













































































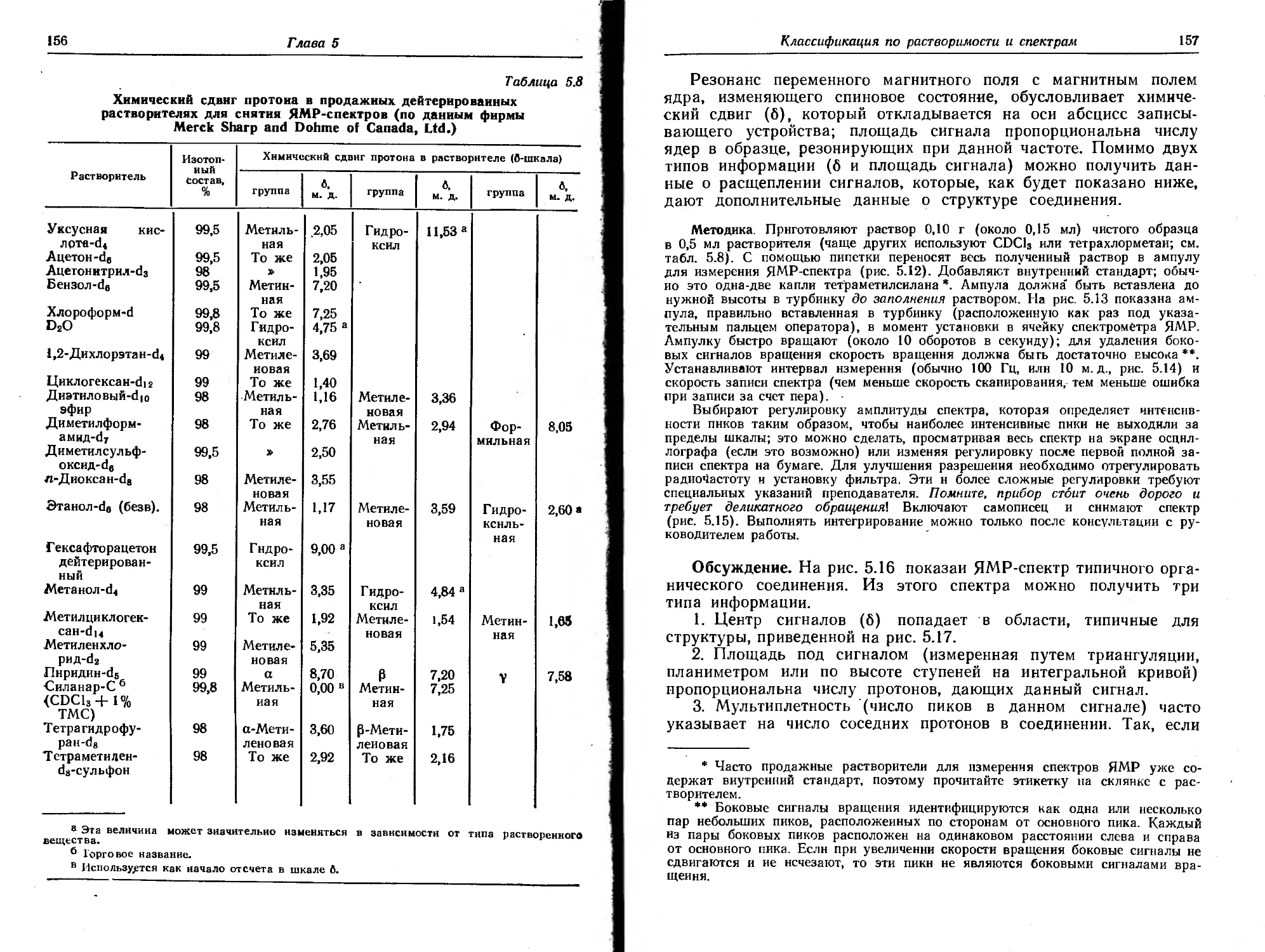


























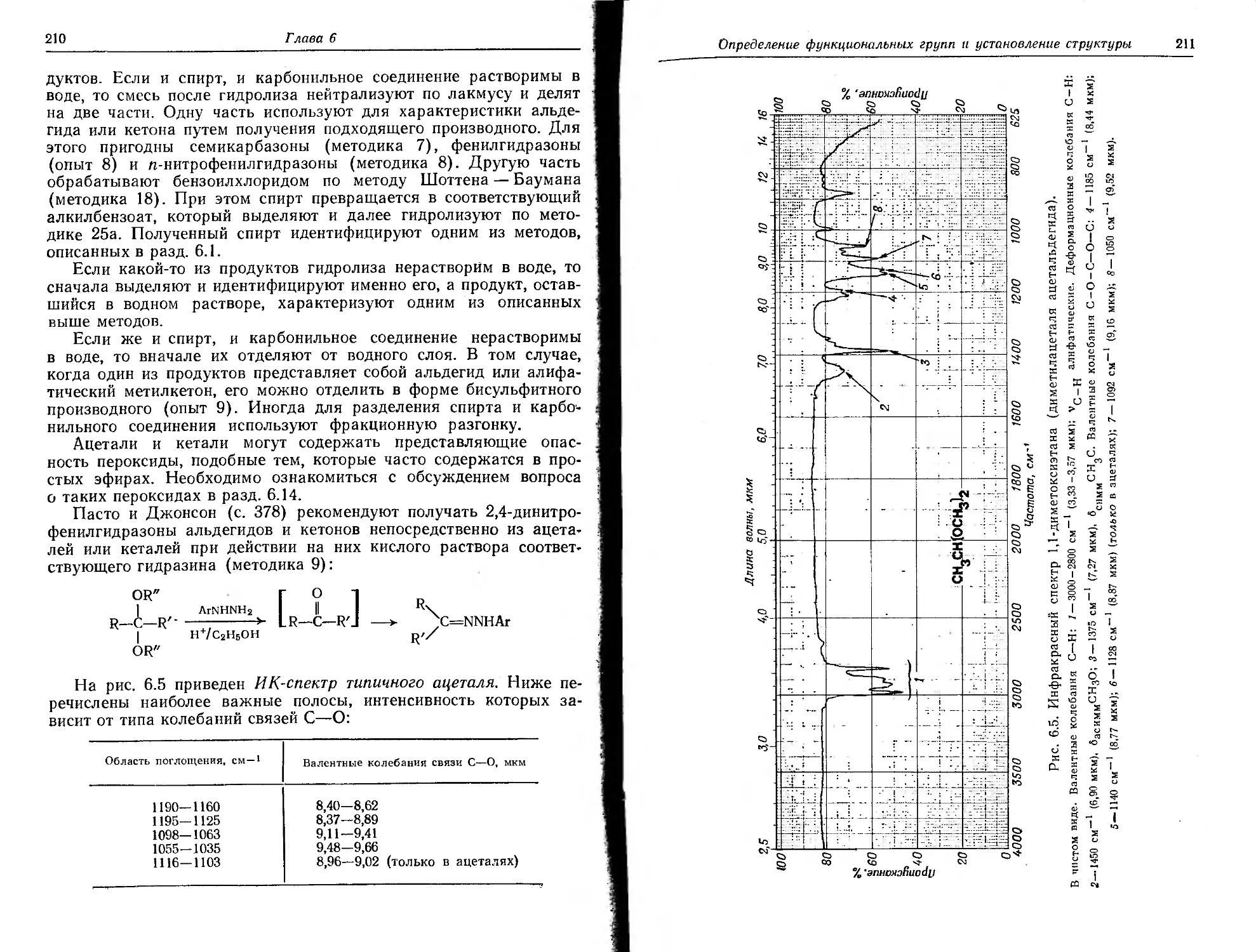






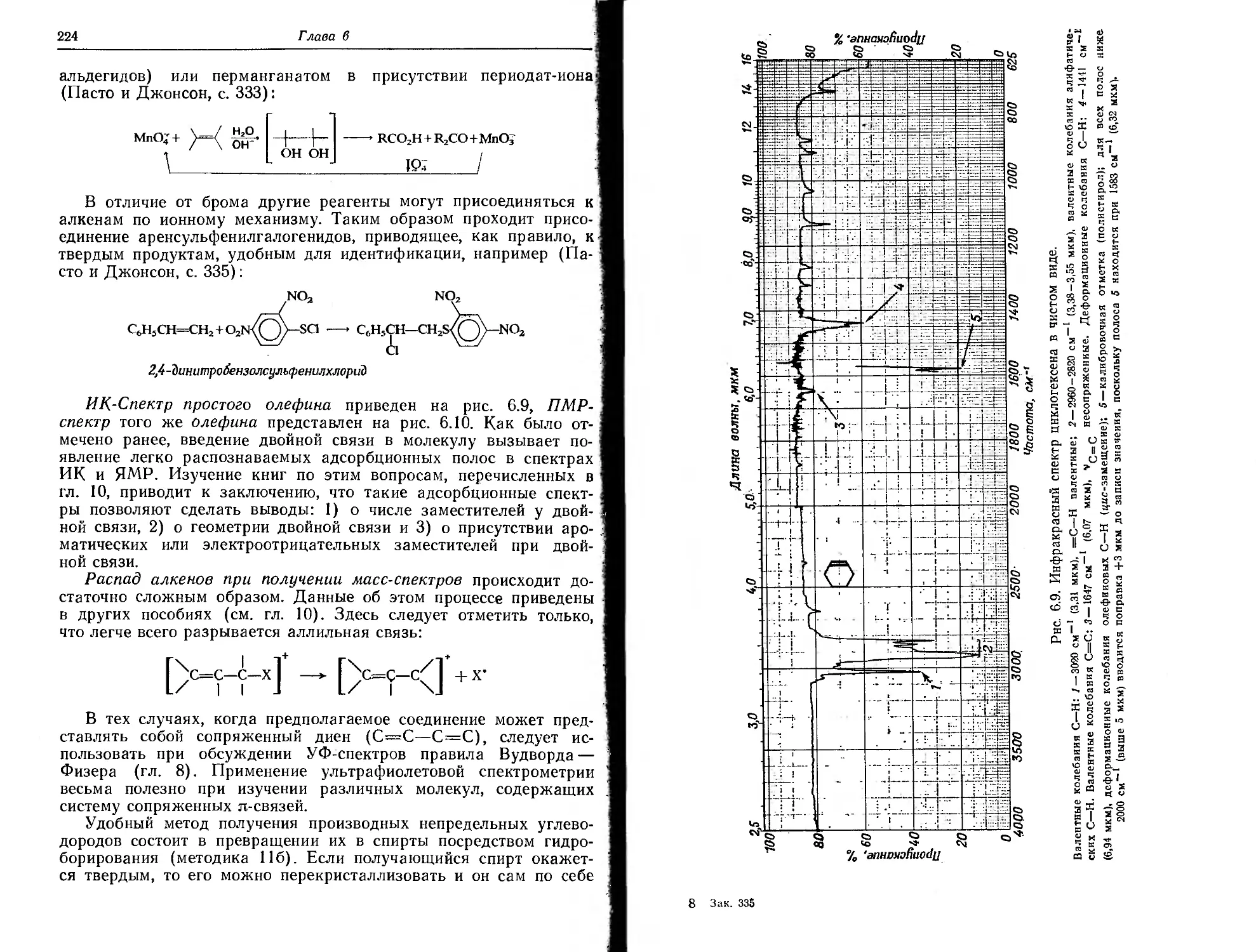















































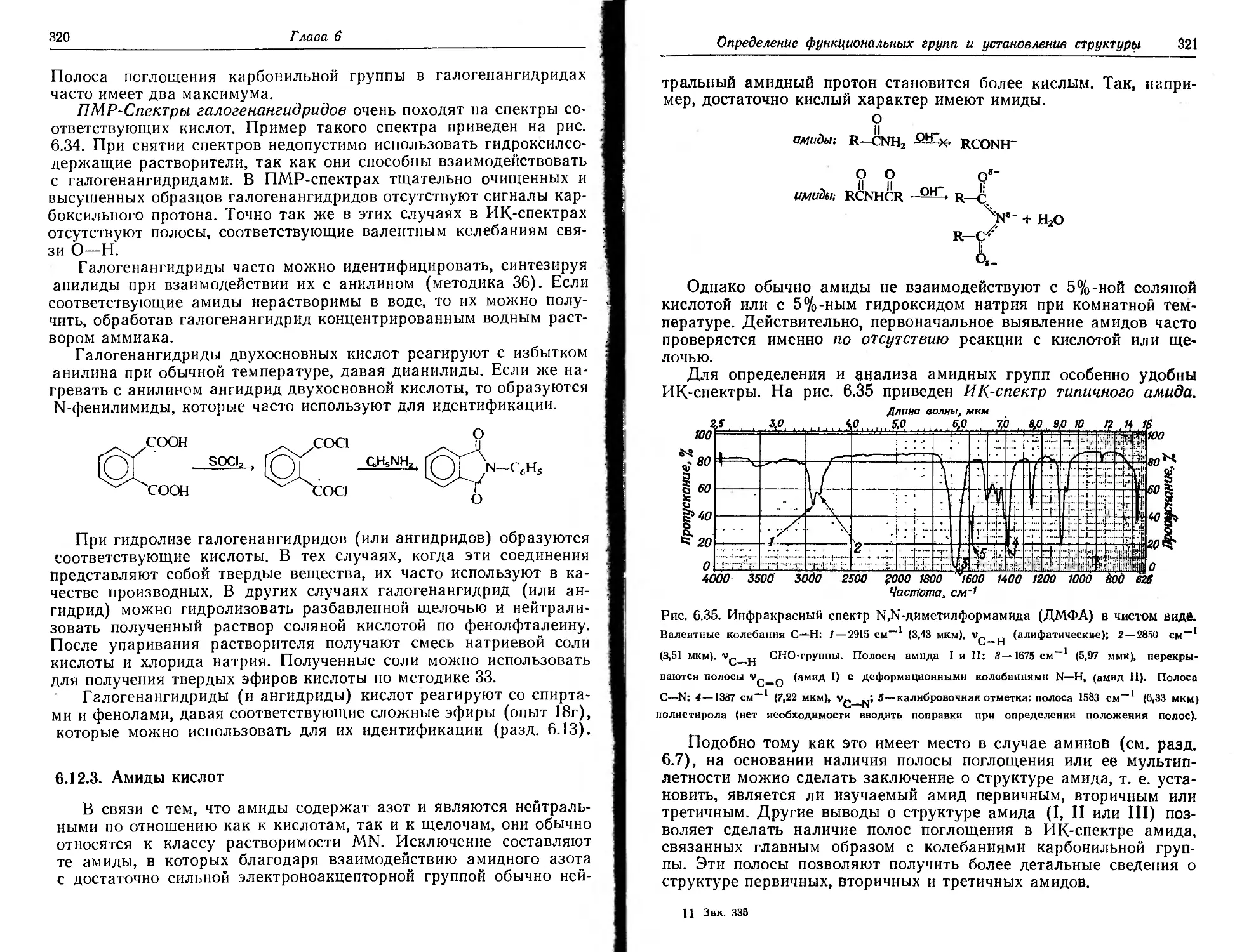




























































































































































































Текст
THE SYSTEMATIC
IDENTIFICATION
OF ORGANIC COMPOUNDS
A LABORATORY MANUAL
6th ed
RALPH L- SHRINER
Visiting Professor Chemistry
Southern Methodist University,
Dallas, Texas 75275
Formerly Professor and Head of the
Department of Chemistry
University of Iowa, Iowa City, Iowa
The late REYNOLD C. FUSON
Formerly Distinguished Visiting Professor
University of Nevada, Reno, Nevada 89507
Formerly Professor of Organic Chemistry and
Member of the Center for Advanced Study
University of Illinois, Urbana, Illinois
DAVID Y. CURTIN
Professor of Chemistry
University of Illinois
Urbana, Illinois 61701
TERENCE C. MORRILL
Professor of Chemistry
Rochester Institute of Technology
Rochester, N. Y. 14623
JOHN WILEY & Sons
New York Chichester Brisbane Toronto
Р. Шрайнер
Р Фьюзон
Д. Кёртин
Т.Моррилл
Идентификация
органических
соединений
Перевод с английского
канд. хим. наук С. С. Юфита
под редакцией
проф. Б. А. Руденко
Москва «Мир» 1983
Больше химической литературы и
прочих полезных материалов для
химиков на https://vk.com/chemzone
More chemistry books and other
useful resources for chemists are
available on https://vk.com/chemzone
СНВйПИЕ
vk.com/chemzone
ББК 24.23
И 29
УДК 5474-643
Идентификация органических соединений: Пер. с англ./
И29 Р. Шрайнер, Р. Фьюзон, Д. Кёртин, Т. Моррилл. — М.:
Мир, 1983. — 704 с., ил.
Книга американских авторов представляет собой лабораторное руководство по
систематической идентификации органических соединений. Отражает все современ-
ные достижения в области исследования органических соединений. Как полный и
необходимый в повседневной работе справочник будет настольной книгой в любой
синтетической лаборатории.
1803000000-080
И 041 (01)-83
86-83,
ч.1
ББК 24.23
547
Редакция литературы по химии
аь-ччы-з.-,
ГОС. ПУБЛИЧНАЯ
БИоЛ'-ОТ J КА
Ji-. : рат
ОЭ
• Copyright 1935, 1940, 1948 by
Ralph L. Shriner and Reynold
C. Fuson
• Copyright © 1956, 1964, 1980 by
John Wiley & Sons, Inc,
All Rights Reserved. Authorized
translation from English language
edition published by John Wiley &
Sons, Inc
© Перевод на русский язык, «Мир», 1983
ПРЕДИСЛОВИЕ РЕДАКТОРА ПЕРЕВОДА
Предлагаемая читателю книга Р. Шрайнера, Р. Фьюзона,
Д. Кёртина и Т. Моррилла «Идентификация органических соеди-
нений» издается на русском языке во второй раз. Первое издание
книги, написанной Шрайнером и Фыозоном, было переведено на
русский язык и выпущено Издательством иностранной литературы
в 1950 г. под названием «Систематический качественный анализ
органических соединений» и долгое время пользовалось призна-
нием химиков-органиков, встречающихся в своей практике с про-
блемой идентификации неизвестных органических веществ. Однако
за тридцать лет со времени выхода в свет этой книги произошли
весьма значительные изменения в методическом оснащении орга-
нической химии. Помимо классических методов исследования со-
става смесей и строения индивидуальных веществ, сохраняющих
и поныне свое значение, появились такие мощные методы, как
масс-спектрометрия органических соединений, методы спектроско-
пии ядерного магнитного резонанса на протонах, ядрах углеро-
да-13, фтора, фосфора, бора и других. Обычными даже для рядо-
вой органической лаборатории стали приборы для спектрометрии
в ультрафиолетовой и инфракрасной областях спектра.
Особое значение для органической химии имело бурное раз-
витие методов анализа и разделения вещества, базирующихся' на
принципах хроматографии, открытой выдающимся русским ученым
М. С. Цветом. Совершенно исключительное значение этого от-
крытия для развития экспериментальной техники во всех направ-
лениях химической науки ныне признается научной обществен-
ностью всего мира. Развитые на его основе методы газожидко-
стной, тонкослойной и жидкостной хроматографии высокого
разрешения внесли поистине революционные преобразования в
мир химической лаборатории, позволяя в считанные часы решать
такие исследовательские задачи, которые раньше требовали мно-
гих лет упорного труда.
Эти выдающиеся достижения экспериментальной техники, одна-
ко, не умаляют значения химических методов изучения вещества,
что требует развития у новых поколений исследователей соответ-
ствующего химического мышления, позволяющего ученому твор-
чески сочетать возможности физико-химических методов с осо-
бенностями химического поведения изучаемых веществ.
в
Предисловие редактора перевода
Задачу систематического подхода к этой проблеме и решает
предлагаемая читателю книга.
В ней поставлена задача систематического подхода к пробле-1
ме идентификации неизвестного или нового органического соеди-
нения на основе комплексного изучения его физических и химиче-
ских свойств. Основное назначение книги — служить учебным по-
собием для студентов высших учебных заведений, полезным при
изучении курса органической химии и при переходе к выполнению
самостоятельных исследовательских задач. В соответствии с этим
и отобраны вопросы, освещаемые в книге. Авторы последователь-
но описывают процесс выяснения строения нового или неизвестно-
го органического вещества, начиная с простейших операций опре-
деления чистоты химического соединения, его физических кон-
стант и молекулярной формулы и кончая выявлением природы
присутствующих функциональных групп и установлением тонких
особенностей пространственного строения молекулы. <
На всех этапах процесса установления строения органическо-
го соединения предполагается применение наиболее передовых
методов исследования, таких, как газовая и жидкостная хромато-
графия, масс-спектрометрия, ИК- и УФ-спектрометрия, ядерный
магнитный резонанс на протонах и ядрах углерода-13.
Однако добрая треть посвящена химическим методам иденти-
фикации органических соединений, несущих те или иные функцио-
нальные группы. Этот раздел охватывает все наиболее важные
классы органических веществ, показывая широкие возможности
чисто химических методов изучения молекулярной структуры.
В целях систематизации анализа в книге используется анали-
тическая классификация органических соединений, основанная на
различиях их растворимости в воде, органических растворителях и
в водных растворах щелочей и кислот, согласно которой все ве-
щества подразделены на шесть основных групп. Более детальная
классификация позволяет разделить все многообразие органиче-
ских веществ на одиннадцать групп. Хотя такой подход к систе-
матизации анализа, может быть, и не оптимальный, он, тем не ме-
нее, создает основу для планомерного, целенаправленного поиска
в почти необъятном море органических соединений.
Большое внимание в книге уделено вопросам определения чи-
стоты вещества и разделения смесей нескольких соединений. Для
советских химиков будет интересным и полезным описание про-
стого и весьма эффективного метода хроматографического разде-
ления веществ в сухих колонках. Этот метод, достаточно широко
применяемый во многих зарубежных лабораториях, относительно
мало известен в СССР.
Большинство методов и приемов, применяемых для иденти-
фикации органических соединений, проверены авторами книги в
многолетней практике преподавания в высшей школе и описаны
с достаточной полнотой. Во многих случаях авторы не ограничи-
Предисловие редактора перевода
7
ваются описанием практических приемов работы и приводят ряд
теоретических сведений, изложенных на достаточно высоком
уровне.
С точки зрения преподавателя органической химии в высшей
школе очень полезны также приведенные в книге примерные
учебные планы, формы отчетов о лабораторных работах, задачи и
упражнения, списки литературных источников и многочисленные
таблицы свойств органических соединений. Последние могут так-
же оказать существенную помощь и в работе квалифицированных
химиков-органиков в исследовательских лабораториях.
Будучи руководством практического характера, данная книга
содержит подробные сведения о технике безопасности при работе
в химической лаборатории. Даются сведения о мерах первой по-
мощи при ожогах, травмах, отравлениях. Имеются достаточно
подробные списки ядовитых веществ, а также противоядий и спо-
собы их применения. Большое внимание к вопросам лабораторной
техники безопасности несомненно является достоинством обсуж-
даемой книги.
Все перечисленные особенности книги позволяют надеяться,
что настоящее пособие будет интересным и полезным достаточно
широкому кругу читателей-химиков, студентов и аспирантов в
высших учебных заведениях, в научно-исследовательских учреж-
дениях и в аналитических лабораториях промышленных пред-
приятий.
Б. Руденко
Посвящение
Рейнольд К. Фьюзон (
(1 июня 1895 — 4 августа 1979)
Мы посвящаем настоящее издание нашему
соавтору Рейнольду К. Фьюзону, скончавше-
муся летом 1979 года. Боб Фьюзон, как назы-
вали его многочисленные друзья, был выдаю-
щимся химиком-органиком, известным своими
оригинальными исследованиями. Кроме того,
он написал несколько книг. В течение всей
многолетней научной деятельности он оказал
неоценимую помощь многим студентам и
аспирантам.
ПРЕДИСЛОВИЕ АВТОРОВ
В течение 43 лет, истекших с момента публикации первого из-
дания этого учебного пособия, появились многочисленные новые
методы разделения, очистки, идентификации и полной характери-
стики органических соединений. Однако наша первоначальная
цель — облегчить изучение основ органической химии — осталась
неизменной. Кроме того, мы полагаем, что органическую химию
ныне следует преподавать с использованием упомянутых новых
методов. В определенной степени те же методы будут использовать
и химики, уже работающие в науке и на производстве, при повы-
шении своей квалификации. Наконец, мы считаем себя обязанны-
ми ознакомить широкие круги химиков с многочисленными опуб-
ликованными работами, которые могут быть использованы либо в
Качестве дополнений к настоящей книге, либо как самостоятель-
ные источники информации.
Главной темой настоящей книги остается развитие методов, по-
зволяющих охарактеризовать и идентифицировать «неизвестное»
органическое соединение. Поэтому читатели получают системати-
ческие указания по идентификации веществ методами, уже опи-
санными в литературе по органической химии. Часто те же про-
цедуры могут быть использованы для работы с такими вещества-
ми, которые ранее не были получены или описаны.
Данная книга написана таким образом, что по качественному
анализу органических соединений, она может быть как учебным
пособием, так и промежуточным звеном для перехода к научно-
исследовательской работе. Это означает, что читателями этой книп
среди прочих будут студенты старших курсов и аспиранты, а по-
этому в ней должны быть рассмотрены наиболее сложные методы,
которые могут применяться для идентификации неизвестных ве-
ществ. С этой целью в книге сохранено рациональное зерно опы-
та преподавания материалов, связанных с проблемой идентифика-
ции неизвестных соединений, которое дополнено более подробным
Предисловие авторов
о
обсуждением различных методов, имеющихся в распоряжении
химика-органика. Эти методы следует обсуждать независимо от
того, что в лабораторных занятиях по первоначальному курсу ор-
ганической химии они могут еще и не использоваться. Представ-
ляется важным, чтобы студенты были осведомлены о том, каким
образом осуществляется идентификация неизвестных соединений
в научно-исследовательских лабораториях и в промышленности.
Эти сведения могут быть приобретены без ущерба для тех знаний,
которые студенты получают при изучении основ органической
химии. Например, может оказаться необходимым объяснить студен-
там, что, хотя масс-спектрометрия и представляет собой исключи-
тельно мощный аналитический метод, она не может быть исполь-
зована для повседневных анализов во всех студенческих лабора-
ториях. Поскольку вопросы качественного анализа начинают
освещаться уже на начальных этапах изучения органической
химии, то мы полагаем, что настоящая книга сможет быть весьма
полезной также и для студентов младших курсов.
Общие направления изложения аналитических методов в
гл. 3—6 кратко охарактеризованы в гл. 2. Особенно полезны
приведенные в разд. 2.6 формы представления результатов. После
того как студент приобретет некоторую информацию о неизве-
стном веществе (например, о его растворимости, содержании в
нем тех или иных элементов, данные его ПК- или ЯМР-спектров,
описанные в гл. 3—5), он сможет определить строение этого со-
единения путем детального анализа ПК- и ЯМР-спектров с вы-
полнением подтверждающих химических реакций (гл. 6).
Классифицикационные химические реакции («мокрые» реак-
ции), детальный анализ ПК- и ЯМР-спектров и получение про-
изводных объединены в одной большой по объему гл. 6. Изложе-
ние материала в этой главе проведено на основе функциональных
групп. При этом последовательно описываются ИК- и ЯМР-спек-
тры, химические («мокрые») реакции и техника получения произ-
водных для каждого большого класса органических соединений
(спиртов, кетонов и т. д.).
Таким образом, суммируя изложенное выше, можно утвер-
ждать, что при выполнении операций, описанных в гл. 3—5, сту-
дент сможет с большой вероятностью определить ряд возможных
структур «неизвестного» соединения. Дальнейшие аналитические
процедуры и обсуждение полученных данных, описанные в гл. 6,
должны дать возможность студенту определить, какая из предпола-
гаемых структур является истинной, и доказать это утверждение
так, чтобы не оставалось и тени сомнения.
В гл. 7 описаны некоторые операции, оказывающиеся полез-
ными при разделении органических соединений. Рассмотренные
в предыдущих изданиях стандартные приемы экстракции рас-
творителями с применением, например, кислот и щелочей допол-
нены описанием различных видов хроматографической техники
10
Предисловие авторов
(жидкостной, тонкослойной, газовой и т. д.). Можно рекомендо-
вать студентам ознакомиться с соответствующими разделами этой
главы в любой момент, когда преподаватель сочтет это целесо-
образным. В гл. 8 описаны некоторые специальные виды аналити-
ческой техники (например, ЯМР на ядрах 13С, дисперсия оптиче-
ского вращения и т. п.). Эти приемы могут быть применены при
Изучении некоторых особых неизвестных веществ. Возможно так-
же использование этого материала особо подготовленными сту-
дентами, желающими попробовать свои силы в более сложных за-
даниях.
В гл. 9 включены все связанные с выбором «путей» анализа
задачи и примеры, использованные в предыдущем издании. В не-
которых случаях добавлены еще спектральные данные. В гл. 10
обсуждаются литературные источники, которые могут быть полез-
ными при изучении органической химии как для установления
структуры неизвестных веществ, так и для решения более общих
исследовательских задач.
В настоящем издании сохранены таблицы температур плавле-
ния и кипения, а также указатели, получившие признание чита-
телей в предыдущих изданиях. Эти таблицы существенно рас-
ширены. Кроме того, приведены таблицы других данных, которые
могут быть полезны в практической работе органических лабора-
торий (таблицы растворителей для хроматографирования
и т. п.).
В книге сохранено описание большинства процедур предвари-
тельной характеристики вещества, опубликованных в предыдущих
изданиях (определение температур плавления и кипения, выясне-
ние характера растворимости и т. п.). Однако при обсуждении этих
операций описаны также соответствующие наиболее современные
приемы (например, проверка чистоты веществ с помощью тонко-
слойной хроматографии и др.). Раздел о качественном элементном
анализе (путем сплавления с натрием) дополнен описанием ис-
пользования масс-спектрометрии и других новейших методов од-
новременно для качественного и количественного анализа. Мы ре-
комендуем определять молекулярную массу веществ с помощью
описанных в настоящей книге методов масс-спектрометрии или
осмометрии в паровой фазе вместо приведенного в предыдущих
изданиях метода Раста, основанного на измерении понижения тем-
пературы замерзания. Этот метод слишком часто приводит к не-
удачным результатам. В соответствии с многочисленными поже-
ланиями читателей в настоящем издании группы растворимости
вновь обозначены буквами латинского алфавита (Si, S2, Aiiit. д.),
как и в четвертом издании. Кроме того, характеристики раство-
римости дополнены указаниями об отношении к органическим
растворителям. Это приводит к результатам, полезным для спек-
трального анализа, хроматографического анализа и для перекри-
сталлизации.
Предисловие авторов
И
ИК-Спектрометрия и метод ЯМР включены в общую схему
идентификации веществ. В гл. 5 изложена техника получения
ИК- и ЯМР-спектров, а в гл. 6 рассказано, каким образом со-
отнести эти спектры со строением вещества и с результатами про-
веденных с ним химических реакций. Особое внимание уделено
изучению ИК- и ЯМР-спектров и корреляции полученной при этом
информации со строением веществ. Теория методов ИК- и ЯМР-
спектроскопии излагается в учебниках по органической химии и
инструментальному анализу, к которым и отсылаются интересую-
щиеся этими вопросами читатели.
Для всех химиков являются исключительно важными вопро-
сы техники безопасности в лаборатории. В гл. 1, представляющей
собой очень краткое введение к настоящей книге, подробно об-
суждены способы оказания первой помощи при несчастных слу-
чаях. Описаны также токсические свойства употребляемых в ла-
бораторной практике реагентов и приведены ссылки на более де-
тальные руководства по технике безопасности. Во многих местах
книги подчеркнуто наличие ядовитых или опасных в других отно-
шениях обсуждаемых веществ, в том числе раздражающих, канце-
рогенных и взрывчатых.
В последние годы развернулась широкая дискуссия по вопро-
су о токсических свойствах бензола. Фактически во многих ла-
бораториях, где может быть использована настоящая книга, упо-
требление бензола запрещено. Было предложено применять в ка-
честве заменителя бензола толуол. Необходимо подчеркнуть, что
вследствие несколько различной растворяющей способности этих
растворителей многие процедуры, легко выполнимые с бензолом,
при использовании толуола проходят гораздо труднее (напри-
мер, медленнее) или не могут быть осуществлены вообще. Кроме
того, при этом следует учитывать токсичность и самого толуола.
В целом следует проявлять осторожность при использовании обоих
названных выше ароматических углеводородов, во всех случаях
работая под вытяжными шкафами и тщательно избегая попадания
этих растворителей на кожу.
В связи со сложностями, связанными с техникой безопасности
и применением опасных для здоровья веществ, в настоящую кни
гу включены сведения о максимально допустимом времени воз-
действия, утвержденные OSHA. Авторы настоящей книги пола-
гают, что каждое научное учреждение должно располагать
сведениями о ныне существующих органичениях, чтобы иметь воз-
можность учесть их в своей работе.
Основная работа по подготовке настоящего издания была вы-
полнена Теренсом С. Морриллом. Главной заслугой Ральфа
Л. Шрайнера было представление хорошо проверенных и улуч-
шенных методик химических реакций. Кроме того, вследствие его
многолетнего преподавательского опыта и тесных связей с органи-
ческой химией и качественным анализом его советы оказали
12
Предисловие авторов
неоценимую помощь для определения общего направления настоя-
щего издания. Весьма значительная часть рукописи этой книги
была подготовлена в период пребывания Теренса С. Моррилла в
качестве приглашенного профессора в Рочестерском университете.
В этой связи надо отметить теплое гостеприимство химиков Ро-
честерского университета и ряд внесенных ими конструктивных
советов и предложений.
В подготовку настоящего издания внесли свой вклад многие
промышленные фирмы. Авторы хотели бы выразить особую бла-
годарность следующим из них: Асе Glass Со., Perkin-Elmer Corp.,
Varian Associates, Sadtler Laboratories и Waters Associates.
Авторы выражают свою признательность перечисленным ниже
химикам, пожертвовавшим свое время и поделившимся своими
идеями для этого издания: С. Ф. X. Аллену, Уильяму Биглеру,
Роберту Э. Джилмену и Томасу П. Уоллесу из Рочестерского
технологического института, Льюису Фрейдриху и Джеку Кэмп-
майеру из Рочестерского университета, Уильяму Клоссону
(SUNY-Albany), Р. Э. С. Смиту (Мичиганский университет) и
Дэвиду Страку (Waters Associates). В подготовке и перепечатке
рукописи и иллюстраций оказали большую помощь Джойс Бен-
нетт, Бэкки Дэвис, Сью Хабрегсон, Черил Кэйн, Гэйлин Моррилл
и Шарон Вальер. Т. С. Моррилл хотел бы выразить особую при-
знательность своей жене Гэйлин Моррилл и редактору Гэри Карл-
сону за их терпение, проявленное при подготовке рукописи на-
стоящей книги.
В заключение авторы выражают надежду, что предлагаемая
книга будет не только учебником в области качественного ана-
лиза органических соединений, но и полезной настольной спра-
вочной книгой. Авторы заранее выражают признательность за
все замечания и предложения. Подготовка этого издания была
существенно облегчена в результате непрерывного и достаточно
широкого обзора всех имеющихся данных. Внесение любых изме-
нений в следующее издание этой книги будет возможным лишь
в том случае, если авторы и впредь будут осведомлены о мнениях
и суждениях ее читателей.
Р. Шрайнер
Т. Моррилл
ПРЕДИСЛОВИЕ АВТОРОВ К ПЕРВОМУ ИЗДАНИЮ
В последние двадцать пять лет возросла популярность курсов
лабораторных работ, направленных на обучение методам иденти-
фикации органических соединений. Это направление было зало-
жено Малликеном, классический труд которого «Идентификация
чистых органических соединений» был издан в 1904 г. После это-
го было опубликовано несколько книг по этому вопросу, что при-
вело к широкому распространению систематической идентифика-
ции органических соединений в программах высших учебных за-
ведений.
В настоящее время общепризнана важность такого курса в
программе подготовки квалифицированных химиков. Однако уме-
ние идентифицировать то или иное вещество, само по себе весьма
ценное для химика-органика, не является основной причиной боль-
шой популярности курсов лабораторных работ по этой теме. Су-
щественное отличие этого направления от других циклов лабора-
торных работ, обычно входящих в программу обучения химии,
заключается в том, что до настоящего времени отсутствует ка-
кая-либо схема, позволяющая свести работу студента к простому
выполнению определенных инструкций. На каждой стадии процес-
са идентификации веществ известными ныне методами студент
должен иметь свое собственное суждение. Достоинством таких цик-
лов лабораторных работ является то, что они позволяют студенту
проявить и развить способность к тщательным наблюдениям, сде-
лать правильные выводы из этих наблюдений и проявить свой
индивидуальность при планировании работы. С этой точки зрений
очевидно, что такой способ обучения представляет наилучшие воз-
можности студентам, готовящимся к научно-исследовательской
работе. В ходе таких занятий студенты не только получают пред-
ставление о необходимости проведения научных исследований, но
и знакомятся с методами, применяемыми в таких работах.
Естественным и важным дополнительным результатом такого
интереса к методам идентификации веществ служит большое чис-
ло исследований, выполненных в данной области в последнее вре-
мя и направленных, в частности, на получение производных,
которые удобны для того, чтобы охарактеризовать и идентифици-
ровать изучаемые вещества. В результате столь интенсивной ра-
боты основной материал таких курсов постоянно нуждается в об-
новлении.
14
Предисловие авторов к первому изданию
Настоящая книга обобщает опыт, накопленный в течение не-
скольких лет педагогической и исследовательской работы в области
идентификации органических соединений. Интерес к этой работе
в Университете Иллинойса возник по инициативе проф. С. Г. Дер-
рика, который первым ввел преподавание соответствующего курса
в' 1908 г. В дальнейшем в развитии этого курса участвовал проф.
Оливер Камм, чей отличный учебник по этому вопросу был опу-
бликован в 1922 г. Введенные тогда лабораторные работы сохра-
нились в Университете Иллинойса и в настоящее время и прово-
дятся в течение одного семестра в форме двух трехчасовых лабо-
раторных занятий в неделю. Выполняемые при этом работы имеют,
однако, такой характер, что они могут быть легко совмещены как
с более короткими, так и с более длительными сроками обучения
путем изменения числа неизвестных веществ, даваемых для иден-
тификации. Обсуждаемый курс рассчитан на студентов, изучав-
ших органическую химию в течение одного учебного года.
При подготовке настоящей книги были использованы многие
методы, которые могут быть найдены в нескольких работах сход-
ного содержания. Основными источниками среди них являлись
следующие книги: Mulliken, «Identification of Pure Organic
Compounds», Kamm, «Qualitative Organic Analysis», Clarke,
«Handbook of Organic Analysis», Staudinger, «Introduction to
Qualitative Organic Analysis». Portes, Stewart, Branch, «Methods
of Organic Chemistry», Bargellini, «Esercizi numerici di chimica
organica», за что авторам этих работ нельзя не выразить глубокую
признательность. Авторы благодарны также многим преподавате-
лям Университета Иллинойса и других высших учебных заведений
за новые приемы и методы, включенные в настоящую книгу. В про-
цессе подготовки рукописи постоянную и неоценимую помощь ока-
зал проф. К. С. Марвел. Авторы рады сообщить о многих ценных
предложениях, внесенных профессорами Джоном Р. Джонсоном,
А. У. Ингерсолом, С. М. Мак-Илвейном, Г. X. Кольманом, Уолле-
сом Р. Бродом, Ральфом Коннором и С. Ф. X. Алленом. Наконец,
авторы должны выразить особую признательность за большую по-
мощь многим студентам Университета Иллинойса, которые
пользовались данной книгой и явились окончательными судьями,
оценившими ее особенности.
Урбана, Иллинойс
сентябрь 1935
Р. Шрайнер
Р. Фьюзон
Глава 1
Введение
1.1. ИДЕНТИФИКАЦИЯ ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ,
РАНЕЕ ОПИСАННЫХ В ХИМИЧЕСКОЙ ЛИТЕРАТУРЕ
Характеристики углеродсодержащих веществ, выделяемых из
живых организмов (например, из растений и животных), из горю-
чих ископаемых (каменного угля, нефти, природного газа, торфа,
лигнита), а также синтезированных в лаборатории, публикуются
в химических журналах в течение более чем 150 лет. К середине
70-х годов число охарактеризованных таким путем соединений уже
превышало 5 млн. Каждый год это число увеличивается еще на
несколько тысяч.
Химик, которому надо установить природу имеющегося у него
некоего вещества, конечно, не может располагать всеми опублико-
ванными сведениями об известных соединениях с тем, чтобы сопо-
ставить с ними свойства «неизвестного» материала. Поэтому весь-
ма существенно наличие систематического подхода к этой пробле-
ме. Такой подход должен позволить с самого начала исключить
возможно большее число структур. Далее число возможных струк-
тур должно быть сведено к минимуму (допустим, к трем-четырем).
Наконец, из этих возможностей должна быть выбрана и оконча-
тельно подтверждена одна структура, отвечающая строению опре-
деляемого вещества.
Совершенно ясно, что преподаватель качественного органиче-
ского анализа не может располагать 5 млн. веществ. Обычно в лю-
бой химической лаборатории в высшем учебном заведении, в
научно-исследовательском институте или на промышленном пред-
приятии имеется лишь ограниченное число веществ. Так, например,
каталог фирмы Eastman насчитывает около 4000 наименований,
а фирма Aldrich Chemical Со. поставляет около 9000 веществ. Мно-
гие другие химические фирмы могут обеспечить получение лишь
100—1000 химических соединений: В химической промышленности
производится от 6000 до 10 000 различных веществ. Таким образом,
список относительно доступных химических веществ охватывает
значительно меньше 5 млн. соединений.
В настоящей книге основное внимание сосредоточено на еще
более ограниченном круге веществ, которые могут быть исполь-
зованы в процессе обучения в качестве «неизвестных». Таблицы
температур плавления и кипения веществ, приведенные в этой кни-
ге, позволяют получить достаточно ясное представление о ее
16
Глава 1
направленности. Преподаватели, пользующиеся этой книгой, конеч-
но могут для расширения круга возможных «неизвестных» объектов
применять и другие источники (например, справочники CRC *, ка-
талог фирмы Aldrich Chemical Со. и т. п.).
Наиболее часто химик-органик сталкивается с одной из двух
следующих проблем:
1 Определение строения ранее не изучавшегося вещества. Ча-
сто такая ситуация возникает при изучении природных соединений,
когда бывает необходимо исследовать вещество, выделенное в
очень малом количестве из растительного или животного мате-
риала.
2 . Как в учебной лаборатории, так и на производстве возни-
кает необходимость проанализировать пробу, содержащую в пре-
обладающем количестве ожидаемые продукты, а также примеси,
наличие которых можно предполагать на основании имеющихся
сведений об исходных реагентах и условиях протекания той или
иной химической реакции. Весьма возможно, что такая проба, про-
исхождение которой хорошо известно, позволит исследователю за-
ранее составить ясное представление о том, каким образом про-
вести ее анализ.
1.2. ВЗАИМОСВЯЗЬ ИЗУЧЕНИЯ МЕТОДОВ
ИДЕНТИФИКАЦИИ ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
С НАУЧНЫМИ ИССЛЕДОВАНИЯМИ
В ОБЛАСТИ ОРГАНИЧЕСКОЙ ХИМИИ
Изучение теории и техники идентификации органических сое-
динений— весьма существенный этап для перехода к научно-ис-
следовательской работе в области органической химии. Курс орга-
нического анализа позволяет накопить и систематизировать зна-
ния о физических свойствах, строении и химической реакционной
способности нескольких тысяч углеродсодержащих соединений,
причем изучение этого материала следует систематической и ло-
гичной схеме процесса идентификации. Хотя первоначальная цель
такого курса — характеристика уже известных соединений, общий
подход к этой проблеме остается тем же и на первых этапах уста-
новления структуры вновь синтезированных органических соеди-
нений.
Так, например, если два известных вещества А и В растворены
в растворителе С и к раствору добавлен катализатор D, то из по-
лученной смеси в соответствующих условиях реакции может воз-
никнуть смесь новых продуктов, содержащая и неизмененные ис-
ходные соединения.
* Например, Handbook of Tables for Organic Compounds Identification, 3rd
ed., Z. Rappoport (cd.), CRC Press, Cleveland, Ohio, 1967.
Введение
17
(2) процессы идентификации
При этом сразу же возникают две новые проблемы:
1. Какие процедуры следует выполнить для того, чтобы раз-
делить полученную смесь на составляющие ее компоненты?
2. Каким образом ’ идентифицировать с полной определенно-
стью индивидуальные компоненты Е—К? Какие из этих компо-
нентов представляют собой непрореагировавшие исходные ком-
поненты? Какие из полученных соединений уже были описаны ра-
нее? Наконец, какие из продуктов впервые получены?
Обычно эти две проблемы тесно связаны между собой. При раз-
делении смесей органических соединений используют как химиче-
ские, так и физические процессы в зависимости от структуры со-
ставляющих компонентов.
В настоящем курсе прежде всего рассмотрен процесс система-
тической идентификации индивидуальных соединений. Отдельные
стадии этого процесса изложены в гл. 2. В дальнейшем в гл. 7 по-
казано применение этих принципов для создания эффективной по-
следовательности операций разделения смесей. Методы, применяе-
мые для этих целей в лабораторной практике, и обсуждение прин-
ципов отдельных этапов идентификации веществ изложены в гл.
3-6.
1.3. НЕКОТОРЫЕ УКАЗАНИЯ ДЛЯ СТУДЕНТОВ
И ПРЕПОДАВАТЕЛЕЙ
Расписание занятий. Точное расписание занятий, применяемое
для всех высших учебных заведений, установить невозможно. Это
связано с тем, что в разных учебных заведениях учебный год де-
лится по-разному — на семестры, триместры или четверти. В ряде
случаев занятия проводятся также и в летнее время. Однако для
семестра продолжительностью 15 недель целесообразно предусмо-
треть два трехчасовых лабораторных занятия и одну «лаборатор-
ную лекцию» в неделю. В конкретных условиях того или иного
учебного заведения предложенное выше расписание может быть
соответствующим образом модифицировано.
18
Глава 1
Лекционный материал. Опыты, методики (гл. 6) и инструкции
по применению инфракрасной спектроскопии и спектроскопии ядер-
ного магнитного резонанса (гл. 5 и 6) описаны таким образом, что-
бы каждый студент мог использовать их непосредственно в ходе
своей работы. В первой лекции целесообразно изложить общую
структуру данного курса, описанную в гл. 2. Понятно, что нет ника-
кой необходимости читать лекции по каким-либо специфическим
«рецептам», изложенным в книге (например, в гл. 6). Каждая сту-
денческая задача по расшифровке структуры неизвестного
вещества представляет собой самостоятельную исследователь-
скую работу, которая должна выполняться независимо от
других.
После того как будут выполнены одна или две задачи с неизве-
стными веществами, было бы полезно проработать на семинарских
занятиях некоторые задачи, приведенные в гл. 9, и обсудить со
студентами взаимосвязи строения веществ с их химическими реак-
циями и со спектральными данными.
Лабораторные работы. Неизвестные вещества. При использо-
вании спектральных данных и химических реакций студенты могут
выполнить в течение одного семестра продолжительностью 15 не-
дель 6—8 задач по расшифровке структуры индивидуальных ве-
ществ и две задачи по анализу смесей, в каждой из которых содер-
жится 2—3 компонента.
Для ускорения начального этапа работы и иллюстрации схе-
мы идентификации целесообразно предложить студентам в
качестве первого неизвестного вещества кислоту, которая может
титроваться щелочью. Студентам сообщают, что неизвестные ве-
щества могут титроваться. Задача обучаемых состоит в проведе-
нии элементого анализа, определении температур плавления или
кипения и эквивалента нейтрализации. На основе этих данных дол-
жно быть рассчитано возможное значение молекулярной массы
вещества. В других случаях студенты могут получать неизвестное
вещество, для которого известны результаты его масс-спектромет-
рического исследования. Далее, если неизвестное вещество содер-
жит галогены или азот, студент должен выбрать и провести две-
три (но не более) классификационные реакции. Затем он должен
составить перечень возможных веществ и их производных, руко-
водствуясь таблицей кислот, приведенной в приложении III. Одно
из этих производных должно быть приготовлено и включено в от-
чет о работе (разд. 2.11). Первая задача должна быть выполнена
в течение двух трехчасовых лабораторных занятий.
Неизвестные соединения, используемые в других заданиях,
должны позволить студенту накопить опыт анализа веществ, со-
держащих самые разнообразные функциональные группы.
Во многих случаях желательно контролировать работу студен-
тов после того, как они провели предварительные реакции, класси-
фикацию по растворимости и элементный анализ. Такую проверку
Введение
19
настоятельно рекомендуется проводить при выполнении первых од-
ной-двух задач.
Чистота неизвестных веществ. Необходимо принимать все меры
для обеспечения высокой степени чистоты проб. Тем не менее сту-
денты и преподаватели не должны забывать о том, что многие
органические соединения при хранении в течение длительного вре-
мени могут разлагаться или реагировать с кислородом, влагой или
углекислым газом, содержащимися в атмосферном воздухе. Такие
пробы могут иметь широкие диапазоны температур плавления или
кипения, которые обычно ниже, чем приводимые в литературе зна-
чения. Поэтому при работе с каждым из неизвестных веществ
студенты прежде всего определяют температуру плавления или ки-
пения пробы и сообщают о ней преподавателю. В случае необхо-
димости преподаватель должен рекомендовать студенту провести
дополнительную очистку исследуемого вещества путем перекри-
сталлизации или перегонки и вновь повторить определение сом-
нительных физических констант. Это позволит избежать потери
времени и нежелательных разочарований, связанных с получением
противоречивых результатов (см. также разд. 2.3).
Количества неизвестных веществ. В качестве общих рекоменда-
ций могут быть предложены следующие количества проб неизвест-
ных веществ:
Неизвестное вещество № 1: кислота, титруемая щелочью, нли вещество с
масс-спектральнымн данными — 4 г твердого вещества или 10 мл жидкости.
Неизвестное вещество № 2: 3 г твердого вещества нли 8 мл жидкости.
Неизвестное вещество № 3: 2 г твердого вещества или 5 мл жидкости.
Неизвестное вещество № 4: 1 г твердого вещества или 5 мл жидкости.
Смеси должны содержать по 4—5 г каждого компонента. Если
потребуется дополнительная очистка пробы, то студент должен по-
лучить соответственно большее количество вещества.
Указанные выше количества, по существу, соответствуют «мак-
роколичествам» образцов. При использовании таких аналитиче-
ских методов, как тонкослойная или газовая хроматография, раз-
мер проб может быть без особых опасений сокращен примерно до
20% указанных выше количеств. При работе с микроколичества-
ми образцов необходимо пересчитать и соответственно уменьшить
количества реагентов в прописях химических реакций и методик
получения производных, описанных в гл. 6.
В конце семестра, когда студенты приобретут достаточные на-
выки работы в химической лаборатории и смогут с большей лег-
костью интерпретировать результаты проводимых химических реак-
ций, можно использовать еще меньшие количества веществ при по-
лучении производных и выполнении классификационных реакций.
Однако при этом должны применяться полумикрометоды проведе-
ния ряда химических операций*. При этом должны быть
* Полумикрометоды подробно описаны в следующих пособиях: Cheronis N. D.,
Entrikin J. В., Semimicro Qualitative Organic Analysis, 2nd ed., Interscience
20
Глава 1
соответственно изменены методики и должна применяться аппа-
ратура для работы с полумикроколичествами. Такие приборы для
данного курса выпускаются фирмой Kontes Glass Со. в наборах,
называемых «Бантамское стекло».
Некоторые указания по вопросу экономии времени. Важно за-
ранее планировать лабораторную работу. Это позволит в течение
одного лабораторного занятия получить результаты элементного
анализа, физические константы, сведения о растворимости, ИК- и
ЯМР-спектры нескольких неизвестных веществ. Эти данные необ-
ходимо тщательно регистрировать в лабораторном журнале. Вече-
ром накануне следующего лабораторного занятия эти данные
следует просмотреть еще раз и попутно обсудить каждую выпол-
ненную операцию. Затем составляют список нескольких класси-
фикационных реакций, которые должны быть проведены, и осу-
ществляют их в лаборатории на следующий день. В некоторых
случаях полезно составить предварительный список возможных
структур изучаемого вещества и желаемых его производных. Ва-
жно отметить, что с любым данным веществом должны быть про-
ведены лишь несколько из 30 описанных в книге классификацион-
ных реакций. Обычно достаточно получить не более двух произ-
водных. Во многих случаях ограничиваются даже одним произ-
водным. Цель лабораторной работы состоит в том, чтобы наиболее
эффективно применить описанную в гл. 2 систематическую по-
следовательность операций.
Преподаватель должен так направлять работу студента, чтобы
правильная идентификация изучаемого вещества являлась бы ре-
зультатом процесса логического дедуктивного размышления. Как
только структура очередного неизвестного вещества установлена,
становится ясным и полным понимание сути тестовых реакций и
спектральных данных. На этом этапе на основании лабораторных
наблюдений студентам легче сделать правильные выводы о строе-
нии изучаемого вещества, если уже в начале курса их ознакомить
с гл. 9. Один из способов ускорить развитие способности делать
умозаключения такого рода состоит в том, что преподаватель пи-
шет на доске структурную формулу вещества и предлагает студен-
там предсказать характер растворимости этого соединения и вы-
брать подходящие классификационные реакции.
Для того чтобы связать проводимые в настоящем курсе зада-
ния по идентификации с настоящей научно-исследовательской ра-
ботой, преподаватель может выбрать несколько типичных приме-
ров природных соединений, таких, как никотин, D-рибоза, хинин,
пенициллин G или витамин Вь и рассмотреть реакции идентифика-
ции, достаточные для решения вопроса о строении этих соединений.
Publishers, New York, 1957; Cheronis N. D., Semimicro Experimental Organic Che-
mistry, John DeGraff, New York, 1958; Cheronis N. D., Ma T. S., Organic Fun-
ctional Group Analysis by Micro and Semimicro Methods, Wiley-Interscience, New
York, 1964.
Введение
21
Много примеров, демонстрирующих важность спектроскопии ПК,
УФ и ЯМР для установления структуры органических соединений,
можно найти в текущей литературе. Изучение механизмов реакции,
используемых в качестве классификационных реакций или приме-
няемых для получения производных, требует знания основных
функциональных групп и их электронной структуры.
Во всех разделах настоящей книги приводятся ссылки на ори-
гинальные статьи, монографии и справочники. В курсе, изучаемом
на протяжении одного семестра, будут использованы далеко не
все эти источники. Однако цитируемые материалы выбраны так,
чтобы студент мог ими воспользоваться на начальных этапах своей
будущей научной деятельности. Поэтому такие источники оказыва-
ются полезными при выполнении исследовательских работ на стар-
ших курсах и в ходе выполнения дипломных и аспирантских ра-
бот.
Пользование настоящей книгой существенно облегчается, если
составить справочные таблицы по каждой главе и по отдельным
разделам глав. Время, затраченное на их подготовку, окупается с
лихвой, поскольку эти таблицы позволяют быстро найти в книге
описание опытов по установлению функциональных групп, мето-
дик получения производных и таблиц их свойств.
На всех этапах работы в лаборатории студенты и преподава-
тели должны соблюдать правила техники безопасности. В лабора-
тории необходимо всегда носить защитные очки и знать меры ока-
зания первой помощи при несчастных случаях.
1.4. ТЕХНИКА БЕЗОПАСНОСТИ В ХИМИЧЕСКОЙ
ЛАБОРАТОРИИ И МЕРЫ ОКАЗАНИЯ
ПЕРВОЙ ПОМОЩИ
Химическая лаборатория представляет собой одно из наиболее
опасных рабочих мест, особенно в тех случаях, когда не уделяет-
ся должное внимание вопросам техники безопасности. В данном
разделе изложены некоторые меры обеспечения безопасности, к ко-
торым необходимо прибегать в тех случаях, когда нет времени об-
ратиться за помощью вне лаборатории.
Весьма важно знать, где помещаются лица и организации, к
которым необходимо обращаться при несчастных случаях, требую-
щих квалифицированной помощи. Преподаватель в лаборатории
должен знать телефоны следующих служб экстренного вызова:
скорой помощи, пожарной команды, врача, отделения милиции,
пункта первой помощи при отравлении, Красного Креста и поли-
клиники.
В химической лаборатории должно быть наготове следующее
оборудование для предупреждения и ликвидации несчастных слу-
чаев: пожарная кошма, огнетушитель, кран-фонтанчик для промы-
вания глаз, аварийный душ, аптечка первой помощи, промывные
22
Глава 1
Таблица 1.1
Некоторые часто встречающиеся яды и симптомы отравления ими
Кислоты и щелочи
Алкоголь
Цианиды
Цианины и моноок-
сид углерода (угар-
ный газ)
Сероводород
Свинец
Ртуть
Метиловый спирт
(метаиол, древес-
ный спирт)
Феиилгидразии
Ппретрин
При попадании на кожу и слизистые оболочки вызы-
вают раздражение и ожоги, могут вызывать пробо-
дение желудка
Сильный депрессант центральной нервной системы
Даже в незначительных дозах вызывают потерю созна.-
ния и быструю смерть в результате паралича дыха-
тельного центра. Цианиды могут попадать в организм
через рот, при вдыхании, через раны и путем вса-
сывания через кожу. (Применяются в быту для уни-
чтожения моли и муравьев)
Вызывают смерть от асфиксии вследствие того, что
вступают в соединение с переносящей кислород си-
стемой крови и тем самым препятствуют поступле-
нию кислорода к жизненно важным органам орга-
низма человека
Горючий ядовитый газ с запахом тухлых яиц. Ощу-
щается обонянием при наличии в воздухе в концен-
трации 0,002 мг/л. Очень опасен: уже после одиого-
двух вдохов достаточной концентрации этого газа
может наступить потеря сознания, кома и смерть.
Сероводород является весьма коварным ядом, так
как при длительном вдыхании человек перестает чув-
ствовать его запах. В низких концентрациях этот газ
вызывает раздражение слизистых оболочек, головную
боль, тошноту и ощущение усталости
Острое отравление свинцом вызывает рвоту, судороги
и необратимые повреждения головного мозга. При
хроническом отравлении наблюдается потеря веса,
слабость и анемия
Чрезвычайно опасна вследствие значительной летуче-
сти [при 25 °C упругость пара 0,27 Па
(2-10-3 мм рт. ст.)] и способности проникать в ор-
ганизм через органы дыхания, неповрежденные кож-
ные покровы и пищеварительный тракт. Острое от-
равление металлической ртутью и ее солями может
вызвать повреждения кожи и слизистых оболочек,
сильную тошноту, рвоту, боли в животе, кровавый
понос, повреждение почек и смерть в течение 10 дней.
При хроническом отравлении наблюдается воспале-
ние слизистых оболочек рта и десен, сильное слюно-
отделение, расшатывание зубов, повреждение почек,
мышечные судороги, спазмы, депрессия и другие пси-
хические нарушения, повышенная возбудимость и
нервозность. Антидотом ртути является димеркапрол
(BAL)
При употреблении внутрь даже в малом количестве
оказывает специфическое разрушающее действие на
глазной нерв, что приводит к его необратимым по-
вреждениям и полной слепоте а
Вызывает гемолиз эритроцитов
Входит в состав инсектицидов. Вызывает резко по-
вышенную возбудимость, нарушение координации и
паралич мышц пли дыхательной мускулатуры
Введение
23
Продолжение табл. 1.1
Нитрат серебра При попадании на кожу или иа слизистые оболочки вызывает раздражение и разъедает их. При приеме внутрь вызывает сильный гастроэнтерит и возможен летальный исход
а В больших количествах приводит к летальному исходу. — Прим, перев.
растворы для обработки ожогов, вызванных кислотами или едки-
ми щелочами.
Все несчастные случаи в лаборатории независимо от послед-
ствий необходимо регистрировать и сообщать о них в установлен-
ном порядке. Если пострадавшие при несчастном случае должны
покинуть лабораторию для дальнейшей обработки в медицинском
учреждении, то к ним обязательно должны быть выделены сопро-
вождающие. Если с пострадавшим случится обморок вне лабора-
тории, последствия несчастного случая могут оказаться существен-
но более тяжелыми.
В большинстве случаев имеются организации, которые проводят
инструктаж по мерам оказания первой помощи (Красный Крест,
добровольные санитарные дружины, медицинские учреждения и
г. п.).
Таблица 1.2
Ядовитые вещества, при отравлении которыми нельзя вызывать рвоту
Нельзя вызывать рвоту, если пострадавший принял внутрь перечисленные ниже
вещества. Следует давать обильное питье, молоко или воду: в возрасте от 1 до
5 лет— 1—2 чашки, в возрасте старше 5 лет — до 1 л
Аммиак а
Бензол
Хлорная известь (гипохлорит натрия) а
Карболовая кислота, феиол (дезинфицирующее средство) 1
Креозот (смесь фенолов)
Моющие средства а
Жидкости для химической чистки одежды
Бензин
Керосин
Негашеная известь (оксид кальция) а
Гашеиая известь (гидроксид кальция) а
Петролейиый эфир
Жидкости для разбавления и удаления красок
Скипидар
Сода (карбонат натрия) °
Сильные кислоты
Стрихнин
а Эти вещества представляют собой щелочи, разрушающие ткани. При отравлении ими
можно давать разбавленную уксусную кислоту, фруктовые соки, молоко или воду.
24
Глава 1
Большинство веществ, применяемых в лабораторной практике,
ядовиты. Поэтому важно знать основные признаки отравления
различными ядами (табл. 1.1)*. Если установлено, какого типа
Рис. 1.1. Инструкция по оказанию первой медицинской помощи.
При сильном кровотечении: 1 — берут стерильную или чистую выстиранную салфетку, на-
кладывают ее иа раиу и крепко прижимают; 2—если возможно, приподнимают поврежден-
ную часть тела как можно выше и держат так до остановки кровотечения; 3—туго забинто-
вывают, обращаются к врачу.
Искусственное дыхание проводят следующим образом: 4 — очищают рот пострадавшего,
запрокидывают назад его голову и зажимают ему нос; 5—плотно прижимают губы ко рту
пострадавшего и вдувают воздух в его легкие до тех пор, пока ие будет видно движение
«го грудной клетки (при оказании помощи детям следует проявлять осторожность); 6—осво-
бождают рот пострадавшего и повторяют вдувание воздуха 12 раз в минуту для взрослых
и 20 раз в минуту для детей. При всех несчастных случаях следует принимать меры против
шока: пострадавшего укладывают, тепло одевают или укрывают одеялом и, если возможно,
приподнимают его ноги на 20-25 см.
яд принял внутрь пострадавший, следует проверить по списку
ядов, допустимо ли вызвать рвоту. В табл. 1.2 перечислены яды,
при отравлении которыми рвоту вызывать нельзя. В большин-
* Дополнительная информация по этому вопросу содержится в книге: The
Toxic Substances List, Eds. E. Christensen, T. Lugenbyhl, U. S., Department of
Health, Education and Welfare, Rockville, Meriland, 1974.
Введение
25
стве таких случаев при рвоте едкие вещества вновь придут в со-
прикосновение с легко поражаемыми тканями организма челове-
ка и вызовут дополнительное их повреждение, поэтому пострадав-
шему необходимо давать обильное питье, чтобы по возможности
разбавить принятый яд (табл. 1.2). В табл. 1.3 перечислены ядо-
витые вещества, при отравлении которыми нужно вызвать рвоту,
и указано, как это сделать.
Таблица 1.3
Ядовитые вещества, при отравлении которыми
нужно вызвать рвоту (путем раздражения
задней стенки гортани)
Этанол
Метанол (древесный
спирт)
Антифриз (этиленгли-
коль)
Бура
Камфора
Формальдегид
Репелленты насекомых
На рис. 1.1 показано, как прекратить кровотечение и провести
искусственное дыхание. В табл. 1.4 кратко изложены основные
меры оказания первой помощи.
Наконец, ниже перечислены несколько руководств по вопросам
техники безопасности, рекомендуемых для использования в хими-
Таблица 1.4
Меры оказания первой помощи
Ранения Необходимо защитить рану от заражения и остановить
кровотечение. Первая помощь (рис. 1.1): наложите на рану стериль- ную повязку, при необходимости давящую для оста- новки кровотечения.
Шок Необходимо удобно уложить пострадавшего. Симптомы: влажная бледная кожа, поверхностное ды- хание, тусклые глаза, слабый пульс. Первая помощь: 1) уложите пострадавшего и, если нет повреждений головы или грудной клетки, приподни- мите ему ноги; 2) укройте одеялом (следите, Чтобы пострадавший не вспотел); 3) напоите водой для утоления жажды.
Искусственное дыха- Необходимо очистить дыхательные пути, держать их
иие свободными, попеременно сжимать и отпускать груд- ную клетку. Симптомы: потеря сознания при ударе электрическим током, при утоплении или отравлении газом. Первая помощь (рис. 1.1): выдвиньте челюсть постра- давшего вперед, а голову откиньте назад. Затем за- жмите нос пострадавшего и с силой вдувайте ртом воздух в рот пострадавшего, отворачивайте лицо в сторону во время выдоха. Повторяйте эту процедуру 15—20 раз в минуту.
26
Глава 1
Продолжение табл. 1.4
Отравления
Переломы
Ожоги
Транспортировка
Потеря сознания
Сердечный приступ
Необходимо разбавить яд и вызвать рвоту, за исклю-
чением тех случаев, которые указаны в табл. 1.2.
Симптомы: ожоги вокруг рта, наличие пустых сосу-
дов от ядовитых веществ.
Первая помощь: давайте воду или молоко, вызовите
рвоту, давая концентрированный раствор соды или
нажимая пальцем на корень языка. Универсальное
противоядие: 1 ч. крепкого чая, 1 ч. молока или
порошка магнезии, 2 ч. пригорелого хлеба (лучше,
если возможно, активированного угля. — Прим, пе-
ред.). Нельзя вызывать рвоту, если пострадавший
проглотил сильную кислоту, керосин или стрихнин.
Проверьте указания о необходимых противоядиях
на этикетках пустых флаконов.
Необходимо обеспечить неподвижность сломанной ко-
сти и соседних суставов.
Симптомы: боль, опухоль, деформация.
Первая помощь: наложите шину, используя твердые
материалы, подушки или одеяла.
Необходимо сиять боль и предохранить от инфекции.
Симптомы: ожог I степени — покраснение кожи, II сте-
пени-пузыри иа коже, III степени — глубокое об-
угливание тканей.
Первая помощь: наложите тонкий слой стерильного
бинта. Химические ожоги промойте водой.
Необходимо при переноске пострадавшего избегать
сгибания, поворотов тела и толчков.
Первая помощь: чтобы избежать новых повреждении
у пострадавшего, уложите его на пальто, одеяло или
ковер; используйте кресло, носилки или переносите
пострадавшего на руках.
Уложите пострадавшего на ровную поверхность или
помогите ему в сидячем положении опустить голову
между коленями и глубоко дышать. Дайте понюхать
нашатырный спирт.
Если у пострадавшего имеются лекарства, помогите
принять их. Уложите его и обеспечьте приток све-
жего воздуха. Вызовите врача.
ческой практике. Все они содержат также сведения о мерах ока-
зания первой помощи:
CRC Handbook of Laboratory Safety, 3rd ed., edited by N. V.
Steere, Chemical Rubber Co., Cleveland, Ohio, 1971.
Merck Index, 9th ed., edited by Martha Windholz, Merck Compa-
ny, Rahway, N.J., 1976.
Dangerous Properties of Industrial Materials, 4th ed., edited by
N. F. Sax, Van Nostrand Reinhold, New York, 1975.
The Toxic Substances List, U.S. Department of Health, Educa-
tion and Welfare, Rockville, Md., 1974.
Safety in the Chemical Laboratory, edited by N.V.Steere, Divi-
sion of Chemical Education of the American Chemical Society, Eas-
ton, Pa., Vol. 1, 1967; Vol. 2, 1971; Vol. 3, 1974.
Введение
27
1.5. ВЗРЫВООПАСНОСТЬ ПРОСТЫХ ЭФИРОВ
Опубликован ряд сообщений о сильных взрывах, происходив-
ших в результате неожиданной детонации пероксидов, образую-
щихся в растворителях. К их числу относятся простые эфиры,
такие, как диэтиловый эфир, диизопропиловый эфир, диоксан,
тетрагидрофуран и многие другие. По-видимому, особо склонен
к образованию пероксидов диизопропиловый эфир. Очевидно, что
наибольшая опасность взрыва существует в том случае, когда эфи-
ры находятся в соприкосновении с воздухом, особенно в течение
длительного времени. Опасность взрыва увеличивается при по-
вышении концентрации пероксидов, например при перегонке эфи-
ра. Любой эфир, используемый в качестве растворителя, в кото-
ром выпал осадок или который кажется более вязким, чем обыч-
но, может содержать пероксиды. С такими растворителями рабо-
тать ни в коем случае нельзя и об этом необходимо немедленно
сообщить преподавателю.
Эфиры, с которыми произошли описанные выше изменения,
нельзя использовать в лаборатории. Ниже описаны пробы на на-
личие малых количеств пероксидов в эфирах и способы их уда-
ления.
Из большого числа существующих реакций на наличие пер-
оксидов здесь будут описаны только две.
Способ А. Реакция иа присутствие пероксидов с тиоцианатом двухвалеитиого
железа. Смешивают 5 мл свежеприготовленного 1%-иого раствора сульфата
двухвалентного железа, 0,5 мл 1 н. серной кислоты и 0,5 мл 0,1 н. раствора
тиоцианата аммония (при необходимости полученную смесь обесцвечивают, до-
бавив щепотку цинковой пыли). Бесцветный раствор встряхивают с равным
объемом проверяемого растворителя. В присутствии пероксидов появляется
красное окрашивание.
Способ Б. Реакция иа присутствие пероксидов с ио ди дом калия. К 10 мл ди-
этилового эфира, помещенного в цилиндр емкостью 25 мл из бесцветного стекла
с притертой стеклянной пробкой, приливают 1 мл свежеприготовленного 10%-
иого раствора иодида калия. Смесь необходимо защищать от действия света.
При просмотре на просвет на белом фоне ни одни из двух слоев, образовав-
шихся в цилиндре, ие должен иметь заметной окраски. Далее, 9 мл испытуе-
мого эфира встряхивают с 1 мл насыщенного раствора иодида калия. Появле-
ние желтой окраски свидетельствует о наличии в проверяемом образце эфира
более 0,005% пероксидов.
Для удаления пероксидов из эфира можно использовать на-
сыщенный раствор сульфата двухвалентного железа. На обработ-
ку 1 л эфира затрачивают 40 г 30%-ного раствора сульфата двух-
валентного железа. Такую обработку следует проводить с
большой осторожностью, так как при наличии в эфире заметных
количеств пероксидов реакция протекает довольно бурно с выде-
лением значительного количества тепла. После обработки эфир
сушат (например, сульфатом магния) и перегоняют.
Простой метод удаления пероксидов из высококачественных
образцов эфира, не требующий перегонки и потому позволяющий
28 Глава 1
избежать значительных потерь эфира, заключается в том, что для
очистки растворитель пропускают через колонки с ионообменной
смолой дауэкс-1. Для удаления пероксидов и следов влаги из ди-
этилового и дибутилового эфиров, диоксана и углеводородных
растворителей можно использовать колонку, наполненную окси-
дом алюминия. Таким путем удаляют пероксиды из тетрагидро-
фурана, декагидронафталина (декалина), 1,2,3,4,-тетрагидронаф-
талина (тетралина), кумола и диизопропилового эфира.
Дополнительные сведения о пероксидах, содержащихся в эфи-
рах, могут быть найдены в следующих книгах: CRC Handbook of
Laboratory Safety (полный список источников на стр. 9) и The
Chemistry of Ether Linkage, ed. S. Patai, Wiley Interscience, New
York, 1967.
Глава 2
Идентификация неизвестных веществ
2.1. ВВЕДЕНИЕ
В настоящей главе описаны два основных направления, в ко-
торых может быть использована общая схема процесса иденти-
фикации и подход к этой проблеме. Первое из них имеет в основ-
ном педагогическое значение: это решение студентами задач на
идентификацию вещества, уже описанных в химической литера-
туре. Второе направление включает идентификацию ранее неиз-
вестного соединения. В этой главе особое внимание уделено пер-
вому направлению, более удобному при выполнении учебных за-
даний и проведении семинарских занятий.
Изложенные ниже рекомендации предназначаются для того,
чтобы должным образом направлять деятельность студентов в
процессе идентификации неизвестного вещества. При этом, ко-
нечно, предполагается, что студент ведет тщательную и система-
тическую регистрацию проведенных наблюдений. Подготовка со-
общения о наблюдаемых результатах экспериментов облегчается,
если придерживаться определенной последовательности операций,
предложенных в этой главе.
Будем исходить из того, что студент получил для исследова-
ния образец, содержащий в основном одно соединение. Следует
подчеркнуть, что при наличии в пробе значительных количеств
двух или более соединений (что вполне возможно) для успеха
выполняемого исследования необходимо проработать материал
гл. 7, посвященной методам разделения различных веществ.
В данной же главе предполагается, что наибольший интерес
представляет установление структуры основного компонента, при-
сутствующего в изучаемом образце в преобладающем количе-
стве. Дальнейшие разделы настоящей главы коротко повторяют
материал, изложенный более подробно в гл. 3—6. В заключение
приведены формы сообщений и отчетов о выполненных работах.
2.2. ПРЕДВАРИТЕЛЬНЫЕ ИССЛЕДОВАНИЯ
Необходимо ознакомиться с разд. 3.1. Следует отметить, яв-
ляется ли исследуемое вещество гомогенным или нет, описать его
физическое состояние (твердое или жидкое), цвет и запах. Далее
выполняют пробу на горючесть (разд. 3.1.4) и записывают резуль-
таты наблюдений.
30
Глава 2
Тонкослойная и газовая хроматография. Следует проработать
разд. 3.2 и 3.4. Тонкослойная хроматография — очень легкий и
прямой способ оценки чистоты вещества. Для твердых проб тон-
кослойную хроматографию можно дополнить определением тем-
пературы плавления (см. ниже) и газовой хроматографией. На-
личие одного пятна на тонкослойных хроматограммах (проявлен-
ных в растворителях с различной полярностью), одного пика на
газовых хроматограммах, полученных на колонках с различной
полярностью, и четкая температура плавления являются вескими
свидетельствами в пользу высокой чистоты исследуемого вещества.
Если проба представляет собой жидкость или твердое вещество,
то применение тонкослойной хроматографии вполне целесообраз-
но. Если проба находится в жидком состоянии, то следует попы-
таться использовать газовую хроматографию. В ряде случаев воз-
можно газохроматографическое исследование и достаточно лету-
чих твердых веществ.
2.3. ФИЗИЧЕСКИЕ КОНСТАНТЫ
Следует изучить разд. 3.3. Если неизвестное вещество твер-
дое, то определяют его температуру плавления (разд. 3.3.1). Если
вещество плавится в интервале температур, превышающем 2°С,
его необходимо перекристаллизовать. Однако некоторые соедине-
ния могут иметь нечеткие температуры плавления независимо от
степени чистоты. Это связано с тем, что вблизи температуры плав-
ления они претерпевают разложение или другие химические из-
менения. Если определяемое вещество представляет собой жид-
кость или твердое тело с очень низкой температурой плавления,
то для этого вещества определяют температуру кипения
(разд. 3.3.2). Диапазон температур, в котором кипит данная
жидкость, не должен быть больше 5°С, за исключением очень вы-
сококипящих веществ. Если температуры кипения указывают на
наличие значительного количества примесей в изучаемом веще-
стве, если оно негомогенно или если есть основания полагать,
что оно изменило свой естественный цвет, то такое вещество под-
вергают перегонке. В том случае, когда при попытке определения
температуры кипения заметны признаки разложения вещества,
перегонку следует проводить при пониженном давлении.
Как было отмечено выше, четкая температура плавления яв-
ляется веским свидетельством в пользу достаточной чистоты опре-
деляемого вещества*. В то же время наличие узких интервалов
температур кипения не означает, что данные вещества достаточно
чисты. Свидетельством в пользу высокой чистоты изучаемой про-
* Могут вводить в заблуждение четкие температуры плавления эвтектиче-
ских смесей. Одиако они встречаются в химической практике довольно редко.
Идентификация неизвестных веществ 31
бы могут быть данные о плотности. Однако на начальном этапе
определения структуры вещества плотность последнего опреде-
ляют лишь в редких случаях. Исключение составляют вещества,
очень неактивные в химическом отношении (например, углеводо-
роды). В таких случаях определение плотности может быть одной
из первых операций при установлении строения веществ. Легко
получить данные о показателе преломления. Его величина позво-
ляет сделать определенные заключения о чистоте вещества и в
дальнейшем может служить подтверждением проведенной иден-
тификации.
2.4. ОПРЕДЕЛЕНИЕ МОЛЕКУЛЯРНОЙ МАССЫ
Помимо молекулярной формулы вещества одной из наиболее
полезных величин при определении структуры органических ве-
ществ является молекулярная масса. По величине молекулярной
массы вещества во многих случаях можно сделать вполне квали-
фицированные заключения о его молекулярной формуле. Класси-
ческим способом определения молекулярной массы в течение дли-
тельного времени был метод Раста (понижение температуры
замерзания растворов). Однако в настоящем издании описание
Метода Раста опущено, так как этот метод не дает точных результа-
тов для довольно широкого круга органических соединений. Для
очень большого числа органических веществ удобно получать мо-
лекулярные массы с помощью метода масс-спектрометрии
(разд. 3.5.2). Однако этот метод может оказаться доступным да-,
леко не во всех учебных лабораториях*. Простым методом, по-
зволяющим получить сведения о молекулярной массе веществ, яв-
ляется осмометрия (разд. 3.5.1). Однако следует опасаться полу-
чения ошибочных слишком высоких значений молекулярной массы
вследствие склонности определяемого вещества к образованию
молекулярных агрегатов. Молекулярные массы или величины, на-
ходящиеся с ними в простых кратных отношениях, можно опреде-
лить на основе эквивалентов нейтрализации или чисел омыления.
Ввиду того что эти показатели связаны с наличием специфических
функциональных групп (кислотных или аминогрупп и сложно-
эфирных групп соответственно), их определение описано в гл. 6.
Для некоторых классов органических соединений применение масс-
спектрального анализа затруднительно, и поэтому более целесо-
образно применять другие методы определения молекулярной
массы.
* В качестве альтернативы преподаватель может снабдить студента масС-
спектрометрическими данными (или значениями осмометрических молекулярных
масс, или сведениями о процентном содержании С, Н, N в определяемом веще-
стве и т. п.). Это позволит студентам приобрести опыт интерпретации таких
Данных и использования их при определении структуры веществ.
32
Глава 2
2.5. ОПРЕДЕЛЕНИЕ МОЛЕКУЛЯРНОЙ
ФОРМУЛЫ ВЕЩЕСТВА
Следует просмотреть гл. 4. Для доказательства наличия в со-
ставе вещества некоторых элементов можно воспользоваться про-
стыми «мокрыми» или «пробирочными» реакциями.
Необходимо проверить изучаемое вещество на наличие в нем
азота, серы, хлора, брома или иода (разд. 4.1.1). Если при испы-
тании горючести вещества образуется твердый остаток, следует
выяснить, какой металл входит в его состав.
Контрольные опыты. Если студенты не знакомы с техникой раз-
ложения веществ или проведения тестов на наличие в них различ-
ных элементов, им следует провести контрольные опыты с изве-
стными веществами параллельно с экспериментами, проводимыми
с неизвестным веществом. Соединение, с которым проводят кон-
трольные опыты, конечно, должно содержать азот, серу и галоге-
ны.
В тех лабораториях, где имеется возможность применять ме-
тод масс-спектрометрии, следует попытаться определить молеку-
лярную формулу изучаемого органического соединения по группе
пиков в масс-спектре, расположенных в области молекулярного
иона. Появление этих пиков связано с наличием вкладов в моле-
кулярный ион разных изотопов элементов, входящих в состав оп-
ределяемого соединения (разд. 4.2.2). Данные масс-спектров мо-
жно также использовать для того, чтобы определить наличие и
число тех атомов в молекуле, которые дают необычно большие
или очень малые вклады в интенсивность пиков, близких к моле-
кулярному иону (разд. 4.2.2).
При определении структуры органических соединений часто
бывает очень полезен анализ методом сожжения и другие коли-
чественные методы. Обычно в органических лабораториях, вы-
полняющих качественные анализы, такие количественные опреде-
ления не проводят. Однако соответствующие данные может пре-
доставить преподаватель.
Последующие операции определения структуры вещества мож-
но разделить на два этапа. Во-первых, нужно оценить раствори-
мость и определить спектральные характеристики вещества
(гл. 5), чтобы отнести «неизвестное» соединение к одному из ос-
новных классов соединений. Во-вторых, представляет интерес оп-
ределение точной структуры изучаемого соединения путем про-
ведения ряда химических реакций, детальной интерпретаций
спектральных данных и, наконец, получения химических произ-
водных (гл. 6).
2.6. ОЦЕНКА РАСТВОРИМОСТИ
См. гл. 5. Определение растворимости неизвестного вещества
может дать полезную информацию при использовании следующих
Идентификация неизвестных веществ
33
растворителей: воды, разбавленной соляной кислоты, разбавлен-
ных растворов гидроксида натрия и бикарбоната натрия, холод-
ной концентрированной серной кислоты (разд. 5.1.1). Если ре-
зультат предварительных экспериментов вызывает сомнения, сле-
дует обработать точную навеску изучаемого вещества точно из-
меренным объемом растворителя.
Рекомендуется также изучить растворимость исследуемого со-
единения в органических растворителях (разд. 5.1.2). Результаты
этих опытов будут полезны при выборе растворителей для спек-
тральных и хроматографических анализов, а также для очистки
вещества перекристаллизацией.
Эксперименты для практики. Строгие и правильные заключения
на основе данных о растворимости требуют некоторой предвари-
тельной практики на известных соединениях. Для этой цели пред-
назначены эксперименты, описанные в разд. 5.1.1 «Определение
растворимости».
При проверке растворимости веществ в воде следует опреде-
лять реакцию раствора или суспензии на лакмус или фенолфта-
леин.
При изучении характера растворимости неизвестного вещества
нужно составлять список химических классов, к которым может
принадлежать исследуемое соединение.
Предварительный отчет. Для того чтобы избежать потери вре-
мени вследствие ошибочных наблюдений, на этой стадии обучения
студентам рекомендуется представить преподавателю полученные
данные о физических константах, элементном составе и характере
растворимости.
2.7. АНАЛИЗ МЕТОДОМ ИК- И ЯМР-СПЕКТРОСКОПИИ
Обычно для установления строения органических соединений
совершенно необходимо применение ИК- или ЯМР-спектроскопии.
Анализ ИК-спектров (разд. 5.2) является превосходным методом
определения функциональных групп. Его можно применять парал-
лельно с проведением химических реакций на те или иные функ-
циональные группы. Такое совместное применение И1\-спектро-
метрии и химических реакций в ряде случаев действительно мо-
жет привести к установлению строения изучаемого вещества. Ча-
сто при выяснении структуры веществ большую помощь оказы-
вает метод ядерного магнитного резонанса. По существу, ЯМР-
спектроскопия представляет собой метод определения относитель-
ного расположения и числа спин-активных ядер (например, про-
тонов).
Предварительный отчет. После предварительной интерпретации
результатов, полученных при изучении растворимости вещества И
его ИК- и ЯМР-спектров, обычно оказывается возможным пред-
ложить для изучаемого соединения одну или несколько достаточно
2 Зак. 335
ГОС. ПУБЛИЧНАЯ
БИБЛИОТЕКА
Ленинград
34
Глава 2
обоснованных структур, после чего перейти к окончательному
установлению его строения. Следует заметить, что было бы не-
плохо, если преподаватель смог просмотреть предварительный от-
чет до того, как студент приступит к последнему этапу работы.
Процесс окончательного установления строения вещества
включает использование «мокрых» классификационных реакций,
детальное изучение ИК- и ЯЛАР-спектров (а возможно, и других
спектров) и, наконец, получение производных изучаемого соеди-
нения. Все эти этапы далее детально рассмотрены в гл. 6.
2.8. КЛАССИФИКАЦИОННЫЕ РЕАКЦИИ
Следует изучить гл. 6. По данным, накопленным в процессе
предварительного исследования неизвестного вещества, необходи-
мо сделать заключение о том, какие функциональные группы при-
сутствуют в молекуле изучаемого вещества с наибольшей вероят-
ностью. Проверку наличия этих функциональных групп следует
провести с помощью соответствующих классификационных реаген-
тов. В гл. 6 приведены более 30 наиболее важных таких реаген-
тов и даны рекомендации по их применению. В табл. 2.1 пере-
числены классификационные реакции, сгруппированные по типам
веществ, наличие которых они помогут установить.
Следует серьезно предостеречь студентов от проведения про-
верок, не вызванных необходимостью. Помимо дополнительных
потерь времени такие проверки увеличивают вероятность ошибок.
Поэтому, например, бессмысленно начинать изучение содержаще-
го азот и обладающего основными свойствами вещества с прове-
дения проверочных реакций на присутствие спиртовых или кетон-
ных группировок. С другой стороны, в этом случае, конечно, це-
лесообразно провести такие реакции, от которых можно ожидать
информации о почти наверняка присутствующих аминогруппах.
Некоторые реакции на кетоны и альдегиды легко осуществ-
ляются и более надежны, чем реакции на другие функциональные
группы, содержащие кислород. Поэтому следует рекомендовать
проведение реакций на карбонильную группу в самом начале ис-
следования нейтральных веществ, в которых можно предполагать
наличие кислорода. Особенно целесообразно проделать такие про-
бы в том случае, если данные ИК-спектра указывают на присут-
ствие карбонильных групп.
В настоящей книге даны рекомендации и указания по съемке
ПК- и ЯМР-спектров. Приведены также примеры спектров орга-
нических веществ, содержащих наиболее типичные функциональ-
ные группы. Однако здесь не предпринимается попыток изложить
теоретические основы и аппаратурные особенности ПК- и ЯМР-
спектрального анализа. Таким образом, для полной интерпрета-
ции ПК- и ЯМР-спектров этих соединений следует обращаться
к помощи других руководств. Эти вопросы рассмотрены в
Идентификация неизвестных веществ
35
Таблица 2.1
Химические реакции функциональных групп
Амины
а) Беизолсульфохлорид (разд. 6.7, методика 16).
б) Азотистая кислота (разд. 6.7, опыт 19)
в) Хлораигндриды кислот (разд. 6.7, опыт 18)
Галогены
а) Раствор нитрата серебра в этаноле (разд. 6.6, опыт 16)
б) Раствор иодида натрия в ацетоне (разд. 6.6, опыт 17)
Альдегиды и кетоны
а) Феиилгидразии (разд. 6.2, опыт 8)
б) 2,4-Дииитрофеннлгидразии (разд. 6.2, опыт 6j
в) Раствор бисульфита натрия (разд. 6.2, опыт 9)
г) Иод и гидроксид натрия (разд. 6.2, опыт 10)
д) Гидрохлорид гидроксиламниа (разд. 6.2, опыт 7)
Альдегиды
а) Раствор Бенедикта (разд. 6.2, опыт 13)
б) Реактив Толлеиса (разд. 6.2, опыт 11)
в) Фуксиновый реактив иа альдегиды (разд. 6.2, опыт 12)
г) Раствор бисульфита натрия (разд. 6.2, опыт 9)
Фенолы
а) Бромиая вода (разд. 6.19, опыт 30)
б) Раствор хлорида железа (III) (разд. 6.19, опыт 29В)
в) Хлораигндриды кислот (разд. 6.7, опыт 18)
Карбоновые кислоты
Специфические тесты отсутствуют. Обычно предполагается, что вещества с
сильно выраженными кислотными свойствами, не являющиеся фенолами или
енолами, представляют собой карбоновые кислоты. При исследовании таких
веществ рекомендуется определять их эквиваленты нейтрализации (разд. 6.12,
методика 32)
Ненасыщенные соединения
а) Раствор перманганата калия (разд. 6.4, опыт 15)
б) Раствор брома в четыреххлористом углероде (разд. 6.4, опыт 14)
Нитросоединения
а) Циик и хлорид аммония (разд. 6.17, опыт 28)
б) Гидроксид железа (II) (разд. 6.17, опыт 27)
в) Раствор гидроксида натрия (разд. 6.17, опыт 29)
Сложные эфиры
а) Раствор гидроксида натрия (разд. 6.13, методика 40), см. также число
омыления (разд. 6.13, методика 41)
б) Гидрохлорид гидроксиламина (разд. 6.2, опыт 7)
Спирты
а) Реактив с хлоридом цинка и соляной кислотой (разд. 6.1, опыт 4)
б) Хлораигндриды кислот (разд. 6.7, опыт 18)
в) Аммонийгексанитратоиерат (разд. 6.1, опыт 2)
г) Металлический натрий (разд. 6.1, опыт 1)
д) Иодная кислота (разд. 6.1, опыт 5)
е) Хромовый ангидрид (разд. 6.1, опыт 3)
2*
36
Глава 2
Продолжение табл. 2.1
Ароматические углеводороды
а) Дымящая серная кислота (разд. 6.9, опыт 21)
б) Безводный хлорид алюминия и азоксибеизол или хлороформ (разд. 6.9,
опыт 22)
Простые эфиры
а) Иодистоводородиая кислота (разд. 6.14, опыт 26)
б) Бромная вода (разд. 6.19, опыт 30)
большинстве лекционных учебников, излагающих основы органи-
ческой химии (например, в учебнике Моррисона и Бойда * или в
книге Соломона). В гл. 10 указаны еще некоторые дополнитель-
ные руководства, которые могут быть при этом полезны.
После того как будут выведены одна или несколько возмож-
ных структур изучаемого соединения, для подтверждения одной
из них необходимо получить производные. Часто для корректного
выбора одной из нескольких возможных структур изучаемого не-
известного вещества может быть достаточно лишь температуры
плавления полученного производного. Иногда может оказаться
полезным охарактеризовать это производное с помощью химиче-
ских и спектральных методов аналогично тому, как это было про-
ведено для неизвестного вещества.
2.9. ПОЛУЧЕНИЕ ПРОИЗВОДНЫХ
Следует ознакомиться с гл. 6. Список возможных соединений,
составленный в результате предшествующих стадий исследования
неизвестного вещества, может включать несколько различных
структур. Следующий шаг в процессе идентификации — подтверж-
дение идентичности одной из этих структур и неизвестного соеди-
нения. Одновременно необходимо показать, что свойства веществ,
соответствующих другим возможным структурам, в том или ином
отношении отличаются от свойств изучаемого соединения. Такие
окончательные доказательства получают путем приготовления
производных.
Конечно, для того чтобы исключить из числа возможных струк-
тур те, которые не отвечают изучаемому веществу, получение
производных не является единственным путем. Для этой цели
можно воспользоваться любыми достаточно характеристичными
свойствами вещества, такими, как плотность, показатель прелом-
ления, оптическое вращение, эквивалент нейтрализации или спек-
тральные данные.
* Есть перевод 2-го издания: Моррисон Р., Бойд Р. Органическая химия.
Пер. с аигл. — М.: Мир, 1974.
Идентификация неизвестных веществ
37
Свойства производных, пригодных для идентификации. Произ-
водные, пригодные для идентификации, должны отвечать пере-
численным ниже требованиям.
1. Их получение не должно вызывать значительных затрудне-
ний и отнимать много времени. Эти вещества должны легко под-
даваться очистке. В большинстве случаев это означает, что такие
производные должны представлять собой твердые вещества. Это
связано с тем, что при выделении и очистке малых количеств
твердых веществ все манипуляции проводить значительно легче,
чем в случае жидкостей. Кроме того, температуру плавления мо-
жно определить точнее, чем температуру кипения. Наиболее под-
ходящие производные должны плавиться выше 50°С, но ниже
250°С. Кристаллизация веществ, имеющих температуру плавления
ниже 50°С, в большинстве случаев связана с трудностями. С дру-
гой стороны, температуры плавления, превышающие 250°С, неже-
лательны, так как при столь высоких температурах может про-
исходить разложение веществ. Кроме того, при измерении высо-
ких температур поправка на выступающий столбик ртути дости-
гает нескольких градусов.
2. Химическая реакция получения производного должна про-
текать с хорошим выходом. Следует избегать таких реакций, ко-
торые сопровождаются перегруппировками и побочными реак-
циями.
3. Свойства полученного производного должны существенно от-
личаться от свойств исходного соединения. Это в основном отно-
сится к температурам плавления исходного вещества и получен-
ного из него производного, которые должны заметно различаться
между собой.
4. Выбранное для получения производное должно достаточно
надежно подтверждать одну из нескольких предполагаемых струк-
тур определяемого вещества, т. е температуры плавления различ-
ных производных, соответствующих этим структурам, должны
различаться не менее чем на 5°С.
Подходящее производное следует выбирать среди веществ, пе-
речисленных в гл. 6. Следует заметить, что имеется много произ-
водных, пригодных для идентификации, однако возможность по-
лучения с их помощью информации о структуре исходных соеди-
нений не очень велика.
При решении вопроса о том, действительно ли наблюдаемые
физические константы отвечают данному соединению, следует учи-
тывать возможность довольно больших экспериментальных ошибок.
Так, например, если температура кипения очень высока, а тем-
пература плавления очень низка, то диапазон температур, в кото-
ром лежат истинные значения этих констант, может оказаться
лишь немного менее 5°С. Для того чтобы исключить возможные
структуры, не отвечающие истинному строению изучаемого со-
единения, можно использовать и другие физические константы.
38
Глава 2
например плотность (разд. 3.3.3), показатель преломления
(разд. 3.3.4) или эквивалент нейтрализации (разд. 6.12). Однако
и при этом возможны большие экспериментальные ошибки. Во
всех случаях необходимо составлять перечень возможных соеди-
нений с указанием производных для каждого из них, даже если
подходящими производными такого рода являются продукты, полу-
ченные при проведении классификационных реакций.
Изучение списка возможных структур идентифицируемых ве-
ществ часто указывает на возможность проведения дополнитель-
ных реакций на другие функциональные группы. Так, например,
если в списке возможных нитросоединений имеется нитрокетон,
то может оказаться полезным провести реакцию на кетогруппу,
особенно если с наличием такой группировки согласуются ИК-
спектры изучаемого вещества.
Если подходящая методика получения производного не описа-
на в данной книге, то следует поискать производные данного
соединения в литературе. Для этого проще всего просмотреть мо-
лекулярные формулы в формульных указателях нижеследующих
изданий в порядке, перечисленном ниже:
Справочник Бейльштейна Handbuch der organischen Chemie,
4-е издание, указатель к 2—4 дополнениям. Эти указатели охва-
тывают литературу до 1959 г. Пользование справочником Бейль-
штейна будет дополнительно обсуждено ниже *.
Chemisches Zentralblatt, Collective Indices: 1922—1924, 1925—
1929, 1930—1934.
Chemisches Zentralblatt, Annual Formula Indices, 1935—1939.
Сведения об источниках, охватывающих литературу после
1939 г., содержатся в гл. 10. Наиболее надежными из них являются
предметный, формульный и авторский указатели (Subject, Aut-
hor and Formula Indexes) реферативного журнала Chemical
Abstracts. Сводные указатели за 10 лет охватывают периоды
1937—1946 и 1947—1956 гг. Более поздние сводные указатели
(Cumulative Indexes) охватывают периоды в 5 лет. Уже изданы
указатели для 1957—1961, 1962—1966, 1967—1971 и 1972—1976 гг.
Сведения о работах последних лет можно почерпнуть в ежегод-
ных указателях. К журналу Chemical Abstracts выпущены свод-
ный формульный указатель (Collective Formula Index) за 1920—
1946 гг. и ежегодные формульные указатели (Annual Formula
Indexes) за последующие годы. Однако эти указатели являются
неполными, и при необходимости провести тщательный поиск ли-
тературных данных полагаться на них не следует.
Важность справочника Бейльштейна Handbuch der organischen
Chemie столь велика, что он заслуживает дополнительного об-
суждения. Основные 27 томов этого справочника, называемые Наи-
* См. гл. 10, где приведен список литературных источников, облегчающих
использование справочника Бейльштейна,
Идентификация неизвестных веществ
39
ptwerk, охватывают литературу по 1909 г. Расположение материа-
ла в справочнике определяется структурой веществ. Поэтому при
достаточном знакомстве с этим справочником любое желаемое
соединение довольно легко найти, не прибегая к помощи указа-
теля. Так, например, ациклические углеводороды, спирты, альде-
гиды и кетоны находятся в т. 1, ациклические органические кис-
лоты— в т. 2, ациклические окси-, альдегиде- и кетокислоты —
в т. 3, сульфокислоты, амины и фосфины —в т. 4. В т. 5 приведены
сведения о циклических (в том числе ароматических) углеводоро-
дах. В последующих томах до 16-го включительно в аналогичном
порядке продолжается представление циклических соединений,
а вт. 17 начинается обсуждение гетероциклических соединений.
Если то или иное конкретное соединение найдено в основных
томах, то его нетрудно найти и в дополнительных выпусках спра-
вочника. В первом дополнении (Erstes Erganzungswerk), охваты-
вающем литературу с 1910 по 1919 г., и в последующих материал
расположен так же, как и в основном справочнике. Таким обра-
зом, соединение, найденное в т. 1 основного справочника, содер-
жится и в т. 1 первого дополнения. Более того, вспомогательные
номера, которые располагаются вверху в середине каждой стра-
ницы дополнений, позволяют соотнести материал, помещенный на
этой странице, с соответствующими страницами основного спра-
вочника. Второе, третье и четвертое дополнения охватывают ли-
тературные данные за 1920—1929, 1930—1949 и 1950—1959 гг.
Предполагается выпуск и дальнейших дополнений.
Encyclopedia of Organic Chemistry, выпущенная издательством
Elsevier, первоначально имела целью столь же полный охват ли-
тературных данных, как и справочник Бейльштейна. К сожалению,
это издание было прекращено после выпуска нескольких первых
томов. Однако опубликованные тома представляют собой весьма
ценное дополнение к справочнику Бейльштейна, так как они вклю-
чают такой материал (например, стероиды), который первоначаль-
ной структурой этого справочника предусмотрен не был.
2.10. СМЕСИ
Следует ознакомиться с материалом гл. 7.
На определенной стадии изучения данного курса могут быть
исследованы одна или несколько смесей. Получив от преподава-
теля смесь веществ, необходимо разделить ее на составляющие
компоненты в соответствии с рекомендациями, изложенными в
гл. 7. Многие смеси содержат летучие компоненты, которые мо-
гут быть удалены путем нагревания смеси на паровой бане. При-
работе со смесями неизвестного состава не рекомендуется про-
водить перегонку при повышенных температурах.
После разделения смеси на составляющие ее компоненты каж-
дый из них идентифицируют таким же путем, как и индивидуаль-
ные неизвестные вещества.
40
Глава 2
2.11. ПРЕДСТАВЛЕНИЕ РЕЗУЛЬТАТОВ ИССЛЕДОВАНИЯ
НЕИЗВЕСТНОГО ВЕЩЕСТВА
После того как идентификация неизвестного соединения пол-
ностью завершена, результаты должны быть оформлены по спе-
циальной форме, которую выдает преподаватель. Ниже приведе-
ны примеры форм отчетов.
Отчет по форме № 1
Соединение: н-бутиловый спирт
Неизвестное вещество № 1
Фамилия, и., о. исполнителя: Джон Смит
Дата: 1 июня 1976 г.
1. Исследование физических свойств.
а) Физическое состояние: жидкость.
б) Цвет: бесцветная.
в) Запах: резкий.
г) Проба на горючесть: горит бесцветным пламенем, сгорает без остатка.
д) Результаты тонкослойной хроматографии.
е) Результаты газовой хроматографии.
2. Определение физических констант.
а) Температура плавления: наблюдаемая
исправленная
Температура кипения: наблюдаемая 114—117°С
исправленная 115—118°С
б) Плотность dl° 0,812
в) Показатель преломления «р 1,3988
3. Элементный анализ
F —; С1 —; Вг —; I —; N —; S —; металлов ие содержит
4. Оценка растворимости.
Н2О NaOH N аНСОз НС1 H2SO4
+
Реакция на лакмус отсутствует.
Реакция иа фенолфталеин отсутствует.
5. Определение молекулярной массы: ие проводилось.
6. П редварительные классификационные реакции.
Реагент Результат Вывод
2,4-Динитрофе- иилгидразии Ацетил хлорид Аммонийгекса- нитратоцерат Реактив Лукаса Осадок не выпадает Происходит реакция с выде- ) лением тепла. Появляется ( фруктовый запах | Красное окоашивапие J Вещество паствоояется в ре- активе, маслянистый слой не отделяется Карбонильная группа отсут- ствует Присутствует гидроксильная группа По-видимому, первичный спирт
Идентификация неизвестных веществ
41
Функциональные группы, выявленные проведенными реакциями: спиртовая
группа, по-видимому, первичная.
7. Результаты спектроскопического исследования.
Тип спектра (растворитель) Характеристичные частоты Вывод
И К (CCU) ЯМР (CDC13) 3600, 3300 СМ-1 1025 см-1 (очень широкая полоса) б 0,95 м.д., ЗН, триплет б 1,25—1,90 м.д., 4Н, муль- типлет б 2,15 м.д.®, 1Н, синглет б 3,65 м.д., 2Н, триплет —о-н -с-о- СН3СН2— Алифатическое соедине- ние ОН —сн2сн2он
в Наблюдалась зависимость величины этого химического сдвига от концентрации.
8. Предварительное изучение литературы.
Возможное соединение Т. пл. или т. кип., °C Предложения о дополнительных испытаниях
Изобутнловый спирт 108
Метнлнзопропнлкарбинол 113 Вторичный метилкарбииол должен давать положитель- ную йодоформную реакцию
Пентанол-3 116
н-Бутнловый спирт 117
Пентанол-2 119 Вторичный метилкарбииол должен давать положитель- ную йодоформную реакцию
Метил-трет-бутилкарбинол 120
9. Дополнительные классификационные и специальные реакции.
Реакция Результат Вывод
Йодоформная реакция Осадок не выпадает Исследуемое вещество ие является вторичным ме- тилкарбииолом
10. Вероятные соединения.
Название Производные н их показатели (температуры плавления, эквиваленты нейтрализации и т. п.) Плотность
3,5-дииитробен- аоат, “С а-нафтил- уретан, °C феиилуретан, “С
н-Бутиловый спирт 64 71 61 0,810
Изобутнловый спирт 86 104 86 0,805
Пентаиол-3 97 71 49 0,820
42
Глава 2
11. Приготовление производных.
Название Т. пл., °C
наблюдаемая по литературным данным
3,5-Динитробензоат 62—63 64
а-Нафтилуретан 68—69 71
Фенилуретан 57—59 61
12. Особые замечания.
Для упрощения ЯМР-спектра может быть использован лантаноидный сдви-
гающий реагент (в частности, Eu(dpm)3; см. Шрайнер и др., гл. 10).
13. Использованная литература.
Таблицы в данной книге; Huntress, Mulliken, Identification of Pure Organic
Compounds, Order 1; The Aldrich Library of Nrnr Spectra.
Отчет по форме № 2
Соединение: 2-амино-4-нитротолуол *
Неизвестное вещество № 2
Фамилия, и., о. исполнителя: Джон Смит
Дата: 13 июля 1976 г.
1. Исследование физических свойств.
а) Физическое состояние: твердый продукт.
б) Цвет: желтый.
в) Запах.
г) Проба на горючесть: горит желтым пламенем, сгорает без остатка.
д) Результаты тонкослойной хроматографии.
е) Результаты газовой хроматографии
2. Определение физических констант.
а) Температура плавления: наблюдаемая 107—108°С
исправленная
Температура кипения: наблюдаемая
исправленная 109—110°С
б) Плотность.
в) Показатель преломления.
3. Элементный анализ.
F —; С1 —; Вг —; I —; N +; S —; металлы отсутствуют
4. Оценка растворимости.
н2о NaOH NaHCO3 НС1 H2SO4
— — +
Реакция на лакмус:
Реакция на фенолфталеин:
* Может быть назван также 2-метил-5-нитроанилином или 2-метил-5-нитро-
фениламином.
Идентификация неизвестных веществ
43
5. Определение молекулярной массы: 150 ±4 (осмометрически в метаноле).
6. Предварительные классификационные реакции.
Реактив Результат Вывод
Хинсберга Азотистая кислота NaOH, чистый раствор; НС1, выпадение осадка Оранжевый осадок с 2-наф- толом Первичный амин Первичный ароматичес кий амин
Функциональные группы, выявленные проведенными реакциями: первичный ароматический амин. 7. Результаты спектральных исследований.
Тип спектра (растворитель) Характеристические частоты Вывод
ЯМР (CDCl3/flMCO-de) й 2,20 м.д., ЗН, триплет й 4,70 м.д., 2Н, уширенный синглет й 6,9—7,6 м.д., ЗН, мульти- плет АгСН3 -nh2 Протоны ароматического кольца
8. Предварительное изучение литературы.
Возможное соединение Т. пл. или т. кип., °C Предложения о других возможных испытаниях
п-Аминоацетофенон 2-Амнно-4-нитротолуол Р-Нафтиламин .и-Нитроанилин 4-Амино-З-нитротолуол 106 107 112 114 116 Реакции на присутствие ме- тилкетона Реакции на присутствие нит- рогруппы Реакция на нитрогруппу Реакция на нитрогруппу. Снять УФ-спектры
9. Дополнительные классификационные и специальные реакции.
Реактив Результат Вывод
2,4-Дпнитрофенилгидразин Иод и гидроксид натрия Цинк и хлорид аммония. Ре- актив применен к бензоиль- ному производному неиз- вестного вещества, затем реактив Толленса — 1 ... — 4 Осадок не выпадает Йодоформ не обра- зуется Образуется серебрян- ное зеркало Исследуемое вещество не является п-амино- аиетофеноном Исследуемое вещество не является метилке- тоном Присутствует нитро- группа
44
Глава 2
10. Вероятные соединения.
Название Т. пл., “С Производные, их температура плавления, эквиваленты нейтрализации и т.п.
беизолсуль- фопамид, "С ацетамид, °C фенол, °C
2-Амино-4-нитротолуол 107 172 150 118
л-Нитроанилин 114 136 155 97
4-Амино-З-нитротолуол 116 102 96 32
11. Приготовление производных.
Производное Т. пл.. °C
наблюдаемая по литературным данным
Бензол сульфонами д 2-Окси-4-нитротолуол 170—171 116-117 172 118
12. Дополнительные замечания.
Имеется сообщение о том, что 4-амино-З-нитротолуол может быть гидроли-
зован до 4-окси-З-нитротолуола действием раствора гидроксида натрия [Л'е-
vill, Winter, Вег., 15, 2893 (1882)]. При обработке неизвестного вещества в
этих условиях выделен только исходный продукт. Неизвестное вещество пе-
реведено в фенол по методу, описанному в работе: Ullmann, Fitzenkam, Вег.,
38, 3790 (1905).
13. Использованная литература.
Таблицы, приведенные в данной книге; статьи, указанные выше; The Aid-
rich Library of Nmr Spectra.
Отчет по форме № 3
Соединение: 0-нафтол
Неизвестное вещество № 3
Фамилия, и., о. исполнителя: Джон Смит
Дата: 13 сентября 1976 г.
1. Исследование физических свойств.
а) Физическое состояние: твердое тело.
б) Цвет: белый.
в) Запах: напоминает запах нафталина.
г) Проба на горючесть: горит коптящим пламенем, сгорает без остатка. Ре-
зультат пробы на горючесть позволяет предположить, что неизвестное ве-
щество является ароматическим соединением.
д) Результаты тонкослойной хроматографии.
е) Результаты газовой хроматографии.
2. Определение физических констант.
а) Температура плавления: наблюдаемая 121—121,5°С
исправленная
Температура кипения: наблюдаемая 284—286°С
исправленная
б) Плотность.
в) Показатель преломления.
Идентификация неизвестных веществ
45
3. Элементный анализ.
F —; С1 —; Вг —; I —; S —; металлы отсутствуют
4. Оценка растворимости.
н?о Na ОН NaHCOs НС1 H2SO4
— + —
Реакция на лакмус:
Реакция на фенолфталеин:
5. Молекулярная масса: 144 (по данным масс-спектрометрии). Молекулярная
формула по пикам М + 1 и М + 2: С|0Н8О.
6. Предварительные классификационные реакции.
Реактив Результат Вывод
Бромная вода Осадок 1 Неизвестное вещество, ве-
Хлорид железа (III) Зеленое окрашивание J роятно, является фенолом
Функциональные группы, выявленные проведенными реакциями: фенол.
7. Результаты спектральных исследований.
Тип спектра (растворитель) Харатеристичиые частоты Вывод
ИК (СНС1з) ЯМР ЯМР (ацетон) 3300 см-1 (широкая полоса) ) 3600 см-1 (узкая полоса) J 1632 см-1 (средняя полоса) 1605 см-1 1200 см-1 (очень широкая полоса) б 7,0—7,8 м.д., 7Н, мультиплет б 8,35 м.д.а, 1Н, уширенный синглет Гидроксильная группа Карбонильная группа? С=С? Ароматическое ядро С-О Протоны ароматического ядра -ОН
а Увеличение концентрации вещества в ацетоне приводит к сдвигу этого сигнала в сла-
бое поле.
8. Предварительное изучение литературы.
Возможное соединение Т. пл. или т. кип., °C Предложения о других возможных испытаниях
п-Оксибензальдегид 115 Следует провести реакцию на карбонильную группу
Монобензиловый эфир гид- рохинона 122 Должен расщепляться при действии кислот
Пикриновая кислота |5-Нафтол 122 122 Содержит азот, сильная кис- лота, маловероятна
Толугидрохинон 124 Должен легко окисляться до хинона, имеющего желтую окраску
46
Глава 2
9. Дополнительные классификационные и специальные реакции.
Реактив Результат Вывод
2,4-Динитрофенилгидра- зин [Ag(NH3)2]+ Осадок ие выпадает Серебряное зеркало Маловероятно, что исследуе- мое вещество является п-ок- сибензальдегидом Толугидрохинон ве исключа- ется
10. Вероятные соединения.
Название Производные, их температуры плавления, эквиваленты нейтрализации и т.п.
бромиды, °C ацетаты, *С
Р-Нафтол Толугидрохинон (1-) 84 (три-) 204 70 (ди-) 52
11. Получение производных.
Производное Т. пл.. °C
наблюдаемая по литературным данным-
Монобромпроизводное Ацетат 84—86 67—68 84 70
12. Дополнительные замечания.
Бромпроизводное было приготовлено обработкой изучаемого вещества
бромом в уксусной кислоте [Smith, J. Am. Chem. Soc., 35, 789 (1913)]
Структура толугидрохинона маловероятна для исследуемого соединения
по следующим причинам: имеются данные о том, что толугидрохинон легко-
растворим в воде. Хлорид железа (III) дает с ним коричнево-красное скра-
шивание в концентрированном растворе и желтую окраску в разбавленном
растворе (Beilstiin, VI, 874). В то же время неизвестное соединение дает
с хлоридом железа (111) зеленую окраску. Такое же окрашивание дает р-на-
фтол (Huntress, Mulliken, р. 234).
Имеется соображение о том, что В-нафтол дает положительную реак-
цию Толленса (Beilstein, VI, 628). Нафталины, замещенные в а- и р-положе-
няях, могут давать поглощение, соответствующее ароматическому кольцу
при высоких волновых числах в области 1650 см-1.
13. Использованная литература.
Таблицы в данной книге; статьи, перечисленные выше; Беллами, Инфра-
красные спектры сложных молекул; Каталог стандартных ЯМР-спектров
Садтлера.
Глава 3
Предварительное исследование, оценка
чистоты и определение физических свойств
органических соединений
К изучению этой главы следует приступить в том случае, если
исследователь считает, что имеющаяся в его распоряжении проба
состоит из одного вещества. Если есть основания предполагать,
что исследуемый образец содержит несколько веществ, следует
проработать гл. 7, посвященную методам разделения смесей. Пред-
положение о том, что в изучаемом образце содержится только
одно вещество, может иметь следующие основания: I) указания
преподавателя, 2) метод синтеза, 3) метод выделения.
3.1. ПРЕДВАРИТЕЛЬНОЕ ИССЛЕДОВАНИЕ
3.1.1. Физическое состояние
Необходимо отметить физическое состояние неизвестного ве-
щества, проверить, является ли оно жидким или твердым. Табли-
цы свойств веществ, приведенные в приложении III, разбиты по
фазовому состоянию соединений. Кроме того, ввиду того, что фа-
зовое состояние вещества связано с его растворимостью и лету-
честью, сведения о том, жидким или твердым является неизве-
стное соединение, могут помочь сделать правильный выбор спо-
соба его очистки. Обычно очистку жидкостей проводят путем
перегонки или с помощью метода газовой хроматографии (гл. 7),
а твердые вещества очищают перекристаллизацией или возгонкой.
3.1.2. Цвет
Необходимо отметить цвет исходной пробы, а также все из-
менения цвета, которые могут произойти при определении темпе-
ратуры кипения (разд. 3.3.2), при перегонке или после хромато-
графического разделения.
Окраска многих веществ связана с наличием примесей. Часто
эти загрязнения являются продуктами медленного окисления ве-
щества кислородом воздуха. Например, анилин обычно имеет крас-
новато-коричневую окраску. В то же время свежеперегнанная
проба является почти бесцветной.
Многие жидкие и твердые вещества окрашены вследствие
наличия в молекуле хромофорных групп. Так, например, окра-
шены многие нитросоединения, хиноны, изосоедцнения, стабильные
48
Глава 3
карбокатионы и карбанионы, а также соединения с достаточно
протяженной системой сопряженных связей. Если неизвестное со-
единение представляет собой бесцветную жидкость или белое кри-
сталлическое твердое вещество, то приведенная выше информа-
ция оказывается полезной, так как позволяет исключить возмож-
ность присутствия хромофорных функциональных групп, а также
многих других групп, превращающихся в хромофорные при окис-
лении.
3.1.3. Запах
Органические соединения многих химических классов имеют
характерный запах. Дать точное описание этого свойства в на-
стоящее время невозможно, однако студент должен ознакомиться
с запахами ряда обычных органических соединений. Спирты обла-
дают запахом, не похожим на запах сложных эфиров, фенолы
пахнут иначе, чем амины, альдегиды — не так, как кетоны. Обыч-
но утверждают, что меркаптаны, изонитрилы и пентаметиленди-
амин имеют неприятные запахи, однако по запаху эти вещества
отличаются друг от друга. Более того, запах обычно сильнее вы-
ражен у более низкомолекулярных членов данного химического
класса вследствие их более высокой летучести. Бензальдегид,
нитробензол и бензонитрил имеют запах горького миндаля. Ха-
рактерными, легко запоминающимися запахами обладают эвге-
нол, кумарин, ванилин, метилсалицилат и изоамилацетат. Разли-
чаются по запаху и углеводороды: специфические запахи имеют
толуол, гексан, изопрен, инден, пинен и нафталин.
Студенты должны тщательно изучать запах обычных органи-
ческих веществ и сравнивать с ним запах неизвестного соедине-
ния. В случае необходимости на этикетках известных веществ
указаны меры предосторожности. При наличии сомнений следует
проконсультироваться в соответствующих справочниках, напри-
мер в Merk Index (см. гл. 10).
3.1.4. Проба на горючесть
Методика проведения. Пробу массой около 0,1 г помещают на
крышку фарфорового тигля (или иную фарфоровую пластинку)
и подносят к краю пламени, чтобы проверить способность веще-
ства к воспламенению. Далее нагревают вещество сначала на не-
большом пламени за защитным щитком. Постепенно нагревание
увеличивают и, наконец, сильно нагревают, чтобы обеспечить пол-
ное сгорание. Отмечают следующие особенности горения веще-
ства: 1) способность воспламеняться и характер пламени (не яв-
ляется ли вещество взрывчатым?); 2) если вещество твердое, то
отмечают, плавится ли оно и каким образом; 3) запах выделяю-
щихся паров и газов {следует соблюдать осторожность'.)', 4) нали-
Предварительное исследование, оценка чистоты
49
чие остатка после сгорания. Плавится ли остаток? Если после
сгорания вещества остался несгораемый остаток, то остужают
крышку тигля, добавляют к остатку каплю дистиллированной
воды и проверяют реакцию образовавшегося раствора на лакмус.
Затем добавляют каплю разбавленной соляной кислоты. Отме-
чают, не выделяется ли при этом газ. Пользуясь платиновой про-
волокой, с полученным солянокислым раствором выполняют про-
бы на окрашивание пламени, чтобы установить присутствие ка-
ких-либо металлов.
При проведении этого испытания, как и ряда других, следует
поставить контрольный опыт, применяя стандартные соединения
известного состава. В тех случаях, когда это возможно, для про-
ведения контрольного опыта следует выбирать известное соеди-
нение, сходное с предполагаемым составом неизвестного вещества.
Для того чтобы оценить диапазон результатов, получаемых при
проведении пробы на горючесть, можно использовать в качестве
контрольных веществ этанол, толуол, бензоат бария, ацетат меди,
виннокислый калий — натрий и обыкновенный сахар.
Обсуждение. Многие жидкости горят характерным пламенем,
что помогает определить природу данного соединения. Так, на-
пример, ароматические углеводороды с довольно высоким содер-
жанием углерода горят желтым коптящим пламенем. Алифатиче-
ские углеводороды также горят желтым пламенем, однако копоти
при этом выделяется много меньше. По мере того как в составе
вещества возрастает содержание кислорода, пламя становится все
более бледным (голубоватым). Если вещество склонно воспла-
меняться, то при дальнейшей работе с ним следует применять со-
ответствующие предосторожности. Проба на горючесть указывает
также, следует ли провести определение температуры плавления
твердого вещества, и позволяет установить, не является ли оно
взрывчатым.
Если после сгорания вещества остается негорючий остаток,
его нужно исследовать на присутствие металлов. Для определе-
ния природы имеющихся металлов обычно достаточно несколько
простых проб*. Если по окрашиванию пламени хотят сделать вы-
вод о присутствии натрия, то пробу определяемого вещества сле-
дует сжечь не на фарфоровой пластинке, а на кусочке платино-
вой фольги. (Почему?)
3.1.5. Выводы и их применение
Описанные в этом разделе пробы очень полезны для решения
вопроса о том, необходима ли дальнейшая очистка вещества и
какие именно методы очистки следует применить. Если различ-
* С этими пробами можно ознакомиться по какой-либо книге по качествен-
ному неорганическому анализу, например: Moeller Т., Qualitative Analysis,
McGraw-Hill, New York, 1958.
50
Глава 3
ные тесты, описанные в этом разделе, указывают на наличие силь-
ного загрязнения исследуемого образца, то почти наверняка по-
требуется применить методы перекристаллизации или адсорбцион-
ной хроматографии (гл. 7). Хотя жидкости очень часто анали-
зируют методом газовой хроматографии (разд. 3.4), в хромато-
граф нельзя вводить те из них, которые сгорают не полностью.
3.2. ТОНКОСЛОЙНАЯ ХРОМАТОГРАФИЯ
Тонкослойная хроматография, по-видимому, представляет со-
бой наиболее быстрый, легкий и наиболее часто применяемый
метод оценки чистоты органических веществ (а также наличия
смеси нескольких компонентов и, часто, природы вещества). Ме-
тод хроматографии можно определить как способ разделения хи-
мических веществ, основанный на различиях в характере их рас-
пределения между двумя фазами, одна из которых неподвижна
(например, поверхность твердого тела), а вторая является транс-
портирующей подвижной средой (например, растворитель или
элюент). Общие вопросы хроматографии.детально разбираются в
гл. 7. В тонкослойной хроматографии неподвижная фаза пред-
ставляет собой тонкий слой адсорбента, распределенный на по-
верхности стеклянной или пластмассовой пластинки. Для связы-
вания частиц сорбента между собой и с подложкой служат суль-
фат кальция или органические полимеры. Небольшое количество
пробы помещают у края пластинки, и этот край опускают в рас-
творитель, налитый тонким слоем в специальный сосуд (см.
рис. 3.2). Расстояние, на которое растворитель, пропитывающий
слой сорбента, продвинет исследуемое вещество, зависит от его
адсорбционной способности в данной системе, а также от многих
других факторов. Достаточно часто удается без особого труда
подобрать такую систему адсорбент — растворитель, которая по-
зволила бы разделить большинство компонентов данной смеси.
Такой метод разделения особенно полезен для работы с термо-
лабильными или нелетучими соединениями, т. е. с такими веще-
ствами, для которых нельзя определить температуру кипения и
которые не могут быть исследованы методом газовой хроматогра-
фии.
Техника выполнения. В продаже имеются готовые пластинки, заранее покры-
тые слоем оксида алюминия (А12О3) или силикагеля (SiOj-xhkO), часто содер-
жащих флуоресцирующие материалы *. Предварительные пробы, помогающие
правильно выбрать систему растворителей, проводят следующим образом. Гото-
вят тонкие стеклянные капилляры (например, вытянутые из более толстых тру-
бок). Под действием капиллярных сил помещают в эти трубки растворы опре-
* Компания Eastman Organic Chemistry Department, 343 State St., Rochester,
N.Y. 14650 Следует также просматривать ежегодные указатели выпускаемого
оборудования и приборов, публикуемые в журналах Science или Analytical Che-
mistry, где можно найти сведения об организациях, поставляющих оборудо-
вание.
Предварительное исследование, оценка чистоты
51
Таблица 3.1
Растворители для хроматографии а (растворители расположены
в порядке возрастания полярности сверху вниз)6
Петролейный эфир
Циклогексан
Четыреххлористый углерод (тетрахлорметан)
Бензол
Метиленхлорнд
Хлороформ (не содержащий спирт)
Диэтиловый эфир
Этилацетат
Пиридин
Ацетон
н-Пропанол
Этанол
Метаиол
Вода
Уксусная кислота
а В качестве проявителей при хроматографических разделениях могут применяться
смеси двух и более растворителей.
® Термин «полярность> здесь имеет значение только для оценки свойств данного раство-
рителя и может отличаться от полярности, оцениваемой, например, путем измерения диэлект-
рической проницаемости данного растворителя.
деляемого вещества в каждом из нескольких подлежащих исследованию рас-
творителей. В табл. 3.1 приведены растворители, которые целесообразно прове-
рить для возможного использования в тонкослойной хроматографии. Далее при-
трагиваются кончиком капилляра к слою адсорбента на пластинке (рис. 3.1) и
Рис. 3.1. Тонкослойная хроматогра-
фия — предварительные операции по
выбору растворителя.
а —хорошее проявление; б — чрезмерное
проявление; в—недостаточное проявление.
1 — тонкий капилляр, заполненный раство-
ром; 2—фронт растворителя; 3-кольца
разделяемых веществ.
Рис. 3.2. Камера для проявления
тонкослойных хроматограмм.
/ — фильтровальная бумага; 2—хромато-
грамма; 3—растворитель (высота 3-4 мм).
позволяют растворителю из капилляра полностью впитаться в слой. Пригодный
для данной пробы растворитель продвигает кольцо исследуемых веществ на
0,3—0,5 радиуса кружка, образованного этим растворителем. В отсутствие ка-
кой-либо информации о других растворителях (например, из опытов по пере-
кристаллизации вещества) целесообразно вначале использовать хлороформ или
растворители сходной полярности.
52
Глава 3
Камеру для проявления хроматограмм (рис. 3.2), обычно закрывающуюся
крышкой, готовят к работе, помещая на дно слой подходящего растворителя
3—4 мм высотой. Вначале готовят раствор исследуемого вещества с концентра-
цией 1 — 10% в любом летучем растворителе. Далее изготовляют тонкие капил-
лярные трубки, например путем вытягивания из капилляров, предназначенных
для измерения температу, плавления. Если концом тонкой капиллярной трубки
прикоснуться к поверхности раствора органического вещества, то под действием
капиллярных сил некоторое количество этого раствора поднимается по трубке.
На хроматографическую пластинку наносят пятно пробы, притрагиваясь к слою
сорбента копчиком заполненного раствором капилляра в точке, отстоящей от
одного из краев пластинки примерно на 0,5 см (рис. 3.3). Нанесенное пятно
пробы должно быть расположено на пластинке так, чтобы в начале анализа
Рис. 3.3. Нанесение пробы на пластинку для тонко-
слойной хроматографии.
1—раствор пробы (1 — 10%) в летучем растворителе.
Рис. 3.4. Камера для вы-
явления пятен на тонко-
слойных пластинках дей-
ствием паров иода.
1 — отметка фронта раство-
рителя.
оно находилось выше уровня растворителя в хроматографической камере. Да-
лее пластинку с нанесенной пробой помещают в камеру для проявления, уста-
навливая ее так, чтобы пятно пробы оказалось в иижпей части камеры. Раство-
ритель должен подняться по слою сорбента до уровня, примерно на 1 см не до-
стигающего его верхнего края (рис. 3.2). В процессе проявления пластинки ка-
мера должна быть закрыта с тем, чтобы атмосфера в ней была бы насыщена
парами растворителя. Для более полного насыщения стенки камеры обклады-
вают фильтровальной бумагой, по которой растворитель поднимается вверх, как
по фитилю. Проявление пластинки размером с предметное стекло для микро-
скопа требует около 5 мин. Пластинки большего размера проявляются в тече-
ние 0,5—1,0 ч. По окончании проявления пластинку извлекают из камеры и
отмечают положение фронта растворителя штрихом на слое сорбента. Все явно
выраженные пятна на хроматограмме также отмечают, процарапывая кружки
каким-либо заостренным предметом *. Затем дают хроматограмме высохнуть
(обычно 5—30 мин) и приступают к визуализации, заключающейся в пере-
воде бесцветных соединений в производные, которые можно наблюдать ви-
зуально.
Существует несколько способов превращения бесцветных веществ в окра-
шенные производные. Если хроматограмма содержит вещества, способные флуо-
ресцировать, то проявленную пластинку рассматривают при освещении ультра-
фиолетовым светом (осторожно, берегите глаза!.). Если разделенные вещества
обладают способностью гасить флуоресценцию индикатора, входящего в состав
* Карандашом, циркулем, стеклянной палочкой с острым концом и т. п.—
Прим, персе.
Предварительное исследование, оценка чистоты
53
слоя сорбента, то в ультрафиолетовом свете наблюдают темные пятна на свет-
лом фоне. Пятна, выявленные в ультрафиолетовом свете или другими спосо-
бами, описанными ниже, следует отметить кружками. Другой способ перевода
пятен в видимую форму заключается в том, что проявленную хроматограмму
помещают в закрытый сосуд, содержащий кристаллический иод (рис. 3.4). По-
чти все вещества (кроме алканов и галогеналканов) образуют с иодом ком-
плексы с переносом заряда. Образование таких комплексов очень часто пред-
ставляет собой обратимый процесс. Поэтому выявленные иодом пятна следует
отметить на пластинке возможно быстрее. Еще один способ перевода пятен в
видимую форму заключается в опрыскивании пятен серной кислотой. При этом
Рис. 3.5. Тонкослойная хроматограм-
ма. Анализ состава реакционной сме-
си.
1 — линия старта; 2—заведомая проба ис-
ходного продукта; 3—реакционная смесь;
4 —заведомая проба продукта реакции;
5—отметка фронта растворителя.
Рис. 3.6. Определение Rt на тонко-
слойной хроматограмме.
/ — линия старта; 2—пятно; 3—отметка
фронта растворителя.
бесцветные вещества темнеют в результате обугливания. Конечно, такой метод
следует рассматривать как деструктивный метод анализа.
Если нельзя воспользоваться имеющимися в продаже хроматографическими
пластинками, их можно без особого труда приготовить в лаборатории из стек-
лянных пластинок подходящего размера. Для этой цели готовят суспензию ад-
сорбента в подходящем растворителе (например, 35 г силикагеля G в 100 мл
хлороформа или 60 г оксида алюминия в 100 мл смеси 67% хлороформа с 33%
метанола). Энергично размешивают суспензию, погружают в нее пару сложен-
ных вместе предметных стекол для микроскопа и медленно вынимают обратно.
Избыток суспензии удаляют с краев пластинок, пластинки разделяют, удаляют
адсорбент, попавший на обратную сторону пластинок, и высушивают их (около
5 мин). Такие пластинки можно использовать сразу после приготовления или
хранить их в эксикаторе.
Обсуждение. Если при проявлении хроматограмм одной и той
же пробы на разных сорбентах (оксид алюминия, силикагель,
кремневая кислота) и в разных растворителях явно получается
54
Глава 3
одно пятно, то это является очень хорошим показателем чистоты
изучаемого вещества. Кроме того, идентичность неизвестного
вещества может быть подтверждена (но не доказана. Почему?)
путем сравнения положения пятен неизвестного соединения и из-
вестных стандартных веществ (рис. 3.5). На рис. 3.5 также по-
казано, как с помощью тонкослойной хроматографии можно на-
блюдать за ходом химической реакции. Наиболее обычным спо-
собом описания результатов тонкослойной хроматографии являет-
ся указание величины Rf (см. также рис. 3.6) на хроматографи-
ческих пластинках данного типа и в данной системе растворите-
лей:
расстояние, на которое продвинулся центр пятна характеризуемого
•___________________________ вещества___________________________
расстояние, на которое продвинулся фронт растворителя
В другом способе представления результатов тонкослойной хро-
матографии вычисляют величину Rx:
расстояние, иа которое продвинулся центр пятна Характеризуемого
___________________________вещества ______________
расстояние, на которое продвинулся центр Пятна стандартного
вещества
Информация, полученная при тонкослойной хроматографии, ока-
зывается полезной при выборе адсорбентов и растворителей для
хроматографии на колонке, в том числе препаративной (см.
гл. 7).
3.3. ОПРЕДЕЛЕНИЕ ФИЗИЧЕСКИХ СВОЙСТВ
3.3.1. Температура плавления*
Термином «температура плавления» обозначают диапазон тем-
ператур, в котором происходит превращение твердого тела в жид-
кость. Ввиду того что этот процесс часто сопровождается разло-
жением вещества, найденная величина может быть просто темпе-
пературой перехода твердое тело — жидкость, а не равновесной
температурой плавления. Если проба на горючесть показывает, что
вещество плавится достаточно легко (между 25 и 300°С), то тем-
пературу плавления следует определять по методу А (см. ниже).
Для более высокого диапазона температур плавения (300—500°С)
необходимо применять специальное оборудование (см., например,
рис. 3.10). Если при определении температуры плавления по ме-
тоду А зарегистрировано разложение вещества или его переход
* Skau Е. L., Arthur J. С., Jr., Wakeham Н., in «Physical Methods of Organic
Chemistry», Ed. A. Weissberger, 3rd ed.. Vol. 1, Part 1, Chapt. 7, Interscience,
New York, 1959.
Предварительное исследование, оценка чистоты
56
из одного кристаллического состояния в другое, то рекомендует-
ся использовать метод Б. Для веществ, плавящихся в пределах
от 0 до 25°С, следует применять метод определения температуры
замерзания (см. ниже в этом разделе).
Температуры плавления большого числа органических веществ
и их производных приведены в таблицах приложения III к настоя-
щей книге. Часто незначительное количество примеси может
вызвать заметное понижение (и увеличение интервала) наблюдае-
мой температуры плавления. Поэтому настоятельно рекомендует-
ся пользоваться методом определения температур плавления сме-
сей, описанным ниже. Если по каким-либо причинам имеющийся
образец загрязнен небольшими количествами примесей, то сле-
дует ознакомиться с разд. 7.3, посвященным перекристаллизации
веществ.
Определение температуры плавления смеси (смешанная проба).
Метод определения температуры плавления смесей (смешанная
проба) позволяет проверить идентичность двух разных проб твер-
дых веществ (конечно, имеющих одинаковые температуры плав-
ления) посредством измерения температуры плавления их меха-
нической смеси. Обычно смесь образцов неидентичных соединений
дает депрессию температуры плавления. Во многих случаях сме-
шанная проба оказывается весьма ценной на определенных эта-
пах процесса идентификации. Тем не менее заключения, сделан-
ные на основании температур плавления смешанной пробы неиз-
вестного вещества с заведомым соединением, взятым с полки, в
этом курсе не будут рассматриваться как доказательства струк-
туры неизвестного соединения.
Имеется несколько пар веществ, не дающих при смешивании
депрессии температуры плавления. Чаще всего отсутствие пони-
жения температуры плавления при смешивании двух веществ на-
блюдается лишь для смесей определенного состава. Измерение
температур плавления нескольких смесей разного состава не тре-
бует усилий, если воспользоваться методом, описанным ниже.
Два изучаемых компонента А и Б насыпают маленькими при-
мерно одинаковыми кучками на ровную поверхность. Смешивают
половину кучки вещества А с половиной кучки вещества Б. Затем
делят полученную смесь на три равные части. К первой из них
прибавляют оставшуюся часть компонента А, к третьей — остав-
шуюся часть компонента Б. Таким образом получаются три сме-
си: 80 и 20% Б, 50% А и 50%Б, 20% А и 80% Б. Температуры
плавления всех трех смесей могут быть определены одновремен-
но по методу, описанному ниже.
Метод А. Во многих случаях при определении температуры плавления при-
меняют капиллярные трубки длиной 6 см и около 1 мм в диаметре. Такие труб-
ки называют капиллярами для определения температуры плавления. Если нет
готовых капилляров, то их можно вытянуть из трубок большого диаметра (око-
ло 15 мм) или из пробирок, изготовленных из мягкого стекла. Для этого чисто
56
Глава 3
вымытые трубки размягчают в пламени газовой горелки. Небольшое количество
вещества, при необходимости измельченного в порошок, помещают иа чистую
твердую поверхность. Постукивая открытым концом капилляра по этой поверх-
ности там, где насыпаны кучки вещества, очень небольшое его количество вво-
дят внутрь капилляра (рис. 3.7, о). Затем капилляр переворачивают открытым
концом вверх и, деожа его вертикально, легко проводят по его краю напильни-
ком или ребристым краем монеты (рис. 3.7,6). Можно также несколько раз
сбросить капилляр открытым концом вверх на твердую поверхность стола вну-
три стеклянной трубки подходящего размера, (рис. 3.7, в). При этом вещество,
Рис. 3.7. Введение вещества в капилляр для определения температуры плавления
(а) и уплотнение его на дне капилляра (б, в).
набранное в открытый конец капилляра, будет сброшено на его дно. Подготов-
ленный таким образом капилляр можно затем установить в прибор для опре-
деления температуры плавления. Некоторые из таких приборов изображены на
рис. 3.8—3.10. Колонка вещества в правильно подготовленном капилляре должна
Предварительное исследование, оценка чистоты
51
иметь длину 2—3 мм. В приборе типа столика Фишера — Джонса (рис. 3.12)
вместо капилляров применяются покровные стекла для микроскопии.
Для экономии времени часто бывает полезно провести пред-
варительное определение температуры плавления при достаточно
Рис. 3.8. Простейший прибор для
определения температуры плавления.
В сосуд помещают минеральное масло (напри-
мер, нуйол), силиконовое или хлопковое масло.
Термометр t-> является вспомогательным и слу-
жит для введения поправки на выступающий
столбик ртути, / — индукционный искробез-
опасный электродвигатель; 2—резиновая проб-
ка № 9, одетая иа ось мешалки; 3—металли-
ческая шайба между пробкой 2 и подшипни-
ком 11; 4 — резиновая пробка № 6. зажатая
в лапку штатива и удерживающая подшипник;
5—кольцо из металла или пластика, «тайгой»
дчя крепления капилляра к термометру /г,
6—стандартный капилляр с пробой для опре-
деления температуры плавления (см. рис. 3.7);
7—кольцо штатива, удерживающее сосуд с на-
гретым маслом 12; «—цилиндрический защит-
ный экран горелки; 9—подача газа; 10—рези-
новая пробка № I, одетая иа ось электродви-
гателя; // — подшипник (например, латунное
пробочное сверло, подходящее по диаметру
к оси мешалки); 12—сосуде нагретым маслом;
13 — к электросети переменного тока; и
t?—термометры.
Рис. 3.9. Прибор Тиле для определе-
ния температуры плавления.
/—термометр (Л). Дополнительную инфор-
мацию см. иа рис, 3.8. Может быть уста-
новлен второй термометр для введения
поправки на выступающий столбик ртути
(см. термометр t2 иа рис. 3.8).
быстром подъеме температуры. После того как установлено при-
мерное значение температуры плавления, проводят второе опреде-
ление, причем температуру быстро поднимают до значения при-
мерно на 5°С ниже приблизительной температуры плавления,
найденной при первом определении. Дальнейший подъем темпера-
58
Глава S
туры осуществляют медленнее. Для каждого определения сле-
дует применять свежую порцию вещества.
Одной из причин получения заниженных значений и широких
интервалов температур плавления является применение недоста-
точно чистых капилляров. Это связано с наличием на их стенках
следов щелочи, катализирующей процессы альдольной конденса-
Рис. 3.10. Прибор Mel-Temp для определения температуры Плавления (воспро-
изведено с разрешения фирмы Laboratory Devices, Cambridge, Mass.).
а—общий вид прибора; б —кривые нагревания при различном напряжении (указаны предель-
ные температуры нагревания при данном напряжении); в — кривые напряжения, необходи-
мого для получения желаемой скорости нагревания вблизи ожидаемой температуры плавле-
ния (цифры на кривых —скорости нагревания, град/мин).
в
60
Глава 3
ции альдегидов и кетонов, мутаротацию сахаров и их производ-
ных и т. п. Например, при определении температуры плавления в
неочищенных трубках a-D-глюкоза размягчается при 133°С и пла-
вится при 142—146°С. В то же время в капиллярах, стенки ко-
торых очищены от следов щелочи, размягчение глюкозы происхо-
дит при 142°С, а плавление — при 146—147°С. Для того чтобы
избежать таких осложнений, следует применять чистые капилляр-
ные трубки, изготовленные из стекла пирекс.
Рис. 3.11. Калибровочная кривая для определения температур плавления.
Исправленные температуры плавления. Применяемый термо-
метр обязательно нужно прокалибровать по наблюдаемым темпе-
ратурам плавления нескольких чистых веществ, например, таких,
как приведенные ниже:
Т. пл. (нспр.), “С Т. пл. (испр.к “С
0 Лед 187 Гиппуровая кислота
53 н-Дихлорбсизол 200 Изатин
90 лиДинитробеизол 216 Антрацен
114 Ацетанилид 238 Карбанилид
121 Бензойная кислота 257 Оксанилид
132 Мочевина 281 Антрахинон
157 Салициловая кислота 332 М,М'-Диацетилбензидин
Если при всех определениях температур плавления использо-
вать один и тот же прибор с одним и тем же термометром, то
удобно построить калибровочную кривую такого типа, как по-
казано на рис. 3.11. Это позволит в дальнейшем сэкономить вре-
мя. Для построения такого графика откладывают на осях коорди-
нат наблюдаемые температуры плавления стандартных соединений
против их исправленных значений и проводят через полученные
Предварительное исследование, оценка чистоты
61
точки плавную кривую АО. При последующих определениях тем-
ператур плавления от оси ординат проводят через точку Б со-
ответствующую наблюдаемой температуре горизонтальную пря-
мую до пересечения с калибровочной кривой. Далее эту точку
пересечения переносят на ось абсцисс, получая исправленное зна-
чение температуры плавления В. Такая калибровочная кривая
включает погрешности градиуровки термометра и поправки на
выступающий столбик ртути. Термометр следует прокалибровать
по наблюдаемым температурам плавления нескольких чистых ве-
ществ.
Весьма важно отмечать диапазон температур, в котором пла-
вится неизвестное вещество, так как это является хорошим пока-
зателем его чистоты. Большинство чистых органических соеди-
нений плавится в пределах 0,5°С или плавится с разложением в
узком интервале температур (около 1°С). Если пределы темпера-
тур, в которых плавится или разлагается данное вещество, слиш-
ком широки, то это соединение следует перекристаллизовать из
подходящего растворителя и повторить определение температуры
плавления. Некоторые органические соединения, такие, как амино-
кислоты, соли кислот или аминов, углеводы и др., плавятся с раз-
ложением в довольно большом интервале температур.
При использовании описанной выше калибровочной кривой мо-
жно ввести поправку на выступающий столбик ртути. Поправку
б вычисляют по формуле
б = + У(Л-/2)(1,54.10’3)
где /V— число градусов, соответствующее длине столбика ртути,
выступающего выше уровня жидкости, — наблюдаемая темпера-
тура плавления и /2— средняя температура столбика ртути над
поверхностью жидкости. Ввиду того что эта последняя величина
может быть оценена лишь приблизительно (способ измерения
температуры t2 показан на рис. 3.8), а также учитывая, что кали-
бровочные кривые обычно включают поправку на выступающий
столбик ртути, описанный способ расчета применяется редко.
Если вещество плавится выше 250°С, его температуру плав-
ления можно определить с помощью блока Макенна *.
На рис. 3.12 показан прибор Фишера — Джонса, состоящий
из нагреваемого электрическим током алюминиевого блока и тер-
мометра на 300°С. Пробу помещают между двумя покровными
стеклами для микроскопии размером 18X18 мм Стекла устанав-
ливают в выточенное в алюминиевом блоке углубление. Темпе-
ратуру блока регулируют с помощью лабораторного автотранс-
форматора, а за процессом плавления вещества наблюдают через
специальное окно с увеличительным стеклом. Для этого прибора
* Конструкция этого блока описана в книгах по лабораторной технике,
указанных в гл. 10.
62
Глава 3
также необходимо получить калибровочную кривую, построенную,
как описано выше, по температурам плавления известных веществ.
Метод Б. Показанный на рис. 3.13 прибор Нальджа-Аксельрода включает
микроскоп с 25-кратным увеличением и поляроидным вкладышем, алюминиевый
блок с электрообогревом, скорость которого регулируется автотрансформатором,
и термометр на 400°С. Пробу помещают в углубление нагревательного блока
между покровными стеклами для микроскопа размером 18 X 18 мм. Углубление
Рис. 3.12. Прибор Фишера — Джонса для определения температуры плавления
(воспроизведено с разрешения фирмы Fisher Scientific Со., Pittsburg, Ра.).
закрывают крышкой с небольшим отверстием, включают осветитель и фокуси-
руют микроскоп на кристаллы исследуемого вещества. Поворачивают тубус так,
чтобы поляроиды были бы почти полностью скрещены, и наблюдают кристаллы
при повышении температуры. Температуру плавления анизотропных кристаллов
при этом заметить очень легко, так как при их плавлении пропадает яркая по-
ляризационная окраска. Сходным образом легко заметить также и переходы из
одной аллотропной модификации в другую. Для описанного прибора также не-
обходимо получить калибровочную кривую (см. выше).
В обычных экспериментах по определению температур плав-
ления (метод А, см. выше) термином «температура плавления»
обозначают интервал температур от первого появления жидкой
фазы до исчезновения последней частички твердой фазы. При ис-
пользовании техники эксперимента с микроскопными покровными
стеклами приложение такого определения наталкивается на не-
которые затруднения. Ввиду того что в этом случае кристаллы
исследуемого вещества хорошо отделены друг от друга, обычно
отсутствует контакт между плавящимися и неплавящнмися кри-
Предварительное исследование, оценка чистоты
63
сталлами. Поэтому во многих случаях расплавление всей пробы
происходит в очень широком интервале температур. Вследствие
этого такой способ определения лучше не применять. Температу-
Рис. 3.13. Прибор Нальджа—Аксельрода для определения температуры плавле-
ния (воспроизведено с разрешения фирмы Nalge Со., Rochester, N. Y.).
ры плавления, определяемые в стеклянных капиллярах, представ-
ляют собой более точный и более значимый критерий чистоты
вещества.
Температура замерзания
Несколько миллилитров жидкости помещают в обычную про-
бирку, снабженную термометром и мешалкой, изготовленной из
медной, никелевой или платиновой проволоки. Пробирку закреп-
ляют на пробке в другой, несколько более широкой пробирке,
погруженной в баню со льдом или с охлаждающей смесью из
льда и соли или из ацетона и сухого льда. В процессе измере-
ния жидкость в пробирке интенсивно перемешивают. Темпера-
туру жидкости отмечают по термометру (рис. 3.14). Как только
64
Глава 3
начнется образование кристаллов, пробирку извлекают из охлаж-
дающей бани и по термометру отмечают температуру. Температу-
рой замерзания является та температура, которая достигается при
ее подъеме после исчезновения эффекта первоначального переох-
лаждения вещества. Температура охлаждающей баии должна быть
лишь немного ниже температуры за-
© мерзания определяемого вещества.
I Температуры замерзания большинства
органических жидкостей, получаемые
в обычных определениях, должны рас-
сматриваться лишь как приближен-
ные. Следует заметить, что это оп-
ределение требует относительно боль-
ших проб вещества.
Описан и более сложный прибор
s'' для определения температур замерза-
/ ния (до—65°С)*.
Рис. 3.14. Простой прибор для
определения температуры за-
мерзания.
ртутный шарик находился чуть
3.3.2. Температура кипения
Применение температуры кипения
веществ для идентификации неизве-
стных соединений описано в разд. 2.3.
Метод А. Собирают небольшую установку
для перегонки жидкостей, сходную с пока-
занной на рис. 3.15. Можно применять для
этой цели также прибор на шлифах, показан-
ный на рис. 3.16 (предпочтительны нормальные
шлифы). Для конденсации паров в качестве
приемников служат пробирки, погруженные
в стакан со льдом. В колбу прибора помещают
10 мл исследуемой жидкости и несколько ку-
сочков пемзы или фаянса для равномерного
кипения и предотвращения толчков. В горло
колбы вставляют термометр, так чтобы его
ниже бокового отвода. Жидкость нагревают до
кипения с помощью маленькой горелки с защитным экраном **. Перегонку жид-
кости ведут по возможности с постоянной скоростью. После отгонки первых
2—3 мл дистиллата, не прерывая отгонки, заменяют приемник и в чистую сухую
пробирку собирают еще 5—6 мл дистиллата. Постоянная температура на термо-
метре устанавливается с довольно большой задержкой, однако при сборе вто-
рой порции отгона уже удается отметить диапазон изменения температуры ки-
пения жидкости. Этот диапазон и регистрируют как температуру кипения ис-
следуемого вещества.
Интервал температур кипения является хорошим показателем
чистоты вещества. Многие органические жидкости гигроскопичны,
* Curtis R. J., Dolenga A., J. Chem. Educ., 52, 201 (1975).
** Если есть возможность, то лучше для нагревания жидкости до кипения
применять нагревательные рубашки (разд. 7.2.1). Это уменьшает опасность по-
жара, связанную с применением горелки с открытым пламенем.
Предварит ельное исследование, оценка чистоты 65
а некоторые из них разлагаются при хранении. Обычно первые не-
сколько миллилитров дистиллата содержат воду или более лету-
чие примеси, и лишь вторая фракция представляет собой чистое
вещество. Если интервал температур кипения окажется слишком
Рис. 3.15. Простейший прибор для перегонки.
В перегонную колбу помещают кусочки пемзы нлн фаянса. Вместо газовой горелки можно
применять электрическую нагревательную рубашку (разд. 7.2.1). Нагревательные рубашки
в общем более безопасны, чем газовые горелки, так как при их использовании уменьшается
возможность воспламенения горючих жидкостей. 1 — колба на 25 мл; 2— асбестовая сетка;
3 —зажим.
большим, то исследуемую жидкость следует подвергнуть повтор-
ному фракционированию на подходящей ректификационной ко-
лонке (см. гл. 7).
Определение температур кипения перегонкой небольших ко-
личеств жидкости, выполняемой, как описано выше, часто приво-
дит к ошибочным значениям. Без специальных мер предосторож-
ности пары перегоняемой жидкости легко перегреть. Кроме того,
наблюдаемые температуры кипения высококипящих жидкостей мо-
гут оказаться слишком низкими, так как требуется достаточное
время для того, чтобы ртуть в шарике термометра нагрелась до
3 Зак. 335
66
Глава 3
температуры окружающего пара. Вторая фракция, полученная при
разгонке исследуемой жидкости по методу, описанному выше, дол-
жна быть использована для более точного определения темпера-
туры кипения по изложенному ниже методу Б. Отдельные порции
Рис. 3.16. Стандартный прибор на шлифах для перегонки жидкостей. Нагрева-
тельные приборы, подводы охлаждающей жидкости и штативы не показаны.
I—термометр; 2—резнноваи муфта или шлиф; 3 —иасадка Вюрца; 4— нагревание; 5—-слив
охлаждающей воды; 6—холодильник; 7—подача охлаждающей воды; 8—приемник.
той же основной фракции должны быть также использованы по
возможности для всех последующих химических и спектральных
анализов и физических измерений.
Метод Б. Пробирку для микроопределення температуры кипения готовят так,
как показано на рис. 3.17. Большую пробирку диаметром 5 мм берут готовую
или изготовляют из подходящей стеклянной трубки. В нее помещают открытым
концом вниз капилляр для определения температуры плавления. Добавляют две
капли жидкости, температуру кипения которой нужно определить, и прикреп-
ляют пробирку к термометру прибора для определения температуры плавления
(рис. 3.8 или 3.9). Медленно поднимают температуру в приборе до тех пор, пока
из отверстия капилляра ие начнет выходить быстрая и равномерная струя пу-
зырьков пара, проходящих через слой жидкости. Тогда нагревание прекращают
и дают бане медленно охлаждаться при непрерывном перемешивании. Замечают
температуру в тот момент, когда пузырьки пара перестанут выходить из отвер-
стия капилляра, т. е. перед тем, как в него войдет жидкость. Эта температура
и есть температура кипения. Обычно результат, полученный таким способом,
оказывается значительно точнее, чем при определении по методу А.
П редварительное исследование, оценка чистоты
67
Таблица 3.2
Изменения температур кипения при небольших изменениях
атмосферного давления
Т. кип., °C Т. кип., К Поправка при разнице давлений 10 мм рт. ст. (1,3 кПа), °C
неассоциированные жидкости ассоциированные а ЖИДКОСТИ
50 323 0,38 0,32
100 373 0,44 0,37
150 423 0,50 0,42
200 473 0,56 0,46
300 573 0,68 0,56
400 673 0,79 0,66
500 773 0,91 0,76
а Ассоциированными называют жидкости, в которых имеются значительные межмолеку-
ляриые ассоциации, обусловленные наличием водородных связей. Примером может служить
метанол.
Влияние давления на температуру кипения. Необходимо реги-
стрировать атмосферное давление во время определения темпера-
туры кипения. Величина таких «барометрических» поправок тем-
пературы кипения для значений атмосферного давления, не отли-
чающихся от 760 мм рт. ст. (101,3 кПа)
более чем на 30 мм (4 кПа), приведена
в табл. 3.2.
Из данных табл. 3.2 очевидно, что при
обычной работе можно пренебрегать лишь
малыми отклонениями [в пределах 5 мм
рт. ст. (650 Па)] атмосферного давления
от 760 мм рт. ст. (101,3 кПа).
Исследователи, работающие в лабора-
ториях, расположенных на больших высо-
тах в условиях низкого атмосферного дав-
ления *, обычно заранее определяют набор
эмпирических поправок, прибавление кото-
рых к наблюдаемым температурам кипения
дает их значения при 760 мм рт. ст.
(101,3 кПа). Эти поправки найдены
в результате перегонки большого числа различных веществ с раз-
ными температурами кипения. Различия между наблюдаемыми и
приведенными в литературе температурами кипения представляют
собой такие поправки.
* На вершине горы Маунт-Эванс в штате Колорадо (США) вода кипит
при 81 °C [среднее давление 460—470 мм рт. ст. (61,2—62,6 кПа), высота
4700 м]. В Университете Колорадо на высоте около 1700 м вода кипит при тем-
пературе около 90 °C,
Рис. 3.17. Микрометод
определения температу-
ры кипения с помощью
капиллярной трубки дли-
ной около 5 см.
3*
68
Глава 3
Разработаны номограммы, связывающие температуры кипения
различных органических веществ с величиной давления. Такие гра-
фики, приведенные в приложении I настоящей книги, могут быть
полезны при проведении перегонки в вакууме.
Для того чтобы читатель получил представление, как меняются
с давлением температуры кипения, в табл. 3.3 приведены данные
Таблица 3.3
Температуры кипения (°C) при пониженных давлениях
Соединение Давленые, мм рт. ст. (кПа) дг а, °C
760 (101,3) 700 (93,2) 650 (86,5) 600 (79,8) 550 (73,2)
м-Гептаи 98 96 94 91 88 10
Пропанол-1 97 95 93 91 89 8
Иодбензол 188 185 182 179 175 13
н-Валериановая кис- 186 183 180 178 175 11
лота
Флуорен 298 294 290 286 282 16
Р-Нафтол 295 292 288 284 280 15
а ДТ равно разности температур кипения при 760 мм рт. ст. (101,3 кПа) и 550 мм рт. ст.
(73,2 кПа).
Таблица 3.4
Температуры кипения и длина цепи для углеводородов
нормального строения
Соединение Т. кип., °C Разность температур кипения соседних членов ряда, °C
Пентан 36 32
Гексан 68 30
Гептан 98 27
Октан 125 24
Нонан 149 24
Декан 173 21
Ундекан 194 21
Додекан 215
Предварительное исследование, оценка чистоты
69
о трех парах ассоциированных и неассоциированных жидкостей.
Согласно этим данным, при снижении давления температуры ки-
пения ассоциированных жидкостей уменьшаются не так сильно, как
температуры кипения неассоциированных жидкостей.
Корреляция температур кипения и структуры веществ. По мере
увеличения молекулярной массы членов данного гомологического
Рис. 3.18. Зависимость температур кипения от молекулярной массы органических
веществ.
ряда их температура кипения возрастает. При этом хотя темпе-
ратура кипения возрастает монотонно (рис. 3.18), ее инкремент на
одну СН2-группу не остается постоянным. В начале любого гомо-
логического ряда температура кипения повышается быстрее, чем
у его последующих членов (табл. 3.4).
Если атом водорода в молекуле насыщенного углеводорода
(алкана) заместить каким-либо другим атомом или группой ато-
мов, то температура кипения повысится. Поэтому алкилгалогениды,
спирты, альдегиды, кетоны, органические кислоты и другие сое-
динения кипят выше, чем углеводороды с таким же углеродным
скелетом.
70
Глава 3
Если природа введенной в углеводород группы такова, что мо-
жет вызывать ассоциацию молекул образующегося при замещении
соединения, то температура кипения может очень сильно увели-
читься. Особенно явно этот эффект выражен у спиртов (рис. 3.18)
и кислот, способных к образованию водородных связей. Например,
различие температур кипения пропана (неассоциированного) и про-
панола-1 (ассоциированного) составляет 142°С. Это различие на-
много больше, чем следовало бы ожидать, исходя из увеличения
молекулярной массы. Введение большего числа гидроксильных
групп в молекулу также влечет за собой увеличение температур
кипения замещенных соединений. Однако это увеличение уже не
столь велико, как вызванное первой гидроксильной группой. Все
же инкремент роста температуры кипения на одну гидроксильную
группу оказывается много большим, чем инкремент на метилено-
вую группу (табл. 3.5).
Таблица 3.5
Температуры кипения веществ при последовательном замещении
атомов водорода иа гидроксильные группы
Соединение Т. кип., 'С Иикремеит температур кипения на одну гидроксильную группу, °C
СН3—СН2—СН3 -45 142
СН3—СН2—СН2ОН 97 119
СН2ОН—СН2—СН2ОН 216 74
СН2ОН—СН(ОН)—СН2ОН 290
Если гидроксильная группа заменяется структурой простого
эфира, ассоциация за счет водородных связей исчезает и темпе-
ратура кипения понижается. Следующий ряд иллюстрирует этот
эффект:
СН2ОН СН2ОС2Н5 СН2ОС2Н5 СН2ОС2Н6
СНОН 1 СНОН 1 СНОН 1 СНОС2Н5
1 СН2ОН 1 СН2ОН 1 СН2ОС2Н5 1 СН2ОС2Н5
290°С 230°С 191°С 185°С
Сравнение кислородсодержащих производных с их сернистыми
аналогами также указывает на то, что ассоциация молекул силь-
нее влияет на температуру кипения, чем увеличение молекулярной
массы. Тиолы RSH лишь слабо ассоциированы. Поэтому они кщ
П редварителоное исследование, оценка чистоты
71
пят ниже, чем их кислородные аналоги, даже несмотря на то, что
последние имеют существенно меньшие молекулярные массы.
Т. кип., “С Т. кип., °C
ион 100 HSH -62
СН3ОН 66 CH2SH 6
СНзСООН 119 CH2COSH 93
Простые эфиры и тиоэфиры неассоциированы. Поэтому имеющие
более высокую молекулярную массу алкилсульфиды кипят выше,
чем соответствующие эфиры:
Т. кип., °C Т. кип., °C
(СН3)2О —24 (CH3)2S 38
(С2Н5)2О 35 (C2H5)2S 92
Эти данные по кислород- и серусодержащим соединениям, а
также по углеводородам, алкилхлоридам, алкилбромидам и ал-
килиодидам иллюстрируют общее правило: замещение любого
атома в молекуле на атом большей молекулярной массы приво-
дит к повышению температуры кипения, если это замещение не
вызывает увеличения или уменьшения степени ассоциации моле-
кул.
Так же как и в случае взаимозависимости строения веществ
и их растворимости (гл. 5), разветвление углеродной цепи и по-
ложение функциональных групп оказывают влияние на темпера-
туры кипения. Из данных температур кипения алифатических
спиртов, приведенных в табл. 3.6, можно сделать следующие вы-
воды. Во-первых, среди изомерных спиртов наибольшие темпе-
ратуры кипения имеют спирты с неразветвленной цепью углерод-
ных атомов. Во-вторых, температуры кипения изомерных спиртов
одного и того же типа тем ниже, чем больше степень разветвле-
ния углеродного скелета молекулы. В-третьих, сравнение темпе-
ратур кипения изомерных первичных и вторичных спиртов с од-
ним и тем же числом атомов углерода в молекуле показывает,
что первичные спирты кипят выше вторичных, а последние в свою
очередь выше третичных спиртов. Знание температур кипения не-
которых относительно простых соединений часто оказывается по-
лезным, так как позволяет исключить из рассмотрения некоторые
72
Глава 3
Таблица 3.6
Зависимость температуры кипения спиртов от степени разветвленности
углеродного скелета молекул
Первичные спирты
Вторичные спирты
Третичные спирты
формула
формула
формула
СН3ОН
СН3СН2ОН
СН3СН2СН2ОН
СН3СН2СН2СН2ОН
СН3СН—СН2ОН
СНз
СН3СН2СН2СН2СН2ОН
СН3СНСН2СН2ОН
vn3
СН3СН2СНСН2ОН
66
78
97
116
108
138
131
129
СН3СНСН2
он
СН3СН2СНСН3
ОН
СН3СН2СН2СНСН3
он
СН3СН2СНСН2СН3
ОН
82
92
119
115
СНз
СНз—С—СНз
ОН
83
S
S
S
СНз
СНз
СНз
СН3—С—СН2ОН
СН3
114
СНз—СН—СНСНз
1з он
111
СН;
!3СНг-С—СН,
ОН
102
типы веществ. При этом могут оказать помощь следующие про-
стые обобщения.
1. Органическое соединение, содержащее хлор и кипящее ни-
же 132°С, должно быть алифатическим. Если такое вещество ки-
пит выше 132°С, оно может быть как алифатическим, так и
ароматическим. Это является следствием того факта, что простей-
ший арилгалогеннд — хлорбензол кипит при 132°С.
2. Аналогично вещества, содержащие бром и кипящие ниже
157°С или содержащие иод и кипящие ниже 188°С, должны быть
алифатическими. Другие соединения, содержащие бром или иод,
могут быть либо алифатическими, либо ароматическими.
Предварителъное исследование, оценка чистоты
73
3.3.3. Плотность
Использование данных о плотности при идентификации не-
известных веществ было рассмотрено в разд. 2.3. Следует вспом-
нить, что в общем виде удельная масса q^ вещества 2 опреде-
ляется следующим образом:
где W2 — масса точно измеренного объема вещества 2 (неизвест-
ного), w\ — масса точно такого же объема вещества 1 (обычно
Рис. 3.19. Микропикяометр.
Рис. 3.20. Сосуд для определения
плотности (миниатюрная мерная кол-
бочка) .
воды), а Т2 и Т\ — температуры этих веществ в момент измерения
их массы. Отсюда легко рассчитать плотность d2 вещества 2:
(<Ч, = ?Ж)Г1
где (rfi)ri — плотность воды или иного стандартного веще-
ства при температуре Л, а №)г2— плотность определяемого
вещества 2 при температуре Т2. Значения плотностей приведены
во многих стандартных химических руководствах.
Величину удельной массы можно определить с помощью ма-
ленького пикнометра.
Методы определения. Если нет готовых пикнометров емкостью 1—2 мл, то
можно воспользоваться одним из двух сосудов, показанных иа рис. 3.19 н 3.20.
Пикнометр, изображенный на рис. 3.19, изготовляют из отрезка стеклянной
капиллярной трубки с внутренним диаметром 1—2 мм. Трубку изгибают, как
показано на рис. 3.19, в ее средней части раздувают маленький шарик, а един
конец вытягивают в тонкий капилляр. Пикнометр подвешивают с помощью тон-
кой нихромовой, алюминиевой или платиновой проволоки и определяют его
массу на аналитических весах с точностью -_t0,5 мг. Затем пикнометр заполняют
74
Глава 3
дистиллированной водой чуть выше метки и подвешивают в бане с постоянной
температурой (например, 20°С). Через 10 мин доводят до метки уровень воды
в пикнометре, отбирая воду из оттянутого кончика капилляра кусочком филь-
тровальной бумаги до тех пор, пока мениск жидкости в другом колене не сов-
падет с меткой. Затем пикнометр вынимают из бани, обсушивают и взвеши-
вают.
Число атомов углерода
Рис. 3.21. Зависимость плотности от молекулярной массы веществ.
На рис. 3.20 изображен сосуд другого типа для определения удельной
массы. Можно использовать имеющиеся в продаже мерные колбочки на 1,00 мл
или несколько большего объема. Определяют массу пустого сосуда. Затем за-
полняют его дистиллированной водой и подвешивают на проволоке в бане с по-
стоянной температурой (например, 20°С). Уровень воды в сосуде доводят до
метки с помощью микропипетки. Затем сосуд извлекают из бани, обсушивают
и взвешивают. Значение массы пустого пикнометра и его массы с дистиллиро-
ванной водой записывают и сохраняют для последующих расчетов. Для опреде-
ления удельной массы неизвестной жидкости заполняют ею пикнометр, тер.мо-
статируют его при 20°С и взвешивают:
масса пробы
масса воды
„20
920
(Зразу после употребления пикнометр тщательно моют и высушивают.
Предварительное исследование, оценка чистоты
75
Следует особое внимание уделять чистоте пробы вещества, применяемого
для определения плотности. Лучше всего использовать для этой цели порцию
средней фракции, собранной при перегонке вещества, или газохрдматографнче-
ской фракции, соответствующей одному пику (гл. 7). Иногда необходимо опре-
делить плотность вещества по отношению к плотности воды, находящейся при
температуре 4°С. Это можно сделать, введя множитель 0,99823:
20= M££££JIP£6iL.o,99823
* масса воды
Обсуждение. Во многих случаях с помощью сведений о плот-
ности исследуемой жидкости можно исключить некоторые веще-
ства из числа возможных структур. Плотность зависит от состава
и строения данного вещества.
Углеводороды обычно легче воды. В данном гомологическом
ряду по мере увеличения молекулярной массы плотность возра-
стает, однако инкремент, соответствующий увеличению молекулы
на одну метиленовую группу, постепенно уменьшается. Измене-
ние плотности в гомологических рядах алканов, 1-алкенов и 1-ал-
кинов показывают кривые I, II и III на рис. 3.21. Следует заме-
тить, что плотность ацетиленовых углеводородов превышает плот-
ность соответствующих олефинов, а последние в свою очередь об-
ладают большей плотностью, чем алканы с тем же числом ато-
мов углерода в молекуле. Положение ненасыщенной связи в мо-
лекуле также оказывает влияние на плотность вещества. Пере-
мещение двойной связи в среднюю часть молекулы влечет за со-
бой увеличение плотности веществ. Эти изменения иллюстрирует
табл. 3.7.
Таблица 3.7
Зависимость плотности непредельных углеводородов от положения
двойных связей в молекуле
Углеводород
Формула
Пентен-1
Пентен-2
Пентадиеи-1,4
Пентадиен-1,3
Пентадиен-2,3
Гексен-1
Гексеи-2
Гексен-3
СН2=СНСН2СН2СН3
СН3СН=СНСН2СН3
СН2=СНСН2СН=СН2
СН2=СНСН=СНСН3
СН3СН=С==СНСН3
СН»=г=СНСН2СН2СН2СН3
СН3СН=СНСН2СН2СН3
СН3СН2СН=СНСН2СН3
0.64625
0,65125
0,659
0,696
0,702
0,673
0,681
0,722
Замещение одного атома в модекуле органического соединения
на атом с большей атомной массой обычно приводит к увеличению
плотности. При этом кривая IV на рис. 3.21, соответствующая ал-
килхлоридам нормального строения, располагается выше кривых
для углеводородов. Следует заметить, что алкилхлориды легче
воды и их плотность уменьшается с увеличением молекулярной
массы.
76
Глава 3
Довольно ограниченные данные по алкилфторидам представ-
лены на рис. 3.21, кривая V. Этот график интересен тем, что ука-
зывает лишь на очень незначительное изменение плотности с уве-
личением числа атомов углерода в молекуле.
Кривые VI и VII на рис. 3.21 показывают, что плотности пер-
вичных алкилбромидов и алкилиодидов превышают единицу и что
в этих гомологических рядах плотность уменьшается при увели-
чении числа атомов углерода в молекуле. Наклон кривых IV, VI и
VII постепенно уменьшается. Это отражает тот факт, что по мере
увеличения молекулярной массы вследствие добавления метилено-
вых звеньев атом галогена составляет все меньшую часть моле-
кулы алкилгалогенида. Относительное расположение кривых на
рис. 3.21 показывает, что плотность возрастает в следующем ряду:
RH < RF < RC1 < RBr < RI
При этом между собой сопоставляются алкилгалогениды, имею-
щие одинаковый углеродный скелет. Вторичные и третичные ал-
килхлориды, алкилбромиды и алкилиодиды образуют аналогич-
ные ряды.
Плотности арилгалогенидов также изменяются по мере воз-
растания атомной массы заместителей (табл. 3.8).
Таблица 3.8
Температуры кипения и плотности арилгалогенидов
Соединение Г. кип., вС .20 d4
Бензол 79,6 0,878
Фторбензол 86 1,024
Хлорбензол 132 1,107
Бромбензол 156 1,497
Иодбензол 188 1,832
Увеличение числа атомов галогенов в молекуле приводит к уве-
личению плотности веществ. Соединения, содержащие в молекуле
два или более атомов хлора либо один атом хлора совместно с
атомом кислорода или ароматической группировкой, обычно име-
ют плотность, превышающую 1,000 (табл. 3.9).
Введение в молекулу функциональных групп, содержащих ки-
слород, приводит к увеличению плотности соединений. Характер
изменения плотности в гомологических рядах некоторых распрост-
раненных органических веществ показывают кривые, приведен
ные на рис. 3.22. Из всех органических соединений, содержащих
кислород, наименьшая плотность у простых эфиров (кривая VIII
на рис. 3.22). Алифатические спирты (кривая IX) тяжелее про-
стых эфиров, но легче воды. Если в молекулу спирта ввести атом
хлора (этиленхлоргидрин), вторую гидроксильную группу (эти-
Предварительное исследование, оценка чистоты
77
Таблица 3.9
Изменение плотности при увеличении числа атомов хлора
и кислорода в молекуле
Соединение .7, dT, тР °C т2 °C
Бензилхлорид 1,1026 18 4
Бензилиденхлорид 1,2557 14 4
Бензотрихлорид 1,3800 20 20
Метиленхлорид 1,336 20 4
Хлороформ 1,4984 15 4
Четыреххлористый углерод 1,595 20 4
Этиленхлоргидрнн 1,213 20 4
Хлорацетон 1,162 16 4
Метил хлорацетат 1,235 20 20
ленгликоль) или ароматическое ядро (бензиловый спирт), то об-
разующиеся соединения имеют плотности, превышающие единицу.
Прогиб на кривой IX связан с тем фактом, что метанол сильнее
ассоциирован, чем этанол. Амины (кривая X на рис. 3.22) менее
ассоциированы, чем спирты, и обладают меньшей плотностью. Ас-
социация молекул приводит к тому, что плотность муравьиной и
уксусной кислот оказывается выше 1,000. Жирные кислоты с бо-
лее высокой молекулярной массой легче воды (кривая XI на рис.
3.22). Простейшие сложные эфиры (кривая XII) и альдегиды
RCHO (кривая XIV) легче воды, тогда как плотность сложных
эфиров многоосновных кислот (кривая XIII), а также сложных
эфиров, содержащих атомы галогенов, гидроксильные группы или
кетогруппы, превышает плотность воды. Введение в молекулу сло-
жных эфиров ароматического ядра также может приводить к то-
му, что их плотность будет выше плотности воды. Примерами
таких сложных эфиров, более тяжелых, чем вода, являются фенил-
ацетат, метилбензоат, бензилацетат, этилсалицилат, н-бутилокса-
лат, триацетин, изопропилтартрат и этилцитрат. Ввиду того что
плотность углеводородов меньше, чем плотность воды, следует
ожидать, что сложные эфиры, содержащие в молекуле длинные
цепи углеводородных радикалов, будут иметь соответственно по-
ниженные значения плотности.
Обычно соединения, содержащие несколько функциональных
групп, особенно таких, которые способствуют ассоциации молекул,
имеют плотность, превышающую 1,0. Сведения о том, тяжелее
или легче воды данное соединение, уже дают некоторое представ-
ление о его сложности. Такие данные могут оказаться весьма цен-
ными, особенно в случае нейтральных жидкостей. Если какое-либо
вещество ие содержит атомов галогенов и имеет плотность меньше
единиц, то, скорее всего, в его молекуле имеется какая-либо одна
функциональная группа в сочетании с углеводородной цепью или
Глава 3
1,250
1,200
1.150
1,100
1,080
1,060
0.880
1,020
ЧГ 1,000
0.980
0.860
0.840
0.820
0,800
0,780
0,760
0.740
0.720
0.700
О 1 2 3 4 5 6 7 8 9 10 11 12 13
Число атомов углерода
Рис. 3.22. Зависимость плотности от молекулярной массы веществ.
фрагментом простого эфира. Если же данное вещество тяжелее
воды, то весьма вероятно, что в его состав входит несколько раз-
личных функциональных групп.
3.3.4. Показатель преломления жидкостей
Показатель преломления жидкостей равен отношению синуса
угла падения к синусу угла преломления луча света, переходя-
Предварительное исследование, оценка чистоты
79
щего через границу раздела воздуха и жидкости (рис. 3.23). При
этом на границе раздела двух фаз происходит изменение скоро-
сти (Увоздух-»-Ужидк) и направления распространения световых
волн. Величина этого изменения зависит от температуры (Т) и of
длины волны X падающего света. Непосредственно измерить угол
падения и угол преломления трудно. Поэтому для измерения по-
казателя преломления были разработаны оптические системы,
позволяющие измерять критический угол отражения на границе
исследуемой жидкости и стеклянной призмы с известным показа-
телем преломления. На этом принципе основан рефрактометр
Аббе. Различные разновидности таких приборов имеются в про-
даже *. Рефрактометры типа Аббе имеют следующие преимуще-
ства. Во-первых, приборы позволяют пользоваться источником бе-
лого света. Тем не менее система призм этих приборов позволяет
определить значения показателя преломления, соответствующие
длине волны желтой линии D в спектре натрия. Во-вторых, для
измерения достаточно нескольких капель жидкости. В-третьих,
приборы снабжены устройствами для термостатирования призм.
* Например, такие приборы поставляет фирма Bausch and Lomb Optical Co.
(США). Списки других организаций, изготовляющих такие приборы, ежегодно
публикуются в журналах по аналитической химии.
80
Глава 3
В-четвсртых, наличие в приборе компенсационных призм Амичи
позволяет определять удельную дисперсию показателя преломле-
ния.
Схема оптической системы рефрактометра Аббе марки 3L, из-
готовляемого фирмой Bausch and Lomb Optical Co., приведена на
рис. 3.24, а общий вид этого прибора изображен на рис. 3.25.
Рис. 3.24. Схема оптической системы рефрактометра Аббе 3L (согласно ин-
струкции к прибору, воспроизведено с разрешения фирмы Bausch & Lomb Opti-
cal Со., Rochester, N. Y.). Объяснения см. в тексте.
Через термостатирующие рубашки 5, окружающие призмы 6 и 7, пропу-
скают воду с температурой 20°С. Маловязкие текучие жидкости вводят в за-
зор 4 между призмами с помощью пипетки через специальный канал. Если ис-
следуемая жидкость очень вязкая, то призму 6 поднимают и распределяют не-
сколько капель жидкости по поверхности призмы 7 с помощью деревянного
шпателя. Затем осторожно опускают верхнюю призму. Избыток жидкости при
этом выдавливается наружу. Включают осветительную лампу 3. Смотря в оку-
ляр 1, вращают верньер грубой регулировки 11 и регулируют положение лампы
3 так, чтобы получить в окуляре равномерно освещенное поле зрения. Затем
фокусируют окуляр 1 на перекрестии волосков 2. После этого вращают верньер
Предварительное исследование, оценка чистоты
81
11 до тех пор, пока граница раздела темной и светлой зон не пройдет точно че-
рез перекрестие волосков в центре поля зрения. Обычно граница освещенной
зоны окрашена. Окраску устраняют, вращая вннт ахроматизирующих диспер-
сионных призм 8 до тех пор, пока в середине поля зрения не будет видна чет-
кая неокрашенная граница раздела светлого и темного полей *. Винт 8 регули-
рует положение двух призм А.мичи 9 и 10, которые компенсируют различия в
Рис. 3.25. Рефрактометр Аббе 3L (воспроизведено с разрешения фирмы Bausch
& Lomb Optical Со., Rochester, N. Y.).
1 — окуляр; 2—барабан; 3—термостат и рующая рубашка; 4— нижняя призма; 5—верхняя
призма; 6—осветительная лампа; 7—термометр; 8 — грубая регулировка; 9—тонкая регули-
ровка; 10— рход охлаждающей воды.
степени преломления лучей света различных длин волн, входящих в состав днев-
ного света. После того как достигнута максимально возможная резкость гра-
ницы раздела светлого и темного полей, вращая микрометрический винт тонкой
регулировки 12, устанавливают границу точно в центре поля зрения на скреще-
нии волосков, как показано па схеме 16. Затем, нажимая кнопку на левой сто-
* Некоторые жидкости с необычно большой дисперсией не дают резкой
границы раздела полей при освещении искусственным светом электролампы.
В таких случаях следует повернуть вниз держатель лампы и подставить к от-
верстию прибора лист белого картона под таким углом, чтобы он отражал на
осветительную призму дневной свет из окна. Обычно это позволяет получать
четкую линию раздела полей при тщательной регулировке положения регули-
рующего винта 8 дисперсионных призм
82
Глава 3
стороне прибора, включают лампу 14, освещающую шкалу 13. Тогда в окуляре
можно прочитать показатель преломления исследуемой жидкости с точностью до
трех знаков после запятой, как показано на схеме 15. Четвертый знак оцени-
вают на глаз. Результат измерения записывают в следующей форме:
n§* = 1,4357
Одновременно с показателем преломления следует зарегистрировать показание
барабана 8. Затем необходимо очистить призмы прибора с помощью ватного
тампона, смоченного толуолом или петролейным эфиром *. Эти растворители
применяют для удаления жидкостей, нерастворимых в воде. Растворяющиеся в
воде вещества удаляют дистиллированной водой. Очистку призм следует выпол-
нять с большой тщательностью, чтобы ие поцарапать их рабочую поверхность.
При нанесении исследуемого вещества лучше избегать применения стеклянных
или металлических шпателей и пипеток. Для чистки призм нужно использовать
только чистую гигроскопическую вату (свободную от пыли). При необходимо-
сти внесения каких-либо изменений в методику измерений следует обращаться
к руководству по эксплуатации прибора. Опубликованы материалы, содержа-
щие детальное обсуждение принципов рефрактометрии и конструкций рефракто-
метров **.
Обсуждение. Значения плотности и показателя преломления
могут быть полезны для того, чтобы исключить из рассмотрения
некоторые вещества при идентификации неизвестного соединения.
Однако необходимо следить за тем, чтобы исследуемый образец
был бы достаточно чистым. Лучше всего определять эти физиче-
ские константы для средней фракции, полученной при перегонке
данного вещества (например, при определении его температуры
кипения). Можно также применять для этой цели образцы, выде-
ленные в процессе препаративной газовой хроматографии.
Две обсуждаемые здесь физические константы могут также
оказаться полезными при последней окончательной проверке после
того, как неизвестное вещество идентифицировано и его структура
установлена. Эти константы полезны и в научно-исследователь-
ской работе при проверке различных возможных структур изуча-
емых соединений. Такую проверку выполняют путем сравнения
наблюдаемых значений констант с величинами, приведенными в
литературе (см. таблицы в приложении III или литературные исто-
чники, перечисленные в гл. 10).
3.4. ГАЗОВАЯ ХРОМАТОГРАФИЯ
Газовую хроматографию раньше называли также парофазной
хроматографией. Для проведения газовой хроматографии необ-
ходима неподвижная фаза, которая обычно представляет собой
термостабильную и нелетучую жидкость. Эту неподвижную фазу
равномерно наносят на инертный твердый носитель (например,
* Для очистки призм нельзя применять ацетон.
** Брауэр Н., Фаянс К., Левин С. Рефрактометрия. — В кн.: Физические ме-
тоды органической химии. Т. 3. Пер. с англ./Под ред. А. Вайсбергера. — М.: ИЛ,
1954.
Предварительное исследование, оценка чистоты
83
на тонкоизмельченный огнеупорный кирпич, отсеянный в виде од-
нородных фракций). Смесь неподвижной фазы и твердого носи-
теля помещают в трубку (металлическую или, реже, стеклянную).
Пробу, подлежащую анализу, вводят в трубку — газохромато-
графическую колонку при температуре, достаточно высокой для
перевода входящих в ее состав компонентов в парообразное со-
стояние. Такая газообразная проба захватывается потоком газа-
носителя, являющегося подвижной фазой. В процессе передвиже-
ния проба вступает в контакт с неподвижной жидкой фазой и
адсорбируется ею. Отдельные компоненты пробы разделяются в
соответствии с их относительной способностью адсорбироваться не-
подвижной жидкой фазой, удерживаемой твердым носителем. Та-
ким образом, степень разделения зависит как от полярности, так
и от летучести компонентов разделяемой смеси *.
Газовая хроматография оказывается полезной для определе-
ния чистоты или анализа компонентного состава достаточно ле-
тучих органических веществ. Если при газохроматографическом
анализе данной пробы в различных экспериментальных условиях
(на разных колонках, при различных температурах и т. п.) наблю-
дается только один газохроматографический пик, то это явля-
ется веским свидетельством в пользу достаточной чистоты иссле-
дуемого соединения. Понятно, что изучаемые пробы должны быть
достаточно термостойкими, чтобы выдерживать условия газохро-
матографического анализа.
Упрощенная схема прибора для газовой хроматографии — га-
зового хроматографа показана на рис. 3.26.
Этот прибор состоит из источника газа-носителя 4 (рис. 3.26), обычно ге-
лия или реже азота. Гелий применяют для этой цели, потому что он достаточно
инертен и обладает высокой теплопроводностью. Для контроля скорости потока
газа-носителя обычно служит редуктор 5, устанавливаемый на баллоне со сжа-
тым газом, и регулирующие устройства в самом приборе, подающие газ с
постоянной скоростью в точке 7. Ввод пробы осуществляется при помощи
устройства 6 (рис. 3.26) или А и В (рис. 3.28). Типичное устройство такого рода
представляет собой штуцер с отверстием, закрытым навинчивающимся металли-
ческим колпачком, прижимающим резиновую или пластиковую прокладку. Через
имеющееся в колпачке отверстие прокладку прокалывают иглой шприца, с по-
мощью которого изучаемую пробу вводят в колонку 2, нагреваемую до желае-
мой температуры с помощью термостата 3. Колонки 2 представляют собой ме-
таллические или стеклянные трубки, заполненные твердым носителем с непо-
движной жидкой фазой. Чаще всего употребляют жидкие фазы, перечисленные
в табл. 3.10. Для удобства размещения в термостате колонка может быть свер-
нута в спираль, после чего ее присоединяют между устройством для ввода про-
бы и детектором /. Детектор — устройство, вырабатывающее электрический
сигнал при выходе из колонки разделенных фракций анализируемой пробы.
Наиболее распространенными видами детекторов являются детектор по тепло-
проводности и пламенно-ионизационный. Детектор по теплопроводности изме-
ряет изменения теплопроводности газа-носителя, окружающего нагреваемую
* Детальный механизм разделения веществ методом газовой хроматографии
весьма сложен. Более подробно этот вопрос обсужден в монографиях по газо-
вой хроматографии, перечисленных в гл. 10.
84
Глава 3
электрическим током металлическую проволоку. Эти изменения теплопроводности
зависят от содержания в газе компонентов анализируемой пробы. Далее измене-
ния теплопроводности преобразуются в электрический сигнал, регистрируемый
самописцем. В пламенно-ионизационном детекторе концентрацию компонентов
пробы в газе-носителе измеряют, сжигая их в небольшом водородном пламени.
Пои этом измеряется количество образующихся ионов.
Методика анализа. Большинство приборов для газовой хроматографии
имеют один прибор для измерения температуры в сочетании с переключателем,
позволяющим выбрать тот узел хроматографа, температура которого должна
Рис. 3.26. Схема газового хроматографа.
1—детектор; 2—хроматографические колонки; 3—термостат колонок; 4—баллон с газом-
носителем; 5—редуктор; 6—устройство для ввода проб; 7—тройник.
быть проконтролирована н данный момент (рис. 3.27). Детектор и устройстно
для внода пробы должны постоянно находиться в нагретом состоянии. Эти уст-
ройства разогреваются до постояииой температуры очень медленно (в течение
ночи). Обычно их поддерживают при температуре, на несколько десятков гра-
дусов более высокой, чем температура колонки. Температуру колонки чаще
всего устанавливают на 10—20°С выше температуры кипения представляющего
наибольший интерес компонента пробы *. Однако температура колонки всегда
должна быть ниже верхнего предела рабочих температур применяемой непо-
движной фазы (табл. 3.10). Обычно температуру колонки устанавливают в пре-
делах от 50 до 200°С. Соответственно температура устройства для ввода проб и
детектора может составлять 80—250°С. Попятно, что выбор температуры ана-
лиза зависит от устойчивости анализируемой пробы. В приборах с детектором
по теплопроводности до начала работы, прежде чем включить ток питания
нитей детектора (моста), путем проверки показаний манометров на баллоне
* Если проба представляет собой твердое вещество, вводимое в хромато-
граф в форме раствора, то следует тщательно продумать, какую температуру
нужно установить в испарителе.
Предварительное исследование, оценка чистоты 85
Таблица 3.10
Краткий перечень неподвижных фаз для газовой хроматографии а
Неподвижная фаза Область применения б Растворитель для приготовления сорбентов Максималь- ная рабочая температура по данным фирмы-изго- товителя, °C
Апиезон L В, S, Р, П—V Бензол, толуол 300
Сквалеи Полидиэтиленгликоль- В, N, Р, III—V, газы III Толуол Ацетон 140 200
адипинат (ДЭГА)
Полидиэтилеигликоль- сукцинат (ДЭГС) Р, S, II—IV Ацетон, хлороформ 200
Т рикрези лфосфат (ТКФ) S, II—V, газы Ацетон, метанол 125
Полиэтиленгликоль Р, S, I—V Хлороформ 250
(карбонакс 20М)
Версамид 900 N, II—V Ацетон ® 350
Силиконовое масло DC 710 N, Р, IV-VI Ацетон, хлороформ 300
Полисилоксан SE-30 N, Р, S, II—VI, газы Хлороформ, толуол 350
FFAPr S, Si, I-VI Метилеихло- рид 275
Порапак (пористый по- лимер) Газы, низкомолекуляр- ные органические соединения, вода, аммиак 200
Нитрат серебра Олефины Метанол 50
а Гордон А., Форд Р., Спутник химика. Физико-химические свойства, методики, библи-
ография. Пер. с англ.—М.: Мир, 1976.
6 Обозиачеиия групп органических соединений: В —борсодержащие соединения; N—азот-
содержащие соединения; Р —фосфорсодержащие соединения; S — серусодержащие соедине-
ния; Si — кремнийсодержащие соединения; I —соединения, способные к образованию сильных
водородных связей (гликоли, аминоспирты, многоосиовные кислоты и т. п.); II — полярные
соединения, способные к образованию водородных связей, и соединения с активным водо-
родом [спирты, фенолы, первичные и вторичные амины, нитрилы (а-Н), нитросоединения
(а-Н)]; III — полярные соединения, способные к образованию водородных связей, но не имею-
щие активных атомов водорода, в том числе и а-Н; IV — соединения, обладающие умеренной
способностью к обмену атомов водорода; V —неполярные соединения, углеводороды, суль-
фиды и др.; VI —различные неорганические и органические соединения.
в По данным Лурье А. А., Хроматографические материалы.—М.: Химия, 1978. — Прим. ред.
г Free Fatty Acids Packing —неподвижная фаза для анализа свободных жирных кислот,
полиэтиленглнкольдинитротерефталат. — Прим. ред.
86
Глава 3
с газом-носителем и на самом хроматографе необходимо убедиться в том, что
через детектор протекает поток гелия *. Кроме того, на выходе газа из детек-
тора необходимо установить измеритель расхода с мыльной пленкой Установив
желаемую скорость потока газа-носителя, включают ток питания моста и под-
держивают его в пределах 100—200 мА (рис. 3.27). Устанавливают иа нулевую
отметку указатель самописца и включают подачу диаграммной бумаги. Затем
с помощью микрошприца вводят пробу в одно из отверстий А или В (рис. 3.28).
Величина пробы составляет примерно 1 мкл чистого вещества илн 10- -100 мкл
раствора в летучем растворителе с концентрацией 1—10%. В момент ввода
Рис. 3.27. Панель управления типичного газового хроматографа с детектором
по теплопроводности (фирма GOW-MAC Instrument Со., США).
пробы делают отметку на диаграммной бумаге. Кроме того, на диаграмме ука-
зывают тип и длину применяемой колонки, температуру колонки, скорость газа-
носителя и другие параметры хроматографического опыта (рис. 3.29). При реги-
страции пиков может оказаться необходимым изменять диапазон чувствитель-
ности прибора (сигнал детектора) гак, чтобы регистрируемый пик целиком
уложился бы на диаграммной ленте Самописца. (На рис. 3.27 видна рукоятка
переключения диапазонов чувствительности, а на хроматограмме рнс. 3.29 ука-
заны моменты переключения.) Время, прошедшее с момента ввода пробы до реги-
страции максимума данного пика, называется временем удерживания соответ-
ствующего компонента. После того как закончилось элюирование всех компонен-
тов анализируемой пробы, выключают хроматограф. При этом сначала отклю-
чают ток питания нитей детектора и лишь после этого снижают до минимума
скорость потока газа-носителя.
Обсуждение. Времена удерживания или объемы удерживания,
равные произведению времен удерживания на объемную скорость
газа-носителя, являются величинами, характерными для анализи-
руемых веществ. Однако следует иметь в виду то обстоятельство,
что точное совпадение таких характеристик с величинами, при-
веденными в литературе, маловероятно, поскольку для этого
* Если при включении тока питания нитей детектора через него не пропу-
скать инертный газ, то весьма вероятно, что нити детектора будут разрушены н
прибор выведен из строя.
Предварительное исследование, оценка чистоты
87
Рис. 3.28. Газовый хроматограф с самопишущим регистратором. Прибор марки
550 фирмы GOW-МАС с самописцем фирмы Honeywell (США).
88
Глава 3
вода (5000 ч. на млн)
К
требуется строгое воспроизведе-
ние слишком большого числа пе-
ременных параметров. Однако
если при вводе смеси вещества
предполагаемого строения с за-
ведомым образцом той же струк-
туры на хроматограмме будет
зарегистрирован лишь один чет-
кий пик, то это является веским
доводом в пользу идентичности
двух сравниваемых соединений.
Такие смеси следует анализиро-
вать как на полярной, так и на
неполярной газохроматографиче-
ской колонке. С помощью газовой
хроматографии удобно наблю-
дать ход той или иной химической
реакции (содержание исходных,
конечных и побочных продуктов
и т. п.), а также контролировать
чистоту различных веществ.
Если данный пик соответству-
ет какому-либо одному компонен-
ту изучаемой смеси, то площадь
под этим пиком пропорциональна
числу молей данного вещества.
Так, например, на' рис. 3.29 эти
площади могут быть определены
путем измерения высот пиков во-
ды и амина и перемножения их на
ширину пиков, измеренную на
середине их высоты *. Площадь
пика воды нужно умножить на 8,
так как он зарегистрирован при
'/8 той чувствительности самопис-
ца, при которой был зафиксиро-
ван пик амина.
Для количественных измене-
ний в газовой хроматографии
* Такой метод измерения площадей
пиков называется триангуляцией. Его
следует применять лишь в том случае,
если эти пики полностью симметричны.
(Хроматографические пики могут быть
приближенно заменены треугольником
также и в том случае, когда они несим-
метричны. — Прим, ред.)
Предварительное исследование, оценка чистоты
89
весьма важен мольный отклик (МО), представляющий собой вели-
чину площади пика на 1 моль соответствующего ему вещества.
Мольный отклик может быть рассчитан по следующей простой
формуле:
__ площадь пика
число молей вещества
Мольный отклик обычно выражают в см2/моль. Во всех возмож-
ных случаях следует определять мольный отклик компонентов,
регистрируемых на газовой хроматограмме. Это можно сделать,
осуществляя хроматографию известных объемов стандартных ра-
створов каждого компонента. В дальнейшем найденные величины
мольного отклика используют для того, чтобы переводить пло-
щади пиков всех компонентов смеси в числа молей этих веществ,
что позволяет далее рассчитать их содержание в смеси в мольных
процентах.
3.5. ОПРЕДЕЛЕНИЕ МОЛЕКУЛЯРНОЙ МАССЫ
Здесь будут рассмотрены пять методов определения молеку-
лярной массы: метод Раста (определение депрессии температуры
замерзания), парофазная осмометрия, масс-спектрометрия, опре-
деление эквивалента нейтрализации и числа омыления. Метод Ра-
ста требует крайне простого оборудования. Кроме того, он часто
оказывается полезен для тех веществ, молекулярную массу кото-
рых невозможно измерить масс-спектрометрически. Результаты,
получаемые по методу Раста, в большинстве случаев оказываются
лишь приближенными, поэтому описание техники проведения из-
мерений по этому способу здесь не приводится *. Осмометрия в
паровой фазе и масс-спектрометрия требуют применения очень
сложных приборов. Наиболее точные значения молекулярной мас-
сы, а часто молекулярная формула и структура вещества, могут
быть получены с помощью масс-спектрометрии. Однако молеку-
лярные массы веществ, термически нестойких, имеющих слишком
малую упругость пара или не образующих стабильных молекуляр
ных ионов, нельзя измерить с помощью масс-спектрометрии и при-
ходится прибегать к другим методам измерения. С помощью ме-
тодов титрования определяют эквиваленты нейтрализации (для
кислот и аминов) и числа омыления (для сложных эфиров). Од-
нако эти методы обязательно требуют информации о числе и ха-
рактере функциональных групп, присутствующих в молекуле дан-
ного неизвестного соединения. Поэтому эти методы обсуждаются
в соответствующих разделах гл. 6. Осмометрия в паровой фазе не
* Установление строения органических соединений физическими и химиче-
скими методами. Пер. с англ./Под ред. А. Вайсбергера.—М.: Химия, 1967, кн. 1,
с. 46. В этой работе подробно обсуждается метод Раста.
90
Глава 3
требует столь высокой термической устойчивости и летучести ве-
щества, как масс-спектрометрия. Эти два метода — осмометрия
в паровой фазе и масс-спектрометрия — обсуждаются ниже.
3.5.1. Осмометрия в паровой фазе
В основе данного метода лежит измерение некоторого свойства
вещества, пригодного для определения молекулярной массы ор-
ганических соединений. Этот метод называется парофазной осмо-
Рис. 3.30. Схема прибора для осмометрии в паровой фазе.
1 — направляющая шприца; 2 — камера; 3 — зеркальное зрительное устройство; 4—термоизо-
ляционный блок; 5—шприц; 6—направляющие штифты; 7—термисторный зонд; 8—сосуд
с растворителем; 9— фитиль.
метрией. Прибор для выполнения таких измерений называется па-
рофазным осмометром, или, более правильно, термоэлектриче-
ским дифференциальным прибором для измерения упругости пара.
В этом приборе использован тот факт, что упругость пара над ра-
створом зависит от моляльной концентрации растворенного в нем
вещества. Схема прибора, имеющегося в продаже, приведена на
рис. 3.30.
Предварительное исследование, оценка чистоты
91
Основным элементом этого прибора являются два бусинковых термистора,
находящиеся в камере, атмосфера которой насыщена парами применяемого рас-
творителя. На дне камеры находится чашка с растворителем и фитиль из пори-
стого материала, обеспечивающие насыщение атмосферы камеры парами раство-
рителя. Оба термистора включены в схему моста Уитстона. В начале опыта мост
сбалансирован, причем каждый термистор смочен каплей растворителя. Затем
одни из термисторов смачивают раствором определяемого вещества в том же
Моляльная концентрация
Рис. 3.31. Калибровочная кривая осмометрии в паровой фазе для бензила в мета-
ноле при 37,5 °C. Продолжительность опыта 500 с.
растворителе. Концентрация раствора должна быть известна. Время от момента
нанесения капли до выполнения измерения должно быть одинаково при всех
определениях. В типичном примере полученных результатов, приведенном на
рис. 3.31, это время равно 500 с. После того как на один из термисторов нане-
сена капля раствора, на этом термисторе начинается процесс конденсации чи-
стого растворителя, так как над раствором упругость пара ниже, чем над чи-
стым растворителем. Происходящее при этом повышение температуры вследствие
выделения теплоты конденсации регистрируется в форме электрического потен-
циала разбаланса моста Уитстона. Прибор и растворитель калибруют по чистым
органическим соединениям известного состава. Хорошим стандартом для калиб-
ровки служит, например, бензил (СбН5СО)2, который легко получить в чистом
виде и легко высушить. Кроме того, это вещество нелетуче и хорошо растворимо
в органических растворителях. При калибровке и при анализе неизвестного ве-
щества должен применяться один и тот же растворитель.
Типичные приборы такого типа, имеющиеся в продаже, могут измерять из-
менения температуры до 10°С с точностью в цеуксдькр стцосительных процентов.
92
Глава 3
Измеряемое напряжение при этом может изменяться от 10 до 105 мкВ. Рабо-
чий диапазон температур прибора может быть выбран от 20 до ~ 130°С. Для
водных и неводных систем необходимы разные пробы. Объем раствора, требую-
щийся для проведения одного измерения, составляет всего 10—100 мкл.
Метод измерения. Готовят стандартные растворы бензила (молекулярная
масса 210) в метаноле с моляльными концентрациями, перечисленными в
табл. 3.11. Приводят осмометр в равновесие при 37,5°С. При этом атмосфера
Таблица 3.11
Результаты калибровки системы для осмометрии
в паровой фазе
Концентрация бензила в метаноле, г/кг растворителя ДУ, мкВ *К.ю-2 т
0,03 236 78,6
0,04 308 77,0
0,07 510 72,9
0,10 680 68,0
(0,00) — (83,1)
камеры должна быть насыщена парами метанола, а каждый из термисторов
смочен каплей того же растворителя. Затем на один из термисторов наносят
каплю одного из калибровочных растворов (например, первого из приведенных
в табл. 3.11, т. е. содержащего 30 мг бензила на 1000 г метанола). Измеряют и
записывают изменение потенциала моста (мкВ). Эту операцию повторяют со
всеми калибровочными растворами. Далее строят график зависимости величины
&V]m от моляльной концентрации раствора т. Экстраполируют этот график к
нулевой концентрации (рис. 3.31). На рис. 3.31 значение kV]m, соответствую-
щее нулевой концентрации, равно для этого примера
( = 83,1-102 мкВ-кг-г-1
Тогда калибровочная постоянная системы прибор — растворитель может быть
найдена по следующему уравнению:
где М — молекулярная масса применяемого для калибровки стандартного ве-
щества (бензила). Отсюда
КсНяОН = 210 г/моль ‘ (83,1 • 102) —? '±L(PaCTBJ.. = 1,75 106 мкВ ' кг (РасТв-)
enjun г (раств в-ва) моль (раств. в-ва )
После этого готовят растворы определяемого вещества в метаноле примерно
с такой же моляльной концентрацией, как и стандартные растворы бензила, опи-
санные выше. Измеряют AV при разных концентрациях неизвестного вещества и
по этим данным строят график зависимости Wlm от т. Экстраполируют гра-
фик до т = 0 и получают (AV/m) ^=0. Теперь можно определить молекуляр-
ную массу неизвестного соединения х по уравнению
М - Ксн3ОН
Х (АР/т)^_0
П редварительное исследование, оценка чистоты
93
Если при калибровке системы используют не бензил, а другое стандартное ве-
щество, то применяют следующую формулу:
А4 К М„ (А1//Щ)"=о
М V = -------- = -------------
(Л17ш)^0 (АУ/щ)^=0
где Мст — молекулярная масса стандартного вещества, а (ДУ/т)^=0 — коорди-
ната точки пересечения с осью ординат графика занисимости LVItn от m для
этого стандартного соединения.
