоторые материалы публикуются впервые.
В книге рассмотрены основные вопросы нового метода силикатного
анализа: аппаратура, теория метода, способы учета матричных эф-
фектов, подготовка проб к анализу и метрология метода.
3
В разделе, посвященном аппаратуре для РФА, приведены тех-
нические данные серийных спектрометров отечественного производ-
ства и стран СЭВ, применяемых для силикатного анализа. Указаны
также основные характеристики приборов, выпускаемых ведущими
фирмами за рубежом.
При изложении способов РСА авторы систематизировали методы
учета матричных эффектов, применяемые к анализу различных си-
ликатных материалов. Детально описаны уравнения связи анали-
тических сигналов с содержанием определяемых элементов в образ-
цах, так как использование уравнений связи лежит в основе автома-
тизации расчета результатов РСА.
Подробно рассмотрен способ подготовки проб к анализу путем
формования стекловидных излучателей, получаемых из распла-
вов силикатных материалов с боратами. Такая пробоподготовка
зарекомендовала себя как наиболее универсальная и поддающаяся
автоматизации.
Чтобы облегчить освоение нового метода в производственных
лабораториях, в книге имеется подробная пропись методики автома-
тизированного силикатного анализа горных пород, разработанная
в лаборатории рентгеноспектрального анализа Института геохимии
им. академика А. П. Виноградова СО АН СССР. Даны метрологи-
ческие характеристики нового способа и фактический материал для
сопоставления с классическим методом силикатного анализа.
Авторы благодарны доктору физико-математических наук
Н. Ф. Лосеву, доктору химических наук С. В. Лонциху и кандидату
физико-математических наук А. Г. Ревенко за ценные замечания и
рекомендации.
Глаза 1
ВОЗБУЖДЕНИЕ РЕНТГЕНОВСКИХ СПЕКТРОВ
И АППАРАТУРА
ДЛЯ РЕНТГЕНОФЛУОРЕСЦЕНТНОГО АНАЛИЗА
Аналитическим сигналом в рентгеноспектральном анализе слу-
жат интенсивности наиболее ярких линий характеристического рент-
геновского спектра, измеренные в относительных единицах. Воз-
буждают рентгеновские спектры бомбардировкой поверхности ана-
лизируемого вещества пучком частиц высокой (десятки килоэлект-
ронвольт) энергии — электронов, рентгеновских квантов, протонов
и попов. Наиболее распространены два первых способа возбужде-
ния. Возбуждение пучком электронов применяется в основном в рент-
геноспектральном микроанализе с помощью электронного зонда.
В РФА образцы возбуждают излучением специальных рентгеновских
трубок. Спектр излучения рентгеновских трубок состоит из харак-
теристического и тормозного компонентов, которые излучаются ма-
териалом анода трубок. Вторичные спектры, или спектры рентге-
новской флуоресценции, возбуждаются, как правило, смешанным
первичным излучением, т. е. путем совместного воздействия на об-
разец тормозного и характеристического спектра излучения рент-
геновской трубки.
1.1. РЕНТГЕНОВСКИЕ ТРУБКИ ДЛЯ РФА
Основной целью, которая преследуется при конструировании
рентгеновских трубок для возбуждения рентгеновской флуоресцен-
ции, является получение мощного потока рентгеновских квантов,
падающих на поверхность анализируемого образца. При этохм важ-
но иметь в спектральном составе излучения трубки достаточно ин-
тенсивный поток квантов низкой энергии для эффективного возбуж-
дения рентгеновской флуоресценции элементов с малыми атомными
номерами. Такое условие накладывает жесткие требования па тол-
щину бериллиевых окон рентгеновских трубок. В мягколучевых труб-
ках используется вакуум-плотный бериллий толщиной до 0,1 мм.
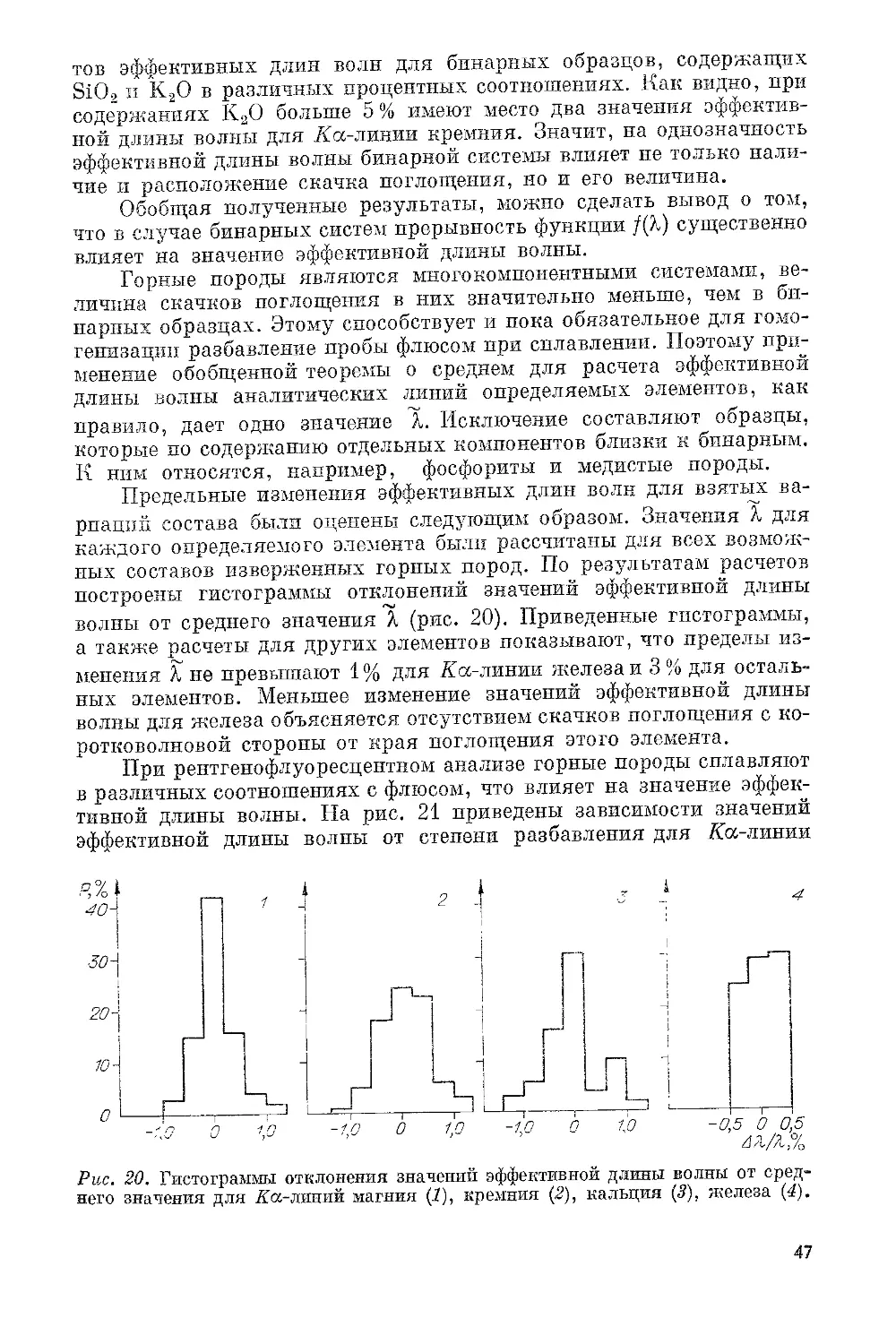
Показанная на рис. 1, а схема является классической. Например,
по ней построены рентгеновские трубки FS-60, применяемые в спек-
трометрах VRA-20 производства фирмы «Карл Цейсс Йена». Схема б
используется в мягколучевых рентгеновских трубках с тонкими
5
Pile. 1. Схемы рентгеновских трубок для РФА.
бериллиевыми окнами. Высокий ускоряющий потенциал в та-
ких трубках в отличие от трубок типа а подается на анодную, а не
на катодную часть. Это резко снижает бомбардировку окна трубки
обратно рассеянными электронами и облегчает установку тонких
окон. По схеме в построены рентгеновские трубки с кольцевым ано-
дом типа БХВ-7, БХВ-9, БХВ-13, применяемые в рентгеновских
квантометрах СРМ-18 и СРМ-20. Анод в трубках БХВ-7 цилиндри-
ческий, а в трубках БХВ-9 и БХВ-13 конический, что увеличивает
угол отбора первичного излучения и снижает поглощение длинно-
волновой части спектра первичного излучения материалом анода.
Толщина бериллиевых окон в этих трубках составляет 0,2—0,3 мм.
В трубках для РФА используют зеркала анодов из следующих
металлов: Сг, Мо, РЛ, Pd, Ag, Re и Ан. Хром, родий, палладий, се-
ребро, рений и золото наносят электролитическим способом на мас-
сивную медную основу слоем толщиной -~0,03 мм.
По схеме г конструируются малогабаритные рентгеновские
трубки, в которых излучающий слой анода наносится непосредст-
венно на внутреннюю поверхность бериллиевого окна. Такие аноды
называют анодами прострельного типа. Мощность малогабаритных
трубок не превышает нескольких ватт, и поэтому охлаждения тру-
бок проточной водой не требуется, что упрощает их эксплуатацию.
Масса самой трубки составляет несколько десятков граммов, масса
блока питания таких трубок не превышает 6 кг, что делает их удоб-
ными для использования в переносной рентгеноспектральной аппа-
ратуре. Основной областью применения маломощных трубок яв-
ляются спектрометры с энергетической дисперсией на базе полу-
проводниковых детекторов рентгеновского излучения. Имеются
примеры удачного применения этих трубок и в кристалл-дифрак-
ционных спектрометрах СПАРК-1, СПАРК-2, АРК-2, СРМ-19.
1.1.1. Тормозной компонент излучения
рентгеновских трубок
При бомбардировке ускоренными электрическим полем электро-
нами материала анода вследствие торможения электронов возникает
непрерывный спектр рентгеновского излучения. Непрерывный спектр
6
занимает определенный интервал энергий фотонов, вплоть до вели-
чин, равных энергии падающих электронов.
Для расчетов интенсивности тормозного рентгеновского излу-
чения часто используется сечение возбуждения, полученное на осно-
ве классической теории Крамерса:
dQ = (8л/3 /3 ) ar2 (Z^mcEE) dExIEv, (1.1)
где а = 1/137; г0 — классический радиус электрона; Z — атомный
номер вещества анода; Е — энергия налетающего электрона; Ev —
энергия возникшего фотона.
В области малых энергий фотонов выражение (1.1) дает зани-
женное значение интенсивности по сравнению с более точным сече-
нием Киркпатрика — Видемана, полученным на основе квантово-
механической теории Зоммерфельда. Однако влияние этого эффекта
на точность расчетов интенсивности рентгеновской флуоресценции
ослабляется поглощением излучения в бериллиевом окне, которое
сильно подавляет длинноволновую область спектра.
Используя сечение (1.1), с учетом обратного рассеяния электро-
нов и поглощения излучения материалом анода для интенсивности
тормозного излучения можно получить выражение
А = ---Г (у-----Д') R фотон/электрон/ср/нм, (1.2)
где L — ln(1166/J0/2J), J = 11,5 эВ, Хо = 1,235/7Г0 нм, R — фактор
обратного рассеяния электронов, /(х) — поправка на поглощение по
Филиберу:
/ (х) = l/[(i + 7» (1 + -~~h (1-3)
h = 1,2Л/22, х = p/sin ф, ф — угол отбора первичного излучения.
По данным Статхэма [301], эффективный коэффициент поглощения
Рис. 2. Спектральное распределение интенсивности тормозного излучения тру-
бок с хромовым и вольфрамовым анодами. Сплошная линия — эксперимент,
штриховая — расчет по формуле (1.2).
7
электронов в случае возбуждения тормозного излучения
а = 4,0 107«6S - А*'85)- (1.4)
На рис. 2 рассчитанные по формуле (1.2) спектральные распре-
деления интенсивности тормозного излучения трубок с хромовым
и вольфрамовым анодами показаны в сравнении с экспериментальны-
ми данными Брауна, Гильфрича и Пекерура [188] для трубок типа
FAQ 60/1 Cr(dBe = 0,3 мм) и FAAQ 60/3,5 W (йВе — 1 мм) при Fo =
= 45 кВ.
1.1,2. Характеристический компонент излучения
рентгеновских трубок
При бомбардировке анода потоком ускоренных электронов од-
новременно с тормозным излучением возникает характеристическое
рентгеновское излучение, обусловленное переходами орбитальных
электронов при заполнении вакансий, образовавшихся за счет уда-
ления электронов с внутренних оболочек атомов вещества анода.
Ионизация наиболее глубокой А-оболочки атома сопровождается
излучением А-спектра, состоящего из совокупности линий, возни-
кающих в результате заполнения вакансий переходами электронов
с L- и М-уровней.
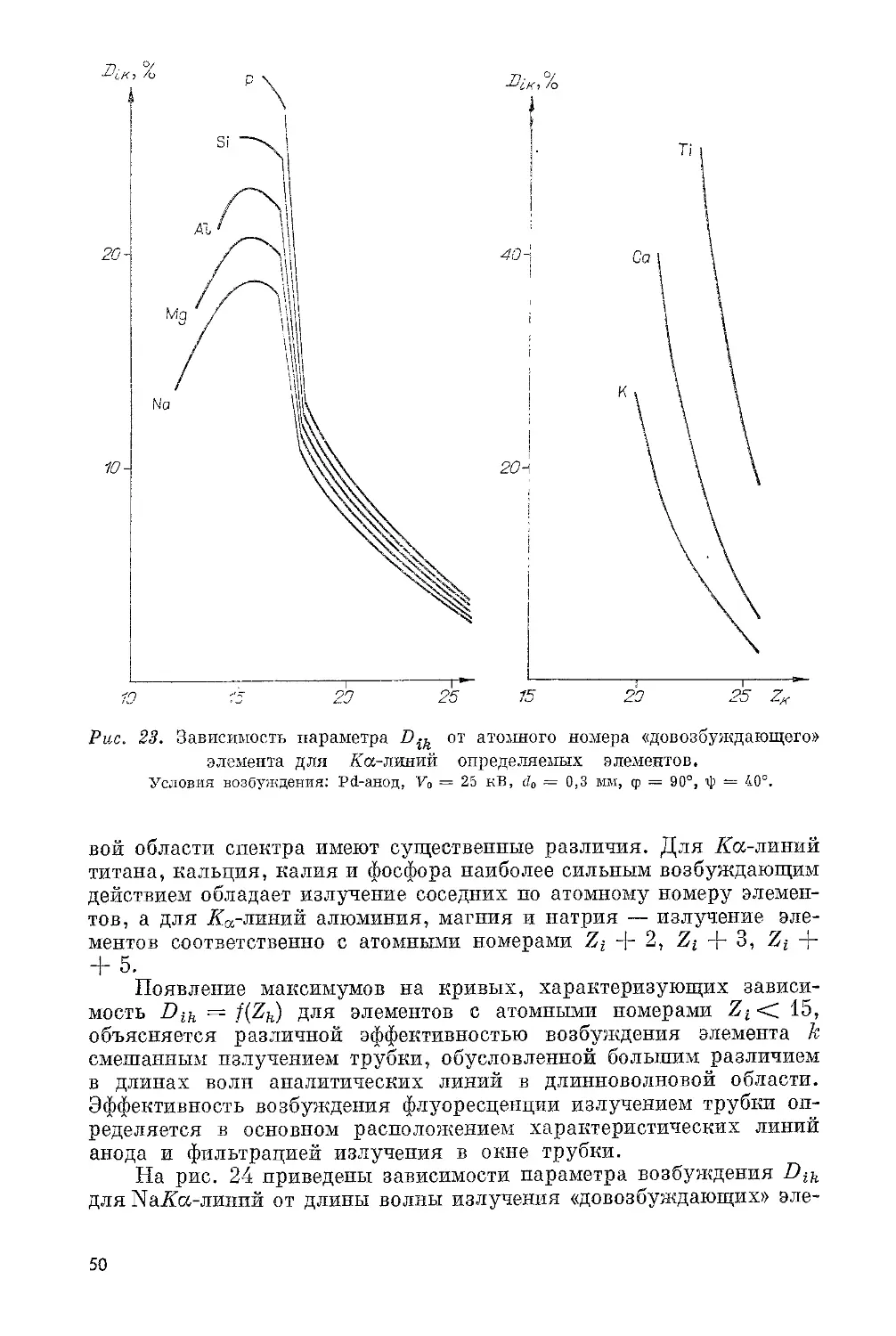
Оболочкам К, L, М, Ал соответствуют электронные орбиты с
последовательно возрастающим средним радиусом и уменьшающейся
энергией связи. А-оболочка включает три подоболочки: — L^;
М — оболочка состоит из пяти подоболочек: — Му. Самая яркая
линия A-серии, обозначаемая символом Acq, возникает при перехо-
де электрона с Ащ-уровня, Аа2-линия связана с переходом Ац-> А,
а Арх-линия — с переходом Мщ -> А. Энергия испускаемых кван-
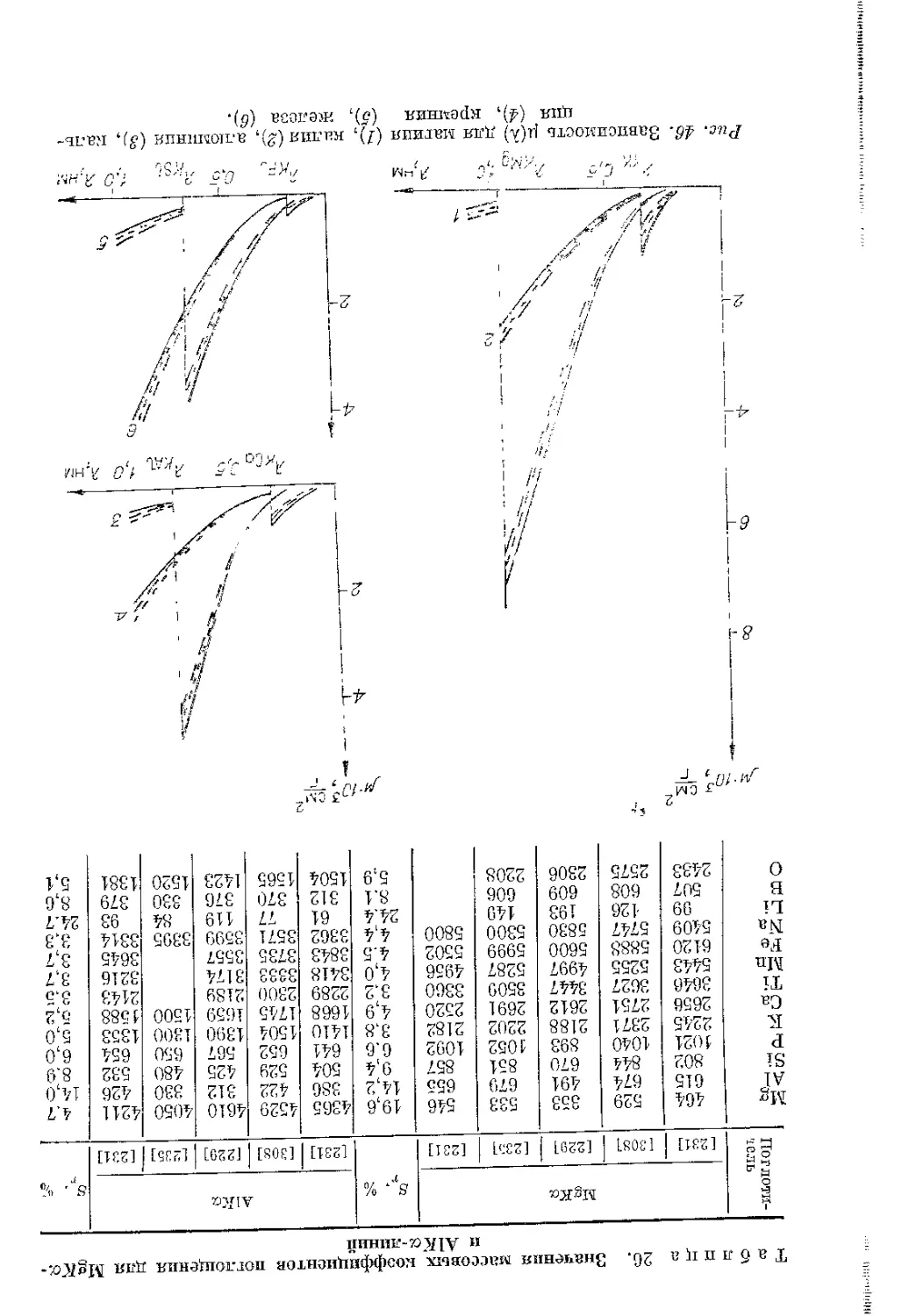
тов рентгеновского излучения равна разности энергий атома в на- чальном и конечном состояниях. L- и М-уровни атома состоят из Таблица 1. Доля характеристического нескольких подоболочек, излучения (%) в спектре для трубок с разными поэтому рентгеновские L- анодами и М-спектры имеют более
V, кВ Сг w сложную структуру, чем
II X л-скекгры. и приложении 11 II приведены длины волн и
20 30 40 50 Тр 45 п расчетнь Тру 70,4 73,7 75,2 77,6 убки FAQ 74,S р и м е ч а е и экспег бка ЕА 75 67,6 71,3 72,6 72,8 60/1 Cr; 1 72,0 н и е. I I имешальнь Cr/W 24,0 34,2 38,7 40,7 A.AQ 60/3 39,8 II — сосг ге значснир относительные интенсив- ности наиболее сильных 26,5 линий А- и £-спектров. 35,6 Для возбуждения данной серии рентгенов- ’ ского спектра энергия 5 W электронов первичного пучка должна превышать 39.0 критическую энергию воз- буждения соответствую- щей оболочки (табл. 11).
8
Вероятность ионизации электронным ударом весьма мала: не более
одного электрона из тысячи создает вакансию на А-оболочке. Ве-
роятность ионизации количественно характеризуется эффективным
поперечным сечением ионизации. Для расчетов обычно используется
квантово-механическое сечение Бете
Qq = ПдЬа(л,е4;ЕЕд)1п(ЕШд), (1.5)
где rig — число электронов на g-оболочке, Ъц =0,61 и bL = 0,032 —
константы.
Число фотонов характеристического излучения в единице телес-
ного угла, приходящихся на один возбуждающий электрон, можно
рассчитать по формуле Грина и Косслетта
(/Д,2/) (1-е)
где U =E/Eq, /(%) — поправка на поглощение по Филиберу, которая
рассчитывается по формуле (1.3). Величина а в поправке на погло-
щение характеристического излучения вычисляется по формуле
о= 4,5-105/(Aj’65-AJ’65). (1.7)
Фактор обратного рассеяния электронов R рассчитывается по Дан-
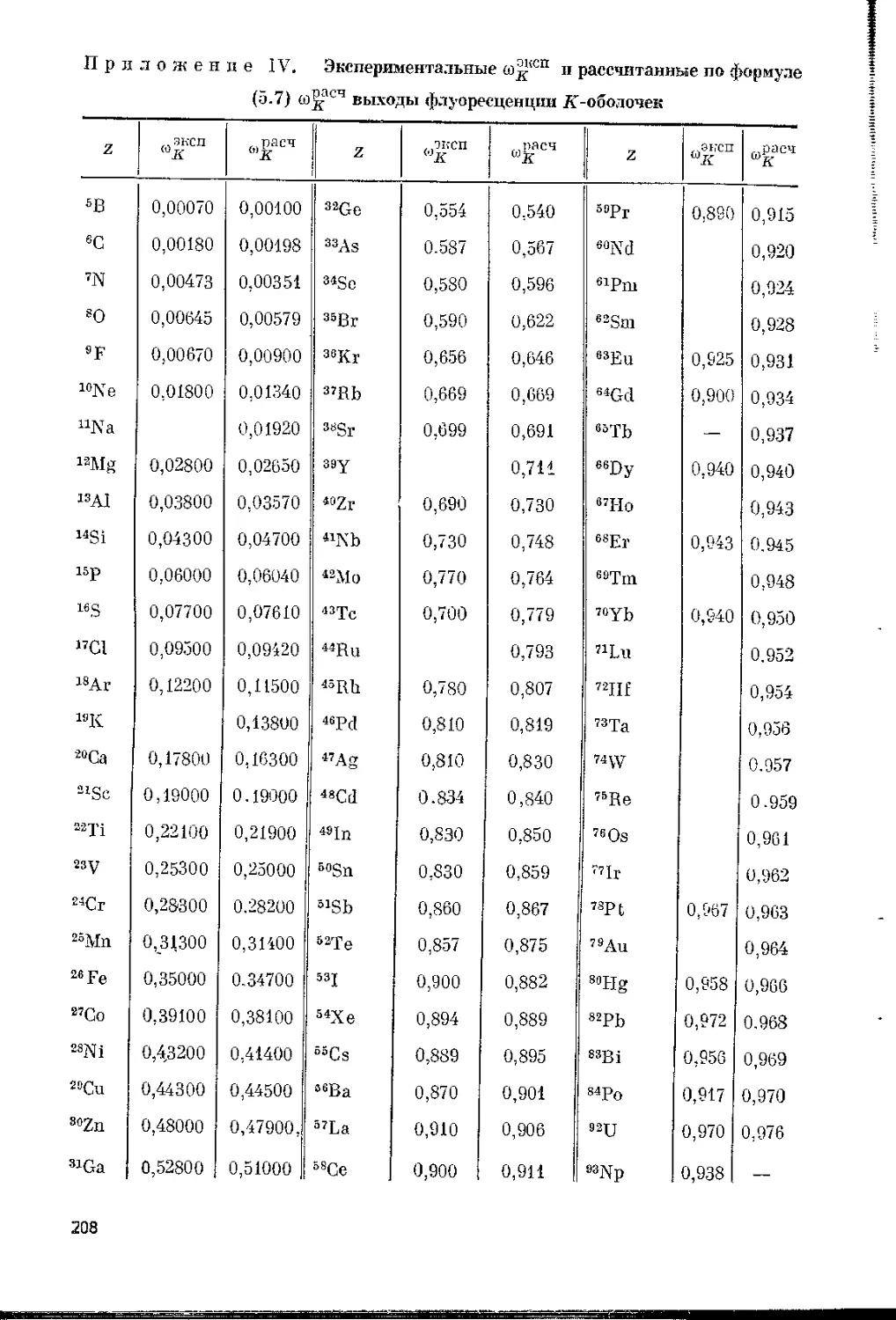
камбу и Риду [44]. Выход флуоресценции равен вероятности ра-
диационного перехода, т. е. доле общего числа ионизаций данной обо-
лочки, сопровождающихся эмиссией рентгеновского излучения.
Для вычисления значений выходов флуоресценции для отдельных
подуровней необходимо учитывать перераспределение вакансий в
результате безрадиационных переходов Костера — Кронига между
подуровнями. В большинстве случаев используются средние значе-
ния со (см. прилож. IV, V), которые можно приближенно рассчиты-
вать по эмпирической формуле [298]
= ZV(ag + Z4), (1.8)
где ак = 1,06-108; aL = 1-Ю8; ам = 1,4-109.
Полное излучение рентгеновской трубки определится суммой
тормозного и характеристического компонентов
I (1) = ехр (— Цц^ЕеРВе) + II СХР (— Ц?е^ВеРВе)- (1-9)
Экспоненциальный член в формуле (1.9) учитывает поглощение из-
лучения трубки бериллиевым окном толщиной d. Величины Д и
Д рассчитываются соответственно по формулам (1.2) и (1.6). Индекс
I относится к Z-линип характеристического спектра трубки.
Важной характеристикой спектрального состава первичного
излучения является соотношение интенсивностей характеристическо-
го и тормозного компонентов. В табл. 1 сопоставлены рассчитанные
по формулам (1.2) и (1.6) соотношения характеристического и тормоз-
ного спектров с экспериментальными величинами, полученными для
трубок ЕА 75 Cr/W, FAQ 60/1 Ст и FAAQ 60/3,5 W [188, 261 ].
9
1.2. АППАРАТУРА ДЛЯ СИЛИКАТНОГО РФА
Для определения содержаний элементов от irNa до 26Fe исполь-
зуются в основном рентгеновские флуоресцентные многоканальные
спектрометры (квантометры). Иногда этот вид анализа может выпол-
няться на рентгеновских спектрометрах последовательного действия
с одним сканирующим каналом. Производительность выполнения
анализов в этом случае существенно ниже. В последние годы появи-
лись рентгеновские спектрометры с энергетической дисперсией, ко-
торые, так же как и квантометры, обеспечивают производительный
многокомпонентный анализ. Все современные рентгеновские спектро-
метры комплектуются управляющими мини-ЭВМ.
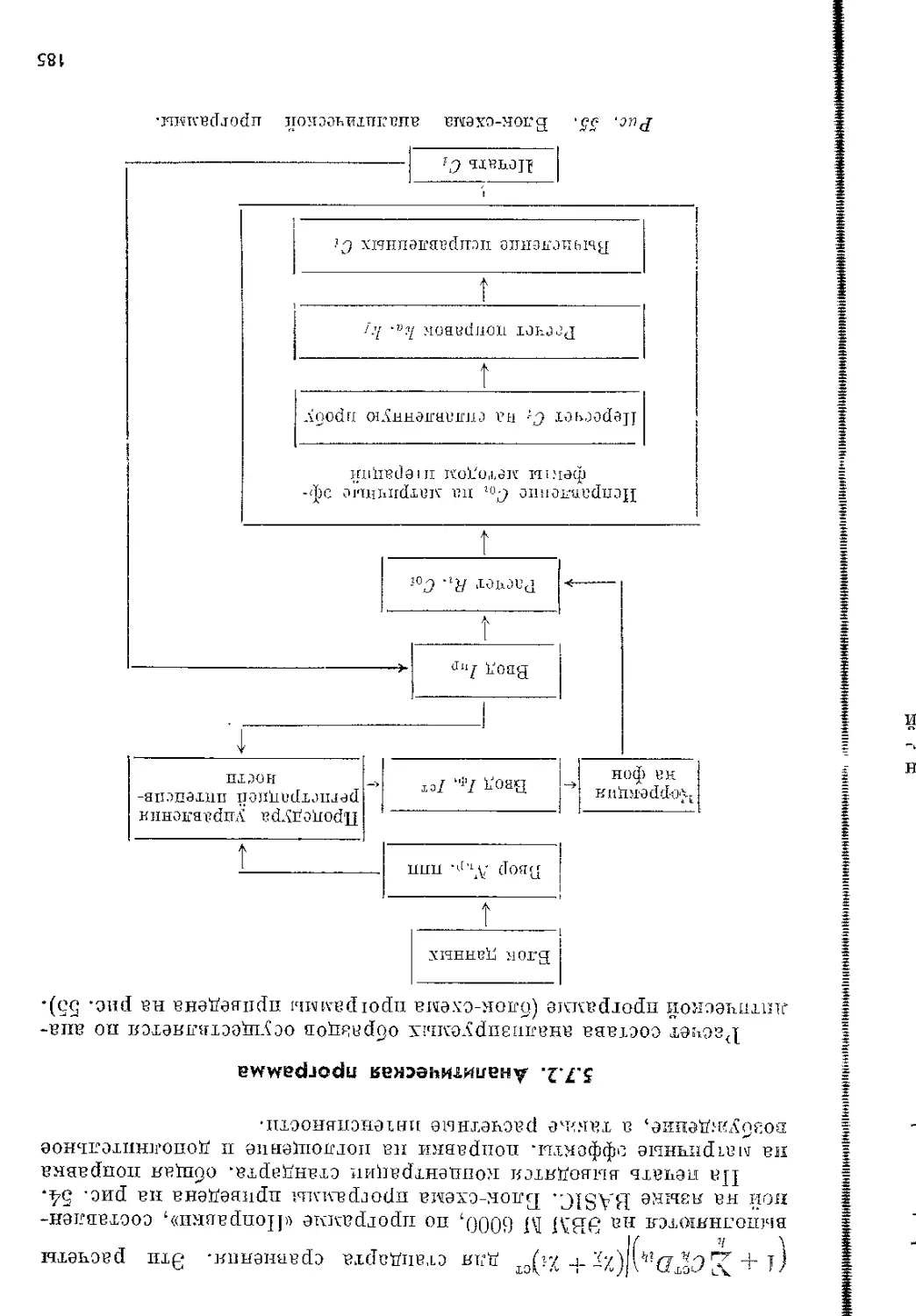
Структурная схема кристалл-дифракционных и энергодисперси-
онных спектрометров представлена на рис. 3. В кристалл-дифрак-
Рис. 3. Функциональная блок-схема кристалл-дифракционных и энерго-
дисперсионных спектрометров.
РТ — рентгеновская трубка, ВИП — источник питания трубки; БПД — блок питания
детектора; БД — блок детектора; \'Ш — усилитель широкополосный; АДД — ампли-
тудный дифференциальный дискриминатор; ПУ — пересчетное устройство; ИН —
интенсиметр; КА — кристалл-анализатор; ГУ — гониометрическое устройство;
КО — камера образцов; О — образец; ЗУ — загрузочное устройство; ВС — вакуум-
ная система; ЭВМ — управляющая электронно-вычислительная машина; ИФ —
интерфейс; АЦПУ —• автоматическое цифропечатающее устройство; ПД — полупро-
водниковый детектор; ХР — хладореагент; ПУ0 — охлаждаемая часть предусилите-
ля; ПУ— предусилитель; АИ — многоканальный анализатор импульсов.
10
ционных спектрометрах используются рентгеновские трубки мощ-
ностью от 1,5 до 3 кВт, в энергодисперсионных — трубки мощностью
от 10 до 200 Вт с анодами прострельного типа.
Загрузочное устройство обеспечивает автоматическую смену
10—12 анализируемых образцов. Некоторые спектрометры снабжа-
ются загрузочными устройствами емкостью 90 и более образцов, что
позволяет проводить анализ без вмешательства оператора в течение
всей рабочей смены.
В рентгеновских квантометрах применяются упрощенные мало-
габаритные спектрометрические каналы, лишенные кинематических
устройств п жестко настраиваемые на аналитическую линию эле-
мента, закрепленного за каждым каналом. Число одновременно уста-
навливаемых каналов может достигать 30. В этом случае кроме ос-
новных элементов могут определяться элементы-примеси. Естествен-
но, в последнем случае необходимы спектрометрические каналы,
обеспечивающие высокую чувствительность.
Спектрометрический канал включает в себя: кристалл-анали-
затор; блок детектора, состоящий из газового пропорционального
млн сцинтилляционного детектора и предварительного усилителя с
коэффициентом усиления до 100; широкополосный усилитель; амп-
литудный дифференциальный анализатор и пересчетиое устройство.
В квантометрах СРМ-18 и СРМ-2 спектрометрические каналы до-
пускают перестройку в достаточно широком диапазоне анализируе-
мых элементов.
Ниже описываются наиболее важные узлы рентгеновских спект-
рометров, от которых в значительной мере зависят их аналитические
возможности.
1.2.1. Детекторы рентгеновского излучения
В современных рентгеновских спектрометрах энергия рентге-
новского излучения с помощью детекторов преобразуется в удоб-
ную для регистрации п последующей обработки форму электрических
импульсов. В основе принципа действия всех типов детекторов ле-
жит способность рентгеновских лучей ионизировать вещество. Все
использующиеся сейчас детекторы являются пропорциональными,
т. е. амплитуда электрических импульсов на выходе этих детекторов
пропорциональна энергии квантов регистрируемого излучения. Од-
нако название «пропорциональные» закрепилось в литературе за
газоразрядными детекторами. Наибольшее практическое применение
находят газоразрядные, сцинтилляционные и полупроводниковые
детекторы.
Газоразрядные пропорциональные детекторы
Этот тип детекторов наиболее распространен в современной рент-
геноспектральной аппаратуре. Конструктивно пропорциональные
счетчики рентгеновского излучения представляют собой газоразряд-
ный двухэлектродный прибор, катодом которого служит металличе-
11
скип корпус, а анодом — тонкая металлическая нить, натянутая
вдоль осп корпуса (рис. 4). В качестве наполнителя счетчика, вообще
говоря, можно использовать любой газ. Однако ионы электроотри-
цательных газов, например кислорода, малоподвижны, легко ре-
комбинируют и не позволяют получить большую амплитуду импуль-
са на выходе счетчика. Поэтому используются инертные газы: крип-
топ. ксенон, аргон и неон, из них чаще всего — аргон. В детекторах
для жесткого излучения предпочтительнее ксенон, так как он более
эффективен вследствие большей поглощающей способности (рис. 5).
Давление газа в счетчике выбирается в пределах от 0,5 до 3 атм и оп-
ределяется диапазоном регистрируемых длин волн. Для жесткого
излучения повышение давления способствует увеличению эффектив-
ности регистрации.
Фотон рентгеновского излучения, взаимодействуя с электронной
оболочкой атомов газа, выбивает один из внешних электронов, в ре-
зультате чего образуется ионная пара Аг-> Ar~ -j- е~~. На образо-
вание одной пары ионов аргона расходуется в среднем 26,4 эВ энер-
гии. Для криптона и ксенона эта величина составляет соответствен-
но 22,8 и 20.8 эВ. Число пар попов, образованных при поглощении
одного кванта энергией е, будет равно
(1.10}
где J — средняя энергия образования одной пары ионов. Количество
пар понов пропорционально энергии падающих фотонов. Поэтому
амплитуда импульса на
выходе счетчика пропор-
циональна энергии реги-
стрируемых рентгеновских
лучей, т. е. счетчик обла-
дает энергетическим раз-
решенном.
ВйМоЗ
Рис. 4. Детектор рентгеновского излучения тина «тандем».
1 — входное бериллиевое окно; 2 — кристалл Xal(TI); з — фотокатод; 4 — динод; 5 — дели-
тель напряжения; 6 — анод; 7 — корпус счетчика; 8 — пить; о — входной коллиматор;
10 — входное окно; 11—выходное окно; 12—дополнительный коллиматор.
12
Под действием ускоряющего потенциала электроны движутся
в направлении к нити, а положительные ионы — в направлении
корпуса детектора. Электроны на своем пути к нити ускоряются
электрическим полем и могут, в свою очередь, ионизировать атомы
наполняющего детектор газа. Таким образом, число электронов
удваивается. Они ускоряются, ионизируют атомы, п удвоение числа
электронов происходит таким путем многократно. Инги достигает
число электронов п п0. Отношение и nQ называется коэффпцпек-
том газового усиления, который составляет обычно величину 101 +
+ 105. Поскольку каждый первичный электрон создает па своем пути
одно и то же сроднее число вторичных электронов, амплитуда сигна-
ла на выходе остается пропорциональной энергии фотонов регистри-
руемого излучения.
При больших коэффициентах газового усиления пропорциональ-
ность между пне нарушается и детектор работает как счетчик Гейге-
ра. Значение коэффициента газового усиления экспоненциально за-
висит от рабочего напряжения на счетчике, поэтому стабильность
высоковольтного питания детектора должна быть не хуже +0,03 %.
Увеличение коэффициента газового усиления позволяет снизить коэф-
фициент усиления электрического импульса радиосхемой и полу-
чить за счет этого выигрыш в отношении епгнал/шум.
Электрический импульс па выходе газоразрядного детектора
асимметричен по форме (рис. б) и имеет длительность порядка 1 мкс.
Крутой фронт импульса обусловлен быстрым продвижением отри-
цательных ионов к аноду, а длительный спад — сравнительно мед-
ленным продвижением положительных попов к катоду. Для более
быстрого гашенпя импульса в инертный газ добавляется немного
гасящего газа. Наиболее распространена смесь Р-10, состоящая из
90 % аргона и 10 % метана. 13 качестве гасящей добавки можно также
использовать пропан пли углекислый газ. Применяются, например,
газовые смеси 97 % Аг -- 3% СО2: 70% Хе 30% СН4; 88% Хе
Н- 12% СО2 и др.
Мягкие рентгеновские лучи поглощаются настолько сильно, что
вся
первичная ионизация пропсхо-
вб.тпзи входного окна, где ела-
поле не препятствует роком-
Рис. 5. Эффективность сцинтилляцион-
ного. полупроводникового и газовых
пропорциональных детекторов с ксено-
новым л аргоновым наполнением.
тектора.
13
бинации ионов. Работу детекторов в длинноволновой области мож-
но улучшить путем уменьшения либо давления газа, либо коэффи-
циента поглощения газовой смеси за счет увеличения содержания
метана до 50 ?zo и более. В области длин волн, больших 1 нм, возмож-
но наполнение счетчика чистым метаном. Эффективность счетчика
зависит от чистоты газа. Особенно неблагоприятны примеси кис-
лорода л водяных паров.
Пропорциональные газовые счетчики выпускаются как в отпа-
янном, так л в проточном вариантах. В отпаянных счетчиках в ка-
честве материала входного окна используют вакуум-плотнып берил-
лий толщиной 0,3—0,2 мм. Срок службы таких детекторов определя-
ется прежде всего непроницаемостью для газа окна детектора. Для
регистрации рентгеновского излучения в длинноволновом диапазоне
нужны очень тонкие входные окна, которые невозможно изготовить
вакуум-плотнымп. Чтобы компенсировать утечку газа, через счетчик
пропускается с малой скоростью струя газа из специального балло-
на. В таких проточных счетчиках окна изготовляются из топких
органических пленок. Используются органические пленки марки
11ЭТФ. терефталата полиэтилена C10HsO4, поликарбоната С^НцО.^
л полипропилена СН2. Полипропиленовые окна особенно прозрачны
для длинноволнового излучения, так как нс содержат кислорода.
Их можно изготовлять путем принудительного растягивания стан-
дартной толстом пленки до толщины 1 мкм. Для этого пленку распо-
лагают над отверстием в камере и камеру откачивают до получения
нужного растя?ксппя пленки. Пленки толщиной 0,1 мкм и менее мож-
но получать из нитроцеллюлозы C22HnO22Nf!, поливинилового фор-
мальдегида С5Н-О2 п аиетата целлюлозы CioHaiOu за счет испарения
растворителя из капель раствора, вылитого па поверхность воды.
Сверхтонкие окна укрепляют на металлической сетке, расположен-
ной с внешней стороны окна. Обычно используют никелевые сетки,
полученные травлением ели электроформовкой. Чтобы избежать
искажения электрического поля в счетчике, внутреннюю поверх-
ность окон напыляют топким слоем алюминия, имеющего контакт с
корпусом счетчика. Рентгеновские кванты большой энергии прохо-
дят через всю толщу газа, достигая задней стенки счетчика и вызы-
вая импульсы малой амплитуды. Этот нежелательный эффект можно
устранить, установив выходное окно напротив входного.
Количество пар ионов, возникающих в процессе первичной по-
пизацип и в последующей лавине, подвержено статистическим флук-
туациям. Это приводит к разбросу величин амплитуд импульсов,
вызванных квантами одной и той же энергии. Пропорциональность
между энергией фотона н амплитудой импульса соблюдается только
по отношению к средней величине амплитуды. Распределение им-
пульсов по амплитудам близко к нормальному закону распределения.
Энергетическое разрешение счетчика характеризуют половиной ши-
рины амплитудного распределения на половине максимума (ПИШИ).
Разрешение счетчика обычно выражают в процентах от средней амп-
литуды имиульса. Так как амплитуда импульса эквивалентна энер-
гии кванта, ширину амплитудного распределения, а следовательно,
14
и разрешение можно выражать в единицах энергии АЕ. Для гаус-
совой кривой среднее квадратичное отклонение о ^У'п. Тогда
ПШПМ = 2,36 Уп.
Поскольку для аргона на образование одной пары ионов рас-
ходуется 0,0264 кэВ, п = е/0,0264. Следовательно, для аргонового
счетчика
ПШПМ = 2,36ст = 2,36/й = 38,3/у? = 35/Г.
Здесь X — длина волны регистрируемого излучения в нанометрах.
На деле разрешающая способность хуже, так как ионизация не но-
сит чисто случайного характера. Этот эффект учитывается так назы-
ваемым фактором Фано, приблизительно равным для аргона 0,8.
Разрешающая способность счетчика зависит от качества нити, кото-
рая чаще всего изготовляется из вольфрама толщиной 25—100 мкм.
Нить должна быть однородной по сечению и иметь гладкую поверх-
ность. Недопустимо провисание нити. Ухудшение разрешения счет-
чика могут вызывать загрязнения, попадающие в счетчик вместе
с потоком газа. Нить следует чистить либо кисточкой, смоченной в
ацетоне, либо путем нагревания, пропуская через нее и последова-
тельно включенное сопротивление ток. Если нить изготовлять из
вольфрама с добавкой 3% рения, то после нагревания до высоких
температур она не теряет гибкости.
Форма катода, т. е. корпуса счетчика, существенного влияния
на разрешение не оказывает. .Поэтому корпус счетчика без потери
разрешения можно изготовлять не только цилиндрической, но и
иной, удобной по конструктивным соображениям формы.
Есть вероятность того, что вторичный фотон выйдет из счетчика
не поглотившимся. Тогда начальная ионизация вторичного фото-
электрона теряется и положение пика смещается в сторону мень-
ших амплитуд. Так возникает «пик потерь», смещенный от основного
пика на величину энергии фотона наиболее яркой линии спектра
атомов газа — наполнителя (рис. 7). Характеристики выпускаемых
в СССР проточных пропорциональных счетчиков приведены в табл. 2.
Пока продолжается разряд, счетчик не может регистрировать
очередной квант рентгеновского излучения. Поэтому при следова-
нии квантов одного за другим чаще чем через одну-две микросекун-
ды детектор будет допускать просчеты. Поправку на «мертвое» время
вводят по формуле
I - Zo/(1 - Zou, " (1.11)
где tM — мертвое время, 10 и I — измеренная и исправленная ин-
тенсивность соответственно. Величина 1 — Е0£м представляет собой
активное время работы счетчика. Мертвое время может значительно
увеличиваться за счет неактивного времени работы регистрирующей
электронной схемы, работающей в комплексе с детектором. Усили-
тель, как правило, не обладает мертвым временем, но работающий с
ним в схеме амплитудный дискриминатор обладает неактивным вре-
менем, равным длительности выходного импульса. Мертвое время
15
Таблица 2, Характеристики газовых пропорциональных детекторов
Параметр 1 СРПП-10 СРШ1-22М
Диапазон регистрируемых длин 0,154-1,2 * 0,154-1,0
волн, нм Диапазон регистрируемых энер- 84-1,0 84-1.2
гий, кэВ Энергетическое разрешение на ли- нии, %: СиХа 154-17 154-17
МпХа 174-20 174-20
Эффективность на линии, %: СиХа 45 30
СаХа 90 90
MgXa 30 30
Собственный фон в канале, охва- 10 10
тывающем 90% излучения линий C.iXa или МпХа, не более имп/мпн Рабочее напряжение, В 15004-2500 15004-2200
Наполнение — газ 90%Аг+Ю%СН4 90%Аг+Ю%СН4
Давление газа, мм рт. ст. 760 760
Внутренний диаметр корпуса, мм 30 20
Входное окно: материал Органическая пленка Органическая пленка
марки ПЭТФ с на- марки ПЭТФ с на-
несенным алюмини- несенным алюмини-
ем (50 нм) ем (50 нм)
толщина, мм 0,004—0,006 0,004—0,006
площадь, мм2 4X15 8X16
Габариты детектора, мм 36X36X107 024X100
Масса, г 450 40
* Диапазон регистрируемых длин волн может быть расширен до 12 нм, для чего ио
наносят тонкую пленку из нитроацетата целлюлозы с высокой прозрачностью в ультра
но перед работой самим потребителем (материал для пленки и инструкция по ее нанесению
электронной схемы может быть удлиняемым. В этих условиях им-
пульс, поступающий в неактивный период, увеличивает его и им-
пульс, пришедший вслед за временем tM, не будет зарегистрирован.
Кроме того, мертвое время может увеличиваться при высокой за-
грузке детектора из-за уменьшения амплитуды импульсов и выхода
их за пределы окна дискриминатора. Поэтому следует избегать вы-
бора слишком узких окон дискриминатора и высоких загрузок де-
тектора.
Существует несколько методов измерения мертвого времени.
Самый простой из них основан на использовании пропорциональной
зависимости интенсивности линий вторичного спектра от силы тока
рентгеновской трубки. Если измерить интенсивности Д и /2 какой-
либо линии спектра при силе тока и то на основании выраже-
ния (1.11) соотношение для расчета мертвого времени можно запи-
сать в виде
(1.12)
16
рентгеновского излучения
СРПО-16 СРПО-100 СРПО-301 БДП-З
0,154-0,25 0,034-0,6 0,0340,6 0,1541,2
84-5 404-2 4042 841,0
154-17 154-17 16419 17420
174-20 174-20 18422 20423
>70 >90 >90 40 90
10 20 150 30 10
160042200 160042200 170042000 15004-2000
90%Хе-НО%СН4 90%Хе+10%СН4 97%Хе-ЬЗ%СО2 90%АгЦ-10%СН4
710 90%Кг+Ю%СН4 90%Аг-|-10%СН4 550 97%Кг+3%СО2 97%Аг+3%СО2 400—700 760
JL О 28 60 30
Бериллий Бериллий Бериллий Органическая пленка
0,2 0,2 0,15 с нанесенным алю- минием (50 нм) 0,006
6X18 6X18 1000 18X25
023X95 035X161 310X70X66 76X90X118
НО 70 700 950
желанию потребителя поставляется дополнительная рамка для входного окна, на которую
мягкой области рентгеновского спектра. Пленка должна наноситься на рамку непосредствен»
входят в комплект поставки счетчика).
Для систем с удлиняемым временем необходимую поправку следует
получать из экспериментальной кривой зависимости скорости счета
на линии от силы тока на рентгеновской трубке.
Сцинтилляционные детекторы
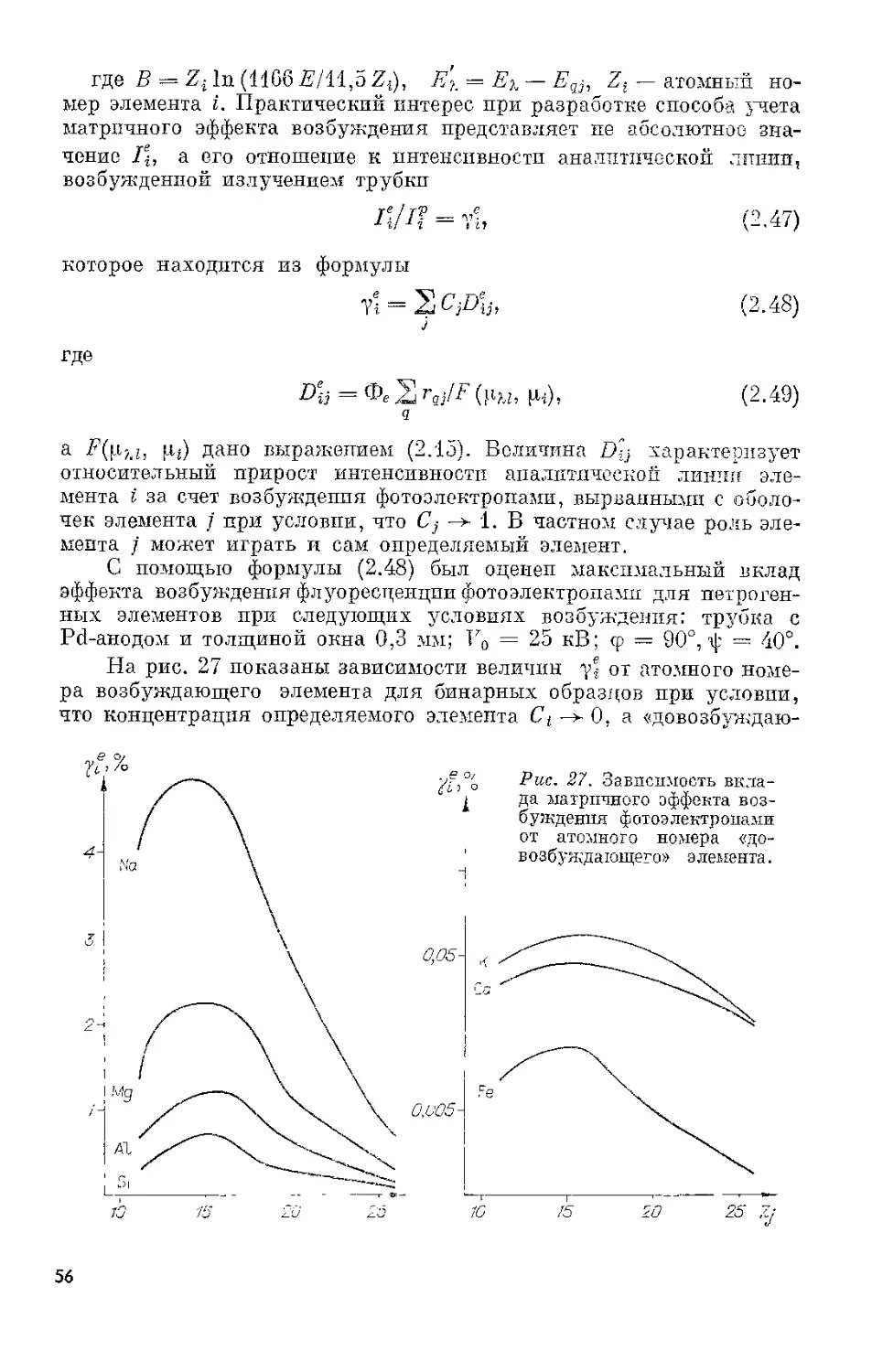
Для регистрации рентгеновского излучения в области длин волн
0,01—0,2 нм часто используются сцинтилляционные детекторы.
В качестве рабочего тела в этих детекторах применяются кристалле-*
фосфоры. Преобразование энергии ионизации в них происходит в
две ступени. Первичная ионизация вызывает вспышку (сцинтил-
ляцию) в видимом диапазоне длин волн. Энергия вспышки преобра-
зуется затем в электрический импульс с помощью фотоэлектронного
умножителя. Сцинтиллятором обычно служит пластинка толщиной
~ 2 мм монокристалла йодистого натрия, активированного таллием.
Ввиду гигроскопичности; кристалл упаковывается в алюминиевый
17
Рис. 7. распре-
деление для FeT^oc-линии.
Рис. 8. Полупроводниковый
детектор с криостатом.
1 — бериллиевое окно; 2 — детек-
тор; з — изолятор; 4 — полевой
транзистор; 5 — держатель; 6—
вакуум; 7 — медный «палец».
контейнер с входным бериллиевым окном толщиной около 0,2 мм
и выходным стеклянным окном. Оптический контакт с входным ок-
ном баллона ФЭУ достигается с помощью различных минеральных и
силиконовых масел или специального оптического клея, которые
наносятся на поверхность колбы ФЭУ.
Максимум интенсивности светового излучения Nal(Tl) лежит
в области 410 нм. В световую энергию преобразуется примерно 6%
энергии рентгеновского излучения, т. е. 50 эВ поглощенной энергии
рентгеновских квантов приводит к образованию одного кванта си-
него цвета. Примерно 90% всех сцинтилляций достигает фотокатода
ФЭУ как напрямую, так и испытав отражение от алюминированного-,
с внутренней стороны бериллиевого окна. Квантовый выход суры-
мяно-цезиевого фотокатода равен ~10%. Таким образом, для по-
лучения одного электрона с фотокатода потребуется затратить около
300 эВ энергии рентгеновских лучей. Сравнивая с энергией обра-
зования одной пары ионов в аргоновом счетчике (26,4 эВ), можно
оценить отличие этих типов детекторов в разрешении: У300/ У26,4=
= 3,4. Следовательно, разрешение сцинтилляционного детектора в
3'—4 раза уступает разрешению газонаполненных пропорциональ-
ных счетчиков. Принимая форму кривой амплитудного распределе-
ния гауссовой и полагая, что ПШПМ = 2,36а, разрешение сцинтил-
ляционного детектора
R = 128//ё = 117/Д
где энергия регистрируемых квантов выражена в килоэлектрон-
вольтах, а длина волны — в нанометрах.
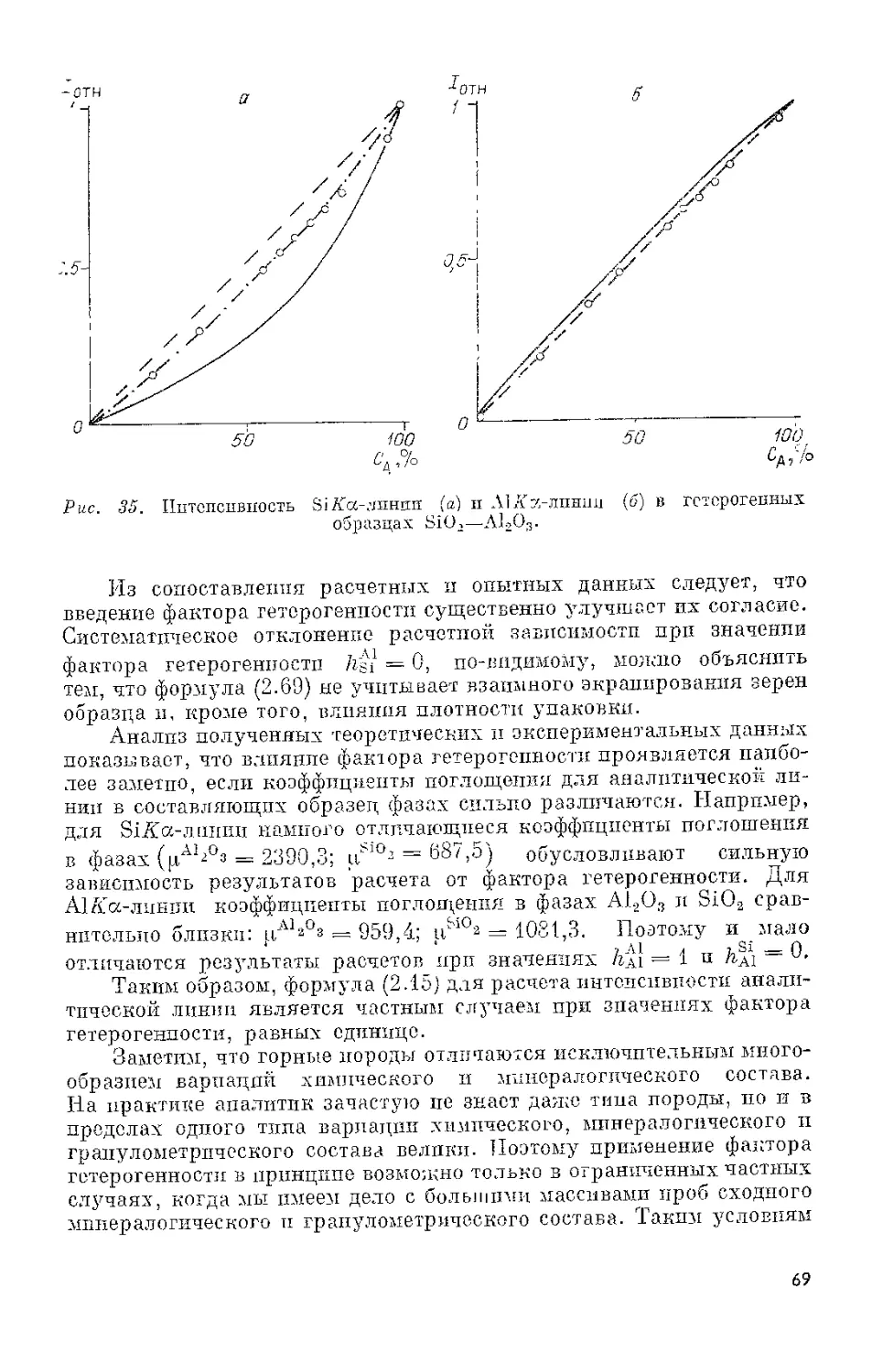
Для FeKa-линии разрешение сцинтилляционного детектора
равно 50%, в то время как для газоразрядного — 15%. Недостаточ-
ное разрешение в какой-то мере компенсируется высокой эффектив-
18
Таблица 3. Характеристики сцинтилляционных детекторов
Параметр БДС-6 БДС-8 БДС-10 БДС-13
Диапазон регистриру- емых длин волн, нм 0,034-0,25 0,034-0,25 0,034-0,25 0,034-0,25
Диапазон регистри- руемых энергий, кэВ Энергетическое раз- решение блока на линии, %: 404-5 404-5 404-5 404-5
СпКа 404-50 404-50 604-80 504-70
МпКа 454-55 454-55 704-90 604-80
Эффективность преоб- разования в диапа- зоне длин волн 0,034-0,02 нм, % Собственный фон бло- ка в канале, охва- тывающем 90% из- лучения линии, имп/мин: >90 >90 >90 >90
СпХа 14-10 14-10 104-50 104-30
МпКа Рабочее напряжение, В: 14-20 14-20 204-80 204-80
ФЭУ предусилитель им- 6004-1000 6004-1000 8004-1500 6004-1000
пульсов Амплитуда сигнала на выходе блока, мВ, на линии: 12 12 12
СиХа 504-500 504-500 104-100 14-5
МпйГа 304-300 304-300 10-4-100 14-5
Длительность импуль- са на уровне 0,5 ам- плитуды, мкс <1,0 <1,0 <1,0 <1,5
Диаметр входного ок- на блока, мм Габариты детектора, мм: 20 20 354-40 16
диаметр 20 20 354-40 16
толщина Особенности конст- рукции: 1 1 1 1
детектор ДРК-201 ДРК-201 ДРК-401 ДРК-161
ФЭУ ФЭУ-85 ФЭУ-85 Посадочное место унифицировано с БДП-3 для вза- имозаменяемости в рентгеновских аппаратах ФЭУ-93 ФЭУ-67Б Работает без пред- усилителя импуль- сов
Габариты блока де- тектора, мм 040X 200 50X80X135 065X225 025X133
Масса, не более кг 1 1,2 1,3 0,25
костью сцинтилляционного детектора в области коротких длин волн
и большой величиной электрического сигнала на выходе ФЭУ. Амп-
литуда сигнала на CuAoc-линии составляет 100—300 мВ, в то время
19
как для газоразрядного счетчика только 1—3 мВ. Поэтому к элек-
тронной схеме, работающей со сцинтилляционными детекторами,
предъявляются менее жесткие требования по коэффициенту усиления
и уровню собственных шумов.
Сцинтилляционный и газоразрядный детекторы удачно допол-
няют друг друга по эффективности регистрации в коротко- и длинно-
волновом диапазонах длин волн (см. рис. 5). Поэтому в некоторых
рентгеновских спектрометрах (VB А-2, VRA-20 и др.) их используют
в виде своеобразного тандема (см. рис. 4). Длинноволновое излуче-
ние регистрируется газоразрядным детектором, коротковолновое
попадает через выходное окно п дополнительный коллиматор во
входное окно сцинтилляционного детектора. В промежуточной об-
ласти длин волн детекторы работают совместно.
Характеристики выпускаемых в СССР сцинтилляционных де-
текторов приведены в табл. 3.
Полупроводниковые детекторы
Применение полупроводниковых детекторов (ППД) в рентгено-
спектральной аппаратуре привело к созданию рентгеноспектральных
аппаратов нового класса. Как и в газоразрядном счетчике, энергия
падающих на твердотельный полупроводник рентгеновских квантов
передается фото- п оже-электронам. В полупроводнике валентная
зона полностью заполнена, а зона проводимости, соответствующая
высоким энергиям, почти свободна и отделена от валентной зоны за-
прещенной зоной. Под воздействием рентгеновских квантов элект-
роны пз валентной зоны переходят в зону проводимости. Каждый
переход сопровождается появлением «дырки» в валентной зоне, ко-
торая в электрическом поле ведет себя как свободный положитель-
ный заряд. Если приложить к полупроводнику разность потенциа-
лов, электроны и дырки будут перемещаться к электродам. Средняя
энергия, затрачиваемая на создание одной пары носителей зарядов
в кремнии (при 100 К), составляет около 3,5 эВ, т. е. почти на
порядок меньше этой энергии для газоразрядного счетчика. Боль-
шое число носителей, создаваемое фотоном данной энергии в ППД,
приводит к малым флуктуациям амплитуды импульсов и хорошему
разрешению.
Для уменьшения шумов необходимо снизить постоянные токи
утечки в детекторе. С этой целью в полупроводник вводят донорные
примесные атомы. Обычно используют литий, атомы которого имеют
малый радиус и легко диффундируют в кремний. Требуемое распре-
деление лития получают посредством «дрейфа» атомов лития в при-
ложенном электрическом поле. На полученный таким образом Si(Li)
материал напыляют тонкий барьерный контактный слой золота.
К этому слою, образующему переднюю часть детектора, подключают
отрицательное напряжение смещения. Положительный электрод
соединяют с входом полевого транзистора (рис. 8). Для уменьшения
уровня шумов за счет поверхностных токов утечки детекторы изго-
товляют с охранной канавкой или в форме охранного кольца. Детек-
20
тор и предусилитель с полевым транзистором монтируют на медном
стержне, второй конец которого погружен в жидкий азот, находя-
щийся при температуре 77 К. Следует заметить, что самые ничтож-
ные изменения емкости между отдельными элементами приводят к
значительному увеличению шумов. Жидкий азот заливают в сосуд
Дыоара емкостью около 5 л и через несколько дней повторяют за-
ливку. При выключенном напряжении смещения можно несколько
раз повысить температуру до комнатной, не повредив детектора.
Поверхность детектора крайне чувствительна к загрязнениям, и в
криостате необходимо поддерживать вакуум до 10-0 тор. Для умень-
шения газоотделенпя применяются сварные детали из нержавеющей
стали и алюминия.
Рентгеновский пучок входит в объем криостата через тонкое
«окно», обычно из бериллия. Бериллий толщиной 8 мкм обеспечивает
50%-ное пропускание фотонов с энергией 1 кэВ, но его надо монти-
ровать на металлической сетке, а толщиной 12 мкм можно исполь-
зовать без сетки при ширине окна не более 6 мм.
Эффективность ППД в коротковолновой области ограничивается
толщиной детектора, а в длинноволновой области — толщиной вход-
ного окна. На рис. 5 показана эффективность Si(Li) детектора толщи-
ной 3 мм с входным бериллиевым окном толщиной 25 мкм. На ре-
гистрации мягких рентгеновских лучей сказывается также плохой
сбор заряда вблизи мертвого слоя.
Энергетическое разрешение ППД для рентгеновских квантов
с энергией е определяется величиной
R = 2,36/Ф//е.
При J 3,5 эВ и факторе Фано Ф = 100 эВ
R& 44//ё ~ 4/Г,
где энергия выражена в килоэлектронвольтах, а длина волны —
в нанометрах. В области длин волн X < 0,07 нм полупроводниковый
детектор позволяет получить разрешение, не уступающее, а то и
превосходящее разрешение кристалл-дифракционного метода (рис. 9).
При анализе низких содержаний элементов, аналитические линии
которых лежат в области 2. > 0,07 им, кристалл-дпфракционный
метод остается незаменимым.
Рис. 9. Разрешающая способ-
ность сцинтилляционного (1),
газоразрядного пропорцио-
нального %), полупроводни-
кового (5) детекторов и крис-
талл-дифракционного спект-
рометра (7).
21
1.2.2. Система регистрации рентгеновских спектров
Системы регистрации в современных спектрометрах в качестве
обязательных имеют следующие блоки: предусилитель, основной
усилитель, амплитудный или многоканальный анализатор, пере-
счетную схему, таймер и управляющий вычислительный комплекс.
Предусилитель благодаря своему низкому выходному сопротив-
лению обеспечивает выходной сигнал, который можно передать без
существенных потерь по длинному коаксиальному кабелю к основ-
ному усилителю. Наилучшими шумовыми характеристиками обла-
дают предусилители с полевым транзистором в первом каскаде. При-
меняются зарядочувствительные схемы, в которых сигнал интегри-
руется на конденсаторе обратной связи и выход пропорционален за-
ряду в импульсе, а не напряжению, зависящему от емкости счетчика
и соединительных проводов, поэтому предусилитель располагают
как можно ближе к счетчику. Импульс на выходе предусилителя
имеет крутой передний фронт и пологий «хвост» экспоненциальной
формы. Такая форма импульса создается дифференцирующей цепоч-
кой для того, чтобы уменьшить наложение соседних импульсов при
высоких скоростях счета. Выходное сопротивление предусилителя
согласуется с импедансом соединительного кабеля, чтобы избежать
потери амплитуды сигнала на пути к основному усилителю и появ-
ления отраженных сигналов.
Основной усилитель увеличивает амплитуду импульсов счетчи-
ка до величины, необходимой для работы амплитудного анализатора.
С выхода главного усилителя снимаются положительные импульсы
с амплитудой до 10 В. Для получения таких импульсов требуется
коэффициент усиления не менее 104. Главный усилитель также фор-
мирует форму импульсов для получения лучшего соотношения сиг-
нал/шум. Формирующие цепочки в главном усилителе представляют
собой частотные фильтры, пропускающие полосу частот, соответ-
ствующую импульсам полезного сигнала, и отсекающие полосу час-
тот, несущую шумы. В простейшем виде цепь формирования состоит
из дифференцирующих и интегрирующих .йС-цепочек, разделенных
усилительным каскадом для уменьшения их взаимного влияния.
Амплитудный анализатор позволяет эффективно выделять полез-
ный сигнал на уровне шумов различной амплитуды. Он включает
два дискриминатора и схему антисовпадений. Дискриминатор исклю-
чает импульсы с амплитудой, меньшей заданной величины, и преоб-
разует поступающие на его вход импульсы в импульсы стандартной
формы для последующей их регистрации пересчетным устройством.
Дискриминатор сравнивает амплитуду входного сигнала с напряже-
нием, устанавливаемым потенциометром порога дискриминации. Ес-
ли импульс превышает заданный порог, запускается ждущий, муль-
тивибратор, на выходе которого формируется импульс стандартной
амплитуды. Этот импульс имеет обычно прямоугольную форму шири-
ной около 0,5 мкс и амплитуду около 5 В, что обеспечивает короткое
мертвое время дискриминатора, хотя мертвое время системы регист-
рации в целом зависит от длительности импульсов, поступающих
22
на вход. В системах детектирования, применяемых в кристалл-ди-
фракционных спектрометрах, «нижний» пороговый дискриминатор ис-
пользуется для отсекания шумов, возникающих в электронных
схемах.
В режиме амплитудного анализа используется «верхний» диск-
риминатор, который позволяет пропускать импульсы с амплитуда-
ми в интервале между нижним и верхним порогами дискриминации.
Импульс на выходе возникает лишь в том случае, если напряжение
сигнала на входе превышает порог нижнего дискриминатора, но не
превышает порога верхнего дискриминатора. Значения верхнего и
нижнего порогов устанавливают потенциометрами. Эту операцию
удобно производить, если в схеме имеются потенциометры регули-
ровки нижнего порога и ширины окна дискриминации. Главным пре-
имуществом амплитудного анализа является возможность суще-
ственного подавления второго и более высоких порядков отражения
от кристалла-анализатора. Следует избегать режимов работы с очень
узкими окнами дискриминации несмотря на то, что при таких режи-
мах достигается наилучшее соотношение сигнал/фон. Дело в том,
что регистрируемая скорость счета очень чувствительна к изменению
амплитуды импульсов вследствие дрейфа режима работы электронных
схем и флуктуаций плотности газа в счетчике.
В приборах со сканирующим каналом вместо непрерывной пе-
рестройки порога амплитудного анализатора при переходе с одной
аналитической линии на другую порог изменяется автоматически с
помощью потенциометра, движок которого связан с кинематическим
механизмом спектрометра. Энергия рентгеновских квантов е и угол
отражения аналитической линии от кристалла-анализатора связаны
соотношением е ~ cosec 0. Поэтому порог дискриминации изменя-
ется таким образом, чтобы амплитуда импульса, соответствующая
середине ширины окна дискриминации, была пропорциональна ве-
личине cosec 0. Указанная система используется в спектрометрах
VBA-2, VBA-20 и др.