3.5.2. Масс-спектрометрия
Масс-спектрометрия — наиболее точный метод определения мо-
лекулярной массы органических соединений. Однако при этом
необходимо, чтобы определяемое вещество было бы достаточно
устойчивым при температуре ввода в масс-спектрограф. Кроме то-
го, структура соединения должна допускать возможность образо-
вания достаточно интенсивного пика молекулярного (первичного)
иона. Если при определении температуры кипения или при газо-
хроматографическом анализе (см. выше) изучаемое вещество про-
являет признаки разложения, то для определения его молекуляр-
ной массы лучше применить другие методы (осмометрию в паровой
фазе, метод Раста * и др.).
Схема типичного масс-спектрометра приведена на рис. 3.32.
В масс-спектрометре с ионизацией электронным ударом пробу пе-
реводят в парообразное состояние и в газовой фазе вводят непос-
редственно в пучок ионизирующих электронов. Реже пробу (осо-
бенно нелетучих веществ) прямо помещают в ионизирующий
пучок электронов. В ходе ионизации вещества наиболее важным
процессом является выбивание одного электрона из его молеку-
лы М с образованием молекулярного иона (катион-радикала М-+):
е
М —> М* + 2е
Такой процесс ионизации не изменяет сколько-нибудь суще-
ственно массу молекул и в то же время позволяет ускорять за-
ряженные частицы в электрическом поле с помощью приложенного
потенциала. Далее ускоренные ионы попадают в магнитное поле,
искривляющее их путь в форме дуги окружности Радиус кривизны
траектории иона пропорционален отношению его массы к заряду
(tn/е). Ввиду того что обычно заряд иона равен 1, величина т/е
является мерой массы частиц. Масс-спектр любого вещества обыч-
но достаточно сложен и включает пики, образующиеся при фраг-
ментации молекулярного иона, например, типа
м* —► f: + f-2
* Установление строения органических соединении физическими и химиче-
скими методами. Пер. с англ./Под ред. А. Вайсбергера.— М.: Химия, 1967, кн. 1,
с. 46.
Предварительное исследование, оценка чистоты
95
Тем не менее во многих случаях при масс-спектрометрии пробы
чистого вещества оказывается возможным идентифицировать мо-
лекулярный ион, который обычно соответствует наибольшему пику
в группе пиков с наибольшими значениями m/е (рис. 3.33). В ряде
Видны молекулярные ноны при т/е = 100, пики М+1 при т/е=101 и (для Б) базовый (наи-
более интенсивный) пик при т/е—43 (соответствует СН3С0+).
случаев молекулярный ион может и не входить в группу сопутству-
ющих ему ионов.
Методика анализа. Основные узлы масс-спектрографа и приемы работы с ним
изображены на рис. 3.34—3.40. После удаления следов предыдущей пробы
(рис. 3.35) закрывают обогреваемый вентиль откачки на устройстве напуска.
Пробу помещают (рис. 3.36) в зонд для ввода образцов н вводят ее в обогре-
ваемое устройство иапуска. Устанавливают такую скорость натекания
(рис. 3.37) из устройства напуска в ионный источник, которая обеспечивает из-
меримое давление паров пробы. Эту скорость натекания подбирают методом
96
Глава 3
Рис. 3.34. Основные части масс-спектрометра ЕМ-600 (воспроизведено с разре-
шения фирмы Varian Instruments Division, Palo Alto, Calif.).
Phc. 3.35. Закрывание крана откачки (воспроизведено с разрешения фирмы
Varian Instruments Division, Palo Alto, Calif.).
Предварительное исследование, оценка чистоты
9?
Рис 3.36. Отбор пробы для масс-спектрометрии (воспроизведено с разрешения
фирмы Varian Instruments Division. Palo Alto, Calif.).
Рис. 3.37. Установка скорости натекания (воспроизведено с разрешения фирмы
Varian Instruments Division, Palo Alto, Calif).
4 Зак. 335
Рис. 3.38. Установка величины энергии ионизирующего луча (воспроизведено с
разрешения фирмы Varian Instruments Division, Palo Alto, Calif.).
Рис. 3.39. Установка начальной точки сканирования (воспроизведено с разреше-
ния фирмы Varian Instruments Division, Palo Alto, Calif.).
П редварительное исследование, оценка частоты
99
проб и ошибок. Устанавливают энергию ионизирующего пучка электронов, рав-
ную, например, 70 эВ (рис. 3.38), начальное значение массы при регистрации
масс-спектра [например, 50 а. е. м. (атомных единиц массы), рис. 3.39] и ско-
рость сканирования (например, 10 а. с. м./мин). Включают самописец и нажи-
мают кнопку начала сканирования (рис. 3.40). Устанавливают амплитуду реги-
стрируемого спектра так, чтобы наиболее интенсивный пик уместился в преде-
лах шкалы прибора. На масс-спектрометрах с линейной зависимостью реги-
Рис. 3.40. Включение самописца и запуск системы сканирования (нажатием стар-
товой кнопки) (воспроизведено с разрешения фирмы Varian Instruments Division,
Paio Alto, Calif.).
стрируемого массового числа от времени отмечают на диаграмме значения мас-
совых чисел в соответствии с выбранной скоростью сканирования. При этом
массу молекулярного иона легко определить прямым подсчетом.
Обсуждение результатов. Если вблизи молекулярного иона на-
блюдается группа пиков, то она может оказаться полезной для оп-
ределения молекулярной формулы. Этот вопрос подробнее обсуж-
дается в гл. 4. Полезны также и массы фрагментных ионов, об-
суждаемые далее в гл. 6.
Глава 4
Определение молекулярной формулы
После того как доказана чистота органического соединения
и определена его молекулярная масса (гл. 3), необходимо уста-
новить молекулярную формулу исследуемого вещества. Даже в
том случае, если определена простейшая эмпирическая формула,
сопоставление ее с молекулярной массой позволяет установить
истинную молекулярную формулу (см. ниже). В настоящей главе
вначале изложены методы, позволяющие выяснить, какие элемен-
ты входят в состав данного органического соединения (качествен-
ный анализ), а затем обсуждаются методы, дающие количествен-
ную информацию о содержании каждого элемента в изучаемом
веществе (количественный анализ).
4.1. КАЧЕСТВЕННЫЙ ЭЛЕМЕНТНЫЙ АНАЛИЗ
Химики-органики обычно не применяют какие-либо химиче-
ские реакции на углерод, водород и кислород. Однако во многих
случаях весьма важно определить присутствие в составе исследу-
емого органического соединения других элементов — азота, серы,
фтора, хлора, брома и иода. Обычно эти сопутствующие элементы
определяют непосредственно с помощью «мокрых» химических ре-
акций после отщепления этих элементов при сплавлении иссле-
дуемого вещества с натрием. Многие из таких химических реак-
ций очень чувствительны. Поэтому все используемые в них водные
растворы должны быть тщательно приготовлены с применением
дистиллированной или, лучше, деионизованной воды. Вещества, ко-
торые при выполнении пробы на горючесть показали признаки
наличия взрывчатых свойств, либо не следует вообще анализи-
ровать путем сплавления с натрием, либо нужно анализировать
микрометодом, описанным ниже. О некоторых веществах досто-
верно известно, что они реагируют с расплавленным натрием со
взрывом; это нитроалканы, органические азиды, диазоэфиры, со-
ли диазония и некоторые органические полигалоидные соедине-
ния (хлороформ, четыреххлористый углерод). При проведении
реакции сплавления с натрием следует обязательно надевать за-
щитные очки с боковыми щитками. При этом необходимо забо-
титься о безопасности соседей по рабочему месту и не направ-
лять в их сторону отверстие реакционного сосуда, в котором про-
изводится сплавление с натрием.
При всех реакциях, в которых определенность результатов
вызывает малейшие сомнения, следует проводить контрольные
опыты. Так, например, если нет уверенности в достоверности на-
Определение молекулярной формулы
101
блюдений, указывающих на наличие азота в исследуемом веще-
стве, следует провести ту же реакцию с веществом, которое заве-
домо содержит азот. В подобных случаях может даже оказаться
целесообразным провести контрольный опыт с таким веществом,
которое заведомо не содержит элемент, представляющий инте-
рес. Наблюдения, сделанные в таком контрольном. эксперименте,
позволяют вывести заключение относительно значимости реакции,
давшей отрицательный результат, или судить о чистоте применяе-
мых реагентов. Знание элементного состава изучаемых органи-
ческих соединений важно по следующим причинам:
1. Эта информация позволяет выбрать подходящие эксперимен-
ты, которые могут служить для доказательства наличия тех или
иных функциональных групп, или предложить методы получе-
ния производных. Таким образом, знание элементного состава
соединения позволяет определить порядок применения рекомен-
даций, содержащихся в гл. 6.
2. Эта информация обеспечивает возможность достаточно
эффективного использования и интерпретации ИК-, УФ-, ЯМР-
и масс-спектров для вывода молекулярной формулы (гл. 4), вы-
явления функциональных групп (гл. 4 и 5) и определения строе-
ния вещества (гл. 6).
В состав органических веществ могут входить почти все эле-
менты периодической системы. Однако в настоящей книге бу-
дут описаны методы определения лишь нескольких элементов,
наиболее часто встречающихся в составе органических веществ.
Детектирование всех других элементов представляет собой за-
дачу, с которой сталкиваются, например, в курсе инструменталь-
ного анализа, включающего атомно-адсорбционный, эмиссион-
ный, пламенно-фотометрический и другие инструментальные ана-
литические методы.
В начале изучения техники идентификации «неизвестных» ор-
ганических соединений рекомендуется ограничить круг возмож-
ных элементов следующими: С, Н, N, О, Р, S, F, С1, Вг и I. Мо-
жно также применять несколько обычных солей, в том числе со-
держащих Na, К и Са (см. описание пробы на горючесть в
разд. 3.1.4).
4.1.1. Сплавление органических соединений
При сплавлении органических веществ с натрием протекают
следующие реакции:
С, Н, О, N, S. X
X = галоген,
F, С1, Вг, 1
| NaX
NaCN
! Na2S
NaSCN
I NaOCN
102
Глава 4
Метод А *. Натрий. Небольшую пробирку из легкоплавкого стекла размером;
100 X 13 мм закрепляют в вертикальном положении с помощью зажима Бун-
зена со снятыми резиновыми обкладками. В пробирку помещают кусочек чистого’
металлического натрия размером около 4 мм. Нижнюю часть пробирки нагре-
вают до тех пор, пока натрий не расплавится и в пробирке начнут подниматься,
его пары. Затем в пробирку вносят одну треть смеси 0,1 г изучаемого соедине-
ния примерно с 50 мг порошкообразной ** сахарозы, после чего пробирку еще
раз подогревают. Добавление указанной выше смеси повторяют еще дважды,
после чего дно пробирки разогревают до темно-красного каления. Затем дают
пробирке остыть и добавляют в нее 1 мл этанола для растворения непрореа-
гировавшего натрия. Еще раз нагревают пробирку до темио-красиого каления
и горячую бросают в небольшой стакан, содержащий 20 мл дистиллированной
воды (осторожно!). При последнем нагревании у выходного отверстия пробирки
могут вспыхнуть пары этанола — на результаты анализа это не влияет. Про-
бирку в стакане с водой разбивают стеклянной палочкой, раствор нагревают до
кипения и фильтруют. Фильтрат должен быть бесцветным; его используют для
проведения специфических реакций иа различные элементы. Эти реакции описаны
ниже.
Метод Б. В небольшую пробирку из стекла пирекс размером 100 X 13 мм
помещают 10 мг твердого неизвестного вещества или около 0,01 мл (10 мкл)
жидкости и свежеотрезаииый кусочек металлического натрия величиной с го-
рошину и массой около 50 мг (осторожно!). Пробирку нагревают, как указано
в описании метода А. Раскаленный обугленный остаток охлаждают до комнат-
ной температуры. Для полного растворения избытка натрия добавляют несколько
капель метанола и перемешивают. Эту операцию повторяют до тех пор, пока
не прекратится дальнейшее выделение пузырьков водорода. К полученному рас-
твору добавляют около 2 мл дистиллированной воды, затем раствор кипятят
(добавить несколько кусочков фаянса, пемзы или стеклянных палочек) и филь-
труют. Фильтрат должен быть бесцветным. Его используют для проведения опи-
санных ниже специфических реакций на различные элементы. Если на этой фазе
анализа имеются признаки неполного разложения пробы (окрашенный филь-
трат), то необходимо повторить весь описанный выше процесс разложения с но-
вой порцией неизвестного вещества.
Если при добавлении первой порции неизвестного вещества при разложе-
нии с натрием происходит бурная реакция или взрыв, процесс разложения пре-
рывают и около 0,5 г неизвестного вещества восстанавливают при осторожном
кипячении с 5 мл ледяной уксусной кислоты и 0,5 г цинковой пыли (осторож-
но!). При выполнении этой операции раствор не следует нагревать очень сильно,
так как это может привести к потере вследствие испарения некоторых веществ,
например ацетатов аминов с низкой молекулярной массой. После растворения
большей части добавленной цинковой пыли смесь выпаривают досуха и сухой
остаток подвергают разложению с помощью описанных выше методов А и Б.
* Здесь описан метод разложения органических соединений натрием по
Лоссеию [см. Campbell К. N-, Campbell В. К., J. Chem. Educ., 27, 261 (1950)].
В этом методе предусматривается осторожное нагревание смесн, добавление не-
сколькими порциями смеси анализируемого вещества с сахарозой и заключитель-
ное прокаливание. В этих условиях реакция протекает достаточно спокойно, без
разбрызгивания и без преждевременной потери вещества из-за его улетучивания.
** Порошок сахарозы, подходящий для проведения этой реакции, продается
в продовольственных магазинах под названием «сахарная пудра». Этот поро-
шок содержит 97% сахара и 3% крахмала. Разложение органических веществ
в смеси с сахарозой способствует обугливанию и созданию в реакционной массе
восстановительных условий. При этом соединения, содержащие азот, например
амиды, иитрозосоединения, иитросоедииения, азосоединения, гидразосоедииеиия
и азотистые гетероциклы, образуют цианид натрия. Серусодержащие соедине-
ния, такие, как сульфиды, сульфоксиды, сульфоны, сульфонамиды и гетероциклы
с атомами серы, образуют сульфид натрия.
Определение молекулярной формулы
103
Для разложения органических веществ можно использовать
также процессы, включающие сплавление со смесями магния и
поташа * или цинка и оксида кальция **.
Специфические реакции на различные элементы
Сера, а) Несколько миллилитров раствора, полученного после
разложения исследуемого органического вещества, подкисляют
уксусной кислотой и добавляют несколько капель раствора аце-
тата свинца(II). Выпадение черного осадка сульфида свинца
указывает на присутствие серы:
Na2S + РЬ(ООССН3)2
—> 4-PbS + 2CH3COONa
черный
осадок
б) К другой порции фильтрата объемом 1 мл, полученного
при разложении с натрием, добавляют 2 капли разбавленного
раствора нитропруссида натрия. Появление глубокой сине-фиоле-
товой окраски указывает на присутствие серы.
Na2S + Na2[Fe(CN)6(NO)J —> Na4[Fe(CN)6(NOS)] + 2NaOH
нитропруссид сине-фиолетовый
натрия «тиоиитрокомплекс»
Азот. Для определения азота необходимы следующие реактивы:
1,5%-ный раствор п-нитробензальдегида в 2-метоксиэтаноле (ме-
тилцеллозольве), 1,7%-ный раствор о-динитробеизола в 2-мет-
оксиэтаноле (метилцеллозольве) и 2%-ный раствор гидроксида
натрия в дистиллированной воде.
Методика определения *. В маленькую пробирку помещают 1 мл раствора
п-нитробензальдегида, 1 мл раствора о-динитробензола и 2 капли раствора гид-
роксида натрия. Затем добавляют 2 капли фильтрата после разложения с на-
трием. В присутствии циаиид-ионов, образовавшихся из азотсодержащих функ-
циональных групп анализируемого вещества, возникает соединение, окрашенное
в интенсивный пурпурно-синий цвет. Желтое или коричневое окрашивание соот-
ветствует отрицательной реакции. Эта реакция на азот значительно более чув-
ствительна, чем реакции образования берлинской лазури или турибулевой сини,
описанные в предыдущих изданиях настоящей книги.
* Baker R. Н., Barkenbus С., Ind. Eng. Chem., Anal. Ed., 9, 135 (1937).
** Bennet E. L., Gould C. W., Swift E. H., Niemann C„ Ind. Eng. Chem,
Anal. Ed., 19, 1035 (1947).
**> Guilbault G. G., Kramer D. N., Anal. Chem., 38, 834 (1966); J. Org. Chem
31, 1103 (1966).
104
Глава 4
Эта реакция может быть проведена в присутствии сульфида натрия или га
логеиидов натрия, которые могут образоваться при разложении органического
вещества, содержащего серу или галогены.
Продукты приведенных выше реакций позволяют объяснить причину вы-
сокой чувствительности такого метода определения азота. Подкисление рас-
твора, в котором присутствует указанный диаииои сине-фиолетового цвета, при-
водит к появлению желтой окраски о-иитрофеиилгидроксиламииа, являющегося
кислотно-основным индикатором:
сине-сриолетовогф
желтого
цвета
цвета
Второй способ обнаружения азота. К 2 мл раствора, получен-
ного после разложения анализируемого вещества, добавляют 2
капли раствора полисульфида аммония и смесь выпаривают на
паровой бане досуха. Затем добавляют б мл разбавленной со-
ляной кислоты, раствор подогревают и фильтруют. К фильтрату
добавляют несколько капель раствора хлорида железа (III). По-
явление красного окрашивания указывает на наличие азота.
NaCN + (NH4)2SX —> NaSCN + (NH4)2SX_1
6NaSCN + FeCl3 —> Na3[Fe(SCN)6] + 3NaCl
красного цвета
В качестве контрольного соединения рекомендуется приме-
нять и-бромбензолсульфонамид (т. пл. 166°С). Это вещество мо-
жет быть подвергнуто разложению с натрием с последующим оп-
ределением азота, брома и серы с помощью реакций, описанных
в разд 4.1.1.
Определение молекулярной формулы
105
Некоторые нитросоединения при обработке гидроксидом на-
трия образуют оранжевые или красные комплексы (разд. 6.17.1).
Амиды и имиды при действии гидроксида натрия могут отщеп-
лять аммиак или амины (разд. 6.12.3). Эти реакции, по существу,
позволяют определить тип функциональных групп, которые могут
содержать азот. Поэтому присутствие этих групп следует ожи-
дать при положительных реакциях на азот.
Галогены, а) Примерно 2 мл раствора, полученного при разло-
жении исследуемого вещества натрием, подкисляют разбавлен-
ной азотной кислотой и осторожно кипятят несколько минут, что-
бы удалить цианистый водород или сероводород, которые могут
присутствовать в растворе*. Затем добавляют несколько капель
0,1 М раствора нитрата серебра. Появление белого или желтова-
того осадка свидетельствует о присутствии хлора, брома или
иода. Хлорид серебра окрашен в белый цвет, бромид серебра —
в бледно-желтый, а иодид серебра имеет желтую окраску. Если
появляется лишь легкая муть или опалесценция, то это может
быть связано с наличием примесей галогенов в реактивах или в
стекле пробирки, использованной для проведения реакции с нат-
рием **.
б) Проба Бейлыитейна. Конец медной проволоки изгибают в
маленькую петлю и прокаливают ее в пламени бунзеновской го-
релки до исчезновения зеленой окраски пламени. Охлаждают
проволоку, погружают в исследуемое вещество и нагревают во
внешней зоне пламени бунзеновской горелки. Зеленое окрашива-
ние пламени свидетельствует о наличии галогенов.
Эта реакция обладает исключительно высокой чувствитель-
ностью. Поэтому ее результат всегда следует перепроверять с по-
мощью реакции с нитратом серебра (образуются галогениды се-
ребра), так как самые незначительные следы содержащих гало-
гены примесей достаточны для того, чтобы дать зеленую окраску
пламени. Очень легколетучие жидкости могут полностью испа-
ряться до того, как проволока будет достаточно разогрета для
их разложения. В таких случаях проба Бейльштейна дает отри-
цательный результат.
Зеленое окрашивание пламени вызывают и некоторые азоти-
стые соединения, не содержащие галогенов. Среди них можно
указать производные хинолина и пиридина, органические кисло-
ты, карбамид и цианид меди. Некоторые неорганические веще-
ства также окрашивают пламя в зеленый цвет.
* Если при этом выделяется хлопьевидный осадок кремневой кислоты, рас-
твор нужно отфильтровать иди отцентрифугировать и далее использовать чистый
фильтрат или цеитрифугат. Кремневая кислота может образоваться из силиката
натрия, который в свою очередь может получиться при взаимодействии стекла
с расплавленным натрием в процессе разложения исследуемого вещества.
** Если описанная реакция на галогены не даст однозначного результата,
следует применить другие реакции, приведенные ниже.
106
Глава 4
в) Бром и иод. В пробирку помещают примерно 3 мл раство-
ра, полученного после разложения органического вещества сплав-
лением с натрием, подкисляют 10%-ной серной кислотой и кипя-
тят несколько минут. После охлаждения раствора добавляют 1 мл
четыреххлористого углерода и 1 каплю свежеприготовленной
хлорной воды *. Пурпурное окрашивание слоя четыреххлористого
углерода указывает на наличие иода.
2Na! + С12(Н2О) —> 2NaCI + 12(СС14)
Продолжают по каплям прибавлять хлорную воду, встряхивая
раствор после каждого прибавления. Пурпурная окраска посте-
пенно исчезает и сменяется красновато-коричневой, что указы-
вает на присутствие брома.
12(СС14) + С12(Н2О) —> 2IC1
2NaBr + С12(Н2О) —> 2NaCl + Вг2(СС14)
г) Бром. Помещают в пробирку 3 мл раствора, полученного
после разложения органического вещества с натрием, и добав-
ляют 3 мл ледяной уксусной кислоты и 0,1 г диоксида свинца. К
отверстию пробирки прикладывают фильтровальную бумагу, смо-
ченную 1%-ным раствором флуоресцеина, и нагревают содержи-
мое пробирки до кипения. При наличии брома его пары вызы-
вают переход желтой окраски флуоресцеина в розовую вслед-
ствие образования эозина. Хлориды и цианиды не мешают про-
ведению этой реакции, а иод вызывает коричневое окрашивание
бумаги.
д) Хлор. Если описанные выше реакции дали отрицательный
результат, а при действии раствора нитрата серебра на раствор,
полученный после разложения с натрием, выпадает обильный бе-
лый осадок, то это указывает на присутствие хлора. Если было
установлено наличие хлора или брома в исследуемом органиче-
ском веществе, то для установления присутствия хлора приме-
няют один из следующих тестов.
е) Хлор, бром и иод. Около 10 мл фильтрата после разло-
жения натрием подкисляют 10%-ной серной кислотой. Раствор
кипятят несколько минут, охлаждают и проверяют на присут-
ствие иода, добавляя к 1 мл раствора 0,5 мл четыреххлористого
углерода и несколько капель раствора нитрита натрия. Пурпур-
ное окрашивание слоя четыреххлористого углерода указывает на
наличие иода. В этом случае оставшийся после отбора пробы на
иод раствор обрабатывают нитритом натрия и экстрагируют иод
четыреххлористым углеродом. Затем раствор кипятят 1 мин. После
охлаждения к 1 мл раствора прибавляют 0,5 мл четыреххло-
* Ввиду того что хлорная вода сохраняется плохо, для проведения этой
реакции можно использовать стабилизированный раствор гипохлорита натри?
(хлорокс). Этот раствор должен иметь кислую реакцию на лакмус.
Определение молекулярной формулы
107
ристого углерода и 2 капли хлорной воды. Коричневое окрашива-
ние слоя четыреххлористого углерода указывает на наличие бро-
ма. Раствор, оставшийся после проведения проб на иод и бром,
разбавляют до объема 60 мл, добавляют 2 мл концентрированной
серной кислоты и 0,5 г персульфата калия (K2S2O8). Затем рас-
твор кипятят 5 мин и после охлаждения добавляют раствор* нит-
рата серебра. Образование белого осадка указывает на наличие
хлора.
Можно также применять аналогичную реакцию, в которой
персульфат калия и серная кислота, применяемые для окисления
хлорид-иона, заменены диоксидом свинца (РЬО2) и ледяной
уксусной кислотой.
ж) Хлор в присутствии азота, серы, брома и иода. Примерно
10 мл фильтрата после разложения органического соединения с
натрием подкисляют разбавленной азотной кислотой и кипятят
.для удаления сероводорода и цианистого водорода. Затем добав-
ляют 0,1 М раствор нитрата серебра в количестве, достаточном
для полного осаждения всех галогенов в форме галогенидов се-
ребра, и осадок отделяют. Если присутствуют и азот и сера, то
осадок кипятят 10 мин с 30 мл концентрированной азотной кис-
.лоты для разложения тиоцианата серебра. Затем смесь разбав-
.ляют 30 мл дистиллированной воды и фильтруют. Далее осадок
галогенидов серебра кипятят с 20 мл 0,1%-ного раствора гидро-
ксида натрия в течение 2 мин. Раствор фильтруют, фильтрат под-
кисляют азотной кислотой и добавляют раствор нитрата сереб-
ра, Образование белого осадка указывает на наличие хлора.
з) Фтор. Примерно 2 мл раствора, полученного после разло-
жения органического вещества натрием, подкисляют уксусной
кислотой. Раствор кипятят, затем охлаждают и одну каплю его
помещают на ализарин-циркониевую индикаторную бумагу. Из-
менение красной окраски бумаги до желтой указывает на при-
сутствие фтора. Индикаторную бумагу готовят, пропитывая ку-
сочки фильтровальной бумаги раствором, составленным из 3 мл
1%-ного раствора ализарина в этаноле и 2 мл 0,4%-ного раство-
ра хлорида или нитрата циркония. Окрашенную в красный цвет
бумагу высушивают, а непосредственно перед употреблением сма-
чивают каплей 50%-ной уксусной кислоты.
4.1.2. Определение металлов и других
неорганических элементов
Металлы и другие неорганические элементы могут быть оп-
ределены стандартными методами качественного анализа *. Эти
методы были модифицированы для проведения элементного
’ Feigl F., Anger V., Spot Tests in Inorganic Analysis. 6th ed., Elsevier,
'New York, 1972.
106
Глава 4
анализа металлов, входящих в состав органических соединений *.
Можно также применять спектральные методы анализа неорга-
нических элементов **. В большинстве случаев спектральные ме-
тоды определения проводить значительно легче, и они дают более
надежные результаты, чем «мокрые» реакции на присутствие
тех же элементов.
4.2. КОЛИЧЕСТВЕННЫЙ ЭЛЕМЕНТНЫЙ АНАЛИЗ
4.2.1. Метод сожжения и другие сходные методы анализа
Обычно для подтверждения структуры новых органических со-
единений приводят данные количественного анализа. Эти данные
оказывают также исключительно большую помощь при опреде-
лении строения неизвестных соединений. Такой микроанализ
обычно осуществляют коммерческие фирмы, располагающие обо-
рудованием для проведения анализа методом сожжения или ины-
ми родственными методами ***. После проверки чистоты подлежа-
щих анализу проб (см. гл. 3) их подсушивают, обычно в сушиль-
ном пистолете Абдергальдена (рис. 4.1). Небольшое количество
подготавливаемого к анализу вещества равномерно рассыпают
тонким слоем в фарфоровой лодочке, которую затем помещают в
горизонтальную часть сушильного пистолета. В боковую колбу
сушильного пистолета помещают свежий безводный высушиваю-
щий агент, например пентаоксид фосфора. Менее эффективны
сульфат или хлорид кальция. Всю систему эвакуируют с помощью
вакуум-насоса. Скорость удаления воды из высушиваемого ма-
териала может быть увеличена, если поместить в нижнюю колбу
толуол или ксилол и обогревать колбу парами этих растворите-
лей, конденсирующихся в обратном холодильнике. При этом, ко-
нечно, проба должна быть устойчива при температуре кипения
этих растворителей. Если предполагается, что проба содержит
следы растворителей углеводородного характера, то вместо высу-
шивающего агента в боковую колбу пистолета Абдергальдена по-
мещают стружки парафина.
Рассматривая пробу через увеличительное стекло, можно убе-
диться в том, что она не содержит волокон фильтровальной бу-
маги или других посторонних примесей такого рода. Обычно для
определения С и Н необходимо около 5 мг пробы. Еще по 5 мг
* Pasto D. 1., Johnson С., Organic Structure Determination, Englewood
Cliffs, Prentice-Hall, New Jersy, 1969, p. 320.
*♦ Willard H. H., Merritt L. L., Dean J. A., Instrumental Methods of Analy-
sis, 5th ed., Van Nostrand, New York, 1974.
**♦ Количественные анализы иа С, Н, N, S и галогены выполняются в сле-
дующих лабораториях США: Galbraith Analytical Laboratories, Knoxville, Tenn.;
Baron Consulting, Orange, Conn.; Huffman Microanalytical Laboratories, Wheatrid-
ge, Colo.
Определение молекулярной формулы
109
вещества требуется для каждого дополнительного анализа (на-
пример, на серу, галоген, дейтерий и т. п.). Типичная схема при-
бора для анализа органических соединений приведена на рис. 4.2.
Водород и углерод определяют по количеству продуктов сгора-
ния анализируемого вещества — воды и углекислого газа. Анализ
4
Рис. 4.1. Сушильный пистолет Абдергальдена (воспроизведено с разрешения
A. Landgrebe, автора книги Theory and Practice of Organic Chemistry и изда-
тельства D. C. Heath).
1— к вакуум-насосу; 2—охлаждающая вода; 3 — высушивающий агент; 4— нагреватель; 5 — вы-
сушиваемый образец.
на кислород обычно не проводят, и его содержание определяют
по разности (см. ниже). Считается, что достаточно уверенное
подтверждение предполагаемой молекулярной формулы изучае-
мого вещества достигнуто, если расхождение между рассчитан-
ным и экспериментально найденным содержанием определяемых
элементов не превышает ±0,3%, например:
Анализ: рассчитано для C|3Hi6O: С 82,93; Н 8,57
найдено: С 82,87' Н 8,67
Если до проведения анализа молекулярная формула вещества
неизвестна, то по данным анализа можно рассчитать простейшую
110
Глава 4
эмпирическую формулу. Способ расчета показан в табл. 4.1. Су-
ществует несколько методов такого расчета, по существу эквива-
лентных друг другу. В большинстве из них вначале делят полу-
ченные значения содержания на атомные массы элементов, взя-
Рис. 4.2. Элементный анализатор, модель 240. Прибор позволяет определять со-
держание С, Н и N из одной навески массой 1—3 мг или меиее. Прибор легко
модифицируется для определения кислорода (воспроизведено с разрешения фир-
мы Perkin-Elmer Corporation, Norwalk, Conn.).
/ — осушители; 2—ввод пробы; 3— зона сжигания; 4 — зона восстановления; 5 — смеситель;
6—переключающие клапаны; 7—пробоотбориый объем; 8 — детектор; 9—ловушки.
тые из таблицы Менделеева. Затем полученные отношения умно-
жают, как показано в табл. 4.1, на корректирующие множители,
которые выбирают так, чтобы получить целочисленные величи-
ны, представляющие собой наименьшие возможные числа атомов
данного элемента, присутствующих в молекуле анализируемого
Таблица 4.1
Расчет эмпирической формулы по данным элементного анализа
Эле- мент Моле- куляр- ная масса Найден- ное содержа- ние, % Отношение Процентного содержания элементов к их молекулярной массе Результаты деления отношений из преды- дущего столбца на их наименьшее значение (приведен иые отношения) Наи- мень- шее возмож- ное число атомов в моле- куле Формула
с 12,011 49,90 49,90/12,011 =4,154 8,99 9 (С9Н9О4С1)Ж,
н 1,008 4,19 4,19/1,008 = 4,157 9,00 9 где х = 1, 2
о 16,000 (29,54) а 29,54/16,000= 1,846 3,996 4 или иное
С1 35,453 16,37 16,37/35,453 = 0,462 1,000 1 целое число
а Найдено по разности (в предположении,
100—(49,90 + 4,19+ 16,3?)=29,54.
что
другие элементы отсутствуют):
Определение молекулярной формулы
111
соединения. Так, простейшей возможной формулой в табл. 4.1 бу-
дет формула C9H9O4CI. При х = 1 эта формула будет молекуляр-
ной. Величину х уточняют по данным независимых измерений мо-
лекулярной массы вещества. Так, например, если с помощью
метода парофазной осмометрии было получено значение молеку-
лярной массы, не более чем на 10% отличающееся от 216,6 а. е. м.
(табл. 4.1, х=1), то молекулярной формулой вещества действи-
тельно является формула C9H9O4CI. Для определения молекуляр-
ной массы веществ идеальным будет метод масс-спектрометрии,
часто позволяющий получить точное значение истинной молеку-
лярной массы изучаемого вещества (например, 216, если в при-
веденном в табл. 4.1 примере учитывать массу преобладающего
изотопа хлора 35С1). Возможно, конечно, что молекулярная мас-
са изучаемого соединения окажется величиной, кратной 216,6.
Так, например, если с помощью какого-либо метода удалось уста-
новить, что молекулярная масса вещества, анализ которого при-
веден в табл. 4.1, равна 435, то этому веществу следует припи-
сать формулу CisHigChCh.
4.2.2. Определение молекулярной формулы с помощью
метода масс-спектрометрии
Как указано в гл. 3, молекулярная масса достаточно термо-
стойкой и летучей пробы, способной образовать достаточно ин-
тенсивный для измерения пик молекулярного иона, может быть
установлена с помощью метода масс-спектрометрии. Кроме того,
использование относительного содержания изотопов (т. е. дан-
ных об интенсивности ионов М-f- 1 и M-f-2), полученных на
масс-спектрографе, разрешающем пики с разницей в 1 а. е. м.,
или точных значений масс, определенных на масс-спектрографе
высокого разрешения, часто позволяет установить молекулярную
формулу неизвестного соединения.
В масс-спектрометре с единичным разрешением (допускаю-
щем определение массы ионов с точностью до ближайших целых
чисел) в области молекулярного иона обычно наблюдается груп-
па ионов с массами, отличающимися на одну единицу. Это связа-
но с наличием заметного содержания нескольких изотопов эле-
ментов, входящих в состав исследуемого вещества (табл. 4.2).
Молекулярный ион обычно составлен из атомов изотопов, присут-
ствующих в наибольшем количестве (например, для метана 12С
и 'Н). Такие изотопы почти во всех случаях имеют и наимень-
шую атомную массу. В образование меньших пиков в этой груп-
пе, соответствующих большим значениям молекулярной массы,
вносят свой вклад молекулы, которые содержат изотопы, присут-
ствующие в меньшем количестве (например, 13С, 2Н). Пики мо-
лекул, содержащих подобные изотопы более чем двух элементов,
обычно слишком незначительны для того, чтобы проявиться в
112
Глава 4
Таблица 4.2
Относительное содержание молекул метана СН4 с различным
изотопным составом
Возможный изотопный состав молекулы Целочисленное значение молекулярной массы Нуклид Относительное содержание по отношению к изотопу, п рису тст ву юще му в наибольшем количестве, %
12сн4 16 >2С (100) а
13С'Н4 17 •3С 1,08
НС’Н4 18 !4С б
12С'Нз2Н 17 2Н 0,016
12СЗД 18 2Н —
12С'Н2Н3 19 2Н —
12С2Н4 20 2Н —.
12С‘Н33Н 18 Зн б
|3С2Н4 21 2Н21аС в
а Значение принято произвольно.
б Зтн соединения содержат радиоактивные изотопы. Их относительное содержание
меняется со временем и, кроме того, обычно слишком мало, чтобы представлять ценность
для масс-спектральиых исследований.
в Крайне маловероятные соединения.
спектре существенным образом. Для определения молекулярной
формулы часто бывают очень полезны данные, получаемые из
соотношения интенсивностей ионов М + 1 и М + 2. Ввиду того
что обычно данной молекулярной массе может соответствовать
большое число различных формул, разработка методов расчета
возможных формул с помощью ЭВМ оказалась исключительно
полезной *. Часть одной из полученных при этом таблиц, относя-
щаяся к метану, представлена в табл. 4.3. Из данных этой таблицы
ясно следует, что если в результате измерения относительной ин-
тенсивности пика М+1 (с т/е 17) будет получена величина
около 1,0%, то это является веским свидетельством в пользу мо-
лекулярной формулы метана.
Понятно, что таблицы, аналогичные табл. 4.3, составленные
для более тяжелых органических веществ, будут довольно гро-
моздкими. К счастью, весьма простые правила позволяют отбро-
сить многие из возможных формул. Это, во-первых, азотное пра-
вило, согласно которому обычное органическое соединение с чет-
ной молекулярной массой не может содержать нечетное число
атомов азота, а соединение с нечетной молекулярной массой долж-
но содержать нечетное число атомов азота. Очевидно, в соот-
ветствии с азотным правилом следует исключить вторую фор-
* Таблицы масс-спектров соединений с молекулярной массой 12—250 можно
найти в книге: Сильверстейн Р., Басслер Г., Моррилл Т. Спектрометрическая
идентификация органических соединений. Пер. с англ. — М.: Мир, 1977,
Определение молекулярной формулы
113
Таблица 4.3
Масса и относительное содержание изотопов всех возможных
комбинаций атомов С, Н, N и О, соответствующих
массовому числу 16 а
Частицы с массовым числом 16 м + 1 М + 2
о 0,04 0,20
nh2 0,41 —
сн4 1,15 —
а Интенсивность молекулярного нона М с т/е 16 принята за 100%. См.
Сильверстейн Р„ Басслер Г., Моррилл Т. Спектрометрическая идентификация
органических соединений. Пер. с англ. — М.: Мир, 1977.
। мулу в табл. 4.3. Менее строгим является правило «наличия смыс-
ла», утверждающее, что молекулярная формула должна соответ-
ствовать стабильной органической молекуле. По этому признаку
следует отвергнуть две первые формулы в табл. 4.3. Конечно,
при установлении молекулярной формулы метана нет необходи-
; мости применять азотное правило и правило «наличия смысла».
Однако эти правила оказываются очень полезными при выборе
возможной формулы для соединений с более высокой молекуляр-
1 ной массой.
На рис. 3.33 показан масс-спектр гексанона-3. Ввиду того что
этот спектр составлен из хорошо образованных пиков, их интен-
сивности считаются пропорциональными высотам этих пиков.
При установлении молекулярной формулы этого кетона ценность
Таблица 4.4
Относительное содержание изотопов некоторых элементов
(число атомов изотопа данного элемента, присутствующего
в наибольшем количестве, принято за 100%)
Элемент Изотоп, содержащийся в наибольшем количестве Относительное содержание изотопов, %
Углерод 12С 13C 1,06
Водород •н 2H 0,018
Азот 14N 15N 0,38
Кислород 16Q ,7O 0,04, 18O 0,20
Фтор isp
Кремний 28Sj 29Si 5,10, 30Si 3,35
Фосфор alp
Сера 32 S 33 S 0,78, 34 S 4,40
Хлор 36C1 37C! 32,5
Бром 79Br 81 Br 98,0
Иод 127}
114
Глава 4
весьма малоинтенсивного пика М +1 {гп]е 101) оказывается до-
вольно ограниченной.
Ясно, что применение масс-спектрометрии во многих случаях
сильно упрощает определение природы и числа присутствующих
Рис. 4.3. Пики в области молекулярного иоиа в масс-спектрах некоторых соеди-
нений, содержащих хлор и бром. Обычно вклады, связанные с С, Н, N и О,
малы по сравнению с вкладами Вг и С1.
в молекуле атомов элементов, других, нежели С, Н, О и N.
Данные, приведенные в табл. 4.4, позволяют разделить все ато-
мы на две группы. В одну из них входят те атомы (например, F,
I, Р), при наличии которых интенсивность пиков М+1 и М + 2
ниже, чем можно было ожидать на основании данных для С,
Н, N и О. Ко второй группе относятся атомы (например, Вг, С1,
S, Si), при наличии которых интенсивность пиков М+1 и М + 2
Определение молекулярной формулы
115
оказывается существенно выше, чем можно ожидать на основа-
нии данных для С, Н, N и О. Атомы первой группы затрудни-
тельно выявить при первоначальном изучении группы ионов,
близких к молекулярному иону. Атомы второй группы обычно
легко и быстро могут быть идентифицированы по масс-спектру.
Один из примеров второй группы заслуживает более подроб-
ного изучения. Алкилхлориды дают масс-спектры, в которых един-
ственный значительный вклад в пик М -f- 2 может быть обуслов-
лен только присутствием изотопа 37С1. Это связано с тем, что со-
держание тяжелого изотопа хлора 37С1 во много раз превышает
содержание тяжелых изотопов углерода и водорода (табл. 4.4).
Таким образом, следует ожидать, что в области молекулярного
иона масс-спектр любого алкилхлорида, содержащего в молеку-
ле один атом хлора, должен выглядеть так, как показано на вто-
рой диаграмме в первом ряду на рис. 4.3.
Для определения молекулярной формулы очень полезна так-
же масс-спектрометрия высокого разрешения. Этот вопрос более
подробно обсуждается во многих руководствах по спектроско-
пии, перечисленных в гл. 10.
Глава 5
Классификация органических соединений
по растворимости, спектрам
ядерного магнитного резонанса
и инфракрасным спектрам
С этой главы мы начнем рассмотрение способов определения
структуры органических соединений. Сходные сведения можно
получить из данных по растворимости и из спектрального ана-
лиза. Например, нерастворимые в воде карбоновые кислоты рас-
творимы в водном гидроксиде натрия. В то же время эти кис-
лоты имеют в ИК-спектре полосы поглощения, характеристичные
для карбоксильных групп. Следовательно, для структурных оп-
ределений очень ценны выводы, сделанные на основе данных как
по растворимости, так и спектрального анализа.
5.1. РАСТВОРИМОСТЬ
Органические соединения можно разделить по растворимости
на два основных типа: вещества, растворимость которых связана
с химическими реакциями, например с кислотно-основными ре-
акциями,
СО2Н
п-нитробензойная
кислота (в воде
нерастворима)
натриевая соль
п-нитробензойной
кислоты ( в воде
растворима)
и вещества, при растворении которых происходит простое сме-
шивание (например, растворение диэтилового эфира в тетрахлор-
метане при ЯМР-анализе). Хотя материалы по растворимости,
рассматриваемые в двух последующих разделах, перекрывают-
ся, все же данные разд. 5.1.1 используются главным образом для
идентификации функциональных групп, а данные разд. 5.1.2—
для выбора растворителя при перекристаллизации, для спек-
трального анализа и проведения химических реакций.
5.1.1. Растворимость в воде, водных кислотах,
водных основаниях и эфире
При исследовании растворимости неизвестного вещества в
воде, 5%-ном растворе гидроксида натрия, 5%-ном растворе би-
карбоната натрия, 5%-ной соляной кислоте и холодной конпент-
Классификация по растворимости и спектрам
117
рированной серной кислоте часто можно получить о нем следую-
щие сведения. Во-первых, указание на наличие функциональных
групп. Например, поскольку углеводороды нерастворимы в воде,
один лишь тот факт, что неизвестное вещество, как, например,
диэтиловый эфир, частично растворимо в воде, показывает, что
в нем имеется полярная функциональная группа. Во-вторых, рас-
творимость в некоторых растворителях часто позволяет получить
более точную информацию о функциональной группе. Например,
бензойная кислота нерастворима в полярных растворителях и
воде, но в разбавленном растворе гидроксида натрия переходит
в соль, хорошо растворимую в воде. Следовательно, в этом слу-
чае растворимость в 5%-ном растворе гидроксида натрия неиз-
вестного вещества, нерастворимого в воде, указывает на наличие
кислотной функциональной группы. В-третьих, в ряде случаев
можно сделать определенные выводы о молекулярной массе ис-
следуемого вещества. Например, во многих гомологических ря-
дах монофункциональных соединений низшие члены с числом ато-
мов углерода менее пяти растворимы в воде, тогда как высшие
гомологи нерастворимы.
В первую очередь проверяют растворимость соединений в
воде. Вещество считается растворимым в воде, если оно раство-
ряется в количестве 3 г на 100 мл растворителя. Такая величина
обусловлена, как будет показано, полуколичественными визуаль-
ными наблюдениями. Необходимо с осторожностью относиться к
применяемым в других работах определениям «растворимый» и
«нерастворимый», поскольку в эти слова часто вкладывают раз-
личный смысл.
При рассмотрении растворимости в разбавленных кислотах
или основаниях большое значение имеет не столько сам факт
растворения вещества в количестве 3% или в иной степени,
сколько увеличение растворимости в водных кислотах и основа-
ниях по сравнению с растворимостью в воде. Такое повышение
растворимости служит хорошим указанием на присутствие кис-
лотных и основных функциональных групп.
Кислотные соединения обычно растворяются в 5%-ном рас-
творе гидроксида натрия. Сильные и слабые кислоты (классы
и А2 соответственно; табл. 5.1 и рис. 5.1) различаются по
растворимости: первые растворяются в слабоосновном раствори-
теле— 5%-ном бикарбонате натрия, а вторые в нем не раство-
ряются. Соединения, являющиеся в водных растворах основания-
ми, будут растворяться в 5 %-ной соляной кислоте; дифференциа-
ция оснований по их силе внутри класса (класс В) не проводит-
ся.
Многие соединения, нейтральные по отношению к 5%-ной
соляной кислоте, ведут себя как основания в более кислых рас-
творителях, например в концентрированной серной или сиропо-
образной фосфорной кислоте. Соединения, содержащие серу или
118
Глава 5
Таблица 5.1
Классы растворимости органических соединений, соответствующие
рис. 5.1 а
S2
SA
Sb
Si
A,
A2
В
MN
N.
N2
I
Соли органических кислот (RCOONa, RSO3Na); гидрохлориды аминов
(RNHHC1); аминокислоты (R—CH(NHj)—COO"); полифуикциональ-
ные соединения с гидрофильными функциональными группами, т. е. угле-
воды (сахара), полиоксисоединеиия, многоосиовные кислоты и т. д.
Одноосновные карбоновые кислоты с пятью или меньшим числом атомов
углерода; ароматические сульфокислоты
Монофункциональные амины с шестью или меньшим числом атомов
углерода
Монофункциональные спирты, альдегиды, кетоиы, сложные эфиры, нит-
рилы и амиды с пятью или меньшим числом атомов углерода
Сильные органические кислоты: карбоновые кислоты более чем с шестью
атомами углерода, фенолы с электроноакцепториыми заместителями в
орто- и паря-положеииях, 0-дикетоиы
Слабые органические кислоты: фенолы, енолы, оксимы, имиды, сульфон-
амиды, тиофенолы, все более чем с пятью атомами углерода, Р-дике-
тоны, нитросоедииеиия с а-водородиым атомом, сульфонамиды
Алифатические амины с восемью или большим числом атомов углерода,
анилины (только одна фенильная группа связана с азотом), некоторые
оксиэфиры
Различные нейтральные соединения, содержащие азот или серу и имею-
щие более пяти атомов углерода
Спирты, альдегиды, метилкетоны, циклические кетоны и сложные эфиры
с одной функциональной группой н с числом атомов углерода более
пяти, но меньше девяти; простые эфиры, содержащие меньше восьми
атомов углерода, эпоксиды
Алкены, алкииы, простые эфиры, некоторые ароматические соединения
(особенно с активированными группами), кетоны (кроме тех, которые
относятся к классу Ni)
Насыщенные углеводороды, алкилгалогеииды, арилгалогениды, диарило-
вые эфиры, дезактивированные ароматические соединения
а Галогенангидриды и ангидриды карбоновых кислот вследствие высокой реакционной
способности не могут быть отнесены ни к одному из перечисленных классов.
азот, имеют атом с несвязанной парой электронов, и следует
ожидать, что они будут растворяться в сильнокислой среде.
Однако при определении растворимости такого типа не получают
никакой дополнительной информации, и поэтому, если элемент-
ный анализ показывает наличие серы или азота, пробы на рас-
творимость не проводят, кроме тех, с помощью которых опреде-
ляют кислотность и основность в водном растворе. Соединения,
содержащие азот или серу и нейтральные в водных кислотах
и основаниях, относятся к классу растворимости MN.
Большинство соединений, содержащих кислород в любой фор-
ме и имеющих нейтральную реакцию в воде, являются сильны-
ми основаниями в концентрированной серной кислоте. Способ-
ность растворяться в серной кислоте или признаки реакции с
серной кислотой указывают на присутствие атома кислорода или
Классификация по растворимости и спектрам
119
UL_ s
S2
вода —
н.р.
эфир
лакмус краснеет
SA
лакмус синеет
лакмус без изменени^
57.-чый
NaHCO3
5%-ный
ЫаОН
н.р.
5%-ная
НС1
Р
МН
85%-ная.---£----N«
Н3РО4
Р
96%-ная
H2SO4
н.р.
Рис. 5.1. Классификация органических соединений по растворимости: определе-
ние растворимости в воде, кислотах, основаниях и простых эфирах (см. табл. 5.1
для соединений, входящих в каждый из классов); р. — растворимо, н. р. — ие
растворимо.
реакционноспособной углеводородной группировки типа олефино-
вой связи или легко сульфирующегося ароматического кольца;
такие соединения относятся к классу N. Дальнейшее подразде-
ление проводят с помощью сиропообразной фосфорной кислоты:
Соединения, растворимые и в фосфорной и в серной кислотах,
120
Глава 5
относятся к классу Nb а вещества, растворяющиеся только в
фосфорной кислоте,— к классу N2. Соединения, имеющие слиш-
ком низкую основность для растворения в серной кислоте, от-
носятся к классу I (инертные соединения).
Ввиду того что растворимость соединений в воде не позво-
ляет получить информацию о присутствии в них кислотных или
основных функциональных групп, такая информация может быть
получена при испытании их водного раствора лакмусом или ин-
дикаторной бумагой. Отметим, что во всех случаях следует про-
верить поведение вещества и по отношению к 5%-ной соляной
кислоте, и к раствору гидроксида натрия, поскольку молекула
может содержать одновременно и кислую и основную функцио-
нальные группы (класс А] — В) или А2— В).
Более детально классы растворимости рассмотрены ниже.
Определение растворимости
Методика определения растворимости в воде. В маленькую пробирку поме-
щают С,2 мл жидкого вещества (или 0,1 г твердого вещества) и добавляют пор-
циями 3 мл воды. После прибавления всего растворителя энергично встряхивают
пробирку, причем смесь должна все время сохранять комнатную температуру.
Если вещество полностью растворится, то оно считается растворимым.
Твердые вещества следует тщательно измельчить для увеличения скорости
растворения. Если твердое вещество не растворяется сразу в воде или эфире, то
иногда рекомендуется осторожно подогреть смесь. Если при этом растворимость
увеличится, то жидкость опять охлаждают до комнатной температуры, встря-
хивая пробирку для предотвращения пересыщения. В таких случаях для уско-
рения кристаллизации полезно добавить в охлажденный раствор кристаллик
твердого вещества. Навеска вещества должна составлять 0,10 г с точностью до
0,01 г.
Жидкости удобнее отмерять градуированной пипеткой, которая обеспечивает
точное измерение прибавленного объема. Если имеются две бесцветные жидкие
фазы, расположенные друг над другом, то часто невозможно увидеть границу
их раздела и создается впечатление, что присутствует лишь одна фаза. Для того
чтобы избежать такой ошибки, энергично встряхивают пробирку; это делают и
в том случае, когда кажется, что неизвестная жидкость растворилась полностью.
Если присутствуют две фазы, то раствор становится мутным. В редких случаях,
когда две бесцветные фазы имеют одинаковые показатели преломления, присут-
ствие второй фазы невозможно определить, даже применяя все меры предосто-
рожности.
Кислотно-осиовиые свойства растворимых в воде соединений определяют
с помощью лакмусовой бумажки; соединения с рКа < 8 попадают в класс Sa
(табл. 5. 1 и рис. 5.1.). Соединения с рКь < 9 относятся к классу SB. Следова-
тельно, фенолы с рДа выше 10 в водных растворах имеют слишком слабую кис-
лотность (pH около 5) для того, чтобы вызвать покраснение лакмуса. Лакмус
краснеет при pH <Z 4,5 и становится синим при pH > 8,3. По той же причине
ароматические амины, такие, как анилин, в водном растворе являются слишком
слабыми основаниями, чтобы вызвать синюю окраску лакмуса. Более точные
определения могут быть сделаны с помощью индикаторной бумаги для измере-
ния pH, одиако удобнее применять дополнительные тесты, обсуждаемые в гл. 6.
Методика определения растворимости в эфире. Используют такие же коли-
чества вещества и растворителя (в данном случае эфира), как описано при
определении растворимости в воде,
Классификация по растворимости и спектрам
121
Методика определения растворимости в водных кислотах или водных осно-
ваниях. Используют такие же количества вещества и растворителя, как описано
в предыдущих методиках.
Для определения растворимости в растворах гидроксида натрия, бикарбо-
ната натрия или в соляной кислоте тщательно встряхивают смесь, отделяют вод-
ный раствор от иерасгворившегося остатка (если надо, фильтруют) и нейтра-
лизуют раствор кислотой или основанием. Очень тщательно исследуют раствор,
чтобы определить, ие выделилось ли неизвестное вещество. Даже легкое помут-
нение нейтрализованного раствора свидетельствует о выделении вещества и дол-
жно рассматриваться как положительная реакция.
При определении растворимости в кислоте или щелочи смесь не подогре-
вают, чтобы не вызвать гидролиза. При тщательном перемешивании смеси не-
обходимое для растворения время составляет 1—2 мии.
Часто одну и ту же порцию вещества можно использовать для испытания
растворимости в нескольких различных растворителях. Так, если обнаружено,
что вещество нерастворимо в воде, то для этой же пробы можно оценить рас-
творимость в разбавленном растворе гидроксида натрия. Для этого к смеси при-
бавляют 1 мл 20%-иого раствора гидроксида натрия и получают в результате
4 мл смеси, содержащей 5% гидроксида натрия. Если вещество совсем не рас-
творяется, то его можно отделить и использовать для пробы на растворимость
в соляной кислоте.
Методика определения растворимости в концентрированных кислотах. При-
меняют способы, описанные выше для определения растворимости в воде; сер-
ную или фосфорную кислоту берут в тех же количествах, что и воду.
При работе с серной кислотой удобнее сначала налить 3 мл растворителя
в пробирку, а затем добавить вещество. С этим реагентом иногда происходят
заметные реакции, и важно наблюдать такие их проявления, как выделение
тепла, изменение окраски, выпадение осадка или выделение газа. Эти наблюде-
ния тщательно фиксируются, поскольку они могут быть очень полезны на после-
дующих стадиях идентификации.
Теория растворимости
Полярность и растворимость. При растворении молекулы или ионы веще-
ства распределяются более или менее равномерно между молекулами раствори-
теля. Например, в кристаллическом хлориде натрия среднее расстояние между
ионами натрия и хлора равно 2,8 А. В 1 М растворе растворитель разделяет
ионы и поэтому ионы натрия и хлора удалены друг от друга на 10 А. На труд-
ность разделения таких ионов указывают высокие температуры плавления
(800°С) и кипения (1413°С) чистого хлорида натрия.
Работа, необходимая для разделения двух противоположно заряженных
пластинок, при введении между ними вещества уменьшается на величину, ко-
торая называется диэлектрической проницаемостью среды. Поэтому не удиви-
тельно, что вода с высокой диэлектрической проницаемостью, равной 80, облег-
чает разделение ионов натрия и хлора и легко растворяет хлорид натрия, тогда
как эфир (диэлектрическая проницаемость 4,4) или гексан (диэлектрическая
проницаемость 1,9) гораздо хуже растворяет соли такого типа. Молекулы воды,
находящиеся между двумя ионами (или заряженными пластинами конденса-
тора), представляют собой маленькие диполи, ориентированные друг к другу
разноименными зарядами («голова к хвосту») так, что они частично нейтрали-
зуют ионные заряды и, таким образом, стабилизируют систему. Поэтому можно
ожидать, что сольватирующая способность и диэлектрическая проницаемость
будут изменяться параллельно. Однако это ие вполне справедливо. Большая ди-
электрическая проницаемость необходима, ио недостаточна для того, чтобы рас-
творитель эффективно растворял ионные соединения. Например, цианистый во-
дород с диэлектрической проницаемостью 116 — очень плохой растворитель для
таких солей, как хлорид натрия. Объяснение этих фактов довольно сложно, ио
одним из основных факторов, обеспечивающим эффективность воды и других
122
Глава в
гидроксилсодержащих растворителей, является их способность к образованию
водородных связей.
Высокая диэлектрическая проницаемость и способность воды к образова-
нию водородных связей, вследствие чего она хорошо растворяет соли, делают
ее плохим растворителем для неполярных соединений. В чистой воде молекулы
ориентированы таким образом, что их положительные центры находятся рядом
с отрицательными. Попытка растворения в воде такого неполярного вещества,
как бензол, по существу, представляет собой попытку разделения противопо-
ложных зарядов в среде с низкой Диэлектрической проницаемостью. В общем
можно ожидать, что полярный растворитель должен легко растворять только
полярные вещества, а неполярный растворитель — только неполярные вещества.
Это соответствует правилу: «Подобное растворяется в подобном».
В табл. 5.1, соответствующую рис. 5.1, включены самые разнообразные со-
единения, которые на основании их структуры относят к различным классам
растворимости. Обсуждение этих тенденций и объяснение такого поведения
даны ниже.
Поскольку вода — полярное соединение, она плохо растворяет углеводо-
роды. Олефиновый, ацетиленовый или бензоидный характер соединения влияет
на его полярность незначительно. Поэтому растворимость ненасыщенных или
ароматических углеводородов в воде мало отличается от растворимости алка-
нов. Изменение растворимости в воде при введении атомов галогенов связано не
с изменением полярности, а обусловлено просто увеличением молекулярной мас-
сы. Так, если вместо водорода ввести галоген, то растворимость в воде умень-
шится. С другой стороны, соли очень полярны, и те, о которых идет речь в этой
работе, обычно растворимы в воде (класс S2).
Растворимость остальных соединений лежит между этими двумя крайними
случаями. Это спирты, сложные и простые эфиры, карбоновые кислоты, амины,
нитрилы, амиды, кетоны, альдегиды и многие другие часто встречающиеся клас-
сы вещее гв.
Как можно было ожидать, кислоты и амины обычно лучше растворимы, чем
нейтральные соединения. Аномально высокая растворимость аминов, вероятно,
обусловлена образованием комплексов с молекулами воды за счет водородных
связей. Эта теория согласуется с тем, что с понижением основности аминов их
растворимость уменьшается. Это также объясняет тот факт, что многие третичные
амииы лучше растворимы в холодной воде, чем в горячей. По-видимому, при
низких температурах следует рассматривать растворимость гидратов, а при вы-
соких температурах гидраты неустойчивы, и растворимость определяется раство-
римостью свободного амина.
Влияние структуры иа растворимость в воде монофункциональных простых
и сложных эфиров, кетонов, альдегидов, спиртов, нитрилов, амидов, карбоновых
кислот и аминов одинаково: в гомологических рядах этих соединений соедине-
ние, еще способное растворяться в воде, соответствует гомологу, содержащему
около пяти атомов углерода.
Поскольку большинство органических молекул содержит и полярные и не-
полярные группы, следует ожидать, что растворимость будет зависеть от соот-
ношения. этих групп. При увеличении углеводородной части молекулы свойства
соединения приближаются к свойствам соответствующего углеводорода, произ-
водным которого является данное соединение. Это означает, что его раствори-
мость в воде уменьшается, а растворимость в эфире увеличивается. Аналогич-
ные изменения в растворимости происходят при увеличении числа ароматиче-
ских колец в соединении. Так, а-нафтол и n-оксибифепил менее растворимы, чем
фенол;
Растворимость в воде:
а-нагртол фенол
п-окси6и<ренил
Классификация по растворимости и спектрам
123
Феиильиая группа, введенная в качестве заместителя в алифатические кис-
лоты, спирты, альдегиды и другие соединения, оказывает примерно такое же
влияние на растворимость, как четырехуглеродный алифатический заместитель.
Например, бензиловый спирт имеет такую же растворимость, как н-амиловый
(или пентиловый) спирт, а гидрокоричная кислота по растворимости аналогична
гептановой кислоте:
Растворимость в воде:
(0V'H2OH & СН,(СН2)3СН2ОН
^ёнзиловыи н-пентиловый
спирт спирт
С6Н5СН2СН2СООН 3 СН3(СН2)5СООН
еептоновая
кислота
Растворимость вещества определяется равновесием между чистым веще-
ством и его раствором. Показано, что такое равновесие обусловлено яе только
взаимодействием растворителя и растворенного вещества, но и силами межмо-
лекулярных взаимодействий в чистом веществе. Эти силы ие зависят от поляр-
ности или других свойств растворителя, и их относительная величина может быть
получена из сравнения температур плавления и кипения Это связано с тем, что
процессы плавления твердого вещества или кипения жидкости приводят к раз-
делению молекул, в некоторой степени сходному с отделением молекул, наблю-
даемым при растворении.
Обратная зависимость между растворимостью и температурой плавления
наблюдается для дикарббновых кислот. Согласно данным табл. 5.2, любая кис-
лота с четным числом атомов углерода имеет более высокую температуру плав-
ления, чем две соседние кислоты ряда, стоящие до и после нее в гомологиче-
ском ряду и, следовательно, имеющие нечетное число атомов углерода. Очевид-
но, что внутрикристаллические силы для членов с четным числом атомов угле-
рода больше, чем для кислот с нечетным числом атомов углерода. Поскольку
предел растворимости твердых веществ определен равным 3,3 г в 100 мл воды,
очевидно, что адипиновая кислота (шесть атомов углерода) нерастворима в
воде, а пимелиновая кислота (семь атомов углерода) в ней растворима.
Таблица 5.2
Растворимость дикарбоиовых кислот НООС—(СН2)Х_2—СООН в воте
Кислоты с четным числом атомов углерода 0 Т. пл., °C Раствори- мость при 20 °C. г/100 г воды Кислоты с нечетным числом атомов углерода а Т. пл., °C Раствори- мость при 20 °C. г/100 г воды
Щавелевая (2) 189 9,5 Малоновая (3) 135 73,5
Янтарная (4) 185 6,8 Глутаровая (5) 97 б4‘
Адипиновая (6) 153 2 Пимелиновая (7) 103 5
Субериновая (8) 140 0,16 Азелаииовая (9) 106 0,24
Себацииовая (10) 133 0,10
а В скобках указано число атомов углерода.
Высокая температура плавления и соответствующая ей низкая раствори-
мость могут быть проиллюстрированы на примере изомерных малеиновой и фу-
маровой кислот. Фумароваи кислота сублимируется при 200°С и нерастворима
в воде. Малеиновая кислота плавится при 130°С и растворима в воде. И для
124 Глава 5
нооссн
II
НССООН
фумаровая кислота
(транс)
С-изомер
CON(CH3)2
CON(CH3)2
т. пл. 80°С
других цис — транс-изомеров цис-соединение более растворимо. Аналогичные за-
кономерности наблюдаются и для полиморфных соединений, таких, как бензо-
фенон *, у которых иизкоплавкая форма лучше растворима.
НССООН
II
НССООН
малеиновая кислота
(цис)
Z-изомер
Диамиды дикарбоновых кислот составляют еще одну группу соединений,
температура плавления которых свидетельствует о наличии сил в кристаллах.
Мочевина (т. пл. 132°С) растворима в воде. Оксамид имеет очень высокую тем-
пературу плавления (420"С), что согласуется с его низкой растворимостью в
воде. Замещение атома водорода амидной группы на метильную группу пони-
жает температуру плавления за счет уменьшения числа межмолекуляриых водо-
родных связей и увеличивает растворимость этого соединения в воде, поэтому
N.N'-диметил- и Й.Н.М'.М'-тетраметилоксамиды растворимы в воде. Амид адипи-
новой кислоты нерастворим в воде, а его М,Ь1,Й',М'-тетраметилпроизводное рас-
творимо в воде.
мочевина оксамид
CONH2 CONHCH3
;с=о | |
H2N^ CONHs CONHCH3
т. пл. 132°С т. пл. 420°С т. пл. 217°С
Амиды типа RCONH2 и RCONHR подчиняются общему правилу, согласно
которому в воде не растворяются соединения, содержащие около пяти атомов
углерода (см. выше). Однако N.N-диалкиламиды (RCONR2) плавятся при более
низкой температуре, чем соответствующие незамещенные амиды, и лучше рас-
творимы в воде. Их граница растворимости приходится иа соединения с 9—
10 атомами углерода. Установлено, что амиды, содержащие группу —CONH2,
ассоциированы за счет того, что эта группа проявляет и донорные и акцептор-
ные свойства при образовании водородных связей. Такая ассоциация невоз-
можна для М,М-дизамещенных амидов (RCONR2), и поэтому для них харак-
терна слабая молекулярная агрегация, на что указывает их низкая температура
плавления и хорошая растворимость.
Ю: Н R
II I S+ «- I
R—С—N—Н О=С—N—Н
•'6 н‘* н
II I : I
R—С— N—Н О=С—N—Н
R
R Н
I I
О=С—N—Н
Й
хорошо растворим в воде, в то время
В общем случае повышение молекулярной массы приводит к увеличению
сил межмолекулярных взаимодействий в твердом веществе. Полимеры и другие
соединения с большой молекулярной массой обычно плохо растворяются в воде
и эфире. Так, формальдегид достаточно
как параформальдегид нерастворим.
СН2О
формальдегид,
растворим в воде
НО(СН2О)ХН
параформальдегид,
нерастворим в воде
Бензофенон существует по крайней мере в четырех формах.
Классификация по растворимости и спектрам
125
Глюкоза растворяется в воде, а ее полимеры — крахмал, гликоген и целлю-
лоза — нерастворимы. Многие аминокислоты растворимы в воде, тогда как их
полимеры — белки — обычно нерастворимы. Некоторые полимеры типа белка,
декстрина и крахмала склонны давать коллоидные растворы, что может при-
вести к ошибкам при оценке их растворимости.
Другой способ увеличения молекулярной массы молекулы состоит во вве-
дении галогенов. Обычно это приводит только к уменьшению растворимости
в воде, благодаря чему некоторые водорастворимые соединения при введении
галогенов переходят в класс веществ, нерастворимых в воде
Верхний предел растворимости в воде, приходящийся на соединения с пятью
атомами углерода, соответствует общему принципу, согласно которому увеличе-
ние структурного подобия между растворяемым веществом и растворителем со-
провождается ростом растворимости. Поскольку вода полярна, то растворимость
в ней других соединений связана с влиянием на растворимость содержащихся
в них полярных групп. При увеличении молекулярной массы членов любого го-
мологического ряда углеводородная (иеполяриая) часть молекулы постепенно
растет, а полярная функция остается неизменной. Это приводит к уменьшению
растворимости в полярном растворителе, таком, как вода.
Тот факт, что верхний предел растворимости в воде одинаков для многих
рядов соединений, связан с близкой полярностью многих функциональных групп.
Одна и та же область (приходящаяся иа гомолог, содержащий пять атомов
углерода) для разных рядов, при которой достигается верхний предел раствори-
мости в воде, целиком определяется произвольно установленными отношениями
количеств растворителя и растворяющегося вещества, принятыми в данной схе-
ме классификации. Если изменить отношение вещества и растворителя, то пре-
дел растворимости может сместиться.
Растворимость в воде некоторых кислородсодержащих соединений может
быть связана с их способностью образовывать гидраты. Устойчивость этих гид-
ратов является фактором, определяющим растворимость в воде и эфире. Вы-
сокая растворимость в воде таких соединений, как хлораль, обусловлена обра-
зованием гидратов.
С13С—СНО С18С—СН(ОН)2 НСООСН2СН3 СН3С(=О)СООСН3
хлораль хлоральгидрат этилформиат метиловый эфир
пировиноградной
кислоты
Низкомолекулярные эфиры муравьиной и пировиноградной кислот гидроли-
зуются водой при комнатной температуре, что подтверждается кислой реакцией
на лакмус водных растворов этих эфиров.
Влияние степени разветвленности цепи иа растворимость. Рассматривая
влияние степени разветвленности углеводородной цепи на температуру кипения
в гомологических рядах углеводородов и спиртов, приводящее к ее понижению,
можно предположить, что разветвление ослабляет межмолекулярные силы и
уменьшает межмолекуляриое притяжение. Тогда не удивительно, что соединения
с разветвленной цепью более растворимы, чем соответствующие соединения с пря-
мой цепью. Это" является общим правилом, особенно для простых алифатических
соединений. Например, растворимость изосоедииений сильно отличается от рас-
творимости нормальных изомеров и приближается к растворимости нормального
изомера соседнего, более низкого члена гомологического ряда. Влияние степени
разветвленности показано в табл. 5.3. В общем случае из двух изомеров более
разветвленный лучше растворим.
Положение функциональной группы в углеродной цепи также влияет на рас-
творимость. Например, пентанол-3 более растворим, чем пентанол-2, который в
свою очередь лучше растворим, чем пентанол-1. Сильное увеличение раствори-
мости наблюдается в том случае, когда в результате разветвления цепи функ-
циональная группа перемещается к центру молекулы, как, например, в случае
2-метилбутанола-2. Обычно у соединений одного типа чем более компактна струк-
тура, тем выше растворимость.
126
Глава 5
Таблица 5.3
Растворимость в воде различных органических соединений
Класс соединений Формула Растворимое соединение Пограничное соединение Нерастворимое соединение
Кислоты RCOOH Пивалиновая кислота (Сб) Изовалериа- иовая кис- лота (С6) «-Валериано- вая кислота (Сб)
Хлорангидриды кислот RCOC1 Хлорангидрид изомасляиой кислоты (С4) Хлорангидрид н-масляпой кислоты (С4)
Спирты ROH Неопентило- вый спирт (С6) 2-Метил- бутанол-3 (С5) н-Амиловый спирт (Сб)
Амиды RCONH2 Амид изо- масляной кислоты (С4) Амид «-мас- ляной кис- лоты (С4)
Сложные эфиры RCOOR' Изопропил- ацетат (Сб) н-Пропил- ацетат (С5)
Кетоиы RCOR' Метил изопро- пилкетон (Сб) Метил-н-про- пилкетои (Сб)
Нитрилы RCN Изобутиро- нитрил (С5) н-Бутиронит- рил (Сб)
Теория кислотно-основной растворимости
Влияние структуры иа кислотность и основность. В общем случае первым
определением примерной силы кислоты или основания являются опыты по рас-
творению в разбавленных кислотах или основаниях неизвестного вещества, не-
растворимого в воде. Структурные особенности, которые стабилизируют органи-
ческий анион (А-), смещают приведенное ниже равновесие* вправо:
НА =?=> Н+-|-А'
[Н*][А~]
[НА]
Это означает, что увеличивается величина Ка:
или уменьшается величина рКа = lg(l/Ka) = —lgKa-
Так же как в большинстве механизмов органических реакций, кислотио-ос-
новиые свойства веществ зависят главным образом от электронного и стериче-
ского эффектов. Ниже эти эффекты рассмотрены более подробно.
Влияние электронных эффектов иа кислотность и основность. Количествен-
ные корреляции структуры с кислотностью и основностью замещенных органи-
ческих соединений изучены достаточно подробно. Эти эффекты объясняются ** с
электронной точки зрения; различные данные указывают на то, что пара- и
* Для краткости часто вместо Н3О+ пишут Н+.
** Более подробно с этим вопросом можно ознакомиться в книгах по физи-
ческой органической химии, приведенных в гл. 10. Результаты исследований ме-
тодом ионного циклотронного резонанса усложняют объяснение.
Классификация по растворимости U спектрам
12?
орто-положения ароматических соединений более чувствительны к электронным
изменениям, чем лета-положения;
б~
Схематически это показано выше иа примере феноксид-иона; согласно тео-
рии резонанса, орто- и пара-положения являются центрами частичного отрица-
тельного заряда, и поэтому водород в этих положениях очень легко замещается
на полярные группы. Однако при орто-замещении всегда следует учитывать сте-
рические факторы (см. ниже).
Большинство органических карбоновых кислот имеют в воде при 25°С кон-
станты диссоциации порядка 10_° или больше, и поэтому они хорошо раство-
римы в 5 %-ном растворе гидроксида натрия. С другой стороны, фенолы, бу-
дучи менее кислыми (константа диссоциации фенола около 10-10), растворяются
в сильном основании — растворе гидроксида натрия, ио нерастворимы в раз-
бавленном растворе бикарбоната натрия (угольная кислота имеет первую кон-
станту диссоциации* 4-10-7). Однако введение заместителей может изменять
кислотность в очень широких пределах. Так, о- и n-иитрофеиолы имеют кон-
станты диссоциации около 6-10-8; другими словами, введение иитрогруппы в
орто- или пара-положения увеличивает кислотность фенола примерно в 600 раз.
Как и следовало ожидать, введение двух нитрогрупп (в 2,4-динитрофеноле) по-
вышает кислотность до такой степени, что соединение становится растворимым
в разбавленном растворе бикарбоната натрия. Увеличение кислотности при вве-
дении нитрогруппы обусловлено стабилизацией феноксид-аииона за счет допол-
нительного распределения отрицательного заряда на нитрогруппу.
Аналогичное повышение кислотности наблюдается при введении в фенол
галогенов. Так, атом брома в орто-положении увеличивает кислотность фенола
примерно в 30 раз, а в пара-положении — примерно в 5 раз. Не удивительно,
что 2,4,6-трибромфенол представляет собой достаточно сильную кислоту для
того, чтобы растворяться в растворе бикарбоната натрия. Изменения, вызывае-
мые введением брома, объясняются индуктивными эффектами.
Электронные эффекты влияют и на основность аминов. Алифатические ами-
ны в водных растворах имеют константы основности порядка 10~3 или 10~4,
что близко к значению для аммиака (10~5). Сопряжение аминогруппы с фениль-
ным кольцом ** понижает основность иа 6 порядков. Так, анилин имеет 7(ь, рав-
ную 5-10~10. Влияние фенильного кольца заключается в стабилизации свободного
* После введения поправки, учитывающей количество растворенного СО2,
первая константа ионизации угольной кислоты равна приблизительно 10~*.
** Свободная электронная пара азота сопрягается с формальными «двой-
ными» связями бензольного кольца, т. е. существует фрагмент —N—С=С.
128
Глава 5
амина, изображенного ниже в левой части уравнения в виде резонансных форм;
фенильная группа уменьшает основность азота также и вследствие индуктив-
ного эффекта.
Не удивительно, что введение второго фенильного заместителя настолько
уменьшает основность амина, что в воде он уже не проявляет заметных основ-
ных свойств. Так, дифениламин нерастворим в разбавленной соляной кислоте.
Введение нитрогруппы в фенильное кольцо анилина понижает его основность
за счет того, что электроакцепторная группа дестабилизирует ион анилиния (со-
пряженную кислоту) и стабилизирует свободное основание.
ион анилиния
Влияние стерических эффектов иа кислотность и основность. орго-Замещен-
ные фенолы имеют очень низкую растворимость в водных щелочах, и, чтобы под-
черкнуть эту особенность их поведения, применяется термин «криптофенол». Для
растворения таких пространственно затрудненных фенолов используют щелочь
Кляйзена (35%-ный раствор гидроксида калия в смеси метанол — вода). Край-
ним примером является 2,4,6-три-трет-бутилфенол, который не растворяется ни
в водном растворе гидроксида натрия, ни в щелочи Кляйзена. Его можно пере-
вести в натриевую соль только обработкой натрием в жидком аммиаке. Так же
необычно ведет себя 2,4,6-три-трет-бутиланилии, который является настолько
слабым основанием, что рКа его сопряженной кислоты слишком мало для изме-
рения в водных растворах. 2,6-Ди-трет-бутилпиридин также имеет основность
более низкую, чем соответствующий диметилпиридии Установлено, что умень-
шение основности аминов вызвано стерическим напряжением, возникающим при
присоединении протона к атому азота; вероятно, неустойчивость 2,5-ди-тр?т-бу-
тилфеноксид-иоиа и упомянутого затрудненного аммониевого иоиа вызвана
главным образом стерическими препятствиями для сольватации ионов.
Стерические затруднения могут и увеличивать и уменьшать кислотность
карбоновых кислот. Например, введение алкильных групп к а-углеродному ато-
му уксусной кислоты приводит к уменьшению кислотности из-за дестабилиза-
ции сопряженного основания за счет стерических препятствий сольватации. В то
же время орто-замещенные бензойные кислоты заметно сильнее, чем соответ-
ствующие пара-изомеры. орто-Заместители выводят карбоксильную группу из
плоскости кольца, что приводит к дестабилизации кислоты в большей степени,
чем иона *. Это может служить примером второго типа стерических эффектов,
очень важного во многих случаях, который называется стерическим ингибиро-
ванием резонанса. В качестве другого примера такого эффекта приведем 3,5-ди-
* Более подробные сведения об этом можно найти в приведенных в гл. 10
книгах по механизмам реакций и физической органической химии.
Классификация по растворимости и спектрам
129
метил-4-нитрофенол; его рКа на 1,6 единицы выше рК, фенола, тогда как рК»
^нитрофенола выше на 2,8 единицы.
рК. прй 25°С: 9,99 7,21
Влияние двух метильных групп на понижение кислотности нитрофенола про-
является в стерическом ингибировании резонанса в анионе. Структура А тре-
бует полной или почти полной копланарности всех атомов нитрогруппы и аро-
матического кольца, а такой копланарности препятствует присутствие в струк-
туре иоиа метильных групп.
Растворимость в разбавленной соляной кислоте. Первичные, вторичные и
третичные алифатические амины образуют при взаимодействии с соляной кис-
лотой полярные ионные соли. Поэтому алифатические амины хорошо раство-
ряются в соляной кислоте (класс В, если вещество нерастворимо в воде).
Сопряжение с арильными группами уменьшает основность атома азота. Хотя
первичные ароматические амины — более слабые основания, чем алифатические
амины, они все же растворяются в разбавленной соляной кислоте, тогда как
диариламины и триариламины уже не растворяются. Нерастворимы, например,
дифениламин, трифениламии и карбазол. Арилалкиламины. содержащие не бо-
лее одной арильной группы, растворимы в разбавленной соляной кислоте,
кароазол
дифениламин
N-^ензилацетамид
Дизамещенные амиды (RCONR2), имеющие достаточно большую молекуляр-
ную массу и нерастворимые в воде, растворимы в разбавленной соляной кис-
лоте. В противоположность им незамещенные амиды (RCONH2) представляют
собой нейтральные соединения *. Большинство монозамещенных амидов
(RCONHR) также нейтрально. Однако N-бензилацетамид проявляет основные
свойства.
Следует отметить, что амины могут реагировать с 5%-ной соляной кисло-
той, давая нерастворимые гидрохлориды. Соединения этого типа могут быть
классифицированы неправильно. Например, некоторые ариламины, такие, как
а-пафтиламин, дают гидрохлориды, умеренно растворимые в разбавленной со-
ляной кислоте. При слабом нагревании смеси и разбавлении ее водой иногда
растворимость улучшается. Появление твердого вещества обычно показывает,
что амин изменился. В сомнительных случаях следует отделить твердое вы-
делившееся вещество и сравнить его температуру плавления с температурой
* Амиды обычно сравнимы с водой по основности, и небольшие структурные
изменения, такие, как алкилирование, должны изменить их рКь только на 2 еди-
ницы для того, чтобы перевести их в категорию «основания».
5 Зак. 335
130
Глава 5
плавления исходного амина. На образование гидрохлорида может также указы-
вать проба со спиртовым раствором нитрата серебра.
NH3C1
гидрохлорид
а-нафтиламина
Исследуемое соединение можно растворить в эфире и затем обработать этот
раствор 5 %-ной соляной кислотой. Образование твердого вещества па границе
раздела фаз указывает на то, что взятое соединение представляет собой основ-
ной амин.
Основные свойства проявляют также некоторые кислородсодержащие со-
единения, способные при действии соляной кислоты давать оксониевые соли.
Растворимость в 5°/о-иом растворе гидроксида натрия и в 5%-ном растворе
карбоната натрия. Перечень различных органических кислот, имеющих отноше-
ние к табл. 5.1, приведен в табл. 5.4. Такую классификацию можно обосновать
с позиций структурного контроля устойчивости сопряженного основания (ани-
она).
Таблица 5.4
Классы растворимости различных органических кислот
Соединение Общая структура Класс растворимости а
Карбоновые кислоты Сульфоновые кислоты RCOOH RSO3H At Ai
Сульфиновые кислоты rso2h Аг
Енолы 1 1 —с=с-он А2
О О Й II
Имиды —С—NH—С— а2
Нитросоединения 6 /СН—no2 А»
Арилсульфопамиды * ArSO2NHR а2
О о
Р-Дикарбонильиые соединения 1 II 1 L —С—СН—С— а2г
Оксимы ^C=N—ОН а2
а Пограничные случаи отмечены в табл. 5.5
® Только первичные (RCH2NO2) и вторичные (R2GHNO2) нитроалкацы.
в Кислотность N—Н-протона используется в пробе Хинсберга (методика
горни относятся сульфонамиды аммиака и первичных аминов.
г При наличии у карбоиильнрй группы сильных электроотрицательных
трифторметильИои, эти соедииення будут относиться к классу Aj.
19). К этой кате-
групп, например
Классификация по растворимости и спектрам
131
Альдегиды и кетоны — достаточно сильные кислоты, чтобы при реакции с
водным раствором щелочи давать анионы, которые служат промежуточными
продуктами в таких процессах, как альдольная конденсация. Одиако их кислот-
ность слишком мала, чтобы они заметно растворялись в растворе гидроксида
натрия. Когда две карбонильные группы присоединены к одному и тому же
атому углерода, как у ацетоуксусного и малонового эфиров и у 1,3-дикетонов,
кислотность сильно увеличивается за счет дополнительной стабилизации аннона
вследствие распределения отрицательного заряда по двум атомам кислорода и
центральному атому углерода.
Следует отметить, что хотя р-дикарбонильные соединения имеют кислот-
ность, близкую к кислотности фенолов, отщепление протона от углерода происхо-
дит довольно медленно и поэтому скорость растворения такого вещества может
быть настолько мала, что оно кажется нерастворимым в щелочи.
Интересно, что нитросоединеиия имеют таутомерную форму, называемую
сщи-формой, кислотность которой близка к кислотности карбоновых кислот. Для
eqtz-формы иитроэтана Ка равно 3,6 Ю~6.
Введение даже одной иитрогруппы делает вещество растворимым в разбав-
ленном растворе гидроксида натрия. Так, нитроэтан имеет Ка 3,5-10-9. Эту ве-
личину можно сравнить с величинами /Са следующих p-дикарбонильиых соеди-
нений:
СН3СОСН2СООС2Н6 2- КГ11
CH3COCH(C2H6)COOC2HS 2- КГ13
СН2(СООС2Н6)2 5-10~14
СН3СОСН2СОСН3 2-10-9
п -----'.II '
Поскольку группа » । ц
О О
обладает кислыми свойствами, имидиая группа —С—NH—С—-
также кислая и имиды растворимы в разбавленном растворе гидроксида на-
трия, ио ие в растворе бикарбоната натрия. В водном растворе я-нитрофениль-
иая группа придает слабые кислотные свойства группе —CONH—. Поэтому
и-иитроацетаиилцд * растворяется в растворе гидроксида натрия. Растворимость
* Соединения этого типа дают аддукты (комплексы Мейзеигеймера) за счет
присоединения гидроксила к атому углерода, связанному с амидной группой.
5*
132
Глава 5
сульфонамидов в основаниях такая же, как и n-нитроацетаиилида. Аналогично
растворяются и оксимы, имеющие гидроксилдиую группу, связанную с атомом
азота. Структуры
fCH3CH—N(—O)2r
[(CH3—)2C—N—Ol-
представляют собой сопряженные основания нитроэтана, бензолсульфонамида,
n-нитроацетанилида и оксима ацетона соответственно.
Сложные эфиры (содержащие пять или шесть атомов углерода), почти пол-
ностью растворимые в воде, гидролизуются при длительном встряхивании с раз-
бавленным раствором гидроксида натрия *. Щелочь ие подогревают, поэтому
растворимость или нерастворимость соединения можно установить через 1 —
2 мии.
Моноэфиры дикарбоновых кислот растворимы в бикарбонате; они тоже быс-
тро гидролизуются в воде даже в присутствии таких слабых оснований, как
бикарбонат натрия.
Жирные кислоты, содержащие 12 и более атомов углерода, медленно реа-
гируют со щелочью, давая соли (мыла). Смесь непрозрачна и представляет со-
бой опалесцирующий коллоидный раствор, вспенивающийся при встряхивании.
Такое поведение позволяет легко определять эти кислоты
Некоторые натриевые соли высоко замещенных фенолов нерастворимы в гид-
роксиде натрия. Это может быть замечено при испытании осадка на раствори-
мость в воде. Некоторые фенолы, нерастворимые в воде, при гидролизе дают
осадок, и поэтому кажется, что они нерастворимы в щелочи.
Растворимость амфотерных соединений. Соединения, содержащие и кислую
и основную группы, являются амфотерными. Низкомолекулярные аминокислоты
существуют в виде диполяриых солей. Они растворимы в воде и дают нейтраль-
ную реакцию на лакмус (относятся к классу S2).
NH2
NH8
RCHCOOH R—CH—COO-
Нерастворимые в воде амфотерные соединения ведут себя и как основания,
и как сильные или слабые килсоты. Это зависит от относительной основности
аминогруппы, поскольку основность определяет степень нейтрализации кислотной
группы за счет образования внутренней соли. Если а-аминогруппа связана
только с алкильными заместителями, соединение растворяется в соляной кис-
лоте и в растворе гидроксида натрия, но ие растворяется в растворе бикарбо-
ната натрия. Такие соединения относятся к классу А2(В):
/СООН
R—СН^
\mr2
Арильная группа у атома азота уменьшает его основность, и соединение
становится растворимым даже в водном растворе бикарбоната. Это можно про-
* Использование гидроксида лития вместо гидроксида натрия часто при-
водит к образованию солей, растворимых в воде.
Классификация по растворимости и спектрам
133
иллюстрировать на примере следующих соединений (класс Ai):
CeH5NHCH2COOH
CeH6NCH3
I
RCHCOOH
Если к атому азота присоединены две арильные группы, то соединение ие
проявляет основных свойств и ведет себя как сильная кислота (класс AJ:
(CeH5)2NCH2COOH
Растворимость в холодной концентрированной серной кислоте. Холодная
концентрированная сериая кислота применяется для проверки растворимости
нейтральных нерастворимых в воде соединений, не содержащих других элемен-
тов, кроме углерода, водорода и кислорода. Если соединение является ненасы-
щенным, легко сульфируется или имеет кислородсодержащие функциональные
группы, то оио растворяется в холодной концеитрироваииой серной кислоте.
Растворение в серной кислоте часто сопровождается такими реакциями, как
сульфирование, полимеризация, дегидратация или присоединение серной кислоты
к олефиновым и ацетиленовым связям; во многих случаях при этом образуются
ионы, которые вновь дают исходное соединение при разбавлении ледяной водой.
Ниже показаны некоторые обычные реакции:
OSO3H
+ HSO4- |
RCH=CHR + H2SO4 —» [RCH2—CHR] ---------> RCH2CHR
rch2oh + 2H2SO4 —> RCH2OSO3H + H3O++ HSO4‘
(Вода, образующаяся в результате реакции, под действием серной кислоты пе-
реходит в ион гидрония.)
(RCH2)2CHOH + 2H2SO4 —> (RCH2)2CHOSO3H + Н3О+ + hso;
(R£H2)3COH + H2SO4 —> (RCH2)2C=CHR + H3O+ + HSO4~
CH3O--X + 2H2SO4 —> CH3O-^—SO3H + H3O+ + hso;
(CeH5)3COH + 2H2SO4 —>
(C6H5)2C=O + H2SO4 —
c6h5c; + h2so4 —>
Njh
(C6HE)3C+ + H3O+ + 2HSO;
(C6h5)2coh + hso; I
OH I н2° исходные
C6H5C^ +HSO; I вещества
X)H I
ch3(2occh3 + 3H2so4 —> 2Ch3c(oh)2 + hso; + hs2o;
134
Глава 5
Таблица 5.5
Границы между классами растворимости
Соединение Класс раствори- мости Соединение Класс. раствори- мости
Кислоты Эфиры простые
Хлоруксусиая S1 S. Этилметиловый Диэтиловый г
а-Хлорпропиоиовая Кротоновая Изовалериановая (Св) St Si Si—А] Этилизопропиловый Диизопропиловый Ди-н-бутиловый Si-N, N, n2
Валериановая (Сз) Ai Эфиры сложные
Спирты Этилацетат Si—N,
1 Метилпропионат St
я-Бутиловый Si я-Пропилформиат Si
трет-Пентиловый Si Изопропилацетат Si
Изопропилметнлкарби- Si-N, н-Пропилацетат S.-N1
НОЛ Метилизобутират Si-N,
Изопентиловый Si—Ni н-Бутилформиат Si— N,
Бензиловый Ni Метилизовалерат (Се) Ni
Циклопентиловый Ni втор-Бутилацетат Ni
н-Бутилаиетат N,
Альдегиды Бензилацетат Nt
Этилкаприлат (С1с) n2
Изомасяяный Si Этилбензоат n2
я-Масляиый Si—N, Метилкарбоиат (Сз) St-N,
Изовалериановый (Cs) Ni Этилоксалат Si-N,
Метилмалонат Si— N,
Амиды Этилкарбоиат (Cs) Si—Ni
Этилсукцинат Ni
Формамид Si—S2 Этил фталат N,
Ацетамид Si—S2 Этилмалонат n2
Пропионамид St—S2 я-Бутилка рбоиат Nt—N2
Амид изомасляной кис- Si—S2 я-Бутилоксалат n2
ЛОТЫ
Амид я-масляной кис- St—M Углеводороды
лоты ( ароматические)
Формаиилид Ацетанилид S,—M M Мезитилен Изодурол n2 n2
Цимол I
Амины 1 л-Ксилол Nr-1
Диэтиламин Si Дифеиилметаи N2—I • N2—I
Изопентиламин я-Пеитиламии Бензиламин Si Si Si о-Ксилол Нафталин N2—2 I
Пиперидин s. Кетоны
Циклогексиламии Si
Ди-я-пропиламии s,— в Этилметилкетон Si
Ди-н-бутиламии в I (зопропилметилкетон S!
Анилин в Метил-я-пропилкетон Si—Ni
Три-н-пропиламии в Пинаколии Si—Ni
Классификация по растворимости и спектрам
135
Продолжение табл. 5.5
Соединение Класс раствори- мости Соединение Класс раствори- мости
Диэтилкетои S.-N, н-Бутиронртрил MN
Циклопентанон S1
Циклогексанои S, Нитросоединения
Ацетофенон Ди-н-бутййкетон Бензил Бензофенон Нитрилы N, N,r-N2 N2 n2 Нитрометан Нитроэтаи Нитробензол Фенолы Si—А2 а2 MN Si Si—Аг Si—a2
Пропноиитрнл Бутироинтрил Сукцинонитрил S, Si—MN Si—Бг—MN Гидрохинон Хлоргидрохинон Флороглюцин Фенол
Глутаронитрил (триме- S2—MN
тиленцианид)
Алкаиы, циклоалканы и их галогенпроизводиые нерастворимы в серией кис-
лоте. Простые ароматические углеводороды и их галогенпроизводиые в этих ус-
ловиях не сульфируются и не растворяются. Однако введение двух или более
алкильных групп * в бензольное кольцо облегчает сульфирование соединений,
поэтому полиалкилбензолы довольно легко растворяются в серной кислоте. Этим
объясняется растворимость изодурола и мезитилеиа. Иногда исследуемое веще-
ство реагирует с образованием нерастворимого продукта Некоторые высокомо-
лекулярные эфиры, например типа дифенилового эфира, сульфируются при ком-
натной температуре настолько медленно, что растворения не происходит.
Многие вторичные и третичные спирты легко дегидратируются концентриро-
ванной серной кислотой и дают олефины, которые затем полимеризуются. Полу-
чающиеся полимеры нерастворимы в холодной концентрированной серной кис-
доте н образуют отчетливый слой на поверхности кислоты. Бензиловый спирт
(и аналогичные спирты) вступают в реакцию с концентрированной серной кис-
лотой с образованием бесцветного осадка.
Граница между классами растворимости. В табл. 5.6 приведен ряд соедине-
ний, выбранных таким образом, чтобы продемонстрировать наиболее важные
границы между классами растворимости. Насколько возможно, эти соединения
сгруппированы по их химической природе. В каждую группу мы включили по-
граничное соединение и одно или несколько соединений по обе стороны гра-
ницы. Для не вошедших в таблицу соединений можно установить класс раство-
римости, рассматривая их положение по отношению к пограничному соедине-
нию, принадлежащему к тому же типу. Так, из таблицы видно, что н-бутиловый
спирт относится к классу Sr, отсюда следует, что другие бутиловые спирты и
более низкомолекулярные гомологи также попадают в этот класс. Аналогично
поскольку изопентиловый спирт относится к классу Ni, то и н-пентнловый спирт
и все более высокомолекулярные спирты относятся к классу Ni или N2.
Таблица растворимости. Обычно класс растворимости соединения можно
предсказать на основании его структурной формулы, однако существует много
исключений. Более того, иногда бывает трудно классифицировать соединения
даже по данным эксперимента; как показано в табл. 5.5, многие соединения за-
нимают пограничные положения. В приложении I приведен ряд наиболее
* Другие активирующие группы также (но не всегда) - облегчают сульфи-
рование.
136
Глава 5
Таблица 5.6
Обычные органические растворители а
Растворитель Формула Применение ® Диэлектри- ческая проница- емость (25 -С)
Ацетон а СН3(С=О)СН3 М, Р, ЯМР в 21
Ацетонитрил CH3CN Р, УФ, ЯМР® 36
Бензол г с6нв Р, ЯМРВ 4,2
Сероуглерод cs2 Р, ПК, ЯМР 2,6
Тетрахлорметан в СС14 Р, ПК, УФ, ЯМР 2,2
Хлороформ я СНС13 М, Р, ПК, УФ, ЯМР в 4,7
N.N-Диметилформамид HC(=O)N(CH3)2 Р, ЯМРВ 37
Диметилсульфоксид
(ДМСО) CH3(S=O)CH3 ЯМРа, Р 49
Этанол в СН3СН2ОН Р, УФ, М, ЯМР ” 24
Диэтиловый эфир7' (СН3СН2)2О Р, м 4
Гектан СН3(СН2)4СН3 УФ, М, Р 2,0
Метанол СН3ОН Р, УФ, ЯМР в, м 33
Метнлеихлорнд СН2С12 Р, М, ИК, ЯМР в 9
"Пиридин О Р, ЯМР в 12
N
Н2С—-сн2
Тетра гидрофураи Н2С\ /СН2 о Р, ЯМРВ 7,3
(Вода) (Н2О) (М, Р, ИК.УФ.ЯМР в) (78,5)
а ик- и ПМР-спектры этих соединений можно найти в каталоге Sadtler'a, а также в книге:
Сильверстейн Р. М., Басслер Г. С., Моррилл Т. С. Спектрометрическая идентификация орга-
нических соединений. Пер. с англ. — М.: Мир, 1977.
б М—для очистки стеклянной посуды; Р — реакционная среда; ИК, ЯМР, УФ — исполь-
зование в качестве растворителя для соответствующего спектрального анализа.
в Для измерения спектров протонного магнитного резонанса используется дейтериро-
ванный растворитель, например ацетои-de (CD3COCD3).
г Бензол можно заменить толуолом, поскольку последний менее токсичен.
д Этот растворитель применяется для предварительного изучения растворимости.
распространенных соединений, для которых трудно предсказать класс раствори-
мости.
В процессе исследования обнаруживаются все новые комбинации функцио-
нальных групп, которые представляют собой исключения из правил. Поэтому
рекомендуется для структур необычного типа обращаться к оригинальной лите-
ратуре.
5.1.2. Растворимость в органических растворителях
Определение растворимости органических соединений в орга-
нических растворителях необходимо для правильного проведе-
ния различных лабораторных операций. Многие растворители, ис-
Классификация по растворимости и спектрам
137
пользуемые химиками-органиками, приведены в табл. 5.6. Эти
растворители применяются для. проведения органических реак-
ций, для растворения веществ при измерении спектров и анали-
зе и для стандартных лабораторных нужд (очистка стеклянной
посуды и т. д.). В табл. 5.7 приведены обычные концентрации
Таблица 5.7
Обычная концентрация растворов при использовании
органических растворителей
Применение а
Концентрация б.
%
Р
ИК
ЯМР»
10
5
25
а См. примечание «б» в табл. 5.6.
6 Масса/объем; только приближенная величина.
в Значительно меньшая концентрация необходима при использовании
приборов высшего класса, при работе с накопителем и компьютерном усредне-
нии н при работе с фурье-преобразователем.
веществ для различных. случаев использования растворителей.
Цифры, приведенные в этой таблице, являются лишь приблизи-
тельными: отклонения от них зависят, например, от сложности
используемых приборов, необходимости более или менее деталь-
ных сведений по поглощению и т. п. Определения с помощью
ультрафиолетовой спектроскопии (гл. 8) обычно выполняются на
более позднем этапе, поэтому здесь они не обсуждаются. Факти-
чески все перечисленные растворители, так же как их смеси, при-
меняются для колоночной и тонкослойной хроматографии, для
перекристаллизации твердых веществ (гл. 7) и для экстракции
в процессе выделения продуктов реакции. Для предварительной
характеристики неизвестного вещества необходимо использовать
только те из перечисленных растворителей, которые не изменяют
вещество; естественно, что из всех этих растворителей образец
может быть опять выделен (путем выпаривания).
Из данных по растворимости в таких растворителях могут
быть сделаны некоторые предварительные заключения о струк-
туре, например динитрофенилбензилсульфид (тиоэфир) нераство-
рим в воде, но удовлетворительно растворяется в полярных орга-
нических растворителях (например, в ДМСО, табл. 5.6, диэлек-
трическая проницаемость* 49), что дает возможность измерить
* Диэлектрическая проницаемость — это одна из количественных характе-
ристик полярности растворителя; см. обсуждение диэлектрических свойств в разд.
«Теория растворимости». Корреляция полярности растворителя с диэлектриче-
ской проницаемостью является очень грубой.
138
Глава 5
спектры ЯМР. Необходимость применения ДМСО-de, а не CDC13
указывает на присутствие в молекуле одной или нескольких поляр-
ных групп. Наконец, поскольку все растворители, указанные в
табл. 5.6, используются для различных целей, то они часто встре-
чаются в виде примесей в исследуемых соединениях.
Методика. Приведите определение растворимости в органиче-
ских растворителях, применяя простые опыты, описанные для
определения растворимости в воде в разд. 5.1.1. Используйте
3 мл растворителя и следующие количества органических соеди-
нений (см. табл. 5.7): Р — 0,3 г (0,20 мл), ИК — 0,15 г (150 мкл),
ЯМР —0,75 г (750 мкл).
Б.2. АНАЛИЗ ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
С ПОМОЩЬЮ ИНФРАКРАСНЫХ СПЕКТРОВ
В данной книге не будут подробно рассматриваться теория
инфракрасных спектров и соответствующие спектрометры. Вве-
дение и углубленная теория ИК-спектров (и приборов) изложе-
ны во многих книгах (гл. 10), начиная от книги Моррисона и
Бойда (глава в курсе органической химии) до книги Беллами и
Конли, посвященной только ИК-спектрам, а также в руковод-
ствах по инструментальному анализу. Следует отметить, что ИК-
спектроскопия — очень чувствительный метод для определения
кратности связи и состава функциональных групп. Таким обра-
зом, ИК-спектроскопия в значительной мере служит в качестве
метода функционального анализа. Часто на основании ИК-спек-
тров можно сделать выводы и о всей структуре соединения.
На рис. 5.2 показана принципиальная схема ИК-спектрометра, из которой
ясно видно, как действует прибор. Раса.ютрим очень кратко оптическую и элек-
трическую схемы инфракрасного спектрометра. Свет от источника излучения,
роль которого, может выполнять раскаленная металлическая нить, с помощью
зеркала направляется через образец и кювету сравнения. Оба этих пучка излу-
чения системой зеркал направляются на прерыватель. Зеркала и прерыватель
формируют одни луч, который представляет собой последовательность импуль-
сов от луча, проходящего через образец, и от луча сравнения. Этот «прерыви-
стый» луч зеркалами фокусируется на входную щель монохроматора. До моно-
хроматора этот луч включает излучение всего диапазона энергии, испускаемой
источником излучения. С помощью дифракционной решетки (или призмы) луч
разделяется в монохроматоре таким образом, что в каждый момент (в соот-
ветствие с положением решетки) на выходную щель и на детектор "направляется
излучение с одной определенной энергией. Если поток света содержит излуче-
ние, которое частично поглощается образцом, то это поглощение воспринимается
детектором как отличное от нуля возмущение. Это возмущение детектор преоб-
разует в электрический импульс и передает иа сервомотор. Последний с помощью
аттенюатора выравнивает интенсивности рабочего луча и луча сравнения. Та-
ким образом, путем детектирования инфракрасного поглощения и компенсацией
луча сравнения, осуществляемой с помощью электрической схемы, детектор все
время поддерживает результирующий соста-виой луч на оптическом нуле. На
самописец подаются два различных сигнала. Один из них отражает изменение
в положении дифракционной решетки (или призмы), связанное электрически
Классификация по растворимости и спектрам
139
Рис. 5.2. Схема двухлучевого инфракрасного спектрометра.
] — регулировка 100% (ручной аттенюатор); 2—самописец; 3 — кювета с образцом; 4 — рабочий луч; 5 —источник излучения; 5—луч сравне-
ния; 7—кювета сравнения; 8—инфракрасное излучение; 9—прерыватель; 10 — аттенюатор; 11 —сервомотор; 12 — к самописцу; 13—усилитель;
/4—регулятор усиления; 15—мотор; /6 —движение пера по осн абсцисс: /7 —дифракционная решетка (или призма); 18 — входная щель
монохроматора; 19—корпус прибора; 20— выходная щель монохроматора; 21 — электрический импульс; 22— детектор.
140
Глава 5
Рис. 5.3. А — инфракрасный спектрометр с решеткой; виден держатель кюветы
с образцом. Б — кюветное отделение двухлучевого инфракрасного спектрометра;
установка кюветы с образцом в держатель. В — парные кюветы для жидких об-
разцов, установленные в спектрометре. Г — газовые кюветы в кюветном отделе-
нии. Обратите внимание на больший размер газовой кюветы по сравнению с
жидкостной кюветой (А — В). Газы дают лучше разрешенные и более детальные
спектры, чем растворы.
Классификация по растворимости и спектрам
141
----------•-------------------------------------------------------------------V
I
142 Глава 5
с разверткой по оси абсцисс (т. е. записывает рассматриваемые энергии или
длины волн), другой связан с положением аттенюатора, т. е. является мерой ве-
личины поглощения (откладываемой по ординате).
В основном мы будем рассматривать здесь вопросы, связан-
ные с подготовкой и снятием ИК-спектров образцов (рис. 5.3).
При двухлучевой технике pei истрации спектров важно соотноше-
ние поглощения кюветы с образцом с поглощением кюветы сравне-
ния, в которых находится чистый растворитель или воздух.
Рис. 5.4. Разборная кювета для ИК-
спектроскопии жидкостей.
Сборку кюветы проводят в следующей по-
следовательности. Устанавливают нижнюю
неопреновую прокладку и ннжнюю пла-
стинку из NaCI в гнездо между винтами
опорной пластины. (Замечание: поверхность
пластинок NaCI нельзя скрести н увлаж-
нять; держать нх нужно осторожно за края.)
Кладут прокладку на нижнее окошко и
капают несколько капель образца; для
сравнительно нелетучих или вязких жидко-
стей прокладку можно не применять.
Снльнолетучне жидкости следует анализи-
ровать в закрывающихся жидкостных кю-
ветах (рнс. 5.5). Затем кладут верхнюю пла-
стинку, верхнюю неопреновую прокладку и
верхнюю металлическую пластину; тща-
тельно затягивают гайки до тех пор, пока
образец не будет равномерно распределен
между окошками. Не следует перетяги-
вать гайки, так как это приводит к рас-
трескиванию солевых пластинок.
Образцы для измерения ИК-спектров (или для других видов
спектрального анализа) должны быть чистыми. Твердые веще-
ства необходимо перекристаллизовать (до постоянной темпера-
туры плавления) или очистить хроматографически, а жидкие ве-
щества — перегнать (или очистить методом газовой хроматогра-
фии, описанным в гл. 7).
Приготовление образцов для анализа проводится одним из
следующих методов. Жидкие вещества исследуются непосред-
ственно в виде тонкой пленки между пластинками («чистыми»)
(рис. 5.4). Растворы веществ в неполярном органическом раство-
рителе, например в тетрахлорметане, дают спектры, несколько ис-
каженные вследствие ассоциации и образования молекулярных
агрегатов типа растворитель — растворенное вещество или веще-
ство— вещество. При измерении спектров в растворах (рис. 5.Б)
обязательно нужна кювета сравнения; в луч сравнения поме-
Классификация по растворимости и спектрам
143
щается кювета с чистым растворителем, имеющая такую же тол-
щину, как и кювета с образцом. Области собственного поглоще-
ния обычных растворителей и веществ, используемых для при-
готовления паст, приведены на рис. 5.6. При приготовлении паст
не обязательно, чтобы исследуемое вещество хорошо смешива-
лось с растворителем. В образцах, приготовленных таким спо-
Рис. 5.5. А — правильный способ заполнения закрытой кюветы; Б — установка
кюветы с раствором.
Для установки кюветы с раствором вынимают обе пробки н шприцем без иглы, как это по-
казано в А, заполняют кювету нужным раствором (около 0,050 мл 5%-ного раствора в кювете
толщиной 0,1 мм). При заполнении кювету держат под углом, чтобы могли выходить пу-
зырьки воздуха. Затем, не вынимая шприца, закрывают одну пробку, вынимают шприц и
закрывают вторую пробку. Кювету промывают чистым растворителем, вводя его шприцем,
и высушивают ее, продувая через кювету сухой воздух (шприцем или вакуумным насосом).
Кюветы хранят в эксикаторе.
собой, сохраняется межмолекулярная ассоциация (например, во-
дородные связи). Основное требование к вспомогательным ве-
ществам состоит в отсутствии собственного поглощения. Самой
хорошей средой для приготовления образцов является бромид
калия, который применяется для приготовления таблеток; одна-
ко приготовление таких таблеток требует специальной .техники
(рис. 5.7) и большой тщательности выполнения всех операций.
Методика. Прежде всего необходимо убедиться, что инфракрасный спект-
рофотометр подготовлен к проведению анализа и предварительно прогрет. Об-
разец для измерения спектра приготавливают в виде тонкой пленки (рис. 5.4),
в растворе (10%-ный раствор для кюветы толщиной 0,1 мм) или в виде сус-
пензии. Для приготовления суспензии смешивают около 1 % вещества со вспомо-
гательной средой и тщательно размельчают смесь пестиком в гладкой агатовой
144
Глава 5
Классификация по растворимости и спектрам
145
Рис. 5.7. Миниатюрный пресс (Wilks Scientific, South Norwalk, Conn.) для при-
готовления пластинок с бромидом калия.
Приготовляют смесь, тщательно растирая пестиком в агатовой ступке около 1,0 мг образца
(эти количества ориентировочные, фактически применяемые количества подбираются экспе-
риментально) с 100 мг высушенного в сушильном шкафу безводного КВг. Устанавливают
в камеру пресса один из винтов 2 (А) и насыпают смесь внутрь камеры (В). Устанавливают
второй вннт 2 и затягивают гайки 3, как это показано на рисунке Б. Полученный таким
образом диск из КВг устанавливают в держатель /. Следует отметить, что отсутствия полос
поглощения воды (валентные колебания О —Н при 3800 — 3000 см-1 и деформационные коле,
бания О—Н при 1750—1520 см-1) можно добиться только при очень тщательном выполнении
всех операций (например, используя вакуумированные пресс-формы, высушенные при нагре-
вании приспособления и т. д.).
Такая специальная техника описана в книге: Conley R- Т., Infrared Spectroscopy, 2nd ed.,
Allyn and Bacon, Boston, Copyright © 1972, chap. 4, p. 47.
от
Длина волны, мкм
Длина волны, мкн
Рис. 5.8. ИК-Спектр полистирола.
А — шкала, линейная в частотах 5— шкала, линейная в длинах волн (мкм).
148
Глава 5
ступке. На рис. 5.7 показано, как приготовляют смесь исследуемого образца с
бромидом калия. Пасты с вазелиновым маслом «нуйол», с перфторированными
углеводородами «флуоролуб» и гексахлорбутадиеном исследуют в кювете, пока-
занной на рис. 5.4, проверяют, соответствует ли шкалам прибора установка бу-
маги на самописце и начальное положение длины волны; точное руководство к
работе для каждого конкретного типа прибора должно быть получено у руково-
дителя. Любые изменения режима усиления и баланса или манипуляции с лу-
чом должны проводиться только под руководством преподавателя.
Рукоятку «Регулировка 100%» (ручной аттенюатор) устанавливают таким
образом, чтобы уменьшить потери разрешения (если процент пропускания Т
слишком низок) или поднять нулевую линию (если величина Т слишком высока).
Часто допустимое начальное значение (при 4000 см-1) процента пропускания
может составлять примерно 85—90%. Выбирают установку щели (обычно нор-
мальную), опускают перо самописца иа бумагу и начинают запись спектра (чем
меньше скорость движения, тем ниже требования к перу и самописцу).
Обсуждение. Для получения хороших ИК-спектров в большинст-
ве случаев приходится проводить измерения неоднократно, дей-
ствуя методом проб и ошибок. Это связано с особенностями под-
готовленного образца (например, с непрозрачностью пасты или
интенсивной окраской раствора). Точное положение полос мож-
но получить только после калибровки спектра по стандартам
(обычно по пленке полистирола). Например, если полоса поли-
стирола при 6,24 мкм (1603 см-1) смещена на определенную ве-
личину (например, 0,05 мкм), то положение полос образца дол-
жно быть исправлено на эту величину. ИК-Спектр полистирола
приведен на рис. 5.8; следует обратить внимание на значительные
различия в спектрах, вызванные переходом от одной линейной
шкалы к другой. Это важно, поскольку дополнительная корреля-
ция калибровки должна делаться только для той же линейной
шкалы, которая была использована при записи спектра образца.
Кроме того, качественная идентификация вещества по области
«отпечатков пальцев» зависит от общего вида спектра. Химик-
органик обычно пользуется длинами волн (Л, мкм), или чаще
частотами, или волновыми числами (v, см-1), интенсивностью
поглощения (с — сильная, ср — средняя, сл — слабая) и в редких
случаях в качестве характеристики применяется ширина полосы.
Для компенсации поглощения растворителя применяют такие
же кюветы, как и для растворов (см. выше). Однако в области
сильного собственного поглощения растворителя (рис. 5.6) нель-
зя регистрировать полосы поглощения образца, так как на при-
емник при этом попадает только очень незначительная часть из-
лучения (после поглощения растворителем). Кроме того, при
этом очень трудно точно подобрать соответствующую кювету с
образцом.
Инфракрасное поглощение связано с валентными и (или) де-
формационными колебаниями различных связей в молекуле изу-
чаемого вещества. Быстрое определение простых классов органи-
ческих молекул часто может быть сделано с помощью корреля-
ционной диаграммы, представленной на рис. 5.9; эти выводы
Длина волны, мкм
4000
3500
3000
2500
^алкильные группы |
•азверяы
илкаяы |
-ароматически, И
^соединения [
простые эфиры |
сп ирты I
А
об
оа-
(связанные)—.
-(широкая) —
^т-г-
ш
(свободные)
(узкая)
кислоты)
f£.
ср
ИГЛ" Г I ,
сложные эфиры\
альдегид^
~кетоны ]
ангидрадбг
~Т~Т
амиды
амилы |
4000
2,75
( широкая)
cUj.—<
ЭД
ж
гтм _Т
згтяг _Т
. н- пропил
1200
№00
2000
1400
юод
600
400см'1
-СНгСНгСНгСН,-
H-cq£p-i
пере CflUpl
втор. |_ -1
трет] _Т
______ «ся-
глоты. _.о-
да
широкая (Эля жидких
I I । аминов)
-CHf-NRj
изопропил
трет-бутил ____
виям-сн-сн»______
бы™.-1,
С-(цае) 5
с5енэд?
. (СООрЯЖ.)
, (сопряж.)
. (соггряж.)
(СОПрЯЗК.)
к-. <сояряж.;
л. монозамет, бензоле
у. оото-дизамеш..
орто-дизамещ,
мета±._\___! _
пара-Г- д _Т_
виц-трозамещ.
несимм.л _ д_
сим лк! _L _Т_
а- нафтол ины -
/з-нифталины—
алшр. эфары.
аром, эфиры.
.карбоновые иаелэти
ионизировании ,.-г.
формиаты!. _
ацетаты
пропионаты- _
бутираты и с.
акрилаты д _.
Фумараты!. _
малеаты ~Г_____
оечзмть,, фтаз~. ~
и—я —ч -амир. альдегиды.
>—£—I- —аром, альдегиды.
--
-сэ,-сыо_
..рстю.
С умер а
(несвязанные ниже)
(несвязанные ниже)
(несвязанные ниже)
(умещает.) (несвязанные ниже)
-(lUUpOKae) •( ^лилапели
'(отсутствует в мономере)
олиф, кетоны.
орем, кетоны
норм, ангщ
цикл, анги!
(широкая) ~’
.лере. амиш —
3500
3000
•и JpU(5bL_
VuJw.I-J____|_.
..ами,—___
. монозамещ. а
дизамеаи амс
2500
2000
180V
1600
1400
1200
1000
800-
600
400 см"
3,00 3,25 3,50 3,75 4,00 4,5 5$
Щ5
6fi
ifi w afi
9,0 10 .11 12 13 14 15
20 25мкм
4000
амины
(продолжение)
3500
—1--I--1
3000
гидрохлориды-
имины | >-
нитрилы]
2500
_втор. амины.
трет, амины____
-J-—I—1 Lc-nh^ci
•7Т
.имины. _ ’.Те "NH .. .
замещ. иминыt
(сопряж. ниже)
разные П
соединения |
ПЧН ijnoiBM ’ — O*f Jt-.»
изоциатды^ —
кумулены
тпгфипЫ'
X=CyXfV3O(iutK
неорг, соли и их произв. |
щае.
ср
Соединения
Соединения со связями угле)
Соединения со связью азот-кис
огпнесеиие\
"сГ-
4000
2.50
ГЛ
КОО
1600
2000
.crtj-NH-сн;
-“-НН-СН
1200
ер
н напряж.
..хлоркар
ЗПОКСидНЫи
цикл - -
400см'1
2,75
исЪ.)
'С-О
SH
яг
-»-Ч PH
9
жвщие"-,
ащие
‘ионные сульфаты (ЗСЦ
Соединения со связью сера-кислород
со связями
>чГоГ
пород
(UU
NH
(ОН им/
вал.
САеал.
-F дама*.;
|--+------г1-;!
СВ$«л СВг,'___
СВг (аткр.)
jC"O (0-лактамы) ь
1ы | с-О
CCli "*£,СС11_ ,
—1 ССТ (&Ш9.)
овал
600
400см'1
800
1000
1400
1600
1800
2000
5,5
5,0
6,5 7,0 7,5 8,0
9,0 10 11 .12 13 14 15
20 25мкм
*С-Н вал.
^С^Свал. .
,НН де<р.
ионные сульфонаты H-SQt-.-
сулырановые космяы и-зКИ
ювадентные сульфаты R-О-SO,- 0-R.
ховалентные ------—‘ -л-^
I I
связями фоарар-кислороо {ковалентные фосфапм
I I । | | I ----
I (ионные карбонаты - *
урод { ковалентные карбонаты о= с(0 - к
___u . I иминокармнаты К" - С
[ионное нитраты ' (HCW7-J-
ковалентные нитраты -о О*
J HUmpO I I । R-NQj.L
Л нитро I | I I I
ковалентные нитриты R-O^-Np
[нитрозо в-мг*
ЫН4‘
-CH, t
__CFiMw
)-(&,₽ uwS)'„
'I 7^7СН|
J—r-iU ЬуС
HN=C(O-
.1 I
R-NO.
несопряж.^
евпряж.--^
3500 3000 2500
I —i_____I----1----L—
3,00 3,25 3.50 3,75 4P0
4сн W
1——|——{он
1200
мС-О вал.
4C-N вал,
‘ С—С вал.
1 С—Свал.
.. ..ГМ маятн
__ ЫН маятн. ।
|7 14_1 1
Рис. 5.10. Корреляционные диаграммы инфракрасного поглощения функциональными группами, составленные
r Н. Б. Колтапом,
152
Глава 5
следует использовать как руководство к выбору химических
реакций, описанных в гл. 6. Можно использовать более подроб-
ные диаграммы, например диаграммы Колтапа, приведенные на
рис. 5.10 (см. ссылки на литературу в гл. 10).
5.3. АНАЛИЗ ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ МЕТОДОМ
ЯДЕРНОГО МАГНИТНОГО РЕЗОНАНСА
Схема спектрометра ядерного магнитного резонанса показана
на рис. 5.11.
Ампулу с образцом 5 помещают в поле электромагнита 4 (вместо электро-
магнита можно применять постоянный магнит). С помощью радиочастотного ге-
нератора генерируется второе поле частотой 60 МГц *, которое создается ка-
тушкой б под прямым углом к первому полю 4. Катушки развертки 8 создают
Рис. 5.11. Схема спектрометра ЯМР.
7— радиочастотный генератор; 2—радиочастотный усилитель; 3— детектор; 4—электромагнит
(нлн постоянный магнит); 5—ампула с образцом; 6—катушкн для наложения переменного
поля; 7 —катушка детектора; 8—катушки развертки; 9— усилитель звуковой частоты; 10—ге-
нератор развертки; 11— осциллограф нлн самописец.
медленно изменяющееся поле, которое при определенных значениях напряжен-
ности резонирует со слабыми магнитными полями ядер (например, протонов)
образца. Частота, при которой наступает резонанс, зависит от величины поля,
создаваемой катушкой б. Это поле дает основной вклад в условия резонанса.
При резонансе происходит изменение ядерного магнитного диполя (или квадру-
поля), индуцирующее напряжение в катушке детектора 7 (расположенной под
прямым углом к полям 6 и 4). Величина поля катушки развертки в условиях
резонанса данного типа протонов образца обозначается vs. Можно представить,
что ядра (протоны) в постоянном магнитном поле прецессируют вокруг осн
постоянного магнитного поля. Приложение переменного магнитного поля под
Эта частота называется частотой спектрометра.
Классификация по растворимости и спектрам
153;
прямым углом к постоянному магнитному полю может привести к изменению
угла прецессии. В тот момент, когда величина переменного магнитного поля
точно соответствует собственному маленькому магнитному полю прецессирую-
щего ядра, происходит изменение ориентации этого ядра (изменение спинового,
состояния). Это явление называют резонансом. Величину переменного поля в
Рис. 5.12. Принадлежности для измерения спектров ЯМР (воспроизведено с раз-
решения Wilmad Class Со., Buena, N. J.).
1—стандартная ампула ЯМР, вмещающая 5—30%-ный раствор вещества в 0,4—0,5 мл раство-
рителя (размеры: А=14,4—19,2 см; В=4 мм; С=около 3,2 мм). 2—микроампула для ЯМР
(минимальное необходимое количество раствора 25 мкл; размеры: А=16,8 см, В=5 мм,
С=1,5 или 2.0 мм, 0=25 мкл; £ = 10 мм). Превосходны ампулы с регулируемой глубиной,
камеры (предполагается, что регулировать форму прямоугольных камер можно с большей,
точностью, чем сферических, и поэтому они лучше вращаются). 3 — ампула из темного стекла
(для светочувствительных образцов); размеры такие же, как в 1. 4—пнпетка разового поль-
зования (длиной 70—100 мм) и груша, применяемые в анализе ЯМР. Сужение наверху может-
быть использовано для ватной пробки, позволяющей фильтровать образец.
этот момент обозначают как резонансное значение для данного ядра. Таким об-
разом, приемная катушка 7 должна быть расположена под прямым углом к ка-
тушке развертки 8 и к постоянному полю б.
Резонансные частоты протонов обычных органических соеди-
нений находятся в диапазоне 0—600 Гц (1 герц соответствует
одному колебанию в секунду), ниже, чем резонансная частота
эталонного соединения vr. В качестве эталона обычно применяют
тетраметилсилан [ТМС, (CHshSi]. Хотя, как правило, изменяю-
щимся параметром является величина напряженности поля, при-
нято выражать сигналы в частотах (Гц). Обычно переводят
частоту положения центра такого сигнала в безразмерный пара-
метр — химический сдвиг, который обозначается буквой 6:
6 = |vs-Vrl . 1О6
^ПрИЛОЖ
154
Глава 5
Рис. 5.13. Установка ампулы с турбинкой в спектрометр ЯМР.
Рис. 5.14. Самописец и панель регулировки спектрометра ЯМР Varian ЕМ-360
(воспроизведено с разрешения фирмы Varian Associates, Palo Alto, Calif.).
Классификация по растворимости и спектрам
155.
Рис. 5.15. Панель регулировки спектрометра ЯМР Varian ЕМ-360 (воспроизведе-
но с разрешения фирмы Varian Associates, Palo Alto, Calif.).
Например, если частота образца находится на 60 Гц ниже,
чем сигнал эталона, химический сдвиг, измеренный на приборе
с рабочей частотой 60 МГц (мегагерц—1 млн. колебаний в се-
кунду), вычисляется как
6 = ео^лГгц ' Ю6== 1>0 м. д. (миллионная доля)
Другая шкала — шкала т — употребляется реже:
т= 10,00 —б
156
Глава 5
Таблица 5.8
Химический сдвиг протона в продажных дейтерированных
растворителях для снятия ЯМР-спектров (по данным фирмы
Merck Sharp and Dohme of Canada, Ltd.)
Растворитель Изотоп- ный Состав» % Химический сдвиг протона в растворителе (0-шкала)
группа б. м. д. группа б, м. д. группа б, м. д.
Уксусная кис- лота-й4 Ацетон-de Ацетонитрил-da Бензол-de Хлороформ-d D2O 1,2-Дихлорэтан^» Циклогекса н-du Диэтиловый-dio эфир 99,5 99,5 98 99,5 99,8 99,8 99 99 98 Метиль- ная То же » Метин- ная То же Гидро- ксил Метиле- новая То же Метиль- ная 2,05 2,05 1,95 7,20 7,25 4,75 а 3,69 1,40 1,16 Гидро- ксил Метиле- новая 11,53а 3,36
Диметилформ- амид-dr Диметилсульф- оксид-de л-Диоксан-dg 98 99,5 98 То же Метиле- новая 2,76 2,50 3,55 Метиль- ная 2,94 Фор- мильная 8,05
Этанол-de (безв). Г ексафторацетон дейтерирован- ный Метанол-d, 98 99,5 99 Метиль- ная Гидро- ксил Метиль- ная 1,17 9,00 а 3,35 Метиле- новая Гидро- ксил 3,59 4,84 а Гидро- ксиль- ная 2,60»
Мети лци клогек- сан-dn Метиленхло- pnfl-d2 99 99 То же Метиле- новая 1,92 5,35 Метиле- новая 1,54 Метин- ная 1,65
Пнридин-dj •Силанар-С 6 (CDC13+ 1% ТМС) Тетрагидрофу- pan-d8 Тетраметиден- da-сульфон 99 99,8 98 98 а Метиль- ная а-Мети- леновая То же 8,70 0,00 8 3,60 2,92 ₽ Метан- ная f-Мети- леиовая То же 7,20 7,25 1,75 2,16 Y 7,58
a Эта величина может значительно изменяться в зависимости от типа растворенного
вегцсст ва.
® Торговое название.
в Используется как начало отсчета в шкале д.
Классификация по растворимости и спектрам
157
Резонанс переменного магнитного поля с магнитным полем
ядра, изменяющего спиновое состояние, обусловливает химиче-
ский сдвиг (б), который откладывается на оси абсцисс записы-
вающего устройства; площадь сигнала пропорциональна числу
ядер в образце, резонирующих при данной частоте. Помимо двух
типов информации (б и площадь сигнала) можно получить дан-
ные о расщеплении сигналов, которые, как будет показано ниже,
дают дополнительные данные о структуре соединения.
Методика, Приготовляют раствор 0,10 г (около 0,15 мл) чистого образца
в 0,5 мл растворителя (чаще других используют CDCls или тетрахлорметаи; см.
табл. 5.8). С помощью пипетки переносят весь полученный раствор в ампулу
для измерения ЯМР-спектра (рис. 5.12). Добавляют внутренний стандарт; обыч-
но это одна-две капли тет'раметилсилана *. Ампула должна" быть вставлена до
нужной высоты в турбинку до заполнения раствором. На рис. 5.13 показана ам-
пула, правильно вставленная в турбинку (расположенную как раз под указа-
тельным пальцем оператора), в момент установки в ячейку спектрометра ЯМР.
Ампулку быстро вращают (около 10 оборотов в секунду); для удаления боко-
вых сигналов вращения скорость вращения должна быть достаточно высока**.
Устанавливают интервал измерения (обычно 100 Гц, илн 10 м. д., рис. 5.14) и
скорость записи спектра (чем меньше скорость сканирования, тем меньше ошибка
при записи за счет пера).
Выбирают регулировку амплитуды спектра, которая определяет интенсив-
ности пиков таким образом, чтобы наиболее интенсивные пики не выходили за
пределы шкалы; это можно сделать, просматривая весь спектр на экране осцил-
лографа (если это возможно) или изменяя регулировку после первой полной за-
писи спектра на бумаге. Для улучшения разрешения необходимо отрегулировать
радиочастоту и установку фильтра. Эти н более сложные регулировки требуют
специальных указаний преподавателя. Помните, прибор стоит очень дорого и
требует деликатного обращениях Включают самописец и снимают спектр
(рие. 5.15). Выполнять интегрирование можно только после консультации с ру-
ководителем работы.
Обсуждение. На рис. 5.16 показан flMP-спектр типичного орга-
нического соединения. Из этого спектра можно получить три
типа информации.
1. Центр сигналов (6) попадает в области, типичные для
структуры, приведенной на рис. 5.17.
2. Площадь под сигналом (измеренная путем триангуляции,
планиметром или по высоте ступеней на интегральной кривой)
пропорциональна числу протонов, дающих данный сигнал.
3. Мультиплетность (число пиков в данном сигнале) часто
указывает на число соседних протонов в соединении. Так, если
* Часто продажные растворители для измерения спектров ЯМР уже со-
держат внутренний стандарт, поэтому прочитайте этикетку на склянке с рас-
творителем.
** Боковые сигналы вращения идентифицируются как одна или несколько
пар небольших пиков, расположенных по сторонам от основного пика. Каждый
из пары боковых пиков расположен на одинаковом расстоянии слева и справа
от основного пика. Если при увеличении скорости вращения боковые сигналы не
сдвигаются и ие исчезают, то эти пикн не являются боковыми сигналами вра-
щения.
158
Глава 5
Рнс. 5.16. Спектр протонного магнитного резонанса этилбензола (ЯМР-спектро-
метр Varian ЕМ-360, (воспроизведено с разрешения фирмы Varian Associates,
Palo Alto, Calif.). Отнесение протонов сделано на основании данных, приведен-
ных на рис. 5.17, и обсуждения ЯМР-спектров в гл. 6.
Алифатические алициклические
I----------------------1
р-Замещенные алифатические
Ацетиленовые
I-----1
а-Монозамещвнные алифатические
|-------------------------Н
1 а, а-Дизамещенные алифатические
1 . Олефиновые।
Ароматические или гетероароматические
I--------------------1
Альдегидные
I------1
I______1_______1_______I______I______1_______1______1_______J______1------1_________
ТО е 87 6 6 4 3 2 1 0 в
О 1 23 4 Б в 7 В 9 Ют
Рис. 5.17, Положение сигналов протонов различных классов органических соеди-
нений.
Классификация по растворимости и спектрам
159
мультиплет состоит из п + 1 линий, то данные протоны имеют п
эквивалентных соседей. Более подробно с этим вопросом можно
познакомиться в книгах, перечисленных в гл. 10.
5.4. ВЫВОДЫ
В гл. 3 было описано определение простых свойств, чистоты
и молекулярной массы неизвестного соединения. В гл. 4 мы по-
казали, как установить элементный состав образца и его моле-
кулярную формулу. В настоящей главе рассмотрено, как опреде-
лить растворимость и спектральные свойства (ИК, ЯМР) веще-
ства, структура которого еще не известна. Заметим, что для всех
образцов обычно проводят почти все предварительные исследо-
вания; однако следует очень строго подходить к выбору допол-
нительных тестов (например, описанных в гл. 6) для того, чтобы
полностью установить и доказать структуру соединения. По-
этому очень важно знать, как планировать дальнейшие исследо-
вания (используя гл. 6 и другую информацию). Это будет по-
казано ниже на примере.
Неизвестное соединение № 1
Название соединения:
Исследователь: Джон Смит
Дата: 1 июня 1975 г.
Определение структуры:
1. Физические исследования.
а) Состояние: жидкость; б) цвет: бесцветное; в) запах: удушливый; г) проба
на сожжение: горит голубоватым пламенем без остатка.
2. Тонкослойная или газовая хроматография.
а) Адсорбент: б) элюат:
й) наблюдения: г) условия:
3. Физические константы.
Т. кип. 114—117°С (неиспр.), Ид 1,3988
4. Качественный элементный анализ.
а) Галогены: нет; б) гетероатомы: нет (кроме, возможно, О); в) металлы- нет.
5. Определение молекулярной массы.
ф) Метод: осмометрический. б) Результат: 87, 82 (второй опыт с меньшим коли-
чеством вещества).
6. Класс растворимости: Si.
7. Реакция на фенолфталеин: нет.
8. Растворимость в органических растворителях (л. р.—легко растворим):
Растворитель Ацетон CCL CHC1, Гексан Этанол Эфир
Растворимость л.р. л.р. л.р. Л.р. л.р. Л.р.
9. ИК-Спектр.
Прибор: Perkin-Elmer 237.
Фаза: тонкая пленка (чистое вещество).
Примечание: была использована прокладка между солевыми пластинами.
160
Глава 5
Линейная шкала: см~' или мкм.
Полоса, см-' Интенсив- ность Вид полосы Предварительное отнесение Окончатель- ное отнесение
3600 3300 3000—2850 1025 Слабая Сильная » Средняя Узкая Широкая Широкая О—Н валентное То же С—Н валентное С—О валентное
10. ЯМР-Спектр.
Ядро: протон-, растворитель: CDC13; частота поля: 60 МГц; концентрация: около
10%; прибор: Varian А60.
б, м.д. Интегриро- вание Мультнплет- ность Предварительное отнесение Окончательное отнесение
0,91 зн Триплет (иска- женный) сн3—сн2
Около 1,20-1,95 4Н Мультиплет сн2—сн2—сн2
3,47 2Н Т риплет —сн2—сн2—он
4,58 III Синглет —он
0,00 — тмс тмс
Даже студент, мало знающий органическую химию, может интерпретировать
имеющиеся данные следующим образом. Согласно предварительным данным, не-
известное вещество представляет собой очень простое соединение. Данные о его
растворимости свидетельствуют о том, что соединение обладает гидрофильными
и липофильными свойствами. Спектральный анализ отчетливо указывает на али-
фатический спирт. По ИК-спектру идентифицируется группа ОН, по спектру
ЯМР — прямая цепь углеродных атомов. Данные по растворимости также по-
казывают, что имеется цепь из четырех атомов углерода, поэтому можно утвер-
ждать, что изучаемое соединение представляет собой н-бутиловый спирт:
СН3СН2СП2СН2ОН
Хотя структура соединения и установлена, однако необходи-
мо провести более тщательное исследование. Для полной харак-
теристики вещества следует уточнить определенную ранее мо-
лекулярную массу. При осмометрическом определении могла
иметь место агрегация молекул, вызванная образованием водо-
родных связей. Опытный химик немедленно обратится к гл 6,
где приведены химические реакции, характерные для спиртов
(например, реакция с металлическим натрием), а также допол-
нительные спектральные характеристики (например, концентра
ционная зависимость поглощения ОН-группы в ИК- и ЯМР-спек-
трах). Обычно проводят химические реакции, приводящие к
производным спиртов (гл. 6). Те производные, для которых в
литературе отсутствуют данные о физических свойствах (напри-
мер, температура плавления), также должны быть полностью
охарактеризованы теми способами, которые были описаны выше.
Глава 6
Определение функциональных групп
и установление структуры изучаемых
соединений
В результате проделанной работы по установлению чистоты
вещества (гл. 3), его физических (гл. 3), химических (гл. 3, 4 и
5) и спектральных (гл. 5) характеристик студент, конечно, уже
имеет возможность сделать некоторые вполне логичные предпо-
ложения о структуре неизвестного соединения. Теперь можно
провести дополнительное более основательное изучение этого ве-
щества для того, чтобы выбрать из этих структур истинную.
С помощью химических и спектральных исследований, описан-
ных в этой главе, можно окончательно охарактеризовать боль-
шую часть органических соединений. Данные, которые получены
при изучении веществ методами, описанными в предыдущих гла-
вах, в большинстве случаев оказываются достаточными, чтобы
позволить студенту выбрать тот (или те) из последующих раз-
делов, в котором описаны методы идентификации, характери-
зующие именно данный конкретный класс соединений. Эти ме-
тоды расположены ниже как по типу определяемых функциональ-
ных групп, так и по типу применяемого реагента.
Каждый раздел, описывающий способ выявления определен-
ной функциональной группы, состоит из следующих частей:
1. Химические реакции.
2. Описание характерных особенностей ИК- и ЯМР-спектров,
а также подробное рассмотрение соответствующих масс-спек-
тров.
3. Методы получения производных.
4. Рассмотрение нетривиальных физических или химических
методов исследования.
Несмотря на огромную важность и легкость проведения спек-
трального анализа, для полной характеристики вещества необ-
ходимо провести характерные для него химические реакции. Дей-
ствительно, для идентификации вещества почти всегда прово-
дят разного рода «мокрые» реакции. Так, например, с помощью
спектрального анализа довольно трудно идентифицировать двой-
ную связь в 1,2-диметилциклогексене. В то же время химические
реакции присоединения (например, при действии брома или пер-
манганата калия) к двойным связям такого рода обычно прохо-
дят быстро. Итак, не следует придавать чрезмерно большого зна-
чения спектральному анализу в ущерб химическим методам
6 Зак. 336
162
Глава 6
идентификации, так как это может привести к определенным
трудностям при попытках идентифицировать данное соединение.
В предыдущих главах была описана только техника получе-
ния ИК- и ЯМР-спектров. Подробное изложение теоретических
положений, необходимых для интерпретации таких спектров,
можно найти в других руководствах. Тем не менее в дальнейшем
будут приведены многие спектральные данные (например, поло-
сы в ЯМР-спектрах), характерные для данного класса молекул,
без подробного обсуждения теоретических вопросов (например,
спин-спинового взаимодействия в ЯМР-спектрах).
С появлением спектрометрии органических молекул значение
методов идентификации органических соединений путем получе-
ния их производных несколько уменьшилось. Тем не менее хотя
в настоящее время, конечно, нет необходимости получать, напри-
мер, для альдегида и семикарбазон, и динитрофенилгидразон,
однако сама процедура получения производных дает представ-
ление как о некоторых физических характеристиках (например,
температуре плавления), так и о химических особенностях ново-
го соединения. Химикам следует также помнить, что некоторые
методы «получения производных» являются, по существу, мето-
дами «превращения» одного известного органического соединения
в другое. Такое превращение (например, окисление вторичного
спирта в кетон) может приводить к новому соединению, ко-
торое также должно быть охарактеризовано со всей тщатель-
ностью. По существу, большая часть методов получения произ-
водных является в действительности синтезами или препаратив-
ными методиками.
В тех случаях, когда известна температура плавления полу-
ченного производного, можно воспользоваться таблицами, при-
веденными в литературе. Если же данные о температуре плавле-
ния вещества отсутствуют, то у химика в руках оказывается, по
существу, новое неизвестное соединение, которое необходимо оха-
рактеризовать. Часто для установления химической структуры
веществ могут оказаться полезными таблицы физических кон-
стант, например показателей преломления. Для более точной
идентификации спектр неизвестного соединения необходимо со-
поставить с имеющимися в литературе сведениями об ИК-,
ЯМР- и (в ряде случаев) масс-спектрах. Спектральные данные
сравнивают так же, как отпечатки пальцев.
Однако при обсуждении спектрального анализа внимание бу-
дет уделено не сравнению «отпечатков пальцев», а главным
образом интерпретации спектров. Мы будем рассматривать те
полосы в спектре, которые могут быть полезны для выявления
функциональных групп и структурных единиц, присутствующих
в молекуле. Затем для полной характеристики изучаемого образца
эти данные будут сопоставлены с результатами химических ис-
следований.
Определение функциональных групп и установление структуры 168
Наконец, интуитивно представляется целесообразным исполь-
зовать специальные характеристики для некоторых классов ор-
ганических соединений. Например, можно ожидать, что для
идентификации аминокислот, часто имеющих хиральные центры,
окажется полезным удельное вращение [а].
Надо подчеркнуть, что серьезный химик-органик должен при-
влекать дополнительную литературу (гл. 10) для поиска новых
способов идентификации и дополнительных данных для харак-
теристики изучаемых соединений.
Трудно предполагать, что какой-либо учебник, рассчитанный
на студенческую аудиторию, может содержать все характерные
химические реакции. Поэтому в настоящей главе для многих хи-
мических реакций, описанных в следующих монографиях, указа-
ны номера страниц: Pasto D. J., Johnson С. Р., Organic Structu-
re Determination, Prentice-Hall, Englewood Cliffs, N. J., 1969;
Cheronis N. D., Entrikin J. B., Hodnett E. M., Semimicro Qualita-
tive Organic Analyses, 3rd ed., Wiley-Interscience, New York,
1965. Еще раз необходимо рекомендовать обращаться за новы-
ми дополнительными методиками к упомянутым выше моногра-
фиям и к приведенным в гл. 10 книгам, а также к научным жур-
налам.
Ниже перечислены в алфавитном порядке классы соединений
и указаны разделы, в которых описаны методики определения
характерных для этих соединений функциональных групп:
Класс соединений Раздел Класс соединений Раздел
Азотистые соединения 6.17 Простые эфиры 6.14
Алифатические галогениды 6.6 Сахара 6.11
Алканы (парафины) 6.3 Сераорганические соединения 6.18
Алкены (олефины) 6.4 Сложные эфиры 6.13
Алкины (ацетилены) 6.5 Солн карбоновых кислот 6.12.4
Альдегиды 62 Спирты 6 1-
Амиды 6.12.3 Сульфаты 6 18.11
Аминокислоты 6.8 Сульфиды 6.18.2
Амины (включая анилины) 6.7 Сульфиновые кислоты 6.18.6
Ангидриды кислот 6.12.1 Сульфоксиды 6.18.4
Ароматические галогениды 6.10 Сульфонамиды 6.18.9
Ароматические соединения 6.9 Сульфонаты 6.18.10
Ацетали 6.22 Сульфоновые кислоты (сульфо- 6.18.7
Галогенангидриды кислот 6.12.2 кислоты)
Дисульфиды 6.18.3 Сульфоны 6.18.5
Карбоновые кислоты 6.12 Сульфонилхлориды (сульфо- 6.18.8
Кетали 6.2.2 хлориды)
Кетоны 6.2 Углеводы 6.11
Меркаптаны 6.18.1 Фенолы 6.19
Многоатомные спирты 6.1.1 Эпоксиды 6.15
Нитрилы 6.17.2 Прочие соединения (библио- 6.16
Нитросоединения 6.17.1 графия)
Полифункциональные карбо- 6.21
нильные соединения
6*
164
Глава б
Реагенты (в алфавитном порядке), используемые в методиках
химической классификации веществ и при получении производных
Опыты
Реагент Номер опыта Определяемые соединения
Азоксибензол 22 Ароматические соединения
Азотистая кислота 19 Амины, анилины
Бенедикта раствор 29а Меркаптаны, тнофенолы
13 Альдегиды, кетоны
Бром в четыреххлористом уг- 296 Меркаптаны, тиофенолы
14 Алкены, алкины
лероде Бромная вода 30 Фенолы, енолы
Динитрофенилгидразин 6 Альдегиды, кетоны
Гидроксиламина гидрохлорид 7 То же
Железа (II) гидроксид 27 Нитросоединения
Железа(III) хлорид в пири- 29в Фенолы, енолы
дине Иодистоводородная кислота 26 Простые эфиры
(проба Цейзеля) Иодная кислота б Вицинальные диолы, полиолы
Йодоформная проба (Iz в KI) 10 Метилкетоны
Калия перманганат 15 Алкены, алкины, вицинальные дио-
Лукаса проба (Zn/HCl) 4 лы Спирты
Натрий металлический 1 Спирты и другие соединения с ак-
Натрия бисульфит 9 тивным водородом Альдегиды, кетоны
Натрия гидроксид 25 Солн аммония и аминов
29 Полиннтр осоединения
Натрия иодид в ацетоне 17 Галогены
Нингидрин 20 Аминокислоты
Серебра нитрат (в этаноле) 16 Подвижные галогены
Серная кислота дымящая 21 Ароматические соединения
Толленса проба 11 Альдегиды
Фенилгидразин (и л-нитрофе- 8 Альдегиды и кетоны
нилгидразин) 24 Углеводы
Фуксин 12 Альдегиды
Хлорангидрнды кислот 18 Спирты, амины, енолы Ароматические соединения
Хлороформ — AlCls 23
Хрома триоксид 3 Первичные и вторичные спирты
Церий (IV), окисление 2 Спирты
Цинк/аммонийхлорид 28 Нитросоединения
Методики получения производных
Соединение (в алфавитном порядке) Производное Номер мето- дики
Альдегиды Продукты окисления 6
Продукты окисления перманганатом 6а
Продукты окисления пероксидом водо- рода 66
Продукты реакции с димедоном 10
Определение функциональных, групп и установление структуры
165
Продолжение
Соединение (в алфавитном порядке) Производное Номер мето- дики
Альдегиды, кетоны Семнкарбазоны 7
л-Ннтрофенилгидразоны 8
2,4-Дннитрофенилгидразоны 9
Оксимы 11
Продукты восстановления диарилкетонов боргидридом натрия Из
Амиды 9-Ацнламидоксантены 38
Г ндролиз 38
Аминокислоты л-Толуолсульфонаты 24
М-2,4-Динитрофенильиые производные 25
Амины Продукты реакции Хинсберга 16
Амины (первичные, вто- Амиды (включая получаемые по методу 17
ричные) Шоттена — Баумана)
Амиды, получаемые действием хлорангид- ридов 35
Сульфонамиды 18
Фенилтиоурета ны 19
Амины (ArNRz) Ннтрозосоедннення 21
Амины (третичные) Четвертичные аммониевые соли Пнкраты Хлорплатинаты 20 22 23
Анилины см. Производные аминов
Анилиды 36
Анилины (нитропроизвод- ные) Ароматические соединения (ArR) Ароматические соединения Продукты гидролиза 37
Продукты окисления боковой цепи 27
Продукты нитрования 26
Ароилбензойные кислоты 28
Продукты ацилирования по Фриделю — Крафтсу 29
Хлорсульфонаты 30
Сульфонамиды (после методики 30) 31
Пикраты 22
Г алогеипроизводные Продукты реакции Гриньяра, ртутьорга- ннческие соединения 12
Арилуретаны (после методики 12) 13
Нитрилы 14а
а-Нафтиловые эфиры 14
Пнкраты S-алкнлтиурониевых производ- ных 15
Карбоновые кислоты " Амиды 35
Анилиды, л-толуидиды и л-броманилиды 36
Фенациловые и замещенные фенациловые эфиры 33
Фенплгидразнды и соли фенилгидразония 34
Карбоновые кислоты (и их соли) Арилтнурониевые соли 39
Кетоны См. Альдегиды, кетоны
Нитрилы Продукты гидролиза 37
Продукты кислотного гидролиза 51
Продукты осторожной гидратации до амидов 51а
166 Глава 6
П родолжение
Соединение (в алфавитном порядке) Производное Номер мето- дики
Нитросоеди нения Нитросоединения (много- атомные) Олефины (алкены) Простые эфиры (феноль- ные) Сложные эфиры 8 Сложные эфиры (арома- тические) Спирты Спирты (многоатомные) Сульфонамиды Сульфонилгалогениды Сульфоновые кислоты и их соли Сульфоновые кислоты (аминозамещенные) Фенолы Эфиры фенолов Продукты восстановления натрием в спирте Продукты окисления до альдегидов Гидрохлориды имидоилтиоуксусной кис- лоты Продукты восстановления до аминов дей- ствием олова в соляной кислоте Продукты конденсации с бензальдеги- дом под влиянием щелочей Продукты селективного восстановления до нитроаминов Продукты окислительного гидроборирова- ния Пикраты Омыление и кислотный гидролиз N-Бензнламиды Гидразиды кислот 3,5-Динитробензоаты Продукты бромирования Хлорсульфонаты Сульфонамиды (после методики 47) Арилуретаны 3,5-Динитробензоаты Бензоаты, л-нитробензоаты З-Нитрофталаты Ацетаты Продукты гидролиза N-Ксантилсульфонамнды Сульфонамиды Сульфонаты бензилтиурония Сульфохлориды и сульфонамиды л-Толуидинсульфонаты Хлорарилсульфонамиды и сульфонанили- ДЫ Арилоксиуксусные кислоты Продукты бромирования Фенилуретаны Пикраты 52 52а 53 50 506 50а 116 49 40 42 43 44, 45 46 47 48 1 2 3 4 5 57 58 48 53а 54 55 56 59 60 61 49
а Карбоновые и сульфоновые кислоты можно идентифицировать но эквивалентам нейт-
рализации (методика 32).
® Сложные эфиры можно идентифицировать по числу омыления (методика 41).
6.1. СПИРТЫ
Химический и спектральный анализ спиртов основан на на-
личии в их молекулах сильно полярных гидроксильных групп. Ха-
рактерными свойствами гидроксильной группы являются способ-
ность к образованию водородной связи и к химическому замеще-
нию ее водорода.
Определение функциональных врупп и установление структуры
167
Опыт 1. Определение активного водорода при действии метал-
лического натрия
Металлический натрий, особенно его свежеотрезанные кусоч-
ки, легко реагирует с гидроксильной группой с выделением во-
дорода
2ROH + 2Na —> 2RO'Na++ Н2 (газ)
Известно, что реакционная способность спиртов по отноше-
нию к натрию уменьшается при увеличении молекулярной массы
алкильного радикала. Многочисленные ограничения этой методи-
ки требуют соблюдать осторожность при интерпретации получае-
мых результатов.
Техника выполнения опыта. К 1 мл бутанола-1 (н-бутилового спирта) до-
бавляют тонко нарезанные кусочки металлического натрия до тех пор, пока он
не перестанет растворяться. Раствор охлаждают и наблюдают происходящие из-
менения. Добавляют равный объем эфира. Что представляет собой осадок? При-
мените эту пробу к ацетону, ди-н-бутиловому эфиру и толуолу.
Эту реакцию можно проводить с твердыми или очень вязкими вещертрамц,
если их растворить в таких инертных растворителях, как безводный лигроин или
бензол.
Обсуждение. Наибольшее значение эта реакция имеет при
исследовании нейтральных соединений, содержащих группы С
подвижным атомом водорода. Газообразный водород выделяет-
ся при действии натрия на функциональные группы, которые со-
держат атомы водорода, связанные с кислородом, азотом или
серой,
2ROH + 2Na —> 2RONa +
2R2NH + 2Na —> 2R2NNa + На
2RSH + 2Na —> 2R8Na + H2
Такую пробу чаще всего используют при исследовании спиртов
не очень большой молекулярной массы, содержащих от трех до
восьми атомов углерода. Спирты более низкой молекулярной
массы трудно получить в безводном состоянии. В присутствии
следов влаги такая проба всегда оказывается положительной.
Спирты с высокой молекулярной массой реагируют с натрием
очень медленно. При этом выделение газа происходит столь вяло,
что применение этой пробы становится нецелесообразным. Если
резать металлический натрий на воздухе, он адсорбирует влагу
воздуха. Адсорбированная вода реагирует с натрием с выделе-
нием водорода даже в том случае, если натрий помещают в со-
верщенно сухой растворитель, Цапрцмер в бензол.
Атом водорода, связанный с атомом углерода, замещается на
металл только в том случае, если рядом с ним находится функ-
циональная группа, способная его активировать. С натрием
168
Глава 6
будут реагировать соединения с активной метиновой группой, та-
кие, как ацетилен или монозамещенные ацетилены:
НС=СН + 2Na —> NaC==CNa + Н2
2R&=CH + 2Na —> 2RC=CNa + H2
Часто выделение газообразного водорода не наблюдается, так
как скорость его взаимодействия с ненасыщенными функциональ-
ными группами оказывается выше скорости его образования.
Атом водорода метиленовой группы может замещаться на на-
трий лишь в том случае, если рядом расположена какая-либо
активирующая группа. Особенно легко такое замещение происхо-
дит, если метиленовая группа расположена между двумя акти-
вирующими заместителями. В этом случае также бывает трудно
наблюдать выделение водорода из-за последующей реакции с не-
насыщенными связями в исходном соединении.
2СН3СОСН2СООС2Н5 + 2Na —► 2(CH3COCHCOOC.H5]'Na+ + Н2
В некоторых соединениях, особенно в метилкетонах, например
ацетоне и ацетофеноне, также имеются реакционноспособные ме-
тильные группы. Эти кетоны реагируют с натрием, давая соот-
ветствующие натриевые производные кетонов и смесь продуктов,
образующихся при их восстановлении и конденсации. Так, на-
пример, ацетон дает ацетонид натрия, изопропоксид натрия, пи-
наколят натрия, окись мезитила и форон.
Из сказанного выше становится очевидным, что металличе-
ский натрий является полезным реагентом для установления типа
соединений, содержащих реакционноспособный водород, но недо-
статочно активных, чтобы продуцировать водородные ионы в
ионизирующих растворителях. Очевидно, что нет никакой необ-
ходимости пытаться воздействовать натрием на соединения, о
которых уже известно, что они являются кислотами.
Наличие «активного» водорода гидроксильной группы во мно-
гих случаях можно выявить с помощью другого метода, вклю-
чающего применение галогенангидридов кислот. Такой метод
описан ниже (опыт 18).
Контрольные вопросы. 1. Что будет происходить при действии натрия на фе-
нол, бензойную кислоту, оксимы, нитрометан и бензолсульфонамид? Почему
применение этой пробы к этим веществам будет ошибкой? Какое влияние ока-
зывает присутствие влаги на результаты этой пробы?
2. Что является принципиальным недостатком металлического натрия как
реагента для классификации веществ?
Опыт 2. Окисление аммонийгексанитратоцератом
—CH—OH + 2Ce(lV) —> \>=О 4-2Се(Ш) + 2Н+
Реактив. К 400 мл дистиллированной воды добавляют 13 мл концентриро-
ванной азотной кислоты. В разбавленной кислоте растворяют 109,6 г желтой
Определение функциональных групп и установление структуры 169
соли (МН4)2[Се(МОз)б]. После того как растворение будет закончено, объем
смесн доводят до 500 мл. Реакцию проводят при комнатной температуре (20—
25°С). Горячий раствор (50—100°С) Ce(IV) окисляет многие типы органических
соединений. Приготовленный, как указано выше, раствор остается пригодным
для использования около месяца.
Техника выполнения опыта, а) Для водорастворимых соедине-
ний. К 1 мл аммонийгексанитратоцератного реактива прибавляют 4—5 капель
неизвестной жидкости или 0,1—0,2 г твердого вещества. Смесь тщательно пере-
мешивают и наблюдают изменение окраски (желтый цвет реактива изменяется на
красный). Если раствор стал красным, то за ним тщательно наблюдают и отме-
чают время, за которое смесь из красной становится бесцветной. Если в течение
15 мин изменения цвета не наблюдается, то пробирку с пробой закрывают и
оставляют на несколько часов или на ночь. При проведении этой пробы следует
также отмечать, выделяются или нет пузырьки углекислого газа.
Контрольные опыты. Проведите описанную пробу с метанолом, пропано-
лом-2, глицерином, глюкозой и молочной кислотой (85%-ной).
б) Для веществ, нерастворимых в воде. Добавьте 4 мл чи-
стого диоксана * к 2 мл реактива. Если смесь краснеет или становится бесцвет-
ной, то диоксан необходимо подвергнуть дополнительной очистке. Если же
смесь остается желтой или приобретает лишь бледно-оранжевый оттенок, то ее
можно использовать для исследования нерастворимых в воде соединений. Раз-
деляют 6 мл раствора пополам, причем 3 мл оставляют для контроля, а к дру-
гим 3 мл диоксанового раствора добавляют 4—5 капель неизвестного жидкого
соединения или 0,1—0,2 г твердого. Тщательно перемешивают и проводят те же
наблюдения, что и в методике (а), описанной выше.
Контрольные опыты. Проведите эту пробу с гептанолом-1, бензиловым спир-
том, d, /-миндальной кислотой.
Обсуждение. Этот реактив представляет собой окрашенный в
желтый цвет раствор аммонийгексанитратоцерата в разбавлен-
ной азотной кислоте и образует красные комплексы с соедине-
ниями, содержащими спиртовую гидроксильную группу. Положи-
тельную реакцию с этим реактивом дают первичные, вторичные и
третичные спирты, содержащие до 10 атомов углерода. Кроме того,
красное окрашивание характерно для всех типов гликолей, поли-
олов, углеводов, оксикислот, оксиальдегидов и оксикетонов.
Показано, что окрашенный в красный цвет комплекс является
промежуточным соединением при окислении спиртов раствором
Се(IV). Следовательно, второй фазой этой реакции будет исчез-
новение красного окрашивания в результате окисления координи-
рованного спирта и превращения окрашенного комплекса Ce(lV)
в бесцветный анион Се(III).
Последовательность реакций для первичного спирта следую-
щая:
а) (NH4)2[Ce(NO3)6] + RCH2OH —> [спирт + реагент]
желтый красный комплекс
* Диоксан должен быть проверен реакцией с раствором церия(IV) для
того, чтобы быть уверенным, что он не дает положительной пробы. Диоксан,
который продается под маркой «для гистологических исследований», обычно
бывает достаточно чистым и его можно использовать для проведения этой реак-
ции. Чистый диоксан не образует красного комплекса. Продажный диоксан
иногда содержит в качестве стабилизаторов гликоли или антиоксиданты и тре-
бует очистки.
170
Глава 6
б) [комплекс] —> RCH2O" + (NH4)2[Ce(NO3)5J + HNO3
бесцветный
в) RCH2O'+ (NH4)2[Ce(NO3)eJ —> RCHO + (NH4)2[Ce(NO3)5J + HNO3
желтый бесцветный
Скорость окисления на стадиях «б» и «в» зависит от структуры
гидроксильного соединения. В табл. 6.1 приведены значения вре-
мени превращения красного комплекса Се (IV) в бесцветный комп-
лекс Се(Ш) при 20° С, определенные при выполнении пробы ука-
Таблица 6.1
Приблизительное время восстановления красного комплекса Ce(lV)
В бесцветный ннтрато-анион Се(Ш) при окислении спиртов
в альдегиды и кетоны
Соединение Время реакции а, ч Соединение Время реакции а
Первичные спирты Пропиленгликоль 15 мин
Аллиловый 6 мин Диэтиленгликоль 3 ч
Метилцеллозольв 1,2 Этиленгликоль 5 ч
Пропанол-1 3,6 Бутандиол-1,4 1 ч
Бензиловый 4,0 Бутиндиол-1,4 36 мин
Бутанол-1 4,1 Бутендпол-1,4(в основ- 3 мин
2-Метилпропанол-1 4,1 ном цис)
.Гептанол-1 Этанол 2-Метилбутанол-1 Метанол Деканол-1 5,0 5,5 7,0 7,0 12,0 Углеводы Глюкоза Фруктоза Г алактоза Лактоза 1 мин 30 с 1 мин 5 мин
Вторичные спирты Мальтоза 8 мнн
Циклогексанол 3,7 Сахароза 12 мин
Пропаиол-2 6,0 Целлюлоза Нераство-
Бутанол-2 9,0 рима
Пентанол-2 17,0 Крахмал Не дает
’Октанол-2 16,0 красного
Дифенилкарбинол 12,0 окрашива-
Третичные спирты грет-Бутанол > 48 Оксикислоты 6 ння
трет-Амиловый спирт > 48 Молочная кислота 15 с
З-Мети лбутин-1 -ол-3 36 Яблочная кислота 30 с
Диолы, триолы, поли- Винная кислота 1 мнн
Миндальная кислота 1 мин
ОЛЫ Пинакон 5 с Лимонная кислота 1 мнн
Маннит 38 с Оксикетоны
Бутандиол-2,3 1 мин З-Оксибутанон-2 15 с
Глицерин 10 мин З-Метилбутанон-2-ол-З 10 с
а Время реакции окисления может изменяться в зависимости
реагента н срока его изготовления.
6 Реакция с оксикнслотами сопровождается выделением СOg.
от размеров капель
Определение функциональных групп и установление структуры
171
занным выше образом. Время окисления может несколько изме-
няться в зависимости от используемого объема реактива и срока
его приготовления.
В случае других гидроксилсодержащих соединений могут про-
текать следующие реакции:
RaCHOH —> R2CO4-H2O
RCH(OH)CH(OH)R —> 2RCHO + H2O
R2C—CR2 —► 2R2CO Н2О
I I
ОН он
RCH(OH)COOH —> RCHO4-CO2 + H2O
Наиболее глубокое красное окрашивание из монооксисоеди-
нений дает метанол. По мере увеличения молекулярной массы
спирта цвет смеси становится все менее интенсивным, приобретая
коричнево-красный оттенок.
Красное окрашивание при этой пробе дает также 40%-ный вод-
ный раствор формальдегида (формалин), что обусловлено нали-
чием в формалине метанола. В случае ацетальдегида красное ок-
рашивание часто появляется вследствие присутствия альдоля
СН3СН(ОН)СН2СНО. Кроме того, такие альдегиды гидратиру-
ются в водных растворах с образованием геминальных диолов,
например ЕСН(ОН)г, также способных к окислению.
Проба с цериевым реактивом отрицательна, т. е. не образуется
красный комплекс и сохраняется желтый цвет реактива, в случае
всех чистых альдегидов, кетонов, предельных и непредельных ки-
слот, сложных и простых эфиров, двух- и трехосновных кислот.
Двухосновные кислоты — щавелевая и малоновая — не дают крас-
ного окрашивания (отрицательная проба), но восстанавливают
желтый Се (IV) в бесцветный Се (III).
Фенолы с этим реактивом не дают характерного красного ок-
рашивания, однако в диоксановом растворе окисляются, в резуль-
тате чего образуются продукты коричневого или черного цвета
Высокоосновные алифатические амины вызывают выпадение
белого осадка гидроксида церия. Если амины растворить в раз-
бавленной азотной кислоте (при этом образуются нитраты аминов)
и этот раствор испытать цериевым реагентом, то красное окра-
шивание появляется только в том случае, если в молекуле амина
присутствует гидроксильная группа. При наличии таких групп
раствор соединения в разбавленной азотной кислоте окрашивается
в красный цвет при действии цериевого реактива. Например, по-
ложительную пробу дают растворы в разбавленной азотной кис-
лоте следующих аминоспиртов: HOCH2CH2NH2, (НОСНгСНг^ЫН,
(HOCH2CH2)3N.
Для спиртов, содержащих галогены, проба также положитель-
на. Например, красный комплекс образуют следующие спирты:
172
Глава 6
С1СН2СН2ОН, ВгСН2СН2ОН, С1СН2СН2СН2ОН и СНзСН(ОН)-
•СН2С1.
Очень плохо растворимые спирты высокой молекулярной мас-
сы, такие, как гексадеканол-1, трифенилкарбинол или бензпинакон,
не реагируют с цериевым реактивом и не дают красного окраши-
вания даже в диоксановом растворе.
Спирты с длинной цепью углеродных атомов от С]2 до Cjs мо-
гут давать положительную пробу, если проводить реакцию с ам-
монийгексанитратоцератом в ацетонитрильном растворе при его
температуре кипения (82°С). Специально для таких длинноцепо-
чечных спиртов разработана описанная ниже методика.
Применяемый в данной методике реагент представляет собой раствор 21,5 г
аммонийгексанитратоцерата в 100 мл ацетонитрила. Помещают 2 мл этого рас-
твора в пробирку и прибавляют около 0,1 г неизвестного соединения. Смесь пе-
ремешивают стеклянной палочкой и нагревают почти до кипения. При наличии
гидроксильных групп в течение от 1 до 6 мин желтый цвет смеси должен изме-
ниться на красный. Красное или оранжевое окрашивание дает даже холестерин
С27Н45ОН. После окисления спиртовой группы красный цвет исчезает.
Все спирты более низкой молекулярной массы и гликоли так-
же дают красное окрашивание с ацетонитрильным раствором при
комнатной температуре, однако, судя по изменению окраски, ско-
рость их окисления сильно отличается от скоростей, указанных
выше.
Опыт 3. Окисление хромовым ангидридом (триоксидом хрома,
окисление по Джонсу)
Этот метод позволяет установить присутствие гидроксильной
Группы, которая находится у атома углерода, соединенного по
крайней мере с одним атомом водорода и поэтому способного к
дальнейшему окислению:
3RCH2OH + 4СгО3 + 6H2SO4 —> 3RCOOH + 9Н2О + 2Cr2(SO4)3
3R2CHOH + 2СгОз + 3H2SO4 —> 3R2CO + 6Н2О + Cr2(SO4),
3RCHO + 2СгО3 + 3H2SO4 —> 3RCOOH + ЗН2О + Cr2(SO4)3
Техника выполнения опыта. К 1 мл ацетона в маленькой пробирке добав-
ляют 1 каплю жидкости или около 10 мг твердого вещества, затем прибавляют
1 каплю реактива — раствора триоксида хрома (хромового ангидрида) в серной
кислоте — и отмечают изменения, которые происходят в течение 2 с. Контроль-
ную пробу проводят с ацетоном и сравнивают результаты. Положительная проба
с первичными или вторичными спиртами состоит в помутнении раствора и появ-
лении зеленого или голубого окрашивания. Третичные спирты не дают видимой
реакции в течение 2 с, раствор остается оранжевым. Никакие изменения окраски
и общего вида раствора спустя 2 с не следует принимать во внимание.
Реактив. Суспензию 25 г хромового ангидрида в 25 мл концентрированной
серной кислоты медленно прибавляют при перемешивании к 75 мл воды. Перед
употреблением раствор, окрашенный в оранжево-красный цвет, охлаждают до
комнатной температуры.
Для проведения пробы можно использовать продажный ацетон высших сте-
пеней очистки. Некоторые образцы ацетона при этой реакции иногда темнеют че-
Определение функциональных групп и установление структуры
173
рез 20 с, однако это не мешает ее проведению, поскольку в первые секунды рас-
твор остается желтым. Если ацетон дает положительную пробу, то его надо
очистить обработкой небольшим количеством перманганата калия с последую-
щей перегонкой.
Обсуждение. Эта реакция позволяет быстро отличать первичные
и вторичные спирты от третичных. Положительную реакцию дают
все первичные и вторичные спирты независимо от их молекуляр-
ной массы. Даже с холестерином C27H4SOH проба положительна.
Альдегиды также дают положительную пробу, однако их можно
отличить от спиртов, применяя другие реакции. Олефины, аце-
тилены, амины, простые эфиры и кетоны дают отрицательную про-
бу при проведении реакции в течение 2 с, если только они не за-
грязнены небольшими количествами спиртов. Енолы также могут
давать положительную пробу, а фенолы окрашивают раствор в
темный цвет, совершенно не похожий на характерную зелено-го-
лубую окраску при положительной пробе. (Препаративное приме-
нение см. Пасто и Джонсон, с. 363.)
Опыт 4. Реакция с хлоридом цинка в соляной кислоте (проба
Лукаса)
Этот метод чаще используют не для определения гидроксиль-
ной группы, а для классификации спиртов. В реакцию вступают
такие соединения, в которых связанный с гидроксильной группой
атом углерода легко приобретает положительный заряд. С первич-
ными спиртами, например с этанолом, эта реакция не идет.
ZnCI2
R2CHOH + НС1 -----> R2CHC1 4- H2O
ZnCl2
R3COH + HCl ------> R3CCI + H2O
Реактив. Растворяют при охлаждении 136 г (1 моль) безводного хлорида
цинка в 105 г (1 моль) концентрированной соляной кислоты.
Техника выполнения опыта, а) К 1 мл спирта в пробирке при 26—27°С при-
бавляют 10 мл раствора хлорида цинка в соляной кислоте. Пробирку закрывают
и встряхивают, после чего отмечают время, требующееся для образования алкил-
галогеннда (указанием служит образование нерастворимого слоя жидкости нли
появление эмульсии). Проведите эту пробу с каждым из перечисленных ниже
спиртов и отметьте время, требующееся для реакции: бутанол-1, пентанол-2, про-
панол-1, трет-бутиловый спирт, пентанол-1, аллиловый спирт и бензиловый спирт.
б) К 1 мл спирта в пробирке прибавляют 6 мл концентрированной соляной
кислоты. Смесь встряхивают и внимательно наблюдают в течение 2 мин. Прове-
дите пробу с каждым из перечисленных ниже спиртов и запишите результаты:
пропанол-1, пентанол-2, трет-бутиловый и бензиловый спирты.
Обсуждение. Поскольку проба Лукаса основана на образовании
алкилгалогенида, который выделяется в виде второй жидкой фазы,
то использовать ее в обычном варианте можно только в случае тех
спиртов, которые способны растворяться в реактиве. В результате
174
Глава 6
этого ограничения данную пробу можно применять только к одно-
атомным спиртам низкой молекулярной массы (меньше С6) и к
некоторым полифункциональным молекулам.
Взаимодействие спиртов с галогеноводородными кислотами
представляет собой реакцию замещения, в которой активной час-
тицей является сопряженная кислота спирта R—ОНг- Можно пред-
полагать, что этот процесс будет аналогичен реакциям замеще-
ния атомов галогенов в органических галогенпроизводных при
действии нитрата серебра и иодид-иона (опыты 16 и 17). Влияние
структуры молекулы на реакционную способность органических
соединений в этих реакциях совершенно одинаково. Так, первич-
ные спирты не реагируют в заметной степени с соляной кислотой
при обычной температуре даже в присутствии хлорида цинка. Это
связано, с одной стороны, с тем, что хлорид-ион— слишком пло-
хой нуклеофильный агент для того, чтобы эффективно участвовать
в сопряженной реакции замещения, и, с другой стороны, со слиш-
ком малой стабильностью первичного карбониевого иона — про-
межуточного соединения при замещении по карбоний-ионному ме-
ханизму. Бромистый и иодистый водород, имеющий более актив-
ные нуклеофильные анионы, реагирует с первичными спиртами зна-
чительно энергичнее. При этом иодистый водород оказывается
более сильным нуклеофильным агентом. Именно такое соотноше-
ние способности к нуклеофильному замещению следует ожидать
для этих веществ в гидроксилсодержащих растворителях.
Третичные спирты реагируют с концентрированной соляной ки-
слотой так быстро, что даже при комнатной температуре уже че-
рез несколько минут наблюдается выделение алкилгалогенида вна-
чале в виде эмульсии, затем и в виде слоя масла. При добавлении
безводного хлорида цинка (сильной кислоты Льюиса) кислотность
среды возрастает и скорость реакции в значительной мере увели-
чивается. В этом случае реакция представляет собой не нуклео-
фильное замещение, как это было в случае первичных спиртов, а
скорее всего, протекает через промежуточное образование карбо-
ниевого иона. Высокая реакционная способность третичных спиртов
обусловлена относительно высокой стабильностью промежуточного
карбониевого иона. Аллиловый спирт, хотя и является первичным
спиртом, также дает сравнительно устойчивый карбониевый ион,
поскольку заряд в нем распределен равномерно на двух концевых
атомах углерода.
н+
СН2=СН—СН2ОН -----> [CH2=CHCHJ *-> СН2СН=СН2]
Этот спирт реагирует с реактивом Лукаса очень быстро с вы-
делением тепла. Аллилхлорид может быть выделен при разбав-
лении смеси ледяной водой.
Вторичные спирты по реакционной способности занимают
промежуточное место между первичными и третичными спиртами.
Определение функциональных групп и установление структуры 175
С чистой концентрированной соляной кислотой они не реагируют,
однако в присутствии безводного хлорида цинка реакция идет до-
статочно быстро, и уже через 5 мин наблюдается помутнение, а
через 10 мин обычно выделяется маслянистый слой алкилгалоге-
нида.
Более подробное обсуждение влияния структуры на реакцион-
ную способность в реакциях замещения такого типа проведено при
рассмотрении реакции с нитратом серебра (опыт 16).
Контрольные вопросы. 1. Напишите структурные формулы и названия изо-
мерных насыщенных спиртов, содержащих пять атомов углерода, которые не-
пригодны для проведения этой пробы. Как они будут реагировать с этим реа-
гентом?
2. Объясните различие в поведении аллилового спирта и пропанола-1, бен-
зилового спирта и пентанола-1.
Инфракрасные спектры спиртов оказываются полезными не
только для идентификации спиртовой группы, но также и для по-
лучения дополнительной информации о структуре молекулы. Хотя
в ИК-спектре спирта, приведенном на рис. 6.1, Л, сделано довольно
много отнесений полос поглощения, наиболее важное значение для
установления структуры молекулы имеют валентные колебания
связей О—Н и С—О. Если ИК-спектры спиртов снимают при
сильном разбавлении, то широкая полоса гидроксильных групп,
связанных водородной связью (рис. 6.1,А), исчезает и вместо нее
появляется новая узкая полоса О—Н, сдвинутая в более коротко-
волновую область спектра (иногда в спектре присутствуют обе по-
лосы). Эта новая полоса относится к колебаниям О — Н свободной
гидроксильной группы, которая не входит в ассоциаты, связанные
водородной связью (рис. 6.1,БД
«- «+ в-
RO---Н—О—R
I
Н
Анализ ПМР сигналов протонов в молекуле спирта также слу-
жит ценным источником информации о структуре и функциональ-
ных группах, присутствующих в молекуле изучаемого соединения.
На рис. 6.2,Л приведены ПМР-спектры простого спирта и указаны
типичные взаимодействия протонов. Величина вицинального вза-
/С—С\
имодействия I | | ) протонов соседних хр3-гибридизованных ато-
MI Н/
мов углерода составляет приблизительно 7 Гц. Поскольку скорость
обмена гидроксильных протонов друг с другом очень велика, то сиг-
нал группы ОН часто выглядит как синглет. Если из растворителя
исключить следы кислот или снять ПМР-спектр в ДМСО-г/6 (или
в ДМСО), то сигнал гидроксильной группы часто превращается
Поглощение
С5
Perkin-Elmer 521
Рис. 6.1. Инфракрасные спектры метанола (Д) и этанола в газовой фазе (5), измеренные на приборе
(© Sadtler Research Laboratories, Inc., Philadelphia, Pa. 1966).
Л-метанол CH4O, мол. масса 32,04, т. кип. 64,7 °C. df 0.7915, 1,3292 (лит.), получен от фирмы The Matheson Со., Inc.),
тонкая пленка. Валентные колебания О —Н: 1 — 3340 см"”1 (2,99 мкм), _ _
(3,40 мкм), vCH3 5 — 2830 см-1 (3,53 мкм), vCHs , Деформационные колебания: 4—1450 см-1 (6,90 мкм), 6^8
асимм СИММ dcnwiM
<7,04 мкм). плоские деформационные колебания О-Н. Валентные колебания С—О: 6—1060 см” (9,43 мкм),
капиллярная кювета,
Vq__pj, ассоциированная. Валентные колебания С—Н: 2—2940 см
5 группы СНзО: 5—1420 см”1
гасимм« ~ ....>* симм‘ г~'"г~г ' асимм
<7,04 мкм). плоские деформационные колебания О —Н. Валентные колебания С—О: 6—1060 см” (9.43 мкм), ______q’ Деформационные колебания:
7 — 650 см”1 (15,39 мкм), неплоскне деформационные колебания О-Н. Б —этанол СгНвО, мол. масса 46,069, т. кип. 78,4 °C (лит.), получен от
Фиомы Sadtler Research Labs., Inc., в газообразном состоянии в газовой 10-сантиметровой кювете (см. IR 188 в жидкой фазе по атласу Садтлера).
Валентная полоса колебаний О-Н сравнительно малоинтенсивна. При анализе в растворе (см. спектр Л) спирты обычно дают интенсивную
полосу валентных колебаний О—Н. Валентные колебания О— Н: 8 - 3660 см-1 (2,73 мкм), vo_H, свободная. Валентные колебания С-Н:
5—2970 см-1 (3,37 мкм), vCc^3 м! 10—2900 см-1 (3.45 мкм), Деформационные колебания С —Н: И -1400 см-1 (7.15 мкм), 12-1250 см-
(8,00 мкм), СНг (веерные). Валентные колебания С —О: /3—1060 см”1 (9,43 мкм),
178
Глава 6
Рис. 6.2. Спектр протонного магнитного резонанса этанола (60 МГц, развертка 600 Гц).
А—растворитель ДМСО-dt; В —растворитель CDC1».
Определение функциональных групп и установление структуры
17©
в мультиплет (рис. 6.2,Б), структура которого зависит от харак-
тера спирта:
Спирт Первичный (RCHjOH) Вторичный (R2CHOH) Третичный (R3COH)
Мультиплетность сиг- нала ОН Триплет или квад- руплет при R = Н Дублет Синглет
При проведении масс-спектрометрического анализа спиртов
очень часто возникают трудности при детектировании молекуляр-
ного иона. В общем в зависимости от структуры спирта интенсив-
ность пика меняется следующим образом:
Спирт
Первичный
Вторичный
Третичный
Интенсивность пика
молекулярного нона
Низкая
»
Обычно отсутствует
Молекулярную массу спиртов можно определить масс-спект-
рометрическим методом, если предварительно перевести их в си-
лильные эфиры (см. Пасто и Джонсон, с. 368). Для установления
структуры спиртов типа R1R2R3COH часто бывает полезно иденти-
фицировать ряд осколочных пиков:
Структура фрагмента ml» Примечание
[СН2ОН]+ [RiR2COH]+ [М—Н2О] 31 [29 + R, + R2] [М—18] Обычно наиболее интенсивный пик в случае первичных спиртов (М—R3). Чаще всего происходит от- рыв наиболее тяжелого радикала Rs Образование этого иоиа облегчает- ся при увеличении длины цепи и протекает как 1,4-элимииирование
Наиболее общими процессами трансформации спиртов является
либо получение сложных эфиров — ОН * — O(C = O)R (методики
1—3), либо окисление /СНОН^>^С=О (литературные ссылки
приведены ниже).
Третичные спирты не имеют а-водородного атома и поэтому
окисляются только в очень жестких условиях.
180
Глава 6
Методика 1. Получение фенил- и а-иафтилуретанов
В пробирку помещают 1 г безводного спирта или фенола и добавляют
0,5 мл фенилизоцианата (Осторожно] Изоцианаты слезоточивы) или а-нафтил-
изоцианата. При проведении реакции с фенолами необходимо добавить 2—3 кап-
ли безводного пиридина или триэтиламина в качестве катализатора. Если реакция
не начинается самопроизвольно, то раствор необходимо нагревать на водяной
бане около 5 мин. Затем смесь охлаждают в стакане со льдом и стеклянной па-
лочкой трут стенки пробирки, чтобы ускорить образование кристаллов. Полу-
ченные уретаны очищают, растворяя в 5 мл петролейного эфира или тетрахлор-
метана. Горячий раствор фильтруют и охлаждают фильтрат на ледяной бане.
Выпавшие кристаллы отделяют фильтрованием, сушат на керамической пла-
стине и определяют температуру плавления.
Методика 2. Получение 3,5-динитробеизоатов
NO
NO2
+ НС1
Около 0,5 г 3,5-динитробензоилхлорида смешивают в пробирке с 2 мл спир-
та и смесь осторожно кипятят 5 мин. Затем добавляют 10 мл дистиллирован-
ной воды и охлаждают раствор на ледяной бане до тех пор, пока продукт не
затвердеет. Выпавший осадок отделяют фильтрованием, промывают 10 мл 2%-
ного раствора карбоната натрия и перекристаллизовывают из 5—10 мл смеси
этанола и воды, составленной в такой пропорции, чтобы образовавшийся эфир
растворялся в горячем растворе, но выделялся из него при охлаждении. Вы-
павшие кристаллы отделяют фильтрованием, сушат на пористой пластинке и
определяют температуру плавления.
При отсутствии 3,5-динитробензоилхлорида его можно приготовить. Для
этого 0,5 г 3,5-динитробензойной кислоты и 1 г пентахлорида фосфора смеши-
вают в пробирке. Смесь осторожно нагревают до начала реакции. Вначале
реакция протекает довольно бурно, затем постепенно затихает, и для ее полного
завершения смесь необходимо нагревать в течение примерно 4 мин так, чтобы
происходило энергичное выделение пузырьков газа. По окончании реакции смесь
выливают на часовое стекло. Когда масса затвердеет, ее переносят на чистую
керамическую пластинку н растирают шпателем для удаления оксихлорида
фосфора. Оставшийся хлорангидрид кислоты немедленно используют для полу-
чения эфира, как описано выше.
O2N
Определение функциональных групп и установление структуры 181
3,5-Динитробензоаты можно получать также и в присутствии пиридина,
как описано в методике За.
Методика 3. Получение бензоатов и я-нитробензоатов
О
II
ROH + ArCCl
Аг = С6П5 или n NO2C6H4
а) Растворяют 1 мл спирта в 3 мл безводного пиридина и добавляют 0,5 г
бензонлхлорида или л-нитробензоилхлорида. После того как энергично проте-
кающая вначале реакция замедляется, смесь нагревают на небольшом пламени
около 1 мин и выливают ее при сильном перемешивании в 10 мл воды. После
выпадения осадка жидкость декантируют. Осадок тщательно перемешивают с
5 мл 5%-ного раствора карбоната натрия, фильтруют и очищают перекристалли-
зацией из спирта.
б) Смешивают 1 мл спирта с 0,5 г бензонлхлорида или п-ннтробензоил-
хлорида и кипятят на небольшом пламени несколько минут. Смесь выливают в
воду и проводят очистку, как описано в методике «а».
Ниже приведены некоторые важные реакции спиртов.
Окисление трет-бутилгипохлоритом (Пасто и Джонсон, с. 363,
препаративная методика):
I (СНз)зСОС! \
Н—С—ОН ------------> ,С=О
(СН3)3СОН + NaOCl —> (СН3)3СОС1
Окисление реагентом Саретта (Пасто и Джонсон, с. 362; необ-
ходимо строго соблюдать порядок смешивания реагентов):
СгО:
С=О
Хорошо известно применение лантаноидных сдвигающих реа-
гентов для упрощения спектров ЯМР (гл. 8). Спирты — один из
наиболее удобных объектов для применения этой техники.
В ультрафиолетовых спектрах спиртов имеются только слабые
полосы поглощения, и поэтому эти спектры лишь в редких слу-
чаях используются для установления их структуры.
Методика 4. Получение 3-нитрофталатов
По описанным ниже методикам не все спирты дают твердые про-
изводные. Однако наличие карбоксильной группы в получаемых
продуктах придает им ряд полезных свойств. При выделении этих
182
Глава 6
производных можно применять кислотно-основную экстракцию
(гл. 7), а эфиры оптически активных спиртов во многих случаях
можно разделить взаимодействием с хиральными аминами (гл. 7).
Весьма полезным применением этих производных является оп-
ределение эквивалентов нейтрализации (методика 32) кислых эфи-
ров. В ряде случаев это дает возможность определить молекуляр-
ную массу изучаемого соединения.
а) Получение эфиров спиртов, кипящих ниже 150°С. В пробирке, снабжен-
ной стеклянной трубкой в качестге обратного холодильника, нагревают смесь
0,4 г 3-нитрофталевого ангидрида и 0,5 мл спирта в течение 5—10 мин при
слабом кипении до тех пор, пока смесь не станет однородной. Полученную жид-
кость бхлаждают, разбавляют 5 мл воды и нагревают до кипения. Если смесь
полностью не растворится, то добавляют еще 5—10 мл горячей воды. По охла-
ждении раствора эфир выделяется в виде кристаллов. Иногда образуются масло-
образные продукты. В этом случае смесь нужно оставить на ночь для кристал-
лизации. Продукт один или два раза перекристаллизовывают из горячей воды.
б) Получение эфиров спиртов, кипящих выше 150°С. Смесь 0,4 г 3-нитро-
фталевого ангидрида, 0,5 г спирта и 5 мл сухого толуола кипятят до полного
растворения ангидрида и затем еще 15 мин. Толуол удаляют в вакууме водо-
струйного насоса, остаток дважды экстрагируют 5 мл горячей воды, не раство-
рившееся в воде масло растворяют в 10 мл 95%-ного спирта и нагревают до ки-
пения. Если горячий раствор непрозрачен, его надо профильтровать. К горя-
чему раствору добавляют воду до тех пор, пока не начнет появляться муть,
исчезающая при добавлении одной-двух капель спирта. Раствор медленно охла-
ждают и оставляют стоять. Многие 3-нитрофталатные производные моноалкиль-
ных эфиров этиленгликоля и диэтиленгликоля с достаточно большими алкиль-
ными группами выделяются в виде масла и твердеют только через несколько
дней. Иногда для перекристаллизации можно вместо водно-спиртопой смеси ис-
пользовать толуол. Желательно определять не только температуру плавления,
ио также и эквивалент нейтрализации полученных кислых фталатов.
В книге Черониса, Энтрикина и Ходнетта описаны дополни-
тельные реакции на спирты, в том числе реакции с вападий-ок-
сином (с. 368), гидроксаматом железа (с. 368), получение ксанта-
тов (с. 368) и реакция с N-бромсукцпнимидом (с. 369).
6.1.1. Многоатомные спирты
Химия некоторых представителей этого класса спиртов отли-
чается от химии одноатомных спиртов из-за присутствия в них гид-
роксильных групп у соседних атомов углерода:
Методы химической идентификации и получения производных поз-
воляют установить точные места расположения этих групп. Полу-
ченные производные очень часто менее устойчивы, чем исходные
многоатомные спирты. Эти методы особенно полезны для иден-
тификации и характеристики полиольных фрагментов углеводов и
других аналогичных соединений.
Определение функциональных групп и установление структуры
183
Опыт 5. Идентификация вицинальных диолов и сходных соеди-
нений с помощью иодной кислоты
RCHOH + HIO4 —► 2RCHO + Н2О + НЮз
I
RCHOH
RCHOH + HIO4 —> RCHO + R'COOH + НЮз
I
R'C=O
RC=O + HIO4 4-Н3О+ —> 2RCOOH + HIO3
I
RC=O
RCH(OH)CH(NH2)R' + HIO4 —> RCHO + R'CHO + NH3 + НЮз
В маленькую пробирку помещают 2 мл иодной кислоты, добавляют 1 каплю
(не больше!) концентрированной азотной кислоты и сильно встряхивают. Затем
добавляют 1 каплю пли маленький кристаллик исследуемого соединения. Смесь
встряхивают в течение 10—15 с и добавляют 1—2 капли 5%-ного водного рас-
твора нитрата серебра. Мгновенное образование белого осадка йодата серебра
указывает на окисление органического вещества перйодатом, восстанавливаю-
щимся при этом до йодата.
НЮз + AgNO3 —-> HNO3 + AgIO3 (тв.)
Образование такого осадка является признаком положительной пробы. При от-
рицательной пробе осадок или не выпадает вообще, или имеет коричневую
окраску. Коричневый осадок растворяется при встряхивании пробирки.
Для облегчения реакции с нерастворимыми в воде полиолами к реакцион-
ной смеси можно добавлять диоксан.
Проделайте эту пробу со следующими веществами: пропанол-2, ацетон, эти-
ленгликоль, глицерин, формалин, винная кислота и молочная кислота.
Реактив. Раствор иодной кислоты для проведения пробы готовят, растворяя
0,5 г парапериодной кислоты (Н5Ю6) в 100 мл дистиллированной воды.
Обсуждение. Иодная кислота очень селективно окисляет 1,2-
гликоли, а-оксиальдегиды, а-оксикетоны, 1,2-дикетоны, а-оксикис-
лоты и а-аминоспирты. Скорость реакции уменьшается в том же
порядке. Иногда в описанных выше условиях а-оксикислоты дают
отрицательную пробу. 0-Дикарбонильные соединения и другие ве-
щества с активной метиленовой группой также реагируют с иод-
ной кислотой.
При проведении этой реакции важно точно соблюдать соотно-
шение между реактивом и азотной кислотой. Проба основана на
том, что иодат серебра очень плохо растворим в разбавленной азот-
ной кислоте, в то время как перйодат серебра растворяется в ней
очень хорошо. Однако если в растворе присутствует много азотной
кислоты, то иодат серебра может в осадок не выпасть.
Олефины, вторичные спирты, 1,3-гликоли, кетоны и альдегиды
в этих условиях с иодной кислотой не реагируют. Проба с иодной
кислотой лучше всего подходит для соединений, растворимых §
воде.
184
Глава 6
Был предложен
диолов:
следующий механизм окисления вицинальных
ОН ОН
I I
—с—с—+ню4
нс/ /ж
перенос двух
протонов
Н°\/°
НО—1—ОН
НО—I—он
Н3Ю4 —> НТО3+Н2О
Методика 5. Получение ацетатов полиоксисоединений
Эта методика, так же как и некоторые другие, описанные ниже
(например, триметилсилилирование), используется для изменения
летучести углеводов и сходных соединений.
а) Смешивают 3 г безводного полиоксисоединения с 1,5 г измельченного
плавленого ацетата натрия и 15 мл уксусного ангидрида. Смесь нагревают около
2 ч па паровой баие, периодически встряхивая. После этого теплый раствор,
энергично перемешивая, выливают в 100 мл ледяной воды. Оставляют стоять,
периодически встряхивая, до тех пор, пока не закончится гидролиз избытка ук-
сусного ангидрида. Кристаллы отделяют фильтрованием, тщательно промывают
водой и очищают перекристаллизацией из спирта.
б) Прибавляют 2 г полиокспсоединения к 20 мл безводного пиридина.
Встряхивают, добавляют 8 г уксусного ангидрида. После того как бурная вна-
чале реакция затихнет, раствор кипятят 3—5 мин с обратным холодильником.
Смесь охлаждают и выливают в 50—75 мл ледяной воды. Отфильтровывают
ацетильное производное, промывают холодной 2%-ной соляной кислотой и затем
водой, после чего перекристаллизовывают из спирта.
Методы идентификации и характеристики полиоксисоединений,
описанные в книге Пасто и Джонсона, приведены ниже:
Образование боратов:
L I J
анион 'сложного эфира борной
Ийслотъ! При ион тетраалкилбората
Определение функциональных групп и установление структуры 185
Качественная проба на кислые бораты приведена в книге Па-
сто и Джонсона, с. 365.
Получение ацетонидов (препаративная методика см. Пасто и
Джонсон, с. 367):
Н3СХ ZCH3
ОН ОН о/ \о
। । n-толуолсульфокислота । |
кеталь ацетона
(1,3-диоксолан)
Триметилсилилирование (препаративная методика см. Пасто
и Джонсон, с. 368):
2ROH+ (CH3)3SiNHSi(CH3)3 —> 2ROSi(CH3)3 + NH3
гексаметилдисилазаи триметилсили-
(фирмеиное иазва- ловый эфир
ние TRL-SIL)
6.2. АЛЬДЕГИДЫ (RCHO) И КЕТОНЫ (RCOR)
Химическая и спектральная идентификация альдегидов и кето-
нов основана на наличии в их молекулах карбонильной группы
(С = О). Обычно альдегиды более реакционноспособны, чем ке-
тоны. Кроме того, связь С—Н в альдегидной группе СНО позво-
ляет с помощью спектрального анализа отличить альдегиды от ке-
тонов, не содержащих такой связи.
Ниже сначала обсуждаются химические методы, позволяющие
обнаружить присутствие карбонильной группы почти в любых ке-
тонах и альдегидах. Далее приводятся более селективные методы,
в том числе и такие, которые пригодны только для обнаружения
альдегидов.
Опыт 6. Реакция с 2,4-динитрофенилгидразином
Эта реакция относится, вероятно, к наиболее изученным и ус-
пешно применяемым среди всех качественных реакций и методик
получения производных. Более того, общие методические приемы,
разработанные для этой реакции, служат моделью для проведения
ряда других сходных химических процессов, таких, как получе-
ние озазонов, семикарбазонов, оксимов и других арилгидразонов
о II С + O2N/£j^-NHNH2 NNH{0\n02 H,S04 II Спирт NOa
186
Глава 6
Растворяют 1 или 2 капли испытуемого вещества в 2 мл 95%-ного этанола
н смешивают с 3 мл реактива, содержащего 2,4-динитрофеиилгидразин. Смесь
энергично перемешивают и, если осадок сразу не выпал, оставляют раствор иа
15 мии.
Реактив (иногда называемый реактивом Брэди) получают растворением 3 г
2,4-динитрофенилгидразина в 15 мл коицентрированиой серной кислоты. Затем
при перемешивании полученный раствор приливают к смеси 20 мл воды и 70 мл
95%-ного этанола, тщательно перемешивают и фильтруют.
Обсуждение. Почти все альдегиды и кетоны дают твердые не-
растворимые динитрофенилгидразоны. Выпадающий осадок вна-
чале может представлять собой масло, однако при стоянии оно
затвердевает. Все же многие кетоны образуют маслообразные
динитрофенилгидразоны. Так, например, не дают твердых динитро-
фенилгидразонов метил-н-октилкетон, ди-н-пентилкетон и другие
подобные соединения.
Другое затруднение при проведении этой реакции связано с воз-
можностью окисления этим реагентом некоторых производных ал-
лильных спиртов в альдегиды или кетоны с последующим образо-
ванием соответствующих динитрофенилгидразонов (положительная
проба). Такие производные были получены из коричного спирта,
4-фенилбутен-3-ола-2 и из витамина А! с выходами от 10 до 25%.
Бензгидрол (дифенилкарбинол) с небольшим выходом дает динит-
рофенилгидразон бензофенона. Кроме того, всегда есть опасность,
что положительную пробу могут дать образцы спиртов, в которых
могут присутствовать примеси соответствующих карбонильных со-
единений, образовавшихся в результате окисления кислородом во-
здуха. Если динитрофенилгидразон образуется в очень малом ко-
личестве, то следует провести реакцию в таких условиях, чтобы
можно было установить выход продукта, например, как указано
в описании методики 9. Выход рассчитывают по молекулярной мас-
се карбонильного соединения, которую можно достаточно надежно
оценить на основе предположений о возможных структурах неиз-
вестного соединения по данным, приведенным в таблицах. Необ-
ходимо проверять температуру плавления получаемых твердых ве-
ществ, чтобы быть уверенным, что она отличается от температуры
плавления самого. 2,4-динитрофенилгидразина (198°С с разложе-
нием).
При необходимости полученный гидразон можно перекристал-
лизовать из таких растворителей, как, например, этанол. Не сле-
дует использовать растворители, содержащие активную карбониль-
ную группу (например, ацетон), так как при этом возможно об-
разование другого гидразона.
По окраске 2,4-динитрофенилгидразона можно сделать неко-
торые выводы о структуре исходного альдегида или кетона. Динит-
рофенилгидразоны альдегидов и кетонов, в которых карбонильная
группа не сопряжена с другой функциональной группой, имею!?
желтый цвет. Сопряжение с двойной углерод-углеродной связью
или с бензольным кольцом сдвигает максимум поглощения в ви-
Определение функциональных групп и установление структуры
187
димую область, что легко устанавливается по ультрафиолетовому
спектру *. Однако такой сдвиг способствует также изменению цве-
та соединения от желтого до оранжево-красного. В общем можно
считать, что желтые динитрофенилгидразоны образуются из несо-
пряженных карбонильных соединений. Однако при получении оран-
жевых или красных гидразонов выводы необходимо делать с осто-
рожностью, так как их окраска может быть обусловлена наличием
окрашенных примесей (например, сам 2,4-динитрофенилгидразин
имеет оранжево-красную окраску).
В трудных случаях можно попытаться получить динитрофенил-
гидразон в диметиловом эфире этиленгликоля (диглиме), моно-
метиловом эфире этиленгликоля (метилцеллозольве),диметилфор-
мамиде или диметилсульфоксиде. Однако при этом возникают труд-
ности, связанные с удалением нелетучего растворителя. Вместо
этанола можно применять метанол, однако более летучий раство-
ритель может затруднять очистку получаемого продукта.
Опыт 7. Реакция с гидрохлоридом гидроксиламина
Эта реакция протекает по механизму, сходному с тем, который
был рассмотрен выше для получения арилгидразонов. В резуль-
тате образования оксимов выделяется хлористый водород, что мо-
жно установить с помощью подходящего индикатора.
rcho + h2noh-hci —-> RCH=NOH + HC1 + H2O
r2co +h2noh-hci —> R2C=NOH + HC1 + H2O
а) Для нейтральных соединений. К. 1 мл реактива добавляют 1 каплю или
небольшой кристалл испытуемого соединения и наблюдают за изменением окра-
ски. Если при комнатной температуре никаких изменений не происходит, то
смесь следует нагреть до кипения. Проба считается положительной, если цвет
реакционной смеси изменится из оранжевого в красный. Примените эту пробу к
н-масляиому альдегиду, ацетону, бензофенону и глюкозе.
б) Для кислых или щелочных соединений. К 1 мл раствора индикатора в
пробирке добавляют около 0,2 г испытуемого соединения и, прибавляя по кап-
лям разбавленные (1%-ные) растворы гидроксида натрия или соляной кислоты,
добиваются такого изменения окраски смеси, чтобы она соответствовала цвету
1 мл реагента, налитого в пробирку такого же объема. Получившийся раствор
приливают к 1 мл реагента и отмечают, появляется ли красная окраска. Прове-
дите эту реакцию с салициловым альдегидом и л-диметиламинобензальдегидом.
Проведите пробу по методикам «а» и «б» с винной кислотой.
Реактив. К раствору 5 г гидрохлорида гидроксиламина в 1 л этанола добав-
ляют 3 мл универсального кислотно-основного индикатора (например, универ-
сальный индикатор фирмы Bogen или Grammercy). Прибавляя по каплям раз-
бавленный 5%-ный спиртовой раствор гидроксида натрия, добиваются ярко-
оранжевой окраски раствора, что соответствует pH 3,7—3,9. Реактив стабилен
течение нескольких месяцев.
Раствор индикатора готовят, растворяя 3 мл любого из указанных выше
индикаторов в 1 л 95°/о-иого этанола.
* Rappoport Z., Sheradsky Т., J, Chem. Soc. (В), 1968, 277; Jones L. A.,
Holmes J. C., Seligman R. B., Anal. Chem., 28, 191 (1956).
188
Глава 6
Обсуждение. Изменение цвета индикатора обусловлено образо-
ванием хлористого водорода в результате реакции карбонильного
соединения с гидрохлоридом гидроксиламина. Получающийся при
этом оксим не обладает достаточной основностью, чтобы связы-
вать хлористый водород. Почти все альдегиды и большая часть ке-
тонов сразу же изменяют цвет реактива. Некоторые кетоны высо-
кой молекулярной массы, такие, как бензофенон, бензил, бензоин
и камфора, реагируют при нагревании. Сахара, хиноны и прост-
ранственно затрудненные кетоны (как, например, о-бензоилбен-
зойная кислота) дают отрицательную пробу.
Многие альдегиды самоокисляются на воздухе и поэтому со-
держат заметное количество кислот:
воздух
RCHO -------> RCOOH
В связи с этим необходимо всегда проверять их реакцию с водным
раствором или суспензией лакмуса. Если раствор оказывается кис-
лым, то следует применять методику «б». Так же поступают в тех
случаях, когда характер растворимости изучаемого вещества ука-
зывает на то, что оно может быть кислым или щелочным.
Механизм реакции карбонильных соединений с гидроксилами-
ном, по-видимому, очень напоминает соответствующий механизм
взаимодействия этих соединений с фенилгидразином, динитрофе-
нилгидразином и семикарбазидом. Наиболее подробно изучена ре-
акция образования семикарбазонов. Ниже приведен механизм этой
реакции в водном растворе в присутствии катализатора — кисло-
ты НА.
Из рассмотрения этого механизма следует, что реакция силь-
но замедляется как в очень кислых, так и в очень щелочных сре-
дах. Это происходит вследствие того, что в реакцию вступает сво-
бодный семикарбазид и источник протонов (НА). В сильнокислых
средах скорость реакции падает, так как почти весь семикарбазид
превращается в неактивную сопряженную кислоту, а в сильноще-
лочных средах реакция тормозится из-за перехода кислоты НА в
ее неактивное сопряженное основание. Наиболее подходящими ус-
ловиями для проведения этой реакции будут такие, при которых
произведение концентраций НА и NH2NHCONH2 будет максималь-
ным,
R2c=o+НА+H2NNHCONH2f^R2C=OH А+H2NNHCONH2
л
4 ОН - он
R,c^ +HAi=»R2C^ +а~
NHNHCONH2 NH2NHCONHa
+
OH-HA
RaG f^R2C=NHNHCONH2+H2O+A~
' NHNHCONHj. И
R,C=NNHCONH3,+Н2О+Н А
Определение функциональных групп и установление структуры
189
Эта реакция обратима. В слабокислых растворах равновесие
может быть сдвинуто почти целиком вправо, однако сильные кис-
лоты сдвигают его в сторону образования свободного кетона из-за
превращения семикарбазида в его сопряженную кислоту.
Препаративное получение семикарбазонов описано ниже в ме-
тодике 7.
Опыт 8. Реакции с фенилгидразином и п-нитрофенилгидразином
Внимание! Арилгидразины канцерогенны, поэтому обращаться
с ними надо осторожно.
^С=о + H2NNHC6H5 —► /C=NNHC6H5 + Н2О
а) Смешивают 0,5 мл фенплгидразина пли 0,5 п-нитрофенплгпдразина с
5 мл воды и прибавляют по каплям уксусную кислоту до полного растворения
гидразина. После этого прибавляют 0,5 мл ацетона и встряхивают. Если осадок
не образуется, то осторожно нагревают несколько минут на небольшом пламени,
добавляют 2 мл воды и охлаждают.
б) Растворяют 0,5 мл ацетофенона в 2 мл этанола и добавляют по каплям
воду до появления легкой мути, исчезающей при встряхивании. Если воды при-
бавлено слишком много, то надо добавить немного спирта, пока раствор не ста-
нет прозрачным. К этому прозрачному раствору прибавляют 0,2 мл чистого фе-
нилгидразина или 0,2 г л-нитрофенилгидразина. Если спустя несколько минут
раствор остается прозрачным, то катализируют реакцию каплей уксусной кис-
лоты, осторожно нагревают несколько минут и охлаждают. Какие преимуще-
ства имеет эта методика по сравнению с описанной выше для ацетона?
Обсуждение. Хотя в приведенных выше условиях большая часть
альдегидов и кетонов образует фенилгидразоны, однако многие
из них оказываются маслообразными, что затрудняет визуальное
определение карбонильных соединений. я-Нитрофенилгидразин в
значительной степени свободен от этого недостатка, что позволяет
рекомендовать его для проведения такой пробы. Механизм этой ре-
акции, вероятно, сходен с механизмом образования семикарбазо-
нов, который обсуждался выше.
По крайней мере один источник трудностей, возникающих при
использовании фенплгидразина, состоит в том, что образующийся
вначале фенилгидразон не только может существовать в двух раз-
личных стереохимических формах, но способен также превращаться
в растворе в изомерное азосоединение. Наличие столь различных
форм весьма осложняет процесс разделения продуктов реакции.
Присутствие нитрогрупп в ароматическом ядре способствует ста-
билизации гидразонной формы, подавляя таутомерное превращение.
Опыт 9. Реакция образования продуктов присоединения с би-
сульфитом натрия
Выпадение осадка комплекса, возникающего при присоедине-
нии бисульфита, является указанием на присутствие различных
190
Глава в
карбонильных соединений:
ОН
RCHO + NaHSO3 —> R—С—SOJ Na+
I
Н
Ввиду того что эта реакция, по-видимому, очень сильно зависит
от стерического окружения карбонильной группы, такие компле-
ксы образуют не все кетоны (см. ниже).
Спиртовой раствор бисульфита натрия готовят, добавляя 3 мл этанола к
12 мл 40%-ного водного раствора соли. Некоторое количество соли осаждается
спиртом. Этот осадок необходимо удалить декантацией или фильтрованием,
после чего реактив готов для употребления.
В пробирку помещают 1 мл реактива, добавляют 0,3 мл бензальдегида, про-
бирку закрывают и сильно встряхивают. Проведите эту пробу с гептаналем и
ацетофеноном.
Обсуждение. Образование продуктов присоединения с бисуль-
фитом, представляющих собой, как было показано, а-алкансуль-
фонаты, является общей реакцией альдегидов. Подобным же
образом ведет себя большинство метилкетонов, циклических кето-
нов низкой молекулярной массы (до циклооктанона) и ряд дру-
гих соединений, содержащих очень активную карбонильную группу.
Однако некоторые метилкетоны образуют такие соединения очень
медленно или не образуют их вообще. Примером могут служить
арилметилкетоны, пинаколин и окись мезитила. С другой стороны,
коричный альдегид образует продукт присоединения, содержащий
две молекулы бисульфита.
Бисульфитный продукт присоединения находится в равнове-
сии с карбонильным соединением. Ввиду того что бисульфит на-
трия разрушается как кислотами, так и щелочами, такой продукт
присоединения стабилен только в нейтральных растворах. Сое-
динения, полученные из карбонильных соединений малой моле-
кулярной массы, растворимы в воде. Эти соединения очень по-
лезны, так как они представляют собой твердые вещества, легко
поддающиеся очистке. Ввиду того что продукты присоединения
бисульфита легко разлагаются под действием кислот или щело-
чей, из них легко регенерировать исходные карбонильные соеди-
нения в чистом виде.
Бисульфит натрия также легко присоединяет азотистые ана-
логи альдегидов — имины (или основания Шиффа). Образующие-
ся продукты идентичны тем соединениям, которые получаются при
действии первичного амина на бисульфитное производное соответ-
ствующего альдегида.
RCH=N—R'+NaHSOj R—CH—NUR' •
/ SO3Na
RCHOH+R’NHj
SOaNa
Определение функциональных ерупп и установление структуры
191
Связь углерод — сера в этом соединении реакционноспособна. По-
этому сульфонатная группа может замещаться при реакции с та-
кими анионами, как С№.
Контрольные вопросы. 1. Объясните, почему циклогексанон легко реагиру-
рует с бисульфитом натрия, а диэтилкетон не реагирует.
2. Как объяснить, почему пииаколин не реагирует с бисульфитом? Сравните
этот случай с реакцией ацетофенона.
3. Объясните поведение коричного альдегида.
4. Почему используют спиртовой раствор бисульфита? Попытайтесь про-
вести реакцию с ацетоном, используя водный раствор бисульфита.
Опыт 10. Йодоформная реакция
Реакцией на метилкетоны является образование йодоформа
(йодоформная реакция). Эта проба дает положительный резуль-
тат и с соединениями, которые могут окисляться до метилкето-
нов в условиях проведения реакции образования йодоформа.
О
II
RCHOHCHs + I2 + 2NaOH —> R—С—СН3 + 2NaI + 2Н2О
О
II
RCOCHs + 3I2 + 3NaOH —> R—С—CI3 + 3NaI + ЗН2О
|иа°н
RCOONa + СН13
Примените эту пробу к пропанолу-2, ацетону, этилацетату,
этилацетоацетату, ацетофенону и чистому метанолу. (Следует
учесть, что применение продажного метанола низкого качества
может ввести в заблуждение, так как он дает положительную
реакцию — образует йодоформ — вследствие наличия примесей.)
Помещают 4 капли исследуемой жидкости (0,1 г твердого вещества) в про-
бирку диаметром 1Б мм. Добавляют В мл диоксана* и встряхивают до полного
растворения образца. Затем добавляют 1 мл 10% -ного раствора гидроксида на-
трия и встряхивают, прибавляя раствор иода и иодида калия (см. ниже) до
тех пор, пока небольшой избыток реактива не даст хорошо заметной темной
окраски иода. Если обесцветилось меньше 2 мл раствора иода, то пробирку
надо нагреть на водяной баие до 60°С. Если при этом небольшой избыток иода
обесцветится, то добавление раствора иода и иодида калия продолжают, под-
держивая температуру диоксаиового раствора при 60°С и встряхивая пробирку
до тех пор, пока небольшой избыток иодного раствора вновь не окрасит смесь
в темный цвет. Добавление иода продолжают при 60°С таким же способом, пока
темная окраска не перестанет исчезать в течение 2 мин после прибавления оче-
редной порции раствора.
Избыток иода удаляют, приливая несколько капель 10%-ного раствора гид-
роксида натрия и встряхивая. Затем добавляют в пробирку воду и оставляют
иа 15 мин. Если образуется дурио пахнущий желтый осадок йодоформа, то проба
* Диоксан используют при проведении реакции с нерастворимыми в воде
соединениями; для растворимых в воде веществ вместо диоксаиа добавляют
2 мл воды.
192
Глава 6
считается положительной. Осадок отделяют фильтрованием, высушивают и оп-
ределяют температуру плавления. Йодоформ (CH3I) имеет характерный не-
приятный запах и плавится при 119—121°С с разложением. Если образовав-
шийся йодоформ имеет красноватую окраску, то его растворяют в 3—4 мл дио-
ксана, добавляют I мл 10%-ного раствора гидроксида натрия и встряхивают
до тех пор, пока раствор ие станет лимонно-желтого цвета. Затем смеси разбав-
ляют водой и фильтруют.
Реактив. Раствор иода и иодида калия готовят, добавляя 200 г иодида ка-
лия и 100 г иода к 800 мл дистиллированной воды. Смесь встряхивают до пол-
ного растворения добавленных веществ.
н2о
la + KI ---> Юз
Раствор имеет темно-коричневый цвет, обусловленный присутствием трииодид-
аниона (13).
Обсуждение. Положительную йодоформную реакцию дают со-
единения, содержащие группировки СН3СО—, СН21СО— или
СН12СО—, которые присоединены к атому водорода или к атому
углерода, не связанному с высокоактивными атомами водорода
или с группировками, создающими стерические препятствия. Эта
проба будет положительной, конечно, и для таких соединений,
которые при взаимодействии с реактивом дают продукты, содер-
жащие одну из перечисленных выше группировок. Напротив, со-
единения, которые содержат такие группировки, не будут давать
йодоформа, если они разрушаются в результате гидролитического
действия реагента до завершения иодирования.
Ниже приведены основные типы соединений, дающие поло-
жительную йодоформную реакцию:
СН3СНО СН3СН2ОН CH3COR CH3CH(OH)R
rcoch2cor rchch2chr
он он
где R— любой алкильный или арильный радикал (за исключе-
нием ди-орто-замещенных арильных радикалов). Однако очень
большие радикалы R стерически ингибируют эту реакцию. Опи-
санная проба оказывается отрицательной для соединений сле-
дующего типа:
CH3COCH2COOR CH3COCHaCN CH3COCH2NO2 и т. д.
В таких соединениях реагент отщепляет ацетильную группу и
превращает ее в уксусную кислоту, которая устойчива и в дан-
ных условиях не иодируется *.
Для того чтобы отличить метилкетоны от метилкарбинолов,
предложен модифицированный реактив **, представляющий собой
* Более детально эта реакция обсуждается в работе Fuson R. С., Bull В. А.,
Chem. Rev., 15, 275 (1934).
** Rothlin Е., Arch. Escuela Farm. Fac. ci. Med. Cdrdoba [R. A] Secc. ci.,
1039, № 10, p. 1 [C. A., 35, 5091 (1941)].
Определение функциональных групп и установление структуры
193
раствор 1 г цианида калия *, 4 г иода и 6 мл концентрирован-
ного раствора аммиака в 50 мл воды:
О
I 12
Снз-С-К KCN/N^ СН1з
•г
CH3CH(OH)R 1 л,» реакция ие идет
г\СГч/JN Г14U г!
Этот реактив образует йодоформ только с метилкетонами, с ме-
тилкарбинолами реакция не протекает.
Вторая стадия образования йодоформа — расщепление трига-
логенкетонов щелочью — сходна с реакцией, обратной конден-
сации Кляйзена. И в том и в другом случае реакция происходит
благодаря стабильности образующегося аниона:
О г ° -1
II I
RC—С13 + ОН’ —> R—С—СГ3
I
L ОН -1
О О
II II
RC—CH2CR + ОН’ —->
Г °’ О 1
I II
R—С—СН2—CR
I
L ОН -I
RC< + ’С13
ХОН
О
II
+ ’CH2CR
Вторичные спирты и кетоны типа
RCH2CH(OH)CH2R и RCH2COCH2R
не образуют йодоформа, хотя метиленовая группа, соседняя с
карбонильной группой, может галогенироваться. Иногда продаж-
ный диэтилкетон дает слабую положительную йодоформную ре-
акцию. Это обусловлено наличием в нем примесей, например пен-
танона-2. Положительную йодоформную реакцию дают бифункцио-
нальные спирты и кетоны следующих типов:
СН3СН(ОН)СН(ОН)СН3 СН3СОСН(ОН)СН3
СНзСОСОСНз СН3СОСН2СН2СОСН3
Р-Кетоэфиры не образуют йодоформа по этому методу, однако их
щелочные растворы реагируют с гипоиодитом натрия.
Ацетоуксусная кислота нестабильна, в кислых водных раст-
ворах она распадается, давая СО2 и ацетон:
н+
СН3СОСН2СООН ----> сн3сосн3 + со2
Образующийся ацетон дает положительную йодоформную реак-
цию. Такая реакция очень полезна в тех случаях, когда одним из
* Цианид калия чрезвычайно опасен; работать с ним можно только с раз-
решения преподавателя. Не смешивать с кислотой!
7 Зак. 336
194
Глава 6
возможных предполагаемых веществ является 0-кетоэфир. Это
связано с тем, что такие эфиры гидролизуются при кипячении с
5%-ной серной кислотой (кислотно-катализируемая реакция рас-
пада, обратная конденсации)
н+
RCOCH2COOR' + Н2О ------> RCOCH3 + R'OH + СО2
Техника выполнения опыта. В колбу для перегонки емкостью 100 мл поме-
щают 0,5 г образца p-кетоэфира, добавляют 50 мл 5%-ной серной кислоты, кла-
дут несколько кусочков пористого фарфора или пемзы («кипелок») и смесь осто-
рожно нагревают до кипения. Собирают 6—8 мл дистиллата и делят его на три
части. Одну часть обрабатывают гипоиоднтом натрия (опыт 10), вторую — 2,4-
динитрофенилгидразнном (опыт 6), а третью проверяют на присутствие спирта
(R'OH) действием аммонийгексапитратоцератного реагента (опыт 2).
Легко гидролизующиеся этиловые или изопропиловые эфиры
дают положительную йодоформную пробу.
Часто в тех случаях, когда химические и спектральные данные
указывают на наличие в изучаемом веществе карбонильной груп-
пы, химик должен установить, является ли это вещество альдеги-
дом или кетоном. Поэтому далее описаны некоторые пробы, даю-
щие положительный результат с альдегидами и отрицательный
— с кетонами.
Простой химической пробой на альдегиды является реакция
с триоксидом хрома. Эта реакция, описанная применительно к
спиртам в опыте 3 (разд. 6.1), может быть использована в слу-
чае альдегидов, окисляющихся достаточно легко:
3RCHO 4-2СгО3 4-3H2SO4 —> 3RCOOH 4-ЗН2О 4-2Cr2(SO4)3
Кетоны в этих условиях обычно не реагируют. Не окисляются они
и перманганатом калия (разд. 6.4), поскольку в них отсутствуют
связи С — Н, соединенные непосредственно с карбонильной груп-
пой.
Опыт 11. Реакция с реактивом Толленса
RCHO 4- 2Ag(NH3)2OH —> 2Ag (осадок) 4- RCOONH4 4- Н2О 4- 3NH3
Приготовьте реактив Толленса, как описано ниже, и проведите
реакцию с формалином, ацетоном, бензальдегидом, глюкозой,
гидрохиноном и n-аминофенолом. Опыты следует проводить, при-
бавляя лишь небольшое количество реактива. Если реакция не
идет на Холоду, то раствор слегка нагревают.
Реактив. В оченц чистую пробирку помещают 2 мл 5 %-него раствора нит-
трата серебра и добавляют каплю разбавленного (10%-иого) раствора гидро-
ксида натрия. По каплям прибавляют разбавленный раствор аммиака (прибли-
зительно 2%-ный). Раствор непрерывно встряхивают до полного растворения
осадка оксида серебра. Для того чтобы реагент был более чувствителен, необ-
ходимо избегать большого избытка аммиака.
Этот реактив должен быть приготовлен непосредственно перед употребле-
нием. Его не следует хранить, так как при стоянии раствор в результате разло-
жения образует весьма взрывчатый осадок.
Определение функциональных групп и установление структуры
195
Контрольный вопрос. 1. Будет ли мешать проведению этой реакции наличие
в молекуле активного атома галогена?
Обсуждение. Следует заметить, что ацилоины, дифениламин и
другие ароматические амины, а также а-нафтол и некоторые дру-
гие фенолы также дают положительную реакцию с реактивом
Толленса. а-Алкокси- и а-диалкиламинокетоны также восстанав-
ливают аммиачный раствор нитрата серебра. Кроме того, поло-
жительную реакцию дает стабильный гидрат трифторацетальде-
гида.
Часто в результате этой реакции на внутренних стенках про-
бирки осаждается гладкая пленка металлического серебра, обла-
дающая зеркальным блеском. Потому эту реакцию часто назы-
вают реакцией «серебряного зеркала». Однако в некоторых
случаях, особенно если стеклянные стенки пробирки недостаточно
чисты, серебро выделяется только в виде комковатого осадка
серого или черного цвета.
Реакция является автокаталитической: она ускоряется обра-
зующимся металлическим серебром и часто имеет индукционный
период, длящийся несколько минут.
Опыт 12. Реакция с фуксинсернистой кислотой
К 2 мл функспнсернистой кислоты в пробирке добавляют 1 каплю масля-
ного альдегида, осторожно встряхивают пробирку и наблюдают за изменением
окраски реактива в течение 3—4 мин. Повторите эту реакцию с бензальдегидом,
раствором формальдегида, ацетофеноном и ацетоном.
Для успешного проведения этой реакции реактив нельзя нагревать, а ис-
пытуемый раствор не должен быть щелочным. В тех случаях, когда эту пробу
проводят с неизвестным веществом, предварительно нужно в тех же условиях
провести для сравнения реакцию с каким-либо известным альдегидом.
Реактив. Растворяют 0,5 г чистого фуксина (гидрохлорида поро-розанилина)
в 500 мл дистиллированной воды; полученный раствор фильтруют. Насыщают
500 мл дистиллированной воды сернистым газом и смешивают ее с профильтро-
ванным раствором фуксина. Смесь оставляют стоять в течение ночи. Эта прак-
тически бесцветная смесь представляет собой очень чувствительный реактив на
альдегиды.
Обсуждение. Фуксин — розовый трифенилметановый краситель,
дающий с сернистой кислотой бесцветную лейкосульфоновую кис-
лоту. По-видимому, при этом происходит 1,6-присоединение сер-
нистой кислоты к хиноидному ядру красителя.
розовый раствор
бесцветное основание Шиффа
7*
196
глава 6
Эта лейкосульфоновая кислота нестабильна и при взаимодействии
с альдегидом теряет сернистую кислоту, образуя пурпурно-фиоле-
товый краситель:
пурпурно-фиолетовый раствор
Важно отметить, что цвет этого красителя отличается от цвета
исходного фуксина. Вместо светло-розовой окраски он имеет тем-
но-голубой цвет, граничащий с фиолетовым или пурпурным. Не-
которые кетоны и непредельные соединения реагируют с сернистой
кислотой, восстанавливая розовую окраску, характерную для ис-
ходного фуксина. Поэтому появление светло-розового окрашива-
ния не следует рассматривать как положительную пробу на аль-
дегиды. Тот факт, что некоторые соединения вызывают появле-
ние розового окрашивания, свойственного фуксину, может быть
положен в основу соответствующей качественной реакции. Так,
имеются сведения о том, что в присутствии специально приготов-
ленного реактива при проведении реакции в течение 1 ч альдозы
дают розовое окрашивание, а кетозы и дисахариды (кроме маль-
тозы) его не дают. Эту модификацию реакции Шиффа следует
использовать с осторожностью, так как при встряхивании на воз-
духе с этим реактивом многие органические соединения дают ро-
зовое окрашивание. Другие вещества, например а,|3-непредельные
кетоны, взаимодействуют с сернистой кислотой и тем самым сдви-
гают влево равновесие первой из приведенных выше реакций.
Опыт 13. Реакция с раствором Бенедикта
Для соединений, не содержащих серу:
2Си2+ +
цитратный
комплекс
RCHO —> RCOOH+Cu2O
RCHOH —> R—CO + Cu2O
I I
RCO R—CO
ArNHNHAr —► ArN=NAr + Cu2O
ArNHNH2 —» ArH + ArOH + N2 + Cu2O
ArCHO —> реакция не идет
К раствору или суспензии 0,2 г изучаемого соединения в 5 мл воды добав-
ляют 5 мл раствора Бенедикта и замечают цвет осадка (желтый пли желто-зе-
леный). Затем нагревают смесь до кипения и отмечают, образуется ли осадок, и
если он образуется, то какого он цвета. Проведите эту реакцию с н-масляным
альдегидом, ацетоином, бензоином, глюкозой, сахарозой, глицерином, бутано-
лом-2, ацетоном и фенилгидразином.
К раствору 0,2 г сахарозы в 5 мл воды прибавляют 2 капли концентрирован-
ной соляной кислоты. Раствор кипятят 1 мин, затем его охлаждают, нейтрали-
зуют кислоту разбавленным раствором гидроксида натрия и действуют реакти-
вом Бенедикта. Объясните полученные результаты.
Определение функциональных групп и установление структуры
197
Реактив Бенедикта получают путем растворения в дистиллированной воде
17,3 г гидратированного сульфата меди (кристаллического), 173 г цитрата на-
трия и 100 г карбоната натрия (безводного). Цитрат и карбонат растворяют
при нагревании в 800 мл воды. После этого доводят общий объем раствора до
850 мл. Сульфат меди растворяют в 100 мл воды. Полученный раствор мед-
ленно при перемешивании приливают к раствору цитрата и карбоната. Добавляя
воду, доводят общий объем смеси до 1 л.
Обсуждение. Реактив Бенедикта содержит медь в форме комп-
лексного аниона и действует как селективный окисляющий агент.
Он был предложен для восстановления сахаров вместо сильноще-
лочной фелинговой жидкости. С помощью этого реагента можно
определять 0,01 % глюкозы в воде. Цвет осадка должен быть крас-
ным, желтым или желто-зеленым в зависимости от природы и ко-
личества присутствующего восстановителя.
Реактив Бенедикта восстанавливает а-оксиальдегиды, а-окси-
кетоны и а-кетоальдегиды. Простые ароматические альдегиды
этим реактивом не окисляются так же, как и молекулы, содержа-
щие только спиртовую группу (первичные, вторичные или третич-
ные спирты или гликоли) или только кетогруппу.
Этот реактив окисляет также производные гидразина, напри-
мер фенилгидразин и гидразобензол. Реактив Бенедикта восста-
навливается также и другими соединениями, способными к легко-
му окислению, например, такими, как фенилгидроксиламин, ами-
нофенолы и подобные им реактивы, применяемые в качестве фо-
тографических проявителей.
Меркаптаны (тиолы) мешают проведению реакции с реакти-
вом Бенедикта. Применение этой пробы для определения —SH-
групп описано ниже (опыт 296).
В книге Черониса, Энтрикина и Ходнетта приведены дополни-
тельные пробы на карбонильные соединения с 3,5-динитробензой-
ной кислотой (с. 393), N-оксибензолсульфонамидом * (с. 394) и
2-гидразинобензотиазолом (с. 396).
Инфракрасные спектры простых альдегидов приведены на
рис. 6.3, Л. Для группы —СН=О характерны сильная полоса по-
глощения при 1725 см-1 (5,80 мкм), соответствующая группе
С=О, и две полосы при 2800 и 2700 см-1 (3,57 и 3,70 мкм), обу-
словленные связями С—Н. Спектр, приведенный на рис. 6.3, А, сле-
дует сравнить со спектром на рис. 6.3, Б, где полоса при 1660 см-1
(6,02 мкм) относится к дважды сопряженной карбонильной группе.
Такое уменьшение частоты (увеличение длины волны) является
типичным результатом сопряжения карбонильной группы с л-элек-
тронной системой.
На рис. 6.3, В представлен ИК-спектр простого кетона. Сопря-
жение кетонной карбонильной группы с л-электронной системой
* Этот реактив называют также бензолсульфошидроксамовой кислотой, или
кислотой Пилоти.
198
Глава 6
Рис. 6.3. Инфракрасный спектр дифенилацетальдегида (Л), бензофенона (5) и
ацетона (В).
д— раствор в четыреххлористом углероде в парных кюветах. Валентные колебания С—Н:
/ — 3050 см”1 (3,28 мкм), V_ ароматические; 2—2890 см-1 (3.57 мкм), 2700 см”1 (3,70 мкм).
с—н
VC Н’ альдегиднЬ1Й дублет за счет резонанса Фермн с обертоном полосы 1380 см”1. Валент-
ные колебания С=О: 3—1725 см""1 (5,80 мкм), Валентные колебания О=С: 4— 1690 см”1
(6,25 мкм), 1490 см”1 (7,15 мкм), 1450 см”1 (6,90 мкм). Деформационные колебания
(С—О)—Н: 5 —1380 см”1 (7,25 мкм), Деформационные колебания С—С неплоские:
6—700 см”1 (14,3 мкм), неплоские деформационные колебания С«=С. Ь —2%-ный раствор
в четыреххлористом углероде. Валентные колебания С—Н: 7—3050 см”1 (3.28 мкм),
Валентные колебания С=«О: 8 — 1660см”1 (6,02 мкм), Валентные колебания О*С:
р— 1600 см”1 (6,25 мкм), 1450 см”1 (6,90 мкм), vc с- Валентные колебания С—(С—О)—С:
10—1275 см”1 (7,85 мкм). Деформационные колебания С=*=С: // — 700 см”1 (14.3 мкм). В — аце-
20
тон в чистом виде CsHgO, мол. масса 58,08, т. кип. 56,5 °C, т. пл. -94° С, Пр 1,3591, получен
от фирмы United States Testing Со., Inc., капиллярная кювета, измерено иа приборе Perkin-
Elmer 521 (© Sadtles Research Laboratories, Inc., Philadelphia, Pa., 1966). Валентные колеба-
НИЯ С—H: 72 — 3005 см”1 (3,33 мкм), 2960 см-1 (3,38 мкм), 2920 см-1 (3.42 мкм), V , vCH3
асимм снмм»
обертон соответственно. Валентные колебания С=О: /3—1715 см”1 (5,83 мкм), V
Деформационные колебания С—Н: 14—1420 см”1 (7,04 мкм), 6аснммСН3-группы СН3СО;
/5—1363 см”1 (7,33 мкм), 6симмСН3-группы СН3СО; 16—1220 см”1 (8,20 мкм), валентные н
деформационные колебания С—С(=О)—Q.
Определение функциональных групп и установление структуры
199
% 'anHOHsRuodu
202
Глава 6
приводит к такому же сдвигу положения карбонильной полосы,
как и в рассмотренном выше случае (рис, 6.3,Л и Б).
Поглощение циклических кетонов в значительной степени за-
висит от величины цикла. В малых циклах карбонильная группа
более напряжена и поэтому поглощает при больших частотах:
циклогексанон при 1715 см-1 (5,83 мкм), циклопентанон при
1740 см~* (5,75 мкм), а циклобутанон при 1780 см-1 (5,62 мкм).
На рис. 6.4, А представлен ПМР-спектр простого альдегида.
Интересно сравнить положение сильно дезэкранированного про-
тона альдегидной группы в области низких частот на рис. 6.4, А
с сигналами ПМР-спектра простого кетона в области высоких
частот на рис. 6.4, Б. Кроме того, следует обратить внимание на
малую величину взаимодействия (/ = 2—3 Гц, не разрешенное
на рис. 6.4, А) между вицинальными протонами в том случае, ко-
гда один из них связан с s/Агибридизованным атомом, а другой —
С зр3-гибридизованным атомом углерода. Такая малая величина
вицинального взаимодействия резко отличается от обычно на-
блюдающейся величины, которая характерна для вицинальных
протонов, находящихся у двух s/Агибридизованных атомов угле-
рода, и приблизительно равна 7 Гц.
При распаде альдегидов и кетонов в масс-спектрометре часто
образуются следующие фрагменты, полезные для идентификации:
Фрагмент Масса Источник
RCO+ АгСО+ /ОН R + 28 Аг+ 28 Расщепление связей R—COR' или R—СНО Расщепление связей Аг—COR' или Аг—СНО
R-C'- ^CRiRj 41 R + Ri Rg Расщепление Мак-Лафферти
(СНО)+ 29 Расщепление связи R—СНО (толь- ко для альдегидов)
За исключением фрагментов, образующихся при расщеплении
Мак-Лафферти, все остальные фрагменты образуются при раз-
рыве связей, соединяющих их с карбонильной группой;
При отрыве фрагмента Gi образуется заряженный фрагмент
G2CO+. Способность различных групп стабилизировать заряд и
вследствие этого давать большой пик фрагментного иона возра-
стает в ряду Аг > R > Н. Так, ацетофенон дает очень большой
Определение функциональных групп и установление структуры
203
пик с т/е =105 (С6Н5СО+) и очень маленький пик с mfe — 43,
соответствующий СН3СО+. Расщепление по Мак-Лафферти вклю-
чает перегруппировку и перенос протона, который должен для
этого находиться в у-положении.
Действительно, те системы, которые содержат карбонильную груп-
пу и водород в у-положении и обладают достаточной гибкостью
для образования шестичленного цикла, могут претерпевать пере-
группировку Мак-Лафферти.
Хотя пик, соответствующий СНО+(/н/е = 29), обычно мал, он
оказывается очень полезным для идентификации альдегида.
Очень часто для характеристики карбонильного соединения
полезно использовать ультрафиолетовую (УФ) спектрометрию.
Информация, полученная из УФ-спектров, оказывается полезной
при изучении дисперсии оптического вращения, при выборе длин
волн для фотохимических превращений карбонильного соединения
и для установления структурных деталей а,р-непредельных кар-
бонильных соединений (правило Вудворда — Физера). Особенно
полезны УФ-спектры в тех случаях, когда карбонильная группа
сопряжена с двойной связью или с ароматическим кольцом. Об-
суждение этих вопросов можно найти в гл. 8.
Производные альдегидов обычно получают, превращая кар-
бонильную группу в какую-либо другую функцию, например
/C=N—NHR, или окисляя СНО-группу до карбоксильной. По-
добным же образом кетоны можно превращать в замещенные гид-
разоны или оксимы. Альдегиды и кетоны можно восстанавливать
до первичных и вторичных спиртов соответственно, которые затем
можно идентифицировать, получая производные, как описано в
разд. 6.1.
Методика 6. Окисление альдегидов до соответствующих кислот
a) RCHO КМПО4 н+
- • > ксион
ОН"/Н2О
б) RCHO Н2О2 RCOO‘Na+ н+ -=-* RCOOH
он~/н2о
AgNO3 HNO3
в) RCHO — . - > NaOH [RCOO'Na*] > RCOOH
204
Глава 6
а) Перманганатный метод. К раствору или суспензии 1 г альдегида в 10—
20 мл воды добавляют несколько капель 10%-ного раствора гидроксида натрия,
затем при энергичном встряхивании прибавляют такое количество насыщенного
раствора перманганата калия, чтобы появилась пурпурная окраска. Смесь под-
кисляют разбавленной серной кислотой и добавляют раствор бисульфита натрия
до тех пор, пока весь перманганат калия и диоксид марганца не превратятся
в сульфат марганца, о чем можно судить по исчезновению пурпурного окра-
шивания. Образовавшуюся кислоту отделяют фильтрованием и перекристаллизо-
вывают из смеси ацетона и воды. Если кислота не выделяется, ее можно экс-
трагировать хлороформом или эфиром.
б) Окисление пероксидом водорода. В колбу емкостью 500 мл помещают
20 мл 5%-ного раствора гидроксида натрия и 30 мл 3%-ного раствора перо-
ксида водорода. Смесь нагревают до 65—70°С и прибавляют 1 г альдегида.
Раствор встряхивают и нагревают при 65°С 15 мин. Если альдегид ие раство-
ряется, то добавляют несколько миллилитров этанола. Для окончания реакции
добавляют еще 10 мл пероксида водорода и дополнительно нагревают 10 мин.
Раствор подкисляют до кислой реакции по конго красному. Образовавшуюся
кислоту отделяют фильтрованием.
Если полученная кислота растворима в воде, то лучше всего нейтрализо-
вать раствор по фенолфталеину и упарить его досуха. Полученную таким обра-
зом натриевую соль превращают в подходящее производное по методикам 33,
34 или 39.
в) Окисление одновалентным серебром *. В стакане емкостью 50 мл раст-
воряют 3,4 г нитрата серебра в 10 мл дистиллированной воды. При сильном
перемешивании добавляют по каплям 10%-ный раствор гидроксида натрия до тех
пор, пока не прекратится выделение осадка оксида серебра (примерно 4 мл).
Продолжая перемешивание, добавляют 1 г альдегида и дополнительно 5 мл
10%-него раствора гидроксида натрия. В результате окисления реакционная
смесь обычно разогревается и оксид серебра превращается в металлическое се-
ребро. После перемешивания в течение 10—15 мии выпавший осадок серебра и
ие вошедший в реакцию оксид серебра отфильтровывают. Фильтрат подкисляют
разбавленной азотной кислотой (20% -ной) и охлаждают раствор на ледяной
бане. Выделившуюся твердую кислоту собирают на фильтре, высушивают и оп-
ределяют температуру плавления. В случае необходимости ее можно перекри-
сталлизовать из небольшого количества горячей воды или из смеси равных объ-
емов воды и пропаиола-2. Если кислота настолько растворима в воде, что не
выпадает при подкислении, то ее можно выделить, упаривая водный слой и экс-
трагируя его хлороформом или эфиром.
Методика 7. Получение семикарбазонов
—А=о + H2NNHCONH2 —> —c=nnhconh2 + н2о
а) Для водорастворимых соединений. В пробирке растворяют 1 мл альдегида
или кетона, 1 г гидрохлорида семикарбазида и 1,5 г ацетата натрия в 10 мл
воды. Смесь сильно встряхивают, пробирку ставят в стакан с кипящей водой и
дают остыть. Затем пробирку помещают в стакан со льдом и трут о стенки про-
бирки стеклянной палочкой. Кристаллы семикарбазона отделяют фильтрованием
и перекристаллизовывают из воды или 25—50-ного водного раствора этанола.
б) Для нерастворимых в воде соединений. В 10 мл этанола растворяют 1 мл
альдегида или кетона и прибавляют воду до получения раствора. Муть устра-
* Thomason S. С., Kubler D. G., J. Chem. Educ., 45, 546 (1968). Эти авторы
описывают применение оксидов одновалентного и двухвалентного серебра.
Определение функциональных групп и установление структуры 205
ияют, добавляя несколько капель спирта. После этого прибавляют 1 г гидро-
хлорида семикарбазида и 1,5 г ацетата натрия. Далее проводят опыт, как опи-
сано в пункте «а».
Методика 8. Получение п-нитрофенилгидразонов
—C=O+ArNHNH2 —> —C=NNHAr + H2O
Смесь 0,5 г п-нитрофенилгидразина, 0,5 г альдегида или кетона и 10—15 мл
этанола нагревают до кипения и прибавляют 1 каплю ледяной уксусной кис-
лоты. В течение нескольких минут смеси не дают остывать, а затем, если это
необходимо, добавляют спирт до тех пор, пока раствор не станет прозрачным.
Смесь охлаждают и выделившийся п-нитрофенилгидразон собирают па фильтре.
Полученный продукт может быть перекристаллизован из небольшого количе-
ства этанола. Если производное не выделяется из раствора при охлаждении,
то смесь нагревают до кипения, прибавляют воду до помутнения раствора и
затем добавляют 1—2 капли спирта, чтобы раствор вновь стал прозрачным.
Выпавший при охлаждении гидразон перекристаллизовывают из водного эта-
нола.
Методика 9. Получение 2,4-динитрофеиилгидразонов
—С=О + ArNHNH2 —> —C=NNHAr + Н2О
Для приготовления раствора 2,4-динитрофенилгидразина в конической колбе
емкостью 25 мл смешивают 0,4 г 2,4-динитрофенилгидразина и 2 мл концентри-
рованной серной кислоты. Затем по каплям прибавляют около 3 мл воды, вра-
щая или встряхивая колбу, пока не закончится растворение. К еще теплому
раствору приливают 10 мл 95%-ного этанола.
Раствор карбонильного соединения в спирте готовят, растворяя 0,5 г ве-
щества в 20 мл 95%-ного этанола. К этому раствору прибавляют свежеприго-
товленный раствор 2,4-динитрофенилгидразина и смесь оставляют стоять при
комнатной температуре. Обычно уже через 5—10 мин начинают выпадать кри-
сталлы 2,4-динитрофеиилгидразона. Если осадок не образуется, то смесь остав-
ляют стоять на ночь.
Перекристаллизацию проводят чаще всего следующим образом. Смесь 2,4-
динитрофеиилгидразона с 30 мл 95%-ного этанола нагревают на кипящей во-
дяной бане. Если растворение произойдет сразу, то постепенно прибавляют воду,
пока не появится муть или пока не будет добавлено 5 мл воды (не больше).
Если динитрофенилгидразон не растворяется, то к горячей смеси медленно до-
бавляют этилацетат до тех пор, пока не будет достигнуто полное растворение.
Горячий раствор фильтруют через складчатый фильтр и оставляют стоять при
комнатной температуре до тех пор, пока кристаллизация не закончится (около
12 ч).
Реакцию в диметиловом эфире диэтиленгликоля (диглиме) проводят с рас-
твором 4 г 2,4-динитрофенилгидразина в 120 мл диглима при нагревании. Рас-
твор оставляют при комнатной температуре на несколько дней. К 5 мл этого
раствора прибавляют при комнатной температуре 0,1 г карбонильного соедине-
ния в 1 мл 95%-иого этанола или в 1 мл диметилового эфира диэтиленгликоля.
После этого смесь подкисляют тремя каплями концентрированной соляной кис-
лоты. Если гидразон не выпадает немедленно, то разбавляют раствор водой и
оставляют стоять.
206
Глава 6
Методика 10. Получение производных димедона и октагидроксантенов
метон (или
димедон)
производное метона
или бис-метон
а) Продукты конденсации альдегидов с двумя молекулами димедона. В слу-
чае алифатических альдегидов на 0,1 г альдегида в 50%-ном этаноле берут 0,4 г
димедона *, если же альдегид ароматический, то достаточно 0,3 г димедона. При-
бавляют одну каплю пиперидина и смесь осторожно кипятят в течение 5 мин.
Если по истечении этого времени горячий раствор остается прозрачным, то при-
бавляют по каплям воду до помутнения раствора. Затем смесь быстро охлаж-
дают, выпавшее производное отделяют фильтрованием, промывают его 2 мл
холодного 50 %-ного этанола и перекристаллизовывают из смеси метанола и
воды.
6) Циклизация с образованием замещенных октагидроксантенов. Если жела-
тельно получить другое димедоновое производное, кроме описанного выше, то
полученный по способу «а» продукт можно превратить в замещенный октагид-
роксантен, для чего 0,1 г его кипятят в 3—6 мл 80%-ного этанола, к которому
добавлена одна капля концентрированной соляной кислоты. Циклизация закан-
чивается за 5 мин, после чего к раствору добавляют воду до помутнения. Смесь
охлаждают и выпадающий замещенный ксантен отфильтровывают. Обычно он
представляет собой достаточно чистый продукт, однако при необходимости его
можно перекристаллизовать из водного метанола.
Кетоны. Реагенты на карбонильную группу, описанные выше,
могут быть использованы также для получения твердых производ-
ных кетонов. Кетоны низкой молекулярной массы можно охарак-
теризовать путем получения 2,4-динитрофенилгидразонов (мето-
дика 9), п-ннтрофенилгидразонов (методика 8) или семикарбазо-
нов (методика 7). Для кетонов высокой молекулярной массы
больше подходит получение гидразонов, фенилгидразонов
(опыт 8) и оксимов (методика 11).
Метилкетоны можно селективно окислять гипохлоритом нат-
рия **. Этот реагент особенно удобен для окисления непредельных
кетонов, так как обычные окислители могут затрагивать двойную
связь.
RCH==CHCOCH3 + 3NaOCl —> RCH=CHCOONa + СНС13 + 2NaOH
* Название «димедон» — сокращение от диметилцнклогександион.
** Van Arendonk А. М., Cupery М. Е., J. Am. Chem. Soc., 53, 3184 (1931);
Hurd C. D., Thomas C. L., ibid., 55, 1646 (1933).
Определение функциональных групп и установление структуры
207
В качестве производных, особенно для кетоэфпров *, можно
рекомендовать гидантоины, образующиеся при нагревании карбо-
нильного соединения со смесью цианида калия или натрия и кар-
боната аммония.
rcor<nSoTr1
Методика 11. Получение
:с=о
оксимов
NH2OH-HC1
---------->
основание
C=NOH + Н2О + соль
а) Пиридиновый метод. Смесь 1 г альдегида или кетона, 1 г гидрохлорида
гидроксиламина, 5 мл пиридина и 5 мл абсолютного этанола нагревают 2 ч на
кипящей водяной бане с обратным холодильником. Растворители удаляют вы-
париванием в токе воздуха под тягой. Остаток тщательно растирают с 5 .мл хо-
лодной воды и фильтруют. Оксим перекристаллизовывают из метанола, этанола
или из смеси этанола и воды.
б) Около 0,5 г гидрохлорида гидроксиламина растворяют в 3 мл воды, к
раствору прибавляют 2 мл 10%-ного раствора гидроксида натрия и 0,2 г аль-
дегида или кетона. Если карбонильное соединение нерастворимо в веде, то в
смесь добавляют такое количество этанола, чтобы раствор стал прозрачным.
Смесь нагревают 10 мин на кипящей водяной бане и затем охлаждают льдом.
Чтобы ускорить кристаллизацию, трут стенки колбы стеклянной палочкой. Ино-
гда выделению оксима способствует прибавление нескольких миллилитров ди-
стиллированной воды. Продукт может быть перекристаллизован из воды или из
разбавленного этанола.
Некоторые циклические кетоны, например камфора, требуют применения из-
бытка щелочи и более длительного нагревания. Если кетон не образует оксима
ни по одному из описанных выше способов, то 1 г этого кетоиа следует обра-
ботать 1 г гидрохлорида гидроксиламина, 4 г гидроксида калия и 20 мл 95% -
иого этанола. Смесь кипятят с обратным холодильником 2 ч и выливают в
150 мл воды. Суспензию перемешивают и оставляют стоять до выделения ие
вошедшего в реакцию исходного кетона. Раствор фильтруют, подкисляют соля-
ной кислотой и оставляют стоять до кристаллизации оксима. Продукт перекри-
сталлизовывают из этанола или из смеси этанола с водой.
Диарилкетоны восстанавливаются боргидридом натрия до ди-
арилкарбинолов. Эта реакция проходит в существенно более мяг-
ких условиях, чем восстановление алюмогидридом лития или на-
трием в спирте.
Методика На. Восстановление диарилкетонов до диарилкарбинолов
с помощью боргидрида натрия
NaBH4 NaOH
4Ar’CO NaOH/tCHahCHOH^ I^CHO^B4Ar2CHOH
В колбу емкостью 50 мл помещают суспензию 0,4 г боргидрида натрия в
15 мл изопропилового спирта и прибавляют 3 г диарилкетоиа. Компоненты тща-
тельно смешивают, в колбу вносят несколько кусочков пористого фарфора или
Henze Н. R., Speer R. J., J. Am. Chem. Soc., 64, 522 (1942).
208
Глава 6
пемзы и закрывают ее обратным холодильником. Смесь кипятят 30 мин на ки-
пящей ёодййой бане, охлаждают дб комнатной температуры н при сильном Пе-
ремешивании добавляют 20 мл 10%-кого раствора гидроксида натрия. Раствор
охлаждают до комнатной температуры и экстрагируют тремя порциями метй-
леихлорида. Экстракт сушат, добавив 1 г безводного сульфата магния, мети-
ленхлорид отгоняют на водяной бане. Остаток перекристаллизовывают из смеСи
метаиоАа или пропанола-2 с водой. Кристаллы собирают иа фильтре, Йяфйй-
вают й определяют температуру плавления полученного продукта.
Боргидрид натрия в изопропиловом спирте селективно вос-
станавливает карбонильную группу в альдегидах и кетонах. Этот
метод удобен для таких кетонов, которые дают при восстановле-
нии твердые спирты. Альдегиды проще идентифицировать, полу-
чая их 2,4-динитрофенилгидразоны.
Ввиду того что по этой методике двойные углерод-углеродные
связи, даже сопряженные, не восстанавливаются, ее можно ис-
пользовать в сложных случаях:
NaBH4
с6н6сн=сн—сно faai^ko* свН5сн=сн—сн2он
NaBH4
С6Н5СН=СН—COR и # g6h5ch=.chchr
NaOH-f-HsO ।
он
Нитро- и циангруппы, амиды и сложные эфиры в этих условиях
не восстанавливаются. Следует, однако, помнить, что амиды и
сложные эфиры при кипячении со спиртовым раствором гидро-
ксида натрия гидролизуются. Поэтому щелочной раствор необ-
ходимо подкислять, прежде чем проводить экстракцию метилен-
хлоридо^г. Во многих случаях оксикислота (или соответствующий
лактон *) выпадает в виде осадка и может быть отфильтрована.
NaBH4 НС1
6вН«СОС6Н4СООСН3СвН5СН—C6H4COONa -------------
JNaOH, Hgo ।
он
—> CeH5CHC6H4COOH
I
он
Литература: Brown Н. С., Boranes in Organic Chemistry, Cornell
University Press, Ithaca, N. Y., 1972, Chaps. XII, XIII.
Ряд методов идентификации простых карбонильных соедине-
ний приведен в книге Пасто и Джонсона, с. 393. Для получения
водорастворимых производных карбонильных соединений можно
использовать следующие методы (метод с использованием соли
* о
С6Н5
Определение функциональных групп и установление структуры
209
п-толуолсульфокислоты см. Allen С. F. Н., Gates J. W., lr., J. Org.
Chem., 6, 596 (1941):
V-NNHCoftcH,), О
реактив Жирара Т s -
<^N^-CH2C0NHNH,Cr \
Cl-
pediitnue Жерара Р
сн3
соль п-тслуолсумфокислоты /
Большую часть реакций, описанных выше, можно провести и в
обратном направлении.
6.2.1. Полифункциональные карбонильные соединения *
Методики, приведенные выше, а также те, которые будут рас-
смотрены в последующих разделах, пригодны для характеристи-
ки сахаров и других групп углеводов. С этой целью часто при-
меняют реакцию с иодной кислотой (опыт 5), которая дает хо-
рошие результаты не только с соединениями, содержащими ви-
цинальные карбонильные группы (например, с а-дикетонами), но
с равным успехом реагирует с соединениями, содержащими ви-
цинальные гидроксильные группы.
6.2.2. Ацетали и кетали
Ацетали можно идентифицировать на основе сведений о том,
какой спирт и какой альдегид или кетон образуются при их гид-
ролизе. Гидролиз проводят нагреванием ацеталей с разбавленны-
ми кислотами. При этом происходят следующие реакции:
ZOR' R4
X/ +Н2о 2R'OH+ V=O
(R)HZ X)R' (R)H/
Низкомолекулярные ацетали легко гидролизуются при их ки-
пячении с обратным холодильником в присутствии 1—2%-ной со-
ляной кислоты в течение 3—5 мин. Ацетали с более высокой мо-
лекулярной массой требуют более продолжительного нагревания
(30—60 мин). Гидролиз ацеталей, образованных из нерастворимых
в воде спиртов и альдегидов или кетонов, протекает легче при
использовании в качестве растворителя 50%-ного диоксана.
Выбор тех или иных из приведенных ниже более подробных
методик зависит от характера получающихся При гидролизе про-
* Особую группу этого класса соединений составляют углеводы, подробно
рассматриваемые в разд. 6.11.
210
Глава 6
дуктов. Если и спирт, и карбонильное соединение растворимы в
воде, то смесь после гидролиза нейтрализуют по лакмусу и делят
на две части. Одну часть используют для характеристики альде-
гида или кетона путем получения подходящего производного. Для
этого пригодны семикарбазоны (методика 7), фенилгидразоны
(опыт 8) и n-нитрофенилгидразоны (методика 8). Другую часть
обрабатывают бензоилхлоридом по методу Шоттена — Баумана
(методика 18). При этом спирт превращается в соответствующий
алкилбензоат, который выделяют и далее гидролизуют по мето-
дике 25а. Полученный спирт идентифицируют одним из методов,
описанных в разд. 6.1.
Если какой-то из продуктов гидролиза нерастворим в воде, то
сначала выделяют и идентифицируют именно его, а продукт, остав-
шийся в водном растворе, характеризуют одним из описанных
выше методов.
Если же и спирт, и карбонильное соединение нерастворимы
в воде, то вначале их отделяют от водного слоя. В том случае,
когда один из продуктов представляет собой альдегид или алифа-
тический метилкетон, его можно отделить в форме бисульфитного
производного (опыт 9). Иногда для разделения спирта и карбо-
нильного соединения используют фракционную разгонку.
Ацетали и кетали могут содержать представляющие опас-
ность пероксиды, подобные тем, которые часто содержатся в про-
стых эфирах. Необходимо ознакомиться с обсуждением вопроса
о таких пероксидах в разд. 6.14.
Пасто и Джонсон (с. 378) рекомендуют получать 2,4-динитро-
фенилгидразоны альдегидов и кетонов непосредственно из ацета-
лей или кеталей при действии на них кислого раствора соответ-
ствующего гидразина (методика 9):
OR"
| ArNHNH2
R-C-R'- —--------->
I н+/с2нЕон
OR"
О
II
R—C—R'.
R\
V=NNHAr
Rz/
На рис. 6.5 приведен ИК-спектр типичного ацеталя. Ниже пе-
речислены наиболее важные полосы, интенсивность которых за-
висит от типа колебаний связей С—О:
Область поглощения, см—1
Валентные колебания связи С—О, мкм
1190—1160
1195-1125
1098-1063
1055-1035
1116—1103
8,40—8,62
8,37—8,89
9,11—9,41
9,48-9,66
8,96—9,02
(только в
ацеталях)
Определение функциональных групп и установление структуры
211
Длина волны, мкм
Рис. 6.7. Инфракрасный спектр вазелинового масла (в чистом виде), полученного от фирмы Plough, Inc., и снятый на при-
боре Perkin-Elmer 521 (© Sadtler Research Laboratories, Inc.).
Вазелиновое масло (нуйол) состоит из насыщенных углеводородов. Валентные колебания С —Н: 1 — 2950, 2920, 2850 см 1 ( 3,39, 3,42, 3,51 мкм),
VC Н (алканы)' Деформационные колебания С—Н: 2—1460, 1375 см""1 (6,86, 7,28 мкм), (алканы).
214
Глава 6
500. . 400 300 . 200 • । 100 , 0Г«
Рис. &.8. Спектр протонного магнитного резонанса октана (60 МГц, развертка 600 Гц, растворитель CDClj).
Определение функциональных групп и установление структуры
215
ПМР-спектр простого ацеталя приведен на рис. 6.6. В даль-
нейшем будет показано, что для этих и различных других функ-
циональных групп во многих случаях приходится иметь дело с
интерпретацией спектральных данных, которые относятся к ато-
мам углерода и водорода, связанным с атомами кислорода или
расположенным в непосредственной близости от них.
При получении масс-спектров ацетали и кетали распадаются,
легче всего отщепляя фрагменты, расположенные рядом с ато-
мами кислорода. Основные правила фрагментации можно устано-
вить, рассматривая фрагменты, образующиеся из обычных ацета-
лей:
Фрагмент из RCH(ORZ)2 Масса Примечание
xOR' RC/ + ^OR’ [HC(OR')2]+ 44 + R + 2R' 45 + 2R' В кеталях легче отщепляется боль- ший радикал R Процесс меиее важный, чем приве- денный выше
Химические реакции, приведенные выше для идентификации
ацеталей и кеталей, часто могут быть применены и к углеводам,
поскольку последние часто содержат ацетальные или кетальные
группировки.
сн2он
а-о-(+)-глмиола
(жирным шрифтом. .OR
выделена полу-
ацефальная / \OR
группа )
метил-р-Р-фуранозиЪ
(жирным шрифтом
выделена кетальная
группа)
Эти соединения можно охарактеризовать с помощью описан-
ных выше реакций для спиртов и полиоксисоединений. Кроме то-
го, можно использовать реакции на карбонильную группу
(разд. 6.2), поскольку в растворах углеводы, являющиеся цикли-
ческими ацеталями и циклическими кеталями, могут находиться
в равновесии с ациклическими формами, содержащими кетонные
или альдегидные группы. Добавление кислоты ускоряет наступ-
ление равновесия.
6.3. АЛКАНЫ (ПАРАФИНЫ)
Как правило, для идентификации алканов (углеводородов) не
прибегают к химическим методам, так как в обычных условиях
проведения качественных реакций эти соединения крайне инертны.
216
Глава 6
Исключение составляет реакция с сульфурилхлоридом, которая
будет рассмотрена ниже. При идентификации углеводородов
химики обычно полагаются на физические и спектральные
(рис. 6.7 и 6.8) методы. В дополнение к ним могут быть прове-
дены такие исследования, которые покажут, что данное соедине-
ние не относится к алканам, так как является более реакционно-
способным (например, таким способом можно исключить алке-
ны).
Вследствие исключительно высокой инертности насыщенных
углеводородов трудно подобрать для них какую-либо реакцию по-
лучения производных. Для этой цели удается использовать лишь
небольшое число химических реакций, протекающих легче дру-
гих. Так, например, сильно напряженные циклические алканы мо-
жно прогидрировать*;
При действии хлористого сульфурила на
лучить алкилхлориды **:
углеводороды можно пр-
SO2C12
RH ---------► RC1
Однако эти методы имеют существенные ограничения. Первый
требует строгого соблюдения условий проведения реакций и иден-
тификации образующегося сложного продукта. Второй метод при-
водит к смеси нескольких веществ (например, с большими выхо-
дами образуются продукты многократного замещения). Реакции
такого рода, как правило, непригодны для получения производных.
Более полезны для идентификации алканов таблицы физических
свойств (температур кипения и плавления, «о, [а] и др.), а так-
же спектры ПК и ЯМР ***. При получении масс-спектров фраг-
ментация углеводородов протекает весьма сложным образом, по-
этому для расшифровки таких спектров необходима специальная
подготовка (гл. 10). Однако некоторые пики, постоянно присут-
ствующие в масс-спектрах углеводородов, могут быть без труда
идентифицированы. Разветвление углеродной цепи способствует
ее расщеплению. Эти данные позволяют установить структуру
изучаемого углеводорода.
* Hall Н. К-, Jr., Smith С. D., Baldt J. И., J. Am. Chem. Soc.,95, 3197 (1973).
** Markgraf H., J. Chem. Educ., 46, 610 (1969); Adcluci J. M., Dayton J. H.
Eaton D. C„ J. Chem. Educ., 48, 313 (1971).
*** В последние годы достигнуты большие успехи при идентификации угле-
водородов методом масс-спектрометрии. — Прим. ред.
Определение функциональных групп и установление структуры
217
6.4. АЛКЕНЫ (ОЛЕФИНЫ)
Двойную углерод-углеродную связь в алкенах легко идентифи-
цировать с помощью ПК-спектров (полосы поглощения связей
С=С и =С—Н см. гл. 5) и ЯМР-спектров (химический сдвиг
олефиновых протонов см. гл. 5). Наличие двойной связи легко
установить и с помощью характерных для нее химических реак-
ций. Описанные ниже весьма характерные для связи С=С хи-
мические реакции включают присоединение по двойной связи.
Опыт 14. Реакция с раствором брома в четыреххлористом угле-
роде
\ / ВГ2 \ /
,с=с' -----> ,с-с;
/ \ /\ |\
Вг Вг
В этом опыте 0,1 г твердого или 0,2 мл жидкого исследуемого соединения
смешивают с 2 мл четыреххлористого углерода. К этой смеси при перемешива-
нии прибавляют по каплям 5%-ный раствор брома в четыреххлористом угле-
роде до тех пор, пока пе перестанет исчезать окраска брома. Проведите эту
реакцию с пентеном-2, гексаном, бензолом, фенолом, муравьиной кислотой, бенз-
альдегидом, этанолом, аллиловым спиртом, ацетофеноном и анилином.
Обсуждение. Этот реагент широко применяют для открытия
двойной и тройной связей. Его следует использовать одновремен-
но с реакцией окисления перманганатом калия (опыт 15).
Четыреххлористый углерод — хороший растворитель для бро-
ма и многих органических соединений, однако он не растворяет
выделяющийся бромистый водород. Поэтому выделение бромистого
водорода указывает на то, что происходит реакция замещения, а
не присоединения. Применение этого реагента для обнаружения
непредельных связей может привести к ошибочным выводам по
двум причинам. Во-первых, не все олефиновые соединения спо-
собны присоединять бром. Во-вторых, присутствие отрицательно
заряженных группировок у атомов углерода двойной связи тормо-
зит его присоединение, а в некоторых случаях даже препятствует
этой реакции. Это можно проиллюстрировать следующими при-
мерами:
С6НБСН=СН2 + Вг2 бЫСТР-°> С6НБСНВгСН2Вг
С6НБСН=СНСООН + Вг2 медле-°>. с6Н6СНВгСНВгСООН
Положительной пробой на присутствие непредельных связей
можно считать только исчезновение окраски брома без выделе-
ния бромистого водорода.
Четыреххлористый углерод представляет собой прекрасный
растворитель для проведения качественных реакций, однако для
изучения механизма реакций он непригоден, поскольку в неполяр-
ных растворителях ход реакций усложняется. В некоторых слу-
чаях реакция проходит гетерогенно — на стенках сосуда. Кроме
218
Глава 6
того, этот процесс очень сильно ускоряется под влиянием следо!
воды или кислот и ингибируется кислородом. Все это, естествен-
но, затрудняет получение воспроизводимых результатов и зача-
стую не позволяет правильно истолковать отрицательные резуль-
таты качественной реакции.
В полярных растворителях, таких, как вода, метанол или уксус-
ная кислота, реакция протекает как двухстадийный процесс. На
первой стадии в результате взаимодействия олефина с бромом
образуется бромониевый ион, который на второй стадии взаимо-
действует с ионом бромида, образуя продукт реакции. Поскольку
бромониевый ион раскрывается с обращением конфигурации, то
в конечном результате оказывается, что два атома брома присо-
единились к двойной связи с разных сторон *:
/Вг\ Г
R2C=CR2 + Br—Br -> R2C——CR2 — R2C—CR2
Br" Br
Заместители, стабилизирующие положительно заряженный бро-
мониевый ион, увеличивают скорость реакции, а заместители, де-
стабилизирующие этот ион, тормозят реакцию. Например, введе-
ние алкильной группы к атому углерода, связанному двойной
связью, увеличивает скорость реакции присоединения в растворе
уксусной кислоты в 30—40 раз.
Исчезновение окраски брома, сопровождающееся выделением
бромистого водорода, т. е. протекание реакции замещения, ха-
рактерно для многих соединений. К ним относятся енолы, многие
фенолы и соединения, способные к енолизации. Метилкетоны бо-
лее активны в этой реакции, чем другие кетоны, однако, как это
характерно и для других карбонильных соединений, при их бро-
мировании наблюдается индукционный период. Это связано с
тем, что выделяющийся бромистый водород-действует как ката-
лизатор процесса енолизации.
Сложные эфиры простого состава не дают этой реакции. Аце-
тоуксусный эфир тотчас обесцвечивает раствор, в то время как
для реакции с малоновым эфиром может потребоваться более
1 мин. Ряд соединений с активной метиленовой группой, не обес-
цвечивающих раствор брома при комнатной температуре, быстро
реагирует при 70°С. К таким соединениям относятся пропионовый
альдегид и циклопентанон. Подобным же образом ведут себя и
простые ароматические эфиры. Для реакции бензилцианида даже
при 70°С требуется несколько минут.
Ароматические амины представляют собой исключение, так
как первый моль образующегося бромистого водорода не выде-
* См. де ла Мар П., Болтон Р. Электрофильное присоединение к ненасы-
щенным системам. Пер. с англ. — М.: Мир, 1968.
Определение функциональных групп и установление структуры
219
ляется, а реагирует с амином, давая соль. В результате эту ре-
акцию часто рассматривают как простое присоединение.
Необычным соединением является бензиламин, который легко
реагирует с бромом. В этом случае бром, по-видимому, вначале
замещает атомы водорода в аминогруппе, после чего происхо-
дит распад бромамина с образованием бензонитрила:
3C6H5CH2NH2 + 2Вг2 —> [CeH6CH2NBr2] + 2C6H5CH2NHJ Вг’
CeHsCN + 2НВг
Некоторые третичные амины, например пиридин, при бромиро-
вании образуют N-бромпроизводные:
+ Вг2
Сходным образом все алифатические амины также обесцве-
чивают раствор брома.
Опыт 15. Взаимодействие с раствором перманганата калия*
2КМпО4 + Н2О —> 2КОН + 2МпО2 + 3(0)
ч / \ / дальнейшее \ /
хс=сх + (О) + н2о —> с' -----------> с=о + о-с;
\ / | I \ окисление \
он он
Метод А. Реакция Байера в водном растворе. К 2 мл воды или этанола при-
бавляют 0,1 г (или 0,2 мл) испытуемого вещества. Смесь встряхивают, прибав-
ляют по каплям 2%-ный раствор перманганата калия, пока не перестанет ис-
чезать пурпурная окраска раствора перманганата. Проведите эту реакцию с
пентеном-2, толуолом, фенолом, бензальдегидом, анилином, муравьиной и корич-
ной кислотами, глюкозой. Если окраска перманганата не изменяется в течение
0,5—1 мни, то оставьте пробирки на 5 мин, периодически сильно их встряхивая.
Следует проявлять осторожность в выводах в тех случаях, когда реакция идет
медленно, так как это может быть связано с присутствием примесей.
Хотя ацетон чаще, чем этанол, применяют в качестве растворителя для не-
растворимых в воде соединений, однако оказалось, что некоторые тщательно
Ritter F. О., J. Chem. Educ., 30, 395 (1953).
220
Глава 6
очищенные олефины не обесцвечивают перманганат в ацетоне, но реагируют с ним
в спирте. Этанол при 20°С не реагирует с нейтральным разбавленным раствором
перманганата по крайней мере в течение 5 мин.
Водный уксуснокислый раствор перманганата калия используют для того,
чтобы отличать первичные и вторичные спирты ст третичных. При проведении
реакции по описанной выше методике первичные и вторичные спирты реагируют
с данным реактивом, а третичные — не взаимодействуют. Поэтому трет-бутило-
вый спирт можно использовать в качестве добавочного растворителя в реакции
Байера вместо этанола или ацетона.
Метод Б. Использование четвертичных аммониевых солей в качестве ката-
лизаторов фазового переноса. Реактив «пурпурный бензол». В делительную во-
ронку на 125 мл помещают раствор 10 г хлорида натрия и 0,05 г перманганата
калия в 50 мл дистиллированной воды и прибавляют 50 мл бензола. Смесь
слегка перемешивают и оставляют стоять для разделения слоев. При этом бен-
зольный слой остается неокрашенным, а водный слой имеет пурпурный цвет.
Добавляют к смеси 0,1 г тетрабутиламмонийбромида, перемешивают и остав-
ляют для разделения слоев. Бензольный слой окрашивается в глубокий пурпур-
ный цвет в результате переноса аниона перманганата из водной фазы в бензол
в форме ионной пары с катионом тетрабутиламмония. Перенос происходит не
полностью, и устанавливается определенное равновесие. После разделения слоев
наблюдают за изменением окраски пурпурного бензола в течение 5 мил. Появ-
ление бурого окрашивания указывает на наличие примесей в четвертичной ам-
мониевой соли. В этом случае раствор не следует употреблять, а перед приготов-
лением новой порции четвертичную аммониевую соль нужно перекристаллизо-
вать. Бензол не окисляется разбавленным раствором перманганата при 20°С,
поскольку он относится к ароматическим соединениям и не содержит изолиро-
ванных непредельных связей.
Для проведения пробы на наличие непредельных соединений 5 мл пурпур-
ного бензола помещают в пробирку, прибавляют 1 каплю дистиллированной
воды и 0,1 г неизвестного соединения. Закрывают пробирку пробкой и встряхи-
вают, после чего перевертывают пробирку то вверх, то вниз дном и на фоне
листа белой бумаги внимательно следят за изменением окраски от пурпурной
до коричневой. На это требуется от 30 с до 5—15 мин. Проведите эту реакцию
с циклогексеном, стильбеном, 1,1-дифенилэтиленом, трифенилэтиленом, триолеа-
том глицерина (кукурузное или хлопковое масло) и дифенилаиетилеиом. Срав-
ните время, требуемое для взаимодействия перманганата с этими веществами
по методам А н Б.
Помимо тетрабутиламмонийбромида можно использовать доступные в до-
статочно чистом виде хлорид или бромид тетрагексиламмония. Применяют и дру-
гие галогеноводородные соли четвертичных аммониевых оснований с длинной
цепью атомов углерода.
Обсуждение. Метод А. Раствор перманганата калия обесцвечи-
вается соединениями, содержащими этиленовые или ацетилено-
вые связи. Эту реакцию называют пробой Байера на непредель-
ность. В холодном разбавленном растворе перманганата калия
основным продуктом реакции олефинов являются соответствую-
щие гликоли. Если реакционную смесь нагреть, то происходит
дальнейшее окисление, приводящее в конце концов к разрыву
углеродной цепи:
RCH=CHR —> RCH—CHR —> R—С=О + О=С—R
II I I
ОН ОН ОН ОН
Метод Б. Применяемая в этом методе техника фазового пере-
носа с использованием солей длинноцепочечных четвертичных
Определение функциональных групп и установление структуры
221
аммониевых оснований или краун-эфиров является удобным спо-
собом проведения многих типов реакций. Для рассматриваемой
здесь качественной пробы Байера хорошие результаты дает при-
менение солей четвертичных аммониевых оснований, в то время
как использование доступных в настоящее время краун-эфиров
приводит к ошибкам. За последние 10 лет опубликовано большое
число работ по этим вопросам. Хорошее обсуждение процессов
фазового переноса и обширную библиографию можно найти в
следующих книгах: Вебер В., Гокель Г. Межфазный катализ в
органическом синтезе. Пер. с англ.— М.: Мир, 1980. Starks С. М.,
Liotta С., Phase Transfer Catalysis, Academic Press, New York,
1978.
Ацетиленовые связи обычно при окислении также расщепляют-
ся с образованием кислот:
R—С=С—R'+ Н2О + 3[О] —> RCOOH + R'COOH
Скорость, с которой непредельные соединения обесцвечивают
перманганат, зависит от растворимости органического соедине-
ния. Если соединение плохо растворимо в воде, его либо следует
растереть в порошок и сильно взбалтывать несколько минут, либо
растворить в таком растворителе, который не взаимодействует с
перманганатом. Некоторые тетразамещенные олефины, например
С6Н5СВг=СВгСбН5 и (С6Н5)2С=С(С6Н5)2, не дают положитель-
ной реакции ни с раствором брома в СС14, ни с растворами пер-
манганата, описанными выше.
Из приведенного ниже уравнения следует, что по мере про-
текания реакции раствор становится все более щелочным:
3ХчС=С/ + 2КМпО4 + 4Н2О —> З^С— + 2МпО2 (тв.) + 2КОН
/ \ /\ |\
• ОН он
Однако необходимо избегать применения сильнощелочного рас-
твора, так как при этом изменяется характер протекающей реак-
ции. Например, в растворе карбоната натрия даже ацетон дает с
перманганатом положительную реакцию. Выпадение осадка ди-
оксида марганца во многих случаях не наблюдается, просто пур-
пурный цвет раствора постепенно переходит в красновато-бурый.
Однако возможно применение перманганата и в строго ней-
тральной среде. Так, при использовании перманганата цинка в
результате реакции образуется гидроксид цинка, который очень
слабо растворим в воде. Поэтому раствор остается практически
нейтральным К такому же результату приводит применение пер-
манганата калия в присутствии сульфата магния. В этом случае
гидроксильный ион осаждается в форме нерастворимого гидрок-
сида магния.
Проба Байера является более удачной химической реакцией
на непредельные соединения, чем реакция с бромом, хотя при ее
222
Глава 6
использовании наблюдаются некоторые осложнения. Эту реакцию
дают все легкоокисляющиеся соединения. Карбонильные соеди-
нения, которые обесцвечивают растворы брома, обычно не всту-
пают в реакцию Байера. Хорошим примером может служить аце-
тон. Хотя он быстро обесцвечивает раствор брома, при проведе-
нии реакции Байера его можно использовать в качестве раство-
рителя. Альдегиды дают положительную реакцию Байера, однако
многие из них, например бензальдегид и формальдегид, не обес-
цвечивают растворы брома. Муравьиная кислота и ее эфиры также
восстанавливают перманганат вследствие того, что они содержат
группировку О=СН—.
Другим важным классом соединений, обесцвечивающих рас-
творы перманганата, но не реагирующих с бромом, являются
спирты. В чистом виде эти вещества неохотно вступают в реак-
цию, однако они часто содержат легкоокисляющиеся примеси.
Другие соединения также могут содержать небольшие количе-
ства примесей, которые могут обесцвечивать перманганат. Имен-
но поэтому обесцвечивание одной или двух капель перманганат-
ного раствора не может рассматриваться как положительная
проба.
Фенолы и ариламины также восстанавливают раствор перман-
ганата, давая хиноны. Под влиянием избытка реактива эти соеди-
нения могут претерпевать дальнейшее окисление, превращаясь в
смесь продуктов окисления, среди которых найдены малеиновая
и щавелевая кислоты и диоксид углерода.
ОН о
Сераорганические соединения, в которых сера находится в
восстановленном состоянии, также восстанавливают перманга-
нат, претерпевая окисление. Меркаптаны окисляются в дисуль-
фиды, а тиоэфиры и тиокетали — в сульфоны:
2RSH + (O) —-> RSSR + H2O
(О) (О)
RSR -----> R2SO ------> RSO2R
(О)
R2C(SR)2 -----> R2C(SO2R)2
Окисление тиосоединений легче проходит в кислой среде. По-
этому если по данным элементного анализа в исследуемом ве-
ществе содержится сера, то растворяют 0,1 г этого вещества в
2 мл ледяной уксусной кислоты и прибавляют по каплям разбав-
Определение функциональных групп и установление структуры 223
ленный раствор перманганата калия. Если при этом наблюдает-
ся исчезновение пурпурной окраски, то изучаемое соединение мо-
жет содержать способную окисляться серусодержащую группу.
Перманганат не восстанавливается в тех случаях, когда сера в
соединении находится в форме сульфона, алкилсульфата или не-
замещенной сульфокислоты. Однако некоторые замещенные суль-
фонаты, например бисульфитные соединения альдегидов и кето-
нов, а также фенолсульфокислоты, восстанавливают перманга-
нат.
На основании того факта, что образующиеся из олефинов гли-
коли представляют собой продукты присоединения двух гидро-
ксильных групп с одной стороны двойной связи (этот процесс на-
зывают цис- или смн-присоединением), было сделано предположе-
ние, что на одной из промежуточных стадий этой реакции обра-
зуется циклический эфир марганцевой кислоты, который анало-
гичен описанному выше промежуточному соединению, возникаю-
щему при окислении спиртов иодной кислотой (опыт 5).
Контрольные вопросы. 1. Какие функциональные группы вступают в реак-
цию и с бромом, и с перманганатом?
2. Какая из этих реакций лучше для открытия непредельных связей?
3. В каких случаях полезно использовать обе реакции?
Ряд специфических реакций для идентификации олефинов
описан в книге Пасто и Джонсона. Одна из них — реакция обра-
зования комплексов с двойными связями (Пасто и Джонсон,
с. 328 и 332 соответственно):
+C(NO2)4 СНСА- C(NO,)4
Цвет образующегося комплекса сходен с цветом, который дают
простые эфиры и арены при обработке их иодом.
+ 12 --♦
жидкий
окрашенный комплекс
с переносом заряда
Другая состоит в расщеплении двойной связи при озонолизе:
R'CHO+R2CO
/ Г12'-'
R’ ,R . 0-0
УЦ -°3-R'CH CR2
Н R \ н о
R'CO2H + R2CO
Расщепление алкенов можно провести также, окисляя их пер-
манганатом в щелочной среде (методика 6, описанная выше для
224
Глава 6
альдегидов) или перманганатом в присутствии периодат-иона
(Пасто и Джонсон, с. 333):
rco2h+r2co+MnOj
J9a
В отличие от брома другие реагенты могут присоединяться к
алкенам по ионному механизму. Таким образом проходит присо-
единение аренсульфенилгалогенидов, приводящее, как правило, к
твердым продуктам, удобным для идентификации, например (Па-
сто и Джонсон, с. 335):
2,4-Ъинитробензолсульфенилхлорид
ИК-Спектр простого олефина приведен на рис. 6.9, ПМР-
спектр того же олефина представлен на рис. 6.10. Как было от-
мечено ранее, введение двойной связи в молекулу вызывает по-
явление легко распознаваемых адсорбционных полос в спектрах
ПК и ЯМР. Изучение книг по этим вопросам, перечисленных в
гл. 10, приводит к заключению, что такие адсорбционные спект-
ры позволяют сделать выводы: 1) о числе заместителей у двой-
ной связи, 2) о геометрии двойной связи и 3) о присутствии аро-
матических или электроотрицательных заместителей при двой-
ной связи.
Распад алкенов при получении масс-спектров происходит до-
статочно сложным образом. Данные об этом процессе приведены
в других пособиях (см. гл. 10). Здесь следует отметить только,
что легче всего разрывается аллильная связь:
:с=с—С—Х
:с=с—с:
В тех случаях, когда предполагаемое соединение может пред-
ставлять собой сопряженный диен (С=С—С=С), следует ис-
пользовать при обсуждении УФ-спектров правила Вудворда —
Физера (гл. 8). Применение ультрафиолетовой спектрометрии
весьма полезно при изучении различных молекул, содержащих
систему сопряженных л-связей.
Удобный метод получения производных непредельных углево-
дородов состоит в превращении их в спирты посредством гидро-
борирования (методика 116). Если получающийся спирт окажет-
ся твердым, то его можно перекристаллизовать и он сам по себе
Длит волны, мкм
г
* 3
° ।
& *
<u S
8 Зак. 335
226
Глава 6
Рис. 6.10. Спектр протонного магнитного резонанса циклогексена (60 Мгц, развертка 600 Гц, растворитель CDCls).
600 , SOO
Определение функциональных групп и установление структуры
227
будет служить производным исходного олефина. Однако, как пра-
вило, спирты при 25°С представляют собой жидкости. Поэтому
для получения твердых производных приходится превращать их
в 3,5-Динитробензоаты или в а-нафтилуретаны (методики 2 и 13
соответственно).
Методика 116. Перевод алкенов в спирты
методом окислительного гидроборирования
№ВН4/ВРз-О(С2Нб)2
ТГФ. Ng. 25°с
Н2О2
--------->
NaOH. НгО
Н ОН
I I
—> —С—С— (—» производные спиртов)
I I
Первую стадию этой реакции следует проводить таким образом, чтобы ис-
ключить попадание влаги и кислорода воздуха, поэтому вся стеклянная посуда
должна быть тщательно вымыта и высушена в сушильном шкафу. Применяемый
Рис. 6.11.
f—ток азота: 1—трубка с осушителем (CaSO4); 3—ТГФ + BF3 .OlCjHsh; 4—выравниватель
давления: 5—ТГФ 4- ИаВЩ 4- олеф ш; 6—водяная баня: 7 — водяное охлаждение
прибор, показанный на рис. 6.11, состоит из трехгорлсй колбы на 100 мл, об-
ратного холодильника, маленькой капельной воронки, термометра и тонкой, до-
ходящей до дна трубки, по которой подается ток сухого азота *. Ток азота не-
обходим для удаления кислорода и для перемешивания реагирующих веществ.
При проведении реакции устанавливают ток сухого азота и вносят в колбу 20 мл
сухого тетрагидрофурана (ТГФ), 0,25 г NaBH4 и 1,2 мл олефина. Раствор 1,2 г
ВГз‘(СгНьЪО** в 3 мл сухого ТГФ наливают в маленькую капельную воронку.
* Скорость газа ие должна быть слишком велика, чтобы во время прове-
дения реакции ие происходило уноса жидкости.
•• Эфират BFa лучше всего очищать перегонкой в вакууме,
8*
228
Глава 6
С помощью водяной бани устанавливают температуру 25°С и тетрагидрофурано-
вый раствор прикапывают из капельной воронки с такой скоростью, чтобы тем-
пература держалась около 25°С. Прибавление проводят в течение 3—5 мин.
Смесь оставляют на 1 ч, пропуская слабый ток сухого азота. После этого не во-
шедший в реакцию гидрид разлагают, добавляя по каплям 10 мл воды. Увели-
чив ток азота так, чтобы смесь энергично перемешивалась, прибавляют 3,2 мл
3 М раствора гидроксида натрия, а затем 3,2 мл 30%-кого раствора пероксида
водорода. Смесь нагревают до 30—40°С на водяной баие. Спустя 15 мин насы-
щают водную фазу 4 г хлорида натрия, что приводит к отделению раствора
спирта в ТГФ. Смесь охлаждают до 20°С, ток азота прекращают и для облегче-
ния расслаивания прибавляют 5 мл абсолютного эфира. Органический слой отде-
ляют, промывают 5 мл насыщенного раствора хлорида натрия, сушат 1 г без-
водного сульфата магния и фильтруют через небольшой сухой бумажный фильтр
в пробирку. Растворители (ТГФ и эфир) удаляют, направляя ток сухого азота
прямо в пробирку. Оставшийся жидкий спирт можно превратить в а-нафтилуре-
тан (методика 1) либо в 3,5-динитробеизоат (методика 2). При перекристалли-
зации следует применять только минимально необходимое количество раствори-
теля, так как удается выделить только около половины взятого вещества.
Обсуждение. Гидроборирование может проходить последова-
тельно, давая моно-, ди- и триалкилбораны:
№вн4, тгф рСН2СН2ВН2
RCH=CH2 b~f3.(C2h5)^ (RCH2CH2)2BH
t (RCH2CH2)3B
Количество вступающих в реакцию связей В—Н в значительной
степени контролируется объемом групп R: простые алкены с ра-
дикалами R умеренного размера обычно дают триалкилбораны
(х = 3, у - 0, см. ниже).
Окислительное расщепление алкилборанов щелочным раство-
ром пероксида водорода с образованием спиртов — одна из наи-
более хорошо изученных реакций борорганических соединений:
H2O2/NaOH
(RCH2CH2)xBH„ ----——> xRCH2CH2OH + Na3BO8
а H2C)
*4-0 = 3
Борорганические соединения можно превратить не только в
спирты, но также в алканы, амины, галогениды и др. Литерату-
ру по этим вопросам можно найти в книге Брауна (см. ниже).
Гидроборирование проходит как антимарковниковское цис-
присоединение. Например, 1-метилциклопентен по методике На
дает почти чистый транс-2-метилциклопентанол:
жОН
СХсн
Симметричные 1,2-дизамещенные алкены или циклоолефины
превращаются во вторичные спирты, которые можно далее окис-
Определение функциональных групп и установление структуры
229
лить в кетоны (опыт 15) и охарактеризовать в виде 2,4-динитро-
фенилгидразонов (методика 9):
[01
I. вн3
2. Н2О2
NNHAr
Несимметричные дизамещенные олефины (RCH=CHR') об-
разуют смесь двух вторичных спиртов в близких концентрациях.
Поэтому гидроборирование в этих случаях не применяется. Оле-
фины такого типа легче идентифицировать на основании каких-
либо других свойств (температура кипения, плотность, nD, ИК- и
ЯМР-спектры).
1. вн«
СН3СН=СНСН2СН3 9 —
2. Н2О2
CHSCHOHCH2CH2CH3 4- СНзСН2СНОНСН2СН3
При гидроборировании олефинов с концевой двойной связью
атом бора присоединяется к концевому атому углерода, давая на
90—99% продукт присоединения против правила Марковникова.
С выходом 1—10% образуются продукты присоединения бора к
замещенному атому углерода.
Г СНз 1
।
Lrch— ъвну
Н2®; NaOH
---------->
H2O2
СНз
I
RCHOH
При окислительном расщеплении образующегося продукта полу-
чается вторичный спирт. Этот спирт также будет давать а-наф-
тилуретан или 3,5-динитробензоат, которые будут понижать тем-
пературу плавления основных получаемых производных. Однако
при использовании большого количества растворителя для кри-
сталлизации полученного производного с тем, чтобы концентра-
ция примеси в растворе была минимальной, можно получить про-
дукт, достаточно чистый для идентификации.
С помощью реакции гидроборирования можно превратить со-
пряженные диены в 1,4-гликоли, из которых затем могут быть
получены твердые производные*;
СНз СНз
I I
сн2=с—с=сн2
СНз СНз
I I
сн2—сн—сн—сн2
I I
он он
Литература. Zweifel G., Brown Н. С., Org. Reactions, 13, 1
(1963). Эта статья содержит превосходный обзор, посвященный
* Эти диолы часто настолько полярны, что для их выделения необходимы
особые приемы экстракции органическими растворителями.
230
Глава 6
гидратации олефинов с помощью реакции гидроборирования. См.
также: Brown И. С., Hydroboration, Benjamin, N. Y., 1962.
Brown H. C., Boranes in Organic Chemistry, Cornell University
Press, Ithaca, N. Y., 1972. Brown H. C., Organic Syntheses via Bo-
ranes, Wiley, N. Y., 1975.
6.5. АЛКИНЫ (АЦЕТИЛЕНЫ)
Поскольку в данном разделе рассмотрены соединения с трой-
ной связью (CsC), которая во многом сходна с двойной связью
в алкенах, то не удивительно, что многие реакции, описанные в
предыдущем разделе для алкенов, могут быть использованы и
для веществ, содержащих тройную связь.
Приведенные ниже реакции считаются (Пасто и Джонсон,
с. 339) весьма полезными для идентификации алкинов. Ввиду
того что нитроароматические соединения и вещества, содержа-
щие ртуть *, взрывчаты, работать с ними необходимо чрезвычай-
но осторожно. Совершенно недопустимо применение больших ко-
личеств реагентов и высоких температур при перегонке.
№С—H+HgCl2/KI - кон -> (RC=C)2Hg
Сравните эту реакцию с реакцией с металлическим натрием
(опыт 1)
На рис. 6.12 и 6.13 приведены ИК- и ПМР-спектры ацетилено-
вых соединений. Симметрично замещенная связь CssC в ИК-
спектре определяется с трудом. Обычно тройная связь в ПК-
спектре проявляется только в тех случаях, когда при ней имеется
заместитель только с одной стороны, т. е. в ацетиленах с конце-
вой тройной связью. Ввиду того что находящаяся в середине цепи
углеродных атомов тройная связь не соединена с протонами,
при интерпретации ПМР-спектров следует полагаться на протон,
н—с—с~с— у
Конечно, это менее удобно для анализа адсорбционных
находящийся в положении 2 к кратной связи
* Внимание. Ацетилениды меди, серебра в ртути обладают взрывчатыми
свойствами.
Определение функциональных групп и установление структуры
231
Длина волны, мкм
232
Глава 6
Рис. 6.13, Спектр протонного магнитного резонанса фенилацетилена (60 МГц, развертка 600 Гц, растворитель CDC13)
Определение функциональных групп и установление структуры
233
спектров, чем, например, использование сигналов протона при
атоме углерода тройной связи ( = С—Н).
Расщепление алкинов при получении масс-спектров проходит
так же, как и в случае олефинов.
6.6. АЛИФАТИЧЕСКИЕ ГАЛОГЕНИДЫ
Поскольку алифатические галогениды первоначально иденти-
фицируются по данным элементного анализа на галогены (гл. 4),
то естественно, что для их последующего изучения используют
способность атома галогена к замещению. Ниже обсуждаются
две реакции замещения галогена (X), дополняющие друг друга.
Они очень часто используются для установления структуры алкил-
галогенидов.
увеличение реакционной способности
Nal в ацетоне: -------------------------•*
R3cci r2chci rch2ci
AgNO3 в этаноле: ----------------~~--------*
& уменьшение реакционной способности
После обсуждения экспериментальных методик и получаемых
при их применении результатов приведен краткий очерк теории
реакций замещения галогенов.
Опыт 16. Раствор нитрата серебра в этаноле
RX + AgNO3 —> AgX (тв.) 4- RONO2
Этот реактив применяют для выявления соединений, содержащих атомы га-
логена. Прибавляют 1 каплю галогепзамещенного соединения к 2 мл 2%-ного
раствора нитрата серебра в этаноле. Если в течение 5 мин при комнатной тем-
пературе никакой реакции не происходит, нагревают раствор до кипения и от-
мечают, выделяется ли осадок. Если осадок образуется, то следует обратить
внимание на его окраску. Прибавляют 2 капли 5%-иой азотной кислоты и отме-
чают, растворяется лн осадок. Галогениды серебра нерастворимы в разбавлен-
ной азотной кислоте, тогда как серебряные соли органических кислот в ней легко
растворяются.
Алкилгалогениды часто содержат небольшие количества изо-
мерных примесей, поэтому в некоторых особых случаях полезно
собрать и взвесить сухой осадок галогенида серебра, полученный
из предварительно взвешенного образца неизвестного соединения.
Обычно на основании сопоставления физических констант и рас-
смотрения возможных структур неизвестного вещества можно сде-
лать приблизительную оценку его молекулярной массы. Рассчи-
танный теоретически выход галогенида серебра можно сравнить
с полученным количеством. Если выход составляет лишь несколь-
ко процентов от теоретического, то пробу считают отрицательной.
Для того чтобы отличить медленно реагирующий алкилгалоге-
ннд, дающий лишь небольшое количество осадка галогенида се-
ребра, от смеси инертного галогенида с небольшим количеством
234
Глава 6
активной примеси, надо отделить выпавший осадок и прибавить
к фильтрату новую порцию нитрата серебра.
Проведите эту реакцию с бензоилхлоридом, бензилхлоридом,
этилбромидом, бромбензолом, хлороформом и хлоруксусной кис-
лотой.
Обсуждение. Многие галогенсодержащие вещества реагируют с
нитратом серебра, давая нерастворимые галогениды серебра.
Скорость этой реакции может служить мерой реакционной спо-
собности атома галогена. Эти данные представляют большой ин-
терес, так как позволяют сделать выводы о структуре молекулы.
Активность галогена тем больше, чем сильнее его способность к
ионизации. Наиболее ярким примером могут служить соли ами-
нов и галогеноводородных кислот [КМНз]+Х~. Реже приходится
сталкиваться с оксониевыми и карбониевыми солями, содержа-
щими ионы галогенов.
- —|+
R:Q:R Г~\ +
r’ X (CH3)2N/ } С С1
L д3
К' = алкил, Н кристаллический
фиолетовый
Водные растворы этих солей с водным раствором нитрата сереб-
ра тотчас образуют осадок галогенида серебра.
Ниже просуммированы результаты, которые могут получиться
при использовании спиртового раствора нитрата серебра.
1. Растворимые в воде вещества, тотчас же дающие осадок с
водным раствором нитрата серебра.
а) Соли аминов и галогеноводородных кислот:
(RNH3)+X~ + Ag*NO3 —> AgX (тв.) + (RNH3)+NO3‘
б) Оксониевые соли.
в) Карбонийгалогениды.
г) Хлорангидриды низкомолекулярных кислот, многие из ко-
торых гидролизуются в воде с образованием ионов галогена:
RCOC1 + Н2О —> RCOOH + НС1
2. Соединения, нерастворимые в воде, условно можно разбить
на три группы по их отношению к спиртовому раствору нитрата
серебра.
а) Вещества первой группы немедленно образуют осадок при
комнатной температуре:
RCOCl RCHC1OR R3CC1
RCH=CHCH2X RCHBrCH2Br RI
б) Вторая группа включает вещества, которые при комнатной
температуре реагируют медленно или не реагируют вообще, но
Определение функциональных групп и установление структуры 235
легко образуют осадок при более высокой температуре:
RCH2C1 R2CHC1
RCHBt2
в) Третью группу образуют вещества, устойчивые к действию
спиртового раствора нитрата серебра:
АгХ RCH=CHX НСС13
Циклогексилгалогениды по сравнению с нециклическими вто-
ричными галогенидами менее реакционноспособны по отношению
к нитрату серебра. Циклогексилхлорид неактивен, а циклогексил-
бромид хотя и дает осадок со спиртовым раствором нитрата се-
ребра, но реагирует медленнее, чем 2-бромгексан. 1-Метилцикло-
гексилхлорид также значительно менее реакционноспособен, чем
нециклические третичные хлориды. Однако как 1-метилциклопен-
тилхлорид, так и 1-метилциклогептилхлорид более активны, чем
их аналоги с открытой цепью.
Как уже указывалось выше, реакционная способность по от-
ношению к спиртовому раствору нитрата серебра резко отличает-
ся от активности галогенидов по отношению к раствору иодида
натрия в ацетоне (опыт 17). Поэтому при исследовании неизвест-
ных галогенсодержащих соединений следует применять обе эти
реакции.
Опыт 17. Раствор иодида калия в ацетоне
RCl + Nal —> RI + NaCI (тв.)
RBr + Nal —» RI + NaBr (тв.)
В пробирке к 1 мл раствора иодида калия в ацетоне прибавляют 2 капли
соединения, в котором элементным анализом было установлено присутствие хлора
или брома. Если это соединение представляет собой твердое вещество, то при-
мерно 0,1 г его растворяют в возможно малом количестве ацетона и этот рас-
твор прибавляют к реактиву. Пробирку встряхивают и оставляют при комнат-
ной температуре на 3 мин. Отмечают, выделился ли осадок и появилось ли
красно-бурое окрашивание от выделяющегося свободного иода. Если при ком-
натной температуре никаких изменений ие произошло, то пробирку ставят в ста-
кан с водой, нагретой до 50°С. Спустя 6 мин охлаждают смесь до комнатной
температуры и смотрят, прошла или не прошла реакция. Примените эту реакцию
к я-бутилбромиду, втор-бутилбромиду, трет-бутилхлориду, этилеибромиду, беи-
зилхлориду, бензоилхлориду, бензолсульфонилхлориду и а-хлорацетофеиоиу.
Реактив. Растворяют 15 г иодида натрия в 100 мл чистого ацетона. Бес-
цветный раствор становится постепенно лимоино-желтым. Его следует хранить
в темной склянке и при появлении красно-бурого окрашивания заменять новым.
Обсуждение. Эта реакция основана на том, что хлорид и бромид
натрия очень плохо растворимы в ацетоне. Как и следовало ожи-
дать, реакционная способность простых галогенидов уменьшает-
236
Глава 6
ся в ряду: первичные < вторичные < третичные. Первичные бро-
миды дают осадок с иодидом натрия через 3 мин при 25°С. Хло-
риды в этих условиях не дают осадка, и, чтобы реакция про-
шла, требуется нагревание до 50°С. Вторичные и третичные
бромиды реагируют при 50°С. Однако третичные хлориды реа-
гируют очень медленно — при стоянии в течение 1—2 дней.
Циклопентилхлорид реагирует с раствором иодида натрия в
ацетоне с такой же скоростью, как ациклические вторичные хло-
риды, а циклогексилхлорид — значительно медленнее. Действи-
тельно, циклогексилхлорид, циклогексилбромид, борнилхлорид и
другие подобные соединения не реагируют с иодидом натрия при
50°С в течение 6 мин. Такое торможение реакции в случае цикло-
гексильных соединений обусловлено особенностями геометрии
циклогексанового кольца, влияющего на структуру переходного
состояния.
Бензилгалогениды (АгСНгХ) и аллилгалогениды
-С—X
чрезвычайно активны в реакции с иодидом натрия в аце-
тоне и дают осадок хлорида натрия при 25°С всего за 3 мин.
Причины столь высокой активности будут обсуждаться ниже.
Можно было бы предполагать, что объемистые заместители в
трифенилметилхлориде будут препятствовать реакции замещения
иодид-ионом, однако в действительности это соединение реаги-
рует с раствором иодида натрия в ацетоне значительно быстрее,
чем бензилхлорид. При проведении этой реакции наблюдается вы-
деление свободного иода. Следовательно, она не является простой
реакцией замещения с образованием третичного иодида. Этот
пример показывает, что при истолковании результатов качествен-
ных реакций такого типа необходимо соблюдать осторожность.
Следует помнить, что некоторые представители изучаемого клас-
са соединений могут реагировать с применяемым реагентом не-
стандартно и что влияние одних и тех же структурных факторов
на реакционную способность может быть совершенно различным
в зависимости от типа реакции.
В литературе имеются сообщения о том, что пикрилхлорид при
обработке раствором иодида калия в этаноле дает пикрилиодид.
Однако при действии иодида калия в кислой среде, например в
кипящей уксусной кислоте или даже в ацетоне, содержащем
уксусную кислоту, при комнатной температуре образуются толь-
ко тринитробензол и свободный иод.
Соединения, содержащие несколько атомов брома, например
бромоформ и 1,1,2,2-тетрабромэтан, реагируют с иодидом натрия
при 50°С с образованием осадка и выделением свободного иода.
Тетрабромметан реагирует уже при 25°С.
Определение функциональных групп и установление структуры
237
Сульфонилхлориды сразу же дают осадок и выделяют иод.
По-видимому, свободный иод получается при действии иодида
натрия на образующийся сульфонилиодид:
ArSOjCl + Nal —> ArSO2I + NaCl (тв.)
]NaI
ArSO2Na + I2
Бензолсульфонилхлорид дает с выходом 60% бензолсульфо-
нат натрия; кроме того, образуются дифенилсульфон (27%) и фе-
нилтиосульфинат (10%).
Алкильные эфиры сульфокислот также вступают в эту реак-
цию, образуя осадки соответствующих натриевых солей сульфо-
кислот:
ArSO2OR + Nal —> ArSO2ONa + RI
Возможность протекания этой реакции следует учитывать в тех
случаях, когда одна из замещающих групп в сложном эфире суль-
фокислоты содержит атомы галогенов.
1,2-Дихлор- и 1,2-дибром производные также не только дают
осадок хлорида или бромида натрия, но и выделяют свободный
иод:
Вт Вг
I I
RCH—CHR + 2NaI --> RCH=CHR + 2NaBr+I2
Г<’вг
Ступенчатая
реакция: R—CH—CH—R ------> RCH=CHR + IBr+Br
^Вг
1Вг+Г ---> 12 + Вг*
Сравнение активностей этиленгалогенидов приводит к следующим
результатам:
Этиленгалогенид
ВгСН2СН2Вг
ВгСН2СН2С1
С1СН2СН2С1
Время, за которое выпадает осадок
при 25 °C, мин
1.5
3
Не реагирует (осадок образуется за
2,5 мии при 50 °C)
Теория реакций замещения
Ввиду того что реакция со спиртовым раствором серебра и замещение иона
галогена при действии иодида натрия тесно связаны между собой, их следует
рассматривать совместно. Реакции нуклеофильного замещения, в которых участ-
вуют галогенсодержащие соединения и другие аналогичные вещества, удобно
238
Г лава 6
разделить на две группы в зависимости от их механизма. Первый из них —
Sn2 (substitution nucleophilic bimolecular, бимолекулярное нуклеофильное заме-
щение), «вытеснение», или «N» (нуклеофильный механизм), а второй —SN1,
«сольволитнческпй», или «лим», т. е. механизм с «лимитирующей стадией».
В этих двух механизмах совершенно по-разному проявляются различные осо-
бенности галогенсодержащего соединения. Поэтому при любом рассмотрении
влияния строения галогенсодержащего соединения на его реакционную способ-
ность необходимо принимать во внимание механизм изучаемого процесса.
Подробное обсуждение этого вопроса выходит за рамки этой книги. Опуб-
ликованы отличные обзоры по всем аспектам данной проблемы *. Однако крат-
кое изложение особенностей этих двух механизмов может существенно облег-
чить понимание процессов, протекающих при действии растворов нитрата се-
ребра в спирте и иоднда натрия в ацетоне.
Механизм Sn2, или механизм прямого замещения, представляет собой со-
гласованную реакцию, в которой нуклеофильный реагент, например иодид или
гидроксид-ион, вытесняет реагирующий галоген. Вытеснение происходит в ре-
зультате подхода нуклеофильной частицы к углеродному атому со стороны,
противоположной по отношению к уходящему галогену.
R\ V
Г + Н—С—Х->1 - С- -X
hZ \t
переходное
состояние
В переходном состоянии отрицательный заряд в значительной степени распре-
делен между входящей и уходящей группами. Поскольку реакция происходит
с образованием ковалентной связи с 1~. то тыльная сторона атома углерода,
у которого происходит замещение, не должна иметь значительных стерических
препятствий. Это необходимо для того, чтобы облегчить приближение атакующего
атома или группы. Такое протекание реакции определяет ее высокую чувстви-
тельность к стерическим затруднениям у реакционного центра и малую чувстви-
тельность к электронным влияниям. Механизм SnI, или карбокатионный, вклю-
чает две стадии. На первой стадии происходит ионизация реагирующего гало-
генида.
R-X —► R+ + X-
R++ Y~ —> R-Y
Очевидно, что этот механизм не будет чувствителен к стерическим затрудне-
ниям в тыльной части атома углерода в R, но, напротив, будет весьма чувстви-
телен к электронным влияниям, т. е. реакция будет очень сильно ускоряться при
введении в R таких заместителей, которые облегчают стабилизацию положитель-
ного заряда. В общем галогенсодержащие соединения реагируют с иодидом
натрия в ацетоне по механизму Sn2, а со спиртовым раствором нитрата сереб-
ра — по механизму SK1. Эти выводы могут быть сделаны на основе рассмотре-
ния свойств этих реагентов. В растворе иодида натрия в ацетоне содержится
иодпд-ион, который является хорошим вытесняющим агентом (иными словами,
сильным нуклеофилом). Ацетон представляет собой очень плохой растворитель
для процессов ионизации. Таким образом, этот реактив весьма подходит для
проведения реакций, идущих по N-механизму. Нвтрат-ион обладает очень сла-
быми нуклеофильными свойствами, а этанол является растворителем с умерен-
ной, но достаточно явно выраженной ионизирующей способностью. Кроме того,
* Streitwieser A., Chem. Rew., 56, 571 (1956) и Solvolytic Displacement Reac-
tions, McGraw-Hill, New York, 1962; Ингольд К. Теоретические основы органи-
ческой химии. Пер. с англ. — М.: Мир, 1973.
Определение функциональных групп и установление структуры
239
ион серебра благодаря его особой склонности к координации с уходящим га-
логеном оказывается превосходным агентом, способствующим его ионизации. Все
эти факторы приводят к тому, что реакция с этим реактивом проходит по ме-
ханизму SNt.
Предельные галогениды. Третичный карбонпевый ион стабильнее вторич-
ного, который в свою очередь более устойчив, чем первичный. Одной из причин
такого различия является имеющаяся у третичного иона возможность рассре-
доточения положительного заряда. У вторичного иона эта возможность меньше,
у первичного — совсем невелика.
третичный вторичный первичный
Вторым фактором, благоприятствующим образованию третичного карбо-
пневого иона из соответствующего галогенида по сравнению с образованием пер-
вичных или вторичных ионов, является наличие стерической напряженности в
объемистом третичном галогениде (угол между валентными связями у атома
углерода равен 109°), которая заметно уменьшается при образовании плоского
нона (угол между связями 120°). Как и следовало ожидать, предсказанный тео-
рией порядок уменьшения реакционной способности алкилгалогенидов при реак-
ции с нитратом серебра согласуется с найденным экспериментально:
третичный > вторичный > первичный
Интересно отметить, что с такой чрезвычайно разветвленной молекулой, как
три-трет-бутилметилхлорид, сольволитическая реакционная способность которой
очень велика, реакция может идти более чем в 40 000 раз быстрее реакции с
грег-бутилхлоридом.
Как и следовало предполагать, порядок реакционной способности простых
галогенидов в реакции замещения при действии щелочных иодидов будет об-
ратным, поскольку стерические препятствия для подхода иодид-нона будут воз-
растать в ряду галогенидов следующим образом: первичный > вторичный >
> третичный.
Бициклические галогениды. Галоген, стоящий у мостикового углеродного
атома в некоторых бициклических системах, обладает крайне малой реакцион-
ной способностью как в реакциях сольволиза, так и в реакциях замещения. На-
пример, апокамфилхлорид(1) не реагирует пи при обработке 30%-ным раство-
ром гидроксида калия в 80%-ном этаноле за 24 ч, ни с нитратом серебра в вод-
ном этаноле при нагревании за 48 ч. В этом случае нуклеофильная атака по ме-
ханизму S№ невозможна, поскольку подход нуклеофила с задней стороны коль-
ца блокирован. В то же время образование карбокатиона (1а) ингибируется из-за
сопротивления циклического углеродного скелета переходу к копланарному рас-
положению атомов Ci, С2, С6 и С7, что является необходимым геометрическим
требованием для ар2-гибридизованного карбокатионного центра у Ср
I la II
1-Бромбнцикло-[2,2,1]-гептан превращается в соответствующий карбинол
при нагревании до 150°С в течение 2 дней с водным раствором нитрата серебра.
Если же взять бромид бицикло-[2,2,2]-октана(II), то он превращается в спирт
уже через 4 ч при комнатной температуре. Большая реакционная способность
240
Глава 6
[2,2,2]-системы по сравнению с системой [2,2,1] объясняется, вероятно, тем, что
бром по сравнению с хлором удаляется значительно легче и что циклическая
система в соединении II обладает существенно большей подвижностью колец,
что позволяет связям у С] расположиться в одной плоскости и тем самым спо-
собствует образованию карбаниона.
Арил- и винилгалогениды. Винилгалогениды и те арилгалогениды, в которых
галоген связан непосредственно с ароматическим кольцом, как правило, не всту-
пают в реакцию замещения ни по механизму Sn2, ни по сольволитическому ме-
ханизму.
СН2=СН—X
Hal/ацетон
AgNO3
С2Н6ОН
• реакция
не идет
Ингибирование этих процессов происходит из-за высокой прочности связи С—X
и нестабильности потенциального карбокатиона, который образуется на проме-
жуточной стадии реакции *.
Бензнл- н аллилгалогениды. Поскольку, как показано ниже, бензильный и
аллильные карбокатионы способны к резонансной стабилизации, то не удиви-
тельно, что оин реагируют с нитратом серебра быстрее, чем соответствующие
насыщенные галогениды. Бензил- и аллилгалогениды активны также и по отно-
шению к раствору иодида натрия в ацетоне.
аллил: сн2=сн—сн2—х
сн2=сн—сн;
сн2-сн=сн2
СН2СН-СН2—Y
Тот факт, что эти соединения одинаково хорошо реагируют по двум разным
механизмам, можно объяснить их строением — они представляют собой первич-
ные галогениды, но в то же время способны давать стабильный карбокатион.
Введение арильных заместителей, как правило, увеличивает скорость реак-
ций, идущих по механизму SN1. Так, бензгидрилхлорид быстро реагирует с теп-
лым этанолом даже без добавления нитрата серебра, а трифенилметилхлорид
при растворении в диоксиде серы (этот растворитель не принимает участия в
реакции, но эффективно сольватирует ионы) почти полностью ионизируется, да-
вая трифенилкарбоиийхлорид (С6Н6)зС+С1-.
Влияние соседних групп. Следующий раздел целесообразно изучать лишь
после того, как читатель достаточно глубоко познакомился с реакциями основ-
ных функциональных групп, рассматриваемых в этой книге. Изложенный ниже
материал более подходит для тех студентов, которые изучали курс органиче-
ской химии хотя бы в течение одного семестра.
* Другие данные см. в работе; Rappoport Z., Accts. Chem. Res., 9, 265
(1976).
Определение функциональных групп и установление структуры 241
Можно было бы ожидать, что присутствие любой электроноакцепгорной
группы или атома у того углеродного атома, где будет происходить сольволиз,
приведет к понижению скорости реакции, поскольку наличие частичного поло-
жительного заряда на том месте, где должен появиться полный положительный
заряд, будет препятствовать этому процессу. Иллюстрацией такого предположе-
ния может служить стабильность в этой реакции р-алкоксигалогенидов, а-гало-
генкетопов (например, а-хлорацетофеиона), p-хлоралкилтолуолсульфонатов н
вицинальных дихлоридов.
<Х-хлорацетофенон
Однако следует заметить, что а-галогенкетоны, сложные эфиры, амиды и ни-
трилы весьма реакционноспособны по отношению к иодид-иону и при обработке
раствором иодида натрия в ацетоне при 25°С уже через 3 мин дают осадок га-
логенида натрия.
Nal
ацетон
Ускорение реакций замещения под влиянием карбонильной группы, располо-
женной в a-положении, можно объяснить стабилизацией переходного состояния
в результате частичного распределения отрицательного заряда на карбонильный
атом кислорода:
С6Н5 О*'
"ч'-Л -сг’
Скорость сольволиза увеличивается также и в присутствии групп
R2N — RS—, : I—, если они находятся в р-голожении к уходящему атому га-
логена. Эти группы оказывают внутримолекулярное содействие замещению га-
логена, как показано на схеме:
В тех случаях, когда рядом с реакционным центром присутствуют такие
группы, как бром или ацетоксильный радикал, скорость реакции сольволиза по
сравнению со скоростями реакций сольволиза незамещенных соединений умень-
шается незначительно. Это объясняется тем, что понижение скорости реакции
242
Глава 6
в результате индуктивного влияния (аналогично снижению скорости реакции,
вызываемому атомом хлора, находящимся по соседству с реакционным цент-
ром) компенсируется, хотя и не в полной мере, внутримолекулярным содейст-
вием сольволизу.
: Вг: СНзСОО 1 1 н 1 1
I 1 —С—С— и 1 I —с—с— < 1 1 : —с—с— 1 1
1 1 X X 1 1 X
Поскольку при внутримолекулярном замещении атома галогена X соседней
группой, т. е. в результате атаки с тыла по отношению к уходящей группе, долж-
но проходить вальденовское обращение, то заместители, находящиеся в цис-
положении в циклических системах, не способны компенсировать снижение ско-
рости, вызываемое индуктивным эффектом. Действительно, наличие цнс-ацетокси-
группы в положении 2 циклогексил-п-бромбензолсульфоната снижает скорость
его сольволиза в ледяной уксусной кислоте почти в 5000 раз (индуктивный эф-
фект). Тот же заместитель, но находящийся в трпнс-положении, замедляет реак-
цию только в 5 раз, поскольку его индуктивное влияние компенсируется тем,
что этот заместитель проявляет высокую активность при внутримолекулярном
содействии замещению и имеет удачное пространственное расположение для
гронс-атаки при этой реакции.
Другие галогенсодержащие соединения. Большая часть хлорангидридов и
бромангидридов кислот быстро реагирует с этанолом даже в отсутствие ни-
трата серебра. Поэтому со спиртовым раствором нитрата серебра немедленно
образуется осадок соответствующего галогенида. Так же быстро эти соедичения
реагируют с иодидом натрия в ацетоне; реакция проходит, вероятно, по меха-
низму присоединения к карбонильной группе.
О
II AgNO3
RCX ТТДДГ RCOOC2H5 + AgCl (тв.)
C2ti5<Jri j
о
Nal
ацетон
RCI + NaX (тв.)
а-Галогенэфиры быстро подвергаются сольволизу, что обусловлено образо-
ванием сравнительно устойчивого карбокатиона (его можно рассматривать как
эфир сопряженной кислоты кетона):
I
R-О—С-------> [R—О—С— R—О=С—]
Соединения такого типа часто очень быстро реагируют со спиртовым раствором
нитрата серебра, немедленно давая осадок соответствующего галогенида.
Накопление атомов хлора у одного атома углерода в алифатических соеди-
нениях снижает их способность реагировать с нитратом серебра. Примерами та-
ких веществ могут служить хлороформ, четыреххлористый углерод, 1,1,2,2-тетра-
хлорэтан и трихлоруксусная кислота. Однако это правило не распространяется
на бромпроизводные: четырехбромистый углерод реагирует с нитратом серебра
при 25°С, а бромоформ и тетрабромэтан дают осадок с кипящим раствором ни-
трата серебра в спирте.
В соединениях типа аллилгалогенидов накопление атомов хлора у одного
углеродного атома не только не приводит к уменьшению скорости, но даже не-
сколько увеличивает ее. Бензотрихлорид (а,а,а-трихлортолуол) гидролизуется
Определение функциональных ерупп и установление структуры
243
легче, чем бензальхлорид (а,а-дихлортолуол, или бензилиденхлорид), который в
свою очередь более активен, чем бензилхлорид:
СС13 >
СНС12 >
СН2С1
Простые полихлорированные соединения, в которых атомы хлора находятся
у одного атома углерода, например, такие, как хлороформ, четыреххлористый
углерод и трихлоруксусная кислота, не реагируют с иодидом натрия в ацетоне.
Однако бензальхлорид и бензотрихлорид вступают в эту реакцию, поскольку
могут давать стабильный бензильный катион:
Галогениды RX могут быть охарактеризованы в виде производ-
ных N-замещенных фталимидов:
По-видимому, для получения производных соединений, содержа-
щих один атом галогена *, более всего подходят соли тетрахлор-
фталимида, а для соединений, имеющих два атома галогена **, мо-
гут применяться соли незамещенного фталимида. Методы полу-
чения таких производных описаны в литературе.
На рис. 6.14 и 6.15 представлены типичные И К- и ПМР-спек-
тры алифатических галогенидов. Присутствие галогенов в молеку-
ле исследуемых соединений приводит к появлению в ПК-спектрах
полос умеренной интенсивности в следующих областях:
Связь Область поглощения валентных колебаний связи С—X (мкм). см~1
C—F C—Cl C—Br C—I 1250—960 (8,00-10,42) 830—500 (12,04—20,0) 667-290 (14,99-34,5) 500—200 (20,2—50,0)
Allen С. F. Н„ Adams IF. R., Myers С. L., Anal. Chem., 37, 158 (1965).
Allen C. F. H., Glauser 1. P., Anal Chem., 44, 1694 (1972).
to
a
05
a
Рис. 6.14. Инфракрасный спектр хлороформа в чистом виде, измеренный на приборе Perkin-Elmer 521, © Sadtler Research
Laboratories, Inc., Philadelphia, Pa., 1966.
СНС1з, мол. масса 119,39, т. кип. 61—62 °C, 1,4476, 1,484 (лит.), получен от фирмы United States Testing Со.. Inc., капиллярная
кювета. Валентные колебания С-Н: 7-3010 см-1 (3,32 мкм), vc_H (валентные колебания С—Н метиновой группы мало характеристичны).
Деформационные колебания С—Н: 2—1215см-1 (8,23 мкм), 6с—Н- Валентные колебания С— С1: 3 — 755 см-1 (13,25 мкм), Vc—cb
246
Глава 6
Ввиду того что многие из применяемых спектрографов не мо-
гут работать в области частот ниже 625 см-1, полосы валентных
колебаний связей С—Вг и С—I обычно не используются. Если в
молекуле имеется только одна связь галоген — углерод, то в
спектре наблюдается одна полоса поглощения. Возрастание числа
таких связей приводит к увеличению интенсивности и усложне-
нию спектров в этой области.
Химический сдвиг протона в ПМР-спектре алифатических га-
логенидов зависит от природы галогена и от типа разветвления
у того атома углерода, с которым этот протон связан:
Галоген б (ТМС-СО,00 м. д. а)
СНзХ rch2x r2chx R3CX
F 4,25 4,50 4,05 б
С1 3,05 3,45 4,05 б
Вг 2,70 3,40 4,10 б
I 2,15 3,15 4,25 б
8 В СС14; ± 0,05 м. ц.
6 Сигнал g-протона обычно имеет в 2,22 м. д. или сдвинут еще дальше в сильное поле.
Распознавание органических галогенидов по масс-спектрам
очень удобно проводить по числу и типу присутствующих ато-
мов галогенов. Эти вопросы обсуждались в гл. 4.
Все алифатические галогениды, кроме фторидов, могут реаги-
ровать с реактивами Гриньяра. Например, из циклогексилброми-
да можно получить циклогексилмагнийбромид при действии маг-
ния в сухом эфире (см. методику 12):
Mg
---------->
(СН3СН2)2О
Полученный реактив Гриньяра необходимо немедленно ввести в
реакцию с подходящим реагентом для получения производного,
которое можно было бы выделить и охарактеризовать.
Методика 12. Реактив Гриньяра и алкилртутьгалогениды
Mg
RX > RMgX
эфир
HgX2
RMgX ------> RHgX
Реактив Гриньяра получают, обрабатывая в чистой и сухой пробирке 0,3 г
магния в 15 мл сухого эфира 1 мл алкилгалогенида. Для ускорения начала реак-
ции прибавляют кристаллик иода. После окончания реакции раствор фильтруют
через небольшой комочек стеклянной ваты в пробирку, в которой находится 4—
Определение функциональных групп и установление структуры
247
5 г хлорида, бромида или иодида ртути (в зависимости от природы галогена
в исходном алкилгалогениде). Реакционную смесь при энергичном встряхивании
нагревают несколько минут на кипящей водяной бане и затем выпаривают до-
суха. Остаток кипятят с 20 мл 95%-ного этанола и фильтруют. Фильтрат раз-
бавляют 10 мл воды и охлаждают льдом. Выделившийся алкилртутьгалогенид
собирают на фильтре и перекристаллизовывают из 60%-ного этанола.
Методика 13. Получение анилидов, толуидидов и N-нафтиламидов
из алкилгалогенидов
RX + Mg
RMgX
Реактив Гриньяра получают, как списано в методике 12, и действуют на
него раствором 0,5 мл фенил-, n-толил- или а-нафтилизоцианата в 10 мл аб-
солютного эфира. Смесь встряхивают и оставляют на 10 мин. После этого при
сильном перемешивании прибавляют 25 мл 2%-ной соляной кислоты. Эфирный
слой отделяют и сушат сульфатом магния. Эфир отгоняют, а остаток перекри-
сталлизовывают из метанола, эфира или петролейного эфира.
Методика 14. Получение алкил-Р-нафтиловых эфиров
К раствору 0,6 г гидроксида натрия в 25 мл этанола прибавляют 2 г (5-на-
фтола и 2 г алкилгалогеннда. В случае хлорида добавляют еще 0,5 г иодида
калия. Смесь нагревают с обратным холодильником 30 мин, выливают в 75 мл
холодной воды, подщелачивают по фенолфталеину и сильно перемешивают. Ал-
кил-р-нафтиловый эфир отфильтровывают и перекристаллизовывают из этанола
или из смеси этанола и воды.
Эту методику иногда используют для получения производных
дигалогенидов типа Х(СН2)ПХ. Если соединение представляет со-
бой 1,2-дигалогенид (п = 2), добавлять иодид калия не следует.
Почему?
248
Глава 6
Методика 14а. Замещение хлорида в первичных и вторичных алкилхлоридах
на циаигруппу *
дмсо
R—Cl + NaCN -------> R—CN + NaCl
Получение нитрилов из первичных хлоридов. В трехгорлую колбу, снабжен-
ную мешалкой, обратным холодильником и термометром, помещают 40 мл су-
хого диметилсульфоксида (ДМСО) и 7,5 г сухого порошкообразного цианида
натрия * **. Полученную суспензию нагревают на водяной бане до 90°С, после чего
баню убирают и при хорошем перемешивании медленно прибавляют через верх
холодильника 12 г алкилхлорида (или 6 г дихлорида). Температура реакционной
смеси обычно поднимается до 150—160°С. После добавления всего количества
галогенида (что занимает около 10 мин) смесь перемешивают еще 10 мин и за-
тем охлаждают до комнатной температуры.
Если желательно получить мононитрил, смесь выливают в 100 мл холодной
воды и нитрил экстрагируют двумя порциями метиленхлорида объемом 25 мл
каждая. Экстракт трижды промывают порциями по 25 мл насыщенного рас-
твора хлорида натрия, сушат, добавив 3 г безводного хлорида кальция, и отго-
няют метиленхлорид на водяной бане. Маслянистый остаток нитрила можно пре-
вратить в амид (методика 51а) или в 2,4-динитрофенилгидразон альдегида (ме-
тодика 52а).
Если желаемым продуктом является динитрил, то в реакционную смесь пос-
ле охлаждения прибавляют 50 мл хлороформа и получившуюся смесь выливают
в 25 мл насыщенного раствора хлорида натрия. Добавляют около 50 мл воды,
слой хлороформа отделяют, промывают его тремя порциями насыщенного рас-
твора хлорида натрия объемом по 25 мл каждая и сушат, добавляя 3 г безвод-
ного хлорида кальция. Хлороформ отгоняют, а остаток маслообразного дини-
трила превращают либо в диамид, либо в динитрофенилгидразон (методика 51а
или 52а соответственно).
Получение нитрилов из вторичных хлоридов ***. Для получения нитрила сус-
пензию цианида натрия нагревают с помощью электрической колбоиагреватель-
ной рубашки до 90°С и при сильном перемешивании через верх холодильника
медленно прибавляют вторичный галогенид в течение 30 мин. Температуру по-
степенно повышают до 150°С и смесь перемешивают 3 ч. Реакционную массу ох-
лаждают и затем обрабатывают тем же способом, что описан выше для первич-
ных нитрилов.
Обсуждение. Алкилхлориды и алкилендихлориды обычно до-
статочно легкодоступны. Однако превращению этих хлоридов в
другие полезные продукты мешает малая скорость реакции заме-
щения атомов хлора на нуклеофильный заместитель (реакция
Sn2) в гидроксилсодержащих растворителях. Реакция идет с
низким выходом, требует много времени и осуществляется под
давлением в автоклавах. Использование в качестве растворителя
♦ Smiley R. A., Arnold С. Д., J. Org. Chem., 25, 257 (1960); для проведения
этой реакции достаточно брать одну десятую от указанного в статье количества
реагентов.
** Соли цианистой кислоты чрезвычайно ядовиты и поэтому очень опасны.
Нельзя смешивать их с кислотами. Уничтожение остатков этих солей проводится
строго по указаниям преподавателя.
*** Эта методика аналогична описанной для первичных хлоридов, за исклю-
чением тех случаев, которые указаны в этом разделе.
Определение функциональных групп и установление структуры 249
диметилсульфоксида позволяет провести такое замещение очень
быстро и с высоким выходом:
NaCN
RCH2C1 RCH2CN + NaCl
дмсо
NaCN
R2CHC1 - „—>• R2CHCN + NaCI
ДМ с о
2NaCN
C1CH2CH2C1 ———> NC—CH2CH»—CN + 2NaC!
дмсо
Выходы нитрилов составляют 70—90%. Поскольку полученные
нитрилы представляют собой при 25°С жидкости или легкоплав-
кие вещества, их необходимо далее перевести в твердые амиды
(методика 51а) или в арилгидразоны (методика 52а):
методика 14а методика 51а
NaCN а) ВГа-2СН3СООН
«, N.OH/H.O ' ' * RC0NH-
NI Ренея ArNHNH2
I—--------77-777* RCHO -------* RCH=NNHAr
75%-ная HCOOH
методика 52a
Диметилсульфоксид необходимо тщательно высушить *. При
атмосферном давлении ДМСО кипит при 189°С; при 2,6 кПа
(20 мм рт. ст.) его температура кипения равна 85—87°С, df 1,1014,
nD 1,477, дипольный момент 3,96 Д, диэлектрическая проницае-
мость 49.
Первичные и вторичные бромиды также реагируют с циани-
дом натрия в ДМСО с образованием нитрилов. Однако обычно
для сокращения затрат времени бромиды идентифицируют, пере-
водя их в соответствующие реактивы Гриньяра и далее в заме-
щенные уретаны (методики 12 и 13).
Будучи высокополярным растворителем, ДМСО способен рас-
творять соединения различных типов, что позволяет проводить
другие реакции Бм2-замещения. Имеются сообщения ** о том, что
в водном растворе ДМСО ускоряется щелочное омыление слож-
ных эфиров. Продажный препарат часто имеет характерный
запах. Следует избегать его попадания на кожу. Фармакологиче-
ская активность чистого ДМСО полностью не изучена.
* Этот растворитель очень гигроскопичен. Его можно высушить перегонкой
над гидридом кальция (т. кип. 64°C при давлении 530 Па, или 4 мм рт. ст.).
** Vison J. A., Hocker Е. К-, J. Chem. Educ., 46, 245 (1969).
250
Г лава 6
Методика 15. Получение пикратов S-алкилтиомочевии
H2N—С—nh2
II
S
RX
CjH6OH
В пробирке смешивают 0,5 г растертой в порошок тиомочевииы, 0,5 г алкил-
бромида или алкилиодида и 5 мл 95%-ного этанола и кипятят 2 мин. В другой
пробирке растворяют 0,4 г пикриновой кислоты в возможно малом количестве
кипящего этанола. Оба раствора смешивают и оставляют до охлаждения. Пи-
крат S-алкилтиомочевины отфильтровывают и перекристаллизовывают из эта-
нола.
Для проведения реакции с алкилхлоридами иногда оказывается необходимо
прибавить в первую смесь 1 г иодида калия. В этом случае необходимо доба-
вить такое количество спирта или воды, чтобы при кипячении получился про-
зрачный раствор. Последующая обработка проводится так же, как указано
выше.
Пасто и Джонсон приводят еще две методики для получения
производных алкилгалогенидов (с. 351 и 352 их книги соответ-
ственно):
Реактив Гриньяра:
Mg in со2
RX ----> RMgX -------* RCOOH
(2) Н3О+
Диоксид углерода можно вводить как в твердом виде (сухой
лед), так и в газообразном состоянии.
В книге Пасто и Джонсона на с. 353 обсуждаются также и
необычные свойства фторированных углеводородов. Наилучшим
способом идентификации этих соединений является спектромет-
рия flMP-l9F, конечно, если этот метод оказывается доступным *.
Однако даже при отсутствии соответствующих приборов опреде-
ление атома I9F можно провести с помощью протонного магнит-
ного резонанса, так как атом фтора и находящийся рядом с ним
протон очень сильно взаимодействуют между собой.
* Mooney Е. F., Ап Introduction to 19F NMR Spectroscopy, Heyden, London —
Sadtler, Philadelphia, 1970; Dungan С. H., van Wazer U. R. Compilation of l9F
NMR Chemical Shifts, Wiley-Interscience, New York, от 1951 г. до середины
1967 г.
I
Определение функциональных групп и установление структуры
251
6.7. АМИНЫ
Нуклеофильный атом азота в аминах (R3N;) благодаря на-
личию свободной пары электронов легко вступает в реакции с
электрофильными реагентами, такими, как кислоты Бренстеда,
Н4 или хлорангидриды кислот.
Опыт 18. Определение активного атома водорода с помощью хлор-
ангидрида кислоты
а) Осторожно прибавляют несколько капель ацетилхлорида к 1 мл воды
в пробирке. Пробирку держат в руке, чтобы почувствовать разогрев реакционной
смеси. Аналогично проводят реакцию с бензоилхлоридом. Какие различия на-
блюдаются в ходе этих реакций?
О
О
II
RC—С1 + Н2О —> RC—ОН + НС1
Хлорангидриды, как правило, обладают сильными слезоточивыми свой-
ствами, поэтому после окончания реакции их необходимо нейтрализовать разбав-
ленным раствором гидроксида аммония.
б) Оба опыта повторяют, взяв вместо воды 0,5 мл анилина. Какое соедине-
ние получается в результате этой реакции? Вылейте реакционную смесь в 5 мл
воды. Что выпадает в осадок и что остается в растворе?
в) Обрабатывают 0,5 мл пиридина или хинолина несколькими каплями аце-
тилхлорида. Объясните выделение тепла при этой реакции. Не противоречит ли
RC—С1 +
выделение тепла тому факту, что при разбавлении смеси водой и последующей
нейтрализации исходный амии выделяется обратно?
г) Прибавляют по каплям 1 мл ацетилхлорида к 1 мл этанола и к 0,5 г фе-
О
О
RC—Cl + R'OH —*• RC—OR' 4- НС1
нола. В обоих случаях реакционной смеси дают постоять 1—2 мии и затем
осторожно выливают в 5 мл воды Во втором опыте жидкость Сливают и про-
веряют растворимость остатка в холодном разбавленном растворе гидроксида
натрия.
252
Глава 6
д) Реакция Шопена — Баумана
О О
11 , II
RC—Cl + R'ОН + NaOH —> RC—OR' + NaCI + Н2О
О О
II II
RC—Cl + R'NH2 + NaOH —> RC—NHR' + NaCI + H2O
В небольшую склянку с притертой пробкой помещают 5 мл этанола, 10 мл
воды и 2 мл бензоилхлорида. К этой смеси прибавляют по частям при сильном
встряхивании 10 мл 20%-ного раствора гидроксида натрия. Смесь встряхивают
несколько минут, а затем раствор проверяют лакмусовой бумажкой, чтобы быть
уверенным, что он остается щелочным. Что образуется в результате этой реак-
ции? Повторите этот опыт, но вместо этанола возьмите 2 мл анилина. Каким
преимуществом обладает эта реакция по сравнению с реакцией бензоилирования
без добавления щелочи, как это описано в пункте «б»? От каких факторов за-
висит успех реакции Шоттеиа — Баумана?
Обсуждение. Нуклеофильные агенты, например спирты и ами-
ны, могут реагировать с электрофильной карбонильной группой
ацилхлорндов по механизму присоединения — отщепления:
о о* о
„ Н I в II .
R—С—X + RNH, > R—С—X R—CNHR'+X' + BH*
S+ । — Мл
+nh2
R' Ъ=основание
*O О' О
II I „ II
RC—X + R'OH R—С—X R—COR'+X' + BH"
Ci [ •—MX
r/O\H
Реакция Шоттена — Баумана представляет большой интерес,
так как присутствующие в реакционной смеси вода и гидроксиль-
ные ионы, как и следовало ожидать, конкурируют со спиртом и
амином в реакции ацилирования, заметно снижая выход. Успех
реакции зависит от правильного подбора условий ее проведения.
Реакция проходит в гетерогенной системе. При этом бензоилхло-
рид и органический реагент находятся в одной фазе. Можно пред-
полагать, что все органические реагенты взаимодействуют в ор-
ганической фазе, а в водной фазе проходит только реакция ней-
трализации кислот и оснований.
Опыт 19. Действие азотистой кислоты
а) Диазотирование. Растворяют 1 мл анилина в 3 мл концентрированной
соляной кислоты, разбавленной 5 мл воды, и охлаждают раствор до 0°С в ста-
кане с кусочками льда. Растворяют 1 г нитрита натрия в 5 мл воды и медленно
Определение функциональных групп и установление структуры
253
при встряхивании прибавляют этот раствор к холодному раствору гидрохлорида
анилина. Прибавление продолжают до тех пор, пока смесь не даст положитель-
ной реакции на азотистую кислоту. Для этого помещают каплю раствора иа
(ГС
<^2^/N2Cl + 2H2O
Н20 + нагревание
H + N2 + HC!
иодкрахмальную бумажку. Появление голубого окрашивания указывает на при-
сутствие азотистой кислоты. Помещают 2—3 мл раствора в другую пробирку и
осторожно нагревают, следя за выделением таза. Какие продукты образуются
в этой реакции? Как называется последняя реакция?
6) Реакция азосочетаиия. Прибавляют вторую порцию холодного диазоние-
вого раствора (около 2 мл) к раствору 0,1 г f-нафтола в 2 мл 10%-ного рас-
твора гидроксида натрия и 5 мл воды. Отмечают образование оранжево-крас-
ного диазокрасителя.
iNa + NaOH
оранжево-красный
азокраситель
в) Растворяют 2 мл метиланилина в 5 мл концентрированной соляной кис-
лоты, разбавляют, добавив 5 мл воды, и охлаждают льдом R2NH + HONO —>-
R2NN = О + Н2О. Медленно при встряхивании прибавляют раствор 1,5 г ни-
трита натрия в 5 мл воды. Наблюдают происходящие изменения *. Какой продукт
получается в этой реакции? Имеются ли какие-нибудь различия между вторич-
ными ароматическими и алифатическими аминами в этой реакции?
г) Проводят реакцию (в) с диметиланилином. Отмечают цвет и характер
образовавшегося продукта. Так же ли проходит эта реакция с третичными али-
фатическими аминами?
+ Н2О
д) Кристаллик нитрита натрия вносят в 2 мл концентрированной серной
кислоты и встряхивают до растворения. После этого прибавляют 0,1 г фенола
* Осторожно! Некоторые нитрозамины проявляют сильные канцерогенные
свойства. Следует избегать вдыхания паров этих соединений и попадания их
на кожу или одежду.
254
Глава 6
и отмечают изменение цвета Раствор выливают в 20 мл ледяной воды и снова
обращают внимание на окраску полученной смеси. Прибавляют раствор гидро-
ксида натрия до щелочной реакции и вновь наблюдают за изменением окраски.
красный синий (HSO,)
NaOH
синиц
Появление в этой реакции синего окрашивания обусловлено фенолиндофе-
полом, который образуется в результате взаимодействия получающегося на пер-
вой стадии реакции и-нитрозофенола (монооксима хинона) с избытком фенола.
Эта реакция, известная как «реакция нитрозирования Либермана», характерна
для фенолов, не замещенных в орто- и пара положениях. Ее можно использо-
вать для обнаружения иитрозогрупп. Для этого смешивают равные количества
нитрозосоедипеиия и фенола, смесь прибавляют к серной кислоте и проводят
дальнейшую обработку, как описано выше.
Обсуждение. Реакции первичных аминов. Как алифатические,
так и ароматические первичные амины реагируют с азотистой
кислотой, давая в качестве первоначального продукта соответ-
ствующие диазониевые ионы. Как можно было предвидеть, али-
фатические диазониевые соединения существенно менее стабиль-
ны, чем ароматические. Их существование не имеет прямых
подтверждений. При распаде они образуют азот, спирт, олефин и
другие продукты, а также соединения, получающиеся в резуль-
тате тех или иных реакций карбониевого иона. На схеме R'+
представляет собой карбониевый ион, образующийся в результате
1,2-перегруппировки.
ROH+олефин+ RC1 + ROR
R—Ni
RNHj bGNQ.
R'OH +мефин + R'Cl + R’OR
Ароматические соли диазония обычно устойчивы в растворах
при 0°С. При нагревании водных растворов они теряют азот, пре-
вращаясь в арильный катион Аг+, который быстро реагирует с
водой с образованием фенола.
Определение функциональных групп и установление структуры
255
Показано, что реакция азосочетания между солью диазония и
фенолом включает взаимодействие диазониевого иона с феноксид-
ионом *. Если раствор слишком кислый, то феноксид-ион превра-
щается в фенол и реакции не происходит. Если же раствор слиш-
ком щелочной, то ион диазония реагирует с ионом гидроксида,
давая диазотат ArN2O_, не способный вступать в реакцию азосо-
четания. Поэтому для того, чтобы успешно провести реакцию со-
четания, необходимо применять буферные растворы.
Диазотирование антраниловой кислоты приводит к образова-
нию цвиттер-иона или циклического ацилдиазотата. Это соедине-
ние неустойчиво и распадается с выделением азота и диоксида
углерода. При этом образуется высокореакционноспособное про-
межуточное соединение, так называемый «бензин», или «дегидро-
бензол» (А). Активный дегидробензол реагирует с этанолом, да-
вая фенетол, с диэтиловым эфиром малеиновой кислоты обра-
зуется замещенный бензоциклобутен и с антраценом—триптицен.
о
А
Наиболее высокие выходы получаются при проведении реакции
диазотирования с амилнитритом, а реакции замещения — в апро-
тонном растворителе типа метиленхлорида, тетрагидрофурана
или ацетонитрила **.
Реакции вторичных аминов. Алифатические и ароматические
вторичные амины реагируют с азотистой кислотой, образуя N-
нитрозосоединения, обычно называемые нитрозамидами ***. Они
представляют собой желтоватые масла или твердые соединения:
HONO
R2NH ----► R2N—NO
нитрозамин
Реакции третичных аминов. Химизм реакций третичных амийов
с азотистой кислотой крайне сложен ****. В некоторых условиях
* Ингольд К. Теоретические основы органической химии. Пер. с англ. —*
М.: Мир, 1973; см. также Frigero N. A., J. Chem. Educ., 43, 142 (1966).
** Будьте осторожны! о-Диазоиийбензоат (Б) в твердом состоянии взры-
вается. Не допускайте полного удаления растворителя.
*** Внимание! Многие из этих соединений канцерогенны, поэтому работать
с ними нужно с большой осторожностью.
**** Hein G. Е., J. Chem. Educ., 40, 181 (1963).
256
Г лава 6
они, по-видимому, не претерпевают никаких изменений, однако
это бывает только при низком pH, при невысоких температурах
и в достаточно разбавленных растворах. В таких мягких усло-
виях амины только протонируются, давая соли:
н* +
R3N: — * R3NH
основание
Образование таких солей легко доказать путем превращения их
в исходные амины действием щелочей.
При более высокой температуре и меньшей кислотности среды
третичные амины могут взаимодействовать с азотистой кислотой,
давая различные продукты. Характерны следующие реакции ами-
нов:
Алифатические амины:
R3N: + HONO —> R2N—NO + ROH
Ароматические амины:
C6H5NAr2+HONO ——> NAr.
Смешанные жирно-ароматические амины:
NO
C„H5NR2+HONO —- C6H5—N—R +ON—ZQ)\-nr2
Хотя азотистую кислоту чаще всего применяют для реакций
С аминами, ее можно использовать и для реакций с другими
функциональными группами, например можно превратить мети-
леновую группу, соседнюю с карбонильной группой, в оксимино-
группу *. Алкилмеркаптаны с азотистой кислотой дают красные
S-алкилтионитриты.
HONO
СН3СОСН2СН3 ------> СН3СО—С—СНз + Н2О
N—ОН
HONO
rch2sh--------> rch2sno + h2o
красный
Очень простую пробу на присутствие аминов можно прове-
сти капельным методом с водным раствором сульфата двухва-
лентной меди. Амины с этим раствором часто дают интенсивное
синее или сине-зеленое окрашивание из-за образования комплек-
са Cu(II):
Н2О
xRNH2 + C11SO4 ---> [Cu(NH2R)J2t + SOf
Semon IV'. L., Damrell V. R., Org. Syntheses, Coll. Vol. 11, 204 (1943).
Определение функциональных групп и установление структуры
257
Техника проведения этой реакции описана в книге Пасто и
Джонсона, с. 415.
ИК- и ПМР-Спектры типичных аминов приведены на рис. 6.16
и 6.17. В принципе (и очень часто на практике) на основании
ИК-спектров аминов можно сделать вывод о числе атомов водо-
рода в аминогруппе. Связь N—Н в аминогруппе, если протон не
участвует в водородной связи, поглощает в области 3550—3320
см-1 (2,82—3,01 мкм). Образование водородной связи сдвигает
эту полосу в область 3300—3000 см-1 (3,03—3,33 мкм). В пер-
вичных аминах из-за взаимодействия двух связей N—Н наблю-
даются две полосы поглощения:
ХН
—N симметричные валентные колебания N—Н, 3400 см-1 (2,94 мкм)
%
—N антисимметричные валентные колебания N—Н, 3490 см-1 (2,87 мкм)
Вторичные амины, имеющие только одну связь N—Н, дают в
спектре одну полосу поглощения. Чистые третичные амины (R3N)
не имеют колебаний, соответствующих связи N—Н.
К сожалению, иногда встречаются исключения из этих правил.
Так, если спектры первичных аминов измеряют в условиях, не
позволяющих получить необходимое разрешение, то наблюдают
только одну полосу поглощения валентных колебаний N—Н. Кро-
ме того, в некоторых условиях в ИК-спектре вторичных аминов
наблюдается несколько полос поглощения связи N—Н. Именно
поэтому при установлении структуры аминов для получения дей-
ствительно достоверных данных совершенно необходимо исполь-
зовать и спектральные, и химические методы.
ИК-спектры гидрохлоридов аминов имеют широкую полосу по-
глощения валентных колебаний N—Н в области 3175—2381 см-1
(3,15—4,20 мкм). В этой области возможно появление нескольких
полос.
ПМР-Спектры первичных и вторичных аминов, снятые в обыч-
ных условиях, имеют нерасщепленный широкий сигнал в области
60,5—3,0 м.д. (т9,5—7,0 м.д.). Размытость этого сигнала вы-
звана как быстрым обменом протонов N—Н-группы, так и квад-
рупольным моментом атома азота. При этом сигнал протона груп-
пы N—Н не расщепляется на протоне соседней СН-группы
(HN—СН). Если этот обмен затормозить путем тщательного
высушивания амина сплавом Na—К или превращением аминогруп-
пы в ацетамидогруппу (CH3CONH), то удается наблюдать спин-
спиновое взаимодействие во фрагменте НС—NH, измеряя ПМР-
спектры С—Н-протона (7 = ~5 Гц). Однако сигнал протона
9 Зак. 335
а
сь
а
Рис. 6.16. Инфракрасный спектр втор-бутиламина в чистом виде.
Валентные колебания N—Н: / — 3347 см-1 (2,99 мкм), NH,; 2—3285 см-1 (3,04 мкм), (1 и 2 перекрываются). Валентные
□(.НИМ л симм 4
колебания С—Н: 3 — 3000—2850 см-1 (3,33—3,51 мкм), V_„ , v„TI . Деформационные колебания N—Н: 4— 1590 см-1 (6,29 мкм), в.,„ (нож-
бпз Cnj NHz
ничиые). Деформационные колебания С—Н: 5— 1457 см-1 (6,86 мкм), (ножничные); 3—1375—1348 см-1 (7,27, 7,42 мкм),
L>H3 СНз’
^СНа* Азот: 7—1200—800 см-1 (8,33-12,50 мкм), валентные С—N и веерные N—Н (для чистого образца), в основном веерные N—Н.
Определение функциональных групп и установление структуры
260
Глава 6
связи N—Н остается уширенным вследствие наличия уже упоми-
навшегося квадрупольного момента атома азота.
В литературе имеются сообщения * о том, что идентификацию
аминов можно облегчить, снимая ПМР-спектры в трифторуксус-
ной кислоте. В этом растворителе амины полностью превращают-
ся в соли трифторуксусной кислоты (RNH3 OOCCF3). Мульти-
I 1 +
плетность сигнала На во фрагменте На—С—N — во многих слу-
чаях позволяет установить число протонов у атома азота. При
Рис. 6.18. Инфракрасный спектр л-толуиднна в чистом виде.
колебания N-Н (ассоциированная): 7—3257, 3145 см-1 (3,07. 3,18 мкм), V (рас-
1ЧП2
Валентные колебания С—Н: 2 — 2959 см-1 (3,38 мкм), v„ (ароматические):
С—н
(алифатические). Валентные колебания С=С: 4—1613 см-1
(ароматические). Деформационные колебания N—Н: 5—1587 см-1 (6,30 мкм).
(ножничные). Валентные колебания С=С: 6—1481, 1458 см —1 (6,75, 6,86 мкм), V
Валентные
щеплены).
3— 2849 см~' (3,51 мкм), V„ __
С—-Н
(6,20 мкм), V(
6N—Н iiTiiuitp иадсл 1 note KU.'icuan/iTi и---Iiui, *-х>>и vm u,uv niixm/,
ароматические). Веерные .колебания N—H: 7 — 870 см-1 (11,5 мкм). Деформационные колеба-
ния а=С—Н (неплоские): 8— 775 см —1 (12.9 мкм). Деформационные колебания С—С (иеплоскне):
9 — 690 см-1 (14,5 мкм).
обработке бензиламина СбН5СН2ПН2 трифторуксусной кислотой
в ПМР-спектре появляется сигнал протонов СН2-группы при
6 4,4 м. д. (т 5,6 м. д.), представляющий собой квадруплет (/ —
= 7 Гц). Поскольку протоны бензольного кольца не взаимодей-
ствуют с метиленовой группой, то эта мультиплетность обусловле-
на расщеплением на трех протонах, связанных с атомом азота в
образовавшейся соли СсН5СН2МНз.
Масс-спектры аминов могут быть очень полезны. Интенсив-
ность пика молекулярного иона в масс-спектрах ациклических
Anderson W. R., Jr., Silverstein R. M., Anal. Chem., 37, 1417 (1965).
Определение функциональных групп и установление структуры
261
алифатических аминов часто очень слаба, а в спектрах аромати-
ческих и циклических аминов — весьма велика.
Очень полезно знать величину m/е молекулярного иона ами-
нов и других соединений азота: как было показано в гл. 4,
азотсодержащие соединения с четной массой молекулярного иона
не могут иметь нечетное число атомов азота. Азотсодержащие
соединения с нечетной молекулярной массой должны содержать
нечетное число атомов азота.
Наиболее важным направлением расщепления аминов являет-
ся то, которое облегчается наличием атома азота:
-С- +
।
C=N +
Такая фрагментация дает перечисленные ниже ионы, сигналы
которых имеют достаточную интенсивность и могут служить для
установления структуры изучаемого соединения.
Фрагмент Масса (гп/е) Примечания
ch2nh; 30 Если пик интенсивный, то он соот- ветствует первичному амину, не имеющему разветвления в а-поло- жении
RCHNHJ 29+ R Образуется главным образом при от- щеплении самого большого заме- стителя R', находящегося в a-по- ложении; массы R и R' позволяют делать заключения о структуре изучаемого соединения
c2h4n+ 42 Соответствует циклическому амину
[М—HCN]+ 66 Соответствует анилиновым основа- ниям
[М—HCN—Н]+ 65 То же
[Mz—HCN]+ 66 + R Соответствует анилинам с замести- телем R в бензольном кольце
[М'—HCN—Н]+ 65+ R То же
Сопряжение свободной электронной пары в амине с аромати-
ческим кольцом анилина приводит к тому, что его основность
существенно меньше основности алифатических аминов (см. гл. 5)
Тем не менее основность анилина достаточно велика для того,
чтобы он растворялся в минеральных кислотах:
ArNH2 + НС1 —> ArNHjCr
Многие из описанных выше химических реакций, применяемых
для идентификации алифатических аминов, пригодны и для иден-
тификации анилина. Спектры ароматических аминов в той части,
262
Глава б
200 , । 100 , РГ«
10 987654321 Ом.Ъ.
Рис. 6.19. Спектр протонного магнитного резонанса п-хлоранилина (60 Мгц, развертка 600 Гц, растворитель CDCI3).
Определение функциональных групп и установление структуры
263
которая относится к самой аминогруппе, также очень схожи со
спектрами алифатических аминов. Однако наличие ароматиче-
ского кольца создает некоторые особые возможности идентифи-
кации с помощью спектров. Типичные И К- и ПМР-спектры анили-
нов приведены на рис. 6.18 и 6.19 соответственно. Практически все
описанные ниже методы получения производных аминов могут
быть использованы для получения производных анилинов.
Весьма полезны для идентификации анилинов ультрафиолето-
вые спектры. Наличие ароматического кольца приводит к появле-
нию интенсивных и довольно хорошо определяемых полос. На-
дежным подтверждением присутствия в молекуле аминогруппы,
связанной с ароматическим кольцом, может служить типичный
для таких соединений сдвиг в более длинноволновую область
(батохромный сдвиг):
Соединение \<акс нм (емакс)
полоса Е2 полоса В
С6Н5—NH2 (в воде) 230 (8600) 280 (1430)
С6Н5—NHJ (в водных растворах кис- лот) 203 (7500) 254 (160)
При получении производных первичных и вторичных аминов
чаще всего используют присутствие в них реакционноспособной
связи N—Н. Наиболее распространено в этом случае получение
бензолсульфонамидов. Приготовление таких производных являет-
ся промежуточным этапом при идентификации аминов по Хйнс-
бергу (методика 16). В тех случаях, когда получение бензолсуль-
фонамида оказывается почему-либо неудобным, можно восполь-
зоваться многочисленными другими производными, каждое из ко-
торых пригодно для идентификации определенного типа амина.
Можно рекомендовать получение n-толуолсульфонамида (мето-
дика 18а), /г-бромбензолсульфонамида (методика 186), л^-Нитро-
бензолсульфонамида (методика (186), метансульфонамида (ме-
тодика 186) и а-нафталинсульфонамнда (методика 186).
Для характеристики низкомолекулярных водорастворимых
аминов особенно подходят фенилуретаны, образующиеся при
взаимодействии аминов с фенилизоцианатами (методика 19):
RNH2 + C6H5NCS —> C6H5NHCSNHR
r2nh +C6H6NCS —> c6h5nhcsnr2
Сам фенилизоцианат не реагирует с водой, поэтому данную ре-
акцию можно проводить с разбавленными водными растворами
аминов.
Амиды уксусной (методика 17а), бензойной (методика 176)
или n-нитробензойной (методика 17в) кислот удобно получать,
264
Глава 6
обрабатывая соответствующие амины уксусным ангидридом, бен-
зоилхлорндом или п-нитробензоилхлоридом.
RNH2 + (СН3СО)2О -> RNHCOCH3 + СНзСООН
2RNH2 + С6Н5СОС1 -> RNHCOC6H5 + RNH3CI
2RNH2+O2n/(Q/COC1 -» O2n/Q\cONHR+RNH3C1
Для целей идентификации очень полезны ацетильные и бензоиль-
ные производные, поскольку их дают почти все первичные и вто-
ричные амины.
З-Нитрофталевый ангидрид реагирует с первичными и вторич-
ными аминами с образованием моноамида фталевой кислоты.
Это соединение, полученное из первичного амина, при нагрева-
нии до 145°С дегидратируется и превращается в алкил-3-нитро-
фталимпд. Производные вторичных аминов устойчивы к действию
высокой температуры.
no2
Как уже было упомянуто выше, получение таких производных
фталевой кислоты (методика 4) и хиральных соединений типа
оптически активных аминокислот позволяет прийти к соедине-
ниям, удобным с экспериментальной точки зрения. Эти вещества
можно выделять с помощью кислотно-основной экстракции, ис-
пользуя реакции карбоксильной группы. Применение названного
реагента позволяет отличить друг от друга первичные, вторичные
и третичные амины и дает возможность приготовить характерные
производные первичных и вторичных аминов.
Для характеристики различных аминов могут оказаться полез-
ными и другие производные, такие, как 2,4-динитробензилидено-
вые производные, азосоединения (опыт 19), соли п-толуолсуль-
фокислоты, молекулярные соединения с фенолом, соединения с
n-нитробензилгалогенидами и пикраты (методика 22).
Первичные и вторичные амины взаимодействуют с арилизо-
цианатами и ацилазидами, образуя замещенные производные кар-
бамида:
ArN=C=O + RNH2 —> ArNHCONHR
ArN=C=O + R2NH —» ArNHCONR2
Д rnh2
ArCON3----> ArN=C=O ----->- ArNHCONHR
Определение функциональных групп и установление структуры
265
Описаны также фенил-, а-нафтил-, р-нафтил-, лг-бромфенил-, п-
бромфенил-, n-хлорфенил-, льнитрофенил-, /г-нитрофенил- и 3,5-
динитрофенилкарбамиды.
При пропускании сухого хлористого водорода в эфирный или
бензольный раствор амина можно получить ряд гидрохлоридов
аминов. Некоторые из них имеют достаточно четкую температуру
плавления или разложения и могут служить в качестве произ-
водных изучаемых веществ.
Гидрохлориды аминоспиртов легче всего получаются при ней-
трализации растворов аминоспиртов в пропаноле-1 сухим хлори-
стым водородом.
HCI (газ)
RNH2 -------► RNHjCl’
Методика 16. Взаимодействие аминов с беизолсульфохлоридом
(метод Хинсберга для отличия первичных, вторичных и третичных аминов)
Ниже описана техника проведения качественной реакции.
Если необходимо получить большое количество производного, до-
статочное для выделения в чистом виде, следует брать реагентов
в 5—50 раз больше, чем указано ниже.
К 0,3 мл анилина в пробирке прибавляют 5 мл 10%-ного раствора гидро-
ксида натрия и 0,4 мл бензолсульфохлорида. Пробирку закрывают пробкой и
сильно встряхивают. Необходимо проверить, остается ли раствор щелочным. По-
сле того как прореагирует весь бензолсульфохлорид, раствор фильтруют нли
сливают с остатка А. Следует отметить, является ли остаток А твердым или жид-
ким веществом и легче он или тяжелее, чем раствор щелочи. Какие выводы мо-
жно сделать из этих наблюдений? Проверяют растворимость остатка А в воде
и в разбавленной соляной кислоте. Растворимость А в соляной кислоте свиде-
тельствует о том, что исходный продукт был третичным амином. Натриевые соли
некоторых сульфонамидов с высокой молекулярной массой могут оказаться не-
растворимыми в растворе щелочи *. Обычно эти соединения растворимы в воде.
Если в процессе реакции реакционная смесь нагреется, ее следует
охладить. Некоторые К,М-диалкиланилины образуют прн слишком сильном
* Бензолсульфонамиды, полученные из циклических соединений от цнклогек-
силамина до циклодециламинов и из некоторых аминов с высокой молекулярной
массой, нерастворимы в 10%-ном растворе гидроксида натрия. См. Fanta Р. Е.,
Wang С. S., J. Chem. Educ., 41, 280 (1964). Некоторые первичные амины могут
давать нерастворимые в щелочи дисульфонильные производные. Их можно гид-
ролизовать кипячением в течение 30 мин с 5%-ны.м раствором этилата натрия
в абсолютном этаноле.
266
Глава б
разогревании смеси пурпурное красящее вещество. Образования окрашенных
веществ можно избежать, если проводить реакцию при 15—20°С.
Прозрачный фильтрат подкисляют и для того, чтобы ускорить кристаллиза-
цию продукта Б, трут стеклянной палочкой стенки пробирки. Этот опыт повто-
ряют с метил- и диметиланилином вместо анилина. Некоторые вторичные амины
реагируют медленно, в этих случаях реакционную смесь иногда приходится по-
догревать.
Написав схемы реакций, покажите как, применяя описанные выше методики,
можно отличить друг от друга первичные, вторичные и третичные амины.
В тех случаях, когда метод Хинсберга применяется для разделения смеси
аминов, их необходимо выделить в чистом виде в форме индивидуальных ами-
нов. Бензолсульфонамиды могут быть прогидролизованы согласно следующим
уравнениям;
/QYso^nhr+НгО+НС1 -» +RNH3C1
NRj+HjO+HCl-»
Сульфонамиды гидролизуются при нагревании в колбе с обратным холо-
дильником 10 г вещества со 100 мл 25%-ного раствора соляной кислоты. Для
гидролиза сульфонамидов первичных аминов требуется кипячение в течение
24—36 г, а для сульфонамидов вторичных аминов достаточно 10—12 ч. После
полного растворения смесь охлаждают, добавляют 20%-ный раствор гидроксида
натрия до щелочной реакции и экстрагируют 3 раза эфиром порциями по 50 мл.
Эфирный раствор высушивают и после удаления эфира амин перегоняют. Если
амины кипят при очень низких или очень высоких температурах, то в ряде слу-
чаев удобнее получать их в форме гидрохлоридов, пропуская сухой хлористый
водород в сухой эфирный раствор амина.
Многие сульфонамиды гидролизуются с большим трудом. В методике, даю-
щей наилучшие результаты, используют 48%-ную бромпстоводородную кислоту
и фенол *. Эта реакция не является простым гидролизом. Она включает восста-
новительное расщепление, при котором бромистый водород окисляется до брома,
а сульфонамид восстанавливается в дисульфид. Основным назначением фенола
является связывание брома с образованием л-бромфенола.
2ArSO2NR2 + 5НВг + 5С6Н6ОН —> ArSSAr + 2R2NH + 5BrCcH4OH + 4Н2О
Обсуждение. Арилсульфохлориды могут быть полезны для ха-
рактеристики первичных и вторичных аминов. Для разделения
аминов по способу Хинсберга используют различную раствори-
мость сульфамидов первичных и вторичных аминов в щелочах:
первые растворимы в них, вторые — нет. Ввиду того что третич-
ные амины не образуют сульфонамидов, этот метод позволяет
идентифицировать и разделять все три типа аминов. Однако при
определении аминов нельзя полагаться только на методику Хинс-
берга. Следует также учитывать растворимость исследуемого со-
единения. Если это вещество является амфотерным, т. е. раство-
ряется и в кислотах и в щелочах, то метод Хинсберга для
его идентификации непригоден. Например, n-N-метиламинобензой-
ная кислота реагирует с бензолсульфохлоридом и со щелочью,
* Snyder Н. Р., Heckert R. Е., J. Am. Chem. Soc., 74, 2006 (1952); Sny-
der H. R., Geller H. C., ibid. 74, 4864 (1952),
Определение функциональных групп и установление структуры 267
давая раствор натриевой соли N-бензолсульфонильного произ-
водного:
СООН COONa
+ C6H5SO2C1 + 2NaOH —> + NaCl + 2H2O
I I
CH3NH CH3NSO2C6H5
При подкислении этого раствора образуется осадок свободной
кислоты. Уже сам по себе этот факт указывает на то, что исход-
ное соединение было не первичным, а вторичным амином.
Методика 17. Получение замещенных амидов из аминов
a) RNH2 (или R2NH) НС1, NaOH (СН3СО)2О ” RNHCOCH3 (или R2NCOCH3) с. Hgx-iV/ Vz in a
б) RNH2 (или R2NH) CeH6COCl C6H5CONHR (или C6H5CONR2) C5H5N, CgHe
в) RNH2 (или R2NH) ArCOCl s- ArCONHR (или ArCONR2) основание Ar = C6H5, w-O2NC6H4
а) Получение замещенных ацетамидов из аминов, нерастворимых в воде.
Готовят раствор 0,5 г амина в 25 мл 5%-ной соляной кислоты. Небольшими пор-
циями прибавляют 5%-ный раствор гидроксида натрия до помутнения и затем
2—3 мл 5%-ной соляной кислоты до исчезновения мути. Добавляют несколько
кусочков льда и затем 5 мл уксусного ангидрида. Смесь энергично перемеши-
вают палочкой или вращением сосуда и немедленно добавляют заранее приго-
товленный раствор 5 г ацетата натрия (тригидрата) в 5 мл воды. Если продукт
не кристаллизуется сразу, то смесь оставляют на ночь на холоду.
Перекристаллизацию полученного ацетамида проводят из циклогексана или
из смеси циклогексана и бензола. После перекристаллизации из этих раствори-
телей амид необходимо тщательно высушить. Можно проводить перекристалли-
зацию также и из водно-спиртовой смеси.
6) Получение замещенных бензамидов. Пиридиновый метод. К раствору
0,5 г вещества в 5 мл сухого пиридина и 10 мл сухого бензола прибавляют по
каплям 0,5 мл бензоилхлорида. Полученную смесь нагревают 30 мин на водяной
бане при 60—70°С и затем выливают в 100 мл воды. Бензольный слой отделяют,
а водный промывают один раз 10 мл бензола. Объединенный бензольный раст-
вор промывают водой, затем 5%-ным раствором соды н высушивают неболь-
шим количеством сульфата магния. Осушитель удаляют фильтрованием через
складчатый фильтр и сухой бензольный раствор упаривают до объема 3—4 мл.
При перемешивании к смеси прибавляют около 20 мл гексана, выпавшее кри-
сталлическое бензоильное производное отфильтровывают и промывают гекса-
ном. Перекристаллизацию проводят обычно из смеси гексана и циклогексана или
из смеси циклогексана и этилацетата. Многие соединения можно кристаллизо-
вать из этанола или из его смеси с водой.
в) Получение замещенных бензамидов и п-нитробензамидов. Для получения
этих соединений можно применять реакцию Шоттена — Баумана в ее обычной
форме, описанной в методе 18д. Ниже приведено описание двух модификаций
этой реакции.
268
Глава 6
1. Смесь 20 мл 5%-ного раствора гидроксида натрия, 5 мл хлороформа,
0,5 г исследуемого вещества и 0,5 мл бензоилхлорида перемешивают или встря-
хивают около 20 мин и оставляют на 12 ч. Хлороформный слой отделяют, а вод-
ный слой промывают еще 10 мл хлороформа. Объединенные хлороформные
растворы промывают водой, высушивают безводным сульфатом магния и упари-
вают до объема 2—3 мл. К полученному раствору при размешивании прибав-
ляют 20—25 мл гексана, выпавшее производное отфильтровывают и промывают
гексаном.
2. К раствору 1 г бензоилхлорида или л-нитробензоилхлорида в 20 мл су-
хого бензола прибавляют около 1 мл амина. Полученную смесь кипятят около
15 мин с обратным холодильником и затем дают охладиться. Раствор фильтруют
и осадок промывают 10 мл теплого бензола. Этот бензольный раствор объеди-
няют с основным фильтратом, промывают 10 мл 2%-ного раствора карбоната
натрия, затем 10 мл 2%-ного раствора соляной кислоты и, наконец, 10 мл ди-
стиллированной воды. Бензол испаряют, а остаток перекристаллизовывают из
разбавленного этанола.
Методика 18. Получение сульфонамидов из аминов
R2NH (или RNH2)
ArSOsCI
-------------->
(ПЛИ CH3SO2CI)
о
R2N—S—Аг
I
О
о
или RNH—S-—Аг
I
О
а) Бензолсульфонамиды и п-толуолсульфонамиды. Эти производные полу-
чают по способу, описанному в методике 17; при этом берут такие количества
исходных веществ, которые достаточны для того, чтобы провести дополнитель-
ную перекристаллизацию из 95%-ного этанола.
б) Бензил-, n-бромбензол-, лг-нитробензол-, а-нафтил- и метаисульфонамиды.
Готовят раствор 1 г сульфохлорида в 25 мл сухого бензола и прибавляют к
нему 2 мл амина. Раствор встряхивают и оставляют на 10 мин. Гидрохлорид
амина отфильтровывают и фильтрат упаривают. Остаток перекристаллизовы-
вают один или два раза из разбавленного этанола.
Методика 19. Получение фенилтиокарбамидов из аминов
S S
Л 11
С—CNR-
Равные количества амина и изотиоцианата смешивают в пробирке и смесь
встряхивают 2 мин. Если реакция не начинается самопроизвольно, то смесь на-
гревают на небольшом пламени. Алифатические амины обычно реагируют не-
медленно, а ароматические амины требуют предварительного нагревания. Затем
смесь охлаждают в стакане со льдом, пока она не затвердеет. Затвердевшее
вещество растирают в порошок и промывают лигроином и 50%-ным этанолом
для удаления не вошедших в реакцию реагентов. Остаток перекристаллизовы-
вают из 95%-ного этанола.
Описаны и другие реакции, в которых участвует связь N—Н
в первичных и вторичных аминах. Для идентификации NH2-rpyn-
пы, находящейся в вицинальном положении к гидроксильной
/НО NH2\
группе I —С—С— I, можно использовать реакцию с иодной
Определение функциональных групп и установление структуры 269
кислотой (опыт 5). Этот реагент можно также использовать и
для расщепления таких аминоспиртов с целью получения других
производных.
Обработка первичных и вторичных аминов 2,4-динитробензол-
сульфенилхлоридом приводит к получению соответствующих арен-
сульфенамидов. Подробное описание соответствующей методики
приведено в книге Пасто и Джонсона, с. 421.
Ar—SNR2 + R2NH2cr
аренсульфенамид
В некоторых случаях производные тиокарбамида претерпевают
сложную обратимую реакцию диспропорционирования с исход-
ным амином:
R S
II II
C6H5NHCNHR + RNH2 RNHCNHR + C6H5NH2
S S
II II
C6HeNHCNHR + C6H5NH2 C6H5NHCNHC6H5 + RNH2
Этих осложнений можно избежать, если не допускать чрезмерно
длительного нагревания смеси.
Третичные амины. Третичные амины столь сильно различаются
между собой по своей природе, что для них не удалось найти
какой-либо общий тип производных. По-видимому, наиболее при-
годными производными этих веществ являются четвертичные ам-
мониевые соли, образующиеся при взаимодействии амина с метил-
иодидом (методика 20а), бензилхлоридом (методика 206) или
метил-д-толуолсульфонатом (методика 206).
RSN + CHSI —> (R3NCH3)+r
R3N + C6H5CH2C1 —-> (R3NCH2C6H5)+Cr
R3N + CH3C6H4SO3CH3 —> (R3NCH3)+(CH3C6H4SO3)’
Часто в качестве производных третичных аминов применяют
соли галогеноводородных кислот, пикриновой кислоты (методика
22), п-толуолсульфокислоты и платинохлористоводородной кисло-
ты. По той же методике, что и пикраты (методика 22), можно по
лучать пикролонаты — соли 3-метил-4-нитро-1-п-нитрофенил-5-
пиразолона. Пикролонаты — наиболее обычные производные тре-
тичных аминов, обладающие приемлемыми для идентификации
свойствами. Эти продукты быстро образуются и в тех случаях,
когда получение других производных оказывается затрудни-
тельным. Используются также рейнекаты, соли кислоты
H+[Cr(NH3)2(SCN)4]~.
270
Глава 6
Некоторые N.N-диалкиланилины реагируют с азотистой кис-
лотой, давая n-нитрозосоединения, которые в некоторых случаях
могут служить в качестве производных.
О+Н2О
Методика 20. Получение четвертичных аммониевых солей третичных аминов
(a) R3N: +СН31 —* R3N—СН3 Г
а) Смесь 0,5 г амина и 0,5 мл метилиодида нагревают в пробирке на не-
большом пламени в течение нескольких минут и затем охлаждают во льду.
Чтобы вызвать кристаллизацию, необходимо потереть стеклянной палочкой по
Стенкам пробирки. Продукт реакции очищают перекристаллизацией из абсолют-
ного 31 анола или метанола либо из этилацетата.
б) К раствору 2—3 г бензилхлорида или метил-л-толуолсульфоната в 10 мл
сухого бензола * прибавляют 1 г амина. Раствор кипятят 10—20 мин и охлаж-
дают. Полученные вещества перекристаллизовывают, растворяя их в возможно
малом количестве кипящего этанола, затем прибавляют этилацетат до тех пор,
пока не начнется выпадение кристаллов, после чего смесь охлаждают. Продукт
отфильтровывают и быстро высушивают, растирая на пористой керамической
пластинке, после чего немедленно определяют температуру плавления.
Методика 21. Получение нитрозосоединений из N, N-диалкилаиилинов
Растворяют 2 г амича в 20 мл 10%-ной соляной кислоты и раствор охлаж-
дают в охлаждающей смеси. Медленно при сильном перемешивании приливают
раствор 1,5 г нитрита натрия в 2 мл веды. Смесь оставляют на 15 мин во льду,
а затем отделяют фильтрованием гидрохлорид нитрозосоединения. Желтые кри-
сталлы смешивают с 3 мл воды и помещают в лед. Затем прибавляют разбав-
ленный раствор гидроксида натрия до щелочной реакции. Зеленое нитрозосоеди-
нение экстрагируют эфиром. После испарения эфира это вещество получают в
виде блестящих зеленых кристаллов, которые отфильтровывают и высушивают
на пористой пластинке.
* Вместо бензола можно использовать толуол, этанол, ацетон пли тетра-
хлорэтан. Токсичность бензола, аминов и галогенидов обсуждается в приложе-
нии 4.
Определение функциональных групп и установление структуры
271
Методика 22. Получение пикратов из аминов
и из ароматических углеводородов
АгН
а) Образец изучаемого соединения (0,3—0,5 г) прибавляют к 10 мл 95%-
ного этанола. Если все вещество не растворится, то смесь встряхивают до обра-
зования насыщенного раствора, после чего фильтруют. Фильтрат приливают к
10 мл насыщенного раствора пикриновой кислоты в 95%-ном этаноле и смесь
нагревают до кипения. Раствор медленно охлаждают, выпавшие желтые кри-
сталлы пикрата отфильтровывают и перекристаллизовывают из этанола. Некото-
рые пикраты (особенно пикраты углеводородов, приведенные в приложении 3
в таблице) диссоциируют при нагревании, и поэтому их нельзя перекристаллизо-
вать. В таких случаях выпавший осадок промывают очень небольшим количест-
вом эфира и высушивают на пористой керамической пластинке, после чего опре-
деляют температуру плавления. При работе с пикратами необходимо проявлять
большую осторожность, так как некоторые из них при нагревании взрываются.
б) Равные количества исследуемого вещества и пикриновой кислоты сме-
шивают в пробирке и смесь нагревают 10 мин на кипящей водяной бане или на
очень маленьком пламени до тех пор, пока смесь не расплавится Сплав охлаж-
дают и, если это вещество достаточно устойчиво, перекристаллизовывают его
из этанола.
Пикриновая кислота является не только сильной кислотой, но
и активным окислителем. При взаимодействии пикриновой кис-
лоты с некоторыми ароматическими соединениями (обычно бо-
гатыми л-электронами) происходит образование аддуктов — да-
тивных комплексов (л — л-комплексов). Такие аддукты могут быть
использованы для установления молекулярной массы неизве-
стных соединений, как описано ниже. В методике 49 (разд. 6.14)
описан способ получения пикратов ароматических эфиров.
Потенциально очень ценным применением пикратных ком-
плексов с аминами является использование их для микроопреде-
ления молекулярной массы неизвестных соединений *. Эти пикраты
имеют максимум поглощения в ультрафиолетовом свете при-
близительно около 350 нм. Молекулярная масса комплекса мо-
жет быть установлена с точностью ±2 после определения вели-
чины абсорбции комплекса (1g 1^/1) при 350 нм по уравнению
13,440 С -п
мол. масса = —;—
__________ 1g (/о//)
* Физер Л., Физер М. Реагенты для органического синтеза, Т. 1. Пер.
с англ. — М.: Мир, 1970, Cunnigham К,. G., Dawson W., Spring F. S., J. Chem.
Soc., 1951, 2305.
272
Глава 6
где С — концентрация комплекса (г/л), п—мольное соотношение
пикриновой кислоты и амина или ароматического углеводорода в
комплексе. Используя значение молекулярной массы пикриновой
кислоты (229), можно рассчитать молекулярную массу амина
или ароматического углеводорода.
Методика 23. Получение хлорплатииатов аминов
НС1 +
R3N • + H2PtCl6 • 6Н2О ——> R3NH HPtClj
Г12О
К раствору 0,5 г амина в 10 мл 10%-ной соляной кислоты медленно при
встряхивании прибавляют 10 мл 25%-ного водного раствора платннохлористо-
водородной кислоты (Н2Р1С1б-6Н2О). Кристаллический хлороплатинат отделяют
на фильтре и промывают 10%-ной соляной кислотой. Полученное соединение
можно перекристаллизовать из этанола, к которому добавлена капля концентри-
рованной соляной кислоты для предотвращения гидролиза. Следует избегать
соприкосновения хлороплатинатов с металлическими шпателями, так как это
приводит к выделению металлической платины.
Ниже описан другой метод получения производных аминов —
получение тетрафенилборатных солей:
+ NaB(C6H6)4 ♦
RNH2 + HC1 —> RNH3Cr ------------------> RNH3B(C6H5);
R4N+X* + NaB(C0Hs)4 —> R4N+B(C0H5)4-
Этот метод, применимый не только к аминам, но и к четвертич-
ным аммонийным солям, описан в книге Пасто и Джонсона,
с. 424.
Ниже приведены некоторые дополнительные тесты на амины,
приведенные в книге Черониса, Энтрикина и Ходнетта (см. гл. 10):
Реактив Страница в книге Черониса и др.
Флуоресцеинхлорид 379
Определение основности * 380
Хингидрон 380
N - Галогенсукцинимид 380
3,3,5,5-Тетр абромфенолфталеин 381
Лигнин 384
Хлоранил 385
2,4-Динитрофторбензол 385
* Определение основности позволяет идентифицировать амины по величине нх рКь«
Как уже было отмечено в гл. 5, основность аминов чрезвы-
чайно сильно связана с их структурой (сравните, например,
основность анилина и алифатических аминов). Основность ами-
нов часто является причиной того, что эти соединения оказывают-
Определение функциональных групп и установление структуры
273
ся чувствительными к действию лантаноидных сдвигающих ре-
агентов (гл. 8). Это может оказать помощь при расшифровке
ПМР-спектров.
6.8. АМИНОКИСЛОТЫ
Предварительные сведения о химии аминокислот можно по-
лучить, изучая другие разделы этой главы, где описаны реак-
ции аминов (разд. 6.7) и карбоновых кислот (разд. 6.12). Одна-
ко необходимо помнить, что возможность образования цвиттер-
иона— полярной формы аминокислоты сильнейшим образом
влияет на химические и физические свойства этих соединений.
/СООН /СОСУ
R—С1Д 4=h r—ст/
У\’Н2 '4NH|
цвиттер-ион
В отличие от других органических соединений аминокислоты,
способные образовывать цвиттер-ионы, характеризуются отсут-
ствием четкой температуры плавления, которая обычно является
температурой разложения, и хорошей растворимостью большей
части аминокислот в воде.
Для определения аминокислот существуют разнообразные хи-
мические реакции, которые специфичны для некоторых из них
(табл. 6.2). Эти реакции используются довольно редко, так как
аминокислотный анализатор позволяет легко проводить каче-
ственный и количественный анализ и инструментальное детекти-
рование этих соединений. Этим методом плохо определяется
триптофан (Try) из-за его повышенной чувствительности к кис-
лотам, поэтому его лучше идентифицировать с помощью химиче-
ских реакций (табл. 6.2). Химической основой работы аминокис-
лотного анализатора является реакция с нингидрином, описанная
в опыте 20.
На рис. 6.20 и 6.21 приведены характерные ИК- и ПМР-спек-
тры аминокислот. Особенно важно рассмотреть способы приго-
товления образцов для снятия спектров, поскольку растворимость
аминокислот весьма отличается от растворимости других органи-
ческих соединений.
В тех случаях, когда в молекуле аминокислоты нет аромати-
ческого заместителя, сигнал молекулярного нона в масс-спектре
или невелик, или вообще отсутствует. Это чаще всего бывает при
использовании стандартного ионизационного потенциала, так как
в этих условиях расщепление аминокислоты проходит очень лег-
ко. Кроме того, масс-спектральный анализ аминокислот ограни-
чен низкой летучестью цвиттер-ионных соединений. Часто можно
вводить образец в спектрометр в газообразной форме, проводя
осторожное вакуумное сублимирование при температурах от 15Q
Рис. 6.20. Инфракрасный спектр глицина (таблетки с КВг).
Колебания N—Н и карбоксилатной группы: 7 — 3250—2300 см—| (3,08—4,35 мкм), группы — NH* (валентные колебания С—Н пере-
крываются с /): 2—1608 см-1 (6,22 мкм), асимметричные деформационные колебания N-Н группы —NHt; 3—1570 см-1 (6,37 мкм),
— 1 J
v С(—О)'; 4-1530 см (6,54 мкм), С> . деформационные колебания N—Н группы —NHt; 5—1403 см-1 (7,13 мкм)
ЗСНММ 4 СНММ О
тенсивный сигнал воды, образовавшейся в результате обмена D2O с протонами группы —NH3 аланина.
ю
сл
276
Глава б
Таблица 6.2
Химические реакции для идентификации аминокислот •
Аминокислота Реакция Реактив Окраска
Аргинин Цистеин Гистидин, ти- розин Триптофан Тирозин Тирозин, трип- тофан, фе- нилаланин Реакция Сакагучи Реакция с нитропрус- сидом Реакция Салливана Реакция Паули Реакция Эрлиха Реакция с глиоксило- вой кислотой (реак- ция Гопкинса —Ко- ле) Реакция Фолина — Чиокальти Реакция Миллона Ксантопротеиновая ре- акция а-Нафтол и гипохло- рит натрия Нитропруссид натрия в разбавленном рас- творе КНз 1,2-Нафтохипон-4-суль- фонат натрия и гид- росульфит натрия Диазотированная суль- фаниловая кислота в щелочном растворе н- Диметиламинобенз- альдегид в конц. НС1 Глиоксиловая кислота В КОНЦ. H2SO4 Фосфорном оли бден- вольфрамовая кисло- та HgNOs в азотной кис- лоте со следами азо- тистой кислоты Кипящая конц. HNOs Красная » » » Синяя Пурпурная Синяя Красная Желтая
а Ряд экспериментальных методик для такого рода реакций описан в книге: Гринштейн
Дж., Виниц М. Химия аминокислот и пептидов. Пер. с англ.—М:, Мир. 1965.
до 240 °C. Сублимации можно подвергать непосредственно про-
дукт реакции, поскольку сами сублиматы достаточно чисты. Одна-
ко часто ограничиваются прямым вводом твердого образца в
спектрометр. Летучесть и соответственно удобство в обращении
образцов часто можно очень сильно увеличить, превратив амино-
4- _
кислоты в соли галогеноводородных кислот NH3X~,
R—СНСООН
ацетамиды, трифторацетамиды или в различные эфиры по карбо-
ксильной группе. Можно использовать также триметилсилильные
производные*. Натриевые соли сами по себе неспособны к ис-
парению.
* Митрука Б. М. Применение газовой хроматографии в микробиологии и
медицине. Пер. с англ. — М.: Медицина, 1978.
Определение функциональных групп и установление структуры 277
Все аминокислоты можно разделить на четыре большие груп-
пы. Это деление основано на их кислотно-основных свойствах и
на характере распределения зарядов.
1. Гидрофобные. Аминокислоты, которые существенно менее
растворимы в воде, чем вещества класса 2. Примером таких ве-
ществ может служить фенилаланин
С6Н5СН2СН^
ХЗОО"
Он относится по растворимости (гл. 5) к классу А2(В) и прак-
тически нерастворим в воде.
2. Гидрофильные (полярные незаряженные). Эти аминокисло-
ты имеют полярную функциональную группу ОН, в результате
чего, независимо от наличия положительного или отрицательного
заряда, эти кислоты довольно хорошо растворимы в воде. При-
мером соединений этой группы служит треонин
/NHJ
снзсн—а/
I \соо'
он
Эти соединения отличаются от соединений следующих групп тем,
что присутствующая в них полярная группа не обладает замет-
ной кислотностью или основностью (т. е. способностью принимать
или отдавать протон).
3. Положительно заряженные (основные). Аминокислоты это-
го типа содержат основную, обычно азотную функцию, которая
может протонироваться при значениях pH, характерных для жи-
вой клетки (внутриклеточные pH 6,0—7,0). Типичным представи-
телем веществ этой группы является лизин, содержащий е-амино-
группу, которая может протонироваться в этих условиях.
• » у в
h2n—сн2сн2сн2сн2—сн
""сосу
4. Отрицательно заряженные (кислые). Эти аминокислоты
имеют кислую, т. е. способную к ионизации при внутриклеточ-
ных значениях pH карбоксильную группу. Например, р-карбо-
ксильная группа аспарагиновой кислоты
СОО’
НООС-СН,СН
NH*
в этих условиях превращается в карбоксилатную.
278
Глава 6
Температуры плавления или разложения аминокислот не оп-
ределяются точно. Их значения зависят от скорости нагревания.
Поэтому при использовании этих констант для составления спи-
ска возможных соединений следует делать поправку на их не-
точность.
а-Аминокислоты (кроме глицина), встречающиеся в природе
в растениях и животных или полученные при кислотном или фер-
ментативном гидролизе белков и пептидов, опитически активны
и имеют L-конфигурации. Величины удельного вращения могут
быть использованы в качестве констант при идентификации ами-
нокислот. Список их приведен в табл. 6.3. Величины удельного
Таблица 6.3
Удельное вращение ь-а-аминокислота
Ь-Амнпокислота Раствори- тель Концент- рация, г/100 мл Темпе- ратура. °C («ID. град R/S-Конфи- гурация
Аланин 1,0 н. НС1 5,8 15 +14,7 2(S)
Аргинин 6,0 в. НС1 1,6 23 +26,9 2(S)
Аспарагиновая То же 2,0 24 +24,6 2(S)
Цистин 1,0 н. НС1 1,0 24 —214,4 2 (S),2' (S)
Глутаминогая 6,0 н. НС1 1,0 22 +31,2 2(S)
Гистидин То же 1,5 22 + 13,0 2(S)
Оксипролин 1,0 н. НС1 1,3 20 —47,3 2(S),4(fl)
Аллооксипролин Вода 2,6 18 -58,1 2 (S),4 (S)
Изолейции 6,0 н. НС1 5,1 20 +40,6 2(S),3 (S)
Аллоизолейцин То же 3,9 20 +38,1 2 (S),3 (R)
Лейцин 2,0 26 + 15,1 2(S)
Лизин » 2,0 23 +25,9 2(S)
Метионин 0,2 н. НС1 0,8 25 +21,2 2(S)
Фенилаланин 5,4 н. НС1 3,8 20 —7,1 2(S)
Пролин 0,5 н. НС1 0,6 20 -52,6 2(S)
Серин 1,0 н. НС1 9,3 25 + 14,4 2(S)
Треонин Вода 1,3 26 +28,4 2 (S),3 (R)
Аллотреонин 1,6 26 +9,6 2 (S),3 (S)
Триптофан 1,0 22 -31,5 2(S)
Тирозин 6,3 н. НС1 4,4 20 -8,6 2(S)
Валин 6,0 н. НС1 3,4 20 +28,8 2(S)
а Основная литература о свойствах аминокислот приведена в справочнике: Merck Index,
9th ed.( Merck and Co., Rahway, N. J.. 1976.
вращения в других растворителях и ссылки на литературу мож-
но найти в недавно опубликованном справочнике: Tables for Iden-
tification of Organic Compounds, Chemical Rubber Co., Cleveland,
Ohio.
Определение функциональных групп и установление структуры
279
Опыт 20. Реакция с нингидрином
К раствору 0,2 г нингидрина (моногидрат индантриона-1,2,3) в 50 мл воды
прибавляют 2 мг а-аминокислоты. Смесь нагревают до кипения 15—30 с. Поло-
жительной пробой считается появление снне-фиолетового окрашивания, которое
дают а- и р-аминокислоты. Появление иной окраски (желтой, оранжевой, крас-
ной) означает, что проба отрицательна.
Обсуждение. Важность этой реакции определяется не только
тем, что она представляет собой качественную пробу, но и тем,
что при этом образуется поглощающее свет вещество, которое
можно определить количественно с помощью автоматического
аминокислотного анализатора. Эту цветную реакцию используют
также для установления присутствия и положения аминокислот
после разделения их с помощью хроматографии на бумаге (гл.7).
Пролин, оксипролин, а также о-, м- и n-аминобензойные кис-
лоты не дают синего окрашивания, вместо этого появляется жел-
тый цвет. Положительную пробу дают аммонийные соли, а не-
которые амины (анилин) дают красно-оранжевое окрашивание,
которое считается отрицательным результатом.
Производные аминокислот обычно получают через аминокар-
боксилатные соли, образующиеся при щелочной обработке; при
этом увеличивается скорость нуклеофильных реакций свободной
аминогруппы.
/NHJ в- zNH2
R—С1/ ----► R—Cl/ + ВН
ХТОСГ ХЮО'
Для получения твердых производных аминокислот обычно ис-
пользуют производные по аминогруппе, а не по карбоксильной.
280
Глава б
как в случае кислот. По реакции Шоттена — Баумана получают
бензоильные производные (методика 17в, опыт 18). Та же проце-
дура, но с использованием уксусного ангидрида приводит к аце-
тильным производным (методика 17). Фенилизоцианаты реаги-
руют с аминокислотами, давая соответствующие замещенные
фенилмочевины (методика 1). Хорошие производные многих амино-
кислот получают по реакции Хинсберга с использованием м-то-
луолсульфохлорида (методика 18).
/СОСУ
R—ClZ
xnh:
R'COCl
---------->
основание
zCOO"
R—С1Г
\nhcor
При действии 2,4-дпнитрофторбензола на аминокислоты, пептиды
и белки получаются N-замещенные 2,4-динитроанилины. Эти ди-
нитроарильные производные дают цветное окрашивание при дей-
ствии щелочей (опыт 29).
Контрольный вопрос. 1. Предложите механизм реакции с нингидрином.
Методика 24. Получение n-толуолсульфонильных производных аминокислот
R—СН
Около 1 г аминокислоты растворяют в 20 мл 1 н. раствора гидроксида на-
трия. К этому раствору прибавляют 2 г n-толуолсульфохлорнда в 25 мл эфира
и смесь встряхивают на качалке или энергично перемешивают 3—4 ч. Эфирный
слой отделяют, а водный слой подкисляют разбавленной соляной кислотой до
кислой реакции по конго красному нли до pH около 2 по индикаторной бумаге.
Обычно производное выделяется в виде твердого вещества, которое отделяют
фильтрованием и перекристаллизовывают из 4—5 мл. 60%-ного этанола. Если
после подкисления выделяется масло, то для того, чтобы вызвать кристаллиза-
цию, смесь помещают на ночь в холодильник.
Натриевые соли производных фенилаланина и тирозина слаборастворимы в
воде и поэтому выпадают в осадок уже в начале реакции. При подкислении об-
разовавшейся суспензии соль переходит в раствор. л-Толуолсульфонильное про-
изводное выкристаллизовывается из эфирного слоя и может быть отделено филь-
трованием.
Производные глутаминовой и аспарагиновой кислот, аргинина, лизина, три-
птофана и пролина кристаллизуются с трудом. Если они образуются в виде
масла, то следует приготовить какое-либо другое производное.
Определение функциональных групп и установление структуры
281
Методика 25. Получение 2,4-динитрофенильных производных аминокислот
NO2
'СО,- (2) 02n/QVF
СО2Н
К раствору или суспензии 0,5 г аминокислоты и 1,0 г бикарбоната натрия
в 10 мл воды прибавляют раствор 0,8 г 2,4-динитрофторбензола в 5 мл этанола.
Смесь очень сильно встряхивают и оставляют при комнатной температуре на 1 ч,
периодически сильно встряхивая. Затем прибавляют 5 мл насыщенного раствора
соли и экстрагируют дважды порциями по 10 мл эфира для удаления непро-
реагировавших реагентов. После этого водный слой при сильном перемешивании
выливают в 25 мл холодной 5%-ной соляной кислоты. Смесь должна давать
четкое красное окрашивание конго красного. Иногда продукт выделяется в виде
масла, тогда, чтобы он затвердел, его надо помешать или потереть стеклянной
палочкой. Вещество собирают на фильтре и перекристаллизовывают из смеси
равных объемов воды и спирта. Температуры плавления этих производных приве-
дены в последнем издании Tables for Identification of Organic Compounds, Che-
mical Rubber Co., Cleveland, Ohio.
Этот метод можно применять к аминам, белкам и пептидам,
имеющим в своем составе несвязанную первичную аминогруппу
у одного из атомов углеродной цепи или на конце ее.
Аминокислоты с функциональными группами в боковой цепи
(например, ОН, SH и др.) часто можно идентифицировать, полу-
чая их производные по этим функциональным группам. Для это-
го следует обратиться к соответствующим методикам, описанным
в данной главе.
Для идентификации аминокислот очень удобны тонкослойная
хроматография и хроматография на бумаге (см. литературу в
гл. 10 и описание техники в гл. 7).
Лантаноидные соли (например, нитрат европия) являются хо-
рошими реагентами, сдвигающими сигнал в спектрах ПМР ами-
нокислот (гл. 8 и 10).
6.9. АРОМАТИЧЕСКИЕ СОЕДИНЕНИЯ
Уже результаты предварительных химических реакций на на-
личие ароматических свойств в данном соединении позволяют
сделать некоторые предположения о химической характеристике
этого класса органических соединений. Так, часто используют
введение новых заместителей в ароматическое ядро или модифи-
цируют имеющиеся заместители с целью получения новых про-
дуктов, удобных для идентификации исследуемого соединения.
Если в молекуле уже имеются подходящие заместители, то химик
может обратиться к другим разделам этой главы, где описаны
реакции этих групп (например, карбоновые кислоты, амины,
анилины, сложные и простые эфиры, карбонильные соединения
и т. д.). Кроме того, в этой книге имеются специальные обзоры
реакций некоторых типов ароматических соединений (ароматиче-
282
Глава 6
ские нитросоединения, разд. 6.17.1; арилгалогениды, разд. 6.10;
фенолы, разд. 6.19).
Эти реакции будут рассмотрены ниже, начиная с тех из них,
в которых используются самые жесткие условия, и постепенно
переходя к тем, где условия становятся все мягче и мягче. Неко-
торые наиболее инертные ароматические соединения не вступают
в реакцию даже в самых жестких условиях; такие инертные со-
единения следует идентифицировать по спектральным (ИК-,
ЯМР- и масс-спектры) и физическим (молекулярная масса, тем-
пературы плавления и кипения, [а] и т. д.) характеристикам.
Опыт 21. Дымящая серная кислота
HaSO4SO?
Предостережение. Используйте этот реагент только с относи-
тельно инертными соединениями. Соединения, в которых предва-
рительные реакции показали присутствие активирующих групп
(ОН, NH2 и т. п.), могут разлагаться дымящей серной кислотой.
Методика. В чистую сухую пробирку наливают 2 мл 20%-ной дымящей сер-
ной кислоты и прибавляют 1 мл ароматического соединения. Смесь сильно
встряхивают и оставляют на несколько минут. Следует обратить внимание, про-
изошло ли полное растворение. Повторите этот опыт, используя в качестве аро-
матического соединения бензол, бромбензол, этиленбромид и циклогексан.
Обсуждение. Концентрированная серная кислота представляет
собой замечательный растворитель по двум причинам. Ее ди-
электрическая проницаемость, пожалуй, самая большая среди тех
соединений, для которых она была измерена *. Вследствие этого
силы, связывающие растворенные ионы в разбавленных раство-
рах, настолько малы, что коэффициент активности их можно счи-
тать равным единице. Вторым необычным свойством этого раст-
ворителя является наличие кроме обычного автопротолиза,
характерного и для других гидроксисодержащих соединений
(например, для воды)
2H2so4 4=^ h3so; + hso;
еще и самодиссоциации, в результате которой образуются три-
оксид серы и вода. Однако при таких высоких концентрациях, ко-
торые рассматриваются, каждая из образовавшихся частиц ре-
агирует достаточно полно с молекулами серной кислоты. Таким
образом, общая система равновесий выглядит следующим обра-
зом:
(1) H2SO4 H2O+SO3
(2) SOs + H2SO4 hso3+ + hso4-
(3) H2O + H2SO4 «=* H3O+ + HSO4-
(1) + (2) + (3) = 3H2SO4 HSOt + HSO+ + 2HSO4
* Gillespie R. J., Hughes E. D., Ingold C. K., J. Chem. Soc., 1950, 2473.
Определение функциональных групп и установление структуры
283
Кроме того,
h2so44-hso3 +hso; h3so;+hs2o7-
Концентрированная серная кислота (100%-ная) при взаимодей-
ствии с этиленом дает этилсерную кислоту (этилводородсульфат),
однако при использовании серной кислоты, содержащей прибав-
ленный в нее триоксид серы (дымящая серная кислота), образу-
ется этионовая кислота. Причины таких различий становятся по-
нятны, если учесть, что в 100%-ной серной кислоте содержание
сульфирующих частиц SO3 или HSO3 так мало, что они не мо-
гут конкурировать с реакцией присоединения протона. Поэтому
первая стадия реакции приводит к образованию алкилсульфата:
СН2=СН2 + H2SO4 —> СН3СН2 + hso;
Однако в дымящей серной кислоте присоединение HSO3, SO3 или
других сульфирующих агентов становится важным направлением,
и основной реакцией будет следующая:
СН2=СН2 4-HSOJ —> +СН2—CH2SO3H
И в том и в другом случае вторая стадия реакции будет одина-
ковой: взаимодействие образовавшегося на первой стадии карбо-
ниевого иона с бисульфат-ионом.
ch3ch2+hso; —> сн3сн2—о—so3h
кислый этиловый эфир
серной кислоты
+сн2сн2—so3H 4- hso; —> ho3soch2ch2—so3h
этионовая кислота
Первая стадия образования этионовой кислоты сходна, по край-
ней мере формально, с первой стадией сульфирования аромати-
ческих соединений. Однако истинная структура электрофильного
реагента, по-видимому, является более сложной.
Взаимодействие дымящей серной кислоты с 1,2-дигалогенсо-
единениями протекает достаточно сложно. Смесь темнеет, и ино-
гда выделяется свободный галоген. По-видимому, вслед за поте-
рей галогеноводорода происходит полимеризация образующегося
винилгалогенида. Вероятную схему превращений этиленбромида
можно представить следующим образом:
Вг—СН2—СН2—Вг —> СН2=СНВг + НВг
нСН2=СНВг —> — (СН2—СНВг—)„
2HBr + H2SO4 —> Вг2 4- SO2 4- 2Н2О
Контрольные вопросы. 1. Обратите внимание, что описанная реакция при-
годна только для соединений, нерастворимых в серной кислоте. Почему?
2. Напишите уравнения происходящих реакций. Сравните продукты, получаю-
щиеся при этой реакции, и вещества, которые образуются при определении рас-
творимости какого-либо олефина, например гексена-1, в концентрированной сер-
ной кислоте.
284
Глава 6
Опыт 22. Азоксибензол и хлорид алюминия
CeH6N=N—С6Н,
АгН
AICI
AICI
окрашенный комплекс
В чистую и сухую пробирку помещают 2 мл сухого ароматического соедине-
ния и прибавляют 1—2 кристаллика азоксибензола и около 0,1 г безводного хло-
рида алюминия. Обратите внимание на цвет смеси. Если окраска не появилась
сразу, то следует подогреть смесь в течение нескольких минут. Проверьте, как
реагируют в этих условиях петролейиый эфир, хлорбензол, эгилбромид, нафта-
лин и бензол. Если углеводород представляет собой твердое вещество, то можно
использовать раствор 0,5 г этого вещества в 2 мл сухого сероуглерода.
Обсуждение. Эту реакцию применяют только к тем соединени-
ям, которые нерастворимы в серной кислоте. Появление окраски
обусловлено продуктом присоединения, образующимся из п-арил-
азобензола и хлорида алюминия.
Образование n-арилазобензола можно рассматривать как аро-
матическое замещение в арене (АгН) аддуктом хлорида алюми-
ния с азоксибензолом и последующим элиминированием элемен-
тов Н2О н А1С13:
азоксибензол
А1С13
О—А1С1,“
п-арилазобензол
Производные бензола и соответствующие им галогенпроизводные
дают окрашенные растворы от интенсивно-оранжевых до темно-
красных или образуют осадки такого же цвета. Конденсирован-
ные полиядерные углеводороды, например такие, как нафталин,
антрацен и фенантрен, окрашивают растворы в коричневый цвет.
Алифатические углеводороды не дают никакого окрашивания или
появляется слабо-желтая окраска.
Опыт 23. Хлороформ и хлорид алюминия
AlCls
ЗАгН+СНС13 -----► А13СН
Аг3СП + R+ ---> Ar3C+ + 3HCI + RH
окрашен-
ный катион
Определение функциональных групп и установление структуры
285
К 2 мл сухого хлороформа в пробирке прибавляют 0,1 мл или 0,1 г аро-
матического соединения, сильно перемешивают и, наклонив пробирку, смачивают
ее стенки. После этого прибавляют 0,5—1,0 г безводного хлорида алюминия
так, чтобы часть порошка попала на стенки пробирки. Отметьте цвет порошка
на стенке и цвет раствора. Проведите эту реакцию с петролейным эфиром, хлор-
бензолом, бифенилом и бензолом.
Обсуждение. Ароматические соединения с хлороформом и хло-
ридом алюминия дают чрезвычайно характерное окрашивание.
Алифатические соединения, нерастворимые в серной кислоте, либо
не дают никакой окраски, либо окрашиваются только в очень сла-
бо-желтый цвет. При этой реакции появляются следующие харак-
терные цвета:
Соединение Окраска
Бензол и его гомологи Арплгалогениды Нафталины Бифенил Фепашрен Антрацен От оранжевого до красного То же Синий Пурпурный » Зеленый
С течением времени цвета изменяются, принимая различные от-
тенки коричневого цвета. Такие же цвета получаются, если хло-
роформ заменить четыреххлористым углеродом.
Реакция начинается тремя последовательными реакциями Фри-
деля— Крафтса [(1) — (3) на приведенной ниже схеме] между
ароматическим и хлорированным углеводородами. Эти реакции
алкилирования промотируются кислотами Льюиса и А1С13 и облег-
чаются делокализацией положительного заряда между хлором и
арильной группой:
СНС13+А1С13
[снС12—Cl--A1CI,]
+ А1СЦ-
НСС12++АгН Аг< —н+- ArCHClz
СНС12
н
ArCHCI -Aff ♦ ArCH—Ar
(2) |
CI
АгСН—Аг - А|СЬ. АгСН—Аг
I
С1
н
+1 — н*
АгСН—Аг —» Аг3СН
Аг
UOSWttSHUG
г!э1вэгэЯ I9ftbfi2@) 123 1эгп13-пЫ1эЧ эдоЭндп вн йиннэдэмен ,адна мотэнр а вкокнэя-ж дтяэпэ йинэвдявдфнЫ .22.9 .энЧ
. (9961 ,.вЧ ,BirfglabfiIirf4 ,.эп1 ,гэпо)вюс1вЗ
!Н—3 нннвдовоя эинтнэивЯ .втэясия квндкклнпвх „оп! .BnnswsIoa-nBbuBviO ымднф то нэрукоп ,(.тнк) 0° ееI—881 .пни ,т .VI,901 воовм .ком ,oiHj3
-иле) tH3 у (мям 09.8) 1—мо 0982 — 8 ;(эняоэрнтвфнкв) _НЭ у ,(мям ЕЬ.8) 1—мо 9162—2 • у эняоэрнтвмодв ,(мям 26,6) | —мо 0106 — 1
ММНЭ о ММНЭЕ rl^-
.8191— J :Э—Э нннвдэкоя эинтнэлвЯ .(М.Э .энд ,мо) эннэдюмг.г-отоя. вн таваиевяу ,(мм 00,9—00,9) 1—мо 1991-0002 — ^ :инотдэЭО .(эняоэрнтвф
| —мо ОвЭ ЛЭ7—\ ; 6 ,(мям 29,6 ,61,6 ,99,8) мо 0801 ,8601 ,0Ш — Ь :Н—Э кннвЭэкох энннондвмдофэД „V ,(мям ОТ,9 ,81,9) мо 26М .0281
grl<J J—J
-дофэд эняоокпэн ,(мхм 0,62) *~мо 868—8 :Э—Э кннвЭэкоя эиннондвмдофэД .Н—Э— вннвЭэкоя эиннонивмдофэд энхоокпэн ,(мям 08,81 ,80,61)
.Э—Э нннвЭэкоя эиннондвм
snrta&.eoHsm\j w nn\jqs эмнал.э6эцпО
.(sIOCID алэтлдоатэвд ,ц1 009 вятдэаевд ,д7М 09) вкоукот вэнвнобэд оюнтнндвм отоннотодп дтяэпЭ .02.9 .энЧ
288
Глава 6
Частично замещенные хлориды (например, Аг2СНС1 или
АгСНСЬ) могут реагировать с хлоридом алюминия, давая моно-
арил- или диарил-катионы:
Аг2СНС1 + А1С13 —» Аг2СН++ А1С1Г
Такие катионы могут отрывать гидрид-ион от триарилметана, прев-
ращая его в стабильный триарил-катион:
Аг3СН + Аг2СН+ —> Аг3С++ Аг2СН2
окрашен
Контрольные вопросы. 1. Объясните, почему не образуется тетраарилметан.
На рис. 6.22 приведен типичный ИК-спектр, а на рис. 6.23 —
спектр ПМР ароматических соединений. Для характеристики аро-.
матических соединений можно использовать функциональные груп-
пы, если они имеются в этом соединении. Для этого следует
Таблица 6.4
Химические сдвиги протонов (м. д.) монозамещениых бензолов
(бензол, синглет, 0=7,27 м. д.)
Типы заместителя Соединение Положение заместителя
орто мета пара
С6Н5СНз а 7,10 7,18 7,10
СеНб—сен5 7,45 7,27 7,35
СВН5—SR 7,25 7,3 7,3
Электроиоакцепторные СсН5—no2 8,22 7,44 7,60
заместители СВН6-СНО 7,85 7,48 7,50
СВН5—(C=O)R 7,9 7,45 7,55
свн,—соон 8,17 7,41 7,47
СВН6—COOR 8,2 7,35 7,5
СВН5—(С=О)С1 8,10 7,43 7,6
СВН5—CN 7,54 7,38 7,57
СВН5—NH3+ 7,62 — —
Электронодонорные свн5—nh2 6,52 7,03 6,64
заместители СВН5—ОН 6,77 7,13 6,87
CgHs—OR 6,75 7,2 6,75
Ссн5—О(С=О)СНз 7,15 7,26 7,26
CBH5^OSO2CBH4CHs-n 7,01 7,22 7,22
CBH5—NH(C=O)CH3 7,52 — —
Г алогены c6H5-R 6,97 7,25 7,04
СВН5—CI 7,29 7,21 7,21
Сен5—Вг 7,49 7,14 7,24
Свн5—I 7,67 7,00 7,24
а На приборе с частотой 60 МГц при развертке 600 Гц толуол имеет лишь слегка уши-
ренный синглет ароматических протонов. При этих условиях для разрешения сигнала арома-
тических протонов необходимо разрешение более 0,1—0,2 Гц.
Определение функциональных групп и установление структуры
289
обратиться к тем разделам книги, где описаны реакции соответст-
вующих функциональных групп. Число и относительное распо-
ложение неполярных заместителей в бензольном кольце во мно-
гих случаях можно установить при помощи данных табл. 6.4 и
рис. 6.24. Замена полярных групп на алифатические заместители
в боковой цепи обычно не приводит к заметно-
му изменению химического сдвига сигналов аро-
матических протонов, находящихся в слабом
поле. Однако если заместитель входит в ядро,
то наблюдаются очень существенные изменения
в положении сигналов ароматических протонов
(табл. 6.4).
Масс-спектры ароматических соединений ха-
рактеризуются, как правило, очень интенсивным
пиком молекулярного иона. Таким образом,
если в окрестностях молекулярного иона нет пи-
ков фрагментов или примесей, то интенсивность
пиков М, М-f-] иМф2 достаточно велика для
установления молекулярной формулы (гл. 4).
2000 1667см-’
моно
ДИ
орто
мета
пара
Методика 26. Нитрование ароматических соединений
(a) HNO3
H,SO.
АгН
(6) Зымящая
азотная
кислота
Нитрование ароматических соединений, тем
более неизвестных, следует проводить с осо-
Рис. 6.24. Схематическое изображение ИК-поглоЩения
бензольных соединений в области 5—6 мкм, характер-
ное для каждого типа замещения (Дайер Д. Р. Прило-
жения абсорбционной спектроскопии органических со-
единений. Пер. с англ. — М.: Химия, 1970, с. 62. Пере-
печатано с разрешения Prentice-Hall, Inc., Englewood
Cliffs.).
три
1,2,3-
быми предосторожностями, так как многие из них реагируют весь-
ма бурно. Перед началом реакции следует провести пробу с ма-
лым количеством вещества, пользуясь при этом защитным экра-
ном.