Пересчетное устройство позволяет измерять скорость счета им-
пульсов в десятичной системе в течение выбранного оператором
интервала времени. Основной элемент электронной схемы счета им-
пульсов — мультивибратор с двумя устойчивыми состояниями, пред-
ставляющий собой двоичную цифровую ячейку. Мультивибратор
переходит из одного состояния в другое в момент прихода очередного
импульса. Схема устроена так, что регистрирует лишь 10 входных
импульсов. Путем последовательного соединения десятичных кана-
лов можно вести счет с любым заданным предельным числом импуль-
сов. Обычно приборы имеют не более 8 декад.
Таймер — электронная схема, регистрирующая импульсы, вы-
рабатываемые генератором стандартной частоты, который синхро-
низируется обычно частотой электрической сети. Большую точность
можно получить, если использовать кварцевый генератор. В режи-
ме с заданным временем таймер прекращает счет по установленному
оператором интервалу времени, а в режиме с заданным числом им-
пульсов счет прекращается после набора установленного числа
23
импульсов. Преимуществом последнего режима является постоянство
статистической ошибки измерения, однако на практике более удобен
первый режим. При малой скорости счета набор импульсов можно
повторять многократно для улучшения статистики счета.
Многоканальный анализатор распределения импульсов по амп-
литудам используется в комплекте с полупроводниковым детектором.
Для измерения величины амплитуд импульсов в таких анализаторах
применяется аналого-цифровой преобразователь (АЦП). Входной
импульс заряжает конденсатор током, текущим через диод. На пике
импульса диод перестает проводить, и конденсатор держит постоян-
ный заряд, соответствующий пиковому напряжению. Напряжение
на диоде воспринимает схема, запирающая входной вентиль, после
чего конденсатор начинает разряжаться через источник постоянного
тока. Одновременно метки времени с выхода электронных часов на-
чинают поступать в счетчик адресов памяти анализатора. Счет их
прерывается в тот момент, когда напряжение на конденсаторе упадет
до нуля. Число импульсов генератора, зарегистрированное счетчи-
ком, пропорционально времени заряда конденсатора, а следователь-
но, амплитуде импульса.
Входные амплитуды импульсов для АЦП обычно соответствуют
интервалу напряжений от 0 до 10 В. Этот интервал разделяется на
каналы с помощью переключателя коэффициента преобразования, ко-
торый изменяет ток заряда конденсатора. Достаточно иметь каналы
шириной 10 эВ, т. е. 1024 канала для всего рентгеновского спектра,
занимающего диапазон от 0 до 10 кэВ. В некоторых типах анализа-
торов АЦП непосредственно связывается с вычислительной машиной,
входящей в комплект спектрометра. В этом случае отпадает необхо-
димость в собственной памяти анализатора.
Для визуального наблюдения спектра амплитуд служит экран
электронно-лучевой трубки. Спектр на экране высвечивается в виде
гистограммы с числом каналов вдоль горизонтальной оси и числом
зарегистрированных импульсов, представляемых точками вдоль вер-
тикали для каждого канала. Обработка спектра амплитуд произво-
дится автоматически с помощью миникомпьютера.
Для идентификации пиков считаются номера каналов, соответ-
ствующие каждому пику. Анализаторы, специально разработанные
для рентгеновской спектроскопии, градуируются в энергиях фото-
нов. Ошибку градуировки, связанную с ошибкой в установке коэф-
фициента усиления и нуля отсчета, можно откорректировать по пи-
кам линий рентгеновского спектра известной энергии или по импуль-
сам от калибровочного генератора.
Управляющие вычислительные комплексы на основе ЭВМ ма-
лой мощности стали неотъемлемой частью современных рентгеновских
спектрометров и выполняют самые разнообразные задачи. С помощью
ЭВМ производятся математическая обработка результатов измере-
ний, включая учет фона, наложения спектральных линий, мертвого
времени системы регистрации, вычисление статистической флуктуа-
ции интенсивности, построение градуировочных зависимостей и рас-
чет межэлементных влияний, а также расчет результатов анализа и
выдачу их на цифропечать в сервисном виде.
24
ЭВМ выполняет, кроме того, роль организующего звена в про-
цессе анализа. С ее помощью выбираются оптимальный режим работы
источника возбуждения рентгеновских спектров, длительность из-
мерения аналитического сигнала, управление автоматической сме-
ной проб в заданной оператором последовательности, работы изме-
рительной системы, осуществляется периодический контроль рабо-
тоспособности прибора и выявляются дефекты в работе его узлов.
Миникомпыотер используется также в качестве связующего звена
между автоматизированными системами аналитического контроля
АСАК и АСУ, АСУП и АСУТП. Сюда входят: управление качеством
продукции, выдача сигналов о браке, выдача рекомендаций по ве-
дению технологического процесса в оптимальном режиме.
Отечественные спектрометры комплектуются управляющими вы-
числительными комплексами типов М-6000, СМ-1, Электроника-60.
Объем оперативной памяти в зависимости от комплекта составляет
величину от 16 до 32 К. В некоторых случаях ЭВМ комплектуются
внешней памятью на жестких магнитных дисках. В комплекты при-
боров зарубежных фирм входят мпнпкомпьютеры типов Nova-200,
РДР-11/04, Р-800, Р-855 и другие с оперативной памятью объемом
28—32 К. В качестве внешних носителей используются мягкие маг-
нитные диски (флоппп-диекп) с объемом памяти 128x2 К и более.
Быстродействие современных мпнпкомпьютеров позволяет обеспечи-
вать результаты анализа в реальном масштабе времени и ие являет-
ся лимитирующим фактором. Более сложную проблему представляет
выбор объема оперативной и внешней памяти. Для реализации боль-
шинства алгоритмов вычисления концентраций достаточен объем опе-
ративной памяти 16 К. В таком случае нет необходимости в устрой-
ствах, внешней памяти. Методическая часть алгоритма при этом реа-
лизуется на ЭВМ более высокого класса, например на ЕС-1022,
ЕС-1033 и др. Прп использовании миникомпыотера для решения и
методической части алгоритма требуется оперативная память объ-
емом не менее 32 К. В качестве носителей внешней памяти при этом
удобны мягкие магнитные диски. Такой вариант управляющих ЭВМ
для рентгеноспектрального анализа можно считать оптимальным.
При анализе способом прямого внешнего стандарта и способом стан-
дарта-фона удобны микропроцессоры, встроенные в регистрирующую
часть спектрометра.
1.2.3. Кристалл-дифракционные
спектрометрические каналы
Использование полупроводниковых детекторов с энергетическим
разрешением порядка 150 эВ позволяет выделить аналитический сиг-
нал методом энергетической дисперсии, не прибегая к более громозд-
кому кристалл-дифракционному методу. Однако при анализе проб
сложного химического состава разрешения ППД недостаточно для
того, чтобы избежать многочисленных наложений спектральных ли-
ний элементов. Поэтому кристалл-дифракционные приборы с волно-
25
вой дисперсией, очевидно, еще долго будут находить широкое при-
менение.
Волновая дисперсия основана на использовании уравнения
Вульфа — Бреггов для условий дифракции рентгеновских лучей на
монокристалле
sin 0 — nh/2d, (1.13)
где 9 — угол дифракции рентгеновских лучей с длиной волны X,
п — порядок отражения, d — межплоскостное расстояние в моно-
кристалле.
Из выражения (1.13) следует, что путем изменения угла 0 за
счет поворота кристалла можно последовательно выделять из пучка
рентгеновских лучей излучение с заданной длиной волны X. Устрой-
ство спектрометров позволяет поворачивать кристалл-анализатор в
пределах от 20 до 70°. Ясно, что весь диапазон длин волн рентгенов-
ского спектра одним кристаллом-анализатором перекрыть нельзя.
Поэтому используется набор кристаллов-анализаторов с различны-
ми межплоскостными расстояниями. Каждый кристалл работает в
определенном диапазоне длин волн. Кристаллы с большими меж-
плоскостными расстояниями применяются в области больших
длин волн.
Кристаллы-анализаторы должны удовлетворять следующим тре-
бованиям: обладать высоким коэффициентом отражения, не давать
собственного флуоресцентного излучения в рабочем диапазоне длин
волн, иметь малый коэффициент термического расширения, допус-
кать изгиб по радиусу круга фокусировки. Наилучшими характерис-
тиками обладают синтетические кристаллы фтористого лития (LiF),
пентаэритрита (РЕТ), этилендиаминдитартрата (ЕДДТ) и кислого
фталата таллия (ТАР), которые перекрывают весь диапазон длин
волн для силикатного анализа (табл. 4).
Кристалл-дифр акционные каналы собираются по определенной
оптической схеме, определяющей взаимное расположение анализи-
руемого образца, кристалла-анализатора и детектора. Наиболее
простой рентгенооптической схемой, нашедшей широкое распростра-
нение, является нефокусирующая схема Соллера с плоским крис-
таллом-анализатором.
В схеме Соллера (рис. 10, а) между образцом и кристаллом по-
мещается коллиматор, представляющий собой набор параллельных
пластин длиной I, расположенных друг от друга на расстоянии Ь.
С помощью коллиматора Kt из потока флуоресценции отбирается
параллельный пучок лучей, расходимость которого характеризуется
углом б = Ы1. С уменьшением угла б повышается разрешающая спо-
собность, но снижается светосила. Коллиматор К2, помещенный пе-
ред входным окном детектора, не влияет на разрешение, но снижает
фон рассеянного рентгеновского излучения. Кинематическое ус-
тройство должно обеспечивать поворот кристалла на угол 0 и синх-
ронное с ним перемещение детектора на угол 20. Простота кинема-
тического устройства позволяет применять схему Соллера в спектро-
метрах с автоматически сканирующими по спектру каналами. Хоро-
26
Таблица 4.
Кристаллы-анализаторы
Кристалл Химическая формула Отражающая плоскость 2d, нм Анализи- руемый элемент Коэффициент отражения — 5
К-се- рия L-ce- рпя
LiF (фтористый литий) LiF 220 0.2848 23—68 >58 20
LiF (фтористый литий) LiF 200 0.4028 19—58 >49 40
Кварц SiO, 1011 0.6886 15-46 >40 15
Кварц SiO2 1010 0,850 14—41 >37 3
РЕТ (пентаэритрит) ЕДДТ (этилендиампидп- C(CH2OH)4 C6H14N2O8 002 020 0,8742 0,8808 13—41 13-41 >37 >37 14 12
тартрат)
ТАР (кислый фталат тал- COOIL C0H4COOT1 001 2,590 9—23 >23 20
лия) RAP (кислый фталат ру- COOH-C6H4COORb 1010 2,612 8-23 >23 15
бидпя)
КАР (кислый фталат ка- соон-сдцсоок 1010 2,6632 8-23 >23 10
лия)
Ge (германий) NaF (фтористый натрий) Ge 111 0,6533 13-41 >37 8
NaF 200 0,4638 16—41 >47 40
шей разрешающей способности можно добиться фокусировкой диф-
рагировавшего пучка изогнутыми монокристаллами. Наиболее со-
вершенной схемой фокусировки является схема Иоганссона.
В схеме Иоганссона кристалл изгибается по цилиндру радиусом
R. Кристалл предварительно вышлифовывается по цилиндрической
поверхности радиусом 0,5 R. Окружность радиуса г = 0,57? носит
название фокальной окружности (рис. 10, б). Рентгеновская трубка
и исследуемый образец находятся вне фокального круга. Излучение
образца попадает через вертикальную щель на кристалл-анализатор,,
который фокусирует отраженное излучение на приемной щели де-
тектора. Входная и приемная щели расположены симметрично от-
Рис. 10. Рентгеноопти-
ческие схемы.
РТ — рентгеновская трубка;
О — анализируемый обра-
зец; Ki, — коллимато-
ры; КА — кристалл-анали-
затор, Qi —входная щель;
(?2 — приемная щель; Д —
детектор.
носительно точки пересечения F перпендикуляров к отражающим
плоскостям кристалла. Поэтому дуга AF равна дуге FB, а следова-
тельно, равны между собой углы ANF, FNB, AEF, FEB, AMF,
FMB, а также для лучей AN, АЕ и AM — углы падения и отбора
излучения. Таким образом, для любого участка кристалла в плос-
кости фокального круга выполняется условие отражения. Кинема-
тическое устройство в схеме Иоганссона должно обеспечивать пово-
рот кристалла и синхронное с этим поворотом перемещение кристал-
ла и детектора по окружности круга фокусировки. Последнее усло-
вие значительно усложняет конструкцию кинематических устройств
фокусирующих спектрометрических каналов.
Шлифовка кристаллов и последующий их изгиб представляют
собой сложную технологическую операцию, которую можно выпол-
нить далеко не над каждым монокристаллом. Поэтому часто ис-
пользуют упрощенный вариант изгиба кристалла без предваритель-
ной вышлифовки — в рентгенооптической схеме Иоганна.
В схеме Иоганна (рис. 10, в) кристалл по мере продвижения к
крайним его точкам отстоит от фокального круга на все большее рас-
стояние. Поэтому возникают некоторая дефокусировка и уширение
линии, приводящие к ухудшению разрешающей способности спектро-
метрического канала.
Для коротковолнового излучения согласно уравнению Вуль-
фа — Бреггов дифракция происходит в области малых углов. В та-
ком случае лучшие результаты дает разложение в спектр по схеме
Кошуа. Монокристалл для схемы Кошуа вырезается так, чтобы его
отражающие плоскости были перпендикулярны плоскости пластины
кристалла, которая изгибается по цилиндрической поверхности
радиусом В. Флуоресценция от образца проходит через коллиматор
Кх и при отражении от плоскостей кристалла, расположенных вее-
рообразно вследствие изгиба кристалла, фокусируется на фокальной
окружности радиусом В/2 (рис. 10, г). Многопластинчатый коллима-
тор в схеме Кошуа служит для предотвращения попадания излуче-
ния от образца прямо в приемную щель детектора без дифракции и
не влияет на разрешение спектрометра. Для ограничения рассеяния
на пластинах коллиматора его угловая расходимость не должна быть
велика.
В последние годы широкое распространение стала получать
рентгенооптическая схема с изгибом кристалла-анализатора по ло-
гарифмической спирали (рис. 10, д'). Входная щель помещается
в центре спирали. Рентгеновские лучи, проходящие через центр ло-
гарифмической спирали, в силу его геометрических свойств пересе-
кают поверхность кристалла-анализатора под одним и тем же углом
0, что обеспечивает выполнение условия дифракции. Из рис. 10, д
вытекает, что
ctg 0 = drlrdtf — Ъ,
откуда уравнение логарифмической спирали для обеспечения ус-
ловий фокусировки следует в виде
г = а ехр (Ьф).
28
При выполнении этого условия дифрагировавшие лучи собираются в
точке Q2, где монтируется приемная щель детектора излучения. По
условию фокусировки методом логарифмической спирали для каж-
дой длины волны, а следовательно, для каждого определяемого эле-
мента радиус изгиба кристалла-анализатора рассчитывается инди-
видуально.
Этот метод фокусировки обеспечивает высокие разрешающую
способность и светосилу рентгенооптической схемы. Метод не требу-
ет по сравнению с методом Иоганссона вышлифовки кристалла-ана-
лизатора, что позволяет использовать синтетические металлорга-
нические монокристаллы.
Преимущество фокусирующих рентгенооптических схем перед
схемами с плоским кристаллом заключается не только в лучшей раз-
решающей способности, ио и в значительно меньшем фоне рентгенов-
ского излучения. Особенно важно при этом существенное снижение
диффузного и флуоресцентного компонентов рентгеновского фона.
Величина этих компонентов возрастает пропорционально площади
входного окна детектора. Поэтому эти компоненты фона особенно
сильны при разложении излучения в спектр по методу Соллера.
1.2.4. Спектрометры для силикатного анализа
Многоканальные спектрометры (квантометры)
Многоканальный рентгеновский спектрометр СРМ-18. Для си-
ликатного анализа используется серийно выпускаемая модификация
спектрометра: СРМ-18-12-С-УМ. Прибор обеспечивает одновремен-
ный экспресс-анализ на элементы Na, Mg, Al, Si, P, S, K, Ca, Ti,
Мп и Fe.
Оперативный стол спектрометра (рис. И) включает камеру об-
разцов, крпсталл-дифракционные спектрометрические каналы с бло-
ками детекторов, рентгеновскую трубку и вакуумную систему.
Прибор состоит из следующих функциональных блоков: высо-
ковольтного источника питания, оперативного стола, измерительной
системы, системы управления, устройства вывода информации и уп-
равляющей ЭВМ.
Высоковольтный источник питания ВИП обеспечивает рентге-
новскую трубку стабилизированным выпрямленным напряжением
до 70 кВ и стабилизированным анодным током до 100 мА. Стабили-
зация анодного тока и напряжения проводится по вторичной цепи.
Используются рентгеновские трубки с торцевым выходом излу-
чения и тонкими бериллиевыми окнами типа БХВ-9 и БХВ-13.
Угол падения первичного излучения 90°. Образец облучается сверху.
Спектрометрические каналы расположены вокруг трубки с разными
углами отбора в шахматном порядке. Углы отбора флуоресценции
25 п 35°. Камера образцов вмещает 12 образцов одновременно, ко-
торые в заданном оператором порядке подаются в камеру предва-
рительного вакуумирования. Для определения элементов Fe, Мп и
Са используются перестраивающиеся каналы по Иоганну с кристал-
29
Рис. 11. Многоканальный рентгеновский спектрометр СРМ-18.
2 — оперативный стол; 2 — высоковольтный источник питания; 3 — измерительная систе-
ма; 4 — система автоматического управления; 5 — устройство вывода информации,
лами-анализаторами SiO2 (10 11), для определения элементов Ti,
К, Si, Al, Mg и Na — перестраивающиеся каналы по Соллеру
с кристаллами LiF, NaF, EDDT, RAP и для определения фосфора —
фиксированный канал по Соллеру с монокристаллом германия. При-
менение германия в этом случае вызвано тем, что он не имеет второго
порядка отражения, что исключает наложение на аналитическую
Ао^-линию фосфора АР2-линии кальция во втором порядке
отражения. Для определения натрия требуется дополнительная
установка тонкого окна детектора.
Вакуумная система состоит из двух частей, обслуживающих ос-
новную камеру спектрометрического устройства совместно со спек-
трометрическими каналами и камеру предварительного вакуумиро-
вания. Блок управления вакуумной системы обеспечивает работу
электромагнитных вакуумных клапанов. Питание каждого детектора
обеспечивается по индивидуальной линии. В Fe-канале используется
сцинтилляционный блок детектирования БДС-8, в остальных ка-
налах — пропорциональные блоки детектирования БДП-3.
Электронно-вычислительное устройство ЭВУ построено по блочному
типу, число измерительных каналов соответствует числу анализируе-
мых элементов. Каждый измерительный канал состоит из широко-
полосного усилителя, анализатора амплитуды импульсов и пере-
счетного устройства.
Система автоматического управления САУ подает команды на
исполнительные элементы спектрометров, осуществляющие введение
анализируемого образца в шлюзовую камеру под излучение, ваку-
30
умирование шлюзового объема, измерение интенсивности, вывод ре-
зультатов измерения интенсивности в компьютер или устройство вы-
вода информации, смену анализируемого образца. САУ включает
в себя сменный блок связи с компьютером и блок программ, который
используется при работе без компьютера. В последнем случае ре-
зультаты измерения регистрируются цифропечатающим устройством
и перфоратором на перфоленте в международном телеграфном коде
2 МТК.
Управляющие вычислительные комплексы М 6000 или СМ-1 с
объемом памяти 16К позволяют управлять процессом анализа, кон-
тролировать работу спектрометра, хранить исходную информацию
по анализируемым материалам и элементам, рассчитывать резуль-
таты анализа по уравнениям связи.
Техническая характеристика спектрометра СРМ-18
Диапазон определяемых элементов uNa~--92U
Число спектрометрических каналов 12
Число загружаемых образцов 12
Интервал определяемых концентраций 0,001—100%
Размеры анализируемых образцов:
диаметр 40 мм
высота не более 20 мм
Среднее время анализа 1 мин
Максимальная мощность трубки 3,5 кВ-А
Максимальный ускоряющий потенциал 70 кВ
Максимальный анодный ток 100 мА
Нестабильность анодного тока ±0,1%
Нестабильность напряжения питания детектора
±0,05%
Расход воды 15 л/мин
Питание 220/380 В
Потребляемая мощность 15 кВ'А
Масса 2100 кг
Установочная площадь 30 м2
Многоканальные спектрометры СРМ-2 и СРМ-20. Многоканаль-
ный рентгеновский спектрометр СРМ-2 можно считать усовершен-
ствованной конструкцией спектрометра СРМ-18. Число спектромет-
рических каналов доведено до 16, предусмотрена возможность зна-
чительного увеличения емкости загрузочного устройства, использу-
ется новый тип рентгеновской мягколучевой трубки с высоким по-
тенциалом на аноде. Прибор особенно эффективен при анализе на
легкие элементы.
Многоканальный спектрометр СРМ-20 является улучшенным
вариантом рентгеновского флуоресцентного квантометра КРФ-1Б.
Число спектрометрических каналов 8, смена анализируемых образ-
цов ручная по одному образцу. Спектрометр предназначен главным
образом для анализа материалов цементной промышленности.
Квантометр АРК-2. Перспективным для силикатного анализа
можно считать портативный автоматический рентгеновский кванто-
метр АРК-2. Благодаря светосильной рентгеновской оптике в каче-
стве источника возбуждения рентгеновской флуоресценции в нем
используется маломощная рентгеновская трубка с анодом прострель-
31
ного типа и за счет этого значительно снижена потребляемая мощ-
ность и габариты прибора.
Техническая характеристика квантометра АРК-2
Диапазон определяемых элементов nNa4-82U
Число каналов 12
Емкость загрузочного устройства 10
Интервал определяемых концентраций 0,0014-100%
Потребляемая мощность 0,6 кВ-А
Масса 300 кг
В комплект прибора входит малогабаритный управляющий вы-
числительный комплекс «Искра-1251».
Квантометр ARL-72000S (Швейцарское отделение фирмы
ARL). Прибор комплектуется мощной мягко лучевой рентгеновской
трубкой OEG-75 (RB), собранной по схеме, изображенной на рис 1, б.
Охлаждение трубки осуществляется по замкнутому циклу деиони-
зированной водой. Квантометр снабжен 30 фиксированными канала-
ми, собранными по схеме Иоганссона. Вместо трех фиксированных
каналов может устанавливаться один сканирующий канал, который
записывает спектр излучения анализируемых образцов. Сканер име-
ет два кристалла-анализатора.
Загрузочное устройство может быть исполнено в четырех вари-
антах: вручную по одному образцу, вращающийся диск на 7 образ-
цов для предварительной загрузки и постепенной догрузки, вращаю-
щийся диск на 36 образцов и система конвейерной ленты. Использу-
ются запаянные пропорциональные счетчики. Проточные пропорцио-
нальные счетчики применяются только на каналах фтора и натрия.
Управляющая ЭВМ имеет оперативную память 16К и внешнюю па-
мять на мягких магнитных дисках.
Техническая характеристика ARL-72000S
Диапазон определяемых элементов 9F4-82U
Число спектрометрических каналов 30
Максимальная мощность трубки 3 кВ-А
Емкость загрузочного устройства 36 п более
Масса 2400 кг
Квантометр PW 1600 («Philips», Голландия). Этот квантометр
по своим аналитическим возможностям близок к описанному выше
квантометру ARL-72000S. Прибор снабжен управляющим компью-
тером Филипс Р852М с объемом памяти 32К.
Техническая характеристика PW 1600
Диапазон определяемых элементов SF+S2U
Число спектрометрических каналов 28
Емкость загрузочного устройства 10
Максимальный ускоряющий потенциал 60 кВ
Максимальный ток трубки 85 мА
Максимальная мощность 3 кВ-А
Нестабильность высокого напряжения +0,01%
32
Квантометр MR S 400 («Siemens», ФРГ). Отличительной особен-
ностью квантометра MRS 400 является использование в спектрометри-
ческих каналах рептгенооптической схемы с изгибом кристаллов-ана-
лизаторов по логарифмической спирали. Каналы юстируются в завод-
ских условиях. Предусмотрена возможность точной юстировки крис-
таллов после установки в спектрометр непосредственно на рентге-
новском лучке путем их наворота с помощью специального юстировоч-
ного моторчика. Для регистрации излучения с энергией более 5,5 кэВ
используются сцинтилляционные счетчики, для регистрации более
длинноволнового излучения — проточные пропорциональные счет-
чики. Окна счетчиков в зависимости от рабочей области длин волн
имеют толщины 6,0; 2,0; 1,0 и 0,4 мкм. Счетчики выполнены в двух-
камерном варианте и наполняются в зависимости от длины волны
излучения следующими газовыми смесями: 90% Аг + 10% СН4,
70% Не + 30% СН4, 88% Не + 12% СО2. Таким образом достига-
ются оптимальные условия регистрации излучения во всех диапазо-
нах длин волн аналитических линий.
Квантометр снабжен устройством термостатировання рабочего
объема. Давление газа в счетчиках также стабилизировано. Во из-
бежание потерь скорости счета при больших загрузках детекторов
применяются программно-управляемые ослабители, которые уста-
навливаются перед окном детекторов. Анализируемые пробы могут
достигать диаметра до 60 мм. Предусмотрены бленды, ограничиваю-
щие диаметр проб до 34, 27 или 20 мм. Поверхность бленд покры-
вается химически чистым графитом или золотом.
Рентгеновская флуоресценция возбуждается излучением мощ-
ной мягколучевой трубки с Кй-анодом, собранной по схеме, указан-
ной на рис. 1, б. Оптимальные условия возбуждения п регистрации
излучения обеспечивают низкие значения пределов обнаружения.
Например, для стали они составляют 0,0003; 0,0007; 0,0002; 0,0006;
0,0004% для Al, Si, Р, Ст и Ni соответственно. Предусмотрена воз-
можность установки двух сканирующих каналов. Все блоки прибора
расположены компактно в пыленепроницаемом шкафу. Прибор снаб-
жен управляющим миникомпьютером MDS 310.
Техническая характеристика MRS 400
Диапазон определяемых элементов 9F±92U
Число спектрометрических каналов 28
Емкость загрузочного устройства 10
Максимальный ускоряющий потенциал 60 кВ
Максимальный ток трубки 80 мА
Максимальная мощность 3 кВА
Нестабильность высокого напряжения ±0,01%
Квантометр Симультикс IV («Rigaku Denki», Япония). Этот 28-
канальный квантометр по своим основным техническим характерис-
тикам близок к техническим данным квантометров ARL-72000S,
PW 1600 и MRS 400.
Рентгеновский спектрометр TXRF («Rank Hilger», Великобри-
тания). Отличительной особенностью конструкции спектрометра
33
TXRF является наличие трех типов спектрометрических каналов:
фиксированных, перестраивающихся и сканирующих. Спектрометр
по индивидуальному заказу комплектуется произвольной комбина-
цией этих трех типов каналов. Возможна установка максимально 24
фиксированных каналов. Максимальное число перестраивающихся
и сканирующих каналов может быть доведено до 17. Все каналы со-
браны по схеме Соллера. Переход с одной аналитической линии на
другую вручную на перестраивающихся каналах занимает 2—3 мин.
Загрузочное устройство карусельного типа обладает емкостью 50
образцов. Остальные технические характеристики — типовые для
современной рентгеноспектральной аппаратуры.
Рентгеновские спектрометры
с автоматически сканирующим каналом
Рентгеновские спектрометры с автоматически сканирующим ка-
налом предназначены для последовательного определения произ-
вольного количества и набора элементов в диапазоне атомных номе-
ров от 9 (F) до 92 (U). По сравнению с квантометрами эти приборы
позволяют более гибко перестраиваться на различные аналитические
задания, т. е. они более универсальны. Достоинством приборов
является, кроме того, возможность произвольного выбора аналити-
ческих линий применительно к условиям конкретной задачи, а также
многих других условий возбуждения и регистрации аналитического
сигнала. Рассматриваемые спектрометры позволяют записывать весь
спектр анализируемого образца, что открывает возможность прове-
дения качественного анализа проб неизвестного состава. На спектро-
метрах со сканирующими каналами более точно учитывается фон за
счет измерения его рядом с аналитической линией. Однако произ-
водительность одноканальных спектрометров значительно уступает
производительности квантометров при многоэлементном анализе.
Практически все спектрометры со сканирующим каналом соби-
раются по рентгенооптической схеме Соллера, так как точная авто-
матическая установка на пик линии в схемах с изогнутыми кристал-
лами является чрезвычайно сложной задачей. В качестве источника
возбуждения используются рентгеновские трубки мощностью до
3 кВ - А с боковым окном, собранные по схеме, указанной на рис. 1, а.
Зеркала анодов изготовляются из вольфрама, молибдена, хрома, зо-
лота, родия, палладия, меди, кобальта, серебра и железа. Широкий
выбор спектрального состава первичного излучения позволяет со-
здать оптимальные условия возбуждения различных групп элемен-
тов. Использование плоских кристаллов снижает контраст аналити-
ческих линий, однако за счет более точного учета фона реальные пре-
делы обнаружения элементов на спектрометрах последовательного
действия не хуже, чем на квантометрах с фокусирующей оптикой.
Пределы обнаружения, полученные на спектрометре со скани-
рующим каналом VRA-2 для стандартных образцов горных пород,
руд и рудных концентратов СГ-1А, СГД-1А, GT-1A, ВМ, КН, GM,
34
Рис. 12. Пределы обнаружения элементов
на рентгеновском спектрометре VRA-2.
2 — для Кашляний; 2 — для Ьо^-лпнпй.
СВТ-3, GBT-5, GBT-6, GBT-16A и
GBT-9, представлены на рис. 12.
Измерения выполнены при возбуж-
дении образцов излучением вольфра-
мовой трубки, работающей при ус-
коряющем потенциале 40 кВ. Слиш-
ком высокие значения пределов обнаружения для легких эле-
ментов обусловлены в основном большой толщиной окна детектора
(лавсан, 6 мкм) и бериллиевого окна рентгеновской трубки (1 мм).
Приборы снабжаются шестью кристаллами-анализаторами и двумя
коллиматорами Соллера, которые устанавливаются автоматически по
программе, заданной оператором. Прецизионное гониометрическое
устройство позволяет устанавливать угол дифракции с точностью
+ 0,002° (20). Имеется возможность как непрерывного сканирования
с регистрацией результатов на диаграммной ленте, так и шагового
сканирования по заданной программе с выдачей результатов на цпф-
ропечать.
В качестве детектора используется тандем, состоящий из про-
точного пропорционального и сцинтилляционного детекторов (см.
рис. 4). Пороги дискриминации устанавливаются автоматически при
переходе с одной аналитической линии на другую. Современные спект-
рометры обязательно снабжаются системой термостатирования внут-
реннего объема п системой стабилизации давления газа в проточных
пропорциональных детекторах. Емкость загрузочных устройств может
достигать 80 образцов, что позволяет проводить анализ в течение
длительного времени без вмешательства оператора. Для уменьшения
Таблица 5. Рентгеновские спектрометры с автоматически сканирующим
каналом
Спектрометр Фирма Страна Анод трубки ! Ъг Макснмальное на- пряжение, кВ Емкость за- грузочного устройства
штатного 1 дополпи- [ тельного
VRA-30 ’’Carl Zeiss Je- na“ гдр W, Мо, Ст, Pd 60 10 80
PW 1400 ’’Philips’ ’ Голландия 1 W, Мо, Сг, 100 6 72
PW 1400/20 ’’Philips’ ’ Голландия J Си, Со, Fe, Ag, 60 4 Нет
SRS 200 ’’Siemens’ ’ ФРГ W, Мо, Сг, Аи, ВИ 60 10 80
XRD-8565/75A ARL США W, Мо, Сг, Pt 60 10 Нет
35
влияния неоднородности ооразцов на результаты анализа преду-
смотрено вращение образцов со скоростью 6—60 об/мин. Для анали-
за жидких образцов предусмотрена возможность непрерывной про-
дувки объема спектрометров гелием.
Все операции анализа, включая расчет концентраций по изме-
ренным интенсивностям, автоматизируются с помощью минпкомпью-
теров с оперативной памятью 16—32К, входящих в комплект спектро-
метров.
В табл. 5 приводятся основные технические данные автоматиче-
ских спектрометров со сканирующим каналом.
При анализе горных пород на автоматических спектрометрах со
канпрующим каналом кроме определения основных породообразую-
щие. 18. Сопоставление пределов обнаружения элементов рентгенофлуоресцент-
ным методом п кларковых содержаний элементов в земной коре.
36
щих элементов целесообразно проводить определения элементов-при-
месей, таких как Sc, V, Gr, Со, Ni, Си, Zn, Ga, As, Rb, Sr,. Il, Zr,;
Nb, Mo, Sn, Ba, La, Ge, Nd, Sm, Pb, Th, U. Наглядное представление
о возможности и ограничении применения рентгеновских кристалл-
дифракционных спектрометров в геолого-геохимических исследова-
ниях дает сопоставление пределов обнаружения элементов Cmin и
средних (кларковых) содержаний Ск элементов в земной коре (рис. 13).
Величина Cmin существенно зависит от типа применяемой аппара-
туры, условий возбуждения и регистрации аналитических линий,-
а также от химического состава анализируемых проб. Границы изме-
нения величины Cmin укладываются в основном в «коридор», огра-
ниченный прямыми линиями. Рентгенофлуоресцентным методом мо-
гут определяться кларковые содержания элементов, точки которых
на диаграмме лежат над верхней линией. Анализ на элементы, ле-
жащие внутри «коридора», может выполняться только на светосиль-
ных спектрометрах высокого разрешения.
Рентгеновские спектрометры
с энергетической дисперсией
Появление полупроводниковых детекторов рентгеновского излу-
чения высокого разрешения (см. рис. 8) привело к созданию принци-
пиально нового типа рентгеновских спектрометров, в которых ана-
литический сигнал выделяется не с помощью кристаллов-анализато-
ров, а путем амплитудного анализа электрических импульсов, посту-
пающих с детектора (см. рис. 3). Эти спектрометры обладают следую-
щими основными достоинствами: возможностью определять одновре-
менно практически все элементы, составляющие образец, сколько бы
пх ни было; сравнительно малыми габаритами и малой потребляемой
мощностью; сравнительно низкой стоимостью.
В качестве источников возбуждения в ППД-спектрометрахисполь-
зутотся радиоактивные изотопы, маломощные рентгеновские трубки
с анодами прострельного типа (см. рис. 1, г) и вторичные излучатели,
флуоресцентное излучение которых возбуждается мощными рентге-
новскими трубками, обычно используемыми в рентгеноспектральном
анализе. Наиболее удобны маломощные (до 50 Вт) рентгеновские
трубки с третьим управляющим электродом, работающие в импульс-
ном режиме. Первичное излучение фильтруется для повышения конт-
раста аналитического сигнала с помощью устанавливаемых автома-
тически фильтров по программе, заданной оператором.
Практически все ППД-спектрометры используют 81(Ь1)-детекто-
ры с ультратонкими входными бериллиевыми окнами, которые дают
возможность определять все элементы в диапазоне атомных номеров
от 11 (Na) до 92 (U). В качестве хладореагента применяют жидкий
азот, который периодически заливают в малогабаритный сосуд Дьюа-
ра. К электронике, используемой в ППД-спектрометрах, предъяв-
ляются очень жесткие требования, так как профили линий ампли-
тудного распределения не должны иметь флуктуаций по шкале энер-
3-7
гий и по ширине профиля. Многоканальный амплитудный анализа-
тор интегрируется обычно с миникомпыотером, с помощью которого
обрабатывается спектр амплитудного распределения импульсов,
вносятся поправки на аппаратурные эффекты, рассчитываются ре-
зультаты анализа и осуществляется управление работой всего
прибора.
В методическом отношении анализ на ППД-спектрометрах мало
чем отличается от обычного рентгенофлуоресцентного. Исключение
составляет лишь необходимость обязательного учета взаимных нало-
жений спектральных линий. Такой учет представляет собой весьма
сложную задачу, решаемую с помощью миникомпыотера.
По реально достигаемым пределам обнаружения элементов
ППД-спектрометры в большинстве случаев почти на порядок усту-
пают кристалл-дифракционным спектрометрам.
В практике аналитических лабораторий наибольшее распро-
странение получили следующие ППД-спектрометры с энергетической
дисперсией: Tefa III производства фирмы «Ortec» (США), XR-500
производства фирмы «Link System» (Великобритания), EXAM SIX
производства фирмы «Philips» (Голландия), 8420 XRF производства
фирмы ARL (США).
Глава 2
МАТРИЧНЫЕ ЭФФЕКТЫ
На интенсивность рентгеновской флуоресценции влияет хими-
ческий состав и физическое состояние компонентов анализируемого
образца. Эти влияния принято называть матричными эффектами.
К ним относятся следующие эффекты:
1) поглощение аналитической линии материалом излучателя;
2) поглощение первичного излучения;
3) возбуждение аналитической линии флуоресцентным излуче-
нием некоторых элементов образца (вторичная флуоресценция);
4) возбуждение третичной флуоресценции и флуоресценции более
высоких порядков;
5) возбуждение аналитической линии рассеянным первичным из-
лучением;
6) возбуждение аналитической линии рассеянным вторичным из-
лучением;
7) возбуждение оже-электронами анализируемого образца;
8) возбуждение фотоэлектронами анализируемого образца;
9) сдвиг положения аналитической линии в зависимости от хи-
мической связи атомов определяемого элемента в анализируемом ве-
ществе;
38
10) влияние размера частиц порошковых материалов на интен-
сивность рентгеновского спектра;
1) влияние минералогического и фазового состава на интенсив-
ность аналитической линии.
Кроме того, влияние матричных эффектов сказывается и на
интенсивности рентгеновского фона, на котором регистрируется ана-
литическая линия. Однако проблему учета фона традиционно отно-
сят не к матричным, а к аппаратурным эффектам, так как относи-
тельный вклад различных компонентов фона существенно зависит
от конструкций спектрометрических каналов.
Относительный вклад каждого из перечисленных матричных эф-
фектов зависит от условий возбуждения и области длин волн, в ко-
торой лежат аналитические линии определяемых элементов. Прп ана-
лизе силикатных материалов наиболее сильны матричные эффекты.в
указанные в пп. 1, 2 и реже 3. Матричные эффекты, изложенные в
пп. 10 и И, характеризуют влияние гетерогенности проб, они силь-
ны в длинноволновой области спектра и трудно поддаются корректно-
му учету. Поэтому при силикатном анализе чаще всего не учитывают
эффектов гетерогенности, а устраняют путем гомогенизации проб
различными методами, приведенными в гл. 4.
2.1. ИНТЕНСИВНОСТЬ РЕНТГЕНОВСКОЙ ФЛУОРЕСЦЕНЦИИ
В ПОЛИХРОМАТИЧЕСКОМ ПРИБЛИЖЕНИИ
Пусть на плоскопараллельный идеально гомогенный образец
под углом ф к его поверхности падает поток квантов 1^ первичного
излучения с длиной волны % (рис. 14). Число квантов первичного
излучения, достигших слоя dx, равно
biz) = h exp (—f^p-r/sin ф), (2.1)
где Цх — коэффициент поглощения материалов образца первичного
излучения с длиной волны X. Число ионизаций g-уровня атомов эле-
мента I первичными квантами на пути ds будет равно произведению
поперечного сечения ионизации g-уровня на число атомов элемента
i в элементарном объеме Qqds:
dnqi = CiQqiIx(x)p(N/Ai)ds, (2.2)
где Ci и Ai — соответственно концентрация и атомная масса эле-
мента I, р — плотность образца, N — число Авогадро. Сеченпе ио-
низации путем захвата фотона
равно частичному атомному коэф-
фициенту поглощения элемента i
для излучения с длиной волны К,
т. е.
Qqi = — гN, (2.3)
Рис. 14. К расчету интенсивности рент-
геновской флуоресценции.
39
где i'qi — (Sqi — i}!Sqi, — массовый коэффициент ослабления
излучения с длиной волны А, элементом I. В слое dx путь ds = cWsin <р.
Число возбужденных квантов аналитической линии в слое dx
di?035 = ®q:Jpidnqi. (2.4)
Число квантов аналитической линии от слоя dx, вышедших через
поверхность образца под углом ф, будет
dZj = cZZf 036 exp (— prep,j/sin ф), (2=5)
где [ij — массовый коэффициент поглощения аналитической линии
материалом образца. Подставляя в (2.5) выражения (2.4), (2.3), (2.2)
и (2.1), имеем
dli = kfiiZxPxiexp [—[Tj./sin ср 4~ p,z/sin ф)]р(£с, (2.6)
где
kt = ^qiPiVqi/sin ср. (2.7)
Интегрируя (2.6) по px в пределах от 0 до оо, получим
It = к sin ф + pti/sin ф). (2.8)
Если рентгеновская флуоресценция побуждается непрерывным рент-
геновским спектром, то интенсивность аналитической линии получим
интегрированием (2.8) по длинам волн в пределх от до
Я = н), (2.9)
где
н) = !РмЛАЯрг/зшф + рц/зтф). (2.10)
%
При возбуждении характеристическим спектром интенсивность флуо-
ресценции можно вычислить суммированием выражений вида (2.8)
по всем линиям характеристического спектра излучения трубки
Pi), (2.11)
где
Fx(m, ц{) = ЗРгЙоЛР-гМпф + рц/зшф). (2.12)
г
Интенсивность рентгеновской флуоресценции, возбужденная сме-
шанным (тормозным и характеристическим) излучением трубки, рав-
на сумме выражений (2.9) и (2.11):
Ц = 14) + 14). (2-13)
Учитывая, что
f 2 3 (ж — Xi) f(x)dx = ^f (х^),
i г
выражение для интенсивности рентгеновской флуоресценции, воз-
бужденной смешанным первичным излучением, можно представить
40
в компактном виде:
1г = k^F^, (2.14)
где
F (рм, Pi) = j рм/(Х) </(р?Л1пф + рц/зшф), (2.15)
%
/(^) = Л +2б(Х-Хг)Л. (2.16)
i
Величины /1 и /; определяются выражениями (1.2) и (1.6).
2.2. ИНТЕНСИВНОСТЬ РЕНТГЕНОВСКОЙ ФЛУОРЕСЦЕНЦИИ
В МОНОХРОМАТИЧЕСКОМ ПРИБЛИЖЕНИИ
Формула для расчета интенсивности флуоресценции существен-
но упрощается, если излучение рентгеновской трубки описать эф-
фективной длиной волны. Эффективную Длину волны % при возбуж-
дении флуоресценции тормозным излучением трубки можно рассчи-
тать по обобщенной теореме о среднем. Согласно этой теореме, если
подынтегральную функцию можно представить произведением функ-
ций
Р(х) = Kx)g{x), (2.17)
то справедливо равенство
ь ь
j j(x)g(x)dx = /(£) ^g(x)dxt (2.18)
а а
Подынтегральную функцию Р(х) можно представить в виде произ-
ведения следующих функций:
/(%) = l/(p%/sin ср 4- p;/sin 4), (2.19)
g(X) = Z^,p^,i. (2.20)
Для проверки, какая из этих функций монотонна на участке [%0,
ХдД, построим зависимости полной подынтегральной функции Р(к)
и ее составляющих /(X) и g(X) от длины волны для MgAtz-линии
(рис. 15). Среди трех подынтегральных функций, указанных на ри-
сунке, только функция /(%) монотонная, поэтому использование обоб-
щенной теоремы о среднем в виде (2.18) дает одно значение эффектив-
ной длины волны.
Подставляя (2.19), (2.20) и (2.10) в (2.18), имеем
Kqi
ЛДрь, Pi) = [l/(p^./sin(p + p{/sin4)] J (2.21)
%
Откуда
\(i
p^. = sin ф J I^idK/F^ (p?., pi) — pi sin ф/sin ф. (2.22)
41
Рис. 15. Зависимость подынтегральных
функций Р(к) (7), g(k) (2), /(20 (3) от длины
волны для А?а-линий магния стандартного
образца состава базальта ВМ.
Учитывая, что
= СХ”, (2.23)
с помощью выражений (2.22) и (2.23)
можно рассчитать значение эффек-
тивной длины волны Хг при возбуж-
дении флуоресценции тормозным
компонентом излучения рентгенов-
ской трубки. Коэффициенты С и п
в формуле (2.23) выбираются по дан-
ным приложения I.
При использовании теоремы о среднем точность расчета X су-
щественно зависит от числа интервалов, на которые разбивается ин-
тегрируемый отрезок [Хо, ] при вычислении интеграла методом Сим-
псона. Для всех определяемых элементов удобно применять одина-
ковое число интервалов, поскольку при увеличении атомного номера
хотя и уменьшается величина отрезка интегрирования, но увеличи-
вается крутизна функции, и для правильного расчета необходимо
уменьшать шаг интегрирования. Практика показала, что достаточно
разбивать отрезок [Хо, Х5Д на 400 интервалов.
Для эффективного возбуждения элементов с малыми атомными
номерами обычно используется спектр трубки, состоящий из тормоз-
ного и характеристического компонентов. Метод расчета эффектив-
ной длины волны такого спектра Хг основан на учете относительного
вклада тормозного и характеристического компонентов излучения
трубки в возбуждение вторичного спектра. В качестве весовых мно-
жителей применяются относительные эффективности возбуждения,
которые определяются величинами функций FT(p,x, Pi) и Fx(pi, рг)
для тормозного и характеристического компонентов соответственно.
Значение Хг- находится из выражения
~ Vt (Bv Pi) + S Vx (Pl’ Pi)
______________________i______________
1 Fr(Pv Pi) + Fx(Pl’ Pi)
(2-24)
При использовании эффективной длины волны функция Ft при-
мет вид
F> = h^i/fai +
где
Kfa = Pvi/Sill<P’ & =
(2.25)
(2.26)
42
Согласно формуле (2.24), значение эффективной длины волны
зависит как от условий возбуждения, так и от химического состава
анализируемого материала.
2.2.1. Влияние условий возбуждения
на значение эффективной длины волны
Условия возбуждения флуоресценции элементов с малыми атом-
ными номерами существенно зависят от применяемой аппаратуры.
Например, при использовании квантометра СРМ-18 вторичное излу-
чение возбуждается трубкой с Pd-анодом и толщиной Ве-окна 0,3 мм,
а квантометра ARL-72000 — трубкой с Rh-анодом при толщине Ве-
окна 0,125 мм. В спектрометре VRA-20 флуоресценция возбуждается
трубками с зеркалами анодов из Ст или W при d0 = 1 мм. Напряже-
ние на трубке изменяется от 25 до 50 кВ. Геометрия приборов также
различна: угол падения первичного излучения может изменяться
от 40 до 90°, а угол отбора вторичного излучения — от 15 до 80°.
Следует ожидать, что значение эффективной длины волны в за-
висимости от реальных условий возбуждения будет изменяться в ши-
роких пределах. По формуле (2.24) были рассчитаны эффективные
длины волн для аналитических линий легких элементов при типичных
условиях возбуждения (табл. 6). Из таблицы видно, что для элемен-
тов, возбуждаемых смешанным первичным излучением, значения
близки к длине волны возбуждающей характеристической линии
анода трубки. Поэтому эффективные длины волн для таких элементов
при прочих равных условиях изменяются при смене материала анода
трубки значительнее, чем для элементов, возбуждаемых только не-
прерывным спектром. Например, при смене палладиевого анода хро-
мовым значения Хг для Аа-линий Mg, Al, Si, Р изменяются в
1,6 раза, а для Мп и Fe почти не изменяются.
В длинноволновой области наблюдается довольно сильная зави-
симость значений эффективной длины волны от фильтрации возбуж-
дающего излучения материалом окна трубки. На рис. 16 приведены
Таблица 6. Значения эффективных длин волн (нм) определяемых эле-
ментов стандартного образца базальта ВМ при возбуждении флуоресценции
излучением различных анодов
Материал зеркала анода Длина волны ха- рактеристической линии анода, нм Mg Al Si p К Ca Ti Mn Fe
Ка, Moq
Pd Сг 0,229 0,436 0,395 0,229 0,393 0,229 0,390 0,229 0,385 0,229 0,251 0,229 0,231 0,229 0,197 0,228 0,144 0,144 0,138 0,137
W Длина волны краев по- глощения 0,148 0,697 0,404 0,951 0,395 0,795 0,361 0,674 0,353 0,578 0,247 0,344 0,227 0,307 0,193 0,250 0,145 0,190 0,139 0,174
Примечание. Условия возбуждения: d0 = 0,3 мм, V» = 25 кВ, <р =190°,
ф = 40°,
43
Рис. 16. Зависимость значений
эффективной длины волны пер-
вичного излучения для SiTfa-ли-
нии от толщины окна рентгенов-
ской трубки с Pd-анодом.
Условия возбуждения: Vo = 25 кВ
Ф = 90°, ф = 40°,
Рис. 17. Зависимость значений
эффективной длины волны для
Si-Zfa-линии от напряжения на
рентгеновской трубке с Pd-ано-
дом.
Условия возбуждения: d0 = 0,3 мм.
Ф = 90°, ф = 40°.
зависимости значений эффективных длин волн полного и тормоз-
ного спектров трубки с Pd-анодом от толщины окна трубки при
возбуждении флуоресценции SiTTa-линий. Видно, что при изменении
d0 от 0,2 до 1,0 мм значение эффективной длины волны уменьшается
в 1,6 раза, что связано с возрастанием роли коротковолновой части
спектра за счет более сильной фильтрации длинноволнового компо-
нента.
Пределы изменений значений эффективной длины волны опреде-
ляются также величиной ускоряющего потенциала трубки. На рис. 17
приведены зависимости значений эффективной длины полного и тор-
мозного спектров от напряжения на трубке. Значения эффективной
длины волны незначительно изменяются в функции от напряжения
на трубке. При изменении напряжения от 20 до 60 кВ значение Z
Рис. 18. Зависимость значений
эффективной длины волны для
Si/C«-линии от угла падения пер-
вичного излучения на образец ср
и угла отбора вторичного излу-
чения ф.
Условия возбуждения: Pd-анод, d„ =
= 0,3 мм, Vo = 25 кВ.
44
изменяется на 0,001 нм. С увеличением напряжения на трубке эф-
фективная длина волны тормозного спектра несколько смещается в
коротковолновую область за счет смещения коротковолновой грани-
цы и максимума спектральной интенсивности. При этом эффективная
длина волны всего спектра трубки смещается в длинноволновую об-
ласть, что обусловлено усилением эффективности возбуждения ха-
рактеристического спектра анода трубки.
Величина эффективной длины волны первичного излучения бо-
лее чувствительна к изменению угла отбора вторичного излучения,
нежели угла падения первичного. Это подтверждается графиком
зависимости значений (X) от угла падения возбуждающего излучения
Ф и угла отбора флуоресценции фдля Si/fa-линии (рис. 18). При из-
менении угла ф от 70 до 90° наблюдается «выполаживание» зависи-
мости Х(ф). На основании этого можно сделать важный в практиче-
ском отношении вывод: неопределенность в значениях углов падения
в пределах 70—90°, присущая трубкам с кольцевым анодом, не приве-
дет к заметным погрешностям в вычислении поправки на поглоще-
ние. Следовательно, можно не усложнять модель расчета поправки
учетом расходимости первичного пучка фотонов, а в первом прибли-
жении положить, что ф = 90°.
Согласно приведенным результатам, значение эффективной дли-
ны волны первичного излучения изменяется в зависимости от усло-
вий возбуждения в широких пределах. На практике рентгенофлуо-
ресцентный анализ ведется при постоянных аппаратурных условиях,
поэтому эффективную длину волны нужно заново рассчитывать толь-
ко при смене аппаратуры или типа рентгеновской трубки.
2.2.2. Зависимость значений эффективной длины волны
от химического состава анализируемого материала
При заданных условиях возбуждения эффективная длина волны
зависит от состава анализируемого материала. Это влияние прежде
всего проявляется в скачках поглощения элементов матрицы, нахо-
дящихся в интервале [Хо, Х?г], которые могут привести к нарушению
однозначности X. Такой эффект в коротковолновой области отмечал-
ся в работе [83] для бинарных систем Ni—Са, Ni—Fe.
Функция /(А,), характеризующая зависимость от химического
состава и определяющая расчетные значения X, имеет разрывы пер-
вого рода из-за скачков поглощения, но она интегрируема на всем
участке длин волн [Ло, Лд;], и применение обобщенной теоремы о
среднем для вычисления эффективных длин волн в поправке на по-
глощение в длинноволновой области вполне корректно.
Влияние скачков поглощения на значение X в длинноволновой
области продемонстрируем на примере Kcz-линии кремния для би-
нарных образцов, состоящих из 10% SiO2 и 90% соответственно MgO,
Р2О6, К2О, Fe2O3. В таких системах влияние скачков поглощения,
максимально возможное при реальном анализе, их расположение в
45
Рис. 19. Зависимость фуикиио /(1) для SiKa-линип от длины волны для бинар-
ных систем.
1 — SiO2— MgO; 2 — SiO2—Р2О5; 3 — SiO2—К2О; 4 — SiO2—Ге2О3.
интервале [Xo, Х?] для Ка-линии Si различно и охватывает все воз-
можные аналитические ситуации. Система SiO2—MgO, в которой от-
сутствует скачок поглощения, взята как реперная. На рис. 19 при-
ведены зависимости /(%) для перечисленных бинарных систем и зна-
чения эффективных длин волн, получаемые с помощью теоремы о
среднем. Как видно, наличие скачков поглощения проявляется двоя-
ко. Скачки поглощения фосфора и железа на кривых 2 и 4 смещают
значение %2 и относительно значения эффективной длины волны
для бинарного образца SiO2—MgO. Скачок поглощения калия на
кривой 3 приводит к двум значениям эффективной длины волны Х3.
Из этих графиков можно сделать вывод: прерывность функции /(X)
существенно влияет на значение эффективной длины волны. Скачок
поглощения партнера по бинарному образцу с коротковолновой сто-
роны от края поглощения определяемого элемента обусловливает
появление двух значений эффективной длины волны только в том
случае, если он расположен близко по шкале длин волн к тому зна-
чению X, которое получилось бы при отсутствии скачка поглощения.
Наличие скачка поглощения калия для SiKa-линии не является
единственно определяющим. В табл. 7 приведены результаты расче-
Таблица 7. Зависимость однозначности значений эффективной длины
волны (нм) для SiKa-линии от величины скачка поглощения калия
Состав бинар- ной системы, % Л, л2 Состав бинар- ной системы, % м Кг
SiO2 К2О SiO2 К2о
1 99 0,255 0,458 60 40 0,301 0,390
10 90 0,258 0,442 70 30 0,311 0,381
20 80 0,265 0,429 80 20 0,324 0,372
30 70 0,274 0,419 90 10 0,340 0,365
40 60 0,281 0,408 95 5 0,349
50 50 0,299 0,399 99 1 0,358
Примечание. Условия возбуждения: Pd-анод, d0 = 0,3 мм, Vo = 25 кВ,
(р = 90°, гр = 40°.
46
тов эффективных длин волн для бинарных образцов, содержащих
SiO2 и К2О в различных процентных соотношениях. Как видно, при
содержаниях К2О больше 5 % имеют место два значения эффектив-
ной длины волны для Ка~линии кремния. Значит, на однозначность
эффективной длины волны бинарной системы влияет не только нали-
чие и расположение скачка поглощения, но и его величина.
Обобщая полученные результаты, можно сделать вывод о том,
что в случае бинарных систем прерывность функции /(X) существенно
влияет на значение эффективной длины волны.
Горные породы являются многокомпонентными системами, ве-
личина скачков поглощения в них значительно меньше, чем в би-
нарных образцах. Этому способствует и пока обязательное для гомо-
генизации разбавление пробы флюсом при сплавлении. Поэтому при-
менение обобщенной теоремы о среднем для расчета эффективной
длины волны аналитических линий определяемых элементов, как
правило, дает одно значение X. Исключение составляют образцы,
которые по содержанию отдельных компонентов близки к бинарным.
К ним относятся, например, фосфориты и медистые породы.
Предельные изменения эффективных длин волн для взятых ва-
риаций состава были оценены следующим образом. Значения X для
каждого определяемого элемента были рассчитаны для всех возмож-
ных составов изверженных горных пород. По результатам расчетов
построены гистограммы отклонений значений эффективной длины
волны от среднего значения X (рис. 20). Приведенные гистограммы,
а также расчеты для других элементов показывают, что пределы из-
менения X не превышают 1 % для К а- линии железа и 3 % для осталь-
ных элементов. Меньшее изменение значений эффективной длины
волны для железа объясняется отсутствием скачков поглощения с ко-
ротковолновой стороны от края поглощения этого элемента.
При рентгенофлуоресцентном анализе горные породы сплавляют
в различных соотношениях с флюсом, что влияет на значение эффек-
тивной длины волны. На рис. 21 приведены зависимости значений
эффективной длины волны от степени разбавления для й'а-линии
Рис. 20. Гистограммы отклонения значений эффективной длины волны от сред-
него значения для Ка-линий магния (7), кремния (2), кальция (5), железа (4).
47
Рис. 21. Зависимость значений эффектив-
ной длины волны для Ко,-линий кремния
и железа от степени разбавления тетрабора-
том лития.
кремния и железа в граните. Сте-
пень разбавления изменялась от 1 : 1
до 1 : 10. При этом значение эф-
фективной длины волны для 7£а-ли-
нии кремния увеличивалось на
0,0003 нм, а для линии железа оста-
валось постоянным в пределах пог-
решностей расчета. Слабая зависи-
мость значений X от степени разбав-
ления, по-видимому, объясняется
тем, что добавление таких легких
элементов, как Li, В и О, не изме-
няет формы кривой /(X). Расчеты, выполненные для всех четроген-
ных элементов, позволили сделать вывод о том, что применение од-
ного значения эффективной длины волны для различных степеней
разбавления (от 1 : 1 до 1 : 10) не приведет к заметным погрешностям
в расчете поправки на поглощение.
2.3. МАТРИЧНЫЙ ЭФФЕКТ ВОЗБУЖДЕНИЯ
2.3.1. Оценка эффекта возбуждения
в длинноволновой области
Выражение (2.14) учитывает только прямое возбуждение анали-
тической линии излучением рентгеновской трубки. Аналитическая
линия может возбуждаться косвенно флуоресцентным излучением
элементов, входящих в состав анализируемого образца. В возбужде-
нии такой вторичной флуоресценции принимают участие только те
элементы, энергия вторичного излучения которых превосходит энер-
гию возбуждения аналитической линии.
С учетом дополнительного возбуждения интенсивность аналити-
ческой линии можно представить суммой
Z, = Zf + Zj, (2.27)
/Г) tS О
г и щ—соответственно интенсивности аналитической линии,
возбужденные излучением трубки и излучением матрицы. Выраже-
ние (2.27) удобно записать в виде
Л = Z? (1 + Ti), (2.28)
где
Vi = Ii'/Il (2.29)
4В
Рис. 22. К расчету матричного эффекта
возбуждения.
образом:
Величина Z? может быть рассчи-
тана по формуле (2.14).
Найдем выражение для у.
Согласно схеме (рис. 22), интен-
сивность аналитической линии,
возбужденную излучением эле-
мента к, можно найти следующим
оо оо оо
Isik = j J f j /(X)exp(- p^/sin ср) (1/4лЯ2)
% ^=0 r=o <=0
X exp (— рц-Я) 2aRdrCirqi(i>qixhi exp (— pi^/sin ф) dx2. (2.30)
Полагая xM « рйг и интегрируя по х2, г и х2 в пределах от
0 до оо, получим
lsih = (fix, Pi, pift), (2.31)
где
^‘qk
Ф = f (2.32)
J Ц;/81Пф + {.Ц/Sill Ip’ V 7
^0
LK = (sin <p/pjln(l + p^/pftsinip) + (sini|)/pi)ln(l + pi/tuftsimp). (2.33)
Величина Z(X) определяется выражением (2.16). Тогда
Yu — ChDih,
(2.34)
Dik = 0,5pfer9fe<BgftTftiO(ptx, рг, pfe)/B(p%, рг). (2.35)
Величина уг найдется суммированием по всем возбуждающим эле-
ментам
Yi = S ChDik.
h
(2.36)
Выражение (2.36) получено в предположении, что все излучение
элемента к сосредоточено в его наиболее сильной линии. Если такое
приближение окажется недостаточно точным, то необходимо провес-
ти еще суммирование величины у по всем линиям вторичного спектра
элемента к. Параметр возбуждения характеризует дополнитель-
ное возбуждение элемента i элементом к при Сг->0, Ch -> 1.
Параметры Dih были рассчитаны по формуле (2.35) для А’а-ли-
ний элементов Na, Mg, Al, Si, P, К, Ca и Ti (рис. 23). Вклад матрич-
ного эффекта возбуждения возрастает с увеличением атомного номера
анализируемого элемента. Если для Na/Га-линии максимальное
значение Dih не превышает 20%, то для TiAa-линии оно увеличива-
ется до 50%. Зависимости Dik = /(ZA) для коротко- и длинноволно-
49
Рис. 23. Зависимость параметра Dот атомного номера «довозбуждающего»
элемента для К a-линий определяемых элементов.
Условия возбуждения: Pd-анод, Vo = 25 кВ, й0 = 0,3 мм, <р = 90°, ip = 40°.
вой области спектра имеют существенные различия. Для Ла-линий
титана, кальция, калия и фосфора наиболее сильным возбуждающим
действием обладает излучение соседних по атомному номеру элемен-
тов, а для Ко,-линий алюминия, магния и натрия — излучение эле-
ментов соответственно с атомными номерами + 2, + 3, Zt 4-
+ 5.
Появление максимумов на кривых, характеризующих зависи-
мость Dlh — f(Zh) для элементов с атомными номерами Zt < 15,
объясняется различной эффективностью возбуждения элемента к
смешанным излучением трубки, обусловленной большим различием
в длинах волн аналитических линий в длинноволновой области.
Эффективность возбуждения флуоресценции излучением трубки оп-
ределяется в основном расположением характеристических линий
анода и фильтрацией излучения в окне трубки.
На рис. 24 приведены зависимости параметра возбуждения Dih
для Nafe-линпй от длины волны излучения «довозбуждающих» эле-
50
Этот вывод подтверждается данными, приведенными на рис. 2о,
26. Па кривых, представленных на рис. 25, скачки отсутствуют. Сле-
довательно, наличие скачка на кривых зависимости = f(Zk) для
элементов, возбуждаемых смешанным излучением трубки, обуслов-
лено резким увеличением интенсивности флуоресценции «довозбуж-
дающих» элементов в результате их возбуждения характеристическим
компонентом излучения трубки. Действительно, при возбуждении
флуоресценции NaZfa-лпгшн излучением трубок с хромовым и пал-
ладиевым анодами положение скачков на зависимости Dih = /(Л) со-
ответствует значениям длин воли PdLa и Сг/йа-лнний (рис. 26).
В длинноволновой области в отличие от коротковолновой макси-
мум эффекта матричного возбуждения определяется не только бли-
зостью аналитических линий «возбуждающих» элементов к краю по-
глощения анализируемого элемента, но п эффективностью возбужде-
ния флуоресценции этих элементов первичным излучением. Относи-
тельный вклад дополнительного возбуждения излучением матрицы
существенно зависит от условий возбуждения. Значения для
Na-Ka-линцп в бинарных системах, рассчитанные для различных
условий возбуждения, приведены в табл. 8. Можно заметить, что от-
носительная доля матричного эффекта возбуждения увеличивается
с уменьшением эффективности возбуждения Na/йа-линпп первичным
излучением. Вклад матричного возбуждения повышается при замене
палладиевого анода хромовым, так как ОЛос-линия менее эффектив-
но возбуждает Уа/ба-липию, чем PdZa-лпппя. Наиболее показа-
тельно влияние фильтрации первичного излучения. Увеличение тол-
щины окна трубки с Pd-анодом от 0,1 до 1 мм приводит к возраста-
нию вклада возбуждения Na/fa-линии излучением атомов кремния
в 2 раза, а излучением железа почти в 20 раз. Чем эффективнее воз-
буждение аналитической линии первичным излучением, тем меньше
относительная доля матричного эффекта возбуждения.
В горных породах петрогепные элементы присутствуют в окис-
ной форме в концентрациях, не превышающих 90%. Кроме того,
при массовом анализе горные породы гомогенизируются путем сплав-
Таблица 8. Относительный вклад матричного эффекта воз-
буждения (%) для NaKoc-лшши при различных условиях возбуж-
дения. Уо = 25 кВ, ср 90°, ф — 40°
Состав бинарного образца do. mw
0.1 0.3 0,7 | 1,0 0,3
Z , с ~о z/. c^° Pd- НОД Сг-апод
Na Mg 12.5 13.5 14,7 15,1 18,3 14.5
Al 13.8 15.7 17.4 17,3 19,7
Na Si 13.7 17,4 20,0 21,5
Na P 13,0 18.5 22,4 24,2 21.7
Na к 3.6 8.7 22,5 26.6 18,5
Na Ca 3,0 7,7 19.7 26,0 17,6
Na Ti 2.1 4.4 15,5 22,4 15,4
Na Fe 1.0 2,3 8,3 17,6 2.7
52
Таблиц а 9. Относительный вклад вторичного возбуждения флуоресценции
(%) для аналитических линий петрогенных элементов в горных породах
Порода Na Mg Al Si P К Ca Ti Bin
Габбро-диорит СГД-1 3,5 3,6 3,2 1,4 1,3 4,1 2.1 2,1 0,1
Грани? GAI 4,1 4.4 4,2 0,5 0,5 0,6 0,4 0,6 0,1
Базальт ВМ 3,3 3,3 4.0 2,9 0,9 0,8 3,1 1,2 1,9 2,6 0,1 0,1
Глии, сланец ТВ 3,6 3,4 0,7 0,6 1,4 1,9
Сериевтинит 2,9 3.5 1,9 1,8 1, 4 0,4 1,4 4,6 1,6 2,6 2,6 3,5 0,1
Трапп СТ-1 3.6 3,3 1,3 2,8 1,2 0,9
Извсстаяк Сп-1 3,3 2,9 2,8 2,9 4,5 11,1 16,7 0,1 0,2 0,2 0
Известняк КН 4,5 4,6 4,6 4,5 0,1 0
Диатомит 4,0 5.4 5.9 0,1 0,1 0,2 0,1 0,1 0
Боксит 2,7 2,8 1,7 1,7 1,7 5,8 8,2 10,1 0,5
Фосфор:?! 5.0 5.2 5,4 5,8 3,9 13,6 0,2 0,2 0
лонпя с боратами. Поэтому, хотя максимальный вклад матричного
эффекта возбуждения отдельными элементами может быть достаточ-
но большим, вплоть до 50 %, в образцах горных пород относительный
вклад дополнительного возбуждения обычно на порядок ниже.
В табл. 9 приведены значения относительной доли дополнительного
возбуждения для нескольких типов горных пород. Как следует из
таблицы, для основных элементов изверженных горных пород этот
вклад составляет 1—6%, для карбонатов п метаморфических горных
иород — 10—17 %.
2.3.2. Упрощенное выражение
для расчета эффекта возбуждения
Отношение"функций Ф(ци, рд, [_ц)/77(цхп сц) в формуле (2.35)
можно упростить, если использовать эффективную длину волны воз-
буждающего излучения рентгеновской трубки
Ф (!-’;.! !-Ч’ J-O) (^) 77
(2.37)
Расчеты показали, что с достаточной степенью точности можно за-
писать равенство
т(7.,Г Л«р(-,урА) ’ ;
где и~ — коэффициент поглощения первичного
волны X. в окне рентгеновской трубки, р0 п
плотность и толщина окна трубки. Тогда
излучения с длипой
cl0 — соответственно
ф _ А (УХУ схр (- 4>,рл) (ж+ы ж
г ~ р (Ц)л еч> (- !1ЦрА) (Ж + Ы
(2.39)
S3
Подставляя выражение (2.39) в
уравнение (2.35), имеем
Dil; = 0,5a)qkrql.pltxklJ^h х
х (М:+У)
КУЬмЧ-Мдд) (ц.-нц’
(2.40)
где
ЛГ1 = Л'ЖРФ + Ж.ЛЧ +
+ (I/ZOMI ^ХИЦ). (2.41)
Подынтегральные функции в
выражениях для O(u?v, ц.-, ^7i)
и /’(ci?., jaJ различны. Следова-
тельно, должны быть разными и
значения эффективных длин волн
для этих функций. Данное об-
стоятельство усложняет расчет
поправки па матричное возбуж-
дение. Рассмотрим, насколько ве-
лико отличие эффективных длин
воли Ап Лф в поправках па погло-
щение и возбуждение. В табл. 10
приведены значения эффективных
длин воли А (отмечены звездоч-
кой) и Аф, рассчитанные для хими-
ческого состава стандартного об-
разца базальта ВМ. Как видно, в
основном значения эффективных
длин волн А и Аф достаточно
близки. Поэтому .можно ожидать,
что использование значения А для
расчета функции Ф не приведет к
большим погрешностям в расчете
поправки па возбуждение. В об-
щем случае аналитическая линия
может возбуждаться не только за
счет эффекта вторичной, по и тре-
тичной флуоресценции, а кроме
того, рассеянным в образце пер-
вичным и вторичным излучением,
а также оже- и фотоэлектронами,
возникшими при облучении ана-
лизируемого образца излучением
трубки. Относительный вклад тре-
тичной флуоресценции, по данным
54
работ [74, 106]. составляет 1 — 2 % в коротковолновой обла-
сти. а для элементов с -малыми атомными номерами его величина
пренебрежимо мала. Эффекты рассеяния играют значительную роль
только в случае анализа тяжелых элементов в легкой матрице [245],
а вклад возбуждения оже-электропами незначителен во всем диапа-
зоне атомных номеров [109, 209]. Согласно расчетам Эбеля и др.
[209], величина эффекта возбуждения фотоэлектронами анализируе-
мого образца составляет около 0,1 % от интенсивности, возбужденной
излучением трубки с вольфрамовым анодом. Однако расчеты Г. В. Па-
влппского и В, Т. Гуляева [109], выполненные в монохроматическом
приближении, показали, что в длинноволновой области величина
эффекта возбуждения фотоэлектронами может достигать 30 %. По-
этому важно оценить такой эффект в реальных условиях возбужде-
ния легких элементов смешанным излучением рентгеновской трубки.
2.3.3. Возбуждение рентгеновской флуоресценции
фотоэлектронами анализируемого образца
При облучении поверхности образца потоком фотонов 1(К)
число фотоэлектронов, образовавшихся в элементарном слое dx и
лежащих на глубине х под поверхностью образца, будет равно
СД(%) exp (— pytf/sin. ср) (йж/sin ср) 3 (*$д — (2.42)
q
Энергия фотоэлектрона, вырванного с уровня q элемента /,
равна
(2.43)
Допустим, что всю свою энергию фотоэлектроны теряют в пробе.
Тогда число возбужденных ими вторичных Л’-фотонов элемента i,
вышедших из пробы под углом ф, можно найти как
/; = С,А(икЖпф)2»-5-фс- (2-44)
q
При возбуждении смешанным спектром трубки
Ащах
ч
где
= ^0Agj/(Xgj ' Д)), Атах = 12,39о/(Лд,^ Eqj}.