а) К 4 мл концентрированной серной кислоты прибавляют около 1 г иссле-
дуемого вещества. Затем приливают по каплям 4 мл концентрированной азот-
ной кислоты, встряхивая смесь после прибавления каждой капли. Колбу соеди-
няют с обратным холодильником и помещают ее в стакан с водой на 5 мин при
45°С. Реакционную смесь выливают на 25 г битого льда и собирают осадок на
фильтре. Его можно перекристаллизовать из разбавленного этанола.
10 Зак. 335
290
Глава 6
б) Нитрование проводят по описанной выше методике, однако вместо кон-
центрированной азотной кислоты берут 4 мл дымящей азотной кислоты * и реак-
ционную смесь нагревают на паровой бане в течение 10 мни. В тех случаях, ког-
да исследуемое соединение плохо нитруется, можно вместо концентрированной
серной кислоты использовать дымящую серную кислоту.
Обсуждение. Введение нитрогруппы в ароматическое кольцо
позволяет получать другие замещенные ароматические соедине-
ния. Так, например, полизамещенные бензольные соединения мо-
гут быть идентифицированы с помощью реакции нитрования и по-
следующего восстановления нитросоединения до амина, который
в свою очередь ацетилируется или бензоилируется, давая моно- или
диацетамидо- или бензамидопроизводные.
(1) (2)
АгН —> ArNO2 ------> ArNH2 ----> ArNHCOR
(1) методика 50
(2) опыт 18 и методика 17
По методу «а» можно получать ж-динитробензол из бензола
или нитробензола и n-нитропроизводные из хлорбензола, бром-
бензола, бензилхлорида или толуола. Фенол, ацетанилид, нафта-
лин и дифенил дают динитропроизводные. В случае галогениро-
ванных ароматических соединений лучше использовать методику
«б», так как при этом образуются динитропроизводные, которые
легче очистить, чем мононитропроизводные, получаемые по спо-
собу «а». Мезитилен, ксилолы и псевдокумол дают тринитропро-
изводные..
Методика 27. Окисление боковой цепи в ароматических соединениях
. ,z АгСООН
(a)/ H2SO4, Д *
АгК \н*
КМ"°4--♦ АгСОО'-^
Д, ОН'
а) Окисление бихроматом. В Небольшую колбу помещают 15 мл воды, 7 г
бихромата натрия и 2—3 г окисляемого соединения. Прибавляют 10 мл концен-
трированной серной кислоты И, соединив колбу с обратным холодильником,
сильно встряхивают смесь. Осторожно нагревают колбу до начала реакции, за-
тем пламя убирают и, если это необходимо, колбу охлаждают. После того как
смесь перестанет кипеть за счет теплоты реакции, колбу нагревают с обратным
холодильником еще 2 ч, после чего содержимое колбы выливают в 25 мл воды
и осадок собирают на фильтре. Осадок смешивают с 20 мл 5%-ной серной кис-
поты И нагревают на кипящей водяной бане при энергичном перемешивании.
После охлаждения осадок отделяют и промывают 20 мл воды. Остаток раство-
ряют в 20 мл 5%-ного раствора гидроксида натрия и раствор фильтруют. Филь-
* Такую концентрированную кислоту (~90%) иногда называют «белой»
или «желтой» дымящей азотной кислотой. Этот реактив — сильный нитрующий
агент. Не следует путать эту кислоту с «красной» азотной кислотой, которая со-
держит растворенный диоксид азота NO2 и является мощным окислителем.
Определение функциональных групп и установление структуры
291
трат при сильном перемешивании выливают в 25 мл 10%-ной серной кислоты.
Осадок собирают на фильтре, промывают водой и очищают перекристаллизацией
из этанола или бензола.
б) Окисление перманганатом. Прибавляют 1 г исследуемого соединения к
80 мл водного раствора, содержащего 4 г перманганата калия, добавляют 1 мл
10%-ного раствора гидроксида натрия и смесь нагревают с обратным холодиль-
ником до тех пор, пока не исчезнет пурпурный цвет перманганата (от 0,5 до
3 ч). После этого смеси дают остыть и затем осторожно подкисляют серной кис-
лотой. Подкисленную смесь нагревают 0,5 ч и охлаждают. Избыток диоксида
марганца удаляют прибавлением небольшого количества раствора бисульфита
натрия. Выпавшую кислоту собирают на фильтре и перекристаллизовывают из
бензола или этанола. Если кислота достаточно растворима в воде, то она может
не выпадать из таких кислых разбавленных растворов. В этом случае получен-
ную кислоту можно экстрагировать хлороформом, эфиром или бензолом. При
подкислении иногда может выпасть небольшой осадок кремневой кислоты, по-
этому весьма важно перекристаллизовать полученную кислоту перед определе-
нием температуры плавления.
Обсуждение. Ароматические углеводороды, имеющие боковую
цепь, могут быть окислены до соответствующих кислот. Это пре-
восходный метод для соединений, имеющих только одну боковую
цепь. Лучше всего для этой цели подходит окисление перманга-
натом (методика 276). Ароматические кислоты, имеющие несколько
карбоксильных групп, иногда трудно поддаются разделению. По-
этому применение окислительных методов в этих случаях доволь-
но ограниченно. Однако окисление можно рекомендовать, если
две боковые цепи находятся рядом друг с другом в кольце, так
как при таком расположении заместителей в результате окисления
получается фталевая кислота, которую легко идентифицировать.
о-Диалкилбензолы, так же как и другие соединения, имеющие за-
местители в о-диалкильных цепях, претерпевают полное окисле-
ние при действии Cr(VI) (методика 27а). Таким образом, приме-
нение этого окисляющего агента может приводить к ошибочным
выводам. Поэтому следует использовать окисление пермангана-
том (методика 276). Температуры плавления кислот, получаемых
при окислении алкилбензолов, можно найти в таблицах (см. при-
ложение IV).
Ароматические кольца, имеющие электроноакцепторные заме-
стители (нитрогруппу, галогены и т. п.), не затрагиваются даже
при очень жестких условиях окисления. Напротив, в случае аро-
матических соединений с электронодонорными заместителями окис-
ление с участием углеродных атомов кольца протекает легче, чем
окисление боковой цепи. Например, при окислении замещенного
фенола образуется алифатическая кислота с выходом, достаточно
высоким для ее идентификации.
ТГО2УС> RC°2H
“jw, Zu U
( иВентифици—
руютп, как
указано
в разд. 6.12)
10*
292
Глава 6
Очевидно, что наличие атомов водорода в «-положении к кольцу
(бензильный водород) облегчает такое окисление
’ реакция не идет
Два ароматических заместителя у одной алкильной группы могут
препятствовать окислению ароматического ядра
О
окисление ||
АгСН2Аг' ----------> Аг—С—Аг'
Недавно была предложена очень хорошая методика для окис-
ления боковых цепей с помощью краун-эфиров *. Для препара-
тивного окисления можно также использовать четвертичные ам-
монийные соли в качестве межфазовых реагентов (см. опыт 15Б).
Методика 28. Получение ароилбензойных кислот
К раствору 1 г сухого ароматического углеводорода и 1,2 г фталевого ан-
гидрида в 10 мл сухого сероуглерода прибавляют 2,4 г безводного хлорида
алюминия. Смесь нагревают с обратным холодильником на кипящей водяной
бане в течение 30 мин и затем охлаждают. Сероуглеродный слой декантируют
и к остатку приливают 10 мл концентрированной соляной кислоты и 10 мл воды.
Вначале кислоту следует прибавлять медленно, охлаждая, если это необходимо,
льдом. После прибавления кислоты смесь сильно встряхивают. Если ароилбен-
зойная кислота выделяется в твердом состоянии, то ее собирают на фильтре и
промывают холодной водой. Если же она выделяется в виде масла, то смесь
некоторое время охлаждают льдом, чтобы вызвать кристаллизацию. Если этого
не происходит, то жидкость, находящуюся над маслом, сливают, а масло промы-
вают холодной водой. Сырой продукт, твердый или маслообразный, кипятят
1 мин с 30 мл 10%-иого раствора гидроксида аммония, к которому прибавлено
0,1 г активированного угля (норита). Горячий раствор фильтруют и охлаждают,
после чего к нему прибавляют 25 г толченого льда и подкисляют концентриро-
ванной соляной кислотой. Ароилбензойную кислоту отфильтровывают и перекри-
сталлизовывают из разбавленного этанола (30—80%). В некоторых случаях для
того, чтобы образовались кристаллы, смесь надо оставить на ночь.
* Методы получения 18-крауи-6 см. в книге: Fieser L. F., Williamson К.,
Organic Experiments, 3rd ed., Heath, Lexington, Mass, 1975, pp. 386—388. Окис-
ление и другие реакции с применением краун-эфиров описаны в работе: Gakel G.,
Durst И., Synthesis, 1976, 168, которая представляет собой обширный обзор с
подробным описанием экспериментальных методик, охватывающий 190 ссылок.
Определение функциональных, групп и установление структуры 293
Обсуждение. Ароматические углеводороды и их галогенпроиз-
водные вступают с фталевым ангидридом в реакцию Фриделя —
Крафтса, давая с хорошим выходом ароилбензойные кислоты. В
результате этой реакции образуются производные с карбоксиль-
ной группой, которую легко идентифицировать (разд. 6.12). На
пример, можно определить эквивалент нейтрализации (методика
32) кислоты, на основании полученных данных можно установить
молекулярную массу (мол. масса) кетокислоты и затем рассчи-
тать молекулярную массу исходного ароматического соединения:
мол. масса АгН = (мол. масса кислоты) — 148
(Объясните происхождение числа 148 в этой формуле.)
Методика 29. Получение арилметилкетонов
/ NNHR\
CHsCOCI I ||
АгН ----—-----> АгСОСНз \->АгССН3 7
А1С1з
В коническую колбу емкостью 50 мл, снабженную хлоркальциевой трубкой,
помещают 1,4 г порошкообразного безводного хлорида алюминия, 5 мл серо-
углерода и 0,8 мл ацетилхлорида. Спустя 5 мин при перемешивании прибав-
ляют раствор 2 мл ароматического соединения в 5 мл сероуглерода. Смесь остав-
ляют стоять при комнатной температуре до тех пор, пока весь хлорид алюминия
не растворится (10—1-5 мин), затем выливают на лед, смешанный с 5 мл концен-
трированной соляной кислоты, и, наконец, прибавляют еще 5 мл сероуглерода.
Органический слой тщательно промывают 3 мл 10%-ной соляной кислоты, водой,
5%-ным бикарбонатом натрия и водой Промывку водой повторяют до тех пор,
пока промывные воды не станут нейтральными по лакмусу. Органический слой
высушивают хлоридом кальция, после чего сероуглерод упаривают. Остаток рас-
творяют в этаноле и получают 2,4-динитрофенилгидразон или семикарбазон, как
обычно (методики 8 или I соответственно).
Обсуждение. Описанная методика проведения реакции Фриде-
ля— Крафтса пригодна для умеренно активных ароматических
соединений. Соединения, устойчивые к электрофильному арома-
тическому замещению, часто не реагируют по этой методике, на-
пример нитробензол не ацетилируется. Высокореакционноспособ-
ные ароматические соединения (например, амины или фенолы)
могут реагировать с замещающей группой (т. е. с NH2 или ОН).
Такие активные соединения часто дают сложные или плохо иден-
тифицируемые продукты реакции.
Различные другие методики получения производных, пригод-
ных для охарактеризования ароматических соединений, приве-
дены в других разделах данной главы. Производные пикриновой
кислоты (методика 22) позволяют определить молекулярную мас-
су ароматических соединений. Анилин и замещенные в кольце ани-
лины могут быть идентифицированы с помощью азотистой кислоты
(опыт 19) и реакции Хинсберга (методика 16). Фенольные соеди-
нения можно охарактеризовать действием бромной воды (мето-
дика 60).
294 Глава 6
Для получения комплексов с переносом заряда из богатых эле-
ктронами ароматических соединений можно использовать мето-
дики, описанные в книге Пасто и Джонсона, с. 343—344. Такне
комплексы образуются при взаимодействии ароматических сое-
динений с 2,4,7-тринитрофлуореноном (ТНФ);
NO2
KJI Сд тнф
no2
В книге Черониса, Энтрикина и Ходнетта (с. 351) описана ме-
тодика, позволяющая отличать ароматические соединения от бо-
лее инертных органических соединений:
нсно. н2о
АгН ——s ——> окрашенные полимерные карбокатионы
Механизм этой реакции включает последовательное алкилирова-
ние ароматических систем по Фриделю—Крафтсу. Реакционно-
способные соединения (как полиены, так и ароматические соеди-
нения) дают окрашенные в различные цвета растворы или осадки;
предельные углеводороды в эту реакцию обычно не вступают.
Дополнительную возможность для установления структуры или
для ее подтверждения дают ультрафиолетовые спектры аромати-
ческих соединений. Ароматические системы имеют интенсивные
полосы поглощения, положение максимума которых позволяет уста-
новить тип заместителя в ароматическом кольце. Эти вопросы об-
суждаются в гл. 8.
В случае особо неактивных ароматических соединений (напри-
мер, фторбензола) разумнее полагаться на очень тщательно вы-
полненные измерения простых физических свойств (температуры
кипения и плавления, показатель преломления). Полученные дан-
ные следует сопоставить с данными, приведенными в соответст-
вующих таблицах. Искомым соединением будет то, у которого со-
впадают все эти константы.
6.10. АРИЛГАЛОГЕНИДЫ
Арилгалогенидами называются такие соединения, в которых
атом галогена непосредственно связан с ароматическим ядром.
Они представляют собой разновидность ароматических соединений,
рассмотренных в предыдущем разделе. В то же время они рази-
тельно отличаются по своим свойствам от алкилгалогенидов. Али-
фатические галогениды обычно значительно более реакционноспо-
собны, чем соответствующие арилгалогениды.
Для установления структуры арилгалогенидов полезно ис-
пользовать как качественную реакцию на присутствие галогена
Длина волны, мкм
296
Глава 6
Рис. 6.26. Спектр протонного магнитного резонанса 4-бром-л-ксилола (60 МГц, развертка 600 Гц, растворитель
Определение функциональных групп и установление структуры
297
(сплавление с натрием), так и анализ масс-спектров, описанный
в гл. 4. С помощью масс-спектрометрического анализа часто мож-
но установить число атомов брома или хлора в этих соединениях.
Выбор дополнительных химических методов анализа должен быть
проведен только с учетом высокой устойчивости арилгалогенидов
к типичным реакциям замещения. К таким реакциям относятся
и описанные выше реакции со спиртовым раствором нитрата се-
ребра (опыт 16) и с раствором иодида натрия в ацетоне (опыт 17).
На рис. 6.25 приведен типичный ИК-спектр арилгалогенида, а
на рис. 6.26 — соответствующий спектр ПМР. Установить присут-
ствие галогена (X) во многих случаях можно по наличию в ИК-
спектре полосы валентного колебания С—X (разд. 6.6). В отличие
от простых алкильных заместителей атомы галогенов часто вызы-
вают неэквивалентность сигналов атомов водорода ароматиче-
ского кольца в ПМР-спектре. Влияние галогенов и других заме-
стителей в кольце на химический сдвиг протонов ароматического
ядра описано в разд. 6.9.
Для идентификации арилгалогенидов очень полезно изучить
их масс-спектры. Ароматический характер этих соединений обычно
приводит к довольно стабильным молекулярным ионам, интенсив-
ность пиков которых достаточна для качественного и количествен-
ного определения брома и хлора. Иногда группы пиков, близких
к молекулярному иону арилбромидов и арилхлоридов, позволяют
провести количественное определение С, Н, N и О.
Ряд описанных ниже методов позволяет получать производ-
ные арилгалогенидов. Эти методы рассмотрены также в других
главах книги. Во всех случаях для удобства предполагается, что
в результате реакций образуются пара-замещенные продукты,
хотя на самом деле в некоторых случаях образуются и орто-изо-
меры, а иногда даже небольшие количества лета-замещенных со-
единений. Наиболее общим методом получения производных
X=галоген, R = алкил
СО2Н
(1) ArNCO
(2) Н2О
\ (Х=Вг,1)
\ Mg/сухой эфир
методика 12
Х = Вг
СО2
Пасто и Джонсон,
с. 552
298
Глава 6
арилгалогенидов является нитрование (методика 26). Однако
бром- и хлорпронзводные, имеющие боковую цепь, часто при нит-
ровании окисляются до соответствующих кислот (методики 27а и
276). Арилиодиды и арилбромиды рекомендуется превращать в
соответствующие карбоновые кислоты или анилиды (методика 13).
Арилгалогениды легко реагируют с хлорсульфоновой кислотой,
давая сульфонилхлориды (методика 30), которые при обработке
аммиаком превращаются в сульфонамиды (методика 31).
Методика 30. Получение сульфоиилхлоридов
2CISO3H v Z^?\ У
Х X-\Q/-S-C1 + HC1 + H2SO.
о
В чистую сухую пробирку помещают раствор 1 г арилгалогенида в 5 мл
сухого хлороформа, смесь охлаждают в стакане со льдом до 0°С и прибавляют
по каплям 5 мл хлорсульфоновой кислоты. После того как прекратится выделе-
ние хлористого водорода, пробирку вынимают изо льда и оставляют ее нагре-
ваться до комнатной температуры (около 20 мин). Содержимое пробирки выли-
вают в стакан емкостью 50 мл, наполненный кусочками льда. Отделяют хлоро-
формный слой, промывают его водой и растворитель упаривают. Оставшийся
сырой сульфонилхлорид можно перекристаллизовать из низкокипящего петролей-
ного эфира, бензола или хлороформа.
Примечание. Описанные выше условия проведения реакции подходят для
получения производных большинства простых арилгалогенидов -и алкилбензо-
лов. В случае галогентолуолов желательно нагревать хлороформный раствор
при 50°С около 10 мин и затем вылить смесь на толченый лед.
В случае полигалогенированных соединений требуются более жесткие усло-
вия для проведения реакции: реакцию проводят без растворителя и реакцион-
ную смесь кипятят с обратным холодильником при 100°С около 1 ч.
Обсуждение. В процессе получения сульфоиилхлоридов могут
проходить две «необычные» реакции. При обработке хлорсульфо-
новой кислотой фторбензола, иодбензола и о-дибромбензола при
50° С без растворителя образуются соответствующие сульфоны
о
хО|нО-х
Эти сульфоны представляют собой твердые вещества и поэтому
могут служить в качестве производных для установления струк-
туры указанных галогенбензолов. В других случаях (см. методи-
ку 31) также могут образовываться небольшие количества суль-
фонов, однако ввиду того, что они нерастворимы в щелочах, их
можно отделить от сульфонамидов. Вторая необычная реакция —
хлорирование в ядро. Она наблюдается при получении сульфонил-
хлоридов ц-дииодбензола и 1,2,4,5-тетрахлорбензола. Реакция идет
плохо с арилиодидами.
Определение функциональных групп и установление структуры
299
Арилгалогениды с атомом галогена, активированным нитро-
группой, легко реагируют с пиперидином, давая пиперидильные
производные, которые можно использовать для идентификации.
Арилиодиды присоединяют хлор с образованием иодарил-
дихлоридов, которые могут применяться в качестве производных
или как хлорирующие реагенты:
Ari + С12 —> ArlCh
Литература: Deikel М. К., Piperidyl Derivatives of Aromatic
Halogeno-Nitro Compounds, J. Am. Chem. Soc., 62, 750 (1940);
Lucas H. J., Kennedy E. R., lodoaryl Dichlorides, Org. Syn. Coll.
Vol. Ill, p. 482.
Методика 31. Получение сульфонамидов
Около 0,5 г сульфонилхлорида кипятят с 5 мл концентрированного водного
раствора аммиака примерно 10 мин. Смесь разбавляют водой, охлаждают и
фильтруют. Полученный сульфонамид растворяют в 10 мл 5%-ного раствора
гидроксида натрия, при необходимости осторожно подогревая. Раствор филь-
труют, отделяя сульфон или продукт хлорирования. Фильтрат подкисляют раз-
бавленной соляной кислотой и отфильтровывают сульфонамид. Его перекристал-
лизовывают из разбавленного этанола и сушат при 100°С.
Обсуждение. При использовании первичных и вторичных ами-
нов вместо аммиака эта методика или реакция Хинсберга (мето-
дика 16) может быть использована для получения N-замещенных
сульфонамидов.
6.11. УГЛЕВОДЫ И САХАРА (САХАРИДЫ)
К углеводам (карбогидратам) относятся полиоксиальдегиды и
полиоксикетоны. Более простые из широкого круга этих соеди-
нений называют сахарами (или сахаридами). Название «карбо-
гидраты» происходит от старинного предположения, что эти веще-
ства общей формулы СГ1(Н2О)п (например, глюкоза С6Н120б) пред-
ставляют собой «гидраты углерода». Сахароза (обычный пищевой
сахар) имеет другую формулу Сг2Н220ц, т. е. С„(Н2О)т. Углеводы
похожи друг на друга по своим свойствам, что позволяет их легко
идентифицировать. Обычно они представляют собой растворимые
в воде твердые вещества, плавящиеся с разложением. Эти свой-
ства указывают на присутствие в их молекуле нескольких высоко-
полярных функциональных групп. Таким образом, при изучении
углеводов приходится иметь дело с химией функциональных групп,
300
Глава 6
уже частично рассмотренных в различных разделах этой книги
(табл. 6.5).
Таблица 6.5
Реакции, обычно используемые для идентификации сахаридов 3
Функциональная группа (название) Описание теста Реакция
ОН (спиртовый гидроксил) Методика 5 Ацетилирование
С=О (кетонный карбонил) Опыт 8 Получение фенилгидразонов (или озазонов, опыт 24)
СНО (альдегидный карбо- нил) 6 ОН ОН Опыт 11 Проба Толленса (аммиачный раствор нитрата серебра)
—С—С— (вицинальные диолы) Опыт 5 Окисление перйодатом
СНО (альдегидный карбо- нил) 6 \ Z0R / XpZ (ацетали, кетали, ✓ \ полуацетали) OR Опыт 13 Разд. 6.2.2 Реактив Бенедикта и фелин- гова жидкость
а Можно применять и другие реакции, описанные в разд. 6.1 и 6.2; в случае производ-
ных сахаридов (например, глюконовых кислот) проще использовать реакции новых получен-
ных групп (например, реакции карбоновых кислот, описанные в разд. 6.12), '
б Сахара, дающие положительную реакцию иа эти пробы, называются «восстанавлива-
ющими» сахарами.
Во многих случаях, например при получении производных са-
харов, взаимодействие нескольких функциональных групп приво-
дит к результатам, значительно отличающимся от результатов
реакций монофункциональных соединений. Хорошим примером та-
кого различия может служить получение производных сахаридов
действием на них фенилгидразина. При этом образуются не фенил-
гидразоны, а озазоны, что резко отличает углеводы от простых
карбонильных соединений (опыт 8).
Опыт 24. Получение озазонов
RCHOH
I
о=сн
rc=nnhc6h5
или +3H2NNHC6H6 —> I + C6H6NH2 + NH3 + 2Н2О
ch=nnhc6h5
RC=O
I
CH2OH
Готовят образцы глюкозы, мальтозы, сахарозы и галактозы по 0,2 г каж-
дый. Все четыре сахара испытывают одинаково. Помещают образцы в отдельные
Определение функциональных групп и установление структуры 301
пробирки и прибавляют в каждую из них по 0,4 г гидрохлорида фенилгидразина,
0,1 г кристаллического ацетата натрия и 4 мл дистиллированной воды. Закры-
вают пробирки пробками с отверстиями и помещают все пробирки в один ста-
кан с кипящей водой. Отмечают времч погружения и момент выпадения осадка
каждого озазона *. Чтобы смесь не вскипала, пробирки необходимо периодиче-
ски встряхивать.
Спустя 20 мин пробирки вынимают из горячей водяной бани и охлаждают.
После охлаждения помещают небольшое количество жидкости и кристаллов на
часовое стекло. Покачивая часовое стекло из стороны в сторону, отделяют кри-
сталлы, а маточный раствор удаляют кусочком фильтровальной бумаги, ста-
раясь не повредить кристаллы. Рассматривают полученные кристаллы под ми-
кроскопом (при 80—100-кратном увеличении) и сравнивают с микрофотогра-
фиями.
Образования смолистых продуктов окисления фенилгидразином можно избе-
жать, если прибавить в смесь 0,5 мл насыщенного раствора бисульфита натрия.
Это необходимо делать в тех случаях, когда желательно выделить озазоны и
определить их температуру плавления.
Обсуждение. Время, требующееся для образования озазонов,
может служить для идентификации различных сахаров. Малликен
приводит следующие значения времени выпадения озазонов из го-
рячего раствора: фруктоза 2 мин, глюкоза 4—5 мин, ксилоза 7 мин,
арабиноза 10 мин, галактоза 15—19 мин, раффиноза 60 мин, лак-
тоза— озазон растворим в горячей воде, мальтоза — озазон ра-
створим в горячей воде, манноза 0,5 мин (гидразон), сахароза
30 мин (вначале проходит гидролиз, а затем образуется озазон
глюкозы).
Ниже приведены некоторые известные реакции для открытия
сахаров:
Реакция Реактивы Литература
Реакция Молиша Опрыскивание хрома- тограмм Реакция с антроном Реакция с резорцином а-Нафтол, серная кис- лота Гидрохлорид п-анизи- дина , бутанол-1 Антрон, серная кислота Резорцин, серная кис- лота Пасто и Джонсон, с. 396; Че- ронис, Энтрикин, Ходнетт, с. 390 Пасто и Джонсон, с. 397 Черонис, Энтрикин, Ходнетт, с. 388 Черонис, Энтрикин, Ходнетт, с. 390
Во многих случаях, учитывая растворимость простых саха-
ридов, целесообразно получать ИК-спектры в таблетках с КВг
(рис. 6.27), а ПМР-спектры (рис. 6.28) в тяжелой воде (D2O).
Иногда спектральный анализ сахаридов можно упростить, если
использовать для этого не сами сахариды, а их производные, та-
кие, как ацетониды и гликозиды.
Hassid W. Z., McCready R. М., Ind. Eng. Chem., Anal. Ed., 14, 683 (1942).
Рис. 6.27. Инфракрасный спектр 0-d-(+).глюкозы (таблетки с КВт).
Валентные колебания О И: 2 —3600 - 3000 см-1 (2,78-3,33 мкм), vQ_H (с сильной водородной связью). Валентные колебания С—Н: 2—2915 см,
3.43 мкм, »с_н; з—2000см-1 <5,00 мкм), смена решеток. Валентные колебания С—0:4—1103, 1073, 1008 см (9,07, 9,32, 9,92 мкм), V (первнч-
аые и вторичные спирты).
304
Глава 6
При характеристике сахаридов и их производных (например,
гликозидов) необходимо прежде всего решить вопрос о том, пред-
ставляет ли данное соединение циклический или ациклический
продукт. Если
новить, какую
изучаемое вещество циклическое, то важно уста-
форму имеет его цикл (а- или р-конфигурацию)
снгон
\ .OR
но й /
р—/
гексапиранозы
По своему строению а- и p-изомеры различаются положением
группы OR, находящейся в одном из двух аномерных положений
у атома С-1. Сигнал аномерного протона у С-1 находится в сла-
бом поле и хорошо отделяется от других сигналов. Это связано
с тем, что этот протон испытывает дезэкранирующее влияние сразу
двух атомов кислорода, связанных с атомом С-1, в то время как
экранирование остальных протонов в кольце снижается благодаря
влиянию только одного атома кислорода, находящегося рядом с
ними. С помощью спектров довольно легко отличить а-изомер от
Р-изомера, потому что аномерный протон в сс-изомере находится
в экваториальном положении и поэтому сигнал его обычно слегка
сдвинут в слабое поле по сравнению с сигналом аномерного про-
тона в p-изомере*. Для шестичленных циклов характерно, что эк-
ваториальные протоны дают сигнал, сдвинутый на 0,3—0,9 м. д.
дальше в сторону слабого поля, чем сигнал аксиальных протонов.
Дополнительную информацию о конфигурации цикла можно по-
лучить, изучая расщепление сигнала аномерного протона. Такое
расщепление является общим свойством шестичленных циклов.
Г'ХхУ*_проекция
V I If двугранного
Н,. угла
Joo'=8-ur4
Н.
На ньюменовской проекции шестичленного кольца видно, что ви-
цинальные протоны расположены по отношению друг к другу
под углом либо 180° (а — а'), либо 60° (а — е', е — а’, е — е'). На
основании мультиплетности сигнала аномерного протона можно
установить конфигурацию (е или а) протона у атома С-2. Напри-
мер, если величина расщепления аксиального аномерного протона
составляет около 8 Гц, то, следовательно, гидроксильная группа
у С-2 находится в экваториальном положении.
* Сильверстейн Р., Басслер Г., Моррилл Т. Спектрометрическая идентифи-
кация органических соединений. Пер. с англ.—М.: Мир, 1977, гл. 4.
Определение функциональных групп и установление структуры 305
Как видно из данных табл. 6.5, сахариды содержат обычные
органические функциональные группы, поэтому более подробное
описание их ИК-спектров можно найти в тех разделах книги, где
рассматриваются эти группы.
Масс-спектры углеводов из-за их низкой летучести можно по-
лучить, только применяя специальные методы. Например, по-
скольку углеводы обычно возгоняются с большим трудом, для их
масс-спектрального анализа часто используют метод прямой иони-
зации. Кроме того, с помощью доступных реагентов можно пре-
вратить углеводы в более летучие полиэфиры (например, в три-
метилсилиловые), которые затем можно ионизировать обычными
методами. Анализ масс-спектров затрудняется также и сложным
характером распада, свойственным большей части углеводов и их
производных.
Многие сахариды, а также другие углеводы и их производные
являются хиральными соединениями. Поэтому очень полезно опре-
делить их удельное вращение [а]. Методы определения [а] опи-
саны в гл. 8. Анализ ЯМР-спектров углеводов можно упростить
при использовании лантаноидных сдвигающих реагентов (гл. 8 и 10).
Озазоны в качестве производных можно получать по методике
24; для целей идентификации можно также использовать поли-
ацетаты и другие полиэфиры карбоновых кислот. Методы их по-
лучения описаны в разд. 6.1.1.
6.12. КАРБОНОВЫЕ КИСЛОТЫ
Карбоновые кислоты (RCOOH) обычно относятся по раствори-
мости к группе А]. Кроме того, присутствие водорастворимых ки-
слот можно установить с помощью индикаторов. Для этого исполь-
зуют два индикатора — конго красный и универсальный инди-
катор, применяемые либо в виде раствора, либо в форме инди-
каторной бумаги. Однако кислую реакцию дают не только карбо-
новые кислоты. Сульфокислоты, некоторые нитрозамещенные фе-
нолы и другие органические соединения также могут изменять
цвет индикаторов.
Индикатор Наблюдаемый цвет
Универсальный («гидрион») Конго красный Различный, зависит от величины pH Синий цвет указывает на присут- ствие кислоты
В тех редких случаях, когда возникают трудности при уста-
новлении кислотного характера вещества, можно использовать
следующие реакции, описанные в книге Черониса, Энтрикина и
Ходнетта: иодат-иодидный реагент (с. 359), родамин-Б/уранилаце-
тат (с. 359) и выделение азотистой кислоты (с. 360).
306
Глава 6
Присутствие карбоксильной группы (СООН) в органическом
соединении довольно просто устанавливается с помощью спек-
трального анализа. Для этой цели чаще используют ИК-спектры.
Типичные ИК-спектры карбоновых кислот приведены на рис. 6.29.
Рис. 6.29. Инфракрасный спектр п-хлоркоричнон кислоты (таблетки с КВг *).
Валентные колебания О-Н: 1 — 3226 - 2326 см-1 (3,10-4,30 мкм), (ассоциированная)**.
Валентные колебания С=О: 2—1695 см-1 (5,93 мкм). (сопряженная). Валентные
колебания С=С: 3 — 1634 см~1 (6,12 мкм), v^,_(сопряженная). Валентные колебания
С=="С: 4—1630, 1577, 1493 см-1 (6,25, 6,35, 6,70 мкм), v^, Валентные колебания С—О,
деформационные колебания О —Н (плоские): 5—1425, 1307 см-1 (7,02, 7,65 мкм), V в
С—(J О—гР
связанные. Деформационные колебания «С-Н (плоские): 6—1282, 1227, 1087 см”1 (7,80, 8,15,
9,20 мкм), б (плоские). Деформационные колебания =С-Н (неплоские олефиновые):
—н
7 988 см (10,12 мкм), —Н (неплоские олефиновые). Деформационные колебания О—Н
(иеплоскне): 8—около 930 см”1 (около 10,75 мкм), б_ (неплоские). Деформационные коле*
О — Н
бания к=С—Н (неплоскне): 9—823 см”1 (12,15 мкм), б (неплоские); 10-715 см”1 (13,98 мкм),
н
б_с__(неплоскне).
* Калибровка: поскольку длина волны (мкм) при записи слегка увеличивается, для всех
полос это значение надо уменьшить на 0,1 мкм.
* * Типичная широкая полоса днмеров карбоновых кислот.
Наличие карбоксильной группы проявляется в виде широкой по-
лосы поглощения в области 3,00—4,00 мкм (3333—2500 см-1), часто
с несколькими максимумами. Эта широкая полоса возникает, по
крайней мере частично, из-за поглощения димеров кислот, т. е.
двух молекул кислоты, соединенных водородными связями:
Н-о
R—Ce+ С—R
хонс/
в- 6 +
Кроме того, обнаружению карбоксильной группы помогают силь-
ные полосы валентных колебаний С=О (5,81— 5;,86 мкм, 1720—
Определение функциональных групп и установление структуры
307
1706 см-1) и С—О (7,58—8,26 мкм, 1320—1210 см-1). Колебания
С-0 используются особенно часто ввиду того, что положение этой
полосы зависит от л-сопряжения с карбонильной группой, ана-
логично описанному выше для альдегидов и кетонов (разд. 6.2.).
В частности, валентные колебания С=О в «.^-ненасыщенных кис-
лотах находятся в области 5,85—5,95 мкм (1710—1680 см-1).
Высокая полярность карбоксильной группы приводит к тому,
что карбоновые кислоты крайне малоподвижны в условиях хро-
матографии.
ЯМР-спектры, хотя и не в такой степени, как ИК-спектры, так-
же можно использовать для обнаружения карбоксильной группы.
На рис. 6.30 приведен типичный ПМР-спектр карбоновой кислоты.
Сигнал протона карбоксильной группы обычно находится в области
б 10,0—13,3 м. д. Студентов вначале может привести в замеша-
тельство тот факт, что положение сигнала протона одной и той же
кислоты для разных растворов меняется. Это связано с тем, что
положение сигнала зависит от концентрации кислоты и темпера-
туры. Сигнал протона карбоксильной группы растворов кислот в
ДМСО-de может находиться в существенно более сильном поле,
чем б 10,0. Проверить отнесения сигнала к поглощению карбо-
ксильного протона можно добавлением к образцу D2O. При этом
в спектре исчезает сигнал ОН, соответствующий карбоксильной
группе, и появляется сигнал протонов воды около б 4,8 м.д.
Трудно ожидать, что применение масс-спектрометрии позволит
уточнить молекулярную формулу или. молекулярную массу кар-
боновой кислоты, поскольку пики молекулярного иона в их масс-
спектрах очень слабы.
Фрагментация молекулы кислоты может дать ценную инфор-
мацию о направлении распада:
Фрагмент Масса Примечание
R—С=О+ R+ RiR2C=C(OH)2 M*=RCOOH* М- 17 М — 45 58 + R, + R2 Более заметно для малых R То же Перегруппировка Мак-Лафферти
Одной из наиболее полезных характеристик карбоновых кислот
является эквивалент нейтрализации, который определяется коли-
чественным титрованием кислоты стандартным раствором щелочи
308
Глава 6
Определение функциональных групп и установление структуры 309
(методика 32). Молекулярная масса кислоты в пределах экспери-
ментальных ошибок равна целому числу (обычно 1, 2, 3 и т. д.),
умноженному на эквивалент нейтрализации.
Методика 32. Эквивалент нейтрализации
RCOOH + NaOH —> RCOONa ± Н2О
Пробу исследуемой кислоты около 0,2 г взвешивают с точностью до треть-
его знака после запятой и затем растворяют в 50—100 мл воды или этанола.
Смесь нагревают, если это необходимо, до полного растворения кислоты. Полу-
ченный раствор титруют 0,1 и. раствором гидроксида натрия с фенолфталеином
в качестве индикатора. Титр этого раствора (N) определяют заранее. Эквива-
лент нейтрализации кислоты рассчитывается по формуле
„ масса пробы • 1000
эквивалент нейтрализации = — -----———г—гг
объем щелочи (мл) • N
Обсуждение. Молекулярную массу кислоты (М) можно опреде-
лить, умножая эквивалент нейтрализации на число кислотных
групп (х) в молекуле:
М = х •(эквивалент нейтрализации)
Изменение характера среды, например переход от чистой воды
к чистому этанолу, влияет на рК кислоты и индикатора. Поэтому
наилучшие результаты получаются при титровании в воде или вод-
ном этаноле. При этом берут минимальное количество спирта, не-
обходимое для растворения органической кислоты. В абсолютном
или 95%-ном этаноле часто бывает трудно добиться четкого изме-
нения цвета фенолфталеина. В этих случаях следует в качестве
индикатора использовать бромтимоловый синий. Кислоты можно
титровать и в смешанном растворителе — в смеси спирта с бен-
золом или толуолом.
Во всех случаях следует проводить холостой опыт с применя-
емым растворителем. При этом берут такое же количество фенол-
фталеина, как и при титровании исследуемой кислоты. При обыч-
ной технике выполнения работы эквивалент нейтрализации опре-
деляют с точностью ±1%. Если использовать хорошо очищенные
и высушенные образцы и очень тщательно проводить титрование,
то ошибку определения можно уменьшить до ±0,3%.
Для того чтобы получить точную величину эквивалента нейтра-
лизации, титруемое вещество должно быть чистым и сухим. Если
полученное значение эквивалента нейтрализации заметно отлича-
ется от ожидаемого теоретического значения, то вещество следует
перекристаллизовать из подходящего растворителя и тщательно
высушить.
Таким же способом можно титровать соли аминов и сильных
кислот.
Контрольные вопросы: 1. Рассчитайте эквивалент нейтрализации бензойной
и фталевой кислот.
310
Глава 6
2. Если кислота плохо высушена, то больше или меньше будет ее эквива-
лент нейтрализации?
3. Будет ли сказываться иа величине эквивалента нейтрализации присутст-
вие в молекуле ароматической аминогруппы? Будет ли оказывать такое же влия-
ние алифатическая аминогруппа?
4. Какие типы фенолов можно титровать количественно?
5. Объясните с теоретической точки зрения, какой должна быть константа
ионизации для того, чтобы кислоту можно было титровать с фенолфталеином?
Для характеристики кислот в качестве производных пригодны
амиды и эфиры. Методики получения и превращения этих соеди-
нений в другие продукты описаны в различных разделах данной
главы.
д КСООИ’Гсложнь/е зриры, разд. 6.13)
(RCO)2O ---•—-<---- RCOOH
(ангидриды кислот, I
разд. 6.12.1)
RCONH.V
V (амиды, разд. 6.12.3)
'RCOCI
(хлорангидриды,
разд. 6.12.2)
Для характеристики кислот полезно использовать их твердые
эфиры. Хотя некоторые метиловые эфиры и представляют собой
твердые соединения, однако в большинстве случаев получают
n-нитробензиловые, фенациловые n-хлорфенациловые, д-бромфе-
нациловые и n-фенилфенациловые эфиры обработкой солей кислот
соответствующими галогенидами (методика 33).
RCOONa + «-C1CH2C6H4NO2 —> n-RCOOCH2C6H4NO2 + NaCl
RCOONa 4- BrCH2COAr —> RCOOCH2COAr + NaBr
Этот метод удобен тем, что не требует высушивания кислоты.
Хорошим производным является также продукт взаимодействия
фенилгидразина с исследуемой кислотой. При температуре плавле-
ния фенплгидразина (243°С) простые незамещенные алифатические
одно- и двухосновные кислоты образуют фенилгидразиды (мето-
дика 34).
RCOOH + H2NNC6H5 —> RCONHNHC6H5 + Н2О
При нагревании бензольного раствора фенилгидразина и суль-
фокислот, а-хлоралифатических кислот и двухосновных алифати-
ческих кислот образуются соответствующие соли фенилгидразина.
При обработке тионилхлоридом и аммиаком кислоты можно
превратить в амиды:
RCOOH + SOC12 —> RCOC1 4- SO2 4- HC1
RCOC14-2NH3 —> RCONH2 4-NH4C1
Этот способ особенно удобен в тех случаях когда образую-
щиеся амиды нерастворимы в воде. Амиды можно получить, обра-
Определение функциональных групп и установление структуры 311
батывая хлорангидрид кислоты концентрированным водным раст-
вором аммиака (методика 35).
Очень хорошими производными кислот являются анилиды, то-
луидиды и броманилиды, поскольку их получение и очистка про-
текают без особых затруднений. Эти производные можно получить
как из свободных кислот, так и из их солей (методика 36).
Часто в качестве производных применяются соли органических
кислот и сильных основных аминов, таких, как бензиламин, а-
фенилэтиламин и пиперазин.
RCOO
Карбоновые кислоты и их производные можно также превра-
тить в тиурониевые производные (методика 39).
Методика 33. Получение n-иитробензильных, фенацильных, п -хлорфенацильных,
n-бромфенацильных и n-феиилфенацильных эфиров кислот
СН2С1
или
RCOO'Na'
АгСОСН2Вг
О
II
RCOCH2Af
или
О
II
RCO—СН2САг
+ NaX
О
Аг — феиильиая или замещенная феиильиая группа
В маленькой колбе к 1 г кислоты прибавляют 5 мл воды и осторожно ней-
трализуют 10%-ным раствором гидроксида натрия. Прибавляют небольшое коли-
чество кислоты до кислой реакции иа лакмус. Если исходная кислота взята в
форме натриевой соли, то 1 г этой соли растворяют в 5—10 мл воды. Если полу-
ченный раствор имеет щелочную реакцию, то его подкисляют одиой-двумя кап-
лями разбавленной соляной кислоты. К смеси приливают 10 мл спирта и 1 г га-
логенида (Будьте осторожны} Феиацилгалогеииды слезоточивы) и нагревают с
обратным холодильником 1 ч в случае одноосновной кислоты, 2 ч — для двух-
основной и 3 ч — для трехосиовиой. Если во время кипения образуется осадок,
то следует периодически прибавлять еще несколько миллилитров спирта. Смесь
охлаждают и выпавший эфир кислоты очищают перекристаллизацией из спирта
При проведении этой реакции необходимо следить за тем, чтобы
исходная смесь не имела щелочной реакции, поскольку щелочи
гидролизуют фенацилгалогениды в фенациловые спирты. В тех слу-
чаях, когда в натриевой соли кислоты присутствует заметное ко-
личество хлорида натрия, нельзя использовать для проведения ре-
акции п-бромфенацилбромид.
Образование эфиров кислот облегчается в присутствии краун-
эфиров. Так, из ацетата калия и н-гептилбромида с количествен-
ным выходом образуется н-гептилацетат *.
Gokel G., Durst И., Synthesis, 1976, 178.
312
Глава 6
Методика 34. Получение феиилгидразидов и фенилгидразоновых солей
a) RCOOH + C6H6NHNH2
б) RCOOH + C6H6NHNH2
или
RSO3H + CcH6NHNH2
О
нагревание ||
--------->- RCNHNHCeHE
{О
II
RCNHNHC6H6
или
RSO3 +NH3NHC6H5
а) Растворяют 1 г кислоты в 2 мл фенилгидразина и смесь осторожно ки-
пятят около 30 мин. Выделившийся при охлаждении кристаллический продукт
отсасывают на фильтре и промывают небольшим количеством бензола (бензол
ядовит, см. примечание к методике 20) или эфира, пока кристаллы не приобре-
тут белого цвета. При использовании большого избытка фенилгидразина для
того, чтобы осадок выпал, иногда приходится разбавлять смесь бензолом. Произ-
водные низших одноосновных кислот перекристаллизовывают из горячего бен-
зола, а производные высших и двухосновных кислот лучше всего перекристалли-
зовывать из этанола или водно-спиртовых смесей. Двухосновные кислоты дают
по этой методике бис-В-фенилгидразиды.
б) Смешивают 1 г кислоты с 2 мл фенилгидразина и смесь растворяют
в 5 мл бензола. В некоторых случаях тотчас выпадает белый осадок. Его отде-
ляют и перекристаллизовывают из этанола. Если осадок ие выпадает, то смесь
нагревают с обратным холодильником около 30 мии. Продукт, выпадающий при
охлаждении, отфильтровывают, промывают эфиром и перекристаллизовывают
из бензола или этанола. По этой методике можно получить соли сульфокислот,
галогенозамещеиных алифатических кислот и двухосновных алифатических кис-
лот; простые незамещенные алифатические кислоты дают в этих условиях фе-
нилгидразиды.
Методика 35. Получение амидов из карбоновых кислот
О О
II NH3 ||
RCOOH + SOC12 -у* RCC1 -------> RCNH2
(RCOC1 4- R'NH2 —> RCONHR')
В течение 15—30 мин нагревают с обратным холодильником 1 г кислоты
с 5 мл хлористого тионила. Смесь осторожно выливают в 15 мл охлажденного
льдом концентрированного водного раствора аммиака. Выпавший амид отфиль-
тровывают и перекристаллизовывают из воды или разбавленного этанола.
Методика 36. Получение анилидов, n-толуидидов и n-бромаиилидов
SOC12 ArNH2
a) RCOOH --------> RCOC1 -------RCONHAr
HCl/ArNH2
б) RCOO’Na* -----------> RCOONHAr
а) Смешивают в пробирке 1 г кислоты или ее натриевой соли с 2 мл тио-
нилхлорида и смесь нагревают с небольшим обратным холодильником 30 мин.
После охлаждения прибавляют раствор 1—2 г амина (анилина, n-толуидина или
n-броманилина) в 30 .мл бензола и смесь нагревают на кипящей водяной бане
Определение функциональных групп и установление структуры
313
2 мин. Бензольный раствор декантируют в делительную воронку и последова-
тельно приливают 2 мл воды, 5 мл 50 %-ион соляной кислоты, 5 мл 50%-ного
раствора гидроксида натрия и 2 мл воды. Бензол упаривают, а амид перекри-
сталлизовывают из воды или этанола.
б) В пробирку помещают 0,4 г сухой порошкообразной натриевой соли кис-
лоты, 1 мл ариламина и 0,3 мл концентрированной соляной кислоты. Смесь на-
гревают 45—60 мии иа масляной бане при температуре 150—160°С, после чего
пробирку вынимают из бани и очищают продукт одним из описанных ниже
методов.
1. В том случае, когда исследуемая кислота содержит меньше 6 атомов уг-
лерода, полученный продукт кипятят с 5 мл 95%-иого этанола и раствор декан-
тируют в 50 мл горячей воды. Этот раствор упаривают до объема 10—12 мл и
охлаждают льдом. Выпавшие кристаллы отфильтровывают и перекристаллизо-
вывают из небольшого количества воды или разбавленного этанола.
2. Если исходная кислота содержит больше 6 атомов углерода, то получен-
ный продукт измельчают и промывают 15 мл 5%-ной соляной кислоты, а затем
15 мл холодной воды. Остаток кипятят с 30—40 мл 50%-иого этанола и рас-
твор фильтруют. Фильтрат охлаждают льдом и выпавшие кристаллы замещен-
ного амида отфильтровывают. Продукт можно перекристаллизовать из разбав-
ленного спирта.
Карбоновые кислоты можно изучать с помощью ЯМР-спектро-
скопии с использованием лантаноидных сдвигающих реагентов;
с этой целью лучше применять реагенты с более высокофториро-
ванными лигандами: Eu(fod)s и Eu(tfn)3 (гл. 8).
6.12.1. Ангидриды кислот
При идентификации ангидридов классификация по раствори-
мости может оказаться бесполезной или даже ввести в заблужде-
ние. Это связано с тем, что ангидриды быстро превращаются в воде
в соответствующие кислоты, особенно при использовании в каче-
стве катализаторов минеральных кислот:
н*
(RCO)2O + H2O ---> 2RCOOH
Во многих случаях результаты, полученные при изучении раство-
римости, могут в действительности относиться к соответствующим
кислотам.
Спектральный анализ необходимо проводить с соблюдением
всех предосторожностей, какие только возможны, чтобы избежать
контакта ангидрида с влагой. ИК-Спектр типичного ангидрида
приведен на рис. 6.31. Наиболее существенной особенностью ИК-
спектра ангидридов является наличие в нем двух валентных ко-
лебаний карбонильной группы. Это происходит из-за наложения
симметричных и несимметричных колебаний двух карбонильных
групп *.
* В алифатических ациклических системах полоса, соответствующая анти-
симметричным колебаниям, несколько сильнее. В циклических ангидридах с пяти-
членным циклом, напротив, сильнее полоса симметричных колебаний.
314
Глава 6
Длина волны, мкм
о
I
о
Определение функциональных групп и установление структуры
315
симметричные колебания около 5,19 мкм
(~1738 см-1), сильное поглощение
антисимметричные колебания около 5,48 мкм
(~1825 см-1), сильное поглощение
Сопряжение карбонильных групп с зх-электронной системой при-
водит к некоторому сдвигу полосы поглощения в более длинно-
волновую область (в сторону меньших частот).
ПМР-Спектры ангидридов сходны со спектрами соответству-
ющих кислот. Но конечно, очень чистые и сухие образцы не дают
сигнала карбоксильного протона. Типичный ПМР-спектр ангид-
рида показан на рис. 6.32.
Масс-спектры ангидридов во многих случаях не имеют пика
молекулярного иона. Фрагментацию ациклических ангидридов ча-
сто можно предсказать: они распадаются так же, как и карбоно-
вые кислоты:
—♦ RCO+ R+ [R-HJ+
часто этот фрагмент дает
основной пик
Распад циклических ангидридов более сложен *.
Производные ангидридов карбоновых кислот (RCO)2O могут
быть синтезированы по тем же методикам, что и производные са-
мих кислот. В результате превращения ангидрида в кислоту можно
получить анилиды RCONHAr (методика 366) или амиды RCONH2
(методика 35, в этом случае нет необходимости использовать ти-
онилхлорид). Ниже на примере фталевого ангидрида показано
образование N-фталимида при реакции с анилином
о
о
* Budzikiewicz Н., Djerassl С., Williams D. И., Mass Spectrometry of Organic
Compounds, Holden-Day, San Francisco, 1967,
316
Глава 6
Ю 9 8 1 6 5 4 3 2 1 О М.Ъ.
Рис. 6.32. Спектр протонного магнитного резонанса уксусного ангидрида (60 МГц, развертка 600 Гц, растворитель CDC13).
Определение функциональных групп и установление структуры
317
Реакция Шоттена — Баумана (опыт 18д) приводит к образова-
нию из ангидридов сложных эфиров. Эта реакция может проходить
не только с галогенангидридами, но и при простой обработке ан-
гидридов спиртом:
RCOC1 —I
(RCO)2O -I
R'OH
----—+ RCOOR'(RCOOAr)
(нли АгОН)
При реакции спиртов с галогенангидридами двухосновных
кислот образуются обычные эфиры спиртов, а при реакции с ан-
гидридами двухосновных кислот в качестве основного продукта по-
лучают кислые эфиры (полуэфиры). Фталевый ангидрид со спир-
том дает кислый фталат. Диэфир образуется только при обработке
избытком спирта в присутствии катализатора.
6.12.2. Галогенангидриды кислот
Галогенангидриды, так же как и ангидриды, очень легко гидро-
лизуются с образованием карбоновой и минеральной кислот. Это
может быть причиной ошибочных выводов при изучении их раство-
римости, как это уже было описано в предыдущем разделе при рас-
смотрении реакций ангидридов кислот. При гидролизе галогенан-
гидридов образуется по одной молекуле органической и неоргани-
ческой кислот:
RCOC1+H2O —> RCOOH 4-НС1
Вследствие этого «результаты» изучения растворимости будут
на самом деле характеризовать продукты гидролиза. Хлорангид-
риды, наиболее часто встречающиеся соединения этого класса, мо-
гут быть идентифицированы по реакциям «подвижного» атома
галогена (спиртовой раствор нитрата серебра, опыт 16). Более ле-
тучие галогенангидриды можно распознать просто по их неприят-
ному запаху и слезоточивым свойствам.
Как и в случае ангидридов, при проведении спектральных ис-
следований необходимо предпринимать меры предосторожности,
чтобы исключить попадание влаги в исследуемый образец. ИК-
Спектр типичного хлорангидрида приведен на рис. 6.33. Полоса
валентных колебаний карбонильной группы в алифатических хлор-
ангидридах находится в области 5,51—5,60 мкм (1815—1785 см-1).
Сопряжение карбонильной группы с зт-электронной системой сдви-
гает эту полосу на 0,05 мкм (уменьшает частоту на 15 см-1).
Рис. 6.33. Инфракрасный спектр бензоилхлорида в чистом виде.
Валентные колебания С—Н: 7 — 3070 см“‘ (3,26 мкм), vQ_H (ароматические). Валентные колебания C=Ot 2—1.770 см (5,65. мкм), v(,_o;.
о__1727 см-1 (5 79 мкм) полоса за счет Ферми-резонанса частоты 2' н обертона 872 см-L (11,47 мкм). Валентное колебание С—С1: 4 — 872 см“1‘
' (11,47 мкм), vc_Cj.
Рис. 6.34. Спектр протонного магнитного резонанса бензоилхлорида (60 МГц, развертка 600 Гц, растворитель CDC13).
Определение функциональных групп и установление структуры 319
320
Глава 6
Полоса поглощения карбонильной группы в галогенангидридах
часто имеет два максимума.
ПМР-Спектры галогенангидридов очень походят на спектры со-
ответствующих кислот. Пример такого спектра приведен на рис.
6.34. При снятии спектров недопустимо использовать гидроксилсо-
держащие растворители, так как они способны взаимодействовать
с галогенангидридами. В ПМР-спектрах тщательно очищенных и
высушенных образцов галогенангидридов отсутствуют сигналы кар-
боксильного протона. Точно так же в этих случаях в ИК-спектрах
отсутствуют полосы, соответствующие валентным колебаниям свя-
зи О—Н.
Галогенангидриды часто можно идентифицировать, синтезируя
анилиды при взаимодействии их с анилином (методика 36). Если
соответствующие амиды нерастворимы в воде, то их можно полу-
чить, обработав галогенангидрид концентрированным водным раст-
вором аммиака.
Галогенангидриды двухосновных кислот реагируют с избытком
анилина при обычной температуре, давая дианилиды. Если же на-
гревать с анилином ангидрид двухосновной кислоты, то образуются
N-фенилимиды, которые часто используют для идентификации.
'СООН
,СООН
SOCI,
При гидролизе галогенангидридов (или ангидридов) образуются
соответствующие кислоты. В тех случаях, когда эти соединения
представляют собой твердые вещества, их часто используют в ка-
честве производных. В других случаях галогенангидрид (или ан-
гидрид) можно гидролизовать разбавленной щелочью и нейтрали-
зовать полученный раствор соляной кислотой по фенолфталеину.
После упаривания растворителя получают смесь натриевой соли
кислоты и хлорида натрия. Полученные соли можно использовать
для получения твердых эфиров кислоты по методике 33.
Галогенангидриды (и ангидриды) кислот реагируют со спирта-
ми и фенолами, давая соответствующие сложные эфиры (опыт 18г),
которые можно использовать для их идентификации (разд. 6.13).
6.12.3. Амиды кислот
В связи с тем, что амиды содержат азот и являются нейтраль-
ными по отношению как к кислотам, так и к щелочам, они обычно
относятся к классу растворимости MN. Исключение составляют
те амиды, в которых благодаря взаимодействию амидного азота
с достаточно сильной электроноакцепторной группой обычно ней-
Определение функциональных групп и установление структуры
321
тральный амидный протон становится более кислым. Так, напри-
мер, достаточно кислый характер имеют имиды.
О
Омиды; R—CNH2 — Х> RCONFT
о о
, II II
имиды-. RCNHCR
+ Н2О
Однако обычно амиды не взаимодействуют с 5%-ной соляной
кислотой или с 5%-ным гидроксидом натрия при комнатной тем-
пературе. Действительно, первоначальное выявление амидов часто
проверяется именно по отсутствию реакции с кислотой или ще-
лочью.
Для определения и диализа амидных групп особенно удобны
ИК-спектры. На рис. 6.35 приведен ИК-спектр типичного амида.
Рис. 6.35. Инфракрасный спектр N.N-диметилформамида (ДМФА) в чистом виде.
Валентные колебания С—Н: 1 — 2915 см-1 (3,43 мкм), V„ (алифатические); 2 — 2850 см-1
С —н
(3,51 мкм). v СНО-группы. Полосы амнда I и II: 3—1675 см”"1 (5,97 ммк), перекры-
ваются полосы *с_о (амид I) с деформационными колебаниями N—Н, (амид II). Полоса
С—N: 4—1387 см“' (7,22 мкм), v^_^; 5—калибровочная отметка: полоса 1583 см-1 (6,33 мкм)
полистирола (нет необходимости вводить поправки при определении положения полос).
Подобно тому как это имеет место в случае аминов (см. разд.
6.7), на основании наличия полосы поглощения или ее мультип-
летности можно сделать заключение о структуре амида, т. е. уста-
новить, является ли изучаемый амид первичным, вторичным или
третичным. Другие выводы о структуре амида (I, II или III) поз-
воляет сделать наличие полос поглощения в ИК-спектре амида,
связанных главным образом с колебаниями карбонильной груп-
пы. Эти полосы позволяют получить более детальные сведения о
структуре первичных, вторичных и третичных амидов.
11 Зак. 338
322
Глава 6
Для основных структур амидов, приведенных ниже, характер-
ны следующие особенности в ИК-спектрах:
О rcnh2 первичный амид; имеет две полосы поглощения N—Н ПМР-Спектр типичного О О II II RCNHR RCNR2 вторичный амид; третичный амид; имеет одну полосу полосы поглощении поглощения N—Н N—Н отсутствуют амида приведен на рис. 6.36. Наиболее
часто для характеристики амидов используют следующие сигналы:
С, м. д. Отнесение сигнала
2,0—2,4 а —Ан—CONR2
1,95-2,85 а 5,0-8,5 rconhAh— RCONHR'
а Обычно метильный протон дает сигнал в более сильном поле (меньшие значения 6),
метиленовый—в середине этого интервала, а метиковый—в более слабом поле (большее
значение 6).
Хотя протон в амидной группе N—Н не участвует в быстрых
обменных процессах, подобно протону в аминах, однако сигнал
этого протона в спектре ЯМР также уширен. Такое уширение про-
исходит вследствие взаимодействия с квадрупольным моментом
ядра атома азота. Таким образом, уширенный синглет N—Н-груп-
пы не является неразделенным мультиплетом, что позволяет иден-
тифицировать сигнал атомов водорода, находящихся у связан-
ного с азотом углеродного атома. Сигналы атомов водорода у
такого углерода могут, однако, расщепляться в результате взаимо-
действия с протоном N—Н-группы:
О
~ Я, Hi — уширенный синглет
|| Н2 — разделенный мультиплет
Н2 Н,
Типичное значение Ji2 около 5 Гц.
Вращение амидной группы существенно затруднено из-за боль-
шого вклада резонансной формы Б.
:&) :О-
„. IK |
R c *—' R~C4.
N
R
Б
Определение функциональных групп и установление структуры
323
Ю 981 654321 Ом.д.
Рис. 6.36. Спектр протонного магнитного резонанса ацетанилида (60 МГц, развертка 600 Гц, растворитель CDCla).
11*
324
Глава 6
Подтверждением этого может служить ЯМР-спектр М,М-диметил-
формамида, в котором имеются два отдельных сигнала неэквива-
лентных метильных групп.
‘-о
А сн,
Н .X
CHj
В масс-спектрах амидов обычно имеется ясно выраженный пик
молекулярного иона. Знать молекулярную массу амида очень по-
лезно, так как соединение с четной величиной молекулярной массы
либо должно иметь четное число атомов азота, либо не содержать
азота вообще. Соединение с нечетной величиной молекулярной
массы должно иметь нечетное число атомов азота.
Некоторые важные фрагменты, типичные для масс-спектров
амидов, приведены ниже:
Фрагмент Масса Примечание
[О=С-NR2]+ Г /ОН "I ‘ RaC=c/ L XhJ 42 + 2R 57 4-2R Интенсивный пик у первичных ами- дов (R = H); в случае вторичных и третичных амидов используется для установления массы замести- теля R Результат расщепления по Мак- Лафферти. При R=H масса рав- на 59; основной пик в перввчных амидах, имеющих у-атом водоро- да
Расщепление по Мак-Лафферти, описанное выше, иллюстрирует
важность численных соотношений между массами молекулярных
ионов и образующихся из них фрагментов. Если соединение имеет
четную молекулярную массу, то в результате простого расщепле-
ния обычно образуются фрагменты с нечетной массой. Аналогично
простое расщепление соединений с нечетной молекулярной массой
дает фрагменты с четной массой. Под простым расщеплением по-
нимают разрыв только одной связи в молекуле. Так, бутирамид
(масса 87) дает в качестве основного пик с т/е 59. Очевидно, что
этот фрагмент не мог образоваться в результате простого расще-
пления. Действительно, при расщеплении происходит перегруппи-
ровка, включающая переход протона (перегруппировка Мак-Лаф-
ферти) :
:о;
ch,ch2ch2cnh2
т/е 87
Н
?О:
CH, NH2
ш/е 59
Определение функциональных групп и установление структуры 825
Вторичные и третичные амиды, имеющие у азота разветвленные
алкильные заместители, распадаются более сложным путем. Пути
распада таких амидов описаны в литературе, приведенной в гл. 10.
Наиболее общий метод химической идентификации амидов —
их кислый или щелочной гидролиз (методика 37):
RCONH2 + Н2О + НС1 —> RCOOH+ NH4C1
RCONH2 + NaOH —> RCOO'Na* + NH3
При гидролизе замещенных амидов вместо аммиака образу-
ются первичные или вторичные амины:
RCONHR'+ NaOH —> RCOO'Na* + R'NH2
RCONRz + NaOH —> RCOO"Na+ + R'NH
Идентификация амидов может включать получение производных
продуктов гидролиза, т. е. кислоты, аммиака и первичного или вто-
ричного амина. Соответствующие методики для кислот даны в разд-
6.12, для солей кислот — в разд. 6.12.4 и для аминов —в разд. 6.7.
Хорошими производными для идентификации незамещенных
амидов и имидов являются 9-ациламидоксантены, которые образу-
ются при реакции амидов и имидов с ксантгидролом (методика 38),
Описаны и другие полезные методики получения производных
амидов. Черонис, Энтрикин и Ходнетт приводят ряд капельных ре-
акций:
NOH
(1) 3NH2OH-HC1 II
8RCONH’ --------------* (RCO)sFe
красно-фиолетовое
окрашивание
(реакция с гидроксаматом железа, см. Черонис, Энтрикин и Ход-
нетт, с. 373; N-замещенные амиды этой реакции не дают).
NOH
6%-иый HjOz FeCla ||
ArCONH2 --------[ArCONHOH] -----------> (ArCO)3Fe
сине-красное
окрашивание
(Черонис, Энтрикин и Ходнетт, с. 374; алифатические амиды пе-
роксидом водорода до гидроксамовой кислоты не окисляются).
Полезным твердым производным являются N-ацилфталимиды,
'образующиеся при взаимодействии незамещенных амидов с фта-
'лилхлоридом:
NCOR + 2НС1
J. Аш. Chem. Soc„ 51, 3651 (1929).
826
Глава 6
При реакции оксида ртути (II) с некоторыми амидами обра-
зуются высокоплавкие твердые производные Ы.М'-ртуть-бпс-ацил-
амиды:
HgO + 2RCONH2 —-> Hg(NHCOR)2 4-Н2О
J. Am. Chem. Soe., 64, 1738 (1942)
Некоторые амиды образуют со щавелевой кислотой твердые
соли, которые можно использовать для идентификации:
RCONH2 + (СООН)2 —> соль
Ind. Eng. Chem., Anal. Ed., 12, 737 (1940)
По той же методике, которая используется для получения п-нит-
робензиловых эфиров кислот (методика 33), можно синтезировать
М,1М-/ьнитробензильные производные барбитуратов, вводя их в ре-
акцию с п-нитробензилбромидом.
n-O,NC.H4CH2Br
Na,CO3
R
N—CH2C6H4NO2(n>
N—CH2C6H4NO2(n)
J. Am. Chem. Soc., 66, 1440 (1944)
Методика 37. Гидролиз амидов, нитрилов и нитроанилииов
а) Щелочной гидролиз
RCONH2 4- NaOH —> RCOONa + NHS
RCONHR' 4- NaOH —> RCOONa 4- R'NH2
RCONR£ 4-NaOH —> RCOONa 4- R2NH
RCN 4-NaOH 4-H2O —> RCOONa 4-NH3
В пробирке 0,2 г мочевины обрабатывают 5 мл 10%-ного раствора гидро-
ксида натрия. Смесь встряхивают и наблюдают выделение аммиака. Повторяют
эту реакцию с бензамидом, ацетанилидом, бензонитрилом и 2,4-динитроанили-
ном. После окончания выделения аммиака нагревают каждую из пробирок до
кипения и отмечают запах. Проверяют действие выделяющихся паров на влаж-
ную розовую лакмусовую бумажку.
Охлаждают полученные растворы, подкисляют их соляной кислотой и от-
мечают результат. Что означает образование осадка?
б) Кислотный гидролиз. Многие замещенные амиды легче гидролизуются
при нагревании с 20%-ной серной кислотой (с обратным холодильником):
2RCONHR'4-H2SO4 4-2Н2О —> 2RCOOH 4- (R'NH3)2SO4
2RCONR2 4~ H2SO4 4-2Н2О —> 2RCOOH 4-(R£NH2)2SC)4
Если образующиеся кислоты летучи, то их можно выделить перегонкой.
Если же эти кислоты нерастворимы в воде, то их отделяют фильтрованием.
Амииы могут быть выделены при добавлении щелочи и охарактеризованы в
виде соответствующих аренсульфонильных производных.
Нитрилы, в частности циангидрины, часто подвергают гидролизу, превращая
их в кислоты. При обработке 90—95%-ной серной кислотой или концентрирован-
ной соляной кислотой при температурах от 10 до 50°С нитрилы превращаются в
Определение функциональных групп и установление структуры 327
амиды. Затем амиды могут быть гидролизованы в кислоты. Для этого смесь раз-
бавляют водой и нагревают с обратным холодильником в течение 0,5—2 ч.
н2О н2О
RCN ц * RCONH2 „ RCOOH + NH4HSO4
H2SO4 H2SO4
Обсуждение. Образующиеся при щелочном гидролизе аммиак
или амины могут быть охарактеризованы с помощью реакции
Хинсберга (методика 16). Для этой цели лучше использовать боль-
шие пробы (1 г) и отгонять аммиак или летучие амины из щелоч-
ного раствора в приемник, содержащий разбавленную соляную кис-
лоту. Этот раствор далее нейтрализуют и обрабатывают по мето-
дике 16.
Нерастворимые в воде амины можно экстрагировать эфиром.
Затем эфир отгоняют и идентифицируют амины обычными реак-
циями. Водорастворимые нелетучие амины можно превратить в
аренсульфонильные производные. Эти вещества можно отделить
от органической кислоты, которая является другим продуктом гид-
ролиза, используя различия в растворимости или летучести.
Ариламины с нитрогруппой в орто- или пара-положении к ами-
ногруппе при гидролизе горячими щелочами дают соответствующие
нитрофенолы и аммиак или амины. Нитрозогруппа сходна с нитро-
группой по тому, как она влияет на подвижность заместителей,
находящихся по отношению к ней в орто- или пара-положении.
Например, и-нитрозодиалкиланилины гидролизуются щелочами с
образованием вторичных аминов и натриевой соли л-нитрозофе-
нола (монооксима бензохинона).
Одной из движущих сил реакций такого типа является акти-
вирование нитро- и нитрозогруппами орто- и пара-положений аро-
матического кольца. Это приводит к образованию комплекса Мей-
зенгеймера, который, обладая малой энергией, облегчает реакцию
нуклеофильного замещения.
комплекс
Мейзенг ей мера
328
Глава 6
Методика 38. Получение О-ациламидоксантеиов
Растворяют около 0,5 г ксантгидрола в 7 мл ледяной уксусной кислоты.
Если раствор окажется мутным нз-за присутствия продуктов диспропорциони-
рования ксантгидрола, то его оставляют стоять несколько минут или центри-
фугируют. Прозрачный раствор декантируют в чистую пробирку. К этому рас-
твору прибавляют 0,5 г амида и смесь нагревают при 85°С в стакане с горячей
водой в течение 20—30 мин. После охлаждения ациламидоксантен собирают на
фильтре и перекристаллизовывают из смеси диоксан — вода в соотношении 2:1.
Некоторые амиды плохо растворяются в уксусной кислоте, в этом случае ре-
акцию проводят в смеси, состоящей из 5 мл этанола, 2 мл ледяной уксусной
кислоты и 3 мл воды.
Применение лантаноидных сдвигающих реагентов облегчает
анализ ПМР-спектров амидов (гл. 8); связывание с реагентом про-
исходит, вероятнее всего, по кислороду карбонильной группы. Ре-
зонансные формы, приведенные ниже, увеличивают льюисовскую
основность атома кислорода. Такой кислород может реагировать
с атомом лантаноида, выступающим в качестве кислоты Льюиса.
А
R-<f — R-C^
(_NH2
6.12.4. Соли карбоновых кислот
Соли карбоновых кислот (RCOO~Na+) представляют собой ион-
ные соединения и вследствие этого резко отличаются от органиче-
ских соединений, которые содержат только ковалентные связи.
Обычно соли карбоновых кислот растворимы в воде, но нераство-
римы в эфире. В результате этого они относятся по растворимости
к классу S2, что также является основой для различных схем эк-
стракции карбоновых кислот растворителями (гл. 7).
Ионный характер этих солей особенно наглядно проявляется
в их чрезвычайно малой подвижности при хроматографическом ана-
лизе по сравнению с подвижностью ковалентных органических со-
единений. Кроме того, эти соли имеют обычно очень высокие тем-
пературы плавления, низкую летучесть и дают остаток после про-
каливания. Характер атома металла можно установить, используя
методы, описанные в руководствах по неорганической и аналити-
ческой химии (гл. 10).
На рис. 6.37 и 6.38 представлены соответственно ИК- и ПМР-
спектры типичных солей карбоновых кислот. Обратите внимание,
Определение функциональных групп и установление структуры
329
что для получения спектров применяют специальные приемы, ис-
пользующие физические свойства этих ионных солей.
Характерной полосой в ИК-спектрах солей является полоса,
соответствующая валентным колебаниям группы С (=0)2. Связь
С==0 в таких солях длиннее связи С = О в альдегидах и кетонах
и короче связи С — О в спиртах. В результате этого поглощение
карбоксильной группы оказывается промежуточным между по-
лосами поглощения групп С = О и С — О. Из-за взаимодействия
связей углерод — кислород группа С (=0)г имеет две полосы пог-
лощения в ИК-спектрах:
симметричное валентное колебание около
1400 см-1 (7,15 мкм), из двух основных
полос эта полоса более слабая
асимметричное валентное колебание около
1600 см-1 (6,25 мкм), сильное поглощение
Спектры ПМР солей могут быть получены как в D2O, так и в
различных полярных органических растворителях, например в
ДМСО-бб или ацетоне-de. Чистые безводные образцы солей не дол-
жны давать сигнал в ЯМР-спектре в области 6 10—15 м. д. (об-
ласть протона группы СООН), однако часто в этой области наб-
людается сигнал, вызванный адсорбцией влаги воздуха. Точные
данные о положении протона в таких смесях получить труднее
из-за обмена протона между несколькими различными основными
состояниями, однако сигнал протона воды можно выделить на ос-
новании следующих данных:
Растворитель Химический сдвиг протона Н-О-группы, м. д.
d2o ~ 4,5-5,0
дмсо 3,2—3,4
Ацетон 2,6—2,9
Данные масс-спектрального анализа редко используют для ана-
лиза солей. Это связано с их крайне низкой летучестью, препят-
ствующей проведению такого анализа. Соли можно превратить в
соответствующие кислоты (см. ниже), анализ масс-спектров ко-
торых не представляет труда (разд. 6.12).
Прибавление сильных минеральных кислот к солям карбоно-
вых кислот приводит к выделению свободной кислоты:
. 2RCOONa + H2SO4 —> 2RCOOH + Na2SO4
Свободные карбоновые кислоты можно выделить перегонкой, эк-
стракцией или фильтрованием. Поскольку соли металлов плавятся
Рис. 6.37. Инфракрасный спектр ацетата натрия (таблетки с КВг).
Валентные колебания О—Н: /—около 3450 см-1 (2,90 мкм), (за счет прнмесн уксусной кислоты). Валентные колебания С—Н: 2 — 2990,
5930 см-1 (3,34, 3,41 мкм), (группа СН^у Валентные колебания С (—О)"5 3— 1570 см-1 (6,37 мкм), ',аснммС(—О)"; с“-1
(7,04 мкм), v С(-О),; 5—смена решеток.
СлМ М
Рис. 6.38. Спектр протонного магнитного резонанса ацетата натрия (60 МГц, развертка 600 Гц, растворитель D2O).
Относительные интенсивности (площади) двух синглетов указывают, что отношение ацетат натрия: вода (или ук-
сусная кислота) равно 90:10.
Глава 6________________ .вИ Определение функциональных групп и установление структуры
332
Глава в
только при очень высоких температурах и обычно разлагаются при
Плавлении, то для идентификации и установления их строения сле-
дует использовать физические константы кислот, образующих эти
соли. Кислоты можно либо превратить в подходящие производные,
либо прямо из солей получить анилиды, толуидиды (методика 36),
л-нитробензиловые или л-бромфенациловые эфиры (методика 33).
5-(л-Бромбензил)тиуронийбромид, который получают нагрева-
нием л-бромбензилбромида с тиомочевиной, реагирует с натрие-
выми или калиевыми солями карбоновых кислот, давая соответ-
ствующие 5-(л-бромбензил)тиурониевые соли (методика 39).
Методика 39. Получение бензил-, п-хлорбеизил-
и n-бромбеизилтиурониевых солей
а) Приготовление реагента.
(см. методику 15) 5-(п-6ром<$ензил)тиуронщМромид
б) Получение производных.
Br~ + RCO2H (wiu RCOO*Na+) —»
+
"О OCR
6-(п-£ром^ензил)тиуронийкар6оксилат
К 0,3 г исследуемой кислоты (или 0,5 г натриевой или калиевой соли) при-
бавляют 3—4 мл воды и каплю фенолфталеина в качестве индикатора. Раствор
нейтрализуют, добавляя по каплям 5%-ный раствор гидроксида натрия. Следует
избегать избытка щелочи. Если ее прибавлено слишком много, надо нейтрали-
зовать избыток разбавленной соляной кислотой до тех пор, пока смесь ие ста-
нет бледно-розовой. К этому водному раствору натриевой соли прибавляют го-
рячий раствор 1 г арилалкилтиуронийхлорида или арилалкилтиуронийбромида
в 10 мл 95%-ного этанола. Смесь охлаждают, выделившуюся соль собирают на
фильтре. Некоторые соли (например, соли муравьиной кислоты) не дают осадка;
в этих случаях для того, чтобы соль выпала в осадок, надо упарить часть эта-
нола.
Полученные таким образом тиурониевые соли органических кис-
лот выделяются обычно в очень чистом виде и, как правило, не
требуют перекристаллизации. Если необходимо, то их перекристал-
лизовывают из небольшого количества диоксана.
Температуры плавления многих этих солей близки друг к другу,
и их плавление происходит в узком температурном интервале.
Таким образом, всегда лучше провести подтверждение пдентифи-
Определение функциональных групп и установление структуры
333
нации каким-либо дополнительным способом, например определить
эквивалент нейтрализации.
Аналогичным способом получают S-бензил- и 5-(л-хлорбензил)-
тиурониевые соли. Эти соли также образуются в очень чистом виде,
но обладают слишком близкими температурами плавления.
Аммонийные соли карбоновых кислот по своим химическим и
спектральным характеристикам не отличаются от соответствую-
щих натриевых солей, которые описаны выше. Аммонийные (и
аминные) соли можно определить, обработав их гидроксидом нат-
рия (опыт 25), в результате чего образуется аммиак (или амины).
Кроме того, в ИК-спектре солей аммония и аминов имеются хо-
рошо разделенные и интенсивные полосы валентных и деформаци-
онных колебаний N — Н:
Соль Колебание N—Н Частота, см 1 Длина волны, мкм
NH, Валентное Деформационное 3300—3030 (широкая) 1430—1390 3,0—3,3 7,0—7,2
rnh: Валентное Асимметричное деформацион- ное Симметричное деформационное — 3000 (очень широкая) 1600-1575 1490 ~3,3 6,25-6,35 ~ 6,7
r2nh2 Взаимодействующее валентное Деформационное 2700—2250 (широкая) а 1600-1575 6 3,7—4,4 6,25—6,35
RSNH+ Валентное 2700—2250 в 3,7—4,45
а Эта полоса может иметь сложный максимум.
& Эта полоса обычно средней интенсивности; все остальные приведенные полосы имеют
обычную иитенсивиость.
в Полоса деформационных колебаний N—Н в RgNH* имеет низкую интенсивность и не
может служить для идентификации.
Опыт 25. Взаимодействие аммониевых солей и солей аминов е
гидроксидом натрия
RCOONH< + NaOH —> RCOONa + NHS + Н2О
RNH3C1 + NaOH —► RNH2 + NaCl + H2O
В пробирку помещают 5 мл 10%-ного раствора гидроксида натрия, прибав-
ляют к нему 0,2 г бензоата аммония и сильно встряхивают. Отмечают запах
аммиака.
В пробирке к 0,4 г гидрохлорида анилина прибавляют 5 мл 10%-иого рас-
твора гидроксида натрия. Смесь встряхивают н оставляют стоять несколько ми-
нут. Отмечают выделение маслянистого слоя амина.
6.13. СЛОЖНЫЕ ЭФИРЫ
Обычно сложные эфиры растворимы в органических раствори-
телях (классы растворимости N, и Si). В воде растворимы только
эфиры, содержащие пять или меньше атомов углерода (класс
Рис. 6.39. Инфракрасный спектр этнлацетата в тонкой пленке, измеренный на приборе Perkin-Elmer 521 (© Sadtler Research
Laboratories, Inc., Philadelphia. Pa, 1966).
С<Н8О2, мол. масса 88,11, т. кнп. 77 °C, d20 0,902, n2® 1,3719 (лит.), получен от фирмы Dodge & Olcott, Inc., капиллярная кювета. Валентные коле-
4 D
бания С—Н: 7—2980 см-1 (3,36 мкм), v СН_; 2 — 2930 см"1 (3,41 мкм), v „„ СН„; 3—2910 см-1 (3,45 мкм), V СН . Валентные колебания
асимм и асимм симм з
С=О: 4— 1740 см-1 (5,75 мкм), v_ ~. Деформационные колебания С—Н: 5 —1375 см-1 <7,27 мкм), 6 СН • Валентные колебания С-О:
СИММ и
6—1240 см-1 (8,07 мкм), валентные колебания С(С=О)—О; 7—1048 см-1 (9,54 мкм), валентные колебания О—С—С.
Определение функциональных групп и установление структуры 335'
836
Глава б
растворимости SJ. Результаты, полученные при изучении раствори-
мости эфиров, надо оценивать с осторожностью, так как эфиры
могут гидролизоваться (часто легко), давая соответствующую кар-
боновую кислоту и спирт. Поэтому необходимо принимать во вни-
мание растворимость этих продуктов гидролиза.
......> RCOQH+R'OH
гидролиз т
F
—> RCOO'+R'OH
омыление
Сложные эфиры часто можно идентифицировать по запаху, он
обычно приятный, но иногда отвратительный.
Применение ПК- (рис. 6.39) и ЯМР-спектроскопии (рис. 6.40)
для установления структуры сложных эфиров мало эффективно.
В ИК-спектрах имеются интенсивные полосы валентных колебаний
С = О и С — О. Эта последняя особенность позволяет отличить
спектр сложного эфира от спектра альдегида или кетона.
Положение карбонильной полосы в ИК-спектре сложных эфиров
особенно чувствительно к изменениям их структуры, например к
сопряжению карбонильных групп с а,р-двойной связью. Сложные
эфиры имеют в ИК-спектре две полосы колебаний С — О, по-
скольку содержат две простые С — О-связи: «кислотную» и «спир-
товую». Первая из них сильно взаимодействует с остатком кисло-
ты *, а вторая — с остатком спирта, поэтому эти полосы правильнее
называть полосами колебаний связей С—(С = О)— О и О—С—С
соответственно. Наиболее важные полосы поглощения в ИК-спек-
тре сложных эфиров приведены в табл. 6.6.
Анализ ПМР-спектров сложных эфиров позволяет получить три
типа очень ценной информации о структуре исследуемой молекулы:
1. Химический сдвиг а-протона в спиртовой части молекулы!
О
I
Н *— б 3,2—4,5**
2. Химический сдвиг а-протона в кислотной части молекулы!
О
I II
-С—С—OR'
I
Н *---------- б 2,0—2,5**
3. Группа сигналов ароматических протонов (особенно орто-
протонов), появляющаяся вследствие влияния заместителей в слож-
* Более подробное обсуждение вопроса о взаимодействии колебаний
см. Сильверстейн Р., Басслер Г., Моррилл Т. Спектрометрическая идентификация
органических соединений. Пер. с англ. — М.: Мир, 1977, гл. 3.
** Сигнал протонов метильной группы находится в более сильном поле, мети-
леновой — в середине этой области, а метннной — в более слабом поле.
Определение функциональных групп и установление структуры
337
Таблица 6.6
Характеристические полосы поглощения в ИК-спектрах сложных эфиров
Колебания Положение полосы
см 1 МКМ
Валентные колебания С=О
RCOOR' (простые алифатиче- ские) Около 1750—1735 Около 5,71—5,76
ArCOOR', /С=С—COOR' О II 1 / 1730-1715 5,78—5,83
р с о с—cz Около 1776 Зависит от сопряж Около 5,63 гния, размера цикла
Лактоны
и т. п. (более подробное описание см.
Валентные колебания С—О Валентные колебания С—(С=О)—О в литературе по И К -спектрам в гл. 10)
RCOOR' (простые алифатиче- ские) 1210-1163 8,26—8,60
Ацетаты 1240 8,07
RCOOAr, винилацетаты 1190-1140 8,40-8,77
ArCOOR' 1310—1250 7,63-8,00
Лактоны Валентные колебания О—С—С 1250-1111 8,00-9,00
RCOOR' (R' — первичный, про- стые) 1064-1031 9,40-9,70
RCOOR' (R' — вторичный, про- стые) 1100 9,09
ArCOOR' (R' — первичный, про- стые) 1111 9,00
ном эфире (сравните: у бензола только один синглет с 6 7,27 м.д.,
см. разд. 6.9)
6 ~т;з
Анализ ПМР-спектров сложных эфиров облегчается при ис-
пользовании лантаноидных сдвигающих реагентов (гл. 8). Наи-
более пригоден в этом случае Eu(fod)a.
338
Глава в
Обычно при масс-спектрометрии сложных эфиров пик молеку-
лярного иона выражен достаточно отчетливо. Для эфиров харак-
терны два пути распада. Один включает перегруппировку Мак-
Лафферти, а другой связан с отрывом фрагментов, связанных с
карбонильной группой.
Для того чтобы прошла перегруппировка Мак-Лафферти, вклю-
чающая переход протона, необходимо наличие протона в кислотной
части молекулы эфира в у-положении к карбонильной группе.
Пики образующихся при этом фрагментов обычно очень интен-
сивны и поэтому хорошо идентифицируются. Кроме того, простое
расщепление дает фрагменты с нечетной величиной массы, обра-
зующиеся из молекулярного иона с четным массовым числом. Из
того же самого молекулярного иона в результате перегруппировки
Мак-Лафферти образуется фрагмент с четной величиной массы.
Например, метилкаприлат (М — т/е 158) в результате перегруп-
пировки Мак-Лафферти дает основной пик с т/е 74.
При разрыве связи, находящейся в a-положении к карбониль-
ной группе, могут образовываться четыре приведенных ниже типа
ионов:
Молекулярный иои Наблюдаемый фрагмент (заряженный) Побочный продукт (радикал) Примечание
R—С OR' : (teC-OR' R* Встречается редко. Имеет значение только R'=CHS (m/e 59)
*О; R—С \>R' R+ O=C—OR' •• Обычно малой интен- сивности
R C=O+ R'O’ Появляется, как пра- вило, в тех слу- чаях, когда R не- большой
Z R-C' OR'
or SOS + R C—О Встречается редко
R—С J OR' ••
Определение функциональных групп и установление структуры
339
Наиболее важной реакцией, применяемой для идентификации
сложных эфиров, является реакция омыления, в результате которой
сложный эфир превращается в спирт и соль карбоновой кислоты.
Следует заметить, что, хотя и образующийся спирт, и кислоту мож-
но охарактеризовать порознь, опыт показывает, что лучше всего по-
лучать производные из самого эфира (см. ниже).
Методика 40. Омыление и гидролиз сложных эфиров
Метод А. Кипячение водных растворов.
Метод Б. Кипячение с диэтиленгликолем.
Метод В. Реакция с концентрированной серной кислотой.
(A) NaOH/H,O
RCOONa* +R'OH
(Б) NaOH______
диэтиленгликоль
|н,о+
RCOO
(В) (1) КОНЦ. H,SO,
RCOOH+R'OH
(2) H20
Метод А. В круглодонную колбу с эффективным обратным холодильником
помещают 40 мл 25%-кого раствора гидроксида натрия. Прибавляют 5 мл этил-
беизоата (или другого эфира) и смесь нагрегают до кипения. Для предотвра-
щения вскипания в колбу кладут кусочки пемзы или пористого фаянса. Кипяче-
ние продолжают до исчезновения слоя эфира или его запаха (около 0,5 ч). За-
менив обратный холодильник на нисходящий, отгоняют около 5 мл дистиллята
и насыщают его карбонатом калия. Заметьте, что образуются два слоя (объяс-
ните почему). Очевидно, что величина пробы, используемой для проведения ре-
акции, зависит от молекулярной массы выделяющегося спирта и исходного
эфира.
Оставшуюся в колбе смесь охлаждают и подкисляют разбавленной фосфор-
ной кислотой. Что выпадает в осадок? Как убедиться в том, что это не фосфат
натрия? Если получающаяся кислота летуча, то ее можно отделить перегонкой.
Жидкую кислоту перегоняют следующим образом. Раствор помещают в пе-
регонную колбу и кладут туда несколько кусочков пемзы или пористого фаянса
для облегчения кипения. Термометр применять не следует, и верх перегонной
колбы должен быть закрыт. Колбу соединяют с коротким холодильником, охла-
ждаемым водой, причем боковой отвод перегонной колбы должен достаточно
глубоко входить в суженную часть холодильника. Весь прибор для перегонки
устанавливают таким образом, чтобы холодильник был наклонен под углом 45°
к горизонтали. Колбу нагревают так, чтобы капли дистиллата падали из холо-
дильника с постоянной скоростью. Перегонку не следует вести слишком бы-
стро, чтобы несконденсировавшиеся пары кислоты выходили из холодильника,
или слишком медленно, чтобы между отдельными каплями не были слишком
большие интервалы времени.
Обсуждение. Сложные эфиры сильно отличаются друг от друга
по скорости омыления. Эфиры более простого строения, кипящие
ниже 110°С, полностью омыляются за 0,5 ч при кипячении с
25%-ным раствором гидроксида натрия, как это описано в мето-
де А. Эфиры, кипящие между НО и 200°С, требуют для полного
омыления более длительного времени (1—2 ч).
Щелочной гидролиз нерастворимых в воде эфиров сильно уско-
ряется при прибавлении к реакционной смеси 0,1 г лаурилсульфата
340
Глаеа 6
натрия (гардинола). Смесь энергично перемешивают для эмульги-
рования эфира, а затем нагревают до кипения. Следует исполь-
зовать колбу большого объема, так как в присутствии эмульгатора
образуется много пены.
Высококипящие (выше 200° С) сложные эфиры нерастворимы
в воде и гидролизуются медленно. При этом длительное кипячение
может привести к потере летучего спирта. В методе Б используют
раствор гидроксида калия в диэтиленгликоле (т. кип. 244° С). Ди-
этиленгликоль не только прекрасный растворитель для сложных
эфиров, но также позволяет проводить реакции при более высоких
температурах. При проведении реакции в диэтиленгликоле все про-
дукты, кроме высококипящих спиртов, можно отогнать из реак-
ционной смеси в чистом виде.
Метод Б. В перегонную колбу емкостью 10 или 25 мл помещают 3 мл ди-
этиленгликоля, 0,5 г таблетированного гидроксида калия и 0,5 мл воды. Смесь
нагревают на небольшом пламени, пока щелочь ие растворится, после чего смесь
охлаждают. К раствору щелочи прибавляют 1—2 г сложного эфира и энергично
перемешивают. В насадке перегонной колбы устанавливают термометр, а в ка-
честве приемника используют пробирку, охлаждаемую в стакане со льдом. Вна-
чале, продолжая встряхивать, колбу нагревают на небольшом пламени. В тех
случаях, когда в смеси имеется только одна жидкая фаза или одна жидкая и
одна твердая фазы, смесь можно нагревать сильнее, чтобы отгонялся образую-
щийся спирт. Дистиллят используют для получения твердых производных, на-
пример 3,5-дикарбоксилатов (методика 2).
В колбе остается суспензия или раствор калиевой соли кислоты, входивший
в состав сложного эфира. К остатку прибавляют 10 мл воды и 10-мл этанола и
сильно встряхивают. После этого прибавляют разбавленную серную кислоту
(6 н.) до слабокислой реакции по фенолфталеину. Смеси дают постоять 5 мин
и затем фильтруют. Фильтрат используют непосредственно для получения про-
изводных. Его можно обработать п-нитробензилбромидом или п-фенилфенацил-
бромидом для получения соответствующих твердых эфиров. Если исходный
сложный эфир был настолько высококнпящим, что определить его температуру
кипения оказалось затруднительным, то лучше фильтрат разделить на две ча-
сти и получить два различных производных.
Обсуждение. Омыление чаще всего используется для идентифи-
кации сложных эфиров. Однако необходимо помнить, что горячая
щелочь может действовать также и на другие функциональные
группы в молекуле. Альдегиды, имеющие в a-положении водо-
родный атом, претерпевают альдольную конденсацию и осмоля-
ются. Альдегиды, у которых нет а-водородного атома, участвуют
в реакции Канниццаро и дают спирт и натриевую соль соответ-
ствующей кислоты:
2R3CCHO + NaOH —► RsCCH2OH + RsCCOONa
Полифункциональные соединения, такие, как 0-дикетоны и 0-
кетоэфиры, при действии горячей щелочи расщепляются. Возмож-
ность таких побочных реакций устанавливается с помощью других
классификационных реагентов, рассмотренных в этой главе. Все
изложенное подтверждает тот факт, что для точного доказатель-
ства присутствия какой-либо функциональной группы нельзя ис-
Определение функциональных групп и установление структуры
341
пользовать только один классификационный реагент. Выводы, о
структуре неизвестного соединения необходимо делать на основе
результатов всех проведенных классификационных реакций в их
тесной взаимосвязи.
Метод В. Эфиры стерически затрудненных кислот, например 2, 4,6-триалкил-
бензоаты, с трудом гидролизуются под влиянием щелочей. Однако они легко гид-
ролизуются при растворении их в 100%-ной серной кислоте и последующем
разбавлении смеси ледяной водой. Это происходит вследствие того, что прн рас-
творении эфиров стерически затрудненных кислот в 100%-ной серной кислоте
протекает следующая реакция:
COOR СО
При добавлении воды промежуточный нон превращается в кислоту:
Эта реакция обратима, и стерически затрудненные кислоты можно превра-
тить в эфиры, растворяя их в 100%-ной серной кислоте и обрабатывая получен-
ный раствор спиртом.
Незатрудненные эфиры в эту реакцию ие вступают. После растворения в
100%-ной серной кислоте и обработки ледяной водой они остаются неизменен-
ными. Сложные эфиры, в которых и кислота, и спирт являются стерически за-
трудненными, чрезвычайно устойчивы к гидролизу щелочами, однако легко гид-
ролизуются при кипячении в течение 1 ч с 18%-ной соляной кислотой*.
Другие методики омыления, в которых в качестве растворителя применяется
этанол, описаны в книге Пасто и Джонсона, с. 412.
Контрольные вопросы. 1. Напишите уравнения реакции щелочного гидролиза
n-фенилфенацилацетата, дибензоата этиленгликоля, н-бутилоксалата, полимерного
эфира глицерина и фталевой кислоты. Предложите подходящие методики для
определения образовавшихся при этих реакциях продуктов.
Большую помощь оказывает определение числа омыления слож-
ных эфиров (методика 41), особенно в тех случаях, когда не уда-
ется предварительно провести определение молекулярной массы.
Число омыления равно молекулярной массе эквивалента эфира,
установленной титрованием. Методика титрования аналогична
описанной выше для карбоновых кислот. Молекулярная масса
эфира, с учетом экспериментальных ошибок, равна числу омыления
или является кратной ему величиной, т. е. произведением числа
омыления и некоторого небольшого целого числа х.
Cohen S. О., Schneider A., J. Am. Chem. Soo,, 68, 8882 (1941),
342
Глава 6
Методика 41. Число омыления сложного эфира
а) Получение раствора гидроксида калня в диэтиленгликоле. Реактив по-
лучают, растворяя 3 г химически чистого таблетированного гидроксида калия
в 15 мл днэтиленглнколя. При необходимости ускорить растворение смесь осто-
рожно нагревают. Смесь перемешивают термометром, следя, чтобы ее темпера-
тура не поднималась выше 130 °C. Нагревание до более высокой температуры при-
водит к окрашиванию реактива. После того как весь гидроксид калия раство-
рится, теплый раствор смешивают с 35 мл днэтиленглнколя в колбе с притертой
стеклянной пробкой. Смесь хорошо перемешивают и охлаждают. При этом по-
лучается приблизительно 1,0 н. раствор. Для установления точного титра * от-
бирают 10 мл раствора в колбу, прибавляют 10 мл воды и титруют 0,25 и. соля-
ной кислотой с известным титром.
Омыление сложного эфира. Точно отмеривают пипеткой 10 мл реактива и
вносят его в маленькую колбу Эрленмейера с притертой стеклянной пробкой.
Помещают некоторое количество эфира в небольшую взвешенную колбу, снаб-
женную маленькой пипеткой. Определяют массу колбы, эфира и пипетки и пере-
носят 0,4—0,6 г эфира в колбу Эрленмейера. Пипетку снова помещают во взве-
шенную колбу. Разница масс соответствует взятой для омыления навеске эфира.
Вращая колбу, смешивают исследуемый сложный эфир с реактивом. За-
крепляют надежно пробку и нагревают смесь на водяной бане с такой ско-
ростью, чтобы температура 70—80°С была достигнута за 2—3 мни. Жидкость во
время нагревания перемешивают круговыми движениями колбы **. После дости-
жения нужной температуры колбу извлекают из нагревательной банк н сильно
встряхивают. Когда жидкость стечет со стенок, колбу осторожно открывают
(опоено!), чтобы снять избыток давления. Затем колбу снова закрывают проб-
кой и нагревают ее до 120—130°С. В тех случаях, когда реакцию проводят с
очень высококипящими эфирами, вместо пробки можно установить термометр.
При этой температуре колбу выдерживают 3 мнн и затем охлаждают до
80—90°С. Вынимают пробку и обмывают стенкн колбы дистиллированной водой
(около 15 мл). Содержимое колбы перемешивают и титруют 0,25 н. соляной кис-
лотой с фенолфталеином в качестве индикатора. Число омыления рассчитывают
по уравнению:
,, навеска вещества (мг)
Число омыления = -т—---------------г—г;---:---j—z—1 -——— г—г-;---------=-
[объем щелочи (мл) • NK0H] — [объем кислоты (мл)-МнсЛ
где объем раствора щелочи считается равным 10,0 мл.
Обсуждение. При этой методике достигается полное омыление
нерастворимых в воде сложных эфиров. Полностью омыляются
такие эфиры, как, например, бензилацетат, бутилфталат, этил-
себацинат, бутилолеат, эфиры гликолей и глицерина.
б) Омыление сложных эфиров спиртовым раствором гидроксида натрия.
Приготовление спиртового раствора гидроксида натрия. В 250 мл абсолютного
этанола растворяют 8 г натрия н после полного растворения прибавляют 25 мл
воды. Определяют титр (точную концентрацию) этого раствора, титруя его с по-
мощью навески чистого кислого фталата калия.
Омыление сложного эфира. Эфир помещают во взвешенную колбу, снабжен-
ную маленькой пипеткой. Взвешивают колбу с веществом и с помощью пипетки
переносят 0,2—0,4 г эфира в коническую колбу емкостью 150 мл. Пипетку поме-
щают обратно в колбу с веществом и снова взвешивают. Разница в массе (взве-
шивание проводят на аналитических весах) соответствует навеске эфира. В кол-
* Тнтр обычно определяется с точностью до третьего знака, поэтому 0,25 н.
означает концентрацию в пределах 0,230—0,270.
** Будьте осторожны. Помните, что шарик термометра очень хрупок.
Определение функциональных групп и установление структуры
343
бу, содержащую навеску эфира, из бюретки приливают 15 мл приготовленного,
как описано выше, спиртового раствора гидроксида натрия. Соединяют колбу с
эффективным обратным холодильником, осторожно нагревают смесь до кипения
и кипятят 75—90 мин. После этого оставляют смесь медленно охлаждаться. Хо-
лодильник и насадки ополаскивают водой из промывалки. Промывные воды сли-
вают в колбу с веществом. К полученному раствору прибавляют две капли фе-
нолфталеина и избыток щелочи оттитровывают 0,25 н. соляной кислотой. Конеч-
ную точку определяют по бледно-розовой окраске. Для этого лучше оттитровать
раствор до полного обесцвечивания, а потом провести обратное титрование ис-
ходным раствором щелочи.
Обсуждение. Для того чтобы получить точную величину числа
омыления, необходимо, чтобы эфир был чистым и безводным. Хо-
роших результатов можно достигнуть, соблюдая следующие пре-
досторожности:
1. Нормальность спиртового раствора гидроксида натрия (М)
необходимо определить непосредственно перед применением.
2. Количество титрованного раствора щелочи, вводимого из
бюретки, следует измерять с максимальной точностью (особенно
в случае эфиров с высокой молекулярной массой), так как неболь-
шая ошибка при этом измерении приводит к большой ошибке в ве-
личине числа омыления.
3. Нагревание в течение 90 мин приводит к омылению боль-
шинства сложных эфиров, однако иногда необходимо проводить
нагревание в течение более длительного времени (от 2 до 24 ч).
4. Для соединения колбы и холодильника не следует исполь-
зовать корковые или резиновые пробки, так как пары спирта эк-
страгируют из них примеси, которые понижают силу щелочи. Надо
использовать стеклянные шлифы, тщательно промытые дистилли-
рованной водой.
5. В конечной точке титрования раствор должен быть бледно-
розовым. Эта окраска фенолфталеина соответствует pH 9, что бли-
зко к концентрации иона водорода в растворах натриевых солей
большинства органических кислот.
6. Молекулярная масса сложного эфира равна числу омыле-
ния, умноженному на число сложноэфирных групп (х) в молекуле.
Контрольные вопросы. 1. Определите число омыления для этилового эфира
ацетоуксусной кислоты, н-бутилпропионата, кислого этилового эфира фталевой
кислоты, этилового эфира цнануксусной кислоты и w-бутилфталата.
2. Что произойдет, если бензальдегид обработать по методике 41?
3. Как изменится величина числа омыления, если гидролиз сложного эфира
пройдет не полностью?
Поскольку разделение и очистку продуктов гидролиза провести
довольно трудно, то производные сложных эфиров лучше всего
получать непосредственно из самих эфиров. Сложные эфиры, со-
держащие другие функциональные группы, часто можно иденти-
фицировать, превращая их в твердые производные с помощью раз-
личных реакций, таких, как галогенирование, нитрование или аце-
тилирование.
344
Глава 6
Производные кислотной части сложного эфира можно получить,
используя следующие методы:
I. Реакция с бензиламином в присутствии небольшого количе-
ства хлорида аммония приводит к N-бензиламидам, которые можно
использовать в качестве производных при идентификации (мето-
дика 42):
RCOOR' + C6H5CH2NH2 —> RCONHCH2C6H5 4- R'OH
Реакция хорошо проходит с метиловыми или этиловыми эфи-
рами. Сложные эфиры, содержащие остатки R' более высокомо-
лекулярных спиртов, необходимо сначала превратить в метиловые
эфиры метанолизом (техника такой переэтерификации описана в
конце методики 42):
CH3ONa
RCOOR' + СНзОН--------► RCOOCHs 4- R'OH
Полученный таким образом метиловый эфир можно подвергнуть
аминолизу.
2. Гидразин легко реагирует с метиловыми и этиловыми эфи-
рами с образованием гидразидов кислот, являющихся довольно
хорошими-их производными (методика 43):
RCOOCH3 + H2NNH2 —> RCONHNH2 + СН3ОН
Сложные эфиры высших спиртов необходимо превратить в мети-
ловые эфиры.
3. В качестве производных можно использовать амиды, кото-
рые образуются при взаимодействии некоторых сложных эфиров с
водным или спиртовым раствором аммиака:
RCOOR' + NH3 —> RCONH2 + R'OH
Однако большинство сложных эфиров в эту реакцию вступает
только при нагревании под давлением.
4. С помощью следующих реакций можно получить п-толуиди-
ды:
C2H5MgBr + ArNH2 —» ArNHMgBr 4-С2Н6
RCOOR'4-ArNHMgBr —> RC(NHAr)2 4-R'OMgBr
OMgBr
|2HC1
Ar=CH3CeH4— RCONHAr 4- ArNHjcr + MgBrCl
Твердые производные спиртовой части сложного эфира можно
синтезировать с помощью обменной реакции между сложным эфи-
ром и 3,5-динитробензойной кислотой в присутствии концентриро-
ванной серной кислоты (методика 44):
H2SO4
АгСООН 4-RCOOR' ------> ArCOOR'4-RCOOH
Эту методику можно использовать для большинства сложных
эфиров, за исключением тех, в которых R илн R' могут реагиро-
Определение функциональных ерупп и установление структуры
345
вать с концентрированной серной кислотой, В эту реакцию не
вступают также эфиры с большой (>250) молекулярной массой.
В тех случаях, когда не удается получить производные ни
спирта, ни кислоты непосредственно из самого сложного эфира,
следует провести его гидролиз. Эта операция лучше всего прохо-
дит путем омыления со щелочами по методике 40. Дальнейшие
реакции, которые необходимо провести с образовавшейся после
омыления смесью, зависят от природы кислоты и спирта. Кислота
может быть одноосновной или многоосновной, растворимой или
нерастворимой в воде. Спирт может быть одно- или многоатом-
ным либо представлять собой фенол. Таким образом, необходимо
тщательно обдумать план разделения и идентификации продук-
тов омыления, исходя из возможных предположений об их струк-
туре, и на основании полученных результатов применять необхо-
димые классификационные реакции.
Методика 42. Получение N-бензиламидов из сложных эфиров
О
CH2NH2 ch2nhcr
“таг Я^СНз.СЛ
Если R" =#= R', то необходимо сначала провести реакцию
СНзОН
RC00R" RC00CHs
Смесь 1 г сложного эфира, 3 мл бензиламина и 0,1 г порошкообразного хло-
рида аммония нагревается 1 ч в пробирке из стекла пирекс, которая снабжена
небольшим пальчиковым холодильником. После охлаждения реакционную смесь
промывают водой, чтобы удалить избыток бензиламина н вызвать кристаллиза-
цию. Можно ускорить кристаллизацию добавлением небольшого количества раз-
бавленной соляной кислоты. Необходимо избегать избытка кислоты, так как
она растворяет N-бензиламиды. Иногда кристаллизации может препятствовать
присутствие не вошедшего в реакцию сложного эфира. В этом случае для уда-
ления эфира лучше всего прокипятить раствор несколько минут с водой в чашке
для выпаривания. Твердый амид отделяют на фильтре, промывают небольшим
количеством лигроина и перекристаллизовывают из смеси спирта илн ацетона
с водой.
Сложные эфиры, спиртовой остаток которых содержит спирт, более тяже-
лый, чем этанол, нагревают примерно 30 мин с 5 мл абсолютного метанола, в ко-
тором предварительно растворен небольшой кусочек (0,1 г) металлического на-
трия. После кипячения метанол упаривают, а остаток используют для проведе-
ния реакции по указанному выше способу.
Методика 43. Получение гидразидов кислот из сложных эфиров
RCOOR'+ NH2NH2(85%-Hbift) —> RCONHNH2 + R'OH
R' == СНз нли С2Н5
Смешивают 1 г метилового нли Этилового эфира карбоновой кислоты с 1 мл
85%-ного гндразннгндрата и кипятят смесь 15 мин. Затем через обратный хо-
лодильник приливают абсолютный этанол до образования прозрачного раствора
346
Глава 6
После этого смесь кипятят еще около 2 ч, спирт упаривают, а остаток охлаж-
дают. Кристаллы полученного гидразида отделяют на фильтре и перекристалли-
зовывают из воды илн водно-спиртовой смеси.
Сложные эфиры, содержащие внешне спирты, перед проведением реакции
с гидразином необходимо подвергнуть метанолнзу, как описано выше.
Методика 44. Получение алкил-3,5-дииитробеизоатов из сложных эфиров
+RCOOR
конц. H2S04
д
COOR'
+ RCOOH
Около 2 мл сложного эфира смешивают с 1,5 г 3,5-днннтробензойной кис-
лоты и прибавляют 2 капли концентрированной серной кислоты. Если исходный
эфир кипит ниже 150°С, то смесь слегка нагревают до кипения. Если же эфир
кипит выше 150°С, то смесь нагревают на масляной бане, имеющей температуру
выше 150°С. Необходимое для окончания реакции время колеблется от 30 мин
до 1 ч. В том случае, когда растворение 3,5-динитробензойной кислоты не закан-
чивается за 15 мин, нагревать смесь приходится дольше. После остывания смеси
в нее добавляют 25 мл абсолютного эфира и, чтобы отделить не вошедшие в
реакцию 3,5-диннтробензойную н серную кислоты, дважды экстрагируют пор-
циями по 15 мл 5%-ным раствором карбоната натрия. Слой эфира промывают
10 мл воды. Эфир упаривают, а остаток (обычно в виде масла) растворяют в 5 мл
кипящего этанола. После фильтрования раствора к фильтрату прибавляют воду
до начала помутнения. Чтобы вызвать кристаллизацию производного, смесь ох-
лаждают н перемешивают.
6.14. ПРОСТЫЕ ЭФИРЫ
Простые эфиры обладают несколько большей полярностью и
более высокой реакционной способностью, чем насыщенные угле-
водороды или алкилгалогеннды. Под действием концентрирован-
ной серной кислоты эфирный кислород может быть протонирован:
r2o + h2so4 <=> r2oh+hso; *
••
Эта реакция показывает, почему эфиры по растворимости отно-
сятся к классу Ni (а не к классу I, включающему инертные угле-
водороды и алкилгалогеннды).
Внимание! При стоянии, особенно при действии воздуха и света,
простые эфиры образуют исключительно взрывчатые пероксиды.
С жидкими эфирами, в которых имеется твердый осадок, вообще
не следует работать.
Наличие пероксидов в простом эфире можно обнаружить с
помощью иодкрахмальной бумаги, смоченной разбавленной соля-
ной кислотой: в присутствии пероксидов бумага становится го-
лубой. Методы удаления пероксидов и гидропероксидов описаны
Пасто и Джонсоном, с. 371.
Чистые простые эфиры представляют собой соединения, ко-
торые первоначально легче определить по их химической инертно-
сти, чем по их способности вступать в химические реакции. По-
этому для определения структуры простых эфиров исключительно
Длина волны, мкм
348
Глава 6
большое значение имеют спектры. ИК-Спектр типичного просто-
го эфира показан на рис. 6.41. Для характеристики простых эфи-
ров наиболее важное значение имеют полосы поглощения, об-
условленные колебаниями связей С—О—С. Соответствующие по-
лосы поглощения приведены в табл. 6.7; положение этих полос
поглощения зависит от сопряжения свободной пары электронов
кислорода с л-электронной системой:
” 1 /
—О—С=С\
Таблица 6.7
Полосы поглощения С—О—С в инфракрасных спектрах простых эфиров
Валентные колебания С—О—С Полоса поглощения а
см 1 МКМ
ROR (простые алифатические)
антисимметричные 1150-1085 (с) 8,70—9,23
симметричные ROA.r Около 1125 (сл) Около 8,89
антисимметричные 1275-1200 (с) 7,84-8,33
симметричные Виниловые эфиры 1075—1020 (с) 9,30—9,80
антисимметричные 1225-1220 (с) 8,16-8,33
симметричные 1075—1020 (с) - 9,30—9,80
8 с—сильная, сл—слабая.
ПМР-Спектр типичного эфира показан на рис. 6.42. Наиболее
легко опознаваемой особенностью таких спектров обычно являют-
ся сигналы протонов, присоединенных к атому углерода, находя-
щемуся рядом с кислородом (а-протоны)*:
CHjOR
R'CH2OR
R/CHOR
<Г~3,2-
<Г~3,4-
<Г*з,<
Значения этих химических сдвигов очень сходны с химическими
сдвигами аналогичных протонов в спиртах и сложных эфирах. При
замене R на Аг каждый сигнал сдвигается в слабое поле на не-
сколько десятых миллионной доли:
СНэОАг
V, <5^3,85
R'CHjOAr
R'jCHOAf
А 4,55
* Следует отметить, что сигналы протонов в каждой серии сохраняют обыч-
ный порядок: сигналы метильных протонов находятся в более сильном поле, чем
сигналы метиленовых, которые в свою очередь находятся в более сильном поле
по сравнению с сигналами метиновых протонов,
Определение функциональных групп и установление структуры
349
Рис. 6.42. Спектр протонного магнитного резонанса ди-н-бутнлового эфира (60 МГц, развертка 600 Гн, растворитель
CDClsA
350
Глава 6
Алкоксигруппа в ароматическом кольце обычно сдвигает аромати-
ческие протоны в более сильное поле по сравнению с протонами
бензола (6 7,27 м.д.). Более подробно это явление описано в
разд. 6.9.
Анализ масс-спектров простых эфиров может быть затрудни-
тельным вследствие слабости пика молекулярного иона (М). Од-
нако при увеличении концентрации образца пик М 1 часто
становится более интенсивным. Это происходит в результате пе-
реноса Н« при ион-молекулярных соударениях:
н
RCHjOR' --е---» RCHgOR' RCH^R , RCH2—+О—R'+RCHOR
г!" ’ ’ ...
М М+1
Наиболее обычный путь фрагментации для простых эфиров со
стоит в расщеплении связи между а- и р-атомами углерода:
—с 'сн2 о—R
I <> z • •
la
—С- + СН2=О—R'—* СН2—О—R
В этом примере стабилизация катиона за счет резонанса является
фактором, очень сильно влияющим на место расщепления моле-
кулы при наличии более чем одного пути фрагментации. Другим
фактором, влияющим на направление такого расщепления, будет
размер отщепляющейся группы R*. Обычно легче удаляется боль-
шая группа R *.
При обработке простого эфира иодистоводородной кислотой
происходит расщепление простого эфира. Иодистоводородная кис-
лота— достаточно сильная кислота для протонирования просто-
го эфира, а образующийся иодид-ион обладает достаточной нук-
леофильностью, чтобы произошла необходимая реакция замеще-
ния:
••hi + I Hl
R2O ---► R2OH ---> RI + ROH --->
•• ••
г
ROH2 —► ri + h2o
Г
ROH2 ---► R+ ---► RI
Опыт 26. Иодистоводородная кислота (определение
групп методом Цейзеля)
ROR' + 2HI —> R'I + RI + HjO
HOR' + HI —> R'I + H2O
ArOR' + HI —> R'14-ArOH
RCOOR'4-Hl —> R'l + RCOOH
RCH(OR')2 + 2HI —> 2R'I + RCHO + H2O
алкокси-
Определение функциональных групп и установление структуры
351
В пробирку размером 16 X 150 мм помещают около 0,1 г (или 0,1 мл) веще-
ства. С помощью пипетки осторожно прибавляют 1 мл ледяной уксусной кислоты
и 1 мл 57%-ной иодистоводородной кислоты (плотность 1,7). Вносят небольшой
кусочек неглазурованного фарфора и в отверстие пробирки вставляют тампон нз
марли, сделанный, как описано ниже. Тампон складывают так, чтобы он хорошо
подходил к пробирке, н опускают в пробирку, чтобы верхний край его был на
4 см ниже верхнего края пробирки. Сверху на тампон осторожно помещают не-
большой кусочек негигроскопической ваты, уплотняя ее стеклянной палочкой,
чтобы из нее образовался слой толщиной 2—3 мм. На вату помещают кусочек
фильтровальной бумаги размером 2 X 10 см, сложенный в длину и смоченной
раствором нитрата ртути(П). Пробирку погружают на 4—5 см в масляную баню,
нагретую до 120—130°С.
При кипении реакционной смеси поднимающиеся пары проходят через тампон,
который обычно становится серым. Летучий алкилиодид, проходящий через там-
пон, реагирует с нитратом ртути(II) и окрашивает бумажку в цвета от светло-
оранжевого до ярко-красного вследствие образования иодида ртути(П). Реак-
ция будет положительной, если при нагревании в течение 10 мин индикаторная
бумага станет оранжевой или ярко-красной. Желтая окраска указывает на отри-
цательную или сомнительную реакцию.
Проведите эту реакцию с анизолом, метилбензоатом н а-метилглюкозидом.
Тампоны из марли. Раствор 1 г ацетата свинца в 10 мл воды прибавляют
к 60 мл 1 н. раствора гидроксида натрия и перемешивают до растворения осадка.
К полученному раствору плюмбита натрия прибавляют раствор 5 г кристалличе-
ского тиосульфата натрия в 10 мл воды и около 1 мл глицерина, после чего
объем раствора доводят до 100 мл. Около 5 мл этого раствора пипеткой нано-
сят на сложенные вдвое полоски марли размером 2 X 45 см. Эти полоски высу-
шивают и свертывают так, чтобы они подходили к пробиркам.
Раствор нитрата ртути(П). Приготовляют насыщенный раствор нитрата
ртути (II) в 49 мл дистиллированной воды, к которой прибавлен 1 мл концен-
трированной азотной кислоты.
Обсуждение. Эта реакция основана на классическом методе
Цейзеля для количественного определения процентного содержа-
ния метоксильных или этоксильных групп. Функциональные груп-
пы, содержащие метильный, этильный, н-пропильный или изопро-
пильный радикалы, присоединенные к кислороду, расщепляются
иодистоводородной кислотой с образованием летучего алкилгало-
генида. Алкоксильные производные, содержащие н-бутильную
группу или еще больший радикал, расщепляются с трудом, а
образующиеся иодиды имеют слишком высокую температуру ки-
пения, чтобы испаряться в условиях опыта. Некоторые н-бутокси-
соединения дают положительную реакцию, однако метод оказы-
вается не надежным (температура кипения н-бутилиодида
131°С).
Эта реакция наиболее полезна для простых и сложных эфиров
и ацеталей, в которых алкильными группами являются метиль-
ная или этильная. Метанол, этанол, оба пропанола и даже более
высокомолекулярные спирты, такие, как бутанол-1 и пентанол-2,
также дают положительную реакцию. Эта реакция использова-
лась для определения алкоксильных групп в многочисленных ал-
калоидах и сахарах. Основные осложнения связаны с присут-
ствием функциональных групп, содержащих серу и образующих
сероводород при нагревании с иодистоводородной кислотой.
352
Глава 6
Для разложения некоторых эфиров может потребоваться бо-
лее энергичный реагент. Иногда для разложения 0,1 г вещества
лучше использовать смесь, состоящую из 2 мл иодистоводородной
кислоты, 0,1 г фенола и 1 мл пропионового ангидрида.
Метод, позволяющий получать кристаллические производные
для идентификации небольших количеств простых алифатических
эфиров, заключается в их расщеплении с образованием соответ-
ствующих 3,5-динитробензоилхлоридов в присутствии хлорида
цинка (методика 45):
ArCOCl + ROR —> ArCOOR + RCl
Этот метод непригоден для смешанных простых эфиров, в кото-
рых обе группы эфира алифатические. Алифатические простые
эфиры и арилалкиловые эфиры можно расщепить иодистоводород-
ной кислотой (опыт 26):
R2O + 2H1 —> 2R1 + H2O
ArOR + Hl —> ArOH+RI
При использовании большого количества вещества (от 5 до
10 г) можно выделить алкилиодид и превратить его в производ-
ное (методики 12—15). Также можно выделить фенол из арилал-
килового эфира и перевести его в производное (методики 59—6Г},
Методика 45. Получение алкиловых эфиров 3,5-дииитробензойиой кислоты
из простых эфиров
В пробирку помещают смесь 1 мл простого эфира, 0,15 г безводного хло-
рида цинка и 0,5 г 3,5-дннитробензоилхлорида и соединяют ее1 с обратным холо-
дильником. Смесь кипятят 1 ч, охлаждают н прибавляют 10 мл растворе соды.
Затем смесь нагревают до 90°С на водяной бане, охлаждают и фильтруют. Оса^
док промывают 5 мл 5%-ного раствора карбоната натрия и 10 мл дистиллиро-
ванной воды, после чего растворяют в 10 мл горячего четыреххлористого угле-
рода. Раствор фильтруют в горячем состоянии, фильтрат охлаждают ' ледяной
бане. Если сложный эфир не выделяется сразу, то упаривают четыреххлористый
углерод, остаток высушивают на пористой пластинке и определяю® ево> темпе-
ратуру плавления.
Простые эфиры можно расщеплять реагентами, образующими
активные электрофильные ацилы, которые атакуют эфирный кис-
лород. Так, по одной из методик эфир можно превратить а а-це-
Определение функциональных групп и установление структуры 353
тат при обработке безводным хлоридом железа (III) в уксусном
ангидриде *:
О О
FeCl3 II II
ROR' + (СН8СО)2О -------> ROCCH3 + R'OCCHs
По другой методике активный ацилиевый ион получают из
смешанного ангидрида**. Для этого сначала требуется получить
смешанный ангидрид из соответствующей сульфокислоты или ее
производных. Продуктами, которые обычно достаточно легко оха-
рактеризовать, являются ацетаты.
R2CHCH3COTs — ROCCH3+ROTs
Наиболее часто применяемые производные ароматических про-
стых эфиров получают бромированием (методика 46) и нитрова-
нием (методики 26а и 266). Применяют также пикраты (методика
49). Во всех этих методиках используется склонность ароматиче-
ского кольца подвергаться атаке электронодефицитными части-
цами.
Ароматические простые эфиры, имеющие алкильную боковую
цепь, можно окислить в соответствующие бензойные кислоты (ме-
тодики 27а и 276)
кмпо4
СНзОС^СНгСНз -------> СН3ОС6Н4СООН
Ароматические простые эфиры легко реагируют при 0°С с
хлорсульфоновой кислотой, образуя сульфонилхлориды (методи-
ка 47):
ROCeH6+ CISO3H —> n-ROCeH4SO2Cl
Эти сульфонилхлориды представляют собой маслянистые или
низкоплавкие твердые вещества. Их обрабатывают аммиаком или
* Ganem В., Small V. R., J. Org. Chem., 39, 3728 (1974).
** Karger M. H., Mazur Y., J. Am. Chem. Soc. 90, 3878 (1968).
19 Зак. 335
354
Глава 6
углекислым аммонием (методика 48), получая таким путем суль-
фонамиды, являющиеся полезными производными:
n-ROC6H4SO2Cl + (NH4)2CO3 —> n-ROCeH4SO2NH2
Методика 46. Бромирование
В 15 мл ледяной уксусной кислоты растворяют 1 г вещества н к получен-
ному раствору прибавляют 3—5 г брома. Смесь оставляют на 15—30 мин и за-
тем выливают в 50—100 мл воды. Выделившееся бромпронзводное отфильтровы-
вают и очищают перекристаллизацией нз разбавленного этанола. В некоторых
случаях вместо уксусной кислоты в качестве растворителя можно использовать
четыреххлорнстый углерод. В этом случае четыреххлористый углерод отгоняют
н остаток перекристаллизовывают.
Методика 47. Получение сульфоиилхлоридов
OR OR
+ HC1 + H2SO4
O=S=O
т
ci
Раствор 1 г вещества в 5 мл сухого хлороформа, находящийся в чистой су-
хой пробирке, охлаждают в стакане со льдом до 0°С. К этому раствору по кап-
лям прибавляют около 5 г хлорсульфоновой кислоты. После того как выделение
хлористого водорода почти полностью прекратится, пробирку вынимают из ста-
кана со льдом и дают нагреться до комнатной температуры (приблизительно
20 мин). Содержимое пробирки выливают в стакан емкостью 50 мл, наполнен-
ный кусочками льда, отделяют слой хлороформа и промывают его водой, после
чего хлороформ упаривают, а остающийся сульфонилхлорид перекристаллизовы-
вают нз низкокипящего петролейного эфира, бензола или хлороформа.
Методика 48. Получение сульфонамидов
Смешивают 0,5 г Сульфонилхлорнда с 2,0 сухого порошкообразного углекис-
лого аммония и нагревают при 100°С 30 мин. Смесь охлаждают и трижды про-
мывают холодной водой порциями по 10 мл. Сырой сульфонамид растворяют в
1Q мл 5%-него раствора гидроксида натрия, при необходимости осторожно подо-
Определение функциональных групп и установление структуры
355
гревают. Раствор фильтруют для удаления сульфонов и продуктов хлорирова-
ния. Фильтрат подкисляют разбавленной соляной кислотой и отфильтровывают
сульфонамид, который далее очищают перекристаллизацией из разбавленного
этанола и высушивают при 100°С.
Методика 49. Получение пикратных комплексов простых эфиров фенолов
Раствор 1—2 г эфира в минимальном количестве хлороформа (2—10 мл)
прибавляют к отдельно приготовленному раствору 2 г пикриновой кислоты в
10 мл кипящего хлороформа. Смесь энергично перемешивают и, прекратив на-
гревание, дают ей охладиться. При стоянии смесн выкристаллизовывается пик-
рат, который можно очистить перекристаллизацией из очень малого количества
кипящего хлороформа. Температуру плавления этого вещества следует опреде-
лять как можно скорее после получения, так как некоторые пикраты разлагают-
ся при стоянии.
Эти комплексы пикратов часто можно использовать для определения моле-
кулярной массы простого эфира (см. методику 22, разд. 6.9).
6.15. ЭПОКСИДЫ
Обычно можно предполагать, что по растворимости эпоксиды
относятся к тому же самому классу (Ni), что и простые эфиры.
Однако следует тщательно рассмотреть вопрос, достаточно ли
устойчивы эпоксиды в условиях определения их растворимости.
В эпоксидах разрыв связи углерод — кислород происходит значи-
тельно легче, чем в случае простых эфиров. Эти реакции ката-
лизируются как кислотой, так и основанием:
О О~ он
R—СН^—<Н2 ОН~ > RCHCH,npgmo^ RCH—СН.ОН
। “ реагент
он
он
r£hch2oh rch-ch26h
В таких случаях нужно рассматривать растворимость, а также и
химические свойства образующихся гликолей.
Эпоксиды можно анализировать методами ИК- и ЯМР-спектро-
скопии. Обычно интерпретация спектральных характеристик яв-
ляется достаточно сложной задачей.
12*
356
Глава 6
Наличие напряженного трехчленного цикла приводит к ха-
рактерным особенностям ИК-спектров эпоксидов (табл. 6.8).
Таблица 6.8
Группа Тип колебаний Частота, см-1 Длина волны, мкм
О Rcrf——С Валентные колебания метиковой группы С-Н 3040—3000 3,99-3,33-
О R—С1-£—— СНз (терминаль- ные эпок- сиды) Валентные колебания СНз 3050 3,28
Эпокси Симметричное колеба- ние кольца * Антисимметричное де- формационное коле- бание кольца 6 1250 950-810 840-750 8,00 10,53-12,35 11,90-13,38
а Обычно называют «8 д-полосой».
6 Обычно называют «11 д-полосой».
в Обычно называют «12 ц-полосой».
Ниже приведены данные ПМР для этиленоксида и пропилен-
оксида:
Следует отметить, что в принципе ПМР-спектр эпоксидов мо-
жет быть сложным; это происходит вследствие того, что такие
ABC-системы часто состоят из протонов (АВС), которые слабо
отличаются по химическим сдвигам. Так, например:
1. Химические сдвиги геминальных протонов (А и В) неоди-
наковы, и часто обнаруживается геминальная константа спин-
спинового взаимодействия.
2. Константы спин-спинового взаимодействия каждого из про-
тонов В и А с С неодинаковы.
3. Весьма возможно, что мультиплеты А, В и С нельзя ана-
лизировать по правилам первого порядка.
В масс-спектрах эпоксидов имеются пики молекулярных ионов,
однако пути их фрагментации очень сложны.
Определение функциональных групп и установление структуры
357
Таблица 6.9
Класс соединений (в алфавитном порядке) Общая формула Литература, в которой описаны методы определения а
Азиды rn8 [1], с. 123-124; [5], с. 445—446
Азоксисоеди- нения R—N=N(O)R [3], с. 262; [4], с. 178; [5], с. 443
Азосоединения RN=NR [3], с. 262; [4], с. 224; [5], с. 443
Гидразины RNHNH2 [2], с. 339; [3], с. 289; [4], с. 217; [5], с. 443
Гидразоны R2C=NNHAr [2], с. 406; [4], с. 226, 344; [5], с. 445
Гидразосоеди- RNHNHR [5], с. 443
нения
Диазосоедине- ния RNj [1], с. 123-124; [3], с. 266
Изоиитрилы R—NG [1], с. 124—126; [3], с. 254; [4], с. 164, 354; [5], с. 446
Изотиоцианаты R—NCS [1], с. 124—126; [3], с. 297; [4], с. 178; [5], с. 463
Изоцианаты R—NCO [1], с. 125-126; [3], с. 229; [5], с. 442—445
Имиды, имииы R2C=NR [1], с. 139; [3], с. 229; [5], с. 432—435
Карбодиимиды r2c=n- -n=cr2 [5], с. 442—444
Лактамы 1 ( (CH2), ;=o [1], с. 152, 167, 171, 180; [4], с. 198,
276, 287, 362; [5],
L-—r 1H с. 432—435
Лактоны f :=O [1], с. 152, 161, 177; [3], с. 180; [4}, с. 188, 198, 248,
J 1 (CH2)K
L г 4 317
Нитроамины r2n—no2 [5], с. 446—447
Нитраты rono2 [2], с. 401; [5], с. 447
358
Глава 6
П родолжение табл. 6.9
Класс соединений (в алфавитном порядке) Общая формула Литература, в которой описаны методы определения а
Нитриты RONO [2], с. 401
Нитрозосоеди- иеиия R—NO [1], с. 184, 186; [2J; с. 405; [3], с. 301; [41, с. 226; [5], с. 447
Нитроны R2C=N(O)R [5], с. 448
Оксиды аминов RsN —► О (или RaN—о) [5], с. 447
Оксимы R2C=NOH [2], с. 406; [4], с. 191, 216, 226; [5], с. 445
Пероксиды R—О—О—R [3], с. 204; [5], с. 408—409
Перфторал- каиы [5], с. 353
Семикарба- зоиы R2C=NNH(C=O)NH2 [4], с, 336; [5], с. 445
Сульфениль- иые соедине- ния rsx, rsnh2, rsoh [5], с. 457
Тиокарбониль- ные соедине- ния R(C=O)SH, r2c=s, rch=s, R(C=O)SRZ, R(C=S)ORZ, R(C=S)NH2 [3], с. 297, 355; [4[, с. 177; [5], с. 462—463
Тиоцианаты RSCN [3], с. 348; [4], с. 177
Хиноны О о (^S]r um’a о о [3], с. 212; [4], с. 248; [5], с. 394-395
8 1. Conley R. Т„ Infrared Spectroscopy, 2nd ed., Allyn & Bacon, Boston, 1972.
2. Cheronls N., Enirikin J. B., Hodnett E. M„ The Systematic Identification of Organic
Compounds, 3rd ed„ \Viley-Interscience. New York, 1965.
3. Cheronls N„ Ma T. S., Organic Functional Group Analysis. Wiley-Interscience, New York,
1964.
4. Jackman L. M„ Sternhell S., Nuclear Magnetic Resonance In Organic. Chemistry, 2nd ed.,
Pergamon, Elmsford, N. Y., 1969.
5. Pasto D., Johnson C., Organls Structure Determination, Prentice-Hall, Englewood Cliffs,
N. J.. 1969.
Определение функциональных групп и установление структуры
359
Таблица 6.10
Данные ИК-спектров для различных соединений *
Класс соедине- ний (в алфа- витном порядке) Соединение или общая формула б ' Колеба- ние B Полоса или область поглощений [частота, см“’ (длина волны, мКм)] Примечания
Азиды RN3 уЗНТИСИММ ~21Q0 Сильная
Циклогексилазид Ng Ж.СИММ VNS -.аитисимм (4,76) 1350-1170 (7,41-8,55) 2110(4,76) Средняя или слабая Сильная,
Азосое- RN=NR Na VN-N (380-1570 без раство- рителя Слабая,
диие- ния гранс-СН3СН2Ы=МСН2СН2СН3 VN=N (7,25—6,37) 1563 (6,40) непригодна для опре- деления Слабая
TpaHC-C6H6N=NCeH6 VN=N 1455, 1395 Слабая,
цис- C6H6N=NC6H6 трянс-СбНвК=МС6НБ VN=N VN=N (6,87, 7,17) 1511 (6,62) 1442 (6,93) КВг; поло- жение мало изме- няется от замеще- ния в кольце КР-Спектр,
ArN=NAr VN-N 1463-1380 сильная, пригодна для опре- деления То же
Диазо- r2c=n2 VC=N2 (6,83-7,25) 2200-2000 Сильная,
соеди- нения oh2n2 VC=N2 (4,54-5,00) 2075 (4,82) сопряжение смешает полосу поглощения в сторону ббльщих частот Сильная, tci4
n2chcooch2ch8 vc-n2 2101 (4,76) Сильная, СН2С12 Сильная,
N2C(CN)2 vc=n2 2140, 2225
(4,67, 4,49) КВг
360
Глава б
Продолжение табл. 6.10
Класс соедине- ний (в алфа- витном порядке) Соединение или общая формула б Колеба- ние B Полоса или область поглощения [частота, см“* (длина волны. мкм)[ Примечания
Изонит- рилы Изотио- циа- наты Изоциа- наты RNC CH3CH2CH2CH2NC C6H6NC RNCS, ArNCS —NCS R—NCO —NCO VN=C VN=C V№=C VNCS VNCS ..аитисимм VNCO ..симм VNCO ..анти симм VNCO 2180—2100 (4,59-4,76) 2146 (4,66) 2117 (4,72) 2220—1975 (4,50-5,06) 2180—2053' (4,59-4,87) 2280—2250 (4,39—4,44) 1460—1340 (6,84—7,46) 2257 (4,43) Сильная, сопряже- ние сме- щает полосу поглощения в сторону меньших частот Сильная, без рас- творителя То же Сложные полосы поглощения разной интенсив- ности Четыре полосы поглоще- ния, СС14 Сильная Слабая, непригодна для опре- деления Сильная, ecu
C6H5—NCO ..аитисимм VNCO 2267 (4,41) То же
Имины C6H5(C=O)NCO r2c=nh R2C=NR', R = алкил, H + /R r2c=n; \h „антисимм VNCO *N-H VC=N - VC=N 2225 (4,49) 3400—3000 (2,94-3,33) 1675-1620 (5,97-6,17) 1650—1705 (6,06-5,87) Сильная, без рас- творителя Сильная, непригодна для опре- деления Сильная, средняя, слабая Сильная
Определение функциональных групп и установление структуры
361
Продолжение табл. 6.10
Класс соедине- ний (в алфа- витном порядке) Соединение или общая формула б Колеба- ние Б Полоса или область поглощения [частота» см~! (длина волны, мкм)] Примечания
Карбо- дии- миды Лактамы Лактоны ch8ch2ch=nch2ch2ch3 R—N==C=N—R ch3ch2n=c=nch2ch8 о II ср? х = 2 х = 3 х = 4, 5 О д *C=N vn=c=n Vn=c=n vaccou „свобод VNH связ. VCONH VCO vco VCO vco 1673 (5,98) 2260—2100 (4,42-4,76) 2128 (4,70) 3375-3000 (2,96—3,33) 3450-3400 (2,90—2,94) ~ 3200 (~ 3,12) 1760—1730 (5,68-5,78) 1750—1700 (5,71-5,88) 1650 (~ 6,06) 1748 (5,72) Сильная, СС14 Сильная Сильная без раст- ворителя Сред- няя — силь- ная; слож- ные полосы поглощения, обуслов- ленные водородной связью Сильная, нет водо- родной связи Средняя, смешанная полоса поглощения Сильная, при мень- шей вели- чине цикла частота карбониль- ных валент- ных коле- баний увеличи- вается Сильная Сильная, СС14
362
Глава 6
Продолжение табл. 6.10
Класс
соедине-
ний
(в алфа-
витном
порядке)
Соединение пли общая
формула б
Лактоны
Колеба- ние в Полоса или область поглощения [частота, см"1 (длина волны, мкм)] Примечания
vco 1743 (5,73) Сильная, CCI4, соп- ряженная двойная связь в кольце влияет на величину валентного колебания карбониль- ной группы
vco 1770 (5,65) Сильная, СС14
Нитраты
х = 2, р-лактоны
СО
1818
(~ 5,50)
х = 3, у-лактоны
х = 4, д-лактоны
RONO2
vco
vco
„антиснмм
VNO2
-,СИММ
vno2
1795-1760
(5,57-5,68)
1750-1735
(5,71-5,76)
1660—1625
(6,02-6,15)
1285-1270
(7,78-7,87)
Сильная,
уменьшение
размера
цикла
приводит
к увеличе-
нию
частоты
валентного
колебания
карбониль-
ной группы
Сильная
»
»
Определение функциональных групп и установление структуры
363
Продолжение табл. 6.10
Класс соедине- ний (в алфа- витном порядке) Соединение или общая формула б Колеба- ние в Полоса или область поглощения [частота. см~* (длина волны, мкм)] Примечания
Нитраты CH3CH2ONO2 -.аитисимм VNO3 _,симм VNO3 1634, 1282 (6,12-7,80) Сильная, без раст- ворителя
no; .аитисимм VNO3 1040 (9,53) Неоргани- ческие
соли, в ва-
зелиновом
Нитриты Нитрозо- соеди- нения Оксиды амииов RONO R—NO Ar—NO (R—NO)2 (Ar—NO)2 rno2 „транс VN=O «Ч“с VN=O VN—О VN=O VNO „транс VNO V4UC VNO „транс *NO VNO VNO 1681-1648 (5,95-6,07) 1625-1605 (6,15-6,23) 814-751 (12,29-13,32) 1621—1539 (6,17-6,50) 1513-1488 (6,61-6,72) 1290-1176 (7,75—8,50) 1420—1330 (7,04-7,52) 1344-1323 (7,44-7,56) 1299—1253 (7,70-7,98) ~ 1409, 1397—1389 (~7,10, 7,16-7,20) 970-950 (10,31-10,53) масле (нуйоле) Сильная » > » Водородная связь уменьшает частоту; обычно
сильная
(n->o VNO 1300-1200 (7,69-8,33) полоса То же
(ароматический) N-Оксид пиколина VNO 1250 (8,00)
§64
Глава В
Продолжение табл. 6.10
Класс соедине- . ннй (в алфа- витном порядке) Соединение или общая формула 6 Колеба- ние в Полоса или область поглощения (частота, см-1 (длина волны, мкм)1 Примечания
Оксимы Перо- ксиды Тиокар- боннль- ные соеди- нения r2C=N—ОН (RCO)2O2 ro2h ro2r RCO3H С8НЬСО3Н л-Хлорнадбензойиая кислота CS2 RCS2R' VOH VC=N VN—О VCO vc-o первич- ное VC—о вторич- ное VC—о VOH vc=o ^OH vc—0 vo—0 vc=o Vc=o vc=s 3300-3130 (3,03-3,19) 1690—1620 (5,92-6,17) ~ 930 (~ 10,75) 1816-1787 (5,51-5,60) 1790-1765 (5,59-5,67) ~ 1465 (~ 6,83) 1352—1334 (7,40-7,50) 779-769 (12,84-13,00) ~ 3280 (~ 3,05) ~ 1760 (5,68) ~ 1450 (6,90) ~ 1175 (8,51) ~865 (11,56) 1775 (5,63) 1735 (5,76) 1510 (6,62) 1200-1170 (8,3-8,55) Сильная Слабая Сильная » Сильная; эти валент- ные коле- бания С—О примерно на 30 см~1 больше, чем валент- , ные коле- бания С—О в соответ- ствующих спиртах Сред- няя — силь- ная Активно в КР-спектре Газ » » » » Сильная, СС14 То же Сильная, без раст- ворители То же
Определение функциональных групп и установление структуры
36S
Продолжение табл. 6.10
Класс соедине- ний (в алфа- витном порядке) Соединение или общая формула 6 Колеба- ние B Полоса или область поглощения (частота, см”1 (длина волны, мкм)] Примечания
Тиокар- S vc=s 1187 (8,42) Сильная,
бониль- II без раство-
ные СН3СН2С—SCH2CH3 рителя
соеди- R (C=S)NH2 vc=s 1290—1020 То же
неиия (9,75-9,80)
Тиоциа- R—SCN VteN 2170—2135 Сред-
наты (4,61-4,62) НЯЯ — СИЛЕ-
иая
Фториды RF VC—F 1350—1120 (7,41-8,93) Сильная
ArF VC-F 1100-1270 (9,09-7,87) »
Винилфториды VC=CF 1340-1300 (7,46-7,69)
Хиноны О=/ \=O VC=O 1680—1640 (5,96-6,10)
VC=O 1669 (5,99) Сильная, re.
vc—c 1600 (6,25) Средняя,
CS2
VCH 3077 (3,25) Слабая,
CS2
а Взято из литературных ссылок по ИК-спектроскопни, перечисленных в гл. 10,
и из книги: Dolphin D., Wick A., Tabulation of Infrared Spectral Data, Wiley-Interscience,
New York, 1977.
6 R— алкил, Ar—ароматический радикал (арил).
в v—валентные колебания, б—деформационные колебания.
Ряд методик и реакций для определения эпоксидов химически-
ми способами описан Пасто и Джонсоном, с. 376 и 378.
он он
R— СН—СН2 —RCHO + HCHOtlOj-
Ю3 +Ag+ ------------- Agio, (белый осадок)
ОН С1
I I
R—СН—СН—R
хлоргидрин
(1) L1A1H4
(2) Н2О
ОН
I
R2C—СН,
366
Глава 6
6.16. ПРОЧИЕ СОЕДИНЕНИЯ
Было бы слишком претенциозным пытаться описать методы
определения для всех типов органических соединений. В этом
разделе в виде таблицы приведены ссылки
на
химические
спектральные методы анализа (табл. 6.9), которые слишком спе-
цифичны для того, чтобы их описывать в этой книге. Приведена
также таблица полос поглощения в ИК-спектрах этих соедине-
ний (табл. 6.10).
6.17. СОЕДИНЕНИЯ, СОДЕРЖАЩИЕ АЗОТ
(НИТРОСОЕДИНЕНИЯ, НИТРИЛЫ)
Амины, а также анилины, представляющие собой особый слу-
чай аминов, были описаны в разд. 6.7. Здесь мы рассмотрим
нитросоединения (RNO2, разд. 6.17.1) и нитрилы (RCN), назы-
ваемые также органическими цианидами (разд. 6.17.2).
Многие
другие соединения, содержащие азот, кратко упоминались в
разд. 6.16.
6.17.1. Нитросоединения
Обычно по растворимости нитросоединения относятся к клас-
су MN. Необычная растворимость, характерная для некоторых
нитросоединений, была описана в гл. 5.
ИК-спектр типичного нитросоединения приведен 'на рис. 6.43;
обычно нитрогруппа имеет в спектре две интенсивные полосы по-
глощения, обусловленные связанным взаимодействием высокопо-
лярных связей азот — кислород:
Антисимметричные
валентные коле-
бания N (=“О)а
Симметричные ва-
лентные колеба-
ния N(==O)2
Частота, см—1
1661—1499
Длина волны, мкм
6,02—6,67
1389—1259 7,20—7,94
ПМР-спектр типичного ароматического нитросоединения пока-
зан на рис. 6.44. Нитрогруппа смещает сигналы ароматических
протонов на несколько десятых миллионных долей в слабое поле
по сравнению с сигналами протонов бензола. Другие примеры
этого влияния описаны в разд. 6.9. Химические сдвиги а-протонов
по отношению к нитрогруппе подчиняются обычному порядку,
хотя и с необычно малыми инкрементами, величины которых за-
висят от разветвления у углерода, несущего а-протон (разд. 6.14)
CH3NO;
RCHjNO;
R2CHNO;
4.3
Определение функциональных групп и установление структуры
367
Длина волны, мкм
368
Глава 6
Рнс. 6.44. Спектр протонного магнитного резонанса нитробензола (60 МГц, развертка 600 Гц, растворитель CDCls).
Определение функциональных групп и установление структуры ЗвЭ
В масс-спектрах алифатических нитросоединений пики молеку-
лярных ионов обычно малы или вообще отсутствуют. Стабилизи-
рующее влияние ароматического кольца делает молекулярный
ион ароматических нитросоединений достаточно стабильным. Ча-
сто при анализе масс-спектра нитросоединений требуется исполь-
зование азотного правила (гл. 4). Во многих случаях очень по-
лезна фрагментация в масс-спектрах нитросоединений; вместо
пика молекулярного иона (Л!) алифатических нитросоединений
можно использовать пик (М — NO2). Наиболее важные фрагмен-
ты приведены в табл. 6.11.
Таблица 6.11
Нитросоедииення Фрагмент Масса
Алифатические Ароматические [M-NO2]+ NO+ NOJ [М—NO2]+ [М—NO]+ {М—[NO2 + С2Н2] }* {М—[NO + СО] Г NO+ 30 46 (77 для нитробензола) (93 для нитробензола) 30
Обе химические реакции для нитросоединений и их производ-
ных основаны на восстановлении нитрогруппы в амино- или гид-
роксиламиногруппу. Число присутствующих нитрогрупп можно оп-
ределить по отношению изучаемого соединения к обработке гид-
роксидом натрия (опыт 29).
Опыт 27. Восстановление гидроксидом железа (II)
RNO2 + 6F е(ОН)2 + 4Н2О —> RNH2 + 6Fe(OH)3
Около 10 мг исследуемого соединения прибавляют к находящемуся в про-
бирке 1 мл раствора сульфата железа(II) (реактив А) и затем добавляют 0,7 мл
спиртового раствора гидроксида калия (реактив Б). В пробирку вставляют стек-
лянную трубочку так, чтобы она доходила до дна пробирки, и в течение 30 с
пропускают через нее ток инертного газа для удаления воздуха. Пробирку бы-
стро закрывают и встряхивают. Через 1 мин отмечают цвет осадка. Проведите
эту реакцию с нитробензолом, л-нитроанилином, этанолом н пропанолом-2.
Реактивы. (А) К 500 мл свежепрокипяченной дистиллированной воды при-
бавляют 25 г кристаллов двойной сернокислой соли аммония и двухвалентного
железа и 2 мл концентрированной серной кислоты. В раствор опускают желез-
ный гвоздь для предотвращения окисления кислородом воздуха.
(Б) Растворяют 30 г гидроксида калия в палочках в 30 мл дистиллирован-
ной воды и полученный раствор прибавляют к 200 мл 95%-ного этанола.
870
Глава 6
Обсуждение. Образование осадка красно-бурого или бурого
цвета указывает на положительную реакцию*. Этот цвет обуслов-
лен образованием гидроксида трехвалентного железа при окисле-
нии двухвалентного железа нитросоединением, которое в свою
очередь восстанавливается в амин. Зеленоватый цвет осадка ука-
зывает на отрицательную реакцию. В некоторых случаях может
происходить незначительное потемнение гидроксида двухвалент-
ного железа вследствие частичного окисления.
Практически все нитросоединения дают положительную реак-
цию приблизительно в течение 30 с. Скорость восстановления
нитросоединения зависит от его растворимости. Так, п-нитробен-
зойная кислота, растворимая в щелочном реагенте, дает положи-
тельную реакцию почти сразу же, в то время как а-нитронафта-
лин нужно встряхивать приблизительно в течение 30 с.
Другие соединения, которые окисляют гидроксид двухвалент-
ного железа, также дают положительную реакцию. К таким со-
единениям относятся: нитрозосоединения, хиноны, гидроксилами-
ны, алкилнитраты и алкилнитриты. Для исследования сильно ок-
рашенных соединений эту реакцию применять нельзя.
Опыт 28. Восстановление цинком и хлоридом аммония
N°2
+ 41н1
NHOH
+ Н2О
Растворяют 0,5 мл нитробензола в 10 мл 50%-ного этанола и к раствору
прибавляют 0,5 г хлорида аммония и 0,5 г цинковой пыли. Смесь встряхивают
и нагревают до кипения. Затем оставляют на 5 мин, фильтруют н проверяют дей-
ствие фильтрата на реактив Толленса (опыт 11).
Эта реакция основана на восстановлении исследуемого соеди-
нения в гидразин, гидроксиламин или аминофенол; соединения
окисляются реактивом Толленса, который восстанавливается до
металлического серебра.
C6HSNHOH + 2Ag(NH3)2OH —> CeHsNO + 2Н2О + 2Ag + 4NH3
Реакция неприменима в тех случаях, когда само исследуемое
соединение восстанавливает реактив Толленса.
Для качественного определения нитрогруппы было предложе-
но восстановление нитросоединений в азосоединения под дей-
ствием литийалюминийгидрида **.
* Hearon W. М., Gustravson R. G., Ind. Eng. Chem., Anal. Ed., 9, 352 (1937).
** Gilman H., Goreau T. N., J. Am. Chem. Soc., 73, 2939 (1951),
Определение функциональных групп и установление структуры
371
Опыт 29. Обработка ароматических нитросоединений гидрокси-
дом натрия
К 5 мл 20%-ного раствора гидроксида натрия прибавляют 3 мл этанола,
по каплям добавляют нитробензол и смесь энергично встряхивают. Окраску по-
лученного раствора сравнивают с цветом смеси одной капли нитробензола, 5 мл
воды и 3 мл этанола. Эту реакцию повторяют с л-нитрофснолом и п-нитроани-
лииом.
л-Дннитробензол (0,1 г) растворяют в 10 мл ацетона и при встряхивании
прибавляют 2—3 мл 10%-ного раствора гидроксида натрия. Отмечают образо-
вавшуюся окраску. Эту реакцию проводят также с нитробензолом.
Обсуждение. С этими реагентами мононитроаромэтические со-
единения не дают окраски (или дают очень светлую желтую
окраску). Если к одному циклу присоединены две нитрогруппы, то
появляется синевато-фиолетовая окраска; в присутствии трех
нитрогрупп получается кроваво-красный цвет. Присутствие в мо-
лекуле аминогруппы, замещенной аминогруппы или гидроксиль-
ной группы ингибирует появление характерного красного или
фиолетового цвета.
Из полинитросоединений иногда образуются комплексы Мей-
зенгеймера, которые могут давать окрашенные растворы:
Из нитрофенолов способны образоваться сильно сопряженные и
стабильные фенокси-анионы, которые также могут быть окрашены:
Нитросоединения можно классифицировать по их реакции с
азотистой кислотой: для того чтобы такая реакция прошла, необ-
ходимо наличие протона в a-положении к нитрогруппе. Под дей-
ствием основания из первичных нитросоединений удаляется остаю-
щийся а-протон:
NO г NO 1*
а HONO I ОН" I | I
Первичные: RCH2NO2 ------* RCHNO2 -----* LRCNO2J
(красное
окрашивание)
372
Глава 6
Методика описана в книге Пасто и Джонсона, с. 441.
a HONO
Вторичные: R2CHNO2 -------RCNO2
no
(голубое
окрашивание)
R
I HONO
Третичные: R—С—NO2 -------»- реакция не идет (окраска не появляется)
“I
R
При восстановлении нитросоединений в кислой среде образуют-
ся первичные амины (методика 50):
RNO2 + 6[Н] —> RNH2 + 2Н2О
Их можно превратить в соответствующие производные, такие,
как N-замещенные бензамиды, ацетамиды и арилсульфонамиды
(разд. 6.7).
Ароматические мононитросоединения часто можно идентифи-
цировать превращением в соответствующие динитро- или трини-
тропроизводные (методики 26а и 266).
В некоторых случаях наиболее подходящий способ получения
производных состоит в бромировании кольца (методика 46) или
окислении алкильной боковой цепи (методики 27а и ^76).
Нитроалканы можно идентифицировать восстановлением цин-
ком и соляной кислотой в соответствующий первичный амин
(опыт 28), который затем можно превратить в кристаллическое
производное. Недавно был разработан метод, позволяющий вос-
станавливать одну нитрогруппу в присутствии другой (методика
50а).
Методика 50. Восстановление оловом и соляной кислотой
Sn NaOH
RNO2 RNHjer --------------> rnh2
nLl
В небольшую колбочку помещают 2 г гранулированного олова и прибавляют
1 г соединения, содержащего азот (нитро-, нитрозо-, азо-, азокси- или гидразо-
соединеиия). Колбочку соединяют с обратным холодильником и небольшими
порциями добавляют 20 мл 10%-ной соляной кислоты. После прибавления каж-
дой порции колбочку энергично встряхивают. После окончания прибавления
смесь нагревают на кипящей водяной бане 10 мин. Горячий раствор деканти-
руют в 10 мл воды и прибавляют 40%-ный раствор гидроксида натрия в коли-
честве, достаточном для растворения гидроксида олова. Раствор экстрагируют
несколько раз эфиром порциями по 10 мл, экстракт высушивают и эфир отго-
няют.
Восстановление очень труднорастворимых нитросоединений ускоряется при
добавлении 5 мл этанола.
Первичные амины, остающиеся после отгонки эфира, превращают в одно из
их производных, описанных в разд. 6.7.
Определение функциональных групп и установление структуры 373
Методика 50а. Селективное восстановление полинитроароматических соединений
в нитроанилины
В круглодонной колбе объемом 200 мл с обратным холодильником раство-
ряют 2 г полинитросоединения в 30—50 мл метанола. Поскольку в ходе реакции
восстановления выделяется сероводород, то либо прибор должен находиться в
хорошем вытяжном шкафу, либо трубку холодильника следует вывести в склян-
ку для фильтрования под вакуумом объемом 500 мл, в которой находится 200 мл
20%-него раствора гидроксида натрия. К реакционной смеси прибавляют ку-
сочки пемзы или пористого фарфора и смесь нагревают на водяной бане до лег-
кого кипения. Если иитросоединение не растворилось в 50 мл метанола, то сле-
дует добавить 5—10 мл толуола. Водяную баню убирают и в течение 5 мин при
встряхивании добавляют 40 мл метанольного раствора гидросульфида натрия
(см. ниже). Смесь кипятят 20 мин, затем горячий раствор фильтруют в перегон-
ную колбу и метанол отгоняют на водяной бане. К остатку прибавляют 50 мл
теплой воды. Продукты, не содержащие кислых групп (ОН, СООН), можно со-
брать на фильтре, перекристаллизовать из горячего водного изопропилового
спирта и перед определением температуры плавления высушить. Продукты, со-
держащие кислые группы (нитроаминофенолы или нитроаминокислоты), раство-
ряют в щелочном водном растворе и высаживают подкислением раствора уксус-
ной кислотой до слабокислой реакции по лакмусу. Осадок собирают на фильтре
и перекристаллизовывают из водного пропанола-2. Перед определением темпера-
туры плавления продукт следует высушить.
Реактив. Метанольный раствор гидросульфида натрия получают растворе-
нием 40 г Na2S-9H2O в 100 мл воды. Раствор охлаждают ниже 20°С и медленно
при перемешивании и охлаждении прибавляют 14 г порошкообразного бикарбо-
ната натрия. Поддерживая температуру ниже 20°С, прибавляют 100 мл метанола,
смесь перемешивают 30 мин и отфильтровывают гидратированный карбонат на-
трия. Фильтрат немного подщелачивают по фенолфталеину и оставляют при-
мерно на 1 неделю в коричневой склянке при температуре немного ниже 20°С
(но не в холодильнике). Для селективного восстановления одной нитрогруппы в
2 г образца необходимо приблизительно 40 мл этого реактива.
Обсуждение. Ди- и тринитроароматические соединения селек-
тивно восстанавливаются в ароматические нитроамины с выходом
от 70 до 93% при обработке метанольным раствором гидросуль-
фида натрия. Ниже приведены некоторые примеры:
NaSH
1,3-Динитробензол -------► 3-иитроанилин
NaSH
2,4-Динитротолуол -------*• 4-амиио-2-нитротолуол
NaSH
2,4-Динитробензойная кислота --------*• 4-амиио-2-иитробензойиая кислота
NaSH
2,4-Динитрофеиол --------► 2-амино-4-нитрофеиол
NaSH
2,4,6-Тринитротолуол -------*• 4-амино-2,6-динитротолуол
NaSH
1,3,8-Трииитронафталин -------► 2-амино-4,5-дииитронафталин
NaSH
3,4'-Динитродифеиил --------> 4'-амино-3-иитродифеиил
374
Глава 6
Такие ароматические нитроамины являются кристаллическими
веществами, пригодными для использования в качестве полезных
производных. Далее амины можно охарактеризовать, применяя
методики, описанные в разд. 6 7 для аминов и анилина.
Поскольку механизм реакции полностью не выяснен, то пред-
сказать, какая нитрогруппа в системах с неэквивалентными нитро-
группами будет восстанавливаться, затруднительно.
Приведенную выше методику можно использовать также для
восстановления ароматических мононитропроизводных, например
для восстановления n-нитрофенилуксусной кислоты в п аминофе-
нилуксусную кислоту. Однако обычно предпочитают восстановле-
ние оловом (методика 50), цинком или железом (опыт 27) и со-
ляной кислотой.
В некоторых о-Динитросоединениях происходит замещение
одной нитрогруппы:
Литература. Idoux J. Р. J.Chem. Soc. (С), 1970, 435; Hodg-
son Н. Н., Ward Е. R., J. Chem. Soc., 1945, 794.
Алифатические нитросоединения можно перевести в производ-
ные конденсацией с альдегидами, если в нитросоединении имеют-
ся два атома водорода в a-положении, обладающие кислотными
свойствами Нитрогруппа стабилизирует анион, образующийся
при удалении а-протона:
Затем этот анион может атаковать карбонильное соединение. От-
щепление воды от промежуточно образующегося 0-нитроспирта
приводит к получению нитроолефина; типичная последователь-
ность реакций приведена в методике 506.
Методика 506. Конденсация нитрометана с бензальдегидом
под действием основания
г °н л
NaOH I I I НС1
CHSNO2 + СвН6СНО " Lc6H6—CHCH2NOj -------►
CH3UH
C6H5CH=CHNO2
Смесь 3 мл нитрометана, 5,3 бензальдегида и 10 мл метанола в малень-
кой колбочке погружают в баню со смесью льда и поваренной соли. Когда смесь
хорошо охладится, очень медленно прибавляют (например, при помощи капил-
лярной пипетки) охлажденный раствор 2,1 г гидроксида натрия в 7 мл воды,
поддерживая температуру в течение прибавления ниже 10°С. Через 15 мин до-
бавляют 30' мл толченого льда и воды и слегка мутный раствор выливают в пред-
Определение функциональных групп и установление структуры 375
варительно охлажденный раствор 10 мл концентрированной соляной кислоты в
15 мл воды в небольшом стакане. Мутную плавающую сверху жидкость деканти-
руют, осадок собирают на фильтре и промывают 40—50 мл воды. Большую часть
остающейся воды удаляют путем расплавления продукта нагреванием над теп-
лой (60°С) водой, охлаждая его до затвердевания, и декантации. Сырой продукт
в стакане емкостью 30 мл кристаллизуют из 5 мл этанола, нагревая на паровой
бане. Получают 2—3 г пушистых светло-желтых игл, т. пл. 56—68°С. (Внима-
ние-. 0-иитростирол раздражает кожу!).
6.17.2. Нитрилы
По растворимости нитрилы обычно относятся к классу MN.
Они имеют несколько сладковатый запах, который часто слабо
напоминает запах соответствующего альдегида. Например, запах
бензальдегида (СбНбСНО) и бензонитрила (CeHsCN) напоминает
запах горького миндаля. В качестве примесей нитрилы могут со-
держать следы изонитрилов (RNC), которые пахнут отвратительно.
Поэтому следует с большой осторожностью ориентироваться на за-
пах при идентификации.
Анализ ИК-спектров нитрилов обычно позволяет делать впол-
не определенные выводы. Для циангруппы характерна узкая
полоса поглощения с интенсивностью от умеренной до слабой в об-
ласти 2260—2210 см'1 (4,42—4,52 мкм), которая обусловлена ва-
лентными колебаниями C = N. В этой области имеется всего не-
сколько полос поглощения, обусловленных другими группами;
двумя такими группами являются С=С и —NCO. Полоса погло-
щения C=N очень слаба и в некоторых случаях ее можно не
обнаружить; например, а-окси- и а-аминонитрилы часто не имеют
полос поглощения в этой области. ИК-Спектр типичного нитрила
показан на рис. 6.45.
ПМР-Спектр типичного нитрила приведен на рис. 6.46. Сигна-
лы протонов у атома углерода, связанного с циангруппой *, не-
сколько сдвинуты в слабое поле:
CH3CN RCH2CN R2CHCN
М2,15 <-<£-2,45 ^<£-2,90
Пик молекулярного иона нитрилов в их масс-спектрах очень
слаб. Поэтому для его обнаружения обычно необходимо исполь-
зовать специальную масс-спектрометрическую технику (гл. 3).
При изучении нитрилов часто бывает полезно использовать азот-
ное правило для определения молекулярной массы, описанное в
гл. 4. У нитрилов, имеющих по краней мере один водород в
a-положении к функциональной группе, возможно появление пика
(М—1)+, обусловленного потерей а-атома водорода. Выявление
См. примечание * на стр. 348.
w
Рис. 6.45. Инфракрасный спектр пропионнтрила (без растворителя).
Валентные колебания С—Н: /—3010—2890 см“’ (3,32—3.46 мкм), V- (алифатические). Валентные колебания C=N: 2 — 2240 см“’ (4,46 мкм),
G—г!
v.._M (алифатические). Деформационные колебания С—Н: 3— 1458 см-1 (6,86 мкм), 6 СН-С (алифатические); 4—1427 см-1 (7,01 мкм),
G:—1ч аСНММ о
в CH.CN; 5—781 см-1 (12,80 мкм), метиленовые колебания.
СИМ м
378
Глава 6
этого пика также может быть затруднительным из-за его низкой
интенсивности:
R2CH—CN: + е". H2CH -CN* ---- R2C-^g=N*
Mt н”)
r2c=c=n;
Нитрилы, имеющие по крайней мере один атом водорода в у-по-
ложении, могут претерпевать перегруппировку Мак-Лафферти:
Химическая идентификация нитрилов основана- на превраще-
нии умеренно реакционноспособной циангруппы в более реакцион-
носпособную карбоксильную группу или первичную аминогруппу.
Эти продукты можно использовать в качестве производных или
превратить их в другие производные.
Нитрилы можно гидролизовать в соответствующие карбоно-
вые кислоты действием минеральных кислот (методики 51 и 376)
или щелочей (методика 37а)
RCN + 2H2O + HCI —> RCOOH 4-NH4CI
RCN + Н2О + NaOH —> RCOONa + NH3
Если образующаяся кислота является кристаллической, то она
сама может служить в качестве прекрасного производного. Если
кислота представляет собой жидкость или растворима в воде, то
ее трудно выделить в чистом виде. В таком случае лучше всего
охарактеризовать кислоту в виде n-бромфенацилового эфира (ме-
тодика 33).
При контролируемом гидролизе (методика 51а) образуется
соответствующий амид:
RCN4-H2O —>
[ОН
I
R—C=NH
О
R—^NH2
Восстановление нитрилов натрием в спирте приводит к пер-
вичным аминам (методика 52), которые можно идентифициро-
вать прямым превращением в замещенные фенилтиомочевины:
Na
RCN + 4[Н] RCH2NH2
Определение функциональных групп и установление структуры 379
При обработке нитрилов реактивом Гриньяра и последующем
гидролизе получаются кетоны *, которые можно охарактеризовать,
например, в виде их семикарбазонов:
R'
I
RCN + R'MgX —> RC=NMgX
R' Rz
I I
RC=NMgX + H2O + 2HX —> RC=O + NH«X + MgX2
Однако в случае некоторых нитрилов происходит альтернатив-
ная реакция, в которой циангруппа замещается алкильной или
арильной группой **. К соединениям этого типа относятся: а-кето-,
а-окси-, а-алкокси- и а-аминонитрилы.
В присутствии хлористого водорода меркаптоуксусная кислота
(тиогликолевая кислота) конденсируется с нитрилами ' образо-
ванием гидрохлоридов а-(имидоилтио) уксусных кислот (методика
53):
xNH-HCl
RCN + HSCH2COOH + HC1 —> RC^
\SCH2COOH
Эти соли можно идентифицировать по температурам их разло-
жения и по эквивалентам нейтрализации. При титровании ще-
лочью в присутствии тимолового синего в качестве индикатора
они ведут себя как двухосновные кислоты.
Все эти методы основаны на реакции присоединения к циан-
группе. Для орто-замещенных бензонитрилов, например о-толу-
нитрила или о-хлорбензонитрила, такие методы непригодны Про-
странственно затрудненные нитрилы можно гидролизовать в кис-
лоты нагреванием с 75%-ной серной кислотой (методика 51).
Методика 51. Кислотный гидролиз нитрилов
H,S04/H?0/NaCI
Д
бензонитрил
бензойная
кислота
В круглодонную колбу емкостью 100 мл, снабженную обратным холодиль-
ником, помещают около 25 мл 75% ной серной кислоты и 1 г хлорида натрия.
Колбу нагревают до 150—16О°С на масляной бане и через верхнее отверстие хо-
лодильника прибавляют 5 г нитрила порциями по 0,5 мл. После прибавления
каждой порции колбу энергично встряхивают. Смесь нагревают при перемешива-
нии 30 мин при 160°С и затем 30 мин при 190сС. После этого смесь охлаждают
* Shriner R. L. Turner Т. Л-, J. Am. Chem. Soc. 52, 1267 (1930). Получение
реактива Гриньяра описано в методике 12 в этой книге.
** См. Kharasch М. S.. Reinmuth О., Grignard Reactions of Nonmetallic Subs-
tances, Prentice-Hall, Englewood Cliffs, N. J., 1954, p. 776.
380
Глава 6
н выливают в стакан, содержащий 100 г мелкоистолченного льда. Выделяющий'
ся осадок собирают на фильтре и обрабатывают небольшим избытком 10%-ногс
раствора гидроксида натрия. Нерастворимый амид отфильтровывают. При под.
кислении фильтрата выделяется карбоновая кислота, которую можно очистит!
перекристаллизацией из бензола * или из смеси ацетона с водой.
Для гидролиза некоторых нитрилов соляная кислота более эф-,
фективна по сравнению с серной. Однако для трудно гидролиЧ
зующихся эфиров предпочитают использовать серную кислоту,’
так как она имеет более высокую температуру кипения. Прибавь
ление небольшого количества соляной кислоты (в виде хлористо-
го натрия) увеличивает скорость реакции.
Кроме того, нитрилы можно гидролизовать в соответствующие
карбоновые кислоты действием сильных оснований; методика та-’
кого гидролиза описана в этой книге (методика 37а), а также в
книге Пасто и Джонсона, с. 436.
Нитрилы, в которых циангруппа имеет пониженную рёакцион-j
ную способность, например, вследствие наличия разветвления у,
а-атома углерода, гидролизуются труднее. В этих случаях можно
йсйользойать более высококипящий растворитель.
кон. д нао+
R—CN ----------------> RCOO‘K+ -----► RCOOH
гликоль в качестве
растворителя
Гидролиз нитрилов, катализируемый кислотой, можно остано-1
вить на стадии образования амида. Это бывает полезно в тех слуЧ
чаях, когда образующиеся амиды являются кристаллическими и;
нерастворимы в воде (методика на с. 437 в книге Пасто и Джон-,
сона):
конц. H2SO4 холодная H2O HONO
RCN »• --------> RCONHs (-------► RCOOH)
Методика Sla. Контролируемая гидратация нитрила в амид
[ОН 1 О
I I II
R—C=NHj —> R—С—NH2
ВГЗ" Ы13ЬМ2П
В колбу емкостью 200 мл, снабженную обратным холодильником, помещают
3 г нитрила, 4 г воды и 20 г комплекса трифторида бора с уксусной кислотой **
и смесь нагревают на масляной бане до 115—120°С, выдерживают 10 мин при
этой температуре и затем охлаждают в бане со льдом до 15—20°С. Медленно
прибавляют 6 М раствор гидроксида натрия, поддерживая температуру ниже
20°С, до тех пор, пока смесь не станет слабощелочной по лакмусовой бумажке.
* Вместо бензола можно использовать толуол, этанол, ацетон нли тетра-
хлорэтан. Токсичность бензола, а также аминов и галогенидов обсуждается в
приложении IV.
** Используется жидкий комплекс BF3-2CHSCOOH; он продается фирмой
Harshaw Chemical Со. (1945, Е. 97th St., Cleveland, Ohio). Получение его опи-
сано в книгах: Физер Л., Физер М. Реагенты для ограиического синтеза. Пер.
с аигл.—М.: Мир, 1970, т. 1, с. 112, и Синтезы органических препаратов. Пер.
с аигл. — М.: ИЛ, 1949, т. 2, с. 29.
Определение функциональных групп и установление структуры
381
Для этого необходимо около 90—100 мл раствора гидроксида натрия. Холод-
ный раствор экстрагируют трижды порциями по 100 мл смесью диэтилового
эфира и этилацетата (1 : 1). Объединенные экстракты высушивают 5 г безвод-
ного сульфата натрия, фильтруют в сухую перегонную колбу и смесь раствори-
телей (эфира и этилацетата) отгоняют на водяной бане. Остающийся амид очи-
щают перекристаллизацией из воды или из смеси воды и метанола. Перед опре-
делением температуры плавления амид высушивают на пористой керамической
пластинке в вакуумном эксикаторе.
Такую контролируемую гидратацию нитрила в амид можно провести за ко-
роткое время с выходом 90—95%, используя в качестве катализатора комплекс
трифтсрида бора и уксусной кислоты в присутствии ограниченного количества
воды *.
Диамиды (из динитрилов) можно экстрагировать из холодной щелочной
смеси более эффективно тремя порциями метиленхлорнда или хлороформа объ-
емом по 100 мл каждая.
Методика 52. Восстановление нитрилов в амины с последующим превращением
их в замещенные феиилтиокарбамиды
Na/C2H6OH НС1 NaOH
CH3CH2CH2C=N ----------->• —--------> RNHjer ------
Н=н-бутил
бутироиитрил
—> CH3CH2CH2CH2NH2
н-бутиламин
, CeHsNCS ч
[ rnh2----------> rnhcnhc6h5\
В чистую сухую круглодонную колбу емкостью 200 мл, снабженную обрат-
ным холодильником, помещают 20 мл абсолютного этанола и 1 г алифатического
йитрнла (или 2 г ароматического нитрила). Через верх холодильника прибав-
ляют 1,5 г тонко нарезанного металлического натрия так быстро, как это воз-
можно, чтобы реакция шла не слишком энергично. Когда восстановление закон-
чится (через 10—15 мин), смесь охлаждают до 20°С и через холодильник при
энергичном перемешивании содержимого колбы прибавляют по каплям 10 мл
концентрированной соляной кислоты до кислой реакции по лакмусу. Переносят
смесь в перегонную колбу емкостью 200 мл, соединенную с холодильником, и
отгоняют около 20 мл этанола и воды. Колбу с реакционной смесью охлаждают
и в горло перегонной колбы вставляют маленькую капельную воронку, в которой
находится 15 мл 40%-ного раствора гидроксида натрия. К концу холодильника
присоединяют алонж, конец которого погружают в 3 мл воды, находящиеся в
колбе Эрленмейера емкостью 50 мл. Щелочь прибавляют по каплям при переме-
шивании. Реакция идет энергично, поэтому следует проявлять осторожность и
избегать слишком быстрого прибавления щелочи. После прибавления всей ще-
лочи смесь нагревают до полной отгонки амина. Перегонку прекращают, когда
содержимое колбы станет очень вязким.
К дистиллату ** прибавляют 0,5 мл феннлизотиоцианата н смесь очень энер-
гично встряхивают в течение 3—5 мин. Если производное не выделяется сразу,
то вызывают кристаллизацию охлаждением колбы в бане со льдом и трением
стеклянной палочки о стенки колбы. Осадок собирают на фильтре, промывают
небольшим количеством холодного 50%-ного этанола и дважды перекристалли-
зовыЬают из горячей смеси этанола с водой.
* Hauser С. R., Hoffenberg D. S., J. Org. Chem., 20, 1448 (1955).
** Часть этого дистиллата следует оставить для спектральных исследований
н других химических рейкций.
382
Глава б
Кроме того, нитрил можно восстановить в соответствующий
первичный амин обработкой литийалюминийгидридом (Пасто и
Джонсон, с. 438):
(1) LiAlH,
R~feN (2)H2O > RCH2NH’
Предупреждение. Следует обратить внимание на то, что ли-
тийалюминийгидрид энергично реагирует с водой и другими про-
тонными реагентами; при обработке реакционных смесей с остат-
ками литийалюминийгидрида следует соблюдать исключительную
осторожность. При проведении реакций нужно использовать ма-
лые количества вещества, а протонные реагенты прибавлять очень
медленно при охлаждении.
Восстановление нитрилов менее чувствительно к простран-
ственным затруднениям, чем их гидролиз. Так, например, для иден-
тификации такого соединения, как изобутиронитрил, восстановле-
ние является более подходящим методом.
Н2О
Методика 52а. Восстановление — гидролиз нитрилов в соответствующие альдегиды
[Н
R—C=NH
/о-й-ная муравьиная
кислота
н
| ArNHNH2
—► R—С=О (------------> RCH=NNHAr)
а) Для нитрилов, кипящих выше 125°С. В круглодонную колбу емкостью
100 мл помещают 12 мл 90%-ной муравьиной кислоты, 3 мл воды, 1 г (или 1 мл)
нитрила и 1 г сплава никеля Ренея (Al: Ni) * **. Присоединяют обратный холо-
дильник, содержимое колбы тщательно перемешивают встряхиванием и дают
смесн спокойно кипеть. Реакционная смесь дымнт в результате выделения водо-
рода н углекислого газа. Водород образуется в результате реакции алюминия
с муравьиной кислотой, и мелкодисперсный никель катализирует разложение му-
равьиной кислоты на водород и углекислый газ.
Через 1 ч горячую смесь фильтруют. (Предупреждение-, следует избегать
контакта с муравьиной кислотой нли ее парами.) Оставшийся никель промы-
вают 3—4 мл пропанола-2. Фильтрат и промывной спирт объединяют. Половину
полученного раствора можно использовать для превращения альдегидов в 2,4-
диннтрофенилгидразон по методике 9. Если необходимо, то нз другой половины
можно синтезировать димедоновое производное по методике 10.
б) Для нитрилов, кипящих ниже 12Б°С. В круглодонную колбу емкостью
100 мл помещают 16 мл 90%-ной муравьиной кислоты, 9 мл воды, 1,5 г нитрила
и 1,5 г сплава никеля Ренея (Al: Ni). Содержимое колбы тщательно перемеши-
вают и оставляют стоять на 5 мин, периодически встряхивая. Затем к колбе
присоединяют насадку для отгонкн с холодильником для нормальной перегонки.
Алонж к холодильнику присоединяют так, чтобы конец его трубки был неглу-
боко погружен в 2 мл воды, находящейся в короткой пробирке. Смесь осто-
рожно нагревают до кипения, регулируя нагревание так, чтобы в насадке для
* van Es Т., Staskun В., J. Chem. Soc., 1965, 5775.
** Здесь был использован сплав с соотношением компонентов 1:1. Катализа-
тор никель Ренея поставляет фирма W. R. Grace-Raney Catalyst Division, Chat-
tanooga, Tenn.
Определение функциональных групп и установление структуры
383
отгонки отгон конденсировался полностью. Через 15 мин нагревание усиливают
до такой степени, чтобы происходила медленная отгонка. Альдегид с некоторым
количеством воды и небольшим количеством Муравьиной кислоты отгоняют, соби-
рая отгон в 2 мл воды, помещенной в приемник. Собирают около 3 мл дистил-
лята и половину его используют для получения 2,4-диннтрофенилгидразона по
методике 9. Другую половину можно использовать для получения димедонового
производного по методике 10.
Методика 53. Хлоргидраты а-(имидоилтио)уксусных кислот
[NH 1 NH-HC1
II I сухой НС1 II
R—С—SCH2COOHJ -----------> R—С—SCH2COOH
В чистой сухой пробирке растворяют 1 г нитрила и 2 г меркаптоуксусной
(тноглнколевой) кислоты в 15 мл абсолютного эфира. Раствор охлаждают в
бане со льдом и тщательно насыщают сухим хлористым водородом. Пробирку
плотно закрывают пробкой и оставляют в бане со льдом илн в холодильнике
до тех пор, пока не выделятся кристаллы производного. Алифатические нитрилы
образуют продукты присоединения через 15—30 мин, в то время как для арома-
тических нитрилов обычно требуется стояние в течение ночи в холодильнике.
Кристаллы отфильтровывают, тщательно промывают абсолютным эфиром и по-
мещают в вакуум-экснкатор, на дне которого налита концентрированная серная
кислота, а в верхней части находятся небольшие стаканы с таблетками гидро-
ксида калня и с парафином. Температуру разложения определяют в обычном
приборе для определения температуры плавления. При необходимости опреде-
ляют эквивалент нейтрализации титрованием стандартным раствором щелочи
с использованием в качестве индикатора тимолового синего.
На основании результатов определения эквивалента нейтра-
лизации (методика 32) можно вычислить массу группы R. Сле-
дует помнить, что в таких производных имеются две кислотные
группы.
Поскольку нитрилы имеют слишком малую основность по от-
ношению к лантаноидным сдвигающим реагентам (гл. 8), при ана-
лизе ЯМР-спектров нитрилов следует применять только высоко-
фторированные сдвигающие реагенты.
6.18. ОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ СЕРЫ
Обычно серу в органических соединениях легко обнаружить
как на основании химического (сплавление с Na), так и масс-
спектрометрического (интенсивность пика M-f-2) анализа (гл. 4).
Наличие атома серы в органических веществах приводит к тому,
что большая часть этих соединений по растворимости относится
к классу MN.
Поскольку сера может образовывать двухвалентные соедине-
ния, а также и соединения с более высокой степенью окисления,
существует большое число классов соединений серы. Классы серу-
содержащих соединений, наиболее важные для химиков-органи-
ков, перечислены в табл. 6.12. Поскольку анализ ИК-спектров
обычно важен для точного определения класса органического со-
единения серы, в табл. 6.12 приведены также важные полосы
поглощения в ИК-спектрах.
384
Глава 6
Таблица 6.12
Классы органических соединений серы и данные их ИК-спектров
Раздел Класс соединений Общая структура Валентные колебания связей Частота, см"1 Длина волны, мкм
6.18.1 Меркап- таны (тио- фенолы) Сульфиды RSH ArSH S—н S—н 2600-2550 (сл) а 2560—2550 (сл) 3,85—3,92 3,91—3,92
6.18.2 RSR С—S Исключительно обычно непр определения слабые, и годны дли
6.18.3 Дисуль- фиды RSSR С— S, S—S Обычно непригодны для определения
6.18.4 Сульфо- ксиды О II R—S—R s=o 1070-1030 (с) 9,35-9,71
6.18.5 Сульфоны О II R—S—R II О S(=O)2, антисим- метричные S(=O)2, симмет- ричные 1350—1300 (с) 1160—1120 (с) 7,41—7,70 8,62—8,93
6.18.6 Сульфнно- вые кислоты О R—S—ОН О—н, свободные S=O ~ 3650 (с) ~ 1050 (с) ~ 2,74 ~ 9,52
6.18.7 Сульфоно- вые кис- лоты (сульфо- кислоты) О II R—S—ОН О r—so;m+ (солн) О—н S(=O)2, антисим- метричные S(=O)2, симметрич- ные S(=O)2, антисим- метричные S(=O)2, симметрич- ные 3500—3100 (ср) 1350-1342 (с) 1165-1150 (с) ~ 1175 (с) ~ 1055 (с) 2,86—3,23 7,41—7,45 8,58—8,69 ~ 8,5 ~9,5
6.18.8 Сульфо- нил гало- гениды (сульфо- галогениды) О II R—S—С1 О S— О S(=O)2, антисим- метричные S(=O)2, симметрич- ные 700-610 (с) 1410-1380 (с) 1204-1177 (с) 14,28—16,39 7,09—7,25 8,31—8,49
6.18.9 Сульфона- миды (сульфа- миды) О R—S— NH2 II О S(=O)2, антисим- метричные S(=O)2, симметрич- ные 1370—1320 (с) 1170-1140 (с) 7,30—7,57 8,55-8,77
Определение функциональных групп и установление структуры 386
Продолжение табл. 6.12
Раздел Класс соединений Общая структура Валентные колебания связей Частота, см-1 Длина волны, мкм
6.18.10 Эфиры сульфоно- вых кислот О R—S—OR' S(=O)2, антисим- метричные 1372—1375 (с) 7,29—7,49
(сульфо- эфиры) О (ковалент- ные) S(=O)2, симметрич- ные 1195—1168 (с) 8,37—8,56
6.18.11 Сульфаты (ковалент- ные) О R—О— S—OR II О S(=O)2, антисим- метричные S(=O)2, симмет- ричные 1415—1380 1200—1185 7,06-7,24 8,33-8,44
а Интенсивности полос поглощения: с—сильная, ср—средняя, сл—слабая.
6.18.1. Меркаптаны
Первоначально меркаптаны (или тиолы) можно обнаружить
по их чрезвычайно неприятному запаху, обусловленному доста-
точно большой летучестью этих соединений. Для более точного
химического доказательства химическими и спектральными мето-
дами определяют наличие функциональной группы SH. При об-
работке нескольких капель меркаптана насыщенным спиртовым
раствором ацетата двухвалентного свинца обычно выпадает жел-
тый осадок:
RSH + РЬ(И) + 2Н2О —> 2Н3О++ Pb(SR)2
желтый
осадок
Ниже описан реактив, н-бутилнитрит, при помощи которого
можно отличить меркаптаны от других соединений серы (опыт
29а), а также третичные меркаптаны от первичных и вторичных.
Кроме того, для определения меркаптанов можно использовать
раствор Бенедикта (опыт 296).
Опыт 29а. Нитрозирование меркаптанов
НС1
RSH + H-CiHgONO -----> RSNO + h-C4H9OH
эфир
красное
соединение
Около 10 мг соединения, содержащего серу, прибавляют к 1 мл эфира. За-
тем добавляют 1 каплю 6 н. соляной кислоты и 2 капли н-бутилннтрита * (или
* Предупреждение: н-бутилнитрит, как н другие алкилнитриты, обладает
сильной физиологической активностью (сосудорасширяющим действием).
13 Зак. 335
386
Глава 6
этилнитрита). Осторожно встряхивают смесь и отмечают цвет образовавшегося
раствора. Первичные н вторичные алкилмеркавтаны образуют растворы крас-
ного цвета, что обусловлено протеканием следующих реакций:
НС1
RCH2SH + C4H9ONO -----* RCHzSNO + С4Н9ОН
HCl
R2CHSH + C4H9ONO —> r2chsno + C4H9OH
Окраска эфирного слоя в случае третичных алкилмеркаптанов, R3C—SH, тио-
эфиров и дисульфидов бывает от светло-зеленой до желтой. Сульфоксиды, суль-
фоны, сульфинаты и сульфонаты дают при этой пробе отрицательную реакцию.
Обсуждение. Такие же производные образуются и с азотистой
кислотой (опыт 19). Реакция может осуществляться под дей-
ствием любого реагента, который способен быть эффективным
переносчиком NO+. К таким реагентам относятся N2O4, NOBr и
NOC1.
HONO
RSH--------► RSNO + H2O
Кроме того, меркаптаны обесцвечивают бромную воду. Сле-
дует ознакомиться с описанием метода и обсуждением, приведен-
ным в опыте 30.
Опыт 296. Обработка меркаптанов и тиофенолов раствором Бе-
недикта
RSH + раствор Бенедикта —> (RS)2Cu
д си . е I (ArS)2Cu .
ArSH 4- раствор Бенедикта -—► <
I Ar—S—S—Аг
К раствору илн суспензии 0,2 г исследуемого соединения в 3 мл 10%-ного
раствора бикарбоната натрия прибавляют 2 мл раствора Бенедикта *. Отмечают
окраску смеси и наличие нли отсутствие осадка. Затем смесь кипятят 1 мин и от-
мечают полученный результат. Проведите эту реакцию с лаурилмеркаптаном,
тиофенолом, тиофеном, дн-н-бутнлеульфидом, гидрохлоридом L-цистеина и тио-
мочевиной.
Обсуждение. Алифатические и ароматические меркаптаны
образуют желтый осадок, который превращается при кипячении
в жирные желтые шары. С третичными алкилмеркаптанами по-
лучается только голубой раствор. Такая же окраска возникает
и с соединениями, содержащими сульфидную или дисульфидную
группу. Некоторые соединения, имеющие тиольную группу (тио-
гликолевая кислота, меркаптоэтанол и тиосалициловая кислота),
не дают осадка и получаются почти бесцветные растворы.
ь-Цистеин и l-цистин подвергаются глубокой деградации с
образованием черного сульфида меди (II). Диалкилсульфоны,
диарилсульфоны и сульфонаты с реактивом Бенедикта не ре-
агируют.
Валентное колебание связи S—Н меркаптана можно опреде-
лить в ИК-спектре, однако при этом концентрация образца долж-
Получение реактива описано в опыте 13.
Определение функциональных групп и установление структуры
387
на быть достаточна для того, чтобы обнаружить обычно слабое
валентное колебание S—Н. ИК-Спектр типичного меркаптана по-
казан на рис. 6.47. Данные ИК-спектров меркаптанов представ-
лены в табл. 6.12.
Данные спектров ядерного магнитного резонанса ряда обычно
встречающихся модельных меркаптанов приведены ниже; ПМР-
спектр типичного меркаптана показан на рис. 6.48. Протон груп-
пы SH меркаптанов в отличие от протонов, связанных с гетероато-
мами аминов или спиртов, не способен к быстрому обмену, по-
этому его сигнал обычно наблюдается в виде резкого, хорошо опре-
деляемого мультиплета в относительно узкой области. Константа
спин-спинового взаимодействия SH-протона с протонами у а-атома
углерода равна приблизительно 8 Гц.
V „ ArSH CH,SH RCH2SH
^1,2-1,6 M 2,8-3,6 4----*2,1
RjCHSH
^~2,6 8 -3,05
К сожалению, сигнал протона SH в алифатических меркап-
танах часто закрывается сигналом алифатических (С—Н) прото-
нов.
В масс-спектрах алифатических меркаптанов обычно имеются
достаточно интенсивные пики молекулярного иона (Л4) и пики
М 4- 2, что позволяет без труда идентифицировать эти соединения.
Интенсивность пика М + 2 можно использовать для определения
серы, имея в виду вклад 34S в интенсивность пика М Ц- 2 (см.
гл. 4). На основании интенсивности пика М + 2 можно вывести
молекулярную формулу (гл. 4). Ниже приведены некоторые по-
лезные фрагменты, образующиеся при фрагментации меркапта-
нов:
Фрагмент Масса (т/в) Примечание
[CH2SH] [М—34] 47 Указывает на наличие структуры RCH2SH Интенсивный пик, обусловленный по- терей H2S
Ряд методик получения производных показывает существен-
ную нуклеофильность меркаптанов и меркаптид-аниона (RS~).
Меркаптаны и тиофенолы можно превратить в 2,4-динитрофе-
нилтиоэфиры реакцией с 2,4-динитрохлорбензолом:
RSNa + Cl—<
NO?
•NO2 —► RS
NO2 + NaCl
NO2
Методику см. в книге Пасто и Джонсона, с. 451.
18*
Рис. 6.47. Инфракрасный спектр бензотиола (также называют тиофеиолом или фенилмеркаптаном) (без растворителя).
Валентные колебания С—Н: /—3060 см-1 (3,27 мкм), (ароматические). Валентные колебания S—Н: 2—2580 см-1 (3,73 мкм), Vg
(Эту полосу поглощения часто трудно обнаружить в спектрах других тиолов, потому что она бывает значительно слабее, чем показано здесь).
Ароматические обертоны: 3 - 2000-1667 см-1 (5,00—6,00 мкм); образец, приведенный здесь, показывает монозамещенное ароматическое
кольцо (см. рис. 6.24). Валентные колебания С=^С; 4—1600—1400 см"1 (6,25-7,15 мкм), УСшС'
390
Глава б
Для получения сложных тиоэфиров можно использовать 3,5-
динйтробензоилхлорид:
°х /SR
СОС1
Методику см. в книге Пасто и Джонсона, с. 451.
Меркаптаны реагируют также с 3-нитрофталевым ангидридом:
Наличие в этих производных тиоэфиров кислотной группы позво-
ляет определить массу группы R, используя эквивалент нейтра-
лизации (методика 32).
Меркаптаны можно легко окислить в дисульфиды (или в бо-
лее высокое окисленное состояние); при этом достаточны такие
мягкие условия, как обработка иодом:
2RSH + 12 —> RSSR + 2HI
Исключительная легкость образования дисульфидов и их высо-
кая стабильность заставляют думать, что они присутствуют в
качестве примесей в меркаптанах и других достаточно реакцион-
носпособных соединениях серы.
6.18.2. Сульфиды
Если неизвестное органическое соединение содержит один
атом серы, не содержит атомов кислорода и не доказано наличие
меркаптогруппы, то предполагают, что это соединение является
сульфидом. Анализ ИК-спектров не очень полезен для определе-
ния органических сульфидов. Однако для идентификации их мож-
но использовать следующие данные ПМР-спектров:
(€H3)2S (RCH2)2S (R2CH)2S ch3sc„h,
V(?2,05 X 8 -2,55 k 8 -3,0 k 2,45
CH3CH2SR R'CH2CH2SR R'2CHCH2SR
\ 6 -1,25 8 -1,6 ( 8 -1,9
При добавлении небольшого количества сульфида к насыщен-
ному раствору хлорида ртути(II) в этаноле образуется осадок:
R2S + xHgCl2 —» R2s : (HgCl;)*
ОС 3 ДОК
Определение функциональных групп и установление структуры 391
Сульфиды можно окислить в сульфоксиды (R2SO, разд. 6.18.4)
или сульфоны (R2SO2, разд. 6.18.5); ниже приведены соответ-
ствующие реакции и ссылки на литературу:
кмпо4/сн3соон
R2S ----------------> R2SO2 (Пасто и Джонсон, с. 453)
v2o5
R2S -----> R2SO2 (Пасто н Джонсон, с. 453)
H2O2/CH3COOH
R2S ----------------> R2SO2 (Пасто и Джонсон, с. 453)
NalO4
R2S ц R2SO [J. Org. Chem., 27, 282 (1962)]
G2H5UH
Анализ ПМР-спектров сульфидов можно облегчить, используя
лантаноидные сдвигающие реагенты (гл. 8), однако ясно, что
при этом только высокофторированные реагенты будут давать
существенные химические сдвиги *.
6.18.3. Дисульфиды
Дисульфиды можно обнаружить либо непосредственно по
присутствию в молекуле двух атомов серы (на основании резуль-
татов элементного анализа и особенно масс-спектрометрии, гл.4),
либо косвенным путем на основании отсутствия доказательств
наличия SH-группы (разд. 6.18.1). Для определения дисульфидов
ИК-спектрометрия малопригодна. Анализ дисульфидов при помо-
щи ПМР-спектров аналогичен обсуждавшемуся выше анализу
сульфидов. Ниже приведены некоторые полезные данные:
CH3SSR RCH2SSR
~2,35 S —2,70
Хотя дисульфидная связь сера — сера легко образуется, эту
связь также легко можно разорвать действием обычных реаген-
тов. Такое расщепление сульфидов может происходить с окис-
лением в сульфонаты или с восстановлением в меркаптаны:
КМпО4
RSSR -------> 2RSO3H (методика 27)
Zn/H +
RSSR -------> 2RSH
Полученные при этом продукты далее можно идентифицировать
по методикам, описанным в разд. 6.18.7 (сульфоновые кислоты)
и разд. 6.18.1 (меркаптаны).
Morrill Т. С. et al., Tetrahedron Lett., 1973, 3715; 1975, 397.
6.18.4. Сульфоксиды
Органические сульфоксиды (R2SO), особенно с низкой моле-
кулярной массой, часто бывают очень гигроскопичными соедине-
ниями. Это может легко привести к появлению ложных полос
поглощения О—Н в их ИК-спектрах. Сульфоксиды, как прави-
ло, --имеют отвратительный запах, который может быть обусловлен
примесью органического сульфида. Связь сера — кислород и не-
связанная пара электронов сульфоксида конфигурационно ста-
бильны; например, для приведенного ниже бис-сульфона возмож-
но существование двух геометрических изомеров.
Обычными растворителями для ЯМР-спектроскопии различных
органических веществ являются следующие:
О
II
CH3SCH3
дмсо
днметил сульфоксид
О
//
CD3SCD3
ДМСО-ф,
Внимание. Сульфоксиды реагируют со взрывом с хлорной кис-
лотой! ДМСО или ДМСО-d^ нельзя использовать в качестве рас-
творителя для образцов при съемке ЯМР-спектров в тех случаях,
когда в реакциях получения исследуемых веществ была исполь-
зована в качестве катализатора НС1О4.
ИК-Спектры сульфинильных соединений имеют интенсивные
полосы поглощения валентных колебаний S=O (табл. 6.12), что
оказывает неоценимую помощь при идентификации этих веществ.
При интерпретации ПМР-спектров могут помочь приведенные
ниже значения химических сдвигов:
О
II
СНз—S—R
О
R'CHjSR
<5* ~2.6
<7 ~3,0
Метиленовые протоны в приведенной выше второй структуре
имеют неэквивалентные химические сдвиги; такая система прото-
нов может давать удивительно сложный сигнал в ПМР-спектре.
Сульфоксиды можно окислить в сульфоны (R2SO2) по мето-
дикам, описанным в разд. 6.18.3 (сульфиды).
6.18.5. Сульфоны
Обычно сульфоны не имеют запаха, бесцветны и в химическом
отношении довольно инертны. Для определения сульфонильной
группы полезны две сильные полосы поглощения в ИК-спектрах,
обусловленные связанными валентными колебаниями сера — кис-
лород (табл. 6.12).
W W
К l/ \
симметричные антисимметричные
Часто бывает полезен также анализ ПМР-спектров; ниже приве-
дены данные для типичных соединений:
Одной из важных химических реакций сульфонов является их
восстановление в сульфиды под действием литийалюминийгид-
рида *. Другая реакция заключается в образовании анионов со-
пряженных оснований сульфонов с последующим связыванием
этого аниона электрофильными агентами:
ООО
II В' || - Е+ ||
R—S—СН8 > R—S—СН2' —-> R—S—СН2Е
А А А
6.18.6. Сульфиновые кислоты
Получен ряд ароматических сульфиновых кислот (ArSO2H),
однако в свободном виде эти кислоты неустойчивы и превращают-
ся в соответствующие сульфоновые кислоты и эфиры тиосульфо-
кислот. Эфиры тиосульфокислот гидролизуются с образованием
тиофенолов и сульфоновых кислот:
3ArSO2H —> ArSO3H + ArS—SO2Ar + Н2О
I
ArSH+ ArSO3H
Алифатические сульфиновые кислоты еще менее устойчивы, чем
ароматические. Поэтому оба типа кислот обычно встречаются в
виде натриевых, калиевых или магниевых солей, которые более
Fordwell F. G„ McKellin IF. Н., J. Am. Chem. Soc., 73, 2251 (1951).
394
Глава 6
устойчивы. Наилучшими их производными являются соединения,
получающиеся при алкилировании, причем при этом образуются
главным образом сульфоны, а не эфиры. Например, в реакциях
метилиодида с натриевыми солями аренсульфиновых кислот по-
лучаются метилсульфоны:
ArSO2Na + СН31 —> ArSO2CH3 + Nal
Для синтеза кристаллических 1,2-диалкилсульфонилэтанов из
солей алифатических сульфиновых кислот используют дибром-
этан *:
CH2SO2R
BrCH2CH2Br + 2RSO2Na —> | + 2NaBr
CH2SO2R
В реакциях натриевых солей арил- и алкилсульфиновых кислот
с хлоридом ртути(II) образуются соответствующие арил- и алкил-
меркурийхлориды, представляющие собой кристаллические веще-
ства **.
Наиболее полезный метод количественного анализа сульфи-
новых кислот — титрование стандартным раствором нитрита нат-
рия в кислом растворе ***:
2RSO2H + HNO2 —> (RSO2)2NOH + Н2О
Иодкрахмальная бумага используется в качестве внешнего ка-
пельного индикатора. До появления избытка нитрита на индика-
торной бумаге не наблюдается никакой окраски. В присутствии
нитрита возникает пурпурное окрашивание. Сульфиновые кислоты
можно легко идентифицировать растворением образца в холодной
концентрированной серной кислоте с прибавлением капли анизола
(или фенетола). При положительной реакции появляется голубая
окраска ****.
Кроме того, для анализа сульфиновых кислот могут приме-
няться следующие методы:
ИК- н УФ-спектрометрия
Очистка с помощью солей
Fe(III)
Анализ с помощью солей
Fe(III)
Количественный анализ с по-
мощью окисления гипохлори-
том
Detoni S., Hadzi D., J. Chem. Soc., 19Б5
3163; Bredereck H., Brod G., Hoschele G.,
Chem. Ber., 88, 438 (1955)
Overberger C. G., Godfrey J. J., J. Polymer
Sci., 40, 179 (1959)
Krishna S., Singh H., J. Am. Chem. Soc.,
60, 792 (1928)
Ackerman L., Ind. Eng. Chem., Anal. Ed., 18,
243 (1946)
* Allen C. F. H., J. Org. Chem., 7, 23 (1942).
** Marvel C. S., Adams С. E., Johnson R. S„ J. Am. Chem. Soc., 68, 2735
(1946).
*** Kice J. L., Bowers K. J. Am. Chem. Soc., 84 , 605 (1962); Lindberg B.,
Acta Chem. Scand., 17, 383 (1963); Marvel C. S., Johnson R. S., J. Org. Chem.,
13, 822 (1948).
**** Vogel A. I., Practical Organic Chemistry, 3rd ed., Longmans, London,
1956, p.‘1078.
Определение функциональных групп и установление структуры 395
6.18.7. Сульфоновые кислоты (сульфокислоты)
Поскольку сульфоновые кислоты отличаются от серной кислоты
лишь тем, что одна гидроксильная группа в них замещена на ор-
ганическую группу, то не удивительна высокая кислотность этих
соединений. Обычно сульфокислоты и их металлические соли рас-
творимы в воде О II НО—S—ОН о О О II II R—S—ОН Ar—S—ОН О О
серная кислота алкнлсульфокислота арнлсульфокислота (алкнлсульфоновая (арнлсульфоновая кислота) кислота)
и нерастворимы в органических растворителях. Анализ ИК-спек-
тров может быть осложнен тем, что колебания связи сера — кисло-
род могут давать мультиплет полос поглощения вместо трех хо-
рошо определяемых полос, указанных в табл. 6.12. Анализ ИК-
спектров может осложняться также высокой гигроскопичностью
сульфоновых кислот.
Сульфоновые кислоты и их соли, аналогично карбоновым ки-
слотам и их солям, легко образуют характерные производные с
S-бензилтиуронийхлоридом. Эта реакция представляет собой са-
мый быстрый и прямой способ получения производных этих сое-
динений (методика 53а).
Сульфоновые кислоты и их соли превращаются в сульфонил-
хлориды при нагревании с пентахлоридом фосфора (методика 54):
RSO3Na + РС1В —> RSO2C1 + РОС13 + NaCl
Полученный сульфонилхлорид затем обрабатывают аммиаком или
амином для получения амида (методики 31 и 54):
RSO2C1 + 2NH3 RSO2NH2 + NH,C1
Поскольку сульфоновые кислоты являются сильными кислота-
ми, выделение путем подкисления растворов их солей оказывает-
ся невозможным. Исключение составляют сульфокислоты, очень
плохо растворимые в воде или соляной кислоте. Сульфокислоты,
взаимодействуя с аминами, образуют соли, которые имеют четкие
температуры плавления или разложения и поэтому могут исполь-
зоваться в качестве производных.
При обработке концентрированного раствора натриевой соли
сульфоновой кислоты соляной кислотой и n-толуидином можно вы-
делить n-толуидиновые соли (методика 55), пригодные для иден-
тификации:
RSO3Na + НС1 + ArNH2
ArNH3RSOJ + NaCl
396
Глава 6
Для алифатических сульфокислот используют фенилгидразиновые
соли *.
При реакции серебряных солей сульфокислот с л-нитробензил-
хлоридом и пиридином образуются соответствующие п-нитробен-
зилпиридиниевые соли **. Эти соли легко кристаллизуются, имеют
четкие и характерные температуры плавления и пригодны в ка-
честве производных. Аминосульфокислоты бензольного и нафтали-
нового ряда можно охарактеризовать замещением аминогруппы
на хлор по реакции Зандмейера с последующим превращением
сульфоновой кислоты в сульфонамид или в сульфанилид (мето-
дика 56).
.RSO3~Ag+ArCH2Cl + C,H5N-> /Qn—СН2Аг RS<V+AgCI
Методика 53а. Беизилтиуроииевые соли сульфоновых кислот
RSO3H + NaOH —* RSOJNa*
RSOJNa++CeH5CH2SC(NH2)tCr —> C6H5CH2SC(NH2)2+RSO3 + NaCl
Около 1 г натриевой илн калиевой соли сульфокислоты растворяют в мини-
мально возможном количестве воды. Если необходимо, то раствор подогревают.
В тех случаях, когда исходным веществом является свободная сульфоновая кис-
лота, ее растворяют в 2 н. растворе гндрокснда натрия, а избыток щелочи ней-
трализуют соляной кислотой, используя в качестве индикатора фенолфталеин.
Получение тиурониевого реактива описано в методике 39.
Растворяют 1 г бензнлтиуронийхлорида в минимально возможном количе-
стве воды. Этот раствор н раствор сульфоната охлаждают в банё с ледяной во-
дой, смешивают и хорошо перемешивают. Иногда бывает необходимо потереть
палочкой о стенки пробирки н охладить смесь в бане со льдом, чтобы вызвать
кристаллизацию. Кристаллы бензнлтиурониевой соли сульфокислоты собирают
на фильтре, промывают небольшим количеством холодной воды и перекристалли-
зовывают нз горячего 50%-ного этанола.
Методика 54. Получение сульфонилхлоридов и сульфонамидов
из солей сульфокислот
В чистой сухой колбе смешивают 2 г соли с 5 г пентахлорида фосфора, при-
соединяют обратный холодильник н колбу нагревают на масляной бане в тече-
ние 30 мин при 150°С. Смесь охлаждают н прибавляют 20 мл сухого бензола ***.
Затем смесь нагревают на паровой бане, хорошо перемешивая твердую массу.
Раствор фильтруют через сухой бумажный фильтр, фильтрат промывают дважды
водой порциями по 15 мл. Бензол отгоняют на паровой баие. Остающийся суль-
фонилхлорид можно перекристаллизовать из петролейного эфира илн из хлоро-
форма. Поскольку сульфонилхлориды обычно имеют низкую температуру плав-
* Latimer Р. Н., Bost R. IV., J. Am. Chem. Soc., 59, 2500 (1937).
** Huntress E. H., Foote G. L., J. Am. Chem. Soc., 64, 1017 (1942).
*** Бензол ядовит. См. примечание к методике 20.
Определение функциональных групп и установление структуры 397
.пения, то лучше получить амид, прибавляя при энергичном перемешивании их
бензольный раствор к 20 мл концентрированного раствора аммиака. Иногда
сульфонамид выпадает в осадок и его можно отфильтровать; в других случаях
для получения сульфонамида упаривают бензольный слой. Сульфонамиды можно
перекристаллизовать из этанола.
Методика 55. n-Толуидиновые соли сульфокислот
а) Из свободных сульфокислот. Растворяют 1 г сульфокислоты в минималь-
ном количестве кипящей воды и прибавляют 1 г n-толуидина. Для получения
прозрачного раствора добавляют еще воды нли некоторое количество сульфо-
кислоты. Раствор охлаждают и трением стеклянной палочкой о стенки колбы
вызывают кристаллизацию соли. Соль отфильтровывают и перекристаллизовы-
вают из минимального количества кипящей воды.
б) Из растворимых солей сульфокислот. Около 2 г натриевой, калиевой или
аммонийной соли сульфокислоты растворяют в минимальном количестве кипя-
щей воды и прибавляют 1 г n-толуидина и 2—4 мл концентрированной соляной
кислоты. Если выделяется осадок или если л-толунднн растворился ие полностью,
то добавляют горячую воду и несколько капель концентрированной соляной кис-
тоты до тех пор, пока при кипячении не получится прозрачный раствор. Раствор
охлаждают и трением стеклянной палочкой о стенки колбы вызывают кристал-
лизацию соли. Продукт отфильтровывают и перекристаллизовывают из неболь-
шого количества воды нли разбавленного этанола.
Методика 56. Получение хлорарилсульфонамидов н хлорарилсульфанилидов из аминосульфокислот (о) SO,H SO, Na2CO3 —'J ''— G * nh2 nh; HONO, CuCI I HCI ’ 1 SO,Na* G NH, SO.,~Na+ КЗ-0 N/СГ SO,H .Qh-g 1 Cl
398
Глава 6
а) Около 1,5 аминоарилсульфокислоты растворяют в 10 мл воды, содержа-
щей 0,5 г карбоната натрия. Для диазотирования прибавляют 2 мл концентри-
рованной соляной кислоты, после чего быстро приливают около 5 мл 10%-иого
раствора нитрита натрия. Температуру смеси поддерживают около 10—15°С, до-
бавляя в смесь лед.
В это же время готовят хлорид меди(1), смешивая раствор 2,16 г сульфата
меди и 0,56 г хлорида натрия в 10 мл воды с раствором 0,46 г бисульфита
натрия и 0,33 г гидроксида натрия в 10 мл воды. Затем осадок хлорида меди(1)
растворяют в 10 мл концентрированной соляной кислоты. Раствор охлаждают
во льду до 5°С и при перемешивании быстро прибавляют раствор соли диазония.
Смеси дают медленно нагреться до комнатной температуры, продолжая пере-
мешивать в течение 1 ч, а затем нагревают 30 мин при 60—70°С на паровой
бане. Медь осаждают сероводородом и выделившийся сульфид меди отфильтро-
вывают. Сырую хлорарилсульфокислоту получают, упаривая фильтрат досуха
на паровой бане.
б) В небольшом стакане смешивают сырую сульфокислоту' с двойным по
массе количеством пентахлорида фосфора. После окончания энергичной реакции
стакан нагревают в течение короткого времени на масляной бане при 130—140°С
(под тягой) для удаления хлорокиси фосфора. После охлаждения хлорид про-
мывают декантацией холодной водой. Полученное масло прибавляют к 45 мл
концентрированного водного раствора аммиака и полученный раствор упари-
вают досуха на паровой бане. Сырой сульфонамид перекристаллизовывают из
этанола или воды. Для очистки полученного продукта прибавляют активирован-
ный уголь.
в) Для получения хлорарилсульфаннлида раствор сырого сульфонилхлорида
в 10 мл бензола * смешивают с 2,5 г анилина и полученный раствор кипятят в
течение 1 ч. Затем раствор упаривают до половины объема и охлаждают. Выде-
лнвшнйси твердый осадок собирают на фильтре, тщательно промывают теплой
водой и перекристаллизовывают из хлорбензола.
6.18.8. Сульфоиилхлориды (сульфохлориды)
Сульфонилхлориды имеют ряд характерных полос поглоще-
ния в ИК-спектрах (табл. 6.12). Кроме того, летучие сульфонил-
галогениды обладают проникающим неприятным запахом. Из-за их
высокой реакционной способности сульфонилгалогениды часто
идентифицируют, превращая их в другие соединения, например, в
сульфонамиды, которые обладают меньшей реакционной способ-
ностью и поэтому легче идентифицируются.
Бензол ядовит. См. примечание к методике 20.
Определение функциональных групп и установление структуры 399
Сульфонилхлориды легко превращаются в амиды обработкой
водным аммиаком (методика 54) или карбонатом аммония (ме-
тодика 48). Реакция Хинсберга (методика 16) используется для
получения сульфанилидов или сульфотолуидидов, удобных для
идентификации.
16.18.9. Сульфонамиды (сульфамиды)
Большинство сульфонамидов можно отличить по полосам по-
глощения в ИК-спектрах, обусловленных валентными колебаниями
связей S(=O)2 и N — Н. Сульфонамиды обычно являются ста-
бильными кристаллическими соединениями. Наилучший метод хи-
мического анализа этих соединений — расщепление связи сера —
азот с последующей идентификацией продуктов расщепления.
Основная реакция сульфонамидов состоит в их расщеплении
путем гидролиза (методика 57). Амины (разд. 6.7) и сульфокис-
лоты (разд. 6.18.7) можно идентифицировать по методикам, опи-
санным в указанных разделах.
Методика 57. Гидролиз сульфонамидов
RSO2NHR' + Н2О + НС1 —> RSO3H + R'NH3C1
RSO2NR£+ Н2О + НС1 —> RSO3H+ R£NH2C1
Гидролизуют 10 г сульфонамида кипячением с обратным холодильником со
100 мл 25%-ной соляной кислоты. Для гидролиза сульфонамидов первичных
аминов требуется кипячение в течение 24—36 ч, а сульфонамиды вторичных
аминов могут быть прогидролнзованы за 10—12 ч. После полного растворения
сульфонамида смесь охлаждают, подщелачивают 20%-ным раствором гидроксида
натрия и экстрагируют 3 раза эфиром порциями по 50 мл. Эфирный раствор
высушивают, удаляют эфир, а оставшийся амин перегоняют. Некоторые амины,
имеющие очень низкую или очень высокую температуру кипения, часто более
удобно превратить в гидрохлориды, пропуская сухой хлористый водород в сухой
эфирный раствор.
Амин можно выделить прибавлением щелочи и охарактери-
зовать в виде соответствующего производного (опыт 19). В случае
необходимости сульфоновую кислоту можно выделить в виде ее
натриевой соли (после удаления амина) и превратить в какое-либо
производное (методика 54 или 55).
Более удобный метод состоит в обработке амида 48%-ной бро-
мистоводородной кислотой и фенолом (методика 16):
2ArSO2NHR + 5НВг + 5С6Н5ОН —> ArS SАг + 2RNH2 + 5ВгСвН4ОН + 4Н2О
Эта реакция позволяет не только охарактеризовать амин, но также
выделить диарилдисульфид и использовать его в качестве произ-
водного (разд. 6.18.3).
Амидный протон является достаточно лабильным, поэтому его
можно удалить от атома азота с образованием анионов сульфон-
амидов, способных к нуклеофильной атаке алкилгалогенидов и
карбонильных атомов углерода; приведенные реакции служат при-
мерами такой нуклеофильной атаки.
400
Глава 6
Сульфонамиды, имеющие по крайней мере один атом водорода
у атома азота, можно алкилировать обработкой основанием и ре-
акционноспособными галогенидами или алкилсульфатами:
RSO2NH2 + NaOH + R'X —> RSO2NHR' + NaX + H2O
RSO2NHR'+ NaOH + R"X —> RSO2NR'R" + NaX + H2O
Эти реакции могут быть полезными, если выбрать такую алкиль-
ную группу, чтобы в результате получался известный сульфон-
амид.
Первичные сульфонамиды реагируют с фталоилхлоридом с об-
разованием N-сульфонилфталимидов:
Первичные амиды сульфоновых кислот, аналогично амидам кар-
боновых кислот (разд. 6.12, методика 38), реагируют с ксантгид-
ролом с образованием N-ксантилсульфонамидов, пригодных для
идентификации (методика 58).
Методика 58. Получение N-ксантилсульфонамидов
ксантгидрол
Около 0,2 г ксантгидрола растворяют в 10 мл ледяной уксусной кислоты.
Если смесь не прозрачная, то ее фильтруют или центрифугируют. К прозрач-
ному раствору прибавляют 0,2 г сульфонамида, смесь встряхивают н оставляют
прн комнатной температуре, пока не выделится производное; на это может по-
требоваться до 1,5 ч. N-Ксантилсульфонамид отфильтровывают и перекристал-
лизовывают из смеси диоксана и воды (3: 1).
Сульфонамиды можно легко расщепить восстановлением ани-
он-радикалом нафталиннатрия. При этом по простой методике*
образуются сульфинат-анион и соль амина:
ГСШЬ.- но
RSO2NHR L —’ -----—------* RSO2-Na++R’NH2
* Closson W. D., JI S., Schulenberg S., J. Am. Chem. Soc., 92, 650 (1970).
Сульфонамиды и сульфонаты можно также расщепить методом, описанным в
ссылках [I] и [2], приведенных в этой статье. Там же обсуждается газохро-
матографический анализ аминов, часто представляющий собой трудную задачу,
Определение функциональных групп и установление структуры
401
6.18.10. Эфиры сульфоновых кислот (сульфонаты)
В данном разделе рассматриваются эфиры сульфоновых кислот
RSO3R', в которых имеется ковалентная связь углерод — кислород
с группой R'. Этим они отличаются от солей сульфокислот RSO3M+,
которые имеют ионную связь металл — кислород (разд. 6.18.7).
Эфиры сульфокислот часто довольно устойчивы, фактически эти
эфиры используются вместо сульфонамидов в качестве производ-
ных сульфокислот. Наилучшими для этих целей являются эфиры
фенолов, гликолей и первичных спиртов.
О
RSO3H — рС1 R'OH II
----” RSO2CI -——г” R—s—OR' + C6H5NH-Cr
RSO3‘Na+ - CbH6N И
О
эфир
сульфокислоты
Такие реакции проводят, обрабатывая раствор сульфонилхло-
рида в пиридине (C5H5N) эквивалентным количеством спирта.
Если реакция при комнатной температуре происходит достаточно
быстро, вызывая заметное разложение веществ, то следует исполь-
зовать ледяную баню. Пиридин используется в качестве раствори-
теля; одновременно он служит основанием для связывания мине-
ральной кислоты — побочного продукта реакции. Гидрохлорид
пиридина может выпадать из раствора в виде кристаллов, что
позволяет контролировать ход реакции визуально.
Наиболее обычными примерами этого класса соединений яв-
ляются алкиларилсульфонаты (ArSO3R). Метилтозилат (метило-
вый эфир п-толуолсульфокислоты) представляет собой прекрасный
алкилирующий реагент:
о н
R2NH+CH34(jH~ОСН, >R2NCH, “OTs
о
TsO—
n-Бромбензолсульфонаты (брозилаты), а также и другие заме-
щенные бензолсульфонаты используются для изучения карбока-
тионов, в том числе и карбокатионного характера группы R:
Группа R может быть связана гидридами металлов:
LiAIH«
ArSO,R RH + ArSOj"
402
Глава 6
Получающаяся соль сульфокислоты может быть охарактеризована,
как описано в разд. 6.18.7.
6.18.11. Сульфаты
Органические сульфаты (сложные эфиры серной кислоты) обыч-
но значительно более реакционноспособны, чем, например, сложные
эфиры сульфокислот. Так, например, диметилсульфат широко ис-
пользуется в качестве метилирующего реагента:
О
II
RCOOH + CH3O—S—OCH3 —> RCOOCH3 + CH3OSO3H
II
О
[(CH3)2SO4]
Внимание. Алкилсульфаты — высокотоксичиые соединения и
работать с ними следует с предельной осторожностью и при на-
личии достаточно хорошей вентиляции. Не следует допускать кон-
такта этого вещества с кожей, так как диметилсульфат способен
проникать в организм человека через кожу.
6.19. ФЕНОЛЫ
Фенолы считают слабыми кислотами (класс Аг), так как они
растворимы в растворах гидроксида натрия и не растворяются в
растворе бикарбоната натрия:
ArOH + NaOH —> ArO'Na* + Н2О
Исключения Из этого правила были рассмотрены в гл. 5. Так, на-
пример, пикриновая кислота растворима в растворах более сла-
бого основания — бикарбоната натрия:
Фенолы можно идентифицировать при обработке их хлоридом
железа (III) (опыт 29в). Методика с использованием пиридина в
качестве растворителя позволяет точно идентифицировать феноль-
ные соединения в 90% случаев. При использовании в качестве
растворителей воды или смеси спирта с водой успех достигается
лишь в 50% случаев.
Определение функциональных групп и установление структуру, 403
Опыт 29в. Реактив хлорид железа(Ш) — пиридин
ЗАгОН+ 3(^] + FeCl3 Fe(OAr)3 + 3 <^NH Cl
В чистую сухую пробирку помещают 2 мл хлороформа и прибавляют 30—
50 мг кристаллического исследуемого вещества (или 4—5 капель жидкого ве-
щества). Смесь перемешивают до растворения. Если окажется, что исследуемое
соединение не растворяется (даже частично), то прибавляют еще 2—3 мл хло-
роформа и пробирку осторожно подогревают. Охлаждают до 25°С и прибавляют
2 капли 1%-ного раствора безводного хлорида железа(III) в хлороформе и за-
тем 3 капли пиридина. Пробирку встряхивают, чтобы перемешать смесь, и не-
медленно отмечают образующуюся окраску. Образование раствора голубого,
фиолетового, пурпурного, зеленого или красно-бурого цвета считается положи-
тельной реакцией на фенолы. Часто окраска раствора изменяется в течение не-
скольких минут. Проведите эту реакцию с n-крезолом, салициловым альдегидом,
о-нитрофенолом и л!-бромфенолом.
Реактивы. В качестве растворителя для получения раствора хлорида же-
леза (III) следует использовать чистый хлороформ, не содержащий этанола. Рас-
твор хлорида железа(III) готовят прибавлением 1 г черных кристаллов безвод-
ного хлорида железа (III) к 100 мл чистого хлороформа в колбе емкостью
150 мл. Смесь периодически встряхивают в течение примерно 1 ч и оставляют
стоять, чтобы нерастворившееся вещество осело на дно. Светло-желтый раствор
декантируют в бутыль с завинчивающейся пробкой, снабженную медицинской
капельницей. Аналитически чистый пиридин также наливают в бутыль с завин-
чивающейся пробкой, снабженную медицинской капельницей.
Обсуждение. Описанный реактив пригоден для определения
соединений, содержащих гидроксильную группу, непосредственно
присоединенную к ароматическому ядру. При обработке хлоро-
формных растворов фенолов, нафтолов и их производных, замещен-
ных в кольце, раствором безводного хлорида железа(III) в хлоро-
форме в присутствии пиридина образуются комплексы, окрашен-
ные в характерные цвета: голубой, фиолетовый, пурпурный, зеле-
ный или красно-бурый.
Спирты, простые эфиры, альдегиды, кислоты, кетоны, углево-
дороды и их галогенпроизводиые дают отрицательные результаты.
Растворы этих соединений остаются бесцветными, светло-желтыми
или рыжевато-коричневыми.
Этот метод особенно ценен для идентификации очень плохо
растворимых в воде замещенных фенолов и нафтолов. Даже 2,4,6-
трихлорфенол, 2,4,6-трибромфенол, нонилфенол, фенолфталеин
и тимолфталеин в этих условиях дают положительные, реакции,
если используется достаточное количество хлороформа (около
5 мл), чтобы перевести их в раствор.
К фенольным соединениям, которые не дают положительные
реакции при этом испытании, относятся: пикриновая кислота,
2,6-ди-трет-бутилфенолы, фенол- и нафтолсульфокислоты, гидро-
хинон, d,/-тирозин, n-оксифенплглицин и п-оксибензойная кислота.
Последнее соединение дает вполне определенную желтую окраску
(которую следовало бы классифицировать как отрицательный ре-
зультат). В то же время с салициловой кислотой появляется фиоле-
товое окрашивание. Сложные эфиры о-оксибензойной кислоты
404
Глава 6
дают пурпурную окраску, а м-оксибензальдегид— фиолетово-пур-
пурную.
Интересно, что прекрасная пурпурная окраска появляется с
5,5-диметилцпклогександионом-1,3 (димедоном). Резорцин дает си-
нефиолетовую окраску. Следует отметить, что некоторые таутомер-
ные формы этих соединений по структуре аналогичны таутомерным
формам фенолов:
Салициловый альдегид образует сильно окрашенный комплекс
с хлоридом железа (III):
Следует четко отдавать себе отчет в том, что безошибочных
реактивов на ту или иную функциональную группу не бывает.
Поэтому во многих случаях оказывается необходимым использо-
вать второй или третий реактивы. Например, п-оксибензойная кис-
лота не дает положительной реакции с хлоридом железа(III) на
фенольную гидроксильную группу. Однако с бромной водой (опыт
30) легко образуется 2,4,6-трибромфенол, дающий с хлоридом же-
леза (III) голубое окрашивание.
Кроме того, щелочной раствор n-оксибензойной кислоты легко со-
четается с солями диазония, образуя оранжево-красный азокра-
ситель (опыт 19).
Определение функциональных групп и установление структуры
40В
В водных или в водно-спиртовых растворах некоторые енолы,
оксимы и гидроксамовые кислоты образуют с водным хлоридом же-
леза (III) комплексы, окрашенные в красный, коричневый или крас-
но-фиолетовый цвета. Однако при реакции с безводным хлоридом
железа(III) те же соединения образуют растворы желтого или свет-
лого рыжевато-коричневого цвета, совершенно не похожие на цве-
та, которые дают в этой реакции фенолы.
Поскольку ароматическое ядро фенола существенно более ре-
акционноспособно по отношению к электрофильному ароматиче-
скому замещению по сравнению с бензолом, то бромирование фе-
нолов следует проводить в мягких условиях (опыт 30).
Опыт 30. Бромная вода
+ ЗНВг
Готовят 1%-ные водные растворы фенола, анилина, салициловой кислоты
и n-ннтрофенола; к каждому раствору по каплям прибавляют бромную воду
до тех пор, пока не перестанет исчезать окраска брома.
Обсуждение. Было показано, что при действии бромной водой
на бензол и о-нитроанизол бромирование происходит по сложному
механизму *.
Меркаптаны под действием бромной воды легко превращаются
в дисульфиды:
2RSH + B1-2 —> RSSR + 2HBr2
Преимущество брома в воде над бромом в четыреххлористом
углероде заключается в том, что более полярный растворитель
значительно увеличивает скорость бромирования по ионному ме-
ханизму. Конечно, при использовании этого растворителя невоз-
можно наблюдать выделение бромистого водорода. При избытке
бромной воды трибромфенол дает желтое тетрабромпроизводное,
2,4,4,6-тетрабромциклогексадиенон, которое легко превращается
снова в трибромфенол при промывании 2%-ной иодистоводородной
кислотой.
* Ингольд К. Теоретические основы органической химии. Пер. с англ. — М.:
Мир, 1973, с. 288, 291.
406
Глава 6
Контрольные вопросы. 1. Почему трибромапилин нерастворим в разбавлен-
ной бромистоводородной кислоте? Может ли бромная вода обесцвечиваться из-
за присутствия неорганического соединения? Приведите примеры.
2. Гидролизуется ли бром в воде? Какое действие будет оказывать бромная
вода на растворимую в воде соль нерастворимой в воде кислоты?
Более детально реакция брома с органическими соединениями
обсуждена в опыте 14.
Рис. 6.49. Инфракрасный спектр о-крезола (без растворителя).
Валентные колебания О—Н: 1 — 3390 см-1 (2,95 мкм), v_ (связанные). Валентные колеба-
О—п
ння С—Н: 2— 3021 см-1 (3,31 мкм), v СН3—2924 см-1 (3,42 мкм), v СН •
асимм 3 енмм 3
Валентные колебания С-—С: 4—1587, 1493, 1460 см-1 (6,30, 6,70, 6,85 мкм), v^, Деформа-
ционные колебания О—Н: 5—около 1333 см-1 (~ 7,5 мкм), 6„ „ (внеплоскостные)
и—н
Валентные колебания С-О: 6—1235 см-1 (8,10 мкм), v^_q (арнл-кислород). Деформацион-
ные колебания =С-Н (плоскостные): 7—1205, 1170. 1105, 1042 см-1 (8,30, 8,55, 9,05, 9,60 мкм). Де-
формационные колебания —Н (внеплоскостные): 3 — 848 см-1 (11,80 мкм); 9 — 752 см~1 (13,3
мкм). Деформационные колебания С=гС (внеплоскостные); 70—714 см-1 (14,0 мкм). Калибровка:
все полосы поглощения спектра в области 3,00—4,00 мкм меньше табличных значений на
0,1 мкм. Все полосы поглощения спектра в области 6,00—16,00 мкм меньше табличных значе
пий на 0.05 мкм.
Поглощение фенолов в ПК-спектрах, как правило, очень сходно
с поглощением спиртов; и в том и в другом классе соединений в
молекуле имеется фрагмент С—О—Н. Наиболее важными обла-
стями поглощений являются следующие:
Колебания
Частота, см-1
Длина волны, мкм
Валентные колебания О—Н
Деформационные колебания О—П
Валентные колебания С—О
~ 3610 (с, ш) а
1410—1310 (ср, ш)
~ 1230 (с, ш)
~ 2,77
7,09-7,63
~ 8,13
с—сильная (интенсивность), ср —средняя (интенсивность), ш — широкий.
Определение функциональных групп и установление структуры
407
500
Рис. 6.50. Спектр протонного резонанса о-крезола (60 МГц, развертка 600 Гц, растворитель CDCh).
408
Глава 6
ИК-Спектр типичного фенола показан на рис. 6.49. Интерпре-
тация ПМР-спектров фенолов (например, рис. 6.50) включает рас-
смотрение положения гидроксильного протона. Кроме того, на сиг-
налы ароматических протонов влияет гидроксильный заместитель;
это явление было описано в разд. 6.9.
Особенно полезна при изучении фенолов УФ-спектрометрия
(гл. 8). При этом, в частности, используется сдвиг основного мак-
симума поглощения в более длинноволновую область (батохром
ный сдвиг) при превращении фенолов в соответствующие окси-
анионы, например:
Соединение \ibkc (емакс)
Ег-полоса В~полоса растворитель
ОН 6 210,5 (6200) 270(1450) Вода (pH 3)
О“ 6 235 (9400) 287 (2600) Вода (pH 11)
фенолят -анион
В масс-спектрах фенолов вследствие стабилизации молекуляр
ного иона ароматическим кольцом пик молекулярного иона обычно
является основным. Ниже приведены основные пики фрагментов
для типичных фенолов с указанием групп атомов, отщепляющихся
в процессе фрагментации:
Фрагмент Масса Фрагмент, образующийся из фенола
формула масса
м—со М-28 СМ т/е 66
М—СНО М - 29 см т/е 65
Большая часть фенолов не обнаруживает интенсивного пика
М—1; исключением является интенсивный М — Н-пик крезола,
возникающий за счет легкого расщепления бензильной связи
углерод — водород.
В присутствии щелочи фенолы легко реагируют с хлоруксус-
ной кислотой с образованием арилоксиуксусных кислот. Эти про-
изводные хорошо кристаллизуются из воды и очень полезны для
идентификации фенолов (методика 59):
ArONa + С1СН2СООН —> АгОСН2СООН + NaCl
Определение функциональных групп и установление структуры
409
Арилоксиуксусные кислоты можно сравнивать не только по тем-
пературам плавления, но также и по их эквивалентам нейтрали-
зации. Точное определение эквивалента нейтрализации может при-
вести к установлению суммарной массы заместителей в аромати-
ческом кольце.
Реакцию с бромом можно использовать не только как качест-
венную реакцию на фенолы (опыт 30), но и для получения произ-
водных фенолов (методика 60). Так как фенольное ароматическое
кольцо реакционноспособно по отношению к таким электрофиль-
ным реагентам, то каждый протон в орто- и пара-положениях мо-
жет замещаться бромом. Фактически бром во многих случаях за-
мещает не только протон, но и другие группы. Например, бром
реагирует и с фенолом, и с n-оксибензойной кислотой с образова-
нием 2,4,6-трибромфенола:
Фенолы, аналогично спиртам, образуют уретаны при обработке
их изоцианатами (методика 61). Среди изоцианатов чаще всего
используется для идентификации фенолов а-нафтилизоцианат (ме-
тодика 1а). Эта реакция катализируется прибавлением несколь-
ких капель пиридина.
CI0H7N=C=O 4-АгОН —> CI0H7NHCOOAr
Другим часто применяемым уретаном является дифенилуретан;
его получают из дифенилкарбамоилхлорида (C6H5)2NCOC1 сог-
ласно следующему уравнению (методика 16):
(C6H6)2NCOC1 + АгОН —> (C6H6)2NCOOAr + НС1
К другим легкодоступным производным фенолов относятся
нитропроизводные (методика 26а), пикраты (методики 22а и 226),
ацетаты (методики 5а и 56), 3,5-динитробензоаты (методика 2) и
эфиры сульфокислот.
Методика 59. Получение арилоксиуксусных кислот
33%-ный NaOH ClCHjCOOH
АгОН -------------> ArO*Na+ ----------> ArOCH2COOH
К смеси 1 г фенола с 5 мл 33%-иого раствора гидроксида натрия прибав-
ляют 1,5 г хлоруксусной кислоты. Смесь хорошо встряхивают и, если необхо-
димо, для растворения натриевой соли фенола прибавляют 1—5 мл воды. За-
тем пробирку со смесью помещают на 1 ч в стакан с кипищей водой. Раствор
410
Глава 6
охлаждают, разбавляют 10—15 мл воды, подкисляют разбавленной соляной кис
лотой по конго красному и экстрагируют 50 мл эфира. Эфирный раствор про-
мывают 10 мл холодной воды и затем встряхивают с 25 мл 5%-ного раствора
соды. Содовый раствор подкисляют разбавленной соляной кислотой, затем оса-
док арилоксиуксусной кислоты собирают на фильтре и перекристаллизовывают
из горячей воды.
Методика 60. Бромирование фенолов
Вг2/КВг
АгОН ---------> Аг'(Вг)хОН
Бромирующий раствор получают следующим образом: 15 г бромида калия
растворяют в 100 мл воды и к этому раствору приливают 10 г брома. Получен-
ный раствор медленно добавляют при встряхивании к раствору 1 г фенола в
воде, этаноле, ацетоне или дноксане. Бромирующий раствор прибавляют до ок-
рашивания смеси в желтый цвет. Затем приливают около 50 мл воды н смесь
энергично встряхивают для измельчения комков образовавшегося бромпроиз-
водного. Его отфильтровывают, промывают разбавленным раствором бисульфита
натрия и перекристаллизовывают из этанола или из смеси этанола с водой.
Методика 61. Получение фенилуретанов
О
c5h5n ||
АгОН + C6HSNCO ---------> ArOCNHC6H6
В сухую колбу емкостью 25 мл помещают смесь 0,5 г сухого фенола и
0,5 мл феннлизоцнаната и прибавляют 1 каплю сухого пиридина. Колбу неплотно
закрывают пробкой из марли и нагревают на паровой бане 15 мин. Если в те-
чение этого времени производное не выделяется, то кристаллизацию вызывают
охлаждением колбы и трением стеклянной палочкой о стенки колбы. После об-
разования кристаллов прибавляют 10 мл сухого этилацетата (или сухого бен-
зола *). Смесь нагревают на паровой бане и фильтруют через складчатый фильтр.
К фильтрату приливают гексан до помутнения раствора или до появления кри-
сталлов. Для завершения кристаллизации раствор оставляют на ночь, затем
продукт отфильтровывают, промывают гексаном, содержащим небольшое коли-
чество бензола, и сушат на воздухе.
Если в исходном образце или в применяемом реагенте присутствовала вода,
то продукт будет загрязнен сши-дифенилмочевиной (т. пл. 238°С). Очистку мож-
но осуществить, нагревая продукт с 10 мл четыреххлористого углерода и удаляя
нерастворимую днфеиилмочевину фильтрованием. Фенилуретан получают при
охлаждении фильтрата. В некоторых случаях, чтобы вызвать кристаллизацию,
фильтрат упаривают до объема 2—3 мл.
ПМР-Спектры фенолов можно упростить, используя лантано-
идные сдвигающие реагенты (гл. 8). Следует применять высоко-
фторированные сдвигающие реагенты, чтобы избежать нейтрализа-
ции лигандов сдвигающего реагента умеренно кислыми фенолами.
Бензол ядовит. См. примечание к методике 20.
Глава 7
Разделение органических веществ
7.1. ВВЕДЕНИЕ
Идентификация компонентов смесей включает два основных эта-
па: разделение смеси на отдельные составляющие ее компоненты
и идентификация каждого компонента с помощью методов и прие-
мов, описанных в гл. 6. Лишь в очень редких случаях удается иден-
тифицировать компоненты смеси без ее предварительного разде-
ления. Разделение соединений, входящих в состав смеси, должно
быть по возможности количественным с тем, чтобы можно было
получить определенное представление о процентном содержании
каждого компонента. Однако значительно важнее провести раз-
деление таким образом, чтобы выделить каждое соединение, вхо-
дящее в состав смеси, в возможно более чистом состоянии. Это су-
щественно облегчает дальнейшую идентификацию выделенных ве-
ществ.
Для разделения следует выбрать такой метод, который позво-
лит выделить каждое соединение именно в той форме, в которой
оно присутствует в первоначальной смеси. Производные первона-
чальных компонентов смеси оказываются существенно менее по-
лезными, за исключением тех случаев, когда они без труда могут
быть превращены вновь в исходные соединения. Такой критерий
оценки методов разделения необходим вследствие того, что иден-
тификация соединений базируется в основном на совпадении фи-
зических констант исходных веществ и их производных с литера-
турными данными.
Сведения о происхождении исследуемой смеси во многих слу-
чаях помогают установить, к какой группе веществ принадлежит
данная смесь. Это позволяет выбрать общий принцип разделения.
В последние годы научное направление, связанное с разделе-
нием смесей для целей анализа, интенсивно развивается и широко
используется химиками-органиками. Поэтому для того, чтобы пра-
вильно выбрать метод разделения, наиболее соответствующий ха-
рактеру исследуемой смеси, необходимо иметь представление о
принципах ряда основных и наиболее доступных методов разде-
ления смесей, известных в настоящее время.
Существует несколько проблем, связанных с разделением сме-
сей и часто встречающихся в практике химика-органика. Один из
довольно обычных случаев такого рода следующий: смесь состоит
из нескольких относительно чистых компонентов, каждый из
412
Глава 7
которых присутствует в достаточно большом количестве *. Другая
проблема разделения связана с отделением одного компонента от
большого количества непрореагировавшего исходного вещества
или от нежелательных побочных продуктов. Часто побочные про-
дукты представляют собой не поддающиеся обработке смолы или
полимерные материалы. Для того чтобы выбрать наиболее эффек-
тивный подход к проблеме разделения данной конкретной смеси,
целесообразно отнести каждую вновь возникающую задачу раз-
деления к одной из двух вышеупомянутых категорий.
Прежде чем выбрать приемлемый способ разделения смеси,
следует провести описанные ниже предварительные испытания.
При выполнении этих испытаний необходимо учитывать следую-
щие факторы:
I. Выдержит ли данная смесь предполагаемый метод разделе-
ния, т. е. достаточно ли устойчивы компоненты этой смеси в усло-
виях, необходимых для ее разделения?
2. Является ли выбранный способ разделения наиболее прос-
тым и эффективным для данной смеси?
Устойчивость данной пробы в условиях разделения может ока-
заться неизвестной до первых попыток разделить ее на состав-
ляющие компоненты. При этом всегда следует обращать внимание
на термическую устойчивость веществ. Пробы, которые неустой-
чивы при температурах, требующихся для перегонки при атмос-
ферном давлении, можно перегонять при пониженном давлении.
Хроматографические методы разделения (кроме газовой хрома-
тографии) обычно не требуют нагревания и поэтому их можно
применять для разделения веществ, разлагающихся при перегонке.
Однако вполне возможно, что проба, подвергнутая хроматогра-
фическому разделению, полностью или частично разложится вслед-
ствие реакции с хроматографическим наполнителем или твердым
носителем. Хорошим способом проверки устойчивости пробы в ус-
ловиях хроматографии является быстрый тест с помощью тонко-
слойной хроматографии, описанный в разд. 3.2.
7.1.1. Предварительное исследование смеси
1. Отмечают физическое состояние смеси. Если она представляет
собой суспензию твердого вещества в жидкости, то твердый оса-
док отделяют и исследуют отдельно. Если смесь содержит две не
смешивающиеся друг с другом жидкости, то их также разделяют
и исследуют отдельно.
2. Проверяют, растворима ли исследуемая смесь в воде.
3. При изучении жидких смесей 2 мл жидкости выпаривают
досуха на крышке фарфорового тигля или на часовом стекле. От
* В смесях, предназначенных дли выполнения студенческих лабораторных
работ, обычно присутствует от двух до пяти компонентов.
Разделение органических веществ
413
мечают наличие или отсутствие остатка. При наличии остатка про-
водят пробу на горючесть. Для твердых смесей пробу на горю-
честь проводят прямым сожжением.
4. Для жидких смесей устанавливают наличие в их составе
воды следующими способами: а) проверяют, смешивается ли дан-
ная смесь с эфиром; б) добавляют безводный сульфат меди;
в) проводят определение воды отгонкой *. Определение присутствия
воды последним способом наиболее надежно и выполняется сле-
дующим образом. В небольшую перегонную колбу помещают 5 мл
жидкой смеси и 5 мл безводного толуола. Смесь осторожно нагре-
вают до начала перегонки и собирают 2 мл дистиллата. Наличие в
приемнике двух слоев жидкости или явно выраженных капель,
взвешенных в толуоле, указывает на присутствие воды. Если ди-
стиллат мутный, то это означает, что в смеси имеются лишь сле-
ды воды.
5. Если установлено, что вода в исследуемой смеси отсутст-
вует, то проверяют наличие в смеси легколетучих растворителей.
Для этого 10 мл смеси помещают в перегонную колбу емкостью
25 мл. Колбу погружают в стакан с холодной водой, которую по-
степенно нагревают до кипения. Любая жидкость, отгоняющаяся
при этих условиях, считается легколетучим растворителем. Дис-
тиллат, который может представлять собой смесь легколетучих
веществ, и остаток в перегонной колбе исследуют отдельно.
Часто растворимая в воде смесь при перегонке дает легкоки-
пящпй растворитель и остаток, не растворяющийся в воде. При
разделении таких смесей вначале полностью удаляют летучий рас-
творитель, а остаток далее рассматривают как смесь, нераство-
римую в воде. Если остаток после перегонки представляет собой
растворимую в воде жидкость, то лучше не стараться на этой
стадии удалять растворитель, так как разделение редко бывает
количественным. Если, однако, остаток после перегонки представ-
ляет собой растворимое в воде твердое вещество и растворитель,
по-видимому, отделяется полностью, в этом случае желательно
полностью отделить растворитель и далее исследовать отдельно
дистиллат и остаток в перегонной колбе.
Следует заметить, что не нужно пытаться проводить такие раз-
деления, если в исследуемой смеси присутствует вода.
6. Необходимо установить реакцию водного раствора или сус-
пензии смеси на лакмус или фенолфталеин. Если смесь имеет яв-
но кислую реакцию, то 5 мл смеси титруют 0,1 н. раствором гид-
роксида натрия. Это дает возможность установить, является ли
кислотность смеси результатом наличия значительного количества
свободной кислоты или следствием появления следов кислоты при
гидролизе сложных эфиров. Титрование следует проводить при
* Для количественного определения воды, присутствующей в исследуемой
смеси, можно применить реакцию Карла Фишера, описанную в приложении I,
414
Глава 7
охлаждении раствора льдом. За конечную точку титрования при-
нимают момент появления розового окрашивания фенолфталеина.
7. Подкисляют соляной кислотой 2 мл смеси и раствор охлаж-
дают. Отмечают выделение газа или выпадение осадка. Затем до-
бавляют разбавленный раствор гидроксида натрия и отмечают ре-
зультат.
8. Пробу разделяемой смеси объемом 2 мл подщелачивают ра-
створом гидроксида натрия. Отмечают выделение аммиака, мас-
лянистого или твердого осадка либо изменение цвета раствора.
Смесь нагревают до начала кипения и вновь охлаждают. Сравни-
вают запах охлажденного раствора с запахом первоначальной
смеси. Изменение запаха часто указывает на присутствие в ис-
ходной смеси сложных эфиров. Далее добавляют разбавленную
соляную кислоту и отмечают результат.
9. При наличии смесей, нерастворимых в воде, следует сде-
лать элементный анализ. Когда установлено, что смесь содержит
воду или большое количество летучего растворителя, элементный
анализ излишен. Если растворимая в воде смесь представляет
собой твердое вещество, то провести элементный анализ целесо-
образно.
10. Если изучаемая смесь не содержит воды, то осторожно
проводят две классификационные реакции: а) с металлическим
натрием и б) с ацетилхлоридом.
11. Исследуемую смесь следует подвергнуть действию следую-
щих классификационных реагентов: а) бромной воды, б) раствора
перманганата калня, в) раствора хлорида железа(III), г) спир-
тового раствора нитрата серебра, д) фуксинсернистой кислоты
(реактив на альдегиды) и е) фенилгидразина.
На этой стадии исследования суммируют результаты перечис-
ленных выше испытаний и по данным о поведении исследуемой
смеси в этих условиях пытаются сделать заключение о ее возмож-
ном составе. Описанные выше предварительные испытания пока-
зывают, к какой группе веществ следует отнести данную смесь и,
следовательно, какой из описанных ниже методов целесообразно
использовать для ее разделения.
ФОРМА ОТЧЕТА
Предварительное исследование смеси
Фамилия, и., о. студента.
Дата
Смесь №
Компоненты
1. а) Физическое состояние.
б) Способ механического разделения.
2. Растворимость в воде.
3. а) Проба на наличие сухого остатка.
‘ б) Проба на горючесть.
Разделение органических веществ
415
4. Проба на присутствие воды,
5. Проба на наличие летучего растворителя.
6. Пробы на кислотность (основность).
7. Проба на газообразные компоненты, выделяемые действием кислот или ще-
лочей.
8. Проба на нелетучие компоненты, выделяемые действием кислот или щелочей.
9. Элементный анализ смеси: F, Cl, Вг, I, N, S, металлы. Растворимость в воде.
10. Классификационные реакции.
Результат пробы 4 на присутствие воды.
а) Металлический натрий.
б) Ацетилхлорид.
И. Классификационные реагенты.
а) Бромная вода.
б) Перманганат калия.
в) Хлорид железа (III).
г) Спиртовый раствор нитрата серебра.
д) Фуксинсернистая Кислота (реактив на альдегиды).
е) Фенилгидразнн.
12. Рекомендации по выбору метода разделения.
Перегонка.
Газовая хроматография.
Кристаллизация.
Колоночная хроматография.
Тонкослойная хроматография.
Жидкостная хроматография высокого давления.
Замечания и литературные источники.
7.2. ПЕРЕГОНКА И СУБЛИМАЦИЯ
7.2.1. Перегонка
Предварительные указания по проведению простой перегонки
были даны в разд. 3.3.2 (в связи с определением температуры ки-
пения). В этом разделе были рассмотрены простейшие случаи
применения перегонки для очистки пробы и определения ее тем-
пературы кипения. Ввиду того что перегонка — очень старинный
метод, к настоящему времени разработано большое число раз-
личных вариантов этого процесса. Ниже рассмотрены некоторые
более сложные способы проведения перегонки.
На рис. 7.1 изображен прибор с укороченным путем отгоняе-
мого пара, полезный для перегонки малых количеств веществ.
Хотя простой процесс испарения и конденсации является недо-
статочно эффективным, изображенный прибор позволяет провести
перегонку таких продуктов (например, низкоплавких твердых ве-
ществ), для которых длительное пребывание при повышенной тем-
пературе может привести к разложению. Для того чтобы повысить
эффективность процесса перегонки, между нагреваемым сосудом
с пробой и входной трубкой холодильника можно установить ко-
лонку. На рис. 7.2 показаны колонки с увеличенной внутренней
поверхностью (например, колонки Вигре) и колонки, представ-
ляющие собой модифицированные холодильники, — конденсаторы
с увеличенной внутренней поверхностью и (или) с повышенной
416
Глава 7
Рис. 7.1. Прибор для перегонки с укоро-
ченным путем отгоняемого пара (вос-
произведено с разрешения Асе Glass,
Inc., Vineland, N. J.).
Рис. 7.2. Дефлегматоры и холодильники (воспроизведено с разрешения Асе
Glass, Inc., Vineland, N. J.).
a — колонка Внгре; б—колонка Внгре с насадкой; в —холодильник Аллена; г—спиральный
холодильник; б—холодильник Фридерикса.
Разделение органических веществ
417
площадью охлаждения. Полностью прибор с промежуточной ко-
лонкой изображен на рис. 7.6. Очень эффективное разделение при
перегонке может быть достигнуто с по-
мощью прибора с вращающейся лентой
(рис. 7.3), обеспечивающего получение
достаточно большого числа теоретиче-
ких тарелок *.
Применение более эффективных при-
боров для перегонки часто связано с уве-
личением времени, в течение которого
пары перегоняемой жидкости достигают
верха используемой колонки. Для того
чтобы снизить потери тепла, колон-
ку снаружи покрывают слоем теплоизо-
ляционного материала, например асбе-
ста ** или стеклянной ваты. Кроме того,
экспериментатор должен иметь воз-
можность изменять количество тепла,
подводимого к разным обогреваемым
зонам прибора для перегонки. Два
устройства, обычно применяемые для
этой цели, показаны на рис. 7.4. Нагре-
вательная лента (рис. 7.4, А и Б) позво-
ляет получить очень равномерный нагрев.
Силу электрического тока, подводи-
мого к нагревательной ленте, можно ре-
гулировать с помощью лабораторного
автотрансформатора (рис. 7.5). Для того
чтобы облегчить перегонку особенно
труднолетучих веществ, можно приме-
нять фен с электрообогревом («тепловой
пистолет»), показанный на рис. 7.4,В.
Рис. 7.3. Ректификационная колонка с вращаю-
щейся лентой (Bates R. В., Schaefer J. Р„ Re-
search Techniques in Organic Chemistry, Copyrigth
© 1971, p. 50; перепечатано с разрешения Pren-
tice-Hall, Inc., Englewood Cliffs, N. J.).
/ — электродвигатель; 2—тефлон; 3—уплотнение и под-
шипник; 4—верхний подшипник; 5—термометр; 6—кран
отбора дистиллата; 7—клапан для установки флегмо-
вого числа; 8—вращающаяся лента; 9 — ннжннй подшип-
ник; Ю—тефлоновая мешалка.
Такой тепловой пистолет по виду и по способу применения похож
на ручной фен для сушки волос. С его помощью поток тепла
* Теория процесса перегонки изложена в книгах, перечисленных в гл. 10.
** Асбест обладает токсичными свойствами, поэтому следует избегать его
применения.
14 Зак. 335
Рис. 7.4. Нагревательная лента для колонки (Л), колонка, обмотанная лентой
(Б) (воспроизведено с разрешения Glass-Col Apparatus Со., Terre Haute, Ind.),
и фен «тепловой пистолет» (В) (воспроизведено с разрешения Masters Appliance
Corp., Racine, Wise.).
Разделение органических веществ
419
направляют на те места прибора для перегонки, где необходимо
небольшое повышение температуры для облегчения прохода ди-
стиллата.
Для перегонки жидких и твердых малолетучих веществ, кото-
рые чувствительны к перегреву, обычно применяют приборы для
Рис. 7.5. Лабораторный автотрансформатор (Superior Electric Со., Bristol, Conn.)'.
перегонки в вакууме (рис. 7.6). С помощью лабораторного ваку-
ум-насоса (не изображенного на рис. 7.6) можно создать рабо-
чее давление около 2 кПа (15 мм рт. ст.). Хороший вакуум-насос,
снабженный устройством для натекания (регулятором подачи
газа для бунзеновской горелки), может обеспечить вакуум от 1,33
до 2000 Па (0,01 —15 мм рт. ст.). На рис. 7.6 показано полезное
устройство для подачи газа. На входную трубку 2 одевают рези-
новую трубку, сжатую зажимом *. Изменяя степень сжатия, мож-
но регулировать количество подаваемого в прибор воздуха **. Этот
воздух, поднимаясь пузырьками через слой перегоняемой жидко-
сти, способствует равномерному кипению без толчков и перебросов.
В подписи к рис. 7.6 отмечены и другие полезные особенности при-
боров для перегонки в вакууме.
* В трубку необходимо вставить медную проволоку, которая препятствует
полному закрытию трубки — Прим, персе
** Или какого-либо инертного газа (азота, аргона, гелия и др.).—Прим,
черев.
14*
420
Глава 7
Рис, 7.6, Прибор для перегонки в вакууме (воспроизведено с разрешения Асе
Glass, 1нс., Vineland, N. J.).
В тексте при обсуждении рис. 7.1 см. описание модификации перегонного куба дли уста-
новки магнитной мешалки, что устраняет необходимость подачи воздуха или инертного
газа. 1— перегонный куб (здесь необходимо измерять и регулировать температуру); 2— си-
стема подачи воздуха; 3—колонка Вигре (дефлегматор); 4 — водяной холодильник; 5—си-
стема для удаления дистиллата и откачки приемника; 6 — приемник.
Рис. 7.7. Электрическая нагревательная рубашка (воспроизведено с разрешения
Асе Glass, Inc., Vineland, N. J.j.
Рис. 7.8. Роторный испаритель (Landgrebe J., Theory and Practice in Organic
Laboratory, 2nd ed., D. C. Health, Lexington, Mass., © 1977; перепечатано c
разрешения).
А—общий вид; Б—отдельные составные части. 1 — к вакуум-насосу; 2—приемник; 3—ледя-
ная баня; 4—реакционная смесь.
422
Глава 7
Для успеха процесса перегонки весьма важным является спо-
соб обогрева перегонного куба (например, путем подвода тепла
к одной точке, как на рис. 7.6). Умеренный обогрев удобно осу-
ществлять с помощью паровой бани (до температур, близких к
температуре кипения воды), однако такой источник тепла не
всегда обеспечивает равномерное нагревание. Можно применять
нагревательные бани, содержащие различные масла или иные
инертные и нелетучие вещества. Подвод тепла с помощью таких
нагретых жидкостей является очень равномерным. Этот способ
обогрева можно применять до весьма высоких температур (при-
Рис. 7.9. Прибор для перегонки «Кюгельрор».
1— стеклянная перегонная колба и приемники; 2— инфракрасный нагреватель мощностью
500 Вт; 3—возвратно-поступательный двигатель с углом поворота 300 °; 4 — полая ocS для
присоединения И вакуум-насосу. Штативы, зажимы, кольца и автотрансформаторы на рисунке
не показаны.
мерно до 250—300°С). Весьма удобны для нагревания электриче-
ские нагревательные рубашки (рис. 7.7), использование которых
позволяет исключить применение масел, пачкающих одежду и
оборудование. Силу тока, подводимого к нагревательной рубаш-
ке, можно регулировать с помощью лабораторного автотрансфор-
матора (рис. 7.5).
Следует отметить, что химик-органик часто применяет такой
вариант перегонки, который позволяет просто удалить летучий
растворитель из раствора, полученного, например, при обработке
какой-либо реакционной смеси. В этих случаях оказываются по-
лезными роторные испарители (рис. 7.8,Л). Для регулярной и без-
отказной работы такого оборудования необходимо периодически
проверять уплотнительные кольца (деталь PF-1049 на рис. 7.8, Б)
и заменять их в случае износа или повреждения.
Один из весьма популярных приборов такого типа изображен
на рис. 7.9. В продаже имеются и более простые варианты по-
добных приборов, например не имеющие вращающего привода.
Большим преимуществом приборов такого типа является их спо-
собность удерживать высокий вакуум (менее 13 Па, т. е.
0,1 мм рт. ст.). Особенно хорошо работают в вакууме приборы,
у которых вся стеклянная часть представляет собой единое целое,
а не составлена из неск&льких сбсудов, соединенных с помощью
шлифов.
Разделение органических веществ
423
7.2.2. Сублимация
Для очистки различных веществ с помощью сублимации ис-
пользуют приборы такого типа, как изображенный на рис. 7.10.
Внутренняя камера представляет собой холодильник пальцевого
типа, который можно извлечь вместе с
верхней частью прибора. Внутрь этого
холодильника загружают подходящий
охлаждающий агент (лед, сухой лед или
смесь сухого льда с ацетоном). Подле-
жащий очистке материал помещают на
дно прибора для сублимации и нагре-
вают, обычно с помощью масляной бани.
Постепенная возгонка компонентов раз-
деляемой смеси приводит к осажде-
нию очищенных твердых продуктов на
охлаждаемой поверхности пальцеобраз-
ного холодильника. В ряде случаев при-
ходится периодически прерывать возгон-
ку и очищать холодильник от оседаю-
щего на нем возогнанного продукта.
В некоторых случаях возможно уско-
рить процесс сублимации, создавая в
приборе пониженное давление путем от-
качки через боковой отвод, показанный
на рис. 7.10.
7.2.3. Перегонка с паром
Перегонка с паром представляет со-
бой метод, в котором соединение с от-
носительно низкой летучестью может
быть подвергнуто очистке путем совме-
стной перегонки с водой. Этот процесс
возможен благодаря тому, что оба жид-
ких компонента перегоняемой смеси
вносят свой вклад в общее давление
Рис. 7.10. Аппарат для суб-
лимации (воспроизведено с
разрешения Асе Glass, Inc.,
Vineland, N. J.).
паров и таким образом
процесс перегонки может протекать при температуре несколько
ниже 100°С (при давлении 0,1 МПа, или 1 атм). Перегонку с
паром обычно осуществляют, направляя струю водяного пара
в сосуд, содержащий разделяемую смесь. Отгоняемый дистил-
лат конденсируют в охлаждаемом водой холодильнике и соби-
рают в приемнике. Методика проведения этого процесса и схе-
мы аппаратуры приводятся в начальных руководствах по лабора-
торной технике в органической химии *.
* См., например, Landgrebe J., Theory and Practice in the Organic Labora-
tory, 2nd ed., D. C. Heath, Lexington, Mass., 1977, pp. 85—87. Пар можно гене-
рировать и без отдельного сосуда.
424
Глава 7
Таблица 7.1
Растворимость органических веществ и возможность их перегонки с паром
Растворимость Тип соединения Летучесть Летучесть с водяным паром
Растворимы Низкомолекулярные Легко перегони- Перегоняются
в воде и в эфире спирты, альдегиды, кетоны, карбоновые кислоты, сложные эфиры, амины, нит- рилы и хлорангид- риды кислот ются. Многие соединения этой группы кипят ниже 100 °C с паром
Растворимы Многоатомные спирты, Трудиолетучи. За некоторыми исключениями, не могут пере- гоняться при атмосферном давлении Не перегони-
в воде, ио не- растворимы в эфире диамины, углеводы, соли аминов, соли металлов, миогоос- новные кислоты, ок- сиальдегиды, оксике- тоны, оксикислоты и аминокислоты ются с паром
Нерастворимы в воде, ио раст- воряются в ра- створах NaOH и NaHCO3 Высокомолекулярные карбоновые кислоты и фенолы с отрица- тельно заряженными заместителями Труднолетучи Обычно пере- гоняются с паром. Имеются некоторые исключения
Нерастворимы Фенолы, сульфонами- Соединения с вы- Обычно
в воде и раст- ды первичных ами- сокими темпе- не псрегоия-
воре NaHCO3, но растворимы в растворе NaOH иов, первичные и вторичные нитросо- единения, имиды и тиофенолы ратурами кипе- ния. Многие из них не спо- собны перего- няться ются с паром
Нерастворимы Амины, содержащие ие Высококипящие Многие веще-
в воде, но раст- воримы в раз- бавленной НС1 более одной арома- тической группы у атома азота, органи- ческие гидразины соединения ства этой группы пере- гоняются с водяным паром Некоторые
Нерастворимы Третичные нитросоеди- Высококипящие
в воде и раз- нения, амиды, амины соединения. вещества
бавленныхраст- с отрицательно за- Многие веще- этой группы
ворах NaOH ряженными замести- ства этой перегоняются
и НС1. Содер- телями, сульфонами- группы не спо- с водяным
жат другие элементы, кроме С, Н, О и гало- генов ды вторичных ами- нов, азо- и азоокси- соединения, алифати- ческие и ароматиче- ские нитрилы, нитра- ты, нитриты, сульфа- ты, фосфаты собиы перего- няться паром
Разделение органических веществ
425
П родолжение табл. 7.1
Растворимость Тип соединения Летучесть Летучесть с водяным паром
Спирты, альдегиды, ке-
тоны, сложные эфи-
ры, ненасыщенные
соединения
Высококипящие
соединения
Обычно мо-
гут перего-
няться
с водяным
паром
Перегоняются
с паром
Нерастворимы
в воде и раз-
бавленных раст-
ворах NaOH
и НС1, но раст-
воримы в сер-
ной кислоте
Нерастворимы
в воде, разбав-
ленных раство-
рах NaOH
и НС1 и серной
кислоте
Алифатические и аро-
матические углево-
дороды и их гало-
генпроизводные
Летучи
Различия в полярности веществ, достаточные для того, чтобы
их можно было разделить методом перегонки с паром, обычно
обусловлены наличием в молекуле дополнительных функциональ-
ных групп. Например, таким способом одноатомные спирты могут
быть отделены от двухатомных и многоатомных спиртов. Сход-
ным образом монофункциональные органические кислоты, амины
и многие другие летучие соединения могут быть отделены от со-
ответствующих ди- и полифункциональных производных. Более
того, дополнительная функциональная группа не обязательно дол-
жна иметь тот же характер, что и присутствовавшая первона-
чально. Поэтому аминокислоты, оксикислоты, нитрокислоты, ке-
токислоты, кетоспирты и цианкетоны лишь в редких случаях
удается перегнать с паром. Фактически общим является правило,
согласно которому наличие в молекуле двух или более полярных
функциональных групп делает соединение нелетучим. В табл. 7.1
приведены данные, какие группы соединений перегоняются, а ка-
кие— не перегоняются с водяным паром. Это правило хорошо
иллюстрирует примеры следующих смесей: уксусная и щавелевая
кислоты, этанол и этиленгликоль, бензойная и фталевая кислоты.
В каждой из этих пар первое вещество может быть удалено пе-
регонкой с паром, а второе остается в кубе.
Интересная группа исключений из правила нескольких функ-
ций встречается в ряду ароматических соединений. Веществами,
летучими с паром, являются о-нитрофенол, салициловый альдегид
и многие другие орто-дизамещенные производные бензола. Эта
кажущаяся аномальная потеря полярных свойств объясняется тем,
что все соединения, являющиеся такими исключениями, способны
к образованию внутримолекулярных водородных связей. У таки>{
426
Глава 7
веществ существенно понижена способность к образованию ас-
социатов с водой, и поэтому они относительно легколетучи. Ниже
показаны структуры салицилового альдегида и о-нитрофенола с
внутримолекулярными водородными связями.
08-
Другим важным примером применения перегонки с паром яв-
ляется отделение продуктов реакции от таких растворителей, как
И,М-диметилформамид и диметилсульфоксид. Эти растворители
используются для проведения ряда химических реакций. Однако
очень высокие температуры кипения этих веществ делают крайне
затруднительными их удаление из реакционных смесей. Поскольку
ни диметилформамид, ни диметилсульфоксид не способны пере-
гоняться с паром, во многих случаях оказывается возможным про-
сто разбавить водой реакционную смесь и выделить из нее про-
дукты реакции и непрореагировавшие исходные продукты с по-
мощью перегонки с паром.
7.3. ПЕРЕКРИСТАЛЛИЗАЦИЯ
Метод кристаллизации основан на том, что при понижении
температуры растворимость твердого вещества в каком-либо рас-
творителе или смеси растворителей уменьшается. Таким обра-
зом, для того, чтобы лучше понять теорию метода кристаллиза-
ции, необходимо ознакомиться с теорией растворимости
(разд. 5.1).
В простейшем случае перекристаллизацию проводят, раство-
ряя твердое маслообразное или пастообразное вещество в под-
ходящем растворителе. Обычно перекристаллизация наиболее
эффективна в том случае, когда для полного растворения веще-
ства приходится подогревать растворитель. Затем нагретый рас-
твор охлаждают до комнатной или более низкой температуры.
В «идеальном» случае при этом медленно выпадают однородные
кристаллы. Часто для полного завершения перекристаллизации
приходиться оставлять раствор на ночь или даже на более дли-
тельное время. После отделения кристаллов, например, фильтро-
ванием на воронке Бюхнера и высушивания на воздухе прове-
ряют чистоту очищенного вещества (определяют температуру пла-
вления, удельную массу, исследуют методом газовой или тонко-
слойной хроматографии, снимают спектр ЯМР и т. п.).
В действительности процедура очистки вещества методом пе-
рекристаллизации может быть гораздо более сложной. Обычно в
Разделение органических веществ
427
Таблица 7.2
Растворители, применяемые для перекристаллизации основных классов
органических соединений (приведены в алфавитном порядке)
Кристаллизуемое вещество Растворитель а Т. кип. раствори- теля. °C Возможные компоненты смешанных растворителей
Амиды Ангидриды кис- лот Ароматические соединения Бромпроизводные Карбоновые кис- лоты Комплексы Неполярные сое- динения Низкоплавкие соединения Нитросоединеиия Озазоиы Полярные соеди- нения Сахара Сложные эфиры Соли Углеводороды Уксусная кислота Диоксан Вода Четыреххлористый лерод Бензол Ацетон Этанол Уксусная кислота Вода Бензол Четыреххлористый лерод Диэтиловый эфир Метил енхлорид Ацетон Этанол Ацетон Ацетонитрил Метилцеллозольв Этилацетат Этанол Уксусная кислота Вода Бензол н-Гексан уг- уг- 118 102 100 76,5 80 56 78 118 100 80 76,5 34,5 40 56 78 56 81,6 77 78 118 100 80 69 Вода Вода, бензол, углеводо- роды Ацетон, спирты, диок- сан, ацетонитрил Диэтиловый эфир, бен- зол, углеводороды Диэтиловый эфир, этил- ацетат, углеводороды Вода, диэтиловый эфир, углеводороды Вода, углеводороды, этилацетат Вода Ацетон, спирты, диок- сан, ацетонитрил Диэтиловый эфир, этил- ацетат, углеводороды Диэтиловый эфир, бен- зол, углеводороды Ацетон, углеводороды, этилацетат, бензол, четыреххлористый уг- лерод Этанол, углеводороды Вода, диэтиловый эфир, углеводороды Вода, углеводороды, этилацетат Вода, диэтиловый эфир, углеводороды Вода, диэтиловый эфир, бензол Вода, бензол, диэтило- вый эфир Диэтиловый эфир, угле- водороды, бензол Вода, углеводороды, этилацетат Вода Ацетон, спирты, диок- сан, ацетонитрил Диэтиловый эфир, этил- ацетат, углеводороды Любые растворители, кроме воды, уксусной кислоты и ацетонит- рила
428
Глава 7
Продолжение табл. 7.2
К р иста ллизу емое вещество Растворитель а Т. кип. раствори- теля, °C Возможные компоненты смешанных растворителей
Хлорангидриды кислот Прочие соедине- ния Четыреххлористый ле род Хлороформ Ацетон Хлороформ Этилацетат Диэтиловый эфир Метиленхлорид Этанол уг- 76,5 61,7 56 61,7 77 34,5 40 78 Диэтиловый эфир, бен- зол, углеводороды Углеводороды Вода, диэтиловый эфир, углеводороды Углеводороды, этанол Диэтиловый эфир, угле- водороды, бензол Ацетон, углеводороды, этилацетат, бензол, четыреххлористый уг- лерод Этанол, углеводороды Вода, углеводороды, этилацетат, бромпро- изводные
а Более полные сведения о приведенных в таблице и других растворителях, в том числе
данные об их токсичности и способности к воспламенению, а также рекомендации практиче-
ского характера, приведены в книге: Гордон А., Форд Р. Спутник химика. Физико-химические
свойства, методики, библиография. Пер. с аигл — М.: Мир, 1976.
Предупреждение. Следует помнить, что многие из перечисленных в таблице растворите-
лей, особенно бензол, обладают весьма значительной токсичностью. По этому вопросу см.
примечание к методике 20 и приложение IV.
Таблица 7.3
Некоторые растворители и бинарные Смеси для перекристаллизации
обычно встречающихся производных
Производные Растворители или их смеси а
Амиды Ацетаты Бензиловые эфиры Бензоаты Бромсодержащие соедине- ния Г ндразоны 3,5-Дииитробензоаты 3,5-Дииитрофеиилуретаны Ксаитиламиды а-Нафтилуретаиы л-Ннтробеизиловые эфиры Нитросоединения и-Нитрофенилуретаны Озазоны Пикраты Метанол, этанол То же Метанол — вода, этанол Метанол, этанол Ацетон — этанол, метанол, этанол Метанол — вода, этанол Метаиол, этанол Петролейиый эфир — бензол Диоксан — вода Петролейиый эфир Метанол — вода, этанол Метанол, этанол, ацетон — этанол Петролейиый эфир — бензол Ацетон — этанол Бензол, этанол, метаиол — вода
Разделение органических веществ
429
Продолжение табл. 7.3
Производные Растворители или их смеси а
Семнкарбазоны Сложные эфиры Этанол, метанол — вода Этнлацетат. метанол, этанол
Соли четвертичных аммо- ниевых оснований Этиланетат, диизопропиловый эфир
Сульфонамиды Сульфонилхлориды п-Толуидиды Фенилуретаны Метанол — вода, этанол Хлороформ, четыреххлористый углерод Метанол, этанол Петролейиый эфир
а Более полную информацию о приведенных в табл. 7.3 растворителях можно найти
в табл. 7.1 и 7.2, а также в справочнике, указанном в примечании к табл. 7.2. Следует обра-
тить внимание на предупреждение о токсичности растворителей в примечании к табл. 7.2.
особенно о токсичности бензола (см. приложение IV).
руководствах по лабораторной технике органической химии при-
водятся необходимые примеры упражнений для освоения этого
метода. В качестве одного из таких стандартных лабораторных
упражнений обычно проводят перекристаллизацию ацетанилида,
содержащего примеси. В этом разделе будут изложены некото-
рые принципы и приемы, которые могут оказаться полезными
при решении задач, связанных с проведением перекристаллиза-
ции веществ в ряде непредсказуемых ситуаций, встречающихся в
качественном органическом анализе.
Первое, что следует принять во внимание при проведении
перекристаллизации,— это правильный выбор растворителя. Со-
гласно «ориентировочному правилу», подлежащее очистке веще-
ство должно растворяться в горячем растворителе в 5 раз лучше,
чем в холодном. Иногда очень удобно проверять растворимость
веществ с помощью метода газовой хроматографии, описанного
в разд. 7.4.5. Некоторые растворители, применяемые для пере-
кристаллизации различных веществ, приведены в табл. 7.2, а ча-
сто употребляемые для этой цели смеси растворителей перечис-
лены в табл. 7.3. Даже в том случае, если в табл. 7.2 или 7.3 не
окажется именно той пары растворитель — кристаллизуемое ве-
щество, которая интересует исследователя, все-таки эти таблицы
позволяют получить представление о том, какие общие группы
и комбинации растворителей применимы при проведении кри-
сталлизации различных соединений. Например, если фенилурета-
новое производное не удается перекристаллизовать из петролей-
ного эфира, то может оказаться более подходящей смесь петро-
лейного эфира и бензола. Или если необходимо перекристалли-
зовать еще не описанный в литературе эфир .м-нитробензойной
кислоты, весьма вероятно, что он будет хорошо кристаллизовать-
ся из смеси метанола и воды (почему?).
430
Глава 7
Обычно при проведении кристаллизации из смеси двух раство-
рителей очищаемое вещество растворяют при нагревании в том
растворителе, в котором его растворимость выше. Далее медлен-
но добавляют второй растворитель до тех пор, пока будет можно
ожидать, что при охлаждении раствора начнется кристаллизация
пробы. По достижении нужного соотношения растворителей даль-
нейшее прибавление второго растворителя вызывает помутнение
раствора, которое очень медленно исчезает при его перемешива-
нии. Обычно оптимальные условия приходится подбирать методом
проб и ошибок.
Довольно часто при попытке кристаллизации какого-либо ве-
щества оно выпадает из раствора не в виде желаемых твердых
кристаллов, а в форме масла. Наиболее вероятная причина обра-
зования масел — недостаточная чистота проб. Если имеются осно-
вания предполагать именно эту причину, то вещество растворяют
в растворителе, в котором оно хорошо растворимо, например в
эфире, обрабатывают полученный раствор обесцвечивающими
агентами (например, активированным углем) и высушивают (на-
пример, сульфатом магния). Затем удаляют растворитель на ро-
торном испарителе (рис. 7.8, Л) и, если необходимо, повторяют
перекристаллизацию вещества.
Выпадение масла может повторяться даже при повторных очи-
стках. Это может быть связано с тем, что данная проба особен-
но трудно кристаллизуется, или с тем, что необходимо путем кри-
сталлизации удалить последние следы загрязнений. В таких слу-
чаях рекомендуется применить следующие приемы:
1. В процессе кристаллизации в раствор очищаемой пробы до-
бавляют маленький кристаллик чистого вещества—затравку.
Этот кристаллик вносят после того, как раствор стал пересыщен-
ным. Например, такую затравку добавляют к раствору, охлаж-
денному до комнатной температуры, из которого, однако, еще не
выпали кристаллы.
2. Начало процесса кристаллизации облегчают различные осо-
бенности поверхности, которые могут являться начальной точкой
роста кристаллов. Стеклянная палочка, царапина на внутренней
поверхности колбы, деревянная палочка, пористые кусочки фар-
фора или пемзы, добавляемые для равномерности кипения, могут
инициировать образование кристаллов твердого вещества, под-
вергаемого очистке.
3. Для образования кристаллов может оказаться необходимым
охлаждение раствора до низких температур. Цель такого охлаж-
дения состоит в том, чтобы понизить растворимость пробы, а
не в том, чтобы заморозить раствор или выпавшее в виде жидко-
го масла вещество. Образование кристаллов при низкой темпера-
туре может происходить достаточно эффективно даже при увели-
чении объема раствора на 20—30%• Пониженные температуры
могут быть достигнуты следующими способами (детали см. при-
Разделение органических веществ
431
ложение I): охлаждение до комнатной температуры, охлаждение
в холодильнике, в бане со льдом, в морозильном отделении хо-
лодильника или в смеси льда с солью, в смеси сухого льда с
ацетоном или жидким азотом.
Не следует путать с кристаллами замерзший раствор или за-
мерзшее аморфное масло: при комнатной температуре такое мас-
ло расплавится и вновь станет жидким.
В некоторых случаях для того, чтобы вызвать кристаллизацию,
необходимо размять маслянистую пробу, например, стеклянной
палочкой. Разминать масло следует обязательно в соприкоснове-
нии с маточным раствором. Одновременно можно применять и
другие описанные выше способы облегчения кристализации, на-
пример внесение затравки. В любом случае для начала кристал-
лизации может потребоваться разминание и растирание пробы в
течение очень длительного времени. Имеется детальное описание
техники перекристаллизации маслянистых веществ *.
7.4. ХРОМАТОГРАФИЯ
7.4.1. Введение и общие замечания
Хроматографию можно определить ** как теорию и практику
такого метода разделения, который осуществляется при взаимо-
действии подвижной фазы (например, растворителя в колоночной
хроматографии), протекающей через слой неподвижной фазы (на-
пример, оксид алюминия в колоночной хроматографии). С по-
мощью этого метода можно разделять различные компоненты
смесей органических соединений, что основано на селективной и
предпочтительной сорбции неподвижной фазой этих компонентов,
находящихся в подвижной фазе.
Для химиков-органиков представляют интерес два основных
варианта хроматографии: газовая и жидкостная. Газовая хро-
матография полезна при изучении относительно летучих и терми-
чески устойчивых органических веществ. В этом методе подвиж-
ная фаза является газообразной (обычно для этой цели
* Swlnehard J. S., Organic Chemistry, An Experimental Approach, Appleton-
Century-Crofts, New York, 1969, pp. 81—84.
** Слово «хроматография» происходит от греческого слова chromatismos,
что означает цвет. В ранних вариантах хроматографии разделение проводили иа
бумаге, а объектами разделения обычно были окрашенные природные продукты.
[Приведенное в примечании объяснение причины возникновения слова «хромато-
графия» не является точным. Этот метод открыт в 1903 г. выдающимся рус-
ским ученым-ботаником М. С. Цветом, разделявшим окрашенные компоненты
хлорофилла растений на колонках, заполненных сорбентами белого цвета (крах-
мал, карбонат кальция, тальк и т. п.). Хроматография на бумаге была введена
в практику Мартином и Синджем в 1941 г., т. е. через 35 лет после открытия
хроматографического метода и появления соответствующего термина. — Прим,
ред].
432
Глава 7
применяется гелий, реже — азот), а неподвижная фаза пред-
ставляет собой жидкость. Жидкая фаза равномерно распреде-
лена на поверхности твердого носителя, например измельчен-
ного кирпича. Газовая хроматография уже была описана в
разд. 3.4.
В жидкостной хроматографии применяется жидкая подвижная
фаза (чаще всего это обычно применяемые органические рас-
творители, разд. 3.2). Неподвижная фаза может быть либо твер-
дым адсорбентом (оксид алюминия или силикагель в колоночной
или тонкослойной хроматографии), либо неподвижной жидкостью,
распределенной на поверхности твердого носителя (применяется
в жидкостной хроматографии высокого давления). Примером
жидкостной хроматографии может служить тонкослойная хрома-
тография, описанная ранее в разд. 3.2.
Большинство химиков-органиков склонны подразделять хрома-
тографические методы на аналитические и препаративные. Мето-
ды, предназначенные для аналитических целей, обычно требуют
очень небольших количеств анализируемых материалов. Попытки
переносить разработанные аналитические методики на иное обо-
рудование, позволяющее работать с большими пробами, необхо-
димыми для препаративного разделения, следует предпринимать
с большой осторожностью. Препаративное разделение целесообраз-
но проводить с достаточно большими пробами с тем, чтобы вы-
деленные продукты можно было использовать для нескольких хи-
мических и спектральных анализов или провести с ними какие-
либо химические реакции.
Для того чтобы сделать выбор между газовой и тонкослойной
хроматографией, необходимо принять во внимание следующие со-
ображения.
Газовая хроматография
1. Проба должна обладать хотя бы умеренной летучестью и
достаточной устойчивостью к нагреванию. В частности, иссле-
дуемое соединение должно быть устойчивым в условиях, требую-
щихся для его перевода в газообразное или парообразное состоя-
ние.
2. Простые приборы для газовой хроматографии недороги, не-
сложны в управлении и обычно позволяют быстро получить же-
лаемые результаты. В настоящее время эти приборы легкодоступ-
ны при весьма умеренной стоимости.
Жидкостная хроматография
1. Разделение с помощью жидкостной хроматографии обычно
требует значительных затрат времени. Особенно длительное вре-
мя приходится затрачивать при работе с классическим вариантом
хроматографии, в котором жидкость протекает через колонку под
действием силы тяжести. Много времени требует также проведе-
ние препаративного разделения. Однако с помощью таких вариан-
тов метода, как хроматография в сухих колонках (разд. 7.4.3) и
Разделение органических веществ
433
жидкостная хроматография высокого разрешения (разд. 7.4.4),
анализы могут быть проведены с весьма значительной скоростью.
2. Медленное протекание хроматографического процесса тре-
бует весьма осторожного подхода к вопросу о возможности хими-
ческих изменений пробы во время ее пребывания в колонке. Так,
например, спирты легко подвергаются химическим изменениям
при хроматографии на оксиде алюминия. Однако при правильном
выборе условий хроматографического процесса с помощью мето-
да жидкостной хроматографии можно анализировать практически
любые органические соединения, за исключением солей.
3. Жидкостная хроматография высокого разрешения, являю-
щаяся более новым методом, чем газовая хроматография, требует
несколько больших начальных затрат. Это связано с необходимо-
стью приобретения высококачественных жидкостных насосов и
дорогих наполнителей для колонок.
Возможности метода хроматографии, обсуждавшиеся выше,
используются в повседневной работе почти всеми химиками-орга-
никами. Операции проверки чистоты веществ с помощью методов
газовой и тонкослойной хроматографии вполне обычны (гл. 3).
При этом тонкослойная хроматография может проводиться в до-
статочно большом масштабе, позволяющем осуществлять препа-
ративное разделение смесей.
7.4.2. Жидкостная хроматография
Колоночная хроматография. Колоночную хроматографию
можно непосредственно использовать для препаративного разде-
ления и очистки веществ, поскольку обычно бывает нетрудно, по
крайней мере в принципе, выбрать размер колонок и количество
сорбента таким образом, чтобы они соответствовали объему под-
лежащей разделению пробы. Однако следует помнить, что такой
метод может потребовать больших затрат времени, достигающих
нескольких часов или даже дней *. Прежде чем проводить коло-
ночную хроматографию, необходимо выяснить, как ведет себя дан-
ная проба в условиях тонкослойной хроматографии (разд. 3.2).
Для проведения колоночной хроматографии имеется ряд го-
товых колонок (рис. 7.11). Однако простые устройства для коло-
ночной хроматографии проб массой примерно до 5 г можно со-
брать из деталей, имеющихся в любой химической лаборатории.
Конструкция колонок. Простейшую колонку для жидкостной хроматографии
можно приготовить следующим образом. В один конец стеклянной трубки диа-
метром около 2 см и длиной примерно 45 см помещают тампон из стеклянной
ваты. На этот конец туго одевают отрезок резиновой трубки подходящего диа-
метра. Затем трубку закрепляют строго вертикально открытым концом вверх
* Эти затруднения могут быть в значительной степени преодолены при ис-
пользовании техники хроматографии в сухих колонках (разд. 7.4.3) или жид-
костной хроматографии высокого разрешения (разд. 7.4.4).
434
Глава 7
(например, прикрепив ее к вертикальной водопроводной трубе). Поверх там-
пона из стеклянной ваты насыпают несколько сантиметров чистого песка, вырав-
нивают его слой, постукивая по колонке, и зажимают резиновую трубку подхо-
дящим зажимом. В колонку заливают петролейиый эфир хорошего качества *
с т. кип. 40—60°С. Через воронку осторожно засыпают в колонку 140 г подходя-
щего адсорбента (табл. 7.4). Техника заполнения колонки имеет решающее зна-
чение для успеха выполняемого разделения. Все адсорбенты следует хранить в
Таблица 7.4
Адсорбенты для хроматографии а
(перечислены в порядке уменьшения адсорбционной способности)
Адсорбент Примечание
Активированный уголь
Силикат магния
Оксид алюминия
Силикагель
Сульфат кальция
Сахар
Порошок целлюлозы
Для разделения веществ, способных к образова-
нию водородных связей, применяется уголь с
более слабой адсорбционной способностью
Обычно применяется для устойчивых органиче-
ских соединений
Широко применяется, особенно для менее устой-
чивых органических соединений
Очень слабый адсорбент
а В настоящее время чаще всего применяются оксид алюминия и силикагель. Сладует
отметить, что имеются так называемые обращенно-фазовые адсорбенты: йа колонках, ста-
KiiMii адсорбентами обычно быстрее элюируются более полярные вещества.
тщательно высушенном состоянии, или они должны быть доведены до необходи-
мой степени активности. Адсорбент осторожно засыпают в колонку небольшими
порциями при непрерывном легком постукивании по колонке (например, стек-
лянной палочкой с одетой на конец резиновой пробкой). Весьма важно, чтобы
уровень адсорбента никогда не превышал уровень растворителя в колонке. При
заполнении колонки и при проведении хроматографического разделения нельзя
допускать, чтобы уровень растворителя в колонке опускался ниже верхнего
уровня сорбента. После того как весь адсорбент засыпан в колонку и его по-
верхность выровнена путем постукивания по колонке, осторожно добавляют
сверху слой песка высотой 2—3 см. Выравнивают поверхность слоя песка, кото-
рый также должен быть покрыт растворителем.
Если колонку приходится оставлять без употребления в течение длительного
времени, то ее необходимо заполнить доверху растворителем, оставив место
только для корковой пробки. (Почему в этом случае неприемлемы резиновые
или пластиковые пробки?) Колонку хранят, тщательно закрыв пробкой, чтобы
предотвратить потери растворителя в результате испарения.
Вместо стеклянной трубки в качестве хроматографической колонки можно
использовать бюретку на 50 мл. Наличие крана исключает необходимость при-
менять резиновую трубку с зажимом. В такую бюретку можно поместить 115 г
адсорбента.
* Низкокачественный петролейиый эфир необходимо промыть водным рас-
твором перманганата калия и (или) серной кислотой, затем промыть водой, вы-
сушить и перегнать.
г
Рис. 7.11. Хроматографические колонки.
а—со шлифом в верхней части; б — со шлифом в верхней части и пористой пластинкой,
удерживающей сорбент, но свободно пропускающей растворитель; при использовании ко-
лонки б пробку крана необходимо закреплять резиновым кольцом; в — колонка со шлнфом
в верхней части, пористой пластинкой н стопорным кольцом, предотвращающим выпадение
пробки краиа; г — такая же колонка, как изображена иа рис. в, ио с дополнительным ниж-
ним шлифом; д — колонка с резервуаром для растворителя.
436
Глава 7
Адсорбенты, наиболее часто употребляемые для заполнения хроматографи-
ческих колонок, перечислены в табл. 7.4. Обычно используют примерно 30 г
адсорбента на 1 г пробы.
В продаже имеются хроматографические колонки различного типа
(рис. 7.11). Вместо резервуара, показанного на рис. 7.11, д, часто применяют
обычную делительную воронку (рис. 7.17). Заполненную растворителем и за-
крытую пробкой делительную воронку с закрытым краном закрепляют зажимом
на том же штативе, который удерживает колонку так, чтобы конец трубки во-
ронки был ниже уровня растворителя в колонке (рис. 7.17). Если при закрытой
пробке воронки открыть ее кран, то растворитель будет вытекать из нее лишь
тогда, когда его уровень в колонке опустится ниже конца выходной трубки
крана. При этом через кран в вороику будут проскакивать пузырьки воздуха *.
Таким образом, уровень растворителя в колонке будет автоматически поддер-
живаться постоянным до полного исчерпания растворителя в делительной во-
ронке.
Таблица 7.5
Обычный порядок элюирования органических веществ
(вещества перечислены в порядке снижения скорости элюирования) ‘
Алканы
Алкены
Ароматические соединения
Простые эфиры
Сложные эфиры
Кетоны
Альдегиды
Спирты
Амиды (имеющие по крайней мере одну связь N—Н)
Диолы
Карбоновые кислоты
Полифункииональные соединения
а Отклонения от указанного порядка могут быть связаны с сильными разлнчинми
каких-либо других свойств веществ, например, молекулярной массы. При использовании
обращенно-фазовых адсорбентов порядок элюирования может стать обратным (см. примеча-
ние к табл. 7.4).
В табл. 7.5 перечислены основные группы органических соединений в по-
рядке снижения их скорости элюирования. Наиболее быстро элюируются н-ал-
каиы, так как обычно они слабее других связываются адсорбентом. Высокопо-
лярные соединения, например карбоновые кислоты, элюируются значительно
медленнее, так как они удерживаются адсорбентами намного сильнее (например,
за счет образования водородных связей или полярных взаимодействий).
Причины различной элюирующей способности растворителей, связанные с
различиями в их полярности, были обсуждены в разд. 3.2 (табл. 3.1).
Примеиеиие наполненных хроматографических колонок. Подлежащую ана-
лизу или разделению хорошо высушенную смесь взвешивают и растворяют в ми-
нимально возможном количестве низкокипящего петролейного эфира. Лишь при
необходимости для облегчения растворения пробы можно подогреть смесь или
добавить немного метиленхлорида. Растворитель из колонки выпускают до тех
пор, пока его верхняя граница не окажется примерно в середине слоя песка,
покрывающего адсорбент сверху. Раствор разделяемой смеси по возможности
тщательно переносят в колонку на поверхность слоя песка (например, с по-
мощью пипетки). Свободный объем в колонке осторожно заполняют петролей-
* Для этого трубка и отверстие в пробке крана делительной воронки долж-
ны иметь достаточно большой диаметр, не менее 5—6 мм. — Прим. ред.
Разделение органических веществ
437
ным эфиром, сверху устанавливают в кольце резервуар с петролейным эфиром
(как показано на рис. 7.17). Резервуар позволяет поддерживать постоянную
скорость подачи растворителя в верхнюю часть колонки. Жидкость должна вы-
текать из колонки примерно со скоростью 1 капля в секунду. Подачу раство-
рителя в колонку продолжают до тех пор, пока из нее не будут элюированы
все наиболее медленно перемещающиеся полосы компонентов пробы.
В тех случаях, когда проба очень трудно растворяется в петролейном эфире,
ее растворяют в более полярном, но легколетучем растворителе и наносят на
небольшое количество адсорбента (эта процедура обсуждается в разд. 7.4.3).
Вытекающую из колонки жидкость собирают равными порциями (например,
удобно отбирать порции объемом 50 мл в колбы на 100 мл). Осторожно вы-
паривают растворитель (например, с помощью роторного испарителя, рис. 7.8)
при уменьшенном давлении *. Нагревание при отгонке растворителей должно
быть минимальным. Необходимо установить суммарную массу всех элюирован-
ных компонентов ** и сопоставить ее с массой смеси, первоначально введенной
в колонку. Общее количество введенной пробы и выделенных фракций должно
быть одинаково. Нарушения такого баланса возможны в тех случаях, когда в
верхней части колонки задерживается значительное количество смолы, не под-
дающейся обработке.
В процессе хроматографического элюирования *** состав растворителя по-
степенно изменяется и его полярность увеличивается, например:
Петролейный эфир, об.% Метилеихлорид, об. %
100 0
96 4
90 10
80 20
50 50
20 80
0 100
Для правильного выбора растворителя, применяемого в качестве элюата,
оказываются полезными результаты предварительных анализов, проведенных ме-
тодом тонкослойной хроматографии. Следует проявлять осторожность, чтобы не
вызвать размывания зон и образования вертикальных фестончатых потеков, про-
никающих глубоко в толщу слоя сорбента. Такое размывание может произойти,
если полярность элюента изменяется слишком резко ****.
За элюированием окрашенных проб можно наблюдать невооруженным гла-
зом. Элюирование бесцветных проб регистрируют с помощью какой-либо подхо-
дящей аналитической техники, например ЯМР, газовой или тонкослойной хрома-
тографии, ИК- и УФ-спектрометрии. Эти же методы применяют и для того, чтобы
* Колбы, применяемые в роторном испарителе, могут быть раздавлены
внешним атмосферным давлением. Во избежание их разрушения следует во всех
случаях, когда применяются колбы объемом более 50 мл, использовать сосуды
с круглым или коническим дном.
** После того как приемники элюата перенумерованы, нужно определить
их массу.
*** Ориентировочно следует считать, что через колонку должно пройти 10—
20 мл растворителя на 1 г оксида алюминия до очередного изменения полярно-
сти. Обычно этот объем соответствует одной-двум фракциям элюата.
**** Сложности, связанные с разделением полос (образование потеков, нару-
шение горизонтальности и т. п.), могут быть вызваны многими факторами, среди
которых следует отметить применение недостаточно чистых растворителей, об-
разование каналов в слое адсорбента из-за неравномерного заполнения колонки,
загрязненный (например, влажный) адсорбент, разложение пробы в колонке
и т. п. Если колонка заполнена адсорбентом слишком плотно, то скорость элюи-
рования может ие достичь желаемой величины 1 капля в 1 с.
438
Глава 7
установить идентичность и чистоту окрашенных и бесцветных веществ после их
элюирования из колонки. Достаточно легко контролировать процесс элюирова-
ния проб, способных флуоресцировать при освещении их ультрафиолетовым све-
том. Для этой цели достаточно наличия затемненного помещения и самодельной
ультрафиолетовой лампы с темным фильтром.
Суммируя все вышесказанное, следует отметить, что оба описанных варианта
жидкостной хроматографии — колоночный и тонкослойный — тесно связаны ме-
жду собой. Поэтому при выборе типа и размеров колонки, а также применяе-
мого растворителя следует учитывать результаты, полученные прн тонкослойной
хроматографии данной пробы.
7.4.3. Хроматография в сухих колонках
Метод разделения веществ, называемый хроматографией в су-
хих колонках, в настоящее время описан во всех деталях *. Этот
мощный метод имеет ряд преимуществ перед обычной колоночной
хроматографией. Они связаны с повышением разделяющей спо-
собности и скорости разделения компонентов изучаемой смеси.
Кроме того, этот метод позволяет использовать почти без всяких
изменений результаты тонкослойной хроматографии (разд. 3.2).
Огромные возможности метода хроматографии в сухих колон-
ках были продемонстрированы ** на примере разделения с по-
мощью этого метода на силикагеле изомерных соединений А и Б.
Это достижение является весьма впечатляющим, если учесть, что
соединения А и Б очень мало отличаются по своему строению.
При замене атома хлора в этих соединениях на водород обра-
зуется энантиомерная пара. Таким образом, разделение изоме-
ров А и Б на силикагеле в сухих колонках происходит лишь
благодаря очень малым различиям в их взаимодействии с по-
верхностью адсорбента ***.
В кратком описании суть этого метода заключается в том,
что колонку заполняют сухим адсорбентом, т. е. в процессе при-
* Loev В., Goodman М. М., Chem. Ind., 1967, 2026. Кроме того, см. лите-
ратурные источники, перечисленные в конце настоящего раздела.
** Morrill Т. С., Malasanta S., Warren К-, Greenwald В., J. Org. Chem.
40, 3032 (1975).
*** Таким образом, благодаря наличию атомов хлора эти соединения обра-
зуют диастереомерную пару. Эти атомы хлора удалены от С-3, являющегося
местом диастереомерной дифференциации. Следует заметить, что нортрициклено-
вый углеродный скелет этих соединений имеет ось симметрии Civ, совпадающую
по направлению со связью С—Н передней (на чертеже) метановой группы и
проходящую через центр плоскости циклопропанового кольца.
Разделение органических веществ
439
готовления колонки растворитель не применяется, и вместо того,
чтобы элюировать разделяемые вещества несколькими отдельны-
ми порциями элюента, сухую колонку проявляют всего один раз
одной порцией растворителя. Такая техника заполнения колонки
сильно отличается от применяемой повсеместно традиционной
(или «мокрой») колоночной хроматографии. Колонки для такой
«мокрой» хроматографии готовят, используя суспензионный ме-
тод заполнения (см. ниже). Для того чтобы разделить компонен-
ты изучаемой смеси на традиционных колонках, обычно требует-
ся их длительное и трудоемкое элюирование последовательным
рядом растворителей.
Сухие колонки можно приготовить, заполняя сухим оксидом
алюминия или сухим силикагелем стеклянные колонки * такого
типа, как на рис. 7.11, а—в. Подлежащую разделению смесь рас-
творяют в минимально возможном количестве растворителя, ис-
пользуемого для элюирования. Полученный раствор осторожно
помещают на верх свежеза полнен ной сухой колонки. Часто для
этой цели применяют колонки не из стекла, а из гибкой найло-
новой пленки. Элюирующий растворитель добавляют до тех пор,
пока его фронт не достигнет дна колонки. Проявленную колонку
затем разделяют на несколько частей, содержащих разделенные
фракции. При использовании найлоновых колонок их просто раз-
резают острым ножом. При этом аналитик должен быть очень
внимательным, используя все возможные методы и средства (ви-
зуальные и др.) для того, чтобы определить положение хромато-
графических полос и таким образом наметить линии разреза.
Затем все разделенные органические соединения экстрагируют из
выделенных порций адсорбента, обычно с помощью метилового
спирта или эфира.
Методика проведения разделения. До первых попыток проведения хрома-
тографического разделения следует подумать о следующих факторах: поведе-
ние пробы в тонкослойной хроматографии, адсорбент, материал колонки (стекло,
найлон), способ заполнения колонки и способ проявления колонки.
Поведение пробы в тонкослойной хроматографии. Под-
лежащую разделению смесь следует проанализировать методом тонкослойной
хроматографии, как было описано в разд. 3.2. Если это возможно, желательно,
чтобы хорошее разделение на пластинке было бы проведено с использованием
только одного растворителя. Если приходится применять смесь растворителей, то
предварительно следует обработать адсорбент таким смешанным растворителем
(см. ниже). Обычно на сухой колонке достигается лучшее разделение, чем на
тонкослойной пластинке.
Адсорбент. Как и для других видов адсорбционных хроматографических
процессов, для хроматографии на сухих колонках исключительно важное
* Стеклянные колонки затрудняют окончательное выделение фракций раз-
деляемой смеси. Для удаления их содержимого переворачивают колонку и осто-
рожно выталкивают слой сорбента, постукивая открытым концом колонки о
твердую поверхность или подавая сжатый воздух через трубку крапа. Извле-
ченный из колонки слой адсорбента помещают в лоток и осторожно разрезают
на части, содержащие разделенные фракции.
440
Глава 7
значение имеет содержание влаги в адсорбенте. Для того чтобы разделение в су-
хой колонке можно было бы проводить в условиях тонкослойной хроматогра-
фии, имеющиеся в продаже адсорбенты различного качества должны быть дезак-
тивированы добавлением следующих количеств воды:
Тонкослойная хроматография Сухая колонка
Силикагель Безводный 15% воды
Оксид алюминия Безводный 3—6% воды
Различные особенности наполнения сухих колонок и факторы, связанные
с применением разных адсорбентов, детально обсуждены в статье Loev В., Good-
man М. М., Chem. Ind., 1967, 2026, и в других литературных источниках, ука-
занных в конце данного раздела.
Дезактивированный адсорбент готовят, добавляя к нему нужное количество
воды и перемешивая смесь в роторном испарителе или шаровой мельнице около
3 ч. При этом очень полезно добавить к адсорбенту флуоресцентный индикатор
(см. ниже).
Способ определения активности описан во всех деталях в статье Brock-
тапп Н., Schodder Н., Вег., 74b, 73 (1941), и в других литературных источни-
ках, указанных в конце данного раздела. Ниже рассмотрен простейший способ
оценки активности адсорбентов, предложенный Лоевом и Гудманом и предусма-
тривающий использование лишь одного стандартного красителя и капиллярной
колонки малого размера («мини-колонки»).
Запаянный на одном конце капилляр размером 75 X 1 мм (например, для
определения температуры плавления) заполняют адсорбентом и в открытый ко-
нец пускают одну каплю бензола. Трубку перевертывают запаянным концом
вверх и отламывают запаянный кончик. Затем смоченным бензолом концом ка-
пилляра касаются поверхности стандартного раствора красителя в бензоле (кон-
центрация 0,5%), который заранее готовят в небольшом сосуде. Подготовлен-
ный таким образом капилляр переносят в другой сосуд, содержащий немного
чистого бензола. Оставляют эту миниатюрную колонку для проявления, отме-
чают положение зоны красителя, когда бензол достигнет верхнего конца слоя
адсорбента, и рассчитывают Rf (формулу см. в разд. 3.2). Это позволяет сде-
лать заключение об активности применяемого оксида алюминия. Необходимые
для этого данные приведены в табл. 7.6 и 7.7.
Таблица 7.6
Активность оксида алюминия
(стандартный краситель п-аминоазобеизол)
Rf красителя Степень активности по Брокмаиу Содержание воды. %
0,00 I 0
0,12 II 3
0,24 III 6
0,46 IV 8
0,54 V 10
Легко приготовить, как описано ниже, флуоресцирующий адсорбент для за-
полнения колонок. Такой адсорбент исключительно полезен для наблюдения за
продвижением по колонке полос бесцветных веществ. Для этого достаточно про-
сто смешать тонкоизмельченный флуоресцентный материал * с адсорбентом в
* Люминесцентные материалы можно приобрести через фирму Du Pont
(Photo Products Dept., Towanda, Pa.) (люминесцентный материал № 609) или
через фирму Alupharm Chemicals, New Orleans, La. (зеленый флуоресцентный
индикатор «вёль.м»).
Разделение органических веществ
441
Таблица 7.7
Активность силикагеля
(стандартный краситель n-диметиламиноазобеизол а)
Rf красителя Степень активности по Брокману Содержание воды. %
0,15 I 0
0,22 — 3
0,33 6
0,44 — 9
0,55 II 12
0,65 III 15
а В акте OSHA № 1910.93с это соединение признано опасным для здоровья.
Его можно заменить 1,4-ди-п-толу11динантрахнионом (Allied Chemicals, Special
Chemicals Div.. New York). Коммерческое название указанного красителя
«D & С Green 6».
количестве примерно 0,5%. Операцию эту удобно выполнить прн смешении ад-
сорбента с водой для дезактивации, как описано выше. После этого можно
легко наблюдать за продвижением хроматографических полос, освещая колонку
ручной ультрафиолетовой лампой и отмечая те участки колонки, где имеет ме-
сто гашение флуоресценции. В идеальном случае каждая из таких нефлуоресци-
рующих зон соответствует одному компоненту разделяемой смеси. Ввиду того
что для метода хроматографии в сухих колонках содержание влаги в адсор-
бенте является решающим, необходимо очень тщательно проверять активность
имеющихся в продаже адсорбентов, прежде чем добавить количество воды, нуж-
ное для получения адсорбента с несколько пониженной активностью.
Приготовление колонок
Стеклянные колонки. В этом виде хроматографии можно применять обыч-
ные стандартные колонки со впаянной пористой пластинкой (например, как на
рис. 7.11,6). Заполнение такой колонки адсорбентом показано на рис. 7.12.
Размер колонок выбирают непосредственно по данным предварительных
опытов по тонкослойной хроматографии. Чем труднее разделить компоненты
дайной смеси, тем более длинной должна быть колонка. На рис. 7.13 представ-
лен график, который поможет выбрать необходимую длину слоя адсорбента в
колонке. В «средних» разделениях обычно приходится иметь дело с относительно
подвижными веществами (Rf « 0,4). Если диаметр колонки составляет 50 мм,
то, согласно графику на рис. 7.13, для того, чтобы осуществить разделение 6 г
смеси «средних» соединений, длина колонки должна быть около 300 мм. Если же
исследуемые вещества более трудноразделяемы (7?, « 0,1), то для разделения
6 г смеси, согласно данным рис. 7.13, нужно взять колонку длиной около 600 мм.
Следует заметить, что графики на рис. 7.13 указывают максимально возможное
количество вещества, которое может быть разделено на данной колонке. Если
брать пробы, составляющие всего лишь 0,75—0,5 г от максимальной емкости ко-
лонки, то эффективность разделения может быть существенно увеличена. Так,
например, пробы массой от 3 до 4,5 г в приведенном выше примере будут раз-
деляться существенно лучше, чем проба массой 6 г. В процессе заполнения ко-
лонки ее кран нужно оставлять открытым, чтобы свести к минимуму количество
воздуха, захватываемого адсорбентом. Сухой адсорбент медленно засыпают в
колонку, осторожно постукивая по ней или применяя электрический вибратор,
как показано на рис. 7.12.
442
Глава 7
Рис. 7.12. Заполнение стеклянной
хроматографической колонки с по-
мощью вибратора (перепечатано с
разрешения из работы: Loev В.,
Goodman "
Диаметр
колонки, мм
18
25
37,5
50
Данные, приведенные на рис. 7.13, соответствуют соотношению 10 г адсов-
бента на 1 г смеси для «средних» разделений или 300 г адсорбента на 1 г смеси
для более трудных разделений *. Эти величины отличаются от указанного ранее
соотношения для классической («мокрой»)
колоночной хроматографии.
Найлоновые колонки. В продаже
имеются найлоновые (полиамидные) труб-
ки, прозрачные для УФ-света **. Такие труб-
ки очень полезны для проведения хромато-
графии в сухнх колонках. Сделанные из
таких трубок колонки после проявления
могут быть разрезаны ножом на отрезки,
соответствующие порциям адсорбента, со-
держащим хроматографически разделенные
компоненты исследуемой смеси. Найлон
достаточно прочен и инертен по отношению
к большинству применяемых растворите-
лей. Имеющиеся в продаже трубки можно
выбирать на основе следующих соотноше-
ний:
Ширина трубки, сложенной
в плоскую ленту, мм
25
38
50
75
Найлоновые трубки поставляются в
виде рулонов длиной от 30 до 300 м. Тол-
щина найлоновой пленки обычно составляет
0,04 мм.
Ширину и длину найлоновой трубки
для приготовления колонки обычно выби-
рают на основе данных рис. 7.13. От руло-
на отрезают трубку необходимой длины и
один конец ее запаивают с помощью на-
гретого утюга пли специального ручного
паяльника (артикул S-2020, фирма Bel-Art
Products, Pequannock, N. J.). Паяльник
устанавливают на максимальный нагрев.
Вместо запаивания можно просто подвер-
нуть конец трубки два-три раза в закрепить
его скрепками. Этот закрытый конец явля-
ется дном колонки, куда помещают тампон
нз стеклянной ваты. Вблизи нижнего конца
в стенках трубки проделывают 2—3 не-
больших отверстия, чтобы предотвратить
образование воздушных мешков при за-
колонки (рнс. 7.14). Быстро засыпают в колонку примерно одну треть
количества адсорбента. Частично заполненную колонку уплотняют,
ее 2—3 раза на твердую поверхность стола с высоты около
М., Chem. Ind., 1967,
2026).
полнении i
нужного 1
сбрасывая
* Пониженная емкость (граммы смесн/высота колонки) для силикагеля по
сравнению с оксидом алюминия связана с тем, что плотность силикагеля состав-
ляет всего лишь около половины плотности оксида алюминия.
** Найлоновые трубки размера «С» могут быть приобретены через фирму
Walter Coles and Со., Ltd., Backhouse Works, Surrey Square, Walworth, London,
S.E., U.K.
Разделение органических веществ
443
150 мм (рис. 7.15). Такой процесс заполнения повторяют еще 2 раза. Правильно
приготовленная найлоновая колонка является достаточно прочной н может
быть закреплена в штативе всего одним зажимом (рис. 7.16).
Нанесение пробы. Разделяемую смесь можно вносить в колонку растворен-
ной в минимальном количестве растворителя, используемого в качестве элюента,
как было описано для обычной колоночной хроматографии. Значительно более
Высота колонки, мм
Рис. 7.13. График для определения диаметра и высоты сухой колонки, необходи-
мых для разделения заданного количества вещества. Построение этого графика
По экспериментальным данным описано в работе: Loev В., Godman М., Chem.
Ind., 1967, 2026 (перепечатано с разрешения).
—— обычные разделения 0,4)’»---------очень трудные разделения « 0,4). Цифры на
кривых — диаметр колонки, мм.
Высота колонки, мм
удачным является способ прямого нанесения пробы разделяемой смеси на не-
большое количество сорбента перед загрузкой его в колонку. Смесь растворяют
в минимально возможном количестве легкокипящего растворителя, например
эфира или метиленхлорида. Этот низкокипящий растворитель не обязательно
должен быть тем же самым, который будет применяться для элюирования. В по-
лученном растворе диспергируют количество адсорбента, примерно в 5 раз пре-
вышающее величину пробы. После тщательного перемешивания полученной сме-
си растворитель осторожно удаляют, например выпаривая его в роторном испа-
рителе при 30—40°С, Высушенную таким образом смесь адсорбента и разделяе-
мой пробы распределяют ровным слоем на верхней поверхности адсорбента в
частично заполненной колонке. Затем введенную таким образом пробу покры-
вают тонким слоем чистого песка или стеклянных шариков малого размера тол-
щиной около 3 мм (рис. 7.17).
Проявление колонки. Следует хорошо запомнить, что в сухие хроматографи-
ческие колонки вводят лишь ограниченное количество растворителя, достаточное
444
Глава 7
для того, чтобы достичь дна колонки. В идеальном случае растворитель не
должен быть смешанным, если, конечно, это возможно для данного разделения.
Приблизительное значение необходимого объема растворителя может быть
определено из табл. 7.8. Однако следует приготовить несколько большее его
Рис 7.14. Прокалывание отверстия в дне
найлонового рукава для изготовления
сухой колонки (перепечатно с разреше-
ния из работы: Loev В., Goodman М.,
Chem Ind., 1967, 2026).
Рис. 7.15. Уплотнение сорбента в за-
полненой найлоновой колонке (пере-
печатано с разрешения из работы:
Loev В., Goodman М., Chem. Ind.,
1967, 2026).
количество. Растворитель заливают в колонку так, чтобы над поверхностью ад-
сорбента в процессе разделения сохранялся бы слой растворителя высотой 30—
50 мм. Удобно поддерживать постоянный уровень растворителя автоматически
с помощью делительной воронки с открытым краном, закрытой сверху пробкой,
как показано на рис. 7.18 (см. также обсуждение этого способа подачи раство-
Таблица 7.8
Объем растворителя, необходимый дли проявлении
сухих хроматографических колонок длиной 500 мм
различного диаметра
Диаметр колонки, мм Объем растворителя, мл
12,5 20
25 90
37,5 300
50 500
Разделение органических веществ
445
рителя в тексте при описании рис. 7.11). Как только растворитель достигнет дна
колонки, следует прекратить его добавление, так как проявление в этот момент
заканчивается. Те места, в которых колонка должна быть разрезана, отмечают
фломастером. Если разделяемые вещества бесцветны и для выявления зон при-
менялся флуоресцентный индикатор, смешанный с адсорбентом, то такие метки
Рис. 7.16. Заполненная нанлоновая Рис. 7.17. Сухая колонка с нанесен-
колонка, готовая к употреблению (пе- ной смесью красителей, готовая для
репечатано с разрешения из работы: проявления (перепечатано с разреше-
Loev В., Goodman М., Chem. Ind., ния из работы: Loev В., Goodman М„
1967, 2026). Chem. Ind., 1967, 2026).
наносят, освещая колонку ручной ультрафиолетовой лампой. Затем найлоновую
колонку вместе С ее содержимым аккуратно разрезают по меткам и разделенные
порции адсорбента экстрагируют метанолом или эфиром для выделения желае-
мых компонентов смеси.
Если для разделения была использована стеклянная колонка, то по оконча-
нии проявления ее переворачивают открытым концом вниз и, открыв кран, осто-
рожно извлекают содержимое, слегка постукивая по боковым стенкам колон-
ки или при необходимости подавая через открытый кран сжатый воздух *.
* Следует избегать использования стеклянных колонок, так как при очень
плотной набивке приходится применять слишком большие усилия для выдавли-
вания содержимого.
446
Глава 7
Извлекаемый адсорбент аккуратно размещают на подходящем лотке (например,
на сковороде или большой чашке), чтобы не допустить перемешивания разделен-
ных компонентов. Затем слой адсорбента аккуратно разделяют на отдельные
порции, как описано выше для найлоновой колонки.
Рис. 7.18. Начальная (Л) и заключительная (Б) стадии проявления в сухой
(стеклянной) колонке (перепечатано с разрешения из работы: Loev В., Good-
man М., Chem. Ind., 1967, 2026).
При использовании метода хроматографии в сухих колонках применялись
колонки длиной до 180 см, допускающие одновременное разделение проб мас-
сой до 50 г.
Дополнительные литературные источники по хроматографии в су-
хих колонках
Loev В., Goodman М. М., Dry-Column Chromatography, in
«Progress in Separation and Purification», Vol. 3, eds. E. S. Perry,
C. J. Van Oss, Wiley, New York, 1970, pp. 73—95.
Bohen J. M., Joullie M. M., Kaplan F. A., Loev B., J. Chem.
Educ., 50, 367 (1973).
Loev B., Bender P. E., Smith K., Synthesis, 1973, 362. В этой
статье описано разделение смесей алкенов в сухих колонках с
адсорбентом, обработанным нитратом серебра.
Разделение органических веществ
447
7.4.4. Жидкостная хроматография высокого разрешения
Жидкостную хроматографию высокого разрешения часто на-
зывают жидкостной хроматографией высокого давления, или,
реже, высокоскоростной жидкостной хроматографией. С помощью
этого метода были достигнуты большие успехи в решении таких
проблем, для которых ранее применялась традиционная колоноч-
ная хроматография.
Скорости подвижной фазы в традиционной колоночной жидко-
стной хроматографии обычно довольно низки по сравнению, на-
пример, со скоростями в газовой хроматографии, так как диффу-
зия молекул разделяемых веществ в стационарной фазе жидкост-
ной хроматографии происходит относительно медленно. Это связа-
но с тем, что в традиционной жидкостной хроматографии стацио-
нарная фаза применяется в форме довольно крупных частиц отно-
сительно большого размера (примерно той же величины, что и в
газовой хроматографии). Для того чтобы увеличить скорость диф-
фузии молекул пробы в неподвижной фазе, в жидкостной хрома-
тографии высокого разрешения применяются частицы очень ма-
лого размера. Малые размеры таких мелких частиц создают
определенные затруднения: для того чтобы продавить подвижную
фазу через колонку, плотно заполненную очень мелкими части-
цами, требуется давление, намного превышающее атмосферное.
Начиная с 1968 г. это направление хроматографии развивалось
очень быстро. Для нагнетания подвижной жидкой фазы в колон-
ки, заполненные очень мелкими частицами, применяются насосы,
развивающие давление в сотни килограммов на квадратный сан-
тиметр. Величина частиц современных адсорбентов составляет
всего несколько микрометров. Разработаны специальные непод-
вижные фазы, имеющие непроницаемую для жидкости твердую
сердцевину, что ограничивает диффузию органических соединений
только поверхностным слоем адсорбента. Это облегчает элюиро-
вание разделяемых веществ. Обычно в жидкостной хроматогра-
фии высокого давления применяют детекторы, регистрирующие
элюируемые из колонки вещества по изменению показателя пре-
ломления, по поглощению УФ-света и по возникновению флуорес-
ценции. Это экспериментальное направление развивалось очень
быстро, и сейчас этот высокоэффективный метод разделения стал
доступен химикам-органикам.
Взаимосвязь жидкостной хроматографии высокого давления с
другими хроматографическими методами иллюстрирует рис. 7.19.
Преимуществами жидкостной хроматографии высокого разреше-
ния являются высокая скорость разделения, возможность много-
кратного использования колонок, автоматическая непрерывная
подача растворителя, возможность с высокой воспроизводимостью
осуществлять программы изменения состава смешанного раство-
рителя в процессе разделения (градиентное элюирование) и
448
Глава 7
возможность автоматической и непрерывной регистрации элюируе-
мых проб. Недостатки метода связаны с тем обстоятельством, что
для его осуществления требуется высококачественное оборудова-
I
Подготовка
слоя
адсорбента
Нанесение
пробы
3
Поток
растворителя
Детектирование
и количественная
обработка
Результаты
Номер фракции
появление
цветных
пятен
| насос Н
I
колонка
может быть
использована
повторно
ввод пробы
шприцем или
с помощью
дозирующей
петли
непрерывное
проявление
Непрерывное
проявление
Рис. 7.19. Схема методов обычной жидкостной хроматографии (Л), тонкослойной
хроматографии (Б) и современной жидкостной хроматографии (В) тередеча-
тано с разрешения из книги: Kirkland J. J.t Snyder L., Modern Liquid Chromato-
graphy, Wiley-Interscience, New York, Copyright © 1974).
ние (насосы, колонки и т. п.). В последнее время техника жид-
костной хроматографии высокого разрешения была реализована
в препаративном масштабе, что дало возможность разделять боль-
шие количества вещества. Это позволяет в необходимых случаях
использовать этот метод вместо газовой хроматографии или тра-
Разделение органических веществ
449
диционной колоночной хроматографии. Несмотря на ее ограни-
чения, жидкостная хроматография высокого разрешения представ-
ляет собой исключительно мощный метод анализа и разделения
веществ. В настоящее время этот метод находит повседневное
применение во многих органических лабораториях.
Приборы для жидкостной хроматографии высокого разрешения. Для того
чтобы иметь возможность правильно применять метод жидкостной хроматогра-
фии высокого разрешения, химик должен иметь некоторое представление об ос-
новных элементах наиболее распространенных современных жидкостных хрома-
тографов *. На рис. 7.20 схематически показаны основные составные части обыч-
ной системы для жидкостной хроматографии высокого разрешения, которые
Рис. 7.20. Схема жидкостного хроматографа высокого давления (перепечатано
с разрешения из книги: Kirkland J. J., Snyder L., Modern Liquid Chromatography,
Wiley-Interscience, New York, Copyright © 1974).
1— резервуар для растворителя; 2—нагреватель; 3— магнитная мешалка; 4 — насос высокого
давления; 5—манометр; б—фильтр; 7—гидродинамическое сопротивление (для сглажива-
ния импульсов); 8— форколонкд (наличие ее необязательно); 9—аналитическая колонка;
10—датчик давления для контроля работы насоса (необязателен); 11 — система ввода проб;
12^-термостат колонок (может отсутствовать); 13 — детектор (например, по показателю пре-
ломления или по поглощению в УФ-свете); 14 — самопишущий регистратор системы обработки
данных.
ниже будут обсуждены более подробно. При этом необходимо иметь в виду,
что различные приборы, имеющиеся в продаже, могут весьма существенно раз-
личаться (например, по типу применяемых насосов и т. п.). Типичный прибор
заводского изготовления для жидкостной хроматографии высокого разрешения
изображен на рис. 7.21.
Вначале целесообразно рассмотреть элементы прибора, связанные с резер-
вуаром для растворителя (1—3 на рис. 7.20). Сам резервуар обычно изготов-
ляют из стекла или нержавеющей стали. Резервуар может быть снабжен уст-
ройствами для подогрева или перемешивания растворителя. Обе эти меры весь-
ма полезны, так как способствуют освобождению растворителя от растворенных
в нем газов. Для более полного удаления растворенных газов резервуар может
* Приборы для жидкостной хроматографии высокого разрешения могут
)ыть приобретены через фирмы Waters Associates, Varian Associates и du Pont
nstruments Division.
15 Зак. 333
Б
Рис. 7.21. Общий вид (Л) и схема (S) аналитического прибора для жидкостной
хроматографии высокого давления фирмы Waters Associates, Milford, Mass.
1 — резервуар для растворителя; 2—система подачи растворителя под высоким давлением;
3— устройство для ввода проб в систему, находящеюся под высоким давлением; 4—ввод
пробы; 5—колонка для жидкостной хроматографии; 6— место ввода анализируемой смеси,
содержащей компоненты Л, В, С, в верхнюю часть колонки; 7—элюирование разделенных ком-
понентов; 8—детектор; 9 — хроматограмма.
Разделение органических веществ
451
иметь специальный патрубок для продувки растворителя азотом. Растворители,
применяемые в качестве подвижной фазы, должны быть освобождены от раство-
ренных газов, так как в противном случае кислород может вступать в реакцию
с подвижной или неподвижной фазой. Кроме того, пузырьки выделяющихся из
растворителя газов могут нарушать нормальную работу детектора. Необходимо
принимать во внимание тот факт, что жидкостная хроматография высокого раз-
решения предъявляет довольно жесткие требования ко всем элементам процесса.
Рис. 7.22. Схема двухцилиндрового возвратно-поступательного насоса фирмы
Perkin-Elmer (воспроизведено с разрешения Perkin-Elmer Corp., Norwalk, Conn.).
Каждый цилиндр емкостью 10J мкл поочередно заполняется жидкостью, в то время как из
другого цилиндра происходит нагнетание. Двухшариковые клапаны с малой массой напра-
вляют поток растворителя только в одну сторону —в колонку. Соотношение фаз движения
обоих поршней таково, что в потоке растворителя практически полностью исключены толчки
давления. Скорость потока регулируется цифровым электронным устройством, изменяющие
скорость вращения электродвигателя. I — вход растворителя; 2—обратный клапан; 3— сум-
марный поток растворителя.
Поэтому для его успешного проведения необходимо применять высокостабиль-
ные материалы и с максимальной тщательностью выполнять все необходимые
операции.
Ясно, что одним из наиболее важных факторов, определяющих возможности
всего жидкостного хроматографа, является качество насоса. Для жидкостной
хроматографии высокого разрешения необходим насос, который может продви-
гать подвижную фазу через длинные колонки малого диаметра, заполненные
плотно упакованными очень мелкими частицами. Кроме того, важно, чтобы воз-
никающие при работе насоса импульсы давления были бы сведены к минимуму,
так как такие импульсы вызывают появление градиентов концентрации пробы,
что приводит к нарушению работы автоматических детектирующих устройств.
В особой степени это проявляется при работе с рефрактометрическим детекто-
ром. Для аналитической работы необходимы скорости подвижной фазы около
]—б мл/мин. Точное значение необходимой скорости элюента зависит от диа-
метра колонки и от характера заполняющего ее материала.
Имеются две основных группы «безымпульсных» насосов — пневматические
И механические. Насосы первой группы поддерживают постоянное давление, на-
сосы второй группы — постоянный расход подвижной фазы. Насосы шприцевого
типа с поршнем, продвигаемым с помощью винтового привода, обеспечивают прн
постоянном перемещении винта вытеснение одного и того же количества жидко-
сти. Такне) насосы широко применялись в ранних моделях приборов для
16*
452
Глава 7
жидкостной хроматографии. Для работы с градиентным элюированием (со сме-
шанным растворителем) могут применяться два таких насоса одновременно. Од-
нако все насосы постоянного вытеснения должны
периодически отключаться для повторного запол-
нения.
В большинстве более новых приборов приме-
няются насосы возвратно-поступательного типа.
В таких насосах при каждом ходе поршня неко-
торое количество растворителя вытесняется в
хроматографическую систему. При обратном ходе
поршня из резервуара через шаровой обратный
клапан в полость цилиндра насоса засасывается
новая порция растворителя. В таких системах
можно поддерживать постоянный расход подвиж-
ной фазы независимо от изменений давления
на входе? в колонку. Пример конструкции воз-
вратно-поступательного насоса приведен на
рис. 7.22.
Следует отметить еще несколько общих тре-
бований к системе подачи растворителя. Следует
по возможности избегать устройств для подавле-
ния импульсов давления, не входящих в кон-
струкцию насоса, так как они увеличивают мерт-
вый объем системы. Такое увеличение мертвого
объема затрудняет градиентное элюирование и
рециркуляцию пробы. Для изготовления прокла-
док, уплотняющих колец и других деталей насо-
са необходимо применять тефлон или материал,
сходный с ним по инертности. Это связано с тем,
что наличие высоких давлений в системе уве-
личивает возможность процессов' химического и
физического разрушения названных деталей.
Весьма важно устанавливать фильтры между
Рис. 7.23. Набор принадлежностей для «осветле-
ния проб», т. е. освобождения их от взвешенных
твердых частиц (с любезного разрешения Waters
Associates, Milford, Mass.).
Если в пробах присутствуют такие частицы, то колонка,
устройство для ввода проб нлн применяемые шприцы
могут быть засорены. Набор принадлежностей для «освет-
ления проб» включает переходный штуцер Суиннн, в ко-
тором устанавливается очень мелкопорнстый фильтр. Для
работы с органическими растворителями рекомендуется
тефлоновый фильтр с диаметром пор 0,45 мкм. Для вод-
ных растворов следует применять фильтр на ацетата
целлюлозы также с диаметром пор 0,45 мкм. Фильтрова-
ние образцов до ввода в хроматографический прибор
является операцией первостепенной важности. 1—пор-
шень; 2—шприц на 10 мл; 3 — верхняя часть переходного
штуцера Суинни, удерживающего фильтр; 4—О-образное
уплотнительное кольцо; 5—предварительный фильтр;
6—основной фильтр; 7 — опорная сетка; 8—плоская шайба;
9—нижняя часть переходного штуцера Суиннн; 10—ка-
нюля.
насосом и колонкой. В противном случае колонка, заполненная частицами
очень малого диаметра, может быть полностью засорена нежелательными твер-
дыми примесями. В большинстве случаев для этой цели лучше всего применять
фильтры из пористой спеченной нержавеющей стали, непроницаемой для твердых
частиц величиной более 2 мкм.
Тип разделения и структура сорбента Характеристика частиц сорбента Характеристика колонки
Аналитическое и микро- препаративное разде- ление Частицы малого диа- метра, пористые во всем объеме Очень высокая эффек- тивность, умеренная емкость, высокая ско- рость анализа
Аналитическое разделе- ние Пелликулярные части- цы (стеклянные ша- рики, поверхность ко- торых покрыта тон- ким слоем силика- геля) Высокая эффективность, низкая емкость, уме- ренная скорость
Препаративное разделе- ние Частицы большого диа- метра, пористые во всем объеме Умеренная эффектив- ность, высокая ем- кость, умеренная ско- рость
Функцио- нальная группа Полярность Характер процесса разделения Имеются в продаже
Силаноль- ная Высокая Адсорбцион- ный, нор- мально-фазо- вый ц-Порасил (частицы пористого силикагеля малого размера) Корасил (силикагель, закреплен- ный на по- верхности непористых стеклян- ных шари- ков) Порасил (полностью пористый силикагель)
Рис. 7.24 (начало).
454
Глава 7
Продолжение рис. 7.24.
Функцио- нальная группа Полярность Характер процесса разделения Имеются в продаже
Амино- группа Высокая Адсорбцион- ный, нор- мально-фазо- вый, обра- щенно-фазо- вый, ионо- обменный, распредели- тельный р-Бонда- пак-МН2
Циаигруппа Промежу- точная Адсорбцион- ный, иор- мальио-фазо- вый, обра- щенно-фазо- вый, распре- делительный р-Боида- пак-CN
Фенильная Низкая Распредели- тельный, об- ращенно-фа- зовый р-Бонда- пак-фенил Боидапак- фенил на кора- силе Бондапак- феиил на порасиле
С18 То же р-Бонда- пак-Cig Боида- пак-Cie на кора- силе ‘ Бонда- пак-С18 на порасиле
— Анализ жир- ных кислот Колонка для ана- лиза жир- ных кислот — —
— Анализ угле- водов Колонка для ана- лиза угле- водов — —
'— — Анализ три- глицеридов Колонка для ана- лиза три- глицеридов — —
Сополимер стирола и диви- иилбеи- зо л а Эксклюзия по размеру молекул, гель-прони- кающая хро- матография р.-Стира- гель Стиратель
Эфирные (RQ), ме- тильная Промежу- точная Эксклюзия по размеру молекул |х-Бонда- гель — —
Рис. 7.24. Материалы для заполнения колонок, обычно применяемые в жидкост-
ной хроматографии высокого давления (с любезного разрешения Waters Associa-
tes, Milford, Mass.).
Разделение органических веществ
455
Жидкостная хроматография высокого разрешения даже более, чем обычная
хроматография, чувствительна к технике ввода проб. В слой хроматографиче-
ского сорбента должна быть введена очень тонкая зона разделяемой смеси. До
Рис. 7.25 Хроматограмма смеси хлорзамещенных изомерных анилинов, получен-
ная при жидкостной хроматографии высокого давления (с любезного разреше-
ния Waters Associate., Milford, Mass.).
Колонка размером 300X4 мм заполнена ц-пораснлом, элюент — метнленхлорнд, скорость
9,9 мл/мин. Давление на входе в колонку 130 кг/см2 (13 МПа).
давлений порядка 7,5 МПа (75 кг/см2) можно применять микрошприцы, выдер-
живающие высокое давление. При более высоких давлениях необходимо вводить
пробу, применяя внешние устройства (например, дозировочные краны). Следует
избегать ввода пробы при остановленном потоке элюента, так как такая опера-
ция нарушает работу и колонки, и детектора. При вводе проб следует применять
устройство, изображенное на рнс. 7.23.
456
Глава 7
Колонки для жидкостной хроматографии высокого давления изготовляют
из нержавеющей стали или из стекла. При выборе материала следует учиты-
вать, что эти колонки должны выдерживать давление в несколько сотен кило-
ацетонитрил -вода
Рис. 7.26. Сорбент для обращенно-фазовой жидкостной хроматографии с приви-
тыми на поверхности неполярными алкнлсилильными Ci8 или фенилсилнльными
группами (с любезного разрешения Waters Associates, Milford, Mass.).
Обычно в этом случае разделение происходит в системе жидкость — твердое тело. В боль-
шинстве случаев сорбенты для обращенно-фазовой хроматография работают в сочетании
с подвижными фазами типа смесей воды с ацетонитрилом или метанолом. Так, например,
при разделении нафталина и бензопирена сорбент и разделяемые вещества являются непо-
лярными. Поэтому сорбент склонен удерживать эти соединения. В то же время подвижная
фаза (в данном случае смесь воды с ацетонитрилом) является очень полярной и поэтому
плохо растворяет разделяемые вещества. В результате эти вещества энергично удерживаются
сорбентом. Действительный механизм разделения очень сложен.
Рис. 7.27. Препаративный хроматограф для жидкостной хроматографии высо-
кого давления (с любезного разрешения Waters Associates, Milford, Mass.).
граммов на квадратный сантиметр (несколько десятков мегапаскалей). Соеди-
нительные детали и заглушки должны быть изготовлены из достаточно инерт-
ных материалов и не должны нарушать равномерность потока подвижной фазы.
Разделение органических веществ 4Е7
Обычно длина колонок составляет 25—150 см, а внутренний диаметр аналити-
ческих колонок — от 1 до 8 мм. Колонки диаметром менее 8 мм трудно запол-
нять без достаточного опыта. Перечень сорбентов, применяемых для заполнения
колонок в жидкостной хроматографии высокого давления, приведен на рис. 7.24.
Типичные результаты разделения на колонке с ц-порасилом показаны на
рис. 7.25. На этом рисунке следует отметить, что полное разделение трех изоме-
ров положения достигнуто менее чем за 1 мин. Весьма полезный вариант жид-
костной хроматографии высокого давления представлен на рис. 7.26. Показанный
на этом рисунке сорбент для так называемой обращенно-фазовой хроматогра-
фии позволяет разделять различные соединения неполярного характера.
Рис. 7.28. Препаративное разделение смеси двух преобладающих компонентов
(с любезного разрешения Waters Associates, Milford, Mass.).
Пики этих веществ иа хроматограмме разделены довольно плохо. Заштрихованные лло.
щаци соответствуют частям разделяемых компонентов, выделенным в чистом виде-
Часть пробы, соответствующая центральной зоне хроматограммы, подвергается разделению
повторно. При этом в последующих циклах величина разделяемой в колонке пробы сни-
жается, что приводит к улучшению достигаемого разделения. При проведении препаратив-
ных разделений методом жидкостной хроматографии высокого давления такая методика
оказывается особенно полезной. 1 — отбор чистого компонента Б; 2—отбор чистого компо-
нента А; 3 — часть пробы, подлежащая повторному разделению.
В последнее время довольно широкое распространение приобрели приборы
для препаративной жидкостной хроматографии. Один из приборов изображен
на рис. 7.27. Очень большим преимуществом таких приборов является то обстоя-
тельство, что в колонку можно вводить достаточно большие пробы. Хотя после
первого прохода через колонку пики отдельных компонентов смеси будут при
этих условиях перекрываться между собой, элюат можно вновь направить на
вход колонки весьма большого размера (рис. 7.28). В результате применение
такого режима циркуляции позволяет добиться отличного разделения за относи-
тельно небольшое время.
7.4.5. Газовая хроматография
Газовая хроматография была описана в разд. 3.4 в качестве
метода анализа и проверки чистоты исследуемых соединений.
В настоящем разделе описание метода газовой хроматографии
будет расширено и распространено на препаративное разделение
веществ. В том случае, если исследуемая проба выдерживает
условия газохроматографического разделения, этот метод оказы-
вается одним из наиболее простых, быстрых и полезных аналити-
ческих методов, которые могут быть применены для указанной
цели.
458
Глава 7
Колонки для газовой хроматографии могут быть капиллярны-
ми и наполненными *. Капиллярные колонки представляют собой
длинные тонкие трубки, содержащие только одну неподвижную
фазу. Наполненные колонки имеют больший диаметр. Их запол-
няют сорбентом, полученным путем нанесения неподвижной фазы
на инертный твердый носитель (например, измельченный огне-
упорный кирпич). Аналитические колонки могут иметь длину от
10—15 см до 1—2 км. Наиболее часто применяют колонки дли-
ной от 1,5 до 3—4 м. Для проведения препаративного разделения
во избежание чрезмерно больших значений времени удерживания
обычно предпочитают колонки умеренной длины (1,5—3,5 м).
Хотя существуют приборы, на которых можно работать с колонка-
ми очень большого диаметра, обычно удобнее применять для пре-
паративного разделения приборы, снабженные детектором по
теплопроводности и имеющие колонки диаметром от 6 до 9 мм. Та-
кие колонки достаточно удобны как для аналитической, так и
для препаративной работы. В том случае, если газовый хромато-
граф имеет детектор, разрушающий пробу (например, пламенно-
ионизационный), то в систему коммуникаций прибора включают
делители потока, направляющие меньшую часть пробы к детек-
тору, а остальное — в систему сбора выделенных фракций.
В стремлении облегчить и ускорить проведение препаратив-
ного разделения не следует применять чрезмерно большие пробы.
Когда величина пробы достигает такого значения, при котором
происходит перегрузка хроматографической колонки, пики, реги-
стрируемые самописцем хроматографа, резко искажаются и те-
ряют свою нормальную острую или колоколообразную форму.
Для лучшего понимания метода газовой хроматографии ниже
будет приведен ряд определений и формул. Следует отметить,
что многие из этих определений и формул применимы также и к
другим видам хроматографической техники (тонкослойной, жид-
костной хроматографии высокого давления и т. п.). Таким обра-
зом, изложенная здесь общая теория может быть использована
и при работе с другими видами хроматографии. Хорошее пони-
мание этой теории очень полезно, так как служит основой для
планирования и успешного использования дальнейших хромато-
графических анализов.
Пусть v — скорость подвижной фазы (см/с), vx — скорость
движения данной хроматографической полосы в колонке. Эти две
скорости можно связать между собой с помощью множителя R,
который представляет собой не что иное, как долю (например,
мольную) молекул вещества х, присутствующих в подвижной фазе
* В продаже имеются готовые колонки, однако, применяя колонки собствен-
ного изготовления, можно добиться значительной экономии средств (Bates R. В.,
Schaefer J. Р., Research Techinques in Organic Chemistry, Englewood Cliffs, New
Jersy, Prentice-Hall, 1971, рис. 7.3).
Разделение органических веществ
459
в данный момент времени:
vx = vR.
Если R = 0, то vx = 0 (т. е. молекулы вещества х вообще не про-
двигаются по колонке), а если R = 1,00, то vx — v (т. е. молеку-
лы вещества х движутся в колонке с той же скоростью, что и
подвижная фаза). Таким образом, скорость, с которой полоса дви-
жется в потоке подвижной фазы, снижается. Это происходит
вследствие влияния неподвижной фазы, уменьшающей число моле-
кул в подвижной фазе.
Полезно также определить величину k' — коэффициента емко-
сти или фактора удерживания *. Он равен отношению чисел мо-
лекул вещества х в стационарной и подвижной фазах (мя и пт
соответственно), т. е.
k' = ^
пт
Таким образом, величина k' оказывается большей для той
хроматографической системы, в которой стационарная фаза бо-
лее сильно связывает молекулы пробы.
На основе приведенного выше определения R можно вывести
следующее соотношение:
п __ vx пт
v nm + ns
или, используя предыдущее соотношение,
и
Vx~~ k'+\
Скорость vx можно определить экспериментально, если изве-
стна длина колонки L (см) и время удерживания пробы tR(c):
Аналогичное соотношение можно записать и для скорости движе-
ния £>о и времени удерживания t0 стандартного вещества
Обычно в качестве такого стандарта выбирают какой-либо от-
носительно слабо удерживаемый компонент, например воздух или
растворитель.
Весьма важно хорошо представлять себе, что отношение вре-
мен удерживания при сопоставимых условиях хроматографиче-
ского опыта не зависит от длины колонки, т. е.
t =
io vx
* В советской литературе по хроматографии для этой величины принято
название «отношение распределения». — Прим, перев.
460
Глава 7
Преобразуя отношение времен и заменяя отношение скоро-
стей с помощью уравнения, связывающего v и k', можно пока-
зать, что
k'= tR~to
'о
Это означает, что коэффициент емкости k' можно определить
путем деления длины отрезка на хроматограмме, соответствую-
щего разности времени удерживания исследуемого вещества к
Рис. 7.29. Типичная хроматограмма, получаемая в результате проведения газо-
вой, тонкослойной или жидкостной хроматографии высокого давления (перепе-
чатано с разрешения из книги: Kirkland J. Snyder L., Modern Liquid Chroma-
tography, Wiley-Interscience, New York, copyright ©, 1974).
t —время удерживания компонента Б; t —время удерживания стандартного соединения
а 0
(часто на этом месте может быть зарегистрирован пнк этого соединения); — время, соот-
ветствующее ширине пика Б.
Пик k' (см. текст)
А 0,7
В 2,2
В 5.7
и времени удерживания стандартного вещества, на длину отрез-
ка, отвечающего времени удерживания этого стандартного соеди-
нения (to). Если tR = to, то fe' = 0, т. е. колонка имеет сорбцион-
ную емкость, равную нулю. Если tR = 2t0, то k' = 1,00, если tR =
= 3/0, то k' = 2,00 и т. д. Таким образом, при сопоставимых усло-
виях величина k' для данного вещества постоянна. Определение
величин t0 и tR иллюстрирует рис. 7.29.
Селективность а измеряют как отношение коэффициентов ем-
кости
^2
а = —г =--------
V, - Vo
где Vi, Vz и Vo — объемы удерживания двух сравниваемых соеди-
нений и одного десорбирующегося соединения соответственно.
Разделение органических веществ
461
Величина упомянутых выше времен удерживания зависит от
скорости подвижной фазы, при которой были измерены эти ве-
личины, и от ряда других факторов. Объемы удерживания изме-
ряются достаточно легко и в меньшей степени зависят от усло-
вий эксперимента. Это связано с тем, что в принципе объем удер-
живания Er компонента х с временем удерживания tR не зависит
от скорости подвижной фазы F (мл/с):
К х\
Если to — время удерживания растворителя, то можно рассчитать
его объем удерживания Vm, т. е. объем подвижной фазы, за-
полняющий пустую колонку:
Ет = /0Е.
и отношение объемов удерживания будет связано с коэффициен-
том емкости соотношением
V
-^=1 + ^
у m
Часто величины Vr оказываются более предпочтительными,
чем tR. Поскольку значения VR, по существу, не зависят от ско-
рости подвижной фазы, они очень полезны в тех случаях, когда
скорость подвижной фазы может меняться (например, в жидко-
стной хроматографии высокого разрешения), или в тех случаях,
когда эту скорость трудно измерять.
Из определения коэффициента емкости k' можно вывести, что
эта величина пропорциональна объему неподвижной фазы 14, так
как
__ ns __ 1-*-]дИS
nm [x]mVm
Отношение концентраций К называется коэффициентом распре-
деления
Эта величина определяет равновесное распределение молекул ве-
щества х между неподвижной и подвижной фазами. Легко заме-
тить, что К не зависит от 14.
Ввиду того что коэффициент емкости k' прямо пропорционален
объему стационарной фазы У£, величина этого коэффициента уве-
личивается при повышении содержания жидкой фазы, нанесенной
на твердый носитель. В частности, пелликулярные или поверхно-
стнослойные стеклянные шарики, широко применяемые в жид-
костной хроматографии высокого разрешения, в данных условиях
эксперимента обычно имеют меньшие значения Vs и k' для ко-
лонки заданных размеров.
462
Глава 7
Важным параметром, который можно рассматривать как меру
эффективности хроматографической колонки, является число тео-
ретических тарелок N. Эту величину можно определить по урав-
нению
yv=i6(_k)2
где tw — ширина полосы, выраженная в единицах времени (с).
На рис. 7.29 величина tw была определена графически для компо-
нента Б. В принципе на одной колонке при одинаковых экспе-
риментальных условиях величина N должна быть одной и той
же для любых веществ. При этом хроматографические полосы
сильнее удерживаемых в колонке веществ должны иметь боль-
шую ширину, чем полосы слабее удерживаемых компонентов
(рис. 7.29). Легко видеть, что отношение tR/tw и потому /V, по
существу, представляет собой меру разделяющей способности ко-
лонки, т. е. чем больше в среднем оказывается N, тем шире диа-
пазон времен удерживания хроматографических полос (fff), со-
храняющих относительно малую ширину (малое значение tw).
Число теоретических тарелок N прямо пропорционально дли-
не колонки L. Для оценки эффективности хроматографического
процесса лучше использовать величину, не зависящую от длины
колонки. Такой величиной является высота, эквивалентная теоре-
тической тарелке (ВЭТТ), обычно обозначаемая буквой Н. Эта
величина показывает эффективность колонки на единицу длины.
и L
Главной задачей хроматографии является обеспечение мини-
мальной величины Н, т. е. получение наибольшего числа теорети-
ческих тарелок на колонке возможно меньшей длины.
Имеется пять основных факторов, определяющих величины
Н и N для данного хроматографического процесса: 1) вихревая
диффузия, 2) массопередача в подвижной фазе, 3) продольная
диффузия, 4) массопередача в пограничном слое подвижной фазы
и 5) массопередача в неподвижной фазе. Для лучшего понимания
и сознательного применения метода хроматографии эти факторы
целесообразно рассмотреть более подробно.
Вихревая диффузия возникает вследствие того, что в хрома-
графической колонке поток подвижной фазы разбивается на боль-
шое число более мелких струй, каждая из которых протекает
через слой подвижной фазы своим путем с определенной скоро-
стью. Это напоминает реку с большим количеством островов и
протоков. При этом в более прямых широких каналах течение
может достигать большей скорости, чем в других, более узких
и извилистых. Чем больше различие скоростей в разных каналах
хроматографического слоя, тем большим оказывается расширение
Разделение органических веществ
463
полосы в колонке. Такое расширение приводит к тому, что хро-
матографические полосы различных компонентов перекрывают
друг друга, и их разделение затрудняется. Для снижения влия-
ния вихревой диффузии применяют колонки, равномерно запол-
ненные небольшими, одинаковыми по форме сферическими ча-
стицами, так, чтобы вихревые возмущения, возникающие в по-
токе подвижной фазы, были по возможности одинаковы.
Массопередача в подвижной фазе имеет важное значение, по-
скольку в каждом отдельном потоке подвижной фазы молекулы,
находящиеся в его центральной части, движутся быстрее, чем та-
кие же молекулы, находящиеся в его периферической части (бли-
же к «берегу реки», т. е. к частицам неподвижной фазы). Это
приводит к размыванию хроматографической зоны, по существу,
такому же, как и в случае вихревой диффузии. Для снижения
отрицательного влияния этого фактора применяют частицы мало-
го диаметра, чтобы каналы между ними были бы достаточно
малы.
Продольная диффузия связана с тем, что молекулы стремятся
диффундировать от центра хроматографической полосы вперед
и назад по направлению ее движения в области с более низкой
концентрацией данного компонента. Это явление проявляется
сильнее при больших коэффициентах диффузии разделяемых ве-
ществ в подвижной фазе и при малых скоростях движения по-
следней. Отрицательное влияние продольной диффузии прояв-
ляется в меньшей степени при повышении скорости потока под-
вижной фазы.
Массопередача в пограничном слое подвижной фазы связана
с тем, что отдельные молекулы движущейся хроматографической
полосы могут на некоторое время задерживаться в порах частиц
неподвижной фазы, заполняющей колонку. В результате движе-
ние этих молекул по колонке замедляется, что также приводит
к расширению хроматографической полосы. Эти затруднения мо-
жно уменьшить, применяя в качестве неподвижной фазы либо
частицы с непроницаемой центральной частью и тонким адсорб-
ционным слоем на поверхности, либо частицы очень малого диа-
метра. Это приводит к уменьшению числа закрытых пор и ка-
налов, которые могут приводить к задержке подвижной фазы.
Роль массопередача в неподвижной фазе весьма существенна
и, по существу, аналогична влиянию предыдущего фактора. Пос-
ле того как молекула вещества х проникает в канал внутри ча-
стицы неподвижной фазы, эта молекула должна достичь стенки
этого канала. Этот процесс облегчается при использовании сор-
бентов, в которых на твердый носитель нанесена тонкая пленка
неподвижной жидкой фазы. Таким путем уменьшается отрица-
тельное влияние рассматриваемого фактора.
Проведенные выше рассуждения в основном относились к
газожидкостной хроматографии, в которой обычно получаются
464
Глава 7
пики, имеющие почти идеальную форму гауссовой кривой
(рис. 7.29). При использовании газовой хроматографии для пре-
паративного разделения обычно наблюдаются значительные от-
клонения формы пиков от гауссовой кривой. Для понимания
возникающих при этом проблем вначале следует рассмотреть яв-
ление образования размытых фронтов пиков («хвостов»), неко-
торые виды которых показаны на рис. 7.30.
Рис. 7.30. Пики с размытыми фронтами (имеющие «хвосты») в жидкостной хро-
матографии (перепечатано с разрешения нз книги: Kirkland J. J., Snyder L.,
Modern Liquid Chromatography, Wiley-Interscience, New York, copyrigth © 1974).
A— химическое размывание; Б —размывание, связанное с наличием в пробе большого коли-
чества растворителя; В — пуассоновское размывание; Г —экспоненциальное размывание.
Имеется ряд факторов, вызывающих образование размытых
фронтов («хвостов») газохроматографического пика. При этом
пики могут приобретать несимметричную форму или быть ско-
шенными к той стороне, которая соответствует большим значе-
ниям времени удерживания. Типичные случаи изображены на
рис. 7.30.
Размывание, связанное с химическими факторами (рис. 7.30,Л),
обычно является следствием несоответствия химической природы
разделяемой пробы и, например, неподвижной фазы. Если такое
размывание, типичным проявлением которого является медленное
возвращение пика к нулевой линии, оказывается настолько интен-
сивным, что приводит к ухудшению разделения пиков, то во мно-
гих случаях приходится обращаться к какому-либо другому ва-
рианту хроматографической техники.
Разделение органических веществ
465
Образование несимметричного пика, связанное с растворите-
лем, в котором растворена проба (рис. 7.30,5), наблюдается в
том случае, когда компонент, представляющий интерес, регистри-
руется на фоне размытого заднего фронта очень большого пика,
соответствующего преобладающему компоненту разделяемой сме-
си. Очень часто таким компонентом является растворитель, в ко-
тором проба вводится в хроматографическую колонку.
Пуассоновское и экспоненциальное размывания (рис. 7.30,5 и
Г соответственно) обычно не представляют серьезной проблемы.
Эти виды асимметрии пиков связаны с недостаточной эффектив-
ностью применяемой колонки или с отклонениями формы пиков
Рис. 7.31. Два незначительно перекрывающихся хроматографических пика (пере-
печатано с разрешения из книги: Kirkland J. J., Snyder L., Modern Liquid Chro-
matography, Wiley-Interscience, New York, copyright © 1974).
Разделение i?s уменьшается либо при увеличении tWf, или ip либо при уменьшении
от симметричной кривой нормального распределения. Опублико-
вана работа *, в которой детально обсуждаются явления, связан-
ные с образованием размытых фронтов («хвостов») пиков.
Целью хроматографического процесса обычно является дости-
жение достаточного разделения 7?s компонентов исследуемой сме-
си. Эту величину численно определяют следующим образом:
ti —t\
2"
где fi и t?— времена удерживания разделяемых компонентов 1
2, a t и tWi — значения времени, отвечающие ширине соответ-
ствующих пиков (см. также рис. 7.29 и рис. 7.31). Дробь, опреде-
ляющая величину 7?s, представляет собой отношение разности
времен удерживания двух пиков к их средней ширине. Таким обра-
зом, величина дает определенное представление о возможной
степени перекрывания двух пиков (рис. 7.31). На рис. 7.32 по-
казаны хроматограммы смеси двух компонентов при различном
их соотношении и разных значениях степени их разделения Rs.
Snyder L. R., J. Chromatog. Sci., 10, 200 (1972).
466
Глава 7
Еще одним важным параметром, представляющим интерес,
является время анализа t. Во многих случаях приходится стре-
миться к уменьшению времени данного анализа. Однако следует
учитывать, что время анализа и достигаемое разделение тесно
связаны между собой. Это утверждение можно иллюстрировать,
Рис. 7.32. Некоторые частные случаи хроматографического разделения (перепе-
чатано с разрешения из книги: Kirkland J. J. Snyde' L., Modern Liquid Chroma-
tography, Wiley-Interscience, New York, copyright © 1974).
В верхней части рисунка указано соотношение разделяемых компонентов; цифры слева —
достигаемое разделение; по оси абсцисс отложено время эксперимента (три отдельных от-
резка).
рассмотрев некоторые явления, имеющие место в жидкостной
хроматографии. Время разделения является функцией вязкости
подвижной жидкой фазы, общей пористости колонки, ее длины и
перепада давлений на входе и выходе из колонки. Интенсивное
изучение процесса жидкостной хроматографии высокого разре-
шения (разд 7.4.4) привело к появлению большого числа
сообщений о том, как перечисленные факторы влияют на время
разделения. На рис 7.33 показана зависимость времени удержива-
ния стандартного соединения (/о) от длины колонки для жид-
костной хроматографии высокого разрешения при разных диа-
метрах частиц неподвижной фазы. Из приведенных графиков еле-
Разделение органических веществ
467
дует, что при применении жидкостной хроматографии высокого
разрешения целесообразно использовать частицы малого диамет-
ра *. Данные, представленные на рис. 7.33, получены при прове-
дении анализов на колонках с высококачественным заполнением
мелкими пористыми частицами неправильной формы. Пористые
сферические частицы обладают более высокой проницаемостью и
Рис. 7.33. Зависимость времени удерживания стандартного соединения (to) от
длины колонки (L) при разной величине частиц сорбента (dp) при давлении на
входе 70 кг/см2 (7 МПа) н вязкости подвижной фазы 0,3 сантипуаз (перепеча-
тано с разрешения из книги: Kirkland J. J., Snyder L., Modern Liquid Chroma-
tography, Wiley-Interscience, New York, copyright © 1974).
Цифры на кривых — величина частиц сорбента (мкм).
поэтому позволяют при прочих равных условиях получить зна-
чения времени to, составляющие примерно половину величин, при-
веденных на рис. 7.33. Использование пелликулярных частиц по-
зволяет получить времена удерживания t0, составляющие одну
четверть приведенных на рис. 7.33. Применение в качестве непо-
движных фаз гелей, легче деформируемых и обладающих мень-
шей проницаемостью, приводит к увеличению значений времени
удерживания стандартного компонента t0.
В препаративной хроматографии очень важным вопросом яв-
ляется зависимость достигаемого разделения от величины пробы.
* Преимущество частиц малого диаметра достигает максимума при их раз-
мере около 10 мкм. Более мелкие частицы, размером, например, около 5 мкм,
дают лучшее разделение, но могут засорять коммуникации приборов.
468
Глава 7
Эта зависимость определяет те или иные возможности примене-
ния данного хроматографического метода в анализе. Изменение
характера хроматограммы при увеличении пробы показано на
рис. 7.34. Интересно отметить, что на рис. 7.34,5 хорошо видно,
что при увеличении пробы наблюдается уменьшение времени
Рис. 7.34. Изменение формы пиков при увеличении размера вводимой пробы
(Л — наименьшая проба, Г — наибольшая проба) (перепечатано с разрешения из
книги: Kirkland J. J., Snyder L., Modern Liquid Chromatography, Wiley-Interscien-
ce, New York, copyright © 1974).
удерживания обоих разделяемых компонентов. При этом время
удерживания компонента, элюируемого позже, сокращается в
большей степени. Поэтому значительное увеличение количества
вводимой в колонку смеси приводит к сильному ухудшению раз-
деления. На рис. 7.34, В и Г ясно видно, что пики на хромато-
грамме отделены друг от друга в значительно меньшей степени.
Это происходит вследствие того, что при некоторой величине
пробы исчерпывается сорбционная емкость колонки. Дальнейшее
увеличение количества вводимых веществ приводит к нарушению
линейной связи между концентрациями разделяемых веществ в
неподвижной и подвижной фазах. До этого момента для данной
колонки высота, эквивалентная теоретической тарелке (ВЭТТ),
Разделение органических веществ
469
медленно возрастала с увеличением количества пробы. Далее уве-
личение ВЭТТ протекает очень быстро. Такое увеличение высоты
тарелки означает, что данная колонка содержит меньшее число
теоретических тарелок, и значит, эффективность достигаемого с
ее помощью разделения уменьшится.
Рис. 7.35 Приемники для улавливания фракций, разделенных методом газовой
хроматографии [перепечатано с разрешения из книг: Crippen R. С., The Iden-
tification of Organic Compounds with the Aid of Gas Chromatography, McGraw-
Hill, New York, copyright © 1973 (А и S); Bates R. B., Schaefer J. P., Research
Techniques in Organic Chemistry, Prentice-Hall; Inc., Englewood Cliffs, N. J.,
copyright © 1971 (£?)].
A—капиллярная трубка без охлаждения; Б —капиллярная трубка с охлаждением; В — центри_
фужная пробирка (если не образуется аэрозоль, то тампон из стеклянной ваты можно не уста'
навлпвать). 1 — капиллярная трубка для определения температуры плавления; 2— сухой лед;
3—выходная коммуникация хроматографа; 4—пробка; 5 —бумажный фунтик; 6 — трубка из
стекла пирекс диаметром 4 мм; 7 — вход пробы; 8 — пробка с одним отверстием н боковым
вырезом; 9 — стеклянная вата; 10—центрифужная пробирка; // — боковой вырез в пробке для
выхода газа-носителя; 12— пробка (вид сверху).
Для того чтобы собрать фракции, разделенные в процессе
препаративной газовой хроматографии, нужно применять соот-
ветствующие улавливающие устройства. Ряд таких устройств, в
том числе автоматических, имеется в продаже. Некоторые из них
являются частью хроматографических приборов, а другие могут
быть приобретены отдельно как самостоятельные элементы. При
этом могут применяться поворотные устройства — коллекторы, ко-
торые биохимики используют для сбора фракций таких веществ,
как, например, аминокислоты.
Однако большинство химиков-органиков находят удобным и
вполне достаточным применять простые приспособления для сбора
470
Глава 7
фракций, показанные на рис. 7.35. Следует отметить, что не-
обходимо с большим вниманием подходить к выбору охлаждаю-
щего агента. При попытке конденсировать выходящие из колон-
ки вещества при слишком низких температурах некоторые со-
единения могут образовывать аэрозоли, которые довольно трудно
улавливать в обычных ловушках. Удобно применять для сбора
фракций центрифужные пробирки (рис. 7.35, В). В этом случае
очень малые пробы можно путем центрифугирования перевести на
дно пробирки.
Обычно пробы, собранные после газохроматографического
разделения, отличаются большой чистотой и могут подвергаться
дальнейшему анализу непосредственно. Например, можно смыть
собранную фракцию с помощью CDC13 непосредственно в про-
бирку для ЯМР-спектроскопии. Для получения точных значений
физических констант и других характеристик собранные после
газохроматографического разделения фракции должны быть под-
вергнуты анализу как можно быстрее. Чистоту собранных фрак-
ций проверяют путем повторного ввода небольших проб в газо-
вый хроматограф. Иногда собранные фракции могут быть загряз-
нены летучими компонентами неподвижной фазы, выносимыми из
колонки с потоком газа-носителя. В этом случае собранные фрак-
ции подвергают микроперегонке в приборе соответствующего
объема.
Для препаративного разделения веществ и сбора разделенных
фракций особенно полезной является жидкостная хроматография
высокого разрешения. Этот процесс превосходит традиционные
варианты газовой и жидкостной хроматографии по скорости раз-
деления и удобству работы. Кроме того, при использовании этого
метода снижается возможность разрушения пробы, так как она не
подвергается воздействию высоких температур. Типичный прибор
для препаративной жидкостной хроматографии высокого разре-
шения показан на рис. 7.27.
В некоторых случаях может оказаться затруднительным про-
вести определение класса растворимости исследуемого вещества
(гл. 5). Эти затруднения могут быть связаны с тем, что при опре-
делении растворимости не удается выявить какие-либо изменения
визуально или вследствие того, что образование эмульсий или
изменение окраски растворов мешает оценить, в какой степени
протекает процесс растворения. В таких случаях при изучении
растворимости может оказать помощь газовая хроматография.
Например, с помощью этого метода можно определить очень ма-
лые примеси растворителя и таким образом оценить раствори-
мость вещества в тех случаях, когда она оказывается достаточно
малой. Метод газовой хроматографии можно применять для оп-
ределения растворимости следующим образом. Растворяемое ве-
щество и растворитель подготавливают в таких количествах, ко-
торые были указаны в разд. 5.1 «Определение растворимости»
Разделение органических веществ
471
Таблица 7.9
Отношение массы растворяемого вещества к объему растворителя
Масштаб Твердые вещества Жидкие вещества
Макрометод 100 мг/3 мл 0,2 мл/3 мл
Полумикрометод 30 мг/1 мл 1 капля/15 капель
или приведены в табл. 7.9. Затем стараются растворить исследуе-
мое вещество в растворителе, используя все доступные средства,
в первую очередь интенсивное перемешивание и встряхивание.
Если вещество хорошо растворимо в применяемом растворителе,
так что классификацию по растворимости можно без труда про-
вести на основе визуальных наблюдений, то применять для тако-
го анализа метод газовой хроматографии нет необходимости. Если
это не так, то вначале и для растворителя, и для растворяемого
вещества в одних и тех же экспериментальных условиях опре-
деляют газохроматографические времена удерживания и мольные
отклики детектора (см. ниже). Затем в хроматограф вводят про-
бы объемом 2—10 мкл, отобранные из каждого жидкого слоя,
возникающего при проверке растворимости.
Условия проведения хроматографического разделения должны
быть выбраны таким образом, чтобы площади пиков и раствори-
теля, и растворенного вещества можно было бы измерять при
одних и тех же условиях. Мольные отклики (см2/моль) можно
измерить, вводя в хроматограф известные количества чистого рас-
творителя (с известной плотностью) и стандартного раствора с
известной концентрацией изучаемого вещества. Для вычисления
мольного отклика делят площадь пика (см2) на число молей со-
ответствующего этому пику вещества. Если мольные отклики рас-
творителя и растворенного вещества одинаковы, то отношение
площадей пиков этих веществ позволяет непосредственно опреде-
лять количество изучаемого соединения, перешедшее в раствор.
Если же мольные отклики не равны, то измеренные площади пи-
ков нужно перевести в количества соответствующих компонентов,
используя величины мольного отклика каждого из них. Перевод
отношения площадей пиков (ПП) двух компонентов в соотно-
шение чисел молей каждого из них можно сделать с помощью
приведённого ниже соотношения, используя соответствующие ве-
личины мольного отклика (МО):
число молей компонента 1 (ПП)1/(МО)|
число молей компонента 2 (ПП)2/(МО)2
Так, например, если компонент 1 представляет собой веще-
ство, для которого необходимо определить растворимость, а ком-
понент 2 — растворитель, то приведенная формула позволяет оп-
ределить растворимость компонента 1.
472
Глава 7
Рис. 7.36. Зависимость газохроматографических удельных объемов удерживания
углеводородов и спиртов на колонке с 20% полисилоксана SE-30 от температур
кипения (перепечатано из книги: Crippen R. С., The Identification of Organic
Compounds with the Aid of Gas Chromatography, McGraw-Hill, New York, copy-
right © 1973).
А к-алканы; Q спирты нормального строения; □ метилбензолы.
С помощью газовой хроматографии можно определить нали-
чие воды в исследуемых органических соединениях *. Для этого
пробу раствора изучаемого твердого вещества вводят в газохро-
матографическую колонку с неполярной неподвижной фазой (на-
* Присутствие воды в жидких веществах можно обнаружить, добавляя к
жидкости безводный сульфат меди, имеющий белую окраску, и наблюдая появ-
ление синего цвета, обусловленного гидратом сульфата меди. Удалить воду
можно с помощью молекулярных сит, безводного сульфата кальция или без-
водного сульфата магния. Для обезвоживания твердых веществ применяют
эксикаторы и сушильные пистолеты (рис. 4.1).
Разделение органических веществ
473
пример, с силиконовым маслом DC-710 или полимерным сорбен-
том углеводородного характера «порапак Q»). При этом на хро-
матограмме будет зарегистрирован пик воды. При выполнении
газохроматографического определения воды в органических со-
единениях нельзя использовать пламенно-ионизационный детектор,
а следует применять детектор по теплопроводности. При выборе
Рис. 7.37. Хроматограмма смеси алифатических спиртов, бензилового спирта и
оксикоричной кислоты на колонке размером 4 м X 6 мм с полиэтиленгликолем
«карбовакс-20М» (20%) при 225 °C (перепечатано из книги: Crippen R. С., The
Identification of Organic Compounds with the Aid of Gas Chromatography,
McGraw-Hill, New York, copyright © 1973).
/ — метанол; 2 — этанол; 3—н-пропанол; 4— н-бутанол; 5 — н-пентанол; 6—-н-гексанол; 7 —бензи-
ловый спирт; 8— оксикорнчная кислота-
газохроматографической колонки для анализа определенной груп-
пы веществ нужно учитывать следующее. На относительно инерт-
ных колонках, например, таких, как колонка с полисилоксаном
SE-30 (рис. 7.36), времена удерживания, а значит, и объемы удер-
живания оказываются больше для тех соединений, которые имеют
более высокие температуры кипения. На рис. 7.36 это показано
на примере алканов, алкилбензолов и алифатических спиртов.
На полярных колонках, например, таких, как колонки с поли-
этиленгликолем «карбовакс 20М», форма пиков может изме-
няться в зависимости от характера ассоциации молекул (от на-
личия водородных связей) и от величины молекулярной массы.
Например, на рис. 7.37 заметно, что бензиловый спирт и в еще
большей степени оксикорнчная кислота образуют широкие пики,
что связано с влиянием перечисленных выше двух факторов, а
также с большими величинами времени удерживания.
474
Глава 7
7.5. РАЗДЕЛЕНИЕ ОПТИЧЕСКИХ ИЗОМЕРОВ
Если предполагают, что неизвестное вещество может обладать
строением, придающим ему хиральные свойства, следует измерить
его оптическую активность (разд. 8.1). Сопоставление удельного
вращения измеренного в том же растворителе и в растворе при-
мерно той же концентрации, с литературными данными может
служить для оценки степени чистоты изучаемого соединения и
для доказательства идентичности его строения со строением,
предполагаемым на основании других данных. Отсутствие опти-
ческой активности у данного соединения еще нельзя считать до-
казательством отсутствия у него хиральных свойств, поскольку
данное вещество либо может представлять собой рацемическую
смесь двух оптически активных компонентов, либо наличие в нем
хирального центра обусловливает очень слабую оптическую ак-
тивность Подтверждением хиральной природы неизвестного со-
единения является разделение энантиомеров, входящих в состав
рацемической смеси.
Таблица 7.10
реактивы и методы, используемые для разделения оптических изомеров,
и соответствующие литературные источники
Класс соединений Реактив или метод Литера- тура а
Спирты Амины, фталевые эфиры спиртов 1
Альдегиды, кетоны Комплексы с тартратами и сульфа- тами аминов 2
Ментил-М-амннокарбонаты 1
Алкены Комплексы платины 1
Амины (+)-Винная кислота 2
Аминокислоты а-Фенилэтиламин 2
Хроматография на бумаге 1
Ароматические соединения Колоночная хроматография на таг- 1
(бифенилы) тозе или картофельном крахмале
Карбоновые кислоты а-Фенилэтпламин 2
Эфиры аминокислот Газовая хроматография на непо- движных фазах пептидного ха- 1, з
Соли кислот Ионообменная хроматография 1
а I. Boyle Р. Н., Quart. Rev. (London), 1971, 323; обширный обзор, охватывающий боль
шое число ссылок на литературу и позволяющий получить сведения об оригинальных рабо-
тах по разделению оптических изомеров органических соединений разных классов.
2. Ault A., Org. Syn. Coll., Vol. 5, ed. H. E. Baumgarten, Wiley, New York, 1973, p. 932.
См. также Wilen S. H., Resolving Agents and Resolution in Organic Chemistry, University
ot Notre Dame Press. Notre Dame, Ind., 1971.
3. Забокрицкий M. П., Руденко Б. А., Чижков В. IL, Изв. АН СССР, сер. хим., 1981,
с. 1045; ДАН СССР 263, 1982, с. 1155.
Разделение органических веществ
475
Описан способ разделения оптических изомеров а-фенилэтил-
амина *:
СИ, \/
С6Н5СН—nh2
СНз nh2
рацемат R<+'
[а]о6С= +40“Счистбш)
Н\ Д-6И5
с
н2г/ \нэ
S(-)
(а]п5С = -4СГ(чистый)
Процедура разделения включает обработку рацемата (+)-винной
кислотой, которая имеется в качестве реактива в большинстве
органических лабораторий. В табл. 7.10 перечислены методы раз-
деления оптических изомеров соединений с различными функцио-
нальными группами.
* Ault A., Org. Syn. Coll., Vol. 5, ed. H. E. Baumgarten, Wiley, New York,
1973, p. 932. После публикации этого метода в указанной книге он неоднократно
подвергался независимой проверке. Большинство других методов разделения оп-
тических изомеров, описанных в химических журналах, такой проверке не под-
вергалось.
Глава 8
Специальные методы характеристики
органических веществ
В данной главе мы рассмотрим методы идентификации ве-
ществ, которые обычно не применяются при стандартном анализе
органических соединений. К методам, описанным в этой главе,
прибегают только после того, как соединение проанализировано
методами, приведенными в гл. 6. Результаты реакций, рассмот-
ренных в гл. 6, и общее понимание значения этих методов будут
служить руководством для выбора специальных методов, которые
описаны ниже.
8.1. ОПТИЧЕСКОЕ ВРАЩЕНИЕ
Оптическое вращение определяют только в том случае, если
среди предполагаемых соединений содержатся оптически актив-
ные вещества.
8.1.1. Приготовление раствора
Методика. Тщательно взвешенный образец массой 0,1—0,5 г растворяют
в мерной колбе в 25 мл растворителя. Обычно в качестве растворителей исполь-
зуют воду, этанол и хлороформ. Раствор должен быть прозрачным, не содер-
жать суспендированных частичек пыли или волокон фильтровальной бумаги и
быть по возможности бесцветным. Если раствор непрозрачен, то либо исходное
соединение должно быть перекристаллизовано, либо можно приготовить еще
50 мл раствора и отфильтровать его через небольшой сухой бумажный фильтр.
Первые 25 мл фильтрата отбрасывают, последние 25 мл используют для опреде-
ления.
8.1.2. Заполнение поляриметрической трубки
Методика. На тонкий конец поляриметрической трубки (7, рис. 8.1) навин-
чивают насадку, трубку ставят вертикально и заполняют раствором до тех пор,
пока над верхним концом трубки не образуется округлый мениск. На конец
трубки надвигают стеклянную пластинку так, чтобы в трубке не оставалось пу-
зырьков воздуха, а затем навинчивают латунную насадку.
Предосторожности. 1. Между стеклянной пластинкой и латунной насадкой
должна быть резиновая прокладка. Нельзя класть прокладку между стеклянной
пластинкой и концом трубки. В этом месте должен быть контакт стекло —
стекло.
2. Насадки на концах трубки нельзя завинчивать слишком сильно. Необ-
ходимо только обеспечить достаточно плотный контакт, чтобы раствор не выли-
вался. Если концы завинчены слишком сильно, стеклянные пластинки, закры-
вающие трубку, испытывают напряжение и оптическое вращение наблюдается
Специальные методы характеристики органических веществ 477
даже при пустой трубке. Для веществ с очень малым вращением целесообразно
между измерениями ослаблять насадки и снова завинчивать их перед измере-
нием.
3. Если латунные насадки прилегают к стеклянной трубке неплотно, то они
могут быть укреплены в нужном положении с помощью замазки из оксида свин-
ца с глицерином. При этом необходимо следить, чтобы стеклянная часть находи-
лась на расстоянии 1 мм от латунной насадки.
8.1.3. Работа с поляриметром
Один из распространенных типов поляриметров — прибор Липпиха с двой-
ным полем. Схема прибора показана на рис. 8.1 и 8.2.
Методика. С помощью выключателя 2 включают осветитель 1. Фокусируют
окуляр 12, передвигая его направо или налево до тех пор, пока линия раздела
двух полей не будет четкой.
Для определения нулевого значения ослабляют фиксирующий винт 16 и
выравнивают освещенность полей с помощью рукоятки грубой регулировки
Рис. 8.1. Схема поляриметра.
/-—источник света (натриевое пламя или электрическая натриевая лампа); 2—выключатель;
3—дихроматический фильтр; 4—поляризующая призма Николя; 5— полутеневая призма
Николя; 6—полутеневой регулятор: 7 — трубка с раствором; 8—шкала; 9 —анализирующая
призма Николя; 10—установка нуля; 11 — шкала; 12—окуляр.
14. Для работы с рукояткой 14 необходимо сначала ослабить винт 16 и затем
сдвинуть рукоятку 14 поворотом белой кнопки на конце этой рукоятки. После
примерного выравнивания освещенности полей винт 16 закрепляют, и яркость
полей уравнивают окончательно вращением направо или налево микрометриче-
ского регулятора 15. Включают лампу 13 и считывают показания шкалы. Основ-
ной круг разделен на градусы и 0,25 градуса. Нониус или наружная (внешняя)
шкала разделена на 25 делений, дающих возможность получать значения до
0,01 градуса. Измерения необходимо произвести не менее пяти раз и получае-
мые данные усредняют. Для очень точных работ показания считывают с обеих
шкал 11, а затем их усредняют. При этом исключается ошибка за счет непра-
вильного положения (неточной центровки) кюветы по отношению к окуляру и
призмам Николя.
Для измерения вращения раствора в держатель помещают поляриметриче-
скую трубку. Закрывают крышку и повторяют все операции, описанные для оп-
ределения нулевого значения. Проводят не менее пяти измерений и полученные
данные усредняют. Наблюдаемое вращение вычисляют как разность между по-
лученным и нулевым значением.
478
Глава 8
Замечания и предосторожности. 1. Обычно прибор работает на желтом
свете, соответствующем D-линии натрия. Свет такой длины волны легко полу-
чить с помощью электрической натриевой лампы накаливания, дающей яркий
Рис. 8.2. Поляриметр.
10—установка нуля; // — шкала; 12—окуляр;
13 — лампа; 14— грубая регулировка*»
15 — микрометрический регулятор; 16— фик-
сирующий винт.
4. Рукоятка 14 и контргайка 16
ио. Необходимо только закрепить их,
желтый свет, можно также использо-
вать натриевое пламя, но его яркость
существенно меньше. Для соединений с
небольшим наблюдаемым вращением хо-
рошие результаты получаются при ис-
пользовании белого света, пропущенного
через 3-сантиметровую кювету с раство-
ром-фильтром. Этот раствор состоит из
8,9 г водного сульфата меди, 9,4 г би-
хромата калия и 300 г воды. Раствор
фильтруют и выдерживают некоторое
время для осаждения частичек пыли.
Ртутная дуга дает возможность ис-
пользовать зеленую линию ртути. При
использовании зеленой линии ртути
осветитель 1 удаляют, дихроматический
фильтр 3 отвинчивают, а дугу помещают
на место осветителя 1 и повторяют все
описанные выше операции.
2. После того как прибор установ-
лен и отрегулирован для всех обычных
работ, положение полутеневого регуля-
тора 6 и винта 10 не следует изменять.
3. При считывании показаний лучше
начинать со значений шкалы 11 вблизи
нуля. Если рукоятка 14 сдвинута слиш-
ком сильно, происходит обращение
полей.
должны быть завинчены слишком силь-
прилагая чрезмерных усилий.
8.1.4. Описание полученных результатов
Удельное вращение вещества вычисляется по одной из сле-
дующих формул:
Для чистой жидкости Для раствора
Гп125 °C_ а [п125 °C _]00а
ДЪ id 1аЬ — 1С
где [а]о’С— удельное вращение при 25°С (для D-линии натрия),
а — наблюдаемое вращение, I — длина трубки (дм), d — плотность
(г/мл) и с—-концентрация (г/100 мл раствора).
Следует отметить, что удельное вращение может быть очень
чувствительно к природе растворителя и в некоторых случаях
даже к концентрации исследуемого вещества. Длина волны све-
та, используемого при измерениях, также может оказывать влия-
ние не только на величину, но и на знак вращения. Поэтому
необходимо обращать внимание на точное соблюдение условий,
при которых были проведены измерения вращения, описанные в
литературе.
Специальные методы характеристики органических веществ
479
Правильный способ представления удельного вращения сле-
дующий *:
[а]®6°с= — 40 ± 0,3° (с = 5,44 г/100 мл воды)
Приведенная запись относится к удельному вращению, опре-
деленному при 25°С в воде для света с длиной волны 546 нм при
концентрации раствора 5,44 г на 100 мл. Необходимо определить
наблюдаемое вращение а для двух различных концентраций. В
простейшем случае наблюдаемое вращение уменьшается во
столько раз, во сколько уменьшается концентрация, например:
а, град
-50
-5,0
0,50
Концентрация
х
0,1х
0,01х
Таким образом, в этом случае величина [а], определенная из
трех экспериментов, будет одна и та же, и экспериментатор по-
лучит величину [а], которую можно сравнивать с литературными
значениями [а], определенными при других концентрациях**. Та-
ким путем на основании значений [а] можно идентифицировать
исследуемое вещество. Если эту величину для жидких проб опре-
деляют без растворителя, то удельное вращение записывают в виде
[а]и °с _ 40° (ЧИстое).
8.2. УЛЬТРАФИОЛЕТОВАЯ СПЕКТРОСКОПИЯ
Целесообразность использования ультрафиолетовой спектроско-
пии химиками-органиками станет яснее после сопоставления ме-
тодов ИК-, ЯМР- и УФ-спектроскопии. При характеристике ор-
ганического соединения химик обычно применяет ИК-спектроско-
пию для определения и идентификации функциональных групп
и ЯМР-спектроскопию для определения структурного окружения
протонов и атомов углерода. Составив предварительное представ-
ление о структуре соединения, исследователь решает, будет ли
* Необходимо указывать все эти параметры. Однако часто встречаются
работы, в которых отсутствуют указания о концентрации или применяемом рас-
творителе. Необходимо также сообщать метод определения интервала ошибок
(в данном случае ±0,3°). Например, следует указать, составляет ли погреш-
ность плюс или минус одно стандартное отклонение или оценка ошибки полу-
ченных результатов основана на характеристиках применяемого метода измерений.
Ошибка, приведенная в данном примере, велика по сравнению с ошибками бо-
лее современных приборов.
** В некоторых случаях величина удельного вращения зависит от концен-
трации, как, например, для полярных хпральных субстратов в спиртовых рас-
творителях вследствие образования водородных связей, которое зависит от кон-
центрации. Поэтому очень важно указывать концентрацию образца.
480
Глава 8
УФ-спектроскопия полезна для характеристики соединения. На-
личие кратных связей, особенно сопряженных, является указанием
на целесообразность применения У’Ф-спектроскопии. Изучение ма-
териала данной главы даст представление о том, когда и в каких
случаях следует применять этот метод.
Хотя подробное обсуждение ультрафиолетовой спектроскопии
выходит за рамки этой книги, однако мы укажем типичные за-
дачи, возникающие при изучении строения веществ, которые мо-
гут быть решены с помощью этого метода. Подробно применение
УФ-спектроскопии для структурных исследований изложено в сле-
дующих работах:
Jaffe Н. Н., Orchin М., Theory and Applications of Ultraviolet
Spectroscopy, Wiley, N. Y., 1962.
Гиллем А., Штерн E. Электронные спектры поглощения орга-
нических соединений. Пер. с англ.— М.: ИЛ, 1957.
Штерн Э., Тиммонс К. Электронная абсорбционная спектро:
скопия в органической химии. Пер. с англ.— М.: Мир, 1979.
Наиболее полными и чаще всего используемыми источниками
информации для химиков-органиков являются следующие книги:
Braude Е. A., Determination of Organic Structures by Physical
Methods, edited by Braude E. A. and Nachod F. C., Academic Press,
New York, 1955, chap. 4.
Mason S. F., Quart. Rev., 15, 287 (1961).
Введение в теорию поглощения ультрафиолетового и видимого
излучения хорошо изложено в книгах:
Hershenson Н. М., Ultraviolet and Visible Absorption, Academic
Press, New York, 1955. Эта книга является относительно простым
описанием спектров, опубликованных в 1930—1957 гг.
Стрейтвизер Э. Теория молекулярных орбит. Для химиков-ор-
гаников. Пер. с англ.— М.: Мир, 1965. Здесь ультрафиолетовая
спектроскопия рассматривается с использованием теории молеку-
лярных орбиталей.
Ультрафиолетовая область обычно подразделяется на ближ-
нюю, или «кварцевую», УФ-область (область, где воздух и кварц
прозрачны для излучения), которая охватывает интервал 200—
750 нм (2000—7500 А), и далекую, или «вакуумную», УФ-область.
Спектрофотометры Сагу и Beckman измеряют спектры в ближней
области. Измерения в области длин волн ниже 200—220 нм мо-
гут быть проведены достаточно надежно только в специальных
условиях. Эти доступные приборы могут работать до 175 нм. Ра-
бота в далекой ультрафиолетовой области требует специального
оборудования вследствие сильного поглощения излучения возду-
хом.
Ультрафиолетовый спектр в отличие от инфракрасного или
спектра комбинационного рассеяния (раман-спектра) возникает
вследствие электронного возбуждения молекулы под действием
падающего света. Важным с точки зрения структурных исследо-
Специальные методы характеристики органических веществ
481
ваний следствием этого является тот факт, что по поглощению
в ультрафиолетовой области можно обнаружить в молекуле на-
личие электронов кратных связей или несвязывающих электронов.
За некоторым исключением, это вызвано тем, что только моле-
кулы, содержащие кратные связи, имеют достаточно устойчивые
возбужденные состояния, переход в которые вызывает поглоще-
ние в ближней ультрафиолетовой области. Следовательно, насы-
щенные углеводороды, спирты и простые эфиры прозрачны в
этой области. Более того, монофункциональные олефины, ацети-
лены, карбоновые кислоты, сложные эфиры, амиды и оксимы
имеют максимумы поглощения за пределами ближней ультрафио-
летовой области (ниже 200 нм), и для них наблюдается только
концевое поглощение.
Третий класс соединений, включающий альдегиды, кетоны,
алифатические нитросоединения и эфиры азотной кислоты, харак-
теризуется максимумами поглощения в ближней ультрафиолето-
вой области, но интенсивность поглощения очень мала, так что
обычно его можно наблюдать только в специальных условиях.
Например, молярное поглощение ацетона равно 16 (е — мера ин-
тенсивности поглощения) в максимуме полосы при 270 нм. Для
сравнения сильная полоса имеет е 1000—10 000; не редки вели-
чины е порядка 100000. Тем не менее ультрафиолетовая спектро-
скопия иногда бывает очень полезна и для изучения слабо по-
глощающих функциональных групп. В табл. 8.1 приведены ти-
пичные данные о положении максимума поглощения и моляр-
ного коэффициента поглощения некоторых изолированных (несо-
пряженных) функциональных групп.
Таблица 8.1
Функциональная группа ^макс’ HM e
RCH=CHR 185 8 000
R2C=O 270 16
RCOOH 204 41
RCQC1 235 53
rno2 275 12
r2s 210 1200
r2c=nnhconh2 229 10 900
RN=NR 350 140
RSN ( 210 500
I 270 60
RN=O f 300 t 670 1Q0 20
Можно показать, что использование ультрафиолетовой спек-
троскопии для монофункциональных молекул довольно ограни-
ченно. Однако, к счастью, такие функциональные группы, как
16 Зак. 335
482
Глава 8
олефиновая, ацетиленовая и карбоксильная (и в общем любая функ-
циональная группа, содержащая двойную или тройную связь),
при их взаимном сопряжении сильно поглощают излучение в
ближней ультрафиолетовой области. Таким образом, ультрафио-
летовая спектроскопия является основным методом изучения со-
пряженных систем *. Из изложенного ясно, что использование
ультрафиолетовых спектров для структурных определений силь-
но отличается от метода инфракрасной спектроскопии; в действи-
тельности оба метода часто дополняют друг друга.
Современный двухлучевой УФ-спектрофотометр обычно реги-
стрирует зависимость поглощения (оптической плотности) от дли-
ны волны в нанометрах. Поскольку поглощение (Л) данного рас-
твора является количественной характеристикой в ультрафиолето-
вой спектроскопии и поскольку используемые растворы обычно
достаточно разбавлены и подчиняются закону Бера, при публи-
кации данных целесообразно пересчитывать величину поглощения
в значения молярного коэффициента поглощения (е)**. Эта ве-
личина определяется по следующему уравнению:
А
е— ьс
где b — длина кюветы в сантиметрах (обычно 1 см), а С — кон-
центрация в молях на литр. Ясно, что е является мерой погло-
щения раствора концентрации 1 моль/л в кювете толщиной 1 см,
и значения е различных веществ можно сравнивать друг с другом,
даже если спектры сняты при различных концентрациях.
Таблица 8.2
Растворители, применяемые в УФ-спектроскопии
Растворитель Нижний предел пропускания, нм
Вода 205
Этанол (95%-ный или абсолютный) 210
Гексан 210
Циклогексан 210
Метанол 210
Диэтиловый эфир 210
Ацетонитрил 210
Тетрагидрофуран 220
Днхлорметан 235
Хлороформ Тетрахлорметан Бензол 245 265 280
* Ferguson L. N., Nnadi J. С., J. Chem. Educ., 42, 529 (1965).
** Молярный коэффициент поглощения (е) имеет размерность см®/моль.
Таким образом, эта величина выражается как поперечное сечение молекулы по
отношению к фотонам, энергия которых соответствует данной длине волны.
Специальные методы характеристики органических веществ
483
В ультрафиолетовой спектроскопии в качестве растворителей
обычно используют циклогексан, 95%-ный этанол, абсолютный
этанол (не содержащий бензола) и диоксан. В табл. 8.2 приве-
дены растворители для УФ-спектроскопии, расположенные в по-
рядке уменьшения их практической значимости. Необходимые
для измерения концентрации растворов определяются из ожидае-
мых максимальных значений е. Так как шкала поглощения при-
бора Сагу имеет пределы от 0,0 до 2,4 единицы поглощения, то,
считая, что при максимальном значении е — 104 поглощение дол-
жно быть равно 2,0, можно выбрать правильную концентрацию,
равную 2/10*, или 2-10-*, молярной, при толщине кюветы 1 см.
8.2.1. Идентификация функциональных групп по положению мак-
симумов поглощения
Использование спектроскопии для идентификации функцио-
нальных групп на основании положения максимумов поглощения,
как в инфракрасной области, в ультрафиолетовой области при-
меняется редко по двум причинам. Во-первых, наиболее важные
функциональные группы поглощают слабо или вообще не погло-
щают, а во-вторых, спектры большинства молекул сравнительно
просты. Обычно они имеют только один или два максимума вме-
сто 10 или 20, как это характерно для ИК-спектров. Поэтому не-
избежно, что многие типы функциональных групп поглощают в
одной и той же области. Тем не менее изучение ультрафиолето-
вых спектров в ряде случаев позволяет опытному исследователю
выявить ранее не обнаруженные функциональные группы; обычно
это ароматические и гетероциклические кольца, присутствующие в
природных соединениях неизвестной структуры. Типичным приме-
ром может служить обнаружение с помощью ультрафиолетовых
спектров нитрофенильных групп в хлоромицетине. Однако в об-
щем, прежде чем использовать ультрафиолетовые спектры, необ-
ходимо иметь некоторые сведения о возможных функциональных
группах. Конечно, часто можно однозначно сделать заключение
об отсутствии функциональных групп, поглощающих в ультрафио-
летовой области.
8.2.2. Использование модельных соединений
На первый взгляд может показаться, что ультрафиолетовая
спектроскопия как будто мало полезна для структурных исследо-
ваний, поскольку только лишь отдельные функциональные груп-
пы поглощают в ультрафиолетовой области, а непоглощающие
структурные элементы спектрофотометр не обнаруживает. Одна-
ко несмотря на то, что спектр молекулы почти никогда невозмож-
но предсказать теоретически, часто может быть получен спектр
«модели», представляющей собой более простое вещество, которое
16*
484
Глава 8
отличается по структуре от неизвестного вещества, но таким обра-
зом, что на спектре это не сказывается.
Примером использования модельных соединений для расшиф-
ровки ультрафиолетовых спектров является структурное исследо-
вание дицианпроизводных хинолина *. Хинолин реагирует с бром-
цнаном и цианистым водородом, давая два дицианпроизводных,
Рис. 8.3. Спектр поглощения легкоплавкого дициаиида хинолина (I), феиил-
циаиамида (III) и N-метилфеиилциаиэмида (IV) (перепечатано из Journal of
the American Chemical Society с любезного разрешения Общества и проф.
К. Ноллера).
которым приписывались структуры I и II. Было необходимо по-
лучить подтверждение этого предположения и, кроме того, уста-
новить, какое из полученных дицианпроизводных хинолина имеет
структуру I, а какое — структуру II. Поскольку не было подхо-
дящих синтетических методов и отсутствовали способы разделе-
ния изомеров, были изучены ультрафиолетовые спектры этих ве-
ществ. В качестве модели «дицианида хинолина» (I) был выбран
• Seeley М. G., Yates /?. £., Nailer С. R., J. Am. Chem. Soc., 73, 772 (1951).
Специальные методы характеристики органических веществ
485
фенилцианамид(Ш) и N-метилфенилцианамид (IV). Из рис. 8.3
видно, что общий контур кривых, положение и высота макснму-
Рис. 8.4. Спектр поглощения высокоплавкого дицианида хинолина (И) и N-ме-
тил-о-стирилцианамида (V) (перепечатано из Journal of the American Chemical
Society с любезного разрешения Общества и проф. К. Ноллера).
мов для всех соединений находятся в хорошем соответствии. Сле-
дует отметить, что дицианамид хинолина I содержит структурный
элемент а.р-ненасыщенного нитрила, который отсутствует в мо-
дельных соединениях. Наличием этой группы можно пренебречь,
486
Глава 8
так как ее поглощение намного меньше, чем поглощение других
структурных элементов.
Моделью для другого дицианида хинолина(II) был выбран
Ы-метил-о-стирилцианамид(У). Спектры II и V показаны на
рис. 8.4. Видно, что они хорошо соответствуют друг другу и от-
личаются от спектров, приведенных на рис. 8.3. Таким образом,
с помощью ультрафиолетовых спектров получено четкое дока-
зательство структуры дицианпроизводных хинолина.
8.2.8. Принцип аддитивности
При решении структурных проблем наиболее плодотворным
аспектом ультрафиолетовой спектроскопии является принцип ад-
дитивности, который может быть объяснен следующим образом.
Спектр молекулы, содержащей два поглощающих структурных
элемента, разделенных одним или большим числом изолирующих
атомов (например, группой —СН2—), примерно соответствует
сумме спектров поглощающих структурных элементов *. НиЖе
приведено несколько примеров использования принципа аддитив-
ности.
При установлении структуры террамицина ** важным продук-
том деструкции было соединение VI, часть структуры которого
была установлена, а часть, заключенная в квадратные скобки,
Не была определена. Исследователи предполагали, что
'[СбН2((ЭСНз)з] является остатком одного из изомеров тримет-
оксибензола. Из формулы видно, что два хромофора — нафтали-
новая система и бензольная система — изолированы друг от друга
группой —СИОН— (не сопряжены между собой), и поэтому
спектр молекулы может быть представлен как сумма спектров
обеих этих частей. В соответствии с этим авторы просуммировали
имеющийся спектр вещества VII последовательно со спектрами
1,2,3-, 1,2,4- и 1,3,5-триметоксибензолов. Только в случае 1,2,4-
* Braude Е. A., J. Chem. Soc., 1949, 1902. Хотя одна метиленовая группа
в первом приближении и изолирует два хромофора, для хорошего выполнения
принципа аддитивности необходимы две или большее число метиленовых Групп
(см. ссылки иа литературу, приведенные в начале разд. 8.2).
** Stephens С. R., Conover L. И. Е., Pasternack R., Hochstein F. A., More-
land W. T., Regna P. P., Pilgrim F. J., Brunings K. J., Woodward R. B., J. Am,
Chem. Soc., 76, 3568 (1954),
Специальные методы характеристики органических веществ
487
производного суммарная кривая достаточно хорошо соответство-
вала спектру вещества VI
СН3
СН2ОН
VII
Мак-Кензи *, изучая структуру усниновой кислоты, исследовал
спектры продуктов ее деструкции (VIII и IX). Он заметил, что
если предложенная структура верна, то продукт VIII должен со-
держать хромофор IX, соединенный с изолированным хромофо-
ром ацетилацетона в енольной форме (X). Поэтому он вычел
спектр IX из спектра VIII и получил кривую, которая совпадала
со спектром ацетилацетона. Таким образом предложенная струк-
тура была подтверждена.
СН3С=СНССН3
I
ОН
8.2.4. Влияние геометрии молекулы на ультрафиолетовые спектры
поглощения
При выборе модельного соединения для сравнения его спект-
ра поглощения со спектром неизвестного соединения часто встре-
чаются затруднения, связанные с возможными неожиданными
изменениями в спектре одного из сравниваемых веществ, вызван-
ными стерическими эффектами. Наиболее обычной причиной та-
ких затруднений являются стерические препятствия для резонан-
са. Например, спектр бифенила не является суммой спектров
двух бензольных колец вследствие того, что резонансное взаимо-
действие двух сопряженных бензольных колец стабилизирует воз-
бужденное состояние, возникающее при поглощении света. С другой
Mackenzie S., J. Am. Chem. Soc.. 74, 4067 (1952).
488
Глава 8
стороны, было показано, что такое резонансное взаимодей-
ствие может происходить при копланарном расположении двух
колец в бифениле. Для димезитила (XI), имеющего достаточно
объемистые метильные группы в каждом из четырех орто-положе-
ний, невозможно предположить существование конформации с
двумя копланарными фенильными кольцами. Поэтому резонанс-
ное взаимодействие между двумя бензольными кольцами отсут-
ствует, и вследствие этого спектр димезитила очень похож на
сумму спектров* двух изолированных мезитиленовых колец и от-
личается от спектра бифенила **.
Изучение спектров орто-замещенных нитробензолов показы-
вает, что при увеличении размера орто-заместителей нитрогруппа
все сильнее выводится из плоскости бензольного кольца, что при-
водит к постепенному изменению спектра; в конечном итоге макси-
мум при 265 нм, имеющий у n-трет-аллилнитробензолов е более
10 000, полностью исчезает в о-нитро-трет-бутилбензоле ***.
Аналогичные стерические препятствия резонансу -наблюдались
при исследовании влияния орто-заместителей на спектры произ-
водных анилина ****. Характерным примером служит бензохи-
нуклидин (XII), в котором атом азота, включенный в жесткий
углеродный скелет, находится в таком положении, при котором
сильно затрудняется резонансное взаимодействие между амино-
группой и бензольным кольцом. Как и следовало ожидать, уль-
трафиолетовый спектр бензохинуклидина больше похож на спектр
бензола, чем на спектр диметиланилина *****.
XII
* Напомним, что обычно интенсивность поглощения активного в ультра-
фиолетовой области соединения прямо пропорциональна числу изолированных
хромофоров, вызывающих поглощения. Например, поглощение в области бен-
зольных колец соединения с четырьмя изолированными бензольными кольцами
в 2 раза интенсивнее, чем соединения с двумя изолированными бензольными
кольцами.
** Pickett L. W., Walter G. F„ France H., J. Am. Chem. Soc., 58, 2296(1936);
O'Shaughnessy M. T., Rodebush W. H., ibid., 62, 2906 (1940).
**• Brown W. G., Reagan H., J. Am. Chem. Soc., 69, 1032 (1947).
**** Remington W. R., J. Am. Chem. Soc., 67, 1838 (1945).
***** Wepster В. M., Rec. Trav. Chim., 71, 1159 (1952).
Специальные методы характеристики органических веществ
489
Заметное изменение ультрафиолетовых спектров может проис-
ходить не только из-за стерических препятствий резонансу, но и
вследствие таких стерических факторов, которые изменяют взаим-
ное расположение диполей (полярных групп) в молекуле. По-
добный пример будет приведен в разд. 8.2.6 при обсуждении влия-
ния положения атома хлора в а-хлоркетонах на ультрафиолетовое
поглощение карбонильной группы. Исследование а-дикетонов *
показало, что на их спектры очень сильное влияние оказывает
взаимная ориентация карбонильных групп. В общем при выборе
модельных соединений, используемых в ультрафиолетовой спек-
троскопии, необходимо проявлять осторожность. Другой эффект,
сильно зависящий от геометрии молекулы, обусловлен взаимо-
действием несвязанных хромофоров, расположенных соответ-
ствующим образом в пространстве. Например, дикетоолефин
(XIII) имеет максимумы поглощения при 223 нм (1g е 3,36) и
при 296 и 307 нм (1g е 2,41). Инфракрасные спектры показывают,
что это соединение не енолизовано, и необычно интенсивное по-
глощение объясняется трансаннулярным взаимодействием олефи-
новой двойной связи с карбонильными группами **.
XIII
Это и другие взаимодействия несвязанных хромофоров рас-
смотрены и обсуждены***.
8.2.5. Более тонкие корреляции структуры с ультрафиолетовыми
спектрами
Ряд исследований посвящен влиянию на ультрафиолетовые
спектры числа двойных или тройных связей в системах типа
X—(СН=СН)п—Y и X—(С)„—Y. Поскольку такой материал из-
ложен в книге Гиллема и Штерна (см. список общей литерату-
ры, приведенный в начале разд. 8.2), здесь он обсуждаться не
будет.
Более важна для химиков, изучающих природные вещества,
количественная корреляция ультрафиолетовых спектров со струк-
турой, предложенная Вудвордом ****, который создал эмпириче-
* Leonard N. J., Mader Р. М., J. Am. Chem. Soc., 72, 5388 (1950).
** Grob С. A., Weiss A., Helv. Chim. Acta, 43, 1390 (I960).
*** Wilcox C. F., Winstein S., McMillan W. G., J. Am. Chem. Soc., 82, 5450
(1960).
**** См. Физер Л., M. Стероиды. Пер. с англ. — М.: Мир, 1964, или общие
руководства, приведенные в начале этого раздела.
490
Глава 8
Таблица 8.3
Правило Вудворда для сопряженных диенов (растворитель — этанол)
Ациклические и гетероаннулярные диены Гомоаииулярные диены Прибавляют на каждый заместитель (нм) — R алкил (включая части карбоциклических колец) —OR алкоксигруппа — SR тиоэфириая группа — С1, —Вг — ОС (=О) R ацилоксигруппа — СН=СН—, участвующая в сопряжении Одна двойная связь, экзоцпкличиая к одному кольцу Одна двойная связь, экзоцикличная одновременно к двум кольцам Возможный сдвиг за счет влияния растворителя пренебрежимо мал. еыакс 6000—35 000 215 им < 253 нм 1 +5 . +6 I +30 I +5 i +0 ‘ +30 +5 +10
Таблица 8.4
Правило Вудворда для а, Р-ненасыщенных карбонильных
соединений (растворитель — этанол)
а
I
Кетоны —С=С—СО— ациклические илн 6-члеииые
кольца
б-члениые кольца
I I
Альдегиды — С=С—СНО
Кислоты н сложные эфиры —С=С—COOH(R)
В случае сопряжения прибавляют
дура
1111
—С=С—С=С—СО— н т. д.
В том случае, когда вторая двойная связь гомоан-
нулярна с первой
215 нм
202 нм
207
197
+30 нм
+39 нм
Прибавляют на каждый заместитель, нм а Р V д
—R алкил (включая часть карбоциклического кольца) —OR + 10 35 + 12 30 + 17 17 31
—ОН 35 30 30 50
Специальные методы характеристики органических веществ
491
П родолжение табл. 8.4
-SR -G1 —qEoR (ацетоксигруппа) —NH2, —NHR, —NR2 15 25 6 80 -г 12 12 30 25 6 6 95 - 12 25 6
В том случае, когда одна двойная связь экзоцикличиа +5
к одному кольцу
Если одна двойная связь экзоцикличиа одновременно к двум кольцам Вмакс 4500—20 000 +10
Сдвиг, обусловленный растворителем, нм
Вода +8 Метанол 0
Хлороформ —1 Диоксаи —5
Диэтиловый эфир —7 Гексан —11 Циклогексан — 11
ский способ вычисления положения максимума поглощения со-
Жяженных диенов и а,p-ненасыщенных кетонов (табл. 8.3 и 8.4).
э сих пор при изложении материала предполагалось, что при
замене атомов водорода, даже непосредственно присоединенных
к хромофору, на алкильные группы спектр не изменяется, однако
это справедливо только в первом приближении. Для системы XIV
максимум поглощения исходного незамещенного соединения в
растворе этанола находится при 215 нм.
—с=с—с=о
XIV
Для большого числа а- и Р-алкилзамещенных производных мак-
симум поглощения может быть вычислен, если к значению 215 нм
на каждый a-заместитель прибавлять 10 нм, а на каждый р-за-
меститель—12 нм; если имеется кольцо, в котором двойная связь
является экзоциклической, то прибавляется еще 5 нм (табл. 8.4).
Применение метода Вудворда иллюстрируется следующими при-
мерами.
Задача. Вычислите положение ультрафиолетового максимума следующего
соединения (в растворе этанола):
-СН,
:н,
492
Глава 8
Экзоциклическая двойная связь — это связь, соединенная с атомом углерода
кольца таким образом, как это показано в формуле внизу слева. Двойная связь
в формуле справа является экзоциклической для кольца А и эидоциклической
по отношению к кольцу В. Заместители у а-углеродиого атома отсутствуют, ио
имеются два заместителя у f-углеродного атома, для которых к 215 им должно
быть прибавлено 24 нм (2 X 12). Поскольку двойная связь экзоциклична к коль-
цу А, то к 215 нм нужно прибавить 5 + 24 = 29 нм, и тогда вычисленное поло-
жение максимума исходного соединения составит 244 нм.
Аналогично проводят вычисления для сопряженных диенов. Для выяснения
подробностей необходимо обратиться к цитированным работам. Учитывая про-
стоту метода, удивительно, что он дает такое хорошее согласие с эксперимен-
том. Применение этого метода привело к пересмотру неправильных структур
ряда природных соединений.
Таблица 8.5
Данные для вычисления положения основной полосы поглощения
производных бензола ArCOG (растворитель — этанол)
ArCOR/ArC HO/ArCOOH/ArCOOR Положение заместителя _С2н6он лмакс • им
Исходные хромофоры: Аг = С6Н6—
G — алкил или часть кольца (например, ArCOR) 246
G = H (АгСНО) 250
G = OH, OAlk (ArCOOH, ArCOOR) 230
Инкременты для каждого заместителя в Аг —Алкил или часть кольца орто, мета +3
пара +ю
—ОН, —ОСНг, —OR орто, мета +7
пара +25
—О* (оксианион) орто + 11
мета +20
пара +78 а
—С1 орто, мета +0
—Вг пара +10
орто, мета +2
—ЫН2 пара +15
орто, мета +13
—NHAc пара +58
орто, мета +20
пара +45
—NHCH3 пара +73
-N(CH3)2 орто, мета +20
пара +85
а Эта величина может сильно уменьшаться при язмеивияи копланарности из-за стери-
ческих препятствий.
Специальные методы характеристики органических веществ
493
Вторая корреляция, важная для химии ароматических соеди-
нений,— это корреляция Скотта *, который установил, что влия-
ние заместителей на спектр бензола может быть скоррелировано
со структурой изучаемых соединений (табл. 8.5). Использование
этой таблицы показано на следующем примере:
+25 ^^"+3
о
6-мегпокситетпралон
Вычислено: Л,макс °Н = 246 + 3 + 25 (значение из табл. 8.5) = 274 им
Найдено: ^кс5°Н = 276 НМ
Сведения о других систематических исследованиях изменений
спектров при изменении структуры таких классов соединений, как
сопряженные полиены и ацетилены, семикарбазоны, 2,4-динитро-
фенилгидразоны и сопряженные и несопряженные оксимы, при-
ведены в книгах Гиллема и Штерна, а также Штерна и Тимонса
(ссылка в разд. 8.2). Кроме того, опубликовано обширное иссле-
дование 2,4-динитрофенилгидразонов **.
8.2.6. Поглощение монофункциональных соединений
Некоторые группы, например азо- и нитрозогруппы, обладают
интенсивным поглощением в ближней ультрафиолетовой области.
Однако максимумы поглощения других соединений, таких, как
альдегиды, кетоны и амины, в ближней ультрафиолетовой обла-
сти имеют очень низкую интенсивность. Использование таких по-
лос для идентификации всегда связано с опасностью, которая ста-
новится понятной, если учесть, что примесь с е 15 000 при концен-
трации 0,1% дает такую же интенсивность поглощения, как и
чистый кетон с в, равным 15. Тем не менее из подобных спектров
можно получить ценную информацию.
Поскольку в ультрафиолетовой области, доступной химику-
органику, поглощают многие изолированные функциональные
группы, то ниже приводятся необходимые сведения по этому
вопросу.
Несопряженные олефины. Хотя основной максимум поглоще-
ния несопряженных олефинов лежит в вакуумной ультрафиолетовой
* Scott A. /., Interpretation of the Ultraviolet Spectra of Natural Products,
Pergamon Press, Elmsford, N. Y., 1964.
•* Johnson G. D., J. Am. Chem. Soc., 75, 2720 (1953).
494
Глава 8
области (160—200 нм), число алкильных заместителей у ато-
мов углерода двойной связи может быть установлено по спект-
рам *. Такие корреляции очень ценны, поскольку эта ультрафио-
летовая область более доступна химику-органику **. Кроме того,
оказалось возможно для определения степени замещения углерод-
углеродной двойной связи использовать концевое поглощение в
ближней ультрафиолетовой области (205—225 нм)***.
Карбонильные соединения. Слабое поглощение карбонильной
группы при 260—300 нм в некоторых случаях может служить
полезным дополнением к инфракрасным спектрам, поскольку оно
позволяет отличить кетоны или альдегиды от сложных эфиров.
Например, пятичленные циклические кетоны и алифатические
сложные эфиры поглощают в инфракрасных спектрах около
1740 см-1, однако только первые из них имеют заметное погло-
щение в ультрафиолетовой области выше 210 нм. Максимум по-
глощения карбонильной группы может быть использован и для
других целей ****, однако его положение смещается под влиянием
атомов хлора или брома в a-положении, а у замещенных цикло-
гексанонов величина этого сдвига зависит от того, находится ли
атом галогена в экваториальном или аксиальном положении. По-
добный сдвиг наблюдается и в спектрах а-окси- и а-ацетоксике-
тонов *****. Поэтому такое дополнение к инфракрасным спектрам
является способом определения конфигурации замещенных цикли-
ческих кетонов.
Амины. Исследование спектров простых аминов в вакуумной
ультрафиолетовой области ****** показывает, что расширение прак-
тически применяемого УФ-диапазона в эту область спектра по-
зволяет получить ценный метод идентификации первичных, вто-
ричных и третичных алифатических аминов. Ввиду того что на-
сыщенные третичные амины имеют в ближней ультрафиолетовой
области максимум поглощения (около 214 нм), сдвигающийся в
длинноволновую сторону в случае винильных аминов со структу-
рой R2N—CH=CHR (228—238 нм), ультрафиолетовая спектро-
скопия представляет ценность для отличия этой функциональной
группы от насыщенных или несопряженных аминов, в которых
аминогруппа и олефиновая двойная связь изолированы *******
Серусодержащие функциональные группы. Меркаптаны, тио-
эфиры, ди- и полисульфиды имеют характеристические спектры
* Semenow D., Harrison A. J., Carr Е. Р., J. Chem. Phys., 22, 638(1954);
Turner D. W., J. Chem. Soc., 1959, 30.