Число квантов аналитической линии пе, возбужденных электронами,
найдем, используя метод расчета, предложенный Грином и Косслет-
том [222]:
пе = (0,22/В) - 1)1’67, (2.46)
55
где В = Zi In (1166 .£711,5 Z|), = E)„ — Eqj, Z; — атомный но-
мер элемента i. Практический интерес при разработке способа учета
матричного эффекта возбуждения представляет не абсолютное зна-
чение li, а его отношение к интенсивности аналитической линии,
возбужденной излучением трубки
ГЛ? = т', (2.47)
которое находится из формулы
= 2 см, j (2.48)
где
й« = фе2^//'((1,г, q (2.49)
а ^(Ц/д, f-ij) дано выражением (2.15). Величина Dij характеризует
относительный прирост интенсивности аналитической линии эле-
мента I за счет возбуждения фотоэлектронами, вырванными с оболо-
чек элемента / при условии, что 1. В частном случае роль эле-
мента j может играть и сам определяемый элемент.
С помощью формулы (2.48) был оцеиеп максимальный вклад
эффекта возбуждения флуоресценции фотоэлектронами для потроган-
ных элементов при следующих условиях возбуждения: трубка с
Pd-анодом и толщиной окна 0,3 мм; Го = 25 кВ; ср — 90°, ф = 40°.
На рис. 27 показаны зависимости величин от атомного номе-
ра возбуждающего элемента для бинарных образцов при условии,
что концентрация определяемого элемента Ct -+ 0, а «довозбуждаю-
56
Рис. 23. Завпсплггеть относитель-
ной интенсивности, возбужденной
фотоэлектронами, от атомного но-
мера определяемого элемента при
Z.~ 11.
1 •— о = 2 5 к Ь J 2 — То 10 к В.
Рис. 30. Зависимость относитель-
ного вклада эффекта возбужде-
ния фотоэлектронами для FA’a-
линии при возбуждении флуорес-
ценции излучением трубки с Си-
аподом, d0 = 0,1 мм (кривая 7)
и РЗ-адодом, с?й = 0,3 мм (2).
Рис. 29. Зависимость интенсивности NaZia-
и FZfa-лииий, возбужденных фотоэлект-
ронами, от толщины окна трубки с палла-
диевым анодом; Z. = 14.
щегсн элемента С f -> 1. Для элементов с атомными номерами Z > 15
доля интенсивности, возбужденной фотоэлектронами, чрезвычайно
мала, но с уменьшением атомного помора возбуждаемого элемента
она возрастает п достигает 5 отп. % для Na/Ca-miHini. При условиях
возбуждения. присущих квантометру СРМ-18, наиболее эффективно
возбуждают флуоресценцию фотоэлектроны кремния.
Зависимости = f (Z^ для элементов с атомным номером от 6
до 20 при п.х возбуждении фотоэлектронами кремния при двух на-
пряжениях 25 и 10 кВ на трубке с палладиевым анодом приведены
на рис. 28. Графики демонстрируют очень быстрый рост вклада эф-
фекта возбуждения флуоресценции фотоэлектронами для элементов
с атомными номерами меньше 10. Для фтора эта величина составля-
ет 20%, а для углерода интенсивность аналитической липни, воз-
бужденная фотоэлектронами, в 5 раз превышает интенсивность, воз-
бужденную излучением рентгеновской трубки. Такое поведение функ-
ции у2 можно объяснить падением эффективности возбуждения ана-
литических липли элементов с Z <2. 15 излучением трубки с Pd-
анодом при с7с, --- 0,3 мм. Зависимости значений функции у! для
57
Таблица И. Относительная величина матричных
эффектов возбуждения (%) вторичными фотонами и фото-
электронами
(’остан
бинарного
образца
f
Состав
бинарного
образна
Mg Si 2,19 19,36 Na Mg 4.09 13 58
Л1 Si 1.22 21.23 Na Al 427 15,72
Si Р 0,71 25,34 Na £ 4,28 17.43
Р S 0,41 28,35 Na P 4,20 18.56
S Лг 0,12 17,50 Na К 2,71 854
к Са 0 06 36 75 Na Ca 2.38 7.69
Са Ti 0.03 33,76 Na Ti 1,72 5.70
Ка-липий натрия и фтора от толщины окна трубки подтверждают
это предположение. При уменьшении толщины окна от 0,5 до 0,1 мм
интенсивность Ка-линий Na и F, возбужденная фотоэлектронами,
падает приблизительно в 4 раза (рис. 29).
На рис. 30 приведены зависимости 4i(Zj) для Ка-лшпш фтора,
возбужденной излучением трубок с медным (d0 = 0,1 мм) и палладие-
вым (do = 0,3 мм) анодами. Анализ кривых 1 п 2 показывает, что при
более эффективном возбуждении аналитической линии излучением
трубки с Cu-аподом доля интенсивности, возбуждаемая фотоэлектро-
нами, резко падает по сравнению с возбуждением трубкой с Pd-
аподом.
Следовательно, доля интенсивности. возбужденная фотоэлектро-
нами, достигает наибольшего значения при возбуждении вторичного
рентгеновского спектра жестким первичным излучением. При реаль-
ных условиях возбуждения флуоресценции максимальный вклад воз-
буждения фотоэлектронами не превышает 5 отн. %.
По данным, приведенным в табл. 11, .можно сравнить вклад воз-
буждения вторичными фотонами и фотоэлектронами Д для неко-
торых бинарных образцов, состоящих из петрогенпых элементов.
Для всех определяемых элементов интенсивность. возбужденная
вторичными фотонами, в несколько раз превышает интенсивность,
возбужденную фотоэлектронами.
2.4. ЭКСПЕРИМЕНТАЛЬНАЯ ПРОВЕРКА
ФОРМУЛЫ ИНТЕНСИВНОСТИ РЕНТГЕНОВСКОЙ ФЛУОРЕСЦЕНЦИИ
С учетом матричных эффектов поглощения и возбуждения фор-
мулу интенсивности рентгеновской флуоресценции для гомогенного
образца можно записать в виде
h = СД-(ГДС),
(2.50)
5S
где F/C) = F(ji?J, pz) (1 Выражения для F(pu, pt-) и уг оп-
ределяются формулами (2.1о) и (2.36).
Правильность расчета относительных интенсивностей флуорес-
ценции с использованием описания первичного спектра формулами
(1.2) п (1.6) была оценена в диапазоне длин волн 0,2—1,0 нм. Это об-
ласть аналитических линяй основных породообразующих элементов,
определяемых при рептгеноспектральном флуоресцентном силикат-
ном анализе горных пород минералов и других силикатных материа-
лов. Интенсивность флуоресценции вычислялась по формуле (2.49)
с учетом эффекта возбуждения второго порядка для AlKcc-, SiA'a-,
КАа- и FeAa-линпй. Результаты расчетов сопоставлялись с экспе-
риментальными данными.
Измерения проводились на спектрометрах VRA-2, VRA-20 и
опытном образце квантометра КРФ-11. Спектры флуоресценции воз-
буждались трубками типа. FS п ВХВ-9. Излучение регистрировалось
спектрометрическими каналами по Соллеру с кристаллами-анализа-
торами ADP, PET, LiF л проточными пропорциональными детек-
торами.
Токи трубок выбирались так, чтобы скорости счета не превос-
ходили 101 имн е. Измеренные значения интенсивностей были ис-
правлены на мертвое время. В качестве излучателей использовались
шлифованные диски кварцевого стекла SiO2 и монокристалла Si,
металлического А1 и прессованного порошка А12О3, таблетизировап-
пые сплавы торных пород с тетраборатом лития в соотношении 1:1.
В табл. 12 приведены для различных условий возбуждения рас-
считанные и измеренные значения отношений Аисц/Ам; /д12о /-^аь
В качестве сравнения для трубки с Мо-анодом и У=20кВ представ-
лены результаты расчета («) с использованием распределения Кра-
мерса, исправленного только на поглощение в бериллиевом окне
трубки. Характеристическое излучение А-серип молибдена при дан-
ном напряжении но возбуждается, а излучение В-серии практически
полностью подавлено окном трубки. Заметно улучшение правиль-
ности расчета при введении поправки на поглощение в аноде прибли-
зительно на 3 отн, %.
Т а б .ч и ir а 12. Сравнение расчетных относительных ин-
тенсивностей с экспериментальными данными для различных
условно возбуждения
Тип труйни и сиеьтдо.истда V,™ S 1К'.4 А! Ку.
расч. { икса. ра< ч. опсп.
БХВ-iipd; КРФ-11 25 0.405 0,402 0,370 0,371
FSCr би 35; VRA-2C 45 0,297 0,303 0.278 0,288
FSMo 60 50; VRA-2 20 0.288 0,299“ 0,283 0,273 0,282* 0,265
59
Таблица 13. Сравнение расчетных относительных интенсивностей с эк-
спериментальными данными для стандартных образцов горных порея
Порода АПЫх КЯч Т-Хч
раса. 01,СП. раса. г,ЬСП. рай. гч:сп. расч. аксп.
Гранит GM 1.064 1,091 1,797 1,759 1,450 1.548 0,225 С.213
Гранят СГ-1 1.053 1.057 1,782 1.743 1.327 1.313 0,247 0.248
Трапп СТ-1 0.948 0.942 1.056 1.055 0-234 0.232 1,354 1.325
Глинистый сланец ТВ 1.576 1,570 1.353 1.329 1.266 1.323 0.738 0.730
Базальт ВМ 1,140 1 147 1.078 1.077 0.0/3 0.071 0,992 0.990
Нефелиновая руда CIIC-1 2.042 2.064 0.818 0.836 1.082 1.102 0.332 0-344
Почва СП-1 0,843 0,814 1,838 1.801 0.780 0,79и 0,431 0.445
В табл. 13 рассчитанные п измеренные значения отношений ин-
тенсивностей флуоресценции основных породообразующих элементов
приведены для стандартных образцов горных пород со следующим
диапазоном вариации составов: SiO2 — 40—70%, А12О3 — 13—28%,
К2О = 0,7—4,7%, Ге2О3— 2—15%. Интенсивности рассчитаны и
измерены по отношению к стандартному образцу СГД-i (габбро-
диорит). Расхождение рассчитанных п экспериментальных относи-
тельных интенсивностей характеризуется средним относительным
отклонением 1,5—3%. Эти погрешности содержат воспроизводимость
гомогенизации образцов сплавлением с тетраборатом лития порядка
1% и неопределенности, обусловленные неточностью коэффициентов
поглощения.
По приведенным в табл. 14 данным можно сравнить расчетные
и экспериментальные относительные интенсивности для Ге—Сг- н
Ге—Ni-сплавов. Сроднее относительное отклонение is этим случае
на уровне 2—2,5 отн. %. Такой уровень погрешностей согласуется
Таблица 14. Сравнение расчетных относительных интен-
сивностей с экспериментальными данными для Fe—Ст- и Fe—Ni-
сплавов
Состав геК-ч нВ
(.г Ге Ki расч. иГСП. 1-асч. эасп.
3,53 90,27 0,8983 (1.8970
6.08 93,72 0.8339 0.8270
12.14 87,56 0 7009 0.5974
19,00 80,80 0,5846 0.5739
25.03 74,77 0,4984 0,4748
31,94 67,86 0.4152 0.4048
35,э8 03.22 0,3667 0,3579
14,25 84.45 0.7288 0.6998
18,97 80,41 0.6592 0.6556
21,04 78,37 0 53'90 0,6224
27,65 71.92 0.5449 0.5392
33,60 65.91 0.4757 6,4684
38,83 60,64 0,4204 0.4119
60
с результатами сравнения теоретических и экспериментальных от-
носительных интенсивностей при использовании полуэмппрпческого
способа задания спектра трубкп. Таким образом, описание спектра
рентгеновской трубки формулами (1.2), (1.6) в основном правильно
учитывает спектральное распределение и абсолютную интенсивность
первичного спектра и дает результаты, сопоставимые с полученными
значительно более сложными методами. Использование формул (1.2)
и (1.6) не требует привлечения дополнительной эмпирической кор-
рекции и, допуская простую реализацию на ЭВМ, позволяет рассчи-
тать интенсивность флуоресценции в разнообразных условиях воз-
буждения с удовлетворительной точностью.
2.5. ВЛИЯНИЕ ГЕТЕРОГЕННОСТИ ОБРАЗЦОВ
НА ИНТЕНСИВНОСТЬ АНАЛИТИЧЕСКОГО СИГНАЛА
Выражение (2.15) для расчета интенсивности рентгеновской флуо-
ресценции получено в предположении, что образец идеально гомоге-
нен, излучающий объем является представительным по химическо-
му составу для всего образца и, кроме того, образец имеет идеально
гладкую поверхность.
Поскольку единая формулировка требований к гомогенности
образца в рентгеновском флуоресцентном анализе отсутствует, сфор-
Рис. 31. Распределение 1П170ПСИВН0СТП Siлинии по поверх-
ности образца горной породы.
61
мулпруем эти требования следующим образом. Под гомогенным об-
разцом будем понимать монолитный образец, не имеющий градиентов
концентраций составляющих его элементов. Важно подчеркнуть,
что любой порошковый образен согласно этой формулировке следует
считать гетерогенным, так как пустоты между зернами создают ло-
кальные градиенты концентраций. Если образец состоит из зорен,
имеющих различный химический состав, то степень гетерогенности
образца возрастает. Именно таковыми бывают образцы из измель-
ченных горных пород и других силикатных материалов.
На рпс. 31 показано распределение интенсивности излучения
кремния, полученное электронным мккрозопдированпем. Диаграмма
микрозондпрования снята на рентгеноспектральпом мпкроанализа-
торе MS-46 при диаметре электронного зонда 1 мкм. Проба представ-
ляла собой спрессованный стандартный образец горной породы СГ-1.
Диаграмма наглядно отражает распределение S1 Айх-пзлучения, ис-
пускаемого различными зернами минералов, находящихся вблизи
поверхности образца. Приведенный пример показывает, что описа-
ние аналитическою сигнала, возбужденного в гетерогенных образ-
цах,— сложная задача. Наиболее общий подход к ее решению дают
методы численного моделирования па ЭВМ.
2.5.1. Численное моделирование
процесса возбуждения
рентгеновской флуоресценции
в гетерогенных образцах
Рассмотрим схему расчета (рис. 32), позволяющую применить
метод Мопте-Карло крошению задачи вычисления интенсивности рент-
геновской флуоресценции в кусочно-неоднородной среде, каковыми
являются порошковые образцы.
Пусть в направлении оси х вылетает фотол излучения рентге-
новской трубки. Вероятность того, что фотон испытывает первое
столкновение в интервале (х, х Ах), будет равна
ОпАх -|- О(А,:с), (2.51)
где <тп — полное сечение взаимодействия фотона с атомом — можно
/70; представить в виде
у_ / L / JIS Рис. 32. К расчету интенсивности рентгеновской флуоресценции в гетерогенном образце методом Моптс-Карло. Р{х С у ю “г Ах} = Е(х 4- Аж) — — А’(ж) = [1 — A'(x')J [Ахоу(ж)^ -Н^(Ах)]. (2.52) Здесь Р(х) — функция распределе- ния случайного значения длины сво- бодного пробега К, т. е. Р{).< х} = Fix). (2.53) Вероятность того, что фотон достиг-
62
нет точки х, будет равна
[1 - ОД]. (2.54)
Для функции F(x) можно записать уравнение
dF/dx = [1 ~ ад]пп(У). (2.55)
Интегрируя выражение (2.55), имеем
F (х) = 1 — exp j он » ds J (2.56)
Интеграл в выражении (2.56) представляет собой оптическую длину
интервала (0, х). В однородной среде оя не зависит от .г, поэтому
для такой среды
ад = 1 - ехр(ад. (2.57)
Формулу для оценки длины свободного пробега X можно записать
в виде
X = —1пу/(Уп, (2.58)
где у — случайная величина, равномерно распределенная на интер-
вале (0, 1), 1/оп — средняя длина пробега.
В к\ сочно-однородной среде интеграл для оптической длины
интервала можно разбить на сумму
U (s) ds = У в}у} = , (2.59)
3
где <jj — сечение взаимодействия в у-й области, у j — линейный раз-
мер у-й области.
Фотон испытывает столкновение в 7с-й области тогда, когда Т(Х)
удовлетворяет условию
ад< ад < адг адо)
Для оценки /(X) имеем
ад —Jay. (2.61)
Тогда длину свободного пробега определим по явной формуле
Х = 2 dii + (— Bl у — (2.62)
при
ад = —lay <6 д. (2.63)
В простейшем случае моделирование длины свободного пробега
в среде, имеющей случайный характер расположения однородных
зорей, можно разбить па следующие этапы.
1. Разыгрвыается тип фазы и размер зерен.
2. Проверяется, покинул фотон зерно или нет. Если да — то
процесс повторяется с новым значением размера зерна и с вновь
63
разыгранным типом фазы. Если фотон не покинул зерно, то разыгры-
вается .вид взаимодействия (поглощение, упругое или неупругое
рассеяние) в соответствии с вероятностями каждого процесса
РФ = <7ф /СТП» -Рр = Ср/Сп» (2.64)
где Оф — сечение фотоэлектрического поглощения, о'р —• сечение уп-
ругого и пеупругого рассеяния.
3. Рассматривается, рассеялся ля фотон. Если да, то разыгры-
вается новое направление его движения и ого энергия. Далее про-
цесс расчета повторяется для рассеянного фотона.
Разыграв определенное количество историй фотонов, можно ре-
шить достаточно сложные задачи, связанные с прохождением фотонов
через вещество. Алгоритмы, основанные на аналогичном методе мо-
делирования процесса прохождения фотонов и частиц в веществе,
применяются, например, при расчетах закономерностей прохолще-
нця пучка гамма-квантов и нейтронов в неоднородных средах со слож-
ной конфигурацией поверхностей. Ирл выборе способа расчете! по-
тока рентгеновского излучения разумно сделать некоторые упроще-
ния, сокращающие машинное время.
Основной вид взаимодействия рентгеновских лучей в диапазоне
длин волн, наиболее часто используемых при рентгеновском флуо-
ресцентном анализе,— это фотоэффект. Сечение рассеяния для рабо-
чего диапазона длин волн пренебрежимо мало. Поэтому можно пола-
гать полное сечение взаимодействия первичного фотона с атомами
возбуждаемого вещества равным сечению фотоэффекта. Это прибли-
жение освобождает от необходимости разыгрывать вид взаимодей-
ствия и историю фотона после рассеяния.
Для расчета потока флуоресцентного излучения от толстого об-
разца возбуждаемую среду можно считать полубсскопечпой и интег-
рирование вести лишь до глубины насыщенного слоя. Это позволяет
не учитывать значительной части историй, на розыгрыш которых
обычно травится много машинного времени.
Зерна образца, содержащие определяемый элемент г, будем назы-
вать /-фазами, а зерна, не содержащие этого элемента.— а-фазами.
Вероятность попадания фотона в зерно, принадлежащее /-фазе, чис-
ленно равна объемной концентрации С) о гей фазы в образце. Ана-
логично для произвольной a-фазы эта вероятность будет С\_. Путь,
пройденный фотоном в частице, разыгрывается с помощью случайной
величины у. Для частиц сферической формы формула розыгрыша дли-
ны путп имеет простой вид:
х = d/ГПу. (2.65)
Диаметр частицы может быль разыгран на основании предвари-
тельно изученной для каждого конкретного случая закономерности
распределения частиц по размеру.
Пробег фотона вычисляется в следующем порядке.
1. Разыгрывается случайная величина X
X — —In у.
64
Число у запрашивается из генератора псевдослучайных чисел, кото-
рый имеется в системе математического обеспечения ЭВМ.
2. Разыгрывается тип зерна, в которое попал фотон. Если у <2
<ZC)', то зерно считается принадлежащим /-фазе, иначе — а-фазе.
Для многофазных образцов проверяется система неравенств:
~о зерно принадлежит и-й фазе.
3. Разыгрывается длина пути в зерне и вычисляется оптическая
длина пути
= d У1 — у'-, J\ = Ух-,,
где / принимает значение либо /, либо сх.
4. Проверяется следующее условие: если X Д> J±, то фотон про-
летает частицу п необходимо повторить расчет начиная с п. 2 до
тех пор, пока X либо превысит глубину ненасыщенного слоя, либо
выполнится условие
В этом случае считается, что фотон поглотился в 7с-м зерне.
5. Проверяется, принадлежит ли k-e зерно /-фазе. Если пет, то
история считается законченной и расчет повторяется начиная с п. 1.
Если частица принадлежит /-фазе, то вероятность появления
вторичного фотона вычисляется по формуле
TF = Cir^r^a^pi, (2.66)
где Ci — весовая концентрация элемента i. т — массовый коэффи-
циент поглощения первичного излучения с длиной волны ?« в эле-
менте I, rq (SГ1 — i)‘Sq, S— скачок поглощения. — вероят-
ность перехода с испусканием аналитической линии элемента i.
6. Разыгрывается пробег вторичного фотона в соответствии с
пп. 1—4 со значениями коэффициентов поглощения для вторичного
излучения.
Если вторичный фотон с весом W достигает поверхности, то оп за-
носится в счетчик. Осредняемой ве.’шчшюй является отношение числа
вышедших фотонов к числу первичных фотонов. Блок-схема расчета
по данному алгоритму показана па рис. 33.
Результаты расчетов для однофазной (рис. 34, а) п двухфазной
систем удовлетворительно согласуются с экспериментальными дан-
ными работы [185 ] (сплошные линии — эксперимент, штриховые —
расчет). Проведенное сопоставление следует расценивать лишь как
качественное подтверждение правильности алгоритма. Для более точ-
ного сравнения результатов теории и эксперимента необходимы более
детальное изучение формы частиц минералов и флуктуаций их рас-
пределения по размеру, а также точные значения измеренной плот-
ности исследуемых образцов.
65
Процедура „Пробег”
Рис. 33. Блок-схема расчета интенсив-
ности рентгеновской флуоресцешцш ме-
тодом Моте-Карло.
Рис. 34. Зависимость интенсивности Сийбх-лшпш от размера частиц ми-
нералов.
а _ 100% халькозина; О — в смеси 1,1 % халькозина+ У8,9% кварца.
2.5.2. Влияние фактора гетерогенности
на эффект поглощения
Расчеты, выполненные методом Монте-Карло, показали, что для
образцов, не удовлетворяющих условиям гомогенности, рассчитан-
ные с помощью табличных данных коэффициенты поглощения по
будут соответствовать их действительным значениям, что, в свою оче-
редь. приведет к погрешностям в оценке эффекта поглощения.
Поскольку расчет поглощения для гетерогенных образцов пред-
ставляет собой достаточно сложную задачу и требует знания мно-
гих характеристик образца, которые зависят от физико-химического
состояния слагающих его компонентов и которые не всегда можно
установить с достаточной точностью даже после всесторонних и дли-
тельных исследований, влияние гетерогенности можно приближенно
учесть с помощью соответствующих поправочных множителей-фак-
торов гетерогенности h, стоящих при каждом из вычисляемых коэф-
фициентов поглощения.
Тогда выражение для интенсивности перепишется в виде
h ' h^i), (2.67)
где
'•qi
F = f ----------1 W (2.68)
В принципе, фактор гетерогенности может быть рассчитан мето-
дом Монте-Карло по алгоритму, изложенному выше. Выражение в
элементарных функциях может быть найдено только для простых
частных случаев.
Например, для порошкового образца с толщиной излучаемого
слоя менее диаметра одного зерна при отсутствии экранирования
зерен друг другом по первичному и вторичному излучению можно
67
получить приближенное выражение для фактора гетерогенности
(2.69)
где Ск п Cj — концентрации в k-ii п J-й фазах в образце; С\, С±, Св
п Св — соответственно концентрации элементов А и В з к-в. п у'-й
фазах. В числителе выражения (2.69) суммирование ведется по фр.захг,
содержащим в своем химическом составе элементы А и В одновремен-
но. В знаменателе суммирование ведется по всем фаза;?, составляю-
щим образец.
Из формулы (2.69) следует, что Дд = 0, если элементы А п В не
присутствуют совместно ни в одной из фаз. Если эти элементы на-
ходятся в образце совместно только в одной фазе, то — 1. Если
элементы А и 13 совместно содержатся з нескольких фазах образца,
то 0< /Ж < 1.
Очевидно, что при расчете массовых коэффициентов поглощения
с поправкой на гетерогенность необходимо соблюдать условие нор-
мировки;
(Ай-дсф -- Ср/АиЛ А C,./Au9 А • • • + Сх-/АцдТ
Ми=— , н У А--------------------------- д А . (2.70)
с’A t ~ " т' • • • тАа
Иа фактор гетерогенности в реальных пробах горных пород и
минералов оказывают влияние многочисленные характеристики фи-
зико-химического состояния компонентов, составляющих анализи-
руемый образец. Во многих случаях характеристики не поддаются
строгому математическому описанию. Поэтому рассчитанные тем
пли иным методом факторы гетерогенности следует рассматривать как
грубые приближения, требующие уточнения с помощью уравне-
ний регрессии на основе экспериментальных данных.
На рис. 35 точками показаны экспериментальные зависимости
Для SiAcz- и Л1Аа-линий. Съемки были выполнены на опыт-
ном образце рентгеновского квантометра КРФ-11 при возбуждении
образцов излучением рентгеновской трубки БХВ-9 с палладиевым
анодом. Образцы представляли собой бинарные смеси порошков
окисей SiO2 и А12О3. Средняя крупность зерен составляла 10—15 мкм
для Л12О3 и 30—40 мкм — для SiO2. Смеси порошков были спрессо-
ваны з виде двухслойных дисков диаметром 40 мм при силе давления
20 т. Воспроизводимость результатов измерений характеризовалась
средней относительной ошибкой 0,7%. Сплошной кривой показаны
результаты расчета интенсивности аналитических лшшй без учета
фактора гетерогенности, а штриховой — результаты расчетов по
формуле (2.67) с учетом фактора гетерогенности, который был рассчи-
тан для исследуемых бинарных образцов по формуле (2.69). Получе-
ны следующие значения факторов гетерогенности: А] = 1; Z?si :
= 0; 7гй = i; Ц1 = 0.
68
Рис. 35. Интенсивность Si ЛГа-япнпи (а) и .М/Сз-линии (б) й гетерогенных
образцах Sit),—ALO3.
Из сопоставления расчетных и опытных данных следует, что
введение фактора гетерогенности существенно улучшает их согласие.
Систематическое отклонение расчетной зависимости при значении
фактора гетерогенности Ag/ = 0, по-видимому, можно объяснить
тем, что формула (2.69) не учитывает взаимного экранирования зерен
образца и, кроме того, влияния плотности упаковки.
Анализ полученных теоретически?: и экспериментальных данных
показывает, что влияние фактора гетерогенности проявляется наибо-
лее заметно, если коэффициенты поглощения для аналитической ли-
нии в составляющих образец фазах сильно различаются. Например,
для SiAce-aiiHim намного отличающиеся коэффициенты поглощения
в фазах (рА12°з = 2390,3; иЬ1°2 = 687,5) обусловливают сильную
зависимость результатов расчета от фактора гетерогенности. Для
AlA'a-линии коэффициенты поглощения в фазах А12О3 и SiO2 срав-
нительно близки: цл12°з = 959,4; i.?10* = 1081,3. Поэтому и мало
отличаются результаты расчетов при значениях 7г"Н = 1 п =
Таким образом, формула (2.15) для расчета интенсивности анали-
тической линии является частным случаем при значениях фактора
гетерогенности, равных единице.
Замети?!, что горные породы отличаются исключительным много-
образием вариаций химического и минералогического состава.
На практике аналитик зачастую ire знает даже тина породы, по и в
пределах одного типа вариации химического, минералогического и
гранулометрического состава велики. Поэтому применение фактора
гетерогенности в принципе возможно только в ограниченных частных
случаях, когда мы имеем дело с большими массивами проб сходного
минералогического и гранулометрического состава. Таки?! условиям
69
удовлетворяют, например, некоторые продукты цементного произ-
водства, а также черной и цветной металлургии.
В области анализа геологических объектов единственным об-
щим подходом к решению проблемы следует считать гомогенизацию
образцов путем сплавления с соответствующим флюсом.
2.6. ВЛИЯНИЕ ХИМИЧЕСКОГО СОСТАВА ОБРАЗЦА
НА ИНТЕНСИВНОСТЬ РЕНТГЕНОВСКОГО ФОНА
Фон рентгеновского излучения, на котором регистрируется ана-
литическая линия, имеет сложную структуру п в общем случае состо-
ит из следующих компонентов: рассеянного пробой излучения трубки
рассеянного деталями прибора излучения пробы IJJ; диффузно
рассеянного кристаллом-монохроматором излучения пробы 1$,; тор-
мозного излучения фото- и оже-электронов, которые образовались в
пробе под действием излучения рентгеновской трубки, флуорес-
центного излучения элементов, из которых состоит кристалл-моно-
хроматор, Гф, и, наконец, аппаратурного компонента /ф, включаю-
щего излучение трубки, рассеянное деталями прибора, космическое
излучение и фоновые импульсы, имеющие электрическую природу.
Таким образом,
/ф — /гх1ф 4- к21ф -р А’з/ф 4- к^1ф 4- к51ф 4- кс1ф, (2.71)
где коэффициенты к отражают относительный вклад каждого компо-
нента в общую интенсивность фона, причем = К
Интенсивность первых пяти компонентов зависит от химическо-
го состава пробы п эта зависимость, по существу, представляет собой
матричный аффект.
Интенсивность рассеянного пробой компонента фона можно пред-
ставить формулой
тР >. т d(Td&
Гф = const 1К —--------——•, (2.72)
ф ли.уз1п (р 4- u /sin if v
где — массовый коэффициент поглощения для рассеянного излу-
чения. doldQ, — дифференциальное сечение когерентного и некогс-
рептного рассеяния.
Компонент /ф может быть зарегистрирован не только в первом,
но и в высших порядках отражения, не будучи полностью подавлен-
ным при работе счетно-регистрирующего устройства в дифференци-
альном режиме. В рассеянии па порошках горных пород преобладает
диффузное когерентное рассеяние. В длинноволновой области спект-
ра когерентное рассеяние в значительной мере обусловлено ди-
фракцией, определяемой кристаллической структурой образца.
Влияние межатомной интерференции в условиях расходящегося пуч-
ка излучения рентгеновской трубки приводит к появлению сложной
зависимости не только от химического, по и от минералогического
70
Рис. 36. Спектральный состав фона. Р Ге Си
1— ],ьв1о7, г — Feo, СиО. ' 1 1
Номео
состава образца. Относительный
вклад каждого из компонентов фона
зависит от конкретных аппаратур-
ных условий проведения анализа.
На рис. 36 показан спектраль-
ный состав рентгеновского фона,
зарегистрированного от образцов, состоящих из порошков Ы.2В4О7,
FeO и СнО. Съемка выполнена па спектрометре VRA-2 с кристаллом-
анализатором ADP н рентгеновской трубкой с вольфрамовым анодом,
работающей при напряжении 40 кВ и токе 20 мА. Спектр регистри-
ровался на многоканальном амплитудном анализаторе АИ-256-6.
Угол установки кристалла-анализатора при съемке на образце FeO
составлял 80”, а на образце СнО — 60и. Таким образом исключалось
брэгговское отражение характерно! пческпх линий железа и меди.
Следовательно, зарегистрированные в каналах меди и железа пики
представляют собой диффузно рассеянное характеристическое пзлу-
ченпе этих элементов.
Проба из тетрабората лития не содержит элементов, которые
имеют характеристические линии в используемом диапазоне длин
волн, и ее можно рассматривать как «пустую пробу». Фон, зареги-
стрированный от пустой пробы (кривая 1), обусловлен излучением
рентгеновской трубки, рассеянным пробой, прободержателем, дета-
лями камеры и кристаллом-анализатором.
Пики диффузного рассеяния (кривые 2 п 3) имеют «хвосты»,
которые вносят существенный вклад в фон соседних элементов.
Сильные пики в каналах фосфора обусловлены флуоресцентным излу-
чением этого элемента в кристалле-анализаторе ADP. Из сравнения
кривых 1—3 хорошо видно, что фон резко возрастает при наличии
характеристического излучения, возбуждешюго в образце. Причем
это увеличение обусловлено главным образом диффузным и флуорес-
центным компонентами фона. Интенсивность указанных компонентов
будет пропорциональна интенсивности характеристического излу-
чения, а значит, и концентрации возбуждаемых в образце элементов.
При работе на спектрометрах с фиксированными каналами фон
необходимо измерять па образцах-, не содержащих определяемые эле-
менты, Такие образцы будут иметь химический состав, сильно отли-
чающийся от анализируемых проб, и корректный учет фона в этом
случае весьма сложен. Методика учета фона на рентгеновских квап-
тометрах будет подробно изложена в гл. 5.
Глава 3
МЕТОДЫ УЧЕТА МАТРИЧНЫХ ЭФФЕКТОВ
ПРИ МНОГОКОМПОНЕНТНОМ АНАЛИЗЕ
3.1. УРАВНЕНИЯ СВЯЗИ
В РЕНТГЕНОСПЕКТРАЛЬНОМ АНАЛИЗЕ
Рентгеноспектральный, анализ дает информацию о концентраци-
ях в пробе различных элементов, т. е. является элементным анали-
зом. Первичным источником информации служит излучатель, при-
готовленный из анализируемого материала по определенным прави-
лам, которые обеспечивают определенную степень соответствия соста-
ва и свойств излучателя исходному материалу. Аналитическим сигна-
лом является измеренная электронным регистрирующим устройством
скорость счета импульсов, пропорциональная интенсивности линий
рентгеновского спектра.
Число рентгеновских фотонов, излучаемых атомами в единицу
времени, пропорционально количеству атомов, по в результате погло-
щения, рассеяния, дополнительного возбуждения и других видов
взаимодействия рентгеновского излучения с веществом, а также
вследствие влияния аппаратурных условий анализа регистрируемый
аналитический сигнал £-го элемента в излучателе зависит от многих,
подчас трудно учитываемых факторов и в общем случае имеет вид
I; = F(C, X,Y), (3.1)
где С ~ (С[, С1; . • ., Сцу-1) — вектор концентраций всех элементов,
составляющих излучатель, X — вектор фундаментальных характе-
ристик отдельных атомов (атомных номеров, длин волн излучения,
коэффициентов поглощения и рассеяния, выходов флуоресценции
и т. д.), Y — вектор, определяющий аппаратурные условия возбуж-
дения и регистрации аналитических сигналов.
Построение зависимости типа (3.1) является глобальной проб-
лемой теории репттепоспектралыюго анализа (РСА), решаемой на
основе современной физики рентгеновских лучей. Теория РСА рас-
сматривает величины С, X, Y как переменные, варьирующие в ши-
роких пределах, чтобы учитывать возможные реальные условия ана-
лиза различных материалов. Элементарные физические процессы,
сопровождающие аналитические измерения, изучены в физике рент-
геновских лучей довольно хорошо, но строгое описание всего много-
образия и совокупного влияния этих процессов весьма затрудни-
тельно.
Основная задача теории РСА, заключающаяся в нахождении
неизвестной концентрации в анализируемом образце, является обрат-
ной по отношению к построению зависимости (3.1). Поскольку мно-
гие параметры уравнения (3.1) можно зафиксировать, такая задача
72
обычно носит более частный и конкретный характер, что обусловли-
вает возникновение довольно узких направлений развития теории
РСА. Развитие их связано главным образом с упрощением исходных
физических моделей возбуждения аналитических линий, а зачастую
сводится к замене физических моделей формальными математически-
ми моделями, совсем непохожими на исходные физические модели,
но адекватно их описывающими. Таким образом, современный арсе-
нал методов расчета результатов РСА представляет собой множество
физических и математических модификаций общей зависимости (3.1)
применительно к тем или иным экспериментальным условиям и ти-
пам анализируемых материалов.
В практике РСА наиболее часто используется следующая ис-
ходная зависимость:
Л = (3.2)
являющаяся частным случаем зависимости (3.1) при фиксированных
условиях возбуждения аналитического сигнала. Конкретный вид ка-
либровочной загшетшоетк (3.2) устанавливается предварительно
либо теоретически, либо экспериментально по образцам известного
химического состава. Для калибровки прибора и расчета составов
анализируемых образцов используются интенсивности, измеренные
мо отношению к интенсивностям от стандартных образцов в идентич-
ных условиях. Стандартные образцы представляют собой пробы,
содержание определяемых элементов в которых установлено с дове-
рительными интервалами, значительно более узкими, чем требуемые
для выполнения данного вида анализа. Отличительной особенностью
рентгенофлуоресцентного силикатного анализа является то, что тре-
буемая точность определений сопоставима с точностью аттестации
стандартных образцов-
Успехи развития теории возбуждения рентгеновских спектров
привели к тому, что расчеты стали занимать лидирующее положе-
ние в методах построения калибровочной зависимости (3.2). Автома-
тизированный многокомпонентный рентгенофлуоресцентный анализ
основывается на использовании систем уравнений связи между ин-
тенсивностями аналитических линий и содержаниями элементов в
пробе. К настоящему времени предложено несколько десятков урав-
нений связи, которые позволяют эффективно решать широкий круг
аналитических задач.
Уравнения связи в РФА можно условно разделить на два типа в
зависимости от того, физическая или математическая модель форми-
рования аналитического сигнала положена в основу иолучения урав-
нения. Все уравнения связи, базирующиеся на физической модели
возбуждения, можно вывести из формулы интенсивности рентгенов-
ской флуоресценции (2.49) путем тех пли иных преобразований и
упрощений. Далее, в уравнениях связи можно выделить три класса
в зависимости от способа определения параметров и коэффициентов,
характеризующих взаимные влияния элементов в анализируемом
образце. Параметры и коэффициенты влияния могут быть вычислены
73
теоретически, найдены экспериментально или полуэмиирическпм
путем, т. е. рассчитаны, а затем некоторые из них уточнены по дан-
ным эксперимента па пробах известного химического состава. 13 урав-
нениях связи, основанных на математических моделях, численные
значения коэффициентов влияния обычно устанавливаются экспери-
ментально. Каждый из выделенных таким образом типов и классов
уравнений связи можно затем разделить на семейства, виды и разно-
видности, отличающиеся друг от друга более или менее существен-
ными деталями их практической реализации.
3.2. ФИЗИЧЕСКИЕ МОДЕЛИ И УРАВНЕНИЯ СВЯЗИ
Существует два семейства уравнений связи, полученных па осно-
ве физической модели возбуждения. 13 одном из них используется
выражение для У(С) в явной форме, содержащее такие фундамен-
тальные параметры, как массовые коэффициенты поглощения, скач-
ки поглощения, выходы флуоресценции и т. д. Этот метод коррекции
результатов анализа получил название метода фундаментальных
параметров. Другое семейство уравнений связи базируется па пре-
образовании выражения для интенсивности разложением в ряд Тей-
лора или других преобразованиях к простому виду, не содержащему
массовых коэффициентов поглощения и других фундаментальных
параметров в явном виде. Такие методы получили в отечественной
литературе название методов теоретических поправок, а в зарубеж-
ной литературе — методов коэффициентов влияния или методов
а-коррекцип.
3.2.1. Методы фундаментальных параметров
Записывая выражение (3.2) для анализируемой пробы п стан-
дартного образца, можно получить отношение
Тогда искомая концентрация определяемого элемента равна
с4 = с“(д/Лт)(^ТУ!)- (3-3)
Это уравнение является исходным для всех разновидностей методов
фундаментальных параметров. Уравнения типа (3.3) универсальны и
применимы в широком диапазоне концентраций и атомных номеров
определяемых элементов в гомогенных образцах разнообразного хи-
мического состава. Разновидности уравнений связи в методе фунда-
ментальных параметров отличаются расчетными формулами для
Криссом и Бирксом [201] величины Ft вычислялись по формуле
F = у мчжж
< и (Яй)/Мп<р-Ь ц (i,.)/Sin t '
х p + <з.4)
74
где
г м , sin <р 1ПЛ , НМ V sin 1П f-I । '1(-’ч) 'l П 5)
1,1 (1 + n^singj + и MO + HMsinM ' ’
Спектральная интенсивность I(K) интервалов ДЛ первичного
излучения предварительно вводилась в виде таблицы в память ЭВМ.
Значение 2)г-(Х;е) равпо единице, если элемент i возбуждается участ-
ком спектра с длиной волны Х?г,, и равпо нулю, если пе возбуждается.
Кармановым [60] использовалось для расчета выражение
Лосева [83]
Соотношение интенсивностей характеристического и тормозно-
го компонентов излучения трубки 7а17т определено эксперимен-
тально.
В работах [6, 7 ] величина F, рассчитывалась по формуле
О = М(1 -1-М, (3.7)
где функции f.if) и у; определены выражениями (2.15) и (2.36).
Такое представление функции I'} позволяет получить уравнение
связи, в котором поправки на матричные эффекты вводятся мульти-
пликативным способом. Действительно, подставляя выражение (3.7)
в (3.3), имеем
С1 = с“(д.Дт)
(3.8)
или
Ct =- Coik^k^. (3.9)
Здесь
С^С?(1СГ?) (3.10)
— неисправленное на матричные эффекты значение концентрации
(«пулевое приближение»),
Кг = Л(1Ъл, 14) (3.11)
— поправка па различие матричных эффектов поглощения в пробе
и образце сравнения,
Ы = (1 + 7?)/(1 + Ы (3.12)
— поправка на различие эффектов возбуждения в пробе и образце
сравнения.
75
Разделение поправок в мультипликативном методе фундамен-
тальных параметров позволяет оценить относительный вклад каждой
из них и найти разумные пути упрощения расчетных формул для
поправок на эффекты поглощения и возбуждения.
Поправка на поглощение
Относительный вклад матричного эффекта поглощения при сили-
катном анализе горных пород можно оценить отношением
(3.13)
В табл. 15 приведены результаты расчетов для некоторых типов
горных пород. Как и следовало ожидать, для всех аналитических
линий определяемых элементов вклад эффекта поглощения является
доминирующим. Поэтому при анализе горных пород основной по-
Т а б л и ц а 15. Относительный вклад матричного эффекта поглощения (%)
для Ка-линяй петрогеяных элементов
Порода Na Mg Al Si p К са Ti Мп Ге
Габбро-диорит сгд-1 97,5 97,5 97,7 98,9 98,9 96,6 98.3 97,8 99.9 100
Гранит GM 97.5 97,3 97,4 99.7 99,6 99,5 99,7 99,5 99,9 100
Базальт ВМ 97.8 97,7 97,9 99,3 99.3 97,4 98.4 97.8 99,9 100
Глин, сланец ТВ 97 6 97,4 97.8 99.5 99,5 99,0 98,9 98,4 99.9 100
Серпентинит 98,1 98,6 98.6 99,7 99,7 98.9 98,7 97,8 99,9 100
Трапп СТ-1 97,6 97,b 97,7 99,0 99.0 96,2 97.8 97.0 99,8 100
Нефелин, сиенит СНС 97,8 97.7 98.3 99,3 99,3 98.2 99.5 99,3 99.9 100
Долом, известняк Сп-1 97,1 97,5 97,6 97.5 97,5 90,3 99,9 99.9 100 100
Известняк КП 96,2 96,1 96,1 96,3 96.2 85,7 99,8 99,8 100 100
Диатомит 97,2 96,9 96,6 99,9 99,9 99,8 99.9 99,9 100 100
Боксит 97,8 97.7 98.0 98.5 98.5 94.8 93.6 90.1 99,5 100
Фосфорит 96.2 96.1 96,0 95,7 97.0 88,9 89,9 9.9 100 100
правкой будет поправка на поглощение. Значение поправки на по-
глощение для приведенных составов может изменяться в широких
пределах: от 0,9 до 1.2 для Zfa-лппий элементов Mg, Na, Al, Si, К, Са
и от 0,8 до 1,6 дляЛсс-линий Ti, Мп и Fe (табл. 16). При таких вари-
ациях погрешность за счет приближенного расчета функции погло-
щения должна существенно влиять па результаты анализа.
Поправка на возбуждение
В мультипликативном варианте метода фундаментальных пара-
метров поправка на эффект возбуждения определяется отношением
(3.12). Поэтому величина поправки Ад будет зависеть от химического
состава анализируемого образца в меньшей степени, чем величина
yi, характеризующая сам матричный эффект возбуждения. В табл. 17
76
Таблица 115. Значения поправок на поглощение при анализе горных по-
род способом фундаментальных параметров *
Порода Na Mg Al Ы к Са Т1 Мп Ге
Гранит G6I 0,918 0.931 О.Я77 0.893 1.081 1.040 1,063 0,898 0.837 0,827
Базальт ВМ 0.969 0.987 0,993 1.008 1.031 0,998 0.944 0.882 0.871 0.868
Глия, с танец ТВ 0,952 0.941 0.901 0,968 1.064 1,021 0.949 1.032 0,861 0,834 0,826
Серитглинт 0,923 0.900 1.190 1.047 1.006 0.891 0,739 0,709 0.702
Трапп СГ-1 Долом. известняк 1.024 1,022 1,010 1 -005 1.020 1.006 0.963 0,957 0,976 0.977
Си-1 0,952 0.919 1.089 0,939 0.701 0,774 0,800 0.7'Ы) 1.210 1,137 1,449
Известняк КН i .025 0.939 0.929 0.824 0.661 1.111 1,558 1.477 1,498
Диатомит 0,911 0-S92 0.838 0,771 1.127 1 ,065 1.002 0,840 0.770 0.759
Боксит 1,189 1,155 1.092 1.277 0,938 0,910 0.858 0.721 0,950 0,908
Фосфорит 1.027 1,001 0,952 0.841 0.619;! .196 0,931 1,694 1,516 1.534
='• В качестве образца сравнения здесь г, в табл. 17 использовался стандартный обра-
зец cot газа габбро-диорит СГД-1.
приведены значения поправок на эффект возбуждения потрогенных
элементов для нескольких типов горных пород. Продолы изменения
величины к/ растут с увеличением атомного номера определяемого
элемента и составляют 2—4% для Mg, Na. Al, Р и 5—9% для К и
Са. Это существенно превышает значения допустимых погрешностей
на данные элементы. Следовательно, вопреки выводам работы [13о ]
при анализе горных пород на петрогенные элементы введение поправ-
ки на эффект матричного возбуждения необходимо.
Выражение для поправки на возбуждение фотоэлектронами в
способе фундаментальных параметров можно представить в виде
к?г = (1 + Т'г)С1/(1 + Yi)> (3-^4)
где уб определяется выражением (2.48). По формулам (3.14) и (2.48)
были рассчитаны значения кв для нескольких типов горных пород
при использовании в качестве образца сравнения стандартного
Таблица 17. Значение поправок на эффект возбуждения при анализе
горных пород способом фундаментальных параметров
Порода Na Mg Al Si p К Ca Ti Fe
Гранит GM 0,999 0,998 0,997 1,011 1,007 1,031 1,014 1,018 0.996
Базальт ВМ 1,002 1,002 1,003 1,004 1.004 1,009 1,001 1,000 1,000
Глпн. сланец ТВ 1,001 0,999 1,001 0.996 1,006 1,025 1,005 1,006 1,000
Серпентинит 1,005 1,012 1,010 1,008 1,008 1,024 1,004 1,001 1,000
Трапп СТ-1 1,000 1,000 0,999 1,200 1.000 0,997 0,995 0,992 0,999
Долом, известняк Сп-1 0,996 1.001 0,998 0.986 0.985 0.935 1.016 1.021 1,001
Известняк КН 0.987 0,985 0.984 0.974 0,973 0,888 1,016 1,021 1.001
Диатомит 0.996 0,994 0.989 1.011 1,010 1.034 1,017 1,022 1.001
Боксит 1,002 1,002 l,00S 0.996 0,996 0.982 0.953 0,930 0.996
Фосфорит 0.975 0,985 0,982 0 967 0,980 0,921 1,016 1,021 1.061
77
образца габбро-диорита СГД-1. Расчеты показали, что вклад поправ-
ки ке составляет для горных пород, сплавленных в соотношении
1:1с тетраборатом лития, 0,01—0,25 отн. %. Следовательно, при
массовом анализе горных пород эту поправку можно не учитывать.
3.2.2. Мультипликативный метод AF-коррекции
Способ фундаментальных параметров успешно опробован при
анализе материалов разнообразного состава: сплавов па основе желе-
за и никеля [203], сталей, покрытия сварочных электродов и шлаков
[60], осадочных пород [221]. лунных образцов [274], сплавов на ос-
нове меди и железа [247], сварочных шлаков и флюсов [59], разно-
образных типов горных пород на основные породообразующие эле-
менты [6. 272. 274]. Авторы этих работ отмечают, что способ фунда-
ментальных параметров в принципе не имеет ограничений на область
применения в отношении состава анализируемых объектов. Однако
неопределенность в значениях коэффициентов поглощения, выходов
флуоресценции и спектральном распределении первичного излуче-
ния; применимость только к гомогенным образцам; приближенность
лежащей в основе этого способа физической модели, которая недоста-
точно точно описывает процесс возбуждения и регистрации интенсив-
ности флуоресценции в образце, не позволяют полностью реализо-
вать потенциальные возможности метода фундаментальных парамет-
ров при реальном анализе. Тем не мопсе при анализе образцов с ши-
роко меняющимися содержаниями компонентов он является наибо-
лее гибким, не требующим большого числа стандартных образцов
и во всех случаях допускающим автоматизацию трудоемких аналити-
ческих процедур. Рассматриваемый метод не получил широкого
распространения в аналитической практике также и потому, что для
расчета концентраций таким способом требуются ЭВМ с большой
оперативной памятью, в то время как современные рснтгеносиект-
ральные приборы оснащены управляющими ЭВМ малой мощности.
Поэтому он часто применяется для предварительных оценок. Авто-
рами [3, 12, 13, 115, 116] разработана модификация метода фунда-
ментальных параметров с целью его практического использования
на рсптгенофдуоресцентных спектрометрах, управляемых мини-
компыотерамп.
Упрощенная поправка на поглощение
Для упрощения поправки на поглощение воспользуемся форму-
лой (2.25) для Z-Д полученной в монохроматическом приближении.
Тогда
СТ
(3.15)
Здесь и кД — эффективная длина волны для анализируемой про-
бы и образца сравнения соответственно.
78
Основной трудностью в использовании выражения (3.15) для
количественного анализа является то, что эффективную длину вол-
ны Х; необходимо вычислять для каждой анализируемой пробы в
каждом цикле расчета методом итераций. Такая процедура сводит
на нет все преимущества от упрощения поправки на поглощение.
Однако приведенные в предыдущей главе результаты позволяют
сделать важный в практическом отношении вывод: при постоянных
условиях возбуждения эффективная длина волны слабо изменяется в
зависимости от химического состава. Поэтому при расчетах поправ-
ки на поглощение можно использовать одно значение %, равное эф-
фективной длине волны образца сравнения, т. е.
х = Йт = М-
Тогда поправка на поглощение преобразуется к виду
кц = ('/.j- + 7й)/(7.х + Х1)ет- (3.16)
Это существенно упрощает процесс вычисления концентраций, так
как можно заранее рассчитать значение эффективных длин волн
определяемых элементов для всех используемых в лаборатории
стандартных образцов.
Упрощенная поправка на возбуждение
Значение функции определяется концентрацией «довозбужда-
ющего» элемента и параметром Dih, зависящим от состава всего
образца. Учитывая малый вклад эффекта возбуждения, при массовом
анализе рационально пользоваться матрицей значений для об-
разцов сравнения.
В табл. 18 представлены значения для габбро-диорита СГД-1
п гранита СГ-1.
Использование параметра Dih, рассчитанного для образца срав-
нения, позволяет получать сравнительно верные значения величины
Таблица 18. Значения функции матричного возбуждения уг (%), рас-
считанные при условии, что = Z>^
сгд-i
£
Mg
Al
Si
К
Са
Ti
Мн
0,0247
0,0235
0,0134
0,0328
0,0179
0,0239
0,00106
0,0262
0,0252
0,0144
0,0321
0,0194
0,0280
0,00117
0,0272
0,0262
0,0149
0,0315
0,0204
0,0305
0,00121
0,0258
0,0237
0,0151
0,0335
0,0211
0,0304
0,0014
0,0247
0,0237
0,00445
0,00426
0,00353
0,00449
0,00028
0,0221
0,0209
0,00390
0,00394
0,00326
0,00387
0,00025
0,0235
0,0223
0,00422
0,00414
0,00338
0,00424
0,00027
0,0215
0,0196
0,00436
0,00447
0,00356
0,00458
0,00032
79
аффекта возбуждения. В предположении, что параметры Dik для
пробы и образца сравнения равны, поправка на возбуждение примет
компактный вид:
— / 1 + 2 + 2 С/Л/Л (6.17)
к Л\ /
где Ch п Ch — соответственно концентрация довозбуждающего
элемента в пробе н образце сравнения. Суммирование в формуле
(3.17) производится по всем элементам, составляющим матрицу,
с учетом, что
Dik = 0 при > kqi.
Это условно связано с тем. что возбуждать элемент i могут только
лпппп, расположенные с коротковолновой стороны от края погло-
щения (/-уровня элемента i.
Если аналитическая линия возбуждается всеми линиями (/-се-
рин элемента /г, то
Рк = 1-
Если в возбуждении принимает участие только часть линий (/-серии
элемента к, то
Pk < 1
Конкретные значения рк в этом случае определяются статистическим
весом линий, участвующих в возбуждении.
Таким образом, упростив выражение на основе исполь-
зования эффективной длины волны и полагая 2 == ?4'г, имеем сле-
дующее уравнение связи:
Уравнение (3.18) может использоваться для расчетов результатов
анализа многокомпонентных образцов с помощью управляющих
ЭВМ с оперативной памятью, не превышающей 16 К, без внешних
устройств памяти. Расчет может выполняться в реальном масштабе
времени.
3.2.3. Методы ^-коррекции
Общей чертой рассматриваемого в этом разделе семейства мето-
дов введения поправок на матричные эффекты является то. что
поправка на поглощение аналитической липин Цг вводится в явном
виде, а поправки па эффекты поглощения первичного излучения,
матричного возбуждения и другие эффекты — в неявном, обычно в
виде коэффициентов, численные значения которых находятся экспе-
риментально на пробах известного состава. Методы ц-коррекции
можно рассматривать как промежуточные между методами фунда-
ментальных параметров и методами а-коррекции.
80
Метод Холланда и Бриндли
Полагая в формуле (3.18) и пренебрегая матричным
эффектом возбуждения, имеем
Ci = Со4ц/рГ. (3.19)
Формула (3.19) была положена в основу метода Холланда н Бриндли
[232] для расчета результатов силикатного анализа горных пород.
Вычисленные методом итераций значения концентраций затем уточ-
нялись с помощью регрессионного уравнения
(Ji = Ло “И (L]Ci -[- Qj^Ci' (о.20)
Коэффициенты tz0, а1? а., находились методом наименьших квадратов
по данным эксперимента на группе стандартных образцов. По пра-
вилу Крамера
(3.21)
2Ж? s с? ,£ц
2 с А 2 е? 2 с,
2 C'l 2 С; п
2 с? 2 е? 2 е?
2с? 2с? 2с,
2Ц 2с, »
где п — число измерений величин Ct п С\. С помощью регрессионного
уравнения (3.20) результаты анализа корректируются па матричные
эффекты, не учитываемые соотношением (3.19). Метод Холланда и
Бриндли был исиользоаан Симаковым [135] для анализа минераль-
ного сырья.
81
Метод Андермана
Считая приближенно, что вторичное рентгеновское излучение
возникло на некоторой «эффективной» глубине pzt под поверхностью
анализируемого образца, и учитывая поглощение аналитической
линии на пути к поверхности образца, выражение для интенсивности
аналитической линии можно представить в виде
Ii = Iai exp (—(3.22)
Аналогично интенсивность аналитической линии для стандартного
образца запишется как
Л’ = /йехр(—рг"|цт), (3.23)
т. е.
д = Лоехр(-рЦч) г 9 ,,
Возьмем в качестве стандартного образца бинарную смесь, состоя-
щую из элементов i и к. Пусть концентрация определяемого элемен-
тами этом бинарном образце будет равной его концентрации в анали-
зируемой пробе, т. е. CCiT = Ci. Тогда, учитывая, что pi = У, цу-Су
и принимая приближенно рх = рхст = kt, из (3.24) получим форму-
лу Андермана [1681 для корректировки интенсивности аналитиче-
ской линии на матричные эффекты
Ii = /техр['—kt (|*у— (3.25)
где hi = fjii.
Концентрации определяемых элементов находят по зависимости
Iik — которая строится предварительно по бинарным образцам,
состоящим из элементов i и к. Коэффициент kt находится эксперимен-
тально па искусственных смесях и полагается в дальнейшем не за-
висящим от химического состава анализируемого материала. При та-
ком методе определения коэффициента kt на него падает нагрузка по
учету всех факторов, которые но корректируются с помощью коэф-
фициентов поглощения аналитической линия.
Метод Ильина
В методе Ильина [531, так же как и в методе Андермана, поправ-
ка на поглощение вводится в экспоненциальной форме. Но в отличие
от Андермана Ильин учел поглощение первичного излучения и за-
висимость эффективной глубины образования вторичного спектра
от химического состава.
Учитывая в выражении (3.22) поглощение первичного излуче-
ния, имеем
/г = const Ci exp [— pXj (py/sin ср 4- j.q/sin ф)]. (3.26)
82
Аналогично для стандартного образца
7°т = const С"’ exp рту sin ср 4- рДт/в1п ip)
(3.27)
Тогда
(3.28)
Зависимость величины от поглощающей способности образца опи-
сана показательной функцией вида
pxj - a exp [— b (p-^/sm ср 4- рц/sin ф)] 4- d, (3.29)
где а = 7.5-10“3, Ъ = 2-10—3, d 0,3-10~3 (г/см2). Указанные зна-
чения коэффициентов найдены Ильиным по экспериментальным дан-
ным. В формуле (3.29) используются только абсорбционные характе-
ристики пробы п пет таких параметров, как атомный помер элемента,
его атомная масса п плотность. Эти параметры учитываются косвенно
в коэффициентах поглощения. В качестве образцов сравнения при
анализе методом Ильина используются чистые элементы.
Метод Кузьминой и Туранской
Аналитическим уравнением, описывающим зависимость интен-
сивности рентгеновской флуоресценции от концентрации определя-
емого элемента, является параболическое уравнение вида
Л = bCt (3.30)
очень широко используемое в оптическом эмиссионном спектральном
анализе. В логарифмической записи эта зависимость имеет вид
lg Ц = 1g Ъ 4- a 1g Сг. (3.31)
В качестве параметра, определяющего значение величии а, Ъ, а сле-
довательно, и ход зависимости Ц = /(СЗ), Кузьминой и Туранской
[75, 152, 153] был выбран массовый коэффициент поглощения рг.
Для нахождения аналитической формы зависимостей а(р;) и
5(р,) разложим функцию ]g Ц в ряд Тейлора в окрестности точки
= р^ при Ci = const. Ограничиваясь первыми членами ряда
Тейлора, имеем
lg Ii (1ч) = lg ii (1ч) + [1g Л (|ч)4=ин (1ч — цЛ = 1g Н11™) +
+ а (|ЛН) lg Cl + [1g Ъ (11<)Ы->ЧI (14 — 14i) + [«(идЫ-щ; lg Ci (14— 14i)-
(3.32)
Так как значения функций 5(pj) п а(рг) и их производных в точке
83
= р... являются константами, то, вводя обозначения
Ь (lli) ~ Bp, a (j-lj) = D-i’, [1g Ъ (pi)]|-4=Hii ~ /9
(o.oo)
[a (р?)]^=р,^ =,Pij
выражение (3.32) можно переписать в виде
1g Ц = Bi + Dt 1g Ci -Ь «К1Ч — 14i) 4- Pi lg Сг(рг — liu)
(3.34)
ПЛИ
1, c • = (3 35)
g ' Oi+Mth-M • ( ’
В случае, когда коэффициент поглощения для пробы совпадает
с коэффициентом поглощения для чистого элемента получим
уравнение
Di^Ci = lgIi~ (3.36)
которое должно совпадать с линейной зависимостью интенсивности
рентгеновской флуоресценции от концентрации
1g Л - lg IQ + lg Cit (3.37)
Из (3.36) и (3.37) имеем
Di\%Ci + Bi^Ki + ^Ci. (3.38)
Откуда
Di = l\ В{ = \^Кг = \^11 (3.39)
Здесь 7^— интенсивность апалитпческоп линии в пробе с содержа-
нием определяемого элемента, равпым 1%, и массовым коэффици-
ентом поглощения pij = yiff. Таким образом,
В случае Ci = 1% имеем
lg It = Ig/? + ctj (p-i — i-iji). (3.41)
В выражении (3.40) величина а.г(и^ — pif) представляет собой по-
правку, обусловленную изменением интенсивности аналитической
линии с вариацией коэффициента поглощения пробы при 1 %-пом
содержании определяемого элвхмента, а величина 1 + Рг-(Цг ~ Цгг)
характеризует кривизну зависимости 7г = f(Ci) и концентрацион-
ную чувствительность.
Переходя к натуральному логарифму, имеем
Ь =/’ехр[а;(,ч —(3.42)
Входящие в уравнение (3.42) константы аг, |3j п 7? можно определить
на небольшом количестве образцов с известным химическим составом.
84
Метод стандартов-бинаров
Если пренебречь поглощением первичного излучения и в каче-
стве образца сравнения взять бинарную смесь, состоящую из опреде-
ляемого элемента (с концентрацией, равной его концентрации в ана-
лизируемой пробе) п некоторого к-то элемента, то выражение (3.18)
можно переписать в виде
Откуда получим уравнение для коррекции интенсивности
гг-[1 + - Ы Ci]. (3.48)
При анализе методом стандартов-бинаров [37 J предварительно
па искусственных смесях строятся графики зависимости
ЛТ = /(С,..). (3.49)
Второй компонент бинарных смесей выбирается таким, чтобы сбли-
зить по составу образцы сравнения и анализируемые пробы. Коэф-
фициенты kt определяются по экспериментальным данным на образ-
цах известного состава. На практике обычно используются средние
значения коэффициентов kt.
За начальные значения концентраций в рассматриваемом методе
берутся концентрации, найденные по измеренным интенсивностям с
помощью графиков (3.49). Итерационный процесс в методе стандар-
тов-бинаров можно интерпретировать как использование образца
сравнения переменного состава, содержание определяемого элемента
в котором уточняется и шаг за шагом приближается к концентрации
в анализируемом образце.
85
Метод стандартов-функций
Применение метода стандартов-бинаров ограничено рядом недо-
статков, к числу которых следует отнести необходимость изготовле-
ния большого числа бинарных смесей, определение коэффициентов в
уравнениях связи, построение аналитических графиков и контроль
за их поведением при изменении условий работы.
Указанные трудности могут быть в значительной мере устране-
ны путем сближения химического состава стандартов и анализиру-
емых проб. Для этого введено понятие стандарта-функции [107], в ко-
тором концентрация определяемого элемента равна таковой в иссле-
дуемой пробе, а концентрации у-х элементов наполнителя являются
однозначными функциями Ch т. е.
Cj = f(Ci4 aj), (3.50)
где dj — параметр, который может быть определен с учетом априор-
ной информации о химическом составе анализируемого материала.
Концентрация наполнителя стандарта-функции может меняться
за счет одновременного изменения концентраций С;- всех у-х элемен-
тов, так что
Ci = СД1 - C^a-Ci), (3.51)
где Ci и Cj — концентрация элементов i и у, характеризующая сред-
нее содержание в анализируемом материале. При этом величина
ty.j в выражении (3.50) определяется как
= СД1 - Ц) (3.52)
и имеет смысл концентрации у-го элемента в наполнителе среднего
состава. В таком случае зависимость интенсивности аналитической
линии от концентрации определяемого элемента для стандартов
функций, как и для обычных бинарных систем, является однознач-
ной. Параметры этой зависимости можно рассчитать с помощью фор-
мулы интенсивности рентгеновской флуоресценции (2.49).
Поскольку различия величин матричных эффектов для анализи-
руемого материала и стандартов-функций при правильном их выборе
невелики, коэффициент ki в уравнении (3.43) можно положить рав-
ным единице. Тогда
Лт'Ф = Д(.щ/!1ГФ). (3.53)
Учитывая, что по условию (3.51)
нГ* =С^и + (1-С;)У, (3.54)
имеем
2сдаУ(1-л), (3.55)
где ji;, — массовый коэффициент поглощения наполнителя
стандарта-функции, элементов i и у на линии элемента I соответ-
ственно.
86
С учетом (3.54) и (3.55) выражение (3.53) преобразуется к виду
„ л Г <3'« 5i) ci 1
4*-* = Ц 1 + ^=1--------------I (3.5(5)
L ^(1 - Су nJ
Такие уравнения, записанные для всех определяемых элементов,
позволяют получить систему, которая решается относительно кон-
центраций этих элементов.
Заметим, что когда в качестве наполнителя стандарта-функции
выбран основной элемент (т. е. pj — р^), то выражение в квадрат-
ных скобках формулы (3.56) переходит в формулу (3.48), использу-
емую для коррекции результатов анализа методом стандартов-
бинаров.
Метод Ревенко и Лосева
Выберем в качестве стандарта образец с концентрацией определя-
емого элемента Сг = 100 %. Тогда выражение (3.18) запишется в виде
с 1
С”“% (1-2"Л) "М ’
(3.57)
где п = sin cp/sin ф, или
Ci = jj/j™-") (i +
Обозначая
(3.58)
(3.59)
и учитывая, что р$ = С^рц Ч- У Ст5-р^ н = 1 — У Cj, получим
формулу Ревенко, Лосева и др. [122, 124] для коррекции результатов
анализа на матричные эффекты
с, = [3 +
(3.60)
Коэффициент ki в методе Ревенко и Лосева определяется по данным эк-
сперимента на образцах известного состава, близких по составу анали-
зируемым пробам. Число таких образцов зависит от вариации химиче-
ского состава анализируемых проб. После нахождения зависимости
ki от химического состава коррекция результатов анализа выполня-
ется с использованием соотношения (3.60) .методом последовательных
приближений.
3.2.4, Методы «-коррекции
В рассмотренных выше методах AF- и р-коррекции выражения
для ТфуЛ’г, учитывающие матричные эффекты, получены главным
образом за счет упрощения модели возбуждения рентгеновской
87
флуоресценции. В методах «-коррекции это выражение упрощается
путем разложения величины /Ft в ряд Тейлора в некоторой точке
пространства концентраций в окрестности стандартного образца
~ 1 Zl I F. ОС L 'у '1 + 2 “ A ОС: ОС.
г з^т \ ‘ ' £г__ q ст \_ 1 J н
1 Л'.
-Ц^_АС^+--- Р-И)
Здесь Д^ = С5-С-Т; \Ck = Ck-Ccl. (3.62)
В качестве влияющих рассматриваются все элементы, кроме одного,
произвольно выбранного элемента т. Удобно выбирать т — I.
Обозначая коэффициент влияния j'-го элбхмонта на интенсивность
аналитической линии 4-го элемента как
(3.G3)
и ограничиваясь первым членом разложения (3.61), имеем
F"/Fi = 1 + S a-c&Cj. (3.64)
jri
Подставляя (3.64) в (3.3), получим базовое уравнение для разновид-
ностей методов «-коррекции
с г = С" (li/I") (1 + 2 <+;Асф (.3.65)
Выражение (3.65) может быть получено также разложением в ряд
Тейлора формулы для интенсивности, как это было выполнено Ши-
райвой и Фуджпно [296, 297]. Уравнение (3.65), по существу, явля-
ется приближенной формой (3.3), а выражение (3.63) — дифферен-
циальной формой записи формулы (3.1). Числовые параметры урав-
нения (3.65) представляют собой значения дифференциальных коэф-
фициентов влияния (3.63), рассчитанные для стандартного образца,
т. е. уравнение (3.65) становится строгим равенством при С = ССТ.
Следовательно, использование этого уравнения в общем случае спра-
ведливо при сравнительно малых значениях ДС.
Многочисленные разновидности методов «-коррекции различа-
ются между собой учетом числа членов в разложении (3.61), диффе-
ренциальной или интегральной формой записи коэффициентов влия-
ния «/;, а также записью формулы коррекции на матричные эффекты
либо через концентрации, либо через интенсивности влияющих эле-
ментов. Рассмотрим наиболее распространенные виды методов а-
коррекции.
88
Метод Битти и Брисси
Полагая приближенно
Л = АДр;, (3.66)
имеем
А = A'jCj/pj, (3.67)
(3.68)
где
Н = Нм + sin ф/sin ф. (3.69)
Поделив (3.68) на (3.67), получим
Rt = СрцДсц. (3.70)
Здесь
Ri = (3.71)
Из (3.70) следует
Cj = (3.72)
Учитывая, что
Цг = 2 CjUjj, (3.73)
i=M
имеем
Ci = Ri (Сцтa + Cj Pij'jl P-ii- (3.74)
Тогда
Ci = Ri(ci+ S.C^ (3.75)
или
Ci = [Z?/(l-^)]ZC^, (3.76)
где
= Pzj/pjj. (3.77)
Битти и Брисси записали уравнение (3.75) в впде
-[(/““7a)-i]ci + 2«wc^o. (3.78)
Коэффициенты определялись на искусственных бинарных
смесях, каждая пз которых использовалась для нахождения коэф-
фициентов с.ц и Таким образом, для ^-компонентной пробы тре-
бовалось п(п — 1) бинарных смесей.
Система решалась с помощью определителей исключением из
системы уравнений типа (3.78) уравнения, содержащего интенсив-
ность линии элемента с наивысшей концентрацией. В этом случае
решение зависит от того, какое уравнение исключено из системы.
Барнхэм, Хауэр н Джонс [191] предложили умножать величины,
обратные иптепспвпостям, на корректирующий множитель, полу-
ченный приравниванием к нулю определителя системы (3.78). Тогда
89
решение единственно и нс зависит от того, какое уравнение исклю-
чено из системы. Блохин и Друзь [26] в качестве конечного резуль-
тата брали среднее значение решений, полученных путем поочеред-
ного исключения из (3.74) всех уравнений. Фатеми и Бирке [212]
показали, что способ решения зависит от соотношения величин чле-
нов системы (3.78) и его определителя. Если определитель мепыие па
порядок любого члена, то решение единственно. Если определитель
больше некоторых членов или сопоставим с суммой членов, то урав-
нения не имеют решения. Если определитель меньше членов урав-
нения, но сопоставим с ними по величине, то интенсивности необхо-
димо умножить на постоянную величину для приведения определите-
ля к нулю.
Метод Битти и Брисси существенно развит и усовершенствован
Блохиным, Дуймакаевым и Друзем. В варианте Блохина и Друзя
[26] анализ ведется по стандартному образцу с равным содержанием
исследуемых элементов. При таком выборе образца сравнения вели-
чины cc/j полностью характеризуются коэффициентами поглощения
всех составляющих пробу элементов, поэтому они должны находить-
ся па многокомпонентных пробах. Минимальное число таких проб
должно равняться числу определяемых элементов, а их состав охва-
тывать возможные вариации состава исследуемых проб, чтобы точнее
учитывать матричные эффекты. Чтобы уменьшить влияние ошибки
измерений интенсивности на правильность значений коэффициентов
надо использовать как можно большее число стандартов. При
этом система, записанная относительно коэффициентов ui} п допол-
ненная условием 2 Ci = 1, должна решаться методом наименьших
квадратов.
В варианте Блохина и Дуймакаева [27] анализ ведется по образ-
цу сравнения произвольного состава. Если вариации состава ана-
лизируемых проб несущественны, то за образец сравнения можно
брать пробу, близкую по составу к исследуемым материалам. В этом
случае экспериментальные коэффициенты в уравнении изменяются
весьма мало и определяются на пробах известною состава класса
анализируемых материалов.
Преимущество варианта Блохина и Дуймакаева перед вариантом
Блохина и Друзя заключается в целесообразности выбора стандарт-
ного образца и обусловленной этим лучшей точностью.
Неустойчивость способа решения системы уравнений Битти и
Брисси привела к весьма ограниченному практическому применению
этого метода. Более удобный и общий подход к решению систем урав-
нений связи в методе «-коррекции был развит Лачансом п Тоейлом
[251—253].