** Micheli R. A., Applewhite T. H., J. Огр. Chenj., 27, 345(196?).
*** Bladon P., Henbest H. B., Wood G. W.. J. Chem. Soc., 1952, 2737.
**** Cookson R. C., J. Chem. Soc., 1954, 282.
***** Cookson R. C., Dandegaonker S. H., J. Chem. Soc. 1955, 352.
****** Tannenbaum E., Coffin E. M. Harrison A., J. Chem. Soc., 21,311 (1953).
******* Leonard N. J., Locko D. M., J. Am. Chem. Soc., 77, 437 (1955).
Специальные методы характеристики органических веществ
495
в ближней ультрафиолетовой области *. Сульфоны и сульфокси-
ды также обладают характеристическим поглощением **.
Контрольные вопросы Как можно различить следующие соединения с по-
мощью методов УФ-спектроскопии, используя возможно большее число спосо-
бов? Кратко обсудите, какие различия при этом следует ожидать.
1.
СН3СН2СН=СН—СН=СНСН2СН3 и СНзСН2СН=СНСН2СН=СНСНз
4. Вычислите Хмокс в этаноле для каждого из приведенных ниже изомеров.
Если считать, что полученные значения вычислены с точностью до 5 им, можно
ли, пользуясь этим методом УФ-спектроскопии, различить два приведенных ниже
соединения?
5. Сопоставьте приведенные ниже данные
УФ-спектров с указанными струк-
турами.
^макс e
231 21 000
236 12 000
245 18.000
265 6.400
282 11900
Ненасыщенная алкильная.группа
* Fehnel Е. A., Carmack М., J. Am. Chem. Soc., 71, 84 (1949); Baer J. E.,
Carmack M., ibid., 71, 1215 (1949).
** Fehnel E. A., Carmack M., J. Am. Chem. Soc., 71, 232 (1949); 72, 1292
(1950).
496
Глава 8
6. Используя данные табл. 8.5, отнесите наблюдаемые максимумы (раствор
в этаноле) к соответствующим структурам.
^макс
245 п-аминобензойная кислота
253 n-бромбепзойная кислота
288 л1-метоксибеизойная кислота
8.3. ДИСПЕРСИЯ ОПТИЧЕСКОГО ВРАЩЕНИЯ
Показатель преломления вещества зависит от длины волны
проходящего света. Сходным образом от длины волны зависит
и удельное вращение [а]. Изменение удельного вращения при
Рис. 8.5. Кривые дисперсии оптического вращения (перепечатано из кинги:
Djerassi С., Optical Rotatory Dispersion, McGraw-Hill, New York, copyright ©
1960; есть русский перевод: Джерасси К. Дисперсия оптического вращения. Пер.
с англ. — М.: ИЛ, 1962).
— плавная кривая;-------аномальная кривая (положительный эффект Коттопа).
изменении длины волны называется дисперсией оптического вра-
щения (ДОВ).
Многочисленными исследованиями показано, что зависимость
оптического вращения от длины волны в видимой и ультрафиоле-
товой областях спектра может дать более ценные сведения, чем
величина вращения при одной длине волны. Кривые дисперсии
оптического вращения оказались весьма ценными при определе-
нии конфигураций и конформаций оптически активных соедине-
ний.
В областях поглощения, где происходят электронные перехо-
ды, дисперсия оптического вращения часто обладает некоторыми
аномалиями и соответствующая кривая имет один или несколько
максимумов («пиков») и минимумов («впадин»). Такие аномалии
называются эффектом Коттона (рис. 8.5).
Специальные методы характеристики органических веществ
497
Если на кривой наблюдается только один максимум и один
минимум (рис. 8.5), то говорят о «простом эффекте Коттона».
Кривые с двумя или более пиками и соответствующим числом
впадин в области полосы поглощения относятся к «сложному эф-
фекту Коттона». Эффект Коттона положителен, если пик распо-
ложен при больших длинах волн, чем впадина (рис. 8.5), и от-
рицателен, если пик лежит в более коротковолновой области.
Если один из энантиомеров дает положительный эффект Котто-
на, то другой, имеющий равное по величине удельное вращение
противоположного знака, имеет отрицательный эффект Коттона.
Эти эффекты связаны с ориентацией групп, расположенных в мо-
лекуле рядом с хромофором.
Прибор для измерения дисперсии оптического вращения пред-
ставляет собой комбинацию поляриметра, измеряющего оптиче-
ское вращение, источника монохроматического света (видимого
или ультрафиолетового) с известной длиной волны и фотоэлект-
рического устройства для определения угла минимального про-
пускания: таким образом, это фотоэлектрический спектрополяри-
метр.
Наиболее успешным было применение метода дисперсии опти-
ческого вращения для изучения стереохимии циклогексанонов и
сложных циклических соединений, содержащих циклогексановое
кольцо. В этих исследованиях применяют правило октантов, рас-
смотренное в следующем разделе.
8.3.1. Правило октантов
Эмпирическое правило октантов * позволяет установить одну
из четырех характеристик соединения — структуру, конформацию,
конфигурацию или знак эффекта Коттона, если известны три
другие.
Молекулу, содержащую циклогексаноновое кольцо, ориенти-
руют, как показано на рис. 8.6. При этом карбонильный кислород
принимают за начальную точку и молекулу разделяют координат-
ными плоскостями на октанты и затем проецируют на фронталь-
ную плоскость С, перпендикулярную связи С=О. При этом по-
лучают диаграмму октантов (рис. 8.6). Правило октантов для
случаев, когда все заместители кольца лежат в задних октантах
(удаленных от глаза наблюдателя относительно плоскости С),
заключается в следующем.
Атомы, лежащие на любой из разделяющих молекулу плоско-
стей (А, В, С), не дают вклада в удельное вращение. К ним от-
носятся любые непосредственно связанные атомы в положениях
R2e, L2e, 4а и 4е.
* Moffitt W., Woodward R. В., Moscowitz А., Щупе W., Djerassi С., J. Am.
Chem. Soc., 83, 4013 (1961); Mislow R., Class M. A. W., Moscowitz A., Djerassi C.,
ibid., 83, 2771(1961).
498
Глава 8
Атомы, лежащие в верхнем правом октанте (над плоскостью
В и справа от плоскости А) ив нижнем левом октанте (ниже
плоскости В и слева от плоскости А), дают отрицательный вклад
во вращение. Сюда входят любые группы в положениях R3c, R3e
и L2a, а также группы, находящиеся в положениях L2e, R2e, 4а
и 4е и попадающие в эти октанты.
Атомы, находящиеся в верхнем левом октанте (над плоскостью
В и слева от плоскости А), и атомы, лежащие в нижнем правом
Рис. 8.6. Правило октантов (перепечатано из книги: Djerassi С., Optical Rotatory
Dispersion, McGraw-Hill, New York, copyright © 1960; есть русский перевод:
Джерасси К. Дисперсия оптического вращения. Пер. с аигл. — М.: ИЛ, 1962).
октанте (ниже плоскости В и справа от плоскости А), дают по-
ложительный вклад в удельное вращение. К ним относятся атомы
в положениях L3a, L3e и R2a, а также группы в положениях
L2e, R2e, 4а и 4е, лежащие в этих октантах.
Если заместители в циклогексановом кольце попадают в пе-
редние октанты (слева от плоскости С), то вклад этих групп в
удельное вращение имеет знак, противоположный тому, который
имеют те же группы в октантах, лежащих за плоскостью С.
Знаки вкладов относятся к длинноволновой части кривых эф-
фекта Коттона; следовательно, если сумма вкладов положитель-
на, то эффект Коттона положителен; если сумма вкладов отри-
цательна, то и эффект Коттона отрицателен. Предсказанный эф-
фект Коттона связан с полосой поглощения кетонной группы в УФ-
спектре.
Поскольку одинаковые атомы и группы, пространственно рас-
положенные одинаково по обе стороны разделяющих плоскостей,
компенсируют вклады друг друга, то для оценки общего удель-
ного вращения необходимо рассматривать только несбалансиро-
Специальные методы характеристики органически веществ
499
ванные группы. Они называются значащими группами (или ато-
мами).
Ниже приведены два примера использования этого правила.
Конфигурация. Определение конфигурации показано на при-
мере (+)-трамс-10-метилдекалона-2 (рис. 8.7). Если эта молекула
Рис. 8.7. (+)-тронс-10-Метилдекалон-2 (перепечатано из книги: Djerassi С.. Opti-
cal Rotatory Dispersion, McGraw-Hill, New York, copyright © 1960; есть русский
перевод: Джерасси К. Дисперсия оптического вращения. Пер. с англ. — М.: НЛ,
1962).
имеет изображенную на рисунке конфигурацию, то атомы угле-
рода 8, 7 и 6, расположенные экваториально к Сэ, находятся в
нижнем левом октанте, а атом углерода 5 и ангулярная метиль-
ная группа лежат в плоскости А и не дают вклада в эффект Кот-
тона. Заместители у С] и С3 отсутствуют. Поскольку заместители
вне координатных плоскостей находятся только в нижнем левом
октанте, то эффект Коттона для этой молекулы должен быть по-
ложительным. Так как и на самом деле эффект Коттона для
Рис. 8.8. (R)-(+) -3-Метилциклогексанон.
н,с
ориентация
/ молекулы Ъля
применения
правила
октантов
(см, рис. 8.6)
(-|~)-т’ранс-10-метилдекалона-2 положителен, то абсолютная кон-
фигурация, изображенная на рис. 8.7, является правильной.
Конформация. Известно, что (+)-3-метилциклогексанон имеет
^-конфигурацию (рис. 8.8). Если кетон сориентирован соответст-
вующим образом для использования правила октантов, то он мо-
жет быть представлен либо в экваториальной форме с метильной
группой в верхнем левом октанте (заместитель у L3), либо в ак-
сиальной форме с метильной группой в верхнем правом октанте
(заместитель у R3) (рис. 8.8). Другие заместители отсутствуют,
и поэтому экваториальный конформер должен иметь положитель-
ный эффект Коттона, а аксиальный конформер — отрицательный.
БОО
Глава 8
Тот факт, что в действительности эффект Коттона положителен,
подтверждает предположение (высказанное ранее на основании
других конформационных соображений) о преобладании в смеси
изомеров экваториальной формы.
Итак, основное применение дисперсии оптического вращения
при установлении структуры органических соединений состоит
в определении одного из следующих трех параметров, если из-
вестны два других: 1) положения функциональных групп, 2) кон-
фигурации и 3) конформации молекул.
8.4. ЯДЕРНЫЙ МАГНИТНЫЙ РЕЗОНАНС УГЛЕРОДА-13
Общие положения протонного магнитного резонанса справед-
ливы и для углеродного магнитного резонанса. До появления
ряда усовершенствований, сделанных в последнее время, иссле-
дование атомов углерода в органических соединениях методом
магнитного резонанса представляло большие трудности. Это свя-
зано с тем, что |3С, магнитно-чувствительный изотоп углерода,
не является основным изотопом. Кроме того, ядра 13С имеют зна-
чительно меньшую чувствительность ядерного магнитного резо-
нанса, чем протон, из-за меньшей величины гиромагнитного от-
ношения для углерода-13. Современные приборы ЯМР-13С преодо-
левают эти трудности с помощью ЭВМ, которые дают возмож-
ность работать в режиме импульсного возбуждения .ядер 13С и
позволяют проводить фурье-анализ полученных данных для воз-
бужденных ядер 13С.
Значение информации, полученной методом углеродного маг-
нитного резонанса, трудно переоценить. Возможность идентифици-
ровать почти все неэквивалентные атомы углерода в органических
соединениях методом ЯМР-13С делает его очень мощным орудием,
используемым химиками-органиками для структурных исследова-
ний. До последнего времени спектроскопия ЯМР-13С использова-
лась не очень широко. Теперь приборы ЯМР-,3С могут быть при-
обретены за 50 000—100 000 долл. Однако из-за меньшей стоимо-
сти приборов протонного магнитного резонанса (ПМР) до сих
пор химиками-органиками чаще используется протонный магнит-
ный резонанс *. Тем не менее ясно, что для определения струк-
туры органических соединений ЯМР-13С будет использоваться так
же широко, как протонный магнитный резонанс и ИК-спектро-
метрия.
При рассмотрении спектроскопии ЯМР-13С основное внимание
будет обращено на применение этого метода для установления
структуры органических соединений. Для знакомства с теорией
* Для ЯМР-’Н и ЯМР-|8С применяются сокращения PMR и CMR соответ-
ственно. Предложенное авторами обозначение PMR для фосфорного магнитного
резонанса не является ошибкой, но использование такого обозначения ие одоб-
рено IUPAC.
Специальные методы характеристики органических веществ
501
этого явления следует обратиться к литературе о методе ЯМР-13С,
приведенной в гл. 10.
В спектроскопии ЯМР-13С применяются обычно те же раст-
ворители, что и в протонном магнитном резонансе (т. е. CDC13
и др., см. гл. 5). Для некоторых приборов (например, Varian
CFT-20) необходимо, чтобы хотя бы часть растворителя была дей-
терированным соединением (CDC13, D2O, CD3CN и т. д.) для ста-
билизации поля по дейтерию.
В настоящее время принято измерять все химические сдвиги
в миллионных долях (м. д.) по отношению к сигналу 13С в ТМС
(тетраметилсилан); увеличение положительных значений химиче-
ского сдвига соответствует смещению сигнала в более слабое поле
по сравнению с ТМС, а отрицательные значения химических сдви-
гов соответствуют смещению сигнала в более сильное поле. Во
многих более ранних работах использовались различные внутрен-
ние стандарты, химические сдвиги которых приведены в табл. 8.6.
Таблица 8.6
Химические сдвиги ,3С обычных эталонов и растворителей
по отношению к ТМС
Соединение, растворитель Химический с двига. м. д.
соединение, содержащее протий полностью дейтерированное соединение
Уксусная кислота (карбонильный углерод) Ацетон (метильный углерод) 178,3 30,4 29,2
Бензол 128,5 128,0
Четыреххлористый углерод Хлороформ 96,0 77,2 76,9
CS26 CS2 в капилляре® Циклогексан 192,8 193,7 27,5 26,1
Диметилсульфоксид 40,5 39,6
Диоксан Метилеихлорид 67,4 54,0 53,6
а Все сигналы расположены в более слабом поле по отношению к сигналу внутрен-
него стандарта ТМС (±0,05 м. д. при 38 ’С); см. Levy О. С., Cargloil 1. D., J. Mag. Res.,
143 (1972). Приведенные значения химических сдвигов могут быть использованы в качестве
фактора F^ (см. текст).
б Величины химических сдвигов взяты из книги: St others J. В„ Carbon>!3 NMR Spectros-
copy, Academic Press, New York, 1972.
Например, довольно часто используется CS2 (химический сдвиг
193,7 м. д. в сторону слабого поля от ТМС). Если приведены
значения химического сдвига по отношению к CS2, то они могут
502
Глава 8
быть пересчитаны в химические сдвиги по отношению к ТМС пу-
тем прибавления 193,7 м. д. Следовательно,
Ос = Ос + Fl
где б™с — вычисляемый химический сдвиг по отношению к ТМС,
6j — химический сдвиг по отношению к некоторому другому
внутреннему стандарту, a Fi — фактор, значение которого берется
из табл. 8.6 для соответствующего внутреннего стандарта i.
Для структурных определений методом спектроскопии ЯМР-18С
можно получить информацию из данных о химических сдвигах,
а также о мультиплетности и интенсивности сигналов.
8.4.1. Химический сдвиг
Спектр ЯМР-13С даже сложной органической молекулы срав-
нительно редко не дает отдельных хорошо разрешенных сигналов
для каждого типа атомов углерода в молекуле (рис. 8.9). Напри-
мер, типичный спектр шириной 200 м. д. (с полной спиновой раз-
вязкой от протонов) будет обычно состоять из простых линий,
каждая из которых будет соответствовать определенному типу
атомов углерода (рис. 8.9). Обычно спектр ПМР не имеет та-
кого хорошего разрешения, как спектр углеродного резонанса;
поэтому во многих случаях спектр ПМР не дает такого количе-
ства информации о структуре, как спектр ЯМР. Это только одно
из преимуществ, на котором можно показать огромные возмож-
ности спектроскопии ЯМР-13С при структурном органическом ана-
лизе.
В спектрометрии ЯМР-13С могут быть применены те же общие
принципы, которые используются при корреляции структуры с хими-
ческим сдвигом в ПМР-спектрометрии. Например, можно показать
(рис. 8.10), что алканы имеют сигналы атомов углерода (в интер-
вале б от —10 до +35 м. д.) в более сильном поле, чем сигналы
атомов углерода алкенов (б от 100 до 165 м. д.). Следует вспом-
нить, что протоны алканов дают сигналы в более сильном поле,
чем олефиновые протоны. Далее, электроотрицательные атомы
(например, N, О) вызывают появление сигналов непосредственно
с ними связанных атомов углерода в более слабом поле по срав-
нению с алканами; та же самая общая тенденция существует и
в протонном магнитном резонансе.
Если имеются некоторые предположения о структуре органи-
ческого соединения, то часто их можно очень легко уточнить,
используя углеродный магнитный резонанс атомов углерода раз-
ного типа в органическом соединении. В особой степени это от-
носится к карбонильному углероду; на рис. 8.10 показано, что хи-
мический сдвиг карбонильного углерода зависит как от типа
функциональной группы (т. е. от того, находится ли изучаемая
Специальные методы характеристики органических веществ
503
карбонильная группа в альдегидах, кетонах, сложных эфирах
и т. д.), так и от того, сопряжена ли она с другой л-электронной
системой в молекуле. Резонанс карбонильного углерода наблю-
дается в сравнительно слабом поле (6 примерно 145—210 м. д.,
500Q 4500 4000 3500
3000 2500 2000 1500 1000 500 О Гц
ГГТТТТ1ТГГТТТГП11 IГ П ГПТП Г1 ТТГГ1 П Г11 I 11 I 11 11 I 111 I '1111 H Ц I 11 I 11
а Ъ с c b a
CHpHpHj-NH-CHgCHaCH^
li.i । i.Liu 11111 il н 111111 i l.i । n IJ ii i U1111 i 111 h и i hi и 1111111111 Ji 1 i.lL
' /io led Ito 1бо
Соединение
Образец получен on
Химическая
формула
Молекулярная масса
Номер ПМР-спектра
Т. кип
ISO 140lio 120 lid Ю0908070 60 50
Ъи-н-пропиламин
Kastman Organic Chemicals
Rochester, New York
CeH„N
101,19
17051 М
los-uox
Концентрация раствора
Растворитель
Стандарт
Число прохождений
Время
Начало отсчета
507. пб объему
CDClj
ТМС
400
в Мин
40 30 20 10 О м.Ъ.
Отнесения
А
В
11.9 N
~2&~
К
L
м
Рис. 8.9. Спектр ядерного магнитного резонанса ,3С ди-н-пропиламина, снятый
на приборе Varian НА-100/Digilab FT-NMR-3 (воспроизведено с разрешения
Sadtler Laboratories, Inc., Philadelphia, Pa).
рис. 8.10), что вызвано, по крайней мере отчасти, вкладом резо-
нансной структуры с положительным зарядом на атоме углерода.
Уменьшение электронной плотности на атоме углерода карбониль-
ной группы уменьшает парамагнитное экранирование, что в свою
очередь приводит к смещению сигнала резонанса углеродного
атома в слабое поле.
Спектр ПМР далеко не всегда дает непосредственную инфор-
мацию о карбонильной группе. По спектру ЯМР-13С часто можно
504
Глава 8
определить и идентифицировать присутствующие в молекуле кар-
бонильные группы. Следовательно, для характеристики карбо-
нильных соединений можно использовать совместно спектры ИК
и ЯМР-13С.
М(СО)П
Алкены
шшшш
R,CO
арил С
арил
С-0
ЯСНО
арил
C-N
C=N
RCQOH
с=с Алканы
№ С__I
L(CH2)J
о ।
I п > 3 и “ 3
-С-0
I
CZZZI
I
—С—N
I
С I
-С-Я
RCOOR
с6н6 ссц снс!3 сн3он с6н)2 сн4
—J----------fj--------L... .... I-----1_________।
100 о м.д.
I
н--------► тмс
Рис. 8.10. Области химических сдвигов нейтральных соединений (перепечатано
с разрешения из книги: Stothers J. В., Carbon-13 NMR Spectroscopy, Academic
Press, New York, copyright © 1972).
Обозначение гибридизации углерода:
sp2.
sp.
Углерод карбонильной группы обозначен так:
углерод несопряженной Со-группы,
углерод сопряженной СО-группы
например, углерод карбонильной группы пеитаиона-2 будет обозначаться первым способом,
а углерод карбонильной группы метилвииилкетона —вторым.
Спектр ЯМР-|3С простого углеводорода показан на рис. 8.11.
Как обычно, концевой атом углерода алкана дает сигнал в бо-
лее сильном поле (например, С-1 в октане, табл. 8.7), чем внут-
ренний углерод (например, С-4 в октане, табл. 8.7). Этот эффект
является общим: например, во многих случаях введение углеродов
в положения а, 0, у и т. д. к данному атому углерода вызывает
смещение резонансного сигнала этого атома (табл. 8.8). Из
табл. 8.8 видно, что атомы углерода в а- или 0-положении к не-
Специальные методы характеристики органических веществ
боб
которому атому углерода сдвигают резонансный сигнал послед-
него в слабое поле на величину около 9 м. д., тогда как введение
у-углерода в молекулу приводит к небольшому сдвигу сигнала
(2,5 м. д.) в сильное поле. Этот сдвиг в сильное поле вновь по-
W00 4500 4000 3500 3000 2500 2000 1500 1000 500 О' Гц
п11111111111Л1иттт1Т1П1ПТ]Т1'П11тт1упц1Ггл|1-1111Г11Г|Г1111Н11|П11П1111и11'т11Ц111Г11111111
а (> с о Ь а '
СН^£Н/^СН£Нд
। in I и nJirrtj.Iji 4.lnnUii4b,,uj,lj uxlm |Цц.Н 11 11 Li 11111 и 1.1,111 fill u.U и 11111 iki н 1111
200 190 180 no 160 150 140 130 120 1Ю 100 SO 80 70 6Q SO 40 30 20 10 0M.i.
Соединение гексан
Отнесения-
ofyawi получен on*
Phillips Petroleum Со.
Bartlesville, Oklahoma
f Химическая СбНи
формула
Молещ^рная масса
Номер ПМР-спекмра М
7. ОЛ —95 X
TJjfun. 68,8 X
Концентрация раствора
Растворитель
Стандарт
Число прохождений
Время
Начало отсчета
50% по объему
юоо
20 мин
А
в
с
D
£
Г
G
Н
J
J
К
L
М
zz?*:
____s
____т
____и
____V
____W
____X
V
____Z
Рис. 8.11. Спектр ядерного магнитного резонанса 13С гексана, снятый на приборе
Varian НА-100/Digilab FT-NMR-3 (воспроизведено с разрешения Sadtler Labo-
ratories, Inc., Philadelphia, Pa).
называет, что в качестве правил для спектров ЯМР-13С нельзя
использовать правила, по которым происходят изменения в спект-
ре протонного резонанса при изменении структуры изучаемого
соединения, поскольку такие большие сдвиги в сильное поле не
характерны для ПМР-спектров.
Очень заманчиво использовать данные, приведенные в Табл. 8.8,
для предсказания химических сдвигов в алканах. Однако при
этом необходимо рассматривать большее число параметров, так
как число и тип боковых цепей влияют на химический сдвиг. Ожи-
даемый химический сдвиг в алканах может быть вычислен с
506
Глава 8
Таблица 8.7
Химические сдвиги атомов углерода в разветвленных
и неразветвленных алканах а
Алкай С-1 С-2 с-з С-4 С-5
Неразветвленные цепи Метан —2,1
Этан 5,9 8,9 -
Пропан 15,6 16,1 15,6
Бутан 13,2 25,0 25,0 13,2
Пентан 13,7 22,6 34,5 22,6 13,7
Гексан 13,9 22,9 32,0 32,0 22,9
Гептан 13,9 23,0 32,4 29,5 32,4
Октан 14,0 23,0 32,4 29,7 29,7
Нонан 14,0 23,1 32,4 29,8 30,1
Декан 14,1 23,0 32,4 29,9 30,3
Разветвленные цепи Изобутан 24,3 25,2
Изопентан 22,0 29,9 31,8 11,5
Изогексан 22,5 27,8 41,8 20,7 14,1
Неопентан 31,5 27,9
Неогексан 28,9 30,4 36,7 8,7
З-Метилпентан 11,3 29,3 36,7 18,6
2,3-Диметилбутан 19,3 34,1
2,2,3-Триметилбутан 27,2 $2,9 38,1 15,9
3,3-Днметилпентан 6,8 25,1 36,1
4,4 6
а Данные взяты из работ: Grant D. G., Paul Е. G., J. Am. Chem. Soc., 86, 2984 (1964);
Roberts et al., ibid., 92, 1338 (1970); Spiescke H„ Scheider W. G.. J. Chem. Phys., 35. 722 (1961).
® Для углерода замещающей метильной группы. Химический сдвиг в м. д. в слабое
роле от ТМС. Так как эта величина получена из разных спектров с использованием разных
внутренних стандартов (ТМС, бензол, CSj), то ошибка в химическом сдвиге ие меиее 0,2 м. д
Таблица 8.8
Химический сдвиг, обусловленный введением атома
углерода в различные положения цепи в алканах*
Положение Изменение химического сдвига, м. д.
а +9,1 ±0,1
₽ 4-9,4 ±0,1
V -2,5 ±0,1
й 4-0,3 ±0,1
е 4-0,1 ±0,1
а Данные взяты из работы Grant D. М., Paul Е. G., J. Ат.
Chem. Soc., 86, 2984 (1964).
Специальные методы характеристики органических веществ
507
использованием параметров, приведенных в табл. 8.9, по следую-
щему уравнению:
м-л
6/ = + S (NMAnM) + ynNis + AnNZ4
М=2
Таблица 8.9
Параметры для вычисления химического сдвига С-13 для алканов
Параметр Величина, м. д. Параметр Величина, м. д.
В, 6,80 А» 0,25
Ли 9,56 Вз 23,46
41» 17,83 Л 32 6,60
Л14 25,48 4зз 11,14
Yi —2,99 Л34 14,70
А1 0,49 Уз —2,07
В2 16,34 в4 27,77
4»s 9,75 Л42 2,26
А1з 16,70 л 43 3,96
Л24 21,43 л44 7,35
У» -2,69 V4 0,68
а Данные взяты из работы: Lindeman L. Р.. Adams 1. О., Anal. Chem., 43, 1245 (1971).
где б( — вычисленный химический сдвиг для i-ro атома углерода;
Вп — параметр, зависящий от числа атомов углерода (п), присое-
диненных к атому i; Nm — число атомов углерода, соединенных
t атомом I, к которым присоединено два (или более) атома угле-
рода; А„м — параметр, зависящий от числа атомов углерода (п),
непосредственно связанных с атомом i, и от числа атомов угле-
рода (М), присоединенных к углеродам, непосредственно связан-
ным с атомом i; уп — параметр, зависящий от числа атомов (и),
непосредственно связанных с атомом i; NiP — число атомов угле-
рода, удаленных на Р связей от атома i (учитываются только ато-
мы, удаленные на три и более связей); — параметр, завися-
щий от числа атомов (п), непосредственно связанных с атомом i.
Применение этого уравнения и данных табл. 8.9 показано на
примере 2,2,3-триметилбутана:
^ix ®i + N4A14 + ylNs + A1N4
где Bn = Bi = 6,80, так как п = 1 (имеется один атом углерода,
связанный с атомом углерода 1, и из табл. 8.9 В! = 6,80), и
508
Глава 8
имеется только один член, содержащий МмАпм, так как присут-
ствует только один тип атомов углерода (атом углерода 2), при-
соединенный к атому углерода 1, который связан с двумя или
более атомами углерода. К атому 2 присоединено 4 атома угле-
рода, поэтому NM = Nt. Так как имеется только один атом та-
кого типа, то /V4 = 1. Следовательно, атом углерода 1 имеет толь-
ко один связанный с ним атом углерода и поэтому в индексе А
п = 1. Таким образом,
М-4
£ (A^„M) = A4AI4 = (1) (25,48)
уп — Т1 (так как имеется один атом углерода, соединенный с ато-
мом 1) и Л/з = 1, так как есть всего один атом углерода (атом
углерода 4), отделенный тремя связями от атома 1. Наконец, от-
сутствуют атомы углерода, отделяемые от атома 1 четырьмя свя-
зями, поэтому А4 = 0.
Следовательно,
6аыч = 6,80 + (1) (25,48) + (-2,99) (1) = 29,29
Эту величину надо сравнить с б”абл = 27,2.
Аналогично для других атомов цепи также можно вычислить
химические сдвиги:
Й2ЫЧ = В4 + N3Ai3 -|- y4/V3 + Л4А4 =
— 27,77 + (1)(3,96) + у4(0) + Д4(0) = 31,73; ср. с бГбл = 32,9
$ыч = 23,46 + (1) (14,70) = 38,16; ср. с бзабл = 38,1 м. д.
6°ыч= 15,66 м. д.; ср. с 64абл=15,9 м. д.
Стандартная ошибка для таких расчетов составляет 0,8 м. д.
Задача. Вычислите д4.
Способ расчета с использованием данных табл. 8.9 непригоден для цикли-
ческих и полициклических систем.
Аналогичный расчет может быть проведен для алифатических соединений,
содержащих функциональные группы. Данные табл. 8.10 можно использовать
для вычисления химического сдвига соединений с прямой и разветвленной це-
пями. Например, сравните вычисленные и наблюдаемые химические сдвиги угле-
рода в бутаиоле-2:
СНз—
д®ыч 8,2 33,0 66,0 21,2 м. д.
бнабл 10>0 32 0 692 22>7 м д
При вычислении химических сдвигов величины параметров из табл. 8.10 прибав-
ляются к значению химического сдвига соответствующего атома углерода в не-
замещенном алкане (табл. 8.7). Например:
бзыч = 25,0+ 8,0 = 33,0 м. д.
ОН
СН,—СН—СН,
Специальные методы характеристики органических веществ
509
Таблица 8.10
Химический сдвиг сигнала С-13 (м. д.) при замещении протона
на функциональную группу (R) в алканаха
У Р а
формальные., у''''и
разветвленные
У Р а Р У
R a V
нормаль- ные развет- вленные нормаль- ные развет- вленные
СНз 4-9 +6 +10 +8 —2
соон 4-21 +16 +3 +2 —2
СОО" 4-25 4-20 +5 +3 -2
COOR 4-20 +17 +3 +2 —2
СОС1 4-33 4-28 +2
COR 4-30 4-24 + 1 + 1 —2
сно +31 0 —2
с6н6 +23 + 17 +9 +7 —2
он +48 +41 + 10 +8 -5
OR 4-58 +51 +8 +5 —4
OCOR +51 4-45 +6 +5 -3
nh2 4-29 4-24 +11 + 10 -5
nh? +26 4-24 +8 +6 —5
nhr 4-37 +31 +8 +6 —4
nr2 +42 +6 -3
no2 4-63 +57 +4 +4
CN +4 +1 +3 +3 -3
SH + 11 + 11 + 12 + 11 —4
SR +20 +7 -3
F 4-68 4-63 +9 +6 —4
Cl +31 +32 + 11 + 10 —4
Br +20 +25 + H +10 -3
I -6 +4 + 11 + 12 -1
а Нормальные—соединения с иеразветвлеииой цепью углеродных атомов, разветвлен-
ные—соединения с разветвленной цепью. Влияние замещения а у-положении одинаково
и для разветвленных и для иеразветвлеиных соединений. Перепечатано с раарешеиня
из книги: Wehrli F- W.t WirthlinT., interpretation of Carbon—13 NMR Spectra, Heyden, N, Y.,
1976.
где 25,0 м. д. — химический сдвиг для С-2 в бутане (табл. 8.7), а 4-8,0 —хими-
ческий сдвиг, индуцированный в атоме углерода, находящемся в (3-положении
по отношению ко вторичной ОН-группе. Такой расчет успешно применяется и
для разветвленных алканов. Например, рассмотрим бутандиол-1,3
ОН ОН
I I
СН2—СН2—СН—СН3
6°““ 56.2 43,0 61,0 21,2 м. д.
6набл 60 0 40(6 60 0 23 4 м д_
610
Глава 8
Пример расчета: 61 = 13,2 4-48,0 — 5,0 = 56,2 м.д., где 13,2 — химический сдвиг
С-1 в бутане (табл. 8.7), 4-48,0 — химический сдвиг а-углеродного атома под
влиянием группы ОН на конце цепи и —5,0 — химический сдвиг у-углеродного
атома под действием вторичной ОН-группы.
Данные табл. 8.8—8.10 показывают, что присоединение уг-
лерода или функциональной группы к алкану вызывает дезэк-
ранирование у-углеродного атома в цепи. Это вызвано, по край-
5000
4500 4000 3500 3000 2500 2000 1500 1000 500 0 Гц
П| ITii 111 il | in 111111 pi 111111 П['1Г1111 H'lJI 11111111 [П 111111 гр! 1111 irrp п 1111111 |ттпт| 11 ji 11
JJIllj.ll ll LI 1.1J.I.I I I ll 1 I 1,1,1 I II III I I I 1 I
Яю . 190 180 170 1S0 150 140 I3O
Соединение
ацетофенон
Образец получен от
МСВ Manufacturing Chemists
Norwood, ОЫо
Химическая
формула
Молекулярная масса
Номер ПМР-спехтра
Т. пл
Т. кип
сЛо
/20./5
10480 М
19-20*С
202'С
11111111111111111111111111.111111 н I lb 11 iji 11 ill 111 li I.11111111111
120 no too 90 80 10 SO 50 10 30 2Ь ЮОмЛ
Отнесения
А-Ш-Н--------
--—
ExA—•
6
H
I
Концентрация раствора 507, по объему
Растворитель
Стандарт
Число прохождений
Время
Начало отсчета
CDCI.
ТМС
400
8 мин
"Экспериментальное отнесение
.О
V
ж
.X.
У.
-Z.
К
L
М
Рис. 8.12. Спектр ядерного магнитного резонанса ,3С ацетофенона, снятый на
приборе Varian НА-100/Digilab FT-NMR-3 (воспроизведено с разрешения Sadtler
Laboratories, Inc., Philadelphia, Pa).
ней мере частично, стерическими эффектами; например, в углево-
дородах стерические взаимодействия протонов а-углерода с про
тонами у-углерода вызывают химический сдвиг в слабое поле.
bL Ji""
Это явление называют у-гош-эффектом.
Эмпирические параметры для вычисления химических сдви-
гов в других типах систем (циклоалканы, алкены, алкины и т. д.)
можно взять из работ, цитируемых в конце данного раздела.
Специальные методы характеристики органических веществ
511
Таблица 8.11
Влияние заместителей на химический сдвиг сигнала С-13 (м. д.)
в бензольном кольце•
Заместитель Положение заместителя
С-1 орто мета пара
—Вг —CF3 —СН3 —CN —С=С—Н 1,4-ди —CssC—Н —COCF3 —СОСН3 -СОС1 —СНО —СООН —СОСвНб —С1 —F —Н —NCO —nh2 —no2 -OCHS —ОН -c6H5 -SH —SCH3 -SO2NH2 —О'(окси-анион) —ОСбН5 —ОСОСН5 (ацетокси) -N(CH3)3 —N(CH2CH3)2 —NHCOCHs —СН2ОН ——Vat! С.Г12 —СООСНз -coci —СНО —СОСНгСНз —СОСН(СН3)2 —СОС(СН3)3 -5,5 —9,0 +8,9 -15,4 -6,1 -5,6 -5,6 +9,1 +4,6 +8,6 +2,1 +9,4 +6,2 +34,8 0,0 +5,7 + 18,0 +20,0 +31,4 +26,9 + 13,1 +2,3 + Ю,2 + 15,3 +39,6 +29,2 +23,0 +22,6 + 19,9 + Н.1 + 12,3 —32,0 +13,4 +9,5 +1,3 +5,8 +9,0 +7,6 +7,4 +9,4 +3,4 -2,2 +0,7 +3,6 +3,8 +3,8 + 1.8 +0,1 +2,4 + 1,3 + 1,5 + 1,7 +0,4 — 12,9 -3,6 -13,3 -4,8 -14,4 -12,7 -1.1 + 1,1 -1,8 -2,9 —8,2 -9,4 -6,4 -15,6 -15,3 -9,9 -1,4 + 10,2 +4,4 -2,0 -0,5 +2,6 + 1,2 -1,5 -0,5 -1.1 + 1,7 +0,3 -0,1 +0,6 +0,4 +0,7 0,0 0,0 +0,6 0,0 -0,2 + 1,3 + 1,4 + 1,2 +0,9 +0,9 +1,0 + 1,4 +0,4 +1,1 +0,4 +0,4 + 1,9 + 1,6 +1,3 + 1,0 +1,4 +0,2 -1,4 +2,9 —1,1 +0,2 -0,5 +1,2 + 1,2 -1,5 -0,5 —1,1 —1,6 +3,2 —2,9 +3,9 —0,2 +6,7 +4,2 +6,2 +5,5 +5,1 +3,6 -1,9 -4,5 -2,8 -9,8 +5,8 —7,7 -7,3 — 1,2 -3,1 -3,6 +3,3 -13,6 -5,1 -2,3 -11,5 — 12,2 -5,6 -1.4 + 1,0 -1,1 -0,5 +3,5 +7,4 +6,0 +2,4 +4,0 +1,7
а Приведенные данные получены в различных растворителях (CCI4, ДМФА) и без них
с различными стандартами (ТМС, CSj). Положительные величины означают химический
сдвиг в более слабое поле, а отрицательные—сдвиг в более сильное поле. Стандартная
Ошибка ±0,5 м. д. Химический сдвиг сигнала С-13 в незамещенном бензоле равен 128,7 м. д.
512
Глава 8
Спектр типичного соединения бензольного ряда показан на
рис. 8.12. Отнесение сигналов может быть сделано с использова-
нием корреляций химического сдвига со структурой и интенсив-
ностью сигналов. Второй подход, использующий интенсивность
сигналов, будет рассмотрен позже.
Для предсказания химических сдвигов в бензольных углево-
дородах можно воспользоваться данными табл. 8.11. Например,
согласно этой таблице, можно определить химические сдвиги п-
хлоранизола:
6Г'Ч= 128,7 + 31,4 + (-1,9) = 158,?s ср. о 6^"= 158,2
где 128,7 — химический сдвиг атомов углерода бензола, величину
+31,4 берут из табл. 8.11 и она является химическим сдвигом под
влиянием непосредственно присоединенной метоксильной группы,
а величина —1,9 представляет собой химический сдвиг С-1 под
влиянием хлора в «-положении.
Аналогично
бГ4 = 128,7+ (-12,7)+1,3=117,3; ср. с б”абл= 115,2
Остальные данные для n-хлоранизола следующие:
6|ЫЧ= 130,7 бзабл= 129,2
д?ыч= 127,5 б”абл= 125,4
Предсказанные и вычисленные химические сдвиги для различ-
ных соединений бензольного ряда обычно совпадают с точностью
до 2 м. д. Такое совпадение наряду с тем фактом, что сигналы
неэквивалентных углеродов бензольного кольца представляют со-
бой отдельные хорошо разрешенные сигналы, означает, что струк-
туры многих моно- и дизамещенных бензолов могут быть уста-
новлены анализом спектров ЯМР-13С.
Плохое совпадение часто наблюдается для соединений, имею-
щих два или более заместителей в орто-положениях. Для 2,6-ди-
хлорбензальдегида наблюдаемые и вычисленные результаты сов-
падают плохо, особенно для атома углерода, имеющего макси-
мальное число заместителей в орто-положении (Ci):
Положение атома углерода 6выч диабл
СНО 1 137,9 130,3
с,\ /С| 2 137,3 136,6
11 А и 3 127,6 129,6
4 136,6 133,5
Специальные методы характеристики органических веществ 513
Обычно считают, что в этом случае плохое соответствие свя-
зано со стерическими эффектами; химические сдвиги, приведен-
ные в табл. 8.11, обусловлены в значительной степени электрон-
ными эффектами. Однако в некоторых случаях и для орто-заме-
щенных соединений наблюдается хорошее совпадение, например
для о-ксилола:
Положение атома углерода 6выч диабл
/СН3 1 138,4 136,4
©с 2 129,1 129,9
^СНЭ 3 125,4 126,1
Задача. Два соединения с молекулярной формулой С?Н7С1 имеют различные
спектры ЯМР-13С (приведены химические сдвиги в м. д.)
Соединение А
21,0
125,5
127,1
129,1
129,3
134,0
139,7
Соединение В
20,7
128,3
130,4
131,2
136,2
Определите структуру каждого соединения и сделайте отнесение сигналов.
8.4.2. Мультиплетность сигналов
Для химиков-органиков представляют интерес три типа спект-
ров ЯМР-13С, которые различаются степенью магнитной развязки
от протонов: 1) полная спиновая развязка (нет 13С — 'Н-взаимо-
действия), 2) неполная спиновая развязка от протонов (off-reso-
папсе) и 3) полное взаимодействие. Иллюстрации всех трех типов
даны на рис. 8.13.
Когда ЯМР-спектр неизвестного органического образца полу-
чен, его необходимо сравнить с данными, приведенными в литера-
туре. Наиболее часто в литературе встречаются ЯМР-спектры
с полной спиновой развязкой. Такие спектры измеряют для образ-
цов, которые облучаются широким диапазоном частот, захваты-
вающим всю область резонанса протонов. Это облучение выводит
все протоны из взаимодействия с углеродом, и спектр ЯМР-,3С
(см., например, рис. 8.13, в) состоит из синглетов для каждого типа
17 Зак. 335
514
Глава 8
атомов углерода. Такой спектр часто называется «полностью раз-
вязанным» спектром. (Замечание. Ядро 13С, имеющее спин ‘/г.
может взаимодействовать с другими ядрами 13С. Действительно,
в спектрах соединений, обогащенных 13С, наблюдается взаимодей-
ствие вицинальных атомов |3С с константами взаимодействия от
\ ЛI
Рис. 8.13. Спектр ядерного магнитного резонанса ,3С 3-метилпентана (перепеча-
тано с разрешения из книги: Stothers I. В., Carbon-13 NMR Spectroscopy, Aca-
demic Press, New York, copyright© 1972).
a—без развязки от протонов (спектр с полным спин-спиновым взаимодействием); б—спектр
с неполной развязкой от протонов; в—спектр с полной развязкой от протонов. Спектры из-
мерены при частоте 25,15 МГц.
30 до 170 Гц. В спектрах, измеренных для соединений с природным
содержанием 13С, углерод-углеродное взаимодействие отсутствует.
Объясните этот факт.)
Другой полезный тип спектров ЯМР-13С — это спектры с не-
полной спиновой развязкой от протонов. В таком спектре исполь-
зуемое для развязки облучение составляет несколько сотен герц;
это приводит к меньшему расщеплению сигналов углерода. Расщеп-
ление данного сигнала обычно вызвано только протонами, непос-
редственно присоединенными к углероду, дающему этот сигнал.
Таким образом эта техника обычно приводит к расщеплению
сигнала (рис. 8.13,6), что позволяет получить сведения о числе
протонов, связанных с углеродом.
Специальные методы характеристики органических веществ
515
Структурная единица, содержащая углерод Кажущаяся мульти- плетность при неполной спиновой развязке от протонов
Метил (СН3—) Метилен (—СН2—) Метин (СН—) Квадруплет Триплет Дублет
Четвертичный углерод (—С—) Синглет
Поскольку спектры с неполной спиновой развязкой от протонов
позволяют хорошо идентифицировать мультиплеты, то удобно обо-
значать синглеты в полностью развязанном спектре буквами: к —
квадруплет, т — триплет, с — синглет и т. д. и м — мультиплет, ко-
торый настолько сложен, что не может быть охарактеризован как
к, т, д или с.
Задача. Соединение с молекулярной формулой С4Н6О2 имеет следующие
сигналы в спектре ЯМР-13С:
Мультиплетность
20,2
96,8
141,8
167,6
(положение синглетов
в спектрах с полной
спиновой развязкой
от протонов)
к
т
д
с
(кажущаяся мультиплет-
ность, спектр с неполной
спиновой развязкой от
протонов)
Считая, что соединение не имеет других сигналов в спектре ЯМР-13С, опреде-
лите его структуру. Сделайте отнесение всех сигналов.
Вид спектра ЯМР-13С, полученного без развязки от протонов,
приведен на рис. 8.13, а. Этот тип спектра редко используется для
обычных структурных определений, поскольку величины констант
взаимодействия углерод — водород (от 100 до 300 Гц) такие же,
как и разница в положениях углеродных сдвигов. Поэтому в таких
спектрах с полным взаимодействием компоненты мультиплетов
часто совпадают или перекрываются. Это приводит к трудностям
при определении истинного положения сигналов и их мультиплет-
ности. Кроме того, спектры с «полным взаимодействием» услож-
няются благодаря дальнему взаимодействию углеродов и протонов,
разделенных более чем одной связью. Такие дальние взаимодей-
ствия дают ничтожное растепление в спектрах с неполной спиновой
17*
516
Глава 8
развязкой от протонов. Ясно также, что по ряду причин развязка
от протонов изменяет интенсивность сигналов углерода; так, по
сравнению с ними спектры, снятые «при взаимодействии с прото-
ном» и измеренные в тех же экспериментальных условиях, очень
слабы. По этой причине для рутинных структурных определений
обычно не используются спектры с полным протон-углеродным
спин-спиновым взаимодействием.
8.4.3. Интенсивность сигналов
Интенсивность сигнала углеродного резонанса непропорцио-
нальна числу эквивалентных углеродов в молекуле, вызывающих
этот сигнал. Существует ряд дополнительных более важных факто-
ров, определяющих интенсивность сигнала. Многие из них зависят
от настройки прибора, например от продолжительности импульса,
задержки импульса и т. д., а другие — от структурного окружения
ядра |3С (т. е. от величины времени спин-решеточной релакса-
ции Ti). Однако полезно помнить, что время спин-решеточной ре-
лаксации данного атома углерода зависит от числа связанных
с ним протонов. Обычно наблюдается следующая тенденция в изме-
нениях интенсивностей:
СН3— « —СНз— г» —СН(метнн) > —С— (четвертичный)
I I
и
СНз— « —СН2— яа —сн > \:=о
I /
В частности, приведенная последовательность указывает на по-
рядок уменьшения интенсивности сигналов ЯМР-13С этих групп.
Иногда даже бывает возможно различать эти структурные единицы
более четко: например, СН3-группу можно отличить от—СН.
В некоторых случаях заключения, сделанные на основе интен-
сивностей сигналов в спектрах ЯМР-|3С, могут приводить к ошиб-
кам; например, спектр ЯМР-13С циклогексадиена-1,4 обычно изме-
ряют в таких условиях, когда интенсивность сигнала олефинового
углерода примерно равна интенсивности сигнала метиленового
углерода. (Объясните, почему эти интенсивности одинаковы.) Од-
нако обычно используют тенденции в изменении интенсивностей:
Специальные методы характеристики органических веществ
517
В частности, атомы углерода бензольного кольца, имеющие заме-
стители и поэтому не связанные с протонами, как правило, дают
более слабые сигналы (см. рис. 8.12). Резонанс карбонильного уг-
лерода (за исключением альдегидной группы) дает сравнительно
слабый сигнал, так как в этом случае нет протонов, непосредст-
венно связанных с данным атомом углерода.
Задача. Определите структуру соединения, соответствующего молекулярной
формуле CgHnNO н имеющего следующий спектр ЯМР-|3С:
Ьс Интенсивность а
39,7 С
И 0,8 С
124,9 сл
131,9 с
154,1 сл
189,7 ср
а с — сильная, ср — средняя, сл — слабая. Используйте све-
дения об интенсивностях и данные табл. 8.И.
При некоторых условиях, а именно при добавлении к раствору
органического вещества релаксирующих реагентов, содержащих
ионы Сг(Ш) или Gd(III), в спектре ЯМР-13С можно наблюдать
сигналы с интенсивностями, пропорциональными только числу эк-
вивалентных углеродов, соответствующих данному сигналу. Чита-
телю следует ознакомиться с литературой по ЯМР-13С, приведен-
ной в гл. 10 (разд. 10.2.Ю).
8.5. ЛАНТАНОИДНЫЕ СДВИГАЮЩИЕ РЕАГЕНТЫ
При проведении анализа спектров протонного магнитного резо-
нанса (ПМР) органических соединений часто приходится сталки-
ваться с тем, что сигналы в ПМР-спектре разрешены плохо. Это
происходит в том случае, когда окружение протонов различных
типов не слишком различается, что в свою очередь приводит к очень
небольшим различиям в химических сдвигах сигналов разных про-
тонов. В таких случаях перекрывание сигналов очень затрудняет
отнесение сигналов к соответствующим протонам органического со-
единения и получение информации из величин констант спин-спино-
вого взаимодействия.
В настоящее время стало общепринятым применение способа
упрощения ПМР-спектров многих органических веществ путем об-
работки их соединениями, которые получили название лантано-
идные сдвигающие реагенты (Л СР). Л СР представляют собой
518
Глава 8
комплексы некоторых парамагнитных лантаноидных (Ln) ионов
с р-дикетонами:
Ln
R,'
О-С
•х
СН
7
О С.
Центральный ион лантаноида имеет заряд 4-3 и представляет
собой обычно европий(Ш), празеодим(III) или иттербий(III).
Комплексы, содержащие один из этих трех ионов, имеются в про-
даже (Aldrich Chemical Со., Eastman Organic Chemicals, Merck,
Alpha-Ventron). Наиболее часто используются р-дикетоны с Ri=
Ег=трет-бутил или R^rpez-бутил и Р2=перфтор-н-пропил. Ниже
приведена структура широко применяемого комплекса европия —
трис- (дипивалоилметанато) европия, сокращенно обозначаемого
Eu(dpm)3:
Еи(Щ)
С(СНэ)3'
.О-^С
СН
7
о^с
C(CH3)J3
Для сокращенного обозначения таких комплексов берут не-
сколько букв из латинского названия лиганда, например Еи(брт)з,
Pr(dpm)3, Yb(fod)3 и т. д. Часто применяется также комплекс
Еи(П()
/QCHj),
о—с
•X
СН
/
о—с
cf2cf2cf3
трис-(\, 1, 1, 2, 2, 3, 3-гептафтор-7,7-диметил-3,5-октандионато)евро-
пий, Eu(fod)3.
Сдвигающие реагенты добавляют к раствору органического со-
единения в растворителях, обычно используемых в ЯМР-спектро-
скопии (CDC13, СС14, C6D6). Это приводит во многих случаях к уве-
личению неэквивалентности сигналов в спектре ПМР органических
соединений. Добавление сдвигающих реагентов может привести
к полному разделению сигналов в спектре, что дает возможность
анализировать разделенные сигналы как спектры первого порядка.
Например, в отсутствие сдвигающего реагента спектр ПМР бензи-
лового спирта, полученный на спектрометре с рабочей часто*
Специальные методы характеристики органических веществ
519
той 60 мГц, представляет собой слегка уширенный синглет всех
ароматических протонов.
Н2ОН
бензиловый спирт
При добавлении Eu(dpm)3 в количестве 0,39 моля на каждый моль
спирта ароматические протоны дают три раздельных сигнала *.
Кроме того, сигнал протона, расположенного в пара-положении
к группе СН2ОН, наблюдается в виде триплета. Появление трип-
лета согласуется с закономерностями спектров первого порядка при
заметном взаимодействии только с лгета-протонами. Важно также
отметить, что величина смещения сигналов ароматических протонов
под действием сдвигающего реагента изменяется в следующей по-
следовательности: орто > мета > пара. Это согласуется с уравне-
нием Мак-Коннела—Робертсона, которое применяется для объясне-
ния сдвигов сигналов протонов большинства органических соеди-
нений:
Д// гл Г 3 cos2 0 — 1 х
— —К ( £5 )
где КН/Н — величина сдвига, а К — константа, зависящая от сдви-
гающего реагента и природы изучаемого органического вещества
(т. е. бензилового спирта). Значения R и 6 могут быть оценены при
рассмотрении диполь-дипольного взаимодействия ЛСР, являюще-
гося кислотой Льюиса, с теми частями молекулы органического ве-
щества, которые представляют собой основание Льюиса:
е
'--•Eu(dpm)3--£- ось
Н
где R — расстояние в комплексе от центра лантаноидного атома до
интересующего нас протона. Угол 6 строго определяется как угол
между лучом R и главной магнитной осью комплекса. Обычно
удобно, считать, что эта магнитная ось совпадает с прямой, соеди-
няющей центр лантаноидного атома с атомом органического субст-
рата, координированного с этим лантаноидом и являющегося осно-
ванием Льюиса (т. е. с атомом кислорода бензилового спирта).
Уравнение Мак-Коннела—Робертсона может быть применено
для анализа ЯМР-спектра бензилового спирта, полученного в при-
сутствии ЛСР. Член (3cos26 — 1) для орто-, мета- и /гара-протонов
меняется очень мало. Определяющим фактором является обратная
422.
Sanders J. К. М., Williams D. Н., J. Chem. Soc. (D), Chem. Commun., 1970,
520
Глава 8
пропорциональность сдвига кубу расстояния R. Поэтому протоны,
расположенные в комплексе ближе к лантаноидному атому,, при
действии сдвигающего реагента дают наибольший сдвиг. Такая
интерпретация объясняет порядок сдвигов ароматических протонов
бензилового спирта, вызванных ЛСР, и может быть использована
для отнесения сигналов протонов в других органических соедине-
ниях. Вообще если на основании молекулярной модели можно при-
нять, что значение величины (3cos26— 1) не сильно изменяется для
различных протонов вблизи функциональной группы, то те протоны,
сдвиг которых под действием ЛСР больше, расположены ближе
к лантаноидному атому в комплексе и, следовательно, ближе к
функциональной группе, координированной с лантаноидным ато-
мом. При этом молчаливо предполагается, что с ЛСР в заметной
степени координируется только одна эта функциональная группа.
Сдвигающие реагенты успешно применяются для органических
субстратов, которые имеют в своем составе основные функциональ-
ные группы. Обычно наблюдается следующая последовательность
органических соединений по их сродству к обычным ЛСР:
амины > спирты > альдегиды > кетоны > сложные эфиры > нитрилы
Исключения из этой последовательности наблюдаются в том
случае, когда кроме фактора основности появляются какие-либо
другие факторы, такие, как стерические эффекты. Например, трет-
бутиловый спирт связывается с ЛСР слабее, чем простые кетоны.
Значительные сдвиги наблюдаются при комплексообразовании
с сульфоксидами, сульфитами, сульфинами, N-оксидами, N-нитро-
зосоединениями и азоксисоединениями. Для карбоновых кислот и
фенолов сдвиги наблюдаются при использовании ЛСР с менее ос-
новными лигандами (например, fod). Добавление к кислотам или
фенолам Eu(dpm)3 приводит к нежелательным кислотно-основным
реакциям аниона дипивалоилметана с протонами гидроксильной
группы фенолов.
Комплексы Рг(Ш) и Eu(III) с 0-дикетонами обладают еще и
другими свойствами, заслуживающими обсуждения. Обычно комп-
лексы Рг(Ш) сдвигают сигналы протонов субстрата в сильное
поле, а комплексы Eu(III) —в слабое. Часто более предпочтитель-
ным реагентом является комплекс Eu(III), так как в простых сое-
динениях протоны, имеющие сигналы в слабом поле в отсутствие
сдвигающих реагентов, расположены ближе всего к функциональ-
ной группе. Поэтому при добавлении сдвигающих реагентов их
сигналы сдвигаются сильнее всего. Например, в спектре ПМР
н-бутилового спирта протоны у углерода, находящегося в a-положе-
нии к группе ОН, в отсутствие ЛСР дают сигналы в наиболее сла-
бом поле. Прибавление дикетонатного комплекса Рг(Ш) так
сильно сдвигает сигналы этих a-протонов в сильное поле, что они
попадают в ту же область, где уже находятся сигналы других про-
тонов. Это может привести к перекрыванию сигналов и нежелатель-
Специальные методы характеристики органических веществ
521
ному усложнению спектра. В таких случаях комплексы Ен более
пригодны, так как они увеличивают разницу между химическими
сдвигами протонов в таких веществах, как н-бутиловый спирт. При
этом протоны, сигналы которых находились в слабом поле, сме-
щаются в сторону еще более слабого поля.
В некоторых случаях желательно избежать контактного сдвига,
с тем чтобы величина сдвига, индуцированного ЛСР, могла быть
описана уравнением Мак-Коннела—Робертсона. В тех случаях,
когда определение контактных сдвигов может вызвать затрудне-
ния (т. е. всегда в ЯМР-13С- и ПМР-спектрах сильно основных
аминов), можно использовать вместо соединений Рг или Ей комп-
лекс Yb(dpm)3 и другие иттербиевые сдвигающие реагенты.
Анализ хиральных органических соединений проводят, исполь-
зуя хиральные сдвигающие реагенты. Установлено, что для этого
очень полезны производные камфоры:
Ниже приведены два хиральных сдвигающих реагента и их со-
кращенные торговые названия, под которыми они продаются:
1) Pra-Opt, Ln = Рг, R = CF3, трис- (3-трифторметилоксиметилен-
d-камфорато) празеодим (III); 2) Eu-Opt, Ln = Eu, R = CF3, трис-
(З-трифторметилоксиметилен-й-камфорато) европий (III).
Применение хиральных сдвигающих реагентов можно пояснить
на примере рацемического а-фенилэтиламина, содержащего и (+)-
и (—)-энантиомеры:
СНз
I
С6Н5СН—NH2 + Eu-Opt —>
(±) (+)
образец рацеми-
ческого амина
СН, СН3
I I
—► C6H5CHNH2 • Eu-Opt + C6H6CHNH2 • Eu-Opt
(t) w
Сигналы ПМР соответствующих протонов энантиомеров исход-
ного (±)-амина идентичны, поскольку энантиомеры отличаются
только своей способностью вращать плоскость поляризованного
света. Обработка рацемического амина хиральным оптически ак-
тивным комплексом Eu-Opt [здесь произвольно выбран (+)] дает
522
Глава 8
два диастереомерных комплекса; и (±). Сигналы ПМР соот-
ветствующих протонов в этих диастереомерах не идентичны; ин-
тегрирование неидентичных сигналов дает относительные количе-
ства и (+)-комплексов и, следовательно, относительные ко-
личества (+)- и (—)-амина в исходном образце.
Исследование спектров ЯМР в присутствии сдвигающих реа-
гентов проводится обычно в таких растворителях, как CDCI3, СС14
и СбО6. Эти растворители представляют собой слабые основания
Льюиса и поэтому не занимают координационных мест в сдвигаю-
щем реагенте. Протоны р-дикетонатных лигандов сдвигающих реа-
гентов поглощают в следующих областях:
Eu(fod)3 от 0,4 до 2,0 м. д.
Eu(dpm)3 от 1,0 до —2,0 м. д.
Pr(dpm)3 от 3,0 до 5,0 м. д.
Эти области поглощения в некоторой степени зависят от при-
меняемого растворителя и от органического соединения, с которым
координируется сдвигающий реагент. Используются и дейтериро-
ванные сдвигающие реагенты (фирмы Aldrich и Alpha). При этом
исключаются помехи от резонанса протонов таких лигандов. До-
полнительный материал по этому вопросу можно найти в следую-
щих работах:
Sievers R. Е., N. М. R. Shift Reagents, Academic Press, New York,
1973.
Hofer O., Topics in Stereochemistry, edited by N. Allinger and
E. L. Eliel, vol. 9, Wiley-Interscience, New York, 1976, p. Iliff.
Willcott M. R., Ill, Davis R. E., Science, 190, 850 (1975).
Cockerill A. et al., Chem. Rev., 73, 553 (1973).
Campbell J. R., Aldrichimica Acta, 4, 55 (1971).
Глава 9
Установление структуры: методы
определения и упражнения
Лабораторное исследование органических соединений позво-
ляет получить сведения о физических свойствах, наличии или от-
сутствии элементов, растворимости, спектральных данных, пове-
дении по отношению к некоторым соответствующим классам
реагентов, а также в различных специальных реакциях. Все эти на-
блюдаемые факты должны быть сопоставлены и объяснены, с тем
чтобы на их основе можно было вывести возможные структурные
формулы для исследуемого соединения. Необходимо установить,
какие имеются функциональные группы, определить природу ядер,
к которым они присоединены, и найти места их присоединения.
Цель этой главы—показать на некоторых характерных при-
мерах, как следует приступать к решению таких задач и какие
рассуждения используются для вывода возможной структуры мо-
лекулы из экспериментальных данных.
ПРИМЕРЫ
ЧАСТЬ 1. УСТАНОВЛЕНИЕ СТРУКТУРЫ СОЕДИНЕНИИ,
ОПИСАННЫХ В ЛИТЕРАТУРЕ
Для идентификации таких соединений нет необходимости про-
водить количественный анализ присутствующих элементов, опре-
делять молекулярную массу и вычислять молекулярные формулы.
Их идентификация основана на соответствии физических и хими-
ческих свойств изучаемого вещества и данных о его производных.
Лабораторная работа в этом случае, как описано в предыдущих
главах, заключается в изучении этих данных для ранее описанных
соединений.
Две физические константы, описанные в гл. 6, являются очень
полезными, а именно: эквиваленты нейтрализации кислот и осно-
ваний и числа омыления сложных эфиров. Эти цифровые данные
вместе с определением класса растворимости и поведения по от-
ношению к тем или иным реагентам часто дают ценные указания
о молекулярной структуре соединения. Их использование показано
на следующих приведенных ниже примерах.
Пример 1. Эквивалент нейтрализации органической кислоты
равен 45±1. Как было показано в методике 32, эквивалент
524
Глава 9
нейтрализации.кислоты зависит от числа карбоксильных групп в
молекуле. Если присутствует только одна карбоксильная группа,
то эквивалент нейтрализации равен молекулярной массе. Таким об-
разом, если изучаемое соединение является одноосновным, то его
молекулярная масса * может быть 44, 45 или 46. Молекулярная
масса карбоксильной группы равна 45. Следовательно, если бы
найденная молекулярная масса была равна 45, то к карбоксиль-
ной группе больше ничего не могло быть присоединено. Молеку-
лярная масса 44 явно невозможна, а для молекулярной массы 46
после вычета массы карбоксильного радикала остается остаток 1.
Только один элемент (водород) имеет атомную массу, равную 1.
Исходя из этого, единственно возможным соединением будет му-
равьиная кислота (НСООН).
Однако изучаемое соединение может быть двухосновным, и
тогда его молекулярная масса будет равна 90 ± 2. Молекулярная
масса двух карбоксильных групп составляет 2 X 45 = 90. Следо-
вательно, масса возможного остатка может быть равна 0, 1 или 2.
Двухвалентных атомов с таким значением атомной массы нет,
поэтому единственно возможной двухосновной кислотой будет ща-
велевая кислота, в которой соединены две карбоксильные группы:
СООН
I
СООН
Таким образом, предположив наличие сначала•одноосновной
кислоты, а затем двухосновной на основании одного эквивалента
нейтрализации были выведены две возможные структуры. Для
того чтобы сделать выбор между этими двумя возможностями, по-
лезно рассмотреть физическое состояние и класс растворимости
исследуемого соединения. Если вещество с эквивалентом нейтра-
лизации, равным 45 ± 1, представляет собой жидкость и раство-
римо в воде и чистом эфире (группа растворимости Si), то это
муравьиная кислота. Если исследуемое вещество твердое, раство-
римое в воде, но нерастворимое в эфире (группа S2), то это без-
водная щавелевая кислота.
Аналогичное рассмотрение молекулярных масс показывает,
что исследуемое соединение не может быть трехосновным (моле-
кулярная масса 135 ± 3) или четырехосновным (молекулярная
масса 180 ± 4).
Пример 2. Кислота А имеет эквивалент нейтрализации 136+1.
Она не содержит галогенов, азота и серы. Холодный раствор пер-
манганата калия не обесцвечивается этим соединением. Однако
* В данной главе для иллюстративных целей и вычислений используются
целочисленные значения атомных масс углерода, водорода, кислорода, азота и
брома. Действительные атомные массы (которые должны использоваться во всех
точных количественных анализах) отличаются от этих округленных значений
на величину, меньшую экспериментальной ошибки при определении эквивален-
тов нейтрализации и чисел омыления.
Установление структуры: методы определения и упражнения
525
если щелочной раствор этого вещества нагревать в течение часа
с перманганатом калия, а затем подкислить, то осаждается соеди-
нение Б, которое имеет эквивалент нейтрализации 83 ± 1.
Сначала рассмотрим соединение Б. Предположим, что оно од-
ноосновно
молекулярная масса = 83 ± 1
минус одна группа СООН = 45
остаток = 38 ± 1
Этот остаток, к которому присоединена карбоксильная группа,
должен состоять из углерода, водорода и, возможно, кислорода и
быть устойчивым по отношению к горячему раствору перманганата
калия. Исследование показывает, что это невозможно
остаток = 38 ± I
три атома углерода =3X12 = 36
остаток = 2 ± 1
Остатком может быть группа С3Н, С3Н2 или С3Н3, но ни одна
из них не может соответствовать структуре, устойчивой по отноше-
нию к перманганату. В качестве исходного вещества мог бы быть
углеводород С3Н8, и тогда в исследуемое соединение Б входила
бы алкильная группа С3Н?—; аналогично исходным циклоалканом
мог бы быть С3Н6 и радикалом в соединении Б была бы цикло-
пропильная группа С3Н3—.
Наличие кислорода в этом остатке также исключается. Если
предположить, что кислород все же присутствует, то получатся
следующие данные:
остаток = 38 ± 1 или остаток = 38 ± 1
одни атом кислорода = 16 два атома кислорода = 32
22 ± 1 остаток = 6 ± 1
один атом углерода = 12
остаток = 10 ± 1
Ни один из этих остатков не соответствует какому-либо атому
или группе атомов, которые отвечали бы возможному органиче-
скому соединению. Например, для соединения, соответствующего
первому остатку, следует предположить формулу СНюОСООН,
а для второго — Н6О2СООН, причем обе они бессмысленны. Та-
ким образом, можно с уверенностью заключить, что соединение Б
не может быть одноосновным.
Предположим, что соединение Б двухосновно:
молекулярная масса = 2 X S3 ± 1 = 166 ± 2
две карбоксильные группы = 90
остаток = 76 ± 2
626
Глава 9
Если этот остаток является насыщенным и алифатическим, то он
должен состоять из нескольких групп СН2:
—(СН2)6=5Х 14 = 70
—(СН2)6 = 6Х 14 = 84
Ни один из них не соответствует массе остающегося радикала
76 ± 2.
Другой группой, устойчивой к действию горячего раствора пер-
манганата калия, является бензольное кольцо, представляющее
собой систему из шести СН-групп. Молекулярная масса бензола
равна 6X13 = 78. Если присутствуют две карбоксильные группы,
которые замещают два атома водорода, то масса остатка стано-
вится равной 78 — 2 = 76. Это значение совпадает с вычисленной
выше массой остатка, и, следовательно, возможной структурой
соединения Б является СбН4(СООН)2, т. е. изучаемое соединение
может быть одной из фталевых кислот.
Теперь возникает вопрос, не может ли соединение Б быть трех-
основным. Если это так, то будут получены следующие значения:
молекулярная масса = 3 X 83 ± 1 = 249 ± 3
три карбоксильные группы = 3 X 45 = 135
остаток = 114 + 3
Рассмотрение показывает, что этот остаток не Может быть
ароматическим, поскольку он не соответствует одному или более
бензольным кольцам. Исключена также система бензольного коль-
ца с боковой цепью, поскольку боковая цепь окислялась бы пер-
манганатом. Однако значение 114 ± 3 соответствует восьми груп-
пам СН2 (8X14= 112) при наличии экспериментальной ошибки.
Следовательно, соединение C8Hi5 (СООН) 3, имеющее молекуляр-
ную массу 246, попадает в область 249 ± 3. Хотя эта трикарбоно-
вая кислота представляет собой возможную структуру для соеди-
нения с молекулярной массой 249 ± 3, но его нельзя получить из
соединения А, которое имело эквивалент нейтрализации 136 ±1.
Предположим, что кислота А одноосновна:
молекулярная масса Л = 136 + 1
минус одна группа СООН = 45
остаток = 91 ± 1
Поскольку в веществе Б имеется группа СеШ, устойчивая к дей-
ствию перманганата калия, то она должна присутствовать также
и в кислоте А:
остаток — 91 ± 1
С6Н4 = 76
остаток =15+1
Установление структуры: методы определения и упражнения
527
Этот остаток, равный 15 ± 1, соответствует метильной группе,
присоединенной к ароматическому кольцу.
.СООН /СООН
с6н/ —> с6н/
\СН3 \COOH
соединение А соединение Б
эквивалент эквивалент
нейтрализации 136 нейтрализации 83
Исходное соединение может быть о-, м- или n-толуиловой кис-
лотой, каждая из которых способна дать приведенную реакцию.
Для окончательного выбора необходимы дополнительные данные,
такие, как температура плавления, свойства каких-либо производ-
ных или спектры.
Этот пример показывает также, что при окислении соединение
с определенным эквивалентом нейтрализации почти всегда превра-
щается в продукт с более низким эквивалентом нейтрализации.
Это обобщение естественно следует из увеличения числа карбо-
ксильных групп или расщепления молекулы на более мелкие
фрагменты.
Пример 3. Бесцветное кристаллическое соединение А содержит
азот, но не содержит галогенов и серы. Оно не растворяется в во-
де, разбавленных кислотах и щелочах. При его взаимодействии
с аммонийгексанитратоцератным реагентом образуется комплекс
красного цвета. Однако это вещество не реагирует с фенилгидра-
зином. При растворении соединения А в горячем растворе гидро-
ксида натрия выделяется аммиак и образуется прозрачный раст-
вор. При подкислении этого раствора получается соединение Б,
которое не содержит азота; его эквивалент нейтрализации равен
182 ± 1. Окисление соединения Б горячим раствором перманга-
ната калия дает соединение В, эквивалент нейтрализации которого
равен 98 ± 1. При нагревании соединений А или Б в течение не-
которого времени с бромистоводородной кислотой получается сое-
динение Г. Это вещество содержит бром, но не содержит азота.
С бромной водой оно дает осадок, при добавлении хлорида же-
леза(Ш) окрашивается в фиолетовый цвет, легко восстанавливает
разбавленный раствор перманганата калия и растворяется в раст-
воре бикарбоната натрия.
Все эти реакции можно обобщить в виде следующей схемы:
NaOH, + раствор
, / 1Н"
соединение А. соединение Б
|кМпО., нагревание
соединение Г соединение В
На основании того, что при щелочном гидролизе соединения
А выделяется аммиак, можно предположить наличие в нем
528
Глава 9
нитрильной или амидной группы, поскольку при гидролизе нитри-
лов и амидов выделяется аммиак:
RCN + NaOH + Н2О —> RCOONa + NH3
RCONHs + NaOH —> RCOONa + NH3
Присутствие имидной группы —CONHCO—, которая также
распадается с выделением аммиака, исключается на том основа-
нии, что соединение А не растворяется в растворе гидроксида нат-
рия. Ввиду того что соединение Б не содержит азота, то также ис-
ключаются амины с отрицательно заряженными заместителями,
такие, как 2,4-динитроанилин. Отсутствие азота в соединении Б и
тот факт, что это соединение кислое, а не основное, позволяют
исключить из рассмотрения также замещенные карбамиды, при
гидролизе которых также выделяется аммиак.
rnhconh2 + н2о —> rnh2 + co2 + nh3
Образование раствора красного цвета под действием аммоний-
гексанитратоцератного реагента указывает на наличие спиртовой
группы. Присутствие аминогруппы или фенольной группы исклю-
чается, так как соединение А нейтрально. Дополнительным до-
казательством наличия гидроксильной группы является тот факт,
что соединение Г, образовавшееся при действии бромистоводород-
ной кислоты на вещество А, содержало бром:
ROH + НВг —> RBr + H2O
Свойства соединения Г убедительно свидетельствуют о нали-
чии фенольной гидроксильной группы. Легкость бромирования,
чувствительность к действию перманганата калия, образование
окраски с хлоридом железа (III) являются характерными свойст-
вами замещенных фенолов. Эта фенольная гидроксильная группа
образовалась при действии бромистоводородной кислоты на функ-
циональную группу, имевшуюся в А и Б, так как ни одно из этих
веществ не содержало фенольной группы. Одной из групп соеди-
нений, дающих фенол при действии бромистоводородной кислоты,
являются арилалкиловые эфиры:
ArOR + НВг —> ArOH + RBr
Если алкильная группа имеет низкую молекулярную массу, то
при обработке бромистоводородной кислотой она может улету-
читься в виде алкилбромида. Таким образом, соединения А, Б и
В, вероятно, содержат такую смешанную эфирную группу, а так-
же ароматическое ядро, так как последнее имеется в замещенном
феноле Г. Растворимость соединения Г в растворе бикарбоната
натрия, вероятно, обусловлена наличием карбоксильной группы,
так как и нитрильная и амидная группы гидролизуются в карбо-
ксильные группы под действием как кислот, так и щелочей. Сле-
довательно, соединение Г представляет собой оксибензойную кис-
лоту с бромом в боковой цепи. Эта боковая цепь должна при-
Установление структуры: методы определения и упражнения 529
сутствовать также и в соединении А со спиртовой группой вме-
сто атома брома.
Теперь рассмотрим эквиваленты нейтрализации соединений Б
и В. Следует отметить, что эквивалент нейтрализации соединения
В, образующегося при окислении перманганатом, ниже эквива-
лента нейтрализации соединения Б, которое приобрело свои кис-
лые свойства только после реакции гидролиза. Очевидно, это
окисление затрагивает боковую цепь, и поэтому в соединении В
должно быть больше карбоксильных групп, чем в веществе Б.
Предположим, что соединение В двухосновно:
молекулярная масса = 2 X 98 ± 1 = 196 + 2
две карбоксильные группы = 2 X 45 = 90
106 ± 2
одни атом кислорода эфириой связи = 16
90 ± 2
бензол минус три атома водорода (СбН3) = 75
остаток =15 + 2
Остаток 15 соответствует СНз-группе, и, следовательно, эфир-
ной группой должна быть группа СНзО—.
Теперь необходимо определить длину боковой цепи, к которой
присоединена спиртовая группа в соединениях А и Б.
Если соединение В двухосновно, то вещество Б одноосновное:
эквивалент нейтрализации = молекулярная масса = 182 ± 1
карбоксильная группа = 45
137 ± 1
метоксильная группа = 31
1 )6± I
гидроксильная группа= 17
89 + 1
бензольное кольцо (С6Н3) = 75
остаток = 14 ± 1
Этот остаток представляет собой массу алифатической боковой
цепи и явно соответствует группе —СН2—. Следовательно, воз-
можными структурами веществ А, Б, В и Г являются следующие:
CONHzfcw CN) СООН
С6Н,—СН2ОН NaOH. СДЦ—СН2ОН
\)СН, / Хосн,
соединение А у' соединение Б
I НВг / |
НВг| |кМпО4
СООН s' СООН
С6Н,—СН2Вг С„Н3—СООН
\>Н ХОСН,
соединение Г соединение В
530
Глава 9
Частота, см~
Установление структуры: методы определения и упражнения
531
На этом примере видно, что один и тот же реагент может воз-
действовать более чем на одну функциональную группу. Так, ки-
пящая бромистоводородная кислота подействовала на три функ-
циональные группы в соединении А и на две — в соединении Б.
Пример 4. Кристаллическое соединение с т. пл. 60—61 °C не
содержит серы, азота и галогенов, не растворяется в воде, разбав-
ленной кислоте и щелочи, но реагирует с концентрированной сер-
ной кислотой и по растворимости относится к группе MN. ИК-
Спектр тонкой пленки (расплава) этого соединения представлен
на рис. 9.1.
Следующие полосы поглощения указывают на присутствие не-
скольких функциональных групп в этом соединении:
5,95 мкм (1680 см-1)
6,3 мкм (1590 см-1)
6,65 мкм (1500 см-1)
валентные колебания С=О (сопряженной с л-элек-
тронной системой)
валентные колебания ароматических связей С—С
валентные колебания ароматических связей С=С
Наличие этих функциональных групп установлено на основа-
нии таблиц, приведенных в гл. 5 и 6, а также в гл. 3 книги Силь-
верстейна, Басслера и Моррилла *. Сильные полосы поглощения
при 7,9 мкм (1270 см-1), 8,80 мкм (1140 см-1) и 9,8 мкм
(1020 см"1) указывают на наличие в молекуле фрагмента С—О—С;
такой фрагмент имеется в простой эфирной или сложноэфирной
функциональных группах.
Проба исходного соединения давала оранжево-красный осадок
при взаимодействии с 2,4-динитрофенилгидразином, что подтверди-
ло присутствие альдегидной или кетонной группы, но не исклю-
чало возможности наличия эфирной группы наряду с альдегидной
или кетонной группой.
Студентка, имевшая задание идентифицировать это соединение,
изучила таблицы свойств кристаллических кетонов, альдегидов и
эфиров, температуры плавления которых лежат в области ** от
60 до 61 ± 2°С. При этом были получены соединения, приведенные
на стр. 532.
Необходимо отметить, что в приведенном списке содержатся
несколько соединений, которые не дают полос поглощения связей
С—О—С, по-видимому имеющихся в ИК-спектре. Однако ясно,
что вначале лучше иметь список из большего числа соединений,
чем самонадеянно ограничить этот список и случайно пропустить
правильную структуру.
* Сильверстейн Р., Басслер Г., Моррилл Т. Спектрометрическая идентифи-
кация органических соединений. Пер. с англ.—М.: Мир, 1977.
** Расширение области температур плавления от 58 до 62 °C было сделано
для того, чтобы учесть возможность наличия примесей, различий в технике опре-
деления и в качестве термометров, которые использовались предыдущими хими-
ками. Использование такой более широкой области уменьшает возможность про-
пуска данного соединения.
532
Глава 9
Кристаллические
кетоны Т пл -с
U) С6Н5СОСН2С6Н5 60
Т. пл., °C
(Г) СвН6СН=СНСОСвН6
62
62
(В) С6Н,СОСН2СОСН3 61
п
С6Н5—с=снсосн3
он
60
60
Изучение таблиц свойств кристаллических сложных эфиров
с т. пл. 58—63°С, которые также содержали бы альдегидную или
кетогруппу, не привело к расширению этого списка. После этого
был вновь рассмотрен ИК-спектр совместно с приведенными
структурными формулами. Средние полосы поглощения в области
около 3,4 мкм (2940 см-1) и 3,5 мкм (2860 см-1) указывают на
ароматический альдегид (полосы поглощения альдегидных С—Н-
связей); полосы для колебаний С—О—С (см. выше) предпола-
гают наличие метоксигрупп, соединенных с ароматическим яд-
ром.
Для сокращения числа возможных структур были проведены
два дополнительных исследования. Иодисто во дородная кислота
(проба Цейзеля на СН3О—) дала положительный результат. Это
позволяет исключить из рассмотрения соединения А, Б, В, Г, Ж,
3 и И, так что остаются только структуры Д, Е и К, содержащие
метоксильные группы наряду с карбонильной группой.
Хотя уже на основании ИК-спектра было отдано предпочтение
ароматическому альдегиду, кетон Д был исключен из рассмотре-
ния потому, что исходное соединение окислялось раствором пер-
манганата калия и кислым раствором хромового ангидрида. Чтобы
провести различие между остающимися возможностями (соедине-
ниями Е и К), был получен оксим. Его температура плавления
оказалась равной 95°С, что подтверждает идентичность исходного
соединения с 3,4-диметоксибензальдегидом (соединение Е, верат-
ровый альдегид), поскольку для оксима соединения К в литера-
туре приведена т. пл. 102°С.
Установление структуры: методы определения и упражнения
533
Следует отметить, что при таком исследовании могут быть
полезны данные ЯМР-спектров и другие химические реакции, хотя
они и не были использованы в этом примере. Два синглета в
ЯМР-спектре около б 3,8 м. д. подтвердили бы наличие аромати-
ческих метоксигрупП, а положительная проба Толленса свидетель-
ствовала бы о присутствии альдегидной группы.
Пример 5. Одно из неизвестных соединений плавится при
60—6ГС и не содержит серы, азота или галогенов. По раствори-
мости оно относится к группе MN и образует оранжево-красный
осадок с 2,4-динитрофенилгидразином. На рис. 9.2 приведен ИК-
спектр этого неизвестного соединения.
В ПМР-спектре этого неизвестного соединения (в CDC13,
60 МГц, ТМС б = 0,00) имелись следующие сигналы:
б 3,83 м. д., ЗН, узкий синглет
б 6,95 м. д., 2Н, дублет (линия в сильном поле менее активна)
б 7,3—8,0 м. д., 7Н, мультиплет
Первый сигнал в ПМР-спектре четко указывает на наличие ме-
тильной группы; из величины химического сдвига следует, что
метильный радикал связан с кислородом. Все прочие сигналы
указывают на ароматический характер вещества.
На основании наличия полосы поглощения карбонильной груп-
пы при 6,0 мкм (1667 см-1) был составлен список возможных
кристаллических альдегидов и кетонов с температурами плавле-
ния от 58 до 63°С. Этот список оказался идентичным тому, кото-
рый приведен выше в примере 4. Поскольку соединение, структуру
которого было необходимо установить, не восстанавливало пер-
манганата калия и хромового ангидрида, то можно было исклю-
чить все окисляющиеся соединения, а именно В, Г, Е, Ж, 3, И и К.
Остаются возможные структуры А, Б, Д. Из рассмотрения этих
структур видно, что только в соединении Д имеется метокси-
группа. ИК-Спектр неизвестного соединения [антисимметричные
валентные колебания С—О—С арилалкилового эфира при 7,9 мкм
(1266 см-1), симметричные валентные колебания С—О—С при
9,7 мкм (1030 см-1)] также указывал на возможное присутствие
этой функциональной группы. Исходя из этого, была сделана
проба с иодистоводородной кислотой, которая оказалась поло-
жительной. Окончательная идентификация была проведена путем
получения 2,4-динитрофенилгидразона, который после пере-
кристаллизации плавился при температуре 180—18ГС, что под-
твердило структуру 4-метоксибензофенона для исследуемого сое-
динения.
Пример 6. Соединение неизвестной структуры плавится в пре-
делах от 60 до 61°С, не содержит азота, серы и галогенов, а по
растворимости относится к группе MN. При действии 2,4-динитро-
фенилгидразина образуется осадок; ПК-спектр этого вещества
приведен на рис. 9.3.
Рис. 9.2. Инфракрасный спектр к примеру 5 (перепечатано с разрешения из книги: The Aldrich Collection of IR Spectra
edited by C. J. Pouchert, copyright © 1971, 1975).
536
Глава 9
В ПМР-спектре этого соединения (в CDC13, 60 МГц, ТМС
6 = 0,00) имелись следующие сигналы:
6 6,92 м. д., 2Н, узкий сииглет
6 7,3—7,6 м. д., 6Н, мультиплет
б 7,8—8,1 м. д., 4Н, мультиплет
Вызывало недоумение наличие довольно широкой полосы по-
глощения в ИК-спектре в области от 6,0 до 6,5 мкм (1667—
1538 см-1). Вновь был составлен список кристаллических альде-
гидов и кетонов с температурами плавления от 58 до 63°С. Этот
список возможных структур был идентичен тому, который приве-
ден в примере 4. При обработке неизвестного соединения раство-
ром перманганата калия и хромовым ангидридом оба этих реа-
гента восстанавливались. На этом основании из списка были
исключены неокисляющиеся соединения: А, Б и Д. Затем провели
пробу Цейзеля на присутствие метоксигруппы. Поскольку эта
проба оказалась отрицательной, из списка возможных структур
были исключены соединения Е и К- На том же основании сле-
довало бы исключить соединение Д, отвергнутое ранее. Отсутст-
вие в ПМР-спектре изучаемого вещества сигнала метоксильной
группы подтверждает обоснованность исключения этих веществ.
В качестве возможных структур остались соединения В, Г, Ж и 3.
После этого был снова рассмотрен ИК-спектр. При этом его
полосы поглощения сравнивали с таблицами с учетом соображе-
ний, приведенных в гл. 5 и 6. По-видимому, широкая полоса
поглощения в области 6,2—6,5 мкм относится к валентным С=С-
колебаниям енолизованного кетона. Полоса О—Н валентных ко-
лебаний енолов является широкой и в данном примере распрост-
раняется от 3,1 до 4,0 мкм (от 3200 до 2500 см-1, с низким погло-
щением вследствие уширения). Исходя из этого, была проведена
реакция исследуемого соединения с реактивом хлорид желе-
за (III) — пиридин, при этом образовался раствор голубовато-
красного цвета. Полученный препарат 2,4-динитрофенилгидразона
плавился в интервале температур от 150 до 151°С, что согласует-
ся с литературным значением для этого производного бензоилаце-
тона (соединение В).
Ретроспективно можно несколько подробнее рассмотреть сиг-
налы, имеющиеся в ПМР-спектре. Синглет в наиболее сильном
поле относится к метиленовой группе, а ароматические сигналы
в наиболее слабом поле относятся к орто-протонам бензоильной
группы *.
* Интересно отметить, что исследуемый образец в ПМР-спектре не пока-
зывает заметной енолизации. В то же время, несмотря на это, ИК-спектр этого
образца указывает на весьма сильную енолизацню.
Установление структуры: методы определения и упражнения
537
II II
СЙН5—ССН2С—СН, (В)
и
zH,
о 'о
С6Н,—С /С—СН,
ГН
или
,н.
о' 'о
I: :|
С6Н5—^С— СН,
СН
Пример 7. Сложный эфир содержит только углерод, водород
и кислород и имеет число омыления, равное 74 ± 1.
Первой стадией исследования в этом случае является рассмот-
рение предположения о том, что молекула содержит только одну
эфирную группу. В этом случае молекулярная масса равна числу
омыления. Общая формула сложного эфира R—СОО—R', и по-
этому сначала вычитаем из молекулярной массы массу группы
-СОО-
молекуляриая масса = 74 ± 1
—СОО— = 44
остаток = 30 ± 1
Этот остаток представляет собой суммарную массу радикалов
R и R'. В насыщенных эфирах *, содержащих только углерод,
водород и кислород, этот остаток должен соответствовать
СлНгп+2 и, кроме того, всегда должен быть четным числом **.
Поэтому значения массы остатка 31 и 29 невозможны и число 30
представляет молекулярную массу С„Н2п+2. Простое рассмотрение
в этом случае показывает, что углеводородным остатком будет
СгН6, но в общем для нахождения значения п необходимо умно-
жить его на атомные массы элементов, входящих в формулу, и их
сумму приравнять массе остатка.
12и + 1(2л + 2) = 30
14п = 28 и п = 2
Остаток С2Нб представляет собой сумму радикалов R и R', и
теперь необходимо написать для них все возможные комбинации:
R + R' = С2Н6
Н -j- С2Н5
СН3 “Р СНз
* В эфирах, содержащих двойную связь, он будет равен СпН2п, в ацети-
леновых эфирах — С„Н2п-2.
♦* См. примечание к примеру 1.
538
Г лава 9
Следовательно, если предположить, что исследуемое соединение
было моноэфиром, возможными структурами для него будут сле-
дующие:
НСООС2Н5 и СНзСООСНз
этилформиат метилацетат
Если в молекуле находятся две сложноэфирные группы, то мо-
лекулярная масса будет в два раза больше числа омыления. Две
сложноэфирные группы будут содержать две группы —СОО—,
отсюда
молекулярная масса = 2 X 74 ± 1 = 148 ± 2
две группы —СОО— = 2 X 44 = 88
остаток = 60 ± 2
Значение 60 ± 2 представляет собой сумму всех частей молекулы,
за исключением двух групп —СОО—. Аналогично предыдущему
СпНгп+2 — 60 ± 2 и
12п + 1(2п + 2) = 60 ±2
14и = 58±2
Поскольку п должно быть целым числом, то единственным
числом в правой части уравнения, которое будет удовлетворять
этому требованию, является 56, и, следовательно, п == 4. Остаток
должен иметь состав С4Н10, и его следует распределить между
различными углеводородными радикалами в общих формулах
для соединений с двумя сложноэфирными группами. Некоторые
из возможных общих формул приведены ниже:
COOR COOR RCOO RCOO 1
(СН2)Л 1 (RCH), (CH2)X (FCH),
। COOR COOR 1 RCOO 1 RCOO
Используя эти общие формулы, для сложных эфиров дикарбо-
новой кислоты или гликоля можно написать возможные структу-
ры, в которых четыре атома углерода и десять атомов водорода
распределены соответствующим образом в каждой из вышепри-
веденных формул.
На этом примере показано использование чисел омыления для
вывода возможных структурных формул. При этом также видно,
что число омыления не так надежно, как эквивалент нейтрализа-
ции кислоты. При этом всегда желательно иметь какие-либо до-
полнительные данные относительно кислоты или спирта (или
обоих), образующихся при омылении сложного эфира, чтобы
уменьшить число изомерных эфиров, которые имеют одинаковое
число омыления.
Установление структуры: методы определения и упражнения
539
ЧАСТЬ II. ОПРЕДЕЛЕНИЕ СТРУКТУРЫ СОЕДИНЕНИЙ,
НЕ ОПИСАННЫХ РАНЕЕ В ХИМИЧЕСКОЙ ЛИТЕРАТУРЕ
Установление молекулярных формул
В исследовательской работе после проведения количественных
анализов наряду с определениями молекулярных масс часто мож-
но установить молекулярную формулу любого неизвестного ве-
щества. Большую информацию о наличии функциональных групп
во многих случаях можно получить только на основе знания мо-
лекулярной формулы, и по этой причине уместно рассмотреть от-
дельно значимость молекулярной формулы.
Насыщенный углеводород без каких-либо циклов имеет общую
формулу СлН2п+2. Введение кислорода приводит к спирту, про-
стому эфиру, ацеталю или какому-либо другому насыщенному
ациклическому соединению без изменения соотношения между
углеродом и водородом.
Молекулярная формула этих соединений CnH2n+2Om. Для вве-
дения двойной связи или цикла в насыщенную молекулу необхо-
димо удалить два атома водорода, а при введении тройной связи
удаляются четыре атома водорода. Поэтому на основании изуче-
ния соотношения между углеродом и водородом можно сделать
вывод о возможном числе кратных связей или циклов в молекуле.
Например, соединение С3НеО должно иметь или одну двой-
ную связь (олефиновую или карбонильную), или один цикл, но не
может содержать тройную связь, потому что до структуры насы-
щенного соединения не хватает только двух атомов водорода.
В соединении C8Hi2O должны быть или три двойные связи, или
три цикла, или какая-либо комбинация двойных связей, тройных
связей и циклов, которая будет представлять собой три пары не-
достающих атомов водорода. Однако это соединение не может
иметь в своем составе бензольного кольца, поскольку для него
необходимо отсутствие, по сравнению с насыщенным соединением,
четырех пар атомов водорода. (Таким образом, немедленно исклю-
чается возможность того, что кислородная функция представляет
собой фенольную гидроксильную группу.)
Более того, введение атома галогена в насыщенную молекулу
неизбежно повлечет за собой удаление одного атома водорода, и
поэтому насыщенные ациклические моногалогениды имеют общую
формулу С„Н2„+1Х. С другой стороны, при введении атома азота
с образованием ациклического насыщенного амина необходимо
также добавить один лишний атом водорода, так что общая фор-
мула приобретает вид СяН2п+зМ. Значение этих обобщений, кото-
рые ценны при выводе молекулярной формулы из аналитических
данных по определению углерода и водорода, состоит в том, что
в молекуле, не содержащей других элементов, кроме углерода,
водорода и кислорода, должно быть четное число атомов водорода.
540
Глава 9
Нечетное число атомов галогена или азота требует нечетного чи-
сла атомов водорода, а четное число атомов галогена или азота
требует четного числа атомов водорода. Так, например, такая мо-
лекулярная формула, как С5НцОз, вычисленная из аналитических
данных, явно неправильна; правильной должна быть или формула
С5Н10О3, или формула С5Н12О3.
Примеры задач на определение молекулярных формул
Предыдущее обсуждение можно обобщить в форме уравнений.
В обобщенной молекулярной формуле IyII„I11г1 V* (например,
CxHj/NeOn), в которой
I может быть Н, F, CI, Вг, I, D или любой другой одновалентный атом
II может быть О, S или любой другой двухвалентный атом
III может быть N, Р или любой другой трехвалентный атом
IV может быть С, Si или любой другой четырехвалентиый атом
индекс дефицита водорода равен х— у/2z/2Этот индекс
соответствует «недостающим парам» одновалентных атомов, кото-
рые могут отвечать двойным связям, тройным связям и (или)
Циклическим особенностям в изучаемой структуре. Эту формулу
следует использовать с осторожностью в тех случаях, когда в
изучаемой молекуле элементы из второго и третьего рядов перио-
дической таблицы находятся не в низшем состоянии окисления.
Например, индекс для формулы C3H8OS равен нулю. Для этой
формулы возможны следующие структуры:
||
HOCH2CH2SCH3 HOCH2CH2CH2SH CH3SCH2CH3
Последняя формула, сульфоксид, по-видимому, противоречит най-
денному значению индекса. Однако мы можем исключить это
противоречие, если представим ее в виде полярно-ковалентной ре-
зонансной структуры:
I
СН3—S—СН2СН3
4-
Таким образом все три структуры будут иметь индекс, равный
нулю.
Задача. Напишите все возможные структуры для формулы
С3Н9О4Р.
Пример 8. Вещество А имеет формулу СеДю- При восстанов-
лении над платиной в мягких условиях оно превращается в соеди-
нение Б (С6Нц). При озонировании соединения А (СъНю) и по-
следующем восстановительном расщеплении озонида образуются
два продукта-, ацетальдегид и глиоксаль. Эти известные соедине-
ния были идентифицированы с помощью их 2,4-динитрофенилгщф
разонов.
Установление структуры: методы определения и упражнения
541
Из молекулярной формулы видно, что в молекуле имеются мак-
симум или две двойные связи, или два цикла, или одна тройная
связь. Поглощение 2 молей водорода показывает, что в действи-
тельности в молекуле содержатся или две двойные связи, или
одна тройная связь *. На основании результатов озонолиза, при
котором образуются двухуглеродные фрагменты, можно предполо-
жить, что первоначальный углеродный скелет, содержащий 6 ато-
мов, должен расщепляться в двух местах с образованием трех
двухуглеродных фрагментов, а не в одном месте. Это означает,
что молекула должна содержать две двойные связи, а не одну
тройную связь. И наконец, три двухуглеродные части расположе-
ны так, как показано на схеме:
СН3СН=СНСН=СНСН3
о3|сн2С12
О О
СНзСН \н—СН ''сНСНз
1111
о—о о—о
НгО | + Zn -J- рачб. Н3РО4
СН3СНО О=СН—СН=О ОСНСНз
Рассматривая молекулярные формулы нескольких соединений
и их реакции, часто можно вывести возможные формулы этих сое-
динений. В примере 9 большое количество информации получено
на основе лишь нескольких фактов.
Пример 9. Нейтральное соединение А (С15НцО) дает отрица-
тельную реакцию Байера и не изменяется при действии бромистого
водорода; под действием горячего раствора хромовой кислоты оно
окисляется в кислоту Б (СцН^Оз).
Сначала отметим, что при окислении были потеряны один атом
углерода и четыре атома водорода, но приобретены два атома
кислорода. Необходимо найти функциональную группу или не-
сколько групп, с которыми будут происходить такие изменения.
В связи с этим полезно составить таблицу поведения обычных
функциональных групп при окислении, в которой будут указаны
все изменения в составе соединений. Из табл. 9.1 можно видеть,
что окисление этильной группы в боковой цепи точно соответствует
окислению соединения А в Б; в обоих случаях приобретаются
* В некоторых случаях водород присоединяется к насыщенным циклам
соединений, не содержащих кратные связи; в данном случае, чтобы можно было
выдвинуть первоначальные предположения о структуре неизвестного вещества,
целесообразно допустить, что этого не происходит.
542
Глава 9
Таблица 9.1
Исходное соединение Продукт окисления Число приобретенных атомов Число потерянных атомов
RCHO RCOOH iO
RCH2OH RCOOH 10 2Н
r2choh R2CO 2Н
,он
R2C( r2co 4Н 1С
хсн3
rch=ch2 RCOOH 20 2Н 1С
RC=CH RCOOH 20 1С
ArCH2CH2OH ArCOOH Ю 4Н 1С
АгСОСНз ArCOOH Ю 2Н 1С
АгСН3 ArCOOH 20 2Н
АгСН2СН3 ArCOOH 20 4Н 1С
АгСпН2п+1 ArCOOH 20 2пН (п—1)С
Ar2CHCHs Ar2CO Ю 4Н 1С
АГ2СН2 Ar2CO Ю 2Н
Пример 9
С,6Н14О(А) С14Н1оОз(Б) 20 4Н 1С
два атома кислорода и теряются четыре атома водорода и один
атом углерода.
После вычитания этильной группы из А (С15Н14О) или кар-
боксильной группы из Б (С14Н10О3) остается фрагмент С13Н9О.
Этот радикал является производным некоторого начального соеди-
нения В (С13Н10О), которое устойчиво к окислению.
После этого следует рассмотреть характер функциональной
группы, содержащей атом кислорода. Какие функциональные
группы содержат только один атом кислорода и устойчивы к окис-
лению? Рассмотрение различных функциональных групп приводит
к заключению, что одной такой возможностью является простая
эфирная связь, а другой — соответствующим образом замещенный
кетон. Кетоны, не имеющие атомов водорода у а-углеродного ато-
ма, обычно устойчивы к окислению. Примерами могут служить
диарилкетоны.
На следующем этапе установления структуры исследуемого
соединения рассмотрим соотношение углерода и водорода в сое-
динениях А и Б, а также в гипотетическом исходном соединении
В (С13Н10О). В этих соединениях атомы углерода и водорода
должны быть связаны так, чтобы получающееся соединение было
устойчиво к окислению. Если бы соединение В, содержащее 13 ато-
мов углерода, было бы полностью насыщенным СпНгл-нг, то оно
имело бы формулу С13Н28. Даже при допущении, что два атома
водорода эквивалентны одному атому кислорода, совершенно оче-
Установление структуры: методы определения и упражнения
543
видно, что такое соединение не имеет требуемого соотношения
атомов углерода и водорода. Для алициклического соединения
соотношение должно составлять С,гН2п или СйзНге- Простые оле-
финовые пли ацетиленовые соединения с числом двойных или
тройных связей, достаточным для того, чтобы понизить отношение
водорода к углероду до имеющегося у соединения В, исключают-
ся, поскольку они неустойчивы к окислению.
Единственным большим классом соединений с таким низким
соотношением атомов водорода и углерода является класс арома-
тических соединений. Поскольку в бензоле содержится шесть ато-
мов углерода, а в исходном соединении В их 13, то можно пред-
положить наличие двух бензольных колец. После этого остается
определить положение еще одного атома углерода.
Отнимая две фенильные группы (CeHs—)2 от соединения
В(С1зН]0О), получаем остаток СО. Вспомним при этом, что ди-
арилкетоны устойчивы к окислению. Исходное соединение В, оче-
видно, является бензофеноном CeHsCOCeHs, а соединения А и Б,
вероятно, представляют собой
СвН6СОС6Н4СН2СН3
соединение А
(один из изомеров
этилбензофенона)
С6Н6СОС6Н4СООН
соединение Б
(один из изомеров
бензоилбензойной
кислоты)
ЧАСТЬ III. ЗАДАЧИ
Приведенные ниже задачи предназначены для того, чтобы сту-
денты могли приобрести дополнительный опыт в решении вопро-
сов, аналогичных тем, которые разобраны в приведенных выше
примерах; При этом наиболее важно, чтобы ответы находились
методом систематического анализа. Следует настаивать на том,
чтобы студенты избегали решения задач беспорядочным путем.
После определения структурных формул должны быть напи-
саны уравнения всех реакций и все основные спектральные данные
должны быть отнесены к соответствующим фрагментам структуры.
Все ПМР-спектры сняты на приборах с рабочей частотой
60 МГц, если не приведены другие значения (интегрирование:
1Н — один протон, 2Н — два протона и т. д.).
Интенсивности полос поглощения в ИК-спектрах обозначаются
буквами: с — сильная, ср — средняя, сл — слабая, ш — широкая.
Серия 1
При исследовании неизвестных соединений часто встречаются следующие
типы задач. Для каждого примера приведите выводы, которые можно сделать
относительно природы соединения.
1. Желтое нейтральное вещество содержит только углерод, водород и кис-
лород, превращается в кислоту под действием пероксида водорода. В ПМР-
544
Глава 9
спектре этого желтого вещества наряду с другими сигналами имеется синглет
около б 9,0 м. д.
2. При взаимодействии нейтрального соединения с фенилгндразином обра-
зуется продукт, который отличается от ожидаемого феннлгидразона на моле-
кулу этанола, т. е. конденсация сопровождается отщеплением не только моле-
кулы воды, но и молекулы этанола.
3. Соединение содержит только углерод, водород и кислород, реагирует с
ацетилхлорндом, но не взаимодействует с фенилгндразнном. После обработки
иодной кислотой оно превращается в вещество, которое реагирует с фенилгидра-
знном, но не взаимодействует с ацетилхлорндом.
4. Нейтральное соединение желтого цвета дает производное с о-феннлен-
диамнном. В ИК-спектре исходного вещества имеется полоса поглощения при
1710 см-1 (5,85 мкм).
5. Спирт дает положительную йодоформную реакцию и отрицательную про-
бу Лукаса.
6. Нейтральное соединение содержит только углерод, водород, кислород,
реагирует с ацетилхлорндом, но не реагирует с феннлгидразнном. При нагрева-
нии с минеральными кислотами оно превращается в вещество, которое не реа-
гирует с ацетилхлорндом, но дает положительные реакции с фенилгндразином и
бромом в четыреххлористом углероде. В ПМР-спектре исходного соединения
имеются только два синглета.
7. Соединение содержит азот, дает положительную реакцию с азотистой кис-
лотой на вторичные амины, но его производное с бензолсульфохлоридом раст-
воримо в щелочах.
8. Соединение при обработке этнлортоформиатом в присутствии следов кис-
лоты присоединяет молекулу днэтилового эфира. В ПМР-спектре одного из двух
соединений, которые удовлетворяют приведенному выше требованию, имеется
сигнал около б 9,0—10,0 м.д. до обработки ортоформиатом; после взаимодей-
ствия с ортоформиатом этот сигнал сдвигается приблизительно до б 5,0 м. д.
9. Нейтральное соединение содержит углерод, водород и кислород, димери-
зуется в спиртовом растворе цианида натрия. В ИК-спектре исходного соедине-
ния имеются полосы поглощения около 2800 и 2700 см-1 (3,57 и 3,71 мкм).
10. Соединение со свойствами основания не реагирует с бензолсульфохло-
ридом, но дает производное с азотистой кислотой. В ИК-спектре исходного со-
единения не имеется заметных полос поглощения около 3333 см-1 (3,0 мкм).
В ИК-спектре продукта взаимодействия с азотистой кислотой есть полоса по-
глощения приблизительно при 1550 см-1 (6,45 мкм).
И. Число омыления сложного эфира равно 59 ± 1. В его ПМР-спектре
имеются два синглета с отношением интенсивностей 3:1.
12. Кристаллическая кислота растворяется в воде, и ее эквивалент нейтра-
лизации равен 54 + 1. При нагревании выше температуры плавления из нее вы-
деляется диоксид углерода и образуется новая кристаллическая кислота с экви-
валентом нейтрализации 59+1. В ПМР-спектре исходного соединения сигнал
необменнвающнхся протонов представляет собой мультиплет; в ПМР-спектре
продукта, образующегося после нагревания, сигнал необменивающихся прото-
нов становится синглетом.
13. Кислое растворимое в воде соединение содержит азот и серу и имеет
эквивалент нейтрализации, равный 142 ±1. При прибавлении хлорида бария к
водному раствору этого вещества образуется осадок, нерастворимый в кислотах.
Под действием щелочи выделяется соединение со свойствами основания, гидро-
хлорид которого имеет эквивалент нейтрализации 130 ± 1. В ИК-спектрах и пер-
вого и последнего соединений (кислот) имеются широкие полосы поглощения
в области 3333—2500 см-1 (3,00—4,00 мкм).
14. Растворимое в воде соединение содержит азот и имеет эквивалент ней-
трализации 73 ± 1 при титровании соляной кислотой в присутствии метилового
красного в качестве индикатора. При обработке этого вещества бензолсульфо-
хлоридом и раствором гидроксида натрия образуется осадок. В ПМР-спектре
исходного вещества имеются квадруплет (4Н), триплет (6Н), а также уширен-
ный синглет (1Н).
Установление структуры: методы определения и упражнения 545
15. При обработке 12 г соединения С8Н8О раствором брома в четыреххлори-
стом углероде, содержащем 60 г брома, выделяется 16,2 г бромистого водорода.
После окончания реакции раствор остается красным; найдено, что 12 г брома
остались неиспользованными. Вычислите число атомов брома, введенных в мо-
лекулу путем замещения, а если при этом происходит также присоединение, то
и число присоединенных атомов брома. Используйте следующие значения атом-
ных масс: Вг = 80, Н = 1, С = 12, О = 16. Опишите основные характерные
признаки ПМР- и ИК-спектров, которые вы ожидаете как для исходного веще-
ства, так и для продукта его взаимодействия с бромом; подчеркните важные
изменения, происходящие в спектрах в результате обработки бромом.
Серия 2
При решении задач этой серии, а также серий 3 и 4, используя данные, при-
веденные в задачах, придерживайтесь указанной выше последовательности. Сде-
лайте обычные допущения о величине экспериментальных ошибок при опреде-
лении температур кипения и плавления. Соотнесите спектральные данные с фраг-
ментами структур изучаемых соединений.
I. 1) Белые кристаллы, т. пл. 117—118°С.
2) Элементный анализ на присутствие X, N и S отрицательный.
3) Растворимость: вода (+).
4) Реакции для классификации: C8H5NHNH2 — отрицательная, СНзСОС! —
положительная, КМпО4— положительная, Н1О4—положительная и Вг2 в СС14—
отрицательная.
Эквивалент нейтрализации 151 ± 1.
5) Производное: n-нитробензиловый эфир, т. пл. 123°С.
6) Спектры: ПК (минеральное масло нуйол): 3333—2400 см-1 (3,00—
4,17 мкм, с, ш); 1706 см-1 (5,86 мкм, с); 1449, 1379, 1300, 1200, 1190, 943, 865,
730 см"1 (6,9, 7,25, 7,7, 8,2, 8,4, 10,6, 11,55, 13,7 мкм, все ср). ПМР (ацетон):
6 5,22 м.д., 1Н, синглет; д 6,93—7,88 м.д., 7Н, мультиплет.
II. 1) Бесцветная жидкость, т. кип. 259—261°С.
2) Элементный анализ на Вг положительный, на Cl, I, N и S — отри-
цательный.
3) Растворимость: вода (—), NaOH (—), НС1 (—), H2SO4 (+).
4) Реакции для классификации: H2NOH-HC1— отрицательная, КМпО4 —
отрицательная, СН8СОС1 — отрицательная, . Вг2 в СС14 — отрицательная,
AgNO3 — отрицательная и Nal — отрицательная.
При обработке горячим раствором гидроксида натрия получается прозрач-
ный раствор, из которого при подкислении выпадают белые кристаллы с т. пл.
250°С, содержащие бром. Число омыления исходного соединения 229 ± 2.
5) Производные: а) с гидразином образуются бесцветные кристаллы,
т. пл. 164°С, и б) при обработке 3,5-дииитробензойной кислотой и серной кисло-
той выделены бледно-желтые кристаллы, т. пл. 92°С.
III. 1) Коричневая жидкость, т. кип. 198—200°С.
2) Элементный анализ на X, N и S отрицательный.
3) Растворимость: вода (—), NaOH (+), NaHCOs (—), НС1 (—).
4) Реакции для классификации: СН3СОС1 — положительная, Вг2 в
Н2О — осадок, C8H5NHNH2 — отрицательная, FeCK — фиолетовая окраска и
Се (IV) —положительная.
5) Производное: при обработке хлоруксусной кислотой образуются бе-
лые кристаллы, т. пл. 102—103°С.
6) Спектры: ИК (без растворителя): 3390 см-1 (2,95 мкм, с, ш); 1163,
935, 780, 690 см-1 (8,6, 10,75 12,85, 14,5 мкм, все с). ПМР (CDCl,): б 2,25 м.д.,
ЗН, синглет; 6 5,67 м.д., 1Н, синглет; б 6,5—7,3 м.д., 4Н, мультиплет.
IV. 1) Бесцветная жидкость, т. кип. 194—195°С.
2) Элементный анализ на X, N и S отрицательный.
3) Растворимость: все растворители (—).
4) Реакции для классификации: H2SO4 + SO3 — отрицательная, A1C1S +
+ CHCI3 — бледно-желтая окраска.
18 Зак. 335
546
Глава 9
5) Производных нет.
Плотность d4° 0,8963, 1,4811.
V. 1) Белые кристаллы, т. пл. 187—188°С.
2) Элементный анализ на X, N и S отрицательный.
3) Растворимость: вода (+). Водный раствор является кислым по лак-
мусу.
4) Реакции для классификации: КМпО4 — отрицательная, C6H6NHNH2—
отрицательная, Вг2 в СС14 — отрицательная, СН3СОС1 — отрицательная.
Эквивалент нейтрализации 59. Коэффициент распределения 7,7.
5) Производное: при нагревании с фенилгидразином образуются белые
кристаллы, т. пл. 209—210°С.
6) Спектры: ИК (нуйол): 3333—2222 см-1 (3,0—4,5 мкм, с, ш); 1695,
1418, 1307, 1198, 913 см-‘ (5,90, 7,05, 7,65, 8,35, 10,95 мкм все с). ПМР
(ДМСО-de): б 2,43 м.д., сннглет; б 11,80 м.д., широкий сннглет; отношение пло-
щадей пиков 2: 1 (синглет в сильном поле к синглету в слабом поле).
VI. 1) Красновато-коричневое твердое вещество, т. пл. 65—70°С.
2) Элементный анализ на N положительный, на X и S — отрицатель-
ный.
3) Растворимость: вода (—), NaOH (—), НС1 (+). К раствору веще-
ства в разбавленной соляной кислоте, обесцвеченному активированным углем,
прибавляют щелочь. Очищенное таким образом вещество имеет т. пл. 73°С.
4) Реакции для классификации: КМпО4 — положительная,
C6H6SO2C1 + NaOH — осадок, растворимый в соляной кислоте, Вг2 и СС14 — оса-
док, Fe(OH)2 — отрицательная, бромная вода — осадок, реактив Толленса — по-
ложительная, C6H6NHNH2 — положительная.
Под действием горячего раствора гидроксида натрия соединение разлагается.
После отделения темно-корнчневого осадка фильтрованием фильтрат нейтрали-
зуют и получают светло-коричневый осадок. После перекристаллизации нз вод-
ного спирта он плавится с разложением при 236—24(гС, растворяется в НС1 и
NaOH и не растворяется в растворе бикарбоната натрия. При взаимодействии
исходного вещества с ацетоном в присутствии гидроксида натрия образуется жел-
тый осадок, т. пл. 134—135°С.
5) Производные: оксим, т. пл. 144°С; фенилгидразон, т. пл. 148°С; се-
микарбазон, т. пл. 224°С.
6) Спектры: ИК (нуйол): нет поглощения около 3333 см-1 (3,00 мкм);
полосы поглощения при 1653, 1587 см-1 (6,05, 6,30 мкм, ср, с.). ПМР (CDC18):
б 3,05 м. д., ЗН, синглет; б 6,69 м.д., 2Н, дублет; б 7,71 м.д., 2Н, дублет; б
9,70 м. д., 1Н, синглет.
Серия 3
1. Коричневая жидкость (I), т. кнп. 193—195°С. Она содержит азот, но не со-
держит серы и галогенов. Не растворяется в воде, но растворяется в разбавлен-
ной кислоте. Не реагирует с ацетилхлоридом и бензолсульфохлоридом. При об-
работке солянокислого раствора неизвестного соединения нитритом натрия с
последующей нейтрализацией образуется вещество II с т. пл. 83—84°С. Соедине-
ние 11 не растворяется в щелочах, но растворяется в кипящем концентрирован-
ном растворе гидроксида натрия с выделением газа (III). Этот газ был погло-
щен водой. При обработке водного раствора фенилизотиоцнанатом образуется
соединение IV с т. пл. 134—135°С. При осторожном подкислении полученного ра-
нее щелочного раствора с последующей экстракцией выделено соединение V с
т. пл. 125—126°С.
В ПМР-спектре соединения V (в ацетоне) имеются следующие сигналы: б
6,63 м. д., 2Н, дублет; б 7,67 м.д., 2Н, дублет; б около 8,7 м.д., 1Н, широкий
синглет. Сигнал с б 8,7 м. д. такой широкий, что может быть определен только
электронным интегрированием.
2. Бесцветная жидкость, т. кип. 94—96°С, не содержит галогенов, азота и
серы, растворяется в воде и эфире. Она восстанавливает разбавленный раствор
Установление структуры: методы определения и упражнения
547
перманганата калия, обесцвечивает бром в четыреххлористом углероде, реаги-
рует с ацетилхлорндом и выделяет водород при действии металлического на-
трия. При обработке гипоиодитом натрия йодоформ не образуется. Исследуемое
вещество не реагирует с фенилгидразином. При взаимодействии с 3,5-динитро-
бензоилхлоридом получается соединение с т. пл. 47—48°С.
В ПМР-спектре исходного вещества (в CDC13) имеются следующие сиг-
налы: б 3,58 м.д., 1Н, синглет; б 4,13 м.д., 2Н, мультиплет; б 5,13 м.д., 1Н,
мультиплет; б 5,25 м.д., 1Н, мультиплет; б около 6,0 м.д., 10 линий, 1Н. Поло-
жение сигнала с б 3,58 м. д. изменяется в зависимости от концентрации рас-
твора.
3. Желтое кристаллическое вещество I с т. пл. 113—114°С содержит азот,
но не содержит галогенов, серы и металлов, не растворяется в воде и щелочах,
но растворяется в разбавленных кислотах. При обработке кислого раствора ве-
щества I на холоду нитритом натрия и последующем кипячении образуется со-
единение П, выделенное при охлаждении раствора. Продукт II содержит азот
и плавится при 95—96°С, не растворяется в кислотах и растворе бикарбоната
натрия, но растворяется в растворе гидроксида натрия. Продукты, полученные
при обработке соединений I и II цинком в кипящем растворе хлорида аммония,
легко восстанавливают реактив Толленса. При обработке исходного вещества I
бензолсульфохлоридом и щелочью после подкисления полученного раствора вы-
делено соединение III, которое плавилось при 135—136°С.
В ПМР-спектре соединения II (в ДМСО-de) имеются следующие сигналы:
б 7,0—7,75 м. д., 4Н, мультиплет; б 9,8 м д., 1Н, синглет.
4. Бесцветное кристаллическое соединение I с т. пл. 186—187°С содержит
азот, но не содержит галогенов и серы, не растворяется в воде и разбавленных
кислотах, но растворяется в разбавленном растворе бикарбоната натрия. Его
эквивалент нейтрализации равен 180 zE 2. Это вещество не реагирует с бромом
в четыреххлористом углероде, с разбавленным раствором перманганата калия,
с ацетилхлорндом и фенилгидразином. При обработке в течение некоторого вре-
мени кипящей соляной кислотой после охлаждения реакционной смеси выделено
соединение II, плавившееся при 120—121°С и имевшее эквивалент нейтрализации
121 ± 1. Фильтрат после выделения вещества II упаривают досуха, а остаток
III очищают перекристаллизацией. Он содержит азот и хлор, довольно гигро-
скопичен, разлагается при попытке определения его температуры плавления и не
растворяется в эфире. Из водного раствора этого вещества при прибавлении
раствора нитрата серебра выпадает осадок. При обработке вещества III азоти-
стой кислотой на холоду происходит энергичное выделение газа. При взаимо-
действии соединения III с бензолсульфохлоридом и раствором гидроксида на-
трия после подкисления полученного раствора выделен продукт IV с т. пл. 164—
165°С.
ПМР-Спектры: соединение I (натриевая соль, растворитель D2O): б 4,06 м.д.,
2Н, сннглет; б 4,77 м. д., примесь HDO, синглет; б 7,5—8,0 м. д., 5Н, мультиплет.
Соединение II (растворитель СС14): б 7,3—7,7 м.д., ЗН, мультиплет; б 8,0—
8,25 м. д., 2Н, мультиплет; б 12,8 м.д., 1Н, синглет. Соединение III (раствори-
тель D2O): б 3,58 м. д., синглет; б 4,9 м. д., HDO.
5. Бесцветное кристаллическое вещество I плавится при 162—165°С с раз-
ложением, не содержит азота, галогенов, серы и металлов, растворяется в воде,
но не растворяется в эфире. Соединение I реагирует с ацетилхлорндом, обесцве-
чивает раствор перманганата калия, восстанавливает раствор Фелинга и реактив
Толленса, но не дает окраски с реактивом Шиффа. С фенилгидразином обра-
зуется продукт II, который плавится с разложением при 199—20ГС. При нагре-
вании соединения I с концентрированной азотной кислотой происходит бурная
реакция и после охлаждения реакционной смеси выделено соединение III, кото-
рое не растворяется в воде, но легко растворяется в щелочах и имеет эквива-
лент нейтрализации 104 ± 1. Соединение III реагирует с ацетилхлорндом, но не
реагирует с фенилгидразином и плавится около 212—213°С с разложением. Если
это вещество нагревать в течение некоторого времени выше его температуры
плавления, то оно превратится в соединение IV, которое после перекристаллиза-
ции плавится при 132—133°С. Соединение IV не растворяется в воде, но раство-
18*
548
Глава 9
ряется в растворе бикарбоната натрия и имеет эквивалент нейтрализации
111 ±1. При обработке натриевой соли соединения IV n-бромфен ацил бромидом
образуется соединение V с т. пл. 137—138°С.
Исходное вещество I оптически активно с удельным вращением +81°. Про
дукты его превращений (III, IV, V) оптически неактивны.
В ПМР-спектре раствора калиевой соли соединения IV (в D2O) имеются
следующие сигналы: 6 6,59 м. д., 1Н, мультиплет; б 7,05 м. д., 1Н, мультиплет;
б 7,64 м. д., 1Н, мультиплет. В ИК-спектре вещества IV в нуйоле имеются сле-
дующие полосы поглощения: 3100—2400 см-* (3,22—4,27 мкм, с); 1675 см-1
(5,97 мкм, с); несколько полос в области 1667—1000 см-1 (6,00—10,00 мкм);
932 см-1 (10,73 мкм, ср); 885 см-1 (11,3 мкм, ср); 758 см-1 (13,2 мкм, ср).
Серия 4
1. Соединение с т. пл. 68“С содержит азот, не растворяется в воде, растворе
гидроксида натоия и соляной кислоте, но растворяется в концентрированной сер-
ной кислоте.- При обработке оловом и соляной кислотой оно превращается в
вещество, которое реагирует с бензолсульфохлоридом с образованием производ-
ного, растворимого в щелочи. Прн обработке исходного соединения цинком и го-
рячим раствором гидроксида натрия получается соединение с т. пл. 130°С.
Если продукт реакции с оловом и соляной кислотой нейтрализовать основа-
нием и затем перегнать, то в ПМР-спектре перегнанного продукта (в ССЦ)
имеются следующие сигналы: б 3,32 м. д., 2Н, синглет; б 6,44 м. д., 2Н, мульти-
плет; около б 6,6 м. д., 1Н, мультиплет; б 7,0 м. д., 2Н, мультиплет.
2. Вещество с т. кип. 166—169°С содержит серу, но не содержит азота и га-
логенов. Оно не растворяется в воде и разбавленных кислотах, но растворяется
в растворах гидроксида натрия. При взаимодействии натриевого производного
с 2,4-динитрохлорбензолом получается соединение с т. пл. 118—119°С. При стоя-
нии на' воздухе исходное вещество медленно окисляется в производное, плавив-
шееся при 60—61 °C.
В ПМР-спектре продукта окисления на воздухе (в CDCh) имеется только
мультиплет с б 7,2—7,7 м. д. В ПМР-спектре исходного соединения имеются
следующие сигналы: б 3,39 м. д., 1Н, синглет; б 7,12 м. д., синглет, 5Н.
3. Соединение с т. пл. 141—142“С содержит азот, но не содержит галогенов
и серы, растворяется в воде, разбавленных кислотах и разбавленных щелочах
и не изменяется при обработке оловом и соляной кислотой. Прн продолжитель-
ной обработке горячим раствором гидроксида натрия образуется нерастворимое
масло I. Это масло растворяется в разбавленной соляной кислоте н реагирует
о ацетилхлоридом с образованием твердого вещества, плавящегося при 111—
112°С. При подкислении щелочного раствора после удаления соединения I выде-
лено твердое вещество с т. пл. 120—121°С, эквивалент нейтрализации которого
равен 122 ± 1-
В ПМР-спектре вещества с т. пл. 120—121°С в (CDCh) имеются следующие
сигналы: б 7,4—7,7 м. д., ЗН, мультиплет; 6 8,0—8,3 м. д., 2Н, мультиплет; 6
12,8 м. д., 1Н, синглет. В ПМР-спектре масла I (в CDCI3) имеются следующие
сигналы: б 2,15 м. д., ЗН, синглет; б 3,48 м. д., 2Н, широкий синглет; б 6,45—
6,8 м. д., 2Н, мультиплет; б 6,8—7,15 м. д., 2Н, мультиплет.
4. Соединение с т. кип. 159—161°С содержит хлор, но не содержит азота н
серы, не растворяется в воде, разбавленных кислотах и щелочах и в холодной
концентрированной серной кислоте, однако растворяется в дымящей серной кис-
лоте. При его взаимодействии с горячим спиртовым раствором нитрата серебра
осадок не образуется. При обработке горячим раствором перманганата калия
вещество медленно растворяется. Из полученного раствора после подкисления
серной кислотой выпадает осадок, который плавится при 138—139°С и имеет
эквивалент нейтрализации 157 ± 1.
В ПМР-спектре исходного соединения (в CDC13) имеются следующие сиг-
налы: б 2,37 м..д., ЗН, синглет; б ?,0—7,35 м. д., 4Н, мультиплет. В ИК-спектре
вещества с т. пл. 138—139°С (в нуйоле) имеются следующие полосы поглоще-
Установление структуры: методы определения и упражнения
549
ния: 3333—2381 см*1 (3,0—4,2 мкм, с); 1678 см-1 (5,96 мкм, с); несколько по-
лос в области 1587—1389 см*1 (6,3—7,2 мкм, с); 1316 см-1 (7,6 мкм, с); 1053,
1042 см-1 (9,5, 9,6 мкм, с); 9,13 см~* (10,95 мкм, ср); 742 см-1 (13,47 мкм, с),
5. Бесцветная жидкость с т. кип. 188—192°С содержит только углерод, водо-
род, кислород, не растворяется в воде, разбавленных кислотах и щелочах, но
легко растворяется в холодной концентрированной серной кислоте. Это веще-
ство не реагирует с фенилгидразином и ацетилхлоридом, а также не обесцве-
чивает раствор брома в четыреххлористом углероде; медленно растворяется в
кипящих щелочах. При перегонке полученной смеси с водяным паром в дистил-
ляте содержится соединение, которое после очистки имеет т. кип. 129—130°С
и реагирует с а-нафтилизоцианатом с образованием производного с т. пл. 65—
66°С.
Если щелочной раствор, оставшийся после перегонки с паром, подкислить
фосфорной кислотой и снова перегнать с водяным паром, то дистиллат содер-
жит кислоту, анилид которой плавится при 108—109°С.
При обработке исходного соединения литийалюминийгидридом (с последую-
щей обычной осторожной обработкой) образуется только одно соединение;
ПМР- и ИК-спектры полученного вещества идентичны спектрам, полученным для
описанного выше соединения с т. кип. 129—130°С.
В ПМР-спектре имеются следующие сигналы: б 0,92 м. д., ЗН, дублет; б
1,0—2,0 м. д., ЗН, мультиплет; б 2,13 м. д., 1Н, синглет; 6 36,3 м. д., 2Н, дублет.
6. Твердое вещество розового цвета с т. пл. 109—112°С. После обработки ак-
тивированным углем окраска исчезает и температура плавления поднимается
до 112—114°С. Соединение горит коптящим пламенем и сгорает без остатка.
Согласно данным элементного анализа, оно содержит азот и не содержит серы
и галогенов. Соединение не растворяется в воде и разбавленных щелочах, но
растворяется в эфире и разбавленных кислотах. При взаимодействии с бензол-
сульфохлоридом образуется производное, растворимое в щелочи и плавящееся
при 101—102°С. Ацетильное производное имеет т. пл. 132°С.
В ИК-спектре исходного соединения (5000—1250 см*1 в СНС13, 1250—
650 см-1 в CS2) имеются следующие полосы поглощения: 3400 см-1 (2,94 мкм),
3350 (2,99), 3200 (3,13), 3050 (3,28), 1640 (6,10), 1610 (6,21), 1520 (6,58), 1480
(6,75), 1400 (7,15), 1290 (7,75), 1280 (7,81), 1230 (8,13), 1190 (8,40), ИЗО
(8,85), 970 (10,31), 890 (11,24), 870 (11,50), 850 (11,76), 820 (12,19), 750 (13,33)
и 720 (13,89).
Серия 5
Для каждой из задач этой и следующей серий установите структурную фор-
мулу органического соединения, которая удовлетворяла бы поставленным усло-
виям, и напишите уравнения указанных ниже реакций, происходивших с этими
веществами. Соотнесите все основные спектральные полосы с соответствующими
фрагментами найденной вами структуры.
1. Кислота А содержит только углерод, водород и кислород, имеет эквива-
лент нейтрализации 103 ± 1 и дает отрицательную реакцию с фенилгидразином.
При обработке серной кислотой кислота А превращается в кислоту Б, которая
обесцвечивает растворы перманганата и брома и имеет эквивалент нейтрализа-
ции 87 ± 1. Под действием гипоиодита исходная кислота А превращается в йо-
доформ и кислоту В, эквивалент нейтрализации которой равен 52 ±1.
В ПМР-спектре кислоты Б (в СНС13) имеются следующие сигналы: б
1,90 м. д., ЗН, дублет дублетов (/ = 8,2 Гц); б 5,83 м. д., 1Н, дублет квадрупле-
тов (/= 15,2 Гц); б 7,10 м. д., 1Н, мультиплет; б 12,18 м. д., 1Н, синглет.
2. Кислота имеет эквивалент нейтрализации 97, с трудом бронируется даже
в присутствии трнбромида фосфора, а при энергичном окислении превращается
в новую кислоту с эквивалентом нейтрализации 83.
В ПМР-спектре второй кислоты (в CDC13) имеются следующие сигналы: б
8,08 м. д., синглет; б 11,0 м. д., синглет; отношение интенсивностей сигнала б
8,08 м. д. к сигналу 11,0 м. д. равно 2:1.
550
Глава 9
3. Оптически активный углеводород растворяется в холодной концентри
рованной серной кислоте, обесцвечивает растворы перманганата и легко погло
щает бром. При окислении этот углеводород превращается в кислоту с экви
валентом нейтрализации 66, которая имеет то же число атомов углерода, что и
исходное соединение. На основании масс-спектра было найдено, что молекуляр-
ный нон углеводорода имеет т/е 68.
4. Соединение имеет эквивалент нейтрализации 66, не взаимодействует с
раствором брома в четыреххлорнстом углероде и прн нагревании превращается
в кислоту, эквивалент нейтрализации которой равен 88. В ПМР-спектре послед-
ней кислоты (в CDCI3) имеются следующие сигналы: б 1,20 м. д., 6Н, дублет;
б 2,57 м.д., 1Н, септет; 6 12,4 м.д., 1Н, широкий синглет.
5. Основание имеет эквивалент нейтрализации 121 ± 1, при энергичном
окислении превращается в кислоту с эквивалентом нейтрализации 121 + 1.
В ПМР-спектре кислоты (в СС14) имеются следующие сигналы: б 7,52 м. д., ЗН,
мультиплет; о 8,14 м.д., 2Н, мультиплет; б 12,82 м.д., 1Н, синглет. В ПМР-спек-
тре основания (в CDCI3) имеются следующие сигналы: б 0,91 м.д., 2Н, синглет;
6 2,78, м. д., 4Н, 8 линий; б 7,23 м. д„ 5Н, синглет.
6. Кислота, эквивалент нейтрализации которой равен 166, не взаимодейст-
вует с бромом в четыреххлористом углероде, но дает положительную йодоформ-
ную реакцию.
7. Соединение А не содержит азота, серы, галогенов, не растворяется в воде,
но растворяется в разбавленном растворе гидроксида натрия. Вещество А не
дает окраски с хлоридом железа (III) и не обесцвечивает раствор перманганата
калия.
При обработке концентрированной бромистоводородной кислотой вещество
А превращается в два соединения. Одно из них, растворимое во всех раствори-
телях, содержит бром. Прн действии на это вещество раствора иодида натрия
в ацетоне выпадает осадок. Второе вещество — это новая кислота Б, которая не
содержит галогенов, обесцвечивает растворы брома и дает окраску с хлоридом
железа (1П). Эквиваленты нейтрализации соединений А н Б равны 180 + 2 и
137+1 соответственно.
В ПМР-спектре кислоты Б (в CDC18, содержащем ДМСО-с16) имеются сле-
дующие сигналы: б 6,84 м.д., 2Н, дублет (7 = 9 Гц): б 8,2 м.д., 2Н, широкий
синглет.
8, Оптически активная кислота с молекулярной массой 98.
9. Соединение I дает осадок с 2,4-динитрофенилгндразином. Если вещество I
нагреть с 25%-ным водным раствором гидроксида натрия и смесь частично пере-
гнать, то продукт перегонки содержит соединение, которое реагирует с натрием
и дает положительную йодоформную реакцию. Проба Лукаса отрицательна.
Остаток после перегонки подкисляют фосфорной кислотой и смесь перего-
няют с водяным паром. Выделена низкокипящая кислота. Число омыления ее
n-бромфенацилового эфира равно 257 ± 1.
В масс-спектре исходного соединения I имеются следующие пики (приве-
дены только пики с интенсивностью, равной илн большей 10% от интенсивности
основного пика):
т/е Интенсивность, % от интенсивности основного пика т/е Интенсивность, % от интенсивности основного пика
15 13,7 45 13,2
27 22,6 85 18,0
29 42,7 88 12,3
31 28,9 130 10,4
42 13,5 131 0,8
43 (100, основной пик)
Установление структуры: методы определения и упражнения
651
В ИК-спектре соединения I (без растворителя) имеются следующие полосы
поглощения: 3000 см-1 (3,33 мкм, ср); 2933 см-1 (3,41 мкм, сл); 1715 см-1
(5,83 мкм, с); 1634 см-' (6,12 мкм, сл); 1408 см-1 (7,10 мкм, сл); 1364 см-1
(7’33 мкм, ср); 1316 см-1 (7,60 мкм, с); 1250 см-1 (8,0 мкм, с); 1149 см-1
(8,70 мкм, с); 1042 см-1 (9,60 мкм, с).
Серия 6
1. Кислота I содержит азот и имеет эквивалент нейтралнзацин 197 ± 2. При
взаимодействии с тионилхлоридом и последующей обработке гидроксидом ам-
мония она- превращается в нейтральное соединение И, которое реагирует со ще-
лочным раствором гипобромита с образованием соединения 111, обладающего
свойствами основания. При гидролизе соединения 111 получается вещество IV,
растворимое как в кислотах, так и в щелочах. Его эквивалент нейтрализации
равен 186 ± 2. При обработке этого соединения азотистой кислотой в присут-
ствии серной кислоты получается прозрачный раствор. При прибавлении цианида
меди(1) к этому раствору регенерируется соединение 1.
При гидролизе кислоты I образуется кислота V, эквивалент нейтрализации
которой равен 107 ± 1. При нагревании V превращается в нейтральное соедине-
ние VI, из которого можно получить кислоту V гидролизом. Окисление вещества
IV приводит к новой кислоте VII, эквивалент нейтрализации которой равен
71 ± 1. х
В ПМР-спектре соединения V (в CDCI3, содержащем ДМСО-de) имеются
следующие сигналы (соотношение интенсивностей 1 : 1 : 1 : 1): б 7,6 м.д., мульти-
плет; б 7,9 м. д., мультиплет; б 8,28 м.д., синглет; б 11,8 м.д., широкий синглет.
Химический сдвиг сигнала с б 11,8 м.д. изменяется в зависимости от концентра-
ции. Два мультиплета в сильном поле хотя и сложны, но обладают симметрией:
эти два сигнала представляют собой почти зеркальные изображения один дру-
гого.
2. -Неизвестное вещество I содержит азот и бром, не растворяется в воде, но
растворяется-как в разбавленной кислоте, так и в разбавленной щелочи. Удо-
влетворительное значение его эквивалента нейтрализации получить не удалось.
При обработке уксусным ангидридом это вещество превращается в кислоту II,
эквивалент нейтрализации которой равен 270 ± 3. При взаимодействии соеди-
нения I с холодной азотистой кислотой выделяется газ и получается вещество
III, эквивалент нейтрализации которого равен 230 + 2. При энергичном окисле-
нии соединений I, II и III образуется один и тот же продукт — бромсодержащая
кислота с эквивалентом нейтрализации 199 + 2.
В ПМР-спектре продукта окисления (в CDC13, содержащем ДМСО-бв)
имеются следующие сигналы: б 7,60 м. д., 2Н, дублет; б 7,95 м. д., 2Н, дублет;
б 12,18 м. д., 1Н, широкий синглет. Химический сдвиг сигнала с б 12,18 м.д. из-
меняется в зависимости от концентрации *.
3. Кристаллический сложный эфир I омыляют (число омыления 173 ±2) и
щелочной водный раствор упаривают почти досуха. Дистиллат представляет со-
бой чистую воду. Остаток подкисляют и перегоняют; при этом выделено бесцвет-
ное масло II, которое реагирует с ацетилхлорндом и дает окраску с раствором
хлорида железа (111). Из остатка после перегонки выделено кристаллическое
вещество III, растворимое в щелочи. При нагревании соединения III выше его
температуры плавления оно превращается в соединение IV. Это вещество раство-
ряют при встряхивании в теплом водном растворе щелочи. При подкислении
раствора выделены кристаллы, идентичные соединению Ill.
В ПМР-спектре соединения II (в CDC13) имеются следующие сигналы: б
2,25 м. д., ЗН, синглет; б 5,67 м. д., 1Н, синглет; б 6,5—7,3 м. д., 4Н, мультиплет.
Химический сдвиг сигнала с б 5,67 м. д. изменяется в зависимости от концентра-
ции.
* Два дублета настолько сходны друг с другом, что сигнал можно ошибочно
принять за квадруплет с центром около б 7,75 м. д.
552
Глава 9
В ПМР-спектре вещества III (в CDC13) имеются следующие сигналы: 6
7,4—7,9 м.д., мультиплет, и 6 12.08 м.д., синглет; Площадь синглета в два раза
меньше площади мультиплета. Кроме того, химический сдвиг синглета изме-
няется в зависимости от концентрации.
.4. Кристаллы I содержат азот, растворяются в воде н не растворяются в
эфире. При прибавлении холодного раствора щелочи выделяется растворимое в
воде н эфире вещество II, имеющее запах аммиака, которое при взаимодействии
с бензолсульфохлоридом дает производное, нерастворимое в щелочи. При добав-
лении соляной кислоты к разбавленному водному раствору I получается кристал-
лическая кислота III, эквивалент нейтрализации которой равен 167 ± 2. После
кипячения с цинковой пылью и раствором хлорида аммония соединение III вос-
станавливает реактив Толленса.
В ПМР-спектре соединения II (в СС14) имеются следующие сигналы: 6
0,53 м. д., 1Н, синглет; б 0,90 м.д., 6Н, искаженный триплет; 6 1,2—1,7 м.д., 8Н,
мультиплет; 6 2,52 м. д., 4Н, триплет.
В ПМР-спектре соединения III (в CDCI3/CF3COOH) имеются следующие сиг-
налы: б 7,77 м.д., 1Н, триплет (7 = 7 Гц); около б 8,54 м.д., 2Н, дублет муль-
типлетов (7 = 7 Гц, ~2 Гц и меньшие константы спин-спинового взаимодей-
ствия); б 8,96 м.д., 1Н, триплет (7 = ~2 Гц).
5. Оптически активное соединение СЕНюО слабо растворяется в воде. Раст-
воримость соединения заметно не увеличивается при прибавлении гидроксида
натрия или соляной кислоты. Это вещество дает отрицательные реакции с фенил-
гидразином, реактивом Лукаса и гипоиодитом, обесцвечивает растворы брома и
перманганата н реагирует с ацетилхлорндом. При окислении перманганатом ве-
щество превращается в кислоту с эквивалентом нейтрализации 59 ± 1. При на-
гревании этой кислоты выделяется углекислый газ и образуется новая кислота
с эквивалентом нейтрализации 73 ± 1.
6. Соединение I содержит углерод, водород, кислород и азот, растворяется
в разбавленном растворе гидроксида натрия и разбавленной соляной кислоте,
но не растворяется в растворе бикарбоната натрия. При взаимодействии с из-
бытком уксусного ангидрида получается продукт II, который не растворяется
в воде, разбавленных кислотах и разбавленных щелочах. Соединение I обесцве-
чивает бромную воду; при его растворении в избытке разбавленной соляной кис-
лоты и последующей обработке холодного раствора нитритом Натрия образуется
продукт III. Азот при этом не выделяется.
В ПМР-спектре кислой сернокислой соли соединения I, полученной при дей-
ствии серной кислоты (растворитель CF3COOH), имеются следующие сигналы:
б 3,31 м.д., ЗН, триплет (7 = 5 Гц); б 7,12 м.д., 2Н, дублет (7 = 10 Гц); б
7,49 м.д., 2Н, дублет (7 = 10 Гц); о 8,74 м.д., 2Н, широкий синглет.
Серия 7
1. Кристаллы состава С6Н6С>2(1) при нагревании с разбавленным раствором
поташа превращаются в калиевую соль кислоты С9Н8О3(11). При обработке кис-
лотами из соли регенерируется вещество I, которое реагирует с бромом в четы-
реххлористом углероде с образованием CoHeOjBr^III). Последнее при нагрева-
нии с раствором гидроксида натрия постепенно переходит в раствор. При под-
кислении раствора выпадает вещество состава С9НбОз(1У).
В ПМР-спектре кислоты II (в CDC13, содержащем ДМСО-б8) имеются сле-
дующие сигналы: б 6,4—8,2 м. д., мультиплет; б 10,3 м. д., очень широкий сннг-
лет. Химический сдвиг сигнала с б 10,3 м. д. изменяется в зависимости от кон-
центрации образца; интенсивность этого сигнала в три раза меньше интенсивно-
сти сигнала с б 6,4—8,2 м. д.
2. Соединение C8H8ONBr при обработке кипящим раствором гидроксида ка-
лия дает калиевую соль кислоты С8Н8О3, которую разделяют на d- и 1-формы.
В ПМР-спектре кислоты С8Н8О8 (в ацетоне) имеются следующие сигналы:
б 5,22 м.д., 1Н, синглет; б 7,2—7,7 м.д., 7Н, мультиплет. Химический сдвиг сиг-
нала с б 5,22 м. д. не зависит от концентрации.
Установление структуры, методы определения и упражнения
553
3. Соединение СцН12О4(1) реагирует с горячим раствором гидроксида на-
трия с образованием соли С6Н|зО5На(П). При обработке соли горячей разбав-
ленной серной кислотой она превращается в кислоту С9Н8О4(Ш), которая в
свою очередь при нагревании дает соединение Ci8Hi2O8(IV), плавящееся при вы-
сокой температуре.
4. Соединение С6Н8О2(1) прн кипячении с концентрированной соляной кис-
лотой превращается в С5Н9О2С1(П) и медленно реагирует с раствором гидро-
ксида калия с образованием С5Н90зК(П1). Последнее соединение превращается
в C6II8O3(IV) при обработке щелочным раствором перманганата. В результате
обработки раствором гипохлорита натрия из соединения IV получена кислота
C4H8O4(V) с эквивалентом нейтрализации 58 ±1. При нагревании этой кислоты
образуется соединение состава C4H4O3(VI). Из строго нейтрального водного рас-
твора соединения II, приготовленного нейтрализацией раствором гидроксида на-
трия, медленно выпадает исходное соединение I.
В ПМР-спектре соединения V (в ДМСО-б6) имеются только два синглета
с б 2,43 и 11,8 м.д. Отношение интенсивностей синглетов составляет 2: 1 соот-
ветственно и сигнал с б 11,8 м.д. очень широкий.
5. Жидкое нейтральное соединение имеет формулу C7H7NO3. В ПМР-спектре
этого соединения (в СС14) имеются следующие сигналы: б 3,89 м. д., ЗН, синглет;
б 6,91 м.д., 2Н, дублет (I = 10 Гц); б 8,12 м.д., 2Н, дублет (J = 10 Гц).
6. Соединение CI6HI3N дает соли с сильными минеральными кислотами, ко-
торые гидролизуются водой. В ПМР-спектре этого соединения (в СС14) имеются
следующие сигналы: б 5,61 м.д., 1Н, широкий синглет; б 6,60—7,55 м.д., ЮН,
мультиплет; б 7,80 м.д., 2Н, мультиплет.
7. Соединение С14Н10Оз превращается в С13Н10О3 при окислении щелочным
раствором перманганата. Исходное соединение реагирует с натрием с образова-
нием Ci4H9ONa.
8. Кислота с эквивалентом нейтрализации 57 не изменяется при действии
брома в четыреххлористом углероде.
Серия 8
1. Жидкость CSH4O2(I) обесцвечивает раствор перманганата, а также вос-
станавливает раствор Бенедикта. Прн нагревании с цианидом щелочного ме-
талла соединение I димеризуется, а в результате обработки этилортоформиа-
том в присутствии кислого катализатора превращается в новое вещество:
С9Н i4O8.
В ПМР-спектре соединения I (в CDCI3) имеются следующие сигналы: б
6,63 м. д., 1Н, дублет дублетов (7 = 5,2 Гц); б 7,28 м.д., 1Н, дублет (7 =
= 5 Гц); б 7,72 м.д., 1Н, мультиплет; б 9,67 м.д., 1Н, синглет. Химический
сдвиг сигнала с б 9,67 м. д. не зависит от концентрации.
2. Соединение С4Н4О4(1) растворяется в разбавленном растворе гидроксида
натрия, а при обработке бромом превращается в С4Н4О4Вг2(П). Соединение I ре-
генерируется из соединения II при обработке последнего цинковой пылью. При
нагревании с иодистым водородом вещество II превращается в С4Н8О4(Ш),
имеющее эквивалент нейтрализации 59 и теряющее молекулу воды при нагре-
вании. Ангидрид, полученный таким путем, реагирует с бензолом в присутствии
хлорида алюминия с образованием соединения СюНщОз, которое растворяется
в щелочи, реагирует с фенилгидразином и превращается в бензойную кислоту
при энергичном окислении.
В ПМР-спектре соединения СюНюОз (в CDCU) имеются следующие сигналы:
б 2,8 м. д., 2Н, триплет (7 = 7 Гц); б 3,3 м.д., 2Н, триплет (7 = 7 Гц); б 7,2—
7,6 м. д., ЗН, мультиплет; б 8,0 м.д., 2Н, мультиплет; б 11,7 м.д., 1Н, сииглет.
Химический сдвиг сигнала с б 11,7 м.д. изменяется в зависимости от концен-
трации.
3. Соединение CnHioN2 при энергичном окислении превращается в СцН8М2О.
4. Соединение С5НюО обесцвечивает щелочной раствор перманганата калия,
НО не изменяется при действии брома в четыреххлористом углероде. В масс-
554
Глава 9
спектре этого соединения (т. кип. 75°С) имеются следующие пики с интенсив-
ностью, равной или больше 5% основного пика:
т/е Интенсивность, % от интенсивности основного пика mle Интенсивность, % от интенсивности основного пика
86 34 41 83
57 (100. основной пнк) 39 17
55 7 29 40
43 18 27 11
5. Соединение С14Н|2О превращается при окислении хромовой кислотой в
кислоту с эквивалентом нейтрализации 226. В ПМР-спектре продукта окисления
(в CDC13, содержащем ДМСО-(16) имеются следующие сигналы: б 7,45—
7,95 м. д., 7Н, мультиплет; б 8,19 м.д., 2Н, дублет (7 = 7 Гц); б 10,8 м. д., 1Н,
широкий синглет.
6. Нейтральное соединение С10НбО4(1) при нагревании с раствором гидро-
ксида натрия превращается в соль кислоты(II) с эквивалентом нейтрализации
209 ± 2. Соединение 1 реагирует со щелочным раствором пероксида водорода
с образованием новой кислоты(III), эквивалент нейтрализации которой равен
111 ± 1.
В ПМР-спектре соединения I (в CDC13) имеются следующие сигналы: б
6,64 м.д., 2Н, дублет дублетов (7 = 5 Гц, 2 Гц); б 7,63 м.д., 2Н, дублет (7 =
= 5 Гц); б 7,78 м. д., 2Н, дублет (7 = 2 Гц).
7. Соединение CioH702N(I) при обработке железом и соляной кислотой пре-
вращается в вещество CioHgN(II), которое растворяется в разбавленной соляной
кислоте. При энергичном окислении 1 и II получены C8H5O6N и'С8НбО4 соответ-
ственно. Оба продукта окисления растворяются в щелочи.
В ПМР-спектре соединения 1 (в CDC13) имеется сложный мультиплет, рас-
положенный при б 7,15—8,5 м.д. При нагревании продукт окисления II (С8Н6О4)
теряет молекулу воды.
Серия 9
1. Встречающееся в природе соединение СюНюО2(1) не реагирует с ацетил-
хлоридом и фенилгидразином. Однако нагревание с гидроксидом калия вызывает
изомеризацию. Изомер II также не реагирует с ацетилхлорндом н феннлгидрази-
ном. Оба изомера обесцвечивают растворы брома и перманганата. Под дей-
ствием озона изомер II превращается в соединение III, нз которого получен фе-
нилгидразон. В результате окисления соединений II или III щелочным раствором
перманганата образуется кислота C8H6O4(IV).
В ПМР-спектре соединения I (в CDCI3) имеются следующие сигналы: б
3,30 м. д., 2Н, дублет мультиплетов (7 = 7 Гц, дополнительно незначительные
константы спин-спинового взаимодействия); 6 4,90 м. д., 1Н, мультиплет; б 5,5 м. д.,
1Н, мультиплет; б 5,88 м.д., 2Н, синглет; б около 5,6—6,2 м.д., 1Н, мультиплет
(широкая полоса, сильно расщепленная); б 6,67 м.д., ЗН, синглет (с незначи-
тельным расщеплением).
2. Соединение С8Н5О2С1(1) при нагревании с абсолютным спиртом дает ве-
щество Ci4H2o04(II), которое щелочным раствором перманганата окисляется в
СеН8О4. При взаимодействии соединения I с анилином получено соединение
C20H,eON2. Продукт окисления перманганатом обработкой избытком тионилхло-
рида превращен в вещество 111 (С8М4С12О2). В ПМР-спектре соединения III (в
CDCI3) имеются следующие сигналы: б 7,72 м.д., 1Н, триплет (7 = 7 Гц';
б 8,33 м. д., 2Н, триплет дублетов (7 = 7,2 Гц); б 8,82 м.д., 1Н, триплет (7 =
= ~2 Гц).
Установление структуры: методы определения и упражнения
55В
3. Соединение С8НнОз(1) растворяется в разбавленном растворе гидроксида
натрия. Под действием фенилгидразина оно превращается в Ci2H|4ON2(Il), а в
результате нагревания с 20%-ной соляной кислотой образуется С5НюО(1П). Из
соединения III при обработке семикарбазидом получен кристаллический осадок,
однако с раствором гипоиодита натрия йодоформ не образуется.
В ПМР-спектре соединения, полученного при обработке семнкарбазидом со-
единения III (в CDCh), имеются следующие сигналы: 6 1,09 м.д., 6Н, триплет;
6 2,26 м.д., 4Н, квадруплет; 6 5,89 м.д., 2Н, широкий синглет; б 8,59 м.д., 1Н,
широкий синглет.
4. Соединение CgH2N(I) каталитически восстанавливается в СдНцМ(П).
Если соединение II обработать избытком метнлнодида, затем оксидом серебра и
продукт реакции нагреть, то образуется вещество Ci|HiBN(III), которое при
энергичном окислении превращается в C8H8O4(IV), а при обработке озоном и
последующем гидролизе продукта реакции — в Ci0Hi3ON(V). При нагревании
продукта озонирования с разбавленным раствором цианида калия получено ве-
щество состава C2oH2602N2(VI). Соединение VI при энергичном окислении пре-
вращается в IV.
В ПМР-спектре соединения I (в CDCh) имеются следующие сигналы: 6
7,25—8,1 м.д., 5Н, мультиплет; 6 8,52 м.д., 1Н, дублет (7 = 7 Гц); б 9,26 м.д.,
1Н, синглет.
5. Соединение СюН|0О(1) реагирует с натрием с образованием производ-
ного CioHgONa, которое полностью гидролизуется водой. При обработке соеди-
нения I холодной 80%-ной серной кислотой в присутствии сульфата ртути, а за-
тем водой получено вещество СюН12О2(П). Это соединение медленно растворяет-
ся в растворе гипохлорида натрия. При подкислении раствора получено веще-
ство С9Н1оОз(Ш). Под действием кипящей 48%-ной бромистоводородной кис-
лоты соединение III дает C8H7O2Br(IV), энергичное окисление которого приводит
к С8Н8О4.
В ПМР-спектре соединения С8Н8О4 (в CDCI3, содержащем ДМСО-de) имеют-
ся только два синглета с д 8,08 и 11,0 м.д. с отношением интенсивностей 2: 1.
6. Соединение с молекулярной формулой C)4Hi2 прн окислении перманга-
натом превращается в производное с молекулярной формулой С13НюО, которое
не изменяется при дальнейшей обработке перманганатом.
В ПМР-спектре исходного соединения (в СС14, 100 МГц) имеются следую-
щие сигналы: б 5,35 м.д., 2Н, синглет; 6 7,21 м.д., ЮН, синглет (слабо искажен
у основания).
7. Оптически активное соединение C9HI3N превращается при энергичном
окислении в кислоту с эквивалентом нейтрализации 83. В ПМР-спектре очень
близкого изомера исходного соединения (в CDC13) имеются следующие сигналы:
б 0,95 м. д., 2Н, синглет; б 2,30 м.д., ЗН, синглет; 6.2,5—3,0 м.д., 4Н, мульти-
плет (8 симметричных линий); б 7,05 м.д., 4Н, синглет. Окисление изомера при-
водит к той же кислоте, которая получена при окислении исходного соедине-
ния.
8. Соединение не обесцвечивает бромную воду, но реагирует с натрием с об-
разованием C8H8O3Na2. Эквивалент нейтрализации этого вещества равен 152.
Это соединение оптически неактивно и при энергичном окислении превращается
в кислоту С8Н6О4 с эквивалентом нейтрализации 82 ± 2. В ПМР-спектре этой
кислоты (в CDCI3, содержащем ДМСО-de) имеются следующие сигналы: б
8,08 м. д., 4Н, синглет; б 11,0 м.д., 2Н, широкий синглет.
Серия 10
1. Бесцветное кристаллическое вещество I содержит азот, но не содержит
галогенов и серы, растворяется в воде, но не растворяется в эфире. Его водный
раствор кислый и имеет эквивалент нейтрализации 123 ± 1. При обработке ни-
тритом натрия и соляной кислотой выделяется газ, одна треть которого раство-
рена в растворе гидроксида калия. Раствор, полученный при обработке азоти-
стой кислотой, упаривают досуха; остаток состоит только из хлорида натрия,
556
Глава 9
нитрата натрия и нитрита натрия. При нагревании соединения I с разбавлен-
ным раствором гидроксида натрия выделяется аммиак, а из раствора при под-
кислении выделяется другой газ. При упаривании этого раствора остаются толь-
ко неорганические соли.
При осторожной нейтрализации основанием в мягких условиях из исходного
соединения получены кристаллы с т. пл. 135°С. В УФ-спектре этих кристаллов (в
присутствии гидроксида натрия) отсутствует поглощение выше 220 нм. Для этих
кристаллов получены следующие спектры.
Масс-спектр (приведены только пики с интенсивностью 10% и больше от
основного пика):
т/е Интенсивность, % от интенсивности основного пика
60 (100, основной пик)
44 60
43 18
28 17
ИК-спектр: 3450 см-1 (2,90 мкм); 3350 см-1 (2,99 мкм); 1690 см-1 (5,90 мкм);
1640 см~* (6,10 мкм); 1600 см-1 (6,25 мкм); 1470 см~* (6,80 мкм); 1160 см-1
(8,62 мкм); 1050 см~* (9,52 мкм); 1000 см-1 (10,00 мкм); 790 см-1 (12,66 мкм);
710 см-1 (14,8 мкм).
2. Неизвестное соединение содержит углерод, водород, кислород и азот, не
растворяется в воде, в разбавленных кислотах н в разбавленных щелочах. Оно
не реагирует с ацетилхлорндом и фенилгндразином и с трудом восстанавли-
вается. При обработке горячим водным раствором щелочи это вещество мед-
ленно растворяется. Перегонкой щелочного раствора получен дцстиллат, содер-
жащий вещество, которое высолено поташом. Это соединение дает йодоформ-
ную реакцию, но не реагирует с солянокислым раствором хлорида цинка. Исход-
ный щелочной раствор (остаток после перегонки) подкисляют серной кислотой
н полученный раствор снова подвергают перегонке; в дистиллате получена низ-
кокипящая кислота. Этот отгон восстанавливает перманганат. В результате точ-
ной нейтрализапии жидкого остатка после второй перегонки выделены кристал-
лы, содержащие азот, эквивалент нейтрализации которых равен 137 ± 1.
В ПМР-спектре 100 мг низкокипящей кислоты (в 0,5 мл СС14) имеются сле-
дующие сигналы: б 5,8—6,75 м.д., ЗН, мультиплет; б 12,4 м.д., 1Н, синглет.
В ПМР-спектре 60 мг кристаллов с эквивалентом нейтрализации 137 (в 0,5 мл
ацетона) имеются сигналы: б 6,55 м. д., ЗН, синглет (химический сдвиг изме-
няется в зависимости от концентрации); б 6,75 м.д., 2Н, дублет (/ = 8 Гц);
б 7,83 м.д., 2Н, дублет (/ = 8 Гц). В ПМР-спектре соединения, высоленного
поташом, наряду с другими сигналами имеется сигнал, химический сдвиг кото-
рого зависит от концентрации и который представляет собой синглет в спектре,
снятом в растворе CDCls, и триплет (У = ~7 Гц) в спектре раствора в
ДМСО-de.
3. Кислота С6Н8О4(1) под действием пентахлорида фосфора превращается
в СвНбОаСЬЩ). Из этого хлорпроизводного при обработке бензолом в присут-
ствии безводного хлорида алюминия получено вещество С18Н|вО2(1П), которое
не обесцвечивает перманганат, но легко образует дибромид и диоксим. Послед-
ний под действием пентахлорида фосфора перегруппировывается в Cl8Hi8O2N2
(IV). Из этого соединения гидролизом была получена исходная кислота 1.
В ПМР-спектре соединения II (растворе СС14) имеются следующие сигналы:
б 2,2—2,6 м. д., 4Н, мультиплет; б 3,90 м. д., 2Н, триплет (искаженный, 1 =
= ~7 Гц); б 11,9 м. д., 2Н, широкий синглет.
4. Соединение I содержит углерод, водород, кислород, азот и хлор, раство-
ряется в воде, но не растворяется в эфире. При прибавлении нитрата серебра из
водного раствора немедленно выпадает осадок. В результате точной нейтрализа-
Установление структуры: методы определения и упражнения
557
ции водного раствора соединения I выделено соединение II, не содержащее
хлора. Ойо реагирует с уксусным ангидридом с образованием растворимого в
щелочи, но нерастворимого в кислоте соединения III с эквивалентом нейтрализа-
ции 207 ± 2. Соединение III реагирует с бензолсульфохлоридом с образованием
растворимого в щелочи продукта, а при его взаимодействии с азотистой кисло-
той даже при нагревании не выделяется никакого газа. Продукт этой последней
реакции все еще содержит азот и растворяется в щелочах, но не растворяется
в кислотах. Энергичное окисление соединений I, II или III приводит к не содер-
жащей азота кислоте, которая не растворяется в воде и имеет эквивалент ней-
трализации 82 ± 2.
В ПМР-спектре не содержащей азота кислоты (в CDCI3, содержащем
ДМСО-dg) имеются следующие сигналы: б 7,62 м.д., 1Н, триплет (7 = 7 Гц);
6 8,25 м.д., 2Н, искаженный дублет (7= ~2 Гц); б 8,70 м.д., 1Н, искаженный
синглет; б 12,28 м. д„ 2Н, синглет.
5. Соединение СюНвО3(1) обесцвечивает щелочной раствор перманганата и
реагирует с гидроксиламином. Оно разлагается при перегонке при атмосферном
давлении с образованием соединения С9НвО2(П), которое дает мононатриевое
производное и легко окисляется в СдНзОДП!). Соединение III представляет со-
бой кислоту, которая при нагревании с натронной известью превращается в
С7Н6О2(^). Соединение IV разлагается при нагревании с разбавленной соляной
кислотой под давлением с образованием слабокислого соединения формулы
С6Н6О2.
В ПМР-спектре соединения IV (в CDC13) имеются . следующие сигналы: б
5,90 м. д., 2Н, сииглет; б 6,83 м. д., 4Н, синглет (слабые искажения в основании
этого сигнала). В ПМР-спектре соединения III (в CDCI3) имеются следующие
сигналы: б 6,00 м. д., 2Н, синглет; б 6,8 м.д., 1Н, дублет (7 = 7 Гц); б 7,38 м.д.,
1Н, дублет (7 = 2 Гц); б 7,55 м.д., 1Н, дублет дублетов (7 = 7,2 Гц); б 7,6 м. д.,
1Н, широкий синглет. Химический сдвиг сигнала с б 7,6 м.д. изменяется в зави-
симости от концентрации.
Серия 11
1. Соединение А содержит только углерод, водород, кислород, не раство-
ряется в воде, разбавленных кислотах и разбавленных щелочах, но растворяется
в холодной концентрированной серной кислоте. Оно не реагирует с фенилгидра-
знном н ацетилхлорндом. При нагревании с водным раствором гидроксида
натрия образуется масло (Б) н ннзкокипящая кислота с эквивалентом нейтрали-
зации 59 ± 1. Соединение Б реагирует с ацетилхлорндом, но не реагирует с фе-
нилгидразином. При обработке концентрированным раствором бромистого водо-
рода оно превращается в бромсодержащее соединение В, которое дает положи-
тельные реакции с нитратом серебра, хлоридом железа (III) и бромной водой.
При окислении в мягких условиях из вещества Б получена кислота Г с эк-
вивалентом нейтрализации 164 + 2.
Обработка кислоты Г этиловым спиртом в присутствии следов серной кис-
лоты дает соединение Д. В ПМР-спектре соединения Д (в ССЦ) имеются сле-
дующие сигналы: б 1,35 м.д., триплет (7 = 7 Гц); б 1,40 м.д., триплет (7 =
= 7 Гц); б 3,8—4,5 м. д., 4Н, 6 линий (отношение интенсивностей 1 : 3 : 4 : 4 : 3 : 1,
расположенные с интервалами около 7 Гц друг от друга); б 6,79 м.д., 2Н, дуб-
лет (7 = 9 Гц);'б 7,89 м.д., 2Н, дублет (7 = 9 Гц). Сигналы с б 1,35 и 1,40 м. д.
имеют общую площадь 6Н.
2. Соединение I содержит азот и хлор, не растворяется в воде и соляной
кислоте, но растворяется в растворе бикарбоната натрия. Найденный для него
эквивалент нейтрализации равен 210 ± 2. Соединение I реагирует с ацетнлхло-
ридом, но не реагирует с горячим спиртовым раствором нитрата серебра. Аце-
тильное производное II имеет эквивалент нейтрализации 253 ± 2. При кипяче-
нии соединения I со щелочью выделяется аммиак, а при подкислении получен-
ного раствора выпадает осадок кислоты III с эквивалентом нейтрализации
115+1. Соединение 111 содержит хлор, но не содержит азота. При его кипяче-
нии с раствором перманганата калия образуется новое вещество, имеющее такие
558
Глава 9
же характеристики растворимости, как и соединение I, все еще содержащее хлор
и имеющее эквивалент нейтрализации 81 ± 1.
Из соединения IV (эквивалент нейтрализации 71) хлорированием можно
получить соединение с эквивалентом нейтрализации 81. В ПМР-спектре соеди-
нения IV (растворитель СПС1з/ДМСО-б6) имеются следующие сигналы: б 7,57 м.д.,
1Н, триплет (7 = 7 Гц); б 8,18 м.д., 2Н, дублет (7 = 7 Гц); б 11,2 м.д., ЗН,
широкий синглет.
В ИК-спектре соединения 1 (в нуйоле) наряду с другими полосами погло-
щения имеется сильное поглощение при 1779 см-1 (5,62 мкм) и 1712 см-1
(5,84 мкм).
3. Бесцветные кристаллы нейтрального соединения Л не содержат азота,
серы и галогенов, дают положительную реакцию с фенилгидразином и ацетил-
хлоридом, а при окислении в мягких условиях превращаются в соединение Б,
представляющее собой желтые кристаллы. Последнее реагирует с фенилгидра-
зином с образованием того же производного, которое получено из соединения Л.
При энергичном окислении соединения Б образуется кислота В с эквивалентом
нейтрализации 121 ± 1.
В ПМР-спектре соединения А (в CDCls) имеются следующие сигналы: б
4,5 м.д., 1Н, широкий синглет; б 5,9 м.д., 1Н, широкий сннглет; б 7,2—7,5 м.д.,
8Н, мультиплет (основная часть которого была резким синглетом); б 7,85 м.д.,
2Н, дублет дублетов.
4. Жидкость 1 содержит хлор, не растворяется в воде, разбавленной соляной
кислоте, разбавленном растворе гидроксида натрия и в фосфорной кислоте, но
растворяется в холодной концентрированной серной кислоте. Жидкость I не
дает осадка с теплым спиртовым раствором нитрата серебра и не реагирует с
ацетилхлорндом. При обработке фенилгидразином образуется осадок, но с реак-
тивом Шиффа окрашивания не наблюдается. При кипячении с концентрирован-
ным раствором гидроксида натрия соединение I растворяется. Дистнллат при
перегонке этого щелочного раствора дает йодоформную реакцию. При подкисле-
нии щелочного раствора осаждается соединение II, содержащее хлор, с эквива-
лентом нейтрализации 156+1. На соединение II не действует раствор перман-
ганата. После удаления соединения II часть кислого фильтрата перегоняют; дн-
стиллат является кислым по лакмусу. Число омыления исходного соединения 1
равно 113± 1. Из кислого фильтрата получено соединение с эквивалентом ней-
трализации 61 ± 1.
В ПМР-спектре соединения II (в CDC13/ДМСО-de) имеются следующие сиг-
налы: б 7,1—7,5 м. д., ЗН, мультиплет (главным образом большой синглет); б
7,82 м. д., 1Н, мультиплет; б 9,0 м.д., 1Н, широкий синглет.
5. Жидкое вещество имеет число омыления 163 ± 1. При омылении обра-
зуется масло, которое дает положительную реакцию с хлоридом железа (III), и
кислота с эквивалентом нейтрализации 60± 1.
В ПМР-спектре 49 мг масла в 0,5 мл СС14 имеются следующие сигналы:
б 2,18 м. д., 6 Н, синглет; б 5,73 м.д., 1Н, синглет; б 6,33 м.д., 2Н, синглет; б
6,45 м.д., 1Н, синглет. При прибавлении большего количества СО, сигнал с б
5,73 м. д. смещался в сильное поле.
Серия 12
1. Соединение I содержит азот, не растворяется в эфире и растворяется в
воде с образованием щелочного раствора. Прн титровании этого раствора кис-
лотой получен эквивалент нейтрализации 37 ± 1. При обработке холодного рас-
твора соединения I нитритом натрия и соляной кислотой выделяется газ. К по-
лученному раствору прибавляют щелочь до явно щелочной реакции, а затем
бензоилхлорид. При этом выделяется нейтральное соединение 11, число, омыле-
ния которого равно 142 + 1.
В ПМР-спектре соединения I (в CDCI3) имеются следующие сигналы: б
1,08 м.д., синглет; б 1,58 м.д., 5 линий (7 = 7 Гц, отношение интенсивностей
1:4:6:4:1); б 1,75 м.д., триплет, отношение интенсивностей сигналов равно
2:1:2.
Установление структуры: методы определения и упражнения оа»
2. Бесцветная жидкость I не содержит галогенов, азота, серы и металлов,
не растворяется в воде, разбавленной соляной кислоте, разбавленном растворе
гидроксида натрия и фосфорной кислоте, но растворяется в холодной концен-
трированной серной кислоте. Она не реагирует с ацетилхлорндом и фенилгидра-
зином н не изменяется прн нагревании с гидроксидом натрия. При кипячении
с разбавленной фосфорной кислотой и после охлаждения раствора выделяется
масло (II). Соединение II не реагирует с ацетилхлорндом, но при обработке фе-
нилгидразином и раствором бисульфита натрия выпадает осадок. При энергич-
ном встряхиваннн соединения II с концентрированной щелочью из щелочного
раствора выделяется соединение III. Последнее реагирует с ацетилхлорндом, но
не взаимодействует с фенилгидразином. Прн подкислении щелочного раствора
получается вещество IV с эквивалентом нейтрализации 136 ± 1- При энергичном
окислении соединения IV образуется кислота(V) с эквивалентом нейтрализации
82 + 1.
Фосфорнокислый раствор после выделения соединения II перегоняют. Ди-
стиллят насыщают поташом и получают соединение VI, которое реагирует с на-
трием и ацетилхлорндом и дает желтый осадок с гипоиодитом натрия. С реаген-
том Лукаса это вещество не реагирует.
В ПМР-спектре соединения II (в CDC13) имеются следующие сигналы: 6
2,42 м. д., ЗН, синглет; б 7,18 м.д., 2Н, дублет (/ — 8 Гц); б 7,66 м.д., 2Н, дуб-
лет (/ = 8 Гц); 6 9,81 м. д., 1Н, сннглет.
3. Бесцветная жидкость I не содержит азота, серы и галогенов, растворяется
в воде и эфире, не реагирует с натрием, ацетилхлорндом, фенилгидразином и
разбавленным раствором перманганата, не обесцвечивает бром в четйреххлори-
стом углероде и не изменяется прн кипячении со щелочами. При нагревании со-
единения I с избытком бромистоводородной кислоты выделяется масло(П), ко-
торое содержит бром и легко дает осадок со спиртовым раствором нитрата се-
ребра. Это масло не растворяется в воде, кислотах и щелочах. После высуши-
вания н очистки соединение II обрабатывают магнием в чистом эфире. Реакция
сопровождается выделением газа(1П). Реагент Гриньяра не обнаружен. При об-
работке соединения II спиртовым раствором гидроксида калия выделяется
газ(IV), который дает осадок при пропускании его через аммиачный раствор
нитрата серебра. Соединение III не образует осадка с аммиачным раствором
хлорида меди(1). Оба вещества (III и IV) обесцвечивают бромную воду и вос-
станавливают растворы перманганата. Тщательное изучение взаимодействия со-
единения I с бромнстоводородной кислотой показало, что вещество II — единст-
венное органическое соединение, образующееся при этом, и никакие газы при
этой реакции не выделяются.
В масс-спектре соединения I имеются пики со следующими значениями т/е
(в скобках указана относительная интенсивность): т/е 88 (650), т/е 89 (32),
т/е 90 (3); не обнаружено пиков с большими массовыми числами. В ПМР-спек-
тре соединения I (в CDCh) имеется только один синглет с б 3,69 м. д.
4. Соединение I содержит углерод, водород, азот, кислород, не растворяется
в разбавленных щелочах и кислотах. При нагревании в течение некоторого
времени с соляной кислотой нз соединения I получается кристаллическая кис-
лота (II) с эквивалентом нейтрализации 180+1. При окислении вещества I би-
хроматом калия и серной кислотой образуется кристаллическая, содержащая
азот кислота(III) с эквивалентом нейтрализации 166 ±1. Соединение I реаги-
рует с бензальдегидом в присутствии щелочей с образованием бензального про-
изводного.
Прн обработке соединения I хлоридом олова(II) и хлористым водородом в
сухом эфире, а затем водой получается соединение IV с молекулярной формулой
C8H7N. Определение по способу Церевитинова показало, что соединение IV со-
держит один активный атом водорода. Соединение IV представляет собой сла-
бое основание и осмоляется при действии кислот.
В ПМР-спектре соединения I (в CDC13) имеются следующие сигналы: б
4,25 м. д., 2Н, синглет; б 7,5—8,0 м.д., ЗН, мультиплет; б 8,25 м.д., 1Н, дублет
(слабо искажен). В ПМР-спектре соединения II (в CDC13, содержащем ДМСО-
de) имеются следующие сигналы: б 4,02 м. д., 2Н, синглет; б 7,3—7,65 м. д., ЗН,
560
Глава 9
мультиплет; б 8,0 м.д., 1Н, дублет (искаженный); 0 11,2 м.д., 1Н, широкий
синглет. В ПМР-спектре соединения III (в CDCI3, содержащем ДМСО-с16) имеют-
ся следующие сигналы: б 7,5—8,0 м.д., 4Н, мультиплет; б 12,15 м.д., 1Н, синг-
лет. В ПМР-спектре соединения IV (160 мг в 0,5 мл СС14) имеются следующие
сигналы: б 6,38 м.д., 1Н, мультиплет; б 6,76 м.д., 1Н, триплет (искаженный);
б 6,95—7,10 м.д., 4Н, мультиплет; б 7,4—7,65 м.д., 1Н, мультиплет.
Серия 13
1. Бесцветная маслянистая жидкость I, обладающая приятным запахом, со-
держит только углерод, водород и кислород, реагирует с фенилгидразином, легко
обесцвечивает раствор перманганата калия, но не реагирует с ацетилхлоридом.
При обработке ее концентрированным раствором гидроксида натрия н после-
дующем подкислении реакционной смесн получены два продукта: содержащее
кислород соединение II, которое реагирует с ацетилхлоридом, и кислота(III),
которая реагирует с дымящей серной кислотой, а при 200°С с выделением дио-
ксида углерода превращается в содержащее кислород соединение (С4Н4О). Это
вещество обесцвечивает раствор перманганата, но не реагирует с натрием и фе-
нилгидразином. Прн нагревании соединения I с цианидом калия получается ве-
щество IV, которое дает озазон и восстанавливает щелочные растворы меди, а
при окислении иодной кислотой превращается в исходное соединение I и кис-
лоту(Ш).
В ПМР-спектрах некоторых приведенных выше соединений имеются сле-
дующие сигналы:
Химический сдвиг, м. д. Число протонов Мультиплетность
Соединение / (в CDCl3)
6,63 1Н Дублет дублетов (7 = 5 Гц, '2 Гц)
7,28 1Н Дублет (7 = 5 Гц)
7,72 1Н Мультиплет (7 = ~ 2 Гц и меньшие кон- станты спин-спииового взаимодействия)
9,67 1Н Синглет
Соединение 11 (в CDCl3)
2,83 1Н Синглет (химический сдвиг изменяется в зависимости от концентрации)
4,57 2Н Синглет
6,33 2Н Мультиплет а
7,44 1Н
Соединение HI (в ДМСО-с16)
6,88 1Н
7,78 1Н
8,32 1Н
11,88 1Н Широкий синглет
Соединение CtHtO (в CDCl3)
6,37 2Н Триплет
7,42 2Н
а Все отмеченные сигналы имели только незначительные константы спин-сп «нового
взаимодействия (около 0- -2 Гц).
2. Соединение I со слабокислыми свойствами содержит азот,- но не содержит
я н галогенов, реагирует с ацетилхлоридом, но не взаимодействует с фенил-
Установление структуры: методы определения и упражнения 561
гидразином. При нагревании соединения I с разбавленными кислотами получает-
ся соединение II, выделенное перегонкой из кислого раствора и насыщением ди-
стиллята поташом. Соединение II не реагирует с ацетилхлоридом и реактивом
Шиффа, ио дает положительные реакции с фенилгидразином и бисульфитом
натрия.
Соединение I нагревают с пентахлоридом фосфора и выливают в воду, в
результате чего получают вещество III, все еще содержащее азот, но не содер-
жащее галогенов. Соединение III нейтрально и разлагается щелочами. При до-
бавлении бензоилхлорида к щелочному раствору III получена кислота (IV) с
эквивалентом нейтрализации 220 ± 2. При нагревании соединения IV в течение
некоторого времени с разбавленными кислотами и при последующей перегонке
получены два продукта. Один из них (V) не содержит азота, обладает кислыми
свойствами и имеет эквивалент нейтрализации 120 ± 2. Другой продукт иденти-
чен соединению III. При обработке соединения III холодной концентрированной
соляной кислотой получено соединение VI, которое содержит азот и хлор н рас-
творимо в воде. При обработке соединения IV холодным раствором нитрата
натрия выделяется газ.
В ПМР-спектрах соединений I и II имеются следующие сигналы:
Химический сдвиг, м. д. Число протонов Мультиплетиость
Соединение 1 (в CDClB)
1,77 4Н Мультиплет
2,40 4Н
9,12 1Н Широкий синглет
Соединение III (в CDClB)
1,80 4Н Мультиплет
2,38 2Н
3,32 2Н
7,60 1Н Широкий сииглет
3. Соединение I растворяется в воде, но не растворяется в эфире. При на-
гревании оио разлагается и образуются соединение II, нерастворимое во всех рас-
творителях, и соединение III со свойствами основания. При нагревании смесей
II и III снова образуется вещество I. При обработке соединений I и II нитратом
серебра немедленно выпадает осадок. Соединение III ие дает бензолсульфон-
амида, но дает нитрозопроизводное.
В ПМР-спектрах соединений I—III имеются следующие сигналы:
Химический сдвиг, м. д. Число протонов Мульт и плетиость
Соединение I (D?O; 80 мг/0,5 мл)
3,70 9Н Синглет
7,4 5Н Мультиплет
Соединение 11 (растворитель CDCl3)
2,20 - Синглет
Соединение 111 (растворитель CDCl3)
2,85 6Н Синглет
6,4-6,7 ЗН Мультиплет
7,10 2Н »
562
Глава 9
4. Нейтральное соединение 1 дает положительную реакцию на хлор, бром и
иод. При обработке спиртовым раствором нитрата серебра выпадает белый оса-
док, который легко растворяется в аммиаке. С фенилгндразином образуется
осадок, но с ацетилхлорндом реакция не происходит. Перманганат, а также
бром в четыреххлористом углероде медленно обесцвечиваются. При встряхивании
с холодным разбавленным раствором щелочи в течение некоторого времени со-
единение I растворяется. Подкислением щелочного раствора получено вещество
II, которое дает положительную реакцию на бром и иод и имеет эквивалент
нейтрализации 369 ± 3. При кипячении соединения I с разбавленным раствором
щелочи и последующем подкислении выпадает осадок вещества III, которое дает
положительную реакцию на иод, имеет эквивалент нейтрализации 306 ±3 и реа-
гирует как с фенилгндразином, так и с ацетилхлорндом. При обработке соеди-
нения I или II гипохлоритом натрия после подкисления получена кислота, со-
держащая иод и имеющая эквивалент нейтрализации 146 ± 1.
5. Нейтральное соединение А содержит бром и не содержит других галоге-
нов и азота. Оно не реагирует с горячим спиртовым раствором нитрата серебра,
ацетилхлорндом, фенилгндразином и раствором брома в четыреххлористом уг-
лероде. Это вещество растворяется в кипящем растворе гидроксида натрия, но
дистиллат после отгонки из этого щелочного раствора не содержит никаких ор-
ганических соединений. При подкислении щелочного раствора фосфорной кис-
лотой осаждается соединение Б, которое содержит бром и имеет эквивалент нейт-
трализации 200 ± 2. Дистиллат после перегонки с паром кислого раствора тща-
тельно экстрагируют хлороформом. Оставшуюся после отгонки хлороформа бес-
цветную жидкость В очищают перегонкой. Вещество В не содержит брома, рас-
творяется в растворе бикарбоната натрия и имеет эквивалент нейтрализации
102 ± 1. Раствор, оставшийся после удаления вещества В из дистиллата, после
перегонки с паром подщелачивают до явно щелочной реакции, затем к нему
прибавляют беизоилхлорид и смесь энергично встряхивают. Из щелочного рас-
твора выделяется новое нейтральное соединение Д, не содержащее брома. Его
число омыления равно 135 dr 1.
В ПМР-спектрах соединений Б и В имеются следующие сигйалы:
Химический сдвиг, м. д. Число протонов Мультиплетиость
Соединение Б (в ДМСО-сЦ)
7,3 1Н Широкий синглет (очень широкий)
7,71 2Н Дублет (искаженный)
7,90 2Н То же
Соединение В (в ССЦ)
0,93
1,2—1,8
2,31
11,7
ЗН
4Н
2Н
1Н
Триплет (искаженный)
Мультиплет
Триплет
Синглет
Серия 14
1. Соединение 1 содержит только углерод, водород и кислород и имеет экви-
валент нейтрализации 179 ± 1. При его нагревании с водным раствором гидро-
ксида натрия и последующем подкислении реакционной смеси серной кислотой
выделены кристаллы(II), которые растворяются в щелочах и имеют эквивалент
нейтрализации 138 ± 1. Соединение II обесцвечивает бромную воду и дает окра-
ску с хлоридом железа (III). Перегонкой фильтрата после выделения соедине-
ния II получена кислота(III) с эквивалентом нейтрализации 60 ±1,
Установление структуры: методы определения и упражнения 563
В ПМР-спектре соединения II (в смеси ДМСО-de и CDC13) имеются сле-
дующие сигналы: б 6,7—7,0 м.д., 2Н, мультиплет; 6 7,35 м.д., 1Н, триплет дуб-
летов (7 = 9,3 Гц); б 7,75 м.д., 1Н, дублет дублетов (7 = 9 Гц, 3 Гц);
б 11,55 м. д., 2Н, синглет.
2. Соединение содержит азот, ие растворяетси в воде и разбавленных ще-
лочах, но растворяется в разбавленной соляной кислоте. При обработке бензол-
сульфохлоридом и щелочью получается прозрачный раствор. При подкислении
этого раствора выпадает осадок, который растворяется в избытке кислоты. Ис-
ходное вещество не реагирует с фенилгндразином, но разлагается при кипяче-
нии с раствором гидроксида натрия. Масло (II), выделенное из щелочного
раствора (III), все еще содержит азот и растворяется в разбавленной соляной
кислоте. Соединение II дает осадок с бромной водой и реагирует с ацетил-
хлоридом и натрием. При обработке вещества II бензолсульфохлоридом и ще-
лочью получается масло, растворимое в соляной кислоте. Раствор соединения II
в эфире насыщают хлористым водородом, и при этом выделяется кристалли-
ческое вещество IV, растворимое в воде и имеющее эквивалент нейтрализа-
ции 187 ± 1.
При подкислении полученного ранее щелочного раствора(III) выделяется
осадок (V), который растворяется в избытке кислоты. При прибавлении нитрита
натрия к охлажденному льдом кислому раствору соединения' V получается про-
зрачный раствор, при этом азот не выделяется. При прибавлении этого раствора
к раствору р-нафтолата натрия раствор окрашивается в красный цвет. Соеди-
нение V имеет эквивалент нейтрализации 137 ± 1.
Метилированием (дважды) соединения V с последующим восстановлением
в мягких условиях получено вещество II. В ПМР-спектре соединения V (в аце-
тоне) имеются следующие сигналы: б 6,55 м. д., ЗН, широкий синглет; б 6,76 м. д.,
2Н, дублет (7 = 8 Гц); б 7,83 м. д., 2Н, дублет (7 = 8 Гц).
3. Кристаллы (I) содержат азот, серу и бром, не растворяются в эфире, но
растворяются в воде с образованием кислого раствора с эквивалентом нейтра-
лизации 221 ± 1. При добавлении холодной щелочи выделяется масло (II), кото-
рое содержит бром и азот, но не содержит серы. Соединение II реагирует с
бензолсульфохлоридом и щелочью с образованием прозрачного раствора, из кото-
рого осаждается кислота (III), содержащая бром, азот и серу. Соединение II
не образует осадка с нитратом серебра, ио обесцвечивает как бромную воду,
так и разбавленный раствор перманганата. При обработке соединения I азоти-
стой кислотой на холоду получается раствор, при этом выделения газа не на-
блюдается. Раствор выливают в раствор цианида меди (1). При этом выделяется
соединение IV, которое все еще содержит бром и азот, но не растворяется в раз-
бавленных кислотах и щелочах. При кипячении соединения IV в течение неко-
торого времени с разбавленной серной кислотой образуется соединение V, кото-
рое уже не содержит азота, ио содержит бром. Это вещество не растворяется
в воде и разбавленной соляной кислоте, ио растворяется в растворе бикарбо-
ната натрия. Его эквивалент нейтрализации равен 200 ± 2. Соединение V не реа-
гирует с нитратом серебра и перманганатом.
В П.МР-спектре соединения II (в CDC1») имеются следующие сигналы:
б 3,53 м. д., 2Н, широкий синглет; б 6,57 м.д., 2Н, дублет (7 = 9 Гц); б 7,21 м. д.,
2Н, дублет (7 = 9 Гц).
4. Бледно-желтое кристаллическое соединение I содержит азот и бром, ие
растворяется в воде, разбавленных кислотах и щелочах, не реагирует с фенил-
гидразином, ацетилхлорндом и холодным разбавленным раствором перманганата.
Реакция не наблюдается также и с холодным спиртовым раствором нитрата
серебра, однако если раствор прокипятить в течение некоторого времени, то
образуется осадок бромида серебра. Соединение I нагревают с цинком и раство-
ром хлорида аммония, смесь фильтруют. Фильтрат восстанавливает реактив Тол-
ленса. При энергичном окислении соединения I образуется соединение II, кото-
рое все еще содержит бром и азот, не растворяется в соляной кислоте, но
растворяется в растворе бикарбоната натрия. Его эквивалент нейтрализации
равен 145 ± 1.
564
Г лава 9
Обработка соединения I оловом и соляной кислотой, а затем щелочью при-
водит к соединению III, которое содержит азот и бром и растворяется в раз
бавленной соляной кислоте. Соединение III дает осадок с бромной водой,
реагирует с ацетилхлорндом и при взаимодействии с бензолсульфохлоридом и
щелочью дает прозрачный раствор. При энергичном окислении соединения III
образуется белая кристаллическая кислота (IV), не содержащая брома и азота и
имеющая эквивалент нейтрализации 82 ± 1.
При нагревании соединение IV теряет молекулу воды. В ПМР-спектре со-
единения III (в CDC13) имеются следующие сигналы: б 3,96 м. д., 2Н, широкий
синглет; б 6,45 м.д., 1Н, дублет (1 = 8 Гц); б 7,25—7,75 м.д., 4Н, мультиплет;
б 8,0—8,2 м. д., IН, мультиплет.
5. Светло-желтые нейтральные кристаллы (I) содержат хлор, но не содер-
жат азота. Это вещество не реагирует с горячим спиртовым раствором нитрата
серебра, с ацетилхлорндом и с бромом в четыреххлористом углероде. При вза-
имодействии с фенилгидразином образуется осадок. Соединение I не изменяется
под действием холодных растворов щелочей. При нагревании в течение некото-
рого времени с концентрированным раствором гидроксида натрия получается
прозрачный раствор. Отгон из этого щелочного раствора не содержит никаких
органических соединений. При подкислении щелочного раствора фосфорной кис-
лотой выпадает осадок (II), который отфильтровывают. Из фильтрата при его
перегонке и выпаривании досуха не удается выделить никаких органических со-
единений.
Соединение II содержит хлор, имеет эквивалент нейтрализации 297 ± 2 и
реагирует с уксусным ангидридом с образованием вещества III, которое имеет
эквивалент нейтрализации 340 ± 3. Соединение III ие реагирует с бромной во-
дой, бромом в четыреххлористом углероде и фенилгидразином.
Прв энергичном окислении соединения I щелочным раствором перманганата
с очень хорошим выходом получен продукт IV, который содержит хлор и имеет
эквивалент нейтрализации 156 ±1. Никаких других продуктов окисления обна-
ружить ие удается.
При энергичном окислении соединения II или III бихроматом калия и сер-
ной кислотой также образуется продукт IV, но с очень плохим выходом.
В ПМР-спектре соединения IV (в CDC13, содержащем ДМСО-de) имеются
ЗН-сигнал, состоящий из дублета (/ = 9 Гц) при б 7,37 м. д. и широкого син-
глета при б 7,45 м. д., и 2Н-сигнал при б 7,81 м.д., дублет (/=9 Гц). Обра-
ботка раствора тяжелой водой (D2O) приводит к исчезновению сигнала с б
7,45 м. д. При этом в спектре остаются два дублета.
Серия 15
1. Кислота I имеет эквивалент нейтрализации 151 ± 2 и при обработке аце-
тилхлоридом на холоду превращается в кислоту II, имеющую эквивалент ней-
трализации 193 ± 2. При окислении в мягких условиях холодным раствором
перманганата калия соединение I превращается в кислоту III, имеющую экви-
валент нейтрализации 149 ± 2. Соединение III дает производное с фенилгидра-
зииом. Энергичное окисление соединения I, II или III приводит к кислоте IV
с эквивалентом нейтрализации 122 ±I.
В ПМР-спектре соединения I (в ацетоне) имеются следующие сигналы:
б 5,22 м. д., 1Н, синглет; б 6,93—7,88 м.д., 7Н, мультиплет. В ПК-спектре соеди-
нения I (в минеральном масле нуйоле) наряду с другими полосами поглощения
имеются сильная полоса поглощения при 1706 см~* (5,86 мкм) и широкая по-
лоса поглощения при 3333—2400 см-1 (3,00—4,17 мкм).
2. Соединение I имеет эквивалент нейтрализации 223 и при энергичном окис-
лении превращается в кислоту II с эквивалентом нейтрализации 167. При обра-
ботке соединения I цинком в соляной кислоте получена кислота III с эквива-
лентом нейтрализации 179. Установлено, что соединения I, II и III содержат
азот.
Установление структуры: методы определения и упражнения
565
В ПМР-спектре соединения II (в CDCl3/flMCO-d6) имеются следующие сиг-
налы: 6 7,72 м. д., 1Н, триплет (/ — 8 Гц); 6 8,2—8,55 м.д., 2Н, мультиплет;
6 8,71 м. д., 1Н, триплет (7 = 2 Гц); б 12,98 м.д., 1Н, синглет.
3. Соединение содержит серу, растворяется в сильных щелочах, ио ие рас-
творяется в растворах бикарбоната натрия. При энергичном окислении полу-
чена серусодержащая кислота с эквивалентом нейтрализации 102 ± 1. При обра-
ботке этого соединения перегретым паром образуется кислота, ие содержащая
серы.
В ПМР-спектре 100 мг кислоты, не содержащей серы (в 0,5 мл ацетоиа-de),
имеются следующие сигналы: б 7,00—7,75 м. д., 4Н, мультиплет: б 8,00 м. д., 2Н,
синглет. Химический сдвиг сигнала с б 8,00 м. д. изменяется в зависимости от
концентрации. В ПМР-спектре исходного соединения (в CDC13) имеются сле-
дующие сигналы: б 2,29 м.д., ЗН, синглет; б 3,37, 1Н, синглет; б 7,02 м.д., 4Н,
синглет (несколько уширенный у основания).
4. Соединение I содержит хлор и дает положительные реакции со спирто-
вым раствором нитрата серебра, раствором гипоиодита натрия и ацетилхлори-
дом. При гидролизе бикарбонатом натрия получено соединение II, не содержа-
щее хлора, которое дает положительную йодоформную реакцию и окисляется
иодной кислотой с образованием только одного соединения III. Последнее также
дает положительную йодоформную реакцию.
5. Соединение А содержит только углерод, водород и кислород, реагирует
с ацетилхлорндом, но не реагирует с фенилгидразином. При окислении иодиой
кислотой образуется соединение Б, которое восстанавливает реактив Толленса,
но ие восстанавливает раствора Фелиига. При обработке вещества Б цианидом
калия в водном этаноле получено соединение В, которое дает положительные
реакции с ацетилхлорндом и фенилгидразином. При окислении вещества В рас-
твором Фелинга или азотной кислотой образуется соединение Г желтого цвета,
которое дает производное с о-фенилендиамином. При обработке соединения Г
пероксидом водорода получена кислота Д, имеющая эквивалент нейтрализации
135 ± 1. При каталитическом восстановлении соединения В или Г образуется
исходное соединение А.
В ПМР-спектре соединения Б (в CDCI3) имеются следующие сигналы: б
2,42 м.д., ЗН, сииглет; б 7,18 м.д., 2Н, дублет (7 = 10 Гц); б 7,56 м.д., 2Н, дуб-
лет (7 = 10 Гц); б 9,81 м. д., 1Н, синглет.
6. Кристаллическое нейтральное соединение А содержит азот и остается не-
изменным при гидролизе разбавленными кислотами и основаниями. Это веще-
ство дает отрицательные реакции с ацетилхлорндом, бромом в четыреххлори-
стом углероде, раствором гипоиодита натрия и реактивом Толленса, медленно
реагирует с динитрофенилгидразином и восстанавливает реактив Толленса после
обработки цинком и хлоридом аммония.
При обработке солянокислым гидроксиламииом в пиридине соединение А
медленно превращается в соединение Б. В результате обработки вещества Б пен-
тахлоридом фосфора образуется соединение В. При кипячении последнего с кис-
лотой получена кислота Г, содержащая азот и имеющая эквивалент нейтрализа-
ции 168 ± 2. Одновременно образуется соль основания Д. Хлоргидрат вещества
Д имеет эквивалент нейтрализации (при титровании щелочью) 195 ± 2.
Кислоту Г обрабатывают тионилхлоридом и полученный продукт прибав-
ляют к водному раствору аммиака. Образовавшееся при этом нейтральное веще-
ство обрабатывают бромом и раствором гидроксида натрия. В результате выде-
ляется растворимый в воде продукт Е, который после обработки соляной кис-
лотой и нитритом натрия дает окраску с 0-иафтолом. При обработке соединения
Е оловом и соляной кислотой образуется вещество Ж, которое легко окисляется
при действии окислителей. Соединение Ж не дает кристаллического производ-
ного при обработке дибензилом.
Основание Д, полученное в реакции с пеитахлоридом фосфора, обрабаты-
вают нитритом натрия и разбавленной серной кислотой. Полученный раствор дает
окраску с 0-нафтолом. При кипячении водного раствора образуется не содержа-
щее азота соединение 3, которое растворяется в водном растворе щелочи, но не
растворяется в водном растворе бикарбоната натрия. При окислении как Е, так
566 Глава 9
и 3 образуется кислота (эквивалент нейтрализации 83), которая легко превра-
щается в ангидрид. Вещество 3 не вступает в реакцию сочетания с растворами
фенилдиазониевых солей.
В ПМР-спектре соединения Е (в CDCk, содержащем ДМСО-de) имеются
следующие сигналы: б 5,68 м. д., 2Н, широкий синглет; б 6,58 м. д., 2Н, дублет
(/ = Ю Гц); б 7,88 м.д., 2Н, дублет (7 = 10 Гц). В ПМР-спектре соединения 3
(в CDCh) имеются следующие сигналы: б 2,32 м.д., ЗН, синглет; б 5,12 м.д., 1Н,
широкий синглет; б 7,1—7,55 м.д., 4Н, мультиплет; б 7,6—7,9 м.д., 1Н, мульти-
плет; б 8,0—8,2 м. д., 1Н, мультиплет. Сигнал с б 5,12 м.д. сдвигается в слабое
поле при добавлении соединения 3 в ампулу для снятия ПМР-спектра.
Глава 10
Литература по органической химии
10.1. ВВЕДЕНИЕ И ПОСТРОЕНИЕ ГЛАВЫ
Очень существенным для химика-органика является хорошее знание огром-
ного количества литературы и способность быстро найти тот или иной литера-
турный источник, который будет полезен ему в работе. Данная глава должна
оказать помощь при выполнении этой задачи.
Приведенная здесь литература охватывает разделы органической химии, ко-
торые чаще всего находятся в центре внимания химиков-органиков. При работе
с органическими веществами химики неизбежно сталкиваются с проблемами и
понятиями, традиционно относящимися к другим областям химии, таким, как не-
органическая и аналитическая химия. Поэтому включены разделы, посвященные
этим областям химии, а также, например, руководства по биохимии и физиче-
ской химии.
Как и следовало ожидать, литературе по органической химии уделено иаи- -
большее внимание. При этом выделены такие разделы, как анализ органических
соединений, синтезы, механизмы химических реакций и т. д.
Включены также перечни книг более общего характера. Например, здесь
рассмотрены руководства и другие литературные источники, которые необходимы
для всех категорий химиков. Некоторые аспекты работы с химической литера-
турой, такие, как журналы, машинный поиск и т. д., здесь не рассматриваются.
Их можно найти в книгах, перечисленных в разд. 10.8, как, например, руковод-
ство Вудберна. Chemical Abstracts уже рассмотрен в разд. 2.9.
Следует помнить, что расположение книг по специфическим вопросам в не-
которой степени произвольно. Например, отдельные книги, перечисленные в раз-
деле «Физическая органическая химия», могли быть перенесены в раздел «Ме-
ханизмы химических реакций».
В некоторых случаях названия книг снабжены комментариями, которые по-
зволят читателю получить более конкретное представление об их содержании.
Мы надеемся, что эти примечания дадут читателю больше информации, чем
только одно название.
Подход, который следует использовать начинающим для получения инфор-
мации, содержащейся в этой главе, должен включать, во-первых, попытку опре-
делить раздел или разделы, в которых может быть указан конкретный вид ли-
тературы. Затем следует рассмотреть перечень литературы в этих разделах.
И наконец, можно проконсультироваться с преподавателем или студентом, окон-
чившим университет и имеющим опыт работы в этой области. Такая консульта-
ция может дать ценные дополнительные представления о действительной полез-
ности сделанного выбора.
В ряде других изданий также содержатся перечни книг, которые могут быть
очень полезны химикам-органикам. К ним относятся следующие издания:
Моррисон Р., Бойд Р. Органическая химия. Пер. с англ. — М.: Мир, 1974.
March J., Advanced Organic Chemistry, 2nd ed., McGraw-Hill, New York,
1977, p. 1143.
Автор снова дал ценную «энциклопедическую» информацию для химиков-ор-
гаников.
le Noble W. N., Highlights of Organic Chemistry, v. 3 in The series «Studies
in Organic Chemistry, M. Dekker, New York, 1974. Это очень обширная книга по
органической химии. В примечаниях дано большое число ссылок на различные
литературные источники по органической химии.
568
Глава Ю
Journal of Chemical Education — журнал, хорошо известный своими публика-
циями статей педагогического характера; обычно дает в сентябрьских выпусках
список книг для покупателей. В этом списке перечисляются имеющиеся в про-
даже книги по различным областям химии. В сентябрьском выпуске 1976 г. пере-
числено более 1000 книг по 61 разделу химии.
10.2. ОРГАНИЧЕСКАЯ ХИМИЯ
10.2.1. Beilstein
Справочник по органической химии Beilstein’s Handbuch der Organischen Che-
mie (см. обсуждение в разд. 2.9) был начат с целью описать получение и свой-
ства всех известных органических соединений. Хотя издание продолжается, ка-
жется маловероятным, что удастся когда-либо осуществить первоначальную цель.
Несмотря на то что справочник издается на немецком языке, им могут без за-
труднений пользоваться химики, не изучавшие этот язык. Читателю следует об-
ратиться также к книге: Huntress Е. Н., A Brief Introduction to the use of Beil-
stein’s Handbuch der Organischen Chemie, и к приведенной в разд. 10.1 книге
Марча (с. 1152).
10.2.2. Руководства по технике лабораторных работ. Вводный
курс
Roberts R. М., Gilbert J. С., Rodewald L. В., Wingrove A. S., Modern Expe-
rimental Organic Chemistry, 2nd ed., Holt, Rinehart and Winston, New York, 1974.
Landgrebe J., Theory and Practice in Organic Chemistry, 2nd ed., D.C. Heath,
Lexington, Mass, 1977. Очень обширное и подробное лабораторное руководство
с большим количеством рисунков лабораторных операций, выполняемых хими-
ками-органиками.
Swinehart J. S., Organic Chemistry: An Experimental Approach, Appleton-Cen-
tury-Crafts, New York, 1969.
Fieser L. F., Williamson K. L., Organic Experiments, 4th ed., D.C. Heath, Le-
xington, Mass., 1979.
10.2.3. Руководства по технике лабораторных работ. Повышенный
курс
Wiberg К.. В., Laboratory Technique in Organic Chemistry, McGraw-Hill, New
York, 1960.
Исаакс H. Практикум по физической органической химии. Пер. с англ. — М.:
Мир, 1972.
Bates R. В., Schaefer J. Р., Research Techniques in Organic Chemistry, Pren-
tice-Hall, Englewood Cliffs, N. J., 1971.
Newman M. S., An Advanced Organic Chemistry Laboratory Course, MacMil-
lan, New York, 1972.
10.2.4. Механизмы химических реакций*
Ингольд К. К. Теоретические основы органической химии. Пер. с англ.—М.:
Мир, 1973. Большая по объему и пользующаяся заслуженным признанием моно-
* См. также Матье Ж., Паника Р. Курс теоретических основ органической
химии. Пер. с франц. — М.: Мир, 1975; Гото Т., Хирата И., Стоут Г. Современ-
ная органическая химия в вопросах н ответах. Пер. с англ.—М.: Мир, 1971.—
Прим, per),
Литература по органической химии
569
графия. Эта книга, а также книги Марча и Ле-Ноубла (см. выше) являются наи-
лучшими «энциклопедиями» для тех, кто интересуется исследованиями механиз-
мов органических реакций.