Метод Лачанса и Трейла
Примем, как и в предыдущем разделе,
Ft == 1/Ц.
(3.79)
90
Тогда
Iг — Сг/|-Ч — С J \ С ilia -р S Cjflfj
(3.80)
Учитывая, что Ci : 1 — 2 получим
- (С;/ц jf l + 2 c/iJ~ (3-81>
I \ J=#i 1-Чг I
Обозначая
a-ц = (HiJ — (3-82)
имеем
A = ^CV"7- (3'83’
1 + X auci
Отношение (3.83) к интенсивности чистого элемента равно
W“% = С,Д1+ 2«ЦС;)
Отсюда получаем уравнение Лачанса и Троила
С; = ;Ц1+ S«yC;y
Если образец сравнения имеет произвольный состав, то
т ,тп- = ci ( АД;.)
откуда
с! = Г(ДАт)(1 + ЗсчА),
(3.84)
(3.85)
(3.86)
(3.87)
где iCi = Величина постоянного множителя в уравнении
(3.87) зависит от химического состава образца сравнения.
Легко заметить, что коэффициенты влияния в уравнениях Ла-
чанса и Трейла п в уравнениях Битти и Брисси связаны соотноше-
нием
Коэффициенты влияния в уравнении Лачанса — Трейла являются,
по существу, интегральной формой коэффициентов влияния (3.63)
уравнения (3.65) при разложении величины dF-JdCi в точке, равной
[С} = 1, Ci = 0).
91
Система уравнений Лачанса и Трепла решается методом итера-
ций, что позволяет избежать затруднений с вычислением определи-
телей малой величины в методе Битти и Брисси. Уравнение Лачанса
и Трейла предполагает, что поправки на матричные эффекты адди-
тивны и что величина поправки пропорциональна концентрации
влияющего элемента. Последнее обстоятельство приводит к недоста-
точно корректным результатам при наличии матричного эффекта
возбуждения. Большей аппроксимирующей способностью при нали-
чии разнообразных матричных эффектов обладают уравнения
Расберри — Хайнриха и Клайссе — Квинтипа.
Метод Лукаса-Туса и Прайса
Согласно (3.83) выражение для интенсивности аналитической
линии Ц можно записать в виде
(3'88)
где ац — (цу — Бзяв отношение интенсивности аналитиче-
ской линии, зарегистрированной от анализируемой пробы, к ее ин-
тенсивности, зарегистрированной от стандартного образца, получим
<3-89)
3
Сг = li (рн/рТ) fl + S ’’ Л ) + G'-ii |-Ч ) 2 i I’
\ j I L j J
(3.90)
где Zj = ijl^. Полагая приближенно
Ij = kjCj (3.91)
и учитывая интенсивность фона рассеянного излучения, равного
/ф = (3.92)
из (3.90), (3.91) и (3.92) имеем
Ci = Ц ^(рн/[4т) + (рй/рГ) (3.93)
Обозначая
Ci-zO = = Pn/pi ’ Kij = aij ( Цii/pг ),
получим уравнение Лукаса-Туса и Прайса
Ci = ai0 + Z^cci + ZwijZ^. (3.94)
92
В уравнении (3.94) используются непосредственно измеренные ин-
тенсивности аналитических линий и для вычислений не требуется
решения системы уравнений. Поэтому’ вычисления могут проводить-
ся л без использования ЭВМ. Простота метода обусловила его широ-
кое практическое применение. Вместе с том приемлемость уравнения
Лукаса-Туса и Прайса ограничена теми рамками, в которых спра-
ведливо допущение (3.91). В общем же случае интенсивности Ij за-
висят от химического состава анализируемых образцов, что не учи-
тывалось при выводе уравнения. Это обстоятельство ограничивает
использование метода Лукаса-Туса и Прайса группами проб с не-
большими вариациями химического состава.
Метод Расберри и Хайнриха
Интенсивность аналитической липни определяемого элемента
можно представить согласно (2.27) сулимой двух компонентов:
А = + Л- (3.95)
Пренебрегая поглощением первичного излучения, первый член сум-
мы (3.95) можно представить в виде
// = kiC'j/u-i. (3.96)
Найдем приближенное выражение для второго члена. Согласно
(2.36) и (3.96),
= (3.97)
где
= CiHki(O,3-pkrqi(nqirqh&qk)-cp. (3.98)
Здесь Ф определяется выражением (2.32). Полагая приближенно,
что In (1 — г) х, для Lx из выражения (2.33) получим
1ДЦ. (3.99)
Тогда, считая, что iiz цл. для Dik в монохроматическом прибли-
жении получим
Dik^ (З.ЮО)
тдо
Pih = о,5/jfer(2i0Ijfrtl,l!(0Qj.Z?viiu siii^. (3.101)
Величина Р-1к не зависит от химического состава, следовательно,
для приближенной оценки величины можно записать выражение
(3.102)
Тогда
Г = (Жо)2ЛАим.Ъ- (3.103)
Поскольку Г”!“ г - то
Г = + (СГ(Н) 2 (3.104)
93
Учтем, что
pi — ^ipii + 2 CjPv + 2 (3.105)
[is - С^,л + 3 С^и + з с„ц;л, (3.106)
Ci- l-SC;-2Cs- (3.-107)
j li
Тогда
pi -= pii + 2 Cj (po - pH) + 2 Сй (и» ~ Pii)- (3.108)
Подставляя (3.108) u (3.106) в (3.104), имеем
Здесь
к// - (pi/pii) - 1; Gofe/pu) - i; (3.1Ю)
(3.111)
bhh = (3.112)
Перемножая выражения в скобках в формуле (3.109) и пренебрегая
членами 2 2 aij^j 2 Pj/'Gm 2 а также полагая
k
приближенно, что 2 1, получим уравнение
Расберри — Хайнриха
Первоначально уравнение (3.113) было записано Расберри и Хайнри-
хом [284] путем полуэмпирпческого присовокупления к уравнению
Лачапса — Троила третьего члена в скобках. Вид этого члена был
подобран Эжмппрпческп, исходя из параболического вида зависимо-
сти Ii = /(С) при наличии заметного матричного эффекта возбужде-
ния. При преобладании эффекта поглощения Расберри и Хайнрих
рекомендовали учитывать только второй член уравнения (3.113),
а при преобладании эффекта возбуждения — только третий. В прин-
ципе допускалось также и одновременное использование всех членов
уравнения. При этом коэффициенты а п [3 определялись эмпи-
рически ио образцам известного состава. Значения коэффициентов [3,
как правило, получаются отрицательными (из формулы (3.111)
94
становится ясным, что величины [3 и должны быть по смыслу всегда
отрицательными).
Ограничения метода Расберри — Хайнриха связаны с пренебре-
жением членом Методом Расберри — Хайнриха можно по-
лучить более точные результаты, еслп положить член +
равным не единице, а некоторой средней величине, найден-
ной экспериментально по нескольким образцам известного состава.
Метод Клайссе и Квинтина
Согласно (3.61) для отношения функций F™/Ft имеем выражение
(3.114)
Выбирая точку разложения С, равной
С = (С} = 0,0; Ck = 0,0; Сг = 1,0),
и подставляя полученное выражение в (3.114), получим уравнение
Клайссе — Квинтина
Cl = (сГдтГ) [1 + S «уС; + S S IWA) <3.115)
где
_ 1 dFi I
— м ;.Г — *
li ус;[с-(С_;-о,о;с^=1,о)
й 1 V V ( 2 dFi dFi i д2/}
‘ ~ 2 F2 9Ck F, dC.0Ch
(3.116)
(3.117)
Коэффициент учитывает так называемые перекрестные матрич-
ные эффекты. Вид третьего члена в уравнении (3.115) был получен
Клайссе и Квинтпном путем эмпирического подбора [196]. Коэф-
фициенты fiijk определялись экспериментально на пробах известного
химического состава. Благодаря наличию третьего члена в (3.115)
уравнения Расберри — Хайнриха и Клайссе — Квинтина обладают
лучшей по сравнению с уравнением Лачанса — Трепла аппроксими-
рующей способностью и позволяют проводить анализ прп достаточно
широких вариациях химического состава анализируемых проб.
Уравнение Лачанса — Трепла предполагает, что поправка на
матричные эффекты пропорциональна содержаниям влияющих эле-
ментов, что справедливо только в узком диапазоне изменения кон-
центраций. Уравнение Клайссе — Квинтина, по существу, вводит
линейную зависимость коэффициентов аг} от концентрации влияющих
элементов. Сравнивая (3.115) и (3.87), можно видеть связь между
95
коэффициентами влияния afj к в уравнении Клайссе — Квпптпяа и
а$“т в уравнении Лачанса — Трепла
+ 2 в-улС/г- (3.118)
h
Аналогично из сопоставления (3.87) и (3.113) усматривается связь
коэффициентов влияния (Расберри — Хайнриха) и v-j-T?
(Лачанса — Трейла):
«^<- + 2^5, (З.Н9)
т. е. в этом случае имеет место обратно линейная зависимость коэф-
фициентов влияния от концентрации определяемого элемента.
Учет зависимости коэффициентов влияния от концентрации эле-
ментов существенно снижает систематические погрешности при ана-
лизе многокомпонентных систем, и поэтому как уравнение Клайссе —-
Квпнтина, так и уравнение Расберри — Хайнриха в различных их
модификациях нашли широкое практическое применение.
Метод Мосичева и Першина
Согласно (2.49) интенсивность аналитической липни элемента
А = (jiw, (цг) (1 + у;). (3.120)
Положим
Л(Нл.ь иО = (py/sincp 4- [яЫиф)-*. (3.121)
Согласно (2.36)
Т! = 2с,.ж,
где Dik определяется выражением (2.35). Поскольку Ck = const
Ъ = S (3.122)
где Ргй = const Dtk. Подставляя (3.121) и (3.122) в (3.120), имеем
Ц = k\Ct 4- 2 PifeA^(p^/sin ср 4-pj/sin ф). (3.123)
Обозначая ру/sin ср 4- pi/siu ф = М5, получим М» = 2 —
jV-i J
= CwMim 4- 2 Поскольку Cm=l— У Cj, то
96
где
«ij = (Mil —
Подстановка (3.124) в (3.123) дает
(3.125)
(3.126)
(3.127)
(3.127) на
откуда
где к[ = Умножая числитель и знаменатель
— SfVAj -и пренебрегая членами второго порядка малости,
получим уравнение Мосичева п Першина
+ 2 ^С;-2₽.-Л'|. г (3.128)
к jf-m к. /
Авторы работы [98] рекомендуют в сумму по /^-элементам в
третьем члене уравнения (3.128) включать член $ц1 i даже в том слу-
чае, если матричный, эффект возбуждения отсутствует. Нам представ-
ляется, что такой прием недостаточно обоснован с точки зрения фи-
зики возбуждения рентгеновской флуоресценции, но введение члена
ргг7г значительно повышает аппроксимирующие возможности урав-
нения (3.128).
Изменение поправочных коэффициентов с концентрацией опреде-
ляемого элемента аппроксимировано в методе Мосичева и Першина
линейной зависимостью
«ц = a°ii +
; 1 (3.129)
Pife = Pi7j + C$iik.
Необходимость в использовании поправок (3.129) возникает
сравнительно редко. Теоретические интенсивности в методе Мосичева
е Першина дополнительно согласуются с экспериментальными по
квадратичному уравнению
7ГП = + th (Леор)3 + (3.130)
п только после этой корректировки рассчитываются коэффициенты
atj и рг/< методом наименьших квадратов. Таким образом, для опре-
деления коэффициентов влияния попользуются приемы активного
эксперимента с независимыми вариациями одновременно всех компо-
нентов системы.
Метод Ширайвы и Фуджино
При фиксированных условиях анализа интенсивность аналитиче-
ской линии есть функция N — 1 независимых аргументов. Если за-
фиксировать концентрации всех элементов, кроме определяемого
97
i-ro и ?п-го элемента, за счет которого будет изменяться Ci, то интен-
сивность аналитической линии будет однозначной функцией толь-
ко Ci.
Воспользуемся для разложения в ряд выражения (2.49) форму-
лой Тейлора, которая позволяет вычислять с заданной точностью
значения функции j(x), если известны значения функции/(?n),/'(т),
f"(m) и т. д. в некоторой точке разложения х = т. Тогда
h = I°i+ 2 + 2^фЛС?+"-’ <3.131)
jVi.m Зфг,ти^з
где AC; -Cj — C°j.
Ограничиваясь первыми двумя членами ряда и полагая т = i,
получим упрощенную формулу для исправления интенсивности ана-
литической линии на матричные эффекты в виде
А = + (3.132)
/»И1Р = /«м.1_2а..ДСзл (3.133)
\ )
где
= <3-134)
По исправленным на матричные эффекты интенсивностям стро-
ится калибровочный график, который используется затем для на-
хождения концентраций элементов в анализируемых образцах.
Для удобства расчета на ЭВМ график описывается квадратичной
или линейной зависимостью, параметры последней рассчитываются
теоретически и затем могут быть уточнены по данным эксперимента
на образцах известного состава процедурой множественной ре-
грессии.
Расчет концентраций в анализируемых образцах проводится
следующим образом. С помощью полученных ранее калибровочных
зависимостей по неисправленным интенсивностям определяется на-
чальное приближение для концентраций всех элементов. Вычисля-
ются величины АС/, и по формуле (3.133) исправляются значения
интенсивностей. По этим исправленным значениям находится сле-
дующее приближение для значений концентраций, п итерации про-
должаются до завершения процесса сходимости.
Изложенный метод под названием метода теоретических попра-
вок получил существенное развитие в работах Величко, Калинина,
Ревенко и др. [34, 35, 54—58]. Было предложено учитывать измене-
ние коэффициентов влияния ccjj от химического состава линейной
зависимостью
се;; = «у 4-S^o/iACft, (3.135)
98
где коэффициент «^ характеризует изменение при изменении
концентрации к-то элемента на 1%. Суммирование проводится толь-
ко по элементам, изменяющим величину поправки. Введение кор-
ректировки (3.135) значительно расширяет диапазон возможных
вариаций химического состава анализируемых проб.
Калинин [58] показал, что уравнения типа (3.133) являются
строгими равенствами при произвольном выборе стандартного об-
разца, если первичное излучение монохроматпчно и имеет место
только матричный эффект поглощения. В этих условиях уравнения
типа (3.133) позволяют анализировать гомогенные материалы с про-
извольными вариациями химического состава.
Уравнение Ширайвы и Фуджино можно рассматривать как диф-
ференциальную форму записи уравнения Лачанса и Трейла. Все
уравнения методов «-коррекции, включая уравнения Клайссе —
Квинтина и Расберри — Хайнриха, могут быть записаны как в
дифференциальной, так и в интегральной форме.
Метод Котлярова
Применяя к выражению (3.3) теорему Лагранжа о среднем
(формулу конечных приращений), имеем
С, = (CTI./IT)fl - 2 ay (C°) {Ci - C")]. (3.136)
L J
Здесь C° — «средняя» точка, для которой по теореме Лагранжа
если и Cj < <7° < С/т, если
В этой точке выражение (3.136) становится строгим равепством.
Проблема состоит в том, что теорема Лагранжа не позволяет
отыскать среднюю точку. Поэтому полагается приближенно
С» = С™ (3.137)
и выражение (3.136) становится эквивалентным базовому выражению
(3.65) методов «-коррекции.
В интерпретации физического смысла коэффициентов влияния
уравнений (3.136) и (3.65) имеется принципиальная разница. Выра-
жение (3.65) основано па использовании полных дифференциальных
коэффициентов влияния, найденных для стандартного образца, т. е.
коэффициенты должны рассматриваться как константы, и с учетом
такой физической интерпретации применение зависимостей вида
(3.135) для уточнения «-коэффициентов некорректно. В уравнении
(3.136) численные значения параметров не являются константами
для всех проб, ибо средние точки согласно теореме Лагранжа не
постоянны. Поэтому введением поправок к коэффициенту можно
получить теоретически обоснованно различные модификации мето-
дов «-коррекции, расширяющие их возможности.
Другой путь в расширении возможностей методов «-коррекции
-состоит в увеличении числа выражений, описывающих градуировоч-
99
ную зависимость с одними и теми же численными значениями пара
метров. Котляров [66—69] показал, что уравнение связи, исчерпы-
вающее совокупность выражений (3.3), имеет вид
(3.138)
где у, и, г, s, . . t — различные совокупности влияющих элементов,
не включающие одни и те же элементы, т. е. уравнение (3.138) осно-
вано на представлении, что каждый элемент участвует только в од-
ном матричном эффекте.
Уравнению (3.138) соответствует выражение для интенсивности
Л(С) = СЛ(С) (<?'’), (3.139)
I
где — совокупности влияющих элементов, не включающие одни
и те же элементы.
Функции 7^-^ для мультипликативного расчета матричных
эффектов имеют вид
Н? + S аОЧ'Т1,
\ j-rn !
+ 2 <^с^\
j^m
охр Мп 2 УП1')-
\ j^m ,
(3.140)
(3.141)
(3.142)
Каждая из функций (3.140)—(3.112) отражает совокупное проявле-
ние всех матрпчпых эффектов п весьма условно может быть отож-
дествлена с каким-нибудь из них. Выражение (3.138.) позволяет в
каждом случае при одних и тех же численных значениях коэффи-
циентов подбирать такую зависимость, которая лучше соответствует
конкретным условиям проведения анализа. Для каждого определя-
емого элемента число вариантов уравнения (3.138) равно 3-2Л'-1 — 1.
Прп описании матричных эффектов формулой (3,140) выражение
(3.138) переходит в классическое уравнение (3.65).
Следует заметить, что использование обобщенного выражения
(3.138) делает метод «-коррекции по сложности сопоставимый с мето-
дом фундаментальных параметров и практическое значение имеют
более простые, хотя и менее универсальные частные случаи.
3.2.5. Связь между дифференциальной
и интегральной формой уравнений связи
Как уже отмечалось, разновидности методов «-коррекции отли-
чаются друг от друга учетом числа членов при разложении формулы
интенсивности в ряд Тейлора, формой «-коэффициентов (она может
W0
быть или дифференциальной, пли интегральной) и выражением
корректирующих формул (либо через концентрации, либо через
интенсивности).
Большинство разновидностей методов «-коррекции может быть
сведено к следующей обобщенной формуле
Ci = p“”l'l ++ 2 ДД+ 22?«".са). (3.143)
\ /=-’?' ‘ jrikzpi /
При р = 7 = 0 и Pf = Сг имеем метод Лачанса — Трейла,
р = у = 0 и Pt = Ц Лукаса — Туса,
Р — 0 н Pi = Ci Клайссе — Квинтина.
у = 0 и Pi = Ci Расберри — Хайнриха
и т. д.
Уравнения Лачанса и Трейла, Клайссе п Квинтина, Расберри и
Хайнриха записаны их авторами в интегральной форме. Эти уравне-
ния можно преобразовать в дифференциальную форму, полагая прп
сравнении со стандартным образцом, что
= + AQ, (3.144)
Записывая С?7 как функцию Cf в форме (3.115), с учетом
(3.144) имеем
Ci = СГ’ ([ (1|+ 2 «уС" + 2 «ь-сг +...) + (2 +
+ 2 2 +...)]/(! + 2 ацСТ + 2 aj,cr (3.145)
откуда, пренебрегая членами второго порядка малости, получим
дифференциальную форму записи уравнения Клайссе — Квпптина
с\ = сГ(1 + ЖлсД
где
(3.140)
(3.147)
(3.148)
сТУ1 = cTh'iT,
Дифференциальная форма уравнения Лачанса и Трейла будет
отличаться членами второго порядка в выражении для коэффициен-
та влияния
Записывая С™ как функцию СД в формуле Расберри —
Хайнриха, получим
Сг = С'Г [1 + 2И«(С" + АС;) + 1+ДЙ+Дс. (g + АС'.)]-
(3.150)
101
Разлагая в ряд Тейлора, запишем первые члены
1 1 \С.
______i_ 1__________t /3 <5n
14-су + дс| i с(i + cCiT)2’ v ’
С учетом (3.150) и (3.151) дифференциальная форма уравнения
Расберри — Хайнриха примет вид
С, = СГ'(1 + + 2 а«АС; + 2 «кАС,.), (3.152)
Интегральная форма записи коэффициентов влияния в уравне-
ниях связи может быть получена разложением интенсивности вряд
Тейлора в точке пространства концентраций, равной С = (Ct =
= 0, . . ., 0; Cj — 1, . . 0), т. е. в точке, весьма далекой
по химическому составу от состава анализируемого материала.
Поэтому дифференциальная форма записи уравнений связи дает зна-
чительно меньшую погрешность аппроксимации по сравнению с
интегральной. Величина поправки на матричные эффекты менее
чувствительна к погрешностям определения коэффициентов влияния,
когда они записаны в дифференциальной форме. Однако дифферен-
циальная форма имеет существенный недостаток, заключающийся
в зависимости дифференциальных коэффициентов от химического
состава используемого стандартного образца, что можно видеть из
формул (3.63), (3.148)—(3.150). Интегральные коэффициенты вли-
яния являются более универсальными величинами, не зависящими
от состава образцов сравнения.
3.3. МАТЕМАТИЧЕСКИЕ МОДЕЛИ И УРАВНЕНИЯ СВЯЗИ
Главная трудность использования теоретических уравнений
связи заключается в том, что в основе физической модели возбужде-
ния рентгеновской флуоресценции лежат предположения об идеаль-
но гомогенных образцах с пдеально гладкой поверхностью, а также
о возможности полного устранения аппаратурных эффектов. Такие
идеализированные предпосылки далеко не всегда соответствуют ре-
альным условиям возбуждения. Для многих продуктов переработки
минерального сырья построение физической модели, отображающей
реальные условия возбуждения и регистрации рентгеновских спект-
ров и физико-химическое состояние анализируемого материала, не
представляется возможным. В этом случае целесообразно физические
модели заменять формальными математическими моделями. При эм-
162
лирическом построении уравнений связи находят наиболее удобный в
практическом отношении математический аппарат, адекватно описы-
вающий данные эксперимента на пробах известного состава. Такой
аппарат успешно используется в физике при описании плохо органи-
зованных диффузных систем. Реальные процессы возбуждения ана-
литического сигнала при этом подходе заменяются формальной мате-
матической моделью «черного ящика», преобразующего первичное
излучение во вторичное, так же как и анализируемый образец.
Правильно построенная математическая модель не должна увеличи-
вать «шумы» исходных данных.
При построении математической модели выражение (3.1) для
иптенсивпости рентгеновской флуоресценции рассматривается как
ограниченная непрерывная нелинейная функция состава анализи-
руемого образца, которую по теореме Вейерштрасса с любой наперед
заданной точностью можно представить в виде действительного сте-
пенного ряда
Ii = + 2. + 2 + .. . (3.153)
или в записи через интенсивности
С; = а(£+ 2 + 2 + 2 + • • • (3.154)
Обратная математическая модель (3.154) обосновывается тем, что
решение уравнения относительно концентраций есть непрерывная
ограниченная функция интенсивности и «обратные» уравнения мож-
но рассматривать как полиномиальные приближения решения пря-
мого уравнения.
Уравнения типа (3.153) и (3.154) называют в литературе фено-
менологическими, регрессионными пли статистическими.
Волес компактно математические уравнения связи в РСА можно
записать в векторной форме
Р(С, 7) = 0, (3.155)
где С в I — вектор содержаний и интенсивности соответственно;
Р(С, Z) — функционалы, вид которых надо установить дополнитель-
но. Существование решений (3.155) предполагается в форме решений
системы отдельных самостоятельных уравнений связи для каждого
компонента
С [пл = j(AibJi), (3.156)
где Сщдг — содержание z-го компонента в /ой пробе; z = 1, 2, . . .
..., м; — совокупность интенсивностей компонентов /ой пробы,
которая используется для расчета z-ro компонента; —
функционалы, вид которых устанавливается. Так как природа фи-
зических процессов одинакова, функционалы (3.156) считаются за-
103
данными на одном и том же параметрическом семействе функций
(3.157)
где cc[i] — вектор калибровочных коэффициентов, являющихся чис-
ленными параметрами математической модели. Тогда (3.157) при-
мет вид
Слл = ЛЛгм, ®U1)- (3.158)
Использование математической модели (3.158) разбивается па
два этапа: 1) задание класса функций {f(I\_i].h, а[п)} и выбор урав-
нения связи, адекватно описывающего всю совокупность физических
процессов при анализе конкретных материалов в конкретных усло-
виях возбуждения; 2) расчет калибровочных коэффициентов и по-
строение калибровочного уравнения, соответствующего выбранному
уравнению связи.
На практике кроме уравнений связи (3.153) и (3.154) использу-
ются уравнения следующих видов:
Белова — Дуймакаева [19]
С$ — а0 + 2 + 2 2 ^ijjiljlk; (3.159)
j j h
Мэсю — Джонсона [78]
Ci = Ii + 2 aijlilj + 2 aijlilj (Ii ~~ Ij) +22 ^iihliljl-k', (3.160)
Mi jVi
Слуцкина — Бадера [138]
Ci + Vi + 2; V-ijlij + 2 2. ljl‘-Ijlk't (3.161)
Менцика — Бернебурга — Шорта [270]
Ci ~ CCOj -j- СеД; -J- Ii 2 (3.162)
Белова [5]
ii= 2ccij^; +2 2 v-i^CjCb + 2 2 (Cj —• c'k) +
j j=£ik=j+l j+1
+ s.( 2ij2i«y4Ic’AG+...; (3.163)
Блохина — Белова [24]
ii= Ci + 2 G+c’jCj + 2 я-ijCiCj (Ci — Cj) -j- 2 2. ^nkCiC^сk.
j--i i-i 7?=i A>j
(3.164)
Оптимальную форму уравнений находят последовательным пере-
бором различных сочетаний членов при минимальном среднеквад-
ратичном отклонении получаемых результатов при заданном числе
членов уравнений. Иногда в качестве дополнительных факторов при
104
построении уравнений используют интенсивности рассеянного про-
бой или прошедшего через пробу излучения.
Использование концентраций в качестве контролируемых пере-
менных приводит к ограничениям на возможные значения пере-
менных, п задача нахождения численных значений параметров в
уравнениях калибровки существенно упрощается. G другой стороны,
применение в качестве контролируемых переменных интенсивностей
удобно, так как не требует выполнения условия = 1, которое
трудно выполнимо при анализе минерального сырья и продуктов
его переработки пз-за неопределенности стехиометрических соотно-
шений между элементами. Донец [46] предложил компромиссный
вариант, сочетающий достоинства уравнений (3.153) и (3.154):
^2 ~ f'2 ^2i • • • ’ -^'0’
(3.165)
ОШ, c2, ...,zn).
Интенсивности в системе уравнений (3.165) последовательно
заменяются рассчитанными содержаниями. При этом выбираются
хорошо характеризуемые интенсивностями элементы, влияние кото-
рых на другие элементы лучше описывается их содержанием.
Для определения численных значений коэффициентов в эмпири-
ческих уравнениях чаще всего используется линейный метод наи-
меньших квадратов, при котором рационально минимизировать дис-
персии предсказания средних значений получаемых концентраций.
Следует заметить, что минимум среднеквадратичного отклонения не
является самым надежным критерием, так как более важным может
оказаться устойчивость при плохо обусловленной системе уравнений
связи. Характеристикой обусловленности системы может служить
величина копдминимума сопйЛ, показывающая, во сколько раз
ошибка в определении вектора неизвестных больше ошибки в опре-
делении вектора свободных членов пли матрицы параметров уравне-
ний связи.
Для уравнений вида (3.153) и (3.154) cond А имеет наименьшее
значение, когда матрица параметров уравнений связи ортогональна.
При анализе Л'-компонентного образца на N — 1 компонентов мож-
но построить ортогональную матрицу коэффициентов. При анализе
на все компоненты матрицу можно привести к квазпортогональному
виду. Таким образом, для обеспечения минимальной погрешности
анализа и высокой устойчивости уравнений связи к эксперименталь-
ным ошибкам необходимо, чтобы матрица параметров уравнений свя-
зи была орто- или квазиортогональной и система для определения
этих параметров также имела орто- или квазиортогоналытую матри-
цу концентраций калибровочных составов уравнений.
При анализе минерального вещества возникают жесткие корре-
ляционные связи между элементами, обусловленные минералогиче-
ским составом анализируемых проб и стандартных образцов, приме-
няемых для калибровки. Поэтому при случайном наборе стандартных
образцов для калибровки может возникнуть зависимость коэффици-
105
ентов уравнения друг от друга, от количества и химического состава
использованных стандартных образцов и других случайных факто-
ров. Для того чтобы избавиться от неопределенности в значениях
коэффициентов уравнения, необходимо стандартные образцы для
калибровки выбирать но схеме ортогонального планирования.
Шеффе [293] с этой целью ввел понятие планов симплекс-решетки для
экспериментальных ситуаций, относящихся к стандартным образцам,
где отклик функции зависит только от концентраций компонентов
стандартных образцов. Составы стандартных образцов для калибров-
ки определяются при этом положением точек на спмплекс-решетке.
Интерпретация физического смысла отдельных членов в эмпири-
ческих уравнениях связи довольно затруднительна, так как каждый
из факторов математических моделей несет опосредствованную ин-
формацию о всей совокупности матричных -эффектов. Информация о
матричных эффектах при этом оказывается «размазанной» по всему
факторному пространству п каждому фактору математической модели
невозможно приписать фиксированный физический смысл.
Для выявления факторов, наиболее сильно влияющих на резуль-
таты анализа, проводятся отсеивающие эксперименты методами ис-
ключения или включения. Последний заключается в последователь-
ном включении в уравнение связи факторов в соответствии со стати-
стическими критериями их важности. При этом используется шаго-
вая процедура поиска, например алгоритм Эфропмсона. Для про-
ведения отсеивающих экспериментов применяются массивы реаль-
ных или моделированных проб. Массив проб должен представлять
собой невырожденный план эксперимента для всех используемых
уравнений системы.
Основным недостатком уравнений связи, построенных на осно-
вании математических моделей, является их многомерность и мпого-
параметричность.
В гл. 5 дано развернутое описание алгоритма и методики сили-
катного анализа на основе метода AF-коррекцпи. Подобная методика
анализа может быть в принципе построена на основе большинства
описанных в данной главе методов учета матричных эффектов.
3.4. РЕШЕНИЕ СИСТЕМ УРАВНЕНИИ СВЯЗИ В РСА
Непосредственно использовать выражение (3.3) для расчета ре-
зультатов анализа не представляется возможным, поскольку вели-
чина F является нелинейной функцией всех без исключения элемен-
тов, входящих в анализируемый образец. Поэтому для вычисления
концентраций записывают следующую систему уравнений:
Ci = C„iFT/J'\-,
Cs = C.iFT/Fi-,
Са=Сл1^/рл (3.16G)
= ConF^/Fn
106
или в векторной форме
С = С^(ССУГ(С). (3.167)
Уравнение (3.167) в общем случае нелинейно, и его решение разбива-
ется иа два этапа: нахождение и уточнение приближенного решения
системы. Для определения приближенных значений корней системы
нелинейных уравнений не существует общих методов, и каждая не-
линейная система рассматривается как самостоятельная задача.
В уравнении (3.167) в качестве приближенного значения корней
удобно принимать величину
- сГст7/7т. (3.168)
Волее близкие к истинным начальные значения корней можно полу-
чить, воспользовавшись вспомогательным графиком, построенным
по результатам эксперимента на пробах известного химического со-
става (метод построения такого графика будет подробно рассмотрен
в гл. 5).
Для уточнения корней нелинейных систем разработаны общие
математические методы: простой итерации, Ньютона, секущих и др.
Эти методы прежде всего отличаются итерационными формулами
C„+i = с„- kf(Cny,
С -С ,{С^-
— ---i(C \ - \ 1 (^п)
/ (C7t) ! (bk)
(п =0,1,2,*. к < п);
с 1
2 [Г(^
(3.169)
(3.170)
(3.171)
(3.172)
Решение системы уравнений (3.167) при использовании любой
из приведенных итерационных формул начинают со значений
и строят последовательные приближения
£-[» + !] _ £г[п] |
(3.173)
Различные .методы отличаются выбором направления тг-го шага,
определяемого отношением и величиной
шага, характеризуемого параметром А1"^.
На практике число уравнений может превышать число неиз-
вестных. Такие переопределенные системы могут вообще не иметь
решения в математическом смысле из-за наличия ошибок измерения
аналитического сигнала. В этом случае ищется решение, наплучшим
образом удовлетворяющее данной системе уравнений. Обычно ищут
решение, дающее минимум суммы квадратов остаточных рассогла-
сований:
т
107
Переопределенные системы можно решать методом Ньютона или ме-
тодом скорейшего спуска.
Рассмотрим наиболее существенные с практической точки зре-
ния аспекты решения систем уравнений вида (3.167) стационарными
итерационными способами. Ради простоты и наглядности ограни-
чимся двумерным приближением.
Необходимое условие решения. Анализ всего
множества уравнений, реализующих расчет концентраций по изме-
ренным интенсивностям, показывает, что преимущественно эти урав-
нения однородны. Исключение составляют эмпирические уравнения,
содержащие свободные члены. Поэтому система (3.167) может быть
использована для нахождения только отношений концентраций.
Чтобы при решении системы однородных уравнений найти однознач-
ное решение, необходимо ввести в систему хотя бы одно неоднородное
уравнение. Часто такое уравнение находится из условия
2 С; = const, (3.174)
где: а) 2^1 = S Ci—сумма концентраций всех компонентов образца
и const = 1 при расчете составов проб способом, продусматрпваю-
д
щим расчет поглощающих характеристик; 5) 2 = 2 Ci — сумма
i г =1
концентраций только определяемых элементов и
const = 1 при п — О,
const < 1 при п 1.
если анализ выполняется с использованием эмпирических уравнений.
Необходимость включения в расчет условия (3.174) может при-
вести к следующим изменениям системы (3.166). Если уравнения ли-
нейны, то условие (3.174) заменяет любое уравнение системы (3.166).
Из-за неопределенности в измеренных интенсивностях в зависимости
от того, какое уравнение заменено условием (3.174), может возник-
нуть несовместность аналитических результатов. Для ее исключе-
ния экспериментальные интенсивности исправляются, например,
методом наименьших квадратов. Когда уравнения нелинейны, усло-
вие (3.174) либо дополняет систему (3.166), либо используется в впде
Cs = 1 - 2 Ci (3.175)
в качестве А-го уравнения для нахождения концентрации микро-
элемента. В первом случае при анализе способами с эмпирическими
поправками условие (3.175) используется для нахождения абсолют-
ных значений концентраций по отношениям С, вычисленным с помо-
щью первых N уравнений. При анализе способами, предусматриваю-
щими расчет поглощающих характеристик, оно включается в каж-
дый цикл для нахождения абсолютных концентраций, используемых
в законе аддитивности для массовых коэффициентов поглощения. Во
юз
втором случае макрокомпопентом может быть элемент, интенсивность
аналитической линии которого или не может быть измерена (напри-
мер, кислород прп анализе горных пород на СРМ-18), или измеряет-
ся с большой погрешностью (например, железо в сталях). При этом
в величину Су неизбежно войдут концентрации неопределяемых мпк-
рокомпонентов, которые прп дальнейшем расчете будут учитываться
с поглощающими характерце! икамн, равными характеристикам
макрокомнопента.
Чтобы показать, какие могут быть ошибки пз-за неправильной
организации процесса решения системы, в табл. 19 сопоставлены
концентрации, полученные прп решении системы (3.167) без (I),
с использованием (III), а также при включении условия (3.174) только
в пулевом цикле расчета (II). Измеренные концентрации корректи-
ровались методом фундаментальных параметров. За образец сравне-
ния брался стандартный образец горной породы СГ-1А с аттестован-
ным содержанием Сгт. Интенсивности аналитических линий элемен-
тов Mg, Al, Si, Р, К, Са, Ti, Мп л Ре измерялись на рентгеновском
кваптолютро КРФ-11. Из табл. 19 видно, что игнорирование условия
(3.174) приводит к неверным результатам. Частичное его использо-
вание в нулевом цикле, хотя п улучшает правильность конечных
результатов, не исключает полностью ошибки вследствие неправиль-
ного расчета массовых коэффициентов поглощения в последующих
циклах.
Оценка и т е р а ц и о н н ы х м е т о д о в. Геометрический
смысл уточнения по формулам (3.169). (3.170) и (3.171) применитель-
но к реятгепоспектралыюму микроанализу рассмотрен Шпрингером
[299] с помощью графика функции 1(C). Показано, что пересечение
этой кривой и прямой f(C) = C/Cq дает истинную концентрацию.
Каждой формуле уточнения соответствует секущая, определяющая
движение к этой точке. В рентгсноспектральлом многоэлемечгтном
анализе с учетом используемых способов препарирования образцов
в первом приближении функцию /(С) можно считать монотонной,
поэтому такое рассмотрение эквивалентно самому общему выяснению
Таблица 19. Сравнение концентрации С е концентрация-
ми, полученными при разной организации процесса решения
системы, $6
Оь-сп^ Сат I И III
МаО 7,0 8,10 7,14 7,07
14,88 17.22 15,16 15,03
SiO2 40,39 53,68 47,27 46.84
]ДО-( 1,(11 1,15 1,01 1,00
К,О' 2,90 3,42 3,01 2,98
СаО 10.97 12.68 11.17 11.06
TiO, 1.71 1.94 1.71 1.69
МпО 0,172 0.10S 0,174 0.173
Fe.3O3 11,48 13.24 11.66 11.54
100,23 114,40 101,26 109,21
109
Таблица 20. Число циклов л, требуемое для отыскания корней с заданной
точностью, для различных расчетных схем
Проба Габбро- диорит Гранит Проба Габбро- диорит Гранит
I 11 III I II ш I II III I II III
Гранпт 3 5 5 2 2 А12О3-К2О 3 5 7 3 8 9
Песчаник 5 5 2 4 4 SiO2 2 6 8 4 6 В
Амфиболит 2 4 о 2 4 4 СаО 4 4 4 4 б 5
Габбро-диорит 2 2 2 3 5 5 MgO — КоО 3 о 4 5 5
геометрического смысла способов уточнения корней уравнений.
Оценим расчетные схемы (3.169), (3.170) и (3.171). Известно, что
на сходимость различных методов существенно влияет выбор на-
чального приближения. В зависимости от химического состава об-
разца сравнения начальные приближения могут быть либо близкими,
к истинным и в процессе расчета уточняться несущественно, либо
очень далекими от истинных, и тогда роль уточнения резко воз-
растает.
Сопоставим рассматриваемые методы в равных условиях, исходя
из одних нулевых приближений, равных Со, и задаваясь постоянной
точностью расчета. В табл. 20 приведены значения числа циклов п,
требуемые для отыскания корней с заданной точностью при расчете
проб разнообразного состава методом итераций (I) и методом Ньюто-
на (II). Концентрации Со корректировались методом фундаменталь-
ных параметров. Кроме тою, был применен метод Ньютона в линей-
ном приближении (III).
Чтобы проиллюстрировать изменение скорости сходимости мето-
дов в зависимости от близости приближения к корню, значения п
приведены для двух образцов сравнения существенно различного
состава: гранита и габбро-диорита. Как видно из табл. 20, за исклю-
чением случаев анализа гранита по граниту и габбро-диорита по
габбро-диориту метод Ньютона дает более медленную сходимость,
что можно объяснить чувствительностью последнего к точности за-
дания нулевого приближения. Метод Ньютона обеспечивает быструю
сходимость, если нулевые приближения выбраны в соответствии с
условием
?(Q<p''(Q>0- (3.176)
Используемые нами в качестве нулевых приближений Со, как пра-
вило, не удовлетворяют условию (3.176), так как ф"(^о) или равна
в линейном случае нулю, или отрицательна. Именно по этой причине
метод Ньютона рекомендуется применять после разового исправле-
ния нулевых приближений методом простой итерации. С другой сто-
роны, для всех теоретических методов вычисления поправок 0 <Z
< Cof(Cn)<Z 1. Поэтому при удачном нулевом приближении метод
Ньютона может обеспечивать быструю сходимость.
Данные для метода секущих занимают промежуточное положе-
ние между методом Ньютона и методом простой итерации.
Таким образом, наиболее целесообразно применение метода
простой итерации. Даже при сопоставимой сходимости рассмотрен-
ных методов предпочтение следует отдать методу простой итерации»
как методу с наиболее простой расчетной схемой, и поэтому реали-
зуемому на микрокомпьютерах.
Привлечение к решению систем уравнений в рентгепоспектраль-
ном анализе методов, более сложных, чем простая итерация, было
вызвано неустойчивостью некоторых уравнений из-за чувствитель-
ных к сходимости значений коэффициентов уравнений. Замена урав-
нения (3.167) эквивалентным
= (0<?v<l) (3.177)
гарантирует сходимость итерационного процесса. Более того, этому
способствует условие (3.174), которое полностью исключает расхо-
димость.
Точность расчета. Решение системы с любым заданным
числом десятичных знаков может быть получено только при условии,
что все коэффициенты уравнений являются точными числами. Во
всех остальных случаях решение должно выполняться с числом зна-
ков, па единицу меньшим или равным числу знаков, с которым за-
писаны коэффициенты.
На примере определения породообразующих элементов в горных
породах методом AF-коррекцин оценим величину реальной точности
расчета и выясним, чем она обусловлена.
Коэффициенты системы уравнений имеют вид
(ср + ib/siniM..
у }J ' г Из
(3.178)
Включение в рассмотрение поправки на возбуждение не вносит
изменений в конечные выводы. Точность расчета ег будет зависеть
от точности определения всех коэффициентов в пределах одного
уравнения.
По закону сложения ошибок
+
(3.179)
При анализе методом AF-коррекции погрешность, вносимая
неопределенностью ц, не зависит от абсолютного значения р. Поэто-
му можно считать, что в пределах одного уравнения точность всех
коэффициентов одинакова. Воспроизводимости V% измерения ин-
тенсивностей АД /1г и Д/ГМ определяются аппаратурной ошибкой
111
Таблица 21.
Значения ошибок и точностей расчета. %
Pan И Ошибкой ПРОООПОДГОТОВКИ
НИИ, ЧТО Fan= Ран И Рдр -= Vnp,
личины должна соответствовать
ГпР. Следовательно, в предноложе-
точность расчета £ по порядку ве-
сумме ошибок
С = ]/2^п +- 2Vnp +- (АСС1/Сст)2 + (Дц/ц)2. [ (3.180)
Величины первых двух ошибок зависят от длины волны линии и
для каждого спектрометрического канала своп. Поэтому точность
расчета должна задаваться на каждый элемент индивидуально.
В табл. 21 приведены значения ошибок Van, УдР, и точности
расчета £ для Al, Si, Са и Fe. Величина Др/р для всех элементов по-
лагалась равной 1%. В качестве образца сравнения использовался
стандартный образец горной породы СГД-1А. Как видно, использо-
вание одной точности расчета на все элементы не корректно. Со-
поставление £ и Т7х показывает, что пренебрежение точностью изме-
рений может исказить результаты расчета. Полученные выводы яв-
ляются общими для всех способов рентгеноспектрального анализа,
завершающихся решением системы уравнений связи итерационными
методами.
3.5. УЧЕТ АППАРАТУРНЫХ ЭФФЕКТОВ
И УТОЧНЕНИЕ ПАРАМЕТРОВ
Методы ^.-коррекции, так же как я методы фундаментальных
параметров, требуют раздельного учета аппаратурных и матричных
эффектов. Дело в том, что при расчетах матричных эффектов должны
использоваться не экспериментальные, а теоретические интенсив-
ности. Учитывая функциональную зависимость между измеренными
и расчетными интенсивностями
zrcp = (3.181)
уравнение (3.3) запишем в форме
С, = Cfj' (3.182)
Вид функции / (Д-7”)изм определяется наличием таких аппаратур-
ных эффектов, как фон рассеянного излучения, наложение линий
рентгеновского спектра, нелинейность регистрирующей системы при-
бора и т. д.
112
Функцию / (Zt,7jT)ndI'1 можно описать линейной или квадратич-
ной зависимостью вида
afl + й1Л + «7/. (3.183)
Численные значения параметров зависимости (3.183) устанав-
ливают экспериментально.
Принимая во внимание зависимость (3.183), уравнение связи
методом а-коррекцпи можно записать так:
£*= («0 + 0.^1+ a,Ij) [1 + 2«Л]. (3.184)
Член в круглых скобках, характеризующий аппаратурные эффекты,
является обязательным компонентом в современном программном
обеспечении рентгеновских спектрометров. Только раздельный учет
матричных п аппаратурных эффектов позволяет получать стабильные
значения коэффициентов влияния. При определении коэффициентов
а0, аг и а2 значение члена в квадратных скобках принимается равным
единице [295]. Такой прием недостаточно обоснован. Более коррект-
ный и общий прием введения поправки на аппаратурные эффекты,
предложенный авторами настоящей монографии, будет изложен в
заключительной главе. Надо заметить, что полное разделение аппа-
ратурных п матричных эффектов на практике встречает большие
затруднения.
Численные значения параметров в уравнениях сс-коррекции мо-
гут быть лпбо вычислены теоретически, либо найдены по данным
эксперимента на пробах известного состава с помощью процедур мно-
жественной регрессии. Па практике наилучшим образом зарекомен-
довал себя полуэмппрический способ определения численных зна-
чений коэффициентов влияния. Величины а- п ^-коэффициентов в
уравнениях связи вычисляются теоретически, а затем их значения
уточняются и оптимизируются по результатам эксперимента на стан-
дартных образцах.
Проблема оптимизации состоит в нахождении коэффициентов
а0, |Z2 Для заданного количества стандартных образцов, при ко-
торых разница правой и левой частей уравнения (3.181) будет ми-
нимальной. Это эквивалентно минимизации суммы квадратов
s (р) = У /; (Р), (3.185)
2=1
где функция /Др) есть взвешенная разность
Ш = №АС(1, р)_ Ci\-, (3.186)
C(i, р) — расчетное значение концентрации, Ct — аттестованное
значение. Параметр р7-, /==1,2,.. ., п, собран для з’добства записи
в вектор.
При строгом подходе для минимизации (3.185) должна приме-
няться техника нелинейного программирования, поскольку (3.184)
является в общем случае нелинейной функцией. Минимизация по-
113
очередным варьированием одного параметра дает очень медленную
сходимость. Вблизи своего минимума большинство физических функ-
ций квадратичны. Они минимизируются одним шагом, так как опре-
деляются величинами самих функций, а также первой и второй про-
изводной в одной точке. Тогда шаг не будет произвольным и нет необ-
ходимости двигаться по градиенту. Большинство распространенных:
алгоритмов минимизации базируются на методе Гаусса — Ньютона.
Глава 4
ПОДГОТОВКА ПРОБ К АНАЛИЗУ
Горные породы, поступающие на анализ обычно в виде порош-
ков, представляют собой совокупность породообразующих минера-
лов, т. о. частиц, равных по размеру, конфигурации и химическому
составу. Беглое знакомство с породообразующими минералами по-
казывает, что некоторые элементы, и прежде всего кремний, входят
в состав нескольких минералов, причем содержание их в каждом
существенно отлично. Следует отметить, что процентное соотношение
между минералами редко можно определить однозначно по концент-
рации какого-либо элемента.
В исследуемой области рентгеновского спектра флуоресценция
возбуждается в слое сравнительно малой толщины. Сопоставление
величин толщин излучающих слоев для /<а-липмй породообразую-
щих элементов с реальными размерами частиц порошка показывает,
что флуоресценцию излучают преимущественно верхушки зерен.
Только в случае FeTfa-лпнпи «работают» 3—5 слоев. Гомогенизиро-
вать такие гетерогенные порошки даже самой совершенной техникой
измельчения в настоящее время нс представляется возможным. Для
•осуществления анализа необходимо порошки перевести в раствор
(твердый или жидкий).
4.1. СТЕКЛООБРАЗНОЕ СОСТОЯНИЕ
Понятие «стекло» определяли по-разпому. За рубежом самое
широкое распространение получило предложение Американского
общества по испытаниям и материалам: стекло — неорганический
продукт плавления, охлажденный до твердого состояния без кристал-
лизации [91], или аморфно (т. е. без кристаллизации) застывший ра-
сплав [129]. Комиссия по терминологии при Академии наук СССР
[146] предложила называть стеклами аморфные тела, получаемые
путем переохлаждения расплава независимо от их химического со-
става и температурной области затвердевания, причем процесс
114
перехода из жидкого состояния в стеклообразное должен быть об-
ратимым.
Раньше стекла рассматривали как «переохлажденные» жидко-
сти, в которых процессы кристаллизации при переходе расплав —
стекло должны быть заторможены быстрым нарастанием вязкости
расплава и затрудненностью перестройки структуры жидкости. Од-
нако согласно более новым представлениям, между стеклом и переох-
лажденной жидкостью существуют значительные различия. Приня-
то говорить, что жидкость находится в переохлажденном состоя-
нии, если ее удается охладить до температуры ниже точки ее замер-
зания без образования кристаллов. Состояние переохлажденной
жидкости метастабильно, так как ее свободная энергия больше, чем
у соответствующего ей кристалла, однако структуре переохлажден-
ной жидкости присуща меньшая свободная энергия, чем любой дру-
гой непосредственно близкой ей структуре.
Что же происходит при охлаждении расплава? Кристаллизую-
щееся вещество имеет вполне определенную температуру, при кото-
рой происходит затвердевание, сопровождающееся скачкообразным
изменением объема и выделением теплоты. Для веществ, которые
можно охладить до стеклообразного состояния, внезапного измене-
ния объема и экзотермической реакции не наблюдается. Для них в
интервале нескольких градусов вблизи точки превращения 2> имеет
место сильное изменение удельной теплоемкости, коэффициента рас-
ширения, температурного коэффициента диэлектрической прони-
цаемости и показателя преломления (рис. 37) [129]. При температуре
ниже точки превращения вязкость расплава непрерывно возрастает
и достигает столь большой величины, что вещество ведет себя как
жесткое твердое тело. Стекло, нагретое выше точки превращения,
с дальнейшим повышением температуры медленно и непрерывно пере-
ходит в жидкость, получающуюся при плавлении кристаллического
вещества. Ниже точки превращения свойства стекол лишь немного
отклоняются от свойств, которые проявляют те же вещества в кри-
сталлическом состоянии.
В отличие от переохлажденных жидкостей стекла не находятся
в состоянии внутреннего термодинамического равновесия. При
дальнейшем охлаждении стекла сохраняют существующий при точке
превращения беспорядок (энтропию).
Первую попытку уяснить основные положения, определяющие
условия стеклообразования, сделал Гольдшмидт в 1926 г. [91]. Он
предположил, что способность оксида образовывать стекло связана
с тем, каким путем ионы кислорода располагаются вокруг катиона и
входят в элементарную ячейку кристаллической решетки. Так как
ряд оксидов, включая SiO2, GeO2 и Р2О6, составляют в кристалли-
ческом состоянии четырехгранники, Гольдшмидт предложил считать
эту особенность критерием способности к стеклообразованию.
*) Точкой превращения стекла называют температуру, при которой свой-
ства стекла переходят в свойства переохлажденной жидкости.
115
Рис. 37. Температурная зависимость
удельной теплоемкости вещества в жид-
ком, стекловпщюм и кристаллическом
состояниях.
‘е — точка npesj ащегшЮ ts — точна плавле-
ния.
Рис. 38. Область образования стекла
в системе ГКО—А12О3—SiO2.
Захарпасен в 1932 г. [91J показал, что способность оксидов
составлять четырехгранную конфигурацию нельзя считать абсолют-
ным критерием способности стеклообразования. Действительно, ВеО,
например, позволяет создавать четырехгранные структуры, но оксид
бериллия тем не менее не удастся получить в стеклообразном со-
стоянии.
lio Захаркасену, при образовании бинарным соединением МехО,,
стекла должны соблюдаться следующие условия:
а) атом кислорода не должен быть связан более чем с двумя ато-
мами Me;
б) число атомов кислорода, окружающих Me, должно быть мало;
в) у многогранников, составляемых атомами кислорода, могут
быть общие вершины, ио не ребра или грани.
Сформулированное Захарпасеном четвертое условие, предпола-
гающее, что во всяком кислородном многограннике должно быть не
менее трех общих вершин, мепее важно и не всегда приложимо, так
как известны стекла, в которых это условие не соблюдается. Пере-
численные условия выполнимы только для соединений состава Ме3О3,
МеО2 л Ме2О-, и не выполнимы для соединений состава МеО, МеО3
Ме2<)7, МеО4 и т. д.
] 1аиболее известными стекловидными образователями среди
бинарных соединений являются В2О3, As2O3, Sb2O3, BeF2, SiO2, GeO2,
P2O5 и As3O6. Стеклообразный оксид бора B2O3 — хороший пример
стекла, структура которого состоит из отдельных треугольников.
Подобные треугольники существуют во многих кристаллических
боратах. Стеклообразный кварц SiO2, оксид германия GeO2, оксид
фосфора Р2О5 и оксид мышьяка As2O5 являются стеклами, построен-
ными из отдельных четырехгранников.
Для образования многокомпонентных стекол необходим хотя бы
один оксид, который может находиться в стеклообразном состоянии.
Для горных пород таковыми являются оксид кремния SiO2, оксид бора
В2О3 и оксид фосфора Р2О3.
Кроме стеклообразующих, необходимо отличать по их роли в
структуре стекла оксиды двух других типов: видоизменяющие и про-
116
межуточные. Действие первых сводится к разрыхлению сетки стекла.
Придгером таких модифицирующих оксидов служат оксиды натрия,
лития, калия. Как модифицирующие действуют и такие щелочно-
земельные оксиды, как оксид магния, кальция, бария. Промежуточ-
ный оксид сам обычно не способен образовывать стекло, но участву-
ет в образовании сетки стекла вместе с другими оксидами. Проме-
жуточными являются оксиды алюминия, бериллия я двуокиси ти-
тана, циркония.
В многокомпонентном стекле доля стеклообразующего оксида
должна быть не ниже определенной величины. Ниже указано пре-
дельное содержание для бинарной щелочно-силпкатной системы
(мол. %):
ШО5 - 40
NaoO - 47
К2О - 50
SiO2 — Си
SiO2 — 53
SiO2 — 50
с превышением которого расплавы кристаллизуются или расстекло-
вываютсл при охлаждении. Существование предельного состава
связано со структурными изменениями, происходящими при повы-
шении содержания щелочей. Тенденция лития к расстекловыванию
наблюдается и в бинарных боратных системах 1л2О—В2О3.
Реальные стекла лишь в редких случаях состоят только из двух
оксидов. При большем числе компонентов ситуация резко усложня-
ется. Для примера на рис. 38 приведены границы образования стекол
в тройной системе l_i2O—Д12О3—Si02 [91].
За последние годы стали преобладать взгляды, что стекло нель-
зя считать гомогенным в субмикроскопическом масштабе. Не исклю-
чено, что состав стекла изменяется от точки к точке по всему объему.
Например, в натриево-силлкатном стекле одни мпкроучастки могут
иметь состав чистого кварца, а другие — состав такого натриевого
силиката, как дпепликат J\a2O-2Sid2 [91]. Электронная микроско-
пия некоторых стекол, по-впдпмому, свидетельствует о наличии
включенных в основную .массу глобулярных п линзообразных частиц
в поперечнике ~10 нм.
4.2. ФИЗИЧЕСКАЯ ХИМИЯ
И МЕХАНИЗМ ПРОЦЕССА ПЛАВЛЕНИЯ
ГОРНЫХ ПОРОД И МИНЕРАЛОВ
Многие из проблем, возникающие при препарировании твердых
растворив, обусловлены исключительным многообразием идущих в
расплавах процессов. Зависимое'] ь последних от большого числа фак-
торов делает их. особенно в условиях весьма приближенного знания
качественного состава расплава, трудно предсказуемыми и потому
сложными для контроля.
Возможность стеклообразования и различие физико-химических
свойств стекол непосредственно определяются характером: химиче-
ских связей, существующих в системе. По данным [131 ], соединения,
способные находиться в стеклообразном состоянии, имеют смешанные
117
виды связей, в результате чего направленные и ненаправленные свя-
зи взаимодействуют между собой. Это обусловлено существованием
промежуточного состояния между, например, ионной и ковалентной
связью, и потому одна и та же часть соединения может проявлять
различные виды связи по отношению к различным соседям.
Процесс сплавления начинается с химических реакций между
компонентами смеси, основными из которых являются диссоциация,
термическая и электролитическая, ионизация и ассоциация. Соот-
ношение между последними и их конечные продукты зависят от кон-
кретной системы. В частности, термическая и электролитическая
диссоциация силикатов типа Me2SiO4 идет согласно уравнению [16}
п Ме2 SiO4 -* jn Ме2 SiO4 -> 2/дМеО + ;"zjSiO2,
где
} = 1 4- ат(2/" 4- j' — 1);/ = 1 + аа(к — 1);
<хт — степень термической диссоциации nMe2Si.O4; а0 — степень
электролитической диссоциации ??Me2SiO4; /, f, j" — изотонические
коэффициенты при соответствующих соединениях.
Ролен и Зарзинский установили [147], что в борате лития окси-
ды SiO3, TiO2 не диссоциируют и не ионизируются, а ионизация ок-
сидов типа Ме2О3 (Fe2O3, Al2O3 и др.) в метаборате лития протекает
по схеме
Ме2О3->МеО^ + Ме3+ + О2^,
Насколько сложна расплавленная система можно судить по ме-
таллургическим шлакам. В работе ]160] отмечается, что металлурги-
ческие шлаки в расплавленном состоянии в результате термической
и электролитической диссоциации химических соединений состоят
из простых ионов Me2-** (Са2+, Mg2+, Mn2+, Fe2+), О2~ и недиссоцп-
ированных соединений (силикатов, алюминатов, фосфоратов, фер-
ритов п др.). В первом приближении можно считать, что свободные
окпелы типа МеО в жидких шлаках полностью диссоциируют на ионы
Ме2+ и О2’, а кремнезем, фосфорный ангидрид и глинозем находятся
в них в виде недпесоциированных соединений Me2SiO4, MeSiO3 и SiO2,
Ме3(РО4)2 и Р2О5.
Реальные расплавы горных пород состоят из ионов, пе имеющих
одинаковых зарядов, размеров и поляризуемости. В таких системах,
состоящих, например, из АХ и ВХ, энергетическая неравноценность
ионов одинакового знака приводит к тому, что обычно катион, об-
ладающий большей поляризующей силой, например Ап+, выступает
в роли комплексообразователя. В результате химического взаимо-
действия возникают комплексные анионы типа (AXTO)(m"n> , в то
время как катионы В+ остаются относительно свободными, не свя-
занными в группировки. Следовательно, в чисто ионных системах
прп химическом взаимодействии возникают не молекулы нового
соединения, а комплексные ионы, отвечающие этому соединению..
118
В зависимости от условий протекания реакций комплексообра-
зование носит частный характер. Соли некоторых оксикпслот типа
SiO2, В2О3 и Р2О5 образуют особую группу комплексов. В этих солях
группы SiO4, РО4, ВО3 и ВО4 приводят к возникновению комплекс-
ных ионов различных размеров, связанных общими атомами кисло-
рода. Обычно реакции с образованием таких оксидов описываются
кислотно-основной схемой. Например, реакция между силикатами и
карбонатами в расплаве может быть описала, уравнением
— Si — О — Si — + сот = - Si - (ГСГ - Si - + СО2 (газ),
II II
а реакции образования силпкатов — такими уравнениями:
2Ме2^ 4- 2О2- + SiO2 Me2SiO4,
Ме2+ 4- О2- + SiO2 MeSiO3
Исследование комплексообразования силикатов в области между
метасиликатами и чистой двуокисью кремния показало [147], что
представление о процессе плавления как о возрастании числа вакан-
сий непрпложпмо к плавлению соединений, подобных SiO2. Связь
кремния с кислородом чрезвычайно прочна, а теплота плавления сос-
тавляет только 1,85 ккал. Энтропия плавления SiO2 мала, и, следо-
вательно, расплав SiO2 сохраняет почти ту же степень упорядочен-
ности, что и кристалл кристобалита. Плотность двух форм двуокиси
кремния практически одинакова, так же как и низкотемпературная
теплоемкость кремниевого стекла п кристобалита. Вязкость расплав-
ленного SiO2 высока и резко снижается при добавлении оксидов ще-
лочных металлов. Все это указывает па то, что переход двуокиси
кремния в расплав сопровождается образованием кремнекислород-
ных комплексов
SiO2-j- иО2“ = SiOf^r
в расплаве SiO2 содержатся связанные вершинами тетраэдры Si04 и
структура жидкости мало отличается от структуры кристобалита.
При добавлении окиси щелочного металла типа К2О к расплав-
ленному SiO2 происходит реакция
I ! ’ + + I
— Si—О—Si— + К2О = — Si—О-К+К+О“—Si—
II i 1
Кислородные мостики разрушаются и образуют два несвязанных ато-
ма кислорода.
Изучение фпзико-хпмических свойств силикатных расплавов,
состав которых отвечает формулам Me2O-Me2O-4SiO2, Ме2О МеО-
*4SiO2, где Ме2О — оксид щелочного металла, МеО — оксид щелоч-
но-земельного металла, показало, что взаимодействие в этих распла-
вах тесно связано с состоянием иона кислорода [94] (прочность его
связи в самих оксидах подвержена взаимному влиянию катионов
119
[93, 94]). Исследование структуры трехкомпонентных силикатных
стекол с щелочными оксидами и оксидами двухвалентных металлов
позволило авторам [147] прийти к заключению, что алюминий, ти-
тан, цирконий, олово, железо, ниобий, тантал и т. д. соединяются
с атомами кремния через атомы кислорода, вводимые щелочным ок-
сидом. Это означает превращение анионов Si3O7A Si3Of~ и Si3Os“
в более сложные анноны, содержащие атомы алюминия, титана и
т. д.; например, в случае алюминия образуются анионы Si307(A10_),
Si3Or(AlCT), Si30s(A10-)2,Si30|^(A10-), 813ОГ(А1О“), Si309(A10-)3.
Путем присоединения к таким анионам одно-или двухвалентных по-
нов образуются определенные химические соединения.
Вораты имеют много общего с силикатами. В качестве примера
можно привести совокупность реакций при сплавлении оксида двух-
валентного металла МеО с обезвоженным тетраборатом натрия
Лта2В4О7:
a) Ха2В4О7^ 2NaBO2 + В3О3;
b) NaBO2 + МеО ХаМеВО3;
с) В2О3 Ч- МеО Ме(ВО3)2;
d) Ме(ВО,)2 + 2NaBO2-> Ха2Ме(ВО2)2.
Каждый пз продуктов термической диссоциации тетрабората
натрия реагирует с оксидом металла согласно уравнениям Ъ) и с), п,
кроме того, продукт стадии с) вступает в реакцию с метаборатом на-
трия. Системы с двумя стеклообразующпми оксидами состоят из
двух типов молеку’л, которые дают начало двум рядам сложных ани-
онов [147]. В случае системы Ме2О—В2О3—SiO2 такими молекулами
являются Si3O6H В4О(5, пз которых образуются анионы Si3O7“, Si30f—
Si3or пВ40Г,В40Г.
Возможные компоненты расплава и область составов, которая
будет давать расплав при данной температуре, можно ориентировоч-
но оценить с помощью диаграммы состояния. Хотя фазовые диаграм-
мы относятся к равновесию между твердыми и жидкими фазами, они
дают известные представления о структуре жидкости выше темпера-
туры плавления и позволяют сделать определенные заключения о
взаимодействиях компонентов в жидкой фазе.
На рис. 39 приведены простейшие типы диаграмм плавкости
двойных систем [10]. На каждой из нпх имеются две линии и трп
поля. Верхняя линия — линия ликвидуса, или линия жидкости.
Нижняя линия солидуса, или линия твердого. Поле, расположенное
выше линии ликвидуса,— поле жидкости или расплава. Если фигу-
ративная точка, отражающая состояние системы состав — темпера-
тура, находится в этом поле, система представляется однофазной.
Ниже линии солидуса расположено поле твердого состояния. В от-
лпчие от поля расплава поле твердого не является однофазным и пред-
ставляет собой совокупность подполей. Между линиями солидуса и
ликвидуса расположены поля кристаллизации. Система, фигуратпв-
120
Рис. 39. Простейшие тины диаграмм плавкости двойных систем:
а — эвтектического типа; б — непрерывный ряд твердых растворов, в — с конгруэнт-
но плавящимся химическим соедггнением; г — с шгконгруэнтно плавящимся хими-
ческим соединением; д — лонотектпческого типа.
нал точка которой расположена в любом из этих полей, обязательно
двухфазна, причем одна фаза жидкая, другая — твердая.
Диаграмма эвтектического типа приведена на рис. 39, а. Над
линией ликвидуса имеется раствор веществ А и В друг в друге. В этой
области без нарушения равновесия можно произвольно менять тем-
пературу и состав. В эвтектической точке е, когда в равновесии на-
ходятся трп фазы — одна жидкая и две твердые (А и В), нельзя ме-
нять произвольно ни один параметр состояния без нарушения равно-
весия. В случае неограниченной взаимной растворимости компонен-
тов как в жидком, так и в твердом состоянии (см. рис. 39,6) на ли-
нии моаовариантного равновесия acb гомогенный жидкий раствор
находится в равновесии с гомогенным твердым раствором. Если
компоненты Л и В обладают неограниченной растворимостью в жид-
ком состоянии п образуют химическое соединение АВ в твердом,,
то на линии ликвидуса должно быть три ветви, пересекающиеся
между собой: две ветви кристаллизации чистых компонентов и ветвь
кристаллизации химического соединения. При этом может быть два.
случая: состав жидкой фазы, образующейся при плавлении химиче-
ского соединения, совпадает с составом его в твердом виде или соста-
вы соединения к жидкой фазы не совпадают. В первом случае (см.
рис. 39, в) химическое соединение плавится как чистый компонент и
на диаграмме плавкости ему отвечает максимум. Во втором (г) при
нагревании химического соединения при определенной температуре
происходит распад его на жидкость состава точки е и компонент В.
121
В случае ограниченной растворимости компонентов в жидком состоя-
нии систему в простейшем виде можно представить как эвтектиче-
скую, в которой на одной из ветвей лпнпи ликвидуса имеется рассла-
ивание в жидкой фазе (см. рис. 39, д').
Диаграммы состояния боратных л силикатных систем многих
элементов и оксидов можно найти в работе [45].
Состав тройной системы можно изобразить с помощью равносто-
роннего треугольника. Если из любой точки его восстановить пер-
пендикуляры, пропорциональные температуре начала кристаллиза-
ции сплавов, то концы перпендикуляров окажутся в криволинейных
поверхностях ликвидуса. Таким образом, диаграмма выражается
некоторой объемной фигурой. Обычно поверхность ликвидуса со
всеми линиями равновесия и точками их пересечения проектируется
на плоскость концентрационного треугольника. Па таких проекциях
с помощью изотерм передаются все подробности объемной диаграммы.
Если компоненты не образуют твердых растворов и химических
соединений, то получается простейший тип диаграммы — с тройной
эвтектикой (рис. 40, а). Она состоит из полей первичной кристалли-
зации компонентов, трех линий кристаллизации двойных эвтектик:
еге, е2е и е3е и тройной эвтектической точки е. Если в одной из двой-
ных систем, например А—С (рис. 40, б), образуется конгруэнтное
(устойчивое) химическое соединение S, то всю тройную диаграмму
можно представить в виде двух элементарных или частных тройных
систем.
Движение к нужной части диаграммы состояния всегда может
быть достигнуто за счет изменения массы образца и флюса. Таким
образом, обеспечивается своеобразный, хотя н весьма приближенный
контроль за качественным составом расплава. Например, в серии
реакций для системы МеО—Ма2В4О7 кислород, способствующий пе-
ремешиванию в процессе плавления, можно сделать доминирующим
за счет излишка В2О3 в исходной смеси. На практике это достигается
использованием в качестве флюса пе чистой буры, а смеси буры с
борной кислотой.
После завершения основных химических реакций в результате
дальнейшего повышения температуры резко возрастают скорость
Рис. 40. Простейшие тины диаграмм плавкости тройных систем.
а — с тройной эвтектикой; б — с одиим'ионгруэлтно плавящимся двойным соедине-
нием; в — с двумя конгруэнтно плавящимися двойными соединениями,
122
диффузионных процессов и скорость растворения SiO2 и силикатов.
Как правило, взаимодействия, протекающие при высоких темпера-
турах, являются сложными гетерогенными процессами. Их протека-
ние в значительной мере зависит от скоростей доставки реагирующих
веществ в зону реакции, которые можно изменить, например, враще-
нием образца. В этом случае скорость доставки вещества v к фронту
гетерогенной реакции может быть определена выражением
v = 0,62
где D — коэффициент диффузии, — концентрация транспорти-
руемого вещества, v — кинематическая вязкость, со — угловая ско-
рость вращения образца. В некоторых случаях перемешивание (ус-
корение диффузии) заметно ускоряет реакции, но действие его каж-
дый раз конкретно. В [161] отмечается, что скорость растворения
SiO2 в метасилпкате лития при температурах 1000—1250°С не зави-
сит от числа оборотов образца. Напротив, в указанных условиях
растворение кремнезема в метасиликате натрия, а также в системе
СаО—SiO2—А12О3 возрастает при увеличении со.
Благодаря диффузионным процессам выравниваются концентра-
ции растворов силикатов на различных участках и в первом прибли-
жении образуется относительно однородный расплав. Завершают
процесс дегазация и гомогенизация за счет молекулярной диффузии
при малой вязкости расплава.
4.3. СОСТАВ ПЛАВНЕЙ
В принципе расплавы горных пород могут застывать в виде стек-
ла, так как основной составной частью большинства породообразую-
щих минералов является один пз главных стеклообразующих окси-
дов — SiO2. По температуры перехода природных силикатов из
кристаллического состояния в жидкое относительно высоки [103]:
Минерал Температура
плавления, °C
SiO2 (кварц) 1610
Mg3[SiO4] (форстерит) 1890
CaMg[SiO4 ] (монтичеллит) 1503
Ca2Al[AlSiO7l (галенит) 1584
CaSiOg (волластонит) 1813
CaMg[Si3O6] (цпопспд) 1391
Ca[Ai2Si2O8] (анортит) 1550
Na[AlSiOJ (иефолпп) 1523
Na[AlSi3O8] (альбит) 1118
Для пород они еще выше из-за присутствия таких тугоплавких ми-
нералов, как перовскит CaTiO3 (1970°С), ильменит FeTiO3 (1367’’С),
шпинель Mg(A102)2 (2135°С), периклаз MgO (2800сС) и др. Кроме того,
весьма ограничены области стеклообразованпя (рис. 41). Поэтому
для получения стекол в реально обеспечиваемых условиях горные
породы приходится сплавлять с флюсами.
123
SiC';
Рис. 41. Области образования стекла в системах MgO— А12О3—
—Si02 и CaO—Fe2O3—SiO2 [89, 90].
Флюс не до л жен содержать определяемых элементов, иметь не-
большую поглощающую способность и невысокую вязкость, пол-
ностью растворять пробу при достаточно низких температурах и не
образовывать с компонентами пробы летучих соединений. Конкрет-
ный выбор флюса и степени разбавления пробы зависит от анализи-
руемого материала, требований к чувствительности и точности ана-
лиза, используемой методики учета матричных эффектов.
С точки зрения получения стекла лучшим флюсом должен быть
борный ангидрид В2О3. Это бесцветная хрупкая стекловидная гигро-
скопическая масса, плавящаяся при температуре 450сС и в расплав-
ленном состоянии легко растворяющая оксиды металлов. Ниже для
сравнения приведены точки плавления некоторых силикатов и бо-
ратов:
Соединение
Температура
плавления, °C
Соединение
Na,0-Si0o
Na^O-B2O3
Na;O-2B2O3
Na2O-3B,O3
Na,O-4B2O3
K2O-SiOa
K2O-B2O3
1088
960
741
694
783
966
947
CaO'SiO2
Cat)-B2O3
MgO-SiO2
MgO-B2O3
BaO-SiO2
BaO-B2O3
Температура
плавления. С
1540
1095
1554
1142
1604
1060
Отчетливо видно, что замена SiO2 на В2О3 в большинстве случаев за-
метно понижает температуру плавления большинства оксидов; рас-
ширяется и область стеклообразования [89, 90]. Практически при
любых реальных соотношениях В2О3 : SiO2 из расплавов получаются
стекла.
Поскольку стекловидный оксид бора обычно получают обезво-
живанием борной кислоты 1]3ВО3, которая летуча с водяными пара-
ми, для сплавления используют соли борной п тстраборной (Н2В4О7)
кислот, застывающие подобно В2О3 из расплава в виде стекла. Пре-
имущественно это соли щелочных металлов: лития и натрия — для
них характерны чрезвычайная легкоплавкость и крайняя реакционно-
способность в химическом отношении.
524
Бура Ха2В4О?’ 10 Н2О — натриевая соль тетраборной кислоты —
образует большие бесцветные прозрачные моноклинные кристаллы,
которые выветриваются в сухом воздухе. Плавление буры в собствен-
ной кристаллизационной воде можно избежать при осторожном на-
гревании до 350—400°С. Получающийся при этом безводный тетра-
борат натрия Na2B4O7 плавится при 878СС. Иногда сплавление ведут
со смесью буры с содой (1\та2СО3)(1 : 2) млн пероксидом натрия
(Лта3О2). Спекание выполняют прп 420'С в течение 2 ч в платиновых
(при содержании серы менее 5%) тиглях. Но натриевые плавни мало
удобны, так как натрий приходится определять из отдельной навески.
Более того, поскольку Na3B4O- — соль слабой кислоты п сильного
основания, она недостаточно активна для основных пород. В настоя-
щее время натрий применяется преимущественно для получения
твердых расплавов из рудных материалов [190, 327].
Применительно к природным материалам различного характера
более универсальны бораты лития Li2O • пВ3О3, щелочных металлов,
поведение которых ближе к поведению щелочно-земельных металлов.
Из диаграммы плавкости системы Li2O—В2О3 [45] видно, что при
используемых температурах смесь этих оксидов в любых весовых со-
отношениях является жидкой. Значительно расширяется область стек-
ла по сравнению с системой Ха2О—В2О3 (рис. 42). Для сравнения jia
рис. 42 приведена область стекла для смеси флюсов БцО—Na2O —
В2О3.
Промышленностью выпускаются литий тетраборный L.2B4Or
п литий борнокислый Л(еп?л-2-вод1гый T.iB<J2-2H.2O. Тетраборат ли-
тия плавится при температуре 915СС п разлатает широкую область
материалов. Но при малых степенях разбавления он относительно
слабо взаимодействует с материалами, содержащими в больших
количествах кислотные оксиды, например кислыми породами, и боль-
ше подходит для образцов с высоким содержанием основных окси-
дов (карбонатные породы) [3'13]. Прп температурах, близких к точке
плавления, тетраборат лития является очень вязкой жидкостью.
Для того чтобы получить жидкий расплав, обеспечивающий удовлет-
ворительное перемешивание и выливание, температура плавления
должна быть сравнительно высокой (1200—1300=С).
Повысить щелочность и понизить точку плавления (735°С) про-
мышленного тетрабората лития можно добавкой карбоната лития
2205
Рус. 42. Область стеклообразоваиия в системах Na,О—К,О—В2О3; Li2O—К2О —
-В2О3; Li2O-Na2O—В2О3 [89, 90].
125
Li2CO3. Из-за летучести оксида лития и образования пероксида ли-
тия смесь должна быть предварительно сплавлена. Для приготовле-
ния такого флюса 16] исходные материалы берутся в следующих
количествах (г); Li2B4O7 — 38,0; Li2CO3 — 29,6; La2O5 — 13,2. Ок-
сид лантана добавляется в качестве тяжелого абсорбента. До взве-
шивания соли ли'шя прокаливаются при 500°С, а оксид лантана —
при 900°С. Смесь высыпается в платиновый пли стеклографитовый
тигель и нагревается в течение 10 мин при 1000°С. За это время раст-
воряется оксид лантана и прекращается’ бурное выделение газа. Что-
бы предотвратить разбрызгивание расплава на внутренние части пе-
чи, тигель закрывается крышкой. Расплав выливается на массивную
алюминиевую полированную плиту. Для предотвращения расплав-
ления алюминия и вплавлония в него стекла толщина растекающегося
по плите расплава должна быть не менее 3 мм. Стекло измельчает-
ся в грубый порошок п засыпается в герметическую посуду. За дли-
тельный пролмежуток времени флюс, особенно если он тонко измель-
чен, может поглотить несколько процентов воды. Но она легко уда-
ляется прокаливанием порошка при 450°С в течение 2 ч.
Обезвоженный метаборат лития намного активнее тетрабората
при сплавлении кислотных оксидов, таких как кремнезем и глино-
зем, поэтому он более подходит для получения твердых растворов
из изверженных пород. Он плавится при 854°С и за счет резко выра-
женной у лития склонности к образованию двойных солей быстро
растворяет большинство силикатов и многие неспликаты, давая стек-
ло, механически прочное п негигроскоппческое.
Иногда для повышения разлагающей способности флюса в не-
больших количествах вводятся такие добавки, как углекислый ли-
тий 1п2СО3, нитрат (NaNO3) пли пероксид (Na.2O2) натрия. Они име-
ют относительно низкие температуры плавления (735; 306,8 и 460°С
соответственно) и обладают окислительным действием, в результате
которого элементы с переменной валентностью, содержащиеся в ис-
ходном образце (Сг, Мп), окисляются до наивысшей степени [121].
Если в щелочном материале недостаточно образующих дисперс-
ную структуру элементов Si, А], Р, то щелочность расплава пони-
жают добавлением нескольких процентов SiO2, В.2О3, ВеО [227].
В последнее время появились работы по исследованию возмож-
ностей применения слоя;ных флюсов, состоящих из смеси боратов в
различных соотношениях: Li2B4O7 п LiBO2; Li3B4O7 и Г\а2В4О7
[3131. Авторы дают рекомендации по выбору сложных флюсов для
гомогенизации различных видов минерального сырья.
Эффективность сплавления в значительной степени определяется
соотношением между частями пробы и флюса. Очевидно, что единого
для всех случаев соотношения нет, оно зависит от природы материала
и требований анализа. Для гомогенизации силикатов применяют в
основном умеренные 1 : (5—10) [174, 268, 272, 289, 3131 или низкие 1:
:(1 —2) [40, 151, 227, 167] степени разбавления. Повышение степени
разбавления в указанных пределах, улучшая качество расплава,
снижает чувствительность анализа. Поэтому в каждом конкретном
случае находится компромиссное решение.
«26
В первом приближении расплав можно рассматривать как
раствор различных оксидов и силикатов в SiO2 (растворителе). 0 этой
точки зрения при увеличении отношения образец: флюс выше 1 : 10
из-за малого содержания оксида кремния повзлшается вероятность
получения негомозепных расплавов. Поэтому для силикатов рабочее
соотношение колеблется в интервале 1 : 3 и 1 : 10, для несиликатов.
оксид кремния может быть добавлен в процессе сплавления.
4.4. СПЛАВЛЕНИЕ ГОРНЫХ ПОРОД И МИНЕРАЛОВ
Предварительно тщательно перемешанная, вручную или меха-
нически, смесь тонко истертой пробы и флюса высыпается в тигель.
Как правило, применяемый для сплавления тигель не должен быть
заполнен более чем наполовину. Вначале нагревать следует медлен-
но, иначе могут быть потери за счет разбрызгивания из-за выделения
паров воды и других газов. Для большей надежности тигель следует
закрывать.
Чтобы избежать контакта расплава со стенками тигля, тигель
вращают. При этом расплав собирается в центре и ускоряется на-
ступление равновесия.
4.4.1. Состав атмосферы
Для сплавления применяют следующие среды:
1) нейтральные или защитно безокпслптельные (азот, аргон,
гелий);
2) окислительные (О2, П2О, СО2) — воздух, продукты сгорания
с избытком воздуха (О2 в СО2); продукты неполного сгорания с из-
бытком воздуха и остаточными — углеродом, кислородом и углево-
дородами; ограниченный объем воздух-a, изолированный от расплава;
3) восстановительные — водород, окись углерода, диссоцииро-
ванный аммиак, продукты неполного сгорания природного газа.
Восстановителями могут быть твердые и газообразные вещества. Так
как в общем случае реакция восстановления имеет вид: МеО Ч- В =•
— Me -+- ВО, где В — восстановитель, Me — металл, в качестве вос-
становителей применяют вещества, обладающие в данных условиях
большим сродством к кислороду, чем восстанавливаемый элемент.
Для установления равновесия в системе динамическими метода-
ми через нее постоянно пропускают газовую смесь определенного
состава, из баллона или газгольдера. При статических методах уста-
новления равновесия введенную в систему порцию газа или образо-
вавшуюся в ней атмосфер^7 неоднократно перекачивают, создавая
замкнутый цикл газовой атмосферы. Для этих целей используют
циркуляционные насосы различных конструкций. Восстановитель-
ную атмосферу в печи можно создать путем использования графито-
вого цилиндра, внутри которого на графитовой подставке-блоке по-
мещается тигель. Благодаря окислению графита при нагреве в атмо-
сфере печи содержится некоторое количество СО, предотвращающее
окисление.
127
4.4,2. Температура и время сплавления
Два минерала могут образовать непрерывную серию твердых
растворов только при температуре, выше критической 7’с. При бо-
лее пизких температурах раствор может стать неустойчивым в опре-
деленном диапазоне состава 1'131].
В зависимости от используемого флюса, степени разбавления и
состава анализируемого материала температура сплавления колеб-
лется от 950 до 1300°С. Высокий нагрев гарантирует энергичное
протекание реакций. При реакции карбонатов щелочпых элементов
с кремнеземом образуется двуокись углерода, карбонаты щелочно-
земельных металлов разлагаются, а их окиси переходят в раствор;
разлагаются гидраты и нитраты, а оксиды взаимодействуют с сили-
катным расплавом. Выделяющиеся газы энергично перемешивают
расплав, способствуя быстрому течению реакций. В конечном итоге
достигаются полное растворение п сплавление материалов. Повыше-
ние температуры сплавления уменьшает вязкость расплава и спо-
собствует его гомогенизации, но возникает опасность улетучивания
заметных количеств элементов, особенно щелочных [6]. Использо-
вание температур ниже 950°С приводит к неполному растворению
пробы [272]. Источниками нагревания служат муфельные печи, Мек-
кер-горелки п высокочастотные печи.
Основная задача нагревательного устройства сводится к созда-
нию зопы равномерного нагрева в определенном ограниченном объе-
ме и обеспечению температурного ре,1,има во времени {79]. Наиболь-
шее распространение в лабораторной практике получили электриче-
ские печи. В зависимости от способа нагрева различают печп сопро-
тивления и высокочастотные печп.
Печи сопротивления хорошо поддаются регулированию, удобны
для создания заданного распределения температуры в камере naiре-
ва. Максимально допустимая температура в таких печах зависит
от устройства печи п планируемого срока службы нагревателя. Для
них можно применять нагреватели различной формы. Чтобы полу-
чить температуры 1200—1б00°С, в качестве нагревателей использу-
ют благородные металлы.
Платиновая спираль служит достаточно долго при работе в
области 1400—1500°С. Сплавы платины с родием могут применяться
при нагреве до 1700сС, но срок их службы меньше, чем у платины.
Для более высоких температур преимущественны молибден, воль-
фрам, тантал, графит.
Молибденовую проволоку пли вольфрамовые прутки допускает-
ся применять в вакууме и при создании восстановительной атмосфе-
ры, например водородной. Водород подают в печь непрерывно, под-
держивая в корпусе установки положительное давление, что устра-
няет возможность подсоса воздуха и образования гремучей смеси.
Па выходе из гечп водород сжигают. Нагреватели из дпеилицпда
молибдена можно попользовать в окислительной атмосфере до 1600сС.
Из неметаллических нагревателей наибольшее распространение
получили силитовые нагреватели — готовые стержни, трубы из
128
карбида кремния и стержни из дпсплицпда .молибдена, работающие
в окислительных условиях. В печи стержни могут располагаться как
вертикально, так п горизонтально. Силптовые нагреватели прп пра-
вильной эксплуатации в условиях высоких температур служат в те-
чение нескольких тысяч часов. Большое значение имеет закрепление
стержней в токоподводах, через которые проходят большие токи.
Для плотного закрепления конец нагревательного стержня можно
сделать на 0,25 мм больше по диаметру, чем отверстие в токоподво-
де, п пропилить его вдоль, что обеспечит плотный и надежный кон-
такт.
К достоинствам высокочастотных печей относятся универсаль-
ность бесконтактного нагрева, хорошая регулируемость теплового
режима, возможность noxj чення высоких температур л литания раз-
личных установок от одного генератора. Источником питания слу-
жат высокочастотные ламповые и машинные генераторы, выпускае-
мые промышленностью для лабораторных установок различной
мощности. В лабораторной практике индуктор приходится рассчи-
тывать редко. Чаще его подбирают к имеющемуся генератору и на-
греваемому объекту, т. е. к тиглю с расплавом. Для нагрева графи-
та, платины пригодны частоты до 10 000 Гц. Для материалов с более
высоким электросопротивлением требуются более высокие частоты.
Трудность нагрева неметаллических и немагнитных материалов,
ограничение размеров нагреваемого пространства контуром индук-
тора являются недостатками печей этого тина.
Время бурного протекания реакций в расплаве составляет 10 %
всего времени сплавления, остальные 90% необходимы для подпой
гомогенизации расплава. Общее время сплавления зависит от соста-
ва сплавляемого материала п источника нагревания. При использо-
вании электрического муфеля или Меккер-горелкп длительность
сплавления 10—20 мин для легкоплавких продуктов и до 30 мгш для
материалов, богатых кварцем, а также рудных материалов. Гриме-
нение высокочастотной печи позволяет сократить время сплавления
до 3—5 мин [313]. Чаще всего сплавляют предварительно прока-
ленную пробу, перемешанную с флюсом [272, 309]. Для гомогениза-
ции непрокаленного материала процесс сплавления лучше осущест-
влять поэтапно [218, 268. 290]. Па первом этапе смесь пробы с флю-
сом подвергается нагреванию до температуры разложения смеси.
Из смеси выходят воздух, диоксид углерода и вода. Такая процедура
предупреждает разбрызгивание расплава во время плавления. На
втором этапе расплав нагревают до более высокой температуры
(1200—1300°С)• При этом в результате понижения вязкости расплава
резко возрастает скорость диффузных процессов. Для лучшего раст-
ворения пробы п очищения стенок тигля от капель расплава тигель
с расплавом встряхивают после первого периода сплавления [218,
257] нлп вращают тигель в течение второго периода сплавления
[259].
Замечено [6], что при получении стеклопзлучателей из геологи-
ческих образцов скорость счета некоторых элементов, например Si
и 1?е, изменяется на 2—3% при каждом переплавлепии и становится
129
постоянной после трехкратного переплавлепия. Наиболее вероят-
ной причиной такого поведения стекла является неполное растворе-
ние образна при сплавлении, что легко обнаруживается с помощью
микроаналпзатора по наличию в излучателе фаз. В этом случае долж-
ны быть пересмотрены условия сплавления: подбирается более реак-
тивный флюс, увеличиваются степень разбавления и температура
сплавления 1276].
Качество и чистота расплава зависят от свойств используемых
тиглей.
4.4.3. Материал тиглей
Очевидно, что для зпгля необходимо использовать термостойкий
материал, который по смачивается стеклами, полученными в резуль-
тате сплавления пробы с флюсом, химически инертен к реагентам и
нс загрязняет расплава. Ниже приведены значения температуры плав-
ления соединений и металлов, которые используются или с учетом
их свойств могут быть использованы в качестве материалов тиглей
для сплавления:
Веще- ство Температура плавления, 'С Веще- Температу- стео ра плавле- ния. С Веще- ство Температу- ра плавле- ния. С
С 2500 V 1735 Th 1827
В 2300 Zr I860 Не 3180
В4С 2450 Мо 2625 Та 2990
S1 с 2700 W 3410 Os 27(И)
В У 3000 Pt 1774 Вп З'Л 111
SiO> 1710 Ап 1336 1г 2 4;>4
Сг 1890 Р<1 1823 Ш 211(1
'£i 1725 Ш1 I960 Nb 2415
(3 амые дешевые п доступные — графитовые тигли. сделанные
высверливанием нужной полости в графитовом стержне высокой
чистоты 133, 15(, .194, 272]. Тигли, конечно, должны быть свободны
от выпуклостей и других механических несовершенств. Если графи-
товые тигли предварительно прокалены, то расплав собирается в ко-
ролек и легко без потерь удаляется пз тигля. Многократность ис-
пользования графитовых тиглей зависит от толщины их стенок и
атмосферы мечи. Если воздух циркулирует слишком свободно, тигли
сгорают очень быстро. При подобранных условиях сплавления тигли
обгорают отпосител ыго медленно и каждый .может бы ть использован
несколько раз. Однако полученные в них корольки часто покрыты
тонким рваным слоем графита, который может изменить интенсив-
ность аналитических линий элементов кремния, алюминия и магния
на 3% [272]. Кроме того, при сплавлении в графитовых тиглях суль-
фатсодержащпх пород полностью выгорает сера, частично — фос-
фор и железо 1260]. Использование графитовых тиглей нередко ог-
раничивается тем. что примеси графита переходят в расплав и вьгзы-
1-0
вают его прилипание, особенно при плавлении материалов, содер-
жащих легкоокисляемые компоненты.
Смачивание твердых тел жидкими расплавами веществ с высоким
поверхностным натяжением имеет место в том случае, когда между
веществами жидкой и твердом фаз возникают сильные химические
связи. Ноэтохму влияние примесей определяется в значительной мере
спецификой взаимодействия растворенного вещества с твердым телом,
чаще всего — возмохкностьто образования химических соединений
или твердых растворов. В ряде случаев влияние примесей связано с
тем. что в их присутствии изменяются химический состав и строение
поверхности твердого тела.
Смачивание графита происходит, как правило, в тех случаях,
когда элемент достаточно интенсивно взаимодействует с углеродом
148, 99, 100]. Карбпдообразующие непереходные элементы (алюми-
ний, кремпий, бор) могут давать с углеродом ковалентные соедине-
ния, поэтому расплавы таких веществ могут смачивать графит при
определенных условиях. Важную роль играет температура. Напри-
мер, ниже 1000°С смачивание жидким алюминием отсутствует, но
при более высоких температурах (при контакте с графитом выше
1200°С) происходит полное смачивание [99].
Оксид кальция, взаимодействуя с графитом на стенках тигля,
образует карбид кальция, который затем распадается на границах
раздела с газом. Появляющийся углерод на поверхности образует
кристаллики графита пли переходит в расплав, вызывая его наугле-
роживание. В итоге в прилегающих слоях фаз изменяются концентра-
ции других ионов, и прежде всего кислорода, железа и кремния.
Все переходные металлы (титан, хром, цирконий, марганец и
др.) образуют с углеродом прочные металлоподобные связи, поэтому
они, как правило, хорошо смачивают графит. Этим свойством облада-
ют чистые металлы подгруппы железа (железо, кобальт, никель)
[99]. Характерно, Что смачивание графита непереходными металлами
значительно улучшается при растворении в пих переходных металлов
(ниобия, молибдена, ванадия п др.). Особенно сильно улучшают сма-
чивание добавки хрома и титана [96, 99], в ряде случаев они обеспе-
чивают полное смачивание, поскольку растворяемые элементы ин-
тенсивно взаимодействуют с углеродом.
В системах, в которых смачивание связано с образованием твер-
дых растворов, большую роль играет ускорение диффузии вещества
жидкой фазы в твердую подложку. Характерно, что смачивание
происходит не мгновенно после соприкосновения жидкости с твердой
поверхностью, а лишь через некоторый интервал времени, который
тем короче, чем выше температура [99]. Есть основания считать, что
задержка смачивания связана с диффузией вещества жидкой фазы в
твердое тело.
Известно много экспериментальных данных, свидетельствующих
о том, что геометрия твердой поверхности (микрорельеф) также ока-
зывает весьма существенное, а в ряде случаев — решающее влияние
на величину краевых углов при ограниченном смачивании и на воз-
можность растекания жидкости по твердой поверхности.
131
В [148] отмечается, что полированный графит смачивается рас-
плавами солей и оксидов гораздо хуже, чем грубо обработанный.
Поверхность реальных твердых тел никогда не бывает идеально глад-
кой плоскостью; она покрыта многочисленными неровностями раз-
личной формы. Размеры поверхностей изменяются в очень широких
пределах — от нескольких межатомных расстояний до десятков мик-
рон, а в отдельных случаях — до нескольких миллиметров. Влия-
ние шероховатости па смачивание изучено наиболее подробно для
поверхностей с неровностями, средняя глубина или высота которых
составляет от десятых долей до нескольких десятков микрон. По-
скольку движущая сила растекания растет с увеличением коэффи-
циента шероховатости 2>, можно ожидать, что на более шероховатых
поверхностях средняя скорость растекания будет больше, чем ла
гладких. Царапины, параллельные направлению течения жидкости,
могут ускорять растекание, тогда как поперечные царапины вызы-
вают его замедление.
Скорость растекания можно регулировать и неравномерным на-
гревом жидкого слоя. Физическое обоснование данного метода состо-
ит в том, что поверхностное натяжение жидкости уменьшается при
повышении температуры. Поэтому прп различии температур в раз-
ных участках жидкого слоя возникает движущая сила растекания,
которая пропорциональна градиенту поверхностного натяжения
жидкости. В результате возникает поток жидкости в смачивающей
пленке. Этот эффект называется термокалиллярным. Большую роль в
ряде случаев могут играть п другие физико-химические процессы.
В настоящее время тигли из углерода применяются довольно ред-
ко [328].
Использование стеклографптовых тиглей в значительной сте-
пени исключает перечисленные недостатки [1391. Стеклоуглерод
марки СУ-2000 [76] получают термической обработкой специально
подготовленных термореактпвных полимеров, формованных в раз-
личные изделия. Па 99.99 % он состоит из углерода, в котором со-
держание примесей по 19 элементам по превышает 1 • 10_я% по массе.
Стеклоуглерод отличается изотропностью структуры и свойств, мои-з-
лпгпостыо, малой, преимущественно закрытой пористостью, что обус-
ловливает его практически полную газонепроницаемость. Высокая
термическая стойкость стеклоуглерода позволяет пздоигяи из пего
выдерживать быстрый многократный нагрев и послсдуюпдео. быстрое
охлаждение. Тпг.ш из стеклоуглерода выдерживают многократное
сплавление в них проб различных материалов с кислыми и щелоч-
ными плавнями при температурах не выше 7'.;0—750 С; выдержка
при нагреве не более 15—20 мни с обязательным перемешиванием
расплава.
Посуда из стеклоуглерода практически не згнрязляег анализи-
руемой пробы, так как содержание примесей достаточно мало и они
Под коэффтшедто.ч шероховатости понимают ошо’пгниа фактической
площади тювсрхпос'пт (с учетом площади вьядлп в выступов) к проекция на го-
ризонтальную плескает ь.
132
не диффундируют пз стеклоуглерода, а основным продуктом взаимо-
действия стеклоуглерода с кислородом воздуха является диоксид
углерода, самопроизвольно удаляющийся из анализируемого объ-
екта. Образующийся при сплавлении плав легко удаляется пз тиг-
ля, поскольку стеклоуглерод не смачивается расплавами солей и ще-
лочей. В процессе сплавления посуда ле деформируется.
При сплавлении в платиновых тиглях и тпглях из сплавов на
основе платины (золото-платиновых, платино-родпевых) уровень за-
грязнения резко понижается. К ценным химическим свойствам этого
мягкого тяжелого металла относятся инертность по отношению ко
многим расплавленным солям, стойкость к окислению (даже при по-
вышенной температуре) и очень высокая температура плавления.
Тигли пз этих материалов длительно работают в воздушной и инерт-
ных средах, более кратковременно — в вакууме вследствие избира-
тельного испарения отдельных составляющих, например родия из
сплава платпнородий, и весьма кратковременно — в восстановитель-
ных средах и атмосферах продуктов неполного сгорания за счет вза-
имодействия с восстановленными из окружающей среды парами ме-
таллов и неметаллов, в большинстве своем приводящих к быстро-
му охрупчиванию и последующему разрушению.
Сплавленпе в платиновых тпглях следует выполнять в высоко-
частотной индукционной печи или пламенп горелки, обеспечивающих
скачкообразное изменение температуры. Атмосфера печи должна
быть окислительной; уменьшение ее окисляющих свойств способст-
вует быстрому экстрагированию пз расплава в платину Fe, Си, РЬ,
Со, Мп и ряда других элементов. При сильном нагревании Pt легко
сплавляется с такими металлами, как Ан, Ag, Bi, Zn. Осажденные
металлы не только делают анализ ненадежным, но и при последую-
щих сплавлениях, возвращаясь, загрязняют расплав. Для полной
гарантии исключения каких-либо потерь элементов при сплавлеипи
к флюсу можно добавлять некоторое колпчество (до 2%) нитрата
натрия, который способствует переходу пеустойчпшлх соединений
серы в устойчивые сульфатные.
Необходимо иметь в виду еще ряд присущих платине свойств.
При высокой температуре (~1300°С) платина адсорбирует пары ме-
таллов, а также кремния, серы, фосфора п др. из окружающей среды.
Самым неблагоприятным моментом оказывается наличие в атмосфере
серы, которая с выделившимся при разложении SiO2 кремнием об-
разует соединение SiS2, разлагающееся в контакте с платиной. Отри-
цательно сказывается влияние угольных материалов, так как углерод
является восстановителем оксидов, в которых присутствуют примеси
кремния.
Кремний и бор представляют собой наибольшую угрозу. Они
легко поглощаются платиной с выделением силицидов и боридов
платины, в результате чего происходит ее охрупчивание. Поскольку
температура пх образования ниже используемых температур плавле-
ния, время жизни платиновых тиглей резко сокращается. Когда
сырье растворяется во флюсе при температурах 1000—1300°С, на-
блюдаются восстановление оксидов кремния и бора п образование
эвтектик [162].
133
Реакцию этого процесса можно записать следующим образом:
Pt + SiO2 -г В -> 14 — Si 4- В-оксид.
Как видно, нагревание в воздухе при достаточно высокой темпера-
туре должно тормозить реакцию восстановления кремния за счет
окисления восстанавливающего вещества. Существенно, что эта реак-
ция окисления в платиновом тигле может идти без каких-либо по-
бочных эффектов, приводящих к порче тигля.
Наряду с образованием эвтектик имеет место миграция металли-
ческой платины в стекло. Процесс миграции элементов в стекле соот-
ветствует следующей схеме:
— +
^2Na+ (SiO3).v 4 -SiOr
мигрирует (SiO;!)x I -SiO^
2Na(SiO,X-.SiO2 -P О
2e~
Тигли из чистой платины неудобны по той причине, что расплав
смачивает их поверхность, а следовательно, значительное количество
его остается в тигле после выливания. Масса прилипшего стекла за-
висит от плотности и вязкости расплава.
Твердые металлы хорошо смачиваются расплавами в тех случаях,
когда контактирующие вещества дают химические соединения пли
твердые растворы; в отсутствие химического взаимодействия или
растворимости смачивания обычно не наблюдается [99]. Связь меж-
ду выделением химических соединений (или твердых растворов) и сма-
чиванием подтверждают данные о взаимодействии различных твер-
дых металлов (железо, золото, серебро) с жидкими металлами в ат-
мосфере водорода.
При полной несмешиваемости в жидком и твердом состояниях
смачивание отсутствует [16]. Наличие окисной пленки может ради-
кально изменить картину: определяющими становятся характер вза-
имодействия расплава с оксидом и строение оксидной пленки (поры,
трещины).
Связь оксидный расплав — металл возникает в две стадии. Пер-
вая — образование связи металл — кислород за счет окисления по-
верхности металла в процессе нагрева его в кислородсодержащей
атмосфере печи. Вторая — взаимодействие пленки окисленного ме-
талла с окисным расплавом.
Если расплав реагирует с твердым металлом, то он постепенно
растворяет слои металла, непосредственно прилегающие к окисной
пленке [99]. Далее расплав распространяется между окисным слоем
и металлом, при этом окисная пленка постепенно отделяется; в ре-
зультате поверхность металла оказывается полностью смоченной.
При используемых температурах цдавления платина окисляет-
ся стеклом, в то же время эти оксиды растворяются в последнем.
Прилипание наблюдается тогда, когда доминирующим будет первый
процесс, что имеет место в большинстве случаев из-за высокой актив-
ности боратных расплавов.
134
Явление прилипания имеет электрохимическую природу. Об-
разование химической связи идет параллельно с переходом электро-
нов пз металла в жидкий расплав. Если Me” — заряженный катион,
то вследствие катодного процесса Ме“ ' переходят пз металлического
электрода в окпсвое состояние л затем — в раствор
Me Me’l+ + пе~'
Процесс сопровождается изменением свободной энергии До системы
До — —nFV, где п — число участвующих электронов, V — электро-
движущая сила. F — число Фарадея.
Малое изменение свободной энергии окислительной реакции
п.татины приводит к возникновению потенциала полуячейки жидкое
стекло — платина. Уменьшение оксидов млатипы, сводящее прили-
пание к нулю, легко реализовать, если пропускать постоянный ток
через гальваническую пару платина — жидкое стекло. Поскольку
необходимо исключить любой механический контакт между электро-
дом и поверхностью стекла, применяется высоковольтный разряд.
Действие искры уменьшает окисный слой, и в результате увеличи-
вается электрическое сопротивление в месте удара. Последовательно
проходя участок за участком, искра уменьшает окисный слой по по-
верхности и осуществляет, таким образом, процесс очистки, который
продолжается до тех нор, пока не будут восстановлены все оксиды.
Плотность такой искры должна быть достаточно высокой, чтобы ее
действие могло конкурировать с одновременно идущей реакцией
окисления.
Управлять смачиванием можно и с помощью поверхностно-актив-
ных веществ3). Влияние ПАВ на смачивание определяется прежде
всего химической природой (составом) контактирующих веществ
и самого ПАВ. Вместе с тем большую роль играют п многие другие
факторы: температура, особенности реальной структуры твердой
поверхности и т. и. Влияние ПАВ на смачивание во многом за-
висит от того, на какой поверхности раздела фаз, участвующих в сма-
чивании, происходит их адсорбция. На практике встречаются сле-
дующие случаи.
1. Адсорбция ПАВ на иоьерхпости жидкости, происходящая
до начала ее контакта с твердым толом. Для этой цели заранее раст-
воряют ПАВ в жидкости. В результате снижается поверхностное на-
тяжение жидкости и, как следствие, изменяется краевой угол при
контакте раствора с твердым телом.
2. Адсорбция веществ, растворенных в жидкости, н<\ поверх-
ности твердого тела во время смачивания. В результате изменяется
межфазное поверхлтостпоо натяжение.
В табл. 22 приведены значения поверхностного натяжения при
температуре 7’, несколько большей температуры плавления, и его
3) Новерхностяо-акгпвны.чгг веществами (ПАВ/ называются вещества, ко-
торые могут адсорбироваться на поверхности раздела фаз, снижая ее поверх-
ностное натяя-юппе.
135
Таблица 22
Соединение C s= Я л ® t=: E-1 S V, -1 ДЧН-СМ Т(П
Nal 662 83,4 433,7- 0.793Г 4- 0,437-10-3 7'2
NaBr 750 103.3 164,8 0.(5809 T
KI 685 7 t К 438,7 -0.087'f
КВг 735 88,2 142.2-0.072Г
Bbl 640 79,85 140,2—0,103 Г--0.193-10-32'‘-
llbBr 680 86,2 138,0—0,072?
Na2B4O7 950 230,0 —
NaBO. 996 193,3 359.(3— О.163Г
LiBO2 845 261,0 197.7-',-0,174 Г— l,16-10-Jr2
зависимости от температурь! для иодидов и бромидов щелочных ме-
таллов (Na, К., Rb), которые используются в рентгеноспектральпом
анализе в качестве ПАВ [214, 268, 291, 328]. Для сравнения в нее
включены подобные величины ti для -гетра- п мстаборатов натрия
и лития.
В присутствии окислителя эффективен только бромид. Добавку
желательно делать в конце сплавления, так как все эти соли, за иск-
лючением НВг, летучи при температуре 1200°С. Что касается огра-
ничений, то платина легко растворяется в царской водке, в смесях
хлоридов с окислителями. При повышенных температурах опа раст-
воряется также в расплавах оксидов щелочных металлов, в перокси-
дах и до некоторой степени — в гидроксидах. Платина медленно раст-
воряется прп контакте с расплавленными нитратами, цианидами.,
хлоридами щелочных и щелочно-земельных металлов при температу-
ре свыше 1000°С; при температуре свыше 700°С металл слегка реаги-
рует с сероводородом. Поверхность платины подвержена воздейст-
вию аммиака, хлора, летучих хлоридов, диоксида серы л газов с вы-
соким содержанием углерода. При температуре красного каления
мышьяк, сурьма и фосфор легко реагируют с платиной, придавая ей
хрупкость. Такое же действие на платину оказывают селен, теллур
и в меньшей степени — сера п углерод. Наконец, длительное нагре-
вание при температуре свыше 1500°С способствует значительной по-
тере массы вследствие улетучивания металла.
Основные рекомендации по обращению с платиновой посудой
сводятся к следующему [136, 137].
1. Пользоваться платиновой посудой только в тех случаях, ког-
да нет опасений, что металл будет поврежден. Если состав системы
вызывает сомнения, перед употреблением платиновой посуды не-
обходимо убедиться в отсутствии потенциально вредных для нее ком-
понентов.
2. Избегать резких колебаний температуры: если при охлаж-
дении объем содержимого платинового сосуда увеличивается, сосуд
может деформироваться.
136
3. Из-за склонности к образованию сплавов следует препятст-
вовать контакту платины с другими металлами п их легко восста-
навливающимися оксидами. Раскаленные платиновые изделия пере-
мещать держателями из чистых неглазурованных материалов, квар-
ца или самой платины; щипцами из нихрома или нержавеющей стали
можно пользоваться только после охлаждения платины ниже тем-
пературы белого каления.
4. Сразу же после употребления очистить платиновую посуду
подходящим химическим реагентом: для удаления органических
веществ — горячей хромовой смесью, карбонатов и основных окси-
дов — кипящей соляной кислотой, диоксида кремния, металлов и их
оксидов — расплавленным бисульфатом калия; расплав горной по-
роды можно устранить сплавлением с карбонатом натрия. Чтобы
поверхность изделия была блестящей, следует полировать ее морским
песком.
5. Избегать нагревания платины в восстановительных условиях,
особенно в присутствии углерода.
В настоящее время платиновые тигли применяют только для по-
лучения стекла из легкоплавких материалов с низкой вязкостью, на-
пример цементов [257, 309]. Для сплавления тугоплавких материалов
с реактивными флюсами используются стойкие к смачиванию спла-
вы платины. Смачиваемость тиглей из платины и ее сплавов с 2%
Rh, 40 % Rb. 5% Ан, 5% Ап н с 10% Rh различными боратными
стеклами исследована в работе 1214]. Авторы пришли к выводу, что
сплав платины с 5 % золота обладает хорошей сопротивляемостью
смачиванию. Он имеет точку плавления около 16(Ю°С и примерно та-
кой же жесткий. как д сама платина. Такими же свойствами обладает
сплав платины с 5% Au л 10% Rh, но он более жестким и трудно фор-
муется [214. 276]. Некоторые авторы попользуют для тиглей сплавы
Pt % 20% Pd [273], Pt % 15% Pd Д- 5% Fe [313] пли платину, по-
крытую золотом [272.1. Эти тигли не смачивглотся расплавом, но не
обладают достаточной термостойкостью и применяются до темпера-
туры не выше 1060'С. Ио мнению многих авторов [218, 227, 268, 274.
276], для пол .учения боратных стекол лучшшш являются платино-
золотые тигли сплава ПлЗл5.
Золотые тигли загрязняются меньше, и расплав пе смачивает
их стенки. Полное смачивание па золоте наблюдается только прп до-
статочно шероховатой поверхности. Однако температура плавления
золота так низка, чго имеется опасность разрушения тиглей. Поэто-
му предпочтительнее использовать тигли из платины с гальваниче-
ским покрытием их поверхности золотом, а также тигли из сплава
ла основе платины с добанкамн 5—10% Ан или золотопалладлевого
(80% Ан -б 20% Pd) сплава, плохо смачивающегося расплавами.
Малые остатки последних легко удаляются резким охлаждением
тигля в воде.
Со временем из-за перекристаллизации сплава прп высокой тем-
пературе портится качество поверхности нишей; их нужно периоди-
чески мыть п полировать [28с8].
Металлы п сплавы вольфрама я молибдена способны длительно
работать в вакууме, водороде и нейтральных атмосферах, более огра-
137
ниченное время — в восстановительных средах, особенно содержа-
щих С и СО. Металлы и сплавы иридия, титана н сплавы па их основе
длительно сохраняются в вакууме и инертных газах. В восстанови-
тельной среде иридий быстро охрупчивается. В средах, имеющих даже
незначительное содержание кислорода, сплавы вольфрама, молиб-
дена, иридия и титана служат весьма ограниченно (десятки минут);
они охрупчиваются, окисляются, оксиды их возгоняются и, загряз-
няя керамику, снижают ее электроизоляционные свойства.
Размеры тигля меняются г. соответствии с количеством спл авля-
емой смеси, источником нагревания и необходимой площадью об-
лучаемой поверхности излучателя [276]. Важное значение имеет со-
стояние поверхности тигля, так как это влияет па степень смачи-
ваемости тигля расплавом. Как правило, тигли периодически поли-
руют. По назначению тигли делятся па два типа: комбинированные
и раздельные [276]. Тигли первого типа используются и для сплав-
ления. и для получения стеклянного излучателя путем охлаждения
расплава в самом тигле, а второго — исключительно для сплавления,
после чего расплав выливается в подложку.
Тугоплавкие соединения металлов с бором, азотом, кремнием,
углеродом, фосфором, серой, а именно: бориды, нитриды, силициды,
фосфиды и сульфиды — обладают высокими огнеупорными свойст-
вами и потому применяются в различных высокотемпературных про-
цессах. Как правило, они имеют высокую теплопроводность, значи-
тельный коэффициент термического расширения, электропроводность,
близкую к металлам, высокую стойкость в агрессивных средах при
высоких температурах, обладают большой твердостью, высокой тем-
пературой плавления. Однако их механическая прочность низка
и они плохо переносят тепловые удары. Силициды устойчивы в окис-
лительной среде. Карбиды титана, тантала, гафния п ниобия устой-
чивы в науглероживающих средах нрп высоких температурах.
Непереходные металлы (олово, индий, германий, галлий, медь,
золото, серебро, свинец) имеют малое сродство к бору, азоту и крем-
нию, поэтому они практически не вступают во взаимодействие с ука-
занными материалами. Соответственно смачивание в подавляющем
большинстве указанных систем отсутствует. Напротив, в системах
с интенсивным химическим взаимодействием (при контакте с пере-
ходными металлами, а также с алюминием, бором, кремнием) наблю-
дается интенсивное растекание.
Основные рекомендации по использованию тиглей пз этих мате-
риалов: 1) хранить сухими, чтобы предотвратить раскалывание;
2) перед использованием прокалить при 950°С в течение 2 ч; 3) сплав-
ление выполнять при низкой температуре.
4.4.4. Состояние исходной смеси
Большое значение имеет физическое состояние компонентов сме-
си будущего расилава, при котором ошт загружаются в печь. Рых-
лая порошкообразная смесь будет плавиться иначе, чем брикетиро-
ванная. В последней возрастает теплопроводность, вследствие чего
138
сокращаются потерн щелочей и оксида бора, улучшается гомогени-
зация расплава. На скорость плавления влияет размер зерен кварца.
Поглощаемая при плавлении теплота так велика, что полное расплав-
ление смесп, содержащей крупную фракцию крупнозернистого песка,
при температуре 1200°С наступает через 10 мин. Плавление смеси
с порошкообразным кварцем происходит при 1100°С. Уменьшение
величины зерна кварца от 0,28 до 0,03 мм при всех прочих равных
условиях повышает скорость стеклообразованпя в 8—9 раз [162].
На плавкость особенно сильно влияет уплотненность отдельных
частиц известняка.
Скорость плавления зависит также от влажности флюса. Плав-
ление ускоряется при использовании содержащей кристаллически
связанную воду буры вместо безводной. Смачивание кварцевых зерен
бурой, растворяющейся в собственной кристаллизационной воде,
происходит намного полнее, чем безводным вязким расплавом при
значительно более высоких температурах.
При содержании влаги в пределах 1—4% гомогенизация улуч-
шается. Одпако прп больших количествах воды в исходном материа-
ле образуются конкреции, которые препятствуют гомогенизации.
В процессе плавления из расплавов выделяются газообразные про-
дукты реакции. В основном это оксиды углерода и серы и большое
количество водяного пара. Например, в сульфатсодержащпх поро-
дах происходят’ следующие гетерогенные реакции:
Na2SO4 + SiO3 Na.,SiO3 + SO3, 2SO3 2SO2 -L- O2.
Равновесие этпхТреакций определяется температурой и давле-
нием. Следует отметить, что даже при температуре 1450°С имеется
некоторое количество остаточного сульфата. Частично 'такие газы
в виде маленьких пузырьков могут оставаться в расплаве, способст-
вуя кристаллизации; поэтому дегазация расплава имеет большое
значение.
Выделению водяного пара и газов из расплава способствует нит-
рат аммония, и в результате происходит интенсивное перемешивание
расплава. Расплав можно быстро очистить от газов, обработав
его ультразвуком. Для полного удаления газа следует увеличить те-
кучесть расплава, например, повышением температуры. Плавление
в вакууме тоже можно рассматривать как путь улучшения качества
расплава.
Образование пузырьков предотвращает прокаливание исходного
материала (без флюса) при температуре 1200°С в течение 2 мин. Если
расплав после вытаскивания из печи сразу не выливается из тигля,
наблюдаются мякросегрегацмн. Следствие последних — большие
интенсивности излучения для большинства элементов. Одпако дейст-
вительные шпенсивностн линий заметно зависят от температуры плав-
ления. Отмечается [6]. что при сплавления при 780°С скорость стета
для Ее/<а-лпнии равна 89 000 пмп'с, а при сплавлении при 900°С
п выше — 86 ООО пмы/с. Если после сплавления при 900°С расплав
в течение 45 с держится в lur.'ie, скорость счета становится равной
139
80 000 имп/с. Если ожидаются сегрегации, образец должен быть рас-
плавлен при температуре выше 9503С. Индикатором сегрегации яв-
ляется изменение интенсивности линии, например 'железа.
4.5. УСТОЙЧИВОСТЬ СТЕКЛА
Преимущественным в термодинамическом отношении превраще-
нием расплава в твердое состояние является процесс кристаллиза-
ции. Различают две стадии процесса кристаллизации: образование
зародышей и рост кристаллов. Зародышеобразование заключается
в возникновении областей с более дальним, чем это обычно наблюда-
ется в жидкой фазе, порядком в расположении атомов. Такие не-
устойчивые промежуточные состояния называются дозародышамп,
а дозародышп с минимальной критической величиной, способные
самопроизвольно вырастать в крупные частицы стабильной фазы,
известны как центры кристаллизации. Зародышеобразование может
быть гомогенным или гетерогенным. В первом случае крохотные за-
травки имеют тот же состав, что и вырастающие в пих кристаллы.
При гетерогенном зародышеобразовании химический состав центров
кристаллизации совершенно иной, чем кристаллов.
Кристаллизация — процесс, в ходе которого создастся упорядо-
ченная решетка кристалла пз менее упорядоченной структуры жид-
кости. Протекание этого процесса зависит от характера структур-
ных изменений, которые должны произойти в системе, чтобы в ней
начался рост кристаллов. Кристаллизация будет тем более затруд-
нена, чем больше состав и структура расплава будут отличаться от
состава и структуры кристаллов. Следовательно, наименьшую склон-
ность к кристаллизации будут обнаруживать расплавы, содержащие
различные структурные группировки.
Расплавы, которые можно охлаждением перевести в стеклообраз-
ное состояние, не претерпевают кристаллизации. Это можно объяс-
нить либо очень малой скоростью зародышеобразования, либо не-
достаточной скоростью роста кристаллов прп всех температурах.
Таммаи, который в 1925 г. классически исследовал кристаллизацию
в переохлажденных жидкостях и в неорганических стеклах, показал,
что процессы кристаллизации при переходе расплав — стекло за-
торможены быстрым нарастанием вязкости расплава и затруднен-
ностью перестройки структуры таким образом, чтобы она соответст-
вовала структуре выделяющихся кристаллов. В результате скорости
зародышеобразования и роста кристаллов будут характеризоваться
наличием максимумов (рис. 43). Здесь -У 7'2 — метастабильная
зона переохлаждения, в которой нс происходит образования заро-
дышей с заметной скоростью, но кристаллы способны расти, если
в расплав ввести зародыши. Именно высокая вязкость при пони-
женных температурах, с одной стороны, препятствует перераспре-
делению атомов п возникновению упорядоченной ориентировки ионов,
атомов и групп атомов типа Ме04 или Ме2О7 и образованию мелких
силикатных кристаллов, представляющих собой начальную стадию
140
|ЧЧИВН1И111Н1О11|1Ж|>1ШШ111№1|ИШ11ЙШ1П<
кристаллизации фаз. а с другой — замедляет процессы диффузии,
необходимые для зародышеобразования п роста кристаллов. Таким
образом, процесс стенлообразования проходит следующие стадии
[147]: образование в жидкости все большего количества группиро-
вок усложненного состава, близких по строению к элементам струк-
туры стекла, и связывание их в интервале стеклования в каркас.
Процесс структурирования принципиально может протекать в жид-
кости при любых температурах начиная с критической. С пониже-
нием температуры, особенно в области ниже ликвидуса, процесс
структурирования резко усиливается и достигает своего предела
при температуре стеклования, так как сложные группировки энер-
гетически более предпочтительны по сравнению с простейшими.
Принято считать гетерогенное зародышеобразование маловероят-
ным при охлаждении стекол, поскольку расплавленное стекло, как
правило, хорошо растворяет всякие случайные примеси [91]. По-
добная точка зрения представляется разумной, потому что эффектив-
ные катализаторы зародышеобразования применительно к кристал-
лическим силикатам по своей структуре близки к силикатам, т. е. сами,
вероятно, являются силикатами. При повторном нагреве они быст-
ро растворяются в расплавленном стекле. Гомогенное зародышеобра-
зование в стеклообразующих жидкостях тоже представляется мало-
вероятным, так как энергия активации диффузии даже при высо-
ких температурах имеет сравнительно большую величину, которая
с понижением температуры становится еще больше. При таких усло-
виях расплав можно при охлаждении провести через «опасную»
температурную зону без образования достаточно многочисленных
устойчивых зародышей.
Положение и протяженность «опасной» температурной зоны за-
висят от состава расплава. Изотермы вязкости для расплавов SlO2—
—В3О3 свидетельствуют о том, что изменение в расплаве содержания
141
оксида кремния SiO., существенно влияет на процесс образования
стекла.
Важным фактором является исходная вязкость расплава. Чем
она выше, тем легче осуществить переход жидкого расплава в стекло.
Принимая во внимание сложность процесса, трудно говорить об одно-
значной связи между вязкостью и составом расплава. Каждый из его
компонентов воздействует на вязкость по-своему, в тесной, зависи-
мости от полного состава расплава. Некоторые компоненты пони-
жают вязкость расплава стекла и ценны тем, что позволяют ускорить
процессы плавления и рафинирования (удаления пузырьков газа).
Преимущественно это оксиды щелочных металлов. Оксид лития силь-
нее уменьшает вязкость расплава, чем оксид натрия, который, в свою
очередь, действует сильнее, чем оксид калия. Оксид бора особенно
благоприятен на стадии завершения плавления и рафинирования,
так как увеличение ого содержания в расплаве вызывает понижение
величины поверхностного натяжения. Но есть л такие оксиды, на-
пример оксид алюминия, оксид магния, которые затрудняют плав-
ление, увеличивая вязкость.
На рис. 44 приведены изотермы вязкости системы Na2B4O7 —
—В2О3 от содержания борного ангидрида при температурах от 770
до 1050°С. Отчетливо видно, что максимумы вязкости, образующиеся
в системе при содержании В2О3 около 20,40 и 47% и соответствую-
щие соединениям Na2O-3B2O3 п Ха2О-4В2О3, постепенно сглаживают-
ся с повышенном температуры; однако до последнего сохраняется
тенденция к увеличению вязкости расплава прп возрастании содер-
жания борного ангидрида. Это связано с укрупнением борокислород-
ных комплексов и уменьшением их подвижности.
4.6. ФОРМОВАНИЕ ИЗЛУЧАТЕЛЕЙ ИЗ РАСПЛАВОВ ПОРОД
После завершения процесса рафинирования (выделение пузырь-
ков газа из расплава) стеклу дают остыть от температуры плавления
до рабочей температуры формования, при которой оно обладает по-
вышенной вязкостью. Эта температура может быть на несколько гра-
дусов ниже температуры просветления. Затем стекло формуют. Излу-
чатели нужного размера изготовляют или выливанием расплава в
соответствующую форму (кольцо, диск, шайбу), или резким охлаж-
дением его непосредственно в самом тигле. Фиксация может обеспе-
чиваться различными процессами, но все они должны предотвра-
щать возникновение внутренних напряжений и кристаллизацию.
Успех процесса формования определяется шириной и уровнем
рабочего температурного интервала формования, верхний предел
которого грубо есть температура плавления, а пижннй — темпера-
тура. при которой стекло становится слишком густым для формова-
ния. Чем шире и ниже температурный интервал, тем лсг’Ш выпол-
нить формование. Параметры температурного интервала зависят
от химического состава стекла п варьируют с составом весьма сущест-
венно. Например, оксиды щелочных металлов благоприятствуют рас-
142
искрению п понижению температур формования. Расплавы без щело-
чей и с высоким содержанием оксидов кальция и магния имеют уз-
кий рабочий интервал. Расплавы с высоким содержанием оксида
алюминия из-за высокой вязкости быстро охлаждаются и застывают,
для них желательно высокотемпературное формование. Поэтому выб-
ранный температурно-временной режим формования должен
включать возможные ого вариации с составом и одновременно исклю-
чать любую возможность расстекловывания и последующего раз-
рушения излучателя. Если расстекловывание наступает в процессе
формования стекла, то из-за внезапного и непредвиденного измене-
ния вязкости оно весьма пагубно отражается на работе формующего
оборудования. Вместо с тем образование кристаллической фазы,
заметно отличающейся по своему тепловому расширению от исходно-
го стекла, мо/кет сопровождаться возникновением опасно больших
внутренних напряжений в стекле за счет неодинакового сжатия
этих фаз при охлаждении.
Почти всегда кристаллизация начинается на поверхности. Од-
ной из причин этого может быть отличие химического состава стек-
ла на поверхности от состава внутри. Другими причинами являются
уход летучих компонентов с поверхности стекла в процессе плавле-
ния и тенденция компонентов, понижающих величину поверхност-
ной энергии, сосредоточиваться на поверхности. Однако еще важнее
существование па поверхности неоднородностей, способных действо-
вать в качестве катализаторов зародышеобразования,— либо частиц
ныли, каталитическая способность которых зависит от степени ее
сходства ио кристаллическое структуре с образующейся кристалли-
ческой фазой, либо мельчайших царапин или дефектов на поверх-
ности стекла. Способность царапни к подобному действию была до-
казана Эрнсбергсром в 1962 г. 191]. занимавшимся исследованием
механической прочности стекла. Наличие пузырьков газа или посто-
ронних частиц внутри стекла способно ускорять расстекловывание
в прилегающих к ним областях. Большая неоднородность стекла,
проявляющаяся в наличии участков неустойчивого состава, тоже
благоприятствует расстекловыванию.
Стекла с высоким содержанием оксидов щелочных металлов,
особенно оксидов лития и магния, легко расстекловываются при
операциях обработки стекла. Однако при охлаждении оксиды алю-
миния, бора подавляют эту тендешщю к расстекловыванию. Увели-
чение содержания SiO2 также снижает кристаллизационную способ-
ность стекол. Во многих случаях действие указанных оксидов сво-
дится к понижению температуры ликвидуса стекла. Расстекловыва-
ние, наблюдающееся при повторном нагревании стекол, представляет
собой в значительной мере не поддающийся контролю процесс, завися-
щий от таких случайных явлений, как присутствие на отдельных
участках поверхности посторонних частиц, которые могут стать ка-
тализаторами зародышеобразования.
4) Под расстекловыванием (девитрификацией: подразумевается образова-
ние кристаллического материала в стекле,
143
Как правило, к расстекловыванию приводят операции формова-
ния стекла, требующие длительного выдерживания стекла в зове
критической температуры, когда возможен рост кристаллов. При-
менимы лишь те операции формования, которые позволяют быстро
охлаждать стекла в критической температурной зоне. Быстрое
охлаждение расплава в момент выливания гаргщтнруег выполнение
данного условия. Таким образом, основная задача сводится к тому,
чтобы не проскочить ппжиий предел формования. Это возлагается
на температурный режим формующего устройства, который должен
определять дальнейшие скорость теплопередачи п температурный
градиент стекломассы. Температура формующих поверхностей долж-
на поддерживаться в довольно узком интервале. Учитывая, что вы-
литый расплав уже существенно остыл, она устанавливается в пре-
делах от 280 (для графптовых плунжера и формы) до 400°С (для дюр-
алюминиевых формующих поверхностей). Если поверхность плун-
жера нагрета выше определенной температуры, то расплав прили-
пает к ней, если шике, то расплав быстро охлаждается, вследствие
чего ухудшается поверхность стекла.
При выборе материала формующей поверхности следует руко-
водствоваться его теплоемкостью л теплопроводностью. Он должен
обладать жаростойкостью (продолжительная работа при высокой
температуре); термостойкостью (периодические, часто резкие измене-
ния температуры); химической устойчивостью (коррозионное дейст-
вие стекломассы и атмосферы при высокой температуре); механи-
ческой прочностью: твердостью (истирание стекломассой) и долго-
вечностью.
Чаще всего формующие поверхности изготовляют из металла
(бронзы, латуни, дюралюминия), реже -- из графита. Применяются
п специальные сплавы: перлитовые и аустенитовые чугуны; хромо-
никелевые стали; силаны, содержащие в разных сочетаниях крем-
ний, алюмпшпй, марганец, хром, никель, ванадий, вольфрам, иногда
прп полном отсутствии железа. Поскольку, как правило, сплавы до-
роги, ими покрывают только поверхность формующих деталей, кото-
рые изготавливают из обыкновенной стали. Сопоставление стекол,
полученных в графптовых и дюралюминиевых формах, спресованных
плунжером из соответствующего материала, показало, что в дюр-
алюминиевых формах они обладают лучшей воспроизводимостью,
особенно для легких элементов. Скорее всего это связано с мигра-
цией элементов от верхней к нижней поверхности стекла при его
формовании и охлаждоппп, особенно в графитовых формах. Поэто-
му использование графты а на стадии формования излучателей не
рекомендуется.
Часто при остывании расплава в тигле возникают сегрегации не-
которых элементов, прежде всего Ее 1227, 274, 288]. Оказалось, что
при сплавлении различных материалов с добавлением нитратов,
таких как л\аЛтО3, а также бромидов, ггодцдов и окиси лантана мик-
росегрегацип не наблюдается [272, 274. 288].
Нужную скорость теплопередачи можно обеспечить, если менять
габариты формующих деталей. Jllnpono применяются искусственные
144
средства воздействия па температуру формующих поверхностей: их
обогрев или охлаждение, например воздушное.
Конкретных рецептов получения излучателей из стекла весьма
много. При сплавлении в графитовых подложках расплав формуется
подогретым до ЗОО'С плунжером непосредственно на подложке [33,
151]. Если расплав получают в металлических тиглях, то его выли-
вают в нагретые до 250'С подложки и прижимают плунжером, пока
расп.тав не остынет с 1000 до 300сС [174, 227, 272, 309]. Иногда рас-
плав запрессовывают в металлическое кольцо, расположенное на
подогретой подложке, что увеличивает прочность получаемого излу-
чателя [272, 3131- Поверхность получаемых таким способом излуча-
телей неровная п пх приходится шлифовать [174]. Поскольку фор-
мующие поверхности при используемых температурах легко окис-
ляются, существует опасность загрязнения расплава.
Иесмачиваемость ллатинозолотых тиглей позволяет существен-
но упростить процесс получения стеклянных дисков. При сплавле-
нии в таких тиглях излучатели можно не формовать, а охлаждать
расплав потоком воздуха непосредственно в тигле (комбинированный
вариант) [268, 274, 3281 или выливать в подложку из такого же ма-
териала, подогретую до 700”С (раздельный вариант) [288, 313]. По-
лученные диски анализируются с нижней стороны. При многократ-
ном использовании дно тигля деформируется и поверхность диска
становится неровной, поэтому дно тигля периодически полируют
]268, 274, 288 ]. Обнаружено [288], что добавка малых (5% от полной
навески) количеств бромидов и иодпдов к расплаву не только улуч-
шает свойство спека отделяться от тигля, но также позволяет, изме-
няя поверхностное натяжение п вязкость расплава, сделать верхнюю
поверхность диска плоской. Становится возможным применение при
анализе верхней и нижней поверхности диска.
Если воспроизводимость приготовленпя стеклоизлучателем не-
удовлетворительна, то образцы переплавляют.
Такое многообразие конкретных способов изготовлелпя дисков
вызвано недостаточной изученностью поведения остывающего стекла,
факторов, характеризующих его прочность и химическую стойкость.
В реальных стеклах имеется набор пли «спектр» дискретных уровней
прочности различной природы, каждый из которых обусловлен ка-
ким-то дефектом структуры стекла [146]. В стеклах типичными де-
фектами, определяющими нх прочность, являются микротрещпны,
микроразрывы, включения, а татке резкие нарушения плотности и
химического состава в объемах, значительно превышающих элемен-
ты микропеоднородпостей структуры стекла, вызванные ликвацией,
кристаллизацией или случайными технологическими причинами.
Для примера на рис. 45 приведен спектр уровней прочности силикат-
ного сгекла [146]. Резкие нарушения плотности и состава можно
связать с ограниченной растворимостью различных оксидов в бора-
те. Поэтому, когда один из компонентов переходит предел указан-!
пой растворимости, стекло кристаллизуется тт образуются трещины.
В атмосферных условиях вследствие действия поверхностио-
акшвпой среды — влаги — наиболее опасными являются дефекты
5. П. Афонин, Т, Н. Гуничева, Л. Ф. Пискунова
145
Рис. 45. Спектр уровней прочности силикатного
стекла.
сг? — макротрещины; см — микротрещшш: ъГ— су5-
микротрсщины: си —прочность структур стекла; <>,'—
прочность поверхности стекла.
на поверхности стекла. Они могут быть различного вида, причем
каждый вид дефекта может давать более или менее размытый уровень
прочности. В [1461 приведены зависимости между уровнями проч-
ности стеклянных волокон щелочно-сплпкатного, алюмоборосилпкат-
ного л алюмосиликатного состава п кварцевых волокон от суммар-
ного содержания стеклообразователей SiO2, В3О3 и А12О3. Делается
вывод, что. поскольку прочность связей Si—О, В—О н А1—О прак-
тически одинакова, суммарное их содержание в стекле будет влиять
на высшие уровни прочности примерно так же. как и отдельно взя-
тые стсклообразователп.
Чем больше по размерам дефекты, тем сильнее опп влияют на
процесс разрушения стекла. Наибольшей прочностью в атмосферных
условиях обладают бездефектные стекла. Причиной разрушения мо-
жет явиться холодная деформация стекла, имеющая место при выли-
вании расплава при относительно низкой температуре. Она делает
стекло очень чувствительным к коррозии в воздухе, что ведет к спон-
танному разрушению.
В процессе формования под воздействием разности температур
при охлаждении возникают внутренние напряжения. Чтобы пре-
дотвратить растрескивание, их приходится снимать путем отжига.
Последний осуществляется нагревом до определенной температуры в
пределах «интервала отжига». Напряжение снимается благодаря
вязкому течению, после чего стекло охлаждается со значительно
меньшей скоростью, чтобы предотвратить возникновение в стекле
большого температурного градиента. Допустимая скорость охлажде-
ния определяется толщиной стеклянного диска. Для тонких стекол
весь процесс можно завершить в течение 1 ч; толстые требуют не-
сколько часов, чтобы остаточные напряжения по превышали допусти-
мых пределов.
Механическая прочность стекол существенно зависит от состоя-
ния поверхности. Роль травления сводится к уменьшению толщины
поверхностного слоя, который может оказывать упрочняющее дейст-
вие либо в силу специфической структуры, обусловленной вытяги-
ванием, либо за счет своей бездефектности. Отмечается [154]. что
прочность образцов после абразивной зачистки составляла около
81% прочности неочищенных образцов. Мехапичесжая абразивная обра-
ботка стекла дает материал с заметно пониженной прочностью
146
по сравнению с материалом, наготовленным из стекла, которое не
подвергалось механической абразивной обработке.
В работе [268] приведена оценка влияния способов обработки
поверхности диска на воспроизводимость результатов. Авторы
пришли к выводу, что шлифовка поверхности образца алмазным
кругом 50 мкм контролируется точнее, нем последующая полировка,
поэтому длительная процедура полировки не улучшает воспроизво-
димости препарирования.
Поведение стекла после закрепления формы определяется его
химххческой устойчивостью — способностью его противостоять раз-
рушающему действию воды, влаги и газов атмосферы, а также раство-
ров различных химических реагентов. Устойчивость стекла зависит
от его химического состава и природы действующего реагента. Стек-
ла с высоким содержанием оксидов натрия и калия не обладают
высокой химической стойкостью- Оксиды щелочных металлов, MgO,
СаО, повышают химическую стойкость стекол. Благоприятным обра-
зом влияют на химическую стойкость оксиды алюминия, цин-
ка, бора.
Для силикатных стекол характерна сильная способность к ад-
сорбции воды. Па поверхности стекла откладывается устойчивая
«пленка воды», которая служит причиной многих явлений. Вода в
этой пленке но инертна, она химически связана с поверхностью. Си-
ликаты поверхности стекла, взаимодействуя с водой, гидролизуют-
ся. Щелочные силикаты образуют при этом едкую щелочь it гель
кремниевой кислош. Первая может свободно выливаться пли оста-
ваться на поверхности (в зависимости от условий разрушения), гель
кремниевой кислоты остается на поверхности стекла в виде более
или менее равномерного слоя, толщина л плотность которого опре-
деляют скорость диффузии через пего молекул воды. Этот слой за-
медляет процесс дальнейшего разрушения стекла, скорость которого
будет постепенно замедляться по мере утолщения защитной кремне-
земной пленки.
Химическая устойчивость стекла сильно зависит от изменения
температуры: при повышении температуры на Г'С скорость разруше-
ния стекла возрастает ла 15—30%- Весьма сильное разрушающее
действие оказывает вода при температуре выше 100°С. К аналогич-
ному явлению относится и адсорбция жидких веществ, которая весь-
ма вредно отзывается на качестве поверхности стекла. Обработка
кислотами даже повышает гигроскопичность, что приводит к появ-
лению высокоадсорбирующего слоя геля кремниевой кислоты.
Небезразлично стекло и к окружающей среде. В системе стек-
ло — водород происходит катодная миграция водорода в стекло,
в результате которой вследствие взаимного обмена иопов натрия и
водорода получается замещенный силикат. Если ионы водорода про-
никают в стекло, то сразу выделяется вода. При участии активного
водорода в качестве побочного продукта может образоваться дисилан
8цН(). В стекло могут войш даже ионы NE, которые в результате вто-
ричной реакции даюг оксид азота. В боратных стеклах возможно
образование нитрида бора.
Р)>
147
4.7. АВТОМАТИЗАЦИЯ ИЗГОТОВЛЕНИЯ
СТЕКЛОВИДНЫХ ИЗЛУЧАТЕЛЕЙ
Эффективность выбранного температурно-временного режима
препарирования стекловидных излучателей существенно зависит от
того, с какой стабильностью он выдерживается. Стабильность можно
улучшить, сжав во времени процессы плавления п формирования.
Это достигается, например, за счет использования при плавлении
малых по объему тиглей (соответственно малых количеств сплавляе-
мой смеси) и более высоких температур 1300°С). Допуская формо-
вание и остывание стекла в самом тигле и обеспечивая резкий пере-
пад температур струей сжатого воздуха, можно значительно уско-
рить охлаждение сформованного излучателя. Однако наиболее пол-
ная реализация преимуществ каждой из этих операций возможна
только при автоматизации процесса препарирования. Фирмой
«Philips» создан полуавтоматический агрегат PW-1234 для препари-
рования стекловидных излучателей, рекомендуемый для рентгено-
спектрального анализа цементов, шлаков, стекол, бокситов, геоло-
гических образцов п других материалов. С его помощью один опера-
тор может препарировать гомогенный излучатель за время, соизме-
римое со временем выполнения других операций. Температурные
режимы основных этапов и время их выполнения заранее запрограм-
мированы, чтобы исключить ошибки со стороны оператора.
Работает PW-1234 следующим образом. Предварительно про-
сушенный дозированный порошок исследуемого материала засыпает-
ся в платиновый тигель, расположенный над высокочастотной нагре-
вательной катушкой индукционной печи. Вторая катушка с рефлек-
торными пластипамп находится сверху и, когда нужно, опускается
вниз, пока не окажется пад тиглем. При температуре 1200°С поро-
шок прокаливается в течение 2 мин. При этом пз него уходят СО2,
большая часть кристаллической воды; разлагаются карбонаты, суль-
фиды; оксиды переходят в высшую степень окисления, что в даль-
нейшем предотвращает образование газовых пузырьков в остываю-
щего стекле. После термической обработки исходного материала высо-
кочастотный генератор отключается, катушка поднимается вверх,
в тигель добавляется определенное количество флюса и начинается
плавление. В течение заданного, сравнительно малого времени смесь
нагревается, затем проплавляется при 1300°С. При такой температу-
ре флюс становится достаточно жидким, чтобы растворить образец
за несколько минут и дать гомогенное стекло. Бремя сплавления за-
дается, а температура может варьироваться в соответствии с хими-
ческим составом образца.
При охлаждении через расплав к тиглю пропускается постоян-
ный ток. Оп передается расплаву в виде искрового разряда. Скорость
охлаждения регулируется воздушной струей. Вместе эти две опера-
ции предотвращают прилипание стекла к тиглю и разрушение ею
из-за перенапряжений. После охлаждения до температуры ~ ЗОО'С
излучатель легко удаляется из тигля и может помещаться в спектро-
метр. Полностью процесс препарирования длится около 5 мин для
148
цементного порошка, несколько оольше или меньше — для других
материалов.
Полученные этой аппаратурой воспроизводимости для всех ана-
лизируемых элементов, кроме Si. не выше 0,3—0,4%. Повторное
нереплавление уменьшило ошибку для оксида кремния с 1,4 до 0,4%,
что указывает на тот факт, что сплавления в течение 2 мин иногда
недостаточно — остается некоторая неоднородность.
Более совершенная модель PW-8910 агрегата для препарирова-
ния рекомендуется фирмой «Philips» для изготовления металли-
ческих образцов из стружки, проволоки, осколков п другой подоб-
ной структуры. В этой модели гомогенность расплава улучшается за
счет перемешивания в процессе плавления вихревым током. Быстрое
охлаждение обеспечивается центрифугой, форма изложницы которой
может быть подогнана иод нужный для спектрометра размер излуча-
теля. Чтобы предотвратить окисление, образец при плавлении к
отливке омывается струей аргона. За час агрегат позволяет препа-
рировать 20—30 излучателей.
4-8. ПРИГОТОВЛЕНИЕ
ПОРОШКОВЫХ ИЗЛУЧАТЕЛЕЙ ИЗ РАСПЛАВА
Процессы формования и закрепления формы расплава весьма
продолжительны п требуют особой тщательности. Поэтому, когда
есть необходимость, можно использовать способ препарирования таб-
летизпрованных излучателей из расплава. Резко охлажденный рас-
плав дробится, измельчается до ~ 70 мкм в механической ступке
или вручную, и из порошка прессуется излучатель. Так как порошки
стекол сыпучие, лучшие излучатели получаются при прессовании их
в виде двухслойных дисков. В качестве основы используются полп-
карбонатная пленка, борная кислота, целлюлоза, алюминиевая фоль-
га, резина, пластмасса.
Вариантов способов препарирования двухслойных дисков весь-
ма много. Каждый из них тем или иным способом гарантирует проч-
ность излучателя. В общем случае при условии, что размер частиц
меньше 50 мкм, порошки хорошо прессуются при давлении ~ 2 —
5 т/см2. Когда связующие свойства материала плохи, можно приме-
нять большие давления, вплоть до 50 т/см2.
Изучение влияния давления на интенсивность линий спектра
флуоресценции показало, что в случае хорошо измельченного мате-
риала результаты анализа практически не зависят от давления.
Прессование иод высоким напряжением в пресс-форме часто при сня-
тии напряжения обусловливает образование трещин в таблетке. Это
связано со слабой деформацией пресс-формы под давлением и может
быть исключено прессованием порошка в форме, которая изменяется
необратимо под высоким давлением.
Значимого расхождения в воспроизводимости результатов ана-
лиза при препарировании измельченного и формованного стекла не
149
обнаружено. Однако, учитывая, что, измельчая стекло, мы получаем
порошок, который в общем случае не отвечает всем требованиям
гомогенной среды, предпочтение следует отдать формованию стекло-
видных излучателей.
Глава 5
АВТОМАТИЗИРОВАННЫЙ АНАЛИЗ ГОРНЫХ ПОРОД
Разработка методики рентгенофдуоресцентного силикатного ана-
лиза включает несколько этапов:
1) изучение химического и минералогического состава объекта
анализа п требований к точности и чувствительности выполнения
определений;
2) выбор метода коррекции влияния матричных эффектов с уче-
том характеристики анализируемого материала и возможностей ис-
пользуемой аппаратуры;
3) разработку способа препарирования при выполнении требова-
ний к чувствительности и экспрессности анализа;
4) отработку схемы выделения аналитического сигнала из заре-
гистрированной прибором скорости счета па аналитической липип
вторичного рентгеновского спектра;
5) метрологические исследования, позволяющие выявить и
исключить систематические погрешности па различных этанах ана-
лиза;
6) разработку алгоритма пересчета аналитического сигнала в
концентрации и его воплощение в программах для ЭВМ;
7) определение метрологических характеристик методики на
стандартных образцах состава.
5.1. ХИМИЧЕСКИЙ
И МИНЕРАЛОГИЧЕСКИЙ СОСТАВ ГОРНЫХ ПОРОД
Горные породы — эго минеральные агрегаты, ттз которых сос-
тоит земная кора. Всего двенадцать химических элементов — О, Si.
Al, Fe, Са. Na, К. Mg, Ti, If, P, Mn — слагают 99,39% твердой зем-
ной коры. Их принято называть основными породообразующими или
петрогенными. На все прочие 80 элементов, называемые микроком-
попеитамп, приходится 0,61%. I lei рогелные элементы в земной ко-
ре преимущественно связаны с кислородом, поэтому состав горных
пород определяется в виде суммы оксидов: Na.,О, MgO, SiO2,
Р.2О.-„ И.,О, СаО, К.,О, MnO. Fe2O3, FeO.