Gould Е. S., Mechanism and Structure in Organic Chemistry, Holt, New York,
1958. Меиьшая по объему, чем книга Ингольда; хороший учебник по теории ме-
ханизмов химических реакций, несмотря на то что он издан довольно давно.
Ferguson L. N., The Modern Structural Theory of Organic Chemistry, Prenti-
ce-Hail, Englewood Cliffs, N. J., 1963.
Wheland G. W., Advanced Organic Chemistry, 3rd ed., Wiley, New York, 1960.
Lowry T. H., Richardson K. S., Mechanism and Theory in Organic Chemistry
Harper & Row, New York, 1976. Новое и весьма детальное (748 с.) обсуждение
механизмов реакций и теории органической химии. Рассмотрены механизмы всех
реакций (присоединение, элиминирование и т. д.), а также теория молекулярных
орбиталей и правила орбитальной симметрии.
Alder R. W., Baker R., Brown J. M., Mechanism in Organic Chemistry, Wiley-
Interscience, New York, 1971.
Frost A. A., Pearson R. G., Kinetics and Mechanism, 2nd ed., Wiley, New York,
1961.
Бреслоу P. Механизмы органических реакций. Пер. с аигл. — М.: Мир, 1968.
Краткое введение в рассмотрение механизмов реакций.
Следует также просмотреть список книг в разделе «Физическая органиче-
ская химия» (разд. 10.2.7).
10.2.5. Теория молекулярных орбиталей*
Liberies A., Introduction to Molecular Orbital Theory, Holt, Rinehart and Win-
ston, New York, 1966. Очень простая книга для химиков со слабой подготовкой
или совсем' ие подготовленных в данной области. Математическая часть вклю-
чает только алгебраические операции и очень простые вычисления.
Orchin М., Jaffi Н. Н., Symmetry, Orbitals and Spectra, Wiley-lntersience,
New York, 1971.
Hanna M., Quantum Mechanics in Chemistry, Benjamin, New York, 1965. Бо-
лее строгое и полное изложение по сравнению с книгой Либэрлса, указанной
выше л этом разделе. Книга написана ясным языком. Ее математическая часть
включает только алгебраические операции и простые вычисления.
Дьюар М. Теория молекулярных орбиталей в органической химии. Пер. с
аигл. — М.: Мир, 1972. От читателя требуется достаточно существенная подго-
товка в теории молекулярных орбиталей. Однако имеются главы, включающие
применение данной теории к органическим системам и реакциям, которые легко
понятны для всех химиков-органиков.
Стрейтвизер А. Теория молекулярных орбит для химиков-органиков. Пер. с
аигл. — М.: Мир, 1965. По своему уровню эта книга находится между уже ука-
занными книгами Дьюара и Либэрлса. Разработаны методы, используемые в тео-
рии молекулярных орбиталей Хюккеля, рассмотрено их применение к органиче-
ским системам. Включены также ясные и обширные комментарии к применению
этой теории при исследовании органических реакций и корреляций в органиче-
ской химии. К сожалению, эта книга вышла до развития представлений об ор-
битальной симметрии (правил Вудворда — Хофмана).
Робертс Дж. Расчеты по методу молекулярных орбит. Пер. с аигл. — М.:
Мир, 1963. Прекрасное краткое введение в предмет, указанный в названии. Не
содержит сведений по орбитальной симметрии.
Zimmerman И., Quantum Mechanics for Organic Chemists, Academic Press,
New York, 1975. Учебник для студентов, прослушавших годичные курсы органи-
* См. также: Дьюар М., Догерти Р. Теория возмущений молекулярных ор-
биталей в органической химии. Пер. с англ. — М.: Мир, 1977; Потапов В. М. Сте-
реохимия.— М.: Химия, 1976. — Прим. ред.
570
Глава 10
ческой химии и теории молекулярных орбиталей. Освещены вопросы орбиталь-
ной симметрии.
Реакционная способность и пути реакции. Пер. с англ./Под ред. Г. Клоп-
мана. — М.: Мир, 1977.
Jorgensen W. L., The Organic Chemists Book of Orbitals, Academic Press, New
York, 1973.
10.2.6. Правила орбитальной симметрии
Вудворд Р., Хоффман Р. Сохранение орбитальной симметрии. Пер. с англ. —
М.: Мир, 1971.
Джилкрист Т., Сторр Р. Органические реакции и орбитальная симметрия.
Пер. с англ. — М.: Мир, 1976.
(См. также книги Дьюара и Циммермана, приведенные в разд. 10.2.5.)
10.2.7. Физическая органическая химия
Гаммет Л. Основы физической органической химии. Пер. с англ. — М.: Мир,
1972. Рассмотрены скорости, равновесия и механизм химических реакций. Автора
называют «отцом физической органической химии». Книга охватывает все пред-
меты, перечисленные в заголовке, но не затрагивает теорию молекулярных орби-
талей.
Hine J., Physical Organic Chemistry, 2nd ed., McGraw-Hill, New York, 1961.
Рассмотрены механизмы реакций и другие вопросы, близкие к этой теме, ио в
ней лишь в малой степени затронута теория молекулярных орбиталей.
Wiberg К., Physical Organic Chemistry, Wiley, New York, 1964.
Ferguson L. N., Organic Molecular Structure, Willard Grant Press, Boston,
1977.
Harris J. M., Wamser С. C., Fundamentals of Organic Reaction Mechanisms,
Wiley, New York, 1976.
Hirsch J., Concepts of Theoretical Organic Chemistry, Allyn and Bacon, Bos-
ton, 1974.
Liberies A., Introduction to Theoretical Organic Chemistry, Macmillan, New
York, 1968. Введение в физическую органическую химию, написанное в очень
простой форме.
Ritchie С. D., Physical Organic Chemistry: Fundamental Concepts, M. Dekker,
New York, 1975.
Leffler J. E., Grunwald E., Rates and Equilibria of Organic Reactions, Wiley,
New York, 1963. Рассмотрено применение кинетики и термодинамики к исследо-
ванию механизмов органических реакций; особенно ценна для изучения экстратер-
модииамических понятий и представления о «линейных» соотношениях свобод-
ных энергий (например, а — р-уравнение Гаммета).
Темы разделов «Физическая органическая химия» (разд. 10.2.7) и «Меха-
низмы химических реакций» (разд. 10.2.4) перекрываются между собой.. Поэтому
следует просматривать списки книг по обеим темам.
10.2.8. Качественный анализ органических соединений
Cheronis N. D., Entrikin J. В., Hodnett Е. М., Semimicro Qualitative Organic
Analysis, 3rd ed., Wiley, New York, 1965. Приведен еще один прекрасный допол-
нительный список реакций в растворах (см. ниже также книгу Пасто и Джон-
сона). Полезны также более ранние книги Черониса и Энтрикина *.
Paste D. J., Johnson С. R., Organic Structure Determination, Prentice-Hall,
Englewood Cliffs, N. J., 1969. Книга была переработана в 1979 г.
* Черонис Н. Микро- и полумикрометоды органической химии. Пер. с
англ. — М.: ИЛ, 1960.
Литература по органической химии
571
Feigl F., Spot Tests in Organic Analysis, 7th ed., Elsevier, Amsterdam — New
York, 1966. Обширный перечень реакций в растворах для анализа органических
соединений с подробным описанием методики их проведения.
Schneider F. L., Qualitative Organic Microanalysis, Academic Press, New York.
1964.
10.2.9. Реакции и реактивы*
Organic Reactions, Wiley, New York**. Издание серии книг, начатое в
1942 г. и продолжающееся до настоящего времени. Каждой реакции (например,
восстановление по Клемменсену) посвящена отдельная глава, при этом особое
внимание уделяется ее применению в синтезе. Приведены обширные списки лите-
ратурных источников.
Wagner R. В., Zook Н. D., Synthetic Organic Chemistry, Wiley, New York,
1953. В 39 главах рассмотрены 576 типов реакций, методы их проведения, вы-
ходы, ссылки на литературу, физические константы веществ (для соединений,
имеющих не более двух функциональных групп).
Физер Л., Физер М. Реагенты для органического синтеза. Пер. с англ. — М.:
Мир, т. 1—3, 1970; т. 4 и 5, 1971; т. 6, 1975. В т. 1—5 перечислены в алфавит-
ном порядке реагенты, катализаторы, растворители и т. д., используемые для
органических лабораторных методик. Аналогично построен т. 6, включающий
новые соединения и новые данные для соединений, перечисленных в т. 1—5. Из-
дание снабжено обширной практической информацией, такой, как ссылки иа ли-
тературу, источники коммерческого снабжения, методы высушивания и физиче-
ские константы.
Литература данного раздела и разд. 10.2.12 «Синтезы» перекрывается, по-
этому при тщательном литературном поиске следует просмотреть оба раздела.
10.2.10. Спектроскопия (спектрометрия)
Сильверстейн Р., Басслер Г., Моррилл Т. Спектометрическая идентификация
органических соединений. Пер. с англ. — М.: Мир, 1977. Учебник, которым могут
пользоваться студенты с очень слабой подготовкой или совсем не подготовлен-
ные в области ПК-, ЯМР-, УФ- и масс-спектрометрии. Подчеркивается комплекс-
ное использование спектрометрической информации для определения структуры
органического соединения.
Dyer 1. R., Applications of Absorption Spectroscopy of Organic Compounds,
Prentice-Hall, Englewood Cliffs, N. J., 1965. В небольшой по объему книге рас-
сматриваются те же вопросы, что и в предыдущем учебнике.
Colthup N. В., Daly L. Н., Wiberly S. Е., Introduction to Infrared and Raman
Spectroscopy, 2nd ed., Academic Press, New York, 1975.
Conley R. T., Infrared Spectroscopy, 2nd ed., Allyn and Bacon, Boston, 1972.
Подробное описание лабораторной техники ИК-спектроскопии. Приведено много
иллюстраций и большое число таблиц полос поглощений в ИК-спектрах, наличие
которых связано с присутствием различных функциональных групп.
Bellamy L. Л, The Infrared Spectra of Complex Molecules, Vol. 1, 3rd ed., Hal-
sted Press-Wiley, New York, 1975. Эта и все ранее изданные книги Беллами по-
лезны для анализа ИК-спектров органических соединений
* Матье Ж., Панико Р., Вейль-Рейналь Ж. Измеиеиие и введение функ-
ций в органическом синтезе. Пер. с франц.—М.: Мир, 1980.—Прим. ред.
** Имеются переводы на русский язык: Органические реакции. Пер. с
англ./Под ред. Р. Адамса — М.: Издатинлит, сб. 1, 1948; М.: ИЛ, сб. 2, 1950;
66. 3—5, 1950; сб. 6, 1953; сб. 7 и 8, 1956; сб 9, 1959; М.: Мир, сб. 10, 1963; Пер.
с англ./Под ред. А. Коупа—М.: Мир, сб. 11 и 12, 1965; сб. 13, 1966; сб 14
1967.
572
Глава 10
Наканиси К. Инфракрасные спектры и строение органических соединений.
Пер. с анг*.— М.: Мир, 1965.
J ackman- L. М., Sternhell S., Applications of N. M. R. Spectroscopy in Organic
Chemistry, 2nd ed., Pergamon Press, Elmsford, N. Y., 1969. Новое издание суще-
ственно расширено; содержит, по существу, то же введение к основным прави-
лам, как и первое издание, но к этому добавлено обширное количество данных
по спектрам ПМР. Книга не затрагивает данные по теории и спектрам ЯМР-13С.
Будзикевич Г., Джерасси К., Уильямс Д. Интерпретация масс-спектров орга-
нических соединений. Пер. с англ. — М.: Мир, 1966.
Biemann К., Mass Spectra and Organic Chemical Applications, McGraw-Hill,
New York, 1962.
Introduction to Spectroscopic Methods for the fdentification of Organic Com-
pounds, Vols. 1 and 2, edited by Scheinmann F., Pergamon Press, Elmsford, N. Y.
1970.
Stern E. S., Timmons T. C. J., Electronic Absorption Spectroscopy in Organic
Chemistry, St. Martin’s Press, New York, 1971. Полезно также третье издание
ранее опубликованной книги Джиллама и Стериа по УФ-спектроскопии.
Леви Г., Нельсон Г. Руководство по ядерному магнитному резонансу угле-
рода-13 для химиков-оргаииков. Пер. с англ. — М.: Мир, 1975. Краткое введе-
ние в область, а также источник данных по спектрам ЯМР-13С.
Slathers J. В., Carbon 13 N. М. R. Spectroscopy, Academic Press, New York,
1972. Обширное, выполненное на высоком уровне рассмотрение теории. Исклю-
чительно обширный и ценный справочный источник спектральных данных (осо-
бенно химических сдвигов).
Wehr li F. W., Wirthlin Т., Interpretation of Carbon-13 N. M. R. Spectra, Hey-
den, New York, 1976. Полное введение в теорию и технику определения спект-
ров ЯМР-13С. Широкое использование интерпретации спектров ЯМР для опре-
деления структуры дополнено физической химией рассматриваемого спектраль-
ного явления.
Детр IV., Organic Spectroscopy, Halsted-Wiley, New York, 1974.
Sadtler Standard Spectra, Sadtler Research Laboratories, 3316'Spring Garden
St, Philadelphia, Pa. 19104. Описано большое число призм и решеток для ИК-
спектроскопии, дан перечень спектров ПМР (60 и 100 МГц) и ЯМР-13С.
Atlas of Spectral Data and Physical Constants for Organic Compounds, 2nd ed.,
Vols. 1—6, CRC, Press Inc., Boca Raton, Fla., 1973. Перечислены спектральные
данные, температуры кипения и плавления, сведения о растворимости и плот-
ности для 21 000 органических соединений. Приведиы только таблицы данных,
однако имеются ссылки на сборник Садтлера и другие источники.
Aldrich Library of Infared Spectra, 2nd ed., Aldrich Chemical Co., Milwau-
kee, Wise., 1975. Приведены ИК-спектры 10 000 соединений, объединенных по
химическим классам. Ввиду того что на одной странице даио несколько спект-
ров, легко изучать изменения в спектрах, связанные со структурными измене-
ниями внутри одного химического класса.
Johnson L. F., Jankowski W. С., Carbon-13 N. M. R. Spectra, Wiley, New York,
1972. Книга представляет собой сборник спектров ЯМР-13С 500 часто встречаю-
щихся органических соединений.
10.2.11. Стереохимия
Илиел Э. Стереохимия соединений углерода. Пер. с англ. — М.: Мир, 1965.
Полный свод основных правил и обсуждение их применения, включающее кон-
формационный анализ и механизмы органических реакций.
Mislow К., Introduction to Stereochemistry, Benjamin,’ New York, 1965. Изло-
жение предмета, указанного в названии, иа весьма высоком уровне, включая
группы симметрии и механизмы химических реакций.
Илиел Э. Основы стереохимии. Пер. с аигл. — М.: Мир, 1971. Книга может
служить дополнением к учебникам для студентов младших курсов.
Литература по органической химии
573
10.2.12. Синтезы
Organic Syntheses *, Wiley, New York. Сборники проверенных методик син-
теза органических соединений. Этим они отличаются от непроверенных методик
в оригинальной литературе, которые слишком часто нельзя воспроизвести.
Buehler С. A., Pearson D. Е., Survey of Organic Syntheses, 2 Vols., Wiley,
New York, 1970, 1977.
Sandler S. R., Karo W., Organic Functional Group Preparations, Vols. 1—3,
Academic Press, New York, 1968—1977.
Harrison I. T., Harrison S., Compendium of Synthetic Methods, Wiley, New
York, Vol. 1, 1971; Vol. 2, 1974; Vol. 3, 1977, Ed. by L. Hegedus and L. Wade.
Patai S. (Ed.), Chemistry of the Functional Group, Wiley, New York. Серия
книг, в каждой из которых описана химия определенной функциональной группы
(гидроксильной группы, простой эфирной связи и т. д.).
Е. Н. Rodd’s Chemistry of Carbon Compounds, edited by Coffey, Elsevier, Am-
sterdam, 1964 to date.
Houben-Weyl, Methoden der organischen Chemie, Georg Thieme, Verlag, Stutt-
gart, 1952 to date.
House H. 0., Modern Synthetic Reactions, 2nd ed., Benjamin, New York, 1972.
Дополнительные книги no синтезу приведены в разделе «Реакция и реак-
тивы» (разд. 10.2.9).
10.3. АНАЛИТИЧЕСКАЯ ХИМИЯ**
Snyder L. R., Kirkland J. An Introduction to Modern Liquid Chromato-
graphy Wiley-Interscience, New York, 1974.
Mann C., Vickers T., Gulick W., Instrumental Analysis, Harper & Row, New
York, 1977.
Willard H. H., Merritt L- L., Jr., Dean J. A., Instrumental Methods of Analy-
sis, 5th ed., Van Nostrand, New York, 1974.
Karger B. L., Snyder L. R., Horvath C., An Introduction to Separation Science,
Wiley, New York, 1973.
Хроматография в тонких слоях. Пер. с аигл./Под ред. Э. Шталя. — М.: Мир,
1965. Полная и хорошо иллюстрированная книга по этому предмету.
Crippen R. С., Gas Chromatography, McGraw-Hill, New York, 1973. Изложе-
ние предмета аналогично построению настоящей книги, т. е. цель книги Крип-
пена — проведение качественного анализа органических соединений.
Skoog D. A., West D. М., Principles of Instrumental Analysis, Holt, Rinehart
and Winston, New York, 1971.
Скуг Д., Вест Д. Основы аналитической химии. В 2-х томах. Пер. с англ. —
М.: Мир, 1979.
Hejtmann Е., Chromatography: A Laboratory Handbook of Chromatographic
and Electrophoretic Methods, Van Nostrand-Reinhold, New York, 1975.
♦ Имеются переводы на русский язык: Синтезы органических препаратов.
Пер. с англ. — М.: Издатиилит, т. 1 и 2, 1949; М.: ИЛ, т. 3, 1952; т. 4, 1953;
Т.Б, 1954; т. 6 и 7, 1956; т. 8, 1958; т. 9, 1959; т. 10 и 11, 1961; М.: Мир, т. 12
1964.
** См. также Митчелл Дж., Смит Д. Акваметрия. Пер. с англ. — М.: Химия,
1980; Газовая хроматография. Библиографический указатель отечественной и за-
рубежной литературы. В 5-и томах. — М.: Наука, 1961—1982; Жидкостная коло-
ночная хроматография. В 3-х частях. Пер. с англ./Под ред. 3. Дейла, К. Мацека,
Я. Янека. — М.: Мир, 1978; Высокоэффективная тонкослойная хроматография.
Пер. с англ./Под ред. А. Златкиса, Р. Кайзера.—М.: Мир, 1980; Руденко Б. А.
Капиллярная хроматография. — М.: Наука, 1978; Кирхиер., Тонкослойная хро-
матография. В 2-х томах. Пер. с англ. — М.: Мир, 1980. — Прим. ред.
574
Глава 10
Hadden N. et al., Basic Liquid Chromatography, Varian Aerograph, Walnut
Creek, Calif., 1971.
Мак-Нейр Г., Бонелли Э. Введение в газовую хроматографию. Пер. с англ. —
М.: Мир, 1970.
Meites L., Handbook of Analytical Chemistry, McGraw-Hill, New York, 1963.
Peters D. G., Hayes J. M., Heiftje G. M., Chemical Separations and Measure-
ments, Saunders, Philadelphia, 1974. Обширный обзор реакций в растворах и ин-
струментальных аналитических методов.
10.4. БИОХИМИЯ
Ленинджер А. Биохимия. Молекулярные основы структуры и 'функций клет-
ки. Пер. с англ. — М.: Мир, 1976. Фундаментальная книга, содержащая 1104 стр.
Включено, большое число задач, которые могут быть даны студентам.
Colowick S. Р., Kaplan N. D., Methods of Enzymology, Academic Press, New
York. Продолжающаяся серия, состоящая из многих томов.
Таблица 10.1
Класс соединения Метод анализа Литера- тура а
Аминокислоты Аминокислотный анализ (автоматизированная колоночная хроматография) 1
ДНК Колориметрический анализ (реакция с дифенил- амином) 2
Ферменты Центрифугирование в градиенте плотности (се- диментационное поведение) 3
Липиды Метод выделения и очистки липидов из тканей животных 4
Пептидные цепи Гель-электрофорез в присутствии додецилсуль- фата. Определение числа цепей и молекуляр- ной массы 5
Белки DISC-электрофорез в акриламидных гелях 6
Иодирование *251 (поверхности мембран) 7
Сахара Фенольный метод (модификация метода Фоли- на — Чиокалтеу) 8
Колориметрический анализ (сахара и родствен- ные соединения) 9
Меркаптаны Хроматография на бумаге 10
Тиольные группы тканей 11
(тиолы)
а 1. Spackman D. Н„ Stein W. Н„ Moore S., Anal. Chem., 30, 1190 (1958).
2. Burton К-, Biochem. J., 62, 315 (1956).
3. Martin R. 0., Ames B., J. Biol. Chem.. 236, 1372 (1961).
4. Folch I., Lees M.. Stanley G. H. S., J. Biol. Chem.. 226, 497 (1967).
5. Weber K-, Osborn M. J., J. Biol. Chem., 244, 4406 (1968).
6. Davis В. I., Ann. N. У. Acad. Sci., 121, 404 (1964).
7. Marchalonis 1. J., Biochem. J., 113, 299 (1969); Humes R. O. Proc. Nat. Acad. Sci.
(U. S. A), 70. 3170 (1973).
8. Lowry О. H., Rosebrough H. J., Farr A. L., Randall R. I., J. Bioi. Chem., 193, 265
(1951).
9. Dubois M., Gilles K. A., Hamilton W. K.., Rebers P. A.. Smith F„ Anai. Chem., 28,
350 (1956).
10; Trevelyan W. E„ Proctor D. P„ Harrison J. S„ Nature, 166, 444 (1950).
11. Ellman G. L., Arch. Biochem. Blophys.. 82, 70 (1959).
Литература по органической химии 575
Metzler D. Е., Biochemistry, Academic Press, New York, 1977. Книга содер-
жит указатель литературных ссылок, который опущен в большинстве книг, опи-
сывающих обзорные курсы.
Handbook of Biochemistry, 3rd ed. by G. D. Fassman, CRC Press, Cleveland,
Ohio, 1976.
Mahler H. R., Cordes E. H., Biological Chemistry, 2nd ed., Harper & Row, New
York, 1971.
Dawson R. M. C„ Elliott D. C., Elliot W. H., Jones К- M., Data for Biochemi-
cal Research 2nd ed., Oxford University Press, New York, 1969. Обширное опи-
сание используемых биохимиками методов анализа в растворах и инструмен-
тальных методов, снабженное многочисленными литературными ссылками на до-
полнительные методы.
В табл. 10.1 перечислены функциональные группы, важные для биологиче-
ских систем, и даны ссылки на работы, в которых описаны аналитические методы
для этих систем.
10.5. ОБЩАЯ ХИМИЯ
The Merck Index of Chemicals and Drugs, Merck and Co., Rahway, N. J. Де-
вятое издание вышло в 1976 г. Обширный перечень химических соединений, имею-
щих биологическое значение; имеются указатели химических, общих и торговых
названий. Для каждого соединения приведены формула, физические, медицин-
ские и токсикологические свойства. Имеется также перечень названий органиче-
ских реакций.
Handbook of Tables for Organic Compounds, CRC Press, Inc., 2000 N. W. 24th
St., Boca Raton, Fla., 3rd ed., 1967. Приведены обширные данные о температурах
кипения и плавления соединений и о температурах плавления их производных.
Handbook of Chemistry and Physics, CRC Press, Inc., 200 N. W. 24th St., Bo-
ca Raton, Fla., 55-е издание вышло в 1974—1975 гг.
Dictionary of Organic Compounds, 5 vols., Oxford University Press, New
York, 1965. Названия, свойства и ссылки на литературные источники для
40 000 органических соединений.
Драго Р. Физические методы в химии. Пер. с англ.—-М.: Мир, 1981. Очень
удобная книга, содержащая различные данные, таблицы, теорию и т. д. Опи-
саны различные спектральные и другие физико-аналитические методы.
Summers D. В., The Chemistry Handbook: Facts, Figures and Formulas, Wil-
lard Grant Press, Boston, 1970.
10.6. НЕОРГАНИЧЕСКАЯ ХИМИЯ
Коттон Ф., Уилкинсон Дж. Современная неорганическая химия. В 3-х частях.
Пер. с англ.—М.: Мир, 1969; Коттон Ф., Уилкинсон Дж. Основы неорганиче-
ской химии. Пер. с англ. — М.: Мир, 1978. Почти энциклопедический охват экс-
периментальных данных и теории неорганической химии.
Purcell К. F., Kotz I. С., Inorganic Chemistry, Saunders, Philadelphia, 1977.
Huheey J. E., Inorganic Chemistry, Principles of Structure and Reactivity, Har-
per & Row, New York, 1972.
Cotton F. A., Chemical Applications of Group Theory, 2nd ed., Wiley-Inter-
science, New York, 1971. Прекрасное введение в предмет и в простую теорию
молекулярных орбиталей Хюккеля.
Файгль Ф., Ангер В., Капельный анализ неорганических соединений. В 2-х
томах. Пер. с англ. — М.: Мир, 1976.
Pauling L., The Nature of the Chemical Bond, 3rd ed., Cornell University
Press, Ithaca, N. Y., 1960.
Shoemaker D. P., Garland C. U7., Steinfeld J. I., Experiments in Physical Che
mistry, 3rd ed., McGraw-Hill, New York, 1974.
576
Глава 10
10.7. ФИЗИЧЕСКАЯ ХИМИЯ
Barrow G. М., Physical Chemistry, 3rd ed., McGraw-Hill, New York, 1973.
Wilson E. B., Decius J. C., Cross P. C., Molecular Vibrations: The Theory of
Infrared and Raman Vibrational Spectra, McGraw-Hill, New York, 1955.
Castellan G. IV., Physical Chemistry, 2nd ed., Addison-Wesley, Reading, Mass.,
1971.
Даниэльс Ф., Ольберти P. Физическая химия. Пер. с англ. — М.: Мир, 1978.
10.8. ХИМИЧЕСКАЯ ЛИТЕРАТУРА
Woodburn Н. М., Using the Chemical Literature: A Practical Guide, Marcel
Dekker, N. Y., 1974.
Fieser L. F., Fieser M., Style Guide for Chemists, Reinhold, N. Y., 1960.
Приложение I
Таблицы, удобные для использования
в органической лаборатории
Состав и свойства обычных кислот и оснований
Кислота или основание Удельная масса Содержание, масс, % Концентрация
моль/л г/100 мл
Соляная кислота
концентрированная 1,19 37 12,0 44,0
с постоянной температурой ки- пения (252 мл конц. кислоты + 4-200 мл воды, т. кип. НО °C) 1,10 22,2 6,1 24,4
10%-иая (100 мл конц. кисло- ты 4-321 мл воды) 1,05 10 2,9 10,5
5%-иая (50 мл конц. кислоты 4- 4- 380,5 мл воды) 1,03 5 1,4 5,2
1 и. (41,5 мл конц. кислоты, разбавленной до 500 мл) 1,02 3,6 1 3,7
Бромистоводородная кислота с по- стоянной температурой кипения (126 °C) 1,49 47,5 8,8 70,8
Иодистоводородная кислота с посто- янной температурой кипения (127 °C) Серная кислота 1,7 57 7,6 97
концентрированная 1,84 96 18 177
10%-ная (25 мл конц. кисло- ты 4- 398 мл воды) 1,07 10 1,1 10,7
1 н. (13,9 мл коиц. кислоты, раз- бавленной до 500 мл) 1,03 4,7 0,5 4,8
Азотная кислота концентрированная 1,42 71 16 101
Гидроксид натрия, 10%-иый раствор 1,11 10 2,8 11,1
Гидроксид аммония концентриро- ванный 0,90 28,4 15 25,6
Фосфорная кислота концентрирован- ная (сиропообразная) 1,7 85 14,7 144
Состав обычных буферных растворов
pH Компоненты
0,1 1,1 2,2 3,9 5,0 1 н. соляная кислота 0,1 н. соляная кислота 15,0 г винной кислоты на 1 л раствора (0,1 М) 40,8 г калиевой соли фталевой кислоты на 1 л 14,0 г моиокалиевой соли фталевой кислоты 4- 2,7 г NaHCOs на 1 л (нагреть для удаления диоксида углерода, затем охладить)
19 Зак. 335
578
Приложение I
Продолжение
pH Компоненты
6,0 7,0 8,0 9,0 10,0 11,0 12,0 13,0 14,0 23,2 г КН2РО4 + 4,3 NS2HPO4 (безв.) на 1 л 9,1 г КН2РО( + 18,9 г Na2HPO4 на 1 л 11,8 г борной кислоты + 9,1 г буры (Na2B4O7-ЮНгО) 6,2 г борной кислоты + 38,1 г буры на 1 л 6,5 г NaHCOs + 13,2 г Na2COs на 1 л 11,4 г Na2HPO4 + 19,7 г NasPO4 на 1 л 24,6 г NasPO4 на 1 л раствора (0,15 М) 4,1 г гранулированного гидроксида натрия на 1 л раствора (0,1 М) 41,3 г гранулированного гидроксида натрия на 1 л раствора (1 М)
Номограмма зависимости температуры кипения от давления
при перегонке в вакууме а
Наблюдаемая
температура
кипения при
давлении Рмм
•С °F
Температура
кипения при 7$0мм
’с Т
700
600 i М100
: !1000
600^ Г
900
300
600
200 -V 400
100 -4
400 4 ►
; Г 700
а
а Если в литературе указана температура кипения и соответствующее давление, то нуж-
но соединить примой линией эти две величины иа шкалах а и в. Теоретическое значение тем*
пературы кипения при 760 мм получают в точке пересечения этой линии со шкалой б. Затем
соедиияют^прямой линией значение иа шкале б с другим значением давления иа шкале в;
приблизительное значение температуры кипения получают в точке пересечения этой прямой
линии со шкалой а.
Таблицы для использования в органической лаборатории
579
Растворители, применяемые для элюирования в хроматографии
(растворители перечислены в порядке увеличения полярности а)
Адсорбент оксид алюминия Адсорбент силикагель
Фторированные углеводороды Пеитаи Изооктан Легкий петролейный эфир Гексан Циклогексан Циклопентан Тетрахлорметаи Сероуглерод Ксилол Диизопропиловый эфир Толуол 1-Хлорпропан Хлорбензол Циклогексан Гептан Пеитан Тетрахлорметаи Сероуглерод Хлорбензол Этилбензол Толуол Бензол 2-Хлорпропан Хлороформ Нитробензол Диизопропиловый эфир Диэтиловый эфир
Бензол Этилбромид Диэтиловый эфир Диэтилсульфид Хлороформ Метилеихлорид Тетрагидрофуран 1,2-Дихлорэтан Этилметилкетон Этилацетат 2-Бутанол Этанол Вода Ацетон Уксусная кислота Метанол Пировиноградная кислота
1-Нитропропан
(Ацетон)
1,4-Диоксаи
Этилацетат
Метилацетат
1-Пентанол
Диметилсульфоксид
Анилин
Диэтиламин
Нитрометан
Ацетонитрил
Пиридин
Бутилцеллозольв
Пропанол-2
Пропаиол-1
Этанол
Метаиол
Этиленгликоль
Уксусная кислота
а Способность растворителя элюировать вещество нз хроматографической колонки зави-
сит от природы как самого вещества, так н адсорбента, поэтому могут встречаться отклоне-
ния от приведенных рядов. Однако в общем в данной таблице растворители расположены
в порядке увеличивающейся полярности, т. е. в принципе растворители, находящиеся ниже
в списке, легче элюируют более полярные соединения.
19*
580
Приложение I
Твердые носители для газохроматографических колонок
Носитель Фирма-изготовитель а Носитель Фирма-изготовитель а
Аэропак Анакром Огнеупорный кирпич С-22 Целит 545 Хромосорб Диатом Диатопорт S Эмбацел Газ-хром Аиапорт б Хромосорб Т в Флуоропак 80 г Хромосорб 100 (серия про- дуктов) Анапорт Церабедс Корниигбедс Диатомитовая земля Varian Aerograph Ana labs Johns-Manville Johns-Manville Johns-Manville Debton Co. Hewlett-Packard May anb Baker Applied Science Labs. Галогенуг Analabs Johns-Manville Fluorocarbon Co. Пористые Johns-Manville Стекла и duo Analabs 1 Analans Corning Glass Works] и огнеупорный кир GC-32 (целатом) GC Суперсаппорт Гифло S-80 Силоцелбрик Стерхамол Супелкопорт Варапорт (см. Аэропак) певодороды Галопорт в Кель-F б Тисикс в полимерыД Порапак ксид кремния ' Корнингглас ж Порасил Гласпорт пич Eagle-Picher Coast Engineering Lab. Johns-Manville R. Grutzmacher Griffin anb George S terchamol - W erke Supelco Hewlett-Packard 3M Company Analabs Waters Assoc. Corning Glass Works Waters Assoc. Hewlett-Packard
а Адреса компаний см.: Гордон А., Форд Р. Спутник химика. Физико-химические свой-
ства, методики, библиография. Пер. с англ.—М.: Мир» 1976.
б ХлорфторуглеводородиыЙ полимер.
в Тефлон 6 (фирма Du Pont).
г Фторуглеводородный полимер.
А Полимеры стирола используются при хроматографии полярных соединений, таких,
как кислоты и т. п.
е Для хроматографии соединений с высокой молекулярной массой при низких темпера-
турах.
ж Получают из стекла викор 7930.
Охлаждающие смеси солей со льдома
Соль Начальная температура, °C Количество соли иа 100 г воды или льда, г Конечная температура, °C
Na2CO3 — 1 (лед) 20 —2,0
NH«NO3 20 106 -4,0
NaC^HsOz 10,7 85 -4,7
NH4C1 13,3 30 -5.1
NaNO3 13,2 75 -5,3
Таблицы для использования в органической лаборатории
581
Продолжение
Соль Начальная температура, °C Количество соли на 100 г воды или льда, г Конечная температура, °C
№2520з • 5Н2О 10,7 но -8,0
СаС12 6Н2О — 1 (лед) 41 -9,0
К Cl 0 (лед) 30 — 10,9
KI 10,8 140 — 11,7
NH4NO3 13,6 60 -13,6
NH4C1 — 1 (лед) 25 -15,4
NH*NO3 — 1 (лед) 45 -16,8
NH4SCN 13,2 133 -18,0
NaCI — 1 (лед) 33 —21,3
CaCl2-6H2O 0 (лед) 81 -21,5
H2SO4 (66,2%) 0 (лед) 23 —25
NaBr 0 (лед) 66 -28
H2SO4 (66,2%) 0 (лед) 40 -30
C2H6OH (4°) 0 (лед) 105 -30
MgCl2 0 (лед) 85 -34
H2SO4 (66,2%) 0 (лед) 91 —37
CaCl2 • 6H2O 0 (лед) 123 -40,3
CaCl2 • 6H2O 0 (лед) 143 -55
“ Прибавление вещества, указанного в первой колонке, не всегда может понижать тем-
пературу баии до той температуры, которая указана в последней колонке. Это может быть
обусловлено недостаточным дроблением льда и т. д.
Жидкости, используемые в баиях для нагревания
Жидкость Т. пл.. °C Т. кип., °C 1 емпературная область приме- нения а, °C Темп, вспышки, ‘С Примечание
Н2О 0 100 0—80 Нет Идеальная жидкость в ограниченной области
Этиленгликоль -12 197 — 0—180 115 Дешевый, огнеопасный, трудно удаляется из аппарата
Силиконовое масло Доу Корнинг 330 Ниже — 148 — 30—280 290 Очень вязкое при низ- кой температуре
20% Н3РО3 и 80% Н3РО4 Ниже 20 2( )—250 Нет Растворима в воде, ие- огнеопасна, вызывает коррозию, при высо- кой температуре «ды- мит»
Т риэтиленгликоль -5 287 0 —250 156 Растворим в воде, до- статочно стабилен
Силиконовое масло Доу Корнинг 550 -60 0 -250 310 Некорродирующая жид- кость, может исполь- зоваться до 400 °C в атмосфере азота
582
Приложение I
Продолжение
Жидкость Т. пл., °C Т. кип., °C Температурная область приме- нения а, °C S а я Й Q С X «у —< о Примечание
Глицерин 18 290 -20-260 160 Способен к переохлаж- дению, растворим в воде, вязкий
Парафин ~50 — 60—300 199 Огнеопасен
Дибутил фталат — 340 150—320 — Вязкий при низкой тем-
Сплав Вуда (50% Bi, 25% Pb, 12,5% Sn, 12,5% Cd) 70 — 70-350 — Окисляется при дли- тельном использова- нии выше 250 °C
Тетракрезилснликат Ниже -48 ~440 20—400 Не вызывает коррозии, огнестоек, дорогой
® Область используемых температур для открытых бань; все эти банн, за исключением
водяной, следует использовать только в вытяжном шкафу.
Растворители для экстракции из водных растворов
Растворитель Т. кип., °C Огнеопас- ность 6 Токсич- ность б Примечания
Бензол 80,1 3 3 Склонен к образованию эмульсийв, удобен для экстракции алкалои- дов и фенолов из буферных рас- творов
Бутанол-2 99,5 1 3 Высококипящнй растворитель, удо- бен для экстракции полярных, растворимых в воде веществ нз буферных растворов
Тетрахлорметан 76,5 0 4 Легко высушивается, хороший экс- трагент для неполярных веществ
Хлороформ 61,7 0 4 Может образовывать эмульсию, лег- ко высушивается
Диэтнловый эфир 34,5 4 2 Адсорбирует большие количества воды; обычный хороший раствори- тель
Диизопропиловый эфир 69 3 2 Прн длительном стоянии может об- разовывать взрывчатые перокси- ды; хороший экстрагент для из- влечения кислот нз фосфатных бу- ферных растворов
Этилацетат 77,1 3 1 Адсорбирует большое количество воды; хороший экстрагент для по- лярных веществ
Таблицы для использования в органической лаборатории
583
Продолм>сди§
Растворитель Т. кип., °C Огнеопас- ность 6 Токсич- ность б Примечания
Фреон 11 24 0 1 Фреоны — хорошие экстрагенты для легколетучих неполярных соедине- ний; довольно дороги
Фреон ИЗ 47,7 0 1 То же
Метнленхлорид 40 0 1 Может образовывать эмульсин, лег- ко высушивается
н-Пентан 36,1 4 1 Т Углеводороды легко высушиваются;
н-Гексан 69 4 1 > плохие экстрагенты для полярных
н-Гептан 98,4 3 1 ) соединений
а В основном по данным: Гордон А., Форд Р. Спутник химика. Физико-химические свой-
ства, методики, библиография. Пер. с англ. — М.: Мир, 1976.
б Огнеопасность и токсичность оцениваются по четырехбалльной системе (0—иеогне-
опасный).
® Эмульсии могут образоваться при экстракции из водных растворов органическими
растворителями, при этом достичь хорошего разделения очень трудно илн даже вообще не-
возможно. Чаще всего это происходит прн экстракции из щелочных растворов; прибавление
разбавленной серной кнслоты (если это возможно) может разрушать такие эмульсии. Для
разрушения эмульсий существуют следующие общие методы: насыщение вэдиой фазы солью
(NaCl, Na2SO] и т. д.); прибавление нескольких капель спирта нли эфира (особенно, когда
органическим слоем является хлороформ). Одним нз наиболее эффективных методов является
центрифугирование смесн.
Высушивающие агенты умеренной силы для органических растворителей
Высушивающий агент Емкость, моли на 1 моль сорбента Относительная а скорость высушивания Примечания
CaSO4 V2H2O Очень быстро Поступает в продажу под на- званием «драйерит» с цвет- ным индикатором C0CI2 илн без него. Очень эффективен. Сухой CaSO4 с индикато- ром имеет голубую окраску, становится розовым при по- глощении воды (СоС1г мо- жет поглощать 6 молекул воды, СоС12-6Н2О); исполь- зуется прн температурах от —50 до +86 °C. Некоторые органические растворители выщелачивают СоС1г или из- меняют его окраску (аце- тон, спирты, пиридин и др.)
СаС12 6Н2О Очень быстро (мед- леннее, чем CaSO4) Не очень эффективен; исполь- зуется только для углеводо- родов и алкнлгалогенндов (образует сольваты, ком- плексы нли реагирует со многими соединениями, со- держащими азот и кисло- род)
584
Приложение I
Продолжение
Высушивающий агент Емкость, моли на 1 моль сорбента Относительная а скорость высушивания Примечания
Mg so. 7Н2О Быстро (медленнее, Превосходный осушитель об-
Молекулярное Высокая чем СаС12) Быстро (существен- щего применения; очень инертен, но может быть слабокислым (следует набе- гать использования его для осушки соединений, очень чувствительных к кисло- там). Может растворяться в некоторых органических рас- творителях Очень эффективный осуши-
снто 4А NasSO, ЮН2О но медленнее, чем MgSO,) Много медленнее, тель; рекомендуется предва- рительно высушить веще- ства обычным высушиваю- щим агентом (более подроб- но о молекулярных ситах см. ниже). Превосходно вы- сушивает молекулярное си- то ЗА Очень мягкий, малоэффектив-
К2СО3 2Н2О чем MgSO, Быстро ный, медленно высушиваю- щий, недорогой агент с вы- сокой емкостью; удобен для грубого предварительного высушивания; раствор не следует нагревать Этот осушитель удобен для
NaOH, КОН Очень сложных эфиров, нитрилов, кетонов и особенно спиртов, но его не следует использо- вать для кислых соединений Очень эффективны, но нсполь-
H2SO, высокая То же Очень быстро зуются только для инертных растворов, в которых эти агенты нерастворимы; осо- бенно удобны для аминов Очень эффективна, но ее при-
Оксид алюмн- » То же мененне ограничено только насыщенными или аромати- ческими углеводородами или галогенидами (реаги- рует с олефинами и с со- единениями основного ха- рактера) Особенно удобны для углево-
НИЯ (А12О3) или силика- гель (SiOz) а Pearson В. Ollerensha?# Chem. Ind., 1966, 370. дородов. Их следует тща- тельно измельчать; после использования можно реге- нерировать нагреванием (300 °C для SiO2 и 500 °C ДЛЯ AI2O3)
Таблицы для использования в органической лаборатории
585
Более сильные осушители для органических жидкостей
Осушитель а Продукты, образующиеся при действии воды Примечания
Na6 NaOH, Н2 Превосходный осушитель для насыщенных угле- водородов и простых эфиров; его нельзя ис- пользовать для галогенсодержащих соедине- ний
СаН Са(ОН)2, Н2 Один нз иаилучшнх осушителей, высушивает медленнее, чем LiAlH4, но также эффективен и менее опасен. Используется для углеводо- родов, простых эфиров, аминов, сложных эфи- ров, спиртов С4 и выше (нельзя использовать для осушки спиртов Ci, С2, С3). Нельзя ис- пользовать для альдегидов и соединений с ак- тивной карбонильной группой
LiAlH/ LiOH, А1(ОН)3, н2 Используется только для инертных растворите- лей [углеводороды, арил (но не алкил) галоге- ниды, простые эфиры]; реагирует с вещества- ми, содержащими активные атомы водорода, н с большинством функциональных групп (га- логены, карбонильные, ннтро-группы и т. д.). При использовании этого реагента следует со- блюдать осторожность; избыток можно разру- шить медленным прибавлением этилацетата
ВаО или СаО Ва(ОН)2 или Са(ОН)2 Медленно работающие, но эффективные осуши- тели; удобны главным образом для спиртов и аминов; их не следует использовать для со- единений, чувствительных к сильным основа- ниям
р2о5 НРО3, Н3РО4, Н4Р2О7 Очень быстрый и эффективный осушитель, силь- нокислый. Рекомендуется предварительное вы- сушивание менее энергичными осушителями. Используется только для инертных соединений (особенно углеводородов, простых эфиров, га- логенидов, кислот и ангидридов)
Наилучшими осушителями являются те, которые быстро и необратимо реагируют
с водой (и не реагируют с растворителем и растворенными веществами); в то же время оии
наиболее опасны и их следует использовать только после предварительного грубого высуши-
вания менее энергичными высушивающими агентами (см. ниже). Эти агенты почти всегда
применяются только для высушивания растворителей перед или при перегонке. Хотя MgCICU-
однн из наиболее эффективных высушивающих агентов, его не рекомендуют использовать,
потому что неумелое обращение с ним может привести к взрывам. Сведения об эффектив-
ности осушителей см.: Bur field D. R., Lee Smithers R. H., J. Org. Chem., 42, 3060(1977).
6 Фирма J. T. Baker Co. продает сплав 10% Na и 90% Pb под иазваиием Drl-Na; этот
сухой гранулированный агент медленно реагирует с воздухом, но является такиад Же эффек-
тивным высушивающим агентом, как натриевая проволока для высушивание простых эфиров
и др. См. Физер Л., Физер М. Реагенты для органического синтеза. Пер. с англ.—М.: Мир,
1971, т. 5, с. 324.
в Меиее опасный, ио столь же эффективный высушивающий агент NafCHaOCHsCHaOJaAlHz,
известен под названием «внтрид» (Realco Chemical Company, 783 Jersey Avenue, New Brun-
swick, N. J. 08902); может быть получен также через фирму Eastman Kodak, США.
586
Приложение I
Обнаружение воды. Количественное определение малых количеств
Титрование по Фишеру — одни из наиболее доступных чувствительных ме-
тодов определения очень малых количеств воды в органических жидкостях. Реак-
тив Фишера представляет собой обычно раствор в метаноле (как растворитель)
иода, диоксида серы н пиридина. Раствор этого реактива выпускают, например,
фирмы Fischer Scientific, Matheson, А. Н. Thomas, титрованный раствор выпу-
скает фирма А. Н. Thomas. Более подробно метод описан в книге: Cheronis N. D.t
Ma Т. S., Organic Functional Group Analysis, Wiley-Interscience, New York, 1964,’
pp. 472—475. Детальное описание метода можно найтн в книге: Mitchell I.,
Smith D., Aquametry, Wiley-Interscience, New York, 1948.
Более быстрым и универсальным методом определения воды является метод
ГЖХ с использованием колонок, наполненных порапаком [Hogan J., Engel R.,
Stevenson H., Anal. Chem., 42, 249 (1970)]. Детали метода описаны фирмой
Fisher Scientific Company в книге «Trace Water Analysis in Organics»; этот метод
позволяет определять от 1 до 10 м.д. воды с высокой точностью н воспроизво-
димостью.
Обзор других методов анализа воды приведен в книге: Organic Solvents, in:
«Techniques of Chemistry», Vol. 2, Wiley-Interscience, New York, 1971.
Приложение II
Оборудование и реактивы для лаборатории
Ниже приведен типичный список лабораторных принадлежностей и реакти-
вов, используемых для проведения идентификации органических соединений, а
также оборудование н реактивы, которые необходимы для ответа на многие во-
просы, описанные в этой книге. Отличие одной лаборатории от другой по имею-
щемуся оборудованию может потребовать модификации или замены некоторого
описанного здесь оборудования.
ПРИБОРЫ
Оборудование для индивидуального рабочего стола
Удобно закрепить за каждым студентом отдельный стандартный стол в ор-
ганической лаборатории, оборудованный обычным набором приборов, необходи-
мых на первом году практической работы по органической химии. К этому сле-
дует добавить специальный набор приборов, содержащий оборудование для вы-
полнения реакций для идентификации и получения производных в небольших
количествах.
Оборудование, хранящееся в столе
Стаканы химические емкостью 50, 100, 250, 400 и 800 мл — 5 шт.
Горелка с трубкой длиной 1м — 1 шт.
Микрогорелка — 1 шт.
Кожух для защиты пламени горелки — 1 шт.
Запальная горелка — 1 шт.
Насадка для горелкн «ласточкин хвост» — 1 шт.
Зажим трехзубый — 1 шт.
Зажнм удлиненный с держателем — 2 шт.
Зажнм винтовой — 1 шт.
Зажим для пробирок — 1 шт.
Пробковое кольцо диаметром ПО мм — 1 шт.
Мерный цилиндр емкостью 10 мл — 1 шт.
Мерный цилиндр емкостью 100 мл — 1 шт.
Блок для фильтрования — 1 шт.
Колбы Эрленмейера емкостью 25, 50, 125 и 250 мл — по 2 шт., емкостью
500 мл — 1 шт.
Склянкн для фильтрования под вакуумом емкостью 125 мл — 1 шт., ем-
костью 250 мл — 1 шт.
Воронин Бюхнера диаметром 55 мм, пластмассовые — 2 шт.
Воронин Хирша, 000, 0000 — 2 шт.
Делительная воронка емкостью 250 мл — 1 шт.
Воронка с маленькой ручкой диаметром 75 мм — 1 шт.
Воронка с короткой стеклянной трубкой — 1 шт.
Кольца диаметром 4 и 2 см, железные — 2 шт.
Резиновые трубки для повышенного давления длиной 1 м — 3 шт.
Пробирки 13 X 100 мм — 6 шт.
588
Приложение ll
Пробирки 16 X 150 мм —6 шт.
Термометр от —5 до +360°С — 1 шт.
Щипцы тигельные — 1 шт.
Часовые стекла диаметром 100 мм — 2 шт.
Дополнительные принадлежности
Пипетка градуированная емкостью 1 мл — 1 шт.
Пипетка градуированная емкостью 10 мл — 1 шт.
Пипетки неградуированные — 5 шт.
Склянки для фильтрования под вакуумом емкостью 50 мл — 1 шт.
Шпатель из нержавеющей стали, маленький — 1 шт.
Фарфоровый тигель с крышкой № 00 — 1 шт.
Эксикатор малый — 1 шт.
Перегонная колба на 25 мл к прибору для перегонки на нормальных шли-
фах — 1 шт.
Перегонная колба на 10 мл к прибору для перегонки на нормальных шли-
фах — 1 шт.
Пробирки с боковой ручкой нз стекла пирекс, 15X125 мм — 2 шт.
Пробирка с боковой ручкой нз стекла пнрекс, 20 X 150 мм — 1 шт.
Общее лабораторное оборудование
В обшественном пользовании находятся изделия нз железа, в том числе
штативы, все трн типа зажимов и штативные муфты.
Сушильный шкаф электрический с постоянной температурой от 20 до 250°С—
1 шт.
Сушилка с горячим воздухом для сушки химической посуды — 1 шт.
Большой эксикатор вакуумный с вакуумным насосом — 1 шт.
Трехрычажные весы «Охаус», позволяющие взвешивать до 500 г на цен-
тральном рычаге, 0—10 г на переднем рычаге и 0—100 г на заднем ры-
чаге — 1 шт.
Металлический блок с электрообогревом и термометром до 500°С нли блок
для определения температуры плавления «Mel-Temp» с термометром до
500°С — 1 шт.
Стеклянные трубки и палочки, различные.
Специальное лабораторное оборудование
Это оборудование следует хранить в местах, защищенных от воздействия
корродирующих газов и паров.
Аналитические весы. Следует иметь примерно одни весы на каждые пять
студентов.
Рефрактометр фирмы Bausch & Lomb, Abbe type 3L or Valentine (Industro-
Scientific Co., Philadelphia, Pa.) — 1 шт.
Поляриметр фирмы Rudolph лабораторный, модуль 63 с натриевой лампой
и полярнметрнческимн трубками длиной 1 и 2 дм — 1 шт.
Прибор для определения плотности фирмы Fisher — Davidson — 1 шт.
Инфракрасный спектрометр с принадлежностями. Пригодна модель Инфра-
корд 137 Perkin — Elmer Corp. (США) — 1 шт.
Отдельные предметы оборудования и реактивы, получаемые во
временное пользование от руководителя или из хранилища
Платиновая проволока длиной около 5 см (№ 20), впаянная в стеклянную
палочку.
Платиновая фольга (около 2,5 см2), впаянная в стеклянную палочку.
Оборудование и реактивы для лаборатории
589
Поляриметрические трубки длиной 1 и 2 дм.
Ампулы для ЯМР, тетраметилсилан и растворители для ЯМР-спектрометрии
(СС14, CDC13 ацетон-de, ДМСО-й6, D2O и ДСС).
ИД-ячейки, масла и калибровочное окно.
Маленькая агатовая ступка и пестик.
Баллон с сухим азотом.
РЕАКТИВЫ В ЛАБОРАТОРНЫХ ШКАФАХ
Органические соединения
Перечисленные ниже реактивы используются для определения растворимо-
сти, проведения реакций идентификации, а также для получения некоторых (но
не всех) производных. Удобно заранее приготовить серию склянок емкостью
около 100 мл, используя широкогорлые стеклянные склянки для твердых ве-
ществ и узкогорлые стеклянные флаконы для жидкостей. Для таких обычных
растворителей, как ацетон, бензол, хлороформ, тетрахлорметан, диоксан, эфир,
петролейиый эфир (т. кип. 70—90°С) и толуол, следует использовать склянки
большего объема (около 500 мл). Для группы в 20 студентов в эти склянки, на-
ходящиеся на полках, можно поместить от 20 до 50 г органических соединений,
хотя для экономии при покупке их можно приобретать в количествах 50, 100
нли 500 г. Действительные количества, которые необходимы каждому студенту,
естественно, будут изменяться в зависимости от природы неизвестного соедине-
ния, от правильности выбора реакции для идентификации, умения проведения
этой реакции студентом. Следует постоянно напоминать, что многие из этих со-
единений являются токсичными (соответствующие сведения приведены в прило-
жении IV).
Азокснбензол л-Бромфеиилгидразин
Аллиловый спирт
Аллнлхлорид
л-Аминофенол
Анизол
Анилин
Анилин солянокислый
Ацетанилид
Ацетон
Ацетонитрил
Ацетофенон
Бензальдегид
Бензамид
Бензидин
Бензиламин
Бензиловый спирт
S-Бензилтиуронийхлорид
Бензилхлорнд
Бензоат аммония
Бензойная кислота
Бензол
Бензонитрил
Бензофенон
Бнфеиил
л-Броманилин
л-Бромбензилбромид
S- (л-Бромбензил)тиуронийхлорнд
л-Бромбензилхлорид
Бром бензол
л-Бромбензол сульфохлорид
л-Бромфенацилбромид
л-Бромфенилизоцнанат
втор-Бутнлбромид
н-Бутилбромид
трет-Бутнлбромид
втор-Бутиловый спирт
н-Бутиловый спирт
трет-Бутиловый спирт
Винная кислота
Галактоза
Гардинол (натриевая соль лаурил-
сульфата)
Гексаи
н-Гептаналь
н-Гептиловый спирт
Гндроксиламин солянокислый
Гидрохинон
Глицерин
Глюкоза
л,л'-Диаминодифенилметан
л-Дибромбензол
Ди-н-бутнловый эфир
л-Диметиламинобензальдегид
Диметиланилин
Диметилсульфоксид
2,4-Динитроанилин
3,5-Днннтробензоилхлорид
3,5-Динитробеизойная кислота
ж-Динитробензол
о-Динитробензол
2,4-Динитрофенилгидразин
590
Приложение II
3,5-Динитрофеннлизоцианат
2,4-Динитрохлорбензол
п-Диоксан
Дифенил карбамоилхлорид
Диэтиламин
Диэтиленгликоль
Изоамнловый спирт
Изопропиловый спирт
Иодистый метил
Йодноватая кислота, реактив
п-Иодфенацилбромнд
Камфора
Коричная кислота
Ксантгндрол
Ксилол
Лактоза
Лауриловый спирт
Малеиновый ангидрид
Мальтоза
н-Масляный альдегид
Мезнтилен
Меркаптоуксусная кислота (тно-
глнколевая кислота)
Метансульфохлорид
Метиланнлнн
Метилбензоат
Метиловый спирт (метанол)
Метил-п-толуолсульфонат
Метилцеллозольв (2-метоксиэта-
нол)
Метон (днмедон)
Молочная кислота (85%-ная)
Мочевина
Муравьиная кислота
Нафталин
а-Нафталинсульфохлорид
Р-Нафтилгидразин
а-Нафтилизотиоцианат
P-Нафтил изоцианат
Р-Нафтол
а-Нафтохннон
льНнтроанилин
п-Нитроанилин
п-Ннтробензазид
п-Ннтробензальдегид
льНнтробензгидразид
п-Нитробензилхлорид
п-Нитробензоилхлорнд
n-Нитробензойная кислота
Нитробензол
льНитробеизолсульфохлорид
Нитрометан
n-Нитрофенил гидразин
п-Нитрофенилизоцианат
п-Нитрофеиол
З-Нитрофталевый ангидрид
n-Окси бензойная кислота
Пента и
Пентанол-2
Пентен-2
Петролейный эфир (40—60°С)
Петролейный эфир (70—90°С)
Пикриновая кислота
Пиперазин
Пиперидин
Пиридин (над гидроксидом калия)
н-Пропиловый спирт
Псевдосахаринхлорид
Резорцин
Салициловая кислота
Салициловый ангидрид
Сахароза
Семикарбазнда гидрохлорид
Сероуглерод
Сульфаниловая кислота
Тетрагндрофуран
Тетрахлорфталевый ангидрид
Тиомочевнна
п-1 о лн леем нка рб ази д
п-Толуиднн
Толуол
л-Толуолсульфокнслота
1,3,5-Т риннтрохлорбензол
Трнтилхлорид
Триэтаноламин
Триэтиламин
Уксусный ангидрид
л-Фенацилбромнд
Фенилгидразин
Фенилгидразина гидрохлорид
о-Фенилендиамин
Фенилсемикарбазнд
а-Фенилэтиламнн
Фенол
Флороглюцин
Формалин
Фруктоза
Фталевый ангидрид
Фталимид
а-Хлорацетофенон
n-Хлорбенз гидразид
S- (и-Хлорбензил)тиуронийхлорнд
Хлорбензол
п-Хлорнитробензол
Хлороформ
n-Хлорфенацилбром ид
Холестерин
Циклогексан
Четыреххлористый углерод (тетра-
хлорметан)
Щавелевая кислота
Этилацетат
Этилацетоацетат (ацетоуксусный
эфир)
Этилбензоат
Этилбромид
Этиленбромид
Этиленгликоль
Оборудование и реактивы для лаборатории
591
Неорганические соединения
Предлагаемые количества достаточны для группы в 20 студентов, хотя не-
которые сосуды с наиболее часто используемыми реактивами придется попол-
нять несколько раз в семестр. Многие нз приведенных соединений токсичны. Этот
вопрос обсуждается далее в приложении IV.
Аммония карбонат 100 г Натрия гидроксид (грану- 1 КГ
Аммония хлорид 100 г лированный)
Гидразингидрат 25 г Натрия карбонат (безв.) 1 кг
Г идразинсульфат 50 г Натрий металлический 50 г
Железо (II) аммонийные 100 г (под толуолом)
квасцы Натрия нитрат 100 г
Железо, порошок 100 г Натрия нитрит 200 г
Иод (возогнанный) 100 г Натрия нитропруссид 5 г
Иодистоводородная кис- лота (57%-ная) 100 г Натрия сульфат (безв.) Натрия сульфид (крист.) 1 50 кг г
Калня бромид 100 г Натрия сульфид, нонагид- 100 г
Калия гидроксид (грану- лированный) 1 кг рат Натрия тиосульфат 100 г
Калия иодид 100 г (крист.)
Калия карбонат (безв.) 1 кг Натрия хлорид 1 кг
Калия перманганат Калия персульфат 200 г Натрия цианид (токсичен) 500 г
25 г Олово (гранулированное) 2оо г
Калня тиоцианат 100 г Олова хлорид 100 г
Калия цианид 100 г Ртути бромид 10 г
Кальция хлорид (безв.) 1 кг Ртути иодид 10 г
Магний, стружка 100 г Ртути нитрат 100 г
Магния сульфат (безв.) 1 кг Ртути оксид 50 г
Меди сульфат (безв.) 50 г Ртути сульфат 50 г
Натрия ацетат (безв.) 200 г Свинца ацетат 150 г
Натрия ацетат (крист.) 100 г Свинца пероксид 100 г
Натрия бикарбонат 1 кг Фосфорная кислота 1 кг
Нитрия бисульфит 1 кг (85%-ная)
Натрия бисульфит (безв.) 100 г Хромовый ангидрид 100 г
Натрия бихромат 1 кг Цинк (пыль) 2Q0 г
Натрия боргидрид 100 г Цинка хлорид (безв.) 100 г
Другие соединения, реактивы и растворы
Удобно приготовить от 500 до 1000 мл растворов и хранить их на полке
или иа столе в лаборатории. Способы приготовления этих растворов описаны в
гл. 6 и в книгах по количественному и качественному неорганическому анализу.
Алюминиево-никелевый сплав
Азотная кислота разбавленная (5%-ная)
Аммония гидроксид, 2%-ный раствор
Бенедикта реактив
Богена индикатор, раствор
Бора трифторнд, бисацетатный комплекс (см. гл. 6, методика 51а)
Бром в четыреххлористом углероде, 5%-ный раствор
Бромная вода (насыщенная)
Тетра-н-бутнламмоиийбромид
Гидроксиламина гидрохлорид, реактив
Обесцвечивающий уголь («дарко», «норит» и др.)
2,4-Динитрофенилгндразии, реактив
Железо(П)аммонийные квасцы, раствор (50 г/л)
592
Приложение II
Железо (III)хлорид, 5%-ный раствор
Иод — иодид калия, раствор
Иод-крахмальная индикаторная бумага
Иодная кислота, реактив
Калня гидроксид, 10%-ный спиртовый раствор
Калия перманганат, 2%-ный раствор
Калия фторид, 20%-ный раствор
Калия хлорид, раствор (насыщенный)
Кизельгур
Конго красный, индикаторная бумага
Магния — калия карбонаты, смесь
Минеральное масло
Натрия бикарбонат, 5%-иый раствор
Натрия бисульфит, водно-спиртовый раствор
Натрия гидроксид, 20%-ный раствор
Натрия гидроксид, 0,1 и. стандартный раствор (23 л, стеклянная бутыль)
Натрия иодид в ацетоне
Натрия карбонат, 10%-ный раствор
Натрия нитропруссид
Натрия плюмбит
Никеля хлорид — 5-нитросалициловый альдегид, реактив
Никеля хлорид — сероуглерод, реактив
Пероксид водорода, 3%-ный раствор
Ртути нитрат
Свинца ацетат, 1%-ный раствор
Серебра нитрат, 5%-ный водный раствор
Серебра нитрат, 2%-ный спиртовый раствор
Серная кислота разбавленная (10%-ная)
Соляная кислота, 0,25 н. стандартный раствор (23 л, стеклянная бутыль)
Соляная кислота — хлорид цинка, раствор (реактив Лукаса)
Стеклянная вата
Тимол голубой, индикатор, 0,2%-иый раствор в 95%-ном спирте
Универсальный индикатор, раствор (Eastman А 4953)
Фелинга раствор № 1
Фелинга раствор № 2
Фенолфталеин, индикатор, 1%-ный раствор в 95%-ном спирте
Фосфорная кислота, 20%-ный раствор
Фуксин, реагент на альдегиды
Хлопковое масло
Хлопчатобумажные фильтры
Хлорная вода (свежая)
Церий (IV), нитратный комплекс, реактив [необходим (КН4)2Се(ЦО3)б]
Цнрконий-ализариновая индикаторная бумага
Реактивы, которые хранятся в вытяжном шкафу
Азотная кислота (дымящая)
Алюминия хлорид (безв.)
Аммония полисульфид
Ацетилхлорнд
Бензоилхлорид
1?ензолсульфохлорид
Бром
Р-Нафтилизотнрцианат
а-Нафтил изоцианат
Серная кислота (дымящая)
Сероводород (водный раствор)
Тионилхлорид
п-Толилизоцианат
п-Толуолсульфохлорид
Фенацилбромид
Фенилизотиоцианат
Фенил изоцианат
Фосфора пентахлорид
Фосфора трихлорид
Фосфора хлороксид
Фталоилхлорид
Хлорсульфоновая кислота
Оборудование и реактивы для лаборатории
593
Реактивы определенной концентрации в 250-миллилитровых стеклянных
склянках:
Азотная кислота концентрированная, уд. вес 1,42 (70%-ная HNOs)
Аммония гидроксид, уд. вес 0,90 (28% Ь1Нз)
Натрия гидроксид, 10%-ный раствор
Серная кислота концентрированная, уд. вес 1,84 (95% H2SO<)
Соляная кислота концентрированная, уд. вес 1,19 (37% НС1)
Соляная кислота, 10%-ный раствор
Уксусная кислота ледяная
Особые реактивы
Диэтиловый эфир абсолютный
Этанол 95%-ный
Этанол абсолютный
Платинохлорнстоводородная кислота
Неизвестные соединения
Вещества, используемые в качестве неизвестных соединений, следует тща-
тельно отбирать по их чистоте. Типичные примеры можно выбрать из таблиц в
приложении III, однако ограничиваться только соединениями, приведенными
там, нет необходимости. Достаточно 5—10 г кристаллического вещества и 10—
15 мл жидкости. Смесн также должны состоять нз 5—10 г кристаллического и
10—15 мл жидкого вещества, которые разбавляют большими количествами под-
ходящего растворителя. Поскольку к концу семестра у студентов накапливается
опыт и совершенствуется техника, постепенно можно давать значительно мень-
шие количества неизвестных соединений. Прн этом пробы на растворимость, ре-
акции для классификации н получение производных можно выполнять с исполь-
зованием значительно меньших количеств веществ (около одной десятой коли-
честв, приведенных в экспериментах и методиках).
Приложение HI
Таблицы производных
В приведенных ниже таблицах собраны данные о свойствах распространен-
ных органических соединений различных классов; вещества расположены в по-
рядке увеличения их температур кипения нлн плавления. Эти таблицы помогут
читателю подтвердить свои предположения о возможной структуре неизвестного
соединения. После того как одно из них будет выбрано в качестве наиболее ве-
роятного, необходимо обратиться к первоисточникам для рассмотрения пол-
ного перечня производных этого соединения н для обсуждения их свойств
(гл. 1). Таблицы производных не претендуют на полноту. Одна из причин этого
состоит в том, чтобы заставить студента обратиться к литературным источникам
и продолжить изучение специфических путей превращения избранных соединений
в их производные. Такой способ идентификации неизвестных веществ повысит
шансы студентов на успех. Пути исследования, предложенные в гл. 6, являются
компромиссными, предполагающими использование максимально возможного
числа соединений из различных классов. Тем не менее очевидно, что во многих
случаях эти пути неудовлетворительны, н здесь следует применять более подроб-
ные методики, найденные при исследовании соответствующих соединений. Другая
причина неполноты таблиц связана с содержанием самого курса^ который пред-
полагает, что студенты имеют достаточный практический опыт поиска литера-
туры и работы с ней.
Естественно, что прн описании соединений в литературе обычно дается интер-
вал значений температур кипения или плавления. Чтобы избежать излишней за-
груженности таблиц, в них приведены только верхние значения интервала тем-
ператур кипения и плавления, округленные до целых градусов. Если не указано
особо, то значения удельной массы приведены для температуры 20°С относи-
тельно воды при 4°С. Показатели преломления даны для D-линии натрия при
20°С.
Студенты должны помнить, что полученное значение температуры плавления
может в определенных пределах зависеть от наблюдателя и от метода ее опре-
деления, поэтому часто в литературе приводится несколько различных значений
для одной константы. Обычно в таких случаях в таблице приведено наибольшее
значение, хотя это может привести к серьезным ошибкам. Следовательно, для
выяснения деталей студент должен обратиться к оригинальной литературе. Не-
обходимо помнить, что многие нз этих соединений токсичны (см. приложе-
ние IV).
Ацетали (жидкие)
Ацеталь Т. кип., °C Ацеталь Т. кип., ”С
Днметоксиметан (метилаль) 45 1,3-Дн оксан 105
1,1 -Диметоксиэтан 64 2-Метил-1,3-диоксан 110
2-Метнл-1,3-диоксолан 82 1,1 - Диизопропокснэтан 122
Диэтоксиметан (этилаль) 89 1,1 - Диэтоксипроп ан 124
1,1-Диэтоксиэтан (ацеталь) 102 1,1 -Днэтокснпропен-2 126
Таблицы производных
595
Продолжение
Ацеталь Т. кип., °C | Ацеталь Т. кип., °C
Дн-н-пропоксиметан 140 Ди-н-бутоксиметан 181
1,1 -Днэтоксибутан 143 1,1 -Диэтокси-2,2-дихлорэтан 184
1,1 -Дн-н-пропоксиэтан 148 1,1 - Дн-н-бутоксиэтан 186
1,1 -Днэтоксн-2-хлорэтан 157 а,а-Днметокситолуол 199
1,1 -Диэтокси-2-бромэтан 170 Дн-н-пентоксиметан 219
1,1 -Д и-втор-бутоксиэтан 171 а,а-Диэтокснтолуол 222
1,1 -Диизобутоксиэтан 176 1,1 -Дн-н-пентокснэтан 222
Ангидриды и галогенангидриды кислот (жидкие)
Соединение Т. кип., ®С т. пл. производного, °C
амид анилид
Ацетнлфторид 21 82 114
Трифторуксусный ангидрид 39
Пропнонилфторид 45 81 106
Ацетнлхлорнд 55 82 115
Оксалнлхлорид 64 419 (разл.) 245
Бутнрилфторнд 67 115 96
Акрнлилхлорид 76 85 1U5
Пропионилхлорнд 80 79 103
Ацетнлбромнд 81 82 115
Изобутирнлхлорнд 92 129 105
н-Бутирилхлорид 100 115 90
Пропионилбромид 103 81 106
Хлорацетнлхлорид 105 119 134
Дихлорацетнлхлорид 107 98 118
Ацетилиоднд 108 82 114
Метоксиацетилхлорид ИЗ 97 58
Изовалернлхлорид 115 135 109
Трихлорацетилхлорид 115 141 94
Метилэтилацетилхлорид 116
Кротонилхлорид 126 161 118
Пропионилиоднд 127 81 106
н-Валерилхлорид 127 106 63
Хлорацетилбромид 127 119 134
Бромацетнлхлорид 127 91 131
Бутирилбромид 128 115 96
Уксусный ангидрид 138 82 115
Изовалерилбромид 140 135
Изокапроилхлорид 147 121 112
Бутнрилиоднд 148 115 96
Бромацетилбромид 149 91 131
а-Бромпропнонилбромнд 153 123 99
Н-Капроилхлорид 153 98 95
Смешанный ангидрид уксусной и 154
пропионовой кислот
596
Приложение III
Продолжение
Соединение Т. кип., °C Т. пл. производного, °C
амид аннлид
Бензоил фторнд 159 128 160
Фумарилхлорнд 160 267 (разл.) (ди)
Пропионовый ангидрид 168 79 103
н-Гептоилхлорид 176 94 71
Изомасляный ангидрид 182 128 105
Сукцннилхлорид 190 (разл.) 242 226
к-Масляный ангидрид 191 115 90
Бензоилхлорид 197 128 160
Фенацилхлорид 210 154 117
Цитраконовый ангидрид 213 187 (разл.) 175
н-Валериановый ангидрид 215 106 63
Изовалериановый ангидрид 215 135
Дихлоруксусный ангидрид 216 (разл.) 98 118
Бензонлбромид 218 128 160
п-Хлорбензонлхлорид 222 179 194
м-Метоксибензоилхлорид 224
л-Хлорбензоилхлорнд 225 134 122
о-Хлорбензонлхлорид 238 142 114
л-Бромбензоилбромнд 243
Кротоновый ангидрид 248 160 118
о-Метокснбензоилхлорид 254 128 62
Капроновый ангидрид 257 100 95
н-Гептановый ангидрид 258 96 71
Фталоилхлорид (симм) 276 220 169
Ангидриды и галогенангидриды кислот (твердые)
Соединение Т. пл., °C Т. пл. производного, °C
кислота амид анилид
п-Хлорбензоилхлорид 16 242 179 194
Сукциннлхлорид 16 188 242 226
о-Нитробензоил хлорид 20 146 174 155
Стеарилхлорид 22 69 108 33
Анизилхлорид 26 184 162 168
Ангидрид quc-циклогексаи-1,2-ди- 32 192 (разл.)
карбоновой кислоты
л-Нитробензоилхлорид 35 140 143 153
Цинна моилхлори д 36 133 142 153
о-Толуиловый ангидрид 39 105 143
п-Бромбензоилбромид 42 189 197
Бромуксусный ангидрид 42 50 91 131
Бензойный ангидрид 42 121 128 160
Лауриновый ангидрид 42 44 ПО 78
Таблицы производных
597
Продолжение
Соединение Т. пл., °C Т. пл. производного, ®С
кислота амид анилид
Хлоруксусный ангидрид 45 62 120 134
2,4-Диннтробензоил хлорид 46 203
Глутаровый ангидрид 56 97 176 224
Малеиновый ангидрид 56 130 153 187
л-Иодбензоилбромнд 60 217 210
3,5-Динитробензоилбромид 60 183 234
Итаконовый ангидрид 68 165 (разл.) 192 190
Стеариновый ангидрид 70 70 109 95
.«-Толуиловый ангидрид 71 113 94 126
Фенилуксусный ангидрид 72 76 156 116
3,5-Днннтробензонлхлорнд 74 202 183 234
н-Нитробензонлхлорид 75 241 201 204
о-Хлорбензойный ангидрид 79 142 142 114
Дифеннлкарбамндхлорид 85 189
л-Хлорбензойный ангидрид 95 158 134 122
л-Толунловый ангидрид 95 179 160 145
4-Нитрофталевый ангидрид 119 165 200 (разл.) 192
Янтарный ангидрид 120 188 242
Коричный ангидрид 130 133 142 153
Фталевый ангидрид 131 184 149
о-Нитробензойный ангидрид 135 146 176 158
а-Нафтойный ангидрид 146 162 205 161
.м-Нитробензойный ангидрид 160 140 143 154
З-Ннтрофталевый ангидрид 162 218 201 (разл.) (ди) 234 (ди)
1,2-Нафтойный ангидрид 169 175 (разл.) 265 (разл.)
л-Ннтробензойный ангидрид 189 241 201 211
л-Хлорбензойиый ангидрид 194 240 179 194
d-Камфарный ангидрид 221 187 192 210
2,3-Нафтойный ангидрид 246 241 (разл.)
Тетрахлорфталевый ангидрид 256 250 (разл.)
1,8-Нафтойный ангидрид 274
Тетрабромфталевый ангидрид 275 266
Тетраиодфталевый ангидрид 318 327
Карбоновые кислоты (жидкие)
Кислота Т. кип., “С Т. пл. производного, *С
аинлид п-толуи- дид п-иитро- беизило- вый эфир п-бром- фенаци- ловый эфир
Тиоуксусная 93 76 130
Муравьиная 101 47 53 31 135
Уксусная 118 114 147 78 85
598
Приложение III
Продолжение
Кислота Т. кип., “С Т. пл. производного, °C
анилид п-толун- дид п-иитро- беизнло- вый эфир п-бром- фен аци- ловый эфир
Акриловая 140 104 141
Пропионовая 140 103 124 31 63
Пропиоловая 144 (разл.)
Изомасляная 155 105 104 77
Метакриловая 163 87
н-Масляная 163 95 72 35 63
Пнвалиновая 164 129 120 76
Пировиноградная 165 (разл.) 104 109
Кротоновая (цис) 165 (разл.) 102 132 82
З-Бутеновая 169 58
Этилметилуксу сна я 176 ПО 93 55
Изовалериановая 176 109 109 68
Хлоруксусная 185 134 120 105
а-Хлорпропноновая 186 92 124
н-Валериановая 186 63 70 75
2,2-Диметнлбутановая 187 92 83
Днхлоруксусная 189 118 153 99
Днэтилуксусная 193 124 116 66
Изокапроновая 195 111 63 79
d,/-2-Метилпентановая 196 95 81 •
3-Метилпентановая 197 87 75
Тиглиновая 198 77 76 64 68
а-Бромизомасляная 200
Метоксиуксусная 203 58
н-Капроновая 205 95 75 72
а-Бромпропноновая 205 99 125
Этокснуксусная 206 32 105
Бромуксусная 208 131 91 88
2-Этнлпентановая 209 94 129
2-Метилгексановая 210 98 85
а-Бром-н-масляная 217 (разл.) 98 92 49
4-Метнлгексановая 218 77
Гептановая 224 71 80 72
а-Этил-н-капроновая 224
а-Бромнзовалериаиовая 230
Гексагндробензойная 232 142
Днбромуксусная 234
н-Каприловая 236 57 70 67
а-Бром-н-капроновая 240
Левулиновая 250 (разл.) 102 109 61 84
Пеларгоновая 253 57 84 68
а-Феннлпропноновая 265
Каприновая 270 62 78 67
Ундекановая 275 (разл.) 71 80 68
Р-Феннлпропионовая 280 92 36
Ундециленовая 295
Таблицы производных
599
Карбоновые кислоты (твердые)
Кислота Т. пл., °C Т. пл. производного, °C
анилид амид п-нитро- бензи- ловый эфир п-б ром- фен аци- ловый эфир
Олеиновая 16 41 76 40
Метакриловая 16 87 106
rf.Z-Молочная 18 59 74 112
Р-Бромизомасляная 22
Тнобензойная 24
а-Бромпропионовая 24 99 123
Ундециленовая 24 85
Ундекановая 29 71 103 68
Г ексагндробензойная 30 142 186
Каприновая 30 62 108 67
Фторуксусная 32 108
Левулиновая 33 102 108 61 84
Пивалнновая 35 129 154 76
сс-Метилгидрокоричная 37 109
2-Фенилмасляная 42 86
Р-Хлорпропионовая 42 101
Лауриновая 43 76 98 76
Тридекановая 44 80 100 75
а-Бромизовалериановая 44 133
Ангеликовая 46 126 128
Дибромуксусная 48 156
Р-Фенилпропионовая (гидро- 48 92 82 36 104
коричная)
а-Бромизомасляная 49 148
Бромуксусная 50 131 91 88
Элаидиновая 51 93 65
у-Фенилмасляная 52 84
Миристиновая 54 84 102 81
Трихлоруксусная 57 94 141 80
Маргариновая 60 106 49 83
Р-Бромпропионовая 62
Пальмитиновая 62 90 106 42 86
Хлоруксусная 63 134 120 105
Тиглнновая 64 77 76 64 68
а,Р-Дибромпропионовая 64 130
Цнануксусная 66 198 123
Хаульмугровая 68 89 106
Стеариновая 69 93 108 90
Кротоновая (транс) 72 118 160 67 96
Фенил уксусная 76 117 154 65 89
Гликолевая 79 96 120 107 142
а-Оксиизомасляная 79 136 98 80
Р-Иодпропионовая 82 101
Иодуксусная 84 143 95
а-Бензоилмасляная 87 149
Дибензнлуксусная 89 155 128
о-Бензоилбеизойная (гидрат) 90 195 165 100
Цитраконовая 91 175 187 (разл.) 71
Фенокснуксусная 96 99 101 148
600
Приложение III
Продолжение
Т. пл. производного, °C
Кислота Т. пл., °C анилнд амид п-иитро- беизило- вый эфир п-бром- феи аци- ловый эфир
Фталальдегидная 97
Глутаровая 97 224 174 69 137
Гомовератровая (безв.) 99 147
/-Яблочная 100 197 102 (моно) 124 179
156 (ди)
Лимонная (гидрат) 100 199 210 102 148
о-Метокснбензойная 100 131 128 113
Щавелевая (гидрат) 101 257 419 (разл.) 204 242 (разл.)
о-Толунловая 102 125 142 91 57
Пимелиновая 105 155 137
Азеланновая 106 185 175 44 131
л!-Толунловая ПО 125 97 86 108
Этнлмалоиовая 111 150 214 75
Метилянтарная 115 200 165 (моно)
225 (дн)
В-Бензоилпропионовая 116 150 125
п-Изопропилбензойная 116 133
(/./-Троповая 117 169
Бензилмалоиовая 117 (разл.) 217 225 119
d,/-Миндальная 118 151 133 124
льНнтрофеиилуксусная 120 ПО
Бензойная 121 160 128 89 119
Т рнхлормолочная 124 164 145
Аконитовая 125 170 (разл.)
(моно)
200 (разл.)
Диэтилмалоновая 125 224 (ди) 91 (ди)
З-Нитросалициловая (гидрат) 125 145
о-Бензоилбензойная 126 195 165 100
у-Бензоилмасляная 127
2,4-Диметилбензойиая 127 179
Малеиновая 130 187 153 89 168
о-(n-Толунл)бензойная 130
(/./-Яблочная 131 163 (ди)
2,5-Днметилбеизойная 132 186
Фурановая (пнрослизевая) 133 123 141 133 138
Себациновая 133 198 210 72 147
d- или /-Миндальная 133 122
Коричная 133 153 147 116 145
а-Нафтилуксусная 133 155 181
Малоновая 133 (разл.) 224 170 85
Ацетоиднкарбоновая 135 (разл.) 135
Ацетилсалициловая 135 136 138 90
Метнлмалоновая 135 (разл.) 182 217 75
Феннлпропиоловая 136 125 102 83
Глутаконовая (цис) 136 135 (моно)
Глутаконовая (транс) 138 167 (моио)
228 (дн)
Таблицы производных
601
Продолжение
Кислота Т. пл., °C Т. пл. производного, ®С
анилнд амид п-ннтро- бе кай- ловый эфир п-бром- фенаци- ловый эфир
2,6-Дихлорбензойная 139 202
о -X ло рбензойна я 140 114 139 106 106
л-Нитробензойная 140 153 142 142 132
лезо-Винная 140 190
Пробковая 140 187 216 85 144
Фурилакриловая 141 168
о-Нитрофенилуксусиая 141 161
Р-Нафтилуксусная 142 200
З-Ннтросалиц иловая 144 145
Днфенилуксусная 145 180 167
о-Нитробензойная 146 155 174 112 107
Фталоиовая (безв.) 146 176 (моно) 0 179
208 (ди) Р 155
о- (п-Т ол нл) бензойна я 146 176
д-Оксифенилуксусная 148 167
о-Бромбензойная 150 141 155 ПО
Бензиловая 150 175 154 99 152
Адипиновая 152 235 220 106 154
п-Ннтрофенилуксусная 152 198 207
2,5-Дихлорбензойная 153 155
Лимонная 153 210 (разл.) 102 148
л-Бромбензойная 155 146 155 105
2,4,6-Триметил бензойная 155 188 140
Салициловая 157 134 139 96
Хлорбензойная 158 122 134 107 116
2,4-Днхлорбензойная 160 ПО
о-Иодбензойная 162 142 184 111
а-Нафтойная 162 161 205 135
Тиосалициловая 163
2,3-Днхлорбензойная 164 130
3,4-Диметилбензойная 164
4-Нитрофталевая 165 192 200 (разл.) 117
Итаконовая 165 (разл.) 190 192 (ди) 91
Мезитиленовая 166 133
Трикарбаллиловая 166 252 207 (разл.) 138
d,/-Дибромянтарная 167 222 (ди) 211 (ди)
с/,1-Фенилянтарная 167 216
d- или /-Винная 169 180 195 163
3,5-Динитросалицнловая 173 181 197 153
п-Толунловая 177 140 158 104
2,4-Крез отовая 178 294 (разл.) 162
Ацетилендикарбоиовая 179 166
Вератровая (безв.) 181 164
л-Фторбензойная 182 131 154
4-Хлор-З-нитробензойная 182 156 142 158
2,4-Динитробензойная 183 168 203
Анисовая 184 162 132 152
Р-Нафтойная 185 173 195
602
Приложение III
Продолжение
Кислота Т. пл., °C Т. пл. производного, °C
аннлид амид п-ннтро- бензн- левый эфир п-бром- фенаци- ловый эфир
Ацетилаитраниловая 185 167 171
d-Камфарная 187 203 192 66
Гиппуровая 187 208 183 136 151
л-Иодбензойная 187 186 121 128
Янтарная 188 226 242 88 211
3-Нитроанисовая 190 163
Аконитовая 191 250 (разл.) 76 186
Диметилм алоновая 193 (возг.) 269 (ди) 84
Протокатехоловая 194 (разл.) 166 212 188
л-Нитрокоричная 199 196 174 173
2-Хлор-3,5-динитробензойная 199
л-Окси бензойная 201 155 170 106 176
3,5-Дииитробензойная 202 234 183 157
Мезаконовая 202 185 176 134
d.Z-Винная 204 226 147
и-Кумариновая 206 194
Фталевая 206 (разл.) 169 149 155 153
о-Кумариновая 207 209 (разл.) 152
Ванилиновая 207 140
и-Оксибеизойная 213 202 162 198 191
Р-Резорциловая 213 127 222 . 189
Слизевая 213 (разл.) 310 225
и-Цианбензойная 214 179 189
Пипериновая 216 145
З-Нитрофталевая 218 234 201 189
2,4,6-Тринитробензойная 220 (разл.) 264 (разл.)
2-Окси-З-нафтойиая 222 249 218
1 -Окси-2-нафтойная 226 218
5-Нитросалициловая 227 224 225
Метилим ииодиуксусная 227 (разл.) 169
Дифеновая 228 230 212 183
Пиперониловая 229 169
о-Нитрокоричиая 240 185 132 141
Галловая 240 (разл.) 207 245
Мезаконовая 241 186 (ди) 177 (ди) 134
и-Нитробензойная 241 217 201 168 137
и-Хлорбензойиая 242 194 179 129
Тетрахлорфталевая 250 (разл.) 180
п-Бромбензойиая 251 197 189 139
Хелидоновая 262
л-Иодбеизойиая 265 210 217 141 147
п-Нитрокоричная 285 204 186
Меллитовая 288 (разл.)
Изофталевая 300 280 215 186
Терефталевая 300 (возг.) 337 263 225
Фумаровая 302 314 266 (разл.) (ди) 365 (разл.) 151
Тримезиновая 350 120 (разл.) 197
Таблицы производных
603
Спирты (жидкие)
Спирт Т, кнп., °C Т. пл. производного, °C
а-нафтил- уретан фенил- уретан 3,5’дннитро- бензоат
Метиловый (метаиол) 66 124 47 107
Этиловый (этанол) 78 79 52 93
Изопропиловый 83 106 88 122
трет-Бутиловый 83 101 136 142
Бутен-З-ол-2 96
Аллиловый 97 109 70 48
н-Пропиловый 97 80 51 74
втор-Бутиловый 99 97 65 75
трет-Пентиловый 102 71 42 117
Изобутиловый 108 104 86 86
Метилизопропилкарбинол 113 112 68 76
н- Бутиловый 116 71 63 64
Диэтилкарбинол 116 71 48 97
Иеитанол-2 119 76 61
2 -Метилпентанол-2 121 239 72
Этиленгликоль, монометило- 125 113
вый эфир 1 -Хлорпропаиол-2 127 83
З-Метилпентанол-З 128 50
втор-Бутилкарбинол 128 97 51 62
Этилеихлоргидрин 129 101 92
Изопентилов ый 130 67 55 62
4 -Метилпентанол-2 131 88 143 65
2-Хлорпропанол-1 132 76
Этиленгликоль, моиоэтиловый 135 67
эфир Гексанол-3 135 46 77
н-Пентиловый 138 68 46
Циклопеитанол 140 118 132 115
Триэтилкарбинол 142
Ацетоин 145
Оксиацетон (ацетол) 146 51
2-Метилпентанол-1 148 76
З-Этилбутанол-1 149 76 51
2-Бромэтанол 150 86
Гептанол-3 156 64
Ди-н-пропил карбинол 156 80 42
н-Гексиловый 156 59 58
Циклогексанол 160 128 82 112
Гептанол-2 160 54 38 49
Триметиленхлоргидрин 161 (разл.) 76 77
2-Метилциклогексаиол 165 155 103 80
Фурфуриловый 170 129 45
Этиленгликоль, моно-я-бути- 171
левый эфир Пинакои 172 215 130
4-Метилциклогексанол 174 160 125
3 - Мети лци клогекса нол 175 122 96
604
Приложение П/
Продолжение
т. пл. производного, °C
Спирт Т. кип., °C а-иафтил- фенил- 3.5-дни нтро-
уретан уретан беизоат
Диизобутилкарбииол 175
«-Гептиловый 176 62 68 47
1,3-Дихлорпропаиол-2 176 115 73 129
Триметиленбромгидрии 176 (разл.) 73
2-Метилпропандиол-1,2 178 141
Тетрагидрофурфуриловый 178 61 84
Окта иол-2 179 63 114 32
2,2-Дибромэтанол 181
Циклогексилкарбииол 182 110 96
2,3-Дихлорпропанол-1 182 93 73
Бутандиол-2,3 183 201 (ди)
2-Этилгексанол-1 184 61 34
Пропиленгликоль 188 153
Бутироин 190
«-Октиловый 192 66 74 61
Диэтиленгликоль, мономети- 193
ловый эфир Нонанол-5 194
2-Метилпентаидиол-2,4 196
Этиленгликоль 197 176 157 169
Линалоол 197 53 65
Нонанол-2 198 56 43
Днэтиленгликоль, моиоэтило- 202
вый эфир Метилфенилкарбииол 203 106 94 95
Бензиловый 205 134 78 112
Бутандиол-1,3 208 184
Деканол-2 211 69 44
З-Хлорпропандиол-1,2 215 (разл.)
«-Нониловый 215 65 62 52
Триметиленгликоль 216 164 137 164
Бензилдиметилкарбииол 216
л-Толилкарбинол 217 116
Метил-п-толилкарбинол 219 96 108
Р-Феиилэтиловый 219 119 79
2,3-Дибромпропанол-1 219 77 (а, р-дибром-
пропионовая кислота 64°С)
Этилфенилкарбинол 219 102
1,3-Дибромпропанол-2 219 (разл.) 84
й,1-а-Терпинеол 221 152 113 79
Цитронеллол 222 (Р-метиладипи-
новая кис-
лота, 89 °C)
Изопропилфенилкарбинол 224 117 62
Гераниол 229 47
Диэтилеигликоль, моно-н-бу- 231
тиловый эфир
Таблицы производных
605
Продолжение
Спирт Т. кип., °C Т. пл. производного, °C
а-нафтил- уретан феннл- уретан 3,5-ди нитро- бензоат
н-Дециловый 231 71 60 57
З-Фенилпропаиол 237 147 (ди) 45 92
Пента метиленгликоль 238 164 (ди)
Ундециловый 243 62 149
Диэтиленгликоль 245
Этиленгликоль, моиофенило- 245
вый эфир о-Метоксибензиловый 247 136
Коричный 250 114 90 121
Глицерин 290 (разл.) 191 180 (трибензоат, 76 °C) 141
Бензгидрол 297 136 140
Спирты (твердые)
Спирт Т. пл., “С Т. пл. производных, °C
а-нафтил- уретан феннл- уретан различные
Ацетоин 15 Фенилозазон 243 (разл.)
Циклогексаиол 16 128 82 3,5-Динитробеизоат 112
Т рихлорэтиловый 19 120 87
Лауриловый 24 80 74 3,5-Динитробензоат 60
п-Метоксибенз иловый л-Нитробенз иловый 25 27 94 Анисовая кислота 184
Г ексадиеи-2,4-ол-1 31 79 3,5-Динитробензоат 85
Коричный 33 114 90 3,5-Динитробензоат 121
Пинакон 35 215 спльи-Тетраметилдн- хлорэтаи 160
а-Терпинеол ф-Т олилкарбинол 35 36 147 112 79 3,5-Дииитробензоат 78
Миристиловый 39 71 4'-Иодбифенилуретан 146
d,/-Фенхиловый 39 149 104 и-Нитробеизоат 109
(—) -Ментол Пинаконгидрат 42 46 128 111 Бензоат 55
Цетиловый 50 82 73 3,5-Динитробензоат 66
Неопентиловый Пиперониловый п-Толилкарбинол 1,2-Дифенилэтанол 53 58 60 67 100 144 102
Бензгидрол Эритрит о-Нитробензиловый 69 72 74 136 140 Бензоат 88
606
Приложение III
Продолжение
Спирт Т. пл., -с Т. пл. производных, °C
а-нафтил- уретан фенил- уретан различные
2,2,2-Трибромэтиловый 80
Фенациловый 86 и-Нитробеизоат 129
о-Оксибензиловый 87
п-Нитробензиловый 93
(+)-Сорбит 98 Ацетат 99
Терпин 102 Гидрохлорид 50
Терпингидрат 117 Гидрохлорид 50
Бензоин 133 140 165 (см. Кетоны)
Ситостерин 137 Ацетат 127
(—)-Холестерин 148 160 168 Бензоат 150
Т рифеиилкарбииол 162 Трифенилметан 92
Эргостерин 165 Ацетат 180
(+) -Маннит 166 303 Гексаацетат 120
Бензпииакон 186
Дульцит 189
(+)-Борнеол 208 132 138 п-Нитробензоат 137
Инозит 225 Гексаацетат 216
Пентаэритрит 253 Тетраацетат 84
Альдегиды (жидкие)
Альдегид Т. кип., вС Т. пл. производных, °C
оксим I сеиикарбазон фенилгидразон 2,4-дииитрофе- нилгидразон п-нитрофенил- гидразои
Формальдегид —21 Жидк. 169 Жидк. 166 182
Ацетальдегид 21 47 162 63 147 129
99 168
Пропионовый 50 40 89 Жидк. 154 124
154
Глиоксаль 50 178 270 180 328 311
Акролеин 52 171 52 165
(пиразолин)
Изомасляный 64 125 Жидк. 182 132
а-Метилакролеии 73 198 74 206
(пиразолин)
н-Масляный 74 104 Жидк. 122 92
Пивалиновый 75 41 190 209 119
Изовалериановый 92 48 107 Жидк. 123 101
Таблицы производных
607
Продолжение
Альдегид Т. кип., °C Т. пл. производных, °C
оксим в э и в о О. ч S S У фенилгидразон 2,4-динитрофе- нилгидразон п-нитрофенил- гидразон
а-Метил-я-масляный 93
Хлораль 98
я-Валериановый 103
Кротоновый 103
а-Этил-я-масляный 116
Паральдегид 124
я-Капроиовый 128
я-Гептаиовый 156
Фурфураль 161
Гексагидробеизойный 162
а-Этил-я-капроновый 163
1,2,3,6-Тетрагидробензойный 165
Каприловый 171
Бромаль 174
Бензойный 179
Пеларгоновцй 185
5-Метилфурфураль 187
Фенилуксусный 194
Салициловый 196
л-Толуиловый 199
о-Толуиловый 200
я-Толуиловый 204
Цитронеллаль 206
о-Хлорбензойный 208
л«-Хлорбензрйиый 208
Каприновый 209
Феноксиуксусный 215
(разл.)
фксикоричный 224
Цитраль 228
(разл.)
л-Метоксибеизойиый 230
л-Бромбеизойиый 236
Анисовый (п-метоксибензой- 247
ный)
Коричный 252
Вератровый 285
103 120
56 131
52 106
119 199 56 190 184
96 134
51 106 104
57 109 108 73
89 202 97 229
74
91 173
254 121
(разл.)
76 154 163
60 101 106 80
115
35 222 158 237
64 100 100
син 112 211 148 212
анти 52
103 156 58 121
57 231 142 252
(разл.)
60 213 84 211
49 208 101 195
79 221 114 239
НО
Жидк. 82 77
75 225 86 207
70 228 134 256
69 104
95 145 86
94 127 149
Жидк. 164 116
233 76 171
(разл.)
72 205 141
а 45 210 120 254
а' 64 (разл.)
Р 133
138 215 168 255
(разл.)
95 177 121 265
608
Приложение 111
Альдегиды (твердые)
Альдегид Т. пл., 'С Т. кип., °C Т. пл. производного, °C
оксим семи- карбазон фенил- гндразон 2,4-цини- трофенил- гндразон
Пальмитиновый 34 88 109 108
а-Нафтойный 34 292 90 221 80
Фенилуксусный 34 195 99 156 63 121
о- Иод бензойный 37 206 108 206 79
Пиперональ 37 263 НО 230 100 266
(разл.)
о-Метоксибензойный 38 245 92 215 253
Стеариновый 38 89 119 НО
о-Аминобензойиый 40 135 221
о-Нитробеизойиый 44 102 256 156 250
(разл.)
Лауриновый 45 238 78 106 106
п-Хлорбензойный 47 214 110 230 127 270
Хлоральгидрат 53 56 90 131
(разл.)
2,3-Диметоксибеизойиый 54 285 99 231 138 264
(разл.)
Фталевый 56 191
п-Бромбеизойиый 57 с ин 157 228 113
анти 111
л-Иодбензойиый 57 62 226 455
л-Нитробензойный 58 120 246 124 293
(разл.)
Вератровый 60 285 95 177 121 265
Р-Нафтойиый 60 156 245 206 270
(разл.)
2,4-Диметоксибеизойный 69 106
2,4-Дихлорбеизойный 71 136
Ваиилин 80 285 117 229 105 271
(разл.) (разл.)
Феиилглиоксаль Гидрат а 128 а 217 152
91 Ди 168 (разл.) (ди)
льОксибензойный 105 88 199 131 260
(разл.)
и-Нитробеизойиый 106 129 221 159 320
(разл.)
Метальдегид 115
п-Оксибензойный 115 72 224 177 280
(разл.)
Терефталевый 116 245 200 154 278
(ди)
^./-Глицериновый 142 160 132 167
(разл.) (Ди)
3,4-Диоксибеизойный 154 157 230 175 275
(протопирокатехии) (разл.) (разл.)
3,5-Диоксибензойиый 157 223
Таблицы производных
609
Амиды (жидкие)
Амид Т. кнп„ °C Амид Т. кип., ’С
N.N-Диметилформамид N.N-Диэтилформамид Формамид 153 176 195 (разл.) Формилпиперидии Ацетилпиперидин 222 226
Амиды (твердые)
Амид Т. пл., °C Амнд Т. пл., ’С
Олеанилид 41 о-Нитроацетанилид 92
Форманилид 46 а-Фенилпропиоиамид 92
Бензоилпиперидии 48 я-Гешамид 94
Этилкарбамат 49 4-Метил-2-нитроацетанилид 94
Малонамид 50 (моно) М-Метил-N-a нафтилацет- 95
N-я-Пропилацетаиилид 50 амид
Дифторацетамид 52 Анилид я-масляной кисло- 95
Метилкарбамат 52 ты (бутиранилнд)
Фенилкарбамат 53 Иодоацетамид 95
Ацетоацетамид 54 Семикарбазид 96
я-Бутилкарбамат 54 Метоксиацетамид 97
N-Этилацетаиилид 54 Стеараиилид 97
Изобутилкарбамат 55 Т рихлорацетанилид 97
N-Метил-о-ацетотолуид 56 я-Ацетокси-N-метилацет- 98
Метоксиацетаиилид 58 анилид
я-Пропилкарбамат 60 a.a-Дихлорацетамид 98
Этилгиппурат 60 Г идроциннамаиилид 98
я-Валераиилид 63 N.N-Дифенил ацетамид 101
Изоамнлкарбамат 64 Метилмочевина 101
л-Ацетотолуид 65 я-Капроамид 101
Этилоксанилат 66 N-Метилацетанилид 102
Гептананилид 70 Пропионаиилид 103
N.N-Дифеиилформамид 73 Пируванилид 104
В инила цетанилид 73 Акриланилид 105
Олеамид 76 Г идроциннамамид 105
Пропионамид 79 Изобутиранилид 105
а,Р-Диэтилкарбанилид 79 Р-Феиилпропионамид 105
Аллилмочевина 80 сплл-Диметилмочевина 106
й,/-а-Хлорпропионамид 80 Пальмитамид 106
Ацетамид 82 Хаульмуграмид 106
Этоксиацетамид 82 Фторацетамид 108
N -Метил-п-ацетотолуид 83 Стеарамид 109
Ацетоацетаиилид 85 Изовалераиилид 109
Акриламид 85 о-Ацетотолуид 112
Аллилмочевина 85 Ацетанилид 114
о-Хлорацетанилид 88 о-Хлорбеизанилид 114
я-Бутилоксамат 88 Этилоксамат 114
d-Хаульмугранилид 89 я-Бутирамид (амид я-мас- 115
Бромацетамид 91 ляной кислоты)
Изопропилкарбамат 92 Метакриламид 116
20 Зак. 335
610
Приложение III
Продолжение
Амнд Т. пл.. ®С Амнд Т. пл.. ’С
2-Бром-4-метилацетанилид 117 Гексагидробензанилид 146
а-Феиилацетанилид 117 Фенилмочевина 147
Дихлорацетанилид 118 Дифенилгуанидин 147
N-Этил-п-иитроацетанилид 118 4-Метил-З-нитроацетаиилид 148
а-Хлорацетамид 119 Сукцинанилид (аиилид яи- 148 (моно)
«,Р-Диметилкарбаиилид 121 тарной кислоты)
о-Толуанилид 125 Беизилмочевина 149
Сукцинимид (имид яитар- 126 Нитромочевина 150 (разл.)
ной кислоты) п-Ацетотолуид 153
«-Бутилэтилбарбитуровая 12В N-Метил-п-иитроацетани- 153
кислота лид
л«-Толуанилид 129 л-Нитробеизаиилид 153
Этил-н-гексилбарбитуро- 126 Циниаманилид 153
вая кислота Этилизоамилбарбитуровая 154
п-Метоксиацетанилид 127 кислота
а-Ацетил-Р-фенилгидразии 128 и-Фторбензамид 154
Бензамид 128 а-Феиилацетамид 154
Изобутирамид 128 Бензиламид 155
о-Метоксибензамид 129 о-Бромбеизамид 155
Пиперин 129 .и-Бромбензамид 155
Аднпамид 130 2,5-Дихлорбензамид 155
3,4-Диметилбензамид 130 а-Нафтилацетанилид 155
Бромацетанилид 131 л-Нитроацетаиилид 155
о-Метоксибензанилид 131 Дибромацетамид 156
Малонаиилид 132 (моно) N-Фенилсукцинимид ’ 156
Мочевина (карбамид) 132 Сукцинамид 157 (моно)
2,4-Диметилацетанилид 133 п-Бензтолуид 158
п-Изопропилбензам ид 133 N-а-Нафтилацетамид 159
Мезитиленамид 133 и-Толуамид 1§0
Хлорацетаиилид 134 Бензанилид 161
л-Хлорбензамид 134 N-a-Нафтилбеизамид 161
N-0-Нафтилацетамид 134 п-Аминоацетанилид 162
Фенацетин 13В а-Нафтаиилид 163
л-Бромбензаиилид 136 п-Бромацетаиилид 167
л-Этоксибензамид 139 а-Бензоил-р-фенилгидра- 168
Салициламид 139 ЗИН
а,а,а-Трихлорацетамид 14Q Дифенилацетамид 168
о-Толуамид 140 и-Оксиацетанилид 169
о-Иодбензанилид 141 Аллоксаи 170 (разл.)
о-Бромбензаиилид 141 и-Этоксибензанилид 170
л-Толилмочевина 14? Малеамид 170
Фурамид 142 Малонамид 170
Циниамамид (амид корич- 142 Р-Нафтанилид 171
ной кислоты) Этилфенилбарбитуровая 172
о-Беизотолуид 142 кислота
л-Нитробеизамид 143 Диаллилбарбитуровая кис- 172
Иодацетаиилид 144 лота
и-Толуанилид 145 п-Фенетилмочевина 174
а-Трифенилгуаиидин 145 Малеанилид 175
а-Бромизовалерилмочевина 14В о-Нитробензамид 176
Циклогексанкарбоксанилид 146 п-Хлорацетанилид 179
Д иэти л м а л он а м ид 146 (моно) а-Ацетил-р-метилмочевина 180
Таблицы производных
611
Продолжение
Амид Т. пл., °C Амид Т. пл., “С
Дифеиилацетанилид 180 й,/-Фенилсукцинамид 211 (ди)
а-Нафтилацетамид 181 п-Иодбензамид 217
п-Толилмочевина 181 Ацетилмочевина 218
Диметилмочевииа (несимм) 182 симм- Ди-л-толилмочевина 218
п-Иодацетанилид 182 Гидантоин 218
Тиомочевина 182 Фталамид 219 (разл.)
3,5-Динитробензамид 183 Адипамид 220 (ди)
о-Иодбензамид 183 Сахарин 220
М,!\1'-Диацетил-о-фенилен- 185 d.Z-Фенилсукцинанилид 222 (ди)
диамин Диэтилмалонамид 224 (ди)
Циклогексанкарбоксамид 186 Малонаиилид 225 (ди)
Гексагидробензамид 186 Сукцинанилид 226
л-Иодбеизамид 186 Нитрогуанидин 230 (разл.)
Гиппуровая кислота 187 Фталимид 233
Диэтилбарбитуровая кис- 188 Кофеин 234
лота 3,5-Динитробензанилид 234
и-Бромбензамид 189 Терефталанилид 237
Дифенилмочевииа (не- симм) 189 Карбанилид (сгдил-дифе- нилмочевина) 238
Ы.М'-Диацетил-.и-фенилен- 191 п-Окси-М-метилацетанилид 240
диамин Барбитуровая кислота 245
Р-Нафтамид 192 Сукцинамид 250
о-Т олилмочевина 192 саш<-Ди-о-толилмочевииа 250
Биурет 192 (разл.) Оксанилид 257
п-Хлорбензанилид 194 Теофиллин 264
2-Метил-4-иитроацетанилид 196 2,4,6-Т ринитробеизамид 264 (разл.)
Изатин 200 Фумарамид 266 (разл.)
Метилсукцина мид 200 (ди) снлгл-Ди-п-толилмочевина (ди)
Р-Нафтилацетамид 200 268
п-Нитробеизамид 201 симм- Динафтилмочевина 287
Этилизопропилбарбитуро- 201 Креатинин 292 (разл.)
вая кислота N.N'-Диацетил-л-фенилен- 300
п-Этоксибензамид 202 диамин
2,6-Дихлорбензамид 202 Фумаранилид 314
2,4-Динитробензамид 203 Креатин 315 (разл.)
N-Фенилфталимид 205 Теобромин 337 (возг.)
Дициандиамид 207 Ксантин 360
п-Иодбеизаиилид 210 Оксамид 419 (разл.)
п-Нитроацетаиилид 210 Мочевая кислота Разл.
и-Нитробензаиилид 211
Первичные амины (жидкие)
Амин Т. кнп., °C Т. пл. производного, °C
беязол- сульфон- амид беизамнд п-толуол сульфон- амид феиилтио- мочевина
Метиламин -6 30 80 75 113
Этиламии 19 58 71 63 106
Изопропиламии 33 26 101
20*
612
Приложение III
Продолжение
Амин Т. кип., ’С Т. пл. производного, °C
бензол- сульфон- амид бензамид п-толуол- сульфон- амид фенилтно- мочевнна
трет-Бутиламин 46 134 120
н-Пропиламин 49 36 84 52 63
Аллиламии 56 39 64 98
етор-Бутиламин 63 70 76 55 101
Изобутиламин 69 53 57 78 82
н-Бутиламин 77 42 65
Изопентиламин 95 65 102
н-Пентиламин 104 69
Первичные амины (жидкие)
Т. пл. производного, °C
Амнн Т. кип., •с беизол- сульфои- амнд ацет* амид бенз- амид гг-толуол- сульфон- амид феннл тиомо чевина
Этилендиамин 116 168 172 249 160 102
1,2-Диамииопропан 120 139 192 103
н- Гексилам ин 128 96 40 77
Циклогексиламин 134 89 104 149 148
1,3-Диаминопропан 136 96 126 147 148
н-Гептиламин 155 75
1,4-Диамииобутан 160 168
2-Оксиэтиламин 171 135
1,5-Диаминопентан 178 119 148
н-Октиламин 180
Анилин 183 112 114 160 103 154
Бензиламин 184 88 60 105 116 156
а-Фенилэтиламин 185 57 120
п-Фторанилин 188 152 185
Р-Фенилэтиламин 198 69 114 116 135
о-Толуидин 199 124 112 143 108 136
ж-Толуидин 203 95 65 125 114 94
1-Ментиламин 205 145 156 135
о-Хлоранилии 207 129 87 99 105 156
4-Амино-1,3-диметилбензол 212 128 133 192 133
2-Амино-1,4-днметилбензол 215 138 139 140 119 148
«-Этиланилин 215
2-Амино-1,3-димет илбеизол 216 176 168 204
о-Этиланилии 216 111 147
п-Этилаиилин 216 94 151 104
о-Амиио-Ь1,М-диметилаиилин 217
5-Амиио-1,3-диметилбензол 220 136 144 153
2,3-Диметиланилии 222 136 189
о-Броманилин 229 99 116 146
о-Феиетидии 229 102 79 104 137
Таблицы производных
613
Продолжение
Амии Т. кип., °C Т. пл. производного, °C
бензол- сульфон- амид ацет- амид беиз- амнд п-толуол- сульфон- амид фенил- тиомо- чевнна
ж-Хлоранилин 230 121 72 120 124
4-Амино-З-бромтолуол 240 117 149 154
5-Хлор-2-амииотолуол 241 140
2-Амино-4-изопропилтолуол 242 71 102
Фенил гидразин 243 148 128 168 151 172
о-Амино-Ы-метиланилин 245
ж-Фенетидин 248 97 103 157 138
ж-Анизидин 251 81 68
ж-Бромаиилин 251 87 136 143
п-Фенетидин 254 143 135 173 106 136
З-Бром-2-метиланилин 255 163 177
Изодуридии 255 211
З-Бром-4-метиланилин 257
п-Амино-К-метиланилин 258 165
Метилантранилат 260 (разл.) 107 101 100
п-Амиио-М,М-диэтиланилин 262
Этилантранилат 265 (разл.) 92 61 98
аг-Тетрагидро-а-нафтиламни 275 158
аг-Тетрагидро-р-нафтиламин 275 107
Этил-ж-аминобензоат 294 ПО 148
Первичные амины (твердые)
Т. пл. производного, °C
Амин пл., °C кнп., “С >.4 о а и ® а) о X Ч ч 2 ss <У о ч X 2 я со X толуол- •льфонамнд h X У
н н а к КО ё о •&S
Этилантранилат 13 265 (разл.) 92 61 98
2-Амино-1,4-диметилбеизол 15 215 138 139 140 119
ж-Бромаиилин 18 251 87 136 97
Фенилгидразин 19 243 148 128 168 151 172
Метилантранилат 24 260 (разл.) 107 101 100
Изодуридин 24 255 211
4-Амино-З-бромтолуол 26 240 117 149 154
1,4-Диаминобутан 27 159 137 177
(ди) (ди)
ж-Иоданилин 27 119 157 128
5-Хлор-2-аминотолуол 29 241 140
З-Аминобифеннл 30 148
614
Приложение III
Продолжение
Т. пл. производного, °C
Амин О о О ч 1 ч ф 1) □ S ч D X ч X S ч * ° о О га X и
Ч С X X и и я "О Й о а т X ч^ Ч и X о X sr
Ь Ь ч ю е о
о-Бромаиилин 31 250 99 116 146
несилш-Дифенилгидразин 34 184 192
и-Амино-М-метиланилин 36 258 63 165
аг-Тетрагидро-0-нафтиламии 38 275 107
5-Хлор-4-амино-1,2-диметил- 40 148
бензол
п-Амино-N.N-диметилаиилии 41 262 130 228
2-Амино-6-метилпиридии 41 90 90
1,6-Д иа миногексан 42 154 155
2,4'-Диаминобифенил (ли) (Ди)
45 363 20 2 278
и-Толуидии 45 200 120 153 158 117 141
2-Аминобифенил 45 299 119 86
4-Амино-1,2-диметилбензол 49 226 118 99 154
2,5-Дихлоранилии 50 251 132 120
а-Нафтиламин 50 300 167 159 160 157 165
2-Амино-3,5-дихлортолуол 50 186
4-Аминобифенил 53 302 171 230
2-Аминопиридин 56 165
о-Иоданилии 56 109 139
н-Бутил-л-аминобензоат 56
л-Анизидин 57 243 95 127 154 114 154
2-Амиио-5-бромтолуол 59 156
4-Амино-3,5-дихлортолуол 60
2,3-Диаминотолуол 61 255
и-Иоданилин 62 183 222 153
л-Фенилендиамин 63 283 194 191 240 172
n-AMHHO-N-метилацетанилид 63
2,4-Дихлоранилии 63 245 128 145 117 126
З-Аминопиридии 64 252 133 119
2,5-Диамииотолуол 64 120
л-Толилгидразии 65 121 146
л-Бромаиилин 66 245 134 167 204 101 148
(разл.)
1,8-Диамииоиафталин 67 136
Псевдокумидии 68 235 161 167
л-Хлоранилин 70 232 121 179 192 95 152
о-Нитроаиилин 71 104 92 94 142
2-Хлор-3,5-диаминотолуол 73 228
4-Нитромезидин 75 163 191 169
4-Аминодифениламин (безвод- 75 158 203
ный)
4-Амино-5-иитро-1,3-диметил- 76 173 185
бензол
4-Амино-2-иитротолуол 77 160 148 172 164 145
2,4,6-Трихлоранилин 77 262 204 174
3,5 -Дибром -4-а минотолуол 78 183
Таблицы производных
615
Продолжение
Амни Т. пл., *С Т. кип., °C Т. пл. производного, °C
бензолсуль- фонамнд ацетнл- про извод ное бензамид п-толуол- сульфонамид феиилтио- мочевина
2,4-Диброманилин 79 146 134 134
2,4-Диаминофенол 79
(разл.)
2-Амииодифениламин 80 91
З-Амино-6-хлортолуол 83 241
2,6-Диброманилин 83 210 178
2,4-Диаминохлорбеизол 88 , 170 1 (моио) 215 (ди)
1 243
89 < (ди)
Этил-п-аминобеизоат НО 148
3,4-Диаминотолуол 89 265 157 167
2-Метил-6-нитроанилин 91
З-Амииохинолин 94 172
п,п'-Д иа м ино дифен и л мета и 2-Метил-З-нитроанилин 94 228
95 158 192
6-Хлор-2,4-диброманилин 95 227
1,2-Диаминонафталин 95 234 291
2,4-Дииодаиилин 96 181
Семикарбазид 96 194 165 225
8-Нитро-1-иафтиламии 97 191
2-Амиио-4-метилпиридин 98 103 114
4-Хлор-1 -нафтиламин 98 184
2,4-Диаминотолуол 99 280 178 224 224 192
о-Фенилеидиамии 102 256 185 185 301 201
2,6-Диаминотолуол 105 203
(ДИ) 205
n-Аминоацето фенон 106 295 128 167 203
п-Бромфенилгидразин 106 172 150
2-Амино-4-нитротолуол 107 148
N-Фенил-Р-нафтиламин 108 93
р-Нафтиламин 112 300 102 132 162 133 129
л-Нитроаиилии 114 284 136 155 155 138 160
4-Бром-8-нитро-1 -нафтиламии 116 102 202
4-Амино-З-иитротолуол 116 96 148 166
2,4,6-Трибромаиилин 119 300 ( 127 198
J (ди) 1 232
'(моио)
5-Нитро-1 -нафтиламин 119 183 220 280
1,4-Диаминонафталин 120 303 156
л-Аминофеиол 122 . 101 J (Ди) S 148 153 157
(.(моно)
4-Амиио-6-нитро-1,3-диметил- 123 149 159 200
бензол
616
Приложение III
Продолжение
Амин Т. пл., °C Т. кип., “С Т. пл. производного, °C
бензолсуль- фонамид ацетил- производное ; бензамид п-толуол- сульфояамид феиилтио- мочевина
п-Аминобензофенон 124 153 152
п-Аминоазобензол 125 360 144
1-Нитро-2-нафтиламин 126 156 124 159
Бензидин 127 400 232 317 352 243
о-Толидин 129 314 265
2-Амино-5-нитротолуол 130 158 198 174 174
З-Амйно-6-нитротолуол 135 102
Дианизидин 135 242 236
и-Ацетофенетидин 138
2,6-Динитроанилин 138 197
л-Фенилеидиамин 140 267 247 3Q4 300 266
2-Амино-5-нит’ро- 1,4-диметил- 142 162 166
бензол
2-Амино-5-нитронафталин 144 186 182
1 -Амино-2-нитронафталин 144 199 175
1 -Аминофеиантрен 146 220
и-Ннтроанилин 147 139 210 199 191
4-Аминохинолир 154 178 193 ,
и-Нитрофенилгидразин 157 . 205
(разд.)
и-Амииоацетанилид 162 304
4'Амино-3,5-динитротолуол 166
2,7-Диаминоиафталин 166 261 267
(ди) (Ди)
Цикрамовад кислота 4-Амино-2,6-динитротолуол 5-Амино-2-окситолуол 16§ 18 201 ( 103 220 194
J (ди)
] 179 ((моно)
{^Дмицофенол 174 141 201 182 139 146
£4-Динитроадилин 180 120 202 219
п-Амйнофенол 184 125 ( 150 234 150
(разл.) J (ди)
1 168 ((моно)
2,4,6-Тринитроанилин (пикр- 190 211 230 196
амид) 1 ,б-Дннитро-2-нафтпламин 191 201 182
Дйнитромезидин 194 275 185
1 -Амино-4-ннтронафталин 195 158 190 224
2,4-Динитрофенилгидразин 198 (разл.) 197 312 206
4,4'-Драминостильбен 226
л-Аминоацетанилид 279 191
2-Аминоантрахинон 302 271 257 228 304
Таблицы производных
617
Вторичные амииы (жидкие)
Амии Т. кип., °C Т. пл. производного, °C
беизол- сульфбн- аМид бензамид п-толуол- сульфой- амнд фенил- тиомоче- внна
Диметиламин 7 47 41 79 135
N-Метилэтиламин Диэтил а ми и 35 35 42 42 60 34
Диизопропила мин Пирролидин Пипёридин 86 39 105 93 48 123 96 101
Вторичные амины (жидкие)
—___~ __Л:_i
Амин т.г„ -1 - . -- , Т. пл. производного, “С
бензол- сульфон- амид ацет- амид бенз- Амйд п-толуол- ЬульфБн- амйд фенил- тномоче- внна
Ди-я-пропиламии ПО 51 69
Дйаллйламин 111
d.l-2-Метнлпиперидии 116 45 55
4,/-3-МетилпиперИдин 126
Морфолин 130 118 75 147 136
Диизобутила мин 139 55 86 113
Ди-я-бутиламин 160 86
N -Метилбензил а мин 185 95
Диизоамиламйи 187 72
Метиланилин 192 79 102 63 94 87
N-Этилбензиламин 199 50
N-Этилаиилин 205 54 60 67 89
Дй-я-пентиламии 205 72
N-Метил-л-толуидии 206 66
N-Метил-о-толуидии 207 55 66 120
N -Метил-п-толу идин 208 64 83 53 6о 89
N-Изопропилаиилнн 213 39
N-Метил-о-хлораиилин 214
N-Этил-я-толуидин 217 66 40 70
N-Этил-о-толуидии 218 62 72 75
N -Этил-л-толуид ин 221 72
4-Ь1-Метиламино-1,3-диметнл- 2^1
бензол
N-Изобутиланилин 231
Тетрагидроизохинолин 233 154 46 129
N -я-Бутиланйлин 236
N-Метил-п-хлоранилии 240 92
Тетрагидрохинолйн 250 67 75
N-Изопейтиланилин 254
N-Метил-п-броманйлин 260
Ди(2-оксиэтил)амин (диэта- 268
ноламин)
618
Приложение III
Продолжение
Амин Т. кнп., °C Т. пл. производного, °C
бензол- сульфон- амид ацет- амид бенз- амид п-толуол- сульфон- амнд фенил- тномоче вина
N-Метил-а-нафтиламни N-Бензиланилин 293 298 119 95 58 121 107 103
Дибензиламии Дифениламин 300 302 68 124 101 112 180 144 152
N-Метил-Р-нафтиламин N-Этил-Р-нафтиламин N-Этил-а-нафтиламин 309 315 325 107 49 68
Вторичные амины (твердые)
Т. пл. производного, °C
Амин О о ч К О о В X х л ч й я 4 а ° я И Я о а ч Ч я х м Н X <У о ч X S л Я X толуол- льфонамнд 2 « s s £ « §8 X У
н Н О-& я к' ю ё о •&S
Тетрагидрохинолин 20 250 67 75
Ди- (2-оксиэтил) амин (диэта- 28 268
ноламин) N-Бензиланилин 37 298 119 58 107 103
о-Нитро-Ы-метиланилии 37 70
N-Метилтриброманилин 39 237 101
N -Метилпсевдоку м идии 44 128
4-(Ы-Метиламино)-2-нитро- 45
толуол Иидол 52 254 68
Дифениламин 54 302 124 101 180 141 152
4-(№Этиламиио)-3-иитротолуол 59 335 115 152 127
N-Фенил-а-иафтиламии 62
л-Нитро-N -этилаиилин 65 83 89 156
N-Метил-л-иитроанилии 66 95
N -n-Т олил-а-нафтиламин 79 330 83 124 140
Ди-п-толиламин 79 85 125
4- (N-Метиламино-З-нитро- 84 64
толуол п-Окси-N-метнланилин 85 240 (моно) 175
97
(ди) 150
о-Оксн-Ы-метнланилин 87 150 107
N-Этнл-п-нитроаиилин 96 118 98
п-Толил-р-нафтиламин 103 85 139 173
Пиперазин 104 140 282 134 191
N-Этил-2,4-динитроанилии 113 120 153 111
п-Нитро-Ы-метиланилин 152
N-Метил-2,4-диинтроанилии 176 351 69 98
Карбазол 243
Таблицы производных
619
Третичные амины (жидкие)
Амин Т. кип., "С Т. пл. аддукта, °C
с хлоро- платн- новой кислотой с метнл- п-толуол- сульфо- натом с метил- нодндом с пикри- новой кислотой
Триметиламин 3 242 230 216
(разл.) (разл.)
Триэтиламин 89 173
Пиридин 116 241 139 117 167
а-Пиколин 129 195 150 230 169
(разл.)
Р-Диметиламиноэтиловый 133 96
спирт
2,6-Диметилпиридин 143 233 168
Р-Пиколин 143 150
V-Пиколии 143 167
З-Хлорпиридин 149 135
Три-я-пропиламин 153 116
Триаллиламин 155
2,4-Лутидин 159 113 180
2,5-Диметилпиридин 160 169
Р-Диэтиламиноэтиловый спирт 161
2,3-Диметилпиридин 164 188
2-Хлорпиридин 166 120
З-Бромпиридии 170 156 165
2,4,6-Коллидин 172 155
N.N-Диметил-о-толуидии 185 193 210 122
(разл.)
N.N-Диметил бензиламин 185 192 179
Диметиланилин 193 173 161 228 163
(разл.) (разл.)
2-Бромпиридин 194 127
З-Этил-4-метилпиридин 196 205 150
(разл.)
Ы-Этил-Ы-метиланилин 201 125 134
N,N - Диэтил-о-толуидин 206 224 180
(разл.)
о-Хлор-N.N-диметиланилин 207
N.N-Диметил-я-толуидии 210 85 219 127
Три-я-бутиламин 211 186 105
N.N-Диметил-л-толуидин 2Г2 177
N.N-Диметилмезидин 213
N.N - Диэтилаиилин 218 102 142
N.N-Диметилпсевдокумидии 220
N.N-Диэтил-я-толуидин 229 184
я-Хлор-Н,Ы-диметилаиилии 230
N.N-Диэтил-л-толуидин 231 97
Хииолин 239 218 126 ( 72 203
J (гидрат)
1 133
(безв.)
Изохинолин 240 263 163 159 222
(разл.)
d, /-Никотин 242 218
620
Приложение III
Продолжение
Амин Т. кнп., °C Т. пл. аддукта. °C
с хлоро- плати- новой кислотой с метнл- п-толуол- сульфо- натом с метнл- нодидом с пикри- новой кислотой
Ь1,Ы-Ди-н-пропиланилин 245 156
Триизоамиламин 245 125
Хинальдин 247 226 161 195 191
Три-н-амиламин 257 80
(разл.)
6-Метилхинолин 258 154 219 229
3-Бром-М,Ь1-диметилаиилин 259 259
N ,N - Д и-н-бутиланилин 261 180 125
Лепидин (4-метилхинолин) 263 174 212
2,4-Диметил хинолин 264 229 194
2-Фенилпиридин 269
N.N-Дпметил-а-иафтиламии 272 145
6-Бромхинолин 278 273 217
N-Метил дифениламин 296
2,4,6-Трибром-М,М-диметил- 301
анилин
N.N-Диметил-Р-нафтиламин 305
6-Метоксихинолин 305
(разл.)
Ы-Бензил-М-метиланилии 306 164 103
И-Бензил-И-этиланилии 312 161 117
(разл.)
Третичные амины (твердые)
Амин Т. пл., °C Т. кип., "С Т. пл., аддуктов, °C
с метил- п-толуол- сульфо- иатом с метнл- иодндом с пикрн новой кнслото:
Изохинолин 24 240 163 159 222
6-Метоксихннолин 28 305
(разл.)
п-Бром-М,1Ч-диэтиланилин 34 270
N,N-Этилбензиланилин 34 120
2-Хлорхииолин 38 267 122
7-Метилхинолин 39 252 237
6-Хлорхинолин 41 262 143 248
Ы,М-Диметил-2-иафтиламин 47 206
2-Бромхииолин 49 210
8-Метоксихинолин 50 283 160 143
п-Бром-!М,М-диметиланилин 55 264 185
2,6-Диметилхинолии 60 265 175 237 178
N.N-Диметил-л-нитроанилии 60 285 205 119
N-Фенилпиррол 61 234
Таблицы производных
621
Продолжение
Амин Т. пл.. °C Т. кип.. °C Т. пл. аддуктов, “С
с метнл- п-толуол- сульфо- натом с метнл- нодндом с пикри- новой кислотой
а,а'-Дипиридил 69 158
N.N-Дибензиланилин 70 300 135 131
(разл.) (разл.)
п-Диметиламинобензальдегид 73
8-Оксихииолин 75 266 153 204
(разл.)
п-Окси-М,М-диметиланилин 76 201
1Ч,М-Диэтил-2,4-динитроани- 80
ЛИН
N.N-Диэтил-л-нитрозоаиилин 84
ж-Окси-Ы.Ы-диметиланилии 85 268 182
п-Нитрозо-М,Ь1-диметиланилин 87 140
М,М-Диметил-2,4-динитроанп- 87
ЛИИ
Трибензиламии 91 380 184 190
Акридин 108 224 208
3,5-Дибромпиридин 112 222 219 274
Антипирин 113
п- Диметиламиноазобензол 117 174
Трифениламин 127
Метиленаминоацетонитрил 129
6-Нитрохииолин 149 245
N.N-Диметил-п-нитроаиилии 163
6-Нитрохииальдин 164
п,п'-бис- (Д иметиламино) бен- 174 105 156
зофеион
6-Оксихииолин 193 236
2-Оксихинолии 199
Г ексаметилентетр амин 280 205 190
Аминокислоты 8
Аминокислота Т. разл., °C Т. пл. производного, °C
бензонл- производное фенил* мочевина п-толуол- сульфонил* производное разные
N-Фенилглиции 126 63 195
Антраниловая d.Z-Оксиглутамииовая 144 150 182 181 Ацетил 185
л-Аминобензойная 174 248 270 Ацетил 250
п-Аминобензойная ₽-Алаиин 186 196 278 300 174 Ацетил 252
D- или L-Глутаминовая 198 138 117 а-Нафтилмоче- вииа 236
622
Приложение III
Продолжение
Аминокислота Т. разл., °C Т. пл. производного, °C
бензоил- пронзводное феннл- мочевина п-толуол- сульфон ил- ii ронз водное разные
п-Амииофенилуксусная 200 198 Ацетил 170
d, /-Пролин 203 170 Пикрат 137
Саркозин 210 102 Ацетил 135
L-(-)-Пролин 222 170 133 Пикрат 154
D- или L-Лизии 224 184 Дибензоил 145
D- или L-Аспарагин 226 164 Пикрат 180 (разл.)
d,/-Глутаминовая 227 157
</./-Серин 228 171 169 213 Р-Нафталинсульфо-
нил 220
Глицин 232 187 163 147 Ацетил 206
d, /-Треонин 235 148
L-(+)-Аргинин 238 Пикрат 217 (разл.)
(/./-Аргинин 238 315 Пикрат 201
п-Оксифенилглицин 248 117 Ацетил 203
(/./-Аллотреонин 252 177
D- или L-Треонйн 253 148
(моно)
L-(-)-Цистин 260 181 117 203
(разл.)
Глицилглицин 264 176
D- или L-Аспартовая 270 185 162 140 а-Нафтилмочевииа 115
L-(—)-Оксипролии 270 175 153
D- или L-Аллотреонин 272 128
(/./-Метионин 272 151 Ацетил 114
(/./-Фенилаланин 273 188 182 135 Пикролоиат 182
L-(—)-Гистидин 277 230 204 Пикрат 86
D- или L-Изолейцин 279 117 120 132 Пикролонат 170
а-Аминоизомасляная 280
(/./-Аспартовая 280 165
L- (—) -Диоксифенилаланин 280
L-(—)-Метионин 283 а-Нафтилмочевина 186
5-Амииосалициловая 283 252 Ацетил 218
L- (+) -а-Амино-н-масляная 285 121 а-Нафтилмочевина 195
L-(+)-Норлейцин 285
L-(—)-Триптофан 289 166 176 а-Нафтилмочевина 158
Креатинин 292 Пикрат 220
(/./-Изолейцин 292 118 140
(/./-Аланин 295 166 190 139 Пикронолат 214
D- или L-Аланин 297 151 168 133 а-Нафтилмочевина 198
(/./-Валии 298 132 163 ПО Формил 145
(/./-а-Амино-н-масляная 307 147 170 а-Нафтилмочевииа 194
D- или L-Валин 315 147 147 Формил 156
(/./-Тирозин 318 197 226 Пикролоиат 260
D- или L-Фенилаланин 320 146 181 161 Формил 167
(/./-Норлейцин 327 124 Формил 114
(/./-Лейцин 332 141 165 Пикролоиат 150
Таблицы производных
623
Продолжение
Аминокислота Т. разл.. ’С Т. пл. производного, °C
беизоил- производное фенил- мочевяна л-толуол- сульфонил- производное разные
D- или L-Лейцин L-(-)-Тирозин сМ-Ориитии D- или L-Орнитин d.Z-Лизии d,(-Изосерин L-(~—)-Изосерин 337 344 107 166 267 (разл.) 240 (разл.) 249 151 109 115 104 192 190 196 184 122 114 а-Нафтилмочевина 205 Пикрат 195 Пикрат 204 Дибензоил 146
а Оптическое вращение обозначаетсн знаками (+) и (—). Приставки D и L указывают
относительную стереохимическую конфигурацию, d, I обозначает рацемат.
Углеводы и гликозиды*
Соединение Т. разл., °C Удельное вращение в воде при 20°С, град Время, необхо- димое для обра- зования озазона, мин Т. пл. производного, °C
озазои ацетат
Раффиноза (водная) 80 + 104,5 99
Р-Мелибиоза (водная) 85 + 129,5 178
Глюкоза (водная) 90 +47,7 4-5 205 Г 132 (Р)
1 112(a)
D-Рибоза 95 —21,5 166
Мальтоза (водная) 100 + 129,0 206 158
Левулоза (о-фруктоза) 104 -92,0 2 205 (Ю9(Р) 1 70 (а)
L-Рамиоза 105 +9,4 182
L-Ликсоза 105 + 13,5 163
Раффиноза (безводная) 119 + 123 99
D-Манноза 132 + 14,1 0,5 205
132 (димерид)
d, (-Глицеральдегид 142 6 154
D-Ксилоза 145 + 18,7 7 163 141
Глюкоза (безводная) 146 +52,8 4-5 205 132 (Р)
112(a)
Мелезитоза 147 +88
L-Арабиноза 160 + 104 10 166
L-Сорбоза 160 -43,1
d, (-Арабиноза 164 169
а-Метилглюкозид 165 + 157,6 101
Арбутии (водный) 165 -60,3 136
624
Приложение III
Продолжение
Соединение т. ра <л.. Удельное вращение в воде при 20°С, град Время, необхо- димое для обра- зования озазона, мин Т. пл. производного, °C
озазон ацетат
Мальтоза 165 +129 206 158
d,/-Аскорбиновая кислота 169
D-Аскорбиновая кислота -48
L-Аскорбииовая кислота +49
D-Галактоза 170 +81,7 15 201 142 (₽)
95(a)
Гелицин 175 -60,4
Инулин 178 —39,5 25
Популин 180
Сахароза 185 +66,5 30 205 89
Кониферин 18б -66,9 125
а-Метилманнозид 193 +79,2
Салицин 201 -62,4 130
Лактоза 203 +52,5 200 100
(разл.)
а,а-Трегалоэа 203 + 197,1
Амигдалин 214 —41 166
Целлобиоза 225 +35,0 198
Гликоген 240 198
Ацетат целлюлозы Разл.
Целлюлоза »
Кбахмал
а D или L обозначает конфигурацию сахара.
® Плавится без разложения.
Сложные эфиры (жидкие)
Сложный эфир Т. кнп., °C Удельная масса
Метилформиат 32 0,998$
Этилформиат 54 0,938
МетилацеТат 57 0,958
Изопропилформиат 68 0,883
Винилацетат 72 0,9317$°
Метилхлорформ иат 75 1,23б]5
Этилацетат 77 0,924
Метилпропиоиат 79 0,937
н-Пропилформиат 81 0,918
Аллилформиат 83 0,932$7
Метилакрилат 85 0,977$
Таблицы производных
625
Продолжение
Сложный эфир Т. кип., °C Удельная масса
Метилкарбонат 90 1,06б|7
Изопропилацетат 91 0,917
Метилйзобутират 92 0,912$
Этилхлорформиат 93 1,144$5
Изопропеиилацетат 96 0,931^
втор-Бутилформиат 97 0,882
Изобутилформиат 98 0,905
Этилпропионат 98 0,912
трег-Бутилацетат 98 0,867
Метилметакрилат 99 0,936
Метилпивалат 101 0,891$
н-Пропилацетат 101 0,909
Этилакрилат 101 0,925$
Метилортоформиат 101 0,974$3
Метил-к-бутират 102 0,919
Аллилацетат 103 0,938
Изопропилхлорформиат 105 0,911
н-Бутилформиат 107
Этилйзобутират НО 0,890
Хлорметилацетат 111 1,19б|4
втор-Бутилацетат 111 0,870
н-Пропилхлорформиат 118 1,094$5
Изобутилацетат Метилизовалерат 116 116 0,892 0,901
Этил-н-бутират 120 0,900
н-П ропилпропиоиат 122 0,902
Изопептилформиа'Г 123 0,894
н-Бутилацетат 125 0,902
Этилкарбоиат 126 6,976
Изопропил-н-бутират 128 0,879$
Метил-н-валерат 130 0,910
Изобутилхлорформиат 130 1,053$5
Бромметилацетат 130 1,6бв|2
Метилхлорацетат 130 1,238
н-Пеитилформиат 130 0,902
Этилизовалерат 134 0,865
Метилпируват 136 1,154
Изобутилпропиоиат 137 0,888
Этилкротонат 138 0,9175$°
Изопеитилацетат 142 0,884
н-Пропил-н-бутират 143 0,893
н-Бутилпропионат 144 0,895
Метиллактат 144 1,0904
Метилбромацетат 144 (разл.) 1,657*9
626
Приложение IH
Продолжение
Сложный эфир Т. кнп., °C Удельная масса
Этиленгликольмонометиловый эфир, ацетат 144 1,0067“
Этил-н-налерат 145 0,8765“
н-Бутилхлорформиат 145 145 145 145 146 146 1,078
Этилхлорацетат 1,158
Этилортоформиат 0,897
Р-Хлорэтилацетат 1,178
«-Пентилацетат 0,896
Этил-а-хлорпропиоиат 1,087
Бензилхлорацетат 147 1,2221
Изобутилизобутират 147 0,875
Метилизобутилкарбинол ацетат 148 0,8805^
Метил-н-капроат 150 0,904
Этиллактат Этилэтоксиацетат 152 152 1,0311® 1,032{5
Изопеитилхлорформиат 154
н-Гексилформиат 154 0,898®
Этилпируват 155 1.08014
Этилеигликольмоиоэтиловый эфир, ацетат 156 0,9749^
Изобутил-н-бутир ат 157 0,888
Этилдихлорацетат 158 1,282
Этилбромацетат 159 1,506
Изоамилпропионат 160 0,888
Этилгликолат 160 1,08691s
Циклогексилформиат 162 1,010
Этил-а-бромпропионат 162 1,329
Р-Бромэтилацетат 163 1,524
н-Бутил-н-бутират 165 0,888
Этил-н-капроат 166 0,889
Этилтрихлорацетат 167 1,3691s
«-Пропил-м-валерат 167 0,88882
«-Пропил карбонат 168 0.94917
Изопропиллактат 168 0,876
«-Амилпропионат 169
Метилацетоацетат 169 1.08115
Метил -«-гептоат 173 0,898
Циклогексилацетат 175 0,9721®
н-Бутилхлорацетат 175 1,0811s
Фурфурилацетат 176 1,1176
Изопентил-н-бутират 178 0,882
Этил-Р-бромпропионат 179 1,425
Метилфуроат 181 1,17864
Метилмалонат 181 1,16013
Этилацетоацетат 181 1,0311®
Таблицы Производных
627
Продолжение
Сложный эфир Т. кип., °C Удельная масса
Р-Оксиэтилацетат 182
«-Пентил-«-бутират 185 0,88325
н-Пропил-н-капроат 186 0,8844q
Этилоксалат 186 1,082
Этилметилацетоацетат 187 l,024f
Бутиллактат 188 0,984“
Этил-н-гептоат 189 0,888
Этиленгликольдиацетат 190 1,128
Изопропилоксалат 190 i.oiof
Изобутилкарбоиат 190 0,919*5
Изопеитилизовалерат 190 0,870
Метилэтилацетоацетат 190 0,989
Метиллевулииат 191 1,068
н-Гептилацетат 192 0,8891о
Метилкаприлат 193 0,894
Тетрагидрофурфурилацетат 194 1,0624“
Метилсукцииат 195 1,120f
Этил метил м ал он ат 196 1.01915
Фенилацетат 196 1,08 if
Этилмалонат 198 1,077
Метилбеизоат 198 1,103
Этилэтилацетоацетат (ацетоуксусный эфир) 198 0,972
2-Эти л-1 -гексил ацетат 199 0,8733“
Метилцианацетат 200 1,0962“
Беизилформиат 203 l,081f
Винилбеизоат 203 1,065
н-Пентилвалерат 204 0,881®
у-Бутиролактои 204
Этиллевулинат 205 1,016
Этилкаприлат 206 0,887
н-Бутилкарбоиат 207 0,9412
у-Валеролактон 207 l,0465f
о-Крезилацетат 208 l,048f
Т риметилеигликольдиацетат 210 l,070f
Феиилпропионат 211 l,054f
н-Пропилфуроат 211 l,0745f
Этиленгликольдипропиоиат 212 1,045“
м-Крезил ацетат 212
п-Крезилацетат 212 l.O50f
628
Приложение III
Продолжение
С ложный эфир Т, кнп., *С Удельная масса
н-Пропил оксалат 213 1,038
Метилпеларгонат 213 0,892
Этилбеизоат 213 1,066
Метил-о-толуат 213 1,073}5
Метил -л-толуат 215 1,06б15
Этилфумарат 215 1,04721“
Бензилацетат 216 1,0621s
Этилсукцинат 216 1.04915
Метил-п-толуат 217
Диэтиленгликольмоноэтиловый эфир, ацетат 218 1,0114го
Изопропилбензоат 218 1,010|4
Метилфенилацетат 218 1,0441°
н-Пропиллевулииат 221 0,98951°
Метилсалицилат 224 1,1841“
Этилмалеат 225 1,0741s
Этилпеларгоиат 227 0,86б17
Фенил-н-бутират 227 1,0271s
1-Ментилацетат 227 0,91851°
Изоамилкарбонат 228 0.91214
Изобутилоксалат 229 1.00214
Этилфеиилацетат 229 1,031
к-Пропилбензоат 230 1,0251s
Аллилбензоат 230 1,0671
Метил-л-хлорбензоат 231
Метилкапрат 232 0,8761е
Этилглутарат 234 1,022
Метил-о-хлорбензоат 234
Этилсалицилат 234 1,1471
Этилброммалонат 235 1.42бЦ
Изопропилсалицилат 237 1.09519
Бензил-н-бутират 238 1,03316
н-П ропилсалицилат 239 1,0981s
Этил-л-бутил м алонат 240
Изобутилбензоат 241 1,0021s
н-Бутилоксалат 243 1,010
Тимилацетат 244 1,009
Метил-о-бромбензоат 244
Этиладипат 245
Таблицы производных
629
Продолжение
Сложный эфир Т. кип., °C Удельная масса
Этнлкапрат 245 0,8684®
«-Пропилсукцинат 246 1.0161
Диэтиленгликольмоно-н-бутиловый эфир, ацетат 246 0,987120
Изобутилфен илацетат 247 0.999]8
Метилундециленат 248
и-Бутилбензоат 249 1,000
Этилацетоидикарбоксилат 250 1,113
н-Бутилфеиилацетат 254 0,99б]8
Этилпимелат 255 0,9945“
Этилбензоилформиат 257
Г лицерилтриацетат 258 1,161]5
Изоамилоксалат 262 0,968]*
Изоамилбензоат 262 1,004
Изобутилсукцинат 265 0,974
Метиллаурат 268 0,869]9
н-Бутилсалицилат 268 1.07419
Этилаиизат 269 1,119]
Этиллаурат 269 0,8674s
Этилбензоилацетат 270 (разл.) 1.121915
Этилциниамат 271 1,050
Изопентилсалицилат 277 1,0651s
Резорцииолдиацетат 278 l,180]°
Метил-л-нитробензоат 279
d-Этилтартрат 280 1,199
Метилфталат 282 1,19б]°
Этилсуберат 282 0,9822]°
Глицерилтрипропионат 289 1,0831s
Этилазелат 291 0,9766g5
Этилцитрат 294 1,137
Метил миристат 296 0,873]°
Изоамилсукцинат 297 0,961]3
Этилфталат 298 1,117
Этилбензилмалоиат 300 1,0771s
Изопропилфталат 302 1,06 б]9
Этилмиристат 306 0,8651°
Этилсебацат 307 0,9651®
о-Крезилбензоат 307 1.114]9
Глицерилтрибутират 318 1.033*7
630
Приложение Ill
Продолжение
Сложный эфир Т. кип., "С Удельная масса
Беизилсалицилат Бензилбензоат 320 323 1,114^
н-Бутилфталат 338 1.05049
Изоамилфталат 349 1,024|7
Сложные эфиры (твердые) •
Сложный эфир Т. пл., °C Т. кип., °C
Метиллаурат 5 268
Этиланизат 7 269
Этилмиристат 11 306
D-Этилтартрат 17 280
Метилмиристат 18 296
Метил сукцинат 19 195
Фенилпропиоиат 20 211
Метил-я-хлорбензоат 21 231
Цетилацетат 22
D-н-Бутилтартрат 22
Этилпальмитат 24
Бориилацетат 29 221
Метилпальмитат 30
Метил-п-бромбензоат 32
Метил-п-толуат 33 217
Тимилбензоат 33
Этилстеарат 33
Этилфуроат 33
Метилциннамат 36 263
Этилманделат 37 254
Метилсебацат 38 288 (разл.)
Метилстеарат 38
Бензилциннамат 39
Бензилфталат 42
Фенилсалицилат 42
Циклогексил оксалат 42
Метил-п-хлорбензоат 44
Циннамилциннамат 44
Бензилсукцинат 45
Метиланизат 45 255
Этил-З-нитрофталат 46
Этил-я-иитробеизоат 47 296
D-Метилтартрат 48(61) 280
а-Нафтилацетат 49
Метилоксалат 51 163
Метил-о-бензоилбензоат 52 352
Фенилстеарат 52
я-Крезилбензоат 54
Этил-п-нитробеизоат 56
Метилманделат 58
Таблицы производных
631
Продолжение
Сложный эфир Т. пл., °C Т. кип., °C
о-Крезилкарбонат 60
Этилтрихлорлактат 66 162
Метил-4-нитрофталат 66
Кумарин 67 290
Фенилбензоат 68 299
Метил-З-нитрофталат 69
Фен ил фтал ат 70
Метил-л-оксибензоат 70
р-Нафтилацетат 70
Глицерилтристеарат 71
я-Крезилбеизоат 71
Фенилциннамат 72
Этил-л-оксибензоат 72 282
Глицерилтрибензоат 72
Этиленгликольдибензоат 73
Фталид 73 290
Метил-о-нитроциннамат 73
Метил-л-нитробензоат 78
Фенилкарбонат 78 306
Метилцитрат 79 285 (разл.)
Этил-л-нитроциннамат 79
Беизилоксалат 80
Метил-я-бромбензоат 81
Гваяколкарбонат 86
Метил-d,/-тартрат 90 282
Этил-3,5-динитробеизоат 94
Р-Нафтилсалицилат 95
Метил-я-нитробензоат 96
Этил-5-нитросалицилат 102
Флороглюцинолтриацетат 106
Фениладипат 106
Р-Нафтилбензоат 107
Метил -3,5-дииитробензоат 108
Этилоксамат 115
41-Крезилкарбонат 115
Этил-я-оксибензоат 116
Резорцилдибензоат 117
Этил-З-иитросалицилат 118
Метил-5-нитросалицилат 119
Фенилсукцииат 121 330
Г идрохиноидиацетат 123
Метил-л-нитроциниамат 124
Лактид 128 255
Метил-я-оксибеизоат 131
Метил-З-нитросалицилат 132
Этил-я-иитроциниамат 137
Метилтерефталат 140
Пирогаллолтриацетат 161
Метил-я-нитроциниамат 161
а Большое число твердых эфиров приведено средн производных карбоновых кислот
спиртов и фенолов; см. соответствующие таблицы в приложении III. '
632
Приложение III
Эфиры неорганических кислот
Вфир Т. кип., °C Удельная Масса
Нитриты
Этилиитрит 17 0,900?5
н-Пропилнитрит 44 0,998
Изобутилнитрит 67 0,888?
н-Бутилиитрит 75 0,911
Изопентилнитрит 99 0,880?°
н-Пеитил нитрит 104 0,8528?°
Нитраты
Метилнитрат 65 1,232?
Этилнитрат 87 1,130?
м-Пропилнитрат 110 1,075?
Изобутилнитрат 123 1,033?
н-Бутилнитрат 136 1,048
Изопеитилнитрат 147 1,000?
Сульфаты
Диметилсульфат 188 1.321?5
Диэтилсульфат 208 1,172?5
Простые алифатические эфиры (жидкие)
Простой эфир Т. кип., °C Удельная масса Т. пл. алкил-3,5- дини1робейзоата, °C
Метил этиловый 10 0,7252°
Этилеиоксид И 0,882$
Фуран 32 0,9644°
Диэтиловый 35 0,719?® 93
Пропиленоксид 35 0,830
Этилвиииловый 36 0,760
Дихлордиметиловый 59 1,020?°
1,2-Эпоксибутаи 62 0,837
2-Метилфуран 64 0,913
Т етрагидрофураи 65 0,889
Аллилэтиловый 65
Диизопропиловый 68 0,724 122
н-Бутил метиловый 70 0,774
Т етрагидросильван 79 0,855
Хлорметилэтиловый 80
Этиленгликольдиметиловый 85 0,867
Таблицы производных
633
Продолжение
Простой эфир Т. кип.. "С Удельная масса Т. пл. алкил-3,5- дннитробензоата, ’С
Дигидропираи 86 0,923
Т етрагидропиран 88 0,881
Ди-н-пропиловый 90 0,744*° 74
Этил-н-бутиловый 92 0,7522*°
2,5-Диметилфуран 94 0,888
Ди-а-хлорэтиловый 98
н-Пентилэтиловый 99 0,761
Диоксаи 102 1,0353*°
Циклопентил этиловый 10Б 0,862
а,-а'-Дихлорметиловый 105 1,328$5
Ди-Р-хлорэтиловый 107 1,057$
а.а'-Дихлорэтилоный 116 1,138$*
Эпихлоргидрин 117 1,204$
н- Пентилэтиловый 118 0,762
Этилеигликольдиэтиловый 121 0,8424$}
Циклопентил этиловый 122 0,853
Диизобутиловый 122 0,762$5 86
н-Гексилметиловый 126 0,772
Ди-Р-бромэтиловый 127 1,370$
Циклогексилметиловый 134 0,875
Этиленгликольмоноэтиловый эфир 135 0,9311$
Ди-н-бутиловый 140 0,769 64
Ди-а,Р-дихлорэтиловый 140 1,174$3
«- Г ексилэтиловый 142 0,772
Циклогексил этиловый 149 0,864
Дибромдиметилоный 150 2,201
Диэтилеигликольдиметиловый 162 0,9440*°
Бедзилметиловый 167 0,981$
н-Пропил-а-фурфуриловый 168
Этиленгликольмоио-н-бутиловый 171 0,9022$
Диизопеитиловый 172 0,774*5 62
Цинеол 176 0,927
Ди-р,р'-дихлорэтиловый 179 1,23g
Диэтилеигликольдиэтиловый 188 0,909$}
Этилбензиловый 189 0,949$°
Ди-н-пентиловый 190 46
Диэтиленгликольмонометиловый 193 1,0р541$
Дивтиленгликольмоноэтиловый 202 0,9902$}
Ди-н-гексвдовый БензилиЬббутиловый ?ое 212 58
634
Приложение III
Продолжение
Простой эфир Т. кип., «с Удельная масса Т. пл. алкил-3,5- дииитробеизоата, °C
Бензил-н-бутиловый 212
Ди-у,у'-дихлорпропиловый 215 1,140
Диэтил енгликольмоио-н-бутиловый 231 0,995420
Ди-н-гексиловый 229
Этиленгликольмонофениловый 245 l,1094|g
Ди-н-гептиловый 260 0,815° 47
Тетраэтиленгликольдиметиловый 266 1,009
Ди-н-октиловый 288 0,806
Дибензиловый 298 1,03б4в 112
Ароматические эфиры (жидкие)
Простой эфир Т. кип.. °C Удельная масса Т. пл. производного, °C
пикрат другие производные
Анизол 154 0,988 81 Динитро 87
о-Крезилметиловый 171 0,966° 119 Бром 63
Фенетол 172 0,979* 92 Нитро 58
п-Крезилметиловый 176 0,987° 89 Анисовая кислота 184
л-Крезилметиловый 177 0,9854 114 Тринитро 91
Фенил-н-пропиловый 188 0,949
о-Крезил этиловый 192 118 Динитро 51
л-Крезилэтиловый 192 115 .и-Этоксибензойная 137
п-Крезилэтиловый 192 111 и-Этоксибензойная 195
л-Хлоранизол 194
о-Хлоранизол 195 Нитро 95
и-Хлоранизол 200 Нитро 98
Гваякол 205 89
Вератрол 206 1,08б|5 57 Дибром 92
о-Хлорфенетол 208 0,950 Нитро 82
н-Бутилфениловый 210 112
п-Хлорфенетол 212 Динитро 54
Резорциндиметиловый 214 1,080° 58 Дибром 140 Тринитро 124
Метил тимоловый 216 0,954° Тринитро 92
о-Бром анизол 218 Нитро 106
н-Бутил-о-крезоловый 223 0,9444
п Бром ан изол 223 1,494° Нитро 88
о-Бромфенетол 224 Нитро 98
п-Бромфенетол 229 Нитро 47
Таблицы производных
635
Продолжение
Простой эфир Т. кип.. °C Удельная масса Т. пл. производного, °C
пикрат другие производные
Сафрол 232 1,09б|8 105 Пентабром 169
Анетол 232 0,989^8 70 Трибром 108
Резорциидиэтиловый 235 58
о-Иоданизол 240 1,800 Нитро 95
Эвгенолметиловый 244 1.055]5 114 Трибром 78
о-Иодфенетол 245 Нитро 96
Изосафрол 246 1,12б]4 75 Трибром 109
Дифениловый 252 1,073 ПО Дибром 58
Метил-а-иафтиловый 265 1.09614 113 Бром 46
о-Нитрофеиетол 267
о-Нитроанизол 272 1,254 о-Аиизидин 225
Этил-а-нафтиловый 278 1,074 100 4-Бром 48
Этил-0-нафтиловый 282 1,064 44 1-Бром 66
Ароматические эфиры (твердые)
Простой эфир Т. пл., •с Т. пл. производного, °C
пикрат другие производные
Вератрол 21 57 Нитро 95
п-Хлорфенетол 21 Дииитро 54
Анетол 22 70 Трибром 108
н-Пентил-2-иафтиловый 25 67
Изоамил-2-иафтиловый 26 91
п-Иодфенетол 27 Нитро 96
Дифениловый 28 ПО Дииитро 135
Р- Бромэти лфеииловый 32 Дибром 54 Бром 56
Апиол 32 Апиоловая кислота 175
о-Метоксибифенил 32 Нитро 95
Изобутил-2-нафтиловый 33 85
•и-Нитрофенетол 34 л-Фенетидинпикрат 158
втор-Бутил-2-нафтиловый 34 86
н-Бутил-2-иафтиловый 36 67
Этил-Р-нафтиловый 37 44 Бром 66
л-Нитроаиизол 39 л-Анизидиипикрат 169
Пирогаллолтриэтиловый Изопропил-2-нафтиловый 39 40 95 (разл.)
н-Пропил-2-нафтиловый 40 81
2,4,6-Трихлорфенетол 43 Динитро 100
Пирокатехиндиэтиловый 43 71 Тринитро 122
Пирогаллолтриметиловый 47 81
636
Приложение III
Продолжение
Простой эфир Т. пл., •С Т. пл. производного, °C
пикрат другие производные
Г идрохиионмоиометиловый 53
н-Нитроанизол 54
Г идрохинондиметиловый 55 119 Дибром 142
Феноксин 59
2,4,6-Трихлоранизол 60 Динитро 95
п-Нитрофенетол 60 Фенацетин 138
Метил-Р-нафтиловый 72 108 Бром 86
Г идрохииондиэтиловый 72 Трииитро 130
2,4,6-Трибромфенетол 72 Нитро 79
2,4,5-Трибромфеиетол 73
Беизил-а-иафтиловый 77
п-Метоксибифеиил 85
2,4,6-Триброманизол 87
Дибензофуран 87 94 Нитро 182
2,4-Динитроанизол 95
Бензил-2-нафтиловый 102 123
3,Б-Динитроаиизол 106
Гидрохинондибензиловый 127 Нитро 83
Алкеиилгалогеииды
Алкенилгалогеннд Т. кип., °C Удельная масса Т. пл. производного, °C
анилид алкеиилртуть- галогенид
Винилбромид 16 1.51714 104
2-Хлорпропен 30 0,9184
1 -Хлорпропен 36 114
Аллилхлорид 46 0,938 а
2-Бромпропен 48 1,362
Винилиодид 56 2,080$
Хлоропрен 59 0,958^
1 -Бромпропеи 60 1,428 114
Аллилбромид 71 1,398 а
Аллилиодид 103 1,848!| а
Р-Хлорстирол 199 1,112]5 115
Р-Бромстирол 221 1,427 115 91
а Кротонанилид 114“С.
Таблицы производных
637
Алкил-, циклоалкил- и аралкилгалогениды
Галогенид Т. кнп., °C Т. пл. производного, °C
анилид а-иафталид алкилртуть- галогенид
Хлориды
Этил 12 104 126 192
Изопропил 36 103 97
«-Пропил 46 92 121 147
трет-Бутил 51 128 147
втор-Бутил 67 108 129
Изобутил 68 109 125
н-Бутил 77 63 112 127
Неопентил 85 131 118
трет-Пентил 86 92 138
Изопентил 100 108 111 86
н-Пеитил 107 96 112 НО
н-Гексил 134 69 106 125
Циклогексил 142 146 188 164
н- Гептил 160 57 95 119
Бензил 179 117 166
н-Октил 184 57 91
Б-Фенилэтил 190 97
а-Фенилэтил 195 133
н-Нонил 202
Борнил 207
н-Хлорбеизил 214 166
н-Ундецил 241
н-Децил 223
Цетил 289 (разл.) 102
Бромиды
Метил 5 114 160 160
Этил 38 104 126 193
Изопропил 60 103 93
н-Пропил 71 92 121 138
трет-Бутил 72 128 147
втор-Бутил 90 108 129 39
Изобутил 91 109 125 55
н-Бутил 100 63 112 129
трет-Пентил 108 92 138
Неопентил 109 126
Изопеитил 118 108 111 80
2-Пентил 118 93 104
З-Пентил 119 121
н-Пентил 129 96 112 122
Циклопеитил 137
н-Гексил 157 69 106 118
Циклогексил 165 146 188 153
н-Гептил 174 57 95 118
Беизил 198 117 166 119
н-Октил 204 57 91 109
а-Фенилэтил 205 133
2-Этилциклогексил 212
Р-Фенилэтил 218 97 169
н-Нонил 220 109
638
Приложение III
Продолжение
Галогенид Т. кип., ’С Т. пл. производного, °C
аиилид а-иафталид алкилртуть- галогеиид
Йодиды Метил 43 114 160 145
Этил 72 104 126 182
Изопропил 89 103 125
трет-Бутил 98 128 147
н-Пропил 102 92 121 112
втор-Бутл 119 108 129
Изобутил 120 109 125 72
трет-Пеитил 128 92 138
н- Бутил 130 63 112 117
2-Иодпентан 142
Изопентил 148 108 111 122
н-Пентил 156 96 112 ПО
«-Гексил 180 69 106 ПО
«-Гептил 204 57 95 103
Октил 226
Арилгалогениды (жидкие)
Арилгалогеннд Т. кип., “С Продукт нитрования Производное Т. пл. производ- ного, ’С
положе- ние Т. пл., ’С
Фторбензол 85 Сульфон 98
о-Фтортолуол 114
лг-Фтортолуол 115
п-Фтортолуол 117
Хлорбензол 132 2,4 52 а
Бромбензол 157 2,4 75 а-Нафталид 161
о-Хлортолуол 159 3,5 63 о-Хлорбеизойная кислота 140
лг-Хлортолуол 162 4,6 91 л-Хлорбензойиая кислота 158
п-Хлортолуол 162 2 38 n-Хлорбензойиая кислота 242
л-Дихлорбеизол 172 4,6 103
1 -Хлор-2-этилбензол 178 о-Хлорбензойиая кислота 140
о-Дихлорбензол 179 4,5 ПО
о-Бромтолуол 181 3,5 82 о-Бромбеизойиая кислота 147
п-Хлорэтилбеизол 182
лг-Бромтолуол 183 4,6 103 л-Бромбензойная кислота 145
1 -Хлор-З-этилбеизол 184 л-Хлорбензойиая кислота 158
п-Бромтолуол 185 2 47 n-Бромбензойная кислота 251
Иодбеизол 188 4 171 п-Бромиодбензол 91
1 -Хлор-2-изопропилбеи- 191 о-Хлорбензойиая кислота 140
ЗОЛ
2,4-Дихлортолуол 195 3,5 104 2,4-Дихлорбензойная кис- 160
лота
Таблицы производных
639
Продолжение
Продукт ct
нитрования га «
Арилгалогеиид О о и е Производное о ВО
И Я я о ч а> Ч И
f- о S Я Я ь н И
о-Бромхлор бензол 195
1.З-Брдмхлорбензол 196 п-Хлорб^нзойная кислота 242
п-Хлоркумол 198
2,6-Дихлортолуол 199 2,6-Дихлорбеизойная кис- 130
лота
1 -Бром-2-этцлбензол 199 о-Бромбензойная кислота 147
jw-Иодтолуол 204 л-Иодбеизойная кислота 186
1 -Бром-4-этилбеизол 205 n-Бромбензойная кислота 251
1 -Бром-2-изопропилбен- 210 о-Бромбензойная кислота 147
ЗОЛ
о-Иодтолуол 211 6 103 о-Иодбеизойная кислота 162
п-Иодтолуол 211 n-Иодбензойиая кислота 265
1,2,4-Трихлорбензол 213 5 56
ж-Дибромбензол 219 4 61
п-Бромкумол 219 n-Бромбензойная кислота 251
о-Дибромбеизол 224 4,5 114
Броммезитилен 225
2-Бромцимол 234 ?, ? 97 Анилид 143
2,5-Дибромтолуол 236 2,5-Дибромбензойная кис- 157
лота
3,4-Дибромтолуол 241 3,4-Дибромбензойная кис- 233
лота
о-Бромиодбензол 257 4,5 180 Пикрат
а-Хлориафталин 263 137
а-Бромнафталин 279 4 85 о-Бромбензойная кислота
2-Бромбифенил 297 147
1-Иодиафталин 305 Пикрат 128
т-1------
а Известны метастабильиые формы с т. пл. 27 и 43°С.
Арилгалогениды (твердые)
Арилгалогеиид Т. пл., °C Продукт нитрования Производное Т. пл. производ- ного, 'С
положе- ние т. пл., °C
а-Бромтолуол 2-Хлорбифенил 28 34 2 47 n-Бромбензойиая кис- лота о-Хлорбеизойиая кис- лота 251 140
640
Приложение tit
Продолжение
Арилгалогеиид О о ч с Продукт нитрования Производное Т. пл. производ- ного, °C
положе- ние т. пл., °C
п-Иодтолуол а,Р-Дихлорнафталин ж-Дииодбензол 1,2,4-Трибромбеизол 1,2,3,4-Тетрахлорбеизол 1,2,3,5-Тетрахлорбензол 1,2,3-Трихлорбензол п-Дихлорбеизол ₽-Иоднафталии З-Хлорнафталин Р-Бромнафталин 2,2'-Дихлорбифенил Р-Фторнафталин 1,3,5-Т рихлорбензол 1,2-Дибромнафталин п-Бромхлорбензол 2,5-Дихлор-п-ксилол 4-Хлорбифенил 2,2'-Дибромбифенил 1,4-Дибромнафталин л-Дифторбензол Пеитахлорбеизол 1,2,3-Трибромбеизол п-Дифторбензол п-Дибром бензол 4-Бромбифеиил п-Бромиодбензол о-Дифторбеизол 4-Иодбифеиил 1,3,5-Трибромбензол п-Дииодбензол 1,2,4,5-Тетрахлорбензол 4,4'-Дихлорбифенил 4,4'-Дибромбифенил 1,2,4,5-Тетрабромбеизол Нафталинтетрахлорид Г ексахлорбензол 35 36 40 44 46 51 52 53 55 58 59 60 60 63 67 67 68 77 81 82 82 86 87 88 89 89 92 92 114 1?0 129 138 149 169 180 182 229 4 2 1,8 2 2 2 3 56 54 175 68 72 84 168 n-Иодбензойная кисло- та Анилид Пикрат Анилид Пикрат 3,4-Дибромфталевая кислота 2,5-Дихлортерефтале- вая кислота 3,6-Д ибромфталев ая кислота Дихлорид п-Иоднитробензол n-Хлорбензойная кис- лота 1,3-Дихлорнафталин Хлоранил 265 170 81 170 101 196 306 135 102 (разл.) 171 242 61 290
Таблицы производных
641
Полигалогенпроизводные углеводородов (жидкие)
Галогенид Т. кип., °C Удельная масса "D
1,2-Бромфторэтилеи 36 1,693$°
Метиленхлорид 42 1,378$ 1,4237
Дихлорэтилен (цис) 48 1,265$° 1,4490
Перфтор-«-гексан 57 1,699$° 1,2515g2
Дихлорэтилен (транс) 60 1,291 *5 1,4518
Этилиденхлорид 60 1,180$2 1,4166
Хлороформ 61 1.504}2 1,4467
.и-Нитробеизилидеихлорид 65
2,2-Дихлорпропан 70 1,093 1,4093
1,1,1-Трихлорэтан 74 1,325$° 1,4349
Четыреххлористый углерод 78 1,59125 1,4607
(тетрахлорметаи)
п-Нитробензилиденбромид 82
1-Бром-1-хлорэтан 83 1,667$®
Этилеихлорид 83 1,256 1,4443
Трихлорэтилен 90 1.44Оо5 1,4782
Метилеибромид 98 2,498$°
Пропиленхлорид 98 1,16б14 1,4388
Бензилидентрифторид 102
Бромтрихлорметаи 105
1 -Бром -2-хлорэтан 107 1,689$°
Этилидеибромид 112 2,100$7 1,5128
1,1,2-Трихлорэтан 114 1,4571° 1,4711
2,3-Дихлорбутаи 116
Тетрахлорэтилен 121 1,631$ 1,5055
1,2-Дихлорбутан 124
Триметилеихлорид 125 1,190$8
Этилеибромид 130 2,178 1,5379
1,1,1,2-Тетрахлорэтан 131
1,2-Дибромпропеи 132 2,008$°
2,3-Дибромпропеи 140
Пропиленбромид 142 1,933 1,5203
Триметиленхлорбромид 143 1,593$° 1,4708
силж-Тетрахлорэтаи 147 1,614$ 1,4942
Изобутиленбромид 149 1,759 1,509
1,2-Дибромбутеи-1 150 1,887$
Бромоформ 151 2,904$° 1,589
1,2,3-Трихлорпропаи 155 1,417
1,3-Дибромпропен 156 2,097$ 1,538$°
2,3-Дибромбутаи 158 1,830
Пентахлорэтаи 161 1,6931° 1,504
21 Зак. 335
642
Приложение III
Продолжение
Галогенид Т. кнп.» °C Удельная масса nD
1,2-Дибромбутаи 165 1,820g9
Триметиленбромид 165 1,973У 1,523
1,3-Дибромбутен-2 169 1,877^ 1,548
1,2,3-Трихлорбутан 169
1,3-Днбромбутан 174 1,807 1,507
Метилениоднд 180 (разл.) 3,285^ 1,7425
1,1,2-Трйбромэтан 189 2,62114° 1,5933
1,4-Дибромбутан 198 1,8472
силл-Тетрабромэтан 200 (разл.) 2,971^ 1,638
Бензилиденхлорнд 212 1,29б}6 1,5516
о-Х лор бензн лхлор ид 214
п-Хлорбензилхлорид 214
л-Хлорбензилхлорид 216 1,2694°
1,2,3-Трнбромпропан 219 2,43693 1,584
Бензотрнхлорнд 220 1,380^ 1,5573
Пентаметиленбромид 221 1,70б}8
1,8-Дибромоктан 272 1,468^ L501J5
Полигалогенпроизводные углеводородов (твердые)
Галогенид Т. пл.» °C Производное Т. пл. производ- ного, •с
n-Хлорбензнлхлорнд Гептахлорпропан 29 29 п-Хлорбензойная кислота 242
о-Бромбензилбромнд 30 о-Бромбензойная кислота 147
л-Бромбензилбромид 41 л-Бромбензойная кислота 155
п-Бромбензнлхлорнд 50 n-Бромбензойиая кислота 251
n-Бромбе из ил бромид 63 п-Бромбензойная кислота 251
Этиленноднд Тетрабромметан 1,1,1 -Трихлор-2,2-бис- (п-хлор- фенил)этан (ДДТ) 82 92 108 Этнленглнкольди-Р-нафтнло- вый эфир 148
Йодоформ а-Бензолгексахлорнд сйлл-Тетраметнлднхлорэтан силл-Тетраметилдибромэтан Гексахлорэтан Р-Бензолгексахлорид 119 157 160 169 187 310 Продукт присоединения хи- нолина 65
Таблицы производных
643
Ароматические углеводороды (жидкие)
Углеводород Т. кип., °C Удельная масса Продукт нитрования Т. пл. производного, •с
положе* ние т. пл,, °C ароилбеи* зойная кислота пикрат
Бензол 80 0,874 1.3 89 127 84
Толуол 111 0,884 2,4 70 137 88
Этилбензол 135 0,876]° 2,4,6 37 122 96
п-Ксилол 137 0,86б]4 2,3,5 137 132 90
м- Ксилол 139 0.871]2 2.4 83 126 91
о-Ксилол 142 0,8904 4,5 71 178 88
Изопропилбензол (ку- мол) м-Пропнлбензол 153 0,875] 2,4,6 109 133
158 0,861 125 103
Мезнтилен 164 0,869]° 2,4 86 211 97
п-Этилтолуол 162 0,861
о-Этилтолуол 165 0,881 97
Псевдокумол 168 0,895 3,5,6 185
трет-Бутилбензол 169 0,867 2,4 62
Изобутилбензол 173 0,853
втор-Бутилбензол 173 0,862
м-Кумол 175 0,861
п-Кумол 175 0,857 2,6 54 123 91
1,2,3-Т риметн лбензол 176 0,894
Гидринден 177 0,965
о-Кумол 178 0,877
Инден 180 1,000 98
л-Пропнлтолуол 182 0,8623
л<-Днэтил бензол 182 0,860 2,4,6 62 114
н-Бутилбензол 182 0,862 97
п-Пропилтолуол 184 0,859
о-Днэтилбензол 184 0,881
n-Днэтил бензол 184 0,862
Изодурол 195 4,6 157 213
н-Пентил бензол 202 0,86б}|
м- Диизопропилбензол 203 0,856
о-Диизопропнлбензол 204 0,877
Преннтол 204 ОДП20 5,6 176
1,2,3,4-Тетраметнлбензол 205 0,905 5,6 176 95
Тетралин 206 0,971 5,7 95 153
п-Днизопропилбензол 210 0,857
1,3,5-Трнэтилбензол 218 0,863 2,4,6 108 129
н-Гексилбензол 226 0,878
Цикл огексил бензол 237 0,955 4 58
1 -Метнлнафталин 240 1.001]9 4 71 169 141
1-Этилнафталнн 258 1,008 78
1,4-Диметнлнафталин 263 1,008 140
1,1-Дифенилэтан 270 l,003g°
21*
644
Приложение III
Ароматические углеводороды (твердые)
Углеводород Т. пл., °C Продукт нитрования Т. пл. производного, °C
положе- ние т. пл., °C ароилбен- зойная кислота пикрат
Дифеннлметан 26 2,4,2',4' 172
Р-Метнлнафталнн 32 1 81 116
Пентаметилбензол 51 6 154 131
Дибензил 52 4,4' 180
Бнфенил 70 4,4' 233 224
Дурол 79 3,6 205 263
Нафталин 80 1 61 172 149
.м-Дифенилбензол 85
Трифеннлметан 92 4,4',4" 206
Аценафтен 95 5 101 198 161
Ретен 98 124
Фенантрен 100 144
2,3-Диметилнафталин 104 124
Флуорен 115 2,7 199 227 84
Пирен 148 220
а.а'-Бннафтил Г ексаметнлбензол 160 145
162 176 170
Р,Р'-Бинафтил 188 184
силси-Тетрафенилэтан 211 4,4,4,4 143
п-Терфеиил 213
Антрацен 216 214 138
Хризен 251 273
Пицен 364
Алкаиы и циклоалкаиы
Углеводород Т. кип., °C Удельная масса „20
Неопентан 9 0,613° 1,3513 (0°С)
Изопентан 31 0.613*4 1,355
Пентан 36 0,631 1,3750
Цнклопентан 50 0,750 1,4093
2-Метнлпентан 60 0,653 1,3716
3-Мет нлпентан 63 0,664 1,3764
н-Гексаи 68 0,660 1,3754
Метнлцнклопентан 72 0,749 1,4100
Циклогексан 80 0,790 1,4263
1,1 - Д нметнлцнклопентан 88 0,755 1,4136
2-Метилгексан 90 0,679 1,3851
1,2-Днметнлциклопентан (транс) 92 0,751 1,4120
З-Метнлгексан 92 0,687 1,3887
З-Этилпентан 94 0,698 1,3934
н-Гептан 98 0,684 1,385
2,2,4-Трнметилпентаи 99 0,692“ 1,3916
Таблицы производных
645
П родолжение
Углеводород Т. кип., °C Удельная масса 20 D
Метнлциклогексан 100 0,769 1,4235
1,2-Диметилциклопентан (цис) 100 0,773 1,4222
Этилциклопентан 104 0,766 1,4198
1,1,3-Триметилциклопентан 105 0,748 1,4112
4-Метилгептан 118 0,705 1,3985
2-Метилгептан 118 0,698 1,3950
З-Этилгексап 119 0,714 1,4440
1,1 - Диметилциклогексан 120 0,781 1,4290
1,4-Днметилциклогексан (транс) 119 0,763 1,4209
1,4-Диметилциклогексан (цис) 124 0,783 1,4297
я-Октан 125 0,703 1,3890
Этилциклогексан 132 0,788 1,4332
я-Нонан 149 0,717 1,405
я-Пропилциклогексан 157 0,795 1,4370
2,7-Ди м етн локтан 158 0,735]° 1,408
п-Ментан 169 0,79б]° 1,437
Циклононан 172 0,8534]° 1,4328 (16°С),
я-Декан 173 0,730 1,415
Декалин (транс) 185 0,870 1,4697
Декалин (цис) 194 0,896 1,4811
я-Ундекан 194 0,745]° 1,4184
Циклодекан 201 0,8577]° 1,4692
я-Додекан 215 0,755]° 1,4209
я-Тридекан 236 0,7563 1,4256
Тетрадекан 254 0,764 1,4289
Пентадекан 271 0,769 1,4310
Гексадекан 287 0,773 1,4352
Ненасыщенные углеводороды
Углеводород Т. кип., °C Т. пл., °C Удельная масса nD Т. пл. производного, °C
З-Метилбутен-1 21 0,660{|
Пентадиен-1,4 26 0,661 1,3887
Бутин-2 27 0,688 1,3893 Тетрабромид 243
Бутин-1 30 0,641 1,3710
2-Метилбутен-1 31 0,650 1,3778
Изопрен 34 0,681 1,4219
Пеитен-2 36 0,651 1,3789
Т рнметилэтилен 38 0.668]3 1,3855
Пентин-1 40 0,688 1,4079 Меркурид 118 .
Цнклопентадиен 42 0,805]9 1,4470
Пента диен-1,3 42 0,6803 1,4309 Тетрабромид 114
646
Приложение HI
Продолжение
Углеводород Т. кип., °C Т. пл., °C Удельная масса "D Т. пл. производного, °C
Пентадиен-1,2 45 0,693 1,4209
Циклопентен 46 0,774$8 1,4218
Пентаднен-2,3 48 0,695 1,4284
Пентин-2 55 0,710 1,4045
Гексаднен-1,5 59 0,690 1,4010
Гексен-1 66 0,673 1,3858
Гексен-3 67 0,682 1,3942
Гексен-2 68 0,681 1,3928
Гексин-1 70 0,712 1,3990 Меркурнд 99
Тетраметилэтнлен 72 0,728$
Гексатрнен-1,3,5 78 0,718 1,4330
Цнклогексаднен-1,3 80 0,841 1,4740
Гекса диен-2,4 81 0,715 1,4493 Тетрабромид 185
Гекснн-3 81 0,724 1,4115
Циклогексен 84 0,809 1,4492 Адипиновая кнс-
лота 152
Гептен-1 94 0,697 1,3998
Гептин-1 100 0,750$9 1,418 Меркурид 61
Динзобутнлен 101 0,715$° 1,4082
4 -Метилциклогексен 103 0,799 1,4430 0-Метнладипино-
вая кислота 93
З-Метилцнклогексен 105 0,800 1,4426 а-Метнладнпино-
вая кислота 64
1 -Метилцнклогексен 111 0,809 1,4496 Ннтрозохлорнд 97
Циклогептен 115 0,823 1,4580
Октеи-1 121 0,716 1,4088
Октин-1 132 0,747 1,4172
Феннлацетнлен 140 0,930 1,5524 Меркурид 125
Стирол 146 0,925 1,5485 Дибромнд 73
Z-Борн илен 146 113
Нонен-1 147 0,729 1,4154
d- нли Z-Пинен 156 0,858 1,4653 Дибромнд 164
Аллнбензол 157 0,893
/-Камфен 160 42 0,822 1,4621 Дибромнд 89
Изопропеннлбензол 165 0,910 1,5386
Мирцен 167 0,798 1,4706
л-Метилстирол 168 0,900 1,542
п-Метилстнрол 169 0,897 1,541
Децен-1 171 0,741 1,4220
о-Метилстнрол 171 0,916 1,550
Лимонен 176 0,846$8 1,4727 Тетрабромид 104
Сильвеетрен 176 0,851$° 1,4774 Тетрабромнд 135
Пропенил бензол 177 0,914^ 1,5143
Инден 180 1,000 1,5710 Пнкрат 98
Днпентен 181 0,854$° 1,4730 Тетрабромид 124
Ундецен-1 198 0,750 1,4261
Дигидронафталнн 212 15 0,998 1,5740
Таблицы производных
647
Продолжение
Углеводород Т. кип., •С Т. пл., •С Удельная масса "D Т. пл. производного.
Додецен-1 213 0,758 1,4300
1,1 - Д ифенилэтилен Дифеннлацетнлен 277 298 8 1.038]4 1,5967
Стильбен (транс) 1,4-Днфеннлбутадиен (транс) 1,4-Днфеннлбутаднен (цис) 1,1,2-Т рнфеннлэтнлен 306 350 125 148 70 76 0,970[|5 Днбромид 237
Кетоны (жидкие)
Кетон Т. кип., “С Т. пл. производного, ®С
окснм семи- карба- зон фенил- гидра- зон 2,4-Дини- трофеиил- гидразон п-иитро- фенил- гидразон
Ацетон 56 59 187 42 126 152
Этнлметилкетон 80 146 117 129
Метилвнннлкетон 80 141
Диацетнл 88 74 278 245 315
(моно)
245
(Дн)
Изопропилметилкетон 94 113 117
Метил-н-пропнлкетон 102 58 ПО 144 117
Диэтнлкетон 102 69 139 156 141
Пинаколнн 106 74 157 125 139
Изобутилметилкетон 119 58 135 95
Хлорацетон 119 164
(разл.)
а,а- Дихлора цетон 120 163
Динзопропилкетон 125 34 160 95
н-Бутнлметилкетон 129 49 122 106 88
Мезитилоксид 130 49 164 203 134
Циклопентанон 131 56 205 50 142 154
Бромацетон 136 36
Ацетнлацетон 139 149 122
(моно)
209
(Ди)
2-Метилциклопентаион 139 184
Дн-н-пропнлкетон 145 133 75
Ацетон н 145 185 243 315
(разл.)
648
Приложение lit
Продолжение
Т. пл. производного, °C
Кетон т.сТ.. семи- фенил- 2,4-Дини- п-ннтро-
оксим карба- гндра- трофенил- фенил-
ЗОН зон гидразон гидразон
Окснацетон (ацетол) 146 71 196 103 129
я-Пентнлметнлкетон 151 127 207 89 73
Циклогексанон 155 90 166 77 162 146
Ди-втор-бутилкетон 162 84
2-Метнлциклогексанон 163 43 195 137
Диацетоиовый спирт 164 57 159
Днизобутилкетон 168 210 121 92
3-Метилциклогексанон 169 180 155
4-Метцлциклогексаион 169 37 199 130
н-Гексилметил кетон 172 122 58 92
Метйлциклогексилкетон 180 60 177 140 154
Цнклогептанон 181 23 163 148 137
Ди-я-бутилкетон 187 90 41
Ацетонила цетон 188 137 220 120 255
(Дн) (дн) (Дн)
Бутироин 190 99
Форон 198 48 186 112
Ацетофенон 200 59 198 105 250
Р-Туйон 202 55 174 114
/-Ментон 207 59 187 53 146
Изофорон 214 76 191 68 130
Метнл-2-тиенилкетон 214 81 191 95
2-Окснацетофенон 218 117 210 110
Бензилметнлкетон 216 70 198 87 156
Метил-о-толнлкетон 216 61 206 159
Пропиофенон 218 53 174 191
Метн л -я-то лн лкетон 220 57 200 207
Изобутнрофенон 222 f 61 181 73 163
1 94
Пулегон 224 174 142
Изовалерофенон 225 74 210 240
Карвон 225 72 142 106 193
Ди-я-а ми лкетон 226 Масло
Бензйлэтилкетон 226 135
Метил-п-толилкетон 226 86 205 94 260
л-Хлорацетофенон 228 88 232 176
Фенил-я-пропнлкетон 230 50 184 200
«-Хлорацетофенон 232 95 201 114 231
Метил-Б-феиилэтнлкетон 235 85 142
л-Метокснацетофенон 240 196
я-Валерофенон 242 52 166 162 166
о-Метоксиацетофенон 245 83 183 114
Ацетомезитилен 245
2-Ацето-п-цимол 250
Днпнон 345 син 151
134
анти
78
Таблицы производных
649
Кетоны (твердые)
Кетон Т. пл., °C Т. кип., °C Т. пл. производного, РС
оксим семикарбазон фенилгидразон 2,4-дин итрофе- иилгндразон
Бензилметилкетон 27 216 70 198 87 156
Форон 28 198 48 186
л-Метилацетофенон 28 226 88 205 96 258
2-Окснацетофенон 28 218 117 210 ПО
Дигексилкетон 33
а-Нафтнлметнлкетон 34 139 229 146
Днбензилкетон 34 330 125 146 120 100
л-Хлорпропиофенон 36 63 176
л-Метокснацетофенон 38 258 87 198 142 220
Бензнлнденацетон 41 262 115 187 157 223
Инданон-1 42 242 146 233 135 258
а,а'-Днхлорацетон 45 173 120
Тимохннон 45 232 162 202 93 а 180 а
Бензофенон 48 305 141 164 137 239
со-Бромацетофенон (фенацнл- 50 ( 88 146
бромид) t 97
л-Бромацетофенон 51 255 128 208 126 230
Метил-Б-нафтилкетон 53 300 145 237 176 262
Фенил-л-толилкетон 54 326 ( 154 122 109 200
t 136
<в-Хлорацетофенон (фенацил- 59 244 89 156 212
хлорид)
Дезокснбензоин 60 320 98 148 116 204
л-Метнлбензофенон 60 154 122 109 202
Бензоилацетон 61
Бензйлиденацетофенон 62 348 ( 73 ( 168 120 245
I 75 t 180
л-Метоксибензофенон 62 355 f 116 ( 132 180
I 138 t 90
л-Толухинон 68 134 179 130 а 128 •
Лаурой 69 40 179
Диокснацетон 72 84 278 .
(+)-а-Бромкамфора 76 274
Дибензонлметан 78 205
л-Хлорбензофенон 78 163 106 185
л-Нитроацетофенон 81 202 132 257 128 228
Флуоренон 83 341 195 234 151 283
Дн-л-толилкетон 92 334 163 100 229
Бензил 95 347 ( 137 f 182 ( 225 189
1(моно) 1 (моно) 1 (дн)
) 237 1 244
(Ди)
З-Окснацетофенон 96 195
л-Бромфенацилбромид 109 115
650
Приложение Ilf
Продолжение
т. пл. производного, ’С
S о S V я
Кетон и о О о rt о о. ч о о
ч в с X X Я Я о а X а я ч а а Я Ч к а ?1=
н f-J о QJ и е '4* д СЧ Я
Дибензнлиденацетон 112 144 190 180
Бензохинон 115 240 (разл.) 243 152а 231 а
Р-Нафтохинон 120 169 184 138 а
Аценафтенон 121 175 90
а-Нафтохинон 125 207 247 206а 278 а
п-Фенилфенацилбромид 125
Ванилнденацетон 130 128 230
Бензоин 133 343 151 206 (разл.) 106 245
Фуронн 135 161 81 217
п-Окснбензофенон 135 152 194 144 244
2,4-Дноксиацетофенон 147 199 214 159
п-Оксипропнофенон 148 229
4,4'-Днхлорбензофенон Метон (5,5-диметилднгидро- 148 149 135 115
резорцин) (моно)
176
(дн)
(безв.)
Бензантрон 170
Хингидрон 171
Ксантон 173 350 161 152
Г аллацетофенои 173 163 225
d,(-Камфора 178 205 118 235 164
(+)-Камфора р*Метнлантрахннон 179 179 205 118 237 233 177
Камфорхинон 198 / 170 ( 236 170а ( 36 а
J(моно) 1 140 * (дн) ( 158 1 147 t 190
Феиантренхиион 207 360 220 165а 313
1 (моно) 1 202 * (Дн) (разл.) (разл.)
Фторацетофенон 219
Антрахинон 286 224 183 а
Ализарин 289
Хлоранил 290 220
а Приведенные значении отвечают аддуктам хинонов
продукты не являются истинными гидразонами.
и замещенных гидразинов. Эти
Таблицы производных
651
Меркаптаны
Меркаптан Т. кип., °C Т. пл., ’С Т. пл. 2,4-динитро- фенилового эфира. °C
Метил 6 128
Этил 36 115
Изопропил 56 94
н-Пропил 67 81
Изобутнл 88 76
Аллил 90 72
трет-Пентил 97
я-Бутил 97 66
2-Пентил 112
Изопентнл 117 59
н-Пентнл 126 80
Этанднтиол-1,2 147 248
я-Гексил 151 74
Цнклогекснл 159 148
Тиенил 166 119
Феннл (тнофенол) 169 121
Пропан дитиол-1,3 169 194
я-Гептнл 176 82
Бензнл 194 130
о-Тиокрезол 194 15 101
л-Тнокрезол 195 91
п-Тиокрезол 195 43 103
н-Октнл 199 78
а-Феннлэтил 199 89
я-Нонил 220 86
Цетил 170 (3 мм) 50 91
п-Хлортнофенол 53
п-Бромтиофенол 75
.м-Нитротиофенол 75
в-Тнонафтол 81 145
4-Ксенил 111 146
Нитрилы (жидкие)
Нитрил Т. кип., °C Т. пл. производного, ®С
семнкар- базон алкил- феиил- кетона алкил-2,4,6- триокси- феи и лкетон гидрохлорид а-имино- алкнлмер- каптоуксус- иой кислоты
Акрилонитрил Ацетонитрил 78 81 199 218 114
Пропионнтрил 97 174 175 124
Изобутнронитрнл я-Бутироннтрил 108 118 184 181 135
З-Бутенонитрнл (аллнлциа- нид) а-Оксннзобутироннтрил Метокснацетоннтрнл 118 120 120
652
Приложение 111
Продолжение
Нитрил Т. кнп., °C Т. пл. производного, ®С
семикар' базой алкил- феннл- кетона а л кил-2,4,6- трнокси- фенилкетон гидрохлорид а-имино- алкилмер- каптоуксус- ной кислоты
Хлорацетонитрнл Изовалероннтрнл н-Валероннтрил 127 130 141 157 149 , 133 137
Фуроннтрнл Изокапроннтрил 146 155 146 (гидрат 88) 122 128
п-Капронитрил 164 128 (гидрат 104) 121 136
Манделонитрил Р-Хлорпропионнтрил Лактоннтрнл Энантонитрнл Гликольнитрил Бензонитрил у-Хлорбутнронитрнл Каприлоннтрил о-Толу нитрил Этилцианацетат .и-Толунитрнл п-Толунитрил Малоннтрил Р-Окснпропноннтрнл Феннлацетоннтрнл у-Оксибутиронитрил Деканоннтрил Цнннамоннтрил Глутароннтрил Адипонитрил 170 (разл.) 178 182 (разл.) 183 183 (разл.) 191 197 200 205 207 212 217 219 221 234 240 245 254 286 295 (гидрат 96) 133 124 134 168 181 144
Нитрилы (твердые)
Нитрил т. пл.. ®с Т. кип., *С
а-Нафтоннтрил 35 299
.м-Бромбензоннтрил 38 225
п-Толуннтрил 38 217
л-Хлорбензоннтрил 41
о-Хлорбензонитрнл 43 232
о-Бромбензонитрил 53
Сукциноннтрил 54 267 (разл.)
о-Иодбензонитрнл 55
п-Метокснбензонитрил 62 267
₽-Нафтонитрнл 66 306
Цнануксусиая кислота 66 165 (разл.)
Таблицы производных
653
Продолжение
Нитрил Т. пл., 'С Т. кип., °C
п-Хлорбензонитрил о-Ннтробеизонитрил п-Бромбеизонитрил н-Ннтрофенилацетонитрнл л-Ннтробензонитрил Метиленаминацетонитрил п-Нитробензонитрил 92 ПО 112 116 118 129 147
Нитросоединения (жидкие)
Нитросоедииеиие Т. кип., °C Т. пл. производного, °C
ацилпроизводные амина другие производные
бензол- сульфон- амид бензамид
Нитрометан Хлорпикрин 101 113 30 80
Нитроэтан 114 58 71
2-Нитропропан Тетраннтрометан 120 126 26 84 Амин-НС1 140
1-Ннтропропан 130 36 Амин-НС1 157
2-Нитробутан 140 70 76
2-Метнл-1 -ннтропропан 2-Метил-2-ннтробутан 1-Нитробутан 2-Ннтропентан З-Метнл-1 -ннтробутан 1-Нитропентан 141 150 152 154 164 173 53 58
1-Нитрогексан 193 96 40
Нитробензол 209 112 160 л-Днннтробензол 90
о-Ннтротолуол о-Ннтроэтнл бензол 2,6-Диметнлннтробензол 224 224 225 124 143 147 2,4-Днннтротолуол 70 2-Нитронзофта ле ва я кислота 300
Феннлнитрометан 226 (разл.) 88 105
л-Ннтротолуол 231 95 125
2,5-Днметилнитробензол 234 138 140 Триннтро-п-кснлол 137
2,4-Днметнлннтробензол 238 128 192 1,-3-Днметил-4,6-дн- ннтробензол 93
п-Нитроэтнлбензол 2,3-Диметилнитробензол Р-Нитростирол 241 250 260 (разл.) 151 Т рнннтроэтнлбензол 37
2-Нитроцнмол 264 102 2,6-Дннитроцнмол 54
о-Ннтроанизол 265 89 2,4,6-Тринитроанн- зол 68
о-Ннтрофенетол 268 102 2,4-Днинтррфенетол 86
654
Приложение III
Нитросоединеиия (твердые)
Т. ил . производного, ®С
ацилпроиз-
водные аминов
Н нтросоеди пение О
о - S другие производные
Ч С с S *5 2 0*9* « л 5 Ж п т м
Н н Ou rt ю
Трнбромннтрометан 10
Тетранитрометан 13 126
Триннтрометан 15 Соль NH< 200 (разл.)
•«-Нитротолуол 16 231 95 125
л-Ннтробензиловый спирт 27 Бензоат 94
2-Метил-2-ннтропропан 26 127
3,4-Днметилнитробензол 29 258
о-Хлорнитробензол 32 246 129 99 2,4-Днннтрохлорбен-
зол 50
2-Нитробифенил 33
4-Бром-З-нитротолуол 33
2,4-Дихлорнитробензол 33 258 128 115
л-Нитрофенетол 34 103
6-Хлор-2-ннтротолуол 37 238 6-Хлор-2-ннтробензой-
ная кислота 161
4-Хлор-2-интротолуол 38 240 4-Хлор-2-нитробензой-
ная кислота 142
ж-Иоднитробензол 38 •м-Иоданйлнн 33
.м-Нитроаннзол 38 258
о-Бромнитробензол 43 261 116 2,4-Динитробромбензол 79
п-Ннтробензнлнденхлорнд 43 n-Ннтробензойная кнс
лота 242
л-Хлорннтробензол 44 235 121 120 3,4-Диннтрохлорбен-
зол 37
Нитромезитнлен 44 245 204 Дниитромезитилен 86
о-Ннтробензил хлорид 48
о-Нитронодбензол 49
2,4 - Диннтрохлорбензол 52 а 315 178 2,4-Днннтрофенол 114
(разл.)
2,5-Днхлорнитробензол 54 120 2,5-Днхлор-1,3-днннт.
робензол 104
п-Нитроанизол 54 258 95 154 2,4-Дннитроаннзол 89
л-Бромннтробензол 54 257 136 3,4-Днннтробромбен-
зол 59
п-Ннтротолуол 54 238 120 158 2,4-Диннтротолуол 70
4-Иод-З-ннтротолуол 55 4-Иод-З-амннотолуол
л-Нитробензилбромнд 58
6-Нитростирол 58 260
(разл.)
п-Ннтрофенетол 60 283 143 173 2,4-Динитрофенетол 86
З-Нитробнфенил 61 З-Амннобифенил 30
а-Нитронафталнн 61 304 167 160
ж-Ннтробензилиденхлорид 65
Таблицы производных
655
Продолжение
Н итросое д ииеи ие Т. пл., ’С Т. кнп.. °C Т. пл. производного, °C
ацилпропз- водные аминов другие производные
бензол- сульфон- амид бензамид
2,6-Дннитротолуол 2,4,6-Т ринитроанизол 2,4-Динитротолуол л-Нитробензнлхлорид 2-Нитро-л-хлорбромбензол 2,4-Днннтробромбензол о-Нитробензиловый спирт 3,5 - Д имети ли нтробензол 3,5-Динитро-о-ксилол 2,4,6-Т риннтрофенетол 0-Нитроиафталнн 3,4-Диннтро-о-ксилол л-Нитробеизнлнденбромнд 2,4,6-Тринитротолуол 66 68 70 71 72 72 74 75 76 78 78 82 82 82 270 (разл.) 273 136 162 Пикриновая кислота 122 2,4,6-Триннтротолуол 82 л-Толундин 45 2,4-Дннитрофенол 114 Ацетат 35 Нитроксилндин 75 Пнкрнновая кислота 122 2-Ннтро-3,4-днметил- аиилин 66 n-Ннтробензойная кис- лота 241 2,4,6-Триннтробензой- ная кислота 220
л-Хлорннтробензол Пнкрнлхлорид 2,4-Диннтро-л-кснлол 4-Бром-1 -ннтронафта лнн 2,5-Дибромнитробензол 2,4-Динитромезнтилен 2,4-Динитроанизол 83 83 84 85 85 85 89 242 121 211 192 л-Ннтрофенол 114 Пикриновая кислота 122 3-Нитро-2,4-днметнл- аннлин 84 4-Бром-1 -нафтнламин 102 Ацетамид 171 2,4,6-Тринитромезитн- лен 232 2,4-Диннтрофенол 114
ж-Диннтробензол 3,6-Динитро-о-ксилол 2,4,5-Т ринитро-л-кснлол 3,5-Динитротолуол 2,4-Днметнл-1,3-дин нтро- бензол 2,4'-Дннитробифенил 2,3-Диннтро-л-ксилол л-Ннтробензиловый спирт 4-Хлор-2-нитроанизол л-Нитробензилбромид 2,5-Динитро-л-ксилол 2,3'-Динитробифенил 90 60 90 92 93 93 93 93 98 99 101 НО 302 194 240 .и-Нитроанилнн 114 4 -Ннтро-2,3-диметнл- анилнн 114 2,4-Диметил-1,3,5-три- нитробензол 125 2,3-Дннитро-л-толуи- ловая кислота 249 Ацетат 78 4-Хлор-о-аиизндин 84 4-Нитро-2,6-диметнл- анилин 158
656
Приложение III
Продолжение
Ннтросоедииение Т. пл.. ’С Т. кип., ’С Т. пл. производного, *С
апнлпронз- водные аминов другие производные
бензол- сульфон- амнд бензамид
4-Ннтробнфеннл 113 230
о-Днннтробензол 4,5-Д иннтро-о- ксилол 1,3,5-Триннтробензол 2,6-Дннитро-п-ксилол 2,4,6-Трибромнитробензол 118 118 122 123 125 319 186 301 198 о-Ннтроанилии 71 2-Нитро-4,5-диметил- анилин 140 Продукт присоединения нафталина 153 3-Ннтро-2,5-диметил- аннлнн 98 2,4,6-Т ръброманилин 122
п-Бромнитробензол 2,2'-Диннтробифенил 4,5-Динитро-л-ксилол п.п'-Днннтрофеннловый эфир 2,5-Динитро-п-ксилол 1,8-Дннитронафталин п-Нитронодбензол 126 128 132 143 147 170 171 259 134 204 210 п-Ннтрофенол 114 2,2'-Диаминобнфенил 81 2-Ннтро-4,6-днметнл- аннлин 76 4-Нитро-2,5-диметил» анилин’ 145 1,3,8-Трннитронафт а- лин 218
п-Динитробензол 3,3'-Дннитробнфенил 1,5-Динитронафталин 4,4'-Дннитробнфенил 172 200 214 237 299 247 300 п-Ннтрофенол 114 З.З'-Днамннобнфенил 94 1,4,5-Тринитронафта- лин 154 Бензидин 128
а Известны метастабнльные формы с т. пл. 27 и 43"С.