Вся совокупность горных пород делится на типы. Тин горной
породы характеризует способ ее образования, т. е. имеется в виду ее
генезис — магматический, осадочный, метаморфический.
150
Табл и ц а 28. Распределение минеральных видов в земной коре
Минорат XiiMii'iecHiid состав Содер- жание,
Полевые шпаты Пироксен Ca[Al,Si.Osl. К [AlSt3OelNaIAlSi3Os | (Са, Mg,-FL-)t[SiO2] ) 58
Амфиболит Оливии Ca,(Mg. FcMOII/SijOnLs (Mg. Fe)2[SfO4I / 16.5
Кварц SiO2 12.5
Слюда K(Mg. Fe. Mn)3 [(OH)JAlSi3Olrt] 3,5
Глинистые минералы AlJtOHVSIjOjJ 1 3.5
Гематит Магнетит Fe3O. 1 Fe.,O3 J
Кальцит CaCO3 15
Т а б л к Ц а 24. Химический состав (%) основных групп маг-
матических горных пород
Грустна Кислые Средние Основные Уль1! раосно!?- ные
SiO, (54-78 51-65 42-53 34-46
'ПО, 0-2 0,07—4 0,05--5 0.2—6
А1аб, 11 — 22 10—25 0,1—32 0,2-30
FeA 0-13 0.5-10 0,2-50 4-15
FeO 0-7 0,3-10 0.4—25 2-12
MnO 0-1 0 -1.2 0-1,15 0-0.3
MgO 0-4 0.01-8 0,05—35 1-47
CaO 0.03—7 0.2-11 0.2—23 0-35
Na2O 0.5-7 0.3-12 0—17 0-1(5
IGO 0,3—8 0.1 — 20 0-14 0-6
P.,O-, 0,03—0.3 0.05 —14 0-2.5 0—2.5
ICO 0-1.5 0-5 0-5 0-15
Самыми расирострапеннымп в земной коре являются магмати-
ческие горные породы; они составляют 95% всех пород. Как показы-
вают данные табл. 23, более 90% пз них представляют собой кремне-
кислородные соединения Al, Fe. Са. Na, К и Mg, называемые сили-
катами [1631-
Для выделения групп магматических пород (ультраосиовных,
основных, средних, кислых) применяется химический признак —
содержание кремнезема. Используются следующие границы [18]:
кислые породы — более 64% SiO2. средине породы — 51—66%
SiO3, основные породы — 42—53% SiO2. ультраосновные— менее
46% SiO.,. Из табл. 24, в которой приведены средние содержания пет-
рогенных оксидов в основных типах пород, видно, что при переходе
от ультраосновных пород к кислым уменьшается содержание MgO,
CaO, Fe2O3, а содержание А12О3. Na.,O, К2О увеличивается. При этом
содеруканис кремнекислоты, главной составной части этих пород,
может изменяться от 43 до 80%. Содержание других оксидов варьи-
рует практически от нуля до следующих пределов: 30% А12О5, 35%
СаО, 47% MgO. 50% Fe,O3, 17% Na2O, 20% К2О.
151
Химический состав горных пород не является однородным по
всему объему и зависит от распределения различных слагающих их
минералов. Как следует из табл. 23, изверженные горные породы на
50—60% состоят из половых пшатов, содержание других минералов
меняется в широких пределах. Например, свободный кварц появ-
ляется в породах среднего класса и достигает 30% в кислых поро-
дах. По мере уменьшения кислотности кварц заменяется железо-
магнезиальными минералами. Хотя в земной коре в качестве породо-
образующих минералов присутствует незначительное число мине-
ральных видов, при разделении горных пород по содержанию кремне-
зема в одну группу попадают породы, различные по набору минера-
лов (как с кварцем, так л с оливином или фельдшпатоидами) п по их
количественным соотношениям. Следовательно, количественный ми-
неральный состав конкретных горных пород настолько изменчив,
что его трудно охарактеризовать.
5.2. ТРЕБОВАНИЯ К ТОЧНОСТИ АНАЛИЗОВ ГОРНЫХ ПОРОД
Требования к качеству аналитических работ вытекают из постав-
ленных задач, для которых используются полученные результаты,
п возможностей современной аналитической службы. IJ о можно отме-
тить тенденцию к неуклонному повышенно этих требований 151].
В основу допусков при контроле за аналитическим процессом
положены требования к качеству результатов аналитических измере-
ний при определении различных компонентов в минеральном сырье,
предложенные Государственной комиссией по запасам полезных
ископаемых. В них отражен средний достигнутый уровень воспроиз-
водимости аналитических определений, используемых для подсчета
Та'блцца 25. Допустимые огноемтрлидше стандартные отклонения , %
И птервая соде’ uiiii Оп; еде.-;-ег!! 01 сил
Х;-2О МаО л,о, 1 -10, Р/К К,0 СаО ’£10. МпО Ре.Оз
GO—69,99 1.1 0.7 0,7
50—59.9$) 1.4 1.2 1 i S 1 ' 0.8 0.8
40- 4<\уу 1,7 1.0 1.0 1.4 1.0 0,9 1,1
30-39.99 1.8 9. 1 1.3 1.8 1.2
20—29.99 ' -1 2,8 1,9 ‘’.1 1.5 1,4
10—19,99 о ~ а 4 3 5 3,2 л .'> 2.1 1.1 2 1
5 -9,99 ;> -i 4,0 Г> 4 ,i (1 5.4 5,0 ’’ !> 2.0 4,3
2-4,99 8.0 О .) 8,0 а 3 2 8,0 6.8 л .4 2 8 7.0
1-1.99 10.0 9 О 11.0 9 3 10.0 9.0 7.0 3.4 10,0
0,5- 0.99 j /.□ 1 3 !) 15.0 12,0 9.0 12.5 12.0 9,0 5 4 13,5
0,2-0.499 lb 0 16.0 20.0 17.0 8.2 16.5 16,0 11.0 8,0 17.0
0.1— 0.199 20.0 21 0 25,0 -. । . 0 9,3 20.0 21,0 14,0 11,0 21.0
O.U5-0 099 24.9 27 0 28.0 27.0 12,н 23.0 28,0 18,0 17,0 25,0
0.02— СЦ<149 ‘Ж О 30,0 30.0 10.0 28.0 30.0 21,0 21.0 21,0 28,0
0,01—0.019 f 1! 3<).о .'Ю.О 30.0 21,0 30.0 3(1.0 ?7.0 24,0 30.0
0,005—0.099 30,0 39,0 30.0 30,0 25.0 30.0 ЗО.о 29,9 28,0 30,0
152
запасов полезных ископаемых [97 |. Анализ горных пород на нетро-
гепные элементы отнесен ко И категории полных рядовых анализов.
Эта категория накладывает ограничения на точность но только для
отдельных компонентов, но к для их суммы. Относительные стан-
дартные отклонения, характеризующие воспроизводимость анализа,
не должны превышать допусков, приведенных в табл. 25, для всех
интервалов содержаний. Как видно, макрокомпоненты в горных по-
родах необходимо находить с точностью не менее 1 — 1,5 отн.%.
Еслп в пробе определяются все элементы, содержание которых пре-
вышает 0,1%, сумма компонентов должна лежать в интервале 99,5±
±1,5%. Ирм определении всех компонентов, содержания которых в
пробе выше 0,01 %, сумма должна лежать в интервале 99,9±1,5%.
Из характеристики объекта анализа и требований к его точности
следует, что силикатный анализ горных пород является сложной ана-
литической задачей. Это обусловлено экстремальными вариациями в
химическом составе и минералогии горных пород п жесткими требо-
ваниями к точности полных силикатных анализов.
5.3. УЧЕТ МАТРИЧНЫХ ЭФФЕКТОВ
Использование современных рентгсяофлуоресцентных аналити-
ческих. комплексов для силикатного анализа, таких как СРМ-18/М
6000, позволяет рассчитывать концентрации многокомпонентных
систем с помощью теоретических уравнений связи.
Для учета матричных эффектов при определении петрогепных
элементов целесообразно применение упрощенного метода AF-кор-
рекцпи, в котором поправки на поглощение п дополнительное воз-
буждение вводятся мультипликативным способом. Прп сохранении
главного достоинства метода фундаментальных параметров — при-
менимости к широкой области изменения состава анализируемых
материалов — он обладает простой схемой реализации. приемлемой
для автоматизации анализа с помощью малой ЭВМ.
Расчет концентраций определяемых элементов в анализируемой
пробе упрощенным методом AF-коррекции осуществляется с по-
м ощ ью выр а ж ения
ct = C^kf, (5.1)
С„ =,С'М;!1?,
где
(5.2)
= (хх + Х1)/(Хх+Х,)“
(5.3)
2г" = X: = u./siritf, J*y - I1: =~ ±чА
‘/-(П-2ИМ?) /(1 + 2АА), (5.4)
153
Dik =
L7.k = (Il'/-~.I,)111 (1 + ХХл/**л) + С^х»)ln (1 + Xi/fM-
(5.5)
В выражение (5.1) входят фундаментальные параметры, такие
как массовые коэффициенты поглощения, скачки поглощения, выхо-
ды флуоресценции. Численные значения этих параметров постоянно
уточняются, по даже по данным последних лет экспериментальные
значения существуют но для всех элементов, и расхождения между
ними значительны. Следовательно, необходимо выбрать рабочие
интерполяционные формулы для расчета фундаментальных парамет-
ров и оценить степень влияния неопределенности в их значениях на
правильность расчета поправок на матричные эффекты.
Уравнение (5.1) получено путем упрощения физической модели
возбуждения аналитической линии, которое может привести к систе-
матическим ошибкам в результатах определений. Необходимо оце-
нить погрешности в значениях поправок на матричные эффекты при
их расчете по упрощенным формулам.
5.3.1. Влияние неопределенности
фундаментальных параметров
Точность вычисления поправки на поглощенно существенно за-
висит от неопределенности значений массовых коэффициентов погло-
щения. Расхождения между значениями, приведенными в различ-
ных таблицах, особенно в области скачков поглощения, составляют
десятки процентов. Наиболее остро эта проблема стоит в длинно-
волновой области, так как с увеличением длины волны неопределен-
ность значений массовых коэффициентов поглощения растет.
В табл. 26 приведены коэффициенты поглощения Mg7fa- п
AIAa-линий элементами от лития до железа, предложенные различ-
ными авторами, и относительные стандартные отклонения, характе-
ризующие рассеяние относительно среднего значения. Как и следова-
ло ожидать, наибольшее рассеяние наблюдается для элементов с ма-
лыми атомными номерами, таких как Li и В, п элементов, аналити-
ческие линии которых расположены в непосредственной близости от
края поглощения рассматриваемой Хк-лпнии. Л,ля Mg/fcc-лттпии
такими элементами являются Mg, Al, Si, для AlA'a-линии — Si и
Al. Для остальных элементов рассеяние значений не превышает 5%.
Наиболее полными и точными таблицами массовых коэффициен-
тов поглощения в настоящее время можно считать таблицы Хайнри-
ха, Леру и Тайзела [229, 258, 308]. Чтобы выбрать рабочие таблицы,
построены зависимости ц(%) для Mg, Al, Si, К, Са и Fe, рассчитанные
по данным, приведенным в названных таблицах. Эти зависимости
показаны на рпс. 46. Максимальное расхождение значений массовых
коэффициентов поглощения, рассчитанных по разным таблицам,
наблюдается в области краев поглощения. С уменьшением длины
154
Таблица 2G. Значения массовых коэффициентов поглощения для MgKa-
и AlTi'a-лшцш
ё S* gs MgJfe sr, % AlKa sr. %
[231] 1308] [220] [233] [231] [231] [308] [229] 1235] [231]
Mg 4G4 529 353 533 546 19,6 4365 4529 4610 4050 4211 4.7
Al 615 674 491 679 6bD 14,2 38G 422 312 330 426 14,0
Si 802 844 670 851 857 9,4 504 529 425 480 532 8.9
P 1021 1040 893 1052 1092 G.6 641 652 567 650 654 6,0
К 2245 2371 2188 2202 2182 3.8 1410 1504 1390 1300 1353 5,0
Са 2658 2751 2612 2691 2520 4,9 1668 1745 1659 1500 1Э88 5,2
Ti 3646 3G27 3447 3509 3360 3.2 2289 230(1 2189 2143 3.5
Мп 5443 5255 4997 5287 4956 4,0 3418 3333 3174 3216 3,7
Fe 6120 5888 5600 5999 5502 4.5 3843 3735 3557 3645 3,7
Na 5409 5747 5830 5300 5800 4,4 33G2 3571 3599 3395 3314 3.3
Li 99 126 193 149 24.4 61 7/ 119 <84 93 24.7
В 507 608 609 G06 8.1 312 370 376 330 379 8,G
0 2433 2575 2306 2208 5.9 1504 1565 1423 1520 1381 5,1
Рис. 46. Зависимость р(л) для магния (2), калия (2), алюминия (3), каль-
ция (4), кремния (5), железа (6).
Таблиц а 27. Погрешности в поправке на поглощение (%) за счет неопре-
деленности значений массовых коэффициентов поглощения
Порода М? AI Si р К С а Т1 Мп Ре
Гранит СГ-1 Граподпорпт «Ры- 0,43 0,21 0,55 0,62 -0,22 —0,27 —0 -51 0,20 0,27
ЖШ(» 0,31 0,22 0,46 0,45 -0.14 -0.19 — 0,40 0,23 0,23
Нефелиновый сие-
ИНГ 0.37 0,00 0,32 0.11 -0,10 -9.12 — О.иО 0.20 0,21
Базальт ВМ (1.21 0,12 0.11 0.07 -0,17 -0.20 —0.24 0.21 9,21
Глин, сланец ТМ 0.28 0.29 0.51 0,34 -0.1" -9.30 —0.57 0,31 0,33
Диабаз ДТШ-1 0.06 0,01 0.19 Ш26 -0.16 — 9.22 -0,20 0.12 0,13
Миаскпт МИВ-1 0,45 0,13 О 43 ।) ?3 -0.13 -0.17 —0.64 0,27 0,28
Трапп СТ-1 -0.11 - O.ji) —0,04 0,02 -0.13 -0.16 О.05 0,02 0,02
волны поглощаемого излучения и увеличением атомного номера
поглотителя расхождение уменьшается. Сравнение зависимостей
p(?i) показывает, что таблицы Хайнриха и Тайзена сопоставимы. Од-
нако предпочтение мы отдаем таблицам Тайзена, поскольку его ин-
терполяционная формула более корректна, чем формула Хайнриха.
Погрешности, вносимые неопределенностью значений массовых
коэффициентов поглощения в расчет поправки па поглощение, вы-
численные для различных типов горных пород, приведены в табл. 27.
В качестве образца сравнения использовался стандартный образец
состава габбро-дпорпта СГД-1. Согласно табл. 26 неопределенность
в массовых коэффициентах поглощения Др/ц равна J0 ?6 для элемен-
тов, края поглощения которых расположены вблизи края поглоще-
ния ТГа-лшшп анализируемого элемента, и 5% для всех остальных
элементов, хотя в ряде случаев величины значительно меньше. Для
Li, В п О значения отношения Дц;р брались равными 20%. Значение
систематической погрешности в поправке на поглощение за счет
неопределенностей значений и для горных пород в среднем равно
0,2—0,3 отн. ?6 п не превышает 0,6 отн. % (см. табл. 27).
Правильность расчета поправки на матричное возбуждение за-
висит от неопределенностей значений фундаментальных параметров,
входящих в формулу (5.5). Прежде всего к таким параметрам отно-
сятся значения выходов флуоресценции и скачков поглощения, так
как погрешность в расчете параметра Dik пропорциональна неточ-
ности значений со9 и rq. Возможны два варианта: использование
экспериментальных значений tof, и rq или рассчитанных по эмпири-
ческим формулам. Поскольку экспериментальные значения выходов
флуоресценции п скачков поглощения имеются не для всех элемен-
тов, на наш взгляд, удобнее использовать для расчета песложвые вы-
ражения.
Многие авторы рассчитывают выход флуоресценции, пользуясь
формулой Шпрингера [298]:
сох = Z4, (Z1 -К йь-). 1де й7; = 1.06-КВ. (5.6)
Таблица 28. Экспериментальные и расчетные значения выходов флуо-
ресценции для К-серпи определяемых элементов
По ДЛШГЫ.М ,яксп и) Це формулам ,„Г>асч ° И Погрешности при расчете по формулам, %
г [64] [215] (5.6J (3.71 (5.0) Е
nNa 0,022 0.019 0,0136
0,030 0.028 0.027 0.0192 7,'14 30
+А1 0.036 0.038 0.036 0.026 7,89 28
0,047 0.043 0.047 0,035 9,3 25
1ьр 0,060 и .060 U.OGO 0.046 0 23
§ По данным °к'сп По форму- лам Погрешности при расчете по форму- ,1<Ш, %
[64] 12(5] (5.Л (о. 1J
1УК 0,138 __ 0.138 0,116
20Са 0,163 0.178 0.163 0,13 8/ 18
22Т; 0,219 0.221 0.219 0,18 1,4 18
0.314 0,313 0,314 0,27 0,3 13
2«Fe 0.347 0.350 0-347 0,301 0,8 13
В работе 164] показано, что наилучшее согласие с эксперименталь-
ными данными дает формула Б урона с коэффициентами Финка [179]
/(1 ~ tM]1/4 = Ак + Вк Z -% C,:Z\ (5.7)
1де Дк = 0,015+0,01, Вк — 0,0327+0,000-э, Ск ~ —(0,64=ь
+0,07) • 10“7.
В табл. 28 приведены значения оА-, вычисленные по формулам
Шпргшгера (5.6) п Буроиа (5.7), а также последние эксперименталь-
ные данные из работ [64] и [215]. Сопоставление эксперименталь-
ных данных с расчетными позволяет сделать вывод, что формула
Шпрингера для расчета (<ук в длинноволновой области непригодна,
так как погрешность расчета достигает 30 отн. %. Если использовать
для вычисления &к формулу (5.7) рационально, то она аппроксими-
рует экспериментальные данные с точностью до 10 %.
Сравнение экспериментальных значений о)к, определенных раз-
личными авторами, показывает, что неопределенность значений вы-
ходов флуоресценции может достигать 20 % для элементов от Na до
J> и менее 10% для элементов с большими атомными номерами.
В формуле (5.5) относительную долю фотонов, поглощенных д-
уровнем, .можно рассчитать, пользуясь экспериментальными значе-
ниями скачков поглощения, или по эмпирической формуле [298]
[rq = bq — cqZ.
(5.8)
Для Л'-уровия -= 0,924, ск = 1,44-10-3. Значения г,г вычислен-
ные по экспериментальным значениям Sq и по эмпирической форму-
ле (5.8), можно сравнить по данным, приведенным в табл. 29. Как
видно, эмпирическая формула (5.8) достаточно хорошо аппроксими-
рует значения относительной долл фотонов, поглощенной К-уров-
пем. Точность аппроксимации для определяемых элементов не хуже
3 отн. %.
Значения погрешностей в расчете поправки на возбуждение,
обусловленных неопределенностью значений и rq, оценивали пу-
157
том расчета величин kf для
средних составов несколь-
Таблица 29. Сопоставление эксперимен-
тальных и расчетных значений rq для К-
уровня ких типов пород, приве- денных в [73], со значе- ниями н>к и гл-, вычислен- ными по формулам (5.7) п (5.8). Затем эти же но-
Эде- ме ЕГТ SK „ы:сп r К _Т‘асч ТК Расхождение ••
Na 13.8 0.928 0.908 2.17
Si 11.2 0.911 0.904 0,66 правки вычислялись при
Ti 8.47 0.880 0.892 1.48 изменении сок п тк на 20
и 10 % соответственно. В
качестве образца срав-
нения использовался стандартный образец габбро-диорита СГД-1.
Относительное расхождение значении поправок на возбуждение
&kr!kf для определяемых элементов приведено в табл. 30. Хотя изме-
нение значений параметра Dih пропорционально изменению входных
параметров, т. е. равно 22%, благодаря тому, что поправка на воз-
буждение — величина относительная, значение &kjlk; не превышает
0,3 отн. %.
Можно ожидать, что влияние неопределенности значений массо-
вых коэффициентов поглощения на величину поправки на возбужде-
ние должно быть невелико, так как в формуле для расчета Dih они
Таблица 30. Максимальные погрешности в значениях поправки на воз-
буждение, обусловленные неопределенностью значений фундаментальных пара-
метров <ок н для различных типов пород
порода ДЬ //(., %
Nil Mg Al Si p к Cd Ti I’p
Гранит 0.08 0,16 0,18 0.14 0,15 0.20 0.20 0,30 0,01
Песчаник 0.04 0,06 0.07 0,09 0.09 0.32 0,13 0.15 0,01
Трапп 0.02 0.01 0,01 0,03 0,02 0,04 0,09 0.15 0,01
Дунит 0,07 0.26 0.22 0,15 — 0.30 0,03 — 0.01
Таблиц а 31. Значения массовых коэффициентов поглощения ц, выходов;
петрогенных
Аналити- ческая линия Значения ц
Na Mg Al Si p К Ga
УаА’а- MgA'a- Alka- SiA'a- PAa- KAa- CaA’a- TiAa- MnKa~ Fctfa- 682,30 5742,50 3571.35 2302.6 1531.87 381.63 282,35 161,42 76.37 60,72 888,14 529.03 4528.93 2919.99 1942,6 483,96 358.06 207.7 96,84 7 ( 1131.94 674,24 422,20 3633,14 2417.04 602,16 445,51 254,7 120.5 95,81 1416,93 843.98 52.8,92 343,71 2959,02 737,18 545,4 311.81 147.52 117,29 1746.41 1040.21 651,90 423.71 283.89 889.97 658.44 376.43 178.09 141,6 3923,92 2371.18 1504,02 983,53 662.98 172.74 1127,69 661,71 324.17 260.52 4553.33 2751,47 1745,24 1141.28 769,31 200.45 149.75 751.09 367,95 304.24
158
присутствуют как в числителе, так и в знаменателе. Как отмечалось
выше, неопределенность в массовых коэффициентах поглощения со-
ставляет 20 % для элементов Na, Mg, Al, Si, P и 10 % для остальных
элементов. Расчеты показали, что такая неопределенность в массо-
вых коэффициентах поглощения может изменить величину поправки
Ау на 0,001 отп. %.
Таким образом, погрешности в значениях поправок на матрич-
ные эффекты, обусловленные неопределенностью фундаментальных
параметров, не превышают допустимых.
Приведенные исследования позволили выбрать рабочие интер-
поляционные формулы для расчета массовых коэффициентов погло-
щены,ч, выходов флуоресценции и скачков поглощения. Рассчитанные
значения этих параметров для определяемых элементов приведены
в табл. 31.
S.3.2. Погрешности за счет упрощения
модели возбуждения
Использование упрощенного выражения (3.3) для расчета по-
правки па поглощение приводит к неслучайным погрешностям. По-
следние обусловлены двумя приближениями: заменой эффективности
возбуждения полного спектра трубки эффективностью возбуждения
излучения с длиной волны л и использованием одного значения к
при расчете функции поглощения пробы и образца сравнения. Вели-
чина погрешности, вносимая в расчет поправки на поглощение за
сштг введения эффективной длины волны, была оценена путем срав-
нения поправок, рассчитанных по полной (2.19) и упрощенной (5.3)
(формулам. Расчеты проведены для средних составов нескольких тп-
иов пород, приведенных в [73]. В качестве образца сравнения ис-
пользовался стандартный образец состава габбро-диорит СГД-1
(табл. 32). Как видно из (аблццы, предложеппая методика расчета
эффективной длины волны позволяет рассчитывать поправку на по-
глощение с погрешностью 0.03—0.07 оттт.% п примерно одинаковой
для всех определяемых элементов. В редких случаях опа достигает
флуоресценции и относительной доли поглощенных А'-уровнем фотонов
элементов
Мп Ге гл в 0 к К
б;юз.1б 8697.56 9745.39 218.40 1045,63 4417,15 0.019 0.908
3627.47 5255,38 5888,45 126.70 608,42 2575.45 0.027 0.907
2390,88 3333,45 3735 77.06 369,77 15(55,23 0.036 0.905
15O4.G3 2179,86 2442.45 48,68 237,58 988.74 0.047 0.904
1014.24 1469,4 1646.4 31.78 152,47 645.41 0.060 0.902
264,2(5 382,86 428.98 . 4 35.6 150.71 0.138 0.897
4"-1 286.02 320.48 5.41 25,97 109.95 0.163 0.895
114.92 166.5 186.55 3.02 14.47 а т 24 0.219 0,892
465,62 80.69 90.41 1.38 6.61 27.98 0.314 <6888
374.2 64.81 1.08 0.4 22.1 о 347 0.887
159
Таблица 32. Погрешности АА-ДА- (%) за счет введения эффективной
длины волны
Порода Mg А! м К Са Ге
Анортит -0,05 —0,03 -0.03 —0,0-4 -0,04 -0.08
Трапп —0,05 —0.06 -0,16 0.0/ — 0.20 -0,15
Гранодиорит 0.03 0,00 0,13 —('.02 -0.04 —0,08
Диорит -0.03 -0.09 0,01 0,11 0,02 -0,09
Гранит -0.04 —0,05 -0.02 —0,02 0.05 0,18
Перидотит -0.03 —0,02 —0.07 ('.('О 0,09 0,02
Кимберлит -0,08 —0,10 0.02 0.02 -0,07 0.04
Кварц, диорит -0,04 —0,08 ('.03 (>.03 0,02 0,09
Габбро -0,08 -0.03 -0,02 -0.02 —0.05 0,05
0.2 отн. % Эта погрешность на порядок меньше допустимо!! и не
должна сказаться па правильности результатов анализа.
Погрешности за счет использованпя одного значения /, для рас-
чета функции поглощения пробы и образца сравнения приведены в
табл. 33. Использование одного значения к для пробы п образца
сравнения дает различные погрешности для определяемых элемен-
тов. В среднем погрешность растет с увеличением атомного номера
анализируемого элемента. Сравнение погрешностей, приведенных в
табл. 32 п 33, показывает, что ошибка за счет использования Л образ-
ца сравнения для расчета функции поглощения пробы значительно
больше, чем за счет введения эффективной длины волны, особенно
для К, Са п Fe. Следовательно, величина суммарной погрешности в
поправке на поглощение будет зависеть от вариаций поправки на
поглощение, т. е. от выбора образца сравнения.
Были изучены распределения поправок на поглощение и погреш-
ностей при их расчете по упрощенной формуле (5.3) в зависимости
от выбора образца сравнения. Для этого по формуле (3.11) рассчитаны
значения полных поправок ка при определении петрогенных элемен-
тов для всех возможных типов изверженных горных пород, состав
Таблица 33. Значения погрешностей (%) при расчете поправки на погло-
щение за счет предположения л — ?.ст
Порода Mg AI S1 к Са Ге
Анортит 0.08 0,06 0,12 —0.61 -0,29 0.30
Трапп 0.09 0,10 0,13 —0.74 -0,33 0.05
Гранодиорит 0,01 -0.06 -0,10 0,14 0 0.23
Диорит о.об -0.14 -0.24 0,20 —0,06 0.42
Гранит 0,03 -0,02 —0,08 —0.0, —ОЛЗ 0.45
Перидотит 0.17 0.22 0.28 0.82 0.27 0.14
Кимберлит 0,04 0.09 0,08 0.15 0.69 0,43
Кварц, диорит 0,1'2 0.04 —0,07 0,10 0,05 0.06
Габбро 0,02 0.02 -0.27 0,13 0,03 -0,02
160
которых приводен в работе J73J (всего 82 пробы). Расчеты выполня-
лись для четырех составов образца сравнения, принадлежащих к
различным типам пород, результаты представлены в виде гистограмм
отклонений поправки ka от единицы (рис. 47, 7). Как видно, поправ-
ка па поглощение в зависимости от выбора образца сравнения может
изменяться в довольно широких пределах: до 14% для /Са-линии
Si, Са, до 22% для алюминия и до 44% для FeXa-линии. Распределе-
ние полных погрешностей при расчете поправки на поглощение
по разработанной .методике было построено тем же способом, что и
распределение самой поправки. Погрешности за счет всей совокуп-
ности приближений оценивались как разность поправок, рассчитан-
ных по полной (3.11) и приближенной (5.3) формулам (рис. 47, 2).
Гистограммы распределения ошибок показывают, что в боль-
шинстве случаев закон распределения ошибок близок к нормальному,
п с 95?о-ноп вероятностью можно утверждать, что относительное
стандартное отклонение, характеризующее погрешность A/ia'/ra, не
превышает 0,4% для магния и алюминия, 0,5% для кремния и желе-
за и 1 % для калия и кальция.
Увеличение погрешности для калия и кальция объясняется тем,
что дли этих элементов значения массовых коэффициентов поглоще-
ния для первичного и вторичного излучений сближаются и погреш-
ность в расчете массовых коэффициентов поглощения первичного из-
лучения сказывается сильнее. Меньшую величину погрешности для
железа по сравнению с калием и кальцием можно отнести за счет
отсутствия скачков поглощения с коротковолновой стороны от края
поглощения.
Если в качестве образца сравнения применять образцы близкого
пли среднего для всех типов пород состава, то погрешности значи-
тельно уменьшаются (см. заштрихованные части гистограмм на
рис. 47). Приведенные результаты позволяют сделать вывод, что по-
лученное выражение для поправки на поглощение позволяет с точ-
ностью не хуже 0,3 отн. % учитывать различие поглощающих харак-
теристик пробы п образца сравнения. При этом необходимо, чтобы
образец сравнения принадлежал к среднему типу пород.
Неслучайную погрешность, вносимую в поправку7 на эффект
матричного возбуждения при се расчете по формуле (5.4). можно
разделить на две составляющие: погрешность, обусловленную при-
ближенным расчетом параметра D^., и погрешность за счет примене-
ния одного значения Dih для пробы и образца сравнения.
Первая из них оценивалась как разность поправок, вычислен-
ных с помощью Dih, полученных из выражений (2.35) и (5.5). Зна-
чения ошибок AD ^,‘D ih, Аурфг и А7щ kf для стандартных образцов
гранита СГ-1, габбро-диорита СГД-l при использовании в качестве
образца сравнения траппа СТ-1 приведены в табл. 34. Хотя относи-
тельная погрешность в расчете для калия, кальция я 'пттала
составляет 30%, тем не менее благодаря структуре поправки на воз-
буждение величина ААщ'/су для Mg, Al, Si, Р равна 0.1%, для К —
0,01, Са — 0,2 и Ti —0,44%. Несмотря па резкое отлитие хими-
ческих составов стандартных образцов СГ-1 и СГД-1, погрешности в
11 В. П. Афонин, Г. Н. Гуничева. Л. Ф. Пискунова
161
Рис. 47. Распределение поправок на поглощение (1) я ошибок в лх
расчете по упрощенной формуле (2) для Ко.-линий кремния (а),
алюминия (б), калыщя (а), железа (г).
Та б лид a 34, Значения ошибок в параметрах Д Л .Л/Л ft, относительной
величине матричного возбуждения и в поправке па возбуждение при использо-
вании упрощенной модели их расчета, %
Образец сравнения Определяе- мый элемент Довозбуждающнй элемент Ду. ДЬ
А( Si Р К Са Т1 Мп Ее 7 г
СГД-1 Mg Al Si Р К Са Ti Мп 0,3 0,3 0,2 0.5 0,5 0,4 12.2 9,8 9,6 6,4 17.2 14А 11.6 10.5 0,3 19.2 15.7 12,3 11,2 3.0 9,2 12,0 14,9 16.0 2,9 32.2 29,9 1.7 1,6 4.8 5,9 24J 22,4 9,0 7,9 9,3 18,6 17.7 0,3 9,6 14,0 11,0 0,09 0.12 0,10 0,10 0,01 0.19 0,43 0
СГ-1 Mg Al SI Р К Са Ti Мп 0,7 0,9 0,6 0,1 0.2 0,2 20.8 17,2 14,2 15,6 17,0 13,0 9,8 11.3 7,8 11,3 6,8 3.2 5,0 16,0 10,6 1,4 5,7 9.2 7.6 2,9 25.2 19,3 1,2 3,2 7,0 5.0 2,8 23,9 18.5 1,04 2.9 3.5 29,4 28.3 0,6 3.5 8,1 23,0 0,09 0.10 0,11 0,11 0,01 0,20 0,44 0
определении поправки к5 для них примерно одинаковы. Следова-
тельно, упрощенное выражение для расчета матричного эффекта воз-
буждения достаточно хорошо учитывает влияние состава матрицы.
Выясним теперь величину ошибки в расчете к,- за счет предпо-
ложения, что параметры Dih для пробы и образца сравнения равны.
Для этого по полной формуле (2.35) были рассчитаны значения Dik
для стандартных образцов гранита СГ-1, габбро-диорита СГД-1 и
серпентинита SW. Поправки на возбуждение для анализируемого
образца вычислялись при использовании значений D{h = D^,. По-
грешности в поправке к/ при таком расчете для различных комби-
наций анализируемой пробы н образца сравнения приведены в
табл. 35.
Сравнение погрешностей, приведенных в табл. 34 и 35, показы-
вает, что ошибки за счет приближения D.th = DcJt пе превышают
ошибок за счет упрощенной модели расчета Dik. Следовательно, пра-
вильность вычисления поправки на возбуждение по упрощенной фор-
муле будет в меньшей степени зависеть от выбора образца сравне-
ния, чем правильность расчета поправки на поглощение.
В связи с этим при оценке полной погрешности за счет упрощен-
ной поправки на возбуждение в качестве образца сравнения исполь-
зовался один образец среднего состава. Для 70 проб горных пород в
пределах указанных выше вариаций состава были построены гисто-
граммы отклонений поправки на возбуждение от единицы, а также
гистограммы распределения полных погрешностей за счет прибли-
11*
163
Рис. 48. Распределение поправок па возбуждение (Z) и погрепшостей за счет
упрощенного расчета (2) для А.’с'-лпшй .магния (а), кремния (6), кальция (<з)?
титана (а).
Табл и ц а 35. Значения погрешностей ДАуД- (96) в поправке на матричное
возбуждение при использовании одного значения для пробы и образца
сравнения
Образец Образец срав- нения Mg Al Si P к TI Ге
сгд-1 СГ-1 SW 0,24 0,09 0,26 0.01 0,14 0,15 0,06 0,19 0,04 0,06 0.24 0.21 0.36 0.33 0,01 0,03
СГ-1 СГД-1 SW 0,25 0.31 0.27 UA1 0.05 0.01 0,03 0,01 0.02 0,0 0,06 0.0 0,18 0,06 0,0 6,0
SW СГД-1 СГ-1 0,06 0,10 0,0 (1.20 0.07 0,02 0,10 0,06 0,07 0.06 <1.21 0,0 0,34 0,29 0.0 0.0
желной модели расчета к; (рис. 48). Полные погрешности оценива-
лись как разность между значениями поправок, рассчитанных по
полной (2.35) и приближенной (5.4) формулам.
Вариации поправок на возбуждение невелики: 1% для Mg. Л1,
Si, Р и до 3 96 для остальных элементов. Если использовать в качест-
ве образца сравнения образец среднего состава, то закон распределе-
ния ошибок близок к нормальному, и с 95%-пой вероятностью можно
утверждать, что относптельное стандартное отклонение, характе-
ризующее погрешность расчета поправки на возбуждение, не превыша-
ет 0,3 % для Mg, Al, Si, Р, К, 0/ для Са п 0,5% для Ti. Расчеты
выполнены для следующих условий возбуждения: рентгеновская
трубка с Pd-анодом и толщиной Бе-окна 0.3 мм, Ро - -- 25 кВ, ф =
=== 90’, ф --- 40’. Сделанные выводы справедливы и для друшх усло-
вий возбуждения.
Таким образом, оценка систематических погрешностей при рас-
чете концентраций по уравнению (5.1) показала, что при использо-
вании образца сравнения среднего состава для всех типов горных
пород эти погрешности но превышают 0,3% допустимых. Следова-
тельно, упрощенный метод AF-поправок вполне приемлем для сили-
катного анализа горных пород.
5.4. ПОЛУЧЕНИЕ ГОМОГЕННЫХ ИЗЛУЧАТЕЛЕЙ
Использование уравнения (5.1) для расчета концентраций пет-
рогенных элементов в юрных породах позволяет вводить поправки
на матричные эффекты с высокой точностью, поэтому правильность
анализа методом AF-коррекцип зависит прежде всего от точности
экспериментального определения начального приближения концент-
раций Со. Возможности метода фундаментальных параметров можно
реализовать только при условии выполнения предпосылок, при ко-
торых получено уравнение (5.1), 'г. е. излучатель должен быть гомо-
генным и иметь плоскую поверхность.
165
Универсальным способом гомогенизации силикатов является
получение твердого раствора путем их сплавления с боратами. Опти-
мальные условия для приготовления излучателей зависят от вариа-
ций состава и физико-химических свойств анализируемого материа-
ла, требований, предъявляемых к чувствительности и экспрессиости
анализа.
5.4.1. Выбор условий гомогенизации
Решающая роль в получении твердого раствора принадлежит
выбору флюса и степени разбавления. Наиболее активным плавнем
по отношению к силикатным структурам является метаборат лития,
вследствие повышенного содержания щелочного окисла он позволяет
использовать меньшие разбавления и температуры сплавления по
сравнению с Ы3В4О7. Однако область существования прозрачных
незакристаллизованных стекол, полученных с помощью метабората,
еще уже, чем с тетраборатом, так как он содержит в 2 раза меньше
стеклообразующего оксида в В2О3 [92]. Поэтому были опробованы
оба флюса при сплавлении горных пород различного состава (грани-
тов, габбро, серпентинитов и известняков) в соотношении 1 : 2 в зо-
лото-платиновых тиглях при температуре 120043. Сплавление с тет-
раборатом даст прозрачные стекла для всей области составов. Распла-
вы, полученные сплавлением карбонатных пород с метаборатом, прп
охлаждении кристаллизуются. Следовательно, 1лВО2 непригоден
для гомогенизации карбонатных пород.
Стекла, полученные из силикатных пород, изучались на гомоген-
ность с помощью микроанализатора MS-46 «Камека». Уровень гомо-
генности образцов оценивался следующим образом. Сначала под
микроскопом определялось присутствие фаз и включений. Затем ре-
гистрировались интенсивности рентгеновского излучения элементов
в образце в зависимости от его положения прп механическом скани-
ровании относительно падающего пучка электронов. Таким образом,
можно выявить любые неоднородности более 10 отн. %. Эксперимент
должен быть объемным, т. с. необходимо исследовать как поверх-
ность диска, так и его поперечный разрез.
Изучение дисков под микроскопом с 400-кратным увеличением
показало отсутствие фаз во всех типах пород, сплавленных как с
LiBO2, так и с Li2B4O7. Распределение интенсивности рентгеновско-
го излучения основных компонентов равномерно, за исключением
Si в граните, сплавленном с LigB.jO?. На рис. 49 приведены типичные
диаграммы распределения интенсивности Si/ia-линии в излучателях
габбро-диорита СГД-1 (кривая 7), серпентинита SW (?) и гранита
СГ-1 (5), сплавленных с различными флюсами. Распределение крем-
ния во всех образцах, кроме гранита СГ-1 (кривая 3, б), равномерно.
Проверка показала, что неравномерное распределение кремния наб-
людается только в стеклах пз пород, содержащих кварц, поскольку
тетраборат растворяет кварц медленнее, чем другие силикаты, п
расплав кварца в борате более вязкий. Величину вязкости и повер-
хностного натяжения расплава можно уменьшить малой добавкой
166
2.
50 мкм
Рис. 49. Распределение интенсивности SiKa-линии крезшля по поперечному
разрезу излучателей из стандартного образца сравнения габбро-диорита СГД-1
(7), серпентинита SW (5), гранита СГ-1 (5), сплавленных в соотношении 1:2с
ЫВО2 (а), Ы2В4О7 (б) и Li2B4O7+ 5% KI (в).
ПАВ, таких как подиды Nal, KI [214, 828] и бромиды NaBr, КВт,
ПВг [268, 291].
Экспериментальная проверка показала, что лучшую воспроизво-
димость сплавления обеспечивают добавки KI и CsI, так как среди
использованных солей они обладают наименьшей вязкостью.
167
Полученные с добавкой KI стекла из стандартных образцов
состава гранита СГ-1, габбро-дпорпта СГД-1 и серпентинита SW ис-
следовались на гомогенность по описанной схеме. Диаграммы рас-
пределения интенсивности характеристического излучения кремния
вдоль поперечного разреза излучателей, приведенные на рис. 49, в,
показывают, что добавка йодида кадия в расплав способствует его
гомогенизации, особенно для пород, расплавы которых имеют высо-
кую вязкость.
Диаграммы, представленные на рис. 49. позволяют сделать вы-
вод, что использование в качестве флюса Ы2В4О7 5% KI, так же
как и ПВО», дает гомогенные излучатели из силикатных горных
пород. Но сплавлеппе с добавками подидов имеет ряд недостатков.
Поскольку они летучи, трудно контролировать количество оставшей-
ся в расплаве соли и ввести соответствующую поправку в результаты
анализа. Страдает правильность определения калия, усложняется
процесс препарирования. Следовательно, при гомогенизации магма-
тических пород целесообразнее использовать в качестве флюса мета-
борат лития.
Степень гомогенизации расплава существенно зависит от степени
разбавления пробы флюсом. При анализе горных пород желательно
применять низкие «епени разбавления, что понижает предел обна-
ружения легких элементов и дает возможность использовать для
определения -макро- н микрокомпонентов один излучатель. Были
опробованы две степени разбавления флюсом 1 : 1 и 1 : 2 пород раз-
личного минерального состава. Оказалось, что сплавление с метабо-
ратом лития обеспечивает приемлемую воспроизводимость как в пер-
вом, так н во втором случае. По при разбавлении 1 : 1 усложняется
формование стекла из-за сильно отличающихся значений вязкости
расплавов различных горных пород.
Следует отметить, чю гомогенные стекла из расплавов горных
пород с боратами лития в соотношении 1 : 1 п 1 : 2 можно получить
только при условии, что перед сплавленном смесь пробы с флюсом
тщательно перемешивается. В противном случае в твердом растворе
будут включения нерасплавленной породы.
Температура н время сплавления определяются свойствами ис-
пользуемого флюса и .материала образца, степенью разбавления и
способом нагревания. Высокочастотные печи обеспечивают более вы-
сокую температуру’ н позволяют сократить время сплавления, поэто-
му для сплавлении использовалась высокочастотная печь 13ЧП-4-10
с рабочей температурой 1200‘С. Время сплавления завист от мине-
рального состава сплавляемого материала к можег изменяться от 1
до 5 мин. Сплавление осуществляется в два этапа. На первом смесь
пробы с флюсом нагревается, из нее выходят воздух, двуокись угле-
рода л вода п образуется расплат Длительность первого периода
определяется оператором путем визуального наблюдения за распла-
вом. Ла втором этапе после получения прозрачного расплава он пере-
мешивается путем встряхивания тигля л снова нагревается до
120()'С в течение такого же периода времени, что необходимо для пол-
ной гомогенизации расплава. Ультраосповпые породы сплавляются
168
в среднем 2 мин, основные — 3 мин, а пробы, богатые кварцем,—
3—5 мин. Чистота расплава во многом зависит от свойств используе-
мых тиглей.
Как отмечалось в гл. 4, для гомогенизация силикатов исполь-
зуются стойкие к смачиванию сплавы платины и углерод, включая
гтеклографпт.
По мнению многих авторов, для получения боратных стекол луч-
шими являются тигли из сплавов плачмпы с золотом и родием.
Проверка показала, что тигли из сплавов ПлЗлй и ПлЗлйРдо
несколько смачиваются вязкими расплавами горных пород в боратах
1л л расплав не выливается полностью. Смачиваемость тигля распла-
вом уменьшается при сплавлении с добавками иодидов. Добавка 5 96
1\1 к расплаву обеспечивает несмачлваемость тиглей из указанных
сплавов. !4о со временем из-за перекристаллизации сплава при высо-
кой температуре портится поверхность тиглей, и они смачиваются
расплавом. Поэтому тигли нужно периодически мыть н полировать,
что сокращает срок их службы. К сожалению, тигли из сплавов пла-
тины с золотом и родием но выпускаются очечестненной промышлен-
ностью, и возможности их практического применения очень ограни-
чены.
Тигли из платпно-золотого сплава можно заменить стеклографи-
товыми. Их стенки слабо смачиваются расплавом, а остающиеся кап-
ли легко удаляются со стенок после охлаждения. Эю дает возмож-
ность проводить сплавление без добавок иодидов. Сплавление в стек-
лографнтовьих тиглях рекомендуется проводить в инертной среде.
На воздухе через 8—10 плавок они сгорают.
5.4.2. Приготовление излучателей
Излучатели из твердого раствора готовят непосредственно фор-
мованием расплава [257, 272, 276, 288, 313]. Различают два ва-
рианта приготовления стсклопзлучателей: расплав охлаждается по-
током воздуха непосредственно в тише (комбинированный вариант)
или формуется на подогретой подложке (раздельный вариант).
При сплавлении в стеклографитовых тиглях без добавки ПАВ
возможен только раздельный вариант. Обычно расплав выливается
в нагретые до 250—ЗОО'С подложки и прессуется плунжером до тех
пор, пока не остынет от 1000 до 300:С [250. 313, 328]. Поверхность
получаемых таким образом излучателей неровная, и их необходимо
шлифовать [172, 268]. Эксперимент показал, что шлифовка поверх-
ности образца алмазным кругом 50 мкм улучшает воспроизводимость
препарирования. Но это процедура длительная, и существует опас-
ность заражения поверхности диска используе.мыми пастами. Чтобы
избежать операции шлифования, способ формования стеклянных
дисков был модифицирован. На рис. 50 представлен эскиз формующе-
го устройства. Расплав выливается в углубление стальной плиты
с хромированной поверхностью, подогретой до 300 С. В углубление
опускается шайба, которая держится на хромированном плунжере
169
Рис. 50. Формующее устройство.
1 — стальная плита; 2 — полированный шпн-
жер; 3 — шайба; 4 — капля расплава; 5 —
постоянный магнит.
с помощью магнита. Вращатель-
ным движением плунжера капля
расплава впрессовывается в шай-
бу. Затем плунжер поднимается,
а расплав остывает на воздухе.
Эта операция позволяет получать
диски с гладкой поверхностью.
Латунная шайба не позволяет
расплаву собираться в каплю и
увеличивает прочность получае-
----------------------------- мо г о и з л уч а те л я.
Для определения воспроизводимости разработанного способа
препарирования было приготовлено по 12 излучателей для трех раз-
личных типов пород. Тщательно перемешанная смесь пробы с мета-
боратом лития в соотношении 1 : 2 общей навеской в 6 г сплавлялась
в стеклографитовых тиглях марки СУ 2000 в индукционной печи при
температуре 1200°С. Время сплавления зависит от типа пород и ко-
леблется от 1 до 5 мин. Стеклянные диски с гладкой поверхностью
получали описанным способом.
Интенсивности аналитических линий петрогенных элементов из-
мерялись дважды от каждого излучателя на спектрометре СРМ-18,
С помощью одноступенчатого дисперсионного анализа были выявле-
ны погрешности, характеризующие воспроизводимости работы аппа-
ратуры и препарирования (табл. 36). Из таблицы видно, что погреш-
ности препарирования примерно одинаковы для всех приведенных
составов горных пород. Относительные стандартные отклонения,
Т а б л ид а 36. Результаты дисперсионного анализа для оценки ошибок пре-
парирования стеклянных излучателей
Обра- зец S Na Mg Al Si P j К Ca Ti Mn Fe
ПМ sr ,ап 1,81 0,98 0,31 0,20 0,60 0,16 0,27 0,37 0,54 0,20
б-,пр 1,31 0,65 0,37 0,30 0,60 0,70 0,35 0.33 0,44 0.61
sr,S 2,21 1,17 0,47 0,35 0,83 0,70 0,45 0,49 0,69 0.62
сгд sr,an 1,78 0,72 0,29 0,17 0,56 0,26 0,32 0,26 0,76 0,15
S/',np 1,75 0.80 0,58 0,54 0.39 0,47 0,51 0,79 0,40
sr,2 2,46 1,05 0,63 0,55 0,56 0,46 0,55 0,56 1,07 0,42
СГ-2 an 2,56 2.43 0,29 0,17 2.98 0,12 0,50 0,57 8,17 0,09
sr,np 0,70 0,55 0,51 0,54 0,35
V7,S 2,56 2.43 0,73 0.56 2,98 0.51 0.73 0,57 8,17 0.35
170
характеризующие погрешности препарирования и воспроизводимость
работы аппаратуры, соизмеримы. Суммарная погрешность для эле-
ментов с содержаниями выше 1 % на уровне 0,5—1 отн. %. Исключе-
ние составляет натрий, для которого эта величина достигает 2,5%.
Сравнение относительного стандартного отклонения, характеризую-
щего суммарную воспроизводимость измерений, с допусками
(см. табл. 25) показывает приемлемость разработанного способа пре-
парирования.
5.5. КОРРЕКЦИЯ ИЗМЕРЕННЫХ ИНТЕНСИВНОСТЕЙ
Точность определений методом фундаментальных параметров
существенно зависит от правильности процедуры выделения анали-
тического сигнала из измеренной прибором интенсивности аналити-
ческой лишит, так как аппаратурные эффекты не учитываются после-
дующей процедурой коррекции результатов на матричные эффекты.
Для получения точных значений относительных интенсивностей не-
обходимо учесть наложение линий, статистику процесса, мертвое
время, возможное загрязнение анализируемого образца п образца
сравнения и вклад фона в регистрируемую интенсивность.
5.5.1. Поправка на фон
В работе [123] отмечается, что при рентгеноспектральпом анали-
зе геологических проб на петрогенные элементы из-за некорректной
поправки на фон для различных линий в ряде случаев относительное
стандартное отклонение может превышать 10 отн. %. Такие погреш-
ности выходят за пределы допускаемых II категорией классифика-
ции лабораторных методов анализа минерального сырья.
Поправка на фон есть вычитание из измеренной на пике линии
интенсивности той ее части, которая не связана с аналитической ли-
нией. Используемые в настоящее время частичные методики нахож-
дения этой интенсивности, как правило, основаны на измерениях
рядом с линией. На многоканальных спектрометрах такие измерения
для фиксированных каналов недоступны, для перестраивающихся —
нерациональны, так как отсутствие автоматической установки угла
дифракции делает переход с линии в положение измерения фона
при массовом анализе весьма трудоемким п приводит к потере про-
изводительности.
Авторами предложено проводить измерения интенсивности фона
на фоновом образце, а фон пробы рассчитывать путем ввода в изме-
ренные интенсивности поправок на различие спектрального состава
фола пробы и «фонового образца»5). Исследования фона в длинно-
волновой области рентгеновской флуоресценции [80, 81] показали,
5) Под «фоновым образцом» понимается излучатель из такого материала,
зависимое от химического состава фоновое излучение Хчоторого можно с доста-
точной точностью аппроксимировать рассеянным им излучением трубки.
171
что для набора кристаллов-анализаторов в отечественных спектро-
метрах такими материалами .могут быть углерод С. борная кислота
Н3ВО3, углокислый Li2CO3 и тетраборнокислый 1я2В jO7 литий, фто-
рид лития LiF. Некоторые из них используются при анализе для го-
могенизации исследуемых материалов. Такие фоновые образцы бу-
дут частично моделировать химический состав анализируемых об-
разцов. Рассмотрение практического фона от излучателей этих ма-
териалов показало, что тормозным излучением возникают их в них
фотоэлектронов из-за низкого уровня можно пренебречь [17, 273].
Аналитические линии элементов, входящих в состав перечисленных
материалов, не могут внести сколько-нибудь существенный вклад в
фон, так как расположены вне области обычно используемых для
анализа длин воли и при возможных условиях возбуждения, если и
возбуждаются, то неэффективно. Поэтому для интенсивности фона
фонового образца будет справедливо соотношение [170]
(ЧЭо = const Гл".У- <5-В 9)
где — интенсивность излучения, падающего на образец; о0 и
— соответственно массовый коэффициент рассеяния и ослабления
излучения с длиной волны л для фоновых образцов. Принимая во
внимание, что в области длин волн > 0,2 нм интенсивность значи-
мых компонентов фона, кроме связанных с рассеянием излучения
трубки, пропорциональна интенсивности падающей па кристалл-ана-
лизатор линии, для интенсивности фона пробы на месте аналити-
ческой линии элемента I можно записать следующее выражение:
Тфг = const -Ь S Zj (5.10)
где «j:;, — коэффициент, определяемый параметрами регистрирую-
щей системы; о п р — соответственно массовый коэффициент рассея-
ния и ослабления анализируемого образца излучения с длиной вол-
ны Ху I,ц — интенсивность к-й линии элемента у. Суммирование вы-
полняется но интенсивным линиям всех присутствующих в пробе
элементов у. Каждый член ail!t /у выражения (5.10) эквивалентен
сумме компонентов фона, обусловленных линиями элемента у, из
которых превалирующими являются их диффузное рассеяние и воз-
бужденная ими флуоресценция атомов кристалла-анализатора, ког-
да флуоресценция интенсивна и относительно близка по шкале длин
волн к аналитической линии элемента i.
Разделив (5.10) на (5.9), получим следующее соотношение, свя-
зывающее интенсивности фона пробы п фонового образца:
^фг (ЗД0 [ Одо + Jd X 0 / о hk 1 • (5.11)
L /‘ i j А ° /-г J
В первом приближении Ijh ~ Характерная для каждого
элемента относительная близость по шкале длин волн интенсивных
линий в К- и Z-сериях позволяет с приемлемой погрешностью, во-
первых, ограничиться одним значением ai} для всех лпппй одной се-
172
рпи элемента / и, во-вторых, допустить u7-?; =- ну. Тогда соот-
ношение (5.11) можно переписать как
^ф? — (Л1н)о
Vf'i , у 1
о7в" Т ;
(5.12)
где bij — коэффициент, зависящий от параметров регистрирующей
системы и эффективности возбуждения линий элемента j. Значение
bij определяет дополнительный к рассеянному излучению пробы фон,
даваемый в расчете па 1 % элемента j в образце, не содержащем дру-
гих элементов. Обозначив
О, U; б’;/и,-
0-13)
о /а/ о /д '
запишем (5.12) в сжатом виде
Л!)£ = (Ж)<Л; !,')
(jAi)
Как правило, практический фон содержит нс зависимую от хими-
ческого состава составляющую (ифД-ш, которая кроме аппаратурно-
го фона будет учитывать возможное загрязнение фонового образца.
Считая, что (пф£)а11-= const п пф; = /фг 2- (пфг)ап, ("$i)o =” (Ц jo
(^фОап, из соошошепття (5.14) получим выражение, позволяющее
рассчитать скорость счета фона для анализируемого образца (Иф,),
исходя из измеренной скорости счета фонового образца О/фДд
«Ф. Жфф[(1 - П(с + Л) + Л (5.15)
где I =- (Нфг)ап ' (н,1п)о — доля аппаратурного компонента в измерен-
ной скорости счета фонового образца.
Из выражения (5.13) видно, что .s-z есть поправка на различное
рассеяние излучения трубки пробой и фоновым образцом; Д учиты-
вает компоненты фона, отсутствующие в фоновом образце. Конкрети-
зация характеристик фонового образца позволяет сгруппировать
компоненты фона но ответственным за них линиям, что упрощает
учет сложных для разделения компонентов фона и унифицирует
схему ьктада в фон всех присутствующих в пробе элементов. Исклю-
чение из значений Ьц характеристик фонового образца о°/ц" и цу
делает матрицу этих коэффициентов единой для возможных фоновых
образцов и существенно сужает пределы изменения значений
Последнее в значительной степени снижает ограничения на вариации
химического состава проб, по которым они определяются. Коэффи-
циенты b:j находятся .методом наименьших квадратов но чистым
элехМентам и нескольким несильно разбавленным смесям / с материа-
лом, удовлетворяющим определению «фонового».
Данный способ учета фона был апробирован в области длин волн
0,2—1.0 им при определении петрогенных элементов в горных поро-
дах. гомогенизированных сплавлением с тетраборатом лития в соот-
ношении 1 : 1. па комплексе СРМ-18/М 6000. Анализ аппаратурных
условий показал, что заметную флуоресценцию кристалла-анализа-
тора следует ожидать только на NaA'a- и MgTfc'-.Tininax. Так как
173
Таблица 37.
Сопоставление измеренных и рассчитанных
Fe
Ti
Са
К
Si
Al
Mg
2,91
3.28
13,72
2,48
0.83
1,09
2.74
3,32
12,00
2.48
0,86
1,12
Пр имела н л е.
Состав образца: 50% оксида + 50% тетрабората лития.
большинство каналов соллеровские, должна быть значительной диф-
фузная составляющая фона.
В табл. 37 для аналитических линий петрогенных элементов
приведены рассчитанные значения поправок для оксидов этих эле-
ментов, разбавленных 1 : 1 тетраборатом лития. Из-за малых кон-
центраций в горных породах оксиды Мп п Р в рассмотрение не вклю-
чались. Также даны отношения измеренных фоновых интенсивностей
7ф;/(7фг-)0 образца и «фонового образца». «Фоновый» излучатель прес-
совался из порошка углерода. Постоянная составляющая фона
определялась из соотношения
(° /.1Ч)-(°/Иг)1
где (и и Рг)х и (^ф,)1 — соответственно рассеивающие п абсорбционные
характеристики и измеренные скорости счета образца 1, удовлетво-
ряющего определению «фонового». Соотношение (5.16) получается пз
выражения (5.14), записанного для образца 1.
Сопоставление значений аг- и 7ф;/(/ф£)о показывает, что в за-
висимости от кристалла-анализатора, геометрии канала и состава
излучателя доля рассеянного излучения в зарегистрированном фоне
может изменяться от ~ 100% на РеА’а-линип для всех представлен-
ных составов до ~ 10% на MgA’a-линии для оксида кальция. Оче-
видна тенденция уменьшения вклада рассеянного первичного излу-
чения в практический фон при смещении в область больших длин
волн (в направлении от FeKa- к MgAa-липии). Таким образом, для
всех линий, кроме FeAxz-, поправки только на различное рассеяние
первичного излучения недостаточно. Необходимо введение поправки
Д-, даже когда ее численное значение определяется только диффуз-
ным рассеянием линпй присутствующих в пробе элементов.
По определению, величинаД должна учитывать разницу между
7фг/(7ф£-)о и Sj. Из табл. 37 видно, что в пределах одной линии в за-
висимости от элемента / эта разница изменяется в широких преде-
174
фоновых интенсивностей
СаКа TiKa MnKa FeKa
J<i o •ч / Гф/ Тф(1 *-rf s s-f 1ф/1фО s s+f
1,00 0.85 0,44 0,48 0.52 0,56 0,62 0.73 0.19 0,34 0,36 0-38 0,93 0,85 0.38 0,41 0,53 0.58 2,15 0,34 0,30 0,91 0.45 0,71 0,61 0,17 0,18 0,33 0.35 0.38 2,12 0,34 0,30 0,90 0,42 0,61 1.13 1,19 0.22 0,24 0,51 0.50 0,51 0,60 0,14 0,23 0,17 0,32 0.34 0,37 0,93 0.14 0.23 0,24 0,55 0,50 0.61 0.15 0.21 0,17 0,45 0,35 0,39 0,14 0,16 0,17 0.32 0,34 0,37 0.14 0.16 0,17 0,32 0,34 0,37
лах, особенно для Mg'Z^a-линии, где из-за флуоресценции атомов
кристалла-анализатора, эффективно возбуждаемой линиями каль-
ция, титана и железа, наблюдается более чем на порядок изменение
в значениях ^фг/(/ф/)о- Концентрация оксидов всех элементов в пред-
ставленных образцах одинаковая, поэтому предположение, выска-
занное в работе [125], о возможности использования одного значе-
ния Ь, для всех элементов, возбуждающих флуоресценцию атомов
кристалла-анализатора, в данном случае не выполняется. Коррект-
ное определение /7- возможно только при конкретизации вклада в
фон линии каждого элемента j через значения коэффициентов Ъц.
Эксперимент по определению коэффициентов планировался
таким образом, чтобы найти сами значения коэффициентов, оценить
зависимость Ьи = Ь^(С7-) и устойчивость Ъ{1 к выбору фонового об-
разца. Необходимые излучатели прессовались из оксидов петроген-
ных элементов и смесей оксидов с углекислым литием в соотношениях
1:1,1:2,113,1: 4, 1 : 10, 1 : 19. При подборе химических реак-
тивов предпочтение следует отдавать реактивам марки ОСЧ, так
как пики элементов примесей, особенно если это примеси определяе-
мых элементов, могут существенно исказить истинную ситуацию.
На рис. 51 для Mg^a-линии приведены графики зависимости
величин Ь и т от концентраций окспдов Са и Ti -- элементов, линии
Рис. 51. Зависимости коэффициентов Ъ^„^{1) и ??гьг>г(2)от концент-
рации элемента /(г).
а — СаО; б —TiO,.
175
'Г аблнца 38. Значения коэффициентов Ъ^-
ПИШИ! 1 Э.К'Меи'1 j
Ха Mg И Si к С.» I Т1 )’е
NaAct- (',() 11.0 11.5 2.8 4.5 3,8 2,8 1,4
МйАа- 1.0 0.0 31.5 2.0 26.3 90.7 15.9 4,3
AlA'a- л л / 7 В 0.0 12.6 1 г 0.2 2,7 0.2
SiKa- 0,2 о 14.0 0,0 5.5 О.8 0.8 0.3
1>К<Х- 0.0 л л 13.3 29.2 12.9 6.6 5.7 1,7
К Act- 0.0 0.5 1.0 2,0 0,0 46,3 6.0 7 5
СаАа 0.0 0.1 12.0 2.0 21,1 0.0 5,0 29,4
TiAa- 0,0 0.1 1.8 1.5 1.0 / .2 0.0 11,4
МпА’а- 0,0 0,0 0.0 0Л) 0.0 0.2 1.0 23.8
FeAa- 0,0 о.о (',0 0.0 0.0 0.0 0.0 0,0
которых дают наибольший вклад в фоновое излучение на данной ли-
нии. Для сравнения приведена аналогичная зависимость для коэф-
фициентов т^.г в члене ^т^гСг 1'125], ответственном за тот же
эффект. Для удобства сопоставления величины ДЛ1е7 и тж.. нор-
мированы к соответствующим величинам для излучателя, состоящего
из 100% оксида. Пз рис. 51 видно, что по сравнению с т1г ----- mir{Cr)
концентрационная зависимость Ъц — Ьц(С/), определяющая рамки
применимости частных значений Ь,,-, в предложенном алгоритме рас-
чета фона в значительной степени снята. На всю концентрационную
область состава можно пользоваться одними значениями коэффи-
циентов. Поэтому при анализе для нахождения коэффициентов bi}-
достаточно измерить интенсивности фона /ф£ па аналитических ли-
ниях от излучателей, приготовленных из смесей 1 : 2 определяемых
элементов с углекислым литием, по отношению к интенсивностям от
фонового образца (/фг)о- Значения Ъ^, рассчитанные по формуле
(5.12), приведены в табл. 38. Анализ значений коэффициентов bif
показывает, что при наличии флуоресценции атомов кристалла-ана-
лизатора наибольший вклад в фон вносит коэффициент для элемента
7, линия которого эффективно возбуждает эту флуоресценцию. При
ее отсутствии максимальное значение имеют линии ближайшего
п наиболее эффективно возбуждаемого элемента /.
Проверка устойчивости матрицы коэффициентов к выбору
фонового образца показала, что для линий, где постоянная состав-
ляющая фона мала, расхождение между коэффициентами ^£-у. полу-
ченными по двум фоновым образцам, не превышает 15—20%. J> об-
ласти л > 0,4 нм, где скорость счета (ПфД-ш существенная, из-за
большой погрешности в ее определении независимость йц от фоново-
го образца надежно подтвердить не удалось.
Приведенные в табл. 37 величины суммарных поправок (х£ -|- /)
рассчитаны с использованием указанных в табл. 38 значений
Расхождение их с отношениями измеренных интенсивностей для всех
линий не превышает 15—20%, тогда как расхождение с интенсив-
ностью фонового образца (отклонение от единицы) — нередко по-
рядка 200—300'%.
176
5.5.2. Учет аппаратурных погрешностей
Существует два основных источника аппаратурных погреш-
ностей: мертвое время регистрирующей системы п нестабильность ра-
боты прибора. Обычно регистрируемая скорость счета корректирует-
ся ла потери, обусловленные мертвым временем, с помощью следую-
щего выражения:
Л' - Лт',(1 у- Л'Ч),
где N — истинная скорость счета, Л7' — измеренная скорость счета,
т — мертвое время счета.
Значения мертвого времени счета для спектрометрических кана-
лов СРМ-18 определялись ио методике, удобной для спектрометров с
фиксированными каналами 1288]. Для каждого канала подбирались
два образца с такими содержаниями, которые обеспечивают интен-
сивности аналитических линий, отличающиеся примерно на порядок.
Скорости счета на аналитической линии измерялись при нескольких,
значениях тока. Мертвое время счета рассчитывалось но формуле
т „________________________мж-мж_________________ (517>
-ТЖ (Ж -- -''и) - Vn ж - Ж ’ ’
где Na и Лг6 — скорости счета двух образцов при одном значении
тока; Ха3 и ]\'ъх — те же величины при другом значении тока. На-
пример, для определения мертвого времени счета спектрометрическо-
го канала на SiA’a-лннпп использовались излучатели двух стандарт-
ных образцов: гранит GM и известняк Си-1 с содержаниями SiO2
73,5 п 2,73% соответственно. Скорости счета измерялись прп двух
значениях напряжения на трубке и пяти значениях тока. Расчет
мертвого времени производился для всех возможных сочетаний зна-
чений тока. Значения мертвого времени счета приведены в табл. 39.
Проверка показала, что прп скоростях счета п X Ю3 имп/с погреш-
ность в определении относительной интенсивности без введения по-
правки на .мертвое время счета .меньше 1 %, но при скоростях счета
3-101 она достигает 5%. При измерении интенсивности AlA'a-, SiZCa-,
CaZ£a- и РеАа-лнний на квантометре СРМ-18 достигается скорость
счета п-104 пмп.'с, поэтому для указанных элементов введение по-
правки па мертвое время счета при требуемой точности анализа обя-
зательно.
Другой источник экспериментальных погрешностей связан с
нестабильностью работы аппаратуры. Чтобы уменьшить влияние ап-
паратурного дрейфа при определении интенсивностей, используют
реперные образцы. .В способе фундаментальных параметров им может
одновременно служить образец сравнения. Применение образца
сравнения среднего состава для горных пород обеспечивает мини-
мальную погрешность в определении относительной интенсивности
из-за статистических флуктуаций числа регистрируемых импульсов,
так как он содержит все определяемые элементы в достаточном ко-
личестве. Приготовление излучателей в виде стеклянных дисков га-
рантирует постоянство интенсивностей флуоресценции образца срав-
В. П. Аф >нин, Т. Н. Гумилева. Л. ф. Пискунова
177
Таблица 39. Результаты определения мертвого времени счета для Si-ка-
нала спектрометра СРМ-18
Напря- жение, кВ Я Мертвое время счета т, мкс Среднее т Напря- жение, кВ Я Мертвое время счета т, мкс Среднее т
20 70 60 50 40 30 3,3 3,0 2.9 2,5 2,7 2.7 2.8 2,8 2,8 3,4 2,9±0,5 25 70 60 50 40 30 2,9 2.4 2,6 2,9 2,8 2,9 3,1 3,4 2,5 3,6 2,9±0,6
нения во времени. Условия регистрации интенсивностей на кванто-
метре СРМ-18 были выбраны следующие: рабочее напряжение 25 кВ,
ток трубки 70 мА. Экспозиция в 100 с обеспечивает для реперного
образца следующие статистические ошибки счета, %: Na — 1,56;
Mg - 0,81; Al - 0,28; Si - 0,18; P - 0,4; К - 0,15; Са — 0,16;
Ti — 0,18; Мп — 0,53; Fe — 0,06. Регистрация интенсивностей фо-
нового и реперного образцов проводилась через каждые 10 проб.
Воспроизводимость измерения относительных интенсивностей
определялась на стандартных образцах и контрольных пробах, при-
готовленных пз этих образцов путем разбавления тетраборатом лития
Рис. 52. Зависимость относительной квадратичной погрешности 5 от относи-
тельной интенсивности J!lcr и концентрации С определяемого элемента.
178
в соотпсптсниях 1 : 2, 1 : 5, 1 : 10. Из каждого образца было приго-
товлено но шесть излучателей. Зарегистрированное число импульсов-
аналитических линий исправлялось на фон и мертвое время счета
согласно выражения?! (5.15) и (5.17) соответственно. Относительные
интенсивности рассчитывались по отношению к интенсивностям об-
разца сравнения габбро-диорита СГД-1. Измерения проводились
независимо три раза через несколько дней, чтобтя учесть временной
дрейф прибора. На рис. 52 приведены зависимости относительного-
стандартного отклонения, характеризующего воспроизводимость из-
мерений, от концентрации определяемых элементов для Mg, Al. Si,
К, Са и Fe. Установлено, что для большинства типов горных пород
содержания определяемых элементов лежат в области, где Sr(C) =
---- const. Относительные стандартные отклонения, характеризующие
суммарную воспроизводимость препарирования и работы аппарату-
ры, в этой области не превышает 1% для всех определяемых эле-
ментов.
5.6. КОРРЕКЦИЯ НАЧАЛЬНЫХ ПРИБЛИЖЕНИЙ
Как отмечалось в гл. 3, при использовании теоретического спо-
соба учета матричных эффектов необходима коррекция измеренных
интенсивностей с рассчитанными по используемой модели. Это по-
зволяет за счет оптимизации параметров при определении Со мини-
мизировать влияние не учтенных уравнением (5.1) эффектов. К ним
относятся: остаточная неоднородность излучателя, систематические
погрешности при выделении аналитического сигнала из зарегистри-
рованной интенсивности линии вторичного рентгеновского спектра,
а также неполнота используемой модели учета матричных эффектов.
Учет взаимных влияний элементов методом фундаментальных
параметров и исправление измеренных интенсивностей на аппаратур-
ные эффекты позволяют при анализе магматических пород функцию
/(/г/Дт) представить в виде линейной, тогда
= + (5.18)
где коэффициенты н аг находятся следующим образом. Из образ-
цов известного химического состава готовятся излучатели путем
сплавления с флюсом в различных соотношениях. Измеряются от-
носительные интенсивности, в которые по описанной методике вво-
дятся поправки на фон п мертвое время счета. По формуле (5.1) с по-
мощью программы «Поправки» рассчитываются теоретические ин-
тенсивности. По значениям и (7//ст)расч методом наи-
меньших квадратов вычисляются величины a0, а2.
Для нахождения корректирующих зависимостей использовались
стандартные образцы горных пород гранита СГ-1, известняка Си-1,
почв СП-1, СП-2, СП-3 и оксиды определяемых элементов, сплавлен-
ные в соотношении 1 : 1, 1 : 2, 1 : 4, 1 : 5, 1 : 10 с метаборатом ли-
12*
179'
Рис. -5S. Корректирующие зависимости для определения начальных прибли-
жений.
тпя. Полученные контрольные пробы охватывают следующие вариа-
ции состава: М^О — до 50%, А12О3 — до 40, SiO, — до 100, СаО —
до 50, Fe2O3 — до 25%. Значения относительных интенсивностей
найдены трехкратным измерением интенсивностей флуоресценции
контрольных образцов (по три излучателя для каждого) на кванто-
метре СРМ-18 с j четом поправок на фон п мертвое время счета.
Проверка гипотезы линейности показала, что для названных
элементов в пределах указанных вариаций состава корректирующие
зависимости в области относительных интенсивностей, для которой
справедливо условие 5r(C) - const, можно представить прямой
(рис. 53). Значения параметров <70 п о1? найденные методом наимень-
ших квадратов, и стандартных отклонений 5(|, характеризующих
рассеяние точек относительно прямой, приведены в табл. 40.
Проведено сопоставление результатов расчета концентрации в
стандартных образцах' горных пород разнообразного состава по об-
разцу сравнения СГД-1 для двух способов определения величин С’о:
по выражению (5.2) — I и с помощью формулы (5.18) -- II (габл. 41).
Сравнение найденных содержаний Ср,с с аттестованными пока-
зывает, что использование корректирующей зависимости (0.18)
позволяет получать правильные значения концентраций во всем диа-
Таблица 40. Значения параметров уравнения (5.18) и стандартное
_____________________ отклонение _______________________________
Определя- емый эле- мент “о So Определя- емый эле- мент «0 So
Мд А1 Si 0.009 —0.017 —0,051 0.970 1.022 1.062 0.020 0.015 0,013 Са Fe —0.050 —0,019 1.060 1.043 0.015 0.014
180
Таблица 41. Сопоставление результатов расчета кон-
центраций при двух способах определения величины Со
Стандартный оортгеи Иоицснзрацгит ЫО_,, % Довери- тельный 1- s интер- вал Р—0.95
ciL СР с
I и
Гранит Альбит, гранит Гранит Алевролит Серпентин Вазальт Трапп Образец сравнения ''ЗАО 73,36 72.1 60.6 39,05 49,51 49,42 46,39 71,26 71.34 70.20 59,49 39.53 49.23 48.78 73,41 73:60 72.32 60,56 39,07 49.28 48.93 0,31 0,31 U,21 0.31 0.32 0.38 0.31
п&'зоте изменения состава горных пород (расхождение между С\гг и
С{_.х (II) находшс-я в пределах экспериментальных погрешностей).
Использование выражения (5.2) дает сопоставимые результаты толь-
ко в 'гом случае, если содержания определяемых элементов в пробе
is образце сравнения близки (ВМ н СТ-1). Следовательно, примене-
ние корректирующей зависимости при определении величины Cfl
необходимо для тех элементов, концентрации которых в анализируе-
мом хМатериале изменяются в широких пределах. К таким элементам
в горных породах относятся Mg, Al, Si, Са, Be.
5.7. АВТОМАТИЗАЦИЯ АНАЛИЗА ГОРНЫХ ПОРОД
Разработанная на базе метода AF-коррекцШ! .методика анализа
горных пород реализована на рештеиофлуоресцентном аналптн-
песком комплексе квантометр СРМ-18/М 6000, позволяющем автома-
тизировать все операции после загрузки излучателей в спектрометр.
К ним относятся: процедуре! управления автоматической сменой об-
разцов и процессом регистрации интенсивностей по заданной схеме,
периодический контроль работоспособности измерительной системы
л выявление дефектов в работе узлов, процедура выделения анали-
тического сигнала и расчет концентраций но измеренном,у значению
интенсивностей (аналитическая программа), а также предваритель-
ные расчеты, необходимые на этапе разработки методики (методи-
ческая программа).
Математическое обеспечение выполнено на языке BASIC. Работа
в этой системе обеспечивает удобный диалоговый режим, оператив-
ную замену исходных данных, простоту операций вызова, переза-
пуска программы, внесения корректив в вычислительный процесс.
Стандартный вариант интерпретатора BASIC был скомпонован с про-
цедурами управления п приема информации с кваптометра, выпол-
ненными на машинном ориентировании.
181
5.7.1. Методические расчеты
Для реализации метода AF-коррекцпп необходимо выбрать стан-
дарт сравнения оптимального состава. Состав некоторых наиболее
часто применяемых в рентгеноспектральном силикатном анализе
стандартных образцов горпых пород приведен в табл. 42. Как пока-
зали предыдущие расчеты, в качестве образца сравнения можно
попользовать стандартные образцы, принадлежащие к среднему плп
основному типу пород, например габбро-диорит СГД-1, базальты
ВМ, BCR-1, андезит AGV-1, гранодиорит GSP-1. Для выбранного-
образца сравнения вычисляются значения эффективных длин волн
первичного излучения определяемых элементов. Поскольку обеъм
оперативной памяти М 6000 и СМ-1 (16К) недостаточен для такой за-
дачи, эффективная длина волны рассчитывалась по программе, со-
ставленной па языке АЛГОЛ-60 для ЭВМ БЭСМ-6. Интенсивность
тормозного и характеристического спектра трубки находилась соот-
ветственно по формулам (1.2) и (1.6). Интеграл в функции l‘t вычис-
лялся методом Симпсона. Эффективная длина волны при возбужде-
нии флуоресценции тормозным компонентом излучения трубки А.
рассчитывалась по теореме о среднем (формула (2.22)).
При использовании теоремы о среднем точность расчета А, су-
щественно зависит от числа интервалов, на которые разбивается ин-
тегрируемый отрезок [Ао, при вычислении интеграла методом
Симпсона. Удобно для всех определяемых элементов использовать
одинаковое число интервалов. Это оправдало тем, что при увеличении
Таблица 42.
Стандартные
Определя- емый ком- понент Состав, %
Андезит AGV-1 Эи 3 3. о Перидотит РСС-1 Диабаз W-1 Терригенная. глина СДО-1 Вулк.-тер- риг. ил СДО-2 j Известняк, пл СДО-3
Институт прпк-
ИГУ
Геологическая служба США (USGS)
Na90 4.31 3,29 0,01 4,06 2,80 0.01 2.15 4 47 4,03 1.88
MgO 1,55 3,49 49,83 0.77 0,96 43,63 6(63 3’34 4,67 3.48
Д]2О3 17,22 13,68 0.29 15,35 15,19 0.73 14,87 12,65 14,33 3,55
SiO, 59,72 54,85 40,68 69,19 67.31 42,15 52,72 41,82 43,61 11.87
р2о5 0,50 0.33 0,00 0.14 0,28 0.60 6.14 0,25 0,28 0.24
К2О 2,93 1.68 0.00 4.52 5.53 0.00 0,64 1.34 1,36 0,48
СаО 5,00 6,98 0,15 1,98 2,02 0,03 10,98 9.24 7,81 39,35
тю2 1,05 2,22 0.01 0.50 0.66 0,01 1.07 0J3 2,32 0.29
МпО 0,10 0,19 0,11 0.04 0.04 0.12 0,17 1.05 0.27 0.22
Fe2O3 6,88 13,54 8,61 2,62 4,34 8,24 11,10 7.00 11,97 2.41
S 0,01 0,04 0,00 0,00 0,04 0,01 0,01 0'28 0,01 0.19
F 0,04 0.05 0,00 0,13 0,38 0,00 0,02
Н2(У 0,82 0,73 0.46 0.46 0,57 4,64 0,53
со2 0,01 0,03 0,06 0,06 0,12 0,16 0,06 17,44 9,22 36,59
182
атомного номера хотя и уменьшается величина отрезка [Хо, но
увеличивается крутизна функции, л для правильного расчета необ-
ходимо уменьшать шаг интегрирования. Практика показала, что
стабильные значения X можно получать при числе интервалов не ме-
нее 350— 400.
Эффективная длина волны для элементов, возбуждаемых сме-
шанным излучением трубки, вычислялась по формуле (2.24). При
использовании трубки с Pd-анодом к таким элементам относятся Na,
Mg. Al, Si, P. Значения X, рассчитанные для состава стандартного
образца СГД-1, сплавленного в соотношении 1:2с 1и2ВрЭ?, и усло-
вий возбуждения, типичных для СРМ-18 (трубка с Pd-анодом. напря-
жение 25 кВ, (р = 90°, ф =- 35 и 25е). следующие (А):
Na Mg Al Si Р К Са Ti Мп Fe
4,23 4,24 4.22 4.21 4,20 2,29 2,17 1.91 1,41 1.36
В гл. 2 показано, что значения л слабо зависят от состава гомо-
генизированных сплавлением горных пород. Следовательно, приве-
денные значения эффективных длин волн можно использовать и для
других образцов горных пород среднего состава (например, ВМ,
BCR-1. AGV-1) и при различных степенях разбавления. Как показа-
ла практика, при этом не вносится заметной погрешности в резуль-
таты анализа.
Для сокращения времени расчета концентраций непосредствен-
но при массовом анализе необходимо предварительно рассчитать зпа-
образцы состава
ладной физики (СССР) Институт петрографических и гео- химических исследований (ГДР) Центральным гео- логический инсти- тут (Франция)
0.8 1,15 1,16 4,64 3,76 1.31 0,013 0.08 3.07 3,55 3,85
1,02 0.77 1,95 7,46 0.38 1.94 38,50 0.03 13.35 0,95 0.03
10.37 9,57 12,61 16,23 13,55 20,60 0.66 14,70 10,25 14,51 12.51
69,53 78,33 65,72 49,51 73,50 60,24 39.05 71.45 38,39 69,96 75,85
0.17 0.08 0.21 0,101 0,063 0,10 0,02 0.02 1,05 0,12 0.01
2,29 2’47 2,51 0,20 4,74 3.85 0.01 2,63 1.41 4,03 4,76
1.63 0,81 2,86 6.46 1,04 0.33 0,18 0.61 13,87 2,45 0,69
0,75 0,84 0,73 1,14 0.21 0.93 0,016 0Д2 2,61 0,38 0,08
0.08 0,07 0,09 0.14 0,04 0.05 0,083 0,17 0,20 0,09 0,05
3.81 2.98 4,91 9,69 2,02 6,94 7,40 5.92 12,94 2.83 1,34
0.07 0,06 0,03 0.015 0.016 0,04
0,02 0.07 0,07 0,007 3,36 0,10 0,05 0,35
3,62 0,35 3,82 13.6 1.8 2,31 0.87 0.46
9,17 2,59 6,7 1,34 0,28 0.13 0,29 0.04 0,86 0.11 0,14
183
Окончание табл. 42
Состав, %
Определя- емый ком- понент Альбит, гранит СГ-1 Габбро СГД-1 Трани (Л-1 Алевролит СА-1 Алясьлт. гри;шт С.Г-2 Нефели- нов. ргцу С.НС-1 Доломят, известняк С,п-1
Институт прикладной физики ИГУ (СССР)
Na2O 5 46 2.82 2,49 2.3 2.50 12.2 0.08
Мь?О 0,05 7.09 5,74 2 2, 0.21 1,01 20.8
А120з 13,84 14.88 14,23 18,2 14.0 28.5 0.43
SiO2 73.37 46.39 49.12 60,6 72.1 40.18 2.73
Р3О5 0,01 Н)1 0.21 0.18 0.08 0.51 0,02
К2О 4,14 2.96 0,69 3.45 7.2 3,3 0.38
СаО 0,14 10.97 10,20 0.48 0.73 7,13 29.6
Т1О2 0.07 1,71 1.82 0.95 0,23 0.24 0,03
МпО 0.19 0.17 0.21 0.04 0,(12 0.04 О.05
Fe2O-) Ц2Э 11,48 15.32 7,2 2’30 3,25 0.47
S 0.02 0.02 0.07 0.09 0.02 0.08 0.02
F 0,30 0.12 0.03 0,06 0.03 0.03
Н2О+ 0,21 0.83 0.97 4,1 0.35 1,9 0.57
СО2 0.07 0.13 0,10 0,2 0.13 1,6 45.1
чепия массовых коэффициентов поглощения эффективных длин волн
в элементах, присутствующих в анализируемых образцах, парамет-
ров возбуждения а также значения поправочных функций
Рис. 54. Блок-схема программы «Поправки»).
184
4- У -г Х-;)ст для стандарта сравнения. Эти расчеты
выполняются на ЭВМ М 6000, по программе «Поправки», составлен-
ном на языке BASIC. Блок-схема программы приведена на рис. 54.
Па печать выводятся концентрации стандарта, общая поправка
па матричные эффекты, поправки на поглощение п дополнительное
возбуждение, а также расчетные интенсивности.
5.7.2. Аналитическая программа
Расчет состава анализируемых образцов осуществляется по ана-
литической программе (блок-схема протраммы приведена на рис. 55).
Рис. 55. Блок-схема аналитической программы.
185
Она выполняет три функции: управляет сменой образцов по задан’
пой схеме п процессом регистрации интенсивностей аналитических
линий, выделяет аналитический сигнал из зарегистрированной ско-
рости счета, переводит аналитический сигнал в состав анализируе-
мого образца.
Процедура управления и регистрации интенсивностей обеспечива-
ет регистрацию интенсивностей от фонового образца, образца сравпе-
ппя и 10 анализируемых проб, а также исправление зарегистрирован-
ного числа импульсов на мертвое время счета.
В блоке коррекции на фон интенсивность фона, измеренного на
фоновом образце, исправляется па зависимость фона от состава ана-
лизируемой пробы. Первоначально полагается, что интенсивности
фона на аналитической линии пробы и образца сравнения равны. По
полученному отношению интенсивностей рассчитывается прибли-
женное значение Со. Найденные величины Со используются для рас-
чета интенсивностей фона аналитических линий анализируемого об-
разца. По уточненным с помощью корректирующей зависимости
(5.18) значениям относительных интенсивностей находятся началь-
ные приближения концентраций определяемых элементов. В экспе-
риментально найденные значения 6'0 вводятся поправки на различие
матричных эффектов анализируемой пробы и образца сравнения
/та и Ау.
Исправленные значения концентрации находятся с помощью
решения системы уравнений методом итераций. Процесс итераций
продолжается до достижения наперед заданной точности по каждому
из определяемых элементов. Обычно при анализе силикатов доста-
точно 3—4 итераций. Полученные концентрации пересчитываются па
исходный материал.
Па печать выводятся: интенсивность от фонового образца и об-
разца сравнения, пулевые приближения концентраций, концентра-
ции в исходном материале и сумма концентраций.
5.8. МЕТРОЛОГИЧЕСКИЕ ХАРАКТЕРИСТИКИ МЕТОДИКИ
Общепринятыми метрологическими характеристиками, опреде-
ляющими качество методики анализа, являются воспроизводимость,
правильность и предел обнаружения. Опп устанавливались согласно
требованиям, утвержденным Научным советом по аналитическим .ме-
тодам Мпнгео СССР (НСАЛ1) для анализа геологических образцов.
5.8.1. Воспроизводимость
Воспроизводимость результатов определений — это метрологи-
ческая характеристика случайной погрешности методики анализа.
Прп рентгенофлуоресцсптнОхМ анализе ее основными компонентами
являются погрешности приготовления излучателей и регистрации
интенсивности, которые оценивались на этапе разработки методики.
Суммарная воспроизводимость методики анализа на петрогенпые
186
iumMHiliiHluHMHlttHH<w<liH«H«HHilii'«iHii<...........t«HiiH«Hi«tHiHH<iliii»<iiii'<ilHrtWHi»«iiHt>Hii'i»".......i**i ii < ши и i hi I и >'><<) u t»n и tiiHi i ни и 11 м 11 ii ttm и ни и к <i iii «ни и n HU u t-1 и ill < tn iihh IIIIHIIfHHUHttHtttWmi НИНЖНМ
187
Таблица 43. Зависимость воспроизводимости анализа от содержания определяемых компонентов
Интервал содержа- ний ХсиО MgO А1гО3
а sr Z а г S? Z сг^ sr Z
20-39,99 2,6 2,0 1,3 2,4 0,8 3,0
10 -10.99 3,5 5,0 0,7 3,4 2,3 1// 3.5 0,8 4,4
5 -9,99 5,4 5,0 1,1 4,6 2,4 1,9 5,4 0,9 6,0
1-4.99 0,0 9,5 0,9 7,8 10,0 0,8 9,5 1,7 5,6
0,5--0,99 12,5 12,0 1,0 13,0 14,0 0,9 15,0 5,5 2,7
0.2—0,499 16.5 26,0 0,6 16,0 17,0 0,9 20,0 10,0 2,0
0,1- 0.199 21,0 28,0 0,8 25.0 12,5 2.0
0,05—0,099 28,0 20,0 1,4
0,02-0,049 30,0 29,0 1,0
С °тш’ '° 0,1 0,08 0,01
188
11 род о л ж сипе т а б л. 43
Интервал содержа- ний Si(),
<j т s z <т ,
60-79,99 0,7 0,7 1,0
40—59,99 0,9 0,7 1,3
20—39,99 1,6 0.8 2,0
10 19,99 3,2 1.0 3,2
5 9.99 5,0 1,2 4,2
1- 4,99 8,00 4,0 2.0 4.3
0.5-0,99 12,0 6.0 2,0 6.0
0,2-0.499 8,2
0,1 -0,199 9,3
0,05- 0,099 12,0
0,02- -0,049 16,0
0,01—0,019 21.0
^mjn’ 0/0
P.O. K.,0
sr z z
0,9 6,0
1,0 4.3 9,0 0.9 10
1,5 4,0 12,5 3.0 4,2
2,0 4.1 J 6,5 3.5 4,7
4,0 2.3 20,0 11,0 1,8
8.0 1,5 23,0 18,0 1.3
10.2 1.6 28,0 20,0 1,4
26,0 0.8 30,0 32,0 0,9
0,00'5 0.005
элементы оценивалась одним
из способов, предложенных
в работе |971. Из стандарт-
ных образцов горных пород
и приготовленных из них
контрольных проб, а также
проб известного состава бы-
ло изготовлено по три излу-
чателя. Концентрации опре-
делялись на аналитическом
комплексе СРМ-18/М 6000 не
менее трех раз. Результаты
объединялись в группы по
интервалам содержания. Вы-
борочная дисперсия рассчи-
тывалась по формуле
si=[i i с» -
(5.19)
где n — число параллельных
определений, т — число
проб, С-,, — концентрация
определяемого элемента.
Число степеней свободы
для каждого интервала со-
держаний колебалось от 18
до 30.
Относительное стандарт-
ное отклонение:
Sr - S- 100/С, (5.20)
где с —- среднее содержа-
ние анализируемого элемента
в пробах данного интерва-
ла. которое находится по
формуле
Чтобы выяснить, удовлетво-
ряет ли данная методика тре-
буемой категории анализа,
189
вычисляется так называемый «запас точности»:
Z == ог/£г,
(5.22)
где ог — допустимое значение относительного стандартного откло-
нения для данного интервала содержаний. Если Z У> 0,7, то данная
методика удовлетворяет требуемой категории анализа. Зависимость
воспроизводимости, рассчитанной по предложенной методике, от
концентрации элементов приведена в табл. 43. Видно, что для всех
элемептов значения Z > 0,7. Следовательно, разработанная методи-
ка силикатного анализа удовлетворяет требуемой для полных анали-
зов И категории классификации. Причем определение оксидов всех
элементов, кроме натрия и магния, выполняется с большим «запасом
точности».
5.8.2. Предел обнаружения
Предел обнаружения в рентгенофлуоресцентном методе зависит
от множества факторов. Па величину предела обнаружения влияют
эффективность возбуждения флуоресценции первичным излучением,
условия регистрации аналитических линий, а также методика опреде-
ления фона. Для нахождения предела обнаружения использовались
стандартные образцы: граниты С1-2 и GM, почвы СП-1, СП-2, СП-3,
серпентинит SW, известняк Си-1. Предел обнаружения рассчитывал-
ся по формуле
Cmin = —С. (5,23)
где I — разность между средним значением интенсивности, измерен-
ным па месте аналитической линии, и интенсивностью фона; С —
концентрация элемента; З’ф — стандартное отклонение измерений
фона; п — число измерений фона, по которым рассчитывалось З’ф.
Выражение (5.23) справедливо, если разброс значений холостого
опыта соответствует случайной погрешности .методики анализа вбли-
зи предела обнаружения. Поэтому для каждой аналитической линии
выбиралось по два стандартных образца с такими содержаниями
определяемого элемента, для которых контраст аналитической линии
не превышал двух. Значения 1 к 1$ рассчитывались ио 15 измере-
ниям. Измеренная па фоновом образце интенсивность фона пересчи-
тывалась па интенсивность фона от образца в соответствии с описан-
ной методикой. Пределы обнаружения, рассчитанные с доверитель-
ной вероятностью 99 %, для всех определяемых элементов, кроме
кремния и железа, приведены в табл. 43. Для большинства петрогеп-
ных элементов предел обнаружения па уровне содержаний 0,002—
0.005%, только для оксидов магния и натрия они пиже: 0,08 и 0,1%
соответственно. Такие значения пределов обнаружения вполне удов-
летворительны для силикатного анализа.
190
Таблиц а 44. Сопоставление данных рентгеносиектрального определения концентрации С с в стандартных образцах
горных пород с аттестованными содержаниями С , %
Na,0 MgO Ai2O3
Стандартный обрааед Сдт Ср.е Cl,r СР с СР-с
Гранит GM 3.76+0,05 3,91+0,23 0,38+0.025 0,30+-0,05 13,55+0,07 13,5+0.11
Грейзен GnA 0,<>8+0,012 - 0,03+0,007 - 14,7+0,1 14,7+0,09
Гл. сланец ТВ 1,31 +0,03 1,26+0,11 1,94+0.06 1.87+0,17 20,6+0,08 20,62±0,10
Ъазульт ВМ 4,64+0.06 4,65+0,34 7,46+0,05 7,54+0,11 16,23+0.10 16,15+0,08
Сериснтшпп SW 0,013+0,001 - 3,85+0,12 38,1+0,51 0,66 +0,04 0,65+0,03
Гранит СГ-1 5,46+0,05 5,53+0,31 0,054+0,014 - 13,84+0,04 13,69+0,08
Гранит СГ-2 2,50+0,10 2.,)3 +0,1 0,21+0,03 0,17+0,04 14,0+0,2 13,9+0,10
Алевролит СА-1 2,3+0,1 2,3+0,22 2,2+0,1 2,3+0,15 18,2+0,03 18,1+0,03
Неф. сиенит CHG 12,2+0,2 12,0±-0,39 1,01±0,02 1,01+0,08 28,5+0,1 28,4+0,15
Трапп СТ-1 2,49±:0,03 2,44±0,19 5,47±0,07 5,62±0,13 14,23±0,09 14,19±0.09
Известняк Ся-1 0,079+0,03 — 20,8+0,04 21.11+0,41 0,43+0,10 0,45+0,04
II р о д о л ж с и и с т а б л. 44
с сГ 4,79+0.05 g g 11 о ю ю 1! ст
+1 о 11 11 L~- 11 4м
-1! •<* CU 11
ст со 1Л о со ст -Ст <
cl * 11 1 11 11 Л1 О 11 1! 11
-е' of со о' ст ст о- °1
сГ 11 g 10+0,005 11 11 +1 5 >1+0,004 1 £00‘ОТО( 1 1 18+0,002 1
О ° °. ° о- О со> О °.
с о Сг S о !а =£ со — 1О о
1 о 11 11 1 4, 11 11 11 11 11
X- О = ст_ 5
£ О м со г? сч ст ст ст ст
ч +1 11 +1 11 11 4 11 11
о С5 <ч z- си и1 со ст”
1*
сс СП у- от
п “ +1 11 +1 +1 11 11 11 О 11
£ ЧГ. г- О £ м io S ? 3 ст
щ
О
К
а га ч: О с 32 S ZT1 и и g и 5
5
192
5.8.3. Правильность
Окот аггпо табл, 44
о |i+hhi МО -- СО £ О
o'3 +1 л+1+1+i+i+1+1+i 3+1 сч с-а сч со м* ю о оi1Л Т; г- о с: ©о (Ц’сз i~ сг cs C'f од со о
ч ?Н1?+Н1+Н1+1+Н'^ со мо со -!* со мо -ч1 ’Н
ТЮ; о" ОТ с-л от ото +1+1 +1+1 +1+1 +1+1+1+Й СО со СО 'З1 О сч СО МО -<г СО МО Cl Ci — t-- М Cl Cl X) Ы со = о- о о о- ж
о
о'
Стандартны!! образец GM GnA ТВ вм sw С Г-1 СГ-2 СА-1 СПС СТ-1 Си-1
Правильность — метрологиче-
ская характеристика, опреде-
ляющая близость к нулю систе-
матической погрешности. При раз-
работке методики анализа на пет-
рогенпые элементы особое внима-
ние было уделено оптимизации
систематических погрешностей
как на этапе рассмотрения мето-
да учета матричных эффектов,
так и при измерении относитель-
ных интенсивностей.
Правильность методики си-
ликатного анализа оценивалась
на стандартных образцах горных
пород. Выполнялось по девять па-
раллельных определений для каж-
дого образца на аналитическом
комплексе — многоканальном спе-
ктрометре СРМ-18/М 6000. Ре-
зультаты определения петроген-
ных элементов Ср,с. в сравнении
с аттестованными содержаниями
С’ат приведены в табл. 4-4. Дове-
рительный интервал для средних
значений концентрации установ-
лен с доверительной вероятностью
95%. Для всех определяемых эле-
ментов в проверяемом диапазоне
концентраций систематические
погрешности оказались незначи-
мыми. Таким образом, предложен-
ный способ определения петро-
генных элементов для всех про-
веряемых интервалов содержаний
удовлетворяет по правильности
и воспроизводимости требовани-
ям к методам анализа .11 катего-
рии классификации.
13 В. П. Афонии, Т. Н. Гунииева, Л. Ф. Пискунова
193
Приложение I. Значения коэффициентов С и п в формуле ц = б'л’1
[259, 230, 315]
Поглощаю- 1 « j Поглощаю- z. <
щий элемент
C n c n
1 2 3 4 5 c
3Ы 0,157 2,92 43Rli 404,52 2.83
4Ве 0.380 2,92 46Pd 428,12 2,83
БВ 0.755 2,92 47Aa 452.94 2,83
6С 1,322 2,92 48Gd 478.56 2.83
7N 2,122 2,92 49In 504.71 2.83
8О 3.198 2,92 50Sn 532,36 2,83
эр 4,591 2,92 51Sb 560.90 2,83
10Ne 6.344 2.92 52Tc 590.24 2,83
lxNa 9,613 2.79 53 J 628,00 2,83
]2Mg 12,191 2.79 54Xe 651,81 2,83
13А1 15.168 2,79 55Gs 683,94 2,83
14Si 18,569 2,79 56Ba 717,20 2,83
15p 22,418 2,79 57La 752,38 2.66
16g 26,737 2,79 58Ge 787,75 2,66
17GI 31,550 2,79 59pr 824,10 2,66
18Ar 36.878 2,79 60Nd 861,36 2,66
19K 44,944 2.66 61Pm 900.18 2,66
20Ga 51.015 2.66 62Sm 939,70 2,66
21Sc 57,548 2,66 ЙЗЕн 980,05 2,66
22Tj 64.556 2,66 64Gd 1021.8 2,66
23y 72,048 2,66 ti5Tb 1065,2 2,66
24Gr 80,034 2,66 esDy 1109,4 2,66
25Mn 88,525 2,66 67l-lo 1154.8 2,66
26 Fe 97,530 2.66 C8E1- 1201,6 2,66
27Go 107,058 2.66 69Tm 1248.3 2,66
28Ni 117,121 2,66 -оуь 1297,9 2,66
29Gu 127,725 2,66 71Lu 1349,0 2,66
30Zn 138.881 2,66 72Hf 1400,6 2,66
31Ga 150,597 2.66 73Ta 1454,6 2,66
32Ge 162,882 2,66 74W 1509,2 2.66
33As 175,745 2,66 75Re
34Se 189,194 2.66 ! 760s
35Br 203,236 2.66 77Ir
36 Kl. 217,882 2,66 ] 7<Pt
37P.b 235,500 2,68 79Au
3sSr 251.300 2,68 80Hg
зэу 268.100 2.67 siTi
40Zr 297,82 2,83 82Pb
41Nb 317,56 2,83 S3Bi
42Mo 338,07 2,83 j 84Po
43Tc 359,48 2,83 ! 3’At
44Ru 381,48 2,83 I S6Rn
87Fr
194
Продолжение приложения I
гоглошаю- % V 4i Si < X LIII
c n c 74 c n
1 2 3 5 6 7
“No 0.762 2,74
12Mg 0,992 2.74
13А1 1,264 2,74
14Si 1,583 2.74
lap 1,951 2,74
18й 2,372 2,74
1'С1 2,850 2.74
l.SAj. 3,390 2.74
1SK 4,900 2.70
20Qi 5,685 2,70
21Sc 6,550 2,70
22T1 7,496 2.70
23у 8.527 2,70
24Cr 9,647 2,70
25Mn 10.860 2,70
28 Fe 12,168 2.70
27Co 13,576 2.70
2feNi 15.086 2,70
2sCu 16,702 2,70 14,06 2,73
3°Zn 18,427 2.70 15,60 2,73 11.20 2,73
31Ga 20,265 2,70 17.25 2,73 12,40 2,73
S2Gc 22.220 2,70 18,95 2,73 13,60 2.73
33A s 24,294 2,70 20,75 2,73 14,90 2,73
34Se 26,491 2.70 22,55 2,73 16,20 2,73
35Br 28,814 2,70 24.60 2.73 17,70 2.73
36Kr 31,267 2,70 26,70 2,73 19,15 2,73
37Rb 33,826 2,70 28,80 2,73 20,80 2,73
38Sr 36,526 2,70 31,20 2,73 22,40 2,73
-39y 39.364 2,70 33,50 2,73 24,10 2,73
40Zr 42,341 2.70 36,15 2,73 25,95 2,73
41Nb 45,462 2,70 38.90 2.73 27,90 2,73
42Mo 48,729 2,70 41,45 2.72 29,75 2,72
43Tc 52,146 2,70 44,50 2,72 31,95 2,72
44Ru 55,716 2,70 47,50 2,72 34,10 2,72
45Rh 59,441 2,70 50,70 2,72 36,40 2,72
48Pd 63,325 2,70 53,95 2,72 38,70 2.72
4,Ag 67,371 2,70 57,50 2,71 41,30 2,71
48Cd 71,583 2,70 60,70 2,71 43,60 2.71
4!lIn 75.962 2,70 64.50 2,71 46.30 2,71
5“Sn 80,513 2,70 68,00 2,71 48,80 2.71
48 b 85,239 2,70 71,95 2,70 51,70 2,70
13*
195
Продолжение приложения I
1 2 3 5 7
62Те 90.141 2,70 75.65 2.70 54,30 2.70
53 J 95.225 2,70 80,35 2.70 57,70 2.70
54Хс 100,491 2,70 84,00 2,69 60,30 2,69
55Cs 104,387 2,70 88,40 2,69 64,94 2,62
6вВа 109,848 2,70 93,15 2,69 68.33 2,62
57La 115,490 2,70 97,80 2,68 71,83 2,62
58Се 121.317 2,70 102.50 2.68 75,44 2.62
5»Pj. 127.330 2.70 107,30 2,68 79,16 2,G2
"Nd 133,533 2.70 112.80 2,67 83,00 2.62
61Pm 139.928 2,70 118,50 2.67 86.97 2,62
62Sm 146,517 2,70 122,50 2,67 91,05 2,62
63Eu 153,304 2.70 128.00 2,66 95,25 2.62
64Gd 160,291 2.70 134.00 2,66 99,57 2,62
65Tb 167,481 2,70 140,00 2.65 104,02 2,62
««Dy 174,876 2,70 146,00 2.65 108,60 2,62
67Ho 182,479 2,70 151,00 2,65 113,30 2,62
68Er 190,292 2.70 157,00 2,64 118,14 2,62
6STm 198,319 2,70 165.00 2,64 123,10 2.62
70Yb 206,561 2,70 170.00 2.63 128.20 2,62
71Ln 215,021 2,70 176,00 2,63 133.43 2.62
72Hf 195.263 2,50 183.00 2.62 131,00 2.62
73Ta 204,582 2,50 190,00 2,62 136,00 2,62
74\V 214,210 2.50 197,00 2,61 142,00 2.61
75Re 224,152 2,50 204,00 2,61 147.00 2.61
780s 234,415 2,50 211.00 2.60 152,00 2,60
771r 245.005 2,50 219,00 2,60 157,00 2.60
78pt 255,927 2,50 225.00 2.59 161,00 2.59
79Au 267,187 2,50 232.00 2,59 167,00 2,59
8°Hg 278,792 2,50 240,00 2,58 172.00 2.58
81T] 290,747 2,50 247,00 2.58 177,00 2,58
82pb 303,059 2,50 255,00 2,57 183.00 2.57
"Bi 315,733 2,50 262,00 2.56 188,00 2,56
"Po 328,776 2,50 270,00 2.56 194,00 2.56
"At 342,194 2,50 278.00 2,55 199,00 2,55
88Rn 355,993 2,50 286,00 2.55 205.00 2,55
87 Fr 418,40 2,66 323,81 2.69 210,00 2,55
8sRa 432,51 2,66 335.61 2,69 232,54 2.58
89Ac 446,95 2,66 347,72 2,69 240,23 2,58
"Th 461,68 2,66 360,10 2,69 248,07 2.5S
91Pa 476,73 2,66 372,78 2,69 256,08 2,58
92U 492,23 2,66 400,00 2.69 264,26 2.58
93Np 507,81 2,66 410.00 2.69 272,61 2.58
S4pu 523,84 2,66 420,00 2,69 281,14 2,58
196
Продолжение приложения I
Поглоща- ющий .'лемент <S'n ЧЧ 44’^ <l«v <?,<
c n c n c л c n c n c Ti
1 2 3 i Ь 6 7 8 9 10 11 12 ia
ик 20Са 21SC 22Ti 23у 24Сг 25Мп 26 Ре 27Со 2SNi 29Сп ^Zn 31Ga 32Ge 33As 34Sc 35Br 3GKr 37Rb 38Sr зэу 40Zr 41Nb «Mo «Тс «Bu «Rh 46Pd 4\g «Cd 49jn 50Sn 51Sb E‘2Te 53J 5JXc 55Cs 5eBa 57La 5sCe 0,747 0,892 1,057 1,242 1,449 1,680 1,936 2,218 2,529 2,869 3,241 3,645 4,084 4,560 5,074 5,628 6,224 6,863 6,10 6.62 7,18 7,78 8,39 9,05 9,74 10,47 11.24 12.03 12.87 13,76 14,68 15,65 16.66 17,72 18,82 19,97 24,262 25.601 26,987 28,423 2,44 2,44 2,44 2.44 2.44 2,44 2,44 2,44 2,44 2,44 2,44 2.44 2,44 2,44 2,44 2,44 2.44 2,44 2,60 2,60 2.60 2,60 2.60 2,60 2,60 2,60 2.60 2,60 2,60 2,60 2,60 2,60 2.60 2,60 2,60 2,60 2,50 2,50 2,50 2,50 18,2 19,3 20,4 21,6 2,60 2,60 2,60 2,60 16,0 1G.9 17,9 2.60 2,60 2,60 15,5 2,60
197
Окончание приложения I
1 2 3 4 5 6 1 8 !) 10 11 12 13
59ру 29,908 2.50 22,8 2,60 18,9 2,60 16,3 2,60
OyNd 31,444 2,50 24,1 2,60 20,0 2,60 17-2 2.60
33,032 2.50 25,4 2,60 21,0 2.60 18,2 2.60
34,672 2,50 26,7 2,60 22,2 2.60 19,1 2,60
€3 J.vt 36,365 2,50 28,1 2.60 23.3 2.60 20.1 2.60 14,50 2,26 13,85 2,22
38,113 2,50 29,6 2,60 24,5 2,60 21,2 2,60 15.02 2,26 14,30 2,22
b5Tf! 39,915 2,50 31,1 2,60 25,8 2,60 22,3 2,60 15,75 2,26 14,80 2,22
6G£)y 41,773 2,50 32,6 2,60 27,0 2,60 23,4 2.60 16.45 2,26 15,30 2.22
67] J ) 43,687 2,50 34,3 2,60 28,5 2,60 24,5 2,60 17,00 2,26 15,85 2,22
ssKr 45,659 2.50 35.9 2,60 29,8 2,60 25.7 2,60 17.70 2,26 16,30 2,22
®Tai 47,690 2.50 37.7 2,60 31,2 2,60 27.0 2,60 18,40 2,26 16,85 2,22
*°Yb 49,779 2.50 39,6 2.60 32,7 2.60 28,3 2,60 19.10 2,26 17,45 2,22
'^Lu 51,928 2,50 41,3 2,60 34,2 2,60 29,6 2,60 19,80 2,26 18,00 2,22
51,501 2.55 43.2 2,60 35.8 2.60 30,9 2,60 20,50 2,26 18,55 2.22
33Ta 53,744 2,55 45,2 2.60 37.5 2,60 32,4 2,60 21,30 2,26 19.05 2,22
?«W 56,052 2,55 47,2 2,60 39,1 2,60 33,8 2.60 22,10 2,26 19,60 2,22
?5Re 58,425 2,55 49,3 2,60 40,9 2,60 35,3 2,60 23,00 2,26 20,10 2,22
Os 60,866 2,55 51,5 2,60 42,6 2,60 36,9 2,60 23,85 2,26 20,45 2,22
Щг 63,375 2,55 53.7 2,60 44.5 2.60 38,4 2,60 24,60 2,26 21,35 2.22
65,953 2,55 56,0 2,60 46.4 2,60 40,1 2,60 25,50 2,26 21,95 2,22
?9Au 68,601 2,55 58,3 2,60 48,3 2.60 41,8 2,60 26.45 2,26 22,50 2,22
S°IIg 71,320 2.55 60,8 2.60 50,4 2.60 43,6 2,60 27,15 2,26 23,10 2,22
81T1 74.111 2,55 63,5 2,60 52.6 2,60 45,4 2,60 28.15 2,26 23,80 2,22
83pb 76,975 2,55 66,1 2.60 54.7 2,60 47,3 2,60 29,00 2,26 24,40 2,22
S3Bi 79,912 2,55 68,9 2,60 57.1 2,60 49,3 2,60 30,00 2.26 25,10 2,22
espo 82,925 2,55 72,0 2,60 59,7 2,60 51,6 2,60 31,00 2.26 25,80 2,22
85 At 86,014 2,55 75,2 2,60 62.3 2,60 53,9 2,60 31,95 2,26 26,60 2,22
66Rh 89.179 2.55 78,5 2,60 65.2 2,60 56,3 2,60 33.10 2,26 27,40 2,22
87 Fr 105.383 2.63 82,2 2,60 68,1 2.60 58,9 2,60 34,00 2,26 28,30 2,22
88Ra 110,641 2,63 86,0 2,GO 71,2 2,60 61,5 2,60 35.00 2,26 29,60 2,22
8yAc 116,097 2,63 89,9 2,60 74,5 2,GO 64,4 2,60 36,10 2,26 30,80 2,22
°°Th 121,757 2,63 94,3 2,60 78.1 2.60 67,5 2,60 37,10 2,26 31,30 2,22
91Pa 127,625 2,63 98.7 2,60 81,8 2,60 70,7 2,60 38.20 2,26 32,40 2,22
92y 133.708 2,63 104,0 2,60 86,2 2,60 74,5 2,60 39,30 2,26 33,60 2,22
93Np 140,010 2.63 109,5 2.60 90.8 2,60 78,5 2,60 40,50 2,26 34,90 2,22
94Pu 146,536 2,63 116,5 2,60 96,7 2.60 83,6 2.60 41.85 2,26 36.40 2,22
?98
Приложение II. Потенциалы возбуждения (кВ) и длины воли краев
поглощения н диаграммных линий К-серии рентгеновского спектра элементов (А)
Элемент Линия a0 а. а2 6, 62 04 — K-iipaii поглощении Потенциал позбуж- деник
1 [среход 1 K^UIII т К1 сГ
Стат, вес 0,867 0,578 0,289 0,0 7 0,029 0,006 0,006 0,006
1 2 3 4 0 6 7 8 9 10 Н 12
3Li 4Во *В 6С 7N 8О °F i°Ne J1Na 13Л1 i^Si 15 р 16S 17С1 18ДГ 19К 20Са 215с 22Т1 23у 21Сг 25Мп 26 Ре 27Со 240, ИЗ, 67, 44, 31,603 23,707 18,307 14,615 11.909 9.889 8,339 7,126 6,155 5,373 4,729 4,192 3,744 3.360 3.032 2,750 2,505 2,291 2,103 1,937 1.791 8.338 7,125 6.154 5,372 4,728 4.191 3.742 3-359 3,031 2,749 2.503 2.290 2,102 1.936 1.789 8,341 7,127 6.157 5.375 4.731 4,194 3,745 3,362 3,034 2,753 2,507 2,294 2,105 1,940 1.793 14 ,460 11.617 9,558 7,981 6,769 5,804 5,032 4,403 3,886 3,454 3,089 2,780 2, 2,С 1,910 1,757 1,621 14 85 85 3,442 3.074 2.764 2.498 2,270 2.071 1,897 1.757 1,609 226,953 107,200 65,604 43,767 31,052 23,367 17,897 14,170 11,475 9.512 7.951 6,744 5,787 5.018 4,397 3,871 3.437 3,070 2,758 2,497 2.269 2.070 1,896 1.743 1,608 0,055 0,116 0,192 0,283 0.399 0,531 0,687 0,874 1,080 1,303 1,559 1,838 2,142 2.470 2,819 3,203 3,607 4,038 4,496 4,964 5.463 5,988 6.537 7,111 7,709
28Ni 1.659 1.658 1,661 1,500 1,489 1.489 1,488 8,331
2SCu 1,542 1,540 1,544 1,393 1,393 1,381 1,382 1,380 8.980
30Zn 1.437 1,435 1,439 1,296 1,284 1.285 1,284 9.660
31Ga 1,341 1,340 1,344 1,207 1,208 1,196 1.197 1,195 •10.368
32Ge 1,256 1,255 1.258 1,129 1,129 1 ,117 1,119 1,116 11,103
S3As 1,177 1,175 1,179 1.057 1,058 1,045 1,049 1,045 11,863
3*Se 1.106 1,105 1.109 0,992 0.903 0,980 0.984 0.980 12,652
S5Br 1,041 1.040 1.044 0.933 0,933 0,921 0.926 0,920 13,475
36Kr 0.981 0.980 0,984 0,879-0,8790,866 0.866 0.871 0,866 14,323
3?Rb 0,927 0,926 0,930 0,82910,830[0.817 0,816 15,201
199
Окончание приложения II
1 2 3 I 4 5 6 | 7 | 8 9 10 11 .2
abSr 0.877 0.875.0,880 0.783 0,784 0,771 0.770 16,106
0,831 0.829 0,833 0.740 0,741 0.728 0,727 0,727 17.037
*°Zr 0,788 0.786 0,791 0.701 0.702 0.690 0,688 17,998
41 \T 0,748 0,747 0,751 0,665 0,666 0,654 0,653 18.987
*2Мэ 0.710 0,709 0,713 0,632'0,633 0,621 0,620 0,620 20,002
«3'1 с 0,674 0,673 0,676 0,6(12 0,589 21,054
41,г;‘Л 0,644 0.643 0,647 0,572 0.573 0,562 0,560 22.118
i!5H а 0,614 0,613 0,617 0.546 0,546 0,535 0.534 23,224
««Pd 0,587 0.585 0,590 0,521 0.521 0.510 0,509 24.347
17 Ль, 0,561 0,559 0,564 0,497 0.498 0,487 0,486 25,517
;isCd 0,536 0,535 0,539 0,475 0.476 0,465 0,464 26,712
19In 0,514 0,512 0,517 0,455 0.455 0,445 0,444 27,928
',0Sn 0.492 0,491 0,495 0,435 0,436 0.426 0,425 29,190
5iSb 0.472 0.470 0.475 0,417 0,418 0-408 0,407 30,486
52Te 0,453 0.451 0,456 0,400 0.401 0,391 0,390 31,809
S3J 0.435 0.433 0,438 0,384 0,385 0.376 0,374 33,164
54Xe 0,418 0,416 0,421 0,369 0,360 0.359 34,579
35Cs 0,402 0,401 0,405 0,355 0.355 0,346 0,345 35,959
3GBa 0,387 0.385 0,390 0,341 0,342 0,333 0.332 37 .410
*7La 0,373 0,371 0,376 0.328 0,329 0,320 0,319 38,931
58Ce 0,359 0.357 0,362 0.316 0,317 0,309 0,307 40,449
59P1‘ 0,346 0.344 0,349 0,305 0.305 0,297 0,296 41,998
aaNd 0,334 0,332 0,337 0,294 0.294 0,287 0,285 43,571
«Pm 0.322 0.321 0,325 0,283 0,274 45,207
32Sm 0,311 0,309 0.314 0.274 0,274 0,267 0,265 46,846
saEu 0,301 0.299 0,304 0,264 0,265 0-258 0,256 48,515
34Gd 0,291 0,289 0,294 0,255 0,250 0,249 0,246 50,229
«5T5 0,281 0.279 0,284 0.246 0,246 0,239 0,238 51.998
36Dy 0.272 0,270 0,275 0,237 0,238 0,231 0.230 53,789
O7Ho 0.263 0,261 0,266 0,223 55,615
68Er 0,255 0.253 0,258 0.222 0.223 0,217 0,215 57.483
fi5Tm 0,246 0,244 0.250 0,215 0,216 0,209 59,335
?°Yb 0.238 0,236 0,241 0,208 0.209 0,203 0,202 61,303
"1Ln 0.231 0,229 0.234 0.202 0,203 0.197 0,195 63,304
73JU 0,224 0,222 0,227 0,195 0,196 0,190 0,189 65.313
73Ta 0,217 0.215 0,22C 0,190 0,191 0,185 0,184 67,40(
7J\V 0,211 0.209 0,213 0,184 0,185 0.179 0,178 69,508
76Re 0.204 0.202 0.207 0,179 0,179 0,174 0,173 71,662
7fiOs 0,198 0,196 0.201 0,173 0.174 0,169 0,168 73,86(
77Ir 0.193 O.191O.19G 0,168 0.169 0,164 0,163 0,167 0,163 0,163 76.097
78Pt 0.187 0,1850.196 0,163 0,164 0-159 0.159 0,162 0,158 0,158 78,379
7aAu 0,182 0.180^0-185 0,159 0,160 0.155 0.154 0.158'0,153 0,153 80.713
200
Приложение Ш. Потенциалы возбуждения (кВ), длины волн краев
поглощения и диаграммных линий Z-серии рентгеновского спектра элементов (А)
Элемент Линия Pa 04 0. 0!O v2 b ?4 Lj-край поглоще- ния Потенциал возбужде- ния
Переход ьГмш sT гГ Lr°in
Стат, вес 0,064 0,043 <0,002 <0,002 0,021 0,021 0,021
1 2 3 4 5 6 7 8 9 1C
17С1 52,100 0.238
18Аг 43.200 0.287’
I 19К 36,400 0.341
! 20Са 30.700 0,399
I 21SC 26,800 0,462
I 22Ti 23,400 0.530
i азу 19,803 0.604
I 24Cr 17,840 0,670
i 25Mn 16,138 0,762
1 26 Fe 14,650 0,849
1 27Co 13,333 0,920
1 2SNi 12.201 1,015
I 29C*j 11,172 1.100
I 30Zn 10,262 1,200
I 3JGa 9,416 1.30O
i 32Ge 8,692 1.420
I 33As 8,102 1,520
f 3JSe 7,505 1,652
I 35Br 6,920 1.794
1 36Kr 6,444 1,934
I 27Rb 6,788 6,821 6,045 5,997 2,067
i 38Sr 6,367 6,403 5,644 5.582 2,221
1 say 5,983 6,094 5,283 5,233 2,369
40Zr 5,632 5,668 4,953 4,867 2.547
1 41Nb 5,310 5,346 4,654 4,581 2,706
I *2Mo 5,013 5,048 4,380 4,299 2,884
43Tc 4,068 3,054
44Ru 4,487 4,523 3,897 3,837 3,236
45Rh 4,253 4,289 3,685 3,626 3.419
48Pd 4,034 4,071 3,792 3,799 3,489 3,428 3,617
17AK 3,834 3,870 3,605 3,611 3,307 3,254 3,8Ю
1 48Gd 1 i!fn 3.644 3,681 3,430 3,437 3,137 3,084 4.019
3,470 3,507 3,268 3,274 2,980 2,926 2,926 4.237
1 50Sn 3,306 3,344 3,115 3,121 2,835 2,778 2,778 4,464
201
Продолжено приложения III
1 2 3 4 5 | 6 8 9 10
51Sb 3,152 3,190 2,973 2,979 2,695 2,639 2,639 4,697
52Те 3,009 3,046 2,839 2,847 2,567 2,511 2.510 4,938
53J 2,874 2,912 2,713 2,720 2,447 2,391 2.389 5,190
5iXe 2,274 5,452
55Cs 2,628 2.666 2,478 2,492 2,237 2,233 2,174 2,167 5,720
5SBa 2,516 2,555 2,376 2,387 2,138 2,134 2,075 2,068 5,995
57La 2,410 2,449 2,282 2,290 2,046 2,041 1,983 1,973 6,283
58Ce 2,311 2,349 2,188 2,195 1,960 1,955 1,899 1.890 6,561
59?r 2,216 2,255 2,100 2,107 1,879 1,874 1,819 1,811 6,846
6|Ш 2,126 2,166 2,016 2,023 1,801 1,797 1,745 1,735 7,144
slPm 1,675 7,448
®2Sm 1,962 2.000 1,862 1,870 1.659 1,655 1,606 1,598 7,754
^Eu 1,887 1,926 1.792 1,800 1,597 1,591 1,544 1,536 8,069
34Gd 1,815 1,853 1,731 1,534 1,529 1,485 1,477 8,393
SoTb 1.747 1,785 1,667 1,477 1,471 1,427 1.421 8,724
66Dy 1,681 1.720 1,423 1,417 1,374 1,365 9,083
S7Ho 1,619 1,658 1,371 1.364 1,323 1,318 9,411
««Er 1.561 1,601 1,485 1,494 1,321 1,315 1,276 1,269 9,776
«Tin 1,505 1,544 1,274 1,268 1,222 10,144
1,452 1.491 1,384 1,392 1.228 1,222 1,185 1,181 10,486
71Lu 1,402 1.441 1,336 1.343 1,185 1,179 1,143 1.140 10,867
1,353 1.392 1,291 1,299 1,144 1,138 ’ 1,103} 1,099 11,264
33Ta 1,307 1,346 1.247 1.254 1.105 1.099 1,065 1,061 11,676
54\V 1.263 1,302 1,204 1,212 1,068 1.062 1,028 1,024 12,090
75He 1,220 1,260 1,165 1,172 1.032 1,026 0,993 0.990 12,522
VsOs 1,179 1,218 1,126 1,133 0,998 0,992 0.959 0.956 12,965
77b 1,141 1,179 1.090 1.097 0.966 0.959 0.928 0,923 13.413
’8Pt 1,104 1,142 1,054 1,062 0.934 0,928 0,897 0.893 13,878
79Au 1,068 1,106 1,021 1,028 0.905 0,898 0,867 0,864 14,353
80Hg 1,034 1,072 0,986 0,996 0,876 0.869 0,839 0,836 14,841
siT1 1,001 1,039 0,957 0,964 0.848 0.842 0,812 0,808 15,346
82pb 0,969 1,007 0,927 0,934 0,822 0,815 0,782 15,870
83Bi 0,939 0.977 0,898 0,905 0.796 0,790 0,761 0,757 16,393
8 Фо 0,909 0,948 0,765 0,734 16,935
87 Fr 0,665 18,638
88Ra 0,803 0,841 0,769 0,776 0,682 0,675 0,649 0,644 19,233
39Th 0,755 0,793 0,723 0,730 0,642 0,635 0,611 0,606 20,460
9ipd 0,732 0,770 0,701 0,708 0,624 0.617 0,594 0,587 21,102
93U 0,710 0,748 0,681 0.687 0,605 0,598 0,577 0,569 21,753
93Np 22,417
202
Продолжение приложения III
Линия Pl 6? Vi Vr, Ve Vs V
Элемент t( H ® T a S<T sT гм 7 cT cT Ljj-край поглоще- ния Потенциал возбужде- ния
Стат, вес 0,170 <0,002 0,085 <0,002 0.043 <0,002 0,021
1 2 3 4 5 6 7 8 9 10
17С1 1аА1’ 19К 29Са 21 Sc 22Ti 23у 24СГ 25Мп 2$г е 27Со 28Ni 29Cu 30Zn 31Ga S2Ge 53As 34gp 35Br 3«Kr 37Rb 38gf 39Y 43Zr 4iNb 42Mo 43Tc 41Ru 45Rb 46pd 47Ag •18Cd 49In 50Sn 51Sb 52Te 53] 54Xc 33Cs 36.022 31.072 27,074 23,898 21,323 19,158 17,290 15,698 14.308 13.079 12.009 11.045 10.194 9.414 8,735 8,126 7,075 6,623 6,211 5,836 5.492 5,176 4,620 4,374 4.146 3,935 3.739 3,555 3,385 3,226 3,077 2.937 2,683 5.384 5.036 4,726 4,182 3,944 3.725 3.523 3,336 .3.162 3.001 2,852 2,712 2,582 2,348 6,754 6.297 5.875 5,497 5,151 4,837 4,288 4,045 3,822 3,616 3,426 3,249 3,085 2,932 2,790 2,657 2,417 67.25 56,813 47,325 40,542 35,200 30,942 27,375 24,339 21,864 19.73 17,86 16,304 14,940 13,719 12,620 11,608 10.732 9.959 9,253 8.042 7,517 7,040 6.606 6.210 5,847 5,204 4,922 4.660 4.418 4,193 3,983 3.789 3,607 3.438 3,280 2,994 60,739 50.025 41,754 35,200 30.943 26,936 23.877 17,9 19,086 17,188 15,545 14.104 13,010 11,861 10,728 9.840 9.124 8,417 7.721 7,158 6.643 6,172 5,756 5.378 3,031 4,719 4.431 4,179 3.942 3.724 3.514 3,326 3,147 2,982 2,830 2,687 2.553 2.429 2,314 0.203 0.247 0.297 0.352 0.411 0,460 0.519 (',583 0.650 0.721 0,794 0.871 0.953 1,045 1.134 1.248 1.359 1.473 1.599 1.727 1.866 2,008 2,154 2.305 2,467 2.627 2,795 2.966 3,145 3,329 3,528 3.737 3,939 4.157 4,381 4,613 4,856 5,104 5.358
203
'Проц олженпе приложения III
1 2 3 4 5 6 7 8 9 10
^8Ва 2,567 2,242 2.300 2,222 2,862 2,204 5,623
57La 2,458 2,141 2,205 2,740 2,103 5,894
58Се 2,356 2,048 2,110 2,023 2,620 2,022 6,165
лорг 2,259 1,961 2,020 1,936 2,512 1,924 6,443
'S8N 1 2,166 1,878 1,935 1,855 2,409 1,843 6,727
31Р1П 2,081 1,775 7,018
*2Sm 1,998 1,726 2,218 1,702 7,281
1,920 1,657 1,708 1,632 1,626 7,624
-чн 1,847 1,592 2,049 1,561 7,940
ч-ЗЦ) 1,777 1,530 1,501 8,258
^Dv 1,710 1,473 1,518 1,898 1,438 8,621
37По 1,647 1,417 1,462 1,826 1,390 8,920
3SEr 1,587 1,364 1,406 1.757 1,339 9,263
*9Tni 1,530 1,316 1,355 1,695 1,288 9,628
™Yb 1.476 1,268 1,307 1,243 1,250 1,635 1,243 9,977
1.424 1,222 1,260 1,198 1,204 1,478 1.198 10,345
W£ 1.374 1,437 1.179 1,215 1.155 1,161 1,523 1,154 10,734
изта 1,327 1,138 1.173 1,114 1,120 1,471 1,113 11,130
tivv 1,282 1,339 1,098 1.132 1,074 1,081 1,421 1,074 11,535
''•’Re 1,238 1,293 1,061 1,094 1,037 1,044 1,374 1,037 11,955
7«0s 1,197 1.025 1,057 1,001 1,008 1,328 1,001 12,383
7?ie 1,158 0,991 1,022 0,967 0,974 1.285 0,967 12,819
yspt 1,120 1,166 0,938 ,0.988 0,934 0,941 1.243 0,934 13,268
78Au 1,083 1,128 0.927 0,956 0,903 0,910 1,202 0,903 13,733
1.049 1,090 0.897 0,925 0,873 0,880 1,164 0,872 14,212
«Т1 1,015 1,056 0,863 0,895 0,845 0,852 1,127 0,844 14,697
ззрЬ 0,982 1,022 0,840 0,867 0,817 0,824 1,092 0,815 15,207
S3Bi 0,952 0,989 0,814 0.840 0,791 0,799 1,058 0,789 15,716
«‘Po 0,922 0,788 0,765 0.762 16,244
S7Fr 0,840 0,716 0,692 17,904
Wa 0,814 0,844 0,694 0,717 0,673 0,680 0,908 0,670 18,481
aoTh 0,766 0,653 0,675 0,632 0,640 0,855 0,630 19,688
Sip ( 0,742 0,634 0,655 0,613 0,830 0,610 20,311
92U 0,720 0,615 0,635 0.595 0,601 0,806 0,592 20,943
S3Np 0,698 0,597 21,596
204
' ..................... U.XHII' ..........в,...... „в,mi....................... , ..............................Uh.
Линия cc «1 с/, lb fh
Элемент llBlfCXlhl I, ->M f 111 IV Л11Г’л/у «Jiv Li.[i"-Vv гиГ -'°iv,v Г1И^Л1 L
Стат, вес 0,234 0,213 0,021 0.128 0,004 <0,002
1 2 3 4 5 6 7
A7CI
lsAr
29Ca 36,393
21Sr 31,363
22Tj 27,445
23V 24,309
24Cr 21,713
2Г-МП 19,489
26 Fe 17,602
27Co 16,000
2«Nj 14,595
29Cti 13,357
30Zn 12,282
31 Ga 11 313
32Gp 10.456
33As 9,671
31Se 8,990
['7 (V. 1 8 1 К 4‘ Е
иГ°1 IX Ы1 МП1 ми
<0,002 <гО,(Л12 0,364 <0,002 <0,002 41 kJ'-1 g $ £ о В Се
s <) 10 И J2 13 14
07,84 61,222 0,202
56.212 50,446 0,245
47,835 42,179 0,294
41,042 35,561 0,34 0
35.671 31,296 0,406
31.423 27,290 0,454
27,826 24,229 0,512
2-4,840 20,7 0,574
22,315 19,417 0,639
20,201 17,504 0,708
18,358 15,843 0,779
16,693 14,387 0,853
15,297 13,289 0,933
14,081 12,130 1.022
12,976 10,930 1,117
11.944 10,089 1,217
11,069 9,367 1,323
10,293 8,645 1,434
206
Окончание приложения Ш
1 2 3 4 5 6 7
jf’Br 3GKr 8,375
>7вь 7,318 7,325 6,984
38Sr 6,863 6,870 6,519
зэу 6,449 6,456 6,094
«Zr 6,070 6,077 5,586 5,710
4Wb 5,725 5,732 5,238 5,361
42Мо «Тс 5,406 5,414 4,923 5,048
44Ru 4,846 4,854 4,372 4,487
45RL 4,597 4,605 4,130 4,242
46Pd 4,368 4,376 3,909 4,016
"Ag 4,154 4,162 3,703 3,808
«Cd 3,956 3,965 3,514 3,614
49jn 3,752 3,781 3,339 3,436
50Sn 3,600 3,609 3,175 3,270
^Sb 3,439 3,448 3,023 3,115
62Te 3.290 3,299 2,882 2,971
531 B4Xe 3,148 3,157 2,751 2,837
55Cs 2.892 2,902 2.511 2,593
56Bu 2,776 2,785 2,404 2,482
57La 2,665 2,674 2,303 2,379
68Ce 2,561 2,570 2,208 2,282
8 !) 10 11 12 13 14
9,583 7,952 1,552
7,377 1,675
8,363 6,864 1,806
7,836 6,387 1,941
7,356 5,962 2,079
6,918 5,583 2,220
6,517 5,223 2,374
6,150 4,913 2,523
4,623 2,677
5,503 4,369 2,837
5,217 4,129 3,002
4,952 3,908 3,172
4,707 3,698 3,352
4,480 3,504 3,538
4,269 3,325 3,729
3,155 4,071 3,156 3,928
3,005 3,888 3,000 4,132
2,863 3,716 2,856 4,341
2,730 3,557 2,719 4,559
2,592 4,782
2,485 3,267 2,474 5,011
2,382 3,135 2,363 5,247
2,275 3,006 2,259 5,489
2,180 2,892 2,164 5,729
J9pr
eoNd
filPm
62Sm
63Eu
e4Gd
"'Tb
6RBy
u7Ho
d8Er
G9fm
7oYb
71Lu
?2Hf
7STa
7HV
75Be
7,i0s
77lr
7SPt,
7bAu
80Hg
81T{
B2pb
83Bi
s“Po
87 Er
88Ba
a°Th
«Pa
92U
§ 93Np
2.463 2,473 2,119 2,190 2.091 2,784 2,077 5,968
2.370 2,382 2,035 2,103 2.009 2,675 1.995 6,215
2.283 1,928 6.406
2, J 99 2,210 1,882 1,779 1,946 1,856 2,482 1,845 6,721
2,120 2,131 1,812 1,875 1,788 2.395 1,776 6,983
2.046 2.057 1.746 1,807 1.723 2,312 1,709 7,252
1,970 1,986 1,682 1,577 1,742 1.659 2,234 1,648 7,519
1,909 1,920 1,623 1.681 1,599 2,158 1,579 7,850
1,845 1,856 1,567 1,622 2,086 1,535 8,074
1,785 1,796 1,514 1,567 1,494 2,019 1,482 8.364
1,726 1,738 1,463 1,515 1,955 1,433 8,652
1,672 1,682 1,416 1,387 1,466 1,395 1,894 1,831 1,386 8,943
1,619 1,630 1,370 1,342 1,419 1,350 1,372 1,836 1,776 1,342 9.241
1,569 1,580 1.327 1,298 1,374 1,306 1,328 1,782 1,663 1,723 1,298 9,556
1,522 1,533 1,285 1,256 1,331 1,264 1.287 1,728 1,612 1,672 1,256 9,876
1,476 1,487 1.245 1,215 1,290 1,224 1,247 1,678 1,215 10,198
1.433 1,444 1,206 1.177 1.252 1,186 1,208 1,630 1,177 10,531
1,391 1.402 1.169 1.140 1,213 1,149 1,171 1.585 1,140 10,869
1,352 1.363 1,135 1,106 1,179 1,115 1,137 1,541 1,105 11,211
1,313 1,325 1,102 1,072 1.143 1,082 1,499 1,072 11,559
1,277 1,288 1,070 1,040 1.111 1,050 1.072 1,460 1.352 1,414 1,040 11,919
1,242 1,253 1,040 1,010 1,080 1,019 1,041 1,422 1,009 12,285
1,207 1.218 1,010 0,981 1,050 0,990 1,012 1,385 1,279 1,342 0.979 12.657
1,175 1,186 0.983 0,953 1,021 0,962 0,984 1,350 1,244 1,308 0,950 13.044
1,144 1,155 0,955 0,926 0.993 0,935 0,957 1,317 1,210 0,924 13.424
1,114 1,125 0,929 0,900 0.9G7 0,931 0,897 13,817
1,030 0,858 0,825 15,028
1,005 1,017 0,836 0,807 0,871 0,817 0,838 1,167 0,803 15,442
0,956 0,968 0,794 0,765 0,828 0,775 1,115 1,011 1,080 0,761 16.296
0,933 0,945 0,774 0,746 0,808 0.755 1,091 0,741 16.731
0,911 0,923 0,755 0,726 0,789 0,736 1,067 0,964 1,035 0,722 17,163
0,889 0,735 17,164
Приложение IV. Экспериментальные w^<cn и рассчитанные по формуле
(5.7) Ых'1'4 выходы флуоресценции А'-оболочек
Z ,,эксп aK , П0СЧ ЫК z ЭКСП <3K вксч z ,,эксп слрасч
5В 0,00070 0,00100 33Ge 0,554 0,540 oOpji 0,890 0,915
6С 0,00180 0,00198 33As 0.587 0,567 60Nd 0,920
7N 0,00473 0,00351 34Se 0,580 0,596 61Pm 0,924
SO 0,00645 0,00579 35Br 0,590 0,622 63Sm 0,928
9F 0,00670 0,00900 30Kr 0,656 0,646 G3Eu 0,925 0,931
10Ne 0,01800 0,01340 37Rb 0,669 0,669 64Gd 0,900 0,934
41Na 0,01920 3BSr 0,699 0,691 eoTb — 0,937
Ч1й 0,02800 0,02650 1 39Y 0,711 8фу 0,940 0,940
13AI 0,03800 0,03570 : «Zr 0,690 0,730 : G7Ho 0,943
14Si 0,04300 0,04700 0,730 0,748 6sEr 0,943 0.945
15p 0,06000 0,06040 42Mo 0,770 0,764 69Tm 0,948
16S 0,07700 0,07610 43Tc 0,700 0,779 70Yb 0,940 0,950
*7Cl 0,09500 0,09420 44Ru 0,793 71Lu 0.952
1SAr 0,12200 0,11500 45Rb 0,780 0,807 72IIf 0,954
19K 0,13800 46Pd 0,810 0,819 73Ta 0,956
29Ca 0,17800 0,16300 47Ag 0,810 0,830 W 0.957
21Sc 0,19000 0.19000 48Cd 0.834 0,840 75Re 0.959
22Ti 0,22100 0,21900 49In 0,830 0,850 76 0s 0,961
23у 0,25300 0,25000 50Sn 0,830 0,859 77Ir 0,962
24Cr 0,28-300 0.28200 ^Sb 0,860 0,867 7SPt 0,967 0,963
25Mn 0,31300 0,31400 &2Te 0,857 0,875 79Au 0,964
26 Fe 0,35000 0.34700 53J 0,900 0,882 80Hg 0,958 0,966
87Co 0,39100 0,38100 54Xe 0,894 0,889 82pb 0,972 0.968
23N1 0,4,3200 0,41400 53Cs 0,889 0,895 83Bi 0,956 0,969
29Cu 0,44300 0,44500 sfiBa 0,870 0,901 84p0 0,917 0,970
80Zn 0,48000 0,47900, 57La 0,910 । 0,906 92U 0,970 0,976
«Ga 0,52800 0,51000 58Ce 0,900 ; 0,911 93Np 0,938 —
208
Пр и л ож е п п е V.
Выходы флуоресценции 7,-подоболочек
Z w 7 “3L
1 2 3 i 5
22Tj 2,80-10"4 1,18-10-3 2,4-10'3
23у
24Сг 2,97-lir4 3,29-10-3 3,0-10-3
25 Мп
26 Fe 3,84-10"4 5.59-10-3
1,43-IO'3 1,49-10-3
2SNi 4,63-IO’4 2,69-10-3 8,02-10-3
2ЭСи 3,57-10-3 3,83-10-3 6,0-10-3
30Zn 5,23-IO"4 — l,08-10-2 6,4-10"3
34Ga
82Ge 7,70-IO-4 7,72-Ю-з 1,44-10-2
33As 1,40-IO"3 8,85-Ю-з 9,74-10-3
34Se 1,30- 1СГ3 9,94-IO’3 1,78-10-2
35Br l,09-10-2
36Kr 1,85-10"3 2,20-10-2 2,36-10-2 1,0-10-2
37Rb 1,32-IO'2 1,0-10-2
38Sr 3,00-10‘3 2.24-10-2 2,43-10-2
3Sy 3,2-10-2
40Zr 3,97-10“3 2,94-10-2 2,95-10-2
3,96-I0-3 1,89- IO'2 2,01-10-2
42Mo 5,75-lC)-3 3.50-10-2 3.73-10-2 6,7-10-2
G,34-10-3 2,45-10-2 2,59-10-2
44Ru 7,74-IO-3 4,18-10'3 4,50-10-2
«Ag l,02-10-2 5.47-10-2 6,02-10'2 5.6-10-2
l,01-10-3 4,30-10-2 4,49-10'2
50Sn 1,30-ю-2 6.56-10-2 7,37-10-2
&1Sh 3,11-10-2 6,16-10-2 6,33-10-2 1,2-IO’1
52Te 1.2-10-1
64Xe 5,84-10-2 9,12-10-2 9,70-10-2 1,1-IO’1
55Cs 8,9-10-2
14 В. П, Афонин, Г. Н. Гуцн--сва. Л. Ф. Пискунова
209
Окончание приложения V
1 2 3 1 4 1 5
66Ва 4,46-10~2 9,07-10-2 8,99-10-2 9,3.10-2
&7La 1,0-10"1
б8Се 1,6-10-1
6vpr 1.7-IO-1
eoNd. 7,46-10-2 0,133 0,135 1,7-Ю-1
62Sm 1,9-Ю'1
63Eu 1,7-Ю-1
61Gd 2,0-Ю'1
65ТЪ 0,166 0.160 2.0-10"1
6cDy 1,4-IO-1
67Ho 0,112 0,302 0.201
7оуь 0.112
71Lu 2,9-IO-1
72IIf 2,6-Ю-1
73Ta 2.3-10-1
74\V 0.115 0,287 0,268 3,0-IO-1
0,138 0,271 0,253
76Os 3.5-IO-1
77 If 3,0-КГ1
78Pt 3,3-Ю-1
79Au 0,105 0.357 0,327 3,9-10-1
80Hg 0,098 0,352 0,321 3,9-ICr1
81T1 4,6-Ю-1
82Pb 3,8-Ю-1
"Bi 0,120 0,417 0,389 4,1- Ю’1
85At 0,129 0,422 0.380
fiOTh 0,197 0,529 0,401
92(J 5,2.IO’1
93Np 0,460 0.472
210
ЛИТЕРАТУРА
1. Алимарин И. И. Современное состояние и некоторые перспективы разви-
тия методов аналитической химии.— Журн. аналит. химии, 1970, т. 25,
№ 4. с. 599—609.
2. Алимарин IT. П., Золотов 10. А. Перспективы развития аналитической хи-
мии.— Жури, аналлт. химии, 1975, т. 30, № 7, с. 1253—1259.
3. Афонии В. П. Теория рентгеносиектрального флуоресцентного анализа
пород и минералов.— В кн.: Фундаментальные исследования. Науки о
Земле. Новосибирск: Паука, 1977. с. 1.37—142.
4. Афонин В. П. Успехи рептгонофлуореснентного анализа.— Журн. апалпт.
химии, 1980, т. 35, Аг 12, с. 2428—2440.
5. Афонии В‘. П. Рентгенофлуоресцентпый метод анализа.— Журн. ВХО
или Д. И. Менделеева, 1980, т. 25, А1 6, с. 610—615.
6. Афонин В. П., Гуничева Т. Н. Ренггсносиектральпый флуоресцентный
анализ горных пород п минералов. Новосибирск: Наука, 1977. 256 с.
7. Афонин В. II., Гуничева Т. IT-, Харченко А. М., Пискунова Л. Ф. Расчет
поправок иа поглощение п возбуждение при рентгеноспектральном флуо-
ресцентном анализе горных пород.— Зав. лаб., 1974, т. 40. А» 6, с. 655 —
657.
8. Афонии В. И.. Ложкин В. II. Завпсммость предела обнаружения рентге-
новским флуоресцентным методом от атомного номера определяемого эле-
меита.-- Зав. лаб., 1978. т. 44, № 9. с. 1086—1088.
9. Афонин В. П.„ Ложкин В. II., Гуничева Т. Н., Пискунова Л. Ф. Автома-
тизация расчета концентрации при рентгсцоспекгральиом силикатном ана-
лизе_____В кн.: Ежегодник-1973 Ин-та геохимии им. А. П. Виноградова
СО АН СССР, Новосибирск: Наука. 1974, с. 334—336.