Нитрозо-, азокси-, азо- и гидразосоединения
Соединение Т. пл., °C Соединение Т. пл.. “С
Н итрозосоединения N-Ннтрозоднфеннламин 66
1 -Нитрозо-2,4-днметнлбен- зол и-Ннтрозотолуол N-Ннтрозоацетанилнд 41 Нитрозобензол о-Нитрозотолуол 68 72
48 51 (разл.) п-Ннтрозо-М-этнланнлин п-Ннтрозо-Ь1,М-днэтнлани- 78 84
л-Нитрозотолуол 53
Таблицы производных
657
П родолжение
Соединение Т. пл.. *с Соединение Т. пл.. -С
п-Ннтрозо-М,Н-диметил- 87 1 -Бензолазонафталин 70
анилин о-Оксназобензол 83
1 -Нитрозонафталнн 98 Бензолазоднфениламин 96
5-Ннтрозо-о-анизидин 107 n-Д иметнла миноазобензол 117
1 -Ннтрозо-2-нафтол 109 п-Амнноазобензол 125
п-Нитрозо-М-метнланнлин 116 Бензолазо-о-крезол 128
п-Нитрозофенол 125 (разл.) 1 -Бензолазо-2-нафтол 131
5-Нитрозо-о-крезол 135 о-Азофенетол 131
п-Ннтрозодифеннламнн 144 2- Бензолазон афта лнн 131
2-Нитрозо-1 -нафтол 152 (разл.) 1,2'-Азонафталин 136
Ннтрозотнмол 162 2-Бензолазо-2-нафтол 138
п-Ннтрозоанилнн 174 п-Азотолуол 145
4-Нитрозо-1 -нафтол 194 (разл.) о-Азобифенил 145
п-Оксназобензол 152
Азоксисоединения п-Азофенетол 160
Азокснбензол 36 Бензолазорезорцнн 170
ж-Азокситолуол 39 1,1'-Азонафталин 190
о-Азокснтолуол п-Азокситолуол 59 75 4-Бензолазо-1 -нафтол 2,2'-Азонафталин 206 (разл.) 208
1, Г-Азоксинафта лин 127
п,п'-Дихлоразоксибензол 154 Гидразосоединения
о-Азоксибнфенил 158
2,2'-Азоксинафталнн 168 м-Гндразотолуол 38
4,4'-Диннтроазбксибензол 200 Гидразобензол 130
п-Азокснбнфеннл 212 п-Гндразотолуол 134
Азосоединения 2,2'-Г ндразонафталнн 1,1 '-Гидразонафталин 140 153
о-Азотолуол 55 о-Гндразотолуол 161
ж-Азотолуол 55 п-Гндразобифенил 170
Азобензол 68 о- Г н дразобифени л 182
Фенолы (жидкие)
Фенол Т. кип., °C Т. пл. ’С Т. пл. производного, °C
а-нафтил- уретан бромпроизводное арилокси- уксусная кислота
о-Хлорфенол 175 7 120 145
Фенол 180 42 133 Трнбром 95 99
о-Крезол 190 31 142 Днбром 56 152
о-Бромфенол 195 129 Трибром 95 143
З-Хлор-п-крезол 197 108
658
Приложение III
П родолжение
Фенол Т. кип., °C Т. пл., °C Т. пл. производного, °C
а-нафтил- уретан бром про изво диое арилокси- уксусная кислота
п-Крезол 202 36 146 Дибром 49 136
л-Крезол 202 3 128 Трибром 84 103
Гваякол 205 28 118 Трибром 116 121
о-Этнлфенол 207 141
2,4-Днхлорфенол 209 43 Бром 68 138
2,4-Диметилфенол 212 27
л-Хлорфенол 214 33 158 НО
п-Хлорфенол 217 43 166 156
л-Этилфенол 217 77
о-Этоксифенол 217 28
п-Пропилфенол 232 22
п-Изобутилфенол 236 125
л-Бромфенол 236 32 108
Карвакрол 237 0 116 Бром 46 151
2,4-Днбромфенол 238 36 Трибром 95 153
Монометиловый эфир 243 129 Трибром 104 118
резорцина
п-Бутилфенол 248 22 81
Эвгенол 250 122 Тетрабром 118 80
п-Амнлфенол 253 23 90
Изоэвгенол 267 150 Дибром 94 94
Фенолы (твердые)
Фенол Т. пл., °C Т. пл. производного, °C
а-нафтнл- уретан бромпроизводиое сложный эфир
2,4-Диметнлфенол 26 135 Бензоат 38
Гваякол 28 118 Трнбром 116 Бензоат 57
о-Этоксифенол 28 Бензоат 31
о-Крезол 31 142 Дибром 56 3,5-Дннитробен-
зоат 133
л-Бромфенол 32
2-Бром-4-хлорфенол 34 Бензоат 100
З-Нитро-п-крезол 34 Бензоат 102
п- Крезол 36 146 Тетрабром 108 Бензоат 71
2.4-Дибромфенол 36 Трибром 95 Бензоат 97
л-Иодфенол 40
Фенол 42 133 Трибром 95 Бензоат 68
Таблицы производных
659
Продолжение
Фенол Т. пл. производного, °C
а-иафтнл- уретан бромпронзводное сложный эфир
n-Хлор фенол 43 166 Бензоат 93
2,4-Дихлорфенол 43 Бром 68 Бензоат 97
о-Иодфенол 43
о-Нитрофенол 45 113 4,6-Днбром 117 3,5-Дннитробен-
зоат 142
6-Хлор-З-метилфенол 46 128 Бензоат 31
п-Этилфенол 47 Бензоат 60
5-Хлор-о-крезол 48 Бензоат 71
3,5-Дибром-п-крезол 49 176 Бензоат 95
2,6-Днметилфенол 49 Бром 79
Тнмол 50 160 Бром 55 Бензоат 32
2-Хлор-З-метилфенол 50 Бензоат 56
о-Цнклогексилфенол 53
Г ндрохинон, мономети- 53 Бензоат 87
ловый эфир о-Бензилфенол 54
3,5-Днбром-о-крезол 57 п-Ннтробен-
зоат 137
2,3-Днхлорфенол 57 Днбром 90
2-Окси-3,5-днбромто- 57
луол Орснн (гидрат) (3,5-ди- 58 160 Трнбром 104 Дибензоат 88
окситолуол) о-Феннлфенол 58 Динитро 207
4-Хлортимол 59
2,5-Днхлорфенол 59 Бензоат 69
п-Бромфенол 63 169 Трнбром 95 Бензоат 102
3,4-Днметнл фенол 63 142 Трнбром 171 Бензоат 58
2-Хлор-5-окснтолуол 66 154 Бензоат 86
2,4,6-Трихлорфенол 67 188 Бензоат 70
3,5-Дихлорфенол 68 Трнбром 189 Бензоат 55
2,4,5-Трнхлорфенол 68 Бензоат 93
3,5-Диметил фенол 68 Трнбром 166 3,5-Дннитробен-
зоат 195
2,4,6-Триметилфенол 69 Днбром 158
4-н-Гекснлрезорцин 69
Мезнтол 70 Дибром 158 Бензоат 62
Псевдокуменол 71 Бром 35 Бензоат 63
2,4-Дииодфенол 72 Бензоат 98
2,5-Диметилфенол 74 173 Трибром 178 Бензоат 61
2,3-Диметнлфенол 75
З-Оксибифенил 78 Бензоат 61
4-Хлор-2-нодфенол 78 Бензоат 88
Изодурол 81 Бензоат 72
п-Бензилфенол 84 Бензоат 87
2-Оксн-3,5-днннтрото- 86 Бензоат 132
луол сс-Нафтол 94 152 2,4-Днбром 105 Ацетат 46
п-Иодфенол 94 Бензоат 119
660
Приложение III
Продолжение
Фенол Т. пл., “С Т. пл. производного, °C
а-нафтнл- уретан бромпроизводное сложный эфир
п-трет-Бутнлфенол 95 Бром 50 Бензоат 83
2,4,6-Трнбромфенол 95 153 Тетрабром 120 Ацетат 82
п-трет-Амилфенол 96 Бензоат 61
.«-Нитрофенол 97 167 Днбром 91 Бензоат 95
Пирокатехин 104 175 Тетрабром 192 Диацетат 63
Хлоргндрохинон 106 Ди ацетат 72
Орсин (3,5-днокснто- 107 160 Трнбром 104 Днбензоат 88
луол) 1,2-Диоксннафталнн 2,2'-Днокснбнфеннл 108 Ацетат 106
ПО Ацетат 95
Резорцин 110 4,6-Дибром 112 Днбензоат 117
3-Оксн-2,4,6-тринитро- НО Ацетат 135
толуол Бромгндрохннон ПО Днбром 186 Диацетат 72
п-Ннтрофенол 114 151 2,6-Днбром 142 Ацетат 81
2,4-Диннтрофенол 114 6-Бром 118 Ацетат 72
п-Окснбензальдегид 115 3,5-Днбром 181 Бензоат 90
4-Хлор-3,5-днбромфенол 121 Бензоат 93
Гидрохинон, монобензи- 122
левый эфир Пикриновая кислота 122 Ацетат 76
Р-Нафтол 122 157 Бром 84 Ацетат 70
Толугидрохинон 124 Диацетат 52
п-Цнклогексилфенол 132 Беизоат 118
Пирогаллол 133 Днбром 158 Триацетат 165
2,4-Диннтронафтол 138 Бензоат 174
1,8-Диоксннафталин 140 Днацетат 148
1,2,4-Триокснбензол 140 Триацетат 97
2,4-Дннитрорезорцин 148
2,4,6-Трииодфенол 159 Бензоат 137
п-Феннлфенол 165 Ацетат 89
Гидрохинон 169 247 Днбром 186 Диацетат 123
Галлацетофенон 173
1,4-Дноксинафталин 176 220 Диацетат 128
Стифннновая кислота 180
2,7 - Дноксинафта лин 186 Дибеизоат 139
Пентахлорфенол 190 Ацетат 150
Карбостирнл 200
Тетрабром-о-крезол 210 Ацетат 154
4-Хлор-2,4,5,6-тетра- бромфенол Флороглюцин 215 Бензоат 203
218 Трибром 151 Триацетат 105
Дн-Р-нафтол 218
2,3,5,6-Тетрахлоргидро- 237 Днацетат 245
хннон
1,5-Диоксинафталнн 258 Диацетат 160
Фенолфталеин 261 Диацетат 146
п.п'-Диокснбнфенил Дн-а-нафтол 272 Диацетат 160
300
Таблицы Производных
661
Сульфонамиды (твердые)
Сульфонамид Т. пл.. 'С Сульфонамид Т. пл., °C
З-Метнл-1-бутан- 3 о-Амннобеизол- 153
2-Метнл-1 -пропан- 16 л-Бромбеизол- 154
1-Бутан- 45 2-Иод-1 -нафталин- 154
1-Пропан- 52 2,6-Днхлор-4-метилбензол- 155
Этан- 58 2,4-Днннтробензол- 157
2-Пропан- 60 2,3-Днхлор-5-метилбензол- 158
4-Изопропил-2-метил бензол- 73 л-Нитробензил- 159
1-Гептан- 75 5-Хлор-2-ннтробензол- 159
2,4,5-Трнметокснбензол- 76 3-Хлор-2-метнл-5-ннтробен- 161
З-Этнлбензол- 86 ЗОЛ-
Метан- 90 п-Аминобензол- 163
2,6-Диметил бензол- 96 4-Хлор-2-нитробензол- 164
Цетил- 97 3,6-Днхлор-2,5-днметилбен- 165
2-Этилбензол- 100 ЗОЛ-
З-Метил-4-фторбензол- 105 5-Хлор-З-нитробензол- 165
Бензил- 105 л-Нитробензол- 166
л-Толуол- 108 2-Метил-5-бромбензол- 166
4-Этилбензол- 109 п-Бромбензол- 166
1 -Метил-6-нафталин- 116 2,3-Диметнл бензол- 167
Имид о-сульфобензойиой кис- 121 2,4-Днметоксибензол- 167
лоты (сахарин) 5-Хлор-2-метил-3-нитробен- 167
2-Фенил-1-этаи- 122 ЗОЛ-
п-Фторбензол- 125 2-Метил-4-бромбензол- 168
4-Метнл-З-хлорбензол- 128 2,4-Днхлор-6-метил бензол- 168
4-Хлор-З-метилбензол- 128 2,5-Днметнл-З-нитробензол- 173
З-Хлор-2-метокси бензол- 131 2-Метнл-4-фторбензол- 173
(+)-Камфор-10- 132 1 -Метнл-4-нафталин- 174
3,5-Днметилбензол- 135 3,4-Дибромбензол- 175
3,4-Днхлорбензол- 135 2,4-Дихлор-5-метнлбензол- 176
(+)-Камфор-8- 137 4-Хлор-З-ннтробензол- 176
2,4-Днметил бензол- 137 4-Бром-З-ннтробензол- 177
п-Толуол- 137 Фенол-4- 177
о-Ннтробензнл- 137 2-Хлор-5-метнл-6-нитробен- 177
2-Бром-1-нафталнн- 140 ЗОЛ-
2-Метил-5-фтор бензол- 141 п-Ннтробензол- 180
л«-Амннобензол- 142 2,4-Днхлорбензол- 180
2-Метил-5-хлорбензол- 142 2,4,5-Т рнметилбензол- 181
2,4,6-Тр имети лбензол - 142 3-Хлор-2-метнл-6-ннтробен- 181
(+)-Камфор-3- 143 ЗОЛ-
5-Хлор-2-метилбензол- 143 2,5-Днхлорбензол- 181
3,4-Диметилбензол- 144 5-Хлор-4-метнл-2-ннтробен- 181
«-Хлорбензол- 144 ЗОЛ-
1 -Метил-З-нафталнн- 144 5-Хлор-4-метил-3-ннтробен- 182
4-Метил-З-бром бензол - 146 ЗОЛ-
2,5-Днметилбензол- 148 5-Хлор-2-метил-6-нитробен- 183
л-Хлорбензол- 148 ЗОЛ-
1-Нафталин- 150 1-Хлор-4-нафталнн- 185
4,6-Днхлор-2,5-диметилбен- 150 4 -Хло р -2,5 - дн метнл бензол - 185
ЗОЛ- 4-Хлор-2-метилбензол- 185
о-Толуол- 153 , 6-Хлор-З-ннтробензол- 186
Бензол- 153 о-Бромбензол- 186
662
Приложение III
Продолжение
Сульфонамид Т. пл.. •с | Сульфонамид Т. пл., -С
4-Хлор-1-нафталин- 186 З-Метил бензол-1,5-ди- 216
4,5-Дихлор-З-метилбензол- 186 2-Нафталин- 217
2,3-Дихлор-6-метилбеизол- 186 2,4,6-Трибромбензол- 220
3,5-Дихлор-2-метилбензол- 186 (разл.)
2-Хлор-4-метил бензол- 186 3-Хлорбензол-1,5-ди- 224
2,4-Днаминобеизол-1,5-ди- 187 2-Метилбензол-1,4-ди- 224
8-Иод-1 -нафталин- 187 2,3,4-Трихлорбеизол- 226
2-Метил-5-нитробеизол- 187 (разл.)
о-Хлорбеизол- 188 3,4-Дихлор-2-метилбеизол- 228
2,6-Дихлор-З-метилбеизол- 188 1,3-Бензолди- 229
2-Хлор-5-метил-4-иитробеи зол- 188 4-Дифенил- 230
л-Бромбеизил- 188 1-Иод-5-иафталин- 236
1 -Метил-7-нафталин- 189 4-Метилбеизол-1,2-ди- 237
4-Этилбензол-1,3-ди 190 2,3-Дихлор-4-метилбеизол- 237
2,4 - Дибром бензол- 190 4-Хлор-З-иитробеизол- 237
о-Нитробензол- 191 1,2-Диметилбеизол-3,5-ди- 239
4-Метилбензол-1,3-ди- 191 4 -Метоксибеизол-1,3-ди- 240
2,5-Диметил-6-иитробеизол- 192 2,7-Нафталинди- 242
2,7-Аитрахинонди- 192 1,5-Антрах инонди- 246
1 -Бром-4-иафталии- 193 2,4-Диметилбензол-1,5-ди- 249
2,5-Дибром бензол- 194 2,З-Д иметилбеизол-1,4-ди- 251
2-Хлор-5-метил-3-иитробеи зо л- 196 1,2-Беизолди- 254
2,5-Диметил-4-иитробеизол- 198 З-Бром бензол-1,5-ди- 256
8-Хлор-1 -нафта л ни- 199 2-Амиио-5-метилбензол- 1',3-ди 257
п-Нитробензил- 200 2-Метил бензол-1,3-ди- 260
2,4,5-Т рихл орбензол- > 200 Аитрахиион-Р- 261
3,4-Дихлор-2,5-диметилбензол- 201 1,4-Нафталинди- 273
4-Хлор-3-метил-5-иитробензол- 201 2,4-Дихлорбензол-1,5-ди- 276
1 -Иод-4-нафталин- 202 1,4-Беизолди- 288
2,4-Дихлор-З-метилбеизол- 203 2,5-Диметилбензол-1,3-ди- 295
п-Иодбеизил- 206 1,6-Нафталинди- 298
2-Метил-6-нафталин- 206 Дифеиил-4,4'-ди- 300
2,6-Диметил-8-иафталин- 207 1,5-Нафталинди- 310
4-Окси-2,6-диметилбензол-1,3- 208 2,5-Диметилбензол-1,4-ди- 310
ди- 1,3,5-Бензолтри- 312
2,4,6-Трихлорбензол- 212 1 -Циаи-8-иафталин- 336
1 -Нитро-2-иафталин- (разл.) 214 1,8-Антрахиионди- 340
Сульфоиилхлориды (жидкие)
Сульфоннлхлорид Т. кип., °С/мм „25 "D Производное
кислота. т. кип. (®С/мм) амид, т. пл. ГС» анилид, т. пл. (®С)
Метай- 60/21 1,4490 167/10 90 99
Этаи- 70/20 1,4506 58 58
2-Пропан- 61/9 1,4522 60 84
Таблицы производных
663
П родолжение
Сульфонилхлорид Т. кип., •С/мм „25 "D Производное
кислота. т. кип. (°С/мм) амид, т. пл. °C анилид, т. пл. (®С)
1-Пропан- 67/9 1,4518 136/1 52 -10
2-Метил-1 -пропан- 73/11 1,4520 16 38
1-Бутан- 75/10 1,4517 147/0,5 45
З-Метил-1-бутаи- 1-Гептан- 88/9 125/9 1,4530 3 75 42
Сульфонилхлориды (твердые)
Сульфонилхлорид Т. пл.. °C Т. пл. производного, °C
кислота амид анилнд
о-Толуол- 10 57 153 136
л-Толуол- 12 108 96
2-Этилбензол- 12 100
4-Этилбеизол- 12 110
Бензол- 14 66 153 110
Гептан- 19 75
3,4-Дихлорбеизол- 19 135
2,6-Дихлор-З-метил бензол- 19 188
2-Метил-5-хлорбензол- 21 142
2,5-Диметилбензол- 25 48 148
о-Хлорбеизол 28 188
2-Феиил-1-этан- 33 91 122 77
2,4,5-Трихлорбеизол- 34
2,4-Днметилбензол- 34 62 137 ПО
3,4-Дибромбензол- 34 175
2-Метил-5-бромбензол- 35 166
п-Фторбеизол- 36 125
З-Хлор-4-метилбеизол- 38 134 96
2,5-Дихлорбеизол- 38 181 160
2,6-Д иметилб ей зол 39 98 96
2,4,6-Трихлорбензол- 40 212 (разл.)
2,3-Дихлор-4-метилбеизол- 41 237
4-Хлор-З-нитробензол- 41 176
2,4 - Д ихлор-6-мети лбензол 43 168
2-Метил-5-нитробеизол- 44 187 148
2-Хлор-4-метилбензол- 46 186
2,3-Диметилбензол- 47 167
2,3-Дихлор-6-метил бензол- 49 186
2-Метил-4-бромбензол- 50 168
4-Хлор-2,5-диметилбензол- 50 100 185 155
4-Бром-З-метилбензол- 50 146
о-Бромбеизол- 51 186
3,4-Диметилбензол- 52 64 144
3,4-Дихлор-2-метилбензол- 52 228
664
Приложение III
Продолжение
Сульфоиилхлорид Т. пл., °C Т. пл. производного, °C
кислота амид анилид
2,4-Дихлорбензол- 53 180
л-Хлорбензол- 53 68 144 104
2-Метил-4-хлорбензол- 53 185
3,5-Д их лор -2- мети лбенз ол- 54 186
4-Хлор-2-метилбензол- 54 185
Цетил- 54 97
2,6-Дихлор-4-метилбеизол- 56 155
4-Метил бензол-1,3-ди- 56 191 189
2,4,6-Триметилбеизол- 56 77 142 109
2,4-Дихлор-З-метил бензол- 59 203
5-Хлор-2-метил-3-иитробензол- 60 167
4-Метил-З-бромбензол- 60 146
3-Хлор-2-метил-6-нитробеизол- 60 181
2,4,6-Трибромбензол- 60 220 (разл.)
2,4,5-Триметилбензол- 61 112 181
2,5-Диметил-З-иитробензол- 61 200 173 144
3,4-Дихлор-2,5-диметилбензол- 62 201 157
ж-Нитробеизол- 63 166 126
4-Метил-З-хлорбензол- 63 128
1,3-Бензолди- 63 229
2-Хлор-5-метил-3-нитробеизол- 63 196
2,3-Дихлор-5-метилбеизол- 64 158 .
3-Хлор-2-метил-5-иитробеизол- 64 161
2,3,4-Трихлорбеизол- 65 226 (разл.)
(+)-Камфор-10- 67 193 132 121
1-Нафталин- 68 90 150 112
о-Нитробензол- 68 70 191 115
л-Толуол- 69 92 137 103
3,5-Дихлор-4-метилбеизол- 69 191
5-Хлор-4-метил-3-иитробеизол- 70 182
2,4-Диметоксибензол- 71 192 167
2,5-Дибромбензол- 71 194
3,6-Дихлор-2,5-диметилбензол- 71 165 171
2,4-Дихлор-5-метилбеизол- 72 176
2,5-Диметил-4-нитробензол- 75 140 198 131
л-Бромбензол- 75 103 166 119
2-Нафталин- 76 91 217 132
2,4-Дибромбензол- 79 190
1,2-Д иметилбеизол-3,5-ди- 79 239 200
л-Нитробензол- 80 95 180 171
2,5-Диметилбензол-1,3-ди- 81 295 174
4,6-Дихлор-2,5-диметил бензол- 81 150 175
1 -Метил-4-нафталии- 81 174 158
З-Хлор-4-метоксибеизол- 82 131
1 -Бром-4-иафталии- 83 193
л-Иодбеизол- 85 183 143
4-Метоксибеизол-1,3-ди- 86 240 209
(+)-Камфор-3- 88 143 124
t-Метил-б-нафталии- 88 116
Таблицы Производных
665
Продолжение
Сульфонилхлорид Т. пл., •С Т. пл. производного, °C
кислота амид аиилид
4,5-Дихлор-З-метилбензол- 88 186
2-Метилбензол-1,3-ди- 88 260 162
6-Хлор-З-нитробензол- 90 104 186
1,2-Этанди- 91
Беизнл- 92 105 102
2-Хлор-5-метил-4-иитробензол- 92 188
3,5-Диметил бензол- 94 135 119
Этанди- 95 104 69
З-Метилбеизол-1,5-ди- 95 216 153
4-Хлор-1 -нафталин- 95 186 143
2-Метилбеизол-1,4-ди - 98 224 178
5-Хлор-4-метил-2-нитробензол- 99 128 181
3,5-Дннитробензол- 99 235
я-Нитробеизнл- 100 159 (разл.)
З-Бромбензол-1,5-ди- 100 256
8-Хлор-1 -нафталин- 101 199
2,4-Дииитробензол- 102 157
З-Хлорбеизол-1,5-ди- 106 100 (разл.) 224
2,5-Диметил-6-нитробензол- НО 145 192 182
2-Иод-1-нафталин- ПО 154
4-Метилбеизол-1,2-ди- 111 237 190
1 -Иод-5-иафталии- ИЗ 236
2,5-Дихлорбензол-1,3-ди- 114 217
8-Иод-1-нафталин- 115 187 140
4-Бифеиил- 115 230 125
4-Окси-2,6-диметил бензол-1,3-ди- 119 208 207
1 -Иод-4-нафталин- 121 202 133
1 -Нитро-2-нафталии- 121 105 214 202
2-Хлор-5-метил-6-иитробеизол- 122 177
1,7-Нафталинди- 122
2,4-Дихлорбензол-1,5-ди- 123 276
1 -Метил-7-нафталии- 123 189
Дифенилметан-4,4'-ди- 124 59 178
1 -Метил-З-иафталин- 125 144
1,6-Нафталинди- 128 125 298 196
2,4-Диметилбензол-1,5-ди- 129 249
2,4,5-Триметоксибензол- 130 76 170
1,3-Нафталииди- 138
Н-Камфор-8- 138 137
1,4-Бензолди- 139 288
1,2-Бензолди- 143 254 241
1,3,5-Нафталиитри- 5-Хлор-2-метил-о-нитробеизол- 146 154 183
1,4,5-Нафталинтри- 156 257 192
2-Амино-5-метилбензол-1,3-ди- 156
2,7-Нафталинди- 162 242 223
2,5-Диметилбеизол-1,4-ди- 164 310
1,4-Нафталииди- 166 273 179
2-Хлор-7-антрахинон- 176
1,5-Нафталииди- 183 245 310 249
666
Приложение III
Продолжение
Сульфоиилхлорид Т. пл., °C Т. Пл. производного. °C
кислота амид аиилид
2,7-Аитрахииоиди- 186 192
1,3,5-Беизолтри- 187 100 312 237
(разл.)
1,3,6-Нафталинтри- 193
Аитрахииои-₽- 197 261 193
1,6-Антрахииоиди- 198 217 227
(разл.)
2,3,6-Нафталиитри- 200
2-Хлор-6-антрахииои- 202
Дифенил-4,4'-ди- 203 72 300
Антрахиион-а- 214 218 216
9,10-Дихлорантрацен-2- 221 279 248
1,8-Аитрахииоиди- 223 294 340 238
2,6-Нафталииди- 225
Антрацен-1,8-ди- 225 333 224
1,7-Антрахинонди- 232 120 238
2,6-Антрахинонди- 250 321
1,5-Ант рахинонди- 265 310 246 270
(разл.) (разл.) (разл.)
2,4-Диаминобензол-1,5-ди- 275 187 236
1,3,5,7-Нафталннтетра- 310
Приложение IV
Токсичность органических соединений
В последнее время вопросу токсичности органических соединений уделяется
много внимания. Поэтому мы включили в книгу четыре приведенные ниже таб-
лицы. Хотя их содержание едва ли хоть сколько-нибудь полно и всесторонне от-
ражает суть вопроса, мы надеемся, что эта попытка обращения к такой серьез-
ной проблеме будет полезной.
Табл. IV. 1 представляет собой список нескольких сотен органических соеди-
нений и соответствующих средних значений допустимых концентраций для них.
Эти средние значения указывают максимально допустимые концентрации дан-
ного вещества в помещении при 8-часовом рабочем дне и 40-часовой рабочей не-
деле по нормам OSHA. Они могут быть определены по формуле, где время воз-
действии Е связано с концентрацией вещества Ci, с которым работающий нахо-
дится в контакте в течение некоторого временного периода i продолжительностью
h часов:
с t + c.t. + ... + С t
Е , а а г Ъ Ъ ' ' п п
8
Из формулы следует, что хотя и можно некоторое время работать с ацет-
альдегидом в помещении с концентрацией его, большей 200 м. д., однако этот
период должен быть компенсирован работой в течение другого периода времени
при концентрации ацетальдегида, меиыцей 20 м.д. Соотношение между этими
временами должно быть таким, чтобы за 8-часовой рабочий день средняя кон-
центрация была ие выше 200 м. д.
В табл. IV. 2 приведен ряд соединений и их предельные концентрации, ко-
торые недопустимо превышать при работе с ними. Эти вещества часто относятся
к тем классам органических соединений, которые перечислены в табл. IV. 4.
В табл. IV. 3 приведены средние за 8 ч и предельно допустимые концентра-
ции для сильно ядовитых соединений, таких, как бензол. Кроме того, в таб-
лицу включены максимально допустимые времена воздействия на человека при
работе с этими веществами в больших концентрациях. Например, химик может
работать с метиленхлоридом не более 5 мин за 2 ч при его концентрации
2000 м.д.; при концентрации 1000 м.д. он может работать дольше, но среднее
время работы с этим веществом должно быть таким, чтобы его средняя кон-
центрация была ниже 500 м. д. за 8-часовой рабочий день.
В табл. IV. 4 приведены либо названия, либо структурные формулы, либо
аббревиатуры веществ, преимущественно органических, хорошо известных своей
токсичностью. Одна из главных целей этой таблицы — развить у химиков чутье
иа такие классы соединений, которые очень часто оказываются токсичными.
С любыми представителями этих классов необходимо обращаться с крайней
осторожностью.
Данные, приведенные в таблицах, получены от OSHA и NIOSH; одиако со
временем они подвергаются коррекции, поэтому для получения последних дан-
ных о предельно допустимых концентрациях токсичных веществ необходимо об-
ращаться в эти правительственные ведомства. Хорошим справочником по всем
этим вопросам является книга: Green М. Е., Turk A., Safety in Working with
Chemicals, MacMillan, New York, 1978. В ией приведены данные ие только о воз-
можных химических отравлениях, но также и об опасных физических воздей-
ствиях, с которыми можно встретиться при работе в химических лабораториях.
668
Приложение IV
Таблица IV. 1
Допустимые концентрации обычных соединений *
Соедииеиие Концентрация в воздухе
ч. на млн. мг/м3
Акриламид, через кожу 0,3
Акрилонитрил, через кожу 20 45
Акролеин 0,1 0,25
Аллиловый спирт, через кожу 2 5
Аллилпропилдисульфид 2 12
Аллилхлорид 1 3
Альдрии, через кожу 0,25
«-Амилацетат 100 525
етор-Амилацетат 125 650
2-Аминопиридин 2-Амииоэтанол см. Этаноламин 0,5 2
Аммиак 50 35
Аиизидии (о,л-изомеры), через кожу 0,5
Аиилии, через кожу 5 19
Ацетальдегид 200 360
Ацетилен, тетрабромид 1 14
Ацетон 1000 2400
Ацетонитрил 40 70
Бензилхлорид 1 5
Бензин 500 2000
Бензоилпероксид 5
л-Бензохинон (хинон) 0,1 0,4
Бифенил 0,2 1
Бортрифторид 1 3
Бромоформ, через кожу 0,5 5
Бутадиен-1,3 1000 2200
Бутанои-2 (метилэтилкетон) 200 590
н-Бутилацетат 150 710
етор-Бутилацетат 200 950
трет-Бутилацетат 200 950
н-Бутилглицидиловый эфир (БГЭ) 50 270
Бутилмеркаптаи 10 35
н-Бутиловый спирт (бутанол-1) 100 300
втор-Бутиловый спирт (бутанол-2) 150 450
трет-Бутиловый спирт (2-метилбутаиол-2) 100 300
n-трет-Бу тилтол у ол 10 60
2-Бутоксиэтанол (бутилцеллозольв), через кожу 50 240
Винилтолуолы 100 480
н-Гексан 500 1800
Гексанои-2 (гексои, метилизобутилкетон) 100 410
втор-Гексилацетат 50 300
Гексахлорнафталии, через кожу 0,2
Гексахлорэтаи, через кожу 1 10
н-Гептаи 500 2000
Г идрохинон 2
2.4-Д 10
ДДТ, через кожу 1
Диазометаи 0,2 0,4
Токсичность органических соединений
669
Продолжение
Соединение Концентрация в воздухе
ч. на млн. мг/м’
1,2-Диамииоэтаи, см. Этилендиамии Диацетоиовыи спирт (4-окси-4-метилпеитанон-2) 50 240
Дибутилфталат 5
Диизобутилкетои 50 290
Диизопропиламин, через кожу 5 20
Диизопропиловый эфир 500 2100
Дильдрии, через кожу 0,25
Диметиламин 10 18
Диметиланилин (N.N-диметилаиилин), через ко- 5 25
жу Диметилацетамид, через кожу 10 35
Диметилбензол см. Ксилол 2,6-Диметилгептаион-4 см. Диизобутилкетои 1,1-Диметилгидразии, через кожу 0,5 1
Диметилсульфат, через кожу 1 5
Диметилформамид, через кожу 10 30
Диметилфталат 5
Диметоксиметаи см. Метилаль Динитробензол (все изомеры), через кожу 1
о-Дииитрокрезол, через кожу 0,2
Дииитротолуол, через кожу 1,5
Диоксаи, через кожу 100 360
Дифениловый эфир, пары 1 7
Дифтордибромметан 100 860
п-Дихлорбеизол 75 450
Дихлордифторметаи 1000 4950
Дихлор метай см. Метилеихлорид 1,2-Дихлорпропаи см. Пропилендихлорид Дихлортетрафторэтан 1000 7000
Дихлорфторметан 1000 4200
1,1-Дихлорэтан 100 400
1,2-Дихлорэтилеи 200 790
Диэтиламии 25 75
2-Диэтиламииоэтаиол, через кожу 10 50
Диэтиловый эфир 400 1200
Изоамилацетат 100 525
Изоамиловый спирт (З-метилбутаиол-1) 100 360
Изобутилацетат 150 700
Изобутиловый спирт (2-метилпропанол-1) 100 300
Изопропиламин 5 12
Изопропилацетат 250 950
Изопропиловый спирт (пропанол-2) 400 980
Изофорон 25 140
Каменноугольная смола (фракция, растворимая 0,2
в бензоле; антрацен, бензо[а]пирен, фенантрен, акридин, хризен, пиреи) Камфора 2
Крезол (все изомеры), через кожу 5 22
Кротоновый альдегид 2 6
Ксилол 100 435
670 Приложение IV
Продолжение
Соединение Концентрация в воздухе
ч. на мли. мг/м3
Кумол, через кожу 60 245
Малеиновый ангидрид 0,25 1
Мезитилоксид 25 100
Метаиол (метиловый спирт) 200 260
Метилакрилат, через кожу 10 35
Метилаль (диметоксиметаи) 1000 3100
Метил-н-амилкетои (гептаион-2) 100 465
Метиламин 10 12
Метилацетат 200 610
Метилацетилен (пропин) Метилбутилкетон см. Гексанон-2 1000 1650
Метилизобутилкарбинол, через кожу 25 100
Метилизоцианат, через кожу 0,02 0,05
Метилиодид, через кожу 5 28
Метилметакрилат Метиловый спирт см. Метаиол Метилпропилкетон см. Пеитанон-4 100 410
Метилформиат 100 250
Метилцеллозольв, через кожу 25 80
Метилцеллозольвацетат, через кожу 25 120
Метилциклогексан 500 2000
Метилциклогексанол 100 470
Метилэтилкетои см. Бутанон-2
Морфолин, через кожу 20 70
Муравьиная кислота 5 9
Нас )талии 10 50
а-Нафтилтиомочевииа 0,3
Неоть (из угля) 100 400
Никель, карбонил 0,001 0,007
Никотин, через кожу 0,5
и-Нитроанилии, через кожу 1 6
Нитробензол, через кожу 1 5
Нитроглицерин, через кожу 0,2 2
Нитрометан 100 250
1-Нитропропаи 25 90
2-Нитропропаи 25 90
Нитротолуолы, через кожу Нитротрихлорметаи см. Хлорпикрин 5 30
п-Нитрохлорбеизол, через кожу 1
Нитроэтан 100 310
Октаи 500 2350
Октахлорнафталин, через кожу 0,1
Оловоорганические соединения 0,1
Пентаи 1000 2950
Пеитанон-2 200 700
Пеитахлорфенол, через кожу 0,5
Перхлорметилмеркаптан 0,1 0,8
Пикриновая кислота, через кожу 0,1
Пиридин 5 15
Платина, растворимые соли (пересчитано на Pt) 0,002
Токсичность органических соединений 671
Продолжение
Соединение Концентрация в воздухе
ч. на млн. мг/м3
Пропан 1000 1800
Пропаргиловый спирт, через кожу 1
н-Пропилацетат 200 840
Пропилен, окись 100 240
Пропилендихлорид 75 350
Пропиленимин, через кожу 2 5
н-Пропилиитрат 25 ПО
н-Пропиловый спирт (пропанол-1) 200 500
Родий, растворимые соли 0,001
Селен, соединения (пересчитано на Se) 0,2
Серебро, растворимые соединения 0,01
Стрихнин 0,15
2,4,5-Т 10
Таллий, растворимые соединения, через кожу 0,1
(пересчитано иа Т1) Тетрагидрофураи 200 590
Тетраметилсукцинонитрил, через кожу 0,5 3
Тетранитрометаи 1 8
1,1,1,2-Тетрахлор-2,2-дифторэтан 500 4170
1,1,2,2-Тетрах лор-1,2-дифторэтан 500 4170
Тетраэтилсвинец, через кожу (пересчитано на 0,075
РЬ) о-Толуидин, через кожу 5 22
Тринитротолуол, через кожу 1,5
1,2,3-Трихлорпропан 50 300
1,1,2-Трихлор-1,2,2-трифторэтаи 1000 7600
1,1,1-Трихлорэтаи 350 1900
1,1,2-Трихлорэтан, через кожу 10 45
Триэтиламии 25 100
Уксусная кислота 10 25
Уксусный ангидрид 5 20
Уран, растворимые соединения 0,05
Феиилгидразин, через кожу 5 22
п-Феиилеидиамии, через кожу 0,1
Фенол, через кожу 5 19
Фосген (карбонилхлорид) 0,1 0,4
Фталевый ангидрид 2 12
Фтортрихлорметан 1000 5600
Фурфураль, через кожу 5 20
Фурфуриловый спирт 50 200
Хинон см. и-Беизохиион Хлор 1 3
а-Хлорацетофеион (феиацилхлорид) 0,05 0,3
о-Хлорбеизилиденмалоиитрил (ОХЬМ) 0,05 0,4
Хлорбензол 75 350
Хлорбромметан 2-Хлорбутадиен-1,3 см. Хлоропрен 200 1050 0,5
Хлорированный камфеи, через кожу
Хлорированный дифенилоксид 0,5
1 -Хлор-1 -нитропропаи 20 100
6?2
Приложение 1V
Продолжение
Соединение Концентрация в воздухе
ч. на млн. мг/м3
Хлордан, через кожу 0,5
Хлоропрен (2-хлорбутадиен-1,3), черва кожу 25 90
Хлорпикрин 0,1 0,7
1-Хлор-2,3-эпоксипропан см. Эпихлоргидрин
2-Хлорэтанол см. Этиленхлоргидрин
Циклогексан 300 1050
Циклогексаиол 50 200
Циклогексанон 50 200
Циклогексен 300 1015
Циклопеитадиеи 75 200
Щавелевая кислота 1
Эпихлоргидрин, через кожу 5 19
Этанол (этиловый спирт) 1000 1900
Этаиоламии 3 6
Этилакрилат, через кожу 25 100
Этил-етор-амилкетон (5-метилгептанон-З) 25 130
Этиламин 10 18
Этилацетат 400 1400
Этилбензол 100 435
Этилбромнд 200 890
Этилбутилкетон (гептанои-3) 50 230
Этилеидиамин 10 25
Этилеиимии, через кожу 0,5 , 1
Этиленоксид 50 90
Этиленхлоргидрин, через кожу 5 16
Этиловый спирт см. Этанол
Этилформиат 100 300
Этилхлорид 1000 2600
2-Этоксиэтанол, через кожу 200 760
а Содержание примесей выражено в частях газа на миллиом частей загрязненного воз-
духа при 25 °C и 760 мм рт. ст. нли в миллиграммах примеси на I м’ воздуха. Данные
в таблице взяты из таблицы Z-l, OSHA 2077, General Industry Standards.
Таблица IV, 2
Предельные концентрации органических соединений *
Содержание Концентрация в воздухе
ч. на млн. мг/м3
Аллилглицидиловый эфир 10 45
Бутиламин 5 15
Винилхлорид 500 1300
Диглицидиловый эфир 0,5 2,8
о-Дихлорбензол 50 300
Токсичность органических соединений
673
Продолжение
Содержание Концентрация в воздухе
ч. на млн. мг/м’
1,1 -Дихлор-1 -нитроэтан 10 60
Дихлорэтнловый эфир, через кожу 15 90
Метилгидразин 0,2 0,35
Метилмеркаптан 10 20
Терфенил 1 9
Хлор а цета ль дегид 1 3
Хлороформ 50 240
Этилмеркаптан 10 25
а См. примечание к табл. IV. 1.
Таблица IV. 3
Допустимые концентрации, предельные концентрации
И максимальная продолжительность кратковременного воздействия
особо токсичных органических соединений а
Соединение Концентрация в воздухе, ч. на млн. Продолжительность кратковре- менного воздействия при макси- мально допустимой концентрации
средняя аа 8 ч предельно допустимая концентрация, ч. на млн. максимальное время, мнн
Бензол 1 5 2000 5 (за 2 ч)
Метиленхлорид 500 1000 300 5 (за 3 ч)
Метилхлорид 100 200 1 мг/10 м3
Ртуть Ртутьорганические со- единения (алкил) Свинец и его неоргани- ческие соединения Сероводород 0,01 мг/М3 0,2 мг/м3 0,04 мг/м3 20 50 10 (если не+
Сероуглерод 20 30 100 других токсиЧ* ных веществ) 30
Стирол 100 200 600 5 (за 3 ч)
Тетрахлорметан 10 25 200 5 (за 4 ч)
Тетрахлорэтилен 100 200 300 5 (за 3 ч)
Толуол 200 300 500 10
Трихлорэтилен 100 200 300 5 (за 2 ч)
Формальдегид 3 5 10 30
Четыреххлористый угле- род см. Тетрахлорме- та в Этилендибромид 20 30 50 5
Этилендихлорид 50 100 200 5 (за 3 ч)
22 Зак. 335
674
Приложение IV
Таблица IV. 4
Классы органических соединений,
представители которых могут быть сильными ядамн
Азотистые иприты [RN(CH2CH2C1)2]
Алифатические амины
Ароматические углеводороды
бензол, толуол, ксилол, нафталин, антрацен и т. д.
Ароматические амииы
анилины, иафтиламииы, бензидины и т. д.
Гидразины
NH2NH2, арилгидразииы, алкилгидразины
Диазосоединения
Метилфторсульфонат («магический метил»)
Неорганические соединения
соединения элементов: As. Be, Bi, Cd, Co, Cr,
галогены, Hg, Mn, Ni, P, Pb, Se, Те, Tl, Th, V
4-Нитробифенил
N-Нитрозосоедииения
Хлорированные алифатические углеводороды
СНС13, СС14, СН3СС13, СНС12СН2С1, С12С=СНС1, СН2=СНС1
Хлорированные ароматические углеводороды
ДДТ, ПХБ, 2,4,5-Т, 2,4-Д
Предметный указатель
Цифры в круглых скобках означают температуры кипения, цифры в квадратных скоб-
ках — температуры плавления; цифры без скобок — номера страниц.
В указателе приведены как тривиальные, так и систематические названия. Студент
должен усвоить, что в повседневном обращении находятся многие варианты названий, и
должен уметь наводить справки в учебнике илн указателях Chemical Abstracts для уста-
новления структур, соответствующих названиям.
В указатель включены основные соединения из таблиц приложения III (с температу-
рами кипения и плавления), но производные не индексированы. Методики получения про-
изводных, описанные в гл. 6, в указателе приведены, однако многие специальные произ-
водные и реакции опущены. Они могут быть найдены с помощью ссылок, которые
приведены для каждого класса соединений, обсуждаемых в гл. 6.
Аббе рефрактометр 78—82
Абдергальдена пистолет для сушки 109
Абсорбции максимумы 477
Адипамид (ди) [22*j|, 611
Адипамид (моно) [130], 6Ю
Адипиновая кислота [152], 601
Адипонитрил (295), 652
Адсорбенты, хроматография 50
ГХ 84
Азеланновая кислота [1061, 600
Азиды 358
ИК-спектры 360
Азобензол [68], 657
о-Азобнфенил [145], 657
Аз окси бензол [36], 284, 657
о-Азоксибнфеннл [158], 657
п-Азоксибифеннл [212], 657
I, Г-Азокси нафталин [127], 657
2,2'-Азоксннафталин [1681, 657
Азоксисоединения, производные 358, 657
таблица 657
м-Азокситолуол [39], 657
о-Азокситолуол [59]. 657
п-Азокситолуол [75], 657
1,Г-Азоиафталин [190], 657
1,2'-Азонафталнн [136], 657
2,2'-Азоиафталин [2081, 657
Азосоединення 358
ИК-спектры 360
таблица 657
Азот, проба ЮЗ, 104
Азотистая кислота 252, 371
Азотистые соединения органические 366
Азотная кислота 284, 288
дымящая 284, 288
м-Азотолуол [551, 657
о-Азотолуол [55], 657
л-Азотолуол [1451, 657
о-Азофенетол [131], 657
л-Азофеиетол [160], 657
Аконитовая кислота [1951, 602
гидрат [125], 600
Акридин [1081, 621
Акриламид [851, 609
Акрилаинлид [1051, 609
Акриловая кислота (140), 598
Акрилоил хлорид (76), 595
Акрилонитрил (78), 651
Акролеин (52), 606
Акролеинацеталь (126), 594
D-Аланин [297, разл.], 622
L-Алании [297, разл.], 622
спектр ЯМР 275
d,/-Аланин [295, разл.], 622
[3-Аланин [196], 621
Ализарин [289], 650
Алканы 215 см. Парафины
спектры ЯМР-,3С 504, 506
температуры кипения 215
удельная масса 73
Алкен ил галоген иды, таблица 636
Алкеиы 217
бромирование 217—219
гидроборирование 227
ИК-спектры 224
комплексы 223—224
масс-спектры 224
окисление 219
спектры ЯМР 226 и сл.
УФ-спектры 224
Алкилгалогениды 233
алкнлртутьгалогеннды 246
анилиды 247
ИК-спектры 243
масс-спектры 114, 115, 246
пробы 233, 235
реактивы Гриньяра 246
реакции замещения, теория 237
спектры ЯМР 245—246
таблица 636
температуры кипения 70
толуидиды 246
удельная масса 75
S-Алкилнзотиурония пнкраты 250
Алкил-₽-нафтиловые эфиры 247
Алкилртутьгалогениды 246
Алкины 230
ИК-спектры 230
масс-спектры 233
меркурироваиие 230
присоединение 2,4-динитробензолсульфе-
ннлхлорида 230
Аллиламин (56), 612
Аллилацетат (ЮЗ), 625
Аллилбензоат (230), 628
Аллил бензол (157), 643
Аллнлбромид (71), 636
Аллилнодид (ЮЗ), 636
Аллилмеркаптан (90), 651
Аллилмочевина [801, [851, 609
Аллиловый спирт (97), 603
Аллилформиат (83), 624
Аллилхлорид (46), 636
Аллилцианнд (118), 651
Аллилэтиловый эфир (65), 632
D-Аллотреонин [272, разл.], 622
L-Аллотреонин [272, разл.], 622
d,bАллотреонин [252, разл.], 625
Аллоксан [170, разл.], 610
Альдегиды 77, 78, 285
ИК-спектры 197
масс-спектры 202
окисление 194, 203
производные 203
спектры ЯМР 202
таблица 606—608
Алюминийхлорид, безводный 284
Алюминия оксид, хроматография 50
Амнгдалнн [214, разл.], 624
Амнды 320
гидролиз 326
ИК-спектры 320
масс-спектры 322
получение 263, 266, 312, 315
спектры ЯМР 321—322
таблица 609—611
676
Предметный указатель
н-Амнламин (104), 612
«-Амилацетат (146). 626
к-Амилбензол (202), 643
«-Амилбромнд (129), 637
трет-Амилбромид (108), 637
«-Амил-«-бутират (185), 627
н-Амилвалерат (204), 627
«-Амнлиодид (156), 638
трет-Амнлиодид (128), 638
«-Амилмеркаптан (126), 851
втор-Амилмеркаптан (112), 651
трет-Амнлмеркаптан (97), 651
«-Амилметилкеюн (151), 648
н-Амнл метиловый эфир (99), 633
«-Амил-2-нафтиловый эфир [25], 635
«-Амилнитрит (104), 632
«-Амиловый спирт (138), 603
втор-Амиловый спирт (119), 603
трет-Амиловый спирт (102), 603
н-Амилпропионат (169), 626
л-Амилфенол (253), 658
л-трет-Амнлфеиол (96), 600
н-Амилформиат (130), 625
«-Амилхлорид (107), 637
трет- А мил хлор ид (86), 637
«-Амилэтнловый эфир (118), 625
л-Аминоазобензол (360), (125], 657
2-Аминоан трах икон (302], 616
л-Аминоацетанилид [279], 611
л-Аминоацетанилид [162], 610
л-Аминоацетофенон (295), [106], 615
о-Ам и нобенз альдегид [40], 608
л-Аминобензойная кислота [174, разл.], 601
л-Аминобензойиая кислота [186, разл.], 602
л-Аминобеизол сульфон амид [142], 661
о-Аминобеизолсульфонамид [153], 661
л-Аминобензблсульфонамид [163], 661
л-Аминобензофенон [124], 616
2-Аминобифенил (299), [45], 616
4-Аминобифенил (302), [53], 616
2-Амино-5-бром толуол [59], 616
4-Амино-З-бромтолуол (240), 613
Аминов оксиды 358
ИК-спектры 360
o-AMHHo-N.N-диметиланилии (217), 620
л-Амнно-Ы,Ы-димСтиланилин (262), [42], 620
2-Амино-1,3-дим етилбензол (216). 612
2-Амино-1,4-дим етилбензол (215), 612
4-Амино-1,2-диметилбензол (226), 612
4-Амино-1,3-диметнлбензол (212), 612
5-Амино-1,3-ди метил бензол (220), 612
4-Амино-2,6-динитротолуол [169], 612
4-Амнно-3,5-динитротолуол [165], 612
2-Аминодифениламин [80], 615
4-Аминодифеннламни [75], 614
2-Амино-3,5-дихлортолуол (501. 614
4-Амнно-3,5-дихлортолуол [60], 614
л-Амино-М.Ы-диэтиланилнн (262), 613
а-Аминоизомаслявая кислота [280, разл.],
622
2-Амино-4-изопропил толуол (242), 613
Аминокислоты 273
ИК-спектры 273—274
конфигурация 276
масс-спектры 275
нингидриновая проба 279
оптическое вращение 278
производные 279
спектры ЯМР 272, 275
таблица 278, 621—623
л-толуолсульфонильные производные 280
удрлчнч* масса 77
L-cc-Амино-н-м 1сляная кислота [285, разл.],
U22
d.l-OL-Амнно-н-масляная кислота [307, разл.],
622
о-Амнно-М-метиланилнн (245), 618
л-Амино-N'MeT ил анилин (258), [36], 618
л-Амино-Ы-метил ацетанилид [63], 609
2-Амнно-5-метилбензол-1,3-дисульфонамид
1257], 662
2-Ам ино-5-м етилбензол -1,3-дисул ьф ои ил-
хлорид [1551, 665
2-Амино-4-метилпиридин [98], 615
2-Амнно-6-метилпнридин [41], 614
2-Амиио-5-нитро-1.4-диметилбензол [142], 616
4-Амнно-5-ннтро-1,3-диметнлбензол [76], 614
4-Амино-6-нитро-1,3-днметилбензол [123], 615
1-Амино-2-нитронафталнн [144], 616
1-Амиио-4-нитронафталин [195], 616
2-Амино-5-ннтронафталин [144], 616
2-Амино-4-иитротолуол [107], 615
2-Амино-5-нитротолуол [130], 616
3-Амино-6-нитротолуол [135], 616
4-Амино-2-нитротолуол [77], 614
4-Амино-З-иитротолуол [116], 615
5-Амино-2-окситолуол [173], 616
2-Амниопирндин [56], 614
З-Амииопнридин [64], 614
5-Аминосалициловая кислота [283, разл.],
602
1-Амниофенантрен [146], 616
л-Аминофенилуксусная кислота [283, разл.],
602
л-Аминофеиол [122], 615
о-Амннофенол [174], 616
л-Аминофенол [184], 616
З-Аминохинолин [94], 615
4-АмиИохинолин [154], 616
З-Амино-6-хлортолуол (241), 616
Амины 76, 77, 250
амидные производные 266
бензолсульфониламндный тест 264
ИК-спектры 256, 257
масс-спектры 260
нитрозосоедннения 270
первичные, вторичные производные 261
пикраты 270
различные тесты 272
спектры ЯМР 257, 259
сульфонамидные производные 268
таблицы 611—621
тест с азотистой кислотой 252
третичные производные 261
УФ-спектры 261
хлор ангидридный тест 250
хлорплатииаты 271
четвертичные аммониевые соли 269
Аммониевые солн, инфракрасные спектры
332
обработка гидроксидом натрия 332
Аммония гексанитратоцерат 169, 171
Амфотерные соединения, растворимость 132
Ангеликовая кислота [46], 599
Аиетол 232 [22]. 635
л-Анизидин (251), 613
л-Анизидии (243), 613
Аиизнлхлорид [26], 596
Анизол (154), 634
Анилиды 246, 312, 315, 609—611
Анилин (183)
основность 127
растворимость 137
Анилины, бррмирование 217
Анисовая кислота [184]. 601
Анисовый альдегид [247], 607
Анисовый спирт [25], 605
Антипирин [113], 546
Антраниловая кислота [144, разл.], 601
Антрахинон [286], 650
1,5-Антрахииондисульфонамид [246], 662
1,8-Антрахииоидисульфонамнд [340], 662
1,5- Антрахинондисульфоннлхлорид [265,
разл.], 666
1,6- Аитрахинондисульфонилхлорид [198], 666
Предметный указатель
677
1,7- Антрахннондисульфонилхлорид (232], 666
1,8- Аитрахинондисульфонилхлорид (223], 666
2,6-Антрахннондисульфоиилхлорид [250], 666
2,7-Аитрахинондисульфоиилхлорид [186], 666
Антрахипон-Р-сульфонамид [261], 662
Антрахинон-а-сульфонилхлорид [214], 666
Антрахинон-0-сульфон ил хлор ид [197], 665
Антрацен [216], 644
окрашивание с А1С13—СНС13 284
Антроновый тест на сахара 300
Дпиол [32], 635
/-Арабиноза [160, разл.], 623
ri.Z-Арабиноза [164, разл.], 623
Арбутии [165, разл.], 623
L-(+)-Аргинин [238, разл.], 622
d,l-Аргинин [238, разл.], 622
тест 273
Арилгалогениды 294, 638—640
Арилметнлкетоны 293
Арилоксиуксусные кислоты 409
Аронлбензойные кислоты 292
Ароматические соединения 281
ИК-спектры 289
проба с дымящей серной кислотой 281
производные 297
спектры ЯМР-,3С 513
— ЯМР, химические сдвиги 289
удельная масса 76
Ароматические углеводороды, таблица 643,
644
D-Аскорбиновая кислота 624
L-Аскорбиновая кислота 624
d.Z-Аскорбнновая кислота [169, разл.], 624
D-Аспарагнн [226, разл.], 622
L-Аспарагин [226, разл.], 622
D-Аспартовая кислота [270, разл.], 622
L-Аспартовая кислота [270, разл.], 622
d.Z-Асиартовая кислота [280, разл.], 622
Аценафтен [95], 644
Ацеиафтенон [121], 650
Ацетали, производные 210
масс-спектры 210
таблица 594—595
Ацеталь (102). 594
Ацетальдегид (21), 606
Ацетальдоль, окисление Се(IV) 171
Ацетамид [82], 609
растворимость 137
Ацетанилид [114], 609
растворимость 137
спектр ЯМР 322
Ацетаты 183
полиоксисоединений 183
Ацетилантранилловая кислота [185], 602
Ацетила цетон (139), 647
Ацетилбромид (81), 595
Ацетнленднкарбоновая кислота [179], 601
Ацетилены см. Алкииы
Ацетилиодид (108), 595
сс-Ацетил-Р-метилмочевина [180], 610
Ацетилмочевина [212], 611
Ацетилпиперидин (226), 611
Ацетилсалициловая кислота [123], 600
а-Ацетил-Р-феиилгидразин [128], 610
Ацетилфторнд (21). 595
Ацетил хлорид (55), 595
проба иа активный водород 251
Ацетоацетамнд [54], 609
Ацетоацетанилид [85], 609
Ацетоин (145). [15], 603
л-Ацетокси-М-метилацетанилнд [98], 609
Ацетол (146), 603
Ацетомезитнлен (245), 648
Ацетон (56), 647
ИК-спектры 202
как растворитель 137
Ацетондикарбоиовая кислота [135, разл.1,
600
Ацетониды 184
Ацетонил ацетон (188). 648
Ацетонитрил (81), 651
как растворитель 137
Ацетоноксим, сопряженное основание 131
л-Ацетотолунд [85], 609
о-Ацетдтолуид [112], 609
п-Ацетотолуид [153], 610
Ацетофенетидин [138], 616
Ацетофенон (200), 648
растворимость 137
спектр ЯМР-13С 512
2-Ацето-л-цимол (250), 648
9-Ацил амидоксантены 328
Барбитуровая кислота [245], 601
Бенедикта раствор 196, 386
Бензальдегид (179), 607
Бензальхлорид (212), 642
Бензамид [128], 610
Бензанилид |161], 6Ю
Бензантрон [170], 650
Бензгидрол (297), [69], 605
Бензидин (400), [127], 616
Бензил (347), [95], 649
растворимость 137
БенЗиламид [155], 610
N-Бензиламнды
Бензиламин (184), 612
бромирование 217
растворимость 137
Бромгидрохинон [НО], 658
N-Бензиланилин (298), [37], 618
N-Беизилацетамид 618
Бензил ацетат (216), 628
Бензилбензоат (323), 630
Бензил бромид (198), 637
Беизил-н-бутиловый эфир (212), 634
Бензил-н-бутират (238), 628
Бензилдиметилкарбинол (216), 604
Бензилиден ацетон (262), [41], 649
Беизилиденацетофенон (348), [62], 649
Бензнлизобутиловый эфир (212), 633
Беизнлмалоновая кислота [117, разл.], 600
Бензилмеркаптан (194), 651
Ь1-Бензил-1Ч-метиланилин (306), 620
Бейз ил мети л кетон (216), [27], 649
Беизилметиловый эфир (167), 633
Бензнлмочевина [149], 610
Бензил-а-нафтиловый эфир [77], 636
Бензил-2-иафтиловый эфир [102], 636
Бензиловая кислота [150], 601
Бензиловый спирт (205), 605
окисление Се (IV) 170
растворимость 122
Бензилоксалат [80], 631
Бензилсалицилат (320), 630
Бензил сукцинат [42], 630
Бензилсульфонамнд [105], 661
Бензилсульфонилхлорид [92], 665
Бензилтнуронийалкилов кислые сульфаты
181
Бензилтиуронийкарбоксилаты 329
Беизнлтиуроиийсульфонаты 395
о-Бензилфенол [54], 659
n-Бенз ил фенол [84], 659
Беизнлформиат (203), 627
Бензилфталат [42], 630
Бензилхлорацетат (147), 626
Беизилхлорид (179), 637
Бензилциниамат [39], 630
М-Беизил-Ы-этиланилин (312, разл.), 620
Бензилэтилкетон (226), 648
Беизилэтиловый эфир (189), 627
Бензоаты 180
Бензоил а цетон [61], 649
ИК-Спектр 533
678
Предметный указатель
о-Бензоилбензойная кислота (126], 600
о-Бензоилбензойная кислота, гидрат [90],
599
Беизоилбромид [218], 597
а-Беизоилмасляная кислота [87], 599
V-Бензоилмасляная кислота [127], 600
Беизоилпиперидин [48], 609
Р-Беизоилпропионовая кислота [1161, 000
а-Беизонл-р-феиилгидразин [168]
Бензоилфторид (159), 596
Беизоил хлорид (197), 596
ИК-спектры 319
спектры ЯМР 320
Беизоии (343), [133], 650
Бензойная кислота [121], 600
Бензойный ангидрид [42], 596
Бензол (80), 643
окрашивание с А1С1з—СНС1з 284
как растворитель 138
Бензолазодифеииламии [96], 657
Бензол аз о-о-к резол [128], 657
1-Бензолазонафталин [70], 657
2-Беизолазонафталин [131], 657
2-Бензолазо-1-нафтол [138], 657
4-Бензолазо-1-нафтол [206, разл.], 657
1-Бензолазо-2-нафтол [131], 657
Бензолазорезорцин [170], 657
а-Бензол гекс а хлорид [157], 642
Р-Беизол гексахлорид [310], 642
1,2- Бензолдисульфонамид [254], 662
1,3- Бензолдисульфонамид [229], 662
1,4- Бензол дисул ьфои амид [288], 662
1,2- Бензолдисульфоннлхлорид [143], 665
1,3- Беизолдисульфонилхлорид [63], 664
1,4- Бензолдпсульфонилхлорид [139], 665
Беизолсульфоиамид [153], 661
сопряженное основание 131
Бензолсульфоннлхлорид [14], 264, 663
1,3,5-Беизолтрисульфоннлхлорид [187], 666
Бензонитрил (191), 652
бромирование 217
Беизопииакон [186], 650
о-Бензотолуидид [142], 610
л-Бензотолуидид [158], 610
Беизотрихлорид (220), 642
Бензофенон (305), [48], 649
ИК-спектры 202
Бензохинон [115], 650
а,а'-Бинафтил [160], 644
Р.Р'-Бинафтил [188], 644
Бисметон (205)
Бифенил [70], 644
окрашивание с А1С1з—СНС1з 284
Биурет [192, разл.], 611
Боратный тест 184
структурные эффекты 69—72
(-Ь)-Борнеол [208], 606
Борнил ацетат (221), [29], 630
L-Бориилен (146), [113], 646
Бориилхлорид (207), 637
Борогидридное восстановление 207
Бортрифторида эфират 224
Бромаль (174), 607
Бром в четыреххлористом углероде 217
о-Броманизол (218), 634
л-Бромаиизол (223), 634
n-Бром анилиды 312 и сл.
л-Броманилии (251). 613
о-Бромаиилин (229), [31], 613
л-Броманилин (245, разл.), [66], 614
Бром ацетамид [91]. 609
а-Бромацетанилид [131], 610
л-Бромацетанилид [167], 610
Бромацет ил бромид (149), 595
Б ром ацетил хлор ид (127), 595
Бромацетон (136), 647
©-Бромацетофенон [50], 649
Л-Бром бензальдегид (236), 607
л-Бромбензальдегид [57], 608
л-Бром бензамид [155], 610
о-Бромбензамид [155], 610
л-Бромбензамид [189], 611
л-Бромбензанилид [136], 6Ю
о-Бромбензаиилид [141], 610
л-Бром бенз ил бромид [41], 642
о-Бромбензилбромид [30], 642
л-Бромбензилбромид [63], 642
л-Бромбензилсульфоиамид [188], 662
л-Бромбензилтиурониевые соли 327
л-Бром бенз ил хлорид [50], 642
л-Бромбензоилбромид (243), 596
л-Бромбензоилбромид [42], 596
л-Бромбензойиая кислота [155], 601
о-Бромбензойная кислота [150] 601
л-Бромбензойная кислота [251], 602
Бром бензол (157), 638
3-Бромбензол-1,5-дисульфонамид [256], 662
3-Бром бензол-1,5-дисул ьфои ил хлорид [100],
665
л-Бромбензолсульфонамид [154], 661
о-Бром беизолсульфоиамид [186], 661
л-Бромбеизолсульфоиамид [166], 661
о-Бром бензол сульфоннлхлор ид [51], 663
л-Бром бензоле ульфон ил хлорид [75], 664
л-Бромбензонитрил [38], 662
о-Б ром бензонитрил [53], 652
л-Бромбензонитрил [112], 653
2-Бромбифенил (297), 639
4-Бромбифенил [89], 640
Бромгидрохинон [110], 658
З-Бром-Ы.М-диметил анилин (259), 620
л-Бром-Ы.М-днметиланилии (264), 620
л-Бром-N.N-диэтил анилин (270), [34], 620
а-Бромизовалернановая кислота (230), [44],
599
а-Бромизовалерилмочевнна [145], 610
а-Бромизомасляная кислота (200), [49], 599
Р-Бромизомасляная кислота [22], 599
1-Бром-2-изопропилбензол (210), 639
о-Бром иод бензол (257), 642
л-Бром иод бензол [92], 642
(4-)-а-Бром кам фор а (274), [76], 649
а-Бром-н-капроновая кислота (240), 598
4-Б ром-л-ксилол, спектр ЯМР 297
л-Бром кумол (219) 639
а-Бром-н-масляная кислота (217, разл.), 598
Броммезитилен (225), 639
З-Бром-2-метиланилин (255), 618
З-Бром-4-метиланилин (257), 618
2-Бром-4-метилацетанилид [117], 610
Б ром мети л ацетат (130). 625
а-Бромнафталин (279), 639
В-Бромнафталин [59], 640
1-Бром-4-нафталннсульфонамнд [193], 662
2-Бром-1-нафталинсульфоиамид [140], 661
1-Бром-4-иафталинсульфоинлхлорид [83], 664
Бромная вода 405
л-Бромнитробензол (257), [54], 640
о-Бромнитробензол (261), [43], 540
л-Бромнитробензол (259), [126], 640
4-Бром-1-нитронафталин [85], 640
4-Бром-8-иитро-1-нафтиламии [116], 640
4-Бром-З-иитротолуол [33], 639
Бромоформ (151)» 641
2-Бромпиридин (194). 619
З-Бромпнридин (170). 619
1-Бромпропен (60), 636
2-Бромпропен (48). 636
а-Бромпропионилбромид (153), 595
а-Б ром пропионовая кислота (205), [24], 599
Р-Бромпропиоиовая кислота [62], 599
Р-Бромстнрол (221), 639
р-Бромтиофенол [75], 651
л-Бромтолуол (183). 638
о-Бромтолуол (181), 638
л-Бромтолуол (185), [28], 639
Предметный указатель
679
Бромтрихлорметан (105), 641
Бром уксусная кислота (208), [50], 599
Бром уксусный ангидрид [42], 596
п-Бромфеиацилбромид (109], 597
о-Бромфеиетол (224), 634
л-Бромфенетол (229), 634
л-Бромфеиилгидразин [106], 615
л-Бромфенол (236), [32], 658
о-Бромфенол (195), 657
л-Бромфенол [63], 659
1,2-Бромфторэтилен (36), 641
2-Бромхинолнн [49], 620
6-Бромхинолин (278), 620
1,3-Бром хлорбензол (196), 639
о-Бромхлорбензол (195), 639
л-Бромхлорбензол [67], 640
2-Бром-4-хлорфенол [34], 659
1-Бром-1-хлорэтаи (83), 641
1-Бром-2-хлорэтан (107), 641
2-Бромцимол (234), 639
2-Бромэтанол (150), 603
Р-Бромэтил ацетат (163), 626
1-Бром-2-этилбензол (199), 639
1-Бром-4-этилбеизол (204), 639
Р-Бромэтнловый эфир (127), 633
p-Бромэтилфениловый эфир [32], 635
Брэди реагент 186
Бутаидиол-1,3 (208), 604
спектр ЯМР-13С 509
Бутандиол-2,3 (183), 604
окисление Ce(IV) 171
Бута идиол-1,4, окисление Ce(IV) 171
1-Бутансульфонамид [45], 661
1-Бутансульфонилхлорид (75/10 мм), 663
Бутенднол-1,4, окисление Ce(IV) 171
З-Бутеновая кислота (169). 598
Бутен-З-ол-2 (96), 603
Р-Бутеноиитрил (118), 651
я-Бутиламин (77), 612
втор-Бутиламин (63), 612
ИК-спектр 257
трет-Бутилам ин (46), 612
н-Бутил-л-амииобензоат [56], 630
N-я-Бутиланилин (236), 617
я-Бутилацетат (125), 625
растворимость 137
втор-Бутилацетат (111), 625
растворимость 137
н-Бутнлбензоат (249). 629
я-Бутилбензол (182), 643
втор- Бут нл беи зол (173). 643
трет- Бутил бензол (169), 643
я-Бутилбромид (100), 637
вгор-Бутилбромид (90), 637
трет-Бутилбромид (72), 647
я-Бутнл-я-бутират (165), 626
я-Бутнлиодид (130), 638
втор-Бутилиодид (119). 638
трет-Бутил иодид (98), 638
я-Бутилкарбамат [54], 609
втор-Бутилкарбинол (128), 603
н-Бутилкарбоиат (207), 627
растворимость 137
я-Бут ил -о- крезоловый эфир (223), 634
я-Бутиллактат (188), 627
я-Бутилмеркаптаи (97), 651
я-Бутилметилкетои (129), 647
я-Бутилметиловый эфир (70), 632
я-Бутнл-2-нафтиловый эфир [36], 635
втор-Бутил-2-нафтнловый эфир [34], 635
я-Бутилнитрат (136). 632
я-Бутилнитрнт (75), 632
я-Бутиловый спирт (116), 603
окисление Ce(lV) 17!
спектр ЯМР 160
справка 39
втор-Бутилевый спирт (99). 603
окисление Ce(iV) 171
спектр ЯМР-|3С 508
трет-Бутиловый спирт (83), 603
окисление Ce(IV) 171
я-Бутилоксалат (243), 628
растворимость 137
я-Бутилоксамат [88], 609
я-Бутилпропионат (144), 626
я-Бутилсалицилат (268), 629
d-я-Бутилтартрат [22], 630
я-Бутилфеинлацетат (254), 629
я-Бутилфениловый эфир (2Ю). 634
л-Бутилфенол (248), [22], 658
л-трет-Бутилфенол [95], 600
я-Бутилформиат (107), 625
втйр-Бутилформиат (97), 625
я-Бутилфталат (338), 630
спектр ЯМР 337
н-Бутилхлорацетат (175), 626
я-Бутилхлорид (77), 637
спектр ЯМР 246
втор-Бутилхлорид (67), 637
трет-Бутилхлорид (51), 637
н-Бутилхлорформиат (145), 626
я-Бутилэтилбарбитуровая кислота [125], 610
Бутин-2 (27), 645
Бутиидиол-1,4, окисление Се (IV) 171
н-Бутирамид [115], 609
растворимость 137
я-Бутиранилид [95], 609
я-Бутирилбромид (128), 595
я-Бутирил иод ид (148), 595
я-Бутирилфторид (67), 595
я-Бутирилхлорид (100), 595
Бутироин (190), 648
V-Бутиролактон (204), 627
я-Бутиронитрил (118), 651
растворимость 137
Буферные растворы, состав
Вакуумная перегонка, номограмма давле-
ние — температура 578
я-В ал ер а и ил ид [63], 609
я-Валериаиовая кислота (186), 598
растворимость 137
я-Валериановый
альдегид (103), 607
ангидрид (215), 596
я-Валерил хлорид (127), 595
V-Валеролактов (207), 627
я-Валеронитрил (141), 651
D-Валин [315, разл.], 622
L-Валнн [315, разл.], 622
d,/-Валии [298, разл.], 622
Ванилидинацетои [130], 650
Ванилии (285, разл.), 608
Ванилиновая кислота [207], 602
Вератровая кислота, безводная [181], 601
Вератровый альдегид (285), [58], 608
ИК-спектр 532
Вератрол (206), [21], 635
Винилацетаиилид [73], 609
Винилацетат (72), 624
Винилбензоат (203). 627
Вииилбромид (16), 636
Винилиодид (56), 636
Виннлуксусная кислота (169), 598
d-Винная кислота [169], 601
/-Виииая кислота [169]. 601
d,Z-Вниная кислота [204], 602
яезо-Винная кислота [140], 601
Вода
растворимость, граничные случаи 126, 135,
137
как растворитель 137
Газовая хроматография 30, 82—89
Газовый хроматограф 83, 86, 87
680
Предметный указатель
D-Галактоза 1170, разл.], 624
Галлацетофенон [173], 650
Галловая кислота [240, разл,], 602
Галогена содержание, масс-спектрометрия
24
Галогены, пробы 105—107
Гваякол (205), [28]. 634
Гексагндробензальдегид (162), 607
Гексагидробензамид [186], 611
Гексагидробензанилид [146], 610
Гексагидробензойная кислота (232), 598
Гексадекан (287), 645
Гексадиен-2,4 (81), 646
Гексадиен-2,4-ол-1 [31], 605
Гексаметил бензол [162], 644
Гексаметилентетрамин [280], 621
Н-Гексан (68), 644
как растворитель 137
спектр ЯМР-13С 506
Гексанол-3 (135), 604
Гексатриен-1,3,5 (78), 646
Гексахлорбензол [229], 640
Гексахлорэтан [187], 642
Гексен-1 (66), 645
Гексен-2 (68), 645
Гексен-3 (67), 645
н-Гексиламин (128), 612
н- Гексил бензол (226), 643
н-Гексил бромид (157), 637
н-Гексйлиодид (180), 637
н-Гексилмеркаптан (151), 651
н-Гексил метилкетон (172), 648
н-Гексилметиловый эфир (126), 633
н-Гексиловый спирт (156), 603
н-Гексилрезорцин [69], 659
н-Гекснл формиат (154), 626
н-Гексилхлорид (134), 637
н-Гексилэтиловый эфир (142), 633
Гекснн-1 (70), 646
Гекснн-3 (81), 646
Гёлиции [175, разл.], 624
н-Гептальдегид (166), 607
н-Гептамид [94], 609
н-Гептан (98), 644
Гепт ан ил ид [70], 609
Гептановая кислота, как растворитель 122
н-Гептановая кислота (224), 598
Гептанол-2 (160), 603
Гептанол-3 (156), 603
Гептахлорпропан [29], 642
Гептен-1 (94), 646
н-Гептиламин (155), 612
н-Гептилацетат (192), 627
н-Гептнлбромид (174), 637
н-Гептнлиодид (204), 637
н-Гептилмеркаптан (176), 651
н-1?ептиловый спирт (176), 604
окисление Ce(IV) 171
Гептин-1 (100), 646
Гептоилхлорнд (176), 596
Гептойный ангидрид (258), 596
Гераниол (229), 604
Гидантоин [218], 611
Гидантоины 206
Гидразиды 345
Гидразины 357
Гидразобензол [130], 657
о-Гидразобнфеиил [182], 657
/г-Гидразобифенил [170], 657
1,1'-Гндразонафталнн [153], 657
2,2'-Гидразонафтални [140], 657
Гидразоны 357
Гидразосоедннеиия 357
производные 372
таблица 657
л{-Гн др азотолуол [38], 657
о-Гндразотол.уол [161], 657
н-Гндразотолуол [134], 657
Гидрннден (177), 643
Гидроборирование 224
Гидрокоричная кислота [48], 599
Гидрокоричный альдегид (224). 607
Гидролиз 325, 339
Гидрохинон [169], 660
диацетат [123], 631
ди бензиловый эфир [127], 636
диметиловый эфир [55], 636
диэтиловый эфир [72], 636
моиобеизиловый эфир [122], 660
монометилевый эфир [53], 659
растворимость 137
Гидроциниамамид [105], 609
Гидроциииамаиилид [98], 609
Гиппуровая кислота [187], 602
L-Гистидин [277, разл.], 622
Гликоген [240, разл.], 624
Гликозиды, таблица 623
Гликолевая кислота [79], 599
Гликольнитрил (183, разл.), 652
Глиоксаль (50), 606
Глицерилтриацетат (258), 629
Глицерилтрибензоат [72], 631
Глицерилтрибутнрат (318), 629
Глицерилтрипропиоиат (289), 629
Глицерилтристеарат [71], 631
Глицерин (290, разл.), 605
окисление Ce(lV) 171
«/./-Глицериновый альдегид [142], 608
Глицил гл ици и [264, разл.], 622
Глиции [232], 622
ИК-спектр 272
трянс-Глутаконовая кислота [138], 600
х^нс-Глутакоиовая кислота [136], 600
D-Глутаминовая кислота [198, разл.], 621
L-Глутаминовая кислота [198, разл.], 621
dJ-Глутамнновая кислота [227, разл.], 622
Глутаровая кислота [97], 600
растворимость 137
Глутаровый ангидрид [56], 595
Глутаронитрил (286), 652
растворимость 137
Глюкоза [90, разл.; 146, разл.], 623
окисление Ce(IV) 171
Р-0-(+)-Глюкоза
ИК-спектр 301
спектр ЯМР 301
Гомовератровая кислота [99], 600
Гриньяра реактивы 246
Дезоксибензоин (320), [60], 649
тракс-Декалин (185), 645
цис- Декад им (194), 645
н-Де кай (173), 645
Декаиол-2 (211), 604.
Децен-1 (171), 646
н-Дециловый спирт (231), 605.
окисление Ce(IV) 171
н- Децил хлорид (223), 63Z
Диазосоединения 358
ИК-спектры 359
Диазотирование 253
Диаллил (59), 646
Дналлиламнн (II 1>, 617
Диаллилбарбитуровая кислота [172], 610
Ди-н-амиламин (205), 617
Ди-н-амилкетон (226), 648
Ди-н-амиловый эфир (190), 633
2,4-Диамииобензол-1,5-дисульфонамид [187].
662
2,4-Диаминобензол-1,5-дисульфонил хлорид
[275], 666
2,4'-Диаминобнфенил (363), 614
1,4-Днамниобутан (160), 612
1,6-Диамнногексан [42], 614
л,п'-Днаминодифеннлметан (94), 6)5
1,2-Диамннонафталин [95]. 615
Предметный указатель
681
1,4-Диамииопафталин [120], 615
1,8-Диаминонафталин [67], 614
2,7-Диамннонафталнн [166], 616
1,5-Диамииопентан (178), 612
1,2-Диаминопропан (120), 612
1,3-Днамииопропан (136), 612
4,4'-Диаминостильбен [226], 616
2,3- Дн аминотолуол (255), [61], 614
2,4- Диамииотолуол (280), 615
2,5- Диамннотолуол [64], 614
2,6- Диамииотолуол [105], 615
3,4-Диаминотолуол (265), 615
2,4-Диаминофеиол [79, разл.], 615
2,4-Диамииохлорбеизол [88], 615
Дианизидин [135], 616
Диацетил (88), 647
Ы,ЬГ-Диацетил-л<-фенилендиамии [191], 611
N.N'-Диацетнл-о-фенилендиамнн [185], 611
N.N'-Диацетил-п-феннлендиамии (300], 611
Диацетоновый спирт (164), 648
Дибензальацетон [112], 650
Дибеизил [52], 644
Дибеизиламин (300), 618
М,Ь1-Дибензиланнлин (300. разл.), [70], 621
Дибензилкетон (330), [34], 649
Днбензиловый эфир (298), 634
Дибензилуксусная кислота [89], 599
Ди бен зоил метан [78], 649
Днбензофураи [87| 636
3,5-Дибром-4-амниотолуол [78], 614
2,4-Днброманилии [79], 615
2,6-Дибром анилин [83]. 615
Дибромацетамид [156]. 610
ти-Днбром бензол (219). 639
о-Ди бром бензол (224), 639
n-Днбром бензол [89], 640
2,4-Дибромбензолсульфонамнд [190], 662
2,5-Дибромбеизолсульфопамнд [194], 662
3,4-Ди бром бензол сульфон а мид [175], 661
2,4-Дибром бензолсульфонилхлорид [79], 664
2,5-Дибром бензолсульфонилхлорид [71], 664
3,4-Дибром бен зол сульфонил хлор ид [34], 663
2,2'-Дибромбнфеиил [81], 640
4,4'-Дибром бифеиил [169], 640
1,2- Ди бром бут ан (165), 642
1,3- Дибром бутан (174), 642
1,4- Дибром бут ан (198), 642
2,3-Дибром бутан (158), 641
1,3-Дибром бутен-2 (169), 642
Дибромдиметиловый эфир (150), 633
3,5-Дибром-о-крезол [57], 659
3,5-Дибром-/г-крезол [49], 659
1,2-Дибромнафталин [67], 640
1,4-Дибром нафталин [82], 640
2,5-Ди бром нитробензол [85], 655
1,8-Дибромоктан (272), 642
3,5-Дибром пиридин (222), [112], 621
1.3~Днбромпропаиол-2 (219, разл.), 604
2,3-Дибром пропанол-1 (219), 604
1,2-Дибромпропен (132), 641
1,3-Дибромпропен (156), 641
2,3-Дибромпропен (140), 641
а, P-Дибром пропионовая кислота [64], 599
3,4-Ди бром толуол (241), 639
Ди бром уксусная кислота (234), [48], 599
2,4-Ди б ром фенол (238), [36], 658
2,2-Дибромэтанол (181), 604
dJ-Дибромянтариая кислота (167], 601
Ди-н-бутиламнн (160), 617
растворимость 137
N.N-Ди-н-бутил анилин (261), 620
Ди-н-бутилкетон (187), 648
растворимость 137
Ди-втор-бутнлкетон (162), 648
Дн-н-бутиловый эфир (140), 633
ИК-спектр 348
Ди-н-бутоксиметан (181), 595
Ди-вгор-бутоксиэтан (171), 595
Ди-н-бутоксиэтаи (186), 595
Дигалогеналканы 243
Ди гексил кетон [33], 649
Ди-н-гексиловый эфир (229), 634
Ди-н-гептиловый эфир (260), 634
1,2-Дигидробензол (80), 646
Дигидрокоричиый альдегид, спектр ЯМР
Дигидроиафталин (212). [15], 646
Ди гидропир ан (86), 633
Ди-а,а'-дихлорэтиловый эфир (116), 633
Ди-а,р-дихлорэтиловый эфир (140), 633
Ди-р,Р'-дихлорэтиловый эфир (179), 633
Диизоамиламин (187), 617
Диизоамиловый эфир (172), 633
Диизобутилкарбинол (175), 604
Диизобутилкетои (168), 648
Диизобутиловый эфир (122), 633
1,1-Диизобутоксиэтан (176), 595
Диизопропилам ин (86), 617
jw-Ди изопропил бензол (203), 643
о-Диизопропил бензол (204). 643
n-Диизоироп ил бензол (210), 643
Ди изопропил кетон (125), 647
Диизопропиловый эфир (68), 137, 632
1,1-Диизопропокснэ7ан (122), 594
2,4-Дииоданилии [96], 618
М-Дииодбензол [40], 640
n-Дииодбензол [129], 640
2,4-Динодфенол [72], 659
Димедон, реагент 205
Диметиламин (7), 617
n-Диметнл аминоазо бензол [117], 657
п-Диметиламинобеизальдегид [73], 621
п,n'-бисАДиметиламнно)бензофенон [174], 621
Р-Диметнламиноэтиловый спирт (133), 619
Диметиланилин (193), 619
2,3-Диметиланилин (222), 612
Диметилацеталь (64), 594
ИК-спектр 210
спектр ЯМР 215
2,4-Диметилацетанилид [133], 610
3,4-Диметилбензамид [130], 610
Ы,Ы-Диметилбензиламин (185), 612
2,4-Диметилбензойная кислота [127], 600
2.5-Диметилбеизойная кислота [132], 600
3,4-Диметилбензойная кислота [164], 601
1,2-Дим етил бензол-3,5-днсул ьфон а м нд [239],
662
2,4-Дим етил бензол-1,5-д исул ьфон амнд [2491;
662
2,5-Диметил бензол -1,3-дисульфои а мид [295].
662
2,5-Дим етил бензол -1,4-дисул ьфон ам ид [310],
662
1,2-Дим етил бензол-3,5-дисул ьфон ил хлорид
2,4- Днметилбензол-1,5-дисул ьфон ил хлорид
[129], 665
2,5-Диметилбензол-1,3-дисульфонилхлорид
[81], 664
2,5- Димел ил бензол-1,4-дисульфонилхлорид
[164], 665
2,3- Дим етил бензол сульфонам ид [167], 661
2,4- Диметнл бензол сульфона мид [137], 661
2,5- Диметилбеизолсульфоиамид [148], 661
2,6- Диметилбензолсульфонамид [96], 661
3,4-Диметилбензолсульфонамид [144], 661
3,5-Дим етил бензол сульфон амид [135], 661
2,3- Дим етил бензол сульфонил хлор ид [47], 663
2,4- Дим етил бензол сульфонил хлор ид [34], 663
2,5- Днметилбензосульфонилхлорнд [25], 663
2,6- Диметнлбензолсульфоннлхлорид [39], 663
3,4-Диметилбензолсульфонилхлорид [52], 663
3,5-Днметилбензосульфонилхлорид [94], 665
N.N-Диметил-п-бром анилин (264), [55], 620
5,5-Диметилднгидрорезорцин [149], 650
Ы,М-Диметил-2,4-дииитроанилин [87], 621
682
Предметный указатель
2,4-Диметил-1,3-дииитробензол [93], 655
а,р-Диметилкарбанилид [121], 610
Диметилмалоиовая кислота [193], 602
N.N-Диметилмезидин (213), 619
несилсм-Диметилмочевина [182]. 611
силл-Диметилмочевииа [106], 609
1,4-Диметилнафталии (263), 643
2,3-Днметилнафталин [104], 644
2,6-Диметил-8-нафтсалннсульфонамид
662
N.N-Диметил-а-нафтиламин (272), 620
N.N-Диметил-р-иафтиламин (305), 620
2,3- Диметилнитробензол (250), 653
2,4-Диметилиитробеизол (238), 653
2,5-Диметилнитробензол (234), 653
2,6-Диметилиитробензол (225), 653
3,4-Диметилиитробензол (258), 653
3,5-Диметилнитробеизол (273), 655
2,5-Диметил-З-нитробеизолсульфонаМид
661
2,5-Днметил-4-иитробензолсульфонамид
662
2,5-Диметил-6-нитробензолсульфоиамид
[207],
[173],
[198],
[192],
[75],
[НО],
137
620
[75], 659
(212), [27], 658
[74], —*
[49],
[63],
[68],
659
659
659
659
610
662
2,5-Диметил-З-нитробеизолсульфоннлхлорид
[61], 664
2,5-Диметил-4-иитросульфоиил хлорид
664
2,5-Диметил-6-нитросульфоиилхлорид
665 ™
3,5-Диметил-4-нитрофеиол, растворимость 129
2,3-Диметилпиридин (164), 619
2,5-Диметилпиридин (160), 619
2,6-Диметилпиридин (143), 619
N.N-Диметилпсевдокумидии (220), 619
Диметилсульфоксид как растворитель
N.N-Диметил-л-толуидии (212). 619
N.N-Диметил-о-толуидии (185), 619
Ы,Ы-Днметил-я-толуидин (210), 619
Г4,Ь1-Диметил-2,4,6-трибромаинлии (301)
2,3-Дим етил феиол 'чгл
2,4- Дим етил феиол
2,5-Дим етил феиол
2,6-Диметилфеиол
3,4- Дим етил ф еиол
3,5-Диметилфенол ...
N.N-Диметилформамид (ДМФА) (153),
ИК-спектр 321
как растворитель 137 .
2,5- Диметилфуран (94), 633
2,4-Диметилхинолин (264), 620
2,6- Диметилхинолин (265), 620
N.N-Диметил-о-хлораиилии (207), 619
N.N-Диметил-л-хлоранилин (230), 619
1,1-Диметилцнклогексан (120), 645
транс-1,4-Думетилциклогексаи (119), 645
цис-1,4-Диметилциклогексан (124), 645
1,1-Диметилциклопеитан (88), 644
транс-1,2-Дим етилциклопентаи (92), 644
цис-1,2-Дим етил циклопент ан (100), 645
а,а-Диметиляитарный ангидрид, ИК-спектр
315
2,3- Диметокси бенз альдегид [54], 608
2,4-Днметоксибеиз альдегид [69], 608
3,4-Дим етоксибензальдегид, ИК-спектр
2,4-Дим етоксибеиз ол сульфон амид [167],
2,4-Диметоксибензолсульфонилхлорид
664
Диметоксиметан (45)
1,1-Диметоксиэтан (64), 594
ИК-спектр 210
спектр ЯМР 215
Ди-а-иафтол [300]
Ди-[3-иафтол [218]
4,4'-Динитроазоксибеизол [200], 657
2*4-Динитроанизол [95], 636
3,5-Динитроанизол [106], 636
2,4-Динитроаннлин [180], 616
529
661
[71],
601
602
2,6-Дииитроанилии [138], 616
2,4- Динитробензамид [203], 611
3,5-Динитробеизамид [183], 611
3,5-Дииитробензанилид [234], 611
3,5-Дииитробензоаты 180, 345, 352
3,5-Динитробензоилбромид [60], 597
2,4-Дииитробензоилхлорид [46]. 597
3,5-Динитробеизоилхлорид [74], 597
2,4-Дниитробеизойная кислота [183],
3,5-Динитробеизойиая кислота [202],
л-Динитробеизол (302), [90], 655
о-Дииитробензол (319), [118], 656
п-Динитробензол (299), [172], 656
2,4-Диннтробензолсульфенил хлорид,
едииеиие к алкеиам 224
2,2'Динитробифен ил [128], 656
2,3-Дннитробифенил [ПО], 656
2,4'-Динитробнфенил [93], 656
3,3'-Динитробифен ил [200], 656
4,4'-Динитробифенил [237], 656
2,4-Динитробромбензол [72], 655
2,4-Динитро-л-ксилол [84], 655
2,5-Дииитро-л-ксилол [101], 655
4,5-Дииитро-л-ксилол [132], 656
3,4-Дииитро-О-ксилол [82], 655
3,5-Дииитро-о-ксилол [76], 655
3,6-Дииитро-о-ксилол [90], 655
4,5-Динитро-о-ксилол [118], 656
2,3-Дииитро-п-ксилол [93], 655
2,5-Дииитро-я-ксилол [147], 656
2,6-Диинтро-л-ксилол [123], 656
Дииитромезидин [194], 616
2,4-Динитромезитилен [85], 655
1,5-Дннитроиафталин [214], 656
1,8-Дииитронафталин [170], 656
1,5-Дииитро-2-нафтиламин [191], 616
2,4-Дииитронафтол [138], 660
2,4-Динитрорезорции [148], 660
3,5-Динитросалициловая кислота [173], 601
2,4-Динитротолуол [70], 655'
2,6-Динитротолуол [66], 655
3,5-ДиннтротОлуол [92], 655
2,4-Динитрофеиилгидразни [198, разл.], 616
реагент для классификации 185
производные 205
2,4-Дииитрофенилгидразоиы
я, я'-Динитрофен иловый эфир [143], 656
2,4-Динитрофеиол [114], 660
2,4-Дниитрохлорбензол (315, разл.), [52], 654
Диоксан (102), 633
1,3-Диоксан (105), 633
Диоксиацетон [72], 649
2,4-Диоксиацетофенон [147], 650
3,5-Диоксибензальдегид [157], 608
2,2'-Диоксибнфеиил ' ’
п,п'- Диоксибифенил
1,2-Дноксинафталин
1,4-Диоксин афталии
1,5-Диоксннафталии
1,8-Диоксин афта лин
2,7-Дноксииафталин _ . „
L-Диоксифенилалании [280, разл.]. 622
Ди-(2-оксиэтил)амин (268), [28], 618
Ди-я-октиловый эфир (288), 216, 634
Дипеитеи (181), 646
1,1-Ди-я-пентоксиметан (219), 595
1,1-Ди-я-пентоксиэтан (222), 595
а,а'-Дипиридил [69], 621
Дипнои (345), 648
Ди-я-пропиламин (ПО), 617
спектр ЯМР-,3С 504
растворимость 137
N.N-Ди-я-пропиланилин (245). 620
Ди-я-пропилкарбинол (156), 603
Ди-я-пропилкетон (145), 647
Ди-н-пропиловый эфир (90), 633
Ди-я-пропоксиметан (140), 595
Дн-я-пропоксиэтан (148), 595
присо-
[1101. 660
[272], —
[108],
[176],
[258],
[140],
[186],
660
660
660
660
660
660
Предметный указатель
683
Дистилляция 415
в вакууме 422
вращающаяся лента 422
колонки 417
нагревание колонок 422
с паром 423
Дисульфиды 391
ИК-спектры 386, 391
производные 391
спектры ЯМР 391
Ди-п-толиламин (330), [79], 618
Ди-п-толилкетон (334), [92], 649
сшсж-Дн-лс-толилмочевина [218], 611
сижж-Ди-о-толилмочевина [250], 611
сыжж-Ди-л-толилмочевина [268], 611
Дифениламин (302). [54], 618
основность 128
Дифенилацетальдегид, ИК-спектр 202
N.N-Дифенилацетамид [101], 609
Дифенилацет амид [168]. 610
Дифен ила цетаннл ид [180], 611
Днфенил ацетилен (298), 647
и-Дифеиилбеизол [85], 644
транс-1,4-Дифенилбутадиен (350), [148], 647
цас-1,4-Дифенилбутадиен [70], 647
нссажж-Дифенилгидразин [34], 614
Дифенилгуанидин [147], 610
Дифенил-4.4'-дисульфоиамид [300], 662
Днфенил-4,4'-дисульфонилхлорид [203], 666
Дифеи ил карбамил хлорид [85], 597
Дифенилкарбииол, окисление Ce(lV) 171
Дифенилметаи [26], 644
растворимость 137
Дифенилметан-4,4-дисульфонилхлорид [124],
665
несимм-Дифенил мочевина [189], 611
сижж-Дифенил мочевин а [238], 611
Дифениловый эфир (252), [28], 635
Дифенилуксусная кислота [145], 601
N.N-Дифенилформамид [73], 609
1,1-Дифенилэтаи (270), 643
1,2-Дифенилэтанол ]67], 605
1,1-Дифеи ил эти лен (277), [8], 647
Дифеновая кислота [228], 602
Дифторацетамид [52], 609
jf-Дифторбензол [82]. 640
о-Днфторбеизол [92]. 640
п-Дифторбензол [88], 640
п,rf-Дихлоразоксибензол [154], 657
2,4- Дихлораиилин (245), [63], 614
2,5- Дихлораиилии (251), [50], 614
а,а-Дихлорацетамнд [98], 609
а,а-Дихлорацетаиилид [118], 610
Дихлорацетилхлорид 107), 595
а.а-Дихлорапетон (120), 647
а,а'-Дихлор ацетон (173), 648
2,4-Дихлорбензальдегид [71], 608
2,6- Дихлорбензальдегид, спектр ЯМР-13С 512
2,5- Дихлорбензамид [155], 610
2,6- Дихлорбензамид [202], 611
2,3- Дихлорбензойная кислота [164], 601
2,4- Днхлорбензойиая кислота [160], 601
2,5- Дихлорбензойная кислота [153], 601
2,6-Д ихлорбензойиая кислота [139], 601
м-Дихлорбензол (172), 638
О-Дихлорбензол (179), 638
л-Дихлорбензол [53], 640
2,4-Дихлорбеизол-1,5-дисульфоиамид [276],
662
2,4-Дихлорбеизол-1,5-дисульфоиилхлорид
[123], 665
2,5-Дихлорбензол-1,3-д исульфон илхлорид
[114], 665
2,4-Дихлорбензолсульфоиамид [180], 661
2,5-Дихлорбензолсульфонамид [181], 661
3,4-Дихлорбензолсульфонамид [135], 661
2,4-Дихлорбензолсульфоиилхлорнд [53], 664
2,5-Дихл орбензолсульфон ил хлорид [38], 663
3,4-Дихлорбензолсульфонилхлорид [19], 663
4,4-Дихлорбензофенон [148], 650
2,2'-Дихлорбифенил [60], 640
4,4'-Дихлорбифенил [149], 640
1,2-Дихлорбутан (124), 641
2,3-Дихлорбутаи (116), 641
3,4-Дихлор-2,5-дим етил беизолсульфоиамид
[201], 662
3,6-Дихл ор-2,5-диметил беизолсульфоиамид
[165], 661
4,6-Дихлор-2,5-диметилбензолсульфоиамид
[150], 661
3,4-Дихлор-2,5-диметилбеизолсульфонилхло-
рид [62]. 664
3,6-Дихлор-2,5-диметилбеизолсульфонил хло-
рид [71], 664
4,6-Дихлор-2.5-диметилбеизолсульфоиилхло«
рид [81], 664
Дихлордиметнловый эфир (59), 632
2,5-Дихлор-п-ксилол [68], 640
2,3-Дихлор-4-метилбензолсульфоиамид [2371.
662
2,3-Дихлор-5- м ет и л бенз о л с у л ьфон а м ид [158],
661
2,3-Дихлор-6-метилбензолсульфонамид [186],
661
2,4-Дихлор-З-метилбензолсульфоиамид [203],
662
2,4-Дихлор-5-метилбеизолсульфонамнд [176],
661
2,4-Днхлор-6-метилбеизолсульфоиамид [168].
661
2,6-Дихлор-З-м етил беизолсул ьфон амид [188],
662
2,6-Дихлор-4-метилбензолсульфонамид [155],
661
3,4 -Дихл ор-2 - м ет и л беиз ол сул ьфон а м ид [2281,
662
3,5-Дихлор-2-метилбеизолсульфонамид [186],
661
4,5-Дихлор-З-метилбеизолсульфонамид [186],
661
2,3-Дихлор-4-метилбензолсульфонилхлорид
[41], 663
2,3-Д их лор -5-м ет ил бензоле у л ьфои и л хлорид
[64], 664
2,3- Д и хлор -6- м етил бензол с ул ьфон нл хлор ид
]49], 663
2,4-Дихлор-З-метилбензолсульфонилхлорид
[59], 664
2.4- Д нхл ор -5-м етил бенз ол с у л ьфон ил хлорид
[72], 664
2,4- Дихл ор-6- м ет и л бенз ол с у л ьфон ил хлор ид
[43], 653
2,6-Дихлор-З-метилбеизолсульфонилхлорид
|19], 663
2,6- Ди хл ор -4-м етил бенз о л с ул ьфон ил хлорид
[56], 664
3,4-Дихлор-2-метилбензолсульфонилхлорид
[52]. 663
3,5-Днхлор-2-метилбензолсульфоиилхлорид
[54], 664
4,5-Дихлор-З-метилбензолсульфоиилхлорид
[88], 664
а,а'-Днхлорметиловый эфир (105), 633
а,₽-Днхлориафталии [36], 640
2,4- Днхлорнитробеизол (258), 653
2,5- Дихлоринтробензол [54], 654
2,2- Дихлорпроп ан (70), 641
1,3-Дихлорпропаиол-2 (176), 604
2,3- Дихлорпропаиол-| (182), 604
•у/р'-Дихлорпропиловый эфир (215), 634
2,4- Дихлортолуол (195), 639
2,6-Дихлортолуол (199), 639
Ддхлоруксусиая кислота (189), 598
Дихлоруксусиый ангидрид (216, разл.), 596
2,3- Дихлорфенол [57]. 659
2,4- Дихлорфенол (209) [43], 659
684
Предметный указатель
2,5- Дихлорфеиол [59], 659
3,5-Дихлорфеиол [68], 659
транс-Дихлорэтилен (60), 641
цпс-Дихлорзтилеи (48), 641
Ди-сс-хлорэтиловый эфир (98), 633
Ди-0-хлорэтиловый эфир (107), 633
Дициандиамид [207], 611
Диэтаноламин (268), [28], 618
Диэтилам ин (55), 617
растворимость 137
3-Диэтил аминоэтиловый спирт (161), 619
Ы,Ы-Диэтиланилнн (218), 619
Диэтилбарбитуровая кислота [188], 611
м-Диэтил бензол (182), 643
о-Диэтилбензол (184), 643
л-Диэтилбензол (184), 643
М,М-Диэтил-2,4-дииитроанилии [80], 621
Диэтиленгликоль (245), 605
диметиловый эфир (162), 633
диэгиловый эфир (188), 633
моно-н-бутиловый эфир (231), 634
моно-н-бутилевый эфир, ацетат (246), 629
моиомет иловый эфир (193), 633
моноэтиловый эфир (202), 633
моиоэтиловый эфир, ацетат (218), 628
растворимость 137
а,Р-Диэтилкарбаиилид [79], 609
Днэтилкарбинол (116), 603
Диэтилкетон (102), 647
окисление Ce(IV) 171
Диэтилмалонамид(ди) [224], 611
Диэтилмалонамид(моно) [146], 610
Диэтилмалоиовая кислота [125], 600
Диэтиловый эфир (35), 603
ИК-спектр 348
растворимость 137
как растворитель 137
N.N-Диэтил-лг-толуидин (231), 619
Ь1,К-Диэтйл-о-толуидин (206), 619
N.N-Диэтил-л-толуидин (229), 619
Диэтил уксусная кислота (193), 598
N.N-Диэтилформамид (176), 610
1,1-Диэтокси-2-бромэтан (170), 595
1,1-Диэтоксибутаи (143), 595
1,1-Диэтокси-2,2-дихлорэтан (184), 595
Диэтоксиметаи (89), 594
1,1-Днэтоксипропан (124), 594
1,1-Диэтоксипропен-2 (126), 594
а,а-Диэтдкситолуол (222), 595
1,1-Диэтоксй-2-хлорэтаи (157), 595
н-Додекан (215), 645
ДОдецеи-1 (213), 647
Дульцит [189], 604
Дурол [79], 644
Дымящая серная кислота 282, 604
Еиолы
бромирование 218
окисление Cr(VI) 173
Железа
гидроксамат, проба на амиды 325
(II) гидроксид 369
(III) хлорид, раствор 403
Жерара реагент
Р 209
Т 209
Замерзания точки 64
Запах 48
Иза гии [200], 611
Изоамиламин (95), 612
Изоамиланилнн (254), 618
Изоамилацетат (142), 625
Изоамилбензоат (262), 629
Изоамнлбромид (118), 637
Изоамил-н-бутират (178), 626
Изоамилизовалерат (190), 627
Изоамилиодид (148), 638
Изоамилкарбамат [64], 609
Изоамилкарбоиат (228), 628
Изоамилмеркаптан (117), 651
Изоамил-р-иафтиловый эфир [26], 635
Изоамилиитрат (147), 632
Изоамилнитрит (99), 632
Изоамиловый спирт (130), 603
Изоамилоксалат (262), 629
Изоамил пропион ат (160), 626
Изоамилсалицилат (277), 629
Изоамилсукцинат (297), 629
Изоамилформиат (123), 625
Изоамилфталат (349), 630
Изоамил хлорид (100), 637
Изоамилхлорформиат (154), 626
Изобутиламии (69), 612
N-Изобутилаиилин (231), 617
Изобутил ацетат (116), 625
Изобутил бензоат (241), 628
Изобутилбензол (173), 643
Изобутил бром ид (91), 637
Изобутил-н-бутират (157), 626
Изобутилизобутират (147), 626
Изобутилиодид (120), 638
Изобутилкарбамат (55), 609
Изобутилкарбоиат (190), 627
Изобутилмеркаптан (88), 651
Изобутнлметилкетои (119), 647
Изобутил-2-иафтиловый эфир [33], 635
Изобутилиитрат (123), 632
Изобутилиитрит (67), 632
Изобутиловый спирт (108), 603
окисление Ce(lV) 171
Изобутилпропионат (137), 625
Изобутил сукцинат (265), 629
Изобутилфенилацетат (247), 629
Изобутилформиат (98), 626
Изобутйлхлорид (68), 637
Изобутилхлорформиат (130), 625
Изобутирамид [128]. 610
растворимость 137
Изобутиранилид [105], 609
Изобутирилхлорид (92), 595
Изобутиронитрил (108), 651
растворимость 137
Изобутирофеион (222), 648
И?овалеранилид [109], 609
Изовалернановая кислота (176), 598
Изовалериаиовый альдегид (92), 606
Изовалероилбромид (140), 595
Изовалероилхлорид (115), 595
Изовалероиитрил (130), 652
Изовалерофеион (225), 648
Изодуренол [81], 658
Изодуридин (255), 613
Изодурол [81], 644
растворимость 137
Изокапроилхлорид (147), 595
Изокапроиитрил (153), 652
Изокапроиовая кислота (195), 598
D-Изолейции [279, разл.], 622
L-Изолейцин [279, разл.], 622
d.Z-Изолейции [292, разл.], 622
Изомасляная кислота (155), 598
Изомасляиый альдегид (64), 606
растворимость 137
Изомасляиый ангидрид (182), 596
Изонитриты 359
Изопеитаи (31), 644
Изопеитиламин, растворимость 137
N-Изопеитиланилин (254), 617
Изопеитилацетат (142), 625
Изопеитилбензоат (262), 629
Предметный указатель
685
Изопентилбромид (118), 637
Изопентил-я-бутират (178), 626
Изопентнлизовалерат (190), 627
Изопентнлиодид (148), 638
Изопентилкарбамат [64], 609
Изопентилкарбонат (228), 628
Изопентилмеркаптан (117), 651
Изопентил-Р-нафт иловый эфир [26], 635
Изопентилнитрат (147), 632
Изопентнлнитрит (99), 632
Изопентиловый спирт (130), 603
растворимость 137
Изопентилоксалат (262), 629
Изопентилпропионат (160), 626
Изопеитилсалицнлат (277), 629
Изопентилсукцинат (297), 629
Изопентилформиат (123), 625
Изопентилфталат (349), 630
Изопентилхлорнд (100), 637
Изопентилхлорформиат (154), 626
Изопрен (34), 645
Изопропенил ацет ат (96), 625
Изопропеиилбензол (1'65), 643
Изопропиламин (33), 611
N-Изопропиланилин (213), 617
Изопропил ацетат (91). 625
растворимость 137
я-Изопропилбензамид [133], 610
Изопропнлбеизоат (218), 628
я-Изопропилбензойная кислота [116], 600
Изопропилбензол (153), 643
Изопропил бром ид (60). 637
Изопропил-н-бутират (128), 625
Изопропилиодид (89). 638
Изопропилкарбамат [92], 609
Изопропиллактат (168), 626
Изопропилмеркатан (56), 651
4-Изопропил-2-метилбензолсульфонамид [73],
661
Изопропилметилкетон (94), 647
растворимость 137
Изопропил-2-нафтиловый эфир [40], 635
Изопропиловый спирт (83), 603
окисление Ce(IV) 171
Изопропилоксалат (190), 627
Изопропилсалицилат (237), 629
Изопропилфенилкарбинол (224), 604
Изопропилформнат (68), 624
Изопропилфтал ат (302), 629
Изопропил хлор ид (36), 637
Изопропилхлорформиат (105), 625
Изосафрол (246), 635
L-Изосерин (разл.), 623
dJ-Изосерин (разл.), 623
Изотиоцианаты 358
ИК-спектры 360
Изофорон (214), 648
Изофталевая кислота [300], 629
Изоцианаты 358
ИК-спектры 360
Изоцианиды (изонитрилы) 358
ИК-спектры 360
Изоэвгенол (267), 658
а-(Имидоилтио)уксусиые кислоты, хлоргид-
раты 383
Имиды 357
производные 325
таблица 357, 360
а-Иминоалкилмеркаптоуксусные кислоты,
хлоргидраты 382
Имииы 190, 358
ИК-спектры 360
Ииданои-1 (242). [42], 649
Инден (180), 646
Индикаторная бумага (pH) 305
Иидол (254). [52]. 618
Инозит [225], 606
Инулин [178, разл.], 624
Инфракрасная спектроскопия 33, 138
диаграммы Колтуна 148
области поглощения 148
образцы 142
пластинки 145
приборы 117, 118
растворители 148
Иод, проба иа алкены 223
0-Иоданизол (240), 635
ж-Иодаиилии [27]. 613
о-Иоданилин [56], 614
я-Иоданилин [62], 614
Иодат-иодный реагент, проба иа кислоты
305
Иод ацетамид [95], 609
сс-Иодацетанилид [144], 610
я-Иодацетаиилид [182], 611
.я-Иодбензальдегид [57], 608
о-И од бенз альдегид [37], 608
л-Иодбензамид [186], 611
о-Иодбеизамид [183], 611
я-Иодбензамид [217], 611
о-Ибдбеизанилид [141], 610
я-Иодб^нзаиилид [210], 611
я-Иодбеизилсульфонамид [206], 662
я-Иодбеизоилбромид [60], 597
.м-Иодбензойная кислота [187], 602
о-Иодбензойная кислота [162], 601
я-Иодбензойная кислота [265], 602
И од бензол (188), 638
о-Иод бензонитрил |55], 652
Иодистоводородная кислота ”350
1-Иоднафталин (305), 639
(З-Иодиафталин [55], 640
1-Иод-4-нафталинсульфоиамид [202], 662
1-Иод-5-нафталйнсульфоиамид [236], 662
2-Иод-1-нафталиисульфонамнд [154], 662
8-Иод-1-нафталинсульфоиамид [187], 662
1-Иод-4-иафталинсульфоиилхлорнд [121], 665
1-Иод-5-нафталинсульфоннлхлорид [113], 665
2-Иод-1-нафталинсульфонилхлорнд [ПО], 665
8-Иод-1-нафталинсульфонилхлорид [115], 665
л-Иодиитробензол [38], 654
4И6д-3-нитротолуол [55], 654
Йодоформ [119], 642
2-Иодпентан (142), 638
Р-Иодпропионовая кислота [82], 599
Л{-Иодтолуол (204), 639
о-Иодтолуол (211), 639
я-Иодтолуол (211), [35], 639
Иодуксусиая кислота [84], 599
о-Иодфенетол (245), 635
я-Иодфенетол [27], 635
м-Иод фенол [40], 658
Итаконовая кислота [165, разл.], 598
Итаконовый ангидрид [68], 597
/-Камфен (160), [42], 646
( + )-Камфора (206), [179], 650
d,/-Камфора (205), [178], 650
d-Камфорная кислота [187], 602
ангидрид [221], 597
( + )-Камфор-3-сульфоиамид [143], 661
( + )-Камфор-8-сульфоиамид [137], 661
( + )-Камфор-10-сульфонамид [132], 661
( + ) Камфор-3-сульфоиилхлорид [88], 664
( + )-Камфор-8-сульфонилхлорид [132], 665
( + )-Камфор-10-сульфонил хлорид [67], 664
Камфорхинон [198], 650
н-Каприловая кислота (236), 598
Каприловый альдегид (171), 607
Каприлоиитрил (200). 652
Каприновая кислота (270), [30], 599
Каприновый альдегид (209), 607
н-Кап роил хлорид (153), 596
н-Капронамид [110], 609
686
Предметный указатель
н-Капронитрил (164), 652
н-Капроновая кислота (205), 602
н-Капроновый
альдегид (128), 607
ангидрид (257), 596
Карбазол (351), [243], 618
основность 128
Карбамид [132], 610
Карбанилид [238], 611
Карбодиимиды 358
ИК-спектры 360
Карбоновые кислоты, соли 328
ИК-спектры 328
масс-спектры 329
производные 332
спектры ЯМР 329
Карбостирил [200], 660
Карвакрол (237), 658
Карвон (225), 648
Качественный анализ 161
Кетали 209
Кетоиы 185
восстановление 207
ИК-спектры 197—202
масс-спектры 197—203
производные 203
таблица 647—651
спектры ЯМР 197—202
Р-Кетоэфиры 193
Кипения температура 30
влияние давления 68
Кислот ангидриды 313
ИК-спектры 313
масс-спектры 315
таблица 595—597
спектры ЯМР 315
Кислот галогенангидриды, производные 317
ИК-спектры 317
спектры ЯМР 320
Кислот гидразиды 345
Кислотность
влияние структуры 126
пространственные эффекты 127
электронные эффекты 127
Кислот хлорангидриды 595—597
Кислоты карбоновые 76, 77, 305
ИК-спектры 305, 306
масс-спектры 307
спектры ЯМР 307
Количественный элементный анализ, сожже-
ние 84
Конго красный, индикаторная бумажка 305
Кониферин [185, разл.], 624
Концентрации вредных веществ, предельно
допустимые 668—672
Коричная кислота [133], 600
Коричный
альдегид (252), 607
ангидрид [130], 597
спирт (250), [33], 605
Кофеин [234], 611
Креатин [315, разл.], 611
Креатииии [292, разл.], 622
я-Крезил ацетат (212), 627
о-Крезилацетат (208), 627
л-Крезил ацетат (212), 627
ж-Крезилбензоат [54], 630
о-Крезилбензоат (307), 629
л-Крезилбензоат [71], 631
ж-Крезилкарбоиат [115], 631
о-Крезилкарбоиат [60]. 631
ж-Крезилметиловый эфир (177), 634
о-Крезилметиловый эфир (171), 634
л-Крезилметиловый эфир (176), 634
м- Крез ил эти левый эфир (192), 634
О-Крезилэтиловый эфир (192), 634
л-Крезилэтиловый эфир (192), 634
ж-Крезол (202), [3], 658
о-Крезол (190), [31|, 657
ИК-спектр 408
спектр ЯМР 408
л-Крезол (202), [36]. 658
2,4-Крезотовая кислота [178], 601
Кротон ил хлор ид (126), 595
транс-Кротоиовая кислота [72], 599
класс растворимости 137
Члс-Кротоиовая кислота (165, разл.). 598
Кротоновый
альдегид (103), 607
ангидрид (248), 596
N-Ксаитилсульфоиамиды 400
Ксаитин [360], 611
Ксантои (350), [173], 650
4-Ксенилмеркаптан [111], 651
D-Кснлоза [145, разл.], 623
ж-Ксилол (139), 643
ИК-спектр 289
о-Ксилол (142), 643
спектр ЯМР-,3С 513
л-Ксилол (137), 643
растворимость 137
Кумарии (290), [67], 631
о-Кумаровая кислота [207], 602
л-Кумаровая кислота [206], 602
Кумол (153), 643
Лабораторное оборудование 587—593
Лактамы 358
ИК-спектры 361
Лактид (255), [128]. 631
Лактоза [203, разл.], 624
окисление Ce(IV) 171
Лактоиы 358
Лактоионитрил (182, разлД, 611
Лантаноидные сдвигающие реагенты 517
хиральиые 521
Лауриловый спирт [24], 605
Лауриновая кислота [43], 599
Лауриновый
альдегид [45], 608
ангидрид [42], 596
Лаурон [69], 649
Левулиновая кислота (250, разл.), 599
Левулоза [104, разл.], 624
D-Лейцин [337, разл.], 623
L-Лейцин [337, разл.], 623
d.Z-Лейции [332, разл.], 623
Лепидин (263), 620
Летучесть с паром 117
Либермана нитрозореакция 253
L-Ликсоза [105, разл.], 623
Линалоол (197), 604
Липиды, проба 574
D-Лизии [224, разл.], 622
L-Лизии [224, разл.], 622
d,/-Лизин [разл.], 622
Литература 38, 557
Лукаса
проба 172
реагент 173
2,4-Лутидин (159), 619
Малеамид [170], 610
Малеанилид [175], 610
Малеиновая кислота [130], 600
Малеиновый ангидрид [56], 597
Малонамид [170], 610
Малонамид (моно) [50], 609
Малоианилид (ди) [225], 611
Малонаиилид (моно) [132], 610
Малоновая кислота [133, разл.], 600
Малоионитрил (219), 652
Предметный указатель
687
Мальтоза [100, разл., 165. разл,], 623
окисление Ce(IV) 171
Маиделонитрил (170, разл.), 652
D-Маннит [166], 624
окисление Ce(IV) 171
D-Маииоза [132], 623
Маргариновая кислота [60], 599
н-Масляиая кислота (163), 598
растворимость 137
н-Масляный
альдегид (74), 608
— растворимость 137
ангидрид (191), 596
Масс-спектр 95
Масс-спектрометр 93
Масс-спектрометрия 31
Мезаконовая кислота [202, возг.], 602
Мезитила окись (130), 647
Мезитилен (164), 643
растворимость 137
Мезитиленамид [133], 610
Мезитиленовая кислота [166], 601
Мезитол [70], 659
Мелезитоза [147, разл.], 623
Р-Мелибиоза гидратированная [85], 623
Меллитовая кислота [288, разл.], 602
л-Меиган (169), 645
(—)-Ментиламин (205), 612
j—)-Ментил ацет ат (227), 628
(—)-Ментол [42], 605
L-Меитон (207), 648
Меркаптаны 385
ИК-спектры 386
масс-спектры 387
окисление 222
производные 385—387
спектры ЯМР 386
таблица 651
Метакриламид [116], 609
Метакриловая кислота (163), [16], 599
Металл, проба 107
Метальдегид [115], 608
Метансульфоиамид [90], 661
Метансульфоннлхлорид (60/21 мм), 663
а-Метилакролеин (73). 606
Метиламин (26), 611
4-(Г4-Метиламино)-1,3-диметилбензол (221),
617
4-( N- Метил амино)-2-иитротолуол [45], 618
4-(К-Метиламиио)-3-иитротолуол [84], 618
Метил анизат (255), [45], 630
Метиланилии (192), 617
Метилантранилат (260, разл.), [24], 630
р-Метилаитрахиион [179], 650
N-Метилацетаиилид [102], 609
N-Метил-о-ацетотолуид [56], 609
Ы-Метил-п-ацетотолуид [83], 609
N-Метилбензиламии (185), 617
2-Метилбензол-1,3-дисульфоиамид [260],
2-Метилбензол-1,4-дисульфоиамид [224],
3-Метил бензол-1,5-дисульфои а мид [216], __
4-Метилбензол-1,2-днсульфонамид [237], 662
4-Метилбензол-1,3-дисульфонамид [191],
2-Метил бензол-1,3-дисульфоиилхлорид
664
2-Мети л бензол-1.4-дисульфонил хлорид
665
З-Метил бензол-1,5-дисульфоиилхлорид
665
4-Метил бензол-1,2-дисульфонилхлорид
665
4-Метнл бензол-1,3-дисульфоиилхлорид
664
Метилбеизоат (198), 627
Метил-о-беизоилбензоат [52], 630
п-Метилбеизофенои [60], 649
Ы-А4етил-л-броманилин (260), 617
Метилбромацетат (144, разл.), 626
662
662
662
662
[88],
[98].
195].
[Ill],
[56].
[168],
[166].
[146],
598
598
Метил-л-бром бензоат [32], 630
Метнл-о-бромбензоат (244), 628
Метил-п-бромбеизоат [81], 631
2-Метил-4-бромбензолсульфонамид
2-Метил -5- бром бен зол су л ьфон ам ид
4-Метил-З-бромбеизолсульфонамид
2-Метил-4-бром бензолсульфонилхлорид
663
2-Метил-5-бромбензолсульфоиилхлорид
663
4-Метнл-З-бром бензолсульфонилхлорид
664
Метилбромид (5), 637
2-Метилбутанол~1. окисление Ce(IV) 171
2-Метилбутаиол-2, растворимость
З-Метил-1 -бутаисульфоиил хлорид
663
2-Метилбутеи-1 (31), 645
3-Метил бутен-1 (21), 645
Метнл-н-бутират (102), 625
Метил-н-валерат (130), 625
Метилвинилкетон (80), 647
2-Метилгексаи (90), 644
З-Метилгексан (92), 644
2-Метилгексаиовая кислота (210),
4-Метилгексановая кислота (218),
2-Метилгептаи (118), 645
4-Метнлгептаи (118), 645
Метил-н-гептоат (173), 627
а-Метнлгидрокоричиая кислота [37], 599
а-Метнл глюкоз а [165, разл.], 624
Г4-Метил-2,4-динитроанйлин [176], 618
Метил-3,5-дииитробензоат [108], 625
2-Метил-1,3-диоксан (ПО), 594
2-Метил-1,3-дноксолан (82), 594
N-Метилдифеииламин (296), 620
Метиленамииоацетоиитрил [129], 653
Метилен бром ид (98), 641
Метилениодид (180, разл.), 641
Метиленхлорид (42), 641
как растворитель 137
Метилизобутил карбинол (131), 603
Метилизобутил карбинолацетат (148), 626
Метилизобутират (92), 625
растворимость 137
Метилизовалерат (116), 625
растворимость 137
Метилизопропилкарбинол (113), 603
Мети л им ииоди у кс у си а я кислота [227, разл.],
602
Метнлиодид (43), 638
Метилкаприлат (193), 627
Метил-н-капрон ат (150), 626
Метилкапринат (232), 628
Метилкарбамат [52], 609
Метилкарбонат (90). 625
Метилкетоны 191
Метиллактат (144), 625
Метиллаурат (268), [5], 630
Метиллевулниат (191), 627
Метилмалонат (181), 626
растворимость 137
Метилмалоиовая кислота [135, разл.], 600
Метилманделат [58], 630
а-Метилмаииозид [193, разл.], 624
а-Метил-н-масляный альдегид (93), 607
Метилмеркаптаи (6), 651
Метилмиристат (296), [18], 630
Метилмочевина [101], 609
1-Метилиафталии (240), 643
Р-Метилнафталин [32], 644
1 -Метил-З-иафталинсульфонамид
1-Метил-4-нафталинсульфонамид
1 -Метил-6-нафталинсул ьфон ам ид
1 -Метил-7-нафталиисульфонам ид
2 Метил-6-нафталиисульфонамид
1 Метил-З-наф'
661
661
661
[50],
135],
[60].
в воде 137
(88/9 мм).
144], 661
174], 661
116], 661
189], 662
________________ 206], 662
____________,>талиисульфоиилхлорид [125], 665
1Метил-4-нафталинсульфоиилхлорид [81], 664
688
Предметный указатель
1-Метил-б-нафталинсульфонилхлорид [88]. 664
1-Метил-7-нафталинсульфонилхлорид [123],
665
Ы-Метил-а-нафтиламин (293), 618
N-Метил-^-иафтиламин (309), 618
ТЧ-Метил-Ы-а-нафтил ацетамид [95], 609
Метил-р-нафтилкетон (300). [53], 649
Метил-а-нафтиловый эфир (265), 635
Метил-Р-иафтиловый эфир [72], 636
Метилнитрат (65), 632
N-Метил-лс-нитроанилин [66], 618
2-Метил-З-нитроаиилии [95], 615
2-Метил-6-нитроанилин [91], 615
N-Метил-п-иитроацетанилид [153], 611
2-Метил-4-нитроацетанилид [196], 611
4-Метил-2-нитроацетаиилид [94], 609
4-Метнл-З-нитроацетаннлид [148], 610
Метил-уи-нитробензоат (279), [78], 631
Метил-п-нитробензоат [96], 631
2-Метил-2-нитробутаи (150), 653
З-Метил-1-нитробутан (164), 653
2-МеТил-1-нитропропан (141), 653
2-Метил-2-иитропропаи (126), 653
Метил-З-нитросалицилат [132], 631
Метил-5-нитросалицилат [119], 631
Метил-З-иитрофталат [69], 631
Метил-4-нитрофталат [66], 631
Метил-я-нитроциннамат [124], 631
Метил-о-нитроциниамат [73], 631
Метил-п-нитроциннамат [161], 631
Метиловый спирт (66), 603
ИК-спектр 176
окисление Ce(IV) 171
как растворитель 137
Метилоксалат (163), [51], 630
Метил-л<-оксибензоат [70], 631
А4етнл-л-оксибензоат [131], 631
З-Метил-З-оксибутаион-2, окисление Ce(IV),
171
З-Метил-З-оксибутин-1, окисление Ce(IV) 171
Метилортоформиат (101), 625
Мет ил пальмитат [30], 630
Метилпеларгонат (213), 628
2-Мет_илпентаи (60), 644
З-Метилпеитаи (63), 644
2-Метилпеитандиол-2,4 (196), 604
d,l-2- Метил пентановая кислота (196), 598
З-Метилпеитановая кислота (197), 598
2-Метилпеитанол-1 (148), 603
2-МеТилпентаиол-2 (121), 603
З-Метилпентанол-З (128), 603
4-Метилпеитанол-2 (131), 603
с/,/-2-МетилпипеЬйдин (116), 617
d.Z-3-Метилпиперидин (126), 617
Метилпируват (136), 625
растворимость в воде 137
2-Метилпропандиол-1,2 (178), 604
2-Метилпропанол-1, окисление Ce(IV) 171
2-Метил-1-пропансульфонамид [16], 661
2-Метил-1-пропансульфонилхлорид (73/11 мм),
663
Метил-н-пропилкарбинол (119), 603
Метил-н-проп ил кетон (102), 647
Метил пропион ат (79), 625
растворимость 137
Ы-Метилпсевдокумндин (237), [44], 618
Метнлсалицилат (224), 628
Метнлсебацат (228, разл.), [38], 630
Д4етилстеарат [38], 630
м-Мети л стирол (168), 643
о-Метилстирол (171), 643
«-Метилстирол (169), 643
Метилсукцинамид (ди) [200], 611
Метилсукцинат (195), [19], 630
ДЪетиЛсуЛЬфаТ (188). 632
D-Метилтартрат (280), [48], 630
d.Z-Метнлтартрат [90], 631
Метилт ерефталат [140], 631
Метил-2-тиенилкетон (214), 648
Метилтимиловый эфир (216), 634
Метил-п-толилкарбинол (219), 604
Метил-л-толил кетон (220), 648
Метил-о-толилкетон (216), 648
Метил-п-толилкетон (226), 648
Метил-лс-толуат (215). 628
Метил-б-толуат (213), 628
Метил-л-толуат (217), [33], 628
И-Метил-м-толуидин (206), 617
N-Метил-о-толуидин (207), 617
N-Метил-л-толуидин (208), 617
N-Метилтрибррманилин [39], 618
Метил ундецил ин ат (248), 629
Метилфеннлкарбинол (203), 604
Метил-[З-фенилэтилкетон (235), 648
Метилформиат (32), 624
Метилф’алат (282), 629
2-Метнл-4-фторбензолсульфонамид [173], 661
2-Д4етил-5-фторбензолсульфонамид [141]. 661
З-Метил-4-фторбензолсульфонамид [105], 661
2-Метилфуран (64), 632
Метнлфуроат (181), 626
5-Метилфурфурол (187), 607
4-Метилхинолин (263), 620
6-Метилхииолин (258), 620
7-Метилхииолин (252), [39], 620
N-Метил-о-хлораиилин (214), 617
N-Метил-л-хлораиилин (240), 617
Метил хлорацетат (130), 625
Метил-ти-хлорбензоат (231), 628
Метил-о-хлорбеизоат (234), 628
Метил-п-хлорбеизоат [44], 630
2-Метнл-4-хлорбензолсульфонамид [185], 661
2-Метил-5-хлорбензолсульфонамид [142], 661
.............. ‘ '""], 661
[53],
603
604
603
648
648
648
(165),
(175),
(174),
(163),
(169).
(169),
4- Метил -3- хлор бенз о л с ул ьфон а м ид [ 128],
2-Метил-4-хлорбензолсульфонилхлорид
664
2-Метил-5-хлорбензолсульфонилхлорид
663
4-Метил-З-хлорбензолсульфонил хлорид
664
Метилхлорформиат (75). 624
Метил целлозольв 171
окисление Ce(IV) 171
растворимость 137
Метилцианоацетат (200), 627
Метилциклогексан (100), 645
2-Метнлциклогексанол
З-Метилциклогексаиол
4-Метилциклргексаиол
2-Метилциклогексанон
3-Метил циклогексанон
4-Метилциклогексанои . ,.
11-Метилциклогексеи (111), 646
З-Метнлциклогексен (105), 646
4-Метнлциклогексен (103), 646
Метилциклогексилкетон (180). 648
Мет н л циклопен тан (72), 644
2-Метилцнклопентанон (139), 647
Метил цини ам ат (263), [36], 630
Метилцитрат (285, разл.), [79], 631
N-Метилэтиламин (35), 617
Метил этил ацетил хлорид (116), 695
Метилэтилацетоацетат (190), 627
Метилэтиловый эфир (10), 137, 632
Метилянтариая кислота [115], 600
L-Метнонин [283, разл.], 622
d,/-Метионин [272], 622
Метокснапетамид [971. 609
Метоксиацетаиилид [58], 609
л-Метоксиацетаннлид [[27], 610
Метоксиацетилхлррид (113), 595
Метокснацетонитрнл (120), 651
Л4-МетойСйацётофенон (245). 648
n-Метоксй ацетофен он (258), 648
уи-Метоксибензальдегнд (230), 607
р-Метоксибензальдегид [38], 608
[211,
[63],
Предметный указатель
689
о-Метоксибензамид [129], 610
о-Метоксибензанилид [131], 610
0-Метоксибензиловый спирт (247), 605
n-Метокси бенз иловый спирт [25], 605
лс-Метоксибензоилхлорид (224), 596
о-Метоксибензоилхлорид (254), 596
о-Метоксибеизойная кислота [100], 600
и-Метоксибензофенон (355), [62], 649
Метоксиуксусиая кислота (203), 598
Метон [149], 650
производные 205
D-Миидальная кислота [133], 600
L-Миндальная кислота [133], 600
d,/-Миндальная кислота [118], 600
Миристиловый спирт [39], 605
Миристиновая кислота [54], 599
Мирцен (167), 646
Молярный отклик 86
Молекулярная масса 89
Молекулярные формулы 538
Молиша проба на сахара 300
d,/-Молочная кислота [18], 599
окисление Ce(IV) 171
Морфолин (130), 617
Мочевая кислота [разл.], 611
Мочевина [132], 610
Муковая кислота [213, разл.], 602
Муравьиная кислота (101), 597
[169], 597
[274], 597
[246], 597
Нагревательные бани 581—582
Нальге — Аксельрода прибор для определе-
ния температуры плавления 63
Натрий 167
Натриевый плав 191. 192
Натрия
ацетат, ИК-спектр 328
— спектр ЯМР 329
бисульфид как восстановитель 372
бисульфит, раствор 189
борогидрид 207
гидроксид, раствор 333, 339
иодид, раствор в ацетоне 234
1,2- Нафталевый ангидрид ' ’
1,8- Нафталевый ангидрид
2,3-Нафталевый ангидрид
Нафт а л иды 247
а-Нафтальдегид [34], 608
6-Нафтальдегид [60], 608
Нафталин [80], 644
растворимость 137
1,4-Нафталиндисульфоиамид
1,5-Нафталиндисульфонамид
1,6- Нафталиидисульфонамид
2,7-Нафталиндисульфонамид
1,3-НаГ
1,4- Hai. ... . ..
1,5- Нафталиндисульфоиилхлорид
1,6- Нафталнндисульфоннл хлорид
1,7-Н афталиндисульфонилхлорид
2,6-Нафталиндисульфоннл хлорид
2,7-Нафталиндисульфонилхлорид
1-Нафталинсульфонамид [150], 661
2-Нафталинсульфонамид [217], 662
ЬНафталинсульфоиилхлорид [68], 664
2-Нафталннсульфонилхлорид [76], 664
1,3,5,7-Нафталннтетрасульфонилхлорид
656
[273], 662
[310], 662
[298], 662
______ _ _ . _ .... _ [242], 662
кЬталиндисульфонилхлорид 138],
крталиндисульфоиилхлорид 166],
665
665
665
665
665
666
665
183].
128],
122],
225],
162],
[ЗЮ],
Нафталннтетрахлорид [182], 640
1,3,5-Нафт ал интрисульфонилхлорид [146], 665
1,3,6-Нафталинтрисульфонилхлорид [193], 666
1,4,5-Нафталинтрисульфонилхлорид [156], 665
2,3,6-Нафталинтрисульфонилхлорид [200], 666
^-Нафтамид [192], 611
а-Йафт^нилид [163], 610
^-Нафтанилид [171], 610
а-Нафтиламин (300), [50], 614
основность J29
6-Нафтиламин (300), [112], 615
а-Нафтилацетамид [181], 611
P-Нафтил ацетамид [200], 611
N-a-Нафтилацетамид [159], 610
N-fJ-Нафтилацетамид [134], 610
a-Нафтилацетанилид [155], 610
a-Н а фтил ацетат [49], 630
(З-Нафтилацетат [70], 631
N-a-Нафтилбензамид [161], 610
6-Нафтилбензоат [107], 631
а-Нафтилметилкетон [34]. 649
[З-Нафтиловые эфиры
[З-Нафтилсалицилат [95], 631
а-На фтил сульф он амиды 267
а-Нафтилу ксусиая кислота [133]. 600
₽-Нафтилуксусная кислота [142], 601
a-Нафтил уретаны 179
a-Нафтойиая кислота [162], 601
6-Нафтойная кислота [185], 601
a-Нафтойиый ангидрид [146], 597
а-Нафтол [94], 659
6-Нафтол [122], 660
а-Нафтонитрил (299), [35], 652
Р-Нафтонитрил (306), [66], 652
а-Нафтохинои [125], 650
₽-Нафтохинон [120], 650
Нейтрализации эквивалент 309
Ненасыщенные углеводороды, производные,
таблица 645—647
Неопентан (9), 644
Неопентил бром ид (109), 637
Неопентиловый спирт (53), 605
Неопентилхлорид (85), 637
d,/-Никотин (242), 619
Нитрамины 358
Нитраты органические 359
ИК-спектры 362
Нитрилы 366
восстановление 380
восстановление — гидролиз 381
гидролиз 325, 380
из галогенидов 247
ИК-спектры 375
масс-спектры 375
производные 378, 379
спектры ЯМР 377
таблица 651—653
Нитриты органические 359
л«-Нитроаиизол [39], 635
о-Нитроаиизол (272), 635
п-Нитроанизол [54], 636
ж-Нитроаиилии (284), [114], 615
о-Нитроаиилин [71], 614
п-Ннтроанилин [71], 614
растворимость 137
Ннтроанилины, гидролиз 326
З-Нитроаиисовая кислота [190], 598
м-Нитроацетанилид [155], 610
о-Нитроацетанилид [92], 609
л-Нитроацетаиилид [210], 611
льНитроацетофенон (202), [81], 649
л-Нитробеизальдегид [58], 608
о-Нитробензальдегид [44], 608
n-Нитробензальдегид [106], 608
м-Нитробензальхлорид [65]. 641
п-Нитробензальхлорид [43], 641
л-Нитробензамид [143], 610
о-Нитробензамид [176], 610
я-Нитробензамид [201], 611
я-Нитробенз амиды 266
ти-Нитробеизаиилид [153], 610
я-Нитробензанилид [211], 611
м-Нитробензил бром ид [58], 654
я-Нитробеизилбромид [99], 655
я-Нитробейзилидеибромид [82], 641
я-Нитробензнловые эфиры 311
ж-Нитробензиловый спирт [27], 654
о-Нитробеизиловый спирт [74], 655
690
Предметный указатель
л-Нитробеиз иловый спирт [93], 655
л-Нитробензилсульфонамид [159]. 661
О-Нитробеизилсульфоиамид [137], 661
л-Нитробеизнлсульфоиамид [200], 662
о-Нитробензилхлорид [48], 654
л-Нитробензилхлорид [71], 655
л-Нитробензоаты 180
л«-Нитробеизоилхлорид [35], 596
о-Нитробеизоилхлорид [20], 596
л-Ннтробензоилхлорид [75], 596
ж-Нитробензойная кислота [140], 601
О-Нитробензойиая кислота [146], 601
л-Нитробензойная кислота [241], 602
л«-Ннтробензойный ангидрид [160], 597
о-Нитробензойный ангидрид [135], 600
л-Нитробеизойный ангидрид [189], 602
Нитробензол (209), 653
ИК-спектр 366
растворимость 137
спектр ЯМР 366
ж-Нитробеизолсульфонамид [166], 661
о-НитробензолсульФоиамнд [191], 662
л-Нитробензолсульфонамид [180], 661
м-Нитробеизолсульфонамиды 267
лс-Нитробензолсульфоиилхлорид [63], 664
о-Нитробензолсульфоиилхлорид [68]. 661
л-Нитробензолсульфоннлхлорид [80], 664
ж-Нитробеизоиитрил [118], 653
о-Нитробензоиитрил [ПО], 653
л-Нитробензоиитрил [147], 653
2-Нитробифеиил [33], 654
З-Нитробифеиил [61], 654
4-Нитробифенил [113], 656
1-Нитробучаи (152), 653
2-Нитробутан (140), 653
Нитрование 289
1-Нитрогексаи (193). 653
Нитрогуанидин [230, разл.], 611
ж-Нитро-К.ЬГдиметиланилин (285), 620
л-Нитро-Ь[,М-диметиланилин [163], 621
Нитрозироваиие 383
5-Нитрозо-о-анизидин [108], 657
л-Нитрозоанилии[174], 657
N-Нитрозоацетанилнд [51, разл.], 656
Нитрозобеизол [68], 656
л-Нитрозо-Ы.Ы-диметнл анилин [87], 657
1-Нитрозо-2,4-диметилбензол [41], 656
N-Нитрозодифениламин [66], 656
л-Нитрозодифениламин [144], 657
л-Нитрозо-Н.Н-диэтилаиилии [84], 656
5-Нитрозо-о-крезол [135], 657
л-Нитроз о-N-м етил анилин [116], 657
1-Нитрозоиафталин [98], 657
1-Ннтрозо-2-иафтол [109], 657
2-Нитрозо-1-нафтол [152, разл.], 657
4-Нитрозо-1 -нафтол [194, разл.], 657
Нитрозосоединеиия 359
восстановление 372
из N.N-диалкиланилинов 271
ИК-спектры 361
получение 253. 271
таблица 656—657
Нитрозотимол [162], 657
ж-Нитрозотолуол [53], 656
о-Нитрозотолуол [721, 656
п-Нитрозотолуол [48], 656
л-Нитрозофеиол [125, разл.], 657
л-Нитрозо-И-этил анилин [78], 656
О-Ннтроиодбензол [49], 654
л-Нитроиодбеизол [171], 656
ти-Нитрокоричная кислота [199], 602
О-Нитрокоричиая кислота [240], 602
л-Ннтрокоричиая кислота [285], 502
З-Нитро-л-крезол [34], 658
4-Нитромезидин [75], 614
Нитромезитилен (245), [44], 654
Нитрометан (101), 653
конденсация 374
растворимость 137
о-Нитро-ГЧ-метнланилин [37], 618
л-Нитро-Ы-метилаиилии [152], 618
Нитромочевина [150, разл.], 610
а-Нитронафталин (304), [61], 654
₽-Нитронафталин [78], 655
1-Нитро-2-иафталинсульфонамид [214], 662
I -Н итро-2-н афтал иисул ьфонил хлорид [121],
565
1-Нитро-2-иафтиламии [126], 616
5-Нитро-1-нафтиламии [119], 615
8-Нитро-1-нафтиламин [97], 615
Нитроны 359
1-Нитропентаи (173), 653
2-Нитропеитан (154), 653
1-Нитропропан (130), 653
2-Нитропропан (120), 653
З-Нитросалнциловая кислота [144], 601
З-Нитросалициловая кислота, гидрат [125],
600
5-Ннтросалициловая кислота [227], 602
Н итросоедин ения
восстановление 369. 370
ИК-спектры 364
масс-спектры 364
производные 366
селективное восстановление 372
спектры ЯМР 364
таблица 653—656
[З-Нитростирол (260, разл.), [58], 654
ти-Нитротиофенол [75]
лг-Нитротолуол (231). [16], 654
о-Ннтротолуол (224), 653
«-Нитротолуол (238), [54], 654
ж-Ннтрофенетол [34], 654
о-Нитрофеиетол (267), 653
л-Нитрофеиетол [60], 654
л-Нитрофеиилацетонитрил [116], 653
л-Нитрофеиилгидразии [157* разл.], 616
л-Нитрофенилгидразиновая проба 187
л-Ннтрофенилгидразоны 188, 189
л-Нитрофенилуксусиая кислота [120], 600
о-Нитрофеиилуксусная кислота [141], 601
л-Нитрофеиилуксусная кислота [152], 601
.«-Нитрофенол [97], 660
о-Нитрофенол [45], 659
л-Нитрофенол [114], 660
Нитроформ [15], 654
З-Нитрофталаты 181
З-Нитрофталевая кислота [2181, 602
4-Нитрофталевая кислота [165], 601
З-Нитрофталевый ангидрид [162], 597
- 4-Нитрофталевый ангидрид [119], 597.
6-Ннтрохин альдин [164], 621
6-Нитрохииолин [149], 621
2 Нитро-л-хлорбром бензол [72], 655
2-Нитроцимол (264), 653
Нитроэтаи (114), 653
растворимость 137
сопряженное основание 131
м-Нитро-Ы-этилаиилин [65], 618
о-Нитроэтилбеизол (224), 653
л-Нитроэтилбеизол (241), 653
н-Нонан (149), 645
Нонановая кислота, спектр ЯМР 307
Нон анол-2 (198), 604
Нонанол-5 (194), 604
Нонен-1 (147), 646
н-Нонилбромид (220), 637
н-Нонилмеркаптаи (220), 651
н-Ноииловый спирт (215), 604
н-Ноиилхлорид (202), 637
Ё-Норлейции [285, разл.], 622
d,/-Норлейцин [327, разл.], 622
Нуйол, ИК-спектр 216
Озазоны, образование 299
Озонолиз 223
Предметный указатель
691
Окисление
бихроматом 290
молекулярной формулы изменение 541
перманганатом 290
Оксалилхлорид (64), 595
Оксамид [419, разл.], 611
Оксаиилид [257], 611
о-Окси азобензол [83], 657
л-Оксиазобензол [152], 657
n-Окси ацет анилид [169], 610
Оксиацетон (146), 648
2-Окси ацетофенон (218), [28], 649
З-Оксиацетофенон [96], 649
м-Оксибензальдегид [105], 608
л-Оксибеизальдегид [115], 608
о-Оксибензиловый спирт [871, 606
л-Оксибензойиая кислота [201], 602
л-Оксибензойная кислота [213], 602
З-Оксибифеннл [78], 659
л-Оксибифенил, растворимость в воде 137
З-Оксибутанои, окисление Ce(lV) 171
V-Оке и бутиронитрил (240), 652
а,/-Оксиглутаминовая кислота [150, разл,],
621
2-Окси-3,5-дибромтолуол [57], 659
л-Окси-К.Й-диметиланилин (268), [85], 621
л-Окси-Ы,Ы-диметиланилин [76], 621
4-Окси-2,6-диметилбензол-1,3-дисул ьфон амид
[208], 662
4-Окси-2,6-дим ет ил бензол-1,3-дисульфонил-
хлорид [119], 665
2-0 кси-3,5-ди нитротолуол [86], 659
а-Оксиизобутироиитрил (120), 651
а-Оксиизомасляиая кислота [79], 599
о-Окси-Ы-метил анилин [871, 618
л-Окси-И-метил анилин [85], 618
л-Окси-Ы-метилацетанилид [240], 611
Оксимы 259
ИК-спектры 363
1-Океи-2-иафтойная кислота [2261, 602
2-Окси-З-иафтойная кислота [222], 602
L-Оксипролнн [270, разл.], 622
р-Оксипропионитрил (221), 652
л-Оксипроп иофеи он [148], 650
3-Окси-2,4,6-тринитротолуол [НО], 660
л-Оксифенилглицин [248, разл.], 652
л-Оксифеиил уксусная кислота [148], 601
2-Оксихинолии [199], 621
6-Оксихинолин [193], 621
8-Оксихниолин (266), [75], 621
2-Оксиэтиламни (171), 612
0-Оксиэтилацетат (182), 627
Октагидроксаитеиы 205
н-Октан (125), 645
Октаиол-2 (179), 604
окисление Ce(lV) 171
Октантов правило 496
Октен-1 (121). 646
н-Октнламии (180), 612
н-Октилбромид (204), 637
н-Октилиодид (226), 638
н-Октилмеркаптан (199), 651
н-Октиловый спирт (192), 604
н-Октилхлорид (184), 637
Октии-1 (132), 646
Олеинамид [76], 609
Олеиианилид [41], 609
Олеиновая кислота [16], 599
Олефины см. Алкены
Олово как восстановитель 372
Омыления число 341, 537
Оптического вращения дисперсия 474
Оптическое
вращение 475
расщепление 474
Органические кислоты, общие структуры 130
Органические растворители 135, 137
D-Орнитии [разл.], 623
L-Оринтин [разл.], 623
d.Z-Орнитин [разл.], 623
Орсин [58, 107], 660
Осмометр парофазный 90
Осмометрия парофазиая 90—93
Основность
влияние структуры 127
пространственные эффекты 128
электронные эффекты 127
Осушители 583 —585
Охлаждающие бани, состав 580—581
Пальмитамид [106], 609
Пальмитиновая кислота [62], 599
Пальмитиновый альдегид [34], 608
Паральдегид (124), 607
Парафины (алканы), производные 215
таблица 644—645
Пеларгоновая кислота (253), 598
Пеларгоновый альдегид (185), 607
Пентадекан (271)
Пент адиен-1,2 (45), 645
Пентадиен-1,3 (42), 645
Пеитадиеи-1,4 (26), 645
Пеитаметилбензол [51], 644
Пеитаметиленбромид (221), 642
Пентаметилеигликоль (238), 605
Пентан (36), 644
Пентаиол-1 (138). 603
растворимость 137
Пентанол-2 (119), 603
окисление Ce(IV) 171
растворимость 137
Пентанол-3, растворимость в воде 137
Пента хлорбензол [86], 640
Пейта хлорфенол [190], 660
Пентахлорэтаи (161), 641
Пентаэритрит [253] 606
Пентен-1 (30). 645
Пентен-2 (36), 645
н-Пеитиламнн, растворимость 137
2-Пентил бром ид (118), 637
З-Пентилбромид (119), 637
трет-Пентиловый спирт (102). 603
окисление Ce(IV) 171
растворимость 137
Пентин-1 (40), 645
Пентин-2 (55), 646
Пептиды, анализ
Перекристаллизация 428
Перманганат калия, раствор 203
Пероксид водорода как окислитель 203
Пероксиды 358
ИК-спектры 364
в эфирах 27
Перфторалканы 358
Перфтор-н-гексан (57), 641
Пивалиновая кислота (164), 598
Пивалиновый альдегид (75), 606
Пикнометр 73
а-Пиколин (129), 619
(З-Пиколин (143), 619
V-Пиколии (143), 619
Пикрамид [190], 611
Пикрамнновая кислота [158], 601
Пикраты 271, 355
Пикрнлхлорид [83], 655
Пикриновая кислота [122], 600
Пимелиновая кислота [105], 600
ПинаколиН (106), 647
Пииакои (172), [35], 603, 605
окисление Ce(lV) 171
Пинаконгндрат [46], 605
d-Пннен (156), 646
Z-Пинен (156), 646
Пиперазин (140), [104], 618
692
Предметный указатель
Пиперидин (105), 617
растворимость 137
Пиперин [129], 610
Пиперовая кислота [216], 602
Пиперональ (263), [37], 608
Пиперониловая кислота [229], 602
Пиперониловый спирт [58], 605
Пиреи [148], 644
Пиридин (116), 619
бромирование 218
как растворитель 138
Пировиноградная кислота (165, разл.), 601
Пирогаллол [133], 660
триацетат [161], 631
триметиловый эфир [47], 635
триэтил овый эфир [39], 635
Пирокатехин [104], 660
диэтиловый эфир [43], 635
Пиром у коновая кислота [133], 600
Пирролидин (89), 617
Пируваиилид [104] 609
Пицеи [364], 644
Плавления температуры 30
Пламя как проба на элементы 49
Показатель преломления 78
Полигалогеиные производные небензоидных
углеводородов, производные, таблица
641 -642
Полин ит роаром этические соединения, изби-
рательное восстановление 372
Полиоксисоединения, характеристика 181
Поляриметр 478
Поп ул ин [180, разл.], 624
Преиитол (204), 643
Пробковая кислота [140], 601
Производные 36
методики получения, таблица 164
L-Пролии [222, разл.], 622
d.Z-Пролин [203, разл.], 622
Пропандитиол-1,3 (169), 651
спектр ЯМР 387
1-Пропансульфоиамид [52], 661
2-Пропаисульфонамид [60], 661
1-Пропансульфонилхлорид (67/9 мм), 663
2-Пропансульфонилхлорид (61/9 мм), 663
Пропеиилбеизол (177). 643
я-Пропиламии (49), 612
N-я-Пропилацетанилид [50], 609
я-П роли л ацетат (101), 625
Н-Пропилбензоат (230), 628
Н-Пропил бензол (158), 643
я-Пропилбромид (71), 637
я-Пропил-я-бутират (143), 625
я-Пропил-я-валерат (167), 626
Пропиленбромид (142), 641
Пропилен гл и коль (188), 604
Пропилен оксид (35), 632
Пропилен хлор ид (98). 541
я-Пропилиодид (102), 638
я-Пропил-я-капроат (186), 627
я-Пропил карбонат (168), 626
я-Пропиллевулинат (221), 628
я-Пропилмеркаптан (67), 651
я-Пропил-2 нафтиловый эфир [40], 630
я-Проп ил нитрат (ПО). 632
я-Пропилнитрит (44), 632
я-Пропиловый спирт (97), 603
окисление Ce(IV) 171
я-Пропилоксалат (213), 628
я-Пропнлпропионат (122), 625
я-Пропвлсалицилат (239), 628
я-Пропилсукцииат (246), 629
м-Пропил то луол (182), 643
я-Пропилтолуол (184), 643
я-Пропилфенол (232). [22], 658
я-Пропилформиат (81), 624
растворимость 137
я-Пропилфуроат (211). 627
я-Пропнл а-фурфуриловый »фир (168), 633
я-Пробилхлорид (46), 637
я-Пропилхлорформиат (113), 625
я-Пропилциклогексан (157), 645
Пропиоловая кислота (144, разл ), 598
Пропионамид [79], 609
растворимость 137
Пропионаиилйд [103], 609
Пропионилбромид (103). 595
Пропионилиоднд (127), 595
Пропионнлфторнд (45), 595
Пропионил хлорид (80), 595
Пропионитрил (97). 651
ИК-спектр 375
растворимость 137
спектр ЯМР 375
Пропионовая кислота (140), 598
Пропионовый
альдегид (50), 606
ангидрид (168), 596
Пропиофенон (218)
Протокатеховая кислота [194, разл.], 602
Протокатеховый альдегид [154], 608
Протонный магнитный резонанс 152
Псевдокуменол [71], 659
Псевдокумол (168) 643
Пулегон (224), 648
«Пурпурный бензол» 220
L-Рамноза [105, разл,], 623
Раста метод определения молекулярной мас-
сы 92
Растворимость 116—138
в воде 120
в концентрированной серной кислоте 132—
135
в разбавленном растворе бикарбоната
натрия 121
----едкого натра 121
в разбавленной соляной кислоте 120
диаграмма 119
классы 118
теория 121—135
Растворители для хроматографирования 50
Раффниоза [80, разл.; 119, разл.], 523
Реагенты для определения классов органи-
ческих соединений 34, 164
алюминия хлорид 283
Бенедикта раствор 196, 386
бензолсульфонилхлорнд 264
бром в четыреххлористом углероде 217
бромиая вода 404
гндроксиламина хлоргидрат 187
2,4-динитрофенилгидразин 186
железа(П) гидроксид 369
железа(П1) хлорид 403
иод — едкий натр 191
иодистоводородная кислота 350
йодоформная проба 191
кислот хлор ангидриды 317
натр едкий 332.
натрий 167
натрия бисульфит 189
— иодид в ацетоне 235
я-нитрофенилгидразнн 189
перманганата калия раствор 203. 219
серебра нитрат 233
серная кислота дымящая 281
соляная кислота — хлорид цинка 173
Толленса реактив 194
фенилгидразин 189
фуксии 195
Хинсберга проба 265
хрома триоксид 172
церий(1У) 169
цинк — хлорид аммония 370
(4-Резорцнловая кислота [213], 602
Предметный указатель
699
Резорцин [НО], 660
диацетат (278), 629
днбензоат [117], 631
диметиловый эфир (214), 634
диэтиловый эфир (235), 635
монометиловый эфир (243), 658
проба иа сахара 300
Ретен [98], 644
D-Рнбоза [95, разл.], 623
Родамин В — уранилацетат, проба на кис-
лоты 306
Роторный испаритель 422
Салициламид [139], 610
Салициловая кислота [157], 601
Салициловый альдегид (196), 607
Салицин [201, разл.], 624
Саркозин [210, разл.], 622
Саретта реагент, окисление 180
Сафрол (232), 635
Сахара (см также Углеводы) 299
анализ
ИК-спектр 300
производные 304
спектр ЯМР 300, 304
удельное вращение 305
Сахарин [220], 611
Сахароза [185, разл.], 624
Себациновая кислота [133], 600
Семикарбазид [96], 609
Сем и карбазоны 204
Сера, проба 103
Сераоргаиические соединения 401
окисление перманганатом 385
Серы соединения, температуры кипения 70
Серебро
нитрат, спиртовый раствор 233
оксид как окислитель 204
d.Z-Серин [228, разл.], 622
Серная кислота дымящая 281
Сероуглерод как растворитель 137
Сжигание как проба 108
Силикагель в ЖХ и ТСХ 50
Силилированне 184
Сильвестрен (176), 646
Ситостерин [137], 606
Смеси 39
нерастворимые в воде 413
перегонка с паром 425
предварительное изучение 412
растворимые в воде 413
синтетические 16
температуры плавления 47
Смешанного плавления проба 47
Соляная кислота — хлорид цинка, реактив
172
( + )-Сорбит [98], 606
Соседние группы, участие 240
Сочетание диазониевых иоиов 252
Спектроскопические методы 203
ИК-спектры 203
ПМР-спектры 221
УФ-спектры 237
Спин-спииовое взаимодействие 230
Спирты 166
ИК-спектры 175
масс-спектры 175, 179
окисление Ce(IV) 171
производные 164. 179
растворимость 137
спектры ЯМР 175
таблица 603—606
температуры кипения 72
удельная масса 76—77
Стационарные фазы (ГХ) 84
Стеарамид [109], 609
Стеаранилид [97], 609
Стеарилхлорид [22], 596
Стеариновая кислота [69], 599
Стеариновый
альдегид [38], 606
ангидрид [70], 597
Стнфниновая кислота [180], 660
Сублимация 422
Сукцинамид (моно) [157], 610
Сукцинимид [125], 610
Сукциноннтрнл (267, разл.), 652
растворимость 137
Сульфаты органические 401
ИК-спектры 385
Сульфенильные соединения 358
Сульфиновые кислоты 393
Сульфонамиды
гидролиз 399
ИК-спектры 385
получение 267. 298
расщепление 265
таблица 661—662
Сульфонаты 400
ИК-спектры 386
Сульфоксиды 391
ИК-спектры 386
масс-спектры 391
производные 391
спектры ЯМР 391
Сульфон илхлориды
ИК-спектры 386
получение 309, 354, 395
таблица 662—666
Сульфоновые кислоты 394
ИК-спектры 395
производные 386
эфиры 393
Сульфоны 298. 393
ИК-спектры 386
масс-спектры 386
производные 393
спектры ЯМР 393
Теобромин [337, возг.], 611
Теофиллин [264], 611
Терефталанилид [237], 611
Терефталевая кислота [300, возг.], 602
Терефталевый альдегид (245), 607
Терпин [102], 606
Терпингидрат [117], 506
d.Z-a-Терпинеол (221), 604
а-Терпииеол [35], 605
п-Терфеиил [213], 644
1,2,4,5-Тетрабромбензол [180], 640
Тетрабром-о-крезол [210], 660
Тетрабромфталевый ангидрид [275], 597
сплш-Тетрабромэтаи (200, разл.), 555
Тетра-н-бутиламмонийбромид, использова-
ние в «пурпурном бензоле» 220
1,2,3,6-Тетрагидробензальдегид (165), 607
Тетрагидроизохинолин (233), 617
аг-Тетрагидро-а-нафтиламин (275), 613
аг-Тетрагидро-р-нафтиламии (275), 613
Тетрагидропиран (88), 633
Тетрагидросильван (79), 632
Тетрагидрофуран (65), 632
как растворитель 137
Тетрагидрофурфурил ацетат (194), 627
Тетрагидрофурфуриловый спирт (178), 604
Тетрагидрохииолин (250), [20], 618
Тетрадекаи (254), 645
Тетраиодфталевы й ангидрид [318], 597
Тетралин (206), 643
1,2,3,4-Тетраметилбензол (205), 643
ситил-Тетраметилдибромэтаи [169], 642
сгмои-Тетраметилдихлорэтан [160], 642
Тетраметилэтилеи (72), 646
694
Предметный указатель
Тетранитрометан (126), [13], 653
проба иа алкены 222
с«л(ж-Тетрафеннлэ1ан [2111, 644
1,2,3,4-Тетрахлорбеизол [46], 640
1,2,3,5-Теграхлорбензол [51], 640
1,2,4,5-Тетр а хлорбензол [138], 640
2,3,5,6-Тетра хлоргидрохинон [237], 660
Тетрахлорфталевая кислота [250, разл.], 602
Тетрахлорфталевый ангидрид [256], 597
1,1,1,2-Тетрахлорэтаи (131), 641
сижж-Тетрахлорэтан (147), 641
Тетрахлорэтилен (121), 641
Тетраэтилеигликоль, диметиловый эфир
(266), 634
Тиглииовая кислота (198), [64], 599
Тиеиилмеркаптан (166), 651
Тимнлацетат (244), 628
Тимилбеизоат [33], 630
Тимол [50], 659
Тимохинон (232), [45], 649
Тиобензойиая кислота [24], 599
Тиокарбонильиые соединения 358
ИК-спектры 366
ж-Тиокрезол (195), 651
о-Тиокрезол (184), 651
л-Тиокрезол (195), 651
Тиомочевина [182], 611
₽-Тиоиафтол [81], 651
Тиосалициловая кислота [163], 599
Тиоуксусная кислота (93), 599
Тиофенол (169), 651
ИК-спектр 387
Тиоцианаты 358
L-Тирозин [344, разл.], 623
d.Z-Тирозин [318, разл.], 622
Токсичность, проба на 667 и сл.
Токсичные соединения 22—26, 667—674
о-Толидин [129], 616
л-Толилгидразин [65], 614
ж-Толилкарбинол (217), 604
о-Толилкарбииол [36], 605
л-Толилкарбинол [60], 605
л-Толилмеркаптан (195), [43], 651
ж-Толилмочевина [142], 610
о-Толилмочевина [192], 611
л-Толилмочевина [181], 611
N-л-Толил-а-нафтиламин [79], 618
N-л-Толил-р-иафтиламин [103], 618
Толленса реактив 194
о-Толуамид [140], 610
л-Толуамид [160], 610
ж-Толуаннлид [126], 610
о-Толуаиилид [125], 610
л-Толуанилид [145], 610
Толугидрохииои [124], 660
лг Толуидин (203), 612
ИК-спектр 263
О-Толуидин (199), 612
л-Толуидин (200), [45], 614
о-(л-Толуил)бензойная кислота [130], 600
гидрат [146]
ж-Толуиловая кислота [НО], 600
о-Толуиловая кислота [102], 600
л-Толуиловая кислота [177], 600
ж-Толуиловый
альдегид (199), 607
ангидрид [71], 597
о-Толуиловый
альдегид (200), 607
ангидрид [39], 596
л-Толу иловый
альдегид (204), 607
ангидрид [95], 597
ж-Толунитрил (212), 652
о-Толуиитрил (205). 652
л-Толунитрил (217), [38], 652
Толуол (111), 643
спектр ЯМР 289
ж-Толуол сульфон амид [108], 661
о-Толуолсульфонамид [153], 661
л-Толуолсульфоиамнд [137], 661
ж-Толуолсульфонилхлорид [12], 663
О-Толуолсульфонилхлорид [10], 663
л-Толуолсульфоиилхлорид [69], 664
л-Толухинон [68], 649
Тонкослойная хроматография 30
методика 50
определение 49
проявление 51
D-Треоиии [253. разл.], 622
L-Треонии [253, разл.], 622
d.Z-Треоиин [235, разл.], 622
Триаллиламин (155), 619
Три-н-амиламин (257), 620
Трибензил амии (380), [91], 621
2,4,6-Трибромаиизол [87], 636
2,4,6-Триброманилин (300), [119], 615
1,2,3-Трибром бензол [87], 640
1,2,4-Трибромбензол [44], 640
1,3,5-Трнбромбензол [120], 640
2,4.6-Трибромбензолсульфонамид [220, разл ]
662
2,4,6-Трибромбензолсульфонилхлорид [60],
664
2,4,6-Трибромнитробеизол [125], 656
Трибромнитрометаи [10], 654
1,2,3-Трибромпропан (219), 642
2,4.5-Трибромфеиетол [73], 636
2,4,6-Трибромфенетол [72], 636
2,4,6-Трибромфенол [95], 660
1,1,2-Трибромэтан (189), 642
2,2,2-Трибромэтиловый спирт [80], 606
Три-н-бутиламии (211), 619
н-Тридекаи (236), 645
Тридекановая кислота [44], 599
Триизоамиламии (245), 620
2,4,6-Трииодфенол [159], 660*
Трнкарбаллиловая кислота [166], 601
Тримезииовая кислота [350], 602
Триметиламии (3), 619
2,4,6-Триметилбеизойная кислота [155], 601
2,4,5- Тримебилбензолсульфонамид [181], 661
2,4,6- Триметилбензолсульфонамид [142] 661
2,4,5- Триметнлбензолсульфонилхлорид [61],
664
2,4,6- Триметилбеизолсульфонилхлорид [56],
664
Триметиленбромгидрин (176, разл.), 604
Триметилен бром ид (165), 642
Трнметиленгликоль (216), 604
диацетат (210), 627
Триметиленхлорбромид (143), 641
Триметилеихлоргидрии (161, разл.), 603
Триметиленхлорид (125), 641
Триметиленцианид (глутаронитрил) (286),
652
2,2,4-Триметилпентан (99), 644
2,4,6-Триметилфенол [69], 659
1,1,3-Трнметилциклопентан (105), 645
Триметилэтилев (38), 645
2,4,6-Тринитроанизол [68], 655
2,4,6-Трииитроаиилии [190], 615
2,4,6-Трииитробензамид [264, разл.], 611
2,4.6-Тринитробензойиая кислота [220, разл.]»
602
1,3,5-Трииитробензол [122]. 656
2,4,5-Тринитро-ж-ксилол [90], 655
Трииитрометаи [15], 654
2,4,6-Тринитротолуол [82], 655
2,4,6-Тринитрофенетол [78], 655
1,2,4-Триоксибензол [140], 660
Три-н-пропиламии (153), 619
растворимость 137
L-Триптофан [289, разл.], 622
Трифениламин [127], 621
Трнфеиилгуаниднн [145], 610
П редметный указатель
695
Трифен ил карбинол [162], 606
Трифенилметан [92], 644
1,1,2-Трифеннлэ1Нлеи [73], 647
Трифторуксусиый ангидрид (39), 595
2,4,6-Трихлоранилии (262), [77], 614
2,4,6-Трихлоранизол [60], 636
а,а,а Трихлорацетамид [140], 610
Трихлорацетанилид [97], 609
Трнхлорацетнлхлорид (115), 595
1,2,3-Трихлорбензол [52], 640
1,2,4-Трихлорбензол (213), 639
1,3,5-Трихлорбензол [63], 640
2,3,4-Трихлорбеизолсульфоиамид [226, разл.],
662
2,4,5-Трихлорбеизолсульфонамид [>200], 662
2,4,6-Трихлорбеизолсульфоиамид [212, разл.],
662
2,3,4-Трихлорбензолсульфонилхлорид [65],
664
2,4.5-Трихлорбензол сульфонил хлорид [34],
663
2,4,6-Трихлорбеизолсульфоиилхлорид [40],
663
1,2,3-Трихлорбутаи (169), 642
Трихлормолочиая кислота [124], 600
1,2,3'Трихлорпропан (165), 641
Трихлоруксусиая кислота [57], 599
2,4,6-Трихлорфенетол [43], 635
1,1,1 -Трихлор-2,2-бис-(л-хлорфеиил) этан
(ДДТ) [108], 642
2,4,5-Трихлорфеиол [68], 659
2,4,6-Трихлорфенол [67], 659
1,1,1-Трихлорэтан (74), 641
1,1,2-Трихлорэтан (114), 641
Трихлорэтилен (90), 641
Трихлорэтиловый спирт [19]. 605
Триэтиламин (89), 619
1,3,5-Трнэтилбензол (218), 643
d.Z-Троповая кислота [117], 600
Р-Туйои (202), 648
Углеводы см. Сахара
оптическое вращение 623, 624
таблица 623—624
Углерод четырехбромистый (92)
Углерод четыреххлористый (78), 641
растворимость 137
Удельная масса 30
Удельное вращение 478
Удерживания
времена 86
объемы 86
Уксусная кислота (18), 597
Уксусиопропионовый ангидрид (154), 595
Уксусный ангидрид (138), 595
спектр ЯМР 315
Ультрафиолетовая спектроскопия 479
аддитивности прицип 486
амины 493
Вудворда правила 490
геометрия молекулы, определение 486
карбонильные соединения 493
молярное поглощение 481
монофункциональные соединения 481, 493
иесопряжеииые олефииы 493
растворители 482
сопряженные системы 493
структура по модельным соединениям 485
н-Ундекаи (194), 645
Ундекановая кислота (275, разл.), 598
Ундецен-1 (193), 646
Уидециленовая кислота (295), [24], 599
Ундециловый спирт (243), 605
Ундецилхлорид (241), 637
Уретаны 180
Фенантрахинои (360), [207], 650
фенантрен [100], 644
Фенацетин [135], 610
Феиацилбромид [50], 597
Фенациловые эфиры 311
Фенациловый спирт [86], 606
Феиацилхлорид (244), [59], 649
л/’Феиетидин (248), 613
о-Феиетидин (229), 612
л-Фенетидин (254), 613
и-Фенетилмочевина [174], 610
Фенетол (172), 634
Феииладипат [106], 631
D-Фенилалаиин [320, разл.], 622
L-Фенилалании [320, разл.], 622
d,/-Фенилаланин [273, разл.], 622
Феиилацетальдегид (194), [34], 608
а-Феиилацетамид [154], 610
а-Фенилацетаиилид [117], 610
Феиилацетат (196), 627
Фенилацетилен (140), 646
ИК-спектр 230
спектр ЯМР 233
Фенилацетилхлорид (210), 596
Фенилацетоиитрил (234), 652
Фенилбеизоат (299), 629
Фенил-н-бутират (227), 628
Фенилгидразиды 311
Фенилгидразнн (243), [19], 613
как реагент для классификации 188
Фенил гидразон иевые соли 312
Фенил глиоксаль [91], 608
N-Феиилглицин [126, разл.], 621
л/-Фенилендиамин (283), [63], 614
о-Фенилендиамии (256) [102], 615
л-Фенилеидиамин (267), [140], 616
Фенилкарбамат [53], 609
Феиилкарбонат (306), [78], 631
2-Феиилмасляная кислота [42], 599
у-Феннлмасляиая кислота [52], 599
Фенилмеркаптан (169), 651
Фенилмочевииа [147], 610
N-Фенил-а-иафтиамин (335), [62], 618
Фенилиитрометан (226, разл.), 653
2-Фенилпиридин (269), 620
N-Фенилпиррол (234), 620
3-Фен ил пропан о л (237), 605
Фенил-н-пропилкетон (230), 648
Фенил-н-пропиловый эфир (188), 634
Феиилпропиоловая кислота [136], 600
а-Фенилпропионамид [92], 609
р-Фенилпропионамид [105], 609
а-Фенилпропиоиовая кислота (265), 598
Р-Фенилпропионовая кислота (280), 598
Фенилсалицилат [42], 630
Фенилстеарат [52], 630
d.Z-Фенилсукцииамид (ди) [211], 611
^./-Фенилсукцинанилид (ди) (222), 611
Феиилсукцииат [121], 631
N-Фенилсукциннмид [156], 610
Фенилтиомочевииы 261, 268
Фенил-л-толнлкетон (326), [54], 649
Фенил уксусная кислота [76], 599
Фенилуксусный ангидрид [72], 597
Феиилуретаиы 410
л-Фенилфенацилбромид [125], 650
о-Феи ил фенол [58]. 659
л-Фенилфенол [165], 660
Феиилфталат [70], 631
N-Фенилфталимид [205], 611
Фенилциннамат [72], 631
2-Фенил-1-этансульфоиамид [122], 661
а-Фенилэтиламин (185), 612
Р-Фенилэтиламин (198), 612
а-Фениэтилбромид (205), 637
р-Феиилэтнлбромид (218), 637
а-Феиилэтилмеркаптан (199), 651
а-Фенилэтиловый спирт (203), 604
Р-Феиилэтиловый спирт (219), 604
а-Фенилэтнлхлорид (195), 637
696
Предметный указатель
[J-Феиилэтилхлорид (190), 637
d.Z-Фенилянтарная кислота (167], 601
Фенокси ацетальдегид (215, разл.), 607
Феиоксин (59], 636
Феноксиуксусная кислота [96], 599
Фенол (180), (42], 658
кислотность 128
растворимость 122. 137
Фенолфталеин (261]. 660
Феиолы 402
ИК-спектры 406
масс-спектры 408
окисление перманганатом 222
спектры ЯМР 406
таблица 657—660
УФ-спектры 405
Феиолят-анион 127
^./-Фенхиловый спирт [39], 605
Флороацетофенон [219], 650
Флороглюцин (218], 660
растворимость 137
триацетат (106], 631
Флуореи [115]. 644
Флуоренон (341), [83], 649
Формальдегид (221), 606
Формамид (195, разл.), 609
растворимость 137
Формаиилид [46], 609
растворимость 137
Формилпиперидин (222), 609
Форон (198), [28], 649
D-Фруктоза [104, разл.], 623
окисление Ce(IV) 171
Фтал альдегид [56] 608
Фталальдегидокислота [97], 600
Фталамид [219, разл.], 611
Фталевая кислота (206, разл.], 602
Фталевый ангидрид [131], 597
Фталид (290), [73], 631
Фталимид [233], 611
Фталоилхлорид (276), 596
Фталоиовая кислота [146], 601
безводная [146]
Фторацетамид [108], 609
«-Фторбензамид [154], 610
n-Фторбензойная кислота [182], 601
Фторбеизол (85), 638
ИК-спектр 297
п-ФторбеизолсульФоиамид [125], 661
«-Фторбензолсульфонилхлорид [36], 663
Фториды, ИК-спектры 358
р-Фторнафталии [60], 639
л^-Фтортолуол (115) 638
о-Фтортолуол (114), 638
п-Фтортолуол (117), 638
Фторуксусная кислота [32], 599
Фуксин, реактив на альдегиды 195
Фумарамид (ди) [266, разл.], 611
Фумарамид (моно) [142], 610
Фумаранилид [314], 611
Ф ум арил хлорид (160), 596
Фумаровая кислота [200, возг., 302 в
паянной трубке], 602
Функциональные группы
пробы иа 35
указатель 163
Фурамид [1421, 610
Фуран (32), 632
Фуровая кислота [133], 600
Фуроин [135]. 650
Фуроиитрил (146), 652
Фурфурилацетат (176), 626
фурфуриловый спирт (170), 603
Фурфурол (161). 607
Хаульм уграм ид [106]. 609
d-Хаульмуграннлид [89], 609
Хаульмугревая кислота [68], 599
за-
Хелидоновая кислота [262], 602
Химикалии для качественного анализа 589
и сл.
Химические классы токсических соединений
668 и сл.
Хинальдин (247), 620
Хингидрон [171], 650
Хинолин (239), 619
Хинои 358
ИК-спектры 363
Хинсберга проба 265
Хлораль (98), 607
растворимость 137
Хлоральгидрат [53], 608
растворимость 137
5-Хлор-4-амнно-1,2-диметилбензол [40], 613
5-Хлор-2-аминотолуол (241), [29], 613
лг-Хлор а низ о Л (194), 634
о-Хлораиизол (195), 634
«-Хлоранизол (200), 634
спектр ЯМР-,3С 512
Хлоранил [290], 650
льХлораиилнн (230). 613
о-Хлоранилин (207), 612
ц-Хлоранилнн (232), [70], 613
спектр ЯМР 264
2-Хлор-6-антрахиноисульфонилхлорид [202], 666
2-Хлор-7-антрахинонсульфонилхлорид [1761 665
Хлораренсульфонамнды 397
Хлораренсульфонанилиды 397
а-Хлорацетамид [119], 610
а-Хлорацетанилид [134], 610
о-Хлорацетанилид [88], 609
«-Хлорацетанилид [179], 610
Хлорацетилбромнд (127), 595
Хлорацетилхлорид (105), 595
Хлорацетон (119), 647
Хлорацетонитрил (127), 652
м -Хлорацетофенон (228), 648
п-Хлорацетофенои (232), 648
(0-Хлорацетофенон (244), [59], 649
м-Хлор бенз альдегид (208), 607
о-Хлорбеизальдегид (208), 607
n-Хлорбенз альдегид (214), [47], 608
м-Хлор бенз а мид [134], 610
о-Хлорбензанилид [1141, 609
п-Хлорбензанилид [194], 637
л-Хлорбензилтиуронневые соли 329
л^-Хлорбеизилхлорид (216), 637
о-Хлорбензнлхлорид (214), 637
«-Хлорбензидхлорид (214), [29], 637
.м-Хлорбензоилхлорид (225), 596
о-Хлорбензоилхлорид (238), 596
n-Хлорбеизоил хлорид (222), [16], 596
лг-Хлорбеизойная кислота [158], 601
о-Хлорбензойная кислота [140], 601
«-Хлорбензойная кислота [242], 602
льХлорбензойный ангидрид [95), 597
о-Хлорбензойный ангидрид [79], 597
п-Хлорбеизойный ангидрид [194], 597
Хлорбензол (132), 638
З-Хлорбеизол-1,5-дисульфоиамид [224], 662
3-Хлорбензол-1,5-дисульфонил хлорид [106],
665
л«-Хлорбензолсульфонамид [148], 661
о-Хлорбензолсульфонамид [188], 662
п-Хлорбензолсульфонамид [144], 661
о-Хлорбензолсульфонилхлорнд [28], 663
ц-Хлорбензолсульфоннлхлорид [53], 664
лс-Хлорбеизонитрил [41], 652
о-Хлорбеизонитрнл [43], 652
п-Хлорбеизонитрил [92], 653
ц-Хлорбеизофенон [78], 649
2-Хлорбнфенил [34], 639
4-Хлорбнфенил [77], 640
V-Хлорбутнронитрил (197), 652
Хлоргндрохинон [106], 660
растворимость 137
Предметный указатель
697
2-Хлор-3,5-днаминотолуол [73], 614
6-Хлор-2,4-дибром анилин [95], 615
4-Хлор-3,5-дибромфенол [121], 660
о-Хлор-диметиланилин (207), 619
л-Хлор-М,1Ч-днметнланилин (230), 619
4-Хлор-2,5-диметилбеизолсульфонамид [185],
661
4-Хлор-2,5-дим етил бензолсульфонилхлорид
[50], 663
2-Хлор-3,5-динитробензойная кислота [199],
602
1-Хлор-2-изопропилбензол (191), 638
4-Хлор-2-иодфенол [78], 659
n-Хлоркоричиая кислота, ИК-сйектр 307
5-Хлор-о-крезол [48], 659
З-Хлор-л-крезол (197), 657
п-Хлоркумол (198), 639
2-Хлор-4-метилбензолсульфонамид [186], 662
4-Хлор-2-метилбензолсульфонамид [186], 662
2-Хлор-2-метилбеизолсульфонилхлорид [46],
663
4-Хл о р-2- м етил беизол сул ьф он и л хлорид [54],
664
2-Хлор-5-метил-3-ии1 робензолсульфоиам ид
[196], 662
2-Хлор-5-м етил-4-иит робензолсульфоиам ид
[188], 662
2-Хлор-5-метил-6-нитробеизолсульфоиамиД
[177], 661
З-Хл о р -2- м етил-5-н итробеиз олсул ьф ои ам ид
[161], 661
3-Хлор-2-метил-6-иит робеизолсульфон ам ид
[181], 661
5-Хлор-2-метил-3-ннт робензолсульфоиам ид
[167], 661
5-Хлор-2-метил-6-иитробеизолсульфонамид
[183], 661
5-Хлор-4-метил-2-нитробеизолсульфоиамид
[181], 661
5-Хлор-4-м етил -3-н итробенз олсул ьфои ам ид
[182], 661
2-Хлор-5-метил-3-иитробеизолсульфонил хло-
рид [63], 664
2-Хлоп-5-м е гил-4-ннтробензолсульфонилхло-
рид [92], 665
2-Хлор-5-метнл-6-нитробензолсульфоннлхло-
рнд [122], 665
З^Хлор-2метил-5-иитробензолсульфонилхло-
рид [64], 664
3-Хлор-2-метил-6-нитробеизолсульфоннл хло-
рид [60], 664
5тХл ор -2-м етил -3-н итробеизолсу л ьфон ил хло-
рид [60], 664
5Хлор-2-метил-6-нитробёнзолсульфонилхло-
рид [154], 665
5-Хлор-4-метил-2-нитробензолсульфоиилхло-
рид [99], 665
5-Хлор-4-метнл-3-нитробензолсульфонилхло-
рид [70], 665
2-Хлор-З-метилфенол [50], 659
6-Хлор-З-метилфенол [46], 659
Хлорметилэтиловый эфир (80), 632
З-Хлор-4-метоксибеизолсульфонамид [131],
661
З-Хлор-4-метоксибензолсульфонил хлорид [82],
664
а-Хлорнафталнн (263), 639
p-Хлорн афт алии [56], 640
1-Хлор-4-иафталинсульфоиамид [185], 661
4-Хлор-1-наф1алинсульфоиамид [186], 662
8-Хлор-1-нафталинсульфоиамид [199], 662
4-Хлор-1-иафталинсульфонилхлорид [95], 665
8-Хлор-1-нафталнпсульфонилхлорид [101], 665
4-Хлор-1-нафтиламин [981, 615
4-Хлор-2-иИ1роаннзол [98]. 655
4-Хлор-З-ишробензойная кислота [182]. 601
ж-Хлорнитробензол (235). [441, 654
о-Хлорнитробензол (246). [32]. 654
п-Хлориитробензол (242), [83], 655
4-Хлор-2-нит робензолсульфоиам ид [164], 66Г
4-Хлор-2-нитротолуол (240), [38], 654
6-Хлор-2-нитротолуол (238), [37], 664
2-Хлор-5-окситолуол [66], 659
Хлоропрен (59), 636
Хлороформ (61), 641
ИК-спектр 246
как растворитель 137
Хлорпикрин (ИЗ), 653
2-Хлорпиридин (166). 619
З-Хлорпиридии (149), 619
Хлорплатинаты из аминов 271
З-Хлорпропандиол-1,2 (215, разл.), 604*
1-Хлорпропаиол-2 (127), 603
2-Хлорпропаиол-1 (132), 603
1-Хлорпропеи (36), 636
2-Хлорпропен (30), 636
<2,/-а-Хлорпропионамид [80], 609
Р-Хлорпропионитрил (178), 652
а-Хлорпропионовая кислота (186), 598'
класс растворимости 137
[3-Хлорпропионовая кислота [42], 599
Хроматограмма сахаров, проявление 300,
449
Хроматография 431 и сл.
газовая 82. 457 и сл.
жидкостная 50, 432 и сл.
— высокого разрешения 446 и сл.
колонки 468
— сухие 438
----найлоновые 442
---- наполнение 442
---- проявление 443
----стеклянные 441
теория 458 и сл.
тонкослойная 60
Хромовый ангидрид
окисление 171
проба 171
Цейзеля метод анализа метоксильных rpynff
350
Целлобиоза [225, разл.], 624
Целлюлоза [разл.], 624
ацетат [разл.], 624
окисление Ce(IV) Г7Г
ЦернйЦУ) как окислитель 171
Цетил ацетат [22], 630
Цетилмеркаптан (170/3 мм), [50], 651
Цетиловый спирт [50], 605
Цетилхлорнд (289, разл.), 637
п-Цианбензойная кислота [214], 602
1-Циан-8-иафталиясульфонамид [336], 662
Циануксусная кислота [66], 652
Циклогексадиен-1,3 (80), 646
Циклогексан (80), 644
цис-Циклогексаидикарбоновая кислота, ан-
гидрид [32], 596
Циклогексаикарбоксамид [186], 611
Циклогексаикарбоксаиилид [146], 610
Циклогексанол (160), [16], 603, 605
окисление Се(IV) 171
Циклогексанон (155), 648
класс растворимости 137
Циклогексен (84), 646
ИК-спектр 224
спектр ЯМР 224
Циклогексил амии (134), 612
растворимость 137
Циклогексилацетат (175), 626
Циклогекс ил бензол (237), 643
Циклогекснлбромид (165), 637
Циклогексил карбинол (182), 604
Цнклогекснлмеркаптан (159), 65 Г
Цнклогекснлмет иловый эфир (134), 633
Циклогексилоксалат [42]. 630
698
Предметный, указатель
о-Цнклогекснлфенол [53], 659
л-Циклогексилфенол (132], 660
Циклогексилформиат (162), 626
Циклогексилхлорид (142), 637
Циклогексил этиловый эфир (149), 633
Циклогептанон (181), 648
Циклогептен (115), 646
Циклодекан (201), 645
Циклононан (172), 645
Циклопарафины, таблица 644—645
Циклопентаднен (42), 645
Циклопеитан (50), 644
Циклопентаиол (140), 603
растворимость 137
Циклопеитанон (131), 647
растворимость 137
спектр ЯМР 203
Циклопентен (46), 646
Циклопентил бромид (137), 637
Циклопентилметиловый эфир (105), 633
Циклопентиэтиловый эфир (122), 633
м-Цимол (175), 643
о-Цимол (178), 643
п-Цимол (175), 643
растворимость 137
Цинк — хлорид аммония 370
Цинка соли, окисление перманганатом 221
Циннамамид [142], 610
Циннамаиилнд [153], 610
Циннамилциннамат [441, 630
Циннамоил хлорид [36], 596
Циннамонитрил (254), 652
Цистеин, проба на 272
L-Цистии [260, разл.], 622
Цитраконовая кислота [91], 599
Цитраконовый ангидрид (213), 596
Цитраль (228, разл.), 607
Цнтроиеллаль (206), 607
Цитронеллол (222), 604
Четвертичные аммониевые соли из третич-
ных аминов 269
Шиффа реагент 195
Шиффовы основания 190
Шоттена — Баумана реакция 251
Щавелевая кислота [101], 600
растворимость 137
Эвгенол (250), 658
метиловый эфир (244), 636
Экстракция, растворители 137
Элаидиновая кислота [51]. 599
Элементы, пробы на 31
Энаитоннтрил (183), 652
Эпихлоргидрин (117), 633
1,2-Эпоксибутаи (62), 632
Эпоксиды 355
ИК-спектры 356
масс-спектры 356
производные 356
спектры ЯМР 356
Эргостерин [165], 606
Эритрит [72], 605
1,2-Этандисульфонил хлорид [91], 665
1,2-Этанднтиол (147), 651
Этансульфонамнд [58], 661
Этансульфоннлхлорид (70/20 мм), 663
Этиладипат (245), 628
Этилазелат (291), 629
Этилакрилат (101) 625
Этнлаль (89), 594
Этиламин (19), 611
Этил-лг-аминобензоат (294), 613
Этил-n амииобензоат [89], 615
4-(1Ч-Этиламино)-3-нитротолуол [59], 618
Этиланнзат (269), [7], 629
льЭтиланилин (215), 612
N-Этнланилин (205). 617
о-Этилаинлин (216). 612
л-Этиланилин (216), 612
Этилаитранилат (265, разл.), [13], 613
N-Этилацетаиилид [54], 609
Этилацетат (77), 624
ИК-спектр 337
растворимость 137
Этилацетоацетат (181), 626
бромирование 217
Этил ацетондикарбоксилат (250), 629
N-Этилбеизиламин (199), 617
N.'N-Этилбеизиланилнн [34], 620
Этилбеизилмалонат (300), 629
Этнлбензиловый эфир (189), 633
Этил бензоат (213) 628
растворимость 137
Этилбензоилацетат (270, разл.), 629
Этилбензоилформиат (257), 629
Этилбензол (135), 643
спектр ЯМР 157
4-Этил бензол 1,3-дисульфон амид [190], 662
2-Этилбензолсульфонамид [100], 661
З-Этилбеизолсульфоиамнд [86], 661
4-Этилбензолсульфонамид [109], 661
Этил бром ацет ат (159), 626
Этил бром ид (38), 637
Этилброммалонат (236), 628
Этил-а-бромпропионат (162), 626
Этил-^-бромпропионат (179), 626
2-Этилбутанол-1 (149), 603
Этил-н-бутилмалонат (240). 628
Этил-н-бутиловый эфир (92), 633
Этил-я-бутират (120), 625
Этил-н-валерат (145), 626
Этилвиниловый эфир (36), 632
З-Этнлгексан (119), 645
2-Этил гекса иол-1 (184), 604
2-Этил-1-гексил ацетат (199), 627
Этил-н-гексилбарбитуровая кислота [126], 610
Этил-н-гептоат (189), 627
Этилгинпурат [60], 609
Этилгликолят (160) 626
Этилглутарат (234), 628
Ы-Этил-2,4-динитроанилин [113], 618
Этил-3,5-дииитробеизоат [94], 631
Этилдихлорацетат (158), 626
Этилена оксид (11), 632
Этилен бром гидрин (150), 603
Этиленбромид (130), 641
Этиленгликоль (197), 604
днацетат (190), 627
дибеизоат [73], 631
диметиловый эфир (85), 632
днпропионат (212), 627
диэтиловый эфир (121), 633
моно-н-бутиловый эфир (171), 603, 633
монометиловый эфир (125), 603
монометиловый эфир ацетат (144), 626
монофениловый эфир (245), 605, 634
моноэтиловый эфир (135), 603, 633
моноэтиловый эфир ацетат (156), 626
Этилендиамии (116) 612
Этилеииодид [82]
Этиленхлоргидрин (129), 603
Эгилеихлорид (83) 641
Этилнденбромид (112), 641
Этилиденхлорнд (60). 641
Этилизоамилбарбитуровая кислота [154],
610
Этилизобутира! (ПО). 625
Этилизовалерат (134), 625
Этилизопропилбарбитуровая кислота ]201],
611
Предметный указатель
699
Этилизопропиловый эфир, растворимость 137
Этилиодид (7), 638
Этилкаприлат (206), 627
Этилкапрат (245), 629
Этил-н-капроат (166), 626
а-Этилкапроновая кислота (244), 598
[З-Этилкапроновый альдегид (163), 607
Этилкарбамат [49], 609
Этилкарбоиат (126), 625
Этилкротонат (138), 625
Этиллактат (152), 626
Этиллаурат (269), 629
Этиллевулинат (205), 627
Этилмалеат (225), 625
Этилмалоиат (198), 627
бромирование 217
Этилмалоиовая кислота [111], 600
Этилмаиделат (254), [37], 630
а-Этил-н-масляный альдегид (116), 607
Этилмеркашан (36), 651
N-Этил-Ы-метилаиилии (210), 619
Этилметилкетон (80). 647
растворимость 137
Этнлметилмалоиат (196), 627
Этилметилуксусиая кислота (176), 598
З-Этил-4-метиопиридин (196), 619
Этилмиристат (306), [11], 629, 630
1-Этилнафталин (258), 643
N-Этил-а-иафтиламин (325), 618
N-Этил-р-иафтиламии (315), 618
Этил-а-иафтиловый эфир (278), 635
Этил-З-иафтиловый эфир (282), [37], 635
Этилнитрат (87), 632
Этилнитрит (17), 632
N-Этил-п-иитроаиилин [96], 618
N-Этил-л-иитроацетаиилид [118], 610
Этил-л-нитробензоат (206), [47], 630
Этил-л-иитробеизоат [56], 630
Этил-З-нитросалицилат [118], 631
Этил-л-нитросалицилат [102], 631
Этил-З-нитрофталат [46], 630
Этил-Л1-интроциннамат [79], 631
Этил-л-интроциииамат [137], 631
Этиловый спирт (78), 603
ИК-спектр 179
окисление Ce(IV) 171
растворитель 137
спектр ЯМР 179
Этилоксалат (186), 627
растворимость 137
Этилоксамат [114], 609
Этилоксанилат [66], 609
Этил-л-оксибензоат (282), [72], 631
Этил-л-оксибензоат [116], 631
Этилортоформиат (145), 626
Этилпальмитат [24], 630
Этилпеларгоиат (227), 628
З-Этилпентан (94), 644
2-Этилпеитановая кислота (209), 598
Этилпимелат (255), 629
Этилпируват (155), 626
Этилпропиоиат (98), 625
Этил салицил ат (234), 628
Этилсебацат (307). 629
Этилстеарат [33]. 630
Этилсуберат (282), 629
Этилсукцииат (216); 628
растворимость 137
Этилсульф ат (206). 632
D-Этилтартрат (280), [17], 629
ЬГ-Этил-лг-толуидии (221), 617
N-Этил о-толуидии (218), 617
N-Этил-л-толуидии (217), 617
о-Эт илтолуол (165) 643
л-Этилтолуол (162), 643
Этилтрихлорацетат (167), 626
Этил трихлор лактат (162), [66], 631
Этилфеиилацетат (229) 626
Этилфеиилбарбитуровая кислота [172], 610
Этилфенилкарбинол (219), 604
л-Эт ил фенол (217) 658
о-Этнлфеиол (207) 658
л-Этилфенол [47], 659
Этилформиат (54), 624
растворимость 137
Этилфталат (298), 629
растворимость 137
Этил фумарат (215), 628
Этилфуроат [33], 630
Этилхлорацетат (145), 626
Этил хлор ид (12), 637
Этил-а-хлорпропионат (146), 626
Этилхлорформиат (93), 625
Этилцианоацетат (207), 652
Этилциклогексаи (132), 645
2-Этилциклогексилбром ид (212), 637
Этилциклопеитан (104), 645
Этил циннамат (271), 629
Этилцитрат (294), 629
Этилэтилацетоацетат (198), 627
Этилэтоксиацетат (152), 626
Этокси ацетамид [82], 609
м-Этоксибензамид [139], 610
л-Этоксибеизамид [202], 611
л-Этоксибензаиилид [170], 610
Этоксиуксусная кислота (206), 598
о-Этоксифенол (217), [28], 658
Эфиры простые 76, 346
алифатические, производные 348, 352
— ИК-спектры 348
— масс-спектры 348
— спектры ЯМР 348
— таблица 632—634
ароматические, бромирование 354
— таблица 634—636
Эфиры сложные 77 333
ИК-спектры 339
масс-спектры 338
неорганические, таблица 632
производные 324
спектры ЯМР 339
таблица 624—631
Z-Яблочная кислота [100], 600
d,Z-Яблочная кислота [131], 600
окисление Ce(lV) 171
Ядериый магнитный резонанс 32, 152—160
ампулы 157
аппаратура 153
области поглощения 157
стандарты 157
,3С 500
— алифатические соединения 509
— интенсивность сигнала 515
— мультиплетиость сигнала 513
— полная развязка от протонов 514
— пространственные эффекты 512
— стандарты внутренние 501
— химический сдвиг 502
— аош-эффект 512
— шумовая развязка 514
химические сдвиги 156
Яды 22—25
Янтарная кислота [188], 602
Янтарный ангидрид [120], 597
Содержание
Предисловие редактора перевода ........................................ 5
Предисловие авторов.................. .... .8
Предисловие авторов к первому изданию................................ 13
Глава 1. Введение .....................................................15
1.1. Идентификация органических соединений, ранее описанных в хи-
мической литературе.............................................15
1.2. Взаимосвязь изучения методов идентификации органических со-
единений с научными исследованиями в области органической
химии...........................................................16
1.3. Некоторые указания для студентов и преподавателей .... 17
1.4. Техника безопасности в химической лаборатории и меры оказания
первой помощи...................................................21
1.5. Взрывоопасность простых эфиров..............................27
Глава 2. Идентификация неизвестных веществ............................29
2.1. Введение....................................................29
2.2. Предварительные исследования................................29
2.3. Физические константы........................................30
2.4. Определение молекулярной массы..............................31
2.5. Определение молекулярной формулы веществ....................32
2.6. Оценка растворимости........................................32
2.7. Анализ методом ИК- и ЯМР-спектроскопии......................33
2.8. Классификационные реакции...................................34
2.9. Получение производных.......................................36
2.10. Смеси......................................................39
2.11. Представление результатов исследования неизвестного вещества 40
Отчет по форме № 1........................................ .40
Отчет по форме № 2......................................... 42
Отчет по форме № 3......................................... 44
Глава 3. Предварительное исследование, оценка чистоты и определение фи-
зических свойств органических соединений 47
3.1. Предварительное исследование ...... 47
3.1.1. Физическое состояние .... 47
3.1.2. Цвет 47
3.1.3. Запах............................................... 48
3.1.4. Проба на горючесть...................................48
3.1.5. Выводы и их применение......................... .... 49
3.2. Тонкослойная хроматография..................................50
3.3. Определение физических свойств..............................54
3.3.1. Температура плавления................................54
3.3.2. Температура кипения................................ .64
3.3.3. Плотность............................................73
3.3.4. Показатель преломления жидкостей.....................78
3.4. Газовая хроматография.......................................82
3.5. Определение молекулярной массы..............................89
3.5.1. Осмометрия в паровой фазе............................90
3.5.2. Масс-спектрометрия...................................93
Глава 4. Определение молекулярной формулы ....... . 100
4.1. Качественный элементный анализ.............................100
4.1.1. Сплавление органических соединений ... .101
4.1.2. Определение металлов и других неорганических элементов 107
4,2, Количественный элементный анализ ..........................108
Содержание
701
4.2.1. Метод сожжения и другие сходные методы анализа . . 1С8
4.2.2. Определение молекулярной формулы с помощью метода
масс-спектрометрии......................................... 111
Глава 5. Классификация органических соединений по растворимости, спект-
рам ядерного магнитного резонанса и инфракрасным спектрам 116
5.1. Растворимость...............................................116
5.1.1. Растворимость в воде, водных кислотах, водных основа-
ниях и в эфире............................................. 116
5.1.2. Растворимость в органических растворителях............136
5.2. Анализ органических соединений с помощью инфракрасных
спектров........................... ........................138
5.3. Анализ органических соединений методом ядерного магнитного
резонанса’...............................................152
5.4. Выводы...................................................159
Глава 6. Определение функциональных групп и установление структуры
изучаемых соединений . ........................................161
6.1. Спирты...................................................166
6.1.1. Многоатомные спирты...............................182
6.2. Альдегиды (RCHO) и кетоны (RCOR)........................185
6.2.1. Полифункциональные карбонильные соединения .... 209
6.2.2. Ацетали и кетали............................. .... 209
6.3. Алканы (парафины).......................................215
6.4. Алкены (олефины)........................................217
•6.5. Алкины (ацетилены)......................................230
6.6. Алифатические галогениды................................233
6.7. Амины...................................................251
-6.8. Аминокислоты............................................273
6.9. Ароматические соединения
6.10. Арилгалогениды.............
6.11. Углеводы и сахара (сахариды)
6.12. Карбоновые кислоты ....
6.12.1. Ангидриды кислот . .
6.12.2. Галогена нгидриды кислот
6.12.3. Амиды кислот ....
6.12.4. Соли карбоновых кислот
6.13. Сложные эфиры..............
6.14. Простые эфиры..............
6.15. Эпоксиды...................
Прочие соединения
6.16.
6.17. Соединения,-содержащие азот (нитросоединения, нитрилы)
6.17.1. Нитросоединения............................
6.17.2. Нитрилы....................................
6.18. Органические соединения серы......................
Меркаптаны...................................
Сульфиды......................................
Дисульфиды.....................................
Сульфоксиды...................................
Сульфоны ....................................
Сульфиновые кислоты...........................
Сульфоновые кислоты (сульфокислоты) ....
6.18.8. Сульфонилхлориды (сульфохлориды)...........
6.18.9. Сульфонамиды (сульфамиды).................
6.18.10. Эфиры сульфоновых кислот (сульфонаты) . .
6.18.11. Сульфаты.............................. . .
6.19. Фенолы ...........................................
6.18.1.
6.18.2.
6.18.3.
6.18.4.
' 6.18.5.
’ 6.18.6.
6.18.7.
281
294
299
305
313
317
320
328
333
346
355
366
366
366
375
383
385
390
391
392
393
393
395
398
399
401
402
402
702
Содержание
Главв 7. Разделение органических веществ ............................411
7.1. Введение..................................................411
7.1.1. Предварительное исследование смеси ..................412
Форма отчета...................................................414
Предварительное исследование смеси ........................ 414
7.2. Перегонка и сублимация................................... 415
7.2.1. Перегонка............................................415
7.2.2. Сублимация......................................... 423
7.2.3. Перегонка с паром....................................423
7.3. Перекристаллизация........................................426
7.4. Хроматография.............................................431
7.4.1. Введение и общие замечания.........................431
7.4.2. Жидкостная хроматография...........................433
7.4.3. Хроматография в сухих колонках.................. . 438
7.4.4. Жидкостная хроматография высокого разрешения . . . 457
7.4.5. Газовая хроматография..............................457
7.5. Разделение оптических изомеров............................474
Глава 8. Специальные методы характеристики органических веществ . . . 476
8.1. Оптическое вращение.......................................476
8.1.1. Приготовление раствора...............................476
8.1.2. Заполнение поляриметрической трубки..................476
8.1.3. Работа с поляриметром................................477
8.1.4. Описание полученных результатов......................478
8.2. Ультрафиолетовая спектроскопия............................479
8.2.1. Идентификация функциональных групп по положению мак-
симумов поглощения............................ш,............483
8.2.2. Использование модельных соединений..................483
8.2.3. Принцип аддитивности................................486
8.2.4. Влияние геометрии молекулы на ультрафиолетовые спектры
поглощения.................................................487
8.2.5. Более тонкие корреляции структуры с ультрафиолетовыми
спектрами..................................................489
8.2.6. Поглощение монофункциональных соединений.............493
8.3. Дисперсия оптического вращения............................496
8.3.1. Правило октантов.....................................497
8.4. Ядерный магнитный резонанс углерода-13....................500
8.4.1. Химический сдвиг.....................................502
8.4.2. Мультиплетность сигналов.............................513
8.4.3. Интенсивность сигналов...............................516
8.5. Лантаноидные сдвигающие реагенты .........................517
Глава 9. Установление структуры: методы определения и упражнения 523
Примеры........................................................523
Часть 1. Установление структуры соединений, описанных в лите-
ратуре ......................................................523
Часть II. Определение структуры соединений, не описанных ранее
в химической литературе....................................539
Установление молекулярных формул...........................539
Примеры задач на определение молекулярных формул .... 540
Часть III. Задачи............................................543
Серия 1................................................. 543
Серия 2...................................................545
Серия 3..................................................546
Серия 4..................................................548
Серия 5..................................................549
Содержание 703
Серия 6.......................................................551
Серия 7.......................................................552
Серия 8.......................................................553
Серия 9.......................................................554
Серия 10......................................................555
Серия 11......................................................557
Серия 12......................................................558
Серия 13......................................................560
Серия 14......................................................562
Серия 15......................................................564
Глава 10. Литература по органической химии.............................567
10.1. Введение и построение главы................................567
10.2. Органическая химия ........................................568
10.2.1. Beilstein...........................................568
10.2.2. Руководства по технике лабораторных работ. Вводный
курс........................................................568
10.2.3. Руководства по технике лабораторных работ. Повышен-
ный курс...................................................568
10.2.4. Механизмы химических реакций........................568
10.2.5. Теория молекулярных орбиталей.......................569
10.2.6. Правила орбитальной симметрии.......................570
10.2.7. Физическая органическая химия.......................570
10.2.8. Качественный анализ органических соединений .... 570
10.2.9. Реакции и реактивы..................................571
10.2.10. Спектроскопия (спектрометрия)......................571
10.2.11. Стереохимия........................................572
10.2.12. Синтезы............................................573
10.3. Аналитическая химия........................................573
10.4. Биохимия...................................................574
10.5. Общая химия................................................575
10.6. Неорганическая химия.......................................575
10.7. Физическая химия...........................................576
10.8. Химическая литература......................................576
Приложение I. Таблицы, удобные для использования в органической лабо-
ратории ...........................................................577
Приложение II. Оборудование и реактивы для лаборатории.................587
Приборы............................................................588
Оборудование для индивидуального рабочего стола..................588
Общее лабораторное оборудование..................................588
Специальное лабораторное оборудование .......................... 588
Отдельные предметы оборудования и реактивы, получаемые во вре-
менное пользование от руководителя или из хранилища..............588
Реактивы в лабораторных шкафах.....................................589
Органические соединения..........................................589
Неорганические соединения........................................591
Другие соединения, реактивы и растворы...........................591
Реактивы, которые хранятся в вытяжном шкафу......................592
Особые реактивы................................................. 593
Неизвестные соединения ..........................................593
Приложение III. Таблицы производных . . 594
Приложение IV. Токсичность органических соединений.....................667
Предметный указатель...................................................675