10. Афонпн В. П., Лосев II. Ф., Павлинскнп Г. В. и др. Расчет интенсивности
тормозного излучения рентгеновских трубок.— Зав. лаб., 1970, т. 36, № 4,
с. 431-434.
11. Афонин В. П., Пискунова Л. Ф. Расчет интенсивности рентгеновского
характеристического излучения, возбужденного фотоэлектронами апали-
зпрусмого образца.— Зав. лаб., 1978. т. 44. № 9, с. 1083—1086.
12. Афонии В. П., Пискунова Л. Ф.. Гуничева Т. II., Ложкин В. II. Теоретиче-
ские поправки на матричные эффекты прп рентгепоспектральпом флуо-
ресцентном анализе.— Зав. лаб.. 1976, т. 42, № 6, с. 670—674.
13. Афонин В. П., Пискунова Л. Ф.. Гуничева Т. И., Финкельштейн А. Л.
Рентгенофлуорссцентныи анализ горных пород па петрогенные элементы
упрощенным способом фундаментальных параметров. — Журн. апалнт.
химии, 1982, т. 37. А» 7, с. 1239—1247.
14. Афонии В. П., Ревенко А. Г., Лосев Н. Ф., Харченко А. М. К выбору пер-
вичного излучения при реытгепоспектральном флуоресцентном определе-
нии элементов с малыми атомными номерами. — Зав. лаб., 1969. т. 35, Д» 8,
с. 929—933.
15. Афонин В. П., Ревенко А. Г., Харченко А. М-, Лосев Н. Ф. Влияние филь-
трации первичного излучения окном трубки на эффективность возбужде-
ния рентгеновской флуоресценции элементов с малыми атомными номера-
4* 211
мп.— В кн.: Аппаратура и методы рентгеновского анализа. Выц. 6. Л.:
Машиностроение, 1970, с. 99—105.
16. Байбаков Ю. В., Ветюков М. М. Электролиз расплавленных солей. М.:
Металлургия, 1966. 560 с.
17. Бахтиаров А. В., Блохин М. А., Мейер В. А. Исследование фона ко-
ротковолнового спектрометра по Кошуа с использованием ППД высокого
разрешения.— В кн.: Аппаратура и методы рентгеповскох’о анализа.
Выл. 19. Л.: Машиностроение, 1977. с. 118—134.
18. Бейли Б. Введение в петрологию. М.: Мир, 1972. 279 с.
19. Белов В. Т., Дуймакаев Ш. И. Обобщенные уравнения связи в рентгено-
флуорссцентпом анализе.— Зав. лаб., 1974, т. 40, № 8, с. 958—960.
20. Белов В. Т., Кохмаиьски С. С. Приближенная формула для интенсивности
рентгеновской флуоресценции прп возбуждении первичным тормозным
спектром.— Зав. лаб., 1980, т. 46, № 12, с. 1112.
2J. Берхоер И. Д., Сорин М. И., Шехель А. 3., Шифрин В. Б. Математические
модели и статистические характеристики рентгеновского квантометра
ФРК-1Б.— В кн.: Аппаратура и методы рентгеновского анализа. Выл. 10.
71.: Машиностроение, 1972, с. 114—121.
22. Блохин М. А. Физика рентгеновских лучей. М.: ГПТТЛ, 1957. 518 с.
23. Блохин М. А. Методы рептгеноспектральных исследований. М.: Фкз-
матгпз, 1959. 366 с.
24. Блохин Ы. А.. Белов В. Т.. Дуймакасв Ш. II.. Цопова-Гречихииа А. Н. Фе-
номенологические уравнения связи в рентгеноспектральпом анализе.—
Зав. лаб., 1973, т. 39, № 9, с. 1081—1085.
25. Блохин М. А., Бикс В. А. Рентгенослектральпый анализ многокомпонент-
ных смесей.—Зав. лаб., 1981, т. 27. № 1, с. 31—34.
26. Блохин М. А., Друзь В. В. Рептгсноспектральяый анализ многокомпонент-
ных смесей.— Зав. лаб., 1963, т. 29, № 9, с. 1070—1074.
27. Блохин М. А., Дуямакаев Ш. II. Рентгеноспектральпьш анализ многоком-
понентных смесей.— Зав. лаб.. 1964. т. 31’, Л? 4. с. 425—42(5.
28. Блохин М. А., Швейцер II. Г. Рентгепосиектралыгый справочник. HI.: Нау-
ка, 1982. 376 с.
29. Богданова П. В., Егоров Г. Б.. Кобрина Е. П. Внедрение рептгеноснект-
рального анализа в цементную промышленность.— Цемент, 1970, № 6,
с. 17—19.
30. Бондаренко Б. IO-, Павлинский Г. В. Автоматизация методических иссле-
дований в рентгеноспектральпом флуоресцентном анализе с помощью
ЭВМ.— В кп.: Аппаратура и методы рентгеновского анализа. Вып. 25.
Л.: Машиностроение, 1981, с. 43—55.
31. Боровский 11. Б. Физические основы рептгеноспектральпых исследований.
М.: Пзд-во МГУ, 1956. 463 с.
32. Ванюков А. В., Моптильо И. Изучение дпффузпп кальция, железа, ни-
келя и серы в жидких шлаках.— Изв. вузов. Сер. цветн. металлургия,
1959, № 5, с. 59—68.
33. Васильева Л. Н., Быков В. П. Рентгепоспектральпып флуоресцентный
анализ состава плавленых сварочных флюсов.— В кв.: Новые методы ин-
струментального анализа материалов. М.. 1979, с. 3—6.
34. Величко Ю. II., Калинин Б. Д., Межевич А. II. и др. Исследование зави-
симости величин теоретических поправок от химического состава проб
при рентгеноспектральпом анализе сталей.— Зав. лаб., 1977, т. 43, № 4,
с. 437-442.
35. Величко 10. И., Калинин Б. Д., Павлинский Г. В. и др. Учет взаимных
влиянии элементов при рентгеноспектральном флуоресцентном анализе
легированных сталей.— Зав. лаб., 1974, т. 40, № 6, с. 668—671.
36. Григорьев В. А. Одномерные феноменологические модели рентгенофлуо-
ресцентного анализа.— Зав. лаб., 1982, т. 48, № 6, с. 23—26.
37. Гуничсва Т. Н. Получение н исследование уравнений связи при рентгено-
спектральном определении высоких концентраций элементов с малыми
атомными номерами. Автореф. каяд. дпс. Иркутск: пзд. ИГУ, 1970. 17 с.
38. Гуничева Т. Н., Афонин В. П., Пискунова Л. Ф., Изотова Е. М. Влияние
неопределенности массовых коэффициентов ослабления рентгеновских
212
лучей в поправке на поглощение на правильность результатов анализа.—
Зав. лаб., 1975, т. 41, Ка 11, с. 1325—1330.
39. Гуничева Т. II., Афонин В. П., Финкельштейн А. Л. Учет фона при ана-
лизе на многоканальных рентгенофлуоресцентных спектрометрах.— Журн.
аналит. химии, 1982, т. 37, .V» 7, с. 1157—1164.
40. Гуничева Т. Н., Павлинекий Г. В., Лосев II. Ф., Межевпч А. II. О погреш-
ности приготовления излучателей при рентгеноспектралыюм флуорес-
центном силикатном анализе.— В кн.: Аппаратура и методы рентгенов-
ского анализа. Выл. 4. Л.: Машиностроение, 1969, с. 191—196.
41. Гуничева Т. Н., Павлинекий Г. В., Лосев 11. ф., Пискунова Л. Ф. Опыт
рентгеноспектральлого определения основных породообразующих .эле-
ментов в различных горных породах.— Зав. лаб., 1972, т. 38, № 8, с. 932—
936.
42. Гуничева Т. Н., Пискунова Л. Ф., Павлинекий Г. В.. Лосев II. ф. Резуль-
тат опробования методики рентгеноспектральпого анализа главных поро-
дообразующих элементов на десятиканальпом рентгеновском кванто-
метре.— В кп.: Ежегодник-1970 Ии-та геохимии им. А. П. Виноградова
СО АП СССР. Иркутск, 1971, с. 409—411.
43. Гурвпч 10. М., Межсвич А. II., Плотников Р. II., Рогачев II. М. К выбору
уравнений форм множественной регрессии при рентгеиоспектральном ана-
лизе пульп.— В кн.: Аппаратура и методы рентгеновского анализа.
Вып. 16. Л.: Машиностроение, 1975, с. 28—36.
44. Данкамб П., Рид С. Прогресс в вычислении тормозной способности и фак-
тора обратного рассеяния.— В кн.: Физические основы реятгеноспект-
рального локального анализа. М.: Наука, 1973. с. 117—138.
4. 5. Диаграммы состояния силикатных систем. Справочник. Выл. 1/Торо-
пон Н. А.. Борзаковский А. 11., Лепин В. В., Курцева Н. II. Л.: Наука,
1969. 822 с.
4G. Донец IO. Т. Способ градуировки при рентгеиоспектральном анализе про-
дуктов обогащения.— Зав. лаб., 1978, т. 44, Да 6, с. 674—677.
47. Дуймакаев Ш. II., Вершинин А. С., Никольский А. П.. Шильман В. X.
Получение уравнений связи в рентгеиоспектральном анализе для условий
низкой стабилизации возбуждения спектра.— Зав. лаб., 1976, т. 42, № 8,
с. 943—945.
48. Еременко В. Н., Пайдич 10. В.. Лавриненко 11. А. Спекание в присутствии
жидкой металлургической фазы. Киев: Наукова думка, 1968. 123 с.
49. Есин О. А., Гельц П. В. Физическая химия ппрометаллургпческлх про-
цессов. Ч. II- М.: Метадлургнздат, 1954. 206 с.
50. Загускпн В. Л. Справочник по численным методам решения алгебраиче-
ских и трансцендентных уравнений. М.: фвзматгиз, I960. 216 с.
51. Золотов 10. А. Очерки аналитической химии. М.: Химия, 1977. 239 с.
52. Зоммерфельд А. Волновая механика. М.: ГТТИ, 19,33. 479 с.
53. Ильин Н. П. Количественный рентгенофлуоросцептный локальный анализ
при а галопировании по чистому элементу.— Журн. аналит. химии, 1979,
т. 34. № 4, с. 670—679-
51. Калинин Б. Д., Лобашева II. М., Плотников Р. II. и др. Использование ме-
тода теоретических поправок в программном обеспечении рентгеновского
аналитического комплекса КРФ-18 — М6000.— В ни.: Аппаратура и ме-
тоды рентгеновского анализа. Вып. 25. Л.: Машиностроение, 1981,
с. 153—158.
55. Калинин Б. Д., Межевнч А. И., Плотников Р. II. и др. Применение метода
множественной регрессии в рентгеиоспектральном анализе.— В кн.: Ап-
паратура и методы рентгеновского анализа. Вып. 13. Л.: Машиностроение,
1974, с. 122-128.
56. Капании Б. Д., Мясникова В. М., Баранов А. А. и др. Применение способа
теоретических поправок при рентгеноспектральном анализе сплавов на
основе меди.— Зав. лаб., 1975, т. 41, № 11, с. 1329—1330.
57. Калинин Б. Д., Паиасюк В. А., Плотников Р. И. К методу теоретических
поправок в рентгеиоспектральном анализе при наличии неопределенных
компонентов.— Зав. лаб., 1981, т. 47, № 6, с. 39—40.
58. Калинин Б. Д., Плотников Р. IL, Федорова П. М. К обоснованию метода
213
теоретических поправок в рентгеносиектральном анализе.— Зав. лаб.,
1980, т. 46, № 6, с. 505—507.
59. Карманов В. II., Вопткевич В. Г., Триков В. П., Походил И. К. Рентгено-
спектральный анализ сварочных шлаков.— Жхрн. аналпт. химии 1979,
т. 34, № 7. с. 1329—1334.
60. Карманов В. II., Походня И. К., Марченко А. Е. Рентгеяоспектралытый
анализ с одним эталоном н корректировкой интенсивности на ЭВМ.— Зав.
лаб., 1972, т. 38. № 2, с. 167—169.
61. Карманов В. II., Походня И. К., Марченко А. Е., Демченко В. Ф. Рентгено-
спектральный анализ рудоминералометаллических композиций.— Зав.
лаб., 1970, т. 36, № 6. с. 680—682.
62. Классификация и номенклатура магматических горных пород/Андрее-
ва Е. Д., Богатиков О. А., Бородаевская М. Б. идр. Ы.: Недра. 1981. 160 с.
63. Колесниченко Г. А. Смачивание поверхности алмаза и плрографита спла-
вами Ре—V.— В кп.: Смачиваемость и поверхностные свойства расплавов
и твердых тел. Киев: Наумова думка, 1972. с. 71—75.
64. Коляда В. М., Зайченко А. И., Дмитриенко Р. В. Рептгепоспектральш.ш
анализ с ионным возбуждением. М.: Атомиздат, 1978. 246 с.
65. Корн Г., Корн Т. Справочник по математике. М.: Наука, 1968. 720 с.
66. Котляров Я. Б. К вопросу об одновременной оценке аппаратурных и мат-
ричных эффектов в рентгеноспектральном анализе.— В кн.: Аппаратура
и методы рентгеновского анализа. Выи. 25. Л.: Машиностроение, 1981,
с. 207—211.
67. Котляров Я. Б. Расширение аналитических возможностей способа теоре-
тических поправок в рснтгепоспектральном анализе.— Жхрн. аналпт.
химии, 1982, т. 37. № 6. с. 1122—1132.
68. Котляров Я. Б.. Плотников Р. И. Коррекция матричных эффектов в репт-
геноспектральном анализе с помощью парциальных коэффициентов влия-
ния.— В кн.: Аппаратура и методы рентгеновского анализа. Вып. 25. Л.:
Машиностроение, 1981. с. 211—216.
69. Котляров Я. Б.. Плотников Р. 11., Прошкина II. А. Метод парциальных
коэффициентов влияния в рентгеноспектральном анализе. Л.: ВПИИНП,
1980. 43 ст.. Дек. ВИНИТИ. № 2356-80.
70. Котляров Я. Б., Плотников Р. II., Сербин А. Я. Построение и использова-
ние статистических уравнений связц в рептгопоспектральном анализе.—
В кн.: Аппаратура н методы рентгеновского анализа. Выи. 21. Л.: Машино-
строение, 1978. с. 191—211.
71. Кохов Е. Д., Мамиконян С. В.. Мамыш В. А. Учет матричных эффектов
в многоэлементном рснтгснрадиометрпческом анализе с обработкой ре-
зультатов измерений на ЭВМ.— В кн.: Радиационная техника. Вып. 11.
Атомиздат. 1975, с. 289—295.
72. Кравченко-Бережной Р. А.. Полежаева Л. И. Способ изготовления излуча-
телей для рентгеноспектрального анализа морошковых материалов.— Зав.
лаб., 1968, т. 34. № И, с. 1396—1397.
73. Краткий справочник по геохимпи/Вопткевпч Г. В., Мирошников А. Е.,
Повторенных А. С., Прохоров В. Г. М.: Недра. 1977. 182 с.
74. Кузнецова А. И., Лаврентьев 10. Г. Оценка дополнительного возбужде-
ния при рентуенофлуоресцептном анализе.— Зав. лаб., 1976, т. 42, .У 6,
с. 674—680.
75. Кузьмина Т. Г. Метод рентгеноспектрального флуоресцентного анализа
силикатных пород.— В кп.: Комплексные исследования Миоового океана.
М.: ИОАН, 1975, с. 311—314.
76. Лабораторная посуда из стеклоуглерода. — Журн. аналпт. химии, 1982,
т. 37, № 8. с. 1528.
77. Лаврентьев Ю. Г., Кузнецова А. П. Программа для вычисления относи-
тельных интенсивностей линий прп рентгенофлуоресцентном анализе.—
В кн.: Физико-химические методы анализа минералов. Новосибирск: Нау-
ка, 1977. с. 60—70.
78. Лаврентьев 10. Г., Кузнецова A. II. Уравнения связи в рентгенофлуорес-
центном анализе.— Зав. лаб., 1979, т. 45, № 4, с. 315—326.
79. Липчевский Б. В. Техника металлургического эксперимента. М.: Метал-
лургия, 1979. 256 с.
214
80. Ложкин В. И. Последование фона в длинноволновой области спектра рент-
геновской флуоресценции.— В кн.: Геохимические методы поисков. Ме-
тоды анализа. Иркутск, 1979, с. 88—93.
81. Ложкин В. И., Афонин В. П. Исследование компонента рентгеновского
фона, обусловленного флуоресценцией кристаллов-анализаторов.— Зав.
лаб., 1976, т. 42, № 9, с. 1073—1074.
82. Ложкин В. И., Афонин В. П., Перепелица В. В. Экспериментальное изу-
чение рентгеновского фона, обусловленного флуоресценцией кристалла-
апализатора ADP.— В кн.: Ежегодник-1974 Ин-та геохимии им. А. П. Ви-
ноградова. Новосибирск: Наука. 1976, с. 330—332.
83. Лосев И. Ф. Количественный рентгеноспектральный флуоресцентный ана-
лиз. Мл Наука, 1969. 336 с.
84. Лосев II. Ф., Афонин В. П., Ревенко А. Г. О возбуждении рентгеновской
флуоресценции в длинноволновой области.— Зав. лаб., 1966, т. 32, А» 4,
с. 418—422.
85. Лосев Н. Ф., Глотова А. Н., Афонии В. П. О влиянии крупности частиц
порошковой пробы на интенсивность аналитических линий прп рентгено-
спектральном флуоресцентном анализе.— Зав. лаб.. 1963, т. 29, № 4,
с. 421-426.
86. Лосев Н. Ф.. Смагунова А. Н. Основы рентгеноспектрального флуоресцент-
ного анализа. Мл Химия, 1982. 208 с.
87. Лухминский Б. Е. Перенос излучений в статистически неоднородных гор-
ных породах.— В кн.: Теория функций и применения методов Монте-Кар-
ло. Уфа, 1975, с. 118-119.
88. Любимова С. В., Мамиконян С. В., Светайло Ю. Н.. Щекин К. П. Приме-
нение планирования эксперимента для калибровки многоэлсментпого
реитгенрадиометрического анализа.— Зав. лаб., 1979, т. 45, № 3, с. 252 —
256.
89. Мазурин О. В., Стрельцина М- В., Швайко-Шваиковская Т. П. Свойства
стекол и стеклообразующпх расплавов. Справочник. Т. IV. Ч. 1. Лл Нау-
ка, 1980. 464 с.
90. Мазурин О. В.. Стрельцина М. В., Швайко-Шваиковская Т. П. Свойства
стекол п стеклообразующих расплавов. Справочник. Т. IV. Ч. 2. Лл Наука,
1981. 376 с.
91. Макмиллан П. У. Стеклокерамика. Мл Мир, 1967. 263 с.
92. Максимов В. Н., Богданова И. В., Витошинскии Ю. И., Гордин В. Л. Ис-
следование условий получения гомогенного образца сплавлением для
РФА.— В кн.: Аппаратура п методы рентгеновского анализа. Вып. 24.
Лл Машиностроение, 1980, с. 145 —150.
93. Малкин В. П. О зависимости относительной подвижности катионов от соот-
ношения их координационных чисел в трехкомпонентных силикатных рас-
плавах.— Жури, аналит. химии, 1961, т. 35, № 2, с. 336—341.
94. Малкин В. И., Могутнов Б. М. Самодиффузпя щелочных попов в сили-
катных расплавах.— Докл. АН СССР, 1961, т. 141, № 6, с. 1127—ИЗО.
95. Малкин В. И., Могутнов Б. Мм Рыльникова А. Г. Рефракция иона кисло-
рода в трехкомионенгпых силикатных расплавах.— В кнл физическая
химия расплавленных шлаков. Киев: Наукова думка, 1970, с. 77—83.
96. Медведева Ю. А. и др. Смачивание графита и стали медными припоями.—
Сварочное производство, 1973, № 7, с. 35—36.
97. Метрологические основы исследования химического состава горных пород,
руд и минсралов/Под род. Г. В. Остроумова. Мл Недра, 1979. 400 с.
98 Мосичев В. И., Першин И. В., Ковалева Н. Б., Николаев Г. II. Теоретиче-
ский учет межэлементных влияний на основе нового градуировочного урав-
нения связи.— Зав; лаб., 1981, т. 47, № 1, с. 41—48.
99. Найдич IO. В. Контактные явления в металлических расплавах. Киев:
Наукова думка, 1972. 196 с.
100. Найдич 10. В., Колесниченко Г. А. Взаимодействие металлических распла-
вов с поверхностью алмаза и графита. Киев.: Наукова думка, 1967. 89 с.
101. Налимов В. В. Применение математической статистики прп анализе ве-
щества. М.: Физматгиз, 1960. 430 с.
1('2 . Налимов В. В., Чернова И. А. Статистические методы планирования экст-
ремальных экспериментов. Мл Наука, 1965. 340 с.
215
103. Наумов Г. Б., Рыженко Б. П., Ходаковский II. Л. Справочник термодина-
мических величин. М.: Атомпздат, 1971. 239 с.
104. Никольский А. П., Афонин В. II., Верховский Б. И., Межевпч А. Н Совре-
менное состояние и тенденции развития автоматизации рентгеноспектраль-
пого контроля. Вып. 2. М., 1982. 44 с.
105. Ольховой В. А., Бондаренко А. В. К выбору форм уравнений множествен-
ной регрессии при рентгеиоспектральном анализе пульп продуктов обога-
щения медно-никелевых руд-— Обогащение руд, 1981, Д° 2, с. 33—37.
106. Павлинекий Г. В. Исследование интенсивности рентгеновского спектра
флуоресценции, возбужденного смешанным первичным излучением.
Автореф. канд. дне. Иркутск: изд. ИГУ, 1966. 23 с.
107. Павлинекий Г. В., Бондаренко Б. Ю., Лосев II. ф. Способ стандартов-функ-
ций в рентгеиоспектральном флуоресцентном анализе.— Зав. лаб., 1978,
т. 44, № 2, с. 160—163.
108. Павлинекий Г. В., Величко IO. II., Ревенко А. Г. Программа расчета ин-
тенсивностей аналитических линий рентгеновского спектра флуоресцен-
ции.— Зав. лаб.. 19/7, т. 43, А? 4, с. 433—436.
109. Павлинекий Г. В., Гуляев В. Т. Оценка вклада фото- и оже-электронов
в возбуждена? флуоресцентного рентгеновского излучения.— В кн.: Ис-
следования в области фпзпкп твердого тела. Вып. 2. Иркутск: изд. ИГУ,
с. 230—233.
110. Павлинекий Г. В., Лосев И. Ф. К оценке избирательного возбуждения рент-
геновской флуоресценции в случае смешанного первичного излучения.—
Журн. техн, физики, 1969, т. 39, Д» 9, с. 1664—1675.
111. Павлинекий Г. В., Лосев П. Ф., Гуничева Т. Н., Ревенко А. Г. О методике
рентгеноспекзрального флуоресцентного анализа способен последова-
тельных поправок с помощью стандартов-бпнаров.— В кн.: Аппаратура
и методы рентгеновского анализа. Вьш. 4. Л.: Машиностроение, 1969,
с. 184—190.
112. Паньков С. Д., Смагунова А. II., Хабеев II. А.. Бобров Ю. Д. Построение
регрессионной модели при рентгеноспектральном анализе.— Журн. ана-
лит. химии, 1981. т. 36, До 1, с. 54—64.
ИЗ. Панькова Л. М., Смагунова А. Н., Хренова Г. П. и др. Итерационный ме-
тод расчета концентраций при рентгеиоспектральном определении элемен-
тов в пульпе.— Зав. лаб.. 1977, т. 43, Д'г 4, с. 443—445.
114. Пискунова Л. Ф., Афонии В. П., Гуничева Т. Н. Оценка эффекта дополни-
тельного возбуждения элементов с малыми атомными номерами.— В кн.:
Ежегодник-1975 Ин-та геохимии им. А. П. Виноградова СО АН СССР. Но-
восибирск: Наука, 1976, с. 262—265.
115. Пискунова Л. ф., Афонин В. П., Гуничева Т. И. О методе расчета эффектив-
ной длины волны в поправке на поглощение при реитгеноспектраль-
ном флуоресцентном анализе.— Зав. лаб., 1977, т. 43, № 9. с. 1075—1078.
116. Пискунова Л. ф., Афонин В. П., Гуничева Т. Й. Учет матричного возбуж-
дения при рентгенофлуоресцентном анализе горных пород,— Жури,
аналит. химии, 1980, т. 35, Д? И, с. 2180—2186.
117. Пискунова Л. ф., Емельянов П. Т., Завьялова Л. А. Методика приготовле-
ния гомогенных стеклянных излучателей из горных пород для рентгено-
спектрального флуоресцентного анализа.— В кн.: Геохимические методы
поисков. Методы анализа. Иркутск, 1979, с. 87—91.
118. Плаксин И. П. Металлургия благородных металлов. М.: Металлургиздат,
1958. 366 с.
119. Плотников Р. И., Смагунова А. Н., Ревенко А. Г. Сопоставление различ-
ных способов оценки взаимного влияния элементов в рентгеяоспектраль-
ном анализе.— Зав. лаб., 1974, д. 40, № 6, с. 671—673.
120. Попова В. II., розова О. Ф., Смагунова А. И. и др. Оценка возможности
использования различных устройств при подготовке проб к рентгено-
спектральному анализу.— В кн.: Аппаратура и методы рентгеновского ана-
лиза. Вып. 20. Л.: Машиностроение, 1978, с. 135—140.
121. Посыпайко В. II., Васина Н. А. Аналитическая химия и технический ана-
лиз. М.: Высшая школа, 1979. 384 с.
122. Ревенко А. Г. Исследование и выбор условий рентгеновского флуоресцент-
216
иого анализа элементов с малыми атомными номерами. Автореф. панд.
дас. Иркутск: изд. ИГУ, 1970. 26 с.
123. Ревенко А. Г., Величко Ю. II. Использование теоретических интенсивно-
стей в количественном рептгеноспектральном анализе.— В кн.: Аппара-
тура и методы рентгеноспектрального анализа. Вып. 22. Л.: Машинострое-
ние, 1979, с. 146—161.
124. Ревенко А. Г., Павлинский Г. В., Лосев И. Ф. Новый вариант способа ка-
либровки в рентгеноспектральном флуоресцентном анализе. — Зав. лаб.,
1970, т. 36, № G, с. 675—680.
125. Ревенко А. Г., Павлинский Г. В., Лосев И. ф. Исследование зависимости
интенсивности рентгеновского фона в длинноволновой области от химиче-
ского состава проб.— Зав. лаб., 1974, т. 40, № 11, с. 1334—1338.
126. Ревенко А. Г.. Павлинский Г. В., Лосев II. ф., Афонин В. П. О рентгенов-
ском фоне в длинноволновой области спектра.— Зав. лаб., 1970, т. 36, А'° 2,
с. 166-169.
127. Ревенко А. Г., Павлинский Г. В., Маков В. М.. Лосев II. ф. Рентгенов-
ский флуоресцентный анализ геологических проб с использованием спо-
соба калибровки.— Зав. лаб.. 1975, т. 41, № 12, с. 1451—1456.
128. Ревенко А. Г., Храмченко 3. А., Величко Ю. П. идр. Исследование возмож-
ностей некоторых способов учета взаимных влияний элементов ири ронт-
гедосцетральиом анализе многокомпонентных проб.—' В кн.: Исследо-
вания в области физики твердого тела. Выи. 1. Иркутск: изд. ИГУ, 1974,
с- 212-217.
129. Реми Г. Курс неорганической химии. М.: Мпр, 1972. 824 с.
130. Репев В. К.. Багаев И. В., Тронева 11. В. Применение способа калибровки
при рентгенофлуоресцсптном определении РЗЭ.— Зав. лаб., 1971, т. 37,
А» 4, с. 418—420.
131. Саксена С. Термодинамика твердых растворов породообразующих минера-
лов. М-: Мир, 1975. 205 с.
132. Самсонов Г. В. Тугоплавкие соединения. Справочник по свойствам и при-
менению. М-: Металлургиздат,1963. 398 с.
133. Сербин А. Я., Плотников Р. Й. Применение ступенчатого метода поиска
оптимальных форм регрессионных уравнений в рептгеноспектральном
анализе.— В кн.: Аппаратура и методы рентгеновского анализа. Вып. 17.
Ли Машпностроелнс, 1975. с. 151—155.
134. Сиденко II. М. Измельчение в химической промышленности. 2-е изд. М.:
Химия, 1977. 368 с.
135. Симаков В. А. Учет матричных эффектов поправками на поглощение при
рептгенофлуоресцентпом определении основных элементов в пробах с ши-
рокой вариацией состава.— Журн. аналмт. химии, 1980, т. 33, А» 5, с. 870—
872.
136. Скуг Д., Уэст Д. Основы аналитической химии. 1. М.: Мпр, 1979. 480 с.
137. Скуг Д., Уэст Д. Основы аналитической химии. 2. М.: Мир, 1979. 440 с.
138. Слуциип М. А., Бадер В. II. К выбору упрощенных формул расчета кон-
центрации в рентгеноспектральном анализе.— Зав. лаб., 1974, т. 40, № 12,
с. 1465—1407.
139. Смагунова А. Н., Гуничева Т. Н., Обольяншюва В. Г. и др. Препарирова-
ние проб в рентгеноспектральном флуоресцентном анализе.— В кп.: Ап-
паратура и методы рентгеновского анализа. Вып. 12. Л.: Машиностроение,
1973, с. 243—2G4.
140. Смагунова A. И., Лосев И. Ф. Рентгеносиектральнып флуоресцентный ана-
лиз. Иркутск: изд. ИГУ, 1975. 225 с.
141, Смагунова А. Н., Паньков С. Д.. Лосев И. Ф. и др. Влияние и учет хими-
ческого состава при рептгеноспектральном анализе продуктов производст-
ва.— Жури, аналпт. химии, 1974, т. 29, А» 12, с. 2335—2340.
142. Смагунова А. Н., Паньков С. Д., Лосев Н. Ф., Плотников Р. И. Контроль
корректирующих коэффициентов при рептгеноспектральном анализе спо-
собом калибровки. — Журн. аналпт. химии, 1977, т. 32. As 2, с. 15—20.
143. Смирнов А. С., Иванова II. М., Афонии В. П. Методика экспрессного рент-
юноспектрального анализа сырьевой смеси цементного производства.—
В ки.: Цементная и асбестоцементная промышленность. М., 1975, с. 5—7.
217
144. Сочеваиов В. Г.. Беренштейи Л. Е., Масалович М. С. Обоснование новых
требований к точности полных анализов в горных породах.— В кн.: Экс-
пресс-информация. Лабораторные и технологические исследования и обо-
гащение минерального сырья. Выл. 2. М., 1975, с. 1.
145. Справочник по расплавленным солям. Т. 2/Под ред. А. Г. Морачевского.
Л.: Химия. 1972. 160 с.
146. Стеклообразное состояние. Л.: Наука, 1971. 400 с.
147. Строение расплавленных солей/Под ред. Е. А. Укпге. М.: Мир, 1966. 431 с.
148. Сумм Б. Д., Горюнов Ю. В. Физико-химические основы смачивания и рас-
текания. М.: Химия, 1976. 232 с.
149. Термины, определения п обозначения метрологических характеристик
анализа вещества.— Жури, аналит. химии, 1975, т. 30, № 10, с. 2058—
2063.
150. Технология стекла/Под ред. И. И. Китайгородского. М.: Изд-во лит-ры
по строительству. 1967. 564 с.
151. Тихонов В. U., Гуиичева Т. Н., Павлинский Г. В. Рентгеноспектральное
определение основных породообразующих элементов в гранитах с исполь-
зованием литых дисков.— В кц.: Аппаратура и методы рентгеновского
анализа. Вып. 13. Л.: Машиностроение, 1974, с. 163—166.
152. Туранская Н. В., Кузьмина Т. Г. Методика рептгеноспектральното флуо-
ресцентного анализа в применении к дойным осадкам.— Океанология,
1978, т. 18, № 6, с. 1110—1114.
153. Туранская Н. В., Кузьмина Т. Г., Маслов В. Ю. Метод рептгеноспектраль-
ного флуоресцентного анализа силикатных пород.— Океанология, 1976,
т. 16, № 4, с. 713—716.
154. Физическая химия поверхностных явлений в расплавах. Киев: Наукова
думка, 1971. 297 с.
155. Физическая химия поверхностных явлений при высоких температурах.
Киев: Наукова думка, 1971. 300 с.
156. Физическая химия расплавленных шлаков/Под ред. IO. К. Делимарского.
Киев: Наукова думка, 1970. 270 с.
157. Финкельштейн А. Л., Афошш В. П. Расчет интенсивности рентгеновской
флуоресценции в гетерогенных образцах.— В кп.: Ежегодник-1975 Ип-та
геохимии им. А. П. Виноградова СО АН СССР. Новосибирск: Паука, 1976,
с. 168- 171.
158. Финкельштейн А. Л., Гуничева Т. И., Афонин В. П. и др. Расчет спект-
рального излучения при рентгенофлуоресцентпом анализе.— Зав. лаб.,
1981. т. 47. № И, с. 28—31.
159. Харченко А. М., Афонин В. П. Программа для расчета результатов реят-
геноспемтрального анализа многокомпонентных образцов.— В кп.: Ап-
паратура и методы рентгеновского анализа. Вып. 15. Л.: Машиностроение,
1974, с. 160-164.
160. Чуйко И. М. Химическая связь в расплавленных солях и шлаках и опре-
деление активных концентраций в расплавах.— В кн.: Физическая химия
расплавленных шлаков. Киев: Наукова думка, 1970, с. 94—108.
161. Шурыгин II. М. Исследование кинетики гетерогенных реакций на равно-
доступной поверхности вращающегося образца.— В кп.: физическая хи-
мия расплавленных шлаков. Киев: Наукова думка. 1970, с. 147—153.
162. Эйтель В. Физическая химия силикатов. М.: ИЛ, 1962. 1055 с.
163. Юбельт Р., Шрайтер П. Определитель горных пород. М.: Мир, 1977. 235 с.
164. Яковиц X., Хайнрих К. Неопределенность во введении поправки на по-
глощение.— В кп.: Физические основы рептгепоспектральпого локаль-
ного анализа. М.: Наука, 1973, с. 179—197.
165. Alley В., Myers R. Correction for matrix effect? in X-ray fluorescence analy-
sis by using multiregression methods.— Anal. Chem., 1965, v. 37, X 13,
p. 1685—1690.
166. Alley B., Myers R. Calibration method for the X-ray fluorescence analysis
of multicomponent mixtures.— Norelco Rep., 1968. v. 15, N 3, p. 87—92.
167. Anderman G. X-ray emission analysis of finished cements.— Anal. Chem.,
1961, v. 33, N 12, p. 1687—1695.
168. Andermann G. Semitheoretical approach to interclemcnt correction factors
218
in secondary X-ray emission analysis.— Anal. Chem., 1966, v. 38, N 1
p. 82—86.
169. Andermann G., Jones J., Davidson E. The evaluation of the RXQ for the ana-
lysis of cements and related materials.— Adv. X-ray Anal., 1959 v. 2
p. 215—227.
170. Andermann G., Kemp J. W. Scattered X-rays as Internal Standards ins X-ray
emission spectroscopy.—Anal. Chem., 1958. v. 30, p. 1306—1309.
171. Anderson С. H., Mandler J. E., Zeitner J. X-ray fluorescence analysis of port-
laud cement through, the use of experimentally determined correction fac-
tors.— Adv. X-ray Anal., 1974, v. 17, p. 214—224.
172. Archard G. Back scattering of electrons.—J. Appl. Phys., 1961, v. 32,
p. 1505-1509.
173. Artiz В. B. X-ray fluorescence analysis of catalytic converters using single-
element standards and theoretical corrections for interclement effects.— X-ray
Spectrom., 1977, v. 6, N 3, p. 165—170.
174. Ashley D., Andrews K. Analysis of alnmosiiicate materials by X-ray fluores-
cence spectrometry.— Analyst, 1972, v. 97, X 1160, p. 841—847.
175. Austen C., Steel T. The computer calculation from fundamental parameters
oi influence coefficients from X-rav spectrometry.— Adv. X-ray Anal., 1975,
v. 18, p. 362—371.
176. Baird A., McIntyre D., Welday E. Sodium magnesium fluorescence analy-
sis.— Adv. X-ray Anal., 1963, v. 6, p. 377—384.
177. Ball D. Rapid analysis for some major elements in powdered rock by X-ray
fluorescence spectrography.—Analyst, 1965. v. 90, N 1070, p. 258—265.
178. Ball D., Perkins D. Plant analysis by X-ray fluorescence spectrography.—
Nature. 1962. v. 194, N 4834, p. 1163—1165.
179. Bambinck W.. Craseinann B., Fink R. e. a. X-pay fluorescence yields Auger
and Coster — Kronig transition probabilities.— Rev. Mod. Phys., 1972,
v. 44, N <t, p. 716—814.
180. Bean L. A method of producing specimens of pressed powders for tense X-ray
spectrochemical analysis.— Anal. Spectr., 1966. v. 20, N 3, p. 191—193.
181. Beatty II., Brissey R. Calibration method for X-ray fluorescence spectromet-
ry.— Anal. Chem., 1952, v. 26, N 6, p. 980—989.
182. Bennet H., Oliver P. Loss of cobalt and iron from lithium tetraborate fusions
in graphite crucibles.— Analyst. 1971, v. 96, N 1143, p. 427—431.
183. Bennet H., Oliver P. Development of fluxes for the analysis of ceramic mate-
rials by X-ray fluorescence spectrometry.— Analyst, 1976, v. 101, N 1207,
p. 803—807.
18-4 . Berry P., Furuta T., Rhodes J. Particle size effects in radioisotope X-ray
spectrometry.— Adv. X-ray Anal., 1969, v. 12, p. 612—632.
185. Bertin E. P. Principles and practice of X-ray spectrometric analysis. N. ¥.:
Plenum Press, 1970. 679 p.
186. Blount C., Channel R., Leyden D. Preparation of pellets for X-ray spectro-
graphic analysis in ion-exchange resins.—Anal. Chim. Acta, 1971, v. 56,
N 3, p. 456—458.
187. Blount C., Morgan W., Leyden D. Preparation of pellets from ion-exchange
resins for direct analysis for metal ions by X-ray spectroscopy.— Anal. Chim.
Acta, 1971, v. 53, N 2, p.463—468.
188. Biown D., Gilfrieh J., Peckerur M. Measurement and calculation ol absolute
intensities of X-ray spectra.— J. Appl. Phys., 1975, v. 46, N 10, p. 4537 —
4540.
189. Brown G., Kanaris-Sotirion R. The determination of sulphur in solids by
X-ray fluorescence analysis.— Analyst, 1969, v. 94, N 1122, p. 782—786.
190. Buehel E., Lomm H., Loiber F., Becker W. Automalisiertes Aufschlubgcrat
zur Hersteilung von Schinelstablctten fiir die Ronlgenfluoreszenzanalyse.—
Arch. Eisenhuttenwes., 1977, Bd 48, N 7, S. 391—394.
191. Burnham H., Houier J., Jones L. Generalized X-ray emission spectrographic
calibration applicable to varying composition and sample forms.— Anal.
Chem., 1957, v. 29. N 12, p. 1827—1834.
192. Chamberlain G. T. The calibration of an automatic X-ray spectrometer using
calculated relative intensities.— X-ray Spectrom., 1978, v. 7, N 4, p. 190—
194.
219
193. Ciecarelli M. QUAN — A computer programm lor quantitative X-ray fluo-
rescence analysis.— Anal. Chem., 1977, v. 49, N 2, p. 345—346.
194. Claisse F. Analyse quantitative precice per fluorescence de rayon.— Quebec
Dept. Mines Prelim. Rept., 1956, N 327, p. 16.
195. Claisse F. Overcoming the particle size effect in the internal standard method
of X-ray fluorescence analvsis.— Spectrochim. Acta., 1970, v. 25 B, N 5,
p. 209-218.
196. Claisse F., Quintin 3f. Generalization of the Lachance — Traill method for
the correction of the matrix effects in X-ray fluorescence analysis.— Can.
Spectr., 1967, N 12, p. 129—146.
197. Claisse F., Samson C. Heterogeneity effects in X-rav analvsis.— Adv. X-ray
Anal., 1962. v. 5, p. 335—354.
198. Claisse F., Thinlr T. Differential delta-coefficient method for the correction
of matrix effects in X-ray fluorescence analysis.— Anal. Chem., 1979, v. 51,
N 7, p. 954—957.
199. Clark N., Mitchell R. Intra- and inter- spectrometer correction coefficients
for X-rav emission spectrometric calibration equations.— X-ray Spectrum.,
1976, v. 5, N 4, p. 200—203.
200. Coldwell V. E. A practical method for the accurate analysis of high alloy
steels by X-ray emission.— X-ray Spectrom., 1976, v. 5, N 1, p. 31—35.
201. Criss J.. Birks L. Calculation methods for fluorescent X-ray spectrometry.—
Anal. Chem., 1968, v. 40, N 7, p. 1080—1091.
202. Criss J., Birks L., Gilfrieh J. Versatile X-ray analysis programm combining
fundamental parameters and empirical coefficients.— Anal. Chen.., 1978,
v. 50, N 1, p. 33—37.
203. Dahl M., Karlsson A. A simple calculation model with great versatility for
interelement effects in steel by X-ray fluorescence analysis.— X-ray Spect-
rom., 1973, v. 2. N 2, p. 75—83.
204. Drabseh S. Eine Methode zur eichprobenfreien quantitative?! Rontgcnfluores-
zenzanalyse.— X-ray Spectrom., 1974. v. 3, N 3, p. 120—124.
205. Drummond C. Preparation of glass blanks by fusion with lithium tetraborate
for X-ray spectrographic analysis.— Appl. Spectr,, 1966, v. 2(), N 4,
p. 252—255.
206. Dryer II., Appehnan T. Math model design for X-ray analysis of iron base
alloys.— Adv. X-ray Anal., 1975. v. 18, p. 382—395.
207. Dunn W., Efird C., Gardner R., Verghesc K. A mathematical model for ter-
tiary X-ravs from heterogenous samples.— X-ray Spectrom.. 1975, v. 4, N 1,
p. 18-25."
208. Ebel IL, Katinger T., Ho W. G., Burger K. Die Abhangigkeit der Ergebnisse
der Rontgenfluoreszenzanalyse mit iiusserem Standard von der Spannung
an der Spectroackopierohre.— Mikrochim. Acta, 1968. Suppl. Ill, p. 47—54.
209. Ebel 11., handler F., Dirschmid II. Zur Anregung charackteristischer Ront-
genstrahlung durch Pholoclektronen.—Z. Naturforsch., 1971, Bd 26a, N 5,
S. 927—928.
210. Espen P., Nullens M., Adams F. Radiative Auger effects in X-ray fluorescence
analysis.— Anal. Chem., 1979. v. 51, N 8, p. 1325—1327.
211. Fabbi B. A refined fusion X-ray fluorescence technique and determination
of major and minor elements in silicate standards.— Amer. Miner., 1972,
v. 57, N 1, p. 237—245.
212. Fatenii M., Birks L. On obtaining consistent solution of empirical equations
in X-ray fluorescence.—Anal. Chem., 1973, v. 45, N 8, p. 1443—1447.
213. Fatenii M., Birks L. S. Can regression equations be optimized by finagling
X-ray intensities?— Adv. X-ray Anal., 1974. v. 17. p. 302—308.
214. Foner H. The wetting of platinum alloys by various glasses and some impli-
cations thereof in analytical chemistry.— Lab. Pract., 1977, v. 26, N 8,
p. 625—626.
215. Freud H. Recent experimental value for she! X-ray fluorescence yields.—
X-ray Spctrom., 1975, v. 4, N 2, p. 90—91.
216. Frigieri P., Rossi F., Trucco R. Assessment of various mathematical correc-
tion methods ol matrix effects in X-ray fluorescence analysis.— Spectrochim.
Acta, 1980, v. 35 B, N 6, p. 351—366.
220
217. Gardner R. P., Wowthorne A. R. Monte Carlo simulation of the X-ray fluo-
rescence excited by discrete energy photons in homogeneous samples inclu-
ding tertiary inter—element effects.— X-rav Spectrom., 1975, v. 4, N 3
p. 138—148.
218. Giles H., Holmes G. The X-ray fluorescence analysis of ferroniobium by a fu-
sion method.— X-ray Spectrom., 1978, v. 7, N 1, p. 2—4.
219. Gilfrich J., Birks L. Spectral distribution of X-ray tubes for quantitative
X-ray fluorescence analysis.— Anal. Chem., 1968, v. 40. N 7, p. 1077—1080.
220. Gilfrich J., Burkhalter P., Whitlock R. e. a. Spectral distribution of a thin
window rhodium target X-ray spectrographic tube.—Anal. Chem., 1971,
v. 43, N 7, p. 934—936.
221. Gould R., Bates S. Some application of computer program for quantitative
spectrochemical analysis.— X-ray Spectrom., 1972, v. 1, N 1, p. 29—35.
222. Green M., Cosslett V. The efficiency of production X-radiation in thick tar-
gets of a pure element.— Proc. Phys. Soo., 1961, v. 78, p. 1206—1219.
223. Griffith J., Whitehead И. A simple empirical interelement correction proce-
dure to the X-ray fluorescence analysis of Nickel-base alloys.— X-ray Spec-
trom., 1975, v. 4, N 4, p. 178—185.
224. Harry J., Rose IL, Adler J., Flanagan F. Use of X-ray fluorescence in deter-
mination of selected major constituents in silicates.— U. S. Geol. Prof. Pa-
pers, 1965, v. 525 B, p. 155—159.
225. Harvey P.. Taylor D., Hendry R., Bancroft F. An accurate fusion method Cor
the analysis of rocks and chemically related materials by X-ray fluorescence
spectrometry.— X-ray Spectrom., 1973, v. 2, N 1, p. 33—44.
226. Haukka M. T., Thomas I. L. Total X-ray fluorescence analysis of geological
samples using a low-dilution lithium inelaborate fusion method matrix cor-
rection for major elements.— X-ray Spectrom., 1977, v. 6, N 4, p. 204—211.
227. Haukka M., Thomas I. Practical solutions to matrix effects in XRF analysis
by mathematical methods.—Anal. Chem., 1978, v. 50, N 4, p. 592—596.
228. Hawfhorne A. R., Gardner R. P. Monte Carlo simulation of the X-ray fluo-
rescence from homogeneous multielement samples excited by the continuous
and discrete energy photons from X-ray tubes.— Anal. Chem., 1975, v. 47,
N 13, p. 2220-2224.
229. Heinrich IL X-ray absorption uncertainty.— In: The Electron Microprobe.
N. Y., 1966, p. 296—377.
230. Heinriqh IL, Rasberry S. X-ray fluorescence analysis of high—temperature
superallovs-calibration and standards.— Adv. X-ray Anal., 1974, v. 17,
p. 309—317.
231. Henke B., Elgin R. X-ray absorption tables for the 2 — to — 200 A region.—
Adv. X-ray Anal., 1970, v. 13. p. 639—667.
232. Holland J., Brindle D. A self-concistent mass absorption correction for si-
licate analvsis by X-ray fluorescence.— Spectrochim. Acta, 1966, v. 22.
N 12, p. 2083-2093.
233. Hooper P., Atkins L. Preparation of fused rock samples for X-ray fluorescence
analysis.— Miner. Mag., 1969, v. 37, p. 403—413.
234. Hower J., Sclimittroth L., Perry E., Monatt T. X-ray spectrographic major
constituent analysis in undiluted silicate rocks and minerals.— Geel. Soc.
Amer. Spec. Paper, 1965, v. 82, p. 96—97.
235. Huges G., Woodhouse J., Bucklow J. The determination of some X-ray mass
absorption coefficients.— Brit. J. Appl. Phvs., 1968, Ser. 2, v. 1, N 6,
p. 695—706.
236. Hughes H. Technique for determining matrix correction factors and their ap-
plication to X-ray fluorescence analysis of low-grade iron ores over wide range
of composition.— Analyst, 1972, v. 97, N 1152, p. 161—170.
237. Hunter C., Rhodes J. Particles size effects in X-ray emission analysis; formu-
lae for continuous size distribution.— X-ray Spectrom., 1972, v. 1, N 3,
p. 107—111.
238. Hutchison G. A simple dead-time correction for X-ray counting systems.—
X-ray Spectrom., 1976, v. 5, N 4, p. 194—196.
239. Inganielis C. Lithium metaborale flux in silicate analysis.— Anal. Chim.
Acta. 1970, v. 52, N 2, p. 323—334.
221
240. Jenkins R. A review of empirical influence coefficients in X-ray spectromet-
ry.—Adv. X-тау Anal., 1979, v. 22. p. 281-292.
241. Jenkins R., Croke J., Niemann R., Westberg R. Use of calculated alfa-coef-
i'icients in quantitative X-ray spectrometry.— Adv. X-ray Anal., 1975, v. 18,
p. 372—381.
242. Jongh W. X-ray fluorescence analysis applying theoretical matrix corrections
stainless steels.— X-ray Spectrom., 1973, v. 2, N 4, p. 151—158.
243. Jongh W. The atomic number Z — 0; loss and gain on ignition in XRFA
treated by tire JN-cquations.— X-ray Spectrom., 1979, v. 8, N 2. p. 52—56.
244. Kahkonen H., Suonen P. Determination of a binary calibration curve with
cue or two standard specimens in X-ray fluorescence analysis.— X-ray Spect-
rum.. 1975, v. 4, N 1, p. 11 — 13.
245. Keith И., Loomis T. Correction lor scattering in X-ray fluorescence experi-
ments.—X-ray Spectrom., 1978, v. 7, N 4, p. 225—240.
246. Kirclrnayer M.. Dziunikowski B. A new approach to the intensity correction
for matrix effects in the X-rav fluorescence analysis of ores.— X-ray Spect-
rom.. 1978, v. 7, N 3, p. 164X169.
247. Kis-Varga M. A fundamental parameter method for analysis of alloys by iso-
topo-exci ted X-ray fluorescence.— X-ray Spectrom., 1979, v. 8, N 2, p. 73—
75.
248. Kloyber II., Ebel H.. Manlier H., Koitz S. Quantitative Roiitgenfluoreszenz-
analyse mil Hilfe des linearen und quadratischen Hybridmodells. — X-ray
Spectrom., 1980, v. 9, N 4, p. 170—175.
249. Kodama 11., Brydon J., Stone B. X-ray spectrocheinical analysis of silicates
using synthetic standards with a correction for interclemental effects by a com-
puter methods.— Gcochim. Cosmoehim. Acta, 1967, v. 31, N 4, p. 649 —
659.
2o0. Krudhof II. A general XRF spectrometric technique based on simple correc-
tion for matrix effects. — Anal. Chim. Acta. 1978, v. 102, p. 177—180.
251. Lachance G. R. A fundamental coefficients for X-ray spectrochemical analy-
sis.— Can. Spectr., 1970, v. 15, N 3, p. 64—71.
252. Lachance G. R. A practical relation between atomic numbers and alpha coef-
ficients.— X-ray Spectrom., 1980, v. 9, N 5, p. 195—197.
253. Lachance G., Trail R. Practical solution to the matrix problem in X-ray ana-
lysis.— Can. Spectr., 1966, v. 11, N 2. p. 43—48.
254. Laidlev R. X-ray fluorescence ol rock samples as applied to geological prob-
lem.- Appl. Spectr., 1968, v. 22, X 5, p. 420-422.
255. Larson J.. Winkler R., Gully J. A glass fusion method for X-ray fluorescence
analysis.— Adv. X-ray Anal., 1966, v. 10, p. 489—493.
256. Lee R., McConchie D. Comprehensive major and trace element analysis of
geological material by X-ray fluorescence using low dilution lusions.— X-
ray Spectrom., 1982, V. 11, N 2. p. 55—63.
257. Lehouillier R., Tunnal S. Bead homogeneity in the fusion technique for X-
ray spectrochemical analysis.— Anal. Chora., 1974, v. 46, N 6, p. 734—736.
258. Leroux J. Method for finding mass absorption coefficients by empirical equa-
tions and graphs.— Adv. X-ray .Anal., 1961, v. 5, p. 153—160.
259. Loddiiig W., Rhett D. Sample preparation for X-ray fluorescence analysis
Li-borate glass disks.— Amer. Miner., 1972, v. 57, N 1, p. 281—283.
260. Longobucco R. Determination of major and minor constituents in ceramic
materials bv X-ray spectrometrv.— Anal. Chem., 1962, v. 34, N 9. p. 1263—
1267.
261. Loomis T. C., Keith H. D. Spectral distribution о X-ray produced by a ge-
neral electric EA75 Cr,'W tube at various applied constant voltages.— X-ray
Spectrom., 1976, v. 5, N 2. p. 104—114.
262. Lubecki A., Ilolynska B-. Wasilewska M. Grain size effects in non-dispersive
X-ray fluorescence analysis. — Spccirochiin. Acta. 1968, v. 23 D, p. 465—
479. '
263. Lucas-Tooth H., Price B. A mathematical method for the investigation of in-
terolement effects in X-ray fluorescence analysis.— Metallurgia, 1961, v. 64,
p. 149—152.
264. LucaS’Toath IL, Рупе C. The accurate determination of major constituents
222
by X-ray fluorescent analysis in the presence of large interelement effects.—
Adv. X-ray Anal., 1964, v. 7, p. 526—554.
265. Liischow II., Schafter II. Uber die Vervendung von Graphittiegeln zur Pro-
benpraparation in der Rontgenfluoreszenzanalyse.- Z. Anal. Chem., 1970,
Bd 250, N 5, S. 317-318.
266. Mainardi R., Fernandez J., Bonetto R., Riveros J. A theoretical procedure
to determine coefficients from the Rasberry-Heinrich calibration curve in
XRF spectrometry.— X-ray Spectr., 1981, v. 10, N 2, p. 74—78.
267. Mainardi R., Fernandez J., Notes M. Influence of interelement effects on X-
ray fluorescence calibration curve coefficients for binary mixtures.— X-ray
Spectrom., 1982, v. 11, N 2, p. 70—78.
268. Matocha G. An automated system for X-ray fluorescence analysis of alumi-
nium ores.— Aluminium (BDR), 1976, v. 52, N 8, p. 497—499.
269. McKinney C., Rosenherg A. Simultaneous X-ray emission analysis of P, Si,
Ca, Fe, Al, Mg in phosphate rock using a small computer to correct for mat-
rix variations.— Adv. X-ray Anal., 1970, v. 13, p. 125 — 136.
270. Mencik Z., Berneburg P., Short M. A multiple regression procedure for ele-
mental analysis at low concentrations.— Adv. X-ray Anal., 1975, v. 18,
p. 396—405.
271. Mitchell B., Hopper F. Digital computer calculation and correction of matrix
effect? in X-ray spectroscopy.— Appl. Spectr., 1966, v. 20, N 3, p. 172—176.
272. Norrish K., Ruttun J. Accurate X-ray spectrographic method for the analysis
of a wide range of geological samples.—Geochiin. Cosmochim. Acta, 1969,
v. 33, N 4, p. 431—453.
273. Ogier W. T., Carlson R. D., Knoche J. Electron Bremsstrahlung from Proton-
Excited targets.— Phys. Rev., 1966, v. 142, p. 50—52.
274. Palme C., Jagouts E. Application of the fundamental parameter method for
the determination of major and minor elements on fused geological samples
with X-ray fluorescence spectrometry.— Anal. Chem., 1977, v. 49,
N 6. p. 717—722.
275. Philibert 3. A method for calculating the absorption corrections in electron
probe microanalysis.— In: X-ray Optics and X-ray Microanalysis. N. Y.:
Acad. Press, 1963, p. 379—392.
276. Pietrosz J., Kubon Z. Znusenosti a pretavovanim vzorku sypnych matermlu
pro rentgenovou jluorescencni analyzu.— Hntn. Listy, 1979, v. 34, N 9,
S. 566—662.
277. Plesch R. Eine einfache Matrixkorrektur fur die Rontgcnspcklrometiie.—
Z. Anal. Chem., 1974, Bd 272, N 4. S. 262-267.
278. Plesch R. Die hybride Matrixkorrektur in der Rontgenspektrometrie.— X-ray
Spectrom., 1976, v. 5, N 4, p. 204-207.
279. Plesch R. The error influence of the matrix correction coefficients in X-ray
spectrometry.— Z. Anal. Chem., 1978. Bd 292, S. 378—380.
280. Plesch R. Comparison of different inelhodsfor the matrix correction in X-ray
spectrometry.— X-ray Spectrom., 1981, v. 10, N 4, p. 193—195.
281. Pool A. Application of X-ray fluorescence to silicate analysis.— Proc. Brit.
Ceram. Soc., 1970, N 16, p. 41—51.
282. Pool A., Holloway S. A correction methods for elemental interactions and
physical effects in the X-ray fluorescence analysis of silicate powders,— Adv.
X-ray Anal., 1969, v. 12, p. 534—546.
283. Quinn A. P. Quasi-fundamenlal correction methods using broadband X-ray
excitation.—Adv. X-ray Anal., 1979, v. 22, p. 293—302.
284. Rasberry S., Heinrich H. Calibration for interelement effects in X-ray fluo-
rescence analysis.— Anal. Chem., 1974, v. 46, N 1, p. 81—89.
285. Rinaldi F., Aquzzi P. A simple technique for casting glass disks for X-ray
fluorescence analysis.— Spectrochim. Acta, 1967, v. 23 B, N 1, p. 15—18.
286. Riveros J-, Bonetto R., Mainardi R. Extension of the Rasberry — Heinrich
equation for X-ray fluorescence analysis.— Anal. Chem., 1978, v. 50, N 9,
p. 1386—1388.
287. Rolin M. Cryoscopia dans cryolithe fondue L’ionisation de I'alumina at des
oxydes dessorie Revue de Metallurgic.— J. du four elcctrique at de? Indus-
tries, electrochimiques, 1953, v. 6, p. 157—160.
223
288. Rose H., Adler L, Flanagan Г. X-ray fluorescence analysis of the light ele-
ments in rocks and minerals.— Appl. Spectr., 1963, v. 17, p. 81—85.
289. Rousseau Ji., Claisse Г. Theoretical alfa coefficients for the Claisse — Quin-
ton relation for X-ray spcctrochcmical analysis.— X-ray Spectrom., 1974,
v. 3, N 1, p. 31—36.
290. Sagrera J. Determination of aluminium and silicon in mixtures of silica and
alumina by X-ray fluorescence.— Incion qitiin. analit. pure anl. ind., 1966,
v. 20, N 2, p. 51 -56.
291. Sato tl. Fluorescent X-ray analysis of iron ore and the like by glass bead tech-
niques. — Trans, of the Iron and Steel Inst, of Japan, 1978, v. 18, N 11,
p. 721-724.
292. Schaefer H. Use of graphite crusibles for sample preparation in X-ray fluores-
cence analysis.— Z. Anal. Chem., 1970. Bd 250. X 5, S. 317—318.
293. Scheme H. Experiments with mixtures.— Roval statistical Society J., 1958,
v. 20. N 2, p. 344—349.
294. Schreiner W.. Jenkins R. A non—linear least squares fitting routine for opti-
mizing empirical XRF matrix correction models.—X-ray Spectrom., 1979,
v. 8. N 1. p. 33-41.
295. Shiraiwa T.. Fujino X. Theoretical calculation, of fluorescent X-ray inten-
sities in fluorescent X-rav spectrochemical analvsis.— Jap. J. Appl. Phys.,
1966. v. 5, X 10. p. 886—899.
296. Shiraiwa T., Fujino X. Theoretical correction for coexistent elements in fluo-
rescent X-rav analysis ol alloy steel.— Adv. X-ray Anal., 1968, v. 11, p. 63—
94
297. Siiiraiwa T., Fujino N. Theoretical correction procedures for X-ray fluoros-
conce analysis.— X-ray Specirom., 1974, v. 3, X 2, p. 64—73.
298. Springer G. Die Berechnuiig von Korrekturen fiir die quantitative Ek'ktro-
imnstrahlmikroanalysc.— Fortschr. Miner., 1967, Bd 45, N 1, S. 103—124.
299. Springer G. Iterative procedures in electron probe microanalysis correc-
tions.— X-ray Spectrom., 1976, v. 5, X 2, p. 88—91.
300. Stadia G., Bruck If. Zur Vorbereilung oxidischer Proben fur die Rontgcniluo-
reszenzanalyse.—Z. Anal. Chem., 1970, Bd 250, X 5, S. 289—294.
301. Statham P. J. The generation, absorption and anisotropy of thick-tar-
get bremsstrahlung and implication for quantitative analvsis.— X-rav Spec-
DdDi., 1976. v. 5. N 3. p. 154-168.
302. Stephenson D. A. Theoretical analysis of quantitative X-ray omission data:
glasses, rocks and metals.— Anal. Chem., 1971, v. 43, N 3. p. 1761—1764.
303. Stern W. On trace element analysis of geological samples by X-ray fluores-
cence.— X-ray Spectrom., 1976. v. 5, X 2. p, 50—60.
304. Storm 15. Calculated Bremsstrahlung spectra from thick tungsten targets.—
Phys. Rev.. 1972, v. 5, X 6, p. 2328—2338.
305. Sltasheim A., Brandt ]f. A quantitative X-ray fluorescence method for the de-
termination ot miner and trace elements in geological samples using a cor-
rection technique lor the matrix effect4.— Snectrochirn. Acta, 1968, v. 23 B,
X 8, p. 535-541.
306. Sugizaki M.. Akatsuka J. Correction methods for matrix effects and instru-
mental drift in the X-ray fluorescence analysis of glasses.— Rcpls Res. Lab.
Asahi Glass Co., 1974. v. 24, N 1. p. 37—44.
307. Teisen R., Togel K., Vollath D. Massenschwachungskoeffizientcn von Ront-
genslinicn.— Mikrochim. Acta, 1967. Suppl. 11, X 6, S. 16—24.
308. Teisen R., Vollath D. Tabellen der Massenschwachungskoeffizienten von Ront-
genstrahlen. Dus&eldorl: Verlag Siahleiscn. M. B. IL, 1967. 40 S.
309. Tertian R. Sur la determination quantitative precise des elements legers par
fluorescence X. tone comparaison des methodes de fusion.— Anal. Chini. Acta,
1968, v. 41, p. 554—556.
310. Tertian R. A new approach to the study and control of interelement effects
in the X-ray fluorescence analysis of metal alloy's and of the multicomponent
systems. — X-ray Spectrom., 1973, v. 2, N 3, p. 95—109.
311. Tertian R. Concerning interelemenlal crossed effects in X-ray' fluorescence
analysis.— X-ray Spectrom.. 1974, v. 3. N 3, p. 102—1(>8.
312. Tertian R. A self — consistent calibration method for industrial X-ray speet-
rometric analyses.— X-ray Spectrum.. 1975, \. 4, X 2, p. 52—61.
224
31-3- Tertian R. Nouvelie nietode d’analyse precise des rochos et des maleriaux
apparente.' par spectrometrie X. Application а Г anal lb t —
corn. int. etude bauxites, alumina el alum., 19715. X 1:1. л. • ...>m
314. Tertian R. X-ray -pectronieiric analysis by lusion 11 L к j
endure for determinimg effective influence coefficioms. — .x-rav ;-m ।, t mu..
1979, v. 8, X 3. p. -117—120.
315. Tertian R.. Sage R. Crossed influence coMl'icients 1'c
cence analysis ol multicomponent svsioms.—X-rav Spectrom.. 1977. v. G,
X 3. p. 123—131.
316. Tough W. X-ray fluorescence analysis applying theoretical matrix correc-
tions.— X-ray Spectrom., 1973, v. 2. X 2, p. 151 — 158.
317. Trail R., Lachance G. Practical solution to (lie nnilrix problem in X-ray ana-
lysis.— Can. Spectr., I960, v. 11, X 3, p. 63—71.
.318. Tuergcn O. Lithium tetraborate melts for X-ray fluorescence and atomic
absorption analysis of rocks.— Neues Jahrb. Mineral. Monatsh., 1<>7и, X 12,
S. 531—534.
319. Yolborth A. X-ray spectrographic determination of all major oxides in ig-
neous rocks and precision and accuracy of a direct pelleting method.— Nev.
Bur. Mines Repl.. 1963, v. 6. Part A, p. 1—72.
32U. Vollath D. Masseii=cliwachungskooffizicnten fiir langwellige Rontgenslrah-
len.— Mikrochim. Acta, 1968, Suppl. til. S. 11—18.
321. Vos G. Correction method for the X-ray fluorescent determination of 1'c. Mg,
Ca and Na in light matrices.— Spectrochlm. Acta, 1.972, v. 27 B, N 2r p. 89 —
92.
322. Weber K. Die Korroktur der Probentransparonz in der Rontgcn^pcktralana-
lyse.— X-ray Spectrom.. 1974, v. 3, N 4, p. 159—166.
323- Weber K. Eine vereinfachte Formulierung des Kerugr6j3encinflusses.— X-ray
Spectrom., 1976, v. 5, К 1, p. 7—12.
324. Webleraud G-, Tellema T. Spectrochemical. analysis for some major and mi-
nor elements in rocks.— Appl. Spectr.. 1962, v. 16, N 4, p. 133—136.
325. Willgallis A., Schneider (}. Zur Preparation silikatischer Proben fur die Bont-
genfluoreszenzanalyse.— Z. Anal. Chem., 1969, Bd 246, N 2, S. 115—118.
326. Willigen V., Kruidhof H., Dahmen E. Borax fusion technique for X-ray fluo-
rescence analysis.—Taianta, 1971. v. 18. N 4, p. 450—452.
327. Wittmann A., Chmcleff J., Herrmann II. Preparation automatisec dcperlcs
pour I’analyso par la fluorescence de rayons X.— X-rav Spectrom., 1974,
v. 3, N 3,‘p. 137—142.
32S. Wronka V., Becker W. Mechanisierlo Probenvorbereitung fur die Hontgen-
fluoreszenzanalvse for oxidischen Stoffen.— Arch. Eisenhiittenw., 1971,
Bd 42, N 9, S.' 645—648.
ОГЛАВЛЕНИЕ
Предисловие............................................................ 3
Глава 1. Возбуждение рентгеновских спектров п аппаратура для реш-
1.1. Рентгеновские трубки для РФА............................. —
1.1.1. Тормозной компонент излучения рентгеновских трубок С
1.1.2. Характеристический компонент излучения рентгеновских
трубок .................................................... 8
1.2. Аппаратура для силикатною ]-'ФА......................... 10
1.2.1. Детекторы рентгеновского нзлучотшя.................. Ц
Газоразрядные пропорциональные детекторы................. —
Сцинтилляционные детекторы.............................. 17
Цолуяроводвиковые детекторы............................. 20
1.2.2. Система регистрации рептгеновсклх спектров......... 22
1.2.3. Кристалл-дифракциояные спектрометрические каналы 25
1.2.4. Спектрометры для силикатного анализа............... 29
Многоканальные спектрометры (квантоыетры)................ —
Рентгеновские спектрометры: с автоматически сканирующим
каналом................................................. 34
Рентгеновские спектрометры с энортстическоц дисперсией 37
Глава 2. Матричные эффекты....................................... 38
2.1. Интенсивность рентгеновской флуоресценции в полихромати-
ческом приближении............................................ Зу
2.2. Интенсивность рентгеновский флуоресценции в монохромати-
ческом приближении ....................................... 41
2.2.1. Влияние условий возбуждения на значение аффективной
длины волны............................................... 43
2.2.2. Зависимость значений эффективной длины волны or хи-
мического состава анализируемого материала.......... 45
2.3. Матричный эффект возбуждения............................ 48
2.3.1. Оценка эффекта возбуждения в длинноволновой области —
2.3.2. Упрощенное выражение для расчета эффекта возбуждения 53
2.3.3. Возбуждение рентгеновской флуоресценции фотоэлект-
ронами анализируемого образца ............................ 55
'2.4 . Экспериментальная проверка формулы интенсивности рентге-
новской флуоресценция................................... 58
2,5. Влияние гетерогенности образцов на интенсивность аналити-
ческого сигнала......................................... 61
2.5.1. Численное моделирование процесса возбуждения рент-
геновской флуоресценции в гетерогенных образцах ... 62
2.5.2. Влияние фактора гетерогенности па эффект поглощения 67
2.6. Влияние химического состава образца на интенсивность рент-
геновского фона.............................................. 70
Глава 3. Методы учета матричных эффектов при многокомпонентном
анализе....................................................... 72
3.1. Уравнения связи в рештоносиектральном анализе............ —
3,2. Физические модели и уравнения связи .................... 74
3.2.1. Методы фундаментальных параметров....................—
Поправка на поглощение.................................. 76
Поправка на возбуждение................................. —
3.2.2. Мультипликативный метод AF-коррекцпи........... 78
Упрощенная поправка на поглощение.................... —
Упрощенная поправка на возбуждение..................... 79
3.2.3. Методы ц-коррекции................................ 80
Метод Холланда и Бриндли...................... . . 81
Метод Андермана..................................... 82
Метод Ильина ......................................... —
Метод Кузьминой и Тураиской.......................... 83
Метод стандартов-бинаров............................. 85
Метод стандартов-функций............................. 86
Метод Ревенко и Лосева............................. 87
3.2.4. Методы а-коррекцип .... ..................... —
Метод Битти н Брисси................................. 89
Метод Лачанса и Трейла................................. 90
Метод Лукаса-Туса и Прайса............................. 92
Метод Расберри и Хайнриха.............................. 93
Метод Клайссе и Квинтипа............................. 95
Метод Моспчева и Псршипа ........................ 9б
Метод Ширайвы и Фуджино................................ 97
Метод Котлярова........................................ 99
3.2.5. Связь между дифференциальной п интегральной формой
уравнений связи.......................................... 100
3-3. Математические модели и уравнения связи............... 102
3.4. Решение систем уравнений связи в РСА.................. 106
3.5. Учет аппаратурных эффектов и уточнение параметров .... 112
Глава 4. Подготовка проб к анализу............................... 114
4.1. Стеклообразное состояние ............................... —
4.2. Физическая химия н механизм процесса плавления горных
пород и минералов.......................................... It7
4.3. Состав плавней........................................ 123
4.4. Сплавление горных пород и минералов...................... 127
4.4.1. Состав атмосферы...................................... —
4.4.2. Температура м время сплавления.......................128
4.4.3. Материал тиглей.................................... 130
4.4.4. Состояние исходной смеси............................ 138
4.5. Устойчивость стекла..................................... 14!)
4.6. Формование излучателей из расплавов пород................ 142
4.7. Автоматизация изготовления стекловидных 'излучателей . . 148
4.8. Приготовление порошковых излучателей из расплава . . . 149
Глава 5. Автоматпзпровантхый анализ горных пород................... 1а0
5.1. Химический и минералогический состав горных пород .... —
5.2. Требования к точности анализов горных пород.............. 152
5.3. Учет матричных эффектов.................................. 153
5.3. L Влияние неопределенности фундаментальных параметров 154
5.3. 2. Погрешности за счет упрощения .модели возбуждения 159
5.4. Получение гомогеиных излучателей . . . .................. 165
5.4.1. Выбор условий гомогенизации......................... 166
5.4.2. Приготовление излучателей . ........................ 169
5.5. Коррекция измеренных интенсивностей..................... 171
5.5.1. Поправка па фон................................... —
5-6. Коррезщия начальных приближений........................ 179
5.7. Автоматизация анализа горных пород . ..............181
5.7.1. Методические расчеты ... 182
5.7.2. Аналитическая программа........................... 185
5.8.1. Воспроизводимость.................................. —
5.8.2. Предел обнаружения . 190
5.8.3. Правильность....................................... 193
Приложения .... J94
Литература .... 211